CADTH Reimbursement Review
Mirikizumab (Omvoh)
Sponsor: Eli Lilly Canada
Therapeutic area: Ulcerative colitis
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
5-ASA
5-aminosalicylic acid
AE
adverse event
AESI
adverse event of special interest
anti-TNF
anti–tumour necrosis factor
CI
confidence interval
CrI
credible interval
EMA
European Medicines Agency
EQ VAS
EQ visual analogue scale
EQ-5D-5L
5-Level EQ-5D
ES
endoscopic Mayo subscore
GI Society
Gastrointestinal Society
HEMI
histologic endoscopic mucosal improvement
HEMR
histologic endoscopic mucosal remission
HRQoL
health-related quality of life
IBD
inflammatory bowel disease
IBDQ
Inflammatory Bowel Disease Questionnaire
IgG4
immunoglobulin G4
IL-23
interleukin-23
ITC
indirect treatment comparison
ITT
intention-to-treat
JAK
Janus kinase
LSM
least squares mean
MCS
mental component summary
MID
minimal important difference
mITT
modified intention-to-treat
MMS
Modified Mayo Score
mNRI
modified nonresponder imputation
NMA
network meta-analysis
PCS
physical component summary
PGA
physician’s global assessment
PICOS
Population, intervention, comparators, outcomes, and study design
PP
per-protocol
QoL
quality of life
RB
rectal bleeding
RCT
randomized controlled trial
SAE
serious adverse event
SC
subcutaneous
SD
standard deviation
SF
stool frequency
SF-36
Short Form (36) Health Survey
STRIDE-II
Selecting Therapeutic Targets in Inflammatory Bowel Disease–II
TNF
tumour necrosis factor
UC
ulcerative colitis
UCEIS
Ulcerative Colitis Endoscopic Index of Severity
UNRS
Urgency Numeric Rating Scale
WPAI:UC
Work Productivity and Activity Impairment Questionnaire: Ulcerative Colitis
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information on Application Submitted for Review
Item | Description |
|---|---|
Information on drug submitted for review | |
Drug product | Mirikizumab (Omvoh) is available in 3 different dosing forms:
|
Sponsor | Eli Lilly Canada |
Indication | For the treatment of adult patients with moderately to severely active UC who have had an inadequate response, had a loss of response, or were intolerant to conventional therapy, a biologic treatment, or a JAK inhibitor |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | July 20, 2023 |
Recommended dosage | Induction: 300 mg IV infusion for at least 30 minutes at week 0, week 4, and week 8 Maintenance: 200 mg (given as 2 consecutive SC injections of 100 mg each) every 4 weeks after completion of induction dosing |
JAK = Janus kinase; NOC = Notice of Compliance; SC = subcutaneous; UC = ulcerative colitis.
Introduction
Inflammatory bowel disease (IBD) is a term used to describe disorders that involve chronic inflammation of the digestive tract. Ulcerative colitis (UC) is 1 such disease. UC causes inflammation and ulcers in the digestive tract, affecting the innermost lining of the large intestine (colon) and rectum.1,2 UC is characterized by blood in the stool with mucus, frequent diarrhea, loss of appetite, and tenesmus (a strong urge to use the bathroom without necessarily having a bowel movement), in addition to abdominal pain, rectal bleeding (RB), and weight loss.3-5 The most common initial manifestation of UC is bloody diarrhea with or without mucus. While the etiology of UC is not completely understood, there is growing evidence to suggest genetic and environmental factors may contribute to the irregular immune response that aberrantly recruits activated immune cells to the colon,3 resulting in chronic inflammation that damages the colon and causes UC symptoms. UC generally develops in young adulthood6-8 and persists throughout life, marked by periods of spontaneous remission and relapse.9 Though most patients experience this relapsing-remitting disease course, up to 24% of patients report experiencing continuous UC symptoms.9 The majority of individuals living with UC have a mild to moderate disease course, generally with active disease at diagnosis followed by alternating exacerbations and longer periods of remission.10 However, aggressive disease course is experienced in 10% to 15% of patients, with a cumulative risk of relapse of between 70% to 80% at 10 years postdiagnosis.10 Regardless of severity, UC is associated with a substantial reduction in quality of life (QoL) for patients, with considerable impact on many aspects of their lives, including emotional and psychological functioning, social and physical functioning, and work and academic life.11,12 UC is diagnosed clinically, with endoscopy, biopsy, and stool sampling being common tests used in ruling out other causes of symptoms.13 Treatment strategies for UC are dependent on the presence of active disease, severity and extent of the UC, and patient preference with the goal of achieving complete remission. Conventional therapies for UC include 5-aminosalicylic acid (5-ASA) products, corticosteroids, and immunomodulators (such as azathioprine, 6-mercaptopurine, and methotrexate); advanced therapies consist of adalimumab, golimumab, infliximab, ustekinumab, tofacitinib, ozanimod, and vedolizumab. However, current treatments are unable to meet all current needs of patients in terms of short-term or long-term treatment. Remission with treatment is not universal and a patient’s UC can lose response to treatment after an initial period of improvement and relapse even after long periods of remission on an existing therapy. Accordingly, there is a need for novel therapies targeting alternative pathways. An estimated 322,600 patients in Canada are living with IBD.14 In 2030, the number of people living in Canada with IBD is anticipated to be 470,000, accounting for 1.1% of the population with a prevalence for UC specifically of 0.44%.14,15
Mirikizumab is a humanized immunoglobulin G4 (IgG4) monoclonal antibody that binds with high affinity and specificity to the p19 subunit of human interleukin-23 (IL-23) cytokine and inhibits its interactions with the IL-23 receptor.16 Mirikizumab is indicated for the treatment of adult patients with moderately to severely active UC who have had an inadequate response, had a loss of response, or were intolerant to conventional therapy, a biologic treatment, or a Janus kinase (JAK) inhibitor.
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of mirikizumab, 300 mg/15 mL IV (induction) and 100 mg/1 mL subcutaneous (SC) injection (maintenance) in the treatment of adult patients with moderately to severely active UC who have had an inadequate response, had a loss of response, or were intolerant to conventional therapy, a biologic treatment, or a JAK inhibitor.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient and clinician groups that responded to CADTH’s call for input and from a clinical expert consulted by CADTH for the purpose of this review.
Patient Input
Patient input was collected from the Gastrointestinal Society (GI Society) and Crohn’s and Colitis Canada. Patient input was collected through a variety of questionnaires (n = 4 to 432), focus groups, and individual interviews. In addition, 1-to-1 interviews were conducted with 4 patients with UC who received mirikizumab in a clinical trial. After collating responses, it was noted that UC has a profound effect on daily life — physically, emotionally, and socially — at home and at school or in the workplace. Many patients surveyed by Crohn’s and Colitis Canada revealed that they hid aspects of their diagnosis from their friends, coworkers, and classmates and almost two-thirds of patients (63%) agreed that their family and friends do not know or understand what they are going through. Patients noted that symptoms can be relentless, embarrassing, and scary. Based on the surveys conducted by Crohn’s and Colitis Canada, the most frequently reported UC-related complications reported were mental health and stress (65%), joint inflammation and arthritis (51%), anal fissures and hemorrhoids (40%), anemia (33%), skin conditions (30%), malnutrition (30%), and weight loss (30%). Patients stated that more than anything, sustained remission and/or treatment response was more important than relieving any 1 symptom of UC. The constant concern that there would be future flares, possibly worse than the last, at unpredictable times, was noted as being disastrously disruptive.
Regarding current treatments for UC, it was noted that although there are several available options, most patients have difficulty obtaining remission or adequate symptom relief. Based on survey data from the GI Society, only 24% of patients with IBD found available medications to be adequate, 56% of patients found them to be only somewhat adequate, and 20% of patients found them not at all adequate. More than half of patients (56%) surveyed by Crohn’s and Colitis Canada believed that different treatment options could make them feel better. While steroid use is an important part of symptom management for UC, patients surveyed by Crohn’s and Colitis Canada reported that they were not particularly supportive of the treatment option. Almost all patients (93%) surveyed by Crohn’s and Colitis Canada stated that they only take systemic steroids if absolutely necessary. Patient input from the GI Society stressed that treatment response varies across patients, and in some cases response to medication may stop after prolonged use. For these reasons, patients noted it is important to have a variety of treatment options for UC. Patients noted that there is a need for new and effective options to achieve mucosal healing and reduce the debilitating symptoms of UC, as well as provide good QoL. Patients interviewed by Crohn’s and Colitis Canada added that any new treatment must be able to protect a patient’s ability to work productively, attend school and social events, and conduct basic necessities such as leaving the home to run errands. However, patients interviewed by Crohn’s and Colitis Canada also added that potential risks and side effects, especially those related to heart and liver function, are a major source of concern when considering new treatment options. The 4 patients interviewed by the GI Society who received mirikizumab in clinical trials reported at the time they were interviewed that they continue to take the medication. All 4 patients experienced improved gut healing and expressed improvement in QoL following treatment with mirikizumab. Regarding the administration of mirikizumab, 2 patients reported that the initial induction of treatment by infusion was exhausting and time consuming, and although 2 patients did not particularly like the SC administration of mirikizumab, they were willing to tolerate it and described it as manageable.
Clinician Input
Input From Clinical Experts Consulted by CADTH
The clinical expert noted that there is an unmet need for treatments that are better tolerated, improve convenience and compliance, and take into consideration special populations such as those with previous or current malignancies. The clinical expert stated that having available treatments with different administration methods is also important to patients (e.g., IV, SC, oral) as well as having multiple treatment options, given that a patient’s UC will often lose response to treatment and require another therapy. The clinical expert noted that not all patients’ UC will respond to available treatments and patients’ disease often becomes refractory to current treatment options. According to the clinical expert, mirikizumab is an anti–IL-23 drug for UC and would offer a novel treatment mechanism for the disease that is more targeted compared to other therapies such as ustekinumab. The clinical expert anticipated that the place in therapy of mirikizumab would be similar to other biologics and would be recommended as a second-line therapy after 5-ASA, instead of immunomodulators. It was the opinion of the clinical expert that patients do not need to initiate other therapies and have their UC fail to respond to these other therapies before being prescribed mirikizumab, given the limitations and risks of other therapies. The clinical expert noted that patients whose UC is most likely to respond to treatment with mirikizumab would be those with moderate to severe UC (who are biologic-naive or biologic-experienced) that has not responded to conventional therapy. Patients described as biologic-experienced had tried at least 1 biologic therapy and/or tofacitinib. The clinical expert noted that patients least suited for treatment with mirikizumab are patients with active infections, malignancy, and/or severe hepatic impairment, and patients who are pregnant. The clinical expert felt that the patients in greatest need of mirikizumab would be those whose UC had failed to respond to first-line therapy with 5-ASA.
According to the clinical expert, the outcomes used in clinical practice align with those used in clinical trials, such as clinical remission and clinical response (measured by Partial Mayo Score), endoscopic remission and response, and biomarkers (e.g., fecal calprotectin). The clinical expert noted that clinicians routinely schedule a colonoscopy to check for endoscopic healing 6 months to 9 months after a patient has been started on a biologic therapy or a small molecule drug. If concerned about patient response, the expert indicated that some physicians may try to book a flexible sigmoidoscopy soon after the induction period has been completed.
According to the clinical expert, a clinically meaningful response to treatment would be no further RB, no rectal incontinence, rectal urgency that has been reduced or no longer exists, bowel movement frequency that has been reduced or is normal, stools becoming more solid, and abdominal pain that has been reduced or no longer exists. The clinical expert would expect clinical improvement within 4 weeks and clinical remission within 12 weeks; however, depending on the severity of disease and previous medication exposure, the clinical expert noted that patients may have a slower response or a delay to remission. In this case, the expert indicated that they would be comfortable with an extended induction period of 12 weeks for those with disease that does not respond to treatment, which is aligned with the product monograph. The clinical expert noted that most gastroenterologists use standard clinical scores (e.g., Partial Mayo Score, Modified Mayo Score [MMS]) for UC in clinics with an endoscopic component if performing colonoscopy.
Regarding the discontinuation of treatment, the clinical expert suggested that mirikizumab should be discontinued in the event of serious adverse events (SAEs), disease progression, or the inability to taper off steroids. The clinical expert would consider stopping treatment after 24 weeks of therapy if the patient’s UC does not respond. This would include an extended induction phase if the patient’s UC was not responding to initial induction (e.g., 12 weeks). According to the clinical expert, it would be expected that approximately 30% of patients who did not have an initial induction response might have a delayed response to induction treatment.
Clinician Group Input
Clinician group input was received by a group of gastroenterologists in Canada. Input from the clinician group was compiled by 9 gastroenterologists recognized as experts in the management of IBD. Based on input from the clinician group, the goals of UC therapy are multifaceted, ranging from controlling symptoms to preventing disease progress, surgery, and disability with early intervention and a treat-to-target approach. The clinician group identified the following unmet needs in a therapy that treats moderate to severe UC: a therapy that induces and maintains symptomatic remission, is safe with long-term use, and can rapidly improve endoscopic appearance of the bowel and maintain this in the long term. The clinician group emphasized that none of the currently available therapies for UC meet all of the current needs of patients in the short term or long term. Remission with treatment is not universal and a patient’s UC can lose response to therapy after an initial period of improvement and relapse even when in deep remission on an existing therapy. Accordingly, the clinician group advocated for the need for novel therapies targeting alternative pathways. Overall, the clinician group found that mirikizumab has the potential for a broad range of uses in clinical practice from first-line advanced therapy to the treatment of patients with inadequate response or intolerance to multiple advanced therapies.
With regard to treatment with mirikizumab, the clinician group suggested that the aim of treatment should be remission. The clinician group suggested that a meaningful improvement in symptoms as measured by the resolution of stool frequency (SF) and RB should be demonstrated in the first 3 months of therapy. The clinician group expected patients to be in symptomatic remission and off corticosteroids by 6 months after the initiation of mirikizumab. The clinician group added that symptomatic improvement should be accompanied by a decrease in biomarkers of inflammatory activity (C-reactive protein and fecal calprotectin) in the first 3 months after initiating mirikizumab. The clinician group suggested discontinuing treatment with mirikizumab in the event of worsening symptoms or inadequate response. In circumstances where there was an inadequate response to mirikizumab as a first-line biologic, the clinician group indicated that a switch to another class of drugs, such as an anti–tumour necrosis factor (anti-TNF) drug, is warranted. Based on clinical experience, the clinician group suggested that mirikizumab be administered in a clinic by a trained health care professional during the induction phase.
Drug Program Input
Input was obtained from the drug programs that participate in the CADTH reimbursement review process. The following were identified as key factors that could potentially impact the implementation of a CADTH recommendation for mirikizumab:
relevant comparators
consideration for the initiation of therapy
consideration for the continuation or renewal of therapy
consideration for the prescribing of therapy
care provision issues
system and economic issues.
The clinical expert consulted by CADTH provided advice on the potential implementation issues raised by the drug programs. Refer to Table 4.
Clinical Evidence
Pivotal Studies and Randomized Controlled Trial Evidence
Description of Studies
Two double-blind, multicentre, parallel-arm, randomized placebo-controlled trials, the LUCENT-1 and LUCENT-2 trials, were submitted by the sponsor.
The LUCENT-1 trial (N = 1,281) was a 12-week induction trial in which patients were randomized 3:1 to either mirikizumab 300 mg IV every 4 weeks or placebo. The aim of the study was to assess whether mirikizumab 300 mg IV would induce clinical remission at week 12 in adult patients with moderately to severely active UC. Major secondary objectives included alternate clinical remission, clinical response, clinical response in patients who are biologic-experienced, endoscopic remission, symptomatic remission, bowel urgency improvement, and histologic endoscopic mucosal improvement (HEMI), all at week 12. Health-related quality of life (HRQoL) was also evaluated at week 12 using the Inflammatory Bowel Disease Questionnaire (IBDQ), the 5-Level EQ-5D (EQ-5D-5L), and the Short Form (36) Health Survey (SF-36). The Work Productivity and Activity Impairment Questionnaire: Ulcerative Colitis (WPAI:UC), for patients with UC, was also evaluated at week 12 among patients employed at baseline. At baseline, patients had a mean age of 42.5 years (standard deviation [SD] = 13.92), with the majority being male (59.8%) and white (72.3%). There was an equal number of patients with moderate UC and severe UC based on MMSs. The proportion of patients reporting prior biologic or tofacitinib failure was also similar between treatment groups (41.6% and 40.1% of patients randomized to mirikizumab and placebo, respectively).
The LUCENT-2 trial (N = 544 in the primary analysis) was a 40-week maintenance trial in which patients were randomized 2:1 to either mirikizumab 200 mg SC every 4 weeks or placebo. The aim of the trial was to assess whether mirikizumab 200 mg SC would achieve clinical remission from baseline to week 40 in adult patients with moderately to severely active UC who had previously attained a clinical response at week 12 of the LUCENT-1 trial. Major secondary objectives included alternate clinical remission, corticosteroid-free remission, durable clinical remission, endoscopic remission, bowel urgency remission and improvement, and histologic endoscopic mucosal remission (HEMR), all at week 40 (i.e., 52 weeks of treatment in total). The WPAI:UC was also evaluated at week 40 among patients employed at baseline. The baseline characteristics in the LUCENT-2 trial were similar to that of the LUCENT-1 trial. The majority of the patients in the main cohort had a mean age of 42.3 (SD = 13.5) years, and were male (58.4%) and white (71.3%). Based on the MMS, approximately half of the patients in each treatment arm were categorized as moderate UC severity and 35.1% of patients in the mirikizumab group and 35.8% of patients in the placebo group had a history of biologic or tofacitinib failure. Overall, the baseline characteristics were well-balanced between treatment arms.
The LUCENT-2 study also enrolled 405 patients from the LUCENT-1 study whose UC had not responded to 12 weeks of induction dosing with either mirikizumab or placebo. These patients received open-label mirikizumab (300 mg administered intravenously) for 12 weeks. This was referred to as an extended induction period for patients who had previously received 12 weeks of induction dosing (i.e., 24 weeks of continuous therapy).
Efficacy Results
A summary of key efficacy results from the LUCENT-1 and LUCENT-2 trials are available in Table 2.
Induction Period: LUCENT-1 Trial
Clinical Response
In the LUCENT-1 trial, clinical response was evaluated using the MMS. After 12 weeks of treatment, a greater proportion of patients on mirikizumab 300 mg IV than placebo attained clinical response, with a common risk difference of 21.4% (99.875% confidence interval [CI], 10.8% to 32.0%; P < 0.00001). Results of the sensitivity analyses in the intention-to-treat (ITT) population were consistent with the modified intention-to-treat (mITT) population results. In both the biologic-naive and biologic-experienced subgroups, more patients attained clinical response on mirikizumab than on placebo, with a common risk difference of 19.8% (95% CI, 11.3% to 28.3%; P < 0.001) and 23.9% (95% CI, 14.3% to 33.5%; P < 0.001), respectively. The biologic-naive subgroup consisted of patients who did not have experience with biologic therapy and tofacitinib, while the biologic-experienced subgroup consisted of patients whose UC had failed to respond to at least 1 biologic therapy or tofacitinib. The magnitude of effect for both subgroups was consistent with the primary analysis. In the subgroups of patients using corticosteroids or immunomodulators at baseline, the observed effect sizes for clinical response in the mirikizumab group compared to the placebo group were numerically positive but small when compared to the overall population. No subgroup differences were observed.
Clinical Remission
Clinical remission was assessed using 2 different outcomes in the LUCENT-1 study: clinical remission and alternate clinical remission. Clinical remission was based on the MMS and defined as the following: SF subscore equals 0 or SF subscore equals 1 with at least a 1-point decrease from baseline; RB subscore equals 0; and endoscopic subscore (ES) equals 0 or 1 (excluding friability). Alternate clinical remission used the same definition except that it excluded the need for at least a 1-point decrease from baseline in the SF subscore. These were considered appropriate measures by the clinical expert.
Clinical Remission Rate
A greater proportion of patients on mirikizumab 300 mg IV (24.2%) versus placebo (13.3%) experienced clinical remission at week 12, with a common risk difference of 11.1% (99.875% CI, 3.2% to 19%; P = 0.00006). Results of the analyses in the per-protocol (PP) and ITT populations were consistent with the mITT population results. Results of the sensitivity analyses assessing the impact of attrition and missing data were consistent with the results from the primary analysis. In terms of the tipping point analysis, there was no significant difference between groups when imputing missing data as “responder” for the placebo group and as “nonresponder” for the mirikizumab group.
In both the biologic-naive and biologic-experienced subgroups, more patients experienced clinical remission on mirikizumab than on placebo with a common risk difference of 15.1% (95% CI, 8.3% to 21.9%; P < 0.001) and 6.8% (95% CI, 0.5% to 13.0%; P = 0.065), respectively. The magnitude of effect for the biologic-naive subgroup was consistent with the primary analysis. In the subgroups of patients using corticosteroids or immunomodulators at baseline, the observed effect sizes for clinical remission in the mirikizumab group compared to the placebo group were numerically positive but small when compared to the overall population.
Alternate Clinical Remission Rate
The results for alternate clinical remission, a slightly less stringent definition of remission, were aligned with the results for clinical remission.
Endoscopic Remission
At week 12 of the LUCENT-1 study, 36.3% of patients on mirikizumab experienced endoscopic remission versus 21.1% of patients on placebo, with a common risk difference of 15.4% (99.875% CI, 6.3% to 24.5%; P value < 0.00001). Results of the analyses in the ITT population were consistent with the mITT population results.
In both the biologic-naive and biologic-experienced subgroups, more patients experienced endoscopic remission at week 12 on mirikizumab than on placebo, with a common risk difference of 17.9% (95% CI, 9.8% to 25.9%; P < 0.001) and 12.3% (95% CI, 5.2% to 19.4%; P = 0.003), respectively.
In the subgroups of patients using corticosteroids or immunomodulators at baseline, the observed effect sizes for endoscopic remission in the mirikizumab group compared to the placebo group were numerically positive but small when compared to the overall population. No subgroup differences were observed.
Ulcerative Colitis Endoscopic Index of Severity Score of 1 or More
The Ulcerative Colitis Endoscopic Index of Severity (UCEIS) is a physician-reported measure of the endoscopic disease activity of UC on flexible sigmoidoscopy or colonoscopy, which calculates a score ranging from 0 to 8 based on vascular pattern (scored 0 to 2); bleeding (scored 0 to 3); and erosions and ulcers (scored 0 to 3), with higher scores indicating worse outcomes. || |||| || || ||||||||| ||||| || |||||||| || ||||||||||| ||| || ||||| ||||| ||| || |||| |||||| ||| || |||||||| || |||||||| |||||||||| |||| |||||||||| || ||||| |||||||| ||| ||| || ||||| |||||||||
Symptomatic Remission
At week 12 of the LUCENT-1 study, 45.5% of patients on mirikizumab and 27.9% of patients on placebo experienced symptomatic remission, with a common risk difference of 17.5% (99.875% CI, 7.5% to 27.6%; P < 0.00001). Results of the analyses in the ITT population were consistent with the mITT population results.
In both the biologic-naive and biologic-experienced subgroups, more patients experienced symptomatic remission at week 12 on mirikizumab than on placebo, with a common risk difference of 17.1% (95% CI, 8.7% to 25.4%; P < 0.001) and 18.8% (95% CI, 10.1% to 27.4%; P < 0.001), respectively.
In the subgroups of patients using corticosteroids or immunomodulators at baseline, the observed effect sizes for symptomatic remission in the mirikizumab group compared to the placebo group were numerically positive but small when compared to the overall population. No subgroup differences were observed.
Bowel Urgency Improvement: Urgency Numeric Rating Scale
The Urgency Numeric Rating Scale (UNRS) is an instrument used to assess patient-reported severity of bowel urgency in adults with UC with a 24-hour recall period.17 Using the UNRS, the least squares mean (LSM) change from baseline at week 12 in the mirikizumab group was –2.59 points and –1.63 points in the placebo group, a difference of –0.95 points (99.875% CI, –1.5 points to –0.4 points; P value < 0.00001). Results of the analyses in the ITT population were consistent with the mITT population results.
In the biologic-naive subgroup, greater improvement in UNRS was seen in the mirikizumab group (–2.7 points) versus the placebo group (–2.1 points), with an LSM mean difference of –0.6 points at week 12 (95% CI, –1.0 points to –0.2 points; P = 0.002). Similar results were seen in the biologic-experienced subgroup, but with a larger LSM mean difference of –1.5 points at week 12 (95% CI, –2.0 points to –1.0 points; P < 0.001).
In the subgroups of patients using corticosteroids or immunomodulators at baseline, the observed effect sizes for bowel urgency improvement in the mirikizumab group compared to the placebo group were numerically positive but small when compared to the overall population. No subgroup differences were observed.
Health-Related Quality of Life
HRQoL was assessed in the LUCENT-1 trial based on the IBDQ score, EQ-5D-5L score, and SF-36 score.
IBDQ Score
The IBDQ consists of a 32-item list subdivided into 4 dimensions: systemic symptoms, bowel symptoms, emotional function, and social function. Total scores range from 32 to 224, with a higher score indicating a better HRQoL. The IBDQ has been consistently shown to have good internal consistency and test-retest reliability, as well as showing responsiveness to change in IBD.18-20 Available studies have suggested that an improvement of 30 points from baseline or an improvement of at least 15 points above placebo may constitute a minimal important difference (MID).21-24 In the LUCENT-1 trial, the mean change from baseline to week 12 in the IBDQ score was 38.4 points for patients in the mirikizumab group and 25.2 points for those in the placebo group, representing a difference of 13.2 points (P < 0.001) in favour of mirikizumab during the induction phase. The MID for the IBDQ score was defined as a change greater than 30 points from baseline as well as an MID of greater than 15 points over placebo. While the improvement within the mirikizumab group demonstrated clinical benefit (exceeding the MID threshold of 30 points), it fell short of meeting the MID threshold of at least 15 points compared to the placebo group as defined by previous studies.21-24 Nevertheless, the clinical expert believed that the observed change was clinically meaningful in terms of improving QoL.
EQ-5D-5L Score
In the LUCENT-1 study, the mean change from baseline to week 12 in the EQ visual analogue scale (EQ VAS) score was 14.6 points in the mirikizumab group and 9.4 points in the placebo group, with an LSM difference of 5.2 points (95% CI, ||| || |||; P < 0.001). The change in the mirikizumab group was clinically important, but the clinical importance of the difference between groups was uncertain.25
SF-36 Score
In the LUCENT-1 study, the mean change from baseline to week 12 in the SF-36 physical component summary (PCS) score was 5.97 points in the mirikizumab group and 3.90 points in the placebo group, with an LSM difference of 2.07 points (95% CI, 1.21 points to 2.93 points; P < 0.001). The mean change from baseline to week 12 in the SF-36 mental component summary (MCS) score was 5.02 points in the mirikizumab group and 3.42 points in the placebo group, with an LSM mean difference of 1.60 points (95% CI, 0.56 points to 2.63 points; P = 0.002). The change in the mirikizumab group was clinically important, but the clinical importance of the difference between groups was uncertain (i.e., MID = 3 points to 5 points).
Mucosal Healing: HEMI
In the LUCENT-1 study, mucosal healing was assessed based on the HEMI outcome, which considers both histologic and endoscopic outcomes. At week 12, 27.1% of patients on mirikizumab attained HEMI versus 13.9% of patients on placebo, with a common risk difference of 13.4% (99.875% CI, 5.5% to 21.4%; P < 0.00001). The results of the analyses in the ITT population were consistent with the mITT population results.
In both the biologic-naive and biologic-experienced subgroups, more patients attained HEMI at week 12 on mirikizumab than on placebo, with a common risk difference of 17.1% (95% CI, 9.8% to 24.3%; P < 0.001) and 8.4% (95% CI, 2.5% to 14.3%; P = 0.022), respectively. The magnitude of the difference in the biologic-naive subgroup was consistent with the mITT population.
In the subgroups of patients using corticosteroids or immunomodulators at baseline, the observed effect sizes for HEMI in the mirikizumab group compared to the placebo group were numerically positive but small when compared to the overall population. No subgroup differences were observed.
Work Productivity
In the LUCENT-1 study, work productivity was assessed using the WPAI:UC score at week 12. The WPAI:UC is a self-administered, disease-specific scale aimed at measuring the level of work impairment due to UC.26 WPAI:UC considers the 4 domains of absenteeism, presenteeism, overall work performance, and nonwork activities.27 The 4 domains comprise a total of 6 items. The final scores for each domain are a percentage of total impairment, ranging from 0 to 100%, with a higher number indicating greater impairment in that domain. The WPAI:UC is a valid and responsive instrument for use in UC.27
Among those employed at baseline (n = 566), patients on mirikizumab experienced a mean change in WPAI:UC of –20.65, compared to –14.91 for patients in the placebo group (LSM difference = –5.74 points; 95% CI, –10.06 points to –1.42 points; P = 0.009).
Maintenance Period: LUCENT-2 Study
Clinical Remission
Clinical remission was assessed using 3 different outcomes in the LUCENT-2 trial: clinical remission, alternate clinical remission, and durable clinical remission. The same definitions of clinical remission and alternate clinical remission used in the LUCENT-1 trial were used in the LUCENT-2 study. Durable clinical remission was attained if patients who experienced clinical remission at week 12 in the LUCENT-1 trial had ongoing remission at week 40 in the LUCENT-2 trial (i.e., 52 weeks of continuous clinical remission). These were considered appropriate measures by the clinical expert.
Clinical Remission
A greater proportion of patients on mirikizumab 200 mg SC (49.9%) versus placebo (25.1%) experienced clinical remission after 40 weeks of maintenance therapy (common risk difference = 23.2%; 95% CI, 15.2% to 31.2%; P < 0.001). Analyses in the PP and ITT populations were consistent with the mITT population results. In addition, results of the sensitivity analyses were consistent with the results from the primary analysis. In terms of the tipping point analysis, the difference between groups did not reach statistical significance when imputing missing data as “responder” for the placebo group and “nonresponder” for the mirikizumab group.
In terms of subgroups, more patients on mirikizumab than on placebo were clinical remitters at the end of the LUCENT-2 study for both the biologic-naive subgroup (51.5% versus 30.7%) and the biologic-failed subgroup (46.1% versus 15.6%; P < 0.001) with a common risk difference of 20.8% (95% CI, 10.2% to 31.5%; P < 0.001) and 30.5% (95% CI,18.1% to 42.9%; P < 0.001), respectively. The magnitude of the effect in both subgroups was consistent with the primary analysis. Note that the biologic-failed subgroup consisted of patients whose disease had an inadequate response to or loss of response to biologic therapy for UC, or they were intolerant to biologic therapy for UC.
Subgroup results for baseline corticosteroid use (yes and no), immunomodulator use (no), and patients with severe UC at baseline were consistent with the results of the primary analysis. Subgroup results for immunomodulator use (yes) and patients with moderate UC at baseline were numerically positive but small when compared to the overall population. No subgroup differences were observed.
Alternate Clinical Remission
Results for alternate clinical remission, a slightly less stringent definition of remission, were very similar to those of clinical remission.
Durable Clinical Remission
Of patients who experienced clinical remission at week 12 of the LUCENT-1 study, 63.6% who were randomized to mirikizumab 200 mg SC were still in clinical remission at week 40 of the LUCENT-2 trial, compared to 36.9% of those randomized to placebo SC, with a common risk difference of 24.8% (95% CI, 10.4% to 39.2%; P < 0.001). Analyses in the ITT population were consistent with the mITT population results.
In the biologic-naive subgroup, a greater number of patients in the mirikizumab group (62.5%) versus the placebo group (46.8%) attained durable clinical remission at the end of the LUCENT-2 study, with a common risk difference of 15.7% (95% CI, –1.3% to 32.7%; P = 0.078), although the effect size was small compared to the overall population. In the biologic-failed subgroup, a greater proportion of patients in the mirikizumab group versus the placebo group (66.7% versus 11.1%) experienced clinical remission at week 40, with a common risk difference of 55.6% (95% CI, 34.4% to 76.7%; P < 0.001). However, sample sizes for this subgroup were quite small.
Subgroup results for baseline corticosteroid use (no), baseline immunomodulator use (no), and patients with moderate and severe UC at baseline were consistent with the results of the primary analysis. Subgroup results of patients with baseline corticosteroid use (yes) and immunomodulator use (yes) were numerically positive but small when compared to the overall population. No subgroup differences were observed.
Corticosteroid-Free Remission
Corticosteroid-free remission was defined as clinical remission at week 40, symptomatic remission at week 28, and no corticosteroid use for at least 12 weeks before week 40.
A greater number of patients randomized to mirikizumab experienced corticosteroid-free remission at week 40 (44.9%) than those randomized to placebo (21.8%) (common risk difference = 21.3%; 95% CI, 13.5% to 29.1%; P < 0.001).
In both the biologic-naive and biologic-failed subgroups, more patients experienced corticosteroid-free remission on mirikizumab than on placebo, with a common risk difference of 20.4% (95% CI, 10.1% to 30.8%; P < 0.001) and 26.6% (95% CI, 14.5% to 38.6%; P < 0.001), respectively. The magnitude of effect in both subgroups was consistent with the primary analysis.
Subgroup results for baseline corticosteroid use (yes and no), immunomodulator use (yes and no), and patients with moderate and severe UC at baseline were consistent with the results of the primary analysis.
Endoscopic Remission
At week 40 of the LUCENT-2 study, 58.6% of patents on mirikizumab experienced endoscopic remission versus 29.1% of patients on placebo, with a common risk difference of 28.5% in favour of mirikizumab (95% CI, 20.2% to 36.8%; P < 0.001). Analyses in the ITT population were consistent with the mITT population results.
In both the biologic-naive and biologic-failed subgroups, more patients experienced endoscopic remission at week 40 on mirikizumab than on placebo, with a common risk difference of 28.2% (95% CI, 17.5% to 39.0%, P < 0.001) and 30.5% (95% CI, 17.3% to 43.6%, P < 0.001), respectively.
Subgroup results for baseline corticosteroid use (yes and no), immunomodulator use (yes and no), and patients with moderate and severe UC at baseline were consistent with the results of the primary analysis.
Bowel Urgency
In the LUCENT-2 study, bowel urgency outcomes consisted of bowel urgency remission and bowel urgency improvement as measured by the UNRS. The UNRS is an instrument used to assess patient-reported severity of bowel urgency in adults with UC with a 24-hour recall period.17
Bowel Urgency Remission
Of the patients with a UNRS score of at least 3 at the LUCENT-1 study baseline, 42.9% of patients on mirikizumab and 25% of patients on placebo at week 40 experienced bowel urgency remission, with a common risk difference of 18.1% in favour of mirikizumab (95% CI, 9.8% to 26.4%; P < 0.001). Analyses in the ITT population were consistent with results in the mITT population.
In both the biologic-naive and biologic-failed subgroups, more patients experienced bowel urgency remission at week 40 on mirikizumab than on placebo, with a common risk difference of 17.9% (95% CI, 7.0% to 28.8%; P = 0.002) and 30.5% (95% CI, 17.3% to 43.6%; P < 0.001), respectively. The magnitude of the effect was similar with the primary analysis.
Subgroup results for baseline corticosteroid use (yes and no), immunomodulator use (yes and no), and patients with moderate and severe UC at baseline were consistent with the results of the primary analysis.
Bowel Urgency Improvement (Change in UNRS)
At week 40 of the LUCENT-2 trial, patients on mirikizumab experienced a –3.80-point change in UNRS versus the LUCENT-1 study baseline, while patients randomized to placebo had a –2.74-point change in score from the LUCENT-1 study baseline (LSM difference = –1.06; 95% CI, –1.51 to –0.61; P < 0.001). Patients on mirikizumab experienced a clinically significant improvement in bowel urgency from baseline while those on placebo did not meet the MID threshold (MID = 3 points from baseline)28 for clinically significant improvement.
Health-Related Quality of Life
HRQoL was assessed in the LUCENT-2 study based on the IBDQ score, EQ-5D-5L score, and SF-36 score.
IBDQ Score
The IBDQ consists of a 32-item list subdivided into 4 dimensions: systemic symptoms, bowel symptoms, emotional function, and social function. Total scores range from 32 to 224, with a higher score indicating a better HRQoL. The IBDQ has been consistently shown to have good internal consistency and test-retest reliability, as well as showing responsiveness to change in IBD.18-20 Available studies have suggested that an improvement of 30 points from baseline or an improvement of at least 15 points above placebo may constitute an MID.21-24 In the LUCENT-2 study, the LSM mean change from the LUCENT-1 study’s baseline to week 40 in the IBDQ score was 49.8 points and 25.4 points for those in the mirikizumab group and placebo group, respectively, representing a statistically significant difference of 25.2 points in favour of mirikizumab (95% CI, 19.2 points to 31.3 points; P < 0.001).The difference between groups was considered clinically meaningful as the difference was above the MID of at least 15 points above placebo.21-24
EQ-5D-5L Score
In the LUCENT-2 study, the LSM difference at week 40 between groups in the EQ VAS score was 20.1 points in the mirikizumab group and 8.8 points in the placebo group, representing a statistically significant difference of 11.3 points between groups (MID = 14.6 points) (95% CI, ||| || ||||; P < 0.001).The change from baseline in the mirikizumab group appeared clinically important (with an MID of 14.6 points on the EQ VAS),25 but the clinical importance of the difference between groups was uncertain.
SF-36 Score
Only the LUCENT-2 study evaluated change in the health outcome SF-36. At week 40, patients randomized to mirikizumab experienced an LSM change in SF-36 PCS of 9.0 points, compared to 6.7 points in patients randomized to placebo, a 2.3-point difference between the groups (P < 0.001). In the MCS of SF-36, mirikizumab patients had an LSM change of 7.0 points, compared to 5.5 points in the placebo group (LSM change difference between groups = 1.5; P = 0.031). The change from baseline in the mirikizumab group appeared clinically important (MID threshold of at least 3 points),29 but it was unclear whether the difference between groups was clinically important.
Mucosal Healing: HEMR
A greater proportion of patients randomized to mirikizumab attained HEMR (a stricter outcome than HEMI) more often than those randomized to placebo at week 40: 43.3% versus 21.8%, respectively (common risk difference = 19.9%; 95% CI, 12.1% to 27.6%; P < 0.001). Analyses in the ITT population were consistent with the mITT population results.
In both the biologic-naive and biologic-failed subgroups, HEMR occurred more often in patients on mirikizumab than in patients on placebo, with a common risk difference of 20.8% for patients who were biologic-naive (95% CI, 10.5% to 31.2%) and 21.2% for the biologic-failed population (95% CI, 10.9% to 31.4%). The magnitude of the effect was consistent with the primary analysis.
Subgroup results for baseline corticosteroid use (yes and no), immunomodulator use (yes and no), and patients with moderate and severe UC at baseline were consistent with the results of the primary analysis.
Work Productivity
The WPAI:UC is a self-administered, disease-specific scale aimed at measuring the level of work impairment due to UC.26 WPAI:UC considers the 4 domains of absenteeism, presenteeism, overall work performance, and nonwork activities.27 The 4 domains comprise a total of 6 items. The final scores for each domain are a percentage of total impairment, ranging from 0 to 100%, with a higher number indicating greater impairment in that domain. The WPAI:UC is a valid and responsive instrument for use in UC.27 At week 40 of the LUCENT-2 trial, patients randomized to the mirikizumab group had an LSM change of –31.72 points from the LUCENT-1 trial baseline, and placebo patients had an LSM change of –22.59 points from the LUCENT-1 trial baseline, equating to an LSM difference of –9.13 points between the groups (95% CI, –14.26 points to –4.01 points; P < 0.001). An MID was not identified for this outcome.
LUCENT-2 Study Extended Induction
Patients whose UC did not respond to mirikizumab or placebo during the 12-week induction period in the LUCENT-1 study went on to the LUCENT-2 study to receive extended induction (an additional 12 weeks) with open-label mirikizumab 300 mg IV for 3 doses. Of the mirikizumab induction nonresponders from the LUCENT-1 study, 272 patients entered the open-label extended induction arm of the LUCENT-2 study in the mITT population. Of these, 146 patients (53.7%) attained a delayed clinical response (95% CI, 47.8% to 59.6%) at week 12 of the LUCENT-2 trial (i.e., after 24 weeks of continuous mirikizumab 300 mg IV every 4 weeks, for a total of 6 doses). Additionally for this cohort of 272 patients, at week 12 of the LUCENT-2 trial, the rates of clinical remission, endoscopic remission, and symptomatic remission were 11.4% (95% CI, 7.6% to 15.2%), 16.5% (95% CI, 12.1% to 21.0%), and 37.1% (95% CI, 31.4% to 42.9%), respectively. When considering clinical response at the end of the initial 12-week induction period and the extended induction period, it can be noted that 80% of patients (697 of 868 patients) on mirikizumab 300 mg IV attained a clinical response by the end of the 24 weeks.
A total of 146 patients were considered delayed responders at week 12 in the LUCENT-2 study. Of the 146 patients, 144 (99%) entered the open-label maintenance period, and 104 (72.2%) maintained clinical response at week 40 versus || ||| || || ||||||| of patients from the placebo group who entered the maintenance period. Clinical response at week 40 was not evaluated in the LUCENT-2 study for induction responders from the LUCENT-1 study. Hence, no comment can be made on the difference in treatment effects between these 2 cohorts at week 40.
Harms Results
The key harms results from the pivotal trials are summarized in Table 2.
For both the LUCENT-1 and LUCENT-2 trials, the overall rate of adverse events (AEs) was similar between groups, though numerically slightly higher in the placebo groups compared to the respective mirikizumab treatment groups. In the LUCENT-1 trial, 44.5% and 46.1% of patients reported an AE in the mirikizumab and placebo groups, respectively. In the LUCENT-2 trial, 64.5% and 68.8% of patients reported an AE in the mirikizumab and placebo groups, respectively. In the LUCENT-1 study, the most common AEs for patients on mirikizumab 300 mg IV included nasopharyngitis (mirikizumab = 4.1%; placebo = 3.1%), anemia (mirikizumab = 3.3%; placebo = 5.9%), and headache (mirikizumab = 3.3%; placebo = 2.8%). In the LUCENT-2 study, the most common AEs for patients on mirikizumab 200 mg SC included nasopharyngitis (mirikizumab = 7.2%; placebo = 5.7%), arthralgia (mirikizumab = 6.7%; placebo = 4.2%), and UC (mirikizumab = 6.7%; placebo = 20.8%).
The rate of SAEs in the LUCENT-1 study was found to be lower in patients treated with mirikizumab than in those treated with placebo (2.8% versus 5.3%); however, this was due to UC being included as a harm. In the LUCENT-2 study, 3.3% and 7.8% of patients reported an SAE in the mirikizumab and placebo groups, respectively. In the LUCENT-1 trial, the most common SAEs in those on mirikizumab IV included UC (mirikizumab = 0.8%; placebo = 3.1%) and pneumonia (mirikizumab = 0.2%; placebo = 0%). In the LUCENT-2 trial, no SAE (at the “preferred term” level) occurred in more than 1 patient on mirikizumab SC.
Withdrawals due to AEs occurred at a lower rate in mirikizumab-treated patients compared to placebo-treated patients in the LUCENT-1 and LUCENT-2 trials. In the LUCENT-1 study, 1.6% and 7.2% of patients withdrew from the trial due to an AE in the mirikizumab and placebo groups, respectively. In the LUCENT-2 study, 1.5% and 8.3% of patients withdrew from the trial due to an AE in the mirikizumab and placebo groups, respectively. |||||||||| ||||||| ||||||||||||||||| ||||||||||||| |||||||||||||||| |||||||||||||||| ||||||||||||||||| ||||||||||| ||| ||||||||||| ||||||||||||||||| |||||||||| |||| ||| |||| |||||| || ||||| || ||||||||||| || || ||||||||| |||||||||| ||||||| ||| ||| |||| || ||||||||| |||| |||| ||||||| ||||||||||| |||||| |||||| ||||| || ||||||||| ||| |||| || ||||||||||| || |||||||| ||| |||| || ||||||| |||||||| ||||||||| |||| || || |||||| ||| |||||||| ||||||||||
In the LUCENT-1 study, no deaths were recorded. In the LUCENT-2 study, 1 (0.5%) death was recorded in the placebo group due to COVID-19.
Most adverse events of special interest (AESIs) occurred at a similar rate between mirikizumab and placebo patients in the LUCENT-1 and LUCENT-2 studies. One exception was the rate of injection site reactions in the LUCENT-2 trial, where 8.7% of patients on mirikizumab SC experienced this AESI compared to 4.2% of patients on placebo SC. The rates of opportunistic infection, cerebrocardiovascular events, malignancy, depression, suicide/self-injury, and hepatic-related AEs were low overall and similar between groups for both the LUCENT-1 study and the LUCENT-2 study.
Table 2: Summary of Key Results From Pivotal Studies and RCT Evidence (mITT Population)
Outcome | LUCENT-1 (induction trial, week 12) | LUCENT-2 (maintenance trial, week 40) | ||
|---|---|---|---|---|
Mirikizumab 300 mg IV q.4.w. N = 868 | Placebo IV q.4.w. N = 294 | Mirikizumab 200 mg SC q.4.w. N = 365 | Placebo SC q.4.w. N = 179 | |
Clinical response | ||||
Patients contributing to the analysis, n | 868 | 294 | NA | NA |
Patients with clinical response, n (%) | 551 (63.5) | 124 (42.2) | NA | NA |
Common risk difference, % (99.875% CI) for LUCENT-1 study and % (95% CI) for LUCENT-2 study | 21.4 (10.8 to 32.0) | NA | ||
P value | < 0.00001 | NA | ||
Clinical response, biologic-failed population | ||||
Patients contributing to the analysis, n | 361 | 118 | NA | NA |
Patients with clinical response, n (%) | 197 (54.6) | 35 (29.7) | NA | NA |
Risk difference, % (95% CI) | 24.9 (15.2 to 34.6) | NA | ||
P value | < 0.001 | NA | ||
Clinical remission | ||||
Patients contributing to the analysis, n | 868 | 294 | 365 | 179 |
Patients with clinical remission, n (%) | 210 (24.2) | 39 (13.3) | 182 (49.9) | 45 (25.1) |
Common risk difference, % (99.875% CI) for LUCENT-1 study and % (95% CI) for LUCENT-2 study | 11.1 (3.2 to 19.1) | 23.2 (15.2 to 31.2) | ||
P value | 0.00006 | < 0.001 | ||
Alternate clinical remission | ||||
Patients contributing to the analysis, n | 868 | 294 | 365 | 179 |
Patients with alternate clinical remission, n (%) | 222 (25.6) | 43 (14.6) | 189 (51.8) | 47 (26.3) |
Common risk difference, % (99.875% CI) for LUCENT-1 study and % (95% CI) for LUCENT-2 study | 11.1 (3.0 to 19.3) | 24.1 (16.0 to 32.2) | ||
P value | < 0.001 | < 0.001 | ||
Corticosteroid-free remission | ||||
Patients contributing to the analysis, n | NA | NA | 365 | 179 |
Patients with corticosteroid-free remission, n (%) | NA | NA | 164 (44.9) | 39 (21.8) |
Common risk difference, % (95% CI) | NA | 21.3 (13.5 to 29.1) | ||
P value | NA | < 0.001 | ||
Durable clinical remission | ||||
Patients contributing to the analysis, n | NA | NA | 143 | 65 |
Patients with durable clinical remission, n (%) | NA | NA | 91 (63.6) | 24 (36.9) |
Common risk difference, % (95% CI) | NA | 24.8 (10.4 to 39.2) | ||
P value | NA | < 0.001 | ||
Endoscopic remission | ||||
Patients contributing to the analysis, n | 868 | 294 | 365 | 179 |
Patients with endoscopic remission, n (%) | 315 (36.3) | 62 (21.1) | 214 (58.6) | 52 (29.1) |
Common risk difference, % (99.875% CI) for LUCENT-1 study and % (95% CI) for LUCENT-2 study | 15.4 (6.3 to 24.5) | 28.5 (20.2 to 36.8) | ||
P value | < 0.00001 | < 0.001 | ||
Bowel urgency improvement (change in UNRS score) | ||||
Patients contributing to the analysis, n | 868 | 294 | 316 | 104 |
LSM change from baseline (CI) | –2.59 (–2.9 to –2.3) | –1.63 (–2.1 to –1.2) | NA | NA |
LSM change from baseline (SE) | NA | NA | –3.80 (0.139) | –2.74 (0.202) |
LSM difference in change from baseline, (99.875% CI) for LUCENT-1 study and (95% CI) for LUCENT-2 study | –0.95 (–1.5 to –0.4) | –1.06 (–1.51 to –0.61) | ||
P value | < 0.00001 | < 0.001 | ||
Bowel urgency remission (among those with UNRS ≥ 3 at LUCENT-1 study baseline)a | ||||
Patients contributing to the analysis, n | NA | NA | 336 | 172 |
Patients with bowel urgency remission, n (%) | NA | NA | 144 (42.9) | 43 (25.0) |
Common risk difference, % (95% CI) | NA | 18.1 (9.8 to 26.4) | ||
P value | NA | < 0.001 | ||
|||| || ||||| || | ||||
|||||||| |||||||||||| || ||| ||||||||| | ||| | ||| | NA | NA |
|||||||| |||| |||| || ||||| |||||| | ||| |||||| | || |||||| | NA | NA |
|||||| |||| ||||||||||||||| ||| | |||| |||| || ||||| | NA | ||
||||| | |||||| | NA | ||
Symptomatic remission | ||||
Patients contributing to the analysis, n | 868 | 294 | NA | NA |
Patients in symptomatic remission, n (%) | 395 (45.5) | 82 (27.9) | NA | NA |
Common risk difference, % (99.875% CI) for LUCENT-1 study and % (95% CI) for LUCENT-2 study | 17.5 (7.5 to 27.6) | NA | ||
P value | < 0.00001 | NA | ||
IBDQ scorea | ||||
Patients contributing to the analysis, n | 868 | 294 | 365 | 179 |
LSM change from baseline (SE) | 38.4 (1.1) | 25.2 (1.8) | 49.8 (2.1) | 24.5 (2.8) |
LSM difference in change from baseline (95% CI) | 13.2 (9.3 to 17.2) | 25.2 (19.2 to 31.3) | ||
P value | < 0.001b | < 0.001b | ||
HEMI (LUCENT-1 study) or HEMR (LUCENT-2 study) | ||||
Patients contributing to the analysis, n | 868 | 294 | 365 | 179 |
Patients with HEMI or HEMR, n (%) | 235 (27.1) | 41 (13.9) | 158 (43.3) | 39 (21.8) |
Common risk difference, % (99.875% CI) for LUCENT-1 study and % (95% CI) for LUCENT-2 study | 13.4 (5.5 to 21.4) | 19.9 (12.1 to 27.6) | ||
P value | < 0.00001 | < 0.001 | ||
WPAI:UC (overall work impairment) scorec (among those who were employed at baseline) | ||||
Patients contributing to the analysis, n | 429 | 137 | 196 | 107 |
LSM change from baseline (SE) | –20.65 (1.163) | –14.91 (1.985) | –31.72 (1.726) | –22.59 (2.261) |
LSM difference in change from baseline, (99.875% CI) for LUCENT-1 study and (95% CI) for LUCENT-2 study | –5.74 (–10.06 to –1.42) | –9.13 (–14.26 to –4.01) | ||
P value | 0.009b | < 0.001b | ||
Harms, n (%) | ||||
Patients contributing to harms analysis, N | 958 | 321 | 389 | 192 |
Any TEAEs | 426 (44.5) | 148 (46.1) | 251 (64.5) | 132 (68.8) |
Serious TEAEs | 27 (2.8) | 17 (5.3) | 13 (3.3) | 15 (7.8) |
Withdrawal from treatment due to TEAEs | 15 (1.6) | 23 (7.2) | 6 (1.5) | 16 (8.3) |
Death | 0 | 0 | 0 | 1 (0.5) |
Notable harms | ||||
All infections | 145 (15.1) | 45 (14.0) | 93 (23.9) | 44 (22.9) |
Hepatic-related | 15 (1.6) | 5 (1.6) | 12 (3.1) | 4 (2.1) |
Immediate hypersensitivity reaction | 10 (1.0) | 1 (0.3) | 7 (1.8) | 2 (1.0) |
Infusion or injection site reaction | 4 (0.4) | 1 (0.3) | 34 (8.7) | 8 (4.2) |
Depression | 4 (0.4) | 2 (0.6) | 4 (1.0) | 0 |
Malignancies | 2 (0.2) | 0 | 1 (0.3) | 1 (0.5) |
Cerebrocardiovascular events | 1 (0.1) | 2 (0.6) | 0 | 1 (0.5) |
Suicide or self-injury | 0 | 0 | 1 (0.3) | 0 |
CI = confidence interval; EQ-5D-5L = 5-Level EQ-5D; HEMI = histologic endoscopic mucosal improvement; HEMR = histologic endoscopic mucosal remission; IBDQ = Inflammatory Bowel Disease Questionnaire; LSM = least squares mean; MCS = mental component summary; mITT = modified intention-to-treat; NA = not applicable; PCS = physical component summary; q.4.w. = every 4 weeks; RCT = randomized controlled trial; SC = subcutaneous; SE = standard error; SF-36 = Short Form (36) Health Survey; TEAE = treatment-emergent adverse event; UNRS = Urgency Numeric Rating Scale; WDAE = withdrawal due to adverse event; WPAI:UC = Work Productivity and Activity Impairment Questionnaire: Ulcerative Colitis.
Notes: Details in Table 2 have been taken from the sponsor’s Summary of Clinical Evidence.30
Results for the LUCENT-1 trial outcomes are at 12 weeks while the LUCENT-2 trial outcomes are at 40 weeks post–LUCENT-2 trial baseline, unless otherwise specified. Biologic-experienced was defined as patients whose UC had failed to respond to at least 1 or more biologic therapy or tofacitinib.
aIn the LUCENT-2 study, UNRS, IBDQ, EQ-5D-5L, and WPAI:UC scores were reported as change from the LUCENT-1 study baseline to week 40 (e.g., 52 continuous weeks).
bThe ANCOVA model included treatment, baseline value, prior biologic or tofacitinib failure (yes or no), baseline corticosteroid use (yes or no), baseline disease activity (modified Mayo score of 4 to 6 vs. 7 to 9), and region (North America, Europe, or other).
cThe outcome, WPAI:UC, was the overall work impairment score that combined absenteeism and presenteeism.
Sources: Clinical Study Report for the LUCENT-1 study and the LUCENT-2 study.31,32
Critical Appraisal
Internal Validity
Overall, the LUCENT-1 and LUCENT-2 trials were well conducted. They were adequately powered to detect a difference between mirikizumab and placebo in the primary end point and employed an appropriate prespecified graphical multiple testing approach to control key secondary outcomes for multiplicity. Many of the primary and secondary outcomes, including clinical remission, alternate clinical remission, clinical response, symptomatic remission, bowel urgency remission and improvement, HRQoL, and work productivity, may have been at risk of reporting bias and recall bias, due to the subjective nature of the patient electronic reporting diary. However, the direction and magnitude of the bias is unknown. As well, there was a risk of attrition bias against mirikizumab due to higher attrition in the placebo arm compared with the intervention; however, sensitivity analyses of the primary end point and the key secondary end points of clinical remission and clinical response assessed the impact of missing data and showed that the results were consistent with the primary analysis, increasing certainty of the findings. MIDs were provided by the sponsor for the IBDQ, EQ-5D-5L, and SF-36 (PCS and MCS), which were in line with thresholds reported in the literature. Numerically, statistically significant improvements were observed in the mirikizumab group compared to the placebo group |||||| ||| ||| |||||||||||||| ||||| |||||||||||. Notably, the IBDQ did reach the MID threshold for the change from baseline score (i.e., > 30 points) in the mirikizumab treatment groups in both the LUCENT-1 study and LUCENT-2 study, which the clinical expert acknowledged as a meaningful improvement. As for the between-group treatment difference, in the LUCENT-2 study, there was a greater change in the mirikizumab group versus the placebo group in the IBDQ score, exceeding the MID threshold mentioned in the literature of greater than 15 points over placebo. However, in the LUCENT-1 study, the IBDQ score fell short of reaching this 15-point MID threshold over the placebo group.
External Validity
In general, the clinical expert consulted by CADTH considered the baseline demographic and disease characteristics in the pivotal trials to be reflective of patients with moderate to severe UC seen in Canadian clinical practice. Concomitant medication use was also reflective of Canadian clinical practice except for prednisolone, which is not typically used in Canada. In the LUCENT-2 trial, a corticosteroid taper was trialled on all patients in the main cohort. Patients who did not taper their steroid use were allowed to continue their treatment; however, this is in contrast to the input received from the clinical expert whereby patients would be considered treatment failures and discontinue therapy if they could not taper or stop concomitant corticosteroid use by the time of the maintenance phase (i.e., after the induction or extended induction period).33 Therefore, the efficacy of mirikizumab in the trials may appear to be biased, given that patients who could not taper were included in the primary analysis, even though they would have been considered treatment failures in clinical practice. However, the direction and magnitude of this bias is unknown, given that both groups underwent the same tapering protocol. Furthermore, the generalizability of the results may be limited to Canadian clinical practice, given the discrepancy in tapering protocol. The number of screening failures was quite high in the LUCENT-1 study (35%); however, this is similar to other UC trials.34,35 According to the clinical expert, potential reasons for the higher screening failure rate could be due to how patients were referred to the trial.
To be eligible for enrolment in the primary cohort of the LUCENT-2 trial, patients were required to attain clinical response following 12 weeks of induction treatment in the LUCENT-1 trial. This requirement may have resulted in an enriched patient population that was included in the primary analysis of the maintenance trial as it does not take into consideration delayed responders. As per the product monograph, mirikizumab is indicated for patients who experience delayed response. Hence, by excluding these patients in the primary analysis, there is uncertainty about the efficacy of maintenance treatment in the broader population of patients with moderately to severely active UC. Other UC trials have similar concerns regarding enrichment, given that they have used a similar study design. Patients who entered the LUCENT-2 study as nonresponders received open-label mirikizumab, and therefore the results should be interpreted with caution, considering the potential risk of detection or performance bias due to the open-label nature. The clinical expert noted that the duration of follow-up in the LUCENT-1 trial (12 weeks) was not a sufficient amount of time to see a difference in endoscopic remission. However, the issue of insufficient duration is addressed by the LUCENT-2 trial, which measures end points to week 40 (i.e., 52 weeks of continuous therapy). Long-term data beyond 52 weeks is not available; hence, long-term outcomes (e.g., loss of response, harms) may not be sufficiently captured between the 2 trials.
Long-Term Extension Studies
There are currently no published or unpublished long-term extension phase III or phase IV randomized controlled trials (RCTs) or real-world evidence studies evaluating mirikizumab. The sponsor noted that there is an ongoing phase III, open-label, long-term extension trial enrolling patients from the LUCENT-2 study and the phase II study (NCT02589665) into the LUCENT-3 study (I6T-MC-AMAP), with an expected primary completion date of June 6, 2025.97
Indirect Comparisons
Description of Studies
One sponsor-conducted indirect treatment comparison (ITC) indirectly comparing the treatment effect of mirikizumab to other advanced therapies in adult patients with moderate to severe UC via a network meta-analysis (NMA) was included in the sponsor’s submission. In total, || studies evaluating || different treatment regimens were included in the NMA. Of note, among the comparators eligible for inclusion in the NMA, filgotinib and upadacitinib are currently not approved for use in Canada. Moreover, the ustekinumab maintenance regimen of 90 mg every 12 weeks is not used in Canada. Accordingly, these treatment regimens were not reported in the CADTH clinical review. The outcomes assessed in the NMA efficacy analysis included clinical response, clinical remission, and mucosal healing at induction and maintenance, as well as overall SAEs and all-cause discontinuation of treatment.
Efficacy Results
Efficacy results of the NMA are presented for biologic-naive, JAK inhibitor–naive, biologic-experienced, and JAK inhibitor–experienced populations by time points (i.e., induction and maintenance).
||||||||| |||||||| |||||||| ||| ||||||||| ||| ||| ||||||||||||| ||||| ||||||||||| ||| |||||||| ||| |||||||||||| || |||||||||||||| || ||||||||| |||||||| |||||||| |||| ||||| ||| |||| |||| || ||||| ||| ||||||||| |||||||| ||| |||| |||| || ||||| ||||||| |||||||||| ||| |||||||||||| ||| |||| ||| ||| ||||||||| ||||||||||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || ||||||||| |||||||| |||||||| ||| |||||||| |||||||| ||||||| ||||||||||| ||| ||| |||||||| |||||||| |||||| ||||||||||||||| ||| ||||||||||||| ||||||||||| ||||||||||| ||||||||||| || |||||||| |||||||| |||| ||||| ||| |||| |||| || ||||| ||| |||||||| ||||||||| |||| ||||| ||| |||| |||| || ||||| |||||||| ||||||||||| |||||||| || ||||||||||| ||| |||| ||| ||| ||||||||| ||||||||||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || ||||||||| |||||||| |||||||| ||| |||||||| ||||||||| ||||||| ||||||||||| ||| ||| ||||||||| |||||||| |||||| |||||||||||||||||||||||| |||||||| |||||||| ||| ||||||||| ||| ||| ||||||||||||| ||||| ||||||||||| ||||||||| |||| ||||||||||| ||| |||||||||| |||| |||||||||| |||||||||||||||| |||||||| || |||||||||| |||| ||||| ||| |||| |||| || |||||| |||||||||| |||| ||||| ||| |||| |||| || |||||| ||||||||| || || |||| ||||| ||| |||| |||| || ||||| ||||||||| ||| || |||| ||||| ||| |||| |||| || |||||| |||||||| |||| ||||| ||| |||| |||| || |||||| |||||||||||| |||| ||||| ||| |||| |||| || |||||| ||||||||||| || || |||| ||||| ||| ||| |||| || |||||| ||||||||||| ||| || ||| |||| ||||| ||| ||| |||| || |||||| ||||||||||| ||| || ||| |||| ||||| ||| |||| |||| || |||||| ||| ||||||||||| ||| || ||| |||| ||||| ||| |||| |||| || |||||| ||||||||| |||| ||||||||||| ||| |||||||||| |||| |||||||||| |||||||| ||||||||| |||||||| || |||||||||| |||| ||||| ||| |||| |||| || |||||| |||||||||| |||| ||||| ||| |||| |||| || |||||| ||||||||| || || |||| ||||| ||| |||| |||| || ||||| ||||||||| ||| || |||| ||||| ||| |||| |||| || |||||| |||||||| |||| ||||| ||| |||| || |||||| |||||||||||| |||| ||||| ||| ||| |||| || |||||| ||||||||||| || || |||| ||||| ||| |||| |||| || |||||| ||||||||||| ||| || ||| |||| ||||| ||| |||| |||| || |||||| ||||||||||| ||| || ||| |||| ||||| ||| |||| |||| || ||||||||||||||||||| ||| || ||| |||| ||||| ||| |||| |||| || |||||| ||| |||| ||| ||| ||||||||| ||||||||||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || ||||||||||| |||||||| |||||||| ||| ||||||||| ||||||| ||||||||||| ||| ||||||||||| |||||||| |||||| ||||||||||||||| ||| ||||||||||||| ||||||||||| |||||||||| ||| |||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || ||||||||||| |||||||| |||||||| ||| |||||||| ||||||||| |||||||||||||||||| ||| |||||||| |||||| ||||||||||||
||||||||| ||||||| ||||||||||| ||||||||| ||||||| ||||||| || ||| ||||||||||||| ||||| ||||||||||| ||| |||||||| ||| |||||||||||| || |||||||||||| ||||||| |||||||||| ||| ||||||||||||||| ||||| ||| |||| |||| || |||||| ||| |||| ||| ||| ||||||||| ||||||||||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || ||||||||| ||||||| ||||||| ||||||| ||||||||||| ||| ||| ||||||||| |||||||| |||||| |||||||||||| ||| ||| ||||||||||||| ||||||||||| ||||||||||| ||| |||| ||| ||| |||||||| ||||||||||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || ||||||||| ||||||| ||||||| ||||||| ||||||||||| ||| ||||| |||||||| |||||| |||||||||||||||||||||||| ||||||| |||||||||| ||| ||||||||||||| ||||| ||||||||||| ||| |||| ||| ||||||||||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || ||||||||||| ||||||| ||||||| ||||||| ||||||||||| ||| ||| ||||| |||||| ||||||||||||||| ||| ||||||||||||| ||||||||||| ||||||||||| ||| |||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || ||||||||||| ||||||| ||||||| ||||||| ||||||||||| ||| ||| ||||| |||||||| |||||| ||||||||||||
Harms Results
Harms outcomes were presented for the overall mixed population regardless of prior exposure to biologic and/or JAK inhibitor therapy.
||| ||||| |||||||||||||||||||||||||||| ||||||||| ||| ||||||||||| |||||||||||| ||||| |||| || ||||||||| ||||||||||||||| |||||||| || |||||||| ||||| |||| || ||||||||| ||||||||||||||| ||| |||||||| ||||||||| ||||||||| |||| ||||||||||| |||||| ||||||||||| |||| ||||| ||| |||| |||| || ||||| ||| |||||||||| |||| ||||| ||| |||| |||| || |||||| ||| |||| ||| ||| ||||||||| ||||||||||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || ||| ||||| ||||||||||||||| ||||||| ||||||||||| ||| ||| ||||| |||||||| |||||| |||||||||||| |||||||| ||||||| |||||||||| |||| ||| ||| |||||||| ||||||||||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || |||||||| |||| ||||||| ||||||||||| ||| ||| ||||| |||||||| |||||||||||| ||||||||| ||||||||
Critical Appraisal
The NMA was based on studies identified from a sponsor-conducted systematic literature review of relevant randomized evidence of European Medicines Agency (EMA)-approved and FDA-approved treatments for adult patients with moderately to severely active UC.36 The systematic literature review was based on a PICOS defined a priori and the literature search involved multiple electronic databases, clinical registries, and supplementary manual searches, thereby minimizing error and bias in the study selection and data extraction process. The sponsor identified 3 other sources of heterogeneity across the included UC studies: race, trial design, and prior exposure to biologics and/or JAK inhibitors. To account for racial disparity in UC, the NMA evaluated an “only Asian” subgroup, regardless of prior experience with biologics or JAK inhibitors. The network for this subgroup, however, was small, consisting of 9 and 8 studies at the induction and maintenance periods, respectively, evaluating 6 interventions. To mitigate heterogeneity due to trial design (treat-through design versus rerandomized design), statistical adjustments were employed to make treat-through trials comparable to efficacy data from rerandomized trials, and sensitivity analyses were conducted excluding treat-through study design. However, the CADTH review team was unable to confirm whether the method employed adequately adjusted for differences in trial design without introducing bias. Moreover, follow-up sensitivity analyses excluding studies with treat-through study design were unlikely to account for the potential issues since the network was different and associated with validity issues of its own. To account for the potential for heterogeneity due to treatment history (biologic-naive versus biologic-experienced), the sponsor conducted separate analyses for biologic-naive and biologic-experienced subgroups. However, the CADTH review team determined that the definitions of biologic-naive and biologic-experienced varied across studies (definitions included tumour necrosis factor [TNF]-naive versus TNF-experienced, no biologic or JAK inhibitor failure versus biologic or JAK inhibitor failure, biologic-naive versus biologic-experienced, and no biologic failure versus biologic failure), creating heterogeneity within each classification group. The use of separate analyses and reporting for efficacy results by prior exposure to biologics would not account for these differences. The CADTH review team identified several other sources of heterogeneity that could not be adjusted for in the NMA, including differences in definitions of clinical response and remission, prior biologics exposure (due to time periods in which the studies occurred), permitted concomitant medications, outcome assessment methods and definitions, and the duration of the maintenance period. The inclusion of comparator treatments not relevant to the Canadian setting (i.e., filgotinib, upadacitinib, and maintenance ustekinumab 90 mg every 12 weeks) provided information to the network and was not expected to significantly impact the heterogeneity of the NMA above the other sources of heterogeneity mentioned previously. The violation of the exchangeability assumption for efficacy outcomes is likely due to heterogeneity, and several estimates were affected by wide credible intervals (CrIs) that increased uncertainty. Moreover, network consistency or coherence could not be assessed due to the lack of relevant closed loops when comparing mirikizumab to other active treatments. As a result, the NMA evidence was considered to be indirect, thus reducing certainty in the study findings.
Studies Addressing Gaps in the Pivotal and RCT Evidence
No relevant studies addressing gaps in the pivotal and RCT evidence were submitted.
Conclusions
Two pivotal, multinational, double-blind, randomized placebo-controlled trials — the LUCENT-1 (N = 1,281) and LUCENT-2 (N = 554) trials — and 1 ITC informed the assessment of mirikizumab in this review. Both pivotal trials demonstrated the superiority of mirikizumab over placebo across all end points. The evidence from the LUCENT-1 trial (the induction trial) demonstrated the efficacy of mirikizumab 300 mg IV over placebo in achieving induction clinical remission, alternate clinical remission, clinical response, HRQoL, endoscopic remission, symptomatic remission, bowel urgency improvement, mucosal healing, and work productivity in patients with moderately or severely active UC over 12 weeks. The evidence from the LUCENT-2 study, the maintenance trial, further supported these results after 40 weeks of maintenance dosing. Additionally, the LUCENT-2 trial demonstrated the efficacy of mirikizumab 200 mg SC in achieving corticosteroid-free remission, the maintenance of clinical remission, bowel urgency remission, and mucosal remission at week 40 of the LUCENT-2 study among patients who attained a clinical response by week 12 of the LUCENT-1 study. The results from the primary analysis were considered generalizable to the Canadian landscape; however, it should be noted that the patient population for the LUCENT-2 study may have been enriched, as only responders were rerandomized to the trial, excluding delayed responders who represent a subset of the general population for this indication. Regarding the extended induction, although mirikizumab was able to capture delayed response, the data were considered observational due to the absence of a comparison to the main cohort. Clinically meaningful improvements in HRQoL based on the IBDQ were observed in patients receiving mirikizumab for 40 weeks in the maintenance phase. The NMA comparison between mirikizumab and relevant comparators (i.e., adalimumab, golimumab, infliximab, ozanimod, tofacitinib, ustekinumab, and vedolizumab) did not demonstrate a difference in favour of 1 treatment over another in induction clinical remission and response, mucosal healing, all-cause discontinuation, and SAEs. The apparent benefit of mirikizumab in the maintenance period among patients in the biologic-naive population may be explained by heterogeneity between the trials. Definitive conclusions related to treatment effect and harms of mirikizumab compared to other relevant treatments for UC could not be drawn from the NMA analysis due to substantial heterogeneity in patient characteristics, inclusion criteria (e.g., the definition of prior biologic exposure), prior treatment exposure, and outcome definitions, which likely challenged the underlying exchangeability assumption, and wide CrIs for most estimates. Overall, the LUCENT-1 and LUCENT-2 studies demonstrated clinical efficacy and minimal safety concerns for up to 52 weeks of treatment with mirikizumab in patients with moderate to severe UC and relative to placebo. Evidence of efficacy and safety of mirikizumab beyond 52 weeks, as well as direct comparisons with other treatments for UC, are necessary to further understand the long-term benefits and comparative effectiveness of mirikizumab.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of mirikizumab, 20 mg/mL administered by IV infusion for induction and 100 mg/mL administered by SC injection for maintenance, in the treatment of UC in adult patients with moderately to severely active disease who have had an inadequate response, had a loss of response, or were intolerant to conventional therapy, a biologic treatment, or a JAK inhibitor.
Disease Background
Content in this section has been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the CADTH review team.
IBD is a term used to describe disorders that involve chronic inflammation of the digestive tract. There are 2 main types of IBD: Crohn disease and UC. UC causes inflammation and ulcers in the digestive tract, affecting the innermost lining of the large intestine (colon) and rectum.1,2 UC is characterized by blood in the stool with mucus, frequent diarrhea, loss of appetite, and tenesmus (a strong urge to use the bathroom without necessarily having a bowel movement), in addition to abdominal pain, RB, and weight loss.3-5 The most common initial manifestation of UC is bloody diarrhea with or without mucus. In addition, patients with UC report high rates of fatigue and sleep difficulties.10 While the etiology of UC is not completely understood, there is growing evidence to suggest genetic and environmental factors may contribute to the irregular immune response that aberrantly recruits activated immune cells to the colon,3 resulting in chronic inflammation that damages the colon and causes UC symptoms. UC generally develops in young adulthood6-8 and persists throughout life, marked by periods of spontaneous remission and relapse.9 Though most patients experience this relapsing-remitting disease course, up to 24% of patients report experiencing continuous UC symptoms.9 The majority of individuals living with UC have a mild to moderate disease course, generally with active disease at diagnosis followed by alternating exacerbations and longer periods of remission.10 However, aggressive disease course is experienced in 10% to 15% of patients, with a cumulative risk of relapse of between 70% to 80% at 10 years postdiagnosis.10 Regardless of severity, UC is associated with a substantial reduction in QoL for patients, with considerable impact on many aspects of their lives, including emotional and psychological functioning, social and physical functioning, and work and academic life.11,12 Chronic active UC may lead to structural damage of the colon; this in turn can lead to dysmotility, chronic symptoms, reduced QoL, and the risk of colon cancer, which requires a colectomy.
UC is diagnosed clinically, with endoscopy, biopsy, and stool sampling being common tests used to rule out other causes of symptoms.13 These investigations are typically performed in an outpatient setting, with a gastroenterologist ultimately confirming the diagnosis.9,10 UC has a worldwide annual incidence rate of 1.2 cases to 20.3 cases per 100,000 people and a prevalence of 7.6 cases to 246.0 cases per 100,000 people.3 The highest age-standardized prevalence rate of IBD in 2017 occurred in high-income countries in North America,37 with Canada having 1 of the highest rates in the world.6 The estimated annual incidence rates for UC in Canada range from a low of 8.4 per 100,000 people in Alberta to a high of 21.4 per 100,000 people in Nova Scotia.6-8 There are an additional 15,000 individuals living with IBD in Canada who do not have a confirmed diagnosis of Crohn disease or UC (termed indeterminate colitis).38 In 2030, the Canadian IBD prevalence is anticipated to rise to 0.98%, equating to a 0.44% prevalence for UC specifically.15 The incidence of premature mortality associated with UC is no greater than in the general population, but the condition is associated with significant morbidity and an increased risk of colorectal cancer.13,33 Other potentially severe complications associated with UC that may require hospitalization include severe blood loss, fulminant colitis, perforated bowel, or toxic megacolon.39 About 50% of patients with UC will require hospitalization at some point in their lives, with about a 50% risk of rehospitalization within 5 years for these patients.10
Standards of Therapy
Content in this section has been informed by materials submitted by the sponsor and clinical expert input. The following has been summarized and validated by the CADTH review team.
UC treatment strategies are dependent on the presence of active disease, severity and extent of the UC, and patient preference. Treatments for UC are commonly separated into 2 groups: conventional therapies and advanced therapies. Conventional therapies include 5-ASA products, corticosteroids, and immunomodulators (such as azathioprine, 6-mercaptopurine, and methotrexate). Corticosteroids are recommended as an initial treatment to bring about complete remission for patients with moderate to severe UC; however, due to serious side effects and a lack of long-term efficacy, corticosteroids are mainly used for the acute treatment of flares and are not recommended for long-term use. Immunomodulators can be considered next, but the use of biologic therapy may be indicated immediately after steroid failure (or prolonged steroid dependence) in many cases. Biologics and JAK inhibitors are often grouped together as “advanced therapies.” Advanced therapies used in UC include adalimumab, golimumab, infliximab, ustekinumab, tofacitinib, ozanimod, and vedolizumab. However, ustekinumab and ozanimod are not publicly reimbursed for UC in Canada. Tofacitinib, a JAK inhibitor, is recommended for use only in patients considered biologic-failed.40 For patients whose UC fails on a biologic therapy, a biologic with a different mechanism of action or tofacitinib are reasonable next options.41,42 Clinicians will aim to optimize treatment by confirming adherence, checking drug trough levels, and adjusting the dose if subtherapeutic. If despite optimization the patient continues to flare, the clinician will switch therapies. When advanced therapies have been exhausted, surgery is the next therapeutic option or referral to a clinical trial.33 Though surgical treatment of UC is considered curative,43 it comes with the risk of high complication rates.33 As such, surgery is usually reserved for patients who cannot be managed medically, patients with acute severe UC (toxic megacolon, perforation, and uncontrolled severe hematochezia), or patients who develop colorectal cancers.33 The clinical expert on this review noted that treatment decisions are often dictated by access to currently available therapies via health insurance rather than clinical guidelines or expert recommendations.
According to the 2015 Canadian guidelines, the treatment goal of UC is “complete remission,” defined as symptomatic remission (normal SF and no blood in the stool) and endoscopic healing (Mayo ES of 0 or 1).42 Note that the parameters assessed in determining complete remission (SF, RB, and findings on endoscopy) are the same 3 considered in evaluating the MMS.44 Another important treatment goal according to the international 2021 initiative Selecting Therapeutic Targets in Inflammatory Bowel Disease–II (known as STRIDE-II)45 is clinical response, defined as at least a 50% improvement in RB and SF. Clinical response is the most immediate target. An intermediate target is clinical remission, defined as Mayo RB and SF subscores of 0, or a Partial Mayo Score of less than 3 with no Mayo subscore greater than 1. Suggested long-term targets include endoscopic healing and improvement in QoL. According to the clinical expert consulted on this review, treatment goals vary by timeline. Short-term goals are aimed at improving clinical response, improving symptoms, and preventing progression. Medium-term goals are to reduce inflammatory burden, discontinue corticosteroid use, and improve endoscopic healing. Long-term goals are sustained steroid-free clinical remission, endoscopic healing, and mucosal remission. However, according to the clinical expert, treatment decisions are often dictated by access to currently available therapies via health insurance rather than clinical guidelines or expert recommendations.
According to the clinical expert, treatments are needed that work both quickly and in the long term, improve convenience and adherence, provide a variety of options, and are well tolerated and safe.
Drug Under Review
Key characteristics of mirikizumab are summarized in Table 3, along with other treatments available for UC.
IL-23 is an important driver of mucosal inflammation in UC and affects the differentiation, expansion, and survival of T-cell subsets and innate immune cell subsets, which represent sources of proinflammatory cytokines.16 Mirikizumab is a humanized IgG4 monoclonal antibody that binds with high affinity and specificity to the p19 subunit of human IL-23 cytokine and inhibits its interactions with the IL-23 receptor.16
Mirikizumab is indicated for the treatment of adult patients with moderately to severely active UC who have had an inadequate response, had a loss of response, or were intolerant to conventional therapy, a biologic treatment, or a JAK inhibitor. Mirikizumab received a positive opinion from the EMA for the indication under review.46
In the induction phase, mirikizumab is infused intravenously at 300 mg for at least 30 minutes at week 0, week 4, and week 8, and then administered by SC injection at 200 mg every 4 weeks after completion of induction dosing at week 12. For patients who do not have adequate therapeutic response at week 12 after induction dosing, extended induction dosing may be considered by administering 300 mg mirikizumab by IV infusion at week 12, week 16, and week 20. Patients who do not show evidence of therapeutic benefit from extended induction therapy by week 24 should discontinue mirikizumab. If therapeutic benefit is achieved with the additional IV therapy, patients may initiate mirikizumab SC maintenance dosing every 4 weeks.
Table 3: Key Characteristics of Mirikizumab and Main Comparators
Drug | Mechanism of action | Indicationa | Route of administration and recommended dosage | Serious adverse effects or safety issues |
|---|---|---|---|---|
Mirikizumab | Humanized IgG4 monoclonal antibody that binds with high affinity and specificity to the p19 subunit of human IL-23 cytokine to inhibit its interaction with the IL-23 receptor | Treatment of adult patients with moderately to severely active UC who have had an inadequate response, had a loss of response, or were intolerant to conventional therapy, a biologic treatment, or a JAK inhibitor | Induction: 300 mg IV q.4.w. on week 0, week 4, and week 8. Consider extended induction of 300 mg IV q.4.w. on week 12, week 16, and week 20 in nonresponders at week 12. Maintenance: 200 mg SC q.4.w. | Upper respiratory tract infection, headache and site injection reactions (e.g., rash, rash maculopapular, rash papular, and rash pruritic) were commonly reported AEs during clinical trials. |
S1P receptor modulators | ||||
Ozanimod | S1P receptor modulator. Binds to the S1P1 subtype receptors on lymphocytes, preventing egress from lymph nodes. The mechanism by which ozanimod and its active metabolites exert their therapeutic effects in MS and UC is unknown but may involve reduction in lymphocyte migration into the CNS and intestine. | Treatment of adult patients with moderately to severely active UC who have had an inadequate response, had a loss of response, or were intolerant to either conventional therapy or a biologic drug | Dose escalation to 0.92 mg orally once daily. Induction (day 1 to day 4): 0.23 mg once daily. Dose escalation (day 5 to day 7): 0.46 mg once daily. Maintenance (day 8 and onwards): 0.92 mg once daily. | Malignancies, particularly of the skin, have been reported in patients taking ozanimod in clinical trials. The initiation of ozanimod may result in transient reductions in heart rate and atrioventricular delays. |
Anti-TNF | ||||
Infliximab | Anti-TNF. IgG1k monoclonal antibody that neutralizes the biological activity of TNF alpha by specifically binding to its receptors | Induction and maintenance of clinical remission and mucosal healing, and reduction or elimination of corticosteroid use in adult patients with moderately to severely active UC who have had an inadequate response to conventional therapy | Induction dose of 5 mg/kg IV at 0 weeks, 2 weeks, and 6 weeks followed by 5 mg/kg IV every 8 weeks thereafter | Infections and malignancies have been observed in patients receiving infliximab. |
Golimumab | Anti-TNF. Human monoclonal antibody that binds with p55 or p75 human TNF receptors | Induction and maintenance of clinical response in adults with moderately to severely active UC who have had an inadequate response to, or have medical contraindications for, conventional therapy, including corticosteroids, aminosalicylates, azathioprine, or 6-MP | 200 mg initially administered by SC injection at week 0 followed by 100 mg at week 2 and then 50 mg every 4 weeks thereafter | Upper respiratory infections and reactions at the site injection, but no clinically significant differences compared to placebo |
Adalimumab | Anti-TNF. Human IgG1 monoclonal antibody. Binds and blocks TNF alpha and its interactions with p55 and p75 cell-surface TNF receptors | For the treatment of adult patients with moderately to severely active UC who have had an inadequate response to conventional therapy, including corticosteroids and/or azathioprine or 6-MP, or who are intolerant to such therapies | 160 mg at week 0 followed by 80 mg at week 2 administered by SC injection | Serious infections (pneumonia), malignancies, and neurologic events have been reported more frequently in patients taking adalimumab. |
Integrin blocker | ||||
Vedolizumab | IgG1 monoclonal antibody. Binds to the human alpha 4 beta 7 integrin, acting as a gut-selective anti-inflammatory biologic | Treatment of adult patients with moderately to severely active UC who have had an inadequate response, had a loss of response, or were intolerant to either conventional therapy or infliximab, a TNF alpha antagonist | 300 mg administered by IV infusion at 0 weeks, 2 weeks, and 6 weeks and then every 8 weeks thereafter. The SC maintenance dose is 108 mg every 8 weeks. | Infections and malignancies have been reported in patients taking vedolizumab, but no clinically significant differences have been found. |
IL-12 and IL-23 inhibitor | ||||
Ustekinumab | Human IgG1 monoclonal antibody. Neutralizes cellular responses mediated by IL-12 and IL-23 by binding with specificity to the shared p40 protein subunit | Treatment of adult patients with moderately to severely active UC who have failed or were intolerant to treatment with immunomodulators or corticosteroids, but have never failed treatment with a biologic, or have failed or were intolerant to treatment with a biologic | IV infusion, single-use, weight-based dose (approximately 6 mg/kg): 260 mg for those weighing ≤ 55 kg; 390 mg for those weighing > 55 kg to ≤ 85 kg; or 520 mg for those weighing > 85 kg for induction therapy, followed by maintenance therapy of 90 mg SC infections every 8 weeks | Immunomodulating drugs have the potential to increase the risk of infections and malignancy. No clinically significant differences have been found in terms of malignancies. |
JAK inhibitors | ||||
Tofacitinib | Selective JAK inhibitor. Blocks several cytokine pathways and lymphocyte activation | For the treatment of adult patients with moderately to severely active UC with an inadequate response, a loss of response, or intolerance to either conventional UC therapy or a TNF alpha inhibitor | 10 mg orally (as tofacitinib citrate) twice daily | A Health Canada warning indicated an increased risk of thromboses (pulmonary and deep vein thrombosis) and death, and an increased risk of serious infection, including herpes zoster infections. Of note, tofacitinib is not recommended in combination with biological UC therapies or with potent immunosuppressants such as azathioprine and cyclosporine. |
6-MP = 6-mercaptopurine; AE = adverse event; CNS = central nervous system; IgG1 = immunoglobulin G1; IgG1k = immunoglobulin G1k; IgG4 = immunoglobulin G4; IL = interleukin; JAK = Janus kinase; MS = multiple sclerosis; S1P = sphingosine-1-phosphate; SC = subcutaneous; TNF = tumour necrosis factor; UC = ulcerative colitis.
aHealth Canada–approved indication.
Sources: Product monographs for mirikizumab (Omvoh),16 ozanimod (Zeposia),47 ustekinumab (Stelara),48 infliximab (Remicade),49 vedolizumab (Entyvio),50 golimumab (Simponi),51 tofacitinib (Xeljanz),52 and adalimumab (Humira).53
Stakeholder Perspectives
Patient Group Input
This section was prepared by the CADTH review team based on the input provided by patient groups. The full original patient input received by CADTH has been included in the stakeholder section at the end of this report.
Input from patients with moderate to severe UC was received from the GI Society and Crohn’s and Colitis Canada. Patient input from the GI Society was collected using a series of surveys conducted between 2015 and 2022 (n = 54 to 432), focus groups, and 1-to-1 contact with patients with IBD at lectures, patient roundtables, support groups, and via phone, email, and social media interactions, including individual interviews with 3 patients with UC who received mirikizumab in a clinical trial. Patient input from Crohn’s and Colitis Canada was compiled from 2 online surveys conducted in 2022 (n = 354), from individual interviews with 2 patients with UC who participated in the Rinvoq clinical trial, and from 1 phone interview with a patient who participated in the mirikizumab clinical trial.
It was noted that UC has a profound effect on daily life — physically, emotionally, and socially — at home and at school or in the workplace. Many patients surveyed by Crohn’s and Colitis Canada revealed that they hid aspects of their diagnosis from their friends, coworkers, and classmates. Almost two-thirds (63%) of respondents agreed that their family and friends do not understand what they are going through. Patients noted that symptoms can be relentless, embarrassing, and scary. Based on the surveys conducted by Crohn’s and Colitis Canada, the most frequently reported UC-related complications were mental health and stress (65%), joint inflammation and arthritis (51%), anal fissures and hemorrhoids (40%), anemia (33%), skin conditions (30%), malnutrition (30%), and weight loss (30%). Patients stated that sustained remission and/or treatment response is more important than relieving any 1 symptom. The constant concern that there will be future flares, possibly worse than the last, at unpredictable times, was noted as being disastrously disruptive. As expressed by 1 patient surveyed by the GI Society, “The worse part is fear of irreversible permanent damage that will affect your day-to-day life forever.”
Regarding current treatments for UC, it was noted that although there are several available options, most patients have difficulty obtaining remission or adequate symptom relief. Based on survey data from the GI Society, only 24% of patients with IBD found available medications to be adequate, 56% of patients found them to be only somewhat adequate, and 20% of patients found them not at all adequate. More than half of patients (56%) surveyed by Crohn’s and Colitis Canada believed that different treatment options could make them feel better. While steroid use is an important part of symptom management for UC, patients reported not being particularly supportive of the treatment option. Patient input from the GI Society stressed that a variety of treatment options is important because response varies across patients, and there may be a loss of response after prolonged use. Patients noted that there is a need for new and effective options to achieve mucosal healing and reduce the symptoms of UC, and to provide good QoL. Patients interviewed by Crohn’s and Colitis Canada added that any new treatment must be able to protect a patient’s ability to work, attend school and social events, and leave the home to run errands. Patients interviewed by Crohn’s and Colitis Canada also added that potential risks and side effects, especially those related to heart and liver function, are a major source of concern when considering new treatment options.
Among the 4 patients who received mirikizumab in clinical trials, all experienced improved gut healing and continued to take the medication. The initial induction of treatment via infusion was found to be exhausting and time consuming by 2 patients interviewed by the GI Society. Although 2 patients did not particularly like the SC administration of mirikizumab, they added that the injections were manageable. All 4 patients expressed having improved QoL. As expressed by 1 patient interviewed by the GI Society, “My experience has been really good with this drug, and I recommend it. I have no anxieties on going out to public events. I’m definitely a lot more comfortable leaving the house and going about my normal day to day activities.” The 1 patient interviewed by Crohn’s and Colitis Canada stated that treatment with mirikizumab was “[…] a life changer. It's hard sometimes to know what success necessarily looks like, but I would say if I had. If someone asked me, I absolutely have had great success on this medication. I’m thrilled that I have no side effects to deal with. And I would do it all over again in a second. And I'm hoping that it will be available for me to continue and that it continues to work for me.”
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, providing guidance on the potential place in therapy). The following input was provided by 1 clinical specialist with expertise in the diagnosis and management of UC.
Unmet Needs
The clinical expert noted that there is an unmet need for treatments that are better tolerated, improve convenience and compliance, and take into consideration special populations such as those with previous or current malignancies. The clinical expert stated that having available treatments with different administration methods is also important to patients (e.g., IV, SC, oral) as well as having multiple treatment options, given that patients will often lose response to treatment and require another therapy. The clinical expert noted that not all patients’ UC responds to available treatments and patients often become refractory to current treatment options. Treatment success (e.g., clinical response, clinical remission) varies from 15% to 60%. The clinical expert stated that it is difficult to predict which patients with UC will respond to treatment, as there is currently no available clinical test. According to the clinical expert, no treatments are available to reverse the progression of IBD or UC (e.g., damage to the colon). However, if UC is treated and controlled early in the disease course, it is possible to stop or delay progression. The expert provided the example of achieving endoscopic remission, which may be able to prevent structural damage of the colon and reduce risk of dysplasia and/or colon cancer.
Place in Therapy
According to the clinical expert, mirikizumab is an anti–IL-23 drug for UC and would offer a novel treatment mechanism for the disease that is considered more selective. The clinical expert stated that mirikizumab’s place in therapy would be similar to other biologics and would be recommended as a second-line therapy after 5-ASA, instead of immunomodulators. For patients whose UC has failed on all available therapies but require a biologic therapy for another condition, mirikizumab may be used as a dual therapy with a biologic. For patients who are biologic-naive, the expert felt that many clinicians may recommend mirikizumab for induction and maintenance as it may be even more targeted than other therapies. Given the available treatments, mirikizumab may be recommended before other treatments that have more side effects or risks, such as tofacitinib. It was the opinion of the clinical expert that patients do not need to initiate and experience treatment failure with other therapies before being prescribed mirikizumab, given the limitations of other therapies. For example, azathioprine and methotrexate have side effect profiles that include risks to the liver and the risk of lymphoma and take longer to act. In addition, the clinical expert noted that previous trials have shown that patients who are biologic-naive have a better chance of achieving a response to treatment compared to patients who are biologic-exposed. Hence, the clinical expert recommends that mirikizumab be offered both to patients with UC who are biologic-naive and who are biologic-experienced.
Patient Population
The clinical expert noted that patients whose UC is most likely to respond to treatment with mirikizumab would be those with moderate to severe UC (biologic-naive or biologic-experienced) that has failed to respond to conventional therapy. In the opinion of the clinical expert, patients who are in most need of mirikizumab would be those whose UC has failed to respond to first-line therapy with 5-ASA. The clinician indicated that patients who have UC and comorbid psoriasis may have further benefit from mirikizumab as research has demonstrated its efficacy in treating psoriasis.54 According to the clinical expert, these patients can be identified through clinical examination, lab tests, and colonoscopy. No clinical tests are available to determine whether a patient will be more likely to exhibit a response to mirikizumab. Patients least suited for treatment with mirikizumab, according to the clinical expert, are patients with active infections, malignancy, and severe hepatic impairment, and patients who are pregnant.
Assessing the Response Treatment
According to the clinical expert, the outcomes used in clinical practice align with those used in clinical trials such as clinical remission and response (measured by Partial Mayo Score), endoscopic remission and response, and biomarkers (e.g., fecal calprotectin). The clinical expert noted that clinicians routinely schedule colonoscopy 6 months to 9 months after starting biologics or small-molecule drugs to check for endoscopic healing. The clinical expert indicated that it is difficult to schedule or expect a patient to return for a colonoscopy to assess response in the induction phase due to the short time span (e.g., 12 weeks); therefore, surrogate markers such as fecal calprotectin may be used to approximate objective response. If concerned about patient response, the expert indicated that some physicians may try to book a flexible sigmoidoscopy soon after the induction period has been completed.
According to the clinical expert, a clinically meaningful response to treatment would be no further RB, no rectal incontinence, rectal urgency that has been reduced or no longer exists, bowel movement frequency that has been reduced or is normal, stools becoming more solid, and abdominal pain that has been reduced or no longer exists. The clinical expert would expect clinical improvement within 4 weeks, and clinical remission within 12 weeks; however, depending on the severity of disease and previous medication exposure, the clinical expert noted that patients may have slower response or a delay to remission. In this case, the expert indicated that they would be comfortable with an extended induction period of 12 weeks for nonresponders, which is aligned with the product monograph. The clinical expert stated that there will not be much variation in assessing response among physicians as most gastroenterologists treating IBD use the standard clinical scores (e.g., Partial Mayo Score, MMS) for UC in clinics with an endoscopic component if performing a colonoscopy. Additionally, physicians will monitor with C-reactive protein and fecal calprotectin.
Discontinuing Treatment
According to the clinical expert, mirikizumab should be discontinued in the event of SAEs, disease progression, or the inability to taper off steroids. The clinical expert would consider stopping treatment after 24 weeks of treatment if the patient’s UC does not respond. This would include an extended induction phase if the patient’s UC was not responding to initial induction (e.g., 12 weeks). According to the clinical expert, it might be expected that approximately 30% of patients who did not have an initial induction response would have a delayed response to induction treatment. If a patient had some response from the first induction period (e.g., 12 weeks), but perhaps could not completely taper off steroids, they could be given an extended induction (e.g., additional 12 weeks). At the end of this additional 12-week period, if their UC continues to not respond or they are unable to stop steroids, they would be considered nonresponders. At that point, the clinical expert stated that the patient would be changed to another therapy.
Prescribing Considerations
The clinical expert noted that the community, hospitals (outpatient clinics), and specialty clinics are all appropriate settings for treatment administration. The clinical expert stated that specialists, such as gastroenterologists, should prescribe mirikizumab. In rural settings, a general practitioner or endoscopist trained for IBD management may also prescribe mirikizumab.
Clinician Group Input
This section was prepared by the CADTH review team based on the input provided by clinician groups. The full original clinician group input received by CADTH has been included in the stakeholder section at the end of this report.
One clinician group input submission was received from a group of gastroenterologists in Canada. The clinician group consists of gastroenterologists in Canada who are recognized nationally and internationally as experts in the management of IBD. Input from the clinician group was compiled by 9 clinicians and was based on the literature, clinical practice guidelines for the management of UC, consultation with Canadian and international expert groups, and a review of individual product monographs and primary trial publications, and information from manufacturers of current and future treatments.
Based on input from the clinician group, the goals of UC therapy have evolved from simply controlling symptoms to preventing disease progression, surgery, and disability with early intervention and a treat-to-target approach. The clinician group noted that treatment for UC is influenced by disease characteristics and may involve multiple medications, including 5-ASA, corticosteroids, immunosuppressants, biologics (which include anti-TNF therapy, anti-integrin therapy, and anti–IL-12 and anti–IL-23 therapy) and advanced small molecule drugs (which include JAK inhibitors and S1P receptor modulators). The clinician group stressed that treatment with corticosteroids is reserved for periods of disease flare-up and not prescribed for long-term use due to side effects and poor effectiveness for maintaining remission. The clinician group noted that surgery is necessary in UC when medications prove ineffective, for fulminant disease which is medically refractory, or if complications arise, such as dysplasia or strictures. The clinician group emphasized that the ongoing need for surgery in patients with UC reflects the ongoing need for medical therapies that can prevent complications and meet treatment goals. Accordingly, the clinician group identified unmet needs for the treatment of moderate to severe UC. This includes the need for therapy that induces symptomatic remission, can be used to maintain symptomatic remission, is safe with long-term use, and can rapidly improve endoscopic appearance of the bowel and maintain this in the long term. The clinician group emphasized that none of the currently available therapies for UC meet all current needs of patients in terms of short-term or long-term treatment. Remission with treatment is not universal and patients can lose response after an initial period of improvement and relapse, even after long periods of remission on an existing therapy. Accordingly, the clinician group stressed the need for novel therapies targeting alternative pathways.
Overall, the clinician group suggested that mirikizumab has the potential for a broad range of uses in clinical practice, from first-line advanced therapy to treatment of patients with inadequate response or intolerance to multiple advanced therapies. The clinician group anticipates that mirikizumab may become a treatment of choice for most patients with UC as it appears to have a favourable benefit-risk ratio. The general consensus among the clinician group members was that mirikizumab may be a good candidate to help address unmet needs as a potential first-line option or for individuals whose UC has failed to respond to other advanced therapies. The clinician group recommended that management strategies using mirikizumab aim for remission, which is defined as both symptomatic and endoscopic remission and aligns with Canadian consensus guidelines. The clinician group suggested that a meaningful improvement in symptoms as measured by the resolution of SF and RB should be demonstrated in the first 3 months of therapy. The clinician group expected patients to be in symptomatic remission and off corticosteroids by 6 months. In addition, the clinician group indicated that symptomatic improvement should be accompanied by a decrease in biomarkers of inflammatory activity (C-reactive protein and fecal calprotectin) in the first 3 months after initiating mirikizumab. The clinician group suggested that it would be appropriate to discontinue treatment with mirikizumab in the event of worsening symptoms or inadequate response. In circumstances where there was an inadequate response to mirikizumab as a first-line biologic, the clinician group indicated that a switch to another class of drugs, such an anti-TNF drugs, may be warranted. Based on clinical experience, the clinician group suggested that it would be appropriate for a trained health care professional to administer mirikizumab in a clinic during the induction phase.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Considerations for initiation of therapy | |
Are patients eligible for re-treatment if their UC has failed to respond to initial mirikizumab treatment? | According to the clinical expert, yes, patients are eligible for re-treatment if they lose response to the drug. However, they should not be re-treated if their UC fails to respond after re-treatment. In the LUCENT-2 trial (the maintenance trial), extended doses for an additional 12 weeks produced a clinical response in up to 50% of patients who did not have a clinical response to 12 weeks of induction doses. Patients would not be re-treated with mirikizumab if their UC failed to respond to extended induction and clinicians would not re-treat with the drug later in the patient’s therapeutic journey after this point. |
In the LUCENT-1 and LUCENT-2 trials, patients were excluded if they had had surgery or were to have surgery to treat their UC. Given that recurrence may happen even after surgery, would patients who have a recurrence of UC postsurgical treatment be eligible for mirikizumab? | The clinical expert noted that for UC, surgery refers to a colectomy (remove the whole colon and leave the rectal stump). According to the clinical expert, typically, the operating physician will add either a permanent ostomy bag or J pouch. Patients will stop medications postsurgery and would not normally have recurrence of UC if they have a J pouch. However, there is a possibility of developing inflammation of the J pouch, which could develop into a Crohn-like phenotype of the pouch that is refractory to antibiotics. Only in this rare case would patients need to go back onto a biologic, which may be mirikizumab if they had not been on it before. |
Should patients be trialled on other biologics before eligibility to mirikizumab? Current criteria proposed by the sponsor is a trial of at least conventional therapy. | In the opinion of the clinical expert, patients do not need to trial other biologics to be eligible for the reimbursement of treatment with mirikizumab. The clinical expert would expect that mirikizumab can be initiated after conventional therapy with 5-ASA. |
Considerations for continuation or renewal of therapy | |
Will response to induction therapy be the requirement for the reimbursement of maintenance therapy in a manner similar to Stelara? | The clinical expert advised that a patient should have attained a clinical response to induction therapy within 24 weeks (an extended induction period) for the reimbursement of treatment with mirikizumab to continue to maintenance therapy. CADTH noted that this is aligned with the draft product monograph. |
Considerations for prescribing of therapy | |
Who should be able to prescribe mirikizumab? Can this be extended to internists in remote and rural areas? | According to the clinical expert, a gastroenterologist should prescribe the treatment, but in the case of rural and remote areas, a general internist or endoscopist trained for IBD management may also prescribe mirikizumab. |
System and economic issues | |
Mirikizumab requires IV administration for induction and would likely be administered in an outpatient IV clinic. This is an additional cost that should be taken into consideration. Once on maintenance, the drug will be administered subcutaneously. Need to consider the cost of IV administration: clinic, staff, materials, and more. | For CDEC consideration. |
5-ASA = 5-aminosalicylic acid; CDEC = CADTH Canadian Drug Expert Committee; IBD = inflammatory bowel disease; UC = ulcerative colitis.
Clinical Evidence
The objective of CADTH’s Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of mirikizumab, 300 mg/15 mL IV (induction) and 100 mg/1 mL SC injection (maintenance), in the treatment of adult patients with moderately to severely active UC who have had an inadequate response, had a loss of response, or were intolerant to conventional therapy, a biologic treatment, or a JAK inhibitor.The focus will be on comparing mirikizumab to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of mirikizumab is presented in 2 sections, and CADTH’s critical appraisal of the evidence is included after each section. The first section, the systematic review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. The second section includes indirect evidence from the sponsor.
Included Studies
Clinical evidence from the following is included in the CADTH review and appraised in this document:
two pivotal studies (placebo-controlled RCTs)
one systematic review with an NMA.
Pivotal Studies and RCT Evidence
Content in this section has been informed by materials submitted by the sponsor. The following has been summarized and validated by the CADTH review team.
Description of Studies
The LUCENT-1 and LUCENT-2 trials were pivotal phase III trials evaluating the safety and efficacy of mirikizumab for UC in the induction phase (the LUCENT-1 study) and maintenance phase (the LUCENT-2 study). Characteristics of the included studies are summarized in Table 5.
Table 5: Details of Pivotal Studies and RCT Evidence Identified by the Sponsor
Characteristic | LUCENT-1 (induction trial) | LUCENT-2 (maintenance trial) |
|---|---|---|
Designs and populations | ||
Study design | Phase III, multicentre, randomized, double-blind, parallel, placebo-controlled induction study | Phase III, multicentre, randomized, double-blind, parallel, placebo-controlled maintenance study |
Locations | 163 centres in North America, Europe, Asia, South America, and Australia | 368 centres in North America, Europe, Asia, South America, and Australia |
Patient enrolment dates | Start date: June 18, 2018 End date: NR | Start date: October 19, 2018 End date: NR |
Randomized (N) | 1,281 | 1,178 (including 544 LUCENT-1 study blinded mirikizumab responders) |
Inclusion criteria | Patients aged 18 years to 80 years with a diagnosis of UC (MMS of 4 to 9 with endoscopic subscore of at least 2), who have had an inadequate response, had a loss of response, or were intolerant to a conventional UC therapy and/or biologic and/or tofacitinib | Patients from the LUCENT-1 study who had received at least 1 dose of study drug and had all necessary evaluations to assess MMS at the end of the LUCENT-1 study |
Exclusion criteria |
| If a patient developed any of the exclusion criteria outlined in the LUCENT-1 study, they would be excluded from participation in the LUCENT-2 study. |
Drugs | ||
Intervention | Mirikizumab 300 mg IV q.4.w. for 12 weeks | For responders to blinded mirikizumab from LUCENT-1 study: Mirikizumab 200 mg SC q.4.w. for 40 weeks; if LOR during week 12 to week 28 of the LUCENT-2 study, participants would be changed to open-label mirikizumab 300 mg IV q.4.w. for 3 doses For responders to blinded placebo from LUCENT-1 study: Placebo SC q.4.w. for 40 weeks; if LOR during week 12 to week 28 of the LUCENT-2 study, participants would be changed to open-label mirikizumab 300 mg IV q.4.w. for 3 doses For nonresponders in LUCENT-1 study: Open-label mirikizumab 300 mg IV for 3 doses; those who responded to mirikizumab would continue with open-label mirikizumab 200 mg SC q.4.w. for the rest of the LUCENT-2 study and nonresponders would discontinue |
Comparator(s) | Placebo IV q.4.w. for 12 weeks | For responders to blinded mirikizumab from LUCENT-1 study: Placebo SC q.4.w. for 40 weeks; if LOR during week 12 to week 28 of the LUCENT-2 study, participants would be changed to open-label mirikizumab 300 mg IV q.4.w. for 3 doses For responders to blinded placebo from LUCENT-1 study: NA For nonresponders to blinded mirikizumab or blinded placebo: NA |
Study duration | ||
Screening phase | 4 weeks | None |
Run-in phase | None | Visit 1 (week 0) was to occur within 10 days of visit 5 (week 12) of the LUCENT-1 trial. |
Treatment phase | 12 weeks | 40 weeks |
Follow-up phase | Patients had the option to complete 16 weeks of posttreatment follow-up or enter the LUCENT-2 study, the maintenance study. | Patients had the option to complete 16 weeks of posttreatment follow-up or enter the LUCENT-3 study, a long-term extension study. |
Outcomes | ||
Primary end point | The proportion of patients in clinical remission at week 12, based on MMS | The proportion of patients in clinical remission at week 40, based on MMS |
Secondary and exploratory end points | Secondary end points at week 12
Other secondary end points
| Secondary end points at week 40
Other secondary end points
|
Publication status | ||
Publications | Dubinsky et al. (2023)55 | Dubinsky et al. (2023)55 |
EQ-5D-5L = 5-Level EQ-5D; ES = endoscopic Mayo subscore; HEMI = histologic endoscopic mucosal improvement; HEMR = histologic endoscopic mucosal remission; IBDQ = Inflammatory Bowel Disease Questionnaire; LOR = loss of response; MCS = mental component summary; MMS = Modified Mayo Score; NA = not available; NR = not reported; q.4.w. = every 4 weeks; PCS = physical component summary; RCT = randomized controlled trial; SC = subcutaneous; SF-36 = Short Form (36) Health Survey; UC = ulcerative colitis; UCEIS = Ulcerative Colitis Endoscopic Index of Severity; UNRS = Urgency Numeric Rating Scale; WPAI:UC = Work Productivity and Activity Impairment Questionnaire: Ulcerative Colitis.
Note: Details in Table 5 have been taken from the sponsor’s Summary of Clinical Evidence.20,30
Sources: Clinical Study Reports for the LUCENT-1 and LUCENT-2 studies.31,32
LUCENT-1 Study
The LUCENT-1 trial (N = 1,281) was a 12-week, multicentre, randomized, double-blind, parallel-arm induction trial evaluating the safety and efficacy of mirikizumab 300 mg IV versus placebo in adults with moderately to severely active UC. The primary objective of the LUCENT-1 study was to evaluate the ability of mirikizumab to induce clinical remission at week 12. Major secondary objectives included the ability of mirikizumab to induce alternate clinical remission, clinical response, endoscopic remission, symptomatic remission, clinical response in patients who are biologic-experienced, bowel urgency improvement, and HEMI, all at week 12, as well as symptomatic remission at week 4. The LUCENT-1 study consisted of a 4-week screening period and a 12-week induction dosing period. At the end of the induction period, patients had the choice of transitioning to the LUCENT-2 study (a maintenance trial) or discontinuing study treatment and completing the 16-week posttreatment follow-up period. A study design flow diagram of the LUCENT-1 and LUCENT-2 studies is outlined in Figure 1.
The LUCENT-1 study consisted of patients from North America, Europe, Asia, South America, and Australia, and included patients from 9 Canadian sites. Patients were randomized to receive either blinded mirikizumab 300 mg IV or placebo IV every 4 weeks in a 3:1 ratio. Randomization was stratified by biologic-experienced status (yes or no), baseline corticosteroid use (yes or no), baseline disease activity (MMS of 4 to 6 or 7 to 9), and region (North America, Europe, or other). The LUCENT-1 study data discussed is based on a database lock date of June 11, 2021. Patients who completed the LUCENT-1 trial were given the option to enrol in the LUCENT-2 trial.
LUCENT-2 Study
The LUCENT-2 study (N = 554, the primary analysis) was a 40-week, multicentre, randomized, double-blind, parallel-arm maintenance trial evaluating the safety and efficacy of mirikizumab 200 mg SC in adults with moderately to severely active UC, in the main cohort analysis. The primary objective of the LUCENT-2 trial’s main cohort was to evaluate the ability of mirikizumab to induce clinical remission at the LUCENT-2 study’s week 40 (week 52 of continuous therapy). Major secondary objectives included the ability of mirikizumab to induce alternate clinical remission, durable clinical remission, endoscopic remission, corticosteroid-free remission, HEMR, bowel urgency improvement, and bowel urgency remission, all at week 40. In addition to the main cohort analysis, subcohort and other cohort analyses in the LUCENT-2 study were aimed at determining the safety and efficacy of extended induction with mirikizumab IV in mirikizumab induction nonresponders from the LUCENT-1 and LUCENT-2 trials, and the safety and efficacy of mirikizumab IV reinduction in the loss of response cohort originally randomized in the LUCENT-2 trial to mirikizumab SC. Of note, treatment of the reinduction cohort is not aligned with the recommended dosing in the Health Canada–approved product monograph and therefore will not be discussed further in this report.
Figure 1: Study Design of LUCENT-1 and LUCENT-2 Studies, Including All Cohorts
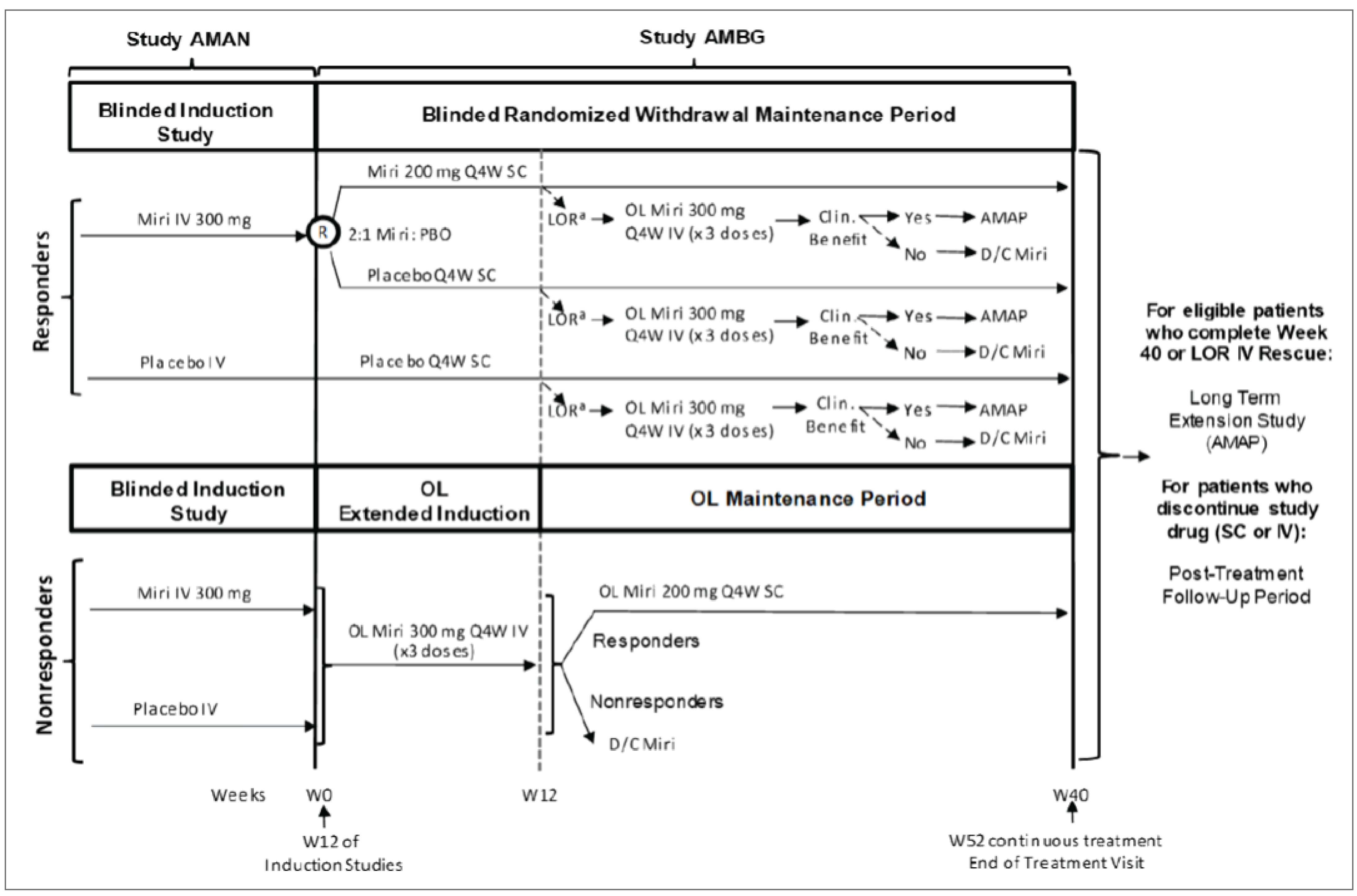
AMAN = LUCENT-1 study; AMAP = LUCENT-3 study; AMBG = LUCENT-2 study; clin. = clinical; D/C = discontinue; LOR = loss of response; miri = mirikizumab; OL = open-label; PBO = placebo; Q4W = every 4 weeks; R = randomization; RB = rectal bleeding; SC = subcutaneous; SF = stool frequency; W0 = week 0; W12 = week 12; W40 = week 40; W52 = week 52.
Note: If patients had clinical benefit with OL rescue therapy with mirikizumab after a loss of response during the treatment period, those patients were eligible to enter the LUCENT-3 study. However, if patients did not experience a clinical benefit after a loss of response during the treatment period, those patients discontinued mirikizumab and entered the posttreatment follow-up period. Similarly, if delayed clinical response was lost during the OL maintenance period after OL extended induction, those patients discontinued mirikizumab and entered the posttreatment follow-up period.
a Loss of response at or after week 12 and up to and including week 28. LOR is defined as a 2-point or greater increase from the maintenance baseline in the combined SF plus RB scores, and a combined SF plus RB score of at least 4 on 2 consecutive visits and confirmed by an endoscopic subscore of 2 or 3.
Source: Clinical Study Report for the LUCENT-2 study.32
The main cohort of the LUCENT-2 study included 544 patients from the LUCENT-1 study who received and whose UC responded to blinded mirikizumab IV (i.e., induction dosing) at 12 weeks. These patients were randomized to receive either blinded mirikizumab 200 mg SC or placebo SC in a 2:1 ratio. Randomization was stratified by biologic-experienced status (yes or no), baseline corticosteroid use (yes or no), region (North America, Europe, or other), and induction remission status (yes or no). Although the LUCENT-2 study main cohort included 544 patients, the total number of patients who entered the LUCENT-2 study was 1,178, including patients from the same 9 Canadian clinics as in the LUCENT-1 trial. Additional cohorts of patients included the following.
Placebo induction responders: Patients who received blinded placebo IV in the LUCENT-1 trial and whose UC responded to placebo were continued on blinded placebo SC every 4 weeks throughout the LUCENT-2 study.
Extended induction: Patients whose UC did not respond to mirikizumab or placebo during the 12-week induction period in the LUCENT-1 study went on to the LUCENT-2 trial to receive extended induction (an additional 12 weeks) with open-label mirikizumab 300 mg IV for 3 doses. Those whose UC responded 12 weeks after the first extended induction dose were referred to as “delayed clinical responders” and received open-label mirikizumab 200 mg SC every 4 weeks (e.g., maintenance dosing) for the remainder of the LUCENT-2 study. Those whose UC did not respond after 24 weeks of induction therapy discontinued mirikizumab.
After completing the LUCENT-2 trial, patients from any cohort who had not yet discontinued mirikizumab were given the option of moving to the long-term extension study (the LUCENT-3 study), or of discontinuing treatment and entering the 16-week posttreatment follow-up period. The LUCENT-2 study data discussed is based on a database lock date of December 6, 2021.
Populations
Inclusion and Exclusion Criteria
Key inclusion criteria for the LUCENT-1 study included being aged 18 years to 80 years, and having an established diagnosis of UC at least 3 months before baseline, endoscopic evidence of UC, a histopathology report that supported UC, an MMS of 4 to 9 with an ES of at least 2 within at least 14 days from baseline, UC extending beyond the rectum, and an inadequate response, a loss of response, or an intolerance to a conventional therapy (corticosteroids and/or immunomodulators), a biologic (approved anti-TNF and/or anti-integrin antibodies), or tofacitinib. Patients were excluded from the LUCENT-1 trial if they had UC limited to the rectum, any other forms of IBD, an immunodeficiency syndrome that would cause UC-like colonic inflammation, extensive colonic resection, stricture or stenosis in the small bowel or colon, toxic megacolon, colonic adenoma not yet removed, dysplasia of colonic mucosa, gastrointestinal cancer, and/or an adequate trial of 3 or more biologic therapies for UC (excluding tofacitinib).
Key inclusion criteria for the LUCENT-2 study included patients who completed the LUCENT-1 trial, received at least 1 study drug dose in the LUCENT-1 study, and participated in all necessary evaluations to assess MMS at the end of the LUCENT-1 study. If patients met any of the LUCENT-1 trial exclusion criteria during or at the end of the LUCENT-1 trial, they would be excluded from the LUCENT-2 trial.
Interventions
In the LUCENT-1 study, patients received either mirikizumab 300 mg IV or placebo IV every 4 weeks for 3 doses, infused over 30 minutes, at the study site (either a hospital or infusion centre) by study personnel. Mirikizumab IV was supplied to study sites in a 15 mL vial at a concentration of 20 mg/mL. Placebo IV was also supplied in 15 mL syringes.
In the LUCENT-2 study main cohort, patients received either mirikizumab 200 mg SC or placebo SC every 4 weeks. SC administration for placebo or mirikizumab was done by study personnel at the study site via 2 sequential injections to different body sites, with each injection containing 100 mg of drug in 1 mL. Self-administration at home was allowed in the case of COVID-19–related restrictions.
In both studies, pharmacists who prepared the study drug and nurses and pharmacists who administered the study drug were all blinded. IV mirikizumab and the placebo were visually indistinguishable from each other. Dose modifications were not permitted in either pivotal trial. During the LUCENT-2 study, if a main cohort patient or placebo induction responder lost response between week 12 and week 28 of the LUCENT-2 trial, they were given open-label mirikizumab 300 mg IV every 4 weeks for 3 doses as a rescue therapy, interchangeably referred to as “reinduction.” Patients were stopped from taking the drug if toxicity developed. Patients who completed the LUCENT-2 trial through week 40 (with an acceptable safety profile) were eligible to participate in the long-term extension study (the LUCENT-3 study).
Study treatment could be permanently discontinued due to AEs or patient decision. Patients who discontinued study treatment early underwent early termination procedures, which included an early termination visit and posttreatment follow-up visits.
Concomitant Medications
Concomitant UC therapies that were permitted in both the LUCENT-1 study and the LUCENT-2 study included oral 5-ASA, oral corticosteroids, azathioprine, 6-mercaptopurine, and methotrexate. Patients who required and used a prohibited medication had to discontinue the study drug. The use of nonprohibited concomitant medication was documented at each patient visit.
In the LUCENT-2 trial, a corticosteroid taper was trialled on all patients in the main cohort (i.e., mirikizumab induction responders) depending on the steroid and formulation they were using, as follows (beginning at baseline):
oral budesonide multimatrix system 9 mg per day — reduce to 9 mg every other day for 2 weeks, then 9 mg every third day for 2 weeks, then discontinue
oral beclomethasone dipropionate 5 mg per day — 5 mg every other day for 4 weeks, then discontinue
all other oral steroids:
if dose is greater than 10 mg per day prednisone or equivalent — reduce daily dose by 5 mg per week until 10 mg per day, then continue tapering daily dose by 2.5 mg per week until 0 mg per day
if dose is less than 10 mg per day prednisone or equivalent — taper daily dose by 2.5 mg per week until 0 mg per day.
If patients could not tolerate the taper without the return of clinical symptoms, the taper was paused or the dose was increased to the original LUCENT-1 study baseline dose. If this occurred, an attempt was made to reinitiate the taper within 2 weeks of taper interruption. The suggested goal was to complete tapering by week 12 of the LUCENT-2 study.
Amendments
One amendment was made to the protocol of the LUCENT-1 and LUCENT-2 trials, referred to as Amendment (a), on September 12, 2019. Major changes included the addition, modification, and deletion of major secondary and other secondary end points, as well as clarifications to the objectives and end points. In addition, updates were made to the schedule of activities (expanding the time window for assessments), the permitted and prohibited medications list, and entry criteria (e.g., criteria excluded patients requiring systemic corticosteroids for non-UC indications except for premedication for study drug infusion or locally). These were minor changes.
A second amendment was made during the LUCENT-1 study on August 21, 2020. It included extending the window for the week-12 endoscopy assessment and study drug administration, permitting the use of local laboratories if a central laboratory was not available, permitting the use of virtual telephone visits, and lastly, allowing a patient missing the week-12 endoscopy to continue to the maintenance study.
Outcomes
A list of efficacy end points assessed in this Clinical Review Report are summarized in Table 6. The summarized end points are based on those included in the sponsor’s Summary of Clinical Evidence30 as well as any end points identified as important to this review according to stakeholders — for example, the clinical expert, clinician groups, or patient groups.
Table 6: Outcomes Summarized From Pivotal Studies and RCT Evidence Identified by the Sponsor
Outcome | LUCENT-1 (induction trial) | LUCENT-2 (maintenance trial) | ||
|---|---|---|---|---|
Time point | Outcome type | Time pointa | Outcome type | |
Clinical response | ||||
Clinical response rate | At week 12 | Key secondaryb | At week 12 (only in the extended induction cohort) | Other secondary |
Clinical remission | ||||
Clinical remission rate | At week 12 | Primaryb | At week 40 | Primaryb |
Alternate clinical remission rate | At week 12 | Key secondaryb | At week 40 | Key secondaryb |
Durable clinical remission rate | NA | At week 40 | Key secondaryb | |
Corticosteroid-free remission rate | NA | At week 40 | Key secondaryb | |
Endoscopic outcomes and disease activity | ||||
Endoscopic remission rate | At week 12 | Key secondaryb | At week 40 | Key secondaryb |
Endoscopic disease activity: Rate of UCEIS score ≤ 1 | At week 12 | Other secondary | NA | |
Symptoms | ||||
Symptomatic remission | At week 12 | Key secondaryb | NA | |
Bowel urgency | ||||
Urgency remission (UNRS score) | NA | At week 40 | Key secondaryb | |
Urgency improvement (UNRS score) | At week 12 | Key secondaryb | At week 40 (vs. start of LUCENT-1 study) | Key secondaryb |
HRQOL | ||||
IBDQ score | At week 12 | Other secondary | At week 40 (vs. start of LUCENT-1 study) | Other secondary |
EQ-5D-5L score | At week 12 | Other secondary | At week 40 (vs. start of LUCENT-1 study) | Other secondary |
SF-36 score (PCS and MCS only) | At week 12 | Other secondary | At week 40 (vs. start of LUCENT-1 study) | Other secondary |
Mucosal healing (histologic and endoscopic outcomes) | ||||
HEMI rate | At week 12 | Key secondaryb | NA for LUCENT-2 study | |
HEMR rate | NA | At week 40 | Key secondaryb | |
Work productivity and impairment | ||||
WPAI:UC score | At week 12 | Other secondary | At week 40 (vs. start of LUCENT-1 study) | Other secondary |
EQ-5D-5L = 5-Level EQ-5D; HEMI = histologic endoscopic mucosal improvement; HEMR = histologic endoscopic mucosal remission; IBDQ = Inflammatory Bowel Disease Questionnaire; MCS = mental component summary; NA = not applicable; PCS = physical component summary; RCT = randomized controlled trial; SF-36 = Short Form (36) Health Survey; UCEIS = Ulcerative Colitis Endoscopic Index of Severity; UNRS = Urgency Numeric Rating Scale; vs. = versus; WPAI:UC = Work Productivity and Activity Impairment Questionnaire: Ulcerative Colitis.
Note: Details in Table 6 have been taken from the sponsor’s Summary of Clinical Evidence.20,30
aThe LUCENT-2 study time points mentioned assume that week 0 is the start of the LUCENT-2 trial, unless otherwise indicated. Week 40 of the LUCENT-2 trial represents 52 weeks of continuous therapy from the LUCENT-1 study’s baseline.
bStatistical testing for primary and key secondary end points was adjusted for multiple comparisons via a prespecified graphical testing approach.
Sources: Clinical Study Reports for the LUCENT-1 and LUCENT-2 studies.31,32
Efficacy Outcomes
A description of the efficacy outcome measures and their measurement properties that were used in both the LUCENT-1 study and the LUCENT-2 study are presented in Table 7.
Many outcomes in the LUCENT-1 and LUCENT-2 trials were defined by subscores of the Mayo scoring system. The pivotal trials used the MMS as opposed to the overall Mayo Score. Scoring of the MMS subscores are further detailed in Table 7.
Clinical Response
Clinical response was measured in the LUCENT-1 trial at week 12 and in the extended induction cohort of the LUCENT-2 trial at week 12. Clinical response was based on the MMS and defined as:
a decrease in the MMS total score (the sum of the SF subscore, RB subscore, and ES) of at least 2 points and at least a 30% decrease from baseline
a decrease of 1 point or less in the RB subscore from baseline or an RB score of 0 or 1.
The SF and RB subscores were recorded by patients in an electronic diary daily during the treatment period. Patient diary adherence reviews were conducted at each treatment visit. For SF, a stool was defined as a trip to the toilet when the patient had either a bowel movement, passed blood alone, passed blood and mucus, or passed mucus only. The total number of stools passed in a 24-hour period was recorded by the patient in a daily electronic diary.
For RB, patients recorded their subscore daily and an average of the subscores was used to calculate the MMS score and RB subscore at the end-of-treatment visit.
The ES is a physician-reported measure that reports the worst appearance of the mucosa on flexible sigmoidoscopy or colonoscopy on a 4-point scale. Endoscopy was used to determine the Mayo ES at screening and week 12 (or end-of-treatment visit) in the LUCENT-1 study and week 40 in the LUCENT-2 study. A flexible sigmoidoscopy or colonoscopy was performed on all patients at each time point by a licensed physician. Site and blinded central reading of endoscopies were used to determine ES. Disagreement between the site and central read were adjudicated by an additional blinded central reader. Scoring of the MMS subscores are further detailed in Table 7.
Clinical Remission
Clinical Remission
Clinical remission was the primary end point in the LUCENT-1 study at week 12 and the LUCENT-2 study at week 40. Clinical remission was based on the MMS and defined as:
SF subscore equals 0, or SF equals 1 with at least a 1-point decrease from baseline
RB subscore equals 0
ES equals 0 or 1 (excluding friability).
The MMS subscores were calculated using the same procedures described for the clinical response.
Alternate Clinical Remission
Alternate clinical remission was measured in the LUCENT-1 study at week 12 and the LUCENT-2 study at week 40. Alternate clinical remission was defined as:
SF subscore equals 0, or SF equals 1
RB subscore equals 0
ES equals 0 or 1 (excluding friability).
The MMS subscores were calculated using the same procedures described for the clinical response.
Durable Clinical Remission
Durable clinical remission at week 40 was assessed in the LUCENT-2 study only. Durable clinical remission was attained if patients who experienced clinical remission at week 12 in the LUCENT-1 study had ongoing remission at week 40 in the LUCENT-2 study (i.e., 52 weeks of continuous clinical remission).
Corticosteroid-Free Remission
Corticosteroid-free remission at week 40 was assessed in the LUCENT-2 study only. Corticosteroid-free remission was defined as:
clinical remission at week 40
symptomatic remission at week 28
no corticosteroid use for at least 12 weeks before week 40.
Endoscopic Outcomes
Endoscopic Remission
Endoscopic remission was assessed in the LUCENT-1 trial at week 12 and the LUCENT-2 trial at week 40. Endoscopic remission was defined as an ES of 0 or 1 (excluding friability) on the MMS at week 12 and week 40 for the LUCENT-1 trial and the LUCENT-2 trial, respectively. The endoscopies were performed by a licensed physician qualified to perform colonoscopies. The site endoscopist determined the Mayo ES at each endoscopy and recorded the result in the electronic case report form. The ES was calculated using the same procedures described for the clinical response.
Endoscopic Disease Activity
Endoscopic disease activity, as measured by the UCEIS score, was assessed in the LUCENT-1 study only. The UCEIS is a physician-reported measure of the endoscopic disease activity of UC on a flexible sigmoidoscopy or colonoscopy. The UCEIS consists of the following 3 descriptors calculated as a simple sum: vascular pattern (scored 0 to 2), bleeding (scored 0 to 3), and erosions and ulcers (scored 0 to 3).56,57 The 3 descriptors are summed, resulting in a range of UCEIS scores of 0 to 8, with higher scores indicating worse outcomes (i.e., 0 = remission; 8 = severe).57 Only blinded central reading of endoscopies was used to determine the UCEIS score for each endoscopy. The proportion of patients with a UCEIS score less than or equal to 1 at week 12 was reported.
Symptoms
Symptomatic Remission
The symptomatic remission rate at week 12 was assessed in the LUCENT-1 study only. The symptomatic remission rate was defined as:
SF equals 0, or SF equals 1 with at least a 1-point decrease from baseline
RB equals 0.
The MMS subscores were calculated using the same procedures described for the clinical response.
Bowel Urgency Remission (in Patients With UNRS Score of 3 or More at Baseline)
Bowel urgency remission at week 40, as measured by the UNRS, was assessed in the LUCENT-2 study only. Bowel urgency remission was defined as a UNRS score of 0 or 1 among patients who experienced clinical response and who had a baseline UNRS score of at least 3 points. The UNRS is a patient-reported, validated, 11-point numeric rating scale aimed at measuring the degree of bowel movement urgency.17 Scores range from 0 (no urgency) to 10 (worst possible urgency). Analyses of patients in the LUCENT-1 study suggested that a within-patient 3-point improvement or decrease in the UNRS score from baseline would be considered clinically important.58 Responses were collected using a patient electronic diary daily. Refer to Table 7 for further details on the UNRS.
Bowel Urgency Improvement
Bowel urgency improvement as measured by UNRS was assessed in the LUCENT-1 study at week 12 and the LUCENT-2 study at week 40. This end point was defined as the change in UNRS score from induction baseline to the end of study treatment.
Health-Related Quality of Life
Inflammatory Bowel Disease Questionnaire
The IBDQ59 was used to assess disease-specific HRQoL in the LUCENT-1 trial at week 12 and the LUCENT-2 trial at week 40. IBDQ questionnaires were self-administered, with responses recorded electronically using a tablet device at appropriate visits. The IBDQ consists of a 32-item list subdivided into 4 dimensions: systemic symptoms, bowel symptoms, emotional function, and social function. Total scores range from 32 to 224, with a higher score indicating a better HRQoL. The IBDQ has been consistently shown to have good internal consistency and test-retest reliability, as well as showing responsiveness to change in IBD.18-20 Available studies have suggested that an improvement of 30 points from baseline or an improvement of at least 15 points or greater above placebo may constitute an MID.21-24
5-Level EQ-5D
The EQ-5D-5L was used as a generic HRQoL measure in the LUCENT-1 study at week 12 and the LUCENT-2 study at week 40. The EQ-5D-5L is a widely used patient-reported measure. Its 5 dimensions include mobility, self-care, usual activities, pain/discomfort, and anxiety/depression, with each dimension including 5 levels ranging from “no problems” to “extreme problems.”60 A 1-digit score is assigned for each dimension, which is then combined into a 5-digit score code to represent the total score on the EQ-5D-5L. This 5-digit score code is then converted to a utility from 0 to 1, where 1 represents perfect health and 0 represents death. The second, complementary piece of the EQ-5D-5L is a patient-reported visual analogue scale (the EQ VAS); with the EQ VAS, the patient ranks their overall health on a continuum, converted to a score from 0 to 100, where a higher number suggests better self-reported health.60 Though the EQ-5D-5L is a generic scale, it has been validated specifically for the IBD population, which includes the UC population.25,61,62 The EQ-5D-5L index score (i.e., the utility) was shown to have an MID ranging from 0.050 to 0.076 in patients with IBD.25 The EQ VAS MID was found to be a 10.9-point change from baseline in patients with IBD.25 According to the evidence, the type of IBD (Crohn disease or UC) did not have an impact on the MID threshold.25
Responses were completed at baseline and week 12 (or the end-of-treatment visit) in the LUCENT-1 study and week 40 in the LUCENT-2 study.
Table 7: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
Modified Mayo Score | The Modified Mayo Score is the modified version of the Mayo Score. In this version, the definition of ES of 1 no longer includes mucosal friability and the PGA is excluded. The components of the Modified Mayo Score include:
Scale components are scored on a scale from 0 to 3, with the score of 0 indicative of normal and a higher score indicative of more severe symptoms. The Modified Mayo Score is a sum of the Mayo SF subscore, the RB subscore, and the ES, giving a maximum score of 9. | Validity: In a cross-sectional survey of 2,608 patients with UC and their treating gastroenterologist, increases in the Modified Mayo Score were associated with increased odds of adverse outcomes, including a current flare (OR = 1.52; SE = 0.10), a higher number of flares in the past year (OR = 1.17; SE = 0.03), deterioration in clinical status (OR = 1.48; SE = 0.10), and patient-reported overall WPAI (score = 6.94; SE = 0.888).65 A 1-point increase in the Modified Mayo Score was associated with a 0.02-unit decrease in the EQ-5D-5L and a 2.73-point decrease in the SIBDQ, suggesting a change in score of > 4 might be associated with a clinically meaningful reduction in HRQoL.65 Reliability and responsiveness: No studies of the reliability and responsiveness of the Modified Mayo Score were identified. | Evidence of an MID for the Modified Mayo Score in patients with UC was not identified. |
Geboes Score | The Geboes Score is a histologic index for assessing disease severity and/or activity in UC.66,67 It consists of 6 grades — with 4 subgrades each — that are meant to be progressive. Grading is performed on hematoxylin-eosin–stained sections from biopsies obtained in the colonic mucosa. The grades are defined as follows:
Subgrades are assessed based on the worst area of the biopsy. The higher the grade or subgrade, the greater the inflammation. | Validity: Criterion validity of the Geboes Score was supported by a strong correlation between the Geboes Score and a global disease activity, assessed using a VAS (r = 0.66; 95% CI, 0.57 to 0.72).68 Construct validity was supported by strong correlations between the Geboes Score and the Mayo ES, endoscopic activity index, and clinical activity index (Spearman rank correlation range = 0.54 to 0.80).69,70 Reliability and responsiveness: The Geboes Score was found to have substantial to almost perfect intrarater agreement (ICC range = 0.77 to 0.84) and moderate interrater agreement (ICC range = 0.51 to 0.60).68 The Geboes Score was found to be responsive to treatment as evaluated using an analysis of SES and GRS. The responsiveness to change was moderate based on treatment assignment (SES = 1.87; 95% CI, 1.54 to 2.20; GRS = 1.23; 95% CI, 0.97 to 1.50) and a Mayo subscore of at least 2 points (SES = 1.05; 95% CI, 0.78 to 1.31; GRS = 0.84; 95% CI, 0.59 to 1.09).71 Histological activity, defined as a Geboes Score ≥ 3.1, was found to be an independent risk factor for clinical relapse in patients with UC (OR = 4.31; 95% CI, 1.52 to 12.21; P = 0.006).72 | Evidence of an MID for the Geboes Scale was not identified. Histological healing: Histological healing was empirically defined in specimens of endoscopically uninflamed tissue as the average Geboes Score below 2.66 |
SF-36 | The SF-36 is a generic self-reported HRQoL questionnaire consisting of 8 domains:
The SF-36 also provides 2 component summaries: the PCS and the MCS, which are scores created by aggregating the 8 domains. The SF-36 PCS and MCS and individual domains are each measured on a scale of 0 to 100, with an increase in score indicating improvement in health status.63 | Validity: Construct validity in UC was demonstrated through moderate-to-strong correlations (r > 0.4) between the 8 subscales of the SF-36 and the corresponding domains of 5 patient-reported clinical constructs. The scale showed evidence of discriminant validity (for disease activity and symptom status).64 Reliability and responsiveness: The SF-36 was found to have good internal consistency for all 8 subscales (Cronbach alpha > 0.7) and good test-retest assessments for 6 of the 8 subscales (ICC > 0.7).64 The scale and its subscores were found to be responsive to treatment-related changes to disease activity and symptoms.64 | For the PCS and MCS summaries, an MID of 3 points to 5 points was identified.64 An absolute score increase of 2 points to 4 points was determined as the MID to capture improvement in disease activity and symptoms.64 |
EQ-5D-5L | The EQ-5D-5L is a generic preference-based HRQoL instrument comprising 5 dimensions: mobility, self-care, usual activities, pain/discomfort, and anxiety and/or depression. Each domain has 5 levels: no problem, slight problems, moderate problems, severe problems, and extreme problems. The EQ-5D-5L also includes a 20 cm EQ VAS that has end points labelled 0 and 100, with respective anchors of “worst imaginable health state” and “best imaginable health state.” | Validity: Construct validity was supported by the correlation of the EQ-5D-5L index score and EQ VAS scores to disease activity indices (Spearman rank r = –0.67 to –0.75; P < 0.0001) and the difference between active disease and remission groups.25 Reliability and responsiveness: Test-retest reliability was generally moderate for all domains of the EQ-5D-5L (kappa = 0.41 to 0.58), except for the anxiety and/or depression domain (kappa = 0.28).62 Based on a standardized response means, the EQ VAS was observed to be more responsive to deterioration in health than to improvement and more responsive than the index score, while the index scores were most responsive to deterioration in health in patients in remission and to improved health in patients with active disease.25 | An EQ VAS score of 10.9 and EQ-5D-5L index score of 0.5 to 0.076 in patients with IBD that includes UC25 (the MID did not change depending on the type of IBD) |
IBDQ | The IBDQ is a disease-specific questionnaire used to assess disease-specific HRQoL in patients with IBDQ.59 The IBDQ is a 32-item Likert-based questionnaire divided into 4 dimensions:
Responses are graded on a scale from 1 (worst situation) to 7 (best situation). Total IBDQ scores range between 32 and 224, with higher scores representing better HRQoL. Scores ranging from 170 to 190 are indicative of remission. | Validity: The emotional function dimension of the IBDQ was found to be strongly correlated with the emotional function dimension of the Rand questionnaire (r = 0.76; P < 0.001), the systemic symptoms dimension of the IBDQ was weakly correlated to change in the disease activity index (r = 0.036; P = 0.442), and patients’ global rating of change in emotional function was moderately correlated to the emotional function dimension (r = 0.52; P < 0.001) and the bowel symptom dimension (r = 0.42; P = 0.003) of the IBDQ.59 The IBDQ was found to detect changes in the social and emotional state of patients.18 Reliability and responsiveness: The IBDQ was shown to be highly reliable through evaluation of internal consistency (Cronbach alpha = 0.7) and test-retest assessment (ICC = 0.9 to 0.99 or Pearson’s r ≥ 0.8). The IBDQ was also shown to be responsive to change in patients with IBD (P < 0.05).19,20 | An established MID for the IBDQ in patients with UC was not identified. While some suggest that an increase between 16 points and 32 points may be considered a clinically relevant improvement in HRQoL for patients with Crohn disease and UC, evidence from clinical trials suggests that a change of more than 30 points is associated with clinical benefits and an improvement of 15 points or greater above placebo is required.21-24 |
UNRS | The UNRS is an instrument used to assess patient-reported severity of bowel urgency in adults with UC with a 24-hour recall period.17 Patients self-report the immediacy of their UC symptoms on an 11-point numeric rating scale ranging from 0 (no urgency) to 10 (worst possible urgency). Higher scores are indicative of worse urgency severity. | Validity: UNRS scores were strongly correlated to average PGR-S scores (r = 0.8). The UNRS was found to be moderately correlated to the average number of stools (r = 0.5), suggesting bowel urgency being distinct from stool frequency.17 Reliability: Test-retest reliability of the UNRS using bootstrapped samples was found to be high (ICC = 0.877; 95% CI, 0.770 to 0.947).17 | An analysis of patients with UC in the LUCENT-1 study indicated that a decrease of 3 or more points indicates meaningful improvement in bowel urgency.28 |
WPAI:UC | The WPAI:UC is a self-administered 6-item questionnaire with a 7-day recall period used to measure work impairment.26 The questionnaire consists of 6 items including:
The items are grouped into 4 domains:
Scores from all 4 domains are expressed as percentages of impairment (0% to 100%), with higher values indicating greater impairment due to the health problem and less productivity.64 | Validity: Convergent validity was demonstrated for all WPAI domains between the SIBDQ bowel symptoms (Spearman rank order coefficient = 0.47 to 0.68) and SF-12, version 2, bodily pain (0.52 to 0.55) subscores, and between the WPAI and measures of disease activity (median = 0.45).27 Known-group validity, a form of construct validity, demonstrated that patients with worse health outcomes scored worse on the WPAI than patients with better health outcomes, based on Partial Mayo Score, SCCAI, UCDAI, and FACIT-F disease severity measures.27 Reliability and responsiveness: Established reliability of the WPAI:UC was not identified. Responsiveness to effective treatment was demonstrated with an approximate 20% decrease in presenteeism, OWI, and activity impairment, and an 8% decrease in absenteeism.27 Patients with active UC disease who experienced remission at week 8 reported a 25% to 30% decrease in presenteeism, OWI, and activity impairment, and a 9% decrease in absenteeism. | Evidence of an MID for WPAI:UC was not identified. |
CI = confidence interval; EQ VAS = EQ visual analogue scale; EQ-5D-5L = 5-Level EQ-5D; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; GRS = Guyatt’s responsiveness statistic; HRQoL = health-related quality of life; IBD = inflammatory bowel disease; IBDQ = Inflammatory Bowel Disease Questionnaire; ICC = intraclass correlation; MCS = mental component summary; MID = minimal important difference; OR = odds ratio; OWI = overall work impairment; PCS = physical component summary; PGA = Physician’s Global Assessment; PGR-S = Patient Global Rating of Severity; RB = rectal bleeding; SCCAI = Simple Clinical Colitis Activity Index; SE = standard error; SES = standardized effect size; SF = stool frequency; SF-12 = Short Form (12) Health Survey; SF-36 = Short Form (36) Health Survey; SIBDQ = Short Inflammatory Bowel Disease Questionnaire; UC = ulcerative colitis; UCDAI = Ulcerative Colitis Disease Activity Index; UNRS = Urgency Numeric Rating Scale; VAS = visual analogue scale; WPAI = Work Productivity and Activity Impairment Questionnaire; WPAI:UC = Work Productivity and Activity Impairment Questionnaire: Ulcerative Colitis.
Short Form (36) Health Survey
The SF-36 was used as another generic HRQoL measure in the LUCENT-1 study at week 12 and the LUCENT-2 study at week 40. The SF-36 is a non–disease-specific, self-reported questionnaire.63 The 8 domains evaluated by the SF-36 are physical function, role limitations due to physical health, role limitations due to emotional health, bodily pain, general health, vitality, social functioning, and mental health. The output is 2 separate summary scores — the physical and mental component summaries, measured on a scale from 0 to 100, with a higher score indicative of a better health state.63 Using data from 43 studies, a systematic review indicated that SF-36 has acceptable validity and reliability (for most scales) for UC.64 For the PCS and MCS summaries, an MID of 3 points to 5 points was identified.64 An absolute score increase of 2 points to 4 points from baseline was determined as the MID to capture improvement in disease activity and symptoms.64
Mucosal Healing
Histologic Endoscopic Mucosal Improvement
HEMI at week 12 was assessed in the LUCENT-1 trial only. HEMI was defined using the Geboes scoring system (refer to Table 7 for details) with neutrophil infiltration in fewer than 5% of crypts; no crypt destruction; no erosions, ulcerations, or granulation tissue; and endoscopic remission, defined as ES equals 0 or 1 (excluding friability) on the MMS. Biopsies were obtained at each endoscopy to support assessment of the histopathology end points and scoring was performed by a blinded central reader. The ES was calculated using the same procedures described for the clinical response.
Histologic Endoscopic Mucosal Remission
HEMR with resolution of mucosal neutrophils at week 40 was assessed in the LUCENT-2 study only. HEMR was defined as achieving both endoscopic remission and histologic remission. Endoscopic remission was defined as an ES of 0 or 1 (excluding friability). Histologic remission was characterized as the resolution of mucosal neutrophils, defined using the Geboes scoring system with subscores of 0 for grades:
2b (lamina propria neutrophils)
3 (neutrophils in the epithelium)
4 (crypt destruction)
5 (erosion or ulceration).
Work Impairment
Work Productivity and Activity Impairment Questionnaire: Ulcerative Colitis
Work impairment, as evaluated by the WPAI:UC, was assessed in the LUCENT-1 trial at week 12 and the LUCENT-2 trial at week 40. The WPAI:UC is a self-administered, disease-specific scale aimed at measuring the level of work impairment due to UC.26 WPAI:UC considers the 4 domains of absenteeism, presenteeism, overall work performance, and nonwork activities.27 The 4 domains comprise a total of 6 items. The final scores for each domain are a percentage of total impairment, ranging from 0 to 100%, with a higher number indicating greater impairment in that domain. The WPAI:UC is a valid and responsive instrument for use in the assessment of UC.27 An established MID for WPAI:UC was not identified in this patient population.
Harms Outcomes
AEs were predefined and reported in both studies, including SAEs, suspected unexpected serious adverse reactions, AESIs, withdrawal due to AEs, and deaths due to AEs. AESIs for the LUCENT-1 and LUCENT-2 studies were defined as, but not limited to, opportunistic infections, hypersensitivity events, injection site reactions, cerebrocardiovascular events, malignancies, depression and/or suicidal ideation/behaviour, and hepatic AEs.
Safety data were captured during the treatment periods — during a 16-week follow-up after the LUCENT-1 study (for those who did not go on to the LUCENT-2 trial) and during a 16-week follow-up after the LUCENT-2 trial (for those who did not go on to the LUCENT-3 trial). Given that most patients from the LUCENT-1 study went on to the LUCENT-2 study and most patients from the LUCENT-2 study transitioned to the LUCENT-3 trial, few patients completed the 16-week posttreatment follow-up periods. As such, safety data for mirikizumab from the LUCENT-1 and LUCENT-2 trials is mostly limited to 52 weeks of therapy.
Statistical Analysis
The statistical analysis of efficacy end points in the pivotal trials assessed in this Clinical Review Report are summarized in Table 8.
Sample Size and Power Calculation
For the LUCENT-1 study, a sample size of 1,160 patients was deemed appropriate by the sponsor based on the following power calculation assumptions:
about 50% of randomized patients will be considered conventional-failed and 50% of patients will be considered biologic-failed
at week 12, the predicted clinical remission rate is expected to be 23% for mirikizumab and 7.8% for placebo
for patients who are considered conventional-failed, this is expected to be 30% for mirikizumab versus 12% for placebo
for patients who are considered biologic-failed, this is expected to be 16% for mirikizumab versus 3.5% for placebo
desired power was greater than 90% with a chi-square test with a 2-sided significance level of 0.00125 for the end point of clinical remission at 12 weeks.
For the LUCENT-2 study, a sample size of 470 patients in the main cohort was expected to satisfy the following power calculation assumptions:
at week 40, clinical remission rates for patients randomized to mirikizumab and placebo would be 47% and 27%, respectively
desired power was greater than 95% with a chi-square test with a 2-sided significance level of 0.05 for the end point of clinical remission at 40 weeks
desired power for 2 key secondary end points, endoscopic remission and corticosteroid-free remission, at week 40 was greater than 80% with a 2-sided significance level of 0.05.
In addition, in the LUCENT-2 study’s statistical analysis plan, the following assumptions about the LUCENT-1 study were made:
the LUCENT-1 study would randomize at approximately 1,160 patients, 90% of whom (1,044 patients) would complete the LUCENT-1 study and enrol in the LUCENT-2 study (i.e., there would be a 10% dropout rate from the LUCENT-1 trial to the LUCENT-2 trial)
50% of patients in the LUCENT-1 study would be biologic-experienced
the LUCENT-1 study would have a clinical response rate of 60% and clinical remission rate of 23% for patients randomized to mirikizumab
75% of LUCENT-1 study patients would receive mirikizumab (as per the 3:1 randomization ratio).
Statistical Test or Model
In both the LUCENT-1 trial and the LUCENT-2 trial, all primary analyses of efficacy end points were based on the mITT population, described in Table 9, while some sensitivity analyses may have used other analysis populations, as outlined in Table 8.
For the primary end point in the LUCENT-1 and LUCENT-2 trials, as well as other binary efficacy and health outcome end points, the Cochran-Mantel-Haenszel test was used. For continuous efficacy and health outcome end points, comparisons were made using mixed-effects model of repeated measures analysis. An analysis of covariance was used for continuous efficacy and health outcome end points with a single postbaseline time point. For each end point, adjustment factors including specific covariates as well as sensitivity analyses are listed in Table 8.
Sensitivity Analyses
In the primary and sensitivity analyses, various techniques were used to handle dropouts or missing data. Nonresponder imputation was used for missing data for analyses of binary efficacy and health outcome variables, whereby patients with missing clinical efficacy data at a time point of interest were deemed to be nonresponders. Some sensitivity analyses in Table 8 discuss an “alternative estimand.” This is a conservative estimate that considers each of the following to be a treatment failure: failing to meet protocol-defined primary end point criteria, missing data to calculate primary end point criteria, initiating the use of systemic corticosteroids, increasing systemic corticosteroid dose beyond baseline, switching between corticosteroid therapies, or having a UC surgery.
End points with continuous variables that used the mixed model of repeated measures to make comparisons assumed that missing data were missing at random. For patients who discontinued the study drug, the modified baseline observation carried forward approach may have been used. With modified baseline observation carried forward, those who discontinue due to AEs had their baseline observation carried forward for all missing observations after discontinuation. For discontinuation not due to AEs, the last nonmissing, postbaseline observation before discontinuation was carried forward. Some sensitivity analyses applied a modified nonresponder imputation (mNRI) method for dealing with missing data, which included the imputation of data for patients who discontinued treatment due to COVID-19 reasons, were lost to follow-up, or had a protocol deviation. Patients with sporadically missing data or who had had an endoscopy outside of day 71 to day 113 for the LUCENT-1 study or day 267 to day 337 for the LUCENT-2 study had data imputed. With mNRI, patients who discontinued for other reasons, including AEs, were considered nonresponders.
A final method of dealing with missing data was tipping point analysis, conducted as a sensitivity analysis on primary end points in the LUCENT-1 and LUCENT-2 studies. The tipping point analysis assumed the most extreme conservative case, imputing the worst possible outcomes for missing data in patients randomized to mirikizumab, but the best possible outcomes for patients with missing data randomized to placebo. Table 8 highlights where each of these missing data techniques was used.
Multiplicity Adjustments
To control for multiplicity for the primary and key secondary end points, a prespecified graphical multiple testing approach was used, controlling the type I error rate at a 2-sided alpha of 0.00125 for the LUCENT-1 trial and 0.05 for the LUCENT-2 trial. Figure 2 and Figure 3 show the graphical testing approach to control type I error rate in the LUCENT-1 study and the LUCENT-2 study, respectively.
Subgroup Analyses
Subgroup analyses in both the LUCENT-1 study and the LUCENT-2 study were prespecified in the statistical analysis plans and were conducted for primary and key secondary efficacy end points. No analysis for the comparability of baseline covariates across subgroups was performed. Subgroup analyses did not control for multiplicity, except for those specified as key secondary end points (i.e., clinical response in the biologic-failed population at week 12 in the LUCENT-1 trial). In the LUCENT-2 trial, subgroup analyses were conducted for the same factors as the LUCENT-1 trial as well as by country, number of failed biologics or tofacitinib (≤ 2 or > 2), and prior failure of conventional therapies but not biologics or tofacitinib (yes or no).
The following subgroups, planned a priori in the statistical analyses plans, aligned with the subgroups deemed relevant for this CADTH review: patients treated previously with conventional therapy (i.e., steroids or immunomodulators), patients treated previously with advanced UC therapy (i.e., biologics, TNF, or JAK inhibitors), baseline MMS (moderate versus severe), and disease extension (extensive colitis versus nonextensive colitis). Only the subgroups identified as relevant are reported here.
Figure 2: Graphical Testing Approach to Control Type I Error Rate in LUCENT-1 Study
Source: Clinical Study Report for the LUCENT-1 study.31
Figure 3: Graphical Testing Approach to Control Type I Error Rate in LUCENT-2 Study
AMAN = LUCENT-1 study.
Source: Clinical Study Report for the LUCENT-2 study.32
Table 8: Statistical Analysis of Efficacy End Points
End point | Statistical model | Analysis population | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|---|
LUCENT-1 study (induction trial) | |||||
Clinical remission rate | CMH chi-square test | mITT | None | NRI; mBOCF for those who discontinue |
|
| CMH chi-square test | mITT | None | NRI; mBOCF for those who discontinue |
|
HRQoL:
| ANCOVA (for change from baseline) CMH (for IBDQ response and remission and SF-36) | mITT | None | NRI; mBOCF for those who discontinue | None |
UNRS score | MMRM | mITT |
| MAR assumption; mBOCF for those who discontinue |
|
HEMI rate | CMH chi-square test | mITT | None | NRI; mBOCF for those who discontinue | None |
WPAI:UC | ANCOVA | mITT in patients with baseline employment status of yes | None | NRI; mBOCF for those who discontinue | None |
LUCENT-2 study (maintenance trial) | |||||
Clinical remission rate | CMH chi-square test | mITT | None | NRI; mBOCF for those who discontinue |
|
| CMH chi-square test | mITT | None | NRI; mBOCF for those who discontinue |
|
HRQoL:
| ANCOVA (for change from baseline) CMH (for IBDQ response and remission and SF-36) | mITT | None | NRI; mBOCF for those who discontinue | None |
UNRS score | MMRM | mITT |
| MAR assumption; mBOCF for those who discontinue |
|
WPAI:UC | ANCOVA | mITT in patients with baseline employment status of yes | None | NRI; mBOCF for those who discontinue | None |
ANCOVA = analysis of covariance; CMH = Cochran-Mantel-Haenszel; EQ-5D-5L = 5-Level EQ-5D; HEMI = histologic endoscopic mucosal improvement; HEMR = histologic endoscopic mucosal remission; HRQoL = health-related quality of life; IBDQ = Inflammatory Bowel Disease Questionnaire; ITT = intention-to-treat; MAR = missing at random; mBOCF = modified baseline observation carried forward; MCS = mental component summary; mITT = modified intention-to-treat; MMRM = mixed model of repeated measures; MMS = Modified Mayo Score; mNRI = modified nonresponder imputation; NR = not reported; NRI = nonresponder imputation; PCS = physical component summary; PP = per-protocol; SF-36 = Short Form (36) Health Survey; UNRS = Urgency Numeric Rating Scale; vs. = versus; WPAI:UC = Work Productivity and Activity Impairment Questionnaire: Ulcerative Colitis.
Note: Details in Table 8 have been taken from the sponsor’s Summary of Clinical Evidence.20,30
Source Clinical Study Reports for the LUCENT-1 and LUCENT-2 studies.31,32
Analysis Populations
Analysis populations of the LUCENT-1 and LUCENT-2 studies are summarized in Table 9.
Note that the safety populations in the LUCENT-1 and LUCENT-2 studies refer to those who were randomized and received at least 1 dose of any study treatment. For the LUCENT-1 trial, this includes all patients randomized to mirikizumab IV or placebo, while in the LUCENT-2 trial, it refers to main cohort patients randomized to mirikizumab SC or placebo.
Study | Population | Definition | Application |
|---|---|---|---|
LUCENT-1 (induction trial) and LUCENT-2 (maintenance trial) | Screening population | All patients who signed informed consent | Used for disposition analysis |
mITT population | All patients randomized who received at least 1 dose of any study treatment, regardless of whether it was the treatment they were randomized to or not | Used for efficacy and health outcome analyses | |
Safety population | Used for safety-related analyses | ||
ITT population | All patients randomized; patients were analyzed according to the treatment group they were assigned | Used for sensitivity analyses for primary and key secondary end points | |
PP population | All mITT patients who had no significant protocol deviations, were not deemed noncompliant to treatment, and whose investigator site did not have significant GCP deviations requiring reporting to regulatory agencies |
GCP = good clinical practice; ITT = intention-to-treat; mITT = modified intention-to-treat; PP = per-protocol.
Note: Details in Table 9 have been taken from the sponsor’s Summary of Clinical Evidence.20,30
Sources: Clinical Study Reports for the LUCENT-1 and LUCENT-2 studies.31,32
Results
Patient Disposition
A summary of patient disposition in the LUCENT-1 study and the LUCENT-2 study is in Table 10. In addition, the patient disposition of the mirikizumab group in the induction nonresponders (extended induction cohort) from the LUCENT-2 study is presented in Table 11.
LUCENT-1: Induction Trial
Of the 2,079 patients screened in the LUCENT-1 trial, 730 (35%) patients were screen failures and 33 (1.6%) patients were patient withdrawals. In the LUCENT-1 trial, 959 patients were randomized to the mirikizumab arm and 322 patients were randomized to the placebo arm. Study treatment discontinuation occurred in 39 (4.1%) patients in the mirikizumab arm and 37 (11.5%) patients in the placebo arm. The most common reason for treatment discontinuation was AEs in both the mirikizumab arm (1.6%) and the placebo arm (7.1%).
LUCENT-2: Maintenance Trial
From the LUCENT-1 trial, 1,178 patients entered the LUCENT-2 trial. Of the responders in the mirikizumab and placebo groups from the LUCENT-1 study, 581 patients were randomized to the primary efficacy analysis cohort in the LUCENT-2 study. Of these patients, 389 patients were randomized to the mirikizumab arm and 192 patients were randomized to the placebo arm in the LUCENT-2 trial. Study treatment discontinuation occurred in 42 (10.8%) patients in the mirikizumab arm and 73 (38.0%) patients in the placebo arm. The most common reason for treatment discontinuation was loss of response (i.e., the patient was switched to open-label rescue IV mirikizumab) in both the mirikizumab arm (4.9%) and the placebo arm (21.9%). Of the patients receiving rescue therapy, 4 (1.0%) patients in the mirikizumab arm and 4 (2.1%) patients in the placebo arm discontinued rescue mirikizumab.
Of the mirikizumab nonresponders from the LUCENT-1 study in the ITT population, 313 patients (272 patients according to the mITT population) entered the open-label extended induction period; of these, 171 (54.4%) patients (146 [53.7%] patients in the mITT population) responded and entered the maintenance period with open-label 200 mg mirikizumab SC. Of the placebo nonresponders, ||| |||||||| entered the open-label extended induction period; of these patients ||| ||||||| responded and entered the maintenance period with open-label 200 mg mirikizumab SC.
In the LUCENT-2 trial, 135 patients from the placebo induction responders in the LUCENT-1 study continued placebo SC in the extended induction period. Of these, 90 (66.7%) patients completed blinded placebo SC maintenance.
Table 10: Summary of Patient Disposition From Pivotal Studies Submitted by the Sponsor
Patient disposition | LUCENT-1 (induction trial) | LUCENT-2 (maintenance trial) | ||
|---|---|---|---|---|
Mirikizumab | Placebo | Mirikizumab | Placebo | |
Screened, N | 2,079 | 1,178 | ||
Discontinued before randomization, N (%) | 798 (38.4) | 1 (< 1.0) | ||
Screen failure | 730 (NR) | 0 | ||
Withdrawal by patient | 33 (NR) | 1 (NR) | ||
Other | 19 (NR) | 0 | ||
Adverse event | 12 (NR) | 0 | ||
Physician decision | 4 (NR) | 0 | ||
Randomized, N | 959 | 322 | 389 | 192 |
Discontinued from study, N (%) | 39 (4.1) | 37 (11.5) | 42 (10.8) | 73 (38.0) |
Adverse event | 15 (1.6) | 23 (7.1) | 6 (1.5) | 16 (8.3) |
Withdrawal by patient | 5 (0.5) | 8 (2.5) | 8 (2.1) | 7 (3.6) |
Lack of efficacy | 5 (0.5) | 5 (1.6) | 6 (1.5) | 4 (4.2) |
Protocol deviation | 5 (0.5) | 1 (0.3) | 1 (0.3) | 0 |
Loss to follow-up | 3 (0.3) | 0 | 1 (0.3) | 0 |
COVID-19–related disruptions | 2 (0.2) | 0 | 0 | 0 |
Site terminated by sponsor | 1 (0.1) | 0 | 0 | 0 |
Loss of response (switched to open-label rescue IV mirikizumab) | NA | NA | 19 (4.9) | 42 (21.9) |
Discontinued rescue mirikizumab | 0 | 0 | 4 (1.0) | 4 (2.1) |
Other | 3 (0.3) | 0 | 0 | 0 |
mITT, N | 868 | 294 | 365 | 179 |
Safety, N | 958 | 321 | 389 | 192 |
ITT, N | 959 | 322 | 389 | 192 |
PP, N | ||| | ||| | ||| | ||| |
ITT = intention-to-treat; mITT = modified intention-to-treat; NA = not applicable; NR = not reported; PP = per-protocol.
Sources: Clinical Study Reports for the LUCENT-1 and LUCENT-2 studies.31,32
Table 11: Summary of Patient Disposition of Induction Nonresponders and Placebo Induction Responders From LUCENT-2 Study
Patient disposition | LUCENT-2 study | |
|---|---|---|
Mirikizumab | Placebo | |
All entered from LUCENT-1 study, N | 1,178 | |
Discontinued before randomization, N | 1 | |
Withdrawal by patient | 1 | |
Induction nonresponders | ||
Entered open-label extended induction period, N | 313 | ||| |
Completed open-label extended induction and entered open-label mirikizumab SC maintenance, N (%) | 171 (54.6) | ||| |||||| |
Discontinued open-label mirikizumab IV, N (%) | ||| |||||| | || |||||| |
Adverse event | 10 (3.2) | ||||| |
COVID-19–related disruptions | ||||| | ||||| |
Lack of efficacy | ||| |||||| | || |||||| |
Physician decision | ||||| | || |||||| |
Protocol deviation | ||||| | ||||| |
Withdrawal by patient | ||||| | ||||| |
Other | || |||||| | ||||| |
Placebo induction responders | ||
Entered open-label extended induction period, N | NA | 135 |
Completed blinded placebo SC maintenance, N (%) | NA | 90 (66.7) |
Discontinued open-label mirikizumab IV, N (%) | NA | 45 (33.3) |
NA = not applicable; SC = subcutaneous.
Notes: Details in Table 11 have been taken from the sponsor’s Summary of Clinical Evidence.20,30
No comparisons were drawn between the mirikizumab induction nonresponders and placebo induction nonresponders.
Source: Clinical Study Report for the LUCENT-2 study.32
Baseline Characteristics
A summary of baseline patient demographics, disease characteristics, and medication history in the pivotal trials is shown in Table 12. The baseline characteristics outlined in Table 12 are limited to those that are most relevant to this review or were felt to impact the outcomes or interpretation of the study results.
LUCENT-1: Induction Trial
The study population in the LUCENT-1 trial had a mean age of 42.5 (SD = 13.92) years. The majority of patients were male (59.8%) and white (72.3%). Based on the MMS, patients’ UC severity was either categorized as moderate (46.5% of mirikizumab patients and 47.1% of placebo patients) or severe (53.3% of mirikizumab patients and 52.9% of placebo patients). In terms of prior UC therapies, the proportion of patients reporting prior biologic or tofacitinib failure was similar between treatment groups (41.6% and 40.1% of patients randomized to mirikizumab and placebo, respectively). Loss of response to a biologic or tofacitinib was present in 22.6% of mirikizumab patients and 22.1% of placebo patients. Baseline corticosteroid use was present in 40.4% of patients in the mirikizumab arm and 38.4% of patients in the placebo arm. Overall, the baseline characteristics were well-balanced between treatment arms.
LUCENT-2: Maintenance Trial
The study population in the LUCENT-2 study main cohort had a mean age of 42.3 (SD = 13.5) years. The majority of patients were male (58.4%) and white (71.3%). Based on the MMS, approximately half of the patients in each treatment arm was categorized as moderate UC severity (MMS score of 4 points to 6 points). In terms of prior UC therapies, 35.1% of patients in the mirikizumab group and 35.8% of patients in the placebo group had a history of biologic or tofacitinib failure. Loss of response to a biologic or tofacitinib was reported in 20% of mirikizumab patients and 13.7% of placebo patients. Baseline corticosteroid use was present in 37% of patients in the mirikizumab arm and 38% of patients in the placebo arm. Overall, the baseline characteristics were well-balanced between treatment arms.
Table 12: Summary of Baseline Characteristics of Pivotal Studies and RCT Evidence Submitted by the Sponsor (mITT Populations)
Characteristic | LUCENT-1 (induction trial) | LUCENT-2 main cohort (maintenance trial) | ||
|---|---|---|---|---|
Mirikizumab 300 mg IV q.4.w. (N = 868) | Placebo IV q.4.w. (N = 294) | Mirikizumab 200 mg SC q.4.w. (N = 365) | Placebo SC q.4.w. (N = 179) | |
Age, mean years (SD) | 42.9 (13.9) | 41.3 (13.8) | 43.4 (14.2) | 41.2 (12.8) |
Male, n (%) | 530 (61.1) | 165 (56.1) | 214 (58.6) | 104 (58.1) |
BMI category, n (%) | ||||
Normal (≥ 18.5 kg/m2 to < 25 kg/m2) | 451 (52.0) | 149 (50.7) | 196 (53.7) | 97 (54.2) |
Overweight, obese, or extreme obese (≥ 25 kg/m2) | 362 (41.7) | 117 (39.8) | 143 (39.2) | 74 (41.3) |
Race, n (%) | ||||
White | 614 (71.5) | 219 (74.7) | 261 (71.9) | 125 (70.6) |
Asian | 223 (26.0) | 68 (23.2) | 93 (25.6) | 51 (28.8) |
Other | 22 (2.6) | 6 (2.1) | 3 (0.8) | 1 (0.6) |
Disease duration, mean years (SD) | 7.2 (6.7) | 6.9 (7.0) | 6.9 (7.1) | 6.7 (5.6) |
Disease location, n (%) | ||||
Left-sided colitis | 544 (62.7) | 188 (64.2) | 234 (64.1) | 119 (66.5) |
Total Mayo Score category, n (%) | ||||
Moderate: 6 to 9 points | 519 (62.9) | 186 (66.0) | 224 (64.4) | 108 (63.2) |
Severe: 10 to 12 points | 297 (36.0) | 93 (33.0) | 119 (34.2) | 61 (35.7) |
Modified Mayo Score category, n (%) | ||||
Moderate: 4 to 6 points | 404 (46.5) | 138 (47.1) | 181 (49.6) | 77 (43.0) |
Severe: 7 to 9 points | 463 (53.3) | 155 (52.9) | 184 (50.4) | 102 (57.0) |
Mayo endoscopic subscore = 3 points (severe disease), n (%) | 574 (66.1) | 200 (68.3) | 235 (64.4) | 106 (59.2) |
Prior UC therapy, n (%) | ||||
Prior biologic or tofacitinib exposure | 376 (43.3) | 123 (41.8) | 136 (37.3) | 65 (36.3) |
Biologic or tofacitinib failure | 361 (41.6) | 118 (40.1) | 128 (35.1) | 64 (35.8) |
Inadequate response to a biologic or tofacitinib | 203 (23.4) | 70 (23.8) | 61 (16.7) | 38 (21.2) |
Loss of response to a biologic or tofacitinib | 196 (22.6) | 65 (22.1) | 73 (20.0) | 31 (13.7) |
Intolerance to a biologic or tofacitinib | 51 (5.9) | 14 (4.8) | 18 (4.9) | 9 (5.0) |
Prior biologic failure | 360 (41.5) | 117 (39.8) | 128 (35.1) | 64 (35.8) |
Prior anti-TNF failure | 325 (37.4) | 97 (33.0) | 112 (30.7) | 58 (32.4) |
Prior vedolizumab failure | 159 (18.3) | 59 (20.1) | 47 (12.9) | 23 (12.8) |
Prior tofacitinib failure | 34 (3.9) | 6 (2.0) | 8 (2.2) | 8 (4.5) |
Baseline UC therapy, n (%) | ||||
Corticosteroids | 351 (40.4) | 113 (38.4) | 135 (37.0) | 68 (38.0) |
Immunomodulators | 211 (24.3) | 69 (23.5) | 78 (21.4) | 39 (21.8) |
Aminosalicylates (e.g., 5-ASA) | 646 (74.4) | 217 (73.8) | 278 (76.2) | 134 (74.9) |
Bowel urgency severity (UNRS score), median (Q1 to Q3) | 6.0 (5.0 to 8.0) | 7.0 (5.0 to 8.0) | 6.0 (5.0 to 8.0) | 6.0 (5.0 to 8.0) |
Fecal calprotectin, mcg/g, median (Q1 to Q3) | 1,559.0 (634.0 to 3,210.0) | 1,471.5 (626.5 to 2,944.5) | 1,482.0 (558.0 to 3,045.0) | 1,750.0 (754.0 to 3,519.0) |
C-reactive protein, mg/L, median (Q1 to Q3) | 4.1 (1.5 to 9.6) | 4.2 (1.2 to 9.5) | 3.8 (1.4 to 8.7) | 3.0 (1.0 to 7.7) |
5-ASA = 5-aminosalicylic acid; BMI = body mass index; mITT = modified intention-to-treat; q.4.w. = every 4 weeks; Q1 = first quartile; Q3 = third quartile; RCT = randomized controlled trial; SC = subcutaneous; SD = standard deviation; TNF = tumour necrosis factor; UC = ulcerative colitis; UNRS = Urgency Numeric Rate Score.
Note: Details in Table 12 have been taken from the sponsor’s Summary of Clinical Evidence.20,30
Sources: Clinical Study Reports for the LUCENT-1 and LUCENT-2 studies.31,32
Exposure to Study Treatments
Treatment exposure from the pivotal trials is summarized in Table 13. Study treatment durations occurred at 4-week intervals. In the LUCENT-1 study’s mITT population, 98.6% of patients randomized to placebo and 98.0% of patients randomized to mirikizumab were deemed adherent, defined as missing no study doses. || |||||||||| |||| |||||| |||| ||||||||||| ||||||| ||||||||| ||| ||||| || || ||||| ||| |||||||| |||||||||| || ||||||||||| ||| ||||| ||| ||||| |||||||||| || |||||||| ||||||| || ||||||||| ||| ||||||||| |||| |||| || ||||| |||||||||| ||| || ||||| ||| || ||||||| In the LUCENT-2 study, a greater proportion of patients in the mirikizumab group (90.5%) compared to the placebo group (64.6%) had at least 32 weeks of treatment exposure. Fewer patients had a treatment exposure of at least 40 weeks, with a greater proportion in the mirikizumab group (67.6%) compared to the placebo group (47.4%).
Table 13: Summary of Patient Exposure From Pivotal Studies and RCT Evidence Submitted by the Sponsor (Safety Populations)
Weeks of exposure, n (%) | LUCENT-1 (induction trial) | LUCENT-2 main cohort (maintenance trial) | ||
|---|---|---|---|---|
Mirikizumab 300 mg IV q.4.w. (N = 958) | Placebo IV q.4.w. (N = 321) | Mirikizumab 200 mg SC q.4.w. (N = 389) | Placebo SC q.4.w. (N = 192) | |
> 0 weeks | 958 (100.0) | 321 (100.0) | 389 (100) | 192 (100) |
≥ 4 weeks | 952 (99.4) | 315 (98.1) | 389 (100) | 187 (97.4) |
≥ 8 weeks | 938 (97.9) | 301 (93.8) | 386 (99.2) | 185 (96.4) |
≥ 12 weeks | 725 (75.7) | 211 (65.7) | 383 (98.5) | 178 (92.7) |
≥ 16 weeks | — | — | 374 (96.1) | 170 (88.5) |
≥ 24 weeks | — | — | 360 (92.5) | 144 (75.0) |
≥ 32 weeks | — | — | 352 (90.5) | 124 (64.6) |
≥ 40 weeks | — | — | 263 (67.6) | 91 (47.4) |
Q.4.w. = every 4 weeks; RCT = randomized controlled trial; SC = subcutaneous.
Note: Details in Table 13 have been taken from the sponsor’s Summary of Clinical Evidence.20,30
Sources: Clinical Study Reports for the LUCENT-1 and LUCENT-2 studies.31,32
Concomitant Medications and Cointerventions
Concomitant medication use in the pivotal trials is summarized in Table 14 ||||||||||| |||||||||| ||| ||| ||||||||| ||||||| |||||| ||||||||| |||| ||| ||||||| ||||||.
||||||||||| ||||||||||||| | ||||||||||||||||||| |||||| | |||||||| |||| ||||||||||||||||||| |||||| | ||
|---|---|---|---|---|
||||||||||| ||| || | ||||||| || ||||||| |||| | ||||||||||| ||| || || ||| | ||||||| || ||||||| |||| | |
||| |||||||||||||| | ||| |||||| | ||| |||||| | ||| |||||| | || |||||| |
|||||||||||| | ||| |||||| | || |||||| | || |||||| | || |||||| |
|||||||||| | ||| |||||| | || |||||| | || |||||| | || |||||| |
|||||||||||||||||| | || ||||| | || ||||| | || ||||| | || |||||| |
|||||||||| | || ||||| | || ||||| | ||||| | || ||||| |
||||||||||||| | ||||| | ||||| | ||||| | ||||| |
|||||||||||||| | ||||| | ||||| | ||||| | ||||| |
||||||||||||| | ||||| | ||||| | ||||| | ||||| |
||||||||||||| | ||||| | ||||| | ||||| | ||||| |
||||||||| | ||||| | ||||| | ||||| | ||||| |
|||||||||||| | ||||| | ||||| | ||||| | ||||| |
||| ||||||||||||||| | ||||| | ||||| | ||||| | ||||| |
|||||||||||| | ||||| | ||||| | ||||| | ||||| |
|||||||||||||| | ||||| | ||||| | ||||| | ||||| |
|||||||||||| | ||||| | ||||| | ||||| | ||||| |
|||||||||| | ||||| | ||||| | ||||| | ||||| |
||| |||||||||||| |||| ||||||||||| ||| ||||||||||||| |||||||| |||||||| ||||| ||||||| ||| |||||||| ||| ||||||||31,32 |||||||||| |||| ||| ||||| |||| |||| ||||| |||| ||| ||||||||| ||||||| || |||||||| |||||||||20
Protocol Deviations
Protocol deviations in the pivotal trials for the mITT population are summarized in Table 15.
|| ||||||||| ||| |||||||||| || |||||||| |||| || |||||||||||||| |||||||| ||||||||| ||| ||| ||||||| || ||| ||||||||||| ||| ||| || ||||||| || ||| ||||||| |||| ||| |||| |||||||| |||||||| |||||||||| || ||| ||||||| ||||| |||| |||||||||||||||||||| |||| ||| |||||||| ||||||||||| |||||||| ||||||||| ||||| ||| ||||||||| |||||| |||||| |||||||| ||| || |||| |||| || |||||| |||||||| |||||| |||||| ||| ||||||||| |||||||||||||| || |||||||| |||||| |||||| || ||| ||||||||||||||||||||| |||||| ||| |||| |||||||| |||||||| |||||||||| |||| |||||||| ||| || |||| |||| || |||||| |||||||| |||||| |||||| |||||||||||||||||||| |||| ||| |||||||| |||||||||| ||||||||| |||||| |||||||||| ||||||||||| ||| ||||||||| |||||||||||||| || |||||||| |||||| ||||||||| ||||||||| ||||| ||||||||||| ||||||||| ||||||||||| ||| ||||||||||| || |||||||| |||| || |||||||||||||| |||||||| ||||||||| ||||| ||||| |||||| ||| ||| ||||||||||| ||| ||| ||||| |||||| ||| ||| ||||||| |||| ||| |||||||| || ||| ||||||| |||||| ||| |||| |||||||| |||||||| ||||||||| |||||||||| ||||| |||||||||||||||||||| ||| |||||||| ||| ||||||| ||| ||||||||| ||| ||||| || || || |||||||||||||||| |||||| || ||||| ||| |||||| |||||| |||||||| ||||||||| ||||| ||| ||||||||| || || || |||||||||||||||| ||||||||| || |||||||| |||||| |||||| ||| |||||| || |||||||| || ||| || || ||||| |||| |||||||| ||| ||| ||||||||||| || ||| |||||| |||||||||| ||||||| ||||| |||| |||| || ||| |||||||| |||||||||| |||||||| |||||||| |||||||| ||| ||||| ||||||| || |||||||||||||| || ||| ||||| ||||||| || |||||||||||| ||
|||||||| ||||||||||||||| | ||||||||||||||||||| ||||||| | ||||||||||||||||||||| |||||| | ||
|---|---|---|---|---|
||||||||||| ||| || || ||||||||||| | ||||||| || ||||||||||| | ||||||||||| ||| || || ||| | ||||||| || ||||||||||| | |
|||||||| |||| || ||||||||| |||||||| ||||||||| | ||| |||||| | || |||||| | || |||||| | || |||||| |
||||| ||||||||| | ||| |||||| | || |||||| | || |||||| | || |||||| |
||||||||||| | || |||||| | || |||||| | || |||||| | || |||||| |
||||||||||||||| ||||||| | || |||||| | || |||||| | || |||||| | || |||||| |
||||||||| ||||||||||||||||||||||| ||||| | || |||||| | || |||||| | || |||||| | || |||||| |
|||||||| ||||||| | || |||||| | || |||||| | || |||||| | || |||||| |
|||||| | || |||||| | || |||||| | || |||||| | || |||||| |
|||||||||||||||||||||||| | || |||||| | || |||||| | || |||||| | || |||||| |
||||| |||||||| |||||||| | || |||||| | || |||||| | || |||||| | || |||||| |
||||||| |||| |||||||||| ||| ||| ||| ||| | || |||||| | || |||||| | || |||||| | || |||||| |
|||||||||||| ||||||||||||||||||||||| |||||||| ||||| ||||||| ||| |||||||| ||| |||||||||31,32
Transcription Error in the Electronic Clinical Outcome Assessment Devices in Poland and Turkey Impacting Outcomes Assessment
In the LUCENT-2 trial, an error was identified in the way that the electronic clinical outcomes assessment devices worded daily self-reported SF and RB assessments for patients in Turkey and Poland, respectively. The error was corrected when noticed. Overall, 104 impacted patients (4 patients from Turkey and 100 patients from Poland), whose LUCENT-1 study baseline data were collected using incorrectly transcribed patient-reported outcome questions on the electronic clinical outcomes assessment devices, were excluded from the primary efficacy analysis. Impacted patients were, however, still included in the primary safety analysis. Sensitivity analyses for efficacy were performed on primary and selected key secondary end points at week 40 by including impacted patients using the ITT population with the mNRI method, with RB score imputed for impacted patients in Poland and SF scores imputed for impacted patients in Turkey.
Efficacy
Summarized end points are based on those included in the sponsor’s Summary of Clinical Evidence30 as well as any end points identified as important to this review according to stakeholders — for example, the clinical expert, clinician groups, or patient groups. These end points are summarized in Table 16 for the LUCENT-1 trial and Table 17 for the LUCENT-2 trial. Results are presented by induction period (the LUCENT-1 study) and maintenance period (the LUCENT-2 study).
LUCENT-1 Study: Induction Period
Clinical Response
A greater proportion of patients on mirikizumab 300 mg IV (63.5%) than on placebo (42.2%) experienced clinical response at week 12 (common risk difference = 21.4%; 99.875% CI, 10.8% to 32.0%; P < 0.00001). Sensitivity analyses in the ITT population were consistent with the mITT population results.
In the biologic-naive subgroup, a greater proportion of patients on mirikizumab 300 mg IV (70.1%) versus placebo (50.3%) experienced clinical response (common risk difference = 19.8%; 95% CI, 11.3% to 28.3%; P < 0.001). Similarly, patients in the biologic-experienced subgroup also demonstrated greater clinical response on mirikizumab (54.6%) versus placebo (30.9%), with a common risk difference of 23.9% (95% CI, 14.3% to 33.5%; P < 0.001).
In terms of baseline conventional therapy use, of the 464 patients receiving corticosteroids and 280 patients receiving immunomodulators at baseline in the mITT population, the observed effect sizes for the mirikizumab group compared to the placebo group were numerically positive but small when compared to the overall population. The subgroup of patients without baseline corticosteroid and immunomodulator use had similar results to patients in the overall mITT population. The interaction P values did not suggest a difference between the groups. The LUCENT-1 study baseline therapy subgroup data are in Appendix 1.
The subgroup analysis for baseline MMS score was prespecified in the LUCENT-1 study; however, the analysis was not available from the sponsor.
Clinical Remission
Clinical remission was assessed using 2 different outcomes in the LUCENT-1 trial: clinical remission and alternate clinical remission. A summary of key remission outcomes is presented in Table 16.
Clinical Remission Rate
A greater proportion of patients on mirikizumab 300 mg IV (24.2%) versus placebo (13.3%) experienced clinical remission at week 12, with a common risk difference of 11.1% (99.875% CI, 3.2% to 19.1%; P = 0.00006). Analyses in the PP and ITT populations were similar to the mITT population results. In addition, results of the sensitivity analyses using different imputation methods were consistent with the results from the primary analysis. In terms of the tipping point analysis, there was no significant difference between groups when imputing missing data as “responder” for the placebo group and as “nonresponder” for the mirikizumab group.
In terms of the biologic-naive subgroup, a greater number of patients in the mirikizumab group (30.9%) versus the placebo group (15.8%) were clinical remitters at the end of the LUCENT-1 trial (common risk difference = 15.1%; 95% CI, 8.3% to 21.9%; P < 0.001). The magnitude of the effect was consistent with the primary analysis. Similar results were seen in the biologic-experienced subgroup between patients receiving mirikizumab (15.4%) versus placebo (9.8%), although the effect was small (common risk difference = 5.7%; 95% CI, –0.7% to 12.1%; P = 0.135).
In the subgroups of patients using corticosteroids or immunomodulators at baseline, the observed effect sizes for clinical remission in the mirikizumab group compared to the placebo group were numerically positive but small when compared to the overall population. The subgroup of patients without baseline corticosteroid and immunomodulator use had similar effect sizes to patients in the overall mITT population. The interaction P values did not suggest a difference between the groups.
Appendix 1 includes the LUCENT-1 study baseline therapy subgroup data. The subgroup analysis for baseline MMS score was prespecified in the LUCENT-1 trial; however, data were not available in the submission provided by the sponsor.
Alternate Clinical Remission Rate
Alternate clinical remission used a definition similar to that of clinical remission except it did not require a 1-point decrease in the SF subscore. Results for alternate clinical remission were very similar to those of clinical remission. At week 12 of the LUCENT-1 study, 25.6% of patients on mirikizumab versus 14.6% of patients on placebo experienced alternate clinical remission, with a common risk difference of 11.1% in favour of mirikizumab (99.875% CI, 3.0% to 19.3%; P < 0.001). Analyses in the ITT population were consistent with the mITT population results.
In the biologic-naive subgroup, a greater number of patients in the mirikizumab group (32.5%) versus the placebo group (18.1%) experienced alternate clinical remission at the end of the LUCENT-1 study (common risk difference = 14.4%; 95% CI, 7.3% to 21.5%; P < 0.001). Results were similar in the biologic-experienced subgroup between patients receiving mirikizumab (15.4%) versus placebo (9.8%), although the effect was small (common risk difference = 6.7%; 95% CI, 0.3% to 13.2%; P = 0.079).
|| ||| ||||||||| || |||||||| ||||| ||||||||||||||| || |||||||||||||||| || ||||||||| ||| |||||||| |||||| ||||| ||| ||||||||| |||||||| ||||||||| || ||| ||||||||||| ||||| |||||||| || ||| ||||||| ||||| |||| ||||||||||| |||||||| ||| ||||| |||| |||||||| || ||| ||||||| ||||||||||| ||| |||||||| || |||||||| ||||||| |||||||| |||||||||||||| ||| ||||||||||||||| ||| ||| ||||||| |||||| ||||| || |||||||| || ||| ||||||| |||| ||||||||||| ||| ||||||||||| |||||||| ||| ||| ||||||||||||||||| ||||||| ||| ||||||| Appendix 1 |||||||| ||| |||||||| |||||||| ||||||| |||||||| ||||.
The subgroup analysis for baseline MMS score was prespecified in the LUCENT-1 study; however, data were not available from the sponsor.
Endoscopic Remission
At week 12 of the LUCENT-1 trial, 36.3% of patients on mirikizumab experienced endoscopic remission versus 21.1% of patients on placebo (common risk difference = 15.4%; 99.875% CI, 6.3% to 24.5%; P value < 0.00001). Analyses in the ITT population were consistent with the mITT population results.
In the biologic-naive subgroup, a greater proportion of patients on mirikizumab (45.9%) versus placebo (28.1%) experienced endoscopic remission at week 12 (common risk difference = 17.9%; 95% CI, 9.8% to 25.9%; P < 0.001). Similarly, within the biologic-experienced subgroup, a greater proportion of patients on mirikizumab (23.7%) versus placebo (11.4%) experienced endoscopic remission at week 12 (common risk difference = 12.3%; 95% CI, 5.2% to 19.4%; P = 0.003).
In the subgroups of patients using corticosteroids or immunomodulators at baseline, the observed effect sizes for endoscopic remission in the mirikizumab group compared to the placebo group were numerically positive but small when compared to the overall population. The subgroup of patients without baseline corticosteroid and immunomodulator use had similar effect sizes to patients in the overall mITT population. The interaction P values did not suggest a difference between the groups.
Appendix 1 includes the LUCENT-1 study baseline therapy subgroup data. The subgroup analysis for baseline MMS score was prespecified in the LUCENT-1 trial; however, data were not available in the submission provided by the sponsor.
|||||||| |||| || || ||||||||| ||||| || |||||||| || ||||||||||| ||| || ||||| ||||| || || ||||||||||| ||| || |||||||| || |||||||| ||||||||| |||| |||||||||| || ||||| |||||||| ||| ||| || ||||| |||||||||
Symptomatic Remission
At week 12 of the LUCENT-1 study, 45.5% of patients on mirikizumab and 27.9% of patients on placebo experienced symptomatic remission, with a common risk difference of 17.5% (99.875% CI, 7.5% to 27.6%; P < 0.00001). Analyses in the ITT population were consistent with the mITT population results.
In the biologic-naive subgroup, a greater proportion of patients on mirikizumab (50.4%) versus placebo (33.3%) experienced symptomatic remission at week 12 (common risk difference = 17.1%; 95% CI, 8.7% to 25.4%; P < 0.001). Similarly, within the biologic-experienced subgroup, a greater proportion of patients on mirikizumab (38.6%) versus placebo (18.8%) experienced endoscopic remission at week 12 (common risk difference = 18.8%; 95% CI, 10.1% to 27.4%; P < 0.001).
|| ||| ||||||||| || |||||||| ||||| ||||||||||||||| || |||||||||||||||| || ||||||||| ||| |||||||| |||||| ||||| ||| ||||||||||| ||||||||| || ||| ||||||||||| ||||| |||||||| || ||| ||||||| ||||| |||| ||||||||||| |||||||| ||| ||||| |||| |||||||| || ||| ||||||| ||||||||||| ||| |||||||| || |||||||| ||||||| |||||||| |||||||||||||| ||| ||||||||||||||| ||| ||| ||||||| |||||| ||||| || |||||||| || ||| ||||||| |||| ||||||||||| ||| ||||||||||| |||||||| ||| ||| ||||||||||||||||| ||||||| ||| |||||||
Appendix 1 includes the LUCENT-1 study baseline therapy subgroup data. The subgroup analysis for baseline MMS score was prespecified in the LUCENT-1 study; however, data were not available from the sponsor.
Bowel Urgency Improvement: UNRS
The LSM change from baseline at week 12 in the mirikizumab group was –2.59 points and –1.63 points in the placebo group, a difference of –0.95 points (99.875% CI, –1.5 points to –0.4 points; P < 0.00001). Analyses in the ITT population were consistent with the mITT population results.
In the biologic-naive subgroup, greater improvement in UNRS was seen in the mirikizumab group (–2.7 points) versus the placebo group (–2.1 points), with an LSM mean difference of –0.6 points at week 12 (95% CI, –1.0 points to –0.2 points; P = 0.002). Similar results were seen in the biologic-experienced subgroup, but with a larger LSM mean difference of –1.5 points at week 12 (95% CI, –2.0 points to –1.0 points; P < 0.001).
|| ||| ||||||||| || |||||||| ||||| ||||||||||||||| || |||||||||||||||| || ||||||||| ||| |||||||| |||||| ||||| ||| ||||| ||||||| ||||||||||| || ||| ||||||||||| ||||| |||||||| || ||| ||||||| ||||| |||| ||||||||||| |||||||| ||| ||||| |||| |||||||| || ||| ||||||| ||||||||||| |||||||| ||||||| |||||||| |||||||||||||| ||| ||||||||||||||| ||| ||| ||||||| ||||||| || |||||||| || ||| ||||||| |||| ||||||||||| ||| ||||||||||| |||||||| ||| ||| ||||||||||||||| ||||||| ||| |||||||
Appendix 1 includes the LUCENT-1 study baseline therapy subgroup data. The subgroup analysis for baseline MMS score was prespecified in the LUCENT-1 study; however, data were not available from the sponsor.
Health-Related Quality of Life
HRQoL was assessed in the LUCENT-1 trial based on the IBDQ score, EQ-5D-5L score, and SF-36 score (PCS and MCS). A summary of HRQoL outcomes is presented in Table 16.
IBDQ Score
In the LUCENT-1 study, the mean change from baseline to week 12 in the IBDQ score was 38.4 points for patients in the mirikizumab group and 25.2 points for those in the placebo group, representing a difference of 13.2 points (95% CI, 9.3 points to 17.2 points; P < 0.001) in favour of mirikizumab. The change from baseline in the mirikizumab group met the MID of at least 30 points, representing a meaningful improvement from baseline to week 12. However, the difference between groups was not considered to be clinically important as it did not meet the suggested MID of at least 15 points over placebo.21-24
EQ-5D-5L Score
In the LUCENT-1 study, the mean change from baseline to week 12 in the EQ VAS score was 14.6 points in the mirikizumab group and 9.4 points in the placebo group, with an LSM difference of 5.2 points (95% CI, ||| || |||; P < 0.001). The change from baseline in the mirikizumab group met the MID of 10.9 points on the EQ VAS;25 however, it was not clear whether the difference between groups was clinically important given that no MID was identified for between-group differences.
SF-36 Score
In the LUCENT-1 study, the mean change from baseline to week 12 in the SF-36 PCS score was 5.97 points in the mirikizumab group and 3.90 points in the placebo group, with an LSM difference of 2.07 points (95% CI, 1.21 points to 2.93 points; P < 0.001). The mean change from baseline to week 12 in the SF-36 MCS score was 5.02 points in the mirikizumab group and 3.42 points in the placebo group, with an LSM mean difference of 1.60 points (95% CI, 0.56 points to 2.63 points; P = 0.002). The change from baseline in the mirikizumab group appeared to meet the identified MID threshold (i.e., MID = 3 points to 5 points) for the PCS and MCS. However, it was not clear whether the difference between groups was clinically important given that no MID was identified for between-group differences.
Mucosal Healing: HEMI
In the LUCENT-1 study, mucosal healing was assessed based on the HEMI outcome, which considers both histologic and endoscopic outcomes. At week 12, 27.1% of patients on mirikizumab experienced HEMI versus 13.9% of patients on placebo, with a common risk difference of 13.4% (99.875% CI, 5.5% to 21.4%; P < 0.00001). Analyses in the ITT population were consistent with the mITT population results.
Within the biologic-naive subgroup, the percentage of patients who experienced HEMI was higher in the mirikizumab treatment group (35.8%) compared to the placebo group (18.7%), with a common risk difference of 17.1% (95% CI, 9.8% to 24.3%; P < 0.001). The magnitude of the difference was consistent with the mITT population. Within the biologic-experienced subgroup, the percentage of patients who experienced HEMI was greater in the mirikizumab treatment group (15.7%) compared to the placebo group (7.3%), with a common risk difference of 8.4% (95% CI, 2.5% to 14.3%; P = 0.022).
|| ||| ||||||||| || |||||||| ||||| ||||||||||||||| || |||||||||||||||| || ||||||||| ||| |||||||| |||||| ||||| ||| |||| || ||| ||||||||||| ||||| |||||||| || ||| ||||||| ||||| |||| ||||||||||| |||||||| ||| ||||| |||| |||||||| || ||| ||||||| ||||||||||| |||||||| ||||||| |||||||| |||||||||||||| ||| ||||||||||||||| ||| ||| ||||||| ||||||| || |||||||| || ||| ||||||| |||| ||||||||||| ||| ||||||||||| |||||||| ||| ||| ||||||||||||||||| ||||||| ||| |||||||
Appendix 1 includes the LUCENT-1 trial subgroup data. The subgroup analysis for baseline MMS score was prespecified in the LUCENT-1 study; however, data were not available from the sponsor.
Work Productivity
In the LUCENT-1 study, work productivity was assessed using the WPAI:UC score at week 12. Among those employed at baseline (n = 566), patients on mirikizumab experienced a mean change in WPAI:UC of –20.65, compared to –14.91 in the placebo group (LSM difference = –5.74 points; 95% CI, –10.06 points to –1.42 points; P = 0.009). An MID was not identified for the WPAI:UC in patients for UC; hence, the clinical importance of the results was uncertain.
Table 16: Summary of Key Efficacy Results From LUCENT-1 Study (mITT Population at Week 12)
Outcome | LUCENT-1 (induction trial) | |
|---|---|---|
Mirikizumab 300 mg IV q.4.w. N = 868 | Placebo IV q.4.w. N = 294 | |
Clinical response | ||
Patients contributing to the analysis, n | 868 | 294 |
Patients with clinical response, n (%) | 551 (63.5) | 124 (42.2) |
Common risk difference, % (99.875% CI) | 21.4 (10.8 to 32.0) | |
P value | < 0.00001 | |
Clinical response, biologic-experienced population | ||
Patients contributing to the analysis, n | 376 | 123 |
Patients with clinical response, n (%) | 206 (54.8) | 38 (30.9) |
Risk difference, % (95% CI) | 23.9 (14.3 to 33.5) | |
P value | < 0.001 | |
Clinical remission | ||
Patients contributing to the analysis, n | 868 | 294 |
Patients with clinical remission, n (%) | 210 (24.2) | 39 (13.3) |
Common risk difference, % (99.875% CI) | 11.1 (3.2 to 19.1) | |
P value | 0.00006 | |
Alternate clinical remission | ||
Patients contributing to the analysis, n | 868 | 294 |
Patients with alternate clinical remission, n (%) | 222 (25.6) | 43 (14.6) |
Common risk difference, % (99.875% CI) | 11.1 (3.0 to 19.3) | |
P value | < 0.001 | |
Endoscopic remission | ||
Patients contributing to the analysis, n | 868 | 294 |
Patients with endoscopic remission, n (%) | 315 (36.3) | 62 (21.1) |
Common risk difference, % (99.875% CI) | 15.4 (6.3 to 24.5) | |
P value | < 0.00001 | |
|||| || ||||| || | ||
|||||||| |||||||||||| || ||| |||||||| | ||| | ||| |
|||||||| |||| ||||| |||||| | ||| |||||| | || |||||| |
|||||| |||| ||||||||||||||||||| ||| | |||| |||| || ||||| | |
||||| | |||||| | |
Bowel urgency improvement (UNRS score) | ||
Patients contributing to the analysis, n | 868 | 294 |
LSM change from baseline (CI) | –2.59 (–2.9 to –2.3) | –1.63 (–2.1 to –1.2) |
LSM difference in change from baseline (99.875% CI) | –0.95 (–1.5 to –0.4) | |
P value | < 0.00001 | |
Symptomatic remission | ||
Patients contributing to the analysis, n | 868 | 294 |
Patients with symptomatic remission, n (%) | 395 (45.5) | 82 (27.9) |
Common risk difference, % (99.875% CI) | 17.5 (7.5 to 27.6) | |
P value | < 0.00001 | |
IBDQ score | ||
Patients contributing to the analysis, n | 868 | 294 |
LSM change from baseline (SE) | 38.4 (1.1) | 25.2 (1.8) |
LSM difference in change from baseline (95% CI) | 13.2 (9.3 to 17.2) | |
P value | < 0.001a | |
EQ-5D-5L score | ||
|||||||| |||||||||||| || ||| |||||||| | ||| | ||| |
LSM change from baseline (SE) | 14.6 (|||) | 9.4 (|||) |
LSM difference in change from baseline (95% CI) | 5.2 (||| || |||) | |
P value | < 0.001a | |
SF-36 score (PCS) | ||
Patients contributing to the analysis, n | 868 | 294 |
LSM change from baseline (SE) | 5.97 (0.242) | 3.90 (0.393) |
LSM difference in change from baseline (95% CI) | 2.07 (1.21 to 2.93) | |
P value | < 0.001a | |
SF-36 score (MCS) | ||
Patients contributing to the analysis, n | 868 | 294 |
LSM change from baseline (SE) | 5.02 (0.291) | 3.42 (0.472) |
LSM difference in change from baseline (95% CI) | 1.60 (0.56 to 2.63) | |
P value | 0.002a | |
HEMI | ||
Patients contributing to the analysis, n | 868 | 294 |
Patients with HEMI, n (%) | 235 (27.1) | 41 (13.9) |
Common risk difference, % (99.875% CI) | 13.4 (5.5 to 21.4) | |
P value | < 0.00001 | |
WPAI:UC (overall work impairment) score (among those who were employed at baseline) | ||
Patients contributing to the analysis, n | 429 | 137 |
LSM change from baseline (SE) | –20.65 (1.163) | –14.91 (1.985) |
LSM difference in change from baseline (95% CI) | –5.74 (–10.06 to –1.42) | |
P value | 0.009a | |
CI = confidence interval; EQ-5D-5L = 5-Level EQ-5D; HEMI = histologic endoscopic mucosal improvement; IBDQ = Inflammatory Bowel Disease Questionnaire; LSM = least squares mean; MCS = mental component summary; mITT = modified intention-to-treat; PCS = physical component summary; q.4.w. = every 4 weeks; SE = standard error; SF-36 = Short Form (36) Health Survey; UCEIS = Ulcerative Colitis Endoscopic Index of Severity; UNRS = Urgency Numeric Rating Scale; WPAI:UC = Work Productivity and Activity Impairment Questionnaire: Ulcerative Colitis.
Notes: Details in Table 16 have been taken from the sponsor’s Summary of Clinical Evidence.20,30
The 2-sided alpha was 0.00125 for the LUCENT-1 trial.
aP value has not been adjusted for multiple testing.
Source: Clinical Study Report for the LUCENT-1 study.31
LUCENT-2 Study: Maintenance Period
Clinical Remission
Clinical remission was assessed using 4 different outcomes in the LUCENT-2 study: clinical remission, alternate clinical remission, corticosteroid-free remission, and durable clinical remission. A summary of key remission outcomes is presented in Table 17.
Clinical Remission
A greater proportion of patients on mirikizumab 200 mg SC (49.9%) versus placebo (25.1%) experienced clinical remission after 40 weeks of maintenance therapy (common risk difference = 23.2%; 95% CI, 15.2% to 31.2%; P < 0.001). Analyses in the PP and ITT populations were similar to the mITT population results. In addition, results of the sensitivity analyses were consistent with the results from the primary analysis.
In the tipping point analysis, all missing data for the placebo group was imputed as a responder and all missing data for the mirikizumab group were imputed as a nonresponder. When considering this extreme case, there was no difference between the mirikizumab group and placebo the group.
Clinical remission was analyzed by subgroups. In the biologic-naive subgroup, 51.5% of patients on mirikizumab and 30.7% of patients on placebo experienced clinical remission at week 40 of the LUCENT-2 trial, corresponding to a common risk difference of 20.8% (95% CI, 10.2% to 31.5%; P < 0.001). In the biologic-failed subgroup, 46.1% of patients on mirikizumab and 15.6% of patients on placebo experienced clinical remission with a common risk difference of 30.5% (95% CI,18.1% to 42.9%; P < 0.001).
Subgroup results for baseline corticosteroid use (yes and no), immunomodulator use (no), and patients with severe UC at baseline were consistent with the results of the primary analysis. Subgroup results for immunomodulator use (yes) and patients with moderate UC at baseline were numerically positive but small when compared to the overall population. ||| ||||||||||| |||||||| ||| ||| ||||||||||||||||| ||||||| ||| ||||||. Appendix 1 contains the LUCENT-2 study subgroup data.
Alternate Clinical Remission
Alternate clinical remission used a definition similar to that of clinical remission except it did not require a 1-point decrease in the SF subscore. Results for alternate clinical remission were very similar to those of clinical remission. At week 40 of the LUCENT-2 study, 51.8% of mirikizumab patients and 26.3% of placebo patients experienced alternate clinical remission with a common risk difference of 24.1% (95% CI, 16.0% to 32.2%; P < 0.001). |||||||| || ||| ||| |||||||||| |||| |||||||||| |||| ||| |||| |||||||||| |||||||.
Alternate clinical remission was analyzed by subgroups. In the biologic-naive subgroup, 54.1% of patients on mirikizumab and 32.5% of patients on placebo experienced alternate clinical remission, corresponding to a common risk difference of 21.7% (95% CI, 10.9% to 32.4%; P < 0.001). ||||| ||| ||||||||||||||| ||||||||| ||||| || |||||||| || ||||||||||| ||| ||||| || |||||||| || ||||||| ||||||||||| ||||||||| |||||||| ||||||||| |||||||||| |||| |||||||||| || ||||| || |||||| || ||||||||||| |||| ||| |||| || ||||| ||||||||| ||| ||||||||| || |||||| ||| |||||||||| |||| ||| ||||||| |||||||||
Subgroup results for baseline corticosteroid use (yes and no), immunomodulator use (yes and no), and patients with moderate and severe UC at baseline were |||||||||| |||| ||| ||||||| || ||| ||||||| ||||||||. Appendix 1 contains the LUCENT-2 study subgroup data.
Durable Clinical Remission
Of patients who experienced clinical remission at week 12 of the LUCENT-1 study (n = 210), 63.6% of patients who were randomized to mirikizumab 200 mg SC were still in clinical remission at week 40 of the LUCENT-2 trial compared to 36.9% of those randomized to placebo SC, with a common risk difference of 24.8% (95% CI, 10.4% to 39.2%; P < 0.001). Analyses in the ITT population were consistent with the mITT population results.
In the biologic-naive subgroup, 62.5% of patients on mirikizumab and 46.8% of patients on placebo experienced durable clinical remission at week 40 of the LUCENT-2 study, with a common risk difference of 15.7% (95% CI, –1.3% to 32.7%; P = 0.078), although the effect size was small compared to the overall population. In the biologic-failed subgroup, 66.7% of patients in the mirikizumab group and 11.1% of patients in the placebo group experienced durable clinical remission at week 40, with a common risk difference of 55.6% (95% CI, 34.4% to 76.7%; P < 0.001). However, sample sizes for this subgroup were quite small.
Subgroup results for baseline corticosteroid use (no), baseline immunomodulator use (no), and patients with moderate and severe UC at baseline were consistent with the results of the primary analysis. Subgroup results of patients with baseline corticosteroid use (yes) and immunomodulator use (yes), were numerically positive but small when compared to the overall population. ||| ||||||||||| |||||||| ||| ||| ||||||||||||||||| ||||||| ||| ||||||. Appendix 1 contains the LUCENT-2 study subgroup data.
Corticosteroid-Free Remission
A greater number of patients randomized to mirikizumab experienced corticosteroid-free remission than those randomized to placebo at week 40 — 44.9% versus 21.8%, respectively (common risk difference = 21.3%; 95% CI, 13.5% to 29.1%; P < 0.001). This between-group difference was consistent in both the biologic-naive and biologic-failed subgroups (P < 0.001 for both).
In the biologic-naive subgroup, 46.7% of patients on mirikizumab and 26.3% of patients on placebo experienced corticosteroid-free remission at week 40 with a common risk difference of 20.4% (95% CI, 10.1% to 30.8%; P < 0.001). In the biologic-failed subgroup, 40.6% of patients on mirikizumab and 14.1% of patients on placebo experienced corticosteroid-free remission at week 40 with a common risk difference of 26.6% (95% CI, 14.5% to 38.6%; P < 0.001), respectively. The magnitude of effect was consistent with the primary analysis.
Subgroup results for baseline corticosteroid use (yes and no), immunomodulator use (yes and no), and patients with moderate and severe UC at baseline were consistent with the results of the primary analysis. ||| ||||||||||| |||||||| ||| ||| |||||||||||||||| ||||||| ||| ||||||. Appendix 1 contains the LUCENT-2 study subgroup data.
Endoscopic Remission
At week 40 of the LUCENT-2 trial, 58.6% of patents on mirikizumab experienced endoscopic remission versus 29.1% of patients on placebo, with a common risk difference of 28.5% in favour of mirikizumab (95% CI, 20.2% to 36.8%; P < 0.001). Analyses in the ITT population were consistent with the mITT population results.
In the biologic-naive subgroup, 62.4% of patients on mirikizumab and 34.2% of patients on placebo experienced endoscopic remission at week 40, with a common risk difference of 28.2% (95% CI, 17.5% to 39.0%; P < 0.001). In the biologic-failed subgroup, 50.8% of patients on mirikizumab and 20.3% of patients on placebo experienced endoscopic remission at week 40, with a common risk difference of 30.5% (95% CI, 17.3% to 43.6%; P < 0.001).
Subgroup results for baseline corticosteroid use (yes and no), immunomodulator use (yes and no), and patients with moderate and severe UC at baseline were consistent with the results of the primary analysis. Appendix 1 contains the LUCENT-2 study subgroup data.
Bowel Urgency
In the LUCENT-2 study, bowel urgency outcomes consisted of bowel urgency remission and bowel urgency improvement. The results are presented in Table 17.
Bowel Urgency Remission
Of the patients with a UNRS score of at least 3 at the LUCENT-1 study baseline (n = 420), 42.9% of patients on mirikizumab and 25% of patients on placebo at week 40 experienced bowel urgency remission, corresponding to a common risk difference of 18.1% (95% CI, 9.8% to 26.4%; P < 0.001). Analyses in the ITT population were consistent with results in the mITT population.
In the biologic-naive subgroup, 46.6% of patients on mirikizumab and 28.7% of patients on placebo experienced bowel urgency remission at week 40, with a common risk difference of 17.9% (95% CI, 7.0% to 28.8%; P = 0.002). In the biologic-failed subgroup, 35.2% of patients on mirikizumab and 19% of patients on placebo experienced bowel urgency remission at week 40, with a common risk difference of 30.5% (95% CI, 17.3% to 43.6%; P < 0.001).
Subgroup results for baseline corticosteroid use (yes and no), immunomodulator use (yes and no), and patients with moderate and severe UC at baseline were consistent with the results of the primary analysis. Appendix 1 contains the LUCENT-2 study subgroup data.
Bowel Urgency Improvement (Change in UNRS)
At week 40 of the LUCENT-2 study, patients on mirikizumab experienced a –3.80-point change in UNRS versus the LUCENT-1 study baseline while patients randomized to placebo had a –2.74-point change in score from the LUCENT-1 study baseline (LSM difference = –1.06; 95% CI, –1.51 to –0.61; P < 0.001). Patients on mirikizumab experienced a clinically significant improvement in bowel urgency from baseline while those on placebo did not meet the MID threshold (MID = 3 points from baseline)28 for clinically significant improvement.
Health-Related Quality of Life
HRQoL was assessed in the LUCENT-2 study based on the IBDQ score, EQ-5D-5L score, and SF-36 score.
IBDQ Score
In the LUCENT-2 study, the LSM mean change from the LUCENT-1 study baseline to week 40 in the IBDQ score was 49.8 points and 25.4 points for those in the mirikizumab and placebo groups, respectively; this represented a difference of 25.2 points in favour of mirikizumab (95% CI, 19.2 points to 31.3 points; P < 0.001).The difference between groups was considered clinically meaningful as the difference was above the MID of at least 15 points or greater above placebo.21-24
EQ-5D-5L Score
In the LUCENT-2 study, the LSM difference at week 40 between groups in the EQ VAS score was 20.1 points in the mirikizumab group and 8.8 points in the placebo group, representing a difference of 11.3 points between the groups (95% CI, ||| || ||||; P < 0.001). The change from baseline in the mirikizumab group was clinically important based on an MID of 10.9 points on the EQ VAS.25 It is unclear whether the difference between groups was clinically important given that no MID was identified for between-group differences.
SF-36 Score
At week 40 of the LUCENT-2 study, patients randomized to mirikizumab experienced an LSM change from induction baseline in SF-36 PCS of 9.0 points compared to 6.7 points in patients randomized to placebo, a 2.3-point difference between groups (95% CI, 1.1 points to 3.5 points; P < 0.001). In SF-36 MCS, mirikizumab patients had an LSM change from induction baseline of 7.0 points compared to 5.5 points in the placebo group (LSM change difference between groups = 1.5 points; 95% CI, 0.1 points to 2.8 points; P = 0.031). The change from baseline was clinically important in the mirikizumab group based on an MID threshold at least 3 points.29 The clinical importance of the difference between groups is uncertain given that no MID was identified for between-group differences.
Mucosal Healing: HEMR
A greater proportion of patients randomized to mirikizumab experienced HEMR (a stricter outcome than HEMI) than those randomized to placebo at week 40 — 43.3% versus 21.8%, respectively (common risk difference = 19.9%; 95% CI, 12.1% to 27.6%; P < 0.001). Analyses in the ITT population were consistent with the mITT population results.
HEMR was analyzed by subgroups. In the biologic-naive subgroup, 47.2% of patients on mirikizumab and 26.3% of patients on placebo experienced HEMR at week 40, with a common risk difference of 20.8% (95% CI, 10.5% to 31.2%). In the biologic-failed subgroup, 36.8% of patients on mirikizumab and 13.8% of patients on placebo experienced HEMR at week 40, with a common risk difference of 21.2% (95% CI, 10.9% to 31.4%). The magnitude of the effect was consistent with the primary analysis.
Subgroup results for baseline corticosteroid use (yes and no), immunomodulator use (yes and no), and patients with moderate and severe UC at baseline were consistent with the results of the primary analysis. Appendix 1 contains the LUCENT-2 trial subgroup data.
Work Productivity
At week 40 of the LUCENT-2 study, patients randomized to the mirikizumab group had an LSM change from induction baseline of –31.72 points from the LUCENT-1 study baseline and placebo patients had an LSM change from induction baseline of –22.59 points from the LUCENT-1 study baseline, equating to an LSM difference of –9.13 points between groups (95% CI, –14.26 points to –4.01 points; P < 0.001). An MID was not identified for the WPAI:UC in patients for UC; hence, the clinical importance of the results is uncertain.
LUCENT-2 Study Extended Induction
Of the mirikizumab induction nonresponders from the LUCENT-1 trial, 272 patients entered the open-label extended induction arm of the LUCENT-2 trial in the mITT population. Of the 272 patients who underwent extended induction in the LUCENT-2 trial, 146 (53.7%) patients (95% CI, 47.8% to 59.6%) experienced a delayed clinical response after 24 weeks of continuous therapy with mirikizumab 300 mg IV, for a total of 6 doses. Additionally for this cohort of 272 patients, the rates of clinical remission, endoscopic remission, and symptomatic remission were 11.4% (95% CI, 7.6% to 15.2%), 16.5% (95% CI, 12.1% to 21.0%), and 37.1% (95% CI, 31.4% to 42.9%), respectively, at week 12 of the LUCENT-2 study. Furthermore, of the 146 mirikizumab induction nonresponders who experienced delayed clinical response at week 12 in the LUCENT-2 study, 144 patients entered the open-label maintenance period and 72.2% (n = 104) of patients maintained clinical response at week 40 versus ||||| ||||||| of placebo induction nonresponders who entered the maintenance period. Clinical response at week 40 was not evaluated in the LUCENT-2 trial for induction responders from the LUCENT-1 trial. Hence, no comment can be made on the difference in treatment effects between these 2 cohorts at week 40.
Table 17: Summary of Key Efficacy Results From LUCENT-2 Study (mITT Population at Week 40)
Outcome | LUCENT-2 (maintenance trial) | |
|---|---|---|
Mirikizumab 200 mg SC q.4.w. N = 365 | Placebo SC q.4.w. N = 179 | |
Clinical remission | ||
Patients contributing to the analysis, n | 365 | 179 |
Patients with clinical remission, n (%) | 182 (49.9) | 45 (25.1) |
Common risk difference, % (95% CI) | 23.2 (15.2 to 31.2) | |
P value | < 0.001 | |
Alternate clinical remission | ||
Patients contributing to the analysis, n | 365 | 179 |
Patients with alternate clinical remission, n (%) | 189 (51.8) | 47 (26.3) |
Common risk difference, % (95% CI) | 24.1 (16.0 to 32.2) | |
P value | < 0.001 | |
Durable clinical remission | ||
Patients contributing to the analysis, n | 143 | 65 |
Patients with durable clinical remission, n (%) | 91 (63.6) | 24 (36.9) |
Common risk difference, % (95% CI) | 24.8 (10.4 to 39.2) | |
P value | < 0.001 | |
Corticosteroid-free remission | ||
Patients contributing to the analysis, n | 365 | 179 |
Patients with corticosteroid-free remission, n (%) | 164 (44.9) | 39 (21.8) |
Common risk difference, % (95% CI) | 21.3 (13.5 to 29.1) | |
P value | < 0.001 | |
Endoscopic remission | ||
Patients contributing to the analysis, n | 365 | 179 |
Patients with endoscopic remission, n (%) | 214 (58.6) | 52 (29.1) |
Common risk difference, % (95% CI) | 28.5 (20.2 to 36.8) | |
P value | < 0.001 | |
Bowel urgency improvement (change in UNRS score) | ||
Patients contributing to the analysis, n | 316 | 104 |
LSM change from baseline (SE) | –3.80 (0.139) | –2.74 –(0.202) |
LSM difference in change from baseline (95% CI) | –1.06 (–1.51 to –0.61) | |
P value | < 0.001 | |
Bowel urgency remission (among those with UNRS ≥ 3 at the LUCENT-1 study’s baseline)a | ||
Patients contributing to the analysis, n | 336 | 172 |
Patients with bowel urgency remission, n (%) | 144 (42.9) | 43 (25.0) |
Common risk difference, % (95% CI) | 18.1 (9.8 to 26.4) | |
P value | < 0.001 | |
IBDQ scorea | ||
Patients contributing to the analysis, n | 365 | 179 |
LSM change from baseline (SE) | 49.8 (2.1) | 24.5 (2.8) |
LSM difference in change from baseline (95% CI) | 25.2 (19.2 to 31.3) | |
P value | < 0.001b | |
EQ-5D-5L scorea | ||
|||||||| |||||||||||| || ||| ||||||||| | ||| | ||| |
LSM change from baseline (SE) | 20.1 (|||) | 8.8 (|||) |
LSM difference in change from baseline (95% CI) | 11.3 (||| || ||||) | |
P value | < 0.001b | |
SF-36 score (PCS) | ||
Patients contributing to the analysis, n | 365 | 179 |
LSM change from baseline (SE) | 9.0 (0.4) | 6.7 (0.5) |
LSM difference in change from baseline (95% CI) | 2.3 (1.1 to 3.5) | |
P value | < 0.001b | |
SF-36 score (MCS) | ||
Patients contributing to the analysis, n | 365 | 179 |
LSM change from baseline (SE) | 7.0 (0.5) | 5.5 (0.6) |
LSM difference in change from baseline (95% CI) | 1.5 (0.1 to 2.8) | |
P value | 0.031b | |
HEMR | ||
Patients contributing to the analysis, n | 365 | 179 |
Patients with HEMR, n (%) | 158 (43.3) | 39 (21.8) |
Common risk difference, % (95% CI) | 19.9 (12.1 to 27.6) | |
P value | < 0.001 | |
WPAI:UC (overall work impairment) scorea (among those who were employed at baseline) | ||
Patients contributing to the analysis, n | 196 | 107 |
LSM change from baseline (SE) | –31.72 (1.726) | –22.59 (2.261) |
LSM difference in change from baseline (95% CI) | –9.13 (–14.26 to –4.01) | |
P value | < 0.001b | |
CI = confidence interval; EQ-5D-5L = 5-Level EQ-5D; HEMR = histologic endoscopic mucosal remission; IBDQ = Inflammatory Bowel Disease Questionnaire; LSM = least squares mean; MCS = mental component summary; mITT = modified intention-to-treat; PCS = physical component summary; q.4.w. = every 4 weeks; SC = subcutaneous; SE = standard error; SF-36 = Short Form (36) Health Survey; UNRS = Urgency Numeric Rating Scale; WPAI:UC = Work Productivity and Activity Impairment Questionnaire: Ulcerative Colitis.
Notes: Details in Table 17 have been taken from the sponsor’s Summary of Clinical Evidence.20,30
The 2-sided alpha was 0.05 for the LUCENT-2 study.
aIn the LUCENT-2 trial, UNRS, IBDQ, EQ-5D-5L, and WPAI:UC scores are reported as change from the LUCENT-1 study’s baseline to week 40 (e.g., 52 continuous weeks).
bP value has not been adjusted for multiple testing.
Source: Clinical Study Report for the LUCENT-2 study.32
Harms
A summary of harms reported in the LUCENT-1 and LUCENT-2 trials is provided in Table 18.
Adverse Events
For both the LUCENT-1 trial and the LUCENT-2 trial, the overall rate of AEs was similar between groups. In the LUCENT-1 study, 44.5% and 46.1% of patients reported an AE in the mirikizumab and placebo groups, respectively. In the LUCENT-2 study, 64.5% and 68.8% of patients reported an AE in the mirikizumab and placebo groups, respectively. Over 12 weeks of treatment in the LUCENT-1 study, the most common AEs for patients on mirikizumab 300 mg IV included nasopharyngitis (mirikizumab group = 4.1%; placebo group = 3.1%), anemia (mirikizumab group = 3.3%; placebo group = 5.9%), and headache (mirikizumab group = 3.3%; placebo group = 2.8%).
Over 40 weeks of treatment in the LUCENT-2 study, the most common AEs for patients on mirikizumab 200 mg SC included nasopharyngitis (mirikizumab group = 7.2%; placebo group = 5.7%), arthralgia (mirikizumab group = 6.7%; placebo group = 4.2%), and UC (mirikizumab group = 6.7%; placebo group = 20.8%).
Serious Adverse Events
Overall, the rate of SAEs in the LUCENT-1 and LUCENT-2 studies was found to be lower in patients treated with mirikizumab than with placebo; however, this was due to UC being included as a harm. In the LUCENT-1 study, 2.8% and 5.3% of patients reported an SAE in the mirikizumab and placebo groups, respectively. In the LUCENT-2 study, 3.3% and 7.8% of patients reported an SAE in the mirikizumab and placebo groups, respectively. In the LUCENT-1 trial, the most common SAEs in those on mirikizumab IV included UC (mirikizumab group = 0.8%; placebo group = 3.1%) and pneumonia (mirikizumab group = 0.2%; placebo group = 0%).
In the LUCENT-2 trial, no SAE (at the “preferred term” level) occurred in more than 1 patient on mirikizumab SC.
Withdrawals Due to Adverse Events
In the LUCENT-1 study, 1.6% and 7.2% of patients withdrew from the trial due to an AE in the mirikizumab and placebo groups, respectively. In the LUCENT-2 study, 1.5% and 8.3% of patients withdrew from the trial due to an AE in the mirikizumab and placebo groups, respectively. Withdrawals due to AEs occurred at a lower rate in mirikizumab-treated patients compared to placebo-treated patients in the LUCENT-1 and LUCENT-2 trials, ||||||| |||||||||| ||||||| |||||| ||| |||||||||| || || || ||||||| || ||||||||||||||||||| ||||||| |||||||||||||||| ||||||||||||| |||||||||||||||| |||||||||||||||| ||||||||||||||||| ||||||||||| ||| ||||||||||| |||||||||||||||| ||||||||| |||| ||| |||| |||||| || ||||| || ||||||||||| || || |||||||||
|||||||||| ||||||| ||| ||| |||| || ||||||||| |||| |||| ||||||| ||||||||||| |||||| |||||| ||||| || ||||||||| |||| |||| || ||||||||||| || |||||||| ||| |||| || ||||||| |||||||| ||||||||| |||| || || |||||| ||| |||||||| ||||||||||
Mortality
In the LUCENT-1 trial, no deaths were recorded. In the LUCENT-2 study, 1 (0.5%) death was recorded in the placebo group due to COVID-19.
Notable Harms
Most AESIs occurred at a similar rate between mirikizumab and placebo patients in the LUCENT-1 and LUCENT-2 studies. One exception was the rate of injection site reactions in the LUCENT-2 study, where 8.7% of patients on mirikizumab SC experienced this AESI compared to 4.2% of patients on placebo SC.
The rates of opportunistic infection, cerebrocardiovascular events, malignancy, depression, suicide/self-injury, and hepatic-related AEs were low overall and similar between groups for both the LUCENT-1 trial and the LUCENT-2 trial.
Table 18: Summary of Harms — Pivotal and RCT Evidence (Safety Population)
Harms, n (%) | LUCENT-1 (induction trial, week 12) | LUCENT-2 main cohort (maintenance trial, week 40) | ||
|---|---|---|---|---|
Mirikizumab 300 mg IV q.4.w. (N = 958) | Placebo IV q.4.w. (N = 321) | Mirikizumab 200 mg SC q.4.w. (N = 389) | Placebo SC q.4.w. (N = 192) | |
Most common TEAEs, n (%)a | ||||
Patients with ≥ 1 TEAE | 426 (44.5) | 148 (46.1) | 251 (64.5) | 132 (68.8) |
Nasopharyngitis | 39 (4.1) | 10 (3.1) | 28 (7.2) | 11 (5.7) |
Arthralgia | 20 (2.1) | 4 (1.2) | 26 (6.7) | 8 (4.2) |
Ulcerative colitis | 17 (1.8) | 24 (7.5) | 26 (6.7) | 40 (20.8) |
Injection site pain | NA | NA | 17 (4.4) | 6 (3.1) |
Headache | 32 (3.3) | 9 (2.8) | 16 (4.1) | 2 (1.0) |
Rash | 5 (0.5) | 2 (0.6) | 14 (3.6) | 0 |
Pyrexia | 14 (1.5) | 3 (0.9) | 13 (3.3) | 5 (2.6) |
Anemia | 32 (3.3) | 19 (5.9) | 8 (2.1) | 9 (4.7) |
SAEs, n (%)b | ||||
Patients with ≥ 1 SAE | 27 (2.8) | 17 (5.3) | 13 (3.3) | 15 (7.8) |
Ulcerative colitis | 8 (0.8) | 10 (3.1) | 0 | 6 (3.1) |
Pneumonia | 2 (0.2) | 0 | 0 | 0 |
AEs resulting in treatment discontinuation, n (%)b | ||||
Patient WDAEs | 15 (1.6) | 23 (7.2) | 6 (1.5) | 16 (8.3) |
|||||||||| ||||||| | ||||| | || ||||| | ||||| | || ||||| |
|||||||||||||||| |||||||||||||||| | ||||| | || ||||| | || ||||| | || ||||| |
||||||||||| | ||||| | || ||||| | || ||||| | || ||||| |
Deaths, n (%) | ||||
Patients who died | 0 | 0 | 0 | 1 (0.5) |
COVID-19 infection | 0 | 0 | 0 | 1 (0.5) |
Notable harms, n (%) | ||||
All infections | 145 (15.1) | 45 (14.0) | 93 (23.9) | 44 (22.9) |
Serious infections | 7 (0.7) | 2 (0.6) | 3 (0.8) | 3 (1.6) |
Opportunistic infections | 5 (0.5) | 1 (0.3) | 5 (1.3) | 0 |
Candidiasis | 1 (0.1) | 0 | 1 (0.3) | 0 |
Cytomegalovirus disease | 2 (0.2) | 0 | 0 | 0 |
Herpes zoster | 1 (0.1) | 1 (0.3) | 4 (1.0) | 0 |
Tuberculosis | 1 (0.1) | 0 | 0 | 0 |
Hepatic-related | 15 (1.6) | 5 (1.6) | 12 (3.1) | 4 (2.1) |
Immediate hypersensitivity reaction | 10 (1.0) | 1 (0.3) | 7 (1.8) | 2 (1.0) |
Infusion/injection site reaction | 4 (0.4) | 1 (0.3) | 34 (8.7) | 8 (4.2) |
Depression | 4 (0.4) | 2 (0.6) | 4 (1.0) | 0 |
Malignancies | 2 (0.2) | 0 | 1 (0.3) | 1 (0.5) |
Cerebrocardiovascular events | 1 (0.1) | 2 (0.6) | 0 | 1 (0.5) |
Suicide/self-injury | 0 | 0 | 1 (0.3) | 0 |
AE = adverse event; NA = not applicable; q.4.w. = every 4 weeks; RCT = randomized controlled trial; SAE = serious adverse event; SC = subcutaneous; TEAE = treatment-emergent adverse event; WDAE = withdrawal due to adverse event.
aFrequency of 3% or more of patients in any treatment group during the LUCENT-1 or LUCENT-2 study.
bFrequency of 2 or more patients in any treatment group during the LUCENT-1 or LUCENT-2 study.
Note: Details in Table 18 have been taken from the sponsor’s Summary of Clinical Evidence.20,30
Sources: Clinical Study Reports for the LUCENT-1 and LUCENT-2 studies.31,32
Critical Appraisal
Internal Validity
The LUCENT-1 and LUCENT-2 trials were parallel-arm, multicentre, double-blind, placebo-controlled randomized trials. Both trials employed appropriate methods for blinding, treatment allocation, and randomization. Randomization was stratified appropriately by important effect modifiers and used an interactive web response system for concealment of the randomization assignment. There were no notable differences between treatment arms for most baseline characteristics. The use of separate induction and maintenance studies was appropriate and consistent with other studies assessing other medications for the treatment of UC.73 Statistical analyses were appropriate. In addition, a prespecified graphical multiple testing approach was used to control key secondary outcomes for multiplicity. The study was powered to detect a difference in the primary end point between treatment arms and the enrolled sample size was adequate. A hierarchical testing procedure was appropriately used to account for multiplicity in the primary and key secondary outcomes.
Many of the primary and major secondary end points as well as patient-reported outcomes (UNRS, IBDQ, EQ-5D-5L, and SF-36), were dependent on the complete and accurate daily recording of the Mayo SF and RB subscores and other data points by patients in their electronic diary. Daily reporting from patients is subjective and subject to error, and can lead to possible reporting bias and recall bias potentially in favour of mirikizumab if patients became unblinded. However, blinding was appropriate; hence, it is difficult to predict the direction of bias. The sponsor noted that there was a patient adherence review every 2 weeks at each study visit to ensure adherence to diary recordings. However, no details were provided on how adherence was reviewed. In addition, details were not provided on how patients were trained to use the electronic diary or tablet device that was used for the diary recordings to mitigate user error. In 2 of the study countries, Poland and Turkey, there was also a transcription error that led to 104 patients being excluded from the LUCENT-2 study’s primary efficacy analysis (but not the safety analysis). Overall, using multiple imputation to impute missing data due to transcription errors, COVID-19, loss to follow-up, or a protocol deviation did not have an impact on the primary or secondary end points.
Both pivotal trials used the mITT population for the main analysis rather than the ITT population. While the preferred approach would be to use the ITT population, results of the sensitivity analyses of the ITT population were aligned with the mITT results across all end points, which increases the certainty of the findings. The methods used for the sensitivity analyses to assess impact of attrition and missing data were valid and appropriate.
In both the LUCENT-1 study and the LUCENT-2 study, a notably higher proportion of patients in the placebo arm (11.5% and 38%, respectively) discontinued from the study compared with the mirikizumab arm (4.1% and 10.8%, respectively). The main reason for discontinuation was ||| || |||| |||||||||| ||| || ||| ||||||||||| || || ||||||. Prespecified sensitivity analyses were conducted to account for attrition and missing data and the results were consistent with the main analysis, increasing the certainty of the findings.
HRQoL and work productivity were identified as an important outcome by the patient and clinician groups providing input for this review. The IBDQ, EQ-5D-5L, and SF-36 were valid and reliable measuring instruments of HRQoL for patients with UC. MIDs were provided by the sponsor for the IBDQ, EQ-5D-5L, and SF-36 (PCS and MCS), which were in line with thresholds reported in the literature. MID estimates for these instruments have been established in patients with UC, although in the case of the EQ-5D-5L, sometimes the literature was based on IBD generally. An MID was provided for the WPAI:UC in patients with Crohn disease; however, this was not taken into consideration given the alternative patient population.
The LUCENT-2 trial enrolled 405 patients from the LUCENT-1 trial who had not responded to 12 weeks of induction dosing with either mirikizumab or placebo. These patients received open-label mirikizumab (300 mg administered intravenously) for 12 weeks. This was referred to as an extended induction period for patients who had previously received 12 weeks of induction dosing (i.e., 24 weeks of continuous therapy). As patients in the extended induction cohort received open-label mirikizumab, results should be interpreted with caution given the potential risk of detection or performance bias due to the open-label nature. In terms of subgroup analyses, the LUCENT-1 trial had included 1 subgroup analysis as part of its statistical testing hierarchy: clinical response in the biologic-experienced group. The results of the subgroup analysis were consistent with the results of the primary analysis, with mirikizumab being superior to placebo. No testing was done to compare between subgroup differences; hence, no conclusions could be drawn on whether mirikizumab is more beneficial in biologic-naive versus biologic-experienced populations. In terms of the other relevant prespecified subgroup analyses |||||| |||||||||||| ||||||||||| || |||||||| ||| ||| |||||||| || |||||||||| || ||||||||||| ||| || ||||| ||| || ||| |||| || |||||| |||| ||||||||||||| ||| |||| || ||||||| ||| |||||||||||| ||| |||||||| || ||| ||||||||| Hence, subgroup results should be considered exploratory.
External Validity
The clinical expert consulted by CADTH considered the baseline demographic and disease characteristics in the pivotal trials to be reflective of patients with moderate to severe UC seen in Canadian clinical practice. In addition, the clinical expert noted that the measurement instruments used were appropriate. Inclusion and exclusion criteria aligned with the selection criteria for candidates for mirikizumab treatment. However, the criteria that excluded patients who had received or whose UC had failed to respond to 3 or more biologic therapies may have excluded some patients unnecessarily, given that some patients may have received a biologic but stopped taking it due to reasons other than clinical failure (e.g., financial reasons). Hence, low socioeconomic patients may have been systemically excluded as a subgroup. Furthermore, there was a high number of screening failures (35%) in the LUCENT-1 study; hence, there may have been potential for selection bias in terms of how patients were being referred to the trial, according to the clinical expert.
Concomitant medication use was reflective of medications used in Canadian clinical practice with the exception of a few drugs, such as prednisolone, which is not typically used in Canada. In the LUCENT-2 trial, the maintenance trial, a corticosteroid taper was trialled on all patients in the main cohort. Patients who could not taper corticosteroids were not considered to be treatment failures; rather, tapering was reset or paused. This criterion contrasts with the input received from the clinical expert who indicated that patients would be considered treatment failures and discontinue treatment with the drug if they could not taper or stop concomitant corticosteroid use by the time of the maintenance phase (i.e., after the induction or extended induction period).33 Therefore, the efficacy of mirikizumab in the trials may appear to be biased given that patients who could not taper were included in the primary analysis, even though they would have been considered treatment failures in clinical practice. However, the direction and magnitude of this bias is unknown, given that both groups underwent the same tapering protocol. Furthermore, the generalizability of the results may be limited to Canadian clinical practice due to the discrepancy in tapering protocol.
The trial criterion in the LUCENT-1 study of the achievement of clinical response, which directed entry into the primary cohort of the LUCENT-2 study, may have created an enriched patient population in the maintenance trial as it did not take into consideration delayed responders and only included patients who showed a timely response. As per the product monograph, extended induction dosing of mirikizumab may be considered in patients who experience delayed response (i.e., in adequate therapeutic response at week 12 after induction dosing); hence, by excluding these patients in the primary analysis, there is uncertainty about the efficacy of maintenance treatment in the broader population of patients with moderately to severely active UC.
The clinical expert noted that the duration of follow-up in the LUCENT-1 trial (i.e., 12 weeks) may not have been sufficient to adequately assess certain end points, such as endoscopic remission. The clinical expert noted that it was unlikely to see a difference in endoscopic outcomes by week 12 in the trial. However, the issue of insufficient duration was addressed by the LUCENT-2 trial, which measured end points at the week-40 time point (i.e., 52 weeks of continuous therapy). Long-term efficacy data beyond 52 weeks was not available; therefore, long-term outcomes such as durable clinical remission or endoscopic remission may not have been sufficiently captured between the 2 trials.
Long-Term Extension Studies
Content in this section has been informed by materials submitted by the sponsor. The following has been summarized and validated by the CADTH review team.
There are currently no published or unpublished long-term extension phase III or phase IV RCTS or real-world evidence studies. The sponsor did make note that there is an ongoing phase III, open-label, long-term extension trial — the LUCENT-3 study (I6T-MC-AMAP) — which is enrolling patients from the LUCENT-2 study and the phase II I6T-MC-AMAC study (NCT02589665).97 The objective of the LUCENT-3 trial is to determine the efficacy and safety of mirikizumab 200 mg SC every 4 weeks in various clinical, patient-reported, and health outcomes at various time points up to 160 weeks (i.e., 160 weeks in the LUCENT-3 study, which represents up to 212 weeks of continuous mirikizumab therapy for patients coming from the LUCENT-2 trial). The expected primary completion date for the LUCENT-3 trial is June 6, 2025.
Indirect Evidence
Content in this section has been informed by materials submitted by the sponsor. The following has been summarized and validated by the CADTH review team.
Objectives for the Summary of Indirect Evidence
In the absence of direct head-to-head trials evaluating the comparative efficacy and safety of mirikizumab versus relevant comparators for moderately to severely active UC in adults, the sponsor submitted a systematic review with an NMA.73 The sponsor-conducted NMA was used to inform the sponsor-submitted economic model for mirikizumab.
Description of Indirect Comparison
The sponsor submitted 1 NMA comparing the relative efficacy of induction and maintenance treatment with mirikizumab versus other advanced therapies on clinical response rate, clinical remission rate, and mucosal healing in adult patients with moderately to severely active UC. The NMA also assessed the relative safety of induction treatment with mirikizumab versus other advanced therapies on all-cause discontinuation and SAEs in the indication population.
ITC Design
Objectives
The objective of the sponsor-submitted NMA was to evaluate the relative efficacy and safety of mirikizumab versus relevant advanced therapies for moderately to severely active UC in adults.
Study Selection Methods
The studies eligible for inclusion in the sponsor-submitted NMA were selected according to a sponsor-conducted systematic review.36 The sponsor’s systematic review was defined by the relevant population, intervention, comparators, outcomes, and study design (PICOS) described in Table 19. The scope of the systematic review included RCT evidence for adult patients undergoing treatment for moderately or severely active UC defined by a Mayo Score or by the Ulcerative Colitis Disease Activity Index.
Clinical evidence for the systematic review was identified using multiple electronic databases and trial registries as listed in Table 20, along with handsearching of conference proceedings. The reference lists of the 5 most relevant systematic reviews and meta-analysis were also scanned. The literature search is current to June 9, 2022. In addition to articles identified in the systematic review, the unpublished LUCENT-1 and LUCENT-2 trials were considered in the NMA. Articles were screened by 2 independent reviewers; a third reviewer was consulted to resolve any discrepancies. Data extraction was conducted by a single reviewer with a quality check by another. An assessment of risk of bias of included articles was conducted using the checklist for RCTs from CRD’s Guidance for Undertaking Reviews in Health Care (2009), courtesy of the Centre for Reviews and Dissemination.74 Critical appraisals were not conducted for conference proceedings due to insufficient methodological data to assess the study quality. No studies were excluded based on the risk of bias assessment. The methods used for risk of bias assessment were not reported.
Table 19: PICOS for Sponsor-Conducted Systematic Literature Review
PICOS | Inclusion criteria |
|---|---|
Population | Adult patients older than18 years with moderate to severe UC |
Intervention | Approved targeted therapies and biosimilars:
Emerging therapies:
|
Comparator |
|
Outcomes | Induction phase:
Maintenance phase:
Additional outcomes:
Safety outcomes:
|
Study design | RCT |
AE = adverse event; IBDQ = Inflammatory Bowel Disease Questionnaire; PGA = physician’s global assessment; PICOS = population, intervention, comparators, outcomes, and study designs; RB = rectal bleeding; RCT = randomized controlled trial; SAE = severe adverse event; SF = stool frequency; UC = ulcerative colitis; UCDAI = Ulcerative Colitis Disease Activity Index.
Source: Systematic literature review technical report.36
The study selection and methods for inclusion in the NMA are summarized in Table 20. For the purposes of the NMA, the population of interest was based on the population recruited in the LUCENT-1 and LUCENT-2 trials. All EMA-approved and FDA-approved doses and regimens of targeted therapies for the treatment of moderately to severely active UC were included as comparators in the NMA. Different dosing arms of the same medication were treated as individual comparators. Studies from the systematic review that did not meet the approved therapies, doses, and regimens criteria were not included in the NMA. Of note, among the comparators eligible for inclusion in the NMA, filgotinib and upadacitinib are currently not approved for use in Canada. Moreover, the ustekinumab maintenance regimen of 90 mg every 12 weeks is not used in Canada; the approved maintenance dose used in the Canadian practice setting is 90 mg SC every 8 weeks.
Outcomes evaluated in the NMA included clinical response and remission and mucosal healing and endoscopic remission for the induction and maintenance phases, as well as all-cause discontinuation (lack of efficacy and AEs) and the incidence of SAEs (grade 3 and grade 4 AEs) during the induction phase. The definitions of clinical response and remission and the definitions for safety end points were consistent with the definitions used in the included clinical trials. For the purposes of the NMA, the definition of mucosal healing was aligned with the LUCENT trials as being an ES of 0 or 1. Of note, due to different trial designs for the maintenance phase, the placebo safety populations in the maintenance phases across studies were not comparable. For example, mirikizumab patients in the LUCENT-1 study whose UC responded to blinded placebo were continued on blinded placebo in the LUCENT-2 study. However, in the tofacitinib phase III OCTAVE trial, placebo responders were rerandomized in the maintenance phase. Accordingly, only safety outcomes reported during the induction phase were analyzed.
Table 20: Study Selection Criteria and Methods for NMA Submitted by Sponsor
Characteristics | Indirect comparison |
|---|---|
Population | Aligned with patients enrolled in the LUCENT-1 and LUCENT-2 studies: Adults (≥ 18 years) with moderately to severely active UC, defined by Mayo Score (full Mayo Score from 6 to 12 points with endoscopic subscore ≥ 2) or UCDAI |
Intervention | Induction: Mirikizumab 300 mg IV q.4.w. Maintenance: Mirikizumab 200 mg SC q.4.w. |
Comparator | Comparator dosing was based on EMA-approved and FDA-approved dosing.
|
Outcome | Efficacy End of inductionb and maintenancec:
Safety Induction onlyb:
|
Study design | Published RCTs (unpublished for mirikizumab only) |
Exclusion criteria |
|
Sources searched | Full publications from databases:
Conference proceedings:
Ongoing trials:
|
Search limits | Years of publication: 1990 to June 9, 2022 English language |
Selection process | Title-abstract and full-text screening conducted by 2 independent reviewers, with a third reviewer consulted if needed |
Data extraction process | Independent extraction by a single reviewer with verification of outcome data by another. A project manager performed a quality check for 10% of included studies. |
Quality assessment | Checklist for RCTs from CRD’s guidance for undertaking reviews in health care (2009), courtesy of the Centre for Reviews and Dissemination. Methods not reported. |
b.i.d. = twice a day; EMA = European Medicines Agency; NMA = network meta-analysis; p.o. = orally; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; q.8.w. = every 8 weeks; q.12.w. = every 12 weeks; RCT = randomized controlled trial; SC = subcutaneous; UC = ulcerative colitis; UCDAI = Ulcerative Colitis Disease Activity Index.
Note: Details in Table 20 have been taken from the sponsor’s Summary of Clinical Evidence.30
aHere, 1 mg of ozanimod refers to 1 mg of hydrochloride salt. 1 mg of ozanimod hydrochloride equals 0.92 mg of molecular ozanimod, which aligns with the dose form available in Canada.
bCaptured at the end of the induction period of the respective study.
cCaptured at the end of the maintenance period of the respective study.
Sources: Network meta-analysis technical report and systematic literature review technical report.21,36
ITC Analysis Methods
Model Structure
Efficacy analyses were conducted for biologic-naive, biologic-experienced, and Asian-only subgroups of patients. Safety analyses were conducted on the overall population in the induction phase only. The NMA was powered by a Bayesian model using the Markov chain Monte Carlo method. A noninformative prior was used for unknown data. For the treatment effect prior, a normal density distribution was used (mean = 0; variance = 100). A normal prior (mean = 0; variance = 10,000) was used for the study-specific intercepts and for random-effects models, a half-normal prior (mean = 0; SD = 5) was used for the between-study SD.
A multinomial model with a probit link was used to account for the mutually exclusive nature of the relationship between response and remission outcomes (i.e., those who do not experience response cannot be in remission). For other dichotomous outcomes, a binomial model with a logit link was used.
A common baseline was estimated using all placebo arms, with the mean estimated using a normal prior (mean = 0; variance = 10,000). The between-study SD was estimated with a half-normal prior (mean = 0; SD = 5). Both fixed-effects and random-effects models were considered, and the deviance information criterion was used to select the most appropriate model.75 Various graphical and statistical methods were used to assess model performance and facilitate selection.
Heterogeneity
There are 2 main trial designs related to the transition from induction to maintenance dosing: treat-through trial design and rerandomized responder design. To mitigate the heterogeneity caused by comparing patients from different trial designs, the treat-through trial results were adjusted for both response and remission end points. The adjustment incorporated these key assumptions:
The total number of responders in treat-through trials during induction acted as a proxy for the number of patients entering maintenance.
The adjusted clinical response in maintenance was based on the proportion of patients achieving durable or sustained response during maintenance in treat-through trials (prevents counting induction nonresponders).
The adjusted clinical remission in maintenance was based on the number of patients achieving remission during the maintenance phase in treat-through trials (this assumed those in remission at maintenance were at least in response at induction).
To account for heterogeneity in patients and trial factors that are known to have a measurable impact on placebo response rates, baseline risk–adjusted models were considered that included study location, trial duration, disease status, disease duration, and prior exposure to biologic therapy.76-80
Heterogeneity was assessed statistically by evaluating the posterior distribution for the between-study SD. If statistical heterogeneity was present, meta-regression, subgroup analysis, or random-effect modelling were considered.
Consistency was evaluated by comparing direct and indirect evidence for conflicting results. Tests using the unrelated mean effects model (i.e., the inconsistency model) were conducted, with results compared to that of the standard NMA using model fit statistics and the deviance contribution.
Sensitivity Analysis
Sensitivity analyses were conducted to explore the influence of potential outlier studies on the overall NMA result. Of note, data from treat-through design trials were excluded from the analysis of maintenance phase clinical response and remission in the sensitivity analysis. Given that the treat-through and rerandomized responder trial designs are so distinct from each other, the sensitivity analysis was used to provide insight on whether the approach in the base case NMA to standardize treat-through trials was sufficient in mitigating heterogeneity.
Table 21: NMA Analysis Methods
Methods | Description |
|---|---|
Analysis methods | NMA powered by a Bayesian model using MCMC. Multinomial model with probit link or binomial model with logit link. |
Priors | Noninformative priors used |
Assessment of model fit | Various: DIC, convergence by Gelman-Rubin Rhat (potential scale reduction factor)and Monte Carlo Standard Error |
Assessment of consistency | Comparison of direct and indirect evidence from closed loops; comparison to unrelated mean effects model |
Assessment of convergence | Gelman-Rubin Rhatand Monte Carlo Standard Error |
Outcomes | Clinical response rate, clinical remission rate, mucosal healing, all-cause discontinuation rate, and rate of any serious adverse event |
Follow-up time points | At the end of the induction phase and maintenance phase for efficacy, and only at the end of induction phase for safety |
Construction of nodes | Each node represents an FDA-approved or EMA-approved dosing regimen for UC. A different approved dose, dosing frequency, or route of administration would be represented by its own node. Drugs of the same mechanism of action are not grouped into the same node. |
Sensitivity analyses | Exclusion of treat-through design trial data to evaluate the impact on statistical heterogeneity |
Subgroup analyses | Biologic-naive, biologic-experienced, and Asian-only subgroup analyses for efficacy. No subgroup analysis for safety (mixed population only) |
Methods for pairwise meta-analysis | Not conducted |
DIC = deviance information criterion; EMA = European Medicines Agency; MCMC = Markov chain Monte Carlo; NMA = network meta-analysis; UC = ulcerative colitis.
Note: Details in Table 21 have been taken from the sponsor’s Summary of Clinical Evidence.30
Sources: Network meta-analysis technical report and systematic literature review technical report.21,36
Results of NMA
Summary of Included Studies
In total, || publications reporting on || unique studies were included in the systematic literature review. The inclusion of the unpublished LUCENT trials resulted in || unique studies eligible for inclusion in the NMA. Of these, || studies were excluded from the NMA. In || of those, the reason for exclusion was the inclusion of a non–EMA-approved or non–FDA-approved dosing regimen. ||| ||||| was excluded due to not meeting the inclusion criteria for the study population and another for not presenting outcome data by subgroups for prior exposure to biologics and/or JAK inhibitors. In total, || studies were included in the NMA. An overview of the included studies is summarized in Table 21.
Of the included studies, || were induction studies (|| reported on induction clinical response; || on induction clinical remission and || on induction mucosal healing) and || maintenance studies (|| reported on maintenance clinical response, || reported maintenance clinical remission, and 12 on maintenance mucosal healing). At least 1 safety outcome for the overall population was reported in || studies. All included studies were double-blinded studies with sample size ranging from || || ||| patients. The duration of therapy ranged from | ||||| || || ||||| for induction and || ||||| || || ||||| for maintenance.
Baseline patient characteristics across the studies included in the NMA were not provided by the sponsor. The following details pertain to the studies included in the NMA feasibility assessment, part of the sponsor’s systematic literature review. Across the studies evaluating induction response, mean age ranged from |||| || |||| |||||, mean disease duration ranged from ||| || |||| |||||||| || ||||| of patients were male, and the mean total Mayo Score values ranged from ||| || |||. Across the studies evaluating maintenance response, mean age ranged from |||| || |||| years, mean disease duration ranged from ||| || |||| years, ||| || ||||| were male, and the mean total Mayo Score values ranged from ||| || |||.
Potential sources of heterogeneity across the included studies are summarized in Table 23. Two major sources of clinical and methodological heterogeneity identified by the sponsors were the differing rates of placebo group response across trials (I2 = |||) and inconsistent maintenance phase treatment allocation (i.e., treat-through versus rerandomization responders) as detailed earlier in the Heterogeneity section.
Additional sources of heterogeneity were identified that were related to the definitions of clinical response, clinical remission, and prior experience with a biologic and/or JAK inhibitor applied across studies. The definition of induction and maintenance clinical response used in the LUCENT-1 trial differed from the commonly reported definition in the NMA (Table 24). Two definitions of induction and maintenance clinical remission were applied in the LUCENT-1 trial. Both these definitions differed from the most commonly reported definitions across studies included in the NMA (Table 25). Finally, the definition of prior experience with a biologic and/or JAK inhibitor varied across studies, as well as the prior therapies that patients may have been exposed to, due to the time period across which the studies occurred. Prior experience with a biologic and/or JAK inhibitor may be defined as TNF-experienced, TNF failure, biologic-experienced, biologic failure, or biologic and/or JAK inhibitor failure. As a result, the categorization of biologic-naive and JAK inhibitor–naive versus biologic-experienced and JAK inhibitor–experienced subgroups across studies was not consistent. Furthermore, permitted concomitant mediations differed across the included studies.
||||| | |||| | ||||||||| | ||||||||||| | ||| || | ||||||||| ||| | ||||||| | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|||81 | ||| | |||| | ||||||||||| | |||||| | |||| | |||||||| | |||| |||| | |||||| | ||||||| | ||||||| | ||||| | ||| |
||||||| |||||||||| |||||||||| |||||||||| |||||||| || ||| |||||||| |||||||| ||||||| | ||||||||||||
|||81 | ||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| || | ||| ||||| |
|||81 | ||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| || | ||| ||||| |
||||||82 | ||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| || | ||| ||||| |
||||||||83 | ||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| || | ||| ||||| |
||||||||83 | ||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| || | ||| ||||| |
||||| ||||84 | ||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| || | ||| ||||| |
|||||||| ||||85 | ||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| || | ||| ||||| |
|||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| || | ||| ||||| | |
|||||| ||||86 | ||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| || | ||| ||||| |
|||||| |87 | ||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| || | ||| ||||| |
|||||| |87 | ||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| || | ||| ||||| |
|||||| |||||||87 | ||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| || | ||| ||||| |
||||||| ||||88 | ||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| || | ||| ||||| |
|||||||||| |89 | ||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| || | ||| ||||| |
|||||||||| |89 | ||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| || | ||| ||||| |
|||||||||90 | ||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| || | ||| ||||| |
||||||||| 91 | ||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| || | ||| ||||| |
|||||||| ||||92 | ||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| || | ||| ||||| |
|||||| ||||93 | ||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| || | ||| ||||| |
|||| |||||94 | ||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| || | ||| ||||| |
|||||95 | ||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| || | ||| ||||| |
||||96 | ||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| || | ||| ||||| |
|||||97 | ||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| || | ||| ||||| |
|||||||98 | ||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| || | ||| ||||| |
||| | ||| ||||| | ||| ||||| | ||||||||||
|||||||99 | ||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| || | ||| ||||| |
||||||| |||||||||| |||||||||| |||||||||| ||| |||||||| || ||| |||||||| |||||||| ||||||| | ||||||||||||
|||||||||100 | ||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| || | ||| ||||| |
|||||||||100 | ||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| || | ||| ||||| |
|||||||| | ||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| || | ||| ||||| |
||||||||||101 | ||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| || | ||| ||||| |
|||||||||101 | ||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| || | ||| ||||| |
||||||||| ||||102 | ||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| || | ||| ||||| |
|||||97 | ||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | ||| || | ||| ||||| |
|||||| ||||| |||||||||||||||| |||||||||||||| ||||||||||||| ||||||||||||| |||||||||||||| |||||||||||||||| |||| ||||||||||| |||||||||| |||||||||||||| ||||| ||||||||| |||||||||||| |||||| |||||| |||||||| || |||||| ||||||||||||| |||||||||||| |||||||||||||| |||||||||||||||| ||||| ||||||| ||||||| ||||| |||||||| |||||||||| |||||||| |||||||||||||||| |||||||||||||| ||||||||||||||||||| ||||||||||||||||| ||| |||||||||||||| |||||||| || || ||||||||||| ||| ||| ||||| |||||||||||| ||||| || ||||||||| || |||| ||| ||||||||| ||| ||||||||||| ||||||||||||| |||||||||| |||| ||||||| || ||||| ||||||||||||||||||| ||||| ||||| ||||||| |||||| |||||||| ||||| || |||| |||| || ||| ||| || ||||||||||| |||||||| ||| |||||||| || ||| |||| |||| |||||||| || |||| |||||||| ||||||| ||| |||||||||||||||| ||| ||| ||| || |||||||||| ||||||||||| |||||| |||||| ||||||| ||| |||||| || ||||| || ||| ||||||||||| |||||| || ||| |||||| |||| ||||| || ||||||||| || |||| ||| ||||||||| ||| ||||||||||| |||||||||||||||| ||| ||||||||| |||||||21
Table 23: Assessment of Homogeneity for Studies Included in the NMAs
Characteristics | Description and handling of potential effect modifiers |
|---|---|
Racea | To account for racial disparity of UC phenotypes across races, the NMA evaluated an “only Asian” subgroup, regardless of prior experience with biologics or JAK inhibitors. The network for this subgroup, however, was small, consisting of 9 studies at induction and 8 studies at maintenance. |
Treatment historya | Prior exposure to biologic therapy at enrolment is a known effect modifier in trials of patients with UC. To account for the potential for heterogeneity due to treatment history, separate analyses were performed for patients who were biologic-naive and biologic-experienced. |
Definition of biologic and/or JAK inhibitor experienceb | The definitions for biologic-naive, JAK inhibitor–naive, biologic-experienced, and JAK inhibitor–experienced populations varied across studies; they also permitted concomitant use. There may be differences within each definition as well. For example, some trials included biologic exposure under biologic-naive as long as the patient did not lose remission. Due to when comparators entered the market, the type of biologics that patients were exposed to would differ. The previously failed (and potentially number of failed) treatments would differ based on the time period during which each study was conducted. The different classifications of biologic-naive and biologic-experienced groups across the studies represents a major source of heterogeneity, which could not be accounted for in the NMA. |
Placebo responsea | Significant heterogeneity was observed in event rates between placebo groups. This was most significant in the maintenance setting for “response (no remission),” with I2 = |||| In an attempt to mitigate the impact of this substantial heterogeneity, meta-regression on this baseline risk was included in “adjusted” models; however, the adjusted model was not always identified as the best fit model and in some comparisons, the unadjusted model was used.21 |
Definitions of end pointsb | Most reported definitions of clinical remission and response differed from what was used in the LUCENT-1 and LUCENT-2 studies. While most comparator studies used the same definition for clinical remission and response based on Mayo Scores, some studies used the full Mayo Score while some evaluated the Modified Mayo Score. The definition of mucosal healing was consistently applied, regardless of the terminology used to describe the outcome in individual publications (endoscopic Mayo subscore of 0 or 1). However, some studies relied on central vs. local endoscopy readings. |
Duration of induction and maintenance periods and timing of end point evaluationb | The induction period ranged from | || ||||| across studies, while the maintenance period ranged from || || || |||||. To mitigate issues related to length of maintenance period, analysis was restricted to outcomes assessed from 52 weeks to 60 weeks. A longer duration of treatment allows more time for treatment to achieve a desirable treatment response; however, longer maintenance duration may lead to an increase in AEs or loss of remission due to prolonged use of a treatment. |
Study designa | Study design (i.e., treat-through vs. rerandomized responders) may be a significant source of heterogeneity. Only some infliximab and adalimumab trials were treat-through, while the vast majority of included trials were of the rerandomized responder design. This was mitigated via 2 mechanisms: standardizing arms from treat-through trials such that they are more similar to rerandomized responder trial arms, and performing sensitivity analysis to remove any treat-through trial arms from the analysis. |
JAK = Janus kinase; NMA = network meta-analysis; UC = ulcerative colitis; vs. = versus.
Note: Some details in Table 23 have been taken from the sponsor’s Summary of Clinical Evidence.30
aIdentified by the sponsor.
bIdentified by the CADTH clinical review team.
Sources: Network meta-analysis technical report and systematic literature review technical report.21,36
Table 24: Definition of Clinical Response Applied Across Studies Included in the NMA
Definition | Na |
|---|---|
Induction phase | |
Decrease in the Modified Mayo Score of ≥ 2 points and ≥ 35% from baseline, as well as a reduction in the RBS of ≥ 1 point or an absolute RBS of 0 or 1b | |||||| |
Decrease in the total Mayo Score ≥ 3 points and ≥ 30%, with an accompanying decrease in the RBS of ≥ 1 point or an absolute RBS of 0 or 1 | |||||| |
Decrease from baseline in the adapted Mayo Score of ≥ 2 points and ≥ 30%, with an accompanying decrease in RBS of ≥ 1 point or an absolute RBS of 0 or 1 | |||||| |
Decrease from baseline in the adapted Mayo Score of ≥ 1 point and ≥ 30%, with an accompanying RBS of ≥ 1 point or an absolute RBS of 0 or 1 | |||||| |
Decrease in the Modified Mayo Score of ≥ 2 points and ≥ 35% from baseline, as well as a reduction in the RBS of ≥ 1 point or an absolute RBS of 0 or 1 | |||||| |
Not reported | |||||| |
Maintenance phase | |
Decrease in the Modified Mayo Score of ≥ 2 points and ≥ 30% from baseline, with an accompanying decrease in the RBS of ≥ 1 point or an absolute RBS of 0 or 1b | |||||| |
Decrease in the total Mayo Score ≥ 3 points and ≥ 30%, with an accompanying decrease in the RBS of ≥ 1 point or an absolute RBS of 0 or 1 | |||||| |
Decrease in the Modified Mayo Score of ≥ 2 points and ≥ 35% from baseline, with an accompanying RBS of ≥ 1 point or an absolute RBS of 0 or 1 | |||||| |
Decrease in the partial adapted Mayo Score of ≥ 1 point and ≥ 30% from baseline, with an accompanying decrease in the RBS of ≥ 1 point or an absolute RBS of 0 or 1 | |||||| |
Decrease in the partial adapted Mayo Score of ≥ 1 point and ≥ 30% from baseline | |||||| |
Not reported | |||||| |
NMA = network meta-analysis; RBS = rectal bleeding subscore.
aThe SELECTION A and SELECTION B studies and the U-ACCOMPLISH phase III study reported results using multiple definitions of response. Thus, the sum of the number of studies exceeds the total number of studies reporting clinical response.
bThis was the definition used in the LUCENT-1 study.
Source: Network meta-analysis technical report.21
Table 25: Definition of Clinical Remission Applied Across Studies Included in the NMA
Definition | Na |
|---|---|
Induction phasea | |
RBS of 0, stool frequency subscore ≤ 1 and decrease from baseline ≥ 1, and endoscopic subscore ≤ 1 (excluding friability)b | |||||| |
RBS of 0, stool frequency subscore ≤ 1, and endoscopic subscore ≤ 1 (excluding friability)b | |||||| |
Total Mayo Score of ≤ 2 points, with no individual subscore > 1 and RBS of 0 | |||||| |
Total Mayo Score of < 2 points, with no individual subscore > 1 and RBS of 0 | |||||| |
Adapted Mayo Score: Stool frequency subscore of ≤ 1 and not greater than baseline, RBS of 0, and endoscopic subscore of ≤ 1. Evidence of friability during endoscopy in patients with otherwise mild endoscopic activity will confer an endoscopic subscore of 2. | |||||| |
Partial Mayo Score ≤ 2 points, with no individual subscore > 1 | |||||| |
Not reported | |||||| |
Maintenance phasec | |
RBS of 0, stool frequency subscore ≤ 1 and decrease from baseline ≥ 1, and endoscopic subscore ≤ 1 (excluding friability)b | |||||| |
RBS of 0, stool frequency subscore ≤ 1, and endoscopic subscore ≤ 1 (excluding friability)c | |||||| |
Total Mayo Score of ≤ 2 points, with no individual subscore > 1 | |||||| |
Total Mayo Score of ≤ 2 points, with no individual subscore > 1 and RBS of 0 | |||||| |
Adapted Mayo Score ≤ 2, stool frequency subscore ≤ 1 and not greater than baseline, RBS of 0, and endoscopic subscore ≤ 1 (excluding friability) | |||||| |
RBS of 0, stool frequency subscore ≤ 1 and decrease from baseline ≥ 1, and endoscopic subscore ≤ 1 | |||||| |
RBS of 0, stool frequency subscore ≤ 1, and endoscopic subscore ≤ 1 | |||||| |
RBS of 0, stool frequency subscore of 0, and endoscopic subscore ≤ 1 (excluding friability) | |||||| |
Not reported | |||||| |
NMA = network meta-analysis; RBS = rectal bleeding subscore.
aThe SELECTION A and SELECTION B studies, the U-ACHIEVE phase IIb study, and the U-ACCOMPLISH phase III study reported results using multiple definitions of remission. Thus, the sum of the number of studies exceeds the total number of studies reporting clinical remission.
bThis was the definition used in the LUCENT-1 study.
cThe LUCENT-1 and LUCENT-2 studies, the SELECTION studies, and the VISIBLE 1 study reported results using multiple definitions of remission. Thus, the sum of the number of studies exceeds the total number of studies reporting clinical remission.
Source: Network meta-analysis technical report.21
Results
|||||||| ||||||| || ||| ||| ||| ||||||||| ||| |||||||| |||||| |||| ||||| ||| ||||||||||| ||||||||||| || |||| |||||| |||||| ||||||||| ||| ||||||||||||| ||||||| ||||||| || |||| |||||||| ||||||||||| ||| ||||||||| || Appendix 1|||||||| |||||||||| |||| |||| ||||| |||| ||| ||| |||||||| ||||||||| ||||| || ||||||||| |||||| ||| ||||||||| |||||||| ||||||||| ||||||||| ||| ||||||| ||||||| ||||||| ||||||||||| ||| ||||||||||| ||| |||||||||| || Table 26 |||||||| |||||||||| |||| || ||| ||| ||| || ||||||||||| |||||||| ||||||||| ||||||||| ||| ||||||| ||||||| ||| |||||||||| || Table 27||||||||||| |||||||| |||||||| ||| |||||||||||||||||||||| ||||| |||||||||||||| ||| |||||||| ||| ||||||||| |||||||| |||||||| ||| ||||||||| || |||||||| ||| |||| ||||||||||||| ||||| ||||||||| || || ||||||| |||||||||| || |||||||||||||| ||||||||| ||||||||||| ||||||||||| |||||||||| ||||||||||| |||||||||||| ||||||||| |||||||||||| ||||||||||||| ||||||||||| ||| |||||||||||| ||||||||| || ||| |||||||||| || |||||||| ||| |||||| |||| ||| |||||| ||||||| ||| |||||||||| || ||||||||||| ||||||||||| ||| ||||||| ||||||||| || |||||||| || |||||| ||||| ||||| || ||||| || |||||| || ||| |||||| |||| ||||||||| ||| ||||| || |||| ||| ||| |||||||||| || || ||| |||||| ||||||| ||||||||||||||| ||||| ||| ||||||||||||| |||||||| |||||||||| ||||||| |||||||| |||| ||||||||||| ||| ||| ||||||||||||| ||||| ||||||||||| ||| |||||||| ||||||||||||| |||| |||||||| |||| |||||||| ||| |||||||| ||| |||||||||||| || |||||||||||||| || |||||||| |||||||| |||| ||||| ||| |||| |||| || ||||| ||| |||||||| ||||||||| |||| |||| ||||| || |||||| ||||||| ||||||||||| ||| ||||||||||| ||| |||| ||| ||| ||||||||| ||||||||||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || ||||||||| |||||||| |||||||| ||| |||||||| ||||||||| ||||||| ||||||||||| ||| ||| |||||||| |||||||| |||||| ||||||||||| ||||| |||||||| ||| |||| ||||| || |||||||||||||||||||||||||||| ||||||||||| |||||||||||||| ||| |||||||| ||| ||||||||| |||||||| |||||||| ||| ||||||||| || |||||||| ||||||||||| |||| |||||||||||||| ||||||||| || || ||||||| |||||||||||||||||||||| ||||||||| ||||||||||| ||||||||||| |||||||||||| ||||||||| |||||||||||| ||||||||||||| ||||||||||| ||| |||||||||||| ||||||||| || ||| |||||||||| || |||||||| ||| |||||| |||| ||| |||||| ||||||| ||| |||||||||| || ||||||||||| ||||||||||| ||| ||||||| |||||| || ||||||||| |||||| |||| ||||||||| ||| ||||| || |||| ||| || ||| ||||||||||||| ||||||||||| |||||||||| ||| ||| ||||| |||||| ||||| ||||||| |||||||| |||| |||||||||||||| ||| ||||||||||||| ||||||||||| ||||||||||| ||| |||||||||||||| |||||| ||| ||||||||||| |||| |||||||| |||| |||||||| ||||||||||| || |||||||| |||||||| |||| ||||| ||| |||| |||| || ||||| ||| |||||||| ||||||||| |||| ||||| ||| |||| |||| || ||||| |||||||| ||||||||||| |||||||| || ||||||||||| ||| |||| ||| ||| ||||||||| ||||||||||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || ||||||||| |||||||| |||||||| ||| |||||||| ||||||||| ||||||| ||||||||||| ||| ||| ||||||||| |||||||| |||||| ||||||||||| ||||| |||||||| ||| ||| ||||| ||||||||||| |||| |||||||||||||||||||| |||| |||||||||||||||||||| || ||||||||| |||||||| |||||||| ||| ||||||||| |||||||| ||||||||||| |||| |||||||||| ||| |||| |||| |||| ||||| || ||||| ||| |||| ||||| || ||||| ||| |||||||| |||||||| ||| |||||||||| ||||||||||||| ||| |||||||||| ||||| ||||| || ||||| ||| |||| ||||| || |||||| |||||||||||| || ||| ||||| |||| ||||||||| ||| |||| ||| ||| ||||||||| |||||||| ||||||||||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || ||||||||| |||||||| |||||||| ||| |||||||| ||||||||| || |||| ||||||||||||||||||||| |||||||| |||||||| ||| ||||||||||||||||||||| ||||| |||||||||||||| ||| |||||||| ||| ||||||||||| |||||||| |||||||| ||| ||||||||| || |||||||| ||| |||| ||||||||||||| ||||| ||||||||| || || ||||||| |||||||||| || ||||||||||||| ||||||||||| ||||||||||| |||||||||| ||||||||||| |||||||||||| ||||||||| |||||||||||| ||||||||||||| ||||||||||| ||| ||||||||||| |||||| || |||||| ||||||||| ||||||||| || ||| |||||||||| || |||||||| |||| |||||| ||||| |||| |||||| ||||||| ||| ||||||||||| ||| ||| ||||||||| || ||| ||||||||| ||| || ||| ||||||| |||||||||| ||| ||||||||||| ||| ||||||||||| || || ||||| ||||||||||| || ||| ||| |||||||| ||| ||||||||||| ||||||||||| ||| |||| ||||||||||| ||| || |||| ||||||||||| ||| ||| ||| |||||||| ||| |||||||||||| || ||| ||||||||||| || || ||| |||||||| ||| ||| ||||||||||||| ||||||||||| || || ||| |||||||| ||| ||||| || |||| ||| ||| |||||||||| || || ||| ||||| |||||| ||||| |||| |||||||| |||| ||||||||||| |||||| |||||||| || ||| ||||||||||||| ||||| ||||||||||| ||| ||||||||| |||||||||||||||||||| |||||||| |||||||| ||| ||||||||| |||||||| || ||||||| |||||| ||| ||||||||||| ||||||| ||| ||| ||||||||||||| ||||| ||||||||||| ||||||||| |||| ||||||||||| ||| |||||||||| |||| |||||||||| |||||||| |||||||| |||||||| || |||||||||| |||| ||||| ||| |||| |||| || |||||| |||||||||| |||| ||||| ||| |||| |||| || |||||| ||||||||| || || |||| ||||| ||| |||| |||| || ||||| ||||||||| ||| || |||| ||||| ||| |||| |||| || |||||| |||||||| |||| ||||| ||| |||| |||| || |||||| ||||||||||||| |||| ||||| ||| |||| |||| || |||||| ||||||||||| || || |||| ||||| ||| ||| |||| || |||||| ||||||||||| ||| || ||| |||| ||||| ||| ||| |||| || |||||| ||||||||||| ||| || ||| |||| ||||| ||| |||| |||| || |||||| ||| ||||||||||| ||| || ||| |||| ||||| ||| |||| |||| || |||||| ||||||||| |||| ||||||||||| ||| |||||||||| |||| |||||||||| |||||||| ||||||||| |||||||| || |||||||||| |||| ||||| ||| |||| |||| || |||||| |||||||||| |||| ||||| ||| |||| |||| || |||||| ||||||||| || || |||| ||||| ||| |||| |||| || ||||| ||||||||| ||| || |||| ||||| ||| |||| |||| || |||||| |||||||| |||| ||||| ||| |||| || |||||| |||||||||||| |||| ||||| ||| ||| |||| || |||||| ||||||||||| || || |||| ||||| ||| |||| |||| || |||||| ||||||||||| ||| || ||| |||| ||||| ||| |||| |||| || |||||| ||||||||||| ||| || ||| |||| ||||| ||| |||| |||| || ||||| ||| ||||||||||| ||| || ||| |||| ||||| ||| |||| |||| || |||||| ||| |||| ||| ||| ||||||||| ||||||||||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || ||||||||||| |||||||| |||||||| ||| ||||||||| ||||||| ||||||||||| ||| ||| ||||||||| |||||||| |||||| ||||||||||| || ||| ||||||||||||| ||||| ||||||||||||||||||||||||| ||||||||||| |||||||||||||| ||| |||||||| ||| ||||||||||| |||||||| |||||||| ||| ||||||||| || |||||||| |||| ||||| |||||||||| |||| |||||||||||||| ||||||||| || || ||||||| |||||||||||||||||||||| ||||||||||| ||||||||||| |||||||||||| ||||||||| |||||||||||| ||||||||||||| |||||||||||| ||| |||||||||||| |||||| || |||||| |||||||||| ||||||||| || ||| |||||||||| || |||||||| |||| |||||| ||||| |||| |||||| ||||||| ||| ||||||||||| ||| ||| ||||||||||| || || ||||| ||||||||||| || ||| ||| |||||||| ||| ||||||||||| ||||||||||| ||| || |||| ||||||||||| ||| |||| ||| ||||||||||| ||||||||||| ||| |||| ||||||||||| ||| || ||| ||| |||||||| ||| ||| ||||||||||||| ||||||||||| || || ||| |||||||| ||| ||||| ||||| ||| |||| |||||||| ||| ||||| || || ||| ||||| |||||| ||||| ||||||| |||||||| |||| ||||||||||| ||| ||| ||||||||||||| ||||||||||| ||||||||||| ||| |||||||| ||||||||||||| |||||||||||| |||||||| ||||||||||| |||||||| |||||||| ||| ||||||||| |||| |||||||| ||| |||| ||| ||| |||||||| ||||||||||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || ||||||||||| |||||||| |||||||| ||| |||||||| ||||||||| ||||||| ||||||||||| ||| ||| |||||||| |||||||| |||||| ||||||||||| ||| ||| ||||| |||||||||| |||| ||||||||||||||||||| |||| |||||||||||| ||| |||||||| ||| ||| ||||| |||| |||||||| ||||||| |||| ||| |||| || |||| ||| |||||||||| || ||| ||||||||| || ||||||||||| |||||||| |||||||| ||| ||||||||| ||||||| ||||||||||| ||| ||| ||||| |||||||| |||||| ||||||||||||||||||||||| |||||||||||||||||||| |||||||| || |||||||| |||||||| ||| ||||||||| ||||||||| |||| ||||||||||||| ||||||| |||||||| || ||| ||||||||| || |||||||||| ||| ||||||||||| || |||| |||| ||| ||||||||| || ||| ||||||||||||| ||||||||| |||||| ||||||| ||| ||||||||||| |||||||| ||||||| ||| ||| ||||||||||||| ||||| |||||||||| ||| |||||||| |||||||| ||| ||||||||| ||| ||||||||| || || ||||||| |||||||||||||||||||||||| ||| ||||||||||| |||||||| ||||||| ||| ||| ||||||||||||| ||||||||||| |||||||||| ||| |||||||| |||||||| ||| ||||||||| ||| ||||||||| || || ||||||| ||||||||||||||||||||||||||| ||| ||||||||||||| ||||| ||||||||||| ||| |||| ||| ||| ||||||||||| |||||| ||||||||||||| |||||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || ||||||||||| |||||||| |||||||| ||| |||||||| ||||||||| ||||||| ||||||||||| ||| ||| |||||||| |||||| |||||||||||| ||| ||| ||||||||||||| ||||||||||| ||||||||||| ||| |||| ||| ||| ||||||||| ||||||||||| |||||| ||||||||||||| |||||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || ||||||||||| |||||||| |||||||| ||| ||||||||| ||||||| |||||||||||| ||| ||||| |||||| |||||||| |||||||||||| |||||||||| ||||||| ||||||||||||||||||||| ||||| |||||||||||||| ||||||| |||||||| ||| ||||||||| ||||||| ||||||| ||| ||| ||||||||||||| ||||| |||||||||| ||||||||| || || ||||||| |||||||||| || |||||||||||||| ||||||||| ||||||||||| ||||||||||| |||||||||| ||||||||||| |||||||||||| ||||||||| |||||||||||| ||||||||||||| |||||||||||| ||| |||||||||||| ||||||||| || ||| |||||||||| || |||||||| || |||||| ||||| |||| |||||| ||||||| ||| ||||||||||||||||| ||||||| ||||| ||||||||||||||| ||||| ||| ||| ||||||||||||| |||||||| ||||||||| |||| |||||||| |||| |||||||||| ||| |||||||||| |||| || |||||||| ||| |||||||| || ||||| ||||||| |||| ||||||||| ||||||| ||||||| || ||| ||||||||||||| ||||| ||||||||||| ||| |||||||| ||| |||||||||||| || |||||||||||| ||||||| ||||||||||| ||| |||||||||| |||| ||||| ||| |||| |||| || |||||| ||| |||| ||| ||| ||||||||| |||||||| ||||||||||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || ||||||||| ||||||| ||||||| ||||||| ||||||||||| ||| ||| ||||||||| |||||||| |||||| ||||||||||| || ||| ||||||||||||| ||||| ||||||||||||||||||||||| |||||||||||||| ||||||| |||||||| ||| ||||||||| ||||||| ||||||| ||| ||| ||||||||||||| ||||||||||| |||||||||| ||||||||| || || ||||||| |||||||||||||||||||| ||||||||| ||||||||||| ||||||||||| |||||||||||| |||||||||||| ||||||||||||| ||||||||||| ||| |||||||||||| ||||||||| || ||| |||||||||| || |||||||| || |||||| ||||| |||| |||||| ||||||| ||| |||||||||||| ||| |||||||| ||||| |||||| ||||| ||||||| |||||||| |||| |||||||||| ||| |||||||||| || || ||| |||| |||| ||| ||| ||||||||||||| ||||||||||| ||||||||||| ||| |||| ||| ||| |||||||| ||||||||||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || ||||||||| ||||||| ||||||| ||||||| ||||||||||| ||| ||||| |||||||| |||||| |||||||||||||||| |||| |||||||||||| ||| |||||||| ||| ||| ||||| |||| |||||||| ||||||| ||| ||| |||| || |||| ||| |||||||||| || ||| ||||||||| || ||||||||| ||||||| ||||||| ||||||| ||||||||||| ||| ||||| |||||||| |||||| |||||||||||||||||||||||| ||||||| ||||||||||||||||||||| ||||| |||||||||||||| ||| |||||||| ||| ||||||| ||||||| || ||| ||||||||||| |||||| ||| ||||||||||||| ||||| |||||||||| ||||||||| || || ||||||| ||||||||||||||||||||||| ||||||||| ||||||||||| ||||||||||| |||||||||| ||||||||||| |||||||||||| |||||||||||| ||||||||||||| ||||||||||| ||| |||||||||||| ||||||||| || ||| |||||||||| || |||||||| |||| |||||| ||||| |||| |||||| ||||||| ||| ||||||||||| ||| ||| ||||||||||| || || |||| ||||||||||| |||| ||| ||||||| |||||| ||||||| ||| |||||||||||| || ||| ||||||||||| || || ||| ||||||| |||||||||| |||||| ||| |||||||||||| ||| || |||| ||||||||||| ||| |||| ||| ||||||| |||||||||| |||||| |||| ||||||||| ||| ||||||||||| ||| ||| |||||||||| ||| ||||||| ||||||| || |||||| ||||| |||||| |||| ||| ||||| || ||| ||||||| ||||||||| ||| ||| ||||||||||||| || ||| ||||||| ||||||| ||||||| ||||||| ||| |||||||| |||||| ||||||| ||||| ||| ||||||||| ||| ||| ||||||||||||| ||||| ||||||||||| ||| |||| ||| ||||||||||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || ||||||||||| ||||||| ||||||| ||||||| ||||||||||| ||| ||| ||||| |||||||| |||||| |||||||||||||||||||||||||| ||||||||||| |||||||||||||| ||| |||||||| ||| ||||||| ||||||| || ||| ||||||||||| |||||| ||| ||||||||||||| ||||||||||| |||||||||| |||||||| |||||||| |||||||||||||||||||||| ||||||||| ||||||||||| ||||||||||| |||||||||||| |||||||||||| ||||||||||||| |||||||||||| ||| |||||||||||| ||||||||| || ||| |||||||||| || |||||||| |||| |||||| ||||| |||| |||||| ||||||| ||| ||||||||||| ||| ||| ||||||||||| || || |||| ||||||||||| |||| ||| ||||||| |||||| ||||||| ||| |||||||||||| || ||| ||||||||||| || || ||| ||||||| |||||||||| |||||| ||| |||||||||||| ||| || |||| ||||||||||| ||| |||| ||| ||||||| |||||||||| |||||| |||| ||||||||| ||| ||||||||||| ||| || |||| |||||||||| ||| ||||||| ||||||| || |||||| ||||| |||||| |||| ||| ||||| || ||| ||||||| ||||||||| ||| ||| |||||||||||||| |||||||||||| ||| ||||||| ||||||| ||||||| ||||||| ||| |||||||||| ||||| |||||| ||||| ||| |||||||||||| ||| ||||||||||||| ||||||||||| ||||||||||| ||| |||| ||| ||| ||||||||| ||||||||||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || ||||||||||| ||||||| ||||||| ||||||| ||||||||||| ||| ||| ||||| |||||||| |||||| |||||||||||||||||| |||| |||||||||||| ||| |||||||| ||| ||| ||||| |||| |||||||| ||||||| ||| ||| |||| || |||| ||| |||||||||| || ||| ||||||||| || ||||||||||| ||||||| ||||||| ||||||| ||||||||||| ||| ||||| |||||||| |||||| ||||||||||||
Table 26: Pairwise OR With 95% CrI of Treatment Effect for Induction Clinical Response, Clinical Remission, and Mucosal Healing Between Mirikizumab and Comparators in Patients with UC
||||||||||| | |||||||||||| ||||| | |||||||||| ||||||||||| | ||||
|---|---|---|---|---|---|---|
||| | |||||||| |||||||| | |||||||| ||||||||| | ||||||| ||||||| | |||||||| |||||||| | |||||||| ||||||||| | ||||||| ||||||| |
||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
||| | |||| ||||| || ||||| | |||| ||||| || ||||| | || | |||| ||||| || ||||| | |||| ||||| || ||||| | || |
||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | || | || | || |
||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | || | || | || |
||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
|||||||||||||| |||||||||||||| |||||||||||| ||||||||||||| ||||||||||||||| |||| |||||||||||||| |||||||||||||| |||||||||||| ||||||||||||| ||||||||||||||| |||| |||||||| || ||| |||||||| |||||||||||| ||||||||||| |||||||||||||| |||||||||||||||| |||||||||||||| ||||||||||||||| ||||||||| ||||||| |||||||||||||| |||||||||| || |||||| || ||||| ||| ||| ||||||||||||| |||||||| |||||||||| ||||||| |||||||| |||| |||||||||||||||| ||||||||| ||||||| |||| |||||||| |||| ||||||||||||||| |||||||||||| |||||||||| || |||||| || ||||| ||| ||| ||||||||||||| |||||||| ||||||||||| ||||||||| ||||||| ||||||| |||||||| |||| |||||||||||||| ||||||||| ||||||| |||| |||||||| |||| ||||||||||||||||||||| || |||||||||| |||||| || || |||||| || ||||||||||||||||| ||| ||| ||| ||| ||| |||||||| ||| ||| || |||||| ||| ||||||||| ||| |||||||| ||| ||||||||||||| || ||| ||||| |||||||||||||| ||| ||||||||| ||||||21
Table 27: Pairwise OR With 95% CrI of Treatment Effect for Maintenance Clinical Response, Clinical Treatment, and Mucosal Healing Between Mirikizumab and Comparators in Patients with UC
||||||||||| | |||||||||||| ||||| | |||||||||||| ||||||||||| | ||||
|---|---|---|---|---|---|---|
|||||||| ||||||| | |||||||| |||||||| | ||||||| |||||| | |||||||| ||||||| | |||||||| |||||||| | ||||||| |||||| | |
||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
||| ||| || ||| | |||| ||||| || ||||| | |||| ||||| || ||||| | || | |||| ||||| || ||||| | |||| ||||| || ||||| | || |
||| ||| || ||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
||| ||| || ||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
||| || || | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
||| | |||| ||||| || ||||| | |||| ||||| || ||||| | || | |||| ||||| || ||||| | |||| ||||| || ||||| | || |
||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | || | || | || |
||| || || | |||| ||||| || ||||| | |||| ||||| || ||||| | || | || | || | || |
||| ||| || | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | || | || | || |
||| || || ||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
||||||||||||| |||||||||||||| |||||||||||| ||||||||||||| ||||||||||||||| |||| |||||||||||||| |||||||||||||| |||||||||||| ||||||||||||| ||||||||||||||| |||| |||||||| || ||| |||||||| |||||||||||| ||||||||||| |||||||||||||| |||||||||||||||| |||||||||||||| ||||||||||||||| ||||||||| ||||||| |||||||||||||| |||||||||| || |||||| || ||||| ||| ||| ||||||||||||| |||||||| |||||||||| ||||||| |||||||| |||| |||||||||||||||| ||||||||| ||||||| |||| |||||||| |||| ||||||||||||||| |||||||||||| |||||||||| || |||||| || ||||| ||| ||| ||||||||||||| |||||||| ||||||||||| ||||||||| ||||||| ||||||| |||||||| |||| |||||||||||||| ||||||||| ||||||| |||| |||||||| |||| ||||||||||||||||||||| || |||||||||| |||||| || || |||||| || ||||||||||||||||| ||| ||| ||| ||| ||| |||||||| ||| ||| || |||||| ||| ||||||||| ||| |||||||| ||| ||||||||||||| || |21
Harms
||||| |||||||| |||| ||||||||| ||| ||| ||||||| ||||| |||||||||| |||||||||| || ||||| |||||||| || |||||||| |||||| |||| |||||||| ||||||| ||||||| ||| ||| ||||| ||||||||||||||| ||| |||| ||| ||||||||| || Appendix 1 |||||||| ||||||||||| || ||| ||||| ||||||||||||||| ||| |||| |||| || ||| ||| ||| ||||||| ||||||||||| ||| ||||||||||| ||| |||||||||| || Table 28 ||||| ||||| ||||||||||||||||||| ||| |||||||| ||| ||| ||||| ||||||||||||||| |||||| ||| ||||||||| |||||| ||| ||| ||||||| ||||| |||||||||| ||||||||| || || ||||||| |||||||||| || |||||||||||||| ||||||||| ||||||||||| ||||||||||| |||||||||| ||||||||||| |||||||||||| ||||||||| |||||||||||| ||||||||||||| |||||||||||| ||| |||||||||||| ||||||||| || ||| |||||||||| || |||||||| || |||||| ||||| |||| |||||| ||||||| ||| ||||||||||| ||| ||||| |||||| ||||| ||| |||||||| || |||||| ||| |||| |||| ||||||||||||| ||||||||| ||| ||||||||||||||||||||||| ||||| |||| || ||||||||| ||||||||||||||| |||||||| || |||||||| ||||| |||| || ||||||||| ||||||||||||||| ||| |||||||| ||||||||| ||||||||| |||| ||||||||||| |||||| ||||||||||| |||| ||||| ||| ||| |||| || ||||| ||| |||||||||| |||| ||||| ||| ||| |||| || |||||| ||| |||| ||| ||| ||||||||| ||||||||||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || ||| ||||| ||||||||||||||| ||||||| ||||||||||| ||| ||| ||||| |||||||| |||||| |||||||||||| |||||||| ||||||| |||||||||| ||| || |||| |||||| ||| ||||||||| |||||| ||| ||| ||||||| ||||| |||||||||| ||||||||| || || ||||||| |||||||||| || |||||||||||||| ||||||||| ||||||||||| ||||||||||| |||||||||| ||||||||||| |||||||||||| ||||||||| |||||||||||| ||||||||||||| |||||||||||| ||| |||||||||||| ||||||||| || ||| |||||||||| || |||||||| || |||||| ||||| |||| |||||| ||||||| ||| ||||||||||| ||| |||||| ||||||| ||||| ||| |||||| |||| |||||||||||| |||| |||| ||| ||| |||||||| ||||||||||| |||| ||| |||| || |||| ||| ||||||||||| || ||||||||| || |||||||| |||| ||||||| ||||||||||| ||| ||| ||||| |||||||||||| ||||||||| ||||||||
Table 28: Pairwise OR With 95% CrI of All-Cause Discontinuation and SAEs During Induction Treatment Between Mirikizumab and Comparators in Patients With UC (Mixed Biologic-Naive and -Experienced Populations and JAK Inhibitor–Naive and –Experienced Populations)
|||||||||| | ||| ||||| |||||||||||||| | ||||||| ||||||| |||||| |
|---|---|---|
||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
||| | || | |||| ||||| || ||||| |
||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
||||||||||||| |||||||||||||| |||||||||||| ||||||||||||| ||||||||||||||| |||| ||||||||||||| |||||||||||||| |||||||||||| ||||||||||||| ||||||||||||||| |||| |||||||| || ||| |||||||| |||||||||||| ||||||||||| |||||||||||||| |||||||||||||||| |||||||||||||| ||||||||||||||| ||||||||| ||||||| |||||||||||||| |||||||||| || |||||| || ||||| ||| ||| ||||||||||||| |||||||| |||||||||| ||||||| |||||||| |||| |||||||||||||||| ||||||||| ||||||| |||| |||||||| |||| ||||||||||||||| |||||||||||| |||||||||| || |||||| || ||||| ||| ||| ||||||||||||| |||||||| ||||||||||| ||||||||| ||||||| ||||||| |||||||| |||| |||||||||||||| ||||||||| ||||||| |||| |||||||| |||| ||||||||||||||||||||| || |||||||||| |||||| || || |||||| || ||||||||||||||||| ||| ||| ||| ||| ||| |||||||| ||| ||| || |||||| ||| ||||||||| ||| ||||||||21
Critical Appraisal
The NMA was based on studies identified from a sponsor-conducted systematic literature review of relevant randomized evidence of EMA-approved and FDA-approved treatments for adult patients with moderately to severely active UC.36 The systematic literature reviewed was based on a PICOS defined a priori. The search was comprehensive, involving multiple electronic databases, clinical registries, and supplementary manual searches. Error and bias in the study selection and data extraction process were minimized. While the risk of bias of the comparator trials was assessed, this rating was not detailed in the systematic review, and this was done at the study level rather than the outcome level.
The sponsor conducted a feasibility assessment to evaluate potential areas of heterogeneity based on study design and baseline patient characteristics; no studies were excluded based on heterogeneity. The sponsor noted several population characteristics that impact placebo response rate (or baseline risk). To manage heterogeneity due to the use of different definitions for what qualifies as “placebo,” which could not be adjusted for by restricting NMA inclusion criteria without reducing the evidence base or removing comparators of interest, the sponsor employed where possible the adjustment of baseline risk via a meta-regression model with baseline risk as a covariate to explore the possible influence of heterogeneity in placebo across studies. However, the baseline risk–adjusted model was not used in all cases (e.g., if another model had better fit).
The sponsor identified 3 other sources of heterogeneity across the included UC studies. First, race is known to impact placebo response in UC trials. As noted by the clinical expert consulted by CADTH, there appears to be racial disparity to the incidence and severity of UC across races — namely, people of Asian origin presenting with less extensive disease and more perianal disease than the general population, people of South Asian ethnicity tending to have a higher risk of developing UC compared to the general population, and people of Hispanic or Asian descent tending to have a higher risk of pancolitis.103-105 To account for this known racial disparity, the NMA evaluated an “only Asian” subgroup, regardless of prior experience with biologics or JAK inhibitors. The network for this subgroup, however, was small, consisting of |||||||||| at induction and maintenance, respectively, evaluating ||| interventions. Second, as described earlier, 2 main trial designs are employed across UC studies (i.e., treat-through trial design versus rerandomized trial design). To mitigate heterogeneity due to trial design, statistical adjustments were employed to make treat-through trials comparable to efficacy data from rerandomized trials. The sponsor noted that strong assumptions were applied to some studies in the adjustment process (i.e., if sustained response and remission were not reported from a trial, the value from another trial with the same treatment was extrapolated). To investigate whether these assumptions affected the efficacy analyses, sensitivity analyses were conducted excluding treat-through study design. However, the CADTH review team was unable to confirm whether the method employed adequately adjusted for differences in design trial without introducing bias, and suspects that simulation studies are required to confirm the validity of such adjustments. Moreover, follow-up sensitivity analyses excluding studies with treat-through study design were unlikely to account for the potential issues since the trials included in the network were different and limited by heterogeneity issues of their own, such as differing definitions of prior experience with biologics, and differing definitions of clinical response and remission and length of induction and maintenance periods across trials. Third, prior exposure to biologic therapy at enrolment is a known effect modifier in trials of patients with UC. To account for the potential for heterogeneity due to treatment history — as biologic-naive and biologic-experienced patients’ UC would be expected to respond to treatment differently — separate analyses were performed for patients who were biologic-naive and patients who were biologic-experienced. However, the definition of biologic-naive and biologic-experienced varied across studies (definitions included TNF-naive versus TNF-experienced, no biologic and/or JAK inhibitor failure versus biologic and/or JAK inhibitor failure, biologic-naive versus biologic-experienced, and no biologic failure versus biologic failure). Furthermore, the clinical expert consulted by CADTH for this review noted that there may be differences within each definition. For example, some trials included biologic exposure under biologic-naive as long as the patient did not lose remission. The clinical expert also noted that due to when comparators entered the market, the type of biologics that patients were exposed to would differ. For example, the only biologic failure that patients in trials evaluating adalimumab may have experienced would be failure with infliximab. As noted by the clinical expert, exposure to certain mechanisms of action may have affected response to subsequent mechanisms of action. Thus, the different classifications of biologic-naive and biologic-experienced across the studies represented a major source of heterogeneity.
The CADTH review team identified several other sources of heterogeneity that could not be adjusted for in the NMA, including differences in patient characteristics, definitions, and assessment methods for clinical response and remission; permitted concomitant medications; and the duration of the maintenance period. The clinical expert consulted by CADTH noted that the difference in the definitions of clinical response and remission applied in the LUCENT trials versus the most commonly applied definitions across the comparative studies included in the NMA may have a meaningful impact on the NMA results since it is unclear what subscores were driving the treatment effect (ES versus SF). Regarding the duration of the maintenance period, the clinical expert noted that longer duration allows more time for a treatment to achieve a desirable treatment response; however, longer maintenance duration may lead to an increase in AEs or a loss of remission due to longer follow-up time. Based on input from the clinical expert, it is uncertain how the duration of the maintenance period would influence treatment response, loss of response, and AEs. The apparent benefit of mirikizumab in the maintenance period among patients in the biologic-naive population may be explained by heterogeneity between the trials, which was supported by feedback from the clinical expert consulted by CADTH.
The inclusion of comparator treatments not relevant to the Canadian setting (i.e., filgotinib, upadacitinib, and maintenance ustekinumab 90 mg every 12 weeks) provided information to the network and was not expected to significantly impact the heterogeneity of the NMA over the other sources of heterogeneity described earlier.
Violation of the exchangeability assumption for efficacy outcomes cannot be ruled out due to heterogeneity and NMA efficacy results should be interpreted with uncertainty. Moreover, network consistency or coherence could not be assessed due to the lack of relevant closed loops when comparing to other active treatments. As a result, the NMA evidence was considered indirect, thus reducing certainty in the study findings.
Based on input from the clinical expert consulted by CADTH, the characteristics of patients included in the NMA — age, disease duration, Mayo score, and sex — is reflective of the Canadian clinical practice setting. However, the clinical expert noted that certain racial and ethnic populations — namely, Asian, South Asian, and Hispanic populations — may have a different phenotype of IBD. Given the reduced number of studies eligible for inclusion in the Asian-only subgroup analysis, generalizability of the NMA results may be limited in this population.
Studies Addressing Gaps in the Pivotal and RCT Evidence
No studies addressing gaps in the pivotal and RCT evidence were submitted.
Discussion
Summary of Available Evidence
This report summarizes the evidence for the safety and efficacy of mirikizumab in the treatment of adults with moderately to severely active UC based on 2 phase III, parallel-arm, multicentre, double-blind, placebo-controlled randomized trials, as well as 1 ITC.
LUCENT-1 (N = 1,281) was a 12-week induction trial in which patients were randomized 3:1 to either mirikizumab 300 mg IV every 4 weeks or placebo. The LUCENT-1 study evaluated whether mirikizumab 300 mg IV would induce clinical remission at week 12 in adult patients with moderately to severely active UC. Major secondary objectives included the ability of mirikizumab to induce alternate clinical remission, clinical response, clinical response in patients who were biologic-experienced, endoscopic remission, symptomatic remission, bowel urgency improvement, and HEMI, all at week 12. HRQoL was also evaluated at week 12 using the IBDQ, EQ-5D-5L, and SF-36 (PCS and MCS). At baseline, patients had a mean age of 42.5 (SD = 13.92) years, with the majority being male (59.8%) and white (72.3%). There was an equal number of patients with moderate UC and severe UC based on MMS. The proportion of patients reporting prior biologic or tofacitinib failure was also similar between treatment groups (41.6% and 40.1% of patients randomized to mirikizumab and placebo, respectively).
LUCENT-2 (N = 544 in the primary analysis) was a 40-week maintenance trial in which patients were randomized 2:1 to either mirikizumab 200 mg SC every 4 weeks or placebo. The aim of the trial was to assess whether mirikizumab 200 mg SC would maintain clinical remission from baseline to week 40 in adult patients with moderately to severely active UC who experienced a clinical response at week 12 of the LUCENT-1 study. Major secondary objectives included the ability of mirikizumab to maintain alternate clinical remission, corticosteroid-free remission, durable clinical remission, endoscopic remission, bowel urgency remission and improvement, and HEMR, all at week 40. The baseline characteristics in the LUCENT-2 study were similar to those of the LUCENT-1 study. The majority of the patients in the main cohort had a mean age of 42.3 (SD = 13.5) years, and were male (58.4%) and white (71.3%). Based on the MMS, approximately half of the patients in each treatment arm were categorized as moderate UC severity and 35.1% of the patients in the mirikizumab group and 35.8% of the patients in the placebo group had a history of biologic or tofacitinib failure.
In the absence of an active comparator, 1 sponsor-submitted ITC was summarized and critically appraised. The sponsor performed an NMA to estimate the comparative treatment effect of mirikizumab to other advanced therapies in patients with moderately to severely active UC. The NMA included || double-blind RCTs. The outcomes assessed in the NMA efficacy analysis included clinical response, clinical remission, and mucosal healing at induction and maintenance, as well as overall SAEs and all-cause discontinuation of treatment. Several sources of heterogeneity across the included UC studies were identified, including trial design, differences in definitions of clinical response and remission, prior biologics exposure (due to time periods in which the studies occurred), permitted concomitant medications, outcome assessment methods and definition, and duration of the maintenance period.
Interpretation of Results
Efficacy
Evidence from the LUCENT-1 and LUCENT-2 studies demonstrated the efficacy of mirikizumab compared to placebo in treating patients with moderately to severely active UC when used to achieve induction of remission (assessed after 12 weeks) and maintenance of remission (assessed after 40 weeks). All prespecified primary and key secondary end points in both the LUCENT-1 study and the LUCENT-2 study were achieved, thus addressing outcomes such as clinical remission and clinical response that patients and clinicians had noted were important. Clinical remission, as identified by STRIDE-II, is a pivotal intermediate target in the management of active UC and was successfully attained with mirikizumab treatment in both these trials.45 The MMS was used to measure the primary and major secondary end points and is considered a valid measure for use in patients with UC and consistent with clinical practice. The magnitude of benefit observed in both the LUCENT-1 and LUCENT-2 trials was deemed clinically meaningful, substantiated by the input received from the clinical expert consulted for this review.
Furthermore, the durability of clinical remission was demonstrated in the LUCENT-2 study, where after 40 weeks of treatment with mirikizumab 200 mg SC every 4 weeks, a larger proportion of patients maintained clinical remission compared to those on placebo. This finding is particularly promising as it indicates the potential for the sustained effectiveness of mirikizumab, as noted by the clinical expert. Additionally, the relatively low occurrence of loss of response observed in the LUCENT-2 trial further supports the clinical significance of mirikizumab treatment. Another noteworthy benefit was corticosteroid-free remission, which was achieved in more than 90% of patients randomized to mirikizumab in the LUCENT-2 trial who experienced clinical remission after maintenance. The ability to taper steroids is a very important outcome emphasized by both patients and clinicians in the management of UC.
Notably, improvements in endoscopic remission and endoscopic mucosal healing were observed in greater proportions in the mirikizumab-treated patients versus placebo-treated patients in both pivotal trials. Unlike the other outcomes that rely on patient reporting, such as symptomatic remission, endoscopic remission is based on colonoscopy and provides objective insight into a patient's treatment recovery. The clinical expert highlighted that it is uncommon to observe endoscopic remission by week 12 of the treatment period, indicating that endoscopic remission and healing at week 40 may provide more reliable indicators of these outcomes.
QoL is impacted in all facets of the lives of patients with UC, including their personal, school, and work lives, as noted by patient groups. The measures of HRQoL — IBDQ, EQ-5D-5L, and SF-36 — are valid and reliable for use in UC, with established MIDs; these measures were secondary end points in both trials. In particular, the clinical expert affirmed that the IBDQ is an appropriate instrument for evaluating HRQoL in patients with UC. ||| ||||| ||||||||| resulted in clinically meaningful improvements in HRQoL and numerically greater improvements with 52 weeks of mirikizumab therapy compared to placebo. The change in IBDQ score for the mirikizumab group compared to the placebo group did not meet suggested thresholds for a clinically important difference during the induction phase but was considered clinically important at the end of the maintenance phase (52 weeks of continuous therapy). Overall, HRQoL outcomes were not adjusted for multiplicity and should be interpreted with consideration of potentially increased type I error. The WPAI:UC, a valid and responsive measure of work productivity in UC, demonstrated improved work productivity for those patients working at baseline compared to placebo.
Bowel urgency is a troubling symptom experienced by patients with UC. An improvement in bowel urgency was assessed in the LUCENT-1 and LUCENT-2 studies using the UNRS, which is an appropriate instrument used to measure changes and improvement in bowel urgency. In the LUCENT-2 trial, patients who received 52 weeks of continuous treatment (i.e., patients who completed both the LUCENT-1 and LUCENT-2 studies) experienced a clinically significant improvement in bowel urgency compared to patients who received placebo. In terms of bowel urgency remission, an end point measured only in the LUCENT-2 trial, mirikizumab 200 mg SC was more likely to induce bowel urgency remission at week 40 of the LUCENT-2 trial versus placebo.
In terms of subgroups, mirikizumab demonstrated clinical efficacy in the biologic-naive and biologic-experienced subgroups similar to the primary analysis of the induction and maintenance trials. Results of the other prespecified subgroup analyses (i.e., baseline corticosteroid use, baseline immunomodulator use, and baseline MMS score) in the LUCENT-2 study appeared to align with the overall results of the trials. No difference was seen between mirikizumab and placebo across the end points in the LUCENT-1 study for patients with baseline corticosteroid use or immunomodulator use. However, no definitive conclusions can be drawn on these analyses due to the lack of sample size consideration to detect a difference and control for multiplicity.
The study designs of the LUCENT-1 and LUCENT-2 trials also provided insight into the ability of mirikizumab to achieve extended induction in patients with UC who did not have an initial response. The LUCENT-2 study demonstrated that an extended induction period of mirikizumab by an additional 12 weeks in nonresponders at week 12 of the LUCENT-1 study resulted in more than half of these initial nonresponders (53.7%) achieving delayed response at week 24 of continuous IV mirikizumab therapy. Overall, 80% of patients from the LUCENT-1 study (initial responders and delayed responders) experienced clinical response by week 24. The clinical expert noted that this number was higher than expected based on their experience. Due to the lack of comparison to the main cohort, the data from the extended induction were observational. There is a gap in evidence in terms of detecting a difference in clinical outcomes between delayed responders and induction responders in maintaining clinical remission and response. Long-term efficacy data beyond 52 weeks was not available; hence, long-term outcomes may not have been sufficiently captured between the 2 trials. Of note, a long-term extension trial, the LUCENT-3 study, is under way and is expected to be completed by June 2025.
Overall, the NMA comparison between mirikizumab and relevant comparators (i.e., adalimumab, golimumab, infliximab, ozanimod, tofacitinib, ustekinumab, and vedolizumab) did not demonstrate a difference in favour of 1 or the other in induction clinical remission and response, mucosal healing, all-cause discontinuation, and SAEs. The apparent benefit of mirikizumab in the maintenance period among patients in the biologic-naive population may be explained by heterogeneity between the trials, which was supported by feedback from the clinical expert consulted by CADTH. Definitive conclusions related to treatment effect and the harms of mirikizumab compared to other relevant treatments for UC could not be drawn from the NMA analysis due to substantial heterogeneity in patient characteristics, inclusion criteria (e.g., the definition of prior biologic exposure), prior treatment exposure, and outcome definitions, which likely challenged the underlying exchangeability assumption, and wide CrIs for most estimates. The validity of the analytical techniques used to attempt to account for differences in study design in the maintenance phase is uncertain.
Harms
Overall, mirikizumab appeared to be well tolerated based on up to 52 weeks of treatment observed in the pivotal trials. In both the LUCENT-1 and LUCENT-2 trials, the overall rates of treatment-emergent AEs, SAEs, AEs resulting in treatment discontinuation, and AESIs were lower in the mirikizumab group or similar between mirikizumab and placebo patients. The AEs that were more common among mirikizumab patients than in those on placebo include nasopharyngitis, headache, hypersensitivity reaction, and injection site reaction. However, the difference was very small (commonly less than 1%). According to the clinical expert consulted by CADTH for this review, mirikizumab has an acceptable safety profile based on data from the LUCENT-1 and LUCENT-2 studies.
The sponsor-submitted ITC included an analysis of all-cause discontinuation and SAEs. However, the ITC had multiple sources of heterogeneity between trials that challenged the underlying exchangeability assumption and therefore, most estimates were affected by substantial imprecision (wide CrIs). As such, conclusions cannot be drawn with regard to the safety of mirikizumab compared to other drugs for patients with UC.
Conclusion
Two pivotal, multinational, double-blind, randomized placebo-controlled trials, the LUCENT-1 (N = 1,281) and LUCENT-2 (N = 554) trial, and 1 ITC informed the assessment of mirikizumab in this review. Both pivotal trials demonstrated the superiority of mirikizumab over placebo across all end points. The evidence from the LUCENT-1 trial (the induction trial) demonstrated the efficacy of mirikizumab 300 mg IV over placebo in achieving induction clinical remission, alternate clinical remission, clinical response, HRQoL, endoscopic remission, symptomatic remission, bowel urgency improvement, mucosal healing, and work productivity in patients with moderately or severely active UC over 12 weeks. The evidence from the LUCENT-2 trial (the maintenance trial) further supported these results after 40 weeks of maintenance dosing. Additionally, the LUCENT-2 study demonstrated the efficacy of mirikizumab 200 mg SC in achieving corticosteroid-free remission, maintenance of clinical remission, bowel urgency remission, and mucosal remission at week 40 of the LUCENT-2 trial among patients who experienced a clinical response by week 12 of the LUCENT-1 study. The results from the primary analysis were considered generalizable to the Canadian landscape; however, it should be noted that the patient population for the LUCENT-2 study may have been enriched, as only responders were rerandomized to the trial, excluding delayed responders who represent a subset of the general population for this indication. Regarding the extended induction, although mirikizumab was able to capture delayed response, the data were considered observational due to the absence of a comparison to the main cohort. Clinically meaningful improvements in HRQoL based on the IBDQ were observed in patients receiving mirikizumab for 40 weeks in the maintenance phase. The NMA comparison between mirikizumab and relevant comparators (i.e., adalimumab, golimumab, infliximab, ozanimod, tofacitinib, ustekinumab, and vedolizumab) did not demonstrate a difference in favour of 1 treatment over another in induction clinical remission and response, mucosal healing, all-cause discontinuation, and SAEs. The apparent benefit of mirikizumab in the maintenance period among patients in the biologic-naive population may be explained by heterogeneity between the trials, Definitive conclusions related to treatment effect and the harms of mirikizumab compared to other relevant treatments for UC could not be drawn from the NMA analysis due to substantial heterogeneity in patient characteristics, inclusion criteria (e.g., the definition of prior biologic exposure), prior treatment exposure, and outcome definitions, which likely challenged the underlying exchangeability assumption, and wide CrIs for most estimates. Overall, the LUCENT-1 and LUCENT-2 studies demonstrated clinical efficacy and minimal safety concerns for up to 52 weeks of treatment with mirikizumab in patients with moderate to severe UC and relative to placebo. Evidence of the efficacy and safety of mirikizumab beyond 52 weeks, as well as direct comparisons with other treatments for UC, are necessary to further understand the long-term benefits and comparative effectiveness of mirikizumab.
References
1.Schroeder KW, Tremaine WJ, Ilstrup DM. Coated oral 5-aminosalicylic acid therapy for mildly to moderately active ulcerative colitis. A randomized study. N Engl J Med. 1987;317(26):1625-1629. PubMed
2.Stenson W. Inflammatory bowel diseaes. In: Goldman I, Bennett J, eds. Cecil Textbook of Medicine. 21st ed. Philadephia (PA): WB Saunders Co; 2000: p. 722-729.
3.Danese S, Fiocchi C. Ulcerative colitis. N Engl J Med. 2011;365(18):1713-1725. PubMed
4.Travis SP, Stange EF, Lémann M, et al. European evidence-based consensus on the management of ulcerative colitis: current management. J Crohns Colitis. 2008;2(1):24-62. PubMed
5.Rubin DT, Sninsky C, Siegmund B, et al. International perspectives on management of inflammatory bowel disease: opinion differences and similarities between patients and physicians from the IBD GAPPS survey. Inflamm Bowel Dis. 2021;27(12):1942-1953. PubMed
6.Ng SC, Shi HY, Hamidi N, et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet. 2017;390(10114):2769-2778. PubMed
7.Molodecky NA, Soon IS, Rabi DM, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. 2012;142(1):46-54.e42; quiz e30. PubMed
8.Benchimol EI, Fortinsky KJ, Gozdyra P, Van den Heuvel M, Van Limbergen J, Griffiths AM. Epidemiology of pediatric inflammatory bowel disease: a systematic review of international trends. Inflamm Bowel Dis. 2011;17(1):423-439. PubMed
9.Cosnes J, Gower-Rousseau C, Seksik P, Cortot A. Epidemiology and natural history of inflammatory bowel diseases. Gastroenterology. 2011;140(6):1785-1794. PubMed
10.Fumery M, Singh S, Dulai PS, Gower-Rousseau C, Peyrin-Biroulet L, Sandborn WJ. Natural history of adult ulcerative colitis in population-based cohorts: a systematic review. Clin Gastroenterol Hepatol. 2018;16(3):343-356.e343. PubMed
11.Dubinsky MC, Watanabe K, Molander P, et al. Ulcerative Colitis Narrative global survey findings: the impact of living with ulcerative colitis-patients' and physicians' view. Inflamm Bowel Dis. 2021;27(11):1747-1755. PubMed
12.López-Sanromán A. Perceived emotional and psychological impact of ulcerative colitis on outpatients in Spain: UC-LIFE Survey. Dig Dis Sci. 2017. PubMed
13.Lynch WD, Hsu R. Ulcerative colitis. StatPearls. Treasure Island (FL): StatPearls Publishing; 2022: https://www.ncbi.nlm.nih.gov/books/NBK459282/. Accessed 2022 Dec 29.
14.Impact of inflammatory bowel disease in Canada. Toronto (ON): Crohn's Colitis Canada; 2023: https://crohnsandcolitis.ca/Crohns_and_Colitis/documents/reports/2023-IBD-Report-English-LR.pdf?ext=.pdf. Accessed 2023 Jun 2.
15.Coward S, Clement F, Benchimol EI, et al. Past and future burden of inflammatory bowel diseases based on modeling of population-based data. Gastroenterology. 2019;156(5):1345-1353.e1344. PubMed
16.Omvoh (mirikizumab): 100 mg/mL solution for subcutaneous injection; 20 mg/mL solution for intravenous infusion [product monograph]. Toronto (ON): Eli Lilly Canada; 2023 Jul 17.
17.Dubinsky MC, Irving PM, Panaccione R, et al. Incorporating patient experience into drug development for ulcerative colitis: development of the Urgency Numeric Rating Scale, a patient-reported outcome measure to assess bowel urgency in adults. J Patient Rep Outcomes. 2022;6(1):31. PubMed
18.Pallis AG, Mouzas IA, Vlachonikolis IG. The inflammatory bowel disease questionnaire: a review of its national validation studies. Inflamm Bowel Dis. 2004;10(3):261-269. PubMed
19.Chen XL, Zhong LH, Wen Y, et al. Inflammatory bowel disease-specific health-related quality of life instruments: a systematic review of measurement properties. Health Qual Life Outcomes. 2017;15(1):177. PubMed
20.Alrubaiy L, Rikaby I, Dodds P, Hutchings HA, Williams JG. Systematic review of health-related quality of life measures for inflammatory bowel disease. J Crohns Colitis. 2015;9(3):284-292. PubMed
21.Irvine EJ, Feagan B, Rochon J, et al. Quality of life: a valid and reliable measure of therapeutic efficacy in the treatment of inflammatory bowel disease. Canadian Crohn's Relapse Prevention Trial Study Group. Gastroenterology. 1994;106(2):287-296. PubMed
22.Pallis AG, Vlachonikolis IG, Mouzas IA. Assessing health-related quality of life in patients with inflammatory bowel disease, in Crete, Greece. BMC Gastroenterol. 2002;2(1):1. PubMed
23.Gregor JC, McDonald JW, Klar N, et al. An evaluation of utility measurement in Crohn's disease. Inflamm Bowel Dis. 1997;3(4):265-276. PubMed
24.Irvine EJ. Development and subsequent refinement of the inflammatory bowel disease questionnaire: a quality-of-life instrument for adult patients with inflammatory bowel disease. J Pediatr Gastroenterol Nutr. 1999;28(4):S23-27. PubMed
25.Stark RG, Reitmeir P, Leidl R, Konig HH. Validity, reliability, and responsiveness of the EQ-5D in inflammatory bowel disease in Germany. Inflamm Bowel Dis. 2010;16(1):42-51. PubMed
26.Reilly MC, Zbrozek AS, Dukes EM. The validity and reproducibility of a work productivity and activity impairment instrument. Pharmacoeconomics. 1993;4(5):353-365. PubMed
27.Yarlas A, Maher SM, Bayliss MS, Lovley A, Cappelleri JC, DiBonaventura MD. Psychometric validation of the work productivity and activity impairment questionnaire in ulcerative colitis: results from a systematic literature review. J Patient Rep Outcomes. 2018;2(1):62. PubMed
28.Dubinsky MC, Shan M, Delbecque L, et al. Psychometric evaluation of the Urgency NRS as a new patient-reported outcome measure for patients with ulcerative colitis. J Patient Rep Outcomes. 2022;6(1):114. PubMed
29.Yarlas A, Bayliss M, Cappelleri JC, et al. A systematic review of the SF-36® Health Survey for measuring health-related quality of life in patients with ulcerative colitis. Value Health. 2016;19(7).
30.Clinical evidence report for Omvoh™ (mirikizumab) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: mirikiziumab, 100 mg/mL solutions for subcutaneous injection; 20 mg/mL solution for intravenous infusion. Toronto (ON): Eli Lilly Canada, Inc.; 2023.
31.Clinical Study Report: I6T-MC-AMAN. A phase 3, multicenter, randomized, double-blind, parallel, placebo-controlled induction study of mirikizumab in conventional-failed and biologic-failed patients with moderately to severely active ulcerative colitis. LUCENT-1 [internal sponsor's report]. Toronto (ON): Eli Lilly Canada; 2021.
32.Clinical Study Report: I6T-MC-AMBG. A phase 3, multicenter, randomized, double-blind, parallel-arm, placebo-controlled maintenance study of mirikizumab in patients with moderately to severely active ulcerative colitis. LUCENT-2 [internal sponsor's report]. Toronto (ON): Eli Lilly Canada Inc.; 2022.
33.Rubin DT, Ananthakrishnan AN, Siegel CA, Sauer BG, Long MD. ACG Clinical Guideline: Ulcerative colitis in adults. Am J Gastroenterol. 2019;114(3):384-413. PubMed
34.Feagan BG, Rutgeerts P, Sands BE, et al. Vedolizumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2013;369(8):699-710. PubMed
35.Sandborn WJ, Su C, Sands BE, et al. Tofacitinib as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2017;376(18):1723-1736. PubMed
36.SLR of efficacy and safety of treatments for patients with moderate to severely active ulcerative colitis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: mirikizumab (Omvoh), 100 mg/mL solutions for subcutaneous injection; 20 mg/mL solution for intravenous infusion. York (UK): Eli Lilly and Company Ltd.; 2022 Nov 20.
37.Alatab S, Sepanlou SG, Ikuta K, et al. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol Hepatol. 2020;5(1):17-30. PubMed
38.Coward S, Clement F, I BE, al e. The rising prevalence of inflammatory bowel disease in Canada: analyzing the past to predict the future. Gastroenterol. 2018;1:2.
39.Pola S, Patel D, Ramamoorthy S, et al. Strategies for the care of adults hospitalized for active ulcerative colitis. Clin Gastroenterol Hepatol. 2012;10(12):1315-1325.e1314. PubMed
40.Newton L, Randall JA, Hunter T, et al. A qualitative study exploring the health-related quality of life and symptomatic experiences of adults and adolescents with ulcerative colitis. J Patient Rep Outcomes. 2019;3:66. PubMed
41.Feuerstein JD, Isaacs KL, Schneider Y, et al. AGA clinical practice guidelines on the management of moderate to severe ulcerative colitis. Gastroenterology. 2020;158(5):1450-1461. PubMed
42.Bressler B, Marshall JK, Bernstein CN, et al. Clinical practice guidelines for the medical management of nonhospitalized ulcerative colitis: the Toronto consensus. Gastroenterology. 2015;148(5):1035-1058.e1033. PubMed
43.Kühn F, Klar E. Surgical principles in the treatment of ulcerative colitis. Viszeralmedizin. 2015;31(4):246-250. PubMed
44.Raine T, Bonovas S, Burisch J, et al. ECCO Guidelines on Therapeutics in Ulcerative Colitis: Medical Treatment. J Crohns Colitis. 2022;16(1):2-17. PubMed
45.Turner D, Ricciuto A, Lewis A, et al. STRIDE-II: an update on the Selecting Therapeutic Targets in Inflammatory Bowel Disease (STRIDE) initiative of the International Organization for the Study of IBD (IOIBD): determining therapeutic goals for treat-to-target strategies in IBD. Gastroenterology. 2021;160(5):1570-1583. PubMed
46.Committee for Medicinal Products for Human Use (CHMP). Summary of opinion (initial authorisation): Omvoh (mirikizumab). EMA/CHMP/138535/2023. Amsterdam (Netherlands): European Medicines Agency; 2023 Mar 30: https://www.ema.europa.eu/en/documents/smop-initial/chmp-summary-positive-opinion-omvoh_en.pdf. Accessed 2023 Nov 10.
47.Zeposia (ozanimod): 0.23 mg, 0.46 mg and 0.92 mg capsules [product monograph]. Saint-Laurent (QC): Celgene Inc; 2022 Apr 7: https://pdf.hres.ca/dpd_pm/00065373.PDF. Accessed 2022 Apr 29.
48.Stelara (ustekinumab): 45 mg/0.5 mL, 90 mg/1.0 mL solution for subcutaneous injection; 130 mg/26 mL (5mg/ML) solution for intravenous infusion [product monograph]. Toronto (ON): Janssen Inc; 2020 Jan 23: https://pdf.hres.ca/dpd_pm/00054738.PDF. Accessed 2022 Mar 29.
49.Remicade (infliximab): 100 mg/vial powder for solution, sterile, lyophilized [product monograph]. Toronto (ON): Janssen Inc; 2019 Jun 06: https://pdf.hres.ca/dpd_pm/00051606.PDF. Accessed 2022 Mar 29.
50.Entyvio (vedolizumab): 300 mg / vial powder for concentrate for solution for intravenous injection; 108 mg/0.68 mL prefilled syringe or pen solution for subcutaneous injection [product monograph]. Toronto (ON): Takeda Canada Inc; 2015 Jan 29: https://pdf.hres.ca/dpd_pm/00058905.PDF. Accessed 2022 Mar 29.
51.Simponi (golimumab): 50 mg/0.5 mL, 100 mg/1.0 mL solution for subcutaneous injection; 50 50 mg/4.0 mL solution for intravenous infusion [product monograph]. Toronto (ON): Janssen Inc; 2019 Jun 20: https://pdf.hres.ca/dpd_pm/00052895.PDF. Accessed 2022 Mar 29.
52.Xeljanz (tofacitinib): 5 mg, 10 mg oral tablets; 11 mg extended-release oral tablets [product monograph]. Kirkland (QC) Pfizer Canada ULC; 2019 Oct 24: https://pdf.hres.ca/dpd_pm/00053735.PDF. Accessed 2022 Mar 29.
53.Humira (adalimumab injection): 40 mg in 0.8 mL sterile solution (50 mg/mL) subcutaneous injection, 10 mg in 0.1 mL sterile solution (100 mg/mL) subcutaneous injection, 20 mg in 0.2 mL sterile solution (100 mg/mL) subcutaneous injection, 40 mg in 0.4 mL sterile solution (100 mg/mL) subcutaneous injection, 80 mg in 0.8 mL sterile solution (100 mg/mL) subcutaneous injection [product monograph]. St-Laurent (QC): AbbVie Corporation; 2021 Apr 21: https://pdf.hres.ca/dpd_pm/00061690.PDF. Accessed 2022 May 2.
54.Blauvelt A, Kimball AB, Augustin M, et al. Efficacy and safety of mirikizumab in psoriasis: results from a 52-week, double-blind, placebo-controlled, randomized withdrawal, phase III trial (OASIS-1). Br J Dermatol. 2022;187(6):866-877. PubMed
55.Dubinsky MC, Clemow DB, Hunter Gibble T, et al. Clinical effect of mirikizumab treatment on bowel urgency in patients with moderately to severely active ulcerative colitis and the clinical relevance of bowel urgency improvement for disease remission. Crohn's & Colitis 360. 2023;5(1):otac044. PubMed
56.Arai M, Naganuma M, Sugimoto S, et al. The Ulcerative Colitis Endoscopic Index of Severity is Useful to Predict Medium- to Long-Term Prognosis in Ulcerative Colitis Patients with Clinical Remission. J Crohns Colitis. 2016;10(11):1303-1309. PubMed
57.Ikeya K, Hanai H, Sugimoto K, et al. The Ulcerative Colitis Endoscopic Index of Severity more accurately reflects clinical outcomes and long-term prognosis than the Mayo Endoscopic Score. J Crohns Colitis. 2016;10(3):286-295. PubMed
58.Dubinsky MC, Delbecque L, Lewis J, Hunter Gibble T, Shan M. Psychometric validation and interpretation of a patient-reported outcomes instrument to assess bowel urgency among adults with moderate to severe ulcerative colitis [conference poster]. Poster presented at: 17th Congress of ECCO, virtual, 2022 Feb 16-19. Vienna (Austria): European Crohn's and Colitis Organisation (ECCO); 2022: https://www.ecco-ibd.eu/publications/congress-abstracts/item/p138-psychometric-validation-and-interpretation-of-a-patient-reported-outcomes-instrument-to-assess-bowel-urgency-among-adults-with-moderate-to-severe-ulcerative-colitis.html. Accessed 2023 Nov 9.
59.Guyatt G, Mitchell A, Irvine EJ, et al. A new measure of health status for clinical trials in inflammatory bowel disease. Gastroenterology. 1989;96(3):804-810. PubMed
60.Herdman M, Gudex C, Lloyd A, et al. Development and preliminary testing of the new five-level version of EQ-5D (EQ-5D-5L). Qual Life Res. 2011;20(10):1727-1736. PubMed
61.Stark RG, Reitmeir P, Leidl R, König H-H. Validity, reliability, and responsiveness of the EQ-5D in inflammatory bowel disease in Germany. Inflamm Bowel Dis. 2010;16(1):42-51. PubMed
62.Panés J, Domènech E, Aguas Peris M, et al. Association between disease activity and quality of life in ulcerative colitis: Results from the CRONICA-UC study. J Gastroenterol Hepatol. 2017;32(11):1818-1824. PubMed
63.Bernklev T, Jahnsen J, Lygren I, Henriksen M, Vatn M, Moum B. Health-related quality of life in patients with inflammatory bowel disease measured with the short form-36: psychometric assessments and a comparison with general population norms. Inflamm Bowel Dis. 2005;11(10):909-918. PubMed
64.Yarlas A, Bayliss M, Cappelleri JC, et al. Psychometric validation of the SF-36® Health Survey in ulcerative colitis: results from a systematic literature review. Qual Life Res. 2018;27(2):273-290. PubMed
65.Naegeli AN, Hunter T, Dong Y, et al. Full, partial, and modified permutations of the Mayo Score: characterizing clinical and patient-reported outcomes in ulcerative colitis patients. Crohn's & Colitis 360. 2021;3(1):otab007. PubMed
66.Geboes K, Riddell R, Ost A, Jensfelt B, Persson T, Löfberg R. A reproducible grading scale for histological assessment of inflammation in ulcerative colitis. Gut. 2000;47(3):404-409. PubMed
67.Bryant RV, Winer S, Travis SP, Riddell RH. Systematic review: histological remission in inflammatory bowel disease. Is 'complete' remission the new treatment paradigm? An IOIBD initiative. J Crohns Colitis. 2014;8(12):1582-1597. PubMed
68.Mosli MH, Feagan BG, Zou G, et al. Reproducibility of histological assessments of disease activity in UC. Gut. 2015;64(11):1765-1773. PubMed
69.Lemmens B, Arijs I, Van Assche G, et al. Correlation between the endoscopic and histologic score in assessing the activity of ulcerative colitis. Inflamm Bowel Dis. 2013;19(6):1194-1201. PubMed
70.Kim DB, Lee K-M, Lee JM, et al. Correlation between histological activity and endoscopic, clinical, and serologic activities in patients with ulcerative colitis. Gastroenterol Res Pract. 2016;2016:5832051-5832051. PubMed
71.Mosli MH, Feagan BG, Zou G, et al. Development and validation of a histological index for UC. Gut. 2017;66(1):50-58. PubMed
72.Lobatón T, Bessissow T, Ruiz-Cerulla A, et al. Prognostic value of histological activity in patients with ulcerative colitis in deep remission: a prospective multicenter study. United European Gastroenterol J. 2018;6(5):765-772. PubMed
73.Network meta-analysis for moderately to severely active ulceritis colitis (2019-8488) report [internal sponsor's report]. Drug Reimbursement Review sponsor submission: mirikiziumab, 100 mg/mL solutions for subcutaneous injection; 20 mg/mL solution for intravenous infusion. Toronto (ON): Eli Lilly Canada, Inc.; 2023.
74.Centre for Reviews and Dissemination. Systematic reviews: CRD's guidance for undertaking reivews in health care. Layerthorpe, York (UK): CRD, University of York; 2009: https://www.york.ac.uk/media/crd/Systematic_Reviews.pdf. Accessed 2023 Nov 9.
75.Hoaglin DC, Hawkins N, Jansen JP, et al. Conducting indirect-treatment-comparison and network-meta-analysis studies: report of the ISPOR Task Force on Indirect Treatment Comparisons Good Research Practices: part 2. Value Health. 2011;14(4):429-437. PubMed
76.Garud S, Brown A, Cheifetz A, Levitan EB, Kelly CP. Meta-analysis of the placebo response in ulcerative colitis. Dig Dis Sci. 2008;53(4):875-891. PubMed
77.Jairath V, Zou GY, Parker CE, et al. Placebo response and remission rates in randomised trials of induction and maintenance therapy for ulcerative colitis. Cochrane Database Syst Rev. 2017;9(9):CD011572. PubMed
78.Sedano R, Hogan M, Nguyen TM, et al. Systematic review and meta-analysis: clinical, endoscopic, histological and safety placebo rates in induction and maintenance trials of ulcerative colitis. J Crohns Colitis. 2022;16(2):224-243. PubMed
79.Elsenbruch S, Enck P. Placebo effects and their determinants in gastrointestinal disorders. Nat Rev Gastroenterol Hepatol. 2015;12(8):472-485. PubMed
80.Macaluso FS, Maida M, Ventimiglia M, Renna S, Cottone M, Orlando A. Factors affecting clinical and endoscopic outcomes of placebo arm in trials of biologics and small molecule drugs in ulcerative colitis: a meta-analysis. Inflamm Bowel Dis. 2019;25(6):987-997. PubMed
81.Rutgeerts P, Sandborn WJ, Feagan BG, et al. Infliximab for induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2005;353(23):2462-2476. PubMed
82.Feagan BG, Rutgeerts P, Sands BE, et al. Vedolizumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2013;369(8):699-710. PubMed
83.Rubin DT, Raine T, Finney-Hayward T, et al. S854. Upadacitinib is effective at inducing clinical remission and response in ulcerative colitis patients regardless of baseline corticosteroid use: results from two phase 3 studies [conference abstract]. Am J Gastroenterol. 2021;116:S398. 2021 ACG Annual Meeting Abstracts.
84.Jiang XL, Cui HF, Gao J, Fan H. Low-dose infliximab for induction and maintenance treatment in Chinese patients with moderate to severe active ulcerative colitis. J Clin Gastroenterol. 2015;49(7):582-588. PubMed
85.Kobayashi T, Suzuki Y, Motoya S, et al. First trough level of infliximab at week 2 predicts future outcomes of induction therapy in ulcerative colitis-results from a multicenter prospective randomized controlled trial and its post hoc analysis. J Gastroenterol. 2016;51(3):241-251. PubMed
86.Motoya S, Watanabe K, Ogata H, et al. Vedolizumab in Japanese patients with ulcerative colitis: a Phase 3, randomized, double-blind, placebo-controlled study. PLoS One. 2019;14(2):e0212989. PubMed
87.Sandborn WJ, Su C, Sands BE, et al. Tofacitinib as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2017;376(18):1723-1736. PubMed
88.Probert CS, Hearing SD, Schreiber S, et al. Infliximab in moderately severe glucocorticoid resistant ulcerative colitis: a randomised controlled trial. Gut. 2003;52(7):998-1002. PubMed
89.Sandborn WJ, Feagan BG, Marano C, et al. Subcutaneous golimumab induces clinical response and remission in patients with moderate-to-severe ulcerative colitis. Gastroenterology. 2014;146(1):85-95; quiz e14-15. PubMed
90.Hibi T, Imai Y, Senoo A, Ohta K, Ukyo Y. Efficacy and safety of golimumab 52-week maintenance therapy in Japanese patients with moderate to severely active ulcerative colitis: a phase 3, double-blind, randomized, placebo-controlled study-(PURSUIT-J study). J Gastroenterol. 2017;52(10):1101-1111. PubMed
91.Sandborn WJ, Feagan BG, Marano C, et al. Subcutaneous golimumab maintains clinical response in patients with moderate-to-severe ulcerative colitis. Gastroenterology. 2014;146(1):96-109.e101. PubMed
92.Sandborn WJ, van Assche G, Reinisch W, et al. Adalimumab induces and maintains clinical remission in patients with moderate-to-severe ulcerative colitis. Gastroenterology. 2012;142(2):257-265.e251-253.
93.Suzuki Y, Motoya S, Hanai H, et al. Efficacy and safety of adalimumab in Japanese patients with moderately to severely active ulcerative colitis. J Gastroenterol. 2014;49(2):283-294. PubMed
94.Sandborn WJ, Feagan BG, D'Haens G, et al. Ozanimod as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2021;385(14):1280-1291. PubMed
95.Reinisch W, Sandborn WJ, Hommes DW, et al. Adalimumab for induction of clinical remission in moderately to severely active ulcerative colitis: results of a randomised controlled trial. Gut. 2011;60(6):780-787. PubMed
96.Sandborn WJ, Ghosh S, Panes J, et al. Tofacitinib, an oral Janus kinase inhibitor, in active ulcerative colitis. N Engl J Med. 2012;367(7):616-624. PubMed
97.Sands BE, Sandborn WJ, Panaccione R, et al. Ustekinumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2019;381(13):1201-1214. PubMed
98.Sands BE, Peyrin-Biroulet L, Loftus EV, Jr., et al. Vedolizumab versus adalimumab for moderate-to-severe ulcerative colitis. N Engl J Med. 2019;381(13):1215-1226. PubMed
99.Sandborn WJ, Baert F, Danese S, et al. Efficacy and safety of vedolizumab subcutaneous formulation in a randomized trial of patients with ulcerative colitis. Gastroenterology. 2020;158(3):562-572.e512. PubMed
100.Feagan BG, Danese S, Loftus EV, Jr., et al. Filgotinib as induction and maintenance therapy for ulcerative colitis (SELECTION): a phase 2b/3 double-blind, randomised, placebo-controlled trial. Lancet. 2021;397(10292):2372-2384. PubMed
101.Danese S, Vermeire S, Zhou W, et al. Upadacitinib as induction and maintenance therapy for moderately to severely active ulcerative colitis: results from three phase 3, multicentre, double-blind, randomised trials. Lancet. 2022;399(10341):2113-2128. PubMed
102.Sandborn WJ, Ghosh S, Panes J, et al. Efficacy of upadacitinib in a randomized trial of patients with active ulcerative colitis. Gastroenterology. 2020;158(8):2139-2149.e2114. PubMed
103.Barnes EL, Loftus EV, Jr., Kappelman MD. Effects of race and ethnicity on diagnosis and management of inflammatory bowel diseases. Gastroenterology. 2021;160(3):677-689. PubMed
104.Dhaliwal J, Tuna M, Shah BR, et al. Incidence of inflammatory bowel disease in South Asian and Chinese people: a population-based cohort study from Ontario, Canada. Clin Epidemiol. 2021;13:1109-1118. PubMed
105.Afzali A, Cross RK. Racial and ethnic minorities with inflammatory bowel disease in the united states: a systematic review of disease characteristics and differences. Inflamm Bowel Dis. 2016;22(8):2023-2040. PubMed
Appendix 1: Detailed Outcome Data
Note that this appendix has not been copy-edited.
Induction Period: LUCENT-1 Study (Subgroup Results)
Table 29: Primary and Key Secondary Outcome Results at Week 12 for LUCENT-1 Study, by Baseline Corticosteroid Use (mITT Population)
Outcome | Baseline corticosteroid use (yes) | Baseline corticosteroid use (no) | |||
|---|---|---|---|---|---|
Mirikizumab 300 mg IV q.4.w. | Placebo IV q.4.w. | Mirikizumab 300 mg IV q.4.w. | Placebo IV q.4.w. | ||
Clinical response | |||||
Patients with event of interest, n/Ns (%) | 207/351 (59.0) | 53/113 (46.9) | 344/517 (66.5) | 71/181 (39.2) | |
Risk difference, % (95% CI) | 12.1 (1.5 to 22.6) | 27.3 (19.1 to 35.5) | |||
P value | 0.029 | < 0.001 | |||
Clinical remission | |||||
Patients with event of interest, n/Ns (%) | 71/351 (20.2) | 19/133 (16.8) | 139/517 (26.9) | 20/181 (11.0) | |
Risk difference, % (95% CI) | 3.4 (–4.7 to 11.5) | 15.8 (9.9 to 21.8) | |||
P value | 0.495 | < 0.001 | |||
Alternate clinical remission | |||||
Patients with event of interest, n/Ns (%) | 76/351 (21.7) | 21/113 (18.6) | 146/517 (28.2) | 22/181 (12.2) | |
Risk difference, % (95% CI) | 3.1 (–5.3 to 11.4) | 16.1 (9.9 to 22.2) | |||
P value | 0.595 | < 0.001 | |||
Endoscopic remission | |||||
Patients with event of interest, n/Ns (%) | 111/351 (31.6) | 30/113 (26.5) | 204/517 (39.5) | 32/181 (17.7) | |
Risk difference, % (95% CI) | 5.1 (–4.4 to 14.6) | 21.8 (14.8 to 28.8) | |||
P value | 0.348 | < 0.001 | |||
Symptomatic remission | |||||
Patients with event of interest, n/Ns (%) | 143/351 (40.7) | 38/113 (33.6) | 252/517 (48.7) | 44/181 (24.3) | |
Risk difference, % (95% CI) | 7.1 (–3.0 to 17.2) | 24.4 (16.8 to 32.0) | |||
P value | 0.185 | < 0.001 | |||
HEMI | |||||
Patients with event of interest, n/Ns (%) | 83/351 (23.6) | 19/113 (16.8) | 152/517 (29.4) | 22/181 (12.2) | |
Risk difference, % (95% CI) | 6.8 (–1.4 to 15.0) | 17.2 (11.1 to 23.4) | |||
P value | 0.151 | < 0.001 | |||
Bowel urgency improvement (change in UNRS)a | |||||
Patients with event of interest, n/Ns | 327/351 | 98/113 | 502/517 | 160/181 | |
LSM change from baseline (SE) | –2.7 (0.13) | –2.2 (0.23) | –2.6 (0.10) | –1.3 (0.17) | |
LSM difference between groups (95% CI) | –0.5 (–1.0 to 0) | –1.2 (–1.6 to –0.8) | |||
P value | 0.075 | < 0.001 | |||
CI = confidence interval; HEMI = histologic endoscopic mucosal improvement; q.4.w. = every 4 weeks; SE = standard error; UNRS = Urgency Numeric Rating Scale
Note: 2-sided alpha at 0.05 significance level was used for the LUCENT-2 study.
Source: Clinical Study Report for the LUCENT-1 study.31
Table 30: Primary and Key Secondary Outcome Results at Week 12 for LUCENT-1 Study, by Baseline Immunomodulator Use (mITT Population)
||||||||| | |||||||| ||||||||||||||| ||| ||||| | |||||||| ||||||||||||||| ||| |||| | |||
|---|---|---|---|---|---|
||||||||||| ||||| || |||| | ||||||| || |||| | ||||||||||| ||||| || |||| | ||||||| || |||| | ||
|||||||| |||||||| | |||||
|||||||| |||| ||||| || ||||||||| |||| ||| | ||||||| |||||| | ||||| |||||| | ||||||| |||||| | |||||| |||||| | |
|||| ||||||||||||||| ||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||
|||| | ||||| | |||||| | |||
|||||||| ||||||||| | |||||
|||||||| |||| ||||| || ||||||||| |||| ||| | |||||| |||||| | ||||| |||||| | ||||||| |||||| | |||||| |||||| | |
|||| |||||||||||||| ||| | ||| ||||| || ||||| | |||| |||| || ||||| | |||
|||| | ||||| | |||||| | |||
||||||||| |||||||| ||||||||| | |||||
|||||||| |||| ||||| || ||||||||| |||| ||| | |||||| |||||| | ||||| |||||| | ||||||| |||||| | |||||| |||||| | |
|||| ||||||||||||||| ||| | ||| ||||| || ||||| | |||| |||| || ||||| | |||
||||| | ||||| | |||||| | |||
|||||||||| ||||||||| | |||||
|||||||| |||| ||||| || ||||||||| |||| ||| | |||||| |||||| | ||||| |||||| | ||||||| |||||| | |||||| |||||| | |
|||| ||||||||||||||| ||| | ||| ||||| || ||||| | |||| ||||| || ||||| | |||
||||| | ||||| | |||||| | |||
||||||||||| ||||||||| | |||||
|||||||| |||| ||||| || ||||||||| |||| ||| | |||||| |||||| | ||||| |||||| | ||||||| |||||| | |||||| |||||| | |
|||| ||||||||||||||| ||| | ||| ||||| || ||||| | |||| ||||| || ||||| | |||
||||| | ||||| | |||||| | |||
||||||||||| ||||||||| | |||||
|||||||| |||| ||||| || ||||||||| |||| ||| | |||||| |||||| | ||||| |||||| | ||||||| |||||| | |||||| |||||| | |
|||| |||||||||||||| ||| | ||| ||||| || ||||| | |||| |||| || ||||| | |||
|||| | ||||| | |||||| | |||
||||| ||||||| ||||||||||| ||||||| || |||| |||||| | |||||
|||||||| |||| ||||| || ||||||||| |||| | ||||| | ||||||| | ||||||| | ||||||| | |
||| |||||| |||| |||||||| |||| | |||| |||||| | |||| |||||| | |||| |||||| | |||| |||||| | |
||| |||||| |||||||||| ||||||| |||||| |||| ||| | |||| ||||| || |||| | |||| ||||| || ||||| | |||
|||| | ||||| | |||||| | |||
||||||||||||| |||||||||||||| |||||||||||| ||||||||||||| ||||||||||||||| |||| |||||||| || ||| |||||||| |||||||||||| ||||||||||| |||||||||||||| |||||||||||||||| |||||||||||||| ||||||||||||||| ||||||||| ||||||| |||||||||||||| |||||||||| || |||||| || ||||| ||| ||| ||||||||||||| |||||||| |||||||||| ||||||| |||||||| |||| |||||||||||||||| ||||||||| ||||||| |||| |||||||| |||| ||||||||||||||| |||||||||||| |||||||||| || |||||| || ||||| ||| ||| ||||||||||||| |||||||| ||||||||||| ||||||||| ||||||| ||||||| |||||||| |||| |||||||||||||| ||||||||| ||||||| ||||31
Maintenance Period: LUCENT-2 Study (Subgroup Results)
Table 31: Primary and Key Secondary Outcome Results at Week 40 for LUCENT-2 Study, by Prior Advanced Therapy Failure (mITT Population)
Outcome | Biologic-naive subpopulation | Biologic-faileda subpopulation | ||
|---|---|---|---|---|
Mirikizumab 200 mg SC q.4.w. N = 229 | Placebo SC q.4.w. N = 114 | Mirikizumab 200 mg SC q.4.w. N = 128 | Placebo SC q.4.w. N = 64 | |
Clinical remission | ||||
Patients with event of interest, n/Ns (%) | 118/229 (51.5) | 35/114 (30.7) | 59/128 (46.1) | 10/64 (15.6) |
Risk difference, % (95% CI) | 20.8 (10.2 to 31.5) | 30.5 (18.1 to 42.9) | ||
P value | < 0.001 | < 0.001 | ||
Alternate clinical remission | ||||
|||||||| |||| ||||| || ||||||||| |||| ||| | ||||||| |||||| | |||||| |||||| | |||||| |||||| | ||||| |||||| |
|||| ||||||||||||||| ||| | |||| ||||| || ||||| | |||| ||||| || ||||| | ||
|||| | |||||| | |||||| | ||
Corticosteroid-free remission | ||||
Patients with event of interest, n/Ns (%) | 107/229 (46.7) | 30/114 (26.3) | 52/128 (40.6) | 9/64 (14.1) |
Risk difference, % (95% CI) | 20.4 (10.1 to 30.8) | 26.6 (14.5 to 38.6) | ||
P value | < 0.001 | < 0.001 | ||
Durable clinical remission | ||||
Patients with event of interest, n/Ns (%) | 65/104 (62.5) | 22/47 (46.8) | 24/36 (66.7) | 2/18 (11.1) |
Risk difference, % (95% CI) | 15.7 (–1.3 to 32.7) | 55.6 (34.4 to 76.7) | ||
P value | 0.078 | < 0.001 | ||
Endoscopic remission | ||||
Patients with event of interest, n/Ns (%) | 143/229 (62.4) | 39/114 (34.2) | 65/128 (50.8) | 13/64 (20.3) |
Risk difference, % (95% CI) | 28.2 (17.5 to 39.0) | 30.5 (17.3 to 43.6) | ||
P value | < 0.001 | < 0.001 | ||
HEMR | ||||
Patients with event of interest, n/Ns (%) | 108/229 (47.2) | 30/114 (26.3) | 46/128 (35.9) | 9/64 (14.1) |
Risk difference, % (95% CI) | 20.8 (10.5 to 31.2) | 21.2 (10.9 to 31.4) | ||
P value | < 0.001 | < 0.001 | ||
Bowel urgency remission (among those with UNRS ≥ 3 at baseline) | ||||
Patients with event of interest, n/Ns (%) | 96/206 (46.6) | 31/108 (28.7) | 43/122 (35.2) | 12/63 (19.0) |
Risk difference, % (95% CI) | 17.9 (7.0 to 28.8) | 16.2 (3.3 to 29.1) | ||
P value | 0.002 | 0.027 | ||
Bowel urgency improvement (change in UNRS score)b | ||||
LSM change from baseline (SE) [Ns] | –3.82 (0.153) [229] | –2.69 (0.233) [114] | –3.60 (0.228) [128] | –2.66 (0.346) [64] |
LSM change difference between groups (95% CI) | –1.13 (–1.68 to –0.58) | –0.93 (–1.75 to –0.11) | ||
P value | < 0.001 | 0.026 | ||
CI = confidence interval; HEMR = histologic endoscopic mucosal remission; q.4.w. = every 4 weeks; SC = subcutaneous; SE = standard error; UNRS = Urgency Numeric Rating Scale.
Notes: Details in Table 31 have been taken from the sponsor’s Summary of Clinical Evidence.20,30
A 2-sided alpha at 0.05 significance level was used for the LUCENT-2 study.
aBiologic-failed refers to patients who have failed at least 1 biologic and/or tofacitinib.
bFor UNRS, change is reported at week 40 vs. the LUCENT-1 study’s baseline.
Source: Clinical Study Report for the LUCENT-2 study.32
Table 32: Primary and Key Secondary Outcome Results at Week 40 for LUCENT-2 Study, by Baseline Corticosteroid Use (NRI) (mITT Population)
||||||||| | |||||||| |||||||||||||| ||| ||||| | |||||||| |||||||||||||| ||| |||| | ||
|---|---|---|---|---|
||||||||||| ||| || || || | ||||||| || |||||| ||| | ||||||||||| ||| || || || | ||||||| || |||||| ||| | |
|||||||| ||||||||| | ||||
|||||||| |||| ||||| || ||||||||| |||| ||| | ||||| |||||| | ||||| |||||| | ||||||| |||||| | |||||| |||||| |
|||| ||||||||||||||| ||| | |||| ||||| || ||||| | |||| ||||| || ||||| | ||
||||| | |||||| | |||||| | ||
||||||||| |||||||| ||||||||| | ||||
|||||||| |||| ||||| || ||||||||| |||| ||| | ||||| |||||| | ||||| |||||| | ||||||| |||||| | |||||| |||||| |
|||| ||||||||||||||| ||| | |||| ||||| || ||||| | |||| ||||| || ||||| | ||
||||| | |||||| | |||||| | ||
||||||||||||||||||| ||||||||| | ||||
|||||||| |||| ||||| || ||||||||| |||| ||| | |||||| |||||| | ||||| |||||| | ||||||| |||||| | |||||| |||||| |
|||| ||||||||||||||| ||| | |||| ||||| || ||||| | |||| ||||| || ||||| | ||
||||| | |||||| | |||||| | ||
||||||| |||||||| ||||||||| | ||||
|||||||| |||| ||||| || ||||||||| |||| ||| | ||||| |||||| | |||| |||||| | ||||| |||||| | ||||| |||||| |
|||| ||||||||||||||| ||| | |||| |||| || ||||| | |||| |||| || ||||| | ||
||||| | ||||| | ||||| | ||
|||||||||| ||||||||| | ||||
|||||||| |||| ||||| || ||||||||| |||| ||| | |||||| |||||| | ||||| |||||| | ||||||| |||||| | |||||| |||||| |
|||| ||||||||||||||| ||| | |||| ||||| || ||||| | |||| ||||| || ||||| | ||
||||| | |||||| | |||||| | ||
||||| ||||||| ||||||||| |||||| ||||| |||| |||| || || ||||||||| | ||||
|||||||| |||| ||||| || ||||||||| |||| ||| | |||||| |||||| | ||||| |||||| | |||||| |||||| | |||||| |||||| |
|||| ||||||||||||||| ||| | |||| |||| || ||||| | |||| |||| || ||||| | ||
||||||||||||||| ||| | ||||| | ||||| | ||
|||||||||| ||||||||| | ||||
|||||||| |||| ||||| || ||||||||| |||| ||| | |||||| |||||| | ||||| |||||| | ||||||| |||||| | |||||| |||||| |
|||| ||||||||||||||| ||| | |||| |||| || ||||| | |||| ||| || ||||| | ||
||||| | |||||| | |||||| | ||
||| |||||||||| ||||||||| ||||| ||||||||||||||||||||| ||||||| |||||||||||||||| || |||||||| |||||| |||| |||||||| ||||||||| |||| || |||||||| |||||| ||| |||||||| ||||||||| ||| ||||||||||||| ||| |||||||| |||||| ||||| ||||||| ||||||| |||||| ||||| |||||| ||||||| ||||| || |||| |||||||||||| ||||| ||| |||| ||| ||||||||||||||||| |||||||| ||||| |||||| ||| ||||||||32
Table 33: Primary and Key Secondary Outcome Results at Week 40 for LUCENT-2 Study, by Baseline Immunomodulator Use (NRI) (mITT Population)
||||||||| | |||||||| ||||||||||||||| ||| ||||| | |||||||| ||||||||||||||| ||| |||| | ||
|---|---|---|---|---|
||||||||||| ||| || || || | ||||||| || |||||| ||| | ||||||||||| ||| || || || | ||||||| || |||||| ||| | |
|||||||| ||||||||| | ||||
|||||||| |||| ||||| || ||||||||| |||| ||| | ||||| |||||| | ||||| |||||| | ||||||| |||||| | |||||| |||||| |
|||| ||||||||||||||| ||| | |||| |||| || ||||| | |||| ||||| || ||||| | ||
||||| | ||||| | |||||| | ||
||||||||| |||||||| ||||||||| | ||||
|||||||| |||| ||||| || ||||||||| |||| ||| | ||||| |||||| | ||||| |||||| | ||||||| |||||| | |||||| |||||| |
|||| |||||||||||||| ||| | |||| |||| || ||||| | |||| ||||| || ||||| | ||
||||| | ||||| | |||||| | ||
||||||||||||||||||| ||||||||| | ||||
|||||||| |||| ||||| || ||||||||| |||| ||| | ||||| |||||| | |||| |||||| | |||||| |||||| | |||||| |||||| |
|||| ||||||||||||||| ||| | |||| |||| || ||||| | |||| ||||| || ||||| | ||
|||| | ||||| | |||||| | ||
||||||| |||||||| ||||||||| | ||||
|||||||| |||| ||||| || ||||||||| |||| ||| | ||||| |||||| | |||| |||||| | |||||| |||||| | ||||| |||||| |
|||| |||||||||||||| ||| | ||| |||||| || ||||| | |||| ||||| || ||||| | ||
||||| | |||||| | |||||| | ||
|||||||||| ||||||||| | ||||
|||||||| |||| ||||| || ||||||||| |||| ||| | ||||| |||||| | ||||| |||||| | ||||||| |||||| | |||||| |||||| |
|||| ||||||||||||||| ||| | |||| ||||| || ||||| | |||| ||||| || ||||| | ||
||||| | ||||| | |||||| | ||
||||| ||||||| ||||||||| |||||| ||||| |||| |||| || || ||||||||| | ||||
|||||||| |||| ||||| || ||||||||| |||| ||| | ||||| |||||| | ||||| |||||| | ||||||| |||||| | |||||| |||||| |
|||| ||||||||||||||| ||| | |||| |||| || ||||| | |||| |||| || ||||| | ||
||||||||||||||| ||| | ||||| | ||||| | ||
||||||| |||||||| ||||||||| | ||||
|||||||| |||| ||||| || ||||||||| |||| ||| | ||||| |||||| | |||| |||||| | ||||||| |||||| | |||||| |||||| |
|||| ||||||||||||||| ||| | |||| |||| || ||||| | |||| ||||| || ||||| | ||
||||| | ||||| | |||||| | ||
||| |||||||||| ||||||||| ||||| ||||||||||||||||||||| ||||||| |||||||||||||| || |||||||| |||||| |||| |||||||| ||||||||| ||||||| || |||||||| |||||| ||| |||||||| ||||||||| ||| ||||||||||||| ||| |||||||| |||||| ||||| ||||||| ||||||| |||||| |||| ||||| |||||| || |||||||| || |||| || |||||| |||||||| |||||||||||||| ||||||| ||||| || |||| |||||||||||| ||||| ||| |||| ||| ||||||||||||||||| |||||||| ||||| |||||| ||| ||||||||32
Table 34: Primary and Key Secondary Outcome Results at Week 40 for LUCENT-2 Study, by Baseline MMS Score (NRI) (mITT Population)
||||||||| | |||||||| || |||||| || |||| | ||||||||| |||||| || |||| | ||
|---|---|---|---|---|
||||||||||| ||| || || ||| | ||||||| || |||||| ||| | ||||||||||| ||| || || || | ||||||| || |||||| ||| | |
|||||||| ||||||||| | ||||
|||||||| |||| ||||| || ||||||||| |||| ||| | |||||| |||||| | ||||| |||||| | ||||||| |||||| | |||||| |||||| |
|||| ||||||||||||||| ||| | |||| |||| || ||||| | |||| ||||| || ||||| | ||
||||| | ||||| | |||||| | ||
||||||||| |||||||| ||||||||| | ||||
|||||||| |||| ||||| || ||||||||| |||| ||| | |||||| |||||| | ||||||||||| | ||||||| |||||| | |||||| ||||| |
|||| ||||||||||||||| ||| | |||| |||| || ||||| | |||| ||||| || ||||| | ||
||||| | ||||| | |||||| | ||
|||||||||| ||||||||| | ||||
|||||||| |||| ||||| || ||||||||| |||| ||| | |||||| |||||| | ||||| |||||| | ||||||| |||||| | |||||| |||||| |
|||| |||||||||||||| ||| | |||| |||| || ||||| | |||| ||||| || ||||| | ||
||||| | ||||| | |||||| | ||
||||||||||||||||||| ||||||||| | ||||
|||||||| |||| ||||| || ||||||||| |||| ||| | |||||| |||||| | ||||| |||||| | |||||| |||||| | |||||| |||||| |
|||| ||||||||||||||| ||| | |||| |||| || ||||| | |||| ||||| || ||||| | ||
||||| | ||||| | |||||| | ||
||||||| |||||||| ||||||||| | ||||
|||||||| |||| ||||| || ||||||||| |||| ||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| |
|||| |||||||||||||| ||| | |||| |||| || ||||| | |||| ||||| || ||||| | ||
||||| | ||||| | ||||| | ||
|||| | ||||
|||||||| |||| ||||| || ||||||||| |||| ||| | |||||| |||||| | ||||| |||||| | |||||| |||||| | |||||| |||||| |
|||| ||||||||||||||| ||| | |||| |||| || ||||| | |||| ||||| || ||||| | ||
||||| | ||||| | |||||| | ||
||||| ||||||| ||||||||| |||||| ||||| |||| |||| || || ||||||||| | ||||
|||||||| |||| ||||| || ||||||||| |||| ||| | |||||| |||||| | ||||| |||||| | |||||| |||||| | |||||| |||||| |
|||| ||||||||||||||| ||| | |||| |||| || ||||| | |||| |||| || ||||| | ||
||||| | ||||| | |||||| | ||
||| |||||||||| ||||||||| ||||| ||||||||||||||||||||| ||||||| |||||||||||||||| || |||||||| |||||| |||| |||||||| ||||||||| |||||||| || |||||||| |||||| ||| |||||||| ||||||||| ||| ||||||||||||| ||| |||||||| |||||| ||||| ||||||| ||||||| |||||| ||||| |||||| |||||||| || || ||||||| |||| ||||| |||| || |||||| || || ||||||| ||||| ||||| |||| || |||||| ||||||| ||||| || |||| |||||||||||| ||||| ||| |||| ||| ||||||||||||||||| |||||||| ||||| |||||| ||| ||||||||32
Figure 4: Network Plot for Clinical Response and Remission in the Induction Period for the Biologic-Naive Population

Note: The figure was redacted at the request of the sponsor.
Figure 5: Network Plot for Clinical Response and Remission in the Induction Period for the Biologic-Experienced Population

Note: The figure was redacted at the request of the sponsor.
Figure 6: Network Plot for Clinical Response and Remission in the Maintenance Period for the Biologic-Naive Population

Note: The figure was redacted at the request of the sponsor.
Figure 7: Network Plot for Clinical Response and Remission in the Maintenance Period for the Biologic-Experienced Population

Note: The figure was redacted at the request of the sponsor.
Figure 8: Network Plot for Mucosal Healing in the Induction Period for the Biologic-Naive Population

Note: The figure was redacted at the request of the sponsor.
Figure 9: Network Plot for Mucosal Healing in the Induction Period for the Biologic-Experienced Population

Note: The figure was redacted at the request of the sponsor.
Figure 10: Network Plot for Mucosal Healing in the Maintenance Period for the Biologic-Naive Population

Note: The figure was redacted at the request of the sponsor.
Figure 11: Network Plot for Mucosal Healing in the Maintenance Period for the Biologic-Experienced Population

Note: The figure was redacted at the request of the sponsor.


Pharmacoeconomic Review
Abbreviations
BIA
budget impact analysis
CEF
cost-effectiveness frontier
CUA
cost-utility analysis
IBD
inflammatory bowel disease
ICER
incremental cost-effectiveness ratio
JAK
Janus kinase
NMA
network meta-analysis
pCPA
pan-Canadian Pharmaceutical Alliance
QALY
quality-adjusted life-year
UC
ulcerative colitis
WTP
willingness-to-pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Mirikizumab (Omvoh), 300 mg/15 mL (vial for IV use), 100 mg/1 mL (autoinjector pen for SC use), 100 mg/1 mL (prefilled syringe for SC use) |
Submitted price | Mirikizumab:
|
Indication | For the treatment of adult patients with moderately to severely active ulcerative colitis who have had an inadequate response, had a loss of response, or were intolerant to conventional therapy, a biologic treatment, or a JAK inhibitor |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | July 20, 2023 |
Reimbursement request | As per indication |
Sponsor | Eli Lilly Canada |
Submission history | Previously reviewed: No |
JAK = Janus kinase; NOC = Notice of Compliance; SC = subcutaneous.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Decision-tree with Markov model |
Target population | Adults with moderately to severely active UC who have had an inadequate response, had a loss of response, or were intolerant to either conventional therapy, a biologic treatment, or a JAK inhibitor, or have medical contraindications to such therapies. Two key subgroups were reported separately: patients who were biologic-naive and patients who were biologic-experienced. |
Treatment | Mirikizumab |
Comparatorsa |
|
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (assumed to be 50 years) |
Key data sources | LUCENT-1 study, LUCENT-2 study, and a sponsor-commissioned, unpublished NMA |
Submitted results | Biologic-naive population: Mirikizumab is dominated by upadacitinib 45 mg/30 mg (incremental costs = $3,022; incremental QALYs = –0.74)b Biologic-experienced: Mirikizumab is dominated by upadacitinib 45 mg/15 mg (incremental costs = $20,091; incremental QALYs = –2.37)c |
Key limitations |
|
CADTH reanalysis results |
|
ICER = incremental cost-effectiveness ratio; JAK = Janus kinase; LY = life-year; NMA = network meta-analysis; QALY = quality-adjusted life-year; S1P = sphingosine-1-phosphate; SC = subcutaneous; TNF = tumour necrosis factor; UC = ulcerative colitis; WTP = willingness-to-pay.
aThe comparators were the same for the biologic-naive and biologic-experienced cohorts, with the exception that golimumab and infliximab were excluded from the biologic-experienced cohort due to a lack of data to facilitate a comparison for that population.
bMirikizumab was also dominated by infliximab in the biologic-naive population.
cMirikizumab was also dominated by tofacitinib in the biologic-experienced population.
Conclusions
Evidence from the LUCENT trials demonstrated the efficacy of mirikizumab in achieving induction and maintenance of clinical remission and clinical response in patients with moderately or severely active UC compared with placebo. Although the results were considered generalizable to the Canadian setting, CADTH identified limitations with the trials that introduced uncertainty in the comparative efficacy of mirikizumab relative to placebo. Furthermore, data pertaining to the use of mirikizumab for extended induction and reinduction were observational. Therefore, no conclusions could be reached regarding the comparative efficacy based on mirikizumab’s use in these populations. As there are no trials comparing mirikizumab with other advanced therapies (i.e., biologics, Janus kinase [JAK] inhibitors, and small molecule drugs), comparisons among treatments were based on a sponsor-commissioned network meta-analysis (NMA). Due to the heterogeneity between studies included in the NMA, the comparative effects of mirikizumab with other advanced therapies is uncertain. Evidence of the efficacy and safety of mirikizumab beyond 52 weeks, as well as direct comparisons with other treatments for UC, are necessary to further understand the long-term comparative effectiveness of mirikizumab.
In the base case, CADTH assumed the clinical efficacy, risk of loss of response, and risk of serious events of all advanced therapies were equal to that of mirikizumab; excluded the treatment effect of extended induction with mirikizumab; and excluded upadacitinib as a relevant comparator. However, concerns regarding the transparency of the model output could not be resolved, which limits the confidence that can be placed on the results from the model. The results of the CADTH base case generally aligned with the sponsor-submitted analysis: mirikizumab is not cost-effective at a willingness-to-pay (WTP) threshold of $50,000 per quality-adjusted life-year (QALY) in patients who are biologic-naive or biologic-experienced compared to advanced therapies. In the CADTH base case, conventional therapy, tofacitinib, and mirikizumab were on the cost-effectiveness frontier (CEF) in both the biologic-naive and biologic-experienced populations. Compared with tofacitinib, mirikizumab was not cost-effective at a $50,000 per QALY gained WTP threshold in either population. Based on publicly available list prices for all comparators and the submitted price of mirikizumab, a price reduction for mirikizumab of approximately 65% would be required for mirikizumab to be cost-effective in either the biologic-naive population or the biologic-experienced population at a WTP threshold of $50,000 per QALY gained. Such a price reduction would reduce the annual per patient costs of mirikizumab from $30,895 to $10,751 in the first year and from $30,871 to $10,743 in subsequent years of treatment. When only considering drug acquisition costs, a price reduction of approximately 85% is required for mirikizumab to be no more costly than the publicly available price of the least costly advanced therapy (i.e., tofacitinib, which costs $4,375 to $6,076 per patient per year).
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process — specifically, information that pertains to the economic submission.
Patient input was received from Crohn’s and Colitis Canada and the Gastrointestinal Society, which collected the perspectives of patients with ulcerative colitis (UC) through online surveys, questionnaires, and interviews. Patients with UC reported diarrhea, bowel urgency, incontinence, bloating, abdominal pain, fever, rectal bleeding, anemia, joint inflammation, arthritis, nausea, and a negative impact on mental health. Patients noted that idiopathic flares were disruptive to daily life, productivity, travel plans, and other social activities. Current treatments that were available to patients included systemic steroids, sulfasalazine, 5-aminosalicylates, biologics, and JAK inhibitors. Treatment goals included fewer side effects, a reduced number of hospitalizations, improved quality of life, and increased productivity. The 4 patients interviewed who had had experience with mirikizumab noted that there was slight discomfort with injections, that there was pain at the site of the injection, and that frequent trips to the clinic were needed for infusions. These patients with previous experience with mirikizumab noted that it did not eliminate all UC symptoms.
Clinician input was received from a group of gastroenterologists in Canada specializing in the management of inflammatory bowel disease (IBD). Clinicians noted that treatment goals were to not only control symptoms but also prevent disease progression, surgery, and disability. Clinicians indicated that the guidelines for the international 2021 initiative Selecting Therapeutic Targets in Inflammatory Bowel Disease–II (known as STRIDE-II) were used in clinical practice to determine short-term, intermediate-term, and long-term goals. The clinicians noted that while histologic remission is not listed as a specific target, the STRIDE-II guidelines state that it can be “used as an adjunct to endoscopic remission to represent a deeper level of healing.” Clinical response, measured by the resolution of stool frequency and rectal bleeding in the first 3 months, is a key short-term goal, while clinical remission and not requiring corticosteroids after 6 months is a key intermediate-term goal. The key long-term goal is endoscopic healing, assessed 9 months to 12 months after treatment initiation. Clinicians noted that patients with UC needed treatments that could achieve symptomatic and endoscopic remission quickly, could maintain remission, and were safe for long-term use. The clinicians noted that not all patients’ UC achieves remission or maintain response on current treatments. They observed that mirikizumab could be used as an option for patients with moderate to severe UC whose disease does not respond to advanced therapies. The clinician input recommended mirikizumab be administered in a clinic under the supervision of a trained health care professional during the induction phase; treatment should be discontinued in cases of worsening symptoms or inadequate response, and these patients should switch to another class of drugs.
CADTH-participating drug plans noted the lack of head-to-head comparative evidence of mirikizumab with other IL-23 inhibitors. The drug plans noted that not all advanced therapies are listed in all jurisdictions, and also flagged that the induction dose of mirikizumab requires infusion, which is likely to be administered in outpatient IV clinics and would be associated with additional costs.
Several of these concerns were addressed in the sponsor’s model:
Clinically important outcomes of clinical response and remission were included in the sponsor’s model.
The cost of administering IV infusion and subcutaneous injections was captured in the sponsor’s estimate of drug acquisition costs.
In addition, CADTH addressed some of these concerns as follows:
The sponsor assumed long-term differences in clinical response, clinical remission, and loss of response with mirikizumab compared to advanced therapies based on results of the sponsor-submitted NMA, including studies with a trial duration of up to 60 weeks. However, there is limited evidence to support this assumption. In reanalysis, CADTH assumed no differences in the comparative efficacy and safety of mirikizumab and advanced therapies.
CADTH was unable to address the following concerns raised from stakeholder input:
The time at which response and/or remission is assessed in the model may differ to that used in clinical practice.
There was a lack of long-term comparative evidence on endoscopic remission.
Disease management of the moderate to severe UC population is characterized by treatment-switching until all therapeutic options are exhausted. There is a lack of efficacy data regarding the sequencing of various advanced therapies.
Economic Review
The current review is for mirikizumab (Omvoh) for adult patients with moderately to severely active UC who have had an inadequate response, had a loss of response, or were intolerant to either conventional therapy, a biologic treatment, or a JAK inhibitor, or have medical contraindications to such therapies.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis (CUA) of mirikizumab compared with conventional therapy and advanced therapies. The term “advanced therapies” was used to refer to biologics, JAK inhibitors, and small molecule therapies.1 The modelled population consisted of adult patients with moderately to severely active UC who have had an inadequate response, had a loss of response, or were intolerant to either conventional therapy, a biologic treatment, or a JAK inhibitor, or have medical contraindications to such therapies. The CUA was conducted separately for the biologic-naive and biologic-experienced populations; in this case, the term “biologic” applies to all advanced therapies (i.e., biologics, JAK inhibitors, and small molecule drugs). The sponsor assumed the same comparators were relevant to each subpopulation. The modelled population is generally aligned with the Health Canada indication and the reimbursement request.
Mirikizumab is available as a 300 mg/15 mL solution for IV infusion for use in an initiation phase, and 100 mg/1 mL solution for subcutaneous injection for use in the maintenance phase. The recommended induction dose is 300 mg infused intravenously over at least 30 minutes at week 0, week 4, and week 8.2 If a patient has an adequate response at week 12, they should transition to maintenance dosing. If patients do not have adequate therapeutic response at week 12 after induction dosing, the induction phase may be extended, allowing patients to receive 300 mg infused intravenously at week 12, week 16, and week 20. Mirikizumab should be discontinued in patients who do not show evidence of therapeutic benefit after 24 weeks of induction therapy. If therapeutic response is achieved with the additional IV induction dosing, patients may initiate maintenance dosing every 4 weeks. The recommended maintenance dose is 200 mg, given as 2 consecutive subcutaneous injections of 100 mg each, every 4 weeks. If patients experience loss of response during maintenance therapy, they may receive up to 3 additional doses of mirikizumab IV infusion; if therapeutic benefit is achieved, patients may continue receiving mirikizumab at the maintenance dose. At the sponsor-submitted prices of $2,374.66 per 300 mg/15 mL vial for infusion and $1,187.33 per 100 mg/1 mL for subcutaneous injection, the cost of induction ranged from $7,123.98 per patient (12 weeks of treatment) to $14,247.96 per patient (24 weeks of treatment), while the annual maintenance cost was estimated to be $30,977 per patient.
The comparators for this analysis included tumour necrosis factor inhibitors (i.e., adalimumab, adalimumab biosimilar, infliximab, infliximab biosimilar, and golimumab), JAK inhibitors (i.e., tofacitinib, upadacitinib 45 mg/15 mg, and upadacitinib 45 mg/30 mg), cell adhesion molecule inhibitors (i.e., ustekinumab, vedolizumab IV, vedolizumab subcutaneous), sphingosine-1-phosphate (S1P) receptor (ozanimod), and conventional therapy (consisting of a combination of 5-aminosalicylic acid, corticosteroids, and conventional immunomodulators). The recommended dosing of comparators was sourced from respective product monographs and drug costs were obtained from the IQVIA DeltaPA database.3-10 The sponsor estimated induction costs for the advanced therapy comparators ranged from $4,485 for ozanimod to $8,883 for infliximab (IV).1 The sponsor estimated annual maintenance costs ranged from $17,978 for upadacitinib 45 mg/15 mg to $27,029 for upadacitinib 45 mg/30 mg. These costs were derived from the sponsor’s weighted average of branded products and biosimilars for adalimumab and infliximab, and incorporated the assumption of escalated doses for adalimumab, infliximab, golimumab, vedolizumab, and tofacitinib.
The economic evaluation was conducted over a lifetime time horizon (assumed 50 years) from the perspective of the Canadian public health care payer. Costs and clinical outcomes (life-years and QALYs) are discounted at 1.5% per annum.
Model Structure
The sponsor submitted a Markov model that considered a short-term induction phase (refer to Figure 1) and a longer-term maintenance phase (refer to Figure 2) to evaluate clinical outcomes and costs.1 Patients entered the model with moderately to severely active UC and received induction therapy with 1 of the included treatments. Throughout the user-specified duration of the induction period (up to 26 weeks), patients transitioned through tunnel states (2-week cycles) to determine initial response: remission, response without remission (hereafter termed “response”), or nonresponse. Patients in whom remission or response was achieved entered the maintenance phase of the Markov model in their corresponding health states at the end of the induction phase. Patients whose UC did not respond to mirikizumab (nonresponder group) could receive extended induction (an additional 12 weeks) before entering the maintenance phase. Those whose UC responded to the extended induction entered the maintenance phase in their corresponding health states, while those who UC did not respond entered the maintenance phase in the active UC state and received conventional therapy. The modelled cycle length was 2 weeks during the induction phase and 12 weeks during the maintenance phase. The same model structure was used for both biologic-naive and biologic-experienced patient populations.
In the maintenance phase, patients were modelled through 3 treatment groups (advanced therapy, conventional therapy, and surgery) and 12 health states. Patients on advanced therapies could be in 1 of 3 health states: remission, response, or mirikizumab reinduction (patients receiving mirikizumab only). Patients on conventional therapy could be in 1 of 3 health states: remission, response, or active UC. Patients on advanced therapies who experienced loss of response or did not respond to mirikizumab reinduction transitioned to conventional therapy in the active UC health state. The sponsor did not model downstream treatments for patients who experienced loss of response. All patients switched to conventional therapy after discontinuation from advanced therapy; transitioning from conventional therapy to advanced therapy was not possible. Patients could have surgery from either the active UC or response health states from the advanced therapy or conventional therapy treatment groups. Surgery consisted of 2 base states (emergency surgery and elective surgery) and 4 postsurgery health states (post–first surgical remission, post–first surgical complication, second surgery, post–second surgery remission). Following the first colectomy, patients discontinued treatment, including conventional therapy and advanced therapy, for the remainder of their lifetime. Postsurgery, patients could transition from post–first surgery remission to post–first surgery complications or remain in remission. From the post–first surgery complications health state, patients could remain in post–first surgery complications or transition to the second surgery health state. Once patients experienced a second surgery, they were assumed to experience remission for the remainder of the model’s time horizon. Finally, patients could transition to death from any of the maintenance model health states at any time. Patients could transition between health states every 12 weeks. Figure 1 and Figure 2 depict the model’s induction and maintenance phases, respectively (Appendix 3).
Model Inputs
Baseline patient characteristics were derived from the LUCENT-1 and LUCENT-2 clinical trials,11,12 stratified for biologic-naive and biologic-experienced patients. The average patient in the biologic-naive cohort was aged |||| years, weighed |||| kg, and was more likely to be male (||||%). Likewise, in the biologic-experienced cohort, the average patient was aged |||| years, weighed |||| kg, and tended to be male (||||%). Efficacy data were obtained from a sponsor-commissioned NMA,13 which assessed the induction and maintenance phases separately in the absence of head-to-head trial data comparing mirikizumab with advanced therapies. Bayesian NMAs were performed using random-effects or fixed-effects models, with a focus on the clinical response and clinical remission outcomes in primary analyses. Conventional therapy efficacy was represented by averaging response rates in the placebo arm of the randomized controlled trials included in the NMA.13 Patients on conventional therapy were also stratified, as possible, by whether or not their UC had responded adequately to prior therapy to derive efficacy inputs for the biologic-naive and biologic-experienced groups.
Remission was defined as a Mayo score of 2 points or less with no individual subscore of 1 point or more and response was defined as a decrease from baseline in the total Mayo score of 30% and at least 3 points with either a decrease in the rectal bleeding subscore of 1 point or more or a rectal bleeding subscore of 0 or 1. Patients whose UC did not meet the criteria for remission or response were considered to be in the active UC state. The mean absolute probabilities of remission, response, neither response nor remission, and discontinuation on advanced treatments due to loss of response were derived from the random-effects NMA’s induction and maintenance phases of the clinical trials (Table 13 and Table 14). The sponsor conducted a delayed response analysis for mirikizumab only and derived remission, overall response, and non–remission response rates after the extended induction period using the LUCENT trials. Due to a lack of long-term efficacy data for UC treatments beyond the trial duration of 1 year, the sponsor assumed a constant treatment effect and corresponding loss of response over the lifetime time horizon.
The risk of surgery and postsurgical complications was obtained from published literature.14-17 The general population mortality risk was increased by 2.8% for patients who underwent surgery.16,18,19 The sponsor assumed no additional UC-related risk of death.
Utility values for nonsurgical health states were sourced from Woehl et al. (2008),20 in which the 5-Level EQ-5D questionnaire was used to collect utility scores from 180 patients with active UC in the UK. These utility scores were used to inform the utility values for the remission, response, and active UC (i.e., moderate to severe disease) health states, as well as the post–second surgical remission state. Utility values for the other surgical states were obtained from Arseneau et al. (2006).21 Utility weights reported in this study were obtained from 48 patients with UC using time trade-off and visual analogue rating scale methods. These were applied to all patients alive in each health state using the age-adjusted and sex-adjusted approach suggested by the National Institute for Health and Care Excellence (NICE) decision support unit.22
The model included costs related to drug acquisition, administration, monitoring, disease management, and adverse events. Unit dose and the dosing frequency of treatments included during the induction and maintenance phase were derived from the respective product monographs for mirikizumab and advanced therapies.3-9 For conventional therapy, a weighted cost was estimated based on dose regimens and patient usage of a combination of 5-aminosalicylic acid, corticosteroids, and conventional immunomodulators obtained from published literature.23 During the maintenance phase, a proportion of patients receiving several different advanced therapies was assumed to have received increased doses; this assumption was based on clinical expert opinion and study on dose escalation conducted by the sponsor.24 The sponsor assumed no escalated dose for vedolizumab (IV/subcutaneous; i.e., patients who used vedolizumab IV for induction and subcutaneous for maintenance), ozanimod and upadacitinib. The sponsor modelled a 1-time cost of reinduction instead of escalated doses for mirikizumab, in line with the draft product monograph submitted by the sponsor pre-Notice of Compliance.25 All drug acquisition unit costs were sourced from the IQVIA DeltaPA database.10 The treatment cost of adalimumab and infliximab was estimated assuming 50% of patients were using brand name product and 50% of patients were using the biosimilar. The sponsor assumed no vial sharing in estimating drug acquisition costs.
No administration costs were assumed for treatments with oral, foam, and suppository route of administration. The cost of administration for subcutaneous injection and IV infusion was obtained from the Government of Canada Job Bank and published literature.26,27 Disease management costs included the costs of health care practitioners, laboratory tests, surgery, emergency visits, and hospitalizations as they related to each health state. The sponsor quantified health care resource use for each health state using published literature, which was then validated by clinical expert opinion obtained by the sponsor.28 Disease management unit costs were obtained using the Ontario Ministry of Health and Long-Term Care’s Schedule of Benefits for Physician Services Under the Health Insurance Act, as well as its Schedule of Benefits for Laboratory Services, the Ontario Case Costing Initiative for hospitalizations, and published literature.29-35
The sponsor assumed that the only important adverse event patients experienced was serious infection, and that this would only be experienced during the first treatment cycle. Costs and disutilities associated with serious infections were sourced from published literature.36,37 The proportion of serious infections were obtained from the LUCENT-1 trial for mirikizumab and conventional therapy, from Rutgeert et al. (2005) for infliximab, and respective product monographs for all other advanced therapies.3-9,11,38
Summary of Sponsor’s Economic Evaluation Results
The sponsor conducted the reference case for the biologic-naive and biologic-experienced populations with moderate to severe UC via a probabilistic sensitivity analysis with 5,000 simulations. The deterministic and probabilistic results were not fully aligned. The probabilistic findings are presented as follows.
Base-Case Results
The sequential findings for each population are presented in Table 3 and Table 4. For the biologic-naive population, the CEF was represented by conventional therapy and upadacitinib 45 mg/15 mg and upadacitinib 45 mg/30 mg, while for the biologic-experienced population, the CEF was represented by conventional therapy and upadacitinib 45 mg/15 mg. All other treatments were either strictly or extendedly dominated. Mirikizumab was dominated by upadacitinib 45 mg/30 mg in the biologic-naive population, and by upadacitinib 45 mg/15 mg in the biologic-experienced population, signifying that the intervention represented higher costs and worse health outcomes than upadacitinib.
Table 3: Summary of the Sponsor’s Economic Evaluation Results — Biologic-Naive Population
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY gained) |
|---|---|---|---|
Conventional therapy | 169,405 | 10.69 | Reference |
Upadacitinib 45 mg/15 mg | 282,538 | 13.32 | 43,108 |
Upadacitinib 45 mg/30 mg | 425,546 | 14.59 | 112,034 |
Mirikizumab | 428,568 | 13.85 | Dominated by upadacitinib 45 mg/30 mg |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: Only treatments that are on the cost-effectiveness frontier, as well as the drug under review, are reported. Full results can be found in Appendix 3. As observed in Table 15, mirikizumab was also dominated by infliximab.
Source: Sponsor’s pharmacoeconomic submission.1
Table 4: Summary of the Sponsor’s Economic Evaluation Results — Biologic-Experienced Population
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY gained) |
|---|---|---|---|
Conventional therapy | 167,042 | 10.63 | Reference |
Upadacitinib 45 mg/15 mg | 372,448 | 15.68 | 40,657 |
Mirikizumab | 392,539 | 13.31 | Dominated by upadacitinib 45 mg/15 mg |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: Only treatments that are on the cost-effectiveness frontier, as well as the drug under review, are reported. Full results can be found in Appendix 3. As observed in Table 16, mirikizumab was also dominated by tofacitinib.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor conducted sensitivity and scenario analyses. Pairwise 1-way sensitivity analyses were conducted using the deterministic model to assess the impact of specific parameters on the incremental cost-effectiveness ratio (ICER), incremental QALYs, and incremental costs for the biologic-naive and biologic-experienced populations. The parameters that had the largest impact on the model’s findings were the cost of mirikizumab subcutaneous; the utility values assigned to clinical remission, response, and active UC; loss of response to mirikizumab; and response rates after the mirikizumab induction.
The sponsor provided scenario analyses exploring the impact of adopting alternative discount rates, shortening the time horizon, having an alternative probability of surgery, having alternative health state utility values, excluding administration costs, excluding adverse events, and assuming no extended induction period for mirikizumab. The probabilistic sensitivity analysis was performed with 1,000 simulations. In the biologic-naive population, the CEF consistently showed that conventional therapy, upadacitinib, and mirikizumab remained undominated strategies. In all scenarios, mirikizumab was associated with increased QALYs and costs with ICERs ranging from $170,149 per QALY gained to $259,515 per QALY gained compared with upadacitinib 45 mg/30 mg. In the biologic-experienced population, mirikizumab was strictly dominated in all scenarios.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis.
The comparative efficacy of mirikizumab with the included comparators is uncertain. In the absence of a direct head-to-head comparison between mirikizumab and other advanced therapies, the sponsor commissioned an NMA assessing the comparative efficacy and safety of mirikizumab and relevant comparators. The CADTH clinical review identified several sources of heterogeneity in the submitted NMA, including differences in patient characteristics, treatment history, the definition and method of assessment of efficacy outcomes (clinical response and remission), permitted concomitant medications, and the duration of the maintenance period. Although the sponsor performed statistical adjustments to mitigate some sources of heterogeneity, CADTH noted that there is a lack of evidence to verify the validity of some of the adjustments, which hinged on strong underlying assumptions, and noted that the sponsor was unable to adjust for all sources. Given the methodological limitations with the NMA, the CADTH clinical reviewers noted that interpretation of the comparative efficacy of mirikizumab and advanced therapies is uncertain. Despite the limitations with the indirect evidence, the clinical expert consulted for this review noted that clinical response and remission rates are expected to be similar between mirikizumab and comparators during the induction and maintenance phases.
Due to the limitations with the sponsor-submitted NMA and clinical expert feedback, CADTH assumed equal clinical efficacy among all advanced therapies. Since conventional therapy efficacy was represented by the placebo arms of the clinical trials included in the NMA, it remained unchanged. In accordance with feedback from the clinical expert consulted by CADTH, the difference in the length of induction periods across treatments is generally aligned with clinical practice. As such, CADTH did not revise the induction periods across treatments.
CADTH considered a scenario analysis assuming numerical differences, informed by the sponsor’s NMA, alongside other revisions to the sponsor’s model. In this analysis, mirikizumab was not on the CEF (i.e., it was dominated by golimumab).
The treatment effect of extended induction with mirikizumab is uncertain. The sponsor estimated that 53.7% of patients who received extended induction with mirikizumab responded to treatment, and assumed this would be the same in the biologic-naive and biologic-experienced groups based on the results of the LUCENT-2 trial.12 This response rate for delayed clinical responders is higher than the rate for initial responders in both the biologic-naive population (52.77%) and the biologic-experienced population (42.49%); the assumption of the same effect between subgroups does not align with the observed data from the initial responders. The CADTH clinical review noted that data from the extended induction and reinduction cohorts were observational given the open-label design and absence of comparison to the main cohort in the LUCENT-1 trial. As a result, there is a gap in evidence in terms of detecting a difference in clinical outcomes between delayed responders and induction responders in maintaining clinical remission and response. Thus, the relative treatment effect of extended induction and reinduction with mirikizumab is uncertain in these populations. Additionally, clinical expert feedback obtained by CADTH noted that the observed response rate for delayed responders was higher than expected, and the mirikizumab response, even in the extended induction phase, was likely to be greater in the biologic-naive group compared with the biologic-experienced group.
The sponsor also allowed for reinduction with mirikizumab within the model, which was associated with an incremental cost as well as an efficacy benefit. However, for comparator treatments, it was assumed that a proportion of patients receiving adalimumab (brand and biosimilar), infliximab (brand and biosimilar), golimumab, tofacitinib, and vedolizumab (IV) would receive dose escalation, which was associated with greater costs but did not result in a clinical efficacy benefit.
Given the limitations with interpretation of the extended induction cohort, CADTH excluded the impact of extended induction with mirikizumab on clinical response.
CADTH considered a scenario analysis in which the response rate among delayed clinical responders was assumed to be the same as the response rate after 12 weeks among induction clinical responders in the biologic-naive population (52.77%) and the biologic-experienced population (42.49%). The change did not have a notable impact on the results.
The loss-of-response assumptions are uncertain and do not align with clinical expectation. The model is underpinned by a key efficacy assumption that loss of response differs between treatments, and that this loss of response remains constant throughout the maintenance phase and over the lifetime time horizon. The sponsor estimated the probability of losing response to treatment is lowest with ||||||||||| in the biologic-naive group and with |||||||||||| || ||||| || in the biologic-experienced group based on the results of the sponsor’s submitted NMA. However, the CADTH clinical review team identified concerns regarding heterogeneity between the trials included in the NMA and other methodological concerns that made the relative loss of response between mirikizumab and advanced therapies highly uncertain. Additionally, the clinical expert consulted by CADTH noted that there is an attenuation in the loss of response rates over time based on published literature.39,40 Long-term efficacy data beyond 52 weeks is not available for mirikizumab; thus, relative long-term effectiveness is uncertain.
CADTH noted that the results of this cost-effectiveness analysis are sensitive to the sponsor’s extrapolation assumptions of risk of loss of response, which were based on trial durations of approximately 1 year.
Due to the limitations with the sponsor-submitted NMA, clinical expert feedback, and inability to revise the sponsor’s model structure, CADTH assumed that the probability in loss of response did not differ between comparators.
The relative adverse event rates with mirikizumab treatment compared with advanced therapies are uncertain. The sponsor assumed that mirikizumab was associated with a lower rate of serious infections compared with adalimumab, vedolizumab IV and IV/subcutaneous, upadacitinib 45 mg/15 mg, and upadacitinib 45 mg/30 mg based on information from the LUCENT-1 trial for mirikizumab and data from the product monographs of the respective comparators.3-9,11,38 The sponsor’s use of an unadjusted comparison between treatments is prone to a high risk of bias and confounding. The clinical expert consulted by CADTH also identified relevant adverse events such as infusion-related hypersensitivity, pneumonia, and lymphopenia that were not considered in the sponsor’s analysis. Inflammation of joints also affects about 10% of patients with UC,41 and the clinical expert noted that joint pains impacted patients’ quality of life. As such, the comparative safety of mirikizumab with advanced therapies is uncertain. However, the clinical expert suggested that mirikizumab has a unique mechanism of action that may make it more targeted than other treatments, which may lead to fewer safety concerns.
Due to the lack of comparative evidence, CADTH assumed that the rates of serious infections did not differ between comparators. Based on the results, CADTH noted these inputs were not key drivers in the model.
The unit price of tofacitinib does not reflect formulary prices. The sponsor obtained the price of tofacitinib using the IQVIA DeltaPA database with a source date of November 2022.10 However, the public list price of tofacitinib generics effective in several provinces as of January 2023 was lower than the price used by the sponsor.
CADTH corrected the price of tofacitinib, using the price paid by the Ontario Ministry of Health as stated in the Ontario Drug Benefit Formulary.
Relevant comparators are not included in the analysis of the biologic-experienced population. The sponsor omitted golimumab and infliximab from the analysis of the biologic-experienced population due to a lack of maintenance efficacy data. While the issues with data limitations are plausible, these comparators are relevant to this population. Their exclusion introduces substantial uncertainty into the sponsor’s submitted analysis and limits the validity of the cost-effectiveness analysis in the biologic-experienced population.
CADTH was unable to address this limitation in reanalyses.
A comparator was included that is not currently publicly reimbursed. The sponsor’s model included upadacitinib as a currently reimbursed treatment for moderately to severely active UC. However, upadacitinib is under review by CADTH at the time of this review for the indication of interest,42 and the timeline for upadacitinib to be reimbursed by public plans is uncertain. The therapy is not listed on CADTH-participating public drug plan formularies for this indication, whose perspective guides this economic evaluation. While patients may access upadacitinib through private payers as well as through out-of-pocket payments, these are beyond the scope of the present review.
CADTH conducted a reanalysis that excluded upadacitinib from the list of comparators.
In the scenario analysis, CADTH included upadacitinib as a relevant comparator.
The economic model lacked transparency and flexibility. The economic model submitted by the sponsor lacked transparency as the sponsor hard-coded inputs such as loss of response probabilities, which rendered it difficult to validate inputs that were major drivers of the CUA results. Furthermore, the sponsor included numerous IFERROR statements in defining distributions for the probabilistic analysis and did not model uncertainty around efficacy inputs obtained from the NMA. CADTH noted that the sponsor’s deterministic results did not align with the probabilistic results and that the model ran into an error when CADTH attempted to perform certain reanalyses (such as loss of response and stopping rule). This also prevented CADTH from fully exploring the impact of uncertainty in the cost-effectiveness analysis.
CADTH was unable to address this limitation. All reanalyses were conducted deterministically.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (refer to Table 5).
Table 5: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
The time horizon was assumed to be 50 years based on a starting age of |||| years for the biologic-naive group and |||| years for the biologic-experienced group. | The time horizon in the pharmacoeconomic model was set to 50 years and approximately 81% of the modelled population was in the death health state by this point. As a result, 19% of the population was still alive at 92.5 years in the biologic-naive cohort and 93.2 years in the biologic-experienced cohort, which does not align with the average life expectancy of individuals in Canada.18 |
The sponsor assumed that 2% of patients on conventional therapy experienced an improvement in health status during each model cycle in the maintenance phase (represented by 1% of patients entering the remission health state from the response health state and 1% of patients entering the response health state from the active UC health state). | Acceptable. CADTH explored the impact of assuming no improvement for patients on conventional therapy. For patients on conventional therapy, the total QALYs accrued decreased and costs increased but there was not a notable impact on the relative cost-effectiveness. |
The sponsor assumed that baseline model characteristics were representative of patients in the LUCENT-1 and LUCENT-2 trials, whereas the efficacy parameters were obtained from the sponsor-submitted NMA. | Uncertain. The average baseline characteristics from the sponsor’s NMA were not reported in the pharmacoeconomic evaluation. CADTH noted differences in the mean age, body mass (kg), and sex in the trials and modelled populations, and the CADTH clinical review noted heterogeneity in the patient populations. |
The cost of dose escalation was estimated by multiplying the increase in dose and the proportion of patients on increased dosing. | Uncertain. Dose escalation proved to be a driver of the relative cost-effectiveness in favour of mirikizumab by increasing the cost of comparators. The clinical expert noted that the sponsor’s estimate of the proportion of patients on escalated doses during the maintenance phase for the biologic-naive and biologic-experienced populations seemed reasonable, although the noted dose escalation may be implemented by shortening the interval between doses or increasing the frequency of dosage, and dose escalation may not be continued long-term. |
The sponsor assumed that disease management costs are associated with the health state. | Acceptable. The sponsor assumed that disease management costs did not differ among treatments. However, the clinical expert consulted by CADTH noted that IV treatments may be associated with an increased number of laboratory tests in clinical practice. While this will lead to greater costs with IV treatments (including mirikizumab), the magnitude of impact of the sponsor’s assumption is uncertain, though it is unlikely to impact the overall results. |
In patients who undergo a second surgery, it was assumed their UC achieved remission after the surgery and patients experienced no surgery-related complications. | Acceptable as a simplifying assumption. However, the relapsing-remitting nature of the disease is not accurately captured after the revision surgery. |
Patients who discontinued treatment due to a loss of response were assumed to switch to conventional therapy. | Acceptable as a simplifying assumption. However, the disease management journey for the moderate to severe UC population is characterized by continual treatment-switching until all therapeutic options are exhausted. |
NMA = network meta-analysis; QALY = quality-adjusted life-year; UC = ulcerative colitis.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
CADTH’s reanalysis addressed several limitations within the economic model. The CADTH base case was derived by making changes in model parameter values and assumptions in consultation with clinical experts. The following changes were applied: assuming the clinical efficacy, rate of serious infections, and loss of response among comparators were equal to that of mirikizumab; excluding the treatment effect of extended induction with mirikizumab; and excluding upadacitinib as a comparator. CADTH also corrected the sponsor’s cost of tofacitinib.
Table 6 details each change made to derive the CADTH revised base case, which was conducted in a stepwise approach to highlight the impact of each change. The summary of results from the stepped reanalysis are presented in Table 17 and Table 18.
Table 6: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
1. Unit price of tofacitinib | 5 mg: $23.9589 10 mg: $42.3436 | 5 mg: $5.9897 10 mg: $21.1718 |
Changes to derive the CADTH base case | ||
1. Comparative efficacy | Probabilities of response and remission derived from the NMA indicated numerical effect differences between advanced therapies (Table 13 and Table 14 in Appendix 4) | Clinical efficacy of all biologic treatments were assumed to be equal to mirikizumab |
2. Treatment effect of extended induction with mirikizumab | Included | Excluded |
3. Risk of loss of response | Probabilities of loss of response derived from the NMA indicated numerical effect differences between advanced therapies (Table 14 in Appendix 4) | Probabilities of loss of response were assumed to be equal to that of mirikizumab |
4. Serious infections | Rates of serious infections indicated numerical effect differences between advanced therapies, including mirikizumab | Rates of serious infections were assumed to be equal to those of mirikizumab |
5. Relevant comparator | Upadacitinib was included | Upadacitinib was excluded |
CADTH base case, biologic-naive and biologic-experienced populations | NA | Reanalysis 1 + 2 + 3 + 4 + 5 |
NA = not applicable; NMA = network meta-analysis.
In the CADTH base case, conventional therapy, tofacitinib, and mirikizumab were on the CEF in both the biologic-naive and biologic-experienced populations. Conventional therapy was found to be the most cost-effective therapy for WTP threshold values below $24,236 per QALY gained and $25,346 per QALY gained in the biologic-naive and biologic-experienced populations, respectively (Table 7 and Table 8). Tofacitinib was the most cost-effective advanced therapy for values above these thresholds. Compared with tofacitinib, mirikizumab was associated with estimated total costs of $395,900 and total QALYs of 13.381 in the biologic-naive population and total costs of $293,988 and total QALYs of 11.818 in the biologic-experienced population. The ICER associated with mirikizumab compared with tofacitinib was $3,758,347 per QALY gained and $2,608,809 per QALY gained in the biologic-naive and biologic-experienced populations, respectively. The probability of mirikizumab to be cost-effective at a $50,000 per QALY gained WTP threshold was 0%.
Even though the assumption of clinical efficacy and safety between mirikizumab and advanced therapies was made, there are differences in total QALYs, which are partially attributed to different induction durations of mirikizumab and advanced therapies. The sponsor assumed that patients receiving induction had the same health utility as patients with active UC. However, given that mirikizumab treatment had the longest induction phase, patients on mirikizumab accrued more QALYs due to a longer induction phase. The sponsor’s approach to model health utilities overestimated QALYs for mirikizumab and biased the results in favour of mirikizumab.
Table 7: Summary of the CADTH Reanalysis Results (Deterministic) — Biologic-Naive Population
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY gained) |
|---|---|---|---|
Conventional therapy | 169,888 | 10.607 | Reference |
Tofacitinib | 236,095 | 13.338 | 24,236 |
Mirikizumab | 395,900 | 13.381 | 3,758,347 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: Only nondominated treatments are presented.
Table 8: Summary of the CADTH Reanalysis Results (Deterministic) — Biologic-Experienced Population
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY gained) |
|---|---|---|---|
Conventional therapy | 168,566 | 10.377 | Reference |
Tofacitinib | 204,219 | 11.784 | 25,346 |
Mirikizumab | 293,988 | 11.818 | 2,608,809 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: Only nondominated treatments are presented.
Scenario Analysis Results
CADTH undertook price reduction analyses based on the sponsor’s and CADTH’s base case. Based on the CADTH base case of the sponsor-submitted model, a price reduction of 65.2% would be necessary to achieve cost-effectiveness at a WTP threshold of $50,000 per QALY gained in the biologic-naive population (Table 9), while a price reduction of 66.1% would be required in the biologic-experienced population (Table 10). As the CADTH base case assumes equal comparative efficacy and safety across treatments, CADTH also considered price reductions based on the submitted price for mirikizumab and the publicly accessible list prices of all other biologics (Table 11 in Appendix 1), which indicated that a price reduction of 80% during the first year and 86% thereafter would be required for mirikizumab to be no more costly than tofacitinib, which is the least costly advanced therapy for moderately to severely active UC.
CADTH undertook a series of scenario analyses to determine the impact of alternative assumptions on the cost-effectiveness of mirikizumab. These analyses are outlined as follows:
CADTH assumed numerical differences in clinical efficacy and safety between mirikizumab and its comparators.
CADTH assumed the response rate among delayed clinical responders to be the same as the response rate after 12 weeks of induction.
CADTH included upadacitinib as a comparator.
The results of these analyses are presented in Table 21 and Table 22. Mirikizumab was not cost-effective at a WTP of $50,000 per QALY gained, or was dominated by other advanced therapies in all scenarios.
Table 9: CADTH Price Reduction Analyses — Biologic-Naive Population
Analysis | ICERs for mirikizumab vs. comparators ($/QALY gained) | |
|---|---|---|
Price reduction | Sponsor base case | CADTH reanalysis |
No price reduction ($30,871) | WTP threshold < $42,309 = conventional therapy $42,309 < WTP threshold < $142,065 = upadacitinib 45 mg/15 mg $142,065 < WTP threshold = upadacitinib 45 mg/30 mg | WTP threshold < $24,236 = conventional therapy $24,236 < WTP threshold < $3,758,347 = tofacitinib $3,758,347 < WTP threshold = mirikizumab |
10% ($27,784) | WTP threshold < $42,309 = conventional therapy $42,309 < WTP threshold < $132,159 = upadacitinib 45 mg/15 mg $132,159 < WTP threshold = mirikizumab | WTP threshold < $24,236 = conventional therapy $24,236 < WTP threshold < $3,189,295 = tofacitinib $3,189,295 < WTP threshold = mirikizumab |
20% ($24,697) | WTP threshold < $42,309 = conventional therapy $42,309 < WTP threshold < $105,139 = upadacitinib 45 mg/15 mg $105,139 < WTP threshold = mirikizumab | WTP threshold < $24,236 = conventional therapy $24,236 < WTP threshold < $2,620,243 = tofacitinib $2,620,243 < WTP threshold = mirikizumab |
30% ($21,610) | WTP threshold < $42,309 = conventional therapy $42,309 < WTP threshold < $78,119 = upadacitinib 45 mg/15 mg $78,119 < WTP threshold = mirikizumab | WTP threshold < $24,236 = conventional therapy $24,236 < WTP threshold < $2,051,191 = tofacitinib $2,051,191 < WTP threshold = mirikizumab |
40% ($18,523) | WTP threshold < $42,309 = conventional therapy $42,309 < WTP threshold < $51,099 = upadacitinib 45 mg/15 mg $51,099 < WTP threshold = mirikizumab | WTP threshold < $24,236 = conventional therapy $24,236 < WTP threshold < $1,482,139 = tofacitinib $1,482,139 < WTP threshold = mirikizumab |
50% ($15,436) | WTP threshold < $36,565 = conventional therapy $36,565 < WTP threshold = mirikizumab | WTP threshold < $24,236 = conventional therapy $24,236 < WTP threshold < $913,088 = tofacitinib $913,088 < WTP threshold = mirikizumab |
60% ($12,348) | WTP threshold < $28,052 = conventional therapy $28,052 < WTP threshold = mirikizumab | WTP threshold < $24,236 = conventional therapy $24,236 < WTP threshold < $344,036 = tofacitinib $344,036 < WTP threshold = mirikizumab |
70% ($9,261) | WTP threshold < $19,539 = conventional therapy $19,539 < WTP threshold = mirikizumab | WTP threshold < $20,415 = conventional therapy $20,415 < WTP threshold = mirikizumab |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus; WTP = willingness-to-pay.
Note: All analyses are based on the publicly available prices of comparators and may not reflect confidential negotiated prices. Only treatments that are on the cost-effectiveness frontier, as well as the drug under review, are reported. Costs are reported as annual maintenance costs.
Table 10: CADTH Price Reduction Analyses — Biologic-Experienced Population
Analysis | ICERs for mirikizumab vs. comparators ($/QALY gained) | |
|---|---|---|
Price reduction | Sponsor base case | CADTH reanalysis |
No price reduction ($30,871) | WTP threshold < $40,887 = conventional therapy $40,887 < WTP threshold < $223,346 = upadacitinib 45 mg/15 mg $223,346 < WTP threshold = upadacitinib 45 mg/30 mg | WTP threshold < $25,346 = conventional therapy $25,346 < WTP threshold < $2,608,809 = tofacitinib $2,608,809 < WTP threshold = mirikizumab |
10% ($27,784) | WTP threshold < $40,887 = conventional therapy $40,887 < WTP threshold < $223,346 = upadacitinib 45 mg/15 mg $223,346 < WTP threshold = upadacitinib 45 mg/30 mg | WTP threshold < $25,346 = conventional therapy $25,346 < WTP threshold < $2,221,361 = tofacitinib $2,221,361 < WTP threshold = mirikizumab |
20% ($24,697) | WTP threshold < $40,887 = conventional therapy $40,887 < WTP threshold < $223,346 = upadacitinib 45 mg/15 mg $223,346 < WTP threshold = upadacitinib 45 mg/30 mg | WTP threshold < $25,346 = conventional therapy $25,346 < WTP threshold < $1,833,913 = tofacitinib $1,833,913 < WTP threshold = mirikizumab |
30% ($21,610) | WTP threshold < $40,887 = conventional therapy $40,887 < WTP threshold < $223,346 = upadacitinib 45 mg/15 mg $223,346 < WTP threshold = upadacitinib 45 mg/30 mg | WTP threshold < $25,346 = conventional therapy $25,346 < WTP threshold < $1,446,465 = tofacitinib $1,446,465 < WTP threshold = mirikizumab |
40% ($18,523) | WTP threshold < $40,887 = conventional therapy $40,887 < WTP threshold < $223,346 = upadacitinib 45 mg/15 mg $223,346 < WTP threshold = upadacitinib 45 mg/30 mg | WTP threshold < $25,346 = conventional therapy $25,346 < WTP threshold < $1,059,017 = tofacitinib $1,059,017 < WTP threshold = mirikizumab |
50% ($15,436) | WTP threshold < $38,405 = conventional therapy $38,405 < WTP threshold < $46,181 = mirikizumab $46,181 < WTP threshold < $223,346 = upadacitinib 45 mg/15 mg $223,346 < WTP threshold = upadacitinib 45 mg/30 mg | WTP threshold < $25,346 = conventional therapy $25,346 < WTP threshold < $671,569 = tofacitinib $671,569 < WTP threshold = mirikizumab |
60% ($12,348) | WTP threshold < $29,530 = conventional therapy $29,530 < WTP threshold < $65,106 = mirikizumab $65,106 < WTP threshold < $223,346 = upadacitinib 45 mg/15 mg $223,346 < WTP threshold = upadacitinib 45 mg/30 mg | WTP threshold < $25,346 = conventional therapy $25,346 < WTP threshold < $284,121 = tofacitinib $284,121 < WTP threshold = mirikizumab |
70% ($9,261) | WTP threshold < $20,654 = conventional therapy $20,654 < WTP threshold < $84,031 = mirikizumab $84,031 < WTP threshold < $223,346 = upadacitinib 45 mg/15 mg $223,346 < WTP threshold = upadacitinib 45 mg/30 mg | WTP threshold < $22,274 = conventional therapy $22,274 < WTP threshold = mirikizumab |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus; WTP = willingness-to-pay.
Note: All analyses are based on the publicly available prices of comparators and may not reflect confidential negotiated prices. Only treatments that are on the cost-effectiveness frontier, as well as the drug under review, are reported.
Issues for Consideration
The modelled price of advanced therapies is based on publicly accessible list prices and does not reflect the existing confidential pricing that has been negotiated by CADTH-participating public drug plans. When existing confidential discounts on advanced therapies are considered, greater price reductions than those referenced in this report may be required to achieve cost-effectiveness.
CADTH notes that the negotiation process with the pan-Canadian Pharmaceutical Alliance (pCPA) for ustekinumab concluded on July 28, 2021, without agreement,43 while ozanimod is under active ongoing negotiations with pCPA.44 Furthermore, upadacitinib is currently under review at CADTH for the indication of UC.42 As observed in the sponsor’s analysis, the inclusion of upadacitinib may impact the cost-effectiveness of mirikizumab. Decision-makers should take into account the negotiated prices of comparator products when ultimately considering the funded price of mirikizumab, should it become available.
The duration of the induction phase varies among advanced therapies, and mirikizumab treatment has the longest induction phase (particularly with the possibility of extended induction). Given that mirikizumab dose infusions are to be administered in infusion clinics and the maintenance dose is administered as subcutaneous injections, the administration of mirikizumab may be associated with additional health care resources and costs as well as indirect costs to patients and caregivers.
Overall Conclusions
Evidence from the LUCENT trials demonstrated the efficacy of mirikizumab in achieving the induction and maintenance of clinical remission and clinical response in patients with moderately or severely active UC compared with placebo. Although the results were considered generalizable to the Canadian setting, CADTH identified limitations with the trials that introduced uncertainty in the comparative efficacy of mirikizumab relative to placebo. Furthermore, data pertaining to the use of mirikizumab for extended induction and reinduction were observational and no conclusions could be reached regarding the comparative efficacy based on mirikizumab’s use in these populations. As there are no trials comparing mirikizumab with other advanced therapies (i.e., biologics, JAK inhibitors, and small molecule drugs), comparisons among treatments were based on a sponsor-commissioned NMA. Due to the heterogeneity between studies included in the NMA, the comparative effects of mirikizumab with other advanced therapies is uncertain. Evidence of the efficacy and safety of mirikizumab beyond 52 weeks, as well as direct comparisons with other treatments for UC, are necessary to further understand the long-term benefits and comparative effectiveness of mirikizumab.
CADTH attempted to address the limitations identified with the economic analysis submitted by the sponsor in the CADTH base case by making the following changes in model parameter values and assumptions in consultation with clinical experts: assuming the clinical efficacy, risk of loss of response, and risk of serious events of all advanced therapies to be equal to that of mirikizumab; excluding the treatment effect of extended induction with mirikizumab; and excluding upadacitinib as a comparator. CADTH also corrected the price of tofacitinib using the publicly available price in a public drug plan formulary. However, there were concerns regarding the transparency of the model output, which limited the confidence that can be placed on the results from the model.
In the CADTH base case, conventional therapy, tofacitinib, and mirikizumab were on the CEF in both the biologic-naive and biologic-experienced populations. Compared with tofacitinib, mirikizumab was associated with higher costs and QALYs. The ICER for mirikizumab was $3,758,347 per QALY gained and $2,608,809 per QALY gained in the biologic-naive population and the biologic-experienced population, respectively. The incremental QALYs were partially attributed to the way utility values were calculated in the induction phase and the difference in induction duration for mirikizumab compared with other advanced therapies. CADTH’s base case was aligned with the sponsor’s results; that is, mirikizumab was not cost-effective at a $50,000 per QALY gained WTP threshold in both populations. Based on publicly available list prices for all comparators and the submitted price of mirikizumab, a price reduction for mirikizumab of approximately 65% would be required for mirikizumab to be cost-effective in either the biologic-naive or biologic-experienced populations at a WTP threshold of $50,000 per QALY gained. Such a price reduction would decrease the annual per patient costs of mirikizumab from $30,895 to $10,751 in the first year and from $30,871 to $10,743 in subsequent years of treatment.
Given the uncertainty in the available indirect comparative clinical evidence, there is insufficient economic evidence to justify a higher drug cost for mirikizumab compared to advanced therapies. CADTH allowed for the consideration of dose escalation of comparators in line with feedback received by CADTH’s consulted clinical expert. When only considering drug acquisition costs, a price reduction of approximately 85% is required for mirikizumab to be no more costly than the publicly available price of the least costly advanced therapy (tofacitinib, which is $4,375 to $6,076 per patient per year).
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Omvoh (mirikizumab). Toronto (ON): Eli Lilly Canada, Inc.; March 7, 2023.
2.Omvoh (mirikizumab): 100 mg/mL solution for subcutaneous injection, 20 mg/mL solution for intravenous infusion [product monograph]. Toronto (ON): Eli Lilly Canada, Inc.; 2022.
3.REMICADE (Infliximab): Powder for Solution, Sterile, Lyophilized, 100 mg/vial [Product monograph]. Toronto (ON): Janssen Inc.; 2022.
4.HUMIRA (adalimimab): 50 mg/mL and 100 mg/mL subcutaneous injection [Product monograph]. AbbVie Corp.: St-Laurent (QC); 2022: https://www.abbvie.ca/content/dam/abbvie-dotcom/ca/en/documents/products/HUMIRA_PM_EN.pdf.
5.SIMPONI (golimumab): 50 mg/0.5 mL 100 mg/1.0 mL solution for injection [Product monograph]. Toronto (ON): Janssen Inc.; 2022.
6.ENTYVIO (vedolizumab): Powder for concentrate for solution for intravenous infusion [IV] (300 mg / vial), Solution for subcutaneous injection [SC] (108 mg / 0.68 mL pre‐filled syringe or pen) [Product monograph]. Toronto (ON): Takeda Canada Inc; 2022.
7.PrXELJANZ (tofacitinib): 5 mg and 10 mg tablets, 11 mg extended-release tablets, [Product Monograph] Kirkland (QC): Pfizer Canada ULC; 2022.
8.ZEPOSIA (ozanimod): capsules, 0.23 mg, 0.46 mg and 0.92 mg, oral [Product monograph]. Saint-Laurent (QC): Celgene Inc., a Bristol Myers Squibb company; 2022.
9.RINVOQ (Upadacitinib): Extended-release tablets, 15 mg and 30 mg [Product monograph]. St-Laurent (QC): AbbVie Corporation; 2022.
10.DeltaPA. [Ottawa (ON)]: IQVIA; 2022: https://www.iqvia.com/. Accessed 2022 Nov 28.
11.Eli Lilly Company. LUCENT-1 Clinical Study Report. Data on file; 2021.
12.Eli Lilly Company. LUCENT-2 Clinical Study Report. Data on file; 2022.
13.Sponsor's NMA report title [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Mirikizumab, 100 mg/mL solution for subcutaneous injection, 20 mg/mL solution for intravenous infusion. Toronto (ON): Eli Lilly Canada, Inc.; Sponsor didn't specify.
14.Misra R, Askari A, Faiz O, Arebi N. Colectomy Rates for Ulcerative Colitis Differ between Ethnic Groups: Results from a 15-Year Nationwide Cohort Study. Can J Gastroenterol Hepatol. 2016;2016:8723949. PubMed
15.Silverberg MS, Satsangi J, Ahmad T, et al. Toward an integrated clinical, molecular and serological classification of inflammatory bowel disease: report of a Working Party of the 2005 Montreal World Congress of Gastroenterology. Canadian Journal of Gastroenterology = Journal Canadien De Gastroenterologie. 2005;19 Suppl A:5A-36A.
16.Excellence UNIfHaC. Tofacitinib for moderately to severely active ulcerative colitis. Technology appraisal guidance [TA547]. 2018.
17.Excellence UNIfHaC. Ustekinumab for treating moderately to severely active ulcerative colitis. Technology appraisal guidance [TA633]. 2020.
18.Canada S. Life Tables, Canada, Provinces and Territories 2023. https://www150.statcan.gc.ca/n1/en/catalogue/84-537-X.
19.Group UIAS. Report of the results for the national clinical audit of adult inflammatory bowel disease inpatient care in the UK. Round 3. 2012.
20.Woehl A. HA, P. M. The relation between disease activity, quality of life and health utility in patients with ulcerative colitis. Gut. 2008.
21.Arseneau KO, Sultan S, Provenzale DT, et al. Do patient preferences influence decisions on treatment for patients with steroid-refractory ulcerative colitis? Clin Gastroenterol Hepatol. 2006;4(9):1135-1142. PubMed
22.Hernandez Alava M, Pudney S, Wailoo A. Estimating the relationship between EQ-5D-5L and EQ-5D-3L: results from a UK population study. Pharmacoeconomics. 2023;41(2):199-207. PubMed
23.Dan A, Boutros M, Nedjar H, et al. Cost of Ulcerative Colitis in Quebec, Canada: A Retrospective Cohort Study. Inflamm Bowel Dis. 2017;23(8):1262-1271. PubMed
24.Drug Reimbursement Review pharmacoeconomic report: Mirikizumab (Omvoh) for adult patients with moderately to severely active ulcerative colitis (UC) who have had an inadequate response with, lost response to, or were intolerant to either conventional therapy (CT), a biologic treatment, or a Janus Kinase (JAK) inhibitor, or have medical contraindications to such therapies. Ottawa (ON): CADTH; 1800: URL. Accessed 1800 Jan 1.
25.Inc ELC. Product monograph: mirikizumab 2018.
26.Canada Go. Registered Nurse (R.N.) in Canada | Wages - Job Bank.
27.Canada S. Consumer Price Index, annual average, not seasonally adjusted 2022.
28.Tsai HH PY, Morris J, Fortun P. A model of the long-term cost effectiveness of scheduled maintenance treatment with infliximab for moderate-to-severe ulcerative colitis. Aliment Pharmacol Ther. 2008. PubMed
29.Ontario Case Costing Initiative (OCCI). Toronto (ON): Ontario Health and Long-Term Care; 2017: https://data.ontario.ca/dataset/ontario-case-costing-initiative-occi. Accessed sponsor not specified.
30.Schedule of benefits for laboratory services: effective July 1, 2020. Toronto (ON): Ontario Ministry of Health; 2020: http://www.health.gov.on.ca/en/pro/programs/ohip/sob/lab/lab_mn2020.pdf. Accessed 2022 Jan.
31.Schedule of benefits for physician services under the Health Insurance Act: (January 25, 2022 (effective July 1, 2022)). Toronto (ON): Ontario Ministry of Health; 2022: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf. Accessed 1800 Jan 1.
32.Bernstein CN NZ, Targownik LE, Singh H, Snider C, Witt J. The Cost of Use of the Emergency Department by Persons With Inflammatory Bowel Disease Living in a Canadian Health Region: A Retrospective Population-Based Study. J Can Assoc Gastroenterol. 2020. PubMed
33.Coward S HS, Clement F, Hubbard J, Proulx MC, Zimmer S, et al. Ulcerative colitis-associated hospitalization costs: a population-based study. Can J Gastroenterol Hepatol. 2015. PubMed
34.LeBlanc K HC, Martins L, Butt B, Wiesenfeld S, Woo K. The Financial Impact of Living in Canada With an Ostomy: A Cross-sectional Survey. J Wound Ostomy Continence Nurs. 2019. PubMed
35.Ilona H. Canadian Family Physician. 2015.
36.Fenu E LV, Acs A, Radu X, Stypa S, Fischer A, et al. Cost Effectiveness of Subcutaneous Vedolizumab for Maintenance Treatment of Ulcerative Colitis in Canada. Pharmacoecon Open. 2022.
37.Stevenson M AR, Tosh J, Simpson E, Everson-Hock E, Stevens J, et al. Adalimumab, etanercept, infliximab, certolizumab pegol, golimumab, tocilizumab and abatacept for the treatment of rheumatoid arthritis not previously treated with disease-modifying antirheumatic drugs and after the failure of conventional disease-modifying antirheumatic drugs only: systematic review and economic evaluation. Health Technol Assess Rep. 2016.
38.Rutgeerts P SW, Feagan BG, Reinisch W, Olson A, Johanns J, et al. Infliximab for induction and maintenance therapy for ulcerative colitis. The New England journal of medicine. 2005. PubMed
39.Schultheiss JPD, Mahmoud R, Louwers JM, et al. Loss of response to anti-TNFα agents depends on treatment duration in patients with inflammatory bowel disease. Aliment Pharmacol Ther. 2021;54(10):1298-1308. PubMed
40.Singh H, Wilson L, Tencer T, Kumar J. Systematic Literature Review of Real-World Evidence on Dose Escalation and Treatment Switching in Ulcerative Colitis. Clinicoecon Outcomes Res. 2023;15:125-138. PubMed
41.Canada CsaC. JOINT INFLAMMATION AND ARTHRITIS. 2019; https://crohnsandcolitis.ca/About-Crohn-s-Colitis/IBD-Journey/Complications-and-Extraintestinal-Manifestations/Joint-Inflammation-and-Arthritis.
42.CADTH. upadacitinib. 2022; https://www.cadth.ca/upadacitinib-2.
43.pCPA. Stelara (ustekinumab). 2023; https://www.pcpacanada.ca/negotiation/21272.
44.pCPA. Zeposia (ozanimod) for UC. 2023; https://www.pcpacanada.ca/negotiation/22096.
45.Adalimumab (Amgevita): 50 mg/mL prefilled syringe for subcutaneous injection, 50 mg/mL autoinjector for subcutaneous injection [Product monograph]. Mississauga (ON): Amgen Canada Inc; 2020: https://www.amgevita.ca/en/assets/pdf/patient-medication-information.pdf.
46.INFLECTRA(infliximab for injection): Powder for Solution, Sterile, Lyophilized, 100 mg/vial [PRODUCT MONOGRAPH]. Kirkland (QC): Pfizer Canada Inc; 2017: https://pdf.hres.ca/dpd_pm/00042037.PDF.
47.RENFLEXISTM (infliximab): Powder for Solution, Sterile, Lyophilized, 100 mg /vial [PRODUCT MONOGRAPH]. Kirkland (QC): Merck Canada Inc.; 2017: https://crohnsandcolitis.ca/Crohns_and_Colitis/images/living-with-crohns-colitis/RENFLEXIS-MONOGRAPH.PDF.
48.Inc J. PRODUCT MONOGRAPH: REMICADE®. 2017.
49.CADTH. CADTH Reimbursement Review: Ozanimod (Zeposia). 2023.
50.Saskatchewan Drug Plan: search formulary. 2022; http://formulary.drugplan.ehealthsask.ca/SearchFormulary. Accessed 2023 May.
51.STELARA (ustekinumab): 45 mg/0.5 mL and 90 mg/1.0 mL Solution for Subcutaneous Injection, 5 mg/mL Solution for Intravenous Infusion [PRODUCT MONOGRAPH]. Toronto (ON): Janssen Inc; 2017: https://crohnsandcolitis.ca/Crohns_and_Colitis/images/living-with-crohns-colitis/STELARA_MONOGRAPH.PDF.
52.Exceptional Access Program (EAP). Toronto (ON): Ontario Ministry of Health; Ontario Ministry of Long-Term Care; 2022: http://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx. Accessed 2023 May.
53.Inc G. MESASAL (5-Aminosalicylic Acid): Enteric Coated Tablets 500 mg [Product Monograph]. Mississauga (ON): GlaxoSmithKline Inc; 2016: https://pdf.hres.ca/dpd_pm/00033758.PDF.
54.MEZAVANT (mesalamine): 1.2g Delayed- and Extended-Release Tablets, [PRODUCT MONOGRAPH). Toronto (ON): Shire Pharma Canada ULC; 2017: https://pdf.hres.ca/dpd_pm/00040692.PDF.
55.PENTASA (Mesalazine*): Extended-release tablets 500 mg and 1 g, Rectal suspension, 1 g and 4 g/100 mL (enema), Suppository 1 g, (*also called 5-aminosalicylic acid, 5-ASA or mesalamine) [product monograph]. North York (ON): Ferring Inc.; 2018: https://ferring.ca/media/1035/pentasa-pm-may-2-2018.pdf.
56.SALOFALK (Mesalamine): Suppositories 500 mg and 1000 mg [Product Monograph]. Mont-Saint-Hilaire (Québec): APTALIS PHARMA CANADA INC.; 2014: https://pdf.hres.ca/dpd_pm/00028806.PDF.
57.SALOFALK (Mesalamine): delayed Release Tablets USP, 500 mg [product monograph]. Mont-Saint-Hilaire (Quebec): APTALIS PHARMA CANADA INC.; 2014: https://allergan-web-cdn-prod.azureedge.net/allergancanadaspecialty/allergancanadaspecialty/media/actavis-canada-specialty/en/products/pms/salofalk-tablets-pm_eng_30dec2014.pdf.
58.SALOFALK (mesalamine): rectal suspension [product monograph]. not listed; https://www.abbvie.ca/content/dam/abbvie-dotcom/ca/en/documents/products/SALOFALK_SUSPENSION_PI_EN.pdf.
59.DIPENTUM (olsalazine sodium): 250 mg Capsules [product monograph]. Montreal (Quebec): Searchlight Pharma Inc; 2015: https://searchlightpharma.com/app/uploads/2016/08/Dipentum-PM.pdf.
60.SULFASALAZINE (Sulfasalazine): tablets USP 500 mg, delayed-release tablets USP 500 mg [PRODUCT MONOGRAPH]. Montréal (Québec): PHARMASCIENCE INC.; 2018: https://pdf.hres.ca/dpd_pm/00049093.PDF.
61.Clinic M. Drugs and Supplements - Corticosteroid (Rectal Route). 2023; https://www.mayoclinic.org/drugs-supplements/corticosteroid-rectal-route/proper-use/drg-20070430.
62.CORTEF (Hydrocortisone): Tablets, 10 mg, 20 mg, Oral [PRODUCT MONOGRAPH]. Kirkland (Quebec): Pfizer Canada ULC; 2022: https://pdf.hres.ca/dpd_pm/00064868.PDF.
63.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Mirikizumab, 100 mg/mL solution for subcutaneous injection, 20 mg/mL solution for intravenous infusion. Toronto (ON): Eli Lilly Canada, Inc.; Sponsor didn't specify.
64.Kaplan GG, Bernstein CN, Coward S, et al. The Impact of Inflammatory Bowel Disease in Canada 2018: Epidemiology. J Can Assoc Gastroenterol. 2019;2(Suppl 1):S6-s16. PubMed
65.Drug Reimbursement Review sponsor submission: drug name, doses and administration [internal sponsor's package]. City (PROV): Sponsor name; 1800.
Appendix 1: Cost Comparison Table
Table 11: CADTH Cost Comparison Table for Severe-to-Moderate Active Ulcerative Colitis
Treatment | Strength / concentration | Form | Price ($) | Recommended dosage | Average monthly cost ($) | Average annual cost ($) |
|---|---|---|---|---|---|---|
Mirikizumab (Omvoh) | 300 mg/15 mL 100 mg/1 mL 100 mg/1 mL | Vial for IV infusion Autoinjector pen for SC injection Prefilled syringe for SC injection | 2,374.6600a 1,187.3300a 1,187.3300a | Induction: 300 mg IV infusion at week 0, week 4, and week 8b Maintenance: 200 mg SC injection every 4 weeks after inductionb | Year 1: 2,574.58 Thereafter: 2,572.55 | Year 1: 30,895 Thereafter: 30,871 |
Comparators: Biologics | ||||||
Adalimumab (Humira) | 40 mg/ 0.8 mL | Prefilled syringe or autoinjector for SC injection | 794.1000 | 160 mg at week 0, 80 mg at week 2, then 40 mg every other week thereafterc | Year 1: $1,919.16 Thereafter: $1,726.46 | Year 1: $23,894 Thereafter: $20,718 |
Adalimumab (biosimilars) | 20 mg/0.4 mL 40 mg/0.4 mL 40 mg/0.8 mL 80 mg/0.8 mL | Prefilled syringe or autoinjector for SC injection | 235.6350 471.2700 942.5400 | 160 mg at week 0, 80 mg at week 2, then 40 mg every other week thereafterd | Year 1: $1,181.68 Thereafter: $1,024.59 | Year 1: $14,180 Thereafter: $12,295 |
Golimumab (Simponi) | 50 mg/ 0.5 mL 100 mg/1 mL | Prefilled syringe or autoinjector for SC injection | 1,555.1700 1,555.1700 | 200 mg at week 0, 100 mg at week 2, then 50 mg every 4 weeks thereaftere | Year 1: $1,765.13 Thereafter: $1,555.17 | Year 1: $21,182 Thereafter: $18,662 |
Infliximab (Inflectra) | 100 mg | Vial Powder for IV infusion | 525.0000 | 5 mg/kg at week 0, 2, and 6, then every 8 weeks thereafterf | Year 1: $1,700.20 Thereafter: $1,426.76 | Year 1: $20,402 Thereafter: $17,121 |
Infliximab (Avsola, Renflexis) | 100 mg | Vial Powder for IV infusion | 493.0000 | 5 mg/kg at week 0, 2, and 6, then every 8 weeks thereafterg | Year 1: $1,596.56 Thereafter: $1,339.79 | Year 1: $19,159 Thereafter: $16,078 |
Infliximab (Remicade) | 100 mg | Vial for IV infusion | 987.5600 | 5 mg/kg at week 0, week 2, and week 6, then every 8 weeks thereafterh | Year 1: $3,198.18 Thereafter: $2,683.33 | Year 1: $38,378 Thereafter: $32,206 |
Ozanimod (Zeposia) | 0.25 mgi 0.5 mgi 1 mgi | Capsule | 68.4929j | 0.25 mg daily on day 1 to day 4, 0.5 daily on day 5 to day 7, then 1 mg daily thereafterk | Year 1: $2,073.34 Thereafter: $2,084.75 | Year 1: $24,880 Thereafter: $25,017 |
Tofacitinib (Xeljanz, generic) | 5 mg 10 mg | Tablet | 5.9897 21.1718 | 10 mg twice daily for at least 8 weeks, then 5 mg twice daily thereafterl | Year 1: $506.32 Thereafter: $364.62 | Year 1: $6,076 Thereafter: $4,375 |
11 mg | ER tablet | 49.5467 | $3,016.16 | $36,194 | ||
Ustekinumab (Stelara) | 130 mg/ 26.0 mL 45 mg/ 0.5 mL 90 mg/ 1.0 mL | Vial for IV infusion Prefilled Syringe for SC injection | 2,080.000m, n 4,593.1400n | 6 mg/kg IV at week 0, then 90 mg SC every 8 weeks thereaftero | Year 1: $2,980.40 Thereafter: $2,496.49 | Year 1: $35,765 Thereafter: $29,958 |
Upadacitinib (Rinvoq) | 15 mg 30 mg 45 mg | Tablet | $51.6810 $74.0000p $101.8100a | 45 mg once daily for 8 weeks, then 15 mg or 30 mg once daily thereaftera | Low dose maintenance Year 1: $1,743.55 Thereafter: $1,498.13 | Low dose maintenance Year 1: $20,923 Thereafter: $17,978 |
High dose maintenance Year 1: $2,382.16 Thereafter: $2,252.38 | High dose maintenance Year 1: $28,586 Thereafter: $27,029 | |||||
Vedolizumab (Entyvio) (IV) | 300 mg | Vial for IV infusion | 3,401.8600q | 300 mg at week 0, 2, 6, then every 8 weeks thereafterr | Year 1: $2,203.36 Thereafter: $1,849.00 | Year 1: $26,440 Thereafter: $22,188 |
Vedolizumab (Entyvio) (SC) | 108 mg/ 0.68 mL | Prefilled syringe or pen for SC injection | 850.4600 | Following 300 mg IV infusions at weeks 0 and 2, 108 mg SC injection is administered every 2 weeks as maintenance only (from week 4 onwards)r | Year 1: $2,274.22 Thereafter: $1,848.99 | Year 1: $27,291 Thereafter: $22,188 |
Comparators: Aminosalicylates | ||||||
5-ASA (Mesasal) | 500 mg | Enteric tablet | $0.6559 | Active: 1.5 to 3 g tabs daily in divided dosess Maintenance: 1.5 g daily in divided doses | $59.85 to $119.70 | $718 to $1,436 |
5-ASA (Mezavant) | 1.2 g | Delayed ER tablet | 1.7805 | Active: 2 to 4 tabs once dailys Maintenance: 2 tabs daily | $38.57 | $471 |
5-ASA (Pentasa) | 500 mg | Delayed ER tablet | 0.6458 | 0.5 g to 1 g, 4 times daily (2 g daily dose)s | $78.57 to $157.14 | $943 to $1,886 |
1,000 mg | ER tablet | 1.2913 | ||||
1 g | Suppository | 2.3548 | 1 g daily | $71.63 | $860 | |
1 g/100 mL 4 g/100 mL | Enema | 5.6501 7.1212 | 1 g to 4 g daily | $171.86 to $216.60 | $2,062 to $2,599 | |
5-ASA (Salofalk) | 500 mg | Delayed ER Tablet | 0.6714 | Active: 3 g to 4 g daily in divided dosess Maintenance: 1.5 g to 3 g per day in divided doses | Year 1: $122.53 to $163.37 Thereafter: $61.27 | Year 1: $1,470 to $1,960 Thereafter: $735 |
500 mg 1,000 mg | Suppository | 1.6510 2.4250 | 1 g to 1.5 g/days | $73.76 to $147.52 | $885 to $1,770 | |
4 g/60 g | Rectal Suspension | 8.7714 | Active: 4 g nightly Maintenance: 2 g nightly or 4 g every 2 nightss | Year 1: 64.89 to 194.68 Thereafter: $64.89 | Year 1: $779 to $2,336 Thereafter: $779 | |
Olsalazine (Dipentum) | 250 mg | Capsule | 0.5330 | Active: 1 g to 3 g daily in divided dosest Maintenance: 1 g daily in divided doses | Year 1: 64.89 to 194.68 Thereafter: $64.89 | Year 1: $779 to $2,336 Thereafter: $779 |
Sulfasalazine (Salazopyrin, generics) | 500 mg | Tablet | 0.2533 | Active: 1 g to 2 g, 3 to 4 times dailyu Maintenance: 1 g, 2 to 3 times daily | Year 1: $46.26 to $123.36 Thereafter: $30.84 to $188.13 | Year 1: $555 to $1,480 Thereafter: $370 to $555 |
500 mg | Enteric tablet | 0.3863 | Year 1: $70.55 to $123.36 Thereafter: $47.03 to $70.55 | Year 1: $847 to $2,258 Thereafter: $564 to $847 | ||
Comparators: Corticosteroids | ||||||
Betamethasone enema (Betnesol) | 5 mg/100 mL | Enema | 14.3357 | 5 mg nightlyj | $436.34 | $5,236 |
Budesonide (Entocort) | 3 mg | Capsule | 2.0992m | 3 mg, 3 times per day up to 8 weeks, followed by 6 mg daily for up to 3 monthsj | $141.07 | $705v |
Hydrocortisone (Cortenema) (Cortifoam) | 100 mg/ 60 mL | Enema | $8.2541 | 100 mg nightly 2 to 3 weeksw | $125.53 to $251.06 | $1,506 to $3,013 |
15 g/pack (14 doses) | 10% Rectal Aerosol | $117.8800 | One dose once or twice daily for 2 to 3 weeksj | $117.88 to $235.80 | $1,415 to $2,830 | |
Hydrocortisone (Cortef) | 10 mg 25 mg | Tablet | 0.2259 0.4078 | 20 mg to 240 mg dailyx | $13.75 to $124.12 | $165 to $1,489 |
Prednisone (generic) | 5 mg 50 mg | Tablet | 0.0220 0.1735 | 40 mg to 60 mg daily to induce remission, then lower dosej | $5.28 to $10.56 | $63 to $127 |
Comparators: Immunomodulators | ||||||
Azathioprine (generic, Imuran) | 50 mg | Tablet | 0.2405 | up to 2.5 mg/kg dailyj | $36.60 | $439 |
Mercaptopurine (Purinethol and generic) | 50 mg | Tablet | 2.8610 | 1.5 to 2.5 mg/kg dailyj | $261.25 to $435.41 | $3,135 to $5,225 |
Methotrexate (generic) | 2.5 mg 10 mg | Tablet | 0.2513 2.7983m | 10 to 25 mg weeklyj | $13.07 to $109.13 | $52 to $437 |
ER = extended release.
The comparators presented in this table have been deemed to be appropriate based on feedback from clinical expert(s). Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Note: All prices are from the Ontario Drug Benefit Formulary (effective May 2023), unless otherwise indicated, and do not include dispensing fees. Annual period assumes 12 months, 365.25 days.
aBased on price submitted by sponsor.1
bProduct monograph mirikizumab (Omvoh).2 The dose for extended induction: 300 mg IV infusion at week 0, week 4, week 8, week 12, week 16, and week 20, followed by 200 mg maintenance treatment every 4 weeks if therapeutic benefit is achieved. The dose for reinduction following loss of response in the maintenance setting is 3 additional doses of 300 mg IV infusion every 4 weeks, followed by 200 mg every 4 weeks if therapeutic benefit is achieved.
cProduct monograph Adalimumab (Humira).4 The product monograph listed a concentration 100 mg/1 mL but no price was available.
dProduct monograph Adalimumab.45
eProduct monograph Simponi golimumab injection.5
fProduct monograph infliximab (Inflectra).46
gProduct monograph infliximab (Renflexis).47
hProduct monograph infliximab (Remicade).48
IReports dose of ozanimod hydrochloride (HCl); a 0.25 mg, 0.5 mg, and 1 mg of ozanimod HCl equivalents to 0.23 mg, 0,46, and 0.92 mg of ozanimod, respectively.
jOzanimod CADTH CDR PE Report.49
kProduct monograph Ozanimod.8
lProduct monograph Tofacitinib.7
mPrice obtained from Saskatchewan Drug Benefit (May 2023).50
nUstekinumab is not covered for UC by public plans.
oProduct monograph ustekinumab (Stelara).51
pIQVIA DeltaPA database (accessed May 2023).10
qPrice obtained from Ontario Exceptional Access Program (accessed May 2023).52
rProduct monograph vedolizumab (Entyvio).6
sThe recommended dosages for 5-ASA were obtained from respective product monographs.53-58
tProduct monograph Olsalazine (Dipentum).59
uProduct monograph Sulfasalazine (Salazopyrin, generics).60
vThe treatment cost is over a period of 5 months.
wHydrocortisone patient information.61
xProduct monograph Hydrocortisone (Cortef).62
Note that this appendix has not been copy-edited.
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes or no | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | Yes | Generally acceptable, however the sponsor included a comparator that is not currently reimbursed by the public health care payer and excluded comparators relevant to subgroups. For more information, refer to CADTH appraisal points on relevant comparators. |
Model has been adequately programmed and has sufficient face validity | No | Refer to CADTH Appraisal for limitations with model programming and validity of the model. |
Model structure is adequate for decision problem | Yes | Generally acceptable, however, the relapsing-remitting nature of the disease and treatment-switching is not captured. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | The uncertainty around some input parameters is not described. For more information, refer to CADTH appraisal points on model programming. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | The reporting In the pharmacoeconomic and budget Impact submissions is clear and consistent with the respective Excel models. Technical documentation regarding the sponsor-commissioned NMA reported the comparative efficacy findings in detail. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
Figure 1: Model Structure, Induction Phase
CT = conventional treatment; nr-Response = non-remission response; UC = ulcerative colitis.
Note: The induction period is implemented using a Markov model. The decision-tree like structure is only included to illustrate the patient flow between decision nodes. From the start of the model to the end of the induction period (maximum 26 weeks), patients transition through 2-week tunnel health states to decision nodes. Death health state is not shown.
Source: Sponsor’s pharmacoeconomic submission.1
Figure 2: Model Structure, Maintenance Phase
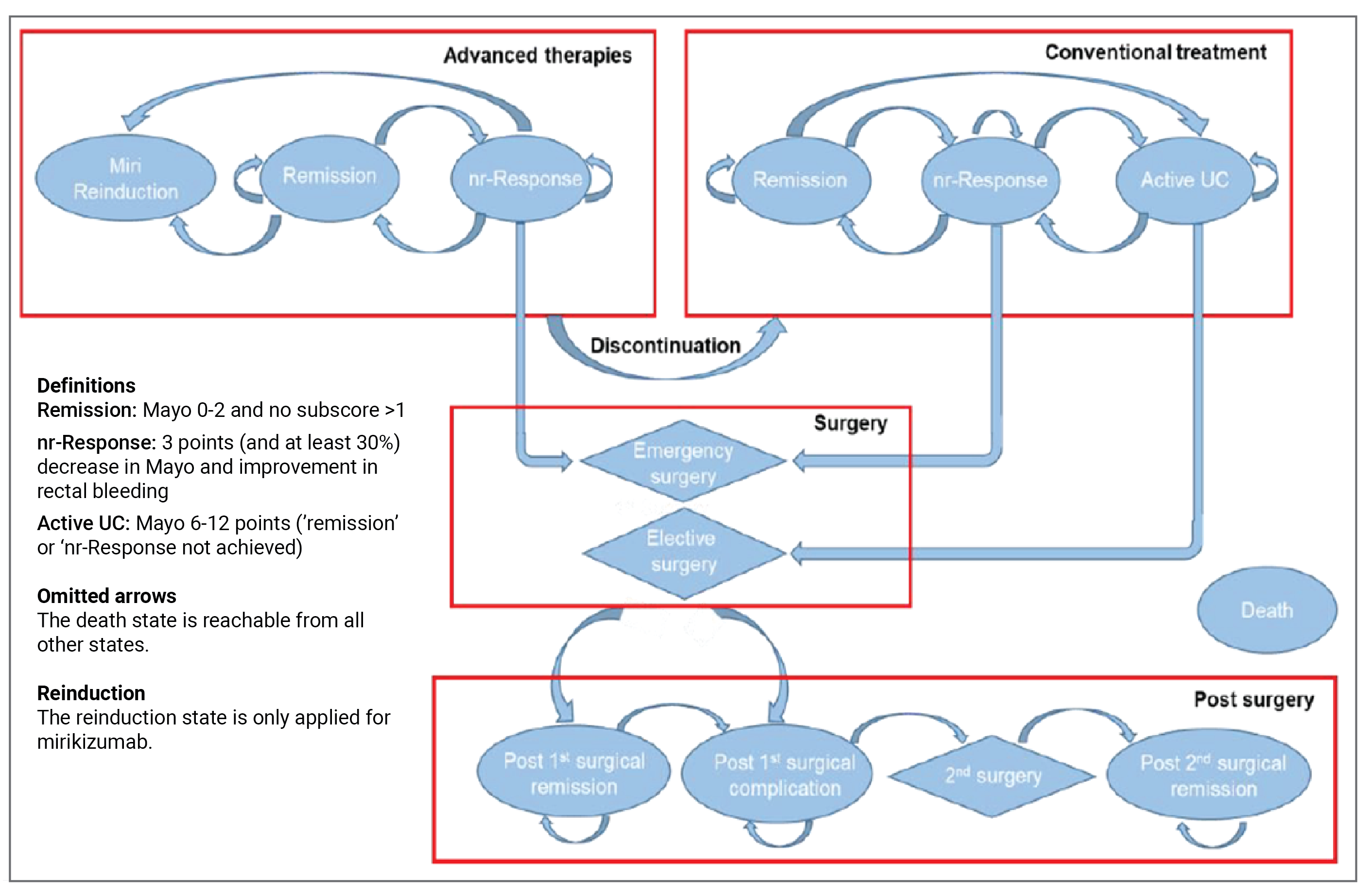
nr-Response = non-remission response; UC = ulcerative colitis.
Note: diamond-shaped health states reflect tunnel states.
Source: Sponsor’s pharmacoeconomic submission.1
Detailed Results of the Sponsor’s Base Case
Table 13: Base-Case Efficacy Inputs, Induction Phase
Drug | Biologic-naive | Biologic-experienced | ||||
|---|---|---|---|---|---|---|
No response | Response | Remission | No response | Response | Remission | |
Mirikizumab | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
Infliximab | |||||| | |||||| | |||||| | ||||||| | ||| |||| | ||| |||| |
Adalimumab | |||||| | |||||| | |||||| | |||||| | |||||| | ||||| |
Golimumab | |||||| | |||||| | |||||| | ||||||| | ||| ||| | ||| ||| |
Vedolizumab IV | |||||| | |||||| | |||||| | |||||| | |||||| | ||||| |
Vedolizumab IV/SC | |||||| | |||||| | |||||| | |||||| | |||||| | ||||| |
Tofacitinib | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
Upadacitinib 45 mg/15 mg | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
Ozanimod | |||||| | |||||| | |||||| | |||||| | |||||| | ||||| |
Upadacitinib 45 mg/30 mg | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
Conventional therapy | |||||| | |||||| | ||||| | |||||| | |||||| | ||||| |
SC = subcutaneous.
Table 14: Base-Case Efficacy Inputs, Maintenance Phase
Drug | Biologic-naive | Biologic-experienced | ||||
|---|---|---|---|---|---|---|
Overall (remission and response) | LOR | Remission | Overall (remission and response) | LOR | Remission | |
Mirikizumab | |||||| | ||||| | |||||| | |||||| | |||||| | |||||| |
Infliximab | |||||| | |||||| | |||||| | ||| ||||||||| | ||| ||||||||| | ||| ||||||||| |
Adalimumab | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
Golimumab | |||||| | |||||| | |||||| | ||| ||||||||| | ||| ||||||||| | ||| ||||||||| |
Vedolizumab IV | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
Vedolizumab IV/SC | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
Tofacitinib | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
Upadacitinib 45 mg/15 mg | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
Ozanimod | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
Upadacitinib 45 mg/30 mg | |||||| | ||||| | |||||| | |||||| | ||||| | |||||| |
Conventional therapy | |||||| | ||||| | |||||| | |||||| | ||||| | |||||| |
LOR = loss of response; SC = subcutaneous.
Sponsor’s Base-Case Results
Table 15: Sponsor’s Economic Evaluation Results, Biologic-Naive Population
Drug | Total costs | Total QALYs | Sequential ICER ($/QALY gained) |
|---|---|---|---|
Conventional therapy | $169,405 | 10.69 | Reference |
Adalimumab | $276,387 | 12.69 | Extendedly dominated |
Upadacitinib 45 mg/15 mg | $282,538 | 13.32 | $43,108 |
Golimumab | $291,502 | 12.70 | Strictly dominated |
Vedolizumab IV/SC | $295,914 | 12.91 | Strictly dominated |
Tofacitinib | $300,773 | 12.93 | Strictly dominated |
Vedolizumab IV | $329,096 | 13.22 | Strictly dominated |
Ozanimod | $334,860 | 13.36 | Extendedly dominated |
Infliximab | $369,614 | 14.06 | Extendedly dominated |
Upadacitinib 45 mg/30 mg | $425,546 | 14.59 | $112,034 |
Mirikizumab | $428,568 | 13.85 | Strictly dominated |
CEF = cost-effectiveness frontier; QALY = quality-adjusted life-year; SC = subcutaneous.
Note: Probabilistic results are presented.
Source: Sponsor’s pharmacoeconomic submission.1
Table 16: Sponsor’s Economic Evaluation Results — Biologic-Experienced Population
Drug | Total costs | Total QALYs | Sequential ICER ($/QALY gained) |
|---|---|---|---|
Conventional therapy | $167,042 | 10.63 | Reference |
Adalimumab | $250,537 | 12.03 | Extendedly dominated |
Ozanimod | $281,932 | 12.32 | Extendedly dominated |
Vedolizumab IV | $285,908 | 12.38 | Extendedly dominated |
Vedolizumab IV/SC | $296,321 | 12.79 | Extendedly dominated |
Tofacitinib | $337,172 | 13.54 | Extendedly dominated |
Upadacitinib 45 mg/15 mg | $372,448 | 15.68 | $40,657 |
Mirikizumab | $392,539 | 13.31 | Strictly dominated |
Upadacitinib 45 mg/30 mg | $485,474 | 15.56 | Strictly dominated |
CEF = cost-effectiveness frontier; QALY = quality-adjusted life-year; SC = subcutaneous.
Note: Probabilistic results are presented.
Source: Sponsor’s pharmacoeconomic submission.1
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Table 17: Summary of the Stepped Analysis of the CADTH Reanalysis Results — Biologic-Naive
Stepped analysis | Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY gained) |
|---|---|---|---|---|
Sponsor’s base case, probabilistic | Conventional therapy | 169,405 | 10.69 | Reference |
Upadacitinib 45 mg/15 mg | 282,538 | 13.32 | 43,108 | |
Upadacitinib 45 mg/30 mg | 425,546 | 14.59 | 112,034 | |
Sponsor’s base case, deterministic | Conventional therapy | 169,888 | 10.607 | Reference |
Upadacitinib 45 mg/15 mg | 267,925 | 12.924 | 42,309 | |
Upadacitinib 45 mg/30 mg | 364,881 | 13.606 | 142,065 | |
Mirikizumab | 437,600 | 13.990 | 189,637 | |
Sponsor’s base case, corrected | Conventional therapy | 169,888 | 10.607 | Reference |
Tofacitinib | 215,442 | 12.433 | 24,938 | |
Upadacitinib 45 mg/15 mg | 267,925 | 12.924 | 107,000 | |
Upadacitinib 45 mg/30 mg | 364,881 | 13.606 | 142,065 | |
Mirikizumab | 437,600 | 13.990 | 189,637 | |
CADTH reanalysis 1 | Conventional therapy | 169,888 | 10.607 | Reference |
Tofacitinib | 212,981 | 12.324 | 25,088 | |
Mirikizumab | 437,600 | 13.990 | 134,872 | |
CADTH reanalysis 2 | Conventional therapy | 169,888 | 10.607 | Reference |
Tofacitinib | 215,442 | 12.433 | 24,938 | |
Upadacitinib 45 mg/15 mg | 267,925 | 12.924 | 107,000 | |
Upadacitinib 45 mg/30 mg | 364,881 | 13.606 | 142,065 | |
Mirikizumab | 395,900 | 13.381 | Dominated by upadacitinib 45 mg/30 mg | |
CADTH reanalysis 3 | Conventional therapy | 169,888 | 10.607 | Reference |
Tofacitinib | 241,304 | 13.570 | 24,100 | |
Upadacitinib 45 mg/15 mg | 315,680 | 14.174 | 23,138 | |
Mirikizumab | 437,600 | 13.990 | Dominated by upadacitinib 45 mg/30 mg | |
CADTH reanalysis 4 | Conventional therapy | 169,888 | 10.607 | Reference |
Tofacitinib | 215,500 | 12.432 | 24,986 | |
Upadacitinib 45 mg/15 mg | 267,812 | 12.926 | 105,886 | |
Upadacitinib 45 mg/30 mg | 364,738 | 13.609 | 141,888 | |
Mirikizumab | 437,600 | 13.990 | 191,507 | |
CADTH reanalysis 5 | Conventional therapy | 169,888 | 10.607 | Reference |
Tofacitinib | 215,442 | 12.433 | 24,938 | |
Mirikizumab | 437,600 | 13.990 | 142,735 | |
CADTH base case, deterministic | Conventional therapy | 169,888 | 10.607 | Reference |
Tofacitinib | 236,095 | 13.338 | 24,236 | |
Mirikizumab | 395,900 | 13.381 | 3,758,347 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: Reference product is least costly alternative.
Table 18: Summary of the Stepped Analysis of the CADTH Reanalysis Results — Biologic-Experienced
Stepped analysis | Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY gained) |
|---|---|---|---|---|
Sponsor’s base case, probabilistic | Conventional therapy | 167,042 | 10.63 | Reference |
Upadacitinib 45 mg/15 mg | 372,448 | 15.68 | 40,657 | |
Sponsor’s base case, deterministic | Conventional therapy | 168,566 | 10.377 | Reference |
Upadacitinib 45 mg/15 mg | 279,883 | 13.100 | 40,887 | |
Upadacitinib 45 mg/30 mg | 367,564 | 13.492 | 223,346 | |
Mirikizumab | 321,990 | 12.231 | Dominated by Upadacitinib 45 mg/15 mg | |
Sponsor’s base case, corrected | Conventional therapy | 168,566 | 10.377 | Reference |
Tofacitinib | 207,849 | 11.967 | 24,710 | |
Upadacitinib 45 mg/15 mg | 279,883 | 13.100 | 63,589 | |
Upadacitinib 45 mg/30 mg | 367,564 | 13.492 | 223,346 | |
Mirikizumab | 321,990 | 12.231 | Dominated by Upadacitinib 45 mg/15 mg | |
CADTH reanalysis 1 | Conventional therapy | 168,566 | 10.377 | Reference |
Tofacitinib | 204,234 | 11.787 | 25,296 | |
Upadacitinib 45 mg/15 mg | 249,289 | 12.210 | 106,609 | |
Upadacitinib 45 mg/30 mg | 308,616 | 12.424 | 276,775 | |
Mirikizumab | 321,990 | 12.231 | Dominated by upadacitinib 45 mg/30 mg | |
CADTH reanalysis 2 | Conventional therapy | 168,566 | 10.377 | Reference |
Tofacitinib | 207,849 | 11.967 | 24,710 | |
Upadacitinib 45 mg/15 mg | 279,883 | 13.100 | 63,589 | |
Upadacitinib 45 mg/30 mg | 367,564 | 13.492 | 223,346 | |
Mirikizumab | 293,988 | 11.818 | Dominated by vedolizumab IV | |
CADTH reanalysis 3 | Conventional therapy | 168,566 | 10.377 | Reference |
Tofacitinib | 207,780 | 11.964 | 24,713 | |
Upadacitinib 45 mg/15 mg | 251,400 | 12.358 | 110,632 | |
Mirikizumab | 321,990 | 12.231 | Dominated by upadacitinib 45 mg/15 mg | |
CADTH reanalysis 4 | Conventional therapy | 168,566 | 10.377 | Reference |
Tofacitinib | 207,898 | 11.966 | 24,757 | |
Upadacitinib 45 mg/15 mg | 279,754 | 13.103 | 63,224 | |
Upadacitinib 45 mg/30 mg | 367,417 | 13.496 | 223,093 | |
Mirikizumab | 321,990 | 12.231 | Dominated by upadacitinib 45 mg/15 mg | |
CADTH reanalysis 5 | Conventional therapy | 168,566 | 10.377 | Reference |
Tofacitinib | 207,849 | 11.967 | 24,710 | |
Mirikizumab | 321,990 | 12.231 | 432,938 | |
CADTH base case, deterministic | Conventional therapy | 168,566 | 10.377 | Reference |
Tofacitinib | 204,219 | 11.784 | 25,346 | |
Mirikizumab | 293,988 | 11.818 | 2,608,809 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: Reference product is least costly alternative.
Detailed Results of CADTH Base Case
Table 19: Disaggregated Summary of CADTH’s Economic Evaluation Results — Biologic-Naive (Deterministic)
Parameter | Conventional therapy | Tofacitinib | Mirikizumab |
|---|---|---|---|
Discounted LYs | |||
Total | 29.781 | 29.778 | 29.793 |
Discounted QALYs | |||
Total | 10.607 | 13.338 | 13.381 |
By health state | |||
Treatment | 0.089 | 5.607 | 5.713 |
Conventional therapy | 9.828 | 7.198 | 7.140 |
Surgery | 0.716 | 0.560 | 0.556 |
Adverse events | –0.026 | –0.028 | –0.028 |
Discounted costs | |||
Total | $169,888 | $236,095 | $395,900 |
Health state cost | $139,775 | $126,239 | $126,010 |
Surgery cost | $2,006 | $1,706 | $1,696 |
Treatment cost | $26,848 | $106,821 | $266,861 |
AE costs | $1,258 | $1,330 | $1,332 |
Societal costs | $0 | $0 | $0 |
LY = life-year; QALY = quality-adjusted life-year.
Table 20: Disaggregated Summary of CADTH’s Economic Evaluation Results — Biologic-Experienced (Deterministic)
Parameter | Conventional therapy | Tofacitinib | Mirikizumab |
|---|---|---|---|
Discounted LYs | |||
Total | 29.427 | 29.419 | 29.433 |
Discounted QALYs | |||
Total | 10.377 | 11.784 | 11.818 |
By health state | |||
Treatment | 0.085 | 2.954 | 3.045 |
Conventional therapy | 9.608 | 8.228 | 8.175 |
Surgery | 0.710 | 0.629 | 0.625 |
Adverse events | –0.026 | –0.027 | –0.027 |
Discounted costs | |||
Total | $168,566 | $204,219 | $293,988 |
Health state cost | $138,493 | $131,585 | $131,379 |
Surgery cost | $1,993 | $1,854 | $1,845 |
Treatment cost | $26,838 | $69,500 | $159,482 |
AE costs | $1,243 | $1,280 | $1,283 |
Societal costs | $0 | $0 | $0 |
LY = life-year; QALY = quality-adjusted life-year.
Scenario Analyses
CADTH undertook 3 scenario analyses: scenario 1 assumed numerical differences in clinical efficacy and safety between mirikizumab and its comparators, scenario 2 assumed response rate among delayed clinical responders to be the same as response rate after 12 weeks of induction, and scenario 3 included upadacitinib as a comparator.
Table 21: Summary of Scenario Analyses Conducted on CADTH Base Case — Biologic-Naive
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY gained) |
|---|---|---|---|---|
CADTH base case, deterministic | Conventional therapy | 169,888 | 10.607 | Reference |
Tofacitinib | 236,095 | 13.338 | 24,236 | |
Mirikizumab | 395,900 | 13.381 | 3,758,347 | |
CADTH scenario analysis 1 | Conventional therapy | 169,888 | 10.607 | Reference |
Tofacitinib | 241,395 | 13.568 | 24,145 | |
Infliximab | 360,425 | 13.871 | 392,375 | |
Mirikizumab | 395,900 | 13.381 | Dominated by golimumab | |
CADTH scenario analysis 2 | Conventional therapy | 169,888 | 10.607 | Reference |
Tofacitinib | 236,095 | 13.338 | 24,236 | |
Mirikizumab | 443,938 | 14.097 | 273,879 | |
CADTH scenario analysis 3 | Conventional therapy | 169,888 | 10.607 | Reference |
Tofacitinib | 236,095 | 13.338 | 24,236 | |
Mirikizumab | 395,900 | 13.381 | 3,758,347 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Table 22: Summary of Scenario Analyses Conducted on CADTH Base Case — Biologic-Experienced
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY gained) |
|---|---|---|---|---|
CADTH base case, deterministic | Conventional therapy | 168,566 | 10.377 | Reference |
Tofacitinib | 204,219 | 11.784 | 25,346 | |
Mirikizumab | 293,988 | 11.818 | 2,608,809 | |
CADTH scenario analysis 1 | Conventional therapy | 168,566 | 10.377 | Reference |
Tofacitinib | 207,829 | 11.963 | 24,760 | |
Mirikizumab | $293,988 | 11.818 | Dominated by tofacitinib | |
CADTH scenario analysis 2 | Conventional therapy | $168,566 | 10.377 | Reference |
Tofacitinib | $204,219 | 11.784 | 25,346 | |
Mirikizumab | $317,905 | 12.176 | 289,977 | |
CADTH scenario analysis 3 | Conventional therapy | $168,566 | 10.377 | Reference |
Tofacitinib | $204,219 | 11.784 | 25,346 | |
Mirikizumab | $293,988 | 11.818 | 2,608,809 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Appendix 5: Submitted Budget Impact Analysis and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 23: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
The sponsor stated that submitted budget impact analysis (BIA)63 assessed the expected budgetary impact of reimbursing mirikizumab for the treatment of adults with moderately to severely active UC who have had an inadequate response, had a loss of response, or were intolerant to either conventional therapy, a biologic treatment, or a JAK inhibitor, or have medical contraindications to such therapies, which generally aligns with the Health Canada-indicated population. The BIA was conducted from the public drug program perspective over a 3-year time horizon.
A claims-based approach was taken to estimate the total market size, in terms of claims, of patients currently receiving publicly reimbursed biologic drugs and JAK inhibitors with a Health Canada–approved indication for an IBD using IQVIA Pharmastat database. These drugs included adalimumab (brand and biosimilar), infliximab (brand and biosimilar), golimumab, tofacitinib, and vedolizumab. The sponsor commissioned IQVIA to conduct an unpublished real-world evidence study to identify the number of patients with an inferred indication of an IBD based on patient’s medical history and physician specialty using the IQVIA Private Drug Plan and Ontario Drug Benefit databases. ||| ||||||| |||||||| ||||||||||| || |||||||| || ||| ||||||||||| |||||||||| ||||||||| |||| ||||||| |||| ||| ||||| ||| ||| ||| |||||||||| ||| ||||||| ||||||||| |||| ||||| || ||||| |||||| |||| |||||||| || || ||| |||||||||| |||||||.64 ||||| |||| |||||||||||| ||| ||||||| ||||||| || |||||||||| |||||||||| |||||| || |||||||| ||| |||||| || |||||| || ||| ||||| |||||||||| |||||||| ||| || ||| |||| ||||||||||| ||| ||||| ||| ||| ||| ||||||||| |||| |||| |||||||| || |||||| ||||| || ||| |||| || ||||| ||||| ||| ||| |||| || |||||||| || ||||||||| || ||| ||||||| ||| ||||| |||||| ||| ||| |||| || |||||||| || ||||| |||||||| ||||| ||| ||| ||||||||||| ||||| || |||| ||||||||| ||| ||||||| ||||||||| |||| ||| || ||| || |||||| |||| || |||||||||||||| |||||||| ||| ||| ||| |||||||||||||||||||| ||||||||. The sponsor adopted best fitting trend curves on a product-by-product basis to forecast the number of claims over the time horizon. In the absence of data, the sponsor used claims data from Prince Edward Island for Nova Scotia and claims data from New Brunswick for British Columbia, adjusted for population size of the respective province. As the days per claim for each product can vary, the sponsor standardized the duration of all claims to a common duration (28 days).
The BIA included costs related to drug acquisition and administration. Drug prices were obtained from each drug programs’ respective formulary. The drug prices were multiplied by the average number of units dispensed, days per treatment cycle, and administrations per cycle to calculate cost per claim, standardized over 28 days. Dosing during induction and maintenance phase was obtained from respective product monographs.3-9 The cost calculations were adjusted for dose escalation during the maintenance therapy of products based on estimates derived from an unpublished real-world evidence study conducted by the sponsor.65 The cost per claim of each product was multiplied by the proportion used for induction therapy and the proportion used for maintenance therapy, also obtained from the sponsor’s real-world evidence study,65 to estimate treatment costs of biologics and JAK inhibitors. Mark-up charges and dispensing fees were excluded. Key inputs to the BIA are documented in Table 24.
Table 24: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
Proportion of claims for ulcerative colitis | 44.4% |
Number of patients eligible for drug under review | ||||||| / ||||||| / ||||||| |
Market uptake (3 years) | |
Uptake (reference scenario) Adalimumab Adalimumab biosimilar Infliximab Infliximab biosimilar Golimumab Ozanimoda Tofacitinib Upadacitinib Vedolizumab | ||||||||||||||||||||| ||||||||||||||||||||| ||||||||||||||||||||| ||||||||||||||||||||| ||||||||||||||||||||| ||||||||||||||||||||| ||||||||||||||||||||| ||||||||||||||||||||| ||||||||||||||||||||| |
Uptake (new drug scenario) Mirikizumab Adalimumab Adalimumab biosimilar Infliximab Infliximab biosimilar Golimumab Ozanimod Tofacitinib Upadacitinib Vedolizumab | ||||||||||||||||||||| ||||||||||||||||||||| ||||||||||||||||||||| ||||||||||||||||||||| ||||||||||||||||||||| ||||||||||||||||||||| ||||||||||||||||||||| ||||||||||||||||||||| ||||||||||||||||||||| ||||||||||||||||||||| |
Cost of treatment (per patient per 28 days)b | |
Cost of treatment per claim Mirikizumab Adalimumab Adalimumab biosimilar Infliximab Infliximab biosimilar Golimumab Ozanimod Tofacitinib Upadacitinib Vedolizumab | $2,375 $2,056 $1,221 $2,493 $1,245 $3,753 $1,918 $2,054 $2,753 $2,014 |
aAlthough there was no claims data for ozanimod and upadacitinib, the sponsor assumed market share of these comparators was proportionately captured from total number of claims.
bThe cost per claim for Ontario is shown. The BIA included formulary prices from each jurisdiction was used to estimate cost per claim of each drug.
Summary of the Sponsor’s BIA Results
The sponsor estimated the net 3-year budget impact of introducing mirikizumab for treatment of patients aged 18 years and older with moderately to severely active UC who have had an inadequate response, had a loss of response, or were intolerant to either conventional therapy, a biologic treatment, or a JAK inhibitor, or have medical contraindications to such therapies to be $15,384,071 (year 1: $1,738,158; year 2: $4,940,409; year 3: $8,705,504).
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications for the results of the BIA:
Use of claims-based approach to estimate market size and treatment costs introduced uncertainty with the anticipated budget impact of mirikizumab: The sponsor estimated market size for mirikizumab was based on claims data for the relevant comparators, however, due to limitations with the available data, and the lack of validation possible by CADTH, it is unclear whether the population size estimated as part of the sponsor-commissioned study is accurate. ||| ||||||| ||||||| || |||||||||| |||||||||| |||||| || |||||||| ||| |||||| || |||||| ||| |||||||||| || |||||||| |||||| || || |||||||| ||||| || |||||||| |||||||||| |||||||||. However, compared to a published BIA using an epidemiological approach to estimating the number of individuals with moderate to severe UC, the sponsor may have overestimated the market size.49 Given the claims database does not provide the indication and the proportion of claims pertaining to use for other indications is unknown, using a claims-based approach to estimate market size introduces significant uncertainty in the estimated market share.
The sponsor also did not convert the claims data into the number of users; instead, the sponsor assumed unit to unit and claim to claim displacement between mirikizumab and comparators. This is inappropriate because comparators have difference cycle lengths during the induction and maintenance phase. The sponsor estimated treatment costs using a cost per claim approach; treatment cost was weighted by proportion of cost attributed to induction phase and maintenance phase. It was unclear if all patients who received induction also accrued the cost of maintenance treatment in the sponsor’s adopted approach. For transparency and completeness, claims data-based models should provide an estimate of the number of active beneficiaries based on the number of claims. Otherwise, given that the prevalence and incidence of UC is available, the sponsor should have used epidemiological data to estimate the number of individuals eligible for treatment with mirikizumab.
CADTH was unable to address the limitations of a claims-based approach in reanalyses.
Unit price of tofacitinib does not reflect formulary prices: The sponsor obtained the price of tofacitinib using IQVIA DeltaPA database,10 which does not reflect the publicly listed price of tofacitinib covered by publicly drug plans.
CADTH corrected the price of tofacitinib using the Ontario Drug Benefit Formulary price. However, the incremental budget impact did not change as uptake of mirikizumab was assumed not to impact the market share of tofacitinib.
Market capture of mirikizumab is uncertain: The sponsor assumed mirikizumab has a market share of 9.9% by year 3 in individuals with moderate to severe UC. Clinical expert feedback obtained by CADTH noted the potential for more rapid uptake of mirikizumab among patients who have used all treatment options and need new treatments in the biologic-experienced population. The clinical expert consulted by CADTH for this review indicated that the market share of mirikizumab will be dependent on relative efficacy of mirikizumab and comparators, and anticipated a higher uptake of mirikizumab in the biologic-naive population if the clinical efficacy of mirikizumab is established in clinical practice. The clinical expert also noted that the market capture of mirikizumab may be higher from tumour necrosis factor inhibitors (adalimumab, infliximab and golimumab) in the biologic-naive population. This is not aligned with the sponsor’s estimate that most of the market share of mirikizumab would be captured from vedolizumab in the total population. As the sponsor’s budget impact was not estimated separately for biologic-naive and biologic-experienced populations, the sponsor’s estimates of market displacement and the resulting budget impact is highly uncertain.
CADTH was unable to adequately address this area of uncertainty. If mirikizumab uptake is higher than expected, it is likely to result in a greater incremental cost to drug plans, given it is more costly than all comparators except for branded infliximab.
Market share of comparators is uncertain: The sponsor allocated market share to upadacitinib and ozanimod, however, these drugs are not currently reimbursed by public drug plans. Given ozanimod is currently under ongoing negotiations with pCPA,44 it is reasonable to assume a market share of ozanimod in year 1 (2024). However, ozanimod should not have a market share in base year (2023). Further, upadacitinib is still under consideration at CADTH for the indication of interest,42 and the timeline for upadacitinib to be reimbursed by public plans is uncertain. CADTH explored the impact of assuming ozanimod does not have a market share in year 1 and excluding upadacitinib as a relevant comparator. However, these changes did not impact the incremental budget impact because the sponsor assumed mirikizumab does not capture market share from ozanimod and upadacitinib. If the market share of mirikizumab is captured from comparators that are more costly than the least costly advanced therapy, the incremental budget impact is expected to decrease.
CADTH was unable to address this limitation.
Model lacked transparency and flexibility: The BIA model submitted by the sponsor lacked transparency as the sponsor hard-coded some inputs, which resulted in a lack of face validity when making changes to model inputs. The model also had limited flexibility to allow the reviewers to assess the impact of changing the sponsor’s base assumptions.
As CADTH did not present reanalyses, this did not greatly impact the results, though limited testing that could occur.
CADTH Reanalyses of the BIA
CADTH did not undertake a base-case reanalysis, as CADTH could not address the limitation of the claims-based approach to estimate the incremental budget impact. CADTH conducted additional scenario analysis assuming a price reduction of 65% for mirikizumab. The results of the sponsor’s submission and CADTH scenario analysis are presented in Table 18.
Table 25: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | 3-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $166,365,520 | $171,417,038 | $182,276,005 | $191,415,935 | $545,108,978 |
New drug | $166,365,520 | $173,155,196 | $187,216,413 | $200,121,440 | $560,493,049 | |
Budget impact | $0 | $1,738,158 | $4,940,409 | $8,705,504 | $15,384,071 | |
CADTH scenario analysis, price reduction of 65% | Reference | $166,365,520 | $171,417,038 | $182,276,005 | $191,415,935 | $545,108,978 |
New drug | $166,365,520 | $172,202,441 | $184,472,868 | $195,236,559 | $551,911,868 | |
Budget impact | $0 | $785,403 | $2,196,863 | $3,820,624 | $6,802,889 |
BIA = budget impact analysis.
Stakeholder Input
Patient Input
Gastrointestinal Society
About Gastrointestinal Society
The GI (Gastrointestinal) Society is committed to improving the lives of people with GI and liver conditions, supporting research, advocating for appropriate patient access to healthcare, and promoting gastrointestinal and liver health.
We are a national charity formed in 2008 on the groundwork of its partner organization, the Canadian Society of Intestinal Research (CSIR), which was founded in Vancouver in 1976. We receive national and international attention, simply because we have earned the respect of both the gastrointestinal medical community and Canadians who battle GI and liver issues daily. Our English and French websites received 6,903,208 pageviews by 5,174,016 unique visitors in 2022.
All our programs and services focus on providing Canadians with trusted, commercial-free, medically-sound information on gut (including obesity) and liver diseases and disorders in both official languages. Our BadGut® lectures, quarterly Inside Tract® newsletter, pamphlets, support groups, and educational videos arm Canadians with the information they require to better understand and manage their specific needs. We also work closely with healthcare professionals and governments at all levels toward system-wide improvements in care and treatment.
Information Gathering
The information we used to complete this submission was obtained primarily through questionnaires and interviews:
2015 survey on biologics and biosimilars (then called subsequent entry biologics) completed by 423 Canadians (English: 317 and French: 106) with inflammatory bowel disease (IBD), including Crohn’s disease and ulcerative colitis.
2018 survey on the unmet need in IBD completed by 432 Canadians with IBD.
2020 survey completed by 579 respondents regarding the unmet needs of IBD.
2020 survey on biosimilars with 145 respondents, most of whom had IBD (some had other inflammatory conditions).
2022 survey about the IBD patient journey with 54 Canadian respondents with IBD.
one-to-one interviews with 3 individuals with ulcerative colitis who participated in a clinical trial for mirikizumab and received the trial drug.
2022 focus group with several persons living with ulcerative colitis so we could map the patient journey and animate it (refer to Figure 1), which is available on our website at www.badgut.org/patient-journeys, and we encourage your reviewers to watch this short video.
We also had contact with patients affected by IBD through one-to-one conversations at our BadGut® Lectures, a patient roundtable, recent phone/email/social media interactions with individuals who have IBD, our support group, and stories submitted over time from patients.
Disease Experience
Ulcerative colitis is an inflammatory bowel disease (IBD) that can arise at any age, often among children and young adults, or even during middle age. There is an increased risk for those who have a family member with the condition. Currently, Canada has among the highest prevalence and incidence yet reported in the world, with approximately 120,000 diagnosed individuals.
Diarrhea, bowel urgency, incontinence, abdominal pain, fever, rectal bleeding, and nausea are common symptoms of ulcerative colitis. Inflammation decreases the intestine’s absorptive surfaces, triggering watery stools that can lead to fecal urgency and poor control of bowel function. Low red blood cell count (anemia) can result from blood loss due to ulcerations in the intestine and from general malnutrition due to decreased nutrient absorption and the debilitating effects of the disease.
Some patients have extra-intestinal manifestations, including fever, inflammation of the eyes or joints (arthritis), ulcers of the mouth or skin, tender and inflamed nodules on the shins, and numerous other conditions. Anxiety, stress, and mental health are major factors.
Ulcerative colitis often has a profound effect on an individual’s life – physically, emotionally, and socially, both at home and at school or in the workplace. Symptoms can be relentless, embarrassing, and scary. The severity of the disease can fluctuate, making it necessary to go through routine testing, reassessments, and medication changes. It is particularly difficult for children and young adults, since it often affects a person’s sense of self.
More than anything, patients have told us that sustained remission/treatment response is more important than relieving any one symptom of ulcerative colitis. As a chronic disease, it is never just one flare that dominates the impact of the disease, but the constant concern that there will be future flares, possibly worse than the last, at unpredictable times, which can disastrously disrupt their lives.
The following quotes are from individuals describing what it feels like during an ulcerative colitis flare, and what their biggest concern is, in their own words:
“Your gut aches and burns and there is often blood in the toilet. You lose your appetite and weight, unhealthily! My biggest concern is I'm going to run out of meds to help!”
“It’s like I can’t control anything, I feel weak and can barely get up. My biggest concern is usually when I see blood and determining at what point to go to the ER.”
“The pain is worse than childbirth... and I have 3 kids...1 labour without drugs.”
“Worst flu symptoms, fatigue, lethargy, like swallowing glass and chili and then having constipation and diarrhea at the same time. Gut cramps and hunger cramps at the same time. Want to die. Biggest concern is needing a toilet at all times with zero minutes waiting time.”
“It feels like my guts are in a vise. The nausea can be so bad I can't move or even vomit and the diarrhea is so painful I'll be literally screaming in the bathroom.”
“The worst part is fear of irreversible permanent damage that will affect your day-to-day life forever.”
“It is so exhausting and feels like it will never end. You start to question if you can still live the life you planned. And no-one gives you a break.”
“A flare can come out of nowhere and completely disrupt your life. Pain can sometimes be so bad that it keeps you in bed. You mostly spend life either asleep or on the toilet. My biggest concern during a flare is being able to keep up with my responsibilities (work, school, social, etc.).”
“It feels like your body is betraying you. You can’t plan anything in advance because you don’t know how your body will feel on a day-to-day basis.”
“There’s a huge element of fear and worry and being faced with mortality at such a young age.”
It’s one thing to read a list of common symptoms or data on how ulcerative colitis affects patients, but it is the individual stories of these patients, as summarized above, which astound us and motivate us to support patients’ need for more diversity in effective treatments. In addition, treatments should improve quality of life, not cause more symptoms, pain, frustration, or hardship.
Experiences With Currently Available Treatments
The treatment of ulcerative colitis is multi-faceted; it includes managing the symptoms and consequences of the disease along with therapies targeted to reduce the underlying inflammation. Typically, a patient starts on one type of treatment and, if there is inadequate response, then switches to another type.
5-ASA helps to settle acute inflammation and, for some patients, keeps the inflammation inactive when taken on a long-term basis (maintenance). To reduce inflammation in for the short-term in ulcerative colitis, corticosteroids can help. For topical relief in the colon, corticosteroids are available in rectal formulations. These are inconvenient therapies that make it difficult for patients to keep a normal routine. Also, if a patient has significant diarrhea, then the rectal medications may be difficult to hold in place for sufficient time to be effective. Immunosuppressive agents reduce dependence on steroids and help patients who have steroid-resistant disease, but it could take up to six months or more of therapy to see results.
Biologics treat ulcerative colitis when older medications fail to relieve symptoms. There are a variety of mechanisms through which they work. On March 29, 2023, the Institut national d’excellence en santé et en services sociaux recommended eliminating the requirement of trialing through conventional therapy before patients living with ulcerative colitis can receive biologics. This provides patients with more effective tools to tackle this debilitating disease early on in their care. It can also lead to savings in healthcare resources. We applaud this recommendation and encourage all jurisdictions in Canada to follow.
A newer class of medication for IBD, Janus kinase (JAK) inhibitors, typically work faster than other immunosuppressive medications, pose no risk for immunogenicity, unlike biologics, and are easier and more convenient to take since they are in pill form. However, they have a risk for side effects of infections, serious heart-related problems, fatal blood clots, and cancer.
While there are a few options available, patients still have a lot of difficulty obtaining remission or adequate symptom relief. In one of our surveys, we asked patients if the currently available medications are adequate to control their disease. Only 24% of those with IBD thought that the available medications are adequate, 56% found them to be only somewhat adequate, and 20% not at all adequate. Patients are still suffering, and they need new and effective options to achieve mucosal healing and reduce the debilitating symptoms of ulcerative colitis.
Medications help avert removal of all or part of the colon. Since ulcerative colitis is a systemic disease, not only the colon is involved. Therefore, if a surgeon removes the colon (colectomy) and then brings the end of the remaining intestine through a new surgical opening in the abdominal wall (ostomy) to which the patient can attach a removable appliance to collect stool, the patient still experiences symptoms. With the loss of colon function, bowel movements can occur frequently and have high liquid content. Another surgical treatment is to remove diseased tissue and create a pouch from remaining tissue so defecation can occur via the rectum. However, one complication that can occur is pouchitis, which is inflammation within the surgically created pouch. This means that even after surgery, patients could face troublesome gastrointestinal symptoms. Living without your colon is very difficult.
Improved Outcomes
Patients affected by ulcerative colitis need access to medications that work and give them a good quality of life. Inadequate access to medication results in preventable patient suffering (e.g., continual, debilitating disease symptoms; secondary illnesses such as depression and anxiety disorders; and loss of family/social interactions). It also leads to unnecessary usage of healthcare resources (e.g., hospital stays, surgeries, diagnostic procedures, other medications) and a ripple effect of financial burden on the government and taxpayers (e.g., through inability to work, long-term disability claims, biologic-related debt, and even bankruptcy).
When a patient receives the right medication at the right time and for the right duration – as determined between physician and patient – these individuals can live full, rewarding lives as productive, valuable citizens who participate in the workforce and community. However, since patients are unique, they respond differently to various medications, and in some cases stop responding to medications after using them for some time, it is important to have a variety of options available.
Experience With Drug Under Review
We interviewed three patients who participated in clinical trials for mirikizumab in Canada. All continue to take the medication.
Patient 1
She received a diagnosis of ulcerative colitis when she was 15-16 years old. From there, she went on a four-week trial of a variety of 5-ASAs, from enemas, suppositories, and pills, including Mezavant®. She was on and off medications after and found it difficult to remember to take her medications, especially in her later teenage years. She then experienced a bad flareup and was hospitalized for a few days, where she experienced about 20 bowel movements a day, rectal bleeding, and was vomiting every single meal that she tried to consume. She then moved to an outpatient clinic for transfusions and was taking prednisone, as well as several other corticosteroids. She said, “Having the active disease, it’s so tiring on my mental and physical health.”
She had just finished her first year of university when her doctor informed her about a clinical trial for mirikizumab. She started with transfusions of intravenous fluids over three hours, which she described was taxing but understandable. Her doctor also prescribed her steroids, which she took before and during the trial. After the first infusion, she stopped experiencing symptoms and she also needed to renew her prescription. Guessing that she was on the trial drug, she stopped taking her other medication and experienced a flareup after 1-2 months. She then learned that she was in the control group and the trial team moved her to the cohort receiving mirikizumab.
After one year of transfusions over three hours, the trial team dropped them to a one-hour duration of transfusions, and then subcutaneous administration. She has been on remission without steroids for two and a half years now and has not experienced negative side effects. She is in nursing school, so she has no issues with needles. She finds that it stings a bit more than other needles she’s worked with but, “it’s worth it,” she said, especially since she only has to take it once a month. “It’s definitely easier to use than suppositories and enemas.”
“It’s affected every aspect of my life positively. Now, I’m only taking mirikizumab. Energy wise, waking up feeling refreshed in the morning. I have my appetite back, no pain the day and overall, so much less stress and anxiety because it can get tiring worrying about your state of health. I’ve always loved sports and keeping in shape and having active disease, it prevents you from doing that. [The drug] got me back to working out and being healthy.”
Patient 2
She struggled with ulcerative colitis symptoms for a year and half before she finally received a diagnosis. Her treatment plan consisted of a biologic, then prednisone for two months and, unfortunately, neither offered any benefits. Instead, she experienced various side effects from these medications and since her body was under stress, her symptoms worsened as well. Her doctor then connected her to the study and, after six months of being on mirikizumab, the comparison between how she felt before the study and after was “day and night.” “It was incredible with how quickly I started to feel, and it was fairly consistent.” Her colonoscopies had shown improvements, and she described it as “it wouldn’t look like someone has colitis.” The only side effects she noted is feeling exhausted leading up to her next injection. Currently, her treatment regimen consists of mirikizumab, iron infusions, and Mezavant®.
When she started the trial, she found that “it was exhaust[ing] since it was a lot on my body and appointments and infusions.” She is also anemic, and she was still getting bad nausea and dizziness, so she received iron infusions simultaneously. However, her doctors had ruled out that these side effects were from the anemia and not mirikizumab.
She’s been on mirikizumab for two years. She’s comfortable with the subcutaneous injections now, but it did take her a while with a few practices at the clinic to learn how to administer them herself. When asked what improvements she’d like to see in a new drug, she said she couldn’t think of any and then suggested that they could be “less frequent than once a month but even that is not a big deal. The earlier treatments were easy, but they didn’t improve my ulcerative colitis.” “My experience has been really good with this drug, and I recommend it.” “I have no anxieties on going out to public events. I’m definitely a lot more comfortable leaving the house and going about my normal day to day activities.”
Patient 3
About 20 years ago, she received a diagnosis of ulcerative proctitis and suffered with it on and off since then. She then received a diagnosis of ulcerative colitis and prescribed a treatment plan consisting of suppositories, enemas, and Mezavant®. “After a while, I noticed that my hair was coming out and my gums were bleeding, and it was a drug reaction. I took it for about a year and then stopped.” She then tried a corticosteroid, which helped, but she had a big sore on her face and needed to get an antibiotic. Her doctor then suggested that she try mirikizumab and sign up for the clinical trial.
“At one point, I was on a ‘no food’ diet, no dairy, no meat, no fruit, whole grains… almost everything except white bread, rice, eggs, and chicken. It was painful with the bleeding and bowel movements. When you’re on the toilet all the time and your gut is bleeding, you’re just tired and you want to take a nap, like a bunch of naps. I would try to go jogging and I go halfway around the track, and I have to go to the toilet. It’s embarrassing and unpleasant.”
After being on mirikizumab, she lives a better quality of life. She has been taking it for about two and a half years, and her colonoscopies have shown that her “gut is healing.” She has not experienced any side effects. “I’m not a big fan of needles but it’s manageable. There are no reactions on the site of injection, and it hurts but then goes away. No reaction any or ever to this stuff, No rashes.” “I like it very much that I only need to take mirikizumab once a month,” she said, “I’m the type of person that doesn’t like taking long term medication at all.” She goes to the hospital once every three months for checkups and gets blood work from time to time. She doesn’t find it to be burdensome, since she can take the bus and the appointments are not long.
She struggles with a lot of stress from personal relationships and believes in the brain-gut connection as stress triggers her gut from time to time. To manage this, she prays, walks her dog in the forest, which she can now do without being anxious on needing toilets nearby, goes to church, and has developed a social support circle. When it comes to her diet, she said “Now I just eat what I want to eat.”
Companion Diagnostic Test
Not applicable.
Anything Else?
The patients we interviewed on mirikizumab clearly show that a variety of treatment options, with diverse methods of administration and dosing schedules, are critical to have available, as ulcerative colitis presents differently in each person.
Conflict of Interest Declaration — Gastrointestinal Society
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
Yes. We are incredibly grateful for the time and input that we received from the patients who had direct experience with mirkizumab. We also have a wide range of individuals from across the country who respond to our surveys and requests for real information on what it is like to live with ulcerative colitis.
Did you receive help from outside your patient group to collect or analyze data used in this submission?
No.
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
Table 1: Financial Disclosures for Gastrointestinal Society
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Eli Lilly Canada Inc. | — | — | X | — |
Crohn’s and Colitis Canada
About Crohn’s and Colitis Canada
Crohn’s and Colitis Canada is the only national, volunteer-based health charity focused on finding the cures for Crohn’s disease and ulcerative colitis, the two main forms of inflammatory bowel disease (UC) and improving the lives of children and adults affected by these diseases.
Crohn’s and Colitis Canada is one of the top health charity funders of Crohn’s and colitis research in the world, investing over $140 million in research since our founding in 1974. The organization also delivers on its promise through patient programs, advocacy and awareness. We help improve the quality of lives today by:
Sharing accurate and reliable information on treatments, research and issues related to life with Crohn’s and colitis through website, print materials, webinars and live events.
Increasing public washroom access through the GoHere program.
Raising awareness about these Canadian diseases with bilingual public communication.
Offering kids with Crohn’s or colitis camp experience.
Providing a peer support program to newly diagnosed people; and
Advocating on behalf of the patients and caregivers on priority concerns and needs.
Crohn’s and Colitis Canada is comprised of approximately 65,000 supporters including volunteers, donors or individuals interested in engaging with the organization. There is no paid membership. Crohn’s and Colitis Canada is governed by a national volunteer Board of Directors. The organization has a network of volunteer-led Chapters in 46 communities across the country, offering information, events, fundraising opportunities and encouragement. There are thousands of volunteers from coast-to-coast supporting Crohn’s and Colitis Canada’s mission.
Crohn’s and Colitis Canada website (https://crohnsandcolitis.ca/
Information Gathering
Information summarized in this submission was compiled from two online surveys undertaken in 2022 and one phone interview with a patient who participated in the Mirikizumab clinical trial.
Survey 1: Our first survey was deployed to our community to better understand unmet needs and priority concerns. The survey included responses from 1706 Canadians, of which 354 had moderate to severe ulcerative colitis.
Survey 2: The goal of our second survey, also deployed last year, was to capture the experience of ulcerative colitis patients who participated in the Rinvoq clinical trial. We received a total of four responses, of which two participated in the Rinvoq clinical trial.
Our answers below are based on the responses from the respondents with moderate to severe ulcerative colitis.
Disease Experience
The results from the patient survey provide a window into how moderate to severe ulcerative colitis (UC) patients live and manage their symptoms. 78% of the respondents were female, 21% male and 1% non-binary.
When asked what UC related complications they are experiencing currently or within the past year, most frequently reported were mental health and stress (65%), followed by joint inflammation & arthritis (51%), anal fissures and hemorrhoids (40%), anemia (33%), and skin conditions and malnutrition and weight loss both at ~ 30%. Other complications include strictures, adhesions (scar tissue), bowel obstruction, eye inflammation, perianal or anal fistulas and abscesses, internal (or intra-abdominal) fistulas or abscesses, stricture, ankylosing spondylitis (arthritis of the spine), liver conditions, and cancer. 13% of the respondents were currently experiencing at least one complication of UC.
Thinking back to when they were first diagnosed, patients noted that they hid aspects of their diagnosis from friends, coworkers and classmates. There is a general misunderstanding of what UC is, which could impact how patients navigate social situations. Nine-in- ten agree that most people don’t know what UC is. This is further compounded by the fact that almost two thirds (63%) of patients agree that their family and friends don’t understand what they are going through. In spite of their medications, two thirds of the patients continue to experience at least one symptom of UC, the most frequent of which are bloating and urgent and frequent need to use the washroom. Over half (56%) believed that different treatment options could make them feel better. At least half of patients felt they could not be open about their UC, felt isolated due to their UC, and believe that their UC has had a negative impact on their romantic relationships with their spouse or partner.
A significant proportion of patients have adjusted their lifestyle and expectations. 72% agreed that they have changed the expectations they had of themselves or that they are always adapting their lifestyle to account for their UC. Two in five patients reported that they changed their travel plans and one in five changed their career aspirations.
The patient we spoke to for this survey, for example, reports that prior to getting on to Mirikizumab, her UC was “was increasingly really interfering with [her] work life and…general life as well” due to what she described as “many, many trips a day to the washroom.”
The patient continues:
“I live outside of Windsor and would go to Toronto and London fairly frequently and would be a worried from this stretch from, you know, Windsor to London about whether there were going to be washrooms along the way… I don't think I’d say that I had a lot of pain, but certainly discomfort. And that desperation of getting to a bathroom often. I tried different diets, and I tried getting rid of foods and doing those sorts of things. I have dogs who I love to walk, and I just couldn't do that because I just didn't know if there would be a washroom nearby and it would come on quite suddenly with very little warning. The impact on the quality of life is substantial, I think. [It] left me just feeling like, oh my gosh, can I travel?
Things ramped up for me early in the pandemic and so it was a little bit of a blessing… that I didn't have to worry about getting back and forth to an office every day. But I do often have to, you know, leave calls or mute myself, or whatever the case may be, and so it did get to the point where I actually took a leave from work. And that was, of course, financially significant because again, it felt like, you know, even with a very support supportive employer, I didn't want to be taking advantage of a of a sick leave.
I think the one last thing I would say would definitely affect some of my family life with my partner with my husband and my children just because they don't really get it either, right. Again, it's a very personal thing to talk about… it certainly has impacted my you know my sexual life with my husband because you know you're always feeling uncomfortable… so feeling intimate, being intimate is not something that has a really good link to colitis.”
Experiences With Currently Available Treatments
Disease management is incredibly important to ensuring patients can live a life of normalcy. Many patients have used a combination of medications to manage their disease, with systemic steroids and biologics being the most common ways (85% and 76% have taken these medications at least once). One third are currently on sulfasalazine & 5-aminosalicylates. Well over half are currently taking a biologic/biosimilar to help manage their UC, though it’s far more likely to be a biologic than a biosimilar. More than one in five are currently taking steroids (30% within last year). Roughly one third of the UC patients have also tried medical cannabis, anti- anxiety medications, and antidepressants to manage their symptoms.
Prior to getting on the study drug, the patient interviewed was taking Pentasa, Prednisone and Salofalk, which she reports having helped “but not to the degree of at all, even remotely close to” the way the study drug helped. “Additionally, having to, you know, administer an enema every single night was not fun. That's an understatement, to say the least. Especially if I was traveling or if I was at someone else's house or I had people over or whatever the case may be, I just found it very frustrating to try and think about the future of doing that. I never really noticed an effect per se of the Pentasa. They keep me on it, so it must have some maintenance kind of a of effect, but I couldn't say that it had changed things much for me.” While other patients we’ve spoken to for previous drug submissions have mentioned the size of the pill being a problem with Pentasa, this patient said “I’m fortunate to be one of those people who can take great big pills.” Of the prednisone, the patient had this to say: “I was put on a super high dose of Prednisone…And I found that really difficult. I felt horrible when I was on it, so we petered off a fair bit quicker than we might have otherwise.”
Steroid use is also an important aspect in symptom management and patients aren’t particularly supportive of this treatment option. Almost all patients surveyed agree that they only take systemic steroids if absolutely necessary (93%) with four in five in agreement that they wish they could eliminate systemic steroids from the list of medications they use. Half of respondents say that systemic steroids is/was a burden in their UC management. This is particularly true among those with moderate to severe forms of UC, and among women. Those under the age of 55 are more likely to agree that they have had side effects from systemic steroids. Those with a severe state of UC indicate that they have also experienced side effects from systemic steroid use (90%).
Among those who are using steroids 84% have been on systemic steroids for less than 12 months; with 42% less than 3 months. 13% of the respondents have been on steroids for over 1 year. Two thirds of the respondents feel that systemic steroids are a burden to their UC treatment, with 71% indicating that they have experienced side effects of the steroids.
Among patients who say managing medication use is important, having enough of their treatment options, understanding side effects, and minimizing steroid use were most important. Women are more likely than men to find it important to ensure they have enough treatment options, understand the side effects of long-term use, and minimize the use of steroids.
Improved Outcomes
Patients seek any treatments that can mitigate these symptoms to protect a patient’s ability to work productively, attend school and social events, and even basic daily necessities like leaving the house to run errands or have the energy to maintain a household or raise children. Quality of life could be greatly improved in UC patients if their flares are brought into remission. Based on our survey results, the majority of patients with moderate to severe UC continue to experience symptoms with current treatment options.
In terms of trade-offs, the patient we interviewed main concern in considering whether or not to participate in the clinical trial was around potential risks and side effects, “particularly side effects that could have long term health impacts, whether that was, you know, your heart or your liver or any of those things... minimizing side effects is a big one.” The patient was especially concerned about health risks as her mother “ended up dying in her mid-sixties” from “heart disease” that was in part “related to the many arthritis drugs she was on over time”.
Experience With Drug Under Review
Asked about the study drug Mirikizumab, the patient we interviewed said “It's been life changing for me.” While the drug hasn’t eliminated all of the symptoms of her UC and “every once in a while, [she] will get a bit frustrated”, she notes that she’s once again able to enjoy all the things she couldn’t do because of her UC:
“I wouldn't go on a vacation. I wouldn't travel 3 hours to see my closest friend. I wouldn't have done any of those things. It would be awkward to sit in a morning meeting at work prior to being on the drug. I can do those things now.
I do have to still do a little bit of planning and mornings are not my best time, but before it would be much worse in the mornings and then unpredictable throughout the course of the day… There’d be good days. There'd be days where I was only in the bathroom.
Yeah, six or seven times. And then there'd be days where I'd be in the bathroom 20 times or more. And so now, it's not like that at all. It's it might still be a few times in the morning, but then I know I can go. I can take my dogs out for a walk in the afternoon. I can take them out for a walk. In the evening. You know, there's lots of days I can take them out in the for a walk in the late morning, too.
There's a lot of times I've gotten in my car and, you know, driven to go get my treatment in the morning and before I would have to, you know, when I drive my 2 hours to London, I would probably stop three to five times on that drive. And there have been many times over the last year that I don't stop at all. Yeah, I go before I leave. I go when I get there. And yeah, that's again, I don't have to plan an extra hour into my 2-hour trip to make sure that I can, you know, get somewhere on time.
I've had no side effects, right? And that's like amazing, right? We can plan some vacations. We can go for a walk. I'm not laying in the in the bed all day and trying to, you know, work from my bed or you know, whatever the case may be. So I think they just have, you know, having someone who doesn't feel unwell for many days a week is a much nicer mom and wife. They understood, but there were many times when I would I'd get frustrated pre again pre premiere that that they didn't understand or didn't recognize what I was going for through or you know. But again, being a mom and a wife.”
Companion Diagnostic Test
It took a bit of time for me to get my first colonoscopy. That was pre COVID, so that wasn't a factor. So, it did take me longer than I think it probably should have. But it wasn't an undue delay. I had to have another colonoscopy but that actually happened in a more timely manner.
Anything Else?
Asked if there was anything else she’d like to add the patient said:
“Miri has been a life changer for me. It's hard sometimes to know what success necessarily looks like, but I would say if I had. If someone asked me, I absolutely have had great success on this medication. I’m thrilled that I have no side effects to deal with. And I would do it all over again in a second. And I'm hoping that it will be available for me to continue and that it continues to work for me. I would like to think that I'm gonna have a nice, healthy long life and not to do damage to my body that is avoidable so.
Conflict of Interest Declaration — Crohn’s and Colitis Canada
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission?
No.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
The first survey was conducted in collaboration with Leger who performed the initial analysis of the data.
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
Crohn’s and Colitis Canada receives grants, sponsorships and scholarship funding from pharmaceutical companies involved in the treatment of Crohn’s disease and ulcerative colitis. These funds are used to sponsor patient education events, community programs, research and medical conferences, educational brochures, kid’s camps, post-secondary scholarships as well as outreach and advocacy activities on behalf of Canadians living with Crohn’s and colitis. The vast majority of Crohn’s and Colitis Canada’s funding comes from individual donors contributing to fundraising events such as the Gutsy Walk. Crohn’s and Colitis Canada is participating in this review as part of our advocacy for Canadians living with inflammatory bowel disease and does not endorse or recommend the use of specific products or treatment or attribute of any product. No sponsor was involved in developing the content of this submission.
Table 2: Financial Disclosures for Crohn’s and Colitis Canada
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Lilly Canada | — | — | — | X |
Clinician Input
Canadian Gastroenterologists
About Canadian Gastroenterologists
Our clinician group (below referred to as the Group) consists of Canadian gastroenterologists who are specialists in the management of Inflammatory Bowel Disease (IBD), with representation from across the country. The Group is comprised of national and internationally recognized experts who collectively have published more than 1500 articles in the IBD field, as well as individuals who have authored multiple clinical trials for pivotal medications in IBD. Several of the authors have been involved in the Canadian Association of Gastroenterology (CAG) Consensus Guidelines on the treatment of Ulcerative Colitis (UC), have been part of international consensus publications on the management of UC and/or were contributors to the 2018 Crohn’s and Colitis Canada Impact of IBD in Canada report (2018).
Information Gathering
The Group reviewed a variety of sources in the literature for the summary of the UC disease state in Canada and consulted clinical practice guidelines for the management of UC, including those from Canadian and International expert groups. For each of the therapies discussed in the context of the current treatment landscape, the individual product monographs and primary clinical trial publications were reviewed. For the treatment under review (mirikizumab), the Group requested and was provided extensive clinical trial information from the manufacturer (Eli Lilly Canada, Inc.). The information from these published sources were supplemented with the Group’s clinical judgement and real-world experience in the treatment of patients with UC.
Current Treatments and Treatment Goals
Ulcerative Colitis in Canada
Approximately 230,000 Canadians suffer from IBD, which includes ulcerative colitis (UC) and Crohn’s disease (CD). Canada has the highest prevalence of IBD in the world [Crohn’s and Colitis Canada 2018]. By 2030, the number of people living with IBD is expected to rise to more than 400,000, or approximately 1% of the population [Coward 2018].
The focus of this review is UC, for which there are ~ 4,500 new cases of diagnosed each year. [Canadian Digestive Health Foundation 2023] UC is a chronic, idiopathic relapsing and remitting IBD characterized by contiguous areas of mucosal inflammation primarily affecting the rectum and colon. [Hanzel 2022, Danese 2019] The main symptom resulting from the inflammatory process is diarrhea, which may be associated with blood, cramping and urgency. [Feuerstein 2019] Severity of symptoms can range from mild disease, with fewer than four stools per day with or without blood, to severe disease with six or more bloody stools per day with severe cramps and systemic symptoms (fever, tachycardia, anemia, or elevated CRP). [Feuerstein 2019] As UC is a chronic but fluctuating disease, patients will go through periods in which the disease flares up, is active and causes disabling symptoms. Flares may be accompanied by decreased appetite, weight loss, fatigue and loss of work productivity. [Crohn’s and Colitis Canada 2018] Collectively these symptoms can lead to markedly impaired quality of life. Endoscopy is the gold standard in diagnosing and detecting active disease. [American Society for Gastrointestinal Endoscopy Standards of Practice Committee 2015] Extent of disease may be defined as involving the rectum only (proctitis), disease distal to the splenic flexure (left-sided UC) or extending proximal to the splenic flexure (extensive UC). [Bressler 2015, Raine 2022]
For the purposes of this review, the focus will be on moderate-to-severe UC. UC can be diagnosed at any age but often presents at a time when patients are pursuing employment, building families, and reaching key milestones. [Feuerstein 2019] People with UC may face several other challenges such as the stigma of a chronic illness that affects toileting habits interfering with activities of daily living, reduced employment opportunities, absenteeism, strained relationships and inequitable access to specialist care and delay to diagnosis. [Becker 2015, Argyriou 2017]
Historically, disease activity in UC has been assessed in clinical trials using tools that measure signs and symptoms of the disease and in clinical practice by subjective assessment of signs and symptoms. In clinical trials, disease activity is measured by the Modified Mayo Clinic Score (MCS). [Cooney 2007] This incorporates patient reported symptoms of rectal bleeding and stool frequency as well as objective measurement of disease activity through endoscopic evaluation.
However, the MCS is a limited point-in-time assessment and does not truly capture the severity of the disease. The score is not associated with prognosis or disease course (such as the need for hospital admissions or surgery). Risk factors that have been associated with a higher incidence of relapse or a more aggressive/complicated disease course include clinical factors (younger age, non-smokers, prior hospitalization for UC), laboratory markers (low hemoglobin, high C-reactive protein (CRP) and high fecal calprotectin levels) and disease location (extensive colitis). [Monstad 2021, Qiu 2019]
Costs of Treatment
Costs can be considered as direct costs as well as indirect costs. Direct costs of IBD are mainly related to the costs of medications, diagnostic testing, in-patient care and outpatient consultations. Collectively, direct costs of IBD were estimated to be at least CAD $1.28 billion in 2018. [Crohn’s and Colitis Canada 2018] A major cost driver is hospitalization, which is often due to incomplete control of the disease with currently available therapy. In a Canadian population-based study, 2.3% of hospitalized IBD patients were re-hospitalized within one month of discharge, while 5.6% were readmitted to hospital within six months and 7.7% within 12 months. [Nguyen 2014] Hospitalization for UC can be associated with the need for emergency colectomy. In a systematic review and meta-analysis of population-based studies, 1-, 5-, and 10-year risks of colectomy in persons with UC was 4.0% (95% CI, 3.3-5.0), 8.8% (95% CI, 7.7-10.0), and 13.3% (95% CI, 11.3-15.5), respectively. [Tsai 2021]
In addition to the aforementioned direct costs, there are several indirect costs associated with IBD. These include caregiver costs, absenteeism, presenteeism, and premature retirement. In a survey study of persons with IBD in Manitoba, reduced workplace productivity during the previous 14 days was reported by 37% of individuals, including a reduction for 1-2 days by 18% of patients, for 3-9 days by 16% of patients, and on most days by 3% of patients. [Shafer 2019] Working persons with IBD may expect to miss an additional 3.5 to 7.5 days from work annually due to illness compared to non-IBD persons. In 2018, it was estimated that there are 97,809 Canadian working-age adults (age 18- 64) with CD. [Statistics Canada 2017] Based on an estimated workforce rate of 68% among persons with CD living in Canada, roughly 66,510 persons would be eligible to experience medical absenteeism. [Rogala 2008]
Based on sick days, short-term disability, premature retirement, and out-of-pocket expenses, the total indirect health-related cost to the Canadian economy due to IBD was estimated to be close to CAD $1.3 billion in 2018, or roughly CAD $4,781 per person with IBD. Importantly, this estimate does not consider presenteeism costs, caregiver costs, and costs of reduced professional development. The estimated annual cost due to medical absenteeism is speculated be as high as CAD $1.6 billion. [Crohn’s and Colitis Canada. 2018]
Current Treatments for Moderate-to-Severe UC
UC is treated with various therapies influenced by disease characteristics. These include 5-aminosalicylates (5-ASA), corticosteroids, immunosuppressants, biologics (which include anti-TNF therapy, anti-integrin, and anti-IL-12/23) and advanced small molecule drugs (SMDs, which include Janus kinase [JAK] inhibitors and sphingosine-1-phosphate receptor [S1PR] modulators). [Rubin 2019, Feuerstein 2020, Paik 2022] Table 1 lists the advanced therapies (biologic agents and small molecules) approved for use in or under Health Canada review for UC in Canada.
Table 3: Advanced Therapies Approved or Under Review in Canada for the Treatment of Moderate-to-Severe Ulcerative Colitis
Drug | Mechanism of action | Approved indication in UC | Route of administration | Efficacy | Common side effects |
|---|---|---|---|---|---|
Anti-TNF | |||||
Adalimumab1 | Binds TNFα and blocks interaction with p55 and p75 cell surface TNF receptors | SC | Significant increase in clinical remission at 8 and 52 weeks with adalimumab compared to placebo | Injection site erythema/pruritus/reaction, malaise, peripheral edema, fatigue | |
Golimumab2 | Binds to soluble and transmembrane TNF, preventing binding to TNF receptors | Treatment of moderate-severe UC with inadequate response to or are intolerance of conventional therapy | Significant increase in clinical remission at 6 and 54 weeks with golimumab compared to placebo | Anemia, injection site reactions, upper respiratory tract infection | |
Infliximab3 | Binds soluble and transmembrane TNFα and inhibits binding to TNF receptors | IV | Significant increase in clinical remission at 8 and 54 weeks with infliximab compared to placebo | Upper respiratory tract infection, arthralgia, abdominal pain | |
Integrin-blocker | |||||
Vedolizumab4 | Binds to α4β7 integrin on pathogenic gut-homing lymphocytes and inhibits binding to MAdCAM-1, interfering with inflammation pathways in the GI tract | Treatment of moderate-severe UC with inadequate response to or intolerance of conventional therapy or infliximab | IV | Significant increase in clinical remission at 6 and 52 weeks with vedolizumab compared to placebo | Nasopharyngitis, arthralgia, headache, upper respiratory tract infection, fatigue, cough |
IL-12/IL-23p40 | |||||
Ustekinumab5 | Binds to the p40 protein subunit of interleukin IL-12 and IL-23, interrupting cytokine pathways in the pathogenesis of UC | Treatment of moderate-severe UC with inadequate response to or intolerance of conventional or biologic therapy | SC | Significant increase in clinical remission at 8 and 52 weeks with ustekinumab compared to placebo | Nasopharyngitis, headache, vomiting, arthralgia, fatigue, dizziness, oropharyngeal pain |
IL-23/IL-17 | |||||
Risankizumab6 | Binds to the p19 subunit of IL-23, inhibiting signalling pathways, including the release of pro-inflammatory cytokine IL-17 | No indication for UC at this time | SC | Trials underway, results expected 20237 | Arthralgia, headache, abdominal pain, nasopharyngitis, nausea, pyrexia7 |
Guselkumab8 | Significant increase in clinical remission at 12 weeks with guselkumab compared to placebo9 | Diarrhea, injection site reaction, upper respiratory infection, gastroenteritis, herpes simplex infection, arthralgia, headache | |||
JAK | |||||
Tofacitinib10 | Inhibits Janus kinases (JAK), blocking cytokine signalling required for lymphocyte activation, proliferation and function | Treatment of moderate-severe UC with inadequate response to or intolerance of conventional therapy or TNFα inhibitor | PO | Significant increase in clinical remission at 8 and 52 weeks with tofacitinib compared to placebo | Nasopharyngitis, arthralgia, headache, upper respiratory tract infection, rash, hypercholesterolemia, herpes zoster, increased blood creatine phosphokinase |
Upadacitinib11 | Under HC review | PO | Significant increase in clinical remission at 8 and 52 weeks with upadacitinib compared to placebo12 | Nasopharyngitis, elevated creatinine phosphokinase, acne, worsening of UC, arthralgia, upper respiratory tract infection12 | |
Sohingosine-1 | phosphate receptor | modulators | |||
Ozanimod13 | Binds to sphingosine 1-phosphate receptors on lymphocytes, preventing egress from lymph nodes and reducing number of lymphocytes in peripheral blood | Treatment of moderate-severe UC with inadequate response to or intolerance of conventional or biologic therapy | PO | Significant increase in clinical remission at 10 and 52 weeks with tofacitinib compared to placebo | Increase in liver function tests, nasopharyngitis, headache |
1AbbVie. Humira. Product Monograph. Sept 24, 2004, updated Sept 16, 2022.
2Janssen. Simponi. Product Monograph. April 7, 2009, updated Sep 9, 2022.
3Janssen. Remicade. Product Monograph. Jun 6, 2001, updated Oct 7, 2022.
4Takeda. Entyvio. Product Monograph. Jan 29, 2015, updated Jul 6, 2022.
5Janssen. Stelara. Product Monograph. Dec 12, 2008, updated January 5, 2023.
6AbbVie. Skyrizi. Product Monograph. Apr 17, 2019, updated Jan 25, 2023.
7Parigi TL, Iacucci M, Ghosh S. Blockade of IL-23: What is in the Pipeline? J Crohns Colitis. 2022 May 11;16(Supplement_2):ii64-ii72.
8Janssen. Tremfya. Product Monograph. Nov 10, 2017, updated Nov 8, 2022.
9The Efficacy and Safety of Guselkumab Induction Therapy in Patients With Moderately to Severely Active Ulcerative Colitis: Phase 2b QUASAR Study Results Through Week 12. Gastroenterol Hepatol (N Y). 2022 Apr;18(4 Suppl 1):12.
10Pfizer. Xeljanz. Product Monograph. April 16, 2014, updated May 9, 2022.
11AbbVie. Rinvoq. Product Monograph. Dec 23, 2019, updated Aug 2, 2022.
12Danese S, Vermeire S, Zhou W, et al. Upadacitinib as induction and maintenance therapy for moderately to severely active ulcerative colitis: results from three phase 3, multicentre, double-blind, randomised trials. Lancet. 2022 Jun 4;399(10341):2113-2128.
13Celgene. Zeposia. Product Monograph. Oct 2, 2020, updated Apr 7, 2022.
Clinical Practice Guidelines
Canadian guidelines in UC were last published in 2015 with The Toronto Consensus. [Bressler 2015] Given the emergence of several new therapeutic options in the past decade, these guidelines will need updating. The American Gastroenterology Association (AGA), the American College of Gastroenterology (ACG), and the European Crohn’s and Colitis Organisation (ECCO) have all published newer guidelines since then. [Feuerstein 2020, Rubin 2019, Raine 2022] The treatment information below draws from these guidelines, supplemented by more recent clinical trials, and is also a reflection of today’s practice in Canada.
The traditional approach is to treat patients with corticosteroids during periods of disease flare to reduce symptoms and induce remission. These drugs are not prescribed on a long-term basis due to side effects and poor effectiveness for maintaining remission. The need for corticosteroids is usually an indication for the need of more advanced therapies. In patients who are refractory to corticosteroids, or corticosteroid dependent, biologics are typically initiated.
Corticosteroids
Prednisone is an effective medication in patients with moderately to severely active UC. Methylprednisolone can be given intravenously (IV) in patients who are hospitalized. [Bressler 2015] These medications non-specifically suppress the immune system. However, corticosteroids do not maintain clinical remission and have not been shown to induce mucosal healing. The Group agreed that these are effective agents for the treatment of symptoms but are associated with significant short- and long-term side effects. They should not be used as a maintenance medication. Moreover, the Group agreed that corticosteroids do not meet modern treatment goals, which include endoscopic remission and mucosal healing.
Immunosuppressants
Immunosuppressants for UC include the purine analogs azathioprine or 6-mercaptopurine (6-MP), which may be used to help decrease corticosteroid dependency and may help maintain disease remission. [Bressler 2015] Meta-analyses of trials investigating these therapies in UC have demonstrated significant improvements compared to placebo for maintenance of remission. [Gisbert 2009, Khan 2011, Timmer 2016] A recent small, randomized, placebo-controlled trial showed that optimized mercaptopurine treatment was superior to placebo for clinical, endoscopic and histological outcomes at one year following corticosteroid induction treatment. [Löwenberg 2023] Thiopurine monotherapies are not authorized by Health Canada for the treatment of IBD. [Marshall 2014]
These immunosuppressant therapies are associated with tolerability issues in the short term and potential for serious toxicity or complications with long-term use. [Bressler 2015] The Group agreed that while these agents may be effective for corticosteroid-dependent disease, they may not be as effective as advanced therapies (i.e., biologics, novel small molecules) at meeting modern treatment goals, which include endoscopic remission and mucosal healing, and have not been shown to decrease the rates of hospitalization or surgeries.
Biologics
Anti-TNF (Infliximab, Adalimumab, Golimumab)
Anti-TNF therapy has been able to effectively induce and maintain remission while sparing corticosteroids. They work in a wide variety of patients including those with extra-intestinal manifestations and hospitalized patients with acute severe colitis. They have also been associated with mucosal healing, and for those who respond a reduction in hospitalization and surgery rates has been demonstrated. The following describes the key results from the pivotal trials in ulcerative colitis with the three anti-TNF agents approved for UC in Canada.
Infliximab: The efficacy and safety of infliximab for UC was established by two pivotal trials, ACT 1 and ACT 2. [Rutgeerts 2005] These were randomized, double-blind, placebo-controlled studies evaluating infliximab for induction and maintenance therapy in adults with moderate-to-severe active UC despite treatment with concurrent medications.
In both studies, infliximab was found to be superior to placebo for clinical response, clinical remission, and mucosal healing at weeks 8 and 30, as well as corticosteroid-free remission at week 30. In ACT 1, which had a longer duration than ACT 2, infliximab was also associated with a significantly higher rate of clinical remission and corticosteroid-free remission at week 54 compared to placebo.
Adalimumab: The second anti-TNF agent to be approved for the treatment of UC in Canada was adalimumab. Its efficacy and safety were established by the pivotal ULTRA I and ULTRA II trials. [Reinisch 2011, Sandborn 2012]
ULTRA I evaluated adalimumab vs. placebo for induction, while ULTRA II evaluated adalimumab vs. placebo for both induction and maintenance. In both studies at 8 weeks, adalimumab was associated with significantly higher clinical remission rates compared to placebo. In ULTRA II, adalimumab was found to be superior to placebo for clinical remission, corticosteroid-free remission and mucosal healing at week 52.
Golimumab: The PURSUIT studies were the pivotal trials establishing efficacy and safety of golimumab in UC. [Sandborn 2014a, Sandborn 2014b.]
In the PURSUIT-SC (induction) study, rates of clinical response, clinical remission and mucosal healing were significantly higher in the golimumab groups relative to placebo at week 6. In the PURSUIT-maintenance study, golimumab was found to be superior to placebo for clinical response, clinical remission and mucosal healing at weeks 30 and 54.
Group Opinion on Anti-TNF Therapy: The group agreed that anti-TNF therapy has been a significant advance in the treatment of UC. However, these medications have limitations, which include immunogenicity, the need for combination with immunomodulators, and loss of response over time. [Bressler 2015; Roda 2016] These agents have also been associated with increased risk of infections, as well as a risk of drug-induced lupus, paradoxical psoriasiform reactions and arthritis. [Yanai 2013, Alivernini 2018, Xie 2022] Furthermore, in clinical practice, the Group agreed that up to half of patients are receiving doses higher than the labelled dosing.
Anti-Integrin (Vedolizumab)
Vedolizumab is a biologic approved for use in moderate to severe UC. Vedolizumab blocks α4β7- integrin on the surface of lymphocytes, thus interrupting their homing to inflamed tissue in the gut. [Soler 2009] By blocking this migration it helps reduce inflammation. Vedolizumab is described as a gut-selective therapy, as α4β7-expressing lymphocytes only home to the gut.
The pivotal placebo-controlled trial establishing the efficacy and safety of vedolizumab in UC was the GEMINI 1 trial. [Feagan 2013] In the induction phase of the study, vedolizumab was superior to placebo for clinical response, clinical remission and mucosal healing at week 6. In the maintenance phase, vedolizumab demonstrated significantly higher rates of clinical remission, corticosteroid-free remission and mucosal healing at week 52.
A post-hoc analysis of the GEMINI 1 study showed that while vedolizumab was superior to placebo for both anti-TNF-naïve and anti-TNF-experienced subjects, the treatment effect was greater among the anti-TNF-naïve subgroup for most outcomes. [Feagan 2017]
Group Opinion on Vedolizumab Therapy: The Group agreed that vedolizumab is a highly effective treatment for biologic-naïve patients with moderately to severely active UC. Its major strength is its safety profile due to its gut selectivity. Limitations include its efficacy in patients with extra-intestinal manifestations, and its reduced efficacy in anti-TNF-exposed patients.
Anti-IL 12/23 (Ustekinumab)
Initially used for the treatment of psoriasis and psoriatic arthritis, ustekinumab is also approved for use in moderate to severe UC. Its efficacy and safety in UC were established by the pivotal, placebo-controlled UNIFI trial. [Sands 2019] In the induction period of this study, ustekinumab demonstrated superiority relative to placebo for clinical response, clinical remission, endoscopic improvement and mucosal healing at weeks 8 and 16. At week 44, ustekinumab demonstrated significantly higher rates of clinical remission, corticosteroid-free remission and mucosal healing relative to placebo. Subgroup analyses showed that the benefit of ustekinumab was seen relative to placebo for both biologic-naïve individuals and those who had been previously treated with anti-TNF therapy and/or vedolizumab.
Group Opinion on Ustekinumab Therapy: The Group agreed that ustekinumab is effective in treating patients with moderately to severely active UC, both those who are naïve to biologic therapy and those exposed to anti-TNF therapy. Its strength is a balance between efficacy and safety. Like anti-TNF therapy, the experience across the Group was that in clinical practice a substantial proportion of patients required dosing higher than the approved regimen, although the proportion is not perceived to be as high in UC as it is in CD.
JAK Inhibitors
JAK inhibitors are SMDs that exert therapeutic effects in UC through the modulation of key inflammatory cytokines. [De Vries 2017] Tofacitinib was the first of these agents to be approved for UC in Canada. A second JAK inhibitor, upadacitinib, is currently (March 2023) undergoing regulatory review by Health Canada.
Tofacitinib: This agent’s efficacy and safety in UC were established by the OCTAVE studies. OCTAVE-1 and -2 were 8-week induction studies and responding patients from both of these trials moved forward into the OCTAVE-Sustain maintenance trial. [Sandborn 2017] In both induction studies, tofacitinib was found to be superior to placebo in terms of clinical remission and mucosal healing at 8 weeks. In the maintenance phase, tofacitinib was associated with significantly greater rates of clinical remission, corticosteroid-free remission and mucosal healing compared to placebo at week 52.
Upadacitinib: The upadacitinib pivotal trial program was similar to that of tofacitinib: two replicate induction studies (U-ACHIEVE induction and U-ACCOMPLISH) feeding into one maintenance study (U-ACHIEVE maintenance). [Danese 2022] In the induction studies, upadacitinib was associated with significantly greater rates of clinical response, clinical remission and bowel urgency at week 8. Notably, there was a large and significant treatment effect as early as week 2, with post hoc analyses demonstrating a significant symptomatic response as early as Day 1. [Loftus 2022] In maintenance, upadacitinib demonstrated superior efficacy compared to placebo for clinical remission, corticosteroid-free remission, endoscopic improvement and mucosal healing at week 52.
Group Opinion on JAK Inhibitor Therapy: The Group agreed that JAK inhibitors are important agents for the management of moderate-to-severe UC. They offer clinicians and their patients an oral option for advanced therapy and are associated with an excellent efficacy profile with a rapid onset of action. The Group also agreed that because these agents are systemic immune modulating small molecules, their off-target side effects and safety concerns (e.g., increased risk of thromboembolism, infection, and hyperlipidemia) are the key limitations of these therapies. [Agrawal 2020]
S1P Receptor (S1PR) Modulator — Ozanimod
Initially used for the treatment of multiple sclerosis, ozanimod is thought to exert its therapeutic effects in UC by preventing the mobilization of lymphocytes to sites of active inflammation. [Choden 2022]
The pivotal clinical trial for ozanimod in UC was the True North study, which included evaluation of ozanimod for both induction and maintenance. [Sandborn 2021a] In the induction period, ozanimod demonstrated superior efficacy relative to placebo for clinical response, clinical remission, endoscopic improvement and mucosal healing at week 10. In maintenance, ozanimod was associated with significantly higher rates of clinical remission, corticosteroid-free remission, endoscopic improvement and mucosal healing at week 52.
Group Opinion on Ozanimod Therapy: The Group agreed that ozanimod is another useful oral advanced therapy option for UC. The Group did, however, agree that a major limitation of this therapy has been limited efficacy in biologic-exposed patients. Also, like the JAK inhibitors, ozanimod’s key limitations are safety concerns (e.g., serious infection, elevated transaminases, need for cardiac assessment and for eye assessment in at-risk individuals). [Khanna 2022]
Surgery
Surgery is necessary in UC when medications prove ineffective, for fulminant disease which is medically refractory, or if complications arise, such as dysplasia or strictures. This typically results in a proctocolectomy with either a subsequent ileo-anal pouch formation or permanent end ileostomy. The Group agreed that surgery should be performed in patients who have developed complications or who have medically refractory disease. Notably, the ongoing need for surgery in patients with UC reflects the ongoing need for medical therapies that can prevent complications and achieve all our treatment goals.
Treatment Goals
The goals of therapy in UC have evolved from simply controlling symptoms to preventing disease progress, surgery, and disability with early intervention and a treat-to-target approach. [Sandborn 2021b]
The STRIDE II guidelines describe the multifaceted goals of contemporary UC management, divided into immediate (short-term), intermediate-term and long-term goals. [Turner 2021]
Immediate (Short-Term) Goals
The STRIDE II group states that clinical response is the short-term goal for individuals with UC. This is defined as a decrease of at least 50% in PRO2 (rectal bleeding and stool frequency). In children, a decrease in the pediatric UC activity index (PUCAI) of at least 20 points is also recommended as a short-term goal.
Intermediate-Term Goals
STRIDE II recommends that the next goal along the timeline of the management of UC is clinical remission. This is defined by PRO2 (rectal bleeding =0 and stool frequency =0) or partial Mayo (<3 and no score >1), and in children PUCAI <10 points. Normalization of key biomarkers (i.e., C-reactive protein and fecal calprotectin) are also listed as intermediate-term goals.
Long-term Goals
Endoscopic healing (assessed by sigmoidoscopy or colonoscopy: Mayo endoscopic subscore = 0 points, or UC endoscopic index of severity [UCEIS] ≤1 points) is a key long-term goal in the management of UC. Additionally, absence of disability and normalized health-related quality of life are long-term treatment targets. In children, restoration of normal growth is also listed as a long-term goal.
While histologic remission is not listed as a specific target, STRIDE II does state that it can be “used as an adjunct to endoscopic remission to represent a deeper level of healing.”
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
Several unmet needs remain in the treatment of moderate to severe UC. There is need for a therapy that rapidly induces symptomatic remission, can be used to maintain symptomatic remission, and is safe with long-term use. In addition, there is need for a therapy that can also rapidly improve the endoscopic appearance of the bowel and maintain this in the long-term. The presently available therapies have inherent limitations, discussed below. The Group also indicated that up to half of patients under their care with currently available agents have undergone dose escalation to levels that are off label to achieve control in UC and that this can extend the time required to achieve an optimal response.
Limitations Associated with Current Treatments
Corticosteroids
Corticosteroids are typically prescribed orally for UC, but safety is a concern, especially with long term use. As such, corticosteroid therapy is not recommended for maintenance therapy. The Group also outlined short-term impacts (e.g., skin thinning, acne, hirsutism, cushingoid or ‘moon face’ appearance) as well as the potential for neuropsychological impact (e.g., hypomania, psychosis). The Group advised against the use of corticosteroids for maintenance therapy but recognized that treatment failure on prednisone is still required on most formularies prior to prescribing a biologic therapy.
Immunosuppressants
Thiopurines are recognized for their steroid-sparing effects. However, they have a slow onset of action (8-12 weeks), which makes them ineffective as induction agents. The Group noted that tolerability issues, inferior efficacy compared to advanced therapies and delayed onset of action are limitations. Added to these are concerns over increased risk of infections, purine pancreatitis, myelosuppression, hepatotoxicity, and malignancy (particularly lymphoma and non-melanoma skin cancer). The Group agreed that immunosuppressants do not meet modern treatment goals for UC, which include endoscopic remission and mucosal healing, and have never been shown to decrease the rates of hospitalization or surgeries.
Biologics
Anti-TNF (Infliximab and Adalimumab): The Group has encountered patient reluctance when prescribing the anti-TNF biologics infliximab and adalimumab due to perceptions of safety concerns. The Group also noted that dose escalation is often required in a substantial proportion of patients to gain disease control, which draws out the management journey for patients. It was also recognized that up to one-third of patients do not respond to this class of therapy at all, and there is a risk for the development of immunogenicity.
The Group agreed that anti-TNF therapy has been a significant advance in the treatment of UC. However, they have limitations which include immunogenicity, the need for combination with immunomodulators, and loss of response over time. Furthermore, the Group identified that between 30-50% of patients in their practices are receiving doses that are outside of the labelled dosing. The Group speculated whether this was due to the limited data available for achieving mucosal healing or whether this is due to a lack of a broad dosing range being evaluated in earlier phases of drug development.
Anti-Integrin (Vedolizumab): The Group agreed that vedolizumab is highly effective and safe in treating bio-naïve patients with moderately to severely active UC. Its biggest strength is its safety profile due to its gut selectivity. The major limitations were considered to be its limited utility in patients who had previously been exposed to anti-TNF therapy, and the substantial number of patients with UC who have other concomitant immune-mediated inflammatory diseases that would not benefit from this gut-specific therapy.
Anti-IL 12/23 (Ustekinumab): The Group agreed that ustekinumab is effective in treating patients with moderately to severely active UC, both in patients naïve to biologic therapy and those previously exposed to anti- TNF. Its strength is a balance between efficacy and safety. However, in the Group’s experience ustekinumab shows sub-optimal efficacy on endoscopic endpoints and is often used at higher off-label doses to achieve remission. The Group noted that, like anti-TNF therapy, the current on-label dosage of this biologic is often inadequate to achieve symptomatic and endoscopic outcomes without a dose escalation. According to the Group, this habitual need for off-label dose escalation occurs in up to 30-50% of patients and can extend the time required to optimize response.
Small Molecule Drugs
JAK Inhibitors: The Group agreed that JAK inhibitors are important agents for the management of moderate-to-severe UC. They offer clinicians and their patients an oral option for advanced therapy and are associated with an excellent efficacy profile with a rapid onset of action. The Group also agreed that because these agents are systemic immune modulating small molecules, their off-target side effects and safety concerns (e.g., increased risk of thromboembolism, infection, and hyperlipidemia) are the key limitations for widespread use.
S1P Modulator — Ozanimod: The Group agreed that ozanimod is another useful oral advanced therapy option for UC, although it was recognized that it has limited efficacy in biologic-exposed patients. Like the JAK inhibitors, ozanimod’s other limitations include safety concerns (e.g., serious infection, elevated transaminases). The Group agreed that safety concerns make this agent undesirable for use among older patients with UC (who represent a significant proportion of the UC patient population).
Summary of Unmet Needs in UC: Clinical development of advanced therapies for the treatment of UC began with anti-TNF agents, which were the main therapeutic agent for moderate-to-severe UC for more than a decade. Later, other biologic agents were approved and came into clinical use, including vedolizumab, a gut-selective antibody to α4β7- integrin; and ustekinumab, an antibody against the p40 subunit of interleukin (IL)-12/23. Development of monoclonal antibodies has been complemented by emerging small molecules, which are conveniently administered orally and avoid immunogenicity, a factor which somewhat limits the efficacy of TNF antagonists. Small molecules approved for the treatment of UC include JAK inhibitors (tofacitinib and upadacitinib) and sphingosine-1-phosphate receptor (S1PR) modulators (ozanimod). None of the available therapies meet all of the current needs of patients in the short or long-term. Remission with treatment is not universal and patients can lose response after an initial period of improvement and relapse even when in deep remission on an existing therapy. Thus, there is a need for novel therapies targeting alternative pathways.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Mechanism of Action
The IL-23 pathway is an important target for drug development in autoimmune disease. Following the approval of ustekinumab, an antibody blocking the p40 subunit of both IL-12 and IL-23, first for CD and subsequently UC, a number of more selective agents only blocking the p19 subunit of IL-23 entered clinical development. Mirikizumab (LY3074828; Eli Lilly) is a humanized IgG4 monoclonal antibody against the p19 subunit of IL-23.
Mirikizumab for Induction in UC
The LUCENT-1 study (NCT03518086) assessed the efficacy and safety of mirikizumab as induction therapy in patients with moderate-to-severe UC [D’Haens 2022] In this Phase 3, multicenter, randomized, double-blind, placebo-controlled study, patients with UC were randomized 3:1 to receive mirikizumab 300 mg intravenously every 4 weeks or placebo. Randomization was stratified by prior biologic failure, baseline corticosteroid use, baseline disease activity by the modified Mayo score, and geographic region. The study excluded patients who had failed three or more biologic therapies or had been exposed to an anti-IL-12/23 agent.
The trial included a total of 1162 patients (868 randomized to mirikizumab, 294 to placebo). The mean disease duration was 7 years, about half of the patients had a baseline modified Mayo score ≥7, and approximately two thirds of patients had endoscopically severely active disease (Mayo endoscopy score of 3). Sixty percent of the patients had not previously failed biologics or tofacitinib.
The primary endpoint of the induction study was clinical remission at week 12, defined by the modified Mayo score. At week 12, clinical remission was significantly higher among patients receiving mirikizumab compared to placebo (24.2% [210/868] vs. 13.3% [39/294], p<0.001). Biologic-naïve patients achieved clinical remission in 30.9%, while patients with previous biologic failure achieved clinical remission in 15.2%. Mirikizumab was also found to be superior to placebo in achieving endoscopic improvement and histo-endoscopic mucosal improvement. Endoscopic improvement was achieved by 36.3% (315/868) in the mirikizumab group and 21.1% (62/294) in the placebo group (p<0.001). Histo-endoscopic mucosal improvement was achieved by 27.1% (235/868) in the mirikizumab group and 13.9% (41/294) in the placebo group (p<0.001).
Mirikizumab for Maintenance in UC
Patients with clinical response to mirikizumab at week 12 were re-randomized 2:1 to mirikizumab 200 mg subcutaneously every 4 weeks or placebo in the LUCENT-2 maintenance study [Dubinsky 2022] The maintenance study included 365 patients randomized to mirikizumab and 179 patients randomized to placebo. Clinical remission was assessed at week 40 of the maintenance study, i.e., at week 52 since the first intravenous induction dose.
Clinical remission was attained by 49.9% (182/365) of patients re-randomized to mirikizumab and 25.1% (45/179) of patients re-randomized to placebo (p<0.001). The corresponding rates of corticosteroid-free remission were 44.9% (164/365) and 21.8% (39/179), respectively. Histo-endoscopic mucosal improvement (the definition was more stringent than in the induction study as no neutrophils were allowed in the epithelium or lamina propria) was achieved by 43.3% (158/365) in the mirikizumab group and 21.8% (39/179) in the placebo group (p<0.001). Rates of both clinical remission and endoscopic improvement were numerically lower in patients with prior failure of advanced therapies, with a numerically greater decrease in endoscopic (62.4% vs. 50.8%) compared to clinical outcomes (51.5% vs. 46.1%).
The results of LUCENT-1 and -2 show that selective blockade of IL-23 with mirikizumab demonstrates efficacy in patients with moderate-to-severe UC refractory to conventional therapy or one or more lines of biologic therapies. The results are notable especially in light of the refractory patient population studied. In addition, there was a large proportion of patients that responded to therapy making them eligible for maintenance therapy. This efficacy did not come at the expense of safety over 52 weeks of therapy.
Overall, the clinical trial outcomes indicate that a broad range of use is possible in clinical practice – from first line advanced therapy to treatment of patients with inadequate response or intolerance to multiple advanced therapies. Most of the Group indicated that it may be a treatment of choice in most patients with UC due to the favorable benefit-risk ratio and its ability to achieve the endoscopic endpoints early and during maintenance.
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
There was general consensus among the Group members that the high efficacy and overall safety of mirikizumab, apparent lack of need for dose escalation, and good endoscopic mucosal healing data, helps to positions this agent in UC as a good candidate to help address unmet needs in the treatment landscape as a potential first-line option or for individuals who have failed other advanced therapies. The lower risk of inadequate response with this agent will help eliminate costs currently incurred from dose adjustments required to optimize response with available biologic therapies. Together with an excellent safety profile and low rate of immunogenicity, this will also reduce the burden on healthcare professionals needing to monitor patients more frequently as is done with many conventional therapies with higher adverse event risk profiles. The relatively fast speed of response with mirikizumab is also key for patient choice.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
The mirikizumab development program aligns closely with what is done in clinical practice and aligns with disease management strategies outlined in STRIDE-II (please refer to section above). The Group recommends that management strategies strive for remission, which is defined as both symptomatic and endoscopic remission and align with Canadian Consensus Guidelines. In the first three months of therapy, a meaningful improvement in symptoms as measured by resolution of stool frequency and rectal bleeding should be demonstrated. Patients would be expected to be in symptomatic remission and off corticosteroids by six months. Symptomatic improvement should be accompanied by a decrease in biomarkers of inflammatory activity (C-reactive protein and fecal calprotectin) in the first three months. Although the LUCENT clinical trial program demonstrated robust endoscopic response with mirikizumab in the first three months, the Group may not assess endoscopic activity until 9-12 months in clinical practice.
What factors should be considered when deciding to discontinue treatment with the drug under review?
The Group would recommend discontinuing treatment with mirikizumab if there is worsening of symptoms or if there is an inadequate response. In circumstances where there is an inadequate response to mirikizumab as a first line biologic, a switch to another class of agents, such an anti-TNF agent is warranted.
What settings are appropriate for treatment with [drug under review]? Is a specialist required to diagnose, treat, and monitor patients who might receive [drug under review]?
In the clinical experience of the Group, mirikizumab would need to be administered in clinic by a trained health care professional during the induction phase.
Additional Information
Canada has one of the highest prevalence rates of UC in the world. [Crohn’s and Colitis Canada 2018] Despite the treatment landscape being revolutionized by the advent of several biologics and small molecules, there remains a significant unmet need. Relapse can occur even in patients with prolonged states of remission. With the anti-TNF agents up to 30% of patients are primary non- responders when they are prescribed as first line therapy, and up to 40% experience a loss of response in the first year during maintenance treatment. [Papamichael 2015] With vedolizumab, the pooled incidence of loss of response is 47.9 per 100 patient-years [Peyrin-Biroulet 2019] There are limited data available for loss of response with ustekinumab in UC; however in CD, up to 34% of patients on ustekinumab experienced a loss of response to maintenance therapy at a median of 47.4 weeks [Ma 2017]. In addition, with currently available biologics there is frequently a need for dose escalation to levels that are off label to achieve treatment response in UC. This is a reality in clinical practice for an estimated 30-50% of patients Canada wide. This results in delays in the patient treatment journey and a deleterious effect on the patient-physician relationship, while driving up costs of care.
The IL-23p19 inhibitor mirikizumab provides sustained clinical and endoscopic outcomes across different patient populations when used as induction and maintenance therapy. This has the potential to improve important outcomes in the care of patients with UC, including reduction in hospitalizations and surgical interventions, as well as reduction in steroid exposure. The favourable efficacy, safety, and durability profile of mirikizumab position it well as an option for first line therapy in moderate-to-severe UC.
Bibliography
Agrawal M, Kim ES, Colombel JF. JAK Inhibitors Safety in Ulcerative Colitis: Practical Implications. J Crohns Colitis. 2020 Aug 1;14(Supplement_2):S755-S760.
Alivernini S, Pugliese D, Tolusso B, et al. Paradoxical arthritis occurring during anti-TNF in patients with inflammatory bowel disease: histological and immunological features of a complex synovitis. RMD Open. 2018 Apr 9;4(1):e000667.
American Society for Gastrointestinal Endoscopy Standards of Practice Committee; et al. The role of endoscopy in inflammatory bowel disease. Gastrointest Endosc. 2015 May;81(5):1101-21.
Argyriou K, Kapsoritakis A, Oikonomou K, et al. Disability in Patients with Inflammatory Bowel Disease: Correlations with Quality of Life and Patient's Characteristics. Can J Gastroenterol Hepatol. 2017;2017:6138105.
Becker HM, Grigat D, Ghosh S, et al. Living with inflammatory bowel disease: A Crohn's and Colitis Canada survey. Can J Gastroenterol Hepatol. 2015 Mar;29(2):77-84.
Bressler B, Marshall JK, Bernstein CN, et al. Clinical practice guidelines for the medical management of nonhospitalized ulcerative colitis: the Toronto consensus. Gastroenterology. 2015 May;148(5):1035-1058.
Canadian Digestive Health Foundation. IBD: Ulcerative Colitis. https://cdhf.ca/en/digestive-conditions/ibd-ulcerative-colitis/ Accessed Feb. 6, 2023.
Choden T, Cohen NA, Rubin DT. Sphingosine-1 Phosphate Receptor Modulators: The Next Wave of Oral Therapies in Inflammatory Bowel Disease. Gastroenterol Hepatol (N Y). 2022 May;18(5):265-271.
Cooney RM, Warren BF, Altman DG, et al. Outcome measurement in clinical trials for Ulcerative Colitis: towards standardisation. Trials. 2007 Jun 25;8:17.
Coward S, Clement F, Benchimol E, et al. The rising prevalence of inflammatory bowel disease in Canada: Analyzing the past to predict the future. Journal of the Canadian Association of Gastroenterology. 2018; 1(Supp 2): A-29.
Crohn’s and Colitis Canada. 2018 Impact of Inflammatory Bowel Disease in Canada. https://crohnsandcolitis.ca/Crohns_and_Colitis/documents/reports/2018-Impact-Report- LR.pdf
Danese S, Allez M, van Bodegraven AA, et al. Unmet Medical Needs in Ulcerative Colitis: An Expert Group Consensus. Dig Dis. 2019;37(4):266-283.
Danese S, Vermeire S, Zhou W, et al. Upadacitinib as induction and maintenance therapy for moderately to severely active ulcerative colitis: results from three phase 3, multicentre, double-blind, randomised trials. Lancet. 2022 Jun 4;399(10341):2113-2128.
De Vries LCS, Wildenberg ME, De Jonge WJ, et al. The Future of Janus Kinase Inhibitors in Inflammatory Bowel Disease. J Crohns Colitis. 2017 Jul 1;11(7):885-893.
D’Haens G, Kobayashi T, Morris N, et al.. OP26 Efficacy and safety of mirikizumab as induction therapy in patients with moderately to severely active Ulcerative Colitis: Results from the Phase 3 LUCENT-1 study. J Crohn’s and Colitis 2022;16(Supplement_1): i028–i029.
Dubinsky MC, Irving PM, Li X, et al.. 867e: Efficacy and safety of mirikizumab as maintenance therapy in patients with moderately to severely active ulcerative colitis: Results from the Phase 3 LUCENT-2 study. Gastroenterology 2022;162(7):S-1393–S-1394.
Feagan BG, Rutgeerts P, Sands BE, et al. Vedolizumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2013 Aug 22;369(8):699-710.
Feagan BG, Rubin DT, Danese S, et al. Efficacy of Vedolizumab Induction and Maintenance Therapy in Patients With Ulcerative Colitis, Regardless of Prior Exposure to Tumor Necrosis Factor Antagonists. Clin Gastroenterol Hepatol. 2017 Feb;15(2):229-239.
Feuerstein JD, Moss AC, Farraye FA. Ulcerative Colitis. Mayo Clin Proc. 2019 Jul;94(7):1357-1373.
Feuerstein JD, Isaacs KL, Schneider Y, Siddique SM, Falck-Ytter Y, Singh S; AGA Institute Clinical Guidelines Committee. AGA Clinical Practice Guidelines on the Management of Moderate to Severe Ulcerative Colitis. Gastroenterology. 2020 Apr;158(5):1450-1461.
Gisbert JP, Linares PM, McNicholl AG, et al. Meta-analysis: the efficacy of azathioprine and mercaptopurine in ulcerative colitis. Aliment Pharmacol Ther. 2009 Jul 1;30(2):126-37.
Hanzel J, Hulshoff MS, Grootjans J, D'Haens G. Emerging therapies for ulcerative colitis. Expert Rev Clin Immunol. 2022 May;18(5):513-524.
Khan KJ, Dubinsky MC, Ford AC, et al Efficacy of immunosuppressive therapy for inflammatory bowel disease: a systematic review and meta-analysis. Am J Gastroenterol. 2011 Apr;106(4):630-42.
Khanna R, Chande N, Marshall JK. Ozanimod for the Treatment of Ulcerative Colitis. Gastroenterology. 2022 Jun;162(7):2104-2106.
Loftus EV Jr, Colombel JF, Takeuchi K, et al. Upadacitinib Therapy Reduces Ulcerative Colitis Symptoms as Early as Day 1 of Induction Treatment. Clin Gastroenterol Hepatol. 2022 Dec 1:S1542-3565(22)01109-0.
Löwenberg M, Volkers A, van Gennep S, et al. Mercaptopurine for the treatment of ulcerative colitis - a randomised placebo-controlled trial. J Crohns Colitis. 2023 Feb 27:jjad022. doi: 10.1093/ecco-jcc/jjad022. Epub ahead of print. PMID: 36847130.
Ma C, Fedorak RN, Kaplan GG, et al. Long-term Maintenance of Clinical, Endoscopic, and Radiographic Response to Ustekinumab in Moderate-to-Severe Crohn's Disease: Real-world Experience from a Multicenter Cohort Study. Inflamm Bowel Dis. 2017 May;23(5):833-839.
Marshall JK, Otley AR, Afif W, et al. Canadian Association of Gastroenterology position statement regarding the use of thiopurines for the treatment of inflammatory bowel disease. Can J Gastroenterol Hepatol. 2014 Jul-Aug;28(7):371-2.
Monstad IL, Solberg IC, Cvancarova M, et al. Outcome of Ulcerative Colitis 20 Years after Diagnosis in a Prospective Population-based Inception Cohort from South-Eastern Norway, the IBSEN Study. J Crohns Colitis. 2021 Jun 22;15(6):969-979.
Nguyen GC, Bollegala N, Chong CA. Factors associated with readmissions and outcomes of patients hospitalized for inflammatory bowel disease. Clin Gastroenterol Hepatol. 2014 Nov;12(11):1897-1904.
Paik J. Ozanimod: A Review in Ulcerative Colitis. Drugs. 2022 Aug;82(12):1303-1313.
Papamichael K, Gils A, Rutgeerts P, et al. Role for therapeutic drug monitoring during induction therapy with TNF antagonists in IBD: evolution in the definition and management of primary nonresponse. Inflamm Bowel Dis. 2015 Jan;21(1):182-97.
Parigi TL, Iacucci M, Ghosh S. Blockade of IL-23: What is in the Pipeline? J Crohns Colitis. 2022 May 11;16(Supplement_2):ii64-ii72.
Peyrin-Biroulet L, Danese S, Argollo M, et al. Loss of Response to Vedolizumab and Ability of Dose Intensification to Restore Response in Patients With Crohn's Disease or Ulcerative Colitis: A Systematic Review and Meta-analysis. Clin Gastroenterol Hepatol. 2019 Apr;17(5):838-846.
Qiu Y, Chen B, Li Y, et al. Risk factors and long-term outcome of disease extent progression in Asian patients with ulcerative colitis: a retrospective cohort study. BMC Gastroenterol. 2019 Jan 10;19(1):7.
Raine T, Bonovas S, Burisch J, et al. ECCO Guidelines on Therapeutics in Ulcerative Colitis: Medical Treatment. J Crohns Colitis. 2022 Jan 28;16(1):2-17.
Reinisch W, Sandborn WJ, Hommes DW, et al. Adalimumab for induction of clinical remission in moderately to severely active ulcerative colitis: results of a randomised controlled trial. Gut. 2011 Jun;60(6):780-7.
Roda G, Jharap B, Neeraj N, et al. Loss of Response to Anti-TNFs: Definition, Epidemiology, and Management. Clin Transl Gastroenterol. 2016 Jan 7;7(1):e135.
Rogala L, Miller N, Graff LA, et al. Population-based controlled study of social support, self-perceived stress, activity and work issues, and access to health care in inflammatory bowel disease. Inflamm Bowel Dis. 2008 Apr;14(4):526-35.
Rubin DT, Ananthakrishnan AN, Siegel CA, et al. ACG Clinical Guideline: Ulcerative Colitis in Adults. Am J Gastroenterol. 2019 Mar;114(3):384-413.
Rutgeerts P, Sandborn WJ, Feagan BG, et al. Infliximab for induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2005 Dec 8;353(23):2462-76.
Sandborn WJ, van Assche G, Reinisch W, et al. Adalimumab induces and maintains clinical remission in patients with moderate-to-severe ulcerative colitis. Gastroenterology. 2012 Feb;142(2):257-65.
Sandborn WJ, Feagan BG, Marano C, et al. Subcutaneous golimumab induces clinical response and remission in patients with moderate-to-severe ulcerative colitis. Gastroenterology. 2014 Jan;146(1):85-95.
Sandborn WJ, Feagan BG, Marano C, et al. Subcutaneous golimumab maintains clinical response in patients with moderate-to-severe ulcerative colitis. Gastroenterology. 2014 Jan;146(1):96-109.
Sandborn WJ, Su C, Sands BE, et al. Tofacitinib as Induction and Maintenance Therapy for Ulcerative Colitis. N Engl J Med. 2017 May 4;376(18):1723-1736.
Sandborn WJ, Feagan BG, D'Haens G, et al. Ozanimod as Induction and Maintenance Therapy for Ulcerative Colitis. N Engl J Med. 2021 Sep 30;385(14):1280-1291.
Sandborn WJ, Feagan BG, Hanauer SB, et al. The Guide to Guidelines in Ulcerative Colitis: Interpretation and Appropriate Use in Clinical Practice. Gastroenterol Hepatol (N Y). 2021 Apr;17(4 Suppl 4):3-13.
Sands BE, Sandborn WJ, Panaccione R, et al. Ustekinumab as Induction and Maintenance Therapy for Ulcerative Colitis. N Engl J Med. 2019 Sep 26;381(13):1201-1214.
Shafer LA, Walker JR, Restall G, et al. Association Between IBD Disability and Reduced Work Productivity (Presenteeism): A Population-Based Study in Manitoba, Canada. Inflamm Bowel Dis. 2019 Jan 10;25(2):352-359.
Soler D, Chapman T, Yang LL, et al. The binding specificity and selective antagonism of vedolizumab, an anti-alpha4beta7 integrin therapeutic antibody in development for inflammatory bowel diseases. J Pharmacol Exp Ther. 2009 Sep;330(3):864-75.
Statistics Canada. Average weekly earnings (including overtime), by province and territory. 2017.
The Efficacy and Safety of Guselkumab Induction Therapy in Patients With Moderately to Severely Active Ulcerative Colitis: Phase 2b QUASAR Study Results Through Week 12. Gastroenterol Hepatol (N Y). 2022 Apr;18(4 Suppl 1):12.
Timmer A, Patton PH, Chande N, et al. Azathioprine and 6-mercaptopurine for maintenance of remission in ulcerative colitis. Cochrane Database Syst Rev. 2016 May 18;2016(5):CD000478.
Tsai L, Ma C, Dulai PS, et al. Contemporary Risk of Surgery in Patients With Ulcerative Colitis and Crohn's Disease: A Meta-Analysis of Population-Based Cohorts. Clin Gastroenterol Hepatol. 2021 Oct;19(10):2031-2045.
Turner D, Ricciuto A, Lewis A, et al. STRIDE-II: An Update on the Selecting Therapeutic Targets in Inflammatory Bowel Disease (STRIDE) Initiative of the International Organization for the Study of IBD (IOIBD): Determining Therapeutic Goals for Treat-to-Target strategies in IBD. Gastroenterology. 2021 Apr;160(5):1570-1583.
Xie W, Xiao S, Huang H, et al. Incidence of and Risk Factors for Paradoxical Psoriasis or Psoriasiform Lesions in Inflammatory Bowel Disease Patients Receiving Anti-TNF Therapy: Systematic Review With Meta-Analysis. Front Immunol. 2022 Mar 1;13:847160.
Yanai H, Shuster D, Calabrese E, et al. The incidence and predictors of lupus-like reaction in patients with IBD treated with anti-TNF therapies. Inflamm Bowel Dis. 2013 Dec;19(13):2778-86.
Conflict of Interest Declarations — Canadian Gastroenterologists
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please refer to the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission?
No.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission?
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician who contributed to the input.
Declaration for Clinician 1
Name: Vipul Jairath, MBChB, DPhil
Position: Professor of Medicine, Departments of Medicine, Epidemiology and Biostatistics; Holder, John and Susan McDonald Endowed Chair in IBD Clinical Research; Director, Advanced IBD Fellowship Program; Associate Chair (Research), Department of Medicine; Schulich School of Medicine & Dentistry I Western University
Date: 28-03-2023
Table 4: COI Declaration for Canadian Gastroenterologists — Clinician 1
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Abbvie | — | — | X | — |
Alimentiv | — | — | — | X |
Arena Pharmaceuticals | X | — | — | — |
Asahi Kasei Pharma | X | — | — | — |
Asieris | X | — | — | — |
Astra Zeneca | X | — | — | — |
Avoro Capital | X | — | — | — |
Bristol Meyers Squibb | — | X | — | — |
Celltrion | X | — | — | — |
Eli Lilly | — | X | — | — |
Endpoint Health | X | — | — | — |
Ferring | X | — | — | — |
Flagship Pioneering | X | — | — | — |
Fresenius Kabi | X | — | — | — |
Galapagos | X | — | — | — |
Gilde Healthcare | X | — | — | — |
Glaxo-Smith-Klein | X | — | — | — |
Genentech | X | — | — | — |
Gilead | X | — | — | — |
Janssen | — | — | X | — |
Merck | X | — | — | — |
Mylan | X | — | — | — |
Metacrine | X | — | — | — |
Pandion Pharma | X | — | — | — |
Pendopharm | X | — | — | — |
Pfizer | — | — | X | — |
Protagonist Therapeutics | X | — | — | — |
Reistone Biopharma | X | — | — | — |
Roche | X | — | — | — |
Sandoz | X | — | — | — |
Second Genome | X | — | — | — |
Sorriso | X | — | — | — |
Takeda Pharmaceuticals | — | — | X | — |
Teva | X | — | — | — |
Topivert | X | — | — | — |
VEntyx | X | — | — | — |
Vividion | — | — | — | — |
Declaration for Clinician 2
Name: Charles N Bernstein
Position: Distinguished Professor of Medicine, University of Manitoba
Date: 27-03-2023
Table 5: COI Declaration for Canadian Gastroenterologists — Clinician 2
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Abbvie | X | — | X | — |
Janssen | X | — | X | — |
Pfizer | X | — | X | — |
Takeda | X | — | X | — |
Bristol Myers Squibb | X | X | — | — |
Sandoz | X | — | X | — |
Amgen | X | — | X | — |
Eli Lilly | X | — | — | — |
JAMP Pharmaceuticals | X | — | — | — |
Roche | X | — | — | — |
Declaration for Clinician 3
Name: Neeraj Narula
Position: Director of IBD Clinic at Hamilton Health Sciences
Date: 27-03-2023
Table 6: COI Declaration for Canadian Gastroenterologists — Clinician 3
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Abbvie | — | — | X | — |
Janssen | — | X | — | — |
Takeda | — | X | — | — |
Eli Lilly | X | — | — | — |
Fresenius Kabi | X | — | — | — |
Pfizer | — | X | — | — |
Viatris | X | — | — | — |
Sandoz | X | — | — | — |
Iterative Health | — | — | X | — |
Innomar Strategies | — | X | — | — |
Declaration for Clinician 4
Name: Jeffrey McCurdy
Position: Gastroenterologist, University of Ottawa
Date: 19-03-2023
Table 7: COI Declaration for Canadian Gastroenterologists — Clinician 4
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Abbvie | — | X | — | — |
Fresenius Kabi | X | — | — | — |
BMS | X | — | — | — |
Ferring | X | — | — | — |
Janssen | — | X | — | — |
Pfizer | X | — | — | — |
Takeda | — | — | X | — |
Declaration for Clinician 5
Name: Cynthia Seow
Position: Professor, Division of Gastroentroenterology and Hepatology, Department of Medicine, University of Calgary, Alberta, Canada
Date: 28-03-2023
Table 8: COI Declaration for Canadian Gastroenterologists — Clinician 5
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Janssen | — | X | — | — |
Abbvie | — | X | — | — |
Takeda | — | X | — | — |
Pfizer | — | X | — | — |
Fresenius Kabi | X | — | — | — |
Bristol Myers Squibb | X | — | — | — |
Declaration for Clinician 6
Name: Remo Panaccione
Position: Gastroenterologist, Professor of Medicine, University of Calgary
Date: 28-03-2023
Table 9: COI Declaration for Canadian Gastroenterologists — Clinician 6
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Abbvie | — | — | X | — |
Alimentiv | — | — | X | — |
Amgen | X | — | — | — |
Arena Pharmaceuticals | X | — | — | — |
Astra Zeneca | X | — | — | — |
Bristol Meyers Squibb | — | X | — | — |
Boehringer Ingelheim | X | — | — | — |
Eli Lilly | — | X | — | — |
Ferring | X | — | — | — |
Fresenius Kabi | X | — | — | — |
Galapagos | X | — | — | — |
Gilead Sciences | X | — | — | — |
Glaxo-Smith-Klein | X | — | — | — |
JAMP Biomed | X | — | — | — |
Janssen | — | — | X | — |
Merck | X | — | — | — |
Mylan | X | — | — | — |
Oppilan | X | — | — | — |
Organon | — | X | — | — |
Pandion Pharma | X | — | — | — |
Pfizer | — | — | X | — |
Progenity | X | — | — | — |
Protagonist Therapeutics | X | — | — | — |
Roche | X | — | — | — |
Satisfai Health | X | — | — | — |
Sandoz | X | — | — | — |
Sublimity | X | — | — | — |
Takeda Pharmaceuticals | — | — | X | — |
Viatris | X | — | — | — |
Declaration for Clinician 7
Name: Yvette Leung
Position: Associate Clinical Professor
Date: 21-03-2023
Table 10: COI Declaration for Canadian Gastroenterologists — Clinician 7
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
BMS | — | X | — | — |
Jannsen | — | X | — | — |
Abbvie | — | — | X | — |
Frenius Kabus | X | — | — | — |
Amgen | X | — | — | — |
Takeda | — | — | X | — |
Pfizer | — | — | X | — |
Lilly | — | X | — | — |
Pendopharm | X | — | — | — |
BioJamp | X | — | — | — |
Declaration for Clinician 8
Name: Waqqas Afif
Position: Associate Professor of Medicine, McGill University Health Center
Date: 28-03-2023
Table 11: COI Declaration for Canadian Gastroenterologists — Clinician 8
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Janssen | — | X | — | — |
Takeda | — | X | — | — |
Abbvie | — | X | — | — |
Pfizer | — | X | — | — |
Eli-Lilly | — | X | — | — |
Amgen | X | — | — | — |
BMS | X | — | — | — |
Sanofi | X | — | — | — |
Declaration for Clinician 9
Name: John Marshall
Position: Professor of Medicine
Date: 31-03-2023
Table 12: COI Declaration for Canadian Gastroenterologists — Clinician 9
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie | — | — | — | X |
Astra Zeneca | X | — | — | — |
Amgen | — | — | X | — |
BMS | — | X | — | — |
Celltrion | X | — | — | — |
Ferring | — | X | — | — |
Fresenius Kabi | — | X | — | — |
Janssen | — | — | X | |
Lilly | — | X | — | — |
Organon | X | — | — | — |
Pfizer | — | — | X | — |
Sandoz | — | X | — | — |
Takeda | — | — | X | — |
Viatris | — | X | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.