CADTH Reimbursement Review
Roflumilast (Zoryve)
Sponsor: Arcutis Biotherapeutics Inc.
Therapeutic area: Plaque psoriasis
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
ACE
angiotensin-converting enzyme
AE
adverse event
BSA
body surface area
CDLQI
Children’s Dermatology Life Quality Index
CI
confidence interval
CrI
credible interval
CSPA
Canadian Skin Patient Alliance
DLQI
Dermatology Life Quality Index
HRQoL
health-related quality of life
IGA
Investigator’s Global Assessment
I-IGA
intertriginous Investigator’s Global Assessment
I-IGA ITT
subset of patients in the ITT population with intertriginous area involvement and an I-IGA of 2 or more at baseline
ITC
indirect treatment comparison
ITT
intention-to-treat
KM
Kaplan-Meier
LS
least squares
MID
minimal important difference
mPASI
modified Psoriasis Area Severity Index
NE
not estimable
NMA
network meta-analysis
OLE
open-label extension
PASI
Psoriasis Area and Severity Index
PGA
Physician’s Global Assessment
PRU4 ITT
subset of patients in the ITT population with a WI-NRS pruritus score of 4 or more at baseline
PSD
Psoriasis Symptom Diary
QoL
quality of life
SAE
serious adverse event
SD
standard deviation
SE
standard error
TEAE
treatment-emergent adverse event
WDAE
withdrawal due to adverse event
WI-NRS
Worst Itch Numeric Rating Scale
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Drug product | Roflumilast cream 0.3% w/w (Zoryve) for topical use |
Sponsor | Arcutis Biotherapeutics Inc. |
Indication | For topical treatment of plaque psoriasis, including treatment of psoriasis in the intertriginous areas, in patients 12 years of age and older |
Reimbursement request | As per the indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | April 28, 2023 |
Recommended dosage | Apply to affected areas once daily |
NOC = Notice of Compliance; w/w = weight by weight.
Introduction
Psoriasis is a chronic, immune-mediated, inflammatory skin disease that is associated with multiple comorbidities, such as psoriatic arthritis, obesity, and metabolic syndrome.1,2 Psoriasis can present similarly in children and adults; the peak ages of onset are between 30 and 39 years and between 50 and 69 years.1 A panel of 11 dermatologists with expertise in psoriasis estimated that the median prevalence of psoriasis is 3% of the general adult population in Canada. The panel estimated that approximately 78% of patients with psoriasis have less than 10% body surface area (BSA) involved (i.e., mild or moderate disease) and 22% of patients have 10% or more BSA involved. The panel further estimated that 50% of patients have less than 3% of BSA involved (i.e., mild disease) and 2% of patients have more than 50% of BSA involved.3 Based on estimates in the US, 50% of patients have facial involvement4 and 21% to 30% of patients have intertriginous area involvement.5
Chronic plaque psoriasis (also known as psoriasis vulgaris)6 is the most common clinical subtype of psoriasis, representing approximately 90% of patient cases in Canada.7 Plaque psoriasis is characterized by well-demarcated, erythematous, cutaneous plaques with overlying, coarse, silvery scales. Plaques can be asymptomatic; however, pruritus and pain are often reported by patients. Common areas of involvement include the scalp, elbows, knees, and gluteal cleft. Additionally, intertriginous areas (inverse psoriasis), ear canals, umbilicus, palms, soles, and nails are possible areas of involvement. Intertriginous psoriasis is characterized by well demarcated, smooth, shiny plaques with no to minimal scales.1
Plaque psoriasis requires lifelong follow-up and treatment. Measures of treatment success in clinical practice may include clearance (absence of signs of disease), control (satisfactory response to therapy as defined by the patient and/or clinician), and remission (maintenance of disease control or suppression of signs and symptoms over time).6
Management of mild plaque psoriasis involving the trunk, limbs, and neck includes topical therapies that can be broadly categorized as corticosteroids, vitamin D3 analogues, retinoids, anthralin (commercial formulations of anthralin are not currently available in Canada) and tars, as well as combination therapy.6,8 The therapeutic options for pediatric patients are generally similar to the options for adult patients.6 The management of intertriginous psoriasis, affecting the groin, axillae, inframammary region, abdominal body folds, gluteal cleft, perianal region, and retroarticular fold areas, can be a challenge, as these areas are at an increased risk of adverse reactions to topical therapy because the skin tends to be thinner in these regions.6 Moreover, there are currently no available treatments indicated for intertriginous psoriasis. Topical calcineurin inhibitors may be an appropriate treatment option for the management of intertriginous psoriasis; however, they are not currently approved for an indication in psoriasis (hence its off-label use).6,8 Treatment selection should be individualized to the patient to improve adherence, support patient satisfaction, and achieve treatment success.6 Management of moderate to severe plaque psoriasis affecting the trunk and extremities that cannot be adequately controlled by the approaches described previously (adequate control is defined by the patient’s perception of the disease and its burdens) include systemic therapy, phototherapy, combination therapy, and topical therapy as adjunct therapy.6
The objective of this report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of roflumilast cream 0.3% for the topical treatment of plaque psoriasis, including treatment of psoriasis in the intertriginous areas, in patients aged 12 years and older.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient and clinician groups that responded to CADTH’s call for input and from the clinical expert consulted by CADTH for the purpose of this review.
Patient Input
Three patient groups submitted a joint patient input submission. The Canadian Skin Patient Alliance (CSPA), Canadian Psoriasis Network, and Canadian Association of Psoriasis Patients are national, not-for-profit organizations that are dedicated to improving the lives of people with psoriasis across Canada.
According to the input, the impact of psoriasis symptoms is considerable in various areas of patients’ lives, such as mental well-being, daily tasks, intimate relationships, and social lives. Most patients considered effectiveness as the most important factor to be addressed in any new treatment for psoriasis. In addition, patients valued lack of side effects, affordability, treatments that are easy to apply, and treatments that are conducive to their schedules.
More than half of survey respondents reported having moderate psoriasis. The body area with the most impact was the scalp, followed by the legs, arms, genitals, hands, torso, skin folds, and palms. Based on the survey, respondents were using topical corticosteroids, topical combination treatments, and/or biological drugs. Most patients with psoriasis have discontinued their treatment at some point during their disease, with the most frequent reasons being that the treatment stopped working, caused side effects, was unaffordable, or was ineffective. Among respondents, 10 patients reported experience with roflumilast, accessed through a clinical trial. Nine of these patients reported benefits of treatment that included clearing of skin, reduced itch and redness, clearing of skin lesions (plaques), and ease of treatment application. All 10 respondents tolerated roflumilast well, except for 1 who experienced some itching. The patient group input emphasized that psoriasis is a chronic and potentially debilitating disease that poses many challenges and is linked to anxiety, depression, and social isolation. This disease can interfere with social and intimate relationships, productivity, and family life and work life. Furthermore, due to the chronicity of this disease, patients are concerned about recurrence and resistance to treatments in the future.
Clinician Input
Input From the Clinical Expert Consulted by CADTH
Unmet Need
The clinical expert indicated there are currently no available treatment options that can permanently reverse the course of plaque psoriasis, but systemic therapies can suppress psoriasis while they are being used.
The clinical expert also indicated that many patients do not respond to currently available topical treatments; in particular, widespread psoriasis and psoriasis of the scalp, palms, and/or soles are often refractory to topical treatment. The clinical expert noted that although topical therapies are generally well tolerated and that topical steroids are used over the long-term for many patients in clinical practice, clinical guidelines caution against the long-term use of topical steroids for plaque psoriasis. As such, the clinical expert agreed that more effective, safer, and better-tolerated treatment options are an unmet need, particularly for long-term use and in patients with facial, genital, and intertriginous involvement.
The clinical expert further suggested that topical formulations that are less messy and more effective may improve adherence, particularly among patients with intertriginous involvement who are offered topical tacrolimus, as it is only available as an ointment and not widely available because it is not indicated (used off-label) for the treatment of psoriasis.
Place in Therapy
For most patients with plaque psoriasis, the clinical expert suggested they would consider roflumilast as an alternative to other first-line topical treatments for psoriasis within the current stepped approach, as this could prevent having to step up patients to systemic therapy. For patients with facial, genital, and intertriginous involvement, the clinical expert anticipated that roflumilast would be a first-line treatment. The clinical expert further suggested that roflumilast may be preferred for long-term use in some patients and by clinicians, given that it is not a steroid.
For mild disease, the clinical expert suggested that roflumilast would be used as monotherapy. Additionally, depending on patient preference, roflumilast could be used simultaneously with other topical treatments. Roflumilast could also be used in combination with systemic therapy or phototherapy, as topical treatments are typically continued as needed.
Patient Population
The clinical expert identified patients with active psoriasis, regardless of whether it is being actively treated or not, as the most in need of an intervention. The clinical expert noted there is a need for an intervention for the hair-bearing scalp area; however, they indicated that this need would not be addressed by roflumilast. Clinician examination and judgment together with shared decision-making with the patient, would determine whether the drug is best suited for the patient. The clinical expert suggested that patients with psoriasis with limited BSA involvement are the most likely to respond to therapy with roflumilast as monotherapy.
Assessing the Response to Treatment
The clinical expert indicated that clearance or near clearance of psoriasis lesions is commonly used to assess psoriasis severity; this is analogous to the Investigator’s Global Assessment (IGA) scales used in clinical trials. The clinical expert also indicated that patient satisfaction is important and is assessed in a gestalt manner that likely differs between physicians.
According to the clinical expert, extensive measurement scales, such as the Psoriasis Area and Severity Index (PASI), Dermatology Life Quality Index (DLQI), Worst Itch Numeric Rating Scale (WI-NRS), and Psoriasis Symptom Diary (PSD), are not typically used in clinical practice; however, PASI and DLQI may be used if mandated by the insurer to fund a drug.
Discontinuing Treatment
According to the clinical expert, intolerance (e.g., stinging at the application site) and ineffectiveness (i.e., minimal improvement after 2 to 8 weeks of application) are factors to consider when deciding to discontinue treatment with roflumilast.
The clinical expert further indicated that roflumilast would be discontinued when lesions have cleared and can be restarted if and when lesions recur. The clinical expert indicated that phototherapy or systemic therapy may be considered for extensive psoriasis regardless of improvement with roflumilast and, if there is substantial improvement with phototherapy or systemic therapy that obviates the need for topical therapy, then roflumilast would be considered for discontinuation.
Clinician Group Input
Three clinician groups provided input: Fraser Health Dermatology (4 clinicians), the Canadian Dermatology Association (3 clinicians), and the Atlantic Provinces Dermatology Association and Dermatology Association of Ontario (12 clinicians).
The clinician groups noted there is a need for a new treatment to substitute existing therapies for plaque psoriasis. The clinician groups noted that the limitations of current treatments include unfavourable effects, poor compliance, difficult application, high costs, limited efficacy, intolerability due to irritation, and inability to be administered to all areas of the body. The treatment goals noted include reducing the severity of symptoms, minimizing adverse events (AEs), improving tolerability and efficacy, increasing patient quality of life (QoL), and reducing the burden on patients and health care systems.
One clinician group recommended the use of roflumilast as a first-line treatment for the management of mild to severe plaque psoriasis and another group suggested roflumilast should be used after treatment with topical steroids has failed, as topical steroids are an inexpensive and usually well tolerated therapy. The input noted that patients best suited for treatment with the drug under review included those with mild to moderate psoriasis, patients with different disease phenotypes, patients with psoriasis that has failed to respond to topical steroids, and patients with intertriginous psoriasis, noting the importance of steroid sparing in these anatomic sites.
One clinician group suggested that the best outcomes to determine treatment response would be Physician’s Global Assessment (PGA) score and level of BSA involvement. The group noted the PASI score is not commonly used in clinical practice, except when applying for drug plan coverage for systemic therapies for patients with moderate to severe disease. The clinician groups noted that assessment of treatment goals is recommended after 8 weeks and that, typically, patients with psoriasis are initially seen every 3 to 6 months to assess response to treatment, and that treatment should be discontinued due to a lack of efficacy or disease progression. The input received stated that patients with psoriasis are diagnosed and treated as outpatients, typically by both specialists and general practitioners or family doctors.
Drug Program Input
Input was obtained from the drug programs that participate in the CADTH reimbursement review process. The following items were identified as key factors that could potentially impact the implementation of a CADTH recommendation for roflumilast:
relevant comparators
consideration for initiation of therapy
consideration for continuation or renewal of therapy
consideration for prescribing of therapy
generalizability
care provision issues
system and economic issues.
The clinical expert consulted by CADTH provided advice on the potential implementation issues raised by the drug programs (Table 4).
Clinical Evidence
Pivotal Studies and Randomized Controlled Trial Evidence
Description of Studies
The sponsor’s submission included 2 phase III, randomized, double-blind, parallel-group, vehicle-controlled trials, the DERMIS-19 (N = 439) and DERMIS-210 (N = 442) trials. The 2 trials assessed success in IGA (defined as an IGA score of clear or almost clear plus an improvement of 2 more grades from baseline at week 8) with roflumilast cream 0.3% compared with matching vehicle in patients with chronic plaque psoriasis involving 2% to 20% of BSA (excluding the scalp, palms, and soles). Patients were excluded if they were unable to discontinue prohibited medications and treatments (defined as systemic, biologic, topical, phototherapy, and investigational treatments) that could affect plaque psoriasis within the prespecified washout period. Patients were randomized in a 2:1 ratio to receive roflumilast or the matching vehicle applied topically once daily for 8 weeks. Secondary outcomes included measures of symptoms and involvement (PASI, IGA, intertriginous IGA [I-IGA], and WI-NRS) and a measure of QoL (PSD). Similarly, exploratory outcomes included measures of involvement (e.g., BSA), measures of health-related quality of life (HRQoL) (e.g., DLQI and Children’s DLQI [CDLQI]), local tolerability, and safety.
At baseline, |||| and |||| of randomized patients in the DERMIS-1 and DERMIS-2 trials, respectively, were in the age category of 12 years to 17 years and ||||| and ||||| of randomized patients in the DERMIS-1 and DERMIS-2 trials, respectively, were aged 18 years or older. The mean age of all randomized patients was 48.0 years (standard deviation [SD] = 14.69) in the DERMIS-1 trial and 47.0 years (SD = 14.72) in the DERMIS-2 trial. Most patients were male (64.9% and 62.4% of randomized patients in the DERMIS-1 and DERMIS-2 trials, respectively), while 33.9% to 39.3% of patients were female. The majority of patients were white (81.5% and 82.8% of randomized patients in the DERMIS-1 and DERMIS-2 trials, respectively), while the remainder of the randomized patients (0% to 7.3%) identified as Asian, Black or African American, American Indian or Alaska Native [wording from original source], Native Hawaiian or other Pacific Islander, other, or more than 1 race. A similar proportion of all randomized patients in the DERMIS-1 and DERMIS-2 trials had facial involvement (27.1% and 26.0%, respectively), genital involvement (16.4% and 14.7%, respectively), and a baseline I-IGA score (23.0% and 19.9%, respectively). The majority of patients had a moderate IGA score at baseline (74.7% and 76.5% of randomized patients in the DERMIS-1 and DERMIS-2 trials, respectively). The mean baseline BSA affected by psoriasis in all randomized patients was 6.66% (SD = 4.538) in the DERMIS-1 trial and 7.30% (SD = 4.918) in the DERMIS-2 trial. The mean baseline PASI score for all randomized patients was 6.5 (SD = 3.35) in the DERMIS-1 trial and 6.7 (SD = 3.33) in the DERMIS-2 trial.
Efficacy Results
The efficacy end points that were noted to be important to patients and clinicians based on stakeholder input are summarized in Table 2.
Investigator’s Global Assessment
The IGA is an investigator-reported, static evaluation of the overall severity of psoriasis of the whole body. The minimal important difference (MID) in IGA has not been estimated. However, achieving a score of 0 (clear) or 1 (almost clear) on the IGA has generally been accepted as clinically meaningful.11,12 Alternatively, or in addition to the achievement of a score of 0 or 1, the responder analysis may also consider the proportion of patients with at least a 2-grade improvement from baseline on the static IGA;11 this was consistent with the definition of IGA success that was used in both trials. The primary end point in both trials was an improvement in the overall severity of psoriasis that was measured by the proportion of patients who experienced treatment success based on the IGA, defined as a score of 0 or 1 plus an improvement of 2 or more grades from baseline at week 8.
The primary end point, IGA success at week 8, was met for both trials in the intention-to-treat (ITT) population. In the DERMIS-1 trial, 42.4% of patients in the roflumilast arm versus 6.1% of patients in the vehicle arm experienced treatment success based on the IGA at week 8; the ratio of the odds of IGA success with roflumilast relative to the odds of IGA success with the matching vehicle was ||||| (95% ||| |||| || |||||; | |||||||) at week 8 from baseline in favour of roflumilast. In the DERMIS-2 trial, 37.5% of patients in the roflumilast arm versus 6.9% of patients in the vehicle arm experienced treatment success based on the IGA at week 8; the ratio of the odds of IGA success with roflumilast relative to the odds of IGA success with the matching vehicle was |||| |||| || |||| || |||||| | | ||||||| at week 8 from baseline, also in favour of roflumilast. In both studies, the results of the primary analyses were generally consistent with the sensitivity analyses of the primary end point. IGA success at week 4 was also reported and was also in favour of roflumilast when compared with the matching vehicle in the DERMIS-1 trial (13.44; 95% confidence interval [CI], 3.72 to 48.58; P < 0.0001) and the DERMIS-2 trial (3.91; 95% CI, 1.76 to 8.70; P = 0.0011).
Intertriginous IGA
I-IGA was defined as the IGA scale but was used to evaluate only intertriginous areas in the trials. In both trials, an improvement in the severity of intertriginous psoriasis was measured by the proportion of patients who experienced treatment success based on the I-IGA, defined as a score of 0 (clear) or 1 (almost clear) plus an improvement of 2 or more grades from baseline at week 8, which was a secondary end point tested in a hierarchical manner and adjusted for multiple comparisons. Note that this analysis was based on the prespecified I-IGA ITT population, a subset of patients in the ITT population with intertriginous area involvement and an I-IGA of 2 or more at baseline; 52 patients (82.5%) in the roflumilast arm and 29 patients (90.6%) in the vehicle arm were available in the DERMIS-1 trial for the analysis at week 8, and 47 patients (88.6%) and 27 patients (87.0%), respectively, were available in the DERMIS-2 trial for the analysis at week 8.
In both trials, the proportion of patients who experienced treatment success based on the I-IGA at week 8 was greater with roflumilast treatment compared with the matching vehicle. More specifically, in the DERMIS-1 trial, the ratio of the odds of I-IGA success with roflumilast relative to the odds of I-IGA success with the matching vehicle was 17.94 (95% CI, 2.33 to 138.20; P < 0.0001) at week 8 from baseline, in favour of roflumilast. In the DERMIS-2 trial, the ratio of the odds of I-IGA success with roflumilast relative to the odds of I-IGA success with the matching vehicle was 11.18 (95% CI, 2.33 to 53.68; P = 0.0004) at week 8 from baseline, also in favour of roflumilast.
Psoriasis Area and Severity Index
PASI is an investigator-reported evaluation of the extent and severity of psoriasis. An MID in PASI has not been estimated. In both trials, an improvement in the extent and severity of disease was measured based on the proportion of patients who had a 75% improvement in PASI (PASI 75) from baseline at week 8, which was a secondary end point tested in a hierarchical manner and adjusted for multiple comparisons.
In both trials, the proportion of patients who had a PASI 75 was greater with roflumilast treatment compared with the matching vehicle. More specifically, in the DERMIS-1 trial, the ratio of the odds of a PASI 75 with roflumilast relative to the odds of a PASI 75 with the matching vehicle was 12.00 (95% CI, 5.15 to 27.93; P < 0.0001) at week 8 from baseline, in favour of roflumilast. In the DERMIS-2 trial, the ratio of the odds of a PASI 75 with roflumilast relative to the odds of a PASI 75 with the matching vehicle was 10.42 (95% CI, 4.49 to 24.19; P < 0.0001) at week 8 from baseline, also in favour of roflumilast. Time to PASI 50 was also reported and was also in favour of roflumilast when compared with the matching vehicle in the DERMIS-1 trial; the median Kaplan-Meier (KM) estimate was 31.0 days (95% CI, 29.0 to 41.0) in the roflumilast arm versus 104.0 days (95% CI, 85.0 to not estimable [NE])) in the vehicle arm [P < 0.0001]) and, in the DERMIS-2 trial, the median KM estimate was 30.0 days (95% CI, 29.0 to 42.0) in the roflumilast arm and NE (95% CI, 71.0 to NE] in the vehicle arm [P < 0.0001]).
DLQI and Children’s DLQI
DLQI and CDLQI are patient-reported tools used to evaluate HRQoL. The estimated within-group MID is 2.2 to 6.9 points in patients with psoriasis and other inflammatory skin disorders.13,14 In both trials, an improvement in HRQoL was measured by change from baseline in DLQI and CDLQI at week 8, which were exploratory end points that were not included in the statistical hierarchy and not adjusted for multiple comparisons.
A decrease in the DLQI score corresponds to an improvement in HRQoL. In the DERMIS-1 trial, the least squares (LS) mean change from baseline in DLQI at week 8 was |||| || |||||| in the roflumilast arm and |||| || |||||| in the vehicle arm. In the DERMIS-2 trial, the LS mean change from baseline in DLQI at week 8 was |||| || |||||| in the roflumilast arm and |||| || |||||| in the vehicle arm.
Not enough data were collected to carry out an analysis of covariance for change in CDLQI at week 8 in both the DERMIS-1 and DERMIS-2 trials.
Worst Itch Numeric Rating Scale
WI-NRS is a patient-reported outcome measure that is used to assess the severity of itch. The MID in WI-NRS has been estimated to be an improvement of 4 or more points in patients with plaque psoriasis,15 which is consistent with the definition of WI-NRS success used in the trials. In the DERMIS-1 and DERMIS-2 trials, a reduction in the severity of itch was measured by the proportion of patients who experienced treatment success based on the WI-NRS, defined as a reduction of at least 4 points from baseline at week 8, which was a secondary end point tested in a hierarchical manner and adjusted for multiple comparisons. This analysis was based on a prespecified subset of patients in the ITT population with a WI-NRS pruritus score of 4 or more (the PRU4 ITT population) at baseline; 191 patients (87.6%) in the roflumilast arm and 97 patients (84.3%) in the vehicle arm were available in the DERMIS-1 trial for the analysis at week 8, and 206 patients (89.9%) and 101 patients (87.0%), respectively, were available in the DERMIS-2 trial for the analysis at week 8.
In both the DERMIS-1 and DERMIS-2 trials, the proportion of patients who reported a reduction in the severity of itch based on WI-NRS success was greater with roflumilast treatment compared with the matching vehicle. More specifically, in the DERMIS-1 trial, the ratio of the odds of WI-NRS success with roflumilast relative to the odds of WI-NRS success with the matching vehicle was 7.84 (95% CI, 3.85 to 15.94; P < 0.0001) at week 8 from baseline, in favour of roflumilast. In the DERMIS-2 trial, the ratio of the odds of WI-NRS success with roflumilast relative to the odds of WI-NRS success with the matching vehicle was 3.59 (95% CI, 2.07 to 6.23; P < 0.0001) at week 8 from baseline, also in favour of roflumilast. WI-NRS success at week 4 was also reported and was also in favour of roflumilast when compared with the matching vehicle in the DERMIS-1 trial (4.36; 95% CI, 2.31 to 8.26; P < 0.0001) and in the DERMIS-2 trial (4.93; 95% CI, 2.65 to 9.18; P < 0.0001). However, improvement in the severity of itch as measured by WI-NRS success was not consistently observed at week 2 across trials.
Psoriasis Symptom Diary
PSD is a patient-reported assessment of the impact of plaque psoriasis on overall QoL. An MID in PSD total score has not been estimated. In both trials, an improvement in QoL was measured based on change from baseline in PSD total score at week 8, which was a secondary end point tested in a hierarchical manner and adjusted for multiple comparisons.
In both trials, a greater improvement in QoL was observed in the roflumilast treatment groups compared with the matching vehicle (lower scores indicate less severe or bothersome symptoms). More specifically, in the DERMIS-1 trial, the LS mean difference between roflumilast and vehicle in change from baseline in PSD total score at week 8 was −30.9 (|||||||| ||||| |||| |||||; 95% CI, −37.2 to −24.6; P < 0.0001), in favour of roflumilast. In the DERMIS-2 trial, the LS mean difference between roflumilast and vehicle in change from baseline in PSD total score at week 8 was −26.5 (| |||||; 95% CI, −33.2 to −19.7; P < 0.0001), also in favour of roflumilast. The LS mean difference between roflumilast and vehicle in change from baseline in PSD total score was also reported at week 4 and was also in favour of roflumilast in the DERMIS-1 trial (−25.8; | |||||; 95% CI, −31.7 to −20.0; P < 0.0001) and in the DERMIS-2 trial (−26.0; ||| |||||| 95% CI, −31.9 to −20.0; P < 0.0001).
Body Surface Area
BSA measures the extent of psoriasis as assessed by an investigator. An MID has not been estimated for BSA. In both trials, an improvement in the extent of disease was measured by percent change from baseline in BSA affected by psoriasis at week 8, which was an exploratory end point that was not included in the statistical hierarchy and not adjusted for multiple comparisons.
A decrease in percent BSA corresponds to an improvement in extent of disease. In the DERMIS-1 trial, the LS mean percent change from baseline in BSA affected by psoriasis at week 8 was |||||| || ||||||| in the roflumilast arm and |||||| || ||||||| in the vehicle arm. In the DERMIS-2 trial, the LS mean percent change from baseline in BSA affected by psoriasis at week 8 was |||||| || |||||| in the roflumilast arm and ||||| || ||||||| in the vehicle arm.
Harms Results
The key harms results are summarized in Table 2.
Adverse Events
The proportion of patients in the roflumilast arm with any treatment-emergent adverse event (TEAE) was 25.2% in the DERMIS-1 trial and 25.9% in the DERMIS-2 trial, while the proportion of patients in the vehicle arm with any TEAE was 23.5% in the DERMIS-1 trial and 18.4% in the DERMIS-2 trial. The most common TEAEs (a frequency of 2% or more of patients in either study) reported in the roflumilast arm were diarrhea (3.5% in the DERMIS-1 trial and 2.8% in the DERMIS-2 trial) and headache (1.0% in the DERMIS-1 trial and 3.8% in the DERMIS-2 trial). All remaining TEAEs were reported in less than 2% of patients in the roflumilast arm in either study.
Serious Adverse Events
The proportion of patients in the roflumilast arm with any serious adverse event (SAE) was 0.7% in the DERMIS-1 trial and no patients in the DERMIS-2 trial, while the proportion of patients in the vehicle arm with any SAE was 0.7% in both the DERMIS-1 and DERMIS-2 trials. The SAEs reported in the roflumilast arm were concussion (1 patient in the DERMIS-1 trial) and foot fracture, thorax deformity, and pneumothorax (1 patient in the DERMIS-1 trial).
Withdrawals Due to Adverse Events
The proportion of patients in the roflumilast arm with any withdrawal due to adverse event (WDAE) was |||| in the DERMIS-1 trial and |||| in the DERMIS-2 trial, while the proportion of patients in the vehicle arm with any WDAE was |||| in the DERMIS-1 trial and |||| in the DERMIS-2 trial. All TEAEs leading to discontinuation of treatment and/or study withdrawal were reported in less than || of patients in the roflumilast arm in either study.
Mortality
Based on information the sponsor provided, no deaths occurred during the DERMIS-1 and DERMIS-2 trials.
Notable Harms
The proportion of patients with application site pain in the roflumilast arm was 0.7% in the DERMIS-1 trial and 1.4% in the DERMIS-2 trial. All remaining TEAEs of special interest (application site pruritis, urticaria, dryness, dermatitis, and irritation) were reported in less than 1% of patients in the roflumilast arm in either study.
Table 2: Summary of Key Results From the DERMIS-1 and DERMIS-2 Trials
Outcome | DERMIS-1 | DERMIS-2 | ||
|---|---|---|---|---|
Roflumilast | Vehicle | Roflumilast | Vehicle | |
Primary outcome | ||||
IGA success,a week 8 (ITT population) | ||||
N | 286 | 153 | 290 | 152 |
n (%) | 255 (89.1) | 132 (86.2) | 264 (91.0) | 131 (86.1) |
IGA success, n (%) | 108 (42.4) | 8 (6.1) | 99 (37.5) | 9 (6.9) |
OR (95% CI)b | ||||| ||||| || |||||| | |||| ||||| || |||||| | ||
P value | < 0.0001 | < 0.0001 | ||
Secondary outcomes | ||||
PASI 75 from baseline, week 8 (ITT population) | ||||
N | 286 | 153 | 290 | 152 |
n (%) | 255 (89.1) | 132 (86.2) | 264 (91.0) | 131 (86.1) |
PASI 75, n (%) | 106 (41.6) | 10 (7.6) | 103 (39.0) | 7 (5.3) |
OR (95% CI)b | 12.00 (5.15 to 27.93) | 10.42 (4.49 to 24.19) | ||
P value | < 0.0001 | < 0.0001 | ||
I-IGA success,c week 8 (I-IGA ITT population) | ||||
N | 63 | 32 | 53 | 31 |
n (%) | 52 (82.5) | 29 (90.6) | 47 (88.6) | 27 (87.0) |
I-IGA success, n (%) | 37 (71.2) | 4 (13.8) | 32 (68.1) | 5 (18.5) |
OR (95% CI)d | 17.94 (2.33 to 138.20) | 11.18 (2.33 to 53.68) | ||
P value | < 0.0001 | 0.0004 | ||
PSD, week 8 (ITT population) | ||||
N | 286 | 153 | 290 | 152 |
n (%) | 250 (87.4) | 129 (84.3) | 257 (88.6) | 127 (83.5) |
Baseline, mean (SD) | 72.1 (42.75) | 73.4 (41.29) | 69.3 (40.66) | 77.4 (41.24) |
Week 8, mean (SD) | 21.4 (30.04) | 50.0 (40.45) | 22.0 (33.25) | 53.6 (44.00) |
LS mean change from baseline (SE) | −50.1 (2.52) | −19.2 (3.14) | −49.3 (2.83) | −22.8 (3.48) |
LS mean difference (95% CI)e | −30.9 (−37.2 to −24.6) | −26.5 (−33.2 to −19.7) | ||
P valuef | < 0.0001 | < 0.0001 | ||
WI-NRS success,g week 8 (PRU4 ITT population) | ||||
N | 218 | 115 | 229 | 116 |
n (%) | 191 (87.6) | 97 (84.3) | 206 (89.9) | 101 (87.0) |
WI-NRS success, n (%) | 129 (67.5) | 26 (26.8) | 143 (69.4) | 36 (35.6) |
OR (95% CI)b | 7.84 (3.85 to 15.94) | 3.59 (2.07 to 6.23) | ||
P value | < 0.0001 | < 0.0001 | ||
Exploratory outcomes | ||||
DLQI, week 8 (ITT population) | ||||
N | 286 | 153 | 290 | 152 |
n (%) | 249 (87.0) | 130 (84.9) | 259 (89.3) | 128 (84.2) |
Baseline, mean (SD) | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| |
LS mean change from baseline (SE) | |||| |||||| | |||| |||||| | |||| |||||| | |||| |||||| |
LS mean difference (95% CI)h | |||| ||||| || ||||| | |||| ||||| || ||||| | ||
P valuei | < 0.0001 | < 0.0001 | ||
BSA, week 8 (ITT population) | ||||
N | 286 | 153 | 290 | 152 |
n (%) | 255 (89.1) | 132 (86.2) | 264 (91.0) | 130 (85.5) |
Baseline, mean (SD) | 6.28 (4.376) | 7.36 (4.762) | 7.08 (4.839) | 7.73 (5.054) |
LS mean percent change from baseline (SE) | |||||| ||||||| | |||||| ||||||| | |||||| ||||||| | ||||| ||||||| |
LS mean percent difference (95% CI)h | |||||| ||||||| || ||||||| | |||||| ||||||| || ||||||| | ||
P valuei | < 0.0001 | < 0.0001 | ||
Harms (safety population) | ||||
N | 286 | 153 | 290 | 152 |
Patients with ≥ 1 TEAE, n (%) | 72 (25.2) | 36 (23.5) | 75 (25.9) | 28 (18.4) |
Patients with ≥ 1 SAE, n (%) | 2 (0.7) | 1 (0.7) | 0 | 1 (0.7) |
Patients with ≥ 1 WDAE (from study treatment and/or study), n (%) | | ||||| | | ||||| | | ||||| | | ||||| |
Deaths, n (%) | 0 | 0 | 0 | 0 |
Notable harmsj (safety population) | ||||
N | 286 | 153 | 290 | 152 |
Application site pain, n (%) | 2 (0.7) | 1 (0.7) | 4 (1.4) | 0 |
Application site pruritus, n (%) | 1 (0.3) | 0 | 2 (0.7) | 1 (0.7) |
Application site urticaria, n (%) | 2 (0.7) | 0 | NA | NA |
Application site dryness, n (%) | 1 (0.3) | 0 | NA | NA |
Application site dermatitis, n (%) | 0 | 1 (0.7) | 0 | 1 (0.7) |
Application site irritation, n (%) | NA | NA | 0 | 1 (0.7) |
ANCOVA = analysis of covariance; BSA = body surface area; CI = confidence interval; DLQI = Dermatology Life Quality Index; IGA = Investigator’s Global Assessment; I-IGA = intertriginous Investigator’s Global Assessment; ITT = intention-to-treat; LS = least squares; NA = not applicable; OR = odds ratio; PASI 75 = 75% reduction in the Psoriasis Area and Severity Index score; PRU4 = patients with a WI-NRS pruritus score ≥ 4 at baseline; PRU4 ITT = subset of patients in the ITT population with a WI-NRS pruritus score of 4 or more at baseline; PSD = Psoriasis Symptom Diary; SAE = serious adverse event; SD = standard deviation; SE = standard error; TEAE = treatment-emergent adverse event; WDAE = withdrawal due to adverse event; WI-NRS = Worst Itch Numeric Rating Scale.
Note: The P value was adjusted for multiple testing of the primary and secondary outcomes using a prespecified hierarchical testing strategy.
aIGA success was defined as a score of 0 (clear) or 1 (almost clear) plus a ≥ 2-grade improvement from baseline.
bOdds ratio, 95% CI, and P value were obtained from the Cochran-Manel-Haenszel test (stratified by study site, baseline IGA, and baseline intertriginous involvement) comparing roflumilast cream 0.3% with vehicle.
cI-IGA success was defined as a score of 0 (clear) or 1 (almost clear) plus a ≥ 2-grade improvement from baseline in patients with intertriginous area involvement and I-IGA ≥ 2 at baseline. This analysis was based on the I-IGA ITT population.
dThe odds ratio, 95% CI, and P value were obtained from the Cochran-Mantel-Haenszel test (stratified by study site and baseline I-IGA) comparing roflumilast cream 0.3% with vehicle.
eEstimates for LS means (change from baseline and difference from vehicle), 95% CIs, and P values are from an ANCOVA with treatment, site, baseline IGA, baseline intertriginous involvement, and baseline PSD score as independent variables.
fThe P value is to test for a zero difference between groups (roflumilast cream 0.3% minus vehicle) in change from baseline.
gWI-NRS success was defined as a ≥ 4-point reduction in WI-NRS pruritus score from baseline in patients with a WI-NRS pruritus score ≥ 4 at baseline. The analysis was based on the PRU4 ITT population.
hEstimates for LS means (change or percentage change from baseline and difference from vehicle) and accompanying 95% CIs, and P values are from an ANCOVA with treatment, site, baseline IGA, baseline intertriginous involvement, and baseline of the variable being analyzed as independent variables.
iThe P value was not adjusted for multiple testing (i.e., the type I error rate was not controlled).
jPatients were counted once for each system organ class and once for each preferred term.
Sources: Clinical Study Reports for the DERMIS-19 and DERMIS-2 trials;10 details from the table were taken from the sponsor’s summary of clinical evidence.16
Critical Appraisal
Internal Validity
In the DERMIS-1 and DERMIS-2 trials, patients were randomized at baseline according to a computer-generated randomization list and randomization was stratified by study site, baseline IGA (2 versus ≥ 3), and intertriginous involvement at baseline (I-IGA ≥ 2, yes versus no). Based on input from the clinical expert consulted by CADTH for this review, the extent and severity of disease as measured by baseline BSA and PASI are additional effect modifiers. Note that the median and mean BSA and PASI scores were slightly higher in the vehicle arm compared with the roflumilast arm in both trials. IGA assesses severity of lesions, while BSA and PASI account for extent and severity of disease. Therefore, stratification by IGA alone may not result in an optimal comparability in disease severity between treatment arms, and this may have introduced bias in the efficacy results against roflumilast due to the aforementioned imbalance (note that the magnitude of this potential bias is not known). No other baseline demographic and clinical characteristics were identified that could have had a potential impact on the results in either study.
The primary efficacy outcome in both studies was IGA success at week 8. Conclusions about the validity and reliability of the 5-point IGA used in both studies are limited due to the use of the 6- and 7-point IGA in the psychometric validation studies; no evidence for responsiveness of IGA was identified. However, the clinical expert suggested that this difference in scales was unlikely to have introduced bias in the results. Although an MID has not been estimated, having a score of 0 (clear) or 1 (almost clear) on the static IGA has been generally accepted as clinically meaningful (i.e., a responder analysis would consider the proportion of patients with psoriasis who had a score of 0 or 1 in a clinical trial as treatment success).11,12 Alternatively, or in addition to having a score of 0 or 1, the responder analysis may also consider the proportion of patients with at least a 2-grade improvement from baseline on the static IGA.11 These outcomes were consistent with the definition of IGA success used in the studies.
The primary and secondary outcomes were controlled for multiplicity in both studies using a prespecified hierarchical testing strategy and the Holm procedure to control the familywise type I error. The planned sample size provided sufficient power to analyze up to the first 5 secondary end points; note that the remaining end points demonstrated statistically significant differences between treatment arms that were consistently in favour of roflumilast, with the exception of WI-NRS success at week 2 in the DERMIS-1 trial. The relatively small sample size of patients available for subgroup analysis, particularly patients aged 12 years to 17 years, significantly limited the interpretation of findings and the assessment of treatment benefit in this subgroup of patients. Moreover, as indicated by the clinical expert, the potential heterogeneity of treatment effect by extent and severity of disease as measured by BSA or PASI was not reported. This further compromised the certainty of evidence on the treatment effect of roflumilast among patients by different extent and severity of disease.
External Validity
Based on clinical expert input, the inclusion and exclusion criteria were considered narrow. For example, patients with an IGA score of 1 and a PASI score of 1 would be considered candidates for treatment with roflumilast in clinical practice in Canada; however, they were excluded from the trials, as a score of at least 2 was required for enrolment. Additionally, the clinical expert suggested that patients with plaque psoriasis involving less than 2% and more than 20% of BSA, excluding the scalp, palms, and soles, would potentially be treated with roflumilast but were also excluded from the trials. As such, the effect of roflumilast in the broader patient population is unknown. Note that the Health Canada indication does not restrict the patient population according to percent of BSA involvement.
In both studies, roflumilast was compared with a vehicle that contained only the excipients of the roflumilast cream. However, given the wide range of topical treatment options currently available in clinical practice for plaque psoriasis, the clinical expert agreed that an active comparator would have been more appropriate; in particular, topical steroids and vitamin D analogues would have been appropriate comparators. However, the vehicle cream may be considered an appropriate comparator, as there are limited options for intertriginous areas, which have been identified as an area of unmet need by the clinical expert and clinician groups.
The clinician groups and the clinical expert agreed that the primary, secondary, and selected exploratory outcomes in the trials were clinically relevant (i.e., to capture the extent and severity of disease and determine treatment response in clinical practice). Moreover, the clinical expert indicated that clearance or near clearance of psoriasis lesions is commonly used in clinical practice to assess psoriasis severity, which is analogous to the IGA used in clinical trials. Both the clinician groups and the clinical expert indicated that tools such as the PASI and DLQI are not commonly used in clinical practice unless mandated by the payers for reimbursement.
Long-Term Extension Study
Description of Study
The DERMIS-OLE (ARQ-151-306)17 trial is an ongoing, phase III, long-term open-label extension (OLE) study conducted to assess the long-term safety of roflumilast in adult and pediatric patients with chronic plaque psoriasis involving up to 25% of BSA. All patients received open-label roflumilast applied once daily to all psoriasis lesions (excluding the scalp) for up to 24 weeks. The study enrolled 267 patients aged 2 years and older, 266 of whom received treatment with roflumilast in the OLE. Patients in cohort 1 (n = 264) successfully completed a prior roflumilast cream study in psoriasis in which they received either roflumilast (n = 171) or vehicle (n = 93). Patients in cohort 2 (n = 2) were naive to treatment with roflumilast and had not yet reached OLE study week 4 at the time of the data cut-off date; therefore, efficacy data were not available for these patients. At the time of data cut-off, a total of 222 patients (83.1%) had completed the OLE study, 32 patients (12.0%) had prematurely discontinued the study, and 12 patients (4.5%) were ongoing in the study.
Efficacy Results
Among patients in cohort 1, at week 24 of the OLE, 50.0% of patients had IGA scores of clear or almost clear. A total of 67.8% of cohort 1 patients had a clear or almost clear IGA status from primary baseline and maintained that status for a median of 93 days. At week 24, a total of 37.1% of cohort 1 patients experienced treatment success based on IGA score, defined by a score of clear or almost clear plus at least a 2-grade improvement from baseline, and this was the case for 54.9% of patients for a median duration of 85 days. For cohort 1 patients with intertriginous area involvement (n = 59), at week 24, 77.8% had I-IGA scores of clear or almost clear and 75.6% experienced treatment success based on I-IGA, defined as a score of clear or almost clear plus at least a 2-grade improvement from baseline. The proportions of cohort 1 patients with PASI 50, PASI 75, and PASI 100 at week 24 of the OLE were 70.5%, 43.8%, and 16.5%, respectively. The proportion of cohort 1 patients who experienced treatment success based on the WI-NRS at week 24, defined as an improvement of 4 or more points, was 62.4%. Overall, the results of the DERMIS-OLE study suggest that efficacy was maintained for up to 24 weeks.
Harms Results
No new safety signals were reported based on the OLE study. No AEs were reported among the patients in cohort 2. Among cohort 1 patients, 26.1% experienced at least 1 AE, the most common of which were sinusitis (2.7%), diarrhea (2.3%), COVID-19 (1.9%), and headache (1.9%). Three patients (1.1%) experienced a total of 5 SAEs, including polycythemia vera, COVID-19 pneumonia, palpitations, dehydration, and syncope, none of which were considered related to the drug. One patient (0.4%) discontinued the study due to an AE of application site irritation. No deaths occurred during the OLE study.
Critical Appraisal
The limitations of the extension study include selection bias, lack of a control group, and a lack of blinding. Reporting of harms and subjective efficacy outcomes such as IGA and I-IGA success may be biased by knowledge of treatment received. As only descriptive statistics were published in this interim report, and without comparator groups, the interpretation of the results is limited. Moreover, there is potential for selection bias, as patients who discontinued the parent studies due to AEs, lack of efficacy, or other reasons were excluded. Furthermore, most patients were white (> 84%), which may limit the generalizability of results to other racial groups. However, the clinical expert consulted by CADTH indicated that the efficacy and safety of roflumilast is not expected to differ by race.
Indirect Comparisons
Description of Studies
One sponsor-submitted indirect treatment comparison (ITC) was included, consisting of a systematic literature review and a Bayesian network meta-analysis (NMA) comparing roflumilast with the other topical therapies available in Canada in patients with plaque psoriasis.18 The primary outcome of interest was IGA treatment response at week 8, which was informed by data from 8 studies, including the DERMIS-1 and DERMIS-2 trials. A subgroup analysis of I-IGA treatment response at week 8 among patients with intertriginous psoriasis was informed by data in 4 studies, including the DERMIS trials.
Efficacy Results
The NMA results for IGA treatment response at week 8 favoured roflumilast versus vehicle, vitamin D analogues, tazarotene, or corticosteroids. IGA treatment response data for roflumilast versus corticosteroids plus vitamin D analogues or corticosteroids plus tazarotene did not clearly favour either treatment, as the 95% credible intervals (Crls) included the null value. For the fixed-effect NMA of I-IGA treatment response at week 8, roflumilast was associated with improved odds of treatment response versus vehicle ||| ||||||| || || |||| || |||||||. The results found that roflumilast versus calcineurin inhibitors did not clearly favour either treatment, as the 95% CrIs included the null value.
No harms or HRQoL outcomes were analyzed in the NMA.
Critical Appraisal
Potential sources of heterogeneity could not be fully assessed in the NMA due to the limited reporting of study design characteristics (i.e., inclusion criteria, frequency of treatment withdrawal, and handling of missing data) and patient baseline characteristics (i.e., disease history duration, prior treatment experience, PASI score, and BSA involvement) and, as such, there is uncertainty in the validity of the results of the NMA. The clinical expert consulted for this review noted there were imbalances across treatment groups in effect modifiers of baseline disease severity. As a result, it is possible that the heterogeneity in this baseline characteristic could result in changing the relative treatment effects. The outcomes were limited to the analysis of IGA and I-IGA treatment response; therefore, other relevant efficacy outcomes such as PASI and HRQoL (i.e., DLQI) were not assessed. Long-term efficacy and safety outcomes were not assessed, limiting the external validity of the results. These limitations result in uncertainty in the relative treatment effect estimates between roflumilast and other comparable topical therapies.
Studies Addressing Gaps in the Pivotal and Randomized Controlled Trial Evidence
No additional studies were included in this report for the review of roflumilast.
Conclusion
Two sponsor-submitted, phase III, randomized, double-blind, parallel-group, vehicle-controlled trials (the DERMIS-1 and DERMIS-2 trials) comparing topical treatment with roflumilast versus a matching vehicle in patients aged 2 years or older with chronic plaque psoriasis involving 2% to 20% (inclusive) of BSA, excluding the scalp, palms, and soles, demonstrated that 8 weeks of treatment with roflumilast reduced the overall severity of psoriasis, as measured by the proportion of patients experiencing treatment success based on the IGA compared with the matching vehicle. Roflumilast also demonstrated a reduction in the severity of psoriasis in intertriginous areas (identified as an area of unmet need), extent and severity of psoriasis, and severity of itch compared with the matching vehicle, as measured by the proportion of patients experiencing treatment success based on the I-IGA, a prespecified reduction in the PASI, and treatment success based on the WI-NRS, respectively, as well as improvement in QoL (specifically impacted by psoriasis) compared with the matching vehicle, as measured by PSD. There was supportive evidence on the overall treatment benefit of roflumilast versus vehicle on HRQoL, as measured by DLQI, and extent of disease, as measured by BSA. Of note, the effect of roflumilast in a broader patient population (e.g., those with < 2% and > 20% of BSA affected) and in combination therapy (and as adjunct therapy) in clinical practice is unknown, the line of therapy that would be supported by the evidence from both trials remains unclear, and the comparative efficacy of roflumilast versus other relevant comparators is uncertain in the absence of direct comparative evidence in the treatment of plaque psoriasis of the whole body. Indirect evidence provided by the sponsor-submitted NMA suggested that roflumilast may offer a benefit over vitamin D analogues, tazarotene, or corticosteroids based on IGA treatment response after 8 weeks of treatment, but the results did not clearly favour either treatment when compared with combination treatments (corticosteroids plus vitamin D analogues or corticosteroids plus tazarotene). Further, there was no clearly favoured treatment when compared with calcineurin inhibitors for I-IGA treatment response at week 8. The results of the NMA must be considered alongside potential uncertainty in the validity of the NMA results, as several potential sources of heterogeneity were not assessed, stratified analyses were not conducted to adjust for effect modifiers, and the similarity assumption of the NMA may have been violated; however, the extent of the potential bias is unknown. There were no safety or tolerability concerns associated with the use of topical roflumilast and no new safety signals were reported in the DERMIS-OLE study.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of roflumilast cream 0.3% for topical use in the treatment of plaque psoriasis, including treatment of psoriasis in the intertriginous areas, in patients 12 years of age and older.
Disease Background
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the CADTH review team.
Psoriasis is a chronic inflammatory skin disease that is associated with multiple comorbidities, such as psoriatic arthritis, obesity, metabolic syndrome, hypertension, diabetes, and atherosclerotic disease.1 Psoriasis is a complex, immune-mediated disease in which dysregulation of the immune system (involving interactions between dendritic cells, T-cells, neutrophils, and cytokines) and keratinocyte hyperproliferation contribute to the manifestation of the disease and the typical clinical findings of scaling, induration, and erythema.1,2 Risk factors for psoriasis include genetics and environmental and behavioural factors. Psoriasis can present similarly in children and adults; the peak ages of onset are between 30 and 39 years and between 50 and 69 years. Of note, children with chronic plaque psoriasis are more likely to present with facial involvement compared with adults.1
Based on a systematic review of worldwide literature, the estimated prevalence of psoriasis in adults ranged from 0.51% to 11.43% and from 0% to 1.37% in children.19 However, the prevalence of psoriasis varies geographically20 and the authors acknowledged a geographic gap in knowledge, as the prevalence data were derived from only 20 countries.19 A panel of 11 dermatologists with expertise in psoriasis estimated that the median prevalence of psoriasis is 3% of the general adult population in Canada. The panel estimated that approximately 78% of patients with psoriasis have less than 10% of BSA involved (i.e., mild or moderate disease) and 22% of patients have 10% or more of BSA involved. The panel further estimated that 50% of patients have less than 3% of BSA involved (i.e., mild disease) and 2% of patients have more than 50% of BSA involved.3 Based on estimates in the US, 50% of patients have facial involvement4 and 21% to 30% of patients have intertriginous area involvement.5
Chronic plaque psoriasis (also known as psoriasis vulgaris6) is the most common clinical subtype of psoriasis, representing approximately 90% of patient cases in Canada.7 Plaque psoriasis is characterized by sharply defined, erythematous cutaneous plaques with overlying, coarse, silvery scales.1 The diameter of plaques can range from less than 1 cm to more than 10 cm. Plaques can occur as single lesions or as generalized disease across wider areas of the body.6 Plaques can be asymptomatic; however, pruritus is commonly reported by patients and painful fissures can be present in palm or sole involvement. The scalp, elbows, knees, and gluteal cleft are common areas of involvement. Intertriginous areas (inverse or flexural psoriasis6), ear canals, umbilicus, palms, soles, and nails are possible areas of involvement. Intertriginous psoriasis is characterized by well demarcated, smooth, shiny plaques with no to minimal scales. Genital involvement, in particular, may have a negative impact on the patient’s QoL.1
For most patients, the diagnosis of chronic plaque psoriasis can be made by history and physical examination. A skin biopsy may be performed, if required, such as in cases where the diagnosis remains uncertain following a history and physical examination of the patient or to rule out other conditions.1
According to the Canadian Guidelines for the Management of Plaque Psoriasis,6 the definitions for mild, moderate, and severe plaque psoriasis based on measures of disease severity differ, depending on the setting (clinical trials versus clinical practice). For example, mild plaque psoriasis may be defined as an upper limit of 5% BSA involvement in clinical trials,21 while mild plaque psoriasis in clinical practice may be described as disease with a minimal impact on QoL (subjective evaluation of the impact of disease on the patient’s life), and the patient is able to have an acceptable level of symptomatic control with routine skin care and/or topical therapy (objective evaluation of the extent and symptoms of the disease).6
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following have been summarized and validated by the CADTH review team.
Plaque psoriasis requires lifelong follow-up and treatment. Measures of treatment success in clinical practice may include clearance (absence of signs of disease), control (satisfactory response to therapy as defined by the patient and/or clinician), and remission (maintenance of disease control or suppression of signs and symptoms over time).6 The clinical expert consulted by CADTH for the purpose of this review identified the treatment goals in clinical practice as sufficient clearing of psoriasis lesions resulting in improvement in HRQoL and reduced symptoms (itch and pain) while minimizing drug-related adverse effects. According to the input from patient groups that was received for this submission, patients with psoriasis have identified the following as key outcomes of new treatments: effectiveness (i.e., treatment of publicly visible body areas to improve mental health, treatment of genital psoriasis to improve intimacy with loved ones, and treatment of symptoms, such as itching and burning, to resume normal daily activities), lack of side effects, and a medication that is conducive to their schedule and has the flexibility to be applied to all affected BSAs.
According to the Canadian Guidelines for the Management of Plaque Psoriasis,6,8 the management of mild plaque psoriasis in clinical practice, specifically disease involving the trunk, limbs, and neck, includes topical therapies that can be broadly categorized as corticosteroids, vitamin D3 analogues, retinoids, anthralin (commercial formulations of anthralin are not currently available in Canada) and tars, as well as combination therapies. According to the clinical expert, anthralin and tars are not commonly used in clinical practice and they were not aware of any that are commercially available in Canada. Corticosteroids are the mainstay of therapy and are widely used for the topical treatment of psoriasis, as they are often effective and well tolerated. However, long-term use of topical corticosteroids and, in particular, high-potency steroids, is limited by their potential to produce side effects, for example, local cutaneous changes and hypothalamic-pituitary-adrenal axis suppression. Corticosteroids are available in a variety of formulations, including creams, ointments, gels, lotions, sprays, foams, and solutions.6,8 Topical vitamin D3 analogues are most commonly associated with mild irritant contact dermatitis.6 The use of topical retinoids as monotherapy is limited by application site irritation (itching, burning, and erythema), a dose-dependent effect.6 Tars are often associated with staining and irritation and their use has declined since the introduction of therapeutic alternatives that are typically more acceptable to patients.6 Compared with monotherapy, combination therapy is generally more effective and can reduce the incidence of adverse effects. The therapeutic options for pediatric patients are generally similar to the options for adult patients.6
Management of intertriginous psoriasis, affecting the groin, axillae, inframammary region, abdominal body folds, gluteal cleft, perianal region, and retroarticular fold areas can be a challenge, as these areas are at an increased risk of adverse reactions to topical treatment, such as corticosteroids, because the skin tends to be thinner in these regions.6 Additionally, there are currently no available treatments indicated for intertriginous psoriasis. Although vitamin D analogues have demonstrated efficacy in facial and intertriginous psoriasis, they are associated with irritation and erythema; calcipotriol is not approved for use on the face or intertriginous areas, and calcitriol carries a warning against facial use. Although topical calcineurin inhibitors may be an appropriate treatment option for the management of intertriginous psoriasis, they are not approved for an indication in psoriasis (off-label use). These treatment options may be supplemented as needed for short periods with topical corticosteroids.6,8
Many of the standard topical therapies for the treatment of mild chronic plaque psoriasis can result in good control of mild psoriatic disease but have distinct profiles of adverse effects, tolerability, and convenience which can have an impact on HRQoL, adherence, and effectiveness of treatment. It should also be noted that psoriatic presentation, personal values, psychosocial health, and treatment expectations can vary between patients.6 Consequently, the Canadian Guidelines for the Management of Plaque Psoriasis indicates that the selection of treatments should be individualized to the patient, with the patient participating in the choice of therapy and with consideration of the type of vehicle that would be acceptable to the patient to improve adherence, achieve treatment success, and support overall patient satisfaction.6
Moderate to severe chronic plaque psoriasis in clinical practice can be defined as psoriasis affecting the trunk and extremities that cannot be adequately controlled by the aforementioned approaches, with adequate control defined by the patient’s perception of the disease and its burdens.6 According to the Canadian Guidelines for the Management of Plaque Psoriasis, management of moderate to severe plaque psoriasis includes systemic therapy with traditional and biologic drugs, phototherapy, combination therapy, and topical therapy as adjunct therapy.6 For some patients with moderate to severe psoriasis, amelioration (short-term improvement and limited long-term disease control) may be considered an adequate treatment goal, while full clearance may be considered an appropriate treatment goal for many patients, particularly with the introduction of biologic therapy.6 The clinical expert consulted by CADTH for the purpose of this review indicated that phototherapy would not typically be used for intertriginous areas.
Of note, the clinical expert consulted by CADTH for the purpose of this review indicated that if lesions are cleared or almost cleared, but QoL continues to be impacted by the need to apply time-consuming and messy topical therapies, then phototherapy or systemic therapy may be considered.
Drug Under Review
Key characteristics of roflumilast are summarized in Table 3 along with other treatments available for plaque psoriasis.
Roflumilast cream 0.3% is indicated for topical treatment of plaque psoriasis, including treatment of psoriasis in the intertriginous areas, in patients 12 years of age and older; the recommended dosage is to apply to affected areas once daily. The Health Canada indication is aligned with the sponsor’s reimbursement request. Roflumilast cream has not been previously reviewed by CADTH. Roflumilast is a selective phosphodiesterase type 4 inhibitor and a nonsteroidal, anti-inflammatory drug, which is thought to exert its therapeutic action through inhibition of phosphodiesterase type 4 and subsequent inhibition of inflammatory markers associated with plaque psoriasis.22
Table 3: Key Characteristics of Roflumilast and Other Relevant Topical Therapies for the Treatment of Plaque Psoriasis
Characteristic | Roflumilast | Corticosteroids (e.g., betamethasone, mometasone, clobetasol propionate) | Vitamin D3 analogues (e.g., calcitriol, calcipotriol) | Retinoids (e.g., tazarotene) | Combination treatments (e.g., calcipotriol and betamethasone dipropionate, halobetasol propionate and tazarotene) |
|---|---|---|---|---|---|
Mechanism of action | Roflumilast is a selective PDE-4 inhibitor and a nonsteroidal, anti-inflammatory drug that is thought to exert its therapeutic action through inhibition of PDE-4 and subsequent inhibition of inflammatory markers associated with plaque psoriasis | Via multiple mechanisms; acts as an anti-inflammatory and immune suppressant | Calcipotriol is a nonsteroidal antipsoriatic drug that, like the active form of vitamin D, regulates cell proliferation and differentiation | Retinoids are thought to modulate keratinocyte proliferation and differentiation, in addition to having anti-inflammatory effects | Calcipotriol and betamethasone: Combination of a vitamin D3 analogue and corticosteroid Halobetasol propionate and tazarotene: Combination of a super potent topical corticosteroid and a retinoid prodrug |
Health Canada indication | For topical treatment of plaque psoriasis, including treatment of psoriasis in the intertriginous areas, in patients 12 years of age and older | Symptomatic relief of acute and chronic skin eruptions where anti-inflammatory, antiallergenic, and antipruritic activity is required Topical corticosteroids are widely used for many other causes of skin inflammation | Topical treatment of psoriasis Combination use with a moderate to very potent topical corticosteroid, cyclosporin A, or acitretin | Topical treatment of plaque psoriasis | Calcipotriol and betamethasone: Topical treatment of psoriasis vulgaris in adults Halobetasol propionate and tazarotene: For improving the signs and symptoms of plaque psoriasis in adult patients with moderate to severe plaque psoriasis |
Route of administration | Topical | Topical | Topical (available as a cream, ointment, and scalp solution) | Topical (gel or cream) | Topical (available as a gel, ointment, foam, or lotion) |
Recommended dosage | Apply to affected areas once daily | Varies between drugs | Varies between drugs | Apply in a thin layer to affected area once a day | Varies between drugs |
Serious adverse effects or safety issues | Contraindicated in patients with moderate to severe liver impairment (Child-Pugh B or C) and patients who are hypersensitive to this drug or to any ingredient in the formulation, including any nonmedicinal ingredient or component of the container | If used under an occlusive dressing, particularly over extensive areas or on the face, scalp, axilla, or scrotum, or when applied to the genitourinary tract or oral mucosa, or when administered rectally, sufficient absorption may take place to give rise to adrenal suppression and other systemic effects Children may be at greater risk of developing systemic complications with the use of topical corticosteroids | Carcinogenesis, serum calcium abnormalities, and renal impairment Contraindicated in patients with hypersensitivity to the drug or formulation, hypercalcemia, systemic treatment of calcium homeostasis, renal impairment or ESRD, or liver dysfunction | Skin irritation Contraindicated in patients with hypersensitivity to retinoic compounds, patients with seborrheic dermatitis, and pregnant patients | Calcipotriol and betamethasone: Safety issues are similar to those listed for topical corticosteroids and vitamin D3 analogues Halobetasol propionate and tazarotene: Reversible HPA axis suppression, infection Contraindicated in pregnant patients and those who are hypersensitive or have viral skin lesions, skin infections, or seborrheic dermatitis |
Other | For topical use only; not for ophthalmic, oral, or intravaginal use | NA | Should not be applied to the eyes, lips, or facial skin (calcitriol) | NA | Not recommended for use on face, axillae, flexures, groin, genitals, scalp, or intertriginous areas Halobetasol propionate and tazarotene should not be used on eczematous skin or with occlusive dressings |
ESRD = end-stage renal disease; HPA = hypothalamic-pituitary-adrenal; NA = not applicable; PDE-4 = phosphodiesterase type 4.
Sources: Canadian Guidelines for the Management of Plaque Psoriasis6 and the product monographs for roflumilast (Zoryve);22 calcitriol (Silkis);23 calcipotriol (Dovonex);24 halobetasol propionate and tazarotene (Duobrii);25 and calcipotriol and betamethasone dipropionate ointment, gel, and aerosol foam (Dovobet, Enstilar).26-28
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups. The full original patient inputs received by CADTH have been included in the stakeholder section at the end of this report.
Three patient groups submitted a joint patient input submission. CSPA, the Canadian Psoriasis Network, and the Canadian Association of Psoriasis Patients are national, not-for-profit organizations that are dedicated to improving the lives of people with psoriasis across Canada. A survey conducted between January and February 2023, in English and French, was hosted by the CSPA and it invited investigators to share the survey with their clinical trial participants. This survey collected a total of 86 responses from across Canada except for Yukon and Nunavut. Respondents were older than 65 years (26%), 55 to 64 years (21%), 25 to 34 years (19%), 35 to 44 years (16%), 45 to 54 years (16%), and younger than 18 years (3%).
A vast majority (94%) of respondents were living with psoriasis and 6% were caregivers or family members. The impact of psoriasis symptoms was considerable in various areas of patients’ lives, such as mental well-being (14 patients; 24%), daily tasks (10 patients; 17%), intimate relationships (8 patients; 13%), and social lives (7 patients; 12%). Patients also described the challenges of treating and living with their psoriasis as expensive and stressful.
Patients who responded to the survey hosted by the CSPA reported living with psoriasis of varying disease severity, with 26% reporting mild disease, 52% reporting moderate disease, and 22% reporting severe disease. Patients reported that the body areas most impacted by psoriasis include the scalp, followed by the legs, arms, genitals, hands, torso, skin folds, and palms, respectively. Most patients (90%) considered effectiveness as the most important factor to be addressed in any new treatment for psoriasis. In addition, 66% of patients valued lack of side effects, 60% prioritized affordability, 53% preferred treatments that are easy to apply, and 23% preferred treatments that are conducive to their schedules. Based on the survey, a substantial proportion of the respondents were using topical corticosteroids (36%), topical combination treatment (32%), and/or biological drugs (27%), which was the most effective current treatment, with 39% and 30% of patients reporting that biologics work well and very well, respectively. More than half of the respondents (53%; n = 33) in a survey reported facing side effects with the existing treatments available for psoriasis. The majority of patients with psoriasis (76%; n = 47) have discontinued their treatment at some point during their disease, with the most frequent reasons being that the treatment stopped working (61%; n = 37), caused side effects (47%; n = 28), was unaffordable (22%; n = 13), or ineffective (14%; n = 7). Among respondents, 10 patients reported experience with roflumilast, accessed through a clinical trial. Nine of these patients reported benefits of treatment, including clearing of skin, reduced itch and redness, clearing of skin lesions (plaques), and the treatment’s ease of application. All 10 respondents tolerated roflumilast well, except for 1 who experienced some itching.
The patient group input emphasized that psoriasis is a chronic and potentially debilitating disease that poses many challenges and is linked to anxiety, depression, and social isolation. This disease can interfere with social and intimate relationships, productivity, and family life and work life. Furthermore, due to the chronic nature of this disease, patients are concerned about recurrence and resistance to treatments in the future.
Clinician Input
Input From the Clinical Expert Consulted By CADTH
All CADTH review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., assisting in the critical appraisal of the sponsor’s summary of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 1 clinical specialist with expertise in the diagnosis and management of plaque psoriasis.
Unmet Needs
The clinical expert indicated that there are currently no available treatment options that can permanently reverse the course of plaque psoriasis, but systemic therapy can suppress psoriasis while it is being used.
The clinical expert also indicated that many patients do not respond to currently available topical treatments; in particular, widespread psoriasis and psoriasis of the scalp, palms, and soles are often refractory to topical treatment. In such cases, patients would need to weigh the associated risks and benefits of treatment with phototherapy or systemic therapy. Furthermore, over time, psoriasis is more likely to become refractory to systemic therapy than to topical therapy. The clinical expert noted that although topical therapies are generally well tolerated and that topical steroids are used over the long-term by many patients in clinical practice, clinical guidelines caution against long-term use of topical steroids for plaque psoriasis. Thus, the clinical expert agreed that more effective, safer, and better-tolerated treatment options are an unmet need, particularly for long-term use and in patients with facial, genital, and intertriginous involvement.
The clinical expert suggested that topical formulations that are less messy and more effective may improve adherence; in particular, in patients with intertriginous involvement who are offered topical tacrolimus, as it is only available as an ointment and not widely available because it is not indicated (used off-label) for the treatment of psoriasis.
Place in Therapy
The clinical expert considered roflumilast to be an anti-inflammatory drug, similar to other currently available topical treatments. For most patients with plaque psoriasis, the clinical expert suggested they would consider roflumilast as an alternative to other first-line topical treatments for psoriasis within the current stepped approach, as this could prevent having to step up patients to systemic therapy. For patients with facial, genital, and intertriginous involvement, the clinical expert suggested roflumilast would be a first-line treatment. The clinical expert further indicated that roflumilast would be preferred in scenarios where steroids should be avoided (e.g., intertriginous or facial involvement and due to patient preference). The clinical expert suggested that roflumilast may be preferred for long-term use in some patients and by clinicians, given that it is not a steroid.
For mild disease, the clinical expert suggested that roflumilast would be used as monotherapy. Additionally, depending on patient preference, roflumilast could be used simultaneously with other topical treatments, for example, roflumilast could be used for intertriginous areas and a separate topical treatment in a foam or lotion formulation could be used for the scalp. For extensive psoriasis, potent topical steroids would not be suggested and roflumilast would be considered a suitable topical alternative. However, most patients with extensive psoriasis will need phototherapy or systemic therapy to reach treatment targets. Roflumilast could also be used in combination with systemic therapy or phototherapy, as topical treatments are typically continued as needed.
For most patients with plaque psoriasis, the clinical expert indicated it would be appropriate to suggest treatment with topical steroids with or without vitamin D analogues before suggesting treatment with roflumilast, as they are considered effective and affordable. However, for patients with intertriginous psoriasis, the clinical expert suggested it is not necessary to offer other treatments before roflumilast, given the limited treatment options available specifically for intertriginous areas affected by psoriasis.
Patient Population
The clinical expert identified patients with active psoriasis, regardless of whether it is being actively treated or not, as the most in need of an intervention. The clinical expert noted there is a need for an intervention for the hair-bearing scalp area; however, they indicated that this need would not be addressed by roflumilast. Clinician examination and judgment, together with shared decision-making with the patient, would determine whether the drug is best suited for the patient. The clinical expert suggested that patients with psoriasis with limited BSA involvement are the most likely to respond to therapy with roflumilast as monotherapy.
Although the diagnosis is straightforward and biopsy tests are not needed for most patients, the clinical expert noted that psoriasis of special sites (scalp, intertriginous areas, palms, and soles) can be difficult to differentiate from other conditions, such as eczema; however, this is uncommon in clinical practice.
The clinical expert also noted it is possible for patients to present with only intertriginous or genital involvement but such patients may be underrecognized in clinical practice, as such presentations are often presented in the context of psoriasis on other areas of the body.
Assessing the Response to Treatment
The clinical expert indicated that clearance or near clearance of psoriasis lesions is commonly used to assess psoriasis severity; this is analogous to the IGA scales used in clinical trials. The clinical expert also indicated that patient satisfaction is important and is assessed in a gestalt manner that likely differs between physicians. Components of patient satisfaction can include improvement in symptoms (e.g., itch), the appearance of lesions, QoL, sexual function (relevant in intertriginous psoriasis), and how convenient and pleasant the treatment is from the patient’s perspective.
According to the clinical expert, extensive outcome scales, such as the PASI, DLQI, WI-NRS, and PSD are not typically used in clinical practice; however, the PASI and DLQI may be used if mandated by the insurer to fund a drug.
Discontinuing Treatment
According to the clinical expert, intolerance (e.g., stinging at the application site, odour, or unpleasant sensation when applying the medication) and ineffectiveness (i.e., minimal improvement after 2 to 8 weeks of application) are factors to consider when deciding to discontinue treatment with roflumilast.
The clinical expert further indicated that roflumilast would be discontinued when lesions have cleared and can be restarted if and when lesions have recurred. The clinical expert indicated that phototherapy or systemic therapy may be considered for extensive psoriasis regardless of improvement with roflumilast and, if there is substantial improvement with phototherapy or systemic therapy that obviates the need for topical therapy, then roflumilast would be considered for discontinuation.
Prescribing Considerations
The clinical expert suggested that roflumilast could be initiated and continued by general practitioners and specialists in any treatment setting, including community and hospital practices.
Clinician Group Input
This section was prepared by CADTH staff based on the input provided by clinician groups. The full original clinician group input received by CADTH has been included in the stakeholder section at the end of this report.
Three different clinician groups provided input, Fraser Health Dermatology (4 clinicians), the Canadian Dermatology Association (3 clinicians), and the Atlantic Provinces Dermatology Association and Dermatology Association of Ontario (12 clinicians).
Clinicians have reached a consensus aligned with the clinical expert consulted by CADTH that there is a need for a new treatment as an alternative to existing therapies for plaque psoriasis, which include topical steroids, topical vitamin D analogues and steroid combinations, vitamin A analogues and steroid combinations, and so forth. The clinician groups mentioned different drawbacks for the currently available treatments, including AEs (e.g., topical corticosteroids), poor compliance (e.g., calcipotriol and betamethasone dipropionate ointment or calcipotriene and betamethasone dipropionate foam), difficult application, high cost for patients and the health care system (e.g., phototherapy and biologic therapies), limited efficacy (e.g., vitamin D analogues and steroid combinations), intolerability due to irritation (e.g., vitamin A analogues and steroid combinations) and inability to be administered to all areas of the body (e.g., topical corticosteroids). Lastly, patient compliance decreases when more than 1 topical therapy is prescribed.
The input from the Canadian Dermatology Association was aligned with the opinion of the clinical expert consulted by CADTH that traditional use of long-term steroids can be limiting, with potential local and systemic side effects. The clinician groups concurred that treatment goals include reducing the severity of symptoms, minimizing AEs, improving tolerability and efficacy, improving QoL, and reducing the burden on patients and health care systems.
Input from the Fraser Health Dermatology clinicians indicated topical roflumilast should be used after the failure of topical steroids (i.e., when a patient’s condition does not respond very well to steroids as a first line of treatment) because topical steroids are an affordable and usually well tolerated therapy, which was consistent with the input provided by the clinical expert consulted by CADTH. Of note, the Fraser Health Dermatology clinicians suggested roflumilast is the first choice for intertriginous areas due to the limited number of available treatments. In contrast, the Atlantic Provinces Dermatology Association and Dermatology Association of Ontario suggested prescribing roflumilast before the use of current combination therapies or as a first-line treatment for the management of mild to severe plaques on any area of the body due to the high clinical efficacy and favourable safety profile of roflumilast. The input noted that patients best suited for treatment with the drug under review included patients with mild to moderate psoriasis, patients with different disease phenotypes, patients with psoriasis that has failed to respond to topical steroids, and patients with intertriginous psoriasis, noting the importance of steroid sparing in these anatomic sites.
According to Fraser Health Dermatology and the Atlantic Provinces Dermatology Association, the best outcomes to determine treatment response would be PGA score and BSA involvement. The groups noted that the PASI score is not commonly used in clinical practice, except when applying for coverage for systemic therapies in patients with moderate to severe disease. Clinician groups noted that assessment of treatment goals is recommended after 8 weeks and that, typically, patients with psoriasis are initially seen every 3 to 6 months to assess response to treatment. The input received stated that patients with psoriasis are diagnosed and treated as outpatients, typically by both specialists and general practitioners or family doctors.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical expert consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
Roflumilast cream 0.3% was compared with the matching vehicle over 8 weeks in 2 phase III studies. Given the wide range of topical treatment options available for plaque psoriasis, is the vehicle an appropriate comparator? | The clinical expert consulted indicated that an active comparator would have been more appropriate. In particular, the expert suggested that topical steroids and vitamin D analogues would have been appropriate comparators. |
Roflumilast is a first-in-class topical selective inhibitor of PDE-4. If roflumilast receives Health Canada approval, it will be the only topical cream indicated for the treatment of plaque psoriasis in the intertriginous areas. | Comment from the drug programs to inform CDEC deliberations. |
Is there any evidence to suggest what contributes to nonadherence with topical therapies, which may contribute to reduced efficacy of plaque psoriasis treatments? | The clinical expert consulted indicated that, anecdotally, patients find topical treatment to be inconvenient due to the messiness, appearance, and time-consuming nature of applying topicals. The clinical expert did not have experience with roflumilast but, based on a survey of patients with eczema,29 they advised that time spent managing the disease was significantly associated with overall disease burden; however, this trial was a cross-sectional study and cannot establish causality. |
Calcineurin inhibitors are not officially indicated for the treatment of plaque psoriasis. Should calcineurin inhibitors be included as a comparator? | The clinical expert consulted suggested that calcineurin inhibitors are a relevant comparator, particularly for intertriginous psoriasis. The clinical expert acknowledged there may be regulatory or other feasibility issues with conducting a study with an off-label comparator. |
Considerations for initiation of therapy | |
Patients with plaque psoriasis lesions on the scalp were excluded from clinical trials. These patients would likely require an alternative topical drug for scalp lesions. | Comment from the drug programs to inform CDEC deliberations. |
Halobetasol propionate and tazarotene (Duobrii) was the product most recently reviewed by CADTH in this treatment space. The patient population may not align with the clinical criteria recommended for Duobrii. Most jurisdictions list a variety of topical corticosteroids as eligible for full benefit. The listing status of retinoids, vitamin D analogues, and fixed combinations vary across jurisdictions. Calcineurin inhibitors may or may not be an appropriate comparator and are not listed for plaque psoriasis in a number of jurisdictions. | Comment from the drug programs to inform CDEC deliberations. |
Considerations for continuation or renewal of therapy | |
Is the IGA used commonly in clinical practice? Are family physicians familiar with using the IGA? | The clinical expert consulted indicated that formal IGAs are not common in clinical practice, particularly in family practice. However, the clinical expert noted that IGAs are a fairly simple way to capture a clinician’s gestalt assessment of disease severity. The clinical expert consulted indicated that physicians (generalists and specialists) commonly use the terms mild, moderate, and severe informally, which can be considered similar to formal IGAs. Additionally, the clinical expert indicated that assessment of response in clinical practice includes a gestalt assessment of disease history, physical examination, and global patient satisfaction. |
Most other restricted drugs in this treatment space do not have assessment criteria for renewal (i.e., no renewal parameters are required to be submitted for evaluation as part of the criteria), with the exception, in a number of jurisdictions, of Duobrii, which uses the IGA. | Comment from the drug programs to inform CDEC deliberations. |
Considerations for prescribing of therapy | |
Regarding concerns related to combination usage, is there any evidence to support combination use with other topical treatments for plaque psoriasis? Is there any evidence to support combination use with biologic therapies for the treatment of plaque psoriasis? | The clinical expert consulted indicated it is common in clinical practice for individual patients to be using multiple topical medications simultaneously (e.g., apply 1 topical medication to the scalp, 1 to intertriginous areas, and 1 to plaques on the body). The clinical expert also indicated that a topical medication is typically offered to patients along with phototherapy or systemic or biologic therapy in clinical practice to treat any residual disease not responding to phototherapy or systemic or biologic therapy. The CADTH review team notes that no evidence for the use of roflumilast in combination with other topical or systemic treatments for psoriasis was identified for this review. |
Generalizability | |
There may be interest in using this product in children younger than 12 years of age. | Comment from the drug programs to inform CDEC deliberations. |
Care provision issues | |
Roflumilast would be available in a 60 g tube ($275.00 per tube) applied once daily (a frequency of administration that would be considered convenient). | Comment from the drug programs to inform CDEC deliberations. |
System and economic issues | |
Consideration should be given to topical therapies that may prevent patients from progressing to more costly or invasive systemic therapies, including biologics. | Comment from the drug programs to inform CDEC deliberations. |
CDEC = CADTH Canadian Drug Expert Committee; IGA = Investigator’s Global Assessment; PDE-4 = phosphodiesterase type 4.
Clinical Evidence
The objective of CADTH’s Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of roflumilast cream 0.3% for topical use in the treatment of plaque psoriasis, including treatment of psoriasis in the intertriginous areas, in patients 12 years of age and older. The focus will be placed on comparing roflumilast with relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of roflumilast is presented in 3 sections, and CADTH’s critical appraisal of the evidence is included after each section. The first section, the systematic review, includes pivotal studies that were selected according to the sponsor’s systematic review protocol. The second section includes sponsor-submitted long-term extension studies. The third section includes indirect evidence from the sponsor.
Included Studies
Clinical evidence from 2 pivotal studies, 1 long-term extension study, and 1 ITC is included in the CADTH review and appraised in this document.
Pivotal Studies
Contents within this section have been informed by materials submitted by the sponsor. The following has been summarized and validated by the CADTH review team.
Description of Studies
Characteristics of the included studies are summarized in Table 5.
The primary objectives of the DERMIS-1 and DERMIS-2 studies were to assess the safety and efficacy of roflumilast cream 0.3% versus vehicle cream. The studies were identically designed phase III, randomized, parallel-group, double-blind, vehicle-controlled studies in which roflumilast or a matching vehicle was administered topically once daily for 8 weeks to patients with chronic plaque psoriasis involving 2% to 20% (inclusive) of BSA, excluding the scalp, palms, and soles (DERMIS-1, N = 439; DERMIS-2, N = 442). Each study was conducted at 43 different centres in the US and Canada; the DERMIS-1 trial included 8 centres in Canada and the DERMIS-2 trial included 10 centres in Canada.
After meeting all inclusion and none of the exclusion criteria, eligible patients were enrolled and randomized at baseline in a 2:1 ratio to roflumilast or matching vehicle according to a computer-generated randomization list. Randomization was stratified by study site, baseline IGA score (2 versus ≥ 3), and intertriginous involvement at baseline (I-IGA ≥ 2, yes versus no). Each kit, which contained tubes of either roflumilast or vehicle, was assigned to each patient using an internet-based randomization system.
The screening period was up to 5 weeks before the baseline visit followed by an 8-week treatment period and a follow-up visit that was scheduled approximately 1 week after treatment was completed for patients who did not enter the OLE study. Upon completing the treatment phase, patients were eligible to enrol in a separate OLE study, the DERMIS-OLE (ARQ-151-306)17 study, for up to 6 months. Patients were assessed at approximately 2-week intervals throughout the treatment period. The first patient was screened on December 9, 2019, in both studies and the last patient completed their last study visit on November 16, 2020, in the DERMIS-1 trial and November 23, 2020, in the DERMIS-2 trial.
Table 5: Details of Pivotal Studies Identified by the Sponsor
Detail | DERMIS-1 | DERMIS-2 |
|---|---|---|
Design and population | ||
Study design | Randomized, double-blind, vehicle-controlled, parallel-group, multicentre phase III studies | |
Locations (number of centres) | US (35) and Canada (8) | US (33) and Canada (10) |
Key dates |
|
|
Randomized (N) | 439 | 442 |
Inclusion criteria |
| |
Exclusion criteria |
| |
Drug | ||
Intervention | Roflumilast cream 0.3% administered topically to lesions of plaque psoriasis once daily | |
Comparator | Vehicle cream administered topically to lesions of plaque psoriasis once daily | |
Study duration | ||
Screening phase | Up to 5 weeks before baseline | |
Treatment phase | 8 weeks | |
Follow-up phase | 1 week or option to enrol in a separate long-term OLE study of up to 6 months (DERMIS-OLE, ARQ-151-306) | |
Outcomes | ||
Primary end point | IGA success (score of 0 [clear] or 1 [almost clear] plus a ≥ 2-grade improvement from baseline) at week 8 | |
Secondary and exploratory end points | Secondary:
Exploratory:
| |
Publication status | ||
Publications | Lebwohl et al.30 ARQ-151-301 NCT04211363 | Lebwohl et al.30 ARQ-151-302 NCT04211389 |
ACE = angiotensin-converting enzyme; BSA = body surface area; CDI-2 = Children’s Depression Inventory 2; CDLQI = Child Dermatology Life Quality Index; C-SSRS = Columbia-Suicide Severity Rating Scale; DLQI = Dermatology Life Quality Index; IGA = Investigator’s Global Assessment; I-IGA = intertriginous Investigator’s Global Assessment; mPASI 50 = 50% reduction in the modified Psoriasis Area and Severity Index score; mPASI 75 = 75% reduction in the modified Psoriasis Area and Severity Index score; mPASI 90 = 90% reduction in the modified Psoriasis Area and Severity Index score; mPASI 100 = 100% reduction in the modified Psoriasis Area and Severity Index score; OLE = open-label extension; PASI 50 = 50% reduction in the Psoriasis Area and Severity Index score; PASI 75 = 75% reduction in the Psoriasis Area and Severity Index score; PASI 90 = 90% reduction in the Psoriasis Area and Severity Index score; PASI 100 = 100% reduction in the Psoriasis Area and Severity Index score; PDE-4 = phosphodiesterase type 4; PHQ-8 = Patient Health Questionnaire depression scale; PHQ-A = Patient Health Questionnaire-9 modified for adolescents; PSD = Psoriasis Symptom Diary; PUVA = psoralen and UV A; SAE = serious adverse event; TEAE = treatment-emergent adverse event; UVB = UV B; WI-NRS = Worst Itch Numeric Rating Scale.
Sources: Clinical Study Reports for the DERMIS-19 and DERMIS-2 trials;10 details from the table were taken from the sponsor’s summary of clinical evidence.16
Populations
Inclusion and Exclusion Criteria
The same inclusion and exclusion criteria were used in the DERMIS-1 and DERMIS-2 studies (Table 5). Patients aged 2 years or older were required to have a clinical diagnosis of psoriasis vulgaris of at least 6 months duration (or 3 months for children) and stable disease for the past 4 weeks. Stable disease was determined by the trained investigator and was intended to indicate no escalation of treatment dose or additional therapy as well as no change in disease severity or extent during the 4 weeks before screening. Patients were required to have psoriasis vulgaris on the face, extremities, trunk, and/or intertriginous areas involving 2% to 20%, inclusive, of BSA (excluding the scalp, palms, and soles) and at least mild disease severity at baseline (IGA score ≥ 2 and PASI score ≥ 2).
Exclusion criteria included the presence of nonplaque or drug-induced forms of psoriasis, known or suspected severe renal insufficiency, and known or suspected moderate to severe liver impairment. Additionally, patients with planned initiation or changes to any concomitant medication that could affect psoriasis vulgaris (e.g., beta-blockers or angiotensin-converting enzyme [ACE] inhibitors) or who were unable to discontinue excluded (prohibited) medications and treatments (defined as systemic, biologic, topical, phototherapy, and investigational treatments) that could affect plaque psoriasis within the prespecified time periods (washout periods) before randomization were excluded from the studies (Table 5).
Interventions
The same interventions were used in the DERMIS-1 and DERMIS-2 studies (Table 5). Patients were randomized to receive roflumilast cream 0.3% or the matched vehicle cream, which were dispensed to the patient or caregiver in identical kits containing 4 45 g tubes. The kits and tubes were labelled in a blinded manner to ensure the patients, investigators, clinical personnel, and the sponsor were not aware of which treatment a patient received until the end of the study. The number of kits dispensed to each patient over the treatment period was based on the BSA involvement of psoriasis.
Patients were directed to apply their assigned treatment once daily topically to psoriatic plaques affecting 2% of BSA up to a maximum application area of 20% of BSA over an 8-week treatment period. Treatment application was in the evening, except on day 1 (baseline) and clinic visit days, when treatment was applied at the study site. Areas of application were all areas affected, including the face, trunk, genitals, intertriginous areas, and limbs, but excluding the scalp. A tar-containing or dandruff shampoo (zinc pyrithione or selenium sulphide) was permitted for treatment of the scalp. Palms and soles were treated but were not counted toward any efficacy measurements. Emollients or moisturizers were not to be applied to treated areas. Treatment of affected areas with roflumilast or vehicle was maintained for the duration of the study, regardless of whether the psoriasis cleared. New lesions that developed during the treatment period were also treated.
The matching vehicle cream contained only the excipients of the roflumilast cream. Of note, a vehicle control arm is distinct from a placebo control arm in that a vehicle control may provide beneficial effects on the signs and symptoms of plaque psoriasis (e.g., reducing scaling, improving skin barrier, and moisturizing the skin).
Treatment was interrupted for up to 1 week if the patient developed an application site reaction with the clinical appearance of an irritation reaction and a severity of a dermal response score of 5 (erythema, edema, and papules) or greater on the Berger and Bowman scale. Treatment was resumed if the reaction had, in the opinion of the investigator, adequately resolved.
Of note, treatment was stopped if, after consultation with the sponsor and medical monitor, a patient required a prohibited treatment or medication during the study period, such as systemic or topical treatments for plaque psoriasis. Any patients with a Columbia-Suicide Severity Rating Scale score indicative of suicidal ideation, a score of 15 or more on the Patient Health Questionnaire depression scale or Patient Health Questionnaire-9 modified for adolescents, a raw total score of 34 on the Children’s Depression Inventory 2, or a weight loss of 5% or more (if not dieting), were considered for treatment discontinuation by a mental health professional (if applicable), the sponsor, and at the discretion of the investigator.
Outcomes
A list of efficacy end points assessed in this Clinical Review Report is provided in Table 6. These end points are further summarized subsequently. The summarized end points comprise those included in the sponsor’s summary of clinical evidence as well as any identified as important to this review according to stakeholders, for example, the clinical expert, clinician groups, or patient groups.
Table 6: Outcomes Summarized From the DERMIS-1 and DERMIS-2 Trials
Outcome measure | Time point | DERMIS-1 and DERMIS-2 |
|---|---|---|
Primary efficacy end point | ||
IGA successa | At week 8 | Primary end pointb |
Secondary efficacy end points, partition 1 | ||
Time to PASI 50 | NA | Secondary end pointb |
PASI 75 | At week 8 | Secondary end pointb |
I-IGA successc | At week 8 | Secondary end pointb |
Change from baseline in PSD total score | At week 8 | Secondary end pointb |
Change from baseline in PSD total score | At week 4 | Secondary end pointb |
IGA successa | At week 4 | Secondary end pointb |
Secondary efficacy end points, partition 2 | ||
WI-NRS successd | At week 8 | Secondary end pointb |
WI-NRS successd | At week 4 | Secondary end pointb |
WI-NRS successd | At week 2 | Secondary end pointb |
Other efficacy end points | ||
IGA score of clear | At week 8 | Exploratory end point |
IGA score of clear | At week 4 | Exploratory end point |
I-IGA successb | At week 4 | Exploratory end point |
PASI 75 | At week 4 | Exploratory end point |
mPASI 75 | At week 8 | Exploratory end point |
PASI 90 and mPASI 90 | At week 8 | Exploratory end point |
PASI 100 and mPASI 100 | At week 8 | Exploratory end point |
Absolute change in DLQI or CDLQI | At week 8 | Exploratory end point |
Percent change in BSA affected by psoriasis | At week 8 | Exploratory end point |
Key harms | ||
TEAEs | NA | Exploratory end point |
SAEs | NA | Exploratory end point |
Mortality | NA | Exploratory end point |
Treatment discontinuation | NA | Exploratory end point |
BSA = body surface area; CDLQI = Children’s Dermatology Life Quality Index; DLQI = Dermatology Life Quality Index; I-IGA = intertriginous Investigator’s Global Assessment; IGA = Investigator’s Global Assessment; NA = not applicable; mPASI 75 = 75% reduction in the modified Psoriasis Area and Severity Index score; mPASI 90 = 90% reduction in the modified Psoriasis Area and Severity Index score; mPASI 100 = 100% reduction in the modified Psoriasis Area and Severity Index score; PASI 50 = 50% reduction in the Psoriasis Area and Severity Index; PASI 75 = 75% reduction in the Psoriasis Area and Severity Index; PASI 90 = 90% reduction in the Psoriasis Area and Severity Index; PASI 100 = 100% reduction in the Psoriasis Area and Severity Index; PSD = Psoriasis Symptom Diary; SAE = serious adverse event; TEAE = treatment-emergent adverse event; WI-NRS = Worst Itch Numeric Rating Scale.
Notes: Upon successful testing of the primary end point, the alpha value was partitioned to test outcomes in partition 1 (alpha = 0.03) and partition 2 (alpha = 0.02).
The definitions of IGA and I-IGA success were identical except that the IGA considered the “whole body” (excluding the scalp, palms, and soles) and the I-IGA considered only the intertriginous regions.
Treatment-free interval and duration of response (remission) were identified as end points of interest by the clinical expert consulted by CADTH for this review, but were not measured in the DERMIS-1 and DERMIS-2 studies.
aIGA success was defined as a score of 0 (clear) or 1 (almost clear) plus a ≥ 2-grade improvement from baseline.
bStatistical testing for these end points was adjusted for multiple comparisons (e.g., hierarchal testing).
cI-IGA success was defined as a score of 0 (clear) or 1 (almost clear) plus a ≥ 2-grade improvement from baseline in patients with intertriginous area involvement and I-IGA ≥ 2 at baseline.
dWI-NRS success was defined as a ≥ 4-point reduction in WI-NRS pruritus score from baseline in patients with a WI-NRS pruritus score ≥ 4 at baseline.
Sources: Clinical Study Reports for the DERMIS-19 and DERMIS-2 trials;10 details from the table were taken from the sponsor’s summary of clinical evidence.16
Efficacy Outcomes
A description of the efficacy outcome measures and their measurement properties that were used in both the DERMIS-1 and DERMIS-2 studies and assessed in this Clinical Review Report are presented in Table 8.
Investigator’s Global Assessment
In the DERMIS-1 and DERMIS-2 studies, IGA success at weeks 8 and 4 were primary and secondary end points, respectively, and I-IGA success at weeks 8 and 4 were secondary and exploratory end points, respectively. IGA and I-IGA (the latter defined as the IGA scale but used to evaluate the intertriginous areas only) success was defined as a score of clear or almost clear plus an improvement of 2 grades or more from baseline. The IGA was completed at screening, at baseline and every 2 weeks thereafter during the 8-week treatment period, and at follow-up. The I-IGA was completed at baseline and every 2 weeks thereafter during the 8-week treatment period and at follow-up.
The IGA is an investigator-reported, static (i.e., an assessment is made at a static moment in time without comparing it with baseline disease severity),12 qualitative evaluation of the overall severity of psoriasis of the whole body. The disease is assessed on an ordinal scale with 5 grades of psoriasis severity: clear skin (0), almost clear (1), mild (2), moderate (3), and severe (4). Each grade is defined by distinct morphologic descriptions of plaque thickening, scaling, and erythema (Table 7).
Of note, the PGA has been referred to as the IGA, both of which are used to evaluate the overall severity of psoriasis. Multiple versions of the static IGA and PGA have been described in psoriasis studies, ranging from a 4-point scale (clear to severe) to an 11-point scale (worst imaginable disease to absence of disease activity). The static IGA and PGA evaluate disease severity at a single point in time and do not depend on recalling baseline disease characteristics. It should be noted that different versions of the IGA and PGA use different descriptions for each grade (e.g., a score of 1 may indicate minimal in 1 version of the scale but indicate almost clear in a different version of the scale), and the criteria for assessment for each descriptor may vary. No single IGA or PGA version has been recognized as the standard scale.11
Table 7: Investigator’s Global Assessment of Disease in the DERMIS-1 and DERMIS-2 Trials
Score | Grade | Morphologic description | ||
|---|---|---|---|---|
Plaque thickening | Scaling | Erythema | ||
0 | Clear | No elevation or plaque thickening over normal skin | No evidence of scaling | No erythema (no residual red coloration but postinflammatory hyperpigmentation may be present) |
1 | Almost clear | No plaque thickening or possible thickening but difficult to ascertain whether there is a slight elevation above normal skin level | No scaling or residual surface drying and scaling | Light-pink coloration |
2 | Mild | Slight plaque thickening but definite elevation | Fine scales partially or mostly covering the lesions | Light-red coloration |
3 | Moderate | Moderate elevation with rounded or sloped edges | Most lesions at least partially covered | Definite red coloration |
4 | Severe | Marked or very marked elevation typically with hard or sharp edges | Nontenacious or thick tenacious scale covering most or all lesions | Very bright red coloration, extreme red coloration, deep-red coloration |
Sources: Clinical Study Reports for the DERMIS-19 and DERMIS-2 trials.10
Psoriasis Area and Severity Index
In the DERMIS-1 and DERMIS-2 studies, time to PASI 50 (a 50% reduction in PASI score) from baseline was a secondary end point. A 75% reduction in PASI score (PASI 75) from baseline at weeks 8 and 4 were secondary and exploratory end points, respectively. A 75% reduction in modified PASI score (mPASI 75), 90% reduction in PASI score (PASI 90), 90% reduction in mPASI score (mPASI 90), 100% reduction in PASI score (PASI 100), and 100% reduction in mPASI score (mPASI 100) from baseline at week 8 were exploratory end points. The PASI and mPASI were assessed at screening, at baseline and every 2 weeks thereafter during the 8-week treatment period, and at follow-up.
The PASI is an investigator-reported, qualitative evaluation of the severity of psoriasis. Disease severity is evaluated by combining the assessment of the severity of lesions and the area affected into a single score ranging from 0 (no psoriasis) to 72 (maximal psoriasis). To determine the PASI score, the percent of disease in the involved area is estimated and transformed into a grade ranging from 0 (0% of involved area) to 6 (90% to 100% of involved area) for the head (10% of skin), arms (20% of skin), trunk (30% of skin), and legs (40% of skin). Within each anatomic area, the severity is estimated on a scale of 0 (none) to 4 (maximum severity) for each clinical sign, erythema (redness), induration (thickness), and desquamation (scaling), and summed. The final PASI score is calculated by combining the severity rating of the lesions of each anatomic area weighted by the area of involvement and the respective proportion of the patient’s skin. To reflect the status of limited disease, an mPASI score was calculated for patients with an anatomic area with less than 10% involvement using the actual percentage of the involved anatomic area.
Of note, the proportion of patients who had a PASI 75 (at least a 75% reduction in PASI score from baseline) is commonly used as an efficacy end point in clinical trials of biologic therapies for psoriasis; PASI 90 (at least a 90% reduction in PASI score from baseline) and PASI 100 (at least a 100% reduction in PASI score from baseline) are occasionally reported, as well.11
Psoriasis Symptom Diary
In the DERMIS-1 and DERMIS-2 studies, change from baseline in the PSD total score at weeks 8 and 4 were secondary end points. The PSD was completed at screening and at baseline and every 2 weeks thereafter during the 8-week treatment period.
The PSD score is a patient-reported assessment of the impact of plaque psoriasis on overall QoL and was completed by adult patients (aged 18 years or older at screening) in the DERMIS-1 and DERMIS-2 studies.31 The patient’s experience of symptom severity, bother, embarrassment, hiding affected skin, and avoiding activities is recorded using a 24-hour recall period. Responses are scored on a scale of 0 (least severe) to 10 (most severe) for each question and the total PSD score is the sum of the scores for all 16 questions, with lower scores indicating less severe or bothersome symptoms.
Worst Itch Numerical Rating Scale
In the DERMIS-1 and DERMIS-2 studies, WI-NRS success at weeks 8, 4, and 2 were secondary end points. WI-NRS success was defined as an improvement of 4 points or more from a baseline score of 4 or more (moderate or worse itch at baseline). The WI-NRS was assessed at screening and baseline and every 2 weeks thereafter during the 8-week treatment period.
The WI-NRS is a patient-reported, single-item tool used to assess the severity of itch and was completed by patients aged 12 years or older in the DERMIS-1 and DERMIS-2 studies. The highest intensity of itch experienced in the previous 24-hour period is rated on a scale of 0 (no itch) to 10 (worst imaginable itch).32
DLQI and Children’s DLQI
In the DERMIS-1 and DERMIS-2 studies, absolute change in the DLQI or CDLQI at week 8 was an exploratory end point. The DLQI or CDLQI were assessed at screening, baseline, and weeks 2, 4, and 8 during the treatment period.
The DLQI and CDLQI are patient-reported tools used to evaluate HRQoL in patients aged 17 years or older and in patients aged 2 to 16 years, respectively. These 10-item questionnaires cover 6 domains (symptoms and feelings, daily activities, leisure, work and school, personal relationships, and bother with psoriasis treatment) that are scored on a 4-point Likert scale (0 = not at all; 3 = very much). The total DLQI score is calculated by summing the scores of each item and ranges from 0 to 30 points, with lower scores indicating greater HRQoL.
Body Surface Area
In the DERMIS-1 and DERMIS-2 studies, percent change in BSA affected by psoriasis at week 8 was an exploratory end point. BSA was assessed at screening, at baseline and every 2 weeks thereafter during the 8-week treatment period, and at follow-up.
The BSA affected by psoriasis was determined by the patient hand method, where the surface area of the patient’s hand (including fingers) was assumed to equal 1% of BSA, as assessed by the investigator.
Harms Outcomes
In the DERMIS-1 and DERMIS-2 studies, AEs were assessed at screening, at baseline and every 2 weeks thereafter during the 8-week treatment period, and at follow-up.
Local Tolerability Assessment
In the DERMIS-1 and DERMIS-2 studies, local tolerability assessments were completed at baseline, week 4, and week 8.
Local tolerability was graded by investigators before treatment application in the clinic using a scale reported by Berger et al.33 Reactions at the application site that could have occurred postbaseline were differentiated from the pre-existing inflammation associated with the patient’s psoriasis. Dermal responses were graded on a scale of 0 (no evidence of irritation) to 7 (strong reaction spreading beyond the application site), with other qualitative effects noted (slight glazed appearance, marked glazing, glazing with peeling and cracking, glazing with fissures, film of dried serous exudates, small petechial erosions and/or scabs, or no other effects).
Local tolerability was also graded by patients or their caregivers in the clinic on a scale of 0 (none, i.e., no sensation) to 3 (severe, i.e., hot, tingling, or stinging sensation that has caused definite discomfort) based on the sensation 10 to 15 minutes after treatment application.
Table 8: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
IGA and I-IGA | A measure of disease severity, usually on a 5-, 6-, or 7-point ordinal scale.34 In the DERMIS trials,9,10 the IGA was measured on a 5-point static ordinal scale allowing clinicians to rate disease severity with 5 grades: clear skin (0); almost clear (1); or mild (2), moderate (3), or severe (4) psoriasis. Success was defined as a score 0 or 1, indicating a clear or nearly clear improvement or complete or minimal resolution. | Validity: The 6-point and 7-point IGA scales have been found to be closely related to other evaluation tools such as the PASI, indicating good correlation (r ≥ 0.77).35,36 A review of the clinimetric properties of psoriasis severity measures shows that the 7-point IGA is considered to have high clinical construct validity, which means it correlates well with other measures of severity.34 Reliability: The reliability of the 6-point and 7-point IGA scales have been assessed indicating moderate interrater reliability (ICC ≥ 70%).36,37 Responsiveness: This has not been identified. | The MID in the IGA has not been estimated. Achievement of a score of 0 (clear) or 1 (almost clear) in the static PGA12 and in the static IGA or PGA11 is generally accepted as clinically meaningful (i.e., a responder analysis would consider the proportion of patients with psoriasis who achieved a score of 0 or 1 in a clinical trial as treatment success). Alternatively, or in addition to the achievement of a score of 0 or 1, the responder analysis may also consider the proportion of patients with at least a 2-grade improvement from baseline on the static IGA.11 |
PASI and mPASI | This tool evaluates psoriasis severity with total scores ranging from 0 (no psoriasis) to 72 (maximal psoriasis). Four subscores were calculated on a scale of 0 (0% involved area) to 6 (90% to 100% involved area) for the head (10% of skin), arms (20% of skin), trunk (30% of skin), and legs (40% of skin). Within each area, the severity was estimated on a scale of 0 (none) to 4 (maximum severity) using 3 clinical signs: erythema (E; redness), induration (T; thickness), and desquamation (S; scaling). A modified version of the PASI score (mPASI) was calculated for patients with < 10% involvement in an anatomic area using the actual percentage of the anatomic area involved (e.g., 0.1 = 1% and 0.9 = 9%), which more precisely reflects the status of limited disease. The mPASI outcome includes all elements of the conventional PASI and allows for the calculation of the conventional PASI score. | Validity: Construct validity was demonstrated through correlation of the PASI and DLQI scores (0.36 ≤ r ≤ 0.54).38 Correlation between the LS-IGA and IGA (Spearman rank correlation of 0.92 and 0.73).36 Reliability: PASI was shown to have good interrater reliability (ICC > 0.75). The coefficient of variation for the PASI score was 36.9, indicating moderate interrater reliability.39 Responsiveness: Responsiveness was found to be low when the affected BSA was < 10%.34,40 | An MID has not been estimated. |
PSD | PSD is a tool for assessment of the effect of plaque psoriasis on overall QoL.31 This tool records the patient’s experience of symptom severity, bother, embarrassment, hiding affected skin, and avoiding activities using a 24-hour recall period to reduce recall bias and error. Responses are scored on a scale of 0 (least severe) to 10 (most severe) for 16 questions and the total PSD score is calculated by summing the subscore for each question. Lower scores indicate less severe or bothersome symptoms. | Validity: Two phase III studies in patients with moderate to severe chronic plaque psoriasis demonstrated construct validity, which was evaluated through correlations with the PASI, IGA, DLQI, EQ-5D, and PGIC. The magnitude of correlation was moderate to strong (ranging from 0.41 to 0.73) by week 12, thus confirming the construct validity.41 Reliability: Test–retest reliability, determined using the ICC on the PSD, yielded a high ICC (> 0.90).41 Responsiveness: Responsiveness was evaluated by estimating mean differences and effect sizes between known groups (PASI and IGA). Results showed that responsiveness effect size estimates were moderate to large (0.6 to 1.5).41 | If a patient’s PSD scores indicate significant improvements, which makes them a “responder,” then it is likely they would demonstrate changes in scores of around 2.0 to 3.0 for minor improvements, and 3.0 to 5.0 for more significant improvements across a range of items.42 |
WI-NRS | Patient-reported itch severity was recorded using the WI-NRS, which is a simple single-item tool to assess the highest intensity of itch experienced during the previous 24-hour period.32 The scale instructions are: “On a scale of 0 to 10, with 0 being no itch and 10 being worst itch imaginable.” | Validity: Data from 1 phase II and 3 phase III randomized clinical studies of patients with moderate to severe plaque psoriasis revealed, for construct validity, that there were significant cross-sectional correlations with DLQI symptoms and feelings domains (r ≥ 60 at baseline, and r ≥ 0.80 at week 12).15 Reliability: Test–retest reliability analyses supported the reproducibility of the measure (intraclass correlation coefficient range 0.71 to 0.74).15 Responsiveness: Large correlations (r ≥ 0.71) between changes in itch scores and changes in DLQI symptoms and feelings domain scores from baseline to week 12 established responsiveness.15 | WI-NRS indicated that an improvement of ≥ 4 points in itch severity was considered clinically meaningful for patients with plaque psoriasis.15 |
DLQI | The DLQI is a 10-item patient-reported questionnaire that covers 6 domains (symptoms and feelings, daily activities, leisure, work and school, personal relationships, and bother with psoriasis treatment). Items were scored on a 4-point Likert scale (0 = not at all, 1 = a lot, 2 = a little, 3 = very much). The total DLQI score was calculated by summing the item scores and ranges from 0 to 30 points, with lower scores indicating greater HRQoL. | Validity: Construct validity of the DLQI in the psoriasis population was based on the correlation of the instrument with either generic, dermatologic, or disease-specific instruments over 37 separate studies.43 The DLQI was most highly correlated with the bodily pain (r = 0.61) and social functioning domains (r = 0.68) of the SF-36, as well as the overall EQ-5D index score (r = 0.71).13 Reliability: Reliability was assessed in the original validation study of the DLQI by Finlay and Khan in a population of various skin diseases.44 The test‒retest reliability correlation coefficients were high for both the overall score (Spearman rank correlation of 0.99) and for individual questions (0.95 to 0.98).44 Slightly lower correlation coefficients (ranging from 0.56 to 0.99) were reported in a later systematic review by Basra et al.14 Responsiveness: Responsiveness to change was measured by comparing DLQI data with PASI and PGA scores.13 The DLQI demonstrated equal responsiveness to the PASI and PGA scores with correlation coefficients of r = 0.69 and r = 0.71, which was not achieved by the general tools, the EQ-5D (r = 0.44) and SF-36 (r = 0.44).13 | The within-group MID is reported to be 2.2 to 6.9 points in patients with psoriasis and other inflammatory skin disorders.13,14 |
BSA | The percentage of BSA affected by psoriasis was estimated using the 1% rule, where the patient’s flat palm represents 1% of total BSA. | Validity: This is not relevant to the evaluation of BSA. Reliability: Interrater reliability was evaluated in 2 studies that determined an ICC of 0.91 and 0.9645,46 when dermatologists used the 1% rule in BSA determination. Interrater variability was determined to be high in 2 separate studies. First, a systematic review conducted by Puzenat et al. determined a coefficient of variation of > 30%.40 Second, Bożek et al. found a coefficient of variation of 57.1 when 10 dermatologists evaluated 9 patients.39 Test–retest reliability was evaluated in 2 separate studies; high test–retest reliability was found in both studies, with ICCs of 0.9845 and 0.96.39 Responsiveness: Currently, there is no evidence regarding the responsiveness to change of the use of the 1% rule in BSA determination. | An MID for BSA reduction has not been identified in the literature for patients with psoriasis. |
BSA = body surface area; DLQI = Dermatology Life Quality Index; HRQoL = health-related quality of life; ICC = interclass correlation; IGA = Investigator’s Global Assessment; I-IGA = intertriginous Investigator’s Global Assessment; LS-IGA = Lattice System Investigator’s Global Assessment; MID = minimal important difference; mPASI = modified version of the PASI score; PASI = Psoriasis Area Severity Index; PGA = Physician’s Global Assessment; PGIC = Patient Global Impression of Change; PSD = Psoriasis Symptoms Diary; SF-36 = Short Form (36) Health Survey; QoL = quality of life; WI-NRS = Worst Itch Numeric Rating Scale.
Statistical Analysis
The statistical analysis of the efficacy end points in the DERMIS-1 and DERMIS-2 studies and assessed in this Clinical Review Report are summarized in Table 9.
Sample Size and Power Calculation
A sample size of approximately 400 patients was planned for each study: approximately 267 patients would receive roflumilast once daily and approximately 133 patients would receive the matching vehicle once daily according to the 2:1 randomization scheme. This sample size would provide more than 99% power to detect a 22.4% difference in the primary end point (IGA success) between treatment groups at an alpha of 0.05 using a 2-sided chi-square test. The intergroup difference was estimated from a phase IIb study (ARQ-151-201, NCT03638258) that compared IGA success rates between roflumilast cream 0.3% and vehicle cream (32.2% versus 9.8%, respectively). The planned sample size would also provide sufficient power to analyze the first 5 secondary end points (Table 5).
Statistical Test or Model
Primary Outcome
The primary efficacy end point (IGA success defined as an IGA score of 0 [clear] or 1 [almost clear] plus an improvement of 2 or more grades from baseline at week 8) was evaluated in the ITT population as the ratio of the odds of IGA success using roflumilast relative to the odds of IGA success using the vehicle after 8 weeks. The primary end point was analyzed using a Cochran-Mantel-Haenszel test with a 2-sided significance level of 5% and stratification by study site, baseline IGA, and baseline intertriginous area involvement. For the analysis of the primary efficacy end point, missing IGA scores were imputed using a regression-based multiple imputation model.
A sensitivity analysis of the primary end point was performed using the original (nonimputed, observed data) dataset, including a repeated-measures logistic regression model with IGA success as the dependent variable and treatment and visit as the independent variables, and a tipping point analysis (post hoc analysis). The primary efficacy analysis and sensitivity analyses were also performed for the primary end point based on the modified ITT population using both observed values and multiple imputation. All other analyses of the primary end point did not adjust for missing data (i.e., used observed data only).
Multiple Testing Procedure
For all primary and secondary efficacy end points, a familywise type I alpha error rate of 0.05 was maintained using a prespecified hierarchical testing strategy. End points were assigned into testing families and testing only proceeded to the next family in accordance with the sequential testing strategy rules.
If the primary end point was met (i.e., a statistically significant improvement in IGA success rate with roflumilast cream 0.3% at week 8), the secondary end points were to be tested inferentially. To control the familywise type I error for multiple comparisons among the secondary end points, the following multiplicity procedure was used.
Upon successful testing of the primary end point (family 1), the alpha was partitioned into 0.03 (partition 1) to sequentially test the secondary end points in families 2 and 3 and into 0.02 (partition 2) to sequentially test WI-NRS time points.
Within the partition 1 alpha of 0.03, the hierarchical testing of family 2 (time to PASI 50, PASI 75 rate at week 8, and I-IGA success at week 8) was performed. Subsequent testing of family 3 (change from baseline in total PSD scores at weeks 8 and 4, and IGA success at week 4) was performed using the alpha available after the testing of family 2, with the Holm procedure used to control for multiple comparisons in family 3.
Within the partition 2 alpha of 0.02, sequential testing was performed for WI-NRS success rates at week 8, then at week 4, and then at week 2.
Subgroup Analyses
Prespecified subgroup analyses were performed for patients with intertriginous area involvement and at least mild severity (i.e., the I-IGA success rate could be assessed) and patients with pruritus who had a WI-NRS score of 4 points or more at baseline (i.e., success in reducing pruritus could be assessed). Both subgroup analyses accounted for multiplicity through alpha partitioning within the hierarchical testing strategy described previously.
Subgroup analyses of the primary end point, which did not account for multiplicity, were also performed using observed data for the following subgroups:
topical corticosteroid — inadequate response, intolerance, or contraindication (yes or no)
topical vitamin D derivatives — inadequate response, intolerance, or contraindication (yes or no)
apremilast — patients with any prior use or no prior use
psoriasis involvement on the elbow (yes)
psoriasis involvement on the knee (yes)
psoriasis involvement on the knee or elbow (either one)
study sites
age group — 2 to 11 years, 12 to 17 years, and 18 years and older
The permitted age range in the DERMIS-1 and DERMIS-2 studies was 2 years old and above, which extends beyond the Health Canada indication in patients 12 years of age and older. Therefore, results from the subgroup analysis for the age group of 2 to 11 years were not summarized in this report.
involvement of face or intertriginous area at baseline
baseline BSA severity — mild (< 5% of BSA), moderate (≥ 5% to < 10% of BSA), and severe (≥ 10% of BSA).
Of the subgroup analyses of the primary end point performed in the studies listed previously, the last 3 subgroups were identified as clinically relevant by the clinical expert consulted by CADTH for this review.
Secondary Outcomes
Binary secondary end points were also analyzed using the Cochran-Mantel-Haenszel test stratified by study site, baseline IGA, and baseline intertriginous area involvement. The binary secondary end points included PASI 75 at week 8, I-IGA success at week 8 (stratified by study site and baseline I-IGA), IGA success at week 4, and WI-NRS success at weeks 8, 4, and 2. Analyses of I-IGA and WI-NRS were performed using the I-IGA ITT and PRU4 ITT populations, respectively; all other analyses were performed on the ITT population.
The continuous secondary end points, change from baseline in total PSD score at week 4 and week 8, were analyzed using an analysis of covariance with the following factors: treatment, study site, baseline IGA, baseline intertriginous area involvement, and baseline of the variable being analyzed. The LS means, standard errors, 95% CIs, and P values were presented. These analyses were performed on the ITT population.
Time to PASI 50 was estimated using the KM method with results calculated as the median and 95% CI for each treatment group, which were compared using the log-rank statistic. A Cox proportional hazards model that included the stratification factors (study site, baseline IGA, and baseline intertriginous area involvement) was used to estimate the hazard ratio and associated 95% CIs. These analyses were performed on the ITT population.
For all secondary efficacy end points other than time to PASI 50, analyses were performed using both multiple imputation for missing data in the primary analysis and observed data. Only observed data were included in the descriptive statistics.
Other Outcomes
Other binary outcomes (e.g., IGA score of clear, mPASI 75, PASI 90, mPASI 90, PASI 100, and mPASI 100) were analyzed using the Cochran-Mantel-Haenszel model and the aforementioned adjustment factors on the ITT population.
Other continuous outcomes (e.g., percent change from baseline in the affected percent of BSA, change in DLQI, and change in CDLQI) were analyzed using an analysis of covariance model and the adjustment factors on the ITT population described previously.
Table 9: Statistical Analysis of Efficacy End Points in the DERMIS-1 and DERMIS-2 Trials
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
DERMIS-1 and DERMIS-2 | ||||
IGA successa at week 8 | CMH test |
| Multiple imputation |
|
Time to PASI 50 |
|
| Only observed data were used | NA |
PASI 75 at week 8 | CMH test |
| Multiple imputation | Original dataset (nonimputed) |
I-IGA successb at week 8 | CMH test |
| Multiple imputation | Original dataset (nonimputed) |
Change from baseline in PSD score at weeks 4 and 8 | ANCOVA |
| Multiple imputation | Original dataset (nonimputed) |
IGA successa at week 4 | CMH test |
| Multiple imputation | Original dataset (nonimputed) |
WI-NRS successc at weeks 2, 4, and 8 | CMH test |
| Multiple imputation | Original dataset (nonimputed) |
IGA score of clear at weeks 4 and 8 | CMH test |
| Multiple imputation | Original dataset (nonimputed) |
I-IGA successb at week 4 | CMH test |
| Multiple imputation | Original dataset (nonimputed) |
PASI 75 at week 4 | CMH test |
| Multiple imputation | Original dataset (nonimputed) |
PASI 90 and PASI 100 at week 8 | CMH test |
| Multiple imputation | Original dataset (nonimputed) |
mPASI 75, mPASI 90, and mPASI 100 at week 8 | CMH test |
| Only observed data were used | NA |
Change from baseline in DLQI or CDLQI at week 8 | ANCOVA |
| Only observed data were used | NA |
Percent change from baseline in affected BSA at week 8 | ANCOVA |
| Only observed data were used | NA |
ANCOVA = analysis of covariance; BSA = body surface area; CMH = Cochran-Mantel-Haenszel; CDLQI = Children’s Dermatology Life Quality Index; DLQI = Dermatology Life Quality Index; IGA = Investigator’s Global Assessment; I-IGA = intertriginous Investigator’s Global Assessment; NA = not applicable; mPASI 75 = 75% reduction in the modified Psoriasis Area and Severity Index score; mPASI 90 = 90% reduction in the modified Psoriasis Area and Severity Index score; mPASI 100 = 100% reduction in the modified Psoriasis Area and Severity Index score; PASI 50 = 50% reduction in the Psoriasis Area and Severity Index; PASI 75 = 75% reduction in the Psoriasis Area and Severity Index; PASI 90 = 90% reduction in the Psoriasis Area and Severity Index; PASI 100 = 100% reduction in the Psoriasis Area and Severity Index; PSD = Psoriasis Symptom Diary; WI-NRS = Worst Itch Numeric Rating Scale.
Note: The definitions of IGA and I-IGA success were identical except that the IGA considered the “whole body” (excluding the scalp, palms, and soles) and the I-IGA considered only the intertriginous regions.
aIGA success was defined as a score of 0 (clear) or 1 (almost clear) plus a ≥ 2-grade improvement from baseline.
bI-IGA success was defined as a score of 0 (clear) or 1 (almost clear) plus a ≥ 2-grade improvement from baseline in patients with intertriginous area involvement and I-IGA ≥ 2 at baseline.
cWI-NRS success was defined as a ≥ 4-point reduction in WI-NRS pruritus score from baseline in patients with WI-NRS pruritus score ≥ 4 at baseline.
Sources: Clinical Study Reports for the DERMIS-19 and DERMIS-2 trials;10 details from the table were taken from the sponsor’s summary of clinical evidence.16
Analysis Populations
The definition and application of each analysis population in the DERMIS-1 and DERMIS-2 studies are summarized in Table 10.
The ITT analysis set included all randomized patients. The modified ITT analysis set included all randomized patients except for those who missed the week 8 disease assessment specifically due to the COVID-19 disruption.
The I-IGA ITT analysis set was a subset of the ITT population and included patients with intertriginous area involvement and with at least mild severity of the intertriginous lesions (I-IGA ≥ 2) at baseline. The PRU4 ITT analysis set was a subset of the ITT population and included patients with a WI-NRS pruritus score of 4 or more points at baseline.
The safety analysis set included all patients who were enrolled and received at least 1 confirmed dose of roflumilast or the vehicle.
Table 10: Analysis Populations in the DERMIS-1 and DERMIS-2 Trials
Population | Definition | Application |
|---|---|---|
ITT | All randomized patients | The primary analysis population for the efficacy analyses |
mITT | All randomized patients except patients who missed the week 8 disease assessment specifically due to COVID-19 disruption | Sensitivity analysis of the efficacy outcomes |
I-IGA ITT | An ITT subset of only patients with at least mild intertriginous lesions at baseline | Analysis of I-IGA success and related outcomes |
I-IGA mITT | The I-IGA ITT population except patients who missed the week 8 disease assessment specifically due to COVID-19 disruption | Sensitivity analysis of I-IGA success and related outcomes |
PRU4 ITT | An ITT subset of patients with a WI-NRS pruritus score of ≥ 4 at baseline | Analysis of WI-NRS success and related outcomes |
PRU4 mITT | The PRU4 ITT population excluding patients who missed the week 8 disease assessment specifically due to COVID-19 disruption | Sensitivity analysis of WI-NRS success and related outcomes |
Safety | All patients who were enrolled and received at least 1 confirmed dose of intervention | All safety analyses |
IGA = Investigator’s Global Assessment; I-IGA = intertriginous Investigator’s Global Assessment; ITT = intention-to-treat; mITT = modified intention-to-treat; PRU4 = patients with WI-NRS pruritus score ≥ 4 at baseline; WI-NRS = Worst Itch Numeric Rating Scale.
Sources: Clinical Study Reports for the DERMIS-19 and DERMIS-2 trials;10 details from the table were taken from the sponsor’s summary of clinical evidence.16
Results
Patient Disposition
Patient disposition was generally consistent between the DERMIS-1 and DERMIS-2 studies (Table 11).
In the DERMIS-1 trial, 539 patients were screened and 439 patients were enrolled; the reasons for screening failure were not reported. A total of 439 patients were randomized in the DERMIS-1 trial, 286 patients to roflumilast cream 0.3% and 153 patients to the vehicle. Similarly, 557 patients were screened and 442 patients were enrolled in the DERMIS-2 trial; the reasons for screening failure were also not reported. A total of 442 patients were randomized in the DERMIS-2 trial, 290 patients to roflumilast cream 0.3% and 152 patients to the vehicle.
The proportion of patients who discontinued from the DERMIS-1 trial was 10.8% and 13.1% of the patients randomized to roflumilast and vehicle, respectively. Similarly, 9.0% and 13.8% of patients randomized to roflumilast and vehicle, respectively, discontinued from the DERMIS-2 trial. Withdrawal by patient in the DERMIS-1 trial was reported in 3.8% versus 7.2% of patients randomized to roflumilast and vehicle, respectively. Similarly, withdrawal by patient in the DERMIS-2 trial was reported in 3.4% versus 7.2% of patients randomized to roflumilast and vehicle, respectively.
Lost to follow-up in the DERMIS-1 trial was reported in 4.2% and 2.6% of patients randomized to roflumilast and vehicle, respectively. Similarly, 5.2% and 4.6% of patients randomized to roflumilast and vehicle, respectively, were lost to follow-up in the DERMIS-2 trial. Study discontinuation as a result of any AEs in the DERMIS-1 trial was reported in 1.7% and 1.3% of patients randomized to roflumilast and vehicle, respectively. Similarly, 0.3% and 1.3% of patients randomized to roflumilast and vehicle, respectively, discontinued the study as a result of any AEs in the DERMIS-2 trial.
Table 11: Summary of Patient Disposition From the DERMIS-1 and DERMIS-2 Trials (All Randomized Patients)
Patient disposition | DERMIS-1 | DERMIS-2 | ||
|---|---|---|---|---|
Roflumilast | Vehicle | Roflumilast | Vehicle | |
Screened, N | 539 | 557 | ||
Reason for screening failure, N | NR | NR | ||
Enrolled, N | 439 | 442 | ||
Randomized, N | 286 | 153 | 290 | 152 |
Discontinued from study, N (%) | 31 (10.8) | 20 (13.1) | 26 (9.0) | 21 (13.8) |
Reason for discontinuation, n (%) | ||||
Withdrawal by patient | 11 (3.8) | 11 (7.2) | 10 (3.4) | 11 (7.2) |
Physician decision | 0 | 1 (0.7) | 0 | 0 |
Noncompliance | 0 | 0 | 0 | 1 (0.7) |
Protocol violation | 1 (0.3) | 0 | 0 | 0 |
Lost to follow-up | 12 (4.2) | 4 (2.6) | 15 (5.2) | 7 (4.6) |
Adverse event | 5 (1.7) | 2 (1.3) | 1 (0.3) | 2 (1.3) |
Pregnancy | 1 (0.3) | 0 | 0 | 0 |
Other | 1 (0.3) | 2 (1.3) | 0 | 0 |
ITT, N | 286 | 153 | 290 | 152 |
I-IGA ITT, N | 63 | 32 | 53 | 31 |
PRU4 ITT, N | 218 | 115 | 229 | 116 |
Safety, N | 286 | 153 | 290 | 152 |
ITT = intention-to-treat; I-IGA = intertriginous Investigator’s Global Assessment; NR = not reported; PRU4 = patients with a WI-NRS pruritus score of ≥ 4 at baseline; WI-NRS = Worst Itch Numeric Rating Scale.
Sources: Clinical Study Reports for the DERMIS-19 and DERMIS-2 trials;10 details from the table were taken from the sponsor’s summary of clinical evidence.16
Protocol Deviations
A summary of the protocol deviations in the DERMIS-1 and DERMIS-2 trials is presented in Table 12.
Overall, the proportion of randomized patients with any protocol deviation was 54.2% in the roflumilast arm versus 62.7% in the vehicle arm in the DERMIS-1 trial, and 59.0% in the roflumilast arm and 60.5% in the vehicle arm in the DERMIS-2 trial. The proportion of patients with any protocol violation related to COVID-19 disruption was numerically greater in the DERMIS-1 trial than in the DERMIS-2 trial (10.3% of all randomized patients versus 4.3% of all randomized patients, respectively), but was generally well balanced between treatment arms within each study.
In the DERMIS-1 trial, major protocol deviations were reported in 26.9% of patients in the roflumilast arm versus 33.3% in the vehicle arm. In the DERMIS-2 trial, major protocol deviations were reported in 22.4% of patients in the roflumilast arm versus 26.3% in the vehicle arm. The most common major protocol deviation reported in both studies was efficacy assessment (14.4% of all randomized patients in the DERMIS-1 trial and 14.9% of all randomized patients in the DERMIS-2 trial); the proportion of randomized patients with a major protocol deviation related to efficacy assessment was generally well balanced between the treatment arms within each study and across studies. This was due to a workflow issue with the electronic clinical outcomes assessment tablet that allowed the study coordinator to remain logged into the tablet while the investigator was performing efficacy assessments. A reconciliation was performed to identify trained and approved investigators who had conducted end point assessments while the study coordinator was logged into the system. At the study sites where this issue was identified, the principal investigators signed affidavits to confirm that the investigator performed the assessment at the time of entry.
Table 12: Summary of Protocol Deviations in the DERMIS-1 and DERMIS-2 Trials (Safety Population)
Category | DERMIS-1 | DERMIS-2 | ||
|---|---|---|---|---|
Roflumilast N = 286 | Vehicle N = 153 | Roflumilast N = 290 | Vehicle N = 152 | |
Patients with any protocol deviation, n (%) | 155 (54.2) | 96 (62.7) | 171 (59.0) | 92 (60.5) |
Patients with any protocol deviation related to COVID-19 disruption, n (%) | 29 (10.1) | 16 (10.5) | 13 (4.5) | 6 (3.9) |
Patients with major protocol deviations, n (%) | 77 (26.9) | 51 (33.3) | 65 (22.4) | 40 (26.3) |
Assessment, efficacy | 39 (13.6) | 24 (15.7) | 43 (14.8) | 23 (15.1) |
Assessment, safety | 3 (1.0) | 1 (0.7) | 1 (0.3) | 3 (2.0) |
Exclusion criteria | 3 (1.0) | 0 | 2 (0.7) | 0 |
Inclusion criteria | 1 (0.3) | 0 | 0 | 0 |
Informed consent | 9 (3.1) | 8 (5.2) | 10 (3.4) | 5 (3.3) |
Laboratory or end point data | 2 (0.7) | 1 (0.7) | 0 | 0 |
Overdose or misuse | 1 (0.3) | 2 (1.3) | 1 (0.3) | 0 |
Prohibited co-medication | 2 (0.7) | 2 (1.3) | 1 (0.3) | 2 (1.3) |
Study druga | 16 (5.6) | 14 (9.2) | 9 (3.1) | 3 (2.0) |
Visit window | 16 (5.6) | 5 (3.3) | 5 (1.7) | 6 (3.9) |
Otherb | 0 | 0 | 0 | 2 (1.3) |
aDeviations related to the study drug due to intervention noncompliance (i.e., less than 80% of doses achieved over the duration of the study or more than 3 consecutive missed doses).47
bOther deviations included application of the long-term extension intervention at week 8 (DERMIS-1) and intervention tubes or kits lost during the study or not returned at the study completion visit.47
Sources: Clinical Study Reports for the DERMIS-19 and DERMIS-2 trials.10
Baseline Characteristics
Note, all patients randomized in the DERMIS-1 and DERMIS-2 trials received at least 1 dose of the intervention; thus, the ITT population and the safety population were identical in each study.
Patients in the DERMIS-1 and DERMIS-2 studies had generally consistent demographic characteristics (Table 13). The proportion of patients in the age category of 12 to 17 years was similar across both studies (|||| || |||| of randomized patients in the DERMIS-1 and DERMIS-2 trials, respectively). The proportion of patients in the age category of 18 years and older was also similar across both studies (||||| || ||||| of randomized patients in the DERMIS-1 and DERMIS-2 trials, respectively). The mean age of all randomized patients was 48.0 years (SD = 14.69) in the DERMIS-1 trial and 47.0 years (SD = 14.72) in the DERMIS-2 trial. Most patients were male (64.9% and 62.4% of randomized patients in the DERMIS-1 and DERMIS-2 trials, respectively), while 33.9% to 39.3% of patients were female. The majority of patients were white (81.5% and 82.8% of randomized patients in the DERMIS-1 and DERMIS-2 trials, respectively), while the remainder of the randomized patients (0% to 7.3%) identified as Asian, Black or African American, American Indian or Alaska Native [wording from original source], Native Hawaiian or other Pacific Islander, other, or more than 1 race.
Patients in the DERMIS-1 and DERMIS-2 studies had generally consistent clinical characteristics (Table 14). The mean baseline of BSA covered (involved) in all randomized patients was 6.66% (SD = 4.538) in the DERMIS-1 trial and 7.30% (SD = 4.918) in the DERMIS-2 trial. Across both studies, the majority of patients had moderate IGA at baseline (74.7% and 76.5% of randomized patients in the DERMIS-1 and DERMIS-2 trial, respectively). A similar proportion of all randomized patients in the DERMIS-1 and DERMIS-2 trials had facial involvement (27.1% and 26.0%, respectively) and genital involvement (16.4% and 14.7%, respectively). Across both studies, the majority of patients had mild to moderate I-IGA at baseline (mild, 11.2% and 8.6% of randomized patients in the DERMIS-1 and DERMIS-2 trials, respectively; moderate, 9.8% and 10.0% of randomized patients in the DERMIS-1 and DERMIS-2 trials, respectively). The mean PASI at baseline for all randomized patients was 6.5 (SD = 3.35) in the DERMIS-1 trial and 6.7 (SD = 3.33) in the DERMIS-2 trial.
Across both studies, the majority of patients had a baseline WI-NRS score of 4 or more points (75.9% and 78.1% of randomized patients in the DERMIS-1 and DERMIS-2 trial, respectively) (Table 14). Note, the mean baseline WI-NRS score was 5.7 (SD = 2.78) in the DERMIS-1 trial and 5.9 (SD = 2.66) in the DERMIS-2 trial across treatment arms. The mean PSD total score at baseline was 72.6 (SD = 42.21) in the DERMIS-1 trial, while in the DERMIS-2 trial, the mean baseline PSD total score was 69.3 (40.66) in the roflumilast arm and 77.4 (41.24) in the vehicle arm. The mean DLQI score at baseline for all randomized patients was 7.3 (SD = 5.47) in the DERMIS-1 trial and 7.2 (SD = 5.61) in the DERMIS-2 trial.
Data on treatment history of psoriasis and data on the number of prior treatments for psoriasis were limited. The majority of patients (85.6% and 84.8% of randomized patients in the DERMIS-1 and DERMIS-2 trials, respectively) reported at least 1 prior medication (Table 14.) Therefore, approximately 15% of randomized patients in each study did not receive any prior medication, irrespective of plaque psoriasis indication (i.e., treatment-naive patients). Most patients across studies, 56.3% and 66.3% of randomized patients in the DERMIS-1 and DERMIS-2 trials, respectively, reported inadequate response, intolerance, or contraindication to topical corticosteroids for the treatment of psoriasis.
In both the DERMIS-1 and DERMIS-2 trials, the demographic and clinical characteristics of patients in the I-IGA ITT and PRU4 ITT populations were generally balanced between treatment arms and similar to the ITT population.
The baseline characteristics outlined in the following tables are limited to those which are most relevant to this review or were felt to impact the outcomes or interpretation of the study results.
Table 13: Summary of Baseline Demographic Characteristics of the DERMIS-1 and DERMIS-2 Trials (Safety Population)
Demographic characteristic | DERMIS-1 | DERMIS-2 | ||
|---|---|---|---|---|
Roflumilast N = 286 | Vehicle N = 153 | Roflumilast N = 290 | Vehicle N = 152 | |
Age, years | ||||
Mean (SD) | 47.6 (14.09) | 48.7 (15.77) | 46.9 (15.07) | 47.1 (14.07) |
Median (minimum, maximum) | 46.0 (9, 86) | 49.0 (13, 88) | 47.0 (6, 82) | 48.5 (8, 82) |
Age category, n (%) | ||||
2 to 11 years | | ||||| | | |||||| | | ||||| | | ||||| |
12 to 17 years | | ||||| | | |||||| | | ||||| | | ||||| |
≥ 18 years | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| |
Sex, n (%) | ||||
Male | 189 (66.1) | 96 (62.7) | 176 (60.7) | 100 (65.8) |
Female | 97 (33.9) | 57 (37.3) | 114 (39.3) | 52 (34.2) |
Ethnicity, n (%) | ||||
Hispanic or Latino | 63 (22.0) | 34 (22.2) | 76 (26.2) | 50 (32.9) |
Not Hispanic or Latino | 223 (78.0) | 119 (77.8) | 213 (73.4) | 102 (67.1) |
Race, n (%) | ||||
White | 234 (81.8) | 124 (81.0) | 240 (82.8) | 126 (82.9) |
Asian | 21 (7.3) | 11 (7.2) | 20 (6.9) | 9 (5.9) |
Black or African American | 8 (2.8) | 8 (5.2) | 13 (4.5) | 9 (5.9) |
American Indian or Alaska Native [wording from original source] | 4 (1.4) | 1 (0.7) | 0 | 1 (0.7) |
Native Hawaiian or Other Pacific Islander | 2 (0.7) | 0 | 3 (1.0) | 1 (0.7) |
Other | 11 (3.8) | 5 (3.3) | 8 (2.8) | 4 (2.6) |
More than 1 race | 2 (0.7) | 1 (0.7) | 1 (0.3) | 0 |
Not reported | 4 (1.4) | 3 (2.0) | 5 (1.7) | 2 (1.3) |
SD = standard deviation.
Sources: Clinical Study Reports for the DERMIS-19 and DERMIS-2 trials;10 details from the table were taken from the sponsor’s summary of clinical evidence.16
Table 14: Summary of Baseline Clinical Characteristics of the DERMIS-1 and DERMIS-2 Trials (Safety Population)
Clinical characteristic | DERMIS-1 | DERMIS-2 | ||
|---|---|---|---|---|
Roflumilast N = 286 | Vehicle N = 153 | Roflumilast N = 290 | Vehicle N = 152 | |
Baseline weight (kg) | ||||
N | 286 | 153 | 290 | 152 |
Mean (SD) | 92.46 (23.769) | 93.47 (24.343) | 89.63 (22.655) | 94.63 (21.654) |
Median | 89.30 | 89.80 | 87.20 | 91.10 |
Minimum, maximum | 28.2, 183.1 | 36.0, 187.1 | 18.1, 204.6 | 28.2, 163.3 |
Baseline BMI, kg/m2 | ||||
N | 286 | 153 | 289 | 152 |
Mean (SD) | 31.98 (9.505) | 31.85 (7.881) | 30.95 (7.004) | 33.46 (13.698) |
Median | 30.35 | 31.10 | 29.90 | 31.30 |
Minimum, maximum | 16.4, 129.9 | 12.3, 61.4 | 13.9, 59.3 | 18.0, 175.0 |
Baseline BSA covered, % | ||||
Mean (SD) | 6.28 (4.376) | 7.36 (4.762) | 7.08 (4.839) | 7.73 (5.054) |
Median | 5.00 | 6.00 | 5.25 | 6.00 |
Minimum, maximum | 2.0, 20.0 | 2.0, 20.0 | 2.0, 20.0 | 2.0, 20.0 |
Baseline IGA, n (%) | ||||
Clear | 0 | 0 | 0 | 0 |
Almost clear | 0 | 0 | 0 | 0 |
Mild | 51 (17.8) | 20 (13.1) | 50 (17.2) | 24 (15.8) |
Moderate | 206 (72.0) | 122 (79.7) | 220 (75.9) | 118 (77.6) |
Severe | 29 (10.1) | 11 (7.2) | 20 (6.9) | 10 (6.6) |
Baseline IGA, numeric | ||||
Mean (SD) | 2.9 (0.52) | 2.9 (0.45) | 2.9 (0.48) | 2.9 (0.47) |
Median | 3.0 | 3.0 | 3.0 | 3.0 |
Minimum, maximum | 2, 4 | 2, 4 | 2, 4 | 2, 4 |
Location of psoriasis involvement, n (%) | ||||
Elbow | 207 (72.4) | 109 (71.2) | 211 (72.8) | 107 (70.4) |
Knees | 171 (59.8) | 85 (55.6) | 169 (58.3) | 86 (56.6) |
Face | 74 (25.9) | 45 (29.4) | 76 (26.2) | 39 (25.7) |
Genitalia | 51 (17.8) | 21 (13.7) | 46 (15.9) | 19 (12.5) |
Baseline I-IGA in all patients, n (%) | ||||
Clear | 0 | 0 | 1 (0.3) | 0 |
Almost clear | 5 (1.7) | 1 (0.7) | 2 (0.7) | 1 (0.7) |
Mild | 33 (11.5) | 16 (10.5) | 25 (8.6) | 13 (8.6) |
Moderate | 27 (9.4) | 16 (10.5) | 27 (9.3) | 17 (11.2) |
Severe | 3 (1.0) | 0 | 1 (0.3) | 1 (0.7) |
Baseline I-IGA in patients with intertriginous area involvement, numeric | ||||
N | 68 | 33 | 56 | 32 |
Mean (SD) | 2.4 (0.70) | 2.5 (0.56) | 2.4 (0.69) | 2.6 (0.62) |
Median | 2.0 | 2.0 | 2.5 | 3.0 |
Minimum, maximum | 1, 4 | 1, 3 | 0, 4 | 1, 4 |
Baseline PSD total score | ||||
N | 282 | 150 | 283 | 148 |
Mean (SD) | 72.1 (42.75) | 73.4 (41.29) | 69.3 (40.66) | 77.4 (41.24) |
Median | 71.0 | 69.5 | 67.0 | 74.0 |
Minimum, maximum | 0, 160 | 3, 158 | 2, 160 | 0, 157 |
Baseline PASI | ||||
Mean (SD) | 6.3 (3.15) | 6.8 (3.70) | 6.5 (3.22) | 7.0 (3.52) |
Median | 5.6 | 6.0 | 5.6 | 6.0 |
Minimum, maximum | 2, 18 | 2, 25 | 2, 19 | 2, 20 |
Baseline mPASI | ||||
Mean (SD) | 4.16 (3.705) | 4.73 (4.308) | 4.26 (3.904) | 4.93 (4.220) |
Median | 2.63 | 3.24 | 2.62 | 3.54 |
Minimum, maximum | 0.3, 18.0 | 0.2, 24.6 | 0.2, 18.9 | 0.3, 18.5 |
Baseline WI-NRS,a n (%) | ||||
0 | 16 (5.6) | 12 (7.8) | 5 (1.7) | 5 (3.3) |
1 | 11 (3.8) | 5 (3.3) | 16 (5.5) | 5 (3.3) |
2 | 14 (4.9) | 11 (7.2) | 19 (6.6) | 7 (4.6) |
3 | 26 (9.1) | 10 (6.5) | 19 (6.6) | 18 (11.8) |
4 | 26 (9.1) | 7 (4.6) | 27 (9.3) | 12 (7.9) |
5 | 32 (11.2) | 14 (9.2) | 35 (12.1) | 8 (5.3) |
6 | 37 (12.9) | 26 (17.0) | 38 (13.1) | 17 (11.2) |
7 | 34 (11.9) | 20 (13.1) | 37 (12.8) | 22 (14.5) |
8 | 49 (17.1) | 23 (15.0) | 49 (16.9) | 25 (16.4) |
9 | 20 (7.0) | 19 (12.4) | 22 (7.6) | 19 (12.5) |
10 | 20 (7.0) | 6 (3.9) | 21 (7.2) | 13 (8.6) |
Baseline WI-NRS,a numeric | ||||
N | 285 | 153 | 288 | 151 |
Mean (SD) | 5.7 (2.75) | 5.7 (2.84) | 5.8 (2.61) | 6.1 (2.75) |
Median | 6.0 | 6.0 | 6.0 | 7.0 |
Minimum, maximum | 0, 10 | 0, 10 | 0, 10 | 0, 10 |
Baseline WI-NRSa ≥ 4 | ||||
n (%) | 218 (76.2) | 115 (75.2) | 229 (79.0) | 116 (76.3) |
Baseline DLQI | ||||
N | ||| | ||| | ||| | ||| |
Mean (SD) | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| |
Median | ||| | ||| | ||| | ||| |
Minimum, maximum | | || | | | || | | | || | | | || | |
Baseline CDLQI | ||||
N | ||| | ||| | ||| | ||| |
Mean (SD) | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| |
Median | ||| | ||| | ||| | ||| |
Minimum, maximum | | || | | | || | | | || | | | || | |
Prior medications | ||||
At least 1 prior medication,b n (%) | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| |
Previous treatment history of psoriasis | ||||
Inadequate response, intolerance, or contraindication by treatment type,c n (%) | ||||
Topical corticosteroids | 161 (56.3) | 86 (56.2) | 196 (67.6) | 97 (63.8) |
Topical vitamin D derivatives | 9 (3.1) | 2 (1.3) | 36 (12.4) | 12 (7.9) |
Apremilast | 11 (3.8) | 7 (4.6) | 11 (3.8) | 8 (5.3) |
Conventional systemic therapy | 11 (3.8) | 8 (5.2) | 9 (3.1) | 6 (3.9) |
Phototherapy | 25 (8.7) | 17 (11.1) | 25 (8.6) | 9 (5.9) |
BMI = body mass index; BSA = body surface area; CDLQI = Children’s Dermatology Life Quality Index; DLQI = Dermatology Life Quality Index; I-IGA = intertriginous Investigator’s Global Assessment; IGA = Investigator’s Global Assessment; mPASI = modified Psoriasis Area and Severity Index; PASI = Psoriasis Area and Severity Index; PSD = Psoriasis Symptom Diary; SD = standard deviation; WI-NRS = Worst Itch Numeric Rating Scale.
aWI-NRS was determined by asking the patient to assess their worst itch over the past 24 hours. The scale was from 0 to 10, which ranged from “no itch” to “worst imaginable itch.”
bPrior medications are all medications that were started before the application of the assigned treatment.
cPatients could be counted more than once if they had received multiple treatment types.
Sources: Clinical Study Reports for the DERMIS-19 and DERMIS-2 trials;10 details from the table were taken from the sponsor’s summary of clinical evidence.16
Exposure to Study Treatments
The extent of exposure to treatment was based on the total number of applications and total weight of treatment; patients were considered compliant if they (or their caregiver) applied 80% or more of the expected applications and they did not miss more than 3 consecutive doses. Treatment compliance rates were generally consistent across studies and were greater than 90% for each treatment arm (Table 15).
The mean number of applications per patient during the treatment period was also generally consistent between the DERMIS-1 (55.1 applications and 54.4 applications in the roflumilast and vehicle arms, respectively) trial and DERMIS-2 trial (54.3 applications and 53.0 applications in the roflumilast and vehicle arms, respectively). Of note, the mean total weight of roflumilast and vehicle applied was 122.73 g (SD = 113.114) and 179.89 g (SD = 183.030), respectively, in the DERMIS-1 trial. Similarly, 122.02 g (SD = 122.465) of roflumilast versus 187.78 g (SD = 180.495) of vehicle was applied in the DERMIS-2 trial (Table 15).
Concomitant Medications
Note, topical and systemic treatments as well as phototherapy for plaque psoriasis were considered prohibited medications and treatments and were not permitted in the DERMIS-1 and DERMIS-2 studies. Eye and ear drops as well as nasal corticosteroid preparations were permitted. Inhaled corticosteroid preparations were permitted if used for a stable condition and at a stable dose for more than 28 days before screening and were continued at the same dose for the duration of the study. Nonmedicated emollients, moisturizers, and sunscreens were permitted as used normally by patients. These could be applied to nontreated areas as needed but should not have been used within 12 hours of a study visit. A tar-containing or dandruff shampoo (zinc pyrithione or selenium sulphide) was permitted for treatment of the scalp.
The addition of new medications, including nonprescription medications, during the study was generally discouraged; however, short-term use of a medication could be authorized by the investigator and chronic medication use was permitted during the study (except as prohibited according to the prespecified list of excluded medications and treatments). At least 1 concomitant medication was used by ||||| of randomized patients in the DERMIS-1 trial (||||| in the roflumilast arm and ||||| in the vehicle arm) and by 67.0% of randomized patients in the DERMIS-2 trial (||||| in the roflumilast arm and ||||| in the vehicle arm). The most common (frequency of 10% or greater) classes of concomitant medications in the DERMIS-1 trial were 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors (|||||), propionic acid derivatives (|||||), and ACE inhibitors (|||||); note ||||| of all randomized patients used ACE inhibitors as a prior medication. Similarly, the most common (frequency of 10% or greater) classes of concomitant medications in the DERMIS-2 trial were propionic acid derivatives (|||||) followed by ACE inhibitors (||||); note, |||| of all randomized patients used ACE inhibitors as a prior medication (Table 16).
Table 15: Summary of Patient Exposure From the DERMIS-1 and DERMIS-2 Trials (Safety Population)
Exposure | DERMIS-1 | DERMIS-2 | ||
|---|---|---|---|---|
Roflumilast N = 286 | Vehicle N = 153 | Roflumilast N = 290 | Vehicle N = 152 | |
Total number of applications with intervention | ||||
N | 286 | 153 | 290 | 152 |
Mean (SD) | 55.1 (12.34) | 54.4 (15.89) | 54.3 (11.24) | 53.0 (14.69) |
Median (minimum, maximum) | 57 (3, 108) | 57 (2, 120) | 56 (1, 86) | 56 (1, 86) |
Total weight of intervention applied,a grams | ||||
N | ||| | ||| | ||| | ||| |
Mean (SD) | |||||| ||||||||| | |||||| ||||||||| | |||||| ||||||||| | |||||| ||||||||| |
Median (minimum, maximum) | |||| ||||| || |||||| | ||||| || || |||||| | |||| ||||| || ||||||| | ||||| ||||| || |||||| |
|||||||||| |||| |||||||| |||||||||||||| ||||b | ||||
> 100% | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| |
≥ 80% to ≤ 100% | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| |
< 80% | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| |
Missed > 3 consecutive doses, n (%) | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| |
Compliant,c n (%) | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| |
SD = standard deviation.
aTotal weight was determined by subtracting returned tube weight from the dispensed tube weight for each tube that was dispensed and summing the weights.
bCompliance was calculated based on number of applications divided by the expected number of applications for each patient multiplied by 100.
cA patient was considered compliant if they (or their caregiver) applied ≥ 80% of the expected applications during the application period and did not miss > 3 consecutive doses.
Sources: Clinical Study Reports for the DERMIS-19 and DERMIS-2 trials;10 details from the table were taken from the sponsor’s summary of clinical evidence.16
Table 16: Summary of Concomitant Medications From the DERMIS-1 and DERMIS-2 Trials (Safety Population)
Concomitant medication | DERMIS-1 | DERMIS-2 | ||
|---|---|---|---|---|
Roflumilast N = 286 | Vehicle N = 153 | Roflumilast N = 290 | Vehicle N = 152 | |
Patients with ≥ 1 concomitant medication, n (%) | ||| |||||| | ||| |||||| | ||| |||||| | | |||||| |
Most commona concomitant medication, n (%) | ||||
HMG-CoA reductase inhibitors | ||| |||||| | ||| |||||| | ||| |||||| | | |||||| |
ACE inhibitors | ||| |||||| | ||| |||||| | ||| |||||| | | |||||| |
Propionic acid derivatives | ||| |||||| | ||| |||||| | ||| |||||| | | |||||| |
ACE = angiotensin-converting enzyme; HMG-CoA = 3-hydroxy-3-methylglutaryl coenzyme A.
Note: Summary of concomitant medications was according to anatomic therapeutic chemical class level 4 and preferred term. Concomitant medications are all medications that were continued or started after the first application of the intervention. Patients were counted only once for each class and preferred term.
aFrequency ≥ 10% of all randomized patients in the study.
Sources: Clinical Study Reports for the DERMIS-19 and DERMIS-2 trials.10
Efficacy
The summarized end points comprise those included in the sponsor’s summary of clinical evidence as well as any end points identified as important to this review according to stakeholders, for example, the clinical expert, clinician groups, or patient groups.
Investigator’s Global Assessment
The primary end point, IGA success at week 8, was met for the DERMIS-1 and DERMIS-2 trials in the ITT population (Table 17). In the DERMIS-1 trial, 42.4% of patients in the roflumilast arm versus 6.1% of patients in the vehicle arm experienced treatment success based on the IGA at week 8; the ratio of the odds of IGA success with roflumilast relative to the odds of IGA success with the matching vehicle was ||||| |||| || |||| || |||||| | | ||||||| at week 8 from baseline in favour of roflumilast. In the DERMIS-2 trial, 37.5% of patients in the roflumilast arm versus 6.9% of patients in the vehicle arm experienced treatment success based on the IGA at week 8; the ratio of the odds of IGA success with roflumilast relative to the odds of IGA success with the matching vehicle was |||| |||| || |||| || |||||| | | ||||||| at week 8 from baseline, also in favour of roflumilast.
In both studies, the results of the primary analyses were generally consistent with the sensitivity analyses of the primary end point (modified ITT dataset, only observed data in the ITT dataset, and fitted point estimates from a generalized estimating equation) (Table 31 in Appendix 1).
The secondary end point, IGA success at week 4, was tested in a hierarchical manner and adjusted for multiple comparisons (Table 17). In the DERMIS-1 trial, the ratio of the odds of IGA success with roflumilast relative to the odds of IGA success with the matching vehicle was 13.44 (95% CI, 3.72 to 48.58; P < 0.0001) at week 4 from baseline in favour of roflumilast. In the DERMIS-2 trial, the ratio of the odds of IGA success with roflumilast relative to the odds of IGA success with the matching vehicle was 3.91 (95% CI, 1.76 to 8.70; P = 0.0011) at week 4 from baseline, also in favour of roflumilast.
The exploratory end points, IGA score of clear at weeks 4 and 8, were not included in the statistical hierarchy and not adjusted for multiple comparisons (Table 19). In the DERMIS-1 trial, 3.4% of patients in the roflumilast arm and no patients in the vehicle arm had an IGA score of clear at week 4; the results were consistent with the results in the DERMIS-2 trial. In the DERMIS-1 trial, 14.1% of patients in the roflumilast arm and 1.5% of patients in the vehicle arm had an IGA score of clear at week 8. Similarly, in the DERMIS-2 trial, 11.0% of patients in the roflumilast arm and no patients in the vehicle arm had an IGA score of clear at week 8.
Subgroup Analyses of the Primary Efficacy End Point
The subgroup analyses were not included in the statistical hierarchy or adjusted for multiple comparisons (Table 18).
Age Group
A prespecified subgroup analysis of the primary efficacy end point by age group (2 to 11 years, 12 to 17 years, and 18 years or older) was performed. In alignment with the Health Canada indication in patients aged 12 years or older, only the results for the 12 to 17 years and 18 years or older age groups are summarized subsequently.
In the DERMIS-1 trial, of the patients who were aged 12 to 17 years, ||||| in the roflumilast arm and ||||| in the vehicle arm, experienced treatment success based on the IGA at week 8. The results among patients who were aged 18 years or older were consistent with the results in the overall ITT population. In the DERMIS-2 trial, of the patients who were aged 12 to 17 years, no patients in either treatment arm experienced treatment success based on the IGA at week 8. The results among patients aged 18 years and older were consistent with the results for the overall ITT population.
Involvement of Intertriginous Area or Face
A prespecified subgroup analysis of the primary efficacy end point by intertriginous or facial involvement was performed.
In the DERMIS-1 trial, among patients with intertriginous or facial involvement, ||||| in the roflumilast arm and no patients in the vehicle arm experienced treatment success based on the IGA at week 8. In the DERMIS-2 trial, among patients with intertriginous or facial involvement, ||||| in the roflumilast arm and 3.9% in the vehicle arm experienced treatment success based on the IGA at week 8.
A post hoc pooled (DERMIS-1 and DERMIS-2) subgroup analysis of patients with facial and/or intertriginous and/or genital involvement was conducted. A numerically greater proportion of patients with plaque psoriasis involving the face and/or genital and/or intertriginous areas in the roflumilast arm experienced treatment success based on the IGA at week 8 compared with the vehicle arm; this was consistent with the results of the DERMIS-1 and DERMIS-2 studies.48
BSA Affected by Psoriasis at Baseline
A prespecified subgroup analysis of the primary efficacy end point by amount of BSA affected by psoriasis at baseline was performed.
In the DERMIS-1 trial, among patients with mild BSA affected (less than 5% of BSA), ||||| in the roflumilast arm and |||| in the vehicle arm experienced treatment success based on the IGA at week 8. In the DERMIS-2 trial, ||||| in the roflumilast arm and |||| in the vehicle arm experienced treatment success based on the IGA at week 8.
In the DERMIS-1 trial, among patients with moderate BSA affected (5% to less than 10% of BSA), ||||| in the roflumilast arm and |||| in the vehicle arm experienced treatment success based on the IGA at week 8. In the DERMIS-2 trial, ||||| in the roflumilast arm and ||||| in the vehicle arm experienced treatment success based on the IGA at week 8.
In the DERMIS-1 trial, among patients with severe BSA (10% and greater BSA), ||||| in the roflumilast arm and no patients in the vehicle arm experienced treatment success based on the IGA at week 8. In the DERMIS-2 trial, ||||| in the roflumilast arm and |||| in the vehicle arm experienced treatment success based on the IGA at week 8.
Intertriginous IGA
Note, the following analyses were based on the prespecified I-IGA ITT population, a subset of patients in the ITT population with intertriginous area involvement and an I-IGA of 2 or more at baseline.
The secondary end point, I-IGA success at week 8, was tested in a hierarchical manner and adjusted for multiple comparisons (Table 17). In the DERMIS-1 trial, the ratio of the odds of I-IGA success with roflumilast relative to the odds of I-IGA success with the matching vehicle was 17.94 (95% CI, 2.33 to 138.20; P < 0.0001) at week 8 from baseline in favour of roflumilast. In the DERMIS-2 trial, the ratio of the odds of I-IGA success with roflumilast relative to the odds of I-IGA success with the matching vehicle was 11.18 (95% CI, 2.33 to 53.68; P = 0.0004) at week 8 from baseline, also in favour of roflumilast.
The exploratory end point, I-IGA success at week 4, was not included in the statistical hierarchy and not adjusted for multiple comparisons (Table 19). In the DERMIS-1 trial, 42.3% of patients in the roflumilast arm and 27.6% of patients in the vehicle arm experienced treatment success based on the I-IGA at week 4. In the DERMIS-2 trial, 52.1% of patients in the roflumilast arm and 17.9% of patients in the vehicle arm experienced treatment success based on the I-IGA at week 4.
Psoriasis Area and Severity Index
The secondary end point, time to PASI 50, was tested in a hierarchical manner and adjusted for multiple comparisons (Table 17). In the DERMIS-1 trial, the median KM estimate of time to PASI 50 was 31.0 days (95% CI, 29.0 to 41.0) in the roflumilast arm versus 104.0 days (95% CI, 85.0 to NE) in the vehicle arm in favour of roflumilast (P < 0.0001). The hazard ratio was 3.867 (95% Wald CI, 2.795 to 5.351). In the DERMIS-2 trial, the median KM estimate of time to PASI 50 was 30.0 days (95% CI, 29.0 to 42.0) in the roflumilast arm and NE (95% CI, 71.0 to NE) in the vehicle arm in favour of roflumilast (P < 0.0001). The hazard ratio was 4.207 (95% Wald CI, 3.029 to 5.844).
The secondary end point, PASI 75 at week 8, was tested in a hierarchical manner and adjusted for multiple comparisons (Table 17). In the DERMIS-1 trial, the ratio of the odds of PASI 75 with roflumilast relative to the odds of PASI 75 with the matching vehicle was 12.00 (95% CI, 5.15 to 27.93; P < 0.0001) at week 8 from baseline in favour of roflumilast. In the DERMIS-2 trial, the ratio of the odds of PASI 75 with roflumilast relative to the odds of PASI 75 with the matching vehicle was 10.42 (95% CI, 4.49 to 24.19; P < 0.0001) at week 8 from baseline, also in favour of roflumilast.
The exploratory end point, PASI 75 at week 4, was not included in the statistical hierarchy and not adjusted for multiple comparisons (Table 19). In the DERMIS-1 trial, 21.8% of patients in the roflumilast arm and 3.0% of patients in the vehicle arm had a PASI 75 at week 4. In the DERMIS-2 trial, 16.2% of patients in the roflumilast arm and 3.6% of patients in the vehicle arm had a PASI 75 at week 4.
The exploratory end point, mPASI 75 at week 8, was not included in the statistical hierarchy and not adjusted for multiple comparisons (Table 32). In the DERMIS-1 trial, 63.1% of patients in the roflumilast arm and 22.0% of patients in the vehicle arm had an mPASI 75 at week 8. In the DERMIS-2 trial, 56.8% of patients in the roflumilast arm and 13.7% of patients in the vehicle arm had an mPASI 75 at week 8.
The exploratory end points, PASI 90 and mPASI 90 at week 8, were not included in the statistical hierarchy and not adjusted for multiple comparisons (Table 19 and Table 32, respectively). In the DERMIS-1 trial, 22.4% of patients in the roflumilast arm and 2.3% of patients in the vehicle arm had a PASI 90 at week 8, and 44.7% of patients in the roflumilast arm and 9.8% of patients in the vehicle arm had an mPASI 90 at week 8. In the DERMIS-2 trial, 17.0% of patients in the roflumilast arm and 2.3% of patients in the vehicle arm had a PASI 90 at week 8, and 35.2% of patients in the roflumilast arm and 8.4% of patients in the vehicle arm had an mPASI 90 at week 8.
The exploratory end points, PASI 100 and mPASI 100 at week 8, were not included in the statistical hierarchy and not adjusted for multiple comparisons (Table 19 and Table 32, respectively). In the DERMIS-1 trial, 13.7% of patients in the roflumilast arm and 1.5% of patients in the vehicle arm had a PASI 100 at week 8; the proportion of patients in each treatment arm who had an mPASI 100 at week 8 was consistent with the results for PASI 100 at week 8. In the DERMIS-2 trial, 11.0% of patients in the roflumilast arm and no patients in the vehicle arm had a PASI 100 at week 8; the proportion of patients in each treatment arm who had an mPASI 100 at week 8 was consistent with the results for PASI 100 at week 8.
DLQI and Children’s DLQI
The exploratory end point, absolute change in DLQI at week 8, was not included in the statistical hierarchy and not adjusted for multiple comparisons (Table 19). In the DERMIS-1 trial, the LS mean change from baseline in DLQI at week 8 was |||| || |||||| in the roflumilast arm and |||| || |||||| in the vehicle arm. In the DERMIS-2 trial, the LS mean change from baseline in DLQI at week 8 was ||| || |||||| in the roflumilast arm and |||| || |||||| in the vehicle arm.
Not enough data were collected in the DERMIS-1 and DERMIS-2 trials to carry out an analysis of covariance for change in CDLQI at week 8 (Table 19).
Worst Itch Numeric Rating Scale
The secondary end points, WI-NRS success at weeks 8, 4, and 2, were tested in a hierarchical manner and adjusted for multiple comparisons (Table 17). Note, the following analyses were based on the prespecified PRU4 ITT population, a subset of patients in the ITT population with a WI-NRS pruritus score of 4 or more at baseline.
In the DERMIS-1 trial, the ratio of the odds of WI-NRS success with roflumilast relative to the odds of WI-NRS success with the matching vehicle was 7.84 (95% CI, 3.85 to 15.94; P < 0.0001) at week 8 from baseline in favour of roflumilast. In the DERMIS-2 trial, the ratio of the odds of WI-NRS success with roflumilast relative to the odds of WI-NRS success with the matching vehicle was 3.59 (95% CI, 2.07 to 6.23; P < 0.0001) at week 8 from baseline, also in favour of roflumilast.
In the DERMIS-1 trial, the ratio of the odds of WI-NRS success with roflumilast relative to the odds of WI-NRS success with the matching vehicle was 4.36 (95% CI, 2.31 to 8.26; P < 0.0001) at week 4 from baseline in favour of roflumilast. In the DERMIS-2 trial, the ratio of the odds of WI-NRS success with roflumilast relative to the odds of WI-NRS success with the matching vehicle was 4.93 (95% CI, 2.65 to 9.18; P < 0.0001) at week 4 from baseline, also in favour of roflumilast.
In the DERMIS-1 trial, the ratio of the odds of WI-NRS success with roflumilast relative to the odds of WI-NRS success with the matching vehicle was 1.76 (95% CI, 0.98 to 3.19; P = 0.1197) at week 2 from baseline (note, this end point failed to reach statistical significance and was potentially underpowered by the planned sample size). In the DERMIS-2 trial, the ratio of the odds of WI-NRS success with roflumilast relative to the odds of WI-NRS success with the matching vehicle was 2.56 (95% CI, 1.43 to 4.58; P = 0.0026) at week 2 from baseline in favour of roflumilast.
Psoriasis Symptom Diary
The secondary end points, change from baseline in PSD total score at weeks 8 and 4, were tested in a hierarchical manner and adjusted for multiple comparisons (Table 17).
Table 17: Summary of Key Efficacy Results From the DERMIS-1 and DERMIS-2 Trials (ITT Population)
Outcome | DERMIS-1 | DERMIS-2 | ||
|---|---|---|---|---|
Roflumilast | Vehicle | Roflumilast | Vehicle | |
Primary outcome | ||||
IGA success,a week 8 | ||||
N | 286 | 153 | 290 | 152 |
n (%) | 255 (89.1) | 132 (86.2) | 264 (91.0) | 131 (86.1) |
IGA success, n (%) | 108 (42.4) | 8 (6.1) | 99 (37.5) | 9 (6.9) |
OR (95% CI)b | ||||| ||||| || |||||| | |||| ||||| || |||||| | ||
P value | < 0.0001 | < 0.0001 | ||
Secondary outcomes | ||||
Sequential testing, partition 1 (alpha = 0.03) | ||||
Time to PASI 50,c days | ||||
N | 286 | 153 | 290 | 152 |
n (%) | ||| |||||| | | |||||| | ||| |||||| | | |||||| |
Events/censors | |||||| | |||||| | |||||| | |||||| |
Mean (SD) | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| |
Median KM estimate (95% CI) | 31.0 (29.0 to 41.0) | 104.0 (85.0 to NE) | 30.0 (29.0 to 42.0) | NE (71.0 to NE) |
P value for median KM estimated | | |||||| | | |||||| | ||
HR,e 95% CI | 3.867 (2.795 to 5.351) | 4.207 (3.029 to 5.844) | ||
PASI 75 from baseline, week 8 | ||||
N | 286 | 153 | 290 | 152 |
n (%) | 255 (89.1) | 132 (86.2) | 264 (91.0) | 131 (86.1) |
PASI 75, n (%) | 106 (41.6) | 10 (7.6) | 103 (39.0) | 7 (5.3) |
OR (95% CI)b | 12.00 (5.15 to 27.93) | 10.42 (4.49 to 24.19) | ||
P value | < 0.0001 | < 0.0001 | ||
I-IGA success,f week 8 (I-IGA ITT population) | ||||
N | 63 | 32 | 53 | 31 |
n (%) | 52 (82.5) | 29 (90.6) | 47 (88.6) | 27 (87.0) |
I-IGA success, n (%) | 37 (71.2) | 4 (13.8) | 32 (68.1) | 5 (18.5) |
OR (95% CI)g | 17.94 (2.33 to 138.20) | 11.18 (2.33 to 53.68) | ||
P value | < 0.0001 | 0.0004 | ||
PSD, week 8 | ||||
N | 286 | 153 | 290 | 152 |
n (%) | 250 (87.4) | 129 (84.3) | 257 (88.6) | 127 (83.5) |
Baseline, mean (SD) | 72.1 (42.75) | 73.4 (41.29) | 69.3 (40.66) | 77.4 (41.24) |
Week 8, mean (SD) | 21.4 (30.04) | 50.0 (40.45) | 22.0 (33.25) | 53.6 (44.00) |
LS mean change from baseline (SE) | −50.1 (2.52) | −19.2 (3.14) | −49.3 (2.83) | −22.8 (3.48) |
LS mean difference (95% CI)h | −30.9 (−37.2 to −24.6) | −26.5 (−33.2 to −19.7) | ||
P valuei | < 0.0001 | < 0.0001 | ||
PSD, week 4 | ||||
N | 286 | 153 | 290 | 152 |
n (%) | 261 (91.2) | 130 (84.9) | 262 (90.3) | 135 (88.8) |
Baseline, mean (SD) | 72.1 (42.75) | 73.4 (41.29) | 69.3 (40.66) | 77.4 (41.24) |
Week 4, mean (SD) | 28.9 (32.47) | 53.1 (39.08) | 27.5 (32.42) | 58.2 (40.78) |
LS mean change from baseline (SE) | −43.5 (2.36) | −17.7 (2.97) | −42.7 (2.56) | −16.7 (3.12) |
LS mean difference (95% CI)h | −25.8 (−31.7 to −20.0) | −26.0 (−31.9 to −20.0) | ||
P valuei | < 0.0001 | < 0.0001 | ||
IGA success,a week 4 | ||||
N | 286 | 153 | 290 | 152 |
n (%) | 262 (91.6) | 132 (86.2) | 267 (92.0) | 139 (91.4) |
IGA success, n (%) | 54 (20.6) | 3 (2.3) | 51 (19.1) | 8 (5.8) |
OR (95% CI)b | 13.44 (3.72 to 48.58) | 3.91 (1.76 to 8.70) | ||
P value | < 0.0001 | 0.0011 | ||
Sequential testing, partition 2 (alpha = 0.02) | ||||
WI-NRS success,j week 8 (PRU4 ITT population) | ||||
N | 218 | 115 | 229 | 116 |
n (%) | 191 (87.6) | 97 (84.3) | 206 (89.9) | 101 (87.0) |
WI-NRS success, n (%) | 129 (67.5) | 26 (26.8) | 143 (69.4) | 36 (35.6) |
OR (95% CI)b | 7.84 (3.85 to 15.94) | 3.59 (2.07 to 6.23) | ||
P value | < 0.0001 | < 0.0001 | ||
WI-NRS success,j week 4 (PRU4 ITT population) | ||||
N | 218 | 115 | 229 | 116 |
n (%) | 201 (92.2) | 100 (86.9) | 212 (92.5) | 105 (90.5) |
WI-NRS success, n (%) | 101 (50.2) | 18 (18.0) | 120 (56.6) | 23 (21.9) |
OR (95% CI)b | 4.36 (2.31 to 8.26) | 4.93 (2.65 to 9.18) | ||
P value | < 0.0001 | < 0.0001 | ||
WI-NRS success,j week 2 (PRU4 ITT population) | ||||
N | 218 | 115 | 229 | 116 |
n (%) | 209 (95.8) | 109 (94.7) | 215 (93.8) | 109 (93.9) |
WI-NRS success, n (%) | 73 (34.9) | 24 (22.0) | 90 (41.9) | 23 (21.1) |
OR (95% CI)b | 1.76 (0.98 to 3.19) | 2.56 (1.43 to 4.58) | ||
P value | 0.1197 | 0.0026 | ||
ANCOVA = analysis of covariance; CI = confidence interval; HR = hazard ratio; IGA = Investigator’s Global Assessment; I-IGA = intertriginous Investigator’s Global Assessment; I-IGA ITT = a subset of patients in the ITT population with intertriginous area involvement and an I-IGA of 2 or more at baseline; ITT = intention-to-treat; KM = Kaplan-Meier; LS = least squares; NE = not estimable; OR = odds ratio; PASI 50 = 50% reduction in the Psoriasis Area and Severity Index; PASI 75 = 75% reduction in the Psoriasis Area and Severity Index; PRU4 ITT = patients in the ITT population with a WI-NRS pruritus score of ≥ 4 at baseline; PSD = Psoriasis Symptom Diary; SD = standard deviation; SE = standard error; WI-NRS = Worst Itch Numeric Rating Scale.
Note: The P value was adjusted for multiple testing using a prespecified hierarchical testing strategy.
aIGA success was defined as a score of 0 (clear) or 1 (almost clear) plus a ≥ 2-grade improvement from baseline.
bOdds ratio, 95% CI, and P value were obtained from the Cochran-Manel-Haenszel test (stratified by study site, baseline IGA, and baseline intertriginous involvement) comparing roflumilast cream 0.3% with vehicle.
cTime to PASI 50 (50% reduction in PASI from baseline) was calculated as date of PASI 50 minus the day 1 date plus 1. Patients who did not have PASI 50 were censored at day of discontinuation or date lost to follow-up, whichever was earlier. Censored values were not included in the descriptive statistics; only patients who had PASI 50 were included in the descriptive statistics.
dP value was obtained from the log-rank test where roflumilast cream 0.3% was compared with the matching vehicle.
eThe HR and 95% Wald CI were obtained from a Cox proportional hazards model comparing roflumilast cream 0.3% with the vehicle with the site, baseline IGA score category, and baseline intertriginous involvement score category as factors in the model.
fI-IGA success was defined as a score of 0 (clear) or 1 (almost clear) plus a ≥ 2-grade improvement from baseline in patients with intertriginous area involvement and I-IGA ≥ 2 at baseline. This analysis was based on the I-IGA ITT population.
gThe odds ratio, 95% CI, and P value were obtained from the Cochran-Mantel-Haenszel test (stratified by study site and baseline I-IGA) comparing roflumilast cream 0.3% with vehicle.
hEstimate for LS means (change from baseline and difference from vehicle), 95% CIs and P values are from an ANCOVA with treatment, site, baseline IGA, baseline intertriginous involvement, and baseline PSD score as independent variables.
iThe P value is to test for a zero difference between groups (roflumilast cream 0.3% minus vehicle) in change from baseline.
jWI-NRS success was defined as a ≥ 4-point reduction in WI-NRS pruritus score from baseline in patients with a WI-NRS pruritus score of ≥ 4 at baseline. The analysis was based on the PRU4 ITT population.
Sources: Clinical Study Reports for the DERMIS-19 and DERMIS-2 trials;10 details from the table were taken from the sponsor’s summary of clinical evidence.16
Table 18: Subgroup Analyses of the Primary Efficacy End Point in the DERMIS-1 and DERMIS-2 Trials (ITT Population)
Outcome | DERMIS-1 | DERMIS-2 | ||
|---|---|---|---|---|
Roflumilast | Vehicle | Roflumilast | Vehicle | |
IGA success in patients aged 12 to 17 years, week 8 | ||||
N | | ||||||| | | ||||||| | | ||||||| | | ||||||| |
n (%) | | ||||||| | | ||||||| | | ||||||| | | ||||||| |
IGA success, n (%) | | |||||| | | |||||| | | |||| | | |||| |
OR (95% CI)a | | |||| | | |||| | ||
P valueb | | ||||||| | | ||||||| | ||
IGA success in patients aged 18 years or older, week 8 | ||||
N | ||| | ||| | ||| | ||| |
n (%) | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| |
IGA success, n (%) | ||| |||||| | | ||||| | | |||||| | | ||||| |
OR (95% CI)a | ||||| ||||| || |||||| | |||| ||||| || |||||| | ||
P valueb | | |||||| | | |||||| | ||
IGA success in patients with intertriginous or facial involvement, week 8 | ||||
N | 120 | 64 | 107 | 58 |
n (%) | 104 (86.6) | 58 (90.6) | 96 (89.7) | 51 (87.9) |
IGA success, n (%) | 45 (43.3) | 0 | 38 (39.6) | 2 (3.9) |
OR (95% CI)a | NE (NE) | 13.45 (2.65 to 68.27) | ||
P valueb | < 0.0001 | 0.0002 | ||
IGA success in patients with psoriasis involving mild baseline BSA (< 5% of BSA), week 8 | ||||
N | ||| | || | ||| | || |
n (%) | ||| |||||| | | |||||| | ||| |||||| | | |||||| |
IGA success, n (%) | | |||||| | | ||||| | | |||||| | | ||||| |
OR (95% CI)a | ||||| ||||| || |||||| | ||||| ||||| || |||||| | ||
P valueb | | |||||| | |||||| | ||
IGA success in patients with psoriasis involving moderate baseline BSA (5% to < 10% of BSA), week 8 | ||||
N | || | || | || | || |
n (%) | | |||||| | | |||||| | | |||||| | | |||||| |
IGA success, n (%) | | |||||| | | ||||| | | |||||| | | |||||| |
OR (95% CI)a | |||| ||||| || |||||| | |||| ||||| || |||||| | ||
P valueb | |||||| | |||||| | ||
IGA success in patients with psoriasis involving severe baseline BSA (≥ 10% of BSA), week 8 | ||||
N | | ||||||| | | ||||||| | | ||||||| | | ||||||| |
n (%) | | ||||||| | | ||||||| | | ||||||| | | ||||||| |
IGA success, n (%) | | ||||||| | | ||||||| | | ||||||| | | ||||||| |
OR (95% CI)a | | |||| | ||||| ||||| || |||||| | ||
P valueb | |||||| | |||||| | ||
BSA = body surface area; CI = confidence interval; IGA = Investigator’s Global Assessment; ITT = intention-to-treat; NE = not estimable; OR = odds ratio.
Note: IGA success was defined as a score of 0 (clear) or 1 (almost clear) plus a ≥ 2-grade improvement from baseline.
aThe odds ratio, 95% CI, and P value were obtained from a Cochran-Mantel-Haenszel test (stratified by site, baseline IGA, and baseline intertriginous involvement) comparing roflumilast cream 0.3% with vehicle.
bThe P value was not adjusted for multiple testing (i.e., the type I error rate was not controlled).
Sources: Clinical Study Reports for the DERMIS-19 and DERMIS-2 trials.10
Table 19: Exploratory Outcomes in the DERMIS-1 and DERMIS-2 Trials (ITT Population)
Outcome | DERMIS-1 | DERMIS-2 | ||
|---|---|---|---|---|
Roflumilast | Vehicle | Roflumilast | Vehicle | |
IGA score of clear, week 4 | ||||
N | 286 | 153 | 290 | 152 |
n (%) | 262 (91.6) | 132 (86.2) | 267 (92.0) | 139 (91.4) |
IGA score of clear, n (%) | 9 (3.4) | 0 | 9 (3.4) | 0 |
OR (95% CI)a | NE (NE) | NE (NE) | ||
P valueb | 0.0691 | 0.0763 | ||
IGA score of clear, week 8 | ||||
N | 286 | 153 | 290 | 152 |
n (%) | 255 (89.1) | 132 (86.2) | 264 (91.0) | 131 (86.1) |
IGA score of clear, n (%) | 36 (14.1) | 2 (1.5) | 29 (11.0) | 0 |
OR (95% CI)b | 11.41 (2.73 to 47.61) | 17.27 (2.51 to 118.80) | ||
P valueb | 0.0001 | 0.0002 | ||
I-IGA success,c week 4 (I-IGA ITT population) | ||||
N | 63 | 32 | 53 | 31 |
n (%) | 52 (82.5) | 29 (90.6) | 48 (90.5) | 28 (90.3) |
I-IGA success, n (%) | 22 (42.3) | 8 (27.6) | 25 (52.1) | 5 (17.9) |
OR (95% CI)d | 1.69 (0.46 to 6.22) | 5.12 (1.40 to 18.69) | ||
P valueb | 0.8234 | 0.0245 | ||
PASI 75 from baseline, week 4 | ||||
N | 286 | 153 | 290 | 152 |
n (%) | 262 (91.6) | 132 (86.2) | 266 (91.7) | 139 (91.4) |
PASI 75, n (%) | 57 (21.8) | 4 (3.0) | 43 (16.2) | 5 (3.6) |
OR (95% CI)a | 10.44 (3.43 to 31.78) | 4.57 (1.77 to 11.83) | ||
P valueb | < 0.0001 | 0.0016 | ||
PASI 90 from baseline, week 8 | ||||
N | 286 | 153 | 290 | 152 |
n (%) | 255 (89.1) | 132 (86.2) | 264 (91.0) | 131 (86.1) |
PASI 90, n (%) | 57 (22.4) | 3 (2.3) | 45 (17.0) | 3 (2.3) |
OR (95% CI)a | 17.16 (4.25 to 69.22) | 8.42 (2.45 to 28.86) | ||
P valueb | < 0.0001 | 0.0002 | ||
PASI 100 from baseline, week 8 | ||||
N | 286 | 153 | 290 | 152 |
n (%) | 255 (89.1) | 132 (86.2) | 264 (91.0) | 131 (86.1) |
PASI 100, n (%) | 35 (13.7) | 2 (1.5) | 29 (11.0) | 0 |
OR (95% CI)a | 10.51 (2.50 to 44.14) | 9.45 (0.69 to 128.93) | ||
P valueb | 0.0003 | 0.0003 | ||
DLQI, week 8 | ||||
N | 286 | 153 | 290 | 152 |
n (%) | 249 (87.0) | 130 (84.9) | 259 (89.3) | 128 (84.2) |
Baseline, mean (SD) | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| |
LS mean change from baseline (SE) | |||| |||||| | |||| |||||| | |||| |||||| | |||| |||||| |
LS mean difference (95% CI)e | |||| ||||| || ||||| | |||| ||||| || ||||| | ||
P valueb | < 0.0001 | < 0.0001 | ||
CDLQI, week 8 | ||||
N | 286 | 153 | 290 | 152 |
n (%) | | ||||| | | |f | | ||||| | | |f |
Baseline, mean (SD) | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| |
LS mean change from baseline (SE) | | |||| | | |||| | | |||| | | |||| |
LS mean difference (95% CI)e | | |||| | | |||| | ||
P valueb | || | || | ||
BSA, week 8 | ||||
N | 286 | 153 | 290 | 152 |
n (%) | 255 (89.1) | 132 (86.2) | 264 (91.0) | 130 (85.5) |
Baseline, mean (SD) | 6.28 (4.376) | 7.36 (4.762) | 7.08 (4.839) | 7.73 (5.054) |
LS mean percent change from baseline (SE) | |||||| ||||||| | |||||| ||||||| | |||||| ||||||| | ||||| ||||||| |
LS mean percent difference (95% CI)e | |||||| ||||||| || ||||||| | |||||| ||||||| || ||||||| | ||
P valueb | < 0.0001 | < 0.0001 | ||
ANCOVA = analysis of covariance; BSA = body surface area; CDLQI = Children’s Dermatology Life Quality Index; CI = confidence interval; DLQI = Dermatology Life Quality Index; IGA = Investigator’s Global Assessment; I-IGA = intertriginous Investigator’s Global Assessment; I-IGA ITT = a subset of patients in the ITT population with intertriginous area involvement and an I-IGA of 2 or more at baseline; ITT = intention-to-treat; LS = least squares; NE = not estimable; OR = odds ratio; PASI 75 = 75% reduction in the Psoriasis Area and Severity Index score; PASI 90 = 90% reduction in the Psoriasis Area and Severity Index score; PASI 100 = 100% reduction in the Psoriasis Area and Severity Index score; SD = standard deviation; SE = standard error.
aThe odds ratio, 95% CI, and P value were obtained from a Cochran-Mantel-Haenszel test (stratified by site, baseline IGA, and baseline intertriginous involvement) comparing roflumilast cream 0.3% with vehicle.
bThe P value was not adjusted for multiple testing (i.e., the type I error rate was not controlled).
cI-IGA success was defined as a score of 0 (clear) or 1 (almost clear) plus a ≥ 2-grade improvement from baseline in patients with intertriginous area involvement and I-IGA ≥ 2 at baseline. This analysis was based on the I-IGA ITT population.
dThe odds ratio, 95% CI, and P value were obtained from a Cochran-Mantel-Haenszel test (stratified by study site and baseline I-IGA) comparing roflumilast cream 0.3% with vehicle.
eEstimates for LS means (change divided by percentage change from baseline and difference from vehicle) and accompanying 95% CIs, and P values are from an analysis of covariance with treatment, site, baseline IGA, baseline intertriginous involvement, and baseline of the variable being analyzed as independent variables.
fNot enough data were collected to carry out an ANCOVA for this parameter.
Sources: Clinical Study Reports for the DERMIS-19 and DERMIS-2 trials.10
In the DERMIS-1 trial, the LS mean difference between roflumilast and vehicle in change from baseline in PSD total score at week 8 was −30.9 (standard error [SE] = 3.22; 95% CI, −37.2 to −24.6; P < 0.0001), in favour of roflumilast. In the DERMIS-2 trial, the LS mean difference between roflumilast and vehicle in change from baseline in PSD total score at week 8 was −26.5 (SE = 3.44; 95% CI, −33.2 to −19.7; P < 0.0001), also in favour of roflumilast.
In the DERMIS-1 trial, the LS mean difference between roflumilast and vehicle in change from baseline in PSD total score at week 4 was −25.8 (SE = 3.00; 95% CI, −31.7 to −20.0; P < 0.0001) in favour of roflumilast. In the DERMIS-2 trial, the LS mean difference between roflumilast and vehicle in change from baseline in PSD total score at week 4 was −26.0 (SE = 3.03; 95% CI, −31.9 to −20.0; P < 0.0001), also in favour of roflumilast.
Body Surface Area
The exploratory end point, percent change in BSA affected by psoriasis at week 8, was not included in the statistical hierarchy and not adjusted for multiple comparisons (Table 19). In the DERMIS-1 trial, the LS mean percent change from baseline in BSA affected by psoriasis at week 8 was |||||| || ||||||| in the roflumilast arm ||| |||||| || ||||||| in the vehicle arm. In the DERMIS-2 trial, the LS mean percent change from baseline in BSA affected by psoriasis at week 8 was |||||| || |||||| in the roflumilast arm and |||| || ||||||| in the vehicle arm.
Harms
A summary of harms from the DERMIS-1 and DERMIS-2 trials is presented in Table 20.
Adverse Events
The proportion of patients in the roflumilast arm with any TEAE was 25.2% in the DERMIS-1 trial and 25.9% in the DERMIS-2 trial, while the proportion of patients in the vehicle arm with any TEAE was 23.5% in the DERMIS-1 trial and 18.4% in the DERMIS-2 trial. The most common TEAEs reported in the roflumilast arm (a frequency of 2% or more of patients in either study) were diarrhea (3.5% in the DERMIS-1 trial and 2.8% in the DERMIS-2 trial) and headache (1.0% in the DERMIS-1 trial and 3.8% in the DERMIS-2 trial). All remaining TEAEs were reported in less than 2% of patients in the roflumilast arm in either study.
Serious Adverse Events
The proportion of patients in the roflumilast arm with any SAE was 0.7% in the DERMIS-1 trial and no patients in the DERMIS-2 trial, while the proportion of patients in the vehicle arm with any SAE was 0.7% in both trials. The SAEs reported in the roflumilast arm were concussion (1 patient in the DERMIS-1 trial) and foot fracture, thorax deformity, and pneumothorax (1 patient in the DERMIS-1 trial).
Withdrawals Due to Adverse Events
The proportion of patients in the roflumilast arm with any WDAE was |||| in the DERMIS-1 trial and |||| in the DERMIS-2 trial, while the proportion of patients in the vehicle arm with any WDAE was |||| in the DERMIS-1 trial and |||| in the DERMIS-2 trial. TEAEs leading to discontinuation of treatment and/or study withdrawal were reported in less than || of patients in the roflumilast arm in either study.
Mortality
Based on information provided by the sponsor, no deaths occurred during the DERMIS-1 and DERMIS-2 trials.
Notable Harms
The proportion of patients with application site pain in the roflumilast arm was 0.7% in the DERMIS-1 trial and 1.4% in the DERMIS-2 trial. All remaining TEAEs of special interest (application site pruritus, urticaria, dryness, dermatitis, and irritation) were reported in less than 1% of patients in the roflumilast arm in either study.
Local Tolerability Assessments
In the DERMIS-1 trial, the majority of patients in the roflumilast arm (||||| investigator-rated; ||||| patient-rated) and in the vehicle arm (97.6% investigator-rated; 90.6% patient-rated) did not have tolerability concerns based on local tolerability assessments at week 8.
In the DERMIS-2 trial, the majority of patients in the roflumilast arm (|||| investigator-rated; ||||| patient-rated) and in the vehicle arm (||||| investigator-rated; ||||| patient-rated) did not have tolerability concerns based on local tolerability assessments at week 8.
Table 20: Summary of Harms From the DERMIS-1 and DERMIS-2 Trials (Safety Population)
Events | DERMIS-1 | DERMIS-2 | ||
|---|---|---|---|---|
Roflumilast N = 286 | Vehicle N = 153 | Roflumilast N = 290 | Vehicle N = 152 | |
Most commona adverse events, n (%) | ||||
Patients with ≥ 1 TEAE | 72 (25.2) | 36 (23.5) | 75 (25.9) | 28 (18.4) |
Diarrhea | 10 (3.5) | 0 | 8 (2.8) | 0 |
Headache | 3 (1.0) | 2 (1.3) | 11 (3.8) | 1 (0.7) |
|||||||| | | ||||| | | ||||| | | ||||| | | ||||| |
|||||| | | ||||| | | ||||| | | ||||| | | ||||| |
Nasopharyngitis | 5 (1.7) | 3 (2.0) | 1 (0.3) | 1 (0.7) |
||||||||||| |||| |||| | | ||||| | | ||||| | | ||||| | | ||||| |
||||||| ||||| ||||||||| | | ||||| | | ||||| | | ||||| | | ||||| |
||||||| |||||||||||||||| ||||||||| | | ||||| | | ||||| | | ||||| | | ||||| |
||||||||| |||||||||||||||| ||||||||| | | ||||| | | ||||| | | ||||| | | ||||| |
SAEs, n (%) | ||||
Patients with ≥ 1 SAE | 2 (0.7) | 1 (0.7) | 0 | 1 (0.7) |
|||||||||| | | ||||| | | ||||| | | ||||| | | ||||| |
|||| ||||||||| ||||||||| ||||||| || |||||||||||| | | ||||| | | ||||| | | ||||| | | ||||| |
|||||| |||| ||||||| | | ||||| | | ||||| | | ||||| | | ||||| |
|||||||| ||||||||||||| | | ||||| | | ||||| | | ||||| | | ||||| |
|||||||| || ||||||| ||||||||| |||||| ||||| || || |||||| |||| | ||||
|||||||| |||| | | |||| | | ||||| | | ||||| | | ||||| | | ||||| |
||||||||||| |||| ||||||||| | | ||||| | | ||||| | | ||||| | | ||||| |
||||||||||| |||| |||||||| ||||||||||| |||| ||||||||| ||||||||||| |||||||||||||||||| ||||||||||||| || ||||| || |||||||| | | ||||| | | ||||| | | ||||| | | ||||| |
|||||||||| | | ||||| | | ||||| | | ||||| | | ||||| |
||||||||||||||| | | ||||| | | ||||| | | ||||| | | ||||| |
||||||| |||||||||||||||| ||||||||| || ||||||||| |||||||||||||||| ||||||||| | | ||||| | | ||||| | | ||||| | | ||||| |
|||||| || ||||||||| | | ||||| | | ||||| | | ||||| | | ||||| |
||||||||||| |||| |||||||||| | | ||||| | | ||||| | | ||||| | | ||||| |
|||||| |||| ||||||| | | ||||| | | ||||| | | ||||| | | ||||| |
||||||||||| |||| |||| | | ||||| | | ||||| | | ||||| | | ||||| |
|||||||||| |||| |||||||| | | ||||| | | ||||| | | ||||| | | ||||| |
|||||||||| |||| | ||||
|||||| | | ||||| | | ||||| | | ||||| | | ||||| |
Notable harms,b n (%) | ||||
Application site pain | 2 (0.7) | 1 (0.7) | 4 (1.4) | 0 |
||||||||||| |||| |||||||| | | ||||| | | ||||| | | ||||| | | ||||| |
||||||||||| |||| ||||||||| | | ||||| | | ||||| | | ||||| | | ||||| |
||||||||||| |||| ||||||| | | ||||| | | ||||| | | ||||| | | ||||| |
||||||||||| |||| |||||||||| | | ||||| | | ||||| | | ||||| | | ||||| |
||||||||||| |||| |||||||||| | | ||||| | | ||||| | | ||||| | | ||||| |
NA = not applicable; NR = not reported; SAE = serious adverse event; TEAE = treatment-emergent adverse event; WDAE = withdrawal due to adverse event.
aTEAEs in > 1% of roflumilast-treated patients in the DERMIS-1 or DERMIS-2 trials.
bPatients were counted once for each system organ class and once for each preferred term.
Sources: Clinical Study Reports for the DERMIS-19 and DERMIS-2 trials;10 details from the table were taken from the sponsor’s summary of clinical evidence.16
Critical Appraisal
Internal Validity
The DERMIS-1 and DERMIS-2 studies were randomized, double-blind, and vehicle-controlled. Patients were randomized at baseline according to a computer-generated randomization list and randomization was stratified by study site, baseline IGA score (2 versus ≥ 3), and intertriginous involvement at baseline (I-IGA ≥ 2, yes versus no). Based on input from the clinical expert consulted by CADTH for this review, the extent and severity of disease as measured by baseline BSA and PASI are additional effect modifiers. Note that the median and mean BSA and PASI scores were slightly higher in the vehicle arm compared with the roflumilast arm in both trials. IGA assesses severity of lesions, while BSA and PASI account for extent and severity of disease. Therefore, stratification by IGA alone may not result in an optimal comparability in disease severity between treatment arms, and this may have introduced bias in the efficacy results against roflumilast due to the aforementioned imbalance (note that the magnitude of this potential bias is not known). No other baseline demographic and clinical characteristics were identified that could have had a potential impact on the results in either study.
In both the DERMIS-1 and DERMIS-2 studies, the duration of the drug washout period specific to each class of excluded medication or treatment, if inappropriately selected, was expected to have a possible carryover effect on psoriasis vulgaris. The duration of the washout period was considered reasonable for each treatment in plaque psoriasis by the clinical expert. Therefore, any continued effect from these treatments on plaque psoriasis was not expected after the washout period and not likely to have an impact on the efficacy assessment of roflumilast.
More than 90% of patients in each treatment arm across studies were compliant with the assigned intervention, defined as a patient (or caregiver) who applied 80% or more of the expected applications during the application period and did not miss more than 3 consecutive doses. Topical and systemic treatments as well as phototherapy for plaque psoriasis were prohibited during the treatment period in both studies. Short-term and chronic medications were permitted during the treatment period; similar proportions of patients who reported use of the most common concomitant medications were observed between treatment arms in both studies. The most common major protocol deviation reported in both studies was efficacy assessment (14.4% of all randomized patients in the DERMIS-1 trial and 14.9% of all randomized patients in the DERMIS-2 trial), which was similar between treatment arms across studies. In both studies, the high rate of treatment compliance, concomitant medication use, and deviation of trial performance were considered unlikely to have introduced bias in the efficacy results.
Sensitivity analyses of the primary efficacy end point were performed using the observed data, modified ITT population, and tabulation of fitted point estimates from generalized estimating equations for binary response on observed data; the results of these sensitivity analyses were generally consistent with the results of the primary analysis. Missing data were imputed for the primary, secondary (with the exception of time to PASI 50), and select exploratory outcomes (I-IGA success, PASI 75, PASI 90, and PASI 100) that were included in this report using a regression-based multiple imputation model. According to the investigator, this approach to imputation was considered superior to other strategies, such as the last observation carried forward approach. However, no sensitivity analysis using the last observation carried forward approach was reported; that method is considered a relatively more conservative approach for a chronic disease such as plaque psoriasis, when missingness is likely at random and nondifferential between treatment arms.
The primary efficacy outcome in both studies was IGA success, defined as an IGA score of 0 (clear) or 1 (almost clear), plus an improvement of 2 or more grades from baseline at week 8. Evidence for the validity34-36 and reliability36,37 of the 6- and 7-point IGA scales was identified in the literature; however, no evidence for the responsiveness of the IGA was identified. Note that conclusions about the validity and reliability of the 5-point IGA used in the DERMIS-1 and DERMIS-2 studies are limited due to the use of the 6- and 7-point IGA in the psychometric validation studies. However, the clinical expert suggested that this difference in scales was unlikely to have introduced bias in the results. Although an MID has not been estimated, achieving a score of 0 (clear) or 1 (almost clear) on the static IGA has been generally accepted as clinically meaningful (i.e., a responder analysis would consider the proportion of patients with psoriasis who experienced treatment success based on the IGA, defined as a score of 0 or 1 plus at least a 2-grade improvement from baseline).11,12 Alternatively, or in addition to a score of 0 or 1, the responder analysis may also consider the proportion of patients with at least a 2-grade improvement from baseline on the static IGA.11These were consistent with the definition of IGA success used in both studies.
The secondary efficacy outcomes in both studies included the PASI, I-IGA, PSD, IGA, and WI-NRS. Evidence for the validity36,38 and reliability39 of the PASI was identified in the literature. The responsiveness of the PASI was found to be low when the affected BSA was less than 10%;34,40 an mPASI score was calculated for patients with an anatomic area with less than 10% involvement to reflect the status of limited disease in the studies. An MID in the PASI has not been estimated. The measurement properties of the mPASI and I-IGA were not identified in the literature. Evidence for the validity, reliability, and responsiveness of the PSD was identified in the literature.41 An MID in PSD total score has not been estimated. Evidence for the validity, reliability, and responsiveness of WI-NRS in patients with plaque psoriasis was identified in the literature.15 The MID in WI-NRS has been estimated to be an improvement of 4 or more points in patients with plaque psoriasis;15 this was used in the definition of WI-NRS success in both studies.
Of note, IGA and PASI are investigator-reported outcome measures, while the PSD, WI-NRS, DLQI, and CDLQI are patient-reported outcome measures. As such, there was a potential risk of bias, likely in favour of roflumilast, due to subjective reporting of patient-reported outcomes and the potential for the unblinding of treatment assignment as a result of patients observing an improvement in the affected area with roflumilast compared with the matching vehicle, which contained only the excipients of the roflumilast cream. Note that this potential for the unblinding of the treatment assignment may not affect investigator-assessed end points, such as IGA, in the same manner. The clinical expert identified treatment-free interval and duration of response (remission) as additional outcomes important to patients that were not captured in the sponsor’s systematic review protocol.
The primary and secondary outcomes were controlled for multiplicity in both studies using a prespecified hierarchical testing strategy and the Holm procedure to control the familywise type I error. The planned sample size provided sufficient power to analyze up to the first 5 secondary end points; note that the remaining end points demonstrated statistically significant differences between treatment arms that were consistently in favour of roflumilast, with the exception of WI-NRS success at week 2 in DERMIS-1. The relatively small sample size of patients available for subgroup analysis, in particular, the number of patients aged 12 to 17 years, significantly limited the interpretation of findings and the assessment of treatment benefit in patient subgroups. Moreover, as indicated by the clinical expert, the potential heterogeneity of treatment effect by extent and severity of disease as measured by BSA or PASI was not reported. This further compromised the certainty of the evidence on the treatment effect of roflumilast among patients by different extent and severity of disease.
External Validity
Based on input from the clinical expert consulted by CADTH for this review, the inclusion and exclusion criteria were considered narrow. For example, patients with an IGA score of 1 and a PASI score of 1 would be considered candidates for treatment with roflumilast in clinical practice in Canada; however, they were excluded from the trials, as a score of at least 2 was required for enrolment. Additionally, the clinical expert suggested that patients with plaque psoriasis involving less than 2% and more than 20% of BSA, excluding the scalp, palms, and soles, would potentially be treated with roflumilast but were also excluded from the trials. As such, the effect of roflumilast in the broader patient population is unknown; however, based on clinical expert input, it is anticipated that roflumilast will be used in the broader patient population in clinical practice despite the lack of evidence. Note that the Health Canada indication also does not restrict the patient population according to percent BSA involvement.
According to the Canadian Guidelines for the Management of Plaque Psoriasis, the definitions for mild, moderate, and severe plaque psoriasis based on disease severity measures differ, depending on the setting (clinical trials versus clinical practice).6 Measures of disease severity in clinical practice include the impact of disease on the patient’s QoL and level of symptomatic control by routine skin care measures and/or topical therapy.6 Similarly, the clinical expert indicated that defining the severity of disease in clinical practice is often a gestalt assessment and would require consideration of clearance or near clearance of psoriasis lesions, treatment history, HRQoL, and indication for topical versus systemic therapy. Measures of disease severity (i.e., various cut-offs and scales) in clinical trials are not consistently defined and can differ, depending on the class of drug being evaluated.6 Note that the IGA assesses the severity of lesions, while the PASI accounts for extent and severity of disease and, as such, the clinical expert agreed the PASI is more clinically meaningful compared with the IGA. Nonetheless, the PASI and IGA are correlated, as both tools assess the severity of lesions.35,36 The lower limit of moderate to severe disease may be set at a PASI score of 8 and the lower limit of severe disease may be set at a PASI score of 106; similarly, the clinical expert estimated a cut-off of 10 on the PASI scale to indicate moderate disease. According to these thresholds for moderate to severe disease, the majority of patients in both studies had mild disease based on the mean baseline PASI score, but based on the baseline PASI range, both studies also included patients with moderate to severe disease (i.e., candidates for systemic therapy).
The intervention used in both studies, roflumilast cream 0.3% administered topically to lesions of plaque psoriasis once daily, was consistent with the dosage recommended in the product monograph.22 The clinical expert indicated that in clinical practice, roflumilast would probably be stopped by most patients when lesions are clear; however, the treatment of affected areas with roflumilast or the vehicle was maintained for the duration of the studies, regardless of whether the psoriasis cleared.
In both studies, roflumilast was compared with a matching vehicle cream that contained only the excipients of the roflumilast cream. However, given the wide range of topical treatment options currently available in clinical practice for plaque psoriasis, the clinical expert agreed that an active comparator would have been more appropriate; in particular, topical steroids and vitamin D analogues would have been appropriate comparators. However, the vehicle cream may be considered an appropriate comparator, as there are limited options for intertriginous areas, which have been identified as an area of unmet need by the clinical expert and clinician groups.
The clinician groups and the clinical expert consulted by CADTH for this review agreed the IGA (otherwise referred to as the PGA in practice), PASI, DLQI or CDLQI, and percent of BSA involvement were clinically relevant and meaningful (i.e., capture the extent and severity of disease and determine treatment response in clinical practice). The clinical expert also indicated that the I-IGA, PSD, and WI-NRS were clinically relevant. Moreover, the clinical expert indicated that clearance or near clearance of psoriasis lesions is commonly used in clinical practice to assess psoriasis severity, which is analogous to the use of the IGA in clinical trials. Both the clinician groups and the clinical expert indicated that tools such as the PASI and DLQI are not commonly used in clinical practice unless mandated by the payers for reimbursement. Additionally, the clinician groups and the clinical expert indicated that patient satisfaction and/or feedback is an important outcome used to determine treatment response in clinical practice. The clinical expert indicated that components of patient satisfaction can include improvement in symptoms (itch), appearance of lesions, QoL and sexual function (relevant in intertriginous psoriasis), and convenience of treatment and pleasantness of treatment formulation from the patient’s perspective. These components are generally consistent with the improved outcomes identified in the input submitted by the patient groups for this review.
The treatment period in each study was 8 weeks, which was considered by the clinical expert to be an appropriate amount of time to assess the efficacy and safety of roflumilast. According to the clinical expert, in the context of topical therapy for plaque psoriasis, 4 weeks is often sufficient to determine whether an adequate response will be achieved; although extensive psoriasis may require more time to achieve an adequate response, it should be well on its way after 4 weeks. Moreover, the Canadian Dermatology Association indicated that treatment goals are assessed after 8 weeks of treatment. The clinical expert also suggested that roflumilast may be preferred for long-term use, as it is a nonsteroidal drug; however, the collection of evidence on the durability of effect and long-lasting remission (based on a longer follow-up period) was limited to the DERMIS-OLE study.
Long-Term Extension Study
The contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the CADTH review team.
Description of Study
This section includes a summary of 1 OLE study, the DERMIS-OLE (ARQ-151-306) study,17 which was included in the sponsor’s submission to CADTH and was considered to provide further information on the long-term safety and efficacy of roflumilast cream 0.3% in patients with chronic plaque psoriasis.
This is a multicentre, open-label, phase III, long-term safety and efficacy study in which patients with chronic plaque psoriasis involving up to 25% of BSA received roflumilast cream 0.3% applied once a day for up to 24 weeks. Patients included those who successfully completed a prior roflumilast cream study in psoriasis (cohort 1) or were treatment-naive with roflumilast cream (cohort 2). A total of 267 patients were enrolled (n = 264 in cohort 1 and n = 3 in cohort 2), of which 266 patients received treatment. The study period was between February 12, 2020, and December 14, 2020.
Of note, 2 out of 3 patients in cohort 2 received treatment but they had not reached study week 4 at the time of the data cut-off. Therefore, efficacy data are not available for those patients. No TEAEs occurred in the 2 treatment-naive patients in cohort 2 as of the data cut-off. As a result, the presentation of efficacy and harms results in this interim report focused on the 264 patients in cohort 1.17
Populations
The eligibility criteria at screening are briefly summarized subsequently.
The DERMIS-OLE study included patients aged 2 years and older with a diagnosis of chronic plaque psoriasis. Patients were enrolled into 1 of 2 cohorts. To be eligible for cohort 1, patients were required to have successfully completed 1 of the following roflumilast cream clinical trials: the phase I trial (ARQ-151-107), a phase II trial (ARQ-151-215 or ARQ-151-216), the DERMIS-1 (ARQ-151-301) trial, or the DERMIS-2 (ARQ-151-302) trial. Patients eligible for cohort 2 were naive to treatment with roflumilast, with a clinical diagnosis of psoriasis vulgaris of at least 3 months duration on the face, extremities, trunk, and/or intertriginous areas involving 2% to 25% of BSA (excluding the scalp, palms, and soles), and with an IGA score of at least mild (score of 2) at day 1. Of note, at the time of the data cut-off, no patient had rolled over from studies ARQ-151-107 (which is an open-label, phase I study in adolescents and adults with plaque psoriasis) or ARQ-151-215 or ARQ-151-216, which are open-label, phase II, 4-week trials in children with plaque psoriasis aged between 2 and 5 years; thus, the cohort 1 data reflect only patients enrolled from the ARQ-151-301 and ARQ-151-302 studies.
Patients were excluded from the study if they experienced a roflumilast treatment–related AE or SAE or were currently taking lithium or an antimalaria drug or medication that can affect psoriasis vulgaris (e.g., beta-blockers, ACE inhibitors). Patients suspected of having severe renal insufficiency or hepatic disorders, HIV infection, or hypersensitivity to the investigational product, were not included in the study. Additionally, patients with a history of severe depression and suicidal ideation or a history of chronic alcohol or drug abuse within 6 months of the initiation of the study medication were excluded.
In cohort 1 (N = 264), the mean age of patients was 47.9 years (SD = 15.79), with ||||| adult, |||| adolescent, and |||| pediatric patients. The proportion of patients who were male was larger than the proportion who were female (||||| versus |||||, respectively). Most patients were white (84.1%), and the mean weight was 92.88 kg (SD = 24.720). In cohort 1, most patients (66.3%) had a moderate IGA score at baseline, the mean baseline BSA affected by psoriasis was 6.49% (SD = 4.48), and the mean baseline PASI score was 6.1 (SD = 3.19).
Interventions
Roflumilast cream 0.3% was applied topically by patients or caregivers once daily to active psoriatic plaques (up to a maximum application area of 25% of BSA) for up to 24 weeks at home.17
Treatment application with roflumilast was to occur in the evening, at least 15 minutes after the patient had bathed and at least 20 minutes before the patient went to bed, and the area was not to be washed until the following morning. Patients applied the roflumilast to all active psoriasis lesions, including any new plaques that developed during the study unless otherwise instructed by the investigator. Application was to all areas affected, including intertriginous and/or genital regions and the face, but excluding the scalp. Patients were not required to continue treatment if the psoriasis plaques completely resolved.
Concomitant Medications
The use of concomitant medication was prohibited for biological therapies, oral corticosteroids, retinoids, apremilast, methotrexate, cyclosporine, and other systemic immunosuppressants. Topical antipsoriasis medication (e.g., corticosteroids), vitamin D analogues, prescription shampoos, psoralen and UV A phototherapy, UV B, and antihistamines were also not allowed during treatment with roflumilast 0.3%.17
Outcomes
The primary outcomes of the DERMIS-OLE study were safety, including TEAEs and SAEs. Secondary efficacy outcomes included the following:
an IGA score of clear or almost clear
IGA success (defined as an IGA score of clear or almost clear plus a 2-grade improvement from baseline)
duration of response, including duration of clear or almost clear status and duration of IGA success (defined as the period from the date of the first observation of an IGA score of clear or almost clear or IGA success until the date of restarting roflumilast)
a 50%, 75%, 90%, or 100% reduction in PASI over time
an I-IGA score of clear or almost clear over time
I-IGA success over time (defined as an I-IGA score of clear or almost clear plus a 2-grade improvement from baseline)
change in WI-NRS score over time.17
Other relevant outcomes assessed included change in BSA score over time, the proportion of patients who met the criteria for disease clearance (i.e., having clear scores on the IGA, I-IGA, PASI, and mPASI and stopping treatment at some time during the study after primary baseline), and the time to restarting study drug (i.e., treatment-free interval), defined as the time when patients experienced disease clearance and stopped treatment to all lesions.
Statistical Analysis
No formal statistical test was performed. Descriptive statistics were used to give a summary of the efficacy and safety results. The analysis was based on the number of patients with measurements available and no imputation was performed. A December 14, 2020, data cut-off date was set for interim reporting because this investigation was ongoing. Of note, the efficacy data for 2 out of the 3 treatment-naive patients (cohort 2) were not available because they had not yet reached trial week 4 at the time of the data cut-off.17
Results
Patient Disposition
At the time of the data cut-off, 264 patients (98.9%) who had participated in parent studies and 3 treatment-naive patients (1.1%), 2 of whom had not reached the week 4 visit, were enrolled into this OLE study. Therefore, 267 patients started the OLE study and 266 (99.5%) of them had received roflumilast. Of note, the patients in the OLE study who had received roflumilast 0.3% in the parent studies received treatment for a total of 32 weeks (8 weeks of treatment in the parent studies plus an additional 24 weeks), whereas patients who received vehicle cream received up to 24 weeks of treatment with roflumilast in the OLE study. At the time of the data cut-off, 222 patients (83.1%) had completed the study, 32 (12%) had discontinued the study, and 12 (4.5%) were ongoing in the study. The most common reasons for discontinuation were lost to follow-up (5.7%) and withdrawal (4.5%). Two patients discontinued due to noncompliance and 1 patient discontinued because of an AE.17 A detailed summary of the patient disposition in the DERMIS-OLE study is available in Table 21.
Table 21: Summary of Patient Disposition From the DERMIS-OLE Study (All Enrolled Patients)
Characteristic | Cohort 1 | Cohort 2 | Total |
|---|---|---|---|
Enrolled, N | 264 | 3 | 267 |
Received treatment, N (%) | 264 (100) | 2 (66.7) | 266 (99.6) |
Completed study, N (%) | 222 (84.1) | 0 | 222 (83.1) |
Ongoing in study, N (%) | 10 (3.8) | 2 (66.7) | 12 (4.5) |
Discontinued study, N (%) | 32 (12.1) | 0 | 32 (12) |
Reasons for discontinuation, n (%) | |||
Withdrawal | 12 (4.5) | 0 | 12(4.5) |
Noncompliance | 2 (0.8) | 0 | 2 (0.8) |
Lost to follow-up | 15 (5.7) | 0 | 15 (5.7) |
Adverse event | 1 (0.4) | 0 | 1 (0.4) |
Other | 2 (0.8) | 0 | 2 (0.8) |
Discontinuation due to COVID-19, N (%) | 2 (0.8) | 0 | 2 (0.7) |
Source: Clinical Study Report for the DERMIS-OLE study.17
Exposure to Study Treatments
In cohort 1, a total of 264 patients received roflumilast cream 0.3% once a day over the course of 24 weeks, of which 171 patients had received roflumilast and 93 patients had received the vehicle in the parent studies. The mean number of product applications for cohort 1 was 182.5 (SD = 54.05), with a mean of 203.5 (SD = 46.36) for patients who received roflumilast in the parent study and 143.8 (SD = 45.31) for patients who received vehicle cream in the parent study. A summary of exposure to study treatment is presented in Table 22.
About 75% of patients in cohort 1 used concomitant medications. The most commonly used types of medications during the study were 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors (about 15% in each group), ACE inhibitors (||| among patients who had previously received roflumilast compared with |||| among those who had received vehicle cream), anilides (|||| among patients who had previously received roflumilast compared with 17.2% among those who had received vehicle cream), and platelet aggregation inhibitors, excluding heparin (||||| among patients who had previously received roflumilast compared with |||| among those who had received vehicle cream).17
Table 22: Summary of Patient Exposure From the DERMIS-OLE Study (Safety Population: Cohort 1)
Category | Cohort 1 | ||
|---|---|---|---|
Roflumilast in prior study N = 171 | Vehicle in prior study N = 93 | Total N = 264 | |
Number of applications with drug | |||
Mean (SD) | 203.5 (46.36) | 143.8 (45.31) | 182.5 (54.05) |
Median (range) | 224 (57 to 273) | 167 (1 to 197) | 197 (1 to 273) |
Compliance rate, N (%) | |||
> 80% to < 100% | 171 (100) | 92 (89.9) | 263 (99.6) |
< 80% | 0 | 1 (1.1) | 1 (0.4) |
> 3 missed consecutive doses N (%) | | ||||| | | ||||| | | ||||| |
OLE = open-label extension; SD = standard deviation.
Source: Clinical Study Report for the DERMIS-OLE study.17
Efficacy
All efficacy analyses were performed on the safety population or a subset of the safety population. It should be noted that the 2 patients in cohort 2 (treatment-naive patients) had not yet reached study week 4 at the time of the data cut-off date. Thus, efficacy data were not available for those patients and results are reported only for cohort 1 patients. Efficacy data were presented using the primary baseline, defined as the last observation recorded before the first dose of roflumilast cream either in the parent study (for patients who received roflumilast in the parent study) or in the OLE study (for patients who received the vehicle cream in the parent study or who were naive to the drug). Refer to Table 23 for efficacy outcomes in the OLE study.17
Investigator’s Global Assessment
In the OLE, among the 171 patients who received roflumilast in a prior study, 39.9% experienced treatment success based on the IGA at week 4, 46.2% at week 12, and 37.2% at week 24. For patients who received vehicle cream in a prior study, the proportions of patients who experienced treatment success based on the IGA in the OLE study were 9.3% at week 4, 38.4% at week 12, and 36.7% at week 24.
At week 12 of the OLE study, the proportions of patients with an IGA of clear or almost clear were 57.1% for patients who received roflumilast cream in the parent study and 60.5% for patients who received vehicle cream. At week 24 of the OLE study, 47.6% who received roflumilast in the parent study and 54.4% who received vehicle cream in the prior study had an IGA score of clear or almost clear.
Intertriginous IGA
At the primary baseline, 59 patients had intertriginous involvement: 33 had received roflumilast cream and 26 had received vehicle cream in a prior study. Among the patients who received roflumilast in a prior study, 79.2% experienced treatment success based on the I-IGA at week 24 of the OLE study. Among the patients who received vehicle cream in a prior study, 71.4% had experienced treatment success based on the I-IGA at week 24 of the OLE study. At week 24 of the OLE study, response rates for I-IGA clear or almost clear ranged from 76.2% to 79.2% of patients in both groups of rollovers.
Duration of Response
In total, 179 out of 264 patients (67.8%) had clear or almost clear IGA status at any time during the study and maintained that status for a median of 93 days; the KM median was 188.0 days. In total, ||| patients (|||||) experienced treatment success based on the IGA from the primary baseline. The overall median duration of success was || days. The KM median was 157 days overall.
Psoriasis Area and Severity Index
The proportion of patients who received roflumilast in the prior study and had a PASI 50 during the OLE study was ||||| at week 24. The proportion of patients who received vehicle cream in the parent study and had a PASI 50 during the OLE study was ||||| at week 24. The proportions of patients who had a PASI 75 at week 24 was 44.1% and 43% for the roflumilast group and the vehicle group in the parent study, respectively. Regarding PASI 90, the proportions of patients were ||||| and ||||| at week 24 for the roflumilast and vehicle groups in the parent study, respectively. The proportions of patients who had a PASI 100 at week 24 was ||||| for the roflumilast group and ||||| for the vehicle group in the parent study.
Worst Itch Numeric Rating Scale
This outcome was defined as experiencing an improvement of 4 or more points from a primary baseline score of 4 or higher on the WI-NRS. Patients experienced treatment success based on the WI-NRS in both rollover groups (i.e., those who received roflumilast cream in the prior study and those who received vehicle cream in the prior study) at week 24 of the OLE study (66.4% and 53.1%, respectively).
Body Surface Area
In the OLE study, the mean percent changes in BSA were comparable in both groups of rollover patients, with mean improvements ranging from |||||| |||||||||||| and |||||| |||||||||||| at week 24 for patients in the roflumilast and vehicle groups, respectively.
Disease Clearance and Treatment-Free Interval
Among all patients in cohort 1, 36 (13.6%) met the criteria for disease clearance (i.e., clear scores for IGA, I-IGA, PASI, and mPASI) and stopped treatment at some time during the study after primary baseline. The mean treatment-free interval among patients who met the criteria for disease clearance was 54.0 days (SD = 47.67).
Table 23: Summary of Efficacy Results From the DERMIS-OLE Study (Safety Population: Cohort 1)
Efficacy outcomes | Roflumilast in prior study (N = 171) | Vehicle in prior study (N = 93) | Total (N = 264) |
|---|---|---|---|
IGA success | |||
Week 12 IGA success, n (%) | 72 (46.2) | 33 (38.4) | 105 (43.4) |
Week 24 OLE, N | 145 | 79 | 224 |
Week 24 IGA success, n (%) | 54 (37.2) | 29 (36.7) | 83 (37.1) |
IGA of clear or almost clear | |||
Week 12 OLE, N | 156 | 86 | 242 |
IGA of clear or almost clear, n (%) | 89 (57.1) | 52 (60.5) | 141 (58.3) |
Week 24 OLE, N | 145 | 79 | 224 |
IGA of clear or almost clear, n (%) | 69 (47.6) | 43 (54.4) | 112 (50) |
I-IGA success | |||
Week 12 OLE, N | 28 | 24 | 52 |
I-IGA of clear or almost clear, n (%) | 23 (82.1) | 20 (83.3) | 43 (82.7) |
I-IGA success, n (%) | 21 (75) | 18 (75) | 39 (75) |
Week 24 OLE, N | 24 | 21 | 45 |
I-IGA of clear or almost clear, n (%) | 19 (79.2) | 16 (76.2) | 35 (77.8) |
I-IGA success, n (%) | 19 (79.2) | 15 (71.4) | 34 (75.6) |
Duration of response | |||
Duration of IGA successa (in days), N | ||| | || | ||| |
Mean (SD) | ||| ||||||| | |||| ||||||| | | ||||||| |
Median | |||| | || | || |
PASI 50 | |||
Week 12 OLE, N | ||| | || | ||| |
PASI 50, n (%) | ||| |||||| | | |||||| | ||| |||| |
Week 24 OLE, N | ||| | || | ||| |
PASI 50, n (%) | ||| |||| | | |||||| | ||| |||||| |
PASI 75 | |||
Week 12 OLE, N | 156 | 86 | 242 |
PASI 75, n (%) | 80 (51.3) | 40 (46.5) | 120 (49.6) |
Week 24 OLE, N | 145 | 79 | 224 |
PASI 75, n (%) | 64 (44.1) | 34 (43) | 98 (43.8) |
PASI 90 | |||
Week 12 OLE, N | ||| | || | ||| |
PASI 90, n (%) | | |||||| | | |||||| | | |||||| |
Week 24 OLE, N | ||| | || | ||| |
PASI 90, n (%) | | |||||| | | |||||| | | |||||| |
PASI 100 | |||
Week 12 OLE, N | ||| | || | ||| |
PASI 100, n (%) | | |||| | | |||||| | | |||||| |
Week 24 OLE, N | ||| | || | ||| |
PASI 100, n (%) | | |||||| | | |||||| | | |||||| |
WI-NRS success | |||
Week 12 OLE, N | 119 | 52 | 171 |
WI-NRS success, n (%) | 86 (72.3) | 29 (55.8) | 115 (67.3) |
Week 24 OLE, N | 116 | 49 | 165 |
WI-NRS success n (%) | 77 (66.4) | 26 (53.1) | 103 (62.4) |
Change from baselineb in BSA | |||
Week 12 OLE, N | ||| | || | ||| |
BSA change, mean (SD) | |||||| |||||| | |||||| ||||||| | |||||| ||||||| |
Week 24 OLE, N | ||| | || | ||| |
BSA change, mean (SD) | |||||| ||||||| | |||||| ||||||| | |||||| ||||||| |
Patients with disease clearance,c n (%) | | |||||| | | |||||| | | |||||| |
Treatment-free intervald (days), mean (SD) | |||| |||||| | |||| |||||| | |||| |||||| |
BSA = body surface area; IGA = Investigator’s Global Assessment; I-IGA = intertriginous Investigator’s Global Assessment; OLE = open-label extension; PASI 50 = 50% reduction in the Psoriasis Area and Severity Index score; PASI 75 = 75% reduction in the Psoriasis Area and Severity Index score; PASI 90 = 90% reduction in the Psoriasis Area and Severity Index score; PASI 100 = 100% reduction in the Psoriasis Area and Severity Index score; SD = standard deviation; WI-NRS = Worst Itch Numeric Rating Scale.
aDuration of IGA success was defined as the last date on which the patient experienced treatment success based on the IGA (before restarting treatment) minus the first date on which the patient experienced treatment success based on the IGA.
bBaseline was defined as the last observation recorded before the first dose of roflumilast cream 0.3% in this extension study (ARQ-151-306).
cPatients who had a score of clear for IGA, I-IGA, PASI, and modified PASI and stopped treatment for all lesions.
dTreatment-free interval is defined as the date on which the investigational product was restarted (end of clear-score period) minus the first date on which the patient had a score of clear in the IGA, I-IGA, PASI, and modified PASI plus 1.
Source: Clinical Study Report for the DERMIS-OLE study.17
Harms
Adverse Events
A summary of the safety results is available in Table 24. There were no TEAEs reported in either of the 2 patients in cohort 2 as of the data cut-off date (December 14, 2020). In total, 26.1% of patients in the OLE study experienced at least 1 TEAE (of total of 129 TEAEs). Among the patients who had received roflumilast in a prior study, 22.8% had at least 1 TEAE, compared with 32.3% of patients who received vehicle cream in the parent study.17
The most common TEAEs overall in the OLE study were sinusitis (2.7%), diarrhea (2.3%), COVID-19 (1.9%), and headache (1.9%). The most common TEAEs among patients who received roflumilast cream in the prior study were sinusitis, COVID-19, and hypertension, each reported in 3 patients (1.8%). The most reported TEAEs among patients who received vehicle cream in the prior study were headache (5.4%), sinusitis (4.3%), and diarrhea (4.3%).
Serious Adverse Events
During the OLE study, 3 patients experienced a total of 5 SAEs. One patient who had received roflumilast in the prior study experienced an SAE of polycythemia vera. Two patients who received vehicle cream in the prior study experienced 4 SAEs, including COVID-19 pneumonia, palpitations, dehydration, and syncope. None of those SAEs was considered related to the treatment.
Withdrawals Due to Adverse Events
During the study, 1 patient discontinued treatment due to an AE. One of the patients who had previously received roflumilast cream 0.3% experienced application site irritation, which was considered likely to be related to the investigation product.
Mortality
Based on the information the sponsor provided, no deaths occurred during the DERMIS-OLE study.
Notable Harms
Among patients in the OLE study, application site exacerbation or worsening of psoriasis was reported in 1.2% of patients. Application site pain, keratoacanthoma, and cellulitis were each reported in 0.4% of patients.
Local Tolerability Assessments
The investigator’s evaluation of application site reaction in this study was assessed up to week 24. Erythema was experienced at week 12 by 4 patients (1.8%) and at week 24 by 8 patients (3.7%). A slight glazed appearance was experienced by 3 patients (1.3%) at week 12 and by 6 patients (2.8%) at week 24. Moreover, 1 patient experienced glazing with peeling and cracking at week 24.
Table 24: Summary of Harms From the DERMIS-OLE Study
Adverse events | Roflumilast in prior study (N = 171) | Vehicle in prior study (N = 93) | Total (N = 264) |
|---|---|---|---|
Most common adverse events, n (%) | |||
Patients with ≥ 1 adverse event | 39 (22.8) | 30 (32.3) | 69 (26.1) |
Sinusitis | 3 (1.8) | 4 (4.3) | 7 (2.7) |
Diarrhea | 2 (1.2) | 4 (4.3) | 6 (2.3) |
COVID-19 | 3 (1.8) | 2 (2.2) | 5 (1.9) |
Headache | 0 | 5 (5.4) | 5 (1.9) |
Anemia | 2 (1.2) | 1 (1.1) | 3 (1.1) |
Nausea | 0 | 3 (3.2) | 3 (1.1) |
Decreased appetite | 1 (0.6) | 2 (2.2) | 3 (1.1) |
Back pain | 2 (1.2) | 1 (1.1) | 3 (1.1) |
Hypertension | 3 (1.8) | 0 | 3 (1.8) |
Serious adverse events, n (%) | |||
Patients with ≥ 1 serious adverse event | 1 (0.6) | 2 (2.2) | 3 (1.1) |
Polycythemia vera | 1 (0.6) | 0 | 1 (0.4) |
COVID-19 pneumonia | 0 | 1 (1.1) | 1 (0.4) |
Palpitations | 0 | 1 (1.1) | 1 (0.4) |
Dehydration | 0 | 1 (1.1) | 1 (0.4) |
Syncope | 0 | 1 (1.1) | 1 (0.4) |
Patients who stopped treatment due to adverse events, n (%) | |||
Patients who stopped treatment due to adverse events | 1 (0.6) | 0 | 1 (0.4) |
Application site irritation | 1 (0.6) | 0 | 1 (0.4) |
Deaths, n (%) | |||
Patients who died | 0 | 0 | 0 |
Notable harms, n (%) | |||
Application site pain | 1 (0.6) | 0 | 1 (0.4) |
Application site exacerbation or worsening of psoriasis | 2 (1.2) | 0 | 2 (1.2) |
Application site keratoacanthoma | 0 | 1 (1.1) | 1 (0.4) |
Application site cellulitis | 0 | 1 (1.1) | 1 (0.4) |
OLE = open-label extension.
Note: Percentages are n divided by the number of patients in the safety population within the actual treatment group at each visit multiplied by 100.
Source: Clinical Study Report for the DERMIS-OLE study.17
Critical Appraisal
Internal Validity
The open-label, single-arm design of the study does not allow conclusions to be drawn about the comparative efficacy or safety of roflumilast. Given that this was an ongoing study, the results were limited to the interim analysis as of December 14, 2020. The treatment-naive cohort 2 (patients without treatment) could not reach week 4; therefore, only patients from the DERMIS-1 and DERMIS-2 studies were enrolled. To meet the eligibility requirements for the OLE study, patients were required to complete the parent studies, which might have introduced some selection bias. Out of a total population of 439 in the parent studies, 264 patients (60.13%) participated in the OLE study. The sponsor did not provide any explanation for why the remaining patients did not participate, which could have further contributed to selection bias in favour of roflumilast.
Additionally, 69 patients (26.1%) had at least 1 major protocol deviation during the study. However, the magnitude and direction of bias are uncertain. Patients who experienced a roflumilast treatment–related AE or SAE during the parent trials were excluded from the DERMIS-OLE study. Therefore, only patients who had high tolerability of the drug may have participated in the OLE study, which may result in an underestimation of safety in the population and overestimation of the efficacy, since the patients who had a benefit from the treatment were more likely to continue in the study. Of note, some of the subjective measurements, such as IGA, might result in performance bias, especially because this is an open-label study and investigators were not blinded.
External Validity
Since the patients who took part in the OLE study were originally from the parent DERMIS-1 and DERMIS-2 trials and the eligibility criteria remained the same, it is reasonable to expect that the same limitations to generalizability present in the DERMIS trials are relevant to this long-term extension study. For example, several exclusion criteria limit the generalizability of the results to a wider population. Finally, despite the longer follow-up in DERMIS-OLE (24 weeks versus 8 weeks for the DERMIS-1 and DERMIS-2 trials), this OLE study might not provide a sufficient period to observe a long-term safety profile for roflumilast.
Indirect Evidence
The contents within this section have been informed by materials submitted by the sponsor. The following have been summarized and validated by the CADTH review team.
Objectives for the Summary of Indirect Evidence
Roflumilast was compared with a matching vehicle in both the DERMIS-1 and DERMIS-2 pivotal trials. In the absence of direct comparative evidence of roflumilast versus other relevant comparators, evidence from the sponsor-submitted NMA has been summarized and critically appraised to fill gaps in the evidence from the pivotal trials.
Description of Sponsor-Submitted ITC
The sponsor-submitted ITC included a review of the literature and an NMA performed using a Bayesian framework that compared roflumilast with other topical therapies for patients with plaque psoriasis.
Methods of Sponsor-Submitted ITC
Objectives
The objective of the sponsor-submitted ITC was to determine the comparative efficacy of roflumilast compared with other topical treatments in patients with plaque psoriasis. A systematic literature search and feasibility assessment were conducted to determine studies for inclusion.
Study Selection Methods
A systematic literature review was conducted by the sponsor, the details of which are presented in Table 25. The databases searched included MEDLINE, Embase, and the Cochrane Central Register of Controlled Trials (CENTRAL) to identify studies of interest on December 7, 2021. Proceedings from the past 2 years from the American Academy of Dermatology and European Academy of Dermatology and Venereology were also screened. Study selection and data extraction were performed by 2 independent reviewers. Following reconciliation, a third reviewer was involved to reach consensus on any remaining discrepancies. The quality assessment of all randomized controlled trials was conducted using the Cochrane risk-of-bias instrument version 2.
Characteristics | Indirect comparison |
|---|---|
|||||||||| |
|
|||||||||||| |
|
|||||||||| |
|
||||||| |
|
||||| ||||||| | |||| |
||||||||||| ||||||||||||||| | ||||||||| | ||||||||||| ||||||| |
||||||||| |||||||| |
|
||||||||| |||||||| |
|
||||||||| ||||||| | |||||||| |||| |||||||| ||||||||||||| || |||||||||||| || |||||| |||||||||| || |||||||| || |||| ||||||| || ||||||||||||| |
|||| |||||||||| ||||||| | ||| ||||||||| ||||||||||||| ||||||||| || |||| || |||||| |||||||||| || |||||||| || |||| ||||||| || ||||||||||||| |
||||||| |||||||||| | ||| ||||||||||| ||||||||| |||| || |||||||| ||||||||||||||| |||| || |||| |||| |||||||| | |
||| ||||| ||||||| ||||| |||| |||||||||||| |||| ||||||| |||||| || ||||||||||||| |||||| ||||||||||| |||| ||||||||| |||| ||||||||| |||||| || |||||||||||| |||||| ||||||||||| || ||||||||| ||||||| |||||||||| ||||| |||||| |||| |||| |||||||||||| || |||||||| ||||||||||||| |||| |||||||| |||||||| |||| |||| || ||||| |||||||||| |||||||| || |||||||| |||||| |||| |||||||||||||||||| ||||||||||||||||| || ||||||||| ||||||| ||||| ||||||| |||| || ||||| |||| |||| ||||| |||| || ||||||||| ||||||| || |||||||| |||||||||
To inform the NMA, the evidence base from the systematic literature review was restricted according to the criteria described in Table 26. ||||| |||||||| |||||||| |||| ||||| |||||||| |||| |||||| ||||||||| | |||||| |||| |||||||||| || || |||||||| || |||| ||||||||||| ||||||||| |||||||| |||| ||||| ||||| || ||||||||| |||||| ||||| ||||||||||||| || ||||| ||||||||||| || ||||||||| |||||||||| |||| || |||||| ||||||| |||||| |||| |||| |||||||||| || |||||||| || |||| ||||||||| ||||||| ||||||||| || |||| ||||||| |||||| |||||| |||||||| |||||||| |||||||||| || || |||| |||||||| |||| |||||||||| | || |||||||||| ||||||| |||| |||||||| || |||| |||||||| |||||||| ||||||| ||||||||||||||| |||||| ||||||| ||||||||| || |||| |||||||||| || || |||||||||||| || ||||||||| |||||| ||||||||| ||||| || ||||||||| || ||||||| |||||| || |||||||| ||||||| |||||||| || |||||||| |||||||| || ||||||||| |||||||| || |||| |||||| |||||||| |||| |||||| ||||||||| || ||||| ||||||||| |||||||| || |||| |||||| |||||||| |||| |||||||||||||| |||||||||| ||||| |||| |||| ||||||||| || || |||||| ||||||| |||||||
Characteristics | Indirect comparison |
|---|---|
|||||||||| |
|
|||||||||||| |
|
|||||||||| |
|
||||||| |
|
||||| ||||||| | 1. |||||||| || |||| || ||||||||| || || |||||||||||||| | |||| || |||||||| |||||||| |||||||||||||||||| || |||| || |||||||| |||||||| ||||||||||||| |||||| || |||| || |||||||||||| |||||||| ||||||||||||||||||| || |||| || |||||||||||| |||||||| ||||||||||||| |
||||||||||| ||||||||||||||| | ||||||||| || ||||||||||| ||||||| |
||||||||| |||||||| |
|
| |||||||||||||| | |||||||||||| |||||| ||||||||||| ||||| |||||||||||||| |||| || |||||||||| |||||||||| |||||| || |||||||| |||||||||||||||||| ||||||||||||||||| || ||||||||| |||||||| ||||| |||||||| |||| || ||||| |||| |||| ||||| |||| || ||||||||| ||||||| || |||||||| |||||||||
||| |||||||| |||||||
||| || |||| || ||||||||| |||||||| || |||| |||| |||||| || || |||| ||||||| || ||||||||| || ||||||||| ||||||||| ||||| ||||||||| |||||||| |||| || |||| |||| |||||||| ||||||| ||||||||||| || ||||||||||| ||||||||||||| || |||||||| |||| |||||||||||||| |||||||||| ||||||||| |||||||| ||||||| || || ||||||||||| || ||||||| |||||| || || ||||||| |||||| |||||| || ||||| | || ||||| ||||||| ||||||||||| || || | ||||| || ||||| |||||| ||||||||||||| |||||||| ||||||||| || ||||||| || |||||||||| | || ||||||||| ||||||||||| || |||||||||| || |||||||||| || ||||||||| | |||||| || |||||||||| | |||||||||| || ||||||||||| || |||||| || |||||||| | ||||||||| ||||||||| || |||| || | |||||||||| || |||||||||| |||||||| || || |||||| ||||||||| |||||||| ||||||||| ||||||| || || ||||||||||| |||||||| |||| ||||||||| || |||| ||||| || || |||||
||||| |||||| || ||||||| |||| ||||||| || |||||||| || ||||||||||||| ||||||||| || ||||||||| |||||| |||||| ||||||||| ||||||||||||| |||||| || |||| ||||||||| ||||| |||| || || ||||||||| |||||||||| || |||||||| ||||||||||| |||| ||||||||| ||||||| || ||||||||| ||||||||| ||||||| || ||||||||
||||||||||||||| |||||||| ||||||||| ||||||||||| |||||||||||||||| |||| ||||||| ||||||||||||||||||||||||| |||| ||||||||||
||| |||||||| |||| ||||||||| ||||| |||||||| |||||||| |||||||||| |||||||||||| | |||||||||||||| |||||| |||| |||||||||| | |||| |||||||| || |||||||| ||||| || || |||||||| ||||||||||| ||||||||| ||||| ||||||||| || ||||||||| || |||||| ||||||| ||||| || ||||| || ||||| || |||| |||| ||||||| || |||||||||| ||||||| ||||||| ||||||| || |||||||||||| ||||| || |||||||| || || || | |||| ||||||| ||||| |||||| |||||| || |||||| || || |||||||||| || ||||||||||| || || |||||||||||| |||||| |||| |||| |||||||||| |||| |||||| ||||||| || |||||||| || |||||||| ||||||||||| || |||| |||||||| |||| |||||||| || |||| | ||||||| |||||||| || ||||||| || |||||| || ||||||||| ||||||| || |||||||||||||| || |||| |||| ||||||||| ||||| || || |||||||||| || |||||||| |||||||||||| || ||||| || |||||||| ||||| ||||||||| |||||||||| ||||| |||| ||||||||| |||||||||| || ||||| ||||| |||||| ||||||||||||||| ||||| ||||||||||||| || ||||| |||||||||| |||| ||||| ||||||| ||||| ||||| ||||| |||||| |||||| || |||| || |||||||| || |||||||||| || || ||||||||| |||||||
| |||||||| |||||||| |||| ||||||||| |||||||||||| |||||||| || ||||||||| || ||||| |||||||| || |||| || ||||| |||||||||||| ||||| ||||||||| |||||||||| || ||||||||| |||||||| ||||||| || ||||||||||| || ||||||||||| || | | |||||| || ||||| || |||| || || ||||||||||| |||||||
||||||||||| || || |||||||||| || ||||||||||| || |||||||||||||| || |||| || ||||||| ||||||| || || ||||||| |||| || |||||| || ||||| |||
Methods | Description |
|---|---|
|||||||| ||||||| | ||| | | ||||||||| ||||| ||||||||| ||||||||| || |||| |||||||||||| | |||||||||||||| |||||| |||| ||||||||||| |
|||||| | |||||| ||||||||||||||| ||||| ||||||||||||| |||| ||||| |
|||||||||| || ||||| ||| | ||||||||||||||| || |||||||| ||||| || |||| |||| ||||| |||||| |||||||||| |||||| ||||| |||| |
|||||||||| || ||||||||||| | ||||||||||| || |||||||| ||||| || || |||||||| ||||||| ||||||||| ||||||||| || ||||||||| |
|||||||||| || ||||||||||| | ||||||||||| || |||||||| ||||| ||||||| ||||| || ||||||||||||||||||| |||||| |
|||||||| | ||| ||||||| || |||| || ||||| ||||||| || |||| ||||||||||||| || ||||||| || ||||||| |||||| |||||| ||||| || |||||||| ||||||||||||| |
||||||||| |||||||||| | ||||| |
|||||||||||| || ||||| | ||||| |||| ||||||| || |||||||| || ||||||||||||| ||||||||| || ||||||||| |||||| |||| || |||||||||| || ||||||| ||||||||| ||||||| |||||| |||| ||||| |
||||||||||| |||||||| | | ||||||||||| |||||||| || ||||| |||||||| || ||||||||| ||||| || |||||||| |||||||||| |||| | |||||||||| |||||| |||||| ||||||||||| || ||||||| || ||||||| |||||| |||||| ||||| ||||||| || |||| || | | ||||||| ||||||||||| || || ||||| ||||||| |
|||||||| |||||||| | ||||| |
||||||| | |||||||| ||||||||||||| | ||||||||| |||||| ||||||||| |||| |||||||| ||||| || || |||||| |||||| || || |||||||| ||||||||| |||||| || |||||| ||||||||||||| ||||||| || |||||||| || |||||||| || ||||||| ||||||| |||| ||||||||| || || |||| ||||| || | |||||||| ||||||||| |
||| |||||||| ||||||||||| |||||||||| | ||||||||||||| |||||| ||||||||||| ||||| |||||||||||||| |||||||||||| |||||| ||||||||||| || |||||||| ||||||||||||||||||||| ||||||||||||||||| |||||||||| |||||||| ||||| ||||||| |||| | ||||| |||| |||| ||||| |||| | ||||||||| ||||||| || |||||||| |||||||||
Results of the Sponsor-Submitted NMA
||||||| || |||||||| |||||||
| ||||| || || ||||||| |||| |||||||||| |||| || |||||||||| |||||||||| |||||| || ||||||||| || |||||||||||| ||||||||||| || ||||| || || | ||||||||| |||||||| || || |||| || |||| ||||||| | ||||| ||||||||| |||||||| |||||| |||||||||||| ||||||||| ||||||||||| || ||||||||| | ||||||||| || || ||||||| || |||||||| || ||||||||| |||| || |||||||| |||| || |||||||| ||||||||||||||| || ||||||||| || |||||||||| |||||||||| |||| |||| || || |||||||| | ||||||||| |||||| || |||||||| ||||||||| || ||||||||| || ||||||||| |||||| ||||||||||| |||||||| || || ||||||||| |||||||| || |||| |||||||| || |||||| || |||||||| |||||||||| || |||||| ||||||| ||||||||||| |||||||| || ||||| ||||||||| |||||||| || |||| |||||| |||||||| |||| |||||||||||||| ||||||||| ||||||| || |||||||| |||||||||| || || |||||||| ||||||| || |||||||||| |||||||| |||| || |||| || | |||||||| |||| ||||||||| || |||||| | ||||||| || || |||||||| |||||||||| |||| |||||||| || |||||||||| || ||||||||||| || || || ||||||| |||||||| || || || || ||||||||| || ||||| | |||||| |||| |||| |||||||| || ||||| | | ||||| | || |||||||| || ||||||| || |||| || | ||||| ||| ||||| | |||||||| || | |||||||||| |||| ||||| || ||||||||| |||||| |||| | |||| |||||||| ||||||| ||||||||||| ||||||||| | ||||||||| ||||||||||||||
| ||||| || || ||||||| |||| |||||||||| |||| || |||||||||| |||||||||| |||||| || ||||||||| || |||||||||||| ||||||||||| || ||||| || || | ||||||||| |||||||| || || |||| || |||| ||||||| | ||||| ||||||||| |||||||| |||||| |||||||||||| ||||||||| ||||||||||| || ||||||||| | ||||||||| || || ||||||| || |||||||| || ||||||||| |||| || |||||||| |||| || |||||||| ||||||||||||||| || ||||||||| || |||||||||| |||||||||| |||| |||| || || |||||||| | ||||||||| |||||| || |||||||| ||||||||| || ||||||||| || ||||||||| |||||| ||||||||||| |||||||| || || ||||||||| |||||||| || |||| |||||||| || |||||| || |||||||| |||||||||| || |||||| ||||||| ||||||||||| |||||||| || ||||| ||||||||| |||||||| || |||| |||||| |||||||| |||| |||||||||||||| ||||||||| ||||||| || |||||||| |||||||||| || || |||||||| ||||||| || |||||||||| |||||||| |||| || |||| || | |||||||| |||| ||||||||| || |||||| | ||||||| || || |||||||| |||||||||| |||| |||||||| || |||||||||| || ||||||||||| || || || ||||||| |||||||| || || || || ||||||||| || ||||| | |||||| |||| |||| ||
| ||||| || || ||||||| |||| |||||||||| |||| || |||||||||| |||||||||| |||||| || ||||||||| || |||||||||||| ||||||||||| || ||||| || || | ||||||||| |||||||| || || |||| || |||| ||||||| | ||||| ||||||||| |||||||| |||||| |||||||||||| ||||||||| ||||||||||| || ||||||||| | ||||||||| || || ||||||| || |||||||| || ||||||||| |||| || |||||||| |||| || |||||||| ||||||||||||||| || ||||||||| || |||||||||| |||||||||| |||| |||| || || |||||||| | ||||||||| |||||| || |||||||| ||||||||| || ||||||||| || ||||||||| |||||| ||||||||||| |||||||| || || ||||||||| |||||||| || |||| |||||||| || |||||| || |||||||| |||||||||| || |||||| ||||||| ||||||||||| |||||||| || ||||| ||||||||| |||||||| || |||| |||||| |||||||| |||| |||||||||||||| ||||||||| ||||||| || |||||||| |||||||||| || || |||||||| ||||||| || |||||||||| |||||||| |||| || |||| || | |||||||| |||| ||||||||| || |||||| | ||||||| || || |||||||| |||||||||| |||| |||||||| || |||||||||| || ||||||||||| || || || ||||||| |||||||| || || || || ||||||||| || ||||| | |||||| |||| |||| |||||||| || ||||| | | ||||| | || |||||||||| |||| || | ||||| ||| ||||| | |||||||| || | |||||||||| |||| ||||| || ||||||||| |||||| |||| | |||| |||||||| ||||||| ||||||||||| ||||||||| | ||||||||| |

||| ||||| ||||||| ||||||| || ||||||||| ||||||| |||| |||||| ||||||||||||| |||||||| ||||||| ||||||||||| |||||||||||||||| ||||||||||||||| |||| ||||||| ||||||||||| ||||||||||| || ||||||||||||||| |||| |||||||||||| || ||||||||||||||| |||||||||||||||| || ||||||||||||| |||||| ||||||||||| || |||||||| |||||||||||||||||| ||||||||||||||||| || ||||||||| ||||||
This figure has been redacted.

||| ||||| ||||||| ||||||| || ||||||||| ||||||| |||||||||| ||||||||||| || ||||||||||| |||||||||| || |||||||| || ||||||| ||||||||| ||||||| || ||||||||||| || ||||||| ||||||||| ||||||| || ||||||||||| ||||||||||| ||||||||||| ||||||||||||||| |||||||||||| |||||| ||||||||||||||||||| ||||||||||||||||| || ||||||||| |||||||
This figure has been redacted.
Table 28: NMA Study Characteristics
Trial | N | Phase | Active interventions | Node label |
|---|---|---|---|---|
DERMIS-1 | 439 | 3 | Roflumilast 0.3% cream | Roflumilast |
Vehicle | Vehicle | |||
DERMIS-2 | 442 | 3 | Roflumilast 0.3% cream | Roflumilast |
Vehicle | Vehicle | |||
Trials evaluating IGA treatment response at week 8 among patients with plaque psoriasis | ||||
|||||| |||| | ||||| | ||||| | Calcipotriol 50 mcg/g + betamethasone 0.5 mg/g gel (Dovobet) | Corticosteroid + vitamin D analogue |
Betamethasone dipropionate 0.5 mg/g gela | Corticosteroid | |||
Calcipotriol 50 mcg/g gela | Vitamin D analogue | |||
||||| ||| | ||||| | ||||| | Halobetasol 0.01% + tazarotene 0.045% lotion (Duobrii) | Corticosteroid + tazarotene |
||||| ||| | ||||| | ||||| | Halobetasol 0.01% + tazarotene 0.045% lotion (Duobrii) | Corticosteroid + tazarotene |
|||||||||| |||||| | | ||||| | ||||| | Halobetasol 0.01% lotion (Bryhali) | Corticosteroid |
|||||||| |||||||| | | ||||| | ||||| | Halobetasol 0.01% lotion (Bryhali) | Corticosteroid |
|||||||| |||| | ||||| | ||||| | Halobetasol 0.01% + tazarotene 0.045% lotion (Duobrii) | Corticosteroid + tazarotene |
Halobetasol 0.01% lotion (Bryhali) | Corticosteroid | |||
Tazarotene 0.045% lotion (Arazlo) | Tazarotene | |||
Trials evaluating I-IGA treatment response at week 8 in the subgroup of patients with intertriginous psoriasis | ||||
||||||| |||| | ||||| | ||||| | Pimecrolimus 1% cream (Elidel) | Calcineurin inhibitor |
||||||| |||| | ||||| | ||||| | Tacrolimus 0.1% ointment (Protopic) | Calcineurin inhibitor |
IGA = investigator global assessment; I-IGA = intertriginous investigator global assessment; NMA = network meta-analysis; NR = not reported.
aNot marketed for use for plaque psoriasis in Canada to date.
Source: Sponsor-submitted NMA technical report.18
Table 29: Assessment of Homogeneity for NMA
Characteristics | Description and handling of potential effect modifiers |
|---|---|
Disease severity | ||| |||||||||| || ||||||| |||||||| |||||| |||||| ||||||| || |||| ||||||||| ||||| || ||||||||| |||||||||| || |||||||||||| || || |||||| |||| |||||| || |||||||||| || |||||||| |||| |
Demographics | |||| || || |||||||| |||||| |||| |||| || |||| |||||| ||||| || ||||| |||| ||||| || ||||| || ||||| |||| |||||| |
Trial eligibility criteria | ||| ||||| |||||||| |||||||| |||| |||| || |||||||| ||||||| ||||||||| |||||||| |||||||| |||||||| |||| |||||||| |||||| ||||||| ||||||||| || |||||| |||||||| |||| |||| || |||||||| ||||||| ||||||| |||||||| ||||| ||||||| |||||||| |||||||| | | ||||| || || || ||||||| |||||||| |||||||| | ||||| || |||| |
Disease and treatment history | |||||||| || |||||||| ||||| ||||||||||| ||||||| || ||||||||||||| ||||| |||||||||| || |||||||||| || |||||||| |||| |||||||||||||| |||| ||||||||||| |||| | ||||| |||||||| |||||| |||||||| |
Vehicle response | ||| |||||||| ||||| || ||||||| |||||| |||| |||| || |||||||||||| |||||||| ||||| || ||||||| |||||| |||| ||||| || |||||| |
Definitions of end points | ||| ||||||| |||| ||||||||| ||||||||| ||||||| |||| | || ||||| |||| || |||| |||||||||| ||||||||||||| ||||| |||||| ||||| |||| | | ||||||| ||||||||||| || | || ||||||| || ||||| |||| |||||||| || |||||| ||||||||||| |||| |||||||| || || |||||||||| || ||||| ||||||||| ||||||| |||||||||||| || || |||||||| ||||||||||| ||||||||||| || ||||| |||| |||||||| ||||| |||||| |
Timing of end point evaluation | ||| || |||||| |||| ||||||||| ||||| ||||| || |||||||||| |
Clinical trial location | ||| |||||| |||| ||||||||| || ||||| |||||||| |
Study design | ||||||| |||| ||||| || |||||| || |||| ||||| || ||||| || |||| || |||||| ||||||| ||||||||| ||||| || |||| || ||||||| |||| ||||||| || ||||||||| || |||||||| |||||||| |
I-IGA = intertriginous Investigator’s Global Assessment; IGA = Investigator’s Global Assessment; NMA = network meta-analysis; PASI = Psoriasis Area Severity Index.
Source: Sponsor-submitted NMA technical report;18 details from the table were taken from the sponsor’s summary of clinical evidence.16
Results
The NMA results for IGA and I-IGA treatment response at week 8 are shown in Table 30. For the fixed-effect NMA of IGA treatment response at week 8, roflumilast was associated with improved odds of treatment response versus vitamin D analogues (| |||||| || |||| |||| || |||||| tazarotene ||| ||||| || |||| |||| || ||||||, corticosteroids ||| ||||| || |||| |||| || |||||, and vehicle ||| |||||| || |||| |||| || ||||||). IGA treatment response data for roflumilast versus corticosteroids plus vitamin D analogues or corticosteroids plus tazarotene did not clearly favour either treatment, as the 95% CrIs included the null value.
For the fixed-effect NMA of I-IGA treatment response at week 8, roflumilast was associated with improved odds of treatment response versus vehicle ||| ||||||| || || |||| || ||||||. The results found that roflumilast versus calcineurin inhibitors did not clearly favour either treatment, as the 95% CrIs included the null value.
Table 30: NMA Results for IGA and I-IGA Treatment Response at Week 8
Roflumilast vs. | OR (95% CrI) for IGA at week 8 | OR (95% CrI) for I-IGA at week 8 | ||
|---|---|---|---|---|
FE model (base case) | RE model | FE model (base case) | RE model | |
||||||| | |||| ||||| || |||||| | |||| ||||| || |||||| | ||||| ||||| || |||||| | ||||| ||||| || ||||||| |
|||||||||||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | || | || |
|||||||||| | |||| ||||| || |||||| | |||| ||||| || |||||| | || | || |
||||||| ||||||||| | |||| ||||| || ||||| | |||| ||||| || |||||| | || | || |
|||||||||||||| |||| ||||||| | | |||| ||||| || ||||| | |||| ||||| || ||||| | || | || |
|||||||||||||| |||| |||||||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | || | || |
||||||||||| |||||||||| | || | || | |||| ||||| || ||||| | |||| ||||| || |||||| |
CrI = credible interval; FE = fixed effects; IGA = Investigator’s Global Assessment; I-IGA = intertriginous Investigator’s Global Assessment; OR = odds ratio; NA = not applicable; NMA = network meta-analysis; RE = random effects.
Note: Bolded values are statistically significant at 0.05 significance level.
Source: Sponsor-submitted NMA technical report.18
Assessment of Consistency
||| || ||||||||| |||||||| || |||| || || |||||||| ||||||| ||||||||| ||||| |||||| |||||| ||||| |||| ||||||||| || ||||||||||||| || || |||||| |||| ||||| |||||||||||| || ||||||||||||||| |||| |||||||||| |||||| || ||||| |||||| ||||| || ||||| || || |||||||||||| || || |||||| |||| ||||||||||| |||| |||||||| |||| || ||||||| || ||||| |||| || |||||| ||||| || || |||||||| ||||||| || ||||| |||||||| || |||||||||||||| ||||||||| ||||| || |||| || |||||||||||| |||| || || ||||||||||
Critical Appraisal
The selection of studies used to inform the sponsor-submitted NMA was based on a systematic review of the literature that was performed using standard methods. The inclusion and exclusion criteria used for screening studies were clear. The authors conducted a search of multiple databases in December 2021, and identified 242 studies for inclusion based on predefined selection criteria. The study selection flow chart was reported, but a list of all excluded studies was not provided and there was no updated literature search conducted to identify potential new evidence. An established risk-of-bias tool (Cochrane risk-of-bias assessment tool, version 2) and acceptable methods were used to assess individual studies. Results were reported, with no studies reported as having a high risk of bias and no studies were excluded based on study quality.
Several efficacy and safety outcomes were evaluated as part of the systematic literature review. However, search results were narrowed to inform the NMA with studies that were consistent with the DERMIS-1 and DERMIS-2 trials. For instance, studies were excluded if they evaluated multiple treatments administered to the same individual or made within-class comparisons of treatments. Assessed outcomes were limited to the analysis of IGA and I-IGA treatment response at week 8 to align with the economic model. As a result, the evidence base in the final networks was small, with 8 studies and 3,093 patients informing the analysis for IGA response and 4 studies and 1,105 patients available for analysis for I-IGA response. Nine of the 12 pairwise comparisons in the network for IGA treatment response at week 8 among patients with plaque psoriasis were informed by a single study; similarly, both pairwise comparisons in the network for I-IGA treatment response in the subgroup of patients with intertriginous psoriasis were informed by 2 studies.
In terms of between-study heterogeneity, the reporting of study design and patient baseline characteristics was limited, restricting the ability to assess for potential sources of heterogeneity. The sponsor provided a description of trial characteristics, including interventions, sample size, study phase, region, and blinding. Other trial characteristics, such as definitions and thresholds for disease severity, frequency of treatment withdrawal, and handling of missing data were not described for all included studies. Furthermore, baseline patient characteristics, such as duration of disease, prior treatment experience, PASI score, and reasons for discontinuing prior treatments were not reported on for all included trials. The sponsor identified 2 patient characteristics a priori that were potential effect modifiers: disease severity (mild, moderate, severe) and type of psoriasis (psoriasis vulgaris versus intertriginous psoriasis). The sponsor stated that baseline disease severity was comparable across treatment groups; however, the definition of disease severity varied across studies and was generally based on different thresholds for combinations of IGA score, PASI score, and percentage of involved BSA. The clinical expert consulted for this review noted there were imbalances in baseline disease severity across treatment groups. As a result, it is possible that the heterogeneity in this baseline characteristic could result in changing relative treatment effects. There were no adjustments made for the effect modifier of disease severity (i.e., a stratified analysis) to explore potential differences in IGA and I-IGA treatment response across patients with mild, moderate, or severe disease. All studies that evaluated IGA treatment response used consistent response definitions. Differences were observed in the definition of I-IGA treatment response in the 2 comparator trials, which did not include the requirement for an improvement of 2 or more points, as was required in the DERMIS pivotal trials. As a result, a sensitivity analysis was conducted using response data from the DERMIS trials based on the less stringent definition used in the comparator trials, the results of which were consistent with the primary NMA results. Two studies used a 6-point IGA or I-IGA scale; all other studies used a 5-point IGA or I-IGA scale. The clinical expert consulted on this review noted that this difference in scales was unlikely to introduce bias into the NMA results.
An assumption was made that all vehicles in the included studies were equally effective. The contents of the vehicle for the included studies were not reported. There are differences in measures of effectiveness between specific vehicles and types of formulations, including between cream and ointment formulations or between branded and generic formulations with the same steroid.55 This may bias the results of the indirect comparison; however, the extent of the potential bias is unknown.
In the NMA, comparative treatments were grouped together by drug class, which the clinical expert found to be appropriate, overall. The clinical expert consulted on this review noted that topical steroids have very different potencies and, therefore, topical steroids of low, moderate, or high potency should not be grouped together to assess comparative efficacy. The analyses for IGA treatment response only assessed 2 topical steroids of high to very high potency (betamethasone dipropionate 0.5 mg/g gel and halobetasol 0.01% lotion), which is unlikely to lead to biased results, according to the clinical expert consulted.
Low heterogeneity was observed for all comparisons informed by more than a single study for IGA treatment response (i.e., I2 = 0% for roflumilast versus vehicle) and I-IGA treatment response (i.e., I2 = 31.1% for calcineurin inhibitor versus vehicle). However, given the small number of studies in the networks (8 studies for IGA and 4 studies for I-IGA), the I2 is subject to bias. The 95% CrIs around many of the efficacy estimates were wide, reflecting a lack of precision, most likely due to the small number of studies that were included in the NMA.
Another relevant limitation of the NMA is that treatment success was evaluated at week 8; therefore, the long-term efficacy of roflumilast versus other treatments in the study population is unknown. The key efficacy and safety outcomes identified in the clinical review protocol (e.g., PASI, HRQoL, infections, SAEs) were not analyzed in the NMA and this is an important gap in the evidence. According to the clinical expert consulted for this review, the NMA would have benefited from including the PASI and DLQI as clinically relevant outcome measures, which were assessed as secondary outcomes in the DERMIS pivotal trials. Furthermore, it was not possible to assess for consistency between a direct and indirect comparison in the network for I-IGA treatment response due to the sparsity of the network, with no closed loops.
Discussion
Summary of Available Evidence
Two phase III, randomized, double-blind, parallel-group, vehicle-controlled trials, DERMIS-19 (N = 439) and DERMIS-210 (N = 442), were included in the sponsor’s systematic review of roflumilast. The DERMIS-1 and DERMIS-2 trials assessed success in IGA, defined as an IGA score of clear or almost clear plus an improvement of 2 or more grades from baseline at week 8 with roflumilast cream 0.3% compared with matching vehicle in patients with chronic plaque psoriasis involving 2% to 20% of BSA (excluding the scalp, palms, and soles). Patients were excluded if they were unable to discontinue prohibited medications and treatments, defined as systemic, biologic, topical, phototherapy, and investigational treatments that could affect plaque psoriasis, within the prespecified washout period. Patients were randomized in a 2:1 ratio to receive roflumilast or vehicle applied topically once daily for 8 weeks. Secondary outcomes included measures of symptoms and involvement (PASI, I-IGA, WI-NRS, and IGA) and a measure of QoL (PSD). Similarly, exploratory outcomes included measures of involvement (e.g., BSA), measures of HRQoL (e.g., DLQI and CDLQI), local tolerability, and safety. At baseline, the mean age of all randomized patients was 48.0 years (SD = 14.69) in the DERMIS-1 trial and 47.0 years (SD = 14.72) in the DERMIS-2 trial. Most patients were male (64.9% and 62.4% of randomized patients in the DERMIS-1 and DERMIS-2 trials, respectively), while 33.9% to 39.3% of patients were female. The majority of patients were white (81.5% and 82.8% of randomized patients in the DERMIS-1 and DERMIS-2 trials, respectively), while the remainder of the randomized patients (0% to 7.3%) identified as Asian, Black or African American, American Indian or Alaska Native [wording from original source], Native Hawaiian or other Pacific Islander, other, or more than 1 race. The majority of patients had moderate IGA at baseline (74.7% and 76.5% of randomized patients in the DERMIS-1 and DERMIS-2 trials, respectively). The mean baseline BSA affected by psoriasis in all randomized patients was 6.66% (SD = 4.538) in the DERMIS-1 trial and 7.30% (SD = 4.918) in the DERMIS-2 trial. The mean baseline PASI score for all randomized patients was 6.5 (SD = 3.35) in the DERMIS-1 trial and 6.7 (SD = 3.33) in the DERMIS-2 trial.
One ongoing, phase III, single-arm, OLE study, the DERMIS-OLE17 study (N = 267), which evaluated the long-term safety of roflumilast applied once daily in patients aged 2 years or older with chronic plaque psoriasis involving 2% to 25% of BSA for up to 24 weeks, was included in this review to address the gap in evidence regarding the safety and efficacy of long-term treatment with roflumilast. The DERMIS-OLE study included patients who completed a prior roflumilast cream study (DERMIS-1 or DERMIS-2) (cohort 1) and patients who were treatment-naive to roflumilast (cohort 2). Note that treatment was stopped when psoriatic plaques completely resolved, in the opinion of the investigator. The primary objective was to assess the long-term safety of roflumilast. Improvement in IGA, I-IGA, PASI, WI-NRS, and BSA were included in the evaluation of efficacy as secondary outcomes. At baseline in cohort 1, the mean age was 47.9 years (SD = 15.79), most patients were male (59.1%), and the majority of patients were white (84.1%). In cohort 1, most patients (66.3%) had moderate IGA at baseline, the mean baseline BSA affected by psoriasis was 6.49% (SD = 4.476), and the mean baseline PASI score was 6.1 (SD = 3.19). Note, 2 of 3 patients in cohort 2 received roflumilast but had not reached week 4 at the time of data cut-off (December 14, 2020) and, as such, efficacy data were not available for those patients.
One NMA,18 which compared roflumilast with other topical therapies available in Canada for patients with plaque psoriasis, was conducted by the sponsor in the absence of direct comparative evidence of roflumilast versus other relevant comparators. Outcomes of interest included IGA treatment response at week 8 among patients with plaque psoriasis and I-IGA treatment response at week 8 among patients with intertriginous psoriasis, which were also evaluated in the DERMIS pivotal trials.
Interpretation of Trial Results
Efficacy
IGA and Intertriginous IGA
According to the patient group submission, patients with psoriasis identified effectiveness (not further defined) as a key outcome of new treatments. Based on the patient group submission, patients with experience with roflumilast stated noticeable benefits with treatment that included clearing of skin lesions, reduced itch, and reduced redness, which can be interpreted as desired outcomes that are important to patients. The clinical expert identified the treatment goals in clinical practice as sufficient clearing of psoriasis lesions resulting in improvement in HRQoL and reduced symptoms (itch and pain), while minimizing drug-related adverse effects. The primary analysis of the results of the primary efficacy end point, IGA success at week 8, and the analysis of the secondary efficacy end point, IGA success at week 4, were in favour of roflumilast versus vehicle in both the DERMIS-1 and DERMIS-2 trials. Note that conclusions about the validity and reliability of the 5-point IGA that was used in the trials are limited due to the use of the 6- and 7-point IGA in the psychometric validation studies. However, the clinical expert suggested that this difference in scales was unlikely to have introduced bias into the results. The MID in the IGA has not been estimated. However, achieving a score of 0 (clear) or 1 (almost clear) on the IGA has generally been accepted as clinically meaningful.11,12 Alternatively, or in addition to the achievement of a score of 0 or 1, the responder analysis may also consider the proportion of patients with at least a 2-grade improvement from baseline on the static IGA;11 this was consistent with the definition of IGA success used in both trials and the clinical expert was in agreement that the definition was clinically meaningful to patients and clinicians. The clinical expert indicated that the proportion of patients who experienced treatment success based on the IGA at week 8 in the roflumilast arm in comparison with the vehicle arm was clinically meaningful and that IGA success with roflumilast was observed as early as week 4. However, in the absence of direct comparative evidence of roflumilast versus other relevant comparators for the treatment of psoriasis on the whole body, the comparative efficacy of roflumilast remains uncertain, as evidence comparing the use of roflumilast with other relevant treatment options for plaque psoriasis was limited to the sponsor-submitted NMA. Results of the prespecified subgroup analyses (by age group, facial and/or intertriginous involvement, and severity of disease according to the amount of BSA affected by psoriasis) of the primary efficacy end point were generally supportive of the primary analysis results.
Clearance (absence of signs of disease) may be used as a measure of treatment success in clinical practice.6 Similarly, the clinical expert indicated that clearance or near clearance of psoriasis lesions is commonly used to assess psoriasis severity in clinical practice; this is analogous to the IGA scales used in clinical trials. Moreover, the Canadian Dermatology Association identified an achievement of an IGA score of 0 (clear) or 1 (almost clear) as a treatment goal. Achieving an IGA score of clear at week 8 was supportive of the benefit of roflumilast treatment in both trials, while an IGA score of clear at week 4 was not supportive of the benefit of roflumilast treatment.
Management of intertriginous psoriasis can be a challenge, as these areas are at an increased risk of adverse reactions to topical treatment, such as corticosteroids, because the skin tends to be thinner in these regions.6 Moreover, the Fraser Health Dermatology Group indicated that an ideal treatment would be a drug that is effective on all areas of the body, without the risk of atrophy and minimal irritation. The results of the secondary efficacy end point, I-IGA success at week 8, was in favour of roflumilast versus vehicle in both trials. The results of I-IGA success at week 4 were not supportive of the benefit of roflumilast treatment in the DERMIS-1 trial but were supportive of the benefit of roflumilast treatment in the DERMIS-2 trial; however, this was an exploratory end point and, as such, the effect was uncertain at week 4. Note that this analysis was based on the prespecified I-IGA ITT population, a subset of patients in the ITT population with intertriginous area involvement and an I-IGA score of 2 or more at baseline. Although the measurement properties of I-IGA were not identified in the literature and an MID has not been estimated, I-IGA was defined as the IGA scale but was used to evaluate intertriginous areas only in the trials, and I-IGA success was consistent with the definition for IGA success. The clinical expert indicated the proportion of patients who experienced treatment success based on the I-IGA at week 8 in the roflumilast arm in comparison with the vehicle arm was clinically meaningful.
Evidence comparing the use of roflumilast with other currently available treatments for plaque psoriasis was limited to the sponsor-submitted NMA. Results for IGA treatment response at week 8 favoured roflumilast versus vitamin D analogues, tazarotene, or corticosteroids. However, results for IGA treatment response did not clearly favour either treatment when compared with corticosteroids plus vitamin D analogues or corticosteroids plus tazarotene. Comparisons of roflumilast with calcineurin inhibitors for I-IGA treatment response at week 8 did not clearly favour either treatment. There is uncertainty in the validity of the results of the NMA, as several potential sources of heterogeneity were not assessed due to the limited reporting of study and patient baseline characteristics. There were no stratified analyses conducted to adjust for effect modifiers, such as baseline disease severity. An assumption was made that all vehicles in the included studies were equally effective, which may violate the similarity assumption of the NMA; however, the extent of the potential bias is unknown. The NMA was limited to IGA and I-IGA treatment responses at week 8, with no comparative evidence for other outcomes of interest to this review, such as PASI and HRQoL.
Psoriasis Area and Severity Index
PASI is a commonly used outcome measure in clinical trials of psoriasis and provides additional context (extent and severity of disease) in comparison with IGA; however, treatment comparisons based on PASI can be challenging, as IGA is more commonly used in the assessment of topical therapy. Of note, the Canadian Dermatology Association identified a reduction in the PASI as a treatment goal for patients with psoriasis. The results of the secondary efficacy end points, PASI 75 at week 8 and time to PASI 50, were in favour of roflumilast versus vehicle in both trials. All other assessments of the PASI (i.e., PASI 75, mPASI 75, PASI 90, mPASI 90, PASI 100, and mPASI 100) were supportive of the benefit of roflumilast treatment in both trials. Evidence for the validity and reliability of PASI was identified in the literature. Although the responsiveness of the PASI to change was found to be low when the affected BSA was less than 10%,34,40 mPASI was calculated for patients in the trials with an anatomic area with less than 10% involvement to reflect the status of limited disease. An MID in the PASI score has not been estimated.
Dermatology Life Quality Index
According to patient groups, patients with psoriasis have identified improved intimacy (treatments targeting genital psoriasis) and medication conducive to their schedule as key outcomes of new treatments. The clinical expert identified the treatment goals in clinical practice as sufficient clearing of psoriasis lesions resulting in improvement in HRQoL; the Canadian Dermatology Association also identified improvement in QoL as a treatment goal for patients with psoriasis. The results of DLQI at week 8 showed an improvement in both trials, although the magnitude of benefit may have been overestimated due to subjective reporting of the outcome when most patients experienced an improvement during the treatment period.
Worst Itch Numeric Rating Scale
According to the patient groups, patients with psoriasis have identified the ability to conduct daily activities (treatments targeting itch and burning) as a key outcome of new treatments. The clinical expert identified the treatment goals in clinical practice as sufficient clearing of psoriasis lesions resulting in reduced symptoms (itch and pain); the Fraser Health Dermatology Group also identified reduction in severity of symptoms as a treatment goal. The results of the secondary end points, WI-NRS success at weeks 8 and 4, were in favour of roflumilast versus vehicle in both trials. Evidence for the validity, reliability, and responsiveness of WI-NRS in patients with plaque psoriasis was identified in the literature. The MID in WI-NRS has been estimated to be an improvement of 4 or more points in patients with plaque psoriasis;15 this was used in the definition of WI-NRS success in the trials. The clinical expert indicated that the proportions of patients who experienced treatment success based on the WI-NRS at weeks 8 and 4 in the roflumilast arm in comparison with the vehicle arm were clinically meaningful. However, benefit as measured by WI-NRS success was not consistently observed as early as at week 2.
Psoriasis Symptom Diary
According to patient groups, patients with psoriasis have identified improvement in mental health (treatments targeting publicly visible body areas in day-to-day lives) as a key outcome of new treatments. The results of the secondary end points, PSD at week 8 and week 4, were in favour of roflumilast versus vehicle in both trials. Evidence for the validity, reliability, and responsiveness of PSD was identified in the literature. An MID in PSD total score has not been estimated. The clinical expert was unable to provide any comment on the clinical meaningfulness of the results, as the PSD is not commonly used in clinical practice.
Body Surface Area
The Canadian Dermatology Association identified reduction in the affected BSA, which considers the extent of disease, as a treatment goal. The results of the amount of BSA affected by psoriasis at week 8 was supportive of the benefit of roflumilast treatment in both trials.
Duration of Response and Treatment-Free Interval
Measures of treatment success in clinical practice may include remission (maintenance of disease control or suppression of signs and symptoms over time).6 Based on the patient group submission, patients are concerned about recurrence and resistance to treatments due to the chronicity of the disease. Moreover, the clinical expert identified duration of response (remission) and treatment-free interval as important outcomes for patients. In the DERMIS-OLE trial, duration of response was defined as either the date from the first observation of an IGA score of clear or almost clear until the date of restarting roflumilast, or the date from the first observation of IGA success until the date of restarting roflumilast. The median duration of response among patients in cohort 1 who had an IGA status of clear or almost clear was 93 days; the KM median was 188 days. The median duration of response among patients in cohort 1 who experienced treatment success based on the IGA was 85 days; the KM median was 157 days. The treatment-free interval was defined as the time when patients met the criteria for disease clearance and stopped treatment to all lesions until treatment was restarted. The mean treatment-free interval among patients in cohort 1 who had disease clearance was 54.0 days (SD = 47.67). Note that the open-label, single-arm study design and the use of only descriptive statistics to summarize the results limit the ability to interpret the safety and efficacy results. Moreover, a relatively large proportion of patients in the DERMIS-1 and DERMIS-2 studies did not enter the DERMIS-OLE study; therefore, there is uncertainty in the results.
Other Considerations
The clinical expert indicated that roflumilast would be used as monotherapy in patients with mild disease and in conjunction with other topical therapies in patients with intertriginous psoriasis as well as psoriasis at different site(s) where the patient may prefer to use a different topical therapy. The clinical expert estimated a cut-off of 20% BSA affected by psoriasis for the use of roflumilast as monotherapy; this cut-off was used as an inclusion criterion and roflumilast was used as monotherapy in the trials. Additionally, the clinical expert suggested roflumilast would be used in combination with systemic therapy or phototherapy in patients with severe psoriasis. This is in line with the recommendations in the Canadian Guidelines for the Management of Plaque Psoriasis regarding the use of adjunct therapy with topical treatment in the management of moderate to severe plaque psoriasis.6 However, other topical and systemic treatments as well as phototherapy for plaque psoriasis were prohibited in both studies, and other evidence of the effect of roflumilast in combination therapy (and as adjunct therapy) was not identified for this review, representing a gap in the evidence for roflumilast. Despite this, the clinical expert consulted by CADTH noted they anticipate that roflumilast will be used in combination with other therapies in clinical practice. Of note, the Health Canada indication also does not restrict the use of roflumilast as monotherapy or combination therapy.
The suggested place in therapy for roflumilast was generally consistent across the input from different stakeholder groups received for this review, with some differences to note and with consideration of intertriginous area involvement. For most patients with plaque psoriasis, the clinical expert suggested they would consider roflumilast as an alternative to other first-line topical treatments for psoriasis within the current stepped approach, as this could prevent having to step up patients to systemic therapy. Similarly, Fraser Health Dermatology Group clinicians suggested they would consider roflumilast after the failure of topical steroids, as topical steroids are affordable and usually well tolerated therapy, and also before combination therapies, as combination therapies cannot be applied to multiple areas of the body, including the face, skin folds, and groin (i.e., facial and intertriginous areas). The Atlantic Provinces Dermatology Association and Dermatology Association of Ontario clinicians suggested they would consider roflumilast in the first line in the management of mild to severe plaques on any area of the body, before systemic or biologic treatment and as an adjunct to systemic or biologic therapy. For patients with facial, genital, and intertriginous involvement, the clinical expert suggested roflumilast would be a first-line treatment. However, it should be noted, and the clinical expert agreed, that treatment selection for plaque psoriasis is based on patient, clinician, and disease factors. Of note, previous treatment history for psoriasis was not prespecified in the inclusion and exclusion criteria and not fully reported in both trials; therefore, the line of therapy (i.e., use of roflumilast as a first or later line of therapy in the treatment of plaque psoriasis in patients with mild to severe disease and specific prior treatment history) that would be supported by the evidence from both studies remains unclear.
The clinical expert, Fraser Health Dermatology Group, and Canadian Dermatology Association indicated that many patients do not respond to currently available topical treatments. In particular, widespread psoriasis and psoriasis of the scalp, palms, and soles are often refractory to topical treatment, as indicated by the clinical expert. Note that both trials excluded patients with plaque psoriasis involving less than 2% and more than 20% of BSA, excluding the scalp, palms, and soles. The clinical expert noted there is a need for an intervention for the hair-bearing scalp area. However, the clinical expert indicated that this need would not be addressed by roflumilast; moreover, the areas of application in both trials did not include the scalp, while palms and soles were treated but were not counted toward any efficacy measurements.
According to the patient groups, ease of application was identified by patients as 1 of the key outcomes of a new treatment for psoriasis. Ease of application was not formally assessed in the trials; however, more than 90% of patients in each treatment arm across the studies were compliant with the assigned intervention, defined as a patient (or caregiver) applying 80% or more of the expected applications during the application period and not missing more than 3 consecutive doses. The clinical expert suggested that topical formulations that are less messy and more effective may improve adherence, particularly among patients with intertriginous involvement who are offered topical tacrolimus, as it is only available as an ointment and not widely available (off-label use in psoriasis). The Canadian Dermatology Association agreed there is an unmet need for treatment for intertriginous areas (difficult to treat optimally, as they are considered sensitive sites) and for nonsteroidal topical therapies (long-term use is limited due to the potential for local and systemic side effects). The Atlantic Provinces Dermatology Association and Dermatology Association of Ontario indicated there is an unmet need for a simplified topical treatment regimen with a new mechanism of action that would provide a fast onset of action (improvement in pruritus), and improved efficacy, tolerability, and safety in 1 topical formulation that can be applied to any affected area on the body, including the skin folds. The Canadian Dermatology Association indicated that 1 drug for multiple affected areas on the body would likely improve adherence to therapy. Roflumilast cream is a nonsteroidal drug that is indicated for the topical treatment of plaque psoriasis, including treatment of psoriasis in the intertriginous areas.
Harms
According to patient groups, patients with psoriasis have identified lack of side effects as a key outcome of new treatments. Of note, some patients have reported they would not be willing to tolerate burning sensations in the skin and headaches as side effects of medications for psoriasis. Additionally, the Fraser Health Dermatology Group and the clinical expert indicated that minimizing drug-related adverse effects, particularly AEs associated with steroid use, is 1 of the treatment goals in clinical practice. The clinical expert noted that although topical therapies are generally well tolerated and that topical steroids are used long-term by many patients in clinical practice, clinical guidelines caution against the long-term use of topical steroids for plaque psoriasis. Thus, the clinical expert agreed that more effective, safer, and better-tolerated treatment options are an unmet need, particularly for long-term use and in patients with facial, genital, and intertriginous involvement.
In both trials, the proportion of patients with TEAEs was generally well balanced between treatment arms. The most common TEAEs reported in the roflumilast arm (a frequency of 2% or more of patients in either study) were diarrhea and headache. All remaining TEAEs were reported in less than 2% of patients in the roflumilast arm in either study. The clinical expert indicated that diarrhea is a known side effect of oral phosphodiesterase type 4 inhibitors and is manageable in clinical practice. The clinical expert speculated that the proportion of patients in the roflumilast arm who reported diarrhea may reflect patients with a greater amount of BSA affected by psoriasis at baseline and, as a result, these patients likely applied a greater amount of roflumilast cream. The expert further noted this would likely be a point of discussion with the patient when selecting treatment. No deaths were reported in the trials. TEAEs leading to discontinuation of treatment and/or study withdrawal were reported in less than 1% of patients in the roflumilast arm in either study. The proportion of patients with application site pain in the roflumilast arm was 0.7% in the DERMIS-1 trial and 1.4% in the DERMIS-2 trial. All remaining TEAEs of special interest relating to local inflammatory reaction were reported in less than 1% of patients in the roflumilast arm in either study. No new safety signals were reported in the DERMIS-OLE study; however, there was potential for selection bias, as patients who discontinued from the parent studies due to AEs, lack of efficacy, or any other reason were excluded. Harms were not measured in the sponsor-submitted NMA; as such, the comparative safety of roflumilast versus other relevant comparators is unknown.
Conclusion
Two sponsor-submitted, phase III, randomized, double-blind, parallel-group, vehicle-controlled trials (the DERMIS-1 and DERMIS-2 trials) comparing topical treatment with roflumilast versus a matching vehicle in patients aged 2 years or older with chronic plaque psoriasis involving 2% to 20% (inclusive) of BSA, excluding the scalp, palms, and soles, demonstrated that 8 weeks of treatment with roflumilast reduced the overall severity of psoriasis, as measured by the proportion of patients experiencing treatment success based on the IGA compared with the matching vehicle. Roflumilast also demonstrated a reduction in the severity of psoriasis in intertriginous areas (identified as an area of unmet need), extent and severity of psoriasis, and severity of itch compared with the matching vehicle, as measured by the proportion of patients experiencing treatment success based on the I-IGA, a prespecified reduction in the PASI, and treatment success based on the WI-NRS, respectively, as well as improvement in QoL (specifically impacted by psoriasis) compared with the matching vehicle, as measured by PSD. There was supportive evidence on the overall treatment benefit of roflumilast versus vehicle on HRQoL, as measured by DLQI, and extent of disease, as measured by BSA. Of note, the effect of roflumilast in a broader patient population (e.g., those with < 2% and > 20% of BSA affected) and in combination therapy (and as adjunct therapy) in clinical practice is unknown, the line of therapy that would be supported by the evidence from both trials remains unclear, and the comparative efficacy of roflumilast versus other relevant comparators is uncertain in the absence of direct comparative evidence in the treatment of plaque psoriasis of the whole body. Indirect evidence provided by the sponsor-submitted NMA suggested that roflumilast may offer a benefit over vitamin D analogues, tazarotene, or corticosteroids based on IGA treatment response after 8 weeks of treatment, but the results did not clearly favour either treatment when compared with combination treatments (corticosteroids plus vitamin D analogues or corticosteroids plus tazarotene). Further, there was no clearly favoured treatment when compared with calcineurin inhibitors for I-IGA treatment response at week 8. The results of the NMA must be considered alongside potential uncertainty in the validity of the NMA results, as several potential sources of heterogeneity were not assessed, stratified analyses were not conducted to adjust for effect modifiers, and the similarity assumption of the NMA may have been violated; however, the extent of the potential bias is unknown. There were no safety or tolerability concerns associated with the use of topical roflumilast and no new safety signals were reported in the DERMIS-OLE study.
References
1.Feldman SR. Psoriasis: epidemiology, clinical manifestations, and diagnosis. In: Post TW, ed. Waltham (MA): UpToDate; 2022: www.uptodate.com. Accessed 2023 Apr 10.
2.Blauvelt A, Ehst, B.D. Pathophysiology of plaque psoriasis. In: Post TW, ed. Waltham (MA): UpToDate; 2021: http://www.uptodate.com. Accessed 2023 Apr 10.
3.Papp KA, Gniadecki R, Beecker J, et al. Psoriasis prevalence and severity by expert elicitation. Dermatol Ther (Heidelb). 2021;11(3):1053-1064. PubMed
4.Canpolat F, Cemil BC, Eskioğlu F, Akis HK. Is facial involvement a sign of severe psoriasis? Eur J Dermatol. 2008;18(2):169-171. PubMed
5.Merola JF, Li T, Li WQ, Cho E, Qureshi AA. Prevalence of psoriasis phenotypes among men and women in the USA. Clin Exp Dermatol. 2016;41(5):486-489. PubMed
6.Canadian Psoriasis Guidelines Committee. Canadian guidelines for the management of plaque psoriasis. 2009 Jun; https://www.dermatology.ca/wp-content/uploads/2012/01/cdnpsoriasisguidelines.pdf. Accessed 2023 Mar 9.
7.Canadian Dermatology Association. Psoriasis. 2023; https://dermatology.ca/public-patients/skin/psoriasis/. Accessed 2023 April 10.
8.Canadian Psoriasis Guidelines Addendum Committee. 2016 addendum to the Canadian guidelines for the management of plaque psoriasis 2009. J Cutan Med Surg. 2016;20(5):375-431. PubMed
9.Clinical Study Report: ARQ-151-301. A phase 3, 8-week, parallel group, double blind, vehicle-controlled study of the safety and efficacy of ARQ-151 cream 0.3% administered QD in subjects with chronic plaque psoriasis [internal sponsor’s report]. Westlake Village (CA): Arcutis Biotherapeutics, Inc; 2021 Sept 14.
10.Clinical Study Report: ARQ-151-302. A phase 3, 8-week, parallel group, double blind, vehicle-controlled study of the safety and efficacy of ARQ-151 cream 0.3% administered QD in subjects with chronic plaque psoriasis [internal sponsor’s report]. Westlake Village (CA): Arcutis Biotherapeutics, Inc; 2021 Sept 14.
11.Langley RGB, Feldman SR, Nyirady J, van de Kerkhof P, Papavassilis C. The 5-point Investigator’s Global Assessment (IGA) Scale: a modified tool for evaluating plaque psoriasis severity in clinical trials. J Dermatolog Treat. 2015;26(1):23-31. PubMed
12.Weisman S, Pollack CR, Gottschalk RW. Psoriasis disease severity measures: comparing efficacy of treatments for severe psoriasis. J Dermatolog Treat. 2003;14(3):158-165. PubMed
13.Shikiar R, Willian MK, Okun MM, Thompson CS, Revicki DA. The validity and responsiveness of three quality of life measures in the assessment of psoriasis patients: results of a phase II study. Health Qual Life Outcomes. 2006;4(1). PubMed
14.Basra MKA, Salek MS, Camilleri L, Sturkey R, Finlay AY. Determining the minimal clinically important difference and responsiveness of the Dermatology Life Quality Index (DLQI): further data. Dermatology. 2015;230(1):27-33. PubMed
15.Kimball AB, Naegeli AN, Edson‐Heredia E, et al. Psychometric properties of the Itch Numeric Rating Scale in patients with moderate‐to‐severe plaque psoriasis. Br J Dermatol. 2016;175(1):157-162. PubMed
16.Roflumilast cream 0.3% w/w (Zoryve) for topical treatment of plaque psoriasis, including treatment of psoriasis in the intertriginous areas, in patients 12 years of age and older: clinical summary [internal sponsor’s report]. In: Drug Reimbursement Review sponsor submission: Zoryve (roflumilast), 0.3% cream. Westlake Village (CA): Arcutis Biotherapeutics; 2023 Feb 1.
17.Clinical Study Report: ARQ-151-306. a phase 3, multicenter, open-label extension study of the long-term safety of ARQ-151 cream 0.3% in subjects with chronic plaque psoriasis [internal sponsor’s report]. Westlake Village (CA): Arcutis Biotherapeutics, Inc; 2021 Jul 28.
18.Indirect treatment comparison of roflumilast versus topical treatments for plaque psoriasis: Technical report of network meta-analysis [internal sponsor’s report]. In: Drug Reimbursement Review sponsor submission: Zoryve (roflumilast), 0.3% cream. Westlake Village (CA): Arcutis Biotherapeutics; 2023 Feb 1.
19.Michalek IM, Loring B, John SM. A systematic review of worldwide epidemiology of psoriasis. J Eur Acad Dermatol Venereol. 2017;31(2):205-212. PubMed
20.Paller A, Broun Lund, E. Psoriasis in children: epidemiology, clinical manifestations, and diagnosis. In: Post TW, ed. Waltham (MA): UpToDate; 2023: www.uptodate.com. Accessed 2023 Apr 27.
21.Pariser DM, Bagel J, Gelfand J.M., et al. National Psoriasis Foundation clinical consensus on disease severity. Arch Dermatol. 2007;143:239−242. PubMed
22.Zoryve (roflumilast): 0.3% w/w topical cream [product monograph]. Burlington (ON): C.R.I; 2023 Apr 11.
23.Silkis (calcitriol): 3 μg/g ointment [product monograph]. Thornhill (ON): Galderma Canada Inc; 2017 Oct 17: https://pdf.hres.ca/dpd_pm/00041695.PDF. Accessed 2023 Mar 10.
24.Dovonex (calcipotriol): 50 mcg/g topical ointment [product monograph]. Thornhill (ON): LEO Pharma Inc; 2021 Dec 13: https://pdf.hres.ca/dpd_pm/00064010.PDF. Accessed 2023 Mar 10.S.
25.Duobrii (halobetasol proprionate and tazarotene lotion): 0.01% w/w halobetasol propionate and tazarotene 0.045% [product monograph]. Laval (QC): Bausch Health, Canada Inc; 2022 Jun 17: https://pdf.hres.ca/dpd_pm/00066402.PDF. Accessed 2023 Mar 10.
26.Dovobet gel (calcipotriol and betamethasone dipropionate gel): 50 mcg/g calcipotriol (as monohydrate) and 0.5 mg/g betamethasone (as dipropionate) gel [product monograph]. Thornhill (ON): LEO Pharma Inc; 2020 Feb 25: https://pdf.hres.ca/dpd_pm/00055197.PDF. Accessed 2023 Aug 10.
27.Dovobet (calcipotriol and betamethasone dipropionate): ointment, 50 mcg/g calcipotriol (as monohydrate) and 0.5 mg/g betamethasone (as dipropionate) [product monograph]. Thornhill (ON): LEO Pharma Inc; 2016 Apr 4: https://pdf.hres.ca/dpd_pm/00034364.PDF. Accessed 2023 Aug 10.
28.Enstilar (calcipotriol and betamethasone dipropionate aerosol foam): 50 mcg/g calcipotriol (as monohydrate) and 0.5 mg/g betamethasone (as dipropionate) [product monograph]. Thornhill (ON): LEO Pharma Inc.; 2022 May 12: https://pdf.hres.ca/dpd_pm/00065877.PDF. Accessed 2023 Mar 10.
29.Elsawi R, Dainty K, Smith Begolka W, et al. The multidimensional burden of atopic dermatitis among adults: results from a large national survey. JAMA Dermatol. 2022;158(8):887-892. PubMed
30.Lebwohl MG, Kircik LH, Moore AY, et al. Effect of roflumilast cream vs vehicle cream on chronic plaque psoriasis. JAMA. 2022;328(11):1073-1084. PubMed
31.Lebwohl M, Swensen AR, Nyirady J, Kim E, Gwaltney CJ, Strober BE. The Psoriasis Symptom Diary: development and content validity of a novel patient-reported outcome instrument. Int J Dermatol. 2014;53(6):714-722. PubMed
32.Naegeli AN, Flood E, Tucker J, Devlen J, Edson-Heredia E. The Worst Itch Numeric Rating Scale for patients with moderate to severe plaque psoriasis or psoriatic arthritis. Int J Dermatol. 2015;54(6):715-722. PubMed
33.Berger RS, Bowman JP. A reappraisal of the 21-day cumulative irritation test in man. J Toxicol Cutaneous Ocul Toxicol. 2008;1(2):109-115.
34.Spuls PI, Lecluse LL, Poulsen ML, Bos JD, Stern RS, Nijsten T. How good are clinical severity and outcome measures for psoriasis?: quantitative evaluation in a systematic review. J Invest Dermatol. 2010;130(4):933-943. PubMed
35.Langley RG, Ellis CN. Evaluating psoriasis with Psoriasis Area and Severity Index, Psoriasis Global Assessment, and Lattice System Physician’s Global Assessment. J Am Acad Dermatol. 2004;51(4):563-569. PubMed
36.Berth-Jones J, Grotzinger K, Rainville C, et al. A study examining inter- and intrarater reliability of three scales for measuring severity of psoriasis: Psoriasis Area and Severity Index, Physician’s Global Assessment and Lattice System Physician’s Global Assessment. Br J Dermatol. 2006;155(4):707-713. PubMed
37.Callis Duffin K, Bushmakin AG, Cappelleri JC, Mallbris L, Mamolo C. A multi-item Physician Global Assessment scale to assess psoriasis disease severity: validation based on four phase III tofacitinib studies. BMC Dermatol. 2019;19. PubMed
38.Simpson MJ, Chow C, Morgenstern H, Luger TA, Ellis CN. Comparison of three methods for measuring psoriasis severity in clinical studies (Part 2 of 2): use of quality of life to assess construct validity of the Lattice System Physician’s Global Assessment, Psoriasis Area and Severity Index and Static Physician’s Global Assessment. J Eur Acad Dermatol Venereol. 2015;29(7):1415-1420. PubMed
39.Bożek A, Reich A. The reliability of three psoriasis assessment tools: psoriasis area and severity index, body surface area and physician global assessment. Adv Clin Exp Med. 2017;26(5):851-856. PubMed
40.Puzenat E, Bronsard V, Prey S, et al. What are the best outcome measures for assessing plaque psoriasis severity? A systematic review of the literature. J Eur Acad Dermatol Venereol. 2010;24 Suppl 2:10-16. PubMed
41.Strober B, Zhao Y, Tran MH, et al. Psychometric validation of the Psoriasis Symptom Diary using phase III study data from patients with chronic plaque psoriasis. Int J Dermatol. 2016;55(3):e147-155. PubMed
42.Strober BE, Nyirady J, Mallya UG, et al. Item-level psychometric properties for a new patient-reported psoriasis symptom diary. Value Health. 2013;16(6):1014-1022. PubMed
43.Basra MK, Fenech R, Gatt RM, Salek MS, Finlay AY. The Dermatology Life Quality Index 1994-2007: a comprehensive review of validation data and clinical results. Br J Dermatol. 2008;159(5):997-1035. PubMed
44.Finlay AY, Khan GK. Dermatology Life Quality Index (DLQI)--a simple practical measure for routine clinical use. Clin Exp Dermatol. 1994;19(3):210-216. PubMed
45.Reolid A, Servitje O, Ginarte M, et al. Validation of an optical pencil method to estimate the affected body surface area in psoriasis. Actas Dermosifiliogr (Engl Ed). 2020;111(2):143-148. PubMed
46.Chandran V, Gottlieb A, Cook RJ, et al. International multicenter psoriasis and psoriatic arthritis reliability trial for the assessment of skin, joints, nails, and dactylitis. Arthritis Rheum. 2009;61(9):1235-1242. PubMed
47.Arcutis Biotherapeutics, Inc. response to January 17, 2023 regarding Zoryve (roflumilast) CADTH review: summary of clinical clarifaxes [internal additional sponsor’s information]. Arcutis Biotherapeutics, Inc.; 2023 Mar 20.
48.Ferris LK, Armstrong AW, Del Rosso JQ, et al. Efficacy and tolerability of roflumilast cream 0.3% in patients with chronic plaque psoriasis involvement on the face, intertriginous, or genital areas: poled results from phase 3 trials (DERMIS-1 and DERMIS-2) [poster]. Presented at the European Academy of Dermatology and Venereology; 2022 Sep 7-11; Milano, Italy. 2022.
49.Sugarman JL, Weiss JS, Tanghetti EA, et al. Safety and efficacy of halobetasol propionate lotion 0.01% in the treatment of moderate to severe plaque psoriasis: a pooled analysis of 2 phase 3 studies. Cutis. 2019;103(2):111-116. PubMed
50.Sugarman JL, Weiss J, Tanghetti EA, et al. Safety and efficacy of a fixed combination halobetasol and tazarotene lotion in the treatment of moderate-to-severe plaque psoriasis: a pooled analysis of two phase 3 studies. J Drugs Dermatol. 2018;17(8):855-861. PubMed
51.Menter A, Gold LS, Bukhalo M, et al. Calcipotriene plus betamethasone dipropionate topical suspension for the treatment of mild to moderate psoriasis vulgaris on the body: a randomized, double-blind, vehicle-controlled trial. J Drugs Dermatol. 2013;12(1):92-98. PubMed
52.Lebwohl M, Freeman AK, Chapman MS, Feldman SR, Hartle JE, Henning A. Tacrolimus ointment is effective for facial and intertriginous psoriasis. J Am Acad Dermatol. 2004;51(5):723-730. PubMed
53.Gribetz C, Ling M, Lebwohl M, et al. Pimecrolimus cream 1% in the treatment of intertriginous psoriasis: a double-blind, randomized study. J Am Acad Dermatol. 2004;51(5):731-738. PubMed
54.Sugarman JL, Gold LS, Lebwohl MG, Pariser DM, Alexander BJ, Pillai R. A phase 2, multicenter, double-blind, randomized, vehicle controlled clinical study to assess the safety and efficacy of a halobetasol/tazarotene fixed combination in the treatment of plaque psoriasis. J Drugs Dermatol. 2017;16(3):197-204. PubMed
55.Stoughton RB. Are generic formulations equivalent to trade name topical glucocorticoids? Arch Dermatol. 1987;123(10). PubMed
Appendix 1: Detailed Outcome Data
Note that this appendix has not been copy-edited.
Table 31: Sensitivity Analyses of the Primary Efficacy End Point in the DERMIS-1 and DERMIS-2 Trials
Outcome | DERMIS-1 | DERMIS-2 | ||
|---|---|---|---|---|
Roflumilast | Vehicle | Roflumilast | Vehicle | |
IGA success (multiple imputation, mITT population), week 8 | ||||
N | 281 | 151 | 285 | 149 |
n (%) | 255 (90.7) | 132 (87.4) | 264 (92.6) | 131 (87.9) |
IGA success, n (%) | 108 (42.4) | 8 (6.1) | 99 (37.5) | 9 (6.9) |
OR (95% CI)a | 20.74 (7.66 to 56.13) | 6.61 (3.18 to 13.75) | ||
P valuea | < 0.0001 | < 0.0001 | ||
IGA success (observed dataset, ITT population), week 8 | ||||
N | 286 | 153 | 290 | 152 |
n (%) | 255 (89.1) | 132 (86.2) | 264 (91.0) | 131 (86.1) |
IGA success, n (%) | 108 (42.4) | 8 (6.1) | 99 (37.5) | 9 (6.9) |
OR (95% CI)a | 16.81 (6.28 to 44.99) | 7.20 (3.48 to 14.90) | ||
P valuea | < 0.0001 | < 0.0001 | ||
IGA success (tabulation of fitted point estimates from generalized estimating equations for binary response, observed dataset, ITT population), week 8 | ||||
N | 286 | 153 | 290 | 152 |
n (%) | 255 (89.1) | 132 (86.2) | 264 (91.0) | 131 (86.1) |
IGA success, n (%) | 108 (42.4) | 8 (6.1) | 99 (37.5) | 9 (6.9) |
OR (95% CI)b | 11.81 (5.60 to 24.88) | 8.97 (4.29 to 18.77) | ||
P valueb | < 0.0001 | < 0.0001 | ||
CI = confidence interval; IGA = Investigator’s Global Assessment; ITT = intention-to-treat; mITT = modified intention-to-treat; OR = odds ratio.
Note: IGA success was defined as a score of 0 (clear) or 1 (almost clear) plus a ≥ 2-grade improvement from baseline.
aThe OR, 95% CI, and P value were obtained from Cochran-Mantel-Haenszel test (stratified by site, baseline IGA, and baseline intertriginous involvement) comparing roflumilast cream 0.3% with vehicle.
bEstimates for OR, 95% CI for the OR, and P value were from a generalized estimating equations for binary response model, with IGA success as the dependent variable and treatment, baseline IGA score, and visit as the independent variables. A Toeplitz covariance structure was used to model the within-patient correlation. The OR is the estimate of the odds of having an IGA response for patients treated with roflumilast cream 0.3% relative to that for patients treated with the matching vehicle.
Sources: Clinical Study Reports for the DERMIS-19 and DERMIS-2 trials.10
Table 32: Exploratory Outcomes in the DERMIS-1 and DERMIS-2 Trials (ITT Population)
Outcome | DERMIS-1 | DERMIS-2 | ||
|---|---|---|---|---|
Roflumilast | Vehicle | Roflumilast | Vehicle | |
mPASI 75 from baseline, week 8 | ||||
N | 286 | 153 | 290 | 152 |
n (%) | 255 (89.1) | 132 (86.2) | 264 (91.0) | 131 (86.1) |
mPASI 75, n (%) | 161 (63.1) | 29 (22.0) | 150 (56.8) | 18 (13.7) |
OR (95% CI)a | 8.89 (4.68 to 16.90) | 6.81 (3.81 to 12.16) | ||
P valueb | < 0.0001 | < 0.0001 | ||
mPASI 90 from baseline, week 8 | ||||
N | 286 | 153 | 290 | 152 |
n (%) | 255 (89.1) | 132 (86.2) | 264 (91.0) | 131 (86.1) |
mPASI 90, n (%) | 114 (44.7) | 13 (9.8) | 93 (35.2) | 11 (8.4) |
OR (95% CI)a | 9.50 (4.44 to 20.32) | 6.14 (2.94 to 12.84) | ||
P valueb | < 0.0001 | < 0.0001 | ||
mPASI 100 from baseline, week 8 | ||||
N | 286 | 153 | 290 | 152 |
n (%) | 255 (89.1) | 132 (86.2) | 264 (91.0) | 131 (86.1) |
mPASI 100, n (%) | 35 (13.7) | 2 (1.5) | 29 (11.0) | 0 |
OR (95% CI)a | 9.50 (2.26 to 40.03) | NE (NE) | ||
P valueb | 0.0002 | 0.0001 | ||
ANCOVA = analysis of covariance; CI = confidence interval; IGA = Investigator’s Global Assessment; ITT = intention-to-treat; LS = least squares; mPASI 75 = 75% reduction in the modified Psoriasis Area and Severity Index score; mPASI 90 = 90% reduction in the modified Psoriasis Area and Severity Index score; mPASI 100 = 100% reduction in the modified Psoriasis Area and Severity Index score; NE = not estimable; OR = odds ratio.
aThe OR, 95% CI, and P value are obtained from Cochran-Mantel-Haenszel test (stratified by site, baseline IGA, and baseline intertriginous involvement) comparing roflumilast cream 0.3% with vehicle.
bThe P value was not adjusted for multiple testing (i.e., the type I error rate was not controlled).
cEstimates for LS means (change divided by the percent change from baseline and difference from vehicle) and accompanying 95% CIs and P values are from an ANCOVA with treatment, site, baseline IGA, baseline intertriginous involvement, and baseline of the variable being analyzed as independent variables.
Sources: Clinical Study Reports for the DERMIS-19 and DERMIS-2 trials.10
Pharmacoeconomic Review
Abbreviations
AE
adverse event
BIA
budget impact analysis
BSA
body surface area
CS-TAZ
corticosteroid plus tazarotene
CS-VDA
corticosteroid plus vitamin D analogue
I-IGA
intertriginous Investigator’s Global Assessment
ICER
incremental cost-effectiveness ratio
IGA
Investigator’s Global Assessment
NIHB
Non-Insured Health Benefits
NMA
network meta-analysis
ODB
Ontario Drug Benefit
OLE
open-label extension
PASI
Psoriasis Area and Severity Index
QALY
quality-adjusted life-year
WTP
willingness to pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Roflumilast (Zoryve), 0.3% w/w cream for topical use |
Submitted price | Roflumilast, 0.3% cream: $275.00 per 60 g tube |
Indication | For topical treatment of plaque psoriasis, including treatment of psoriasis in the intertriginous areas, in patients 12 years of age and older |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | April 28, 2023 |
Reimbursement request | As per indication |
Sponsor | Arcutis Biotherapeutics Inc. (Arcutis) |
Submission history |
|
NOC = Notice of Compliance; w/w = weight by weight.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov model |
Target population | Patients aged 12 years and older with plaque psoriasis, including individuals with intertriginous psoriasis involvement |
Treatment | Roflumilast topical |
Comparators | Topical treatments for plaque psoriasis were considered by class: high-potency CS, VDA, TAZ, CS-VDA, and CS-TAZ |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | 5 years |
Key data source | Pooled results from the sponsor’s DERMIS-1 and DERMIS-2 clinical trials and the sponsor’s NMA |
Submitted results |
|
Key limitations |
|
CADTH reanalysis results |
|
BSA = body surface area; CaIn = calcineurin inhibitor; CS = corticosteroid; CS-TAZ = corticosteroid plus tazarotene; CS-VDA = corticosteroid plus vitamin D analogue; ICER = incremental cost-effectiveness ratio; IGA = Investigator’s Global Assessment; NMA = network meta-analysis; LY = life-year; QALY = quality-adjusted life-year; TAZ = tazarotene; VDA = vitamin D analogue.
Conclusions
The CADTH clinical review found that the proportion of patients with plaque psoriasis experiencing treatment success based on Investigator’s Global Assessment (IGA) success at week 8 was greater in the roflumilast arm compared with the vehicle arm in the DERMIS-1 and DERMIS-2 trials. Roflumilast also demonstrated improvement in the severity of psoriasis in intertriginous areas, compared with vehicle, as measured by intertriginous IGA (I-IGA) success. However, the effect of roflumilast in a broader patient population (e.g., < 2% and > 20% body surface area [BSA] affected) and in combination therapy is unknown and the line of therapy that would be supported by the evidence from both trials remains unclear. The comparative efficacy of roflumilast versus relevant comparators is uncertain in the absence of direct comparative evidence in the treatment of plaque psoriasis. Results from the sponsor-submitted network meta-analysis (NMA) for IGA suggest a clinical benefit for roflumilast versus monotherapies (high-potency corticosteroid, tazarotene, vitamin D analogue); however, the magnitude of the benefit is unknown. No difference in efficacy could be concluded for roflumilast relative to combination therapies such as corticosteroid plus vitamin D analogue (CS-VDA) and corticosteroid plus tazarotene (CS-TAZ).
CADTH undertook reanalyses to address limitations in the sponsor’s pharmacoeconomic evaluation resulting in a CADTH base case where roflumilast was associated with higher quality-adjusted life-years (QALYs) (gain of 0.0005 QALYs or 4 quality-adjusted hours) and higher costs (incremental cost, $506) compared with corticosteroids, resulting in an incremental cost-effectiveness ratio (ICER) of $1,085,171 per QALY gained. Roflumilast had a 0% probability of being cost-effective at a willingness-to-pay (WTP) threshold of $50,000 compared with corticosteroids.
As noted in the CADTH Clinical Review Report, while the sponsor’s NMA favoured roflumilast over monotherapies, including corticosteroids, the magnitude of benefit remains uncertain. The Clinical Review Report also notes there is uncertainty in the validity of the NMA results. In the absence of robust comparative evidence, the predicted incremental gain of 0.0005 QALYs with roflumilast versus corticosteroids may still overestimate the incremental benefits associated with roflumilast, as CADTH could not address the impact of maintenance therapy in the model. Based on CADTH’s reanalysis, in order for roflumilast to be considered cost-effective compared with corticosteroids at a WTP threshold of $50,000 per QALY, the price of roflumilast would need to be less than $1.21 per gram, reflecting a 74% price reduction. As the incremental QALYs gained in CADTH’s reanalysis are associated with uncertainty, further price reductions (e.g., below a price of $1.21 per gram) may be required.
Based on CADTH’s clinical review, there are no differences in clinical efficacy between roflumilast compared with combination therapies for the treatment of plaque psoriasis. As such, there is no evidence to support a price premium for roflumilast over combination therapies. The price for roflumilast would need to be $1.42 per gram to be priced equally to CS-VDA in the sponsor’s model. In the subgroup of patients with intertriginous involvement, the CADTH clinical review concluded there were no differences in efficacy between roflumilast compared with calcineurin inhibitors (CaIn). No assessment was made for roflumilast versus corticosteroids in the I-IGA NMA. As such, there is no evidence to support a price premium for roflumilast over comparators.
The CADTH reanalysis could not address the uncertainty in the clinical evidence or issues with the sponsor’s model structure. CADTH notes that the sponsor and the CADTH base case both included the use of maintenance therapy, which the clinical expert noted was rarely used in their own clinical practice but could not be effectively removed from the analysis. Additionally, the cost-effectiveness of roflumilast in the full Health Canada indication (which is agnostic to the percentage of BSA affected as well as the use of roflumilast as combination versus monotherapy) is unknown, as patients with less than 2% and greater than 20% BSA involvement were excluded from the DERMIS trials and because the trials only studied roflumilast monotherapy.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
Patient input was provided as a joint submission from the Canadian Skin Patient Alliance, the Canadian Association of Psoriasis Patients, and the Canadian Psoriasis Network, which received survey responses from 81 patients in Canada with psoriasis and 5 responses from caregivers. In addition, 2 telephone interviews were conducted with patients who had experience with roflumilast through a clinical trial. The responses indicated that psoriasis symptoms have significant impacts on quality of life related to mental health, daily activities, intimate relationships, and socializing. Patients reported using topical corticosteroids, topical combination treatments, and/or biologic drugs to manage their psoriasis. Survey respondents noted that biologics were the most effective of all current treatments used. Other medications currently used by respondents included medical cannabis, topical retinoids, oral retinoids, apremilast, oral steroids, phototherapy, and other therapies (not specified). More than half of respondents reported side effects with current treatments, with a considerable proportion (76%) of respondents having stopped treatment at some point during their disease due to lack of treatment efficacy, adverse side effects, or financial challenges. Patients expressed that the most important aspects for new treatment options include improved effectiveness, lack of side effects, affordability, ease of application, and medications conducive to their schedule. Patients indicated they would not tolerate side effects such as headaches, burning sensations in the skin, mental disturbances, or fatigue as side effects of treatment for psoriasis. Among all survey respondents, 10 patients reported having used roflumilast before through clinical trials. Nine patients out of 10 reported noticeable benefits during treatment, including significant clearing of skin, reduced itch and redness, and clearing of skin lesions. Patients noted the ease of application and flexibility of treatment by being able to apply roflumilast to all affected areas of the body.
Clinician input received from the Canadian Dermatology Association, Fraser Health Dermatology Group, Atlantic Provinces Dermatology Association, and Dermatology Association of Ontario indicated that current plaque psoriasis treatment includes first-line topical steroid therapy followed by topical vitamin D analogue and steroid combinations. Off-label noncorticosteroid topicals for psoriasis include the phosphodiesterase type 4 inhibitor crisaborole and topical calcineurin inhibitors. If their disease did not respond to second-line therapies, patients would then receive phototherapy or systemic therapy. Off-label systemic therapies commonly include methotrexate and cyclosporine. The clinician input indicated that roflumilast would fulfill the unmet need for more nonsteroidal topical therapies for the treatment of psoriasis and that it could be used following the failure of initial topical steroids in the current treatment paradigm.
The CADTH-participating drug plans noted the lack of an active comparator in the DERMIS trials and the lack of evaluation of the prior therapies required for eligibility. The drug plans commented that there may be interest in using roflumilast in children younger than 12 years of age and also noted a lack of evidence to support combination use with other topical treatments and other biologic therapies for the treatment of plaque psoriasis. The drug plans noted that consideration should be given to topical therapies that may prevent patients from progressing to subsequent therapies that are more costly and invasive (such as systemic or biologic therapies).
Several of these concerns were addressed in the sponsor’s model:
Clinical effectiveness was based on treatment response (IGA success, which incorporates several outcomes important to patients, including plaque thickening, scaling, and redness), with the inclusion of subsequent therapies following nonresponse.
Health-related quality of life was included in the model through health state utility values applied to the response-based health states.
CADTH was unable to address the following concerns raised from stakeholder input:
There is a lack of direct comparative evidence between roflumilast and other currently available topical treatments for plaque psoriasis in the modelled population.
The efficacy of roflumilast following treatment failure could not be evaluated directly due to the lack of clinical data from the pivotal trials.
Economic Review
The current review is for roflumilast 0.3% (Zoryve) for patients aged 12 years and older with plaque psoriasis, including individuals with intertriginous psoriasis involvement.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis assessing roflumilast 0.3% compared with topical treatments for plaque psoriasis reimbursed by at least 1 public drug plan in Canada, grouped by treatment type.1 The model population was aligned with the DERMIS-1 and DERMIS-2 trials and comprised patients aged 12 years and older with chronic plaque psoriasis involving 2% to 20% of BSA, including individuals with intertriginous psoriasis involvement.2,3 The Health Canada indication does not specify amount of BSA involvement; therefore, the modelled population was not aligned with the Health Canada indication.
Roflumilast is available in a 60 g (0.3% weight by weight) aluminum tube for self-administered topical application.1,4 The recommended dosage is application once daily to affected areas of skin.4 The submitted price for roflumilast is $275 per 60 g tube or $4.58 per gram.1 In the model, the sponsor adopted a 4-week cycle cost for roflumilast of $269.50. Drug wastage was not considered in the sponsor’s economic evaluation.
The first-line topical comparator baskets included in the analyses were: high-potency corticosteroids (weighted mix of betamethasone dipropionate, clobetasol propionate, mometasone, furoate, betamethasone valerate, fluocinonide, and amcinonide), vitamin D analogues (weighted mix of calcipotriol and calcitriol), tazarotene (weighted mix of 0.05% and 0.1% tazarotene), CS-VDA (betamethasone dipropionate plus calcipotriol), and CS-TAZ (halobetasol propionate plus tazarotene). An overview of the 4-week cycle costs for each of the comparator regimens adopted in the sponsor’s model is provided in Table 12.
The analysis was conducted from the perspective of the Canadian public health care payer over a 5-year time horizon. Costs and clinical outcomes (life-years and QALYs) were discounted at an annual rate of 1.5% per annum.
Model Structure
The sponsor submitted a Markov model consisting of 9 health states with 4-week cycle lengths (Figure 1).1 Patients enter the model in the “initial treatment 1 flare” state where they receive either roflumilast or a topical treatment from the comparator basket. After 8 weeks, patients whose condition responds to their initial treatment then move to the “responder: no treatment” or “responder: maintenance treatment” health states, depending on whether maintenance treatment was recommended for the initial treatment received. Responders were then at risk of transitioning to a “relapse” health state during each 4-week model cycle. Initial responders who relapse would then restart their initial treatment regimen (regardless of whether they received maintenance treatment) and experience either a response or nonresponse after 8 subsequent weeks of treatment. Patients in the “relapse” state whose condition responds to treatment return to the responder health states. Initial nonresponders and those who relapse and subsequently do not respond to re-treatment move to second-line treatment or the “initial treatment 2 flare” health state where they receive a second-line topical treatment, defined as a market basket of the first-line treatment options. Patients whose condition responds to a second-line topical treatment enter a set of response-based health states similar to those they would enter after the “initial treatment 1 flare.” Patients whose disease does not respond to a second-line topical treatment move to the “nonresponder” health state where they receive a basket of third-line treatments and remain for the duration of the model time horizon. In the model, patients could discontinue treatment due to adverse events (AEs) after the initial 4-week model cycle on treatment based on the discontinuation rates naively derived from the individual trials included in the sponsor’s NMA, after which patients would not incur costs or experience further effects on quality of life for the remainder of the time horizon.
Model Inputs
The pharmacoeconomic model was informed primarily by inputs from the DERMIS trials, 2 phase III randomized, vehicle-controlled trials.2,3 The model’s baseline population characteristics were derived from the DERMIS trials (mean body weight of 92 kg; mean BSA affected of 7%).1-3 Response to initial treatment was based on achievement of IGA success, defined as achievement of a score of clear (0) or almost clear (1) on the 5-point IGA scale and an improvement of at least 2 points on the IGA scale.1 A pooled probability of response at 8 weeks from the DERMIS-1 and DERMIS-2 trials was used to estimate response status for those receiving roflumilast.1 For comparator treatments, response was based on the odds ratios of IGA success at 8 weeks relative to roflumilast from the sponsor’s NMA.1,5
Depending on the topical treatment used, maintenance treatment may or may not be recommended based on Health Canada’s product monograph information. Whether patients received maintenance treatment was assumed to have an effect on the probability of relapse.1 The probability of relapse for responders who do not require maintenance treatment (i.e., those who received a corticosteroid or CS-TAZ) was 47.6%, informed by the PSO-LONG study.6 The sponsor assumed that the relapse probability for patients receiving maintenance treatment with CS-VDA in the PSO-LONG study (30.8%)6 was an adequate proxy for estimating all treatment comparators that require maintenance treatment other than roflumilast (i.e., vitamin D analogue, tazarotene, CS-VDA, and calcineurin inhibitor).1 The maintenance treatment relapse probability of 6.7% for roflumilast was derived from the phase II open-label extension (OLE) trial for roflumilast.7 The sponsor assumed that 50% of patients using maintenance treatment discontinued each cycle, and that patients on maintenance treatment who discontinue restart treatment only upon relapse. The sponsor assumed that responders would have 54% less BSA affected than patients in the flare and relapse health states, which was informed by the DERMIS trials.2,3
The distribution of second-line topical treatment use was informed by data from the IQVIA Ontario Drug Benefit (ODB) claims database for the base-case analysis and differed based on the first-line treatment received.1 As roflumilast and tazarotene were not on the ODB Formulary, it was assumed there would be no utilization of these treatments in the second line. For third-line treatment in the nonresponder health state, patients received a market basket of treatment options that included the previous topical treatments and nontopical treatments, such as biologics and oral systemics (methotrexate, acitretin, and cyclosporine). Utilization of treatments in the nonresponder health state was based on clinical expert opinion and the distribution of psoriasis severity levels from the DERMIS-1 and DERMIS-2 trials.2,3 Nonresponder treatment utilization was estimated by the sponsor to be 42% topicals, 40% oral systemics, and 18% biologics.1
Health state utility values were derived from the PSO-ABLE trial.8 A post hoc utility analysis of EQ-5D values was conducted to obtain utility estimates based on response, which was defined as a 75% reduction in the Psoriasis Area and Severity Index score (PASI 75).8 The utility values for the “initial treatment 1 flare” and “relapse” states reflected the baseline EQ-5D score for the CS-VDA arm (0.80) in PSO-LONG,8 while the utility value for the “responder” state reflected the EQ-5D score of patients who had a PASI 75 at week 4 (0.90).8 The “initial treatment 2” utility value reflected the EQ-5D score of patients who were defined as nonresponders in the PSO-LONG trial (0.82).8 The health state utility value for the “nonresponder” state (0.842) was obtained by calculating a weighted average of the utility values for nonresponders using topical treatment from the PSO-ABLE trial and the utility values from 2 external sources informing nontopical treatment by PASI response.8-10
Costs in the model included drug acquisition costs and health care resource utilization costs. Drug acquisition costs for roflumilast were based on the sponsor’s submitted price.1 Acquisition costs for comparator and subsequent treatments were obtained from the ODB Formulary, except for tazarotene, which was sourced from the British Columbia PharmaCare Formulary.11,12 The distribution of treatments for the mixed comparator treatment groups was informed by IQVIA PharmaStat market-share data and the IQVIA LRx retail sales database.1 Drug costs were calculated in the model by assuming that 0.3 g of topical treatment is required to cover 1% of BSA, and that the average BSA affected among the cohort is 7%.1-3 Health care resource utilization costs included physician visit costs and laboratory costs, including costs related to general practice, dermatology, and rheumatology. The proportion of use was health state–dependent and was informed by clinical expert opinion.1 All unit costs were sourced from the Ontario Schedule of Benefits for Physician Services.13
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (1,000 iterations for the base-case and scenario analyses). The deterministic and probabilistic results were similar. The probabilistic findings are presented subsequently. Additional results from the sponsor’s submitted economic evaluation base case are presented in Appendix 3.
Base-Case Results
In the sponsor’s base-case analysis, roflumilast was associated with an estimated cost of $18,713 and 4.06 QALYs over the 5-year horizon, resulting in an ICER of $28,176 per QALY gained (incremental costs, $158; incremental QALYs, 0.006) compared with CS-VDA in a sequential analysis (Table 3). Other comparators, including corticosteroid, vitamin D analogue, tazarotene, and CS-TAZ, were dominated by CS-VDA (i.e., more costly and equally or less effective). In the sponsor’s base case, roflumilast had a 48% probability of being cost-effective at a WTP threshold of $50,000.
Results were driven by the small incremental QALYs gained with roflumilast versus CS-VDA. The total incremental QALYs gained (0.006) over the 5-year time horizon is equivalent to an additional 2 quality-adjusted life-days of additional health gained for those receiving roflumilast compared with CS-VDA. Approximately 15% of the incremental QALYs for roflumilast were accrued during the DERMIS trial periods of 8 weeks. The incremental benefit associated with roflumilast was accrued during the “initial treatment 1” set of health states, with the majority being accrued in the remission state. The majority of costs (95%) associated with roflumilast are attributed to drug acquisition costs. The sponsor’s results showed that 99% of patients on roflumilast become nonresponders after approximately 2.5 years, while it takes an estimated 1.75 years for 99% of patients to become nonresponders when receiving comparator treatments.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
CS-VDA | 18,555 | 4.0583 | Reference |
Roflumilast | 18,713 | 4.0639 | 28,176 |
Dominated treatments | |||
CS-TAZ | 18,595 | 4.0581 | Reference |
CS | 18,628 | 4.0581 | Dominated |
VDA | 18,973 | 4.0571 | Dominated |
TAZ | 19,005 | 4.0576 | Dominated |
CS = corticosteroid; CS-TAZ = corticosteroid plus tazarotene; CS-VDA = corticosteroid plus vitamin D analogue; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; TAZ = tazarotene; VDA = vitamin D analogue.
Note: The submitted analysis is based on the publicly available prices of all treatments, including comparator treatments.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor conducted a subgroup analysis assessing the cost-effectiveness of roflumilast compared with low-to-medium potency corticosteroids and calcineurin inhibitors in patients with intertriginous involvement, where I-IGA was used to assess response. The sponsor’s NMA examining I-IGA success was used to estimate the probability of response for calcineurin inhibitors.5 Due to a lack of data, the odds ratio from the IGA NMA was used to estimate efficacy for corticosteroids in the I-IGA subgroup (assumed to be equal to IGA).5 In this analysis, roflumilast, dominated low-to-medium potency corticosteroids and calcineurin inhibitors (i.e., roflumilast was associated with lower total costs and higher total QALYs).
The sponsor provided several additional scenario analyses, including various time horizons, lower I-IGA success for corticosteroids, treatment-independent relapse probabilities, removal of treatment discontinuation due to AEs, and alternate assumptions relating to maintenance treatment and utility values. Across all scenario analyses, the ICER comparing roflumilast with CS-VDA remained relatively stable.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
The full Health Canada indication was not adequately captured by the sponsor’s model. The sponsor-submitted analyses reflected the cost-effectiveness of monotherapy with roflumilast among patients with between 2% and 20% BSA affected, with effectiveness informed by the DERMIS trials.2,3 The DERMIS trials excluded patients with an affected BSA of less than 2% and greater than 20%. Given that the Health Canada indication is not restricted by amount of BSA affected, the modelled population is narrower than the Health Canada indication.4 As such, the sponsor’s analyses reflect the cost-effectiveness of roflumilast in only a subset of the indicated population. The clinical expert input received by CADTH indicated that these excluded patients could still be treated with roflumilast in clinical practice.
Furthermore, roflumilast was assessed only as monotherapy in the DERMIS trials, as patients were excluded from the trials if they were receiving any other treatments that could affect plaque psoriasis.2,3 The clinical expert feedback indicated that roflumilast could be used as combination therapy (particularly with systemic therapies and biologic therapies), especially in patients with severe plaque psoriasis. As such, the modelled population is narrower than the Health Canada indication, which is agnostic to whether roflumilast should be used as monotherapy or combination therapy.4 Therefore, there are several important evidence gaps in the trial evidence used to inform the cost-effectiveness analysis.
CADTH was unable to address this limitation owing to a lack of clinical data. As noted in the CADTH clinical review, CADTH was unable to draw conclusions related to the efficacy of roflumilast as combination therapy or in patients with less than 2% and greater than 20% of BSA affected. As such, the clinical effectiveness and cost-effectiveness of roflumilast in patients with less than 2% and greater than 20% of BSA affected as well as the use of roflumilast as combination therapy is unknown, as is the cost-effectiveness of roflumilast in the full Health Canada–indicated population.
The model structure may not accurately reflect the clinical pathway associated with all patients with plaque psoriasis. The sponsor submitted a Markov model with health states defined by response, as determined by IGA success.1 All patients entered the model in the flare state, and movement within the model was based on initial response status assessed at 8 weeks in the DERMIS trials and second- and third-line treatment response and relapse probabilities. This modelling approach is associated with uncertainty for several reasons.
In the sponsor’s model, the nonresponder health state is a nonhomogeneous group of patients with varying disease severity who are receiving different treatments of varying efficacies. Despite this, all occupants of the state are assumed to accrue the same health state costs and utilities. From a methodological perspective, a health state in an economic model should represent a homogenous group of patients who have similar expected costs and quality-of-life considerations; this is not captured by the modelled response-based health states, especially in the nonresponse state. The implications of heterogeneity in health states have been well documented in the literature14 and this does not conform to best practices for health state cohort models.15
The model structure also led to invalid results. To elaborate, when no maintenance treatment was assumed for any comparator in the CADTH base case, tazarotene was associated with the highest incremental benefit. This is because tazarotene had the highest treatment discontinuation rate, meaning that patients progressed faster to, and spent more time in, the nonresponder health state receiving subsequent therapies. According to the clinical expert, it is not clinically expected for a treatment with a higher discontinuation rate to yield the greatest total QALYs.
In addition, the patient transitions through the response-based health states did not meet face validity. Those who relapse while on maintenance treatment are assumed to be re-treated with the same treatment and have an additional opportunity to experience treatment response before moving on to a second line of topical treatment. According to the clinical expert consulted by CADTH, patients who relapsed while receiving maintenance treatment with a given therapy would not be treated with this therapy upon relapse and instead would likely move on to receive a second-line topical therapy.
Clinical expert input also indicated that psoriasis is a chronic condition that fluctuates in severity. This was not sufficiently captured in the 5-year time horizon, and patient experiences with multiple flare-ups and the extended use of topical treatments in nonresponders over a longer time period were not modelled.
CADTH could not fully address this limitation in reanalysis. In the base-case reanalysis, patients who relapse while receiving maintenance treatment were assumed to receive a subsequent topical treatment upon relapse (i.e., no re-treatment with the maintenance therapy). While in the relapse states, patients were assumed to have treatment acquisition costs equivalent to the initial flare state. To test this assumption, CADTH conducted a scenario analysis that assumed no drug acquisition costs in the relapse health states among patients who relapse while receiving maintenance treatment.
CADTH could not create a reliable scenario analysis assessing the removal of maintenance treatment to reflect its variable use in clinical practice and notes that the incremental benefit associated with roflumilast would be further reduced with the removal of maintenance treatment.
The comparative clinical efficacy of roflumilast with other topical psoriasis treatments is highly uncertain. There have been no head-to-head trials of roflumilast versus any topical treatments for patients with plaque psoriasis. To inform the pharmacoeconomic analysis, the sponsor conducted an NMA that included 8 studies to assess IGA treatment response and a second NMA that included 4 studies to assess I-IGA treatment response.5 Like the pharmacoeconomic report, the sponsor’s NMAs estimated roflumilast efficacy relative to grouped comparators.
According to the CADTH Clinical Review Report, there were numerous limitations with the sponsor’s submitted NMAs, including a limited number of studies, heterogeneity that could not be fully assessed, and a lack of precision. Notably, treatment success was evaluated at week 8, and the long-term efficacy of roflumilast versus other treatments is therefore unknown.
Overall, the CADTH clinical review concluded that results from the sponsor’s IGA NMA indicated there appears to be a benefit for roflumilast versus monotherapies (corticosteroids, tazarotene, and vitamin D analogues); however, the magnitude of the benefit is unknown. However, no difference in efficacy could be concluded for roflumilast relative to CS-VDA and CS-TAZ in the IGA NMA or for roflumilast relative to calcineurin inhibitors in the I-IGA NMA. The relative efficacy of roflumilast compared with low-to-medium potency corticosteroids in the I-IGA NMA could not be assessed due to a lack of data. Additionally, since the sponsor used a weighted mix of treatment comparators and did not consider all relevant comparators individually, the cost-effectiveness of roflumilast versus individual treatments is unknown. Due to the uncertainties in the relative clinical efficacy data used in the pharmacoeconomic model, there is a high level of uncertainty in the magnitude of the relative benefit of treatment with roflumilast over other topical psoriasis treatments.
Overall, the IGA NMA only included 2 corticosteroids (betamethasone dipropionate and halobetasol), but the weighted basket comparator in the pharmacoeconomic model includes various formulations (ointment, lotion, cream, solution, gel) of many corticosteroids. The relative efficacy of all corticosteroids compared with roflumilast was assumed to be captured by the 2 corticosteroids of the IGA NMA. The sponsor’s use of a different mix of treatments for cost and efficacy inputs adds uncertainty in the results of the analysis.
Due to the lack of direct evidence and limitations with the comparative evidence utilized by the sponsor in the pharmacoeconomic analysis, the cost-effectiveness of roflumilast compared with topical treatments is highly uncertain.
The conceptualization and parameterization of relapse is highly uncertain. In the sponsor’s model, treatments that do not indicate discontinuation after a certain number of weeks or discontinuation after response were assumed to be used as maintenance treatment (roflumilast, vitamin D analogues, tazarotene, CS-VDA, and calcineurin inhibitors). The clinical expert consulted by CADTH noted that the use of maintenance treatment among responders is variable and may be clinician-dependent, noting that maintenance treatment is rarely used in their own clinical practice. As such, the inclusion of maintenance treatment in the model is uncertain.
Additionally, the data used in the pharmacoeconomic model to estimate relapse probabilities was associated with uncertainty. Relapse was not an outcome formally assessed in the DERMIS trials, and relapse for roflumilast in the model was estimated by transforming the median duration of an IGA score of 0 or 1 from the phase II OLE study into a 4-week relapse probability.7 Of note, roflumilast was not studied as a maintenance treatment in the OLE trial.7 Relapse probabilities for all other maintenance treatments were derived from the PSO-LONG trial,6 which formally assessed the risk of first relapse for patients using maintenance treatment with CS-VDA, which was assumed to apply to all maintenance comparators other than roflumilast. The definitions of relapse from the OLE and the PSO-LONG trials are not equivalent. Further, the benefit associated with roflumilast due to a decreased relapse probability (6.68%) relative to other maintenance treatments (30.81%) is not supported by any robust, formal assessment or direct or indirect comparison with other treatments. Therefore, there is considerable uncertainty regarding the incorporation and parameterization of relapse, particularly with the relative reduction in relapse for roflumilast maintenance treatment that was assumed in the sponsor’s analysis.
Given the lack of comparative data, the relapse probability on maintenance treatment associated with roflumilast was set as equal to that of other maintenance treatments.
Health state utility values did not meet face validity and were associated with uncertainty. The utility values used by the sponsor in the pharmacoeconomic model are uncertain for several reasons. While the maximum observed EQ-5D utility value for a healthy individual in the general population in Canada is 0.885,8 the sponsor’s model used a utility value of 0.9 in the “response to topical treatment” state, meaning the utility for a responder would be higher than that of a healthy person in the general population in Canada.16 The clinical expert input indicated that this did not meet face validity and that responders may actually have a slightly lower utility value than those in the general population in Canada.
In addition, the response-based utility values were derived from a trial that defined response based on PASI 75 rather than IGA or I-IGA success, despite the sponsor’s model using IGA and I-IGA to measure treatment response. The clinical expert input indicated that PASI 75 and IGA success are not equivalent. Furthermore, the data that were used to estimate the health state utility values were derived from a 4-week trial specifically for patients receiving betamethasone dipropionate plus calcipotriol (CS-VDA), although patients were expected to receive multiple treatment comparators from various other treatment categories (i.e., 6 high-potency corticosteroids, 2 vitamin D analogues, tazarotene, and the combination of halobetasol propionate plus tazarotene).8 As such, the short-term evidence may not reflect the experience of patients participating in trials of other treatments over a longer time period, as expected in clinical practice. Finally, as the nonresponder health state represented a nonhomogeneous group of patients (refer to the aforementioned model structure limitation), multiple external sources8-10 were used to estimate a nonresponse utility value. Pooling utility scores across various patient populations to derive a single utility value was not appropriate and introduced additional uncertainty into the cost-effectiveness analyses.
In the CADTH base case, CADTH used the sponsor-provided option to apply a utility value estimate of 0.885 for the general population in Canada for the “response to topical treatment” health state, with all other utility values adjusted to reflect the decrease (−0.015). Owing to unresolved limitations with the sponsor’s model structure, CADTH was unable to adjust the nonresponder health state utility value.
The baseline percentage of affected BSA in the modelled population may not be generalizable to Canadian clinical practice. The baseline percentage of affected BSA in the modelled population was 7% based on the DERMIS trials.2,3 However, the clinical expert input indicated that roflumilast may be used in patients with a higher percentage of affected BSA in clinical practice and the Health Canada indication does not restrict use by the percentage of affected BSA. If patients have a higher baseline percentage of affected BSA requiring more product, roflumilast will be less cost-effective, since it has the highest unit costs of all the topical treatments for plaque psoriasis (Table 8).
CADTH assessed the impact of increasing the baseline percentage of BSA affected to 20% (to reflect the upper limit of the DERMIS trials) and noted that patients with a greater amount of BSA affected would be included in the Health Canada indication for roflumilast.
Additional limitations were identified but were not considered to be key limitations. These limitations are outlined subsequently.
Parameter uncertainty around influential model values is poorly characterized. Several parameters in the model were assumed to have standard error values that were equal to 10% of the mean parameter estimate, including treatment discontinuation, relapse, treatment utilization, utility values, and treatment class utilization. No justification was provided by the sponsor for this approach in characterizing parameter uncertainty, and it is implausible that all the indicated values would have an identical mathematical relationship between mean and standard error. This methodological limitation has an unknown influence on the ICER.
CADTH was not able to address this limitation in reanalysis. CADTH conducted a scenario analysis on the CADTH base case, increasing the standard error to 20%.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
The sponsor assumed that 0.3 g of topical treatment was needed to cover 1% of a patient’s affected BSA. | Likely appropriate. Based on the conventional “fingertip measure” used in clinical practice, it is approximated that an adult male fingertip unit is 0.5 g, which is assumed to cover 2% of affected BSA.17 |
Mortality was not modelled. | Likely appropriate. According to clinical expert input, topical treatments are unlikely to impact mortality. While some patients will experience mortality over the course of the model, this is not expected to impact cost-effectiveness estimates. |
The cost and health impacts of AEs do not substantially impact cost-effectiveness results. | Likely appropriate. According to clinical expert input, differences in AEs across comparators are not expected to be impactful. However, CADTH notes that AEs and safety were not assessed in the sponsor’s NMAs. |
Discontinuation of maintenance therapy was assumed to be 50% of each model cycle. | Likely appropriate. Clinical expert input indicated that maintenance treatment is used infrequently in their practice and noted that these discontinuation rates likely reflect patients’ likelihood of discontinuing once they experience a treatment response. |
Treatment wastage was excluded from the model. | Likely appropriate. Given the sponsor’s assumptions surrounding treatment usage, a small amount of treatment is expected to be wasted per cycle and is not anticipated to be a driver of cost-effectiveness estimates. |
AE = adverse event; BSA = body surface area; NMA = network meta-analysis.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
CADTH undertook reanalyses that addressed limitations in the model, as summarized in Table 5. The CADTH base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts. All CADTH probabilistic reanalyses were based on 1,000 iterations. CADTH was unable to address other key limitations (described previously), including the lack of robust comparative evidence or limitations with the submitted model structure. Due to these key limitations, it is uncertain whether costs and health outcomes have been appropriately captured.
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | — | — |
Changes to derive the CADTH base case | ||
1. Patient treatment pathway for those receiving maintenance treatment and experiencing relapse | Patients who relapse on maintenance treatment are re-treated with the same treatment and either experience response or move on to a subsequent topical treatment. | Patients who relapse on maintenance treatment are not re-treated with the same drug and move on to receive a subsequent topical treatment. |
2. Relapse probabilities for maintenance treatment | The probability of relapse for roflumilast on maintenance treatment is 6.68% and the probability of relapse for all other maintenance treatments (CS-VDA, TAZ, VDA) is 30.81%, based on noncomparative data. | The probability of relapse for all maintenance treatments was set to 30.81%. |
3. Maximum health state utility values | The mean utility value of the responder health state is 0.900. | The mean utility value of the responder health state was decreased to 0.885 to reflect the maximum health utility value of a healthy individual in Canada; other health state utility values were adjusted by the same relative decrease (−0.015). |
CADTH base case | — | Reanalysis 1 + 2 + 3 |
CS-VDA = corticosteroid plus vitamin D analogue; TAZ = tazarotene; VDA = vitamin D analogue.
CADTH undertook a stepped analysis incorporating each change proposed in Table 5 to the sponsor’s base case to highlight the impact of each change (Table 13). In the CADTH base case, CS-TAZ, corticosteroids, and roflumilast remained on the cost-effectiveness frontier. Roflumilast was associated with higher costs (incremental cost, $506) and higher QALYs (incremental QALYs, 0.0005) compared with corticosteroids over a 5-year time horizon, resulting in an ICER of $1,085,171 per QALY gained (Table 6). There is a 0% probability of roflumilast being cost-effective at a WTP of $50,000. The ICER for corticosteroids versus CS-TAZ was $155,855 per QALY gained (Table 6). In the CADTH base case, CS-VDA and vitamin D analogue were dominated by CS-TAZ (i.e., equally or less effective and more costly) and tazarotene was extendedly dominated by roflumilast. Detailed information and disaggregated results are presented in Appendix 4.
The CADTH base-case results were driven by the drug acquisition costs of roflumilast and the small incremental benefit observed between all comparators (Table 14). The total incremental QALYs gained (0.0005) for roflumilast over the 5-year time horizon is equivalent to an additional 4 quality-adjusted life-hours compared with corticosteroid. In 12% of iterations, roflumilast was associated with fewer QALYs than corticosteroids. All of the benefit associated with roflumilast was accrued in the “initial treatment 1” set of health states and the majority of the benefit in those states was accrued in the remission state. Nearly all (99%) of the incremental costs associated with roflumilast were drug acquisition costs.
Table 6: Summary of the CADTH Reanalysis Results
Drug | Total costs ($) | Total QALYs | ICER vs. CS-TAZ ($/QALY) | Sequential ICER ($/QALY) |
|---|---|---|---|---|
CADTH base case | ||||
CS-TAZ | 18,588 | 4.0306 | Reference | Reference |
CS | 18,628 | 4.0308 | 155,855 | 155,855 |
Roflumilast | 19,134 | 4.0313 | 750,018 | 1,085,171 |
Dominated treatments | ||||
CS-VDA | 18,768 | 4.0306 | Dominated | Dominated |
VDA | 19,027 | 4.0305 | Dominated | Dominated |
TAZ | 19,034 | 4.0311 | 925,963 | Extendedly dominated |
CS = corticosteroid; CS-TAZ = corticosteroid plus tazarotene; CS-VDA = corticosteroid plus vitamin D analogue; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; TAZ = tazarotene; VDA = vitamin D analogue; vs. = versus.
Note: The submitted analysis is based on the publicly available prices of the comparator treatments. Given the small QALY differences between treatments, the sequential ICER reported here may be different from the ICER that would have been calculated based on the reported total costs and QALYs within this table, as QALYs were rounded up to only 3 decimal places.
Scenario Analysis Results
CADTH undertook price reduction analyses based on the sponsor’s and CADTH’s base case. A price reduction of 74% would be required for roflumilast to be considered cost-effective relative to the next-most optimal treatment (corticosteroid) at a WTP threshold of $50,000 per QALY gained (Table 7).
Table 7: CADTH Price Reduction Analyses
Price reduction analysis | ICERs for roflumilast vs. comparators ($/QALY) | |
|---|---|---|
Sponsor base case | CADTH reanalysis | |
No price reduction | WTP threshold < $28,176: roflumilast |
|
10% | NA |
|
20% | NA |
|
30% | NA |
|
40% | NA |
|
50% | NA |
|
60% | NA |
|
70% | NA | WTP threshold < $82,328: roflumilast |
80% | NA | Roflumilast is dominant |
CS = corticosteroid; CS-TAZ = corticosteroid plus tazarotene; ICER = incremental cost-effectiveness ratio; NA = not applicable; QALY = quality-adjusted life-year; vs. = versus; WTP = willingness-to-pay.
Given the uncertainty in the magnitude of the benefit of roflumilast versus monotherapy treatments and the absence of clinical evidence to suggest a benefit for roflumilast versus combination therapies or roflumilast for intertriginous use, there is limited evidence to support a price premium for roflumilast. CADTH considered the price at which roflumilast would be equal to the sponsor’s price of comparators used in the model.
Compared with monotherapies, roflumilast would need to be priced at $0.37 per gram to be equal to corticosteroids.
Compared with combination therapies, roflumilast would need to be priced at $1.42 per gram to be equal to CS-VDA.
For intertriginous use, roflumilast would need to be priced at $0.34 per gram to be equal to corticosteroids in the I-IGA subgroup. No price reduction is required to achieve price parity with calcineurin inhibitors.
CADTH undertook scenario analyses to explore the impact of alternate assumptions on the cost-effectiveness of roflumilast versus topical treatments for plaque psoriasis to assess the uncertainty surrounding several drivers of cost-effectiveness estimates:
Applying CADTH base-case changes to conduct a scenario analysis in the I-IGA subpopulation.
Applying 20% affected BSA (reflective of the upper limit of the DERMIS trials) to the baseline patient population.
Increasing the assumed standard error to 20% of the mean parameter estimate to test the sponsor’s arbitrary assumption that the standard error for various influential model values was equal to 10% of the mean parameter estimate.
Assuming no drug acquisition costs in the relapse health states among patients who relapse while receiving maintenance treatment.
The results of these scenario analyses are presented in Table 15. In the subgroup analysis for patients with intertriginous movement, roflumilast was associated with higher costs (incremental cost, $159) and higher QALYs (incremental QALYs, 0.004) compared with calcineurin inhibitors over a 5-year time horizon. The ICER for roflumilast versus calcineurin inhibitors was $35,737 per QALY gained with a probability of being cost-effective at a WTP of $50,000 of 56%. In the scenario analysis assessing the impact of including a baseline of 20% affected BSA, the ICER for roflumilast versus tazarotene was $4,483,392 per QALY gained. Finally, in the CADTH scenario assuming an increased standard error (to 20%), roflumilast was dominated by tazarotene (i.e., more expensive and equally as effective). Additionally, in this scenario, roflumilast had fewer QALYs than the least effective comparator (CS-TAZ) in 43% of iterations.
Issues for Consideration
According to clinical expert input obtained by CADTH, prescribing practices vary across Canada and may contribute to uncertainty regarding the selection of comparators, particularly for topical corticosteroids.
Clinical expert input indicated that roflumilast may be used in patients younger than 12 years of age, although it would not likely be used in infants. Clinical evidence to inform the efficacy of roflumilast in those under 12 was not summarized in the CADTH clinical review, as it was beyond the Health Canada indication and the cost-effectiveness of roflumilast in this population is unknown.
Roflumilast has been previously reviewed by CADTH for chronic obstructive pulmonary disease at a daily cost of $2.10 and was recommended to not be listed by the CADTH Canadian Drug Expert Committee due to the lack of statistically significant differences between roflumilast and placebo in key clinical outcomes.18
The clinical expert consulted by CADTH noted that IGA success is not routinely used in clinical practice to measure treatment response. IGA success further does not adequately capture all outcomes impacting quality of life. The clinical expert further raised concerns with dichotomizing IGA into responders or nonresponders, given the heterogeneity within these groups.
Overall Conclusions
The CADTH clinical review found that the proportion of patients with plaque psoriasis who experienced treatment success based on IGA success at week 8 was greater in the roflumilast arm in comparison with the vehicle arm in the DERMIS-1 and DERMIS-2 trials. Roflumilast also demonstrated improvement in the severity of psoriasis in intertriginous areas compared with vehicle, as measured by I-IGA success. However, the effect of roflumilast in a broader patient population (e.g., < 2% and > 20% BSA affected) and in combination therapy is unknown, and the line of therapy that would be supported by the evidence from both trials remains unclear. The comparative efficacy of roflumilast versus relevant comparators is uncertain in the absence of direct comparative evidence in the treatment of plaque psoriasis. Results from the sponsor-submitted NMA for IGA suggest a clinical benefit for roflumilast versus monotherapies (high-potency corticosteroids, tazarotene, and vitamin D analogues); however, the magnitude of the benefit is unknown. No difference in efficacy could be concluded for roflumilast relative to combination therapies such as CS-VDA and CS-TAZ.
CADTH undertook reanalyses to address limitations in the sponsor’s pharmacoeconomic evaluation by: adjusting the modelled treatment pathway, such that patients were not re-treated with their initial topical treatment upon relapse on maintenance treatment and instead received a subsequent line of treatment; assuming the probability of relapse among all maintenance treatments was equal; and limiting the maximum health utility value to reflect observed Canadian values.
In the CADTH base case, roflumilast was associated with higher QALYs (incremental QALYs, 0.0005) and higher costs (incremental costs, $506) compared with corticosteroids, leading to an ICER of $1,085,171 per QALY gained versus corticosteroids. Roflumilast had a 0% probability of being cost-effective at a WTP of $50,000 compared with corticosteroids. The results of CADTH’s reanalysis were driven by minimal incremental benefit (4 quality-adjusted life-hours gained from roflumilast versus corticosteroid over a 5-year time horizon), as well as roflumilast drug costs.
As noted in the CADTH Clinical Review Report, while the sponsor’s NMA favoured roflumilast over monotherapies, including corticosteroids, the magnitude of benefit remains uncertain. The Clinical Review Report also notes there is uncertainty in the validity of the NMA results. In the absence of robust comparative evidence, the predicted incremental gain of 0.0005 QALYs with roflumilast versus corticosteroids may still overestimate the incremental benefits associated with roflumilast, as CADTH could not address the impact of maintenance therapy in the model. Based on CADTH’s reanalysis, in order for roflumilast to be considered cost-effective compared with corticosteroids at a WTP threshold of $50,000 per QALY, the price of roflumilast would need to be less than $1.21 per gram, reflecting a 74% price reduction. As the incremental QALYs gained in CADTH’s reanalysis are associated with uncertainty, further price reductions (e.g., below a price of $1.21 per gram) may be required.
Based on CADTH’s clinical review, there are no differences in clinical efficacy between roflumilast compared with combination therapies for the treatment of plaque psoriasis. As such, there is no evidence to support a price premium for roflumilast over combination therapies. The price for roflumilast would need to be $1.42 per gram to be priced equally to CS-VDA in the sponsor’s model. In the subgroup of patients with intertriginous involvement, the CADTH clinical review concluded there were no differences in efficacy between roflumilast compared with calcineurin inhibitors. No assessment was made for roflumilast versus corticosteroids in the I-IGA NMA. As such, there is no evidence to support a price premium for roflumilast over comparators.
The CADTH reanalysis could not address the uncertainty in the clinical evidence or issues with the sponsor’s model structure. CADTH notes that the sponsor and CADTH base case both included the use of maintenance therapy, which the clinical expert noted was rarely used in their own clinical practice but could not be effectively removed from the analysis. Additionally, the cost-effectiveness of roflumilast in the full Health Canada indication (which is agnostic to the percentage of BSA affected as well as the use of roflumilast as combination versus monotherapy) is unknown, as patients with less than 2% and greater than 20% of BSA affected were excluded from the DERMIS trials and because the trials only studied roflumilast monotherapy.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Zoryve (roflumilast), 0.3% cream. Westlake (CA): Arcutis Biotherapeutics, Inc; 2023 Feb 1.
2.Clinical Study Report: ARQ-151-302 [DERMIS-2]. A phase 3, 8-week, parallel group, double blind, vehicle-controlled study of the safety and efficacy of ARQ-151 cream 0.3% administered QD in subjects with chronic plaque psoriasis [internal sponsor's report]. Westlake Village (CA): Arcutis Biotherapeutics, Inc; 2021 Sep 14.
3.Clinical Study Report: ARQ-151-301 [DERMIS-1]. A phase 3, 8-week, parallel group, double blind, vehicle-controlled study of the safety and efficacy of ARQ-151 cream 0.3% administered QD in subjects with chronic plaque psoriasis [internal sponsor's report]. Westlake Village (CA): Arcutis Biotherapeutics, Inc; 2021 Sep 14.
4.Zoryve (roflumilast): 0.3% w/w, for topical cream [product monograph]. Burlington (ON): C.R.I; 2023 Apr 27.
5.Precision H. ARC47280 Indirect treatment comparison of roflumilast versus topical treatments for plaque psoriasis: technical report of network meta-analysis [sponsor supplied citation]. 2022.
6.Lebwohl M, Kircik L, Lacour JP, et al. Twice-weekly topical calcipotriene/betamethasone dipropionate foam as proactive management of plaque psoriasis increases time in remission and is well tolerated over 52 weeks (PSO-LONG trial). J Am Acad Dermatol. 2021;84(5):1269-1277. PubMed
7.Clinical Study Report: ARQ-151-202 [Study 202]. A phase 2, multicenter, open-label extension study of the long-term safety of ARQ-151 cream 0.3% in adult subjects with chronic plaque psoriasis who have completed preceding study ARQ-151-201 phase 2 randomized controlled trial (Cohort 1) and non-arq-151-201 subjects (Cohort 2) [internal sponsor's report]. Westlake Village (CA): Arcutis Biotherapeutics, Inc; 2021 Apr 23.
8.Griffiths CE, Stein Gold L, Cambazard F, et al. Greater improvement in quality of life outcomes in patients using fixed-combination calcipotriol plus betamethasone dipropionate aerosol foam versus gel: results from the PSO-ABLE study. Eur J Dermatol. 2018;28(3):356-363. PubMed
9.Pickard AS, Gooderham M, Hartz S, Nicolay C. EQ-5D health utilities: exploring ways to improve upon responsiveness in psoriasis. J Med Econ. 2017;20(1):19-27. PubMed
10.Banken R, Agboola F, A. E, et al. Targeted immunomodulators for the treatment of moderate-to-severe plaque psoriasis: effectiveness and value: condition update: final evidence report. Boston (MA): Institute for Clinical and Economic Review; 2018: https://digirepo.nlm.nih.gov/master/borndig/101745013/ICER_Psoriasis_Update_Final_Evidence_Report_10042018.pdf. Accessed 2023 Apr 24.
11.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2022; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2023 Apr 24.
12.Government BC. BC PharmaCare formulary search. 2022; https://pharmacareformularysearch.gov.bc.ca. Accessed 2023 Apr 24.
13.Schedule of benefits for physician services under the Health Insurance Act: (January 25, 2022 (effective July 1, 2022)). Toronto (ON): Ontario Ministry of Health; 2022: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf. Accessed 2023 Apr 24.
14.Zaric GS. The impact of ignoring population heterogeneity when Markov models are used in cost-effectiveness analysis. Med Decis Making. 2003;23(5):379-396. PubMed
15.Guidelines for the economic evaluation of health technologies: Canada. 4th ed. Ottawa (ON): CADTH; 2017: https://www.cadth.ca/guidelines-economic-evaluation-health-technologies-canada-4th-edition. Accessed 2023 Apr 24.
16.Bansback N, Tsuchiya A, Brazier J, Anis A. Canadian valuation of EQ-5D health states: preliminary value set and considerations for future valuation studies. PLoS One. 2012;7(2):e31115. PubMed
17.Long CC. The rule of hand: 4 hand areas=2 FTU=1 g. Arch Dermatol. 1992;128(8). PubMed
18.CADTH Canadian Drug Expert Committee (CDEC) final recommendation: roflumilast (Daxas - Nycomed Canada Inc.). Ottawa (ON): CADTH; 2011 Jul 27: https://www.cadth.ca/sites/default/files/cdr/complete/cdr_complete_Daxas_August-3-2011_e.pdf. Accessed 2023 April 18.
19.Saskatchewan Drug Plan: search formulary. 2022; http://formulary.drugplan.ehealthsask.ca/SearchFormulary. Accessed 2023 Apr 24.
20.DeltaPA. Ottawa (ON): IQVIA; 2022: https://www.iqvia.com/. Accessed 2023 Apr 24.
21.Government of Alberta. Interactive drug benefit list. 2022; https://idbl.ab.bluecross.ca/idbl/load.do. Accessed 2023 Apr 24.
22.Elmets CA, Korman NJ, Prater EF, et al. Joint AAD–NPF Guidelines of care for the management and treatment of psoriasis with topical therapy and alternative medicine modalities for psoriasis severity measures. J Am Acad Dermatol. 2021;84(2):432-470. PubMed
23.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Zoryve (roflumilast), 0.3% cream. Westlake (CA): Arcutis Biotherapeutics, Inc; 2023 Feb 1.
24.Canadian Dermatology Association. Psoriasis. 2023; https://dermatology.ca/public-patients/skin/psoriasis/#:~:text=Psoriasis%20affects%201%20million%20Canadians,affects%20approximately%2090%25%20of%20patients. Accessed 2023 Apr 24.
25.Eder L, Widdifield J, Rosen CF, et al. Trends in the prevalence and incidence of psoriasis and Psoriatic Arthritis in Ontario, Canada: a population-based study. Arthritis Care Res (Hoboken). 2019;71(8):1084-1091. PubMed
26.Papp KA, Gniadecki R, Beecker J, et al. Psoriasis prevalence and severity by expert elicitation. Dermatol Ther (Heidelb). 2021;11(3):1053-1064. PubMed
27.Petrella RJ, Gregory V, Luciani L, Barbeau M. Characteristics Of chronic plaque psoriasis in Canada: a retrospective database study. Value Health. 2014;17(3).
28.Sutherland G, Dihn T. Understanding the gap: a pan-Canadian analysis of prescription drug insurance coverage. Ottawa (ON): The Conference Board of Canada; 2017: https://www.conferenceboard.ca/e-library/abstract.aspx?did=9326. Accessed 2023 Apr 24.
Appendix 1: Cost Comparison Table
Table 8: CADTH Cost Comparison Table of Topical Treatments for Plaque Psoriasis
Treatment | Strength | Dosage form | Package form | Price per g or mL ($) | Recommended dosage |
|---|---|---|---|---|---|
Roflumilast (Zoryve) | 0.3% | Cream | 60 g | 4.5833a | Apply to affected area once daily. |
High-potency corticosteroids | |||||
Amcinonide (generic) | 0.1% | Cream Lotion Ointment | 60 g 60 mL 60 g | 0.4522 0.2997b 0.3609b | Apply to affected area twice daily, maximum 5 days on face, axillae, scrotum or scalp, 2 to 3 weeks elsewhere. |
Betamethasone dipropionate (generic) | 0.05% | Cream Lotion Ointment | 50 g 75 mL 50 g | 0.2048 0.1980 0.5186 | Apply to affected area twice daily, reassess need at least every 4 weeks. |
Betamethasone valerate (generic) | 0.05% 0.1% | Cream | 450 g | 0.0596 0.0889 | No recommended daily dose. Use as directed by clinicians. |
Clobetasol propionate (generic) | 0.05% | Cream Ointment Solution Lotion Spray | 15 g, 50 g, 450 g 15 g, 50 g 59 mL 59 g 59 mL | 0.2279 0.2279 0.1990 0.1990 1.9259 | Apply to affected area twice daily. Weekly maximum 50 g and limited to 2 consecutive weeks. |
Desonide (generic) | 0.05% | Cream Ointment | 15 g, 60 g, 454 g 60 g | 0.2650 0.2647 | Apply to affected area twice daily; may be increased in refractory cases. |
Desoximetasone (Topicort) | 0.05% 0.25% | Cream | 20 g, 60 g | 0.5537b 0.7790b | Apply to affected area twice daily. |
Desoximetasone (Topicort) | 0.05% 0.25% | Gel Ointment | 60 g 60 g | 0.6060b 0.7812b | Apply to affected area twice daily. |
Fluocinonide (Lyderm, Lidex) | 0.05% | Cream Gel Ointment | 15 g, 60 g, 400 g 15 g, 60 g 15 g, 60 g | 0.2378 0.3076 0.3035 | Apply to affected area twice daily. Weekly 45 g, and limited to 2 weeks. |
Fluocinolone acetonide (Synalar) | 0.01% | Solution | 60 mL, 118 mL | 0.2979b | Solution: Apply 2 to 4 times daily Ointment: Apply 2 to 3 times daily. |
Fluocinonide (Tiamol) | 0.05% | Cream | 25 g 100 g | 0.1980 | Apply 2 to 4 times daily. |
Halobetasol propionate (Ultravate) | 0.05% | Cream Lotion | 50 g 50 g | 1.0445d 0.9816d | Apply to affected area twice daily, limited to 50 g weekly and 2 weeks without re-evaluation. |
Hydrocortisone/hydrocortisone acetate (various)e | 0.2% 0.5% 1% 2.5% | Cream | 15 g, 28 g, 45 g 30 g, 45 g,120 g, 225 g, 454 g, 500 g 45 g, 225 g | 0.1667 0.2087b 0.1718 0.3588f | Use as directed by clinicians. |
Hydrocortisone/hydrocortisone acetate (various)e | 1% | Lotion | 60 mL, 120 mL, 150 mL | 0.1587 | Use as directed by clinicians. |
Hydrocortisone/hydrocortisone acetate (various)e | 0.2% 0.5% 1% | Ointment | 454 g | 0.1667 0.1400 0.0390 | Use as directed by clinicians. |
Hydrocortisone valerate (Hydroval) | 0.2% | Cream Ointment | 15 g, 45 g, 60 g 15 g, 60 g | 0.1667 | Apply to affected area twice daily. Discontinue as soon as lesions heal or if no response. |
Mometasone furoate (generic) | 0.1% | Cream Ointment Lotion | 15 g, 50 g 15 g, 50 g 15 g, 50 g | 0.5542 0.2252 0.3358 | Apply to affected areas twice daily. |
Prednicarbate (Dermatop) | 0.1% | Cream | 20 g 60 g | 2.2420c 1.9772c | Apply to affected areas twice daily. Reassess if no response within a few days to a week. Maximum 2 weeks. |
Triamcinolone acetonide (various) | 0.1% | Cream Ointment | 30 g 15 g | 0.0533 | No recommended daily dose. Use as directed by clinicians. |
Vitamin D analogues | |||||
Calcipotriol (Dovonex) | 50 mcg/g | Ointment | 15 g, 60 g, 120 g, 240 g | 1.0518 | Apply 1 to 2 times daily, maximum of 100 g per week. |
Calcitriol (Silkis) | 3 mcg/g | Ointment | 5 g, 30 g, 100 g | 1.3625 | Apply twice daily. No more than 30 g daily. |
Retinoid | |||||
Tazarotene (Tazorac) | 0.05% 0.1% | Cream/gel Cream/gel | 30 g | 1.4347b | Start with 0.05% once daily, increase to 0.1% if tolerated and medically indicated. Apply once a day in the evening. |
High-potency corticosteroid plus vitamin D analogue | |||||
Betamethasone dipropionate and calcipotriol (Dovobet, Enstilar) | 0.5 mg/g and 50 mcg/g | Foam Gel Ointment | 60 g 30 g, 60 g, 120 g 30 g, 60 g, 120 g | 1.6412 1.3142 1.2545 | Apply to affected area once daily up to 4 weeks. Daily maximum 15 g, weekly maximum 100 g. |
High-potency corticosteroid plus tazarotene | |||||
Halobetasol propionate and tazarotene (Duobrii) | 0.01% and 0.045% | Lotion | 100 g | 1.9300 | Apply to affected area once daily. |
Calcineurin inhibitor | |||||
Pimecrolimus (Elidel) | 1% | Cream | 10 g, 30 g | 2.5627 | Apply to affected area twice daily, discontinue when resolved or after 3 weeks if no improvement or exacerbation. |
Tacrolimus (Protopic) | 0.03% 0.10% | Ointment | 30 g | 2.8884 3.0899 | Apply to affected area twice daily. Discontinue after 6 weeks if no improvement or exacerbation. |
The comparators presented in this table have been deemed to be appropriate based on feedback from clinical experts. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Note: All prices are from the Ontario Drug Benefit Formulary (accessed March 2023),11 unless otherwise indicated, and do not include dispensing fees. Recommended doses from respective product monographs.
aSponsor’s submitted price.1
bSaskatchewan Formulary list price (March 2023).19
cOntario wholesale price, as reported by IQVIA DeltaPA (March 2023).20
dAlberta Formulary list price (March 2023).21
eIncludes compounds with camphor, menthol, pramoxine, and urea.
fBritish Columbia Formulary list price (March 2023).12
Note that this table has not been copy-edited.
Table 9: CADTH Cost Table of Phototherapy Treatments for Plaque Psoriasis
Treatment | Strength | Dosage form | Price per unit ($) | Recommended dosage | Weekly cost ($) |
|---|---|---|---|---|---|
UV light therapy | NA | NA | 7.85 per treatmenta | Administered 2 to 3 times per week. Maintenance therapy may be tapered to once weeklyb | 8 to 24 |
Methoxsalenc (various) | 10 mg 1% | Capsule Lotion | 0.5436 per mg 1.94 per mgd | 30 mgb 1 mL mixed with 2 L of water soaked into hands and feet | 16 to 49 2 to 6 |
NA = not applicable.
Note: All prices are from the Ontario Drug Benefit Formulary (accessed March 2023),11 unless otherwise indicated, and do not include dispensing fees.
aAssumed to be reimbursed private clinic treatment cost: Ontario Schedule of Benefits for Physician Services, code G470 “Ultraviolet Light Therapy” (accessed March 2023). Can also be administered as public outpatient or as home therapy.13
b2022 American Academy of Dermatology and National Psoriasis Foundation guidelines for care for the management and treatment of psoriasis with phototherapy.22
cAdministered as the psoralen component in a psoralen plus UV A light therapy.22
dBritish Columbia Formulary list price (March 2023).12
Note that this table has not been copy-edited.
Table 10: CADTH Cost Comparison Table of Biologic and Systemic Treatments for Moderate to Severe Plaque Psoriasis
Treatment | Strength | Form | Price ($) | Recommended dosage | Daily cost ($) | Annual cost ($) |
|---|---|---|---|---|---|---|
Biologics | ||||||
Adalimumab (Humira) | 40 mg/0.8 mL | Syringe or pen | 471.2700 | 80 mg initial dose, 40 mg every other week starting 1 week after initial dose | First year: 34.86 Subsequent years: 33.57 | First year: 12,724 Subsequent years: 12,253 |
Brodalumab (Siliq) | 210 mg/1.5 mL | Prefilled syringe | 645.0000 | 210 mg at weeks 0, 1, and 2, followed by every 2 weeks thereafter | First year: 47.71 Subsequent years: 45.95 | First year: 17,415 Subsequent years: 16,770 |
Bimekizumab (Bimzelx) | 160 mg/mL | Prefilled syringe or autoinjector | 1,625.0000 | 320 mg at weeks 0, 4, 8, 12, 16 followed by 320 mg every 8 weeks (or every 4 weeks for those ≥ 120 kg) | First year: 80.14 Subsequent years: 57.88 For ≥ 120 kg First year: 115.75 Subsequent years: 115.75 | First year: 29,250 Subsequent years: 21,125 For ≥ 120 kg First year: 42,250 Subsequent years: 42,250 |
Certolizumab pegol (Cimzia) | 200 mg | Prefilled syringe or autoinjector | 664.5100 | 400 mg initial dose at weeks 0, 2, and 4, followed by 400 mg or 200 mg every 2 weeks | First year: 52.80 to 94.67 Subsequent years: 47.33 to 94.67 | First year: 19,271 to 34,555 Subsequent years: 17,277 to 34,555 |
Etanercept (Enbrel)a | 50 mg/mL 25 mg/vial | Syringe or pen Vial | 405.9850 202.9300 | 50 mg twice weekly for 12 weeks, then 50 mg weekly | First year: 71.16 Subsequent years: 57.84 | First year: 25,983 Subsequent years: 21,111 |
Guselkumab (Tremfya) | 100 mg/mL | Prefilled syringe | 3,059.7400b | 100 mg at weeks 0 and 4, followed by every 8 weeks thereafter | First year: 58.68 Subsequent years: 54.49 | First year: 21,418 Subsequent years: 19,888 |
Infliximab (Inflectra) | 100 mg/vial | Vial | 525.0000 | 5 mg/kg/dose, for 3 doses (0, 2, 6 weeks) then 5 mg/kg every 8 weeks | First year: 57.53 Subsequent years: 50.34 | First year: 21,000 Subsequent years: 18,375 |
Infliximab (Renflexis, Avsola) | 493.0000 | First year: 54.03 Subsequent years: 47.27 | First year: 19,720 Subsequent years: 17,255 | |||
Ixekizumab (Taltz) | 80 mg/1 mL | Prefilled syringe | 1,670.4400 | 160 mg initial dose, 80 mg at 2, 4, 6, 8, 10, and 12 weeks; followed by 80 mg every 4 weeks | First year: 82.38 Subsequent years: 59.50 | First year: 30,068 Subsequent years: 21,716 |
Risankizumab (Skyrizi) | 75 mg/0.83 mL 150 mg/mL | Prefilled syringe | 2,467.5000 4,935.0000 | 150 mg at week 0 and 4, followed by 150 mg every 12 weeks thereafter | First year: 67.60 Subsequent years: 58.59 | First year: 24,675 Subsequent years: 21,385 |
Secukinumab (Cosentyx) | 150 mg/mL | Prefilled syringe or pen | 882.5900 | 300 mg at weeks 0, 1, 2, and 3, then monthly injections starting at week 4 | First year: 77.38 Subsequent years: 58.03 | First year: 28,243 Subsequent years: 21,182 |
Tildrakizumab (Ilumya) | 100 mg/mL | Prefilled syringe | 4,935.0000 | 100 mg at week 0 and 4, followed by 100 mg every 12 weeks thereafter | First year: 67.60 Subsequent years: 58.59 | First year: 24,675 Subsequent years: 21,385 |
Ustekinumab (Stelara) | 45 mg/0.5 mL 90 mg/1 mL | Prefilled syringe | 4,593.1400 | < 100 kg patients: 45 mg at weeks 0 and 4, followed by 45 mg every 12 weeks thereafter (same for > 100 kg, at 90 mg) | First year: 62.92 Subsequent years: 54.53 | First year: 22,966 Subsequent years: 19,904 |
Nonbiologic systemic treatments | ||||||
Acitretin (generics) | 10 mg 25 mg | Capsule | 1.2965 2.2770 | 25 mg to 50 mg daily | 2.28 to 4.55 | 831 to 1,662 |
Apremilast (Otezla) | 30 mg | Tablet | 18.7238b | 30 mg twice daily | 37.45 | 13,668 |
Cyclosporine (generics) | 10 mg 25 mg 50 mg 100 mg | Capsule | 0.6818c 0.7870 1.5350 3.0720 | 2.5 mg to 5 mg/kg daily, in 2 divided doses | 6.93 to 13.86 | 2,530 to 5,060 |
100 mg/mL | Oral solution | 5.7410 | 17.22 to 28.71 | 6,286 to 10,477 | ||
Methotrexate (generics) | 2.5 mg 10 mg | Tablet Tablet | 0.2513 2.7983c | 10 mg to 25 mg by mouth or IM weekly | 0.14 to 0.36 | 52 to 131 |
20 mg/2 mL 50 mg/2 mL | Vial Vial | 12.5000 8.9200 | 1.27 | 464 | ||
IM = intramuscular; SC = subcutaneous; SEB = subsequent entry biologic.
Note: All prices are from the Ontario Drug Benefit Formulary (accessed March 2023),11 unless otherwise indicated, and do not include dispensing fees. Recommended doses from respective product monographs unless otherwise indicated. Annual cost assumed 52 weeks or 365 days. Assumed patient weight of 90 kg and wastage of excess medication in vials, if applicable.
aTwo biosimilars of etanercept are currently available in Canada but are not currently approved for the treatment of psoriasis.
bOntario wholesale price, as reported by IQVIA DeltaPA (March 2023).20
cSaskatchewan Formulary (March 2023).19
Note that this table has not been copy-edited.
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes or no | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | Refer to CADTH appraisal regarding the full Health Canada indication not being captured. |
Model has been adequately programmed and has sufficient face validity | No | Refer to CADTH appraisal regarding the model structure. |
Model structure is adequate for decision problem | No | Refer to CADTH appraisal regarding the model structure. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | Refer to CADTH appraisal regarding parameter uncertainty being poorly characterized. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | Refer to CADTH appraisal regarding parameter uncertainty being poorly characterized. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | No | The submission lacked clarity and detail in the technical report (i.e., calculation of roflumilast relapse probabilities, calculations regarding response and pooling of results). Key model inputs were hard-coded and results were repeated across multiple sheets, which added to lack of transparency and clarity. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
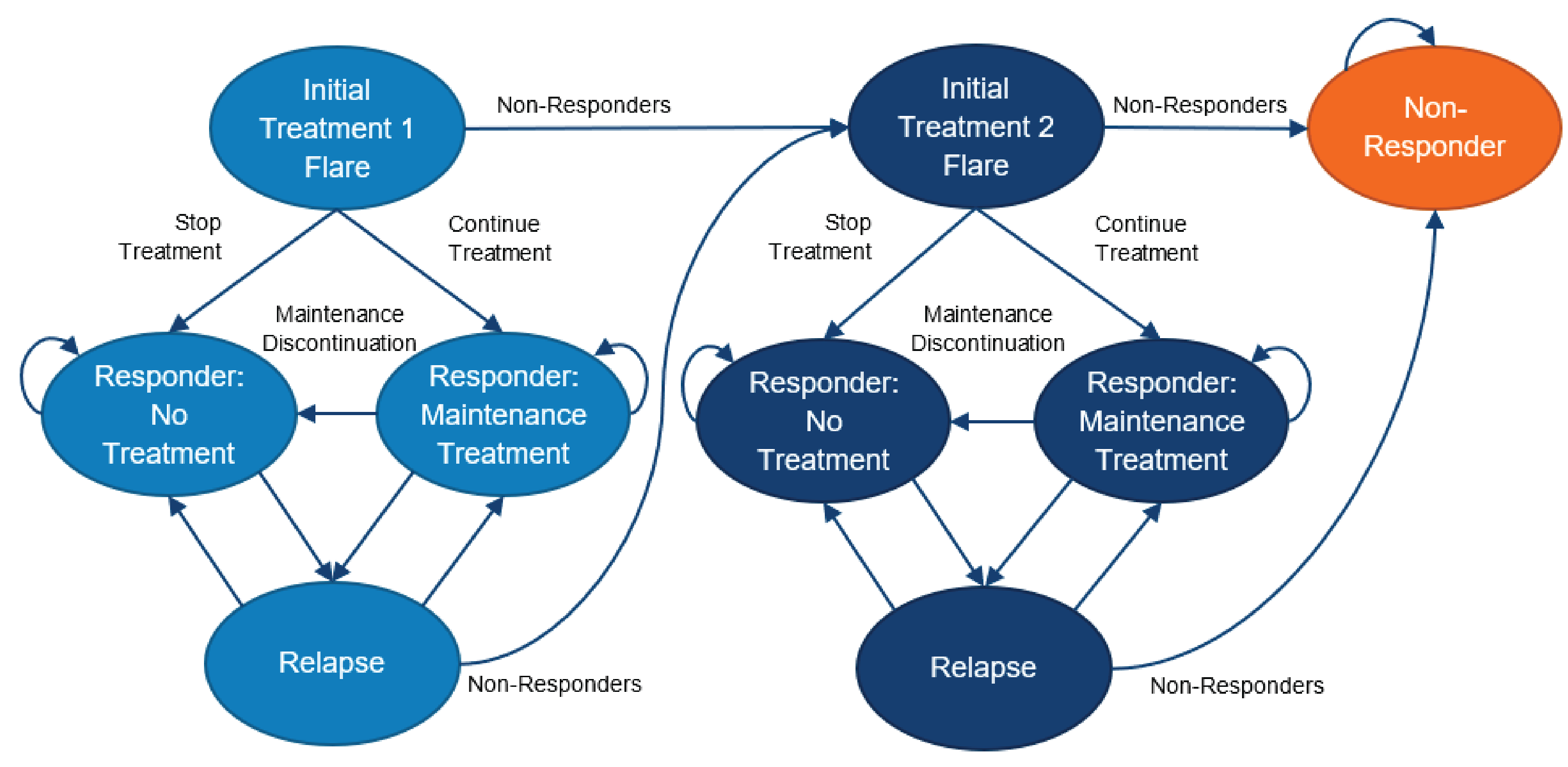
Table 12: Four-Week Model Cycle Costs for Topical Treatments for Plaque Psoriasis
Drug class | Total costs per 4-week cycle ($) | |
|---|---|---|
IGA | I-IGA subgroup | |
Roflumilast | 269.50 | 269.50 |
CS | 21.98 | 19.85 |
VDA | 126.93 | NA |
TAZ | 92.13 | NA |
CS-VDA | 83.25 | NA |
TAZ + VDA | 113.48 | NA |
CaIn | NA | 342.79 |
BSA = body surface area; CaIn = calcineurin inhibitor; CS = corticosteroid; IGA = Investigator’s Global Assessment; I-IGA = intertriginous Investigator’s Global Assessment; NA = not applicable; TAZ = tazarotene; VDA = vitamin D analogue.
Note: The submitted 4-week prices for each comparator are based on the publicly available prices of the comparator treatments.
Source: Sponsor’s pharmacoeconomic submission.1 Costs were calculated based on the sponsor’s assumption of 0.3 g of product required to treat 1% affected BSA, application frequency as per product monograph recommendation, and 7% baseline affected BSA.
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Detailed Results of CADTH Base Case
Table 13: Summary of the Stepped Analysis of the CADTH Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALYs) |
|---|---|---|---|---|
Sponsor’s base case (deterministic) | CS-VDA | 18,603 | 4.0563 | Reference |
Roflumilast | 18,756 | 4.0612 | 31,106 | |
Dominated treatments | ||||
CS-TAZ | 18,644 | 4.0562 | Dominated | |
CS | 18,669 | 4.0561 | Dominated | |
VDA | 19,002 | 4.0551 | Dominated | |
TAZ | 19,028 | 4.0556 | Dominated | |
CADTH reanalysis 1 – subsequent treatment following relapse on maintenance therapy | CS-TAZ | 18,644 | 4.0562 | Reference |
Roflumilast | 19,019 | 4.0586 | 157,466 | |
Dominated treatments | ||||
CS | 18,669 | 4.0561 | Dominated | |
CS-VDA | 18,777 | 4.0555 | Dominated | |
VDA | 19,034 | 4.0550 | Dominated | |
TAZ | 19,043 | 4.0555 | Dominated | |
CADTH reanalysis 2 – relapse probability on maintenance treatment | CS-VDA | 18,603 | 4.0563 | Reference |
Roflumilast | 18,946 | 4.0581 | 185,863 | |
Dominated treatments | ||||
CS-TAZ | 18,644 | 4.0562 | Dominated | |
CS | 18,669 | 4.0561 | Dominated | |
VDA | 19,002 | 4.0551 | Dominated | |
TAZ | 19,028 | 4.0556 | Dominated | |
CADTH reanalysis 3 – health state utility values capped by population maximum | CS-VDA | 18,603 | 4.0212 | Reference |
Roflumilast | 18,756 | 4.0248 | 43,265 | |
Dominated treatments | ||||
CS-TAZ | 18,644 | 4.0209 | Dominated | |
CS | 18,669 | 4.0211 | Dominated | |
VDA | 19,002 | 4.0209 | Dominated | |
TAZ | 19,028 | 4.0214 | Dominated | |
CADTH base case (reanalysis 1 + 2 + 3) –deterministic | CS-TAZ | 18,644 | 4.0209 | Reference |
CS | 18,669 | 4.0211 | 126,606 | |
Roflumilast | 19,135 | 4.0216 | 977,444 | |
Dominated treatments | ||||
CS-VDA | 18,777 | 4.0210 | Dominated | |
VDA | 19,034 | 4.0208 | Dominated | |
TAZ | 19,043 | 4.0213 | Extendedly dominated | |
CADTH base case (reanalysis 1 + 2 + 3) – probabilistic | CS-TAZ | 18,588 | 4.0306 | Reference |
CS | 18,628 | 4.0308 | 155,855 | |
Roflumilast | 19,134 | 4.0313 | 1,085,171 | |
Dominated treatments | ||||
CS-VDA | 18,768 | 4.0306 | Dominated | |
VDA | 19,027 | 4.0305 | Dominated | |
TAZ | 19,034 | 4.0311 | Extendedly dominated | |
CS = corticosteroid; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; TAZ = tazarotene; VDA = vitamin D analogue; vs. = versus.
Note: The submitted analysis is based on the public available prices of the comparator treatments. Given the small QALY differences between treatments, the sequential ICER reported here may be different from the ICER that would have been calculated based on the reported total costs and QALYs within this table as QALYs were rounded up to only 3 decimal places.
Table 14: Disaggregated Summary of CADTH’s Economic Evaluation Results
Treatment | Component | Value | Incremental (vs. reference) | Incremental (sequential) |
|---|---|---|---|---|
Discounted LYs | ||||
CS-TAZ | Total | 4.821 | NA | NA |
CS | Total | 4.821 | 0 | NA |
CS-VDA | Total | 4.821 | 0 | 0 |
VDA | Total | 4.821 | 0 | 0 |
TAZ | Total | 4.821 | 0 | 0 |
Roflumilast | Total | 4.821 | 0 | 0 |
Discounted QALYs | ||||
CS-TAZ | Treatment 1: Initial flare | 0.0881 | NA | NA |
Treatment 1: Remission | 0.0804 | NA | NA | |
Treatment 1: Relapse | 0.0672 | NA | NA | |
Treatment 2: Total | 0.2350 | NA | NA | |
Topicals nonresponder | 3.5599 | NA | NA | |
Total | 4.0306 | NA | NA | |
CS | Treatment 1: Initial flare | 0.08970 | 0.0016 | NA |
Treatment 1: Remission | 0.04667 | −0.0338 | NA | |
Treatment 1: Relapse | 0.03896 | −0.0282 | NA | |
Treatment 2: Total | 0.2546 | 0.0196 | NA | |
Topicals nonresponder | 3.6009 | 0.0411 | NA | |
Total | 4.0308 | 0.0003 | NA | |
CS-VDA | Treatment 1: Initial flare | 0.08934 | 0.0013 | −0.0004 |
Treatment 1: Remission | 0.05675 | −0.0237 | 0.0101 | |
Treatment 1: Relapse | 0.03729 | −0.0299 | −0.0017 | |
Treatment 2: Total | 0.2075 | −0.0276 | −0.0471 | |
Topicals nonresponder | 3.6398 | 0.0799 | 0.0389 | |
Total | 4.0306 | 0.0000 | −0.0003 | |
VDA | Treatment 1: Initial flare | 0.08716 | −0.0009 | −0.0022 |
Treatment 1: Remission | 0.02849 | −0.0520 | −0.0283 | |
Treatment 1: Relapse | 0.01868 | −0.0485 | −0.0186 | |
Treatment 2: Total | 0.2138 | −0.0212 | 0.0063 | |
Topicals nonresponder | 3.6824 | 0.1225 | 0.0426 | |
Total | 4.0305 | −0.0001 | −0.0001 | |
TAZ | Treatment 1: Initial flare | 0.08431 | −0.0038 | −0.0029 |
Treatment 1: Remission | 0.02017 | −0.0603 | −0.0083 | |
Treatment 1: Relapse | 0.01320 | −0.0540 | −0.0055 | |
Treatment 2: Total | 0.2356 | 0.0005 | 0.0218 | |
Topicals nonresponder | 3.6778 | 0.1179 | −0.0046 | |
Total | 4.0311 | 0.0005 | 0.0006 | |
Roflumilast | Treatment 1: Initial Flare | 0.08970 | 0.0016 | 0.0054 |
Treatment 1: Remission | 0.07193 | −0.0085 | 0.0518 | |
Treatment 1: Relapse | 0.04723 | −0.0199 | 0.0340 | |
Treatment 2: Total | 0.2355 | 0.0004 | −0.0001 | |
Topicals nonresponder | 3.5870 | 0.0271 | −0.0908 | |
Total | 4.0313 | 0.0007 | 0.0002 | |
Discounted costs ($) | ||||
CS-TAZ | Drug acquisition | 17,579 | NA | NA |
Physician visit | 857 | NA | NA | |
Testing and monitoring | 152 | NA | NA | |
Total | 18,588 | NA | NA | |
CS | Acquisition | 17,619 | 40 | NA |
Physician visit | 856 | −1 | NA | |
Testing and monitoring | 154 | 2 | NA | |
Total | 18,628 | 40 | NA | |
CS-VDA | Acquisition | 17,753 | 174 | 134 |
Physician visit | 859 | 2 | 3 | |
Testing and monitoring | 156 | 4 | 2 | |
Total | 18,768 | 180 | 140 | |
VDA | Acquisition | 18,014 | 435 | 261 |
Physician visit | 856 | −1 | −3 | |
Testing and monitoring | 157 | 5 | 1 | |
Total | 19,027 | 439 | 259 | |
TAZ | Acquisition | 18,022 | 443 | 8 |
Physician Visit | 855 | −2 | −1 | |
Testing and Monitoring | 157 | 5 | 0 | |
Total | 19,034 | 446 | 7 | |
Roflumilast | Acquisition | 18,120 | 541 | 98 |
Physician visit | 861 | 4 | 6 | |
Testing and monitoring | 153 | 1 | −4 | |
Total | 19,134 | 546 | 100 | |
Treatment | ICER vs. reference ($/QALY) | Sequential ICER ($/QALY) | ||
CS-TAZ | Reference | Reference | ||
CS | 155,855 | 155,855 | ||
CS-VDA | Dominated | |||
VDA | Dominated | |||
TAZ | 750,018 | Extendedly dominated | ||
Roflumilast | 925,963 | 1,085,171 | ||
CS = corticosteroid; ICER = incremental cost-effectiveness ratio; LY = life-year; NA = not applicable; QALY = quality-adjusted life-year; TAZ = tazarotene; VDA = vitamin D analogue; vs. = versus.
Note: The submitted analysis is based on the public available prices of the comparator treatments. Given the small QALY differences between treatments, the sequential ICER reported here may be different from the ICER that would have been calculated based on the reported total costs and QALYs within this table as QALYs were rounded up to only 3 decimal places.
Scenario Analyses
The following scenario analyses of the CADTH base case are presented in the following table.
Table 15: Detailed Results of the CADTH Scenario Analyses
Scenario analysis | Drug | Total costs ($) | Total QALYs | ICER vs. reference | Sequential ICER ($/QALY) |
|---|---|---|---|---|---|
Scenario analysis 1: I-IGA population | CaIn | 17,268 | 4.0106 | Reference | Reference |
Roflumilast | 17,427 | 4.0150 | 35,737 | 35,737 | |
Dominated treatments | |||||
CS | 17,360 | 4.0105 | Dominated | Dominated | |
Scenario analysis 2: Affected BSA 20% | CS | 20,930 | 4.0305 | Reference | Reference |
TAZ | 21,430 | 4.0308 | 2,041,240 | 2,041,240 | |
Roflumilast | 22,515 | 4.0310 | 3,254,381 | 4,483,392 | |
Dominated treatments | |||||
CS-VDA | 20,992 | 4.0303 | Dominated | Dominated | |
CS-TAZ | 21,164 | 4.0303 | Dominated | Dominated | |
VDA | 21,372 | 4.0302 | Dominated | Dominated | |
Scenario analysis 3: SE increased to 20% | CS-TAZ | 18,479 | 4.0183 | Reference | Reference |
CS | 18,527 | 4.0187 | 121,168 | 121,168 | |
TAZ | 18,926 | 4.0191 | 588,306 | 1,100,060 | |
Dominated treatments | |||||
CS-VDA | 18,662 | 4.0186 | Dominated | Dominated | |
VDA | 18,917 | 4.0187 | Dominated | Dominated | |
Roflumilast | 19,021 | 4.0191 | Dominated | Dominated | |
Scenario analysis 4: No drug acquisition costs for people who relapse while receiving maintenance treatment | CS-TAZ | 18,564 | 4.0211 | Reference | Reference |
CS | 18,609 | 4.0214 | 188,797 | 188,797 | |
Roflumilast | 18,904 | 4.0218 | 485,317 | 639,054 | |
Dominated treatments | |||||
CS-VDA | 18,696 | 4.0211 | Dominated | Dominated | |
VDA | 18,963 | 4.0210 | Dominated | Dominated | |
TAZ | 18,992 | 4.0216 | $938,572 | Dominated | |
CaIn = calcineurin inhibitor; CS = corticosteroid; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-years; SE = standard error; TAZ = tazarotene; VDA = vitamin D analogue; vs. = versus.
Note: The submitted analysis is based on the public available prices of the comparator treatments. Given the small QALY differences between treatments, the sequential ICER reported here may be different from the ICER that would have been calculated based on the reported total costs and QALYs within this table as QALYs were rounded up to only 3 decimal places.
Appendix 5: Submitted Budget Impact Analysis and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 16: Summary of Key Takeaways
Key Takeaways of the Budget Impact Analysis |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
The sponsor submitted a budget impact analysis (BIA) estimating the incremental budget impact of reimbursing roflumilast for the topical treatment of plaque psoriasis, including treatment of psoriasis in the intertriginous areas, in patients 12 years of age and older, in line with the Health Canada indication.23 The BIA was undertaken from the perspective of a Canadian public payer over a 3-year time horizon (2024 to 2026) using an epidemiological approach. The sponsor’s pan-Canadian estimates reflect the aggregated results from provincial budgets (excluding Quebec), as well as the Non-Insured Health Benefits (NIHB) Program. Key inputs to the BIA are documented in Table 17.
The sponsor compared a reference scenario in which patients received topical treatments currently used for treatment of plaque psoriasis in Canada to a new drug scenario in which roflumilast was reimbursed. The sponsor’s analysis included drug acquisition costs for roflumilast based on the sponsor’s submitted price. Topical treatment comparators were grouped by treatment class: corticosteroid, vitamin D analogue, tazarotene, CS-VDA (fixed combination), CS-TAZ (fixed combination), and calcineurin inhibitor. Additional combination therapy was included as comparators in the form of CS-VDA (fixed combination) plus corticosteroid, CS-VDA (fixed combination) plus calcineurin inhibitor, CS-VDA, and corticosteroid plus calcineurin inhibitor. Drug acquisition costs for each comparator class were estimated using IQVIA ODB Database data that reported the total annual number of grams or millilitres used per patient in each treatment class. The annual treatment class cost was a weighted average of annual individual treatment costs and their respective utilization.
Table 17: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3, if appropriate) |
|---|---|
Target population | |
Prevalence of psoriasis in Canada per year | |
Proportion of patients with plaque psoriasis | 90%27 |
Proportion of patients diagnosed | 98%27 |
Proportion of patients treated with topical therapy | 82%a |
Proportion of patients with public coverage | Jurisdiction-specific23 |
Proportion of patients 12 years of age or older | 100%a |
Number of patients eligible for drug under review | 174,290 / 176,347 / 178,405 |
Market uptake (3 years) | |
Uptake (reference scenario) Roflumilast CS VDA TAZ CS-VDA (fixed combination) CS-TAZ (fixed combination) CaIn CS-VDA (fixed combination) plus CS CS-VDA (fixed combination) plus CaIn CaIn + CS CS-VDA Otherb | 0% / 0% / 0% 28.5% / 28.5% / 28.5% 8.4% / 8.4% / 8.4% 0.0% / 0.0% / 0.0% 41.4% / 41.4% / 41.4% 0.5% / 0.5% / 0.5% 2.8% / 2.8% / 2.8% 11.4% / 11.4% / 11.4% 1.7% / 1.7% / 1.7% 1.5% / 1.5% / 1.5% 1.5% / 1.5% / 1.5% 2.4% / 2.4% / 2.4% |
Uptake (new drug scenario) Roflumilast CS VDA TAZ CS-VDA (fixed combination) CS-TAZ (fixed combination) CaIn CS-VDA (fixed combination) plus CS CS-VDA (fixed combination) plus CaIn CaIn + CS CS-VDA Other | 4.0% / 11.0% / 16.0% 28.2% / 27.8% / 27.5% 8.1% / 7.8% / 7.5% 0.0% / 0.0% / 0.0% 41.1% / 37.9% / 36.6% 0.4% / 0.3% / 0.2% 2.3% / 1.9% / 1.4% 10.6% / 9.8% / 9.0% 1.2% / 0.8% / 0.3% 1.1% / 0.7% / 0.3% 1.2% / 1.0% / 0.8% 1.7% / 1.1% / 0.4% |
Annual cost of treatment (per patient)c | |
Roflumilast CS VDA TAZ CS-VDA CS-TAZ CaIn Other | $825 $33 $216 $0d $170 $193 $260 $451b |
CS = corticosteroid; CaIn = calcineurin inhibitor; TAZ = tazarotene; VDA = vitamin D analogue.
aBased on sponsor’s assumption, clinical expert opinion, or internal estimates.
bOther treatments included in the model included various combinations of topical comparator treatments that were individually used in less than 1% of patients (e.g., CaIn + VDA; CS-TAZ (fixed combination) + VDA) and were grouped together for simplicity.
cFor each individual treatment class, annual cost of treatment was based on the weighted average of median annual units used per patient multiplied by formulary price per unit and utilization, as reported by the IQVIA ODB Claims database (September 1, 2021, to August 31, 2022).
dThe annual cost of TAZ was assumed to be $0 by the sponsor due to IQVIA ODB Claims Database (September 1, 2021, to August 31, 2022) indicating 0 annual g/mL used per patient.
Summary of the Sponsor’s BIA Results
The sponsor estimated the budget impact of reimbursing roflumilast for the topical treatment of plaque psoriasis, including treatment of psoriasis in the intertriginous areas, in patients 12 years of age and older would be $3,858,697 in year 1, $11,274,322 in year 2, and $16,524,429 in year 3, for a total budget impact of $31,657,448 over 3 years.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Number of patients eligible for treatment is uncertain: The proportion of patients with plaque psoriasis receiving topical treatments was obtained by clinical expert input and was based on the assumption that all patients with mild or moderate disease would receive topical therapy but only 20% of patients with severe disease would be treated with a topical therapy.23 The estimated weighted average of patients treated with topical therapy across all disease severities was estimated to be 82%. Based on clinical expert input obtained by CADTH for this review, the estimated use of topical therapies for patients with severe disease was likely underestimated, with the clinical expert indicating that approximately 90% of patients with more severe disease would be treated with a topical therapy. Given the high drug acquisition costs for roflumilast, the assumption that fewer patients with plaque psoriasis would receive topical therapy led to an underestimation of the budget impact of reimbursing roflumilast for all patients with plaque psoriasis (including mild, moderate, and severe disease).
The sponsor also assumed that 98% of patients would be adults (18+) and 2% would be adolescents (12 to 17) based on the DERMIS trials.2,3 According to clinical expert input, the distribution of patient age from the DERMIS trials is not reflective of the patient population in clinical practice and that the proportion of children with psoriasis is likely underestimated.
CADTH adjusted the proportion of patients with severe plaque psoriasis treated with topical treatments from 20% to 90% to reflect clinical expert input. CADTH tested increasing the proportion of adolescents in the analysis and found that this was not impactful to BIA results.
Uncertainty regarding the anticipated market shares of roflumilast: The sponsor’s base case assumed that 4%, 11%, and 16% of eligible patients would receive roflumilast in year 1, year 2, and year 3, respectively. Clinical expert input obtained by CADTH indicated that the projected uptake for roflumilast was likely underestimated given the nonsteroidal nature of the treatment and the anticipated favourable uptake of use in clinical practice. Uncertainty remains in these estimates and increases in the projected market shares would likely lead to sizable increases in the anticipated budget impact of reimbursing roflumilast.
The sponsor stated that roflumilast was likely to capture higher utilization from combination therapies and calcineurin inhibitors, as well as the market leader CS-VDA (fixed combination), compared with corticosteroids and vitamin D analogues. However, market capture of roflumilast in the submitted model appeared to largely come from CS-VDA (fixed combination) and it was uncertain how the preferential capture from treatment class combination therapies and calcineurin inhibitors was implemented. CADTH notes that there is uncertainty surrounding which comparators roflumilast would capture utilization from, which was supported by clinical expert input. If roflumilast captured greater market from a lower cost comparator than CS-VDA, the expected budget impact will be higher.
In the CADTH base case, the uptake of roflumilast was adjusted to reach 25% by year 3 to reflect the clinical expert input regarding underestimated market shares. The additional uptake of roflumilast in the CADTH base case was captured from CS-VDA (fixed combination). CADTH notes that there is remaining uncertainty surrounding the true capture of utilization from comparator treatments.
Public coverage rates were associated with uncertainty: The sponsor used data from the IQVIA LRx database to estimate the proportion of those covered by public drug plans. CADTH was unable to validate the sponsor’s estimates of public coverage. Public coverage estimates were notably lower than both the percentage of patients eligible and enrolled in the Sutherland and Dinh Understanding the Gap report.28
CADTH adjusted the proportion of patients with public coverage in each CADTH-participating jurisdictions to reflect the proportion of the population enrolled in a public drug plan.28
Assumptions regarding usage were uncertain: Usage of plaque psoriasis medication for comparator treatments was based on IQVIA ODB Claims database.23 For roflumilast, the sponsor assumed that patients will use 3 tubes of product per year, based on the IQVIA ODB Claims database indicating that VDA long-term use was estimated to require 2 tubes of product per year. The sponsor noted that their estimation of roflumilast use was conservative, however there remains uncertainty in the amount of expected product use for roflumilast. CADTH estimated that if the sponsor’s assumptions regarding treatment utilization for responders in the cost-utility analyses were applied, patients would use 2.1 g of product per day, resulting in approximately 13 tubes annually, if patients remained on treatment for a year and applied to the same initial affected BSA, or approximately 6 tubes if patients experienced a 54% reduction in BSA affected while receiving roflumilast.
Additionally, tazarotene was not reported to be used based on IQVIA ODB Claims database information, however this was assumed to apply to all jurisdictions. Based on clinical expert opinion and understanding of Canadian practice, while tazarotene is used to treat plaque psoriasis, it is a less commonly utilized treatment. CADTH notes that although the exclusion of tazarotene utilization reduces the face validity of the sponsor’s BIA, the impact of including tazarotene utilization that is more reflective of clinical practice is not anticipated to be a driver of results because use of tazarotene is generally low.
CADTH notes that there is remaining uncertainty surrounding the amount of roflumilast used per year and that the amount of product used is a major driver of BIA results.
Additional limitations were identified but were not considered to be key limitations. These limitations are outlined subsequently.
NIHB population was inappropriately calculated: The sponsor calculated the total population of CADTH-participating drug plans by adding the population of the provinces as reported by Statistics Canada, excluding Quebec, to the population of NIHB clients. NIHB clients living within the borders of a province are counted within provincial population data; thus, the NIHB population was double-counted in the sponsor’s analysis. Additionally, NIHB clients residing within Ontario are covered primarily by ODB if they are under 25 or over 65 years of age.
To address this limitation, the number of NIHB clients living within each jurisdiction was subtracted from the populations of those provinces.
Uncertainty in the modelling of the basket of comparators for each topical treatment class: The comparator treatments in the submitted cost-utility analysis and BIA are not identical. Combination use of comparator treatments was largely excluded in the economic evaluation, with exception of additional calcineurin inhibitors or corticosteroid use in a proportion of patients for sensitive areas only. However, the BIA includes all combination therapy use as seen in practice as per the IQVIA ODB Database data used. CADTH notes that the inclusion of all combination therapy use is appropriate for the BIA but should have been similarly characterized in the economic model such that both sets of comparator treatments are identical. As such, the costs associated with the weighted baskets differed between the BIA and economic evaluation, but this was not expected to significantly impact the results of either analysis.
CADTH could not address this limitation due to the sponsor’s BIA model structure.
CADTH Reanalyses of the BIA
CADTH corrected the sponsor’s base case by removing the double counting of NIHB clients in the analysis and revised the sponsor’s base case by adjusting the proportion of patients with severe plaque psoriasis receiving topical treatment, adjusting the uptake of roflumilast, and adjusting the public coverage rates (Table 18).
The results of the CADTH stepwise reanalysis are presented in summary format in Table 19 and a more detailed breakdown is presented in Table 20. In the CADTH base case, the 3-year budget impact of reimbursing roflumilast for the topical treatment of plaque psoriasis, including treatment of psoriasis in the intertriginous areas, in patients 12 years of age and older is expected to be $82,850,237 (year 1: $15,487,922; year 2: $28,067,209; year 3: $39,295,106).
Table 18: CADTH Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
1. NIHB population | NIHB clients included in provincial jurisdictions | NIHB clients removed from each provincial jurisdiction, except those in Ontario aged < 25 and 65+ years |
Changes to derive the CADTH base case | ||
1. Proportion of patients with severe plaque psoriasis using topical treatments | 20% | 90% |
2. Market shares of roflumilast in new drug scenario | Roflumilast: 4% / 11% / 16% CS-VDA (fixed): 41.1% / 37.9% / 36.6% | Roflumilast: 10% / 18% / 25% CS-VDA (fixed): 35.1% / 30.9% / 27.6% |
3. Proportion of publicly funded patients | Jurisdiction-specific based on IQVIA LRx data | Jurisdiction-specific based on the proportion of the population enrolled in the public drug plan28 |
CADTH base case | Reanalysis 1 + 2 + 3 | |
CS = corticosteroid; VDA = vitamin D analogue.
Table 19: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | 31,657,448 |
Submitted base case – corrected | 31,422,716 |
CADTH reanalysis 1 – proportion severe patients treated with topicals | 37,295,408 |
CADTH reanalysis 2 – market shares | 56,672,740 |
CADTH reanalysis 3 – public coverage rates | 38,702,016 |
CADTH base case | 82,850,237 |
BIA = budget impact analysis.
Note: Analysis is based on the publicly available prices of the comparator treatments.
CADTH also conducted additional scenario analyses to address remaining uncertainty, using the CADTH base case. Results are provided in Table 20.
Assuming that the price of roflumilast is reduced by 74% (to a price of $1.21 per gram), the price reduction at which roflumilast would be considered cost-effective compared with corticosteroids at a WTP threshold of $50,000 per QALY in the CADTH reanalysis of the cost-utility analysis (Table 7).
Table 20: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 26,713,983 | 27,033,128 | 27,352,272 | 27,671,417 | 82,056,817 |
New drug | 26,713,983 | 30,891,825 | 38,626,594 | 44,195,846 | 113,714,265 | |
Budget impact | 0 | 3,858,697 | 11,274,322 | 16,524,429 | 31,657,448 | |
CADTH base case | Reference | 38,538,638 | 39,095,069 | 39,651,501 | 40,207,933 | 118,954,503 |
New drug | 38,538,638 | 54,582,991 | 67,718,710 | 79,503,039 | 201,804,741 | |
Budget impact | 0 | 15,487,922 | 28,067,209 | 39,295,106 | 82,850,237 | |
CADTH scenario analysis: 74% price reduction | Reference | 38,538,638 | 39,095,069 | 39,651,501 | 40,207,933 | 118,954,503 |
New drug | 38,538,638 | 39,298,923 | 39,815,825 | 40,205,195 | 119,319,942 | |
Budget impact | 0 | 203,853 | 164,324 | −2,738 | 365,439 |
BIA = budget impact analysis.
Note: Analysis is based on the publicly available prices of the comparator treatments.
Stakeholder Input
Patient Input
Canadian Skin Patient Alliance, Canadian Association of Psoriasis Patients, and the Canadian Psoriasis Network
About Canadian Skin Patient Alliance, Canadian Association of Psoriasis Patients, and the Canadian Psoriasis Network
This submission is supported through collaboration between The Canadian Skin Patient Alliance (CSPA), Canadian Psoriasis Network (CPN) and Canadian Association of Psoriasis Patients (CAPP). CSPA is national, not-for-profit organization that improves the health and well-being of people across Canada affected by skin, hair, and nail conditions through collaboration, advocacy, and education. CPN is a national, not-for-profit organization dedicated to improving the lives of people in Canada who live with psoriasis and psoriatic arthritis (psoriatic disease) by providing current information on research and treatment options and by working with others to build awareness and advocacy about the complexity of these conditions. CAPP is a national, not-for-profit organization formed to better serve the needs of psoriasis patients across the country and strives to improve the quality of life for all Canadian psoriasis patients. CAPP’s mission is to be a resource and advocate for psoriatic patients and their families to improve patient care and quality of life.
Information Gathering
Data gathering
Information for this submission was obtained from a patient survey hosted on CSPA’s Survey Monkey and was made available on CSPA’s, CPN’s and CAPP’s communications channels from January 24 to February 12, 2023 in both English and in French. CSPA, CAPP and CPN staff also requested the contact information for Canadian clinical trial site principal investigators and reached out to them inviting them to share a link to the surveys with their clinical trial participants.
In this submission we report on combined English and French survey responses. A total of eighty-six (n=86) survey responses were received, 81 in English and 5 in French. The demographic characteristics of patients that participated in the survey are presented below. Two telephone interviews were also conducted with patients who had experience with roflumilast through a clinical trial.
Regional data
All the survey respondents were from Canada, with the majority being from Ontario (33%, n=28), British Columbia (22%, n=19), and Québec (15%, n=13). A smaller proportion of respondents also came from Nova Scotia (9%, n=8), Alberta (5%, n=4), Manitoba (4%, n=3), New Brunswick (4%, n=3), The Northwest Territories (4%, n=3), Saskatchewan (2%, n=2), Newfoundland and Labrador (2%, n=2), and Prince Edward Island (1%, n=1). There were no survey respondents from Yukon or Nunavut. The two patients who were interviewed by phone were in New Brunswick.
Survey Demographics
Eighty-one of the English survey participants (94%, n=81) identified as living with psoriasis, while five participants (6%, n=5) identified as being a caregiver or family member of a person with psoriasis. Patients were well-distributed across most adult age categories, including >65 years old (26%, n=15), 55-64 years old (21%, n=12), 25-34 years old (19%, n=11), 35-44 years old (16%, n=9), and 45-54 years old (16%, n=9). A smaller proportion of respondents were under 18 (3%, n=2). There were no individuals in the 18-24 age group. Of those who shared their gender (n=57), 79.1% were female (n=45). The most common comorbidities were psoriatic arthritis (22%, n=10) and mental health conditions (20%, n=9) such as depression or anxiety. Of all respondents, 37% (n=23) had a private drug plan, 32% (n=20) used a government drug plan, and 27% (n=17) paid out of pocket; 3% (n=2) were unsure.
Disease Experience
Psoriasis can have significant impacts on several facets of patients’ lives, which were affirmed in the responses to the survey. The majority had diagnosis of psoriasis for more than 20 years (44%, n=27) or 15-20 years (18%, n=11), with 52% (n=30) reporting moderate disease, 26% (n=15) reporting mild, and 22% (n=13) reporting severe psoriasis. The most impacted body areas were the scalp (68%, n=39), legs (65%, n=37), arms (60%, n=34), genitals (40%, n=23), hands (35%, n=20), torso (30%, n=17), skin folds (26%, n=15), and palms (25%, n=14). Symptoms of itching/burning were reported in 22% (n=13), flaking in 17% (n=10), flares in 15% (n=9) and skin pigmentation changes (8.6%, n=6) of participants. The severity of psoriasis symptoms had significant impacts on several aspects of patients’ lives, including mental health (24%, n=14), daily activities (17%, n=10), intimate relationships (13%, n=8), and their social lives (12%, n=7).
When asked to provide any additional information about the challenges of living with psoriasis, participants shared that their disease is “expensive and stressful,” and that it “takes a toll on mental capacity to deal with the physical pain and skin rashes [that] others do not understand.” Participants report that “at my worst, I am unable to work, walk, care for myself,” and “when my fingers flare up, I cannot manage personal hygiene.” Among survey respondents, 31% (n=18) reported impacts on their caregiver or family members. There is significant impact on relationships reported as well: “[my] spouse is constantly vacuuming and is becoming more frustrated over the years,” which may be the result of their skin shedding in flakes, and “I’m always in pain, grumpy, my husband can’t be intimate with me.”
Patient caregivers that also took the survey describe “kids wondering why he’s scratching, thinking maybe he has lice due to scalp psoriasis.” A mother of a teenager with psoriasis notes, “in my daughter's case her psoriasis was mainly on her face. This was especially difficult for her because it's an area that cannot be covered up. As a young teen, this was extremely hard on her self- confidence.” In particular, caregivers noted that they had difficulty encouraging the patient to use their treatments for their psoriasis.
Experiences With Currently Available Treatments
A large proportion of the survey respondents are currently using topical corticosteroids (36%, n=22), topical combination treatment (e.g., Dovobet, Enstilar, etc.) (32%, n=20), and/or biologic agents (27%, n=17). Biologics were reported to be the most effective of all current treatments used by respondents, with 39% reporting biologics work well and 30% reporting biologics work very well. Despite such effectiveness, for 17% of respondents, biologics did not work well and for 13% they did not result in any change to their psoriasis. While over 30% of respondents use topical agents, only 32% report that topical corticosteroids worked well or very well, and 38% report topical combination therapies work well or very well. Other medications currently used by respondents include medical cannabis (5%, n=3), topical retinoids (3%, n=2), oral retinoids (3%, n=2), apremilast (3%, n=2), oral steroids (2%, n=1), phototherapy (2%, n=1) and other therapies (26%, n=16). Table 1 below details patients’ experiences with psoriasis treatments they have tried in the past.
Some medications available to treat psoriasis, such as methotrexate (10%, n=6), were cheaper but not well tolerated by survey participants: “Methotrexate was awful, significant hair loss, upset stomach but not expensive. Can’t believe it is still being used.”
Unfortunately, 53% (n=33) of respondents’ report experiencing side effects with currently available treatments for psoriasis. An overwhelming proportion of patients have stopped a treatment for psoriasis at some point during their disease (76%, n=47), with most common reasons being that it stopped being effective for their psoriasis (61%, n=37), side effects (47%, n=28), financial challenges (22%, n=13), and ineffectiveness (14%, n=7). Affordability of medications for psoriasis has been a long-standing challenge: “As a newcomer is challenging to access biomedicals due to the high prices of the shots.” Given such challenges with currently available treatments for psoriasis, only 35% of survey respondents are satisfied with their current treatment and 69% would be interested in new treatments for psoriasis. A significant barrier to accessing treatments for psoriasis is related to financial challenges (26%, n=16). Nearly three-quarters of respondents (72%, n=44) responded that they wished there was a better treatment for psoriasis.
Table 1: Patient Experience With Past Treatments for Psoriasis
Past Treatment | Did not work at all | Did not work well | No change | Worked well | Worked very well |
|---|---|---|---|---|---|
Topical corticosteroids (e.g., betamethasone, mometasone) n (%) | 7/50 (14.0%) | 24/50 (48.0%) | 3/50 (6.0%) | 15/50 (30.0%) | 1/50 (2.0%) |
Topical Vitamin D Derivatives (eg. Dovonex, Silkis), n (%) | 7/33 (21.2%) | 14/33 (42.4%) | 8/33 (24.2%) | 3/33 (9.1%) | 1/33 (3.0%) |
Topical Combination Treatment (eg. Dovobet, Enstilar), n (%) | 4/39 (10.3%) | 14/39 (35.9%) | 6/39 (15.4%) | 11/39 (28.2%) | 4/39 (10.3%) |
Topical Retinoids (e.g., Tazorac), n (%) | 3/15 (20.0%) | 6/15 (40.0%) | 4/15 (26.7%) | 0/15 (0.0%) | 2/15 (13.3%) |
Apremilast, n (%) | 1/10 (10.0%) | 4/10 (40.0%) | 3/10 (30.0%) | 0/10 (0.0%) | 2/10 (20.0%) |
Cyclosporine, n (%) | 2/9 (22.2%) | 1/9 (11.1%) | 5/9 (55.6%) | 0/9 (0.0%) | 1/9 (11.1%) |
Methotrexate, n (%) | 4/24 (16.7%) | 7/24 (29.2%) | 4/24 (16.7%) | 7/24 (29.2%) | 2/24 (8.3%) |
Oral Retinoids, n (%) | 1/10 (10.0%) | 2/10 (20.0%) | 5/10 (50.0%) | 1/10 (10.0%) | 1/10 (10.0%) |
Oral Steroids, n (%) | 1/13 (7.7%) | 3/13 (23.1%) | 5/13 (38.5%) | 3/13 (23.1%) | 1/13 (7.7%) |
Biologics (e.g., adalimumab, certolizumab pegol, infliximab), n (%) | 0/23 (0.0%) | 4/23 (17.4%) | 3/23 (13.0%) | 9/23 (39.1%) | 7/23 (30.4%) |
Phototherapy, n (%) | 2/23 (8.7%) | 5/23 (21.7%) | 5/23 (21.7%) | 6/23 (26.1%) | 5/23 (21.7%) |
Medical Cannabis, n (%) | 3/13 (23.1%) | 1/13 (7.7%) | 4/13 (30.8%) | 3/13 (23.1%) | 2/13 (15.4%) |
Improved Outcomes
Effectiveness (90%, n=56), lack of side effects (66%, n=41), affordability (60%, n=37), ease of application (53%, n=33) and medications that were conducive to their schedule (23%, n=14) were the key aspects of new treatment for psoriasis identified by survey respondents). Some patients reported that they would not be willing to tolerate headaches, burning sensations in the skin, mental disturbances or fatigue as side effects of medications for psoriasis.
Mental health, ability to conduct daily activities and improved intimacy with loved ones are substantial factors that could be improved with new treatments. Targeting publicly visible body areas in day-to-day life could play a significant role in mental health, while treatments targeting genital regions could improve intimacy. Improving symptoms, such as itch and burning, would be a major factor that could help psoriasis patients resume their normal daily activities.
Experience With Drug Under Review
In our participant cohort, n=10 individuals reported having used roflumilast before, all of whom acquired the treatment through participation in a clinical trial. Nine participants (90% of these respondents) reported noticeable benefits while using roflumilast, which included significant clearing of skin, reduced itch and redness, and clearing of skin lesions (plaques). Moreover, participants noted the treatment’s ease of application and flexibility of being able to apply it to all affected body surface areas. Survey participants provided very positive feedback about roflumilast use: “Roflumilast really helped me and was the first medication I used that felt like it would properly treat my psoriasis. It was easy, convent [sic] and stress free.” Caregivers have also provided positive feedback: “Roflumilast has been life-changing for my daughter. No more cracked, bleeding skin on her face...we're so thankful!”
Roflumilast was tolerated well by all ten individuals, with only one reporting some itching with application of the topical. One of the survey participants reported they would tolerate “almost anything as it (roflumilast) worked wonders.” Another respondent reported that “being invited to join the clinical trial was a blessing in disguise.”
In addition to survey respondents, two people in New Brunswick who had used roflumilast in a clinical trial were interviewed by phone. Both individuals experience moderate-to-severe psoriasis all over their bodies. Both had used various topicals in the past that didn’t clear their skin entirely. Both also had issues accessing phototherapy:
One said that the phototherapy clinic is 60 miles each way and that he had to attend three times per week. He did it for a while and it worked somewhat but it became unaffordable. He also describes being “lucky” because his employer would let him leave for his appointments and make up time at the end of the day. [Please note that this is not always the case for workers.]
The other patient said that he never tried phototherapy because of the time commitment required – he would have had to go to appointments 4-5 days per week and he has elderly parents who he cares for and could not afford that amount of time.
Following the clinical trial for roflumilast, one individual is currently on methotrexate and uses a topical corticosteroid and the other is on a clinical trial for a biologic. The person taking methotrexate has significant apprehensions about this drug. He described it as “taking 6 pills of poison on Sunday that are killing me and spending the rest of the week taking antidotes” (e.g., folic acid). He also talked about the costs associated with the monthly blood work required for the methotrexate treatment – he currently spends $23 for the work up at a clinic because the local hospital had stopped routine blood work due to COVID. He said he will try to resume blood work for free at the hospital but that it is still time-consuming because of the wait at the hospital each time. [Please note that some jurisdictions like NB do not have private laboratories for routine testing and patients need to visit hospital to have their blood taken for testing or pay for this to be done elsewhere and sent to the hospital laboratory for testing.]
Both individuals spoke significantly about the costs of treatments and fears that they won’t be able to afford safe, effective treatments that work for them. One of the patients described challenges with maintaining his previous psoriasis topical treatments because “a tube the size of a lipstick” cost $100. He said he’s looked into the cost of roflumilast in the US and it’s $800 a tube which he would not be able to afford even though it’s the best treatment he has used. In addition, he raised questions about whether family doctors will be able to prescribe roflumilast because it’s difficult to get timely appointments with a dermatologist and he thinks this could be more efficiently managed in primary care.
Both individuals shared that roflumilast cleared their skin entirely in within 1-2 months, with one calling it “magic”. Neither experienced side effects nor had any other negative feedback about the therapy. Both said that though it took time to put on at first (though neither minded this because it was so effective), maintenance was very easy once the skin was cleared (just dabbing on any small areas as needed). One person said that he was "really disappointed” when the clinical trial for roflumilast ended. This time of year is usually worst for him (drier air + less sun) and now that he’s on a new trial, he doesn’t know if this treatment will continue to work for him. Both talked about their concerns regarding safety and risk of treatments with one indicating that they felt safe with roflumilast because this type of treatment has been used to treat COPD and has been used in children.
Companion Diagnostic Test
Not Applicable.
Anything Else?
Psoriasis is a chronic and potentially debilitating disease that poses many challenges, including high prevalence, chronicity, disfiguration, disability, and associated comorbidities. Psoriasis is linked to anxiety, depression, and social isolation, and can interfere with social and intimate relationships, productivity, family life and work life. The physical, psychological, social, and economic impact of psoriasis can significantly burden patients and their families. Access to effective care and appropriate treatment is needed.
Unfortunately, management of psoriasis can be complex with varied patient response to treatments, differences in social determinants of health, lifestyle considerations, and other factors that affect one’s condition. Moreover, due to the chronicity of this disease, patients are concerned about recurrence and resistance in the future to therapies that might be working for them today.
Psoriasis is more than a skin condition. It is an inflammatory disease that can impact several organ systems. It is estimated that up to 30 percent of people with psoriasis develop psoriatic arthritis. People with psoriatic disease also are at greater risk of developing cardiovascular disease, depression and anxiety, diabetes, and cancer.
For more information about the challenges of living with psoriasis, please refer to the following resources:
CPN and CAPP’s joint report, Journey to Stability
CAPP’s report Pso Serious 2018: A Report on Access to Care and Treatment for Psoriasis Patients in Canada
CAPP’s PsoIntimate awareness campaign
CPN and CAPP’s joint infographic on Impact of COVID-19 on the Psoriasis and Psoriatic Community in Canada – Highlights from a National Survey
CPN’s fact sheet for health care providers on Women and Psoriasis: Findings from a Survey of Women-identified People with Psoriatic Disease
Conflict of Interest Declaration — Canadian Skin Patient Alliance, Canadian Association of Psoriasis Patients, and the Canadian Psoriasis Network
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
CSPA worked with three medical student volunteers to compile the survey, analyze the survey results, and draft this submission. The authors of the submission reviewed and edited the submission prior to submitting it to CADTH.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
CSPA worked with three medical student volunteers to compile the survey, analyze the survey results, and draft this submission. CSPA, CAPP and CPN asked Arcutis (the manufacturer) for contact information of the Canadian clinical trial site principal investigators and sent these principal investigators a form letter to let them know about the survey and request that they share the survey with patients who participated in the clinical trial at their site.
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
Table 2: Financial Disclosures for Canadian Skin Patient Alliance
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie Canada | — | — | — | X |
Bausch Health | — | — | X | — |
Bristol Myers Squibb | — | — | X | — |
Boehringer Ingelheim | — | — | X | — |
Janssen Canada | — | — | X | — |
LEO Pharma Canada | — | — | X | — |
Novartis Canada | — | — | X | — |
Pfizer | — | — | X | — |
Table 3: Financial Disclosures for Canadian Association of Psoriasis
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie Canada | — | — | X | — |
Amgen Canada | — | — | X | — |
Arcutis | — | X | — | — |
Bausch Health | — | X | — | — |
Bristol Myers Squibb | X | — | — | — |
Boehringer Ingelheim | — | — | X | — |
Janssen Canada | — | X | — | — |
LEO Pharma Canada | — | — | X | — |
Novartis Canada | — | — | X | — |
Pfizer | — | — | X | — |
Sun Pharma | — | — | X | — |
UCB Canada | — | — | X | — |
Table 4: Financial Disclosures for Canadian Psoriasis Network
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie Canada | — | — | — | X |
Amgen Canada | — | — | X | — |
Arcutis | — | — | X | — |
Bausch Health | — | X | — | — |
Bristol Myers Squibb | — | — | X | — |
Boehringer Ingelheim Canada | — | — | X | — |
Boehringer Ingelheim International | X | — | — | — |
Janssen Canada | — | — | X | — |
LEO Pharma Canada | — | X | — | |
Novartis Canada | — | — | X | — |
Pfizer | — | — | X | — |
Pierre Fabre | — | — | X | — |
Sun Pharma | — | — | X | — |
UCB Canada | — | — | X | — |
Clinician Input
Fraser Health Dermatology Group
About Fraser Health Dermatology Group
Fraser Health dermatology group – we are a group of community dermatologist, some of which also work in part time in tertiary care hospitals. We all also teach medical students and residents.
Information Gathering
We reviewed the clinical trial data for the medication and other topicals for psoriasis. As well some of the groups were involved in the clinical trials for topical roflumilast.
Current Treatments and Treatment Goals
Current first line therapy is topical steroids, low potency for the face, armpits, and groin, and high potency for the rest of the body and scalp. Typically failing topical steroids alone on the body, the next line is topical vitamin D analog/steroid combinations (Dovobet, or Enstilar), or vitamin A analogs/steroid combinations (duborii). Failing second line therapy the patient would be treated with phototherapy (limited access) or systemic therapy. For the face, armpits and groin failing low potency steroids, often off label topical tacrolimus (protopic 0.1% ointment) would be used next. Although on label, monotherapy vitamin D analogs alone are not used particularly often due to significant irritation and clinically efficacy not living up to those in clinical trials. Phototherapy is not used in the groin (due to concerns regarding inducing skin cancer) but could be tried as a third line for the face and axillae. Finally, again for these areas the next step would be to consider systemic therapy. The current treatments act as anti-inflammatories and anti-proliferatives to address the mechanism of action of the disease. They also can help with the itch. The current treatment goals include reducing the severity of symptoms, minimize adverse advents (high potency steroids are a risk for causing atrophy and rapidly cause atrophy when used on the face or in the body folds). As well the other goals of treat are to help patients maintain employment, especially when it is on the hands and inhibit dexterity and causes significant pain with fissuring. Psoriasis on the genitals significantly impacts sexually functioning. Body psoriasis can be itchy, disrupt sleep, cause pain, and inhibit psychosocial functioning due to stigma.
Regarding goals for an ideal treatment, the ideal treatment is one treatment that is effective for all areas, without risk or atrophy, and causes minimal irritation.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
In terms of treatment gaps, there are many patients that do not respond to currently available topical therapies and end up needing to move on to phototherapy and systemic therapies which carry significant burdens to the patient and significant costs to the health care systems. Phototherapy involves coming to a doctor’s office three times per week which can be very disruptive to patients in terms of missed work. Traditional systemic therapies (methotrexate and cyclosporin) need frequent blood work monitoring, and cyclosporine and biologic therapies carry significant costs to the health care system. Regarding topical therapies, post topical steroids some patients cannot tolerate the vitamin A/steroid combination (duborii) due to irritation. In terms of Vitamin D/steroid combos dovobet gel has limited efficacy, and dovobet ointment/Enstilar foam have issues of patient compliance due to the vehicles thickness/greasy texture. On the face and folds, post mild steroids, protopic is used off label but we do not have good data for its efficacy in psoriasis. As well it can sting in a significant minority of patients, and the ointment base limits it use in patients with facial or pubic hair. Lastly many patients are given two different topicals one for the face, folds, groin/genitals, and another for the other areas of the body. Patient compliance decreases when more than one topical therapy is prescribed.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
We would see topical roflumilast being used post failure of topical steroids as topical steroids are an inexpensive, and usually well tolerated therapy. We recommend considering this medication before the current combination therapies as they are limited in, they cannot be used both on the body, as well as on the face, armpits, and groin. As well this medication should be better tolerated than duobrii and have a vehicle that leads to better patient compliance compared to dovobet ointment, and Enstilar foam. Compliance should also be higher from being able to use one medication in all areas and this will eliminate the risk of patients accidentally applying their high potency steroid or combination therapy (contain high potency steroids) to the face, armpits and groin. This should stop the risk of permanent atrophy from the steroids, as well as the risks of steroid induced rosacea, periorificial dermatitis, and addicted steroid syndrome, which then need medical visits and treatments.
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
This medication is best suited for patients with psoriasis who have failed topical steroids. Patients would be identified based on clinical exam. Regarding misdiagnosis the most common misdiagnoses would be atopic dermatitis and seborrheic dermatitis, and this medication has been shown to be effective in clinical trials for those conditions as well.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
The outcome to determine response would be a physician global assessment and body surface area involvement. The pasi score used in trials is not commonly used in clinical practice except when applying for coverage for systemic therapies in moderate to severe patients. For patients with more limited body surface area, the PASI score becomes a less sensitive outcome measure, so I would recommend against the PASI score. Psoriasis patients are typically initially seen every 3-6 months to assess response.
What factors should be considered when deciding to discontinue treatment with the drug under review?
Lack of efficacy after a trial of at least 3 months.
What settings are appropriate for treatment with [drug under review]? Is a specialist required to diagnose, treat, and monitor patients who might receive [drug under review]?
Psoriasis patients are diagnosed and treated as outpatients topically by both specialists and general practitioners/family doctors.
Conflict of Interest Declarations — Fraser Health Dermatology Group
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please refer to the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission?
No.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
Declaration for Clinician 1
Name: Gurbir Dhadwal
Position: Dermatologist
Date: February 27, 2022
Table 5: COI Declaration for Fraser Health Dermatology Group — Clinician 1
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Abbvie | — | — | X | — |
Arcutis | X | — | — | — |
Amgen | X | — | — | — |
Bausch | — | X | — | — |
Boehringer Ingelheim | X | — | — | — |
Bristol Meyers Squibb | X | — | — | — |
Eli-Lilly | — | X | — | — |
Galderma | X | — | — | — |
Janssen | X | — | — | — |
Johnson & Johnson | X | — | — | — |
Leo Pharma | X | — | — | — |
Novartis | X | — | — | — |
Pfizer | X | — | — | — |
Sun Pharma | X | — | — | — |
UCB Pharma | — | X | — | — |
Declaration for Clinician 2
Name: Se Mang Wong
Position: Dermatologist
Date: February 27, 2022
Table 6: COI Declaration for Fraser Health Dermatology Group — Clinician 2
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Abbvie | X | — | — | — |
Amgen | X | — | — | — |
Bausch | — | X | — | — |
Boehringer Ingelheim | X | — | — | — |
Bristol Meyers Squibb | X | — | — | — |
Eli-Lilly | X | — | — | — |
Galderma | X | — | — | — |
Janssen | X | — | — | — |
Johnson & Johnson | X | — | — | — |
Leo Pharma | X | — | — | — |
Novartis | — | X | — | — |
Pfizer | X | — | — | — |
Sun Pharma | X | — | — | — |
UCB Pharma | X | — | — | — |
Declaration for Clinician 3
Name: Aaron Wong
Position: Dermatologist, Clinical Associate Professor, UBC; MEDD 422 Course Director, Faculty of Medicine (UBC)
Date: February 27, 2023
Table 7: COI Declaration for Fraser Health Dermatology Group — Clinician 3
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Abbvie | X | — | — | — |
Janssen | X | — | — | — |
Galderma | X | — | — | — |
Eli Lilly | X | — | — | — |
Novartis | X | — | — | — |
UCB | X | — | — | — |
Amgen | X | — | — | — |
Bausch Health | X | — | — | — |
Sun Pharma | X | — | — | — |
J&J | X | — | — | — |
Declaration for Clinician 4
Name: Gordon Jung, MD, FRCPC, FAAD
Position: Clinical Instructor, Department of Dermatology & Skin Science, University of British Columbia
Date: February 27, 2023
Table 8: COI Declaration for Fraser Health Dermatology Group — Clinician 4
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Atlantic Provinces Dermatology Association and Dermatology Association of Ontario
About Atlantic Provinces Dermatology Association and Dermatology Association of Ontario
Atlantic Provinces Dermatology Association and Dermatology Association of Ontario are physician organizations that include dermatology-certified specialists practicing general dermatology with vast experience managing various skin conditions including psoriasis. These groups previously provided clinician group input for a review on the treatment of moderate-to-severe psoriasis and moderate-to- severe atopic dermatitis. Several members are engaged in clinical research and have extensive experience in clinical trials including the phase II and registrational trials for topical roflumilast.
References
Papp KA, Gooderham M, Droege M, Merritt C, Osborne DW, Berk DR, Thurston AW, Smith VH, Welgus H. Roflumilast Cream Improves Signs and Symptoms of Plaque Psoriasis: Results from a Phase 1/2a Randomized, Controlled Study. J Drugs Dermatol. 2020 Aug 1;19(8):734-740. doi: 10.36849/JDD.2020.5370. PMID: 32845114.
Lebwohl MG, Kircik LH, Moore AY, et al. Effect of Roflumilast Cream vs Vehicle Cream on Chronic Plaque Psoriasis: The DERMIS-1 and DERMIS-2 Randomized Clinical Trials. JAMA. 2022;328(11):1073–1084. doi:10.1001/jama.2022.15632
Lebwohl MG, Papp KA, Stein Gold L, Gooderham MJ, Kircik LH, Draelos ZD, Kempers SE, Zirwas M, Smith K, Osborne DW, Trotman ML, Navale L, Merritt C, Berk DR, Welgus H; ARQ-151 201 Study Investigators. Trial of Roflumilast Cream for Chronic Plaque Psoriasis. N Engl J Med. 2020 Jul 16;383(3):229-239. doi: 10.1056/NEJMoa2000073. PMID: 32668113.
https://www.arcutis.com/wp- content/uploads/aad22_roflumilast_plaque_psoriasis_interim_results.pdf
https://www.arcutis.com/wp-content/uploads/2021/05/Roflumilast-cream-Study-301-302- EADV-presentation_FINAL.pdf
https://www.arcutis.com/wp-content/uploads/2021/03/AU001-20-InnovationsDerm21-Study- 202-LTS-ePoster_MT05.sw-cm_Final.pdf
Information Gathering
Group members gathered relevant scientific and clinical information which was further compiled and circulated for comments and discussion among the larger membership. All members had an opportunity to provide their feedback and opinions. The final document includes group clinician input.
Current Treatments and Treatment Goals
Chronic plaque psoriasis is an immune-mediated, inflammatory skin diseases that varies in severity affecting over 2% - 5% of Canadians. The majority of patients present with mild-to-moderate disease that is often defined by objective clinical measures such as affected body surface area (BSA) and psoriasis area severity index (PASI). It than the BSA number or the PASI score, as evidenced by decreased quality of life (QoL) and higher risks for depression, anxiety and suicide. Although topical corticosteroid (TCS) preparations remain the foundation of treatment and are used across the spectrum of plaque psoriasis, there has been a paucity of clinical innovation in comparison to the advances in biologic therapies made in the past decade.
Despite good treatment responses with TCS preparations, they can be limited for long-term use due to local (e.g., bruising, skin atrophy, and telangiectasia) or systemic (e.g., hypothalamic-pituitary-adrenal [HPA] axis suppression) adverse events (AEs). Topical retinoids and vitamin D analogues, originally introduced in the latter part of the 20th century, have AEs of skin irritation as a common cause for a patients’ dissatisfaction and/or lack of adherence. The only currently available topical phosphodiesterase- 4 inhibitor in Canada, crisaborole and topical calcineurin inhibitors (TCIs), are indicated for atopic dermatitis but have been used for psoriasis with some success in the sensitive intertriginous and facial areas. However, these treatments are also associated with local tolerability concerns such as burning and stinging, limiting their use. There is limited access for these latter topical therapies due to the lack of regulatory approval for psoriasis and limited access depending on patients’ private insurance coverage.
Psoriasis is a heterogeneous disease with varied clinical presentations, with almost 90% of patients experiencing psoriasis in more than one location. Due to the limitations of available topical armamentarium, dermatologists often prescribe 2 or 3 different topicals of varying potencies and/or formulations during one patient visit. For example, the same patient in one visit often prescribed mid- to-high potency TCS lotion for the scalp, high-potency TCS cream for the thicker plaques on the limbs, and a low-potency TCS or TCI for the face and folds. This can be confusing and cumbersome for patients where they have to use different products at the same time. In addition, this multi-topical treatment paradigm often leads to prescriber fatigue and confusion where non-dermatology specialists prescribe either low or high-potency TCS for all affected areas that either associated with poor efficacy or higher rates of AEs.
UPLIFT survey conducted in 2020 included 264,054 patient responders, including patients from Canada, identified reduction in itch as the most important treatment goal. In the same survey, 75% of patients reported current topical treatments burdensome.
As such, there remains a large unmet need to develop a simplified topical treatment regimen that would incorporate a fast improvement in pruritus, high efficacy, improved tolerability and safety, in one topical formulation that can be used on all and anybody location affected with psoriasis.
References
Papp, K.A., Gniadecki, R., Beecker, J. et al. Psoriasis Prevalence and Severity by Expert Elicitation. Dermatol Ther (Heidelb) 11, 1053–1064 (2021). https://doi.org/10.1007/s13555-021-00518-8
Stern RS, Nijsten T, Feldman SR, Margolis DJ, Rolstad T. Psoriasis is common, carries a substantial burden even when not extensive, and is associated with widespread treatment dissatisfaction. J Investig Dermatol Symp Proc. 2004 Mar;9(2):136-9. doi: 10.1046/j.1087- 0024.2003.09102.x. PMID: 15083780.
Reid C, Griffiths CEM. Psoriasis and Treatment: Past, Present and Future Aspects. Acta Derm Venereol. 2020 Jan 30;100(3):adv00032. doi: 10.2340/00015555-3386. PMID: 31971601; PMCID: PMC9128930.
Lebwohl, M., Langley, R.G., Paul, C. et al. Evolution of Patient Perceptions of Psoriatic Disease: Results from the Understanding Psoriatic Disease Leveraging Insights for Treatment (UPLIFT) Survey. Dermatol Ther (Heidelb) 12, 61–78 (2022). https://doi.org/10.1007/s13555-021-00635-4
Armstrong, A., Edson-Heredia, E., Zhu, B. et al. Treatment Goals for Psoriasis as Measured by Patient Benefit Index: Results of a National Psoriasis Foundation Survey.Adv Ther 39, 2657–2667 (2022). https://doi.org/10.1007/s12325-022-02124-2
Armstrong AW, Read C. Pathophysiology, Clinical Presentation, and Treatment of Psoriasis: A Review. JAMA. 2020 May 19;323(19):1945-1960. doi: 10.1001/jama.2020.4006. PMID: 32427307.
Canadian Psoriasis Guidelines Addendum Committee. 2016 Addendum to the Canadian Guidelines for the Management of Plaque Psoriasis 2009. J Cutan Med Surg. 2016 Sep;20(5):375-431. doi: 10.1177/1203475416655705. Epub 2016 Jul 15. PMID: 27421294; PMCID: PMC5014087.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
Psoriasis is a chronic disease that can have significant impact on patients’ QoL. The majority of patients with psoriasis have limited disease that can be treated with topical therapy. Despite the availability of effective topical psoriasis therapies, adherence to treatment remains low. Rates of adherence generally diminish with longer duration of treatment. A survey of 3822 adult patients with psoriasis conducted in Canada, Europe and United States had explored patient expectations and unmet needs in current psoriasis therapy. Eighty percent of patients were eager to try new treatments and 23% of responders agreed with the statement that psoriasis treatments never work. In this study, efficacy, tolerability, convenience and affordability were the main factors affecting adherence to psoriasis treatment and remain unmet needs in psoriasis therapeutic regimens. Similar conclusions were reached in the UPLIFT survey.
The objectives of all therapeutic interventions are appropriate safety, high tolerability, and effective disease control. When considering topical therapies, the objectives are extended to include safety and tolerability appropriate for long-term use, a simplified treatment regimen, and carrier properties than enhance adherence.
Efficacy
Current psoriasis topical regimens include TCS, vitamin D analogues, topical retinoids, and combination therapies. For decades, there has been no true topical innovation and new products represented combination compounds with high-potency TCS, vitamin D analogue and retinoids. There remains an unmet need for topical formulations with new mechanism of action and high efficacy. Current formulations are generally recommended/ restricted to use on a particular body area. For instance, low- potency topical steroids can be used on any body area but are generally not effective for thick psoriatic plaques on the elbows and knees. Although, high-potency topical steroids are effective on the thick psoriatic plaques, this class of agents would not be a suitable treatment for the facial or intertriginous psoriasis. As such, there is an unmet need for a topical formulation with high efficacy that could be effectively utilized on any body surface area, such as thick plaques on the knees and elbows and thin plaques in the body folds.
Pruritus is the most bothersome symptom for patients with psoriasis. High-potency topical steroids are generally helpful to improve this symptom, unlike vitamin D analogues and topical retinoids that do not address this treatment need. There is an unmet need for the new topical formulation to improve pruritus.
Safety and tolerability
TCS formulations represent most commonly utilized topical treatments to manage psoriasis. Unfortunately, adverse events of topical steroids include local effects and systemic side effects. Local adverse events include skin atrophy (telangiectasia, striae, bruising), hypopigmentation, cutaneous infections, and acneiform eruptions. These events are particularly seen with higher potency TCS preparations. As such, efficacy of topical steroids is often proportional to their potency and in turn, their undesirable local effects. This is of particular concern in patients utilizing these high potency preparations on the face, genital area and body folds. Systemic side effects are generally seen in patients utilizing these treatments on a large body surface area and include HPA axis suppression and immunosuppression. Tachyphylaxis (a progressive decrease in efficacy to a given preparation) is commonly seen with TCS preparations used long-term. All of the above limits TCS use to PRN or short- term. There is a need for topical preparations that do not have TCS-associated AEs and can be used safely long-term.
Although vitamin D analogues and retinoids do not have TCS AE profile, their use is generally limited due to lower efficacy and local irritation (burning, stinging, erythema, desquamation). In addition to high efficacy, new topical preparations should have improved tolerability so they can be safely used on anybody surface affected with psoriasis including face and body folds without irritation.
Simplified treatment regimen
Plaque psoriasis has heterogenous clinical presentation with often thick plaques on the elbows and knees and thin irritated plaques in the body folds. In addition, different body areas have different topical absorption (skin folds and genitals have higher absorption, palms and soles have reduced topical absorption) and tolerability (facial skin is more easily irritated than skin on the knees and elbows).
Ideally, new topical preparation could be utilized on any surface area without concerns of absorption/penetration, with equal safety/tolerability and efficacy.
High quality non-greasy preparation
Treatment adherence is complex, with multiple contributing factors. It often incorporates disease- related factors, treatment-related factors and patient-related factors. In clinical practice, patients often report dissatisfaction with greasy preparations and prefer to use preparations that are associated with high tolerability and can be easily spread on the skin. “Steroid-phobia” is also a concern reported by patients, limiting their use of topical therapy.
References
Feldman SR. Disease burden and treatment adherence in psoriasis patients. Cutis. 2013 Nov;92(5):258-263. PMID: 24343214.
Lebwohl, M., Langley, R.G., Paul, C. et al. Evolution of Patient Perceptions of Psoriatic Disease: Results from the Understanding Psoriatic Disease Leveraging Insights for Treatment (UPLIFT) Survey. Dermatol Ther (Heidelb) 12, 61–78 (2022). https://doi.org/10.1007/s13555-021-00635-4
Pinter, A, van de Kerkhof, P. The role of topical therapies along the psoriasis patient journey: An overview from the Symposium ‘Tailoring topical psoriasis treatments to patients' needs and expectations’ of the 30thEADV Congress 2021. J Eur Acad Dermatol Venereol. 2023; 37(Suppl. 1): 3– 8. https://doi.org/10.1111/jdv.18761
Armstrong A, Edson-Heredia E, Zhu B, Burge R, Bell S, Crowley JJ, Smith S. Treatment Goals for Psoriasis as Measured by Patient Benefit Index: Results of a National Psoriasis Foundation Survey. Adv Ther. 2022 Jun;39(6):2657-2667. doi: 10.1007/s12325-022-02124-2. Epub 2022 Apr 11. PMID: 35399114; PMCID: PMC9122869.
Choi JW, Kim BR, Youn SW. Adherence to Topical Therapies for the Treatment of Psoriasis: Surveys of Physicians and Patients. Ann Dermatol. 2017 Oct;29(5):559-564. doi: 10.5021/ad.2017.29.5.559. Epub 2017 Aug 25. PMID: 28966511; PMCID: PMC5597648.
Siegfried Segaert, Piergiacomo Calzavara-Pinton, Pablo de la Cueva, Ahmad Jalili, Dominique Lons Danic, Andrew E. Pink, Diamant Thaçi & Melinda Gooderham (2022) Long-term topical management of psoriasis: the road ahead, Journal of Dermatological Treatment, 33:1,111- 120, DOI: 10.1080/09546634.2020.1729335
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Roflumilast cream 0.3% is once-daily, non-steroidal, topical phosphodiestherase 4 (PDE-4) inhibitor in review for the treatment of plaque psoriasis.
PDE-4 inhibition is not new in psoriasis treatment. Apremilast (Otezla), systemic PD-4 inhibitor was approved for treatment of psoriasis in 2014. Oral roflumilast (Daxas) has been approved for the treatment of COPD in 2010.
The efficacy and safety of roflumilast cream was investigated in Dermis-1 and -2 clinical trials with significant Canadian contribution (19 Canadian sites and approximately 25% of patients in the phase 3 clinical program). These trials included patients with at least mild psoriasis and BSA 2-20%. The primary endpoint of IGA success (IGA 0/1 and >/= 2 grade IGA improvement from baseline) was achieved by 40% of roflumilast-treated patients at week 8 compared to 6.5% of patients in the vehicle arm. High efficacy was also reported in the intertriginous locations where about 60% of patients achieved clear skin at week 8 when treated with roflumilast (~9% vehicle group). PASI-75 improvement was achieved by ~40% of roflumilast-treated patients (6.5% vehicle group) and about 20% of patients had achieved PASI-90% improvement at week 8 (2.3% vehicle group). Most importantly, treatment with roflumilast cream improved patient QoL (Figure on page 9 in the presentation slides presented at the 30th EADV Congress 2021 available at this link here) and rapidly improved pruritus (Figure on page 6 in the presentation slides presented at the 30th EADV Congress 2021 available at this link here). Clinical improvements were maintained for 32 weeks in long-term extension trial (presented at the AAD annual meeting 2022) where at week 32, 54% of patients had clear or almost clear skin and 44.1% of patients had achieved PASI 75. Phase 2b long-term extension trial confirmed long-term efficacy with PRN roflumilast treatment (presented at Innovations in Dermatology conference 2021). Dermis trials revealed that topical roflumilast cream was highly effective on thick psoriatic plaques and in intertriginous sites.
Clinical trials revealed that treatment with roflumilast cream was associated low rates of treatment- emergent AEs and roflumilast cream was well tolerated. There were no treatment-related serious adverse events. Diarrhea was seen in about 3% of patients during the first 8 weeks of treatment. None of the patients discontinued the treatment due to this adverse event. The rates of diarrhea did not increase over time.
Clinicians of this group see this treatment as a game changer in a topical psoriasis management. Some of the clinicians of this group have had first-hand experience with this treatment in clinical trials and found it highly effective and well tolerated by patients. High clinical efficacy and a favorable safety profile of topical roflumilast would place this treatment first line for the management of mild-to-severe plaques on any body area. The clinical trial population included patients with psoriasis affecting 20% BSA. This topical treatment can be used prior to systemic or biologic treatment and as an adjunct to systemic or biologic therapy. In addition, extended indication to an adolescent population would extend the benefits of this treatment to a younger, more vulnerable patient population.
References
Lebwohl MG, Kircik LH, Moore AY, Stein Gold L, Draelos ZD, Gooderham MJ, Papp KA, Bagel J, Bhatia N, Del Rosso JQ, Ferris LK, Green LJ, Hebert AA, Jones T, Kempers SE, Pariser DM, Yamauchi PS, Zirwas M, Albrecht L, Devani AR, Lomaga M, Feng A, Snyder S, Burnett P, Higham RC, Berk DR. Effect of Roflumilast Cream vs Vehicle Cream on Chronic Plaque Psoriasis: The DERMIS-1 and DERMIS-2 Randomized Clinical Trials. JAMA. 2022 Sep 20;328(11):1073-1084. doi: 10.1001/jama.2022.15632. PMID: 36125472; PMCID: PMC9490499.
Lebwohl MG, Papp KA, Stein Gold L, Gooderham MJ, Kircik LH, Draelos ZD, Kempers SE, Zirwas M, Smith K, Osborne DW, Trotman ML, Navale L, Merritt C, Berk DR, Welgus H; ARQ-151 201 Study Investigators. Trial of Roflumilast Cream for Chronic Plaque Psoriasis. N Engl J Med. 2020 Jul 16;383(3):229-239. doi: 10.1056/NEJMoa2000073. PMID: 32668113.
Gold L, et al. Effect of Roflumilast Cream (ARQ-151) on Itch and Itch-related Sleep Loss in Adults with Chronic Plaque Psoriasis: Patient-reported Itch Outcomes of a Phase 2b Trial. Am J Clin Dermatol 2022. https://doi.org/10.1007/s40257-022-00739-3
Gooderham M, et al. Roflumilast Cream 0.3% For the Severity and Impact of Itch in Patients With Chronic Plaque Psoriasis in the Phase 3 DERMIS-1 and DERMIS-2 Studies, presented at EADV 2021
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
One of the advantages of topical roflumilast treatment is that it will likely meet the needs of psoriasis patients with different disease phenotypes. It is known to be highly effective on thick psoriatic plaques on the elbows and knees and thin intertriginous plaques in the folds. This treatment can be utilized on the face and thick palmar-plantar skin. In addition, it can be used in patients with various BSA as first line topical therapy or as an adjunct to systemic or biologic treatment.
Roflumilast cream would be impractical to use for patients with psoriasis affecting large BSA. Topical roflumilast use has not been studied in pregnant and breastfeeding patients.
Psoriasis differential diagnosis includes seborrheic dermatitis and atopic eczema. Topical roflumilast preparations have been studied in clinical trials for these indications (NCT04973228; NCT04773600) revealing efficacy superior to the vehicle arms and a favorable safety profile.
References
Lebwohl MG, Kircik LH, Moore AY, Stein Gold L, Draelos ZD, Gooderham MJ, Papp KA, Bagel J, Bhatia N, Del Rosso JQ, Ferris LK, Green LJ, Hebert AA, Jones T, Kempers SE, Pariser DM, Yamauchi PS, Zirwas M, Albrecht L, Devani AR, Lomaga M, Feng A, Snyder S, Burnett P, Higham RC, Berk DR. Effect of Roflumilast Cream vs Vehicle Cream on Chronic Plaque Psoriasis: The DERMIS-1 and DERMIS-2 Randomized Clinical Trials. JAMA. 2022 Sep 20;328(11):1073-1084. doi: 10.1001/jama.2022.15632. PMID: 36125472; PMCID: PMC9490499.
Lebwohl MG, Papp KA, Stein Gold L, Gooderham MJ, Kircik LH, Draelos ZD, Kempers SE, Zirwas M, Smith K, Osborne DW, Trotman ML, Navale L, Merritt C, Berk DR, Welgus H; ARQ-151 201 Study Investigators. Trial of Roflumilast Cream for Chronic Plaque Psoriasis. N Engl J Med. 2020 Jul 16;383(3):229-239. doi: 10.1056/NEJMoa2000073. PMID: 32668113.
Blauvelt A et al. Efficacy and safety of roflumilast foam 0.3% in patients with seborrheic dermatitis in a phase 3 trial, presented at EADV congress 2022.
Gooderham et al. The Safety and Efficacy of Roflumilast Cream 0.15% and 0.05% in Atopic Dermatitis: Phase 2 Proof-of-Concept Study, presented at EADV congress 2020.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
In clinical practice, response to topical treatment is determined using objective and subjective measures of improvement.
Objective measures of disease severity include physician global assessment (PGA), BSA measurement and PASI score. PASI measurements are rarely utilized in clinical practice with exception of when a clinician is applying for reimbursement for systemic or biologic therapy.
Changes from baseline PGA and BSA are often used as objective measures of treatment response.
Subjective disease improvement is often measured utilizing patient feedback or changes in DLQI. Similar to PASI scoring, DLQI measurements are often used in clinical trials and when clinician applies for systemic or biologic reimbursement, rarely utilized in routine clinical practice.
PASI and DLQI scoring are not utilized in primary care or by specialists with no dermatology expertise.
Clinically meaningful response to treatment would include objective improvement of disease severity as measured by PGA and/or BSA in combination with positive patient feedback.
Assessment of treatment response often varies among clinicians and depends on clinical practice wait times, availability of follow up in primary care and patient-specific factors. Many clinicians reassess treatment response in 3-6 months and then every 3-6 months or on a PRN basis. In general, patients with high disease severity, significant QoL impairment, anxious about AEs or at extremes of age are reassessed more frequently.
What factors should be considered when deciding to discontinue treatment with the drug under review?
Treatment discontinuation was uncommon in clinical trials with roflumilast. Roflumilast topical treatment should be placed on hold for patients with moderate-to-severe diarrhea until recovery and discontinued in patients with persistent moderate- to- severe diarrhea associated with the treatment, pregnant, and breastfeeding patients.
What settings are appropriate for treatment with [drug under review]? Is a specialist required to diagnose, treat, and monitor patients who might receive [drug under review]?
Topical roflumilast treatment is associated with high efficacy, and favorable adverse event and tolerability profiles. It can be used in primary care and specialty dermatology practice.
Additional Information
Topical roflumilast treatment presents a new, highly effective, safe, non-steroidal, once-daily preparation that would benefit many patients with various psoriasis phenotypes. Roflumilast has been shown to improve pruritus early in the course of treatment (2 weeks) and offer convenience of one product that can be used on anybody surface area without irritation. The clinician input includes clinical trial investigators with first-hand experience with this topical formulation, and many positive patient stories with numerous eager patients waiting for regulatory approval.
Conflict of Interest Declarations — Atlantic Provinces Dermatology Association and Dermatology Association of Ontario
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please refer to the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission?
No assistance from outside this clinician group was received to complete this submission.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission?
No help from outside this clinician group was received to collect or analyze any information used in this submission.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
Declaration for Clinician 1
Name: Irina Turchin
Position: Dermatologist, Fredericton, NB, Canada
Date: February 20, 2023
Table 9: COI Declaration for Atlantic Provinces Dermatology Association and Dermatology Association of Ontario — Clinician 1
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Arcutis | — | — | — | X |
Bausch Health | — | — | X | — |
Galderma | — | X | — | — |
Leo Pharma | — | — | — | X |
Pfizer | — | X | — | — |
Declaration for Clinician 2
Name: Kim Papp
Position: Dermatologist, Waterloo, ON
Date: February 20, 2023
Table 10: COI Declaration for Atlantic Provinces Dermatology Association and Dermatology Association of Ontario — Clinician 2
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Arcutis | — | — | — | X |
Bausch Health | — | — | X | — |
Galderma | — | — | X | — |
Leo Pharma | — | — | X | — |
Pfizer | — | X | — | — |
Declaration for Clinician 3
Name: Ronald Vender
Position: Dermatologist, Hamilton, ON; Director, Dermatrials Research Inc.
Date: February 20, 2023
Table 11: COI Declaration for Atlantic Provinces Dermatology Association and Dermatology Association of Ontario — Clinician 3
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Arcutis | — | X | — | — |
Bausch Health | — | — | X | — |
Galderma | X | — | — | — |
Leo Pharma | X | — | — | — |
Declaration for Clinician 4
Name: Marc Bourcier
Position: Dermatologist, Moncton, NB
Date: February 20, 2023
Table 12: COI Declaration for Atlantic Provinces Dermatology Association and Dermatology Association of Ontario — Clinician 4
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Bausch Health | — | — | X | — |
Leo Pharma | — | — | X | — |
Pfizer | — | X | — | — |
Declaration for Clinician 5
Name: Hermenio Lima
Position: Dermatologist, Hamilton, ON; Creator and Director of LEADER Research, Associate Clinical Professor, Department of Medicine, McMaster University,
Date: February 20, 2023
Table 13: COI Declaration for Atlantic Provinces Dermatology Association and Dermatology Association of Ontario — Clinician 5
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Abbvie | — | X | — | — |
Bausch Health | X | — | — | — |
BMS | X | — | — | — |
Leo Pharma | X | — | — | — |
Pfizer | X | — | — | — |
Sanofi | X | — | — | — |
Declaration for Clinician 6
Name: David Adam
Position: Dermatologist, Ajax, ON; President, Dermatology Association of Ontario
Date: February 20, 2023
Table 14: COI Declaration for Atlantic Provinces Dermatology Association and Dermatology Association of Ontario — Clinician 6
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Arcutis | — | X | — | — |
Bausch Health | — | X | — | — |
Galderma | X | — | — | — |
Leo Pharma | — | X | — | — |
Pfizer | — | X | — | — |
Declaration for Clinician 7
Name: Catherine Rodriguez
Position: Dermatologist, Queen Elizabeth Hospital, Charlottetown, PE
Date: February 20, 2023
Table 15: COI Declaration for Atlantic Provinces Dermatology Association and Dermatology Association of Ontario — Clinician 7
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Arcutis | X | — | — | — |
Bausch Health | X | — | — | — |
Leo Pharma | X | — | — | — |
Declaration for Clinician 8
Name: Jensen Young
Position: Dermatologist, Toronto, ON; Assistant professor, Department of Medicine, University of Toronto
Date: February 20, 2023
Table 16: COI Declaration for Atlantic Provinces Dermatology Association and Dermatology Association of Ontario — Clinician 8
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Arcutis | — | — | X | — |
Bausch Health | — | — | X | — |
Galderma | X | — | — | — |
Leo Pharma | — | X | — | — |
Pfizer | — | — | X | — |
Declaration for Clinician 9
Name: Ian Landells
Position: Dermatologist, St. Johns, NFLD
Date: February 20, 2023
Table 17: COI Declaration for Atlantic Provinces Dermatology Association and Dermatology Association of Ontario — Clinician 9
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Arcutis | X | — | — | — |
Bausch Health | — | X | — | — |
Galderma | X | — | — | — |
Leo Pharma | — | X | — | — |
Pfizer | X | — | — | — |
Declaration for Clinician 10
Name: Stacey Northgrave
Position: Dermatologist, Sydney, NS
Date: February 20, 2023
Table 18: COI Declaration for Atlantic Provinces Dermatology Association and Dermatology Association of Ontario — Clinician 10
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Arcutis | X | — | — | — |
Bausch Health | X | — | — | — |
Galderma | X | — | — | — |
Leo Pharma | X | — | — | — |
Pfizer | X | — | — | — |
Declaration for Clinician 11
Name: Lyne Giroux
Position: Dermatologist, Sudbury, ON
Date: February 20, 2023
Table 19: COI Declaration for Atlantic Provinces Dermatology Association and Dermatology Association of Ontario — Clinician 11
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Arcutis | X | — | — | — |
Bausch Health | X | — | — | — |
Galderma | X | — | — | — |
Leo Pharma | X | — | — | — |
Pfizer | X | — | — | — |
Declaration for Clinician 12
Name: Dusan Sajic
Position: Dermatologist, Guelph, ON
Date: February 20, 2023
Table 20: COI Declaration for Atlantic Provinces Dermatology Association and Dermatology Association of Ontario — Clinician 12
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Arcutis | X | — | — | — |
Bausch Health | — | — | — | X |
Galderma | — | — | X | — |
Leo Pharma | X | — | — | — |
Pfizer | — | — | X | — |
Canadian Dermatology Association
About Canadian Dermatology Association
The Canadian Dermatology Association, founded in 1925, is the national medical specialty association that represents Canadian certified dermatologists. The association exists to advance the science and art of medicine and surgery related to the care of the skin, hair and nails; provide continuing professional development for its members; support and advance patient care; provide public education on sun protection and other aspects of skin health; and promote a lifetime of healthier skin, hair and nails.
Clinical review and oversight are provided by the Canadian Dermatology Association’s Pharmacy and Therapeutics Advisory Board and the CDA Board of Directors.
Website: https://dermatology.ca/
Information Gathering
Information was gathered from clinical experience, medical literature, and published trials.
Current Treatments and Treatment Goals
Current treatments for psoriasis include topical therapies such as topical corticosteroids and combination topical products, as well as off-label use of non-corticosteroid topicals such as calcineurin inhibitors and phosphodiesterase 4 inhibitors (particularly for inverse psoriasis). Treatment then progresses to phototherapy, most commonly narrow-band UVB, and systemics. Off-label systemics for psoriasis commonly used include methotrexate and cyclosporine. Health Canada approved systemics for psoriasis include oral small molecules such as the phosphodiesterase 4 inhibitor apremilast, and biologics in the TNFa inhibitor, IL12/23, IL23 and IL17 inhibitor classes.
Treatment goals are to increase quality of life, decrease the affected body surface area and psoriasis area and severity score (PASI), and to reach an investigator global assessment of 0 or 1 (clear or almost clear).
As outlined in the 2016 addendum to the Canadian Psoriasis Guidelines1, Th17 cells are recognized to play a central role in the pathophysiology of psoriasis. Traditionally used treatments such as topical corticosteroids and systemics like methotrexate and cyclosporine take a less targeted approach, decreasing overall T cell function. Biologics take a more targeted approach towards the involved T cells and cytokines. Phosphodiesterase 4 inhibitors work by blocking the degradation of intracellular cAMP, blocking downstream inflammation.
An ideal treatment would address plaque induration, erythema, and scaling and be safe for all treatment sites on the skin. It would lead to improved quality of life, less days of work missed, reduced pruritus, and longer periods of remission between flares.
Reference
Papp et al. 2016 Addendum to the Canadian Guidelines for the Management of Plaque Psoriasis. JCMS. 2016; Vol20(5); 375-431.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
From a clinician’s standpoint, practical points regarding psoriasis topical therapy needs that roflumilast addresses include:
There is a need for FDA approved non-steroidal topical therapies for psoriasis in Canada. There has been much recent innovation and product pipeline availability for FDA approved systemic/biologic treatment for moderate to severe psoriasis. However, from a clinician’s view, topical therapy includes a modern need gap. Many of my patients have been on multiple treatment cycles of topical steroids (including high strength) and off-label steroid sparing topical therapies such as topical calcineurin inhibitors. They have failed these, and many run out of options. Especially for those approaching more moderate to severe disease or impact, we often have to escalate therapy once topical therapy has been maximized and failed. We need a new topical option in our armamentarium, when patients need better psoriasis control. A new topical therapy as a next step option to try may result in reducing the need for systemic therapy in some circumstances.
Roflumilast is once daily use and can be used on multiple areas of the body as long-term monotherapy for all severities of psoriasis in both adolescents and adults.
Often, treatment regimens can prove extensive or overwhelming for the psoriasis patient. Example includes different formulations and strengths of topical products used for body compared to sensitive areas like face and intertriginous areas. Many of our patients get frustrated, and/or confused, and it takes up a lot of my time trying to counsel them on ‘what goes where and how’. One product for many areas can make things easier for patients and save time for clinicians and can improve adherence to therapy.
Steroids and off-label calcineurin inhibitors are traditionally used twice daily, and once daily combined topical psoriasis therapies contain higher potency steroids (e.g., Dovobet, Enstilar). Furthermore, many patients have failed these first line topical therapies.
There is a need for intertriginous areas, and for safety reasons, topical steroid potency use is limited to low doses in folds which are often ineffective. In our experience, intertriginous or genital psoriasis is often quite debilitating and considered a sensitive site that is difficult to treat optimally.
At this point, to relieve burdens on patients and prescribers, we would be willing to do extra paperwork if needed to gain access to this new technology, especially after reviewing clinical trial data and local trial tolerability data.
Roflumilast is steroid free, and overall has excellent local tolerability.
Traditional use of long-term steroids can be limiting, with potential local and systemic side effect potential depending on potency, duration, and body surface area extent of use.
Good tolerability is practically important for both clinicians and patients. Rofumilast carries extremely low application irritation profiles in clinical trials whereas off-label use of calcineurin inhibitors (e.g., tacrolimus, pimecrolimus) and crisaborole 2% is frequently observed in both trials and practice to lead to irritation/stinging, burning, including in folds, which can prohibit patient use and optimized therapy. Often these drugs can be ‘wasted’ if the patient doesn’t tolerate them (e.g., they throw the tube out/unused product that has been paid for). Additionally, ointments are often poorly tolerated in folds due to greasiness as patients have told us it stains their clothing, whereas this is a cream formulation which may be better tolerated.
Topicals are convenient routes of administration.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
There is a great need for non-steroidal topical therapies for adolescents and adults with psoriasis to reduce the severity of itch and pain without the fear of adverse events from topical steroids like steroid atrophy, especially in intertriginous areas. This has the potential to greatly improve quality of life for patients with psoriasis along with many other debilitating inflammatory conditions for which we are very limited in our treatment options including vulvar diseases like erosive lichen planus. This topical medication will allow patients to miss less work/school and be more productive because they are not having to apply creams twice daily but more importantly because they are not itchy. This topical is much more potent than the previously approved PDE-4 inhibitor crisaborole and well tolerated in clinical studies, which is a welcome addition to our very limited non-steroidal treatment options in children.
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
All patients with mild to moderate psoriasis.
Patients with intertriginous psoriasis- particularly important for steroid sparing in these anatomic sites.
Patients would be identified clinically based on exam; sites affected.
No issues related to diagnosis; no diagnostic test required- psoriasis is a clinical diagnosis.
Patients most likely to respond may be those with inverse psoriasis.1
Reference
Lebwohl et al. Trial of Roflumilast Cream for Chronic Plaque Psoriasis. NEMJ. 2020; 383:229-39
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
Based on the clinical trial findings, responses to treatment may be seen as early as six weeks. Assessment of treatment goals is recommended after 8 weeks. Management of potential adverse events can be addressed through patient education.
Clinical Trial Finding 1
The Safety, Efficacy and Pharmacokinetics of ARQ-151 Cream in Subjects With Chronic Plaque Psoriasis (ARQ-151-201) — ClinicalTrials.gov Identifier, NCT03638258
A Phase 2b 12-week parallel group, double-blind trial, vehicle-controlled study in which roflumilast (ARQ-151) cream 0.3%, roflumilast cream 0.15%, or vehicle cream is applied once daily (QD) to 331 subjects with chronic plaque psoriasis involving between 2 and 20% body surface area.
Results: Among 331 patients who underwent randomization, 109 were assigned to roflumilast 0.3% cream, 113 to roflumilast 0.15% cream, and 109 to vehicle cream. An IGA score indicating clear or almost clear at week 6 was observed in 28% of the patients in the roflumilast 0.3% group, in 23% in the roflumilast 0.15% group, and in 8% in the vehicle group (P<0.001 and P=0.004 vs. vehicle for roflumilast 0.3% and 0.15%, respectively). Among the approximately 15% of patients overall who had baseline intertriginous psoriasis of at least mild severity, an IGA score at week 6 indicating clear or almost clear plus a 2-grade improvement in the intertriginous-area IGA score occurred in 73% of the patients in the roflumilast 0.3% group, 44% of those in the roflumilast 0.15% group, and 29% of those in the vehicle group. The mean baseline PASI scores were 7.7 in the roflumilast 0.3% group, 8.0 in the roflumilast 0.15% group, and 7.6 in the vehicle group; the mean change from baseline at week 6 was −50.0%, −49.0%, and −17.8%, respectively. Application-site reactions occurred with similar frequency in the roflumilast groups and the vehicle group.
Conclusions: Roflumilast cream administered once daily to affected areas of psoriasis was superior to vehicle cream in leading to a state of clear or almost clear at 6 weeks.
Reference
Lebwohl et al. Trial of Roflumilast Cream for Chronic Plaque Psoriasis. NEMJ. 2020; 383:229-39
Clinical Trial Findings 2 and 3
Trial of PDE4 Inhibition With Roflumilast for the Management of Plaque Psoriasis (DERMIS-1) - ClinicalTrials.gov Identifier: NCT04211363
A Phase 3, 8-Week, Parallel Group, Double Blind, Vehicle-Controlled Study of the Safety and Efficacy of ARQ-151 Cream 0.3% Administered QD in 439 Subjects with Chronic Plaque Psoriasis involving 2 to 20% body surface area (BSA) of CPP
Twin Trial of PDE4 Inhibition With Roflumilast for the Management of Plaque Psoriasis (DERMIS-2) - ClinicalTrials.gov Identifier: NCT04211389
A Phase 3, 8-Week, Parallel Group, Double Blind, Vehicle-Controlled Study of the Safety and Efficacy of ARQ-151 Cream 0.3% Administered QD in 442 Subjects with Chronic Plaque Psoriasis
Two multicenter, randomized, double-blind, vehicle-controlled trials (DERMIS-1 [NCT04211363] and DERMIS-2 [NCT04211389]) enrolled a total of 881 subjects with mild to severe plaque psoriasis and an affected BSA of 2% to 20%. The study population ranged in age from 6 to 88 years with 4 subjects younger than 12 years of age at baseline. At baseline, 16% of subjects had an Investigator’s Global Assessment (IGA) score of 2 (mild), 76% had an IGA score of 3 (moderate), and 8% had an IGA score of 4 (severe). One hundred seventy-nine (20%) subjects had an intertriginous IGA (I-IGA) score of 2 or higher (mild) at baseline, and 678 (77%) subjects had a baseline Worst Itch-Numeric Rating Score (WI-NRS) score of 4 or higher on a scale of 0 to 10.
Subjects were randomized 2:1 to receive ZORYVE or vehicle applied once daily for 8 weeks. The primary endpoint was the proportion of subjects who achieved IGA treatment success at Week 8 (Table 2). Success was defined as a score of “Clear” (0) or “Almost Clear” (1), plus a 2-grade improvement from baseline. Secondary endpoints included the proportion of subjects that achieved I-IGA success at Week 8 and WI-NRS success sequentially at Weeks 8, 4, and 2. WI-NRS success was defined as a reduction of at least 4 points from baseline in subjects with a baseline WI-NRS score of at least 4.
Results: Among 881 participants (mean age, 47.5 years; 320 [36.3%] female), mean IGA scores in trial 1 were 2.9 [SD, 0.52] for roflumilast and 2.9 [SD, 0.45] for vehicle and in trial 2 were 2.9 [SD, 0.48] for roflumilast and 2.9 [SD, 0.47]) for vehicle. Statistically significantly greater percentages of roflumilast-treated patients than vehicle-treated patients had IGA success at week 8 (trial 1: 42.4% vs 6.1%; difference, 39.6% [95% CI, 32.3%-46.9%]; trial 2: 37.5% vs 6.9%; difference, 28.9% [95% CI, 20.8%-36.9%]; P < .001 for both). Of 9 secondary end points, statistically significant differences favoring roflumilast vs vehicle were observed for 8 in trial 1 and 9 in trial 2, including intertriginous IGA success (71.2% vs 13.8%; difference, 66.5% [95% CI, 47.1%-85.8%] and 68.1% vs 18.5%; difference, 51.6% [95% CI, 29.3%-73.8%]; P < .001 for both), 75% reduction in PASI score (41.6% vs 7.6%; difference, 36.1% [95% CI, 28.5%-43.8%] and 39.0% vs 5.3%; difference, 32.4% [95% CI, 24.9%-39.8%]; P < .001 for both), WI-NRS success (67.5% vs 26.8%; difference, 42.6% [95% CI, 31.3%-53.8%] and 69.4% vs 35.6%; difference, 30.2% [95% CI, 18.2%-42.2%]; P < .001 for both). The incidence of treatment-emergent adverse events was 25.2% with roflumilast vs 23.5% with vehicle in trial 1 and 25.9% with roflumilast vs 18.4% with vehicle in trial 2. The incidence of serious adverse events was 0.7% with roflumilast vs 0.7% with vehicle in trial 1 and 0% with roflumilast vs 0.7% with vehicle in trial 2.
Conclusions and relevance: Among patients with chronic plaque psoriasis, treatment with roflumilast cream, 0.3%, compared with vehicle cream resulted in better clinical status at 8 weeks.
Reference
Lebwohl et al. Effect of Roflumilast Cream vs Vehicle Cream on Chronic Plaque Psoriasis: The DERMIS-1 and DERMIS-2 Randomized Clinical Trials. JAMA. 2022; Sep 20;328(11):1073-1084
Clinical Trial Finding 4
Long-Term Safety of ARQ-151 Cream in Adult Subjects With Chronic Plaque Psoriasis - ClinicalTrials.gov Identifier: NCT03764475
An open-label, long-term safety study of roflumilast (ARQ-151) 0.3% cream in subjects with chronic plaque psoriasis involving up to 25% total Body Surface Area (BSA). Cohort 1 of the study consisted of participants who previously completed study ARQ-151-201 (NCT03638258), and Cohort 2 consisted of participants who were not previously enrolled in ARQ-151-201. Participants applied roflumilast (ARQ-151) cream 0.3% once daily for 52 weeks.
Of the 230 subjects in Cohort 1 who received treatment with roflumilast for a total of 64 weeks, the occurrence of serious adverse events changed from 0.92% in the 12-week study to 3.48% in the 52 week study. The occurrence of other adverse events dropped from 7.34% to 6.52% respectively.
What factors should be considered when deciding to discontinue treatment with the drug under review?
Lack of efficacy/ disease progression
No specific safety signals of concern
What settings are appropriate for treatment with [drug under review]? Is a specialist required to diagnose, treat, and monitor patients who might receive [drug under review]?
Dermatologists best suited to diagnose psoriasis and determine who might receive roflumilast cream. No specific monitoring required apart from response to therapy.
Additional Information
Nothing further at this time.
Conflict of Interest Declarations — Canadian Dermatology Association
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please refer to the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
No.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
Declaration for Clinician 1
Name: Susan Poelman, MD, FRCPC
Position: Chair, CDA Pharmacy and Therapeutics Advisory Board
Date: February 27, 2022
Table 21: COI Declaration for Canadian Dermatology Association — Clinician 1
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Pfizer | X | — | — | — |
Bausch | — | X | — | — |
Declaration for Clinician 2
Name: Alexandra Kuritzky, MD, FRCPC
Position: Member, CDA Pharmacy and Therapeutics Advisory Board
Date: February 22, 2022
Table 22: COI Declaration for Canadian Dermatology Association — Clinician 2
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 3
Name: Rachel Asiniwasis, MD, MD(HS), FRCPC
Position: Expert Advisor, CDA Pharmacy and Therapeutics Advisory Board
Date: February 22, 2022
Table 23: COI Declaration for Canadian Dermatology Association — Clinician 3
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Arcutis (Advisory Board/Consulting Fees) | X | — | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.