CADTH Reimbursement Review
Cenobamate (Xcopri)
Sponsor: Paladin Labs Inc.
Therapeutic area: Epilepsy, partial onset seizures
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
AE
adverse event
ANCOVA
analysis of covariance
ASM
antiseizure medication
CI
confidence interval
CrI
credible interval
CSR
Clinical Study Report
DB
double blind
DRESS
drug reaction with eosinophilia and systemic symptoms
FOS
focal onset seizures
HRQoL
health-related quality of life
IQR
interquartile range
ITC
indirect treatment comparison
mITT
modified intention to treat
mITT-M
modified intention to treat in maintenance period
NICE
National Institute for Health and Care Excellence
NMA
network meta-analysis
OLE
open-label extension
QOLIE-31-P
patient-weighted 31-item Quality of Life in Epilepsy Questionnaire
RCT
randomized controlled trial
SAE
serious adverse event
SD
standard deviation
SUDEP
sudden unexpected death in epilepsy
TEAE
treatment-emergent adverse event
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Information on drug submitted for review | |
Drug product | Cenobamate (Xcopri), 12.5 mg, 25 mg, 50 mg, 100 mg, 150 mg, 200 mg tablet, oral |
Sponsor | Paladin Labs Inc. |
Indication | As adjunctive therapy in the management of partial onset seizures in adults with epilepsy who are not satisfactorily controlled with conventional therapy |
Reimbursement request | As per indication |
Health Canada approval status | Approved |
Health Canada review pathway | Standard review |
NOC date | June 12, 2023 |
Recommended dose | Recommended maintenance dose: 200 mg once daily (titrated up in 2-week intervals over an 11-week period, starting with 12.5 mg once daily) Maximum daily dose: 400 mg once |
NOC = Notice of Compliance.
Introduction
Epilepsy is a chronic neurologic disorder that affects the physical and mental health of patients and significantly interferes with daily activity as well as life expectancy.1 The broad categories of epileptic seizures include partial onset (also known as focal), generalized, combined focal and generalized, and unknown onset.2 In patients with partial onset seizures, only a portion of the brain, typically 1 lobe of 1 hemisphere, is affected by the seizure, while in patients with generalized seizures, large parts of both brain hemispheres are involved.3 The estimated prevalence of active epilepsy is 5.96 per 1,000 population (95% confidence interval [CI], 5.38 to 6.61) based on a meta-analysis of international studies.4 It is estimated that 300,000 people in Canada are living with epilepsy.5
Antiseizure medications (ASMs) are the most common treatments for seizures and are prescribed by physicians after discussing the risks and benefits of the medication with the patient or their caregiver. There are many different ASMs currently available in Canada. These differ in their mechanism of action, potential adverse-effect profiles, the types of seizures they are best at treating, and cost. Although ASMs help to control or reduce seizures, these drugs are not a cure for epilepsy. While the aim of treatment with ASMs is to eliminate seizures with no adverse effects, this may not be achieved in all patients;6 approximately 20% to 40% of patients are at risk of having refractory epilepsy.7,8 Epilepsy is considered to be medically refractory (or drug-resistant) when a patient fails to achieve sustained freedom from seizures after adequate trials of 2 tolerated ASMs, either as monotherapy or in combination.7 Treatment options for patients with refractory epilepsy may be limited.
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of cenobamate oral tablets, 12.5 mg to 200 mg, as adjunctive therapy in the management of partial onset seizures in adults who are not satisfactorily controlled with conventional therapy.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient and clinician groups who responded to CADTH’s call for input and from the clinical expert consulted by CADTH for the purpose of this review.
Patient Input
Six groups, including the Canadian Epilepsy Alliance, Epilepsy Toronto, Epilepsy South Central Ontario, Epilepsy Southwestern Ontario, the Epilepsy Association of Calgary, and the Edmonton Epilepsy Association, provided patient input for this review. These patient groups indicated that uncontrolled seizures and the adverse effects of ASMs affect patients’ daily activities, independence (e.g., they are not legally permitted to drive), and mental health (e.g., they experience higher risks of depression, anxiety, and suicidal ideation). The patient groups noted that whole families are affected. Patients are often unemployed or under-employed and negatively affected by other social determinants of health. The patient groups noted that the most important treatment outcome is seizure freedom, with an alternative expectation of reduced seizure frequency and/or severity. Patients and their families were also highly concerned about adverse effects of ASMs and interactions between these drugs. The patient groups indicated that new drugs could offer hope to patients who are close to giving up, and even a reduction in the absolute number of seizures can potentially improve overall quality of life.
Clinician Input
Input From the Clinical Expert Consulted by CADTH
The expert stated that treatment options are limited for the approximately 30% of patients with focal epilepsy who do not respond to ASMs, and that while ASMs may reduce seizure frequency, none offer a cure for epilepsy. Tolerability of ASMs can be an issue, and a patient’s comorbidities and concomitant medications may contribute to the development of adverse effects. The clinical expert stated that cenobamate is best suited to patients with focal epilepsy who have not responded to conventional ASMs. The expert also anticipates that it will be used as second- or third-line therapy, typically as add-on therapy. Treatment response would be demonstrated by seizure freedom or a reduction in seizure frequency as well as improved quality of life and acceptable tolerability. According to the clinical expert, patients prescribed cenobamate should be under the care of a neurologist or epileptologist in a community or hospital setting.
Clinician Group Input
One clinician group provided input for this review: the Canadian League Against Epilepsy, which has more than 125 health care-related members. The group highlighted that there is an unmet need for more effective treatments for patients with uncontrolled, focal onset seizures. Despite the availability of several ASMs, there has been no meaningful improvement in epilepsy treatment-related outcomes and no significant increase in seizure freedom rates in the past 20 years. The clinician group noted that cenobamate would likely be used in combination with other available treatments (i.e., as an add-on) and is unlikely be used as a monotherapy. However, if cenobamate proves to prevent seizures once added, a physician will occasionally try to minimize a patient’s ongoing treatment by weaning them off other ASMs. There was consistency between the views of the clinical expert consulted by CADTH and the Canadian League Against Epilepsy with regard to how response to treatment is assessed, reasons for discontinuing therapy, the treatment setting, and the specialists required to diagnose, treat, and monitor patients who may receive cenobamate.
Drug Program Input
Input was obtained from the drug programs that participate in the CADTH reimbursement review process. The following were identified as key factors that could potentially affect the implementation of a CADTH recommendation for cenobamate:
relevant comparators
consideration for initiating therapy
consideration for prescribing therapy.
The clinical expert consulted by CADTH provided advice on the potential implementation issues raised by the drug program.
Clinical Evidence
Pivotal Studies and Randomized Controlled Trial Evidence
Description of Studies
The pivotal and randomized controlled trial (RCT) evidence included 2 double-blind (DB), placebo-controlled RCTs of add-on therapy with cenobamate in adults with uncontrolled partial seizures despite ongoing treatment with 1 to 3 ASMs.9,10 In Study C013, 222 patients were randomized to cenobamate 200 mg daily or placebo; in Study C017, 437 patients were randomized to cenobamate 100 mg, 200 mg, or 400 mg once daily or placebo, in addition to the ASMs initiated before enrolment. Each study included an 8-week baseline period before randomization, a 6-week dose titration period, and a 6-week (Study C013) or 12-week (Study C017) maintenance period in which the dose of study drug remained stable. The primary end point was the percent change in seizure frequency per 28 days versus baseline for all simple partial motor, complex partial, and secondarily generalized seizures. The key secondary outcome was the proportion of patients who achieved at least a 50% reduction in partial seizure frequency versus baseline. In both studies, these end points were calculated based on the entire DB treatment period (titration and maintenance phases) and used to support regulatory approval in the US. Study C017 conducted alternate analyses based on seizure data from the maintenance phase only, and these data were used to support regulatory approval in Europe.
The mean age of patients enrolled in the pivotal trials ranged from 36.2 years (standard deviation [SD] = 11.3 years) to 40.9 years (SD = 12.4 years) across treatment groups. There were roughly equal proportions of men (47% to 54%) and women (46% to 53%) enrolled. In Study C013, the median baseline seizure frequencies per 28 days were 5.5 (range = 2 to 237) in the placebo group and 7.5 (range = 0 to 187) in the cenobamate 200 mg group. In Study C017, the median baseline seizure frequencies per 28 days ranged from 8.4 (range = 4 to 704) for placebo to 11.0 (range = 4 to 418) for the cenobamate 200 mg group. On average, the enrolled patients had been diagnosed with epilepsy for more than 20 years. Patients in Study C017 had previously been treated with a median of 3 prior ASMs (range = 1 to 9).
The CADTH review focused on data for the cenobamate 200 mg to 400 mg dosage range, as per the draft product monograph.
Efficacy Results
For the primary end point in Study C013, the cenobamate 200 mg group showed a median 55.6% reduction in partial seizure frequency per 28 days compared with a 21.5% reduction in the placebo group (P < 0.0001) (Table 2). In Study C017, the median percent reductions in seizure frequency per 28 days were 55.0%, 55.0%, and 24.0% in the cenobamate 200 mg, 400 mg, and placebo groups, respectively (with P < 0.001 favouring cenobamate versus placebo for both dosage groups).
For the responder analysis in Study C013, 50.4% of patients in the cenobamate 200 mg group and 22.2% of patients in the placebo group achieved at least a 50% reduction in seizure frequency during the DB treatment period (odds ratio = 3.94; 95% CI, 2.14 to 7.24; P < 0.0001; not controlled for type I error rate). In Study C017, 57.8%, 60.4%, and 21.7% of patients in the cenobamate 200 mg, 400 mg, and placebo groups, respectively, achieved at least a 50% reduction in seizure frequency per 28 days during the DB treatment phase. The analyses favoured the cenobamate groups versus placebo (both P < 0.001; not controlled for type I error rate) (Table 2). The alternate analyses, which were based on treatment response in the maintenance period only, reported that 56.1%, 64.2%, and 25.5% of patients achieved at least a 50% reduction in seizure frequency per 28 days in the cenobamate 200 mg, 400 mg, and placebo groups, respectively. For this alternate primary end point, the differences favoured the cenobamate 200 mg (P < 0.001) and 400 mg groups (P < 0.001) versus placebo.
The proportion of patients who achieved reductions of greater than or equal to 75%, greater than or equal to 90%, and 100% in seizure frequency during the DB period (Table 2) and maintenance period favoured the cenobamate 200 mg and 400 mg groups versus placebo (Appendix 1); however, there was no control of type I error rate for these analyses in either study, and in Study C013, these analyses were conducted post hoc. While these results are generally supportive of the efficacy of cenobamate, these data should be interpreted in light of the potentially inflated risk of type I error and risk of bias associated with post hoc analyses.
Of note, Study C017 also included a 100 mg cenobamate dosage group, which is half the Health Canada–recommended maintenance dose. Both the percent reduction in seizure frequency per 28 days and the proportion of patients who achieved at least a 50% reduction in seizure frequency favoured the cenobamate 100 mg group versus placebo; however, other secondary outcomes, such as the higher responder thresholds, failed to detect a difference between groups (Table 2).
Health-related quality of life (HRQoL) was not assessed in Study C013, and only descriptive data were available in Study C017 for approximately one-quarter of the patients enrolled. No meaningful change in HRQoL was observed in Study C017, based on data from the patient-weighted 31-item Quality of Life in Epilepsy Questionnaire (QOLIE-31-P).
Other outcomes of interest to patients, such as functional status, were not assessed, nor were seizure-free days or treatment retention, which were outcomes specified in the sponsor’s protocol.
Harms Results
During the 12-week to 18-week treatment periods, adverse events (AEs) were reported by 65%, 76%, and 90% of patients who received cenobamate 100 mg, 200 mg, and 400 mg, compared with 63% to 70% of patients who received placebo in studies C013 and C017. The most frequently reported AEs in the cenobamate groups were somnolence (19% to 37%), dizziness (18% to 33%), fatigue (11% to 24%), and diplopia (4% to 15%). In the placebo groups, somnolence was reported in 8% to 12% of patients, dizziness in 14% to 17% of patients, fatigue in 6% to 8% of patients, and diplopia in 2% to 3% of patients.
In Study C013, 5 patients (4%) in the cenobamate group and 3 patients (3%) in the placebo group stopped treatment due to AEs, whereas in Study C017, 11 patients (10%), 15 patients (14%), 22 patients (20%), and 5 patients (5%) stopped treatment due to AEs in the cenobamate 100 mg, 200 mg, 400 mg, and placebo groups, respectively.
No deaths occurred during the pivotal trials. Serious adverse events (SAEs) were reported in 3% to 9% of patients who received cenobamate and in 4% to 6% of patients who received placebo. One patient in the cenobamate 200 mg group of Study C017 developed a drug reaction with eosinophilia and systemic symptoms (DRESS) on day 24 that the investigator considered probably related to the study drug; the study drug was stopped. Other dermatologic reactions that led to study drug withdrawal were reported in 2 patients in the cenobamate 200 mg group. In Study C013, 1 patient who was randomized to the cenobamate group experienced a serious drug hypersensitivity reaction on day 1. The patient stopped cenobamate treatment and recovered after 22 days. In Study C013, suicidal ideation was reported by 1 patient in each treatment group, and in Study C017 by 2 patients in the cenobamate 100 mg group, and by 1 patient in the cenobamate 200 mg group. One patient in the cenobamate 100 mg group in Study C017 attempted suicide.
Table 2: Summary of Key Results From Pivotal Studies and RCT Evidence
Outcome | Study C013 | Study C017 | ||||
|---|---|---|---|---|---|---|
CEN 200 mg (N = 113) | Placebo (N = 109) | CEN 100 mg (N = 108) | CEN 200 mg (N = 110) | CEN 400 mg (N = 111) | Placebo (N = 108) | |
Seizure frequency per 28 days – DB treatment period (mITT population) | ||||||
Number of patients included in analysis | 113 | 108 | 108 | 109 | 111 | 106 |
Baseline rate, median (range) | 7.5 (0 to 186.8) | 5.5 (2.0 to 236.5) | 9.5 (3.5 to 202) | 11.0 (4 to 418) | 9 (4 to 638) | 8.4 (4 to 704) |
End point rate, median (range) | 3.8 (0 to 196.3) | 5.0 (0 to 206.3) | 5.8 (0 to 164.6) | 5.8 (0 to 373.7) | 3.8 (0 to 424.9) | 6.8 (0.7 to 640.8) |
Median percent reduction vs. baseline | 55.6 | 21.5 | 35.5 | 55.0 | 55.0 | 24.0 |
P valuea | P < 0.0001b | Reference | P = 0.007 c | P < 0.001 c | P < 0.001 c | Reference |
≥ 50% reduction in seizure frequency per 28 days – DB treatment period (mITT population) | ||||||
n (%) | 57 (50.4) | 24 (22.2) | 44 (40.7) | 63 (57.8) | 67 (60.4) | 23 (21.7) |
P valued | P < 0.0001 | Reference | P = 0.003 | P < 0.001 | P < 0.001 | Reference |
≥ 75% reduction in seizure frequency per 28 days – DB treatment period (mITT population) | ||||||
n (%) | 32 (28.3) | 11 (10.2) | 18 (16.7) | 23 (21.1) | 39 (35.1) | 9 (8.5) |
P valued,e | P = 0.0007 | Reference | P = 0.099 | P = 0.012 | P < 0.001 | Reference |
≥ 90% reduction in seizure frequency per 28 days – DB treatment period (mITT population) | ||||||
n (%) | 15 (13.3) | 1 (0.9) | 5 (4.6) | 13 (11.9) | 23 (20.7) | 1 (0.9) |
P valued,e | P = 0.0063 | Reference | P = 0.212 | P = 0.001 | P < 0.001 | Reference |
100% reduction in seizure frequency per 28 days – DB treatment period (mITT population) | ||||||
n (%) | 10 (8.8) | 1 (0.9) | 2 (1.9) | 8 (7.3) | 7 (6.3) | 0 (0.0) |
P valued,e | P = 0.0148 | Reference | P = 0.498 | P = 0.007 | P = 0.014 | Reference |
Harms, n (%) – (safety population) | ||||||
Patients with ≥ 1 adverse event | 86 (76) | 69 (63) | 70 (65) | 84 (76) | 100 (90) | 76 (70) |
Patients with SAE | 3 (3) | 4 (4) | 10 (9) | 4 (4) | 8 (7) | 6 (6) |
Patients who stopped treatment due to adverse events | 5 (4) | 3 (3) | 11 (10) | 15 (14) | 22 (20) | 5 (5) |
ANCOVA = analysis of covariance; DB = double blind; CEN = cenobamate; mITT = modified intention to treat; RCT = randomized controlled trial; SAE = serious adverse event.
aIn Study C013, the P value is based on a Wilcoxon rank sum test assessing if the median percent change for CEN is significantly different than for placebo. The Study C017 P value is based on a nonparametric ANCOVA model with terms for ranked baseline seizure rate and treatment group. The mITT population included all randomized patients who received at least 1 dose of the study drug and had any postbaseline seizure data.
bPrimary end point in Study C013; no multiplicity adjustment required.
cPrimary end point for the US regulatory agency in Study C017; the P value for each dosage group has been adjusted for multiple testing.
dIn Study C013, the P value is based on logistic regression model with terms for treatment, country, and baseline seizure frequency (Wald chi-square test). In Study C017, the P value is based on Fisher’s exact chi-square test.
eIn Study C013, greater than or equal to 75%, greater than or equal to 90%, and 100% of responder analyses were conducted post hoc.
Source: Clinical Study Report for Study C013;10 Clinical Study Report for Study C017.9
Critical Appraisal
The risks of bias related to randomization, treatment allocation, and blinding in the pivotal trials were rated as low by the CADTH reviewer. At baseline, the patient characteristics appeared to be reasonably well balanced between groups within the studies. In Study C013, the proportions of patients who discontinued were similar in both groups (10% and 9%), but Study C017 showed differential losses to follow-up, with 12%, 18%, 27%, and 13% of patients stopping treatment in the cenobamate 100 mg, 200 mg, 400 mg, and placebo groups, respectively. As a result, it is not clear if the treatment groups remained balanced throughout the study. Both trials measured the change in partial seizure frequency as well as the proportion of responders (≥ 50%) as primary and secondary end points. The analyses included all simple partial motor, complex partial, and secondarily generalized seizures; the clinical expert agreed that this was appropriate for this population. While the 50% response threshold has been used in other ASM clinical trials and may be accepted as a minimal clinically important difference, the clinical expert indicated that higher response thresholds are desired, and the goal of therapy is seizure freedom. Other seizure response thresholds were tested in both studies, but these analyses were not controlled for type I error rate; in Study C013, these were conducted post hoc. Moreover, the responder analyses were conducted using the last observation carried forward for Study C013 and with no imputation for missing data in Study C017; thus, patients who withdrew early could be considered as treatment responders. Considering that patients who drop out are likely to have worse outcomes than those who continue, it is possible that the results of Study C017 may be biased in favour of cenobamate, due to the extent of the early withdrawals and the differential losses. However, the magnitude of any potential bias and the impact on the overall findings is unclear.
Neither of the pivotal trials was designed to test the impact of cenobamate on HRQoL or on patients’ ability to work or maintain independence. Although Study C017 collected data using the QOLIE-31-P instrument, this information was gathered only from approximately 25% of the patients enrolled and was reported descriptively. Moreover, the trials lacked an active comparator group and were 12 weeks to 18 weeks in duration; thus, the trials can only address short-term efficacy and safety versus placebo. The sample sizes and durations of the studies were insufficient to capture rare AEs.
No major issues were identified by the clinical expert on the generalizability of the pivotal studies. However, it should be noted that both studies had extensive exclusion criteria and were limited to adults up to 70 years of age. The trials used a more rapid titration schedule than has been recommended in the product monograph, which may have affected the occurrence of some AEs.
Long-Term Extension Studies
Description of Studies
Additional longer-term safety data were available from 2 single-arm, open-label extension (OLE) studies: Study C013 OLE and Study C017 OLE.11,12 Patients who completed the randomized phase of Study C013 were eligible to enter the OLE phase (N = 149) and received open-label cenobamate at a daily maximum dose of 400 mg daily for up to 8.6 years. Only safety outcomes were assessed. Patients who completed Study C017 were eligible to enter its OLE phase (N = 356) and were transitioned to open-label cenobamate at a target dose of 300 mg once daily. In this phase, efficacy outcomes of seizure control up to 48 months and safety outcomes up to 6.4 years were assessed.
Efficacy Results
For the Study C017 OLE, Klein et al. (2022) reported interim efficacy outcomes based on a median exposure duration of 53.9 months (range = 1.1 months to 68.7 months), with retention rates at 12 months, 24 months, 36 months, and 48 months of 83%, 71%, 65%, and 62%, respectively.13 Among patients who remained on cenobamate, the treatment effects appear to be maintained for up to 48 months. The median 65.4% reduction in partial seizure frequency versus baseline was reported during the first 6 months of the OLE (interquartile range [IQR] = 52.0%; N = 354), with a 76.1% reduction (IQR = 44.8%) at month 43 to month 48 (N = 213).13 Of the 354 patients who entered the OLE, 10.2% achieved a 100% reduction in partial seizure frequency in the 36-month to 48-month interval.13
Harms Results
In both OLE studies, 89% of patients reported 1 or more treatment-emergent adverse events (TEAEs); dizziness (34% to 36%), somnolence (22% to 24%), and headache (17% to 28%) were the most common. Overall, 9% of patients stopped treatment due to AEs, and 22% to 26% of patients experienced an SAE. There were no cases of DRESS or any serious skin or subcutaneous tissue disorders reported during the OLEs. Four deaths (3%) were reported in the Study C013 OLE; these were due to sudden unexpected death in epilepsy (SUDEP), cardiac arrest, respiratory arrest, and completed suicide. In the Study C017 OLE, 6 patients (2%) died after experiencing myocardial infarction, cardiogenic shock, SUDEP, completed suicide, or sepsis.
Critical Appraisal
Limitations of the OLE studies include selection bias, lack of a control group, and lack of blinding. Given that completion of a pivotal trial was an eligibility criterion for the extension study, patients who discontinued those trials due to AEs or lack of response were excluded. This could have resulted in a population of patients who were more tolerant of cenobamate, which could lead to both a response bias (because those not responding to treatment are less likely to continue) and biased estimates related to AEs, potentially resulting in fewer and less severe AEs being reported. Without comparator groups, the interpretation of the results in relation to an appropriate comparator (e.g., another ASM) is limited. Unblinding of the cenobamate treatment in the OLE can bias the reporting of end points, particularly for any subjective measures, including AEs.
The sample sizes in the Study C013 OLE (N = 149, with 25% completing the study) and Study C017 OLE (N = 356, with 20% completing the study) may not be sufficient to detect rare AEs. In addition, there was wide variance in the follow-up durations for individuals. More common forms of morbidity (e.g., cardiac dysrhythmias) may not be easily identified as related to drug exposure.
Indirect Comparisons
Descriptions of Indirect Treatment Comparisons
Two indirect treatment comparisons (ITCs) were summarized and critically appraised for CADTH’s review of cenobamate. ITC 1 was a sponsor-submitted network meta-analysis (NMA). It was designed to assess the relative efficacy and safety of adjunctive therapy with cenobamate compared to brivaracetam, perampanel, lacosamide, eslicarbazepine acetate, and zonisamide in adult patients with partial onset seizures.14,15 ITC 2 was an NMA conducted by the National Institute for Health and Care Excellence (NICE) to evaluate the relative efficacy and safety of ASMs used as add-on treatments for patients with partial onset seizures.16 Relevant treatments in ITC 2 included brivaracetam, carbamazepine, cenobamate, eslicarbazepine acetate, gabapentin, ganaxolone, lacosamide, lamotrigine, levetiracetam, oxcarbazepine, perampanel, phenytoin, pregabalin, primidone, retigabine, rufinamide, sodium valproate, topiramate, vigabatrin, and zonisamide.16
Efficacy Results
The sponsor-submitted NMA (i.e., ITC 1) included 22 studies for the combined efficacy outcome of a 50% or greater responder rate and seizure freedom.14,15 The results suggest that patients receiving cenobamate may be more likely to achieve a 50% or greater reduction in seizures, or seizure freedom in the short-term, compared to those receiving any of the 5 ASMs in the NMA.14,15
The ITC by NICE (ICT 2) included 99 studies for the analysis of seizure frequency reduction of greater than 50% reduction and 72 studies for the seizure freedom analysis.16 The results suggest that patients who received cenobamate may be more likely to achieve a greater than 50% reduction in seizure frequency in the short-term than patients on most other treatments, including eslicarbazepine acetate, retigabine, gabapentin, lacosamide, lamotrigine, levetiracetam, oxcarbazepine, perampanel, brivaracetam, pregabalin, rufinamide, zonisamide, and primidone.16 Relative treatment effects were not available for seizure freedom due to rarity of the event and insufficient statistical power.16
Harms Results
The sponsor-submitted NMA included 20 studies for the safety outcome of TEAEs leading to treatment discontinuation.14,15 Compared to cenobamate, results for the 5 other ASMs had relatively wide 95% credible intervals (CrIs) that included the threshold of no difference; however, this does not indicate equivalence between ASMs for this outcome.14,15 Due to the lack of precision in the estimates, firm conclusions cannot be made.
Harms data were reported numerically (not comparatively) by treatment in ITC 2 and do not inform relative safety for cenobamate versus other ASMs.16
Critical Appraisal
The main limitations of both ITCs were the inclusion of nonadult patients (cenobamate is indicated for adults), missing relevant comparator ASMs (mainly in ITC 1), and heterogeneity in patient and study characteristics across studies. Moreover, the limited reporting of trial data hindered the CADTH reviewers’ ability to assess the consistency assumption, particularly for ITC 2, and neither NMA conducted analyses to adjust for potential treatment-effect modifiers. Differences across studies in how efficacy outcomes were defined (e.g., seizure freedom) and the treatment period upon which these were based (i.e., the entire treatment period or excluding the dose titration period) were assessed as important potential sources of bias. The durations of the trials were variable (7 weeks to 24 weeks) and limited to a short time frame for this chronic condition. Given these limitations, there was uncertainty in the results of the NMAs that prevented firm conclusions from being drawn from the findings. Furthermore, there was a lack of safety data in the ITCs and no information about HRQoL or long-term outcomes reported; thus, it is not possible to estimate how cenobamate compares to other ASMs for these outcomes.
Studies Addressing Gaps in the Pivotal and RCT Evidence
Description of Studies
The sponsor submitted 1 prospective safety study (Study C021)17 and 3 additional studies to address gaps in the evidence.18-20
Study C021 was a single-arm safety study of cenobamate as adjunctive therapy in patients with uncontrolled partial onset seizures despite treatment with 1 to 3 ASMs (N = 1,340). It consisted of a 12-month open-label treatment period that included a 12-week titration phase followed by an open-label maintenance phase, with a target daily dose of 200 mg and a maximum daily dose of 400 mg.17
The 3 additional studies submitted by the sponsor included Elizebath et al. (2021), a post hoc data analysis of HRQoL outcomes from all the patients enrolled in studies C013, C017 and C021 at a single US centre (N = 49);18 Connor et al. (2022), a real-world evidence study among adults with a developmental disability (N = 28);19 and Elliott et al. (2022), a real-world evidence study in patients with partial epilepsy who received cenobamate (N = 45, 13 of whom were adolescents aged 12 years to 17 years).20
Efficacy Results
Elizebath et al. (2021) reported that the mean of overall QOLIE-31 scores (range = 1 to 100, with higher scores indicating better quality of life) measured for all patients who completed treatment was 67 (SD = 19; range = 32 to 97; n = 37) over a median treatment period of 5.6 years (range = 3 years to 8 years).18 Connor et al. (2022) reported that adjunctive cenobamate treatment, the mean number of focal seizures was reduced from 20.9 seizures per month at baseline to 4.1 seizures per month at 6 months’ follow-up (median = from 3.0 seizures at baseline to 0.5 seizures per month at 6 months’ follow-up); at 6 months, the greater than or equal to 50%, greater than or equal to 75%, greater than or equal to 90%, and 100% responder rates were 92.6%, 81.5%, 55.6%, and 48.2%, respectively.19 Elliott et al. (2022) reported that 60% of all the included patients achieved a reduction in seizures greater than or equal to 50%, and that among the 13 adolescents, 8 (61.5%) achieved a reduction in seizures greater than or equal to 50%.20 No efficacy results were reported in Study C021.
Harms Results
In Study C021, TEAEs occurred in 91% of patients. The most frequently reported TEAEs were somnolence (31%), dizziness (29%), fatigue (19%), and headache (18%). Serious TEAEs occurred in 18% of the patients; the most frequently reported serious TEAE was seizures (2%). One hundred and 83 patients (14%) had at least 1 TEAE leading to discontinuation, most frequently due to dizziness (1.4%) and seizures (0.8%). At the data cut-off date, 10 deaths had been reported in Study C021, with causes of laryngospasm, glioblastoma, subdural hematoma, SUDEP, sudden death, traumatic intracranial hemorrhage, hypovolemic shock, pneumonia viral, status epilepticus, and cardiac arrest.17 One case of sudden death was deemed by the investigator to be remotely related to the study drug; the remainder were deemed to be unrelated.17 No case of DRESS was reported.17
Connor et al. (2022) reported that AEs occurred in 32% of patients and included dizziness (14%), drowsiness (117%), ataxia (7%), and behavioural “acting out” (4%).19 Elliott et al. (2022) reported that the most frequently reported AE was somnolence (18%).20
Critical Appraisal
Limitations of the other studies addressing the evidence gaps include the relatively small sample sizes for the observational studies, selection bias, lack of a control group, lack of blinding, and probable lack of control for confounding in the studies. Due to these factors, CADTH could not determine whether the results of these studies were valid or reliable.
Conclusions
The direct evidence demonstrates that add-on therapy with cenobamate is superior to placebo in reducing the frequency of partial seizures in the short-term in adults whose partial onset seizures are inadequately controlled with up to 3 concomitant ASMs. In addition, a higher percentage of patients who received cenobamate appeared to achieve at least a 50% reduction in seizure frequency compared to placebo. While a 50% reduction in seizure frequency is an accepted end point for clinical trials, it may not be clinically relevant for all patients, given that the goal of therapy is the elimination of seizures. The pivotal studies examined higher seizure reduction thresholds (i.e., ≥ 75%, ≥ 90%, and 100%); however, due to statistical limitations (i.e., lack of control of the type I error rate) and risk of bias (i.e., analyses were conducted post hoc in Study C013), definitive inferences cannot be drawn from these data.
Direct comparative evidence for cenobamate versus other ASMs was unavailable. The indirect evidence from 2 NMAs suggest there may be a short-term benefit favouring cenobamate versus some ASMs for the proportion of patients achieving at least a 50% reduction in seizure frequency; however, the magnitude and clinical relevance of any benefit was unclear because of important limitations in the analyses. There was heterogeneity across the networks in the patient characteristics, in how outcomes were defined and analyzed, and in the durations of follow-up, creating uncertainty in the findings of the NMAs. Due to the uncertainty in the sponsor-submitted NMA, no conclusions could be drawn about the relative effects of cenobamate on seizure freedom.
The impact of cenobamate on HRQoL or other outcomes of importance to patients, such as the ability to work or live independently, is unknown because the placebo-controlled trials were not designed to test for these outcomes. Neither NMA assessed HRQoL or longer-term outcomes.
No conclusions could be drawn regarding the short-term comparative safety of cenobamate versus other ASMs due to the previously described limitations and the lack of precision in the estimates from the NMA. No new safety signals were identified from the longer-term single-arm studies; however, long-term comparative data were not available.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of cenobamate 12.5 mg to 200 mg oral tablets as adjunctive therapy for the management of partial onset seizures in adults who are not satisfactorily controlled with conventional therapy.
Disease Background
The contents of this section have been informed by materials submitted by the sponsor and by clinical expert input. The following paragraphs have been summarized and validated by the CADTH review team.
Epilepsy is a chronic neurologic disorder that affects the physical and mental health of patients and significantly interferes with daily activity as well as life expectancy.1 According to the International League Against Epilepsy, epilepsy is defined by any of the following conditions: at least 2 unprovoked seizures occurring more than 24 hours apart; 1 unprovoked seizure and a probability of further seizures that is similar to the general recurrence risk (i.e., ≥ 60% probability of further seizures) after 2 unprovoked seizures occurring over the next 10 years; or diagnosis of an epilepsy syndrome.21 The broad categories of epileptic seizures include partial onset (also known as focal), generalized, combined focal and generalized, and unknown onset.2 In patients with partial onset seizures, only a portion of the brain, typically 1 lobe of 1 hemisphere, is affected by seizures, while in patients with generalized seizures, large parts of both brain hemispheres are involved.3 A partial onset seizure can be characterized as motor (with muscle activity changes) or nonmotor (without muscle activity changes) type.2 Loss of consciousness (impaired awareness) is 1 of the symptoms in complex partial onset seizures (also known as focal impaired awareness) and generalized seizures.3
As the second most frequent neurologic disorder worldwide (after migraine), the prevalence of epilepsy is high.1 The estimated prevalence of active epilepsy is 5.96 per 1,000 population (95% CI, 5.38 to 6.61), based on a meta-analysis of international studies published from 1985 to 2011.4 Prevalence estimates of active epilepsy in Canada range from 4 in 1,000 people based on the weighted Canadian Community Health Survey data (from 2010 to 2011) to 6.4 per 1,000 based on the Canadian Chronic Disease Surveillance System data (from 2019 to 2020).4,22 It is estimated that 300,000 people in Canada are living with epilepsy.5 Approximately 20,000 Canadians (5,000 children and youths, and 15,000 adults)were diagnosed with epilepsy in 2013 to 14, or on average, 54 people per day.5 The incidence of epilepsy is highest in the first year of life and declines with increasing age.1,4
Epilepsy is considered to be medically refractory epilepsy (also called drug-resistant epilepsy or intractable epilepsy) when a patient fails to achieve sustained seizure freedom after adequate trials of 2 tolerated ASMs, either as monotherapy or in combination.7 Approximately 20% to 40% of patients with epilepsy are at risk of having refractory epilepsy.7,8
Standards of Therapy
Contents within this section have been informed by materials submitted by the sponsor and clinical expert input. The following paragraphs have been summarized and validated by the CADTH review team.
The main goals of therapy are to eliminate seizures or reduce their frequency and to improve patients’ safety, quality of life, and ability to work, attend school, drive, and maintain independence. Other goals are to reduce burden on caregivers, prevent premature death and SUDEP, avoid delayed remission, reduce AEs, and avoid invasive therapies (e.g., surgery).
ASMs are the most common treatment for seizures and are prescribed by physicians after discussing the risks and benefits of the medications with patients or caregivers. There are many different ASMs currently available within and outside Canada. These differ in terms of their mechanism of action, potential adverse-effect profiles, the types of seizures they are best at treating (i.e., focal versus generalized versus epileptic spasms), and cost. ASMs help to reduce seizure frequency, but are not a cure for epilepsy. Although the aim of treatment with ASMs is “no seizures, no adverse effects,” this may not be achieved in all patients.6 Monotherapy with an ASM is ideal; however, some individuals may require more than 1 ASM to control seizures.23,24
The NICE guidelines25 on the management of epilepsy recommend the following:
Use a single antiseizure medication (monotherapy) to treat epilepsy whenever possible.
Review the diagnosis of epilepsy if seizures continue despite an optimal dose of a first-line antiseizure medication.
If first-line monotherapy is unsuccessful and epilepsy diagnosis remains confirmed, try monotherapy with another antiseizure medication, using caution during the changeover period:
Increase the dose of the second medicine slowly while maintaining the dose of the first medicine.
If the second medicine is successful, slowly taper off the dose of the first medicine.
If the second medicine is unsuccessful, slowly taper off the dose of the second medicine and consider an alternative.
If monotherapy is unsuccessful, consider trying an add-on treatment.
When starting an add-on treatment, carefully titrate the additional medicine and review treatment frequently, including monitoring for adverse effects, such as sedation.
If trials of add-on treatment do not result in a reduction in seizures, use the regimen that provides the best balance between effectiveness and tolerability of side effects.25
Approximately half of epilepsy patients will gain control of their seizures with an appropriate first ASM; in general, two-thirds of patients will be seizure-free on ASMs. However, more than one-third do not respond to ASMs and continue to have uncontrolled seizures.26 Of those patients, only 5% per year will enter seizure remission as a result of changing from 1 current ASM to another.27 According to the sponsor and clinical expert input, there are limited treatment options available for patients who are inadequately controlled using current ASMs.28 Moreover, suboptimal seizure control with ASMs exposes these patients to increased risks (associated with uncontrolled seizures) and decreased HRQoL. There is a high risk of SUDEP in patients with uncontrolled seizures, particularly for those with focal to bilateral tonic-clonic seizures.29,30
The clinical expert consulted for this review indicated that all the ASMs shown in Table 3 may be used as adjunctive therapy in patients with focal epilepsy. Other options for adjunctive therapy include off-label use of rufinamide or felbamate and zonisamide. These are not currently approved in Canada, but may be prescribed through the Health Canada Special Access Program. Nonpharmaceutical options for patients with treatment-resistant epilepsy include epilepsy surgery, neurostimulation, or ketogenic diet. Epilepsy surgery is an invasive and costly treatment that can be offered only to select patients with a single identifiable seizure focus located in an area of the brain that can be safely removed.31 Neurostimulation treatments are invasive, costly, palliative options that can reduce seizure frequency, but these rarely lead to seizure freedom.32,33 The ketogenic diet is a palliative, arduous treatment that is poorly tolerated by adults.34
Drug Under Review
Key characteristics of cenobamate are summarized in Table 3 along with other treatments available for partial onset seizures.
The precise mechanism of the therapeutic effects of cenobamate in patients with partial onset seizures is not known.35 Cenobamate functions as a positive allosteric modulator of gamma-aminobutyric acid type A receptors at nonbenzodiazepine-binding sites.36-38 Cenobamate also decreases excitatory currents by inhibiting the persistent component of the sodium current and enhancing the inactivated state of voltage-gated sodium channels.
Cenobamate is approved by Health Canada as adjunctive therapy in the management of partial onset seizures in adults with epilepsy who are not satisfactorily controlled with conventional therapy.35 The usual maintenance dose of cenobamate is 200 mg once daily, and the maximum daily dose — if needed, based on clinical response and tolerability — is 400 mg once daily.35 The dose of cenobamate should be titrated up in 2-week intervals (because of the potential for SAEs), starting with 12.5 mg once daily. It is recommended that discontinuation of cenobamate be undertaken gradually over a period of at least 2 weeks to minimize the potential for rebound seizures, unless safety concerns require abrupt withdrawal.35
The dossiers for cenobamate were submitted to CADTH as a pre-Notice of Compliance submission, with Notice of Compliance received on June 12, 2023. Cenobamate underwent a standard review at Health Canada, and the sponsor has requested reimbursement as per the approved Health Canada indication.39 Cenobamate has not been previously reviewed by CADTH. It received approval from the FDA in November 2019.40
Table 3: Key Characteristics of Cenobamate and Other Drugs for Partial Onset Seizures
Drug name (brand) and year marketed | Mechanism of action41 | Indication in Canada | Recommended dose | Serious adverse effects and/or safety issues41 |
|---|---|---|---|---|
Drug under review | ||||
Cenobamate (Xcopri),35 2023 | Voltage-gated sodium channel blocker and positive allosteric modulator of GABAA | Adjunctive therapy in the management of partial onset seizures in adults with epilepsy who are not satisfactorily controlled with conventional therapy. Cenobamate is not indicated for use in pediatric patients (aged < 18 years) due to unavailability of evidence. | Initial: 12.5 mg oral tablet once daily in weeks 1 and 2 followed by 25 mg, 50 mg, 100 mg, and 150 mg oral tablets once daily at weeks 3 and 4, weeks 5 and 6, weeks 7 and 8, and weeks 9 and 10. Maintenance: 200 mg oral tablets once daily at week 11 and thereafter. Based on clinical response and tolerability, dose may be increased by increments of 50 mg once daily every 2 weeks to a maximum daily dose of 400 mg. | Somnolence, dizziness, fatigue, and headache that are considered as mild or moderate in severity. The safety profile has not been established in the pediatric population. Cenobamate is not recommended for patients with hepatic or renal insufficiency. |
First generation | ||||
Phenytoin (DILANTIN),42 1951a | Voltage-gated sodium channel blocker | Control of generalized tonic-clonic and psychomotor (grand mal and temporal lobe) seizures and prevention and treatment of seizures that occur during or following neurosurgery.42 | Adults: Initial dose of 100 mg oral tablets 3 times daily; lower doses may be required for patients aged > 65 years. The satisfactory maintenance dose is 300 mg to 400 mg daily, and can be increased to 600 mg daily, if necessary. Pediatric patients (aged < 18 years): Initial does of 5 mg/kg/day in 2 or 3 equally divided doses, followed by 4 mg/kg/day to 8 mg/kg/day, with a maximum dose of 300 mg daily. | Enzyme inducer, nonlinear pharmacokinetics. Not useful for absence or myoclonic seizures. Skin hypersensitivity. |
Phenobarbital (PHENOBARB),43 1957a | GABA potentiation | Long-term treatment of generalized tonic-clonic and partial (cortical focal) seizures.43 (Can be used as an anticonvulsant, sedative, or hypnotic, with different dosage recommendations.) | Adults (aged ≥ 18 years): 50 mg to 100 mg oral tablets 2 or 3 times a day. Children (aged < 18 years): 15 mg to 50 mg oral tablets 2 or 3 times a day. | Enzyme inducer. Not useful in absence seizures. Skin hypersensitivity. Drowsiness. |
Ethosuximide (ZARONTIN),44 1960a | T-type voltage-gated calcium channel blocker | Control of absence (petit mal) epilepsy.44 | Initial: Children (aged 3 years to 6 years), 250 mg/day oral tablets; older patients: 500 mg/day in divided doses. Small increments are recommended (e.g., increase daily dose by 250 mg every 4 days to 7 days). The optimal dose for most children is 20 mg/kg/day. Dosages exceeding 1.5 g/day should be administered only under the strictest supervision. | Not useful for other types of seizures. Gastrointestinal adverse effects and insomnia. |
Carbamazepine (TEGRETOL),45 1969a | Voltage-gated sodium channel blockade | Adults and children aged 12 years and older: Initial dose of 100 mg to 200 mg oral tablets once or twice daily followed by 800 mg/day to 1,200 mg/day, with a maximum dose of 1,600 mg/day. Children aged 6 years to 12 years: initial dose of 100 mg in 2 to 4 divided doses on the first day followed by gradually adding 100 mg per day, to a maximum dose of 1,000 mg/day. | Enzyme inducer. Not useful for absence or myoclonic seizures. Skin hypersensitivity. | |
Primidone,46 1977a | GABA potentiation | Control of grand mal and psychomotor seizures. Monotherapy or add-on.46 | Adults and children aged > 8 years: Week 1, 250 mg oral tablets at bedtime; week 2, 250 mg twice daily (morning and evening); week 3, 250 mg 3 times daily; week 4, 250 mg 4 times daily. In patients already receiving other anticonvulsants, the usual dosage is 125 mg to 1,500 mg daily in divided doses. Children under the age of 8 years: Week 1, 125 mg at bedtime; week 2, 125 mg twice daily (morning and evening); week 3, 125 mg 3 times daily; week 4, 125 mg 4 times daily. Dosages exceeding 2 g daily are not recommended. | Enzyme inducer. Not useful in absence seizures. Sedative. Skin hypersensitivity. Less effective than carbamazepine or phenytoin for focal seizures in new onset epilepsy. |
Valproate (DEPAKENE),47 1978a | Multiple (GABA potentiation, glutamate inhibition, sodium channel and T-type voltage-gated calcium channel blockade) | Initial: 15 mg/kg/day oral, increasing at 1-week intervals by 5 mg/kg/day to 10 mg/kg/day, with a maximum dose of 60 mg/kg/day. When the total daily dose exceeds 250 mg, valproic acid should be given in a divided regimen. | Enzyme inhibitor. Substantial teratogenicity. Weight gain. Tremor. | |
Second generation | ||||
Gabapentin (NEURONTIN),48 1994a | Voltage-gated calcium blocker (Alpha2δ subunit) | Adjunctive therapy for the management of patients with epilepsy who are not satisfactorily controlled by conventional therapy.48 | Initial dose of 300 mg oral tablets 3 times daily, adjusted to 300 mg or 400 mg capsules, or 600 mg or 800 mg tablets, 3 times a day up to 1,800 mg/day. The maintenance dosage should be given in 3 equally divided doses daily. To prevent breakthrough convulsions, the maximum time between the 3 daily doses should not exceed 12 hours. | Currently for adjunctive use only. Not useful for absence or myoclonic seizures and can cause weight gain. Not as effective as carbamazepine for new onset focal seizures. Gabapentin is not indicated for use in children under 18 years of age. |
Vigabatrin (SABRIL),49 1994a | GABA potentiation | Treatment of epilepsy in only those patients who respond inadequately to alternative treatment combinations or in whom other drug combinations have not been tolerated and in whom the potential benefits conferred by its use outweigh the risk of ophthalmologic abnormalities.49 | Adults: Initial dose of 1 g/day followed by an adjustment (increase or decrease) of 0.5 g/day, with a maximum dose of 3 g/day. The optimal dose is 2 g/day to 3 g/day. Children (aged 2 years to 16 years): Initial dose of 40 mg/kg/day; maintenance dosage based on body weight:
Infants (for treatment of infantile spasms, i.e., West syndrome): initial dose of 50 mg/kg/day given in 2 divided doses followed by 25 mg/kg to 50 mg/kg increments every 3 days up to a maximum of 150 mg/kg/day. | Not useful for absence or myoclonic seizures. Limited use due to 30% of patients developing a permanent concentric visual field defect. Not as efficacious as carbamazepine for focal seizures. |
Lamotrigine (LAMICTAL),50 1995a | Voltage-gated sodium channel blocker | Adults and children aged > 12 years: Initial dose of a 25 mg oral tablet once daily (weeks 1 and 2) followed by 25 mg twice daily (weeks 3 and 4) and increments of 25 mg to 50 mg every 1 week to 2 weeks (from week 5 onward) to a maintenance dose of 50 mg to 100 mg twice daily. For patients taking medications that induce lamotrigine glucuronidation without valproate acid: An initial dose of 50 mg once daily (weeks 1 and 2) followed by 50 mg twice daily (weeks 3 and 4) and increments of 100 mg every 1 week to 2 weeks (from week 5 onwards) to a usual maintenance dose of 150 mg to 250 mg twice daily. | Enzyme inducer. Skin hypersensitivity. Not as effective as valproate for new onset absence seizures. In pediatric patients (aged < 16 years), the efficacy and safety of lamotrigine have not been established other than for those with Lennox-Gastaut Syndrome. | |
Benzodiazepines
| GABA potentiation | Clonazepam:
Clobazam:
| Sedative. Risk of substantial tolerance (loss of efficacy). | |
Topiramate (TOPAMAX),53 1999a | Multiple (GABA potentiation, glutamate inhibition, and sodium and calcium channel blockade) | Monotherapy for adults and children (aged 6 years and older): week 1, 25 mg oral tablet in evening; weeks 2 and 3, 25 mg twice daily (morning and evening); weeks 3 and 4, 50 mg twice daily. The initial target dose is 100 mg/day and can be increased at weekly intervals in increments of 50 mg/day, if required, to a maximum of 400 mg/day, and should be administered in 2 divided doses. Adjunctive therapy for adults (aged ≥ 17 years): Initial dose of 50 mg/day followed by weekly increments of 50 mg/day (in 2 divided doses) to 200 mg/day to 400 mg/day, with a maximum dose of 800 mg/day. Adjunctive therapy for children (aged 2 years to 16 years): Initial dose of 25 mg (or less, based on a range of 1 mg/kg/day to 3 mg/kg/day) nightly for the first week followed by weekly increments of 1 mg/kg/day to 3 mg/kg/day, up to 5 mg/kg/day to 9 mg/kg/day in 2 divided doses. | Cognitive side effects, kidney stones, speech and other cognitive problems, weight loss. Monotherapy for pediatric patients has been conducted only in clinical trials with limited numbers of patients. | |
Oxcarbazepine (TRILEPTAL),54 2002a | Voltage-gated sodium channel blocker | Monotherapy or adjunctive therapy for partial seizures (aged ≥ 6 years)54 | Adult adjunctive therapy: Initial dose of 600 mg/day (as a twice-daily regimen) followed by weekly increments of ≤ 600 mg/day to 1,200 mg/day. Adult monotherapy: initial dose of 600 mg/day (as a twice-daily regimen) followed by increments of 300 mg/day every third day to 1,200 mg/day. Pediatric (aged 6 years to 16 years) adjunctive therapy: Initial dose of 8 mg/kg/day to 10 mg/kg/day (as a twice-daily regimen), not to exceed 600 mg/day. The target maintenance dose is by body weight and should be achieved over 2 weeks: 20 kg to 29 kg, 900 mg/day; 29.1 kg to 39 kg, 1,200 mg/day; > 39 kg, 1,800 mg/day. Pediatric monotherapy: Initial dose of 8 mg/kg/day to 10 mg/kg/day (as a twice-daily regimen) followed by increments of 5 mg/kg/day every third day to the recommended maintenance dose by body weight, with a range from 20 kg (600 mg/day to 900 mg/day) to 70 kg (1,500 mg/day to 2,100 mg/day). | Enzyme inducer, hyponatremia, skin hypersensitivity. Not useful for absence or myoclonic seizures. For adults using oxcarbazepine as adjunctive therapy, most patients are not able to tolerate the 2,400 mg/day dose, primarily because of central nervous system effects. For pediatric patients (aged < 2 years), the efficacy and safety of oxcarbazepine have not been established. |
Levetiracetam (KEPPRA),55 2003a | Synaptic vesicle glycoprotein (SV2A) modulation | Adjunctive therapy in the management of adult patients (aged > 18 years) with epilepsy who are not satisfactorily controlled by conventional therapy.55 | Initial dose of 500 mg oral tablets twice daily (1,000 mg/day) followed by biweekly increments of 1,000 mg to a maximum daily dose of 3,000 mg. | Psychiatric side effects (irritability and other neuropsychiatric effects). Pediatric patients (aged < 18 years): Health Canada has not authorized an indication for pediatric use. |
Third generation | ||||
Lacosamide (VIMPAT),56 2010a | Enhanced slow inactivation of voltage-gated sodium channels | Monotherapy or adjunctive therapy in the management of partial onset seizures in adult patients.56 | Monotherapy: Initial dose of 100 mg twice daily (200 mg/day) oral tablets, followed by 100 mg/day at weekly intervals to 200 mg/day or 400 mg/day, with a maximum dose of 400 mg/day. Adjunctive therapy: Initial dose of 50 mg twice daily followed by 100 mg twice daily after 1 week. The maintenance dose can be increased by 20 mg twice daily every week to a maximum dose of 200 mg twice daily (400 mg/day). | Most common: Dizziness, headache, nausea, vomiting, diplopia, fatigue, and sedation (more common at higher doses, more likely when used in conjunction with other sodium channel blockers). Pediatric patients (aged < 18 years): The efficacy and safety of lacosamide have not been established. |
Perampanel (FYCOMPA),57 2013a | Glutamate antagonist |
| Initial dose of 4 mg/day oral suspension followed by increments of 2 mg to a maximum dose of 12 mg/day. Dose increments should occur no more frequently than at 1-week intervals. | Currently for adjunctive use only. Boxed warning: serious psychiatric and behavioural reactions (aggression and hostility, with an incidence of 20% at a dose of 12 mg/day). |
Eslicarbazepine acetate (APTIOM),58 2014a | Voltage-gated sodium channel blocker | Adults and children with a body weight of ≥ 40 kg: Initial dose of 400 mg oral tablet once daily followed by 800 mg once daily, with a maximum dose of 1,200 mg once daily. Children aged > 6 years: Initial dose of 10 mg/kg/day once daily followed by weekly or biweekly increments of 10 mg/kg/day up to 30 mg/kg/day, with a maximum dose of 1,200 mg once daily. | Enzyme inducer, hyponatremia. Pediatric patients (aged ≤ 6 years): The efficacy and safety of eslicarbazepine acetate have not been established. | |
Brivaracetam (BRIVLERA),59 2016a | SV2A modulation | Adults: Initial dose of 50 mg oral tablets twice daily (100 mg/day) followed by adjustment between 25 mg twice daily and a maximum dose of 100 mg twice daily (200 mg/day). Children aged ≥ 4 years and adolescents weighing 11 kg to < 20 kg: Initial dose of 1 mg/kg/day to 2.5 mg/kg/day followed by 1 mg/kg/day to 5 mg/kg/day; weighing 20 kg to < 50 kg: initial dose of 1 mg/kg/day to 2 mg/kg/day followed by 1 mg/kg/day to 4 mg/kg/day; weighing 50 kg or more: initial dose of 50 mg/kg/day to 100 mg/day followed by 50 mg/day to 200 mg/day. | Not effective when added to levetiracetam. US: Monotherapy and add-on (no monotherapy trials). For pediatric patients aged ≥ 4 years with hepatic impairment, the lower boundary of the initial doses is recommended. For pediatric patients aged < 4 years of age, the efficacy and safety of brivaracetam have not been established. | |
GABA = gamma-aminobutyric acid; GABAA = gamma-aminobutyric acid type A; SV2A = synaptic vesicle glycoprotein.
aHealth Canada–approved indication(s).
Source: Product monographs.35,41-59
Clinical Evidence
The objective of CADTH’s clinical review is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of cenobamate 12.5 mg, 25 mg, 50 mg, 100 mg, 150 mg, and 200 mg oral tablets as adjunctive therapy for the management of partial onset seizures in adults who are not satisfactorily controlled with conventional therapy. The focus will be on comparing cenobamate to relevant comparators and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of cenobamate is presented in 4 sections. CADTH’s critical appraisal of the evidence is included after each section. The first section, the Systematic Review, includes pivotal studies and RCTs that were selected according to the sponsor’s systematic review protocol. The second section includes sponsor-submitted, long-term extension studies. The third section includes indirect evidence from the sponsor. The fourth section includes additional studies that were considered by the sponsor to address important gaps in the pivotal and RCT evidence.
Included Studies
Clinical evidence from the following are included in the CADTH review and appraised in this document:
Two pivotal trials included in the systematic review
Two long-term extension studies
Two ITCs
One prospective safety study and 3 additional studies addressing gaps in the evidence.
Of note, the sponsor included 3 trials in its summary of the pivotal and RCT evidence (studies C013, C017, and C021). However, Study C021, an open-label single-arm safety study, was not considered a pivotal trial by Health Canada and did not meet the study design criteria for inclusion in the systematic review section of the CADTH report. Study C021 was summarized in the Studies Addressing Gaps in the Pivotal and RCT Evidence section of this report because it provides additional safety data as well as evidence to support the dose titration schedule listed in the product monograph.
Pivotal Studies and RCT Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following information has been summarized and validated by the CADTH review team.
Description of Studies
Characteristics of the included studies are summarized in Table 4.
Table 4: Details of Pivotal Studies and RCT Evidence Identified by the Sponsor
Details | Study C013 | Study C017 |
|---|---|---|
Designs and populations | ||
Study design | A pivotal, phase II, multicentre, randomized, DB, placebo-controlled study | A pivotal, phase II, multicentre, randomized, DB, placebo-controlled study |
Locations | Multicentre study: 40 sites Countries involved: US, India, Republic of Korea, Poland | Multicentre study: 107 study centres Countries involved: US, Australia, New Zealand, South Africa, Bulgaria, Czechia, France, Germany, Hungary, Israel, Republic of Korea, Poland, Romania, Serbia, Spain, Thailand, Ukraine, UK |
Patient enrolment dates | Start date: July 6, 2011 End date: June 15, 2013 | Start date: July 31, 2013 End date: June 22, 2015 |
Randomized (N) | Total N = 222 Cenobamate = 113 Placebo = 109 | Total N = 437 Cenobamate 100 mg/day = 108 Cenobamate 200 mg/day = 110 Cenobamate 400 mg/day = 111 Placebo = 108 |
Inclusion criteria |
|
|
Exclusion criteria |
|
|
Drugs | ||
Intervention | Cenobamate 200 mg oral tablet once daily | Cenobamate 100 mg, 200 mg, or 400 mg oral tablet once daily |
Comparator(s) | Placebo, oral, once daily | Placebo, oral, once daily |
Study duration | ||
Baseline phase | 8 weeks (4 weeks could be retrospective) | 8 weeks |
Treatment phase | 12 weeks (6-week titration phase and 6-week maintenance phase) | 18 weeks (6-week titration phase and 12-week maintenance phase) |
Follow-up phase | 1-week study drug taper period (for patients leaving the study) or a 2-week blinded conversion period (for patients participating in the open-label extension), with a final follow-up visit 3 weeks after the last dose of study drug | 3-week blinded study drug taper period (for patients leaving the study) or a 2-week blinded conversion period (for patients participating in the open-label extension), with a final follow-up visit 2 weeks after the last dose of study drug |
Outcomes | ||
Primary end point | Percent change in seizure frequency per 28 days in the DB treatment period compared to the baseline for all simple partial motor (type B), complex partial (type C), or secondarily generalized (type D) seizures | US and ROW: Percent change from the pre-treatment baseline phase in seizure frequency (average monthly seizure rate per 28 days) of all simple partial motor (type B), complex partial (type C), or secondarily generalized (type D) seizures in the DB treatment period Europe, Australia, New Zealand, and South Africa: The responder rate was defined as a ≥ 50% reduction from baseline in seizure frequency (all type B, type C, or type D seizures) during the maintenance period of the DB treatment period |
Secondary and exploratory end points | Secondary: Responder rate defined as patients who experienced a 50% or greater reduction in partial seizure frequency (all type B, C, and D) in the treatment period of the DB period Exploratory:
| Key secondary: US and ROW: Responder rate defined as a ≥ 50% reduction from baseline in the partial seizure frequency during the DB treatment period Europe, Australia, New Zealand, and South Africa: Percent change in partial seizure frequency from the pretreatment baseline phase compared with the maintenance period of the DB treatment period Additional secondary:
|
Publication status | ||
Publications | Chung et al. (2020)60 | Krauss et al. (2020)61 |
ASM = antiseizure medication; BMI = body mass index; CNS = central nervous system; DB = double blind; EEG = electroencephalogram; QOLIE-31-P = patient-weighted 31-item Quality of Life in Epilepsy Questionnaire; RCT = randomized controlled trial; ROW = rest of world.
Note: Four additional reports were included: FDA Clinical Review,62 FDA Statistical Review,63 Chung et al. (2020),60 and Krauss et al. (2020).61
Source: Clinical Study Report for Study C013;10 Clinical Study Report for Study C017.9
Two pivotal, phase II, DB, randomized, placebo-controlled trials (studies C013 and C017) met the inclusion criteria for the sponsor’s systematic review.
The primary objective of study C013 was to evaluate the efficacy of cenobamate when titrated to 200 mg per day from 50 mg per day in reducing seizure frequency in adults with partial onset seizures whose seizures are not fully controlled despite treatment with 1 to 3 concomitant ASMs. The secondary objectives were to assess the safety and tolerability of cenobamate. Patients who met the enrolment criteria underwent a pre-treatment period to determine the baseline rate of partial seizures. This baseline period could include data from the patient’s own seizure diary completed before enrolment (for 4 weeks) plus another 4 weeks’ prospective data capture, or 8-weeks of prospective follow-up, as part of the baseline study period. Patients were eligible for randomization if they experienced at least 3 seizures per 28 days during the baseline period and must not have a seizure-free interval greater than 21 days despite ongoing therapy with 1 to 3 ASMs. Allocation to treatments was conducted using an interactive web response system based on a computer-generated randomization schedule stratified by country. A total of 222 patients were assigned 1:1 to either cenobamate 200 mg or placebo once daily for 12 weeks (including a 6-week dose titration period and a 6-week maintenance period). The primary outcomes were the percent changes in partial seizure frequency relative to baseline. The study was conducted between July 2011 and June 2013 and included patients from the US, India, Republic of Korea, and Poland.
The objective of Study C017 was to determine the effective dose range and the safety and tolerability of cenobamate as adjunctive therapy for the treatment of partial seizures in adults. The study design for Study C017 is shown in Figure 1. Patients who met the enrolment criteria underwent an 8-week baseline pre-treatment period to assess the frequency of partial seizures. Patients were eligible for randomization if they experienced at least 8 seizures during the baseline period despite ongoing therapy with 1 to 3 ASMs. They must have experienced at least 3 partial seizures during each of the 2 consecutive 4-week baseline periods and must not have had a seizure-free interval greater than 25 days. A total of 437 patients were randomized (1:1:1:1) to placebo, cenobamate 100 mg, cenobamate 200 mg, or cenobamate 400 mg once daily for a total of 18 weeks (a 6-week dose titration period, then a 12-week maintenance period). The computer-generated randomization sequence was based on permuted blocks of 4 stratified by country, with allocation completed centrally using an interactive web response system. The primary outcomes were the percent change in partial seizure frequency relative to baseline and the proportion of patients with at least a 50% reduction in partial seizure frequency. The trial was conducted from July 2013 to June 2015 in Europe, Asia, US, New Zealand, Australia, and South Africa. No patients were enrolled in Canada.
For both studies, patients who completed the trials were eligible to enter an extension phase and receive open-label cenobamate.
Study C017 included a cenobamate 100 mg dosage group. This dosage is lower than the recommended target dose for cenobamate.35 Given that cenobamate was still under review by Health Canada when this report was drafted, and that not all patients may be able to tolerate the 200 mg daily dose, it was determined that a summary of the 100 mg dose group was warranted; thus, these data have been included in CADTH’s clinical review.
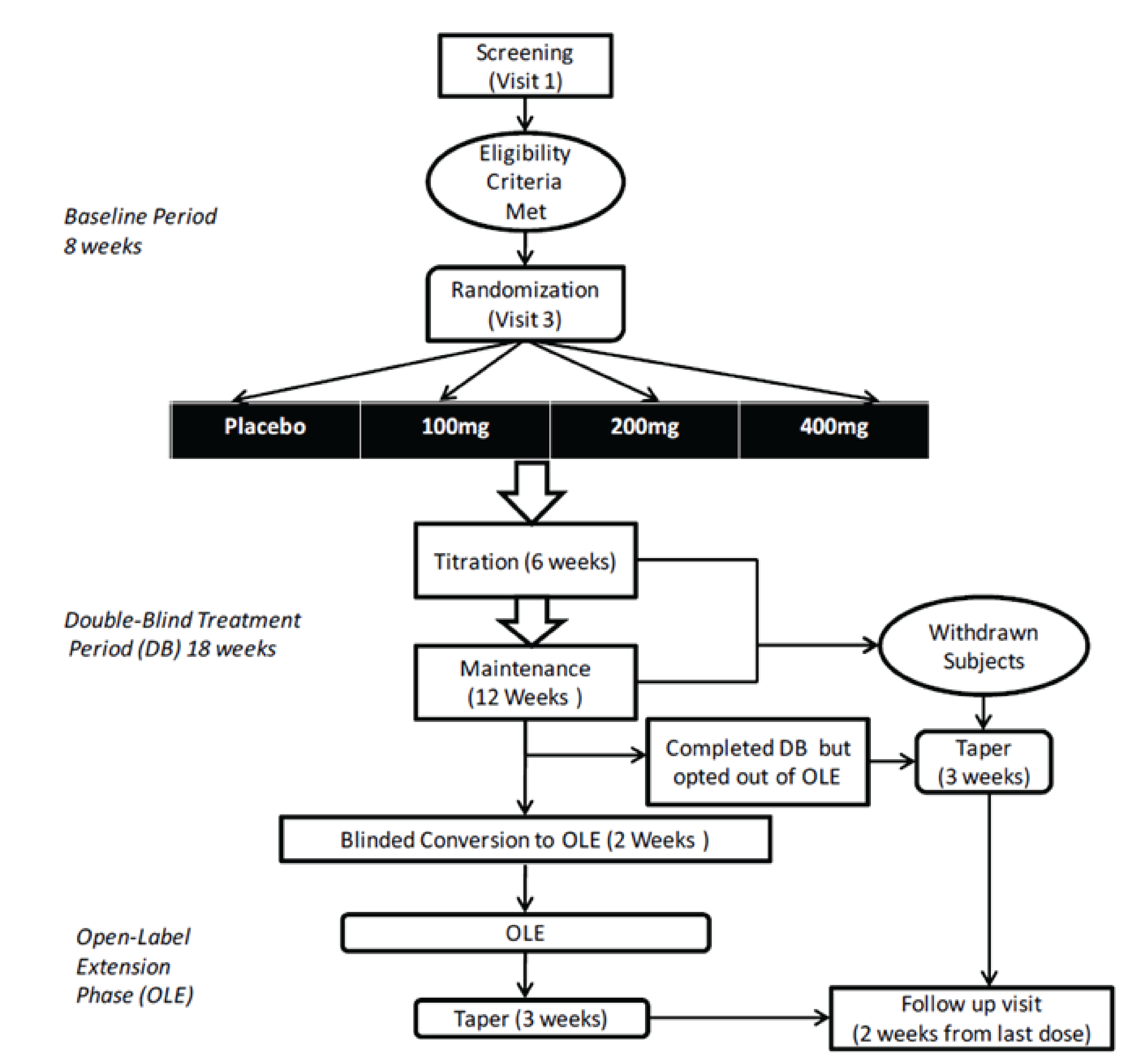
Populations
Inclusion and Exclusion Criteria
Both pivotal trials included adults who were diagnosed with partial epilepsy according to the International League Against Epilepsy’s classification of epileptic seizures and who had uncontrolled seizures despite treatment with 1 to 3 ASMs at stable doses for at least 12 weeks before screening. In Study C013, patients were required to meet the criteria for treatment-resistant partial epilepsy, defined as having failed treatment with at least 2 ASMs at doses that were at least 50% of the WHO–defined daily dose for at least 3 months.
To be eligible for enrolment in the pivotal trials, patients had to experience a minimum number of partial seizures during the 8-week baseline period:
In Study C013, the minimum was at least 3 seizures per month (simple partial with motor component, complex partial, or secondarily generalized), with no 21-day seizure-free period.
In Study C017, the minimum was at least 8 seizures (simple partial with motor component, complex partial, or secondarily generalized), without a seizure-free interval greater than 25 days, and at least 3 seizures per 4-week block.
Patients were excluded from the studies if they had other forms of epilepsy, seizure clusters, or had experienced status epilepticus in the past year. In addition, those with a history of allergic reactions to ASMs, impaired renal or liver function, clinically significant electrocardiogram abnormalities, or cardiac disorders were prohibited from entering the studies. Patients with a neurologic or central nervous system disease, psychiatric or psychotic illness, history of alcohol or drug abuse, or suicidality were also excluded. The prohibited medications are described in the Interventions section.
Interventions
Study Drug
In Study C013, patients were randomized to receive blinded cenobamate 200 mg once daily or placebo for a total of 12 weeks. The study drug was supplied as identical-looking capsules containing cenobamate or placebo in blister packages. The initial dose of cenobamate was 50 mg once daily. This was increased in 2-week intervals until the target dose (200 mg) was reached or until the patient experienced adverse effects (Table 5). Patients who reported adverse effects were not titrated to the higher dose until the next study visit, at which point the investigator had the choice to keep the patient on the same dose or to increase or decrease the dose. Patients who had doses reduced due to adverse effects could not be titrated to a higher dose at the next study visit. The study drug dose could not be titrated up during the maintenance phase, but dose reductions for tolerability were allowed.
In Study C017, patients were randomized to DB treatment with placebo, cenobamate 100 mg, 200 mg, or 400 mg once daily for a total of 18 weeks. All patients and study personnel were blinded to treatment allocation. The study drug was supplied as identical-looking tablets containing cenobamate 50 mg, cenobamate 100 mg, or placebo in blister packs. For the first 6 weeks, daily doses of the study drug were increased each week by 50 mg or 100 mg until the target dose was achieved (Table 5). Patients with significant tolerability issues during week 1 were discontinued from treatment. From week 2 to week 6, if patients did not tolerate the drug, they were allowed to reduce the dose to the previous week’s dosage for the next 7 days to 13 days, after which the upward titration could continue (until week 6) at the discretion of the investigator. If the new upward titration was not tolerated, the daily dose could be reduced by 50 mg (until the end of week 8). No further up-titration was allowed after week 6, and consecutive dose reductions were not allowed.
Those who completed the studies had the option to enter the OLE and were either directly converted to open-label cenobamate or underwent a 2-week DB dose conversion phase. Patients who withdrew or were not continuing in the OLE underwent a 1- or 3-week taper period, with the final visit conducted 2 weeks or 3 weeks after the last dose of study drug.
Concomitant Medications
During the DB treatment period in studies C013 and C017, patients continued taking the background 1 to 3 ASMs that they had been receiving for the 12 weeks before the study. Vagal nerve stimulation was not counted as an ASM; it was allowed to continue during the trial, provided patients’ parameters remained stable. Benzodiazepines taken at least once per week for epilepsy, anxiety, or sleep disorder were counted as 1 ASM.
Patients were allowed to receive intermittent benzodiazepines (other than diazepam) as rescue therapy once during the baseline period and twice during the treatment phase.
During the studies, the doses of the background ASMs were to remain stable. In Study C017, the investigator could modify the timing or amount of an individual dose of background ASMs to improve tolerability, but the total daily dosage and dosage frequency had to remain unchanged.
Prohibited Medications
During the trial and in the 30 days before the baseline phase, patients were prohibited from receiving diazepam, phenytoin, phenobarbital, or metabolites of these drugs, as well as vigabatrin (in the year before screening). Due to potential interactions between drugs, patients were prohibited from taking clopidogrel, fluvoxamine, amitriptyline, clomipramine, bupropion, methadone, ifosfamide, cyclophosphamide, efavirenz, natural progesterone (both trials), and omeprazole (Study C013 only).
Table 5: Dose Titration Schedule — Study C013 and Study C017
Study | Randomized dose group | Daily dose (mg) | ||||||
|---|---|---|---|---|---|---|---|---|
Week 1 | Week 2 | Week 3 | Week 4 | Week 5 | Week 6 | Week 7 and beyond | ||
Study C013 | 200 mg daily | 50 | 50 | 100 | 100 | 150 | 150 | 200 |
Study C017a | 100 mg daily | 50 | 100 | 100 | 100 | 100 | 100 | 100 |
200 mg daily | 50 | 100 | 150 | 200 | 200 | 200 | 200 | |
400 mg daily | 50 | 100 | 150 | 200 | 300 | 400 | 400 | |
aAt the start of Study C017, the titration schedule increased the dose in 100 mg increments. However, following a blinded review of tolerability, the protocol was amended, and doses were increased in 50 mg increments after the first 46 patients were treated.
Source: Clinical Study Report for Study C013;10 Clinical Study Report for Study C017.9
Outcomes
A list of efficacy end points assessed in this clinical review report are provided in Table 6. These end points are further summarized in this section (Table 7). Summarized end points are based on those included in the sponsor’s Summary of Clinical Evidence along with any identified as important to this review according to stakeholders, such as the clinical expert, clinician groups, or patient groups.
In both studies, the seizure events included in the analyses were all simple partial motor (type B, or focal aware motor), complex partial (type C, or focal impaired awareness), and secondarily generalized seizures (type D, or focal to bilateral tonic-clonic), excluding simple partial seizures without a motor component (type A, or focal aware nonmotor). Data on the number and type of seizures experienced were recorded daily by patients using a seizure diary. Seizure diaries were collected and reviewed at each study visit, and new diaries were dispensed. The seizure rate was computed based on days without missing data, assuming that days with missing data would have the same seizure rate as days with nonmissing data. The baseline seizure rate was calculated by counting the number of seizures during the baseline period (56 days) and dividing the result by the number of days in the interval with nonmissing seizure data, then multiplying by 28. The same method was used to calculate the seizure rate during the DB treatment period. The percent change in seizure rate was calculated using the following formula: DB period seizure frequency per 28 days minus the baseline seizure frequency per 28 days, with the result divided by the baseline seizure frequency per 28 days and multiplied by 100. The responder analyses were based on the proportion of patients who achieved at least a 50%, 75%, 90%, or 100% reduction in seizure frequency versus baseline.
The primary outcome in Study C013 was the percent change from baseline in partial onset seizure frequency per 28 days for the DB treatment period (Table 6). The secondary outcome was the proportion of patients who achieved at least a 50% reduction in seizure frequency during the DB treatment period.
In Study C017, 2 sets of primary and key secondary outcomes were defined in the study’s protocol to meet the regulatory requirements of different countries (Table 6). The changes in seizure frequency or responder rate for Europe, Australia, New Zealand, and South Africa were calculated based on the subset of patients who entered the maintenance period (i.e., the modified intention-to-treat-in-maintenance-period [mITT-M] population) based on seizure events that occurred during maintenance therapy only (last 12 weeks). The seizure frequency or responder rates for the US and rest of the world were based on all patients (i.e., the modified intention-to-treat [mITT] population) using seizure data reported during the entire DB treatment period (induction and maintenance).
Both studies also analyzed other seizure response thresholds (defined by cut-offs of ≥ 75%, ≥ 90%, and 100% reduction in partial seizures) for the DB treatment period (mITT population), and for the maintenance period only in the subgroup of patients who took at least 1 dose of cenobamate during the maintenance period (i.e., subgroup 5 in Study C013). In Study C013, these analyses were conducted post hoc.
In Study C017, the QOLIE-31-P version 2.0 was completed by patients at baseline and at the end of the maintenance period. QOLIE-31-P captures overall quality of life, seizure worry, emotional well-being, energy and fatigue, cognitive, medication effects, and social functioning. All subscales and the total score range from 0 to 100, with higher scores indicating better function (Table 7). In Study C017, QOLIE-31-P was completed only by English-speaking patients in the US, UK, and Australia. In addition, patients were prohibited from completing the questionnaire if they were cognitively impaired and unable to understand the scale, were unable to read, or had not fully recovered from a recent seizure and were unable to reschedule the study site visit to complete the questionnaire. The sponsor defined the minimally important change as at an increase of least 11.8 points versus baseline in the weighted overall score.9 No HRQoL data were collected in Study C013.
In both studies, AEs were defined as any symptoms, signs, illnesses, or experiences that developed or worsened in severity during the study and were captured up to 30 days after the last dose of the study drug. In Study C017, seizures were not considered AEs unless these occurred with a measurable increase over the patients’ typical seizure frequency or duration, or included multiple seizures in a pattern that was distinguishable from the usual seizure pattern.
SAEs included events that were fatal, life-threatening, required or prolonged a hospital stay, resulted in persistent or significant disability or incapacity, congenital anomaly, or birth defect, or were an important medical event. The Columbia-Suicide Severity Rating Scale was used to assess for suicidal ideation or behaviour in both studies.
The sponsor listed seizure-free days, treatment retention or adherence, and tolerability as outcomes of interest in its systematic review protocol; however, these outcomes were not measured in either study. In addition, neither study assessed patients’ ability to work, attend school, or maintain independence, which were outcomes of importance to patient groups.
Table 6: Outcomes Summarized From Pivotal Studies and RCT Evidence Identified by the Sponsor
Outcome measure | Time pointa | Description, time point | Study C013 | Study C017 |
|---|---|---|---|---|
Seizure frequencyb | DB treatment period | Percent change from baseline in partial onset seizure frequency per 28 days (mITT population) | Primary | US and ROW: Primaryc |
Seizure frequencyb | Maintenance period | Percent change from baseline in partial onset seizure frequency per 28 days (mITT-M population) | Subgroup analysis | Europe: Key secondary |
≥ 50% seizure responseb | DB treatment period | Proportion of patients who achieved at least a 50% reduction in partial onset seizure frequency vs. baseline (mITT population) | Secondary | US and ROW: Key secondary |
≥ 50% seizure responseb | Maintenance period | Proportion of patients who achieved at least a 50% reduction in partial onset seizure frequency vs. baseline (mITT-M population) | Subgroup analysis | Europe: Primaryc |
Other seizure response thresholdsb | DB treatment period | Proportion of patients who achieved at least a ≥ 75%, ≥ 90%, or 100% reduction in partial onset seizure frequency vs. baseline (mITT population) | Exploratory (post hoc) | Additional secondary |
Other seizure response thresholdsb | Maintenance period | Proportion of patients who achieved at least a ≥ 75%, ≥ 90%, or 100% reduction in partial onset seizure frequency vs. baseline (mITT-M population) | Exploratory (post hoc) subgroup analysis | Additional secondary |
Seizure frequencyb | DB treatment period | Percent change from baseline in partial onset seizure frequency per 28 days, analyzed separately by each type of partial seizure (mITT population) | Other secondary | Additional secondary |
HRQoL | DB treatment period | Change from baseline in QOLIE-P-31 (mITT population in US, UK, and Australia only) | NA | Additional secondary |
DB = double blind; HRQoL = health-related quality of life; mITT = modified intention to treat; mITT-M = modified intention to treat in maintenance period; NA = not applicable; QOLIE-31-P = patient-weighted 31-item Quality of Life in Epilepsy Questionnaire; RCT = randomized controlled trial; ROW = rest of world.
aThe durations of the DB treatment period were 12 weeks in Study C013 and 18 weeks in Study C017. The maintenance periods were 6 weeks in duration for Study C013 and 12 weeks for Study C017, excluding the first 6 weeks of treatment, during which the dose of study drug was being titrated.
bThe analysis of partial seizures was based on the total number of seizures (simple partial with motor component, complex partial [unique seizure types to be combined], and secondarily generalized tonic-clonic). Partial seizures without a motor component (i.e., type A) were not analyzed.
cStatistical testing was adjusted for multiple comparisons using a hierarchical testing procedure for the cenobamate 200 mg, 400 mg, and 100 mg dose groups (in that order) vs. placebo.
Source: Clinical Study Report for Study C013;10 Clinical Study Report for Study C017.9
Table 7: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
Seizure frequency per 28 days | Seizure frequency is represented in terms of the number of seizures occurring in patients over a defined period of time (e.g., 1 month or 3 months).64,65 Seizure counts are normally taken by patients using a diary. Ambulatory EEG devices are also available and widely used to monitor seizures, especially for patients with generalized seizures.64 Seizure frequency is typically considered to refer to seizures counted in a time frame of 28 days.66 Seizure frequency per 28 days is commonly reported in studies that investigate the efficacy and safety of ASMs in adults with partial onset seizures. It enables the comparison across studies. It is calculated as the total number of seizures reported during the period divided by number of days during the same period, multiplied by 28.67 | ILAE suggests that seizure frequency is the most sensitive outcome measure as a continuous variable; it is recommended to be evaluated whenever possible.64 However, seizure events may be under-reported by patients in comparison with EEG-detected events.68-71 Some studies calculated seizure frequency based on the entire treatment period (including the titration and maintenance phases). Others assessed frequency during the maintenance phase only. No information was identified regarding which of these methods was preferred by regulators or others. | Not identified |
Seizure freedom (100% response) was achieved | This refers to the patients experiencing at least a 100% reduction in the frequency of seizures compared with baseline. | To achieve seizure freedom with minimal or no AEs is the goal of ASM treatment. The methods used to analyze and report on seizure freedom in ASM studies might vary.72 Aside from the number or proportion of patients achieving seizure freedom at defined time points, the duration of maintained seizure freedom is another key measurement to be evaluated and reported.73 | NA |
Responder thresholds based on ≥ 90%, ≥ 75%, and ≥ 50% seizure reduction relative to baseline | These refer to the number or proportion of patients experiencing at least a 90%, 75%, or 50% reduction in the frequency of seizures compared with baseline (i.e., reduction between the baseline and a subsequent treatment period), respectively. All are arbitrary cut points.64,65 The 50% responder threshold is a commonly reported outcome measure in studies that investigate the efficacy and safety of ASMs in patients with various types of seizures. As such, it is feasible to use it to compare results across studies.64,74 The 75% and 90% responder thresholds are also reported, but less frequently, and can provide results as sensitivity analyses with different cut points.74 | According to the commission on outcome measurement in epilepsy, ILAE, dichotomization of seizures frequencies (i.e., the proportion of patients achieving a certain percentage reduction in seizure frequencies) offers an alternative outcome for efficacy evaluation. Percentage reduction, or responder threshold, reflects a within-person change; thus, it must be based on a baseline value (or in studies in which a patient acts as their own control, like a crossover design).64 Methodologically, it is desired to prespecify the threshold(s) of cut point(s) for percentage reduction in seizure frequency (i.e., to specify these at the study planning stage and in a publicly accessible protocol).64,75 In several previous CADTH reviews of drugs to treat partial onset seizures76,77 or epilepsy,78 a 50% reduction in seizure frequency from baseline to end of treatment was considered a clinically meaningful effect, according to the clinical experts being consulted. | NA |
QOLIE-31-P | The QOLIE-31 is a survey of health-related quality of life for adults with epilepsy. It is self-administered by patients and comprises 31 items related to the patient’s perception of their health and daily activities, derived from the longer QOLIE-89.79,80 The items in the QOLIE-31 are grouped into 7 subscales: energy fatigue, emotional well-being, social function, cognitive function (thinking, concentrating, memory), medication effects (physical, mental), seizure worry (impact of seizures), and overall QOL.79,81 The QOLIE-31-P is adapted from the original QOLIE-31. It includes an additional item assessing the degree of overall distress in each of the subscales, resulting in an instrument with 38 items for scoring. The subscales and total score range from 0 to 100, with higher scores indicating better function. In the QOLIE-31-P, a final item that asks patients to rank the importance of each subscale topic (where 1 = the very most important topic and 7 = the least important topic) is included, but does not contribute to the total or subscale scores.81 | Validity: Based on data from 304 adults with epilepsy, the QOLIE-31 demonstrates adequate to high internal consistency within each subscale. The internal consistency reliability coefficient measurement using Cronbach alpha value ranges from 0.77 (social functioning) to 0.85 (cognitive functioning).79 Reliability: The intrarater reliability is adequate to high, with person correlation coefficient measures ranging from 0.64 (medication effects) to 0.85 (cognitive functioning).79 No information on the instrument validity of the QOLIE-31-P in patients with partial onset seizures was identified. No information suggesting that the validity reported for the QOLIE-31 is transferable to the QOLIE-31-P was identified. | The MID of the QOLIE-31 instrument using various methodologies and patient population is reported to range from 4.73 to 11.8.80,82,83 The recommended MID of the QOLIE-31 total score in patients with treatment-resistant, partial onset seizures is 5.19.80 A study that assessed 136 consecutive adult patients with medically refractory focal epilepsy reported 11.8 as the MID for QOLIE-31.82 No information on the MID of QOLIE-31-P in patients with POS was identified. No information suggesting that the MID reported for the QOLIE-31 is transferable to the QOLIE-31-P was identified. |
AE = adverse event; ASM = antiseizure medication; EEG = electroencephalogram; ILAE = International League Against Epilepsy; MID = minimal important difference; NA = not applicable; POS = partial onset seizure; QOLIE-31 = 31-item Quality of Life in Epilepsy Questionnaire; QOLIE-89 = 89-item Quality of Life in Epilepsy Questionnaire; QOLIE-31-P = patient-weighted 31-item Quality of Life in Epilepsy Questionnaire.
Statistical Analysis
In both studies, seizure frequency was calculated from the patient-reported seizure diary data based on days without missing data (i.e., both missing interim data from patients who continued in the trial, and missing data from a certain date for patients who dropped out before the end of the trial). It was assumed that days with missing data would have the same seizure rate as days with nonmissing data. The sponsor indicated that there were no missing data for the primary end point of Study C013, whereas in Study C017, end point data were missing for 6 patients (5.6%), 3 patients (2.7%), 6 patients (5.4%), and 4 patients (3.7%) in the cenobamate 100 mg, 200 mg, 400 mg, and placebo groups, respectively.84
In Study C013, the primary outcome was tested based on a nonparametric Wilcoxon rank sum test (i.e., if the median percent change from baseline in the cenobamate was different from the median percent change from baseline in the placebo group). This model was selected after reviewing the results of prespecified model-fitting analyses that were run to understand the distributional characteristics of the seizure frequency data. Due to the nonparametric nature of the data, the Wilcoxon rank sum test was used for the primary analysis. The responder analyses were tested using a logistic regression model that included country and baseline seizure frequency as covariates (i.e., the Wald chi-square test). The study used the last observation carried forward for missing efficacy data. Sensitivity analyses were conducted based on the per-protocol and completer populations. There was no description in the CSR of any steps taken to control the type I error rate for the secondary and other end points.
In Study C017, the percent change in seizure rate per 28 days was analyzed using an analysis of covariance (ANCOVA) model for the ranked values of the primary end point, including the ranked baseline seizure rate as a covariate. Although randomization was stratified by country, country was not included as a covariate in the model. The CSR states that omitting country as a covariate did not affect the validity of the analysis and may not have reduced variability because each region was to capture patients who were relatively homogenous in response. Summary tables for the actual (not ranked) primary efficacy variable were presented, and a sensitivity analysis using a parametric ANCOVA model was performed. The responder rate was summarized using count and percentage of patients achieving at least a 50% response to treatment and analyzed using a Fischer’s exact chi-square test. Alternate responder thresholds (i.e., at least 75%, 90%, or 100% response) were analyzed using the same methods. Sensitivity analyses were conducted for the seizure frequency and responder analyses based on the titration period only and the first and last 6 weeks of the maintenance period only (additional details in Table 8). Sensitivity analyses were also conducted based on predefined regions based on their perceived level of epilepsy care and expertise.
In Study C017, the testing strategy for the primary efficacy end point was to compare each of the cenobamate dosage groups with the placebo group. Due to multiple treatment comparisons, a step-down procedure was used to ensure that the overall type I error rate was controlled at the 5% level. Each of the cenobamate dosage groups was compared with the placebo group at a 2-sided 0.05 level according to the following hierarchy: 200 mg group, then 400 mg group, then 100 mg group. First, the 200 mg dosage group was compared with the placebo group at a 2-sided 0.05 level. If no statistically significant difference was detected between the 200 mg dosage group and the placebo group, the procedure was to stop, and it would be concluded that none of the cenobamate dosages were efficacious. Testing proceeded through the hierarchy if the previous dosage group was statistically significant. The CSR and statistical analysis plan contained conflicting information about whether the key secondary outcomes were included in the hierarchical testing procedure. Based on CADTH’s interpretation of the information available, it was assumed that only the primary outcome was controlled for multiple testing.
An interim analysis was run in both studies, but there were no plans to modify the studies based on these data. These analyses were conducted by independent statisticians, and all study personnel were unaware of the results. The sponsor stated that there was no need to adjust the P value to account for this interim analysis.
Sample Size and Power Calculation
For the pivotal trials, a sample size of 100 patients per treatment group was estimated to provide 80% power to detect a difference of 16% in the percent reduction in seizure frequency between an active dosage group and placebo group at a 2-sided significance level of 0.05, assuming an SD of 40% using a 2-sample Wilcoxon rank sum test (Study C013) or an independent samples t test (Study C017). Potential losses to follow-up were not considered in the power calculations. No sample-size calculations were conducted for the primary responder rate end point of greater than or equal to 50% in Study C017.
Subgroup Analyses
In Study C013, subgroup analyses were conducted based on the completer population, per-protocol population, and 3 groups of patients who received at least 1 dose of the study drug during a specific period of the protocol: seizure frequency data only beyond visit 7 (last 8 weeks of treatment); seizure frequency data only beyond visit 9 (6-week analysis); and seizure frequency data only beyond visit 5 (10-week analysis).
No subgroup analyses were conducted in Study C017.
Table 8: Statistical Analysis of Efficacy End Points
End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|
Study C013 | ||||
Percent reduction in seizure frequency during DB treatment period | Wilcoxon rank sum test (mITT population) | None | Days with missing seizure diary data were excluded from the analysis; LOCF |
|
Proportion of patients with a 50% or greater reduction in seizure frequency during the DB treatment period | Logistic regression model; Wald chi-square test (mITT population) | Country, baseline seizure frequency | Days with missing seizure diary data were excluded from the analysis; LOCF |
|
Responder rate (75%, 90%, and 100%) during DB treatment and maintenance periods | Logistic regression model; Wald chi-square test (post hoc) (mITT population and subgroup 5) | Country, baseline seizure frequency | Days with missing seizure diary data were excluded from the analysis; LOCF | NR |
Study C017 | ||||
Percent reduction in seizure frequency during DB treatment period | Nonparametric ANCOVA model fit to the ranked values of the primary efficacy variable Ties handled using TIES = MEAN (mITT population) Step-down procedure for multiplicity | Ranked baseline seizure rate | Days with missing seizure diary data were excluded from the analysis |
|
Percent reduction in seizure frequency during maintenance period | Nonparametric ANCOVA model fit to the ranked values of the primary efficacy variable Ties handled using TIES = MEAN (mITT-M population) | Ranked baseline seizure rate | Days with missing seizure diary data were excluded from the analysis |
|
Proportion of patients with a 50% or greater reduction in seizure frequency during the DB treatment period | Fisher’s exact chi-square test (mITT population) | None reported | Days with missing seizure diary data were excluded from the analysis |
|
Proportion of patients with a 50% or greater reduction in seizure frequency during the maintenance period | Fisher’s exact chi-square test (mITT-M population) Step-down procedure for multiplicity | None reported | Days with missing seizure diary data were excluded from the analysis |
|
Responder rate (75%, 90%, and 100%) during DB treatment and maintenance periods | Fisher’s exact chi-square test (mITT or mITT-M) | None reported | Days with missing seizure diary data were excluded from the analysis | NR |
Change from baseline in QOLIE-31-P | Descriptive statistics (US, UK, and Australian patients only) | NA | NA | NA |
ANCOVA = analysis of covariance; DB = double blind; LOCF = last observation carried forward; mITT = modified intention to treat; mITT-M = modified intention to treat in maintenance period; NA = not applicable; NR = not reported; QOLIE-31-P = patient-weighted 31-item Quality of Life in Epilepsy Questionnaire.
Source: Clinical Study Report for Study C013;10 Clinical Study Report for Study C017.9
Analysis Populations
The efficacy outcomes in both studies were analyzed based on the mITT population, which included all randomized patients who had taken at lease 1 dose of the study drug and had any postbaseline seizure data (Table 9). Study C017 also analyzed efficacy end points based on the mITT-M population, which included all patients in the mITT population who received at least 1 dose of the study drug and had seizure data during the maintenance period. Subgroup 5 of study C013 included all patients who received at least 1 dose of the study drug during the maintenance period.
The safety population included all randomized patients who received at least 1 dose of the study drug.
Table 9: Analysis Populations of Study C013 and Study C017
Study | Population | Definition | Application |
|---|---|---|---|
Study C013 | mITTa | All randomized patients who have taken at least 1 dose of cenobamate (or placebo) and have any postbaseline seizure data |
|
Subgroup 5 | All randomized patients who have taken at least 1 dose of cenobamate (or placebo) beyond visit 9 (day 43) and who completed or did not complete the DB treatment period | Subgroup for the analysis of seizure data for the last 6 weeks of the study (i.e., maintenance period). This subgroup analysis appears to be comparable to the mITT-M population in C017. | |
Safety-evaluable population | All randomized patients who have taken a single dose of cenobamate (or placebo), analyzed based on dose received |
| |
Study C017 | mITT | All randomized patients who have taken at least 1 dose of cenobamate (or placebo) and have any postbaseline seizure data |
|
mITT-M | All randomized patients who have completed the titration period, have taken at least 1 dose of cenobamate (or placebo) in the maintenance period, and have any maintenance period seizure data |
| |
Safety-evaluable population | All randomized patients who received at least 1 dose of study drug |
|
ITT = intention to treat; mITT = modified intention to treat; mITT-M = modified intention to treat in maintenance period.
aIn the Clinical Study Report for Study C013, this population is described as the ITT population; however, for consistency with Study C017, it has been labelled mITT in this CADTH report.
Source: Clinical Study Report for Study C013,10 Clinical Study Report for Study C017.9
Results
Patient Disposition
In Study C013, 285 patients were screened and 222 patients (78%) were randomized. The reasons for screening failure were not reported. Among the patients randomized, 10% in the cenobamate group stopped treatment compared with 9% in the placebo group. In both groups, 4% of patients stopped due to AEs and 4% stopped due to withdrawal by patient (Table 10).
Of the 533 patients screened for Study C017, 437 patients (80%) were randomized, with 9% excluded because they did not meet the inclusion criteria, 7% excluded due to exclusion criteria, and another 2% not assigned to treatment (no reason reported) (Table 10). The proportion of patients who discontinued treatment varied across groups, with the higher-dose groups reporting more patients who stopped therapy (12%, 18%, 27%, and 13% in the cenobamate 100 mg, 200 mg, 400 mg, and placebo groups, respectively). The most common reasons for stopping therapy were AEs, which were reported by 11%, 14%, 21%, and 5% of patients in the cenobamate 100 mg, 200 mg, 400 mg, and placebo groups, respectively.
Table 10: Summary of Patient Disposition From Pivotal Studies and RCT Evidence Submitted by the Sponsor
Patient disposition | Study C013 | Study C017 | ||||
|---|---|---|---|---|---|---|
CEN 200 mg | Placebo | CEN 100 mg | CEN 200 mg | CEN 400 mg | Placebo | |
Screened, N | 285 | 533 | ||||
Reason for screening failure, N (%) | ||||||
Did not meet inclusion criteria | NR | 48 (9) | ||||
Exclusion criteria | NR | 36 (7) | ||||
Treatment not assigned | NR | 13 (2) | ||||
Randomized, total N (%) | 222 (78) | 437 (80) | ||||
Randomized by group, N | 113 | 109 | 108 | 110 | 111 | 108 |
Discontinued from DB treatment period, N (%) | 11 (10) | 10 (9) | 13 (12) | 20 (18) | 30 (27) | 14 (13) |
Reason for discontinuation, N (%) | ||||||
Adverse events | 4 (4) | 4 (4) | 12 (11) | 15 (14) | 23 (21) | 5 (5) |
Lost to follow-up | 2 (2) | 0 | 0 (0) | 0 (0) | 1 (1) | 0 (0) |
Protocol violation | 0 | 1 (1) | 0 (0) | 1 (1) | 1 (1) | 0 (0) |
Withdrawal by patient | 5 (4) | 4 (4) | 0 (0) | 4 (4) | 3 (3) | 5 (5) |
Lack of efficacy | 0 | 0 | 1 (1) | 0 (0) | 1 (1) | 0 (0) |
Other | 0 | 1 (1) | 0 (0) | 0 (0) | 1 (1) | 4 (4) |
ITT, N (%)a | 113 (100) | 109 (100) | 108 (100) | 110 (100) | 111 (100) | 108 (100) |
mITT, N (%) b | 113 (100) | 108 (99) | 108 (100) | 109 (99) | 111 (100) | 106 (98) |
mITT-M, N (%)c | NA | NA | 102 (94) | 98 (89) | 95 (86) | 102 (94) |
Safety, N (%)a | 113 (100) | 109 (100) | 108 (100) | 110 (100) | 111 (100) | 108 (100) |
DB = double blind; CEN = cenobamate, ITT = intention to treat; mITT = modified intention to treat; mITT-M = modified intention to treat in maintenance period; NA = not applicable; NR = not reported.
aAll randomized patients who received at least 1 dose of study drug.
bAll randomized patients who received at least 1 dose of study drug and had data for at least 1 postbaseline seizure. This population is described as the ITT population in Study C013, but is referred to in this CADTH report as mITT to be consistent across studies.
cAll randomized patients who completed the titration period, received at least 1 dose of study drug, and had data for at least 1 postbaseline seizure during the maintenance period.
Source: Clinical Study Report for Study C013;10 Clinical Study Report for Study C017.9
Baseline Characteristics
The baseline characteristics outlined in Table 11 were limited to those most relevant to this review or that were believed to affect the outcomes or interpretation of the study results.
In general, the patient demographics were similar between groups within the trials. The mean age of patients enrolled in the pivotal trials ranged from 36.2 years (SD = 11.3 years) to 40.9 years (SD = 12.4 years) across treatment groups. There were roughly equal proportions of men (47% to 54%) and women (46% to 53%) enrolled. In Study C017, most patients were white (82% to 86%), with fewer patients who were Asian (8% to 10%) and Black (< 1% to 4%) or who were another race or whose race was unknown (2% to 5%). In Study C013, 50% to 53% of patients were white, 41% to 43% of patients were Asian, 2% to 3% of patients were Black, and 4% of patients were other races or their race was unknown.
Most patients had been diagnosed with epilepsy many years ago, ranging from 22.6 years (SD = 13.7 years) to 25.5 years (SD = 13.4 years) ago. The most common seizure types reported by patients were complex partial (74% to 84% of patients) and partial onset with secondary generalization (55% to 65% of patients). The baseline rate of seizures was highly skewed, and there were some potential imbalances between groups. In Study C013, the median baseline seizure frequency per 28 days was 5.5 (range = 2 to 237) in the placebo group and 7.5 (range = 0 to 187) in the cenobamate 200 mg group. In Study C017, the median baseline seizure frequency per 28 days ranged from 8.4 (range = 4 to 704) for placebo to 11.0 (range = 4 to 418) in the cenobamate 200 mg group. In Study C017, patients had tried 1 to 9 prior ASMs, with a median of 3 ASMs in all groups. Overall, 14% of patients had received 1 ASM before enrolment, while 28%, 24%, and 35% of patients had received 2, 3, or more than 3 ASMs, respectively. In both studies, 1% to 3% of patients had a vagal nerve stimulator implanted.
Table 11: Summary of Baseline Characteristics in Pivotal Studies and RCT Evidence Submitted by the Sponsor
Characteristics | Study C013 | Study C017 | ||||
|---|---|---|---|---|---|---|
CEN 200 mg (n = 113) | Placebo (n = 109) | CEN 100 mg (n = 108) | CEN 200 mg (n = 110) | CEN 400 mg (n = 111) | Placebo (n = 108) | |
Age (years), mean (SD) | 36.2 (11.3) | 37.5 (11.4) | 39.0 (12.1) | 40.9 (12.4) | 39.6 (10.3) | 39.6 (12.4) |
Sex, n (%) | ||||||
Men | 55 (49) | 58 (53) | 57 (53) | 54 (49) | 52 (47) | 58 (54) |
Women | 58 (51) | 51 (47) | 51 (47) | 56 (51) | 59 (53) | 50 (46) |
Race, n (%) | ||||||
Asian | 49 (43) | 45 (41) | 10 (9) | 11 (10) | 11 (10) | 9 (8) |
Black or African American | 3 (3) | 2 (2) | 4 (4) | 3 (3) | 1 (< 1) | 4 (4) |
White | 57 (50) | 58 (53) | 89 (82) | 94 (85) | 96 (86) | 93 (86) |
Other or unknown | 4 (4) | 4 (4) | 5 (5) | 2 (2) | 3 (3) | 2 (2) |
Body mass index (kg/m2), mean (SD) | 25.6 (5.0) | 25.8 (5.0) | 26.0 (5.4) | 26.1 (5.4) | 25.8 (4.9) | 27.4 (7.9) |
Time since diagnosis (years), mean (SD) | 22.6 (13.7) | 23.0 (13.8) | 25.5 (13.4) | 22.8 (13.2) | 24.4 (14.2) | 23.0 (14.2) |
Seizure types by historya | ||||||
Simple partial without motor | 18 (16) | 16 (15) | 23 (21) | 20 (18) | 24 (22) | 24 (22) |
Simple partial with motor | 31 (27) | 25 (23) | 25 (23) | 25 (23) | 22 (20) | 22 (20) |
Complex partial | 83 (74) | 92 (84) | 89 (82) | 84 (76) | 88 (79) | 84 (78) |
Partial onset with secondary generalization | 73 (65) | 67 (62) | 69 (64) | 61 (55) | 72 (65) | 60 (56) |
Generalized | 4 (4) | 5 (5) | 6 (6) | 2 (2) | 4 (4) | 6 (6) |
Clusters | NR | NR | 0 | 1 (1) | 0 | 0 |
Other | 6 (5) | 5 (5) | 0 | 2 (2) | 0 | 2 (2) |
Baseline seizure frequency per 28 days, median (range) b | 7.5 (0c to 187) | 5.5 (2 to 237) | 9.5 (3.5 to 202) | 11.0 (4 to 418) | 9.0 (4 to 638) | 8.4 (4 to 704) |
Number of previous ASM drugs, median (range)d | NR | NR | 3 (1 to 8) | 3 (1 to 7) | 3 (1 to 8) | 3 (1 to 9) |
VNS implant, n (%) | 3 (3) | 2 (2) | 2 (2) | 2 (2) | 1 (1) | 2 (2) |
ASM = antiseizure medication; CEN = cenobamate; NR = not reported; SD = standard deviation; VNS = vagal nerve stimulator.
Note: Data were based on the safety population in both studies.
aPatients may be reported in more than 1 category. Seizure history information was missing for 30 patients in Study C017. Partial onset seizure type may also be described as focal aware nonmotor, focal aware motor, focal impaired awareness, and focal to bilateral tonic-clonic.
bCalculated by the number of seizures over the baseline period divided by number of days in the interval multiplied by 28 (modified intention-to-treat population).
cOne patient with only simple partial seizures with sensory component was randomized and treated in error. This was a major protocol deviation.
dASMs taken any time before the start of the study; these might or might not have been ongoing during the study.
Source: Clinical Study Report for Study C013;10 Clinical Study Report for Study C017;9 additional data from sponsor.84
Exposure to Study Treatments
In Study C013, the mean duration of exposure was similar in the cenobamate group (86.3 days [SD = 21.1 days]) and placebo group (86.3 days [SD = 17.2 days]) (Table 12). The modal dose was not reported for Study C013; however, data on the highest cenobamate dose achieved during the titration period were available for patients who completed the study (N = 102). Most patients achieved a maximum dose of 200 mg daily (67%), with 25%, 5%, and 4% achieving a daily dose of 150 mg, 100 mg, and 50 mg, respectively. Chung et al. (2020) reported that 59 patients out of 102 patients (58%) completed the study on the 200 mg dose.60
In Study C017, the mean duration of exposure was highest for the placebo group (119.6 days [SD = 24.1 days]) and lowest for the cenobamate 400 mg group (108.8 days [SD = 34.8 days]); however, the median duration of exposure was the same for all groups (126 days) (Table 12). Dose reductions were more common in the cenobamate 400 mg group, with 52% of patients requiring a reduction compared with 11%, 22%, and 2% of patients in the cenobamate 100 mg, cenobamate 200 mg, and placebo groups, respectively. The mean modal daily doses were 95.4 mg (SD = 14.6 mg), 184.5 mg (SD = 34.4 mg), and 289.6 mg (SD = 120.1 mg) in the cenobamate 100 mg, 200 mg, and 400 mg dosage groups, respectively.
Table 12: Summary of Patient Exposure From Pivotal Studies and RCT Evidence Submitted by the Sponsor
Exposure to study drug | Study C013 | Study C017 | ||||
|---|---|---|---|---|---|---|
CEN 200 mg (N = 113) | Placebo (N = 109) | CEN 100 mg (N = 108) | CEN 200 mg (N = 110) | CEN 400 mg (N = 111) | Placebo (N = 108) | |
Duration (days), mean (SD) | 86.3 (21.1) | 86.3 (17.2) | 117.7 (27.8) | 111.2 (34.0) | 108.0 (34.8) | 119.6 (24.1) |
Duration (days), median (range) | 91.0 (1 to 137) | 91.0 (2 to 113) | 126.0 (8 to 151) | 126.0 (7 to 140) | 126.0 (10 to 144) | 126.0 (11 to 149) |
Modal dose (mg), mean (SD) | NR | NR | 95.4 (14.6) | 184.5 (34.4) | 289.6 (120.1) | NA |
Modal dose (mg), median (range) | NR | NR | 100 (50 to 100) | 200 (50 to 200) | 300 (50 to 400) | NA |
Adherence, < 80%, n (%) | NR | NR | 7 (7) | 8 (7) | 7 (6) | 2 (2) |
Adherence, 80% to 100%, n (%) | NR | NR | 101 (94) | 102 (93) | 104 (94) | 105 (97) |
CEN = cenobamate; NA = not applicable; NR = not reported; SD = standard deviation.
Note: Data were based on the safety population in both studies.
Source: Clinical Study Report for Study C013;10 Clinical Study Report for Study C017.9
During Study C013, fewer patients were taking 1 concomitant ASM (14%) versus 2 ASMs (47%) or 3 ASMs (39%). In Study C017, 27%, 48%, and 24% of patients were taking 1, 2, or 3 ASMs during the study. Table 13 shows that there was some variation between groups in the distribution of patients receiving 1, 2, or 3 ASMs in Study C017.
In the pivotal trials, the most common ASMs (used by at least 10% of patients) were levetiracetam, lamotrigine, carbamazepine, lacosamide, valproate sodium and valproic acid, oxcarbazepine, topiramate, and clobazam (Table 13).
Table 13: Summary of Concomitant ASMs From Pivotal Studies and RCT Evidence Submitted by the Sponsor
Concomitant ASM | Study C013 | Study C017 | ||||
|---|---|---|---|---|---|---|
CEN 200 mg (n = 113) | Placebo (n = 109) | CEN 100 mg (n = 108) | CEN 200 mg (n = 1 10) | CEN 400 mg (n = 111) | Placebo (n = 108) | |
Number of concomitant ASM drugs, n (%)a | ||||||
1 | 19 (17) | 12 (11) | 28 (26) | 39 (36) | 25 (23) | 26 (24) |
2 | 53 (47) | 52 (48) | 45 (42) | 47 (43) | 62 (56) | 54 (50) |
3 | 41 (36) | 45 (41) | 34 (31) | 23 (21) | 22 (20) | 27 (25) |
> 3 | NR | NR | 1 (< 1) | 1 (< 1) | 2 (2) | 0 |
Concomitant ASM drugs, n (%)b | ||||||
Carbamazepine | 38 (34) | 43 (39) | 29 (27) | 29 (26) | 25 (23) | 39 (36) |
Clobazam | 22 (20) | 16 (15) | 17 (16) | 12 (11) | 17 (15) | 5 (5) |
Lacosamide | 27 (24) | 21 (19) | 16 (15) | 22 (20) | 22 (20) | 20 (19) |
Lamotrigine | 41 (36) | 34 (31) | 44 (41) | 27 (25) | 36 (32) | 31 (29) |
Levetiracetam | 51 (45) | 53 (49) | 47 (44) | 48 (44) | 50 (45) | 41 (38) |
Oxcarbazepine | 24 (21) | 26 (24) | 15 (14) | 17 (16) | 19 (17) | 13 (12) |
Topiramate | 25 (22) | 21 (19) | 11 (10) | 10 (9) | 15 (14) | 9 (8) |
Valproate or valproic acid | 30 (27) | 31 (28) | 23 (21) | 28 (26) | 28 (25) | 31 (28) |
Valproate sodium or semisodium | 17 (15) | 20 (18) | 13 (12) | 15 (14) | 15 (14) | 21 (19) |
Valproic acid | 13 (12) | 11 (10) | 10 (9) | 13 (12) | 13 (12) | 10 (9) |
ASM = antiseizure medication; CEN = cenobamate; NR = not reported.
Note: Data were based on the safety population in both studies.
aASMs ongoing at the start of the study and continued during the study.
bASMs used in 10% or more of all patients.
Source: Clinical Study Report for Study C013;10 Clinical Study Report for Study C017.9
Efficacy
Seizure Frequency
The percent reduction in partial seizure frequency per 28 days for the DB treatment period relative to baseline (mITT population) was the primary outcome in Study C013 and a co-primary outcome in Study C017 (Table 14). In Study C013, the cenobamate 200 mg group showed a median 55.6% reduction in partial seizure frequency per 28 days compared with a 21.5% reduction in the placebo group (P < 0.0001). In Study C017, the median percent reductions in seizure frequency per 28 days were 35.5%, 55.0%, 55.0%, and 24.0% in the cenobamate 100 mg, 200 mg, 400 mg, and placebo groups, respectively. The P values were 0.007 or less, with all cenobamate dosage groups favoured versus placebo.
In Study C017, a secondary analysis of the change in seizure frequency based on the maintenance period (last 12 weeks for the mITT-M population) reported median reductions of 41.5%, 56.5%, 63.0%, and 27.0% in the cenobamate 100 mg, 200 mg, 400 mg, and placebo groups, respectively. The P values for both the cenobamate 200 mg and 400 mg groups versus placebo were less than 0.001, but for the cenobamate 100 mg group, the P value was 0.054. In a subgroup analysis in Study C013, the seizure frequency during the maintenance period (last 6 weeks) showed median 63.4% and 29.7% seizure reductions in the cenobamate and placebo groups, respectively (P < 0.0001). These analyses were not controlled for type I error rate in either study; thus, any statistically significant findings should be interpreted in light of the potential for inflated type I error rate.
For Study C013, sensitivity analyses based on the per-protocol and completer populations showed findings that were consistent with the primary analysis. In Study C017, sensitivity analyses based on region, follow-up periods (titration period, first and last 6 weeks of maintenance period), using a parametric ANCOVA model, and including patients who stopped treatment during the titration period (for the analysis of maintenance period treatment effects) did not consistently detect a difference in seizure frequency between the cenobamate 100 mg group versus placebo. The differences in seizure frequency for the 200 mg and 400 mg dosage groups favoured cenobamate versus placebo.
The percent change in seizure frequency per 28 days for each type of partial seizures is shown in Appendix 1, Table 36. The reductions in seizures (simple partial with motor component, complex partial, and secondary generalized tonic-clonic) favoured the cenobamate 200 mg and 400 mg dosage groups versus placebo. These analyses were controlled for type I error rate.
Table 14: Summary of Seizure Frequency Results From Pivotal and RCT Evidence
Outcome | Study C013 | Study C017 | ||||
|---|---|---|---|---|---|---|
CEN 200 mg (N = 113) | Placebo (N = 109) | CEN 100 mg (N = 108) | CEN 200 mg (N = 110) | CEN 400 mg (N = 111) | Placebo (N = 108) | |
Seizure frequency per 28 days – DB treatment period (mITT population) | ||||||
Number of patients included in analysis | 113 | 108a | 108 | 109 | 111 | 106 |
Baseline rate, mean (SD) | 16.2 (24.7) | 15.4 (29.5) | 21.5 (33.1) | 30.6 (60.9) | 24.1 (63.1) | 25.3 (71.9) |
End point rate, mean (SD) | 12.2 (27.2) | 15.6 (31.8) | 12.3 (20.7) | 22.2 (58.7) | 13.6 (43.9) | 21.6 (65.4) |
Mean (SD) percent reduction | 35.5 (74.5) | 2.0 (91.6) | 33.0 (46.0) | 41.9 (50.1) | 48.3 (46.7) | 17.0 (50.3) |
Baseline rate, median (range) | 7.5 (0 to 186.8) | 5.5 (2.0 to 236.5) | 9.5 (3.5 to 202) | 11.0 (4 to 418) | 9 (4 to 638) | 8.4 (4 to 704) |
End point rate, median (range) | 3.8 (0 to 196.3) | 5.0 (0 to 206.3) | 5.8 (0 to 164.6) | 5.8 (0 to 373.7) | 3.8 (0 to 424.9) | 6.8 (0.7 to 640.8) |
Median percent reduction | 55.6 | 21.5 | 35.5 | 55.0 | 55.0 | 24.0 |
P valueb | P < 0.0001c | Reference | P = 0.007d | P < 0.001d | P < 0.001d | Reference |
Seizure frequency per 28 days – maintenance period (subgroup 5 or mITT-M population)e | ||||||
Number of patients included in analysis | 106 | 102 | 102 | 98 | 95 | 102 |
Baseline rate, mean (SD) | 16.6 (25.4) | 14.6 (27.5) | 21.0 (31.3) | 32.1 (63.9) | 25.8 (68.0) | 25.1 (73.1) |
End point rate, mean (SD) | 11.2 (28.3) | 14.2 (32.7) | 12.9 (21.9) | 26.4 (82.3) | 15.2 (54.9) | 21.3 (64.6) |
Mean (SD) percent reduction | 45.9 (83.4) | 15.9 (99.1) | 33.4 (47.8) | 41.7 (57.6) | 53.1 (50.2) | 17.7 (62.6) |
Baseline rate, median (range) | 7.6 (0 to 186.8) | 5.5 (2.0 to 236.5) | 9.8 (3.5 to 202) | 12.0 (4 to 418) | 9.0 (4 to 638) | 8.1 (4 to 704) |
End point rate, median (range) | 2.8 (0 to 228.1) | 4.6 (0 to 219.2) | 5.7 (0 to 168) | 5.4 (0 to 678.2) | 3.0 (0 to 494.9) | 6.4 (0 to 618.3) |
Median percent reduction | 63.4 | 29.7 | 41.5 | 56.5 | 63.0 | 27.0 |
P valueb | P < 0.0001 | Reference | P = 0.054 | P < 0.001 | P < 0.001 | Reference |
DB = double blind; CEN = cenobamate; mITT = modified intention to treat; mITT-M = modified intention to treat in maintenance period; RCT = randomized controlled trial; SD = standard deviation.
aOne patient who had 0 relevant partial seizures during the baseline period was excluded from the analysis.
bIn Study C013, the P value was based on a Wilcoxon rank sum test assessing whether the median percent change for CEN was significantly different from the median percent change for placebo. In Study C017, the P value was based on a nonparametric ANCOVA model with terms for ranked baseline seizure rate and treatment group.
cPrimary end point in C013; no multiplicity adjustment required.
dPrimary end point for the US regulatory agency in Study C017; the P value for each dosage group has been adjusted for multiple testing.
eSeizure frequency during the maintenance period of Study C013 (last 6 weeks of treatment) was based on subgroup 5 (i.e., patients who received the study drug during the maintenance period and may or may not have completed the study). For Study C017, seizure frequency during the maintenance period (i.e., the last 12 weeks) was based on the mITT-M population (i.e., all randomized patients who completed the titration period, received at least 1 dose of the study drug in the maintenance period, and had any data related to a maintenance period seizure).
Source: Clinical Study Report for Study C013;10 Clinical Study Report for Study C017.9
Seizure-Free Days
This outcome was not reported in either study.
Seizure Freedom
The proportions of patients who achieved a 100% reduction in seizure frequency were lower for the analyses based on the entire treatment period (titration and maintenance; refer to Table 15) than for the analyses that were based on the maintenance period only (Appendix 1, Table 37). In Study C013, 10 patients (8.8%) versus 1 patient (0.9%) achieved a 100% reduction in seizures in the DB treatment period (P = 0.0148). Based on the maintenance period only, 30 patients (28.3%) in the cenobamate 200 mg group and 9 patients (8.8%) in the placebo group reported a 100% reduction in partial seizure frequency per 28 days (P = 0.0001). Both analyses were conducted post hoc, and there was no control for multiple testing.
In Study C017, 0 patients who received placebo, 2 patients in the 100 mg group (1.9%; P = 0.50), 8 patients in the 200 mg group (7.3%; P = 0.007), and 7 patients in the 400 mg cenobamate group (6.3%; P = 0.014) achieved a 100% reduction in seizures for the DB treatment period. The maintenance period analysis reported that 1 patient (1.0%) in the placebo group achieved a 100% reduction in seizure frequency compared to 4 patients (3.9%; P = 0.37), 11 patients (11.2%; P = 0.002), and 20 patients (21.1%; P < 0.001) in the cenobamate 100 mg, 200 mg, and 400 mg groups, respectively. These analyses were not controlled for multiple testing.
Seizure Frequency — Responder Analyses
The proportion of patients who achieved at least a 50%, 75%, or 90% reduction in partial seizure frequency per 28 days was reported based on the DB treatment period (Table 15) and maintenance period only (Appendix 1, Table 37). For both studies, the proportion of responders was generally higher for the analyses based on the maintenance period than the analyses based on the entire DB treatment period.
In Study C017, 40.7%, 57.8%, 60.4%, and 21.7% of patients in the cenobamate 100 mg, 200 mg, 400 mg, and placebo groups, respectively, achieved at least a 50% reduction in seizure frequency per 28 days during the DB treatment phase (Table 15). All analyses favoured cenobamate versus placebo (P = 0.003, P < 0.001, and P < 0.001, respectively; not controlled for type I error rate). In Study C013, 50.4% of patients in the cenobamate 200 mg group and 22.2% of patients in the placebo group achieved at least a 50% reduction in seizure frequency during the DB treatment period (odds ratio = 3.94; 95% CI, 2.14 to 7.24; P < 0.0001; not controlled for type I error rate).
The alternate analyses, which were based on treatment response in the maintenance period only in Study C017, reported that 40.2%, 56.1%, 64.2%, and 25.5% of patients achieved at least a 50% reduction in seizure frequency per 28 days in the cenobamate 100 mg, 200 mg, 400 mg, and placebo groups, respectively. For this co-primary end point, the differences favoured the cenobamate 100 mg (P = 0.036), 200 mg (P < 0.001), and 400 mg groups (P < 0.001) versus placebo. The sensitivity analyses showed results that were consistent with the primary analysis. In Study C013, 50.0% and 21.6% of patients in the cenobamate 200 mg and placebo groups achieved at least a 50% reduction in seizure frequency per 28 days during the maintenance period (P < 0.001; not controlled for type I error rate) (Appendix 1, Table 37).
The proportion of responders who achieved a reduction in seizure frequency of greater than or equal to 75% and greater than or equal to 90% during the DB and maintenance periods favoured the cenobamate 200 mg and 400 mg groups versus placebo, but did not favour the cenobamate 100 mg group versus placebo. Of note, there was no control of type I error rate for these analyses in either study. In Study C013, these analyses were conducted post hoc.
Table 15: Summary of DB Treatment Period Responder Results From Pivotal and RCT Evidence
Outcome | Study C013 | Study C017 | ||||
|---|---|---|---|---|---|---|
CEN 200 mg (N = 113) | Placebo (N = 109) | CEN 100 mg (N = 108) | CEN 200 mg (N = 110) | CEN 400 mg (N = 111) | Placebo (N = 108) | |
≥ 50% reduction in seizure frequency per 28 days – DB treatment period (mITT population) | ||||||
Number of patients included in analysis | 113 | 108 | 108 | 109 | 111 | 106 |
n (%) | 57 (50.4) | 24 (22.2) | 44 (40.7) | 63 (57.8) | 67 (60.4) | 23 (21.7) |
OR (95% CI) | 3.94 (2.14 to 7.24) | Reference | NR | NR | NR | Reference |
P valuea | P < 0.0001 | Reference | P = 0.003 | P < 0.001 | P < 0.001 | Reference |
≥ 75% reduction in seizure frequency per 28 days – DB treatment period (mITT population)b | ||||||
n (%) | 32 (28.3) | 11 (10.2) | 18 (16.7) | 23 (21.1) | 39 (35.1) | 9 (8.5) |
OR (95% CI) | 3.78 (1.76 to 8.15) | Reference | NR | NR | NR | Reference |
P value | P = 0.0007 | Reference | P = 0.099 | P = 0.012 | P < 0.001 | Reference |
≥ 90% reduction in seizure frequency per 28 days – DB treatment period (mITT population)b | ||||||
n (%) | 15 (13.3) | 1 (0.9) | 5 (4.6) | 13 (11.9) | 23 (20.7) | 1 (0.9) |
OR (95% CI) | 17.45 (2.24 to 135.86) | Reference | NR | NR | NR | Reference |
P value | P = 0.0063 | Reference | P = 0.212 | P = 0.001 | P < 0.001 | Reference |
100% reduction in seizure frequency per 28 days – DB treatment period (mITT population)b | ||||||
n (%) | 10 (8.8) | 1 (0.9) | 2 (1.9) | 8 (7.3) | 7 (6.3) | 0 (0.0) |
OR (95% CI) | 13.68 (1.67 to 112.06) | Reference | NR | NR | NR | Reference |
P value | P = 0.0148 | Reference | P = 0.498 | P = 0.007 | P = 0.014 | Reference |
DB = double blind; CEN = cenobamate; CI = confidence interval; mITT = modified intention to treat; NR = not reported; OR = odds ratio; RCT = randomized controlled trial.
aIn Study C013, the OR (95% CI) and P values were based on a logistic regression model with terms for treatment, country, and baseline seizure frequency (Wald chi-square test). In Study C017, the P values were based on Fisher’s exact chi-square test.
bGreater than or equal to 75%, greater than or equal to 90%, and 100% responder analyses were conducted post hoc in Study C013.
Source: Clinical Study Report for Study C013;10 Clinical Study Report for Study C017.9
Health-Related Quality of Life
No data on HRQoL were available in Study C013. Study C017 reported descriptive data for the QOLIE-P-31 instrument that were collected from 116 patients enrolled in the US, UK, or Australia (i.e., 27% of the total study population). The QOLIE-31-P is scored from 0 to 100, with higher scores indicating better quality of life. The CSR stated that an 11.8-point increase in the total score represents the minimal important change. The mean within-group change from baseline ranged from –6.2 to 0.6 points in the cenobamate groups, compared with 3.8 points in the placebo group. The CSR states that the changes observed were not clinically relevant.
Treatment Retention or Adherence
Neither study reported data on treatment retention or adherence.
Harms
Refer to Table 17 for harms data.
Table 16: Change From Baseline in QOLIE-31-P Score — Study C017
Outcome | CEN 100 mg (N = 108) | CEN 200 mg (N = 110) | CEN 400 mg (N = 111) | Placebo (N = 108) |
|---|---|---|---|---|
Change from baseline in QOLIE-31-P score | ||||
N included in analysis | 27 | 27 | 33 | 29 |
Baseline, mean (SD) | 65.6 (13.7) | 57.3 (17.0) | 61.5 (15.3) | 59.4 (17.5) |
End point, mean (SD) | 63.9 (14.8) | 60.8 (16.9) | 55.5 (14.3) | 62.8 (12.7) |
Mean change from baseline (SD) | −0.8 (9.7) | 0.6 (12.0) | −6.2 (17.0) | 3.8 (11.4) |
Median change from baseline | 0.6 | 0.6 | −4.6 | 0.0 |
Patients who achieved at least an 11.8-point increase from baseline, n (%)a | 3 (11.1) | 3 (11.1) | 5 (15.2) | 7 (24.1) |
CEN = cenobamate; QOLIE-31 = 31-item Quality of Life in Epilepsy Questionnaire; QOLIE-31-P = patient-weighted 31-item Quality of Life in Epilepsy Questionnaire; SD = standard deviation.
Note: Data were based on a subgroup of the mITT population in the US, UK, and Australia who completed the QOLIE-31-P questionnaire.
aThe minimally important change was defined as a change from baseline plus or minus 11.8 at the end point for the weighted overall QOLIE-31 score.
Source: Clinical Study Report for Study C017.9
Table 17: Summary of Harms — Pivotal and RCT Evidence
Adverse event | Study C013 | Study C017 | ||||
|---|---|---|---|---|---|---|
CEN 200 mg (n = 113) | Placebo (n = 109) | CEN 100 mg (n = 108) | CEN 200 mg (n = 110) | CEN 400 mg (n = 111) | Placebo (n = 108) | |
Most common adverse events, n (%) | ||||||
Patients with ≥ 1 adverse event | 86 (76) | 69 (63) | 70 (65) | 84 (76) | 100 (90) | 76 (70) |
Somnolence | 25 (22) | 13 (12) | 20 (19) | 23 (21) | 41 (37) | 9 (8) |
Dizziness | 25 (22) | 18 (17) | 19 (18) | 22 (20) | 37 (33) | 15 (14) |
Headache | 14 (12) | 14 (13) | 11 (10) | 12 (11) | 12 (11) | 6 (6) |
Fatigue | 12 (11) | 7 (6) | 13 (12) | 19 (17) | 27 (24) | 9 (8) |
Diplopia | 5 (4) | 3 (3) | 8 (7) | 11 (10) | 17 (15) | 2 (2) |
Balance disorder | 9 (8) | 1 (1) | 3 (3) | 2 (2) | 10 (9) | 0 |
Nystagmus | 11 (10) | 0 | 3 (3) | 4 (4) | 7 (6) | 1 (1) |
Nausea | 13 (12) | 5 (5) | 7 (7) | 1 (1) | 10 (9) | 1 (1) |
Serious adverse events, n (%) | ||||||
Patients with SAEs | 3 (3) | 4 (4) | 10 (9) | 4 (4) | 8 (7) | 6 (6) |
Seizure | 1 (1) | 3 (3) | 2 (2) | 1 (1) | 1 (1) | 0 |
Ataxia | NR | NR | 0 | 0 | 2 (2) | 0 |
Dizziness | NR | NR | 0 | 0 | 2 (2) | 0 |
Nystagmus | NR | NR | 0 | 0 | 2 (2) | 0 |
Suicidal ideation | NR | NR | 2 (2) | 0 | 0 | 0 |
Suicidal attempt | NR | NR | 1 (1) | 0 | 0 | 0 |
Patients who stopped treatment due to adverse events, n (%) | ||||||
Patients who stopped treatment | 5 (4) | 3 (3) | 11 (10) | 15 (14) | 22 (20) | 5 (5) |
Ataxia | 0 | 1 (1) | 0 | 3 (3) | 4 (4) | 0 |
Dizziness | NR | NR | 1 (1) | 1 (1) | 4 (4) | 0 |
Somnolence | 0 | 1 (1) | 1 (1) | 2 (2) | 3 (3) | 0 |
Nystagmus | 1 (1) | 0 | 0 | 1 (1) | 3 (3) | 0 |
Vertigo | NR | NR | 0 | 1 (1) | 3 (3) | 1 (1) |
Deaths, n (%) | ||||||
Patients who died | 0 | 0 | 0 | 0 | 0 | 0 |
Notable adverse events, n (%) | ||||||
DRESS | NR | NR | 0 | 1 (1) | 0 | 0 |
Drug hypersensitivity | 1 (1) | 0 | 1 (1) | 0 | 0 | 0 |
Arrhythmias | 3 (3) | 2 (2) | 0 | 2 (2) | 2 (2) | 2 (2) |
Suicidal ideation | 1 (1) | 1 (1) | 2 (1) | 1 (1) | 0 | 0 |
CEN = cenobamate; DRESS = drug reaction with eosinophilia and systemic symptoms; NR = not reported; RCT = randomized controlled trial; SAE = serious adverse event.
Note: Data were based on the safety population in both studies.
Source: Clinical Study Report for Study C013;10 Clinical Study Report for Study C017.9
Adverse Events
TEAEs were reported by 65%, 76%, and 90% of patients who received cenobamate 100 mg, 200 mg, and 400 mg compared with 63% to 70% of patients who received placebo in studies C013 and C017. The most frequently reported AEs in the cenobamate groups were somnolence (19% to 37%), dizziness (18% to 33%), fatigue (11% to 24%), and diplopia (4% to 15%). In the placebo groups, somnolence was reported in 8% to 12% of patients, dizziness in 14% to 17% of patients, fatigue in 6% to 8% of patients, and diplopia in 2% to 3% of patients.
Serious Adverse Events
SAEs were reported in 3% to 9% of patients who received cenobamate and in 4% to 6% who received placebo.
In Study C013, 2 patients who were receiving cenobamate experienced an SAE (drug hypersensitivity reaction; urinary tract infection), and 1 other patient had status epilepticus just after the end of the taper-off period for cenobamate. Four patients in the placebo group experienced SAEs of status epilepticus (2 patients), major seizure attack, and hospitalization for an abnormal electrocardiogram results detected at screening.
In Study C017, SAEs (reported in at least 2 patients) included suicidal ideation and seizure among those who received cenobamate 100 mg, and ataxia, dizziness, and nystagmus among those who received cenobamate 400 mg. One patient attempted suicide in the cenobamate 100 mg group.
Withdrawals Due to Adverse Events
In Study C013, 5 patients (4%) in the cenobamate group and 3 patients (3%) in the placebo group stopped treatment due to AEs. No specific events were reported in more than 1 patient. In the cenobamate group, the events that led to discontinuation included tachycardia, abdominal pain, gastroesophageal reflux, drug hypersensitivity, nystagmus, aggression, depression, and dyspnea. In the placebo group, the events were altered state of consciousness, ataxia, dyskinesia, grand mal seizure, partial seizures, status epilepticus, somnolence, and tremor.
In Study C017, 11 patients (10%), 15 patients (14%), 22 patients (20%), and 5 patients (5%) stopped treatment due to AEs in the cenobamate 100 mg, 200 mg, 400 mg, and placebo groups, respectively. The most frequently reported AEs leading to withdrawal from the cenobamate groups were ataxia, dizziness, somnolence, nystagmus, and vertigo.
Mortality
No deaths were reported during the DB treatment period of either study.
Notable Harms
CADTH identified DRESS, dermatologic reactions, arrhythmias (short QT syndrome), and suicidal ideation or behaviour as AEs of interest to this review.
One patient in the cenobamate 200 mg group of Study C017 developed a DRESS reaction on day 24 that the investigator considered probably related to the study drug; the study drug was stopped. The patient was also receiving divalproex sodium, levetiracetam, and zonisamide. Other dermatologic reactions that led to study drug withdrawal were reported in 2 patients in the cenobamate 200 mg group. One patient experienced a pruritic rash and pyrexia (concomitant ASMs were clonazepam, clobazam, and lacosamide), and the second patient experienced a rash (concomitant ASMs were valproic acid, levetiracetam, and lacosamide).
In Study C013, 1 patient who was randomized to cenobamate experienced a serious drug hypersensitivity reaction on day 1 of treatment, characterized by reddening of the palms and soles and itching of the ears. The investigator considered this reaction to be definitely related to the study drug. The patient stopped cenobamate treatment and recovered after 22 days.
In Study C013, 3 patients in the cenobamate group reported AEs of atrial flutter, tachycardia, and palpitations, and 1 patient in the placebo group reported palpitations. In Study C017, 2 patients in the cenobamate group reported bradycardia or sinus bradycardia; 2 patients in the 400 mg group reported palpitations; and 2 patients in the placebo group reported sinus bradycardia or tachycardia.
Suicidal ideation was reported by 1 patient in each treatment group in Study C013. In Study C017, suicidal ideation was reported by 2 patients in the cenobamate 100 mg group and by 1 patient in the cenobamate 200 mg group. One patient in the group receiving cenobamate 100 mg in Study C017 attempted suicide.
Critical Appraisal
Internal Validity
The sponsor’s Summary of Clinical Evidence did not meet CADTH’s expectations for clarity, transparency, completeness, or accuracy, and was rated as poor quality by the CADTH reviewer. Moreover, the CSRs for the pivotal trials lacked clarity and had conflicting descriptions of key elements of the study design and methods. Thus, some aspects of the studies’ conduct remain uncertain. This hindered the reviewer’s ability to assess the risk of bias.
Systematic Review
The protocol for the systematic review included the relevant patient population and intervention; however, only 4 drugs plus placebo were included as comparators (eslicarbazepine, brivaracetam, perampanel, and lacosamide). Although the third-generation ASMs are important comparators for cenobamate, the clinical expert consulted by CADTH noted that other drugs listed in Table 3 may also be used as adjunctive therapy in patients with uncontrolled partial seizures. Thus, the focus of the systematic review was considered too narrow.
The outcomes selected were relevant, but the sponsor did not identify any specific AEs of interest. Based on expert input, CADTH identified DRESS, dermatologic reactions, arrhythmias, and suicidal ideation or behaviour as notable AEs.
There were discrepancies in the study design criteria applied in the systematic review. As a result, it is unclear if the criteria were limited to phase II to IV RCTs, phase III RCTs, or phase III and IV RCTs. As noted earlier, the sponsor included Study C021 in its systematic review. However, this study did not meet the study design criteria outlined in the sponsor’s protocol.
Several issues were identified with the literature search conducted by the sponsor. As a result, not all relevant trials may have been identified. Many key search terms were not truncated (i.e., the search used “epilepsy” instead of “epilep*”); thus, the search would have missed terms such as epileptic, epilepsies, and so on. In addition, the search did not include all possible terms for cenobamate. The search included extraneous terms (focal, partial, local, and simple), which likely increased the number of irrelevant results rather than focusing on the population of interest. The description of the methods states that the search was limited to humans, but no terms were included in the search to limit the results. In addition, the reported search strategy contained an error that made the search impossible to execute. Thus, CADTH was unable to verify that the search output would match the numbers reported in the sponsor’s PRISMA chart. Based on these issues, the reported methods were not consistent with CADTH standards, according to the Peer Review of Electronic Search Strategies criteria.
Pivotal Trials
The risks of bias related to randomization, treatment allocation, and blinding in the pivotal trials were rated as low by the CADTH reviewer. There were no major discrepancies in the frequency of specific AEs that may have led to significant unblinding. At baseline, the patient characteristics appeared to be reasonably well balanced between groups within the studies. There was some variation in the median baseline rate of partial seizures, but the impact of these differences is unclear. It was noted that the baseline seizure frequency was highly skewed, with a large range in values. In Study C013, the proportion of patients who discontinued was similar in the cenobamate 200 mg and placebo groups (10% and 9%), but Study C017 showed differential losses to follow-up, with 12%, 18%, 27%, and 13% of patients stopping treatment in the cenobamate 100 mg, 200 mg, 400 mg, and placebo groups, respectively. As a result, it is not clear if the treatment groups remained balanced throughout the study.
Both trials measured the change in partial seizure frequency as well as the proportion of responders (≥ 50%) as primary and secondary end points. The analyses included all simple partial motor, complex partial, and secondarily generalized seizures, which the clinical expert agreed was appropriate for this population. Seizure data were collected from patient diaries. There is evidence that seizures reported by patients were undercounted compared with objective measures, such as electroencephalogram monitoring.68-71 Under-reporting may be related to lack of awareness of an event (e.g., due to the type of seizure or to seizures occurring at night) and to other factors related to the disease itself (as opposed to lack of motivation to record events).68-71,85 However, in the clinical trials, under-reporting is expected to occur in both the active and control groups; thus, it may not bias the findings.
While the 50% response threshold has been used in other ASM clinical trials and may be accepted as a minimal clinically important difference, the expert indicated that generally higher response thresholds are desired and that the goal of therapy is seizure freedom. Other seizure response thresholds were tested in both studies, but these analyses were not controlled for type I error and were conducted post hoc in Study C013. Moreover, the responder analyses were conducted using the last observation carried forward for Study C013 and with no imputation for missing data in Study C017; thus, patients who withdrew early could be considered as treatment responders. Patients who achieved a 100% reduction in seizure frequency were interpreted as achieving seizure freedom. However, only 3 types of partial seizures were included in the analysis. This means that patients could have other types of seizures, including generalized seizures, and still be deemed seizure-free. Similarly, those who stopped treatment early for intolerable AEs could also be classified as responders. The use of a “pragmatic intention-to-treat” analysis, in which patients who withdrew were considered nonresponders, may have been a preferred approach.72
Given the skewed nature of the seizure frequency data, the use of nonparametric statistical testing methods was reasonable. In Study C017, randomization was stratified by country, but country was not included in the nonparametric ANCOVA model. The CSR states that omitting country as a covariate did not affect the validity of the analysis and may not have reduced variability because each region was to capture patients who were relatively homogenous in response. The responder analyses also did not adjust for country or baseline seizure rate. It is unclear if the assumption of homogeneity is valid, but CADTH was unable to assess the potential impact that excluding these covariates may have had on the findings.
The calculation of seizure rates was based on patient-reported diary data, excluding any days with missing data. This method assumed that days with missing data would have the same seizure rate as days with nonmissing data, both for missing interim days and for patients who discontinued the study early. The sponsor indicated that there were limited missing data from the primary efficacy analyses;84 however, it is unclear if this accounts for missing data after dropout for patients who withdrew early. As noted previously, there were differential discontinuation rates across groups in Study C017, with most of the differences explained by withdrawals due to AEs. It is not clear that patients who withdrew from the study early would have had the same seizure frequency as patients who remained in the trial. While the CSR states that several sensitivity analyses were conducted to assess the impact of missing data, these analyses focused on different follow-up periods (i.e., first or last 6 weeks of the maintenance phase), but did not impute any missing data. The sponsor was asked to provide additional information on the rationale for the sensitivity analyses and how these could be used to confirm that the missing data assumption was valid. Based on the information presented,84 the CADTH review team concluded that the analyses did not fully test the missing data assumption because none took a conservative approach (e.g., nonresponder imputation, baseline value carried forward). In general, patients who drop out are likely to have worse outcomes than those who continue; therefore, given the extent of the early withdrawals and the differential losses in Study C017, it is possible that the results may be biased in favour of cenobamate. However, the extent of any potential bias and the impact on the overall findings are unclear.
Study C017 had 2 preplanned statistical analyses with different primary and key secondary outcomes to meet regulator requirements in the US and Europe. The US-based efficacy end points were based on the total DB treatment period (i.e., titration and maintenance, mITT population), whereas the European outcomes were based on the maintenance period only for patients who completed the titration phase (mITT-M). The outcomes based on the DB treatment period were closer to an intention-to-treat approach; however, the clinical expert consulted stated that both methods may be used to assess treatment effects in clinical practice. In Study C017, the multiplicity adjustment appears to apply to the primary end points only; thus, depending on whether the US or European statistical analysis is preferred, only the comparisons for the 3 cenobamate dosage groups versus placebo for the percent change in seizure frequency (mITT population) or the greater than or equal to 50% responder analysis (mITT-M population) would have a family-wise type I error rate controlled at 0.05. In Study C013, there was no control of multiplicity for secondary end points; thus, the family-wise type I error rate may be elevated. An interim analysis was run in both studies, and the CSRs state that there were no plans to modify the studies based on these data; thus, there was no need to adjust the P value to account for the interim analyses. Regarding Study C013, the FDA Statistical Review report states that it was designed as a proof of concept study, and the lack of prespecification of the unblinded interim analysis (e.g., timing, firewall, details of analyses to be conducted) potentially jeopardized the study’s credibility (although the reviewer did not find changes in the study conduct before and after the analysis).63 In addition, the statistical review states that the “ambiguity in the primary analysis adds complexity to the evaluation of the efficacy evidence,” but deemed that the statistical issues did not affect the overall conclusions.63 Given the design and analysis limitations of Study C013, the CADTH review focused on Study C017.
Neither of the pivotal trials was designed to test the impact of cenobamate on HRQoL. Although Study C017 collected data using the QOLIE-31-P, this information was gathered only from approximately 25% of the patients enrolled and was reported descriptively. Moreover, the trials lacked an active comparator group and were 12 weeks to 18 weeks in duration; thus, these can only address short-term efficacy and safety versus placebo. The sample sizes and durations of the studies were insufficient to capture rare AEs.
External Validity
Of the patients screened for entry, 80% were included in the pivotal trials, with limited details available on the characteristics of those not enrolled. Both trials excluded older patients (i.e., aged > 65 years or > 70 years) in addition to patients with comorbid psychiatric or neurologic conditions, substance use disorder, suicidal ideation or behaviour, or a history of drug hypersensitivity reactions, seizure clusters, or status epilepticus. As a result, the treatment effects of cenobamate in these patients is unknown. On average, the patients enrolled were in their late thirties, were predominantly white, and had had a diagnosis of epilepsy for more than 20 years. Those enrolled were required to be experiencing at least 3 to 4 partial seizures per month; however, the baseline seizure frequency was highly variable, with some patients experiencing more than 200 seizures per month. Neither study included patients from Canada, but the clinical expert consulted did not think there were important regional differences that would limit the generalizability of the results to patients in Canada with partial onset seizures.
The pivotal trials used a more rapid titration schedule than has been recommended in the product monograph, which may have affected the occurrence of some AEs. Study C017 included a 100 mg dosage group (i.e., half the recommended maintenance dose), which may be subtherapeutic. But in clinical practice, not all patients may be able to tolerate 200 mg daily and may receive lower doses. In Study C017, 14% of patients had tried 1 ASM before enrolment. For these patients, cenobamate would be considered second-line therapy (prior ASM data were not reported for Study C013). During the trials, it was most common for patients to be receiving 2 or more concomitant ASMs (73% to 86%) versus 1 ASM (14% to 27%), and few patients had a vagal nerve stimulator implanted (1% to 3%). The clinical expert stated that the concomitant ASMs used were consistent with clinical practice. However, no dosing information was reported to determine whether dosages were similar to practice in Canada.
Of note, the studies were 12 weeks to 18 weeks in duration, which the expert considered to be short for a chronic condition that often requires lifelong therapy. Based on these data, the longer-term efficacy and safety are uncertain.
Long-Term Extension Studies
Contents within this section have been informed by materials submitted by the sponsor. The following subsections have been summarized and validated by the CADTH review team.
Description of Studies
Two long-term extension studies were included in this review: Study C013 OLE11,86 and Study C017 OLE.12,13 There were no comparator groups in the OLE studies. Moreover, no efficacy results were reported in the Study C013 OLE. Both studies reported safety results. The CADTH review team summarized the study design and results to provide information as supportive evidence.
The Study C013 OLE was a long-term extension of Study C013, a 2-arm (cenobamate 200 mg once daily or placebo) RCT with a DB treatment period of 12 weeks.86 Patients who completed the randomized phase of Study C013 were eligible to enter the OLE phase and received open-label cenobamate at a daily maximum dose of 400 mg daily. Adults with uncontrolled partial onset seizures (N = 149) were followed-up up to 8.6 years. The only safety outcomes assessed were TEAEs.
The Study C017 OLE was a long-term extension of Study C017, which was a 4-arm (cenobamate 100 mg, 200 mg, or 400 mg once daily or placebo), dose-response trial with a DB treatment period of 18 weeks (a 6-week titration period and a 12-week maintenance period).12 Patients who completed Study C017 were eligible to enter the OLE phase and were converted to open-label cenobamate at a target dose of 300 mg once daily. Efficacy outcomes of seizure control for up to 48 months of the OLE and safety outcomes for up to 6.4 years of cenobamate treatment in adults with uncontrolled partial onset seizures (N = 356) were assessed.
Profiles of the Study C013 OLE and the Study C017 OLE are summarized in Table 18.
Table 18: Summary of Study Characteristics for Study C013 OLE and Study C017 OLE
Study characteristics | Study C013 OLE (N = 149) | Study C017 OLE (N = 356) |
|---|---|---|
Populations | The inclusion and exclusion criteria were the same as for Study C013.60 Patients who completed the DB treatment period and still met the inclusion and/or exclusion criteria (except for seizure frequency) were eligible to continue in the OLE phase. | The inclusion and exclusion criteria were the same as for Study C017.61 Patients who completed the 18-week DB phase and still met all inclusion criteria and none of the exclusion criteria (except for seizure frequency) were eligible to continue in the optional OLE phase. |
Interventions | Cenobamate 100 mg/day, with subsequent dose increases of 50 mg/day every 2 weeks as tolerated. The initial maximum dose during the OLE was 200 mg/day. Following an amendment, this was increased to 400 mg/day (approximately 2 years after the initiation of the OLE). | Patients from the 4 groups in Study C017 underwent a 2-week blinded period of conversion to cenobamate with a target dose of 300 mg/day for the OLE phase (a minimum of 50 mg/day to a maximum of 400 mg/day, depending on tolerability). The initial starting dose was 100 mg/day. This was titrated up by 100 mg/day on a weekly basis. Owing to tolerability, the initial dose was later amended to 50 mg/day, with the titration rate slowed to 50 mg/day increments per week (up to 200 mg/day), then to 100 mg/day increments per week (up to 400 mg/day). |
Outcomes | Incidence of TEAEs Incidence of SAEs Treatment discontinuations | Efficacy outcomes included the percent reduction in seizure frequency (average monthly seizure rate per 28 days) of all seizures during:
Safety outcomes included:
|
Statistical analysis | All patients treated in the DB phase who continued into the OLE and took at least 1 dose of open-label study medication were included in analysis. The results of the OLE were reported separately from the DB phase results using summary tables and data listings. Summaries were presented for demographic and safety information only. | All patients treated in the DB phase who continued into the OLE and took at least 1 dose of open-label study medication were included in analysis. Descriptive statistics were reported. |
DB = double blind; OLE = open-label extension; SAE = serious adverse event; TEAE = treatment-emergent adverse event.
Source: Clinical Study Report for Study C013 OLE;11 Clinical Study Report for Study C017 OLE.12
Populations
The inclusion and exclusion criteria for the OLE studies were the same as for the pivotal studies, but without the specification of seizure frequency at baseline (i.e., adults with uncontrolled partial onset seizures). Of the patients who entered the Study C013 OLE (n = 149), 48.3% were men, which is slightly lower than the proportion of men at baseline in Study C013 (113 patients of 222 patients [50.9%] were men). The mean age of patients in the Study C013 OLE was 37.6 years (SD = 10.9 years), similar to the mean age of 36.9 years (SD = 11.3 years) at baseline in Study C013. The proportion of men among the patients who entered the Study C017 OLE (185 patients out of 356 patients [52.0%]) was slightly higher than that at baseline in Study C017 (221 patients of 437 patients were men [50.6%]). The mean age of the patients who entered the Study C017 OLE (39.6 years; SD = 11.7 years) was similar to that at baseline in Study C017 (mean = 39.8 years; SD = 11.8 years).
Interventions
The initial dose of cenobamate was 100 mg/day. This was followed by increments of 50 mg/day biweekly in the Study C013 OLE11 and by 100 mg/day weekly in the Study C017 OLE (later amended to 50 mg/day),12 as tolerated. The target dose of cenobamate in the Study C017 OLE was 300 mg per day, and the maximum dose was 400 mg/day.11,12 In the OLE for Study C013, the maximum daily dose was initially 200 mg per day. However, 2 years into the study, it was changed to 400 mg per day.
During the OLE periods, investigators could adjust the dosage and add or remove concomitant ASMs as clinically indicated. However, monotherapy with cenobamate was not allowed.
Outcomes
The safety outcomes were same for both OLE studies, including incidences of TEAEs, serious TEAEs, and TEAEs leading discontinuation of the study medication.11,12 No seizure diary data were collected during the OLE of Study C013. The Study C017 OLE also reported the efficacy outcomes as single-arm cohort results without a comparison group, including the incidence of seizure freedom, time to seizure freedom, and responder rates of greater than or equal to 50%, greater than or equal to 75%, and greater than or equal to 90%.12
Statistical Analysis
The numbers and proportion of patients with outcomes were reported using descriptive statistical methods. Outcomes by subgroups were statistically tested and reported when applicable.
Characteristics of the patients who enrolled in the 2 OLE studies are summarized in Table 19.
Results
Patient Disposition
Of the patients initially randomized in Study C013 and Study C017, 67.1% (149 patients of 222 patients in the Study C013) and 81.5% (356 patients of 437 patients in Study C017), respectively, entered the OLE studies. (None of the 43 patients from the study site in India participated in the Study C013 OLE.) Details of patient disposition to cenobamate are summarized in Table 20.
Table 19: Summary of Baseline Characteristics for Study C013 OLE and Study C017 OLE
Characteristics | Study C013 OLE (N = 149) | Study C017 OLE (N = 356) |
|---|---|---|
Age, years, mean (SD) | 37.6 (10.9) | 39.6 (11.7) |
Men, n (%) | 72 (48.3) | 185 (52.0) |
Women, n (%) | 77 (51.7) | 171 (48.0) |
BMI, kg/m2, mean (SD) | 26.3 (5.2) | 26.5 (6.3) |
Race, n (%) | ||
Asian | 37 (24.8) | 32 (9.0) |
Black or African American | 5 (3.4) | 9 (2.5) |
Other or Unknown | 8 (5.4) | 8 (2.2) |
White | 99 (66.4) | 307 (86.2) |
Seizure type by history, n (%)a | ||
Focal aware nonmotor | 37 (18.1) | 75 (21.1) |
Focal aware motor | 35 (23.5) | 77 (21.6) |
Focal impaired awareness | 128 (85.9) | 276 (77.5) |
Focal to bilateral tonic-clonic | 99 (66.4) | 210 (59.0) |
Number of ASMs at baseline, n (%) | ||
0 | 0 | 0 |
1 | 13 (8.7) | 56 (15.7) |
2 | 70 (47.0) | 139 (39.0) |
> 2 | 66 (44.3) | 161 (45.2) |
Background and/or concomitant ASMs (≥ 10% of patients), n (%) | ||
Levetiracetam | 67 (45.0) | 153 (43.0) |
Lamotrigine | 52 (34.9) | 118 (33.1) |
Carbamazepine | 39 (26.2) | 98 (27.5) |
Valproateb | 47 (31.5) | 87 (24.4) |
Topiramate | 39 (26.2) | 63 (17.7) |
Lacosamide | 44 (29.5) | 61 (17.1) |
Oxcarbazepine | 27 (18.1) | 49 (13.8) |
ASM = antiseizure medication; BMI = body mass index; OLE = open-label extension; SD = standard deviation.
Note: Patient characteristics were collected at baseline in the OLEs of Study C013 and Study C017 for the safety population (i.e., patients who entered the OLE phase of the pivotal studies C013 and C017).
aPatients may be reported in more than 1 category.
bIn the Study C013 OLE, valproate included valproate semisodium, valproate sodium, and valproic acid. In the Study C017 OLE, valproate included all forms of valproate, valproic acid, or divalproex sodium.
Source: Clinical Study Report for Study C013 OLE;11 Clinical Study Report for Study C017 OLE.12
Table 20: Patient Disposition for Study C017 OLE and Study C013 OLE
Details | Study C013 OLE (N = 149a) | Study C017 OLE (N = 356) |
|---|---|---|
Entered OLE, n | 149 | 356b |
Analyzed for safety, n | 149 | 355 |
Completed study, n (%) | 37 (24.8) | 5 (1.4) |
Discontinued study, n (%) | 112 (75.2) | 286 (80.3) |
Entered EAP or Navigator | 47 (31.5) | 129 (36.2) |
Lack of efficacy | NR | 67 (18.8) |
Withdrawal by patient | 31 (20.8) | 35 (9.8) |
Adverse event | 16 (10.7) | 28 (7.9) |
Lost to follow-up | 5 (3.4) | 7 (2.0) |
Death | 0 | 7 (2.0) |
Protocol violation | 0 | 3 (0.8) |
Pregnancy | 1 (0.7) | 0 |
Other | 12 (8.1) | 10 (2.8) |
EAP = Extended Access Program; NR = not reported; OLE = open-label extension.
aBecause India did not approve the OLE, all 43 patients from that site were excluded from it.
bOf the 360 patients who completed the DB Study C017,61 356 patients entered the OLE. For the 4 who did not, reasons included adverse events (N = 1), withdrawal by patient (N = 1), withdrawal by sponsor due to noncompliance (N = 1), and reason unspecified (N = 1). One patient did not have any dose data recorded, leaving 355 in the safety population.87
Source: Clinical Study Report for Study C013 OLE,11 Clinical Study Report for Study C017 OLE.12
Exposure to Study Treatments
Details of patient exposure to cenobamate are summarized in Table 21.
Table 21: Patient Exposure for Study C017 OLE and Study C013 OLE
Exposure | Study C013 OLE (N = 149) | Study C017 OLE (N = 356a) |
|---|---|---|
Total, patient-weeks | NR | NR |
Duration, month, mean (SD) | 58.8 (41.2) | 45.9 (27.9) |
Duration, month, median (range) | 87.0 (0.3 to 102.9) | 63.8 (0 to 76.2) |
Modal daily dose, mg, mean (SD) | 199.3 (91.35) | 274.9 (83.2) |
Adherence, % | NR | NR |
NR = not reported; OLE = open-label extension; SD = standard deviation.
aAnalyzed for safety (N = 355); analyzed for efficacy (modified intention-to-treat population, N = 354).
Source: Clinical Study Report for Study C013 OLE,11 Clinical Study Report for Study C017 OLE.12
Efficacy
The efficacy of cenobamate was not analyzed for patients in the Study C013 OLE.
The efficacy of cenobamate was reported for a prospective, single-arm cohort of the study population in the Study C017 OLE. Klein et al. (2022) reported interim efficacy outcomes based on a median exposure duration of 53.9 months (range = 1.1 months to 68.7 months), with retention rates at 12 months, 24 months, 36 months, and 48 months of 83%, 71%, 65%, and 62%, respectively.13 Klein et al. (2022) reported that “the percent of observed patients achieving 100% seizure reduction at consecutive 12-month intervals increased from 13.3% (36/271) during >12-24 months to 16.4% (36/220) during the last 12-month interval, >36-48 months” (observed case data).13 Of the 354 patients who entered the OLE, 10.2%, 9.6%, and 10.2% achieved a 100% reduction in seizure frequency in the intervals of greater than 12 months to 24 months, greater than 24 months to 36 months, and greater than 36 months to 48 months, respectively.13
Kelin et al. (2022) also reported that, “Among the patients in each 12-month interval group, the median IQR durations of 100% seizure reduction for the entire study were 48.0 months (20.1 months), 47.2 months (18.3 months), and 45.1 months (27.4 months). The median modal daily dose for patients with 100% seizure reduction at each 12-month interval was 300 mg (IQRs ranging from 50 to 100 mg).”13 Further, “Median percent reduction in seizure frequency during the first 6 months of the OLE for all cenobamate OLE patients was 65.4% and was similar among patients originally treated with cenobamate or placebo in the double-blind study”13 and the “median percent reduction in seizure frequency over baseline for all cenobamate OLE patients increased with each 6-month OLE interval, up to 76.1% (IQR 44.8%) at months 43–48.”13
During the greater than 36 months to 48 months interval, seizure frequency reductions greater than or equal to 50%, greater than or equal to 75%, and greater than or equal to 90% were achieved in 76.1% of patients (168 patients out of 220 patients), 51.8% of patients (114 patients out of 220 patients), and 39.1% of patients (86 patients out of 220 patients), respectively.13 Among the 354 patients who entered the OLE, 47.5%, 32.2%, and 24.3% achieved seizure frequency reductions of greater than or equal to 50%, greater than or equal to 75%, and greater than or equal to 90% during the 36-month to 48-month interval.13
Harms
According to the CSR for the Study C013 OLE, TEAEs occurred in 88.6% of patients over a median cenobamate treatment duration of 87.0 months, with 22.8% of events (34 out of 149) being mild, 50.3% (75 out of 149) being moderate, and 15.4% (23 out of 149) being severe in severity. SAEs occurred in 26.2% of patients (39 patients out of 149 patients) in the OLE. The most frequently occurring SAEs (experienced by at least 1% of patients) were convulsion (4.0%), inguinal hernia (1.3%), vomiting (1.3%), sepsis (1.3%), and osteoarthritis (1.3%). TEAEs leading to discontinuation were reported in 9.4% of patients (14 patients out of 149 patients); those leading to discontinuation in greater than 1% of patients were fatigue (1.3%) and ataxia (1.3%). Four deaths were reported during the Study C013 OLE. One of these patients had received placebo during the DB treatment period and died after experiencing respiratory arrest on day 2,543; the remaining 3 patients died after experiencing SUDEP on day 267, cardiac arrest on day 697, and completed suicide on day 132, respectively. The SUDEP and completed suicide were deemed by the investigator to be remotely related to the study drug; the respiratory arrest and cardiac arrest were deemed unrelated.11,39
According to the CSR for the Study C017 OLE, TEAEs occurred in 89.0% of patients over a median exposure duration of 63.8 months, with 21.1% of events (75 out of 356) being mild, 45.5% (162 out of 356) being moderate, and 22.5% (80 out of 356) being severe in severity. There were no cases of DRESS or any serious skin or subcutaneous tissue disorders. Thirty-two patients (9.0%) had at least 1 TEAE leading to discontinuation, most frequently (≥ 0.5%) due to dizziness (0.8%), somnolence (0.6%), balance disorder (0.6%), and depression (0.6%). In total, 6 deaths were reported in the Study C017 OLE, with all considered by investigators to be unrelated to the study drug.12 Causes of death were myocardial infarction, cardiogenic shock, SUDEP, completed suicide, and sepsis.
Harms data for Study C013 OLE and Study C017 OLE are summarized in Table 22.
Table 22: Harms Results for Study C013 OLE and Study C017 OLE – Long-Term Extension Studies
Adverse events | Study C013 OLE (N = 149) | Study C017 OLE (N = 356) |
|---|---|---|
Most common TEAEs, n (%) | ||
Patients with ≥ 1 TEAE | 132 (88.6) | 317 (89.0) |
Dizziness | 50 (33.6) | 128 (36.0) |
Headache | 41 (27.5) | 59 (16.6) |
Somnolence | 32 (21.5) | 87 (24.4) |
Nasopharyngitis | 30 (20.1) | 32 (9.0) |
Upper respiratory tract infection | 24 (16.1) | 42 (11.8) |
Diplopia | 14 (9.4) | 52 (14.6) |
Fatigue | 16 (10.7) | 59 (16.6) |
Urinary tract infection | 17 (11.4) | 24 (6.7) |
Serious adverse events, n (%) | ||
Patients with ≥ 1 SAE | 39 (26.2) | 79 (22.2) |
Convulsion | 6 (4.0) | NR |
Inguinal hernia | 2 (1.3) | NR |
Vomiting | 2 (1.3) | NR |
Sepsis | 2 (1.3) | NR |
Osteoarthritis | 2 (1.3) | NR |
Seizure | 2a | 5 (1.4) |
Vertigo | NR | 4 (1.1) |
Patients who stopped treatment due to TEAEs, n (%)b | ||
Patients who stopped treatment | 14 (9.4) | 32 (9.0) |
Fatigue | 2 (1.3) | 0 |
Ataxia | 2 (1.3) | 1 (0.3) |
Dizziness | 1 (0.7) | 3 (0.8) |
Balance disorder | 0 | 2 (0.6) |
Somnolence | 0 | 2 (0.6) |
Depression | 1 (0.7) | 2 (0.6) |
Deaths, n (%) | ||
Patients who died | 4 (2.7) | 6 (1.7) |
Cardiac arrest or cardiac disorders | 1 (0.7) | 2 (0.6) |
Sudden unexplained death in epilepsy | 1 (0.7) | 1 (0.3) |
Completed suicide | 1 (0.7) | 1 (0.3) |
Respiratory arrest | 1 (0.7) | 0 |
Sepsis | 0 | 2 (0.6) |
NR = not reported; OLE, open-label extension; SAE = serious adverse event; TEAE = treatment-emergent adverse event.
aOne event of complex partial seizures and 1 event of seizure cluster were reported. No clear information was provided regarding whether these TEAEs occurred in the 1 same patient or in 2 different patients.
bThe most common TEAEs that led to treatment discontinuation are listed.
Source: Clinical Study Report for Study C013 OLE11 and Clinical Study Report for Study C017 OLE.12
Critical Appraisal
Internal Validity
Neither of the extension studies had an active comparator or placebo group. Thus, the safety data in both studies and the efficacy data in Study C017 OLE could not be used to draw any conclusion in relation to an appropriate comparator, such as another ASM. Furthermore, the open-label study design in the OLE can bias the reporting of end points, particularly subjective measures, including AEs, SAEs, and TEAEs. Given that the completion of a pivotal trial was an eligibility criterion for the extension study, patients who discontinued those trials due to AEs or lack of response were excluded. This exclusion could result in a population of patients who were more tolerant of cenobamate, potentially leading to a response bias because those not responding to treatment are less likely to continue. Having patients who are more tolerant of cenobamate can also lead to biased estimates related to AEs, potentially resulting in fewer and less severe AEs being reported.
The sample sizes in the Study C013 OLE (N = 149) and Study C017 OLE (N = 356) may not be sufficient to detect rare AEs. Approximately 25% of patients in the Study C013 OLE and 20% of patients in the Study C017 OLE completed the studies, and there was wide variance in follow-up durations for individuals. Also, more common forms of morbidity (e.g., cardiac dysrhythmias) may not be easily identified as related to drug exposure versus as “natural” events unrelated to the drug. These factors may affect the internal validity of the study results.
External Validity
The patients enrolled in the 2 OLE studies were from multiple sites in different countries. Although the sample size of Study C013 was relatively small, with a median study drug exposure of 87 months, the OLE provided for longer follow-up for AE assessment versus the DB study. Nevertheless, because the proportion of patients who adhered to the study drug during the longer follow-up period was not reported, the study drug exposure among the patients in the 2 OLE studies was uncertain. There were no study sites in Canada for the OLEs of Study C013 or Study C017. On the other hand, no particular evidence indicating a difference between the study population and patients in Canada was identified in consultation with the clinical expert.
Indirect Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following subsections have been summarized and validated by the CADTH review team.
Objectives for the Summary of Indirect Evidence
The clinical trials included in the pivotal and RCT evidence did not provide direct evidence of the comparative efficacy and safety of cenobamate versus other adjunctive ASMs. The objective of this section is to summarize and critically appraise 1 NMA submitted by the sponsor and 1 NMA conducted by NICE that assessed the relative efficacy and safety of cenobamate versus other therapies in the treatment of adult patients with focal onset seizures (FOS).14-16 This summary also informs the pharmacoeconomic evaluation.
Description of Indirect Comparisons
The sponsor-submitted NMA (ITC 1) was based on a systematic review to assess the clinical efficacy and safety of cenobamate compared to relevant comparator therapies in patients with FOS.14,15 The NMA was originally conducted and submitted as part of the cenobamate evidence package submission to NICE. Treatments included cenobamate, brivaracetam, perampanel, lacosamide, eslicarbazepine acetate, and zonisamide.
The NICE-conducted NMA (ITC 2) was based on FOS and generalized tonic-clonic seizures; the latter are not relevant to the CADTH reimbursement review and will not be further discussed.16 Relevant treatments included brivaracetam, carbamazepine, cenobamate, eslicarbazepine acetate, gabapentin, ganaxolone, lacosamide, lamotrigine, levetiracetam, oxcarbazepine, perampanel, phenytoin, pregabalin, primidone, retigabine, rufinamide, sodium valproate, topiramate, vigabatrin, and zonisamide. Carisbamate, loreclezole, losigamone, selurampanel, and tiagabine were also included as comparators in the NMA, but do not have a Health Canada Notice of Compliance and are not reported on in this review.
The selection criteria and methods for both NMAs are presented in Table 23.
Table 23: Study Selection Criteria and Methods for ITCs Informing the CADTH Review
Characteristics | ITC 1 (sponsor NMA) | ITC 2 (NICE NMA) |
|---|---|---|
Populations | Patients aged ≥ 12 years receiving adjunctive treatment for drug-resistant FOS in epilepsy, where FOS included:
| Patients (children and adults) with FOS who have not responded to 1 or more ASMs or who have refractory focal epilepsy with or without other generalized seizure types (e.g., absence, myoclonus) |
Interventions | New third-generation ASMs:
First- and second-generation ASMs:
| Search was not restricted by treatment and was expected to include the following:
|
Comparators |
|
|
Outcomes | Efficacy:
Safety and tolerability:
| Critical outcomes in the NMA:
Important outcomes separate from the NMA:
|
Study designs |
|
|
Publication characteristics | Studies published in the English language. | Studies published in the English language. |
Exclusion criteria | Population:
Study design:
| Population:
Study design:
|
Databases searched |
|
|
Selection process | Internal validation was performed to confirm that the search strategy was broad enough to identify all relevant records. Search strategies for this systematic review were compared to relevant systematic reviews previously performed in this therapeutic area. Handsearching to identify other studies of interest. Records deduplicated. Study selection performed by 2 independent reviewers, with disagreements resolved through discussion until a consensus was reached or a third reviewer consulted. | Records deduplicated: 2 independent reviewers dual-weeded 10% of references, with disagreements resolved through discussion until a consensus was reached or a third reviewer consulted. The remaining 90% were screened by 1 reviewer. |
Data extraction process | Data from relevant publications were extracted into tables. All data were checked and validated to confirm accuracy. | Data extraction to Excel performed by 2 independent reviewers (who screened the literature), with disagreements resolved through discussion until a consensus was reached or a third reviewer consulted. |
Quality assessment | Quality assessment was performed by 1 reviewer using the Cochrane Risk of Bias 2.0 tool for RCTs and the Cochrane ROBINS-I tool (tailored to assess the study design of OLEs) for OLEs. | Quality assessment was performed by 1 reviewer using the ROBIS tool for systematic reviews and the Cochrane Risk of Bias 2.0 tool for RCTs. |
AE = adverse event; ASM = antiseizure medication; CDSR = Cochrane Database of Systematic Reviews; CENTRAL = Cochrane Central Register of Controlled Trials; DARE = Database of Abstracts of Reviews of Effects; DRESS = drug reaction with eosinophilia and systemic symptoms; FOS = focal onset seizure; HRQoL = health-related quality of life; HTA = Health Technology Assessment; ICTRP = International Clinical Trials Registry Platform; ITC = indirect treatment comparison; ITC 1 = indirect treatment comparison 1; ITC 2 = indirect treatment comparison 2; NICE = National Institute for Health and Care Excellence; NIH = National Institutes of Health; NMA = network meta-analysis; OLE = open-label extension; RCT = randomized controlled trial; ROBINS-I = Risk Of Bias In Nonrandomized Studies of Interventions; ROBIS = Risk of Bias Assessment Tool for Systematic Reviews; SAE = serious adverse event; TEAE = treatment-emergent adverse event.
Note: Details shown in the table have been taken from the sponsor’s Summary of Clinical Evidence.
Source: Sponsor’s Summary of Clinical Evidence; sponsor-submitted ITC report; NICE NMA.14-16
ITC Design – ITC 1 (Sponsor NMA)
Objectives
The objective of ITC 1 was to conduct a systematic review and NMA to estimate the relative efficacy and safety of cenobamate versus comparator ASMs that are relevant to drug plans in Canada for the treatment of FOS in adult patients with epilepsy.14,15
Study Selection Methods
A literature search of the Embase, MEDLINE, Cochrane Central Register of Controlled Trials, National Institutes of Health Clinicaltrials.gov, WHO ICTRP, Cochrane Database of Systematic Reviews, and Epistemonikos databases was conducted for studies published from 1999 to December 2019.14,15 The initial search was updated in October 2020 and again in September 2021. The search plan included both electronic searching and handsearching.
Studies included in ITC 1 may have enrolled patients as young as 12 years old with FOS.14,15 The list of comparators included 4 established third-generation ASMs and zonisamide. The combined efficacy outcome was achieving a 50% or greater responder rate and seizure freedom. The studies used data from the maintenance period (or treatment period, if the former was not reported). The safety outcome was the relative proportion of patients experiencing at least 1 TEAE leading to discontinuation. Both RCTs and OLEs were eligible for inclusion in the NMA. Only articles published in English were included. Patients with primary generalized epilepsy and status epilepticus were excluded from the sponsor’s NMA report.
An internal validation check was conducted to ensure that the search strategy identified all relevant records.14,15 The sponsor’s systematic review search strategy was compared to strategies used in recently published systematic reviews in this therapeutic area. Handsearching was performed to identify other studies of interest and included searching review articles, reference lists of included full-text publications, and free text keyword searching in internet search engines.
Records retrieved from the searches were added to an Endnote library, and duplicate records were removed.14,15 Two reviewers were involved in study selection, and disagreements were resolved through consensus or by a third reviewer. Data were extracted and checked for errors (details not available). One reviewer performed a quality assessment of the included studies using the Cochrane Risk of Bias 2.0 tool for RCTs and the Cochrane Risk Of Bias In Nonrandomized Studies of Interventions tool tailored for OLEs.88,89
ITC Analysis Methods
The analysis methods for ITC 1 are presented in Table 24.
The NMA was conducted using Bayesian Markov chain Monte Carlo methods with both random-effects (base-case) and fixed-effect (supportive analysis) models and noninformative priors.14,15 A multinomial likelihood model with a probit link function was used for the combined efficacy analysis, while a binomial likelihood model with logit function was used for the safety analysis. Model fit was assessed using the deviance information criterion and residual deviance information. Convergence was assessed using trace plots, Gelman-Rubin plots, statistical analyses, and assessment of the effective sample sizes, while correlation was assessed through autocorrelation plots. For the efficacy outcome, the random-effects model was run with 700,000 iterations, 100,000 burn-ins, and a thinning factor of 6, while the fixed-effect model was run with 400,000 iterations, 100,000 burn-ins, and a thinning factor of 4. For the safety outcome, the random-effects model was run with 300,000 iterations, 100,000 burn-ins, and a thinning factor of 70, while the fixed-effect model was run with 100,000 iterations, 25,000 burn-ins, and a thinning factor of 10.
The results formed open-loop networks with ASMs linked through placebo.14,15 All maintenance doses licensed by the European Medicines Agency were pooled into a single treatment arm.
The 2 efficacy outcomes (i.e., 50% or greater responder rate and seizure freedom) were combined, and the analysis was run as a multinomial probit model that included an adjustment for the placebo rate.14,15 Median relative risks with 95% CrIs were reported for the efficacy outcome. The analysis for discontinuations due to TEAEs was run using a binomial model with a logit link. Odds ratios with 95% CrIs were reported for the safety outcome. Data for the maintenance period were reported unless unavailable, in which case the treatment period was used. The last observation carried forward for the mITT maintenance population was used to ensure that the populations between efficacy outcomes aligned for the combined analysis.
To assess homogeneity, a pairwise meta-analysis was performed for each comparison in the network, and the I2 statistic and chi-square test P value were calculated.14,15 A placebo adjustment for the efficacy outcome was included in the model to adjust for cross-study heterogeneity of placebo group response rates using meta-regression methods. A feasibility assessment was conducted to investigate potential sources of heterogeneity and the implications on the NMA. No sensitivity or subgroup analyses were performed.
Because there were no closed loops in the NMA, consistency between direct and indirect comparisons could not be evaluated.
Table 24: ITC 1 Analysis Methods
Methods | Description |
|---|---|
Analysis methods | Efficacy analysis: The NMA was conducted using Bayesian Markov chain Monte Carlo methods. A multinomial likelihood model with a probit link function was used for the combined efficacy analysis. Both random-effects and fixed-effect models were used in the analysis as the base-case and supportive analyses, respectively. The model adjusted for cross-study heterogeneity of placebo response rates using meta-regression methods. The Bayesian formulation uses the “true” baseline, as estimated by the model, as the covariate. For the meta-regression, the placebo covariate was estimated using the baseline model approach. Safety analysis: Similarly, a Bayesian Markov chain Monte Carlo method was used with a binomial likelihood model with logit function. Both fixed-effects and random-effects models were used. A correction of 0.5 was added to all trial arms in which there was at least 1 treatment group with 0 events. Study data were from analyses comparing 2 groups (active vs. placebo). Multiarm adjustments were not made. |
Priors | Noninformative priors applied to both the fixed-effect and random-effects models. |
Assessment of model fit | Model fit was assessed using the DIC and residual deviance information. |
Assessment of consistency | No assessment of inconsistency was undertaken (no closed loops in the network). |
Assessment of convergence | For the efficacy outcome, the random-effects model was run with 700,000 iterations, 100,000 burn-ins, and a thinning factor of 6, while the fixed-effect model was run with 400,000 iterations, 100,000 burn-ins, and a thinning factor of 4. For the safety outcome, the random-effects model was run with 300,000 iterations, 100,000 burn-ins, and a thinning factor of 70, while the fixed-effect model was run with 100,000 iterations, 25,000 burn-ins, and a thinning factor of 10. Convergence was assessed by reviewing trace plots, Gelman-Rubin plots, and statistics. |
Outcomes |
The 2 efficacy outcomes (i.e., ≥ 50% responder rate and seizure freedom) were combined in the ITC. The LOCF for the mITT maintenance population was applied to use the combined analysis approach. |
Follow-up time points | All time points were included in the analyses. There were no sensitivity analyses performed to assess the impact of different follow-up times on the results. For the efficacy analysis, the maintenance period was used unless not reported, in which case the treatment period was used. |
Construction of nodes | Star-shaped, open-loop network, with ASMs linked through placebo. All dosage groups consistent with EMA maintenance doses were pooled into 1 node. |
Sensitivity analyses | Not performed |
Subgroup analysis | Not performed |
Methods for pairwise meta-analysis | The assumption for homogeneity was assessed using a pairwise meta-analysis for each comparison in the network, where the I2 statistic and chi-square test P value were calculated. There was no statistically significant heterogeneity between trial results in the pairwise comparisons. |
ASM = antiseizure medication; DIC = deviance information criterion; EMA = European Medicines Agency; ITC = indirect treatment comparison; ITC 1 = indirect treatment comparison 1; LOCF = last observation carried forward; mITT = modified intention to treat; NMA = network meta-analysis; SD = standard deviation; TEAE = treatment-emergent adverse event; vs. = versus.
Note: Details from the table have been taken from the sponsor’s Summary of Clinical Evidence.
Source: Sponsor’s Summary of Clinical Evidence and sponsor-submitted ITC report.14,15
Results of ITC 1
Summary of Included Studies
The systematic review for ITC 1 identified 2,462 studies. Of these, 269 were retrieved for full-text review, and 56 RCTs and 18 OLEs were considered.14,15 From this group of selected studies, a feasibility assessment was conducted, and 22 phase II and phase III placebo-controlled studies were included in the ITC. All 22 studies were included in the NMA efficacy analysis, and 20 were included in the safety analysis. (One brivaracetam study and 1 lacosamide study reported combined safety results for multiple seizure populations or treatment groups; therefore, these were excluded).
Characteristics of the included studies are presented in Appendix 1, Table 38.
Sample sizes ranged from 157 patients to 768 patients.14,15 There were 2 cenobamate studies (the 2 pivotal trials described in the CADTH main report), 6 brivaracetam studies, 4 lacosamide studies, 4 eslicarbazepine acetate studies, 4 perampanel studies, and 2 zonisamide studies. Most studies were multinational, except for 2 that were conducted in the US only. Where reported, titration periods ranged from 0 weeks to 8 weeks, and maintenance periods ranged from 4 weeks to 18 weeks. Total treatment times (whose definition varied among studies, but was typically the titration and maintenance periods together) ranged from 7 weeks to 24 weeks, where reported.
Baseline patient characteristics are presented in Appendix 1, Table 39. Reporting of baseline characteristic data for studies was limited, and it is unclear what age ranges were included in the studies (e.g., if patients under 18 years were included or not); however, the mean or median age for the studies ranged from 32 years to 40 years.14,15 Proportions of males and females were generally balanced in the studies. The duration of epilepsy ranged from 13.7 years to 25.3 years; it was not reported in 3 studies. Baseline median seizure frequency varied among studies, from 0 seizures to 15 seizures per 28 days; it was not reported in 1 study. The baseline seizure rate was highest in studies of perampanel versus other ASMs. The number of prior ASMs used was rarely reported, and when it was, the definition (e.g., the time period used to define prior ASM use) was inconsistent; therefore, these data could not be assessed. All studies included patients who were using at least 1 ASM at baseline (information was not reported for 2 studies), which was considered a proxy for failed treatment, with most patients being on 2 medications.
The overall risk of bias was assessed using the Cochrane Risk of Bias 2.0 tool for RCTs.88 According to the sponsor, most studies had an overall low risk of bias. The exceptions were 1 lacosamide study and 3 eslicarbazepine acetate studies that had some concerns with risk of bias and 1 zonisamide study that had a high risk of bias.14,15 The main reasons for the concerns were risk of bias arising from the randomization process and missing outcomes data.
The assessment of homogeneity for ITC 1 is presented in Table 25.
Table 25: Assessment of Homogeneity in ITC 1
Characteristics | Description and handling of potential effect modifiers |
|---|---|
Disease severity |
|
Treatment history |
|
Trial eligibility criteria |
|
Dosing of comparators |
|
Placebo response |
|
Definitions of end points |
|
Timing of end point evaluation |
|
Withdrawal frequency |
|
Clinical trial setting |
|
Study design |
|
ASM = antiseizure medication; ITC = indirect treatment comparison; ITC 1 = indirect treatment comparison 1; OLE = open-label extension; RCT = randomized controlled trial; TEAE = treatment-emergent adverse event.
Note: Details from the table have been taken from the sponsor’s Summary of Clinical Evidence.
Source: Sponsor’s Summary of Clinical Evidence and sponsor-submitted ITC report.14,15
Results
Figure 2 presents the network, number of studies, and corresponding populations for each drug contributing to the combined efficacy outcome (i.e., 50% or greater responder rate and seizure freedom). No additional trials were excluded from the analysis. Figure 3 presents the network, number of studies, and corresponding populations for each drug contributing to the safety outcome (i.e., TEAEs leading to discontinuation). Two studies were excluded from the network due to combined reporting of safety results for multiple seizure populations or treatment groups.14,15
For pairwise comparisons, I2 statistic values ranged from | || | for the 50% or greater responder rate outcome, from | || | for the seizure-free outcome, and from | || | for the safety outcome.14,15 P values from the chi-square tests were greater than 0.05 for all comparisons.
Figure 2: Overall Network for Combined 50% or Greater Responder Rate and Seizure Freedom Analysis
ITC = indirect treatment comparison.
Source: Sponsor’s Summary of Clinical Evidence and sponsor-submitted ITC report.14,15
Figure 3: Overall Network for the Proportion of Patients Experiencing at Least 1 TEAE Leading to Discontinuation Analysis
ITC = indirect treatment comparison.
Source: Sponsor’s Summary of Clinical Evidence and sponsor-submitted ITC report.14,15
Efficacy Outcome: 50% or Greater Responder Rate and Seizure Freedom
The results of the models indicated that patients receiving cenobamate were more likely to achieve a 50% or greater reduction in seizures in the short-term compared to any of the 5 ASMs in the NMA using the random-effects model (Table 26).14,15 The fixed-effect model results supported the primary analysis. Similarly, the random-effects model suggested that treatment with cenobamate was more likely to result in seizure freedom compared to the other interventions, a conclusion that was supported by the fixed-effect model results (Table 26). Figure 4 and Figure 5 present the relative risks and 95% CrIs for the random-effects model results as forest plots for the efficacy outcomes.
Table 26: Relative Risk for 50% or Greater Responder Rate and Seizure Freedom
Intervention relative to cenobamate | Median RR (95% CrI) | |
|---|---|---|
Random-effects model | Fixed-effect model | |
≥ 50% responder rate | ||
Brivaracetam | | || || | | | || || | |
Eslicarbazepine acetate | | || || | | | || || | |
Lacosamide | | || || | | | || || | |
Perampanel | | || || | | | || || | |
Zonisamide | | || || | | | || || | |
Placebo | | || || | | | || || | |
Seizure freedom | ||
Brivaracetam | | || || | | | || || | |
Eslicarbazepine acetate | | || || | | | || || | |
Lacosamide | | || || | | | || || | |
Perampanel | | || || | | | || || | |
Zonisamide | | || || | | | || || | |
Placebo | | || || | | | || || | |
Model outputs | ||
Median between-study SD | | || || | | | || || | |
DIC | | || || | | | || || | |
Median total residual deviance | | || || | | | || || | |
Effective number of parameters | | || || | | | || || | |
CrI = credible interval; DIC = deviance information criterion; ITC = indirect treatment comparison; NA = not applicable; RR = relative risk; SD = standard deviation.
Notes: Bolded values indicate a CrI that does not cross 1.0. RR of less than 1 favours cenobamate vs. the comparator. The random-effects model (base case) was run with 700,000 iterations, 100,000 burn-ins, and a thinning factor of 6. The fixed-effect model (supportive analysis) was run with 400,000 iterations, 100,000 burn-ins, and a thinning factor of 4. The mean and precision used in the fixed effect and random effects from the baseline model were | || || || || || |, respectively. The covariate for placebo response was 0.805. Details from the table have been taken from the sponsor’s Summary of Clinical Evidence.
Source: Sponsor’s Summary of Clinical Evidence and sponsor-submitted ITC report.14,15
Figure 4: Forest Plot for 50% or Greater Responder Rate

Figure was redacted per the sponsor request.
Source: Sponsor’s Summary of Clinical Evidence and sponsor-submitted ITC report.14,15
Figure 5: Forest Plot for Seizure Freedom

Figure was redacted per the sponsor request.
Source: Sponsor’s Summary of Clinical Evidence and sponsor-submitted ITC report.14,15
Safety Outcome: Proportion of Patients Experiencing at Least 1 TEAE Leading to Discontinuation
Compared to cenobamate, the results for the comparators had relatively wide 95% CrIs that included the threshold of no difference (1.0) using the random-effects model (Table 27).14,15 The fixed-effect model results were consistent with the primary analysis. Due to the lack of precision in the estimates, firm conclusions cannot be made. Figure 6 presents the ORs and 95% CrIs for the random-effects model results as a forest plot for the safety outcome.
Table 27: Odds Ratio for the Proportion of Patients Experiencing at Least 1 TEAE Leading to Discontinuation
Intervention relative to cenobamate | Median OR (95% CrI) | |
|---|---|---|
Random-effects model | Fixed-effect model | |
≥ 1 TEAE leading to discontinuation | ||
Brivaracetam | | || || | | | || || | |
Eslicarbazepine acetate | | || || | | | || || | |
Lacosamide | | || || | | | || || | |
Perampanel | | || || | | | || || | |
Zonisamide | | || || | | | || || | |
Placebo | | || || | | | || || | |
Model outputs | ||
Median between-study SD | | || || | | | || || | |
DIC | | || || | | | || || | |
Median total residual deviance | | || || | | | || || | |
Effective number of parameters | | || || | | | || || | |
CrI = credible interval; DIC = deviance information criterion; ITC = indirect treatment comparison; NA = not applicable; OR = odds ratio; SD = standard deviation; TEAE = treatment-emergent adverse event.
Notes: Bolded values indicate a CrI that does not cross 1.0. An OR of greater than 1 favours cenobamate vs. the comparator. The random-effects model (base case) was run with 300,000 iterations, 100,000 burn-ins, and a thinning factor of 70. The fixed-effect model (supportive analysis) was run with 100,000 iterations, 25,000 burn-ins, and a thinning factor of 10. The predictive mean and SD used in the fixed effect and random effects from the baseline model were | || || || ||, respectively. Details from the table have been taken from the sponsor’s Summary of Clinical Evidence.
Source: Sponsor’s Summary of Clinical Evidence and sponsor-submitted ITC report.14,15
Figure 6: Forest Plot for the Proportion of Patients Experiencing at Least 1 TEAE Leading to Discontinuation

Figure was redacted per the sponsor request.
Source: Sponsor’s Summary of Clinical Evidence and sponsor-submitted ITC report.14,15
Critical Appraisal of ITC 1
Overall, the methods used in the sponsor’s literature search strategy and screening appeared to be adequate. Study selection was performed in duplicate; however, some details of data extraction were unknown (e.g., the number of reviewers involved and error checking). An internal validation check was performed by comparing the sponsor’s search strategy to those used in published systematic reviews to confirm that all relevant records were captured. An appraisal of the risk of bias conducted using the Cochrane Risk of Bias 2.0 tool found that 4 of the included studies had some concerns for bias (due to missing data or arising from the randomization process), and 1 zonisamide study had a high risk of bias (in the selection of reported results). However, no studies were excluded based on the risk of bias assessment, and there was no plan to investigate the potential impact of the 4 studies in question.
The sponsor conducted a feasibility assessment for the study inclusion and exclusion criteria. Based on input from clinical experts, overall, the approach to this assessment appeared to be reasonable. With 1 exception, no studies were excluded based on their eligibility criteria. Therefore, some studies may have included pediatric patients, who are not included in the proposed Health Canada indication for cenobamate. It was unclear what proportion of patients in the studies were adolescents and what effect their inclusion had on the results; however, the clinical expert consulted by CADTH indicated there would be issues with generalizing from children to adults. ITC 1 included only third-generation ASMs and zonisamide, which the CADTH review team and clinical expert noted was missing other possible treatments for this population and indication. Although zonisamide is not available in Canada, the clinical expert and clinician group input for this review stated that it can be obtained through Health Canada’s Special Access Program for some patients. The clinician group that submitted input for the cenobamate review estimated that 2% to 5% of patients may require drugs accessed through the Special Access Program.
The sponsor stated that seizure freedom was defined differently among studies; this difference has the potential to bias the findings. Some studies used a conservative definition of seizure freedom (i.e., defining it as the absence of any seizures during follow up, and any patients who withdrew early were considered nonresponders) compared to that used in the cenobamate studies. In the cenobamate trials seizure freedom was defined as a 100% reduction in 3 types of partial seizures compared to the baseline rate, based on patients who completed the study as well as those who withdrew early. In addition, it was unclear if the other efficacy and safety outcomes were defined and measured consistently across studies. Results were analyzed from either the maintenance period or the total treatment period, but were not consistent for all studies in ITC 1. This inconsistency is an important source of heterogeneity. The analyses based on the maintenance period may show a greater reduction in seizure frequency than based on the entire treatment period. The maintenance phase analyses exclude the time period when the drug has not yet reached the target dose and may still be subtherapeutic. Another source of heterogeneity was the difference in the durations of the clinical trials. The treatment periods had variable durations across studies, ranging from 7 weeks to 24 weeks. Moreover, the clinical expert consulted by CADTH indicated that the studies were short in duration, considering that epilepsy is a chronic condition that often requires lifelong treatment.
Based on the sponsor’s feasibility assessment, 7 studies were excluded from the NMA with reasonable explanations. As previously mentioned, 1 zonisamide study was excluded for having an entirely Asian population and being relatively small (N = 104). Three dose-escalation studies were also excluded for their short durations at a stable dose (1 eslicarbazepine acetate and 2 perampanel studies), and 2 zonisamide studies were excluded for using dosages outside of the maintenance range approved by the European Medicines Agency. Lastly, 1 zonisamide study was excluded because it was a single-arm study. As noted, most of the exclusions were for zonisamide studies, which reduces the available evidence for comparisons to this drug to 2 studies.
Twenty-two studies contributed to the efficacy analysis, resulting in a sparse network with no closed loops. The sponsor’s report stated that OLEs were included in the network, but it is unclear how the results from these studies could have been included in the analyses. A random-effects model was the base case. The sponsor stated that this was justified was due to potential heterogeneity among studies, and the CADTH review team agreed that it was an appropriate model choice. Results from the fixed-effect model were reported as a supportive analysis and were consistent with those from the random-effects model. The use of a multinomial model to assess different response levels in the same analysis (i.e., 50% response and seizure freedom) was reasonable (and was the method preferred by NICE in its review of cenobamate).90 However, this model assumes a common relative treatment effect for both response thresholds (i.e., the inference for comparator A versus B is the same for a 50% response as for seizure freedom). Whether this assumption is clinically plausible is unclear.
The variability in baseline patient characteristics (particularly the proportion of white patients, mean BMI, duration of epilepsy, seizure frequency per 28 days, and number of ASMs at baseline) suggests that patients differed across studies and that the similarity assumption was likely violated. The duration of epilepsy among the studies in ITC 1 ranged from 13.7 years to 25.3 years, and the clinical expert explained that patients who have had epilepsy for longer have likely been on more ASMs and could have more refractory disease than those more recently diagnosed. The number of prior ASMs was not available due to incomplete reporting in the studies and inconsistent definitions, but the CADTH review team noted this was an important treatment-effect modifier and indicator of disease severity that should be adjusted for in the analysis. Other effect modifiers that the CADTH review team and the clinical expert identified include age, sex, cause of seizures, baseline number of seizures, and duration of active epilepsy. However, these were not adjusted for in any analyses.
The sponsor concluded that there was no statistically significant heterogeneity among pairwise comparisons; however, the results for the I2 statistic and chi-square test P value are inconclusive evidence for homogeneity. There were differences in the definition of drug-resistant epilepsy, baseline patient characteristics, doses used, outcome definitions, and treatment durations that indicated heterogeneity in the populations and study characteristics. Data available for different doses were pooled by drug in the NMA and included only licensed maintenance doses, which may have decreased both the number of studies contributing to the analysis and the heterogeneity between studies. However, sensitivity analyses by dose may uncover possible inconsistencies in treatment effects across dosage groups. The study publication dates covered nearly 15 years, and it is uncertain if standard of care or patients themselves have changed in that time. The model took into account variable placebo responses in an attempt to adjust for cross-study heterogeneity; however, this is only a proxy adjustment, and it is unknown if or how adequately it adjusts for important prognostic factors or effect modifiers in this patient population.
No direct evidence comparing active treatments was identified in ITC 1. Therefore, consistency was not assessable.
Detailed harms data (aside from discontinuations due to AEs) and outcomes for HRQoL were not included in the analysis, yet both of these were noted by the clinical expert and the clinician and patient groups as important to patients with epilepsy. Considering that time on treatment for ASMs must be long enough to sufficiently assess the outcome (for instance, 1-year for seizure freedom, as suggested in the NICE technology appraisal of cenobamate), follow-up times were relatively short in the studies, and long-term comparative efficacy and safety are unknown.90 Of the 22 included studies, only 3 had study locations in Canada, while 3 were “global” (i.e., may have had locations in Canada), which limits the generalizability of the results to clinical practice in Canada. However, the clinical expert was of the opinion that, based on the reported patient characteristics and their breadth, the populations could be similar to patients with epilepsy living in Canada.
Due to the notable heterogeneity in study characteristics and populations, the incomplete list of interventions relevant to FOS treatment in Canada, and the lack of head-to-head studies, there is uncertainty in the sponsor’s NMA results, making it difficult to draw meaningful conclusions.
ITC Design – ITC 2 (NICE NMA)
Objectives
The objective of ITC 2 was to compare the efficacy and summarize the safety data of add-on ASMs in pediatric and adult patients with FOS.16 The NMA conducted by NICE defined add-on therapy as ASMs “prescribed to people that have failed to respond to 1 or more ASM or had refractory epilepsy.”16
Study Selection Methods
The Cochrane Central Register of Controlled Trials, Cochrane Database of Systematic Reviews, Embase, and MEDLINE databases were searched for studies up to February 3, 2021.16 Reference lists from systematic reviews were checked to identify studies not captured in the search.
Studies in ITC 2 enrolled patients with FOS with or without other generalized seizure types, such as absence or myoclonus seizures.16 All ages were eligible except infants younger than 28 days with acute symptomatic seizures. The analysis did not limit by any ASM type as long as the drug was used to treat the population of interest and data for different doses were pooled by drug. The 2 efficacy outcomes were a greater than 50% reduction in seizure frequency and seizure freedom. Data were taken from the treatment or maintenance periods. Safety outcomes were reported for the proportion of patients who experienced a trial-defined AE or SAE or who discontinued treatment due to an AE, and for all-cause mortality. HRQoL and AEs were outcomes of interest, but heterogeneity in data collection and reporting prevented the pooling of results or direct comparisons between drugs. Only RCTs published in English were eligible for inclusion. Studies with mixed populations of patients with epilepsy and patients without epilepsy were excluded (except where there was a subgroup analysis for the former group). Populations that mixed patients with FOS and those with generalized tonic-clonic seizures were included in the analysis because it was reasoned that most patients would have FOS.
Ten percent of the references were dual-weeded by 2 independent reviewers, with disagreements resolved through discussion until a consensus was reached or a third reviewer consulted. The remaining 90% were screened by 1 reviewer.16 Data were extracted by the same 2 reviewers, and disagreements were resolved in a similar manner. Quality assessment was performed by 1 reviewer using the Cochrane Risk of Bias 2.0 tool.88
ITC Analysis Methods
The analysis methods for ITC 2 are presented in Table 28.
The NMA for ITC 2 was conducted similarly to ITC 1 (i.e., Bayesian Markov chain Monte Carlo methods with noninformative priors), but used only random-effects models for the analysis.16 It was believed that the same true effect size was unlikely for all studies; thus, a fixed-effect model would be inappropriate. This choice was also supported by previous NMAs reporting better goodness of fit with random-effects models over fixed-effect models. Goodness of model fit was assessed by comparing the posterior mean of the residual deviance (i.e., the magnitude of difference between model-predicted data and observed data) and the number of data points in the model. The deviance information criterion (equal to the sum of the number of effective parameters and posterior mean deviance) was also used to assess model fit. Lastly, the posterior median for the between-study SD (which measures the heterogeneity of treatment effects) was used to compare models.
For the Bayesian model, 100,000 burn-in simulations were run to allow for model convergence, then 60,000 simulations were run to estimate outputs.16 History and kernel density plots were assessed visually for convergence of the 2 chains.
Each outcome was a separate network, with each treatment a separate node, and results were pooled by treatment.16 Larger nodes indicated more patients randomized to that treatment, and the thickness of the line joining comparators indicated the number of trials for the direct comparison.
The efficacy outcomes were similar to those in ITC 1 (i.e., a 50% or greater decrease in seizure frequency and seizure freedom); however, it is unknown if the outcomes were defined exactly the same way across studies or between the 2 ITCs. Estimates of relative treatment effect were presented as odds ratios with 95% CrIs.16 Summaries for AEs were noncomparative due to heterogeneity in definitions, measurements, and reporting. Follow-up times were not detailed in the analysis, but it was indicated that all data were pooled by treatment.
Study characteristics and baseline patient characteristics were not well reported, and it is unclear if or how homogeneity was assessed among studies in the NMA.
Heterogeneity in the placebo response rate for efficacy outcomes was assessed using Cochran’s Q test of homogeneity.16 Placebo response was investigated using Spearman correlation coefficients between placebo response rate and study publication date (proxy for when a trial began) to detect placebo drift, and between placebo response rate and relative effectiveness to determine the direction of the drift. The sensitivity analysis excluding studies that excluded women of child-bearing age was not conducted because the networks did not include at least 75% of the treatments for the primary analysis. For the same reason, subgroup analyses for previous treatment and patients with treatment-resistant epilepsy were not conducted.
Consistency was assessed using an unrelated mean-effects model (i.e., comparison of fit for a model that assumes consistency against a model that allowed for inconsistency) and deviance plots (i.e., posterior mean deviance of data points from a model with consistency plotted against those from a model with inconsistency).16 Potential inconsistencies were further explored using node-splitting methods. Inconsistency was suspected if the point estimates from the direct evidence fell outside of the 95% CrI of the corresponding indirect estimates. Instances of inconsistency were checked for data accuracy. Loops with inconsistencies had baseline characteristics compared to identify possible differences in treatment-effect modifiers.
Table 28: ITC 2 Analysis Methods
Methods | Description |
|---|---|
Analysis methods | It was suspected that the placebo response in studies has changed over time, affecting the appropriateness of pooling the placebo results. First, a pooled placebo response rate was calculated, then the correlation between placebo response rate and study publication date (proxy for when a trial began) was investigated. Spearman’s rank correlation coefficients were calculated to assess correlation between placebo response rate and relative effectiveness (reported as ORs), then to determine the direction of placebo drift. Meta-regression analyses (covariates of placebo response rate and publication year) were conducted to find significant coefficients (P < 0.05), if any, for relative effectiveness. All active treatments were pooled as 1 comparator. Similar analyses were conducted for baseline seizure frequency of more than 28 days (thought to be correlated with relative effectiveness). Additional placebo response-adjusted NMAs for the primary efficacy outcomes were created to investigate if this was a better model fit for the data. Patients with missing data were considered nonresponders (i.e., not achieving the outcome). Bayesian Markov chain Monte Carlo methods and noninformative priors were used. A binomial likelihood with a logit link model was used for the 2 efficacy outcomes. Only random-effects models were used because the same true effect size was unlikely for all studies. |
Priors | Noninformative priors. |
Assessment of model fit | Comparison of the posterior mean of the residual deviance and number of data points in the model, DIC measures, and the posterior median for the between-study SD were used to compare models and assess model fit. |
Assessment of consistency | Unrelated mean-effects model and deviance plots were used to assess consistency. Node-splitting methods were used to further explore potential inconsistency. Inconsistency was suspected if point estimates from direct evidence were outside the 95% CrI of the corresponding indirect estimates. Instances of inconsistency were checked for data accuracy. Loops with inconsistencies had baseline characteristics compared to identify possible differences in treatment-effect modifiers. |
Assessment of convergence | 100,000 burn-in simulations were run to allow for model convergence, then 60,000 simulations were run to estimate outputs. History and kernel density plots were visually assessed for convergence of the 2 chains. |
Outcomes |
|
Follow-up time points | Data were pooled by drug. Data for maintenance and treatment periods were used. |
Construction of nodes | 1 node per intervention, where the size represented the number of randomized patients and line thickness indicated the number of trials for direct comparison. |
Sensitivity analyses | Conducted only if networks included > 75% of the treatments in the primary analysis.
|
Subgroup analysis | Conducted only if networks included > 75% of the treatments in the primary analysis.
|
Methods for pairwise meta-analysis | RCTs with ≥ 1 pairwise comparison between treatments were included. Interventions not connected to a network were analyzed in a pairwise meta-analysis. ORs with 95% CrIs for pairwise comparisons were reported. |
AE = adverse event; CrI = credible interval; DIC = deviance information criterion; HRQoL = health-related quality of life; ITC 2 = indirect treatment comparison 2; NICE = National Institute for Health and Care Excellence; NMA = network meta-analysis; OR = odds ratio; RCT = randomized controlled trial; SAE = serious adverse event; SD = standard deviation.
Source: NICE NMA.16
Results of ITC 2
Summary of Included Studies
In total, 18,066 studies were identified from the literature search, of which 448 were retrieved for full-text review.16 After screening, 109 RCTs were included: 99 studies (27,686 patients and 26 treatments) contributed to the greater than 50% reduction in seizure frequency analysis, and 72 studies (20,826 patients and 21 treatments) contributed to the seizure freedom analysis. The placebo response meta-analyses included 73 of the 99 studies (25,824 patients) for the greater than 50% reduction in seizure frequency outcome and 41 of 72 studies (13,579 patients) for the seizure freedom outcome. Both cenobamate pivotal trials described in the CADTH main report were included in the analyses.60,61
Study and baseline patient characteristics were not described in the NMA, making it difficult to assess heterogeneity among studies. The majority of studies were placebo-controlled, and 10 studies included an active comparator.16
Overall, 43 studies were judged to have a low risk of bias; 41 studies had some concerns for bias; and 26 had a high risk of bias.16 Greater concerns for bias were attributed mainly to 4 of the 5 domains: bias arising from the randomization process; bias due to deviations from intended interventions; bias due to missing outcome data; and bias arising from measurement of the outcome.
The assessment of homogeneity for ITC 2 is presented in Table 29.
Table 29: Assessment of Homogeneity for ITC 2
Characteristics | Description and handling of potential effect modifiers |
|---|---|
Disease severity |
|
Treatment history | NR |
Trial eligibility criteria |
|
Dosing of comparators | NR (however, data within trials were pooled). |
Placebo response |
Greater than 50% reduction in seizure frequency:
Seizure freedom:
|
Definitions of end points | Unclear how outcomes were defined in the studies and whether these were consistent within the NMA |
Timing of end point evaluation | NR |
Withdrawal frequency | Treatment discontinuations due to AEs were described narratively |
Clinical trial setting | NR |
Study design | RCTs |
AE = adverse event; ASM = antiseizure medication; CI = confidence interval; FOS = focal onset seizure; ITC 2 = indirect treatment comparison 2; NICE = National Institute for Health and Care Excellence; NMA = network meta-analysis; NR = not reported; RCT = randomized controlled trial.
Source: NICE NMA.16
Results
Figure 7 and Figure 8 present the networks for greater than 50% reduction in seizure frequency and seizure freedom, respectively. Results were reported relative to placebo. Less than 10% of all evidence included in the NMA was direct evidence between active comparators.16
Efficacy Outcome: Greater Than 50% Reduction in Seizure Frequency
Odds ratios with 95% CrIs based on adjusted data for treatments compared to cenobamate are presented in Table 30. The results of the analyses indicate that patients who received cenobamate were more likely to achieve a greater than 50% reduction in seizure frequency than those on most other treatments, including eslicarbazepine acetate, retigabine, gabapentin, lacosamide, lamotrigine, levetiracetam, oxcarbazepine, perampanel, brivaracetam, pregabalin, rufinamide, zonisamide, and primidone.16 The relative treatment effects for cenobamate versus other comparators had wide CrIs that contained 1.0 (no difference).
Efficacy Outcome: Seizure Freedom
Relative treatment effects were not available for seizure freedom. It was reported that studies were generally underpowered for this outcome due to the rarity of the event, particularly for patients randomized to placebo, and CrIs were wide for available data.16
Figure 7: Network for Greater Than 50% Reduction in Seizure Frequency
NICE = National Institute for Health and Care Excellence; NMA = network meta-analysis.
© NICE [2022] Epilepsies in children, young people and adults: [F] Add-on therapy for generalized tonic-clonic and focal onset seizures. Available from https://www.nice.org.uk/guidance/ng217/evidence/f-addon-therapy-for-generalisedtonicclonic-and-focal-onset-seizures-pdf-398366282815 All rights reserved.
Subject to Notice of rights.
NICE guidance is prepared for the National Health Service in England. All NICE guidance is subject to regular review and may be updated or withdrawn. NICE accepts no responsibility for the use of its content in this product/publication.
Source: NICE NMA.16
Safety Outcomes
Safety data were limited and reported by treatment, and do not inform relative safety for cenobamate versus other ASMs. For cenobamate, 547 patients (63.4%) experienced an AE during the study; 204 patients (23.6%) discontinued treatment due to an AE; and 3 patients (0.35%) died.16
Critical Appraisal of ITC 2
In general, the ITC 2 search strategy methods were adequate, a prespecified protocol was used for the systematic review, and the choice of databases seemed appropriate. Only 10% of the literature was screened by 2 reviewers, while the remaining 90% was screened by 1 reviewer. The data extraction and verification process was performed by the same 2 reviewers. The Cochrane Risk of Bias 2.0 tool was used to evaluate the selected studies: nearly a quarter had a high risk of bias and more than a third had some concerns for bias. No studies were excluded based on the risk of bias assessment, and there was no reported plan to investigate the potential impact of these studies.
Figure 8: Network for Seizure Freedom
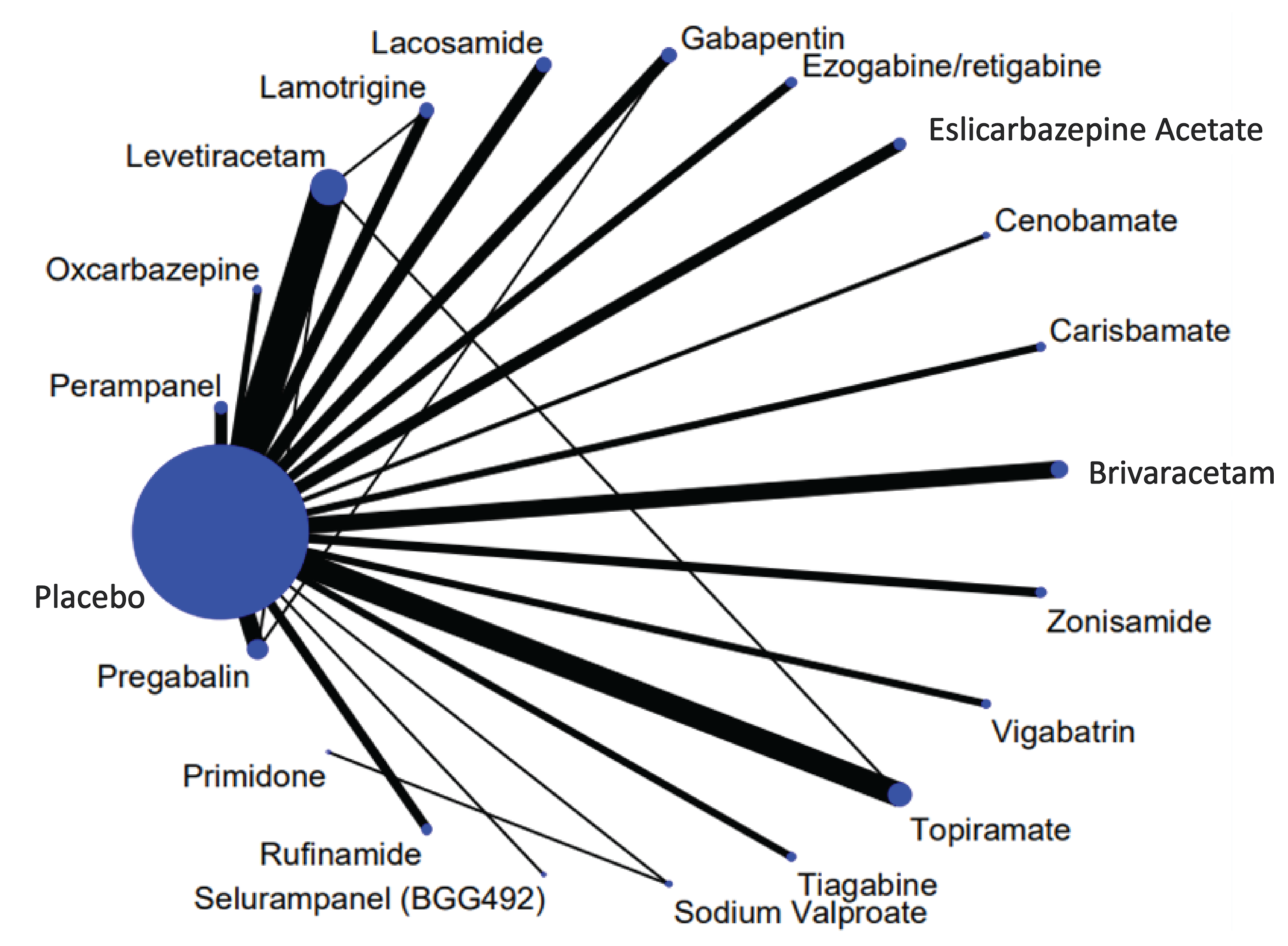
NICE = National Institute for Health and Care Excellence; NMA = network meta-analysis.
© NICE [2022] Epilepsies in children, young people and adults: [F] Add-on therapy for generalized tonic-clonic and focal onset seizures. Available from https://www.nice.org.uk/guidance/ng217/evidence/f-addon-therapy-for-generalisedtonicclonic-and-focal-onset-seizures-pdf-398366282815 All rights reserved.
Subject to Notice of rights
NICE guidance is prepared for the National Health Service in England. All NICE guidance is subject to regular review and may be updated or withdrawn. NICE accepts no responsibility for the use of its content in this product/publication.
Source: NICE NMA.16
Table 30: Comparative Efficacy for Treatments Relative to Cenobamate for Greater Than 50% Reduction in Seizure Frequency
Cenobamate relative to intervention | OR (95% CrI) from adjusted NMAa |
|---|---|
≥ 50% responder rate | |
Placebo | 3.77 (2.43 to 6) |
Eslicarbazepine acetate | 2.44 (1.44 to 4.17) |
Retigabine | 1.97 (1.09 to 3.62) |
Gabapentin | 2.71 (1.53 to 5) |
Lacosamide | 1.81 (1.09 to 3.08) |
Lamotrigine | 2.39 (1.35 to 4.37) |
Levetiracetam | 1.7 (1.01 to 2.83) |
Oxcarbazepine | 1.87 (1.06 to 3.29) |
Perampanel | 2.58 (1.49 to 4.48) |
Brivaracetam | 2.45 (1.43 to 4.33) |
Pregabalin | 2.19 (1.32 to 3.68) |
Rufinamide | 3.27 (1.74 to 6.13) |
Sodium valproate | 2.03 (0.73 to 5.21) |
Topiramate | 1.66 (0.95 to 2.89) |
Zonisamide | 2 (1.1 to 3.66) |
Primidone | 4.29 (1.19 to 13.83) |
Vigabatrin | 1.28 (0.59 to 2.86)b |
Phenytoin | 1.92 (0.68 to 5.56)b |
Carbamazepine | 0.90 (0.29 to 2.70)b |
Ganaxolone | 2.56 (0.79 to 8.33)b |
CrI = credible interval; NICE = National Institute for Health and Care Excellence; NMA = network meta-analysis; OR = odds ratio.
Note: Bolded values indicate a CrI that does not cross 1.0. An OR greater than 1 favours cenobamate vs. the comparator.
aAdjustments made for baseline placebo response rate using meta-regression.
bValues were presented as the intervention relative to cenobamate in the original NICE report.
© NICE [2022] Epilepsies in children, young people and adults: [F] Add-on therapy for generalized tonic-clonic and focal onset seizures. Available from https://www.nice.org.uk/guidance/ng217/evidence/f-addon-therapy-for-generalisedtonicclonic-and-focal-onset-seizures-pdf-398366282815 All rights reserved.
Subject to Notice of rights.
NICE guidance is prepared for the National Health Service in England. All NICE guidance is subject to regular review and may be updated or withdrawn. NICE accepts no responsibility for the use of its content in this product/publication.
Source: NICE NMA.16
ITC 2 excluded newborns aged younger than 28 days, and — as with ITC 1 — some studies likely included nonadults. According to the clinical expert consulted by CADTH, there may be generalizability issues with extrapolating to adults from data that included pediatric patients (given that cenobamate is indicated for adults). Combined FOS and generalized tonic-clonic seizure populations were also included in the analyses. The authors of ITC 2 deemed this inclusion to be acceptable because the latter group of patients would have made up only a small proportion of the mixed populations. ITC 2 focused on “add-on therapy,” which was defined similarly to the expected place in therapy for cenobamate (i.e., not a first-line treatment). Compared to ITC 1, there were no restrictions on the type of ASM in ITC 2, and more interventions were included, including some drugs not available in Canada. The broader evidence base also included direct comparisons between active treatments, something that was lacking in ITC 1. All doses were pooled by treatment, an approach that the authors of ITC 2 justified because it allowed for drug titration and changes in dose for tolerability and AEs. While this better reflects clinical practice, it could also include doses outside of the Health Canada–indicated ranges for these ASMs. No sensitivity analyses were performed to detect potential differences in treatment effects across doses. The clinician group that submitted input for the CADTH review of cenobamate indicated that ganaxolone and zonisamide (included in ITC 2) are not available in Canada. However, given that patients may access them through Health Canada’s Special Access Program, these drugs have been included in the CADTH report. The efficacy outcomes in ITC 2 were similar to those of ITC 1 in name (i.e., a 50% reduction in seizure frequency and seizure freedom), but it is unclear if the exact definitions were consistent between the ITCs or among the included studies. The authors of ITC 2 justified the choice of outcomes based on their common inclusion in studies and because a 50% reduction would likely lead to a meaningful improvement in HRQoL. Additionally, these outcomes are recommended as trial outcomes by the Commission on Antiepileptic Drugs of the International League Against Epilepsy and were confirmed as being important to patients by the clinical expert and based on the patient and clinician group input.16 However, it was noted in the NICE technology appraisal of cenobamate that a 50% reduction in seizure frequency is not as meaningful to patients as seizure freedom. This is because the aim is to regain independence, and a reduction in the number of seizures may not result in complete patient independence.90 As with ITC 1, data were taken from either the maintenance or treatment period, and the same issues apply with measuring seizure reduction depending on whether the titration period is included or not.90
The authors of ITC 2 assessed the certainty of the evidence for each outcome and concluded that it was very low to moderate for the greater than 50% reduction in seizure frequency outcome. The certainty of evidence was downgraded due to indirectness, wide CrIs, and the inclusion of drugs that were used off-label for FOS. There was low to moderate certainty for the seizure freedom outcome (evidence was downgraded due to indirectness [few events and wide CrIs]). Safety outcomes were not formally assessed due to inconsistent definitions and reporting.
In total, more studies were included in ITC 2, making the networks larger than in ITC 1, yet there were still few direct comparisons of active treatments. For the ASMs common to the 2 networks, ITC 2 had 7 studies not included in ITC 1, while ITC 1 had 1 study not included in ITC 2. Therefore, it is possible that important studies were missed from either network of evidence. The authors of ITC 2 reasoned that it was unlikely there would be a single true effect size for all studies; they chose a random-effects model over a fixed-effect model, which the CADTH review team agreed was appropriate. The ITC 2 authors also noted that previous NMAs for FOS reported better fit with a random-effects model. No preplanned subgroup or sensitivity analyses were conducted because their expected networks did not include at least 75% of the original network’s treatments.
Baseline patient characteristics were not reported in ITC 2, and it was not possible to assess the similarity assumption.
Patient and study characteristics were not reported in ITC 2, making it challenging to compare the heterogeneity of the studies. A possible source of heterogeneity is the wide range in study publication dates (1990 to 2020); it is possible that management of FOS and patients with FOS have changed in that time. Statistical heterogeneity between studies was assessed for placebo response rates for each outcome and was significant for the greater than 50% reduction in seizure frequency outcome (P < 0.001), but not for seizure freedom (P = 0.30). The results of the NMA indicated a changing placebo response rate over time. This finding may also suggest that the study populations are different and support the likeliness of heterogeneity.90 Although placebo response was adjusted for in ITC 2, it is unknown if this was an adequate adjustment for patient characteristics and effect modifiers that may bias the results. Moreover, no separate analyses were performed to adjust for the impact of the previously mentioned potential treatment-effect modifiers.
Less than 10% of all evidence in ITC 2 was direct evidence between active comparators. The results of the consistency assessment were not reported.
It was unclear how many studies took place in Canada, and the lack of reporting of patient characteristics makes it difficult to know how applicable the results are to practice in Canada. HRQoL was noted as important to patients, according to the stakeholder input for this CADTH review, but due to the use of various HRQoL instruments and a general lack of outcome reporting, results for HRQoL were not presented in ITC 2. Safety data were reported as number of patients and proportions without comparisons, and the variation in the definitions of harms in the trials makes it difficult to draw conclusions from these results. There is a need for better and long-term HRQoL and safety data as well as direct comparisons between ASMs.
Relative treatment effects for cenobamate had relatively wide 95% CrIs for a number of comparisons, and while several CrIs excluded the threshold of no difference, including those for comparators in ITC 1, there were other comparisons where the 95% CrIs included the null. This uncertainty in the results, along with the previously mentioned limitations in the analysis, makes it challenging to draw any meaningful conclusions with confidence.
Studies Addressing Gaps in the Pivotal and RCT Evidence
Contents within this section have been informed by materials submitted by the sponsor. The following subsections have been summarized and validated by the CADTH review team.
Six studies were submitted by the sponsor to address gaps in the pivotal and RCT evidence, including a study reporting the outcome of quality of life in epilepsy,18 a study among adults with a developmental disability,19 and 4 other real-world evidence studies.20,91-93 Of these, 3 real-world evidence studies are not included in this report because the studies are available only as posters and contain insufficient information to critically appraise these studies.20,91-93
Additionally, 1 sponsor-submitted phase III safety trial has been summarized in this section: Study C021 is an open-label, multicentre, long-term safety and pharmacokinetic study of cenobamate as adjunctive therapy in patients with partial onset seizures, with a larger sample size.17,94
Description of Studies
A summary of the studies from sponsor’s submission is shown in Table 31.
Table 31: Summary of 3 Sponsor-Submitted Studies Addressing Gaps in the Evidence
Gaps in pivotal and RCT evidence | Studies that address gaps | |
|---|---|---|
Study description | Summary of key results | |
Elizebath et al. (2021)18 Outcome: quality of life in epilepsy | Study design: Post hoc, extension phase data analysis of all the patients enrolled in studies C013, C017, and C021 at a single centre Location: Johns Hopkins Hospital, US Patients enrolled: Total N = 49, with the same inclusion and exclusion criteria as studies C013 (n = 10), C017 (n = 19), and C021 (n = 20) Outcomes: in addition to the efficacy and safety outcomes that have been included in the overall results of studies C013, C017, and C021, quality of life in epilepsy (using the QOLIE-31 [scale range = 1 to 100, where a higher score indicates better quality of life]) and changes in independence and epilepsy-linked disability (using a separate survey) were assessed in 37 patients at the end of this study site with a median treatment period of 5.6 years (range = 3 years to 8 years). |
|
Connor et al. (2022)19 Population: adult patients with a developmental disability | Study design: Retrospective observational study (chart review) Location: Not reported Patients enrolled: Total N = 28; all were adults experiencing uncontrolled focal seizures, receiving cenobamate as an adjunctive treatment, living with a developmental disability (including cognitive, social, orphysical), and living in a group home or with parents. Outcomes: Seizure frequency per month was assessed. The proportions of patients achieving seizure freedom, achieving ≥ 50%, ≥ 75%, and ≥ 90% reductions in focal seizure frequency, and experiencing AEs at 6 months were reported. |
|
Elliott et al. (2022)20 Population: adolescents | Study design: Retrospective observational study (chart review) Location: Single-centre patient records from the Le Bonheur Comprehensive Epilepsy Program, US Patients enrolled: Total N = 45 patients experiencing focal epilepsy who received cenobamate. Of these, 13 patients were adolescents (aged 12 years to 17 years) and 32 were adults (aged ≥ 18 years). Outcomes: ≥ 50% responder rates and AEs during the treatment period |
|
AE = adverse event; ASM = antiseizure medication; QOLIE-31 = 31-item Quality of Life in Epilepsy Questionnaire; RCT = randomized controlled trial; SD = standard deviation.
aOne patient reported 2 AEs: drowsiness and ataxia.
Source: Elizebath et al. (2021);18 Connor et al. (2022);19 Elliott et al. (2022).20
Study C021
The objective of Study C021 was to evaluate the safety and pharmacokinetics of cenobamate and concomitant ASMs when administered as adjunctive therapy for the treatment of partial onset seizures. The additional objective of this study was to characterize the rate of DRESS using a lower starting dose and a slower titration rate. According to studies of healthy volunteers receiving phenytoin and phenobarbital, cenobamate increases plasma levels of phenobarbital and phenytoin levels. A portion of this open-label safety and pharmacokinetics study was designed to understand the impact of adding cenobamate to an ASM regimen, including either phenytoin or phenobarbital in patients with partial onset epilepsy. In addition, this open-label and pharmacokinetics study enrolled patients taking concomitant ASMs.39
Study C021 enrolled patients with poorly controlled partial seizures. It consisted of a 21-day screening period and a 12-month, open-label treatment period. The treatment period included a 12-week titration phase followed by an open-label maintenance phase. For patients discontinuing, it consisted of a taper period and a follow-up visit.
Populations
According to the eligibility criteria, patients had to:
be adults aged 18 years to 70 years with a diagnosis of focal (partial onset) epilepsy, according to the International League Against Epilepsy’s classification of epileptic seizures
be experiencing uncontrolled focal seizures despite treatment with at least 1 ASM within the past 2 years
have an electroencephalographic reading consistent with the diagnosis of focal epilepsy and a CT or MRI scan performed within the previous 10 years to rule out a progressive cause of epilepsy
be currently taking a stable dose of 1 to 3 concomitant ASMs for at least 3 weeks.
The first patient in Study C021 was screened on November 3, 2015, and the last patient visit was on March 31, 2021.39
To note, there were no sites in Canada listed as investigation sites for this study.
Interventions
Most patients were supplied with open-label cenobamate 12.5 mg, 25 mg, 50 mg, and 100 mg tablets to be taken orally once daily. Patients in the US were also supplied with 150 mg and 200 mg tablets. The study drug could be taken with or without food. The target dose was 200 mg per day. After reaching the target dose, all patients were allowed to titrate up to a maximum dose of 400 mg per day.
Study patients were supplied with cenobamate at daily doses of 12.5 mg in weeks 1 and 2, 25 mg in weeks 3 and 4, 50 mg in weeks 5 and 6, 100 mg in weeks 7 and 8, 150 mg in weeks 9 and 10, and 200 mg in weeks 11 and 12 during a titration phase; they could receive a maximum dose of 400 mg per day for more than 12 months in the maintenance or dose-optimization phase. The titration phase included 7 study visits (visit 2 to visit 8), and the maintenance phase included up to 6 visits (visit 9 to visit 14). During the first 16 weeks of treatment, visits occurred every 2 weeks. The follow-up visit (visit 15) was to occur 14 days after the last dose. Patients who benefited from treatment may have continued beyond year 1, with visits occurring every 3 months.39,94
Outcomes
The safety outcomes and assessment intervals included:
frequency and severity of reported AEs and SAEs and their relation to cenobamate treatment
physical examinations to identify evidence of hypersensitivity signs (every 2 weeks during the first 4 months of treatment, at year 1, and yearly thereafter)
in-depth reviews of all hypersensitivity reactions monthly to screen for DRESS.
The end point measurement was at 12 months. After 12 months, patients were re-evaluated and could continue at the discretion of investigator.39
Statistical Analysis
The study planned to enrol at least 1,000 patients and expose them to cenobamate for at least 6 months. At least 20 patients taking phenytoin and at least 20 patients taking phenobarbital were to be enrolled. Additional patients taking ASMs other than phenytoin or phenobarbital were also enrolled to expose at least 1,000 patients for 6 months to further evaluate the long-term safety of cenobamate and the drug interactions with concomitant ASMs.39
Descriptive statistics were used to summarize safety data.39
The pharmacokinetic analyses are beyond the scope of the CADTH review; thus, these are not summarized in the current report. Also, post hoc analyses of cenobamate efficacy (seizure outcomes) were conducted among some subgroups of patients. For example, a subset population in Study C021 who had focal aware motor, focal impaired awareness, or focal to bilateral tonic-clonic seizures (N = 240) were evaluated for efficacy data in a post hoc analysis.95 These efficacy outcomes are not included in the current report because these were not prespecified in the study protocol of Study C021.17
Results
Patient Disposition
All patients who successfully met the entry criteria and provided informed consent to participate in the study were considered as enrolled patients. Safety-evaluable patients were defined as all patients enrolled in the study who received at least 1 dose of study drug medication.
Baseline Characteristics
The baseline characteristics of patients at baseline in Study C021 are summarized in Table 32.
Table 32: Summary of Baseline Characteristics of Study C021 (Safety Population)
Characteristics | Study C021a (N = 1,340) |
|---|---|
Age, years, mean (SD) | 39.7 (12.8) |
Men, n (%) | 673 (50.2) |
Women, n (%) | 667 (49.8) |
BMI, kg/m2, mean (SD) | 26.9 (6.0) |
Time since epilepsy diagnosis, years, mean (SD)b | 22.9 (14.4) |
Race, n (%) | |
Asian | 73 (5.4) |
American Indian/Alaska Native | 59 (4.4) |
Black/African American | 47 (3.5) |
Native Hawaiian or other Pacific islander | 6 (0.4) |
Other | 90 (6.7) |
White | 1,065 (79.5) |
Seizure type by history, n (%)c | |
Focal aware nonmotor | 271 (20.2) |
Focal aware motor or observable component | 323 (24.1) |
Focal impaired awareness | 1,038 (77.5) |
Focal to bilateral tonic-clonic | 790 (59.0) |
Number of ASMs at baseline, n (%)d | |
0 | 3 (0.2) |
1 | 242 (18.1) |
2 | 513 (38.3) |
> 2 | 582 (43.4) |
Concomitant ASMs (≥ 10% of patients), n (%)c,e | |
Carbamazepine | 372 (27.8) |
Clobazam | 202 (15.1) |
Lacosamide | 336 (25.1) |
Lamotrigine | 449 (33.5) |
Levetiracetam | 544 (40.6) |
Oxcarbazepine | 176 (13.1) |
Topiramate | 186 (13.9) |
Valproic acid, all forms | 435 (32.5) |
ASM = antiseizure medication; BMI = body mass index; SD = standard deviation.
aThe safety population was defined as patients who received greater than or equal to 1 dose of cenobamate.
bN = 1,336 for this variable.
cPatients may be reported in more than 1 category.
dBaseline ASMs were defined as ASMs that started before and were ongoing at the time of the first dose of cenobamate.
eConcomitant ASMs were defined as ASMs that started before and were ongoing at the time of the first dose of cenobamate or that started after the first dose of cenobamate.
Source: Clinical Study Report for Study C021.17
Exposure to Study Treatments
Details of patient disposition and exposure to cenobamate are summarized in Table 33. Of the 1,484 patients screened for Study C021, 1,345 patients (91%) were enrolled in the study.94
Table 33: Patient Disposition for Study C021
Patient disposition | Study C021 (N = 1,345) |
|---|---|
Enrolled, n | 1,345 |
Completed, n (%)a | 263 (19.6) |
Discontinued from study, n (%) | 1,082 (80.4) |
Reason for discontinuation, n (%) | |
Adverse events | 183 (13.6) |
Withdrawal by patient | 143 (10.6) |
Protocol violation | 12 (0.9) |
Lost to follow-up | 22 (1.6) |
Entered EAP or Navigator Programb | 627 (46.6) |
Pregnancy | 7 (0.5) |
Other | 88 (6.5) |
Safety population,c n | 1,340 |
EAP = Expanded Access Program.
aIncluded patients who completed the study as per end of study status case report form.
bReceived cenobamate through the EAP, which distributes non-commercial drugs outside the US, or the Navigator Program, which is for commercial distribution of drugs to patients inside the US.
cThe safety population was defined as patients who received 1 or more doses of cenobamate.
Source: Clinical Study Report for Study C021.17
Exposure to Study Treatments
Details of patient exposure to cenobamate are summarized in Table 34.
Table 34: Patient Exposure in Study C021
Patient disposition | Study C021 (N = 1,345) |
|---|---|
Total, patient-weeks or patient-years | Not available |
Duration, months, mean (SD) | 29.6 (15.1) |
Duration, months, median (range) | 36.1 (0 to 49.4) |
Modal daily dose, mg, mean (SD) | 218.6 (106.8) |
Adherence, % | Not available |
SD = standard deviation.
Source: Clinical Study Report for Study C021.17
Harms
According to the Clinical Study Report for Study C021, TEAEs occurred in 90.7% of patients. The most frequently reported TEAEs were somnolence (31.3%), dizziness (28.9%), fatigue (19.4%), and headache (17.5%). Among the patients who experienced at least 1 serious TEAE (238 patients out of 1,340 patients [17.8%]), seizures were the most frequently reported event, experienced by 29 patients (2.2% of the overall safety population). One hundred and 83 patients (13.7%) had at least 1 TEAE leading to discontinuation, mostly frequently due to dizziness (19 patients out of 1,340 patients [1.4%]) and seizures (11 patients out of 1,340 patients [0.8%]). By the data cut-off date, 10 deaths had been reported in Study C021.17 The causes of death were reported as laryngospasm, glioblastoma, subdural hematoma, SUDEP, sudden death, traumatic intracranial hemorrhage, hypovolemic shock, pneumonia (viral), status epilepticus, and cardiac arrest. According to the study investigators, 1 case of sudden death was remotely related to the study drug, and all others were unrelated to the study drug.17 No case of DRESS was reported.17
A summary of TEAEs and all-cause mortality is shown in Table 35.17
Table 35: Summary of Adverse Events for Study C021 (Safety Population)
Adverse event | Study C021a (N = 1,340) |
|---|---|
Most common TEAEs, n (%) | |
Patients with ≥ 1 TEAE | 1,215 (90.7) |
Somnolence | 419 (31.3) |
Dizziness | 387 (28.9) |
Fatigue | 260 (19.4) |
Headache | 235 (17.5) |
Most common serious TEAEs, n (%) | |
Patients with ≥ 1 serious TEAE, n (%) | 238 (17.8) |
Seizures | 29 (2.2) |
Epilepsy | 8 (0.6) |
Status epilepticus | 7 (0.5) |
Ataxia | 7 (0.5) |
Appendicitis | 7 (0.5) |
Fall | 6 (0.4) |
Pneumonia | 6 (0.4) |
Hyponatremia | 6 (0.4) |
Postictal paralysis | 6 (0.4) |
Pulmonary embolism | 6 (0.4) |
Patients who stopped treatment due to TEAEs, n (%) | |
Patients who stopped treatment | 183 (13.7) |
Dizziness | 19 (1.4) |
Seizures | 11 (0.8) |
Somnolence | 10 (0.7) |
Rash | 9 (0.7) |
Fatigue | 9 (0.7) |
Headache | 7 (0.5) |
Death, n (%) | |
Patients who diedb | 10 (0.7) |
Adverse events of special interest, n (%) | |
DRESS | 0 |
Drug hypersensitivity | 2 (0.1) |
Suicidal ideation | 23 (1.7) |
Suicidal behaviour | 1 (0.1) |
Suicide attempt | 6 (0.4) |
Respiratory arrest | 1 (0.7) |
Infections and infestationsc | 551 (41.1) |
DRESS = drug reaction with eosinophilia and systemic symptoms; TEAE = treatment-emergent adverse event.
aThe safety population was defined as patients who received greater than or equal to 1 dose of cenobamate.
bReasons for death included laryngospasm, glioblastoma, subdural hematoma, sudden unexplained death in epilepsy, sudden death, traumatic intracranial hemorrhage, hypovolemic shock, pneumonia (viral), status epilepticus, and cardiac arrest.
cInfections and infestations included specific adverse events of ear infection, influenza, otitis media, upper respiratory tract infection, urinary tract infection, and viral upper respiratory tract infection.
Source: Clinical Study Report for Study C021.17
Critical Appraisal
Internal Validity
An active comparator or placebo group is absent in the 3 sponsor-submitted other studies18-20 and in Study C021.17 Thus, it is difficult to determine if the effects observed are attributable to cenobamate, the natural history of the disease, or other factors. No confounding or selection bias adjustment was conducted in the 3 other studies;18-20 hence, there is a high risk of bias in the effectiveness estimates due to selection bias, measurement error, unmeasured confounding, and residual confounding. In Study C021, unblinding of the intervention could have biased the reporting of end points, particularly subjective measures like AEs.17 Therefore, inferences obtained from these studies have low reliability and validity.
External Validity
The results from the 3 sponsor-submitted studies were based on patient data from a single centre18,20 (or unknown numbers of participating centres)19 in the US. Given the small sample sizes (i.e., fewer than 50) in these studies, the applicability of the results to patients in Canada is unknown or low
The results in the published article of Study C021 showed that the enrolled patients were from more than 100 centres in several countries,17 but consisted of a majority white population (79.4%). However, there were no study sites in Canada.17,94 On the other hand, no particular evidence indicating a difference between the study population and patients in Canada was identified in consultation with the clinical expert.
Discussion
Summary of Available Evidence
The pivotal and RCT evidence included 2 DB, placebo-controlled RCTs (studies C013 and C017) of add-on therapy with cenobamate in adults experiencing uncontrolled partial seizures despite ongoing treatment with 1 to 3 ASMs.9,10 In Study C013 and C017, 222 and 437 patients, respectively were randomized to 12 weeks or 18 weeks of treatment with cenobamate 100 mg, 200 mg, or 400 mg once daily or placebo. The primary end point was the percent change in seizure frequency per 28 days versus baseline for all simple partial motor, complex partial, and secondarily generalized seizures.
The mean age of the patients enrolled in the pivotal trials ranged from 36.2 years (SD = 11.3 years) to 40.9 years (SD = 12.4 years) across treatment groups. Roughly equal proportions of men (47% to 54%) and women (46% to 53%) enrolled. In Study C013, the median baseline seizure frequency per 28 days was 5.5 (range = 2 to 237) in the placebo group and 7.5 (range = 0 to 187) in the cenobamate 200 mg group. In Study C017, the median baseline seizure frequency per 28 days varied from 8.4 (range = 4 to 704) for placebo to 11.0 (range = 4 to 418) for the cenobamate 200 mg group.
Also included in this report was indirect evidence from the sponsor-submitted NMA14 and an NMA conducted by NICE.16 Additional longer-term safety data were available from 2 OLE studies11,12 and an open-label, single-arm study (Study C021).17 Sponsor-submitted data addressing gaps in the RCT evidence included a study reporting HRQoL data,18 a study among adults with a developmental disability,19 and a real-world evidence study in patients with partial epilepsy who received cenobamate.20
Interpretation of Trial Results
Efficacy
Study C013 and Study C017 showed that add-on therapy with cenobamate reduced the frequency of seizures compared to placebo. In addition, patients in the cenobamate groups were more likely to achieve at least a 50% reduction in seizure frequency compared to placebo. The findings were consistent whether seizure rates were calculated based on the entire treatment period or on the maintenance period only; reductions were observed for each seizure type (simple partial motor, complex partial, and secondary generalized) in the cenobamate 200 mg and 400 mg groups versus placebo. There is some uncertainty in the efficacy findings due to the approach to missing data (which were assumed to be missing at random). Although the studies did undertake sensitivity analyses, these analyses did not fully test the validity of the missing data assumption. Given the extent of — and differential withdrawal rate in — Study C017, there is a potential for bias; however, the impact on the findings is unclear. Had the studies conducted sensitivity analyses using conservative missing data assumptions, such as a “pragmatic intention-to-treat” approach (in which patients who dropped out were considered nonresponders72), there would be greater confidence in the robustness of the findings. Seizure freedom is an important outcome to patients, but this end point was not part of the statistical hierarchy to control the type I error rate in Study C017 and was not preplanned in Study C013. The same limitations were present for the proportion of patients who achieved at least 75% and 90% reductions in seizure frequency, which — according to the clinical expert consulted — may be more clinically meaningful thresholds than the 50% response rate. While the 50% reduction threshold is often used in clinical trials, it is an arbitrary cut point, and may not be sufficient to restore many activities of daily living (e.g., driving). No literature was found that linked the partial response thresholds to improvements in HRQoL or functional status. The available results suggest that patients who receive cenobamate 200 mg and 400 mg may be more likely to achieve higher seizure reduction thresholds versus placebo, but the limitations of these data prevent drawing definitive inferences from the findings.
Neither study was designed to test the impact on HRQoL, which is an important outcome to patients. A reduction in seizure frequency that does not improve patients’ quality of life may not be clinically important. The clinical expert stated that the trials were short in duration (12 weeks to 18 weeks) for this chronic condition, and while this is not uncommon for epilepsy trials, the longer-term comparative effects of cenobamate remain uncertain. Some longer-term efficacy data were available from 1 OLE, which suggested the treatment effects on seizure frequency may be maintained up to 48 months in patients who were able to tolerate cenobamate and remained on therapy. However, these data were limited by potential selection and reporting bias, and there was no control group.
With respect to external validity, no major concerns were raised by the clinical expert with regards to the generalizability of the patients enrolled in the pivotal trials, although the trials did exclude some patients who might be treated with cenobamate in clinical practice (i.e., older adults and patients with comorbidities). The clinical expert anticipated that cenobamate would be used (at present) as an add-on to existing ASM therapy, but its exact placement in therapy (i.e., as a second- or third-line treatment) may vary in practice settings. The majority of patients enrolled in studies C013 and C017 were receiving 2 or more ASMs during the trials, which is consistent with third-line therapy. Fourteen percent of patients in Study C017 had tried only 1 ASM before enrolment. The clinical expert noted that patients who had received 1 prior ASM may be more likely to show a treatment response than patients whose seizures had not been controlled through multiple drug trials.
In the absence of direct evidence comparing cenobamate to other ASMs, 2 NMAs (1 conducted by the sponsor and 1 by NICE) were included to inform this gap in the evidence. Overall, the results between the NMAs were aligned for the 50% reduction in seizure frequency outcome, which suggests that cenobamate may be more effective than perampanel, eslicarbazepine acetate, lacosamide, brivaracetam, zonisamide, and several other ASMs that were included in the NICE NMA (including retigabine, gabapentin, lamotrigine, levetiracetam, oxcarbazepine, pregabalin, rufinamide, and primidone). Results for the seizure freedom outcome were consistent, but were available only from the sponsor-submitted NMA, which had a narrow scope. These analyses had several limitations, such as the inclusion of studies in children, missing relevant comparators (mainly in the sponsor-submitted NMA), and heterogeneity in the patient and study characteristics of the trials that informed the analyses. Of key concern were differences across studies in how efficacy outcomes were defined (e.g., seizure freedom) and in the treatment periods upon which these were based (i.e., the entire treatment period or excluding the dose titration period). Given that some studies used more conservative estimates of efficacy compared to others, these differences could potentially bias the findings. Moreover, for the sponsor-submitted analyses, the trial durations varied substantially, from 7 weeks to 24 weeks; and differences in the patient populations were noted (e.g., duration of epilepsy, baseline seizure frequency, and number of ASMs at baseline) and characterized as potential effect modifiers by the clinical expert consulted. The number and type of prior ASMs, relevant comorbidities, and the causes of epilepsy were verified by the clinical expert as important treatment-effect modifiers, but were not reported in either NMA. The NMA by NICE provided few details on the patient and trial characteristics, which limited CADTH reviewers’ ability to assess sources of heterogeneity. In addition, the studies were published over a 30-year time frame, across which changes in epilepsy management, study conduct, and patient characteristics would be expected. Although the NMAs accounted for a placebo response, it is unknown if or how adequately this adjusted for important effect modifiers, and neither of the ITCs conducted sensitivity analyses to explore the impact of potential effect modifiers. Due to these limitations, there was uncertainty in the findings of the NMAs, and although the data suggested there may be a short-term benefit favouring cenobamate in terms of the proportion of patients achieving at least a 50% reduction in seizure frequency, the magnitude and clinical relevance of any benefit remain unclear. No conclusions could be drawn about the relative effects of seizure freedom due to the differences in the outcome definitions and the other limitations described. Neither NMA assessed HRQoL. An assessment of longer-term outcomes was not possible due to the lack of comparative trial data.
The 3 additional studies submitted by the sponsor to address gaps in the evidence were small-sample, uncontrolled studies that were potentially subject to selection bias, reporting bias, and confounding. Thus, these cannot provide robust data on the effects of cenobamate for these select patient groups or HRQoL outcomes.
Harms
Most patients in the RCTs who received cenobamate (65% to 90% of patients) or placebo (63% to 70% of patients), or who received cenobamate in the longer-term safety studies (89% to 91%), reported 1 or more AEs. The short-term comparative evidence suggests a possible dose relationship, with the 200 mg and 400 mg cenobamate dosage groups in Study C017 reporting a higher frequency of AEs and a higher rate of discontinuations due to AEs than the groups receiving cenobamate 100 mg or placebo. In studies C013 and C017, 4% to 20% of patients discontinued cenobamate due to AEs during the randomized phase, and 9% of patients stopped during the OLE phase. The open-label safety study, Study C021, used the same dose titration schedule as the Canadian product monograph, and in this study, 14% of patients stopped cenobamate due to AEs. SAEs were reported in 4% to 6% of patients who received placebo, in 3% to 9% of patients who received cenobamate in the short term RCTs, and in 18% to 26% of cenobamate-treated patients in the longer-term studies (i.e., the OLEs and Study C021). Only 1 case of DRESS was reported in the short- and longer-term studies. No new safety signals were detected from the longer-term data.
Direct evidence comparing cenobamate to other ASMs was not available. The sponsor-submitted NMA for discontinuations due to TEAEs showed results that were imprecise, with wide 95% CrIs that overlapped the null. No comparative harms data were available from the NICE NMA. Thus, the comparative safety of c in either the short- or longer-term remains unclear.
The trials enrolled a select patient population and excluded older adults, those with certain comorbidities, and patients receiving drugs that may interact with cenobamate. Some of these patients may have a greater risk of AEs than the patients included in the studies. As such, the frequency of AEs reported in the trials may not be fully reflective of the AEs that would occur in clinical practice.
Conclusion
The direct evidence demonstrates that add-on therapy with cenobamate is superior to placebo in reducing the frequency of partial seizures in the short-term in adults whose partial onset seizures are inadequately controlled with up to 3 concomitant ASMs. In addition, a higher percentage of patients who received cenobamate appeared to achieve at least a 50% reduction in seizure frequency compared to placebo. While a 50% reduction in seizure frequency is an accepted end point for clinical trials, it may not be clinically relevant for all patients, given that the goal of therapy is the elimination of seizures. The pivotal studies examined higher seizure reduction thresholds (i.e., ≥ 75%, ≥ 90%, and 100%); however, due to statistical limitations (i.e., lack of control of the type I error rate) and risk of bias (i.e., analyses were conducted post hoc in Study C013), definitive inferences cannot be drawn from these data.
Direct comparative evidence for cenobamate versus other ASMs was unavailable. The indirect evidence from 2 NMAs suggest there may be a short-term benefit favouring cenobamate versus some ASMs for the proportion of patients achieving at least a 50% reduction in seizure frequency; however, the magnitude and clinical relevance of any benefit was unclear because of important limitations in the analyses. There was heterogeneity across the networks in the patient characteristics, in how outcomes were defined and analyzed, and in the durations of follow-up, creating uncertainty in the findings of the NMAs. Due to the uncertainty in the sponsor-submitted NMA, no conclusions could be drawn about the relative effects of cenobamate on seizure freedom.
The impact of cenobamate on HRQoL or other outcomes of importance to patients, such as the ability to work or live independently, is unknown because the placebo-controlled trials were not designed to test for these outcomes. Neither NMA assessed HRQoL or longer-term outcomes.
No conclusions could be drawn regarding the short-term comparative safety of cenobamate versus other ASMs due to the previously described limitations and the lack of precision in the estimates from the NMA. No new safety signals were identified from the longer-term single-arm studies; however, long-term comparative data were not available.
References
1.Gilmour H, Ramage-Morin P, Wong SL. Epilepsy in Canada: Prevalence and impact. Health Rep. 2016;27(9):24-30. PubMed
2.Scheffer IE, Berkovic S, Capovilla G, et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58(4):512-521. PubMed
3.SD S. Overview of the management of epilepsy in adults. Waltham (MD): UpToDate. 2022;25 Apr 2022.
4.Public Health Agency of Canada. Scope (prevalence and incidence) of neurological conditions. The National Population Health Study of Neurological Conditions. 2018;Ottawa (ON): Public Health Agency of Canada. Available at https://www.canada.ca/en/public-health/services/reports-publications/mapping-connections-understanding-neurological-conditions/mapping-connections-understanding-neurological-conditions-canada-13.html. Accessed 27 Feb 2023.
5.Public Health Agency of Canada. Epilepsy in Canada. Ottawa (ON): Public Health Agency of Canada. 2018. Available at https://www.canada.ca/en/public-health/services/publications/diseases-conditions/epilepsy.html. Accessed 28 Feb 2023. 2018.
6.Canadian League Against Epilepsy. Management of Epilepsy. Available at: https://claegroup.org/Management-of-Epilepsy.
7.JI S. Evaluation and management of drug-resistant epilepsy. Waltham (MA): UpToDate; 2018 Dec 20. 2018.
8.Sheng J, Liu S, Qin H, Li B, Zhang X. Drug-Resistant Epilepsy and Surgery. Curr Neuropharmacol. 2018;16(1):17-28. PubMed
9.Clinical Study Report. A Multicenter, Double-Blind, Randomized, Placebo-Controlled, Dose-Response Trial of YKP3089 as Adjunctive Therapy in Subjects with Partial Onset Seizures, with Optional Open-Label Extension PROTOCOL NUMBER YKP3089C017 [internal sponsor's report]. Fair Lawn (NJ): SK Life Science, Inc; 2018 Feb 27.
10.Clinical Study Report. A Phase 2, Multicenter, Double-Blind, Randomized, adjuctive Placebo-Controlled trial with an open-label extension to evaluate the efficacy and safety of YKP3089 in subjects with treatment resistant partial onset seizures [internal sponsor's report] Fair Lawn (NJ): SK Life Science, Inc; 2018 Jan 26.
11.Clinical Study Report (Final): YKP3089C013 A Phase 2, Multicenter, Double-Blind, Randomized, Adjunctive Placebo-Controlled Trial with an Open-Label Extension to Evaluate the Efficacy and Safety of YKP3089 in Subjects with Treatment Resistant Partial Onset Seizures [internal sponsor's report]. Paramus (NJ): SK Life Science, Inc; 2021 Sep 21.
12.Clinical Study Report: A Multicenter, Double-Blind, Randomized, Placebo-Controlled, Dose-Response Trial of YKP3089 as Adjunctive Therapy in Subjects with Partial Onset Seizures, with Optional Open-Label Extension PROTOCOL NUMBER YKP3089C017 [internal sponsor's report]. Paramus (NJ): SK Life Science, Inc; 2021 Oct 31.
13.Klein P, Aboumatar S, Brandt C, et al. Long-Term Efficacy and Safety From an Open-Label Extension of Adjunctive Cenobamate in Patients With Uncontrolled Focal Seizures. Neurology. 2022;99(10):e989-998. PubMed
14.Cenobamate Indirect Treatment Comparison Technical Report [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: (cenobamate 12.5 mg, 25 mg, 50 mg, 100 mg, 150 mg, 200 mg) tablets, oral. St-Laurent (Quebec): Paladin Labs Inc.; 2022 Oct. 2022.
15.Sponsor's clinical summary report title [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: cenobamate 12.5 mg, 25 mg, 50 mg, 100 mg, 150 mg, 200 mg tablets, oral. St-Laurent (QC): Paladin Labls Inc.; 2022 Oct.
16.Epilepsies in children, young people and adults. Add-on therapy for generalised tonic-clonic and focal onset seizures. NICE guideline NG217: Evidence reviews underpinning recommendations 5.1.5-5.1.8 & 5.2.4-5.2.7 in the NICE guideline. National Institute for Health and Care Excellence; 2022 Apr: https://www.nice.org.uk/guidance/ng217/evidence/f-addon-therapy-for-generalised-tonicclonic-and-focal-onset-seizures-pdf-398366282815. Accessed 2023 Mar 14.
17.Clinical Study Report: CENOBAMATE, YKP3089,YKP3089C021 AN OPEN LABEL, MULTICENTER, SAFETY AND PHARMACOKINETIC STUDY OF YKP3089 AS ADJUNCTIVE THERAPY IN SUBJECTS WITH PARTIAL ONSET SEIZURES: FINAL REPORT [internal sponsor's report]. SK Life Science, Inc. Paramus (NJ)2022 Feb 4.
18.Elizebath R, Zhang E, Coe P, Gutierrez EG, Yang J, Krauss GL. Cenobamate treatment of focal-onset seizures: Quality of life and outcome during up to eight years of treatment. Epilepsy Behav. 2021;116:107796. PubMed
19.Connor GS, Williamson A. Effectiveness and safety of adjunctive cenobamate for focal seizures in adults with developmental disability treated in clinical practice. Epilepsy Behav Rep. 2022;18:100533. PubMed
20.Elliott T, Ridley-Pryor T, Gienapp AJ, Wheless JW. Initial Real-World Experience With Cenobamate in Adolescents and Adults: A Single Center Experience. Pediatr Neurol. 2022;129:19-23. PubMed
21.Fisher RS, Acevedo C, Arzimanoglou A, et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia. 2014;55(4):475-482. PubMed
22.Canadian Chronic Disease Surveillance System (CCDSS) Epilepsy (active), Canadian age-standardized prevalence, trend over time. https://health-infobase.canada.ca/ccdss/data-tool/Index?G=00&V=18&M=5&Y=2017. Accessed May 2, 2023.
23.Iyer A, Marson A. Pharmacotherapy of focal epilepsy. Expert Opin Pharmacother. 2014;15(11):1543-1551. PubMed
24.Ontario Brain Institute. Clinical Guidelines for the Management of Epilepsy in Adults and Children. Available at: ManagementGuidelines_Nov2020.pdf (clinictocommunity.ca).
25.Epilepsies in children, young people and adults. Add-on therapy for generalised tonic-clonic and focal onset seizures. NICE guideline NG217. National Institute for Health and Care Excellence; 2022: https://www.nice.org.uk/guidance/ng217. Accessed 2023 Mar 23.
26.Sisodiya S. Etiology and management of refractory epilepsies. Nat Clin Pract Neurol. 2007;3(6):320-330. PubMed
27.Callaghan B, Schlesinger M, Rodemer W, et al. Remission and relapse in a drug-resistant epilepsy population followed prospectively. Epilepsia. 2011;52(3):619-626. PubMed
28.Kwan P, Arzimanoglou A, Berg AT, et al. Definition of drug resistant epilepsy: consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia. 2010;51(6):1069-1077. PubMed
29.Hesdorffer DC, Tomson T, Benn E, et al. Combined analysis of risk factors for SUDEP. Epilepsia. 2011;52(6):1150-1159. PubMed
30.Strzelczyk A, Griebel C, Lux W, Rosenow F, Reese JP. The Burden of Severely Drug-Refractory Epilepsy: A Comparative Longitudinal Evaluation of Mortality, Morbidity, Resource Use, and Cost Using German Health Insurance Data. Front Neurol. 2017;8:712. PubMed
31.Blais L, Sheehy O, St-Hilaire JM, Bernier G, Godfroid P, LeLorier JJ. Economic evaluation of levetiracetam as an add-on therapy in patients with refractory epilepsy. Pharmacoeconomics. 2005;23(5):493-503. PubMed
32.Englot DJ, Birk H, Chang EF. Seizure outcomes in nonresective epilepsy surgery: an update. Neurosurg Rev. 2017;40(2):181-194. PubMed
33.Laxer KD, Trinka E, Hirsch LJ, et al. The consequences of refractory epilepsy and its treatment. Epilepsy Behav. 2014;37:59-70. PubMed
34.Verrotti A, Iapadre G, Di Francesco L, Zagaroli L, Farello G. Diet in the Treatment of Epilepsy: What We Know So Far. Nutrients. 2020;12(9). PubMed
35.XCOPRI (cenobamate): 12.5 mg, 25 mg, 50 mg, 100 mg, 150 mg, 200mg tablets, oral [product monograph]. Dublin (Ireland): Endo Ventures Ltd.; 2022 Jun 12.
36.Guignet M, Campbell A, White HS. Cenobamate (XCOPRI): Can preclinical and clinical evidence provide insight into its mechanism of action? Epilepsia. 2020;61(11):2329-2339. PubMed
37.Sharma R, Nakamura M, Neupane C, et al. Positive allosteric modulation of GABA(A) receptors by a novel antiepileptic drug cenobamate. Eur J Pharmacol. 2020;879:173117. PubMed
38.White HS, Brown SD, Woodhead JH, Skeen GA, Wolf HH. Topiramate modulates GABA-evoked currents in murine cortical neurons by a nonbenzodiazepine mechanism. Epilepsia. 2000;41(S1):17-20. PubMed
39.Drug Reimbursement Review sponsor submission: cenobamate 12.5 mg, 25 mg, 50 mg, 100 mg, 150 mg, 200 mg tablets, oral [internal sponsor's package]. St-Laurent (QC): Paladin Labs Inc.; 2023.
40.U.S. Food & Drug Administration. FDA approves new treatment for adults with partial-onset seizures. 2019: https://www.fda.gov/news-events/press-announcements/fda-approves-new-treatment-adults-partial-onset-seizures. Accessed 2023 Feb 27.
41.Schmidt D, Schachter SC. Drug treatment of epilepsy in adults. BMJ. 2014;348:g254. PubMed
42.PrDILANTIN®(Extended Phenytoin Sodium Capsules) Product Monograph. Kirkland, QC, Canada: Upjohn Canada ULC; 2020. P. 1-29. 2020. 2020.
43.PHENOBARB (Phenobarbital Tablets USP) Product Monograph.Montréal, Canada-PENDOPHARM, Division of Pharmascience Inc; 2020.p.1-19.2020. 2020.
44.ZARONTIN* (Ethosuximide Capsules USP) Product Monograph. Montréal, QC, Canada: ERFA Inc; 2013. p. 1-13. 2013. 2013.
45.PrTEGRETOL® (carbamazepine tablets) Product Monograph. Dorval, QC, Canada: Novartis Pharmaceuticals Canada Inc; 2021. p. 1-48. 2021. 2021.
46.PrPRIMIDONE (Primidone Tablets USP) Product Monograph.Vaughan, Ontario. Canada: AA PHARMA INC. 2015. p.1-16. 2015. 2015.
47.PrDEPAKENE® (valproic acid) Product Monograph. Etobicoke, ON, Canada: BGP Pharma ULC; 2021. p. 1-59. 2021. 2021.
48.PrNEURONTIN® (Gabapentin) Product Monograph. Kirkland, QC, Canada: Upjohn Canada ULC; 2020. p. 1-29. 2020. 2020.
49.SABRIL® (vigabatrin) Product Monograph. Saint-Laurent, QC, Canada: Lundbeck Canada Inc; 2021. p. 1-44. 2021. 2021.
50.PrLAMICTAL (Lamotrigine) Product Monograph. Mississauga, ON, Canada: GlaxoSmithKline Inc; 2021. p.1-52. 2021. 2021.
51.APO-CLOBAZAM (Clobazam) Product Monograph. Toronto, ON, Canada: Apotex Inc; 2021. p.1-39. 2021. 2021.
52.APO-CLONAZEPAM (Clonazepam Tablets USP) Product Monograph. Toronto, ON, Canada: APOTEX INC; 2021. p.1-29. 2021. 2021.
53.PrTOPAMAX® (topiramate) Product Monograph. Toronto, ON, Canada: Janssen Inc; 2021. p. 1-76. 2021. 2021.
54.PrTRILEPTAL® (Oxcarbazepine) Product Monograph. Dorval, QC, Canada: Novartis Pharmaceuticals Canada Inc; 2021. p. 1-69. 2021. 2021.
55.PrKeppra® (levetiracetam) Product Monograph. Oakville, ON, Canada: UCB Canada Inc; 2021. p. 1-38. 2021. 2021.
56.PrVIMPAT® (Lacosamide) Product Monograph. Oakville, ON, Canada: UCB Canada Inc: 2021. p. 1-49. 2021. 2021.
57.PrFYCOMPA® (Perampanel Tablets) Product Monograph. Mississauga, ON, Canada. Eisai Limited; 2021. p. 1-53. 2021. 2021.
58.PrAPTIOM® (eslicarbazepine acetate) Product Monograph. Mississauga, ON, Canada: Sunovion Pharmaceuticals Canada Inc; 2019. p. 1-45. 2019. 2019.
59.PrBRIVLERA® (brivaracetam) Product Monograph. Oakville, ON, Canada: UCB Canada Inc; 2021. p. 1-46. 2021. 2021.
60.Chung SS, French JA, Kowalski J, et al. Randomized phase 2 study of adjunctive cenobamate in patients with uncontrolled focal seizures. Neurology. 2020;94(22):e2311-e2322. PubMed
61.Krauss GL, Klein P, Brandt C, et al. Safety and efficacy of adjunctive cenobamate (YKP3089) in patients with uncontrolled focal seizures: a multicentre, double-blind, randomised, placebo-controlled, dose-response trial. Lancet Neurol. 2020;19(1):38-48. PubMed
62.Center for Drug Evaluation R. Clinical review(s). XCOPRI (cenobamate) oral. Company: SK Life Science, Inc. Application No.:212839. Approval date: 11/21/2019 (FDA approval package). Silver Spring (MD): U.S. Food and Drug Administration (FDA); 2019: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/212839Orig1s000MedR.pdf. Accessed 2023 Mar 13.
63.Center for Drug Evaluation R. Statistical review(s). Xcopri (cenobamate) administration. Company: SKLife Science Inc. Application No.:212839. Approval date: 11/21/2019 (FDA approval package). Silver Spring (MD): U.S. Food and Drug Administration (FDA); 2018: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/212839Orig1s000StatR.pdf. Accessed 2023 Apr 11.
64.Baker GA, Camfield C, Camfield P, et al. Commission on Outcome Measurement in Epilepsy, 1994-1997: final report. Epilepsia. 1998;39(2):213-231. PubMed
65.Mohanraj R, Brodie MJ. Measuring the efficacy of antiepileptic drugs. Seizure. 2003;12(7):413-443. PubMed
66.Goldenholz DM, Goldenholz SR, Moss R, et al. Does accounting for seizure frequency variability increase clinical trial power? Epilepsy Res. 2017;137:145-151. PubMed
67.Lattanzi S, Trinka E, Zaccara G, et al. Third-Generation Antiseizure Medications for Adjunctive Treatment of Focal-Onset Seizures in Adults: A Systematic Review and Network Meta-analysis. Drugs. 2022;82(2):199-218. PubMed
68.Hoppe C, Poepel A, Elger CE. Epilepsy: accuracy of patient seizure counts. Arch Neurol. 2007;64(11):1595-1599. PubMed
69.Kerling F, Mueller S, Pauli E, Stefan H. When do patients forget their seizures? An electroclinical study. Epilepsy Behav. 2006;9(2):281-285. PubMed
70.Poochikian-Sarkissian S, Tai P, del Campo M, et al. Patient awareness of seizures as documented in the epilepsy monitoring unit. Can J Neurosci Nurs. 2009;31(4):22-23. PubMed
71.Tatum WOt, Winters L, Gieron M, et al. Outpatient seizure identification: results of 502 patients using computer-assisted ambulatory EEG. J Clin Neurophysiol. 2001;18(1):14-19. PubMed
72.Gazzola DM, Balcer LJ, French JA. Seizure-free outcome in randomized add-on trials of the new antiepileptic drugs. Epilepsia. 2007;48(7):1303-1307. PubMed
73.Halford JJ, Edwards JC. Seizure freedom as an outcome in epilepsy treatment clinical trials. Acta Neurol Scand. 2020;142(2):91-107. PubMed
74.Marson AG, Kadir ZA, Chadwick DW. New antiepileptic drugs: a systematic review of their efficacy and tolerability. BMJ. 1996;313(7066):1169-1174. PubMed
75.Hopewell S, Boutron I, Chan A-W, et al. An update to SPIRIT and CONSORT reporting guidelines to enhance transparency in randomized trials. Nat Med. 2022;28(9):1740-1743. PubMed
76.Drug Reimbursement Review clinical guidance report: brivaracetam (Brivlera) for partial-onset seizures in patients with epilepsy. Ottawa (ON): CADTH; 2016 Mar 9: https://www.cadth.ca/sites/default/files/cdr/clinical/SR0484_Brivlera_CL_Report.pdf. Accessed March 10, 2023.
77.Drug Reimbursement Review clinical guidance report: eslicarbazepine acetate (Aptiom) oral tablets for partial-onset seizures in patients with epilepsy. Ottawa (ON): CADTH; 2016 May: https://www.cadth.ca/sites/default/files/cdr/clinical/SR0391_Aptiom_CL_Report.pdf. Accessed March 10, 2023.
78.Drug Reimbursement Review clinical guidance report: levetiracetam (pdp-levETIRAcetam) for epilepsy. Ottawa (ON): CADTH; 2021 March: https://www.cadth.ca/sites/default/files/cdr/clinical/sr0653-pdp-levETIRAcetam-clinical-and-economic-review-report.pdf. Accessed March 10, 2023.
79.Cramer JA, Perrine K, Devinsky O, Bryant-Comstock L, Meador K, Hermann B. Development and cross-cultural translations of a 31-item quality of life in epilepsy inventory. Epilepsia. 1998;39(1):81-88. PubMed
80.Borghs S, de la Loge C, Cramer JA. Defining minimally important change in QOLIE-31 scores: estimates from three placebo-controlled lacosamide trials in patients with partial-onset seizures. Epilepsy Behav. 2012;23(3):230-234. PubMed
81.Cramer JA, Van Hammée G. Maintenance of improvement in health-related quality of life during long-term treatment with levetiracetam. Epilepsy Behav. 2003;4(2):118-123. PubMed
82.Wiebe S, Matijevic S, Eliasziw M, Derry PA. Clinically important change in quality of life in epilepsy. J Neurol Neurosurg Psychiatry. 2002;73(2):116-120. PubMed
83.Cadth. Canadian Drug Expert Committee Final Recommendation: Brivlera (Brivaracetam). 2017; https://www.cadth.ca/sites/default/files/cdr/complete/sr0484_complete_brivlera_jan-27-17.pdf.
84.Paladin Labs Inc. response to April 5, 2023 CADTH request for additional information regarding cenobamage CADTH review: [internal additional sponsor's information]. St, Laurent (QC): Paladin Labs Inc.; 2023.
85.Blachut B, Hoppe C, Surges R, Stahl J, Elger CE, Helmstaedter C. Counting seizures: The primary outcome measure in epileptology from the patients' perspective. Seizure. 2015;29:97-103. PubMed
86.French JA, Chung SS, Krauss GL, et al. Long-term safety of adjunctive cenobamate in patients with uncontrolled focal seizures: Open-label extension of a randomized clinical study. Epilepsia. 2021;62(9):2142-2150. PubMed
87.Smith MC, Klein P, Krauss GL, et al. Dose Adjustment of Concomitant Antiseizure Medications During Cenobamate Treatment: Expert Opinion Consensus Recommendations. Neurol Ther. 2022;11(4):1705-1720. PubMed
88.Sterne JAC, Savović J, Page MJ, et al. RoB 2: a revised tool for assessing risk of bias in randomised trials. BMJ. 2019;366. PubMed
89.Sterne JAC, Hernán MA, Reeves BC, et al. ROBINS-I: a tool for assessing risk of bias in non-randomised studies of interventions. BMJ. 2016;355. PubMed
90.Cenobamate for treating focal onset seizures in epilepsy: NICE Technology appraisal guidance (TA753). National Institute for Health and Care Excellence; 2021 Dec 15: https://www.nice.org.uk/guidance/ta753/resources/cenobamate-for-treating-focal-onset-seizures-in-epilepsy-pdf-82611373757893#:~:text=Cenobamate%20(Ontozry%2C%20Arvelle%20Therapeutics),anti%2Depileptic%20medicinal%20products'. Accessed 2023 Feb 27.
91.Agrawal S, Fulton K, Chitravas N, Noewborn-Palmer L, Foreman P. Outcomes with Cenobamate Use in Clinical Practice. Poster presented at: American Epilepsy Society (AES) 2022. 2022.
92.Kravets HD, Krutoshinskaya Y. The Stony Brook Medical Center Experience with Cenobamate for Patients with Focal Drug Resistant Epilepsy. Poster presented at: American Epilepsy Society (AES) 2022. 2022.
93.Cortopassi J, Szaflarski J. Seizure Control with Cenobamate in an Outpatient Setting. Poster presented at: American Epilepsy Society (AES) 2022. 2022.
94.Sperling MR, Klein P, Aboumatar S, et al. Cenobamate (YKP3089) as adjunctive treatment for uncontrolled focal seizures in a large, phase 3, multicenter, open-label safety study. Epilepsia. 2020;61(6):1099-1108. PubMed
95.Sperling MR, Abou-Khalil B, Aboumatar S, et al. Efficacy of cenobamate for uncontrolled focal seizures: Post hoc analysis of a Phase 3, multicenter, open-label study. Epilepsia. 2021;62(12):3005-3015. PubMed
Appendix 1: Detailed Outcome Data
Note that this appendix has not been copy-edited.
Table 36: Percent Change in Seizure Frequency by Seizure Type From Pivotal and RCT Evidence
Outcome | C013 | C017 | ||||
|---|---|---|---|---|---|---|
Seizure type | CEN 200 mg (N = 113) | Placebo (N = 109) | CEN 100 mg (N = 108) | CEN 200 mg (N = 110) | CEN 400 mg (N = 111) | Placebo (N = 108) |
Seizure frequency per 28 days – DB treatment period (mITT population) | ||||||
Simple partial with motor component | ||||||
Number included in the analysis | 30 | 26 | 22 | 28 | 22 | 19 |
Median percent reduction vs. placebo | 76.3 | 27.8 | 48.0 | 63.0 | 58.5 | 7.0 |
P valuea | 0.045 | Reference | 0.003 | < 0.001 | < 0.001 | Reference |
Complex partial | ||||||
Number included in the analysis | 87 | 89 | 101 | 97 | 100 | 90 |
Median percent reduction vs. placebo | 55.6 | 21.1 | 32.0 | 55.0 | 60.0 | 28.5 |
P valuea | 0.0009 | Reference | 0.077 | < 0.001 | < 0.001 | Reference |
Secondary generalized tonic-clonic | ||||||
Number included in the analysis | 38 | 37 | 37 | 35 | 43 | 45 |
Median percent reduction vs. placebo | 77.0 | 33.0 | 47.0 | 91.0 | 78.0 | 33.0 |
P valuea | 0.012 | Reference | 0.226 | < 0.001 | 0.004 | Reference |
DB = double blind; CEN = cenobamate; mITT = modified intention to treat.
aC013: P value based on Wilcoxon rank sum test assessing if the median % change for CEN is significantly different than the median % change for placebo. C017 P value based on a nonparametric ANCOVA model with terms for ranked baseline seizure rate and treatment group.
Source: Clinical Study Report for Study C013,10 Clinical Study Report for Study C017.9
Table 37: Summary of Maintenance Period Responder Results From Pivotal and RCT Evidence
Outcome | C013a | C017b | ||||
|---|---|---|---|---|---|---|
CEN 200 mg (N = 113) | Placebo (N = 109) | CEN 100 mg (N = 108) | CEN 200 mg (N = 110) | CEN 400 mg (N = 111) | Placebo (N = 108) | |
≥ 50% reduction in seizure frequency per 28 days – Maintenance period (subgroup 5 or mITT-M population) | ||||||
Number of patients included in analysis | 106 | 102 | 102 | 98 | 95 | 102 |
n (%) | 53 (50.0) | 22 (21.6) | 41 (40.2) | 55 (56.1) | 61 (64.2) | 26 (25.5) |
OR (95% CI) | 4.12 (2.18 to 7.76) | Reference | NR | NR | NR | Reference |
P valuea | P < 0.0001 | Reference | P = 0.036c | P < 0.001 | P < 0.001c | Reference |
≥ 75% reduction in seizure frequency per 28 days – Maintenance period (subgroup 5 or mITT population)d | ||||||
n (%) | 41 (38.7) | 21 (20.6) | 17 (16.7) | 30 (30.6) | 44 (46.3) | 10 (9.8) |
OR (95% CI) | 2.88 (1.48 to 5.62) | Reference | NR | NR | NR | Reference |
P value | P = 0.0019 | Reference | P = 0.215 | P < 0.001 | P < 0.001 | Reference |
≥ 90% reduction in seizure frequency per 28 days – Maintenance period (subgroup 5 or mITT population)d | ||||||
n (%) | 36 (34.0) | 9 (8.8) | 9 (8.8) | 17 (17.3) | 27 (28.4) | 3 (2.9) |
OR (95% CI) | 6.30 (2.74 to 14.47) | Reference | NR | NR | NR | Reference |
P value | P < 0.0001 | Reference | P = 0.134 | P < 0.001 | P < 0.001 | Reference |
100% reduction in seizure frequency per 28 days – Maintenance period (subgroup 5 or mITT population)d | ||||||
n (%) | 30 (28.3) | 9 (8.8) | 4 (3.9) | 11 (11.2) | 20 (21.1) | 1 (1.0) |
OR (95% CI) | 5.35 (2.27 to 12.64) | Reference | NR | NR | NR | Reference |
P value | P = 0.0001 | Reference | P = 0.369 | P = 0.002 | P < 0.001 | Reference |
CEN = cenobamate; CI = confidence interval; mITT-M = modified intention to treat in maintenance period; NR = not reported; OR = odds ratio; RCT = randomized controlled trial.
aC013: The proportion of responders based on the last 6 weeks of treatment in patients from subgroup 5 (received study drug during the maintenance period but may or may not have completed the study). OR (95% CI) and P value based on logistic regression model with terms for treatment, country and baseline seizure frequency (Wald chi-square test)
bC017: The proportion of responders based on the last 12 weeks of treatment in patients from mITT-M population (received at least 1 dose of study drug during the maintenance period). P value based on Fisher’s exact chi-square test.
cP value has been adjusted for multiple testing
d≥ 75%, ≥ 90% and 100% responder analyses were conducted post hoc in study C013.
Source: Clinical Study Report for Study C013,10 Clinical Study Report for Study C017.9
Table 38: Placebo-Controlled Studies Meeting the Inclusion Criteria for ITC 1
Study name/NCT number | Phase | Na | Study period (month, year) | Study duration (weeks) | Location (number of centres) | |||
|---|---|---|---|---|---|---|---|---|
Baseline | Titration | Maintenance | Treatment | |||||
Cenobamate | ||||||||
Study C017/ NCT01866111 | 2 | 437 | 07/13 to 06/15 | 8 | 6 | 12 | 18 | Global (107) |
Study C013/ NCT01397968 | 2 | 222 | 07/11 to 01/21 | 4 or 8 | 6 | 6 | 12 | US, India, Republic of Korea, Poland (40) |
Brivaracetam | ||||||||
Study 1114/ NCT00175929 | 2 | 157 | 05/05 to 03/06 | 4 | 3 | 7 | 10 | Europe (FR, DE, UK) (42) |
Study 1193/ NCT00175825 | 2 | 210 | 11/05 to 06/06 | 4 | 0 | 0 | 7 | Brazil, India, Mexico, US (41) |
Study 1252/ NCT00490035 | 3 | 399 | 09/07 to 02/09 | 8 | 0 | 0 | 12 | Europe (FR, DE, UK), India (88) |
Study 1253/ NCT00464269 | 3 | 400 | 09/04 to 12/06 | 8 | 0 | 0 | 12 | Australia, Brazil, Canada, Mexico, US (85) |
Study 1254/ NCT00504881 | 3 | 480 | 10/07 to 12/08 | 4 | 8 | 8 | 16 | Global (74) |
BRITE Study 1358/NCT01261325 | 3 | 768 | 12/10 to 12/13 | 8 | 0 | 0 | 12 | Global (142) |
Lacosamide | ||||||||
Study 0008/ NCT01710657 | 3 | 548 | 09/12 to 08/14 | 8 | 4 | 12 | 16 | China, Japan (72) |
Study 0667 | NR | 418 | 02/02 to 05/04 | 8 | 6 | 12 | NR | Europe, US (68) |
Study 0754/ NCT00136019 | 3 | 405 | 03/04 to 08/06 | 8 | 6 | 12 | 18 | US (72) |
Study 0755/ NCT00220415 | 3 | 485 | 06/04 to 01/06 | 8 | 4 | 12 | NR | Australia, Europe (FR, DE, UK), Russia (75) |
Eslicarbazepine acetate | ||||||||
Study 301/ NCT00957684 | 3 | 402 | 07/04 to 11/05 | 8 | 2 | 12 | 14 | Europe (DE), Russia (40) |
Study 302/ NCT00957047 | 3 | 395 | 09/04 to 12/06 | 8 | 2b | NR | 14 | Argentina, Australia, Brazil, Europe (DE, UK), South Africa (45) |
Study 303/ NCT00957372 | 3 | 253 | 12/04 to 01/07 | 8 | 2 | 12 | 18c | Mexico, Portugal, Spain (39) |
Study 304/ NCT00988429 | 3 | 650 | 12/08 to 01/12 | 8 | 2 | 12 | NR | Argentina, Australia, Brazil, Canada, Europe (FR, DE), India, South Korea, South Africa, Ukraine, US (173) |
Perampanel | ||||||||
Study 304/ NCT00699972 | 3 | 390 | 04/08 to 10/10 | 6 | 6 | 13 | 19 | Argentina, Canada, Chile, Mexico, US (68) |
Study 305/ NCT00699582 | 3 | 386 | 05/08 to 01/12 | 6 | 6 | 13 | 19 | Australia, Europe (FR, DE, UK), India, Israel, Russia, US, South Africa (78) |
Study 306/ NCT00700310 | 3 | 706 | 08/08 to 05/10 | 6 | 6 | 13 | 19 | Europe, Asia, Australia (116) |
Study 335/ NCT01618695 | 3 | 710 | 05/12 to 09/14 | 6 | 6 | 13 | 19 | Australia, China, Japan, Malaysia, Republic of Korea, Taiwan, Thailand (119) |
Zonisamide | ||||||||
Brodie et al. (2005) | NR | 351 | NR | 12 | 6 | 18 | 24 | Europe and South Africa (54) |
Study 922 | NR | Group A (n = 85) Group B1 (n = 60) Group B2 (n = 58) | NR | 4 | NR | NR | 20 | US (20) |
DE = Germany; FR = France; NCT = National Clinical Trial; NR = not reported.
aAll randomized patients.
bTwo-week titration for 1,200 mg dose only.
cThe DB treatment phase also included a tapering off period.
Source: Sponsor’s Summary of Clinical Evidence and sponsor-submitted ITC report.14,15 [Note – details from the table have been taken from the sponsor’s Summary of Clinical Evidence]
Table 39: Summary of Baseline Characteristics of Studies in ITC 1
Study name/ NCT number | N | Daily treatment dose (mg)a | Age (year) | % white | Sex (% male) | BMI (kg/m2) | Duration of epilepsy (year)b | Median baseline seizure frequency per 28 days | Number of ASMs at baseline | Concomitant AEDs |
|---|---|---|---|---|---|---|---|---|---|---|
Cenobamate | ||||||||||
Study C017/ NCT01866111 | 437 | PBO, 100, 200, 400 | 39.8 | 84.8 | 50.8 | 26.3 | 23.9 | 8.4 to 11.0 | 1: 23 to 36% 2: 43 to 56% 3: 22 to 31% | LEV, CBZ, LTG, VPA, OXC, CLB |
Study C013/ NCT01397968 | 222 | PBO, 200 | Median: 37.0 | 57.3 | 55.3 | NR | 20.5 | 5.5 to 7.5 | 1: 11 to 17% 2: 47 to 48% 3: 36 to 41% | LEV, LTG, LCS, CBZ, TOP, OXC, CLB, VPA |
Brivaracetam | ||||||||||
Study 1114/ NCT00175929 | 157 | PBO, 50, 150 | 37.5 | 99.4 | 44.6 | 24.7 | 22.0 | 7.0 to 11.8c | 1: 14 to 25% 2: 66 to 83% | LEV, CBZ, TOP, LTG, VPA, OXC |
Study 1193/ NCT00175825 | 210d | PBO, 50 | 32.3 | 32.9 | 44.4 to 53.8 | NR | 20.4 | 7.8 to 8.9c | 1: 31 to 37% 2: 57 to 65% ≥ 3: 6% | LEV, CBZ, LTG, VPA, OXC, PHT, CLB |
Study 1252/ NCT00490035 | 399d | PBO, 50, 100 | 37.8 | 76.6 | 55.5 | NR | 21.6 | 7.2 to 8.3c | 1: 14 to 20% 2: 77 to 83% | LEV, CBZ, LTG, VPA, OXZ |
Study 1253/ NCT00464269 | 400d | PBO, 50 | 38.2 | 71.8 | 60.1 | NR | 25.3 | 10.4 to 11.6c | 1: 13% 2: 81% ≥ 3: 5% | LEV, CBZ, LTG, VPA, PHT |
Study 1254/ NCT00504881 | 480 | PBO and 50, 100, 150 (single active arm) | 36.5 | 57.5 | 53.2 | NR | 22.0 | 8.8 to 9.2c | 1: 15 to 19% 2: 36 to 49% ≥ 3: 36 to 45% | LEV, CBZ, TOP, LTG, VPA |
BRITE Study 1358/ NCT01261325 | 768 | PBO, 100, 200 | 39.6 | 72.4 | 48.2 | 26.6 | 13.7 | 9.3 to 10.0 | 1: 28.1% 2: 71.3% | LAC, CBZ, TOP, LTG, VPA, OXC |
Lacosamide | ||||||||||
Study 0008/ NCT01710657 | 548 | PBO, 200, 400 | 32.4 | 0 | 54.9 | 22.7 | 17.7 | 10 to 11 over 8-week baseline | 1: 20 to 22% 2: 39 to 45 3: 32 to 39% | LEV, CBZ, TOP, LTG, VPA, OXC |
Study 0667 | 418d | PBO, 200, 400 | 40.0 | 92.0 | 46.7 | NR | 24.8 | 11 to 13 over 8-week baseline | NR | NR |
Study 0754/ NCT00136019 | 405d | PBO, 400 | 38.6 | 81.1 | 49.1 | NR | 24.4 | 11.5 to 15.0 | 1: 17.6% 2: 53.3% | LEV, LTG, CBZ, OXC, PHT, TOP, VPA |
Study 0755/ NCT00220415 | 485 | PBO, 200, 400 | 37.8 | 99.2 | 43.4 to 55.8 | 25.5 | 22.3 | 9.9 to 11.5 | 1: 11 to 16% 2: 48 to 52% 3: 34 to 41% | NR |
Eslicarbazepine acetate | ||||||||||
Study 301/ NCT00957684 | 402 | PBO, 400, 800, 1,200 | 38.6 | 100 | 43.1 to 55.1 | 24.5 | 21.0 | 6.7 to 7.5 | 1: 32 to 39% 2: 60 to 68% | CBZ, TOP, LTG, VPA |
Study 302/ NCT00957047 | 395 | PBO, 400, 800, 1,200 | 36.9 | 87.6 | 40.6 to 53.1 | 25.0 | 23.9 | 8.0 to 9.0 | 1: 15 to 3% 2: 69 to 6% 3: 6 to 10% | LEV, CBZ, TOP, LTG, VPA, PHT, PHB |
Study 303/ NCT00957372 | 253 | PBO, 800, 1,200 | 36.8 | 34.4 | 44.8 | 26.0 | 23.1 | Mean: 11.3 to 11.6 | 1: 15 to 26% 2: 68 to 79% | LEV, CBZ, TOP, LTG, VPA, PHT |
Study 304/ NCT00988429 | 173 | PBO, 800, 1,200 | Median: 38.5 | 63.5 | 50.2 | 26.2 | 21.4 | 8.0 to 9.0 | 1: 28.2% 2: 71.1% | LEV, CBZ, LTG, VPA |
Perampanel | ||||||||||
Study 304/ NCT00699972 | 390 | PBO, 8, 12 | 36.0 | 86.1 | 48.3 | 26.3 | NR | 12.0 to 13.7 | 1: 12 to 20% 2: 53 to 61% 3: 25 to 35% | LEV, CBZ, ZNS, TOP, LTG, VPA, OXC, CLB, PHT |
Study 305/ NCT00699582 | 386 | PBO, 8, 12 | 35.5 | 83.4 | 48.0 | NR | NR | 11.8 to 13.7 | 1: 7 to 13% 2: 47 to 53% 3: 40 to 35% | LEV, CBZ, ZNS, TOP, LTG, VPA, OXC, CLB |
Study 306/ NCT00700310 | 706 | PBO, 2, 4, 8 | 33.9 | 61.0 to 68.6 | 48.9 | 24.1 | 17.7 | 9.3 to 10.9 | 1: 11 to 16% 2: 49 to 51% 3: 36 to 39% | LEV, CBZ, TOP, LTG, VPA, OXC, CLB |
Study 335/ NCT01618695 | 710 | PBO, 4, 8, 12 | 33.4 | NR | 46.0 to 52.0 | NR | 17.3 | NR | 1: 5 to 9% 2: 34 to 42% 3: 51 to 55% 4: 0 to 1% | LEV, CBZ, TOP, LTG, VPA, OXC, PHT, CLB |
Zonisamide | ||||||||||
Brodie et al. (2005) | 351 | PBO, 100, 300, 500 | 35.7 | NR | 51.0 | NR | 19.9 | 0 to 3 | 1: 18 to 30% 2: 38 to 57% 3: 22 to 30% | CBZ, VPA, LTG, CLB, GBP, PHB, PHT, TOP |
Study 922 | Group A (n = 85) Group B1 (n = 60) Group B2 (n = 58) | PBO, 100, 200, 400 | 34.5 | 85.3 | 51.2 | NR | NR | 11.2 to 13 | NR | NR |
ASM = antiseizure medication; BMI = body mass index; CBZ = carbamazepine; CLB = clobazam; European Medicines Agency; GBP = gabapentin; LEV = levetiracetam; LCS = lacosamide; LTG = lamotrigine; NCT = national clinical trial; NR = not reported; OXC = oxcarbazepine; PBO = placebo; PHB = phenobarbital; PHT = phenytoin; TOP = topiramate; VPA = valproate or valproic acid; ZNS = zonisamide.
aExcludes doses not licensed according to the EMA.
bReporting of the duration of epilepsy varied across studies and included time from diagnosis, time from onset or was unclear in which period was considered.
cReported per 7 days and extrapolated over 28 days.
dIncludes patients in excluded study arms due to doses outside the licensed range.
Note: Values reported in table are mean unless stated otherwise.
Source: Sponsor’s Summary of Clinical Evidence and sponsor-submitted ITC report.14,15 [Note – details from the table have been taken from the sponsor’s Summary of Clinical Evidence]
Pharmacoeconomic Review
Abbreviations
ASM
antiseizure medication
LY
life-year
NMA
network meta-analysis
OLE
open-label extension
OR
odds ratio
QALY
quality-adjusted life-year
RR
relative risk
SMR
standardized mortality ratio
VNS
vagal nerve stimulation
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Cenobamate, oral tablets |
Submitted price | Cenobamate, 12.5 mg, 25 mg, 50 mg, 100 mg, 150 mg, 200 mg: $8.80 per tablet |
Indication | Proposed: An adjunctive therapy for the management of partial onset seizures in adults with epilepsy who are not satisfactorily controlled with conventional therapy. |
Health Canada approval status | Under review (pre-NOC) |
Health Canada review pathway | Standard |
NOC date | Anticipated: June 19, 2023 |
Reimbursement request | As per indication |
Sponsor | Paladin Labs Inc. |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov model |
Target population | Adults with partial onset epilepsy who are not satisfactorily controlled with conventional therapy (antiseizure medication) and require adjunctive therapy |
Treatment | Cenobamate |
Comparators |
|
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (100 years) |
Key data sources |
|
Submitted results | Cenobamate dominated (i.e., was less expensive and more effective than) brivaracetam, eslicarbazepine, and perampanel. |
Key limitations |
|
CADTH reanalysis results |
|
LY = life-year; NMA = network meta-analysis; QALY = quality-adjusted life-year; OLE = open-label extension.
aLacosamide was excluded from the sponsor’s base case but considered in a scenario analysis.
Conclusions
In the absence of direct evidence comparing cenobamate with other ASMs used for adjunctive treatment, the sponsor derived estimates of relative clinical efficacy using an NMA. The CADTH clinical review’s appraisal of the submitted evidence suggests there may be a short-term benefit favouring cenobamate with respect to the achievement of a reduction in seizures. However, the magnitude and clinical relevance of this outcome were unclear. No conclusions regarding relative efficacy were drawn for the achievement of seizure freedom or the risk of discontinuation due to treatment-emergent adverse events. In terms of treatment response outcomes, considerable heterogeneity was identified with respect to the patients enrolled in the studies, the follow-up durations between trials, and the definitions of outcomes (for example, some trials defined seizure freedom as the absence of any seizures, whereas the cenobamate studies defined it as a 100% reduction in only 3 types of partial seizures). Therefore, uncertainties remain regarding the clinical benefit of treatment with cenobamate compared to other adjunctive treatments.
The CADTH base case addressed some key limitations in the sponsor’s economic submission. These changes involved making more appropriate assumptions regarding treatment discontinuation and the characterization of uncertainty for the relative risk (RR) of seizure reduction or seizure freedom. Results from the CADTH base case suggest that cenobamate may be associated with lower total costs (driven by fewer predicted seizure events) and greater health benefits (quality-adjusted life-years [QALYs]) compared with the alternative antiseizure medications (ASMs) considered in the economic model. However, these results should be interpreted with caution, given that the CADTH base case failed to address several key limitations of the economic evaluation.
In a scenario analysis, CADTH explored the impact of a lower estimate of baseline seizure frequency on the estimated costs and QALYs. The findings indicated that an estimate more representative of the target population would reduce, and potentially eliminate, any potential cost savings from cenobamate. Meanwhile, reductions in the differences in relative treatment effectiveness are expected from the resolution of issues relating to relative efficacy and the characterization of parameter uncertainty. When considered together, these issues suggest that a price premium for cenobamate relative to the other relevant comparators may not be warranted.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
Patient input for this review was obtained from the Canadian Epilepsy Alliance, Epilepsy Southwestern Ontario, Epilepsy Toronto, Epilepsy South Central Ontario, the Edmonton Epilepsy Association, and the Epilepsy Association of Calgary. These submissions obtained information from a variety of sources, including members of a patient input group, social workers, patient surveys, and group or individual counselling sessions with patients. Specific patient experiences with any of the currently available treatment options were not provided. Two submissions did state that 30% of patients will have drug-resistant epilepsy and that cenobamate could offer an effective alternative for eligible patients. However, none of the submissions included information about whether any patients had trialled cenobamate. The input noted the distinction between seizure control and seizure freedom; the latter is the aim of epilepsy treatment because it allows increased employment opportunities and greater patient independence. Patient input also identified common side effects of ASMs, including cognitive difficulties (e.g., memory challenges, brain frog), changes in personality, suicidal ideations, and depression and anxiety.
Registered clinician input was received from the Canadian League Against Epilepsy. The submission noted that ASMs are used to treat focal and generalized seizures by preventing their occurrence. Patients typically begin on monotherapy and switch to a second ASM if their seizures are not controlled. If an insufficient response to the second ASM is observed, polytherapy will be initiated by using an adjunctive ASM to increase the chance of seizure control. Patients who do not respond to any ASM will be considered for alternative treatments, which may include surgery, neuromodulation, dietary modifications, monthly infusions, and cannabinoids. The patient input stated that because patients are not able to trial all available ASMs, and patient-specific factors determine the appropriateness of prescribing any medication, there is a need for a novel medication that provides a higher probability of attaining seizure control or seizure freedom. The clinical input submission expected that cenobamate would be used as an adjunctive treatment with existing ASMs and was unlikely to be used as monotherapy.
The input from drug plans sought clarification on cenobamate’s place in therapy, its relative effectiveness, and its relative cost-effectiveness. Concerns were raised about the indirect evidence used to establish relative efficacy and the impact on the conclusion of cost-effectiveness.
Several of these concerns were addressed in the sponsor’s model, which explored the use of cenobamate as an adjunctive treatment for adults with epilepsy and partial onset seizure (POS) epilepsy who are not satisfactorily controlled on monotherapy.
CADTH was unable to address the concern, raised in stakeholder input, that in the absence of direct evidence of clinical effectiveness, the comparative conclusions (drawn from the evidence) rely on indirect evidence using a network meta-analysis (NMA). Therefore, this evidence remains a source of uncertainty in the economic model.
Economic Review
The current review is for cenobamate for adults with partial onset epilepsy who are not satisfactorily controlled with conventional therapy and require adjunctive therapy.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted an economic evaluation comparing cenobamate with third-generation ASMs. The target population was defined as adults with POS epilepsy who are not satisfactorily controlled with conventional therapy and require adjunctive treatment. This target population was aligned with the Health Canada indication and the sponsor’s reimbursement request.1
Cenobamate is available as an oral tablet at dosages of 12.5 mg, 25 mg, 50 mg, 100 mg, 150 mg, and 200 mg.2 The submitted price per tablet for every tablet strength was $8.80.1 Following an initial loading dose of 12.5 mg per day for 14 days, the dosage is increased to the next dose strength for another 14-day cycle. This titration continues until the maintenance dose of 200 mg per day is reached.2 At every dose, cenobamate will cost $8.80 per day and $3,214.20 per year.
Comparators considered in the submission were restricted to licensed, third-generation ASMs whose adjunctive indications are similar to those of cenobamate. In the base case, these included brivaracetam, eslicarbazepine, and perampanel, which have annual costs of $3,156, $3,605, and $3,655, respectively. A fourth comparator, lacosamide (with an annual cost of $639 to $1,059) was included as part of a scenario analysis.1
The clinical outcomes modelled included life-years (LYs) and QALYs. The model took the perspective of the Canadian public health care payer over a lifetime horizon (i.e., 100 years). A 28-day cycle length was assumed for the first 5 cycles, after which the cycle length was extended to 84 days. A 1.5% discount rate was applied to both costs and outcomes.1
Model Structure
The sponsor submitted a Markov state transition model that tracked a cohort of patients from their first adjunctive ASM through death. As illustrated in Figure 1, the modelled health states were specific to patients’ treatment statuses (i.e., whether patients were on their initial adjunctive ASM). Movements between health states were determined by time-dependent transition probabilities with respect to time in the model.1
At model entry, patients initiated their first adjuvant ASM (i.e., cenobamate or an alternative). For the duration of time on that treatment, patients could occupy 1 of 5 health states characterized in terms of treatment response. These treatment response health states were defined using asymmetric categories of the percent reduction in seizure frequency. It was assumed that patients would begin in the “no-response” state, which was defined as a reduction in seizure frequency of less than 50%. Other treatment response states included moderate (50% to 75%), high (75% to 90%), very high (90% to 99%), and complete (100%) seizure frequency reduction. After each cycle on treatment, any patient who did not discontinue their first ASM transitioned to the predicted treatment response state for the next cycle. Meanwhile, patients who discontinued treatment as a result of treatment-emergent adverse events transitioned from their respective treatment response state to the subsequent ASM state.1
In the subsequent ASM state, the model assumed that 1 of brivaracetam, eslicarbazepine, or perampanel would be used as an adjunctive ASM. Unlike the initial ASM state, transitions between seizure response states were not considered in this line of therapy. Patients remained in the subsequent ASM state unless they were eligible for 1 of 2 invasive procedures: surgery or vagal nerve stimulation (VNS). Patients who received an invasive procedure transitioned to the corresponding health state for 1 cycle, then switched to the postsurgery or post-VNS states, where they remained until death. Throughout the model time horizon, patients were subject to a risk of death that reflected an all-cause mortality risk that increased with age and was adjusted for the increased risk of death due to seizure occurrence.1
The final component of the model tracked the number of seizures expected to occur in each health state. These predictions were used to estimate the resource use and costs associated with each seizure event. Estimates were obtained by multiplying the baseline frequency of seizures in each treatment response state by the expected change in seizures in every response state in each cycle of the model.1
Model Inputs
Multiple parameters for the economic evaluation were obtained from the submitted pivotal trials and the systematic review of adjunctive treatments for adults with partial onset seizures. The submission obtained data from 3 pivotal trials. The first, Study C017, was a randomized, double-blind, dose-response trial of cenobamate (100 mg, 200 mg, and 400 mg) versus placebo, with an open-label extension (OLE).3 The second, Study C013, was a randomized pivotal efficacy trial of cenobamate (200 mg) versus placebo.4 The third, Study C021, was an open-label, phase III safety trial of cenobamate as adjunctive therapy in patients with partial onset seizures.5 In addition, the trials identified in the systematic review were used to estimate relative treatment effects through Bayesian NMA.6
Data summarizing patients’ baseline demographics were obtained from Study C017 and Study C013.1,3,4 Baseline age (mean = 39.8 years) and gender (49.4% female; 50.6% male) represented the baseline population from Study C017.3 Meanwhile, 28-day seizure frequency before treatment initiation (mean = 6.92; standard deviation = 0.69) was calculated from data reported in Study C013.1,4 This input represented the average baseline frequency of all seizure types (focal aware, focal impaired awareness, and focal to bilateral tonic-clonic) from every patient in the trial.1,4
Estimates of relative treatment effects were obtained from the indirect comparison of short-term trial data using Bayesian NMA. Comparators in each NMA included brivaracetam, perampanel, eslicarbazepine, lacosamide, zonisamide, and placebo.6 Outcomes of interest included treatment response and the occurrence of at least 1 treatment-emergent adverse event leading to discontinuation.1,6 In the treatment-response NMA, the change in seizure frequency was estimated using 2 different response categories: partial response (i.e., a ≥ 50% but < 100% reduction in seizure frequency) and seizure freedom (i.e., a 100% reduction in seizure frequency). This enabled the estimation of the RR of a partial and seizure-free response for each alternative against cenobamate.1,6 Meanwhile, the treatment discontinuation NMA estimated the odds ratio (OR) of treatment discontinuation due to a treatment-related adverse event for each alternative against cenobamate.6 While fixed- and random-effects models were available for each NMA, only median values from the latter were incorporated into the economic evaluation.
Time-dependent transitions between the treatment response states for cenobamate were obtained from Study C017.1,3 The transition probabilities for each cycle were generated from contingency tables that characterized the change in treatment response from distinct points of the trial. Values for the first 5 cycles of the model (with each cycle containing 28 days) represented data from the randomized period of Study C017 (i.e., visits 3, 5, 7, 8, and 9).1 Meanwhile, data from the OLE were used to estimate the treatment response transition probabilities for cycles 6 through 26. The cycle length in this period was extended to 84 days from 28 days to ensure consistency between visits in the OLE. Importantly, the time horizon of the model extended beyond the observed 26 cycles of C017 study and OLE (approximately 5.2 years). In response, it was assumed that the treatment response transition probabilities for cycle 27 onward would reflect the average transition probabilities for the OLE cycles.1
For the alternative ASMs, treatment response transition probabilities were obtained by combining the cenobamate-specific transition probabilities with the results of the treatment response NMA. However, the definition of a partial response in the NMA did not align with the treatment response states considered in the model (i.e., partial response = a moderate, high, or very high response).1,6 In the base case, this was addressed by assuming that the relative effect observed for a partial response in the NMA was the same as the relative effect for the moderate, high, or very high treatment response states.1 A 3-step process was required to implement this assumption and obtain estimates of the RR of a moderate, high, or very high response. First, the sponsor estimated the odds of achieving each response on cenobamate. This was accomplished using the cenobamate-specific probability of each response outcome after 5 cycles in the model, which corresponded to the end of the randomized period of Study C017. Second, the odds of achieving each treatment response for every alternative treatment were calculated by applying the cenobamate odds of treatment response to the median RR obtained from the NMA. Third, estimates of RR for each treatment response were obtained by converting the odds to probabilities. The alternative-specific treatment response transition probabilities were then obtained by applying the treatment-specific RR to the time-dependent treatment response transition probabilities for cenobamate.
The probability of cenobamate discontinuation in each cycle of the model was obtained from a parametric survival model fitted to data obtained from studies C017, C013, and C021.1,3-5 The majority of the events modelled represented discontinuation as a result of treatment-emergent adverse events. Models were fit using the exponential, log-logistic, log-normal, Weibull, Gompertz, and generalized gamma distributions. In the sponsor’s base case, the generalized gamma distribution was selected based on an inspection of model fit statistics, the high rate of patient retention, and the distribution’s consistency with the treatment discontinuation rates observed in the underlying trials.1 Once the extrapolated survival probabilities were obtained for cenobamate, additional transformations were applied to generate the corresponding values for each alternative. A key assumption was that all comparators would have a discontinuation rate similar to that of cenobamate. To achieve this, the relevant survival probabilities were calculated as the difference between the value of the preceding cycle and the proportion of patients who discontinued cenobamate. In addition, the sponsor assumed that treatment discontinuation as a result of adverse events would occur only in the first 5 months of treatment. This was achieved by applying the RR of treatment discontinuation, converted from the median OR in the corresponding NMA, to the first 5 survival probabilities. Afterward, the transition probabilities were obtained by converting the survival probabilities to a discrete time scale from a continuous one.1
The risk of undergoing an invasive procedure was obtained from clinical opinion.1 In the base case, annual probabilities of 2.13% and 0.35% were used for surgery and VNS, respectively. Transition probabilities were obtained by adjusting the annual probabilities to the 28-day and 84-day cycle lengths.1 Treatment response probabilities following surgery or VNS were sourced from an observational cohort study and economic evaluation of French patients as well as a retrospective cohort study from the UK.7,8 This evidence suggested partial response probabilities of 5.2% (postsurgery) and 53.0% (post-VNS) and seizure-free response probabilities of 69.0% (postsurgery) and 6.0% (post-VNS).7,8 Importantly, the uncertainty associated with this evidence was not characterized in the economic evaluation.1
To predict the number of seizures in each health state and cycle, the model combined the baseline frequency of seizures with the relative reduction in seizures by seizure type and response category. The latter were obtained from Study C017, and the average reduction in seizures across seizure type was used to determine the state-specific predictions.1,3
All-cause mortality was incorporated by applying a risk of death during each model cycle. Standardized mortality ratios (SMRs) were used to adjust the all-cause mortality risk and consider the greater risk of death from seizure occurrence. Seizure-free and partial response SMRs were identified from a 30-year prospective cohort study, and the general population mortality risk was obtained from Statistics Canada life tables published in January 2022.9,10 As with the treatment response transition probabilities, the partial response SMRs were assumed to be the same for each of the partial response health states.1
Health-related quality of life was captured in the model through utilities for the treatment response health states. Values were generated through the indirect measurement of patient preferences in Study C017 using the Short Form Six-Dimension (SF-6D) instrument. Averages of the SF-6D utilities by health state in the final 28 days of the trial were subsequently mapped to EQ-5D utilities for inclusion in the economic evaluation.1,3,11 For the post-treatment health states, utility values were calculated as the weighted average between the mean post-treatment response and the existing health state utilities.1 Upon incorporation into the model, the health state utilities were adjusted using EQ-5D norms for the general population in Canada.1,12
The economic evaluation considered costs associated with treatment acquisition and monitoring as well as those incurred as a result of each seizure.1 Treatment acquisition costs were based on the sponsor’s submitted price for cenobamate and the Ontario Drug Formulary for every other medication considered in the model, including concomitant treatments.1,13 As stated previously, subsequent ASMs were restricted to brivaracetam, eslicarbazepine, and perampanel. Meanwhile, concomitant medications were restricted to levetiracetam, lamotrigine, divalproex sodium, carbamazepine, oxcarbazepine, clobazam, topiramate, gabapentin, phenytoin sodium, and vigabatrin. The acquisition costs for both subsequent ASMs and concomitant medications were calculated as weighted averages of the 28-day cycle cost. While weights for the former were assumed to follow their corresponding market share in Ontario, weights for each concomitant medication were informed by clinical expert opinion.1,14 Unit costs for each invasive procedure were obtained from 2 Canadian studies conducted in the pediatric setting.15,16 It was assumed that these costs were generalizable to the adult population.1
Monitoring costs involved the costs of outpatient services used during the titration and maintenance periods associated with each adjunctive ASM. Monitoring was assumed to be conducted by a neurologist, family physician, or primary care nurse. Corresponding unit costs were obtained from the Ontario schedule of benefits for physicians.17 In the titration period, costs were restricted to outpatient neurology visits. In the maintenance period, service utilization was assumed to be a function of a patient’s seizure response status at each cycle of the model. The 28-day monitoring frequency in both time periods was assumed to follow clinical expert opinion.1
Seizure-related costs included visits to the emergency department, hospitalization, and outpatient follow-up. Unit costs associated with the management of each seizure event were obtained from the Ontario Patient Cost estimator published by the Canadian Institute for Health Information.18 The 28-day frequency of utilization stratified by seizure type was assumed to follow clinical expert opinion.1
Summary of Sponsor’s Economic Evaluation Results
The costs and QALYs for each alternative were generated from a Monte Carlo simulation of 1,000 iterations in the base case and each scenario. Despite the nonlinear model structure and the presence of several time- and treatment-dependent simulation parameters, the deterministic and probabilistic results were aligned. The results from the probabilistic base case are summarized in this section.
Base-Case Results
The submitted economic evaluation was based on the publicly available prices of the comparator treatments.
Results from the sponsor’s base case are presented in Table 3. The expected costs and QALYs for cenobamate were $395,675 and 13.35, respectively. Cenobamate was cost-effective, given that it dominated eslicarbazepine, brivaracetam, and perampanel. This means that cenobamate offered more QALYs at a lower cost when compared with each alternative treatment.
Additional results from the sponsor’s base case are reported in Appendix 3. Inspection of the disaggregated results presented in Table 10 revealed 2 key factors that contributed to the dominance exhibited by cenobamate. First, the probability of achieving no response while on the initial adjunctive treatment was much lower for cenobamate than for the alternatives. This resulted in a total of approximately 5 LYs gained for cenobamate in this state versus more than 10 LYs for the remaining alternatives. Second, the costs of seizure events associated with cenobamate were considerably lower ($280,430) than for the other treatments (> $400,000). Both cases suggest that the frequency with which patients experience seizures is much lower with cenobamate than with any other alternative considered in the model.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
Cenobamate | 395,675 | 13.346 | Reference |
Eslicarbazepine | 603,131 | 12.433 | Dominated by cenobamate |
Brivaracetam | 611,674 | 12.344 | Dominated by cenobamate and eslicarbazepine |
Perampanel | 621,163 | 12.354 | Dominated by cenobamate and eslicarbazepine |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
In addition to the base case, several scenario analyses were considered. The sponsor examined the impact of alternate discount rates (undiscounted and 3%, as per CADTH guidelines), a shorter time horizon (10 years), and a broader societal perspective on costs. In addition, the sponsor explored the impact of a standard 28-day cycle as well as a model structure that considered 3 seizure response states rather than 5. The latter scenario was justified to better align the available evidence on seizure response to the model structure.1
The analysis of costs and effects generated for each scenario did not affect the conclusion regarding the cost-effectiveness of cenobamate. In every scenario considered by the sponsor, cenobamate dominated every included alternative.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
Uncertain relative clinical efficacy of cenobamate: In the absence of direct evidence, a Bayesian NMA was used to estimate the RR of achieving a partial or complete response to treatment (versus cenobamate). Findings from the CADTH clinical review noted that the submitted estimates of relative treatment effect failed to consider 4 important sources of heterogeneity between the included trials. First, the included trials relied on inconsistent definitions of seizure freedom. Results from the NMA may have been biased in favour of cenobamate because some of the comparator trials used a more conservative definition of seizure freedom (i.e., absence of any seizures among patients who did not withdraw) compared to the cenobamate trials (i.e., a 100% reduction in 3 types of partial seizures compared to the baseline rate, including among patients with early withdrawal). Second, the data included in each NMA did not represent the same time period from each trial; instead of comparing the entire treatment period, some trials considered only the maintenance phase, which excluded the time period when the drug had not yet reached the target dose. Comparing the maintenance period in some trials with the entire treatment period in others may show a greater reduction in seizure frequency. Third, treatment periods between studies ranged from 7 weeks to 24 weeks, and differences in treatment effect may be observed as the time under observation increases. Fourth, variability in patient characteristics across studies suggested that the similarity assumption was violated, and that the studies may not be comparable. The clinical report concluded that the magnitude and clinical relevance of a 50% reduction in seizure frequency was unclear, and that no conclusions could be drawn as to the relative effects of cenobamate on seizure freedom. The incremental gains in QALYs predicted by the model relative to comparators should be interpreted with caution due to the high degree of uncertainty.
CADTH was unable to address the uncertainty surrounding the comparative data for cenobamate.
Failure to incorporate discontinuation due to nonresponse: In the sponsor’s economic model, treatment discontinuation followed the time-to-treatment discontinuation survival probabilities estimated from studies C017, C013, and C021. The most common reason for discontinuation in the fitted trial data was a treatment-emergent adverse event. Reliance on this evidence alone resulted in situations where patients remained on an initial adjunctive ASM for years, despite continually occupying the no-response state. Clinical experts consulted by CADTH indicated that discontinuation would be considered if a patient did not achieve (or lost) a partial or complete response to treatment. The impact of this omission in the model was significant, given that the no-response state was associated with lower utility values and higher seizure event management costs (from a greater frequency of seizures). Therefore, the sponsor’s base case underestimated the QALYs and overestimated the costs of every treatment in the economic model.
CADTH modified the economic evaluation to allow for discontinuation of the initial ASM due to initial nonresponse or a loss of response. Clinical experts consulted by CADTH stated that discontinuation would be considered at the patient’s next follow-up visit if no response was observed on treatment. In the Canadian context, it was suggested that such follow-up visits would occur every 6 months. Given the change in cycle length after cycle 5, this was implemented in 2 ways. First, given that cycles 1 to 5 contained 28 days each, clinical assessment of initial nonresponse was assumed to occur in cycle 5. Second, it was assumed that treatment nonresponse would be assessed in every cycle after cycle 5, given the extension of the cycle length to 84 days beginning with cycle 6.
Overestimation of seizures prevented: The predicted number of seizures was used to determine the impact of the treatment response on downstream health care costs. Concerns were identified with the assumption that the baseline seizure frequency followed estimates from Study C013. Clinical experts consulted by CADTH confirmed that a baseline seizure frequency of 6.92 seizures every 28 days is greater than what would be observed in practice. This overestimation of the baseline seizure frequency was attributed to a selection bias in the trial recruitment criteria. Trial designs tend to prefer patients with higher baseline seizure frequencies to ensure that a seizure reduction can be detected in the follow-up period. For example, studies C017 and C013 excluded patients with fewer than 8 or 3 seizures, respectively, in the baseline period.3,4 As a result, the predicted number of seizures for every treatment in the sponsor’s base case was overestimated. In absolute terms, this resulted in increased costs associated with subsequent treatment and the management of seizure events, such as emergency department visits and hospitalizations. This overestimation also inflates the number of seizures avoided for cenobamate compared to its alternatives; therefore, CADTH anticipates lower incremental costs between cenobamate and its comparators when baseline seizure frequency estimates are more generalizable to the indicated population.
CADTH conducted a scenario analysis to explore the impact of a reduction in the baseline seizure frequency on the results of the economic evaluation. In the absence of representative published evidence, and in consultation with clinical experts, the baseline seizure frequency from the sponsor’s base case was halved.
Missing comparators: Alternatives to cenobamate considered in the sponsor’s base case were restricted to third-generation ASMs: brivaracetam, eslicarbazepine, and perampanel; a single second-generation ASM, lacosamide, was considered in a scenario analysis. However, CADTH guidelines state that the identification of comparators should not be limited to a specific type or class of interventions. Instead, all interventions that may be used for treatment or displaced by a new technology should be considered in an economic evaluation.19 Clinical experts consulted by CADTH noted that several relevant comparators, such as clobazam, topiramate, and levetiracetam, would also be prescribed as adjunctive treatments for adults with uncontrolled partial onset seizures. Furthermore, the clinical experts noted the absence of a specific treatment algorithm dictating treatment selection. Therefore, the sponsor’s restriction of the comparators to third-line ASMs and lacosamide was inappropriate. The low acquisition costs of some of the missing comparators may influence the decision uncertainty in the economic evaluation, making the results less favourable to cenobamate.
This limitation could not be addressed by CADTH. Incorporating the relevant evidence for the missing and relevant treatments would have involved significant changes to the programming of the economic model.
Mischaracterization of parameter uncertainty for relative treatment effects: To address the fact that the true value of a parameter may not be known, CADTH guidelines require the probabilistic evaluation of economic models. This involves the repeated estimation of costs and QALYs using random values of each parameter from an assumed distribution.19 To enable the estimation of the full range of possible costs and QALYs for each alternative, it is critical to ensure that an economic evaluation considers the imprecision in all model input values. Failure to do so may result in different mean costs and QALYs, and in many circumstances, a different conclusion regarding the cost-effectiveness of the drug under review. A significant limitation of the sponsor’s submission was the failure to consider the uncertainty associated with the estimates of relative treatment effect for treatment response and discontinuation due to adverse events. Given the absence of head-to-head trial data between cenobamate and its comparators, the required estimates of relative effect were established indirectly using Bayesian NMA. While these parameters affected the value of the health state transition probabilities and the predicted number of seizures, these were not treated as uncertain parameters. Instead, the sponsor assumed that the relative treatment effects would always represent the median of the distribution of values generated from each NMA. These medians were subsequently combined with other model inputs to create intermediate parameters, which were then treated as uncertain values. This approach to the characterization of uncertainty was insufficient because it explicitly excluded the full range of possible values that the relative treatment effects may take. As a result, different estimates of the mean cost and QALYs used to calculate the incremental cost-effectiveness ratio would be expected if this omission was appropriately corrected.
CADTH was able to partially address this issue by modifying the economic evaluation to better characterize parameter uncertainty for the relative treatment effects. However, the scope of these changes was restricted to the treatment response outcomes. Standard errors for the partial and complete response outcomes were estimated from the credible intervals reported alongside the NMA results in the submitted spreadsheet. Values for the RR of a partial or complete response were drawn using a log-normal distribution. Concerns persist regarding the failure to consider the imprecision of the estimates of discontinuation due to adverse events and the impact of this omission on the economic evaluation results.
Mischaracterization of parameter uncertainty due to poor dependency management: The goal of a probabilistic analysis is to generate a distribution of model outputs (i.e., costs and QALYs) estimated from each random draw of every independent parameter value. CADTH guidelines state that a probabilistic analysis must propagate each random value through the entire sequence of steps used to calculate the costs and QALYs for each alternative.19 The sponsor failed to satisfy this requirement due to poor dependency management. For example, the time-dependent treatment response probabilities were used to calculate the RR of treatment discontinuation and the RR of each treatment response outcome. However, both RRs were treated as independent random parameters that followed log-normal distributions. This meant that a new value was drawn for each RR for each draw of the cenobamate treatment responses. Instead of replacing each random value with a new distribution, the RRs should have reflected the joint (combined) distribution of the treatment response parameter and the other independent inputs. This mischaracterization of uncertainty suggests that the sponsor’s base-case results would have been different had the dependencies between parameters been appropriately managed. However, the impact of this mischaracterization of parameter uncertainty on the conclusion of cenobamate’s cost-effectiveness is unclear.
CADTH could not address this limitation. Given that this limitation applies to most of the uncertain parameters in the model, significant changes to the programming of the submitted spreadsheet would have been required. Furthermore, the impact of such changes was constrained by the absence of evidence needed to correctly characterize the uncertainty in the economic evaluation. For example, the submitted spreadsheet did not include the information needed to characterize the uncertainty associated with the cenobamate treatment discontinuation survival probabilities or the NMA-estimated OR of treatment discontinuation.
Inconsistent application of treatment discontinuation risk: The sponsor used a time-dependent transition probability to model discontinuation from a patient’s first adjunctive ASM (i.e., cenobamate or an alternative). In the first 5 cycles of the model, patients who discontinued treatment were forced to transition from their respective treatment response states to the subsequent ASM state. From cycle 6 onwards, this transition probability was restricted to patients who occupied the no-response state. As a result, patients in the other treatment response states were not subject to discontinuation unless they transitioned first to the no-response state. However, the survival analysis used to generate the transition probabilities in question modelled time to discontinuation independently from the level of treatment response. Given that the proportion of the cohort on treatment is spread across all the response states, the risk of discontinuation should not have been restricted to the no-response state. This resulted in overestimating the amount of time that patients spent on their initial adjunctive ASMs.
CADTH modified the economic model to correctly apply the risk of treatment discontinuation from cycle 6 onward. The corresponding transition probability for each treatment was applied to all the treatment response states, which reflected the total proportion of the cohort on treatment at a given point in time.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (refer to Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation
Sponsor’s key assumption | CADTH comment |
|---|---|
Patients are at risk of treatment-specific discontinuation from adverse events in the first 5 cycles (140 days, approximately 5 months) of therapy. | Confirmed by the clinical experts consulted by CADTH. |
Clinical expert opinion was generalizable to the population of interest for the following parameters: the proportion of patients eligible for surgery, the proportion of patients eligible for VNS, and the probability of specific seizure event outcomes (e.g., hospitalization, emergency department visits, family doctor follow-up). | Uncertain. Clinical opinion may not always be representative of the actual population estimates. Different results may be realized from more robust sources of evidence, such as cohort studies that source data from medical records or administrative data. |
VNS = vagal nerve stimulation.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
CADTH conducted a reanalysis of the economic evaluation that addressed some of the key limitations identified in the sponsor’s submission. The CADTH base case was derived by making changes in model parameter values and assumptions in consultation with clinical experts. A summary of each independent modification to the submitted economic evaluation is presented in Table 5. The costs and effects for the CADTH base case were generated using a Monte Carlo simulation of 5,000 iterations.
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to the sponsor’s base case | ||
None | — | — |
Changes to derive the CADTH base case | ||
1. Discontinuation due to nonresponse | Does not occur. | Treatment is discontinued following initial nonresponse or the loss of a partial or complete response. Every 6 months, any patient occupying the no-response state transitions to the subsequent ASM state. |
2. Inconsistent application of treatment discontinuation risk | From cycle 6 onward, only patients in the no-response state are at risk of treatment discontinuation. | Any patient on treatment is at risk of treatment discontinuation, as predicted by the parametric survival model. |
3. Mischaracterization of parameter uncertainty for relative treatment effects | The relative risk of partial or complete response to treatment is assumed to be equal to the median of the range of values generated from the corresponding NMAs. | The relative risk of partial or complete response is assumed to follow a log-normal distribution. |
CADTH base case | 1 + 2 + 3 | |
ASM = antiseizure medication; NMA = network meta-analysis.
Results from the CADTH base case are presented in Table 6. Consistent with the sponsor’s base case, the CADTH reanalysis was based on publicly available prices of the comparator treatments. Results from the Monte Carlo simulation are summarized.
The expected costs and QALYs for cenobamate were $468,324 and 13.14, respectively. Cenobamate dominated the alternative treatments included in the economic evaluation. This meant that cenobamate offered more QALYs at a lower cost when compared with each alternative treatment. At a threshold of $50,000 per QALY gained, cenobamate was expected to have a 99.94% probability of being cost-effective.
Additional details summarizing the CADTH base case are included in Appendix 4. Most prominently, the dominance exhibited by cenobamate was influenced by differences in the amount of time spent on the initial adjunctive ASM. While patients on cenobamate remained on the initial adjunctive ASM for 1.54 LYs, their time on alternative treatments ranged from 0.39 LYs to 0.41 LYs. This confirmed that the patients were expected to respond better to (and therefore spend more time on) cenobamate compared to the other ASMs included in the economic evaluation. Despite similar conclusions with the sponsor’s base case, the modifications described in Table 5 resulted in significantly lower incremental costs between cenobamate and each possible alternative ASM.
Table 6: Summary of the CADTH Reanalysis Results
Drug | Total costs | Total QALYs | ICER vs. reference | Sequential ICER |
|---|---|---|---|---|
Cenobamate | $469,983 | 13.1840 | Reference | Reference |
Brivaracetam | $483,042 | 13.1354 | Dominated by cenobamate | Dominated by cenobamate |
Eslicarbazepine | $483,271 | 13.1388 | Dominated by cenobamate | Dominated by cenobamate |
Perampanel | $483,544 | 13.1370 | Dominated by cenobamate | Dominated by cenobamate and eslicarbazepine |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Scenario Analysis Results
CADTH conducted a scenario analysis to explore the impact of a lower baseline seizure frequency on the results of the economic model. In this scenario analysis, the baseline seizure frequency was halved to better reflect the indicated population. As anticipated, this modification to baseline seizure frequency resulted in a meaningful reduction in incremental costs. For example, the incremental cost between cenobamate and brivaracetam fell to $7,011 from $13,059 in the CADTH base case. This pattern was consistent for all pairwise comparisons presented in Table 7. Assuming no change in relative efficacy, the cost savings from cenobamate will decrease as the assumed number of baseline seizures decreases.
Table 7: Summary of the CADTH Scenario Analysis Results
Drug | Total costs | Total QALYs | ICER vs. reference | Sequential ICER |
|---|---|---|---|---|
Cenobamate | $300,141 | 13.1481 | Reference | Reference |
Brivaracetam | $307,152 | 13.0971 | Dominated | Dominated by cenobamate |
Eslicarbazepine | $307,524 | 13.1010 | Dominated | Dominated by cenobamate |
Perampanel | $307,587 | 13.0988 | Dominated | Dominated by cenobamate and eslicarbazepine |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Based on the CADTH base-case results, cenobamate was cost-effective at a threshold of $50,000 per QALY gained; therefore, additional scenarios exploring the impact of price reductions were not considered. However, there remains a high degree of uncertainty in this conclusion because the CADTH base case failed to address several key limitations of the economic evaluation. Given the uncertainty around the comparative efficacy of cenobamate and the absence of relevant comparators in the model, there is a high degree of uncertainty regarding whether cenobamate warrants a price premium over other relevant comparators.
Issues for Consideration
The Health Canada indication for cenobamate was specific to adults with epilepsy who are not satisfactorily controlled with conventional therapy. Based on feedback from the clinical experts consulted by CADTH, cenobamate would qualify as a second line of therapy following failure of a monotherapy ASM.
Overall Conclusions
In the absence of direct evidence comparing cenobamate with other ASMs used for adjunctive treatment, the sponsor derived estimates of relative clinical efficacy using an NMA. The CADTH clinical review’s appraisal of the submitted evidence suggests there may be a short-term benefit favouring cenobamate with respect to the achievement of a reduction in seizures. However, the magnitude and clinical relevance of this outcome were unclear. No conclusions regarding relative efficacy were drawn for the achievement of seizure freedom or the risk of discontinuation due to treatment-emergent adverse events. In terms of treatment response outcomes, considerable heterogeneity was identified with respect to the patients enrolled in the studies, the follow-up durations between trials, and the definitions of outcomes (for example, some trials defined seizure freedom as an absence of any seizures, whereas the cenobamate studies defined it as a 100% reduction in only 3 types of partial seizures). Therefore, uncertainties remain regarding the clinical benefit of treatment with cenobamate compared to other adjunctive treatments.
The CADTH base case addressed some key limitations in the sponsor’s economic submission. These changes involved making more appropriate assumptions regarding treatment discontinuation and the characterization of uncertainty for the RR of seizure reduction or seizure freedom. Results from the CADTH base case suggest that cenobamate may be associated with lower total costs (driven by fewer predicted seizure events) and greater health benefits (QALYs) compared with the alternative ASMs considered in the economic model. However, these results should be interpreted with caution, given that the CADTH base case failed to address several key limitations of the economic evaluation.
In a scenario analysis, CADTH explored the impact of a lower estimate of baseline seizure frequency on the estimated costs and QALYs. The findings indicated that an estimate more representative of the target population would reduce, and potentially eliminate, any potential cost savings from cenobamate. Meanwhile, reductions in the differences in relative treatment effectiveness are expected from the resolution of issues relating to relative efficacy and the characterization of parameter uncertainty. When considered together, these issues suggest that a price premium for cenobamate relative to the other relevant comparators may not be warranted.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Cenobamate tablets, 12.5 mg, 25 mg, 50 mg, 100 mg, 150mg, 200 mg, oral. Montreal (QC): Paladin Labs Inc.; 2023 Feb 7.
2.XCOPRI (cenobamate): Tablets, 12.5 mg, 25 mg, 50 mg, 100 mg, 150mg, 200 mg, oral [product monograph]. Dublin (Ireland): Endo Ventures Ltd.; 2022.
3.Krauss GL, Klein P, Brandt C, et al. Safety and efficacy of adjunctive cenobamate (YKP3089) in patients with uncontrolled focal seizures: a multicentre, double-blind, randomised, placebo-controlled, dose-response trial. The Lancet Neurology. 2020;19(1):38-48. PubMed
4.Chung SS, French JA, Kowalski J, et al. Randomized phase 2 study of adjunctive cenobamate in patients with uncontrolled focal seizures. Neurology. 2020;94(22):e2311-e2322. PubMed
5.Sperling MR, Klein P, Aboumatar S, et al. Cenobamate (YKP3089) as adjunctive treatment for uncontrolled focal seizures in a large, phase 3, multicenter, open‐label safety study. Epilepsia. 2020;61(6):1099-1108. PubMed
6.Cenobamate Indirect treatment comparison technical report [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Cenobamate tablets, 12.5 mg, 25 mg, 50 mg, 100 mg, 150mg, 200 mg, oral. Montreal (QC): Paladin Labs Inc.; 2023 Feb 7.
7.Hamilton P, Soryal I, Dhahri P, et al. Clinical outcomes of VNS therapy with AspireSR®(including cardiac-based seizure detection) at a large complex epilepsy and surgery centre. Seizure. 2018;58:120-126. PubMed
8.Picot MC, Jaussent A, Neveu D, et al. Cost‐effectiveness analysis of epilepsy surgery in a controlled cohort of adult patients with intractable partial epilepsy: A 5‐year follow‐up study. Epilepsia. 2016;57(10):1669-1679. PubMed
9.Trinka E, Bauer G, Oberaigner W, Ndayisaba JP, Seppi K, Granbichler CA. Cause‐specific mortality among patients with epilepsy: results from a 30‐year cohort study. Epilepsia. 2013;54(3):495-501. PubMed
10.Table: 13-10-0114-01. Life expectancy and other elements of the life table, Canada, all provinces except Prince Edward Island. Ottawa (ON): Statistics Canada; 2022: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011401. Accessed 2022 September 7.
11.NICE. Single Technology Appraisal: Cenobamate for focal onset seizures in epilepsy [ID1553]. 2021.
12.Guertin JR, Feeny D, Tarride J-E. Age-and sex-specific Canadian utility norms, based on the 2013–2014 Canadian Community Health Survey. Cmaj. 2018;190(6):E155-E161. PubMed
13.Ontario Ministry of H, Ontario Ministry of Long-Term C. Ontario drug benefit formulary/comparative drug index. 2022; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2023 March 6.
14.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Cenobamate tablets, 12.5 mg, 25 mg, 50 mg, 100 mg, 150 mg, 200 mg, oral. Montreal (QC): Paladin Labs Inc.; 2023 Feb 7.
15.Chambers A, Bowen JM. Electrical stimulation for drug-resistant epilepsy: an evidence-based analysis. Ontario health technology assessment series. 2013;13(18):1. PubMed
16.Bowen JM, Snead OC, Chandra K, Blackhouse G, Goeree R. Epilepsy care in Ontario: an economic analysis of increasing access to epilepsy surgery. Ontario health technology assessment series. 2012;12(18):1. PubMed
17.Schedule of benefits for physician services under the Health Insurance Act: (January 25, 2022 (effective July 1, 2022)). Toronto (ON): Ontario Ministry of Health; 2022: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf. Accessed 2023 March 13.
18.Patient cost estimator. 2022; https://www.cihi.ca/en/patient-cost-estimator. Accessed 2023 March 13.
19.Guidelines for the economic evaluation of health technologies: Canada. 4th ed. Ottawa (ON): CADTH; 2017: https://www.cadth.ca/guidelines-economic-evaluation-health-technologies-canada-4th-edition. Accessed 1800 Jan 1.
20.Hernandez-Ronquillo L. Characterizing the Epidemiology of Epilepsy in Saskatchewan, Canada. 2017.
Appendix 1: Cost Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s). Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CADTH Cost Comparison Table for the Adjunctive Therapy of Partial Onset Seizures
Treatment | Strength | Form | Price ($) | Recommended dosage | Daily cost ($) | Annual cost ($) |
|---|---|---|---|---|---|---|
cenobamate | 12.5 mg 25 mg 50 mg 100 mg 150 mg 200 mg | Tablet | 8.8000 | Loading Dose: 12.5 mg daily for 14 days Titration: 25 mg/ 50 mg/ 100 mg/ 150 mg daily for 14 days each Maintenance: 200 mg twice daily | 8.80 | 3,214 |
Third-generation ASM | ||||||
brivaracetam (Brivlera) | 10 mg 25 mg 75 mg 100 mg | Tablet | 4.3200 | Starting Dose: 50 mg twice daily Maintenance Dose: 20 mg to 100 mg twice daily | 8.64 | 3,156 |
eslicarbazepine (Aptiom) | 200 mg 400 mg 600 mg 800 mg | Tablet | 9.8700 | Starting Dose: 400 mg daily for 14 days Maintenance Dose: 800 mg daily | 9.87 | 3,605 |
perampanel (Fycompa) | 2 mg 4 mg 6 mg 8 mg 10 mg 12 mg | Tablet | 10.0075 | 4 mg to 12 mg daily | 10.01 | 3,655 |
Second-generation ASM | ||||||
clobazam (generic) | 10 mg | Tablet | 0.2197 | 5 mg to 80 mg daily | 0.11 to 1.76 | 40 to 642 |
clonazepam (generic) | 0.5 mg | Tablet | 0.0418 | 8 mg to 10 mg daily | 0.29 to 0.36 | 105 to 132 |
2 mg | 0.0721 | |||||
clonazepam (Rivotril) | 0.5 mg | Tablet | 0.2479 | 8 mg to 10 mg daily | 1.71 to 2.14 | 624 to 781 |
2 mg | 0.4274 | |||||
gabapentin (generic) | 100 mg | Capsule | 0.0416 | 900 mg to 1,800 mg daily in 3 divided doses | 0.30 to 0.61 | 111 to 222 |
300 mg | 0.1012 | |||||
400 mg | 0.1206 | |||||
gabapentin (Neurontin) | 100 mg | Capsule | 0.4941 | 900 mg to 1,800 mg daily in 3 divided dose | 3.55 to 7.09 | 1,295 to 2,590 |
300 mg | 1.1818 | |||||
400 mg | 1.4084 | |||||
lacosamide (generic) | 50 mg | Tablet | 0.6313 | 200 mg to 400 mg daily in 2 divided doses | 1.75 to 2.90 | 639 to 1,059 |
100 mg | 0.8750 | |||||
150 mg | 1.1763 | |||||
200 mg | 1.4500 | |||||
lacosamide (Vimpat) | 50 mg | Tablet | 2.5250 | 200 mg to 400 mg daily in 2 divided doses | 7.00 to 11.60 | 2,557 to 4,237 |
100 mg | 3.5000 | |||||
150 mg | 4.7050 | |||||
200 mg | 5.8000 | |||||
lamotrigine (generic) | 25 mg | Tablet | 0.0698 | 100 mg to 500 mg daily in 2 divided doses. | 0.28 to 1.40 | 102 to 665 |
100 mg | 0.2787 | |||||
150 mg | 0.4107 | |||||
lamotrigine (Lamictal) | 25 mg | Tablet | 0.4549 | 100 mg to 500 mg daily in 2 divided doses | 1.82 to 9.10 | 510 to 3,323 |
100 mg | 1.8172 | |||||
150 mg | 2.6780 | |||||
levetiracetam (generic) | 250 mg | Tablet | 0.3210 | 1,000 mg to 3,000 mg daily in 2 divided doses | 0.78 to 2.17 | 286 to 791 |
500 mg | 0.3911 | |||||
750 mg | 0.5416 | |||||
levetiracetam (Keppra) | 250 mg | Tablet | 1.7836 | 1,000 mg to 3,000 mg daily in 2 divided doses | 4.34 to 12.00 | 1,587 to 4,396 |
500 mg | 2.1725 | |||||
750 mg | 3.0089 | |||||
oxcarbazepine | 150 mg | Tablet | 0.6209 | 600 mg to 2,400 mg daily in 2 divided doses | 1.82 to 7.28 | 665 to 2,660 |
300 mg | 0.9102 | |||||
600 mg | 1.8204 | |||||
topiramate (generic) | 25 mg | Tablet | 0.2433 | 200 mg to 400 mg daily in 2 divided doses | 0.92 to 1.35 | 335 to 493 |
100 mg | 0.4583 | |||||
200 mg | 0.6748 | |||||
topiramate (Topamax) | 25 mg | Tablet | 2.0797 | 200 mg to 400 mg daily in 2 divided doses | 7.80 to 11.50 | 2,849 to 4,207 |
100 mg | 3.9000 | |||||
200 mg | 5.7585 | |||||
vigabatrin (Sabril) | 500 mg | Tablet | 0.9566 | 2,000 mg to 3,000 mg daily in 2 divided doses | 3.83 to 5.74 | 1,398 to 2,096 |
First-generation ASM | ||||||
carbamazepine (generic) | 100 mg | Tablet | 0.1702 | 800 mg to 1,200 mg daily in 2 divided doses | 1.03 to 1.54 | 374 to 562 |
200 mg | 0.1540 | |||||
400 mg | 0.5126 | |||||
carbamazepine (Tegretol) | 100 mg | Tablet | NA | 800 mg to 1,200 mg daily in 2 divided doses | 1.03 to 1.54 | 374 to 562 |
200 mg | 0.2563 | |||||
400 mg | 0.5126 | |||||
divalproex sodium (generic) | 125 mg | Tablet | 0.1539 | 15 mg/kg per day, up to a maximum daily dose of 60 mg/kg | 1.26 to 4.98 | 461 to 1,820 |
250 mg | 0.2767 | |||||
500 mg | 0.5537 | |||||
divalproex sodium (Epival) | 125 mg | Tablet | 0.3483 | 15 mg/kg per day, up to a maximum daily dose of 60 mg/kg | 2.85 to 11.28 | 1,043 to 4,120 |
250 mg | 0.6263 | |||||
500 mg | 1.2532 | |||||
ethosuximide (Zarontin) | 250 mg | Capsule | 0.5000 | 500 mg daily in 2 divided doses | 1.00 | 365.25 |
phenytoin (generic) | 100 mg | Capsule | 0.0665 | 300 mg to 400 mg daily | 0.20 to 0.27 | 73 to 97 |
phenytoin (Dilantin) | 30 mg | Capsule | 0.1514 | 300 mg to 400 mg daily | 0.20 to 0.27 | 73 to 97 |
100 mg | 0.0665 | |||||
phenobarbital (generic) | 15 mg | Tablet | 0.1399 | 50 mg to 100 mg, twice daily | 0.15 to 0.62 | 56 to 226 |
30 mg | 0.1665 | |||||
60 mg | 0.2257 | |||||
100 mg | 0.3088 | |||||
primidone (generic) | 125 mg | Tablet | 0.0564 | 125 mg to 1,500 mg, twice daily | 0.11 to 1.06 | 42 to 389 |
250 mg | 0.0887 | |||||
ASM = antiseizure medication; NA = not available.
Note: All prices are from the Ontario Drug Benefit Formulary (accessed March 2023), unless otherwise indicated, and do not include dispensing fees.13 Annual costs assume 365.25 days per year.
aCosts for divalproex sodium assume a body weight of 75 kg.
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/no | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | Yes | No comment. |
Model has been adequately programmed and has sufficient face validity | No | Poor dependency management resulted in the mischaracterization of parameter uncertainty. Refer to limitations for more information. |
Model structure is adequate for decision problem | No | Tracking of treatment response health states restricted to first adjunctive ASM. Such responses are also considered for every subsequent intervention. However, only those responses on the initial ASM are considered in the estimation of utilities and seizures. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | Important model inputs like relative treatment effects were not treated as random values. Refer to limitations for more detail. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | Random values were not propagated through the entire sequence of steps needed to calculate costs and QALYs. In addition, distributions were assigned to the wrong parameter sets. Refer to limitations for more detail. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
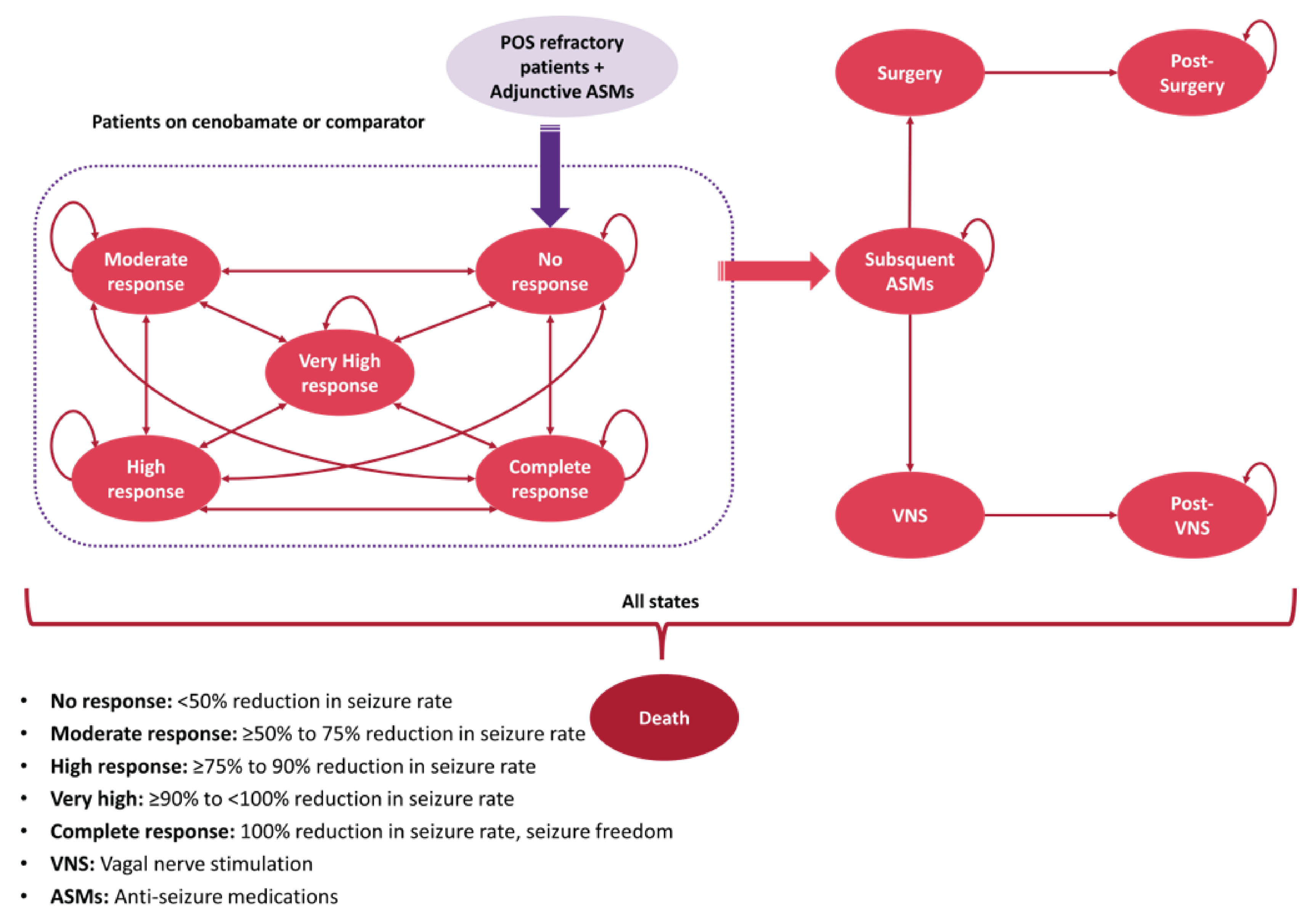
Detailed Results of the Sponsor’s Base Case
Table 10: Disaggregated Summary of the Sponsor’s Base-Case Economic Evaluation
Treatment | Component | Value | Incremental (vs. reference) | Incremental (sequential) |
|---|---|---|---|---|
Discounted LYs | ||||
Cenobamate | Adjunctive treatment | — | NA | NA |
No response | 4.8110 | NA | NA | |
Moderate response | 4.2264 | NA | NA | |
High response | 2.9683 | NA | NA | |
Very high response | 2.7874 | NA | NA | |
Seizure freedom | 5.0578 | NA | NA | |
Subsequent ASM | 4.7316 | NA | NA | |
VNS | 0.0035 | NA | NA | |
Post-VNS | 0.2925 | NA | NA | |
Surgery | 0.0211 | NA | NA | |
Postsurgery | 1.9122 | NA | NA | |
Total | 26.8117 | NA | NA | |
Eslicarbazepine | Adjunctive treatment | — | — | NA |
No response | 10.7583 | 5.95 | NA | |
Moderate response | 2.3337 | −1.89 | NA | |
High response | 0.9373 | −2.03 | NA | |
Very high response | 0.4749 | −2.31 | NA | |
Seizure freedom | 0.2406 | −4.82 | NA | |
Subsequent ASM | 8.1201 | 3.39 | NA | |
VNS | 0.0059 | 0.00 | NA | |
Post-VNS | 0.4892 | 0.20 | NA | |
Surgery | 0.0361 | 0.01 | NA | |
Postsurgery | 3.2010 | 1.29 | NA | |
Total | 26.5972 | −0.21 | NA | |
Brivaracetam | Adjunctive treatment | — | — | — |
No response | 12.0890 | 7.28 | 1.33 | |
Moderate response | 2.5556 | −1.67 | 0.22 | |
High response | 1.0150 | −1.95 | 0.08 | |
Very high response | 0.5102 | −2.28 | 0.04 | |
Seizure freedom | 0.2563 | −4.80 | 0.02 | |
Subsequent ASM | 6.9717 | 2.24 | −1.15 | |
VNS | 0.0050 | 0.00 | 0.00 | |
Post-VNS | 0.4137 | 0.12 | −0.08 | |
Surgery | 0.0309 | 0.01 | −0.01 | |
Postsurgery | 2.7090 | 0.80 | −0.49 | |
Total | 26.5564 | −0.26 | −0.04 | |
Perampanel | Adjunctive treatment | — | — | — |
No response | 11.7755 | 6.96 | −0.31 | |
Moderate response | 2.1836 | −2.04 | −0.37 | |
High response | 0.8339 | −2.13 | −0.18 | |
Very high response | 0.4012 | −2.39 | −0.11 | |
Seizure freedom | 0.1828 | −4.87 | −0.07 | |
Subsequent ASM | 7.6919 | 2.96 | 0.72 | |
VNS | 0.0056 | 0.00 | 0.00 | |
Post-VNS | 0.4598 | 0.17 | 0.05 | |
Surgery | 0.0341 | 0.0130 | 0.0032 | |
Postsurgery | 3.0096 | 1.0974 | 0.3006 | |
Total | 26.5780 | −0.23 | 0.02 | |
Discounted QALYs | ||||
Cenobamate | Adjunctive treatment | — | NA | NA |
No response | 2.0818 | NA | NA | |
Moderate response | 2.0707 | NA | NA | |
High response | 1.5488 | NA | NA | |
Very high response | 1.4540 | NA | NA | |
Seizure freedom | 2.8113 | NA | NA | |
Subsequent ASM | 2.2626 | NA | NA | |
VNS | 0.0015 | NA | NA | |
Post-VNS | 0.1369 | NA | NA | |
Surgery | 0.0091 | NA | NA | |
Postsurgery | 0.9702 | NA | NA | |
Total | 13.3468 | NA | NA | |
Eslicarbazepine | Adjunctive treatment | — | — | NA |
No response | 4.6656 | 2.58 | NA | |
Moderate response | 1.1481 | −0.92 | NA | |
High response | 0.4914 | −1.06 | NA | |
Very high response | 0.2489 | −1.21 | NA | |
Seizure freedom | 0.1345 | −2.68 | NA | |
Subsequent ASM | 3.8767 | 1.61 | NA | |
VNS | 0.0025 | 0.00 | NA | |
Post-VNS | 0.2286 | 0.09 | NA | |
Surgery | 0.0155 | 0.01 | NA | |
Total | 12.4337 | −0.91 | NA | |
Brivaracetam | Adjunctive treatment | — | — | — |
No response | 5.2401 | 3.16 | 0.57 | |
Moderate response | 1.2565 | −0.81 | 0.11 | |
High response | 0.5317 | −1.02 | 0.04 | |
Very high response | 0.2673 | −1.19 | 0.02 | |
Seizure freedom | 0.1432 | −2.67 | 0.01 | |
Subsequent ASM | 3.3254 | 1.06 | −0.55 | |
VNS | 0.0022 | 0.00 | 0.00 | |
Post-VNS | 0.1931 | 0.06 | −0.04 | |
Surgery | 0.0132 | 0.00 | 0.00 | |
Postsurgery | 1.3715 | 0.40 | −0.25 | |
Total | 12.3443 | −1.00 | −0.09 | |
Perampanel | Adjunctive treatment | — | — | — |
No response | 5.1043 | 3.02 | −0.14 | |
Moderate response | 1.0738 | −1.00 | −0.18 | |
High response | 0.4371 | −1.11 | −0.09 | |
Very high response | 0.2103 | −1.24 | −0.06 | |
Seizure freedom | 0.1022 | −2.71 | −0.04 | |
Subsequent ASM | 3.6703 | 1.41 | 0.34 | |
VNS | 0.0024 | 0.00 | 0.00 | |
Post-VNS | 0.2147 | 0.08 | 0.02 | |
Surgery | 0.0146 | 0.01 | 0.00 | |
Postsurgery | 1.5242 | 0.55 | 0.15 | |
Total | 12.3540 | −0.99 | 0.01 | |
Discounted costs ($) | ||||
Cenobamate | Adjunctive treatment | — | NA | NA |
Acquisition | 64,224 | NA | NA | |
Administration | 0 | NA | NA | |
Concomitant ASM | 7,258 | NA | NA | |
Other Resource | 9,772 | NA | NA | |
Subsequent ASM | 20,955 | NA | NA | |
VNS | 1,938 | NA | NA | |
Post-VNS | 1,226 | NA | NA | |
Surgery | 6,916 | NA | NA | |
Postsurgery | 2,955 | NA | NA | |
Seizure events | 280,430 | NA | NA | |
Total | 395,675 | NA | NA | |
Eslicarbazepine | Adjunctive treatment | — | — | NA |
Acquisition | 53,619 | −10,606 | NA | |
Administration | 0 | 0 | NA | |
Concomitant ASM | 5,404 | −1,855 | NA | |
Other resource | 13,142 | 3,370 | NA | |
Subsequent ASM | 35,907 | 14,951 | NA | |
VNS | 3,262 | 1,325 | NA | |
Post-VNS | 2,051 | 825 | NA | |
Surgery | 11,692 | 4,776 | NA | |
Postsurgery | 4,948 | 1,993 | NA | |
Seizure events | 473,107 | 192,677 | NA | |
Total | 603,131 | 207,456 | NA | |
Brivaracetam | Adjunctive treatment | — | — | — |
Acquisition | 52,273 | −11,951 | −1,346 | |
Administration | 0 | 0 | 0 | |
Concomitant ASM | 6,018 | −1,241 | 614 | |
Other resource | 14,350 | 4,578 | 1,208 | |
Subsequent ASM | 30,797 | 9,842 | −5,110 | |
VNS | 2,769 | 831 | −493 | |
Post-VNS | 1,734 | 508 | −317 | |
Surgery | 9,949 | 3,032 | −1,744 | |
Postsurgery | 4,187 | 1,231 | −761 | |
Seizure events | 489,598 | 209,168 | 16,491 | |
Total | 611,674 | 215,998 | 8,542 | |
Perampanel | Adjunctive treatment | — | — | — |
Acquisition | 56,695 | −7,529 | 4,422 | |
Administration | 0 | 0 | 0 | |
Concomitant ASM | 5,635 | −1,624 | −383 | |
Other resource | 13,881 | 4,109 | −469 | |
Subsequent ASM | 33,995 | 13,039 | 3,198 | |
VNS | 3,071 | 1,134 | 303 | |
Post-VNS | 1,927 | 701 | 193 | |
Surgery | 11,024 | 4,108 | 1,075 | |
Postsurgery | 4,652 | 1,696 | 465 | |
Seizure events | 490,282 | 209,853 | 685 | |
Total | 621,163 | 225,488 | 9,489 | |
Treatments | ICER vs. reference ($) | Sequential ICER ($) | ||
Cenobamate | Ref. | Ref. | ||
Eslicarbazepine | Dominated by reference | Dominated by reference | ||
Brivaracetam | Dominated by reference | Dominated by cenobamate and eslicarbazepine | ||
Perampanel | Dominated by reference | Dominated by cenobamate and eslicarbazepine | ||
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
To address some of the key limitations from the sponsor’s submission, a series of changes were implemented to derive the CADTH base case. Each revision listed in Table 5 was implemented independently and the corresponding results are presented in Table 11. All estimates within Table 11 represent expected costs and QALYs calculated from Monte Carlo simulations of 5,000 iterations. The disaggregated summary of the CADTH base-case simulation is presented in Table 12.
Table 11: Summary of the Stepped Analysis of the CADTH Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
Sponsor’s base case | cenobamate | 395,675 | 13.3468 | Ref. |
eslicarbazepine | 603,131 | 12.4337 | Dominated by cenobamate | |
brivaracetam | 611,674 | 12.3443 | Dominated by cenobamate and eslicarbazepine | |
perampanel | 621,163 | 12.3540 | Dominated by cenobamate and eslicarbazepine | |
CADTH reanalysis 1a: Discontinuation due to nonresponse (Cycles 1 to 5) | cenobamate | 425,141 | 13.2607 | Ref. |
perampanel | 510,714 | 12.9491 | Dominated by cenobamate | |
eslicarbazepine | 510,741 | 12.9441 | Dominated by cenobamate | |
brivaracetam | 511,816 | 12.9239 | Dominated by cenobamate | |
CADTH reanalysis 1b: Discontinuation due to loss of response (Cycles 6 to 26) | cenobamate | 446,902 | 12.0987 | Ref. |
eslicarbazepine | 481,902 | 12.9793 | Dominated by cenobamate and eslicarbazepine | |
brivaracetam | 482,103 | 12.9854 | Dominated by cenobamate and eslicarbazepine | |
perampanel | 482,866 | 12.9814 | Dominated by cenobamate and eslicarbazepine | |
CADTH reanalysis 1c: Discontinuation due to loss of response (Cycles 26+) | cenobamate | 453,287 | 13.1438 | Ref. |
eslicarbazepine | 508,492 | 12.8513 | Dominated by cenobamate | |
brivaracetam | 509,467 | 12.8285 | Dominated by cenobamate and eslicarbazepine | |
perampanel | 512,631 | 12.8312 | Dominated by cenobamate and eslicarbazepine | |
CADTH reanalysis 1: Discontinuation due to loss of response (1a + 1b + 1c) | cenobamate | 466,659 | 13.2082 | Ref. |
brivaracetam | 482,612 | 13.1485 | Dominated by cenobamate | |
eslicarbazepine | 482,840 | 13.1520 | Dominated by cenobamate | |
perampanel | 483,224 | 13.1499 | Dominated by cenobamate | |
CADTH reanalysis 2: Inconsistent application of treatment discontinuation risk | cenobamate | 425,331 | 13.1193 | Ref. |
eslicarbazepine | 580,375 | 12.3660 | Dominated by cenobamate | |
brivaracetam | 589,673 | 12.2658 | Dominated by cenobamate | |
perampanel | 598,886 | 12.2749 | Dominated by cenobamate | |
CADTH reanalysis 3: Mischaracterization of parameter uncertainty for the relative treatment effects | cenobamate | 395,675 | 13.3468 | Ref. |
eslicarbazepine | 603,131 | 12.4337 | Dominated by cenobamate | |
brivaracetam | 611,674 | 12.3443 | Dominated by cenobamate | |
perampanel | 621,163 | 12.3540 | Dominated by cenobamate | |
CADTH reanalysis 4: Reduction in baseline seizure frequency | Cenobamate | 254,091 | 13.2593 | Ref. |
Eslicarbazepine | 364,630 | 12.3613 | Dominated by cenobamate | |
Brivaracetam | 364,952 | 12.2701 | Dominated by cenobamate and eslicarbazepine | |
perampanel | 374,018 | 12.2846 | Dominated by cenobamate and eslicarbazepine | |
CADTH base case: Reanalysis 1 + 2 + 3 | cenobamate | 469,983 | 13.1840 | Ref. |
brivaracetam | 483,042 | 13.1354 | Dominated by cenobamate | |
eslicarbazepine | 483,271 | 13.1388 | Dominated by cenobamate | |
perampanel | 483,544 | 13.1370 | Dominated by cenobamate |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; Ref. = reference.
Detailed Results of the CADTH Base Case
Table 12: Disaggregated Summary of CADTH’s Economic Evaluation Results
Treatment | Component | Value | Incremental (vs. reference) | Incremental (sequential) |
|---|---|---|---|---|
Discounted LYs | ||||
Cenobamate | Adjunctive treatment | — | NA | NA |
No response | 0.3051 | NA | NA | |
Moderate response | 0.2850 | NA | NA | |
High response | 0.2413 | NA | NA | |
Very high response | 0.2337 | NA | NA | |
Seizure freedom | 0.4745 | NA | NA | |
Subsequent ASM | 17.1532 | NA | NA | |
VNS | 0.0126 | NA | NA | |
Post-VNS | 1.0878 | NA | NA | |
Surgery | 0.0771 | NA | NA | |
Postsurgery | 7.0994 | NA | NA | |
Total | 26.9697 | NA | NA | |
Brivaracetam | Adjunctive treatment | — | — | NA |
No response | 0.3104 | 0.0052 | NA | |
Moderate response | 0.0506 | −0.2345 | NA | |
High response | 0.0305 | −0.2108 | NA | |
Very high response | 0.0108 | −0.2228 | NA | |
Seizure freedom | 0.0123 | −0.4621 | NA | |
Subsequent ASM | 17.7428 | 0.5897 | NA | |
VNS | 0.0131 | 0.0006 | NA | |
Post-VNS | 1.1573 | 0.0696 | NA | |
Surgery | 0.0805 | 0.0034 | NA | |
Postsurgery | 7.5456 | 0.4461 | NA | |
Total | 26.9539 | −0.0158 | NA | |
Eslicarbazepine | Adjunctive treatment | — | 0.0000 | — |
No response | 0.2970 | −0.0082 | 0.0052 | |
Moderate response | 0.0492 | −0.2359 | −0.2345 | |
High response | 0.0297 | −0.2116 | −0.2108 | |
Very high response | 0.0106 | −0.2231 | −0.2228 | |
Seizure freedom | 0.0118 | −0.4627 | −0.4621 | |
Subsequent ASM | 17.7562 | 0.6030 | 0.5897 | |
VNS | 0.0131 | 0.0006 | 0.0006 | |
Post-VNS | 1.1585 | 0.0707 | 0.0696 | |
Surgery | 0.0805 | 0.0034 | 0.0034 | |
Postsurgery | 7.5530 | 0.4536 | 0.4461 | |
Total | 26.9596 | −0.0101 | −0.0158 | |
Perampanel | Adjunctive treatment | — | 0.0000 | 0.0000 |
No response | 0.3042 | −0.0010 | −0.0134 | |
Moderate response | 0.0429 | −0.2421 | −0.0014 | |
High response | 0.0256 | −0.2157 | −0.0008 | |
Very high response | 0.0085 | −0.2252 | −0.0002 | |
Seizure freedom | 0.0114 | −0.4630 | −0.0005 | |
Subsequent ASM | 17.7550 | 0.6018 | 0.0134 | |
VNS | 0.0132 | 0.0006 | 0.0000 | |
Post-VNS | 1.1590 | 0.0713 | 0.0012 | |
Surgery | 0.0805 | 0.0034 | 0.0000 | |
Postsurgery | 7.5564 | 0.4570 | 0.0075 | |
Total | 26.9568 | −0.0129 | 0.0056 | |
Discounted QALYs | ||||
Cenobamate | Adjunctive treatment | NA | NA | |
No response | 0.1356 | NA | NA | |
Moderate response | 0.1433 | NA | NA | |
High response | 0.1304 | NA | NA | |
Very high response | 0.1260 | NA | NA | |
Seizure freedom | 0.2723 | NA | NA | |
Subsequent ASM | 8.2308 | NA | NA | |
VNS | 0.0055 | NA | NA | |
Post-VNS | 0.5077 | NA | NA | |
Surgery | 0.0335 | NA | NA | |
Postsurgery | 3.5988 | NA | NA | |
Total | 13.1840 | NA | NA | |
Brivaracetam | Adjunctive treatment | — | — | NA |
No response | 0.1382 | 0.0025 | NA | |
Moderate response | 0.0255 | −0.1178 | NA | |
High response | 0.0165 | −0.1139 | NA | |
Very high response | 0.0059 | −0.1202 | NA | |
Seizure freedom | 0.0071 | −0.2652 | NA | |
Subsequent ASM | 8.5297 | 0.2989 | NA | |
VNS | 0.0057 | 0.0002 | NA | |
Post-VNS | 0.5410 | 0.0332 | NA | |
Surgery | 0.0350 | 0.0015 | NA | |
Postsurgery | 3.8308 | 0.2320 | NA | |
Total | 13.1354 | −0.0486 | NA | |
Eslicarbazepine | Adjunctive treatment | — | 0 | — |
No response | 0.1322 | −0.0034 | −0.0060 | |
Moderate response | 0.0248 | −0.1185 | −0.0007 | |
High response | 0.0161 | −0.1143 | −0.0004 | |
Very high response | 0.0057 | −0.1203 | −0.0001 | |
Seizure freedom | 0.0068 | −0.2655 | −0.0003 | |
Subsequent ASM | 8.5362 | 0.3054 | 0.0065 | |
VNS | 0.0057 | 0.0002 | 0.0000 | |
Post-VNS | 0.5415 | 0.0338 | 0.0006 | |
Surgery | 0.0350 | 0.0015 | 0.0000 | |
Postsurgery | 3.8348 | 0.2359 | 0.0039 | |
Total | 13.1388 | −0.0452 | 0.0034 | |
Perampanel | Adjunctive treatment | — | 0 | 0.0000 |
No response | 0.1354 | −0.0002 | 0.0032 | |
Moderate response | 0.0216 | −0.1217 | −0.0031 | |
High response | 0.0139 | −0.1165 | −0.0022 | |
Very high response | 0.0046 | −0.1214 | −0.0011 | |
Seizure freedom | 0.0066 | −0.2657 | −0.0002 | |
Subsequent ASM | 8.5358 | 0.3050 | −0.0004 | |
VNS | 0.0057 | 0.0003 | 0.0000 | |
Post-VNS | 0.5418 | 0.0340 | 0.0003 | |
Surgery | 0.0350 | 0.0015 | 0.0000 | |
Postsurgery | 3.8365 | 0.2377 | 0.0018 | |
Total | 13.1370 | −0.0470 | −0.0018 | |
Discounted costs ($) | ||||
Cenobamate | Adjunctive treatment | — | NA | NA |
Acquisition | 5,199 | NA | NA | |
Administration | 0 | NA | NA | |
Concomitant ASM | 585 | NA | NA | |
Other resource | 701 | NA | NA | |
Subsequent ASM | 76,055 | NA | NA | |
VNS | 7,167 | NA | NA | |
Post-VNS | 4,565 | NA | NA | |
Surgery | 25,377 | NA | NA | |
Postsurgery | 10,976 | NA | NA | |
Seizure events | 339,358 | NA | NA | |
Total | 469,983 | NA | NA | |
Brivaracetam | Adjunctive treatment | — | — | NA |
Acquisition | 1,408 | −3,791 | NA | |
Administration | 0 | $0 | NA | |
Concomitant ASM | 161 | −424 | NA | |
Other resource | 484 | −217 | NA | |
Subsequent ASM | 78,861 | 2,806 | NA | |
VNS | 7,575 | 408 | NA | |
Post-VNS | 4,857 | 292 | NA | |
Surgery | 26,711 | 1,334 | NA | |
Postsurgery | 11,665 | 689 | NA | |
Seizure events | 351,319 | 11,961 | NA | |
Total | 483,042 | 13,059 | NA | |
Eslicarbazepine | Adjunctive treatment | — | 0 | — |
Acquisition | 1,543 | −3,656 | 135 | |
Administration | 0 | 0 | 0 | |
Concomitant ASM | 155 | −430 | −7 | |
Other resource | 726 | 25 | 242 | |
Subsequent ASM | 78,928 | 2,872 | 67 | |
VNS | 7,582 | 415 | 7 | |
Post-VNS | 4,862 | 297 | 5 | |
Surgery | 26,733 | 1,356 | 22 | |
Postsurgery | 11,677 | 701 | 12 | |
Seizure events | 351,066 | 11,708 | −253 | |
Total | 483,271 | 13,288 | 229 | |
Perampanel | Adjunctive treatment | — | 0 | 0 |
Acquisition | 1,536 | −3,663 | −7 | |
Administration | 0 | 0 | 0 | |
Concomitant ASM | 152 | −433 | −3 | |
Other resource | 585 | −115 | −140 | |
Subsequent ASM | 78,932 | 2,876 | 4 | |
VNS | 7,585 | 418 | 3 | |
Post-VNS | 4,864 | 299 | 2 | |
Surgery | 26,743 | 1,366 | 10 | |
Postsurgery | 11,682 | 706 | 5 | |
Seizure events | 351,465 | 12,107 | 399 | |
Total | 483,544 | 13,561 | 273 | |
Treatments | ICER vs. reference ($) | Sequential ICER ($) | ||
Cenobamate | Ref. | Ref. | ||
Brivaracetam | Dominated by reference | Dominated by cenobamate | ||
Eslicarbazepine | Dominated by reference | Dominated by cenobamate | ||
Perampanel | Dominated by reference | Dominated by cenobamate | ||
ICER = incremental cost-effectiveness ratio; LY = life-year; NA = not applicable; QALY = quality-adjusted life-year; Ref. = reference; vs. = versus.
Appendix 5: Submitted Budget Impact Analysis and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 13: Summary of Key Takeaways
Key takeaways of the budget impact analysis |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
The sponsor submitted budget impact analysis (BIA) evaluated the introduction of cenobamate for use as an adjunctive treatment for adult epilepsy patients with POS who are not satisfactorily controlled with conventional therapy.14 Estimates were generated from the perspective of CADTH participating drug plans (all but Quebec) and the results were aggregated into pan-Canadian totals over a 3-year time horizon.14 Key inputs to the BIA are documented in Table 14.
A claims-based approach was used to estimate the eligible population size for this analysis. The sponsor queried the IQVIA PharmaStat database to identify the claims for every ASM by province from 2017 to 2022. Claims were considered relevant if used for the same indication for cenobamate — as an adjunctive therapy for the management of partial onset seizures in adults with epilepsy. For treatments whose indications included pediatric populations, the proportion used in adults were based on epidemiologic data from Saskatchewan.20 Linear regression models fitted for each treatment were used to predict the number of claims in each year of the BIA time horizon. Market share estimates were then calculated as the proportion of claims for each treatment in each year of the BIA model. Total costs were calculated by multiplying the costs per-claim of each treatment by the corresponding market share estimate.14
In the reference scenario, it was assumed that patients would be eligible for 1 of the currently available ASMs indicated for the adjunctive treatment adults with epilepsy and partial onset seizures. These alternatives included: brivaracetam, eslicarbazepine, perampanel, and lacosamide. In the new drug scenario, it was assumed that cenobamate would be included among the currently available ASMs for this indication in Canada.14
Key assumptions of the sponsor’s BIA included:
Claims represented individual patients.
As lacosamide is considered a regular benefit in Nova Scotia and New Brunswick, it was assumed that its use was restricted to the adjunctive treatment of adults with partial onset seizures.
The availability of each treatment was assumed to follow the corresponding coverage status in each province. This meant that brivaracetam, eslicarbazepine, and perampanel availability was not uniform across Canada. Only British Columbia and Manitoba covered all 3 treatments. Brivaracetam was also available in Prince Edward Island and for those with Noninsured Health Benefits. The latter also provided coverage for eslicarbazepine.
In the new drug scenario, the market share for cenobamate was assumed to be 3.12% in year 1, 12.40% in year 2, and 20.00% in year 3. The sponsor assumed that the introduction of cenobamate will capture market share evenly from brivaracetam, eslicarbazepine, and perampanel.
Cenobamate was assumed to take 0% market share from lacosamide. Clinical expert opinion suggested that lacosamide would be used earlier in the treatment pathway.
Table 14: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1/year 2/year 3, if appropriate) |
|---|---|
Target population | |
Number of claims for ASMs in Canada | |
Brivaracetam | 379 / 444 / 509 |
Eslicarbazepine | 194 / 215 / 236 |
Perampanel | 434 / 447 / 461 |
Lacosamide | 2,971 / 3,217 / 3,462 |
% relevant claims | |
Brivaracetam | 100% |
Eslicarbazepine | 100% |
Perampanel | 64.1% |
Lacosamide | 100% |
% claims used for partial onset seizures | |
Brivaracetam | 100% |
Eslicarbazepine | 100% |
Perampanel | 73.0% |
Lacosamide | 100% |
% claims used in adults | |
Brivaracetam | 100% |
Eslicarbazepine | 100% |
Perampanel | 87.8% |
Lacosamide | 100% |
% claims used as adjunctive therapies | 100% |
Number of patients eligible for drug under review | 3,822 / 4,163 / 4,503 |
Market uptake (3 years) | |
Uptake (reference scenario) | |
Brivaracetam | 9.92% / 10.67% / 11.31% |
Eslicarbazepine | 5.06% / 5.16% / 5.24% |
Perampanel | 7.27% / 6.89% / 6.56% |
Lacosamide | 77.74% / 77.28% / 76.88% |
Uptake (new drug scenario) | |
Cenobamate | 3.12% / 12.40% / 20.00% |
Brivaracetam | 8.53% / 4.85% / 1.52% |
Eslicarbazepine | 4.35% / 2.34% / 0.71% |
Perampanel | 6.25% / 3.13% / 0.88% |
Lacosamide | 77.74% / 77.28% / 76.88% |
Cost of treatment (per patient) | |
Cenobamate | $264.00 |
Brivaracetam | $261.79 |
Eslicarbazepine | $297.90 |
Perampanel | $300.50 |
Lacosamide | $59.24 |
POS = partial onset seizure.
Summary of the Sponsor’s BIA Results
In the sponsor’s base case, the budget impact of cenobamate was –$143,063 in Year 1, –$602,201 in Year 2, and –$1,027,859 in Year 3. The 3-year budget impact of cenobamate was –$1,773,123.14
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Use of a claims-based approach to estimate market size introduces uncertainty with the anticipated budget impact of cenobamate: The sponsor relied on public claims data to estimate the market size of the relevant comparators. A key challenge with this approach is its reliance on aggregated data which makes it difficult to distinguish individual patients. Given that multiple refills of an ASM are expected over time, it may not always be appropriate to assume that each claim or prescription will represent a unique patient. As a result, there is a risk that the size of the eligible population and the corresponding market share estimates for each drug were incorrect. The extent to which this limitation will affect the budget impact of cenobamate is unknown.
CADTH was unable to address the limitations of a claims-based approach to estimate the budget impact of cenobamate.
Missing comparators: Alternatives to cenobamate considered in the BIA were restricted to third-generation ASMs: brivaracetam, eslicarbazepine, and perampanel. As with the economic evaluation, lacosamide was included as a comparator in a separate scenario analysis. Clinical experts consulted by CADTH noted that several relevant comparators such as clobazam, topiramate, and levetiracetam would also be prescribed as adjunctive treatments for adults with uncontrolled partial onset seizures. The clinical experts also noted there was no specific treatment algorithm to guide medication selection. Therefore, the sponsor’s restriction of the comparators to third-line ASMs and lacosamide was inappropriate. Given that cenobamate may take market share from any existing adjunctive therapy, the budget impact may be underestimated.
This limitation could not be addressed by CADTH.
The price of drugs paid by public drug plans is uncertain: Both the sponsor’s and CADTH’s analyses are based on publicly available list prices for all comparators.
This limitation could not be addressed by CADTH in our reanalysis. Confidential negotiated prices for cenobamate’s comparators may lead to budgetary savings being limited or eliminated.
Overestimation of lacosamide’s market share: In establishing the market share for cenobamate, the sponsor assumed that cenobamate would take no market share from lacosamide. The sponsor justified this choice as 1 of their clinical experts indicated lacosamide would be used earlier in the treatment pathway than cenobamate or other comparators. This suggested that the total claims for lacosamide represented its use as a monotherapy as well as an adjunctive treatment. However, no steps were taken to identify the proportion of lacosamide claims that represented its use as an adjunctive treatment. The failure to exclude the proportion of lacosamide claims for patients using it as monotherapy means its market share as an adjunctive treatment was overestimated. Clinical experts consulted by CADTH confirmed that the number of lacosamide claims as an adjunctive treatment would be a proportion of the total claims for lacosamide. It was noted there was considerable heterogeneity between clinicians regarding lacosamide’s use at this point of therapy.
Given that cenobamate was assumed to take no market share from lacosamide, this limitation is expected to have no effect on the budget impact results. Furthermore, CADTH was unable to identify reliable evidence summarizing the frequency with which lacosamide is used as an adjunctive treatment.
CADTH Reanalyses of the BIA
In the absence of more reliable estimates to inform the key parameters of the BIA, the sponsor’s submitted base case was maintained. CADTH expects that the budget impact of cenobamate will be sensitive to inputs that may affect the market size calculation.
Table 15: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | 3-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $31,491,287 | $35,917,194 | $40,343,460 | $44,770,426 | $121,031,080 |
New drug | $31,491,287 | $35,774,131 | $39,741,260 | $43,742,567 | $119,257,958 | |
Budget impact | $0 | -$143,063 | -$602,201 | -$1,027,859 | -$1,773,123 |
BIA = budget impact analysis.
Stakeholder Input
Patient Input
Canadian Epilepsy Alliance
About Canadian Epilepsy Alliance
Established over 20 years ago, The Canadian Epilepsy Alliance (CEA) is a Canada-Wide Network of 28 community organizations dedicated to the promotion of independence and quality of Life for people with epilepsy and their families, through support services, information, advocacy and public awareness.
www.canadianepilepsyalliance.org
Information Gathering
Information for this submission has been gathered by the President of the CEA, with input from the membership who are all working in the field providing direct support to patients with epilepsy.
Disease Experience
Epilepsy affects each individual differently. Those whose seizures are not controlled are often placed in dangerous situations should a seizure occur while riding a bus, shopping for groceries or crossing a street, to name a few of the activities that most of us take for granted. Persons with uncontrolled seizures are not permitted by law to operate a motor vehicle, leading to a loss of independence that may be insurmountable, particularly for those living in rural areas Often the medications used to control seizures cause unpleasant side effects that may be completely intolerable to the patient. One side effect of medication often mentioned by patients is impairment of the ability to concentrate or focus. Short term and/or long-term Memory often accompanies a seizure which is problematic in the workforce and in general.
Societal attitudes have a significant impact on persons with epilepsy. Many members of the public are ill informed of the true facts about epilepsy, such that people with the condition often face stigma and discrimination. People experiencing seizures, or in the aftermath of seizures, have been “Tazered” or arrested for being “intoxicated” in public. Parents of children with epilepsy are often afraid to let them participate in the normal activities of childhood, for fear that they will have a seizure and be injured. Children with epilepsy may not be invited to sleep-overs because their friends’ parents are afraid that they won’t know what to do if the child had a seizure while away from home.
The most important & significant outcome for epilepsy treatment is Seizure Freedom. This can be a result of a medication controlling the seizures which provides hope and contributes to the individual living a decent quality of life. Without the social stigma and burden on families, freedom from seizures is an optimal scenario.
Experiences With Currently Available Treatments
Current therapies work for nearly 70% of persons with Epilepsy. The remaining 30% try to remain hopeful that someday a medication will be found that will help them. New drugs like cenobamate bring hope to many who are close to giving up.
When someone has epilepsy, the whole family is affected. Everyone’s life revolves around the seizures. There is anxiety around when and where the next seizure will occur, and what impact it will have. A husband is afraid his wife might have a seizure and drop the baby; parents are nervous if their child is invited to a birthday party; a teenager is anxious as he watches his father leave for work. Is today the day that something terrible will happen? Some caregivers are afraid to leave the person with frequent seizures alone, contributing to a loss of independence and the lack of self-esteem we see so often with this patient population. Compassion fatigue in the care giver is always of concern. Many caregivers are sleep deprived as they either try to stay awake all night in case a seizure happens or go to bed and find they are too anxious to sleep. As well, caregivers often must live with the sometimes highly unpleasant side effects of various medications that their loved ones are taking. Mood swings, sexual dysfunction, suicidal thoughts, memory loss, problems with concentration, fatigue, exhaustion, depression – all can prove devastating to the person involved, and to those around them.
Approximately 30% of people with epilepsy are resistant to the current medications despite trying many and remain drug refractory. A New drug provides hope in alleviating this.
Improved Outcomes
Medication can be very successful in treating seizures however the side-effects can be difficult to deal with. One of the most significant problems is that 30% of people with epilepsy do not obtain seizure control with medication. Side-effects of medication, including effects on cognition, can be greater when multiple drugs and/or higher doses are used to try and improve seizure control which adds to the burden of drug-refractory epilepsy.
The expectation is that the lives of some patients living with uncontrolled seizures will be improved by this new drug if it can help reduce their seizure frequency and potentially lead to seizure freedom compared to other drug treatments. There are many different types of epilepsy disorders, and even among the same epilepsy syndromes people’s response to treatment varies. Some treatments may work for some but not for others. When a treatment is found that controls seizures there can be significant improvements in quality of life.
Experience With Drug Under Review
Each new drug brought to market offers hope to the 30% of epilepsy sufferers whose seizures are not so far controlled. Even a reduction in the absolute number of seizures that these individuals experience can potentially improve overall quality of life. Seizure freedom may become a reality and the person with epilepsy need no longer sit on the sidelines as life passes by. Of course, if this drug is not made available to them, this dream can never come to fruition. People with intractable epilepsy are very often unemployed or under-employed because of the frequency of their seizures. They usually live on very restricted incomes, and because they are not working, or are only working parttime, the majority does not have private insurance plans. If new medications are not placed on the formulary most of our members with intractable epilepsy, the ones who need them the most, will never be given the opportunity to find out if this new drug will work for them.
This new drug, cenobamate, offers hope and that offers improvement in health and well-being.
Companion Diagnostic Test
Not applicable.
Conflict of Interest Declaration — Canadian Epilepsy Alliance
Did you receive help from outside your patient group to complete this submission?
No.
Did you receive help from outside your patient group to collect or analyze data used in this submission?
No.
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
Table 1: Financial Disclosures for Canadian Epilepsy Alliance
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Epilepsy Toronto
About Epilepsy Toronto
Epilepsy Toronto is the place where Torontonians living with epilepsy and other seizure disorders can learn more about their condition, get the help they need and be a part of a family of caring and supporting people. Over the past 60 years, Epilepsy Toronto has been the place where Torontonians living with epilepsy can learn more about their condition, get the help they need and be a part of a family of caring and supporting people.
Epilepsy Toronto prioritizes individual needs, the importance of living as independently as possible and the benefits of community engagement. Our programs address all aspects of epilepsy from the first diagnosis of a child to the struggles that young people face, to adult needs such as employment and relationships. Recently, Epilepsy Toronto has begun to expand our services to address the needs of people living with other seizure disorders, such as functional seizures.
Through Epilepsy Toronto’s programs, individuals can participate in trainings to build confidence and skills, share their medical concerns and challenges, discuss coping methods with people who understand, involve their schools or workplaces in awareness building, enjoy an outing with friends, learn about the latest epilepsy news and enjoy being in a place they can call home.
Our programs include:
Public Education
For more information: https://epilepsytoronto.org/about-us/epilepsy-toronto/our-agency/
Information Gathering
CADTH is interested in hearing from a wide range of patients and caregivers in this patient input submission. Describe how you gathered the perspectives: for example, by interviews, focus groups, or survey; personal experience; or a combination of these. Where possible, include when the data were gathered; if data were gathered in Canada or elsewhere; demographics of the respondents; and how many patients, caregivers, and individuals with experience with the drug in review contributed insights. We will use this background to better understand the context of the perspectives shared.
Epilepsy Toronto collects information from our staff interactions, surveys and data collected from our members. All information was from Canada.
Disease Experience
CADTH involves clinical experts in every review to explain disease progression and treatment goals. Here we are interested in understanding the illness from a patient’s perspective. Describe how the disease impacts patients’ and caregivers’ day-to-day life and quality of life. Are there any aspects of the illness that are more important to control than others?
Epilepsy Toronto has been supporting people living with epilepsy for over 60 years. Though epilepsy affects about 1 in 100 people, the biggest impact on quality of life is due to misinformation and strong myths about epilepsy in the general population. In 2022 our members reported:
62% felt emotional stress; anxiety; depression or other mental health due to their diagnosis.
77% felt like they needed to connect with others in the epilepsy community.
85% want a safe place to share their epilepsy experiences.
66% no longer feel as alone or isolated after meeting with an epilepsy community agency.
Individuals living with epilepsy are more likely to experience mental health concerns, such as depression, anxiety, and suicidal ideations. This reverberates throughout the family unit as concern for family members and the perception of epilepsy in the general population impacts every aspect of someone's quality of life, housing, employment, and education.
Experiences With Currently Available Treatments
CADTH examines the clinical benefit and cost-effectiveness of new drugs compared with currently available treatments. We can use this information to evaluate how well the drug under review might address gaps if current therapies fall short for patients and caregivers.
Describe how well patients and caregivers are managing their illnesses with currently available treatments (please specify treatments). Consider benefits seen, and side effects experienced and their management. Also consider any difficulties accessing treatment (cost, travel to clinic, time off work) and receiving treatment (swallowing pills, infusion lines).
Approximately 70% of people with epilepsy will be able to gain seizure control with one or more anti-seizure medications, while the other 30% are considered to have drug resistant epilepsy and may be treated with surgery or special diets. It is important to note that seizure control is different from seizure freedom, which is the aim of epilepsy treatments.
For those who live outside of surgical centers, they may incur costs for travel and hotels. This may also require taking time off work for the surgery and recovery by both the individual with epilepsy and caregivers.
People with epilepsy often report struggles with their anti-seizure medications which can cause cognitive difficulties such as brain fog, fatigue, and memory challenges, which generally make everyday life more challenging for all involved. Some people also report changes in personality, suicidal ideations, depression and anxiety, or mood swings. People often must take more than one anti-seizure medication, which brings additional side effects.
Improved Outcomes
From the person living with epilepsy to any member of their family circle; seizure freedom is the ultimate goal. The stigma and negative stereotypes about epilepsy impact everyday life and growth. Being seizure free means being able to obtain a driver’s license, increased employment opportunities, and fewer worries about independence.
Companion Diagnostic Test
Not applicable.
Conflict of Interest Declaration — Epilepsy Toronto
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
Canadian Epilepsy Alliance.
Did you receive help from outside your patient group to collect or analyze data used in this submission.
No.
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
Table 2: Financial Disclosures for Epilepsy Toronto
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Epilepsy Southwestern Ontario
About Epilepsy Southwestern Ontario
Epilepsy Southwestern Ontario is a registered not-for-profit, charitable agency dedicated to enhancing the lives of people who are affected by epilepsy through support services, education, advocacy, and community awareness. Our aim is to:
provide information about epilepsy and seizures for individuals with epilepsy and their families, schools, workplaces, and the general public to help reduce the stigma/misunderstanding of epilepsy and make environments safer through knowing how to respond to a seizure.
offer support services, including peer supports, to help improve clients’ mental health and reduce isolation through connections with others.
share social, community, and financial resources for people with epilepsy and their families to help meet their needs.
create opportunities for people with epilepsy to gather as a community to share their experiences with others and to see that they are not alone with their condition.
advocate for those who need additional support (i.e., at school or in the workplace, or due to human rights violations).
This is a link to our Epilepsy Southwestern Ontario website: www.epilepsyswo.ca
Information Gathering
Information has been gathered by the Executive Director from staff members including our Epilepsy Educators and our social worker, our Medical Advisory Board, and from past general feedback from clients. All information was from Canada.
Disease Experience
Though epilepsy is a very common condition, affecting about 1 in 100 people, many people living with epilepsy or who are newly diagnosed report that they don’t know anyone else with epilepsy and that they feel like they are the only one. This may be in part because a societal lack of understanding about epilepsy creates a stigma that makes it difficult to disclose. Additionally, when others don’t know a person has epilepsy or the characteristics of seizures, they may not understand how to respond to seizures, which can create fears for individuals with epilepsy and their families in being able to fully participate in events or activities without a trained or knowledgeable support person. Because seizures can happen suddenly and without warning, living with epilepsy can be quite difficult and unpredictable, causing concerns about safety in everyday activities, especially undertaking activities independently. For example, going for walks, participating in recreational activities, cooking, bathing, or childminding can all be of concern to people with uncontrolled seizures.
People with epilepsy are more likely to experience mental health concerns, such as depression, anxiety, and suicidal ideations, and are more likely to be under employed with lower educational attainment levels. People with epilepsy may also be unable to drive due to uncontrolled seizures, which makes it more difficult to be able to connect with others, to attend appointments, run errands, or to obtain employment. This is a particular challenge for those who are living in rural areas where public transportation is limited or unavailable. All of this can mean that the person with epilepsy and/or their caregivers experience financial stressors due to low income from not being able to work or having to take time off work.
Epilepsy has an impact on everyone in the family because worries about the individual having a seizure are ever present. Caregivers often report difficulty sleeping due to anxiety and fears about seizures and about allowing their person to be independent. The person with epilepsy may also require additional care and support, which can be time consuming for the caregiver who may potentially experience burnout.
Experiences With Currently Available Treatments
Approximately 70% of people with epilepsy will be able to gain seizure control with one or more anti-seizure medications, while the other 30% are considered to have drug resistant epilepsy and may be treated with surgery or special diets. It is important to note that seizure control is different from seizure freedom, which is the aim of epilepsy treatments.
For those who live outside of surgical centers, they may incur costs for travel and hotels. This may also require taking time off work for the surgery and recovery by both the individual with epilepsy and caregivers.
People with epilepsy often report struggles with their anti-seizure medications which can cause cognitive difficulties such as brain fog, fatigue, and memory challenges, which generally make everyday life more challenging for all involved. Some people also report changes in personality, suicidal ideations, depression and anxiety, or mood swings. People often must take more than one anti-seizure medication, which brings additional side effects.
Improved Outcomes
The families and individuals who do find a medication that works for them and can prevent their seizures are extremely grateful for their seizure-free time, especially when they are able to achieve seizure freedom. Seizure freedom is the ultimate goal because it means that more is possible in life, such as being able to obtain a driver’s license, increased employment opportunities, and fewer worries about independence. Caregivers can also begin to relax instead of having to constantly worry about their loved one having a seizure that could cause bodily harm or a potential medical emergency.
Families and individuals with epilepsy have to consider the balance of unpleasant side effects of medications vs. the rate of seizure frequency, with some families recognizing that the side effects cause difficulty with memory, fatigue, or cognition, but the medication does reduce the number of seizures per day which is also very important.
Companion Diagnostic Test
Not applicable
Anything Else?
It’s important to know that because people with epilepsy are often under employed or unemployed and as a result have lower incomes and a lack of employment health benefits, that many people who could benefit from new medications such as this one will not be able to access them unless they are added to the formulary of medications that are covered by government health care plans. Even families who have a middle-class income struggle to be able to afford the high cost of medications when they are not covered by benefits, but we most often see people who do not even have this level of income. People with epilepsy and their families are often desperate to try anything that may work to control seizures and new medication options offer hope for a seizure free future. Seizure reduction, and especially seizure control, make a significant difference in quality of life for the person with epilepsy, as well as their loved ones who support and worry about them.
Conflict of Interest Declaration — Epilepsy Southwestern Ontario
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission?
There was no assistance from outside Epilepsy Southwestern Ontario to complete this submission.
Did you receive help from outside your patient group to collect or analyze data used in this submission?
There was no assistance from outside Epilepsy Southwestern Ontario to collect or analyze data used in this submission.
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
Table 3: Financial Disclosures for Epilepsy Southwestern Ontario
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Epilepsy South Central Ontario
About Epilepsy South Central Ontario
Epilepsy South Central Ontario is a non-profit charitable organization that provides education, information and support to families and individuals. We are dedicated to improving the quality of life for persons living with epilepsy. www.epilepsysco.org
Information Gathering
Epilepsy South Central Ontario gathers client information based on discussions in support groups, one-on-one counselling,
Disease Experience
Epilepsy affects not only the individual, but the entire family. Most people living with epilepsy are dependent on family members to provide day to day support. Most are unable to drive, leaving them to rely on family, friends and neighbours. Their quality of life is highly impacted as most patients never know when a seizure will happen or for how long the seizure and recovery will last.
Experiences With Currently Available Treatments
A new drug could be the difference between becoming seizure free or if not available, to continue with having seizures.
Improved Outcomes
Severity of seizures, decrease in frequency of seizures, becoming seizure free due to new medical availability.
Companion Diagnostic Test
Not applicable.
Anything Else?
Any new drug the comes on the market for Seizures is one more step to the possibility of a patient becoming seizure free or have better control of their seizures. This would be a step in the right direction for the patient and give them hope of having their seizurs under better control allowing them to live a better life, a better quality of life. Unless one lives with having seizures, or works with patients having seizures, this could be life changing for the patient.
Conflict of Interest Declaration — Epilepsy South Central Ontario
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission?
No.
Did you receive help from outside your patient group to collect or analyze data used in this submission?
No.
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
Table 4: Financial Disclosures for Epilepsy South Central Ontario
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Epilepsy Association of Calgary
About Epilepsy Association of Calgary
Epilepsy Association of Calgary provides community-based programming and supports to adults, caregivers (including parents) and youth. Services include: one on one peer connections, registered psycho-educational programs (UpLift, PACES, Hobscotch), support groups, short-term counselling and an annual summer camp (Camp Fireworks). We partner with Edmonton Epilepsy Association to offer a monthly webinar series featuring experts in the field of neuroscience, epilepsy, and wellness.
Information Gathering
Information for this submission has been gathered by the Executive Director of Epilepsy Association of Calgary, drawing on the knowledge and experiences of front-line counselling staff (MSW, RSW), and a Needs and Service Gaps Assessment completed in 2021 drawing on the lived experiences and firsthand knowledge of patients, caregivers, clinicians, volunteers, and Association supporters (donors/funders).
Disease Experience
Current therapies work for nearly 70% of persons with Epilepsy. The remaining 30% try to remain hopeful that someday a medication will be found that will help them. New drugs like Cenobamate bring hope to many who are close to giving up. When epilepsy is not controlled by medications, the impact of the disease on individuals and their families is significant. Individuals with uncontrolled epilepsy can be socially isolated due to stigma due to fear of rejection in social, work, and educational situations. There is a high correlation of mental illness such as depression and anxiety that accompany and initial diagnosis and these can linger when anti-seizure medications do not provide relief.
When someone has epilepsy, the whole family is affected. Everyone’s life revolves around the seizures. There is anxiety around when and where the next seizure will occur, and what impact it will have. A husband is afraid his wife might have a seizure and drop the baby; parents are nervous if their child is invited to a birthday party; a teenager is anxious as he watches his father leave for work. Is today the day that something terrible will happen?
Some caregivers are afraid to leave the person with frequent seizures alone, contributing to a loss of independence and the lack of self-esteem we see so often with this patient population. Compassion fatigue in the care giver is always of concern. Many caregivers are sleep deprived as they either try to stay awake all night in case a seizure happens or go to bed and find they are too anxious to sleep.
As well, caregivers often have to live with the sometimes highly unpleasant side effects of various medications that their loved ones are taking. Mood swings, sexual dysfunction, suicidal thoughts, memory loss, problems with concentration, fatigue, exhaustion, depression – all can prove devastating to the person involved, and also to those around them.
Experiences With Currently Available Treatments
No drug works for everyone, and each new drug brought to market offers hope to the 30% of epilepsy sufferers whose seizures are uncontrolled by one or a combination of existing therapies. Even a reduction in the absolute number of seizures that these individuals experience can potentially improve overall quality of life. Seizure freedom may become a reality. Patients without seizure control are always hopeful that a new therapy lies around the corner and without access to safe, approved therapies, some begin to experiment with alternative medicines or practices (cannabis and other unregulated substances). This experimentation can ultimately prove not only detrimental, but also hazardous to the health of those affected.
Improved Outcomes
People with intractable epilepsy are very often unemployed or under-employed because of the frequency of their seizures. They usually live on very restricted incomes, and because they are not working, or are only working parttime, the majority is without access to employer-funded insurance plans. If new medications are not placed on the Provincial formulary, the majority of those with intractable epilepsy, the ones who need them the most, will never be given the opportunity to find out if this new drug will work for them.
Experience With Drug Under Review
These are questions we cannot answer at this time. We support the concept of a new drug that, through the results of trials, offers hope – that in itself offers improvement in an individual’s overall outlook, health and well-being.
Companion Diagnostic Test
Epilepsy Association of Calgary does not have sufficient data to comment on this at this time.
Conflict of Interest Declaration — Epilepsy Association of Calgary
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission?
Not applicable.
Did you receive help from outside your patient group to collect or analyze data used in this submission
Not applicable.
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
Not applicable.
Table 5: Financial Disclosures for Epilepsy Association of Calgary
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Edmonton Epilepsy Association
About Edmonton Epilepsy Association
Everything about our association can be found at our website: https://edmontonepilepsy.org/ We are a non-profit since 1960 and support the community impacted by epilepsy north of Red Deer, Alberta.
Information Gathering
The executive director of the EEA is completing the form on behalf of our association. She has experience in interacting with members and patients and their needs.
Disease Experience
Epilepsy impacts our members in all sorts of ways, from easy to cope seizures, that are either rare and small, like absence seizures or focal aware seizures that are manageable, to more complex seizures that are treated with medication but still impact the quality of life (QOL) of our members, to fewer cases where our members are in complete need of assistance, from financial support, to housing to daily care. We also help and support numerous families with children living with epilepsy.
Experiences With Currently Available Treatments
Our members do not disclose individual medication and treatments. These are confidential matters with their medical staff and support team. We do offer support in terms of connecting members with the specific providers, and to each other, especially in sharing their stories and experiences with living with seizures. We offer to this end, a mentoring program where we connect members to each other with similar stories to help one another cope better with life with seizures.
We also focus on safe social events for our members, where they do not fear of having a seizure in the middle of the event of with the group. We offer memory coaching programs and will soon start mental health sessions with professionals. We offer monthly education webinars in collaboration with Epilepsy Calgary, available at https://albertaeweb.ca/ – these have included sessions specific to drug managing solutions for epilepsy.
Improved Outcomes
We hear mostly from our members that improvement in side effects is their number one concern and hope. Additionally, interactions between drugs and drug efficacy are also a concern.
Experience With Drug Under Review
Not applicable.
Companion Diagnostic Test
Not applicable.
Conflict of Interest Declaration — Edmonton Epilepsy Association
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission?
Not applicable.
Did you receive help from outside your patient group to collect or analyze data used in this submission?
Not applicable.
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
Table 6: Financial Disclosures for Edmonton Epilepsy Association
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Clinician Input
The Canadian League Against Epilepsy
About The Canadian League Against Epilepsy
The Canadian League Against Epilepsy (CLAE) is a non-profit organization of health care and basic sciences professionals dedicated to the care and education of patients and their families living with epilepsy. CLAE counts with more than 125 members, including physicians, basic scientists, nurses, neuropsychologists, neuroradiologists, students and other health professionals. Our mission is to enable Canadians affected by epilepsy to live a life that is not limited by their condition.
Goals
Our goal is to develop, through research and education, innovative therapeutic and preventative strategies to avoid the consequences of epilepsy. We also aim to support translation of these discoveries into applicable therapies for all Canadians. Finally, we want to promote local, provincial, and national awareness and educate all Canadians about epilepsy and its consequences in collaboration with other entities such as non-professional associations.
Objectives
Maintain our level of excellence in epilepsy research and care by supporting National Priorities. Develop national campaigns to advocate for patients with epilepsy and educated our population; as well as collaborate in international campaigns such as the International League Against Epilepsy “Out of the Shadows” campaign. Realize a national event on a yearly basis to support epilepsy research and care nationwide. Continue to educate physicians and stakeholders across the country through our annual CLAE meeting.
For further information please refer to our website: https://claegroup.org/
Information Gathering
Discussed jointly via meetings and emails, manuscripts reported and experience from colleagues. CLAE Board Members hold frequent teleconferences monthly, and participants agreed to submit a single document to CADTH. The draft was prepared and signed by Dr. Juan Pablo Appendino. Comments and suggestions were incorporated from Board Members.
Current Treatments and Treatment Goals
Anti-seizure medications (ASMs) are the most common treatment used to treat focal and generalized seizures and are prescribed by physicians after a discussion with the patient or their care givers in respect of the risks and benefits of each appropriate medication. There are many different anti-seizure medications currently available in Canada and outside Canada. They differ in the mechanism of action, the potential side-effect profiles, the type of seizures they are best at treating (focal vs generalized vs epileptic spasms), and the costs. The medications do not cure epilepsy; instead, they prevent seizures from occurring. Patients typically start on a monotherapy and if seizures are not controlled, then a second ASM is tried or an adjunctive ASM is added to increase chances of seizure control.
The current available ASMs that could be used on a daily basis or as rescue trial for seizures/epilepsy in Canada are: Acetazolamide (Diamox), Briveracetam (Brivlera), Carbamazepine (Tegretol), Clobazam, Clonazepam (Klonopin), Diazepam (Valium), Divalproex Sodium (Epival), Eslicarbazepine (Aptiom), Ethosuximide (Zarontin), Gabapentin (Neurontin), Lacosamide (Vimpat), Lamotrigine (Lamictal), Levetiracetam (Keppra), Lorazepam (Ativan), Midazolam (Versed), Nitrazepam (Mogadon), Oxcarbazepine (Trileptal), Perampanel (Fycompa), Phenobarbital, Phenytoin (Dilantin), Pregabalin (Lyrica), Primidone (Mysoline), Rufinimide (Banzel), Stiripentol (Diacomit), Topiramate (Topamax),Valproic Acid (Depakene) and Vigabatrin (Sabril). Some are preferred for focal seizures, and some are preferred for generalized seizures.
Felbamate (Felbatol), Fenfluramine (Fintepla), Ganaxolone (Ztalmy), Sulthiame (Ospolot), and Zonisamide (Zonegran) are not approved in Canada, as far as we are aware, but can be obtained through special access programs (SAPs) for some patients. Approximately up to 2 – 5% of patients with drug-resistant epilepsy may require one of these drugs through SAP.
In patients who do not respond to ASMs, other therapeutic avenues are pursued including surgery and alternative measures such as neuromodulation, ketogenic diet, Immunoglobulins monthly infusions, and cannabinoids, each with its own advantages and disadvantages. Not every patient is a candidate for these alternative measures though.
References
Browne TR. Drug therapy reviews: clinical pharmacology of antiepileptic drugs. Am J Hosp Pharm. 1978;35:1048–1056.
Shorvon S, Perucca E, Engel JJ (2020) The treatment of epilepsy. Third edition. In: Shorvon S, Perucca E, Engel JJ (eds) (Oxford: Wiley-Blackwell).
Porter RJ, Dhir A, Macdonald RL, Rogawski MA (2012). Mechanisms of action of antiseizure drugs. In: Stefan H, Theodore WH, editors. Handbook of clinical neurology, vol. 108. Amsterdam: Elsevier. pp. 663–681.
Devinsky O, Vezzani A, O'Brien TJ, Jette N, Scheffer IE, De Curtis M, Perucca P. Epilepsy. Nat Rev Dis Prim. 2018;4:18024. doi: 10.1038/nrdp.2018.24.
The most important goals in treating seizures/epilepsy are:
Increase chances for seizure freedom.
improve health-related quality of life.
increase the ability to maintain employment.
maintain independence.
reduce burden on caregivers.
prevent premature death and SUDEP (sudden, unexpected death of someone with epilepsy)
avoid (or postpone as far possible) delayed remission.
reduce drug load and adverse events.
avoid invasive therapies (surgery)
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
Epilepsy remains a large burden on patients and their families as many patients are still experiencing seizures. Despite the current availability of several ASMs, there has been no meaningful improvement in epilepsy treatment-related outcomes and no significant increase in seizure freedom rates in the past 20 years. More than one-third of treated patients do not respond to ASMs and continue to have uncontrolled seizures, further increasing the risks associated with epilepsy. Recently introduced ASMs into the market have not had a meaningful dent in the seizure burden and recurrence of seizures in patients with epilepsy, although have shown to be better tolerated than old ASMs with a higher compliance rate. Novel ASMs are needed for this group of patients that are still uncontrolled and experiencing frequent seizures. Patients are not able to try all available ASMs in the market and patient-specific factors need to be considered when choosing the correct medication for a particular patient, including etiology, age, biological gender, comorbidities, affordability, professional activities, and concomitant medications; therefore, opting for the best available drug with less side effects and smaller number needed-to-treat (NTT) is perhaps the most critical decision when advising these patients. Lastly, it’s important to find a medication that works for this group of patients because exposing them to multiple ASM regimens frequently increases the risk of experiencing adverse events.
A novel medication with increased chances for patients to improve seizure control or even become seizure free is needed. There is some expectation of reaching this with cenobamate due to its proven efficacy in clinical trials and showing the best NTT number among many new ASMs[1].
Reference
Villanueva, V., Serratosa, J. M., Toledo, M., Ángel Calleja, M., Navarro, A., Sabaniego, J., Pérez-Domper, P., Álvarez-Barón, E., Subías, S., & Gil, A. (2023). Number needed to treat and associated cost analysis of cenobamate versus third-generation anti-seizure medications for the treatment of focal-onset seizures in patients with drug-resistant epilepsy in Spain. Epilepsy & behavior : E&B, 139, 109054. https://doi.org/10.1016/j.yebeh.2022.109054
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Cenobamate has a unique dual complementary mechanism of action. It enhances inhibitory currents through GABA-A receptor modulation and decreases excitatory currents by inhibiting the persistent component of the sodium current and enhancing the inactivated state of voltage-gated sodium channels. Cenobamate is the only ASM which, at clinically relevant concentrations, acts as a positive allosteric modulator of GABA-A receptors at non-benzodiazepine binding sites and preferentially blocks the persistent sodium current [1,2] Cenobamate’s unique, dual mechanism of action (MOA) has the potential to both prevent seizure initiation and limit seizure spread [3-8] offering an important advancement in drug development for the treatment of patients with uncontrolled seizures [9]. Other ASMs with similar dual MOA are not available in Canada.
Cenobamate will likely be used in combination with other available treatments (as an add-on). It is unlikely to be used as a monotherapy; however, if it proves to prevent seizures once added, occasionally physicians will try to minimize on-going treatment and wean off other ASMs. This practice could lead Cenobamate to be a sole treatment for specific patients.
Cenobamate offers a higher degree of effectiveness when compared with other novel ASMs such as Perampanel, Eslicarbazepine, Lacosamide, and Brivaracetam [13]. The expectations are set high in regards of its positive results in clinical trials.
References
Nakamura M, Cho JH, Shin H, Jang IS. Effects of cenobamate (YKP3089), a newly developed anti-epileptic drug, on voltage-gated sodium channels in rat hippocampal CA3 neurons. Eur J Pharmacol. Jul 15 2019;855:175-182. doi:10.1016/j.ejphar.2019.05.007
Sharma R, Nakamura M, Neupane C, et al. Positive allosteric modulation of GABA(A) receptors by a novel antiepileptic drug cenobamate. Eur J Pharmacol. Jul 15 2020;879:173117. doi:10.1016/j.ejphar.2020.173117
Anderson LL, Thompson CH, Hawkins NA, et al. Antiepileptic activity of preferential inhibitors of persistent sodium current. Epilepsia. Aug 2014;55(8):1274-83. doi:10.1111/epi.12657
Piredda SG, Woodhead JH, Swinyard EA. Effect of stimulus intensity on the profile of anticonvulsant activity of phenytoin, ethosuximide and valproate. J Pharmacol Exp Ther. Mar 1985;232(3):741-5.
Stafstrom CE. Persistent sodium current and its role in epilepsy. Epilepsy Curr. Jan-Feb 2007;7(1):15-22. doi:10.1111/j.1535-7511.2007.00156.x
Vreugdenhil M, Hoogland G, van Veelen CW, Wadman WJ. Persistent sodium current in subicular neurons isolated from patients with temporal lobe epilepsy. Eur J Neurosci. May 2004;19(10):2769-78. doi:10.1111/j.1460-9568.2004.03400.x
White HS, Brown SD, Woodhead JH, Skeen GA, Wolf HH. Topiramate modulates GABA-evoked currents in murine cortical neurons by a nonbenzodiazepine mechanism. Epilepsia. 2000;41(S1):17-20.
Smith MC, Klein P, Krauss GL, et al. Dose Adjustment of Concomitant Antiseizure Medications During Cenobamate Treatment: Expert Opinion Consensus Recommendations. Neurol Ther. Dec 2022;11(4):1705-1720. doi:10.1007/s40120-022-00400-5
Guignet M, Campbell A, White HS. Cenobamate (XCOPRI): Can preclinical and clinical evidence provide insight into its mechanism of action? Epilepsia. Nov 2020;61(11):2329-2339. doi:10.1111/epi.16718
Chung SS, French JA, Kowalski J, Krauss GL, Lee SK, Maciejowski M, Rosenfeld WE, Sperling MR, Mizne S, Kamin M. Randomized phase 2 study of adjunctive cenobamate in patients with uncontrolled focal seizures. Neurology. 2020 Jun 2;94(22):e2311-e2322. doi: 10.1212/WNL.0000000000009530. Epub 2020 May 14. PMID: 32409485; PMCID: PMC7357293.
Krauss GL, Klein P, Brandt C, Lee SK, Milanov I, Milovanovic M, Steinhoff BJ, Kamin M. Safety and efficacy of adjunctive cenobamate (YKP3089) in patients with uncontrolled focal seizures: a multicentre, double-blind, randomised, placebo-controlled, dose-response trial. Lancet Neurol. 2020 Jan;19(1):38-48. doi: 10.1016/S1474-4422(19)30399-0. Epub 2019 Nov 14. Erratum in: Lancet Neurol. 2020 Mar;19(3):e3. PMID: 31734103.
Aboumatar S, Biton V, Wechsler R, Ferrari L, Rosenfeld WE. Post hoc analysis of a phase 3 study for treatment of uncontrolled focal seizures: Adjunctive cenobamate dose and seizure reduction by baseline seizure frequency. Epilepsy Res. 2022 Oct;186:107014. doi: 10.1016/j.eplepsyres.2022.107014. Epub 2022 Aug 27. PMID: 36063589.
Villanueva, V., Serratosa, J. M., Toledo, M., Ángel Calleja, M., Navarro, A., Sabaniego, J., Pérez-Domper, P., Álvarez-Barón, E., Subías, S., & Gil, A. (2023). Number needed to treat and associated cost analysis of cenobamate versus third-generation anti-seizure medications for the treatment of focal-onset seizures in patients with drug-resistant epilepsy in Spain. Epilepsy & behavior : E&B, 139, 109054. https://doi.org/10.1016/j.yebeh.2022.109054
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
In clinical trials cenobamate demonstrated a high degree of efficacy in reducing seizures and an unprecedented seizure-free rate in patients regardless of the baseline seizure frequency, epilepsy duration and the number of concomitant ASMs at baseline. Thus, suitable candidates for cenobamate treatment should be any patient older than 18 yo with uncontrolled focal onset seizures. These patients would have most likely tried other ASMs before trying Cenobamate until physicians become more comfortable recommending and managing this medication. In the future, it is likely Cenobamate will be used by pediatric epileptologists in younger patients as it has happened with other ASMs.
It is possible that in the future, if Cenobamate proves to be as effective and well tolerated as clinical trials claim to be, it will be used as a first or second option ASM in the treatment of seizures/epilepsy.
Epilepsy is unusually misdiagnosed as there are clinical and neurophysiological tests (i.e. electroencephalography) to prove the existence of the disease. Therefore, a misdiagnosis and mis-utilization of Cenobamate is unlikely.
References
SK Life Science, Inc. XCOPRI® (cenobamate tablets), for oral use, CV [prescribing information]. Paramus, NJ: SK Life Science, Inc.; April, 2021.
S.S. Chung, J.A. French, J. Kowalski, G.L. Krauss, S.K. Lee, M. Maciejowski, et al.Randomized phase 2 study of adjunctive cenobamate in patients with uncontrolled focal seizures
G.L. Krauss, P. Klein, C. Brandt, S.K. Lee, I. Milanov, M. Milovanovic, et al.Safety and efficacy of adjunctive cenobamate (YKP3089) in patients with uncontrolled focal seizures: a multicentre, double-blind, randomised, placebo-controlled, dose-response trial
W.E. Rosenfeld, A. Nisman, L. Ferrari Efficacy of adjunctive cenobamate based on number of concomitant antiseizure medications, seizure frequency, and epilepsy duration at baseline: a post-hoc analysis of a randomized clinical study Epilepsy Res, 172 (2021
Aboumatar S, Biton V, Wechsler R, Ferrari L, Rosenfeld WE. Post hoc analysis of a phase 3 study for treatment of uncontrolled focal seizures: Adjunctive cenobamate dose and seizure reduction by baseline seizure frequency. Epilepsy Res. 2022 Oct;186:107014. doi: 10.1016/j.eplepsyres.2022.107014. Epub 2022 Aug 27. PMID: 36063589.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
The aim of therapy in patients with epilepsy is total seizure freedom without clinically significant adverse effects. If seizure freedom is not possible, reducing seizure frequency to the lowest level possible will improve patients’ quality of life. Certain types of epilepsy are more dangerous than others, for example, focal to bilateral tonic-clonic seizures are considered one of the major risk factors of injuries related with seizures and even SUDEP, thus controlling any type of seizures but particularly focal onset seizures is extremely important. Reducing the frequency of these dangerous seizures is also a clinically meaningful outcome.
What factors should be considered when deciding to discontinue treatment with the drug under review?
Lack of efficacy, occurrence of unbearable side effects that don’t resolve by dose adjustments.
What settings are appropriate for treatment with [drug under review]? Is a specialist required to diagnose, treat, and monitor patients who might receive [drug under review]?
Cenobamate is a treatment for epilepsy in a community or hospital settings. Neurologists with experience in treating patients with epilepsy can prescribe it.
Additional Information
As a medical provider and as a patient with epilepsy, the more tools we have available in the market to help our patients in controlling their seizures, the more empowered we feel. The availability of Cenobamate into the market has created an expectation among the epilepsy community of a brighter future on seizure control.
Conflict of Interest Declarations for The Canadian League Against Epilepsy
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict-of-interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please refer to the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
Some of the references and content was originally provided by members of the Endo Pharmaceutical Company (https://www.endo.com/) and Paladin Pharmaceutical (https://www.paladin-labs.com/); however, the form was edited in full by Juan Pablo Appendino and reflects what the CLAE thinks about this medication.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
Some of the references and content was originally provided by members of the Endo Pharmaceutical Company (https://www.endo.com/) and Paladin Pharmaceutical (https://www.paladin-labs.com/); however, the form was edited in full by Juan Pablo Appendino and reflects what the CLAE thinks about this medication.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
None.
Declaration for Clinician 1
Name: Juan Pablo Appendino
Position: Pediatric Epileptologist – Associated Professor at University of Calgary. Leader of the Medical Therapeutic Committee at the Canadian League Against Epilepsy
Date: 26-02-2023
Table 7: COI Declaration for The Canadian League Against Epilepsy (CLAE) — Clinician 1
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.