CADTH Reimbursement Review
Foslevodopa-Foscarbidopa (Vyalev)
Sponsor: AbbVie Corporation
Therapeutic area: Parkinson disease
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
AE
adverse event
ANCOVA
analysis of covariance
BMT
best medical therapy
CD
carbidopa
CI
confidence interval
CrI
credible interval
COMT
catechol-O-methyltransferase
CSCI
continuous subcutaneous infusion
C-SSRS
Columbia-Suicide Severity Rating Scale
DBS
deep brain stimulation
DIC
deviance information criterion
EQ-5D-5L
5-Level EQ-5D
FE
fixed effects
HRQoL
health-related quality of life
IQR
interquartile range
IR
immediate release
ITC
indirect treatment comparison
LCIG
levodopa-carbidopa intestinal gel
LD
levodopa
LED
levodopa-equivalent dose
LSM
least squares mean
MAO-B
monoamine oxidase type B
MDS-UPDRS
Movement Disorder Society-Unified Parkinson's Disease Rating Scale
MID
minimally important difference
MMRM
mixed model for repeated measures
MMSE
Mini-Mental State Examination
NMA
network meta-analysis
PD
Parkinson disease
PDQ-39
Parkinson's Disease Questionnaire-39 items
PDSS-2
Parkinson's Disease Sleep Scale-2
PEG-J
percutaneous endoscopic gastrostomy-jejunostomy
PKG
Parkinson's KinetiGraph/Personal KinetiGraph
PRO
patient-reported outcome
QUIP-RS
Questionnaire for Impulsive-Compulsive Disorders in Parkinson’s Disease – Rating Scale
RCT
randomized controlled trial
RE
random effects
SD
standard deviation
SLR
systematic literature review
TEAE
treatment-emergent adverse event
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Table 1: Background Information of Application Submitted for Review
Item | Description |
|---|---|
Information on drug submitted for review | |
Drug product | Foslevodopa-foscarbidopa (Vyalev), 240 mg/mL foslevodopa and 12 mg/mL foscarbidopa solution, subcutaneous infusion |
Sponsor | AbbVie Corporation |
Indication | For the treatment of motor fluctuations in patients with advanced levodopa-responsive Parkinson’s disease who do not have satisfactory control of severe, debilitating motor fluctuations and hyper- /dyskinesia despite optimized treatment with available combinations of Parkinson’s medicinal products |
Reimbursement request | For the treatment of motor fluctuations in patients with Parkinson’s disease who are not adequately controlled on optimized oral therapies (advanced Parkinson’s disease) and who are not candidates for deep brain stimulation |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | May 3, 2023 |
Recommended dose | Dosing is individualized, and the infusion rate of foslevodopa-foscarbidopa is calculated during the titration period and is informed by the patient’s current use of levodopa and other medications for Parkinson disease1 |
NOC = Notice of Compliance.
Introduction
Parkinson disease (PD) is a progressive, neurologic disease2,3 characterized by the dysfunction and loss of dopaminergic cells in the substantia nigra and other brain regions.2 Patients with PD experience motor symptoms such as bradykinesia, tremor, rigidity, and postural instability, as well as nonmotor symptoms including cognitive impairment, mood disorders, and sleep problems.4,5 It is the most common movement disorder, with estimated age-standardized incidence rates ranging from 108 to 212 per 100,000 among patients aged 65 years and older in North America.6 Approximately 10% to 20% of patients with PD do not achieve satisfactory control of their disease despite optimized oral treatment, indicating that their disease has progressed to advanced PD.7,8 There is no universal consensus on a definition of advanced PD; the Delphi-based consensus recommendation9 and a simplified approach known as the “5-2-1” criteria10 are commonly used in clinical practice to guide identification of patients with advanced PD.
Oral therapy, and advanced device-aided therapies, including deep brain stimulation (DBS) and levodopa-carbidopa intestinal gel (LCIG), are currently available for the treatment of motor fluctuations in patients with levodopa-responsive advanced PD.11 Most patients rely on optimization of oral therapy, which typically involves changes in dosage, dose frequency, or combinations of oral medications with different mechanisms of action, to control motor symptoms. While such adjustments can reduce medication response fluctuations, they increase the burden and complexity of oral medication use, and can require patients to take medication every few hours. LCIG, a continuous infusion of levodopa (LD) and carbidopa (CD) suspension into the small intestine, and DBS, which involves implanting electrodes within targeted areas of the brain that control movement, are effective in reducing “off” time (i.e., a period of uncontrolled PD symptoms despite treatment) and dyskinesia (i.e., uncontrolled and involuntary movements due to excess treatment effect) during “on” time (i.e., a period of controlled PD symptoms with treatment);11 however, both are invasive treatments that require a specialized medical team to perform the procedure, monitor, and manage associated adverse events (AEs) and complications (e.g., device failure, infection risk).12 DBS treatment is also only provided at specialized centres and is only appropriate in select patients without contraindications due to associated risks and potentially life-threatening AEs (e.g., intracerebral hemorrhage, stroke, infection, seizure induced by the surgery, and off-target stimulation effects such as changes in speech or freezing of gait).
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of foslevodopa (240 mg/mL) and foscarbidopa (12 mg/mL) solution for subcutaneous infusion in the treatment of motor fluctuations in patients with advanced PD.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient and clinician groups who responded to CADTH’s call for input and from clinical expert(s) consulted by CADTH for the purpose of this review.
Patient Group Input
CADTH received 1 input from Parkinson Association of Alberta and 1 joint input from Parkinson Canada, Parkinson Society British Columbia, and Parkinson Quebec. Parkinson Association of Alberta conducted a survey of 26 patients with PD and care partners or family in Alberta. Parkinson Canada, Parkinson Society British Columbia, and Parkinson Quebec jointly gathered responses from 113 patients with PD and caregivers of patients with PD in Canada via a survey; the majority of respondents were from Ontario (72.6%).
According to both patient groups, “off” periods and motor fluctuations associated with PD substantially impacted quality of life and activities of daily living for patients, led to work absenteeism (and in some cases resulted in early retirement), and caused emotional and financial burden to the caregivers. Respondents from both patient groups noted that symptoms that are most important to control are changes in cognition and memory, fatigue and sleep issues, freezing and unpredictable “off” periods, changes in mood, rigidity, speech and swallowing issues, bladder and bowel issues, impaired balance, slowness, and tremors.
More than half of each patient group experienced side effects with taking oral medications, with fatigue, drowsiness, constipation, and bowel issues the most difficult to endure. More than half of the respondents to the joint input reported that high pill burden (up to 40 pills per day) impacted their lifestyle or quality of life. Difficulties related to medication adherence included difficulty with timing or remembering, swallowing, and storage of medications, and limited improvement of symptoms. Some patients also received some form of rehabilitation (physiotherapy, occupational/speech therapy, or exercise) as a treatment option, but respondents cited cost, lack of motivation, or lack of access as barriers, especially for patients in rural areas. No respondents from either patient group were receiving foslevodopa-foscarbidopa at the time of survey.
Respondents indicated that the most important unmet needs were treatment options that would not increase dyskinesia as time went on, medications that would treat cognitive issues, and longer-lasting medications that would reduce pill burden and OFF periods, eliminating the fluctuations and sleep interruptions caused by medications wearing off. The joint input indicated that a large proportion of patients were very reluctant about undergoing invasive treatment options such as DBS or LCIG, and the majority (65%) would be interested in an injection-based levodopa-carbidopa (LD-CD) treatment; however, only 1 (3.8%) respondent from Parkinson Alberta said they would consider it and 2 (7.7%) were unsure.
Clinician Input
Input From the Clinical Expert Consulted by CADTH
The clinical expert consulted by CADTH noted that there is an unmet need for treatment options that have less resource requirements such that treatment is accessible to patients, especially those residing in rural and remote areas, without the need to travel to major urban centres, as well as treatment options for patients who are ineligible for existing advanced therapies because of existing comorbidities. The clinical expert noted that foslevodopa-foscarbidopa could serve as a treatment option for patients with advanced PD and could fill a treatment gap for patients who cannot travel to access other advanced therapies or who have comorbidities or strong personal aversion to other options.
The clinical expert noted that patients with levodopa-responsive advanced PD would be considered eligible for foslevodopa-foscarbidopa treatment in clinical settings. The clinical expert noted that there is currently no universally agreed-upon definition for advanced PD, and that it would be appropriate to define advanced PD based on the Delphi-based consensus criteria or the “5-2-1” criteria or as “patients with PD who have motor fluctuations inadequately controlled by optimized oral therapy.” Patients with excessive “off” time or “on” time with bothersome dyskinesia are more likely to benefit from treatment, according to the clinical expert, while patients with levodopa-unresponsive symptoms are not expected to benefit from foslevodopa-foscarbidopa treatment as the system of delivery is dopamine precursor treatment. The clinical expert noted that a clinically meaningful response would include improvement in “on” and “off” time measurements and quality of life, which would typically be observed at 3 months after initiation. According to the clinical expert, clinically meaningfulness can be judged differently by treating neurologists and patients, for instance in the predictability of therapy or the flexibility patients have with longer continuous “on” periods; as such, a meaningful response may be best left to the discretion of the treating neurologist. The clinical expert also noted that treatment discontinuation could be considered when patients experience intolerable AEs or significant functional impairments that are not relieved by the treatment. The drug should be prescribed by neurologists who have experience in the treatment of patients with PD and are trained in the use of this drug, as per the clinical expert.
Clinician Group Input
Input was received from the National Movement Disorder Expert Group (11 clinicians), and the BC Movement Disorders Specialist Group (7 clinicians). Overall, the input from both clinician groups aligned with the input given by the clinical expert consulted by CADTH.
The clinician groups agreed about the unmet needs of patients with advanced PD. Patients receiving oral levodopa may have inadequate control of motor fluctuations despite increased dosing frequency over time, and may have contraindications, poor tolerance, or insufficient response to adjunctive medications. They described barriers to accessing advanced therapies for PD (i.e., DBS and LCIG treatments) due to limited numbers of specialists, uneven distribution of resources geographically, intense resource needs, medical contraindications, poor acceptance from patients because of the invasive nature and risks of the treatments, and the impact of PD itself on patients’ ability to travel long distances for DBS or LCIG treatment and to manage at-home aspects of LCIG treatment. The clinician groups also noted that no current treatments exist that address the underlying disease process of PD.
The clinician groups agreed that foslevodopa-foscarbidopa could serve as an additional treatment option for patients with advanced PD and could benefit patients experiencing bothersome end-of-dose “off” periods, unpredictable efficacy of oral therapies as a result of absorption delays, or complex oral medication schedules due to the subcutaneous delivery of foslevodopa-foscarbidopa.
The clinician groups indicated that, as with other existing advanced therapies, eligible patients would include those who have levodopa-responsive PD with bothersome motor and nonmotor fluctuations despite optimized oral therapies. They suggested that eligible patients have advanced PD, according to the “5-2-1” criteria, and that it would be reasonable to recommend first trying at least 1 monoamine oxidase type B (MAO-B) inhibitor and a catechol-O-methyltransferase (COMT) inhibitor, unless contraindicated. In cognitively intact patients aged 70 years or younger, the clinician groups also stated that it would be reasonable to recommend having tried at least 1 dopamine agonist and amantadine (if dyskinesia is bothersome), unless contraindicated. However, they suggested against requiring a previous trial of anticholinergics or apomorphine preparations for reimbursement of foslevodopa-foscarbidopa. The clinician groups agreed with the clinical expert that treatment response would be assessed based on “off” time, presence of disabling dyskinesia, and quality of life. They added that an association with easing of caregiver burden may be considered. The clinician groups agreed that discontinuation could be considered in patients with intolerable AEs (e.g., skin reactions or hallucinations) and those who are unable to use the pump correctly due to cognitive decline as a result of disease progression or lack of caregiver support. The clinician groups agreed with the clinical experts that movement disorder neurologists, general neurologists, and geriatricians with experience in treating PD could be comfortable and qualified to prescribe and maintain treatment with foslevodopa-foscarbidopa.
Drug Program Input
Input was obtained from the drug programs that participate in the CADTH reimbursement review process. The following were identified as key factors that could potentially impact the implementation of a CADTH recommendation for foslevodopa-foscarbidopa:
relevant comparators
consideration for initiation of therapy
consideration for continuation or renewal of therapy
consideration of discontinuation of therapy
consideration for prescribing of therapy
care provision issues
system and economic issues.
The clinical experts consulted by CADTH provided advice on the potential implementation issues raised by the drug programs (refer to Table 5).
Clinical Evidence
Pivotal Studies and Randomized Controlled Trial Evidence
Description of Studies
One pivotal phase III, double-blind, double-dummy randomized controlled trial (RCT) (M15-736,13 N = 141) that assessed whether individualized foslevodopa-foscarbidopa continuous subcutaneous infusion (CSCI) increased change from baseline in average daily normalized “on” time without troublesome dyskinesia compared to oral LD-CD immediate-release (IR) tablet therapy, after 12 weeks in patients with PD who have motor fluctuations inadequately controlled by oral therapy, was included in the sponsor’s submission. Patients with prior DBS or LCIG treatment were excluded, and eligibility for DBS was not a consideration for enrolment. Study-defined key secondary end points included change from baseline in average daily normalized “off” time, Movement Disorder Society-Unified Parkinson’s Disease Rating Scale (MDS-UPDRS) Part II score, and presence of morning akinesia. Secondary end points included “on” time without dyskinesia and other measures of symptoms and health-related quality of life (HRQoL) (Parkinson’s Disease Questionnaire-39 items [PDQ-39], 5-Level EQ-5D [EQ-5D-5L], median and interquartile range [IQR] of bradykinesia and dyskinesia scores assessed using a Parkinson KinetiGraph/Personal KinetiGraph [PKG] device, and Parkinson’s Disease Sleep Scale-2 [PDSS-2]).
At baseline, the mean age of patients was 66.4 years (standard deviation [SD] = 9.5 years) and the majority were male and white. The mean time since PD diagnosis was 8.6 years (SD = 4.9 years). Mean time spent in “off” and “on” without troublesome dyskinesia motor states were 6.13 hours (SD = 2.097 hours) and 9.34 hours (SD = 2.514 hours), respectively.
Efficacy Results
The efficacy end points that were noted to be important to patients and clinicians based on stakeholder input are summarized in Table 2.
“On” Time Without Troublesome Dyskinesia
The least squares mean (LSM) difference between the foslevodopa-foscarbidopa arm and the oral LD-CD arm with respect to change from baseline to week 12 in average daily normalized “on” time without troublesome dyskinesia (primary end point) was 1.75 (95% confidence interval [CI], 0.46 to 3.05; P = 0.0083) hours, in favour of foslevodopa-foscarbidopa. Results of the sensitivity analyses assessing the impact of attrition and results of subgroup analyses of interest (age, duration of PD diagnosis, and levodopa dose intensity) were consistent with the primary analysis.
“Off” Time
The LSM difference between the foslevodopa-foscarbidopa arm and the oral LD-CD arm with respect to change from baseline to week 12 in average daily normalized “off” time (study-defined key secondary end point) was –1.79 (95% CI, –3.03 to –0.54; P = 0.0054) hours, in favour of foslevodopa-foscarbidopa. Results of the sensitivity analyses assessing the impact of attrition and subgroup analyses of interest were consistent with the primary analysis.
PDQ-39 Items (PD-Specific HRQoL Instrument)
The LSM difference between the foslevodopa-foscarbidopa arm and the oral LD-CD arm with respect to change from baseline to week 12 in PDQ-39 summary index (secondary end point) was –4.10 (95% CI, –8.14 to –0.05). The results for this outcome are at increased risk of type I error (false-positive results) because they were tested after failure of the statistical hierarchy.
MDS-UPDRS Part II Score (Motor Experiences of Daily Living)
The LSM difference between the foslevodopa-foscarbidopa arm and the oral LD-CD arm with respect to change from baseline to week 12 in MDS-UPDRS Part II score (study-defined key secondary outcome) was –1.58 (95% CI, –3.65 to 0.48; P = 0.13).
Parkinson’s Disease Sleep Scale-2
The LSM difference between the foslevodopa-foscarbidopa arm and the oral LD-CD arm in change from baseline to week 12 in PDSS-2 total score (secondary end point) was –5.40 (95% CI, –8.03 to –2.78). The results for this outcome are at increased risk of type I error (false-positive results) because they were tested after failure of the statistical hierarchy.
Table 2: Summary of Key Efficacy Results from the M15-736 Trial (FAS)
Treatment arms | Number of patients contributing to the analysis, n (%) | Baseline value, mean (SD) | Change from baseline at week 12, LSM (SE) | Difference in LSM (95% CI) | P value |
|---|---|---|---|---|---|
Average daily normalized “on” Time without Troublesome Dyskinesia (hours)a | |||||
FOS-FOS | 47 (63.5) | 9.20 (2.42) | 2.72 (0.52) | 1.75 (0.46 to 3.05) | 0.0083 |
Oral LD-CD | 62 (92.5) | 9.49 (2.62) | 0.97 (0.50) | Reference | Reference |
Average daily normalized “off” time (hours)a | |||||
FOS-FOS | 47 (63.5) | 6.34 (2.27) | –2.75 (0.50) | –1.79 (–3.03 to –0.54) | 0.0054 |
Oral LD-CD | 62 (92.5) | 5.91 (1.88) | –0.96 (0.49) | Reference | Reference |
PDQ-39 (PD-related HRQoL)b | |||||
FOS-FOS | 45 (60.8) | 29.31 (15.84) | –6.38 (1.83) | –4.10 (–8.14 to –0.05) | 0.047c |
Oral LD-CD | 59 (88.1) | 26.52 (13.89) | –2.28 (1.75) | Reference | Reference |
MDS-UPDRS Part II score (motor experiences of daily living)a | |||||
FOS-FOS | 46 (62.2) | 15.31 (6.93) | –2.65 (0.82) | –1.58 (–3.65 to 0.48) | 0.13 |
Oral LD-CD | 62 (92.5) | 13.27 (6.37) | –1.06 (0.79) | Reference | Reference |
PDSS-2 total score (sleep symptoms)b | |||||
FOS-FOS | 44 (59.5) | 21.7 (9.04) | –7.92 (1.18) | –5.40 (–8.03 to –2.78) | ≤ 0.001c |
Oral LD-CD | 59 (88.1) | 18.7 (8.77) | –2.52 (1.12) | Reference | Reference |
CD = carbidopa; CI = confidence interval; FAS = full analysis set; FOS-FOS = foslevodopa-foscarbidopa; HRQoL = health-related quality of life; LD = levodopa; LSM = least squares mean; MDS-UPDRS = Movement Disorder Society-Unified Parkinson’s Disease Rating Scale; PD = Parkinson disease; PDQ-39 = Parkinson’s Disease Questionnaire-39 items; PDSS-2 = Parkinson’s Disease Sleep Scale-2; SD = standard deviation.
Note: Outcomes summarized in this table includes the primary and secondary end points that were noted to be important to patients and clinicians based on input from patient groups, clinician groups, and the clinical expert consulted by CADTH.
aThe analysis was conducted using a mixed model for repeated measures, adjusted for categorical fixed effects of treatment, country and visit, treatment-by visit and treatment-by-baseline interactions, and baseline measurement (continuous).
bThis analysis was conducted using an analysis for covariate model, adjusted for categorical fixed effects of treatment and country, and baseline score (continuous).
cAlthough the P value is ≤ 0.05, statistical significance cannot be claimed because the results for the second key secondary end point (MDS-UPDRS Part II), a prior end point in the testing hierarchy, were not statistically significant.
Source: M15-736 Clinical Study Report.14 (Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
Harms
The key harms results from the M15-736 trial are summarized in Table 3.
Adverse Events
Treatment-emergent adverse events (TEAEs) were reported for 85.1% of patients in the foslevodopa-foscarbidopa arm and 62.7% of patients in the oral LD-CD arm. The most common TEAEs in the foslevodopa-foscarbidopa arm (at least 10%) were infusion-site erythema, pain, cellulitis, and edema, as well as dyskinesia, all of which were more commonly reported than in the oral LD-CD arm (infusion-site erythema and pain, 1.5% each). The frequency of falls was lower in the foslevodopa-foscarbidopa arm (8.1%) than in the oral LD-CD arm (17.9%).
Serious Adverse Events
Serious TEAE was reported for 6 (8.1%) patients in the foslevodopa-foscarbidopa arm and 4 (6.0%) patients in the oral LD-CD arm.
Withdrawals Due to AEs
Treatment discontinuation due to TEAEs was reported for 21.6% of patients in the foslevodopa-foscarbidopa arm and 1.5% of patients in the oral LD-CD arm. The most common TEAEs leading to treatment discontinuation in the foslevodopa-foscarbidopa arm were infusion-site cellulitis (5.4%), infusion-site pain (4.1%), infusion-site bruising, hemorrhage, and edema (2.7% each).
Mortality
No deaths were reported in the foslevodopa-foscarbidopa arm and 1 (1.5%) death was reported in the oral LD-CD arm.
Notable Harms
The frequencies of infusion-site reactions and infections were notably higher in the foslevodopa-foscarbidopa arm than in the oral LD-CD arm (infusion-site reactions: 62.2% versus 7.5%; infusion-site infections: 28.4% versus 3.0%).
The frequency of hallucination or psychosis was notably higher in the foslevodopa-foscarbidopa arm (14.9%) than in the oral LD-CD arm (3.0%). There were no reports of impulse-control disorder or impulsive behaviour in either treatment arm. There was no notable between-arm difference in the mean change from baseline in score for each impulse-control disorder and related behaviour parameters of the Questionnaire for Impulsive-Compulsive Disorders in Parkinson’s Disease – Rating Scale (QUIP-RS) across almost all time points. Based on the Columbia-Suicide Severity Rating Scale (C-SSRS) assessment, 5 (6.8%) patients in the foslevodopa-foscarbidopa arm and 2 (3.0%) patients in the oral LD-CD arm had suicidal behaviours or ideations. Depression was reported for no patients in the foslevodopa-foscarbidopa arm and 2 (3.0%) patients in the oral LD-CD arm.
Dizziness was reported for 3 patients in each treatment arm. Orthostatic hypotension by preferred term was reported for 1 (1.4%) patient in the foslevodopa-foscarbidopa arm and 2 (3.0%) patients in the oral LD-CD arm. Somnolence was reported for 1 (1.4%) patient in both treatment arms.
Table 3: Summary of Key Harms From the M15-736 Trial (SAS)
Harms, n (%) | Foslevodopa-foscarbidopa arm (N = 74) | Oral LD-CD arm (N = 67) |
|---|---|---|
TEAE | 63 (85.1) | 42 (62.7) |
Serious TEAE | 6 (8.1) | 4 (6.0) |
Withdrawal from treatment due to TEAE | 16 (21.6) | 1 (1.5) |
Death | 0 | 1 (1.5) |
Notable harms | ||
Infusion-site reactionsa | 46 (62.2) | 5 (7.5) |
Infusion-site infections | 21 (28.4) | 2 (3.0) |
Hallucination/psychosisb | 11 (14.9) | 2 (3.0) |
Suicidal behaviours or ideationsc | 5 (6.8) | 2 (3.0) |
Dizziness | 3 (4.1) | 3 (4.5) |
Orthostatic hypotension | 1 (1.4) | 2 (3.0) |
Somnolence | 1 (1.4) | 1 (1.5) |
Depression | 0 | 2 (3.0) |
Impulse-control disorder | 0 | 0 |
Impulsive behaviour | 0 | 0 |
CD = carbidopa; LD = levodopa; SAS = safety analysis set; TEAE = treatment-emergent adverse event.
Note: Unless otherwise specified, safety data are reported using the Medical Dictionary for Regulatory Activities (MedDRA) preferred term.
aAdverse event of special interest, including any adverse event in infusion site–related noninfection reactions company MedDRA query (CMQ).
bAdverse event of special interest, including any adverse event in the hallucinations CMQ or psychosis and psychotic disorder standardized MedDRA query (SMQ).
cAssessed using the Columbia-Suicide Severity Rating Scale.
Source: M15-736 Clinical Study Report.14 (Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
Critical Appraisal
Results of an end-of-study survey aiming to assess the extent of unblinding suggested that the majority of patients were able to infer treatment assignment given the differences in treatment response. There is a risk of reporting bias in patient-reported outcomes (PROs), potentially in favour of foslevodopa-foscarbidopa, due to the subjective nature of these outcomes. However, the extent of bias is unclear. Further, while hierarchical testing procedure was in place to account for multiplicity, no definitive conclusion can be drawn with respect to end points other than the primary end point, and study-defined key secondary end points of “off” time, as well as MDS-UPDRS Part II score, due to a failure of statistical comparison in a prior end point in the testing hierarchy (i.e., MDS-UPDRS Part II score). No conclusion can be drawn on the prespecified subgroup analyses because of the lack of consideration for sample size and statistical power and control for multiplicity. As well, there was a risk of attrition bias in favour of foslevodopa-foscarbidopa due to higher attrition in the foslevodopa-foscarbidopa arm compared with the oral LD-CD arm; however, sensitivity analyses of the primary end point and the key secondary end point of “off” time, which assessed the impact of missing data, showed results consistent with the primary analysis, increasing certainty of the findings.
Patients with cognitive impairment and prior DBS or LCIG treatment were excluded from the study, which represents a gap in evidence; nonetheless, the clinical expert consulted by CADTH did not expect the exclusion of these patients to significantly impact the generalizability of the study population. With respect to outcomes, the clinical expert noted that the PD diary, MDS-UPDRS, and PDQ-39 are clinically relevant instruments that are used in clinical practice, while the relevance of bradykinesia and dyskinesia scores, EQ-5D-5L, and PDSS-2 are limited. Improved cognition was an unmet treatment need according to patients, and reduced caregiver burden is a treatment goal in advanced PD. No conclusion on these outcomes can be drawn from the study because the cognition was not assessed as a stand-alone end point (although it was captured as 1 of the items in MDS-UPDRS scale) and caregiver burden was not measured. The clinical expert noted that the duration of follow-up (12 weeks) was adequate for efficacy assessment, although longer follow-up is required to gain certainty on the maintenance of benefit and safety profile.
Long-Term Extension Studies
The M20-098 trial is an ongoing long-term open-label extension study of the pivotal RCT, M15-736, in which patients received individualized foslevodopa-foscarbidopa CSCI for 24 hours per day for up to 96 weeks. At the time of the submission, no patients had completed the trial, and data were available from fewer than 5 patients from week 24 and beyond for outcomes of interest. Data from M20-098 were therefore too immature to use to draw conclusions.
Indirect Comparisons
Description of Indirect Comparisons
One sponsor-conducted indirect treatment comparison (ITC), which indirectly compared foslevodopa-foscarbidopa with LCIG and best medical therapy (BMT) (oral therapy) with respect to change from baseline in mean “off” time, “on” time without troublesome dyskinesia, and PDSS-2 total score at week 12 in patients with advanced PD via a Bayesian network meta-analysis (NMA), was included in the sponsor’s submission.
Efficacy Results
In the Bayesian fixed-effect NMA, which was based on a total of 4 trials, foslevodopa-foscarbidopa, compared with BMT (oral therapy), was associated with |||||||| |||| |||||| |||| |||||||| at week 12 in average “on” time without troublesome dyskinesia (mean difference, ||||| ||| |||||||| |||||||| |||||||||| || ||||) hours, “off” time (mean difference, |||||| ||| |||| ||||| || |||||) hours, and PDSS-2 total score (mean difference, |||||| ||| |||| |||| || |||||). Compared with LCIG, foslevodopa-foscarbidopa was associated with |||| ||||||||||| |||| |||||| |||| |||||||| at week 12 in PDSS-2 total score (LCIG versus foslevodopa-foscarbidopa: mean difference, ||||| ||| |||| |||| || |||||) and || |||||||||| || “on” time without troublesome dyskinesia (mean difference, ||||| ||| |||| ||||| || ||||) hours, “off” time (mean difference, |||| ||| |||| ||||| || ||||) hours.
Critical Appraisal
The validity of the results of the NMA could not be determined because the key assumptions of the analysis, homogeneity and consistency, could not be determined based on insufficient reporting of study characteristics and a sparse linear network without a closed loop. Based on the available information, there was evidence of heterogeneity between the included studies based on study designs (i.e., blinding, dosing protocol for oral therapies, duration of follow-up), patient populations (i.e., presence of concurrent cognitive impairment and dyskinesia), and patient baseline characteristics (i.e., duration of PD diagnosis, “off” time) that were unaccounted for. These limitations result in uncertainty in the relative treatment effect estimates between foslevodopa-foscarbidopa versus BMT (oral therapy) and LCIG.
Studies Addressing Gaps in the Pivotal and RCT Evidence
M15-741 Trial
One supportive phase III, open-label, single-arm trial (M15-741, N = 244), with the aim of evaluating the safety and tolerability of foslevodopa-foscarbidopa in patients with advanced PD for 52 weeks, was included in the sponsor’s submission. Safety and efficacy were assessed as primary and secondary end points, respectively, most of which were consistent with the end points of M15-736. Baseline patient characteristics were in general similar to the M15-736 trial, although in the M15-741, mean time since PD diagnosis (12.3 years [SD = 5.3 years]) was longer, more patients were at advanced stages of PD (based on the Hoehn and Yahr Scale) and received, on average, more medications from different PD drug classes, suggestive of a patient population with more advanced disease than the patient population included in the M15-736 trial.
Efficacy Results
All efficacy results were not adjusted for multiplicity. Foslevodopa-foscarbidopa was associated with a statistically significant improvement from baseline in average daily normalized “on” time without troublesome dyskinesia ||||| ||| | ||||| ||||||, “off” time |||||| ||| | ||||| ||||||, “on” time without dyskinesia ||||| ||| | ||||| ||||||, PDQ-39 ||||| ||| | |||||||, MDS-UPDRS Part II score ||||| ||| | |||||| and IV score ||||| ||| | ||||||, PDSS-2 total score ||||| ||| | |||||||, and EQ-5D-5L summary index |||||| ||| | ||||||| at 52 weeks. Results did not suggest a difference in change from baseline in MDS-UPDRS Part I and III scores or median and IQR of bradykinesia scores and dyskinesia scores at week 52 with foslevodopa-foscarbidopa treatment.
Harms Results
TEAEs were reported in ||||| || ||||||||| ||| |||| |||||| ||||| ||| ||||| |||| |||| |||||||| |||| ||||||||| ||||||| ||| ||||||||||| ||||||| ||||| |||| |||||||| || ||||| || ||||||||| |||| ||| |||| |||||||||| |||||||| ||| ||||| |||||||| |||| |||||||||| ||||||| ||||||||| ||||||||||||||| ||| || |||| ||| |||||||| || ||||| || ||||||||| |||| |||||||| ||||||| || ||||||||||||| ||||||| |||||||| |||| |||||||| ||| |||||||||| ||||| |||||| ||| |||||||||| ||||||| ||||| |||||| ||||||||| |||| || ||||| ||| |||||||||| || || ||||||| || ||||| |||| || ||| ||| |||||||| The safety profile of foslevodopa-foscarbidopa in this trial was in general consistent with the M15-736 trial, with no new safety signal.
Critical Appraisal
The open-label study design could introduce reporting bias, potentially leading to inflated benefits of foslevodopa-foscarbidopa on PROs and less favourable harms results given the more subjective nature of these outcomes. The noncomparative design means that known and unknown confounding factors were not accounted for and no statistical adjustments were made in the analyses, making it impossible to be certain that the observed treatment benefits are attributable to foslevodopa-foscarbidopa alone. As well, a sizable proportion of patients ||||||| withdrew from study treatment, mostly due to AEs and consent withdrawal. As a result, attrition bias may explain the observed efficacy results as the patients remaining in the study were more likely to be those who experienced benefits and were better able to tolerate the treatment.
The inclusion and exclusion criteria overall aligned with the selection criteria for candidates for advanced therapies used in clinical practice. While patients included in this trial appeared to have more advanced PD than those in the pivotal M15-736 trial, the clinical expert consulted by CADTH noted that the patient population would fit within the spectrum of patients with advanced PD in Canada. The attrition rate was high |||||||, most commonly due to AEs. This could affect the generalizability of the results since patients remaining in the trial tend to be those who are better able to tolerate the AEs of foslevodopa-foscarbidopa. The difference in infusion systems used in the trial versus stated in the product monograph could introduce some uncertainty due to potential differences in treatment interruptions, adherence, and safety.
M15-737 Trial
Early results from the ongoing single-arm long-term open-label extension M15-737 trial were submitted by the sponsor and are summarized in this report. Patients who completed the M15-741 trial could enrol in the M15-737 trial. The objective of M15-737 is to assess the longer-term safety and tolerability of foslevodopa-foscarbidopa delivered by CSCI for 24 hours per day for up to an additional 96 weeks after the 52-week M15-741 trial. The primary outcomes are AEs and safety measures. Efficacy outcomes are also being collected as secondary end points. At the time of this submission, data were limited after 48 weeks, and no patients had completed the study.
Efficacy Results
|||||||| |||| ||||||| |||||||| |||| ||| |||||||| || |||||||||||||||||||||||| |||| |||||||||| |||| |||||||| || |||| || |||| ||||||| || ||||||| ||||| |||||||||| |||| |||| ||||||| ||||||||||| ||||||||||| |||| |||| |||| ||||||||||| ||||||||||| ||||| ||||| |||||||||| ||| |||| |||||| ||||| |||| ||||||| |||||||| ||||| ||| | ||||||||||||| ||||||||||| |||||||| || ||||| |||| |||||||| || |||| || || |||||||| || |||||| ||| ||||||||| |||||| ||||| || |||| || |||| |||||| ||||||| ||||||| |||||||| |||| || || || ||||||||| |||||| |||| ||| |||||| ||||| |||| ||| ||||| || |||| ||||||||||| |||| |||||| | | | || |||||||
Harms Results
In the M15-737 trial, AEs were reported || || |||||||| |||||||| ||| ||| |||||||| |||| |||||||||| ||| |||| || |||||||| || ||||||||| ||| |||| |||||||||| |||||||| ||| || |||| |||| |||||||| |||| ||||||||| |||||| ||| |||||||| |||| ||||||||||| |||| |||||||| |||||| ||| ||| |||| ||| || |||||||||||||||| ||| ||||| || ||||| |||| ||| |||||||||||||||||| ||| ||||||| || |||||| The investigator considered that none of the fatal events had a reasonable possibility of being related to the study drug. No new safety concerns were identified.
Critical Appraisal
The open-label study design could introduce reporting bias, potentially leading to inflated benefits of foslevodopa-foscarbidopa on PROs and less favourable harms results given the more subjective nature of these outcomes. The noncomparative design means that known and unknown confounding factors were not accounted for and no statistical adjustments were made in the analyses, making it impossible to be certain that the observed treatment benefits could be attributed to foslevodopa-foscarbidopa alone. Because patients could only enrol after completion of the parent study, there is a greater likelihood of selection bias given that patients who better tolerated the treatment or who perceived the treatment as benefiting them were more likely to enrol. Finally, the trial is ongoing, and data after week 48 are limited. At the available time points, sample sizes are small. No definitive conclusions could be drawn from the results of this study.
Conclusions
In the pivotal M15-736 trial, foslevodopa-foscarbidopa demonstrated a clinically meaningful improvement in “on” time without troublesome dyskinesia and “off” time compared with oral LD-CD therapy at 12 weeks in patients with advanced PD. Analyses of morning akinesia, HRQoL, bradykinesia, and sleep symptoms also favoured foslevodopa-foscarbidopa, although due to failure of a prior outcome in the statistical testing hierarchy, the results for these outcomes were considered supportive of benefit with foslevodopa-foscarbidopa treatment, but not conclusive. Results did not suggest a difference in motor experiences of daily living, though the MDS-UPDRS instrument may be limited in its utility in assessing certain aspects of motor functioning in patients receiving advanced therapies (e.g., consistent control of motor fluctuations and flexibility in performing daily activities). The pivotal study results were determined to be generalizable overall. The comparative effectiveness and safety of foslevodopa-foscarbidopa relative to comparators other than oral LD-CD could not be determined. There are no direct comparisons with LCIG, and the indirect comparison was inconclusive because of important limitations that prevented verifying whether the underlying assumptions of homogeneity and consistency were met. No direct or indirect comparisons between foslevodopa-foscarbidopa and DBS were submitted. Overall, the safety profile of foslevodopa-foscarbidopa was similar to oral LD-CD therapy, except that infusion-site reactions and infections were more frequent with foslevodopa-foscarbidopa; most reactions and infections were not serious, but some resulted in treatment discontinuation. No new serious safety concerns were identified in the longer-term safety studies.
Introduction
The objective of this report is to review and critically appraise the evidence submitted by the sponsor on the beneficial and harmful effects of foslevodopa (240 mg/mL) and foscarbidopa (12 mg/mL) solution for subcutaneous infusion in the treatment of motor fluctuations in patients with advanced PD.
Disease Background
Disease Overview
PD is an incurable, progressive, neurologic disease2,3 characterized by the dysfunction and loss of dopaminergic cells in the substantia nigra and other brain regions.2 Patients with PD experience motor symptoms such as bradykinesia, tremor, rigidity, and postural instability, as well as nonmotor symptoms including cognitive impairment, mood disorders, and sleep problems.4,5 As PD progresses, patients experience declining mobility, increasingly debilitating symptoms, decreasing independence, and greater impact on their quality of life.9,10,16 Advanced PD also impairs gastrointestinal absorption, decreasing the effectiveness of the standard-of-care medication for PD, oral levodopa, and commonly requiring an increase in the dosage and frequency of administration of this standard of care;17,18 this can lead to complications such as severe dyskinesia and bradykinesia.11
Approximately 10% to 20% of patients with PD do not achieve satisfactory control of their disease despite optimized oral treatment, indicating their disease has progressed to advanced PD.7,8 While there is no universal consensus on a definition of advanced PD, a body of leading specialists from 10 European countries proposed the following 15 clinically important criteria based on a Delphi-panel approach:9
Motor symptoms: moderate level of troublesome motor fluctuations; at least 2 hours of the waking day with “off” symptoms; at least 1 hour of the day with troublesome dyskinesia; moderate level of dyskinesia; troublesome dysphagia; daily oral levodopa doses “at least 5 times a day”
Nonmotor symptoms: mild level of dementia; nontransitory troublesome hallucinations; moderate level of psychosis; nonmotor symptoms fluctuations; moderate level of nighttime sleep disturbances
Functional impacts: repeated falls despite optimal treatment; needs help with activities of daily living at least some of the time; not able to perform complex tasks at least some of the time; moderate impaired mobility
The Delphi-based consensus recommendation was that patients exhibiting 1 of the 6 selected motor indicators or 1 of the 5 selected nonmotor symptoms be classified as having “suspected” advanced PD, depending on the severity of the symptoms.9 These criteria have been further simplified and operationalized as the “5-2-1” criteria, which propose that patients with 5 or more oral levodopa doses daily, 2 or more hours of “off” time (i.e., a period of uncontrolled PD symptoms despite treatment) daily, or 1 or more hours of troublesome dyskinesia (i.e., uncontrolled and involuntary movements due to excess treatment effect) daily should be classified as having suspected advanced PD.10
Incidence, Prevalence, and Mortality
PD is the most common movement disorder and the second most common neurodegenerative disorder globally.2,19,20 According to a global burden of disease study, approximately 6.1 million individuals worldwide were estimated to have PD as of 2016, with approximately 103,903 in Canada (uncertainty interval, 78,532 to 126,685).20 The prevalence of PD increases with age, and PD diagnosis is uncommon in people younger than 50 years.20 PD is approximately 1.4 times more prevalent in men than in women.20 In North America, age-standardized incidence rates were estimated to range from 108 to 212 per 100,000 among people aged 65 years and older, and from 47 to 77 per 100,000 among people aged 45 years or older.6
In 2020, 3,431 Canadians died from PD,21 and age-standardized mortality rate was 9 per 100,000 people.22 The mortality rate among people with PD was 1.2 to 2.4 times higher than in the general population.23 The vast majority of disabilities and morbidities in PD are caused by recurrent falls and postural instability, leading to head trauma and hip fractures.24-26 It is also common for patients with PD to have multiple comorbidities, which become significantly more likely to occur as disease progresses, including hypertension, constipation, heart disease, depression, anxiety, and dementia.27,28
Diagnosis
There is no diagnostic test for PD or advanced PD. Diagnosis is made by clinicians with expertise in movement disorders, based on examination of clinical neurologic features.11,29
Standards of Therapy
Oral therapy and advanced device-aided therapies, including DBS and LCIG, are currently available for the treatment of motor fluctuations in patients with levodopa-responsive advanced PD.11
Levodopa, the cornerstone oral treatment for PD, is often prescribed with adjunctive dopamine agonist, a COMT inhibitor, MAO-B inhibitors, amantadine, or anticholinergics.11,30 While oral treatments are effective in treating symptoms in early stages of PD, response to oral medication tends to become less stable and predictable as the disease progresses, requiring fine adjustments of drug regimen (e.g., dosage, frequency, timing of administration, combinations of oral medications with different mechanisms of action) to minimize medication response fluctuations, incomplete benefit, and dyskinesia. Patients with advanced PD might consider advanced therapies, including DBS or LCIG, if response to oral medication continues to be limited or becomes less predictable or consistent over time.
DBS involves surgically placing stimulating electrodes in the areas of the brain that control movement; it is typically used as an adjunctive treatment to oral therapy.12 LCIG is a gel suspension of levodopa and carbidopa that is continuously infused into the small intestine through a percutaneous endoscopic gastrostomy-jejunostomy (PEG-J) tube connected to a portable pump. Most patients eliminate most or all of other PD medications while on LCIG, according to the clinical expert consulted by CADTH. While both treatments are effective in reducing “off” time and reducing dyskinesia during “on” time (i.e., a period of controlled PD symptoms with treatment),11 each treatment has specific issues in terms of health care resource use and safety that need to be taken into account. Generally, DBS is only done at specialized centres in Canada because the procedure requires an experienced and well-trained multidisciplinary team to conduct presurgical assessments, the surgery, and ongoing device monitoring and programming. Indefinite follow-up to monitor for device failures is also required.31 DBS is associated with risks and potentially life-threatening AEs (e.g., intracerebral hemorrhage, stroke, infection, seizure induced by the surgery, and off-target stimulation effects such as changes in speech or freezing of gait). DBS is only appropriate in some patients, typically those aged 70 years or younger, with severe motor dysfunction and no history of psychiatric disorders, cognitive issues (such as depression), speech disorders, or suicide attempts.11,12,32 The provision of LCIG requires a specialized medical team, including the treating neurologist, a gastroenterologist trained in inserting PEG-J tubes, and nursing care to help in monitoring, device programming, and assessing for device- and tube-related problems (e.g., infection, tube kinking, and dislocations).33 LCIG is associated with AEs of weight loss, abdominal pain, peripheral neuropathy, and diphasic dyskinesia.33
Rescue treatments such as subcutaneous and sublingual apomorphine are available for the acute intermittent treatment of severe motor complications in advanced PD.11,30 Rehabilitative therapies (e.g., physiotherapy, occupational therapy, exercises) are recommended in patients with motor function symptoms and difficulties with activities of daily living.11
Some nonmotor symptoms may respond to levodopa treatment (orally or LCIG) and can benefit from continuous levodopa therapy, as per the clinical expert CADTH consulted. Other treatments for nonmotor symptoms are also available, such as cholinesterase inhibitors for dementia; midodrine, fludrocortisone, and domperidone for orthostatic hypotension; botulinum toxin A for sialorrhea; quetiapine, clozapine, pimavanserin for psychosis.11
According to the clinical expert, treatment choice for motor symptoms is individualized and based on patient and health system factors. Treatment goals include reducing disability from motor and nonmotor symptoms, allowing independent functioning, preserving HRQoL, and reducing caregiver burden.
Drug Under Review
Key characteristics of foslevodopa-foscarbidopa, LCIG, and oral therapies are summarized in Table 4.
Foslevodopa-foscarbidopa is a prodrug combination of levodopa monophosphate and carbidopa monophosphate that is converted in vivo to levodopa and carbidopa. Levodopa relieves symptoms of PD following decarboxylation to dopamine in the brain. Carbidopa, which does not cross the blood-brain barrier, inhibits the decarboxylation of levodopa to dopamine outside the brain, allowing a larger amount of levodopa be available for transportation to the brain and transformation into dopamine.1
This is the first review for foslevodopa-foscarbidopa by CADTH. Foslevodopa-foscarbidopa was granted a Health Canada Notice of Compliance for the treatment of motor fluctuations in patients with advanced levodopa-responsive PD who do not have satisfactory control of severe, debilitating motor fluctuations and hyper-/dyskinesia despite optimized treatment with available combinations of medicinal products for PD. The sponsor is seeking reimbursement of foslevodopa-foscarbidopa for the treatment of motor fluctuations in patients with PD who are inadequately controlled on optimized oral therapies (advanced PD) and who are not candidates for DBS. The sponsor noted that, among other considerations, patients who are not candidates for DBS could be characterized based on 1 or several of the following reasons:34
The patient does not consent due to surgical risk, surgical wait times, hesitancy of undergoing a neurosurgical procedure.
The physician considers the patient is inappropriate for DBS due to ethical concerns
The physician considers that the risks of the procedure outweigh the benefits for the patient.
The patient is older than 70 years of age.
The patient has moderate to severe depression, cognitive decline, other neuropsychiatric disorders, or medical comorbidities that increase surgical risk.
The physician determines that the wait time for access to the DBS procedure is too long for the patient based on their status.
The sponsor noted that, ultimately, physicians will determine which patients are not appropriate candidates for DBS based on their clinical judgment and in consultation with the patient and their caregiver, as appropriate.34
Foslevodopa-foscarbidopa is administered as a continuous subcutaneous infusion, 24 hours per day, using an infusion pump based on an individualized dosing. The starting infusion rate is determined by calculating the levodopa equivalents from all levodopa-containing medications and COMT inhibitors taken during daytime hours, and then increasing it to account for a 24-hour administration. The dose may be adjusted to reach a clinical response that maximizes the functional “on” time and minimizes the number and duration of “off” episodes and “on” episodes with troublesome dyskinesia. The maximum recommended daily dose of foslevodopa is 6,000 mg (or 25 mL of Vyalev per day, equivalent to approximately 4,260 mg levodopa per day). Foslevodopa-foscarbidopa replaces levodopa-containing medications and COMT inhibitors. If required, other classes of medicinal products for PD can be taken concurrently. Foslevodopa-foscarbidopa should only be prescribed by neurologists who are experienced in the treatment of patients with PD.
Table 4: Key Characteristics of Foslevodopa-Foscarbidopa, LCIG, and Oral Therapies
Characteristic | Foslevodopa-foscarbidopa continuous SC infusion | LCIG | Oral PD therapiesa |
|---|---|---|---|
Mechanism of action | Prodrug of levodopa and carbidopa
|
| Levodopa-decarboxylase inhibitor, COMT inhibitor, dopamine agonist, MAO-B inhibitor
NMDA receptor antagonists
Anticholinergics
|
Indicated populationb | Patients with advanced levodopa-responsive PD who do not have satisfactory control of severe, debilitating motor fluctuations and hyper-/dyskinesia despite optimized treatment with available combinations of medicinal products | Patients with advanced levodopa-responsive PD who do not have satisfactory control of severe, debilitating motor fluctuations and hyper-/dyskinesia despite optimized treatment with available combinations of medicinal products and for whom the benefits of this treatment may outweigh the risks associated with the insertion and long-term use of the PEG-J tube required for administration | Patients with PD |
Route of administration | SC infusion (24-hour) | Intestinal infusion (16-hour) via a PEG-J tube | Oral |
Dosing | Individualized, based on previous total dopaminergic drug needs, and adjusted to achieve optimal clinical response | Individualized, based on previous total dopaminergic drug needs, and adjusted to achieve optimal clinical response | Varies by drug; usually involves multiple doses per day adjusted to achieve optimal clinical response |
Serious adverse events or safety issuesc |
|
| Levodopa-decarboxylase inhibitor or dopamine agonist: sudden onset of sleep |
COMT = catechol-O-methyltransferase; LCIG = levodopa-carbidopa intestinal gel; MAO-B = monoamine oxidase type B; NMDA = N-methyl-D-aspartate; PD = Parkinson disease; PEG-J = percutaneous endoscopic gastrostomy-jejunostomy; SC = subcutaneous.
Note: Deep brain stimulation is not summarized in Table 4, but it is also available for the treatment of motor fluctuations in advanced PD in Canada.
aOral therapy includes the following: levodopa-decarboxylase inhibitor and adjunctive COMT inhibitor, dopamine agonists, MAO-B inhibitor, NMDA receptor antagonists, and anticholinergics.
bHealth Canada–approved indication.
cAs per the Health Canada Product Monograph.
Sources: Foslevodopa-foscarbidopa product monograph,1 Duodopa product monograph,35 Compendium of Pharmaceuticals and Specialties – Parkinson Disease.30
Stakeholder Perspectives
Patient Group Input
CADTH received 1 input from Parkinson Association of Alberta and 1 joint input from Parkinson Canada, Parkinson Society British Columbia, and Parkinson Quebec. Parkinson Association of Alberta conducted a survey of 26 patients with PD and care partners or family in Alberta. Parkinson Canada, Parkinson Society British Columbia, and Parkinson Quebec jointly gathered responses from 113 patients with PD and caregivers of patients with PD in Canada via a survey; the majority of respondents were from Ontario (72.6%).
According to both inputs, off periods and motor fluctuations associated with PD substantially impacted quality of life and activities of daily living, led to work absenteeism (and in some cases, early retirement), and caused emotional and financial burden to caregivers. Respondents from both input groups noted that the symptoms that are most important to control were changes in cognition and memory, fatigue, and sleep, freezing and unpredictable “off” periods, changes in mood, rigidity, speech and swallowing issues, bladder and bowel issues, impaired balance, slowness, and tremors.
More than half of each patient group experienced side effects with oral medications, with fatigue, drowsiness, constipation, and bowel issues the most difficult to endure. More than half of the respondents to the joint input reported that high pill burden (up to 40 pills per day) impacted their lifestyle or quality of life. Difficulties related to medication adherence included difficulty to do with timing or remembering, swallowing, and storage of medications, and limited improvement of symptoms. Some patients also received some form of rehabilitation (physiotherapy, occupational/speech therapy, or exercise) as a treatment option, but respondents also cited cost, lack of motivation, or lack of access as barriers, especially for patients in rural areas. No respondents from either patient group were receiving foslevodopa-foscarbidopa at the time of survey.
Respondents indicated the most important unmet needs were treatment options that would not increase dyskinesia as time went on, medications that would treat cognitive issues, and longer-lasting medications that would reduce pill burden and “off” periods, eliminating the fluctuations and sleep interruptions caused by medications wearing off. The joint input indicated that a large proportion of patients were reluctant about undergoing invasive treatment options such as DBS or LCIG and that the majority (65%) would be interested in an injection-based LD-CD treatment; however, only 1 (3.8%) respondent from Parkinson Alberta said they would consider it and 2 (7.7%) were unsure.
Clinician Input
Input From Clinical Expert Consulted by CADTH
Unmet Needs
The clinical expert noted that access to currently available advanced PD therapies can be challenging to patients due to health system and geographical factors. First, the clinical expert noted there is limited access to there are fewer than 80 neurologists specializing in movement disorders in Canada, with most located in urban centres, for more than 100,000 patients with PD.36 Second, the provision of DBS and LCIG treatments requires a complex system of care; DBS requires access to neurosurgical subspecialists and nurses with specialized expertise in device programming and monitoring, a system of care that includes the administration of treatment programs, and a hospital system with operating room staff and resources. As for LCIG, the clinical expert noted that in addition to neurologists and nurses with specialized training, a gastroenterologist trained to insert the PEG-J tube and a system of care that involves support in monitoring of the PEG-J tube and management of the infusion device, pump assistance by the care partner, and in some cases, wound or stoma care for PEG-J insertion site. Because of these resource needs, the clinical expert reported that LCIG and DBS treatments are typically provided in major urban treatment centres, which is a barrier to treatment access considering the inherent difficulties patients with advanced PD have in travelling to these sites. The clinical expert noted that there is an unmet need for treatment options that require less in the way of resources or the need to travel to major urban centres, so that treatment is more easily available, especially for those residing in rural and remote areas.
The clinical expert also noted an unmet need for treatment options for patients who are ineligible for advanced therapies because of comorbidities. DBS is generally not considered for patients with significant nonmotor symptoms (which rarely improve with DBS treatment), severe cognitive impairment, medical contraindications (e.g., long-term anticoagulation), other intracranial lesion, or high surgical risk due to medical comorbidities. Certain medical (e.g., long-term anticoagulation) or gastrointestinal comorbidities preclude eligibility for LCIG.
Place in Therapy
The clinical expert noted that foslevodopa-foscarbidopa and LCIG share a similar mechanism of action (via dopamine precursors) but have different mechanisms of delivery. The clinical expert noted that subcutaneous drug delivery of foslevodopa-foscarbidopa is associated with a lower risk of surgical complications, which addresses a concern expressed by many patients who were reluctant to receive LCIG or DBS because of the risks associated with insertion of PEG-J tube (for LCIG administration) or neurosurgery (for DBS treatment). According to the clinical expert, foslevodopa-foscarbidopa is expected to fill a treatment gap for patients with advanced PD who cannot travel to access other advanced therapy options (e.g., those who reside in rural and remote areas) or who have comorbidities or strong personal aversion to other options (or, specifically, those options that require surgical procedures). The clinical expert noted that foslevodopa-foscarbidopa eliminates the need to coordinate care between movement disorders specialists and a second specialist (e.g., gastroenterologist or neurosurgeon) upon treatment initiation. With these simplifications to advanced therapy for patients with advanced PD, the clinical expert noted that foslevodopa-foscarbidopa could lead to a shift in the current treatment paradigm for advanced PD by improving access to device-aided therapy.
The clinical expert expected that foslevodopa-foscarbidopa would serve as a treatment option in patients with advanced PD. In general, concomitant oral medications are be expected to be stopped at the time of titration of foslevodopa-foscarbidopa, as per the clinical expert.
Patient Population
The clinical expert noted that patients with levodopa-responsive advanced PD would be considered eligible for treatment with foslevodopa-foscarbidopa in clinical settings. Although the lack of a universally agreed-upon definition for advanced PD was noted, “5-2-1” criteria or the Delphi-based consensus criteria are commonly used in clinical practice to assess patients with suspected advanced PD. The clinical expert added that it is accurate to define patients with advanced PD as those who have motor fluctuations that are inadequately controlled with optimized oral therapy; however, this definition hinges on clinicians’ ability to assess motor fluctuations and their familiarity with oral therapy optimization approaches, which would require limiting prescribing to experts in treatment of movement disorders to ensure the treatment is used in appropriate patients. With respect to the definition of “optimized oral therapy,” the clinical expert noted that an individualized optimization approach is typically used; therefore, the decision of what constitutes “optimized oral therapy” is – in their opinion – best left to the judgment of the treating neurologist.
The clinical expert noted that patients with excessive “off” time or “on” time with bothersome dyskinesia are more likely to benefit from treatment with foslevodopa-foscarbidopa, while patients with levodopa-unresponsive symptoms (e.g., axial symptoms such as freezing of gait, aspects of cognitive impairment, or symptoms in a limb that are also affected by neuromuscular weakness or stroke) are not expected to benefit from foslevodopa-foscarbidopa treatment. The clinical expert noted that patients with prior DBS, or LCIG treatment and patients with cognitive impairment could potentially benefit from foslevodopa-foscarbidopa.
Assessing the Response Treatment
The clinical expert noted that in clinical practice, response assessments typically involve subjective discussions of disease management; depending on the clinic model, the target symptoms and the need for advanced therapies, symptom diaries to assess “on” and “off” times, and quality-of-life scales like PDQ-39 may also be conducted intermittently. In the clinical expert’s opinion, a clinically meaningful response would include improvement in “on” and “off” time measurements and quality of life, although the definition of clinical meaningfulness could vary among clinicians. The clinical expert noted that treatment response is typically determined at 3 months after initiation.
Discontinuing Treatment
The clinical expert noted that treatment discontinuation could be considered when patients experience significant functional impairments that are not relieved by the treatment (e.g., becoming dependent for transfers and daily activities) or intolerable AEs (e.g., cutaneous infusion reactions). Functional impairment could occur due to progression of levodopa-unresponsive symptoms, nonmotor symptoms, or other causes (e.g., stroke or other serious medical conditions).
Prescribing Considerations
The clinical expert noted that while specifying prescribing by movement disorders specialists would be appropriate, it was their opinion that some latitude should be given to allow prescribing by neurologists (nonmovement disorders specialists) sufficiently experienced, qualified, and trained to administer and monitor foslevodopa-foscarbidopa treatment in rural or remote areas, where access to movement disorders specialists can be limited. The clinical expert also noted that some community neurologists could be practising in rural or smaller urban settings without a multidisciplinary subspecialty clinic; therefore, prescribing should not be limited to neurologists practising in major urban centres. Taking these into consideration, the clinical expert’s opinion was that it would be appropriate to specify prescribing of foslevodopa-foscarbidopa by neurologists who have experience in the treatment of patients with advanced PD and are trained in the use of this therapy.
Additional Considerations
The clinical expert expressed concerns with limiting eligibility for foslevodopa-foscarbidopa to patients who are not candidates for DBS since this criterion could be interpreted by the jurisdictions to require a neurosurgical consultation to determine DBS eligibility before reimbursement of foslevodopa-foscarbidopa. The clinical expert noted that this would require considering a more invasive treatment option (i.e., DBS) before a less invasive option when the comparative effectiveness of the options has not been determined yet. This criterion, according to the clinical expert, would also remove the advantage of foslevodopa-foscarbidopa for patients living in areas underserved with respect to DBS, particularly rural and remote areas. The clinical expert also noted that patients have long wait times for DBS surgical consultation (e.g., at least 4 years in the clinical expert’s jurisdiction), which is a barrier to timely treatment access in a progressive and debilitating disease. The clinical expert highlighted the fact that not all patients assessed for DBS treatment are eligible to receive it, which also results in significant delays in treatment.
Clinician Group Input
Input was received from the National Movement Disorder Expert Group (11 clinicians), and the BC Movement Disorders Specialist Group (7 clinicians). Overall, the inputs of both clinician groups aligned with that of the clinical expert consulted by CADTH.
The clinician groups concurred on the unmet needs of patients with advanced PD. Patients receiving oral levodopa have inadequate control of motor fluctuations despite increased dosing frequency over time, and may have contraindications, poor tolerance, or insufficient response to adjunctive medications. They described barriers to accessing advanced therapies for PD (i.e., DBS and LCIG treatments) that vary geographically because of limited specialists, uneven distribution of resources geographically, intense resource needs, medical contraindications, poor acceptance by patients because of the invasive nature and risks of the treatments, and the impact of PD itself on patients’ ability to travel long distances for DBS or LCIG treatment and to manage at-home aspects of LCIG treatment. In addition, the clinician groups noted that no current treatments address the underlying disease process of PD.
The clinician groups were aligned in considering that foslevodopa-foscarbidopa could serve as an additional treatment option for patients with advanced PD, and could benefit patients experiencing bothersome end-of-dose “off” periods, unpredictable efficacy of oral therapies because of absorption delays, and/or excessively complex oral medication schedules due to the subcutaneous delivery of foslevodopa-foscarbidopa.
The clinician groups indicated that, as with other existing advanced therapies, eligible patients would include those who have levodopa-responsive PD with bothersome motor and nonmotor fluctuations despite optimized oral therapies. The inputs suggested that eligible patients have advanced PD, identified by the “5-2-1” criteria. They also suggested that it would be reasonable to recommend first trying at least 1 MAO-B inhibitor and a COMT inhibitor, unless contraindicated. It would also be reasonable to recommend that cognitively intact patients aged 70 years or younger first try at least 1 dopamine agonist and amantadine (if dyskinesia is bothersome), unless contraindicated. However, the inputs suggested against requiring a previous trial of anticholinergics or apomorphine preparations for reimbursement of foslevodopa-foscarbidopa. The clinician groups inputs agreed with the clinical expert that treatment response would be assessed based on “off” time, presence of disabling dyskinesia, and quality of life, and added that an association with easing of caregiver burden may be considered. The clinician groups agreed that discontinuation could be considered in patients with intolerable AEs (e.g., skin reactions or hallucinations) and those who were unable to use the pump correctly due to cognitive decline as a result of disease progression or who lack caregiver support. The clinician groups agreed with the clinical expert that movement disorder neurologists, general neurologists, and geriatricians with experience in the treatment of PD could be comfortable and qualified to prescribe and maintain treatment with foslevodopa-foscarbidopa.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 5.
Table 5: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
There are 2 clinical studies: 1. M15-736
2. M15-741
Questions for CADTH and/or the clinical expert: 1. Should LCIG (Duodopa) have been used as a comparator in the clinical studies? 2. Should DBS have been used as a comparator in the clinical studies? | The clinical expert considered LCIG and DBS as relevant comparators for foslevodopa-foscarbidopa because all 3 would generally be considered as treatment options for those who do not have satisfactory control of PD motor symptoms despite optimized treatment with other oral PD medications. The clinical expert had no major concerns with the lack of direct comparison of foslevodopa-foscarbidopa with the other treatments. It was the clinical expert’s opinion that the M15-736 trial of foslevodopa-foscarbidopa had a similar study design as a pivotal trial of LCIG (i.e., double-dummy, active-controlled design, with optimized oral therapy as the comparator) and could similarly provide evidence for the efficacy of foslevodopa-foscarbidopa. |
As a condition for receiving reimbursement for LCIG, British Columbia requires that patients have a contraindication to DBS or be on the DBS waitlist for more than 1 year. The sponsor is also requesting that only patients who are not candidates for DBS be treated with foslevodopa-foscarbidopa. Questions for the clinical expert: 1. If the patient is a candidate for DBS and the procedure is available, should the patient receive DBS rather than foslevodopa-foscarbidopa or LCIG (i.e., the efficacy and safety of DBS is probably superior to drug therapy in most patients)? 2. If the patient does not respond to, or loses response to DBS, would they be an appropriate candidate for foslevodopa-foscarbidopa? 3. Is it reasonable to use foslevodopa-foscarbidopa in a patient who needs to wait for a significant time period (e.g., > 1 year) to receive DBS? 4. If the patient does not respond to, loses response, or is intolerant of LCIG, would they be an appropriate candidate for foslevodopa-foscarbidopa? 5. Is there still a role for LCIG if foslevodopa-foscarbidopa is available? If so, in which patients? | The clinical expert noted that the stated criterion requires clinicians to refer all patients who do not have an absolute contraindication to DBS for a neurosurgical consult before LCIG treatment can be considered, which can pose issues as it requires a more invasive therapy (i.e., DBS) to be considered first when both treatments may be similarly clinically effective. The clinical expert also noted that this criterion could create a barrier for patients residing in areas where access to DBS-related resources may be limited. The issue is compounded by the wait times for DBS surgical consultations. 1. The clinical expert noted that the comparative efficacy of DBS vs. LCIG has not been well established; in the clinical expert’s opinion, the efficacy of these treatments for treating motor symptoms is similar. In the clinical expert’s opinion, efficacy should not be the only clinical factor that guides treatment choice; patient preference and health system factors must also be taken into account. The clinical expert did not agree with criteria that position LCIG and foslevodopa-foscarbidopa as options of last resort (i.e., after assessment of DBS eligibility). 2. The clinical expert noted that patients who do not respond to, or lose response to, DBS could be candidates for foslevodopa-foscarbidopa. 3. The clinical expert noted that patients who have a long wait time for a DBS surgical consult could benefit from treatment with foslevodopa-foscarbidopa for their symptoms and quality of life, and this treatment can be initiated within weeks or months. 4. The clinical expert noted that foslevodopa-foscarbidopa could be considered in patients who respond to LCIG treatment but develop tube complications or a new medical issue that renders LCIG treatment no longer appropriate. 5. The clinical expert noted that while most patients would likely prefer the simplicity of SC infusion of foslevodopa over a PEG-J insertion for LCIG administration, patients with poor tolerability to foslevodopa-foscarbidopa may consider LCIG an option. |
Considerations for initiation of therapy | |
M15-736 inclusion criteria are the following: 1. ≥ 30 years of age 2. diagnosis of levodopa-responsive idiopathic PD that is inadequately controlled by current therapy 3. taking ≥ 400 mg/day levodopa equivalents 4. must have motor fluctuations (“on”/”off”) 5. average ≥ 2.5 hours/day “off” for 3 consecutive days before enrolment 6. ≥ 2 hours/day “off” for 3 consecutive days before randomization 7. MMSE score ≥ 24 8. able to demonstrate correct understanding and use of the delivery system (patient or caregiver) Question for the clinical expert/the CDEC: Should any of the M15-736 inclusion criteria be used as reimbursement criteria? | The clinical expert noted that the following criteria would be reasonable for the reimbursement of foslevodopa-foscarbidopa:
According to the clinical expert, there could be rare scenarios where the onset of PD occurs before the age of 30 years. It would be usual to pursue advanced PD therapies at ≥ 400 mg/day levodopa equivalents as it is a low dose. Patients with cognitive impairment should not be excluded from treatment as cognitive impairment is not a medical contraindication to foslevodopa-foscarbidopa. |
The public drug plans that reimburse LCIG have roughly the same initiation criteria, as follows: 1. The patient has not been able to achieve satisfactory control of severe, debilitating motor fluctuations and hyper-/dyskinesia despite optimized treatment with available combinations of PD treatments including maximally tolerated doses of levodopa in combination with carbidopa, a COMT inhibitor, a dopamine agonist, a MAO-B inhibitor, and amantadine, if not contraindicated. 2. The patient experiences severe disability for at least 25% of their waking day in the “off” state and/or ongoing, bothersome levodopa-induced dyskinesia, despite having tried frequent dosing of levodopa (at least 5 doses per day). 3. The patient has received an adequate trial of maximally tolerated doses of levodopa, with demonstrated clinical response. 4. The benefits of using LCIG treatment outweigh the risks associated with the insertion and long-term use of the PEG-J tube required for administration AND the patient does not have severe psychosis or dementia. Question for the clinical expert/CDEC: Are these initiation criteria (except number 4) for LCIG still clinically appropriate and could they be used for foslevodopa-foscarbidopa? | The clinical expert agreed that the initiation criteria for LCIG stated (except for number 4) would be applicable to foslevodopa-foscarbidopa. |
Considerations for continuation or renewal of therapy | |
The public drug plans that reimburse LCIG have roughly the same renewal criteria, as follows: 1. The patient continues to benefit from the treatment, including significant reduction in the time spent in the “off” state and/or in ongoing, bothersome levodopa-induced dyskinesia, along with an improvement in the severity of the disability in the off state. 2. The duration of approval is 1 year. Question for the clinical expert/CDEC: Are the above renewal criteria for LCIG still clinically appropriate for use with foslevodopa-foscarbidopa? If so, could it be used for foslevodopa-foscarbidopa? | The clinical expert agreed that the renewal criteria for LCIG would be applicable to foslevodopa-foscarbidopa. |
Considerations for discontinuation of therapy | |
Some public drug plans that reimburse LCIG have the following discontinuation criterion: 1. It is expected that physicians will continue to monitor their patients and discontinue LCIG if the patient is no longer benefiting from treatment, as described for renewal criteria, or if LCIG is no longer appropriate. Question for the clinical expert/CDEC: Is the discontinuation criterion for LCIG still clinically appropriate for use with foslevodopa-foscarbidopa? If so, could it be used for foslevodopa-foscarbidopa? | The clinical expert agreed that the discontinuation criterion for LCIG would be applicable to foslevodopa-foscarbidopa. |
Considerations for prescribing of therapy | |
Most of the public drug plans that reimburse LCIG restrict prescribing to movement disorder specialists. Question for the clinical expert/CDEC: Should foslevodopa-foscarbidopa reimbursement be restricted to prescribers specialized in movement disorders? | The clinical expert noted that restricting prescribing of foslevodopa-foscarbidopa to prescribers specialized in movement disorders would be appropriate in most cases. However, neurologists who are sufficiently experienced, qualified, and trained to administer and monitor foslevodopa-foscarbidopa treatment might be available in some areas, e.g., in rural or remote areas, where specifying a movement disorder specialist in the prescribing condition would create a barrier to accessing the treatment for those patients. The clinical expert suggested leaving the prescribing condition broad by allowing prescribing by neurologists who have experience in the treatment of patients with PD to prescribe foslevodopa-foscarbidopa. |
Care provision issues | |
Foslevodopa-foscarbidopa needs to be drawn from a vial using a syringe and then loaded into an AbbVie trademarked pump (Vyafuser) to be continuously infused into the subcutaneous tissue 24 hours a day. Question for the clinical expert: Do you have experience with the administration of this drug? Are patients with PD able to manage this? | The clinical expert did not have experience administering foslevodopa-foscarbidopa; however, they did not foresee the infusion system to be a major barrier to receiving treatment. Based on their experience with LCIG, there is generally adequate trainings involved and movement disorder specialists know that patients and caregivers have to be able to operate the device. Additional support could also be provided to patients or families who could not reliably manage the infusion device. The clinical expert expected the administration of foslevodopa-foscarbidopa to be less of an issue than administration of LCIG, where setting up, cleaning, and flushing the PEG-J tubing and turning on the pump could be difficult when patients are in the “off” state and have poor motor symptoms. |
The longest study was M15-741, at 52 weeks. Question for the clinical expert: Are there side effects with long-term continuous subcutaneous infusion of foslevodopa-foscarbidopa that should be monitored? | The clinical expert noted that there are concerns related to deficiencies in B vitamins with foslevodopa-foscarbidopa (as are monitored for patients on LCIG), although clinicians have had difficulties obtaining approval for ordering some of these laboratory tests. |
System and economic issues | |
LCIG (Duodopa) has undergone negotiation by the pCPA and the negotiated price is kept confidential. | For CDEC consideration. |
CDEC = CADTH Canadian Drug Expert Committee; COMT = catechol-O-methyltransferase; DBS = deep brain stimulation; IR = immediate release; LCIG = levodopa-carbidopa intestinal gel; MAO-B = monoamine oxidase type B; MMSE = Mini-Mental State Examination; pCPA = pan-Canadian Pharmaceutical Alliance; PD = Parkinson disease; PEG-J = percutaneous endoscopic gastrostomy-jejunostomy; SC = subcutaneous; vs. = versus.
Clinical Evidence
The objective of CADTH’s Clinical Review Report is to review and critically appraise the clinical evidence submitted by the sponsor on the beneficial and harmful effects of foslevodopa (240 mg/mL) and foscarbidopa (12 mg/mL) solution for subcutaneous infusion in the treatment of motor fluctuations in patients with advanced PD. The focus is on comparing foslevodopa-foscarbidopa to relevant comparators, and identifying gaps in the current evidence.
A summary of the clinical evidence included by the sponsor in the review of foslevodopa-foscarbidopa is presented in 4 sections, and CADTH’s critical appraisal of the evidence is included after each section. The sections include:
the systematic review, which includes a description of the pivotal studies and RCTs selected according to the sponsor’s systematic review protocol (refer to the Pivotal Studies and RCT Evidence section)
sponsor-submitted long-term extension studies (refer to the Long-Term Extension Studies section)
indirect evidence submitted by the sponsor (refer to the Indirect Evidence section)
additional studies that, according to the sponsor, addressed important gaps in the pivotal and RCT evidence (refer to the Studies Addressing Gaps in the Pivotal and RCT Evidence section).
Included Studies
Clinical evidence from the following is included in the CADTH review and appraised in this document:
1 pivotal phase III double-blind RCT (M15-736)
1 long-term extension study of the pivotal study (M20-098)
1 ITC
2 additional studies, including a phase III, open-label, single-arm study (M15-741), and its long-term extension study (M15-737).
Pivotal Studies and RCT Evidence
Description of Studies
Two phase III trials (M15-73613 and M15-74137) met the inclusion criteria for the systematic review conducted by the sponsor. The pivotal M15-736 trial is presented in this section and its characteristics are summarized in Table 6. Since Health Canada considers the single-arm M15-741 trial to be a supportive trial for the approval of foslevodopa-foscarbidopa,38 M15-741 and its long-term extension trial (M15-737) are summarized in the Studies Addressing Gaps in Pivotal and RCT Evidence section. The M15-741 and M15-737 trials potentially address gaps in evidence by assessing the longer-term efficacy and safety of foslevodopa-foscarbidopa.
The M15-736 trial was a phase III, randomized, multicentre, double-blind, double-dummy, active-controlled trial with the aim of evaluating the safety and efficacy of foslevodopa-foscarbidopa to demonstrate its superiority over oral LD-CD IR tablets for the treatment of motor fluctuations in patients with advanced PD over 12 weeks of treatment (N = 141). The study was conducted in 76 sites in Australia and the US. Patients were enrolled between October 19, 2020, and September 3, 2021. The study is now complete. It consisted of the following study periods:
Screening period (6 to 60 days): Study eligibility assessments.
Oral LD-CD stabilization period (14 to 21 days): All patients received oral LD-CD IR tablets at an individualized dose, and adjustments of dose and schedule were performed over 7 to 14 days to achieve optimal control of motor symptoms. This was followed by a 7-day period where no further adjustment to oral LD-CD IR were allowed.
Double-blind treatment period (12 weeks; day 1 to day 85): Patients were randomized in a 1:1 ratio using interactive response technology to receive either foslevodopa-foscarbidopa plus oral placebo capsules for LD-CD IR (referred to hereinafter as the “foslevodopa-foscarbidopa arm”) or placebo infusion for foslevodopa-foscarbidopa plus oral LD-CD IR tablets (referred to hereinafter as the “oral LD-CD arm”). Randomization was stratified by study site. Between day 1 and day 29 (CSCI optimization phase), the CSCI rate of the study drug solution was adjusted based on clinical response while the same regimen of oral study drug was maintained. After day 29 (maintenance phase), all study drug regimens remained unchanged until the end of the study.
Efficacy and safety results of the double-blind treatment of M15-736 are summarized in this section. For results of the long-term extension study (M20-098),39 refer to the Long-Term Extension Studies section.
Table 6: Details of Pivotal Studies and RCT Evidence Identified by the Sponsor
Detail | M15-736 |
|---|---|
Designs and populations | |
Study design | Phase III, randomized, multicentre, double-blind, double-dummy, active-controlled trial |
Locations | 76 sites in Australia and the US |
Patient enrolment dates: | Start date: October 19, 2020 End date: September 3, 2021 |
Randomized/enrolled (N) | 141 |
Key inclusion criteria |
|
Key exclusion criteria | Had received DBS, LD-CD enteral suspension, or any other PD medication as continuous daily infusion |
Drugs | |
Intervention | Foslevodopa (240 mg/mL) and foscarbidopa (12 mg/mL) solution administered through CSCI over 24 hours/day + placebo for LD-CD IR oral tablets |
Comparator(s) | LD-CD (100 mg/25 mg) IR oral tablets + placebo for foslevodopa-foscarbidopa CSCI solution for infusion |
Study duration | |
Screening period | 6 to 60 days |
Stabilization period | 14 to 21 days |
Treatment period | 12 weeks |
Follow-up phase | Patients who successfully completed the 12-week treatment phase were eligible for inclusion in the M20-098 extension for up to an additional 96 weeks |
Outcomes | |
Primary end point | Change from baseline to week 12 in average daily normalized “on” time without troublesome dyskinesiab |
Secondary and exploratory end points | Key secondary:
Other secondary end points:
Safety (12 weeks)
|
Publication status | |
Publication | Soileau et al. (2022)13 |
AE = adverse event; CD = carbidopa; CSCI = continuous subcutaneous infusion; C-SSRS = Columbia-Suicide Severity Rating Scale; DBS = deep brain stimulation; ECG = electrocardiogram; EQ-5D-5L = 5-Level EQ-5D; HRQoL = health-related quality of life; IQR = interquartile range; IR = immediate release; LD = levodopa; MDS-UPDRS = Movement Disorder Society-Unified Parkinson’s Disease Rating Scale; PD = Parkinson disease; PDQ-39 = Parkinson’s Disease Questionnaire-39 items; PDSS-2 = Parkinson’s Disease Sleep Scale-2; QUIP-RS = Questionnaire for Impulsive-Compulsive Disorders in Parkinson’s Disease – Rating Scale; RCT = randomized controlled trial; VAS = visual analogue scale.
aAs per investigator’s assessment.
bAssessed using the PD diary.
cDefined as a Mini-Mental State Examination score of at least 24.
dAssessed using the Parkinson KinetiGraph/Personal KinetiGraph wearable device.
Source: M15-736 Clinical Study Report.14 (Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
Populations
Patients eligible for inclusion were aged 30 years or older, with a diagnosis of levodopa-responsive idiopathic PD. They were taking a minimum of 400 mg/day levodopa equivalents, were judged by the investigator to have motor symptoms inadequately controlled by current therapy, with recognizable or identifiable “off” and “on” states (motor fluctuations), and with an average “off” time of at least 2.5 hours per day over 3 consecutive PD diary days for a minimum of 2 hours each day. Patients who had previously received DBS, LD-CD enteral suspension, or any other PD medication as a continuous daily infusion were excluded.
Interventions
Oral LD-CD Optimization Period (14 to 21 Days)
All patients received an individualized regimen of oral LD-CD IR tablets that was based on the levodopa dose equivalent calculated from all levodopa-containing medications and COMT inhibitors established before the study using conversion rates published by Tomlinson et al. (2010)40 and guidance from Espay et al. (2017).41 The dose and schedule of oral LD-CD were adjusted over the first 7 to 14 days by the investigator to optimize control of motor symptoms. An optimal clinical response was defined as maximizing functional “on” time and minimizing the number of “off” episodes during the day and “on” time with troublesome dyskinesia. Once stabilized, the dosing regimen remained unchanged for at least 7 days before the start of the double-blind treatment period.
In both oral LD-CD optimization and double-blind treatment periods, the following concomitant PD medications were allowed: nonergolinic dopamine agonists, selective MAO-B inhibitors, amantadine, safinamide, and istradefylline. These medications, if taken, were maintained at the already optimal and stabilized prerandomization dose and schedule for the remainder of the study. No changes were allowed unless, in the investigator’s considered opinion, they were medically necessary (e.g., management of dyskinesia, safety).
Prohibited medication included apomorphine, LCIG, LD-CD enteral suspension, dopamine-depleting agents, MAO-A inhibitor, nonselective MAO-B inhibitors, ergot dopamine agonists, dopamine antagonist or partial agonist, first generation antipsychotics, or antiemetic medications that interact with brain dopamine receptors, oral and/or inhaled medications containing levodopa (other than study drugs), and COMT inhibitors.
Double-Blind Treatment Period (Day 1 to Day 85 [Week 12])
In the CSCI optimization phase (day 1 to day 29), all patients first received 1 loading dose of each of oral LD-CD (100 mg/25 mg) tablets and foslevodopa (240 mg/mL) and foscarbidopa (12 mg/mL) on day 1 at a dose (i.e., number of tablets and volume of infusion) based on the first dose of oral LD-CD of the day at the end of the oral LD-CD stabilization period.
Foslevodopa-foscarbidopa arm: After the dual loading doses, patients started receiving CSCI (24 hours per day) of foslevodopa-foscarbidopa solution via a portable infusion pump plus oral placebo capsules for LD-CD IR. The initial infusion rate of foslevodopa-foscarbidopa was calculated based on the patient’s stabilized oral LD-CD IR therapy at the end of the oral LD-CD stabilization period and a conversion algorithm based on the pharmacokinetic characteristics of foslevodopa-foscarbidopa established from phase I studies. The oral placebo capsules administered matched, in number of tablets and dosing frequency, the therapeutic schedule achieved during the end of the oral LD-CD stabilization period.
Oral LD-CD arm: Patients received CSCI of placebo solution (0.9% weight/volume NaCl solution) in addition to oral LD-CD tablets and the infusion rate, and dose and frequency of oral administration were based on the (conversion of the) dosing of oral LD-CD tablets achieved at the end of the oral LD-CD stabilization period.
In the CSCI optimization phase, only changes to the CSCI rate were permitted in both treatment arms. The investigator adjusted infusion rates after an assessment of PD symptoms and clinical response during each visit to optimize clinical response. The allowable infusion rate ranged from 0.15 mL per hour to 1.04 mL per hour.
In the maintenance phase (day 29 to day 85), patients continued with the same treatment regimen for both CSCI and oral therapies until the end of study, with no dose adjustments allowed.
Training on the correct use of the infusion pump was provided in the screening and oral LD-CD stabilization periods, and all patients had to demonstrate familiarity with the use of the infusion pump to be enrolled in the trial. The Phillips-Medisize Parkinson’s Disease Subcutaneous Pump Delivery System was used (synonymous with the Vyafuser infusion pump approved for use with foslevodopa-foscarbidopa in Canada).
Open-label rescue treatment with oral LD-CD (100 mg/25 mg) IR tablets could be administered as needed in case of sudden deterioration of clinical condition (e.g., motor symptoms) in the double-blind treatment period. The suggested dosing or maximum dose was not stated in the submission.
All patients, caregivers, and investigators were blinded to treatment allocation. LD-CD IR tablets were over-encapsulated to look identical to the placebo capsule and identical packaging was used. Since foslevodopa-foscarbidopa and the placebo solution might not appear identical, and given the nature of the study design, several measures were undertaken to help maintain the blind and prevent functional unblinding. These measures included providing identical packaging of both study drug solutions, requiring separation of site personnel roles, including the use of a separate blinded rater (different from the treating investigator) to perform all in-person efficacy assessments, and disabling the available pump features (extra dose, and high and low infusion rates) designed for the intended commercial pump. In addition, the loading dose was only available during dual loading on day 1; it was then disabled by the investigator for the remainder of the study.
Outcomes
A list of efficacy end points assessed in this clinical review report are provided in Table 7. These end points are further summarized in the following. Summarized end points are based on those included in the sponsor’s Summary of Clinical Evidence15 as well as any identified as important to this review according to stakeholders, for example, the clinical expert, clinician groups, or patient groups.
Table 7: Summary of Outcome Measures in the M15-736 Trial
Outcome measure | Time point | M15-736 |
|---|---|---|
“On” time (assessed based on the PD diary) | ||
Average daily “on” time without troublesome dyskinesia | ||
Normalized value, change from baseline | 12 weeks | Primarya |
Normalized value, % change from baseline | Exploratory | |
Absolute value, change from baseline | Exploratory | |
Average daily “on” time without dyskinesia | ||
Normalized value, change from baseline | 12 weeks | Secondarya |
Normalized value, % change from baseline | Exploratory | |
Absolute value, change from baseline | Exploratory | |
“Off” time (assessed based on the PD diary) | ||
Average daily “off” time | ||
Normalized value, change from baseline | 12 weeks | Key secondarya |
Normalized value, % change from baseline | Exploratory | |
Absolute value, change from baseline | Exploratory | |
Presence of morning akinesiab | At week 12 | Key secondarya |
HRQoL | ||
Change from baseline in PDQ-39 summary index (PD-related QoL) | 12 weeks | Secondarya |
Change from baseline in EQ-5D-5L summary index | Secondarya | |
PD symptoms and daily activities | ||
Change from baseline in MDS-UPDRS scores | ||
Part II score (motor experiences of daily living) | 12 weeks | Key secondarya |
Part I, III, and IV scores | Exploratory | |
Change from baseline in bradykinesia score, median and IQR | 12 weeks | Secondarya |
Change from baseline in dyskinesia score, median and IQR | Secondarya | |
Change from baseline in PDSS-2 total score (sleep symptoms) | 12 weeks | Secondarya |
Safety | ||
AEs, SAEs, AESIs, Infusion Site Evaluation Scale, clinical laboratory values, vital sign measurements, ECGs, C-SSRS, QUIP-RS | 12 weeks | Secondary |
AE = adverse event; AESI = adverse event of special interest; C-SSRS = Columbia-Suicide Severity Rating Scale; ECG = electrocardiogram; EQ-5D-5L = 5-Level EQ-5D; HRQoL = health-related quality of life; IQR = interquartile range; MDS-UPDRS = Movement Disorder Society-Unified Parkinson’s Disease Rating Scale; PD = Parkinson disease; PDQ-39 = Parkinson’s Disease Questionnaire-39 items; PDSS-2 = Parkinson’s Disease Sleep Scale-2; QoL = quality of life; QUIP-RS = Questionnaire for Impulsive-Compulsive Disorders in Parkinson’s Disease – Rating Scale; SAE = serious adverse event.
aThese end points were included in the multiple testing hierarchy.
bMorning akinesia was defined as reporting “off” status as the first morning symptom upon awakening.
Source: M15-736 Clinical Study Report.14 (Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
Efficacy Outcomes
Baseline for PD diary variables was defined as the average of the 3 valid diary entries completed before day –1. Baseline for all other efficacy measures was defined as the last nonmissing assessment before the initiation of double-blinded study drug (i.e., visit 5). Study visits were scheduled at baseline, days 1, 2, 8, 15, 22, 29, 57, and 85 (week 12).
“On” and “Off” Times Outcomes (PD Diary)
Change from baseline at week 12 in average daily normalized “on” time without troublesome dyskinesia (primary end point), “on” time without dyskinesia (secondary), and “off” time (key secondary) were assessed using data collected in the 3 most recent valid PD diary days before each visit (not measured at the day 2 visit). The recorded time spent in each motor state was normalized to a typical waking day of 16 hours (e.g., by dividing the absolute “off” time by the daily awake time, then multiplying by 16 hours). A valid PD diary day was defined as 1 within 7 days before a clinical visit with no more than 2 hours of missing data for the 24-hour diary.
Presence of morning akinesia was a key secondary end point, and morning akinesia was defined as reporting “off” status as the first morning symptom upon awakening and assessed based on the PD diary.
At screening, all patients received a PD diary training to learn about proper completion of the diary. An assessment was conducted to ensure minimum 75% concordance between the patient’s PD diary and the PD diary completed by the investigator before the patient was enrolled in the study. Patients were also required to complete the PD diary for 3 consecutive days before the second screening visit and all subsequent study visits. All patients were reminded to complete their diary entries before each visit to reinforce the importance of PD diary completion. Patients who incorrectly completed their PD diary received retraining.
For a description of the PD diary, as well as its psychometric properties, and minimally important difference (MID) estimates, refer to Table 8.
Movement Disorder Society-Unified Parkinson’s Disease Rating Scale
Changes from baseline to week 12 in MDS-UPDRS Part II score (key secondary end point) and Part I, III, and IV scores (exploratory end points) were assessed. The instrument was administered during the patient’s best “on” time and, if possible, at the same time of day at each study visit, except at day 1 visit. A separate rater (i.e., other than the site personnel who provide the study drug or the treating investigator) was responsible for performing MDS-UPDRS assessments. A description of the MDS-UPDRS instrument, as well as its psychometric properties, and MID estimates are given in Table 8.
Parkinson’s KinetiGraph and Personal KinetiGraph
The secondary outcomes of median and IQR of bradykinesia and dyskinesia scores were measured using the PKG. The PKG is a movement-recording device that the patients wore on their wrists continuously during the double-blind treatment period to collect data during the activities of daily living in the home environment. The PKG is used to assess PD symptoms such as tremor, bradykinesia, and dyskinesia.
For each patient, the PKG collected data continuously, and an algorithm calculated a bradykinesia score every 2 minutes between 9 a.m. and 6 p.m. across multiple days, with the median bradykinesia score calculated at a certain visit for each patient. There was no prespecified range of scores. A higher median score indicated worse bradykinesia. A higher bradykinesia IQR score (calculated as the difference between the third quartile and first quartile bradykinesia scores) indicated a higher degree of variability in bradykinesia score. The scoring of median and IQR of dyskinesia scores was the same.42
Parkinson’s Disease Sleep Scale-2
Change from baseline to week 12 in PDSS-2 total score was a key secondary outcome, and measurements were made at the baseline and day 85 (weeks 12) visits.
The PDSS-2 scale was administered by central raters who did not have access to results of other study assessments or patients’ medical records, and did not participate in the care or management of patients. The instrument is designed to characterize different aspects of nocturnal sleep problems in patients with PD. It consists of 15 items that assesses 3 domains of sleep symptoms: motor symptoms at night, PD symptoms at night, and disturbed sleep. It assesses overall sleep quality, insomnia, sleep fragmentation, restless leg syndrome, hallucinations, sleep apnea, akinesia, pain, nocturnal immobility, and rapid eye movement behaviour disorder. The total score ranged from 0 to 60, where a higher total score indicates more severe nocturnal sleep problems.
Parkinson’s Disease Questionnaire-39 Items
Change from baseline to final visit (week 12) in PDQ-39 summary index was a secondary outcome, and measurements were made at the baseline and day 85 (weeks 12) visits. A description of the PDQ-39 instrument, as well as its psychometric properties, and MID estimates are presented in Table 8.
5-Level EQ-5D
Change from baseline to final visit (week 12) in EQ-5D-5L summary index was a key secondary outcome. The EQ-5D-5L instrument was measured at the baseline and day 85 (weeks 12) visits.
EQ-5D-5L is a generic PRO instrument for measuring HRQoL that can be used in a wide range of health conditions and treatments (including PD). The instrument measures patients’ overall health status by generating an index-based summary score on 5 health dimensions (mobility, self-care, daily activities, pain, and anxiety) based upon societal preference weights. In the study, the health status was converted to an index value that ranged from a worst score of –0.109 to a best score of 1 using the US-specific weighted scoring algorithm.
Harms Outcomes
Safety end points (secondary end points) included local and systemic tolerability based on AEs (at all visits), the Infusion Site Evaluation Scale (at all visits except for baseline and day 1), clinical and laboratory values (at baseline and day 85), electrocardiograms (at baseline, days 1, 15, 29, and 85), C-SSRS scores (all visits), and QUIP-RS scores (at baseline, days 15, 29, and 85).
Infusion Site Evaluation Scale
An investigator or qualified designee evaluated the infusion site using a 2-part (numeric and letter grading) scale to assess irritation. Numeric grades ranged from 0 (no evidence of irritation) to 7 (strong reaction spreading beyond the test site), and letter grading from A (no finding) to G (small petechial erosions and/or scabs). Any score above 2 or C was recorded as an AE.
Columbia-Suicide Severity Rating Scale
The C-SSRS, a patient-administered instrument, was used to assess suicidal behaviours and ideation, track and assess all suicidal events, and assess lethality of attempts. Additional features assessed include frequency, duration, controllability, reason for ideation, and deterrents. Patients were asked to indicate if they had suicidal ideation with plan within the last year by answering yes to questions in the suicidal ideation portion of the instrument or in a clinical interview.
Questionnaire for Impulsive-Compulsive Disorders in Parkinson’s Disease — Rating Scale
The QUIP-RS was administered by an investigator to assess severity of impulse-control disorders and related behaviours reported to occur in PD. The questionnaire uses a 5-point scale for respondents to rate the severity of each symptom based on its frequency, from 0 (never) to 4 (always). The QUIP-RS has 4 primary questions. A higher score indicated greater severity.
Table 8: Summary of Select Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID estimates |
|---|---|---|---|
PDQ-39 | The PDQ-39 is a commonly used, self-administered,a disease-specific HRQoL measure recommended by the MDS.43 It consists of 8 domains (mobility, ADL, emotional well-being, stigma, social support, cognition, communication, and bodily discomfort) and a summary index. Each item within a domain is graded on a 5-point Likert scale (0 = never; 4 = always).44 The summary index score and summary domain scores range from 0 to 100, where lower scores indicate a better perceived health status.15 | Validity: Construct validity of PDQ-39 has been extensively demonstrated via assessments using generic HRQoL scales, disease-specific instruments, “known groups” comparisons, and other health measures.45 Assessments have been conducted in different countries (including Canada) and using different languages,46 with a high completion rate.46,47 Of the 12 areas of HRQoL identified as relevant to patients with PD, the PDQ-39 measures 10 (i.e., all areas except for self-image and sexual function).47 Convergent validity was demonstrated for individual domain score of the scale compared to other patient-reported measures (Columbia-Suicide Severity Rating Scale and Hoehn and Yahr Scale): moderate to strong correlations for physical aspects (Spearman correlation, r > 0.5) but psychosocial aspects had weak correlations (emotions, stigma, social; r < 0.3).46 Concurrent validity has been assessed comparing to other established measures of disease severity, depression, and anxiety.48 Reliability: Adequate internal consistency and test-retest reliability have been documented in several assessments.44,45,49-51 Correlations to related domains of SF-36 were strong across multiple assessments based in different countries (US, UK) with some exceptions (social support subscale in the UK, and social support and cognition subscales in the US).50,52 Responsiveness: Responsiveness of PDQ-39 was confirmed in some of the PDQ-39 subscales in 2 studies according to a published SLR on disease-specific HRQoL instruments in PD.45 One study identified good responsiveness to change in the mobility and ADL subscales (in 51 patients with PD who indicated their situation had worsened over a period of 4 months) based on correlation with self-reported change and change in the SF-3653 and in 4 subscales (mobility, ADL, stigma, and social support) in another study.48 | Total score:54 –1.6 Mobility:54 –3.2 ADL:54 –4.4 Emotional well-being:54 –4.2 Stigma:54 –5.6 Social support:54 –1.4 Cognition:54 –1.8 Communication:54 –4.2 Pain:54 –2.1 Based on a survey of randomly selected members of 13 branches of the Parkinson Society who completed the PDQ-39 on 2 occasions, 6 months apart.54 |
PD diary | The PD diary is a self- or caregiver-administered diary that is used to assess the amount of “on” and “off” time PD patients experience in a 24-hour period. It has 5 categories:
| Validity: Assessed in a single study of 302 patients from 10 countries with idiopathic PD who were experiencing motor fluctuations and dyskinesia and who were capable of accurately completing the diaries, the PD diary was shown to be feasible and simple to use (83% completion rate without duplication or error), although errors and noncompliance were more prevalent after 3 days of use. Acceptable construct (concurrent) validity was also shown: moderate correlation was observed between other VAS measures and corresponding PD diary measures.55 Reliability: Assessed in the same study as validity.55 Mean ICC was 0.71 (mean correlation between any 2 days = 0.74). Test-retest reliability improved as number of diary days increased (Cronbach alpha > 0.8 between any 2 or more days). Responsiveness: No literature was identified. | “Off” time: 1 to 1.3 hours, based on studies of patients with advanced PD receiving pramipexole extended release, pramipexole IR, or rasagiline55-57 |
MDS-UPDRS | The MDS-UPDRS is a revision of the UPDRS originally developed in the 1980s, sponsored by MDS to address problematic areas of the original scale and to provide a more comprehensive assessment across the spectrum of disability.58 The MDS-UPDRS was developed to evaluate nonmotor and motor experiences of daily living in patients with PD. It is an investigator-administered instrument comprising 4 parts, each of which includes several items related to different areas of functioning.58 Each part is meant to be reported separately and not as a combined total MDS-UPDRS summary score.58 Each item is rated on a 5-point scale where 0 = normal and 4 = severe.58 Part I (13 items) assesses “nonmotor experiences of daily living,” including mood, cognitive ability, hallucinations, and sleep disturbances, among other items.58 Score range: 0 to 52.59 Scores < 10 are “mild,” ≥ 22 are “severe.”60 Part II (13 items) assesses “motor experiences of daily living,” including several items related to fine motor skills and daily tasks like eating or dressing.58 Score range: 0 to 52.59 Scores < 12 are “mild,” ≥ 30 are “severe.”60 Part III (33 scores based on 18 items) assesses “motor examination,” including posture and tremor.58 Score range: 0 to 132.59 Scores < 32 are “mild,” ≥ 59 are “severe.”60 Part IV (6 items) concerns “motor complications,” such as time spent with dyskinesia.58 Score range: 0 to 24.59 Scores < 4 are “mild,” ≥ 13 are “severe.”60 | Validity: Assessed in a study of 877 patients with PD.58 Concurrent validity was shown based on high correlations between MDS-UPDRS and the original UPDRS (in whole and when comparing each part). As a measure of internal validity, there was low correlation between the parts (except when comparing Parts II and III, both of which assess motor function), confirming that each part assesses a different aspect of PD. The MDS-UPDRS had excellent factor validity based on the factor analysis confirming that the items cluster in clinically pertinent domains. Similar results have been found in several other studies of validity on the MDS-UPDRS or component parts based on correlations to UPDRS or other HRQoL measures.60-63 Reliability: Internal consistency was adequate for each part (alphas ranged from 0.79 to 0.93 per part).58 Similar results were found in an assessment of the Spanish language MDS-UPDRS.61 Test-retest reliability was also adequate and correlation with related constructs was high.61 Responsiveness: A longitudinal, multicentre study of 362 participants with de novo PD identified that MDS-UPDRS total and individual scores of Parts I, II, and III increased in a linear fashion over 5 years at a similar rate to that observed by the original UPDRS, which has been previously shown to have good sensitivity to change.64 | Part I: Improvement > 2.64 or worsening > 2.45.65 Part II: Improvement > 3.05 or worsening > 2.51.65 Part III: –3.25 for improvement, + 4.63 for worsening.66 Part IV: Improvement > 2.1, worsening > 1.8.67 Part I and II were assessed in 985 paired investigations of 365 patients with PD.65 Part III was assessed in 728 investigations of 260 patients.66 Part IV was assessed in 1,044 paired investigations of 436 patients.67 |
ADL = activities of daily living; HRQoL = health-related quality of life; ICC = intraclass correlation; IR = immediate release; MDS = Movement Disorder Society; MDS-UPDRS = Movement Disorder Society-Sponsored Revision of the Unified Parkinson’s Disease Rating Scale; MID = minimally important difference; PD = Parkinson disease; PDQ-39 = Parkinson’s Disease Questionnaire-39 items; SF-36 = 36-Item Short Form Survey; SLR = systematic literature review; UPDRS = Unified Parkinson’s Disease Rating Scale.
aIn the M15-736 trial, the questionnaire was administered by a blinded central rater via a telephone interview.
Statistical Analysis
Sample Size and Power Calculation
A sample size calculation determined that a total of 130 randomized patients was required to demonstrate statistically significant difference between the foslevodopa-foscarbidopa arm and oral LD-CD arm with respect to the change from baseline to week 12 in average daily normalized “on” time without troublesome dyskinesia (primary end point) at a 2-sided significance level of 0.05 with a power of 90%, assuming a treatment difference between foslevodopa-foscarbidopa and oral LD-CD of 1.86 hours and an SD of 2.9 hours, and 20% of treatment discontinuation. This sample size also has approximately 90% power for key secondary end points of change from baseline in average daily normalized “off” time, MDS-UPDRS Part II score, and presence of morning akinesia at week 12.
Primary Efficacy End Point: Statistical Test and Model
In the primary analysis, the difference in the mean change from baseline to week 12 in average daily normalized “on” time without troublesome dyskinesia between treatment arms in the full analysis set was tested at a 2-sided significance level of 0.05 using a mixed model for repeated measures (MMRM). The model was stratified by categorical fixed effects of treatment, country, and visit, treatment-by visit and treatment-by-baseline interactions, and the continuous fixed covariate of baseline measurement, assuming an unstructured variance covariance matrix.
Primary Efficacy End Point: Subgroup and Sensitivity Analyses
Prespecified subgroup analyses were performed by age, sex, race, country, duration of PD, concomitant dopamine agonist use, and levodopa dose category. Subgroup analyses by age (younger than 65 years versus at least 65 years), duration of PD (less than 10 years versus at least 10 years), and dose category (low versus high levodopa dose) are of interest to this review based on input from the clinical expert. The analyses were performed on the full analysis set using an analysis of covariance (ANCOVA) model adjusted for the effect of treatment, subgroup variable, the treatment-by-subgroup variable interaction, and baseline measurement as a covariate. No adjustment for type I error was made. The treatment-by-subgroup interaction was tested at the significance level of 0.10.
Two sensitivity analyses were performed to account for missing data:
“Jump-to-reference” analytic approach – All missing data in the oral LD-CD arm and nonmonotonic missing data in the foslevodopa-foscarbidopa arm were assumed to be missing at random. For patients in the foslevodopa-foscarbidopa arm with monotonic missing values for reasons other than COVID-19 infections or logistical restrictions, missing values were assumed to be missing not at random and have a profile for visits after discontinuation of treatment that equals the profile of the oral LD-CD arm.
Last available value approach – Missing week 12 data were imputed based on the patient’s last available value in estimating the change from baseline to week 12 based on an ANCOVA model with the categorical fixed effects of treatment and country, and baseline score as a covariate.
Primary Efficacy End Point: Handling of Intercurrent Events and Missing Data
Handling of missing items in efficacy instrument are summarized in Table 9. Intercurrent events related to discontinuation of study drug were handled using the hypothetical strategy, where all data collected after the last dose of blinded study drug were not used in the analyses. Intercurrent events related to rescue medication use were handled using treatment policy strategy, where collected data were used regardless of recue medication use. Missing data were implicitly imputed using a MMRM based on the assumption that data were missing at random.
Secondary Efficacy End Point: Multiple Testing Procedure
The key secondary and other secondary outcomes were included in multiplicity adjustment of the type I error to control the family-wise error rate at a 2-sided significance level of 0.05 in the hierarchal order, as outlined in Table 9.
Secondary Efficacy End Point: Statistical Test and Model
Change from baseline in “off” and “on” times end points, MDS-UPDRS Part II score, and median and IQR of bradykinesia and dyskinesia scores were analyzed using the same statistical approach as the primary analysis of the primary end point. The presence of morning akinesia at week 12 was analyzed using a generalized linear mixed model adjusted for categorical fixed effects of treatment, country, visit, treatment-by-visit interaction, and baseline first morning status upon awakening. The change from baseline to final visit in PDSS-2, PDQ-9, EQ-5D-5L scores were analyzed using an ANCOVA model adjusted for categorical fixed effects of treatment and country, and baseline score. The methods of handling intercurrent events and missing data were the same as the primary end-point analysis.
For all “on” and “off” times outcomes, subgroup and sensitivity analyses were conducted using the same approaches as for the primary end point. For all other key secondary and other secondary end point, only subgroup analyses by dose category were conducted.
Safety End Points
Safety outcomes were compared between treatment arms using descriptive statistics and no hypothesis testing was performed.
Table 9: Statistical Analysis of Efficacy End Points in the M15-736 Trial
Order in testing hierarchy | End point | Statistical model | Adjustment factors | Handling of missing data | Sensitivity analyses |
|---|---|---|---|---|---|
1 | Change from baseline to week 12 in average daily normalized: “On” time without troublesome dyskinesia | MMRM | Categorical fixed effects of treatment, country and visit, treatment-by visit and treatment-by-baseline interactions, and the continuous fixed covariate of baseline measurement | Missing items in efficacy instrument
|
|
5a | Change from baseline to week 12 in average daily normalized: “On” time without dyskinesia | — | |||
2 | Change from baseline to week 12 in average daily normalized: “Off” time | — | |||
3 | Change from baseline to week 12 in MDS-UPDRS Part II score | MMRM | Categorical fixed effects of treatment, country and visit, treatment-by visit and treatment-by-baseline interactions, and the continuous fixed covariate of baseline measurement | Missing items in efficacy instrument
Missing visit data
| Not performed |
— | Change from baseline to week 12 in MDS-UPDRS Part I, III, and IV scores | ||||
9a | Change from baseline to week 12 in: median bradykinesia score | MMRM | Categorical fixed effects of treatment, country and visit, treatment-by visit and treatment-by-baseline interactions, and the continuous fixed covariate of baseline measurement | Missing visit data were imputed using MMRM | Not performed |
10a | Change from baseline to week 12 in: IQR of bradykinesia score | ||||
11a | Change from baseline to week 12 in: median dyskinesia score | ||||
12a | Change from baseline to week 12 in: IQR of dyskinesia score | ||||
4a | The presence of morning akinesia at week 12 | GLMM | Categorical fixed effects of treatment, country, visit, treatment-by-visit interaction, and baseline first morning status upon awakening | No imputation | Not performed |
6a | Change from baseline to final visit in: PDSS-2 total score | ANCOVA | Categorical fixed effects of treatment and country, and baseline score (continuous) | Missing items in efficacy instrument PDSS-2
PDQ-39
EQ-5D-5L
| Not performed |
7a | Change from baseline to final visit in: PDQ-39 | ||||
8a | Change from baseline to final visit in: EQ-5D-5L |
ANCOVA = analysis of covariance; EQ-5D-5L = 5-Level EQ-5D; GLMM = generalized linear mixed model; IQR = interquartile range; MDS-UPDRS = Movement Disorder Society-Unified Parkinson’s Disease Rating Scale; MMRM = mixed model for repeated measures; PDQ-39 = Parkinson’s Disease Questionnaire-39 items; PDSS-2 = Parkinson’s Disease Sleep Scale-2; VAS = visual analogue scale.
aThe results for this outcome should not be interpreted as statistically significant if the P value for the comparison is < 0.05 due to failure of a prior end point in the statistical testing hierarchy (MDS-UPDRS Part II score).
Source: M15-736 Clinical Study Report.14 (Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
Analysis Populations
Analysis populations of M15-736 are summarized in Table 10.
Table 10: Analysis Populations in the M15-736 Trial
Population | Definition | Application |
|---|---|---|
FAS | All randomized patients who received any dose of study drug during the double-blind treatment period and who have baseline and at least 1 post-baseline observation for at least 1 efficacy assessment | All efficacy analyses unless otherwise stated. Patients were included in the analysis according to the treatment group to which they were randomized |
SAS | All patients who received any dose of study drug during the double-blind treatment period | All demographic, baseline, and safety analyses unless stated otherwise. Patients were included in the analysis according to the study drug that they actually received, regardless of randomization |
FAS = full analysis set; SAS = safety analysis set.
Note: The M15-736 trial also included an oral levodopa-carbidopa analysis set, but it was not of interest to this review and was not summarized.
Source: M15-736 Clinical Study Report.14 (Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
Results
Patient Disposition
A summary of patient disposition in the M15-736 trial is shown in Table 11. Of 270 screened patients, 96 (35.6%) were screen failures, primarily because of failure to meet eligibility criteria (18.5%) and consent withdrawal (13.3%). Both the full and the safety analysis sets included 141 patients who were randomized to the foslevodopa-foscarbidopa arm (N = 74; 52.5%) and the oral LD-CD arm (N = 67; 47.5%) in the double-blind treatment period. Of the patients who discontinued study treatment, 26 (35.1%) were in the foslevodopa-foscarbidopa arm and 5 (7.5%) patients in the oral LD-CD arm. The most common reason for study treatment discontinuation in the foslevodopa-foscarbidopa arm was AEs (18.9%).
Table 11: Summary of Patient Disposition in the M15-736 Trial (SAS)
Patient disposition | Foslevodopa-foscarbidopa arm | Oral LD-CD arm | |
|---|---|---|---|
Screened, N | 270 | ||
Primary reason for screening failure, n (%) | 96 (35.6) | ||
Eligibility criteria | 50 (18.5) | ||
Consent withdrawal | 36 (13.3) | ||
Lost to follow-up | 6 (2.2) | ||
Other | 4 (1.5) | ||
Enrolled, N | 141 | ||
Randomized, N (%) | 74 (52.5) | 67 (47.5) | |
Discontinued from study, n (%) | 26 (35.1) | 5 (7.5) | |
Primary reason for discontinuation, n (%) | |||
AEs | 14 (18.9) | 1 (1.5) | |
Consent withdrawal | 5 (6.8) | 3 (4.5) | |
Lost to follow-up | 0 | 0 | |
Lack of efficacy | 1 (1.4) | 0 | |
Difficulty with drug delivery | 4 (5.4) | 1 (1.5) | |
Other | 2 (2.7) | 0 | |
FAS, N (%) | 74 (100) | 67 (100) | |
SAS, N (%) | 74 (100) | 67 (100) | |
CD = carbidopa; FAS = full analysis set; LD = levodopa; SAS = safety analysis set.
Source: M15-736 Clinical Study Report.14 (Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
Baseline Characteristics
A summary of baseline patient demographics, disease characteristics, and medication history in M15-736 trial is shown in Table 12. The baseline characteristics outlined in the table are limited to the ones that are most relevant to this review or that were considered to most likely impact the outcomes or interpretation of the study results.
The mean age of the study population was 66.4 years (SD = 9.5 years). The majority of patients were male (70.2%) and white (92.9%). Mean time since diagnosis was 8.6 years (SD = 4.9 years), and mean time since onset of motor fluctuation was 5.6 years (SD = 4.0 years). The majority of patients had a history of levodopa-induced dyskinesia (62.4%). The mean time spent in “off” states and “on” without troublesome dyskinesia motor states were 6.1 hours (SD = 2.1 hours) and 9.3 hours (SD = 2.5 hours), respectively. Mean dosing frequency was 6.2 times per day (SD = 2.6 times per day). The baseline characteristics were similar in the treatment arms, except that more patients in the foslevodopa-foscarbidopa arm had levodopa response of more than 5 years (77.0%) and received prior dopamine agonist treatment (56.8%) compared with the oral LD-CD arm (68.7% and 46.3%, respectively).
Table 12: Summary of Baseline Characteristics in the M15-736 Trial (SAS)
Characteristic | Foslevodopa-foscarbidopa arm (N = 74) | Oral LD-CD arm (N = 67) |
|---|---|---|
Demographics | ||
Sex, n (%) | ||
Male | 50 (67.6) | 49 (73.1) |
Female | 24 (32.4) | 18 (26.9) |
Age (years), mean (SD) | 66.3 (9.2) | 66.6 (9.8) |
Race, n (%) | ||
White | 70 (94.6) | 61 (91) |
Black/African American | 2 (2.7) | 2 (3) |
Asian | 0 | 3 (4.5) |
American Indian/Alaska Native | 1 (1.4) | 0 (0) |
Native Hawaiian or Pacific Islander | 1 (1.4) | 1 (1.5) |
Multiple | 0 | 0 |
Disease characteristics | ||
Duration of PD since diagnosis (years), mean (SD) | 8.4 (4.2) | 8.8 (5.5) |
History of levodopa-induced dyskinesia, n (%) | 45 (60.8) | 43 (64.2) |
Levodopa response of > 5 years, n (%) | 57 (77.0) | 46 (68.7) |
Baseline PD Hoehn and Yahr Scale stage, n (%) | ||
0 | 0 | 1 (1.5) |
1 | 9 (12.2) | 4 (6.0) |
2 | 43 (58.1) | 45 (67.2) |
3 | 20 (27.0) | 14 (20.9) |
4 | 2 (2.7) | 2 (3.0) |
5 | 0 | 1 (1.5) |
Missing | 0 | 0 |
Duration since onset of motor fluctuation (year), mean (SD) | 5.6 (3.8) | 5.6 (4.1) |
MDS-UPDRS Part III score during “off” state, mean (SD) | NR | NR |
Normalized “on” or “off” time per day (hours),a mean (SD) | ||
“Off” time | 6.3 (2.3) | 5.9 (1.9) |
“On” time without troublesome dyskinesia | 9.2 (2.4) | 9.5 (2.6) |
“On” time without dyskinesia | 7.2 (3.1) | 7.5 (3.7) |
PD treatment history | ||
Prior DBS procedure, n (%) | 1 (1.4) | 0 |
Prior PD medication use, n (%) | 74 (100) | 67 (100) |
Dopa and dopa derivatives | 74 (100) | 67 (100) |
Dopamine agonists | 42 (56.8) | 31 (46.3) |
MAO-B inhibitor | 29 (39.2) | 26 (38.8) |
Amantadine | 17 (23.0) | 19 (28.4) |
COMT inhibitor | 7 (9.5) | 4 (6.0) |
Anticholinergics | 2 (2.7) | 2 (3.0) |
Other | 3 (4.1) | 3 (4.5) |
Number of PD medication class at baseline, n (%) | ||
1 | 19 (25.7) | 23 (34.3) |
2 | 34 (45.9) | 26 (38.8) |
3 | 19 (25.7) | 14 (20.9) |
4 | 2 (2.7) | 4 (6.0) |
> 4 | 0 | 0 |
Baseline oral LD-CD IR dose (mg/day), mean (SD) | 1,203 (572) | 1,134 (583) |
Baseline oral LD-CD IR frequency (times/day), mean (SD) | 6.2 (2.5) | 6.2 (2.9) |
Baseline levodopa-equivalent dose, including other PD medications (mg/day),b mean (SD) | 1,407 (739) | 1,287 (613) |
CD = carbidopa; COMT = catechol-O-methyltransferase; DBS = deep brain stimulation; IR = immediate release; LD = levodopa; MAO-B = monoamine oxidase type B; MDS-UPDRS = Movement Disorder Society-Unified Parkinson’s Disease Rating Scale; NR = not reported; PD = Parkinson disease; SAS = safety analysis set; SD = standard deviation.
aBased on the PD diary.
bRefers to PD medications other than oral LD-CD IR tablets.
Source: M15-736 Clinical Study Report.14 (Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
Exposure to Study Treatments
Treatment exposure from the M15-736 trial is summarized in Table 13. Mean average total daily levodopa dose was 1,667 mg per day (SD = 741 mg per day) in the foslevodopa-foscarbidopa arm and 1,105 mg per day (SD = 583 mg per day) in the oral LD-CD arm. Mean duration of study drug exposure was 62.4 days (SD = 31.2 days) in the foslevodopa-foscarbidopa arm and 81.5 days (SD = 19.0 days) in the oral LD-CD arm. Mean treatment adherence for drug solution and drug capsule was a least 85% in both treatment arms.
Concomitant Medications
Concomitant PD medication use in the M15-736 trial is summarized in Table 14. In the trial, 74.3% of patients in the foslevodopa-foscarbidopa arm and 67.2% of patients received at least 1 concomitant PD medications. The most common class of PD medications (in at least 30% of patients in either treatment arm) were dopamine agonist and MAO-B inhibitors. A higher proportion of patients receiving dopamine agonist in the foslevodopa-foscarbidopa arm (45.9%) than in the oral LD-CD arm (37.3%). No notable between-arm differences were observed for the other PD medications.
Table 13: Treatment Exposure in the M15-736 Trial (SAS)
Exposure in the double-blind treatment period | Foslevodopa-foscarbidopa arm (N = 74) | Oral LD-CD arm (N = 67) |
|---|---|---|
Average total daily levodopa dose (mg) | ||
Mean (SD) | 1,667 (741) | 1,105 (583) |
Median (range) | 1,477 (686 to 3,888) | 964 (237 to 2,637) |
Duration of study drug exposure (days) | ||
Mean (SD) | 62.4 (31.2) | 81.5 (19.0) |
Median (range) | 84.0 (3 to 92) | 85.0 (3 to 106) |
% Adherence, mean (SD) | ||
Drug solutiona | 98.3 (2.7) | 98.7 (1.4) |
Drug capsuleb | 86.5 (18.6) | 92.9 (18.0) |
CD = carbidopa; LD = levodopa; SAS = safety analysis set; SD = standard deviation.
aStudy drug solution adherence (%) = hours of infusion divided by 24, then multiply by 100.
bstudy drug capsules adherence (%) = number of capsules taken divided by number of capsules prescribed, the multiply by 100.
Source: M15-736 Clinical Study Report.14 (Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
Table 14: Concomitant PD Treatment in the M15-736 Trial (SAS)
Concomitant medication (class) | Foslevodopa-foscarbidopa arm (N = 74) | Oral LD-CD arm (N = 67) |
|---|---|---|
Any concomitant PD medications,a n (%) | 55 (74.3) | 45 (67.2) |
Dopaminergic drugs | 54 (73.0) | 43 (64.2) |
Dopamine agonists | 34 (45.9) | 25 (37.3) |
Dopa and dopa derivatives | 3 (4.1) | 0 |
MAO-B inhibitors | 26 (35.1) | 21 (31.3) |
Amantadine | 14 (18.9) | 15 (22.4) |
Anticholinergic drugs | 1 (1.4) | 3 (4.5) |
Other anti-PD drugs | 2 (2.7) | 3 (4.5) |
CD = carbidopa; LD = levodopa; MAO-B = monoamine oxidase type B; PD = Parkinson disease; SAS = safety analysis set.
aNote that patients could have received more than 1 concomitant medication during the treatment period.
Source: M15-736 Clinical Study Report.14 (Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
Rescue Medications
Oral LD-CD was used as a rescue medication. A summary of its use is shown in Table 15. Oral LD-CD tablet as a rescue medication was required by 68.9% of patients in the foslevodopa-foscarbidopa arm and 56.7% oral LD-CD arm. Mean number of doses of rescue medication administered was 44.8 (SD = 52.5) in the foslevodopa-foscarbidopa arm and 77.5 (SD = 125.8) in the oral LD-CD arm. Mean total daily levodopa dose from rescue medications was numerically higher in the oral LD-CD arm than foslevodopa-foscarbidopa arm at most time points.
Table 15: Rescue Medication Use in the M15-736 Trial (SAS)
Rescue medication use (oral LD-CD IR Tablet) | Foslevodopa-foscarbidopa arm (N = 74) | Oral LD-CD arm (N = 67) |
|---|---|---|
Patients requiring ≥ 1 dose of rescue medication, n (%) | 51 (68.9) | 38 (56.7) |
1 rescue dose | 2 (2.7) | 1 (1.5) |
2 rescue doses | 4 (5.4) | 5 (7.5) |
≥ 3 rescue doses | 45 (60.8) | 32 (47.8) |
Number of doses of rescue medication administered | ||
Mean (SD) | 44.8 (52.5) | 77.5 (125.8) |
Median (range) | 19 (1 to 176) | 15.5 (1 to 553) |
Total daily levodopa dose from rescue medications (mg/day), mean (SD) | ||
Week 1 | 174.6 (102.5) | 187.5 (104.2) |
Week 2 | 221.0 (159.1) | 266.7 (193.9) |
Week 4 | 204.9 (127.2) | 240.7 (186.9) |
Week 9 to 12 | 168.5 (98.7) | 287.8 (191.3) |
CD = carbidopa; IR = immediate release; LD = levodopa; SAS = safety analysis set; SD = standard deviation.
Source: M15-736 Clinical Study Report.14 (Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
Protocol Deviations
Protocol deviations in the M15-736 trial are summarized in Table 16. The most common category of protocol deviations was related to study procedure; this occurred in 45.9% of patients in the foslevodopa-foscarbidopa arm and 40.3% of patients in the oral LD-CD arm. Study procedure deviations were most commonly related to extended use of single-use syringe (beyond 24 hours per day) and incomplete recording of PD diary, questionnaire, and rescue dose, although the incidences were not specified.
The sponsor noted that none of the protocol deviations recorded affected the study outcome or interpretation of the study results or conclusions.
Table 16: Protocol Deviations in the M15-736 Trial
Protocol deviations, n (%) | Foslevodopa-foscarbidopa arm (N = 74) | Oral LD-CD arm (N = 67) |
|---|---|---|
Study procedure compliancea | 34 (45.9) | 27 (40.3) |
Wrong treatment or incorrect doseb | 8 (10.8) | 6 (9.0) |
Dosing compliance by patient | 3 (4.1) | 6 (9.0) |
Did not satisfy entry criteriac | 4 (5.4) | 4 (6.0) |
Dispensation or administration of investigation product | 0 | 3 (4.5) |
Expired investigation product | 0 | 3 (4.5) |
Accountability, reconciliation, or destruction of investigation product | 3 (4.1) | 2 (3.0) |
Lack of consent or late consent | 2 (2.7) | 1 (1.5) |
Consent process | 0 | 1 (1.5) |
Inadequate safety reporting | 1 (1.4) | 0 |
Missed visit or visit windows | 1 (1.4) | 0 |
Receive prohibited medication | 1 (1.4) | 0 |
Missing data | 1 (1.4) | 0 |
Received excluded concomitant medication | 1 (1.4) | 0 |
CD = carbidopa; LD = levodopa.
aIn the M15-736 trial, protocol compliance-study procedure deviations included, in decreasing frequency (exact frequencies not stated): single syringe use greater than 24 hours per dosing diary entry, insufficient days completed or incomplete Parkinson disease (PD) diary entries, missed or late questionnaire completion, rescue dose not recorded in dosing diary, study visit completed remotely not due to COVID-19 or before approval of protocol version 4.0, and incorrect loading dose.
bOf the 14 patients who received wrong treatment or incorrect dose in the M15-736 trial, 12 received an incorrect infusion loading dose of 0.5 mL rather than 0.6 mL on day 1 as a result of an Interactive Randomization Technology programming error.
cOf the 8 patients who entered the M15-736 trial but did not satisfy entry criteria, 1 patient’s total daily dose of LD equivalents from LD-containing medications and catechol-O-methyltransferase inhibitors did not meet the minimum 400 mg requirement, 1 patient did not meet the requirement of at least 44 of 48 PD diary entries on 1 of the 3 diary days and also did not have at least 2 hours of “Off” time on 2 of the 3 diary days at baseline, and the other 6 patients did not meet the requirement of at least 44 of 48 PD diary entries on 1 of the 3 diary days at baseline.
Source: M15-736 Clinical Study Report.14 (Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
Efficacy
Unless otherwise specified, efficacy end points from the M15-736 trial that were noted to be important to patients and clinicians based on stakeholder input are summarized in Table 17.
“On” Time Outcomes
“On” Time Without Troublesome Dyskinesia
Change from baseline to week 12 in average daily normalized “on” time without troublesome dyskinesia was the primary end point. In the primary analysis, the LSM difference between the foslevodopa-foscarbidopa arm and the oral LD-CD arm was 1.75 (95% CI, 0.46 to 3.05; P = 0.0083) hours, in favour of foslevodopa-foscarbidopa. Results of the sensitivity analyses (jump-to-reference analytic approach and last available value approach) (refer to Table 46 in Appendix 1) and subgroup analyses of interest (age, duration of PD diagnosis, and levodopa dose intensity) (refer to Figure 4 in Appendix 1) were consistent with the primary analysis.
Results from the analyses of change from baseline in absolute hours and percent change from baseline in normalized hours (Table 47 and Table 48 in Appendix 1) were consistent with the primary analysis of normalized change from baseline.
“On” Time Without Dyskinesia
Change from baseline to week 12 in average daily normalized “on” time without dyskinesia was a secondary end point and is summarized in Table 49 in Appendix 1. The results for this outcome are at increased risk of type I error (false-positive results) because they were tested after failure of the statistical hierarchy. The LSM difference between the foslevodopa-foscarbidopa arm and the oral LD-CD arm was 1.81 (95% CI, 0.46 to 3.16) hours.
“Off” time
“Off” Time
Change from baseline to week 12 in average daily normalized “off” time without dyskinesia was a study-defined key secondary end point and was adjusted for multiplicity. The LSM difference between the foslevodopa-foscarbidopa arm and the oral LD-CD arm was –1.79 (95% CI, –3.03 to –0.54, P = 0.0054) hours, in favour of foslevodopa-foscarbidopa. Results of the sensitivity analyses (jump-to-reference analytic approach and the last available value approach, Table 46 in Appendix 1) and subgroup analyses of interest (age, duration of PD diagnosis, and levodopa dose intensity, Figure 5 in Appendix 1) were consistent with the primary analysis.
Results from the analyses of change from baseline in absolute hours and percent change from baseline in normalized hours (Table 47 and Table 48 in Appendix 1) were consistent with the primary analysis of normalized change from baseline.
Presence of Morning Akinesia
Presence of morning akinesia at week 12 was a study-defined key secondary end point and is summarized in Table 50 in Appendix 1. The results for this outcome are at increased risk of type I error because they were tested after failure of the statistical hierarchy. At baseline, morning akinesia was present in 78.9% of patients in the foslevodopa-foscarbidopa arm, and 76.1% of patients in the oral LD-CD arm. At week 12, morning akinesia was present in 17.0% of patients in the foslevodopa-foscarbidopa arm and 63.3% of patients in the oral LD-CD arm, with an odds ratio of 0.12 (95% CI, 0.04 to 0.31).
Results of subgroup analyses (by levodopa dose intensity) were not reported.
Health-Related Quality of Life
Parkinson’s Disease Questionnaire-39 Items
Change from baseline to week 12 in PDQ-39 summary index total score was a secondary end point. The results for this end point are at increased risk of type I error because they were tested after failure of the statistical hierarchy. The LSM difference between the foslevodopa-foscarbidopa arm and the oral LD-CD arm was –4.10 (95% CI, –8.14 to –0.05).
5-Level EQ-5D
Change from baseline to week 12 in EQ-5D-5L summary index score was a secondary end point. The results for this end point are at increased risk of type I error because they were tested after failure of the statistical hierarchy. Results are summarized in Table 49 in Appendix 1. The LSM difference between the foslevodopa-foscarbidopa arm and the oral LD-CD arm was 0.049 (95% CI, –0.001 to 0.100).
PD Symptoms
Movement Disorder Society-Unified Parkinson’s Disease Rating Scale
Change from baseline to week 12 in MDS-UPDRS Part I (nonmotor experiences of daily living), Part II (motor experiences of daily living), Part III (motor examination), and Part IV (motor complications) were measured.
Part II score was a study-defined key secondary end point and was adjusted for multiplicity. The LSM difference between the foslevodopa-foscarbidopa arm and the oral LD-CD arm in change from baseline to week 12 in MDS-UPDRS Part II score was –1.58 (95% CI, –3.65 to 0.48; P = 0.13).
Part I, III, and IV scores were exploratory end points and were not adjusted for multiplicity. The LSM difference between the foslevodopa-foscarbidopa arm and the oral LD-CD arm in change from baseline to week 12 in Part I, III, and IV scores were 0.00 (95% CI, –1.95 to 1.95), 0.32 (95% CI, –3.62 to 4.26), and –2.22 (95% CI, –3.36 to –1.08), respectively.
Bradykinesia and Dyskinesia Scores Assessed Using the PKG
Change from baseline to week 12 in median bradykinesia score, bradykinesia score IQR (difference between third and first quartile bradykinesia scores), median dyskinesia score, and dyskinesia score IQR, assessed using the PKG wearable device, were secondary end points. The results for these outcomes are at increased risk of type I error (false-positive results) because they were tested after failure of the statistical hierarchy. Results of these analyses are summarized in Table 49 in Appendix 1.
The LSM difference between the foslevodopa-foscarbidopa arm and the oral LD-CD arm in change from baseline to week 12 in median bradykinesia, bradykinesia IQR, median dyskinesia, and dyskinesia IQR scores were 1.72 (95% CI, 0.30 to 3.15), 0.18 (95% CI, –1.20 to 1.55), –2.73 (95% CI, –6.61 to 1.15), and –5.49 (95% CI, –12.71 to 1.73), respectively.
PDSS-2 (Sleep Symptoms)
Change from baseline to week 12 in PDSS-2 total score was a secondary end point and the results for this end point are at increased risk of type I error because they were tested after failure of the statistical hierarchy. The LSM difference between the foslevodopa-foscarbidopa arm and the oral LD-CD arm was –5.40 (95% CI, –8.03 to –2.78).
Table 17: Summary of Key Efficacy Results From the M15-736 Trial (FAS)
Treatment arms | Number of patients contributing to the analysis, n (%) | Baseline value, mean (SD) | Change from baseline at week 12, LSM (SE) | Difference in LSM (95% CI) | P value |
|---|---|---|---|---|---|
Average daily normalized “on” time without troublesome dyskinesia (hours)a | |||||
FOS-FOS | 47 (63.5) | 9.20 (2.42) | 2.72 (0.52) | 1.75 (0.46 to 3.05) | 0.0083 |
Oral LD-CD | 62 (92.5) | 9.49 (2.62) | 0.97 (0.50) | Reference | Reference |
Average daily normalized “off” time (hours)a | |||||
FOS-FOS | 47 (63.5) | 6.34 (2.27) | –2.75 (0.50) | –1.79 (–3.03 to –0.54) | 0.0054 |
Oral LD-CD | 62 (92.5) | 5.91 (1.88) | –0.96 (0.49) | Reference | Reference |
PDQ-39 (PD-related HRQoL)b | |||||
FOS-FOS | 45 (60.8) | 29.31 (15.84) | –6.38 (1.83) | –4.10 (–8.14 to –0.05) | 0.047c |
Oral LD-CD | 59 (88.1) | 26.52 (13.89) | –2.28 (1.75) | Reference | Reference |
MDS-UPDRS Part I score (nonmotor aspects of experiences of daily living)a | |||||
FOS-FOS | 46 (62.2) | 11.35 (6.37) | –1.22 (0.77) | 0.00 (–1.95 to 1.95) | > 0.999d |
Oral LD-CD | 62 (92.5) | 9.37 (5.18) | –1.22 (0.75) | Reference | Reference |
MDS-UPDRS Part II score (motor aspects of experiences of daily living)a | |||||
FOS-FOS | 46 (62.2) | 15.31 (6.93) | –2.65 (0.82) | –1.58 (–3.65 to 0.48) | 0.13 |
Oral LD-CD | 62 (92.5) | 13.27 (6.37) | –1.06 (0.79) | Reference | Reference |
MDS-UPDRS Part III score (motor examination)a | |||||
FOS-FOS | 46 (62.2) | 29.47 (14.97) | –4.25 (1.63) | 0.32 (–3.62 to 4.26) | 0.87d |
Oral LD-CD | 62 (92.5) | 28.23 (13.76) | –4.57 (1.51) | REF | REF |
MDS-UPDRS Part IV score (motor complications)a | |||||
FOS-FOS | 46 (62.2) | 8.91 (3.33) | –2.14 (0.46) | –2.22 (–3.36 to –1.08) | ≤ 0.001d |
Oral LD-CD | 62 (92.5) | 7.73 (3.14) | 0.08 (0.43) | Reference | Reference |
PDSS-2 total score (sleep symptoms)b | |||||
FOS-FOS | 44 (59.5) | 21.7 (9.04) | –7.92 (1.18) | –5.40 (–8.03 to –2.78) | ≤ 0.001c |
Oral LD-CD | 59 (88.1) | 18.7 (8.77) | –2.52 (1.12) | Reference | Reference |
CD = carbidopa; CI = confidence interval; FAS = full analysis set; FOS-FOS = foslevodopa-foscarbidopa; LD = levodopa; LSM = least squares mean; MDS-UPDRS = Movement Disorder Society-Unified Parkinson’s Disease Rating Scale; PD = Parkinson disease; PDQ-39 = Parkinson’s Disease Questionnaire-39 items; PDSS-2 = Parkinson’s Disease Sleep Scale-2; QoL = quality of life; SD = standard deviation; SE = standard error.
Note: Outcomes summarized in this table were noted to be important to patients and clinicians based on input received from patient groups, clinician groups, and the clinical expert consulted by CADTH.
aThe analysis was conducted using a mixed model for repeated measures, adjusted for categorical fixed effects of treatment, country and visit, treatment-by-visit and treatment-by-baseline interactions, and baseline measurement (continuous).
bThis analysis was conducted using an analysis for covariate model, adjusted for categorical fixed effects of treatment and country, and baseline score (continuous).
cAlthough the P value is < 0.05, statistical significance cannot be claimed because the results for the second key secondary end point (MDS-UPDRS Part II), a prior end point in the testing hierarchy, were not statistically significant.
dNot adjusted for multiplicity.
Source: M15-736 Clinical Study Report.14 (Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
Harms
A summary of harms in the M15-736 trial is shown in Table 18. Results of the M15-736 trial referred to findings in the double-blind treatment period.
Adverse Events
TEAEs were reported for 85.1% of patients in the foslevodopa-foscarbidopa arm and 62.7% of patients in the oral LD-CD arm. The most common TEAEs in the foslevodopa-foscarbidopa arm (reported in at least 10% of patients) were infusion-site erythema, pain, cellulitis, and edema, as well as dyskinesia, all of which were more commonly reported than in the oral LD-CD arm (infusion-site erythema and pain, 1.5% each). The incidence of falls was lower in the foslevodopa-foscarbidopa arm (8.1%) than in the oral LD-CD arm (17.9%).
Serious Adverse Events
Serious TEAE was reported for 6 (8.1%) patients in the foslevodopa-foscarbidopa arm (1 each: infusion-site cellulitis, catheter-site cellulitis, dehydration, migraine, psychiatric disorder, prostatomegaly) and 4 (6.0%) patients in the oral LD-CD arm (1 each: COVID-19 pneumonia, cellulitis, contusion or fall, catheter-site cellulitis).
Withdrawals Due to AEs
Treatment discontinuation due to TEAEs was reported for 21.6% of patients in the foslevodopa-foscarbidopa arm and 1.5% of patients in the oral LD-CD arm. The most common TEAEs leading to treatment discontinuation in the foslevodopa-foscarbidopa arm were infusion-site cellulitis (5.4%), infusion-site pain (4.1%), infusion-site bruising, hemorrhage, and edema (2.7% each). One patient in the oral LD-CD arm discontinued treatment due to cellulitis.
Mortality
No deaths were reported in the foslevodopa-foscarbidopa arm, and 1 (1.5%) death was reported in the oral LD-CD arm. The death was related to a serious TEAE of acute respiratory failure that was, according to the sponsor, deemed unrelated to the study drug.
Notable Harms
Infusion-Site Reactions and Infusion-Site Infections
The incidences of infusion-site reactions and infusion-site infections were notably higher in the foslevodopa-foscarbidopa arm than in the oral LD-CD arm (infusion-site reactions: 62.2% versus 7.5%; infusion-site infections: 28.4% versus 3.0%). Six patients (8.1%) in the foslevodopa-foscarbidopa arm and no patients in the oral LD-CD arm had at least 1 incidence of numeric grade at least 5 and letter grade at least D on the Infusion Site Evaluation Scale.
Hallucination or Psychosis, Impulse-Control Disorder, and Suicidality
The incidence of hallucination or psychosis was notably higher in the foslevodopa-foscarbidopa arm (14.9%) than in the oral LD-CD arm (3.0%). There was no report of impulse-control disorder or impulsive behaviour in either treatment arm. There was no notable between-arm difference in the mean change from baseline in score for each impulse-control disorder and related behaviour parameters of the QUIP-RS across almost all time points. Based on the C-SSRS assessment, 5 (6.8%) patients in the foslevodopa-foscarbidopa arm and 2 (3.0%) patients in the oral LD-CD arm had suicidal behaviours or ideations.
Dizziness and Orthostatic Hypotension
Dizziness was reported in 3 patients in each treatment arm. Orthostatic hypotension by preferred term was reported in 1 (1.4%) patient in the foslevodopa-foscarbidopa arm, and 2 (3.0%) patients in the oral LD-CD arm.
Somnolence
Somnolence was reported in 1 (1.4%) patient in both treatment arms.
Depression
Depression was reported in 0 patients in the foslevodopa-foscarbidopa arm, and 2 (3.0%) patients in the oral LD-CD arm.
Table 18: Summary of Harms — Pivotal and RCT Evidence (M15-736) (SAS)
Adverse events | M15-736 double-blind treatment period | |
|---|---|---|
Foslevodopa-foscarbidopa arm (N = 74) | Oral LD-CD arm (N = 67) | |
TEAE, n (%) | ||
Patients ≥ 1 TEAE | 63 (85.1) | 42 (62.7) |
Most common TEAEa, n (%) | ||
Infusion-site erythema | 20 (27.0) | 1 (1.5) |
Infusion-site pain | 19 (25.7) | 1 (1.5) |
Infusion-site cellulitis | 14 (18.9) | 0 |
Fall | 6 (8.1) | 12 (17.9) |
Infusion-site edema | 9 (12.2) | 0 |
Dyskinesia | 8 (10.8) | 4 (6.0) |
Serious TEAE, n (%) | ||
Patients with ≥ 1 serious TEAE | 6 (8.1) | 4 (6.0) |
Infusion-site cellulitis | 1 (1.4) | 0 |
Catheter-site cellulitis | 1 (1.4) | 1 (1.5) |
Dehydration | 1 (1.4) | 0 |
Migraine | 1 (1.4) | 0 |
Psychiatric disorder | 1 (1.4) | 0 |
Prostatomegaly | 1 (1.4) | 0 |
Cellulitis | 0 | 1 (1.5) |
Contusion or fall | 0 | 1 (1.5) |
COVID-19 pneumonia | 0 | 1 (1.5) |
Patients who stopped treatment due to TEAE, n (%) | ||
Patients who stopped treatment due to TEAE | 16 (21.6) | 1 (1.5) |
Most common TEAE leading to treatment discontinuationb, n (%) | ||
Infusion-site cellulitis | 4 (5.4) | 0 |
Infusion-site pain | 3 (4.1) | 0 |
Infusion-site bruising | 2 (2.7) | 0 |
Infusion-site hemorrhage | 2 (2.7) | 0 |
Infusion-site edema | 2 (2.7) | 0 |
Deaths, n (%) | ||
Patients who died | 0 | 1 (1.5) |
Acute respiratory failure | 0 | 1 (1.5) |
Notable harms, n (%) | ||
Infusion-site reactionsc | 46 (62.2) | 5 (7.5) |
Infusion-site infections | 21 (28.4) | 2 (3.0) |
Hallucination/psychosisd | 11 (14.9) | 2 (3.0) |
Suicidal behaviours or ideationse | 5 (6.8) | 2 (3.0) |
Dizziness | 3 (4.1) | 3 (4.5) |
Orthostatic hypotension | 1 (1.4) | 2 (3.0) |
Somnolence | 1 (1.4) | 1 (1.5) |
Depression | 0 | 2 (3.0) |
Impulse-control disorder | 0 | 0 |
Impulsive behaviour | 0 | 0 |
CD = carbidopa; LD = levodopa; SAS = safety analysis set; TEAE = treatment-emergent adverse event.
Note: Unless otherwise specified, all TEAEs were summarized using Medical Dictionary for Regulatory Activities (MedDRA) preferred terms.
a≥ 10% of patients.
b≥ 2% of patients.
cAdverse event of special interest, including any adverse event in infusion site–related noninfection reactions company MedDRA query (CMQ).
dAdverse event of special interest, including any adverse event in the hallucinations CMQ or psychosis and psychotic disorder standardized MedDRA query (SMQ).
eAssessed using the Columbia-Suicide Severity Rating Scale.
Source: M15-736 Clinical Study Report.14 (Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
Critical Appraisal
Systematic Literature Review
A systematic literature review (SLR) was conducted by the sponsor to identify studies for inclusion into the submission. The prespecified selection criteria (population, intervention, comparison, outcomes, and study [PICOS]) were appropriate for identifying relevant studies. The criteria were broad with respect to comparator and study designs, including comparators irrelevant to the Canadian context (e.g., subcutaneous apomorphine injections, levodopa-entacapone-carbidopa intestinal gel) and a variety of study designs (phase II, III, and IV clinical trials and observation studies), although narrower criteria were subsequently applied to tailor the review to include phase II, III, and IV clinical trials of foslevodopa-foscarbidopa with results or data available. It is unclear if these narrower criteria were prespecified, but the risk of missing relevant studies due to these additional criteria appears to be low.
The literature search was comprehensive, including multiple databases (i.e., MEDLINE [In-Process], Embase, and Cochrane Library) and clinical trial registries (ClinicalTrials.gov, International Clinical Trials Registry Platform, Health Canada’s Clinical Trials Database, and European Union Clinical Trials Register) to identify literature published for all time until 6 months before submission to CADTH. Only publications in English were included, for which no justification was provided. (It is recommended that search strategies not be limited by language to minimize publication or language bias, and if such restriction is applied, justification should be provided.68,69) Appropriate study selection and data extraction methods were used. Studies were selected independently by 2 reviewers with a third reviewer to resolve discrepancies. Study selection was appropriately presented using the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) approach, and reasons for article exclusion were documented. Data extraction was performed by 1 reviewer and independently reviewed by a second reviewer, followed by a quality check by a third reviewer. A risk of bias evaluation for the studies included in the SLR was conducted using the Cochrane risk of bias tool. A high-level summary of the risk of bias of included studies was provided, but results for individual included studies were not given.
Pivotal and RCT Evidence
Internal Validity
The M15-736 trial was a randomized, double-blind, double-dummy, active-controlled trial. The methods of randomization, which involved stratification by study site, and interactive response technology for concealment of the randomized assignment, were appropriate. There was no notable difference between treatment arms for most baseline characteristics. The foslevodopa-foscarbidopa arm had a higher proportion of patients who had levodopa response for more than 5 years and had prior dopamine agonist use compared with the oral LD-CD arm, although the clinical expert consulted by CADTH noted that these differences would not be clinically significant in the studied population and were unlikely to have confounded the study results.
As foslevodopa-foscarbidopa and oral LD-CD require a different route of administration, a double-dummy design was used to preserve blinding for patients and investigators. The blinding procedures were in general appropriate; however, an end-of-study survey aiming to identify the extent of unblinding showed that the majority of patients (74.3% in the foslevodopa-foscarbidopa arm; 71.9% in the oral LD-CD arm) were aware of treatment assignment, most commonly for reasons related to change (or absence of change) in PD symptoms. MDS-UPDRS, PDSS-2, and PDQ-39 assessments were conducted by blinded raters who neither participated in the medical management of patients nor had access to study assessment results or medical patient records; however, due to the subjective nature of these outcomes, potential reporting bias in favour of foslevodopa-foscarbidopa might be involved in patients with knowledge of treatment assignment. The same also applies to PD diary outcomes (i.e., “on” and “off” times, presence of morning akinesia) as they were based on patients’ self-report of symptoms. Reporting of harms could also have been biased, potentially in favour of the oral LD-CD arm due to a higher tendency to report harms among unblinded patients in the foslevodopa-foscarbidopa arm. However, the extent of these potential biases is unclear.
Statistical analyses were in general appropriate for the outcomes evaluated. The study was powered to detect a treatment difference in the primary end point between treatment arms and the enrolled sample size was adequate. The sponsor noted that the study was also powered for the key secondary end points; however, the assumptions to support this claim are unclear. A hierarchical testing procedure was appropriately used to account for multiplicity in the key secondary and other secondary outcomes. No conclusion can be drawn on the prespecified subgroup analyses because of the lack of consideration for sample size and statistical power, and control of multiplicity.
A notably higher proportion of patients in the foslevodopa-foscarbidopa arm (35.1%) than in the oral LD-CD arm (7.5%) discontinued from the study. Missing data for the primary and key continuous secondary outcomes were implicitly imputed using an MMRM, which relies on the missing-at-random assumption. This assumption is unlikely to hold as withdrawals in the foslevodopa-foscarbidopa arm were largely driven by AEs. Two prespecified sensitivity analyses were conducted for PD diary-based outcomes to assess the impact of missing data, and their results were consistent with the primary analysis. The analysis based on the “jump-to-reference” approach, which assumed missing values in the foslevodopa-foscarbidopa arm to be missing not at random and allowed missing values after treatment withdrawal to be imputed with values equal to those of the oral LD-CD arm, was considered conservative and had increased certainty of the primary analysis. The analysis using the last available value approach was less informative given that the method relies on the missing-at-random assumption, similar to the primary analysis. The impact of attrition on outcomes other than PD-based outcomes was unclear in the absence of sensitivity analyses.
The trial included outcomes based on PD diary, MDS-UPDRS, and PDQ-39, disease-specific PRO instruments commonly used to assess function and symptoms in clinical trials of PD. There is evidence supporting these instruments’ validity and reliability and their responsiveness in assessing patients with PD. MID estimates for these instruments have been established in patients with PD, although for most estimates (except “off” time assessed based on PD diary), it is unclear if they were derived from patients with advanced disease. Procedures were in place to ensure proper completion of PD diary entries, including trainings on PD diary completion, requiring patients to have at least 75% concordance with the investigators at screening, and retraining on PD diary completion available for patients who incorrectly completed the diary. All patients were also reminded to complete the diary before each visit to ensure adherence.
Protocol deviations related to study procedure adherence was frequently reported (45.9% in the foslevodopa-foscarbidopa arm, and 40.3% in the oral LD-CD arm), and included improper use of syringes for drug administration, incomplete PD diary, missed or late questionnaire completion, missed recording of rescue dose, remote completion of study visits, and incorrect loading doses. According to the sponsor, none of the recorded protocol deviations could have affected the study outcome or study results or conclusion interpretation. The CADTH review team noted that deviations related to PD diary and questionnaire in particular could be a reason for concern for bias; however, results of the sensitivity analyses on PD diary-based outcomes provided some reassurance that the impact of missing data on these outcomes is possibly low.
External Validity
The clinical expert commented that the inclusion and exclusion criteria in general align with the selection criteria for candidates for foslevodopa-foscarbidopa treatment, although patients with cognitive impairment and prior DBS or LCIG treatment would not necessarily be excluded from treatment in clinical practice. Nonetheless, the clinical expert did not expect the exclusion of these patients to significantly impact the generalizability of the patient population in this study. The baseline patient characteristics were also generally reflective of the Canadian patient population with advanced PD, as per the clinical expert.
The infusion pump used in the trial (i.e., Phillips-Medisize Parkinson’s Disease Subcutaneous Pump Delivery System) is the same as the pump intended for use in Canada (brand name Vyafuser for the same device). The clinical expert considered oral therapy to be an appropriate comparator given that most patients with advanced PD currently remain on oral medications. The use of concomitant PD medications was consistent with practice in Canada, as per the clinical expert.
The efficacy outcomes measured in the study were of clinical importance to patients and clinicians, including motor and nonmotor symptoms, functioning, and HRQoL. However, the clinical expert noted that the PKG device used in the study for measuring bradykinesia and dyskinesia scores is currently not available in Canada, and the EQ-5D-5L and PDSS-2 instruments are not used in clinical practice. The clinical expert considered the PD diary, MDS-UPDRS, and PDQ-39 to be clinically relevant as these instruments are sometimes used in clinical practice to assess treatment response. Patients highlighted an unmet need for treatments that can improve cognition. Although this outcome was captured in the MDS-UPDRS Part I (nonmotor aspects of experiences of daily living) scale, the results were pooled with other item scores of nonmotor symptoms, and thus the effect of foslevodopa-foscarbidopa on cognition alone is unclear. The clinical expert noted that a goal of treatment of advanced PD is to reduce caregiver burden; however, the impact on caregiver burden is unclear as it was not a measured outcome. The clinical expert noted that the duration of follow-up (12 weeks in the double-blind treatment period) was adequate for efficacy assessment although longer follow-up is required to gain certainty on the safety profile.
It should be noted that this trial is the only phase III RCT providing direct comparative evidence between foslevodopa-foscarbidopa and standard of care for advanced PD. The clinical expert consulted by CADTH noted that DBS and LCIG therapies are also available for the treatment of advanced PD, although there are unique considerations that guide treatment decisions (e.g., the need for a specialized medical team for DBS and LCIG therapies, provision of DBS at specialized centres only, and selection of patients without contraindications). In patients with advanced PD who are potential candidates for any of the available advanced therapies, the absence of head-to-head evidence between foslevodopa-foscarbidopa versus DBS and LCIG represents an evidence gap.
Long-Term Extension Studies
Description of M20-098 Study
Characteristics of the M20-098 trial are summarized in Table 19. M20-09839 is an ongoing single-arm, multicentre, open-label extension study of the previously described M15-736 trial, with up to 96 weeks of treatment. The objective is to assess the long-term safety and tolerability of foslevodopa-foscarbidopa delivered by CSCI for 24 hours per day. The primary outcomes are AEs and related measures. Efficacy outcomes are also being collected as secondary end points. Patients in M20-098 were previously randomized to either CSCI or oral medications in the parent study M15-736, and as such, the M20-098 long-term extension population had 2 different treatment pathways. Limited follow-up was available for the M20-098 trial at the time of this review.
Table 19: Details of the M20-098 Trial
Category | Description |
|---|---|
Designs and populations | |
Study design | Open-label, single-arm, multicentre extension to M15-736 |
Locations | 44 sites in Australia and the US |
Patient enrolment dates: | First patient first visit: February 18, 2021 Last patient last visit: September 29, 2021 (data cut-off date) |
Enrolled (N) | 103 |
Key inclusion criteria | Completed parent study (M15-736), remained on the study drug and met additional criteria (consent, demographic and laboratory assessments, study participant history, and contraception) |
Key exclusion criteria | None reported |
Drugs | |
Intervention | Individualized foslevodopa (240 mg/mL) and foscarbidopa (12 mg/mL) solution administered via CSCI over 24 hours/day |
Comparator(s) | NA |
Study duration | |
Screening phase | NA |
Stabilization phase | Optimization phase: 4 weeks |
Treatment phase | Up to 96 weeks |
Follow-up phase | NA |
Outcomes | |
Primary end points |
|
Secondary end points | Change from baseline to the end of study of the following:
Percentage of patients with early morning “off,” assessed using the PD diary as the proportion of patients with early morning “off” upon waking up |
Publication status | |
Publication | NA |
AE = adverse event; AESI = adverse event of special interest; CSCI = continuous subcutaneous infusion; ECG = electrocardiogram; EQ-5D-5L = EuroQol 5-dimensions questionnaire; HRQoL = health-related quality of life; MDS-UPDRS = Movement Disorder Society-Unified Parkinson’s Disease Rating Scale; MMSE = Mini-Mental State Examination; NA = not applicable; PD = Parkinson disease; PDQ-39 = Parkinson’s Disease Questionnaire-39 items; PDSS-2 = Parkinson’s Disease Sleep Scale-2; SAE = serious adverse event.
Source: M20-098 Clinical Study Report.39 (Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
Populations
M20-098 included patients with PD who, at completion of the parent study (M15-736), remained on the study drug and met additional criteria (consent, demographic and laboratory assessments, study participant history, and contraception). Patients had to be able to understand the nature of the study and have had the opportunity to have questions answered by the investigator. Patients with decision-making capacity were allowed to sign and date an informed consent form. Patients also had to be willing to comply with procedures required in this study and be considered by the investigator to be a suitable candidate to continue foslevodopa-foscarbidopa. Patients also could not be exhibiting significant suicidal behaviours at the time of the final visit of the parent study. Patients with childbearing potential must have a negative urine pregnancy test, and both female and male patients must have agreed to certain protocol-specified contraception. Patient characteristics at M20-098 study baseline are summarized in Table 20.
Table 20: Summary of Baseline Characteristics in the M20-098 Trial
Characteristic | Oral LD-CD to foslevodopa-foscarbidopa | Foslevodopa-foscarbidopa to foslevodopa-foscarbidopa | Total (safety analysis set) |
|---|---|---|---|
Demographics | |||
N | |||| | |||| | ||||| |
Sex, n (%) | |||
Male | || |||||| | || |||||| | || |||||| |
Female | || |||||| | || |||||| | || |||||| |
Age (years), mean (SD) | |||| ||||||| | |||| |||||| | |||| |||||| |
Race, n (%) | |||
White | || |||||| | || |||||| | || |||||| |
Black/African American | | ||||| | | ||||| | | ||||| |
Asian | | ||||| | ||| | | ||||| |
American Indian/Alaska Native | ||| | | ||||| | | ||||| |
Native Hawaiian or Pacific Islander | | ||||| | | ||||| | | ||||| |
Disease characteristics | |||
Duration of PD since diagnosis (years), mean (SD) | |||| ||||||| | |||| ||||||| | |||| ||||||| |
History of levodopa-induced dyskinesia, n (%) | || |||||| | || |||||| | || |||||| |
Levodopa response of > 5 years, n (%) | || |||||| | || |||||| | || |||||| |
Baseline PD Hoehn and Yahr Scale stage, n (%) | |||
0 | | ||||| | ||| | | ||||| |
1 | | ||||| | | |||||| | | ||||| |
2 | || |||||| | || |||||| | || |||||| |
3 | || |||||| | || |||||| | || |||||| |
4 | | ||||| | | ||||| | | ||||| |
5 | | ||||| | ||| | | ||||| |
Duration since onset of motor fluctuation (years), mean (SD) | |||| ||||||| | |||| ||||||| | |||| ||||||| |
MDS-UPDRS Part III score during “Off” state, mean (SD) | |||| | |||| | |||| |
Normalized “on” or “off” time per day (hours), mean (SD) a | |||
“Off” time | |||| |||||| | |||| |||||| | |||| |||||| |
“On” time without troublesome dyskinesia | ||||| |||||| | ||||| |||||| | ||||| |||||| |
“On” time without dyskinesia | |||| |||||| | ||||| |||||| | |||| |||||| |
“On” time with nontroublesome dyskinesia | |||| |||||| | |||| |||||| | |||| |||||| |
“On” time with troublesome dyskinesia | |||| |||||| | |||| |||||| | |||| |||||| |
PD treatment history | |||
Prior DBS procedure, n (%) | ||| | | ||||| | | ||||| |
Prior PD medication use, n (%) | |||
Dopa and dopa derivatives | || ||||| | || ||||| | ||| ||||| |
Dopamine agonists | || |||||| | || |||||| | || |||||| |
MAO-B inhibitor | || |||||| | || |||||| | || |||||| |
Amantadine derivatives | || |||||| | | |||||| | || |||||| |
COMT inhibitor | |||| | |||| | |||| |
Anticholinergics | | ||||| | | ||||| | | ||||| |
Other | | ||||| | | ||||| | | ||||| |
Number of PD medication class at baseline, n (%) | |||
0 | || |||||| | || |||||| | || |||||| |
1 | || |||||| | || |||||| | || |||||| |
2 | || |||||| | | |||||| | || |||||| |
3 | | ||||| | | ||||| | | ||||| |
4 | ||| | ||| | ||| |
> 4 | |||| | |||| | |||| |
Prior total daily levodopa-equivalent dose, including other PD medications, mean mg/d (SD) | |||||| |||||||| | |||||| |||||||| | |||||| |||||||| |
CD = carbidopa; COMT = catechol-O-methyltransferase; DBS = deep brain stimulation; LD = levodopa; MAO-B = monoamine oxidase type B; MDS-UPDRS = Movement Disorder Society-Unified Parkinson’s Disease Rating Scale; PD = Parkinson disease; SD = standard deviation.
Source: M20-098 Clinical Study Report.39 (Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
Interventions
Patients enrolled in M20-098 received an individualized CSCI (24 hours per day) of foslevodopa-foscarbidopa via an infusion set connected to a pump for up to 96 weeks. Use of concomitant medications reflected the protocol of the parent study.
Outcomes
Efficacy variables were derived from the PD diary, the MDS-UPDRS, the PDSS-2, the PDQ-39, and the EQ-5D-5L and cognitive impairment was assessed using the Mini-Mental State Examination (MMSE). Safety evaluations were the primary objective of this study and included safety and tolerability as evaluated by AEs, the Infusion Site Evaluation Scale, the C-SSRS, and the QUIP-RS.
Statistical Analysis
In M20-098, the full analysis set consisted of all patients who received foslevodopa-foscarbidopa infusion and have a baseline and post-baseline observation for at least 1 efficacy end point and was used for all efficacy analyses. The safety analysis set consisted of all patients who received any foslevodopa-foscarbidopa infusion in the M20-098 study and was used for all safety analyses. Efficacy analyses were conducted by treatment sequence and were evaluated in groups: “oral LD-CD à foslevodopa-foscarbidopa,” “foslevodopa-foscarbidopa à foslevodopa-foscarbidopa,” and overall. Outcomes were summarized descriptively including the number of observations, mean, SD, minimum, median, and maximum; hypothesis testing was not performed. No adjustments were made for missing visit data. For PD diary entries, if no data were available for a visit, the average daily normalized “off” or “on” times values were missing for that visit. The MDS-UPDRS total score and score of each part was calculated as long as no more than 15% of the answers were missing for that assessment. The missing item was imputed as the average of the nonmissing items from the same MDS-UPDRS assessment.
Results
Patient Disposition
Patient disposition is summarized in Table 21. M20-098 was ongoing at the time of submission. No patients had completed the study at the time of the data cut submitted by the sponsors.
The majority of patients who completed the parent study went on to enrol in the long term extension study. Notably more patients discontinued from foslevodopa-foscarbidopa in the M20-098 long-term extension after having received oral LD-CD in the parent study, when compared to patients who were already receiving foslevodopa-foscarbidopa in the parent study.
Exposure to Study Treatments
Prescribed dose by infusion rate and total daily dose, as well as the rates of adherence, are presented in Table 22. The study is ongoing. There were no signals of substantial imbalance between study arms in M20-098. The rates of adherence were high at the time of the available data cut-off.
Table 21: Patient Disposition in the M20-098 Trial
Patient disposition | Oral LD-CD to foslevodopa-foscarbidopa | Foslevodopa-foscarbidopa to foslevodopa-foscarbidopa |
|---|---|---|
Enrolled in parent study M15-736, N | ||| | ||| |
Completed parent study M15-736, N | ||| | ||| |
Enrolled in LTE, N | ||| | ||| |
Ongoing, N (%) | || |||||| | || |||||| |
Completed LTE study | ||| | ||| |
Discontinued LTE study, N (%) | || |||||| | | ||||| |
Reasons for discontinuation, N (%) | ||
AEs | | |||||| | | ||||| |
Infusion site–related infections | | ||||| | | ||||| |
Infusion site–related noninfection reactions | | ||||| | ||| |
Withdrew consent | | ||||| | ||| |
Lost to follow-up | ||| | ||| |
Lack of efficacy | | ||||| | | ||||| |
Difficulty with drug delivery | | ||||| | | ||||| |
Other | ||| | ||| |
Missing | | ||||| | ||| |
FAS, N (%) | || |||||| | || ||||| |
SAS, N (%) | || ||||| | || ||||| |
AE = adverse event; CD = carbidopa; FAS = full analysis set; LD = levodopa; LTE = long-term extension; NR = not reported; SAS = safety analysis set.
Source: M20-098 Clinical Study Report.39 (Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
Table 22: Summary of Patient Exposure in the M20-098 Trial
Exposure | Oral LD-CD to foslevodopa-foscarbidopa | Foslevodopa-foscarbidopa to foslevodopa-foscarbidopa |
|---|---|---|
Initial base prescribed dose, continuous infusion rate (LD mg/hour), mean (SD) | |||| ||||||| | |||| ||||||| |
Final base prescribed dose, continuous infusion rate (LD mg/hour), mean (SD) | |||| ||||||| | | |||| ||||||| | |
Initial totalb daily dose (LD mg/day), mean (SD) | |||||| |||||||| | |||||| |||||||| |
Final totalb daily dose (LD mg/day), mean (SD) | |||||| |||||||| | | |||||| |||||||| | |
Total patient-years | || | || |
Duration of study drug exposure (days) | || | || |
Adherencec | ||
n | || | || |
Mean (SD) | ||||| ||||||| | ||||| |||||||| |
Median (IQR or range) | ||||| ||||||| |||||| | || |
CD = carbidopa; IQR = interquartile range; LD = levodopa; SD = standard deviation.
aEnd of optimization phase.
bTotal dose calculated as “base” (mL/hour) × 24 hours per day × 170.75 mg LD per mL.
cStudy drug solution adherence: (hours of infusion / 24) × 100.
Source: M20-098 Clinical Study Report.39 (Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
Efficacy
At the time of this review, M20-098 is still under way and efficacy results were limited. Each outcome is summarized briefly, but no conclusions can be drawn.
Mean Average Daily Normalized “On” Time Without Troublesome Dyskinesia, “On” Time With Troublesome Dyskinesia, and “Off” Time Based on the PD Diary
||||| ||| |||||| |||||| |||| ||| ||||||| ||||| |||||||| || ||||| |||| ||| || |||| ||||| ||| ||| || |||||||| |||| || ||||| ||||||||||| || |||| | || ||| ||||| ||| ||| |||||||| |||||||||||||||||||||||| || ||| |||||| |||||| |||||||||||| || ||||| |||| ||| || |||| ||||| |||| |||||||| ||| ||| || |||||||| |||| || ||||| ||||||||||| || |||| | || ||| ||||| ||| ||| |||||||| |||| ||||| || ||| |||||| ||||| ||| |||||||||||| || |||||||||||||||||||||||| || ||| ||||||||| |||||| ||| |||||||||| ||||| |||| |||| || ||||| |||| || || ||||| |||||||| |||| ||||||||| |||| || ||||||| || ||| |||| || |||| |||||||
Movement Disorder Society-Unified Parkinson Disease Rating Scale
||| ||||||||| |||| || |||||| || |||| | |||||| |||||| |||||||||| || |||||||| ||| ||| |||||||| |||||||||||||||||||||||| || ||| |||||| |||||| ||| |||| ||||||||||| || |||||||| ||| ||| |||||||| |||| ||||| || ||| |||||| |||||| |||| |||||| ||||||||| |||||| |||| | ||| ||||||||| || ||| |||| || |||| |||||||
Change From Baseline to Each Planned Visit in HRQoL (EQ-5D-5L)
EQ-5D-5L results for the M20-098 trial were not yet available at the time of this review.
Change in Baseline to Each Planned Visit in Cognitive Impairment Measured Using the MMSE
MMSE results for the M20-098 trial were not yet available at the time of this review.
Harms
All AEs presented were treatment emergent and analyzed in the safety analysis set. || ||| ||||||| |||||| ||| |||| |||||||| ||| || |||||||| ||||||| || ||| |||| || ||| |||| |||||||| ||| ||| |||||||| |||| |||||||||| ||| |||| || |||||||| || ||||||||| |||| |||| |||||||| ||| | |||||||| ||||||| || |||||||| ||| |||||
Critical Appraisal
Internal Validity
The main limitation is the absence of comparator arm and the potential biases related to the nonrandomized design. The open-label study design inherently increases risk of bias in the reporting of any subjective outcomes, including harms outcomes, because patients and their clinicians are aware of the treatment received. In addition, because patients could only enrol after completing the parent trial, there is a risk of selection bias given that patients who better tolerated the treatment or perceived the treatment as benefiting them were more likely to enrol in the extension studies.
External Validity
As the extension study consisted of patients who took part in the parent study, it is reasonable to expect that the same strengths and limitations related to generalizability apply to the extension studies, with the additional caveat of potential selection bias due to the nature of the open-label extension study design.
However, little follow-up was available for M20-098 at the time of this review, preventing interpretation of long-term safety or efficacy outcomes.
Indirect Evidence
Objective for the Summary of Indirect Evidence
One ITC comparing foslevodopa-foscarbidopa with alternative treatments for advanced PD was submitted by the sponsor and is included in this report. An ITC was required to address a gap in pivotal and RCT evidence in the absence of studies directly comparing foslevodopa-foscarbidopa with treatments for advanced PD other than oral medications.
Description of Sponsor-Submitted ITC
The sponsor first conducted an SLR to identify evidence for inclusion in the ITC. The relative efficacy of foslevodopa-foscarbidopa in the M15-736 trial was indirectly compared with other treatments for patients with PD who are inadequately controlled with current therapy, via Bayesian NMA. Comparators of interest for the sponsor-submitted NMA included LCIG and BMT (including oral therapy). Outcomes of interest included “off” time, “on” time without troublesome dyskinesia, and sleep symptoms assessed using the PDSS-2 total score.
Methods of Sponsor-Submitted ITC
Objectives
The objective of this study was to estimate the comparative efficacy of foslevodopa-foscarbidopa versus LCIG and BMT, both of which are relevant comparators for foslevodopa-foscarbidopa in Canada, using an NMA.
Study Selection Methods
A SLR was conducted using MEDLINE (In-Process), Embase, and Cochrane Library to identify clinical and observational studies published for all time till the date of search (i.e., June 1, 2021). Two updated searches were conducted in January and June 2022.
Article screening was performed independently by 2 reviewers in 2 stages (titles and abstracts, and then the full texts), with a third reviewer resolving disagreements. Reasons for exclusion were documented. Data extraction was performed by 1 reviewer, with quality check on all data by a second reviewer, and an additional check on 10% of data by a third reviewer. Quality assessment of the selected studies, with the exception of foslevodopa-foscarbidopa clinical studies (not published when the SLR was being conducted), was conducted by 1 reviewer to assess the risk of bias using the Cochrane risk of bias tool.
The initial article search included a broader set of comparators and study designs. More restricted selection criteria, as shown in Table 23, were subsequently applied at the full-text review stage to select trial for inclusion into the NMA.
Table 23: Study Selection Criteria and Methods in ITCs Submitted by the Sponsor
Characteristics | Indirect comparison |
|---|---|
Population | PD patients aged ≥ 18 years who are levodopa-responsive, but inadequately controlled by current therapy |
Intervention |
|
Comparator |
|
Outcome |
|
Study designs | Phase II, III, IV RCT (blinded or open-label) |
Publication characteristics |
|
Exclusion criteria |
|
Databases searched |
|
Selection process | Articles were screened independently by 2 reviewers. Any discrepancies were resolved by a third reviewer |
Data extraction process | Reviewer 1 extracted the data; reviewer 2 independently reviewed for accuracy; reviewer 3 performed an additional 10% quality check of extracted data |
Quality assessment | 1 reviewer using the Cochrane risk of bias tool |
ITC = indirect treatment comparison; LCIG = levodopa-carbidopa intestinal gel; PD = Parkinson disease; PDSS-2 = Parkinson’s Disease Sleep Scale-2; RCT = randomized controlled trial.
Source: Sponsor-submitted Network Meta-Analysis Technical Report.70 (Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
ITC Analysis Methods
A summary of the analysis methods for the NMA is shown in Table 24. All analyses were conducted using a Bayesian analysis framework. Fixed effects (FE) and random effects (RE) models were evaluated for the base-case analyses, and selection was determined based on the deviance information criterion (DIC) model fit statistic. Vague prior distributions on model parameters were used so that model outcomes would be determined primarily by the clinical trial data. These were selected using the recommended priors in the NICE DSU Technical Support Document 2.71 Posterior outcome distributions were based on at least 50,000 simulations after a burn-in of at least 50,000. Adequate convergence was assessed by visual inspection of the autocorrelation and history plots.
Statistical heterogeneity was evaluated based on the I2 statistic derived from direct head-to-head meta-analysis of those treatment comparisons in each network that were reported by more than 1 study.
“On” time without troublesome dyskinesia and “off” time measures are continuous data based on sample means and standard errors that were analyzed using the normal likelihood and the normal link function.71 For studies where measures of uncertainty were not reported, the SD was input using the sample size-weighted average of the available SDs. In this analysis, the majority of studies across all treatments of interest reported a mean change from baseline for the outcomes and not the LSM change. As a result, a decision was made to use mean change as a unit of measure. The mean difference of “on” time without troublesome dyskinesia and “off” time measures for each treatment versus oral standard of care was the NMA output.
The base-case analysis included all 3 comparators for both outcomes: “off” time and “on” time without troublesome dyskinesia. The base-case analysis for “off” time without troublesome dyskinesia included all RCTs. For both of these outcomes, FE and RE models were fitted and compared on DIC to determine the better fitting model (lower DIC values indicate better fit to the data). When DIC differences are small (i.e., less than 3 to 5 points) across different fitted models, common practice is to choose the simplest model because the additional complexity does not result in a better fitted model.71 In addition, while there may be cross-trial heterogeneities in treatment contrasts, the small number of trials made it infeasible to quantitatively examine the level of such heterogeneities. As such, FE models were selected.
An NMA was also run for the outcome PDSS-2 total score. Only 2 RCTs were considered appropriate for potential inclusion into this analysis, and therefore an FE model was fitted. Each treatment was considered a separate node in the evidence network. All analyses assessed the change from baseline at week 12.
No sensitivity or subgroup analysis was conducted.
Assessment for consistency was not possible due to the lack of closed loops.
Table 24: Sponsor-Submitted NMA Analysis Methods
Methods | Description |
|---|---|
Analysis methods | Bayesian framework |
Priors | Recommended priors in the NICE DSU Technical Support Document 271 |
Assessment of model fit | Model fit determined by DIC |
Assessment of convergence | Convergence was assessed by visual inspection of the autocorrelation and history plots |
Outcomes | “Off” time, “on” time without troublesome dyskinesia, PDSS-2 |
Follow-up time points | At 12 weeks |
Construction of nodes | Each treatment was a separate node |
Sensitivity analyses | Not conducted |
Subgroup analyses | Not conducted |
DIC = deviance information criterion; NMA = network meta-analysis; PDSS-2 = Parkinson’s Disease Sleep Scale-2.
Source: Sponsor-submitted Network Meta-Analysis Technical Report.70 (Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
Results of Sponsor-Submitted NMA
Summary of Included Studies
Five total studies were considered for inclusion in the NMA, including 2 foslevodopa-foscarbidopa studies: M15-736 and its extension M20-098. M20-098 could not be included because it was a single-arm trial where all patients received foslevodopa-foscarbidopa. The remaining 3 trials assessed the comparative efficacy between LCIG and oral therapies: Olanow et al. (2014),72 DYSCOVER,73 and INSIGHTS.74
An overview of the assessment of homogeneity of the 4 studies included in the ITC is presented in Table 25. All trials were phase III RCTs; M15-736 and Olanow et al. (2014) were double-blind, while DYSCOVER and INSIGHTS were open-label. All studies were multicentre trials, with sample sizes of between 61 and 141 patients. Year of study was from 2009 to 2022.
All trials included patients with advanced PD with motor fluctuations that were not controlled with optimized oral therapy, although for other inclusion criteria, including exclusion of patients with prior DBS treatment, inclusion of patients with dyskinesia, minimum daily levodopa-equivalent dose requirement, they were only included in some trials. |||| ||| || |||||||| || | ||||| ||||||||| ||| |||||| |||| ||||| ||| | ||||| ||||| ||| || ||||| ||| | ||||| ||||| |||| |||||||||| || ||||||| |||||| ||| ||||| ||||||||| |||| |||||| |||| ||| || |||| Reporting of certain baseline disease characteristics was not consistent across trials and there were variabilities with respect to the duration of PD diagnosis (reported in 3 trials, |||||| |||| |||| ||||| || ||||| |||||| |||| ||||| ||||| |||| ||||||||| || | ||||||| |||||| |||| |||| || |||| ||||| ||| ||||| ||| |||||| |||||| ||||||||| || | |||||||| |||||| ||||||| ||||| || ||||| ||||||), across studies. All treatments were initially dose based on the total levodopa dose established before the studies. Titration to clinical response was allowed for foslevodopa-foscarbidopa and LCIG across trials but was not consistently allowed for oral therapies. No notable differences in the definitions of end points were identified between trials. End points were assessed up to week 12 in all trials, except for INSIGHTS, which measured results up to week 26. The incidence of study withdrawal |||||| ||||||| ||| | || ||||||
The sponsor did not consider the between-trial differences to be clinically meaningful or that they could affect the comparability of the studies’ treatment effect within the NMA, and thus, no adjustments for heterogeneity were made.
Results of the quality assessment of included studies were not reported.
Table 25: Assessment of Homogeneity for Sponsor-Submitted NMA
Characteristics | Description and handling of potential effect modifiers |
|---|---|
Study design |
|
Disease severity |
|
Dosing of comparators |
|
Definitions of end points |
|
Timing of end point evaluation | At week 12 (except for INSIGHTS: 26 weeks) |
Withdrawal frequency | |||||| |||| |||| || ||||| |
LCIG = levodopa-carbidopa intestinal gel; NMA = network meta-analysis; PD = Parkinson disease; PDSS-2 = Parkinson’s Disease Sleep Scale-2; RCT = randomized controlled trial.
Sources: Sponsor-submitted Network Meta-Analysis Technical Report,70 Olanow et al. (2014),72 DYSCOVER,73 and INSIGHT.74 (Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
Results
Three studies were included in the “off” time and “on” time without troublesome dyskinesia networks (Figure 1). Two studies (M15-736, comparing foslevodopa-foscarbidopa versus BMT; INSIGHTS, comparing LCIG versus BMT) were included in the PDSS-2 network. The FE model was selected as the base case for “off” and “on” time without troublesome dyskinesia outcomes, based on the lowest reported DIC ||| |||||| ||||| |||||| ||| || |||||| ||||||. The FE model was selected as the base case for PDSS-2 analysis since only 2 studies were included. Results of the FE model and RE model are summarized in Table 26.
Figure 1: Network Diagram of Studies Included in “Off” Time and “On” Time Without Troublesome Dyskinesia Analyses in Sponsor-Submitted NMA

BMT = best medical therapy; LCIG = levodopa-carbidopa intestinal gel; NMA = network meta-analysis.
Note: This figure has been redacted at the request of the sponsor.
Source: Sponsor-submitted Network Meta-Analysis Technical Report.70 (Note: Details have been taken from the sponsor’s Summary of Clinical Evidence.15)
“Off” Time
For change from baseline in “off” time at week 12, |||||||||||||||||||||||| ||| |||||||| |||| ||| ||||| ||||||||||| ||||| |||| |||| ||||| || ||||||| in the base-case analysis (FE model) but there was insufficient evidence to show a difference in the RE model and the 95% credible interval (CrI) was wide. The evidence was also insufficient to show a difference between LCIG versus foslevodopa-foscarbidopa ||||| ||||||||||| ||| |||| |||| ||||| || ||||||, with a wide 95% CrI in the base-case FE analysis (RE model not reported).
For the pairwise comparisons informed by at least 2 studies (i.e., LCIG and BMT), the I2 statistic was 0%.
“On” Time Without Troublesome Dyskinesia
For change from baseline in “on” time without troublesome dyskinesia at week 12, |||||||||||||||||||||||| ||| |||||||| |||| ||| ||||| ||||||||||| |||| |||| |||| |||| || |||||| in the base-case analysis (FE model) but there was insufficient evidence to show a difference in the RE model and the 95% CrI was wide. The evidence was also insufficient to show a difference between LCIG versus foslevodopa-foscarbidopa ||||| ||||||||||| |||| |||| |||| ||||| || ||||||, with a wide 95% CrI in the base-case FE analysis (RE model not reported).
The I2 statistic was 0% for the pairwise comparisons between LCIG and BMT.
Parkinson’s Disease Sleep Scale-2
For change from baseline in PDSS-2 total score at week 12, |||||||||||||||||||||||| ||| |||||||| |||| ||| (foslevodopa-foscarbidopa versus BMT | |||| ||||||||||| ||||| |||| |||| ||||| || |||||) and LCIG (LCIG versus foslevodopa-foscarbidopa: |||| ||||||||||| |||| |||||||| |||| || |||||).
Table 26: Outcomes of Sponsor-Submitted NMA
Comparisons | Mean difference (95% CrI) | |
|---|---|---|
FE model (base case) | RE model | |
Change from baseline in “off” time at 12 weeks | ||
Foslevodopa-foscarbidopa vs. BMTa | ||||| |||||| || |||||| | ||||| |||||| || ||||| |
LCIG vs. foslevodopa-foscarbidopab | ||| |||||| || ||||| | || |
Change from baseline in “on” time without troublesome dyskinesia at 12 weeks | ||
Foslevodopa-foscarbidopa vs. BMTa | |||| ||||| || ||||| | |||| |||||| || ||||| |
LCIG vs. foslevodopa-foscarbidopab | |||| |||||| || ||||| | || |
Change from baseline in PDSS-2 total score at 12 weeks | ||
Foslevodopa-foscarbidopa vs. BMTa | ||||| |||||| || |||||| | || |
LCIG vs. foslevodopa-foscarbidopab | |||| ||||| || |||||| | || |
BMT = best medical therapy; CrI = credible interval; FE = fixed effects; LCIG = levodopa-carbidopa intestinal gel; NMA = network meta-analysis; NR = not reported; PDSS-2 = Parkinson’s Disease Sleep Scale-2; RE = random effects; vs. = versus.
aThis represents a comparison of treatment effects between foslevodopa-foscarbidopa and BMT. A mean difference (95% CrI) of less than 0 indicates results in favour of foslevodopa-foscarbidopa.
bThis represents a comparison of treatment effects between LCIG and foslevodopa-foscarbidopa. A mean difference (95% CrI) of more than 0 indicates results in favour of foslevodopa-foscarbidopa.
Source: Sponsor-submitted Network Meta-Analysis Technical Report.70 (Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
Critical Appraisal of Sponsor-Submitted NMA
The sponsor-submitted NMA was informed by an SLR using the same methods as described in the Pivotal Studies and RCT Evidence section, and as such, much of the appraisal of the SLR noted previously with respect to the literature search, study selection, and data extraction methods, and risk of bias assessment applies to the SLR informing this NMA. Narrower selection criteria were applied to further distill studies and tailor to the objective of the NMA, although there is no mention of whether the NMA-specific inclusion criteria were determined a priori. Studies that assessed DBS, a relevant comparator of foslevodopa-foscarbidopa as per the clinical expert, was not eligible for inclusion into the NMA. As such, the comparative efficacy of foslevodopa-foscarbidopa and DBS remains unknown. The included outcomes, “on” time without troublesome dyskinesia and “off” time in particular, were relevant to patients and clinicians, but less so for PDSS-2 given that the instrument is not routinely assessed in clinical practice. Other outcomes that are of interest to this review, including MDS-UPDRS and PDQ-39 scores, were not assessed.
The CADTH review team’s assessment of the degree of heterogeneity between the included studies was complicated by the limited reporting of study design and patient characteristics in the sponsor-provided technical report. Aside from description of interventions, sample size, country, duration of follow-up, there is no reporting of other trial characteristics (e.g., the inclusion and exclusion criteria, treatment withdrawal frequency, handling of missing data, and end point definition, and so on) within the report. The sponsor concluded that there was no clinically meaningful difference that may affect the comparability of the studies’ treatment effect within the NMA based on a comparison of select baseline characteristics (i.e., age, duration of PD, baseline “off” time and “on” time without troublesome dyskinesia) between studies; however, the CADTH review team considered there to be notable between-trial differences with respect to duration of PD diagnosis and “off” time. Considering that these factors are potential treatment effect modifiers as per the clinical expert, it is possible that the heterogeneity in these baseline characteristics could result in changing relative treatment effects. No adjustment was made in the modelling of the NMA to account for these differences. Upon reviewing each included trial, the CADTH review team also noted some differences in eligibility criteria across trials. While all trials included patients with advanced PD who have motor fluctuations not controlled on optimized oral therapy, some trials excluded patients with cognitive impairment (M15-736, DYSCOVER), required enrolled patients to have dyskinesia (DYSCOVER), and a minimum duration of “off” per day, assessed using the PD diary (M15-736, Olanow et al., 2014). As well, there were differences in study design with respect to blinding, where 2 studies were open-label and 2 were double-blind. Differences in dosing protocol for oral therapies, follow-up duration, and treatment withdrawal frequency were also noted. Although these difference in study design could not be adjusted for in the analysis, these inherent differences suggests that transitivity assumption of the NMA likely have not been met.
Heterogeneity was statistically assessed based on I2 values. Although the analyses suggest low heterogeneity (I2 = 0) between included studies for “on” time without troublesome dyskinesia and “off” time outcomes, given the small number of studies in the network, the I2 is subject to bias.
The NMA was informed by 4 studies; 3 were included in “on” time without troublesome dyskinesia and “off” time analyses; 2 were included in the PDSS-2 analysis. The resulting networks were sparse with no closed loop; as such, it was not possible to assess consistency of results between direct and indirect comparisons. Results of the NMA were informed solely by indirect evidence, and thus are associated with increased uncertainty.
The NMA was conducted using a Bayesian framework. Fixed-effect models were chosen for the base-case analyses for all assessed outcomes. The decision for “on” time without troublesome dyskinesia and “off” time analyses was based on a lower DIC value, while the decision for the PDSS-2 analysis was determined a priori considering the small number of studies. The fixed-effect model relies on the assumption that there was no between-trial heterogeneity, which unlikely holds true as previously discussed. In the absence of sensitivity analyses that assess the potential impact of trial heterogeneity, there is considerable uncertainty in the relative treatment effect estimates.
Studies Addressing Gaps in the Pivotal and RCT Evidence
The M15-741 trial,37 which Health Canada considers to be a supportive trial for the approval of foslevodopa-foscarbidopa, and its ongoing long-term extension study, M15-737,75 are summarized in this section. The aim of these studies was to assess the long-term safety and efficacy of foslevodopa-foscarbidopa for up to 148 weeks.
Description of the M15-741 Trial
Characteristics of the M15-741 trial are summarized in Table 27. The M15-741 trial was a phase III, open-label, single-arm trial that aimed to evaluate the safety and tolerability of foslevodopa-foscarbidopa in patients with advanced PD for up to 52 weeks of treatment (N = 244). The study was conducted at 60 sites in 13 countries (including 2 sites in Canada). Patients were enrolled between April 29, 2019, and November 5, 2021. The study is now complete. It consisted of a screening period of 10 days to 42 days to assess study eligibility, followed by a 52-week treatment period (day 1 to week 52). In the first 4 weeks (optimization phase), the dose of foslevodopa-foscarbidopa was adjusted to achieve optimal clinical response, and this dose remained unchanged for the remainder of the study (maintenance phase).
Table 27: Details of the M15-741 Trial
Detail | Criteria |
|---|---|
Designs and populations | |
Study design | Phase III, multicentre, open-label, single-arm study |
Locations | 60 sites in Asia, Australia, Europe, and North America (including 2 in Canada) |
Patient enrolment dates: | Start date: April 29, 2019 End date: November 5, 2021 |
Randomized/enrolled (N) | 244 |
Key inclusion criteria |
|
Key exclusion criteria | NA |
Drugs | |
Intervention | Foslevodopa (240 mg/mL) and foscarbidopa (12 mg/mL) solution administered through CSCI |
Comparator(s) | NA |
Study duration | |
Screening period | 10 days to 42 days |
Stabilization period | Optimization phase: 4 weeks |
Treatment period | Maintenance phase: 48 weeks |
Follow-up phase | Eligible patients who completed the 52-week treatment phase can enter a separate extension study (M15-737) for 96 weeks |
Outcomes | |
Primary end point(s) |
|
Secondary and exploratory end points | Secondary: Changed from baseline to end of study in:
Exploratory:
|
Publication status | |
Publication | Not available yet (NCT03781167) |
AE = adverse event; AESI = adverse event of special interest; CSCI = continuous subcutaneous infusion; ECG = electrocardiogram; EQ-5D-5L = 5-Level EQ-5D; HRQoL = health-related quality of life; IQR = interquartile range; MDS-UPDRS = Movement Disorder Society-Unified Parkinson’s Disease Rating Scale; NA = not applicable; PD = Parkinson disease; PDQ-39 = Parkinson’s Disease Questionnaire-39 items; PDSS-2 = Parkinson’s Disease Sleep Scale-2; SAE = serious adverse event.
aAs per investigator’s assessment.
bPatients who had received deep brain stimulation were eligible provided they were stable and still levodopa-responsive.
cAssessed using the PD diary.
dDefined by a Mini-Mental State Examination score of at least 24.
eAssessed using the Parkinson’s KinetiGraph/Personal KinetiGraph wearable device.
Source: M15-741 Clinical Study Report.37 (Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
Populations
The key inclusion criteria were the same as those in the pivotal M15-736 study, except that there was no requirement for minimum daily levodopa-equivalent dose, the criterion for “off” time was based solely on a daily minimum of 2.5 hours each day, and patients who had received DBS therapy were eligible for inclusion if they were stable and still levodopa-responsive.
Interventions
Optimization Phase (Day 1 to Week 4)
Patients received 1 loading dose of IR oral LD tablet in combination with dopa decarboxylase inhibitor (e.g., CD or benserazide), followed by an CSCI (24 hours per day) of foslevodopa (240 mg/mL) and foscarbidopa (12 mg/mL) at an initial infusion rate calculated based on the patient’s oral LD therapy over the 16-hour period prior using the same conversion method, based on levodopa-equivalent dose, as described in the M15-736 trial. The infusion rate was adjusted at the investigator’s discretion to optimize clinical response. The allowable infusion rate ranged from 0.17 to 1.04 mL per hour. The Crono PAR Series III pump was used to deliver the infusion solution. Adjustments of concomitant PD medications were allowed.
Maintenance Phase (Week 5 to Week 52)
Patients continued CSCI of foslevodopa-foscarbidopa at the optimal therapeutic dose and dose adjustments were allowed. Patients maintained a stable regimen of all concomitant PD medications unless changes were considered medically necessary, as per the investigator.
The list of allowed concomitant PD medications was the same as the list in the M15-736 trial. Oral LD (100 mg) in combination with dopa decarboxylase inhibitor, as well as levodopa inhalation powder (84 mg) were allowed as rescue medications. The prohibited medication list was almost identical to that in the M15-736 trial, with the exception that second-generation antipsychotics were included on the prohibited medication list, while LCIG and LD-CD enteral suspension were not.
Outcomes
A list of efficacy end points assessed in this clinical review report are provided in Table 28. These end points are further summarized below. Summarized end points are based on those included in the sponsor’s Summary of Clinical Evidence15 as well as any identified as important to this review according to stakeholders, that is, the clinical expert consulted by CADTH, clinician groups, or patient groups.
Table 28: Summary of Outcomes of the M15-741 Trial
Outcome measure | Time point | M15-741 |
|---|---|---|
“On” time (assessed using the PD diary) | ||
Average daily “on” time without troublesome dyskinesia | ||
Normalized value, change from baseline | 52 weeks | Secondary |
Absolute value, change from baseline | Secondary | |
Average daily “on” time without dyskinesia | ||
Normalized value, change from baseline | 52 weeks | Secondary |
Absolute value, change from baseline | Secondary | |
“Off” time (assessed using the PD diary) | ||
Average daily “off” time | ||
Normalized value, change from baseline | 52 weeks | Secondary |
Absolute value, change from baseline | Secondary | |
HRQoL | ||
Change from baseline in PDQ-39 summary index (PD-related QoL) | 52 weeks | Secondary |
Change from baseline in EQ-5D-5L summary index (HRQoL) | Secondary | |
PD symptoms | ||
Change from baseline in MDS-UPDRS scores | ||
Part II score (motor experiences of daily living) | 52 weeks | Secondary |
Part I, III, and IV scores | Secondary | |
Change from baseline in bradykinesia score, median and IQR | 52 weeks | Exploratory |
Change from baseline in dyskinesia score, median and IQR | Exploratory | |
Change from baseline in PDSS-2 total score (sleep symptoms) | 52 weeks | Secondary |
Safety | ||
AEs, SAEs, AESIs, Infusion Site Evaluation Scale,a clinical laboratory values,b vital sign measurements,b ECGsb | 52 weeks | Primary |
AE = adverse event; AESI = adverse event of special interest; BK = bradykinesia score; DK = dyskinesia; ECG = electrocardiogram; EQ-5D-5L = 5-Level EQ-5D; HRQoL = health-related quality of life; IQR = interquartile range; MDS-UPDRS = Movement Disorder Society-Unified Parkinson’s Disease Rating Scale; PD = Parkinson disease; PDQ-39 = Parkinson’s Disease Questionnaire-39 items; PDSS-2 = Parkinson’s Disease Sleep Scale-2; QoL = quality of life; SAE = serious adverse event.
aThis end point was measured as the proportion of patients with numeric grade ≥ 5 and letter grade ≥ D on the Infusion Site Evaluation Scale in the M15-741 study.
bThis end point was measured as change from baseline to end of study.
Source: M15-741 Clinical Study Report.37 (Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
Efficacy Outcomes
Efficacy end points were secondary in this study; they included the change from baseline to the end of study (week 52) for average normalized daily “off” and “on” times, assessed based on the PD diary; PD symptoms, assessed using the MDS-UPDRS Parts I to IV; sleep symptoms, assessed using the PDSS-2 total score; quality of life, assessed using the PDQ-39; and HRQoL, assessed based on the EQ-5D-5L. The measurement scales were the same as in the M15-736 trial.
Baseline was defined as the last nonmissing assessment before the initiation of foslevodopa-foscarbidopa. Study visits occurred at days 1 and 2 and weeks 1, 2, 3, 4, 6, 13, 26, 39, and 52. All end points were assessed on day 1 and all visits in the maintenance phase. Additional visits were scheduled in the optimization phase for assessing PD diary (week 1) and MDS-UPDRS (all visits) end points.
Harms Outcomes
The harms outcomes were primary end points of this study and included the proportion of patients with AEs, SAEs, adverse event of special interest, numeric grade equal to or higher than 5 and letter grade equal to or higher than D on the Infusion Site Evaluation Scale, and change in clinical laboratory test, vital signs, and ECGs from baseline to end of study.
Statistical Analysis
A sample size calculation determined that approximately 240 enrolled patients were required to obtain exposure data from at least 100 patients treated with foslevodopa-foscarbidopa for at least 12 months and to meet country requirements. With 240 patients receiving foslevodopa-foscarbidopa, the probability of observing an AE with an annual incidence rate of 0.005, 0.01, and 0.02 was 70%, 91%, and 99%, respectively.
All efficacy end points were presented using descriptive statistics. A 2-sided paired-sample t test was performed to assess within-group changes from baseline. No adjustments for multiple statistical testing were performed. The handling of missing items in efficacy instrument was the same as the M15-741 trial but no imputation was performed for missing visit data. Predefined subgroup analyses were performed for all efficacy analyses, with the same subgroup categories as in the M15-736 trial. A sensitivity analysis was conducted for all “on” and “off” times outcomes by modifying the definition of valid PD diary day as a PD diary recording day with no more than 2 hours of missing data (≤ 4 missing entries) for the entire 24-hour diary.
Safety outcomes were compared between treatment arms using descriptive statistics; no hypothesis testing was performed.
Analysis Populations
Analysis populations of the M15-741 are summarized in Table 29.
Table 29: Analysis Populations in the M15-741 Trial
Population | Definition | Application |
|---|---|---|
FAS | All patients who received foslevodopa-foscarbidopa and had baseline and treatment observations for at least 1 efficacy outcome measure | All efficacy analyses |
SAS | All patients who received foslevodopa-foscarbidopa infusion | All safety analyses and some other analyses and evaluations, such as demographics, treatment adherence, and exposure |
FAS = full analysis set; SAS = safety analysis set.
Note: The M15-741 trial also included a treatment-naive analysis set but it was not of interest to this review and was not summarized.
Source: M15-741 Clinical Study Report.37 (Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
Results
Patient Disposition
As summarized in Table 30, ||| |||||||| |||| |||||||| ||| || ||||||| were screen failures. Both the full and the safety analysis sets included 244 patients who entered the 52-week treatment period. Study treatment discontinuation occurred in ||| ||||||| patients. The most common reason for treatment discontinuation was AEs |||||||, similar to the M15-736 trial.
Table 30: Summary of Patient Disposition in the M15-741 Trial (SAS)
Patient disposition | Foslevodopa-foscarbidopa arm |
|---|---|
Screened, N | ||| |
Primary reason for screening failure, n (%) | || |||||| |
Eligibility criteria | || ||||| |
Consent withdrawal | || ||||| |
Lost to follow-up | ||| |
Other | | ||||| |
Enrolled, N | ||| |
Discontinued from study, n (%) | ||| |||||| |
Primary reason for discontinuation, n (%) | |
AEs | || |||||| |
Consent withdrawal | || |||||| |
Lost to follow-up | | ||||| |
Lack of efficacy | || ||||| |
Difficulty with drug delivery | | ||||| |
Other | | ||||| |
FAS, N (%) | ||| ||||| |
SAS, N (%) | ||| ||||| |
AE = adverse event; CD = carbidopa; FAS = full analysis set; LD = levodopa; NA = not applicable; SAS = safety analysis set.
Source: M15-741 Clinical Study Report.37 (Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
Baseline Characteristics
A summary of baseline patient demographics, disease characteristics, and medication history in the M15-741 trial is shown in Table 31. The baseline characteristics outlined are limited to those which are most relevant to this review, or were felt to impact the outcomes or interpretation of the study results.
Patients in the M15-741 trial, compared with those in the M15-736 trial, had a longer mean duration of PD since diagnosis (|||||||| | |||| ||||| || ||| ||| | |||| ||||||, a higher proportion of patients with history of levodopa-induced dyskinesia |||||| || |||||| and of levodopa response of more than 5 years |||||| || ||||||. In addition, the trial had higher proportions of patients with prior DBS procedure ||||| || ||||| and prior oral PD medication other than dopa preparations and higher proportions of patients who had received at least 3 PD medication classes at baseline |||||| || ||||||.
Table 31: Summary of Baseline Characteristics in the M15-741 Trial (SAS)
Characteristic | Foslevodopa-foscarbidopa arm (N = 244) |
|---|---|
Demographics | |
Sex, n (%) | |
Male | ||| |||||| |
Female | || |||||| |
Age (years), mean (SD) | |||| ||||| |
Race, n (%) | |
White | ||| |||||| |
Black/African American | | ||||| |
Asian | || |||||| |
American Indian/Alaska Native | | ||||| |
Multiple | | ||||| |
Disease characteristics | |
Duration of PD since diagnosis (years), mean (SD) | |||| ||||| |
History of levodopa-induced dyskinesia, n (%) | ||| |||||| |
Levodopa response of > 5 years, n (%) | ||| |||||| |
Baseline PD Hoehn and Yahr Scale stage, n (%) | |
0 | ||| |
1 | ||| |
2 | ||| |||||| |
3 | || |||||| |
4 | || |||||| |
5 | || ||||| |
Missing | | |||||| |
Duration since onset of motor fluctuation (year), mean (SD) | ||| ||||| |
MDS-UPDRS Part III score during “off” state, mean (SD) | |||| |||||| |
Normalized “on” or “off” time per day (hours),a mean (SD) | |
“Off” time | ||| ||||| |
“On” time without troublesome dyskinesia | ||| ||||| |
“On” time without dyskinesia | ||| ||||| |
PD treatment history | |
Prior DBS procedure, n (%) | || ||||| |
Prior PD medication use, n (%) | ||| ||||| |
Dopa and dopa derivatives | ||| |||||| |
Dopamine agonists | ||| |||||| |
MAO-B inhibitor | ||| |||||| |
Amantadine derivatives | || |||||| |
COMT inhibitor | || |||||| |
Anticholinergics | | ||||| |
Other | || ||||| |
Number of PD medication class at baseline, n (%) | |
1 | || |||||| |
2 | || |||||| |
3 | || |||||| |
4 | || |||||| |
> 4 | | ||||| |
Baseline levodopa-equivalent dose, including other PD medications (mg/day),b mean (SD) | |||| ||||| |
CD = carbidopa; COMT = catechol-O-methyltransferase; DBS = deep brain stimulation; IR = immediate release; LD = levodopa; MAO-B = monoamine oxidase type B; MDS-UPDRS = Movement Disorder Society-Unified Parkinson’s Disease Rating Scale; NR = not reported; PD = Parkinson disease; SAS = safety analysis set; SD = standard deviation.
aBased on the PD diary.
bRefers to PD medications other than oral levodopa-carbidopa immediate release.
Source: M15-741 Clinical Study Report.37 (Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
Exposure to Study Treatments
Treatment exposure in the M15-741 trial is summarized in Table 32. Mean average total daily levodopa dose was not reported. Mean average levodopa dose (presented by week) ranged from 1,641 mg/day (SD = 667 mg/day) to 1,878 mg/day (SD = 723 mg/day). Mean duration of study drug exposure was ||||| ||| | |||||| days. Mean treatment adherence for drug solution was ||||| ||| | |||||.
Table 32: Treatment Exposure in the M15-741 Trial (SAS)
Exposure in the 52-week treatment period | Foslevodopa-foscarbidopa arm (N = 244) |
|---|---|
Daily levodopa dose (mg) | |
Week 13 | 1,872 (732) |
Week 26 | 1,850 (737) |
Week 52 | 1,878 (723) |
Duration of study drug exposure (days) | |
Mean (SD) | ||||| ||||||| |
Median (range) | ||||| || || |||| |
% adherence, mean (SD) | |
Drug solutiona | |||| ||||| |
SAS = safety analysis set; SD = standard deviation.
aStudy drug solution adherence (%) = hours of infusion divided by 24, then multiplied by 100.
Source: M15-741 Clinical Study Report.37 (Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
Concomitant Medications
Concomitant medication use in the M15-741 trial is summarized in Table 33. In the trial, ||||| of patients receive concomitant PD medications. The most common concomitant PD medication classes (in at least 30% of patients) were |||||||| ||||||||| |||| ||| |||| |||||||||||| ||| ||||| ||||||||||.
Table 33: Concomitant PD Treatment in the M15-741 Trial (SAS)
Concomitant medication (class) | Foslevodopa-foscarbidopa arm (N = 244) |
|---|---|
Any concomitant PD medications,a n (%) | ||| |||||| |
Dopaminergic drugs | ||| |||||| |
Dopamine agonists | ||| |||||| |
Dopa and dopa derivatives | ||| |||||| |
MAO-B inhibitors | ||| |||||| |
Amantadine derivatives | || |||||| |
Other dopaminergic agents | || ||||| |
Anticholinergic drugs | | ||||| |
Other anti-PD drugs | | ||||| |
MAO-B = monoamine oxidase type B; PD = Parkinson disease; SAS = safety analysis set.
aNote that patients could have received more than 1 concomitant medication during the treatment period.
Source: M15-741 Clinical Study Report.37 (Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
Rescue Medications
Mean total daily levodopa dose from rescue medications (by week) ranged between ||||| ||| | ||||| || ||| ||| || ||||| ||| | ||||| || ||| |||.
Protocol Deviations
As summarized in Table 34, protocol deviations were most commonly related to enrolment of patients who did not satisfy the inclusion or exclusion criteria ||||||, and study procedure compliance ||||||. The sponsor noted that none of the protocol deviations recorded could have affected the study outcome or interpretation of the study results or conclusions.
Table 34: Protocol Deviations in the M15-741 Trial
Protocol deviations | Foslevodopa-foscarbidopa arm (N = 244) |
|---|---|
Study procedure compliance | || ||||| |
Wrong treatment or incorrect dose | | ||||| |
Dosing compliance by patient | | ||||| |
Did not satisfy entry criteria | || |||||| |
Dispensation or administration of investigation product | | ||||| |
Expired investigation product | | ||||| |
Inadequate safety reporting | | ||||| |
Missed visit or visit windows | | ||||| |
Receive prohibited medication | | ||||| |
Received excluded concomitant medication | | ||||| |
Medical communication | | ||||| |
aTwenty-three patients did not fully meet eligibility criteria: 5 patients had a low vitamin B12 levels; 7 patients did not have a recognizable/identifiable “off” and “on” state (motor fluctuations) confirmed by the concordance test performed during the screening period, at baseline; 4 patients did not have any valid Parkinson disease (PD) diaries; and 10 patients had PD diaries but at least 1 PD diary day contained < 2.5 hours of “off” time.
Source: M15-741 Clinical Study Report.37 (Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
Efficacy
Unless otherwise specified, the key efficacy results of the M15-741 trial are summarized in Table 35.
“On” Time Outcomes
“On” Time Without Troublesome Dyskinesia
Change from baseline to week 52 in the average daily normalized “on” time without troublesome dyskinesia was a secondary end point and was not adjusted for multiplicity. The mean change from baseline at week 52 was 3.58 (SD = 3.03) hours. Results of the sensitivity analysis were consistent with the primary analysis (Table 46).
“On” Time Without Dyskinesia
|||||| |||| |||||||| || |||| || || ||| ||||||| ||||| |||||||||| |||| |||| ||||||| |||||||||| ||| | ||||||||| |||||||| ||| ||| ||| |||||||| ||| ||||||||||||| ||| |||| |||||| |||| |||||||| || |||| || ||| |||| ||| | ||||| ||||||
“Off” time
Change from baseline to week 52 in the average daily normalized “off” time was a secondary end point and was not adjusted for multiplicity. The mean change from baseline at week 52 was –3.39 (SD = 3.08) hours. Results of the sensitivity analysis were consistent with the primary analysis (Table 46).
Health-Related Quality of Life
Parkinson’s Disease Questionnaire-39 Items
Change from baseline to week 52 in PDQ-39 summary index total score was a secondary end point and was not adjusted for multiplicity. ||| |||| |||||| |||| |||||||| || |||| || ||| |||| ||| | |||||||
5-Level EQ-5D
Change from baseline to week 52 in EQ-5D-5L summary index score was a secondary end point and was not adjusted for multiplicity. Results are summarized in Table 49 in Appendix 1. ||| |||| |||||| |||| |||||||| || |||| || ||| ||||| ||| | |||||||
PD Symptoms
Changes from baseline to week 52 in MDS-UPDRS Part I, Part II, Part III, and Part IV were secondary end points and were not adjusted for multiplicity. ||| |||| |||||| |||| |||||||| || |||| || || |||| || ||| |||| ||| || |||||| |||| |||| ||| | |||||| |||| ||| | |||||| ||| ||| | ||||||| ||| |||| ||| | |||||| |||||||||||||
Bradykinesia and Dyskinesia Scores Assessed Using the PKG
Change from baseline to week 52 in median bradykinesia, bradykinesia IQR, median dyskinesia, and dyskinesia IQR scores were exploratory end points and were not adjusted for multiplicity. Results of these analyses are summarized in Table 49. ||| |||| |||||| |||| |||||||| || |||| || || ||||| |||||||| ||||| ||| ||||||| |||||| ||| ||| | |||||| |||| ||| | |||||| ||| ||| | |||||| ||| ||| ||| | ||||||| ||||||||||||| Results are summarized in Table 49 in Appendix 1.
PDSS-2 (Sleep Symptoms)
Change from baseline to week 52 in PDSS-2 total score was a secondary end point and was not adjusted for multiplicity. ||| |||| |||||| |||| |||||||| || |||| || ||| |||| ||| | |||||||
Table 35: Summary of Key Efficacy Results From the M15-741 Trial (FAS)
Treatment arms | Number of patients contributing to the analysis, n (%) | Baseline value, mean (SD) | Change from baseline at week 52, mean (SD) | P value |
|---|---|---|---|---|
Average daily normalized “on” time without troublesome dyskinesia (hours) | ||||
FOS-FOS | 96 (39.3) | 9.52 (NR) | 3.58 (3.03) | ≤ 0.001a |
Average daily normalized “off” time (hours) | ||||
FOS-FOS | 96 (39.3) | 5.90 (NR) | –3.39 (3.08) | ≤ 0.001a |
PDQ-39 (PD-related QoL) | ||||
FOS-FOS | ||| |||||| | |||| |||| | |||| ||||||| | | |||||| |
MDS-UPDRS Part I score (nonmotor aspects of experiences of daily living) | ||||
FOS-FOS | ||| |||||| | |||| |||||| | |||| |||||| | ||||||| |
MDS-UPDRS Part II score (motor experiences of daily living) | ||||
FOS-FOS | ||| |||||| | |||| |||| | |||| |||||| | | |||||| |
MDS-UPDRS Part III score (motor examination) | ||||
FOS-FOS | ||| |||||| | |||| |||| | ||| ||||||| | ||||| |
MDS-UPDRS Part IV score (motor complications) | ||||
FOS-FOS | ||| |||||| | ||| |||| | |||| |||||| | | |||||| |
PDSS-2 total score summary index (sleep symptoms) | ||||
FOS-FOS | ||| |||||| | |||| |||| | |||| ||||||| | | |||||| |
CI = confidence interval; FAS = full analysis set; FOS-FOS = foslevodopa-foscarbidopa; MDS-UPDRS = Movement Disorder Society-Unified Parkinson Disease Rating Scale; NR = not reported; PD = Parkinson disease; PDQ-39 = Parkinson Disease Questionnaire-39 items; PDSS-2 = Parkinson Disease Sleep Scale-2; QoL = quality of life; SD = standard deviation.
aTwo-sided paired sample t test. End point was not adjusted for multiplicity.
Source: M15-741 Clinical Study Report.37 (Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
Harms
A summary of harms in the M15-741 trial are shown in Table 36. Results of the M15-741 trial referred to the findings in the entire study period (CSCI optimization phase and maintenance phase combined).
|||| ||| |||||||| || ||||| || ||||||||| || |||||||| || ||| |||||| ||||| |||||||| || ||| ||||||| ||||| |||||| |||||||| |||| ||||||||| ||||| |||||||||| ||| ||||||| |||||||| |||| ||||||| |||| ||| ||||||||||||| |||| |||| |||||||| || || ||||| ||| || |||||||| || ||| ||||||| ||||||
Serious Adverse Events
||||||| |||| ||| |||||||| || |||| | || ||||||||| |||| ||| |||| |||||| ||| ||||| |||||||| |||| |||||||||| ||||||| |||||||| || |||||||| |||| ||||||| ||||||| ||||||||||||| ||||||| || ||||||| ||| ||||||||| |||||||| |||||||
Withdrawals Due to Adverse Events
||||||||| ||||||||||||||| ||| || |||| ||| |||||||| || ||||| || ||||||||| |||| |||||||| ||||||| || ||||||||||||| ||||||| |||||||| |||| |||||||| ||| |||||||||| ||||| |||||| ||| |||||||||| |||||||
Mortality
||||| |||||| |||||||| ||| || ||||| ||||||||||||||||||| ||||||| |||||||| |||| |||||| ||| |||||||| ||||||||| ||| ||||||||||||||| |||||||||| ||| |||||| |||||||| |||||| || |||| ||||| ||| |||| |||||||||||||||||||||||| |||||||| ||| || ||||||||| ||||||||| ||||| ||||||||||| |||||||| ||| |||||||||| |||| || ||| |||||| ||| |||||||||| || || ||||||| || ||||| |||| || ||| ||| ||||||||
Notable Harms
Infusion-Site Reactions and Infusion-Site Infections
|||||||| |||| ||||||||| ||| |||||||| |||| |||||||||| |||| |||||||| || ||||| ||| ||||| || ||||||||| ||||||||||||| |||||||||||| |||||||| ||| || ||||| | ||||||||| || ||||||| ||||| || || ||||| | ||| | |||||| ||||| || || ||||| | || ||| |||||||| |||| |||||||||| ||||||
Hallucination or Psychosis, Impulse-Control Disorder, and Suicidality
||||||||||||| || ||||||||| ||| |||||||| || ||||| || ||||||||| ||||||||||||||||| |||||||| ||| ||||||||| ||||||||| |||| |||||||| || | ||||||| ||| | ||||||||||| ||||||||||||| ||| ||||||| |||||| || |||| |||| ||||| |||| ||| ||| |||| ||||||||| ||||||| |||||||| ||| ||||||| |||||||| ||||||||| |||| |||||| ||||| |||| ||||||||| |||| ||||||||| |||||| |||||||||| |||||| |||| || ||||||| |||||||| ||| |||||||| |||||||||| || ||||||||||
Dizziness and Orthostatic Hypotension
||| ||||||||| || ||||||||| ||| ||||||||||| ||||||||||| |||| |||||| ||| ||||| |||||||||||||
Somnolence
|||||||||| ||| |||||||| || || |||||| |||||||||
Depression
|||||||||| ||| |||||||| || | |||||| |||||||||
Table 36: Summary of Harms From the M15-741 Trial (SAS)
Adverse events | M15-741 entire study period — CSCI optimization and maintenance phases combined foslevodopa-foscarbidopa arm (N = 244) |
|---|---|
TEAE, n (%) | |
Patients ≥ 1 TEAE | ||| |||||| |
Most common TEAEa, n (%) | |
Infusion-site erythema | ||| |||||| |
Infusion-site nodule | || |||||| |
Infusion-site cellulitis | || |||||| |
Infusion-site edema | || |||||| |
Fall | || |||||| |
Hallucination | || |||||| |
Infusion-site pain | || |||||| |
Serious TEAE, n (%) | |
Patients with ≥ 1 serious TEAE | || |||||| |
Most common serious TEAEb, n (%) | |
Infusion-site cellulitis | || ||||| |
Infusion-site abscess | | ||||| |
Hallucination | | ||||| |
Parkinson disease | | ||||| |
Psychotic disorder | | ||||| |
Patients who stopped treatment due to TEAE, n (%) | |
Patients who stopped treatment due to TEAE | || |||||| |
Most common TEAE leading to treatment discontinuationb, n (%) | |
Hallucination | || ||||| |
Infusion-site cellulitis | | ||||| |
Infusion-site erythema | | ||||| |
Dyskinesia | | ||||| |
Deaths, n (%) | |
Patients who died | | ||||| |
Cardiorespiratory arrest | | ||||| |
Cerebral mass effect and subdural hematoma | | ||||| |
Cerebrovascular accident | | ||||| |
Notable harms, n (%) | |
Infusion-site reactionsc | ||| |||||| |
Infusion-site infections | || |||||| |
Hallucination/psychosisd | || |||||| |
Suicidal behaviours or ideationse | || |||||| |
Dizziness | || |||||| |
Orthostatic hypotension | || ||||| |
Somnolence | || ||||| |
Depression | | ||||| |
Impulse-control disorder | | ||||| |
Impulsive behaviour | | ||||| |
CSCI = continuous subcutaneous infusion; SAS = safety analysis set; TEAE = treatment-emergent adverse event.
Note: Unless otherwise specified, all TEAEs were summarized using Medical Dictionary for Regulatory Activities (MedDRA) preferred terms.
a≥ 15% of patients.
b≥ 2% of patients.
cAdverse event of special interest, including any adverse event in infusion site–related noninfection reactions company MedDRA query (CMQ).
dAdverse event of special interest, including any AE in the hallucinations CMQ or psychosis and psychotic disorder standardized MedDRA query (SMQ).
eAssessed using the Columbia-Suicide Severity Rating Scale.
Source: M15-741 Clinical Study Report.37 (Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
Critical Appraisal
Internal Validity
The open-label study design could introduce reporting bias, potentially leading to inflated benefits of foslevodopa-foscarbidopa on PROs and less favourable harms results given the more subjective nature of these outcomes. The noncomparative design means that known and unknown confounding factors were not accounted for and no statistical adjustments were made in the analyses, making it impossible to be certain that the observed treatment benefits could be attributed to foslevodopa-foscarbidopa alone. As well, a sizable proportion of patients ||||||| withdrew from study treatment, mostly due to AEs and consent withdrawal. As a result, attrition bias may explain the observed efficacy results as patients remaining in the study were more likely to be those who experienced benefits and able to tolerate the treatment better.
External Validity
The inclusion and exclusion criteria generally align with the selection criteria for candidates for advanced therapies used in clinical practice. While the participants in this trial appeared to have more advanced PD than those in the pivotal M15-736 trial, the clinical expert noted that the patient population would fit within the spectrum of patients with advanced PD in Canada. The infusion pump used in the trial (i.e., Crono PAR Series III pump) was different from the one intended for use in Canada (i.e., Vyafuser or the Phillips-Medisize Parkinson’s Disease Subcutaneous Pump Delivery System), and it is unclear if there is any functional difference between them. Much of the appraisal of the M15-736 trial with respect to appropriate use of concomitant PD medications and relevance of study outcomes applies to the M15-741 study given that the study designs were similar. The clinical expert commented that the duration of follow-up of 52 weeks was adequate for assessing the safety of foslevodopa-foscarbidopa.
Description of the M15-737 Trial
Characteristics of the M15-737 trial are summarized in Table 37. M15-737 is an ongoing single-arm, multicentre, open-label extension study of M15-741, with up to 96 weeks of treatment. The objective is to assess the long-term safety and tolerability of foslevodopa-foscarbidopa delivered by CSCI for 24 hours per day. The primary outcomes are AEs and related measures. Efficacy outcomes are also being collected as secondary end points.
Table 37: Details of the M15-737 Trial
Category | Description |
|---|---|
Design and population | |
Study design | Open-label, single-arm, multicentre extension of M15-741 |
Locations | 41 sites located in Asia, Australia, Europe, North America |
Patient enrolment dates: | First patient first visit: June 8, 2020 Last patient last visit: November 5, 2021 (data cut-off date) |
Enrolled (N) | 105 |
Key inclusion criteria | Completed parent study (M15-741), remained on the study drug and met additional criteria (consent, demographic and laboratory assessments, study participant history, and contraception) |
Key exclusion criteria | NA |
Drugs | |
Intervention | Individualized foslevodopa (240 mg/mL) and foscarbidopa (12 mg/mL) solution administered through CSCI over 24 hours/day |
Comparator(s) | NA |
Study duration | |
Screening phase | NA |
Stabilization phase | Optimization phase: 4 weeks |
Treatment phase | Up to 96 weeks |
Follow-up phase | NA |
Outcomes | |
Primary end points |
|
Secondary end points | Change from baseline to the end of study of the following:
|
Publication status | |
Publication | NA |
AE = adverse event; AESI = adverse event of special interest; CSCI = continuous subcutaneous infusion; ECG = electrocardiogram; EQ-5D-5L = 5-Level EQ-5D; HRQoL = health-related quality of life; MDS-UPDRS = Movement Disorder Society-Unified Parkinson’s Disease Rating Scale; MMSE = Mini-Mental State Examination; NA = not applicable; PD = Parkinson disease; PDQ-39 = Parkinson’s Disease Questionnaire-39 items; PDSS-2 = Parkinson’s Disease Sleep Scale-2; SAE = serious adverse event.
Source: M15-737 Clinical Study Report.75 (Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
Populations
M15-737 included patients with PD who, at completion of the parent study, remained on the study drug and met additional criteria (consent, demographic and laboratory assessments, study participant history, and contraception). Patients had to be able to understand the nature of the study and have had the opportunity to have their questions answered by the investigator. Patients who had decision-making capacity were allowed to sign and date an informed consent form. Patients also had to be willing to comply with the procedures required in this study and not be considered by the investigator to be an unsuitable candidate for foslevodopa-foscarbidopa. Patients also could not be exhibiting significant suicidal behaviours at the time of the final visit of the parent study. Patients of childbearing potential must have had a negative urine pregnancy test, and both female and male patients must have agreed to certain protocol-specified contraception. Patient characteristics at baseline are summarized in Table 38.
Table 38: Summary of Baseline Characteristics in the M15-737 Trial
Characteristic | Foslevodopa-foscarbidopa |
|---|---|
Demographics | |
N | |||| |
Sex, n (%) | |
Male | || |||||| |
Female | || |||||| |
Age (years), mean (SD) | |||| |||||| |
Race, n (%) | |
White | || |||||| |
Black/African American | || |||||| |
Asian | || |||||| |
American Indian/Alaska Native | | ||||| |
Native Hawaiian or Pacific Islander | ||| |
Disease characteristics | |
Duration of PD since diagnosis (years), mean (SD) | |||| ||||||| |
History of levodopa-induced dyskinesia, n (%) | || |||||| |
Levodopa response of > 5 years, n (%) | || |||||| |
Baseline PD Hoehn and Yahr Scale stage, n (%) | |
0 | ||| |
1 | ||| |
2 | || |||||| |
3 | || |||||| |
4 | || |||||| |
5 | || ||||| |
Duration since onset of motor fluctuation (year), mean (SD) | |||| ||||||| |
MDS-UPDRS Part III score during “off” state, mean (SD) | |||| ||||||| |
Normalized “on” or “off” time per day (hours), mean (SD)a | |
“Off” time | |||| |||||| |
“On” time without troublesome dyskinesia | ||||| |||||| |
“On” time without dyskinesia | ||||| |||||| |
“On” time with non-troublesome dyskinesia | |||| |||||| |
“On” time with troublesome dyskinesia | |||| |||||| |
PD treatment history | |
Prior DBS procedure, n (%) | || |||||| |
Prior PD medication use, n (%) | |
Dopa and dopa derivatives | ||| |
Dopamine agonists | ||| |
MAO-B inhibitor | ||| |
Amantadine derivatives | ||| |
COMT inhibitor | ||| |
Anticholinergics | ||| |
Other | ||| |
Number of PD medication class at baseline, n (%) | |
0 | ||| |
1 | ||| |
2 | ||| |
3 | ||| |
4 | ||| |
> 4 | ||| |
Prior total daily levodopa-equivalent dose, including other PD medications, mean mg/d (SD) | ||| |
COMT = catechol-O-methyltransferase; DBS = deep brain stimulation; MAO-B = monoamine oxidase type B; MDS-UPDRS = Movement Disorder Society-Unified Parkinson’s Disease Rating Scale; NR = not reported; PD = Parkinson disease; SD = standard deviation.
Source: M15-737 Clinical Study Report.75 (Note: Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
Interventions
Patients enrolled in M15-737 received an individualized continuous subcutaneous infusion (24 hours per day) of foslevodopa-foscarbidopa via an infusion set connected to a pump for up to 96 weeks. Use of concomitant medications reflected the protocol of the parent studies.
Outcomes
Efficacy variables were measurements of average normalized daily “off” time and “on” time, assessed using the PD diary; PD symptoms, assessed using the MDS-UPDRS Parts I to IV; quality of life, assessed using the PDQ-39 and EQ-5D-5L; and cognitive impairment, assessed using the MMSE. Safety evaluations were the primary end points and included AEs (including serious adverse events and adverse events of special interest), Infusion Site Evaluation Scale scores, vital signs, ECGs, C-SSRS scores, and QUIP-RS scores.
Statistical Analysis
In M15-737, efficacy analyses were performed in the full analysis set using data collected no more than 1 day after the end of the infusion of foslevodopa-foscarbidopa, unless otherwise stated. Two-sided paired-sample t tests were performed to test the change from baseline. Safety analyses were performed in the safety analysis set. All safety variables (except AEs and infusion site evaluations) were evaluated using data collected no more than 1 day after the end of the infusion of foslevodopa-foscarbidopa. For categorical safety outcomes, the number of and percentage of each category were summarized by visit. Hypothesis testing was not performed. Unless otherwise noted, the baseline value for efficacy variables and continuous safety outcomes was collected from the last assessment in the parent study. No adjustments were made for missing visit data. For PD diary entries, if no data were available for a visit, the average daily normalized “off” or “on” times values were missing for that visit. For the MDS-UPDRS, MMSE, and PDQ-39, the score was calculated as long as no more than 15% of the answers were missing for that assessment, and the missing item was imputed as the average of the nonmissing items from the same assessment. EQ-5D-5L summary index was only calculated if answers were provided for all questions.
Results
Patient Disposition
Patient disposition is summarized in Table 39. M15-737 is an ongoing study. No patients had completed the study at the time of the data cuts submitted by the sponsors. The majority of patients who completed the parent study went on to enrol in the long term extension study.
Table 39: Patient Disposition in the M15-737 Trial
Disposition | M15-737 |
|---|---|
Enrolled in parent study M15-741, N | ||| |
Completed parent study M15-741, N | ||| |
Enrolled in LTE, N | ||| |
Ongoing, N (%) | || |||||| |
Completed LTE study | ||| |
Discontinued LTE study, N (%) | | ||||| |
Reasons for discontinuation, N (%) | |
AEs | | ||||| |
Infusion site–related infections | | ||||| |
Infusion site–related noninfection reactions | | ||||| |
Withdrew consent | ||| |
Lost to follow-up | ||| |
Lack of efficacy | | ||||| |
Difficulty with drug delivery | ||| |
Other | | ||||| |
Missing | ||| |
FAS, N (%) | || |||||| |
SAS, N (%) | ||| ||||| |
AE = adverse event; FAS = full analysis set; LTE = long-term extension; NR = not reported; SAS = safety analysis set.
Source: M15-737 Clinical Study Report.75 (Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
Exposure to Study Treatments
Prescribed dose by infusion rate and total daily dose, as well as the rates of adherence, are presented in Table 40. The “final” doses are those given at the end of the currently available data. The rates of adherence were high at the time of the available data cut-off.
Table 40: Summary of Patient Exposure in the M15-737 Trial
Exposure | Foslevodopa-foscarbidopa || | |||| |
|---|---|
Initial base prescribed dose, continuous infusion rate (LD mg/hour), mean (SD) | |||| ||||||| |
Final base prescribed dose, continuous infusion rate (LD mg/hour), mean (SD) | |||| ||||||| |
Initial totalb daily dose (LD mg/day), mean (SD) | |||||| |||||||| |
Final totalb daily dose (LD mg/day), mean (SD) | |||||| |||||||| |
Total patient-years | || |
Duration of study drug exposure (days) | || |
Adherencec | |
n | || |
Mean (SD) | ||||| |||||||| |
Median (IQR or range) | ||||| ||||||| |||||| |
IQR = interquartile range; LD = levodopa; SD = standard deviation.
aEnd of optimization phase.
bTotal dose calculated as “base” (mL/hour) × 24 hours per day × 170.75 mg LD per mL.
cStudy drug solution adherence: (hours of infusion / 24) × 100.
Source: M15-737 Clinical Study Report.75 (Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
Efficacy
At the time of this review, M15-737 is still under way. Early findings are available with relatively small sample sizes, especially after week 48.
Mean Average Daily Normalized “On” Time Without Troublesome Dyskinesia, “On” Time With Troublesome Dyskinesia, and “Off” Time Based on the PD Diary
||| |||| |||||| |||| ||| ||||||| ||||| |||||||| |||| |||| |||||||||| || ||| |||||| ||||| |||||||| || |||| || || |||||||||| ||| ||||| || |||| |||| |||| ||| || |||| ||||||| ||| |||| |||||| ||||||| || |||||||| || ||||||||| |||||| |||| |||| ||||| ||| |||||||| ||| ||||||| |||||||| || |||||| || |||||||||||||||||||||||| |||||||| || ||||||| (Figure 2). Note that the sample size at week 60 onwards is very small.

The percentage of patients in the M15-737 trial who reported early morning nonsleep symptoms from baseline through week 72 are presented in Table 41. Again, the number of patients who had reached the week 60 and week 72 assessments at the cut-off date was small.
Table 41: Distribution of Early Morning Nonsleep Symptoms in the M15-737 Trial (FAS)
Time point | N | Off, n (%) | On without dyskinesia, n (%) | On with nontroublesome dyskinesia, n (%) | On with troublesome dyskinesia, n (%) | Total, n (%) |
|---|---|---|---|---|---|---|
Baseline | || | || |||||| | || |||||| | | ||||| | | ||||| | || ||||| |
Week 12 | || | || |||||| | || |||||| | | ||||| | | ||||| | || ||||| |
Week 24 | || | || |||||| | || |||||| | | ||||| | ||| | || ||||| |
Week 36 | || | | |||||| | || |||||| | | ||||| | | ||||| | || ||||| |
Week 48 | || | | |||||| | || |||||| | | ||||| | ||| | || ||||| |
Week 60 | || | | |||||| | | |||||| | ||| | ||| | | ||||| |
Week 72 | || | | |||||| | | |||||| | ||| | ||| | | ||||| |
FAS = full analysis set.
Source: M15-737 Clinical Study Report.75 (Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
Movement Disorder Society-Unified Parkinson’s Disease Rating Scale
In the M15-737 trial, the MDS-UPDRS scores were consistent from baseline to week 48 (Figure 3).

Change From Baseline to Each Planned Visit in HRQoL (PDQ-39)
||||||| || ||| |||||| ||||||||||| |||| |||||||||| |||| |||||||| || |||| || ||| |||||| | ||||||||||||| ||||||||||| |||||||||| ||| ||||||||| ||||| ||||| |||||| || |||| || (Table 42).
Change From Baseline to Each Planned Visit in HRQoL (EQ-5D-5L)
||||||| || ||| |||||||| ||||||||||| |||| |||||||||| |||| |||||||| || |||| || ||| |||||| | ||||||||||||| ||||||||||| |||||||||| || ||||||||| ||||| ||||| |||||| || |||| || (Table 43).
Change in Baseline to Each Planned Visit in Cognitive Impairment Measured Using the MMSE
|| ||| ||||||| |||||| ||||| ||| || ||||||||||| |||||| || ||| |||| ||||| ||||| |||| ||| |||||||| ||||| |||||||||| || |||||||| || |||| || (Table 44).
Table 42: Change From Baseline to Each Planned Visit in Quality of Life Assessed Using the PDQ-39 in the M15-737 Trial (FAS)
Time point | N | Baseline mean | Visit | Change from baseline | P value | ||
|---|---|---|---|---|---|---|---|
Mean (SD) | Median (range) | Mean (SD) | Median (range) | ||||
Baseline | || | || | |||| ||||||| | |||| ||||| ||||| | || | || | || |
Week 24 | || | |||| | |||| ||||||| | |||| ||||| ||||| | ||| ||||||| | ||| ||||||| ||||| | ||||| |
Week 48 | || | |||| | |||| ||||||| | |||| ||||| ||||| | ||| |||||| | ||| ||||||| ||||| | |||||| |
Week 72 | || | |||| | |||| ||||||| | |||| ||||| ||||| | ||| |||||| | |||| |||||| |||| | |||| |
FAS = full analysis set; PDQ-39 = Parkinson’s Disease Questionnaire-39; SD = standard deviation.
Note: P value obtained from 2-sided paired-sample t test.
*P value ≤ 0.05.
Source: M15-737 Clinical Study Report.75 (Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
Table 43: Change From Baseline to Each Planned Visit in HRQoL Assessed Using the EQ-5D-5L in the M15-737 Trial (FAS)
Time point | N | Baseline mean | Visit | Change from baseline | P value | ||
|---|---|---|---|---|---|---|---|
Mean (SD) | Median (range) | Mean (SD) | Median (range) | ||||
Baseline | || | || | ||||| |||||||| | ||||| ||||||| ||||| | || | || | || |
Week 24 | || | ||||| | ||||| |||||||| | ||||| ||||||| ||||| | |||||| |||||||| | |||||| |||||||| |||||| | |||||| |
Week 48 | || | ||||| | ||||| |||||||| | ||||| ||||||| ||||| | |||||| |||||||| | |||||| |||||||| |||||| | ||||||| |
Week 72 | || | ||||| | ||||| |||||||| | ||||| ||||||| |||||| | |||||| |||||||| | |||||| |||||||| ||||||| | |||||| |
EQ-5D-5L = 5-Level EQ-5D; FAS = full analysis set; HRQoL = health-related quality of life; SD = standard deviation.
Note: P value obtained from 2-sided paired-sample t test.
*P value ≤ 0.05.
Source: M15-737 Clinical Study Report.75 (Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
Table 44: Change From Baseline to Each Planned Visit in Cognitive Impairment Assessed Using the MMSE in the M15-737 Trial (FAS)
Time point | N | Baseline mean | Visit | Change from baseline | P value | ||
|---|---|---|---|---|---|---|---|
Mean (SD) | Median (range) | Mean (SD) | Median (range) | ||||
Baseline | || | ||| | |||| |||||| | |||| |||||| ||||| | ||| | ||| | ||| |
Week 24 | || | |||| | |||| |||||| | |||| |||||| ||||| | |||| |||||| | | ||||||| |||| | |||||| |
Week 48 | || | |||| | |||| |||||| | |||| |||||| ||||| | |||| |||||| | | |||||| |||| | |||||| |
Week 72 | || | |||| | |||| |||||| | |||| |||||| ||||| | |||| |||||| | |||| |||||| |||| | |||||| |
FAS = full analysis set; MMSE = Mini-Mental State Examination; SD = standard deviation.
Note: P value obtained from 2-sided paired-sample t test.
Source: M15-737 Clinical Study Report.75 (Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
Harms
All AEs presented in this section were treatment emergent and analyzed in the safety analysis set.
||| |||| |||||||| || || |||||||| |||||||| ||| ||| |||||||| |||| |||||||||| ||| |||| || |||||||| || |||||||| |Table 45|| ||| ||||||| ||||||||| || ||| ||| ||||||| |||||| |||| |||||||||| ||| |||||||||| ||| |||| |||||||||| |||||||| ||| || |||| |||| |||||||| |||| ||||||||| |||||| ||| |||||||| |||| ||||||||||| |||| |||||||| |||||| ||| ||| |||| ||| || |||||||||||||||| ||| ||||| || ||||| |||| ||| ||||| ||||||| || |||||| |||| || ||| ||||| |||||| |||| |||||||||| || ||| |||||||||||| || |||| | |||||||||| ||||||||||| || ||||| ||||||| || ||| ||||| ||||| || ||| |||||| |||||||| |||| |||||||||||
Table 45: Summary of Harms in the M15-737 Trial
Treatment-emergent adverse events, n (%) | M15-737 (N = 105) |
|---|---|
Any TEAE | || |||||| |
Any SAE | || |||||| |
Any TEAE leading to discontinuation | | ||||| |
Any TEAE leading to death | | ||||| |
Any TEAE related to study drug assessed by investigator | || |||||| |
TEAEs occurring in ≥ 10% of patients | |
Infusion-site erythema | || |||||| |
Infusion-site cellulitis | || |||||| |
Fall | || |||||| |
Hallucination | | ||||| | |
TEAE = treatment-emergent adverse event.
Note: Data cut-off is November 2021. The study is still ongoing.
aIncluded in this table because the proportion with this event was > 10% in the parent study (M15-741).
Source: M15-737 Clinical Study Report.75 (Details in the table have been taken from the sponsor’s Summary of Clinical Evidence.15)
Critical Appraisal
Internal Validity
The main limitation was the absence of a comparator arm and the potential biases related to the nonrandomized design. The open-label study design inherently can increase risk of bias in the reporting of any subjective outcomes, including harms outcomes, because patients and their clinicians are aware of the treatment received. In addition, because patients could only enrol after completing the parent trials, there is an increased likelihood of selection bias given that patients who better tolerated the treatment or perceived the treatment as benefiting them were more likely to enrol in the extension studies.
The trial is ongoing and no patients had completed the trial, so the data are immature. The sample size at each assessment point is small, especially after week 48. No definitive conclusions could be drawn from the results of this study.
External Validity
As the extension M15-737 trial consisted of patients who took part in the parent study (M15-741), it is reasonable to expect that the same strengths and limitations related to generalizability apply, with the additional caveat of potential selection bias given the open-label extension study design.
Discussion
Summary of Available Evidence
This report summarizes the evidence for foslevodopa-foscarbidopa in the treatment of advanced PD based on 1 phase III RCT and 1 phase III single-arm trial, and their respective long-term extension studies, as well as 1 ITC.
Two studies, M15-736 and M15-741, met the inclusion criteria for the systematic review conducted by the sponsor. The M15-736 trial (N = 141) was a pivotal phase III, double-blind, double-dummy RCT with the aim of demonstrating the superiority of foslevodopa-foscarbidopa over oral LD-CD IR tablets for the treatment of motor fluctuations in patients with advanced PD. Patients with prior DBS or LCIG treatment were excluded, but eligibility for DBS was not a consideration for enrolment. In the 12-week randomized treatment period, change from baseline at week 12 in average daily normalized “on” time without troublesome dyskinesia (primary end point), “off” time, MDS-UPDRS Part II score, and presence of morning akinesia (key secondary end points) were assessed. At baseline, mean age was 66.4 years (SD = 9.5 years) and the majority of patients were male and white. Mean time since diagnosis of PD of 8.6 years (SD = 4.9 years). Mean time spent in “off” and “on” without troublesome dyskinesia motor states were 6.1 hours (SD = 2.1 hours) and 9.3 hours (SD = 2.5), respectively.
The M15-741 trial (N = 244) was a supportive phase III, open-label, single-arm trial to evaluate the safety and tolerability of foslevodopa-foscarbidopa in patients with advanced PD for 52 weeks. Safety and efficacy were assessed as primary and secondary end points, respectively, most of which were consistent with the end points of M15-736. Baseline patient characteristics were in general similar to the M15-736 trial, although in the M15-741 mean time since PD diagnosis (12.3 years [SD = 5.3 years]) was longer, and more patients were at advanced stages of PD (based on the Hoehn and Yahr Scale) and received, on average, more medications from different drug classes, suggestive of a patient population with more advanced disease than the patient population included in the M15-736 trial.
Safety and efficacy results from 2 ongoing long-term extension studies, M20-098 (extension of M15-736; N = 103; data cut-off: September 29, 2021) and M15-737 (extension of M15-741; N = 105; data cut-off: November 5, 2021) in which all patients received open-label foslevodopa-foscarbidopa, were also submitted by the sponsor and presented in this report.
One NMA, which compared the efficacy of foslevodopa-foscarbidopa versus LCIG and BMT (oral therapy) on “off” time, “on” time without troublesome dyskinesia, and PDSS-2 total score in patients with advanced PD based on 4 included studies, was conducted by the sponsor in the absence of direct comparative evidence of foslevodopa-foscarbidopa and standard of care other than with oral LD-CD IR.
Interpretation of Results
Efficacy
Evidence from the pivotal phase III M15-736 trial supported the superiority of foslevodopa-foscarbidopa to oral LD-CD IR therapy with respect to the primary end points of change from baseline to week 12 in average daily normalized “on” time without troublesome dyskinesia and the key secondary end point of change from baseline to week 12 in average daily normalized “off” time, thus addressing important outcomes pertaining to motor fluctuations noted by patients and clinicians. The between-arm difference with respect to “off” time met the published MID estimate for patients with advanced PD. The magnitude of benefit in both analyses is considered clinically meaningful based on clinical expert input. Results of average daily “on” time without dyskinesia (secondary end point) and presence of morning akinesia (key secondary end point) were similarly in favour of foslevodopa-foscarbidopa, although failure of a prior end point in the statistical testing hierarchy precludes definitive conclusion on the statistical comparisons of these end points. Results of the prespecified subgroup analyses also supported the overall results of the trial; however, no definitive conclusions can be drawn on these analyses because of the lack of sample size consideration and control for multiplicity.
Quality of life is substantially impacted by motor fluctuations associated with PD, according to patient group input. Two HRQoL end points, including change from baseline to week 12 in PDQ-39 and EQ-5D-5L summary indices, were measured as secondary end points. The PDQ-39 scale is a PD-specific HRQoL instrument for which there is evidence for its validity and reliability in patients with PD. The clinical expert consulted by CADTH indicated that the PDQ-39 may be used in clinical practice. The PDQ-39 analysis showed results in favour of foslevodopa-foscarbidopa versus oral LD-CD IR therapy, while the EQ-5D-5L analysis did not favour either intervention at week 12. No definitive conclusions, however, can be drawn with regards to these HRQoL measures as the hierarchal testing procedure failed at a higher level.
Reducing disabilities from motor and nonmotor symptoms of PD is a key goal of treatment. The MDS-UPDRS, a validated and clinically relevant instrument, assessed the impact of PD on functioning, and captured all symptoms that were noted to be important to control based on the patient group input. The study did not demonstrate superiority of foslevodopa-foscarbidopa with respect to the key secondary end point of change from baseline to week 12 in MDS-UPDRS Part II score, which measures motor experiences of daily living. The clinical expert consulted by CADTH noted that the lack of benefit should be interpreted taking into account how the instrument is constructed. The clinical expert noted that the MDS-UPDRS is designed to assess the level of impairment based on patient’s overall experience in the past week. Based on the clinical expert’s experience, patients with fluctuating motor symptoms tend to plan their activities around when they expect to have a window of “on” time so most patients can still maintain the level of daily activities despite changes in motor symptoms. The clinical expert noted that the likely benefit of foslevodopa-foscarbidopa to provide flexibility, consistency, and freedom for patients to choose when to perform daily activities during the day addresses an aspect of quality of life that the MDS-UPDRS could not measure. Results of MDS-UPDRS Part I (nonmotor aspects of experiences of daily living) and III (motor examination) did not favour either intervention, while results of Part IV (motor complications) did. However, as these were evaluated as exploratory outcomes in the trial and were not adjusted for multiplicity, these results should be interpreted with consideration of potentially increased type I error.
PD symptom-specific secondary end points included changes in bradykinesia and dyskinesia (mean and IQR) scores and PDSS-2 total score, which assessed sleep disruption. The clinical relevance of these outcomes and/or how they were measured is unclear. According to the clinical expert consulted by CADTH, the PKG device measuring bradykinesia and dyskinesia scores is not available in Canada and therefore is not a tool relevant to current clinical practice. Similarly, PDSS-2 is not routinely administered in clinical practice. Results of bradykinesia score and PDSS-2 total score analyses favoured foslevodopa-foscarbidopa, while results of dyskinesia did not favour either intervention. All of these outcomes were lower in the hierarchal testing procedure than the outcome at which the hierarchy failed; therefore, no definitive conclusions on the statistical comparisons can be made.
Patients expressed an unmet need for treatments that can improve cognitive function. Although cognition was captured in the MDS-UPDRS Part I, it was not assessed as a stand-alone end point in the study and, therefore, the effect of foslevodopa-foscarbidopa on cognition was inconclusive in this study. The clinical expert noted that it is reasonable to expect cognitive improvement from a treatment like foslevodopa-foscarbidopa, which provides steady treatment effect, but based on experience with similar treatments the benefit is expected only in select patients with cognitive impairment that fluctuates in parallel with motor fluctuations. In the absence of supporting evidence from the trial, it is also unclear if foslevodopa-foscarbidopa can reduce caregiver burden, a treatment goal that was noted by the clinical expert.
In addition, interpretation of the results should take into account several study limitations. Attrition due to AEs was observed at a higher frequency (difference of more than 17%) in the foslevodopa-foscarbidopa arm than in the comparator arm. Sensitivity analyses assessing the impact of missing data were conducted on “on” and “off” time outcomes only, with results consistent with the primary analyses. The certainty of point estimates for other outcomes is lower due to potential attrition bias that were unaccounted for. Most outcomes were PROs (except bradykinesia and dyskinesia scores) and were potentially subject to reporting bias, especially considering the fluctuating nature of PD and impacts on cognitive function, although the use of central raters for PDSS-2 and PDQ-39 assessments could help reduce the degree of potential bias in differential outcome assessment. As well, there is evidence for the validity and reliability of the PD diary, an instrument commonly used in clinical practice and trials to assess motor fluctuations in patients with PD. Of note, given the apparent differences in treatment response and AE profiles between interventions, there is a risk of reporting bias from patients, potentially toward an inflated efficacy of foslevodopa-foscarbidopa, for PROs due to possible unblinding.
Evidence for the longer-term effects of foslevodopa-foscarbidopa treatment (beyond 12 weeks) is limited in terms of available data and lower quality of the evidence. The M15-741 trial showed improvement from baseline in average daily normalized “on” time without troublesome dyskinesia, “off” time, “on” time without dyskinesia, PDQ-39, MDS-UPDRS Part II and IV scores, PDSS-2 total score, and EQ-5D-5L summary index after 52 weeks of foslevodopa-foscarbidopa treatment. Results did not suggest a difference in change from baseline in MDS-UPDRS Part I and III scores or in bradykinesia and dyskinesia scores. However, these results are subject to uncertainty due to risks of reporting bias and confounding due to the open-label, noncomparative trial design.
Results from M20-098 were too immature to draw conclusions on at the time of this review, given that the sample size was 13 patients or less at week 12 and beyond.
Evidence from M15-737 suggests that the benefits of foslevodopa-foscarbidopa were maintained with respect to average daily normalized “on” time without troublesome dyskinesia, “on” time with troublesome dyskinesia, “off” time, MDS-UPDRS, and MMSE, but decreased with time with respect to HRQoL (PDQ-39 and EQ-5D-5L). Sample sizes in M15-737 were small at week 48, ranging between outcomes, from 24 to 34 patients. Beyond week 48, sample sizes were too small to draw conclusions from (e.g., n = 7 or less). This trial is ongoing; no patient had completed the study as of data cut-off; as such, the findings were considered preliminary.
Overall, results from the long-term extension studies should be interpreted based on important limitations including sample sizes, potential reporting bias and confounding due to the open-label, noncomparative design, and a likelihood of selection and attrition bias.
The results of the sponsor-submitted NMA agreed with the trial comparison that foslevodopa-foscarbidopa was associated with improved average “on” time without troublesome dyskinesia, “off” time, and PDSS-2 total score at 12 weeks compared with BMT (oral therapy). However, the validity of the results of the NMA are uncertain because of important limitations preventing assessment of the key assumptions for the analyses. Because of limited available studies, the network included only 3 interventions, foslevodopa-foscarbidopa connected with LCIG through BMT as the central connection in the linear network. It is not possible to evaluate consistency between direct and indirect comparison in networks with this geometry (a sparse network with no closed loop). As well, the limited number of studies and limited reporting of study characteristics (including baseline patient characteristics) in the technical report hampered assessing homogeneity. Of the available information, there appeared to be important sources of heterogeneity in study designs and patient populations between studies that were unaccounted for, which result in uncertainty in the comparative treatment effect estimates. Therefore, no concrete conclusions could be drawn on the comparative effectiveness and safety of foslevodopa-foscarbidopa and LCIG.
Furthermore, direct or indirect evidence between foslevodopa-foscarbidopa and DBS is not in the submission. The sponsor suggested that DBS is not an appropriate comparator on the basis that DBS has many additional considerations (e.g., patient consent to surgical risk, surgical backlogs, travel issues) that make this comparison inadvisable. The clinical expert noted there is overlap in the patient populations with advanced PD eligible for DBS and foslevodopa-foscarbidopa. This means that both treatments would be presented as options in patients with advanced PD, rendering DBS a relevant comparator in the treatment of these patients. The CADTH review team acknowledges that there are practical considerations that guide treatment choice; however, the lack of any comparative evidence between foslevodopa-foscarbidopa and DBS represents a gap in evidence given the shared place in therapy for advanced PD.
Finally, it is worth noting that there are differences in the existing reimbursement criteria for advanced therapies between jurisdictions in Canada, which could result in the characteristics of the eligible populations to differ between jurisdictions. In most jurisdictions, eligibility for DBS is not a consideration for reimbursement of other treatments in advanced PD, except in British Columbia. A health technology assessment by the British Columbia Health Technology Assessment Committee supported DBS as a more clinically and cost-effective option than LCIG.76 According to the drug plan input for this review, patients in British Columbia are required to have a contraindication to DBS or be on the DBS waitlist for more than 1 year as a condition for receiving reimbursement for LCIG. Given that foslevodopa-foscarbidopa and LCIG have the same expected place in therapy and that DBS eligibility was not a consideration for enrolment in the studies presented in this report, the generalizability of study findings may be lower in British Columbia than in other jurisdictions.
Harms
In the M15-736 trial, foslevodopa-foscarbidopa was associated with a notably higher frequency of TEAEs than the oral LD-CD IR therapy. The most frequently reported TEAEs of foslevodopa-foscarbidopa were infusion-site reactions and infusion-site infections. Although the TEAEs were mostly nonserious, they were the primary reported cause for the higher occurrence of treatment discontinuation in the foslevodopa-foscarbidopa arm. The clinical expert noted that close monitoring for infusion site–related TEAEs is required, although the likelihood of these TEAEs causing significant safety issues is expected to be low. Compared with oral LD-CD therapy, foslevodopa-foscarbidopa was associated with a lower frequency of falls and a higher frequency of hallucination or psychosis. In the clinical expert’s opinion, the higher percentage of patients reporting hallucination would warrant more careful selection of candidates for treatment and conservative dosing when initiating treatment, but hallucinations are not expected to be a major concern as they can likely be managed with dosing adjustment or drugs that are typically used to suppress hallucination. The AE profile of foslevodopa-foscarbidopa was similar during the 12-week phase of the M15-736 trial and the 52-week M15-741 trial phase as well as in the ongoing M15-737 extension trial as of data cut-off. Harms were not measured in the sponsor-submitted NMA, as such, the comparative safety of foslevodopa-foscarbidopa versus LCIG is unknown.
Conclusions
In the pivotal M15-736 trial, foslevodopa-foscarbidopa demonstrated a clinically meaningful improvement in “on” time without troublesome dyskinesia and “off” time compared with oral LD-CD therapy at 12 weeks in patients with advanced PD. Analyses of morning akinesia, HRQoL, bradykinesia, and sleep symptoms also favoured foslevodopa-foscarbidopa, although due to failure of a prior outcome in the statistical testing hierarchy, the results for these outcomes were considered supportive of benefit with foslevodopa-foscarbidopa treatment, but not conclusive. Results did not suggest a difference in motor experiences of daily living, though the MDS-UPDRS instrument may be limited in its utility in assessing certain aspects of motor functioning in patients receiving advanced therapies (e.g., consistent control of motor fluctuations and flexibility in performing daily activities). The pivotal study results were determined to be generalizable overall. The effectiveness and safety of foslevodopa-foscarbidopa relative to comparators other than oral LD-CD could not be determined. There are no direct comparisons with LCIG, and the indirect comparison was inconclusive because of important limitations that prevented verifying whether the underlying assumptions of homogeneity and consistency were met. No direct or indirect comparisons between foslevodopa-foscarbidopa and DBS were submitted. Overall, the safety profile of foslevodopa-foscarbidopa was similar to oral LD-CD therapy, except that infusion-site reactions and infections were more frequent with foslevodopa-foscarbidopa; most reactions and infections were not serious, but some resulted in treatment discontinuation. No new serious safety concerns were identified in the longer-term safety studies.
References
1.Vyalev (foslevodopa/foscarbidopa): 240 mg/mL foslevodopa and 12 mg/mL foscarbidopa solution for subcutaneous infusion [product monograph]. St-Laurent (QC): AbbVie Corporation; 2022 Apr 29.
2.Mhyre TR, Boyd JT, Hamill RW, Maguire-Zeiss KA. Parkinson's disease. Subcell Biochem. 2012;65:389-455. PubMed
3.Jankovic J, Tan EK. Parkinson's disease: etiopathogenesis and treatment. J Neurol Neurosurg Psychiatry. 2020;91(8):795-808. PubMed
4.Clarke CE. Parkinson's disease. BMJ. 2007;335(7617):441-445. PubMed
5.Davie CA. A review of Parkinson's disease. Br Med Bull. 2008;86:109-127. PubMed
6.Willis AW, Roberts E, Beck JC, et al. Incidence of Parkinson disease in North America. NPJ Parkinsons Dis. 2022;8(1):170. PubMed
7.Dahodwala N, Pettit AR, Jahnke J, et al. Use of a medication-based algorithm to identify advanced Parkinson's disease in administrative claims data: associations with claims-based indicators of disease severity. Clin Park Relat Disord. 2020;3:100046. PubMed
8.Malaty I, Jones S, Dahodwala N, et al. Unmet needs and treatment patterns of advanced Parkinson’s disease patients in the United States (2367). Neurology. 2021;96(15 Suppl):2367.
9.Antonini A, Stoessl AJ, Kleinman LS, et al. Developing consensus among movement disorder specialists on clinical indicators for identification and management of advanced Parkinson's disease: a multi-country Delphi-panel approach. Curr Med Res Opin. 2018;34(12):2063-2073. PubMed
10.Santos-García D, de Deus Fonticoba T, Suárez Castro E, Aneiros Díaz A, McAfee D. 5-2-1 criteria: a simple screening tool for identifying advanced PD patients who need an optimization of Parkinson's treatment. Parkinsons Dis. 2020;2020:7537924. PubMed
11.Grimes D, Fitzpatrick M, Gordon J, et al. Canadian guideline for Parkinson disease. CMAJ. 2019;191(36):E989-E1004. PubMed
12.Fabbri M, Rosa MM, Ferreira JJ. Adjunctive therapies in Parkinson's disease: how to choose the best treatment strategy approach. Drugs Aging. 2018;35(12):1041-1054. PubMed
13.Soileau MJ, Aldred J, Budur K, et al. Safety and efficacy of continuous subcutaneous foslevodopa-foscarbidopa in patients with advanced Parkinson's disease: a randomised, double-blind, active-controlled, phase 3 trial. Lancet Neurol. 2022;21(12):1099-1109. PubMed
14.Clinical Study Report: M15-736. A randomized, double-blind, double-dummy, active-controlled study comparing the efficacy, safety and tolerability of ABBV-951 to oral carbidopa/levodopa in advanced Parkinson's disease patients [internal sponsor's report]. Chicago (IL): AbbVie; 2022 Jan 29.
15.Foslevodopa/foscarbidopa (ABBV-951) continuous subcutaneous infusion (CSCI). Treatment of advanced Parkinson's Disease. Clinical summary [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Foslevodopa/Foscarbidopa solution, 240 mg/mL foslevodopa and 12 mg/mL foscarbidopa solution for subcutaneous infusion. St-Laurent (QC): Abbvie Corporation; 2022 Nov 22.
16.Antonini A, Moro E, Godeiro C, Reichmann H. Medical and surgical management of advanced Parkinson's disease. Mov Disord. 2018;33(6):900-908. PubMed
17.Olanow CW, Obeso JA, Stocchi F. Drug insight: Continuous dopaminergic stimulation in the treatment of Parkinson's disease. Nat Clin Pract Neurol. 2006;2(7):382-392. PubMed
18.Wolters E, Lees AJ, Volkmann J, van LT, Hovestadt A. Managing Parkinson's disease with continuous dopaminergic stimulation. CNS Spectr. 2008;13(4 Suppl 7):1-14. PubMed
19.Parkinson's Foundation. Statistics. 2021; https://www.parkinson.org/Understanding-Parkinsons/Statistics. Accessed 2023 Feb 8.
20.Collaborators GBDPsD. Global, regional, and national burden of Parkinson's disease, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018;17(11):939-953. PubMed
21.Statistics Canada. Leading causes of death, total population, by age group. 2022; https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310039401. Accessed 2023 Feb 8.
22.Statista. Death rate for Parkinson's disease in Canada from 2000 to 2020. 2023; https://www.statista.com/statistics/434428/death-rate-for-parkinson-s-disease-in-canada/#:~:text=Death%20rate%20for%20Parkinson's%20disease%20in%20Canada%202000%2D2020&text=In%202020%2C%20around%20nine%20out,at%204.8%20deaths%20per%20100%2C000. Accessed 2023 Feb 8.
23.Macleod AD, Taylor KS, Counsell CE. Mortality in Parkinson's disease: a systematic review and meta-analysis. Mov Disord. 2014;29(13):1615-1622. PubMed
24.Bloem BR, Grimbergen YA, Cramer M, Willemsen M, Zwinderman AH. Prospective assessment of falls in Parkinson's disease. J Neurol. 2001;248(11):950-958. PubMed
25.Martignoni E, Godi L, Citterio A, et al. Comorbid disorders and hospitalisation in Parkinson's disease: a prospective study. Neurol Sci. 2004;25(2):66-71. PubMed
26.Pickering RM, Grimbergen YA, Rigney U, et al. A meta-analysis of six prospective studies of falling in Parkinson's disease. Mov Disord. 2007;22(13):1892-1900. PubMed
27.McLean G, Hindle JV, Guthrie B, Mercer SW. Co-morbidity and polypharmacy in Parkinson's disease: insights from a large Scottish primary care database. BMC Neurol. 2017;17(1):126. PubMed
28.Minar M, Dragasek J, Valkovic P. Comorbidities in Parkinson's disease – the results from national epidemiological study cosmos. J Neurol Sci. 2019;405:211.
29.Postuma RB, Berg D, Stern M, et al. MDS clinical diagnostic criteria for Parkinson's disease. Mov Disord. 2015;30(12):1591-1601. PubMed
30.Grimes D. Parkinson disease. Ottawa (ON): Canadian Pharmacist Association; 2020: https://www.myrxtx.ca/. Accessed 2023 Feb 8.
31.Parkinson's Foundation. Surgical treatment options: deep brain stimulation (DBS). 2021; https://www.parkinson.org/Understanding-Parkinsons/Treatment/Surgical-Treatment-Options/Deep-Brain-Stimulation. Accessed 2023 Feb 07.
32.Deuschl G, Schade-Brittinger C, Krack P, et al. A randomized trial of deep-brain stimulation for Parkinson's disease. N Engl J Med. 2006;355(9):896-908. PubMed
33.Dijka JM, Espayb A, Katzenschlagerc R, de Biea R. The choice between advanced therapies for Parkinson’s disease patients: why, what, and when? J Parkinsons Dis. 2022;10(Suppl 1):S65-S73. PubMed
34.AbbVie Corporation's response to Feb 3, 2023 CADTH request for additional information regarding foslevodopa/foscarbidopa CADTH review: AbbVie Corporation’s characterization of candidacy for deep brain stimulation treatment [internal additional sponsor's information]. St-Laurent (QC): Abbvie Corporation; 2023.
35.Duodopa (levodopa/carbidopa intestinal gel): 20 mg/mL levodopa and 5 mg/mL carbidopa monohydrate [product monograph]. St-Laurent (QC): AbbVie Corporation; 2022 Apr 4: https://pdf.hres.ca/dpd_pm/00065363.PDF. Accessed 2023 Feb 07.
36.Parkinson's Canada. Explorations into health system solutions for Parkinson’s in Canada. 2022 Nov 23; https://www.parkinson.ca/explorations-into-health-system-solutions-for-parkinsons-in-canada. Accessed 2023 Feb 07.
37.Clinical Study Report: M15-741. A 52-week, open-label, single-arm study to evaluate the safety and tolerability of 24-hour daily exposure of continuous subcutaneous infusion of ABBV-951 in subjects with Parkinson's disease [internal sponsor's report]. Chicago (IL): AbbVie; 2022 Jan 25.
38.AbbVie Corporation's response to Jan 17, 2023 DRR request for additional information regarding Foslevodopa/Foscarbidopa DRR review: M15-741 trial status for Health Canada approval [internal additional sponsor's information]. St-Laurent (QC): Abbvie Corporation; 2023.
39.Clinical Study Report: M20-098. An open-label extension of study M15-736 to evaluate the safety and tolerability of 24-hour daily exposure of ABBV-951 in subjects with advanced Parkinson's disease [internal sponsor's report]. Chicago (IL): AbbVie; 2022 Jan 25.
40.Tomlinson CL, Stowe R, Patel S, Rick C, Gray R, Clarke CE. Systematic review of levodopa dose equivalency reporting in Parkinson's disease. Mov Disord. 2010;25(15):2649-2653. PubMed
41.Espay AJ, Pagan FL, Walter BL, et al. Optimizing extended-release carbidopa/levodopa in Parkinson disease: consensus on conversion from standard therapy. Neurol Clin Pract. 2017;7(1):86-93. PubMed
42.AbbVie. NCT04380142: Study comparing continuous subcutaneous infusion of ABBV-951 with oral carbidopa/levodopa tablets for treatment of motor fluctuations in adult participants with advanced Parkinson's disease. ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2022: https://clinicaltrials.gov/ct2/show/study/NCT04380142. Accessed 2023 Feb 07.
43.Martinez-Martin P, Jeukens-Visser M, Lyons KE, et al. Health-related quality-of-life scales in Parkinson's disease: critique and recommendations. Mov Disord. 2011;26(13):2371-2380. PubMed
44.Peto V, Jenkinson C, Fitzpatrick R, Greenhall R. The development and validation of a short measure of functioning and well being for individuals with Parkinson's disease. Qual Life Res. 1995;4(3):241-248. PubMed
45.Marinus J, Ramaker C, van Hilten JJ, Stiggelbout AM. Health related quality of life in Parkinson's disease: a systematic review of disease specific instruments. J Neurol Neurosurg Psychiatry. 2002;72(2):241-248. PubMed
46.Jenkinson C, Fitzpatrick R, Norquist J, Findley L, Hughes K. Cross-cultural evaluation of the Parkinson's Disease Questionnaire: tests of data quality, score reliability, response rate, and scaling assumptions in the United States, Canada, Japan, Italy, and Spain. J Clin Epidemiol. 2003;56(9):843-847. PubMed
47.Damiano AM, Snyder C, Strausser B, Willian MK. A review of health-related quality-of-life concepts and measures for Parkinson's disease. Qual Life Res. 1999;8(3):235-243. PubMed
48.Harrison JE, Preston S, Blunt SB. Measuring symptom change in patients with Parkinson's disease. Age Ageing. 2000;29(1):41-45. PubMed
49.Jenkinson C, Fitzpatrick R, Peto V, Greenhall R, Hyman N. The Parkinson's Disease Questionnaire (PDQ-39): development and validation of a Parkinson's disease summary index score. Age Ageing. 1997;26(5):353-357. PubMed
50.Bushnell DM, Martin ML. Quality of life and Parkinson's disease: translation and validation of the US Parkinson's Disease Questionnaire (PDQ-39). Qual Life Res. 1999;8(4):345-350. PubMed
51.Damiano AM, McGrath MM, Willian MK, et al. Evaluation of a measurement strategy for Parkinson's disease: assessing patient health-related quality of life. Qual Life Res. 2000;9(1):87-100. PubMed
52.Peto V, Jenkinson C, Fitzpatrick R. PDQ-39: a review of the development, validation and application of a Parkinson's disease quality of life questionnaire and its associated measures. J Neurol. 1998;245 (Suppl 1):S10-14. PubMed
53.Fitzpatrick R, Peto V, Jenkinson C, Greenhall R, Hyman N. Health-related quality of life in Parkinson's disease: a study of outpatient clinic attenders. Mov Disord. 1997;12(6):916-922. PubMed
54.Peto V, Jenkinson C, Fitzpatrick R. Determining minimally important differences for the PDQ-39 Parkinson's disease questionnaire. Age Ageing. 2001;30(4):299-302. PubMed
55.Hauser RA, Deckers F, Lehert P. Parkinson's disease home diary: further validation and implications for clinical trials. Mov Disord. 2004;19(12):1409-1413. PubMed
56.Hauser RA, Auinger P. Determination of minimal clinically important change in early and advanced Parkinson's disease. Mov Disord. 2011;26(5):813-818. PubMed
57.Hauser RA, Gordon MF, Mizuno Y, et al. Minimal clinically important difference in Parkinson's disease as assessed in pivotal trials of pramipexole extended release. Parkinsons Dis. 2014;2014:467131. PubMed
58.Goetz CG, Tilley BC, Shaftman SR, et al. Movement Disorder Society-sponsored revision of the Unified Parkinson's Disease Rating Scale (MDS-UPDRS): scale presentation and clinimetric testing results. Mov Disord. 2008;23(15):2129-2170. PubMed
59.Balestrino R, Hurtado-Gonzalez CA, Stocchi F, et al. Applications of the European Parkinson's Disease Association sponsored Parkinson's Disease Composite Scale (PDCS). NPJ Parkinsons Dis. 2019;5:26. PubMed
60.Skorvanek M, Martinez-Martin P, Kovacs N, et al. Differences in MDS-UPDRS scores based on Hoehn and Yahr stage and disease duration. Mov Disord Clin Pract. 2017;4(4):536-544. PubMed
61.Martinez-Martin P, Rodriguez-Blazquez C, Alvarez-Sanchez M, et al. Expanded and independent validation of the Movement Disorder Society-Unified Parkinson's Disease Rating Scale (MDS-UPDRS). J Neurol. 2013;260(1):228-236. PubMed
62.Rodriguez-Blazquez C, Rojo-Abuin JM, Alvarez-Sanchez M, et al. The MDS-UPDRS Part II (motor experiences of daily living) resulted useful for assessment of disability in Parkinson's disease. Parkinsonism Relat Disord. 2013;19(10):889-893. PubMed
63.Skorvanek M, Martinez-Martin P, Kovacs N, et al. Relationship between the MDS-UPDRS and Quality of Life: a large multicenter study of 3206 patients. Parkinsonism Relat Disord. 2018;52:83-89. PubMed
64.Holden SK, Finseth T, Sillau SH, Berman BD. Progression of MDS-UPDRS sores over five years in de novo Parkinson disease from the Parkinson's Progression Markers Initiative cohort. Mov Disord Clin Pract. 2018;5(1):47-53. PubMed
65.Horváth K, Aschermann Z, Kovács M, et al. Minimal clinically important differences for the experiences of daily living parts of movement disorder society-sponsored unified Parkinson's disease rating scale. Mov Disord. 2017;32(5):789-793. PubMed
66.Horváth K, Aschermann Z, Ács P, et al. Minimal clinically important difference on the Motor Examination part of MDS-UPDRS. Parkinsonism Relat Disord. 2015;21(12):1421-1426. PubMed
67.Makkos A, Kovács M, Pintér D, Janszky J, Kovács N. Minimal clinically important difference for the historic parts of the Unified Dyskinesia Rating Scale. Parkinsonism Relat Disord. 2019;58:79-82. PubMed
68.EUnetHTA JA3WP6B2-2 Authoring Team. Process of information retrieval for systematic reviews and health technology assessments on clinical effectiveness. Methodological guidelines. Diemen (NL): EUnetHTA; 2019: https://www.eunethta.eu/wp-content/uploads/2020/01/EUnetHTA_Guideline_Information_Retrieval_v2-0.pdf. Accessed 2023 Feb 16.
69.Lefebvre C, Glanville J, Briscoe S, et al. Chapter 4: Searching for and selecting studies. In: Higgins JPT, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA, eds. Cochrane Handbook for Systematic Reviews of Interventions version 6.3 (updated February 2022). Cochrane, 2022. https://training.cochrane.org/handbook/current/chapter-04. Accessed 2023 Feb 8.
70.The efficacy of treatments for advanced Parkinson's disease: a network meta-analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Foslevodopa/Foscarbidopa solution, 240 mg/mL foslevodopa and 12 mg/mL foscarbidopa solution for subcutaneous infusion. St-Laurent (QC): Abbvie Corporation; 2022 Nov 22.
71.Dias S, Welton NJ, Sutton AJ, Ades AE. NICE DSU Technical Support Document 2: a generalised linear modelling framework for pairwise and network meta-analysis of randomised controlled trials. London (GB): National Institute for Health and Care Excellence (NICE); 2014: https://www.ncbi.nlm.nih.gov/pubmed/27466657. Accessed 2023 Feb 8.
72.Olanow CW, Kieburtz K, Odin P, et al. Continuous intrajejunal infusion of levodopa-carbidopa intestinal gel for patients with advanced Parkinson's disease: a randomised, controlled, double-blind, double-dummy study. Lancet Neurol. 2014;13(2):141-149. PubMed
73.Freire-Alvarez E, Kurca E, Lopez Manzanares L, et al. Levodopa-carbidopa intestinal gel reduces dyskinesia in Parkinson's disease in a randomized trial. Mov Disord. 2021;36(11):2615-2623. PubMed
74.AbbVie. NCT020549092: A study to examine the effect of levodopa-carbidopa intestinal gel (LCIG) therapy relative to that of optimized medical treatment (OMT) on non-motor symptoms (NMS) associated with advanced Parkinson's disease (PD). ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2022: https://clinicaltrials.gov/ct2/show/results/NCT02549092?view=results. Accessed 2023 Feb 07.
75.Clinical Study Report: M15-737. An open-label extension of study M15-741 to evaluate the safety and tolerability of 24-hour daily exposure of continuous subcutaneous infusion of ABBV-951 in subjects with Parkinson's disease [internal sponsor's report]. Chicago (IL): AbbVie; 2022 Jan 27.
76.Deep brain stimulation or Duodopa for advanced Parkinson disease in British Columbia. Evaluation of safety, effectiveness and cost-effectiveness of deep brain stimulation (DBS) or intestinal DUODOPA for treatment of advanced Parkinson disease in British Columbia and budget impact. Vancouver (BC): The Government of British Columbia; 2017: https://www2.gov.bc.ca/assets/gov/health/about-bc-s-health-care-system/heath-care-partners/health-authorities/bc-health-technology-assessments/deep-brain-stimulation.pdf. Accessed 2023 Feb 08.
Appendix 1: Detailed Outcomes Data
Note that this appendix has not been copy-edited.
Table 46: Sensitivity Analyses of Primary and Key Secondary PD Diary Outcomes in the M15-736 and M15-741 Trials (FAS)
Treatment arms | Jump-to-reference analytic approach | Last available value approach (ANCOVA) | Alternative definition of valid PD diary | |||||
|---|---|---|---|---|---|---|---|---|
LSM (SE) | Difference in LSM (SE) | P value | LSM (SE) | Difference in LSM (95% CI) | P value | Change from baseline, mean (SD) | P value | |
Average daily normalized “on” time without troublesome dyskinesia (hours) | ||||||||
M15-736 (at week 12) | ||||||||
FOS-FOS | 2.72 (0.52) | 1.14 (0.44) | 0.0093a | 2.33 (0.49) | 1.53 (0.36 to 2.71) | 0.0111a | NA | NA |
Oral LD-CD | 0.97 (0.50) | REF | REF | 0.79 (0.53) | REF | REF | NA | NA |
M15-741 (at week 52) | ||||||||
FOS-FOS | NA | NA | NA | NA | NA | NA | 3.58 (3.03) | ≤ 0.001a |
Average daily normalized “off” time (hours) | ||||||||
M15-736 (at week 12) | ||||||||
FOS-FOS | –2.75 (0.50) | –1.16 (0.42) | 0.0061a | –2.49 (0.47) | –1.53 (–2.65 to –0.41) | 0.0080a | NA | NA |
Oral LD-CD | –0.96 (0.49) | REF | REF | –0.96 (0.50) | REF | REF | NA | NA |
M15-741 (at week 52) | ||||||||
FOS-FOS | NA | NA | NA | NA | NA | NA | –3.39 (3.08) | ≤ 0.001a |
ANCOVA = analysis of covariance; CD = carbidopa; CI = confidence interval; FAS = full analysis set; FOS-FOS = foslevodopa-foscarbidopa; LD = levodopa; LSM = least squares mean; NA = not applicable; PD = Parkinson disease; REF = reference; SD = standard deviation; SE = standard error.
aNot adjusted for multiplicity.
Sources: M15-736 Clinical Study Report,14 M15-741 Clinical Study Report.37
Figure 4: Subgroup Analyses of Average Normalized “On” Time Without Troublesome Dyskinesia in the M15-736 Trial (FAS)
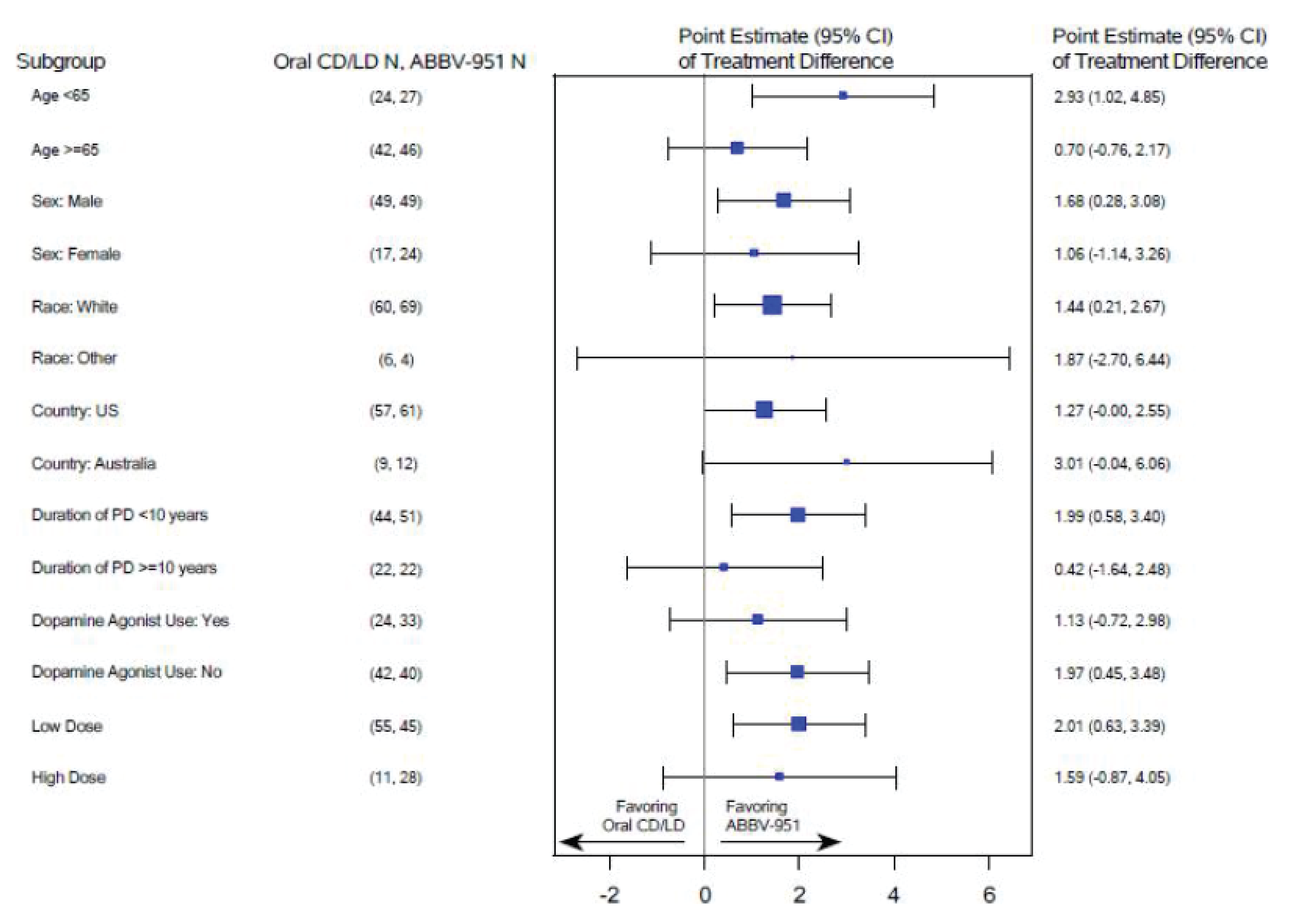
ABBV-951 = foslevodopa-foscarbidopa; CD = levodopa; CI = confidence interval; FAS = full analysis set; LD = levodopa; PD = Parkinson disease.
Source: M15-736 Clinical Study Report.14
Figure 5: Subgroup Analyses of Average Normalized “Off” Time in the M15-736 Trial (FAS)
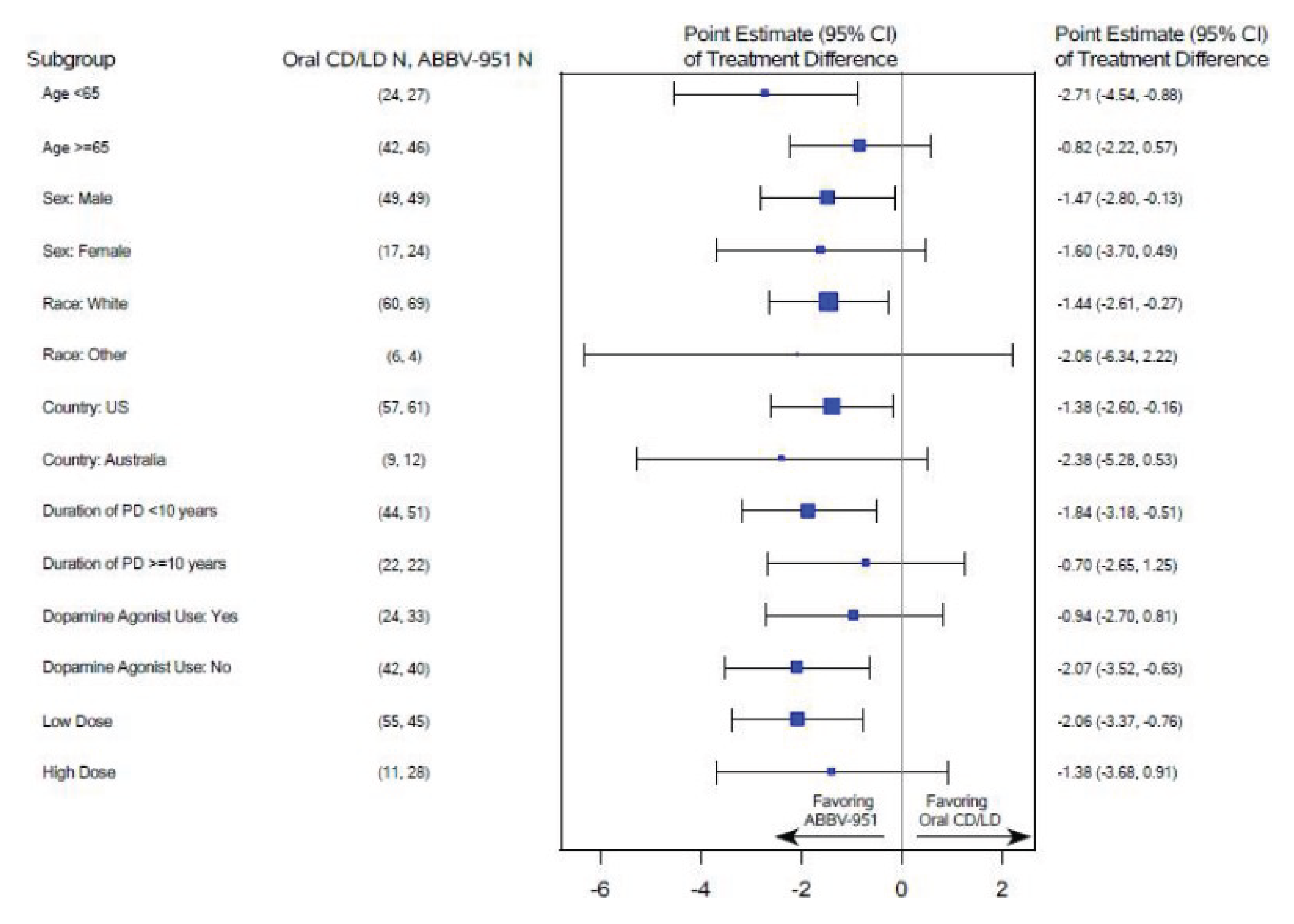
ABBV-951 = foslevodopa-foscarbidopa; CD = carbidopa; CI = confidence interval; FAS = full analysis set; LD = levodopa; PD = Parkinson disease.
Source: M15-736 Clinical Study Report.14
Table 47: Change From Baseline in Average Daily Absolute Time Spent in Motor States in the M15-736 and M15-741 Trials (FAS)
Treatment arms | Number of patients contributing to the analysis, n | Baseline (hours), mean (SD) | Change from baseline, LSM (SE) | Difference in LSM (95% CI) | P value |
|---|---|---|---|---|---|
Average daily absolute “on” time without troublesome dyskinesiaa | |||||
M15-736 (at week 12) | |||||
FOS-FOS | 47 | 9.63 (2.83) | 2.84 (0.54) | 2.03 (0.68 to 3.38) | 0.0035b |
Oral LD-CD | 62 | 9.69 (2.92) | 0.81 (0.53) | REF | REF |
M15-741 (at week 52) | |||||
FOS-FOS | 96 | 9.81 | 3.60 (3.45) | NA | ≤ 0.001b |
Average daily absolute “off” timea | |||||
M15-736 (at week 12) | |||||
FOS-FOS | 47 | 6.47 (2.11) | –2.80 (0.50) | –1.69 (–2.93 to –0.45) | 0.0078b |
Oral LD-CD | 62 | 6.05 (2.05) | –1.11 (0.48) | REF | REF |
M15-741 (at week 52) | |||||
FOS-FOS | 96 | 6.09 | –3.58 (3.11) | NA | ≤ 0.001b |
CD = carbidopa; CI = confidence interval; FAS = full analysis set; FOS-FOS = foslevodopa-foscarbidopa; LD = levodopa; LSM = least squares mean; NA = not applicable; REF = reference; SD = standard deviation; SE = standard error.
aThe analysis was conducted using a mixed model for repeated measures, adjusted for categorical fixed effects of treatment, country and visit, treatment-by visit and treatment-by-baseline interactions, and baseline measurement (continuous).
bNot adjusted for multiplicity.
Source: M15-736 Clinical Study Report,14 M15-741 Clinical Study Report.37
Table 48: Percent Change From Baseline in Average Daily Normalized Time Spent in Motor States in the M15-736 Trial (FAS)
Treatment arms | Number of patients contributing to the analysis, n | Baseline (hours), mean (SD) | % Change from baseline, LSM (SE) | Difference in LSM (95% CI) | P value |
|---|---|---|---|---|---|
Average daily absolute “on” time without troublesome dyskinesiaa | |||||
M15-736 (at week 12) | |||||
FOS-FOS | 46 | 9.20 (2.42) | 29.85 (6.83) | 18.92 (1.53 to 36.31) | 0.0332b |
Oral LD-CD | 61 | 9.49 (2.62) | 10.93 (6.73) | REF | REF |
Average daily absolute “off” timea | |||||
M15-736 (at week 12) | |||||
FOS-FOS | 47 | 6.34 (2.27) | –43.85 (8.53) | –28.63 (–49.68 to –7.57) | 0.0081b |
Oral LD-CD | 62 | 5.91 (1.88) | –15.22 (8.29) | REF | REF |
CD = carbidopa; CI = confidence interval; FAS = full analysis set; FOS-FOS = foslevodopa-foscarbidopa; LD = levodopa; LSM = least squares mean; REF = reference; SD = standard deviation; SE = standard error.
aThe analysis was conducted using a mixed model for repeated measures, adjusted for categorical fixed effects of treatment, country and visit, treatment-by visit and treatment-by-baseline interactions, and baseline measurement (continuous).
bNot adjusted for multiplicity.
Source: M15-736 Clinical Study Report.14
Table 49: Other Outcomes in the M15-736 and M15-741 Trials (FAS)
Treatment arms | Number of patients contributing to the analysis, n (%) | Baseline value, mean (SD) | Change from baseline, LSM (SE)a or mean (SD)b | Difference in LSMa or mean (95% CI)b | P value |
|---|---|---|---|---|---|
Average daily normalized “on” time without dyskinesia (hours)c | |||||
M15-736 (at week 12) | |||||
FOS-FOS | 47 (63.5) | 7.23 (3.14) | 3.13 (0.54) | 1.81 (0.46 to 3.16) | 0.0091d |
Oral LD-CD | 62 (92.5) | 7.47 (3.72) | 1.32 (0.53) | REF | REF |
M15-741 (at week 52) | |||||
FOS-FOS | || |||||| | |||| |||| | |||| |||||| | || | | |||||| |
Median bradykinesia score (BK50) assessed using the PKG wearable devicec | |||||
M15-736 (at week 12) | |||||
FOS-FOS | 34 (45.9) | 25.97 (7.65) | 1.38 (0.56) | 1.72 (0.30 to 3.15) | 0.018d |
Oral LD-CD | 49 (73.1) | 26.63 (7.68) | –0.34 (0.52) | REF | REF |
M15-741 (at week 52) | |||||
FOS-FOS | || | |||| |||| | ||| |||||| | || | ||||| |
IQR of bradykinesia score (BK75 to 25) assessed using the PKG wearable devicec | |||||
M15-736 (at week 12) | |||||
FOS-FOS | 34 (45.9) | 17.13 (4.18) | 0.31 (0.54) | 0.18 (–1.20 to 1.55) | 0.80d |
Oral LD-CD | 49 (73.1) | 17.80 (4.26) | 0.13 (0.49) | REF | REF |
M15-741 (at week 52) | |||||
FOS-FOS | || | |||| |||| | |||| |||||| | || | ||||| |
Median dyskinesia score (DK50) assessed using the PKG wearable devicec | |||||
M15-736 (at week 12) | |||||
FOS-FOS | 34 (45.9) | 4.46 (10.14) | –1.71 (1.41) | –2.73 (–6.61 to 1.15) | 0.17d |
Oral LD-CD | 49 (73.1) | 3.70 (5.45) | 1.02 (1.38) | REF | REF |
M15-741 (at week 52) | |||||
FOS-FOS | || | ||| |||| | ||| |||||| | || | ||||| |
IQR of dyskinesia score (DK75 to 25) assessed using the PKG wearable devicec | |||||
M15-736 (at week 12) | |||||
FOS-FOS | 34 (45.9) | 12.31 (20.36) | –2.77 (2.64) | –5.49 (–12.71 to 1.73) | 0.13d |
Oral LD-CD | 49 (73.1) | 11.31 (16.61) | 2.72 (2.59) | REF | REF |
M15-741 (at week 52) | |||||
FOS-FOS | || | |||| |||| | ||| ||||||| | || | ||||| |
EQ-5D-5L summary indexf | |||||
M15-736g (at week 12) | |||||
FOS-FOS | 44 (59.5) | 0.75 (0.13) | 0.051 (0.022) | 0.049 (–0.001 to 0.100) | 0.057d |
Oral LD-CD | 59 (88.1) | 0.75 (0.13) | 0.002 (0.021) | REF | REF |
M15-741 (at week 52) | |||||
FOS-FOS | ||| |||||| | |||| |||| | ||||| ||||||| | || | | |||||| |
BK = bradykinesia score; CD = carbidopa; CI = confidence interval; DK = dyskinesia; EQ-5D-5L = 5-Level EQ-5D; FAS = full analysis set; FOS-FOS = foslevodopa-foscarbidopa; IQR = interquartile range; LD = levodopa; LSM = least squares mean; PKG = Parkinson KinetiGraph/Personal KinetiGraph; REF = reference; SD = standard deviation; SE = standard error.
aApplicable to the M15-736 trial.
bApplicable to the M15-741 trial.
cThe analysis was conducted using a mixed model for repeated measures, adjusted for categorical fixed effects of treatment, country and visit, treatment-by visit and treatment-by-baseline interactions, and baseline measurement (continuous).
dAlthough the nominal P value is ≤ 0.05, statistical significance cannot be claimed because the results for the second key secondary end point (Movement Disorder Society-Unified Parkinson Disease Rating Scale Part II), a prior end point in the testing hierarchy, were not statistically significant.
eNot adjusted for multiplicity.
fThis analysis was conducted using an analysis for covariate (ANCOVA) model, adjusted for categorical fixed effects of treatment and country, and baseline score (continuous).
gBased on data from the cohort in the US.
Source: M15-736 Clinical Study Report,14 M15-741 Clinical Study Report.37
Table 50: Presence of Morning Akinesia in the M15-736 Trial (FAS)
Outcomes | M15-736 | |
|---|---|---|
Foslevodopa-foscarbidopa arm (N = 74) | Oral LD-CD arm (N = 67) | |
Presence of morning akinesiaa | ||
Baseline, n/N (%) | 56/71 (78.9) | 51/67 (76.1) |
Week 12, n/N (%) | 8/47 (17.0) | 38/60 (63.3) |
Odds ratio at week 12, LSM (95% CI) | 0.12 (0.04 to 0.31) | |
P value | ≤ 0.001b | |
CD = carbidopa; CI = confidence interval; FAS = full analysis set; LD = levodopa; LSM = least squares mean.
aMorning akinesia was defined as reporting “off” status as the first morning symptom upon awakening assessed using the Parkinson disease diary. The analysis was conducted using the generalized linear mixed model, adjusted for categorical fixed effects of treatment, country, visit, treatment-by-visit interaction, and baseline first morning status upon awakening.
bAlthough the nominal P value is ≤ 0.05, statistical significance cannot be claimed because the results for the second key secondary end point (Movement Disorder Society-Unified Parkinson Disease Rating Scale Part II), a prior end point in the testing hierarchy, were not statistically significant.
Source: M15-736 Clinical Study Report.14
Pharmacoeconomic Review
Abbreviations
BIA
budget impact analysis
CD
carbidopa
CMA
cost-minimization analysis
DBS
deep brain stimulation
LCIG
levodopa-carbidopa intestinal gel
LD
levodopa
NMA
network meta-analysis
PD
Parkinson disease
PEG-J
percutaneous endoscopic gastrostomy-jejunostomy
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Foslevodopa-foscarbidopa (Vyalev), subcutaneous infusion |
Submitted price | Foslevodopa-foscarbidopa, 2,400 mg foslevodopa and 120 mg foscarbidopa, solution for subcutaneous infusion: $169.81 per single-use vial |
Indication | For the treatment of motor fluctuations in patients with advanced levodopa-responsive PD who do not have satisfactory control of severe, debilitating motor fluctuations and hyper-/dyskinesia despite optimized treatment with available combinations of medicinal products for PD |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | May 3, 2023 |
Reimbursement request | For the treatment of motor fluctuations in patients with PD that are not adequately controlled on optimized oral therapies (advanced PD) and who are not candidates for deep brain stimulation |
Sponsor | AbbVie Corporation |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance; PD = Parkinson disease.
Table 2: Summary of Economic Information
Component | Description |
|---|---|
Type of economic evaluation | CMA |
Target population | Adult patients with PD who are not adequately controlled on optimized oral therapies (advanced PD) and who are not candidates for DBS |
Treatment | Foslevodopa-foscarbidopa with adjunctive therapy |
Comparator | LCIG with adjunctive therapy |
Perspective | Canadian publicly funded health care payer |
Time horizon | Lifetime (20 years) |
Key data source | A sponsor-commissioned indirect treatment comparison using Bayesian NMA was conducted to compare the relative clinical efficacy between foslevodopa-foscarbidopa and LCIG |
Costs considered | Drug acquisition costs, administration costs, and surgical costs |
Submitted results | Foslevodopa-foscarbidopa is less costly than LCIG after 1 year of treatment, with cost savings of $2,647. The cost savings remained consistent throughout the time horizon. Cost savings of foslevodopa-foscarbidopa were driven by the lack of surgery required for treatment initiation. |
Key limitations |
|
CADTH reanalysis results |
|
CD = carbidopa; CMA = cost-minimization analysis; DBS = deep brain stimulation; LCIG = levodopa-carbidopa intestinal gel; LD = levodopa; NMA = network meta-analysis; PD = Parkinson disease.
Conclusions
The CADTH clinical review concluded that foslevodopa-foscarbidopa demonstrated a clinically meaningful improvement in “on” time without troublesome dyskinesia and “off” time compared with oral levodopa (LD) and carbidopa (CD) therapy in a 12-week randomized treatment period in patients with advanced Parkinson disease (PD). In the absence of direct comparative evidence, the CADTH clinical review team concluded that the results of the sponsor’s submitted network meta-analysis (NMA) were inconclusive due to a sparse network, absence of closed loop, and heterogeneity in study designs and patient populations ||| ||||||||| |||| |||||||||||||||||||||||| ||| |||||||||| |||| || ||||||||||| || |||| |||| ||||||| ||||||||||| ||||||||||| ||||| ||||| ||| ||||| |||||||| |||||||| |||| |||| ||| || ||||||||||| || ||||| |||||||| |||||||| |||| ||||.
CADTH reanalyzed the sponsor’s base case to include updated costing information on the surgical events associated with LCIG treatment. Including drug treatment and administration and surgical event costs, CADTH results were consistent with those of the sponsor. Foslevodopa-foscarbidopa remained less costly than LCIG under the Canadian health care system perspective in the first year because no surgery is required to initiate foslevodopa-foscarbidopa treatment for adult patients with PD who are not adequately controlled on optimized oral therapies and who are not candidates for deep brain stimulation (DBS) (incremental cost difference = –$2,453.03). As LCIG and foslevodopa-foscarbidopa are priced the same, there was no difference in incremental drug acquisition costs. However, the lack of difference in estimated drug acquisition cost between foslevodopa-foscarbidopa and LCIG is based on publicly available list prices and may not reflect actual prices paid by Canadian public drug plans. All cost savings were derived based on the assumption that no surgical events are associated with foslevodopa-foscarbidopa, in comparison with LCIG. As drug acquisition costs are the same and the inclusion of administration and surgical costs resulted in cost savings, no price reduction was completed for this review.
CADTH could not address the limitations to do with uncertainty about the comparative efficacy. If foslevodopa-foscarbidopa confers differential safety or improved efficacy compared to LCIG, a cost-minimization analysis (CMA) would be insufficient to assess the cost-effectiveness of foslevodopa-foscarbidopa and the true cost-effectiveness of foslevodopa-foscarbidopa compared to LCIG would remain unknown. Under the sponsor’s reimbursement request, clinical expert feedback received by CADTH noted that most patients would remain on an oral therapy despite inadequate control of motor symptoms. Furthermore, clinical expert feedback noted potential implementation challenges with the sponsor’s reimbursement request. DBS may be an appropriate comparator for a subset of patients in certain jurisdictions. The cost-effectiveness of foslevodopa-foscarbidopa compared to DBS and advanced PD oral therapies is unknown.
Economic Review
The current review is for foslevodopa-foscarbidopa (Vyalev) for treatment of motor fluctuations in patients with PD who are not adequately controlled on optimized oral therapies (advanced PD) and who are not candidates for DBS.
Economic Information
Summary of Sponsor’s Economic Information
The sponsor submitted a CMA for foslevodopa-foscarbidopa compared with a device-aided therapy that is a duodenal infusion of LCIG, also known as Duodopa, for the treatment of motor fluctuations in patients with advanced PD who are not candidates for DBS.1 In response to an additional information request by CADTH regarding the definition of “not candidates for DBS,” the sponsor noted that it could be characterized based on reasons such as patients not consenting due to surgical risk, wait times, or hesitance toward a neurosurgical procedure; patients aged more than 70 years; patients having moderate to severe depression, cognitive decline, or other medical comorbidities that increase surgical risk; their physician having ethical concerns about the appropriateness of DBS for the patient; their physician considering that the risks of procedure outweigh the benefit for the patient; or their physician determining that the weight time for access to DBS is too long given the patient’s status. This modelled population deviates from the Health Canada indication, in that patients who are candidates for DBS are excluded, but represents the sponsor’s reimbursement request.1
In the absence of head-to-head evidence comparing foslevodopa-foscarbidopa to LCIG, the sponsor submitted an indirect treatment comparison using Bayesian NMA comparing the relative efficacy.2 The trials included in the NMA were phase III randomized controlled trials: M15-736, Olanow et al. (2014), DYSCOVER, and INSIGHTS.3-5 ||||| ||| |||||||| |||||||| |||||||||| || ||| ||||||||| |||| || |||||||||| ||||||| ||||||||| |||||| ||||||| ||||||||||| |||||||||||| ||| |||||2 The sponsor assumed that the efficacy of foslevodopa-foscarbidopa and LCIG was equivalent. The sponsor’s base case only considered drug acquisition costs, administration costs, surgical costs, and adjunctive therapy costs as all other costs were assumed to be equivalent. The analysis was conducted from the perspective of the publicly funded health payer over a time horizon of 20 years and discounting (1.5% per annum) was applied to costs.1
Foslevodopa-foscarbidopa is available in single-dose glass vials, with 10 mL of solution containing 240 mg/mL of foslevodopa and 12 mg/mL of foscarbidopa.6 The submitted price is $169.81 per 10 mL vial. Individual dosing and infusion rate of foslevodopa-foscarbidopa are calculated during the titration period and are informed by the patient’s current use of levodopa and other PD medications.6 Foslevodopa-foscarbidopa is administered as a continuous subcutaneous infusion for 24 hours per day,6 and it is assumed that patients will use approximately 1 vial per day, for a total of 365.25 administrations per year.1 A pump and the ancillaries required to administer foslevodopa-foscarbidopa will be provided by the sponsor at no charge.1
The cost of LCIG was obtained from the Ontario Exceptional Access Program (EAP) at $169.81 per 100 mL cassette. It was assumed that patients would also use approximately 1 cassette per day, for a total of 365.25 administrations per year.
Adjunctive therapy unit costs were informed by Ontario Drug Benefit Formulary.7,8 It was assumed that adjunctive therapy use was the same for foslevodopa-foscarbidopa and LCIG, as informed by the Adelphi Real-World study and clinical input.1,9,10 Administration costs included titration and monitoring, with cost and frequency informed by the Ontario Schedule of Benefits and key opinion leader feedback, respectively.11 Annual administration (i.e., titration and monitoring) costs totalled $714.40 and were included in both treatment arms. Surgical costs, however, were included only in the LCIG analysis and totalled $2,647.30.1 The surgical costs were derived by sourcing the nasogastric and percutaneous endoscopic gastrostomy-jejunostomy (PEG-J) tube insertion inpatient costs from Ontario Case Costing and sourcing frequency from KOL feedback.12
A summary of the sponsor’s economic evaluation is shown in Table 3. The sponsor’s submitted base case estimated that, in adult patients with PD who are not adequately controlled on optimized oral therapies (advanced PD) and who are not candidates for DBS, foslevodopa-foscarbidopa and LCIG were associated with cumulative costs of $63,317.67 and $65,964.97, respectively, in the first year. The predicted cost savings in the first year was $2,647.30, which stems from the expected total cost associated with surgery for LCIG treatment initiation. The sponsor’s base-case analysis results indicated that, after the first year, foslevodopa-foscarbidopa would cost the same as LCIG because surgical nasogastric and PEG-J tube insertion is only required to initiate LCIG treatment. This cost savings therefore remained consistent throughout the 20-year time horizon as foslevodopa-foscarbidopa and LCIG are expected to have equivalent drug costs and the upfront administration and surgical costs are only incurred in the first year.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total drug costs ($) | Incremental drug costs ($) | Total costs ($) | Incremental costs ($) |
|---|---|---|---|---|
Foslevodopa-foscarbidopa | 63,317.67 | Reference | 63,317.67 | Reference |
LCIG (Duodopa) | 63,317.67 | 0.00 | 65,964.97 | –2,647.30 |
LCIG = levodopa-carbidopa intestinal gel.
Note: Negative costs reflect savings for foslevodopa-foscarbidopa.
Source: Sponsor’s economic submission.1
No sensitivity or scenario analyses were conducted by the sponsor.
CADTH Appraisal of the Sponsor’s Economic Information
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
Exclusion of patients who are candidates for DBS is not appropriate: The sponsor’s economic evaluation was specifically conducted in patients with PD who are not adequately controlled on optimized oral therapy (advanced PD) and are not candidates for DBS, which is a subset of the Health Canada indication. The sponsor submitted a request for deviation to focus their analysis on this subpopulation. During the sponsor’s comment period, the sponsor noted that patients who are candidates for DBS refers to patients who are eligible for DBS, able to access DBS, and likely to respond to DBS, and those who have given informed consent to the procedure. In alignment with the CADTH clinical report and clinical expert feedback received by CADTH, DBS is a relevant comparator given that DBS and foslevodopa-foscarbidopa are expected to have the same place in therapy for the treatment of advanced PD. This approach is consistent with the CADTH reimbursement reviews for Duodopa conducted in 2008 and 2018.13,14 Clinical expert feedback obtained by CADTH highlighted that the expected place in therapy for foslevodopa-foscarbidopa would be more closely aligned with its Health Canada indication and noted that patients who are candidates for DBS should not be excluded from receiving foslevodopa-foscarbidopa as DBS candidacy should not impact their eligibility for foslevodopa-foscarbidopa.
CADTH was unable to address this limitation given the lack of data on comparative efficacy of DBS and foslevodopa-foscarbidopa. The cost-effectiveness of foslevodopa-foscarbidopa compared to DBS is unknown.
Exclusion of oral therapies as a comparator was inappropriate: Clinical expert feedback received for this review noted that although some patients with advanced PD may receive advanced therapies such as DBS or LCIG, most patients would remain on oral therapy despite inadequate control of motor symptoms. Therefore, exclusion of LD-CD as a relevant comparator was not appropriate. The CADTH clinical review concluded that foslevodopa-foscarbidopa demonstrated a clinically meaningful improvement in “on” time without troublesome dyskinesia and “off” time compared with oral LD-CD therapy in a 12-week randomized treatment period in patients with advanced PD; however, the cost-effectiveness of foslevodopa-foscarbidopa compared to LD-CD is unknown.
CADTH was unable to address this limitation given the submitted model structure. A CMA is insufficient to assess the cost-effectiveness of foslevodopa-foscarbidopa compared to LD-CD due to expected differences in efficacy and safety.
The assumption of comparative efficacy of foslevodopa-foscarbidopa to other advanced PD treatments is uncertain: In the absence of a direct head-to-head comparison between foslevodopa-foscarbidopa and LCIG, the sponsor submitted an indirect treatment comparison to indirectly compare relative effects. ||||| || |||||||| |||| ||||| |||| the sponsor submitted a CMA based on the assumption of equivalence in efficacy and safety between foslevodopa-foscarbidopa and LCIG. The CADTH clinical review noted that the NMA findings are limited due to a sparse network, absence of closed loop, which rendered a consistency assessment infeasible, as well as heterogeneity in study designs (i.e., blinding, dosing protocol for oral therapies, duration of follow-up), patient populations (i.e., presence of concurrent cognitive impairment and dyskinesia), and patient baseline characteristics (i.e., duration of PD diagnosis, “off” time) that were unaccounted for. As such, the indirect evidence between foslevodopa-foscarbidopa and LCIG should be interpreted with caution.
CADTH was unable to address this limitation.
Missing administration costs and inaccurate surgical costs: The sponsor-submitted CMA highlighted titration and monitoring as the only administrative costs associated with both foslevodopa-foscarbidopa and LCIG. Clinical expert feedback obtained by CADTH highlighted that LCIG would also include additional administration costs beyond those considered by the sponsor. LCIG is expected to require additional resources such as a gastroenterologist consult, an ambulatory care visit for the gastroscopy procedure as well as associated personnel costs. The sponsor-submitted CMA also included inaccurate costing information for the nasogastric tube and PEG-J tube insertion procedures for LCIG. Additional information was requested by CADTH for clarification on the Ontario Case Costing Initiative codes and costs used to inform LCIG surgical costs. From the additional information provided by the sponsor, CADTH noted that included costs were not appropriately inflated.
CADTH was unable to address the limitation on the additional missing administration costs noted above because of the model structure; however, inclusion of the missing administration costs would bias the results in favour of foslevodopa-foscarbidopa as they would increase the cost of LCIG. Surgical costs were recalculated using available Ontario Case Costing Initiative codes and the Ontario Association of Gastroenterology fee guide, all inflated to 2022 costs. Updated costs are reported in the CADTH base case (refer to the CADTH Reanalyses of the Economic Information section).
CADTH Reanalyses of the Economic Information
The CADTH base case included 1 correction to the sponsor’s base case, namely the surgical costs associated with LCIG administration (refer to Table 4). In this reanalysis, foslevodopa-foscarbidopa was associated with cost savings of $2,453.03 in year 1 and these cost savings remained consistent throughout the 20-year time horizon as the cost difference in subsequent years was expected to be $0. As foslevodopa-foscarbidopa and LCIG have equivalent unit prices, with the cost savings driven by the lack of surgery associated with foslevodopa-foscarbidopa, a price reduction analysis was not completed.
Table 4: Summary of the CADTH Reanalysis Results
Drug | Annual drug costs ($) | Incremental drug costs ($) | Year 1 total costs ($) | Incremental costs ($) |
|---|---|---|---|---|
Foslevodopa-foscarbidopa | 63,317.67 | Reference | 64,032.07 | Reference |
LCIG (Duodopa) | 63,317.67 | 0 | 66,485.10 | –2,453.03 |
LCIG = levodopa-carbidopa intestinal gel.
Note: Reanalyses are based on publicly available prices of the comparator treatments. Negative incremental costs represent cost savings.
Issues for Consideration
Comparator pricing based on publicly available prices: The price of LCIG and adjunctive therapies is based on publicly accessible list prices and does not reflect any confidential pricing that may have been negotiated by public plans. The estimated cost savings associated with foslevodopa-foscarbidopa are likely less than any estimated if discounts have been negotiated for LCIG in confidence.
Administration of treatment is heterogenous: The sponsor assumed, for both foslevodopa-foscarbidopa and LCIG, that all patients will use approximately a single vial or cassette per day.1 Clinical expert feedback obtained by CADTH highlighted that a single vial or cassette per day may be sufficient for more than 90% of the instances in which the treatments are required; however, up to 10% of patients may require more than 1 vial or cassette per day. Clinical expert feedback received for this review highlighted that the percentage of patients that would require a higher dosage would be the same for both LCIG and foslevodopa-foscarbidopa.
Availability of pump and ancillaries: As foslevodopa-foscarbidopa is administered subcutaneously, a pump and ancillaries are required. In their submission, the sponsor highlighted that these items will be supplied at no charge;1 however, they gave no additional information on how these would be made available.
Candidacy for DBS treatment: The reimbursement request for foslevodopa-foscarbidopa deviates from the Health Canada indication by excluding patients that are candidates for DBS. Clinical expert feedback and drug plan input obtained by CADTH note that there are issues in implementing this initiation criterion since this criterion could be interpreted by jurisdictions as requiring a neurosurgical consultation for DBS candidacy before foslevodopa-foscarbidopa would be reimbursed, potentially leading to ethical and accessibility issues.
Conclusions
The CADTH clinical review concluded that foslevodopa-foscarbidopa demonstrated a clinically meaningful improvement in “on” time without troublesome dyskinesia and “off” time compared with oral LD-CD therapy in a 12-week randomized treatment period in patients with advanced PD. In the absence of direct comparative evidence, the CADTH clinical review team concluded that the results of the sponsor’s submitted NMA were inconclusive because of a sparse network, absence of closed loop, and heterogeneity in study designs and patient populations ||| ||||||||| |||| |||||||||||||||||||||||| ||| |||||||||| |||| || ||||||||||| || |||| |||| ||||||| ||||||||||| ||||||||||| ||||| ||||| ||| ||||| |||||||| |||||||| |||| |||| ||| || ||||||||||| || ||||| |||||||| |||||||| |||| |||||
CADTH reanalyzed the sponsor’s base case to include updated costing of the surgical events necessary for LCIG treatment initiation. Including drug treatment and administration and surgical event costs, results were consistent with those of the sponsor. Foslevodopa-foscarbidopa remained less costly than LCIG in the first year when including total costs under the Canadian health care system perspective because of the lack of surgery required to initiate foslevodopa-foscarbidopa treatment for adult patients with PD who are not adequately controlled on optimized oral therapies (advanced PD) and who are not candidates for DBS (incremental cost difference = –$2,453.03). As LCIG and foslevodopa-foscarbidopa are priced the same, there was no difference in incremental drug acquisition costs. However, the lack of difference between foslevodopa-foscarbidopa and LCIG in estimated drug acquisition cost is based on publicly available list prices and may not reflect actual prices paid by Canadian public drug plans. All cost savings were derived based on the assumption that no surgical events are associated with foslevodopa-foscarbidopa treatment compared with LCIG treatment. As drug acquisition costs are the same and the inclusion of administration and surgical costs resulted in cost savings, no price reduction was completed for this review.
Limitations related to uncertainty to do with comparative efficacy could not be addressed by CADTH. If foslevodopa-foscarbidopa confers differential safety or improved efficacy compared to LCIG, a CMA is insufficient to assess the cost-effectiveness of foslevodopa-foscarbidopa and the true cost-effectiveness of foslevodopa-foscarbidopa compared to LCIG is unknown. Under the sponsor’s reimbursement request, clinical expert feedback received by CADTH noted that most patients would remain on an oral therapy despite inadequate control of motor symptoms. Furthermore, clinical expert feedback noted potential implementation challenges with the sponsor’s reimbursement request. DBS may be an appropriate comparator for a subset of patients in certain jurisdictions. The cost-effectiveness of foslevodopa-foscarbidopa relative to DBS and advanced PD oral therapies is unknown.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Trikafta, elexacaftor 100 mg/tezacaftor 50 mg/ivacaftor 75 mg tablets and ivacaftor 150 mg tablets; elexacaftor 50 mg/tezacaftor 25 mg/ivacaftor 37.5 mg tablets and ivacaftor 75 mg tablets oral. Toronto (ON): Vertex Pharmaceuticals (Canada) Incorporated; 2023 May 10.
2.Cholon D, Aleksandrov L, Quinney N, Boyles S, Aleksandrov A, Gentzsch M. Potentiator icenticaftor is superior to ivacaftor in rescuing F508del cystic fibrosis transmembrane conductance regulator and does not destabilize corrected mature protein. Journal of Cystic Fibrosis. 2022;21(Supplement 2):S355.
3.Freire‐Alvarez E, Kurča E, Lopez Manzanares L, et al. Levodopa‐carbidopa intestinal gel reduces dyskinesia in Parkinson's disease in a randomized trial. Mov Disord. 2021;36(11):2615-2623. PubMed
4.Olanow CW, Kieburtz K, Odin P, et al. Continuous intrajejunal infusion of levodopa-carbidopa intestinal gel for patients with advanced Parkinson's disease: a randomised, controlled, double-blind, double-dummy study. Lancet Neurol. 2014;13(2):141-149. PubMed
5.AbbVie. NCT02549092: A study to examine the effect of levodopa-carbidopa intestinal gel (LCIG) therapy relative to that of optimized medical treatment (OMT) on non-motor symptoms (NMS) associated with advanced parkinson's disease (PD). ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2022 Nov 22: https://clinicaltrials.gov/ct2/show/NCT02549092. Accessed 2022 Dec 12.
6.Vyalev (foslevodopa/foscarbidopa): 240 mg/mL foslevodopa and 12 mg/mL foscarbidopa solution, subcutaneous infusion [product monograph]. St-Laurent (QC): AbbVie Corporation; 2022 Apr 29.
7.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2023; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2022 Dec 12.
8.Aldred J, Amelin A, Antonini A, et al. Continuous subcutaneous foslevodopa/qfoscarbidopa in advanced Parkinson's disease: results from a 12-month phase 3 study. Eur J Neurol. 2022;29 (Suppl 1):178.
9.Pigliasco F, Cafaro A, Barco S, et al. Therapeutic drug monitoring of cystic fibrosis modulators Ivacaftor, Tezacaftor and Elexacaftor: development and validation of a new LC-MS/MS method and its application to CF patients treated with Kaftrio. Biochim Clin. 2022;46(3):S45-S46.
10.Adelphi Real World PD DSP report 2017-2019 [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: foslevodopa/foscarbidopa (ABBV-951), 10 mL (containing 240 mg/mL foslevodopa and 12 mg/mL foscarbidopa) subcutaneous infusion. Pointe-Claire (QC): AbbVie Corporation; 2022 Nov 22.
11.Schedule of benefits for physician services under the Health Insurance Act: (January 25, 2022 (effective July 1, 2022)). Toronto (ON): Ontario Ministry of Health; 2022: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf. Accessed 2022 Jun 27.
12.Ontario Case Costing Initiative (OCCI). 2017; https://data.ontario.ca/dataset/ontario-case-costing-initiative-occi. Accessed 2022 Dec 12.
13.CADTH Common Drug Review clinical review report: levodopa/carbidopa (Duodopa) for the treatment of patients with advanced levodopa-responsive Parkinson's disease. Ottawa (ON): CADTH; 2018 Sep: https://www.cadth.ca/sites/default/files/cdr/clinical/SR0557_DuodopaFWG_CL_Report.pdf. Accessed 2023 Mar 21.
14.Common Drug Review clinical review report: Levodopa/carbidopa (Duodopa) for treatment of advanced levodopa-responsive Parkinson’s disease in which satisfactory control of severe, disabling motor fluctuations and hyper-/dyskinesia cannot be achieved with available combinations of Parkinson medicinal products. Ottawa (ON): CADTH; 2009 May 20.
15.Exceptional Access Program (EAP). Toronto (ON): Ontario Ministry of Health; Ontario Ministry of Long-Term Care; 2022: https://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx. Accessed 2022 Dec 12.
16.CADTH Common Drug Review pharmacoeconomic review report: apomorphine (Movapo) for the treatment of the acute, intermittent treatment of hypomobility “off” episodes (“end-of-dose wearing off” and unpredictable “on/off” episodes) in patients with advanced Parkinson’s disease. Ottawa (ON): CADTH; 2018 Feb: https://cadth.ca/sites/default/files/cdr/pharmacoeconomic/SR0527_Movapo_PE_Report.pdf. Accessed 2022 Dec 12.
17.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Drug name, doses and administration. City (PROV): Sponsor name; 1800 Jan 1.
18.Statistics Canada. Population estimates, quartely. 2022; https://www150.statcan.gc.ca/t1/tbl1/en/cv.action?pid=1710000901. Accessed 2022 Aug 12.
19.Government of Canada. Non-insured health benefits program: First Nations and Inuit Health Branch: annual report 2018 to 2019. 2020; https://www.sac-isc.gc.ca/eng/1584392581890/1584393350542. Accessed 2022 Dec 12.
20.Wong SL, Gilmour H, Ramage-Morin PL. Parkinson's disease prevalence, diagnosis and impact. Ottawa (ON): Statistics Canada; 2014: https://www150.statcan.gc.ca/n1/pub/82-003-x/2014011/article/14112-eng.htm. Accessed 2022 Dec 12.
21.Hirsch L, Jette N, Frolkis A, Steeves T, Pringsheim T. The incidence of Parkinson's disease: a systematic review and meta-analysis. Neuroepidemiology. 2016;46(4):292-300. PubMed
Appendix 1: Additional Economic Information
Note that this appendix has not been copy-edited.
Cost Comparison Table
The comparators presented in Table 5 have been deemed to be appropriate based on feedback from clinical expert and drug plan. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in Table 5 and as such, the table may not represent the actual costs to public drug plans.
Table 5: CADTH Cost Comparison Table for the Treatment of Adults With Advanced PD
Treatment | Strength | Form | Price ($) | Recommended dosage | Daily cost ($) | Annual cost ($) |
|---|---|---|---|---|---|---|
Foslevodopa-foscarbidopa (Vyalev) | 240 mg foslevodopa / 12 mg foscarbidopa | Vial (10 mL) | 169.8100a | Average: 1 vial daily Max: 3 vials dailyb | Average: 169.81 Max: 509.43 | Average: 62,023 Max: 186,069 |
Anti-Parkinson drug | ||||||
LCIG (Duodopa) | 1 mL (20 mg levodopa / 5 mg carbidopa) | Intestinal gel (100 mL cassette) | 169.8100c | 1 cassette daily | 169.81 | 62,023 |
Dopamine agonists | ||||||
Apomorphine (Movapo) | 10 mg/mL | 3 mL pen | 45.5400c | 0.2 mL to 0.6 mL pre “off” episode, maximum 2 mL dailyd | 4.55 to 22.77 | 8,317 to 11,089 |
Amantadine hydrochloride (generics) | 10 mg/mL 100 mg | Syrup Cap | 0.1098 0.5252 | First week: 100 mg daily Subsequent: 100 mg twice daily | 1.10 0.52 | 795 380 |
Oral Levodopa | ||||||
Levodopa-carbidopa (generics) | 100 mg / 10 mg 100 mg / 25 mg 250 mg / 25 mg | Immediate release tablet | 0.1479 0.2209 0.2466 | 300 mg to 1,500 mg of levodopa in 3 to 4 daily doses | 0.44 to 1.48 | 162 to 540 |
100 mg / 25 mg 200 mg / 50 mg | Controlled release tablet | 0.7974 1.4282 | 200 mg to 1,600 mg of levodopa in 2 to 4 daily doses | 1.60 to 11.43 | 583 to 4,173 | |
Levodopa/benserazide (Prolopa) | 50 mg/12.5 mg 100 mg/25 mg 200 mg/50 mg | Cap | 0.3197 0.5265 0.8839 | 400 to 800 mg of levodopa daily in 4 to 6 doses | 2.11 to 3.54 | 769 to 1,291 |
Non-ergolinic dopamine agonists | ||||||
Rotigotine (Neupro) | 2 mg/24 hour 4 mg/24 hour 6 mg/24 hour 8 mg/24 hour | Patch | 3.5400 6.5000 7.2700 7.2700 | 2 mg to 16 mg daily | 3.54 to 14.54 | 1,293 to 5,307 |
Pramipexole (generics) | 0.25 mg 0.50 mg 1 mg 1.5 mg | Tablet | 0.1950 1.3860 0.3901 0.3901 | 1.5 mg to 4.5 mg in 3 equal doses | 1.17 to 4.16 | 427 to 1,519 |
Ropinirole (generics) | 0.25 mg 1 mg 2 mg 5 mg | Tablet | 0.0710 0.2838 0.3122 1.7450 | 3 mg to 24 mg in 3 equal doses | 0.85 to 3.75 | 310 to 1,369 |
Ergolinic dopamine agonists | ||||||
Bromocriptine (generics) | 2.5 mg 5 mg | Tablet Cap | 1.0188 1.5251 | 2.5 mg to 40 mg daily in 2 to 3 doses | 1.02 to 12.20 | 372 to 4,456 |
COMT inhibitors | ||||||
Entacapone (generics) | 200 mg | Tablet | 0.4010 | 200 mg to 1,600 mg daily in multiple doses | 0.40 to 3.21 | 146 to 1,171 |
Levodopa-carbidopa-entacapone (Stalevo) | 50 mg/12.5 mg/200 mg 75 mg/18.75 mg/200 mg 100 mg/25 mg/200 mg 150 mg/37.5 mg/200 mg | Tablet | 1.7471 | 600 mg to 1,600 mg daily of entacapone in multiple doses | 5.24 to 13.98 | 1,914 to 5,105 |
MAO-B Inhibitors | ||||||
Rasagiline (generics) | 0.5 mg 1 mg | Tablet | 6.1285 | 0.5 mg to 1 mg daily | 6.13 | 2,237 |
Selegiline (generics) | 5 mg | Tablet | 0.5021 | 5 mg twice daily | 1.00 | 367 |
COMT = catechol-O-methyltransferase; LCIG = levodopa-carbidopa intestinal gel; MAO-B = monoamine oxidase type B.
Notes: All prices are from the Ontario Drug Benefit Formulary7 (accessed December 2022), unless otherwise indicated, and do not include dispensing fees. Recommended dosages are derived from the appropriate product monograph, unless otherwise stated.
aSponsor-submitted price.
bIndividual dosing and infusion rate of foslevodopa-foscarbidopa is calculated during the titration period and is informed by the patient’s current use of levodopa and other Parkinson medications. Maximum recommended dose as indicated in the foslevodopa-foscarbidopa product monograph.6
cExceptional Access Program (EAP) price (accessed December 2022).15
dAssumes excess medication disposed of after 48 hours and assumes at least 1 dose required every 48 hours.16
Additional Details on the Sponsor’s Submission
No additional information from the sponsor’s submitted pharmacoeconomic evaluation was considered in the review of foslevodopa-foscarbidopa.
Additional Details on the CADTH Reanalyses and Additional Analyses
CADTH did not conduct any additional pharmacoeconomic analyses in the review of foslevodopa-foscarbidopa.
Appendix 2: Submitted Budget Impact Analysis and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 6: Summary of Key Take-Aways
Key take-aways of the BIA |
|---|
|
BIA = budget impact analysis; DBS = deep brain stimulation; LCIG = levodopa-carbidopa intestinal gel.
Summary of Sponsor’s Budget Impact Analysis
The submitted budget impact analysis (BIA) assessed expected budgetary impact resulting from reimbursing foslevodopa-foscarbidopa for the treatment of motor fluctuations in patients with PD who are not adequately controlled on optimized oral therapies and who are not candidates for DBS.17 The BIA was conducted from the perspective of the pan-Canadian public drug plans over a 3-year time horizon (2024 to 2026) with 2023 as the base year.
The sponsor estimated the eligible population using an epidemiological approach. The target population size was estimated using pan-Canadian (excluding Quebec) populations with estimates of PD incidence and prevalence, the proportion of those patients with advanced disease, and the proportion of patients covered under a public payer.18 Adjustments were made to the provincial populations to remove Non-Insured Health Benefits patients to estimate the provincial public plan population.19
The sponsor’s base-case analysis included drug acquisition costs only (i.e., no markup or dispensing fees). Vial sharing was not included. Data for the model were obtained from various sources included: sponsor-submitted pricing, Ontario Drug Benefit Formulary and the Ontario Exceptional Access Program (EAP).7,15 Additional scenario analyses were performed to include administration costs (i.e., titration and monitoring) sourced from the Ontario Schedule of Benefits as well as surgical event costs sourced from the Ontario Case Costing Initiative.11,12 Key inputs to the BIA are documented in Table 8.
The following key assumptions were made by the sponsor:
The sponsor assumed acquisition costs of adjunctive therapy and monitoring/follow-up will be the same for both foslevodopa-foscarbidopa and LCIG.
The sponsor assumed the “Other” comparator captured advanced PD patients remaining on oral therapies, DBS, or on no treatment (i.e., contraindication to PEG-J/subcutaneous infusion, unable or unwilling to manage a device, with severe psychosis or dementia, or without a needed caregiver). “Other” was not assigned costs as it was assumed to cancel out between the reference scenario and new drug scenario.
Table 7: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
Prevalence of PD Incidence of PD Patients with advanced PD Patients eligible for public payer plan | 19%a 88%b |
Number of patients eligible for drug under review | 10,540 / 11,328 / 12,124 |
Market uptake (3 years) | |
Uptake (reference scenario) LCIG (Duodopa) Other | 2.4% / 2.4% / 2.4% 97.6% / 97.6% / 97.6% |
Uptake (new drug scenario) Foslevodopa-foscarbidopa LCIG (Duodopa) Other | ||||| ||||| ||||| ||||| ||||| ||||| |
Cost of treatmentc (per patient) | |
Cost of treatment over year Foslevodopa-foscarbidopa LCIG (Duodopa) | $63,317.67 $63,317.67 |
LCIG = levodopa-carbidopa intestinal gel; PD = Parkinson disease.
aAssumption.
bSponsor data on file.17
cIncludes both treatment acquisition and adjunctive therapy acquisition costs.
Summary of the Sponsor’s BIA Results
The sponsor’s base case, which considered only drug acquisition costs, reported that the reimbursement of foslevodopa-foscarbidopa for the treatment of patients with advanced PD that are not candidates for DBS would lead to a budget impact of $0. No difference in the budget impact was observed between the 2 scenarios due to foslevodopa-foscarbidopa only capturing market share from a comparator (LCIG) with identical annual treatment costs. The sponsor conducted a scenario analysis that included the costs of administration, surgery, and monitoring. In this analysis that adopts a broader perspective, the reimbursement of foslevodopa-foscarbidopa resulted in an incremental cost savings of $58,515 in Year 1, $110,060 in Year 2, and $151,448 in Year 3 for a 3-year total incremental cost savings of $320,024.
Table 8: CADTH Revisions to the Submitted BIA
Scenario analysis | Sponsor’s value or assumption | CADTH value or assumption | |
|---|---|---|---|
Changes to derive CADTH scenario analyses | |||
Scenario analysis 1: Correction to surgical event costs (under health care system perspective) | NG tube insertion (inpatient) 1 day: $2,303.74 PEG-J tube insertion (inpatient) 1 day: $2,301.74 | NG tube insertion (inpatient) 1 day: $3,586.67 PEG-J tube insertion (inpatient) 1 day: $1,915.03 | |
Scenario analysis 2: Higher market capture for foslevodopa-foscarbidopa | Foslevodopa-foscarbidopa: |||| ||||| ||||| LCIG: |||| ||||| ||||| “Other”: ||||| |||||| ||||| | Foslevodopa-foscarbidopa: 0.5% / 0.9% / 1.2% LCIG: 1.9% / 1.5% / 1.2% “Other”: 97.6% / 97.6% / 97.6 | |
BIA = budget impact analysis; LCIG = levodopa-carbidopa intestinal gel; NG = nasogastric; PEG-J = percutaneous endoscopic gastrostomy-jejunostomy.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Inappropriate exclusion of relevant comparator: The sponsor’s BIA assumed foslevodopa-foscarbidopa will only displace LCIG. It was further assumed that a substantial proportion of the target patients would be receiving treatment from the “Other” category which included advanced PD patients remaining on oral therapies, DBS, or on no treatment (i.e., contraindication to PEG-J/subcutaneous infusion, unable or unwilling to manage a device, with severe psychosis or dementia, or without a needed caregiver).17 While the reimbursement request for foslevodopa-foscarbidopa is indicated for patients with advanced PD that are not candidates for DBS, clinical expert feedback suggested that DBS may be an appropriate comparator for these patients in certain jurisdictions as DBS candidacy should not impact their eligibility for foslevodopa-foscarbidopa.
CADTH could not undertake reanalysis to address this limitation due to inflexibility of the budget impact model and lack of information regarding the number of patients expected to switch from “Other” to foslevodopa-foscarbidopa.
The market uptake of foslevodopa-foscarbidopa is underestimated: The sponsor’s submitted BIA indicated that foslevodopa-foscarbidopa would result in a market uptake || |||| || |||| || |||| || |||| |||| |||| || ||||. These values are driven by foslevodopa-foscarbidopa only capturing market share from LCIG in all 3 years. However, CADTH obtained clinical expert feedback indicating that the market uptake in all 3 years does not align with clinical expectations and indicated that the sponsor likely underestimated foslevodopa-foscarbidopa uptake.
Given the uncertainty surrounding these inputs, CADTH conducted a scenario analysis to explore the impact of foslevodopa-foscarbidopa capturing increased market share. Since no costs are associated with the “Other” category, the scenario analysis explores the impact of 100% capture of LCIG incident cases over the 3-year horizon.
An additional limitation was identified regarding the costs of surgical events but were not considered to be key limitations given the perspective of the BIA’s base case. The costs were corrected in the CADTH scenario analysis, when presenting the BIA under the broader health care system perspective.
CADTH Reanalyses of the BIA
CADTH did not undertake a base-case reanalysis. Scenario analyses were conducted to assess the impact of changing key parameters within the sponsor BIA as outlined in Table 8. The results of the CADTH scenario analysis are presented in Table 9. CADTH accepted the sponsor’s base case but conducted several scenario analyses.
Table 9: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $14,859,605 | $16,048,294 | $17,248,457 | $18,460,388 | $51,757,139 |
New drug | $14,859,605 | $16,048,294 | $17,248,457 | $18,460,388 | $51,757,139 | |
Budget impact | $0 | $0 | $0 | $0 | $0 | |
CADTH scenario 1: including updated surgical costs (health care system perspective) | Reference | $15,095,224 | $16,302,761 | $17,521,954 | $18,753,101 | $52,577,817 |
New drug | $15,095,224 | $16,248,540 | $17,419,971 | $18,612,767 | $52,281,278 | |
Budget impact | $0 | –$54,221 | –$101,983 | –$140,334 | –$296,539 | |
CADTH scenario analysis 2: 100% of LCIG Incidence (health care system perspective) | Reference | $15,095,224 | $16,302,761 | $17,521,954 | $18,753,101 | $52,577,817 |
New drug | $15,095,224 | $16,167,208 | $17,376,264 | $18,597,174 | $52,140,646 | |
Budget impact | $0 | –$135,553 | –$145,690 | –$155,927 | –$437,171 | |
CADTH scenario analysis 2: 100% of LCIG incidence (drug costs only) | Reference | $14,846,898 | $16,034,570 | $17,233,707 | $18,444,601 | $51,712,877 |
New drug | $14,846,898 | $16,034,570 | $17,233,707 | $18,444,601 | $51,712,877 | |
Budget impact | $0 | $0 | $0 | $0 | $0 |
BIA = budget impact analysis; LCIG = levodopa-carbidopa intestinal gel.
Results are presented in Table 9. When accounting for monitoring, administration costs and surgical events, the 3-year incremental budget impact was expected to result in cost savings of $296,539. The exploration of a scenario analysis wherein foslevodopa-foscarbidopa captured 100% of the LCIG incidence cases resulted in a 3-year incremental cost savings of $437,171, in the perspective of the health care system. When only including drug acquisition costs, the scenario analysis that captures 100% of the LCIG incidence results in budget neutrality. As the explored analyses all assumed that the reimbursement of foslevodopa-foscarbidopa would only displace LCIG, the budget impact of foslevodopa-foscarbidopa taking market share from non-LCIG therapies is unknown.
Stakeholder Input
Patient Input
Parkinson Association of Alberta
About the Parkinson Association of Alberta
Parkinson Association of Alberta (PAA) is the voice of Albertans and their families living with Parkinson disease (PD) and Parkinson’s Plus Syndromes.
PAA provides direct programming in three areas – support (mental, emotional & peer), education (information, resources, referrals, webinars) and active (physical, speech/swallowing, cognitive & social) – to the over 10,000 Albertans with PD or a Parkinson’s Plus Syndrome, their families and care partners. We are also providing education and information to health care professionals, community partners and the public at large.
PAA also funds innovative research for a better and brighter future for Parkinson’s.
PAA is a stand-alone Alberta-based registered charitable organization. PAA relies on donations and fundraising initiatives to support the services, resources and programs offered.
Information Gathering
PAA gathered data and perspectives from the Parkinson’s community by conducting a survey. 26 responses were gathered between November 21, 2021, and December 2, 2022.
All respondents were from Alberta. Respondents identified that where they resided was defined as urban (57%), suburban (33%) and rural (10%).
57% of respondents are people with Parkinson disease; 43% are care partners or family members of people with Parkinson disease.
In terms of gender 67% of respondents identified as female, 33% identified as male.
62% of respondents are between the ages of 65-74, 33% were ages of 75-84, and 5% between the ages of 55-64. 100% of respondents are retired.
Disease Experience
The following are areas are what respondents indicated as being most negatively impacted by Parkinson disease. The responses are listed below in order of indicated as most important to least important):
Overall quality of life
Participation in recreational/exercise activities
Participation in social activities
Loss of independence
Managing family obligations
Loss of confidence
Relationships
Work/employment
“Parkinson’s has affected my confidence and sense of who I am. It is hard not to let my disease define me.”
“My husband’s cognitive issues are more stressful than the physical symptoms. I feel like I am always nagging him to take his medication or do his exercises or even get ready for the day. I am always tired, but there is always more to do.”
“I had to resign from work due to Parkinson’s.”
“Our whole lives have changed. We are only two years into my husband’s diagnosis so it’s still manageable, but our plans for our future certainly do not look the same is they did before the diagnosis.”
Respondents ranked ten aspects/symptoms of Parkinson disease in terms of being most important to control and/or manage, they are listed below in order of indicated as most important to least important):
Changes in cognition and memory
Fatigue/Sleep Issues
Freezing/Unpredictable ON-OFF periods
Changes in Mood
Rigidity
Speech and Swallowing Issues AND bladder/bowel issues (tied)
Impaired balance
Slowness and stiffness
Tremors
Experiences With Currently Available Treatments
There is an array of symptomatic treatments available for Parkinson disease. These include medications, surgical/medical procedures (i.e., DBS, Duodopa), other forms of rehabilitation therapy (physiotherapy, occupational therapy, speech therapy, exercise) and psychological follow-up (i.e., counselling). All treatments can have a significant impact on improving quality of life, especially when a personalized combination is utilized as opposed to a one-size fits all approach.
“Medication helps me maintain a better ebb and flow in my day-to-day life.”
“My neurologist and I are struggling to find a medication combination that works well for me. It either seems to be too much and I’m bobbing and weaving all over the place or not enough and I’m freezing.”
Medications
100% of respondents indicated that they or their loved one with Parkinson’s was utilizing oral medication as their primary treatment option. The benefit to utilizing medications is maintaining day-today functioning by way of better management of symptoms.
“I’ve had Parkinson’s for 7 years now, I find I can feel the wearing off start to happen like clockwork. When that “clock” is off I know it is likely time for a medication adjustment.”
In terms of medications being taken to manage Parkinson’s, 58% of respondents have reported that they or the person they are taking care of have experienced side effects. The most noted side effects that respondents indicated were most difficult to endure were fatigue/drowsiness and constipation/bowel issues.
When it comes to experiencing difficulties receiving/taking medication as a treatment for Parkinson’s respondents indicated the following:
Difficulties with timing of and/or remembering to take medications – 49%
Other (includes constipation, sleep issues, nausea, medication interactions) – 19%
Difficulties swallowing medications – 16%
Storage of medications – 3%
Rehabilitation
56% of respondents indicated that they or their loved one with Parkinson’s have included some form of rehabilitation (physio, occupational/speech therapy and/or exercise) as a treatment option.
“I do what I can to keep myself healthy and active despite having PD. I exercise in a group class a couple times a week and then am working out at home for the rest of the days. I’ve also taken part in the Thinking and Memory class Parkinson’s Association runs.”
When it comes to experiencing difficulties receiving rehabilitation as a treatment for Parkinson’s respondents indicated the following:
Lack of motivation/apathy – 37%
Cost – 21%
No access to or wait lists/times to access rehabilitation opportunities – 13%
“Living in rural Alberta means I have limited access to rehab help. Thankfully Parkinson Association of AB offers some classes online, but it isn’t the same as being in person.”
“Everything that can help you costs money. $155/hour for a physiotherapist. $200+/hour for a psychologist. $200+/hour for a speech pathologist. It is ridiculous that I have to pay to get the health services I need to live well.”
Psychological Follow-up
0% have included psychological follow-up.
When it comes to experiencing difficulties receiving psychological follow-up as a treatment for Parkinson’s respondents indicated the following:
Cost – 42%
Difficulties communicating and/or expressing myself – 25%
Surgical /Medical Procedure
0 respondents have undergone a surgical/medical procedure as part of the treatment they are receiving for Parkinson’s.
In terms of accessing treatment for Parkinson disease respondents indicated difficulties with the following:
Access to appropriate healthcare professionals and/or service providers – 38%
“Long waiting lists for everything. If I’m lucky I get to see my neurologist 2x/year and then for only 15 minutes at a time.”
“I was referred to a community neurologist, his lack of understanding about Parkinson’s was astonishing. I’m trying to find a new neurologist, but the wait times are crazy.”
“Unless you live in Calgary or Edmonton, good luck finding any kind of supports for PD. Thank god for PAA.”
Issues with Insurance coverage (25%) and Cost of medications (57%)
“My insurance only covers so much. I’m on a fixed income and it adds up quickly.”
“We need better coverage of medications. My husband’s currently costs us about $1000 per month.”
Improved Outcomes
Survey respondents indicated a variety of improvements that they would like to see that are currently not being achieved. The most reported improvement respondents would like to see is a treatment option that would not increase dyskinesia as time went on. This was followed by medications that would treat cognitive issues. Alternative medication delivery systems were also noted (specifically a patch).
“It’s hard to want to take medication when you know that at some point it’s going to make dyskinesia worse the longer, I take it.”
” My husband’s regular Parkinson’s medications worked just fine, and we had no troubles with them. What our biggest problem is now is his cognitive decline. He has hallucinations and struggles with tasks he used to find easy. I wish there was a medication for cognition troubles.”
“I really like the controlled release pills. I find I have a better day when I take those vs the regular ones.”
“I’m fine with pills, the other options seem too complicated for me.”
“It is getting harder for my husband to remember to take his pills. We looked at the patch version, but it was too expensive. The brain surgery is out of the question and the pump device is too complicated for us. If there was a more affordable patch or a pill that lasts longer, I think that would be great.”
Experience With Drug Under Review
100% of respondents were NOT aware of foslevodopa foscarbidopa as a treatment option for Parkinson disease. No respondents were being treated and/or knew of anyone being treated with foslevodopa foscarbidopa.
Only 1 (of 26) respondent said they would consider (along with their treating physician) trying foslevodopa foscarbidopa to see if it would be beneficial in treating their or their loved one’s Parkinson’s. 85% (22 people) said they would not consider it and 8% (2 people) weren’t sure.
“This seems way too complicated for me.”
“My husband bruises so easily, the thought of having to inject him with medication every single day seems unnecessarily cruel. We would not try this.”
“Too bad it’s not a more affordable patch system.”
“I’m on the fence. I’d certainly talk to my neurologist about it, but needles are not my cup of tea.”
“I’d be worried about infection, bruising and how complicated it could become. My skin is already pretty sensitive, and I think my pain threshold has diminished since my diagnosis.”
Companion Diagnostic Test
Not applicable.
Anything Else?
People with Parkinson’s need access to a variety of options to ensure they can be on a treatment regimen that offers the best possible control of their unique set of symptoms and an improved quality of life. The unpredictability of the disease and loss of quality of life is forcing people with Parkinson’s to withdraw from normal activities too soon (i.e., day-to-day activities (including self-care and household chores) work, travel, maintaining relationships, etc.).
The inability to appropriately control their symptoms on a continuous or predictable basis causes undue stress, anxiety and can lead to depression and/or social isolation.
Furthermore, survey respondents reported it would be a significant financial burden if they did not have coverage to help with their drug costs. Many people with Parkinson’s and care partners report having to leave the workforce early or reduce hours due to the progression of the disease. This limit of incoming resources coupled with increasing expenses for travel to appointments for follow-up/treatment and any incurring drug costs causes a great amount of additional stress and strain on families.
When a disease is not only life-long, but as life limiting as Parkinson’s it is essential to provide coverage to ensure treatments are affordable and accessible for all who need it.
Conflict of Interest Declaration — Parkinson Association of Alberta
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission?
No.
Did you receive help from outside your patient group to collect or analyze data used in this submission?
No.
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
Table 1: Financial Disclosures for the Parkinson Association of Alberta
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie Pharmaceuticals | — | X | — | — |
Parkinson Canada, Parkinson Society of British Columbia, and Parkinson Quebec
About Parkinson Canada, Parkinson Society of British Columbia, and Parkinson Quebec
Parkinson Canada
At Parkinson Canada, people with Parkinson’s are at the centre of everything we do. We empower the Parkinson’s community through tailored programs, innovative research and raising the voice of Canadians impacted by Parkinson’s through our advocacy efforts. A national registered charity, Parkinson Canada fulfils its mission through the generosity of donors.
Parkinson Society British Columbia (PSBC)
PSBC is a non-profit organization governed by a volunteer Board of Directors. The Society receives no government funding and is supported entirely by donations from individuals, members, corporations, foundations and the dedicated efforts of volunteers.
We believe that every person touched by Parkinson's deserves to know that they are not alone in their journey. We are here for the person with Parkinson’s, their care partners, family and friends. Our friendly and knowledgeable staff is committed to offering support, sharing reliable information and raising funds for programs and research.
Parkinson Quebec
Parkinson Quebec has 4 objectives: Advocacy, investing in research, providing information on Parkinson's disease and realizing digital services. Our 9 regional partners have the sole objective of providing local services
Information Gathering
Parkinson Canada gathered patient input via a survey distributed to stakeholders who are either people living with Parkinson’s or care partners. 113 responses were collected in Canada between November 16 and November 29, 2022.
Demographics
0.9% of respondents are from Alberta, 0.9% are from British Columbia, 2.7% are from Quebec, 3.5% are from Manitoba, 3.5% are from Nova Scotia, 4.4% are from New Brunswick, 11.5% are from Saskatchewan, and 72.6% are from Ontario.
48% of respondents identified as female and 52% as male.
11% of respondents are care partners while 89% are persons living with Parkinson’s.
2.6% of respondents are between 35-44 years old, 6.1% are between 45-54 years old, 19.5% are between 55-64 years old, 38% are between 65-74 years old, 28% are between 75-84 years old, and 5% are 85 plus years old.
Disease Experience
In our survey, people with Parkinson’s and their care partners emphasized the overall impact that Parkinson’s has on their quality of life and day-to-day activities:
“Parkinson’s has changed my life greatly for the worse and in many ways. It affects every part of my life. My ability to function, to eat, to sleep, to drive, to think. My relationships. Everything. I want to get a t-shirt that says, ‘Parkinson’s makes everything worse’ but my husband won’t let me”.
“It affects every part of my life from my ability to drive some days, walking, eating, and sleeping. In other words from the time, I wake up to the time I go to bed and at times during my sleep”.
“It just makes what used to be simple tasks very difficult and makes many tasks virtually impossible. It slows you down and seems to result in a constant state of fatigue”.
71% of respondents reported experiencing “off periods” (defined as the return of motor or non-motor symptoms between regularly scheduled doses of dopamine medication), noting that their management is one of the biggest impacts on their quality of life:
“Parkinson’s has resulted in me leaving the workforce early, 5 years ago, and most days now revolve around the medication schedule as I fluctuate between ON and OFF periods. OFF periods tend to limit what I am able to do or am comfortable doing. My ON time is very good but can’t be counted on if I want to plan to do something or go somewhere”.
For some people there is a certain degree of predictability to “off periods” but for others the fluctuations are unpredictable and limit their activities. Respondents mentioned the effect on outings, relationships, and their ability to socialize, which could lead to anxiety and depression:
“I struggle with daily tasks. I have high anxiety and avoid going out or socializing. I don’t sleep well. My balance is poor”.
“Tremor, stiffness, slowness, constipation. Limits my social activity, increases anxiety, reduces physical activity (strength, endurance, and balance), disrupts sleep, interferes with speech”.
“Limited energy, balance problems and risk of falls, tremor in my hands and face creates issues with eating, grooming, etc., challenges with daily caring for my two dogs, embarrassment about my PD symptoms has limited my social contacts and outings”.
Respondents still in the workforce reported having to stop working or reduce their hours due to Parkinson’s:
“It’s impossible to ignore. Some days are better than others, but I feel that I can't rely on being able to do what I have agreed to. I am not able to work and had to take early retirement as my ability to do my job was getting worse. I loved my job. Now, I am tired all the time, with an intense, grinding fatigue”.
“My husband had to go into early retirement, which caused a huge financial strain for us. On a daily basis we need to structure our day around his personal needs, making sure he takes his medication on time, managing his anxiety and his limited ability to help with household chores.”
“Things that require fine motor skills are affected, causes me a great deal of frustration, balance is slightly affected, I lose balance and fall down more often than id like. Frankly, it's a pain in the rear end, client facing work has become more difficult as clients don't understand what is happening.”
Care partners reported on the demands, financial and otherwise, that caring for their loved ones have had:
“I had two caregiver burnouts and now my husband is in long term care because I was unable to take care of him at home. I continue to visit him every day due to staff shortages. I advocate for him daily. I am just as stressed mentally but more relaxed physically”.
“I am the one who controls his pill taking and times, ensures he is eating a well-balanced meal daily, has enough fiber to ensure he uses the washroom daily, walk daily for exercise, play cards daily to keep his hands limber and mind alert, do online banking as he is not able to, do yard and house work as he is limited as to what he can do. I text on his behalf, we use speakers when on the cell phones, I do all of the work on the computer as his hands are not able to, I drive everywhere as he was in an accident 2 years ago and stopped driving then. I pretty much do everything, and he is my helper. I worry about our future and what it will look like. He is 70 and I am 63. We are on a waiting list for assisted living as we know things are going to be changing as our house is on 3 levels and stairs will become harder. We have someone else shovel our driveway. He worries about money, so in turn, I worry about money too as we have no idea how long we are going to live. I am going away on a sewing retreat next week and have his daughter come to stay with him. I am not comfortable with him staying alone and he is not either. Makes it hard for me to be away longer than a few hours. I am trying to find a balance in our lives that works well for both of us ... sure is hard at times. I feel responsible for vaccines, flu shots and his health as I want him and us to keep well and out of harm’s way. I have to think for both of us”.
“As a caregiver I somewhat feel I don't have the freedom of my own life, living in the same home with my brother and supporting and caring in different ways. He is still able to take his own medication, shower and eat or make simple meals”.
Experiences With Currently Available Treatments
54% of respondents reported that the frequency of their oral medication impacts their lifestyle or quality of life. Smoothing out the intake of levodopa-carbidopa could decrease the number of pills required daily:
“Constant medication (5 times a day and over 75 pills per week) is very disruptive. Once I started on the meds, I developed urinary incontinence due to the high volume of water I need to ingest. I have serious, constant back pain, this is being investigated currently. Sleep disturbance, frequent nightmares and falling out of bed are more recent symptoms”.
“An improvement would be taking less pills and having them work longer. Having a more of a combined medicine regime that treats multiple symptoms at once instead of a million pills for one or two symptoms”.
“An injection would be easier than swallowing pills and goes into blood stream and lasts longer”.
“The medication gives only very limited improvement with my PD motor symptoms. Increasing the number of pills per day also increases the side effects”.
The availability of current interventions and medication was ranked on a scale of 1 (not easily accessible) to 5 (very accessible), as follows.
Table 2: Medicine Availability
Accessibility Ranking | Percentage of Respondents |
|---|---|
1 | 6% |
2 | 15% |
3 | 29% |
4 | 32% |
5 | 18% |
“With the many years of research being done you would think that better medications would have been developed by now. People slowly dying from Parkinsons Disease have been using the same medication for 60 years or more!”.
Improved Outcomes
41% of respondents expressed concern over invasive treatment options such as Deep Brain Stimulation or Duodopa and indicated improvements that they would like from new treatments. 33% of respondents referenced the need for longer lasting medications that limit or eliminate “off periods” and lower the number of pills they take daily:
“I would like to be able to eat without worrying about reducing the effectiveness of my medication. I would like to reduce the number of medications and pills I take. I would like to have a better ON/OFF balance and more control over recovering from an OFF period or, if possible, eliminate the fluctuations”.
“Fewer doses needed per day, more exact division of doses, less wearing off”.
“A version of medication that could provide dopamine over a 6- or 12-hour period”.
“Be able to take them less frequently (currently 4x per day)”.
“Each dose would last for longer periods of time rather than 4.5 hours”.
“I am always hopeful that better treatments are on the way”.
Experience With Drug Under Review
Parkinson Canada was unable to connect with patients that have experience with foslevadopa-foscarbidopa, but the survey results showed that respondents take between one and 40 pills per day, that 46% have difficulty sleeping due to their medication wearing off, that the quality of life of 63% of respondents is impacted by the frequency of taking oral medication, and that 65% would be interested in an injection-based levodopa-carbidopa treatment due to its possible benefits:
“I believe injection-based levo-carb would offer more control over ‘off’ period symptoms”.
“Hopefully it would be effective for longer periods of time than the pills, which I take every 4 hours”.
Companion Diagnostic Test
Not applicable.
Anything Else?
People with Parkinson’s need access to a variety of options to ensure the best possible control of their unique set of symptoms and an improved quality of life. The unpredictability of the disease and loss of confidence and independence that one experiences is forcing people with Parkinson’s to withdraw from normal activities too soon (e.g., work, daily chores, socializing, etc.). Many people with Parkinson’s report anxiety and excessive worry over the inability to appropriately control their symptoms on a continuous or predictable basis, which could lead to isolation, anxiety, and depression.
Many people with Parkinson’s and caregivers report having to leave the workforce early or reduce hours due to the progression of the disease. This limit of incoming resources coupled with increasing expenses for travel to appointments for follow-up/treatment and any incurring drug costs causes a great amount of additional stress and strain on families. It is essential to provide coverage for a variety of treatments, as respondents reported taking between one and nine different oral medications per day. This medication may possibly replace or reduce the frequency of use for other medications that bolster the effects of traditional levodopa (i.e., dopamine agonists or monoamine oxidase B (MAO-B) inhibitors) in efforts to minimize “off periods.” Regarding financial limitations, respondents reported the following:
“When I turn 65 in 4 years, I no longer have a private plan to cover Botox injections or any of my medications leaving me to have to choose between food and housing or quality of life”.
Medical treatment for 21% of respondents is covered only by private insurance and 17% pay at least a portion out of pocket. For the latter group, the financial burden to cover medical treatment expenses was ranked on a scale of 1 (no burden at all) to 5 (very high burden), as follows:
Financial Burden Ranking | Percentage of Respondents |
|---|---|
1 | 11% |
2 | 18% |
3 | 26% |
4 | 26% |
5 | 19% |
Conflict of Interest Declaration — Parkinson Canada, Parkinson Society of British Columbia, Parkinson Quebec
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission?
No.
Did you receive help from outside your patient group to collect or analyze data used in this submission?
No.
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
Table 4: Financial Disclosures for Parkinson Canada
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie | — | — | — | X |
Clinician Group Input
National Movement Disorder Expert Group
About the National Movement Disorder Expert Group
We are a group of movement disorder specialists with expertise in the management of Parkinson’s disease (PD). Our group met in Toronto on July 9, 2022, to discuss the unmet needs in the Canadian treatment landscape our patients and their families, we aim to convey the importance that access to novel therapeutic options has on their improved well-being by strongly supporting this review of foslevodopa-foscarbidopa.
Information Gathering
This document contains a summary of the National Expert Group’s meeting conclusions, supporting data on the burden of PD and the need for additional advanced treatment options, with additional input from Canadian movement disorder specialists. The results of phase 3 trials using foslevodopa-foscarbidopa will be briefly reviewed to discuss efficacy and safety of the product.
Current Treatments and Treatment Goals
Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disorder in the world with a prevalence that is expected to keep rising in the following years.1 The disease is characterized by hallmark motor features, including bradykinesia, rest tremor, and rigidity, alongside non-motor features like autonomic dysfunction, cognitive decline, and gastrointestinal impairment which become increasingly burdensome with time.2 To date, no therapies have been shown to delay or reverse the progression of PD, and dopamine replacement therapy is the current standard of care in reducing the burden of motor symptoms.3 Oral levodopa preparation, either levodopa-carbidopa or levodopa-benserazide, is the first-line treatment for PD. However, the progressive loss of endogenous dopaminergic neurons leads to patients becoming dependent on stable levodopa blood levels to avoid prolonged periods of poor symptom control and dyskinesia, which require dose escalation with frequent dosing, along with the addition of other pharmacological treatments by specialized neurologists. Eventually, PD often progresses beyond the ability of oral medications to adequately control fluctuating symptoms, and up to 32% of Canadian patients are no longer able to perform the activities of daily living despite having optimized oral therapeutic regimens.4
Impact on Society
The prevalence of PD increased by more than 20% between 1996 and 2012, and PD affects over 84,000 Canadians over the age of 40 and 50,000 over the age of 65.5-7 Moreover, the prevalence is expected to double by 2031, making PD the most prevalent neurodegenerative movement disorder and the fastest growing neurological disease in the world.1,8 While this increase can be partially attributed to an aging population, epidemiological models suggest that PD rates are increasing beyond what would be expected by age alone, which suggests that multiple factors will likely continue to contribute to the increasing burden of PD in Canada and around the world.9 Age is a significant risk factor for developing PD, with the incidence and prevalence increasing sharply after 60 years.2,5
PD has a multifactorial impact on the health and lives of patients. Overall, PD accounts for 1.1% of disability-adjusted life years (DALYs) in Canada, primarily due to healthy life lost due to disability, which patients typically begin to experience 10 to 15 years after being diagnosed.10,11 Over 70% of the direct costs of PD in Canada can be attributed to long-term disability.11
It is estimated that PD costs the Canadian economy $558.1 million per year, with $470.3 million attributed to the indirect costs of premature mortality and long-term disability.11 An additional $39.7 million is spent on hospital care, $24.1 million on medications, and $23 million on physician care. Additionally, a PD diagnosis increases the relative probability of accessing a medical specialist (PR=1.68 [95% CI: 1.54,1.83]), visiting an emergency department (PR=1.79 [95% CI: 1.45-2.21]), and overnight hospitalization (PR=2.08 [95% CI:1.27,2.05]).12 The National Expert Group underlines that PD patients with symptom fluctuations place a substantial unmeasured burden on the health care system in terms of frequent phone or in-person contact with patients and care partners to assess and address their needs.
Current Oral Treatments and Limitations
Unfortunately, no cure currently exists for PD, nor are there treatments that significantly impact disease progression. As such, pharmacological and behavioural interventions are used to manage the clinical manifestations of PD rather than modifying the disease itself. Once diagnosed, the standard of care to treat motor symptoms is to prescribe dopamine replacement therapy, particularly oral levodopa-carbidopa.2,3,13 Both immediate- and extended-release formulations of oral levodopa are effective in reducing motor symptoms and have acceptable safety profiles.
Early in the disease, most patients have satisfactory relief of their motor symptoms with oral levodopa taken 3 or 4 times a day. However, as dopaminergic neuronal loss progresses over time, most patients experience symptom fluctuations, related to the short half-life of levodopa combined with complex and poorly understood pharmacokinetic and pharmacodynamic mechanisms, including synaptic brain changes along with inadequate gastric absorption of the medication. Four to 6 years into the disease, about 50% of patients experience end of dose wearing off, resulting in a resurgence of motor and/or nonmotor symptoms (tremor, slowness, difficulty walking, gait freezing, bradyphrenia, anxiety, pain, fatigue, etc.) a few hours after taking their levodopa dose. These “off periods” will usually be improved 30 to 45 minutes after the next levodopa dose, leaving patients unwell, with reduced ability to function, for 45 minutes or longer in between each dose. As the disease progresses, 90% of patients will also experience levodopa induced dyskinesia - axial or appendicular involuntary choreiform movements occurring mostly as a peak dose phenomenon but sometimes also during wearing off periods.14,15 Increasingly long durations of “off” periods and medication related dyskinesia lead to more frequent dosing of levodopa and complex medication schedules, with some patients needing medication every every 2 hours. After 5 to 10 years of treatment, the prevalence of motor fluctuations is reported to be as high as 60-90% in PD patients.16
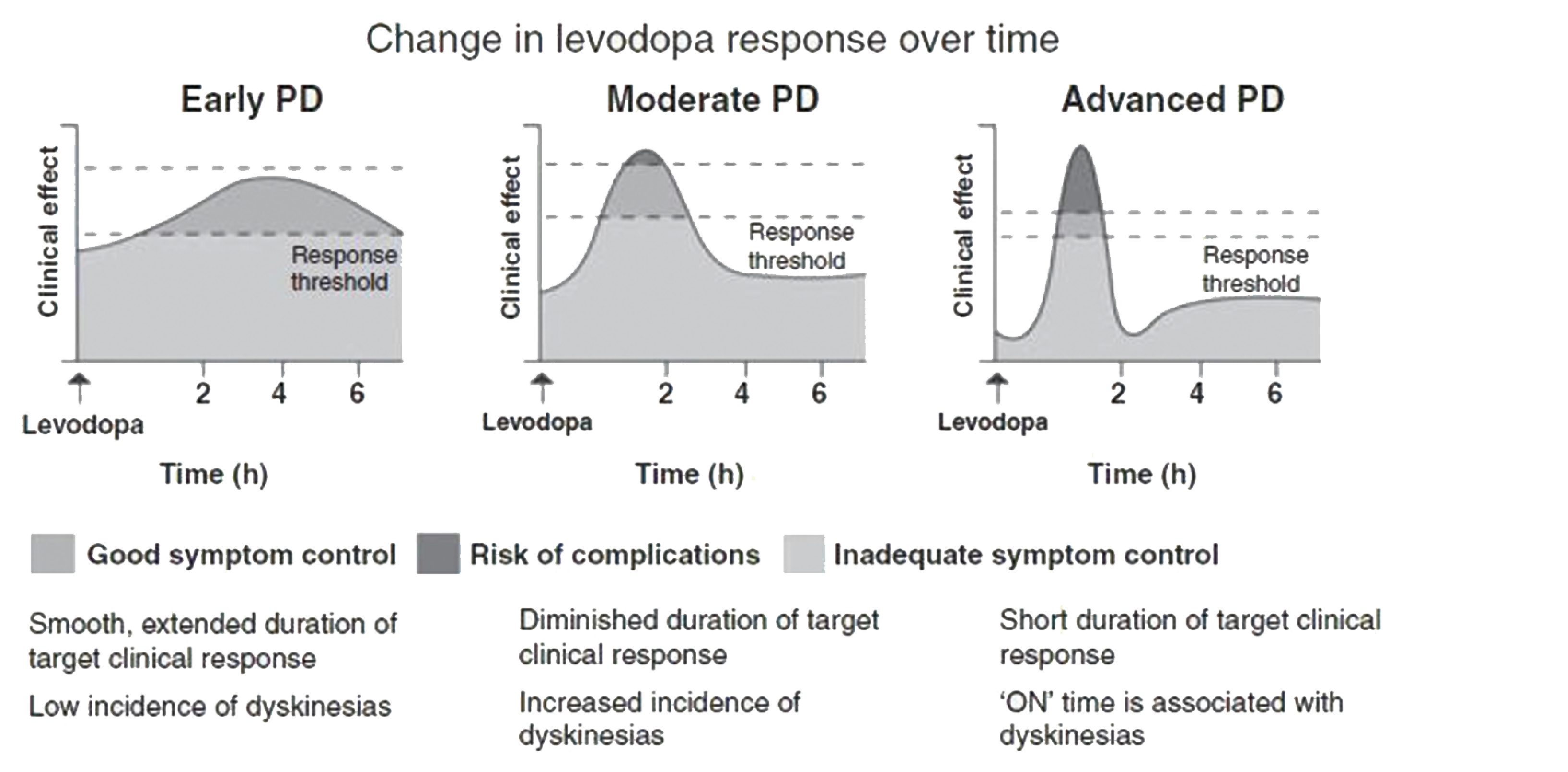
As the disease progresses, the latency and duration of effect of levodopa are reduced and the magnitude of effect is greater, along with a lower threshold for dyskineasia, leading to a reduced therapeutic window. Patients experience end of dose off periods as well as peak dose dyskinesia, leading to frequent levodopa dosing.
Due to the progressive nature of PD, individualized and frequent tailoring of medication classes, formulations, and dosages becomes necessary to reduce off time and treat motor fluctuations and dyskinesia. Dopamine agonists, such as pramipexole, ropinirole and rotigotine are often used as adjuncts to levodopa to reduce off periods, but they carry a higher risk of adverse events including impulse control disorders, excessive sleepiness, cognitive dysfunction and hallucinations, limiting their use.18 Monoamine oxidase B inhibitors (such as rasagiline, selegiline and safinamide) and catechol-O-methyltransferase inhibitors (such as entacapone) can be used alongside levodopa to reduce off time in patients with fluctuating PD, with the potential risk of worsening dyskinesia.13,19 Amantadine is often added for the treatment of dyskinesia, but hallucinations and cognitive dysfunction prohibit its use in older or cognitively impaired patients. Apomorphine injections and sublingual film may help manage severe off periods if administered in specialized clinical settings.13
Despite optimized oral therapies, many patients still have to cope with recurrent off periods. In 2014, an online study with over 3000 PD patients was completed by the Michael J. Fox Foundation and revealed that 64% of patients spent more than 2 waking hours daily in an OFF state, 50% of each off period lasting 45 minutes or longer. Although off periods are often partially predictable, related to levodopa dosing times, off periods can also happen unpredictably, leading to great anxiety and disability. Off periods can also be induced by levodopa absorption delays in the digestive system, leading to dose failures, delay or reduction in levodopa efficacy, especially when taken at mealtime, as protein can reduce levodopa absorption. Over time, patients and caregivers slowly reduce or avoid social activities and reorganize their daily schedule around the recurrent off periods and medication schedule.
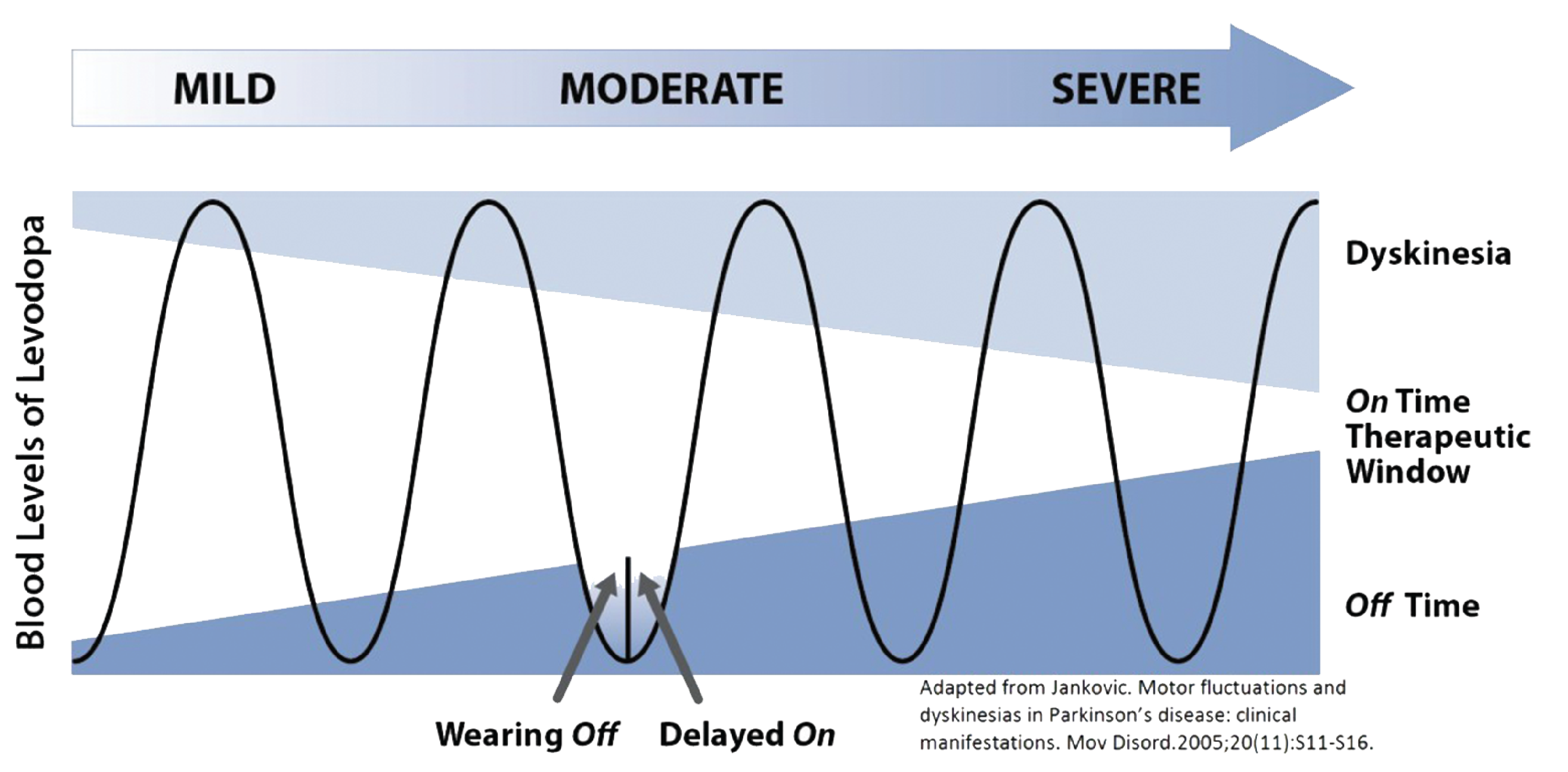
Treatment Goals
Over time, many PD patients require increased dosing frequency of oral levodopa, up to eight times per day or more, but still experience burdensome off periods and dyskinesia due to the short half-life of the drug.3 Although the adjunct of dopamine agonists, monoamine oxidase B inhibitors, catechol-O-methyltransferase inhibitors and amantadine can help reduce motor fluctuations, these agents are often insufficient, poorly tolerated or contra-indicated. As the disease progresses, the medication schedule increasingly results in complex and frequent dosing, further impairing patients’ quality of life and increasing the burden on care providers.
In these patients, continuous dopaminergic stimulation is required to achieve good and sustained relief of their fluctuating motor and nonmotor symptoms.
The Burden of PD
The lives of PD patients are restricted due to their motor and nonmotor symptoms, and many are unable to work due to embarrassment, stigma, and/or reduced functionality brought on by off periods or medication-related dyskinesia. Indeed, a national survey found that between 20% to 50% of PD patients require mechanical support for mobility, and fewer than 20% of PD patients between the ages of 18 to 64 are able to work.1 Moreover, more than 50% of PD patients experience stress, anxiety, and a loss of confidence. As PD progresses and symptom fluctuations become more common, patients’ activities become tightly controlled and/or limited by their oral medication schedule. They tend to avoid situations where they may become off in public.21 Informal care partners, most often the spouses of PD patients, are also negatively impacted by the burden of PD across multiple domains including physical, social, and occupational.22,23 In addition to stress, exhaustion, and guilt, most care partners report feeling anxious, helpless, or frustrated with the person with PD.21 In a mediation analysis of 51 dyads of PD patients and their spouses/care partners, one study found that the severity of the patient’s motor symptoms significantly increases care partner burden through deterioration of the spousal relationship.24 Furthermore, the treatment-specific burden put on care partners (i.e. adhering to frequent and sometimes complicated medication regimens, keeping track of appointments, and burdens associated with travel and the logistics of navigating the allied health environment) can lead to physical and mental exhaustion.25 Care partners also experience significant financial burden as they pay for PD-related services and are less able to work due to the time commitment required to care for the person with PD. Additionally, the health of care partners can rapidly decline as the effects of chronic stress are compounded by a perceived and existent inability to focus on their own needs. This notion is supported by research which indicates that care partners are at risk of accumulating increased financial, mental, and physical burdens over time.26 Importantly, the use of device-aided therapies (DATs) for advanced PD symptoms has been shown to significantly reduce caregiver burden by allowing them more time to engage in family, household, and leisure activities.27
Advanced Treatments Currently Available
Advanced therapies, often referred to as device-aided therapies (DATs), are used to circumvent the pharmacological limitations of oral therapies and have been shown to significantly reduce pill burden for PD patients.28 DATs have been shown to significantly reduce impairment in the activities of daily living, impairment due to motor symptoms, disability related to dyskinesia, and disability related to non-motor symptoms for advanced PD patients.4 To date, there are two DATs approved for the treatment of motor symptoms in fluctuating PD in Canada – deep brain stimulation (DBS) and levodopa/carbidopa intestinal gel (LCIG) infusion.13,19 Although standardized guidelines for the use of one DAT over the other may not be possible,29 the National Expert Group agreed that several factors are considered, including patient age, degree of functional impairment, cognitive status, and patient preferences when determining which patients would be best suited for each DAT.
Deep Brain Stimulation (DBS)
DBS involves the neurosurgical implantation of electrodes, trans-cranially, into the subthalamic nucleus or globus pallidus interna under the control of implanted ‘pacemaker like’ impulse generator.3 A meta-analysis of randomized control trials found that DBS offers a significant advantage against oral therapies alone in terms of UPDRS-measured “on/off” time, dyskinesia, and quality of life.30
Levodopa/Carbidopa Intestinal Gel (LCIG)
As an alternative delivery method for levodopa, LCIG is administered directly to the intestine through percutaneous endoscopic gastrostomy-jejunostomy (PEG-J) tube attached to an external pump.31 Both clinical trial data and real-world evidence show that 16- hour daily LCIG infusion is efficacious in improving PD-related motor symptoms.32-35 In a systematic review, 10/11 studies reported a clinically important improvement in UPDRS-rated motor symptoms following LCIG, 5/7 studies showed improvement in motor complications, and 6/6 studies reported increased quality of life after LCIG.33 Improvements in motor symptoms have been reported in clinical trials after two years of follow up and LCIG has been shown to reduce the duration of “off” time and “on” time with troublesome dyskinesia.34,35 In an observational study after one year, patients on LCIG had reduced dyskinesia-related pain and disability, and experienced significant improvements in overall nonmotor symptoms – sleep, gastrointestinal dysfunction, and mood, specifically.32 In this study, PD patients on LCIG experienced approximately 4 more hours of “on” time, during which their motor symptoms were adequately controlled compared to oral medications alone.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
Advanced therapies for motor and nonmotor fluctuations of PD in Canada currently include DBS and LCIG.13 Both therapies bypass the pharmacological limitations of oral treatments by providing predictable and continuous relief of symptoms and reduce the burden of motor and nonmotor symptoms. Despite the demonstrated benefits, approximately one quarter of advanced PD patients who are eligible decline treatment with advanced therapies, and another 40% remain undecided.4 This suggests that the current options for PD treatment are inadequate for many patients and an unmet need exists for additional DAT options.
Apart from patient’s acceptance of the treatment, limiting factors include the need for multiple specialized physicians (i.e., neurosurgeons and gastroenterologists), the invasiveness of the procedures themselves, and complications related to adverse effects and/or wound management.24,32 The decision to start advanced therapies and select the therapy best suited for each patient is complex and involves the consideration of multiple factors from the perspective of neurologists, patients, and care partners. As such, there are many patients who are poor candidates for, or are unwilling to accept, the current treatment options,4 and access to novel therapeutic mechanisms is necessary to fill this unmet need.
Accessibility: Time and Distance
Unfortunately, only very few centres in Canada offer advanced therapies for PD and many eligible patients don’t have access.
DBS centres require trained neurosurgical specialists as well as movement disorder specialized neurologists: they are therefore limited in number and usually geographically located in major cities. The numerous pre-operative assessments along with post- operative programming and follow-up medication adjustments require frequent and long-term visits to the specialized centre. Many patients are unable or unwilling to repetitively travel a long distance to access this treatment, or simply fear going into the “big city”. It’s important to keep in mind that many patients at this stage of the disease do not drive anymore and need a caregiver to take them to their many appointments.
Although LCIG is geographically more available than DBS, there are still a very limited number of centres offering this treatment in Canada, mostly related to the need for the involvement of trained gastroenterologists for PEG-J placement and digestive complication management. This leads to many good candidates not even being offered this treatment option or not being able or willing to travel to access it.
Moreover, wait times to be seen in specialized centres for possible advanced therapies are very long, up to 2-3 years in some areas. Even once a patient is on the waiting list for DBS, it can take more than a year before the surgery can actually take place. This long wait time leads to anxiety, as some patients will no longer be eligible candidates by the time they get to surgery, because of disease progression. Many patients will not even consider an advanced therapy because of the long wait time.
Eligibility
Selection criteria for DBS are very restrictive, with less than 2% of PD patients being considered good candidates.36 Common contra- indications include age greater than 70, cognitive impairment, balance and speech difficulties or inadequate postoperative support.
The need for anticoagulation or antithrombotic treatment, for example for stroke prevention or coronary artery disease, can also be a limiting factor for DBS surgery.
Medical contra-indications to LCIG are mostly limited to gastrointestinal tract diseases limiting PEG-J placement, but in order to be a good candidate, the patient and/or the caregiver need to have the ability to manipulate the pump and tubes on a daily basis and understand how to deal with common issues, such as tube blockage or leaking.
Risk of Complications
Although adverse events immediately following DBS are rare, it is an invasive brain surgery with a risk of death, cerebral hemorrhage and seizure.37 Long-term adverse events following DBS are likely underreported and include dysarthria, swallowing dysfunction, freezing of gait and balance issues despite using the parameters that are optimal for treating PD motor symptoms.37 Cognitive decline and personality disruptions can also occur following DBS, reducing patients’ acceptance of this therapy.
Unfortunately, due to the invasive nature of the PEG-J administration, adverse events are very common with LCIG, and typically relate to the device, pump, and tubing rather than the medication itself.34 Indeed, the surgical and device-related complications represent a significant barrier to patients accepting and maintaining LCIG treatment. For example, buried bumper syndrome (BBS), a potentially fatal complication caused by the migration of the internal gastrostomy bumper into the PEG tract, occurs more frequently in PD patients on LCIG compared to patients using PEG tubes for feeding.38 In one study, BBS occurred in 17.1% of patients on LCIG compared to 0.8% of non-PD patients with PEG tubes used for enteral feeding and led to LCIG discontinuation in most cases. Daily rotational movement of the tube resulting from stoma care and the activities of daily living can also cause adverse events in patients on LCIG, including the formation of bezoars, ulcers, and significant abdominal pain which may warrant discontinuation.39 In addition, peritonitis can occur in LCIG patients, despite surgical techniques used to minimize the risk of this complication.40
Acceptability
Despite being offered an advanced treatment, more than 50% of patients decline these at the time it is discussed.4 Apart from accessibility, eligibility and the risk of complications, many patients are afraid of DBS surgery because it is invasive and permanent. With regards to LCIG, it is our experience that many patients refuse it because of the embarrassment and stigma related to the PEG- J tube, along with having to carry a cumbersome pump 16 hours a day. This option also makes travelling complicated, as the medication has to be kept in the fridge until use.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Unfortunately, there is currently no treatment to address the underlying disease process in PD. Foslevodopa-foscarbidopa involves 24h subcutaneous infusion of foslevodopa-foscarbidopa, therefore representing a novel delivery system from the current oral tablet form of levodopa. The advantage of this new treatment includes the continuous 24h daily delivery along with bypassing the need for absorption through the digestive system.
The continuous 24h delivery of a short half-life levodopa is critical to effectively treat motor and nonmotor fluctuations in PD patients, preventing end-of-dose off periods, reducing dyskinesia and simplifying the medication schedule. A subcutaneous delivery prevents unpredictable medication response such as dose failures and delayed efficacy caused by absorption delays or competition with amino acids associated with protein intake. Preliminary results of the phase 3 study show an improvement of about 4 hours per day in on time without troublesome dyskinesia (“good” on time) in PD patients using a foslevodopa-foscarbidopa infusion compared to previous oral therapies. The 24h infusion is also critical to help reduce motor symptoms at night and upon awakening, preliminary results showing that morning akinesia prevalence was reduced from 77.7% to 27% with foslevodopa-foscarbidopa treatment.41 During clinical studies, about 1 in 4 patients were on foslevodopa-foscarbidopa alone at the end of the study, therefore achieving the goal of simplifying their complex medication schedule.41
The National Expert Group agreed that such a therapy would provide benefit for patients with bothersome motor and/or non-motor symptom fluctuations and would be superior to their optimized oral therapy. Experience with current advanced therapies along with data from foslevodopa-foscarbidopa phase 3 trials suggest this new treatment would be offered to PD patients with an average disease duration of 8 to 12 years, although some younger patients who tend to have earlier motor fluctuations could benefit as early as 5 to 6 years after diagnosis. Our group feels that, ideally, foslevodopa-foscarbidopa would be offered a few years earlier than we currently offer DBS and LCIG, to maintain social/work activities and quality of life for as long as possible.
Although current advanced treatments (DBS and LCIG) are effective, they are complicated to initiate and maintain, poorly accessible and need the involvement of other specialists (neurosurgeons and gastroenterologists). They are also invasive with significant risk of complications. As a result, most patients refuse these treatments when they are offered, if they are even offered at all.4
The National Expert Group strongly feels foslevodopa-foscarbidopa will be more acceptable for patients due to its reversible and non-invasive nature. The device is completely removable, therefore not affecting body image and the delivery system is small and light, making it easy to hide under the clothes and preventing the stigma associated with the disease and its treatment. Although there have been skin reactions and infections reported with foslevodopa-foscarbidopa, they are usually mild to moderate in intensity and can easily be treated by first-line physicians. Treatment initiation and maintenance with foslevodopa-foscarbidopa is foreseen to be much simpler and thus more accessible than DBS and LCIG, as there is no need for a neurosurgical or gastroenterology team nor any technical procedure, surgery or close monitoring: every step can be easily completed in an outpatient clinic setting and/or with telemedicine. We expect many centres in Canada would offer foslevodopa-foscarbidopa in areas where DBS and LCIG are not available, therefore improving quality of life of patients and caregivers in areas distant from specialized centres.
Treatment with foslevodopa-foscarbidopa should be offered when motor fluctuations are bothersome despite optimized oral therapies. It is important to note that these patients are still responsive to levodopa and would not be considered at the terminal stage of their disease. Candidates for foslevodopa-foscarbidopa will be on multiple daily doses of oral levodopa. It seems reasonable to recommend having tried at least one monoamine oxidase B inhibitor and a catechol-O-methyltransferase inhibitor, unless contraindicated. In patients who are cognitively intact, less than 70 years of age and without contra-indications, it is also reasonable to recommend having tried at least one dopamine agonist, and also amantadine if dyskinesia are bothersome. The National Expert Group recommends not requiring a previous trial of anticholinergics or apomorphine preparations prior to reimbursement of foslevodopa-foscarbidopa. Anticholinergics are usually poorly tolerated, often contra-indicated due to risk of cognitive decline, dry mouth, constipation, somnolence, and are very unlikely to significantly improve motor fluctuations, at this stage of the disease.
Although apomorphine preparations do help treat off periods in PD patients, they are typically used on an “as needed” basis for severe or unpredictable off periods and are available in specialized clinics only. We should aim at preventing off periods as opposed to treating them once they occur.
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
The National Expert Group agreed that foslevodopa-foscarbidopa would benefit levodopa-responsive PD patients with bothersome motor and/or non-motor symptom fluctuations despite optimized oral therapy. Identifying patients at this stage in PD requires expert evaluation by a neurologist using a combination of medication and symptom history. In some patients, symptom diaries or standardized physical examination using the Unified Parkinson Disease Rating Scale (part 3) will be useful, but they are time consuming and seldom necessary, as most patients can easily recognize and describe their fluctuations better than what can be captured by these tools.
Clinician judgement along with patient preference will help choose the right treatment for each patient. No specific diagnostic test or procedure is required. Although preliminary results from phase 3 trials show half the patients were able to manipulate the pump on their own, the other half needed help from a caregiver to do so, presumably due to either cognitive or dexterity issues. The presence of a caregiver will be necessary for some patients.
In 2018, a panel of movement disorder specialists developed criteria for the identification of PD patients who might be good candidates for advanced therapies.42 These “5-2-1” criteria include the presence of one or more hours of troublesome dyskinesia per day, two or more hours of “off” time per day, and having to take five or more doses of oral levodopa per day. These criteria were significantly correlated with physician judgement in a large, international cross-sectional survey, and the National Expert Group agreed that they may be useful in identifying potential candidates for advanced therapies, including foslevodopa-foscarbidopa.4,7
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
When initiating treatment with foslevodopa-foscarbidopa, a titration must be done to achieve optimal personalized symptom control. According to data from the phase 3 trial, patients’ doses were optimized in an outpatient setting after a mean of 3.5 in-person visits.
Although the clinical trial used in-person titration visits, our clinician group expects that many of the titration steps will be feasible to perform remotely. For the first week of titration, experts expect 2 to 4 contacts to be required, either in person or by phone/telehealth, followed by up to 1 contact per week for the next month to ensure patient’s confidence and optimize dosing. Pre-titration teaching for aseptic technique and manipulation of the pump and catheter could be done at the home of the patient by a trained nurse.
Response to treatment is assessed based on history and physical examination, sometimes with the help of symptom diaries. Once a patient’s dose is optimized, outpatient clinic follow up would occur approximately every 6 months depending on associated comorbidities.
A meaningful response to treatment would be a reduction in the bothersome off time and disabling dyskinesia and thus improvement in quality of life for the patient, sometimes associated with an easing of the care partner burden. Although most patients also report improvement in nonmotor symptoms, such a response is variable and should not therefore be a main goal of treatment, unless the patient has clear disabling non-motor fluctuations that also respond to dopaminergic therapy.
What factors should be considered when deciding to discontinue treatment with the drug under review?
Treatment discontinuation with foslevodopa-foscarbidopa is rarely related to a lack of benefit, but rather occurs as a result of either difficulty with manipulation of the device or side effects. In the phase 3 trials, many discontinuations were related to skin reactions. However, it seems these side effects can be reduced with better patient education and with the use of a different cannula than the one used in the first part of the trial. Data from the trial also shows a relatively high rate of hallucinations, which sometimes led to treatment discontinuation. Although disease progression certainly contributes to hallucinations, the National Expert Group strongly believes the 24 hours (as opposed to16 hours) levodopa infusion contributes to a higher incidence of hallucinations. Before deciding to stop the infusion, mitigation strategies could be implemented such as lowering the nighttime dosing or switching to a 16-hour infusion.
With disease progression, many patients will experience cognitive decline, which could lead to discontinuation of foslevodopa- foscarbidopa in some patients, especially if they don’t have a caregiver or if they tend to fidget with the pump and cannula. Patients moving to a nursing home may not be able to continue with foslevodopa-foscarbidopa if the staff is not experienced with manipulating it, therefore leading to discontinuation despite persistent benefit. Ideally this challenge could be overcome by appropriate education for the staff involved.
What settings are appropriate for treatment with [drug under review]? Is a specialist required to diagnose, treat, and monitor patients who might receive [drug under review]?
A physician with interest and experience treating PD patients is required for foslevodopa-foscarbidopa initiation and maintenance. Although such physicians will usually be movement disorder neurologists, some general neurologists or geriatric ians could be comfortable and qualified for this new treatment option. There is no special need for collaboration with other specialty physicians or any technical plateau; an outpatient clinic setting is satisfactory, although some physicians may prefer to initiate treatment in “day medicine” setting for the first 1-3 days. For the initiation period (first 4-6 weeks), a dedicated nurse will be extremely helpful to review technical manipulations with the patient and ensure proper aseptic technique and cannula manipulations, as well as educating them about when to switch flow rates or use extra doses. Once initiated, patients with foslevodopa-foscarbidopa will need standard clinic follow up visits every 6 months or so. If they occur, skin infections could be treated by an experienced neurologist or addressed by first line physicians in the community.
Additional Information
We would like to make this review even more relevant by presenting the imaginary example of a patient. Mr X is a 63 year old patient who was diagnosed with PD 7 years ago. He retired early at 61 due to concentration difficulties, fatigue and socially embarrassing dyskinesia. He likes hunting and fishing but doesn’t go anymore because he is afraid of having an off period while in the forest or on the boat and potentially falling or being unable to get back to the camp. He considers stopping bowling on Thursday nights because he feels embarrassed when others stare at him because of his dyskinesia. His wife still works full time, but she is considering early retirement because she doesn’t like leaving her husband alone at home all day. Although Mr X tries to help with daily chores, he can’t do much in the morning because he is off until 10am. He often has another severe off period between 3pm and 5pm, during which he previously suffered from severe freezing at the grocery store and could not move until a stranger came to help him. Therefore, he can only go out of the house safely between 10am and 3pm, but he needs to carry his medication at all times.
Mr X is now on oral levodopa/carbidopa every 3 hours from 7am to 10pm, along with a dopamine agonist 3 times a day, entacapone 5 times a day and amantadine twice a day. He can’t delay his medication by more than 15 minutes, or he becomes physically and cognitively very slow, feels anxious and tired and cannot walk properly; when this happens, Mr X has to wait 45 minutes after the next levodopa dose to get better and functional again. He has to eat at least 1 hour after or before his medications to ensure proper benefit from them. Because of his PD, he hates having to go to a restaurant in the evening but does it for his wife when she insists.
Although Mr X has heard about DBS surgery, the closest DBS centre is 3 hours away from his home. Unfortunately, he is unable to drive safely for this long. He doesn’t want to impose many appointments to his wife who already misses work too often to help him. He is also afraid of potential complications following the surgery, especially cognitive decline and personality changes. His neurologist offered LCIG, but Mr X is not ready to have a tube through his stomach and expects the pump to be too cumbersome to allow him to go bowling and hunting.
Mr X would be an excellent candidate for foslevodopa-foscarbidopa. After an initial titration phase with a combination of 3-4 in person and phone/telehealth visits, he could see his off time reduced from 5 hours a day to 1 hour a day. He could be functional sooner after awakening and would be unlikely to suffer severe off periods preventing him to go fishing, bowling or running errands. He would likely suffer less from social embarrassment from motor fluctuations and would not avoid social situations as much, potentially leading to a better quality of life for himself and his wife.
References
Public Health Agency of Canada. Mapping Connections: An Understanding of Neurological Conditions in Canada. 2014
Poewe W, Seppi K, Tanner CM, et al. Parkinson disease. Nat Rev Dis Primers. 2017;3:17013.
Armstrong MJ, Okun MS. Diagnosis and Treatment of Parkinson Disease: A Review. JAMA. 2020;323(6):548-560.
Fasano A, C. FVS, Lopiano L, et al. Characterizing advanced Parkinson's disease: OBSERVE-PD observational study results of 2615 patients. BMC Neurology. 2019;19(1):50.
Public Health Agency of Canada. Parkinsonism in Canada, Including Parkinson’s Disease: Highlights from the Canadian chronic disease surveillance system. 2017.
Wong JJ, Kwong JC, Tu K, et al. Time Trends of the Incidence, Prevalence, and Mortality of Parkinsonism. Can J Neurol Sci. 2019;46(2):184-191.
Statistics Canada. Table 13-10-0849-01 Chronic conditions among seniors aged 65 and older, Canadian Health Survey on Seniors, two-year period estimates. Published 2022. Updated April 19, 2022. Accessed August 10, 2022.
Parkinson Canada. Written Submission for the Pre-Budged Consultations in Advance of the 2020 Budget. 2020.
GBD 2016 Parkinson's Disease Collaborators. Global, regional, and national burden of Parkinson's disease, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018;17(11):939-953.
Information CIfH. The Burden of Neurological Diseases, Disorders and Injuries in Canada. Ottawa: CIHI;2007.
Parkinson’s Society Canada. Parkinson’s Disease: Social and Economic Impact. 2003.
Wolfson C, Fereshtehnejad SM, Pasquet R, Postuma R, Keezer MR. High burden of neurological disease in the older general population: results from the Canadian Longitudinal Study on Aging. Eur J Neurol. 2019;26(2):356-362.
Grimes D, Fitzpatrick M, Gordon J, et al. Canadian guideline for Parkinson disease. CMAJ. 2019;191(36):E989-E1004.
Chou KL, Stacy M, Simuni T, et al. The spectrum of "off" in Parkinson's disease: What have we learned over 40 years? Parkinsonism Relat Disord. 2018;51:9-16.
Ahlskog JE, Muenter MD. Frequency of levodopa-related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov Disord. 2001;16(3):448-458.
DeMaagd G, Philip A. Parkinson's Disease and Its Management: Part 4: Treatment of Motor Complications. P T. 2015;40(11):747-773.
Schapira AH, Emre M, Jenner P, Poewe W. Levodopa in the treatment of Parkinson's disease. Eur J Neurol. 2009;16(9):982-989.
Borovac JA. Side effects of a dopamine agonist therapy for Parkinson's disease: a mini-review of clinical pharmacology. Yale J Biol Med. 2016;89(1):37-47.
Fox SH, Katzenschlager R, Lim SY, et al. International Parkinson and movement disorder society evidence-based medicine review: Update on treatments for the motor symptoms of Parkinson's disease. Mov Disord. 2018;33(8):1248-1266.
Jankovic J. Motor fluctuations and dyskinesias in Parkinson's disease: clinical manifestations. Mov Disord. 2005;20 Suppl 11:S11-S16.
McLeod Macey J. People with Parkinson’s face gaps in the availability of health services. https://www.ipsos.com/en- ca/news-polls/parkinson-canada-stakeholder-survey-2018. Published 2018. Accessed August 10, 2022.
Klietz M, von Eichel H, Schnur T, et al. One Year Trajectory of Caregiver Burden in Parkinson's Disease and Analysis of Gender-Specific Aspects. Brain Sci. 2021;11(3).
Wong SL, Gilmour H, Ramage-Morin PL. Parkinson’s disease: Prevalence, diagnosis and impact. Statistics Canada;2014.
Karlstedt M, Fereshtehnejad SM, Aarsland D, Lokk J. Mediating effect of mutuality on caregiver burden in Parkinson's disease partners. Aging Ment Health. 2020;24(9):1421-1428.
Tan QY, Cox NJ, Lim SER, et al. The Experiences of Treatment Burden in People with Parkinson's Disease and Their Caregivers: A Systematic Review of Qualitative Studies. J Parkinsons Dis. 2021;11(4):1597-1617.
Hulshoff MJ, Book E, Dahodwala N, Tanner CM, Robertson C, Marras C. Current Knowledge on the Evolution of Care Partner Burden, Needs, and Coping in Parkinson's Disease. Mov Disord Clin Pract. 2021;8(4):510-520.
Modugno N, Antonini A, Tessitore A, et al. Impact of Supporting People with Advanced Parkinson's Disease on Carer's Quality of Life and Burden. Neuropsychiatr Dis Treat. 2020;16:2899-2912.
Soileau MJ, Pagan F, Fasano A, et al. Comparative Effectiveness of Carbidopa-Levodopa Enteral Suspension and Deep Brain Stimulation on Parkinson's Disease-Related Pill Burden Reduction in Advanced Parkinson's Disease: A Retrospective Real-World Cohort Study. Neurol Ther. 2022;11(2):851-861.
Metta V, Batzu L, Leta V, et al. Parkinson's Disease: Personalized Pathway of Care for Device-Aided Therapies (DAT) and the Role of Continuous Objective Monitoring (COM) Using Wearable Sensors. J Pers Med. 2021;11(7):680.
Bratsos S, Karponis D, Saleh SN. Efficacy and Safety of Deep Brain Stimulation in the Treatment of Parkinson's Disease: A Systematic Review and Meta-analysis of Randomized Controlled Trials. Cureus. 2018;10(10):e3474.
DUODOPA® (levodopa/carbidopa intestinal gel) Product Monograph. In. Saint-Laurent, QC: AbbVie Corporation; April 2022.
Standaert DG, Aldred J, Anca-Herschkovitsch M, et al. DUOGLOBE: One-Year Outcomes in a Real-World Study of Levodopa Carbidopa Intestinal Gel for Parkinson's Disease. Mov Disord Clin Pract. 2021;8(7):1061-1074.
Nyholm D. Duodopa(R) treatment for advanced Parkinson's disease: a review of efficacy and safety. Parkinsonism Relat Disord. 2012;18(8):916-929.
Wirdefeldt K, Odin P, Nyholm D. Levodopa-Carbidopa Intestinal Gel in Patients with Parkinson's Disease: A Systematic Review. CNS Drugs. 2016;30(5):381-404.
Antonini A, Mancini F, Canesi M, et al. Duodenal levodopa infusion improves quality of life in advanced Parkinson's disease. Neurodegener Dis. 2008;5(3-4):244-246.
Morgante L, Morgante F, Moro E, et al. How many parkinsonian patients are suitable candidates for deep brain stimulation of subthalamic nucleus? Results of a questionnaire. Parkinsonism Relat Disord. 2007;13(8):528-531.
Limousin P, Foltynie T. Long-term outcomes of deep brain stimulation in Parkinson disease. Nat Rev Neurol. 2019;15(4):234-242.
Spanaki C, Boura I, Avgoustaki A, et al. Buried Bumper Syndrome: A common complication of levodopa intestinal infusion for Parkinson disease. Parkinsonism Relat Disord. 2021;85:59-62.
Marano M, Pizzicannella M, di Biase L, et al. Jejunal pulling syndrome: A peculiar LCIG complication. Parkinsonism Relat Disord. 2018;52:113-114.
Tsuboi T, Watanabe H, Funasaka K, Takebayashi M, Miyata K, Katsuno M. Peritonitis after percutaneous endoscopic gastrojejunostomy for levodopa–carbidopa intestinal gel treatment despite concomitant use of gastropexy. Neurology and Clinical Neuroscience. 2018;6(2):64-66.
Aldred J, Amelin A, Antonini A, Bergmans B, Bergquist F, Bouchard M, Budur K, Carroll C, Chaudhuri KR, Criswell S, Danielsen EH, Freire Alvarez E, Gandor F, Jia J, Kimber T, Mochizuki H, Robieson WZ, Spiegel AM, Standaert DG, Talapala S, Facheris MF, Fung VSC. Continuous Subcutaneous Foslevodopa/Foscarbidopa in Advanced Parkinson’s Disease: Results From a 12-Month Phase 3 Study. Presented at: European Academy of Neurology 2022 Annual Meeting; June 25−28, Vienna, Austria
Antonini A, Stoessl AJ, Kleinman LS, et al. Developing consensus among movement disorder specialists on clinical indicators for identification and management of advanced Parkinson's disease: a multi-country Delphi-panel approach. Curr Med Res Opin. 2018;34(12):2063-2073.
Conflict of Interest Declarations — National Movement Disorder Expert Group
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation.
Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please refer to the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission?
No.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission?
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
None.
Declaration for Clinician 1
Name: Julius Anang
Position: Consultant (neurologist)
Date: 21-09-2022
Table 5: COI Declaration for National Movement Disorder Expert Group — Clinician 1
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie Canada | — | X | — | — |
Declaration for Clinician 2
Name: Manon Bouchard
Position: Movement disorders specialist and neurologist, Clinique Neuro-Lévis and Hôtel-Dieu de Lévis
Date: Septembre 26th 2022
Table 6: COI Declaration for National Movement Disorder Expert Group — Clinician 2
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Abbviea | — | — | X | — |
Abbvieb | — | X | — | — |
aPayments related to participation in phase 3 clinical trial.
bPayments related to advisory boards and speaker honorarium.
Declaration for Clinician 3
Name: Barbara Connolly
Position: Associate Professor, McMaster University
Date: 22-09/2022
Table 7: COI Declaration for National Movement Disorder Expert Group — Clinician 3
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Abbvie | — | X | — | — |
Declaration for Clinician 4
Name: Douglas Everett Hobson
Position: Movement Disorder Neurologist and co-direct Movement Disorder Program University of Manitoba
Date: 01/11/2022
Table 8: COI Declaration for National Movement Disorder Expert Group — Clinician 4
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 5
Name: Galit Kleiner MD FrC FC
Position: Staff Physician, Baycrest Movement Disorders Clinic, Baycrest Health Sciences, Toronto, ON, Assistant Professor, Dept of Medicine, Division of Neurology, University of Toronto
Date: November 13, 2022
Table 9: COI Declaration for National Movement Disorder Expert Group — Clinician 5
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie | X | — | — | — |
Declaration for Clinician 6
Name: Anthony Lang
Position: Director, Edmond J. Safra Program in Parkinson’s Disease, Toronto Western Hospital
Date: 17-10-2022
Table 10: COI Declaration for National Movement Disorder Expert Group — Clinician 6
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie | X | — | — | — |
BioAdvance | X | — | — | — |
Biogen | — | — | X | — |
BlueRock | X | — | — | — |
Denali | X | — | — | — |
Janssen | X | — | — | — |
Paladin | — | X | — | — |
Roche | X | — | — | — |
Sun Pharma | X | — | — | — |
Sunovion | X | — | — | — |
Declaration for Clinician 7
Name: Tiago Mestre
Position: Scientist, Neuroscience Program, Ottawa Hospital Research Institute; Neurologist, Neurology, Medicine, The Ottawa Hospital; Co-Director, Deep Brain Stimulation Program (Movement Disorders), uOttawa Brain and Mind Research Institute
Date: 15-11-2022
Table 11: COI Declaration for National Movement Disorder Expert Group — Clinician 7
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie | X | — | — | — |
CHDI | X | — | — | — |
Sunovion | X | — | — | — |
Biogen | X | — | — | — |
Medtronic | X | — | — | — |
nQ Medical | X | — | — | — |
Valeo Pharma | X | — | — | — |
Int’l Parkinson and Movement Disorder Society | X | — | — | — |
Declaration for Clinician 8
Name: Alexander Hussain Rajput
Position: Professor, Division of Neurology, University of Saskatchewan
Date: 15-NOV-2022
Table 12: COI Declaration for National Movement Disorder Expert Group — Clinician 8
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 9
Name: Dr. Kerrie Schoffer
Position: Neurologist, QEII Health Sciences Centre, Halifax, NS; Assistant Professor, Dalhousie University
Date: 30 September 2022
Table 13: COI Declaration for National Movement Disorder Expert Group — Clinician 9
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Abbvie – Principle Investigator for M21-471 Trial (Essential Tremor) | X | — | — | — |
Abbvie Market Access Meeting | X | — | — | — |
Declaration for Clinician 10
Name: Oksana Suchowersky
Position: Professor, University of Alberta
Date: 27-Oct-2022
Table 14: COI Declaration for National Movement Disorder Expert Group — Clinician 10
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie | X | — | — | — |
Declaration for Clinician 11
Name: Daryl Wile
Position: Clinical Investigator, UBC Faculty of Medicine Centre for Chronic Disease Prevention and Management, Southern Medical Program ; Clinical Assistant Professor, UBC Department of Medicine, Division of Neurology
Date: 14-11-2022
Table 15: COI Declaration for National Movement Disorder Expert Group — Clinician 11
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Paladin Labs | X | — | — | — |
Clinician Input
BC Movement Disorders Specialist Group
About the BC Movement Disorders Specialist Group
We are a group of movement disorders specialists in British Columbia (BC Movement Disorders Specialist Group) with expertise in the management of Parkinson disease (PD) and are making this submission to advocate on the behalf of patients with Parkinson disease living in British Columbia, to strongly support this review of foslevodopa foscarbidopa.
Information Gathering
This document contains a summary of the BC Movement Disorders Specialist Group and National Expert Group’s conclusions (a national Canadian group of movement disorders specialists, with representation from British Columbia, who met in July 2022 to discuss the unmet needs of patients with advanced PD), as well as supporting data on the burden of PD and the need for additional advanced treatment options. The results of Phase 3 trials using foslevodopa foscarbidopa will be briefly reviewed to discuss efficacy and safety of the product. The contents of this document were reviewed and approved by the clinicians involved in this submission.
Current Treatments and Treatment Goals
Introduction
Parkinson disease (PD) is the second most common neurodegenerative disorder in the world with a prevalence that is expected to double within the next 10 years.1 The disease is characterized by motor features that have a major impact on mobility, including bradykinesia, rest tremor, and rigidity, alongside other non-motor features like autonomic dysfunction, cognitive decline, and gastrointestinal impairment which become increasingly burdensome with time.2 To date, no therapies have been shown to delay or reverse the progression of PD, and dopamine replacement therapy is the current standard of care in reducing the burden of motor symptoms.3 Oral levodopa is the first-line treatment for PD. However, as disease progresses, the continued loss of endogenous dopaminergic neurons leads to prolonged periods of poor symptom control and dyskinesia (uncontrolled excess movements), which requires frequent dosing intervals (for example, requiring medication dosing every 2 hours or less), alongside the addition of other pharmacological treatments by subspecialist neurologists. In spite of this, ultimately PD often progresses to a point that oral medications do not provide adequate control of fluctuating motor and non-motor symptoms, and up to 32% of Canadian patients are no longer able to perform the activities of daily living despite having optimized oral therapeutic regimens.4
Impact on Society
The prevalence of PD increased by more than 20% between 1996 and 2012, and PD affects over 13,300 British Columbians among the more than 84,000 Canadians affected.8-10 Moreover, the prevalence is expected to double by 2031, making PD the most prevalent neurodegenerative movement disorder and the fastest growing neurological disease in the world.1,11 While this increase can be partially attributed to an aging population, epidemiological models suggest that PD rates are increasing beyond what would be expected by age alone, which suggests that multiple factors will likely continue to contribute to the increasing burden of PD in Canada and around the world.12 Age is a significant risk factor for developing PD, with the incidence and prevalence increasing sharply after 60 years.2,8
PD has a multifactorial impact on the health and lives of patients. Overall, PD accounts for 1.1% of disability-adjusted life years (DALYs) in Canada, primarily due to healthy life lost due to disability, which patients typically begin to experience 10 to 15 years after being diagnosed.13,14 In fact, over 70% of the direct costs of PD in Canada can be attributed to long-term disability.14
In 2012-2013, the British Columbia Ministry of Health estimated that $112 million was spent on direct care related to PD including hospital, Medical Services Plan (MSP), and Pharmacare costs. The estimated cost in 2000-2001 was $45 million.14,15 PD patients with advanced disease who experience symptom fluctuations place a substantial unmeasured burden on the health care system in terms of frequent primary and specialist outpatient and inpatient care to assess and address their needs.
Current Oral Treatments and Limitations
No cure currently exists for PD, nor are there treatments that significantly impact disease progression. As such, pharmacological and behavioural interventions are used to manage the clinical manifestations of PD rather than modifying the disease itself. Once diagnosed, the standard of care to treat motor symptoms is to prescribe dopamine replacement therapy, particularly oral levodopa.2,3,5 Both immediate- and extended-release formulations of oral levodopa are effective in reducing motor symptoms and have acceptable safety profiles.
In earlier phases of the disease, most patients have satisfactory relief of their motor symptoms with oral levodopa taken three to four times a day. However, as dopaminergic neuronal loss progresses over time, most patients experience symptom fluctuations, related to the short half-life of levodopa along with complex and poorly understood synaptic brain changes and inadequate intestinal absorption of the medication. Beyond five years into the disease, about 50% of patients experience end-of-dose “wearing off” or “off” periods, where there is a return of motor and/or non-motor symptoms, including but not limited to stiffness, poor mobility, tremor, speech changes, bladder urgency, anxiety, cognitive slowing, pain due to lack of medication efficacy. Repeat medication dosing typically does not provide relief for another 30-60 minutes, such that in spite of regular medication doses patients are experiencing disabling symptoms for at least 45 minutes between dosing intervals, occurring at least three to four times a day. As the disease progresses, 90% of patients will also experience dyskinesia, axial or appendicular involuntary choreiform movements that can occur during “on” periods when medication is otherwise effective (also called peak dose dyskinesia), but also during “off” periods.29,30 Increasingly long durations of “off” periods and dyskinesia lead to more frequent dosing of levodopa and complex medication schedules, some patients needing medication every 2 hours or less.
Due to the progressive nature of PD, individualized and frequent tailoring of medication classes, formulations, and dosages becomes necessary to reduce off time and treat motor fluctuations and dyskinesia. Dopamine agonists, such as pramipexole, ropinirole and rotigotine are often used as adjuncts to levodopa to reduce off periods, but they carry a higher risk of adverse events including impulse control disorders, excessive sleepiness, cognitive dysfunction, and hallucinations, limiting their use.49 Monoamine oxidase B inhibitors (i.e., rasagiline) and catechol-O-methyltransferase inhibitors (i.e., entacapone) can be used alongside levodopa to reduce off time in patients with fluctuating PD, but their efficacy is reduced with disease duration and there is potential risk of increasing dyskinesia.5,48 Amantadine is often added for the treatment of dyskinesia, but hallucinations and cognitive side effects severely limits its use particularly in older or cognitively impaired patients.
Despite optimized oral therapies, many patients still experience disabling recurrent “off” periods. To date, approximately 50% of PD patients can be classified as having refractory motor fluctuations.4,51 In 2014, an online study with over 3000 PD patients was completed by the Michael J. Fox Foundation and revealed that 64% of patients spent more than 2 waking hours daily in an “off” state, 50% of each off period lasting 45 minutes or longer. As noted above, although “off” periods are often partially predictable, related to levodopa dosing times. However, particularly with longer disease duration, “off” periods can also happen unpredictably due to dose failures, leading to great anxiety and disability. Such “off” periods can also occur due to poor or delayed levodopa intestinal absorption, often but not always in relation to mealtimes due to impact of protein in food on absorption, but also due to systemic effects of PD on the gastrointestinal system such as delayed gastric emptying. These factors have major impact on patients and their families, leading them to reduce or avoid social activities, organizing their daily activities around “off” periods and medication schedules, and even limiting food intake.
Treatment Goals
Over time, many PD patients require increased dosing frequency of oral levodopa, up to ten or more times a day (every two hours or less), but still experience burdensome “off” periods and dyskinesia due to the short half-life of the drug.3 As noted above, although the addition of other pharmacological agents including dopamine agonists, monoamine oxidase B inhibitors, catechol-O-methyltransferase inhibitors and amantadine can help reduce motor fluctuations, these agents are often insufficient, poorly tolerated,or contraindicated. Moreover, the medication schedule resulting from complex and frequent dosing further severely impairs quality of life.
In these patients, a continuous dopaminergic stimulation is required to achieve adequate relief of their fluctuating motor and non-motor symptoms. Bypassing the erratic absorption in the digestive tract is also helpful in providing a more predictable response to the treatment.
The Burden of PD
The lives of PD patients are restricted due to their motor and non-motor symptoms, and many are unable to work due to reduced function brought during “off” periods or dyskinesia, in addition to embarrassment and/or stigma of their condition. A national survey found that between 20% to 50% of PD patients require mechanical support for mobility, and fewer than 20% of PD patients between the ages of 18 to 64 are able to work.1 Moreover, more than 50% of PD patients experience stress, anxiety, and a loss of confidence. As PD progresses and symptom fluctuations become more frequent, patients become highly reliant on their frequently dosed oral medication regimen, such that their activities of daily living become tied to and controlled by their medication schedule, and they start to avoid situations where they may experience an “off” period in public.16 Informal care partners, most often the spouses of PD patients, are also negatively impacted by the burden of PD across multiple domains including physical, social, and occupational aspects of life.20,21 In addition to stress, exhaustion, and guilt, most care partners report feeling anxious, helpless, or frustrated with the person with PD. In a mediation analysis of 51 dyads of PD patients and their spouses/care partners, one study found that the severity of the patient’s motor symptoms significantly increases care partner burden through deterioration of the spousal relationship.22 Furthermore, the treatment-specific burden put on care partners (i.e., adhering to frequent and sometimes complicated medication regimens, keeping track of appointments, and burdens associated with travel and the logistics of navigating the allied health environment) can lead to physical and mental exhaustion.23 Care partners also experience significant financial burden as they pay for PD-related services and are less able to work due to the time commitment required to care for the person living with PD. Additionally, the health of care partners can rapidly decline as the effects of chronic stress are compounded by a perceived and existent inability to focus on their own needs. This notion is supported by research which indicates that care partners are at risk of accumulating increased financial, mental, and physical burdens over time.24 Importantly, the use of device-aided therapies (DATs) for advanced PD symptoms has been shown to significantly reduce caregiver burden by allowing them more time to engage in family, household, and leisure activities.25
Advanced Treatments Currently Available
Advanced therapies, often referred to as device-aided therapies (DATs), are used to circumvent the pharmacological limitations of oral therapies and have been shown to significantly reduce oral medication or pill burden for PD patients. DATs have been shown to significantly reduce impairment in the activities of daily living, impairment due to motor symptoms, disability related to dyskinesia, and disability related to non-motor symptoms for advanced PD patients.4 To date, there are two DATs approved for the treatment of motor symptoms in fluctuating PD in Canada – deep brain stimulation (DBS) and levodopa/carbidopa intestinal gel (LCIG) infusion.5,48 Although standardized guidelines for the use of one DAT over the other may not be possible, the BC Movement Disorders Specialists Group in addition to the National Expert Group agree that we would ideally consider several factors, including patient age, degree of functional impairment, cognitive status, and patient preference when determining which patients would be best suited for each DAT.
Deep Brain Stimulation
DBS involves the neurosurgical implantation of wires, trans-cranially, into the subthalamic nucleus or globus pallidus interna.3 A meta-analysis of randomized control trials found that DBS offers a significant advantage against oral therapies alone in terms of UPDRS-measured “on/off” time, dyskinesia, and quality of life.6
Levodopa/Carbidopa Intestinal Gel
As an alternative delivery method for levodopa, LCIG is administered directly to the intestine through a percutaneous endoscopic gastrostomy-jejunostomy (PEG-J) tube using an external pump.56 Both clinical trial data and real-world evidence show that 16-hour daily LCIG infusion is efficacious in improving PD-related motor symptoms.7,57-59 In a systematic review, 10/11 studies reported a clinically important improvement in UPDRS-rated motor symptoms following LCIG, 5/7 studies showed improvement in motor complications, and 6/6 studies reported increased quality of life after LCIG.57 Improvements in motor symptoms have been reported in clinical trials after two years of follow up and LCIG has been shown to reduce the duration of “off” time and “on” time with troublesome dyskinesia.58,59 In an observational study after one year, patients on LCIG had reduced dyskinesia-related pain and disability, and experienced significant improvements in overall nonmotor symptoms – sleep, gastrointestinal dysfunction, and mood, specifically.7 In this study, PD patients experienced approximately 4 more hours of “on” time, when their motor symptoms were adequately controlled, while on LCIG, than they experienced on oral medications alone prior to LCIG initiation.7
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
Advanced therapies for motor and non-motor fluctuations of PD in Canada currently include DBS and LCIG.5 Both therapies bypass the pharmacological limitations of oral treatments by providing predictable and continuous relief of symptoms and reduce the burden of fluctuating symptoms, motor or non-motor. Despite the demonstrated benefits, approximately one quarter of advanced PD patients who are eligible decline treatment with advanced therapies, and another 40% remain undecided. This suggests that the current options for PD treatment are inadequate for many patients and an unmet need exists for additional DAT options.
Apart from patient’s acceptance of the treatment, limiting factors include the need for multiple specialized physicians (i.e., neurosurgeons and gastroenterologists), the invasiveness of the procedures themselves, and complications from adverse effects from the presence of indwelling hardware (i.e., brain electrodes and pacemaker in DBS or PEG-J tube in LCIG), and/or wound management.6,7 The decision to start advanced therapies and select the therapy best suited for each patient is complex and involves the consideration of multiple factors from the perspective of neurologists, allied health care professionals, patients, and care partners. As such, there are many patients who are poor candidates for or are unwilling to accept the current treatment options, and access to novel therapeutic mechanisms is necessary to fill this unmet need.
Accessibility: Geographical Inequities and Waitlist Times
Centres offering advanced therapies in BC are located in urban areas which practically limits access for patients living further from metropolitan areas, particularly in winter months due to impact on travel to specialized centres.
DBS centres require trained neurosurgical specialists as well as movement disorder specialized neurologists: they are therefore limited in number and usually geographically located in the major cities. Currently all DBS procedures are limited in BC within the Lower Mainland. The numerous pre-operative assessments along with post-operative programming and follow-up medication adjustments require frequent and long-term visits to the specialized centre. Many patients, especially when mobility is impacted or when experiencing frequent “off” periods, are unable to repetitively travel long distances to access this treatment, particularly in winter months. It is important to keep in mind that many patients at this stage of the disease are no longer driving and are reliant on a caregiver to take them to their many appointments. Furthermore, in BC, the waitlist for access to DBS treatment is excessively long at greater than 40 months.
Although LCIG is geographically slightly more available than DBS in BC, there are still a very limited number of centres offering this treatment, mostly related to the need for the involvement of a multidisciplinary movement disorders clinic in addition to trained gastroenterologists for PEG-J placement and management of gastrointestinal complications. This again limits the number of otherwise suitable candidates being offered or taking up this treatment option.
Eligibility
Selection criteria for DBS are very restrictive, with less than 2% of PD patients being considered good candidates.66 Common contra-indications include age greater than 70, cognitive impairment, psychiatric symptoms, falls, balance, and speech difficulties.
Medical contra-indications to LCIG are mostly limited to gastrointestinal tract diseases limiting PEG-J placement, but in order to be a suitable candidate, the patient and/or the caregiver need to have the ability to perform daily stoma care and to effectively manipulate the pump and tubes on a daily basis, in addition to understanding how to deal with common issues, such as stoma irritation or hypergranulation, tube leakage, or tube blockage.
Risk of Complications
Although adverse events immediately following DBS are rare, it is an invasive brain surgery with a risk of death, cerebral hemorrhage and seizure.55 Long-term adverse events following DBS are likely underreported and include dysarthria, swallowing dysfunction, freezing of gait significantly increasing falls risk and balance issues.55 Cognitive decline and personality disruptions can also occur following DBS, reducing patients’ acceptance of this therapy.
Unfortunately, due to the invasive nature of the PEG-J administration, adverse events are common with LCIG, and typically relate to the device, pump, and tubing rather than the medication itself.58 Indeed, the surgical and device-related complications represent a significant barrier to patients accepting and maintaining LCIG treatment. For example, buried bumper syndrome (BBS), which is a potentially fatal complication caused by the migration of the internal gastrostomy bumper into the PEG tract, occurs more frequently in PD patients on LCIG compared to patients using PEG tubes for feeding.60 In one study, BBS occurred in 17.1% of patients on LCIG compared to 0.8% of non-PD patients with PEG tubes used for enteral feeding and led to LCIG discontinuation in most cases. Daily rotational movement of the tube resulting from stoma care and the activities of daily living can also cause adverse events in patients on LCIG, including the formation of bezoars, ulcers, and significant abdominal pain which may warrant discontinuation.61 In addition, peritonitis can occur in LCIG patients, even when surgical techniques are used to minimize the risk of this complication.62
Acceptability
Despite being offered an advanced treatment, more than 50% of patients decline it at the time it is discussed. Apart from accessibility, eligibility and the risk of complications, many patients are afraid of DBS surgery because it is invasive and permanent. With regards to LCIG, it is our experience that many patients, particularly those who are younger or still maintain any social life, refuse it because of the embarrassment and stigma related to the PEG-J tube, along with having to carry a cumbersome pump during their entire waking hours on a daily basis. This option also makes travelling complicated, as the medication has to be refrigerated at all times prior to use.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
There is currently no treatment addressing the underlying disease process in PD. Foslevodopa foscarbidopa involves 24-hour subcutaneous infusion of foslevodopa/foscarbidopa, therefore representing a novel delivery system for an existing drug, namely levodopa. The advantages of this new treatment include the continuous 24-hour delivery of levodopa, along with bypassing the digestive system for improved efficacy.
The continuous 24-hour delivery of levodopa, due to its short half-life, is critical to effectively treat advanced PD where frequent motor and non-motor fluctuations occur, preventing “off” periods, reducing dyskinesia, simplifying frequent and complex medication schedules, and reducing pill burden for patients. A subcutaneous delivery combats unpredictable medication responses which cause either delayed medication efficacy or complete dose failures, particularly in relation to protein intake at mealtimes or delayed gastric emptying due to systemic effects of PD. Preliminary results of the Phase 3 study show an improvement of 4 hours per day in achieving good “on” time without troublesome dyskinesia in PD patients using a foslevodopa foscarbidopa infusion compared to previous oral therapies. The 24-hour infusion is also critical to help reduce motor symptoms at night and upon awakening in the morning, preliminary results showing that morning akinesia was reduced from 77,7% to 27% with foslevodopa foscarbidopa treatment.67 During clinical studies, about 1 in 4 patient was on foslevodopa foscarbidopa monotherapy at the end of the study, therefore achieving the goal of simplifying a complex medication schedule.
The BC Movement Disorders Specialist Group in addition to the National Expert Group agrees that such a therapy would benefit patients who experience bothersome motor and/or non-motor symptom fluctuations despite optimized oral therapy. Experience with current advanced therapies along with data from ABBV-951 Phase 3 trials suggest this new treatment would be offered to PD patients with an average disease duration of 8 to 12 years, although some patients could benefit at even 5 to 6 years into the disease. Particularly due to its less invasive nature, it would be ideal to offer foslevodopa foscarbidopa a few years sooner than we do for DBS and LCIG, aiming at maintaining social, and occupational activities and quality of life before these significantly decline.
Although current advanced treatments (DBS and LCIG) are effective, as noted above they are poorly accessible due to long wait times and geographical limitations, along with the need of involvement of other specialists (neurosurgeons and gastroenterologists). They are also invasive with significant risk of complications. As a result, many patients refuse these treatments when they are offered, if they are even offered.
The BC Movement Disorders Specialist Group in addition to the National Expert Group strongly feels foslevodopa foscarbidopa will be more acceptable for patients due do its reversible and non-invasive nature. The device is completely removable, therefore less prone to the significant complications related to the delivery route in both DBS and LCIG. Additionally, because of its removable and non-invasive nature and a delivery system that is small and light, it is easy to hide beneath clothing, thereby making it much less impactful on body image and social stigma associated with PD. Although there have been skin reactions and infections reported with foslevodopa foscarbidopa, they are usually mild to moderate in intensity and can easily be treated by a primary care physician. Treatment initiation and maintenance with foslevodopa foscarbidopa is therefore reasonably expected to be more accessible than DBS and LCIG, as the need for other specialist teams including neurosurgery or gastroenterology. There is no technical bottleneck expected to limit access to foslevodopa foscarbidopa as seen in DBS and LCIG -- every step can be easily completed in an outpatient clinic setting and/or with telemedicine. This would allow patients with advanced PD in BC who otherwise do not have practical access to DBS or LCIG to have access to another advanced therapy, namely foslevodopa foscarbidopa. This would significantly improve disease burden and quality of life of patients and caregivers in areas distant from specialized centres, as well as the burden of care due to uncontrolled symptoms on their local healthcare resources.
Treatment with foslevodopa foscarbidopa should be offered when motor and non-motor fluctuations are bothersome despite optimized oral therapies. This is similar to when other existing advanced therapies (DBS and LCIG) are offered. It is important to note that these patients are still responsive to levodopa and would not be considered at the terminal stage of their disease. Candidates for foslevodopa foscarbidopa will be on multiple daily doses of oral levodopa. It seems reasonable to recommend having tried at least one monoamine oxidase B inhibitor and catechol-O-methyltransferase inhibitor. In patients who are cognitively intact, less than 70 years of age, and do not have a prior psychiatric history concerning for the risk of impulse control disorder, it is also reasonable to recommend having tried at least one dopamine agonist. Amantadine should only be considered in specific patients where disabling dyskinesia (rather than “off” periods) are the main concern, with the additional caveats due to poor tolerability and risk of cognitive and psychiatric effects of the drug, particularly in older patients: (1) those who are cognitively intact, (2) less than 70 years of age, and (3) without other contra-indications. The BC Movement Disorders Specialist Group, in addition to the National Expert Group, recommends not to require previous trial of anticholinergics or apomorphine preparations for reimbursement of foslevodopa foscarbidopa. Anticholinergics are usually poorly tolerated, often contra-indicated at this stage due to cognitive decline, dry mouth, constipation, somnolence, and are very unlikely to significantly improve motor fluctuations at this stage of the disease. Although apomorphine preparations do help treat specific types of “off” periods in PD patients, they are typically used on an “as needed” basis for severe or unpredictable off periods, whereas standing therapy should aim at preventing “off” periods as opposed to treating them once they occur.
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
The BC Movement Disorders Specialist Group, in addition to the National Expert Group, agrees that foslevodopa foscarbidopa would benefit levodopa-responsive PD patients with bothersome motor and/or non-motor symptom fluctuations despite optimized oral therapy. Identifying patients at this stage in PD requires expert evaluation with a neurologist including a combination of medication and symptom history, symptom diaries, and occasionally in person observation of a patient during transitions between “on” and “off” states. Expert clinician judgement along with patient preference will help choose the right treatment for each patient. No specific diagnostic test or procedure is required. Although preliminary results from Phase 3 trials show half the patients were able to manipulate the pump on their own, the other half needed help from a caregiver to do so, presumably due to either cognitive or dexterity issues. The presence of a caregiver could therefore be necessary for some PD patients.
In 2018, a panel of movement disorder specialists developed criteria for the identification of PD patients who might be good candidates for advanced therapies.52 These “5-2-1” criteria include the presence of one or more hours of troublesome dyskinesia per day, two or more hours of “off” time per day, and having to take five or more doses of oral levodopa per day. These criteria were significantly correlated with physician judgement in a large, international cross-sectional survey, and the BC Movement Disorders Specialist Group, in addition to the National Expert Group, agrees that they may be useful in identifying potential candidates for advanced therapies.4, 10
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
When initiating treatment with foslevodopa foscarbidopa, a titration must be done to achieve optimal personalized symptom control. For the first week, experts expect 2 to 4 assessments to be required, either in person or by phone/telehealth, followed by up to 1 visit every 1-2 weeks for the next month to ensure patient’s confidence and optimize dosing. According to data from the Phase 3 trial, most patients were optimized within 3.5 visits. Response to treatment is assessed based on history and physical examination, sometimes with the help of symptom diaries. Once a patient’s dose is optimized, outpatient clinic follow up would occur every 6 months, as typical for most patients with PD, including those not on any advanced therapies.
A meaningful response to treatment would be a reduction in fluctuating bothersome dyskinesia and/or “off” time.
What factors should be considered when deciding to discontinue treatment with the drug under review?
Treatment discontinuation with foslevodopa foscarbidopa is rarely related to a lack of benefit, but rather derives from either difficulty with manipulation of the device or side effects. In the Phase 3 trials, many discontinuations were related to skin reactions. However, it is likely that these side effects can be reduced with better patient education. Data from the trial also shows a relatively high rate of hallucinations, which sometimes led to treatment discontinuation. Although disease progression certainly contributes to hallucinations, the National Expert Group strongly believes the 24 hours (as opposed to the shorter 16 hours typically used in LCIG) levodopa infusion contributes to this observation of a higher incidence of hallucinations. Before deciding to stop the infusion, mitigation strategies could be implemented such as lowering the nighttime dosing or switching to a 16-hour infusion.
With disease progression, many patients will experience cognitive decline, which could lead to discontinuation of foslevodopa foscarbidopa in some patients, especially if they do not have a caregiver.
What settings are appropriate for treatment with [drug under review]? Is a specialist required to diagnose, treat, and monitor patients who might receive [drug under review]?
A physician with interest and experience treating PD patients is required for foslevodopa foscarbidopa initiation and maintenance. Although such physicians will usually be movement disorder neurologists, some general neurologists or geriatricians could also be qualified for this new treatment option. There is no special need for collaboration with other specialist physicians or any technical bottleneck. An outpatient clinic setting is adequate, although some patients and physicians may prefer to initiate treatment in a “day medicine/infusion clinic” setting for the first 1-3 days. For the initiation period (first 4-6 weeks), a dedicated nurse with PD expertise will be extremely helpful to review technical manipulations with the patient and ensure proper aseptic technique and cannula manipulations, as well as educating them about when to switch flow rates or use extra doses. Once initiated, patients with foslevodopa foscarbidopa will need standard clinic follow up visits every 6 months or so. If they occur, skin infections could be treated by an experienced neurologist or addressed by primary care physicians in the community.
Additional Information
Not applicable.
Conflict of Interest Declarations — BC Movement Disorders Specialist Group
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please refer to the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
Yes, as detailed above, this document also includes input from the National Expert Group (a national Canadian group of movement disorders specialists, with representation from British Columbia, who met in Toronto on July 9th, 2022, to discuss the unmet needs of patients with advanced PD).
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
Yes, as detailed above, this document also includes input from the National Expert Group (a national Canadian group of movement disorders specialists, with representation from British Columbia, who met in Toronto on July 9th, 2022, to discuss the unmet needs of patients with advanced PD).
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
Declaration for Clinician 1
Name: Tara Rastin
Position: Clinical Co-director, Vancouver Coastal Health (VCH) Movement Disorders Clinic, Clinical Assistant Professor of Neurology, University of British Columbia
Date: 08-12-2022
Table 16: COI Declaration for BC Movement Disorders Specialist Group — Clinician 1
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie (advisory board) | X | — | — | — |
Sunovion (advisory board) | X | — | — | — |
Declaration for Clinician 2
Name: A Jon Stoessl
Position: Professor & Head, Neurology, University of British Columbia
Date: 08-12-2022
Table 17: COI Declaration for BC Movement Disorders Specialist Group — Clinician 2
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Voyager/Neurocrine (Chair DSMB, no relevant conflict) | X | — | — | — |
AskBio (Member, DSMB, no relevant conflict) | X | — | — | — |
SioGene (Advisor on interpretation of imaging findings, no relevant conflict) | X | — | — | — |
Capsida (Advisor on trial design, biomarkers including imaging, no relevant conflict) | X | — | — | — |
Declaration for Clinician 3
Name: Martin J. McKeown
Position: Professor, and Head, Pacific Parkinson’s Research Centre
Date: 08-12-2022
Table 18: COI Declaration for BC Movement Disorders Specialist Group — Clinician 3
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie (advisory board) | X | — | — | — |
Merz (advisory board) | X | — | — | — |
Ipsen | X | — | — | — |
Declaration for Clinician 4
Name: Melissa Mackenzie
Position: Movement Disorders Specialist, Vancouver Coastal Health (VCH) Movement Disorders Clinic, Clinical Assistant Professor of Neurology, University of British Columbia
Date: 08-12-2022
Table 19: COI Declaration for BC Movement Disorders Specialist Group — Clinician 4
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 5
Name: Claire Hinnell
Position: Movement Disorders Neurologist, Clinical Co-director of the Fraser Health Movement Disorders Clinic, Clinical Assistant Professor of Neurology (University of British Columbia)
Date: 08-12-2022
Table 20: COI Declaration for BC Movement Disorders Specialist Group — Clinician 5
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 6
Name: Anish Kanungo
Position: Movement Disorders Neurologist, Clinical Co-director of the Fraser Health Movement Disorders Clinic, Clinical Assistant Professor of Neurology (University of British Columbia)
Date: 08-12-2022
Table 21: COI Declaration for BC Movement Disorders Specialist Group — Clinician 6
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Abbvie | X | — | — | — |
Sunovion | — | X | — | — |
Merz | — | X | — | — |
Paladin Labs | X | — | — | — |
Declaration for Clinician 7
Name: Keiran Tuck
Position: Director of the Parkinson and Movement Disorder Clinic at Royal Jubilee Hospital, Vancouver Island Health Authority
Date: 08-12-2022
Table 22: COI Declaration for BC Movement Disorders Specialist Group — Clinician 7
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
References
Public Health Agency of Canada. Mapping Connections: An Understanding of Neurological Conditions in Canada. 2014.
Poewe W, Seppi K, Tanner CM, et al. Parkinson disease. Nat Rev Dis Primers. 2017;3:17013.
Armstrong MJ, Okun MS. Diagnosis and Treatment of Parkinson Disease: A Review. JAMA. 2020;323(6):548-560.
Fasano A, C. FVS, Lopiano L, et al. Characterizing advanced Parkinson's disease: OBSERVE- PD observational study results of 2615 patients. BMC Neurology. 2019;19(1):50.
Grimes D, Fitzpatrick M, Gordon J, et al. Canadian guideline for Parkinson disease. CMAJ. 2019;191(36):E989-E1004.
Bratsos S, Karponis D, Saleh SN. Efficacy and Safety of Deep Brain Stimulation in the Treatment of Parkinson's Disease: A Systematic Review and Meta-analysis of Randomized Controlled Trials. Cureus. 2018;10(10):e3474.
Standaert DG, Aldred J, Anca-Herschkovitsch M, et al. DUOGLOBE: One-Year Outcomes in a Real-World Study of Levodopa Carbidopa Intestinal Gel for Parkinson's Disease. Mov Disord Clin Pract. 2021;8(7):1061-1074.
Public Health Agency of Canada. Parkinsonism in Canada, Including Parkinson’s Disease: Highlights from the Canadian chronic disease surveillance system. 2017.
Wong JJ, Kwong JC, Tu K, et al. Time Trends of the Incidence, Prevalence, and Mortality of Parkinsonism. Can J Neurol Sci. 2019;46(2):184-191.
Statistics Canada. Table 13-10-0849-01 Chronic conditions among seniors aged 65 and older, Canadian Health Survey on Seniors, two-year period estimates. Published 2022. Updated April 19, 2022. Accessed August 10, 2022.
Parkinson Canada. Written Submission for the Pre-Budged Consultations in Advance of the 2020 Budget. 2020.
GBD 2016 Parkinson's Disease Collaborators. Global, regional, and national burden of Parkinson's disease, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018;17(11):939-953.
Information CIfH. The Burden of Neurological Diseases, Disorders and Injuries in Canada. Ottawa: CIHI;2007.
Parkinson’s Society Canada. Parkinson’s Disease: Social and Economic Impact. 2003.
Parkinson Society BC. Five Point Plan for Enhanced Support for Parkinson's Disease in BC. 2017.
McLeod Macey J. People with Parkinson’s face gaps in the availability of health services. https://www.ipsos.com/en-ca/news-polls/parkinson-canada-stakeholder-survey-2018. Published 2018. Accessed August 10, 2022.
Dahodwala N, Li P, Jahnke J, et al. Burden of Parkinson's Disease by Severity: Health Care Costs in the U.S. Medicare Population. Mov Disord. 2021;36(1):133-142.
Seppi K, Ray Chaudhuri K, Coelho M, et al. Update on treatments for nonmotor symptoms of Parkinson's disease-an evidence-based medicine review. Mov Disord. 2019;34(2):180-198.
Chaudhuri KR, Antonini A, Robieson WZ, et al. Burden of non-motor symptoms in Parkinson's disease patients predicts improvement in quality of life during treatment with levodopa-carbidopa intestinal gel. Eur J Neurol. 2019;26(4):581-e543.
Klietz M, von Eichel H, Schnur T, et al. One Year Trajectory of Caregiver Burden in Parkinson's Disease and Analysis of Gender-Specific Aspects. Brain Sci. 2021;11(3).
Wong SL, Gilmour H, Ramage-Morin PL. Parkinson’s disease: Prevalence, diagnosis and impact. Statistics Canada;2014.
Karlstedt M, Fereshtehnejad SM, Aarsland D, Lokk J. Mediating effect of mutuality on caregiver burden in Parkinson's disease partners. Aging Ment Health. 2020;24(9):1421- 1428.
Tan QY, Cox NJ, Lim SER, et al. The Experiences of Treatment Burden in People with Parkinson's Disease and Their Caregivers: A Systematic Review of Qualitative Studies. J Parkinsons Dis. 2021;11(4):1597-1617.
Hulshoff MJ, Book E, Dahodwala N, Tanner CM, Robertson C, Marras C. Current Knowledge on the Evolution of Care Partner Burden, Needs, and Coping in Parkinson's Disease. Mov Disord Clin Pract. 2021;8(4):510-520.
Modugno N, Antonini A, Tessitore A, et al. Impact of Supporting People with Advanced Parkinson's Disease on Carer's Quality of Life and Burden. Neuropsychiatr Dis Treat. 2020;16:2899-2912.
Chen YC, Chen R-S, Weng Y-H, et al. The severity progression of non-motor symptoms in Parkinson's disease: a 6-year longitudinal study in Taiwanese patients. Sci Rep. 2021;11(1):14781.
Jankovic J, Tan EK. Parkinson's disease: etiopathogenesis and treatment. J Neurol Neurosurg Psychiatry. 2020;91(8):795-808.
Mantri S, Morley JF, Siderowf AD. The importance of preclinical diagnostics in Parkinson disease. Parkinsonism Relat Disord. 2019;64:20-28.
Chou KL, Stacy M, Simuni T, et al. The spectrum of "off" in Parkinson's disease: What have we learned over 40 years? Parkinsonism Relat Disord. 2018;51:9-16.
Ahlskog JE, Muenter MD. Frequency of levodopa-related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov Disord. 2001;16(3):448- 458.
Luquin MR, Kulisevsky J, Martinez-Martin P, Mir P, Tolosa ES. Consensus on the Definition of Advanced Parkinson's Disease: A Neurologists-Based Delphi Study (CEPA Study). Parkinsons Dis. 2017;2017:4047392.
Pennington S, Snell K, Lee M, Walker R. The cause of death in idiopathic Parkinson's disease. Parkinsonism Relat Disord. 2010;16(7):434-437.
Moscovich M, Boschetti G, Moro A, Teive HAG, Hassan A, Munhoz RP. Death certificate data and causes of death in patients with parkinsonism. Parkinsonism Relat Disord. 2017;41:99-103.
Mestre TA, Eberly S, Tanner C, et al. Reproducibility of data-driven Parkinson's disease subtypes for clinical research. Relat Disord. 2018;56:102-106.15
De Pablo-Fernandez E, Lees AJ, Holton JL, Warner TT. Prognosis and Neuropathologic Correlation of Clinical Subtypes of Parkinson Disease. JAMA Neurol. 2019;76(4):470-479.
Dauer W, Przedborski S. Parkinson's disease: mechanisms and models. Neuron. 2003;39(6):889-909.
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388(6645):839-840.
MacMahon Copas AN, McComish SF, Fletcher JM, Caldwell MA. The Pathogenesis of Parkinson's Disease: A Complex Interplay Between Astrocytes, Microglia, and T Lymphocytes? Front Neurol. 2021;12:666737.
Sanjari Moghaddam H, Zare-Shahabadi A, Rahmani F, Rezaei N. Neurotransmission systems in Parkinson's disease. Rev Neurosci. 2017;28(5):509-536.
Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24(2):197-211.
Borsche M, Pereira SL, Klein C, Grünewald A. Mitochondria and Parkinson's Disease: Clinical, Molecular, and Translational Aspects. J Parkinsons Dis. 2021;11(1):45-60.
Klingelhoefer L, Reichmann H. Pathogenesis of Parkinson disease--the gut-brain axis and environmental factors. Nat Rev Neurol. 2015;11(11):625-636.
Mao X, Ou MT, Karuppagounder SS, et al. Pathological alpha-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science. 2016;353(6307).
Marek K, Chowdhury S, Siderowf AD, et al. The Parkinson's progression markers initiative (PPMI) - establishing a PD biomarker cohort. Ann Clin Transl Neurol. 2018;5(12):1460- 1477.
Rizzo G, Copetti M, Arcuti S, Martino D, Fontana A, Logroscino G. Accuracy of clinical diagnosis of Parkinson disease: A systematic review and meta-analysis. Neurology. 2016;86(6):566-576.
Postuma RB, Berg D, Stern M, et al. MDS clinical diagnostic criteria for Parkinson's disease. Mov Disord. 2015;30(12):1591-1601.
Goetz CG, Tilley BC, Shaftman SR, et al. Movement Disorder Society-sponsored revision of the Unified Parkinson's Disease Rating Scale (MDS-UPDRS): scale presentation and clinimetric testing results. Mov Disord. 2008;23(15):2129-2170.
Fox SH, Katzenschlager R, Lim SY, et al. International Parkinson and movement disorder society evidence-based medicine review: Update on treatments for the motor symptoms of Parkinson's disease. Mov Disord. 2018;33(8):1248-1266.
Borovac JA. Side effects of a dopamine agonist therapy for Parkinson's disease: a mini- review of clinical pharmacology. Yale J Biol Med. 2016;89(1):37-47.
Emre M, Aarsland D, Albanese A, et al. Rivastigmine for dementia associated with Parkinson's disease. N Engl J Med. 2004;351(24):2509-2518.
Marsili L, Bologna M, Miyasaki JM, Colosimo C. Device-aided therapies for advanced Parkinson disease: insights from an international survey. Neurol Sci. 2021;42(7):2961- 2964.
Antonini A, Stoessl AJ, Kleinman LS, et al. Developing consensus among movement disorder specialists on clinical indicators for identification and management of advanced Parkinson's disease: a multi-country Delphi-panel approach. Curr Med Res Opin. 2018;34(12):2063-2073.16
Soileau MJ, Pagan F, Fasano A, et al. Comparative Effectiveness of Carbidopa-Levodopa Enteral Suspension and Deep Brain Stimulation on Parkinson's Disease-Related Pill Burden Reduction in Advanced Parkinson's Disease: A Retrospective Real-World Cohort Study. Neurol Ther. 2022;11(2):851-861.
Metta V, Batzu L, Leta V, et al. Parkinson's Disease: Personalized Pathway of Care for Device-Aided Therapies (DAT) and the Role of Continuous Objective Monitoring (COM) Using Wearable Sensors. J Pers Med. 2021;11(7):680.
Limousin P, Foltynie T. Long-term outcomes of deep brain stimulation in Parkinson disease. Nat Rev Neurol. 2019;15(4):234-242.
DUODOPA® (levodopa/carbidopa intestinal gel) Product Monograph. In. Saint-Laurent, QC: AbbVie Corporation; April 2022.
Nyholm D. Duodopa(R) treatment for advanced Parkinson's disease: a review of efficacy and safety. Parkinsonism Relat Disord. 2012;18(8):916-929.
Wirdefeldt K, Odin P, Nyholm D. Levodopa-Carbidopa Intestinal Gel in Patients with Parkinson's Disease: A Systematic Review. CNS Drugs. 2016;30(5):381-404.
Antonini A, Mancini F, Canesi M, et al. Duodenal levodopa infusion improves quality of life in advanced Parkinson's disease. Neurodegener Dis. 2008;5(3-4):244-246.
Spanaki C, Boura I, Avgoustaki A, et al. Buried Bumper Syndrome: A common complication of levodopa intestinal infusion for Parkinson disease. Parkinsonism Relat Disord. 2021;85:59-62.
Marano M, Pizzicannella M, di Biase L, et al. Jejunal pulling syndrome: A peculiar LCIG complication. Parkinsonism Relat Disord. 2018;52:113-114.
Tsuboi T, Watanabe H, Funasaka K, Takebayashi M, Miyata K, Katsuno M. Peritonitis after percutaneous endoscopic gastrojejunostomy for levodopa–carbidopa intestinal gel treatment despite concomitant use of gastropexy. Neurology and Clinical Neuroscience. 2018;6(2):64-66.
Olanow CW, Kieburtz K, Odin P, et al. Continuous intrajejunal infusion of levodopa- carbidopa intestinal gel for patients with advanced Parkinson's disease: a randomised, controlled, double-blind, double-dummy study. The Lancet Neurology. 2014;13(2):141- 149.
Szasz JA, Constantin VA, Orban-Kis K, et al. Levodopa-Carbidopa Intestinal Gel in Advanced Parkinson's Disease: Observations and Dilemmas after 10 Years of Real-Life Experience. Pharmaceutics. 2022;14(6).
Poewe W, Antonini A. Novel formulations and modes of delivery of levodopa. Mov Disord. 2015;30(1):114-120.
Morgante L, Morgante F, Moro E, et al. How many parkinsonian patients are suitable candidates for deep brain stimulation of subthalamic nucleus? Results of a questionnaire. Parkinsonism Relat Disord. 2007 Dec;13(8):528-31.
Aldred J, Amelin A, Antonini A , et al. Continuous Subcutaneous Foslevodopa/Foscarbidopa in Advanced Parkinson’s Disease: Results From a 12-Month Phase 3 Study, European Academy of Neurology 2022 Annual Meeting, June 25−28, Vienna, Austria
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines.
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.