CADTH Reimbursement Review
Selumetinib (Koselugo)
Sponsor: Alexion Pharma GmbH
Therapeutic area: Neurofibromatosis type 1
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Ethics Review
Stakeholder Input
Clinical Review
Abbreviations
6MWT
6-minute walk test
AE
adverse event
AHI
Apnea-Hypopnea Index
BOR
best objective response
BSA
body surface area
CALM
café-au-lait macules
CI
confidence interval
CMT
clinically meaningful threshold
CORD
Canadian Organization for Rare Disorders
CPBTC
Canadian Pediatric Brain Tumour Consortium
CPK
creatine phosphokinase
CR
complete response
CTCAE
Common Terminology Criteria for Adverse Events
DCO
data cut-off
DOR
duration of response
DVQ
Dysfunctional Voiding Questionnaire
ECOG
Eastern Cooperative Oncology Group
FAS
full analysis set
FEV0.75
forced expiratory volume in the first 0.75 seconds
FEV1
forced expiratory volume in first second
GIC
Global Impression of Change
HR
hazard ratio
HRQoL
health-related quality of life
ICR
independent central review
IPTW
inverse probability of treatment weighting
ITC
indirect treatment comparison
logMAR
logarithm of the minimum angle of resolution
MEK
mitogen-activated protein kinase
MID
minimally important difference
MMRM
mixed model repeated measures
MPNST
malignant peripheral nerve sheath tumour
NCI
National Cancer Institute
NE
not evaluable
NF1
neurofibromatosis type 1
NH
Natural History
NIH
National Institutes of Health
NRS-11
Numerical Rating Scale-11
ORR
objective response rate
PedsQL
Pediatric Quality of Life Inventory
PFS
progression-free survival
PFT
pulmonary function test
PII
Pain Interference Index
PN
plexiform neurofibromas
POB
Pediatric Oncology Branch
PR
partial response
PRO
patient-reported outcome
PROMIS
Patient-Reported Outcomes Measurement Information System
QoL
quality of life
RECIST 1.1
Response Evaluation Criteria in Solid Tumours Version 1.1
REiNS
Response Evaluation in Neurofibromatosis and Schwannomatosis
ROM
range of motion
SAE
serious adverse event
SD
standard deviation
TEAE
treatment-emergent adverse event
TFBC
Tumour Foundation of BC
TTR
time to response
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Selumetinib (Koselugo), 10 mg and 25 mg oral capsules |
Indication | For the treatment of pediatric patients aged 2 years and above, with neurofibromatosis type 1 who have symptomatic, inoperable plexiform neurofibromas |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | August 31, 2022 |
Sponsor | Alexion Pharma GmbH |
NOC = Notice of Compliance.
Introduction
Neurofibromatosis type 1 (NF1) is an autosomal dominant genetic disorder associated with progressive cutaneous, neurologic, skeletal, and neoplastic manifestations.1-3 Approximately half of all NF1 cases are familial, while half arise from spontaneous mutations in the NF1 gene.3,4 Currently, the incidence of NF1 in Canada is unknown, although it is estimated to occur in 1 in 2,500 to 3,000 births.2,4-6 Patient group input received by CADTH for this review noted that there are currently more than 12,000 cases of NF1 in Canada.
The most common manifestations of NF1 include abnormally coloured patches of skin (café-au-lait macules [CALMs]), freckling under the arms and in the inguinal region, and benign tumours predominantly in the skin and nerves known as neurofibromas. Other manifestations may include bone dysplasia, scoliosis, ocular problems, and neurologic complications with impacts such as cognitive impairments and learning disabilities. Neurofibromas are histologically benign nerve sheath tumours that typically originate in the terminal nerve branches of the skin. Plexiform neurofibromas (PNs) are the most common type of tumour in patients with NF1, occurring in up to 50% of patients.7-10 One or multiple PNs may grow along large nerves and plexuses anywhere in the body, with varying manifestations that continue to develop to early adulthood, and multiple PNs may be both symptomatic and asymptomatic in the same individual.11-13 Additionally, PNs have a complex shape and can reach large sizes, resulting in clinical symptoms such as disfigurement, motor dysfunction (weakness and restricted range of motion [ROM]), pain, and neurologic dysfunction. The severity of symptoms from PNs may range from mild to severe; however, the presence of symptoms may depend on their location and impact on surrounding structures. PNs grow most rapidly during early childhood, although growth rates vary among patients.14-16
Treatment and clinical management options for NF1-associated PNs are extremely limited and depend on symptomatology. For symptomatic patients, treatments aim to relieve symptoms caused by the individual PNs. Currently, the only available options to treat and manage NF1-associated PNs are pain management and surgical excision to remove as much of the tumours as possible. However, surgery is not a viable option for many patients as most PN are not amenable to complete resection due to encasement of, or proximity to, vital structures.7,17,18
Selumetinib (Koselugo) is an orally available, selective inhibitor of mitogen-activated protein kinases (MEK) 1 and 2. Selumetinib blocks MEK activity and inhibits growth of RAF-MEK-ERK pathway–activated cell lines, thereby leading to an inhibition of cellular proliferation and PN growth. Selumetinib is available as 10 mg or 25 mg oral capsules. The recommended dosage of selumetinib is 25 mg/m2 twice daily based on body surface area (BSA). The Health Canada indication for selumetinib is for the treatment of pediatric patients with NF1 aged 2 years and older who have symptomatic, inoperable PNs. The notice of compliance was granted on August 31, 2022. Selumetinib has not been previously reviewed by CADTH.
The objective of the current report is to review the beneficial and harmful effects of selumetinib 10 mg and 25 mg twice daily for the treatment of pediatric patients with NF1 aged 2 years and older who have symptomatic, inoperable PNs.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient groups who responded to CADTH’s call for patient input and from clinical experts consulted by CADTH for the purpose of this review.
Patient Input
CADTH received input from 2 patient groups: the Tumour Foundation of BC (TFBC) and the Canadian Organization for Rare Disorders (CORD). The TFBC provides essential information and support services to patients with neurofibromatosis and their families. CORD works with governments, researchers, clinicians, and industry to promote research, diagnosis, treatment, and services for all rare disorders in Canada.
Both patient groups conducted online surveys in November 2022, recruiting patients with NF1 and their caregivers. Additionally, the TFBC conducted a Zoom videoconference focus group. The TFBC group recruited 25 patients and caregivers, and CORD recruited 8 caregivers. Key themes identified by patients and caregivers included limitations on daily living, functional, and social activities; moderate to severe chronic pain; dependency on caregivers into adulthood; financial stress because of the diagnosis; and the lack of treatment options, which negatively affects the emotional well-being of patients and families.
Respondents from both groups described difficulties obtaining a diagnosis of NF1, as well as significant impacts on both affected children and their families in terms of managing physical and mental disability. Additionally, substantial negative mental health impacts were reported, with most patients living with anxiety and fear over their diagnosis, and some patients experiencing suicidal feelings or actions. Respondents to surveys and interviews were surprised and disappointed with the lack of available treatment options and support, with 46% not having been offered any kind of treatment, and only 17% of patients who were offered treatment experiencing minimal improvement in symptoms.
No patients in the TFBC survey had experience with selumetinib. The half (n = 4) of the CORD respondents who had experience with selumetinib through clinical trials described it as “miracle drug” that was “life-changing” due to substantial improvements in pain level, functional abilities including speaking clearly and chewing food, and softening and shrinking tumours that were previously disabling and/or disfiguring.
Numerous outcomes were identified as important to patients, reflecting the heterogenous nature of the disease, but common themes included an overall improved quality of life (QoL), a desire for reduction in pain, reduction or prevention of tumour size or growth, improved function and emotional well-being, greater independence from caregivers, and fewer health care visits.
Clinician Input
Input From Clinical Experts Consulted by CADTH
The information in this section is based on input received from a panel of 6 clinical specialists consulted by CADTH for the purpose of this review.
The clinical experts indicated that the main limitations and unmet needs of pediatric patients with NF1 with symptomatic PNs is the lack of access to disease-modifying medical interventions that can reduce the burden of disease or stabilize symptomatic PNs. The clinical experts noted that there are currently no established practice guidelines for this heterogenous disease. For patients with symptomatic PNs, the only available treatment option, surgery, is either aimed at excising tumours if possible, or debulking if complete excision is not achievable. The experts noted that surgery is not curative for most large or extensive PNs and is associated with significant risks of secondary injuries depending on the number, location, size, and vascularity of tumours, particularly for PNs involving large arteries or nerves. The experts added that multiple invasive surgeries may be required, as tumours may regrow or increase in size, further increasing the risks to patients. Aside from surgery, current treatment strategies consist of “watch and wait” for patients with PNs that are not currently symptomatic. Otherwise, treatment for patients with symptomatic PNs focuses on relieving pain, reducing functional impairment, and improving overall QoL. The panel emphasized the availability of MEK inhibitors through managed access programs, noting that selumetinib is the first and only Health Canada–authorized MEK inhibitor available for the treatment of PNs outside of clinical trials. The panel concluded that selumetinib is expected to cause a shift in the current treatment paradigm given the absence of other medications available for this population. The experts stated that, should selumetinib be recommended for reimbursement, it would likely be the initial therapy of choice.
The clinical experts noted that only a minority of NF1 patients have symptomatic PNs. Patients with NF1 are diagnosed based on standard, well-established, and recently updated clinical diagnostic criteria, including clinical characteristics such as CALMs, and the presence of neurofibromas. Although recently updated diagnostic criteria include genetic testing, the experts noted that genetic testing is not required for diagnosis of NF1, and that the results of genetic testing do not affect treatment decisions once a clinical diagnosis of NF1 has been established. The clinical diagnosis of NF1 is relatively straightforward in older children, adolescents, and adults, but can be challenging in younger children due to the absence of clinical characteristics such as CALMs or PNs. However, the updated diagnostic criteria, which include genetic testing in patients without a family history, have improved confirmatory diagnosis in young children before they manifest other clinical features of NF1. Diagnosis of large, extensive, or rapidly growing PNs generally requires more clinical expertise, and possibly more complex tumour characterization, including MRI, and sometimes a biopsy if there is concern about malignant transformation.
The experts also highlighted that, in terms of natural history, there is a trend for tumours to appear and grow rapidly in early childhood (before the age of 6 to 8 years), and then slow down or remain static in adulthood. Rapid growth of a PN and transformation to a malignant peripheral nerve sheath tumour (MPNST) is a concern. The experts also discussed the uncertainty regarding treatment decisions for asymptomatic patients, as none have been established. In addition, no evidence is available regarding whether treatment with MEK inhibitors such as selumetinib can prevent growth of new PNs.
The experts emphasized the heterogeneity of the disease in NF1 patients, with cutaneous neurofibromas and PNs often occurring throughout the body and ranging in severity from asymptomatic to severely debilitating due to pain, functional impairment, or disfigurement. One clinical expert highlighted that disfigurement due to large, visible PNs is a source of anxiety and concern due to public fear and social stigmatization. The panellist also highlighted the potential for ongoing problems to persist into adulthood due to large PNs that may result in severe disfigurement and displacement of joints and bones; however, there is no clear evidence that treating asymptomatic PNs with selumetinib in children will prevent the development of symptoms in adults. Other concerns for the NF1 population raised by the experts include deficient social skills, frequent learning disabilities, autism, and attention-deficit/hyperactivity disorder, further highlighting the vulnerability and marginalization of these patients.
Treatment with selumetinib is the only available medical treatment for NF1 patients whose extensive inoperable PNs are causing significant pain, functional impairment, and/or disfigurement. Although it is difficult to determine which patients are most likely to respond to treatment, 1 clinical expert currently treating pediatric patients via compassionate access to selumetinib stated that about 80% of patients will respond to treatment. The experts noted that most NF1 patients with PNs are asymptomatic, and the benefit of treatment for these patients has not yet been established. Clinical trials of selumetinib are currently being conducted in the adult population, and these should provide insight into similarities or differences in effectiveness by age. The experts agreed that the lack of knowledge about both the potential benefits and harms associated with long-term selumetinib treatment is a concern, given that NF1 is a life-long disease. The experts also noted that the life expectancy of NF1 patients has been reported to be reduced by 10 to 15 years, although estimates of life expectancy with currently available medical management are unknown.
The clinical experts noted that current clinical trials aim to address important outcomes; however, given the heterogeneity of the disease, standardizing subjective measures (such as pain perception) across this population is problematic, and interpreting the results relies heavily on clinical judgment. The clinical experts agreed that the most important outcomes in the management of pediatric patients with NF1 and symptomatic, inoperable PNs is the reduction or improvement of symptoms (i.e., reduced pain and improved function), as well as overall improvements in QoL and disease stabilization. The experts noted that volumetric MRI, although used in the clinical trial to define disease progression, is only used by the National Institutes of Health (NIH) for research purposes and is not available in Canadian clinical practice. The experts considered a change in planar tumour size of 20% to 25% to be indicative of response to treatment. One expert discussed the potential for symptomatic disease progression despite no evidence of progression on imaging studies and for improvement in symptoms without reduction of tumour size on imaging studies. In addition, the panellists emphasized that it is not always clear which tumours are the cause of symptoms when patients have large numbers of PNs, making it difficult to know when the disease is progressing. The experts also noted that tumours are frequently irregular in shape, making measurements of changes in tumour size difficult. As a result, the panel agreed that response to treatment is multidimensional and must consider reductions in tumour sizes, changes in symptoms, and improvements in function and disfigurement.
The experts stated that young children with NF1 and symptomatic PNs may initially be followed with an MRI every 3 months, in addition to annual follow-ups with NF1 specialists to assess other features of the disease. According to the panel of experts, upon initiating treatment, patients would be seen weekly for a month, then monthly, and if treatment is well tolerated or disease stabilizes, follow-up intervals would be extended to 6 months. The experts also noted that imaging in young children often requires a general anesthetic. While no firm treatment duration for selumetinib has been determined, the experts suggested that, similar to the SPRINT phase II trial, in clinical practice patients would continue treatment until disease progression or toxicity. The experts expected that the initial treatment authorization period for selumetinib should be 18 months. The clinical experts agreed that selumetinib would be discontinued in patients who are not responding (i.e., tumour growth, lack of stabilization, or improvement of symptoms), or in patients with severe adverse events (AEs) that are cannot be managed. The experts also noted that the need for surgery to further debulk tumours could indicate that treatment is not working and should therefore be discontinued. One clinical expert suggested that selumetinib may be used in conjunction with debulking surgery, although there is currently no evidence for this approach.
The experts indicated that expertise in the use of selumetinib in Canada is limited to pediatric oncologists and neurooncologists in tertiary care hospitals. Currently, only pediatric oncologists are prescribing treatment with selumetinib, as they have the experience and know-how to manage these patients. However, the experts highlighted that, with further insight and growing experience, NF1 experts who are pediatricians could manage this oral treatment. Given the heterogeneity in the disease and the individualized approach to treatment, decisions often involve a multidisciplinary team of pediatricians, NF1 experts, neurooncologists, and nurse practitioners. The experts also emphasized the importance of consulting other specialists, including surgeons, cardiologists, ophthalmologists, dermatologists, and pharmacists, on the management of selumetinib, adverse effects, and drug interactions. The expert panel also anticipated access to specialty clinics may be a limiting factor for patients in remote areas, as patients would be required to attend in-person appointments for treatment initiation and imaging follow-up as well as to assess safety.
Clinician Group Input
Input for this review was received through shared clinical experiences from 1 clinician group, the Canadian Pediatric Brain Tumour Consortium (CPBTC), which included 27 pediatric neurooncologists across Canada.
Overall, the clinician group input was aligned with that given by the clinical expert panel convened by CADTH, highlighting that no systemic therapies exist for treating NF1-associated PNs, which represents the major unmet need in this patient population, and that surgical resection, if feasible, is the only option currently available for patients. The clinician group emphasized that selumetinib has clearly shifted the current treatment paradigm and emerged as the standard-of-care, first-line therapy for patients with inoperable, symptomatic PNs. They described the patients most in need of intervention as those in whom PNs are invading critical structures, causing a deformity, or causing functional impairment in activities of daily living such as walking, swallowing, or eating. The clinician group also noted that, in Canada, treatment initiation with selumetinib is currently limited to pediatric oncologists, neurooncologists, or pediatric neurologists with an expertise in neurooncology. The CPBTC suggested that treatment with selumetinib in Canada could be initiated by oncologists and followed up remotely in conjunction with local clinicians.
Finally, the CPBTC group noted that many parents of children with NF1 also have NF1 themselves and are likely to have lower socioeconomic standing in part because of the disease. It was therefore the CPBTC’s opinion that many patients and parents of patients are more likely to lack private insurance that covers selumetinib, which could result in unequitable access in some parts of the country. The CPBTC group emphasized that children without private insurance who are also not eligible for the provincial public drug plans will need special consideration and that the drug in question urgently requires reimbursement and equitable access as a standard-of-care treatment for patients with NF1 and symptomatic PNs.
Drug Program Input
The drug programs identified the following jurisdictional implementation issues: relevant comparators, considerations for initiation of therapy, considerations for continuation or renewal of therapy, considerations for discontinuation of therapy, considerations for prescribing of therapy, generalizability, care provision issues, and system and economic issues. Table 4 provides more details.
Clinical Evidence
Pivotal Studies and Protocol-Selected Studies
Description of Studies
SPRINT phase II is a phase II, open-label, single-arm, multicentre study that aimed to evaluate the efficacy of 25 mg/m2 selumetinib twice daily in 50 pediatric patients with NF1 and inoperable PN. The primary outcome of the SPRINT phase II study was the objective response rate (ORR) determined by change in PN volumes through volumetric MRI. Secondary outcomes included patient-reported outcomes (PROs) and functional evaluations to determine the effect of selumetinib on pain, motor function, and health-related quality of life (HRQoL). Two data cut-offs (DCOs) were submitted for the SPRINT phase II trial.19,20 The primary DCO occurred on June 29, 2018, and an updated DCO occurred on March 31, 2021, providing a maximum follow-up of 5.6 years.19
At baseline, patients included in the SPRINT phase II trial were mostly white (42 [84.0%]) and male (30 [60.0%]), with a mean age of 10.3 years (standard deviation [SD] = 3.92 years). The median number of target PNs causing morbidity was 3 (range = 1 to 4), and the mean target PN volume was 837.11 mL (SD = 925.011), ranging from 5.6 mL to 3,820.0 mL. Pain was present in the target PNs in 26 patients (52.0%). The most common locations of target PNs were the neck and trunk, and the trunk and extremity (12 [24.0%], each), and || ||||||| patients had at least 1 prior PN- or NF1-related surgical procedure.19,20
Efficacy Results
Key results of the efficacy analyses of the SPRINT phase II trial are summarized in Table 2.
Pain
Evaluation of pain was a secondary end point of the SPRINT phase II trial. Pain intensity was measured by the Numeric Rating Scale-11 (NRS-11); a self-evaluation of pain in patients aged 8 years and older consisting of 4 questions scored on a scale of 0 (no pain) to 10 (worst pain imaginable). A threshold of 2 points was suggestive of clinically meaningful change according to the literature. The interference of pain on daily functioning was measured by the Pain Interference Index (PII), a 6-item scale that assesses the extent to which pain has interfered with daily activities in the past 7 days (0 = not at all to 6 = completely). Higher scores for both scales indicate greater impact of pain on patients.
At the June 29, 2018, DCO, the mean adjusted change from baseline score for target tumour pain intensity measured by the NRS-11 was reduced at precycle 13 by −2.07 points (95% confidence interval [CI], −2.84 to −1.31).19 At the March 31, 2021, DCO, representing a longer follow-up period, the NRS-11 target tumour pain was reduced at precycle 13 with an adjusted mean change from baseline of ||||| points (95% CI, ||||| || |||||).20
For the PII, the self-reported adjusted mean change from baseline score at precycle 13 was reduced by −0.65 points (95% CI, −0.89 to −0.42), and the adjusted mean change from baseline in parent-reported PII scores at precycle 13 was reduced by −0.82 points (95% CI, −1.17 to −0.47) at the June 29, 2018, DCO.19 At the March 31, 2021, DCO, results were consistent with the primary analysis, with a reduction in the adjusted mean change from baseline at precycle 13 of ||||| (95% CI, ||||| || |||||) for the self-reported total score, and ||||| (95% CI, ||||| || |||||) for the parent-reported score.20
Motor Function
Motor function was evaluated in patients with motor morbidity using the strength of muscle groups and ROM tests, as well as the Patient-Reported Outcomes Measurement Information System (PROMIS) mobility and upper extremity domains. The PROMIS was completed by both the patient and the parent. Higher scores indicate better physical functioning.
The baseline score for the self- and parent-reported assessments in the mobility domains of PROMIS were 46.57 (SD = |||||) and 37.43 (SD = |||||), while the baseline scores for the self- and parent-reported assessments in the upper extremity domain were 45.95 (SD = ||||||) and 38.15 (SD = ||||||), with higher scores indicating better physical functioning. At the March 31, 2021, DCO, self-reported mobility and self-reported upper extremity improved, with adjusted mean changes from baseline at precycle 13 of |||| points (95% CI, ||||| || ||||), and |||| points (95% CI, ||||| || ||||), respectively. In the parent-reported assessments, the adjusted mean change from baseline at precycle 13 improved in the mobility and upper extremity domains by |||| points (95% CI, |||| || ||||), and |||| points (95% CI, ||||| || ||||), respectively.20
Strength using the manual muscle test (a Medical Research Council 5-point Likert scale) was assessed in the 33 patients who had motor morbidity in any body quadrant at enrolment. At the March 31, 2021, DCO, || patients had evaluable strength assessments at baseline and precycle 13, with a mean strength score of |||| (SD = |||||) at baseline, and an adjusted mean change from baseline of |||| points (95% CI, |||| || ||||). For ROM, the mean ROM sum of all joints was |||||| degrees (SD = |||||||), and the adjusted mean change from baseline at precycle 13 was an increase of ||||| degrees (95% CI, ||||| || ||||||).20
Health-Related Quality of Life
HRQoL, a secondary end point of the SPRINT phase II study, was measured using the Pediatric Quality of Life Inventory (PedsQL) tool, which assesses function in 4 domains: physical (8 items), emotional (5 items), social (5 items), and school (5 items) on a 5-point Likert scale (0 = never a problem; 4 = almost always a problem), with scores reverse-transformed to a 0-to-100 scale, with higher scores indicating better HRQoL. No minimally important difference threshold was identified in the literature. The observed mean score at baseline was 73.91 (SD = ||||||) in the self-reported version, and 60.79 (SD = ||||||) in the parent-reported version. At the March 31, 2021, DCO, the adjusted mean changes from baseline in the self-reported version of the PedsQL were |||| points (95% CI, |||| || |||||) and ||||| points (95% CI, |||| || |||||) in the parent-reported version, suggesting improvements in HRQoL.20
Objective Response Rate
The ORR was the primary end point of the SPRINT phase II study. At the June 29, 2018, DCO, 33 patients (66.0% [95% CI, 51.2 to 78.8]) achieved an ORR, according to the Response Evaluation in Neurofibromatosis and Schwannomatosis (REiNS) criteria. The ORR achieved in the sensitivity analysis based on an independent central review (ICR) was |||||. Differences in the ORR between the primary central analysis and the ICR analysis were primarily due to differences in categorization of confirmed partial response (PR) versus stable disease (based on the chosen threshold of 20% shrinkage to determine response), where |||||| patients were considered to have a confirmed PR despite reductions in tumour size being slightly below the threshold of 20%.19 At the later, March 31, 2021, DCO, the ORR was 68.0% (95% CI, |||| || ||||).20 At both DCOs, the ORR was based on confirmed PRs.
An exploratory ICR analysis using modified Response Evaluation Criteria in Solid Tumors Version 1.1 (RECIST 1.1), which used a 30% volume reduction for PR as opposed to 20% with REiNS, was also conducted at the June 29, 2018, DCO. Based on the RECIST 1.1 assessment, the ORR was only ||||, with | |||||| patients having an unconfirmed PR, and || ||||||| patients having stable disease.19
Change in target PN volume was also assessed as part of the volumetric MRI and application of the REiNS criteria. At the March 31, 2021, DCO, the mean percent change from baseline in target PN volume at precycle 13 was ||||||| (SD = ||||||), corresponding to a mean absolute change of ||||||| mL (|||||||). The proportion of patients with a maximum reduction from baseline of 20% or greater was identical to the June 29, 2018, DCO, at 77.1%, and | ||||||| with a maximum reduction from baseline of 40% or greater.20
Harms Results
Nearly all patients (49 [98.0%]) in the SPRINT phase II trial experienced a treatment-emergent adverse event (TEAE). The most frequent TEAEs reported at the March 31, 2021, DCO were vomiting (|| |||||||), increased blood creatine phosphokinase (CPK) (|| |||||||), diarrhea (|| |||||||), nausea (|| |||||||), and dry skin (|| |||||||). Grade 3 or higher TEAEs were reported in || ||||||| patients, with the most frequent being diarrhea || ||||||||| hypoxia || |||||||| and pyrexia || |||||||. Overall, || ||||||| patients had at least 1 TEAE leading to a dose interruption.20
At the March 31, 2021, DCO, || ||||||| patients experienced serious adverse events (SAEs), the most frequent being infections and infestations || ||||||||| and gastrointestinal disorders || ||||||| constipation, abdominal pain, and diarrhea).20
A total of | ||||||| patients discontinued selumetinib due to AEs; |||||| of which were grade 3 (acute kidney injury, diarrhea| |||||| paronychia, and increased weight) and |||||| were grade 4 |||||| |||||||||| ||||||||| and skin ulcer), with ||| patient experiencing a grade 3 and grade 4 AE leading to withdrawal.20
No AEs with a fatal outcome were reported in the SPRINT phase II study; however, after the March 31, 2021, DCO, |||||| patients died due to progressive neurofibrosarcoma after selumetinib treatment was terminated. These deaths were not attributed to treatment with selumetinib.20
The most frequent notable harm associated with selumetinib was paronychia, occurring in || ||||||| patients. The majority of cases were grade 1, |||||| were grade 2, and |||||| was grade 3. One patient discontinued treatment due to grade 3 paronychia at the earlier (June 2018) DCO. Other notable harms included ||||||| |||||| ||| |||||||| ||| |||||||||||||| |||||| ||| |||||||||20
Table 2: Summary of Key Results From Pivotal and Protocol-Selected Studies
Key results | SPRINT phase II selumetinib 25 mg/m2 twice daily (N = 50) | |
|---|---|---|
June 29, 2018, DCO | March 31, 2021, DCO | |
Pain | ||
NRS-11, physician-selected target tumour (N = 34) | ||
N (%) at baseline | 26 (76.5) | |
Observed mean (SD) score at baseline | 3.15 (|||||) | |
N (%) at precycle 13 | 24 (70.6) | |
Observed mean (SD) score at precycle 13 | 1.00 (|||||) | |
Adjusted mean (95% CI) CFB at precycle 13a | −2.07 (−2.84 to −1.31) | ||||| ||||||| |||||| |
Pain Interference Index, total score | ||
Self-report (N = 34) | ||
N (%) at baseline | 33 (97.1) | |
Observed mean (SD) score at baseline | 1.22 (|||||) | |
N (%) at precycle 13 | 29 (||||) | |
Observed mean (SD) score at precycle 13 | 0.56 (|||||) | |
Adjusted mean (95% CI) CFB at precycle 13a | ||||| ||||||| |||||| | ||||| ||||||| |||||| |
Parent-report (N = 48) | ||
N (%) at baseline | 47 (97.9) | |
Observed mean (SD) score at baseline | 1.50 (|||||) | |
N (%) at precycle 13 | 43 (89.6) | |
Observed mean (SD) score at precycle 13 | 0.67 (|||||) | |
Adjusted mean (95% CI) CFB at precycle 13a | ||||| ||||||| |||||| | ||||| ||||||| |||||| |
Motor function | ||
Patient-Reported Outcomes Measurement Information System | ||
Mobility: self-report (N = 24) | ||
N (%) at baseline | 23 (95.8) | |
Observed mean (SD) score at baseline | 46.57 (|||||) | |
N (%) at precycle 13 | 20 (83.3) | |
Observed mean (SD) score at precycle 13 | 48.02 (|||||) | |
Adjusted mean (95% CI) CFB at precycle 13a | |||| ||||||| ||||| | |||| ||||||| ||||| |
Mobility: parent-report (N = 33) | ||
N (%) at baseline | 32 (97.0) | |
Observed mean (SD) score at baseline | 37.43 (|||||) | |
N (%) at precycle 13 | 29 (87.9) | |
Observed mean (SD) score at precycle 13 | 41.14 (|||||) | |
Adjusted mean (95% CI) CFB at precycle 13a | |||| |||||| ||||| | |||| |||||| ||||| |
Upper extremity: self-report (N = 24) | ||
N (%) at baseline | 22 (91.7) | |
Observed mean (SD) score at baseline | 45.95 (||||||) | |
N (%) at precycle 13 | 20 (83.3) | |
Observed mean (SD) score at precycle 13 | 47.38 (||||||) | |
Adjusted mean (95% CI) CFB at precycle 13a | |||| ||||||| ||||| | |||| ||||||| ||||| |
Upper extremity: parent-report (N = 33) | ||
N (%) at baseline | 31 (93.9) | |
Observed mean (SD) score at baseline | 38.15 (||||||) | |
N (%) at precycle 13 | 29 (87.9) | |
Observed mean (SD) score at precycle 13 | 40.58 (||||||) | |
Adjusted mean (95% CI) CFB at precycle 13a | |||| ||||||| ||||| | |||| ||||||| ||||| |
Strength manual muscle test (N = 33) | ||
N (%) at baseline | 31 (93.9) | |
Observed mean (SD) score at baseline | |||| ||||||| | |
N (%) at precycle 13 | 27 (81.8) | |
Observed mean (SD) score at precycle 13 | |||| ||||||| | |
Adjusted mean (95% CI) CFB at precycle 13a | |||| |||||| ||||| | |
Range of motion (N = 33) | ||
N (%) at baseline | 33 (100.0) | |
Observed mean (SD) score at baseline | 848.73 (426.933) | |
N (%) at precycle 13 | 26 (78.8) | |
Observed mean (SD) score at precycle 13 | |||||| ||||||||| | |
Adjusted mean (95% CI) CFB at precycle 13a | ||||| ||||||| ||||||| | ||||| ||||||| ||||||| |
Health-related quality of life | ||
Pediatric Quality of Life Inventory | ||
Self-report (N = 34) | ||
N (%) at baseline | 33 (97.1) | |
Observed mean (SD) score at baseline | 73.91 (||||||) | |
N (%) at precycle 13 | 29 (85.3) | |
Observed mean (SD) score at precycle 13 | 79.56 (||||||) | |
Adjusted mean (95% CI) CFB at precycle 13a | |||| |||||| |||||| | |||| |||||| |||||| |
Parent-report (N = 50) | ||
N (%) at baseline | 50 (100.0) | |
Observed mean (SD) score at baseline | 60.79 (||||||) | |
N (%) at precycle 13 | 45 (90.0) | |
Observed mean (SD) score at precycle 13 | 73.34 (||||||) | |
Adjusted mean (95% CI) CFB at precycle 13b | ||||| |||||| |||||| | ||||| |||||| |||||| |
ORR, n (%) (full analysis set) | ||
Objective response rate | 33 (66.0) | 34 (68.0) |
95% CI | 51.2 to 78.8 | ||||| |||| |
Complete response | 0 (0.0) | 0 (0.0) |
Confirmed partial response | 33 (66.0) | 34 (68.0) |
Unconfirmed partial response | 4 (8.0) | | ||||| |
Stable disease | 11 (22.0) | 11 (22.0) |
Not evaluable | 2 (4.0) | 2 (4.0) |
Harms, n (%) (safety analysis set) | ||
Adverse events | 49 (98.0) | 49 (98.0) |
Serious adverse events | || |||||| | || |||||| |
Withdrawal (from study treatment) due to adverse events | | |||||| | | |||||| |
Deaths | 0 (0.0) | | ||||| |
Notable harms, n (%) | ||
Cardiac events | 18 (36.0) | || |||||| |
Ophthalmologic events | 8 (16.0) | || |||||| |
Paronychia | 23 (46.0) | || |||||| |
CFB = change from baseline; CI = confidence interval; DCO = data cut-off; NRS-11 = Numeric Rating Scale-11; SD = standard deviation.
aThe model included terms for precycle, baseline score, age, the number of morbidities at baseline and baseline × precycle interaction.
bThe model included terms for precycle, baseline score, age, the number of clinical complications at baseline and baseline × precycle interaction.
Sources: SPRINT Phase II Stratum 1 Clinical Study Report (June 29, 2018, DCO)19 and SPRINT Phase II Stratum 1 Clinical Study Report (March 31, 2021, DCO).20
Critical Appraisal
SPRINT phase II was a phase II, open-label, single-arm, multicentre study. The choice to conduct a single-arm study has implications for the overall strength and interpretability of the results. As a single-arm study, there is an increased risk of bias in the estimation of treatment effects due to the potential for confounding related to natural history, and other unidentified prognostic factors that could affect all study outcomes. The noncomparative design of the SPRINT phase II trial precludes an assessment of the therapeutic benefit or safety of selumetinib. In a single-arm trial, because all patients received the same treatment, treatment effect on time-to-event end points are uninterpretable and were only considered exploratory and supportive. Awareness of treatment assignment by both patients and parents or caregivers increases the risk of detection bias and performance bias and may lead to systematic overestimates or underestimates of the overall treatment effect. As such, the open-label trial design limits interpretability of the clinical outcome assessments such as the PRO and functional end points, as well as AEs. The already small sample size (N = 50) was further restricted for secondary end points, including PROs and functional evaluations, as these were based on patients with target PNs in specific locations or limited to patients of a certain age. The outcome of the SPRINT phase II study was the ORR and was considered appropriate by the clinical experts consulted by CADTH and the CADTH review team as an objective measure to assess the activity of selumetinib. Secondary clinical outcome assessments (PROs and functional evaluations) were considered appropriate to evaluate the wide range of PN-related morbidities; however, based on the design of the SPRINT phase II study, and the lack of statistical tests or imputation of missing data, the results should only be viewed as supportive of the overall effect of selumetinib.
There is a lack of standardized end points for trials in NF1. As previously noted, multiple outcomes were included in the SPRINT phase II trial, including response and time-to-event outcomes based on volumetric MRI using the REiNS imaging criteria. The clinical experts consulted by CADTH noted that volumetric MRI is not used in routine clinical practice as it is not standard of care in Canada, and that evidence of disease progression is multifactorial, based on standard imaging techniques, although they emphasized the importance of clinical symptomatology and physical assessment in determining progression and response. As such, patients in Canadian clinical practice would be evaluated for progression slightly differently than in the SPRINT phase II trial, potentially affecting the generalizability of the results. Patient-reported outcomes (NRS-11, PII, PROMIS, and PedsQL) and functional outcomes (strength and ROM) were also evaluated in the SPRINT phase II trial. The clinical experts consulted by CADTH noted that the outcome scales reported in the trial were not used in routine clinical practice and may not be generalizable to the typical patient in Canada. The clinical experts noted that a gestalt-type approach is considered in clinical practice for overall improvement or deterioration in symptomatology overall, as opposed to specific changes in certain domains (e.g., grooved pegboard test and key pinch grip), although variation and heterogeneity by patients and caregivers is significant in this population.
Indirect Comparisons
No appropriate comparators are available to conduct a standard indirect treatment comparison (ITC), and a placebo-controlled trial design was considered unethical by National Cancer Institute (NCI) Pediatric Oncology Branch (POB) investigators due to significant PN-related morbidity and promising results shown in the phase I trial.21 Indirect comparisons were therefore necessary to estimate the relative benefit of selumetinib. The NCI POB conducted 2 additional studies, a Natural History (NH) study to better understand and quantify NF1 manifestations and to allow more sensitive end points to be developed for clinical studies, and Study 01-C-0222, a randomized, crossover, double-blinded, placebo-controlled phase II study of tipifarnib in children and young adults with NF1 and progressive PN. Given the lack of direct comparative evidence for selumetinib, the sponsor conducted naive qualitative comparisons of the results from the SPRINT phase II trial, with the NH study and the placebo arm of Study 01-C-0222 serving as external control arms. The sponsor also conducted a propensity score modelling analysis of progression-free survival (PFS) compared to the NH study.
Description of Studies
The sponsor conducted a naive, side-by-side comparison of results from stratum 1 of the SPRINT phase II trial versus patients with PNs from the NH study using the outcomes of tumour growth (absolute and annual rates) based on the full NH cohort as well as an age-matched NH cohort. The “age-matched” NH cohort included patients who were aged 3 to 18 years and had at least 1 volumetric MRI within this age range and at least 1 subsequent volumetric MRI. A naive, side-by-side qualitative comparison was also conducted for the outcome of PFS between stratum 1 of the SRINT phase II trial and the placebo arm of Study 01-C-0222.
In the propensity scoring analysis, PFS from stratum 1 of the SRINT phase II trial was compared to the age-matched cohort of the NH study. Prognostic factors were identified based on data from the NH study. The univariate and multivariate Cox models (covariates: study, sex, race, target PN location, PN status, age, weight, height, and target PN volume) were fitted to estimate an unadjusted and adjusted hazard ratio (HR), respectively. Age, weight, height, and target PN volume were kept as continuous variables in the model. Three different matching algorithms were explored (matching 1:1 without replacement, inverse probability of treatment weighting [IPTW], and matching 1:2 with replacement).
Efficacy Results
Plexiform Neurofibromas Growth Rate, Naive Comparison: SPRINT Phase II Stratum 1 Versus Natural History Study
Data on the natural history of NF1-related PNs, based on the patients from the selected external controls, demonstrated that the majority of PNs grow continuously over time or, at best, remain stable in size (i.e., < 20% increase in volume from baseline). In contrast to the median annual volume changes of |||||| and ||||| seen in SPRINT (2018 and 2021 DCOs, respectively), the median annual volume changes in the NH study (an age-matched cohort with maximum follow-up aligned to each DCO of SPRINT) were |||||| and ||||||| respectively.
Over the full duration of the studies, the mean percentage change from baseline in SPRINT was |||||| compared to ||||||| in the NH study. The follow-up duration and included patients differed notably in these populations.
Patients who enrolled in the NH study and later went on to participate in stratum 1 of the SRINT phase II trial (n = ||||||) experienced PN growth before selumetinib (median = ||| per year; maximum = ||| per year), and a median volume reduction of |||||| per year after selumetinib treatment (median follow-up = ||| years; range = ||| || |||). Of these patients, |||||| had a reduction of at least 20% in their target PN and the response was sustained for |||||| patients at the latest DCO.
Progression-Free Survival, Naive Comparison: SPRINT Phase II Stratum 1 Versus NH Study
At the time of the March 32, 2021, DCO, disease progression was experienced by ||||| of patients in the NH study compared to ||||| of patients in stratum 1 of the SRINT phase II trial over a 5.6-year period. Median PFS in the NH study’s age-matched cohort was ||| years (95% CI, 1.1 to 1.6) and was ||| ||||||| in stratum 1 of the SRINT phase II trial. The probability of remaining without progression in stratum 1 of the SRINT phase II trial and the NH study was ||||| (95% CI, ||||| || |||||) and ||||| (95% CI, ||||| || |||||), respectively.
Progression-Free Survival, Naive Comparison: SPRINT Phase II Stratum 1 Versus Study 01-C-0222
Because Study 01-C-0222 required progressive disease for enrolment, a subgroup analysis was conducted for the earlier DCO (i.e., 2018) of stratum 1 of the SRINT phase II trial, including only those with progressive PNs at enrolment. In this subgroup, the probability of remaining without progression at 2 years was 94.7% (95% CI, 80.6% to 98.7%), compared to 20.6% (95% CI, 7.7% to 37.8%) in the placebo arm of Study 01-C-0222. The sponsor did not update this comparison for the 2021 DCO.
Progression-Free Survival, Propensity Scoring Analysis: SPRINT Phase II Stratum 1 Versus NH Study
The univariate Cox analysis identified age, weight, height, and PN status at baseline (i.e., progressive, nonprogressive, or unknown) as associated with PFS; younger patients with progressive PN at baseline had a higher risk of progression. The multivariate analysis identified only PN status as correlated with PFS.
After matching, the sample sizes were small, and some standardized differences remained unbalanced (> 0.1 to > 0.2) in the 1:1 and 1:2 matching analyses. In the IPTW analysis, no baseline characteristics differed by a standardized difference of more than 0.1. However, the effective sample size after IPTW was not reported. Across all 3 methods of propensity scoring analysis, the HR for PFS ranged from |||| || |||| in favour of selumetinib, with P values of less than 0.001.
Harms Results
Safety outcomes were not assessed in the ITCs.
Critical Appraisal
Because the NCI POB investigators deemed it unethical to conduct placebo-controlled trials in this population, only unanchored ITCs were possible. Unanchored naive comparisons are subject to substantial inherent limitations as there is no method of controlling for inherent differences in the study design and patient populations, and differences seen in clinical outcomes may be confounded by underlying differences in the compared trials.
In the naive comparison, results were only reported for mean annual change in target PN volume, absolute and percent change in target PN volume from baseline, and PFS. In the propensity scoring analysis, only PFS was assessed. Patient and clinician input suggests that tumour volume or change in volume does not always correlate directly with symptomatology, in part because it is highly dependent on the location of the PNs with respect to important structures. Outcomes related to symptoms, morbidity, disability, HRQoL, and disfigurement were not assessed. No safety outcomes were evaluated.
Notable differences were evident between the patient populations of the 2 external controls in comparison to the SPRINT trial with regards to baseline age, race, target PN location, PN status (i.e., progressive, nonprogressive, or unknown), target PN volume, and treatment history. The study designs also differed with respect to follow-up and frequency of imaging. The risk of bias and imprecision is inherently high due to small study sizes, observed clinical heterogeneity, and the unanchored and naive approach to the comparison, but the direction of potential bias as a result of these differences is unknown. The sample size of the before-and-after analysis (n = |) of patients participating in both the NH study and SPRINT was particularly small, limiting the interpretation of results.
Propensity scoring analysis was conducted using 3 standard methods. Although this was an appropriate approach to mitigate the impact of between-trial differences in baseline patient characteristics, it is unknown whether all key treatment effect modifiers and prognostic factors were accounted for. The methodology for selecting baseline characteristics was not explained or justified. Of the 3 methods of propensity score analysis, only IPTW demonstrated balance in every baseline characteristic examined, while in the 1:1 and 1:2 matching analyses some standardized differences were still greater than 0.1 or greater than 0.2 in important characteristics. The sample size of all analyses were small as a result of the studies informing the comparisons, but the 1:1 and 1:2 matching analyses also resulted in further drops in sample size. The effective sample size of IPTW was not reported, limiting interpretation.
Overall, interpretation of the ITCs is substantially compromised by important limitations. From the naive comparisons and the propensity scoring analyses, the results suggest selumetinib confers a benefit in terms of reduction in the rate of tumour growth and improvement in PFS. However, the magnitude of the benefit is uncertain, and no conclusions can be drawn from the ITCs regarding other clinically important outcomes or harms.
Other Relevant Evidence
No long-term extension studies or other relevant studies were included in the sponsor’s submission to CADTH.
Conclusions
There is an unmet need for disease-modifying treatment options for the rare population of patients with NF1-associated, symptomatic, inoperable PNs. Patients and clinicians highlighted the need for treatments that reduce pain and disfigurement and improve function, while also preventing the growth of new PNs and shrinking existing PNs. One ongoing, phase II, open-label, single-arm, multicentre study (SPRINT phase II) was included in this review. Notable concerns were associated with the internal and external validity of the SPRINT phase II study, driven primarily by the single-arm, open-label design, which precludes the ability to attribute the study results to treatment with selumetinib as opposed to disease natural history or concomitant interventions, and introduces significant bias to all subjective clinical outcome assessments evaluated. These are considered of critical importance given that measurement of disease progression and treatment response in clinical practice relies on available imaging techniques coupled with clinical symptomatology, which may vary from the methods and outcomes used in the SPRINT phase II trial.
The data submitted to CADTH were clinically relevant in this setting, given the variability of location and extent of PNs between patients. Clinical outcome assessments, including PROs and functional evaluations, were supportive overall of the primary imaging findings of the SPRINT phase II trial, reducing PN-associated morbidity and improving HRQoL. However, given their evaluation as secondary outcomes, small sample sizes, lack of statistical testing, and heterogeneity in the location and size of target PNs, results for PROs and functional evaluations can only be interpreted as supportive of the overall effect of selumetinib. For the primary end point in the SPRINT phase II trial, the clinical experts consulted by CADTH agreed that the ORR of 68.0% was clinically meaningful, although the clinical experts contended the observed responses were underestimated, based on their experience and the definitions used for response and progression. While selumetinib treatment also resulted in reductions in PN volume, the correlation between PN volume changes and improvements in symptoms or function remains uncertain, and the experts noted that tumour size may not always reflect morbidity. While the time-to-event end points, duration of response (DOR) and PFS, appeared to be supportive of the observed ORR, the nonrandomized design of the SPRINT phase II trial made attributing these events to selumetinib challenging.
ITCs included a naive side-by-side comparison and propensity scoring analysis against external controls as representations of natural history. The results suggest selumetinib confers a benefit in terms of reduction in the rate of tumour growth and improvement in PFS. However, due to important between-trial differences in study design and populations, and major uncertainties inherent in the methodologies applied, the magnitude of the benefit is uncertain. The relative efficacy of selumetinib was not assessed with regard to any other important clinical outcomes, such as HRQoL, morbidity, and disfigurement, which may not be directly correlated with changes in tumour volume. No safety outcomes were assessed in the indirect comparisons.
Aside from the AEs known to be associated with MEK inhibitors, selumetinib was generally well tolerated in the SPRINT phase II trial, with limited grade 3 or serious AEs, and an overall toxicity profile that can generally be managed with supportive care or dose interruptions. Although the results of the SPRINT phase II trial were generally positive, it is difficult to draw firm conclusions about the magnitude and the generalizability of the clinical benefit and safety of selumetinib given the identified limitations in the available evidence, which is inherent in the complexity of the disease and trial conduct.
Introduction
Disease Background
Of the 3 distinct forms of neurofibromatosis, the most common is NF1. An autosomal dominant genetic disorder associated with progressive cutaneous, neurologic, skeletal, and neoplastic manifestations,1,2 NF1 affects members of each sex and each ethnic group equally.3 The incidence of NF1 in Canada is unknown, although it is estimated to occur in 1 in 2,500 to 3,000 births.2,4-6 The patient group input received by CADTH for this review estimated that there are currently over 12,000 cases of NF1 in Canada.
The disease is caused by germline mutations in the NF1 tumour suppressor gene (17q11.2), which encodes the tumour suppressor protein neurofibromin 1. An activating protein that promotes the conversion of active RAS GTP proteins to inactive RAS guanosine 5′-diphosphate, neurofibromin 1 is a negative regulator of the RAS proto-oncogene, a key signalling molecule controlling cell growth, resulting in overactivation of the RAS/RAF/MEK/ERK mitogen-activated protein kinase cascade pathway, leading to abnormal cell growth.22-24 Approximately half of all NF1 cases are familial, while half arise from spontaneous mutations in the NF1 gene.3,4
Neurofibromatosis type 1 is a highly heterogenous disease with hallmarks and clinical features that may be evident from birth and can affect a wide range of organ systems. The most common manifestations of NF1 include abnormally coloured patches of skin (CALMs), freckling under the arms and in the inguinal region, and benign tumours predominantly in the skin and nerves, known as neurofibromas. Other possible manifestations include bone dysplasia, scoliosis, ocular problems, and neurologic complications with impacts such as cognitive impairments and learning disabilities. Neurofibromas are histologically benign nerve sheath tumours, typically originating in the terminal nerve branches of the skin. PNs are the most common type of tumour in patients with NF1, occurring in up to 50% of patients.7-10 One or multiple PNs may grow along large nerves and plexuses anywhere in the body, with varying manifestations continuing to develop to early adulthood, and multiple PNs may be both symptomatic and asymptomatic in the same individual.11-13 PNs have a complex shape and can reach large sizes, resulting in clinical symptoms such as disfigurement, motor dysfunction (weakness and restricted ROM), pain, and neurologic dysfunction. The severity of symptoms from PNs range from mild to severe; however, the presence of symptoms may depend on their location and impact on surrounding structures. PNs grow most rapidly during early childhood, although growth rate is highly variable between patients.14-16 In children, rapid PN growth cannot be attributed to the anticipated growth rate of a child, as the PN growth rate does not correlate with increases in body weight or body mass index.14,16 Spontaneous shrinkage of PNs over time has been reported, but mainly in adults.13,15,25
PNs have the potential for malignant transformation.23,26 An MPNST is a type of cancer that forms in the cells of the sheath that covers and protects the peripheral nerves. The risk of developing MPNSTs is greater in patients with NF1, with 1 study citing an incidence of MPNSTs in patients with NF1 of 4.6% compared to 0.001% in the general population,27 and the lifetime risk estimated to be between 8% and 15.8%.28-30 Other tumours associated with NF1 include low-grade gliomas, with optic pathway gliomas occurring in approximately 15% of NF1 patients.23
Diagnostic criteria for NF1 were established in 1987 and updated most recently in 2021.31 Diagnostic criteria are based predominantly on clinical manifestations, although the revised criteria incorporate additional clinical features and genetic testing. A diagnosis of NF1 in patients who do not have a parent diagnosed with NF1 requires the presence of 2 or more of the following:
6 or more CALMs larger than 5 mm (by greatest diameter) in prepubertal individuals and 15 mm in postpubertal individuals
freckling in the axillary or inguinal region
2 or more neurofibromas of any type or 1 PN
optic pathway glioma
2 or more iris Lisch nodules identified by slit lamp examination or 2 or more choroidal abnormalities, defined as bright, patchy nodules imaged by optical coherence tomography/near-infrared reflectance imaging
a distinctive osseous lesion such as sphenoid dysplasia, anterolateral bowing of the tibia, or pseudarthrosis of a long bone
a heterozygous pathogenic NF1 variant with a variant allele fraction of 50% in apparently normal tissue such as white blood cells.31
A child of a parent who meets the diagnostic criteria merits a diagnosis of NF1 if 1 or more of these criteria are present.
Standards of Therapy
Treatment and clinical management options for NF1-associated PNs are extremely limited and depend on symptomatology. Treatment strategies consist of “watch and wait” with frequent monitoring for patients with asymptomatic PN, which generally does not require treatment. For symptomatic patients, treatments aim to relieve symptoms caused by the individual PNs. Currently, the only available options to treat and manage NF1 include pain management and surgical excision to remove as much of the tumours as possible. However, for many patients, surgery is not a viable option as most PNs are not amenable to complete resection due to encasement of, or proximity to, vital structures.7,17,18 Several phase I and II clinical studies for progressive PN in children and young adults with NF1 have failed to demonstrate consistent or durable decreases in PN volume.13,32
Drug
Selumetinib (Koselugo) is an orally available, selective inhibitor of MEK 1 and 2. Selumetinib blocks MEK activity and inhibits growth of RAF-MEK-ERK pathway–activated cell lines, leading to inhibition of cellular proliferation and PN growth.33
Selumetinib is available as 10 mg or 25 mg oral capsules. The recommended dosage of selumetinib is 25 mg/m2 twice daily. Recommended dosages are summarized in Table 3. Dosing is individualized based on BSA (mg/m2) and rounded to the nearest achievable 5 mg or 10 mg dose (up to a maximum single dose of 50 mg). Selumetinib capsules of different strengths may be combined to attain the desired dose.33
Table 3: Recommended Dosage of Selumetinib Based on Body Surface Area
Body surface area (mg/m2) | Recommended dose |
|---|---|
0.55 to 0.69 m2 | 20 mg in the morning; 10 mg in the evening |
0.70 to 0.89 m2 | 20 mg twice daily |
0.90 to 1.09 m2 | 25 mg twice daily |
1.10 to 1.29 m2 | 30 mg twice daily |
1.30 to 1.49 m2 | 35 mg twice daily |
1.50 to 1.69 m2 | 40 mg twice daily |
1.70 to 1.89 m2 | 45 mg twice daily |
≥ 1.90 m2 | 50 mg twice daily |
Source: Selumetinib product monograph.
Treatment with selumetinib should continue as long as a clinical benefit is observed, or until PN progression or unacceptable toxicity.33
The reimbursement request for selumetinib is in line with the approved Health Canada indication: for the treatment of pediatric patients aged 2 years and older with NF1 who have symptomatic, inoperable PNs. Health Canada granted a Notice of Compliance on August 31, 2022.34 Selumetinib has not previously been reviewed by CADTH. Selumetinib has also been approved by other major regulatory bodies including the US FDA (2020), European Medicines Agency (2021), and Australia’s Therapeutic Goods Administration (2021).
To support patients requiring treatment with selumetinib, the sponsor indicated that the Alexion OneSource Patient Support Program provides necessary resources and support by phone and/or email across Canada to patients and caregivers on follow-up touch points for disease education, appointment reminders, and/or adherence support, as well as reimbursement navigation.34
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups. The full original patient inputs received by CADTH are included in the stakeholder section at the end of this report.
CADTH received input from 2 patient groups: the TFBC and CORD. The TFBC provides essential information and support services to patients with neurofibromatosis and their families. CORD works with governments, researchers, clinicians, and industry to promote research, diagnosis, treatment, and services for all rare disorders in Canada.
Both patient groups conducted online surveys in November 2022, recruiting patients with NF1 and their caregivers. Additionally, the TFBC conducted a Zoom focus group. All patients had a diagnosis of NF1. The TFBC group recruited 25 patients and caregivers, and CORD recruited 8 caregivers. A total of 8 patients (32%) included in the TFBC survey were adults, while all patients represented in the CORD survey were younger than 18. Key themes identified by patients and caregivers with NF1 included limitations on daily living, functional, and social activities; moderate to severe chronic pain; dependency on caregivers into adulthood; financial stress because of the diagnosis; and lack of treatment options, which negatively affects the emotional well-being of patients and families.
Respondents from both patient and caregiver groups described difficulties obtaining a diagnosis of NF1, as well as significant impacts on both affected children and their families in terms of managing physical and mental disability, with 96% of TFBC survey respondents indicating that they live with chronic pain rated at 5 or greater on a 0-to-10 pain scale. Additionally, substantial negative mental health impacts were reported, with most patients living with anxiety and fear over their diagnosis, and some patients experiencing suicidal feelings or actions. Respondents also cited the financial burden of out-of-pocket expenses and time lost from work and school, the negative toll of multiple surgeries, and hospitalizations with limited benefits. Respondents who had previous treatment experience described out-of-pocket expenses for psychological and physical supports (e.g., scoliosis braces) and treatment or diagnosis options accessed in the US. Some respondents had experience with repeated surgeries that provided minimal or temporary improvement in key outcomes with substantial recovery time. A total of 92% of TFBC respondents reported expenses related to the care of their neurofibromatosis (such as prescription or nonprescription drugs, medical equipment, physiotherapy, counselling, or travel for medical care), and 40% indicated that they fund their own medical expenses without any public or private benefits. Respondents to both surveys and interviews indicated they were surprised and disappointed by the lack of treatment options or support available, with 46% not having been offered any kind of treatment, and only 17% of patients who were offered treatment experiencing minimal improvement in symptoms.
Numerous outcomes were identified as important to patients, reflecting the heterogenous nature of the disease, but common themes included an overall improved QoL, a desire for reduction in pain and reduction or prevention in tumour size or growth, improved function and emotional well-being, greater independence from caregivers, and a reduced number of health care visits.
No patients in the TFBC survey had experience with selumetinib, however, all of them indicated that they would consider taking selumetinib if given the opportunity to access it. The half (n = 4) of the CORD respondents who had experience with selumetinib through clinical trials described it as “miracle drug” that was “life-changing” due to substantial improvements in pain level, functional abilities including speaking clearly and chewing food, and softening and shrinking tumours that were previously disabling and/or disfiguring. The respondents described either no side effects or mild side effects, such as sensitive skin around the toenails that can be easily infected. Although interviews conducted by CORD pointed out that the long-term benefits of selumetinib have yet to be demonstrated, the patient organizations felt that selumetinib should be available to all appropriate patients.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). In addition, as part of the selumetinib review, a panel of 6 clinical experts from across Canada was convened to characterize unmet therapeutic needs, help identify and communicate situations in which gaps in the evidence could be addressed through the collection of additional data, promote the early identification of potential implementation challenges, acquire further insights into the clinical management of patients living with a condition, and explore the potential place in therapy of the drug (e.g., potential reimbursement conditions). A summary of this panel discussion is presented in the following section.
Unmet Needs
The clinical experts indicated that NF1 is a highly heterogeneous, multisystem, genetic condition manifesting from early childhood with numerous needs that are currently not being met. In patients with NF1 with symptomatic, inoperable (i.e., that cannot be completely removed surgically) PNs, tumours are often located throughout the body, may cause pain or discomfort, and result in impairment in mobility, vision, breathing, or other functions. Moreover, the experts also highlighted that, in terms of natural history, there is a trend for tumours to appear and grow rapidly in early childhood, particularly before ages of 6 to 8 years, and then for growth to become much slower or stop in adulthood.
The experts agreed that the main limitations and unmet needs of pediatric patients with NF1 with and symptomatic PNs that cannot be completely removed surgically is the lack of access to disease-modifying medical interventions that can reduce the burden of disease or stabilize symptomatic PNs. Given the multisystemic, heterogenous nature of the disease, the expert panel emphasized the need for systemic treatment of multiple PNs that cause issues in this population.
Place in Therapy
The clinical experts noted that there are currently no established practice guidelines for this heterogenous disease. Recently, a panel of multidisciplinary NF1 experts published consensus recommendations for the management of NF1-associated PNs (Fisher et al. [2022]). For patients with symptomatic PNs, surgery is the only available treatment option, which is either aimed at excising tumours if possible, or debulking if complete excision is not achievable. The experts noted that surgery is not curative for most large or extensive PNs and is associated with significant risks of secondary injuries based on many factors, including the number, location, size, and vascularity of tumours, particularly for PNs involving large arteries or nerves. Additionally, the experts pointed out that multiple invasive surgeries may be required, as tumours can regrow or increase in size, adding to the risks to patients.
Aside from surgery, current treatment strategies consist of “watch and wait” for patients with PNs who are not currently symptomatic. Otherwise, treatment for patients with symptomatic PNs focuses on relieving pain, reducing functional impairment, and improving overall QoL. The panel emphasized the availability of MEK inhibitors through managed access programs, noting that selumetinib is the first, and only Health Canada–authorized MEK inhibitor available outside of clinical trials for the treatment of PNs. The panel concluded that selumetinib is expected to cause a shift in the current treatment paradigm, given the absence of other medications available for this population. The experts stated that, should selumetinib be recommended for reimbursement in this population, it would likely be the initial therapy of choice, and there is no evidence supporting the use of other treatments before initiation of selumetinib for PNs that cannot be completely excised surgically in children with NF1.
Patient Population
The clinical experts noted that only a small minority of NF1 patients have symptomatic PNs. Patients with NF1 are diagnosed based on standard, well-established, and recently updated clinical diagnostic criteria, including clinical characteristics such as CALMs, and the presence of neurofibromas. Although the recently updated diagnostic criteria include genetic testing, the experts noted that genetic testing is generally not required for a diagnosis of NF1. The availability of genetic testing varies by province, and it may not be funded through provincial health plans for many patients. The experts also noted that the results of genetic tests do not affect treatment decisions once a clinical diagnosis of NF1 has been established.
The diagnosis of NF1 is relatively straightforward in older children, adolescents, and adults, but can be challenging in younger children. However, the updated diagnostic criteria, which include genetic testing in patients without a family history, have improved confirmatory diagnosis in young children before they manifest other clinical features of NF1. The diagnosis of large, extensive, or rapidly growing PNs generally requires more clinical expertise, and may require more complex tumour characterization, including MRI, and sometimes a biopsy if there is concern for malignant transformation. The experts added that, in terms of natural history, there is a trend for tumours to appear and grow rapidly in early childhood (before the age of 6 to 8 years), and then slow down or remain static in adulthood. Rapid growth of a PN is a concern for transformation to a MPNST, which can be metastatic and often fatal despite treatment. The experts also discussed the uncertainty regarding treatment decisions for asymptomatic patients, which have not been established. In addition, there is no evidence available regarding whether treatment with MEK inhibitors such as selumetinib can prevent growth of new PNs.
The experts emphasized the heterogeneity of the disease in NF1 patients, with cutaneous neurofibromas and PNs often occurring throughout the body and ranging in severity from asymptomatic to severely debilitating due to pain, functional impairment, or disfigurement. One expert noted that disfigurement due to large, visible PNs can be a source of anxiety and concern due to public fear and social stigmatization. The panellist also highlighted the potential for continuous problems into adulthood due to large PNs, which may result in severe disfigurement and displacement of joints and bones, but added that there is no clear evidence that treating asymptomatic PNs in children will prevent the development of symptoms in adults. Other concerns raised by the experts for the NF1 population include deficient social skills, frequent learning disabilities, autism, and attention-deficit/hyperactivity disorder, further highlighting the vulnerability and marginalization of these patients.
According to the experts consulted for the review, the patients who are most likely to benefit from treatment with selumetinib are those whose extensive inoperable PNs are causing significant pain, functional impairment, and/or disfigurement. Although it is difficult to determine which patients are most likely to respond to treatment, 1 expert currently treating pediatric patients via compassionate access to selumetinib stated that about 80% of patients will respond to treatment.
Regarding patients least suitable for treatment, the experts noted that most NF1 patients with PNs are asymptomatic, and the benefit of treatment for these patients has yet to be established. Almost all PNs persist throughout childhood and into adulthood, and the panellists noted that other MEK inhibitors may turn out to be just as effective as selumetinib in both children and adults. Clinical trials in the adult population currently being conducted for selumetinib should provide insight into similarities or differences in effectiveness by age. The experts agreed that the lack of knowledge about both the potential benefits and harms associated with long-term selumetinib treatment is a concern, given that NF1 is a life-long disease. The experts also noted that the life expectancy of NF1 patients has been reported to be reduced by 10 to 15 years, although estimates of life expectancy with currently available medical management are unknown.
Assessing Response to Treatment
The clinical experts noted current clinical trials aim to address important outcomes that are used in clinical practice. However, given the heterogeneity of the disease, 1 expert highlighted that standardizing subjective measures (such as pain perception) across this population is an issue; thus, interpreting the results relies heavily on clinical judgment.
The clinical experts agreed that the most important outcomes in the management of pediatric patients with NF1 and symptomatic, inoperable PNs is the reduction or improvement in symptoms (i.e., reduced pain, improved function), as well as overall improvements in QoL and disease stabilization due to the potential for rapid, progressive growth of the tumours. One expert added that, due to disfigurement, cosmetic improvements are also likely important; however, the panel noted that current surgical management for cosmetic removal of most neurofibromas is not funded by provinces, except in special cases.
The clinical experts noted that volumetric MRI measurements are used in clinical trials to define disease progression; however, volumetric MRI is only used by the NIH for research studies, and is not available in Canadian clinical practice. The experts considered a change in planar tumour size of 20% to 25% to be indicative of response to treatment. One expert discussed the potential for symptomatic disease progression despite the lack of imaging evidence of progression and improvement in symptoms without reduction of tumour size. In addition, the experts emphasized that it is not always clear which tumours are the cause of symptoms when patients have large numbers of PNs, making it difficult to know when the disease is progressing. The experts also highlighted that tumours are frequently irregular in shape, making measurements about changes in tumour size difficult. As a result, the panel noted that response to treatment is multidimensional and must consider reductions in tumour sizes, changes in symptoms, and improvements in function and disfigurement. One clinical expert also emphasized the additional challenges of conducting MRI on young children. This often requires an anesthetic and can pose difficulties when determining clinically important growth based on child size compared to adolescent or adult size.
The experts stated that young children with NF1 and symptomatic PNs may initially be followed with MRI imaging every 3 months, in addition to annual follow ups with NF1 specialists to assess other features of the disease. Upon initiating treatment, the experts stated that patients would usually be seen weekly for a month, then every month, and if treatment is well tolerated, or disease stabilizes, then follow ups would be prolonged to every 6 months. The clinical experts also noted that imaging in young children often requires a general anesthetic.
Discontinuing Treatment
When deciding to discontinue treatment, the clinical experts agreed that treatment would be discontinued in patients who are not responding (as indicated by tumour growth, lack of stabilization, or lack of improvement of symptoms), or in patients with severe AEs that cannot be managed. The experts identified the risk of cardiotoxicity and retinal issues associated with the use of selumetinib as examples of significant AEs that would result in discontinuation. The experts also noted that the need for surgery to further debulk tumours could indicate that the treatment is not working and should therefore be discontinued. However, 1 clinical expert pointed out that selumetinib may be used in conjunction with debulking surgery, although the available evidence does not support this approach.
The experts noted that no firm treatment duration or date of discontinuation for selumetinib has been determined, but suggested that, similar to the SPRINT phase II trial, in clinical practice, patients would continue treatment until disease progression or toxicity. The experts agreed that the initial treatment authorization period for selumetinib should be 18 months.
Given the approved Health Canada indication for use in patients aged 2 to 18 years, the clinical experts discussed the use of selumetinib in patients aged 18 or older. Currently, 1 clinical expert has been providing selumetinib and other MEK inhibitor treatments to adults with PNs on an off-label basis through a philanthropic clinic and compassionate access to treatment. As a result, the clinical experts felt that patients may continue to receive benefits beyond the age of 18 years, although this requires further study.
Prescribing Conditions
The experts indicated that expertise in Canada in using selumetinib is sparse and limited to pediatric oncologists and neurooncologists in tertiary care hospitals, although the clinical setting is still evolving. The experts reported that selumetinib should be restricted to use in patients who are being followed in specialized centres, and that the usual approach to disease management begins with discussions with families about natural history, and shared decision-making in creating individualized treatment approaches.
According to the clinical experts, in Canada, only pediatric oncologists are prescribing selumetinib, as they have the experience and know-how to manage these patients. However, the experts noted that, with further insight and growing experience, NF1 experts who are pediatricians could likely continue to manage this oral treatment. Given the heterogeneity in the disease and the individualized approach to treatment, decisions often involve a multidisciplinary team of pediatricians, NF1 experts, neurooncologists, and nurse practitioners. The experts emphasized the importance of consulting other specialists, including surgeons, cardiologists, ophthalmologists, dermatologists, and pharmacists, to manage adverse effects and drug interactions.
The expert panel added that access to specialty clinics may be a limiting factor for patients in remote areas, emphasizing that patients would be required to attend in-person appointments for treatment initiation and imaging follow-up, as well as to assess safety. However, the experts considered the potential for remote monitoring, local bloodwork, or eye exams to be acceptable.
Clinician Group Input
This section was prepared by CADTH staff based on the input provided by clinician groups. The full original clinician group input received by CADTH is included in the stakeholder section at the end of this report.
Input for this review was received input from 1 clinician group, the CPBTC, which included 27 pediatric neurooncologists across Canada representing 16 children’s hospital who provide oncological care for children with neoplasms of the central nervous system and peripheral nerves. Information was gathered through shared clinical experiences of the included clinicians.
Overall, the clinician group input aligned with that given by the clinical expert panel convened by CADTH. It pointed out that no systemic therapies exist for treating NF1-associated PNs, which represent the major unmet need in this patient population, with surgical resection, if feasible, the only option currently available for patients. The clinician group emphasized that selumetinib has clearly shifted the current treatment paradigm and emerged as the standard of care as first-line therapy for patients with inoperable, symptomatic PNs. In terms of patients most in need of intervention, the clinician group agreed with the clinical expert panel that these patients include those in whom PNs are invading critical structures, causing a deformity, or causing functional impairment in activities of daily living such as walking, swallowing, or eating, although the clinician group noted it is impossible to determine which patients will and will not respond.
The clinician group also noted that, in Canada, treatment initiation with selumetinib is currently limited to pediatric oncologists, neurooncologists, or pediatric neurologists with an expertise in neurooncology. However, the CPBTC noted that general neurologists and pediatricians in jurisdictions outside of Canada have been able to prescribe selumetinib, and suggested that this practice would improve access for patients who live in remote areas. The CPBTC suggested that treatment with selumetinib in Canada could be initiated by oncologists and followed remotely in conjunction with local clinicians.
Finally, the CPBTC noted that many parents of children with NF1 also have NF1 themselves and are likely to have lower socioeconomic standing in part because of the disease. It was therefore the CPBTC’s opinion that many patients and parents of patients are more likely to lack private insurance to cover selumetinib, which could result in unequitable access in some parts of the country. The CPBTC group emphasized that children without private insurance who are also not eligible for the provincial public drug plans will need special consideration and that the drug in question urgently requires reimbursement and equitable access as a standard-of-care treatment for patients with NF1 and symptomatic PNs.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
Treatment options include pain management (supportive care) and surgical excision to reduce or remove PN tumours (current standard of care). There is a lack of appropriate comparators in this population. Are there medications marketed in Canada that are used for this condition off-label that would have been appropriate comparators in this population? | Surgical excision is the only treatment option to reduce the number and size of PNs; however, it is associated with numerous risks due to the number, size, location, and vascularity of PNs. Most large or extensive PNs cannot be completely excised. Although other MEK inhibitors are available, they have not been used to treat PNs. As no other treatment options are available to patients, selumetinib monotherapy would be the first and only medication available to treat NF1 patients with symptomatic inoperable PNs. |
Is there evidence to suggest that monotherapy with selumetinib will prevent or successfully treat MPNST? | There is no evidence to support the use of selumetinib in preventing the transformation to or treating MPNST. |
Considerations for initiation of therapy | |
Is NF1 genetically screened in newborns? How is it diagnosed? | NF1 is currently not part of any newborn screening program in Canada. Genetic testing results would not affect treatment decisions for PNs once a clinical diagnosis of NF1 has been established. NF1 is diagnosed via established clinical and/or symptomatic features, including the presence of CALMs, axillary and inguinal freckling, and the presence of neurofibromas. A diagnosis of PNs must often be confirmed by clinicians with expertise and may be aided by imaging (e.g., MRI), and in some cases, a biopsy is necessary to rule out malignancy. |
What is the proportion of operable vs. inoperable PNs seen in your practice? In an individual patient, would there be PNs that are operable and those that are inoperable? Another criterion in the SPRINT study was the ability to swallow intact capsules. What are your thoughts regarding this criterion and practicality? What happens when younger patients are unable to swallow capsules? Would you use this medication in asymptomatic, growing, inoperable PN? | Most large or extensive PNs cannot be completely excised surgically. Surgery is associated with many complications (e.g., bleeding risk, proximity to vital structures, and secondary injury), and due to the extensive and progressive nature of the disease, multiple surgeries may be required. While there may be many PNs in a patient, most do not cause symptoms, most are not eligible for treatment via surgery, and it is not always clear which PNs are symptomatic. Selumetinib is currently provided as capsules that must be swallowed; however, another formulation (oral suspension) is currently being developed for patients who are unable to swallow capsules. The majority of NF1 patients with PNs are asymptomatic, and the role of selumetinib in these patients remains unknown. However, this population remains of critical importance because of the progressive nature of NF1. There are many implications regarding the scope of selumetinib use in this population given the greater number of patients with asymptomatic PNs (e.g., resource use and monitoring). Selumetinib is currently not being used in these patients. |
Considerations for continuation or renewal of therapy | |
How widely is centrally read volumetric MRI accessible and available in the jurisdictions? | Access to volumetric MRI is not available as a standard of care for patients with NF1 in Canada and is limited worldwide. Volumetric MRI currently limited to clinical trials. |
Considerations for discontinuation of therapy | |
Are there situations in which selumetinib is discontinued, and then restarted? How long should patients be on this medication to see response clinically and/or radiographically? How else would clinical benefit defined apart from PN volume (e.g., improvement in pain, airway, or motor function in PN)? How is radiographic benefit defined? When is clinical or radiographic response seen in patients while on this medication? Are there any predictors of response for this medication? Is there an ideal treatment duration, or treatment range for patients? Is there any information on acquired resistance while on this medication? | Treatment with selumetinib may be discontinued in the presence of AEs, and then restarted once resolved. Patients and clinicians may also choose to discontinue treatment following evidence of disease stabilization, and then restart treatment at tumour progression or return of symptoms. Typically, in clinical trials, a 20% to 25% reduction in tumour volume is considered a response to treatment. However, given the lack of availability of volumetric MRI, measurement of treatment response is difficult and multidimensional, considering tumour growth on imaging, the worsening of symptoms such as pain, or deterioration in function (e.g., motor, airway, or bowel). Although there are no predictors of response in this population, it is estimated that up to 80% of patients will respond, while 20% will not, for unknown reasons. No end date for selumetinib treatment has been determined; as is inherent in phase II trial concepts, treatment continues until progression or unacceptable toxicity. In the absence of clinical benefit or toxicity, selumetinib could be initially given for 18 months. The decisions about stopping treatment are discussed on a case-by-case basis with patients and families. No evidence or information is available on acquired resistance to selumetinib. |
While on this medication, is there any time during the treatment course that a “drug holiday” could happen? Can a patient restart or resume this medication and obtain benefits after a treatment interruption for whatever reason? | There is no evidence to support the benefits of a drug holiday. However, the experts were of the opinion that treatment could be stopped in patients who achieve disease stabilization and then restarted at radiographic progression or worsening of symptoms. There are no biologic markers to determine whether the treatment needs to be continued; clinical judgment and discussion with patients and families will be used. |
Considerations for prescribing of therapy | |
The product monograph states that selumetinib should be discontinued if patients are unable to tolerate treatment after 2 dose reductions for AEs. What is the prevalence of discontinuation in practice with this medication? Are there any alternate dosing schedules in patients using this medication (e.g., intermittent dosing)? | The experts noted that approximately 20% of patients will not respond to treatment and will discontinue due to nonresponse. Dose reductions will occur according to the product monograph, and if patients are still not tolerating treatment after 2 dose reductions, treatment will be discontinued. There is no evidence regarding intermittent dosing of selumetinib. |
The product monograph states that treatment should be initiated by a physician experienced in the diagnosis and treatment of patients with NF1-related tumours. However, based on the potential toxicities, the patient would be managed in a multidisciplinary team (i.e., specialized settings), for optimal management. How are pediatric patients with this condition screened and managed, including follow-up, monitoring, and evaluating toxicities with regards to access points to the health care system? | Patients with NF1 may be under the care of specialists in the management of NF1 but prescribing of selumetinib is currently limited to pediatric oncologists and neurooncologists. The clinical setting for administering and monitoring patients is still evolving. Given the heterogeneity in the disease, and the individualized approach to treatment required, decisions often involve a multidisciplinary team of pediatricians, surgeons, NF1 experts, neurooncologists, dermatologists, nurse practitioners, cardiologists, ophthalmologists, and pharmacists to properly monitor the safety and toxicity of selumetinib. Remote monitoring, blood work, eye exams, and other follow-up is possible; however, patients would be required to attend in-person appointments for treatment initiation and imaging needs. |
Are there any situations in which selumetinib is combined with any other medication for this indication? | There is no evidence to support the use of selumetinib in combination with other therapies for this indication. |
Generalizability | |
The clinical trials presented included patients aged 2 to 18 years. Can selumetinib be started in patients aged > 18 years? Although not as common as NF1, would patients with NF2 and schwannomatoses related to genetic variants other than NF1 benefit from selumetinib? | Given that the SPRINT trial was conducted in patients aged 2 to 18 years, there is currently no evidence for using selumetinib in patients older than 18. However, selumetinib is currently provided off-label via compassionate access in Ontario through philanthropic efforts. There is an ongoing randomized controlled trial to determine the efficacy of selumetinib in patients who are older than 18. Other MEK inhibitors are available, although not necessarily for the treatment of NF1 and PNs. There is also no evidence to support the use of selumetinib in patients with NF2 or schwannomatosis. NF2 and schwannomatosis are rare genetic conditions that are entirely distinct from NF1. Alternative treatments are available for NF2 patients, and schwannomatosis is rarely, if ever, diagnosed in children. |
Care provision issues | |
Are supportive medications continued while on therapy? | Selumetinib is provided as monotherapy; however, supportive medications to manage the side effects of treatment (e.g., diarrhea and paronychia) would be used, as needed. |
How often do patients undergo MRIs for PNs? How often is imaging conducted for screening, and follow-up? | Imaging is generally conducted every 3 months initially, based on local standards, although they may be extended to every 6 months, or annually based on response to treatment. |
Selumetinib also includes vitamin E (e.g., 10 mg capsules contain 32 mg vitamin E as the excipient, TPGS, while 25 mg capsules contain 36 mg of vitamin E as TPGS). Is there a clinical relevance (e.g., bleeding risk) for this excipient in this population? | The use of natural health supplements is high in patients with NF1 and symptomatic PNs, and there is a risk of inadvertent toxic levels of natural supplements. Consultation with a pharmacist for patient counselling is required due to the numerous drug interactions associated with selumetinib to ensure patients do not take contraindicated medications. |
System and economic issues | |
Given the incidence of NF1 is 1 in 2,500 to 3,000 births: Maximum dose: 50 mg b.i.d., cost $306.50 × 2 × 365 days/year = $223,745/patient Minimum dose: 10 mg b.i.d., cost $122.60 × 3 × 365 days/year = $134,247/patient Plus cost of supportive care. | This was a comment from the drug programs to inform CDEC deliberations. |
Patient support program available by the company | This was a comment from the drug programs to inform CDEC deliberations. |
AE = adverse event; b.i.d. = twice daily; CALM = café-au-lait macule; CDEC = CADTH Canadian Drug Expert Committee; MEK = mitogen-activated protein kinase; MPNST = malignant peripheral nerve sheath tumour; NF1 = neurofibromatosis type 1; NF2 = neurofibromatosis type 2; PN = plexiform neurofibroma; TPGS = alpha-tocopherol polyethylene glycol succinate.
Clinical Evidence
The clinical evidence included in the review of selumetinib is presented in 3 sections. The first section, the systematic review, includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those studies that were selected according to an a priori protocol. The second section includes indirect evidence from the sponsor and indirect evidence selected from the literature that met the selection criteria specified in the review. The third section includes sponsor-submitted long-term extension studies and additional relevant studies that were considered to address important gaps in the evidence included in the systematic review.
Systematic Review (Pivotal and Protocol-Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of selumetinib 10 mg and 25 mg twice daily for the treatment of pediatric patients aged 2 years and above with NF1 who have symptomatic, inoperable PNs.
Methods
Studies selected for inclusion in the systematic review included pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria listed in Table 5. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
Table 5: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Population | Pediatric patients with NF1 aged 2 years and above who have symptomatic, inoperable PN. Subgroups:
|
Intervention | Selumetinib (10 mg and 25 mg oral capsules) 25 mg/m2 based on body surface area, twice daily |
Comparator |
|
Outcomes | Efficacy outcomes:
Harms outcomes:
|
Study designs | Published and unpublished phase III and IV randomized controlled trials |
NF1 = neurofibromatosis type 1; PN = plexiform neurofibroma; vs. = versus.
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy according to the PRESS Peer Review of Electronic Search Strategies checklist.35
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946–) via Ovid and Embase (1974–) via Ovid. All Ovid searches were run simultaneously as a multifile search. Duplicates were removed using Ovid deduplication for multifile searches, followed by manual deduplication in EndNote. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. Three component searches were conducted. The first used the search concepts Koselugo and NF1. The second search used the concepts of Koselugo and pediatrics. A third search was conducted on Koselugo and CADTH-developed search filters were applied to limit retrieval to randomized controlled trials or controlled clinical trials. Clinical trials registries searched included the US National Institutes of Health’s clinicaltrials.gov, WHO’s International Clinical Trials Registry Platform search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. Appendix 1 provides detailed search strategies.
The initial search was completed on November 16, 2022. Regular alerts updated the search until the meeting of the CADTH Canadian Drug Expert Committee on March 22, 2022.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool For Searching Health-Related Grey Literature checklist.36 Included in this search were the websites of regulatory agencies (US FDA and European Medicines Agency). Google was used to search for additional internet-based materials. Appendix 1 provides more information on the grey literature search strategy.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to include in the review, and differences were resolved through discussion.
Findings From the Literature
One study from the literature was selected for inclusion in the systematic review (Figure 1). The included studies are summarized in Table 6. A list of excluded studies is presented in Appendix 2.
Table 6: Details of Included Studies
Study detail | SPRINT phase II (stratum 1) |
|---|---|
Designs and populations | |
Study design | Phase II, open-label, single-arm, multicentre study |
Locations | 4 centres in the US |
Patient enrolment dates | First patient enrolled: August 12, 2015 Last patient enrolled: August 22, 2016 |
Enrolled (N) | 50 |
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | 25 mg/m2 selumetinib twice daily orally |
Comparator(s) | NA |
Duration | |
Phase | |
Screening | NR |
Treatment | 28-day cycles, with no rest period between cycles until disease progression or unacceptable toxicity |
Follow-up | 5 years after completion of selumetinib, or total duration of 7 years, whichever was longer |
Outcomes | |
Primary end point | Objective response rate (CR or PR, with PR defined as target PN volume decrease ≥ 20% compared to baseline) |
Secondary and exploratory end points | Secondary:
|
Notes | |
Publications | Gross et al. (2020) |
CPAP = continuous positive airway pressure; CR = complete response; CTCAE v4 = Common Terminology Criteria for Adverse Events Version 4; DCO = data cut-off; IOP = intraocular pressure; MEK = mitogen-activated protein kinase; MPNST = malignant peripheral nerve sheath tumour; NA = not applicable; NCI = National Cancer Institute; NF1 = neurofibromatosis type 1; NR = not reported; NYHA = New York Heart Association; PN = plexiform neurofibroma; POB = Pediatric Oncology Branch; PR = partial response; PROMIS = Patient-Reported Outcomes Measurement Information System; QTc = corrected QT interval; QTcF = QT interval corrected using Fridericia’s correction formula; ULN = upper limit of normal.
Source: SPRINT Phase II Stratum I Clinical Study Report (June 29, 2018, DCO).19
Description of Studies
The SPRINT phase II study is a phase II, open-label, single-arm, multicentre study that evaluated the efficacy of selumetinib in pediatric patients with NF1 and inoperable PN. The primary outcome was the ORR (confirmed PR and complete response [CR]) determined by change in PN volumes through volumetric MRI.19
For the SPRINT phase II trial, patients were enrolled in 1 of 2 strata based on their PN-related morbidity as determined by the clinical team at the time of study entry:19
stratum 1: PN-related morbidity present at enrolment
stratum 2: no significant PN-related morbidity present at enrolment, but potential for development of PN morbidity.
Based on the reimbursement request and approved Health Canada indication, only results for stratum 1 are summarized in this report, which includes only patients with symptomatic PNs. A total of 50 patients were enrolled into stratum 1 and received 25 mg/m2 selumetinib twice daily on a continuous 28-day cycle.19
Two DCOs were submitted for the SPRINT phase II trial.19,20 The primary DCO occurred on June 29, 2018, and an updated DCO occurred on March 31, 2021, providing a further 2 years and 9 months of follow-up, 4 years and 7 months after the last patient enrolled in stratum 1 of the SRINT phase II trial, for a maximum follow-up of 5.6 years.
The SPRINT phase II study was sponsored by the Cancer Therapy Evaluation Program, conducted by the NCI POB at 4 study centres in the US, and supported by AstraZeneca through a cooperative research and development agreement.19 No Canadian investigative sites were included,19 and it was unclear if any patients from Canada were enrolled.
Populations
Inclusion and Exclusion Criteria
The inclusion and exclusion criteria for stratum 1 of the SPRINT phase II trial are summarized in Table 6. Briefly, eligible patients were those between the ages of 2 and 18 years with a diagnosis of NF1 and inoperable PNs, defined as those that could not be completely surgically excised without risk substantial morbidity due to proximity to vital structures, invasiveness, or high vascularity. Patients in stratum 1 were those with at least 1 clinically relevant PN-related morbidity as determined by the clinical team at the time of study entry, which was defined as (but not limited to) head and neck PN that could have compromised the airway or great vessels, paraspinal PN that could have caused myelopathy, brachial or lumbar plexus PN that could have caused nerve compression and loss of function, PN that could have resulted in major deformity (e.g., orbital PN) or were significantly disfiguring, PN of the extremity that could have caused limb hypertrophy or loss of function, and painful PNs. Volumetric MRI was used to classify patients’ PNs as typical (nodular component of PN was < 30%), nodular (nodular component of PN was ≥ 30%), or solitary nodular (target PN was a single nodular PN). Patients with volumetric MRI scans within 3 years before enrolment were to have these submitted to the NCI POB to estimate the target PN rate of growth status at enrolment (progressive, nonprogressive, or unknown). Patients were also required to be able to swallow whole capsules.19
Baseline Characteristics
Baseline characteristics for patients enrolled in stratum 1 of the SPRINT phase II are summarized in Table 7. The mean age of patients was 10.3 years (SD = 3.92), ranging from 3.5 to 17.4 years of age. Most patients were white (42 [84.0%]) and male (30 [60.0%]). The mean time between diagnosis of NF1 to start of selumetinib treatment was 9.03 years (range = 2.0 to 16.5 years), and the mean time between diagnosis of PN to start of selumetinib treatment was 7.55 years (range = 0.7 to 16.5 years). The target PN was chosen at study entry by the investigator as the most clinically relevant PN causing morbidity at baseline. Most target PNs were classified as typical (45 [90.0%]), and the mean target PN volume was 837.11 mL (SD = 925.011), with values ranging from 5.6 to 3,820.0 mL. The median number of morbidities arising from each target PN was 3 (range = 1 to 4), with || patients having | morbidities. Disfigurement as a result of PNs was observed in 44 patients (88%), with 33 patients (66.0%) also having motor dysfunction. The most common location of target PNs was the neck and trunk, and the trunk and extremity (12 [24.0%], each). At baseline, 21 patients (42.0%) and 15 patients (30.0%) were classified as having progressive and nonprogressive PN status, respectively, while 14 patients (28.0%) had unknown PN status.19,20
Forty-seven patients (94.0%) had at least 1 disease-related symptom at baseline. The most commonly reported symptoms at baseline were muscular weakness (34.0%), limb asymmetry (32.0%), tumour pain (32.0%), joint ROM decreased (26.0%) and scoliosis (26.0%). In total, || ||||||| of patients had previous disease-related treatment modalities, primarily interferons and imatinib, and || ||||||| patients had at least 1 prior PN- or NF1-related surgical procedure.19,20
Table 7: Summary of Baseline Characteristics (Full Analysis Set)
Characteristic | SPRINT phase II (selumetinib 25 mg/m2 twice daily), N = 50 |
|---|---|
Demographic characteristics | |
Age (years), n (%) | |
Mean (SD) | 10.3 (3.92) |
Median (range) | 10.2 (3.5 to 17.4) |
Sex, n (%) | |
Male | 30 (60.0) |
Female | 20 (40.0) |
Race, n (%) | |
White | 42 (84.0) |
Black or African American | 4 (8.0) |
Asian | 1 (2.0) |
Multiple or unknown | 3 (6.0) |
Body surface area, m2 | |
Mean (SD) | 1.127 (0.3401) |
Median (range) | 1.040 (0.67 to 1.93) |
Disease and clinical characteristics | |
Lansky performance status scorea | |
Mean (SD) | 86.8 (8.10) |
Median (range) | 90 (70 to 100) |
Time from diagnosis of NF1 to start of selumetinib (years) | |
Mean (SD) | |||| ||||||| |
Median (range) | |||| |||||||||| |
Time from diagnosis of PN to start of selumetinib (years) | |
Mean (SD) | |||| ||||||| |
Median (range) | |||| |||||||||| |
Target PN volume, mL | |
Mean (SD) | 837.11 (925.011) |
Median (range) | 487.50 (5.6 to 3,820.0) |
Target PN classification,b n (%) | |
Typical | || |||||| |
Nodular | | ||||| |
Solitary nodular | | ||||| |
Target PN status, n (%) | |
Progressive | 21 (42.0) |
Nonprogressive | 15 (30.0) |
Unknown | 14 (28.0) |
Target PN morbidity assignment, n (%) | |
Disfigurement | 44 (88.0) |
Motor dysfunction | 33 (66.0) |
Airway | 16 (32.0) |
Bowel and/or bladder dysfunction | 10 (20.0) |
Orbital (vision) | 10 (20.0) |
Other dysfunctionc | 12 (24.0) |
Number of target PN morbiditiesd | |
Median (range) | 3.0 (1 to 4) |
Target PN location, n (%) | |
Neck/trunk | 12 (24.0) |
Trunk/extremity | 12 (24.0) |
Head | 9 (18.0) |
Head/neck | 8 (16.0) |
Trunk | 5 (10.0) |
Extremity | 4 (8.0) |
Target PN pain present, n (%) | |
Yes | 26 (52.0) |
No | 22 (44.0) |
Missing | 2 (4.0) |
Prior treatment, n (%) | |
Patients with previous disease-related treatment modalities | || |||||| |
Medical therapye | || |||||| |
Surgeryf | || |||||| |
Radiation | | ||||| |
DCO = data cut-off; NF1 = neurofibromatosis type 1; PN = plexiform neurofibroma; SD = standard deviation.
aLansky performance status was assessed in patients who were aged 16 years or younger and Karnofsky performance status was assessed in patients who were older than 16. A total of 47 patients were less than 16 years, while only 3 patients were 16 or older.
bClassification based on imaging.
cThe “other dysfunction” category included patients with PN pain, swallowing, disfigurement and sensory neuropathy. Patients with PN pain were captured under the pain morbidity category (i.e., pain present = yes) and sometimes also captured in the “other dysfunction” field.
dDoes not include symptoms recorded as “other.” Pain was included as a PN-related morbidity.
eMedical therapy directed at other NF1 tumours may also be included.
fSurgery included 4 patients who had 1 or 2 biopsies only and no other prior PN-related surgical procedures. Other NF1-related surgeries may also be included.
Source: SPRINT Phase II Stratum I Clinical Study Report (June 29, 2018, DCO).19
Interventions
The recommended phase II dose for selumetinib was determined during the SPRINT phase I trial. In the SPRINT phase II trial, selumetinib was administered orally at a dosage of 25 mg/m2 twice daily (approximately every 12 hours) based on BSA, continuously for 28-day cycles with no rest periods between cycles. Selumetinib was supplied as 10 mg (white) and 25 mg (blue) capsules. Patients were instructed to take the dose of selumetinib on an empty stomach (no food or drink other than water for 2 hours before and 1 hour after dosing) with water only. The capsules were not to be crushed or broken and were to be swallowed whole. Doses were rounded to the nearest 5 to 10 mg using a dosing nomogram. Selumetinib dosing was capped at 50 mg when BSA was 1.9 m2 or greater. Selumetinib dosing was adjusted for changes in BSA according to the dosing nomogram.19
The duration of treatment with selumetinib was determined to ensure that patients had the opportunity to derive a benefit but was limited based on the following:19
For patients with documented disease progression within approximately 1.5 years before study entry (defined as ≥ 20% increase in the size of PN or ≥ 13% increase in the product of the longest 2 perpendicular diameters, or ≥ 6% increase in the longest diameter), there was no limit to the duration of treatment if the patient met the requirements for further treatment.
For patients with no previous documented history of disease progression within the 1.5 years before study entry, the duration of the study was to be limited to 2 years if no imaging response (i.e., volume decreased by ≥ 20%) was observed.
Patients who were removed from treatment after 2 years for reasons other than toxicity or progression and who had stable disease were continued to be monitored with a volumetric MRI analysis every 4 to 6 months. If the PN demonstrated some growth (volume increase ≥ 15%) within approximately 2 years of stopping selumetinib, treatment with selumetinib could be restarted with the goal of stopping further PN growth. Patients were re-consented on the study and had to meet all eligibility criteria with the exception of prior treatment with selumetinib or another specific MEK 1 or 2 inhibitor before restarting therapy. In these patients, treatment could continue as long as the PN remained stable or responsive (< 20% increase in the PN volume).
For patients who showed an imaging response, the treatment duration was not to be limited unless the patient experienced subsequent disease progression or met other off-treatment criteria. In all cases, treatment could be discontinued earlier at the discretion of the institutional principle investigator if this was considered to be in the best interest of the patient.
In all cases, treatment could be discontinued earlier at the discretion of the institutional principle investigator if this was considered to be in the best interest of the patient. Patients were to be followed up for 5 years after completing selumetinib, or for a total duration of 7 years, whichever was longer.19
Dose Modifications, Reductions, or Interruptions
Selumetinib was to be withheld in patients with toxicities requiring dose modification. Selumetinib doses held while recovering from toxicity were not made up and the cycle remained 28 days or until recovery from toxicity. If the toxicity resolved to meet study parameters or grade 1 or higher on the Common Terminology Criteria for Adverse Events (CTCAE) Version 4 within 21 days of drug interruption, selumetinib was resumed at a dose reduced by 25% to 33%. Doses reduced for toxicity were not re-escalated, with the exception that the dose could be increased to account for an increase in BSA at the time of on-study evaluations (dosing was rounded to the nearest 5 to 10 mg). If toxicity did not resolve to meet study parameters within 21 days, the patient was removed from selumetinib treatment, except in the event that the patient was receiving clear clinical benefit and recovery occurred within 3 months of discontinuation.19
If target PN volume measurements subsequent to a dose reduction demonstrated an increase of 10% or greater from best response in target PN volume, but had not met criteria for progressive disease, and selumetinib was well tolerated at this reduced dose, dosing could be resumed at the dose level before the dose reduction. In this instance, selumetinib was administered twice daily every 5 days, followed by 2 days of rest.19
If toxicity recurred in any patient who had resumed treatment at the reduced dose, the dose could be reduced a second time using the same criteria, with the exception of cardiotoxicity, for which only 1 dose reduction was allowed. If toxicity recurred after 2 dose reductions, the patient was removed permanently from selumetinib. Patients removed from selumetinib for toxicity were followed until resolution of toxicity and off-study criteria were met.19
Treatment Discontinuation and Withdrawal From Study
Patients in the SPRINT phase II trial could discontinue treatment and assessments at any time, or at the discretion of the investigator. Patients could be discontinued from selumetinib in the following situations:19
nonmedical or administrative reasons:
patient refusal
investigator decision in the interest of the patient
serious protocol violation as determined by the investigator
noncompliance with study requirements
toxicity requiring dose modification, which did not resolve per study parameters within 21 days of selumetinib interruption, except when the patient had shown clear clinical benefit
patients with clinical or imaging evidence of progressive disease on treatment following any treatment cycle
patients who developed a concurrent serious medical condition that could have precluded or contraindicated further administration of selumetinib
pregnancy was to result in immediate removal from selumetinib
patients who underwent complete surgical resection of their PN (thus rendering them with no evidence of disease)
other reasons:
death
lost to follow-up
withdrawal of consent for any further data submission
completion of long-term safety evaluations
screen failure.
Concomitant Medications
Restricted, prohibited, and permitted concomitant medications in the SPRINT phase II study included:
Corticosteroids were allowed for control of symptoms related to the underlying NF1 or for other reasons.
Throughout the study, patients were instructed to avoid changes to, or the addition of concomitant medications. Unless considered clinically indicated, patients were to avoid taking other additional nonstudy medications that could interfere with selumetinib. Patients were to avoid medications that are known to either induce or inhibit the activity of hepatic microsomal isoenzymes CYP1A2, CYP2C19, and CYP3A4, as this may interfere with the metabolism of selumetinib.
Appropriate antibiotics, blood product support, anti-emetics and general supportive care were to be used as indicated.
Selumetinib was to be administered with caution in patients who were also receiving concomitant coumarin anticoagulant medications (e.g., warfarin). These patients were to have their international normalized ratio monitored or anticoagulant assessments conducted more frequently, and the dose of the anticoagulant adjusted accordingly.
No other cancer chemotherapy, radiation, immunotherapy, biologic therapy, hematopoietic growth factors, or investigational agents was permitted while receiving selumetinib.
Use of supplemental vitamin E, including any multivitamin containing vitamin E, was also restricted.19
Outcomes
A list of efficacy end points identified in the CADTH review protocol that were assessed in the clinical trials included in this review is provided in Table 8 and summarized in the following section. A detailed discussion and critical appraisal of the outcome measures is provided in Appendix 4.
Table 8: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure | SPRINT phase II |
|---|---|
Objective responses rate | Primary |
Duration of response | Secondary |
Progression-free survival | Secondary |
Time to response | Secondary |
Numeric Rating Scale-11 | Secondary |
Pain Interference Index | Secondary |
Patient-Reported Outcomes Measurement Information System | Secondary |
Quality of Life (Pediatric Quality of Life Inventory) | Secondary |
Global Impression of Change scale | Secondary |
Functional outcomes (motor function [strength, range of motion, 6MWT], airway function [PFTs], bladder/bowel function [DVQ], orbital function [acuity tests, exophthalmometry]) | Secondary |
6MWT = 6-minute walk test; DVQ = Dysfunctional Voiding Questionnaire; PFT = pulmonary function test.
Primary Efficacy End Point
The primary efficacy end point of the SPRINT phase II study was the ORR, defined as the percentage of patients with a CR or confirmed PR (defined as target PN decrease ≥ 20% compared with baseline; confirmed when observed again within 3 to 6 months). The ORR was based on the NCI POB central analysis (i.e., the primary analysis) of volumetric MRI of the target PN. Response was assessed before cycles 5, 9, 13, 17, 21, 25 and then after every 6 cycles (precycles 31, 37, and 43) until discontinuation of selumetinib.19 Response was measured using the REiNS criteria. Definitions of response per REiNS criteria were:37
CR: disappearance of the target PN
PR: decrease in the volume of the target PN by 20% or more compared with baseline; the PR was considered unconfirmed at the first detection and confirmed when observed again within 3 to 6 months
Stable disease: insufficient volume change to qualify for either PR or progressive disease
Progressive disease: increase in the volume of the target PN by 20% or more compared with baseline or the time of best response after documenting a PR; the appearance of new PN (with the exception of new discrete subcutaneous neurofibromas) or unequivocal progression of existing clinically relevant nontarget PN was also considered progressive disease; the clinical appearance of new discrete subcutaneous neurofibromas did not qualify for disease progression.
Additional assessments of ORR included best objective response (BOR), defined as the best response recorded from the start of treatment until progression or the last evaluable volumetric MRI assessment in the absence of progression, which was summarized by category (confirmed PR, unconfirmed PR, stable disease, progressive disease or not evaluable [NE]), and ORR by PN status at enrolment (progressive, nonprogressive, or unknown).19
Secondary Efficacy End Points
Secondary efficacy outcomes of the SPRINT phase II trial included DOR, PFS, and time to response (TTR), and were based on the NCI POB central analyses of volumetric MRIs of target PN.19
Other secondary end points included clinical outcome assessments or PROs to evaluate pain, motor function, bowel and bladder function, HRQoL, and functional measures for pain, respiratory function, visual function, physical function, and changes in pain intensity or other morbidities.19 A summary of PROs, and respondents for specific measures is provided in Table 9, with a detailed discussion of these outcomes supplied in Appendix 4.
Duration of Response
Only patients who had a CR or confirmed PR were included in the analysis of DOR. The DOR was derived from precycle volumetric MRI assessments and was defined as the first documented response (subsequently confirmed) until documented progression on treatment or death in the absence of disease progression (i.e., precycle volumetric MRI assessment of PFS event or censoring — precycle volumetric MRI assessment of first response, where each cycle was 28 days). If a patient did not progress following a response, then their DOR used the PFS-censoring MRI assessment.19
Progression-Free Survival
PFS was a secondary outcome of the SPRINT phase II trial and was defined as the time from initiation of treatment until the precycle volumetric MRI assessment of objective disease progression on treatment or death. Patients who did progress or died by the time of analysis were censored at their last evaluable precycle MRI assessment. However, if the patient progressed or died after 2 or more missed precycle MRI assessments, the patient was censored at the time of the latest evaluable precycle MRI assessment.19
Time to Response
TTR was a secondary outcome of the SPRINT phase II trial and was defined as the time from study treatment initiation until the precycle volumetric MRI assessment of the first documentation of CR or a subsequently confirmed PR. The precycle volumetric MRI assessment of the first documented response was to coincide with that used for the DOR end point. Only patients who had achieved a CR or a confirmed PR were evaluated for TTR. Patients who had not progressed at the time of analysis were censored at their last evaluable precycle MRI assessment.19
Clinical Outcome Assessments
Table 9: Summary of PROs and Functional Evaluations by Respondent and Age
Outcome measure | Respondent | Age (years) | ||
|---|---|---|---|---|
2 to 4 | 5 to 7 | 8 to 18 | ||
PRO questionnaires | ||||
Pediatric Quality of Life Inventory | Self-reporta | — | — | Y |
Parent/guardian reportb | Y | Y | Y | |
Numerical Rating Scale-11 | Self-report | — | — | Y |
Pain Interference Index | Self-report | — | — | Y |
Parent/guardian report | — | Y | Y | |
Global Impression of Change | Self-report | — | - | Y |
Parent/guardian report | — | Y | Y | |
Mobility and upper extremity (PROMIS) | Self-report | — | - | Y |
Parent/guardian report | — | Y | Y | |
Bowel/bladder questionnaire | Self or parent/guardian report | Y | Y | Y |
Functional evaluations | ||||
Vision | ||||
Acuity testing | — | Y | Y | Y |
Exophthalmometry | — | Y | Y | Y |
Airway | ||||
Sleep study | — | Y | Y | Y |
Pulmonary function test | — | Y | Y | Y |
Endurance | ||||
6-minute walk test | — | — | Y | Y |
Motor | ||||
Strength | — | Y | Y | Y |
Range of motion | — | Y | Y | Y |
Leg length evaluation | — | Y | Y | Y |
Grip and key pinch strength | — | Y | Y | Y |
Grooved pegboard | — | — | Y | Y |
DCO = data cut-off; PRO = patient-reported outcome; PROMIS = Patient-Reported Outcomes Measurement Information System; Y = yes.
aSeparate child: 8 to 12 and adolescent: 13 to 18 forms.
bSeparate toddler: 2 to 4; young child: 5 to 7; child: 8 to 12; and adolescent: 13 to 18 forms.
Source: SPRINT Phase II Stratum 1 Clinical Study Report (June 29, 2018, DCO).19
Pain: The effect of selumetinib on pain was assessed through self-evaluation of pain intensity on the tumour selected by the physician, on the tumour selected by the patient, overall tumour pain, and other pain, as assessed by the NRS-11, and the interference of pain with daily functioning as rated by the patient and by the parent, using the PII.19
Numerical Rating Scale-11: The NRS-11 is a self-evaluation of pain intensity and consists of 4 questions scored on a scale of 0 (no pain) to 10 (worst pain imaginable). The NRS-11 was only used in patients aged 8 years and older. The primary outcome for the self-report NRS-11 was the rating of pain in 1 specific PN (e.g., the “target tumour”). Patients who had their baseline evaluation using the earlier version of the NRS-11 (Version 1), which did not specifically indicate the physician-selected target PN, were included in the primary outcome analysis if the self-selected PN and physician-selected PN matched. The revised NRS-11 (Version 2) was used for all patients enrolled from November 2015. Pain was rated across all study precycles for the same PN each time. Pain palliation was also defined for the primary outcome and was based on reduction in pain intensity, and stability or reduction in analgesic use.19
Clinically meaningful thresholds (CMTs) of change for the NRS-11 were based on literature for other populations and were defined as a decrease of 1 or 2 points.34,38-41 No published data focusing on the reliability, validity, responsiveness to change, or minimal important difference (MID) for the NRS-11 was identified for pediatric patients with NF1. Additional information on the measurement properties in NRS-11 is provided in Table 39 of Appendix 4.
Pain Interference Index: The PII is a 6-item scale that assesses the extent to which pain has interfered with an individual’s daily activities in the past 7 days. Items are rated on a 7-point Likert scale (0 = not at all to 6 = completely), and the total score is the mean of the completed items. The total score was computed if at least 50% of the items were answered. Higher scores indicated more interference with daily activities.19
Reliability and validity of the PII were assessed in patients with NF1 or cancer, as well as in caregivers.42 Cronbach alphas were 0.84 for the patient PII and 0.94 to 0.96 for the parent PII. Additionally, patient and parent scores on the PII were correlated (r = 0.62). The PII scale demonstrated good validity based on correlation with other pain scales. In the subgroup of NF1 patients (N = 31), the correlation between PII and NF1 Disease Severity Scale ratings was assessed. Patients with pain rated as moderate to severe scored significantly higher on PII than did those with pain rated as mild. Responsiveness to change was measured in a small study of adolescents and young adults with NF1 (N = 12), although patient-reported pain did not significantly decrease.43 No published data focusing on the MID was identified. Additional information on the measurement properties in PII is provided in Table 39 of Appendix 4.
Motor function: Analysis of motor function included only patients with motor morbidity at enrolment, with the exception of the leg length discrepancy analysis and the grooved pegboard analysis. These analyses included only patients with lumbosacral plexus or lower limb PN and patients aged 5 years and older at enrolment with cervical, upper thoracic, or upper limb PN. Motor function in all patients with motor morbidity was assessed using the PROMIS Pediatric Short Form Version 1.0 – Mobility 8a and PROMIS Pediatric Short Form Version 1.0 – Upper Extremity 8a, and included a recall period of 7 days.19
Patient-Reported Outcome Measurement Information System: The PROMIS was completed by both the patient and the parent. Physical functioning scales assessed the domains of mobility and upper extremity function and included mobility items such as “I could walk upstairs without holding on to anything” and upper extremity items such as “I could button my shirt or pants.” The short forms consisted of 8 items using a 5-point Likert scale (0 = unable to do, 4 = can do without any difficulty), with higher scores indicating better physical functioning. Raw scores were converted to t scores, which were based on reference data from the US general population (mean = 50 and SD = 10).19 CMTs used for the analysis of PROMIS were:
mobility self-report: 2.26 (raw score), 3.27 (transformed)
mobility parent-report: 3.36 (raw score), 3.66 (transformed)
upper extremity self-report: 3.73 (raw score), 6.45 (transformed)
upper extremity parent-report: 4.64 (raw score), 6.18 (transformed).
No published data focusing on the reliability, validity, responsiveness to change, or MID for the PROMIS were identified for pediatric patients with NF1. Additional information on the measurement properties of the PROMIS are described in Table 39 of Appendix 4.
Functional Evaluations
The primary outcomes for assessing motor function were strength for each muscle group in all patients with PN causing motor dysfunction, weakness, or cord compression were strength evaluations (Medical Research Council 5-point Likert scale) for all muscle groups, and ROM (measured in degrees) of all joints at baseline. Each joint (ROM) and muscle group (strength) was allocated to a location quadrant based on the anatomic location of the PN (upper or lower; right, left, or bilateral). A strength score was calculated as the average strength of all muscles in the same body quadrant as the target PN. Similarly, the ROM score was calculated as the sum of all the degrees of movement for each of the joints in the same quadrant as the target PN; higher ROM scores indicate more degrees of movement.19
Airway function: Analysis of airway function included only patients with airway morbidity at enrolment. All patients with airway PN (upper airway or extrathoracic, and lower airway or intrathoracic) underwent functional evaluations, including sleep studies, evaluation of endurance using the 6-minute walk test (6MWT) and pulmonary function tests (PFTs). The primary outcome for this analysis was the Apnea-Hypopnea Index (AHI) as measured in events per hour, forced expiratory volume in the first second (FEV1) measured in litres, and respiratory resistance at 20 Hz (R20). For pre-school children, forced expiratory volume in the first 0.75 seconds (FEV0.75) was used in place of FEV1.19
A change of 12% or more in FEV1 or FEV0.75 was classified as an improvement, as recommended by the REiNS functional group.44 Functional improvement was defined as a decrease of 20% or more in R20. (The REiNS functional group recommended the threshold of 20% or more for R10, but R20 was used in the SPRINT phase II study.19)
Bowel and bladder function: Analysis of bowel and bladder function included only patients with bowel and/or bladder morbidity at enrolment. Bowel and bladder functionality was measured with the Dysfunctional Voiding Questionnaire (DVQ) and was only completed by patients aged 8 years or older or by the parent or guardian of a patient with bowel and/or bladder morbidity.45 The DVQ consists of 14 items measuring bowel and/or bladder dysfunction on a 5-point Likert scale. The last question requested feedback on the ease of completing the questionnaire and was not included in the total score. Scores ≥ 11 (out of 52) were the threshold for bowel and bladder dysfunction. The absolute change in DVQ score from baseline was calculated as each post-baseline value minus the baseline value.19,45
Vision function: Analysis of vision function included only patients with vision morbidity at enrolment and included measurements of visual acuity and the extent of exophthalmos. Visual acuity was measured using the HOTV chart, or Teller acuity cards if the patient was too young to reliably perform HOTV testing. The HOTV results were reported as the logarithm of the minimum angle of resolution (logMAR), and Teller acuity was recorded in cycles per centimetre, which was converted to a logMAR by the study team.19
Exophthalmos was measured using exophthalmometry (in millimetres). Change from baseline in exophthalmometry and using the logMAR were classified as improvement, no change, or deterioration according to the score at each precycle visit.
A decrease in the logMAR of more than 0.2 and a decrease in exophthalmos of more than 2 mm were considered clinically meaningful improvements, as recommended by the REiNS functional group.19
Global Impression of Change
Global impression of change (GIC) in tumour pain, overall pain, and tumour-related morbidities compared to baseline were measured by the GIC scale, consisting of 3 questions scored on a 7-point scale (1 = very much improved to 7 = very much worse), and was performed by patients and parents. The patient self-report and parent-report ratings for each of the 3 items were analyzed and reported separately.19
Pediatric Quality of Life Inventory
General HRQoL was measured using the PedsQL Version 4.0. The PedsQL assesses function in 4 domains:19
physical functioning (8 items)
emotional functioning (5 items)
social functioning (5 items)
school functioning (5 items).
Each item is scored on a 5-point Likert scale (0 = never a problem; 4 = almost always a problem). For patient-reported and parent-reported measures, items were reverse-scored and linearly transformed to a 0-to-100 scale, with higher scores indicating better HRQoL. Scale scores were computed as the sum of the items divided by the number of items answered. If more than 50% of the items in the scale were missing, the scale score was not computed.46 A total scale score was also derived from the sum of all the items divided by the number of items answered on all the scales. The primary outcomes for HRQoL were the total scale score of the patient-reported PedsQL for children older than 8 years and the total scale score of the parent-reported PedsQL administered to parents of children aged 2 years and older. Secondary outcomes for HRQoL were the mean scores of the 4 domains (physical, emotional, social, and school) of the patient-reported scale completed by children older than 8 years and the 4 domain mean scores from the parent-reported PedsQL administered to parents of children aged 2 years and older.19
Patients were classified with impaired global HRQoL (yes/no) at each precycle visit, using the transformed scores if their total or domain scores fell 1 SD below the population sample mean as reported by Varni et al. (2003).19,46 No published data focusing on the reliability, validity, responsiveness to change, or MID for the PedsQL were identified for pediatric patients with NF1. Additional information on the measurement properties of the PedsQL are described in Table 39 of Appendix 4.
Quality of Life Background Form
A QoL Background Form was completed by the parent of all patients aged 2 to 18 years to document information about demographics, use of pain medication, and NF1 disease severity. Descriptive statistics (counts and percentages) were provided for each question asked.19
6-Minute Walk Test
All patients aged 5 years or older at enrolment, with a lower extremity PN, cord compression, or airway PN (including patients with tracheostomy, providing they can walk independently) underwent endurance testing using the 6MWT. The absolute change from baseline was calculated as each post-baseline value minus the baseline value. CMT was defined as a change of 30 m (improvement or deterioration) or no change (otherwise).19
General PN Symptoms
General PN symptoms were measured by the PN symptoms checklist, which consisted of 36 symptoms scored on a 5-point Likert scale (0 = not at all; 1 = a little; 2 = some; 3 = pretty much; 4 = a lot) for each patient at each assessment.19
Harms Outcomes
Safety and tolerability were assessed in terms of AEs, SAEs, deaths, laboratory data, vital signs, electrocardiogram, left ventricular ejection fraction, physical examination, and performance status (Lansky and Karnofsky), ophthalmology, and bone density examination. Adverse events and SAEs were summarized by system organ class and preferred term. Severity of AEs was defined using the Medical Dictionary for Regulatory Activities (Version 21.0) and CTCAE Version 4.0.19
Statistical Analysis
Sample Size and Power Calculation
The sample size for the primary objective of ORR in patients with PN-related morbidity was based on a target response rate to exclude 15% with a lower 2-sided 95% CI. With 50 total evaluable symptomatic patients, an exact binomial test with a nominal 1-sided 2.5% significance level had 90% power to detect the difference between a null hypothesis response rate of 15% and an alternative hypothesis response rate of 36%. With 14 or more responses out of the 50 patients, the lower limit of the exact 2-sided 95% CI for the response rate would be 16.2% or greater. Thus, 14 or more responses among 50 evaluable patients was consistent with results that would statistically significantly exceed a 15% true response rate based on a 2-sided CI.19
Statistical and Analytical Plans
At baseline, the most clinically relevant tumour was selected by the treating physician as the target lesion and was used to determine response to treatment. All volumetric MRIs were sent to the NCI POB expert reader for central review, which was the primary analytical method. The NCI POB central analysis was carried out on all volumetric MRIs by a single, expert reader. No site-investigator review was performed.
Disease progression was evaluated according to REiNS criteria, which involved the analysis of 1 target PN and up to 2 nontarget PNs, for which disease progression could be based solely on progression of a clinically relevant nontarget PN. However, because no clinically relevant nontarget PN were reported for stratum 1 of the SRINT phase II trial, the use of target PNs only for assessment of disease progression was referred to as “modified REiNS criteria.”19
Following a regulatory agency request, a retrospective ICR of the SPRINT phase II stratum 1 volumetric MRI scans was conducted, according to modified REiNS (sensitivity analysis) and modified RECIST 1.1 criteria (exploratory analysis).19 The ICR analysis was conducted using the same modified REiNS criteria used by the NCI POB central review. The modification in standard REiNS and RECIST 1.1 criteria was based on use of only the target PN for derivation of response. The ICR analysis was performed with volumetric MRIs assessments performed by either an independent whole-body radiologist or a neuro-radiologist (2 radiologists and 1 neuro-radiologist were available). The ICR was used as a sensitivity analysis for the primary objective of ORR and secondary objective of DOR.19 Analyses based on the ICR of the volumetric MRI images were not performed in the March 31, 2021, DCO. Interreader variability analyses were conducted to determine the margin of error for target PN measurements.
A summary of outcomes and analytical methods for the SPRINT phase II study is outlined in Table 10.
Table 10: Summary of Statistical Analysis of Efficacy End Points in SPRINT Phase II
Outcome | End point measure | Analytical method | Adjustment factors | Sensitivity analyses | CMT approach |
|---|---|---|---|---|---|
Time-to-event outcomes | |||||
Objective response rate | Volumetric MRI | Clopper-Pearson method | None | ICR using modified REiNS and RECIST criteria | NA |
Duration of response | Volumetric MRI | Kaplan-Meier method | None | ICR using modified REiNS and RECIST criteria | NA |
PFS | Volumetric MRI | Kaplan-Meier method | None | None | NA |
Time to response | Volumetric MRI | Kaplan-Meier method | None | None | NA |
Patient-reported outcomes and functional evaluations | |||||
Pain | NRS-11 |
| Precycle number, baseline score, age, morbidities at baseline, baseline × precycle interaction | None | |
PII | — | — |
| ||
Motor function | PROMIS |
| Precycle number, baseline score, age, morbidities at baseline, baseline × precycle interaction | None |
|
Strength/ROM |
| — | — |
| |
Airway function | PFTs | Descriptive statistics | NA | None | Literature44 |
Bowel/bladder function | DVQ | Descriptive statistics | NA | None |
|
Vision function |
| Descriptive statistics | NA | None | Literature48 |
Health-related quality of life | PedsQL |
| Precycle number, baseline score, age, morbidities at baseline, baseline × precycle interaction | None |
|
Physical functioning | 6MWT |
| Precycle number, baseline score, age, morbidities at baseline, baseline × precycle interaction | None | CMTs (literature49) |
6MWT = 6-minute walk test; CMT = clinically meaningful thresholds; DCO = data cut-off; DVQ = Dysfunctional Voiding Questionnaire; ICR = independent central review; MMRM = mixed model for repeated measures; NA = not applicable; NRS-11 = Numeric Rating Scale-11; PedsQL = Pediatric Quality of Life Inventory; PFS = progression-free survival; PFT = pulmonary function test; PII = Pain Interference Index; PROMIS = Patient-Reported Outcomes Measurement Information System; RECIST = Response Evaluation Criteria in Solid Tumors; REiNS = Response Evaluation in Neurofibromatosis and Schwannomatosis.
Sources: SPRINT Phase II Stratum 1 Clinical Study Report (June 29, 2018, DCO)19 and SPRINT Phase II Stratum 1 Clinical Study Report (March 31, 2021, DCO).20
Efficacy Analyses
All efficacy analyses were performed on the full analysis set (FAS).
Objective response rate: For the primary end point of ORR assessed by NCI POB volumetric MRI, results were presented with corresponding 2-sided exact 95% CIs based on the Clopper-Pearson method. The overall response (at each DCO) was also summarized by category (unconfirmed PR, confirmed PR, CR, stable disease, progressive disease, or NE).34
The BOR, defined as the best response recorded from the start of the treatment until progression or the last evaluable MRI assessment in the absence of progression, was also be summarized by category (confirmed PR, CR, stable disease, progressive disease, or NE).34 Patients in the FAS with no imaging assessments were counted as nonresponders.19
Subgroup analysis: The ORR was also summarized by PN status at enrolment (progressive, nonprogressive, or unknown). Progressive disease at trial entry was defined as an increase of 20% or greater in neurofibroma volume no more than 15 months before enrolment.
Sensitivity analyses: A sensitivity analysis based on the ICR of volumetric MRI data was also performed for the primary end point of ORR using the REiNS criteria to assess the robustness of the central single reviewer on analysis of ORR. Cross-tabulation summaries of PN response by the NCI POB central analysis versus the ICR analysis were produced to assess the concordance between the NCI POB central analysis target PN responses and the ICR analysis responses. Finally, the ICR review, based on REiNS, was used to evaluate the interreader agreement in the volume measurements at baseline and in changes in target PN volume over time.
The modified RECIST 1.1 assessments conducted by the ICR were used as an exploratory analysis of ORR.
Plexiform neurofibroma volume: Changes in PN growth were evaluated descriptively by summaries of percentage and absolute change in PN volume from baseline to scheduled precycle assessments. The average tumour volume and 95% CI were displayed graphically across time (precycle assessments). The best percentage change from baseline was summarized descriptively and presented graphically using waterfall plots. For patients who had PN progression at study entry, the type of PN (“typical,” “nodular,” or “solitary”) was marked on the waterfall plot.
Duration of response: DOR was summarized by the Kaplan-Meier method. Plots of DOR were presented, along with median DOR and 95% CI, to summarize the number and percentage of patients remaining in response for at least 4, 8, 12, 16, 20, and 24 cycles. The Kaplan-Meier method was also used to calculate the median (and 95% CI) time to onset of response from initiation of study treatment and summarize the number and percentage of responding patients with an onset by or within at least 4, 8, 12, 16, 20 and 24 cycles. Swimmer plots showing the profile of each patient, classified by PN status at enrolment (progressive, nonprogressive, or unknown), were produced.
Sensitivity analysis: Sensitivity analyses for DOR included assessment based on the ICR according to REiNS criteria (as for the outcome of ORR) using the same statistical method used for the NCI POB central analysis. A second sensitivity analysis using the actual dates of the NCI POB–based volumetric MRI assessments or death was conducted to assess the impact of dose interruptions on the DOR.
The modified RECIST 1.1 assessments conducted by the ICR were used as an exploratory analysis of DOR.
Other secondary end points (PFS and TTR): PFS and TTR were summarized using the Kaplan-Meier method. The median PFS and TTR (and 95% CIs) were calculated from the Kaplan-Meier plot. The percentage of patients who are progression-free at cycles 5, 9, 13,17, and 25, and the percentage of patients not in response at cycles 5, 9, 13, 17, and 25 were summarized.
Interreader variability: For outcomes based on volumetric MRI (ORR, DOR, PFS, and TTR), interreader variability was assessed by 2 independent radiologists to determine the amount of agreement between the target PN volume measurements at baseline and changes in volume over time using the intraclass correlation coefficient.
Clinical Outcome Assessments
For analysis of the PROs that include patient-reported and parent-reported questionnaires (PII, PROMIS, DVQ, GIC, general PN symptom checklist, and PedsQL), results were analyzed and presented separately. Summary measures of disposition and compliance at each scheduled assessment were derived for all PROs. The primary analysis of the PROs and functional outcomes were based on the descriptive statistics and mixed model for repeated measures (MMRM) to summarize changes over time. Change from baseline in the PRO and functional evaluation scores at precycle 13 was the primary analysis.19 Evaluation at precycle 13 was chosen based on the results of SPRINT phase I, which suggested that the majority of patients achieve their first response within the first 12 months of treatment.19,21 Changes from baseline were analyzed using a restricted maximum likelihood–based MMRM analysis, including terms for precycle visit, baseline score, age, the number of morbidities at baseline and baseline visit × precycle interaction. The model presented least squares mean estimates, standard errors, 95% CIs and P values for mean changes from baseline to each precycle visit. At each post-baseline assessment, the absolute change in scores from baseline was calculated as the post-baseline value minus the baseline value for each item, domain, and primary PRO outcome as applicable. The change from baseline values were classified as worsening (≥ 3 points, 2, or 1 points compared to baseline), stable, or improved (1, 2 or ≥ 3 points compared to baseline):19
In addition, supportive analyses using CMTs were conducted to help with interpretation of clinical benefit using published literature, and both distribution- and anchor-based approaches. For the anchor-based approach, the GIC was used as an anchor as it asks patients (or parents) to assess changes in pain and other morbidities that can be linked to relevant questions or outcomes from the PROs or functional evaluations. The correlation between the anchor and the PRO or functional evaluation was reported. An anchor was considered adequate if the correlation coefficient was greater than 0.30.50 For the distribution-based approach, the half value of the SD of the baseline scores was used.19
Correlation Analysis (Exploratory)
Correlation and association analyses between changes in PROs and functional outcomes and percent target PN volume change from baseline to precycle 13 were explored to characterize the effect of selumetinib on percent target PN volume change and key PRO and functional outcomes. Correlations between clinical outcome assessments and PN volume at baseline were explored using scatterplots and Spearman rank correlation coefficients (r), 95% CIs, and P values. The Spearman rank correlation coefficients were assessed according to Cohen (1988):51
r = 0 to 0.3 equates to a weak correlation
r = 0.3 to 0.5 equates to a moderate correlation
r = 0.5 to 1.0 equates to a strong correlation.
A positive correlation was defined as both an improvement in the clinical outcome assessment and a reduction in target PN volume. In addition, the association between PN response at precycle 13 and clinically meaningful change in clinical outcome assessments (yes or no) at precycle 13 were evaluated by a Fisher exact test.19
Missing Data
The PRO and functional end points were analyzed using the MMRM under a missing-at-random assumption. In addition, the rates of missing data by precycle assessment for individual items, domains, and end points were summarized. If, after inspection of the patterns and reasons for these missing data, the data were believed to be not missing at random, the impact of missingness on the analyses was explored by making assumptions about the missing items, domains, or days.19,34
Safety Analyses
Safety data were not formally analyzed but summarized descriptively by count and percentage.
Analysis Populations
The following analysis populations of interest were defined in the SPRINT phase II trial:
FAS: all patients who received at least 1 dose of selumetinib. The FAS was used for all efficacy analyses, unless otherwise reported
safety analysis set: the same as the FAS in this study, consisting of all patients who received at least 1 dose of selumetinib
Pharmacokinetic analysis set: patients in the FAS with at least 1 postdose sample taken for pharmacokinetic analysis.
Protocol Amendments, Deviations, and Changes to Planned Analyses
Protocol Amendments
The original clinical study protocol was finalized on April 6, 2011. A total of 18 protocol amendments were reported. The first 8 amendments (A to H) were for the phase I portion of the study (SPRINT I). Amendment I was implemented to expand the SPRINT study to include a phase II evaluation of selumetinib in the same population. Nine additional protocol amendments were recorded for the SPRINT phase II trial, though Amendments O to R occurred following the DCO for the SPRINT phase II trial. A list of protocol amendments and key changes for the SPRINT phase II study are summarized in Table 11.
Table 11: Protocol Amendments and Key Changes to SPRINT Phase II
Amendment (date) | Key changes |
|---|---|
Amendment J (December 28, 2015) |
|
Amendment K (April 26, 2016) | The CAEPR was updated. The list of AEs, which reported on selumetinib studies for which there is insufficient evidence to suggest that there was a reasonable possibility that selumetinib caused the AE, was also updated. |
Amendment L (August 10, 2016) |
|
Amendment M (September 28, 2016) |
|
Amendment N (June 18, 2018) | The CAEPR and SPEER grades were updated. The list of risks and AEs reported on selumetinib studies for which there is insufficient evidence to suggest that there was a reasonable possibility that selumetinib caused the AE was also updated. |
Amendments made after the data cut-off for this Clinical Study Report (June 29, 2018) | |
Amendment O (November 14, 2018) |
|
Amendment P (January 14, 2019) | The CAEPR was updated. |
Amendment Q (August 14, 2019) |
|
Amendment R (April 17, 2020) |
|
AE = adverse event; BiPAP = bilevel positive airway pressure; CAEPR = Comprehensive Adverse Event and Potential Risks; CPAP = continuous positive airway pressure; CSP = clinical study protocol; CTEP = Cancer Therapy Evaluation Program; DCO = data cut-off; ERK = extracellular signal related kinase; IOP = intraocular pressure; PN = plexiform neurofibroma; QTc = corrected QT interval; QTcF = QT interval corrected using Fridericia’s correction formula; SPEER = Specific Protocol Exceptions to Expedited Reporting.
Source: SPRINT Phase II Stratum 1 Clinical Study Report (June 29, 2018, DCO).19
Protocol Deviations
The number of patients with important protocol deviations in each treatment group are summarized in Table 12. Prior to database lock for the June 29, 2018, and March 31, 2021, DCOs, at least 1 important protocol deviation occurred in 2 patients (4.0%), and 4 patients (8.0%) in the SPRINT phase II trial, respectively.
Table 12: Important Protocol Deviations (Full Analysis Set)
Protocol deviations | Frequency Selumetinib 25 mg/m2 twice daily (N = 50) | |
|---|---|---|
June 29, 2018, DCO | March 31, 2021, DCO | |
Patients with at least 1 important deviation, n (%) | 2 (4.0) | | ||||| |
Protocol-required procedure not adhered to | 1 (2.0) | | ||||| |
Received incorrect investigational treatment or dose | 0 (0.0) | | ||||| |
Other | 1 (2.0) | | ||||| |
DCO = data cut-off.
Sources: SPRINT Phase II Stratum 1 Clinical Study Report (June 29, 2018, DCO)19 and SPRINT Phase II Stratum 1 Clinical Study Report (March 31, 2021, DCO).20
Changes to Planned Analyses
Changes from the planned analysis in the protocol before the final outcome results included:
The statistical analysis plan (version 4.0) describes the analysis of phase I and stratrum 1 of the phase II SPRINT trial.
Analysis of the SPRINT phase II secondary objective “To determine the effect of selumetinib on the PN growth rate based on volumetric analysis of MRI studies obtained prior to enrolment (if available and amenable to volumetric analysis)” was not performed.
Analysis of the SPRINT phase II secondary objective “In patients who enroll on this study with presence of an optic pathway tumour or other glioma not requiring treatment with chemotherapy or radiation therapy, to evaluate the effect of selumetinib on changes in the size of the optic pathway tumour or other glioma” was not performed.
Changes from the planned analysis in the protocol after the availability of the final, validated outcome results included:
The threshold for clinically significant improvement for exophthalmometry was corrected from greater than −2 mm to less than −2 mm.
The threshold for clinically significant improvement for HOTV or Teller acuity cards was amended from greater than or equal to 0.2 logMAR to less than −0.2 logMAR.
The threshold for clinically significant deterioration for HOTV or Teller acuity cards was amended from equal to or greater than −0.2 logMAR to greater than 0.2 logMAR.
The margin of error and intraclass correlation coefficient were assessed both for the PN volume at baseline, and for the percentage change in PN volume postbaseline.
The association between post-baseline longitudinal changes in PRO or functional outcomes and changes in tumour volume was assessed explaining the change in clinical outcome by change in tumour volume rather than the opposite.
Growth rates associated 95% CI were not reproduced based on the bootstrap percentile interval technique as individual patient data were available, allowing for a precise estimate of the standard error and 95% CI.
Use of local laboratory ranges instead of project ranges as the local reference ranges were markedly different from the project reference ranges for certain parameters (increased lipase, increased CPK, and conversion to the latter) would have resulted in erroneous grade shifts.
Dose reductions are not presented according to the statistical analysis plan as there was no specific case report form field in the clinical database to explicitly record them.
Results
Patient Disposition
The disposition of patients enrolled in stratum 1 of the SPRINT phase II study is summarized in Table 13. A total of 50 patients were enrolled and received at least 1 dose of selumetinib. At the June 29, 2018, DCO, 16 patients (32%) discontinued selumetinib, primarily due to AEs (6 [12%]), disease progression (3 [6.0%]), and investigator discretion (3 [6.0%]). Of the 3 patients with disease progression, treatment was discontinued at |||| years, |||| years, and |||| years. Four patients who discontinued treatment also terminated the study, primarily due to voluntary discontinuation by patient (2 [4.0%]), loss to follow-up (1 [2.0%]) and “other” unspecified reasons (1 [2.0%]). Three of the patients who discontinued treatment due to AEs also discontinued from the study.19
At the March 31, 2021, DCO, |||||| additional patients discontinued treatment (for a total of || |||||||), with no new additional discontinuations due to AEs, and |||||| additional discontinuations due to disease progression (total of | |||||||) and investigator discretion (a total of 6 [12.0%]). ||||| additional patients who discontinued treatment also terminated the study (total of || |||||||), primarily due to “other” reasons (total of | |||||||) which were not specified, voluntary discontinuation by the patient (total of | ||||||, switch to alternative treatment (total of | ||||||), disease progression (total of | |||||]), and loss to follow-up (total of | ||||||).20
Disposition | SPRINT phase II (stratum 1) | |
|---|---|---|
June 29, 2018, DCO | March 31, 2021, DCO | |
Enrolled, N (%) | 50 | |
Patients ongoing selumetinib at DCO | 34 (68.0) | 24 (48.0) |
Discontinued from study treatment, N (%) | 16 (32.0) | 26 (52.0) |
Reason for discontinuation, N (%) | ||
Adverse event | 6 (12.0) | 6 (12.0) |
Disease progression on studya | 3 (6.0) | | |||||| |
Investigator discretion | 3 (6.0) | 6 (12.0) |
Treatment period completeda | 2 (4.0) | | ||||| |
Patient not willing to continue treatment | 1 (2.0) | | ||||| |
Severe noncompliance to protocol | 1 (2.0) | 1 (2.0) |
Complicating disease/Intercurrent illness | 0 (0.0) | 2 (4.0) |
Patients ongoing study | 46 (92.0) | 38 (76.0) |
Patients who terminated study | 4 (8.0) | || |||||| |
Voluntary discontinuation by patient | 2 (4.0) | | ||||| |
Lost to follow-up | 1 (2.0) | | ||||| |
Disease progression | 0 (0.0) | | ||||| |
Other | 1 (2.0) | | |||||| |
Switched to alternative treatment | 0 (0.0) | | ||||| |
Full analysis set, N | 50 | |
Safety analysis set, N | 50 | |
DCO = data cut-off.
aIn the June 29, 2018, DCO, 2 patients discontinued selumetinib treatment due to a completed treatment period and disease progression on study. Since the June 29, 2018, DCO, both patients have been re-treated with selumetinib and were therefore no longer included in these categories.
Sources: SPRINT Phase II Stratum 1 Clinical Study Report (June 29, 2018, DCO)19 and SPRINT Phase II Stratum 1 Clinical Study Report (March 31, 2021, DCO).20
Exposure to Study Treatments
Exposure in the safety population of SPRINT phase II is summarized in Table 14. At the June 29, 2018, DCO, the total treatment duration of selumetinib was ||||| days (range = || || |||| days; approximately |||| months or ||| years). The actual treatment duration (sum of days study dose was administrated) of selumetinib was |||||| days (range = || || |||| days).19
At the March 31, 2021, DCO, the total treatment duration of selumetinib was |||||| days (range = || || |||| days; approximately |||| months or ||| years). The median treatment duration was |||||| days (range = |||||| to |||| days; approximately |||| months or ||| years).20
Dose Interruptions
At the June 29, 2018, DCO, all patients had at least 1 dose interruption, primarily due to patient compliance ||| ||||||||| “other” ||| ||||||||| AEs ||| ||||||||| or medication error || |||||||. Single missed doses were counted as interruptions. One patient had ||| interruptions recorded; however, interruptions did not exceed |||||| days per interruption. For patients with interruption due to AEs, | |||||| required more than 1 dose reduction. “Other” reasons for interruption included logistical issues such as surgery, travel issues, and participation in a sleep study.19
Table 14: Duration of Exposure Including Both Initial Treatment and Re-Treatment (Safety Analysis Set)
Exposure | SPRINT phase II (stratum 1) selumetinib 25 mg/m2 twice daily (N = 50) | |
|---|---|---|
June 29, 2018, DCO | March 31, 2021, DCO | |
Total treatment duration (days)a,b | ||
Mean (SD) | 725.7 |||||||| | 1,282.7 |||||||| |
Median (range) | 801.50 ||||||||| | 1,583.0 ||||||||| |
Total treatment-years | |||| | ||||| |
Actual treatment duration (days)c | ||
Mean (SD) | ||||| |||||||| | |||||| |||||||| |
Median (range) | |||||| ||||||||| | |||||| ||||||||| |
Total treatment-years | |||| | ||||| |
DCO = data cut-off; SD = standard deviation.
aTotal treatment duration = (last dose date – first dose date + 1).
bFor patients without re-treatment, total treatment duration = (last dose date of initial treatment – first dose date of initial treatment + 1). For patients with re-treatment, total treatment duration = (last dose date of initial treatment – first dose date of initial treatment + 1) + (last dose date of re-treatment – first dose date of re-treatment + 1).
cActual treatment duration = sum of days of study dose administered.
Sources: SPRINT Phase II Stratum 1 Clinical Study Report (June 29, 2018, DCO)19 and Phase II Stratum 1 Clinical Study Report (March 31, 2021, DCO).20
At the March 31, 2021, DCO, the numbers of interruptions due to “other” reasons, patient compliance, AEs, and medical error were || |||||||, || |||||||, || |||||||, and | |||||||, respectively. Between the June 29, 2018, and the March 31, 2021, DCOs, |||||| additional patients had dose interruptions due to missed doses, which were recorded as medication errors, and |||||| additional patients had dose interruptions due to AEs. One patient who had a medication error at the time of the June 29, 2018, DCO experienced an additional medication error as of the June 29, 2018, DCO, which resulted in study treatment overdose.20
Concomitant Therapy
As of the June 29, 2018, DCO, all patients received at least |||||| concomitant medication for the treatment of medical conditions or AEs. The most common concomitant medications ||||| || ||||||||| |||| ||||||||||| |||||||| ||||||||||| |||||||| ||||||||| |||||||| ||||||||||| |||||||| ||||||||| ||||||| ||| ||||||||| |||||||. In total, || ||||||| patients received || ||||| ||||||||||| |||| ||| ||| ||||||| ||| ||| ||||| ||||||||| ||| |||| |||||| ||||| |||||||||||| ||||||||19
At the time of the March 31, 2021, DCO, the most common concomitant medications were ||||||||||| |||||||| ||||||||||| |||||||| ||||||||| |||||||| ||||||||||| |||||||| ||||||||| |||||||| ||||||||| |||||||| |||||||||| |||||||| ||| ||||||||||||| |||||||.20
Efficacy
Only efficacy outcomes and analyses of subgroups identified in the review protocol are reported here. Appendix 3 provides detailed efficacy data.
Symptom Improvement
Pain
Evaluation of pain intensity measured by the NRS-11 and interference of pain on daily functioning as measured by the PII were secondary end points of the SPRINT phase II trial and are summarized in Table 15 (June 29, 2018, DCO) and Table 16 (March 31, 2021, DCO).
NRS-11: At the June 29, 2018, DCO, 34 patients were eligible to complete the NRS-11 based on age (aged ≥ 8 years); however, only 26 had baseline data on the physician-selected target tumour. Eight patients did not have baseline data as they either received the first version that did not contain this item or did not complete the physician-selected target tumour question in the second version, or there was a difference between the self-selected and physician-selected tumours. Overall, only 20 patients (58.8%) completed all questions of the NRS-11 at both baseline and precycle 13, while 33 patients (97.1%) and 29 patients (85.3%) completed at least 1 question of the NRS-11 at baseline and precycle 13, respectively. In total, 24 patients had sufficient data for NRS-11 for physician-selected target tumour pain, at baseline and cycle 13. The mean adjusted change from baseline score for target tumour pain intensity at precycle 13 was −2.07 (95% CI, −2.84 to −1.31). Of the 24 patients with physician-selected target tumour scores who completed both baseline and precycle 13 assessments, 12 (50.0%) reported an improvement of 2 or more points on the NRS-11. No patients reported deterioration at precycle 13. A total of 14 patients with a baseline NRS-11 score of 2 or more had a reduction in pain intensity by at least 2 points without increased analgesic use.19
At the March 31, 2021, DCO, the adjusted mean change from baseline at precycle 13 was ||||| (95% CI, ||||| || |||||), which was maintained throughout the analysis period.20
Sensitivity Analysis: Using the predefined sensitivity analysis threshold of 1 point for the NRS-11, the adjusted mean change from baseline was deemed to be clinically meaningful from precycle 3 through precycle 25 at the June 29, 2018, DCO.19
The anchor-based approach to derive a CMT resulted in a threshold of |||| points. However, the sponsor noted that the results of the NRS-11 were poorly correlated with the GIC and were therefore not presented. The distribution-based approach resulted in a threshold of |||| points, which was not included in the results as it lies between the primary CMT of 2 points, and the original sensitivity analysis of 1 point.19
Sensitivity analyses were not conducted at the March 31, 2021, DCO.
Pain Interference Index: At the June 29, 2018, DCO, 34 patients were eligible to complete the PII self-report based on age (≥ 8 years); however, because 1 patient had severe cognitive impairment, the self-report was not administered. In total, 32 patients (94.1%) and 28 patients (82.4%) completed all questions of the self-reported PII at baseline and precycle 13, respectively, while 33 (97.1%) and 29 (85.3%) completed at least 50% of the questions. The adjusted mean change from baseline in self-reported PII score at precycle 13 was −0.65 (95% CI, −0.89 to −0.42). Of the 29 patients with precycle 13 data, || ||||||| reported an improvement greater than the CMT of 0.75 points or greater,|| ||||||| reported no change, and | |||||| patient-reported deterioration.19
For the parent-reported PII, 48 parents were eligible to complete the parent-reported based on their child’s age (≥ 5 years), although there was no baseline value for 1 patient. In the parent-report PII, 46 parents (95.8%), and 42 parents (87.5%) completed all PII questions and precycle 13 questions, respectively, and 47 (97.9%) and 43 (89.6%) completed at least 50% of the questions. The adjusted mean change from baseline in parent-reported PII score at precycle 13 was −0.82 (95% CI, −1.17 to −0.47). Of the 42 parents who completed both baseline and precycle 13 assessments, || ||||||| patients reported an improvement greater than the CMT of 1.78 points or greater, || ||||||| reported no change, and | |||||| reported deterioration.19
At the March 31, 2021, DCO, results were consistent with the primary analysis, with an adjusted mean change from baseline at precycle 13 of ||||| (95% CI, ||||| || |||||) for the self-report total score, and ||||| (95% CI, ||||| || |||||) for the parent-report total score. Improvements from baseline in self-reported and parent-reported PII scores were observed as early as precycle 3 and were maintained through precycle 49.20
Table 15: Change From Baseline for NRS-11 Pain Intensity and PII Primary Outcomes, MMRM (Full Analysis Set; June 29, 2018, DCO)
Statistic | SPRINT phase II (stratum 1) selumetinib 25 mg/m2 twice daily | |||||
|---|---|---|---|---|---|---|
Baseline | Precycle 3 | Precycle 5 | Precycle 9 | Precycle 13 | Precycle 25 | |
NRS-11 | ||||||
Physician-selected target tumour pain (N = 34) | ||||||
N | 26 | 25 | 25 | 25 | 24 | 18 |
Observed score, mean (SD)a | 3.15 (3.146) | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| |
Observed mean CFB (SD)a | — | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| |
N (CFB)b | NR | 25 | 25 | 25 | 24 | 18 |
Adjusted mean CFB (SE)b | — | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | −2.07 (0.368) | ||||| ||||||| |
95% CI | — | |||||| ||||| | |||||| ||||| | |||||| ||||| | −2.84 to −1.31 | |||||| ||||| |
PII | ||||||
Self-report total score (N = 34) | ||||||
N | 33 | 31 | 31 | 31 | 29 | 23 |
Observed score, mean (SD)a | 1.22 (1.499) | |||| ||||||| | |||| ||||||| | |||| ||||||| | 0.56 ||||||| | |||| ||||||| |
Observed mean CFB (SD)a | — | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| |
N (CFB)b | NR | 31 | 31 | 31 | 29 | 23 |
Adjusted mean CFB (SE)b | — | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| |
95% CI | — | |||||| ||||| | |||||| ||||| | |||||| ||||| | |||||| ||||| | |||||| ||||| |
Parent-report total score (N = 48) | ||||||
N | 47 | 46 | 44 | 46 | 43 | 35 |
Observed score, Mean (SD)a | 1.50 (1.478) | |||| ||||||| | |||| ||||||| | |||| ||||||| | 0.67 ||||||| | |||| ||||||| |
Observed mean CFB (SD)a | — | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| |
N (CFB)b | NR | 45 | 43 | 45 | 42 | 33 |
Adjusted mean CFB (SE)b | — | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| |
95% CI | — | |||||| ||||| | |||||| ||||| | |||||| ||||| | |||||| ||||| | |||||| ||||| |
CFB = change from baseline; CI = confidence interval; DCO = data cut-off; MMRM = mixed model for repeated measures; NR = not reported; NRS-11 = Numeric Rating Scale-11; PII = Pain Interference Index; SD = standard deviation; SE = standard error.
aObserved values have not been adjusted.
bThe model included terms for precycle, baseline score, age, the number of morbidities at baseline and baseline × precycle interaction.
Source: SPRINT Phase II Stratum 1 Clinical Study Report (June 29, 2018, DCO).19
Table 16: Change From Baseline for NRS-11 Pain Intensity and PII Primary Outcomes, MMRM (Full Analysis Set; March 31, 2021, DCO)
Statistic | SPRINT phase II (stratum 1) selumetinib 25 mg/m2 twice daily | ||||||||
|---|---|---|---|---|---|---|---|---|---|
Baseline | Precycle 3 | Precycle 5 | Precycle 9 | Precycle 13 | Precycle 25 | Precycle 37 | Precycle 49 | ||
NRS-11 | |||||||||
Physician-selected target tumour pain (N = 34) | |||||||||
N | 26 | 25 | 25 | 25 | 24 | 18 | 18 | 16 | |
Observed score, mean (SD)a | 3.15 (3.146) | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |
Observed mean CFB (SD)a | — | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | |
N (CFB)b | NR | 25 | 25 | 25 | 24 | 18 | 18 | 16 | |
Adjusted mean CFB (SE)b | — | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | |
95% CI | — | |||||| ||||| | |||||| ||||| | |||||| ||||| | |||||| ||||| | |||||| ||||| | |||||| ||||| | |||||| ||||| | |
PII | |||||||||
Self-report total score (N = 34) | |||||||||
N | 33 | 31 | 31 | 31 | 29 | 23 | 21 | 18 | |
Observed score, mean (SD)a | 1.22 (1.499) | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |
Observed mean CFB (SD)a | — | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | |
N (CFB)b | NR | 31 | 31 | 31 | 29 | 23 | 21 | 18 | |
Adjusted mean CFB (SE)b | — | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | |
95% CI | — | |||||| ||||| | |||||| ||||| | |||||| ||||| | |||||| ||||| | |||||| ||||| | |||||| ||||| | |||||| ||||| | |
Parent-report total score (N = 48) | |||||||||
N | 47 | 46 | 44 | 46 | 43 | 36 | 30 | 26 | |
Observed score, Mean (SD)a | 1.50 (1.478) | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |
Observed mean CFB (SD)a | — | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | |
N (CFB)b | NR | 45 | 43 | 45 | 42 | 34 | 28 | 24 | |
Adjusted mean CFB (SE)b | — | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | |
95% CI | — | |||||| ||||| | |||||| ||||| | |||||| ||||| | |||||| ||||| | |||||| ||||| | |||||| ||||| | |||||| ||||| | |
CFB = change from baseline; CI = confidence interval; DCO = data cut-off; MMRM = mixed model for repeated measures; NRS-11 = Numeric Rating Scale-11; PII = Pain Interference Index; SD = standard deviation; SE = standard error.
aObserved values have not been adjusted.
bThe model included terms for precycle, baseline score, age, the number of morbidities at baseline and baseline × precycle interaction.
Source: SPRINT Phase II Stratum 1 Clinical Study Report (March 31, 2021, DCO).20
Motor Function
Motor function was evaluated using the strength of muscle groups and ROM in patients with motor morbidity.19 Additional assessments of motor function included the PROMIS physical functioning scales, grooved pegboard, grip and key pinch test and leg length evaluations. Results for PROMIS and overall strength and ROM are summarized in the following section, with results for grooved pegboard, grip and key pinch test, and leg length evaluations included in Appendix 3.
Patient-Reported Outcomes Measurement Information System (PROMIS): Results for the PROMIS mobility and upper extremity domains as assessed by the patient and the parent at the June 29, 2018, and March 31, 2021, DCOs are summarized in Table 17. In total, 33 patients had a motor PN-related morbidity and were aged 5 to 18 years. A total of 16 patients (66.7%) completed all questions and at least 50% of questions at baseline, and 20 patients (83.3%) completed all questions and at least 50% of questions at precycle 13 of the PROMIS mobility self-report. For the PROMIS upper extremity self-report, 19 patients (79.2%) completed all questions at both baseline and precycle 13 assessments, while 22 (91.7%) and 20 (83.3%) completed 50% of questions at baseline and precycle 13, respectively. For parent-report mobility, 25 (75.8%) and 29 (87.9%) completed all questions and at least 50% of questions at baseline and precycle 13, respectively. For parent-reported upper extremity, 27 (81.8%) and 29 (87.9%) completed all questions at baseline and precycle 13, while 31 (91.2%) and 29 (87.9%) completed at least 50% of questions at baseline and precycle 13.19,20
At both DCOs, the baseline scores for the self- and parent-report assessments in the mobility domain were 46.57 (SD = |||||) and 37.43 (SD = |||||), while the baseline scores for the self- and parent-report assessments in the upper extremity domain were 45.95 (SD = ||||||) and 38.15 (SD = ||||||). The adjusted mean change from baseline at precycle 13 in the self-reported mobility domain was 1.75 points (95% CI, −0.70 to 4.19) at the June 29, 2018, DCO, and |||| points (95% CI, ||||| || ||||) at the March 31, 2021, DCO, neither of which met the CMT of |||| points. In the parent-reported assessment, the adjusted mean change from baseline at precycle 13 in the mobility domain was |||| points (95% CI, |||| || ||||) at the June 29, 2018, DCO, and |||| points (95% CI, |||| || ||||) at the March 31, 2021, DCO, which also did not meet CMT of |||| points.19,20
The adjusted mean change from baseline at precycle 13 in the self-reported upper extremity domain was |||| points (95% CI, −|||| || ||||) at the June 29, 2018, DCO, and |||| points (95% CI, ||||| || ||||) at the March 31, 2021, DCO, while the adjusted mean change from baseline at precycle 13 in the upper extremity domain was |||| points (95% CI, ||||| || ||||) at the June 29, 2018, DCO, and |||| points (95% CI, ||||| || ||||) at the March 31, 2021, DCO, neither of which met the CMTs of |||| points and |||| points.19,20
Table 17: Change From Baseline for PROMIS, MMRM (FAS With Motor PN-Related Morbidity)
Statistic | SPRINT phase II (stratum 1) selumetinib 25 mg/m2 twice daily (N = 33) | |||||||
|---|---|---|---|---|---|---|---|---|
Baseline | Precycle 3 | Precycle 5 | Precycle 9 | Precycle 13 | Precycle 25 | Precycle 37 | Precycle 49 | |
June 29, 2018, DCO | ||||||||
Mobility: self-reporta | ||||||||
N | 23 | 21 | 22 | 22 | 20 | 14 | — | — |
Observed score, mean (SD)b | 46.57 ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | 48.02 ||||||| | ||||| ||||||| | — | — |
Observed mean CFB (SD)b | — | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | — | — |
N (CFB)c | NR | 21 | 22 | 22 | 20 | 14 | — | — |
Adjusted mean CFB (SE)c | — | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | — | — |
95% CI | — | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | — | — |
Mobility: parent-reportd | ||||||||
N | 32 | 31 | 31 | 32 | 29 | 20 | — | — |
Observed score, mean (SD)b | 37.43 ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | 41.14 ||||||| | ||||| ||||||| | — | — |
Observed mean CFB (SD)b | — | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | — | — |
N (CFB)c | NR | 30 | 30 | 31 | 28 | 19 | — | — |
Adjusted mean CFB (SE)c | — | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | — | — |
95% CI | — | ||||| |||| | |||||| |||| | ||||| |||| | ||||| |||| | ||||| |||| | — | — |
Upper extremity: self-reporta | ||||||||
N | 22 | 21 | 22 | 22 | 20 | 14 | — | — |
Observed score, mean (SD)b | 45.95 ||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | ||||| ||||||| | — | — |
Observed mean CFB (SD)b | — | |||| ||||||| | ||||| ||||||| | ||||| |||||||| | |||| ||||||| | |||| ||||||| | — | — |
N (CFB)c | NR | 21 | 21 | 21 | 19 | 13 | — | — |
Adjusted mean CFB (SE)c | — | |||| ||||||| | ||||| ||||||| | ||||| ||||||| | |||| ||||||| | |||| ||||||| | — | — |
95% CI | — | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | — | — |
Upper extremity: parent-reportd | ||||||||
N | 31 | 31 | 31 | 32 | 29 | 20 | — | — |
Observed score, mean (SD)b | 38.15 ||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | 40.58 |||||||| | ||||| |||||||| | — | — |
Observed mean CFB (SD)b | — | ||||| ||||||| | |||| ||||||| | ||||| ||||||| | |||| ||||||| | |||| ||||||| | — | — |
N (CFB)c | NR | 30 | 29 | 30 | 27 | 18 | — | — |
Adjusted mean CFB (SE)c | — | ||||| ||||||| | |||| ||||||| | ||||| ||||||| | |||| ||||||| | |||| ||||||| | — | — |
95% CI | — | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | ||||| |||| | — | — |
March 31, 2021, DCO | ||||||||
Mobility: self-reporta | ||||||||
N | 23 | 21 | 22 | 22 | 20 | 14 | 12 | 10 |
Observed score, mean (SD)b | 46.57 ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | 48.02 ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| |||||||| |
Observed mean CFB (SD)b | — | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| |||||||| |
N (CFB)c | NR | 21 | 22 | 22 | 20 | 14 | 12 | 10 |
Adjusted mean CFB (SE)c | — | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| |
95% CI | — | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| |
Mobility: parent-reportd | ||||||||
N | 32 | 31 | 31 | 32 | 29 | 20 | 16 | 12 |
Observed score, mean (SD)b | 37.43 |||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | 41.14 ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| |
Observed mean CFB (SD)b | — | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| |
N (CFB)c | NR | 30 | 30 | 31 | 28 | 19 | 15 | 11 |
Adjusted mean CFB (SE)c | — | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| |
95% CI | — | ||||| |||| | |||||| |||| | ||||| |||| | ||||| |||| | ||||| |||| | ||||| |||| | ||||| |||| |
Upper extremity: self-reporta | ||||||||
N | 22 | 21 | 22 | 22 | 20 | 14 | 12 | 10 |
Observed score, mean (SD)b | 45.95 (12.908) | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | 47.38 ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| |
Observed mean CFB (SD)b | — | |||| ||||||| | ||||| ||||||| | ||||| |||||||| | |||| ||||||| | |||| ||||||| | |||| |||||||| | |||| |||||||| |
N (CFB)c | NR | 21 | 21 | 21 | 19 | 13 | 11 | NR |
Adjusted mean CFB (SE)c | — | |||| ||||||| | ||||| ||||||| | ||||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | || |
95% CI | — | |||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | ||||| |||| | ||||| ||||| | || |
Upper extremity: parent-reportd | ||||||||
N | 31 | 31 | 31 | 32 | 29 | 20 | 16 | 11 |
Observed score, mean (SD)b | 38.15 (12.355) | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | 40.58 ||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| |
Observed mean CFB (SD)b | — | ||||| ||||||| | |||| ||||||| | ||||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| |
N (CFB)c | NR | 30 | 29 | 30 | 27 | 18 | 14 | 10 |
Adjusted mean CFB (SE)c | — | ||||| ||||||| | |||| ||||||| | ||||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| |
95% CI | — | ||||| |||| | |||||| |||| | |||||| |||| | |||||| |||| | ||||| |||| | ||||| |||| | ||||| |||| |
CFB = change from baseline; CI = confidence interval; DCO = data cut-off; FAS = full analysis set; MMRM = mixed model for repeated measures; NR = not reported; PN = plexiform neurofibroma; PROMIS = Patient-Reported Outcomes Measurement Information System; SD = standard deviation; SE = standard error.
aPatients aged 8 to 18 years at enrolment were expected to complete self-report measures of the PROMIS (N = 24).
bObserved values are for transformed scores and have not been otherwise adjusted.
cThe MMRM included terms for precycle, baseline score, age, the number of morbidities at baseline and baseline × precycle interaction.
dParents or legal guardians of patients aged 5 to 18 years at enrolment expected to complete the parent proxy PROMIS (N = 33).
Sources: SPRINT Phase II Stratum 1 Clinical Study Report (June 29, 2018, DCO)19 and SPRINT Phase II Stratum 1 Clinical Study Report (March 31, 2021, DCO).20
Strength and Range of Motion: A total of 33 patients had motor morbidity in any body quadrant at enrolment; however, only 31 patients had baseline values. Results for the MMRM analysis including patients with motor morbidity at enrolment, regardless of the location of their target PN at the June 29, 2018, and March 31, 2021, DCOs were nearly identical; thus, only the results for the most recent DCO (March 31, 2021) are summarized in Table 18. At the March 31, 2021, DCO, 27 patients had evaluable strength assessments at baseline and precycle 13, with an adjusted mean change from baseline of |||| (95% CI, |||| || ||||), which was the same as the June 29, 2018, DCO.19,20
Table 18: Change From Baseline for Strength MMT, MMRM (Full Analysis Set With Motor PN-related Morbidity; March 31, 2021, DCO)
Strength MMTa (N = 33) | SPRINT phase II (stratum 1) selumetinib 25 mg/m2 twice daily | ||||||
|---|---|---|---|---|---|---|---|
Baseline | Precycle 5 | Precycle 9 | Precycle 13 | Precycle 25 | Precycle 37 | Precycle 49 | |
N | 31 | 30 | 29 | 27 | 20 | 16 | 10 |
Observed score, mean (SD) | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| |
Observed mean CFB (SD) | NA | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| |
N (CFB) | NR | 30 | 29 | 27 | 20 | 16 | 10 |
Adjusted mean CFB (SE)b | NA | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| |
95% CI | NA | ||||| |||| | |||||| |||| | ||||| |||| | ||||| |||| | ||||| |||| | ||||| |||| |
CFB = change from baseline; CI = confidence interval; DCO = data cut-off; MMRM = mixed model for repeated measures; MMT = manual muscle test; NA = not applicable; NR = not reported; PN = plexiform neurofibroma; SD = standard deviation; SE = standard error.
aAverage strength score from all muscles in the same body quadrant (right upper, right lower, left upper, left lower, upper bilateral, lower bilateral) as the target PN.
bThe model included terms for precycle, baseline score, age, the number of morbidities at baseline and baseline × precycle interaction.
Source: SPRINT Phase II Stratum 1 Clinical Study Report (March 31, 2021, DCO).20
Results of the MMRM analysis for change from baseline in ROM in patients with target PN in any body quadrant are summarized in Table 19. At the June 29, 2018, DCO, at baseline (N = 33), the mean ROM sum of all joints was |||||| degrees (SD = |||||||). There was an increase in ROM from baseline at precycle 13 of ||||| degrees (95% CI, ||||| || ||||||).19 At the March 31, 2021, DCO, the adjusted mean change from baseline at precycle 13 was ||||| degrees (95% CI, ||||| || ||||||).20
Table 19: Change From Baseline for ROM MMT, MMRM (Full Analysis Set With Motor PN-Related Morbidity)
Range of motiona | SPRINT phase II (stratum 1) selumetinib 25 mg/m2 twice daily (N = 33) | ||||||
|---|---|---|---|---|---|---|---|
Baseline | Precycle 5 | Precycle 9 | Precycle 13 | Precycle 25 | Precycle 37 | Precycle 49 | |
June 29, 2018, DCO | |||||||
N | 33 | 28 | 29 | 26 | 20 | — | — |
Observed score, mean (SD) | |||||| ||||||||| | |||||| ||||||||| | |||||| ||||||||| | |||||| ||||||||| | |||||| ||||||||| | — | — |
Observed mean CFB (SD) | NA | ||||| ||||||||| | ||||| ||||||||| | ||||| ||||||||| | ||||| ||||||||| | — | — |
N (CFB)b | NR | 28 | 29 | 26 | 20 | — | — |
Adjusted mean CFB (SE)b | NA | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | — | — |
95% CI | NA | |||||| |||||| | |||||| |||||| | |||||| |||||| | |||||| |||||| | — | — |
March 31, 2021, DCO | |||||||
N | 33 | 28 | 29 | 26 | 20 | 17 | 11 |
Observed score, mean (SD) | |||||| ||||||||| | |||||| ||||||||| | |||||| ||||||||| | |||||| ||||||||| | |||||| ||||||||| | |||||| ||||||||| | |||||| ||||||||| |
Observed mean CFB (SD) | NA | ||||| ||||||||| | ||||| ||||||||| | ||||| ||||||||| | ||||| ||||||||| | ||||| ||||||||| | ||||| ||||||||| |
N (CFB) | NR | 28 | 29 | 26 | 20 | 17 | 11 |
Adjusted mean CFB (SE)b | NA | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| |
95% CI | NA | |||||| |||||| | |||||| |||||| | |||||| |||||| | |||||| |||||| | ||||||| ||||| | ||||||| |||||| |
CFB = change from baseline CI = confidence interval; DCO = data cut-off; MMRM = mixed model for repeated measures; NA = not applicable; NR = not reported; PN = plexiform neurofibroma; ROM = range of motion; SD = standard deviation; SE = standard error.
aSum of all the degrees of movement for each of the joints in the same body quadrant (right upper, right lower, left upper, left lower, upper bilateral, lower bilateral) as the target PN.
bThe model includes terms for precycle, baseline score, age, the number of morbidities at baseline and baseline × precycle interaction.
Sources: SPRINT Phase II Stratum 1 Clinical Study Report (June 29, 2018, DCO)19 and SPRINT Phase II Stratum 1 Clinical Study Report (March 31, 2021, DCO).20
Grooved Pegboard, Key Pinch Grip, and Leg Length Evaluations: Results for grooved pegboard, key pinch grip, and leg length evaluations were only available at the June 29, 2018, DCO and are summarized in Figure 6, Figure 7, Figure 8, and Figure 9 of Appendix 3. Overall, results for these measures were consistent with the PROMIS and overall strength findings.19,20
Airway Function
In stratum 1 of the SRINT phase II trial, 16 patients had airway dysfunction at baseline. Results of tests of airway function, including FEV1 or FEV0.75 (in pre-school children), R20, and AHI, for the June 29, 2018, and March 31, 2021, DCOs are summarized in Table 20.
The mean change from baseline at precycle 13 in FEV1 or FEV0.75 was ||||| L (SD = ||||||), representing a mean percent change from baseline of |||||| (SD = |||||||| || ||) at both DCOs, where | ||||||| patients showed improvement, | ||||||| patients showed no change and no patients showed deterioration in FEV1 or FEV0.75.19,20
At precycle 13 for both DCOs, the mean change from baseline in R20 was |||||| (SD = ||||||), representing a ||||| (SD = ||||||) change from baseline.19,20
At baseline, no patients in SPRINT phase II had an AHI of more than 5 events per hour. The mean baseline AHI was ||||| (SD = ||||||). At both DCOs, the mean change from baseline in AHIs at precycle 13 was ||||| (SD = ||||||), with a mean number of AHI at precycle 13 of ||||| (SD = ||||||).19,20
Table 20: Airway Function Test Scores Over Time and Change From Baseline Over Time (Full Analysis Set With Airway PN–Related Morbidity)
Test | SPRINT phase II (stratum 1) selumetinib 25 mg/m2 twice daily (N = 16) | ||||||
|---|---|---|---|---|---|---|---|
Baseline | Precycle 5 | Precycle 9 | Precycle 13 | Precycle 25 | Precycle 37 | Precycle 49 | |
June 29, 2018, DCO | |||||||
FEV1 or FEV0.75 | |||||||
N | 11 | 11 | 11 | 11 | 9 | — | — |
Mean (SD) | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | — | — |
Mean absolute CFB (SD) | NA | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | — | — |
Mean % CFB (SD) | NA | |||| |||||||| | |||| |||||||| | ||||| |||||||| | ||||| |||||||| | — | — |
% missing | |||| | |||| | |||| | |||| | |||| | — | — |
R20 (resistance) | |||||||
N | 10 | 10 | 10 | 10 | 8 | — | — |
Mean score (SD) | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | — | — |
Mean absolute CFB (SD) | NA | ||||| |||||||| | ||||| |||||||| | |||||| |||||||| | |||||| |||||||| | — | — |
Mean % CFB (SD) | NA | |||| |||||||| | |||| |||||||| | |||| |||||||| | ||||| |||||||| | — | — |
% missing | |||| | |||| | |||| | |||| | |||| | — | — |
Apnea-Hypopnea Index (events/hour) | |||||||
N | 14 | 7 | 6 | 13 | 6 | — | — |
Mean score (SD) | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | — | — |
Mean absolute CFB (SD) | NA | ||||| |||||||| | |||||| |||||||| | ||||| |||||||| | |||||| |||||||| | — | — |
% missing | |||| | |||| | |||| | |||| | |||| | — | — |
March 31, 2021, DCO | |||||||
FEV1 or FEV0.75 | |||||||
N | 11 | 11 | 11 | 11 | 11 | 11 | 6 |
Mean (SD) | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| |
Mean absolute CFB (SD) | NA | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| |
Mean % CFB (SD) | NA | |||| |||||||| | |||| |||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| |
% missing | |||| | |||| | |||| | |||| | |||| | |||| | |||| |
R20 (resistance) | |||||||
N | 10 | 10 | 10 | 10 | 10 | 9 | 5 |
Mean (SD) | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| |
Mean absolute CFB (SD) | NA | ||||| |||||| | ||||| |||||| | |||||| |||||||| | |||||| |||||||| | |||||| |||||||| | |||||| |||||||| |
Mean % CFB (SD) | NA | |||| |||||||| | |||| |||||||| | |||| |||||||| | ||||| |||||||| | |||||| |||||||| | |||||| |||||||| |
% missing | |||| | |||| | |||| | |||| | |||| | |||| | |||| |
Apnea-Hypopnea Index (events/hour) | |||||||
N | 14 | 7 | 6 | 13 | 8 | 8 | 4 |
Mean (SD) | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| |
Mean absolute CFB (SD) | NA | ||||| |||||||| | |||||| |||||||| | ||||| |||||||| | |||||| |||||||| | ||||| |||||||| | ||||| |||||||| |
% missing | |||| | |||| | |||| | |||| | |||| | |||| | |||| |
CFB = change from baseline; DCO = data cut-off; FEV1 = forced expiratory volume in the first second; FEV0.75 = forced expiratory volume in the first 0.75 seconds; PN = plexiform neurofibroma; R20 = respiratory resistance at 20 Hz; SD = standard deviation.
Sources: SPRINT Phase II Stratum 1 Clinical Study Report (June 29, 2018, DCO)19 and SPRINT Phase II Stratum 1 Clinical Study Report (March 31, 2021, DCO).20
Bowel and Bladder Function
Results for bowel and bladder function as assessed by the parent-reported DVQ are summarized in Table 21. A total of 10 patients had bowel and bladder PN–related morbidity at baseline, of whom, only 2 had baseline data for self-reported scores. The mean change from baseline at precycle 13 was therefore not calculable at both DCOs.
In the parent-reported assessment of bowel and bladder function, 6 ||||| and 7 ||||| parents completed all questions at baseline and precycle 13, respectively. Insufficient data were available to determine the adjusted mean change from baseline at both DCOs for the parent-reported assessment. The observed mean change from baseline at precycle 13 was |||| (SD = ||||) at both DCOs.19,20
Table 21: Bowel and Bladder Function Parent-Report Scores Over Time and Change From Baseline Over Time (Full Analysis Set With a Bowel and/or Bladder PN–Related Morbidity)
Bowel and bladder function total scores (N = 10) | SPRINT phase II (stratum 1) selumetinib 25 mg/m2 twice daily | ||||||
|---|---|---|---|---|---|---|---|
Baseline | Precycle 5 | Precycle 9 | Precycle 13 | Precycle 25 | Precycle 37 | Precycle 49 | |
June 29, 2018, DCO | |||||||
N | 8 | 9 | 9 | 8 | 4 | — | — |
Mean (SD) | |||| |||||| | |||| |||||| | |||| |||||| | ||| |||||| | ||| |||||| | — | — |
Observed mean CFB (SD) | NA | |||| ||||||| | |||| ||||||| | |||| |||||| | |||| |||||| | — | — |
March 31, 2021, DCO | |||||||
N | 8 | 9 | 9 | 8 | 5 | 4 | 3 |
Mean (SD) | |||| |||||| | |||| |||||| | ||| || ||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| |
Observed mean CFB (SD) | NA | |||| ||||||| | |||| ||||||| | |||| |||||| | |||| |||||| | |||| |||||| | |||| |||||| |
CFB = change from baseline; DCO = data cut-off; NA = not applicable; PN = plexiform neurofibroma; SD = standard deviation.
Sources: SPRINT Phase II Stratum 1 Clinical Study Report (June 29, 2018, DCO)19 and SPRINT Phase II Stratum 1 Clinical Study Report (March 31, 2021, DCO).20
Vision Function
As of the June 29, 2018, DCO, 10 patients had visual dysfunction at enrolment, || || of whom had no possibility of improvement (enucleation || |||, no light perception || |||, or limited to light perception and position on || || |||). Only || || patients had baseline and precycle 13 assessments, whereas at precycle 13 assessments, the mean change from baseline was |||| points (SD = |||||), meeting the REiNS-defined meaningfulness of 0.2 units. In total, || || patients showed no changes, and || || patients showed deterioration in visual acuity. For exophthalmometry, at precycle 13, || || patient showed improvement, || || patients showed no change and || || patients showed deterioration.19
No assessment of visual function or exophthalmometry was conducted at the March 31, 2021, DCO.
Disfigurement
At the June 29, 2018, DCO, 44 patients (88.0%) had disfigurement as measured by standardized photography. Disfigurement could not be assessed on a population level due to the difficulty evaluating the physical impact of disfigurement posed by the age of the included patients, as well as difficulties anonymizing images and videos. Results for disfigurement were therefore not available in the sponsor-submitted material.19
No assessment of disfigurement was conducted at the March 31, 2021, DCO.
Global Impression of Change
Results for self-reported (N = 34) and parent-reported (N = 48) distribution of GIC item responses for tumour pain, overall pain, and tumour-related morbidities are summarized in Table 22. As the results at the June 29, 2018, and March 31, 2021, DCOs were identical up to those at precycle 25, only results reported at the 2021 DCO are presented.
At precycle 13, |||| ||||||| ||||||| reported a minimally worse change from baseline in self-reported tumour pain and self-reported tumour-related morbidity. Patients most often reported improvements or no change from baseline across categories. While results for the parent-reported GIC were consistent, for the domain of overall pain, | |||||| parent each considered their child’s overall pain to be minimally worse and much worse compared to baseline at precycle 13.20
Table 22: Distribution of GIC Item Responses Over Time (FAS; March 31, 2021, DCO)
Response category | SPRINT phase II (stratum 1) selumetinib 25 mg/m2 twice dailya,b | ||||||
|---|---|---|---|---|---|---|---|
Precycle 3 | Precycle 5 | Precycle 9 | Precycle 13 | Precycle 25 | Precycle 37 | Precycle 49c | |
Self-report (N = 34) | |||||||
Tumour pain, n (%) | |||||||
N | 26 | 30 | 30 | 29 | 23 | 20 | 5 |
Very much improved | | |||||| | | |||||| | || |||||| | || |||||| | || |||||| | | |||||| | | |||||| |
Much improved | | |||||| | | |||||| | | |||||| | | |||||| | | |||||| | | |||||| | | |||||| |
Minimally improved | | |||||| | | |||||| | | |||||| | | |||||| | | ||||| | | |||||| | | |||||| |
No change | | |||||| | || |||||| | | |||||| | | |||||| | | |||||| | | |||||| | | |||||| |
Minimally worse | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| |
Much worse | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| |
Very much worse | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| |
Overall pain, n (%) | |||||||
N | 30 | 30 | 30 | 29 | 23 | 21 | 5 |
Very much improved | | |||||| | | |||||| | | |||||| | | |||||| | | |||||| | | |||||| | | |||||| |
Much improved | | |||||| | | |||||| | | |||||| | | |||||| | | |||||| | | |||||| | | |||||| |
Minimally improved | | |||||| | | |||||| | | |||||| | | |||||| | | |||||| | | ||||| | | |||||| |
No change | || |||||| | || |||||| | | |||||| | || |||||| | | |||||| | | |||||| | | |||||| |
Minimally worse | | |||||| | | ||||| | | |||||| | | ||||| | | ||||| | | ||||| | | ||||| |
Much worse | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| |
Very much worse | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| |
Related morbidity, n (%) | |||||||
N | 23 | 29 | 30 | 29 | 23 | 21 | 5 |
Very much improved | | |||||| | | |||||| | | |||||| | || |||||| | | |||||| | | |||||| | | |||||| |
Much improved | | |||||| | | |||||| | | |||||| | | |||||| | | |||||| | | |||||| | | |||||| |
Minimally improved | | |||||| | | |||||| | | |||||| | | |||||| | | |||||| | | |||||| | | ||||| |
No change | | |||||| | | |||||| | | |||||| | | |||||| | | |||||| | | |||||| | | |||||| |
Minimally worse | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| |
Much worse | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| |
Very much worse | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| |
Parent-report (N = 48) | |||||||
Tumour pain, n (%) | |||||||
N | 38 | 44 | 45 | 43 | 35 | 28 | 11 |
Very much improved | | ||||| | | |||||| | || |||||| | || |||||| | || |||||| | | |||||| | | |||||| |
Much improved | | |||||| | | |||||| | | |||||| | || |||||| | | |||||| | | |||||| | | |||||| |
Minimally improved | || |||||| | | |||||| | | |||||| | | ||||| | | |||||| | | |||||| | | ||||| |
No change | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | | |||||| | | |||||| |
Minimally worse | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| |
Much worse | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| |
Very much worse | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| |
Overall pain, n (%) | |||||||
N | 44 | 44 | 45 | 43 | 35 | 28 | 11 |
Very much improved | | ||||| | | |||||| | | |||||| | || |||||| | || |||||| | | |||||| | | |||||| |
Much improved | | |||||| | || |||||| | || |||||| | || |||||| | | |||||| | | |||||| | | |||||| |
Minimally improved | || |||||| | | |||||| | | |||||| | | |||||| | | |||||| | | |||||| | | ||||| |
No change | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | | |||||| |
Minimally worse | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| |
Much worse | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| |
Very much worse | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| |
Tumour-related morbidity, n (%) | |||||||
N | 34 | 43 | 45 | 43 | 35 | 28 | 9 |
Very much improved | | ||||| | | ||||| | | |||||| | || |||||| | || |||||| | | |||||| | | |||||| |
Much improved | | |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | | |||||| |
Minimally improved | || |||||| | || |||||| | || |||||| | | |||||| | | |||||| | | |||||| | | |||||| |
No change | || |||||| | | |||||| | | |||||| | | |||||| | | ||||| | | ||||| | | |||||| |
Minimally worse | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | |||||| |
Much worse | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| |
Very much worse | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| |
DCO = data cut-off; FAS = full analysis set; GIC = Global Impression of Change.
aPatients aged 8 to 18 years at enrolment were expected to complete self-reported measures of the GIC. Parents or legal guardians of children aged 5 to 18 years at enrolment were expected to complete the parent proxy measures of the GIC.
bPercentages were based on the number of patients with a nonmissing score at each analysis visit.
cPatient numbers at precycle 49 are relatively low because Amendment N (dated June 18, 2018) stipulated that data should be collected annually up to the third year (before cycle 37) if feasible. However, any data that had been collected at later cycles before Amendment N are summarized up to and including precycle 49.
Source: SPRINT Phase II Stratum 1 Clinical Study Report (March 31, 2021, DCO).20
6-Minute Walk Test
The 6MWT was conducted in patients aged 5 or older with lower extremity PNs, cord compression, or airway PNs (N = 34). Results for the 6MWT at the June 29, 2018, DCO are summarized in Table 23. The largest adjusted mean changes from baseline in distance achieved in the 6MWT were reported at precycles 9 (||||| m [95% CI, ||||| || ||||||) and 25 (||||| m [95% CI, |||||| || ||||||).19
No results for the 6MWT were available at the March 31, 2021, DCO.
Table 23: Change From Baseline for 6MWT, MMRM (Full Analysis Set; June 29, 2018, DCO)
Statistic | SPRINT phase II (stratum 1) selumetinib 25 mg/m2 twice daily | ||||
|---|---|---|---|---|---|
Baseline | Precycle 5 | Precycle 9 | Precycle 13 | Precycle 25 | |
6MWT distance achieved, m (N = 34) | |||||
N | 30 | 31 | 31 | 29 | 22 |
Observed distance, mean (SD) | |||||| ||||||||| | |||||| ||||||||| | |||||| ||||||||| | |||||| ||||||||| | |||||| ||||||||| |
Observed mean CFB (SD) | NA | |||| ||||||||| | ||||| |||||||| | ||||| ||||||||| | ||||| ||||||||| |
N | NR | 27 | 28 | 25 | 19 |
Adjusted mean CFB (SE)a | NA | |||| |||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| |
95% CI | NA | ||||||| ||||| | |||||| ||||| | ||||||| ||||| | ||||||| ||||| |
6MWT = 6-minute walk test; CFB = change from baseline; CI = confidence interval; DCO = data cut-off; MMRM = mixed model for repeated measures; NA = not applicable; SD = standard deviation; SE = standard error.
aThe model included terms for precycle, baseline score, age, the number of morbidities at baseline and baseline × precycle interaction.
Source: SPRINT Phase II Stratum 1 Clinical Study Report (June 29, 2018, DCO).19
Subgroup Analysis: Analyses evaluating the effect of airway PNs on patients were conducted. In patients with lower extremity PNs or cord compression but no airway PNs (N = 28), no trends in improvement were observed (cycle 5, ||||| m [95% CI, −6.96 to 54.26]; cycle 9, ||||| m [95% CI, ||||| || |||||]; cycle 13, ||||| m [95% CI| |||||| || |||||]; cycle 25, ||||| m [95% CI, ||||| || |||||]).19
In patients with airway PNs but no lower extremity PNs or cord compression (N = 16), no trends in improvement were observed (cycle 5, ||||| m [95% CI, |||||| || |||||]; cycle 9, ||||| m [95% CI, |||||| || |||||]; cycle 13, |||||| m [95% CI, |||||| || |||||]; cycle 25, |||| m [95% CI, |||||| || |||||]).19
General PN Symptoms
General symptoms related to PNs were evaluated using a PN symptom checklist that was completed by either the patient or parent. At baseline, all (50 patients or parents [100.0%]) completed all questions. At cycle 13, 44 patients or parents (88.0%) completed all questions. At cycle 25, 33 patients or parents (66.0%) completed all questions. A significant drop-off was observed at cycle 37, with only 5 patients or parents (10.0%) completing all questions.19
At the June 29, 2018, DCO, the majority of patients || |||| reported no problems (i.e., “Not at all”) at baseline (N = 50) in all domains except for fatigue (||||| || ||||), as well as sleep problems (||||| || ||||). At cycle 13 (| |||), decreases in patients reporting no problems were observed in the following domains compared to baseline: decreased hearing (|| ||||||| at baseline versus || ||||||| at cycle 13), mouth sores (|| ||||||| versus || |||||||), chest pain (|| ||||||| versus || |||||||), swelling in hands/feet (|| ||||||| versus || |||||||), nausea (|| ||||||| versus || |||||||), diarrhea (|| ||||||| versus || |||||||), constipation (|| ||||||| versus || |||||||), stool incontinence (|| ||||||| versus || |||||||), and dizziness (|| ||||||| versus || |||||||).19
No results for general PN symptom checklist were available at the March 31, 2021, DCO.
Health-Related Quality of Life
Pediatric Quality of Life Inventory
Results for PedsQL at the June 29, 2018, and March 31, 2021, DCOs of the SPRINT phase II study are summarized in Table 24. A total of 27 patients (79.4%) and 24 patients (70.6%) completed all questions of the self-reported PedsQL at baseline and precycle 13, respectively, while 33 (97.1%) and 29 (85.3%) completed at least 50% of questions at baseline and precycle 13, respectively. In the parent-reported PedsQL, 41 (82.0%) and 38 (76%) parents completed all questions at baseline and precycle 13, and 50 (100%) and 45 (90.0%) completed at least 50% of questions at baseline and precycle 13. The observed mean score at baseline was 73.91 (SD = ||||||) in the self-reported version, and 60.79 (SD = ||||||) in the parent-reported version. As of the June 29, 2018, DCO, the adjusted mean change from baseline at precycle 13 was 6.68 points (95% CI, |||| || |||||) in the self-reports, while the adjusted mean change from baseline was 12.73 points (95% CI, |||| || |||||) in the parent-reports.19 Results were similar at the March 31, 2021, DCO, with the adjusted mean change from baseline in the self-reported version of |||| points (95% CI, |||| || |||||), and ||||| points (95% CI, |||| || |||||) in the parent-reported version.20
Table 24: Change from Baseline for PedsQL Primary Outcomes Total Score, MMRM (FAS)
Statistic | SPRINT phase II (stratum 1) selumetinib 25 mg/m2 twice daily | |||||||
|---|---|---|---|---|---|---|---|---|
Baseline | Precycle 3 | Precycle 5 | Precycle 9 | Precycle 13 | Precycle 25 | Precycle 37 | Precycle 49 | |
June 29, 2018, DCO | ||||||||
Self-reported total score (N = 34)a | ||||||||
N | 33 | 31 | 31 | 31 | 29 | 23 | — | — |
Observed score, mean (SD) | 73.91 (||||||) | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | 79.56 (||||||) | ||||| |||||||| | — | — |
Observed mean CFB (SD) | NA | |||| |||||||| | |||| |||||||| | |||| |||||||| | |||| |||||||| | |||| |||||||| | — | — |
N (CFB) | NR | 31 | 31 | 31 | 29 | 23 | — | — |
Adjusted mean CFB (SE)b | NA | |||| ||||||| | |||| ||||||| | |||| ||||||| | 6.68 ||||||| | |||| ||||||| | — | — |
95% CI | NA | ||||| ||||| | ||||| |||| | ||||| |||| | ||||| ||||| | ||||| ||||| | — | — |
Parent-reported total score (N = 50)c | ||||||||
N | 50 | 47 | 47 | 48 | 45 | 35 | — | — |
Observed score, mean (SD) | 60.79 (||||||) | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | 73.34 |||||||| | ||||| |||||||| | — | — |
Observed mean CFB (SD) | NA | |||| |||||||| | |||| |||||||| | |||| |||||||| | ||||| |||||||| | ||||| |||||||| | — | — |
N (CFB) | NR | 47 | 47 | 48 | 45 | 35 | — | — |
Adjusted mean CFB (SE)d | NA | |||| ||||||| | |||| ||||||| | |||| ||||||| | ||||| ||||||| | ||||| ||||||| | — | — |
95% CI | NA | ||||| ||||| | ||||| ||||| | ||||| ||||| | ||||| ||||| | ||||| ||||| | — | — |
March 31, 2021, DCO | ||||||||
Self-reported total score (N = 34)a | ||||||||
N | 33 | 31 | 31 | 31 | 29 | 23 | 20 | 20 |
Observed score, mean (SD) | 73.91 |||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | 79.56 |||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| |
Observed mean CFB (SD) | NA | |||| |||||||| | |||| |||||||| | |||| |||||||| | |||| |||||||| | |||| |||||||| | ||||| |||||||| | |||| |||||||| |
N (CFB) | NR | 31 | 31 | 31 | 29 | 23 | 20 | 19 |
Adjusted mean CFB (SE)b | NA | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | ||||| ||||||| | ||||| ||||||| |
95% CI | NA | ||||| |||| | |||||| |||| | ||||| |||| | ||||| ||||| | ||||| ||||| | ||||| ||||| | ||||| ||||| |
Parent-reported total score (N = 50)b | ||||||||
N | 50 | 47 | 47 | 48 | 45 | 36 | 29 | 25 |
Observed score, mean (SD) | 60.79 |||||||| | ||||||||||||| | ||||| |||||||| | ||||| |||||||| | 73.34 |||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| |
Observed mean CFB (SD) | NA | |||| |||||||| | |||| |||||||| | |||| |||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| |
N (CFB) | NR | 47 | 47 | 48 | 45 | 36 | 29 | 25 |
Adjusted mean CFB (SE)d | NA | |||| ||||||| | |||| ||||||| | |||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| | ||||| ||||||| |
95% CI | NA | ||||| ||||| | ||||| ||||| | ||||| ||||| | ||||| ||||| | ||||| ||||| | |||||| ||||| | ||||| ||||| |
CFB = change from baseline; CI = confidence interval; DCO = data cut-off; FAS = full analysis set; MMRM = mixed model for repeated measures; NA = not applicable; NR = not reported; PedsQL = Pediatric Quality of Life Inventory; SD = standard deviation; SE = standard error.
aChildren aged 8 to 18 years at enrolment were expected to complete self-report measures of the PedsQL.
bThe model included terms for precycle, baseline score, age, the number of morbidities at baseline and baseline × precycle interaction.
cParents or legal guardians of children aged 2 to 18 years at enrolment completed the parent proxy measures of the PedsQL.
dThe model included terms for precycle, baseline score, age, the number of clinical complications at baseline and baseline × precycle interaction.
Sources: SPRINT Phase II Stratum 1 Clinical Study Report (June 29, 2018, DCO)19 and SPRINT Phase II Stratum 1 Clinical Study Report (March 31, 2021, DCO).20
Correlation Analysis
Results for the exploratory correlation analysis between clinical outcome assessments and target tumour volume as of the June 29, 2018, DCO in SPRINT phase II are summarized in Table 25. Most correlations between PROs and functional evaluations and target tumour volume were weak to moderate, with absolute Spearman rank coefficients ranging from 0.10 to 1.00 at baseline and from 0.06 to 0.52 for the change from baseline at precycle 16, and correlations between 0.3 and 0.5 considered moderate.19
Clinical Response
Objective Response Rate
The ORR was the primary end point of the SPRINT phase II trial. Results for the ORR at the June 29, 2018, and March 31, 2021, DCOs are summarized in Table 26. At the June 29, 2018, DCO, 33 patients (66.0%; 95% CI, 51.2 to 78.8) achieved an ORR according to the NCI POB central analysis, all of which were confirmed PRs.19 At the March 31, 2021, DCO, an ORR was observed in 34 patients (68.0%; 95% CI, 53.3 to 80.5).20
Subgroup Analyses: Results for ORR by PN status at enrolment were identical at the June 29, 2018, and March 31, 2021, DCOs. In patients with progressive PN at the time of the June 29, 2018, DCO (N = 21), || |||||| (95% CI, |||| || |||||| had a confirmed PR. In patients with nonprogressive PN (N = 15), || |||||| (95% CI, |||| || ||||) had a response. In patients with unknown PN status (N = 14), || |||||| (95% CI, |||| || ||||) had a response. No additional subgroup analyses were conducted.19
Sensitivity Analyses: At the June 29, 2018, DCO, sensitivity analyses of ORR based on the ICR analysis were conducted. According to the ICR analysis, the ORR was 44.0% (95% CI, 30.0 to 58.7); 22 patients (44.0%) had a confirmed PR, 5 (10.0%) had an unconfirmed PR, 21 (42.0%) had stable disease, 2 (4.0%) were not evaluable, and no patient had progressive disease.19
No sensitivity analyses of ORR by ICR were conducted at the March 31, 2021, DCO.
Table 25: Correlation Between Clinical Outcome Assessments and Percent Change in Tumour Volume at Baseline and From Baseline to Precycle 13 (FAS, June 29, 2018, DCO)
Outcome | Spearman rank coefficient (r) | |
|---|---|---|
Baseline | Change from baseline (precycle 13), 95% CI | |
Pain | ||
Numeric Rating Score-11 | |||| | 0.13 (NR to NR) |
Pain Interference Index | ||
Self-report | |||| | −0.40 (−0.67 to −0.02) |
Parent-report | |||| | 0.09 (−0.23 to 0.39) |
Motor function | ||
Strength (manual muscle test) | ||||| | |||| ||||||| ||||| |
Range of motion | |||| | −0.24 ||||||| ||||| |
Patient-Reported Outcomes Measurement Information System | ||
Mobility: Self-report | ||||| | |||| ||||||| ||||| |
Mobility: Parent-report | ||||| | ||||| ||||||| |||||| |
Upper extremity: Self-report | ||||| | ||||| ||||||| ||||| |
Upper extremity: Parent-report | ||||| | ||||| ||||||| ||||| |
Airway function | ||
Apnea-Hypopnea Index | ||||| | |||| ||||||| ||||| |
Bowel and bladder function | ||
Dysfunctional Voiding Questionnaire | ||
Self-report | ||||| | || |
Parent-report | |||| | ||||| ||||||| ||||| |
Vision function | ||
Visual acuity (HOTV) | ||
Affected eye | ||||| | |||| ||||||| ||||| |
Nonaffected eye | ||||| | |||| ||||||| ||||| |
Exophthalmometry | ||
Affected eye | |||| | ||||| ||||||| ||||| |
Nonaffected eye | ||||| | ||||| ||||||| ||||| |
Health-related quality of life | ||
Pediatric Quality of Life Inventory | ||
Total score: Self-report | ||||| | |||| ||||||| ||||| |
Total Score: Parent-report | ||||| | ||||| ||||||| |||||| |
Physical function: Self-report | ||||| | |||| ||||||| ||||| |
Physical function: Parent-report | ||||| | ||||| ||||||| ||||| |
Other | ||
6-minute walk test | ||||| | |||| ||||||| ||||| |
CI = confidence interval; DCO = data cut-off; FAS = full analysis set; HRQoL = health-related quality of life; NC = not calculable; NR = not reported.
Source: SPRINT Phase II Stratum 1 Clinical Study Report (June 29, 2018, DCO).19
Table 26: Confirmed ORR and BOR: NCI POB Central Analysis (Full Analysis Set)
BOR | SPRINT phase II (stratum 1) selumetinib 25 mg/m2 twice daily (N = 50) | |
|---|---|---|
June 29, 2018, DCO | March 31, 2021, DCO | |
ORRa | 33 (66.0) | 34 (68.0) |
95% CIb | 51.2 to 78.8 | ||||| |||| |
Complete response | 0 (0.0) | 0 (0.0) |
Confirmed partial responsec | 33 (66.0) | 34 (68.0) |
Unconfirmed partial responsed | 4 (8.0) | | ||||| |
Stable diseasee | 11 (22.0) | 11 (22.0) |
Disease progressionf | 0 (0.0) | 0 (0.0) |
REiNS progression | 0 (0.0) | 0 (0.0) |
Death | 0 (0.0) | 0 (0.0) |
Not evaluableg | 2 (4.0) | | ||||| |
BOR = best objective response; DCO = data cut-off; FAS = full analysis set; NCI = National Cancer Institute; ORR = objective response rate; PN = plexiform neurofibroma; POB = Pediatric Oncology Branch; REiNS = Response Evaluation in Neurofibromatosis and Schwannomatosis.
aFor partial responses, a response required consecutive confirmation within 3 to 6 months after the criteria for first response were met.
bCalculated using the Clopper-Pearson exact method for binomial proportions.
cResponse required consecutive confirmation within 3 to 6 months after the criteria for first response were met. Partial response = a decrease in the volume of the target PN by 20% or more compared with baseline.
dPartial response was achieved but either no confirmation assessment was performed or a confirmation assessment was performed but the response was not confirmed.
eInsufficient volume change from baseline to qualify for either partial response or progressive disease.
fIncrease in the volume of the target PN by 20% or more compared with baseline or the time of best response (maximal PN shrinkage) after documenting a partial response.
gTwo patients did not contribute to efficacy analyses as they did not have any scheduled post-baseline volumetric MRI scans.
Sources: SPRINT Phase II Stratum 1 Clinical Study Report (June 29, 2018, DCO)19 and SPRINT Phase II Stratum 1 Clinical Study Report (March 31, 2021, DCO).20
Concordance between assessment of target PN BOR in the primary and ICR analyses at the June 29, 2018, DCO using REiNS criteria was evaluated, but no evaluation of concordance was conducted for the March 31, 2021, DCO given that ICR analyses were not conducted. There was agreement between the primary and ICR analyses that no patients had a BOR of progressive disease. Of the 33 patients with a confirmed PR in the primary analysis, there was agreement that 21 patients had a confirmed PR. For the remaining 12 patients, 3 had an unconfirmed PR and 9 had stable disease, according to the ICR. Of the 4 patients with an unconfirmed PR in the primary analysis, there was agreement that 1 patient had an unconfirmed PR, 2 patients had stable disease, and 1 patient had a confirmed PR according to the ICR. Of the 11 patients with stable disease in the primary analysis, there was agreement that 10 patients had stable disease. Differences in categorizations of BOR between the primary analysis and the ICR analysis were primarily in assignments of confirmed PR versus stable disease (based on the chosen response threshold of 20% shrinkage), with the ICR analyses determining that the change in target PN volume, although representing a reduction, was just below 20% (range = |||||| || ||||||||19
An exploratory ICR analysis using modified RECIST 1.1 criteria was also conducted at the June 29, 2018, DCO. Based on the RECIST 1.1 assessment, the ORR was only |||||, with | |||||| patients having an unconfirmed PR and || ||||||| patients having stable disease.19
Target PN volume: Results for percent change from baseline in target PN volume over time at the June 29, 2018, and March 31, 2021, DCOs are summarized in Figure 2, Figure 3, and Figure 4. A total of 48 patients were included in the descriptive summary of change in target PN volume from baseline. At the June 29, 2018, DCO, 37 patients (77.1%) had a maximum reduction from baseline in target PN volume of 20% or greater and, of these, 3 patients (6.3%) had a maximum reduction from baseline of 40% or greater, with the largest change being −54.5% at precycle 37.19
Figure 2: Percent Change From Baseline in Target PN Volume (Full Analysis Set; NCI POB and ICR Analyses)
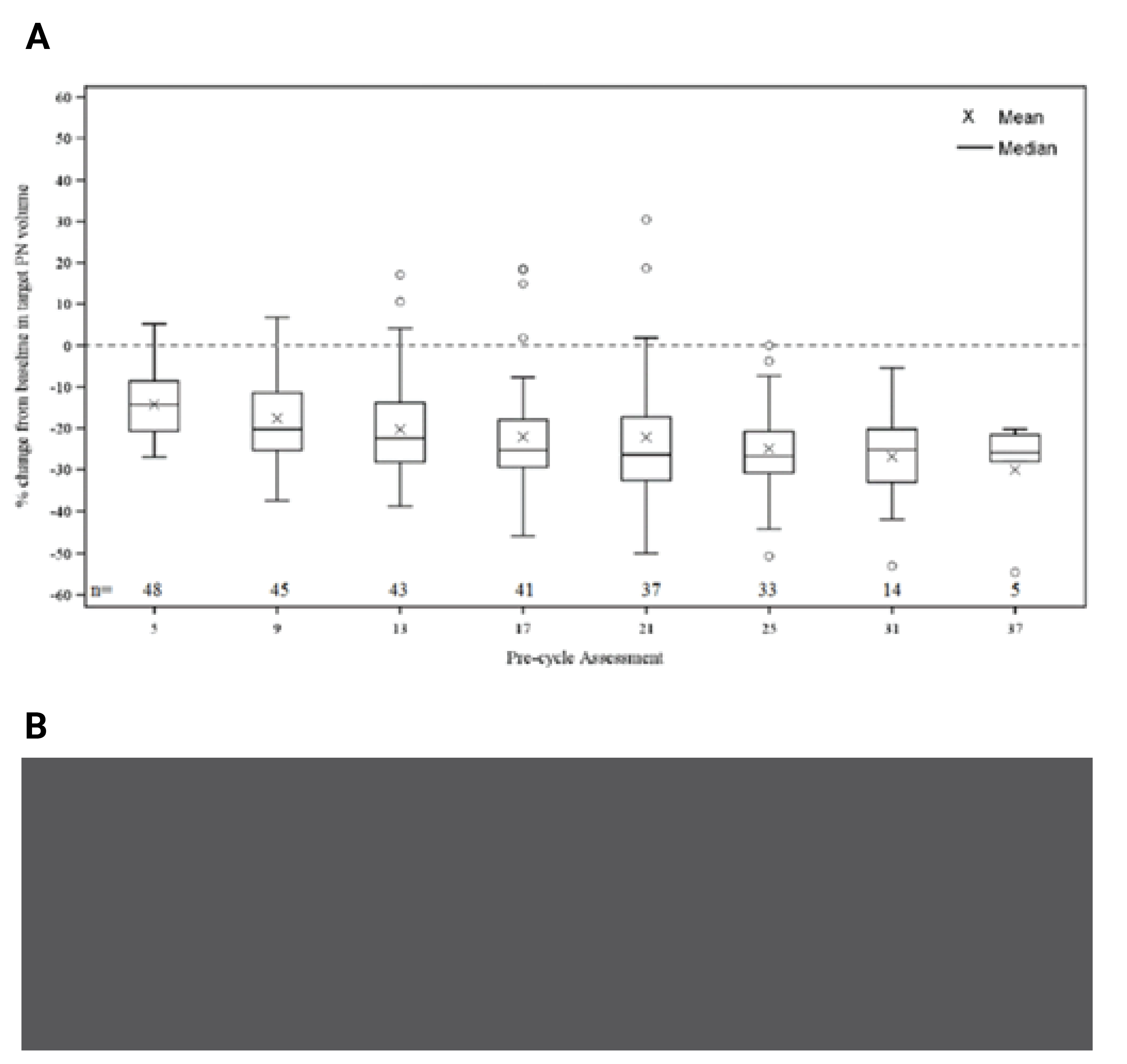
A) June 29, 2018, DCO; B) March 31, 2021, DCO
DCO = data cut-off; ICR = independent central review; NCI = National Cancer Institute; PN = plexiform neurofibroma; POB = Pediatric Oncology Branch.
Note: Figure 2b was redacted at the request of the sponsor.
Sources: SPRINT Phase II Stratum 1 Clinical Study Report (June 29, 2018, DCO)19 and SPRINT Phase II Stratum 1 Clinical Study Report (March 31, 2021, DCO).20
At the March 31, 2021, DCO, 95.8% of patients had a reduction in target PN volume, and the mean percent change from baseline at precycle 13 in the FAS was ||||||| (SD = ||||||), corresponding to a mean absolute change of ||||||| || ||||||||). The proportion of patients with a maximum reduction from baseline of 20% or greater was identical to the June 29, 2018, DCO (77.1%), and 5 (1.04%) had a maximum reduction from baseline of 40% or greater. One patient (2.1%) had a maximum reduction from baseline of 60% or greater, which represented the largest change from baseline (−60.3%) at precycle 55.20
Duration of Response
DOR was a secondary efficacy end point of the SPRINT phase II trial. Results for DOR at both DCOs are summarized in Table 27. At the June 29, 2019, DCO, 32 of 33 patients with a confirmed response had been followed up for at least 12 months from the onset of response, while at the March 31, 2021, DCO, all 34 patients with a confirmed response had been followed up for at least 12 months from the onset of response. At both DCOs the median DOR from onset of response was not reached (95% CI, NE to NE).19,20
The minimum durations of response at the June 29, 2018, and March 31, 2021, DCOs were 4 and 8 cycles, respectively, while the maximum durations of response were 32 and 64 cycles, respectively. After 24 cycles, the proportion of patients remaining in response was estimated to be 91.6% (95% CI, 70.1 to 97.8) at the June 29, 2019, DCO, and 90.3% (95% CI, 72.9 to 96.8) at the March 31, 2021, DCO. After 48 cycles of treatment, the estimated proportion of patients remaining in response was 69.6% (95% CI, 47.9 to 83.7).19,20
Table 27: Duration of Confirmed Response in Patients With Objective Response: NCI POB Central Analysis (REiNS; FAS) (Redacted)
|||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
|---|---|---|
|||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
|||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
b.i.d = twice daily; CI = confidence interval; DCO = data cut-off; FAS = full analysis set; NC = not calculated; NCI = National Cancer Institute; NE = not evaluable; NR = not reported; POB = Pediatric Oncology Branch; REiNS = Response Evaluation in Neurofibromatosis and Schwannomatosis.
Note: This table has been redacted at the request of the sponsor.
aProgressed after duration of response of 16 and 18 cycles.
bDuration of response was defined as the time from the precycle volumetric MRI assessment of the first documented (which was subsequently confirmed) response until the precycle volumetric MRI assessment of documented progression or death in the absence of disease progression (i.e., precycle volumetric MRI assessment of progression event or censoring — precycle volumetric MRI assessment of first response where each cycle was 28 days). Response required consecutive confirmation within 3 to 6 months after the criteria for first response was met.
cCalculated using Kaplan-Meier technique. The denominator is the number of patients with objective response.
Note: This table has been redacted for confidential information.
Sources: SPRINT Phase II Stratum 1 Clinical Study Report (June 29, 2018, DCO)19 and SPRINT Phase II Stratum 1 Clinical Study Report (March 31, 2021, DCO)20
Figure 3: Waterfall Plot of Best Percentage Change From Baseline in Target PN Volume — NCI POB Versus ICR (Full Analysis Set; June 29, 2018, DCO)
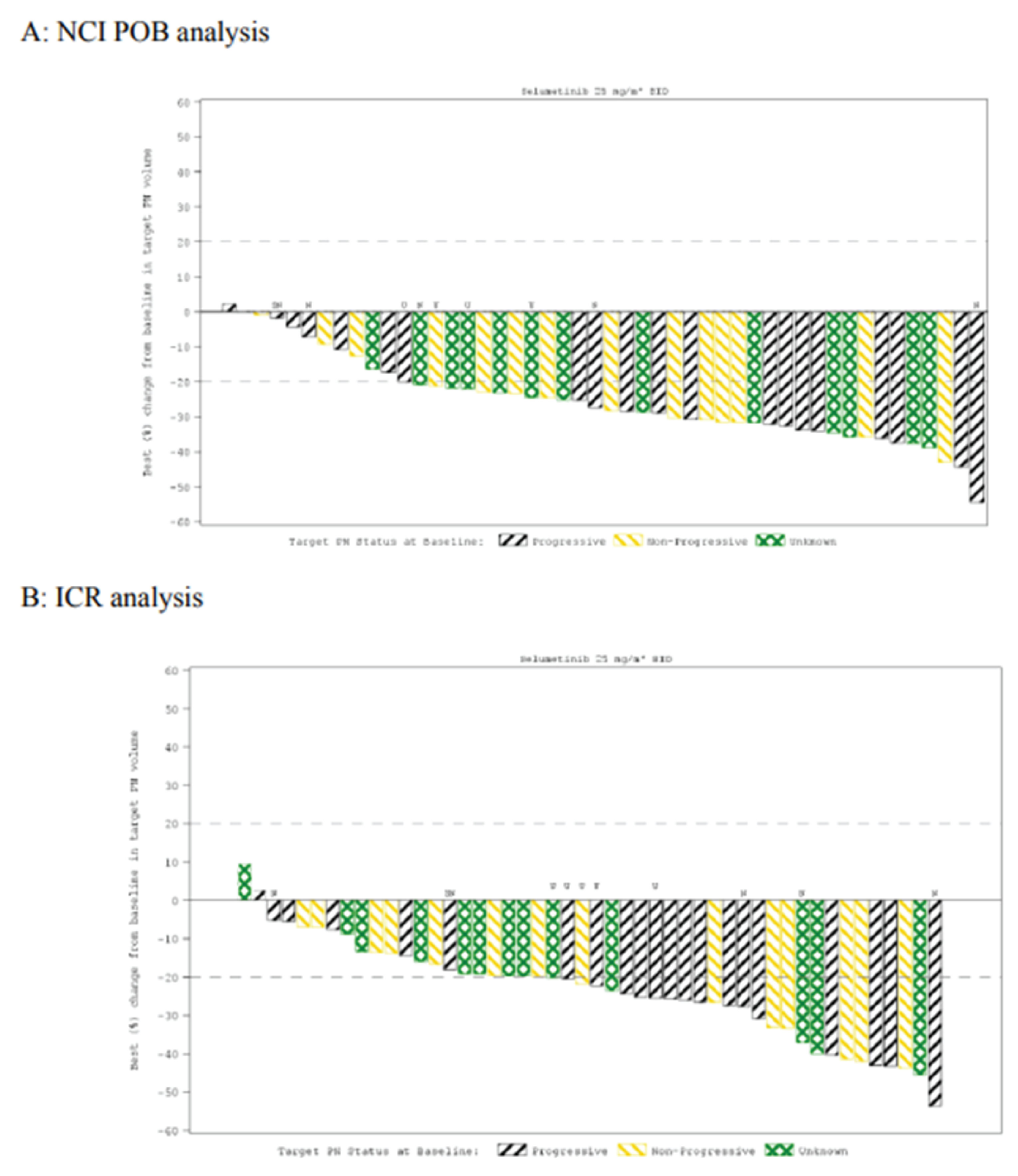
DCO = data cut-off; ICR = independent central review; NCI = National Cancer Institute; PN = plexiform neurofibroma; POB = Pediatric Oncology Branch; N = nodular plexiform neurofibroma; SN = solitary nodular plexiform neurofibroma; U = unconfirmed response.
Source: SPRINT Phase II Stratum 1 Clinical Study Report (June 29, 2018, DCO).19
Figure 4: Waterfall Plot of Best Percentage Change From Baseline in Target PN Volume (Full Analysis Set; March 31, 2021, DCO)
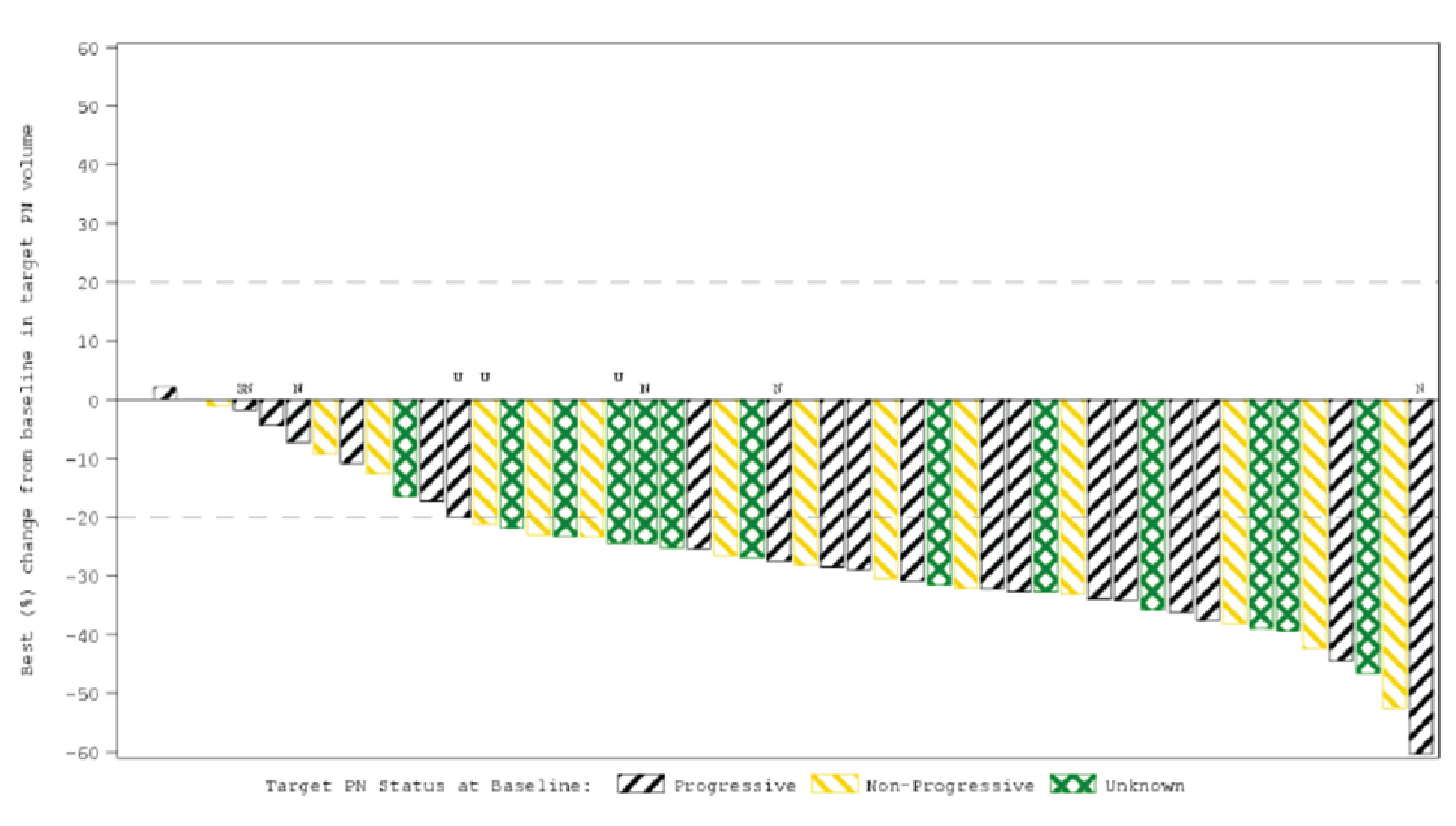
DCO = data cut-off; N = nodular plexiform neurofibroma; PN = plexiform neurofibroma; SN = solitary nodular plexiform neurofibroma; U = unconfirmed response.
Source: SPRINT Phase II Stratum 1 Clinical Study Report (March 31, 2021, DCO).20
Sensitivity Analyses: Because the primary analysis does not account for any delays between cycles or interruptions due to dose modifying toxicities, investigations, or other reasons (e.g., surgery), a sensitivity analysis using the actual dates of the volumetric MRI assessments was performed to assess the robustness of the findings based on precycle assessments. Results of this sensitivity analysis were consistent with those of the primary analysis at both DCOs.19,20
An additional sensitivity analysis using the ICR analysis of DOR was conducted. Based on the ICR assessment, || ||||||| patients had confirmed PR and || ||||| patients subsequently had disease progression. At the June 29, 2018, DCO, the median DOR was reached at the last event; however, the data were not provided as they were considered immature.19 No sensitivity analysis per ICR assessment was conducted at the March 31, 2021, DCO.
An exploratory ICR analysis using modified RECIST 1.1 was also conducted at the June 29, 2018, DCO. Based on RECIST 1.1 assessment, only patients were considered responders, and only || ||||| patient remained in response at 12 cycles or later, although the median DOR was not calculable.19
Time to Response
Results for TTR at the June 29, 2018, DCO and March 31, 2021, DCO are summarized in Table 28. At both DCOs, the median time to onset of response from the first dose was 8 cycles (95% CI, 4.0 to 8.0). In the 33 responders at the June 29, 2018, DCO, almost half (14 patients [42.2%]) had a response detected by 4 cycles from first dose, with most patients (24 [72.7%]) demonstrating a response by 8 cycles from first dose.19 At the time of the March 31, 2021, DCO, the results were consistent with the initial DCO, and were sustained for up to 44 cycles.20
Table 28: TTR in Patients With ORR: NCI POB Central Analysis (Full Analysis Set)
TTR | SPRINT phase II (stratum 1) selumetinib 25 mg/m2 twice daily (N = 50) | |
|---|---|---|
June 29, 2018, DCO | March 31, 2021, DCO | |
Number of patients with ORR | 33 | 34 |
Median time (cycles) to onset of response from first dose (95% CI)a, b | 8.0 (4.0 to 8.0) | 8.0 (4.0 to 8.0) |
≤ 4 cycles | 14 (42.4) | 14 (41.2) |
≤ 8 cycles | 24 (72.7) | 24 (70.6) |
≤ 12 cycles | 32 (97.0) | 32 (94.1) |
≤ 16 cycles | 32 (97.0) | 32 (94.1) |
≤ 24 cycles | 33 (100) | 33 (97.1) |
≤ 32 cycles | NE | 33 (97.1) |
≤ 44 cycles | NE | 34 (100) |
CI = confidence interval; DCO = data cut-off; NE = not estimable; NCI = National Cancer Institute; ORR = objective response rate; POB = Pediatric Oncology Branch; TTR = time to response.
aTTR was defined as the time from study treatment initiation until the precycle volumetric MRI assessment of the first documentation of complete response or a subsequently confirmed partial response.
bCalculated using the Kaplan-Meier technique. The denominator is the number of patients with objective response.
Sources: SPRINT Phase II Stratum 1 Clinical Study Report (June 29, 2018, DCO)19 and SPRINT Phase II Stratum 1 Clinical Study Report (March 31, 2021, DCO).20
Progression-Free Survival
PFS was a secondary outcome of the SPRINT phase II trial, and results for the June 29, 2018, and March 31, 2021, DCOs are summarized in Table 29. At the June 29, 2018, DCO, the median PFS was not reached as only 3 patients (6.0%) had progression events. As of the March 31, 2021, DCO, the median PFS remained unreached as only || ||||||| patients had a PFS event. The PFS rate at cycle 36 was ||||| (95% CI, |||| || ||||), and ||||% (95% CI, |||| || ||||) at the June 29, 2018, and March 31, 2021, DCOs, respectively.19,20
Table 29: Progression-Free Survival by NCI POB Central Analysis (FAS)
Progression-free survival | SPRINT phase II (stratum 1) selumetinib 25 mg/m2 twice daily (N = 50) | |
|---|---|---|
June 29, 2018, DCO | March 31, 2021, DCO | |
Events, n (%)a | 3 (6.0) | 10 (20.0) |
Progression, n (%) | 3 (6.0) | 10 (20.0) |
Death, n (%) | 0 (0.0) | 0 (0.0) |
Censored, n (%) | 47 (94.0) | 40 (80.0) |
Median PFS (cycles)b | NC | NC |
PFS rate, % (95% CI)b | ||
Cycle 16 | 100 (NE to NE) | ||| ||||||| |
Cycle 24 | 94.7 (80.6 to 98.7) | |||| ||||||||||| |
Cycle 30 | 88.8 (66.4 to 96.6) | |||| ||||||||||| |
Cycle 36 | 88.8 (66.4 to 96.6) | |||| ||||||||||| |
Cycle 48 | NE | |||| ||||||||||| |
Median follow-up for PFS (cycles) for censored patients onlyc | 24.0 (0.0 to 36.0) | 48.0 (0.0 to 72.0) |
CI = confidence interval; DCO = data cut-off; FAS = full analysis set; NC = not calculated; NCI = National Cancer Institute; NE = not evaluable; NR = not reported; PFS = progression-free survival; POB = Pediatric Oncology Branch.
aProgression includes deaths in the absence of progression.
bCalculated using the Kaplan-Meier technique.
cCalculated as the median time from study treatment initiation to precycle of censoring (last evaluable precycle volumetric MRI assessment known to be nonprogression) in censored (nonprogressed) patients only.
Sources: SPRINT Phase II Stratum 1 Clinical Study Report (June 29, 2018, DCO)19 and SPRINT Phase II Stratum 1 Clinical Study Report (March 31, 2021, DCO).20
Harms
Only those harms identified in the review protocol are reported here. Table 30 provides detailed harms data.
Adverse Events
Throughout the SPRINT phase II trial, 49 patients (98.0%) experienced AEs as of both the June 29, 2018, and March 31, 2021, DCOs. At the June 29, 2018, DCO, the most frequently reported AEs during the SPRINT phase II trial were vomiting (|| |||||||), increased blood CPK (|| |||||||), diarrhea (|| |||||||), nausea (|| |||||||), and dry skin (|| |||||||). Grade 3 or higher AEs were reported in ||||| of patients, and the most frequent grade 3 or higher AEs included diarrhea (| |||||||), hypoxia (| ||||||), and pyrexia (| ||||||). Generally, grade 3 or higher AEs resulted in dose modification of selumetinib. All events except 1 case of grade 3 diarrhea were considered nonserious. In total, || ||||||| patients had at least ||| AE leading to a dose reduction, and 40 patients (80.0%) had at least ||| AE leading to a dose interruption.19
At the March 31, 2021, DCO, the most frequently reported AEs were the same as at the prior DCO and included vomiting (||| |||||||), increased blood CPK (|| |||||||), diarrhea (|| |||||||), nausea (|| |||||||), and dry skin (|| |||||||). Three additional grade 3 or higher AEs were reported at the March 31, 2021, DCO (|| |||||||), the most frequent being diarrhea (| |||||||), hypoxia, pyrexia, paronychia, and weight gain (| |||||| ||||). At this DCO, ||| additional patients had an AE leading to a dose reduction of selumetinib (|| |||||||), and ||| additional patients had an AE leading to a dose interruption of selumetinib (|| |||||||). The number of patients discontinuing selumetinib due to AEs remained the same between DCOs (| |||||||).20
Serious Adverse Events
As of the June 29, 2018, DCO, 12 (24.0%) patients experienced a total of 29 SAEs, the most frequently reported being infections and infestations (6 [12.0%]; bacterial tracheitis, Clostridium difficile colitis, influenza, osteomyelitis, skin infection, and urinary tract infection), followed by gastrointestinal disorders (3 [6.0%]; constipation, and diarrhea). A total of 6 SAEs led to discontinuation of selumetinib.19
At the March 31, 2021, DCO, || ||||||| patients experienced SAEs, the most frequently reported being infections and infestations (|||||||||||, and gastrointestinal disorders (| ||||||; constipation, abdominal pain, and diarrhea).20
Withdrawals Due to Adverse Events
At the June 29, 2018, DCO, a total of 6 patients (12.0%) had an AE leading to discontinuation of selumetinib. Five withdrawals were due to grade 3 AEs (acute kidney injury, diarrhea, MPNST, paronychia, and weight increased) and 2 were due to grade 4 AEs (blood creatinine increased and skin ulcer). One patient had 2 AEs leading to discontinuation (acute kidney injury and blood creatinine increased).19
At the March 31, 2021, DCO, there were no new patients with AEs that led to discontinuation of selumetinib since the June 29, 2018, DCO.
Mortality
No AEs with a fatal outcome were reported in the SPRINT phase II study at either DCO.19,20 Following the March 31, 2021, DCO, ||| patients died due to progressive neurofibrosarcoma after selumetinib treatment was terminated, although these deaths were not attributed to treatment with selumetinib.20
Notable Harms
Notable harms of interest to this review included cardiac events, ophthalmologic events, and paronychia.
Cardiac Events
Cardiac events as a medical concept under the Medical Dictionary for Regulatory Activities query of cardiac failure were included as AEs of special interest in the SPRINT phase II study. At the June 29, 2018, DOC, cardiac events were reported in |||||| patients: decreased ejection fraction (|| |||||||| all grade 2), peripheral edema (| |||||||| all grade 1), peripheral swelling (| ||||||| grade 1), and decreased right ventricle ejection fraction (| ||||||| grade 1).19
At the March 31, 2021, DCO, 21 patients (42.0%) had cardiac events, with increases from the previous DCO in decreased ejection fraction (13 [26.0%]) and peripheral edema (9 [18.0%]). Both additional decreased ejection fraction events were grade 2, while the additional peripheral edema cases were mostly grade 1, with a single grade 2 event.20
Ophthalmologic Events
Ophthalmologic events were a notable harm of interest to this review. Retinal events as a medical concept were also an AE of special interest in the SPRINT phase II study. A total of 8 patients (16.0%) reported retinal events at the June 29, 2018, DCO, including a chorioretinal scar (1 [2.0%]), photophobia (2 [4.0%]), blurred vision (4 [8.0%]), and vitreous disorder (1 [2.0%]). All ophthalmologic events were grade 1 with the exception of 1 grade 2 AE of blurred vision.19
At the March 31, 2021, DCO, || ||||||| patients had ophthalmologic events; | |||||||||| |||||||| ||| ||||| ||||||| || ||||||| |||||| || ||||||||, which were nonserious and did not require treatment, dose interruption, dose reduction, or discontinuation. ||| additional ophthalmologic events of retinal tear and visual field defect were reported in | ||| patient each.20
Paronychia
Paronychia is a known adverse drug reaction for selumetinib and was an AE of special interest in the SPRINT phase II trial. At the June 29, 2018, DCO, paronychia was reported by 23 patients (46.0%), 2 were grade 1, 18 were grade 2, and 3 were grade 3. All cases were nonserious, and most were managed with symptomatic or supportive treatment. Seven patients with paronychia required a dose interruption, and 3 had an interruption followed by a dose reduction. One patient discontinued treatment with selumetinib due to grade 3 paronychia following 2 dose reductions.19
At the March 31, 2021, DCO, || ||||||| patients reported paronychia; || || cases were grade || ||, and ||| patient had a grade || || event. All additional paronychia events were considered nonserious. One patient required a dose interruption. All patients recovered from paronychia; however, 1 patient had unknown outcome status at the DCO.20
Table 30: Summary of Harms (Safety Analysis Set)
Harm | SPRINT phase II selumetinib 25 mg/m2 twice daily (N = 50) | |
|---|---|---|
June 29, 2018, DCO | March 31, 2021, DCO | |
Treatment-emergent adverse eventsa | ||
Patients with any AEs, n (%) | 49 (98.0) | 49 (98.0) |
Vomiting | 41 (82.0) | || |||||| |
Diarrhea | 35 (70.0) | || |||||| |
Nausea | 33 (66.0) | || |||||| |
Stomatitis | 25 (50.0) | || |||||| |
Abdominal pain | 22 (44.0) | || |||||| |
Abdominal pain upper | 20 (40.0) | || |||||| |
Constipation | 17 (34.0) | || |||||| |
Increased blood creatine phosphokinase | 38 (76.0) | || |||||| |
Increased aspartate transaminase | 23 (46.0) | || |||||| |
Dry skin | 30 (60.0) | || |||||| |
Dermatitis acneiform | 25 (50.0) | || |||||| |
Pruritus | 23 (46.0) | || |||||| |
Maculo-papular rash | 18 (36.0) | || |||||| |
Hypoalbuminemia | 25 (50.0) | || |||||| |
Pyrexia | 28 (56.0) | || |||||| |
Fatigue | 28 (56.0) | || |||||| |
Paronychia | 23 (46.0) | || |||||| |
Oropharyngeal pain | 24 (48.0) | || |||||| |
Cough | 20 (40.0) | || |||||| |
Nasal congestion | 17 (34.0) | || |||||| |
Headache | 24 (48.0) | || |||||| |
Anemia | 21 (42.0) | || |||||| |
SAEs | ||
Patients with any SAE, n (%) | || |||||| | || |||||| |
Bacterial tracheitis | | ||||| | | ||||| |
Clostridium difficile colitis | | ||||| | | ||||| |
Influenza | | ||||| | | ||||| |
Osteomyelitis | | ||||| | | ||||| |
Skin infection | | ||||| | | ||||| |
Urinary tract infection | | ||||| | | ||||| |
Constipation | | ||||| | | ||||| |
Diarrhea | | ||||| | | ||||| |
Abdominal pain | | ||||| | | ||||| |
Anemia | | ||||| | | ||||| |
Fracture | | ||||| | | ||||| |
Procedural hypotension | | ||||| | | ||||| |
Increased blood creatine phosphokinase | | ||||| | | ||||| |
Increased blood creatinine | | ||||| | | ||||| |
Hypoxia | | ||||| | | ||||| |
Pyrexia | | ||||| | | ||||| |
Peripheral edema | | ||||| | | ||||| |
Dehydration | | ||||| | | ||||| |
Hyperkaliemia | | ||||| | | ||||| |
Hyperuricemia | | ||||| | | ||||| |
Hypocalcemia | | ||||| | | ||||| |
MPNST (neurofibrosarcoma) | | ||||| | | ||||| |
Acute kidney injury | | ||||| | | ||||| |
Hematuria | | ||||| | | ||||| |
Proteinuria | | ||||| | | ||||| |
Skin ulcer | | ||||| | | ||||| |
Hematoma | | ||||| | | ||||| |
Depression | | ||||| | | ||||| |
WDAEs | ||
Any WDAEs, n (%) | 6 (12.0) | | |||||| |
Increased blood creatinine | 1 (2.0) | | ||||| |
Weight increased | 1 (2.0) | | ||||| |
Diarrhea | 1 (2.0) | | ||||| |
Paronychia | 1 (2.0) | | ||||| |
MPNST | 1 (2.0) | | ||||| |
Acute kidney injury | 1 (2.0) | | ||||| |
Skin ulcer | 1 (2.0) | | ||||| |
Notable harms | ||
Cardiac events, n (%) | 18 (36.0) | || |||||| |
Decreased ejection fraction | || |||||| | || |||||| |
Edema peripheral | | |||||| | | |||||| |
Peripheral swelling | | ||||| | | ||||| |
RVEF | | ||||| | | ||||| |
Ophthalmologic events, n (%) | 8 (16.0) | || |||||| |
Chorioretinal scar | | ||||| | | ||||| |
Photophobia | | ||||| | | ||||| |
Vision blurred | | ||||| | | |||||| |
Vitreous disorder | | ||||| | | ||||| |
Retinal tear | | ||||| | | ||||| |
Visual field defect | | ||||| | | ||||| |
Paronychia, n (%) | 23 (46.0) | || |||||| |
AE = adverse event; DCO = data cut-off; MPNST = malignant peripheral nerve sheath tumour; RVEF = right ventricular ejection fraction; SAE = serious adverse event; TEAE = treatment-emergent adverse event; WDAE = withdrawal due to adverse event.
Note: For TEAEs, patients with multiple events in the same preferred term (PT) were counted only once in that PT. Patients with events in more than one PT were counted once in each of those PTs. Includes AEs with an onset date on or after the date of first dose and up to and including 30 days following the date of last dose of selumetinib.
aFrequency of greater than 40% of patients at the March 31, 2021, DCO.
Sources: SPRINT Phase II Stratum 1 Clinical Study Report (June 29, 2018, DCO)19 and SPRINT Phase II Stratum 1 Clinical Study Report (March 31, 2021, DCO).20
Critical Appraisal
Internal Validity
The SPRINT phase II trial was the only study included in this review. It was a phase II, open-label, single-arm, multicentre study. The choice to conduct a single-arm trial was justified considering the rarity of the indication (NF1-associated PNs), and the lack of other treatment options. The SPRINT phase II trial enrolled patients from 2015 to 2016, and marketing authorization was not granted until 2020. The sponsor noted that the NCI considered conducting a placebo-controlled trial to be unethical based on the results of its phase I study; however, it is unclear if the off-label use of selumetinib also supported the single-arm design. The decision to conduct a single-arm study also has implications for the overall strength and interpretability of the results. As a single-arm study, there is an increased risk of bias in the estimation of treatment effects due to the potential for confounding related to natural history, and other unidentified prognostic factors could affect all study outcomes. Additionally, in a single-arm trial, because all patients receive the same treatment, the effect of treatment on time-to-event end points from the SPRINT trial, such as PFS, DOR, and TTR, is uninterpretable and was only considered as exploratory and supportive.
Awareness of treatment assignment by both patients and parents or caregivers increases the risk of detection bias and performance bias and may lead to systematic overestimation of the overall treatment effect. As such, the open-label trial design limits interpretability of the clinical outcome assessments such as the PRO and functional end points, as well as AEs. The potential for bias in data evaluated via volumetric MRI was reduced by using an ICR, following a request from the FDA. Results of the independent review of volumetric MRI data were generally consistent with those of the primary NCI POB expert reader evaluation, although, according to the ICR, the ORR was reduced (66.0% [June 2018 DCO] versus |||||). Discordance between the NCI POB and IRC assessments of response was attributed to the 5 responders who had responses just below the cut-off of 20% which defined a PR in the trial. It is possible that the ICR assessment of response provided more unbiased assessments compared with the investigator assessments, given that the investigator assessments were conducted by a single expert in volumetric MRI; however, there is potential for human error with both methods.
The original study protocol was amended 18 times, of which 8 were for the phase I portion of the study, and 9 were for the phase II portion. None of the protocol amendments or deviations were believed to affect the conduct or integrity of the study.
Aside from the primary end point of ORR, no inferential statistical testing was performed for the secondary efficacy outcomes of the SPRINT phase II trial and outcomes were not controlled for multiplicity. They therefore should be interpreted with consideration of the increased risk of type I error. Point estimates with 95% CIs were reported to estimate the magnitude of treatment effect. The threshold for observing a positive study outcome for stratum I of the SPRINT phase II trial was 15%, although the basis for this estimate was unclear.
The primary end point of the SPRINT phase II study was the ORR and was considered by the clinical experts consulted by CADTH and the CADTH review team to be an appropriate objective measure to assess the activity of selumetinib. In oncology, an ORR is a direct measure of a drug’s antitumour activity. In the SPRINT phase II trial, ORR, and other tumour-related outcomes (DOR, TTR, and PFS) were evaluated using the REiNS criteria based on volumetric MRI. An objective response is often defined as the proportion of patients with reductions in tumour size of a predefined amount. Using REiNS criteria, this predefined reduction was 20%, which the clinical experts consulted by CADTH considered appropriate as they use a threshold of 20% to 25%. However, the experts noted that it can be difficult to determine the size of PNs in clinical practice and they rely heavily on symptomatology to determine progression. In an exploratory correlation analysis between clinical outcome assessments and target tumour volume, absolute Spearman rank coefficients were mostly considered poorly correlated for the changes from baseline of each variable, with r values ranging from 0.06 to 0.52. Additionally, no sensitivity analyses were conducted, and the clinical experts noted that age may be a critically important confounding factor when assessing changes in tumour size, as growth tends to be more rapid in younger patients. No formal subgroup analyses were defined in the SPRINT phase II trial. Instead, at study enrolment, patients were classified according to their PN status as progressive (i.e., growth ≥ 20% in the 12 to 15 months before enrolment), nonprogressive, or unknown based on volumetric MRI, although only descriptive results were reported. Other secondary end points of the SPRINT phase II study consisted of clinical outcome assessments of PROs and functional evaluations, which were considered appropriate to evaluate the wide range of PN-related morbidities. However, only the PII was validated for use in the NF1 population and the results should only be viewed as supportive based on the design of the SPRINT phase II study.
A limited number of patients were included in the analyses, with only 50 making up the FAS. The small sample size was further restricted for secondary end points, including PROs and functional evaluations, as these were based on patients with target PNs in specific locations or limited to patients of a certain age (e.g., NRS-11 in patients aged ≥ 8 years); the evaluable population was therefore based on a subset of the FAS that was further reduced for these outcomes. Furthermore, these outcomes were subject to a high level of missing data, with 4.0% to 50.0% of data across outcomes (including both parent- and self-reported questionnaires) missing from precycle 13. Additionally, because no statistical test or imputation of missing data was conducted on these outcomes, the results should only be viewed as supportive of the overall effect of selumetinib. MIDs in PROs and functional evaluations were derived from the literature and through anchor-based methods from the results of the GIC analysis, and distribution-based using one-half SD.
External Validity
The SPRINT phase II pivotal study is an open-label, noncontrolled, single-arm, multicentre trial of the efficacy and safety of selumetinib in children (aged 2 to 18 years old) with NF1 and a PN that could not be surgically completely removed without a substantial risk of morbidity due to encasement of, or proximity to, vital structures, invasiveness, or high vascularity of the PN. As previously noted, the noncomparative design of the SPRINT phase II trial precludes the ability to assess the relative therapeutic benefit or safety of selumetinib in Canadian clinical practice.
In discussion with the clinical experts consulted by CADTH, the inclusion and exclusion criteria for SPRINT phase II study were generally as expected for patients with NF1 with symptomatic, inoperable PNs who would require treatment with selumetinib. However, the experts noted that the requirement of a Karnofsky or Lansky performance level of 70% or greater is indicative of patients who are high-functioning and could exclude some with another significant morbidity (i.e., neurocognitive deficits), which may represent a more well-off, or higher-functioning population.
The clinical experts also described the baseline characteristics as generally reflective of the type of patients eligible for selumetinib in Canada. According to the protocol of the SPRINT phase II study, the target PN was selected by investigators at study entry. While no information on the method, process, or selection of target PNs was provided, the clinical experts consulted by CADTH emphasized that identification of the problematic PN is generally not of concern, although multiple PNs often cause symptoms, disfiguration, or loss of function. In the SPRINT phase II study, an ICR was required to assess the same target PN selected by the investigators; however, it remains unclear whether there were differences in selection of target PNs between the NCI POB central reviewer and ICR team.
There is a lack of standardized end points for trials of patients with NF1. As previously noted, multiple outcomes were included in the SPRINT phase II trial, including response (ORR) and time-to-event outcomes (DOR, TTR, and PFS) based on volumetric MRI and the REiNS criteria. The clinical experts consulted by CADTH highlighted that volumetric MRI is not used in routine clinical practice as it is not standard of care in Canada, and that evidence of disease progression based on standard imaging techniques is multifactorial, although they emphasized the importance of clinical symptomatology and physical assessment in determining progression. As such, patients in Canadian clinical practice would be evaluated for progression slightly differently than in the SPRINT phase II trial, potentially affecting the generalizability of the results. Additionally, the SPRINT phase II trial applied the REiNS criteria, which is an internationally recognized effort to standardize response criteria for determining treatment response in patients with NF1-associated PN. While pain, motor function, and QoL are included in the evaluation of treatment response according to the REiNS criteria, it is unclear how these imaging and other assessment criteria are applied to patients in Canadian clinical practice, and the generalizability of results, using the REiNS imaging response criteria, is uncertain, given the unavailability of volumetric MRI. A sensitivity analysis of ORR and DOR was conducted using modified RECIST 1.1 criteria, producing an ORR of only 2.0%. The threshold for response in RECIST 1.1 was 30%, which has limited applicability in NF1.
Patient-reported outcomes (NRS-11, PII, PROMIS, and PedsQL) and functional outcomes (strength and ROM) were also evaluated in the SPRINT phase II trial. The clinical experts consulted by CADTH noted that the outcome scales reported in the trial are not used in routine clinical practice and may not be generalizable to the typical patient in Canada. For example, the experts pointed out that, given the subjectivity in pain perception, a simple visual-based scale using faces is used to measure pain due to its simplicity for the pediatric population, as opposed to questionnaires and numbered scales such as the NRS-11 or PII. Additional outcomes were included in the SPRINT phase II study to quantify the changes due to selumetinib administration, such as the grooved pegboard test, the key pinch grip, PFTs, sleep studies, and bowel and bladder function, among others. However, the experts noted that a gestalt-type approach is considered in clinical practice, and, although measures specific to location or body quadrant may be individually important and despite the significant variation and heterogeneity among patients and caregivers in this population, the general consensus was that overall improvement or deterioration is 1 of the most important considerations to patients and families. Although the results of the PRO and functional outcomes were generally sustained over the long-term throughout the study period, the large amount of missing data in the PRO and functional outcomes at the predefined time of assessment (precycle 13) and beyond restricts the generalizability of these outcomes.
Considering the age of the enrolled population, as well as the complexity of the disease and multidisciplinary approach to disease management, it is possible that patients included in the SPRINT phase II trial may have received additional background care or monitoring that they would be unable to receive in the real world. While no analysis of these factors was conducted in the SPRINT phase II trial, the experts consulted by CADTH emphasized that the clinical setting for administering and monitoring these patients is still evolving, and access to additional monitoring, supportive care, or follow-up in the SPRINT phase II trial may affect generalizability.
The inability to swallow capsules was an exclusion criterion for the SPRINT phase II study, and, although patients as young as 3 years of age were enrolled, it was considered unlikely that many patients would be unable to swallow the capsules. In most cases however, the experts noted that most PN growth occurs during early childhood, around 6 to 8 years of age, when patients may be more likely to swallow capsules. Additionally, given that the mean age at baseline (10.3 years), it is unlikely that patients would not be able to swallow capsules. Still, this remains a concern for the younger population.
Indirect Evidence
Objectives and Methods for the Summary of Indirect Evidence
The objective of this section is to summarize and appraise the indirect evidence for selumetinib for the treatment of pediatric patients aged 2 years or older with NF1 and symptomatic, inoperable PNs.
A focused literature search for ITCs dealing with Koselugo and NF1 was run in MEDLINE All (1946–) and Embase (1974‒) on November 16, 2022. Retrieval was not limited by publication date or language. Five articles were identified in the search; however, none met the prespecified inclusion criteria for this review.
Description of Indirect Comparisons
As there are no appropriate comparators to conduct a standard ITC, and a placebo-controlled trial was considered unethical by NCI POB investigators due to significant PN-related morbidity and promising results shown in the phase I trial,21 the sponsor submitted qualitative comparisons using 2 external controls, which will together be summarized as the “sponsor-submitted ITCs:”
An ongoing prospective NH study conducted by NCI POB (NCT00924196), which was intended to develop a better understanding and quantification of NF1 manifestations, and to allow more sensitive end points to be developed for clinical studies (hereafter the NH study).19,20
The placebo arm of the NCI POB-coordinated tipifarnib randomized controlled trial in patients aged 3 to 25 years with NF1 and progressive PN (hereafter Study 01-C022).52
The sponsor also submitted a propensity score modelling analysis of PFS compared with the NH study that hereafter is described as the “sponsor-submitted propensity score analysis.”
Methods of the Sponsor-Submitted Indirect Treatment Comparisons
Objectives
The objective of the study was to qualitatively evaluate the relative efficacy of selumetinib against 2 external controls representing the natural history of patients with NF1 and symptomatic PNs who would qualify for selumetinib treatment.
Study Selection Methods
No systematic review or study selection was described.
The NH study was considered a relevant external control by the sponsors because it was conducted by the same investigators as the SPRINT trial. The placebo arm of Study 01-C-0222 was considered an appropriate external control because it was also conducted by the same investigators as the SPRINT trial, and the placebo arm of this study was planned by the investigators to serve as a historical control group for any future phase II studies of new drugs for NF1-related PN.
Sponsor-Submitted Indirect Treatment Comparison Analysis Methods
Natural History Study: Analysis Sets
The comparison to the NH study included a side-by-side comparison of target PN growth rate (annual percent change), total volume change in target PN, and PFS compared to results from 3 different analysis sets.
The full NH study analysis set: all patients in the NH study with NF1-related PN who had at least 2 volumetric MRI scans, including adult patients. For patients who received a MEK inhibitor, such as selumetinib, data collected during the MEK inhibitor treatment period of the NH study have been excluded from all analyses.
Age-matched cohort: a subset of the full NH study analysis set where the first scan done within the age range of 3 to 18 years was considered the baseline, and who had at least 1 subsequent volumetric scan. The age matching was not 1:1, but rather intended to only include patients first treated as pediatric patients for their NF1-related PN.
Natural history subset: a subset of patients in the full NH study analysis set who were also enrolled into stratum 1 of the SPRINT phase II trial and for whom continuous target PN volume data were available.
For this comparison with selumetinib, the NCI POB team selected for each patient the typical PN with the longest follow-up volumetric data, which was used to analyze PN growth rates. Only patients with NF1-related PNs were considered relevant for use as external controls in comparison with SPRINT.
Data on PN-related symptoms (pain, motor, and vision) are also being collected in the NH study but were not reanalyzed by the sponsor for this report due to critical differences that affected comparisons across studies. These differences were not further described.
The comparisons included natural history data at the full duration of follow-up, as well as those aligned with the maximum available follow-up from stratum 1 of the SRINT phase II trial at the DCO in question. As only the maximum value was aligned, the median follow-up available still differed.
No statistical comparisons were made, no adjustments were made for covariates, and there was no description of how missing data or censoring were handled in the ITCs.
Study 01-C-0222: Analysis Sets
The sponsor also compared the stratum 1 of the SPRINT phase II trial to the placebo arm of Study 01-C-0222. Because Study 01-C-0222 was a crossover trial, only patients assigned to placebo at the beginning of the study (i.e., in phase A) were included in the comparison. Subgroup analyses were also conducted using the patients from stratum 1 who had progressive PNs at baseline to align with this requirement in Study 01-C-0222.
No statistical comparisons were made, no adjustments were made for covariates, and there was no description of how missing data or censoring were handled in the ITCs.
Outcome Definitions
Crude plexiform neurofibroma growth rate:
For stratum 1 of the SPRINT phase II trial, the crude PN growth rate was defined as the percent change in target PN volume from the baseline volumetric MRI to the last volumetric MRI assessment over the time period in years. The time period was defined from the baseline volumetric MRI assessment date to the last evaluable assessment date up to the DCO or treatment discontinuation (whichever occurred first).
For the NH study, the crude PN growth rate was defined as the percent change in PN volume from the first to the last volumetric MRI assessment over the time period in years, where the time period was defined from the first to the last available volumetric MRI assessment or last volumetric MRI assessment date before the first use of an MEK inhibitor, including selumetinib. This estimate was also derived for the time period aligned to the maximum follow-up duration observed in stratum 1 of the SPRINT phase II trial.
Progression-free survival:
In the SPRINT trial, PFS was defined as the time from study treatment initiation to the precycle of documented progression or death in the absence of disease progression. Patients not known to have progressed or died at the time of analysis were censored at the last evaluable volumetric MRI assessment. Progression was defined as PN growth 20% or greater from baseline or best response if a PR had been achieved. In the NH study, PFS was defined as the time from first volumetric MRI assessment to the date of documented progression or death in the absence of disease progression. Patients not known to have progressed or died at the time of analysis were censored at the last available MRI assessment date or last MRI assessment date before the first use of a MEK inhibitor, including selumetinib. In Study 01-C-0222, a PN volume increase of 20% or greater in at least 1 PN compared with baseline was defined as progressive disease. The volumetric MRI analyses from Study 01-C-0222 and stratum 1 of the SPRINT phase II trial were performed by the same central reviewer at the NCI POB.
Sponsor-Submitted Indirect Treatment Comparisons:
Summary of Included Studies
Design
The NH study is a prospective study sponsored and conducted by the NCI POB in patients with NF1. In the NH study, any patients receiving medical treatment and/or radiation for NF1-related manifestations are eligible to participate. A maximum of 250 patients with any NF1-related manifestations are planned for enrolment. The study site is the NIH Clinical Center in Bethesda, Maryland, and the dates of recruitment were not reported. As part of participation in the NH study, patients have serial evaluations of their PN with the same image-acquisition protocol and the same volumetric MRI analysis method, typically scheduled yearly until the age of 18 and at least every 3 years thereafter.
Study 01-C-022252 was a phase II, randomized, flexible crossover, double-blinded, placebo-controlled trial of tipifarnib in children and young adults with NF1 and progressive PNs. The target enrolment was not reported. The placebo arm was intended to serve as a historical control group for future phase II single-arm trials directed at progressive PNs. After central randomization to the tipifarnib arm or placebo arm, patients were followed on the first treatment (phase A) until PN progression, based on volumetric MRI analysis, at which time they were crossed over to the other treatment arm (phase B) following a washout period. Patients were monitored until PN progression was documented in phase B, at which time they were removed from the study. Only data from patients who received placebo in phase A of Study 01-C-0222 are relevant for the purposes of this ITC.
Patient Eligibility Criteria
The patient eligibility criteria for stratum 1 of the SPRINT phase II trial, the NH study, and Study 01-C022 are summarized in Table 31. In contrast to the SPRINT trial, the NH study and Study 01-C-0222 included adult patients.
In the NH study, patients must have an Eastern Cooperative Oncology Group (ECOG) performance status of no more than 3. Patients who have not previously been evaluated at the NIH are eligible for enrolment if aged 35 years or younger; however, there is no upper limit for patients previously enrolled in clinical studies at the NIH, patients with MPNST, or with clinical concern for MPNST, or for infrequent or unusual NF1-related manifestations. There were no restrictions as to whether patients received any prior treatment for NF1-related benign or malignant tumours. Although the NH study included patients who may not have had NF1-associated PNs, data were available for the subset of patients with PNs (although not specified as unresectable), and this subset was utilized for the comparison with the SPRINT trial. For patients who received an MEK inhibitor such as selumetinib during the NH study, data collected during the MEK-inhibitor treatment period have been excluded from all analyses. Analyses were also conducted against an “age-matched” cohort of patients with PNs from the NH study; the age-matched cohort includes patients who are aged 3 to 18 years and have at least 1 volumetric MRI within this age and at least 1 subsequent volumetric MRI. Some patients participating in the NH study had also enrolled in stratum 1 of the SPRINT phase II trial, and their historical volumetric data of the target PN were analyzed separately as the NH study subset to determine growth rates before and after initiation of selumetinib.
Table 31: Key Patient Inclusion and Exclusion Criteria of Included Studies
Criteria | SPRINT phase II stratum 1 | NH study | Study 01-C-0222 |
|---|---|---|---|
Key inclusion criteria |
|
|
|
Key exclusion criteria |
|
|
|
CLIA = Clinical Laboratory Improvement Amendments; DCO = data cut-off; ECOG PS = Eastern Cooperative Oncology Group Performance Status; MEK = mitogen-activated protein kinase; MPNST = malignant peripheral nerve sheath tumour; NF1 = neurofibromatosis type 1; NF1-GIST = NF1-associated gastrointestinal stromal tumours; NH = Natural History; NIH = National Institutes of Health; PN = plexiform neurofibroma.
aStratum 2 is not relevant for this comparison and included only patients with no significant PN-related morbidity at enrolment who were identified as having the potential to develop PN morbidity. All SPRINT phase II trial data here are from stratum 1 only (i.e., a significant PN-related morbidity was present in all included patients).
Sources: SPRINT Phase II Stratum I Clinical Study Report (June 29, 2018, DCO),19 SPRINT Phase II Stratum I Clinical Study Report (March 31, 2021, DCO),20 and Widemann et al. (2014).52
Study 01-C-0222 did not require PNs to be symptomatic but rather to have the potential for significant morbidity. Children and young adults (between the ages of 3 and 25 years, inclusive) with unresectable NF1-associated PN were eligible for enrolment. Patients with prior surgical intervention for their progressive PNs were eligible provided the residual tumour was measurable. No prior medical therapy was required, given that there is no standard medical treatment for PNs.
Patient Baseline Characteristics
Baseline patient characteristics of the included studies are summarized in Table 32. As expected, the full NH study cohort had a higher maximum age than did the SPRINT trial; however, the mean age was similar. The age-matched NH study cohort included patients who are aged 3 to 18 years and have at least 1 volumetric MRI within this age and at least 1 subsequent volumetric MRI. The mean and median age of the age-matched NH study cohort was younger than in the SPRINT trial cohort, but the range of ages was similar. In Study 01-C-0222, mean age was not reported, but the median age was also younger than in the SPRINT trial. However, even though the eligibility criteria allowed adult patients, the maximum age at baseline was 17.7 years and was similar to that of the SPRINT trial.
Table 32: Key Patient Baseline Characteristics of Included Studies
Characteristic | SPRINT phase II stratum 1, selumetinib 25 mg/m2 twice daily | NH study (patients with PNs) | NH study subset (patients with PNs later enrolled in SPRINT) | Study 01-C-0222 placebo arm | ||
|---|---|---|---|---|---|---|
Full NH cohort | Age-matched cohorta | During NH study | During SPRINT | |||
Demographic data | ||||||
Nb | 50 | 111 | 92 | | |||||| | | |||||| | 29 |
Age, mean years (SD) | 10.3 (3.92) | |||| |||||| | ||| |||||| | ||| |||||| | |||| |||||| | NR |
Age, median years (minimum to maximum) | 10.2 (3.5 to 17.4) | ||| |||| || || | ||| |||| || || | ||| |||| || || | |||| |||| || || | 8.2 (3 to 17.7) |
Sex, % male | 60 | | |||||| | | |||||| | | |||||| | | |||||| | 58.3 |
Disease characteristics | ||||||
Target PN volume, mean mL (SD) | 837.11 (925.011) | |||||| ||||| | |||||| |||||| | | |||||| | | |||||| | NR median (range): 316 (39.6 to 4,896) |
PN status, n (%) | ||||||
Progressive | 21 (42.0) | | |||||| | | |||||| | | |||||| | | |||||| | Only patients with progressive disease were eligible |
Nonprogressive | 15 (30.0) | | |||||| | | |||||| | | |||||| | | |||||| | |
Unknown | 14 (28.0) | | |||||| | | |||||| | | |||||| | | |||||| | |
Target PN location, n (%)c | ||||||
Trunk | 5 (10.0) | 43 (38.7) | 36 (39.1) | | |||||| | | |||||| | 11 (35.5)d |
Trunk/extremity | 12 (24.0) | 21 (18.9) | 17 (18.5) | | |||||| | | |||||| | 3 (9.7) |
Head | 9 (18.0) | 13 (11.7) | 13 (14.1) | | |||||| | | |||||| | 3 (9.7)e |
Neck/trunk | 12 (24.0) | 16 (14.4) | 13 (14.1) | | |||||| | | |||||| | 9 (29.0)f |
Extremity | 4 (8.0) | 7 (6.3) | 7 (7.6) | | |||||| | | |||||| | 1 (3.2) |
Head/neck | 8 (16.0) | 5 (4.5) | 5 (5.4) | | |||||| | | |||||| | 4 (12.9) |
Whole body | 0 | 6 (5.4) | 1 (1.1) | | |||||| | | |||||| | NR |
PN-directed medical history | ||||||
Number of patients with any PN-directed medical treatment, n (%) | 31 (62.0) | || |||||| | | |||||| | | |||||| | | |||||| | 4 (13.79) |
Pegylated interferong | || |||||| | || |||||| | | |||||| | | |||||| | | |||||| | 0 |
Imatinib | || |||||| | | ||||| | | |||||| | | |||||| | | |||||| | NR |
Sirolimus | | |||||| | || |||||| | | |||||| | | |||||| | | |||||| | NR |
Thalidomide | | ||||| | | ||||| | | |||||| | | |||||| | | |||||| | 0 |
Multi-tyrosine kinase inhibitorsh | | ||||| | | ||||| | | |||||| | | |||||| | | |||||| | NR |
Other chemotherapyi | | ||||| | | ||||| | | |||||| | | |||||| | | |||||| | 3 (10.34) |
Pirfenidone | | ||||| | || |||||| | | |||||| | | |||||| | | |||||| | 0 |
Celecoxib | | |||||| | | |||||| | | |||||| | | |||||| | | |||||| | NR |
Interferon alfa-N3 | | |||||| | | |||||| | | |||||| | | |||||| | | |||||| | NR |
Interferon gamma | | |||||| | | |||||| | | |||||| | | |||||| | | |||||| | NR |
Tipifarnib | | |||||| | | |||||| | | |||||| | | |||||| | | |||||| | NR |
Cis retinoic acid | | |||||| | | |||||| | | |||||| | | |||||| | | |||||| | 0 |
DCO = data cut-off; NH = Natural History; NR = not reported; PN = plexiform neurofibroma; SD = standard deviation.
aThe age-matched cohort includes patients who are aged 3 to 18 years and have at least 1 volumetric MRI within this age and at least 1 subsequent volumetric MRI.
bFull NH study cohort: age at baseline volumetric MRI assessment of target PN; age-matched cohort: age at first volumetric MRI assessment, where the patient is 3 to 18 years; SPRINT phase II stratum 1: age at informed consent.
cStudy 01-C022: Only the target PNs are counted here. Other observed PNs that were not targeted are not quantified in this table but are reported in the study publication. Percentages were not reported and were therefore derived by CADTH based on the absolute numbers reported (target PNs total N = 31).
dSummed from the reported counts: pelvis: n = 6; abdomen: n = 2; back: n = 3.
eFace.
fNeck and chest.
gNH study: includes peg-interferon alfa-2a, peg-interferon alfa-2b. Study 01-C-0222: Includes only peg-interferon alfa 2b.
hNH study: includes sorafenib, sunitinib, PLX3397.
iNH study: includes methotrexate, carboplatin, vinblastine, vincristine. Study 01-C-0222: includes methotrexate, vinblastine.
Data as of February 19, 2019, (imaging data) and 19 March 19, 2019, (subset data); DCO: June 29, 2018 (SPRINT Phase II Stratum 1).
Sources: SPRINT Phase II Stratum I Clinical Study Report (June 29, 2018, DCO)19 and Widemann et al. (2014).52
At baseline, patients in stratum 1 of the SPRINT phase II trial had larger mean PNs compared with patients in the NH study and Study 01-C022, and the distribution of target PN location was different across studies. In the SPRINT trial, the most common target PN location was the trunk and extremity or the trunk and neck, whereas in the NH study and Study 01-C-0222 the most common location was the trunk (alone). Across studies, the least common locations were “whole body” (this was not reported as a category in Study 01-C-0222) and extremity (alone). Prior treatment history differed across the studies. The breakdown of ECOG scores was not reported for SPRINT or the NH study, but in the Study 01-C-0222 placebo arm patients had ECOG scores of 0 (n = 24, 82.76%), 1 (n = 13, 44.83%), or 2 (n = 2, 6.90%). Study 01-C-0222 did not report the proportion of patients whose PNs were symptomatic.
Limited baseline data were available for the NH study subset of patients who had enrolled in both the NH study and subsequently in the SPRINT trial. As expected, these patients were younger than all other cohorts described.
Results of Sponsor-Submitted Indirect Treatment Comparisons
PN Growth Rate: Comparison With Natural History Study
The data from stratum 1 of the SPRINT phase II trial was contrasted with those from the age-matched cohort from the NH study, either with all available follow-up from the NH study (up to 17.7 years) or with the maximum follow-up truncated to align with data from the SPRINT trial (up to a maximum of 2.6 years in the 2018, DCO, and 5.6 years in the 2021, DCO). Note that the median follow-up still differed after aligning the maximum follow-up.
The NH study shows that the vast majority of PNs grow continuously over time, or at best, remain stable in size (i.e., < 20% change in PN from baseline) in contrast to the median annual volume change of −10.2% seen in the SPRINT trial at the 2018 DCO or −5.1% in the 2021 DCO (Table 33). There was no spontaneous PN shrinkage of 20% or greater within 1 year in the NH study (Figure 5).
The median annual volume change for all studies and cohorts was more pronounced in the earlier DCO. Based on an examination of Figure 5, it appears that, for patients in the NH study, the most profound growth may happen earlier during follow-up and level off over time. Similarly, for patients who experienced a reduction in tumour volume in the SPRINT trial, it appears that this reduction happens most profoundly early on and levels off over time, and therefore the average annual rate of change appears to be lower than the maximum rate of change.
Table 33: PN Growth Rate in SPRINT and Age-Matched NH Study in 2018, DCO
Group | SPRINT phase II stratum 1a | Age-matched NH studyb | |
|---|---|---|---|
Maximum follow-up aligned to SPRINT | All available follow-up | ||
2018 DCO | |||
n | 48 | 90 | 92 |
Time period, median yearsc (range) | 1.8 (0.3 to 2.8) | ||| ||||| |||| | ||| ||||| ||||| |
PN volume % change/year,d median (range) | −10.2 ||||||| ||||| | |||| |||||| |||||| | |||| |||||| |||||| |
2021 DCO | |||
n | 48 | 92 | 92 |
Time period, median yearsc (range) | 4.0 (0.3 to 5.6) | ||| ||||| |||| | ||| ||||| ||||| |
PN volume % change/year,b median (range) | −5.1 ||||||| ||||| | |||| |||||| |||||| | |||| |||||| |||||| |
CI = confidence interval; DCO = data cut-off; MEK = mitogen-activated protein kinase; NH = Natural History; PN = plexiform neurofibroma.
aIncluded patients with baseline and at least 1 subsequent volumetric MRI assessment.
bThe full NH study analysis set includes all patients with NF1-related PN (all ages) who have at least 2 volumetric MRI scans. The age-matched cohort includes a subset of the full NH study analysis set including patients with at least 2 volumetric MRI scans in whom the first scan was done within the age range of 3 to 18 years was considered the baseline. Data for the full NH study analysis set is not included here.
cNatural history: Time period was defined from the first to the last available volumetric MRI assessment or last volumetric MRI assessment date before the first use of a MEK inhibitor, including selumetinib. For stratum 1 of the SPRINT phase II trial, the time period was defined from the baseline volumetric MRI assessment date until the last evaluable assessment date up to data cut-off or treatment discontinuation (whichever occurred first).
dPercentage PN volume change from the first volumetric MRI assessment (baseline assessment for stratum 1 of the SPRINT phase II trial) to the last volumetric MRI assessment over that time period in years.
Sources: SPRINT Phase II Stratum I Clinical Study Report (June 29, 2018, DCO)19 and SPRINT Phase II Stratum I Clinical Study Report (March 31, 2021, DCO).20
The mean absolute and mean percentage volume change from baseline to final post-baseline assessment are compared between the SPRINT trial (as of the 2021 DCO) and the NH study (FAS; not age-matched) in Table 34. Over the full duration of the studies, the mean percentage change from baseline in the SPRINT trial was |||||| compared to ||||||| in the NH study. The follow-up duration and included patients differ notably in these populations.
PN Growth Rate: Before-and-After Analysis of NH Patients Later Enrolled in SPRINT Phase II Stratum 1
Nine of the patients originally enrolled in the NH study went on to participate in stratum 1 of the SPRINT phase II trial. Of these patients, all experienced growth in their PN before selumetinib treatment at a median rate of 18.0% per year, with the most rapid growth being 52.0% per year.
The target PN followed may differ between the NH study and stratum 1 of the SPRINT phase II trial.
After initiation with selumetinib, at the March 31, 2021, DCO with a median follow-up of 2.3 years (range = 0.6 to 5.6), the median PN volume change in these 9 patients was ||||| per year (range = −|||| || ||||).
At the earlier DCO on June 29, 2018 (median follow-up = 1.8 years [range = 0.6 to 2.8]), 6 of the 9 patients had a reduction of at least 20% in their target PN during treatment with selumetinib. At the addendum DCO on 31 March 2021 (median follow-up 2.3 years; range = 0.6 to 5.6), the response was sustained at a reduction of 20% or greater in target PN volume from baseline for 5 of those patients while on selumetinib treatment.
Figure 5: Percentage Change in Target PN Volume, Natural History Study (Age-Matched) and SPRINT Phase II Stratum 1 in 2021 DCO
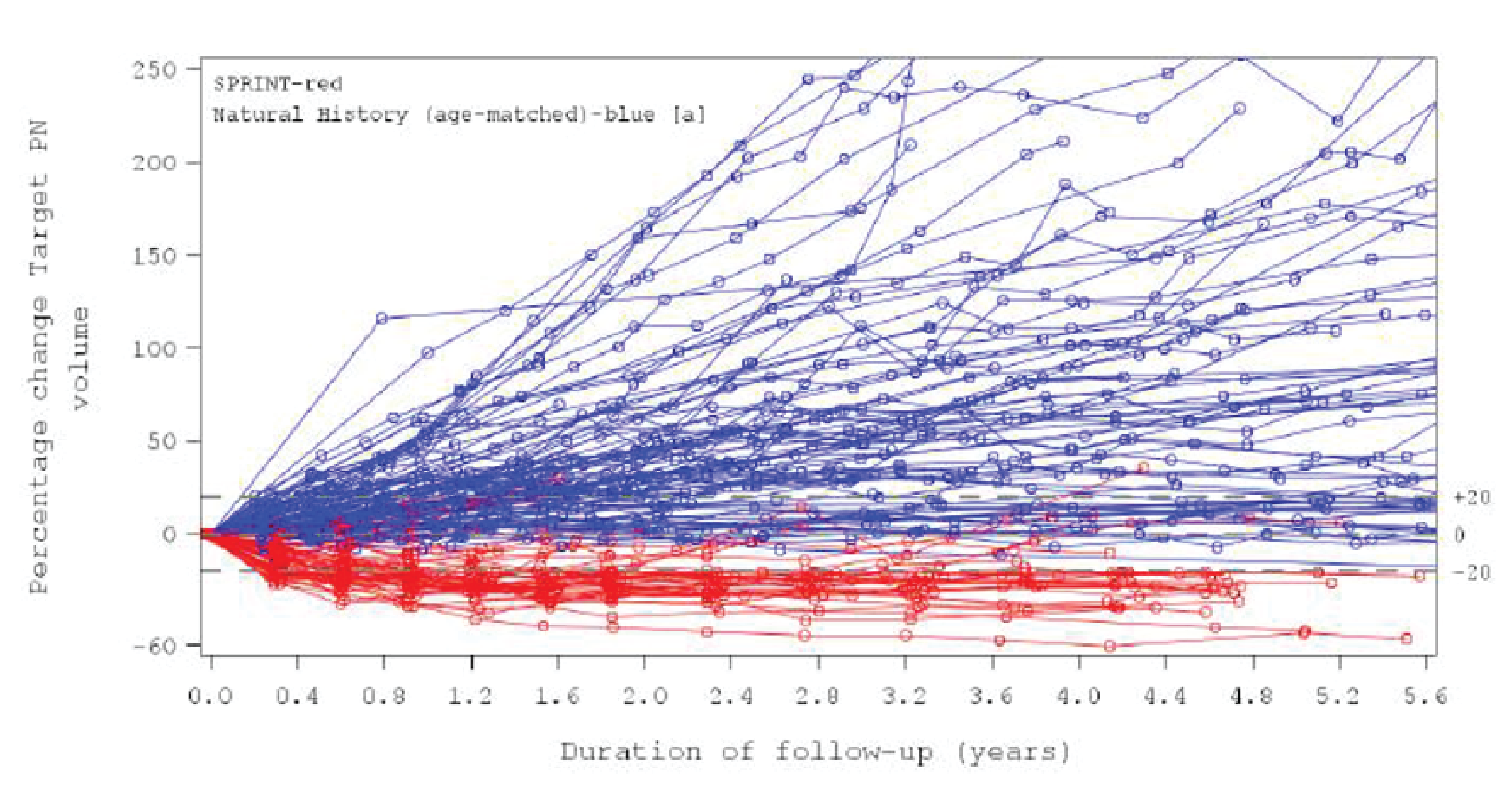
DCO = data cut-off; NH = Natural History; PN = plexiform neurofibroma.
Note: Patients with at least 2 volumetric MRI assessments in each study (including baseline stratum 1 of the SRINT phase II trial) are displayed in this figure. Age-matched NH study patients include those aged 3 to 18 years with at least 1 volumetric MRI within this age and 1 subsequent volumetric MRI. A total of 92 patients are presented in this plot for the NH study and 48 patients for stratum 1 of the SPRINT phase II trial. The figure includes data up to the maximum SPRINT follow-up duration.
Source: SPRINT Phase II Stratum I Clinical Study Report (DCO March, 31, 2021).20
Table 34: Absolute and Percentage Change in Volume of Target PN From Baseline to Final Post-Baseline Assessment in SPRINT (2021 DCO) and NH Study (FAS)
Group | SPRINT phase II stratum 1 | NH Study | ||||
|---|---|---|---|---|---|---|
N | Mean (SD) | Median (range) | N | Mean (SD) | Median (range) | |
Absolute volume change (mL) | 49 | −158.8 |||||||| | −70.0 (−964 to 366) | ||| | ||||| |||||||| | ||||| |||||| ||||| |
Percent volume change (%) | 48 | −15.6 ||||||| | −21.6 (−57 to 35) | ||| | ||||| |||||||| | |||| ||||| ||||| |
DCO = data cut-off; FAS = full analysis set; NH = natural history; PN = plexiform neurofibroma; SD = standard deviation.
Note: A negative change denotes a reduction in target PN volume.
Source: SPRINT Phase II Stratum I Clinical Study Report (DCO March 31, 2021) additional reference document provided by sponsor.20
Progression-Free Survival: Comparison With NH Study
At the time of the March 31, 2021, DCO, disease progression was experienced by ||||| of patients in the NH study compared to ||||| of patients in stratum 1 of the SPRINT phase II trial over a 5.6-year period. The median PFS in the NH study age-matched cohort was 1.3 years (95% CI, ||| || |||) and was not reached in the SPRINT trial. The probability of remaining without progression in the SPRINT trial and the NH study was ||||| (95% CI, |||| || ||||) and ||||| (95% CI, |||| || ||||), respectively.
Progression-Free Survival: Comparison With Study 01-C-0222
Of the 29 patients enrolled with progressive PN in the placebo arm of Study 01-C-0222, || ||||||| had progressed during the study, according to the comparison made in the sponsor’s Clinical Study Report.19 The duration of follow-up was not reported.
At the earlier DCO of June 29, 2018, 6.0% of patients in stratum 1 of the SPRINT phase II trial had progressed over a 2.3-year period; at the later DCO of March 31, 2021, ||||| had progressed over a 5.6-year period.
Because Study 01-C-0222 required progressive disease for enrolment, a subgroup analysis was conducted for the earlier DCO (2018) of stratum 1 of the SPRINT phase II trial. The subgroup of patients with progressive PN at enrolment had a probability of remaining without progression at 2 years of 94.7% (95% CI, 80.6 to 98.7), compared to 20.6% (95% CI, 7.7 to 37.8) in the placebo arm of Study 01-C-0222. The sponsor did not update this comparison for the 2021 DCO.
Critical Appraisal of the Sponsor-Submitted Indirect Treatment Comparisons
The submitted comparisons were descriptive in nature and did not include generating comparative estimates or statistical evaluations. Because the NCI POB deemed it unethical to conduct a placebo-controlled trial, in the absence of any comparative data and particularly due to the absence of any suitable comparators, side-by-side comparisons against studies of the disease’s natural history were appropriate. However, no justification was provided for why adjusted or matched analyses were not also performed for most of these outcomes to mitigate potential bias caused by differences in study design or patient populations.
One comparison was conducted against a prospectively collected study of real-world patients with NF1 (the NH study, using the subgroup with PNs), and the other was against the placebo arm of a randomized controlled trial (Study 01-C-0222). Both comparisons have limitations inherent to the nature of unanchored, unadjusted comparisons: there is no method of controlling for inherent differences in the study design and patient populations of the compared studies, and differences in outcomes cannot be said with certainty to exclusively reflect treatment effects. The results may be biased by underlying differences in the compared studies.
The eligibility criteria of the external control studies (the NH study and Study 01-C-0222) both included adult patients, who tend to have larger but slower-growing PNs, compared to younger patients, who may have less-advanced disease but a generally faster rate of PN growth. However, the mean and median ages in the age-matched cohort of the NH study and the median age of Study 01-C-0222 were lower than patients in stratum 1 of the SPRINT phase II trial, and the maximum age at baseline in Study 01-C-0222 was younger than 18. As the age distribution of each study is unknown, the potential magnitude of bias is uncertain.
In this naive analysis, the proportion of patients in the NH study with progressive or nonprogressive target PN was not reported; however, in the propensity scoring analysis described in the Sponsor-Submitted Propensity Scoring Analysis section, these data are reported for a smaller subset of patients (n = 75) from the NH study’s age-matched cohort, and the between-trial difference is noted as important. Relative to the NH study (n = 75, age-matched cohort with nonmissing baseline data), stratum 1 of the SPRINT phase II trial had a higher proportion of patients categorized as having a “progressive” or “unknown” PN status, and a lower proportion were categorized as “nonprogressive.” As PN status (progressive versus nonprogressive) at baseline is likely a prognostic factor for PFS, this represents an important source of potential bias in naive comparisons.
The proportion of patients with target PNs in the trunk was appreciably lower and the proportion with neck and trunk PNs was higher in the SPRINT phase II stratum 1 patients compared to both the NH study and Study 01-C-0222 placebo arm. The mean volume of target PNs was also larger in the SPRINT phase II stratum 1 patients compared to both comparator studies. Clinical experts were consulted regarding the clinical importance of these differences, but the potential presence and magnitude of bias was uncertain. The differences were not considered to preclude meaningful comparisons. Study 01-C-0222 included only patients who had progressive (and not necessarily symptomatic) PNs at baseline, although a subgroup analysis of the SPRINT trial was conducted using patients with progressive PNs at baseline. Additionally, the before-and-after comparison of NH patients who later participated in SPRINT had a small sample size (n = 9).
The assessment schedule of each study also differed. In stratum1 of the SPRINT phase II trial, in which each cycle was 28 days with no rest period, PN disease evaluation occurred before cycles 5, 9, and 13, then every 4 cycles until cycle 25, then every 6 cycles, until 2 years, at which point it was evaluated every 4 to 6 months. In Study 01-C-0222, in which each cycle is 28 days (including 21 days of treatment and 7 days of rest), MRI was performed for 3-dimensional analysis before cycles 1, 4, 7, and 10, and then after every 6 cycles. In the NH study, volumetric MRI analysis was conducted “typically yearly” until age 18 but this was not defined precisely and could have varied. More frequent MRI analyses may produce a bias in favour of earlier detection of disease progression; Study 01-C-0222 included the most frequent analyses in the earliest cycles, and the NH study had the least frequent MRI analyses.
For patients who received an MEK inhibitor such as selumetinib during the NH study, data collected during the MEK-inhibitor treatment period have been excluded from all analyses; however, no method of control was described for the data associated with these patients after cessation of MEK inhibitors. If there are lasting benefits of MEK inhibitors, this may cause a bias in which these patients had less growth than they otherwise would have. However, if this dataset would select for patients who failed to respond to MEK inhibitors because those who remained on treatment would have been excluded, it may instead bias the NH study data toward a more treatment-resistant disease population. The magnitude of potential bias is unknown.
The duration of follow-up was not reported in Study 01-C-0222. More than a decade of additional follow-up was available for the NH study compared to the SPRINT trial; however, analyses were conducted for the full duration of the follow-up, and data had been capped to reflect the maximum available follow-up of the SPRINT trial at the DCO in question. After capping the follow-up to reflect the maximum available from the SPRINT trial, the median follow-up available from the NH study still exceeded the median available from the SPRINT trial by 0.7 to 0.8 years. Because tumours are expected to progress over time, this additional follow-up may bias the results in favour of selumetinib as less time was available for selumetinib-treated patients to progress.
The outcomes evaluated were limited. Side-by-side comparisons were made against the NH study for PN growth rate, absolute growth, and PFS. Against Study 01-C-0222, only PFS was assessed. Although outcomes and progression related to tumour growth are outcomes of interest, patient and clinician input suggests that tumour volume or change in volume does not always directly correlate with symptomatology, in part because it is highly dependent on the location of the PNs with respect to important structures. Outcomes related to symptoms, morbidity, disability, HRQoL, and disfigurement were not assessed. No safety outcomes were evaluated.
Ultimately, based on naive comparisons to 2 sources of natural history data, the results suggest selumetinib confers a clinical benefit to the majority of patients with NF1 and symptomatic PNs with regards to the rate of target PN growth, but the magnitude of the benefit is uncertain and other important efficacy outcomes have not been evaluated. As there was no evaluation of safety, no conclusions can be made from the ITCs about the harms of selumetinib.
Methods of the Sponsor-Submitted Propensity Score Analysis
Objectives
The objective was to apply propensity score methods to account for imbalances in prognostic factors between patients presenting with NF1 with PN in the SPRINT phase II study versus the NH study external control group to estimate the treatment effect of selumetinib on PFS.
Study Selection Methods
Patients in stratum 1 of the SPRINT phase II trial (March 31, 2021, DCO) were compared with patients from the NH study (previously described) who had PNs and at least 1 PN volumetric MRI scan was done when they were between 3 and 18 years of age and at least 1 subsequent scan (i.e., the age-matched cohort, N = 92).
Patients from the NH study who subsequently enrolled in stratum 1 of the SPRINT phase II trial were excluded from the propensity score analysis to maintain independence between the studies, and 10 patients with missing weight and height data at baseline MRI assessment were excluded. Ultimately, 75 NH patients were eligible for inclusion in the analysis.
Analysis Methods
Identifying Prognostic Factors
Data from the NH study were used to assess the magnitude of the prognostic effect of demographic and disease-related baseline characteristics on PFS. Kaplan-Meier curves were used to visually compare PFS for patients with different characteristics. The continuous variables of age, weight, height, and PN volume were recategorized in low, median, and high groups using the 33rd and 67th percentiles of the aggregated population for the visual presentation of the data. A univariate Cox model was fitted to obtain unadjusted HRs.
Additionally, a multivariate Cox model including all covariates was fitted. The model included SPRINT and NH data, with study, sex, race, target PN location, PN status, age, weight, height, and target PN volume as covariates. Age, weight, height, and target PN volume were kept as continuous variables in the model.
Propensity Scoring
The propensity score for selumetinib treatment was estimated using multivariate logistic regression, with the study (SPRINT for selumetinib treatment, NH for treated with other PN-directed treatment or untreated) fitted as the dependent variable and all baseline covariates (age, race, sex, PN status, weight, height, PN volume, and target PN location) fitted as independent variables, as recommended by the Committee for Medicinal Products for Human Use. Age, weight, height, and target PN volume were kept as continuous variables in the model. Three propensity score matching methods were used:
Matching 1:1 without replacement: Each SPRINT trial patient was caliper-matched by propensity score to an NH study patient using a greedy matching algorithm (i.e., the treated patient is matched with a control patient that has the closest propensity score without accounting for the quality of matching over the entire population). A caliper width of 0.2 of the pooled SD of the logit of the propensity score was used.
IPTW: Each SPRINT (selumetinib-treated) and NH patient was assigned a weight based on the inverse of the propensity score. Stabilized weights were used to preserve the sample size of the original data, to produce appropriate estimation of the variance of main effect, and to maintain an appropriate type I error rate.
Matching 1:2 with replacement: As a sensitivity analysis, each patient from the SPRINT study was matched to up to 2 patients from the NH study, with replacements using the propensity scores.
The baseline characteristics after matching were compared between the studies using standard differences to identify whether balance was achieved. The characteristics assessed were sex, race, age, weight, height, target PN volume, target PN location, and PN status (progressive, nonprogressive, or unknown).
Derivation of Hazard Ratios for Progression-Free Survival
HRs for PFS were derived using Cox models comparing the SPRINT and NH study populations based on either the prematched (i.e., naive) study data or the matched data from each of the propensity scoring approaches. The Cox model was used to derive HRs, either adjusting for only the study as a covariate, or adjusting for several covariates (study, sex, race, age, weight, height, PN location, PN status [progressive, nonprogressive, or unknown], and PN volume).
Results of the Sponsor-Submitted Propensity Score Analysis
Prognostic Factors Identified in the NH Study
According to the univariate Cox model, age, weight, height and PN status at baseline (i.e., progressive, nonprogressive, or unknown) were associated with PFS. Younger patients with progressive PN at baseline had a higher risk of progression.
In the multivariate Cox model, PN status at baseline (i.e., progressive, nonprogressive, or unknown) was the only covariate that remained associated with PFS.
Distribution of Propensity Score and Weights
The distribution of propensity scores by study shows a wide overlap, suggesting that propensity score matching and weighting is not contraindicated. Only 1 weight was greater than 3 for the SPRINT trial, no weights were greater than 2 for the NH study, and no capping of the largest weights was required.
Baseline Characteristics Before and After Matching
The baseline characteristics before and after matching are presented in Table 35 for each method. In summary:
Before matching:
Fifty patients from stratum 1 of the SPRINT phase trial were included.
Seventy-five patients from the NH study’s age-matched cohort were included, representing only patients with nonmissing baseline characteristics, aged 3 to 18 years, with at least 1 MRI in this age range and at least 1 subsequent MRI. Natural history data for patients enrolled in both studies in excluded.
Matching 1:1 without replacement:
Thirty-seven patients from the SPRINT trial were matched to 37 patients from the NH study.
The standard difference between baseline characteristics after matching ranged from 0.000 to 0.247.
IPTW:
The effective sample size was not reported. The sum of weights after IPTW was 51.6 for stratum 1 of the SPRINT phase II trial and 73.3 for the NH study’s age-matched cohort.
The standard difference between baseline characteristics after matching ranged from 0.005 to 0.034.
Matching 1:2 with replacement:
Forty-six patients from the SPRINT trial were matched to 43 unique patients from the NH study (patients from NH could be used multiple times).
The standard difference between baseline characteristics after matching ranged from 0.000 to 0.158.
Progression-Free Survival After Matching
Across the naive and all propensity scoring methods, whether adjusting for multiple covariates or not, the HR values for PFS ranged from |||| || |||| with statistically significant P values of less than 0.001.
Critical Appraisal of the Sponsor-Submitted Propensity Score Analysis
In general, similar limitations apply to the sponsor-submitted propensity score analysis as discussed with regards to the sponsor-submitted ITCs regarding differences in the trial designs and the lack of comparative clinical evidence that precludes anchored comparisons. For this reason and the inherent uncertainty of unanchored ITCs, the HRs should be interpreted with caution. The propensity scoring methodology accounts for differences in known or suspected prognostic factors between the studies, but cannot account for differences in study design nor differences in unknown prognostic factors or treatment effect modifiers that would be mitigated if there were a common comparator.
Overall, nearly all of the critical appraisal concerns for the naive analysis also apply here as this is an unanchored comparison between trials of substantially different design, with different lengths of follow-up and different timing and definitions of assessing progression.
Additionally, no description or justification was provided for the selected baseline characteristics that were evaluated as potential prognostic factors and adjusted in the analyses. The list of characteristics adjusted was relatively comprehensive, including demographics as well as disease characteristics, but it is unknown whether all known and unknown important factors were accounted for. The studies differ in terms of the years in which they were conducted, and there was no description regarding whether treatment or diagnostic practices have varied substantially in that time.
Table 35: Baseline Characteristics of SPRINT Phase II Stratum 1 and the NH Study (Age-Matched Cohort) Before and After Matching
Variable | Before matching | After 1:1 matching | After IPTW | After 1:2 matching | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
SPRINTa | NHb | St Dif | SPRINTa | NHb | St Dif | SPRINTa | NHb | St Dif | SPRINTa | NHb | St Dif | |
N | 50 | 75 | NA | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Sex, n (%) | ||||||||||||
Female | 20 (40.0) | || |||||| | ||||| | || |||||| | || |||||| | ||||| | || |||||| | || |||||| | ||||| | || |||||| | || |||||| | ||||| |
Male | 30 (60.0) | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | ||||
Race, n (%) | ||||||||||||
White | 42 (84.0) | || |||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Asian | 1 (2.0) | | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||
Unknown/other | 3 (6.0) | || |||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||
Black or African American | 4 (8.0) | | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||
Recategorized race, n (%) | ||||||||||||
White | 42 (84.0) | || |||||| | ||||| | || |||||| | || |||||| | ||||| | || |||||| | || |||||| | ||||| | || |||||| | || |||||| | ||||| |
Other | 8 (16.0) | || |||||| | | |||||| | | |||||| | || |||||| | || |||||| | | |||||| | | |||||| | ||||
Target PN location, n (%) | ||||||||||||
Head | 9 (18.0) | | |||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Head/neck | 8 (16.0) | | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||
Neck/trunk | 12 (24/0) | || |||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||
Trunk | 5 (10.0) | || |||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||
Trunk/extremity | 12 (24.0) | || |||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||
Extremity | 4 (8.0) | | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||
Whole body | 0 | | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||
Recategorized target PN location, n (%) | ||||||||||||
Head, head/neck, neck/trunk | 29 (58.0) | || |||||| | ||||| | || |||||| | || |||||| | ||||| | || |||||| | || |||||| | ||||| | || |||||| | || |||||| | ||||| |
Trunk, trunk/extremity, extremity, whole body | 21 (42.0) | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | ||||
PN status, n (%) | ||||||||||||
Progressive | 21 (42.0) | || |||||| | ||||| | || |||||| | || |||||| | ||||| | || |||||| | || |||||| | ||||| | || |||||| | || |||||| | ||||| |
Nonprogressive | 15 (30.0) | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | ||||
Unknown | 14 (28.0) | | |||||| | | |||||| | | |||||| | | |||||| | || |||||| | || |||||| | || |||||| | ||||
Age, years | ||||||||||||
Mean (SD) | 10.3 (3.92) | |||| ||| | ||||| | |||| |||||| | |||| |||||| | ||||| | ||| |||||| | |||| |||| | ||||| | |||| |||||| | |||| ||| | ||||| |
Median (range) | 10.2 (4 to 17) | ||| ||| | | |||| ||| ||| | |||| ||| | | ||| ||| ||| | ||| ||| | | ||| ||| ||| | ||| ||| | ||||
Recategorized age in years, n (%) | ||||||||||||
< 8 | 16 (32.0) | || |||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
≥ 8 to < 13 | 19 (38.0) | || |||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||
≥ 13 | 15 (30.0) | || |||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||
Weight (kg) | ||||||||||||
Mean (SD) | 34.9 (16.48) | |||| |||| | ||||| | |||| ||||||| | || ||||||| | ||||| | |||| ||||||| | |||| ||| | ||||| | |||| ||||||| | |||| ||| | ||||| |
Median (range) | 29.6 (16 to 89) | |||| |||| | |||| |||| ||| | |||| |||| | |||| |||| ||| | |||| |||| | |||| |||| ||| | |||| |||| | ||||
Recategorized weight,d n (%) | ||||||||||||
0 | 16 (32.0) | || |||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
1 | 16 (32.0) | || |||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||
2 | 18 (36.0) | || |||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||
Height, cm | ||||||||||||
Mean (SD) | ||||| ||||||| | ||||| ||| | ||||| | ||||| ||||||| | ||||| |||| | ||||| | ||||| ||||||| | ||||| ||| | ||||| | ||||| ||||||| | ||||| || | ||||| |
Median (range) | ||||| ||||| |||| | |||| |||| | ||||| ||||| | ||||| |||| | ||||| ||||| | ||||| |||| | ||||| ||||| | | ||||| ||| | ||||
Recategorized height,e n (%) | ||||||||||||
0 | || |||||| | || |||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
1 | || |||||| | || |||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||
2 | || |||||| | || |||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||
Body mass index, kg/m2 | ||||||||||||
Mean (SD) | |||| |||||| | |||| |||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
Median (range) | |||| |||| ||| | |||| |||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | |
Target PN volume, L | ||||||||||||
Mean (SD) | ||| ||||| | ||| ||||| | ||||| | ||| |||||| | ||| |||||| | ||||| | ||| |||||| | ||| |||||| | ||||| | ||| |||||| | ||| |||| | ||||| |
Median (range) | ||| ||| |||| | ||| ||| | | ||| |||||| | ||| |||||| | ||| |||||| | | ||| |||||| | ||| |||||| ||| | ||| |||| | ||||
Recategorized target PN volumef, n (%) | ||||||||||||
0 | || |||||| | || |||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
1 | || |||||| | || |||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||
2 | || |||||| | || |||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||
IPTW = inverse probability of treatment weighting; NA = not applicable, NH = natural history; NR = not reported, PN = plexiform neurofibroma, SD = standard deviation, St Dif = standard difference (absolute).
aSPRINT phase II stratum I.
bNH study, age-matched cohort, only patients with nonmissing baseline characteristics, aged 3 to 18 years with at least 1 MRI in this range. NH data for patients enrolled in both studies in excluded.
cSum of weights, SPRINT: 51.6; NH Study: 73.3.
dRank 0 contains patients with weight || || ||||||| |||| ||| || |||||| ||| |||| ||| || ||||||.
eRank 0 contains patients with height || || |||||||| |||| ||| || ||||||| ||| |||| ||| || |||||||.
fRank 0 contains patients with PN volume || || |||||| |||| ||| || ||||| ||| |||| ||| || |||||.
Source: Propensity Score Modelling Report (provided by sponsor).53
Table 36: Progression-Free Survival Hazard Ratio Before and After Making the SPRINT and NH Study Populations Comparable
Cox model analysis | Not adjusted for covariatesa | Adjusted for covariatesb | ||||
|---|---|---|---|---|---|---|
HR | 95% CI | P value | HR | 95% CI | P value | |
Naive (i.e., no propensity score matching) | |||| | ||||| |||| | |||||| | |||| | ||||| |||| | |||||| |
Matched 1:1 (robust variance estimator)c,d | |||| | ||||| |||| | |||||| | |||| | ||||| |||| | |||||| |
Weighted by stabilized IPTW | |||| | ||||| |||| | |||||| | |||| | ||||| |||| | |||||| |
Weighted by IPTW (robust variance estimator) | |||| | ||||| |||| | |||||| | |||| | ||||| |||| | |||||| |
Matched patients 1:2 (robust variance estimator)d,e | |||| | ||||| |||| | |||||| | |||| | ||||| |||| | |||||| |
CI = confidence interval; HR = hazard ratio; IPTW = inverse probability of treatment weighting; NH = natural history.
aHR obtained using Cox regression including study as the only covariate.
bHR obtained using Cox regression including study, sex, race, age, weight, height, PN location, PN status (progressive, nonprogressive, or unknown), and PN volume as covariates.
cGreedy matching algorithm used without replacement.
dThe difference in the logit of the propensity score for a match must be less than or equal to 0.2 times the pooled estimate of the common standard deviation of the logits of the propensity scores.
eEach treated patient is matched up to 2 controls. Matching is performed with replacement.
Source: Propensity Score Modelling Report (provided by sponsor).53
The sponsor used 3 standard methods of propensity score matching, which was appropriate, and conducted thorough and appropriate diagnostics to evaluate the performance and results of each approach, with the exception of effective sample size, which was not reported for the IPTW analysis. The distributions of propensity scores and weights showed wide overlap with few extreme weights, suggesting reasonable precision.
The sample size of the NH study age-matched cohort was slightly reduced due to baseline missing variables, but not to a concerning degree. Patients from the NH study’s age-matched cohort who subsequently enrolled in stratum 1 of the SPRINT phase II trial were also excluded from all analyses, which was appropriate to maintain independence between the studies. The sample sizes of the studies informing these analyses were already small and were further reduced to approximately 75% (for the SPRINT trial) and 50% (for the NH study’s age-matched cohort) in the 1:1 matching analysis. The sample size was preserved somewhat more in the 1:2 matching analysis. The effective sample size after IPTW was not reported.
After matching, the baseline characteristics were more balanced than before matching, although some standardized differences still exceeded 0.1 or 0.2. In 1:1 matching, the variables for which standardized differences were greater than 0.1 included PN volume and location, and the standardized difference for PN status was greater than 0.2. In 1:2 matching, the variables with standardized differences greater than 0.1 were sex, PN volume, PN location, PN status, and age, although none were greater than 0.2. No standardized differences remained greater than 0.1 or greater than 0.2 after IPTW, which suggests this method achieved the most balance in important characteristics.
Only PFS was evaluated as an outcome, and it was defined based on relative change in size of PNs. Although tumour growth is a clinical outcome of interest, patient and clinician input suggests that tumour volume or change in volume does not always directly correlate with symptomatology, in part because it is highly dependent on the location of the PN(s) with respect to important structures. Important outcomes related to symptoms, morbidity, disability, HRQoL, and disfigurement were not assessed. No safety outcomes were evaluated.
Ultimately, based on the propensity scoring analysis that compared stratum 1 of the SPRINT phase II trial to the age-matched cohort of the NH study, the results suggest selumetinib confers a benefit in PFS as defined by relative tumour growth. However, the magnitude of the benefit is uncertain and other important efficacy outcomes have not been evaluated. As there was no evaluation of safety, no conclusions about the harms of selumetinib can be drawn from the propensity scoring analysis.
Other Relevant Evidence
No long-term extension studies or other relevant studies were included in the sponsor’s submission to CADTH.
Discussion
Summary of Available Evidence
One ongoing, phase II, open-label, single-arm, multicentre study (SPRINT phase II) was included in this review. The SPRINT phase II trial consisted of 50 patients, aged 2 to 18 years with NF1 and symptomatic, inoperable PN who were treated with selumetinib at the recommended phase II dosage of 25 mg/m2 twice daily. The primary end point of the SPRINT phase II study was the ORR with secondary time-to-event end points of DOR, change in PN volume, TTR, time to progression, and PFS determined by change in PN volumes based on volumetric MRI. Other secondary end points included clinical outcome assessments or PROs to evaluate pain (NRS-11 and PII), motor function (strength, ROM, and PROMIS), bowel and bladder function (DVQ), HRQoL (PedsQL), respiratory function (PFTs), visual function (visual acuity [HOTV] and exophthalmometry), physical function (6MWT), and changes in pain intensity or other morbidities (GIC).
At baseline, patients included in the SPRINT phase II trial were mostly white (42 [84.0%]) and male (30 [60.0%]), with a mean age of 10.3 years (SD = 3.92). The median number of target PNs causing morbidity was 3 (range = 1 to 4), and the mean target PN volume was 837.11 mL (SD = 925.011), ranging from 5.6 to 3,820.0 mL. Pain was present in the target PNs in 26 patients (52.0%). The most common location of target PNs was the neck and trunk, and the trunk and extremity (12 [24.0%] each), and ||||||| patients had at least 1 prior PN- or NF1-related surgical procedure.
Three sponsor-submitted ITCs were summarized and critically appraised, which consisted of 2 comparisons of the SPRINT phase II trial to external control arms derived from a NH study and the placebo arm of Study 01-C022 evaluating the efficacy of tipifarnib, and 1 propensity scoring analysis of the SPRINT phase II trial and the NH study.
Interpretation of Results
Efficacy
The overall interpretation of the efficacy results from the SPRINT phase II trial was limited given the internal and external validity issues identified, led primarily by the single-arm, open-label design, which precludes the ability to attribute the study results to treatment with selumetinib as opposed to the natural history of the disease or other interventions, as well as the incorporated bias for the subjective clinical outcome assessments due to the knowledge of treatment assignment by both patients and parents and caregivers.
While NF1 is not a rare disease, symptomatic NF1-associated PNs may be rare, although estimates of incidence and prevalence are lacking. According to the input received from patient groups, clinician groups, and the clinical experts consulted by CADTH, the main goal of treatment in patients with NF1-associated PNs is the reduction in pain, and improvement in overall function. PNs can occur throughout the body and, depending on their location, may cause significant pain and result in impaired mobility, vision, breathing, or other functions. The outcomes included in the SPRINT phase II trial were of clinical importance to patients and clinicians, with secondary end points focusing on clinical outcome assessments of selumetinib on pain, motor function, orbit function, airway function, bowel and bladder function, and HRQoL.
Standardizing subjective measures in NF1 across a heterogenous population presents an inherent challenge when measuring and interpreting clinical outcomes in NF1-associated PNs. Given the lack of a comparator, the small sample size of patients included, patient and parent knowledge of treatment assignment, and the high degree of heterogeneity in PN size, location, and symptoms, the results of the clinical outcome assessments can only be considered supportive. The effect of selumetinib on pain was assessed through self-evaluation of pain intensity (NRS-11) and how pain interferes with daily functioning (PII). The adjusted mean change from baseline in NRS-11 score at approximately 1 year of treatment at the June 29, 2018, DCO suggested improvements in pain, and met the defined threshold for clinically meaningful change (i.e., a change of 2 points). Although this threshold was not met at the later, March 31, 2021, DCO, results of the sensitivity analyses using a threshold of a change from baseline of 1 point were suggestive of clinically meaningful change at both DCOs. For the PII pain measure, results were suggestive of improvements in pain according to both the self-reported and parent-reported versions; however, they did not meet the defined thresholds for clinically meaningful change. Motor function was also a secondary clinical outcome assessment included in the SPRINT phase II trial, and was measured using strength tests, ROM tests, and the PROMIS tool. The results of the strength and ROM tests suggested improvements in these domains after 1 year of treatment, and the results for change from baseline at precycle 13 for PROMIS were also suggestive of improvements in mobility and upper extremity function. However, these results were associated with wider 95% CIs, indicating imprecise results. In the PII and PROMIS measures, parent-reported assessments included greater changes from baseline than did the self-reported measures. While other analyses were conducted for outcomes related to PN location, such as airway function or bowel and bladder function, these analyses were limited to a smaller number of patients who had PNs in these locations (N = 16 and 10, respectively). The experts noted that the low number of patients in the trial with PNs in these locations is reflective of clinical practice. The experts also emphasized that patients with airway PNs are often the youngest patients, who cannot undergo PFTs, and results for FEV0.75 in pre-school children and FEV1 in older children were grouped together, although the effect of summarizing these 2 scores is unknown. The results for this cohort may therefore not be generalizable to a larger population.
ORR as measured by volumetric MRI was the primary end point of the SPRINT phase II trial. The NCI POB–assessed ORR of 66% and 68% at the respective June 29, 2018, and March 31, 2021, DCOs (||||| according to the ICR assessment at the June 29, 2018, DCO) were considered clinically meaningful by the clinical experts consulted by CADTH, while also being underestimated due to the definitions of response and progression used in the trial. Response and time-to-event outcomes were measured and assessed using volumetric MRI and REiNS imaging criteria, and while they are appropriate for clinical trials, volumetric MRI and imaging alone are not the only determinants of response used in Canadian clinical practice, and similar changes may not be observed in clinical practice. The clinical experts consulted by CADTH, and the clinician group input highlighted that the assessment of disease progression is multifaceted and relies heavily on clinical symptomatology in combination with imaging, as changes in PN size are difficult to determine using standard imaging practices, and changes in tumour size are not always reflective of changes in disease-related symptoms, and vice versa. As such, measured clinical response and time-to-event outcomes must be interpreted with caution and should only be viewed as supportive of the overall effect of selumetinib.
At baseline, target PN sizes in the enrolled population ranged from 5.6 mL to 3,820.0 mL. Percent change in target PN volume was assessed in the SPRINT phase II trial, although results were only summarized descriptively. At the March 31, 2021, DCO, the mean percent change from baseline at precycle 13 was |||||||, corresponding to a mean absolute change of ||||||| mL. Given the variability in target PN size, both proportional and absolute change should be considered when interpreting changes in PN volume. However, the clinical experts consulted by CADTH emphasized that PN size is often not the sole determinant of pain or dysfunction, and changes in size may not translate to improvements in disease-related symptoms or functionality. Additionally, because tumours tend to grow most often in early childhood, during periods of natural rapid growth, it is difficult to determine whether any change in size reflects regular growth or disease progression. The REiNS criteria define response as a decrease in the volume of the target PN by 20% or more compared with baseline. Given the variability in PN sizes at baseline, a 20% decrease varies by tumour, and by patient, as does the resulting symptomatologic or functional change. Moreover, the clinical experts stated that a 20% reduction in tumour volume is unlikely to occur spontaneously, and the NH study did not demonstrate any reductions of at least 20%. Comparability of tumours by size was not considered and may result in some additional uncertainty in the results of the SPRINT phase II study, as it currently is unclear what impact tumour size has on outcomes. Overall, how the reductions in percentage and absolute change in target PN volume observed with selumetinib will translate into a reduction in clinically significant morbidity (i.e., pain or dysfunction) remains uncertain.
Maintained or improved HRQoL was cited as an important outcome to patients. The PedsQL tool was used to assess QoL. As with other clinical outcome measures, the results of the PedsQL self- and parent-reported evaluations suggest improvements in QoL. However, due to the small sample size included in the analyses, and a decline in patients available to provide assessments over time, the effect of selumetinib on HRQoL remains inconclusive.
As of the March 31, 2021, DCO, the median follow-up of the SPRINT phase II study was 5.6 years. Secondary time-to-event end points including median DOR and PFS remained unreached at this length of follow-up; however, considering the limitations with the design of the study, results for these end points were considered uninterpretable. Although the length of follow-up provides consistent and mature data on the longer-term efficacy and safety of selumetinib, in a chronic, life-long disease, further long-term follow-up is required to understand the overall effect of treating patients before adulthood, and what impact long-term exposure to selumetinib may have on patients.
In the absence of comparative evidence, and to further contextualize the results of the SPRINT phase II study, the sponsor submitted side-by-side comparisons with 2 external controls: the placebo arm of the tipifarnib study and the NH study. Additionally, a propensity score analysis of PFS between the SPRINT phase II trial and the NH study was submitted. There were notable differences in study design between the SPRINT trial and the external control studies, and there were differences in populations, particularly the location and size of target PNs, with the proportion of patients with target PNs located in the trunk lower and the proportion with neck or trunk PNs higher in the SPRINT phase II trial compared to the NH study and Study 01-C-0222, and the mean target PN volume was higher in the SPRINT phase II trial compared to both comparator studies. Due to the lack of statistical testing and adjustment, the impact of the clinical heterogeneity between populations remains unknown. Overall, the results of the ITC suggest that selumetinib confers a clinical benefit to the majority of patients with NF1 and symptomatic PNs in terms of PFS and the rate of tumour growth, as confirmed by the clinical experience of the expert group.
Harms
Analysis of safety was based on the FAS, which included all 50 patients enrolled in the SPRINT phase II study. The overall frequency of harms reported in the SPRINT phase II trial for selumetinib were consistent with those of other MEK inhibitors, according to the clinical experts consulted by CADTH, although they noted that some specific-AEs reported in the trial, such as nausea (36 patients [72.0%]) and vomiting (43 patients [86.0%]) had a higher frequency than expected based on clinical practice. MEKs have been available to treat many cancers in Canada for approximately 10 years, and the experts cited their experience with other MEK inhibitors, although in different populations. At the time of this review, the clinical experts consulted by CADTH emphasized their experience with selumetinib in the pediatric population through compassionate access, noting that the majority of AEs experienced by patients are grade 1 and 2, and dermatologic toxicities were most frequent, ranging in severity.
Known adverse drug reactions for selumetinib and AEs of special interest for this review included cardiac events, ophthalmologic events, and paronychia. In the SPRINT phase II study, cardiac events (decreased ejection fraction, peripheral edema, peripheral swelling, and decreased right ventricle ejection fraction) occurred in 42% of patients, and, while these may be concerning, most were grade 1 or 2 and not considered serious. Blurred vision is a known adverse drug reaction for selumetinib, and was the most frequently occurring ophthalmologic event. However, in the SPRINT phase II trial, the majority of blurred vision events were grade 1, requiring only a brief dose interruption. Paronychia occurred in 56% of patients and is also a known reaction to selumetinib. In the SPRINT trial, all cases were considered nonserious, and the clinical experts noted that consultation with dermatologists and supportive treatment are appropriate to manage events of paronychia. The experts added that increased CPK levels can be observed with MEK inhibitors, although they are most often asymptomatic. The experts were not concerned with the occurrence of increased CPK in the SPRINT phase II trial and noted that this is manageable.
Within the results of the SPRINT phase II study, the sponsor noted that several AEs, including headache, abdominal pain, and nasal congestion, are commonly reported in any pediatric population. As a result, there is a potential for bias in the reporting of AEs due to the ages of the population, and the long duration of follow-up, as it is likely that children will experience abdominal pain, constipation, diarrhea, fever, or other common AEs throughout the treatment period. Additionally, given the noncomparative nature of the SPRINT study, and the lack of safety outcomes assessed in the ITCs, the true frequency of selumetinib-induced AEs in this population is uncertain.
Overall, selumetinib was generally well tolerated in the SPRINT phase II trial, and results were consistent across DCOs, with no new safety signals identified. In addition, the clinical experts consulted by CADTH indicated that, in their experience, the AEs associated with selumetinib are manageable by a multidisciplinary team of cardiologists, ophthalmologists, dermatologists, and pharmacists, and can generally be controlled by supportive care or dose interruptions or adjustments.
Conclusions
There is an unmet need for disease-modifying treatment options for the rare population of patients with NF1-associated, symptomatic, inoperable PN. Patients and clinicians highlighted the need for treatments that reduce pain, disfigurement and improve function, while also preventing the growth of new PNs and shrinking existing PNs. One ongoing, phase II, open-label, single-arm, multicentre study (SPRINT phase II) was included in this review. Notable concerns associated with the internal and external validity of the SPRINT phase II study, driven primarily by the single-arm, open-label design, preclude the ability to attribute the study results to treatment with selumetinib as opposed to disease natural history or concomitant interventions, and introduce significant bias to all subjective clinical outcome assessments evaluated. These are considered of critical importance, given that measurement of disease progression and treatment response in clinical practice relies on available imaging techniques coupled with clinical symptomatology, which may vary from the methods and outcomes used in the SPRINT phase II trial.
The data submitted to CADTH was considered clinically relevant in this setting, given the variability of location and extent of PNs between patients. Clinical outcome assessments including PROs and functional evaluations were overall supportive of the primary imaging findings of the SPRINT phase II trial, reducing PN-associated morbidity and improving HRQoL. However, given their status as secondary outcomes, along with the small sample sizes, lack of statistical testing, and heterogeneity in the location and size of target PNs, results for PROs and functional evaluations can only be interpreted as supportive of the overall effect of selumetinib. For the primary end point in the SPRINT phase II trial, the clinical experts consulted by CADTH felt that the ORR of 68.0% was clinically meaningful, although they noted that, based on their experience and the definitions used for response and progression, the observed responses were underestimated. While selumetinib also resulted in reductions in PN volume, the correlation between PN volume changes and improvements in symptoms or function remains uncertain, and the experts noted that tumour size may not always be reflective of morbidity. While the time-to-event end points of DOR and PFS appeared to be supportive of the observed ORR, the nonrandomized design of the SPRINT phase II trial makes attributing these events to selumetinib challenging.
ITCs included naive side-by-side comparisons and propensity scoring analysis against external controls as representations of natural history. The results suggest selumetinib confers a benefit in terms of reduction in the rate of tumour growth and improvement in PFS. However, due to important between-trial differences in design and populations and major uncertainties inherent in the methodologies applied, the magnitude of the benefit is uncertain. The relative efficacy of selumetinib was not assessed with regard to any other important clinical outcomes such as HRQoL, morbidity, and disfigurement, and may not be directly correlated with changes in tumour volume. No safety outcomes were assessed in the indirect comparisons.
Aside from the AEs known to be associated with MEK inhibitors, selumetinib was generally well tolerated in the SPRINT phase II trial, with limited grade 3 or serious AEs, and the overall toxicity profile can generally be managed with supportive care or dose interruptions. Although the results of the SPRINT phase II trial were generally positive, the ability to draw firm conclusions about the magnitude and the generalizability of the clinical benefit and safety of selumetinib was limited given the identified limitations in the available evidence which is inherent in the complexity of the disease and trial conduct.
References
1.Korf B, Lobbous M, Metrock LK. Neurofibromatosis type 1 (NF1): Management and prognosis. UpToDate. 2022.
2.Ferner RE, Huson SM, Thomas N, et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet. 2007;44(2):81-88. PubMed
3.National Organization for Rare Disorders. Neurofibromatosis 1. 2022; https://rarediseases.org/rare-diseases/neurofibromatosis-type-1-nf1/. Accessed 2022 Nov 17.
4.Evans DG, Howard E, Giblin C, et al. Birth incidence and prevalence of tumor-prone syndromes: estimates from a UK family genetic register service. Am J Med Genet A. 2010;152a(2):327-332. PubMed
5.Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol. 1988;45(5):575-578. PubMed
6.Huson SM, Compston DA, Clark P, Harper PS. A genetic study of von Recklinghausen neurofibromatosis in south east Wales. I. Prevalence, fitness, mutation rate, and effect of parental transmission on severity. J Med Genet. 1989;26(11):704-711. PubMed
7.Korf BR. Plexiform neurofibromas. Am J Med Genet. 1999;89(1):31-37. PubMed
8.Mautner VF, Asuagbor FA, Dombi E, et al. Assessment of benign tumor burden by whole-body MRI in patients with neurofibromatosis 1. Neuro Oncol. 2008;10(4):593-598. PubMed
9.Solares I, Vinal D, Morales-Conejo M, Rodriguez-Salas N, Feliu J. Novel molecular targeted therapies for patients with neurofibromatosis type 1 with inoperable plexiform neurofibromas: a comprehensive review. ESMO open. 2021;6(4):100223. PubMed
10.Waggoner DJ, Towbin J, Gottesman G, Gutmann DH. Clinic-based study of plexiform neurofibromas in neurofibromatosis 1. Am J Med Genet. 2000;92(2):132-135. PubMed
11.Williams VC, Lucas J, Babcock MA, Gutmann DH, Korf B, Maria BL. Neurofibromatosis type 1 revisited. Pediatrics. 2009;123(1):124-133. PubMed
12.Hannema SE, Oostenbrink R. Endocrine Aspects of Neurofibromatosis Type 1. Reference Module in Biomedical Sciences: Elsevier; 2017.
13.Fisher MJ, Blakeley JO, Weiss BD, et al. Management of neurofibromatosis type 1-associated plexiform neurofibromas. Neuro-Oncology. 2022;24(11):1827-1844. PubMed
14.Dombi E, Solomon J, Gillespie AJ, et al. NF1 plexiform neurofibroma growth rate by volumetric MRI: Relationship to age and body weight. Neurology. 2007;68(9):643-647. PubMed
15.Nguyen R, Dombi E, Widemann BC, et al. Growth dynamics of plexiform neurofibromas: a retrospective cohort study of 201 patients with neurofibromatosis 1. Orphanet J Rare Dis. 2012;7(1):75. PubMed
16.Tucker T, Friedman JM, Friedrich RE, Wenzel R, Fünsterer C, Mautner VF. Longitudinal study of neurofibromatosis 1 associated plexiform neurofibromas. J Med Genet. 2009;46(2):81-85. PubMed
17.Needle MN, Cnaan A, Dattilo J, et al. Prognostic signs in the surgical management of plexiform neurofibroma: the Children's Hospital of Philadelphia experience, 1974-1994. J Pediatr. 1997;131(5):678-682. PubMed
18.Packer RJ, Gutmann DH, Rubenstein A, et al. Plexiform neurofibromas in NF1: toward biologic-based therapy. Neurology. 2002;58(10):1461-1470. PubMed
19.Clinical Study Report: A Phase I/II Study of the Mitogen Activated Protein Kinase Kinase (MEK) 1 Inhibitor Selumetinib (AZD6244; HYD-Sulfate) in Children with Neurofibromatosis Type 1 (NF1) and Inoperable Plexiform Neurofibromas (PN) (SPRINT Phase II Stratum 1 DCO 29 June 2018) [internal sponsor's report]. Cambridge (UK): AstraZeneca; 2018.
20.Clinical Study Report: A Phase I/II Study of the Mitogen Activated Protein Kinase Kinase (MEK) 1 Inhibitor Selumetinib (AZD6244; HYD-Sulfate) in Children with Neurofibromatosis Type 1 (NF1) and Inoperable Plexiform Neurofibromas (PN) (SPRINT Phase II Stratum 1 DCO 31 Mar 2021) [internal sponsor's report]. Cambridge (UK): AstraZeneca; 2021.
21.Dombi E, Baldwin A, Marcus LJ, et al. Activity of Selumetinib in Neurofibromatosis Type 1-Related Plexiform Neurofibromas. N Engl J Med. 2016;375(26):2550-2560. PubMed
22.Anderson JL, Gutmann DH. Neurofibromatosis type 1. Handb Clin Neurol. 2015;132:75-86. PubMed
23.Gutmann DH, Ferner RE, Listernick RH, Korf BR, Wolters PL, Johnson KJ. Neurofibromatosis type 1. Nat Rev Dis Primers. 2017;3:17004. PubMed
24.Gutmann DH, Parada LF, Silva AJ, Ratner N. Neurofibromatosis type 1: modeling CNS dysfunction. J Neurosci. 2012;32(41):14087-14093. PubMed
25.Akshintala S, Baldwin A, Liewehr DJ, et al. Longitudinal evaluation of peripheral nerve sheath tumors in neurofibromatosis type 1: growth analysis of plexiform neurofibromas and distinct nodular lesions. Neuro Oncol. 2020;22(9):1368-1378. PubMed
26.Higham CS, Dombi E, Rogiers A, et al. The characteristics of 76 atypical neurofibromas as precursors to neurofibromatosis 1 associated malignant peripheral nerve sheath tumors. Neuro Oncol. 2018;20(6):818-825. PubMed
27.Ducatman BS, Scheithauer BW, Piepgras DG, Reiman HM, Ilstrup DM. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer. 1986;57(10):2006-2021. PubMed
28.Evans DG, Baser ME, McGaughran J, Sharif S, Howard E, Moran A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet. 2002;39(5):311-314. PubMed
29.Nguyen R, Kluwe L, Fuensterer C, Kentsch M, Friedrich RE, Mautner VF. Plexiform neurofibromas in children with neurofibromatosis type 1: frequency and associated clinical deficits. J Pediatr. 2011;159(4):652-655.e652. PubMed
30.Uusitalo E, Leppävirta J, Koffert A, et al. Incidence and mortality of neurofibromatosis: a total population study in Finland. J Invest Dermatol. 2015;135(3):904-906. PubMed
31.Legius E, Messiaen L, Wolkenstein P, et al. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: an international consensus recommendation. Genet Med. 2021;23(8):1506-1513. PubMed
32.Gutmann DH, Blakeley JO, Korf BR, Packer RJ. Optimizing biologically targeted clinical trials for neurofibromatosis. Expert Opin Investig Drugs. 2013;22(4):443-462. PubMed
33.Koselugo (selumetinib): capsules, 10mg and 25mg, oral use [product monograph]. Mississauga (ON): AstraZeneca Canada, Inc.; 2022.
34.Drug Reimbursement Review sponsor submission: Koselugo (selumetinib), 10mg and 25mg capsules. [internal sponsor's package]. Vaughan (ON): Alexion Pharmaceuticals, Inc.; 2022.
35.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. J Clin Epidemiol. 2016;75:40-46. PubMed
36.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2022 May 6.
37.Dombi E, Ardern-Holmes SL, Babovic-Vuksanovic D, et al. Recommendations for imaging tumor response in neurofibromatosis clinical trials. Neurology. 2013;81(21 Suppl 1):S33-40. PubMed
38.Farrar JT, Portenoy RK, Berlin JA, Kinman JL, Strom BL. Defining the clinically important difference in pain outcome measures. Pain. 2000;88(3):287-294. PubMed
39.Kendrick DB, Strout TD. The minimum clinically significant difference in patient-assigned numeric scores for pain. Am J Emerg Med. 2005;23(7):828-832. PubMed
40.Salaffi F, Stancati A, Silvestri CA, Ciapetti A, Grassi W. Minimal clinically important changes in chronic musculoskeletal pain intensity measured on a numerical rating scale. Eur J Pain. 2004;8(4):283-291. PubMed
41.Voepel-Lewis T, Burke CN, Jeffreys N, Malviya S, Tait AR. Do 0-10 numeric rating scores translate into clinically meaningful pain measures for children? Anesth Analg. 2011;112(2):415-421. PubMed
42.Martin S, Nelson Schmitt S, Wolters PL, et al. Development and validation of the English Pain Interference Index and Pain Interference Index-Parent report. Pain Med. 2015;16(2):367-373. PubMed
43.Martin S, Wolters PL, Toledo-Tamula MA, et al. Acceptance and commitment therapy in youth with neurofibromatosis type 1 (NF1) and chronic pain and their parents: A pilot study of feasibility and preliminary efficacy. Am J Med Genet A. 2016;170(6):1462-1470. PubMed
44.Plotkin SR, Davis SD, Robertson KA, et al. Sleep and pulmonary outcomes for clinical trials of airway plexiform neurofibromas in NF1. Neurology. 2016;87(7 Suppl 1):S13-20. PubMed
45.Afshar K, Mirbagheri A, Scott H, MacNeily AE. Development of a symptom score for dysfunctional elimination syndrome. J Urol. 2009;182(4 Suppl):1939-1943. PubMed
46.Varni JW, Burwinkle TM, Seid M, Skarr D. The PedsQL 4.0 as a pediatric population health measure: feasibility, reliability, and validity. Ambul Pediatr. 2003;3(6):329-341. PubMed
47.Thissen D, Liu Y, Magnus B, et al. Estimating minimally important difference (MID) in PROMIS pediatric measures using the scale-judgment method. Qual Life Res. 2016;25(1):13-23. PubMed
48.Fisher MJ, Avery RA, Allen JC, et al. Functional outcome measures for NF1-associated optic pathway glioma clinical trials. Neurology. 2013;81(21 Suppl 1):S15-24. PubMed
49.Harmatz P, Cattaneo F, Ardigò D, et al. Enzyme replacement therapy with velmanase alfa (human recombinant alpha-mannosidase): Novel global treatment response model and outcomes in patients with alpha-mannosidosis. Mol Genet Metab. 2018;124(2):152-160. PubMed
50.Coon CD, Cook KF. Moving from significance to real-world meaning: methods for interpreting change in clinical outcome assessment scores. Qual Life Res. 2018;27(1):33-40. PubMed
51.Cohen J. Statistical Power Analysis for the Behavioral Sciences. 2 ed: Lawrence Erlbaum Associates; 1988: https://www.utstat.toronto.edu/~brunner/oldclass/378f16/readings/CohenPower.pdf. Accessed 2023 Jan 14.
52.Widemann BC, Dombi E Fau - Gillespie A, Gillespie A Fau - Wolters PL, et al. Phase 2 randomized, flexible crossover, double-blinded, placebo-controlled trial of the farnesyltransferase inhibitor tipifarnib in children and young adults with neurofibromatosis type 1 and progressive plexiform neurofibromas. Neuro Oncol. 2014;16(5):707-718. PubMed
53.Non-randomised comparison of PFS in the SPRINT study versus the Natural History study using propensity score modelling. In: Drug Reimbursement Review sponsor submission: Koselugo (selumetinib), 10mg and 25mg capsules. Vaughan (ON): Alexion Pharmaceuticals, Inc.; 2022 Dec 5.
54.Bailey B, Daoust R, Doyon-Trottier E, Dauphin-Pierre S, Gravel J. Validation and properties of the verbal numeric scale in children with acute pain. Pain. 2010;149(2):216-221. PubMed
55.Wolters PL, Martin S, Merker VL, et al. Patient-reported outcomes of pain and physical functioning in neurofibromatosis clinical trials. Neurology. 2016;87(7 Suppl 1):S4-S12. PubMed
56.Castarlenas E, Miró J, Sánchez-Rodríguez E. Is the verbal numerical rating scale a valid tool for assessing pain intensity in children below 8 years of age? J Pain. 2013;14(3):297-304. PubMed
57.Varni JW, Magnus B, Stucky BD, et al. Psychometric properties of the PROMIS ® pediatric scales: precision, stability, and comparison of different scoring and administration options. Qual Life Res. 2014;23(4):1233-1243. PubMed
58.Varni JW, Burwinkle TM, Seid M. The PedsQL as a pediatric patient-reported outcome: reliability and validity of the PedsQL Measurement Model in 25,000 children. Expert Rev Pharmacoecon Outcomes Res. 2005;5(6):705-719. PubMed
59.Nutakki K, Varni JW, Swigonski NL. PedsQL Neurofibromatosis Type 1 Module for children, adolescents and young adults: feasibility, reliability, and validity. J Neurooncol. 2018;137(2):337-347. PubMed
Appendix 1: Literature Search Strategy
Note that this appendix has not been copy-edited.
Overview
Interface: Ovid
Databases:
MEDLINE All (1946-present)
Embase (1974-present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: November 16, 2022
Alerts: Biweekly search updates until project completion
Search filters applied: randomized controlled trials; controlled clinical trials.
Limits:
Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
MeSH | Medical Subject Heading |
.fs | Floating subheading |
exp | Explode a subject heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
# | Truncation symbol for one character |
? | Truncation symbol for one or no characters only |
adj# | Requires terms to be adjacent to each other within # number of words (in any order) |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Keyword heading word |
.dq | Candidate term word (Embase) |
.pt | Publication type |
.mp | Mapped term |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
.yr | Publication year |
.jw | Journal title word (MEDLINE) |
.jx | Journal title word (Embase) |
freq = # | Requires terms to occur # number of times in the specified fields |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
cctr | Ovid database code; Cochrane Central Register of Controlled Trials |
Multi-Database Strategy
(koselugo* or selumetinib* or AZD6244 or AZD 6244 or ARRY 142886 or ARRY142886 or AR142886 or AR 142886 or MK5618 or MK 5618).ti,ab,kf,rn,nm,hw,ot.
6UH91I579U.rn,nm.
or/1-2
exp Neurofibroma/ or genes, neurofibromatosis 1/
(neurofibroma* or neuro fibroma* or multiple neuroma* or neurofibrosit* or neuromatos* or neuro fibrosit*).ti,ab,kf.
(NF1 or NF 1 or Recklinghausen*).ti,ab,kf.
or/4-6
and/3,7
8 use medall
*Selumetinib/
(koselugo* or selumetinib* or AZD6244 or AZD 6244 or ARRY 142886 or ARRY142886 or AR142886 or AR 142886 or MK5618 or MK 5618).ti,ab,kf,dq.
or/10-11
exp Neurofibroma/
(neurofibroma* or neuro fibroma* or multiple neuroma* or neurofibrosit* or neuromatos* or neuro fibrosit*).ti,ab,kf,dq.
(NF1 or NF 1 or Recklinghausen*).ti,ab,kf,dq.
or/13-15
and/12,16
17 use oemezd
conference abstract.pt.
18 not 19
or/9,20
(koselugo* or selumetinib* or AZD6244 or AZD 6244 or ARRY 142886 or ARRY142886 or AR142886 or AR 142886 or MK5618 or MK 5618).ti,ab,kf,rn,nm,hw,ot.
6UH91I579U.rn,nm.
or/22-23
Pediatrics/ or Hospitals, Pediatric/ or Intensive Care Units, Pediatric/ or Adolescent/ or exp Child/ or exp Infant/ or Pediatric Nursing/ or Child, Hospitalized/ or Adolescent, Hospitalized/
(child* or infant* or baby or babies or newborn* or newborns or neonate or neonates or neonatal or preemie? or infancy or paediatric* or pediatric* or toddler* or girl? or boy? or kid? or teen or teens or teenage* or youngster? or youth* or preteen* or adolescent* or adolescence or preschooler* or pre-schooler* or nursery school* or daycare* or school age? or (months adj2 age) or (month? adj2 old) or preadolescen* or juvenile* or prepubescen* or prepubert* or pre-pubescen* or pre-pubert* or pre-adolescen*).ti,ab,kf.
(pediat* or paediat* or child* or adolescen* or juvenile*).jw.
or/25-27
and/24,28
29 use medall
*Selumetinib/
(koselugo* or selumetinib* or AZD6244 or AZD 6244 or ARRY 142886 or ARRY142886 or AR142886 or AR 142886 or MK5618 or MK 5618).ti,ab,kf,dq.
or/31-32
exp pediatrics/ or pediatric hospital/ or pediatric intensive care unit/ or exp adolescent/ or exp child/ or exp pediatric nursing/
(child* or infant* or baby or babies or newborn* or newborns or neonate or neonates or neonatal or preemie? or infancy or paediatric* or pediatric* or toddler* or girl? or boy? or kid? or teen or teens or teenage* or youngster? or youth* or preteen* or adolescent* or adolescence or preschooler* or pre-schooler* or nursery school* or daycare* or school age? or (months adj2 age) or (month? adj2 old) or preadolescen* or juvenile* or prepubescen* or prepubert* or pre-pubescen* or pre-pubert* or pre-adolescen*).ti,ab,kf.
(pediat* or paediat* or child* or adolescen* or juvenile*).jx.
or/34-36
and/33,37
38 use oemezd
(conference abstract or conference review).pt.
39 not 40
or/30,41
(koselugo* or selumetinib* or AZD6244 or AZD 6244 or ARRY 142886 or ARRY142886 or AR142886 or AR 142886 or MK5618 or MK 5618).ti,ab,kf,rn,nm,hw,ot.
6UH91I579U.rn,nm.
or/43-44
45 use medall
*Selumetinib/
(koselugo* or selumetinib* or AZD6244 or AZD 6244 or ARRY 142886 or ARRY142886 or AR142886 or AR 142886 or MK5618 or MK 5618).ti,ab,kf,dq.
or/47-48
(conference abstract or conference review).pt.
49 use oemezd
51 not 50
(Randomized Controlled Trial or Controlled Clinical Trial or Pragmatic Clinical Trial or Equivalence Trial or Clinical Trial, Phase III).pt.
Randomized Controlled Trial/
exp Randomized Controlled Trials as Topic/
“Randomized Controlled Trial (topic)”/
Controlled Clinical Trial/
exp Controlled Clinical Trials as Topic/
“Controlled Clinical Trial (topic)”/
Randomization/
Random Allocation/
Double-Blind Method/
Double Blind Procedure/
Double-Blind Studies/
Single-Blind Method/
Single Blind Procedure/
Single-Blind Studies/
Placebos/
Placebo/
Control Groups/
Control Group/
(random* or sham or placebo*).ti,ab,hw,kf.
((singl* or doubl*) adj (blind* or dumm* or mask*)).ti,ab,hw,kf.
((tripl* or trebl*) adj (blind* or dumm* or mask*)).ti,ab,hw,kf.
(control* adj3 (study or studies or trial* or group*)).ti,ab,kf.
(Nonrandom* or non random* or non-random* or quasi-random* or quasirandom*).ti,ab,hw,kf.
allocated.ti,ab,hw.
((open label or open-label) adj5 (study or studies or trial*)).ti,ab,hw,kf.
((equivalence or superiority or non-inferiority or noninferiority) adj3 (study or studies or trial*)).ti,ab,hw,kf.
(pragmatic study or pragmatic studies).ti,ab,hw,kf.
((pragmatic or practical) adj3 trial*).ti,ab,hw,kf.
((quasiexperimental or quasi-experimental) adj3 (study or studies or trial*)).ti,ab,hw,kf.
(phase adj3 (III or “3”) adj3 (study or studies or trial*)).ti,hw,kf.
or/53-83
or/46,52
and/84-85
21 or 42 or 86
remove duplicates from 87
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
[Search -- Studies with results | neurofibroma*]
WHO ICTRP
International Clinical Trials Registry Platform, produced by the WHO. Targeted search used to capture registered clinical trials.
[Search terms -- neurofibroma*]
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
[Search terms -- neurofibroma*]
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
[Search terms -- neurofibroma*]
Grey Literature
Search dates: November 10—November 12, 2022
Keywords: selumetinib, Koselugo, neurofibroma*
Limits: None
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Appendix 2: Excluded Studies
Note that this appendix has not been copy-edited.
Reference | Reason for exclusion |
|---|---|
Baldwin A, Dombi E, Fischer MJ, et al. Occurrence of Fractures in Children with Neurofibromatosis Type 1 on the MEK Inhibitor Selumetinib for Inoperable Plexiform Neurofibroma. Presented at 2021 NF Virtual Conference; June 14-16, 2021, p. 93 | Study design |
Christensen JA, Gross AM, Dombi E, et al. Longitudinal Assessment of Hearing in Children with Neurofibromatosis Type 1 (NF1) and Facial/Head Plexiform Neurofibromas on the Phase 2 Selumetinib SPRINT Trial. Presented at 2021 NF Virtual Conference; June 14-16, 2021, p. 99 | Duplicate |
Dombi E. Factors Contributing to the Response of Children with NF1 and Plexiform Neurofibromas to Selumetinib. Children's Tumour Foundation NF Conference 2020, 2020, p. 49 | Study design |
Gross A. Assessment of Pulmonary Function in Patients with Neurofibromatosis Type 1 and Airway Associated Plexiform Neurofibromas Before and After Treatment with Selumetinib. Children’s Tumor Foundation NF Conference 2019, 2019, p. 48 | Duplicate |
Gross AM, Baldwin A, Brofferio A, et al. Incidence of Ocular and Cardiac Adverse Events in Children with Neurofibromatosis Type 1 on a Phase 1/2 Study of Selumetinib for Inoperable Plexiform Neurofibromas. Presented at 2021 NF Virtual Conference; June 14-16, 2021, p. 106. | Study design |
Gross AM, Baldwin A, Dombi E, et al. LongTerm Safety and Efficacy of Selumetinib in Children with Neurofibromatosis Type 1 on a Phase 1 Study for Inoperable Plexiform Neurofibromas. Presented at 2021 NF Virtual Conference; June 14-16, 2021, p.107 | Duplicate |
Gross AM, Wolters P, Baldwin A, et al. SPRINT: Phase II study of the MEK 1/2 inhibitor selumetinib (AZD6244, ARRY142886) in children with neurofibromatosis type 1 (NF1) and inoperable plexiform neurofibromas (PN). Journal of Clinical Oncology. 2018;36(15_suppl), p. i143. | Duplicate |
Hampton C. Lack of Retinal Toxicity in Children with Neurofibromatosis Type 1 (NF1) and Inoperable Plexiform Neurofibromas (PN) Treated on SPRINT: A Phase II Trial with the MEK Inhibitor Selumetinib. Joint Global Neurofibromatosis Conference 2018, 2018, p.193. | Duplicate |
Ibeku A, Dombi. E, Baldwin A, et al. Progression of Scoliosis in Children with Neurofibromatosis Type 1 on a Clinical Trial of Selumetinib for Inoperable Plexiform Neurofibroma. Presented at 2022 NF Virtual Conference; June 18-21, 2022, p. 33. | Duplicate |
Jackson S, Baker E, Gross A, Whitcomb T, Baldwin A, Mills J, et al. The effect of selumetinib on spinal neurofibromas in patients with NF1. Neuro-Oncology, 2018 Nov; 20(Suppl 6):vi237. doi: 10.1093/neuonc/noy148.984. Epub 2018 Nov 5. | Duplicate |
Jackson S, Baker EH, Gross AM, Whitcomb P, Baldwin A, Derdak J, et al. The MEK inhibitor selumetinib reduces spinal neurofibroma burden in patients with NF1 and plexiform neurofibromas. Neurooncol Adv. 2020; 2(1):vdaa095. | Duplicate |
Pichard D. Cutaneous Adverse Events in SPRINT: A Phase 2 Trial of the MEK Inhibitor Selumetinib for Pediatric Patients with Neurofibromatosis Type 1 (NF1) and Inoperable Plexiform Neurofibromas (PN). Joint Global Neurofibromatosis Conference 2018, 2018, p. 225 | Duplicate |
Rhodes A, Wolters P, Baldwin A et al. Long-Term Medication Adherence in Children and Adolescents with Neurofibromatosis Type 1 (NF1) on the SPRINT Trial for Selumetinib. Presented at 2022 NF Virtual Conference. June 18-21, 2022, p. 99. | Duplicate |
Wolters P. Prospective Patient-Reported Outcomes (PROs) Document Clinical Benefit in Children with Neurofibromatosis Type 1 (NF1) and Inoperable Plexiform Neurofibromas (PNs) on SPRINT: a Phase II Trial of the MEK 1/2 Inhibitor Selumetinib. Joint Global Neurofibromatosis Conference 2018, 2018, p. 22. | Duplicate |
Wolters PL, Gross A, Martin S, et al. Prospective Patient-Reported Outcome (PRO) Measures Document Long-Term Clinical Benefit in Children with Neurofibromatosis Type 1 (NF1) and Inoperable Plexiform Neurofibromas (PNs) on SPRINT: a Phase II Trial of the MEK 1/2 Inhibitor Selumetinib (AZD6244, ARRY142886). Presented at 2022 NF Virtual Conference. June 18-21, 2022, p. 34. | Duplicate |
Dombi E, Baldwin A, Marcus LJ, Fisher MJ, Weiss B, Kim A, et al. Activity of selumetinib in neurofibromatosis type 1-related plexiform neurofibromas. New England Journal of Medicine. 2016; 375(26), 2550-2560. | Duplicate |
Baldwin A, Dombi E, Fischer MJ, et al. Occurrence of Fractures in Children with Neurofibromatosis Type 1 on the MEK Inhibitor Selumetinib for Inoperable Plexiform Neurofibroma. Presented at 2021 NF Virtual Conference; June 14-16, 2021, p. 93. | Duplicate |
Dombi E. Factors Contributing to the Response of Children with NF1 and Plexiform Neurofibromas to Selumetinib. Children's Tumour Foundation NF Conference 2020, 2020, p. 49. | Duplicate |
Gross AM, Baldwin A, Brofferio A, et al. Incidence of Ocular and Cardiac Adverse Events in Children with Neurofibromatosis Type 1 on a Phase 1/2 Study of Selumetinib for Inoperable Plexiform Neurofibromas. Presented at 2021 NF Virtual Conference; June 14-16, 2021, p. 106. | Duplicate |
Al-Mulla, A. Neurofibromatosis Type 1 Patients with Plexiform Neurofibromas Treated with Selumetinib. Pediatric Blood and Cancer. 2022. 69(SUPPL 2):S37. | Study design |
Baldo F, Grasso AG, Cortellazzo Wiel L, et al. Selumetinib in the Treatment of Symptomatic Intractable Plexiform Neurofibromas in Neurofibromatosis Type 1: A Prospective Case Series with Emphasis on Side Effects. Pediatric Drugs 2020;22:417-423 | Study design |
Coltin H, Perreault S, Larouche V, et al. Selumetinib for symptomatic, inoperable plexiform neurofibromas in children with neurofibromatosis type 1: A national realworld case series. Pediatr Blood Cancer. 2022 Aug;69(8):e29633. | Study design |
Santo VE, Passos J, Nzwalo H, et al. Selumetinib for plexiform neurofibromas in neurofibromatosis type 1: a single-institution experience. Journal of Neuro-Oncology 2020;147:459-463. | Study design |
Passos J, Nzwalo H, Azevedo M, et al. Dramatic Improvement of a Massive Plexiform Neurofibroma After Administration of Selumetinib. Pediatric Neurology 2020;105:69-70. | Study design |
Sharawat IK, Panda PK, Sihag RK, Panda P, Dawman L. Efficacy and safety profile of Selumetinib in symptomatic inoperable plexiform neurofibromas: a systematic review and meta-analysis. J Neurosurg Sci. 2022;17:17. | Review article (SLR) |
Hwang J, Yoon HM, Lee BH, Kim PH, Kim KW. Efficacy and Safety of Selumetinib in Pediatric Patients With Neurofibromatosis Type 1: A Systematic Review and Meta-analysis. Neurology. 2022;98(9):e938-e946. | Review article (SLR) |
Clinical Study Report: A Phase I/II Study of the Mitogen Activated Protein Kinase Kinase (MEK) 1 Inhibitor Selumetinib (AZD6244; HYD-Sulfate) in Children with Neurofibromatosis Type 1 (NF1) and Inoperable Plexiform Neurofibromas (PN) (SPRINT Phase I) [internal sponsor's report]. Cambridge, UK: AstraZeneca; 2019. | Study design |
Appendix 3: Detailed Outcome Data
Note that this appendix has not been copy-edited.
Figure 6: Mean Change From Baseline of Grooved Pegboard Test Scores by Affected vs. Nonaffected Hand — Unilateral (FAS ≥ 5 Years With Cervical, Upper Thoracic, Upper Limb, and PN-Related Morbidity [N = 17]) (Redacted)

CI = confidence interval; DCO = data cut-off.
Note: This figure was redacted at the request of the sponsor.
Source: SPRINT Phase II Stratum 1 Clinical Study Report (June 29, 2018, DCO).19
Figure 7: Mean Change From Baseline of Grooved Pegboard Test Scores by Dominant Versus Nondominant Hand — Bilateral (FAS ≥ 5 Years With Cervical, Upper Thoracic, Upper Limb, and PN-Related Morbidity [N = 8]) (Redacted)

CI = confidence interval; DCO = data cut-off.
Note: This figure was redacted at the request of the sponsor.
Source: SPRINT Phase II Stratum 1 Clinical Study Report (June 29, 2018, DCO).19
Figure 8: Mean Change From Baseline of Grip Strength Test Scores by PN Affected Laterality (FAS With Motor PN-Related Morbidity [N = 19]) (Redacted)

CI = confidence interval; DCO = data cut-off.
Note: This figure was redacted at the request of the sponsor.
Source: SPRINT Phase II Stratum 1 Clinical Study Report (June 29, 2018, DCO).19
Figure 9: Mean Change From Baseline of Leg Length Discrepancies (FAS With Lumbosacral Plexus or Lower Limb PN [N = 30]) (Redacted)

CI = confidence interval; DCO = data cut-off.
Note: This figure was redacted at the request of the sponsor.
Source: SPRINT Phase II Stratum 1 Clinical Study Report (June 29, 2018, DCO).19
Figure 10: Mean Change From Baseline of PedsQL Self-Report Scores Over Time — Transformed Scores (FAS; March 31, 2021, DCO) (Redacted)

DCO = data cut-off; FAS = full analysis set.
Note: This figure was redacted at the request of the sponsor.
Source: SPRINT Phase II Stratum 1 Clinical Study Report (March 31, 2021, DCO).20
Figure 11: Mean Change From Baseline of PedsQL Parent-Report Scores Over Time —Transformed Scores (FAS; March 31, 2021, DCO) (Redacted)

DCO = data cut-off; FAS = full analysis set.
Note: This figure was redacted at the request of the sponsor.
Source: SPRINT Phase II Stratum 1 Clinical Study Report (March 31, 2021, DCO).20
Figure 12: Kaplan-Meier Plot of Progression-Free Survival – NCI POB Central Analysis (FAS)
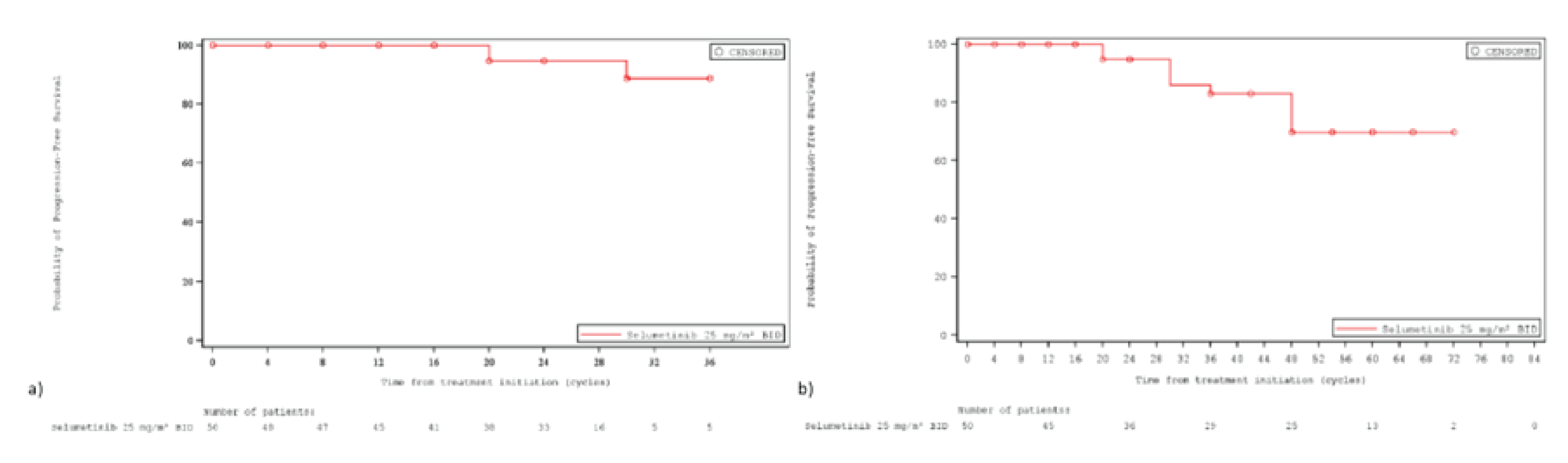
BID = twice daily; DCO = data cut-off; FAS = full analysis set; NCI = National Cancer Institute; POB = Pediatric Oncology Branch.
a) Kaplan-Meier curve of PFS at the June 29, 2018, DCO; b) Kaplan-Meier curve of PFS at the March 31, 2021, DCO
Source: SPRINT Phase II Stratum 1 Clinical Study Report (June 29, 2018, DCO);19 SPRINT Phase II Stratum 1 Clinical Study Report (March 31, 2021, DCO)20
Appendix 4: Description and Appraisal of Outcome Measures
Note that this appendix has not been copy-edited.
Aim
To describe the following outcome measures that were secondary outcome measurements in the SPRINT trial (Phase II Stratum 1) and to review their measurement properties (validity, reliability, responsiveness to change, and minimal important difference [MID]):
Numeric Rating Scale 11 (NRS-11)
Pain Interference Index (PII)
Pediatric Quality of Life Inventory (PedsQL)
Patient-Reported Outcomes Measurement Information System (PROMIS)
Response Evaluation in Neurofibramatosis and Shwannomatosis (REiNS) Criteria
Findings
Table 39: Summary of Outcome Measures and Their Measurement Properties
Measure | Description | Conclusions about measurement properties | MID |
|---|---|---|---|
NRS-11 | A self-completed scale measured from 0 (no pain) to 10 (worst pain you can imagine). Higher scores indicate higher intensity of pain. | Reliability: Reliability of NRS-11 has been demonstrated in children presenting to a pediatric emergency department with acute pain,54 but there are no published data in NF1 populations to date.55 Validity: There is preliminary validity data regarding NRS-11 for children 6 to 7 years based on a population sample of schoolchildren,56 but there is no published data in NF1 populations to date.55 Responsiveness to change: The NRS-11 has demonstrated good sensitivity to change over time in children,54 but there are no published data in NF1 populations to date. | Unknown |
PII | A self-reported or parent proxy form that consists of a 6-item scale to assess the extent to which pain has interfered with a patient’s daily activities in the recent past. The total score is the mean of completed items. The PII is recommended by the REiNS PRO group for assessing pain interference in NF trials of pediatric patients.55 Higher scores indicate more interference with daily activities. | Reliability: Reliability was assessed in patients with NF1 or cancer and their caregivers.42 Cronbach coefficient alpha was 0.84 (unstandardized) for the patient PII and 0.94 and 0.96 for the parent PII when assessed by mothers or fathers, respectively. Patient and parent scores on the PII were significantly correlated (r = 0.62, P < 0.0001). More data are needed regarding the test-retest reliability of PII. Validity: Construct validity was assessed in the same population as above.42 The PII scale demonstrated good validity based on correlation with other pain scales (MBPI, PRS, PRS-P). In the subgroup of NF1 patients (N = 31), the correlation between PII and NF1 Disease Severity Scale ratings was assessed, and patients rated as moderate/severe scored significantly higher on PII than did those rated as mild severity (P = 0.004). The difference in parent-reported PII scores between these groups was not statistically significant (P = 0.37). There was a trend observed wherein patients with higher NF1 Disease Complications Scale scores had higher PII scores, but this was not statistically significant (r = 0.33, P = 0.07). Responsiveness to change: A small pilot study (n = 12, 10 who completed the study) of adolescents and young adults with NF1 assessed the pre-post differences in pain interference after behavioural/therapy-based intervention and observed that parent-reported pain interference significantly declined, as did patient-reported MBPI and patient-reported VAS ratings of pain intensity, but patient-reported PII did not significantly decrease.43 | Unknown |
PROMIS | PROMIS was developed to standardize PRO measures across patient populations with chronic diseases in a variety of health domains.55,57 Pediatric self-report items have been developed for patients age 8 to 17 years across 5 health domains (physical functioning, pain, fatigue, emotional health, and social health) which have been further delineated into 8 latent constructs (depressive symptoms, anxiety, anger, pain interference, peer relationships, fatigue, mobility, and upper extremity functioning).57 Items are rated on a 5-point Likert scale and the measurements yield standardized t scores.55 | Reliability: In general patient populations with a variety of chronic conditions, PROMIS-PI and PROMIS-PF were shown to have very good and moderate internal consistency in pediatric patients, respectively. In children, test-retest reliability was low for PROMIS-PI and moderate for PROMIS-PF. No NF1-specific data are available. No data are available for PROMIS as a whole, nor other domains of PROMIS. Validity: In general patient populations with a variety of chronic conditions, PROMIS-PI and PROMIS-PF have demonstrated validity. No NF1-specific data are available. No data are available for PROMIS as a whole, nor other domains of PROMIS. Responsiveness to change: The PROMIS-PI and PROMIS-PF both failed to show responsiveness to change in pediatric studies based on limited data, none of which was specific to NF1.55 There was no other NF1-specific data located regarding the responsiveness to change of PROMIS as a whole or any other domains of PROMIS. | Unknown |
PedsQL | PedsQL™ 4.0 Generic Core Scales assesses function in 4 domains, each of which are scored on a 5-point Likert scale where 0 = never a problem and 4 = almost always a problem. Scores are computed as the sum of items divided by the number of items answered. The domains include physical functioning (8 items), emotional functioning (5 items), social functioning (5 items), and school functioning (5 items). Higher scores indicate better HRQoL. | Reliability: The generic PedsQL scales have demonstrated reliability in child self-report for ages 5 to 18 years and parent proxy report for ages 2 to 18 years.58 However, there was no evidence located that reported on the reliability of PedsQL in patients with NF1 and symptomatic PNs. Validity: The generic PedsQL scales have demonstrated reliability in child self-report for ages 5 to 18 years and parent proxy report for ages 2 to 18 years.58 However, there was no evidence located that reported on the validity of PedsQL in patients with NF1 and symptomatic PNs. Responsiveness to change: The generic PedsQL scales have demonstrated responsiveness to change in child self-report for ages 5 to 18 years and parent proxy report for ages 2 to 18 years.58 However, there was no evidence located that reported on the responsiveness to change of PedsQL in patients with NF1 and symptomatic PNs. | Unknown |
REiNS | The REiNS criteria with respect to tumour imaging refer to recommendations set forth by the REiNS International Collaboration in the context of NF clinical trials.37 The REiNS criteria recommended, based on consensus, that response evaluations for future studies of NF1 use volumetric analysis to sensitively and reproducibly evaluate changes in tumour size in clinical trials, and selected a 20% volume change as indicating a meaningful change in tumour size. | Reliability: There was no evidence located that reported on the reliability of using the REiNS criteria in patients with NF1 and symptomatic PNs. A correlation between imaging response and overall survival and clinical improvement has not been established for most benign NF tumours.37 Validity: There was no evidence located that reported on the validity of using the REiNS criteria in patients with NF1 and symptomatic PNs. Responsiveness to change: A trial of patients with PNs compared results from volumetric MRI to 2-dimensional (WHO criteria) and 1-dimensional (RECIST criteria) measurements. Volumetric analysis detected tumour progression much earlier than either linear measurements; the median time to progression by volumetric MRI was 14.3 months compared to 52.2 months using WHO criteria. The median time to progression could not be determined using RECIST criteria.37 | Complete response: disappearance of the target lesion. Partial response: at least 20% decrease in volume based on volumetric MRI Progressive disease: at least 20% increase in volume on volumetric MRI compared to baseline or compared to time of best response after a partial response. Stable disease: insufficient change in volume to qualify for either progressive disease or partial response. Can only be considered a response when observed in patients with documented imaging progression of a tumour for which linear growth is expected. |
NRS-11
Description
The NRS-11 is a self-report form for patients aged ≥ 8 years, and measures pain intensity based on 4 questions scored on a scale from 0 (no pain) to 10 (worst pain you can imagine).55 It is recommended by the REiNS PRO group for assessing pain intensity in trials of NF.55
Reliability
Reliability of NRS-11 has been demonstrated in children presenting to a pediatric emergency department with acute pain,54 but there are no published data in NF1 populations to date.55
Validity
There is preliminary validity data regarding NRS-11 for children 6 to 7 years based on a population sample of schoolchildren,56 but there is no published data in NF1 populations to date.55
Responsiveness to Change
The NRS-11 has demonstrated good sensitivity to change over time in children,54 but there are no published data in NF1 populations to date.
MID
There was no evidence located that estimated MID for NRS-11 in patients with symptomatic NF1-associated PNs.
PII
Description
The PII measures pain interference using a 6-item scale to assess the extent to which pain has interfered with an individual’s daily activities in the past 7 days19 or 2 weeks.42 Items are rated on a 7-point Likert scale (where 0 means not at all, and 6 means completely), and the total score is the mean of the completed items. Higher scores indicated more interference with daily activities.19 The PII exists as a self-report adult form (≥ 18 years), self-report pediatric form (6 to 24 years), and a parent proxy form (6 to 18 years). The REiNS PRO group recommended the PII for assessing pain interference in NF trials of pediatric patients.55
Reliability
Martin et al.42 assessed the reliability of the patient PII and parent PII in 60 youth with NF1 or cancer (solid tumour or leukemia) who were enrolled on a medical research protocol at a government research institute and their caregivers (46 mothers, 18 fathers, 1 grandmother). Thirty-one of the patients had NF1. The patients were 58% male, with a mean age 14.7 years (SD 4.3 years, range 6.6 to 24.1 years). The Cronbach coefficient alpha was 0.84 (unstandardized) for the patient PII and 0.94 and 0.96 for the parent PII when assessed by mothers or fathers, respectively. Patient and parent scores on the PII were significantly correlated (r = 0.62, P < 0.0001) but patients reported significantly less pain interference than parents regardless of age group, gender, or disease. The specific items contributing to this difference were the impact of pain on mood and sleep.
Martin et al.42 noted that future longitudinal research is needed to determine the PII test-retest reliability.
Validity
Construct validity was examined by Martin et al.42 in the population of 60 youths with NF1 or cancer and their caregivers previously described. The PII scale demonstrated good validity based on correlation with Modified Brief Pain Inventory (MBPI) scores (r = 0.81, P < 0.0001) and Pain Rating Scale (PRS) scores (r = 0.54, P < 0.0001). Patient PII scores were significantly correlated with their mothers’ PRS scores (r = 0.43, P = 0.003) but not their fathers’ (r = 0.48, P = 0.051). Parent PII (PII-P) and parent PRS (PRS-P) scores were also correlated (r = 0.68, P < 0.0001). Patients taking prescription pain medication reported significantly higher pain interference scores on the PII than those not taking pain medication according to both the patient-reported and parent-reported PII (P < 0.01 for each).
In the subgroup of NF1 patients (N = 31),42 the correlation between PII and NF1 Disease Severity Scale ratings was assessed, and patients rated as moderate/severe scored significantly higher on PII than did those rated as mild severity (P = 0.004). The difference in parent-reported PII scores between these groups was not statistically significant (P = 0.37). There was a trend observed wherein patients with higher NF1 Disease Complications Scale scores had higher PII scores, but this was not statistically significant (r = 0.33, P = 0.07).
Responsiveness to Change
Martin et al.43 conducted a pilot study of Acceptance and Commitment Therapy in adolescents and young adults with NF1 and assessed the pre-post differences using the PII. The study recruited 12 patients, and of the 10 who completed the study, the mean age was 16.9 years (SD = 2.9, range 12 to 20). Nine mothers and 3 fathers completed the workshop.
Parent-reported pain interference on the parent PII significantly declined from baseline to 3 months (P = 0.02) but this was not reflected in the self-report PII (P = 0.14). In contrast, patient-reported MBPI and patient VAS ratings of pain intensity were significantly lower at 3 months.43
The study by Martin et al.43 was a single-arm pilot study of a behavioural intervention with very limited sample size and a follow-up only at 3 months post-baseline. The interpretation of this is inconclusive due to the limited evidence.
MID
There was no evidence located that estimated MID for the PII in patients with NF1 and symptomatic PNs.
PROMIS
Description
PROMIS was developed to standardize PRO measures across patient populations with chronic diseases in a variety of health domains.55,57 Pediatric self-report item banks have been developed for patients aged 8 to 17 years across 5 health domains (physical functioning, pain, fatigue, emotional health, and social health) which have been further delineated into 8 latent constructs of depressive symptoms, anxiety, anger, pain interference, peer relationships, fatigue, mobility, and upper extremity functioning.57 Items are rated on a 5-point Likert scale and the measurements yield standardized t scores.55
The PROMIS scales are considered appropriate for NF trials by the REiNS PRO group because they assess relevant domains in a wide age range, have good psychometric properties, and are translated into multiple languages.55 Specifically, the REiNS PRO group recommended the PROMIS for physical functioning (PROMIS-PF) in NF trials including pediatric, adult, or mixed age populations. However, the PROMIS for pain interference (PROMIS-PI) is recommended only in adult NF populations; in pediatric populations, the PII is recommended instead.
PROMIS-PI and PROMIS-PF will be discussed here in more detail. Both include self-report adult forms (≥ 18 years of age), self-report pediatric forms (8 to 18 years), and parent proxy forms (5 to 18 years).55 Data on the other domains of PROMIS or on PROMIS overall was not available in NF1 populations.
Reliability
In patient populations with a variety of conditions, the PROMIS-PI was shown to have very good internal consistency in pediatric patients. Test-retest reliability was higher in adults but lower in children. However, no NF1-specific data were available.55
The PROMIS-PF showed moderate internal consistency and moderate test-retest reliability in children with various conditions. Again, no NF1-specific data were available.55
No NF1-specific data were available on the reliability of PROMIS as a whole or any other domains of PROMIS.
Validity
The validity of PROMIS-PI and PROMIS-PF have been demonstrated in adults and youth with various conditions and chronic pain, but no data were NF1-specific.55
There were no NF1-specific data located regarding the validity of PROMIS as a whole or any other domains of PROMIS.
Responsiveness to Change
The PROMIS-PI and PROMIS-PF both failed to show responsiveness to change in pediatric studies based on limited data, none of which was specific to NF1.55 There was no other NF1-specific data located regarding the responsiveness to change of PROMIS as a whole or any other domains of PROMIS.
MID
No evidence is available that MIDs for PROMIS or any domains of PROMIS have been estimated in patients with NF1 and symptomatic PNs.
Pediatric Quality of Life Inventory
Description
General HRQoL was measured using the PedsQL 4.0 Generic Core Scales.19 The generic PedsQL assesses function in 4 domains:
Physical Functioning (8 items)
Emotional Functioning (5 items)
Social Functioning (5 items)
School Functioning (5 items)
Each item is scored on a 5-point Likert scale (0 = never a problem; 1 = almost never a problem; 2 = sometimes a problem; 3 = often a problem; 4 = almost always a problem). For patient-reported and parent/guardian-reported measures, which will be analysed separately, items are reverse-scored and linearly transformed to a 0 to 100 scale (0 = 100, 1 = 75, 2 = 50, 3 = 25, 4 = 0), so that higher scores indicate better HRQoL. Scale scores are computed as the sum of the items divided by the number of items answered to account for missing data. If more than 50% of the items in the scale were missing, the scale score was not computed.19
A 104-item PedsQL NF1 Module has also been developed,59 but this was not used in the trials of selumetinib, so it will not be discussed further.
Reliability
The generic PedsQL scales have demonstrated reliability in child self-report for ages 5 to 18 years and parent proxy report for ages 2 to 18 years.58 However, there was no evidence located that reported on the reliability of PedsQL in patients with NF1 and symptomatic PNs.
Validity
The generic PedsQL scales have demonstrated reliability in child self-report for ages 5 to 18 years and parent proxy report for ages 2 to 18 years.58 However, there was no evidence located that reported on the validity of PedsQL in patients with NF1 and symptomatic PNs.
Responsiveness to Change
The generic PedsQL scales have demonstrated responsiveness to change in child self-report for ages 5 to 18 years and parent proxy report for ages 2 to 18 years.58 However, there was no evidence located that reported on the responsiveness to change of PedsQL in patients with NF1 and symptomatic PNs.
Minimal Important Difference
There was no evidence located that reported on MID for PedsQL in patients with NF1 and symptomatic PNs.
Response Evaluation in Neurofibromatosis and Schwannomatosis
Description
The REiNS criteria with respect to tumour imaging refer to recommendations set forth by the REiNS International Collaboration in the context of NF clinical trials.37 The REiNS criteria recommended, based on consensus, that response evaluations for future studies of NF1 use volumetric analysis to sensitively and reproducibly evaluate changes in tumour size in clinical trials, and selected a 20% volume change as indicating a meaningful change in tumour size. Previous measurement approaches used criteria by RECIST or WHO but encountered difficulties with the complex shape, large size, and slow growth of PNs which resulted in highly variable linear measurements requiring long time-periods to detect noticeable change.
Reliability
There was no evidence located that reported on the reliability of using the REiNS criteria in patients with NF1 and symptomatic PNs. A correlation between imaging response and overall survival and clinical improvement has not been established for most benign NF tumours.37
Validity
There was no evidence located that reported on the validity of using the REiNS criteria in patients with NF1 and symptomatic PNs.
Responsiveness to Change
A trial of patients with PNs compared results from volumetric MRI to 2-dimensional (WHO criteria) and 1-dimensional (RECIST criteria) measurements. Volumetric analysis detected tumour progression much earlier than either of the linear measurements; the median time to progression by volumetric MRI was 14.3 months compared to 52.2 months using WHO criteria. The median time to progression could not be determined using RECIST criteria.37
Minimal Important Difference
A consensus agreement was reached by the REiNS International Collaboration that a 20% change in tumour size using volumetric MRI constituted an increase or decrease in tumour size.37
The collaboration37 further defined CR, PR, progressive disease, and stable disease:
A CR was defined as disappearance of the target lesion.
A PR required a decrease in the volume of 20% or more compared to baseline (unconfirmed at first detection; confirmed when observed again within 3 to 6 months; sustained when the response is maintained for 6 months or longer).
Progressive disease was defined as the increase in volume of the target lesion by 20% or more compared to baseline or compared to the time of best response after documenting a PR. The appearance of new lesions or unequivocal progression of existing nontarget lesions is also considered progressive disease.
Stable disease was defined as insufficient volume change to qualify for either progressive disease or PR. The collaboration further noted that disease stability can only be considered a response when in patients with documented imaging progression of a tumour for which linear growth is expected.
Pharmacoeconomic Review
Abbreviations
AE
adverse event
BSA
body surface area
BSC
best supportive care
HRQoL
health-related quality of life
ICER
incremental cost-effectiveness ratio
NF1
neurofibromatosis type 1
PFS
progression-free survival
PN
plexiform neurofibroma
QALY
quality-adjusted life-year
TTD
time to treatment discontinuation
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Selumetinib (Koselugo), oral capsules |
Submitted price | Selumetinib: $122.60 per 10 mg capsule or $306.50 per 25 mg capsule |
Indication | For the treatment of pediatric patients aged 2 years and above with neurofibromatosis type 1 who have symptomatic, inoperable plexiform neurofibromas |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | August 31, 2022 |
Reimbursement request | As per indication |
Sponsor | Alexion Pharma GmbH |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis State-transition model |
Target population | Pediatric patients 2 years of age and older with neurofibromatosis type 1 who have symptomatic, inoperable PN |
Treatment | Selumetinib with BSC |
Comparator | BSC, defined as medication used for pain relief and symptomatic disease management; may include analgesics, antidepressants, and anxiolytics |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs |
Time horizon | Lifetime (97 years) |
Key data source | SPRINT phase I and II trial |
Submitted results | ICER = $123,098 per QALY gained (incremental costs = $752,073; incremental QALYs = 6.11) |
Key limitations |
|
CADTH reanalysis results |
|
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; PN = plexiform neurofibroma; QALY = quality-adjusted life-year.
Conclusions
The CADTH Clinical Review concluded that selumetinib resulted in reductions in plexiform neurofibroma (PN) volume; however, the correlation between PN volume changes and improvements in symptoms or function remains uncertain as the clinical experts consulted by CADTH for this review noted that tumour size may not be reflective of morbidity. The magnitude of the benefit in terms of progression-free survival (PFS) was unclear. Given the nonrandomized design of the SPRINT trial and the many confounding prognostic factors that were not accounted for, this makes interpreting the results challenging and the comparative efficacy of selumetinib also remains uncertain.
Given the identified issues with the sponsor’s model structure and the magnitude of uncertainty in the comparative efficacy of selumetinib against best supportive care (BSC), CADTH was unable to derive a robust base-case estimate of cost-effectiveness. The clinical experts consulted by CADTH noted that a residual benefit may be possible following treatment discontinuation. However, the magnitude of the residual benefit in terms of PFS once a patient is off treatment is unclear due to a lack of trial data. The sensitivity of the model to this assumption was explored in 2 reanalyses, which differed in terms of the magnitude of the assumed residual benefit following treatment discontinuation. In 1 analysis (CADTH reanalysis A), a smaller residual benefit was considered by assuming PFS would follow a log-logistic distribution. In contrast, another analysis modelled a larger residual benefit by using the sponsor’s selected exponential distribution to describe PFS (CADTH reanalysis B). Both reanalyses incorporated changes to address limitations associated with the probability of disease progression with selumetinib, the time required to return to the “progressed” utility value following disease progression, and assumptions surrounding drug acquisition costs.
Both CADTH reanalyses were consistent with the sponsor’s base case: selumetinib is not cost-effective at a willingness-to-pay threshold of $50,000 per QALY gained. In CADTH reanalysis A, which assumed smaller residual benefit (i.e., the mean duration of the progression-free state for patients on selumetinib would be 22.19 years), the incremental cost-effectiveness ratio (ICER) for selumetinib plus BSC was $426,286 per quality-adjusted life-year (QALY) gained compared to BSC alone ($1,177,024 in incremental costs and 2.76 incremental QALYs). CADTH reanalysis B made the same changes as in reanalysis A with the exception that the residual benefit modelled reflected the sponsor’s assumption. In this reanalysis, patients on selumetinib would stay in the progression-free state for an average of 33.96 years. The ICER for selumetinib plus BSC was $294,751 per QALY gained compared to BSC alone ($1,177,024 in incremental costs and 3.99 incremental QALYs). Both analyses assume selumetinib substantially delays disease progression. In the absence of direct evidence, the true comparative impact of selumetinib in delaying progression relative to BSC is highly uncertain. The majority of the incremental QALY gains (i.e., 82.5% and 83.5% in reanalysis A and B, respectively) occurred outside of the trial period, and there is significant uncertainty as to whether such clinical benefits would be realized. Under both scenarios, price reductions of 88.5% or 83.2% would be required for selumetinib to be cost-effective at a willingness-to-pay threshold of $50,000 per QALY.
The price of the submitted drug is high and represents a majority of the total expected costs associated with selumetinib. In a hypothetical patient with a body surface area (BSA) of 1.8 m2, the annual cost of treatment is estimated to be $403,016 per patient. While the sponsor’s assumption that selumetinib would not confer improvements in survival was reasonable according to clinical experts consulted by CADTH, the expected QALYs predicted by the model are highly uncertain. The clinical benefits modelled with selumetinib are based on time spent progression-free (with progression defined by changes in PN volume); however, tumour volume is poorly correlated with health-related quality of life (HRQoL) and the selected utility values used by the model lack validity. Clinical outcomes (i.e., symptom relief and pain management) most relevant to patients and clinical experts consulted by CADTH were not considered within the model. Furthermore, as direct evidence comparing selumetinib and BSC is lacking, the model assumed that all patients on BSC would automatically enter the “progressed” state. This structural assumption would favour selumetinib. As this limitation could not be addressed by CADTH, any analyses performed by CADTH or the sponsor would likely underestimate the true ICER. Given the clinical uncertainty and issues with the model structure, the CADTH reanalyses results should be viewed with extreme caution.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
Patient input for this review was obtained from the Tumour Foundation of BC. Information from the perspective of patients with neurofibromatosis type 1 (NF1) and their caregivers was collected from a focus group held in November 2022 and a health experience survey. All respondents were over the age of 18 and resided in British Columbia. Most patients with NF1 experienced moderate to severe chronic pain, limiting their daily activities and social engagement. This can result in a dependency on caregivers into adulthood. Caregivers reported emotional impact from the long-term responsibilities and financial obligations in caring for these patients. Nearly half of patients in the survey had never been offered any treatment options and, among patients who did receive treatment, minimal symptom improvement was achieved. All respondents expressed a desire for treatments that offer improvements in quality of life, decreases in pain, increases in functionality, and a reduction in the number of health care visits. None had experience with selumetinib.
Clinician group input was received from the Canadian Pediatric Brain Tumour Consortium. The current pathway of care for patients with NF1 currently involves supportive care or large, aggressive surgeries. Patients continue to live with disability and functional impairment as a result of the risk or ineffective nature of current therapies (e.g., radiation therapy). Most PNs are inoperable due to their proximity to vital structures and their intimate involvement with peripheral nerves. Given the significant quality of life benefits from selumetinib, it is expected to transform the treatment pathway and become the first-line therapy in this population. Based on the SPRINT studies and Canadian experience, treatment with selumetinib should lead to cytoreduction in a significant number of patients, alleviate symptoms related to the PN, reduce pain, restore facial deformities, and reduce caregiver burden. This response to treatment is expected to be consistent with outcomes observed from another mitogen-activated protein kinase inhibitor, trametinib.
Input from drug plans included questions relating to treatment eligibility and treatment continuation and renewal. The plans noted that access to specialists may be limited and that multidisciplinary teams in specialized settings may be required for optimal patient management. Plans sought a better understanding of how clinicians would assess and monitor treatment response. In addition, clarification was requested on whether selumetinib would be initiated or continued in patients aged 18 and older. Last, drug plans raised concerns about the expected budget impact of reimbursing selumetinib.
One of these concerns were addressed in the sponsor’s model: BSC was an appropriate comparator, as further verified by CADTH in consultation with clinical experts.
CADTH was unable to address 2 concerns raised in stakeholder input:
The effectiveness of selumetinib relative to BSC could not be established given the lack of comparative evidence.
The sponsor’s model structure failed to characterize the key clinical outcomes noted to be most relevant to patients and the potential benefits expected from selumetinib in terms of decreases in pain and increases in functionality.
Economic Review
The current review is for selumetinib (Koselugo) for the treatment of pediatric patients 2 years of age and above with NF1 who have symptomatic, inoperable PNs.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted an economic evaluation comparing selumetinib plus BSC against BSC alone.1 BSC was defined as therapies used for symptomatic disease management, which may include pain medication, antidepressants, and anxiolytics.1 The target population considered by the model included children 2 years and older with NF1 and symptomatic, inoperable PNs based on the SPRINT trial. This target population aligned with the Health Canada indication and the sponsor’s reimbursement request.1,2
Selumetinib is available as 10 mg and 25 mg oral capsules at submitted prices of $122.60 and $306.50 per capsule, respectively.1 At a recommended dosage of 25 mg/m2 twice daily,3 a hypothetical patient with a BSA of 1.12 m2 would require 56 mg of selumetinib per day (i.e., 6 10 mg capsules) at a cost of $735.60 per day ($268,677 per year). In combination with BSC, the annual cost for selumetinib for this hypothetical patient was estimated to be $268,913 per year, based on the sponsor’s model. As patients in the comparator arm did not receive any disease-directed treatment, the annual costs of BSC were estimated to be $357.
Clinical effects in the model were measured as QALYs. Both costs and effects were estimated from the perspective of the publicly funded health care payer, assuming a 97-year time horizon with an annual discount rate of 1.5% applied to both costs and QALYs.1
Model Structure
The sponsor submitted a state-transition model that tracked a homogeneous cohort of patients across 3 health states: progression-free, progressed, and dead (Figure 1). Progression was defined as in the SPRINT trial as PN growth of at least 20% from baseline or an increase of at least 20% from the best response by volumetric analysis from MRI.1,4 In the selumetinib arm, all patients entered the model in the progression-free health state. From this state, patients could transition to 1 of the remaining states as informed by transition probabilities for all-cause mortality risk and the probability of tumour progression. The approach used to determine which patients entered the progressed health state was a function of age. Prior to age 18, it was calculated as the proportion of the cohort who had progressed in the previous cycle and remained alive, and as the proportion of the cohort who were progression-free in the previous cycle and had progressed. After age 18, the sponsor assumed no patients in the progression-free health state would experience disease progression. Once patients entered the progressed state, they remained in that state until death.1 Discontinuation from selumetinib was modelled independently and only affected cost calculations. In the BSC arm, it was assumed patients would enter the model in the progressed state.1 In effect, as BSC was a simple 2-state model, membership in the progressed state was calculated as the proportion of patients who did not occupy the death state.1
Model Inputs
Baseline characteristics of interest included age (mean = 10.3 years), sex (40% female; 60% male), and BSA (mean = 1.127 m2). These characteristics were sourced from the SPRINT trial, a phase II, open-label, single-arm study of selumetinib treatment for children between the age of 2 to 18 with NF1 who had inoperable PNs.1,4 In its submitted model, the sponsor assumed annual age- and sex-specific linear increases in BSA until stabilization at age 18.1 These baseline characteristics affected estimates of all-cause mortality risks and the dose of selumetinib.4
In the absence of trial data relating to overall survival, the sponsor assumed that selumetinib would not result in any survival benefits when compared to BSC. As such, a time-dependent transition probability for mortality risk associated with NF1 was applied to both arms of the model.1 This parameter, which was based on a French cohort study, was calculated by applying a standardized mortality ratio of 2.02 to age- and gender-specific general-population mortality risks from Statistics Canada Life Tables.1,5,6
Data from the SPRINT trial were used to fit parametric survival models describing PFS and time to treatment discontinuation (TTD) curves.1,4 Both survival models were fitted using exponential, log-logistic, log-normal, Weibull, and generalized gamma distributions.1,7 In the sponsor’s base case, PFS and TTD were assumed to follow the exponential and Weibull distributions, respectively, based on model fit statistics and clinical consultation.1 Derivation of the PFS curve was then used to calculate the annual probability of tumour progression adjusted by removing those who had died during the preceding cycle.1
The model also relied on data from the SPRINT trial to track the occurrence of adverse events (AEs) associated with the use of selumetinib.1,4 AEs were restricted to those classified as grade 3 or higher, such as diarrhea, vomiting, fever, hypoxia, paronychia, and dermatitis acneiform. It was assumed that treatment-related AEs would only occur once per year and would only affect costs. The occurrence of these AEs did not directly affect treatment discontinuation or utilities.1
Health-state utilities were incorporated into the economic evaluation with the “progression-free” state associated with a utility value of 0.74 while the “progressed” state was associated with a utility value of 0.51.1 These values were obtained from the direct elicitation of population preferences in a time trade-off study commissioned by the sponsor involving 100 adult participants from the UK (i.e., aged 18 years and older). The time trade-off exercise was focused on deriving utility estimates with health vignettes based on whether patients were on or off selumetinib treatment.1,8 At model entry, it was assumed all patients would have a utility value of 0.51 and, among those who remain progression-free on selumetinib, their utility would increase to 0.74 by the next year. Among patients younger than 18 who transitioned to the progressed state, the submitted model assumed that it would take 5 years for utilities to return to the progressed utility value, applying an annual linear decline in utility.1 For patients who were in the progressed state after the age of 18, a utility value of 0.51 was applied. No other utility values were considered in the economic evaluation as the sponsor assumed that treatment-emergent AEs would have a minimal impact on HRQoL.
Costs captured within the economic evaluation included those associated with treatment acquisition, monitoring, and the management of treatment-related AEs. Drug acquisition costs for selumetinib arm were based on the sponsor’s submitted price, and public list prices from provincial formularies informed all other drug costs.9-11 Overall treatment costs were calculated based on the TTD curve and further adjusted by 92.3% based on the dose interruptions reported in the SPRINT trial.1 In addition to the cost of selumetinib, other drug-related costs related to selumetinib treatment included anti-emetic medication (ondansetron) (i.e., 62% of patients) and analgesics, as observed in the SPRINT trial.1,4 Based on findings from the SPRINT trial, it was assumed that patients on BSC would consume 67.5% more analgesics than those on selumetinib.1,4 It was further assumed that patients on selumetinib would receive 2 additional MRI scans each year, with costs based on the Ontario Case Costing Initiative, updated to 2021 dollars.12 The same source was used to identify costs related to management of AEs.1,12
Summary of Sponsor’s Economic Evaluation Results
The sponsor conducted a probabilistic analysis based on a 5,000-iteration Monte Carlo simulation.1 The submitted deterministic and probabilistic results were similar. Results from the probabilistic base case are presented in the following section.
Base-Case Results
Results from the base case of the sponsor’s economic evaluation are presented in Table 3. The submitted analysis was based on publicly available prices of the comparator treatments.
In the sponsor’s base case, selumetinib plus BSC was associated with an additional 6.11 QALYs at an additional cost of $752,073 when compared to BSC alone. This resulted in an ICER of $123,098 per QALY gained relative to BSC alone, suggesting that selumetinib plus BSC would not be cost-effective at a threshold of $50,000 per QALY.
Two-thirds of the costs in the selumetinib arm were expected to be incurred in the first 3 years of the model. In contrast, 93% of the costs of BSC were incurred during the extrapolated period of the 100-year time horizon. Similarly, more than 90% of the incremental QALYs accrued in the extrapolated period, although the clinical evidence does not support these modelled benefits. The amount of time patients spent on selumetinib, based on the TTD curve, was the key driver of incremental costs, while the amount of time patients remained progression-free, based on the PFS curve, was the key driver of incremental QALYs.
Table 3: Summary of Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. reference ($ per QALY) |
|---|---|---|---|---|---|
BSC | 14,365 | Reference | 18.35 | Reference | Reference |
Selumetinib plus BSC | 766,438 | 752,073 | 24.46 | 6.11 | 123,098 |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
In addition to the base case, the sponsor considered several scenario analyses. While the ICER was affected in each scenario, none led to a different conclusion regarding the cost-effectiveness of selumetinib.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis:
The submitted model lacked face validity: An appropriate model structure for economic evaluation should capture all relevant and meaningful underlying clinical or biologic processes. The model submitted by the sponsor considered 3 health states for those on selumetinib: progression-free, progressed, and dead; while a 2-state model was applied to those on BSC alone: progressed and dead. Progression was defined as PN growth of 20% or greater from baseline or, if patients had a partial response, an increase of at least 20% from the best response. This does not align with the feedback from either the patient groups or the clinical experts consulted by CADTH. Although this disease has a heterogeneous clinical presentation depending on the location of PN growth, all feedback suggested that symptom relief and pain are more important outcomes. Furthermore, the sponsor assumed that tumour volume size has a direct impact on HRQoL. According to the clinical experts consulted by CADTH, tumour volume size is poorly correlated with HRQoL, given the heterogeneous clinical manifestation of the condition. Rather, as suggested in a Canadian study, more relevant predictors of quality of life would be pain and symptom relief.13
For patients receiving selumetinib, the sponsor further assumed that progression and time on treatment were independent (i.e., treatment effectiveness was not dependent on whether patients remained on treatment). The PFS curve informed the proportion of patients in the progression-free and progressed health states, which affected the estimates of utilities, while the TTD curve affected the estimates of costs only. To ensure consistency, estimation of costs and benefits should be informed by the same patient cohorts. However, the sponsor’s approach implied that costs would be identical among those who have discontinued treatment regardless of whether they occupied the progression-free or progressed health state. This lacks validity according to the clinical experts consulted by CADTH. The sponsor justified that this would capture the residual benefit from selumetinib treatment (i.e., patients will benefit from treatment even following treatment discontinuation). Furthermore, the sponsor assumed that, after the age of 18 years, patients in the progression-free state would remain progression-free for the remainder of their lifetime, regardless of their treatment status. The age 18 cut-off was selected because it was thought to correspond to the time point when PN volumes are expected to plateau as growth rates slow. While the clinical experts consulted by CADTH suggested that a residual benefit in terms of PFS following treatment discontinuation was plausible, the magnitude remains highly uncertain.
Last, in the sponsor’s model, 100% of patients who receive selumetinib start in the “progression-free” health state and 100% of patients who receive BSC alone start in the “progressed” health state. This would imply that patients who receive BSC start the model with an instantaneous 20% increase in tumour volume. This assumption is inconsistent with the available evidence on the natural history of PN progression. Data submitted by the sponsor suggest that, among patients treated with BSC, volume size may increase, remain the same, or, in a small proportion of patients, decrease.14
This limitation could not be addressed by CADTH as it would require a complete redevelopment of the model. The sponsor’s model overestimates the benefits of selumetinib associated with progression given the assumption that 100% of patients on BSC start in the “progressed” health state. Due to the uncertainty in the magnitude of the residual benefit, CADTH conducted 2 distinct reanalysis of 2 plausible scenarios. CADTH reanalysis A assumed a smaller residual benefit by selecting a log-logistic distribution for the PFS curve. In CADTH reanalysis B, the sponsor’s exponential distribution remained to inform the PFS curve. For both reanalyses, CADTH removed age-specific disease-progression rates and assumed that disease progression would continue beyond the age of 18.
There was no direct or indirect evidence regarding the relative effectiveness of selumetinib compared with BSC: As noted in the previous discussion, the effectiveness of selumetinib with respect to PFS was derived from the SPRINT trial. As this was a single-arm study, the sponsor assumed that 100% of patients on BSC entered the model in the progressed health state, eliminating the need to derive a PFS curve for the BSC cohort. The experts consulted by CADTH suggested that this assumption was inconsistent with their clinical experience. This also conflicts with the clinical data submitted by the sponsor. In Study 01-C-0222, a phase II, randomized, double-blind, placebo-control trial on tipifarnib in children and young adults with NF1 and progressive PNs, 20.6% of those in the placebo-arm remained progression-free at 2 years. Patients in the BSC arm should have entered the model in the progression-free state and experienced transitions similar to those described for the selumetinib arm. This use of assumptions to inform the natural history of patients in the BSC arm introduces a high degree of uncertainty into the sponsor’s analysis. The CADTH clinical report concluded that the sponsor’s indirect comparison of selumetinib and a natural history cohort indicated that a clinical benefit may exist, but the magnitude of the benefit is unknown. In the absence of direct evidence comparing selumetinib and BSC, all comparative conclusion between selumetinib and BSC remain highly tenuous.
This limitation could not be addressed by CADTH. The assumptions and evidence used to inform PFS in the submitted economic model likely biased results in favour of selumetinib (see first key limitation).
The model had technical issues: The method used to calculate the probability of tumour progression from the PFS curve was incorrect. The survival probabilities derived from the PFS curve should represent the probability that a patient would remain progression-free between 2 discrete time points. However, the approach used by the sponsor failed to convert the values of the PFS curve to a discrete time scale. Furthermore, an adjustment was made to derive the probability of progression accounting for mortality. However, when using this probability of progression to derive the proportion of patients who have progressed, mortality was accounted for again, resulting in the double-counting of the mortality risk.15
In addition, although the sponsor’s probabilistic results appeared to align with its deterministic results, re-running the analyses resulted in larger differences between the probabilistic and deterministic results than originally reported by the sponsor. Upon further exploration, it was found that individual probabilistic simulations produced outliers in both the expected costs and QALYs. For example, across the individual 5,000 probabilistic simulations of the sponsor’s submitted model, the expected costs associated with selumetinib ranged from $321,299 to $1,478,827, while incremental QALYs ranged from 0.83 to 11.82. The minimum and maximum ICERs ranged from $33,551 to $1,005,589 per QALY gained across the 5,000 simulations.
CADTH used the correct method to calculate the probability of progression in a discrete time period (i.e., 1 minus the ratio of 2 survival probabilities corresponding to the time period of interest). CADTH further revised the calculation to remove the double-counting of mortality and conducted a deterministic analysis for all results.
The assumptions for health-state utilities were problematic: The sponsor conducted a direct utility elicitation exercise to derive the utility estimates used by the model for progression-free and progressed health states. This exercise resulted in the estimation of utilities specific to selumetinib treatment status (on versus off selumetinib treatment) rather than progression status. When estimating health-state utilities, the sponsor assumed that the utility value for “on selumetinib treatment” corresponded with the “progression-free” health state while the utility values for “off selumetinib treatment” corresponded with the “progressed” health state. This was concerning because it assumed that HRQoL would be affected by treatment status and this perfectly correlated with progression.
The sponsor further assumed that, following disease progression, patients under the age of 18 would take 5 years to revert to the utility estimates associated with the progressed health state. This appeared to extend the residual benefit of selumetinib to delay the impact of progression on HRQoL. This approach relied on several unfounded assumptions relating to the amount of time it would take for utilities to reach the value associated with progressed disease and the relationship between utility decline and time. The evidence used to form each assumption was not described and no justification was provided. Although there may be validity to applying utilities based on patients’ proximity to disease progression given the time-dependent nature of tumour volume growth (i.e., time-to-progression utilities), there is limited validity to assuming that the utilities in 2 identical health states (i.e., progression) should differ.
The CADTH reanalysis partly addressed this limitation by assuming that, following disease progression, patients would return to utilities of 0.51 at the earliest time point possible given the model’s structural constraints (i.e., after the first year). The CADTH reanalysis did not address the limitation associated with the use of treatment-specific utility values to inform the utility estimates associated with disease progression. The appropriate utility estimates for the progression-free and progressed health states remain uncertain. As such, CADTH explored the effect of varying the utility value for the progressed health state in scenario analyses.
The costs of selumetinib were underestimated: Several concerns were noted regarding the sponsor’s approach to calculating the costs of treatment. First, the model assumed that patients on selumetinib would experience the same degree of dose interruption as observed in the SPRINT trial. This meant that there would be no drug costs when dose interruption occurs. However, for oral treatments, Canadian pharmacies are likely to fill and dispense each prescription in full. It is unlikely that any unused tablets will lead to lower prescription costs as excess unused tablets are unlikely to be retrieved or redistributed. As such, adjusting treatment costs by dose interruption is likely to underestimate treatment costs. In addition, the sponsor assumed that the TTD curve would follow a generalized gamma distribution. This distribution would result in almost every patient discontinuing treatment by age 18. The clinical experts consulted by CADTH expected that some patients would remain on treatment beyond age 18 unless reimbursement restrictions are in place.
Two changes were made to address this limitation in the CADTH reanalyses. First, the assumption regarding dose interruption was removed to ensure the full treatment acquisition costs were captured. Second, it was assumed that the TTD curve would follow an exponential distribution based on feedback provided by CADTH’s clinical experts.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation
Sponsor’s key assumption | CADTH comment |
|---|---|
Selumetinib does not result in survival benefits compared to BSC. | Appropriate. No clinical studies suggest a survival benefit associated with selumetinib. Based on the input received by clinician groups and from clinical experts consulted by CADTH, selumetinib is expected to mainly affect disease morbidity. |
BSC is required chronically and the definition of BSC (in terms of the composition and proportions) was assumed to be consistent with baseline values over time. | Although an unrealistic simplification, this is unlikely to affect the model’s cost estimates given that the cost of BSC is minimal compared to the drug acquisition costs associated with selumetinib. Few studies have documented the longitudinal change in the composition of BSC and it may be challenging to generalize across studies given the use of BSC would depend on individual patient’s symptom presentation for which the presentation of this disease is heterogeneous.) |
Adverse impacts will not affect HRQoL. | Inappropriate, but unlikely to affect the model. The majority of the adverse impacts considered by the model had a short duration, and, if captured, would likely result in lower expected utilities. |
No difference is expected in monitoring or service utilization between BSC and selumetinib except for additional costs of monitoring for MRI to assess PN progression in patients actively receiving selumetinib. | This assumption was inconsistent with the expectations and experience of the clinical experts consulted by CADTH. Care equivalent to BSC is currently offered to patients by pediatricians, while the administration and monitoring of selumetinib is expected to be provided in a medical oncology clinic. Patients treated with selumetinib are expected to consume more health care resources due to an increased frequency of follow-up during their first year of treatment. These costs are likely underestimated but, relative to the cost of treatment, are minor. |
BSC = best supportive care; PN = plexiform neurofibroma.
CADTH Reanalyses of the Economic Evaluation
Reanalysis Results
Given the magnitude of uncertainty surrounding the residual benefits and concerns raised with the submitted model structure, CADTH was unable to derive a robust base-case estimate of cost-effectiveness. CADTH conducted separate reanalyses involving different assumptions for the magnitude of the residual benefits, together with additional changes to address several key limitations associated with selumetinib, including correctly calculating the probability of progression, revising the assumptions surrounding time to return to progressed utility value, and revising the drug acquisition costs. A summary of the changes applied to the submitted economic evaluation is provided in Table 5. None of the changes included in the CADTH reanalyses addressed the limitations associated with the invalid model structure.
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Changes to derive the CADTH Reanalysis | ||
1a. PFS curve | Exponential | Log-logistic |
1b. Age-specific disease progression Worksheet: _Engine NOTE: Column X = % in progression-free state Column Y = % in progressed state Column t = probability of death Column H = age Column U = probability of progression | Calculation for the proportion of patients who have progressed. Patients over 18 assumed a probability of progression of 0 (i.e., do not progress). Two-step process: 1. Range: Y19:Y116 Sample Formula (i.e., Cell Y19): = IF($L$3 = 7, Y18 × (1 − T18) + X18 × IF(H19 > 18,0,$Q$11) × A19 × (1 − T18), Y18 × (1 − T18) + X18 × IF(H19 > 18,0,$U19) × A19 × (1 − T18)) 2. Formula for the first value within the following columns AA to AO: Sample formula (i.e., cell AA20) = IF($L$3 = 7, IF($H20 > 18,0,$X19 × $Q$11 × $A20 × (1 − $T19)), IF($H20 > 18,0,$X19 × $U20 × $A20 × (1 − $T19))) | Calculation for the proportion of patients who have progressed. Logic (i.e., IF statement) replacing 0 probability of progression at ages 18+ removed. Patients continue to progress after 18 years of age, depending on the PFS curve Two-step process: 1. Range: Y19:Y116 Sample Formula (i.e., Cell Y19): = IF($L$3 = 7, Y18 × (1 − T18) + X18 × ($Q$11) × A19 × (1 − T18), Y18 × (1 − T18) + X18 × ($U19) × A19 × (1 − T18)) 2. Formula for the first value within the following columns AA to AO: Sample formula (i.e., cell AA20) = IF($L$3 = 7, $X19 × $Q$11 × $A20 × (1 − $T19), $X19 × $U20 × $A20 × (1 − $T19)) |
2. Probability of progression Worksheet: _Engine Column S: Probability of progression Column T: Probability of death | Incorrect calculation, which further double-counted mortality when applied to derive the proportion of patients who have progressed. Sponsor calculated the probability of progression by calculating the difference in probability of progression between the current and preceding cycle, subtracting from the joint probability of death and progression. If the calculation was less than 0, replace with 0. Applied to following range of cells: U20:U116 Sample Formula (i.e., Cell U20): = IF((S19 − S20) − S20 × T19 < 0,0,(S19 − S20) − S20 × T19) | Calculated as a time-dependent transition probability, converted from the PFS curve. CADTH took a 3-step process: 1. To calculate probability of progression, subtract the probability of remaining progression-free in each cycle (i.e., converted PFS curve to discrete time scale by dividing the probability of progression in current cycle with that of preceding cycle) from: 1. IFERROR ensured the formula returned 0 instead of returning divide by 0 errors. Applied to following range of cells: U20:U116 Sample Formula (i.e., Cell U20): = IFERROR(1 − ($S20/$S19),0) 2. Update formula to determine membership in progression-free state (applied across all ages). Applied to following range of cells: X20:X33 Sample Formula (i.e., Cell X20) = IF($L$3 = 7,X19 × (1 − ($Q$11 × A20)) × (1 − T19),1 − Y20 − AP20) 3. Remove age logic at age 18 to be consistent with Stepped Analysis 2a. Applied to following range of cells: X34:X116 Sample formula (i.e., cell X34) = IF($L$3 = 7,X33 × (1 − ($Q$11 × A34)) × (1 − T33),X33 × (1 − ($U34 × A34)) × (1 − T33)) |
3. Time to return to progressed utility (selumetinib arm only) | After progression, patients will experience an annual linear utility decrement over 5 years before returning to a utility value of 0.510. | 1 year to return to utility value of 0.510. |
4a. Dose interruption | 92.3% | 100% |
4b. Time-to-discontinuation curve | Weibull | Exponential function |
CADTH reanalysis A | 1a + 1b + 2 + 3 + 4a + 4b | |
CADTH reanalysis Ba | 1b + 2 + 3 + 4a + 4b | |
PFS = Progression-free survival.
aCADTH reanalysis B assumes a larger magnitude of the residual benefit upon treatment discontinuation.
The CADTH reanalysis (Table 6) used publicly available prices of the comparator treatments. The results of the stepwise reanalyses highlight the issues CADTH noted regarding the model structure. For example, changes to the probability of progression had no impact on costs as costs were dependent on the TTD curve, and TTD was independently modelled from disease progression. As the proportion of patients in the progression-free and progressed states varied (i.e., reanalysis 1a), the expected costs within the model were observed to not change. The majority of stepwise changes led to an increase in the ICER relative to the sponsor’s base case, suggesting that the assumptions in the sponsor’s base case were optimistic.
The conclusions from the 2 CADTH reanalyses (i.e., A and B) were consistent: selumetinib plus BSC is not cost-effective at a willingness-to-pay threshold of $50,000 per QALY compared to BSC alone. In CADTH reanalysis A (in which a smaller residual benefit was assumed), selumetinib plus BSC was associated with an ICER of $426,286 per QALY gained compared to BSC (incremental costs = $1,177,024 and incremental QALYs = 2.76). In CADTH reanalysis B (which assumed a larger residual benefit), selumetinib plus BSC was associated with an ICER of $294,751 per QALY gained compared to BSC ($1,177,024 in incremental costs and 3.99 incremental QALYs). The key difference between these 2 reanalyses was the estimated life-years spent in the progression-free state (i.e., the extent to which selumetinib delays disease progression). A larger residual benefit would increase the amount of time patients on selumetinib would spend progression-free, leading to higher utility estimates. As reported in Table 10 and Table 11, a mean of 22.19 life-years were spent in the progression-free state from reanalysis A, while a mean of 33.96 life-years were spent in the same health state from reanalysis B.
In both reanalyses, results were driven by the high drug acquisition costs associated with selumetinib. Incremental QALYs were driven by the amount of time spent in the progression-free health state and are vulnerable to uncertain predictions of PFS outside of the trial period. The majority of the incremental QALY gains (i.e., 82.5% and 83.5% in reanalysis A and B, respectively) in the model occurred outside of the trial period, during which there is significant extrapolation uncertainty. Given that the model structure assumed all patients immediately enter the “progressed” health state and this structural assumption could not be addressed within the CADTH reanalyses, both the CADTH and sponsor analyses are expected to overestimate the benefits of selumetinib to an extent, although the magnitude of overestimation is uncertain.
Table 6: Summary of the Stepped Analysis of the CADTH Reanalysis Results (Deterministic Unless Stated Otherwise)
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($ per QALY) |
|---|---|---|---|---|
Sponsor’s base case (probabilistic) | BSC | 14,365 | 18.35 | Reference |
Selumetinib + BSC | 766,438 | 24.46 | 123,098 | |
Sponsor’s base case | BSC | 14,413 | 18.39 | Reference |
Selumetinib + BSC | 723,099 | 24.47 | 116,544 | |
CADTH reanalysis 1a | BSC | 14,413 | 18.39 | Reference |
Selumetinib + BSC | 732,099 | 23.85 | 129,816 | |
CADTH reanalysis 1b | BSC | 14,401 | 18.38 | Reference |
Selumetinib + BSC | 723,092 | 24.47 | 116,336 | |
CADTH reanalysis 2 | BSC | 15,198 | 19.12 | Reference |
Selumetinib + BSC | 723,616 | 23.07 | 179,698 | |
CADTH reanalysis 3 | BSC | 14,413 | 18.39 | Reference |
Selumetinib + BSC | 723,099 | 23.95 | 127,326 | |
CADTH reanalysis 4a | BSC | 14,413 | 18.39 | Reference |
Selumetinib + BSC | 782,460 | 24.47 | 126,306 | |
CADTH reanalysis 4b | BSC | 14,413 | 18.39 | Reference |
Selumetinib + BSC | 1,100,617 | 24.47 | 178,628 | |
CADTH reanalysis A (1a + 1b + 2 + 3 + 4a + 4b): smaller residual benefit | BSC | 14,413 | 18.39 | Reference |
Selumetinib + BSC | 1,191,437 | 21.15 | 426,286 | |
CADTH reanalysis B (1b + 2 + 3 + 4a + 4b): larger residual benefit | BSC | 14,413 | 18.39 | Reference |
Selumetinib + BSC | 1,191,437 | 22.38 | 294,751 |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Scenario Analysis Results
CADTH conducted 2 sets of scenario analyses to address the uncertainty in the utility values. These scenarios applied different utility values for the progressed health state. Results of the scenario analyses using CADTH reanalysis A and B are presented in Table 12. In the “higher utility value” scenario (i.e., smaller difference in utilities expected between the progression-free and progressed health states), the ICERs increased under both scenarios (i.e., scenario A = $1,106,772 per QALY gained; scenario B = $802,552 per QALY gained). Under a “lower utility value” scenario (i.e., larger difference in utilities expected between the progression-free and progressed health states), the ICERs decreased (i.e., scenario A = $263,981 per QALY gained; scenario B = $180,526 per QALY gained).
CADTH conducted additional scenario analyses to explore the impact of incremental reductions in the price of selumetinib. The results are presented in Table 8. Price reductions of 88.5% or 83.2% would be required for selumetinib to achieve cost-effectiveness relative to BSC at a $50,000 per QALY threshold under CADTH reanalysis A and B, respectively (Table 7).
Table 7: CADTH Price-Reduction Analyses
Analysis | ICERs for selumetinib + BSC vs. BSC alone | ||
|---|---|---|---|
Price reduction | Sponsor base case | CADTH reanalysis A | CADTH reanalysis B |
No price reduction | $123,098 | $426,286 | $294,791 |
10% | $104,892 | $383,737 | $265,331 |
20% | $93,241 | $341,188 | $235,911 |
30% | $81,589 | $298,639 | $206,491 |
40% | $69,937 | $256,091 | $177,071 |
50% | $58,285 | $213,542 | $147,651 |
60% | $46,663 | $170,993 | $118,231 |
70% | $34,982 | $128,444 | $88,811 |
80% | $23,330 | $85.895 | $59,391 |
90% | $11,678 | $43,346 | $29,971 |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio.
Issues for Consideration
Potential comparator: Trametinib is another mitogen-activated protein kinase inhibitor that has been studied in this patient population and was mentioned in the clinician group input included within the Clinical Review report. However, trametinib does not currently have a Health Canada indication for patients with NF1 and symptomatic NF1 and, as such, has not been reviewed by CADTH for this indication.
Risk of off-label use: According to clinical experts consulted by CADTH, there is a potential for off-label treatment with selumetinib. This includes the continued use of selumetinib in adult patients who were treated as pediatric patients, the use of this treatment in patients with NF1 with asymptomatic PNs whose tumours are at risk of becoming symptomatic, and the use in adult patients. The submitted model only captures the first consideration (i.e., the continued use of selumetinib past the pediatric age into adulthood). The other off-label use was not modelled and was considered outside the scope of this review.
Patient support program: The sponsor noted in its response to CADTH that the Alexion OneSource patient support program will be available to Canadian patients eligible for selumetinib and their caregivers. This program provides resources and support by telephone or email on disease education, appointment reminders, adherence, and reimbursement navigation.16
Re-treatment or intermittent treatment: The submitted model assumes a single course of treatment with selumetinib. Upon discontinuation, and as informed by the TTD curve, patients would not be eligible for another course of selumetinib. This does not align with how clinicians consulted by CADTH noted how this drug would be used, as patients in real-world practice may receive intermittent treatment or re-treatment with selumetinib. The cost-effectiveness of re-treatment or intermittent treatment is unknown as there is no evidence regarding either the efficacy or the safety of this practice. If considered, this is expected to affect the cost-effectiveness estimates of selumetinib.
Caregiver impact: Although the patient input received by CADTH noted a large impact on caregivers for patients with this condition, the Clinical Review did not identify any outcomes collected that measured the impact of selumetinib on caregivers. Although an attempt was made by the sponsor to incorporate caregiver disutilities within a submitted scenario analyses based on a series of assumptions, this was not explored further by CADTH given the lack of evidence to support the sponsor’s assumptions surrounding their approach to capture caregiver disutilities.
Overall Conclusions
The CADTH Clinical Review concluded that selumetinib resulted in reductions in PN volume. However, the correlation between PN volume changes and improvements in symptoms or function remains uncertain as the clinical experts consulted by CADTH noted that tumour size may not be reflective of morbidity. The magnitude of the benefit in terms of PFS was unclear. The nonrandomized design of the SPRINT trial and the many confounding prognostic factors that were not accounted for make interpreting the results challenging, and the comparative efficacy of selumetinib remains uncertain.
Given the identified issues with the sponsor’s model structure and the magnitude of uncertainty in the comparative efficacy of selumetinib against BSC, CADTH was unable to derive a robust base-case estimate of cost-effectiveness. Feedback CADTH received from clinical experts noted that a residual benefit may be possible following treatment discontinuation. However, the magnitude of the residual benefit in terms of PFS once a patients is off treatment is unclear due to a lack of trial data. The sensitivity of the model to this assumption was explored in 2 reanalyses that differed in terms of the magnitude of the assumed residual benefit following treatment discontinuation. In the first analysis (CADTH reanalysis A), a smaller residual benefit was considered by assuming PFS would follow a log-logistic distribution. In contrast, another analysis modelled a larger residual benefit by using the sponsor’s selected exponential distribution to describe PFS (CADTH reanalysis B). Both reanalyses also incorporated changes to address limitations associated with the probability of disease progression with selumetinib, the duration to return to “progressed” utility value following disease progression, and assumptions surrounding drug acquisition costs.
Both CADTH reanalyses were consistent with the sponsor’s base case: selumetinib is not cost-effective at a willingness-to-pay threshold of $50,000 per QALY gained. In CADTH reanalysis A, which assumed a smaller residual benefit (i.e., the mean duration of the progression-free state for patients on selumetinib would be 22.19 years), the ICER for selumetinib plus BSC was $426,286 per QALY gained compared to BSC alone ($1,177,024 in incremental costs and 2.76 incremental QALYs). CADTH reanalysis B made the same changes as in reanalysis A with the exception that the modelled residual benefit reflected the sponsor’s assumption. In this reanalysis, patients on selumetinib would stay in the progression-free state for an average of 33.96 years. The ICER for selumetinib plus BSC was $294,751 per QALY gained compared to BSC alone ($1,177,024 in incremental costs and 3.99 incremental QALYs). Both analyses assume selumetinib substantially delays disease progression. In the absence of direct evidence, the true comparative impact of selumetinib in delaying progression relative to BSC is highly uncertain. The majority of the incremental QALY gains (i.e., 82.5% and 83.5% in reanalysis A and B, respectively) occurred outside of the trial period, for which there is significant uncertainty as to whether such clinical benefits would be realized. Under these scenarios, a price reduction of 88.5% or 83.2%, respectively, would be required for selumetinib to be cost-effective at a willingness-to-pay threshold of $50,000 per QALY.
The price of the submitted drug is high and represents a majority of the total expected costs associated with selumetinib. In a hypothetical patient with a BSA of 1.8 m2, the annual cost of treatment is estimated to be $403,016 per patient. While the sponsor’s assumption that selumetinib would not confer improvements in survival was reasonable, according to clinical experts consulted by CADTH, the expected QALYs predicted by the model are highly uncertain. The clinical benefits modelled with selumetinib are based on time spent progression-free (with progression defined as changes in PN volume); however, tumour volume is poorly correlated with HRQoL, and the selected utility values used within the model lacks validity. Clinical outcomes (i.e., symptom relief and pain management) most relevant to patients and clinical experts consulted by CADTH were not considered by the model. Furthermore, as direct comparative evidence on selumetinib compared to BSC alone is lacking, the model assumed that all patients on BSC would automatically enter the “progressed” state. This structural assumption would favour selumetinib. As this limitation could not be addressed by CADTH, any analyses performed by CADTH or the sponsor likely underestimated the true ICER. Given the clinical uncertainty and issues with the model structure, the CADTH reanalyses results should be viewed with extreme caution.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Koselugo (selumetinib), 10mg and 25mg capsules. Vaughan (ON): Alexion Pharmaceuticals, Inc.; 2022 Oct 28.
2.Notice of Compliance: Selumetinib (Koselugo). 2022; https://health-products.canada.ca/noc-ac/nocInfo?no=28862. Accessed 2022 Nov 11.
3.Koselugo (selumetinib): capsules, 10mg and 25mg, oral use [product monograph]. Mississauga (ON): AstraZeneca Canada, Inc.; 2022.
4.Gross AM, Wolters PL, Dombi E, et al. Selumetinib in Children with Inoperable Plexiform Neurofibromas. N Eng J Med. 2020;382(15):1430-1442. PubMed
5.Table: 13-10-0114-01. Life expectancy and other elements of the life table, Canada, all provinces except Prince Edward Island. Ottawa (ON): Statistics Canada; 2022: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011401. Accessed 2022 Nov 11.
6.Duong TA, Sbidian E, Valeyrie-Allanore L, et al. Mortality Associated with Neurofibromatosis 1: A Cohort Study of 1895 Patients in 1980-2006 in France. Orphanet Journal of Rare Diseases. 2011;6(1):18. PubMed
7.Guidelines for the economic evaluation of health technologies: Canada. 4th ed. Ottawa (ON): CADTH; 2017: https://www.cadth.ca/guidelines-economic-evaluation-health-technologies-canada-4th-edition. Accessed 2022 Nov 11.
8.Rowen D, Brazier J, Wong W, Wailoo A. Measuring and valuing health-related quality of life when sufficient EQ-5D data is not available. (NICE DSU Report). Sheffield (UK): Decision Support Unit, ScHARR, University of Sheffield; 2020: https://www.sheffield.ac.uk/nice-dsu/methods-development/measuring-health-related-quality-life. Accessed 2022 Nov 24.
9.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2022; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2022 Nov 24.
10.Government of Nova Scotia. Nova Scotia Pharmacare: Formulary. 2022; https://novascotia.ca/dhw/pharmacare/formulary.asp. Accessed 2022 Nov 30.
11.Yukon Territory. Yukon Drug Formulary. 2022; https://yukon.ca/en/health-and-wellness/medical-professionals/find-drug-coverage-information. Accessed 2022 Nov 30.
12.Ontario Case Costing Initiative (OCCI). Toronto (ON): Ontario Health and Long-Term Care; 2017: https://data.ontario.ca/dataset/ontario-case-costing-initiative-occi. Accessed 2022 Nov 24.
13.Hamoy-Jimenez G, Kim R, Suppiah S, Zadeh G, Bril V, Barnett C. Quality of life in patients with neurofibromatosis type 1 and 2 in Canada. Neuro-Oncol Adv. 2020;2(Supplement_1):i141-i149. PubMed
14.Copley-Merriman C, Yang X, Juniper M, Amin S, Yoo HK, Sen SS. Natural history and disease burden of neurofibromatosis type 1 with plexiform neurofibromas: a systematic literature review. Adolesc Health Med Ther. 2021:55-66. PubMed
15.Briggs A, Sculpher M, Claxton K. Decision modelling for health economic evaluation. Oxford (UK): Oxford University Press; 2006.
16.Alexion. OneSource: Personalized Patient Support from Alexion. 2022; https://alexiononesource.com/. Accessed 2022 Nov 24.
17.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Koselugo (selumetinib), 10mg and 25mg capsules. Vaughan (ON): Alexion Pharmaceuticals, Inc.; 2022 Oct 28.
18.Friedrich RE, Schmelzle R, Hartmann M, Fünsterer C, Mautner V-F. Resection of small plexiform neurofibromas in neurofibromatosis type 1 children. World J Surg Oncol. 2005;3(1):1-6. PubMed
19.Prada CE, Rangwala FA, Martin LJ, et al. Pediatric plexiform neurofibromas: impact on morbidity and mortality in neurofibromatosis type 1. J Pediatr. 2012;160(3):461-467. PubMed
20.Kallionpää RA, Uusitalo E, Leppävirta J, Pöyhönen M, Peltonen S, Peltonen J. Prevalence of neurofibromatosis type 1 in the Finnish population. Genet Med. 2018;20(9):1082-1086. PubMed
21.Lammert M, Friedman JM, Kluwe L, Mautner VF. Prevalence of neurofibromatosis 1 in German children at elementary school enrollment. Arch Dermatol. 2005;141(1):71-74. PubMed
22.Korf BR. Plexiform neurofibromas. Am J Med Genet. 1999;89(1):31-37. PubMed
23.Mautner V-F, Asuagbor FA, Dombi E, et al. Assessment of benign tumor burden by whole-body MRI in patients with neurofibromatosis 1. Neuro-oncol. 2008;10(4):593-598. PubMed
24.Solares I, Vinal D, Morales-Conejo M, Rodriguez-Salas N, Feliu J. Novel molecular targeted therapies for patients with neurofibromatosis type 1 with inoperable plexiform neurofibromas: a comprehensive review. ESMO Open. 2021;6(4) (no pagination).
25.Waggoner DJ, Towbin J, Gottesman G, Gutmann DH. Clinic‐based study of plexiform neurofibromas in neurofibromatosis 1. Am J Med Genet. 2000;92(2):132-135. PubMed
Appendix 1: Cost-Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s) and drug plans. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: Cost-Comparison Table for Pediatric Patients With Neurofibromatosis Type 1
Treatment | Strength | Form | Price | Recommended dosage | Daily cost | Annual cost |
|---|---|---|---|---|---|---|
Selumetinib (Koselugo) | 10mg | Capsule | $122.5998a | 25mg/m2 twice daily3 | $735.60 | $268,677 |
25mg | $306.4995a |
Note: Prices do not include dispensing fees or markups. Costs assume a body surface area of 1.12 m2 and 365.25 days per year.
aSponsor’s submitted price.1
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | See key limitations. The modelled outcomes were based on tumour volume only which is poorly correlated to quality of life. |
Model has been adequately programmed and has sufficient face validity | No | See key limitations. |
Model structure is adequate for decision problem | No | The model structure was inadequate to support decision-making. See key limitations. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | The model failed to incorporate all the available evidence to support decision-making in a context of uncertainty. See key limitation above. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | Limitations associated with the model structure meant that the model could not be flexibly modified to characterize parameter or structural uncertainty. See key limitation. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | No | Spreadsheet contained many erroneous cells which distorted the results. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
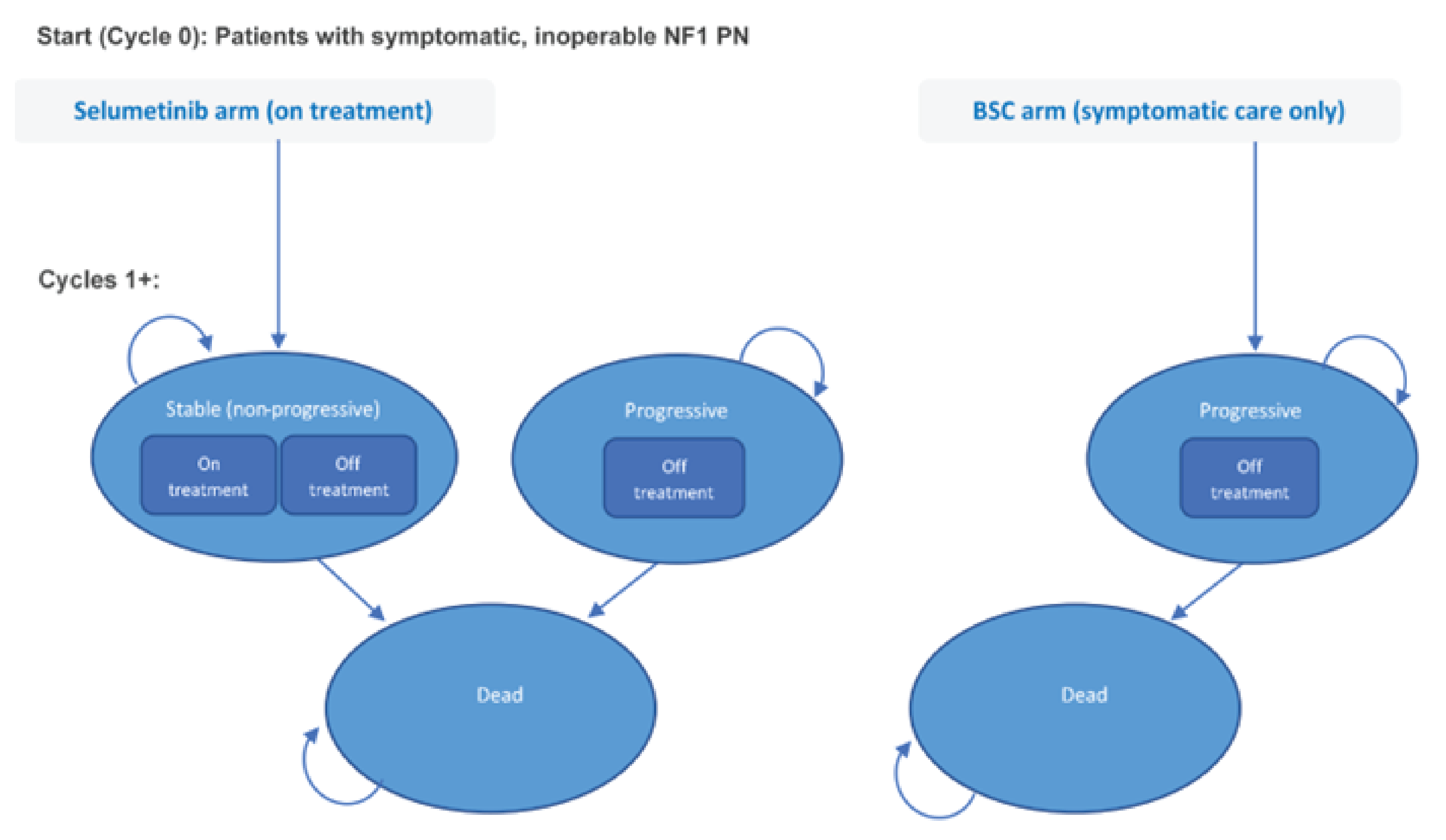
BSC = best supportive care; NF1 = neurofibromatosis type 1; PN = plexiform neurofibroma.
Source: Sponsor’s pharmacoeconomic submission.1
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Table 10: Disaggregated Summary of CADTH Reanalysis A (Deterministic)
Parameter | Selumetinib + BSC | BSC | Incremental |
|---|---|---|---|
Undiscounted LYs | |||
Total | 64.06 | 64.06 | 0.00 |
By health state or data source | |||
Progression-free | 22.19 | 0.00 | 22.19 |
Progressed | 41.87 | 64.06 | −22.19 |
Death | 0.00 | 0.00 | 0.00 |
Discounted QALYs | |||
Total | 21.15 | 18.39 | 2.76 |
By health state or data source | |||
Progression-free | 10.14 | 0.00 | 10.14 |
Progressed | 11.01 | 18.39 | −7.38 |
Death | 0.00 | 0.00 | 0.00 |
Discounted costs ($) | |||
Total | 1,191,437 | $14,413 | $1,177,024 |
Acquisition (primary treatment) | 1,174,823 | $0.00 | $1,174,823 |
Administration (on treatment) | 0.00 | $0.00 | $0.00 |
Concomitant medications (on treatment) | 3,443 | $0.00 | $3,443 |
Monitoring (on treatment) | 3,993 | $0.00 | $3,993 |
Adverse events (on treatment) | 584 | $0.00 | $584 |
Off treatment (postprogression) | 8,654 | $14,413 | −$5,759 |
ICER ($ per QALY) | 426,286 | ||
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year.
Table 11: Disaggregated Summary of CADTH Reanalysis B (Deterministic)
Parameter | Selumetinib + BSC | BSC | Incremental |
|---|---|---|---|
Undiscounted LYs | |||
Total | 64.06 | 64.06 | 0 |
By health state or data source | |||
Progression-free | 33.96 | 0.00 | 33.96 |
Progressed | 30.09 | 64.06 | −33.96 |
Death | 0.00 | 0.00 | 0.00 |
Discounted QALYs | |||
Total | 22.38 | 18.39 | 3.99 |
By health state or data source | |||
Progression-free | 15.10 | 0.00 | 15.10 |
Progressed | 7.28 | 18.39 | −11.11 |
Death | 0.00 | 0.00 | 0.00 |
Discounted costs ($) | |||
Total | 1,191,437 | $14,413 | $1,177,024 |
Acquisition (primary treatment) | 1,174,823 | $0.00 | $1,174,823 |
Administration (on treatment) | 0.00 | $0.00 | $0.00 |
Concomitant medications (on treatment) | 3,443 | $0.00 | $3,443 |
Monitoring (on treatment) | 3,993 | $0.00 | $3,993 |
Adverse events (on treatment) | 584 | $0.00 | $584 |
Off treatment (postprogression) | 8,654 | $14,413 | −$5,759 |
ICER ($ per QALY) | 294,751 | ||
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year.
Scenario Analyses
Table 12: Results From CADTH Scenario Analyses
Analysis | Drug | Total costs ($) | Total QALYs | ICER ($ per QALY) |
|---|---|---|---|---|
1.a. Lower Progressed Utility (Reanalysis A)a | BSC | 14,413 | 14.24 | Reference |
Selumetinib + BSC | 1,191,437 | 18.70 | 263,981 | |
1.b. Lower Progressed Utility (Reanalysis B)a | BSC | 14,413 | 14.24 | Reference |
Selumetinib + BSC | 1,191,437 | 20.76 | 180,526 | |
2.a. Higher Progressed Utility (Reanalysis A)a | BSC | 14,413 | 22.53 | Reference |
Selumetinib + BSC | 1,191,437 | 23.60 | 1,106,772 | |
2.b. Higher Progressed Utility (Reanalysis B)a | BSC | 14,413 | 22.53 | Reference |
Selumetinib + BSC | 1,191,437 | 24.00 | 802,552 |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: Deterministic results reported to facilitate comparison. Higher Progressed Utility = 0.625; Lower Progressed Utility = 0.395.
aDeterministic results reported.
Appendix 5: Submitted Budget Impact Analysis and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 13: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
The sponsor-submitted budget impact analysis (BIA) evaluated the introduction of selumetinib for the treatment of pediatric patients age 2 years and above with neurofibromatosis type 1 and symptomatic, inoperable PNs.17 Estimates were generated from the perspective of CADTH participating drug plans (all but Québec) and the results were aggregated into pan-Canadian totals over a 3-year time horizon. An epidemiological approach was used to estimate the eligible population size for the analysis and is summarized in Figure 2.17 New patients were added to the BIA based on jurisdictional specific population growth rates. Key inputs to the BIA are documented in Table 15.
In the reference scenario, patients were offered BSC. Consistent with the economic evaluation, BSC was defined as a range of topical treatments, analgesics, and other noncurative disease management strategies used to help control PN-related morbidities. In the new drug scenario, it was assumed that selumetinib would obtain market share quickly in the absence of any comparator products.17 As with the economic evaluation, the new drug incorporated concomitant therapies used to manage symptoms and treat AEs.1,17
Key assumptions:
The eligible population was restricted to patients between 2 and 18 years of age and a BSA greater than 0.55 m2.
It was assumed that 90%, 72%, and 53% of patients would remain on treatment after 12, 24, and 36 months of selumetinib treatment respectively. These estimates were obtained directly from the parametric survival function for TTD curves based on the sponsor’s base-case economic evaluation.1,17
A 90% compliance rate was applied to selumetinib treatment.17
Table 14: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as Year 1 / Year 2 / Year 3 if appropriate) |
|---|---|
Target population | |
Pan-Canadian population, aged 2 to 18 (excluding Québec) | 6,871,327 |
NF1 Prevalencea | 2,290 (0.03%) |
Proportion with PN18 | 687 (30%) |
Proportion of symptomatic PNs13 | 440 (64%) |
Inoperable PNb19 | 396 (90%) |
Actively Managedc | 336 (85%) |
Eligible for public coveraged | 336 (100%) |
Number of patients eligible for drug under review | 343/ 349/ 355 |
Market Uptake (3 years) | |
Uptake (reference scenario) | |
BSC | 100% / 100% / 100% |
Uptake (new drug scenario) | |
BSC | 60% / 35% / 15% |
Selumetinib + BSC | 40% / 65% / 85% |
Annual cost of treatment (per patient) | |
BSC | $354.75 |
Selumetinib + BSC | $283,604.04 |
BSC = best supportive care; NF1 = neurofibromatosis type 1; PN = plexiform neurofibroma.
aPrevalence rate of 1 in 3,000.17,20,21
bSponsor market research data on file.
cAssumed that 15% of patients who are not actively managed.17
dAssumption.17
Table 15: CADTH Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Correctionsa to sponsor’s base case | ||
Incorrect Calculation of Net Benefit | Reference Scenario + New Scenario | Reference Scenario – New Scenario |
Changes to derive the CADTH base case | ||
1. Proportion of patients with symptomatic PN | 64% | 32% |
CADTH base case | 1 | |
PN = plexiform neurofibroma; NF1 = neurofibromatosis type 1.
aCorrections are minor errors (e.g., transcription errors between report and model, misapplication of distributions or SEs in probabilistic analyses) that are not identified as limitations.
Summary of the Sponsor’s BIA Results
In the sponsor’s base case, the net annual budget impact of selumetinib for the treatment of pediatric patients with NF1 and symptomatic, inoperative PNs was estimated to be $31,689,517 in Year 1, $45,984,384 in Year 2, and $52,473,862 in Year 3. The 3-year net-budget impact of selumetinib was $130,386,454.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Over estimation of patients with PN: The sponsor assumed that 30% of NF1 patients will have a PN. Clinical experts consulted by CADTH were concerned that this may be an overestimation of what is seen in practice. However, CADTH was unable to verify this estimate given the limited epidemiological literature for the pediatric NF1 population. The budget impact is sensitive to this parameter given it would inform the expected number of patients eligible for this drug: if this parameter is expected to be smaller in the Canadian setting, a smaller budget impact would be expected; similarly, if this parameter value is larger, a larger budget impact would be expected.
CADTH explored this limitation by conducting 2 scenario analyses. One assumed 15% of NF1 patients would have a PN as per clinical expert feedback while the second scenario aligned with the CADTH Clinical Review in which identified several publications that reported 50% of NF1 patients had a PN.22-25
Ambiguous definition of symptomatic PN: It was assumed that 64% of pediatric NF1 patients would have symptomatic PN. This input to the BIA was obtained from a cross-sectional study of adults in Canada with NF1 and NF2 published in 2020.13,17 The extent to which this estimate is generalizable to the pediatric population is unclear. Consultation with clinical experts revealed that there is considerable heterogeneity with regards to the identification of patients with symptomatic PN. They further commented that the proportion of patients with symptomatic PN may have been over-estimated in the sponsor’s submission.
The impact of this assumption was explored in the CADTH reanalysis in the BIA. It was assumed that the prevalence of symptomatic PN was half the value considered in the sponsor’s base case (32%).
Treatment compliance: Of note, the sponsor claimed to have incorporated treatment compliance into the budget impact model and assumed that patients on selumetinib would have a compliance rate of 90%. This was justified based on the 94.13% adherence rate observed in the SPRINT trial. As noted above, adjusting treatment costs by compliance may not be reasonable given selumetinib is an oral medication and Canadian pharmacies are likely to fill and dispense the prescription in full. However, in further exploration of the sponsor’s submitted BIA model, CADTH noted that the sponsor failed to program this feature (i.e., treatment costs was not adjusted by compliance).
CADTH Reanalyses of the BIA
A summary of the CADTH stepwise reanalyses is presented in Table 16 and a more detailed breakdown is presented in Table 17. All CADTH reanalyses were based on publicly available prices of the comparator or background therapies. In the CADTH base-case reanalysis, the budget impact of selumetinib was estimated to be $64,702,506 over the 3-year period. This increase is attributable to the reduction in the market size as a result of changes related to the number of patients with symptomatic PN.
Several scenario analyses were conducted exploring alternate proportions of NF1 patients with PN and assuming different price reduction (i.e., 88.5% or 83.2%). CADTH notes that the budget impact estimates were highly sensitive to the epidemiological inputs to derive the proportion of patients eligible for treatment.
Table 16: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | 3-year total |
|---|---|
Submitted base case | $130,386,454 |
Sponsor corrected base case | $129,405,013 |
CADTH reanalysis 1/ base case | $64,702,506 |
Table 17: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 1 | Year 2 | Year 3 | 3-year total |
|---|---|---|---|---|---|
Submitted base case | Reference | $121,541 | $123,778 | $126,056 | $490,720 |
New drug | $31,567,975 | $45,860,606 | $52,347,806 | $129,895,733 | |
Budget impact | $31,689,517 | $45,984,384 | $52,473,862 | $130,386,454 | |
Sponsor corrected base case | Reference | $121,541 | $123,778 | $126,056 | $490,720 |
New drug | $31,567,975 | $45,860,606 | $52,347,806 | $129,895,733 | |
Budget Impact | $31,446,975 | $45,736,828 | $52,221,751 | $129,405,013 | |
CADTH base case | Reference | $60,771 | $61,889 | $63,028 | $245,360 |
New drug | $15,783,988 | $22,930,303 | $26,173,903 | $64,947,867 | |
Budget impact | $15,723,217 | $22,868,414 | $26,110,875 | $64,702,506 | |
CADTH scenario analysis: 15% of NF1 patients have a PN | Reference | $30,385 | $30,944 | $31,514 | $122,680 |
New drug | $7,891,994 | $11,465,152 | $13,086,952 | $32,473,933 | |
Budget impact | $7,861,608 | $11,434,207 | $13,055,438 | $32,351,253 | |
CADTH scenario analysis: 50% of NF1 patients have a PN | Reference | $101,285 | $103,148 | $105,046 | $408,934 |
New drug | $26,306,646 | $38,217,172 | $43,623,172 | $108,246,444 | |
Budget impact | $26,205,362 | $38,114,023 | $43,518,126 | $107,837,511 | |
CADTH scenario analysis: 88.5% price reduction (CADTH reanalysis A) | Reference | $60,771 | $61,889 | $63,028 | $245,360 |
New drug | $1,895,125 | $2,725,541 | $3,097,614 | $7,777,952 | |
Budget Impact | $1,834,354 | $2,663,653 | $3,034,586 | $7,532,592 | |
CADTH scenario analysis: 83.2% price reduction (CADTH reanalysis B) | Reference | $60,771 | $61,889 | $63,028 | $245,360 |
New drug | $2,726,887 | $3,935,544 | $4,479,584 | $11,201,687 | |
Budget Impact | $2,666,116 | $3,873,655 | $4,416,556 | $10,956,327 |
Ethics Review
Abbreviations
NF1
neurofibromatosis type 1
PN
plexiform neurofibroma
Summary
Patient group, clinician group, clinical expert, and drug program input gathered in the course of this CADTH review, as well as relevant published literature, were reviewed to identify ethical considerations relevant to the use of selumetinib for the treatment of pediatric patients aged 2 and older with neurofibromatosis type 1 (NF1) who have symptomatic, inoperable plexiform neurofibromas (PNs).
The current lack of disease-modifying treatment options for pediatric patients with NF1 who have symptomatic, inoperable PNs beyond surgical intervention leads to challenges in pediatric patient quality of life related to pain, motor function, cognition, and psychosocial functioning, and the potential for social stigma due to the appearance of PNs.
Several challenges arose in the evidence used to evaluate selumetinib, including the contrast between the study design of the pivotal SPRINT trial and clinical practice, and challenges related to the use of volumetric MRI to measure tumour size and response to treatment. As this drug is expected to be administered for life, questions remain about long-term effectiveness, safety, and risks to patients, given the absence of long-term data.
Given the lack of long-term effectiveness and safety data, the use of selumetinib raises ethical considerations related to long-term consent to a novel treatment for patients who are potentially incapable of providing consent, as well as their caregivers, who may have similar limitations. Access to appropriate NF1 and selumetinib expertise (e.g., pediatric neurooncologists and multidisciplinary care teams) also raises challenges, as such expertise is required to diagnose and treat NF1, and also to prescribe, monitor, and follow patients receiving selumetinib. This need for multidisciplinary and specialized care and monitoring raises the potential for equity challenges within the vast geographical disparities of Canada.
The use of selumetinib for patients with NF1 raises several health system and resource considerations relating to how selumetinib will be equitably delivered across Canada, as well as potential challenges in the treatment of patients who are asymptomatic.
Objective
To identify and describe the ethical considerations associated with the use of selumetinib for the treatment of neurofibromatosis type 1 (NF1) in pediatric patients who have symptomatic, inoperable plexiform neurofibromas (PNs), including those related to NF1, the evidentiary basis and use of selumetinib, and health systems.
Research Questions
What ethical considerations arise in the context of NF1 in pediatric patients and their caregivers?
What ethical considerations arise related to the evidence (e.g., clinical and economic data) used to evaluate selumetinib?
What ethical considerations arise in the use of selumetinib for clinicians, patients, and their caregivers?
What ethical considerations for health systems are involved in the context of selumetinib?
Methods
To identify ethical considerations relevant to the use of selumetinib in the treatment of NF1, this Ethics Review was driven by relevant questions identified in the Ethical Analysis domain of EUnetHTA Core Model 3.0,1 and supplemented by relevant questions from the Equity Checklist for HTA (ECHTA).2 These guiding questions were organized to respond to the research questions posed, and investigated ethical considerations related to:
patients living with NF1 and their caregivers (i.e., disparities in incidence, treatment, or outcomes; challenges related to diagnosis or clinical care; factors that might prevent patients from gaining access to therapies)
the evidence used to demonstrate the benefits, harms, and value of selumetinib (i.e., ethical considerations in relevant clinical trials, including their representativeness, choice of outcome measures, appropriateness of analytical methods, and models to all population groups; ethical considerations related to the data or assumptions in the economic evaluation)
the use of selumetinib, including considerations related to benefits and harms to patients, relatives, caregivers, clinicians, or society, and considerations related to accessing this therapy
the uptake of selumetinib in health systems, including considerations related to the distribution of health care resources.
Data Collection: Review of Project Inputs and Literature
Data to inform this Ethics Review drew from the identification of ethical considerations (e.g., values, norms, or implications related to the harms, benefits, and implications for equity, justice, resource allocation, and ethical considerations in the evidentiary basis) in the patient input, drug program input, clinician group input, and clinical expert input gathered during this review, as well as a complementary search of the published literature. Ongoing collaboration and communication with the CADTH reviewers working on the clinical and economic reports for this submission also assisted in the clarification and identification of ethical considerations raised.
Review of Project Inputs
Six main sources of inputs collected during this CADTH review were reviewed by a single reviewer to inform the Ethics Review:
The sponsor submission was reviewed for ethical considerations, noting relevant information and external references or sources relevant to each of the research questions driving this report.
Patient input received by CADTH, compiled by the Tumour Foundation of BC and the Canadian Organization for Rare Disorders (CORD), was reviewed for ethical considerations relevant to each of the research questions driving this report.
Drug program input, collected from provincial drug programs across Canada, was reviewed for ethical considerations relevant to each of the research questions driving this report.
Clinician group input, provided by the Canadian Pediatric Brain Tumour Consortium, was reviewed for ethical considerations relevant to each of the research questions driving this report.
Clinical experts (n = 6) were engaged by CADTH over the course of this reimbursement review for 2 teleconference discussions (3 clinical experts) and 1 panel discussion (6 clinical experts). These clinical experts were active in relevant clinical roles in Canada, all had experience treating or managing patients with NF1, and 2 of the experts had experience with patient(s) receiving selumetinib. During each of the 3 interactions with clinical experts, notes were taken on ethical considerations as they arose. As well, targeted questions corresponding to the research questions driving this Ethics Review were asked of the experts at each of these input calls.
Collaboration with the CADTH clinical and economic reviewers identified domains of ethical interest arising in their reviews.
Literature Search Methods
A literature search was conducted by an information specialist on key resources including MEDLINE via Ovid and Philosopher’s Index via Ovid. Duplicates were removed by manual deduplication in Endnote. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The search consisted of 2 main concepts to retrieve citations related to selumetinib (Koselugo) or to NF1 in pediatrics.
CADTH-developed search filters were applied to limit retrieval to citations related to empirical and normative ethical considerations. The initial search was completed on December 8, 2022, and limited to English-language documents published since January 1, 2002.
Literature Screening and Selection
The selection of relevant literature proceeded in 2 stages. In the first stage, the titles and abstracts of citations were screened for relevance by a single reviewer. Articles were retrieved for full-text review if they identified, or provided normative analysis (i.e., focusing on “what ought to be” through argumentation), or presented empirical research (i.e., focusing on “what is” through observation) of ethical considerations related to the incidence, diagnosis, treatment, or outcomes of NF1, or implications arising in the evidence used in the evaluation or use of selumetinib.
As a parallel process, other sources drawn from relevant bibliographies or in consultation with experts or other CADTH reviewers were retrieved and reviewed following the previously noted selection criteria.
Data Analysis
Data analysis for this Ethics Review included the collection, coding, and thematic analysis of data drawn from the literature and project inputs, driven by the 4 research questions guiding this report. The reviewer conducted 2 cycles of coding to abstract, identify, and synthesize relevant ethical considerations in the literature and from relevant project inputs.
In the initial coding phase, publications and input sources were reviewed for ethical content (e.g., claims related to potential harms, benefits, equity, justice, resource allocation, and ethical issues in the evidentiary basis). Once identified, claims related to ethical content were coded using methods of qualitative description.3 Initial descriptive coding of the reports focused broadly on categories concerning what ethical considerations were described. In the second coding phase, major themes and subcodes were identified through repeated readings of the data3 and summarized into the thematic categories within each domain or research question. Where ethical content emerged that did not fit into the categories or domains outlined in the research questions, this was noted.
Results
Description of Included Sources
Data to inform this Ethics Review drew from a review of the patient and clinician group input and drug program input, as well as consultation with the clinical experts engaged for this review. A description and summary of these sources are included in the Clinical Review.
A total of 168 citations were identified in the search of the published literature. Following screening of titles and abstracts, 90 citations were excluded and 78 potentially relevant publications from the electronic search were retrieved for full-text review. Of the potentially relevant publications, 67 publications were excluded as they did not discuss ethical considerations arising in the context of selumetinib for the treatment of NF1. In total, 11 publications met the inclusion criteria and were included in this report. In addition, 1 relevant publication was retrieved from other sources, including the grey literature search.
A total of 12 publications were used to inform this report. Ten publications examined ethical considerations related to the diagnosis, treatment, and outcomes of patients with NF1; and 2 publications examined ethical considerations related to selumetinib as a treatment for NF1. Details regarding the characteristics of the included publications are reported in Table 1.
Key Ethical Considerations
Disease Burden of NF1
NF1 is an incurable, complex genetic disorder with a prevalence of approximately 1 in 3,000 people. In Canada, there are approximately 12,000 people affected with NF1, according to patient input from the Tumour Foundation of BC. There is currently a lack of disease-modifying treatment options for pediatric patients with NF1 who have symptomatic PNs beyond surgical intervention and social supports.
PNs are the most common tumour type in NF1, affecting around half of patients. The development of PNs can occur in a wide variety of locations in or on the body with unpredictable manifestations such as tumour growth, skin freckling, or café au lait macules, bone dysplasia, scoliosis, ocular problems, and neurologic complications.4 The clinical experts indicated that PNs can ultimately reduce average life expectancy by about 8 years to 10 years. Depending on their location and size, PN can cause pain, disfigurement, motor function deficits, and, in the most severe cases, can be life-threatening due to compression of vital structures (e.g., great vessel compression, spinal cord compression, and airway obstruction).
The clinical experts identified that there is a trend for PNs to appear and grow rapidly in early childhood, from ages 6 to 8 years, before slowing down in adulthood.5 The Clinical Review notes that spontaneous shrinkage of PNs over time has been reported, but mainly in adults. The experts also emphasized that if tumours persist into adulthood, that there is concern for malignant transformation to malignant peripheral nerve sheath tumours, for which treatment options would differ, thereby emphasizing the importance of timely access to diagnosis.
Patient group input describes how the effects of PNs have a substantial and detrimental impact on quality of life, affecting emotional well-being, sleep, physical activity, social functioning, and mental health (e.g., anxiety, panic attacks, depression, suicidal ideation). Children and adolescents with moderate to severe complications of NF1, including the presence PNs, tend to have lower overall quality of life scores than those with mild or no complications. Indeed, PN growth is associated with increases in the number and severity of morbidities over time, and a corresponding decrease in quality of life, and children with more visible complications such as large PNs show worse quality of life scores.6
Diagnosis
Clinician group input and the clinical experts noted how the majority of patients with NF1 are diagnosed between the ages of 6 and 8. Patients with NF1 are diagnosed based on standard, well-established, and recently updated clinical diagnostic criteria that include symptoms related to café au lait macules, presence of neurofibromas (cutaneous and/or plexiform), and presence of NF1 in the patient’s family. Although the updated diagnostic criteria include a genetic testing component, the clinical experts and the clinician input noted that genetic screening and confirmation are generally not required for diagnosis, though genetic testing is widely available in most provinces. The experts also highlighted that results of genetic testing would not impact treatment decisions.
According to clinician group input and the clinical experts, diagnosis of PNs is typically made by either a neurooncologist or a pediatrician, with treatment initiated by the former. Equitable access to appropriate expertise for PN diagnostic assessment may thus be a challenge for patients and families in rural or remote areas as pediatric oncologists and neurooncologists or pediatric neurologists with an expertise in neurooncology almost all work exclusively in large academic pediatric hospitals located in urban centres in Canada. Timely access to appropriate expertise is an ethical and equity consideration due to the irreversible effect of PN growth on normal physical development as it impacts the patient in terms of motor function, pain, and quality of life; the potential for PNs to develop malignancy; and the fact that disparities in access to appropriate diagnosis may impact some groups more than others.
Current Treatment of NF1
The clinical experts noted that there are currently no established practice guidelines for the treatment of patients with NF1 with symptomatic, inoperable PNs. Currently, for patients with PNs, surgery aimed at debulking or excising tumours is the only treatment option available. The clinical experts noted that essentially all PNs are seen as inoperable as surgery is not curative. Surgical debulking of PNs is associated with significant risks based on many factors such as number, location, size, and vascularity of tumours, which may result in secondary injury, particularly for PNs that encase large arteries or nerves. Additionally, the experts cited that multiple invasive surgeries may be required, as tumours may regrow, or increase in size, thereby further increasing the risks to patients.
Aside from surgery, current management strategies consist of monitoring for patients with asymptomatic neurofibromas that may grow or become symptomatic and/or malignant. For patients with symptoms, interventions focus on those caused by individual PNs, including relieving pain, reducing functional impairment and discomfort, and improving overall quality of life, rather than providing disease-modifying or curative effects. For the improvement of psychosocial functioning, the literature suggests the use and further research of “mind-body interventions,” which are therapeutic programs that focus on helping patients with NF1 develop coping skills.7,8 The literature shows positive results when virtual programs can connect patients with NF1 via the internet to work on skills related to stress management, as well as physical and mental health-related mindfulness practices.8
Patient and Caregiver Experience and Vulnerability
All pediatric patients are inherently vulnerable due to their developing autonomy and decision-making capacity and must depend on adult guardians and clinicians for care and environmental navigation. Patients with NF1 can further be made vulnerable due to the need for physical and intellectual disability accommodations that may arise as a result of NF1. Patients with symptomatic NF1 can experience physical disability as a result of pain, lack of motor function, and cognitive deficits, which can affect travel and navigation. Due to these physical disabilities, patient group input described how patients with NF1 can be encumbered by canes, crutches, and wheelchairs in environments where disability accommodations are not prioritized. Based on the unpredictable disease progression and heterogeneity, patients with ophthalmologic considerations, for example, may experience loss of vision and may be further unable to navigate their environment without accommodation.
Some patients with NF1 can be further encumbered by the disease’s impact on cognition, which can affect their autonomy and educational capacity, as well as create an increased dependence on caregiver assistance.5,9 The clinical experts and clinician input shared how a high proportion of patients with NF1 experience learning disabilities, attention-deficit/hyperactivity disorder, and autism spectrum disorder, which can each impact how they navigate the education system, and later on, the work force.10 The lack of educational attainment can result in further marginalization through financial dependence.9
Beyond the physical and cognitive impact of PNs, patients can also experience adverse mental health effects and social stigma associated with the visible manifestation of PNs.11 Literature suggests that 1 of the most impactful factors to patients with NF1’s quality of life is the role of appearance.6 The clinical experts described how PNs on patients can be misunderstood to be a contagious, infectious disease. In the aftermath of COVID-19 and monkeypox concerns, the clinical experts shared how patients with NF1 with PNs were asked to leave certain areas for fear of contagion. School-age children with NF1 can experience considerable bullying and social stigma due to the physical manifestation of PNs,12 and patients with NF1 frequently disengage from social relationships.6,11 Patients described in both the patient group input and literature identified how the unpredictability of their future appearance causes significant distress and often needs to be managed with surgical care, which also poses risks to these patients.6 The clinical experts noted that cosmetic improvements are also likely important for a patient’s self-esteem; however, current surgical management for cosmetic removal of PN tumours is currently not easily funded by the provinces.
NF1 is an autosomal dominant disease, and while half of all cases are spontaneous mutations, approximately half of patients with NF1 patients may have parents with the disease, which can further limit a pediatric patient’s access to adequate care. Since the physical, cognitive, and behavioural symptoms of NF1 continue into adulthood, parents with NF1 can experience difficulty in caring for a child with NF1. The clinical experts discussed how travelling to care appointments, and communication with their child’s clinical team can require additional supports and resources that the family may not have as a result of disabilities caused by NF1, as well as related difficulties in sustaining employment.
Ethics of Evidence and Evaluation of Selumetinib
Clinical Trial Evidence
Several challenges and uncertainties arose in the evidence used to evaluate selumetinib, which is further detailed in the Clinical Review. The clinical experts noted some concern about the absence of long-term safety, as well as comparative effectiveness data, especially as selumetinib is expected to be administered for life, which raises questions about adverse effects and unknown risks to patients. The clinical experts also expressed concerns about the contrast between the study design of the SPRINT trial and actual clinical practice, as volumetric tests and measures of pain and motor functioning in the study differed from the current clinical practices for patients in Canada with NF1 with symptomatic, inoperable PNs.
The SPRINT study is an ongoing, phase II, open-label, single-arm, multicenter study consisting of 50 patients, aged 2 to 18 years of age with NF1 and symptomatic, inoperable PNs. At trial initiation, a placebo arm was deemed to be unethical, and the choice to conduct a single-arm trial was justified based on the proposed rarity of NF1-associated PNs and the lack of other treatment options. Indirect comparisons were therefore deemed necessary to estimate the relative benefit of selumetinib. As detailed further in the Clinical Review, the overall interpretation of the efficacy results from the SPRINT trial was deemed to be limited, given internal and external validity issues, as well as challenges in attributing the study results to treatment with selumetinib rather than the natural history of the disease or other interventions.
The SPRINT study used reduction in growth or shrinking of PNs as an end point, measured by conducting volumetric assessments via volumetric MRI. Yet, in clinical practice, the clinical experts noted that they are most concerned with severity of symptoms associated with PN size and location, rather than volume. Volumetric assessments may be a more objective unit of measure to study, while measures of pain and movement may be subjective. Yet, the clinical experts discussed how, in practice, emphasis is put on the severity of symptoms (i.e., pain and reduced function) experienced by patients rather than the volume of the PN tumours. The clinical experts consulted by CADTH and the clinician group input highlighted that the assessment of disease progression is multifaceted and relies heavily on clinical symptomatology in combination with imaging, as changes in PN size are difficult to determine using standard imaging practices, and changes in tumour size are not always reflective of changes in disease-related symptoms, and vice versa. The clinical experts asserted that the aim of treatment should be to reduce the severity of pain and symptoms. Though the SPRINT trial demonstrated that selumetinib may result in reductions in PN volume, some clinical uncertainties may persist about how and whether this correlated to changes in symptoms or function, given the heterogeneity of symptomatic, inoperable NF1 PNs.
Additionally, the clinical experts discussed how volumetric MRI assessments are not routinely conducted in Canada, first due to the clinical focus on symptom severity, and second, due to several barriers in the use of MRI (e.g., the potential need and risk of anesthetic in pediatric patients, and the cost to health system resources). Beyond cost considerations, there is limited to no availability of volumetric MRI beyond access via clinical trials in Canada. In response to drug program input, the clinical experts noted that access to volumetric MRI is not available as a standard of care for patients with NF1 in Canada and is overall limited worldwide. Given the lack of availability of access to volumetric MRI, measuring treatment response in alignment with the SPRINT trial is difficult, if not impossible, in the Canadian real-world setting.
Economic Analysis
The sponsor-submitted economic analysis faced challenges of modelling a disease such as NF1 due to NF1’s heterogeneity, and the design of the SPRINT trial. As is further detailed in the Pharmacoeconomic Review of this CADTH Reimbursement Review, issues concerning the validity of the model structure in the sponsor’s submitted analysis precluded CADTH from being able to conduct a thorough assessment. Most notable to the Ethics Review, and as previously discussed, the model did not capture clinical outcomes that are most relevant to patients (e.g., reductions in symptoms and improvement of motor function). As a result of these and several other limitations detailed in the Pharmacoeconomic Review, the results of economic models related to the use of selumetinib should be interpreted with caution.
Ethical Considerations in the Use of Selumetinib
Long-Term Consent
While guidelines exist for clinician use on the pediatric long-term consent process, the novelty of selumetinib in the NF1 context can pose questions about long-term informed consent. While the SPRINT trial identified adverse events related to gastrointestinal or dermatological effects, the long-term effects and impacts of these as well as other adverse events or harms remain uncertain. Informed consent for a proposed lifelong therapy requires the patient to understand and appreciate the consequences of accepting or refusing treatment, but in the context of NF1 and selumetinib, not only might patients have difficulty understanding the treatment proposal, but the proposal itself may be filled with several evidentiary uncertainties. If a patient is not capable of giving informed consent, a substitute decision-maker is asked to do so, and in the pediatric context, this duty usually falls to parents. As noted previously, due to the hereditary nature of NF1, many patients with NF1 may have parents with NF1, who may also be incapable of giving substituted informed consent for similar reasons as the patients themselves.
Access to Selumetinib
The clinical experts noted that in Canada, currently only neurooncologists are prescribing treatment with selumetinib through special or compassionate access programs, and have the infrastructure in place to handle these patients; however, they highlighted that with further insight and experience, pediatricians with experience treating NF1 could likely also prescribe this treatment. Given the heterogeneity in the disease, and the individualized approach to treatment, decisions often involve a multidisciplinary team of pediatricians, NF1 experts, neurooncologists, and nurse practitioners. This expertise and these resources are unlikely to be available across the country, and are usually limited to major urban centres in Canada. When considering equity of care access for patients with NF1, it is important to recognize that care for these patients may require additional resources, time, and expertise over a large geographic area.
The clinical experts discussed how for patients in remote areas, access to specialty clinics and multidisciplinary care teams may be a limiting factor, emphasizing that patients would be required to attend in-person appointments for treatment and imaging given the general requirement for sedation for younger children, as well as to assess safety. However, the experts considered the potential for remote monitoring, local bloodwork, or eye exams to be acceptable. Regardless, for follow-up in-person check-ups, proximity to a large health care institution is an important factor and may cause equity challenges.
As pediatric patients age into the adult patient cohort, they will likely require assistance in transitioning from care provided by a pediatrician or a pediatric neurooncologist to an adult neurooncologist and care team. When a young patient changes age cohorts, navigation into a new care team can be difficult; this is further amplified by the potential for cognitive difficulty in NF1 patients, caregiver challenges, and vast geographical access disparities in Canada.
Access to selumetinib itself may be limited for patients who are unable to swallow the oral drug. According to the sponsor submission and clinician group input, the inability to swallow the oral drug is a contraindication to treatment. In some patients, PNs can develop in the throat and restrict the ability to swallow. The development of other modes of drug administration can increase access for patients who may benefit from the drug but are unable to ingest it.
Health System Considerations
The use of selumetinib for patients with NF1 raises several health system and resource considerations, many of which are dependent on the costs of selumetinib and its implementation into health systems, including any other resources needed to implement and support this therapy. Questions also arise about health systems obligations to facilitate equitable access to novel therapies where there is substantial unmet need and where these therapies often require access to specialists or specialized treatment centres.
Additionally, while selumetinib is currently only indicated for patients with symptomatic, inoperable PNs, the potential for the use of genetic testing to identify patients who are asymptomatic may hold additional health system implications. The clinical experts raised questions related to the appropriateness of population screening for NF1. Currently, NF1 is not part of the Newborn Screening Program, nor is genetic screening for NF1 routinely accessed for newborns. However, some literature suggests that there may be some value to routine genetic screening to attain an earlier firm diagnosis.13 Routine screening is also suggested to help guide health policy with more accurate incidence data,13 which may have implications for decision-making around NF1 infrastructure and access, as well as patient social supports.
The use of population screening for NF1 may also identify patients who are asymptomatic, and the clinical experts noted their considerations regarding treatment options for these patients (before PNs become symptomatic) and discussed the potential to use selumetinib as a preventive measure. The potential for demand in access for patients who are asymptomatic and the associated expansion of the scope of the use of selumetinib to include patients who are asymptomatic is an important consideration for health systems. Though currently not part of the indication for selumetinib, the expansion to include patients who are asymptomatic would result in a much higher budget impact of selumetinib.
Limitations
The heterogeneity of NF1-associated PNs, in combination with the novelty of selumetinib and lack of other NF1-specific therapies available on the market, meant that the published literature that raised ethical considerations in this domain was limited. However, augmenting this limited literature with inputs from patient and clinician groups, drug programs, and the clinical experts collected in the course of this reimbursement review provided a more fulsome picture of ethical considerations in the context of selumetinib for the treatment of NF1.
Though this Ethics Review drew and extracted from patient and clinician group, clinical expert, and drug program inputs, it is possible that more directed engagement (such as direct interviews with patients, caregivers, or family members) on their specific experiences with selumetinib would have yielded more relevant domains of analysis.
Conclusion
NF1 is an incurable, complex genetic disorder with a prevalence of approximately 1 in 3,000 people. The lack of treatment options for pediatric patients with NF1 who have symptomatic, inoperable PNs beyond surgical intervention leads to challenges in pediatric patient quality of life related to pain, motor function, cognition, psychosocial functioning, and social stigma due to the appearance of PNs. NF1 treatment is currently focused on symptom management as there are no established treatment guidelines or disease-modifying therapies. All pediatric patients are inherently vulnerable due to their developing autonomy and decision-making capacity; however, patients with NF1 can be further made vulnerable by the need for physical and intellectual disability accommodations, and the caregiver challenges of parents who may also have NF1.
Several challenges arose in the evidence used to evaluate selumetinib, including the contrast between the study design in the pivotal clinical trial and clinically meaningful outcomes; the use of volumetric assessment to measure tumour size and response to treatment, including the lack of availability of volumetric MRI in Canada; and the lack of long-term data on efficacy and safety. Relatedly, ethical considerations in selumetinib treatment arise in challenges associated with long-term consent of a novel treatment, and access to appropriate NF1 and selumetinib expertise. Access to specialist and multidisciplinary care and monitoring due to geography or patient and caregiver functional impairments may be a limiting factor, and further challenges may arise in transitioning from pediatric to adult care, especially given the vast geographical disparities of Canada.
Selumetinib may pose the potential for addressing the underserved, vulnerable population of pediatric patients with NF1 with symptomatic, inoperable PNs who face severe and burdensome disease impacts. However, there are several equity challenges in the context of selumetinib for NF1, including those related to the challenges of accessing multidisciplinary and specialist care and monitoring, especially for an already vulnerable and encumbered population who may also have caregivers who lack capacity. As well, challenges related to clinical and patient decision-making without long-term safety and efficacy data may disproportionately affect this population due to the generational presence of NF1 and associated disease burden.
Table 1: Details of Included Publications
First author (year) | Publication type | Objective | Key ethical considerations | Funding source |
|---|---|---|---|---|
Barke (2014) | Exploratory qualitative interview study | To explore the day-to-day experience of young people living with NF1 in the UK, focusing on the role that appearance plays in this experience | Patient quality of life is affected considerably by the heterogeneity of NF1, the impact on social circle and relationships, and the trust necessary for circle of care. | Health and Life Sciences of the University of the West of England, Bristol |
Barke (2016) | Survey study | To research the psychosocial impact of the appearance changes associated with NF1 during adolescence | Appearance for patients with NF1 is extremely impactful to their quality of life. | None identified |
Chisholm (2018) | Systematic review and meta-analysis | To review findings from research into social function and ASD in children and adults with NF1 and integrate these findings with the Socio-Cognitive Integration Abilities Model (SOCIAL) | Children and adults with NF1 exhibit significantly higher prevalence and severity of social dysfunction, ADHD, and ASD symptomatology. | None identified |
Copley- Merriman (2021) | Systematic literature review | To identify data on the natural history, disease burden, and treatment patterns among patients diagnosed with NF1 and PNs, as well as to identify evidence gaps in these areas | The wide range of PN-related complications creates a substantial QOL burden for patients that includes pain, social functioning, physical function impact, stigma, and emotional distress. The severe burden of NF1 with PNs on the QOL of patients demonstrates the high unmet need for an effective treatment option that can reduce tumour burden and improve QOL. The heterogeneity of measurement tools used to evaluate QOL and the gap in data evaluating the health economic burden of PNs should be the focus of future research. | Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, and AstraZeneca |
Domon-Archambault (2018) | Literature review | To document the psychosocial features of NF1 and to report the interventions described to address the needs of pediatric patients with NF1 | There is a need to develop and assess psychosocial interventions for patients with NF1. | L’Association de Neurofibromatose du Quebec |
Geoffray (2021) | Correlational study | To determine the role of demographic and environmental factors such as age, sex, socioeconomic status, parental NF1 status, and neurologic complications on the cognitive, behavioural, and academic outcomes in NF1 | Cognitive and behavioural phenotypes emerge commonly via a complex interplay between genes and environmental factors, and this is true also of a monogenic condition such as NF1. Early interventions and remedial education may be targeted to risk groups such those with familial NF1, families with lower SES, and those with associated neurologic comorbidities. | None identified |
Holland (2019) | Survey study | To examine the rate (i.e., percentage of participants) and frequency of bullying victimization in a school-age sample of individuals with NF1 | Rates of bullying in NF1 are very high, which may be undervalued among adults and medical professionals, given the lack of research on bullying toward youth with NF1. School psychologists are uniquely positioned to implement programs and interventions to address the high rate of bullying toward the school-age NF1 population. | Texas Neurofibromatosis Foundation |
Lai (2017) | Survey study | To better conceptualize the experience of patients with PNs, this qualitative study sought to identify the most important treatment outcomes to assess from the perspective of patients, families, and clinicians | The most frequently reported concerns raised by patients across all age groups included pain, appearance and disfigurement, social activity and role participation, stigma, and anxiety. For parents, physical functioning was the primary concern, followed by pain, social activity and role participation, appearance and disfigurement, and social relationships. The resulting conceptual framework included 5 domains to represent the most important identified symptoms and/or concerns: pain, social functioning, physical function impact, stigma, and emotional distress. | Neurofibromatosis Therapeutic Acceleration Program |
Nutakki (2018) | Survey study | To report on the measurement properties of the PedsQL NF1 Module for pediatric patients aged 5 to 25 from the perspectives of patients and parents | The PedsQL NF1 Module scales demonstrated acceptable to excellent measurement properties, and may be used as standardized metrics to assess NF1-specific symptoms and problems in clinical research and practice in children, adolescents, and young adults. | Neurofibromatosis Therapeutic Acceleration Program |
Reichman (2020) | Randomized control trial protocol | To present the study design and protocol for the first RCT of a mind-body intervention for adolescents with NF1, resilient youth with NF1, vs. an educational control group | This study examined the clinical and public health implications for the psychosocial functioning of adolescents with NF1. It provides a model for efficient delivery of virtual psychosocial care for adolescents with rare diseases. | US Department of Defense |
Shahzad (2014) | Case report | Report of a rare occurrence of PNs in the genital system (uterus) | Implies significant impact on reproductive system and potential of patients with NF1. | None identified |
Tsang (2012) | Report | Discussing the value of genetic testing | There may be some value in routine genetic screening to attain earlier firm diagnosis. It may also be helpful to accurately identify incidence, to help guide health policy decision-making. | UBC Faculty of Medicine and Child and Family Research Institute |
ADHD = attention-deficit/hyperactivity disorder; ASD = autism spectrum disorder; NF1 = neurofibromatosis type 1; PedsQL = Pediatric Quality of Life Inventory; PN = plexiform neurofibroma; QOL = quality of life; RCT = randomized controlled trial; SES = socioeconomic status; UBC = University of British Columbia; vs. = versus.
References
1.European Network for Health Technology Assessment. EUnetHTA Joint Action 2 Work Package 8, HTA Core Model version 3.0 2016; https://www.htacoremodel.info/BrowseModel.aspx. Accessed 2023 Jan 13.
2.Benkhalti M, Espinoza M, Cookson R, Welch V, Tugwell P, Dagenais P. Development of a checklist to guide equity considerations in health technology assessment. Int J Technol Assess Health Care. 2021;37(1). PubMed
3.Sandelowski M. Whatever happened to qualitative description? Res Nurs Health. 2000;23(4):334-340. PubMed
4.Shahzad R, Younas F. Plexiform neurofibroma of uterus: a rare manifestation of neurofibromatosis 1. J Coll Physicians Surg Pak. 2014;24 Suppl 1:S22-23. PubMed
5.Copley-Merriman C, Yang X, Juniper M, Amin S, Yoo HK, Sen SS. Natural History and Disease Burden of Neurofibromatosis Type 1 with Plexiform Neurofibromas: A Systematic Literature Review. Adolesc Health Med Ther. 2021;12:55-66. PubMed
6.Barke J, Coad J, Harcourt D. The Role of Appearance in Adolescents' Experiences of Neurofibromatosis Type 1: A Survey of Young People and Parents. J Genet Couns. 2016;25(5):1054-1062. PubMed
7.Domon-Archambault V, Gagnon L, Benoit A, Perreault S. Psychosocial Features of Neurofibromatosis Type 1 in Children and Adolescents. J Child Neurol. 2018;33(3):225-232. PubMed
8.Reichman M, Riklin E, Macklin E, Vranceanu AM. Virtual mind-body treatment for adolescents with neurofibromatosis: Study protocol for a single-blind randomized controlled trial. Contemp Clin Trials. 2020;95:106078. PubMed
9.Geoffray MM, Robinson L, Ramamurthy K, et al. Predictors of cognitive, behavioural and academic difficulties in NF1. J Psychiatr Res. 2021;140:545-550. PubMed
10.Chisholm AK, Anderson VA, Pride NA, Malarbi S, North KN, Payne JM. Social Function and Autism Spectrum Disorder in Children and Adults with Neurofibromatosis Type 1: a Systematic Review and Meta-Analysis. Neuropsychol Rev. 2018;28(3):317-340. PubMed
11.Barke J, Harcourt D, Coad J. 'It's like a bag of pick and mix--you don't know what you are going to get': young people's experience of neurofibromatosis Type 1. J Adv Nurs. 2014;70(7):1594-1603. PubMed
12.Holland AA, Stavinoha PL, Swearer SM, Solesbee C, Patel S, Klesse LJ. Rate and frequency of bullying victimization in school-age children with neurofibromatosis type 1 (NF1). Sch Psychol. 2019;34(6):687-694. PubMed
13.Tsang E, Birch P, Friedman JM. Valuing gene testing in children with possible neurofibromatosis 1. Clin Genet. 2012;82(6):591-593. PubMed
Stakeholder Input
Patient Input
Tumour Foundation of BC
About the Tumour Foundation of BC
The Tumour Foundation of BC is a registered charitable organization. www.tumourfoundation.ca
The Tumour Foundation of BC has been providing essential information and support services for individuals with neurofibromatosis and their families for 38 years. The mission of the Tumour Foundation of BC is to improve the lives of individuals with NF. Our vision is optimize the health and well-being for all British Columbians affected by NF. Funded primarily by individual donations, fundraising events and a provincial grant, the organization offers a range of programs and services which include: a consultative virtual medical clinic, one-to-one support, community events, educational scholarships, vital resource publications, and an annual symposium that attracts specialists and attendees from around the world.
Neurofibromatosis (NF) encompasses a set of three distinct genetic disorders (NF Type1, NF Type 2, and Schwannomatosis) that share the manifestation of uncontrollable tumour growth.
In NF1, which is the most common form of NF, tumours develop along nerves throughout the body, and can affect the development of non-nervous tissues such as bones and skin. NF1 can cause additional complications such as disfigurement, bone deformities, learning disabilities, and cancer. NF2 is characterized by the development of benign tumours on the nerve that carries sound and balance information from the inner ear to the brain. These tumours affect both ears, often leading to partial or complete hearing loss. People with NF2 may also develop other types of benign brain or spinal tumours. Finally, Schwannomatosis causes the development of benign tumours — called schwannomas — usually on spinal and peripheral nerves. These tumours develop when Schwann cells, which form the insulating cover around nerve fibres, grow abnormally.
NF1 is considered a rare genetic disorder with an incidence of one in 2,500 to 3,000 births. (NF2 has an incidence rate of 1 in 25,000 and the rate for Schwannomatosis is 1 in 40,000). However, rare is a relative term – there are more than 1700 people in BC, over 12,000 in Canada and two million worldwide affected with this disorder. NF is more common than cystic fibrosis, Duchenne's muscular dystrophy and Huntington’s disease combined. Knowledge of NF within the community and the medical profession, however, falls well below that of less common disorders. As a result, the quality of healthcare available to adult NF patients in BC is severely lacking, highly inconsistent and dependent on the engagement of referring family doctors. There are no NF specialists serving adult patients in BC.
Information Gathering
The staff at the Tumour Foundation of BC sent out invitations to individuals with NF1, and NF1 and plexiform neurofibromas, to share experiences living with the disorder. Invitations were sent via the organization’s email newsletter and posted on various social media platforms. We sought the community’s opinion not only about their experience of living with neurofibromatosis but also on the value of having selumetinib (Koselugo) approved for use in Canada. Participants were invited to join a focus group and/or complete an online health experience survey.
Patients and caregivers affected by NF1 participated in a Zoom focus group in November 2022. The data submitted also reflects the input of 25 individuals who participated in the online health experience survey in November 2022. The respondents included adult patients with NF1 (32%), adult patients with NF1 and a plexiform neurofibroma (24%), and caregivers of a patient with NF1 (44%). 64% of the respondents identified as women and 32% identified as men. Age of the individuals who completed the survey and participated in the focus group ranged from 18 to over 65 and resided in communities across British Columbia.
We have included patients’ and caregivers’ quotes to ensure that the voice of those affected by neurofibromatosis are captured beyond numerical representation. A report sharing all patient comments is also available for review.
Disease Experience
In response to the survey and focus group questions on how NF1 impacts quality of life, five key themes were identified:
NF1 limits daily living and social activities
Patients with NF1 experience moderate to severe chronic pain
NF1 can result in a dependency on caregivers into adulthood
Families experience financial stress as a result of the NF1 diagnosis
Living with NF1, and limited treatment options, negatively impacts the emotional well-being of patients and families.
The quotes that follow from individuals affected by NF1 throughout British Columbia highlight the impact of the disease that goes well beyond the physical symptoms of tumour manifestation.
NF1 Limits Daily Living and Social Activities
“NF1 and a plexiform neurofibroma impacts all of life-continued severe impacts to bones: hip dislocation again; increased scoliosis; life-changing mobility issues already, so fear of being bed ridden; dependent on others already for everyday tasks, so fear of not being able to do anything for oneself; and utter decline in physical and mental health.”
“I have many symptoms with a plexiform tumour on my ankle (which has been waiting for surgery in Vancouver for years now). Shaking and tremors in my hands, Migraines, Temporal lobe seizures (focal impaired) causing emotion swings, memory loss and odd behaviours, Major anxiety, and sleep problems.”
“Being the only one in the family [NF1] affects me greatly. Lately there is major weakness in my legs and bad back pain near the bottom of my spine.”
“Our son has an optical glioma, so he has limited vision. So, for him leaving the house is an issue. He's supposed to use an awareness cane so that people can tell that he can't necessarily see their vehicle coming. He won't use it, he's a 20-year-old young man so, when he leaves the house, there are safety issues certainly for crossing streets.”
“NF has impaired his ability to have more friends, his ability to study and advance more quickly in education.”
“Tumours are not the only effect NF1 has on me. I also have tibial dysplasia of my right leg, which bothers me every day. I also have vascular problems because of NF1. I also had a kidney removed because of renal artery stenosis. This kidney resulted in malignant hypertension.”
“NF impacts us tremendously. My son was just in hospital for five months with continuous hip dislocation.”
“I worry about it [plexiform tumour] growing and affecting my son’s mobility. That is my biggest concern.”
“He has an extreme fear of “standing out” in a crowd, so won’t use his awareness cane.”
“He used to walk without aid and now he has to use crutches, walker and wheelchair. The doctor does not know when or how to lift these restrictions and have asked us to decide for ourselves…”
“He spends so much of his life focused on what he can’t do and on how NF limits him (poor fine motor skills, learning disabilities affected his high school experience and negatively affected his perspective on higher education).”
“NF has given my son more challenges and hardship than any 17-year-old should ever have to endure both physically and mentally, and the most unfortunate thing is, I am not sure it is going to be any easier as time goes on. In fact, he has some serious bone issues with some serious decisions ahead.”
“There is a leg length discrepancy, there's scoliosis as well, and it's all in where the tumours are and it's all impacting those bone issues and we are continuing to have appointments.”
“After our son had two major surgeries to correct scoliosis caused by NF tumours, and a year in recovery where he had to be extremely careful and limit his physical activities (he wasn’t allowed to go out on the playground, etc.), he told us that experience made him very aware of the potential effects of NF in the future and how it could shorten his life.”
“Our son was bed-ridden, barely speaking and could not move or do anything for himself, so we could not leave him on his own.”
Patients with NF1 Experience Moderate to Severe Chronic Pain
96% of survey respondents live with chronic pain which was rated 5 or greater on the 0-10 pain scale.
“The pain feels likes I am being stabbed.”
“The pain is 8/10 on bad days…”
“NF1 disease is often overlooked as it not always presents visually but internally there is a lot of pain.”
“People don't seem to understand the impact of NF1. They question your pain as you may look healthy.”
“Gabapentin was the only thing offered for pain.”
“...would just like to feel normal, be pain free.”
NF1 Can Result in a Dependency on Caregivers into Adulthood
“My daughter has suffered 47 years and we are 74 years old and still need to take care of her.”
“I am 45 years old…my parents are still involved a lot to help me, like doctors, dentist, hair dressing, nail cutting appointments because I don't understand what the doctor or dentist convey to me. I cannot cook because I am only one handed and I don't see well.”
“He cannot fix a meal for himself, cannot bring a bowl of soup to the table for example. We help with dressing, showering, etc. We are his carers. We do not leave him at home by himself for long; I have had his brother miss school to sit in with him.”
“My daughter is 40 now but as far as I'm concerned, I still am a caregiver in that she couldn't possibly support this treatment herself.”
“… I am saddened when I see my son's face drop as we start conversations about how he will have to learn how to start to manage his own care as he becomes independent now that he's an adult….we witness his efforts to make appointments and organize what should be his absolute prime years of independence and care-free fun times so that instead of that he's setting up appointments for tests and check-ups that no one else cares to help him with if we do not.”
“If our son didn't have NF I would probably retire early, being in a position to do so, and enjoy travel and hobbies that I will put off for many, many years or not get to do due to a deep need to provide for him in case his health deteriorates and he can't take care of himself in what should be for him his productive middle years.”
“If our son were a typical child, he would likely be living independently with friends, in 3rd year university, thinking about career plans. My husband and I would be thinking about retirement in a year or two, possibly relocating to a smaller town with less expensive real estate. Instead [our son] is at home with us, taking a single course in his first year at a local college. We have to consider the possibility of him facing life-threatening health challenges and needing our financial help in the future and needing to stay in a large centre with access to good medical care.”
“One of my daughters may not be able to navigate her future needs without my help. As a mom, this is very scary to me because she may miss screenings, forget about annual check-ups, or not follow-up on problems quickly enough. If I am incapacitated, she will not be as effective in dealing with these medical appointments and as a result, she may be at heightened risk for cancer, depression, anxiety, mortality.”
Families Experience Financial Stress as a Result of the NF1 Diagnosis
92% of patients with NF1 incur expenses related to the care of their NF (such as prescription or non-prescription drugs, medical equipment, physiotherapy, counselling, or travel for medical care). 40% of respondents indicated they completely fund their own medical expenses without any public or private benefits.
“I would say I probably spent fifty thousand dollars over my daughter's care. Had I not been able to financially help her find treatment early on, I don't think she would have survived.”
“I researched everything I could find on NF hoping there would be a way to treat the condition. We grew very frustrated with the medical system and found the only way to seek any kind of treatment that would give some hope was by going to private clinics.”
“We have spent money over the course of our NF journey. Two trips to the US for care make up the bulk of that expenditure but it seemed the only way to be able to obtain access to doctors with experience and willingness to help. It has affected us because these are our savings that are being spent and we make lifestyle choices and changes because of it. It is frustrating to have to spend one's savings in this manner and makes me angry that our medical system hasn't supported treatments for NF1.”
“There are no options for complex plexiform neurofibromas. Many with NF cannot afford to self-fund expensive drug therapies if those become available.”
“Without my financial help she would not have had the emotional strength or financial means to obtain the treatments that have improved her physical appearance. Again, the main feeling of this is frustration at our medical systems lack of support for NF patients.”
“I get angry that we live in an immensely rich country in the most medically advanced time in human history, we spend billions on legitimate health concerns that have far less impacts on people's lives, and can't find the resources to adequately care for citizens of this country... I don't get paid to assist my son when my province drops the ball on funding adequate case management. We were told we can't claim tax deductions for caring for someone who is disabled because he's not disabled enough.”
“Because I may be gone when our son is middle-aged, I cannot take that chance and need to build up a nest egg to leave to him…. And in so doing I am sacrificing quality of life for myself now. My stress and workload affect my happiness in my family life, but I feel very strongly compelled to carry on doing what I can for a future I cannot know and may not see myself.”
“Non-stop juggling and constantly going - I used up all my holidays from work, got two weeks sick leave from the GP, then I had to return to work along with sharing shifts at hospital with husband. Everything was on hold - had to cancel husband's eye surgery date. He had been waiting for almost two years.”
“When we went to the US for our care it was disruptive and tiring, but we had a very experienced surgeon who has since retired: Always had to plan for a successful trip, taking time off work, missing school and having to catch up, money for expenses.”
Living with NF1, and Limited Treatment Options, Negatively Impacts the Emotional Well-being of Patients and Families
“It’s just too much to face every day.”
“Depression from NF1 led to a suicide plan and alcoholism.”
“It was dreadful and devastating for all when our son was in hospital for months. We were consumed by stress, anxiety, anger and exhaustion (and if I am honest, I am still all those things at random because there is a huge amount to consider and put in place which takes all my time and money).”
“I live with anxiety, depression, isolation, complete dependency, debilitating physical and mental health, the anger, no peace of mind, no joy or quality of life.”
“NF is a very scary illness. Effects cannot be obvious to others. They therefore do not understand your struggles.”
“We do not have a clinical coordinator and because of that we feel this has resulted in numerous errors and increased stress in our family.”
“This is not a ‘cosmetic’ condition. It really affects our mental health, relationships, etc., and it can be debilitating. The mental issues may be combatted as often patients feel like they have no hope for this progressive disease.”
“NF has taken away normalcy.”
“I think the unknown has always been a challenge for me. Because you don't know everybody is so different. It's so random and there's no there's no rhyme or reason to NF. There's no sort of like timeline of what to expect. I worry about a lot of things just because it is so unpredictable and random it's hard not to let your head go there and then you kind of worry you know how they are going to look after themselves?”
“I live with constant worry and anxiety.”
“I live in constant fear that I will miss the date to call a specialist to make a follow-up appointment and that will result in a delay in seeing the correct specialist or getting the correct diagnostic test. I fear that will result in a drastic negative outcome for our son’s health. I am aware of the potential for NF tumours to sometimes change and become cancerous. The NF community is small, and it feels like I am always hearing of the death of someone we met at a previous NF symposium, or of a former Tumour Foundation board member.”
“I worry terribly that he may have a very difficult end time in his life as we don't know what the ever-growing tumours may bring for pain or disability including motor function, vision loss, mental impairment, breathing or swallowing difficulties…”
“I live in fear of one of the effects of NF’s potential for creating serious consequences for our son’s life. Case management for a person with a complex medical condition is a tremendous burden that I am not equipped to handle, particularly as I struggle with symptoms of ADHD.”
“I've been in therapy and that has helped me somewhat to cope but I still sometimes have waves of panic related to these worries and feeling of uncertainty and my son's NF has definitely affected my enjoyment of my own life.”
“NF1 is a source of great sadness for me and creates a lot of anxiety. I sometimes have bad dreams about him being in pain or being lonely, or him just being sad and I worry in my waking life quite often...”
“I get dizzy spells and vertigo from the pressure of keeping everything organized and in order. A huge amount of my time is, and has always been, spent researching, trying to understand, asking, emailing, organizing, liaising, deciding, planning, making provisions, filling in forms….and not enough time for breaks and enjoying life.”
“NF is a monkey on your back that will never get off. There is constant anxiety of what is coming next, will there be someone willing to help, will I be able to continue to financially help, what if my daughter gives up hope? There is no sense of well-being, it is a constant concern.”
Experiences With Currently Available Treatments
46% of patients with NF1 and plexiform neurofibromas who responded to the survey were never presented with a treatment option. 32% of individuals with NF1 have never had treatment options presented. Of those who have received treatment only 17% experienced minimal improvement in their symptoms.
“Our doctor has never suggested anything to help neurofibromatosis patients.”
“When the plexiform neurofibromas were diagnosed, we were told surgery likely wasn’t an option because they are close to the spine, but at the time, no other options were offered. I was stunned that we were being told about a potentially serious problem and being offered zero solutions.”
“There were no answers and only failed procedures and operations (6 failed ones in total) until the last operation (#7) before the bone would stay in.”
“No one is checking on his NF, nobody’s checking if an intervention should be happening now... It’s all on us the parents or him to say there is a problem. No one is ordering regular scans or mapping the plexiform tumour or whatever that’s on us to manage. And it seems off to me because if somebody had cancer you know there would oncologists that would be sort of tracking all the time.”
“My son has a plexiform tumour the size of a dinner plate on his back, and nobody ever wants to look at it unless we go, ‘hey, check this out’. It’s bizarre to me.”
“Made my own edibles…no other options...”
Improved Outcomes
Improvements in new therapies that patients and caregivers would like to see are treatments, which would:
improve quality of life
decrease pain
increase functionality
reduce the number of health care visits.
“Anything that would improve patients’ lives physically, mentally and socially.”
“Koselugo is the only approved drug treatment in Canada for NF1 in children. There have been many studies that have shown drastic reduction in tumour size resulting in better quality of life. Patients are able to re-enter the workforce, pain drastically reduced, mobility increased, no longer requiring surgery when surgery was the only other option.”
“If tumours were shrunken...it would mean less visits to health care practitioners. There would be proper control over the disease and not intervening with archaic management of the disease (surgeries, off label chemo), which are costly and burdensome to the Canadian health care system.”
“…an oral treatment option would assist in reducing the number visits to a multitude of health care practitioners. For example, in 6 months we are visiting: (1) Ophthalmologist (2) Oncologist (3) NF specialist (4) Neurologist (5) Occupational therapists (6) Speech Therapists (7) Physiotherapists (8) Psychologists (7) Cardiologist (8) CT scans/MRIs (9) Spinal Surgeon (10) Neurosurgeon (11) Oral Surgeon (12) Special Orthodontist at Rehab Hospital.”
“There is always the fear of the unknown and the hope of new research/discoveries and that someday I will see treatments that can improve the quality of life and if not in my lifetime hopefully for future generations.”
Experience With Drug Under Review
Not one of the individuals who participated in the survey, or the focus group has been offered selumetinib as a treatment option for their plexiform neurofibromas. However, 100% of individuals indicated that they would consider taking selumetinib if given the opportunity to access it.
One caregiver in the focus group shared that they were aware of the benefits of selumetinib and had attempted to access it for their child who lives with multiple plexiform neurofibromas. However, they were informed they had to try a less effective drug first.
“A neurologist was ready to do the paperwork for us to trial selumetinib. But then she said that the government had a change of heart and or change in process and said that we had to jump over and try trametinib first. They said that the government wasn't going to fund selumetinib because that they didn't want to fork out all that money. I was in shock. Exhausted and defeated, that I am constantly at the mercy of others for help.”
“There aren't any choices. So, let's pay for the one thing that is an option.”
“…we're not asking for the Cadillac of drugs; we're asking for a drug.”
“…shouldn't have to suffer for [a drug].”
“… to hear there is a potentially very effective solution is truly the only positive news we have heard in nearly 20 years.”
Companion Diagnostic Test
There are no comments to add to 7. Companion Diagnostic Test, as BC residents with NF1 and plexiform neurofibromas are not accessing selumetinib at the time of this submission.
Anything Else?
“When there is no other solution, the hope and possibility of success must trump the possibility of side effects.”
“We need treatment options for these patients now.”
“…NF1 disease is often overlooked as it not always presents visually but internally there is a lot of pain and mental issues encountered. …patients feel like they have no hope for this progressive disease.”
“Having Koselugo accessible to Canadian NF1 children with the government support would assist with giving those hope where hope was not previously possible.”
“My son's NF has made my outlook bleaker, my own relationship with the world around me poorer, and I am forever heartbroken for him.”
“Canada should be at the forefront of providing effective new drugs to those with no other treatment options, rather than not providing and having NF1 patients continue to cycle in and out of the health care system only managing symptoms. This would be a waste of money and time on the health care system to continue managing symptoms rather than effectively treating with Koselugo.”
“It is very important to understand that there are many faces of NF. Each individual has their own unique story and is living with the condition daily that affects many different layers. Approving this drug will change lives today, tomorrow and for future generations. Looking at the NF population as a group that has gone through a lot and continues to suffer. The time has come to make the decision in favour of moving ahead to publicly fund the drug.”
“Patient care in our NF population has been affected for too long with no treatment solution of any sort. Personally, my son is only 17 years of age - this drug would give him a fighting chance.”
“I was disappointed that the GP who I had been seeing for many years would not take me back [after his leave] and made excuses. He was well aware of my condition and when was seeing me took interest and knew how to recommend/refer to the appropriate specialists and for radiology follow-up. Finding a new GP was difficult...”
“It is far less cost to the Canadian taxpayer to prevent the disfiguring and disabling symptoms than dealing with the resulting loss of employment, emotional toll on entire families of the affected patients, hospitalizations and loss of quality of life. This is the first proven drug to slow and reduce tumours leading to better health outcomes rather than doing nothing and waiting for the patient to be disfigured, disabled, and require extensive medical supports.”
“It is very unfortunate when a medical professional tells a patient that he/she has a cosmetic disorder, not fully understanding what the individual goes through and will continue to. It comes down to the training in medical schools and how much time and investment a GP wants to spend on a patient diagnosed with NF. Many times, its due to the knowledge gap of fully understanding the condition.”
“For anyone, having a child changes their life. But having a child with a rare disease changes it further.”
“Knowing there is a potential drug available to treat one of the effects of NF that my son lives with is a huge weight off.”
“To hear that there is a possibility the drug may not be funded is mindboggling; how could this hope be offered, then for families without the means, for that hope to be dashed is simply cruel.”
“…in Canada there could be there could be thousands of people who should access this drug because taking it now will prevent, from the government's perspective, further expensive problems down the line, from the patient's perspective you know life issues...”
“The lack of care shown to sufferers of NF by a massive industry of health care in this province and this country is appalling. It's shameful and enrages me when my tax bill comes along and I read political messages received around election time telling me all the efforts being made and money being spent on making sure ‘no Canadian gets left behind’ and ‘health care for everyone regardless of where they live.”
“Every occurrence of NF is individual, which means a diagnosis for your child means spending a life feeling like you’re standing with your child on the edge of a precipice with your toes hanging just off the edge. What you say out loud to your friends, family and to your child is, ‘Lots of people with NF go through life almost completely unaffected’. But your inner voice says, ‘And some die of cancer in their 20s and 30s. And some are crippled or terribly disfigured by tumour growth’. My emotional and mental health have been negatively affected; I live with anxiety and depression, and I carry a constant concern for our son’s future.”
“…if the government is just interested in watching the bottom line this is kind of like putting on your seat belt instead of waiting for the car to crash.. if it's just about money, it's cheaper to prevent a problem than to try to fix it
Conflict of Interest Declaration — Tumour Foundation of BC
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
None.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
None.
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
Table 1: Financial Disclosures for Tumour Foundation of BC
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Alexion | X | — | — | — |
Note: $4,991 in support of educational initiative; 2022 NF symposium.
Canadian Organization for Rare Disorders
About Canadian Organization for Rare Disorders
The Canadian Organization for Rare Disorders (CORD) is Canada’s national network for organizations representing all those with rare disorders. CORD provides a strong common voice to advocate for health policy and a healthcare system that works for those with rare disorders. The Canadian Organization for Rare Disorders works with governments, researchers, clinicians and industry to promote research, diagnosis, treatment and services for all rare disorders in Canada. Website: www.raredisorders.ca
Information Gathering
This submission is intended as a supplement to the submission from the Tumour Foundation of British Colombia which has conducted a separate survey to all members. This submission draws upon the experiences of patients and their parents who have received or are seeking to access the drug under assessment, selumetinib.
Recruitment: Participants were recruited through one physician in Toronto, Canada (Toronto Hospital for Sick Children) who has been conducting clinical trials. CORD provided an interview protocol and online survey which the physician used to reached out to clinical trial participants to request their agreement to be interviewed and/or to be provided with the survey. Patients were provided with no compensation or any other form of inducement to participate. Three patients provided contact information to receive link to the survey and agreement to be interviewed. In addition, one participant reached out to other NF1 parents (with and without clinical trial experience to complete the online survey.
The survey consisted of open-ended questions about the patient/family’s experience of living with NF1, experience with therapeutic interventions (including drug treatments), and awareness of and experience with the submitted drug (selumetinib).
Respondents: There were 8 parents who completed the online survey, and five who also took part in an interview; each interview lasted approximately 20 minutes. All parents and patients resided in Ontario, Canada. All of the patients represented were under 18 years of age, with five in age range of 2 to 10 years and three who are currently “teenagers.” Seven patients were female and one male. All patients represented were diagnosed with Neurofibromatosis Type 1 (NF1).
Disease Experience
“He has attempted suicide twice due to the anger and hate he has for the disease and how it interferes with his life. He was so sad to hear that over the years it was impossible to have the tumour removed. Since he was little (now a teenager) he continues to request a leg amputation - the tumour has caused so much physical, emotional, social, and personal trauma…”
Why could having Neurofibromatosis Type 1 drive a teenager to suicide? And cause families to separate? NF1 is genetic but in many families, there is no known family history of the disease.
Delayed and challenging road to a diagnosis:
All parents interviewed recounted seeing, often in infancy or early childhood, distinctive “café au lait” pigmentation or spots; these are common “early signs” of NF1 but the pathways to diagnosis varied considerably.
In only one case did the pediatrician recommend a genetic test upon presentation of the telltale spots (which also occur in general population in the absence of NF1). However, the family chose not to wait for diagnosis through the Canadian healthcare system. “The process of getting her test done and results had a window of 6-7 months which we found unacceptable. We decided to send her blood to the United States and pay out of pocket. It was a stressful process but allowed us to get her diagnosis of NF1 (gene deletion not a mutation) within a few weeks.’
Another family also chose the USA-route, noting the impact that a delayed diagnosis had an impact not only on the family but on the unnecessary use of health resources. “Our family made the choice to coordinate in 2015 with Canadian doctors and SickKids to collect blood from our 2-week-old infant and pay out of pocket to fly a blood sample for genetic testing in the United States of America (Lab in Mass, USA). The turnaround time for the results was 2 weeks and we have a confirmed diagnosis at 1 month of age. The Canadian medical system was reporting at the time that it would take up to 6 months for this genetic testing. The 6-month TAT for genetic testing is long and testing not readily supported by the Canadian Medical system.”
One family self-diagnosed based on family history of NF1. “When my son was 13 months old, we noticed his left foot/heel seemed swollen. MRI was ordered and plexiform neurofibroma was identified. We were certain of the diagnosis due to his father who has NF1 and his older brother also NF1.”
Several other families did not get genetic testing until the child had developed additional symptoms, those bumps or tumours, called neurofibromas, forming on or under the surface of the skin. “As soon as the doctor gave us the diagnosis, I immediately knew he was right, so many things that had never made sense about my daughter all of a sudden did. We found out when she was about four years old.”
“Since my daughter has a mutated form of NF #1, it took 3 years and a hospital in Boston USA to finally diagnose her.”
“Consideration should be given as to the long-term financial impact of not having a proper genetic diagnosis and balancing the number of doctors' visits and surgeries on the Canadian medical system.”
NF1 has serious impact on life of the child
Some neurofibromas are benign and have no medical consequences. However, all parents reported, post diagnosis, significant impact of the disease on the child and also on the whole family worsening as the child grows older and the disease progresses.
“NF is a horrible disease. although the degree of impact on one's life can vary drastically, it affects not only the person with it but every aspect of your life, your family's, your future, and mental health, career and lifestyle.”
“The plexiform has caused a loss of teeth on the side of her mouth, she also had a tumour in her ear which caused about 10 ear infections a year, and a loss of hearing.”
Children have endured many interventions including multiple surgeries and chemotherapy, often starting from a very young age.
“My daughter did two years of chemotherapy when she was in kindergarten and grade one, during that time She was always sick and would have extended stays at the hospital because a flu or a cold would be very serious.
Parents reported that tumours on the face interfere with the child’s speech, ability to eat, and can result in loss of hearing and balance. Neurofibromas at the base of the skull and others growing around the nerves (called plexiform neurofibromas) can have a tremendous physical and psychological impact (loss of short-term memory, social shyness, and anxiety.
“My daughter who is now 7 has had two major surgeries, countless appointments, many MRIs and learning difficulties. She is very strong but has been going through things no child should have to.”
NF1 has serious impact on the family
All parents spoke about the significant impact of NF1 on the entire family, including the time and attention required for medical appointments, for extra schooling, social, and psychological support, and the financial strain.
“It has affected us as a family because my daughter is on the spectrum so socially that can be difficult, she also has some learning disabilities, so she requires a lot of help with her schoolwork.”
“Socially and psychologically, our family has reached out to mental health professionals for assistance on how to support our family through a difficult diagnosis and how to cope with a child who has learning disabilities. We have made a personal choice sacrificing many significant financial burdens (out-of-pocket expenses) in order to intervene early (early detection) such as: (1) Full psychological work up which detected significant multiple learning disabilities (2) Speech Therapy (5) Occupational Therapy (4) Physical Therapy (4) Private School (to support learning disability). Financially, our family has had to leave the work force temporarily (leave of absences) resign from the workforce to support surgeries, general health, and learning issues.”
Experiences With Currently Available Treatments
All parents reported that their children had experienced multiple treatment interventions, including surgeries, chemotherapy, speech therapy, and psychological support. Importantly, all parents also spoke about the horrific negative toll on the child (and family) of undergoing multiple surgeries, often with limited and/or short-term benefits.
“Chiari decompression surgery, furlow z plasty oral surgery to help with hyper nasal speech, speech therapy, scoliosis support.”
“Chiari 2 Malformation: underwent necessary neurosurgery to stop progression of syrinx, scolosis, and other serious complications.”
Speech issues (hypernasality): weekly speech therapy at rehab hospital to support speech issues; VPI surgery to support correcting speech issues.
“… although the results and reaction of the PF could never be fully guaranteed or for how long it would help him. We got to a point where as parents, we even questioned the value of surgical intervention over choosing amputation for our child. It would have saved him years of missing out on life, pain, mental health issues - everything! Doctors should really listen to the families and patients- sometimes enough is enough. and a road to a better life may come in a different form than non-stop surgeries and medications.”
“Speech impediment surgery helped slightly and continuing with speech therapist. Base of skull surgery was very scary to relieve pressure/fluid on spine - appears to be stable so far.”
“Initially meds (Lyrica, nortriptyline) always seemed to work for a while for different symptoms or at least ease the intensity somewhat, however, as he grew and nerve pain would increase due to PF growth, there would be no effect. we spent years adjusting dosages, dealing with psychological health and supporting our son; an angry child, suicide attempts, no sleep, financial difficulties etc.”
“Psychological testing was great and was a thorough assessment of our daughters cognitive abilites. (cost of around 5000$). Scoliosis brace we paid for out of pocket and was not used at all as it was quickly noted that her scoliosis cannot be helped with a brace. (3000$) At the moment the spinal treatments are disappointing as we are told to wait and watch. We can see a potential major issue with her spine and there feels like we have very little treatment options or support.”
“Majority of the treatments have been considered successful. However, the current unmet needs/negative experience are: (1) availability/ support in proper diagnosis or early diagnosis and (2) lack of treatment options for inoperable tumors (in our family's case tumors in the brain and spine) within Canada (3) experience of Health Canada's review timelines for orphan drug submissions. Consideration should be given to the financial impact on the Canadian Medical system of no intervention or surgery (hospitalization, surgery, time families have to take away from workforce to care post-surgery etc.) for this progressive disease.”
Improved Outcomes
All of the parents who responded to the survey or interviews were aware of the new therapy, selumetinib and its potential impact. Half (50%) had direct experience with the drug. Their expectations centred on eliminating the impact of tumours, those that were currently present and reduction of appearance and impact of future tumours.
“My expectation is to actually have a treatment. Right now, all we have are measures to deal with tumors after the fact. There is so much pressure on parents to notice all issues by the time our doctors find things its already gone too far. It would be great to have treatment to deal with NF1 tumors before they become a problem. There are no real preventative treatments.”
“In the long term, this drug may save Canadians taxpayers in the long term (i.e., having families leaving the workforce, and rudimentary treatment options). CIHRs national framework is to promise obvious benefits to medical practice as well as the healthcare system, including prevention and screening strategies targeting high-risk individuals, avoidance of serious adverse outcomes, and better matching of therapies to disease and individual profiles.”
"…supporting the development of an evidence base on how to assess and eventually integrate these discoveries and therapeutic approaches into health policy and practice." I would hope that any new treatment and government reimbursement support for treatments would align with the CIHR framework.”
“Our expectation of the drug is to hopefully see a decrease in the size of the PF, as well as a decrease in body pain. Hopefully increasing the quality of life.”
“To become available as treatment at no cost to person with NF1.”
Experience With Drug Under Review
Of the eight respondents, four (50%) had experience with selumetinib through clinical trials in Canada. The response was overwhelmingly positive; the impact on the disease was described as “life changing.” If there is any such thing as a “miracle drug” that significantly improves quality of life without actually “curing” the disease, selumetinib is that miralcle for patients living with NF1.
“Selumetinib: this was offered to our son via trial. It has been the most positive part of our journey in 15 years.”
“The current treatment of Selumetinib is the only thing that has really made a big impact on my daughter. It has made her gain weight [she has failure to thrive] we have tried everything and nothing worked. She has a tumor in her face, and it made the tumor soften so she could swallow. The tumor was making her choke on everything including liquid. With this treatment she rarely has a problem swallowing. It has also stopped the brain tumors from growing any bigger.”
“She has been on Selumetinib for 2.5 years and it has been life changing. She now has teeth growing in on that side of her mouth. The plexiform in her ear has gone down so much that she can now hear perfectly from that ear, and she no longer gets ear infections which has increased the quality of her life so much. She also visibly looks so much better, and she used to have so much pain that she could not even chew on that side of her mouth and if you even lightly touched her cheek she would scream in pain. She can now chew on that side of her mouth and feels no pain when her cheek is touched. Even if you press down hard.”
“She was in chronic pain all the time and visibly deformed. I forgot to mention earlier, before when she spoke it hurt her to speak and nobody could understand what she was saying because her face was so frozen with the tumour that she would slur her words. Now she is able to enunciate all of her words and people understand her. That is life changing for her socially. She no longer has any pain, and it looks so much better. For a teenager this is so important.”
“Selumetinib has changed our son's life! Within 5-6 weeks of the initial start of the medication, there were massive decreases in his body pain. He started to show signs of better sleep, and less stabbing nerve pain in his legs/body, he was able to increase activity such as running and walking for longer durations and not experience the same post-activity pain flares or the inability to participate in activities as before. As the weeks passed, these changes only increased for the better. He sleeps well, which increases his daily focus at school or in his everyday life, his pain is managed very well unless there is physical contact made to his body. He doesn't feel anything negative about being on the drug nor did he experience any harsh or bad side effects.”
“At one point while in the hospital with an infection, the drug was held back for a few days. By the time day 5 rolled around, he noticed increased pain levels returning to his body and restlessness.”
“The only side effect he has experienced, which was shortly after the start of the medication, and continues to hang around is sensitive skin around the nailbeds of his toes, sometimes causing infection.”
Companion Diagnostic Test
The therapy would be available only to patients with a confirmed diagnosis of NF1 through a genetic test, which is routine and therefore would require no additional testing or costs.
Anything Else?
As noted previously, one is always hesitant to call any therapy a miracle but selumetinib for NF1 comes as close to that designation as any drug, short of an outright cure which would eliminate the disease. It seems to work for all patients; it works quickly and efficiently; there are few experienced side effects. While the (very) long-term benefits are yet to be demonstrated, the available evidence is compelling and certainly calls for the therapy to be available to all appropriate patients.
Conflict of Interest Declaration — Canadian Organization for Rare Disorders
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
No help was received from any outside person to complete this submission.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
The survey and interview questions were developed by CORD; the interviews were carried out by the staff of CORD; and the analysis and summary of data were carried out by CORD.
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
Table 2: Financial Disclosures for Canadian Organization for Rare Disorders
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Astra Zeneca/Alexion | — | — | X | — |
Clinician Input
Canadian Pediatric Brain Tumour Consortium
About Canadian Pediatric Brain Tumour Consortium
The Canadian Pediatric Brain Tumour Consortium (CPBTC) is a group of all the pediatric neuro-oncologists across Canada representing all 16 children’s hospital who provide oncological care for children with neoplasms of the central nervous system and peripheral nerves. Our group meets monthly for the past 20 years, and has run several clinical trials particularly in the realm of low grade glioma’s. Within our respective practices, we treat and follow children with neurofibromatosis type I (NF-1), including those with optic pathway glioma’s, plexiform neurofibroma’s and general surveillance of patients with NF-1. The composition of this consortium are primarily pediatric oncologists with additional training in neuro-oncology, or pediatric neurologists with additional training in neuro-oncology. Almost all children in Canada who would be treated with selumetinib would be treated by one of neuro- oncologists who are part of this consortium, with a small minority of patients treated by pediatric oncologists who treat primarily solid tumours – clinicians from that group have also provided input into this document.
Information Gathering
The information gathered in the submission are from shared clinical experiences through this monthly call, meetings in person at conference venues, as well as a publication we had last year where we report the collective Canadian experience with selumetinib provided by the expanded access program. This publication confirmed the results of the SPRINT study that nearly all patients had clinical benefit (Coltin et al, Pediatric Blood and Cancer, 2022). Our group has extensive experience in the management and treatment of patients with NF-1 including both plexiform neurofibroma’s and optic pathway glioma’s.
Current Treatments and Treatment Goals
Treatment for plexiform neurofibroma’s prior to the advent of selumetinib was very limited. No systemic therapy prior to this has been shown to confer any benefit including chemotherapy and a trial of imatinib. Several studies out of the NIH were run prior to selumetinib and did not demonstrate any benefit. As such the only treatment was supportive care in the case of pain, or in extreme instances large aggressive surgeries, which were either heroic or palliative in nature. Most plexiform neurofibroma’s are inoperable due to their proximity to vital structures and their intimate involvement with peripheral nerves. Radiation is not an option as there is a significant risk of malignant transformation due to the germline nature of the disease, second malignancies, and a ve large field of therapy. Moreover, radiation therapy is not particularly effective treatment for this condition. Both the SPRINT studies and the Canadian experience has confirmed that selumetinib can clearly lead to cytoreduction in a significant subset of patients, alleviation of symptoms related to the plexiform neurofibroma, reduction in pain, restoration of facial deformities and reduces the burden on caregivers. In addition to selumetinib, there is an extensive experience in Canada with another MEK inhibitor Trametinib (Novartis) through either the TRAM-01 trial or the Novartis manage access programme, with similar responses, particularly in young infants who require a liquid formulation. In some patients, selumetinib has led to sufficient cytoreduction where large abdominal surgeries or facial reconstructive surgeries have no longer been necessary. It has been very well tolerated in almost all patients, including within Canada, where the majority of patients were treated by the authors of this submission. Selumetinib is the first systemic agent to show any activity in plexiform neurofibroma’s and in many patients has significantly improved their quality of life and ability to live.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
As described above, there have been no disease-directed therapies for plexiform neurofibroma’s prior to the introduction of selumetinib. The only option available currently is surgical resection in a small subset of patients. Although pain medications have been used, they have not been effective in the majority of patients, and patients lived with disabilities and functional impairment. The limitation of large surgeries is that in the majority a complete resection cannot be achieved, and in most patients it comes with significant morbidity, and is reserved as a palliative measure.
As such there has been a huge unmet need for effective systemic therapies in patients with NF-1 associated plexiform neurofibromas which has been met with the introduction of selumetinib.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Selumetinib has clearly emerged as the standard of care as first line therapy for patients with inoperable, symptomatic plexiform neurofibromas. The drug under review would be the first-line treatment, and within Canada the current practice is to either obtain selumetinib through the managed access program. The drug under review has already led to a shift in current treatment paradigm. If selumetinib is not approved for reimbursement, this would lead to major financial hardship for families as they would need to find the funds to pay for this medication, and/or it would need to be covered by fundraising events.
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
The SPRINT study as well as the Canadian experience suggest that the majority of patients benefit from selumetinib, with only a very small subset not achieving an objective clinical response. The patients in need of intervention are those where the plexiform neurofibroma is invading critical structures, is causing a deformity, or causes functional impairment in activities of daily living such as walking, swallowing (tongue plexiform), or eating/swallowing (throat plexiform). As such, it is impossible to determine this small number of patients who do not respond, and the spectacular response rates observed in both clinical trials and real-world experiences would suggest that this would have to be the standard of care for these patients. There are currently no issues related to diagnosis, as these symptomatic plexiform neurofibroma’s are obvious clinically and can be followed radiologically and/or clinically (for example, a plexiform of the tongue can be serially photographed). Plexiform neurofibromas have characteristic features on MRI and clinical exam - the incidence of misdiagnosis in the population with NF1 is thus very low overall.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
Outcomes currently being used by the Canadian Pediatric Neuro-Oncology group include imaging assessment of response using MRI coupled with a clinical examination. A clinically meaningful response to treatment would be an improvement in function, for example we have treated patients with a throat plexiform causing swallowing dysfunction, and the clinical response is ability to eat solid food. The overall Canadian experience published earlier this year in Coltin et al, suggest that objective clinical responses can be assessed and used in real world clinical practice. These improvements in symptoms and activity of daily living have been associated with cytoreduction of the plexiform, and as such the two outcomes routinely used are clinical examination and response evaluation.
What factors should be considered when deciding to discontinue treatment with the drug under review?
The reasons for discontinuation of drug include a lack of clinical benefit, growth of the plexiform neurofibroma as determined through response assessment or intolerability of side effects. The major side effect are dermatological including paronychia, with Grade I toxicity occurring in about half of all patients, and in the Canadian experience has not required discontinuation, but was reported in some patients enrolled on the SPRINT study. A less common side effect is elevation of CK which in the SPRINT study required discontinuation in a small subset of patients. Patients on selumetinib are followed by pediatric dermatologists, and most centres have developed expertise in the prevention and treatment of skin toxicity.
What settings are appropriate for treatment with [drug under review]? Is a specialist required to diagnose, treat, and monitor patients who might receive [drug under review]?
Within Canada, initiation of treatment with selumetinib has been limited to pediatric oncologists/neuro-oncologists or pediatric neurologists with an expertise in neuro-oncology, who almost all work exclusively at one of the academic pediatric hospitals in Canada. The diagnosis of plexiform neurofibroma’s are typically made by either neuro-oncologists or pediatricians, with treatment initiated by the former. Dermatologists are consulted routinely during the course of therapy, and monitoring includes echocardiograms and ophthalmological assessments. In other jurisdictions, general neurologists and pediatricians prescribe the drug, but this has not been the case so far in Canada, although there would be ample support from oncologists with an expertise in treatment to help support this for those patients who live remotely, where treatment can be initiated by an oncologists and followed remotely in conjunction with local clinicians
Additional Information
From a physician perspective, the advent of MEK inhibitors has been a game changer for patients and families. Currently in Canada, there is widespread expertise and experience with the use of MEK inhibitors for plexiform neurofibroma’s, including selumetinib the drug under review or trametinib, which has been accessed through clinical trials or directly from Novartis. This class of medication has clearly changed the life of many patients, and provided substantial improvements in quality of life, decrease in days off work for parents and allowed these patients to attend school normally. As oncologists, we have not seen a drug with this level of activity, where essentially all patients respond. Without MEK inhibitors, these patients have progressive symptoms which continue to worsen their disability, particularly in the case of plexiform neurofibromas of the abdomen, trachea and face where there is progressive deterioration of function which cannot be halted with other means. The availability of a drug that is publically reimbursed will provide equitable access to treatment, in a population where the parents frequently are of lower socioeconomic standing due to their neurofibromatosis type 1. Selumetinib is an oral outpatient medication, which is a daily take home medication. As an outpatient take home drug. even if reimbursed, many children without private insurance will not be eligible depending on provincial drug coverage, as mechanisms such as OHIP+ are not present across the country. Many parents of children with NF-1, have NF-1 themselves do not have private insurance and as such, in terms of equity, it would be an important consideration that universal coverage be available for this group of marginalized children. Children without private insurance that are also not eligible for their provincial public drug plans will also need special consideration. It is our collective opinion that the drug under review urgently requires reimbursement to ensure equitable access to what we consider the standard of care therapy for all patients irrespective of their socioeconomic, ethnic or geographic barriers.
Conflict of Interest Declarations — Canadian Pediatric Brain Tumour Consortium
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please refer to the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission?
No outside help was received.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission
No outside help was received.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician who contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
Declaration for Clinician 1
Name: Vijay Ramaswamy
Position: Staff Neuro-Oncologist, Hospital for Sick Children, Toronto, ON
Date: 15-11-2022
Table 3: COI Declaration for Canadian Pediatric Brain Tumour Consortium — Clinician 1
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Alexion | X | — | — | — |
Declaration for Clinician 2
Name: Craig Erker
Position: Staff Oncologist/Neuro-Oncologist, IWK Health Centre, Halifax, NS
Date: 15-11-2022
Table 4: COI Declaration for Canadian Pediatric Brain Tumour Consortium — Clinician 2
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 3
Name: Bruce Crooks
Position: Staff Oncologist/Neuro-Oncologist, IWK Health Centre, Halifax, NS
Date: 15-11-2022
Table 5: COI Declaration for Canadian Pediatric Brain Tumour Consortium — Clinician 3
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 4
Name: Valérie Larouche
Position: Staff Oncologist/Neuro-Oncologist, CHU de Québec-Université Laval, Quebec, QC
Date: 15-11-2022
Table 6: COI Declaration for Canadian Pediatric Brain Tumour Consortium — Clinician 4
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Alexion | X | — | — | — |
Declaration for Clinician 5
Name: Samuele Renzi
Position: Staff Oncologist/Neuro-Oncologist, CHU de Québec-Université Laval, Quebec, QC
Date: 15-11-2022
Table 7: COI Declaration for Canadian Pediatric Brain Tumour Consortium — Clinician 5
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 6
Name: Sébastien Perreault
Position: Staff Neuro-Oncologist, Hospital Ste Justine, Montreal, PQ
Date: 15-11-2022
Table 8: COI Declaration for Canadian Pediatric Brain Tumour Consortium — Clinician 6
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Alexion | X | — | — | — |
Declaration for Clinician 7
Name: Hallie Coltin
Position: Staff Oncologist/Neuro-Oncologist, Hospital Ste Justine, Montreal, PQ
Date: 15-11-2022
Table 9: COI Declaration for Canadian Pediatric Brain Tumour Consortium — Clinician 7
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 8
Name: Nada Jabado
Position: Staff Oncologist/Neuro-Oncologist, Montreal Children’s Hospital, Montreal, PQ
Date: 15-11-2022
Table 10: COI Declaration for Canadian Pediatric Brain Tumour Consortium — Clinician 8
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 9
Name: Geneviève Legault
Position: Staff Neuro-Oncologist, Montreal Children’s Hospital, Montreal, PQ
Date: 15-11-2022
Table 11: COI Declaration for Canadian Pediatric Brain Tumour Consortium — Clinician 9
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 10
Name: Donna Johnston
Position: Staff Oncologist/Neuro-Oncologist, Children’s Hospital of Eastern Ontario, Ottawa, ON
Date: 15-11-2022
Table 12: COI Declaration for Canadian Pediatric Brain Tumour Consortium — Clinician 10
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Jazz Pharmaceuticals | X | — | — | — |
Declaration for Clinician 11
Name: Adam Fleming
Position: Staff Oncologist/Neuro-Oncologist, McMaster Children’s Hospital, Hamilton, ON
Date: 15-11-2022
Table 13: COI Declaration for Canadian Pediatric Brain Tumour Consortium — Clinician 11
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 12
Name: Shayna Zelcer
Position: Staff Oncologist/Neuro-Oncologist, London Health Sciences Centre, London, ON
Date: 15-11-2022
Table 14: COI Declaration for Canadian Pediatric Brain Tumour Consortium — Clinician 12
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 13
Name: Chantel Cacciotti
Position: Staff Oncologist/Neuro-Oncologist, London Health Sciences Centre, London, ON
Date: 15-11-2022
Table 15: COI Declaration for Canadian Pediatric Brain Tumour Consortium — Clinician 13
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 14
Name: Eric Bouffet
Position: Staff Oncologist/Neuro-Oncologist, Hospital for Sick Children, Toronto, ON
Date: 15-11-2022
Table 16: COI Declaration for Canadian Pediatric Brain Tumour Consortium — Clinician 14
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Novartis | X | — | — | — |
Alexion | X | — | — | — |
Declaration for Clinician 15
Name: Uri Tabori
Position: Staff Oncologist/Neuro-Oncologist, Hospital for Sick Children, Toronto, ON
Date: 15-11-2022
Table 17: COI Declaration for Canadian Pediatric Brain Tumour Consortium — Clinician 15
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 16
Name: Annie Huang
Position: Staff Oncologist/Neuro-Oncologist, Hospital for Sick Children, Toronto, ON
Date: 15-11-2022
Table 18: COI Declaration for Canadian Pediatric Brain Tumour Consortium — Clinician 16
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 17
Name: Julie Bennett
Position: Staff Oncologist/Neuro-Oncologist, Hospital for Sick Children, Toronto, ON
Date: 15-11-2022
Table 19: COI Declaration for Canadian Pediatric Brain Tumour Consortium — Clinician 17
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 18
Name: Magimairajan Vanan
Position: Staff Oncologist/Neuro-Oncologist, Cancer Care Manitoba, Winnipeg, MB
Date: 15-11-2022
Table 20: COI Declaration for Canadian Pediatric Brain Tumour Consortium — Clinician 18
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Alexion | X | — | — | — |
Declaration for Clinician 19
Name: Lucie Lafay-Cousin
Position: Staff Oncologist/Neuro-Oncologist, Alberta Children’s Hospital, Calgary, AB
Date: 15-11-2022
Table 21: COI Declaration for Canadian Pediatric Brain Tumour Consortium — Clinician 19
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Alexion | X | — | — | — |
Declaration for Clinician 20
Name: Bev Wilson
Position: Staff Oncologist/Neuro-Oncologist, Stollery Children’s Hospital, Edmonton, AB
Date: 15-11-2022
Table 22: COI Declaration for Canadian Pediatric Brain Tumour Consortium — Clinician 20
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 21
Name: Sylvia Cheng
Position: Staff Oncologist/Neuro-Oncologist, BC Children’s Hospital Vancouver, BC
Date: 15-11-2022
Table 23: COI Declaration for Canadian Pediatric Brain Tumour Consortium — Clinician 21
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 22
Name: Juliette Hukin
Position: Staff Neuro-Oncologist, BC Children’s Hospital Vancouver, BC
Date: 15-11-2022
Table 24: COI Declaration for Canadian Pediatric Brain Tumour Consortium — Clinician 22
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Alexion | X | — | — | — |
Declaration for Clinician 23
Name: Roona Sinha
Position: Staff Oncologist, Royal University Hospital, Saskatoon, SK
Date: 15-11-2022
Table 25: COI Declaration for Canadian Pediatric Brain Tumour Consortium — Clinician 23
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 24
Name: Laura Wheaton
Position: Staff Oncologist, Kingston Health Sciences Centre, Kingston, ON
Date: 15-11-2022
Table 26: COI Declaration for Canadian Pediatric Brain Tumour Consortium — Clinician 24
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 25
Name: Jack Brezynski
Position: Staff Oncologist (Solid Tumour Specialist), Hospital for Sick Children, Toronto, ON
Date: 15-11-2022
Table 27: COI Declaration for Canadian Pediatric Brain Tumour Consortium — Clinician 25
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 26
Name: Paul Gibson
Position: Staff Oncologist (Solid Tumour Specialist), McMaster Children’s Hospital, Hamilton, ON
Date: 15-11-2022
Table 28: COI Declaration for Canadian Pediatric Brain Tumour Consortium — Clinician 26
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 27
Name: Nirav Thacker
Position: Staff Oncologist/Neuro-Oncologist, Children’s Hospital of Eastern Ontario, Ottawa, ON
Date: 15-11-2022
Table 29: COI Declaration for Canadian Pediatric Brain Tumour Consortium — Clinician 27
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.