CADTH Reimbursement Review
Fostemsavir (Rukobia)
Sponsor: Viiv Healthcare ULC
Therapeutic area: Human immunodeficiency virus type 1
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
AE
adverse event
ANCOVA
analysis of covariance
ARV
antiretroviral
CBRC
Community-based Research Centre
CCR5
C-C chemokine receptor 5
CD4+
cluster of differentiation 4
CDC
Centres for Disease Control and Prevention
CI
confidence interval
CRF
case report form
CSR
Clinical Study Report
DB
double-blind
DCO
data cut-off
EQ-5D-3L
3-Level EQ-5D
FAHI
Functional Assessment of HIV Infection
gp
glycoprotein
GSS
genotypic susceptibility sore
HRQoL
health-related quality of life
HTE
heavily treatment-experienced
INSTI
integrase strand transfer inhibitor
IPD
individual patient data
IRIS
immune reconstitution inflammatory syndrome
ITC
indirect treatment comparison
ITT-E
intention-to-treat, exposed
MAIC
matching-adjusted indirect comparison
MID
minimally important difference
M-MASRI
Modified-Medication Adherence Self-Report Inventory
NNRTI
nonnucleoside reverse transcriptase inhibitor
NRTI
nucleoside reverse transcriptase inhibitor
OBT
optimized background therapy
OL
open-label
OR
odds ratio
OSR
overall susceptibility rating
OSS
overall susceptibility score
PDVF
protocol-defined virologic failure
PHAC
Public Health Agency of Canada
PI
protease inhibitor
PSS
phenotypic susceptibility score
RCT
randomized controlled trial
SAE
serious adverse event
SD
standard deviation
SLR
systematic literature review
ULN
upper limit of normal
VAS
visual analogue scale
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Fostemsavir (Rukobia), 600 mg, extended-release tablets, oral |
Indication | For adults with HIV-1 who are heavily treatment-experienced and have multidrug-resistant HIV-1, and for whom it is otherwise not possible to construct a suppressive antiviral regimen due to resistance, intolerance, or safety considerations |
Reimbursement request | Per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Priority |
NOC date | October 1, 2021 |
Sponsor | Viiv Health care ULC |
HIV-1 = HIV, type 1; NOC = Notice of Compliance.
Introduction
HIV consists of 2 subtypes, HIV-1 and HIV-2, and is transmitted via bodily fluids, including blood, semen, genital secretions, and breast milk. Infection with HIV-1 selectively destroys cluster of differentiation 4 (CD4+) immune cells, resulting in a gradual weakening of the immune system that, over time, leaves the patient in an immunocompromised state, susceptible to opportunistic infections. HIV-1 can progress to AIDS, which is ultimately fatal if untreated.1 According to the Public Health Agency of Canada (PHAC), in 2020, there were an estimated 62,790 patients living with HIV in Canada.2 Among those with HIV, it is estimated that 90% were diagnosed, and of those diagnosed, 87% were on treatment and 95% had a suppressed viral load.2 There are specific populations that appear to be disproportionately impacted by HIV, such as Indigenous people and those who inject drugs.2
HIV-1 is treated using combinations of antivirals; combination therapy is necessary to achieve sustained control of HIV-1 viremia, because resistance occurs quickly when HIV-1 is exposed to insufficient treatment regimens, according to the clinical expert consulted by CADTH. There are 4 main drug classes used in these combination regimens, and typically 2 or 3 of these classes are used in each antiretroviral (ARV) regimen, according to the clinical expert. Infection control is achievable in most patients with combinations that involve these classes, according to the clinical expert; however, there are 2 additional drug classes that can be used as rescue therapies in patients who experience issues with resistance to the conventional 4 classes. The goal of therapy, according to the clinical expert consulted by CADTH on this review, is to control viral replication and/or viremia, which in turn prevents HIV disease progression, prolongs life, prevents transmission, reduces the incidence of HIV-affected chronic diseases, and improves quality of life. According to the clinical expert consulted by CADTH on this review, patients with HIV are defined as being heavily treatment-experienced (HTE) if they have 2 or fewer drug classes of fully active medications available (i.e., with expected ability to treat that patient). These classes tend to be administered in the second line, according to the clinical expert, because of their lower tolerability, higher burden of side effects, and because they present challenges with administration, all of which complicate the safety and stability of long-term therapy.
Fostemsavir (Rukobia) is a first-in-class inhibitor of HIV-1 attachment. After being converted to its active form, temsavir, it inhibits the glycoprotein (gp)120 subunit within the gp160 envelope glycoprotein, preventing attachment and viral entry. It is indicated for adults with HIV-1 who are HTE and have multidrug-resistant HIV-1, and for whom it is otherwise not possible to construct a suppressive antiviral regimen due to resistance, intolerance, or safety considerations. Fostemsavir is administered orally at a dose of 600 mg twice daily.3 The sponsor’s reimbursement request is identical to the indication.4 Fostemsavir underwent the priority review process through Health Canada.
The objective of this report is to perform a systematic review of the beneficial and harmful effects of fostemsavir (extended-release tablets, 600 mg) in combination with other AVRs for the treatment of HIV-1 infection in HTE adults with multidrug-resistant HIV-1 infection.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient groups that responded to CADTH’s call for patient input and from the clinical expert consulted by CADTH for the purpose of this review.
Patient Input
One patient group submitted input for this review: the Community-Based Research Centre (CBRC). The CBRC, a nonprofit charitable organization that promotes the health of people of diverse sexualities and genders, based in Vancouver, British Columbia, provided input to this submission. CBRC conducted 2 surveys: 1 in 2021 (n = 325) and the other in 2022 (n = 144).
Respondents described how the stigma associated with HIV impacts their lives, as well as challenges associated with maintaining adherence to therapy and the way housing and food insecurity can make it even more challenging. They also described the limited treatment options available for the HTE population, and noted that it would be unethical for fostemsavir to not be available, given the risk of harm from untreated HIV and the risk of transmission. According to responses from the 2022 survey, injectable ARVs are preferred by 47% of respondents, whereas 19% prefer orally administered drugs.
Clinician Input
Input From Clinical Experts Consulted by CADTH
The major unmet need in the HTE population, according to the clinical expert consulted by CADTH on this review, is the limited availability of treatment options that are safe, effective, and easily administered, and the clinical expert noted that patients who are resistant to ARVs experience dramatically worse clinical outcomes.
The clinical expert consulted by CADTH on this review noted that fostemsavir would be used for HTE patients or other patients for whom there are limited options for treatment as a result of an underlying disease state or resistance. The clinical expert noted that HIV specialists would identify the patients most likely to respond to fostemsavir, based on clinical history, treatment history, and resistance testing.
Viral load is the most important test to determine response to treatment, according to the clinical expert, and clinical responses, such as resolution of disease-related symptoms, immune reconstitution, rate of opportunistic infections, and survival, add supplemental evidence of response. The clinical expert noted that treatment should be discontinued if there is a lack of response or evidence of resistance (based on phenotypic or genotypic resistance testing), intolerable adverse effects that can lead to safety issues, or patient preference.
Clinician Group Input
There was no clinician group input provided for this submission.
Drug Program Input
Drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The drug plans identified implementation issues related to considerations for initiation of therapy, discontinuation of therapy, and prescribing of therapy. The clinical expert consulted by CADTH weighed evidence from the BRIGHTE study and other clinical considerations to provide responses to the drug programs’ implementation questions. Refer to Table 4 for more details.
Clinical Evidence
Pivotal Studies and Protocol-Selected Studies
Description of Studies
The BRIGHTE study consisted of an initial double-blind (DB) phase that lasted 8 days and a subsequent open-label (OL) phase, currently ongoing, that will last 240 weeks. In the DB phase, 272 patients with HIV-1 who were eligible to receive at least 1 fully active, approved ARV in 1 or 2 ARV classes at baseline were randomized, in a 3:1 ratio, to either fostemsavir 600 mg twice daily or placebo, plus their baseline ARV regimen, for 8 days. The primary analysis was conducted after 8 days, and consisted of the primary outcome: the mean change from baseline to week 8 in HIV-1 RNA. Secondary outcomes, none of which were formally assessed, included the percentage of patients with a decrease in HIV-1 RNA of greater than 0.5 log10 copies/mL and greater than 1.0 log10 copies/mL at day 8, whereas in the OL phase, virologic response (HIV-1 RNA level of < 40 copies/mL at week 24, 48, and 96), resistance testing for patients experiencing virologic failure, mean change in CD4+ count through week 96, and events resulting in a diagnosis of AIDS (using the Centers for Disease Control and Prevention [CDC] classification system) were assessed. In addition to this randomized cohort, there was a nonrandomized cohort that consisted of patients who had no other options for fully active and approved ARVs, and these patients received fostemsavir plus optimized background therapy (OBT), determined based on resistance testing and treatment history. In the randomized cohort, after day 8, patients entered an OL phase, during which they all received fostemsavir plus OBT. The study was expected to last at least 96 weeks, and to continue until an additional option, a rollover study, or marketing approval was in place.
Patients in the randomized cohort were approximately (mean) 44 years of age, and the majority were male (74% of patients) and white (68% of patients). Most patients (89%) had a baseline viral load of 1,000 copies/mL or |||||| ||| |||| |||| |||| of patients) had a baseline viral load of 30,000 copies/mL or more. Approximately one-quarter of patients had a CD4+ count greater than 20 cells/mm3 and a similar percentage had a baseline CD4+ count of 200 cells/mm3 or more. Approximately one-third of patients had been treated for HIV for more than 20 years, and 85% of patients, overall, had a positive AIDS history, meaning that they either had a nadir CD4+ count lower than 200 cells/mm3 or a response of yes to the question, “Does participant have AIDS?” on the disease history component of the case report form (CRF). Most patients (90% or more) had prior exposure to a nonnucleoside reverse transcriptase inhibitor (NNRTI), nucleoside reverse transcriptase inhibitor (NRTI), or protease inhibitor (PI), whereas 75% had prior exposure to an integrase strand transfer inhibitor (INSTI). Other ARVs that patients had prior exposure to included entry (or fusion) inhibitors (39%), C-C chemokine receptor 5 (CCR5) antagonists (26%), and ||||| ||||||||||||||| ||| |||||. The most common ARV classes in the failing regimen were NRTIs (81%), PIs (67%), INSTIs (44%), and NNRTIs (28%), whereas other classes included CCR5 antagonists (12%) and entry inhibitors (4%).
Efficacy Results
During the OL phase, after 96 weeks, 4% of patients had died in the randomized cohort and 17% of patients had died in the nonrandomized cohort. Overall, 2% of patients had a cause of death that was considered to be AIDS-related. The definition of an AIDS-related death, which was investigator-determined,5 was not provided; however, the identification of AIDS-related events, in general, in the BRIGHTE study was based on the CDC list of AIDS-defining events. After 240 weeks, 6% of patients in the randomized cohort and 20% of patients in the nonrandomized cohort had died, and || of patients overall had a death that was AIDS-related. Patients with HIV who are HTE and physicians both highlighted the high risk of mortality in this population.
The percentage of patients progressing to AIDS was not specifically reported in the BRIGHTE study; however, the percentage of patients with AIDS-related events was reported. In the DB phase, after 8 days there were 2 patients in the fostemsavir group who had an AIDS-related event (grade 3 serious adverse event [SAE] of recurrent pneumonia; grade 2 adverse events [AEs] of herpes simplex virus, gastrointestinal other than mouth, throat, perirectal), and 1 patient in the placebo group (grade 3 SAE of Candida esophagitis). After 96 weeks in the OL phase, || of patients who were originally assigned to the fostemsavir group and || of patients who were originally assigned to the placebo group had an AIDS-related event. In the nonrandomized cohort, ||||| || |||||| ||| had an AIDS-related event. ||||| ||| |||||| || of patients originally assigned to the fostemsavir group and || originally assigned to the placebo group had an AIDS-related event, whereas ||| of patients in the nonrandomized cohort had an AIDS-related event. Patients with HIV who are HTE and physicians both highlighted the importance of reducing the risk of AIDS-related morbidities in this population. Hospitalizations were not reported in either the DB or OL phase, and this was an outcome from our protocol that would have provided further context on the impact of adding fostemsavir to OBT on important clinical outcomes in this population.
The mean change from baseline to day 8 in plasma HIV-1 RNA log10 copies/mL was –0.791 log10 copies/mL (95% confidence interval [CI], –0.885 to –0.698 log10 copies/mL) in the fostemsavir group and –0.166 log10 copies/mL (95% CI, –0.326 to –0.007 log10 copies/mL) in the placebo group, for a difference between groups of –0.625 log10 copies/mL (95% CI, –0.810 to –0.441 log10 copies/mL; P < 0.0001). There were ||| of fostemsavir patients and ||| of placebo patients who achieved a decrease in HIV-1 RNA of greater than 0.5 log10 copies/mL by day 8 and ||| of fostemsavir patients and ||| of placebo patients who achieved a decrease in HIV-1 RNA of greater than 1.0 log10 copies/mL by day 8. In the OL phase, the percentage of patients with an HIV-1 RNA greater than 40 copies/mL remained consistent from week 24 (56%) to week 48 (57%) to week 96 (61%), and levelled off by week 240. ||||||||||| |||||||| |||| ||| |||||||| ||| ||| ||||||| ||||||| || |||||||| ||||| |||| ||| || |||||||| ||||| | |||| ||||||||| || |||||||| || ||| ||||||||. For patients with a |||||||| |||||| ||||| ||| ||||| ||||||||| || |||||, there was a mean (standard deviation [SD]) ||||||||| |||| |||||||| || |||||| ||||| ||| ||||| || |||||| ||||||| with fostemsavir and ||||| ||||||| with placebo, and for patients with baseline ||||| ||| || |||| |||| |||| ||||| ||||||||| there was a mean (SD) ||||||||| || |||||| ||||| ||| || |||||| ||||||| ||||| ||||||||| with fostemsavir and ||||| ||||||| ||||| ||||||||| with placebo. Subgroup data for ||||| |||| ||||| || |||||||| |||| ||||| were also reported. Patients with a baseline |||| || ||| ||||||||| ||| | ||||||||| || |||||| ||||| ||| || |||||| (95% CI, |||||| || ||||||| ||||| ||||||||| with fostemsavir and |||||| (95% CI, |||||| || |||||| ||||| ||||||||| with placebo, while patients with baseline |||| |||| || || ||| ||||||||| had a ||||||||| || |||||| ||||| ||| || |||||| (95% CI, |||||| || ||||||| ||||| ||||||||| in the fostemsavir group and ||||| (95% CI, |||||| || |||||) ||||| ||||||||| in the placebo group. For patients with baseline |||| || || || |||| ||||||||| the mean change from |||||||| || ||| | || |||||| ||||| ||| ||| |||||| (95% CI, |||||| || ||||||) ||||| ||||||||| with fostemsavir and ||||| (95% CI, |||||| || |||||) ||||| ||||||||| with placebo, and for patients with a baseline |||| || ||| || |||| ||||||||| the adjusted mean change from baseline to day 8 was |||||| (95% CI, |||||| || ||||||| ||||| ||||||||| with fostemsavir and |||||| (95% CI, |||||| || |||||) ||||||||| with placebo. Finally, in patients with a baseline |||| || ||| || |||| |||||||||, the adjusted mean change from |||||||| || ||| | || ||||| ||| ||| |||||| (95% CI, |||||| || ||||||) ||||| ||||||||| with fostemsavir and |||||| (95% CI, |||||| || |||||) ||||| ||||||||| in the placebo group.
At day 8, the mean (SD) change from day 1 in CD4+ counts was |||| |||||| ||||||||| in the fostemsavir group (from a baseline of ||||| ||||||| ||||||||| and |||| |||||| ||||||||| in the placebo group (from a baseline of ||||| ||||||| |||||||||. In the OL phase, the mean (SD) change from baseline to week 96 in CD4+ counts was ||||| ||||||| ||||||||| in the randomized cohort and ||||| ||||||||| |||||||| in the nonrandomized cohort. After 240 weeks, the mean (SD) change from baseline in CD4+ counts was 296.4 (227.5) cells/mm3 in the randomized cohort and 240.0 (318.5) cells/mm3 in the nonrandomized cohort.
Health-related quality of life (HRQoL) was not assessed in the DB phase ||| ||| |||||||| || ||| || |||||| || || |||| ||| ||| |||| |||| |||||||| ||||||| || |||| | |||||||||| | |||||| |||||| ||| |||||| |||||| ||||| ||||| ||||| ||||||||| |||| |||||||| || |||| || || |||| ||| |||||||||| ||||||| || ||| |||||| ||||||| ||| || ||| |||||||||||||| ||||||| || ||| |||||| |||||| ||| || ||||||| ||||| |||||| ||||||||| |||||||||| || ||||| ||||||||| |||||| || ||| |||||||||| |||||| ||| || |||||| ||||||||| |||||| || ||| |||||||||||||| ||||||| The mean (SD) Functional Assessment of HIV Infection (FAHI) total score increased from baseline to week 96 in both cohorts, by 5.3 (24.0) points in the randomized cohort and by 4.9 (26.4) points in the nonrandomized cohort.
Harms Results
In the OL phase, after 96 weeks, AEs had been experienced by 92% of patients in the randomized cohort and by 99% of patients in the nonrandomized cohort, and after 240 weeks, AEs had been experienced by 95% of patients in the randomized cohort and 99% of patients in the nonrandomized cohort. The most common AEs occurring after 96 weeks (randomized and nonrandomized cohorts) were diarrhea (||| ||| ||||| nausea (||| ||| |||), and upper respiratory tract infection (||| || |||| |||||), ||||| ||| |||| |||||| || ||||| ||| ||||| |||| |||||||| |||| ||| ||||| |||||| |||| || |||| |||||||| ||| ||||| ||||||||||| ||||| ||||||||| |||| ||| ||||.
In the 8-day DB phase, 2% of patients in the fostemsavir group and 3% of patients in the placebo group experienced an SAE. The only SAE that occurred in more than 1 patient in either group was pneumonia, which occurred in 2 patients (< 1%) in the fostemsavir group and in 0 patients in the placebo group. During the OL phase, after 96 weeks, 34% of patients in the randomized cohort and 48% of patients in the nonrandomized cohort experienced an SAE, whereas after 240 weeks, 45% of patients in the randomized cohort and 56% of patients in the nonrandomized cohort experienced an SAE. The most common SAE was pneumonia, occurring in 4% of patients in the randomized cohort and 3% of patients in the nonrandomized cohort after 96 weeks and, after 240 weeks, in 8% of patients in the randomized cohort and 4% of patients in the nonrandomized cohort. In the OL phase, after 96 weeks, 5% of patients in the randomized cohort and 12% of patients in the nonrandomized cohort discontinued treatment due to an AE and, after 240 weeks, 6% of patients in the randomized cohort and 13% of patients in the nonrandomized cohort did. The most common reason was infections and infestations, which occurred in 2% of patients in the randomized cohort after 96 weeks and after 240 weeks, and in 5% of patients after 96 weeks and 6% of patients after 240 weeks in the nonrandomized cohort. Notable harms were infrequent during the DB phase, with the following events occurring in less than 1% of fostemsavir-treated patients: immune reconstitution inflammatory syndrome (IRIS), QTc prolongation, and increased blood alkaline phosphatase. After 96 weeks in the OL phase, IRIS had occurred in 2% of patients, and this was unchanged at the 240-week follow-up. QTc prolongation occurred in 4% of patients after 96 weeks, and the percentage of patients experiencing QTc prolongation was not reported for the 240-week follow-up. There were 1% of patients who reported an alanine transaminase (ALT) level greater than 3 × the upper limit of normal (ULN) and a total bilirubin level greater than 2 × ULN after 96 weeks and after 240 weeks.
Table 2: Summary of Key Results From the Pivotal Study
Characteristic | Randomized cohort | |
|---|---|---|
Fostemsavir (N = 203) | Placebo (N = 69) | |
Viral load | ||
Plasma HIV-1 RNA log10 (c/mL) change from day 1 to day 8 | ||
N | 201 | 69 |
Adjusteda mean, log10 c/mL (95% CI) | –0.791 (–0.885 to –0.698) | –0.166 (–0.326 to –0.007) |
Difference between groups (95% CI) | –0.625 (–0.810 to –0.441) | Reference |
P value | < 0.0001b | Reference |
HIV-1 RNA decrease > 0.5 log10 c/mL | ||
Achieved HIV-1 RNA outcome, n (%) | 131 (65) | 13 (19) |
Difference between groups (95% CI) | 46 (32.95 to 55.45) | Reference |
HIV-1 RNA decrease > 1.0 log10 c/mL | ||
Achieved HIV-1 RNA outcome, n (%) | 93 (46) | 7 (10) |
Difference between groups (95% CI) | 36 (24.16 to 44.25) | Reference |
|||||| || ||||| ||||||||| | ||
||| || | | ||| | ||| |
|||| |||| |||||||| ||||| | ||||| |||||||| | ||||| |||||||| |
|||||| ||||||| | ||||| ||| ||||| | ||||| ||| |||| |
||| || | | ||| | ||| |
|||| |||| |||||| |||| ||| | | |||| ||||||| | |||| ||||||| |
|||||| ||||||| | ||| |||||| |||| | ||| |||||| |||| |
||||||||||| || |||| | ||
||||||||||||| |||||||| ||||||||| | | ||| | ||| |
|||||||||| |||| ||||||||| |||||||||| ||||||||||| | ||
|||||||| |||| |||| ||||| ||||||| || ||||| ||| || ||| | | ||
|||||||| | ||| | ||||||| |||| | ||| |
||| |||||| || ||||| ||||| ||||| || |||| | ||||| |||| | ||| |
|| || ||| || ||| ||||| ||||| | ||||| |||| | ||| |
|||||||||| || |||| |||||||||||| |||||| |||||| ||||||| | ||||| |||| | ||| |
Harms (DB phase, day 8) | ||
Patients with an AE, n (%) | 83 (41) | 22 (32) |
Patients with an SAE, n (%) | 4 (2) | 2 (3) |
Patients who stopped treatment due to an AE, n (%) | 4 (2) | 1 (< 1) |
Notable harms | ||
IRIS, n (%) | 1 (< 1) | 0 |
QTc prolongation, n (%) | 1 (< 1) | 0 |
Blood alkaline phosphatase increased, n (%) | 1 (< 1) | 0 |
AE = adverse event; c/mL = copies per millilitre; CD4+ = cluster of differentiation 4; CI = confidence interval; DB = double-blind; EN = envelope; HIV-1 = HIV, type 1; IRIS = immune reconstitution inflammatory syndrome; RAP = resistance-associated polymorphisms; SAE = serious adverse event; SD = standard deviation.
aMean adjusted by day 1 log10 HIV-1 RNA.
bEstimated by 1-way analysis of covariance (ANCOVA) in the randomized cohort, with log10 HIV-1 RNA change from day 1 to day 8 as the dependent variable, treatment (fostemsavir or placebo) as an independent variable, and log10 HIV-1 RNA at day 1 as a continuous covariate.
cAIDS-defining events were based on the Centers for Disease Control and Prevention (CDC) class C classification system, which includes events, typically infectious diseases, that tend to be characteristic of AIDS.
Source: Clinical Study Report for BRIGHTE,6 week 96 follow-up.
Critical Appraisal
For internal validity, the BRIGHTE study appeared to be reasonably well conducted with respect to measures taken to ensure adequate blinding during the 8-day DB phase and to maintain allocation concealment during randomization. Assessment of HRQoL, an important outcome for the HTE population, was subject to considerable bias due to lack of blinding during the OL phase, and the data are difficult to place into context due to the lack of a control group. The FDA snapshot analysis was used to report results for virologic response. This is a conservative approach that counts missing samples as failures, and may have confounded the results because attrition increased from week 96 to week 240. The use of OBT in the OL phase means that the background therapy that patients received, in addition to fostemsavir, was not standardized, and it assumes that all patients were optimized for their specific clinical situation. Disposition for the 8-day DB phase was not reported in the Clinical Study Report (CSR); therefore, we do not know whether there was a difference in withdrawals between the fostemsavir and placebo groups for this phase, which could potentially impact interpretation of efficacy and harms.
With respect to external validity, although the 8-day DB phase followed FDA guidance for assessing ARVs, this short duration of follow-up limited the ability to assess any outcomes outside of viral load in a comparative manner. For example, CD4+ counts typically take several months to increase in response to a reduction in viral load, and 1 would not expect to see differences in the risk of AIDS-related death or progression to AIDS in 8 days. The HTE population is at much higher risk of experiencing AIDS-related complications, such as opportunistic infections and death; therefore, there remains a gap in knowledge regarding the impact of fostemsavir on these important outcomes in these patients.
Indirect Comparisons
The BRIGHTE trial included an 8-day randomized phase in which fostemsavir plus OBT was compared to placebo plus OBT, followed by a single-arm phase in which all patients received fostemsavir plus OBT. Indirect comparisons were therefore required to estimate comparative effectiveness for any outcomes beyond 8 days.
Description of Studies
The sponsor submitted 1 matching-adjusted indirect comparison (MAIC)7 and CADTH identified 1 published MAIC.8
The objective of the sponsor-submitted MAIC was to generate long-term comparative efficacy estimates for fostemsavir plus OBT versus OBT alone for the management of HTE patients with HIV using individual patient data (IPD) from the BRIGHTE study. The data for OBT alone were populated using outcomes from the VIKING-3 study, which was identified through a systematic literature review (SLR) and feasibility assessment to be the most closely aligned with the BRIGHTE study in terms of patient eligibility criteria regarding treatment history, resistance status, and available treatments remaining. The VIKING-3 study was also identified as the most relevant in the context of treatment practices and patients in Canada, based on a sponsor-conducted feasibility assessment and consultation with physicians.
The published MAIC included the same analysis submitted by the sponsor, alongside analyses comparing the BRIGHTE study to the TMB-301 study and to the BENCHMRK studies, which were considered to be less relevant for the purpose of this review. The TMB-301 study evaluated ibalizumab, which is not currently available or marketed for use in Canada;7 additionally, nearly half of the patients in the TMB-301 study used fostemsavir in their OBT, and subgroup data were not available to exclude these patients. The BENCHMRK studies began in 2006, and the ARV regimens used in the OBT-alone arm did not closely reflect the combination of regimens used in the BRIGHTE study or in current Canadian practice; most notable was the lack of dolutegravir.8
In the sponsor-submitted MAIC comparing the BRIGHTE and VIKING-3 studies as a representation of OBT alone, efficacy was assessed in terms of:
change (from baseline) in CD4+ cell count
rates of virologic suppression
rates of protocol-defined virologic failure (PDVF)
rates of treatment discontinuation.
Secondary analyses included an assessment of the relative safety profile of fostemsavir, based on the rates of SAEs, discontinuation due to AEs, and death.
Efficacy Results
|| ||| ||||||| |||| |||||||| ||| |||||||| ||||||||| ||||||| |||| |||||||||||| ||| ||| |||| |||||| || |||| |||| ||||| |||| |||||||| ||||| ||||||||||| |||||| ||| ||| ||||| || |||||| | | |||||| ||| ||| ||| ||||||| |||| ||||||||| ||||||||||| ||||| || ||||| ||| ||| ||||||||| ||||| ||||| |||| | ||||| ||| ||| |||| || ||||| | | |||||| ||| || ||| |||| |||||||| ||| |||| ||||||| ||||||| |||| |||| ||| |||||||||||| || ||| ||||| |||||||||| ||| | ||||| ||| ||| |||| || ||||| | | |||||| ||| ||| || |||||| || ||||||||||| | ||| |||||| ||| ||||| ||| || |||||| || ||||||||| || ||| ||||||| |||| |||||||| ||| | ||||| ||| ||| |||| || ||||| | | ||||||| ||| ||||||| ||| ||||||||| ||||||||||||||| |||| || |||||| || ||||||||||| | ||| |||||| ||| ||||| || |||| ||| ||||| ||| | ||||| ||| ||| |||| || ||||| | | |||||| ||| |||| |||||||| ||| | ||||| ||| ||| |||| || ||||| | | |||||||
Harms Results
Results of the safety-related MAICs (patients with any SAE, cellulitis, dehydration, pneumonia, pyrexia, acute kidney injury, death, or discontinuation due to AEs) were inconclusive due to wide 95% CIs that included the null value.
Critical Appraisal
The VIKING-39 single-arm study of dolutegravir-containing regimens had the most comparable HTE HIV population, and patients were treated with ARV regimens that were most closely reflective of those in the BRIGHTE study and in Canadian clinical practice, primarily including dolutegravir, darunavir, and the combination of tenofovir disoproxil fumarate plus emtricitabine. Because 82% of patients in the BRIGHTE study received dolutegravir as part of their OBT, the VIKING-3 study was an appropriate trial to select as comparator.
Although adjustments conducted for the MAICs were generally appropriate and the sponsor followed a comprehensive and expert-guided process to identify prognostic factors and treatment-effect modifiers, it is unknown whether all relevant variables were captured. Unanchored MAICs require very strong assumptions about the data and require that all known and unknown prognostic factors and treatment-effect modifiers are accounted for. This threshold may be particularly difficult to meet for discontinuation and safety-related outcomes.
The distribution of the overall susceptibility score (OSS)new in the VIKING-3 study at baseline had to be recalculated to account for patient exposure to dolutegravir throughout the trial. Although multiple assumptions were explored, it is unknown which assumption is the most appropriate, and the magnitude and direction of potential bias is uncertain.
In adjusting the population of the BRIGHTE study to match the population of the VIKING-3 study for MAIC, there was a drop in sample size of nearly 80%, reflecting poor overlap between the trials. The BRIGHTE study allowed patients without fully active ARVs remaining (in the nonrandomized cohort), whereas the VIKING-3 study required at least 1 remaining ARV. The adjusted population primarily represents participants in the BRIGHTE study with more treatment options remaining (i.e., to reflect the distribution of OSSnew in the VIKING-3 study), and is therefore not representative of the full population eligible for fostemsavir, especially those with highly resistant disease and without fully active ARVs remaining.
Although there were statistically significant results for the MAICs of discontinuation and PDVF, interpretation is compromised by the limitations of unanchored MAICs, substantial sample size reduction, and differences in the definition of PDVF between the trials.
The results for change in CD4+ cell count and proportion with virologic suppression (HIV-1 RNA < 50 copies/mL) were inconclusive due to CIs that included the null value.
The results for safety outcomes were generally imprecise, and the interpretation is compromised by substantial differences in study-drug exposure in both the primary analysis (comparing the 48-week data cut-off [DCO] of each trial) and the sensitivity analysis (24-week DCO in the BRIGHTE study compared to 48-week DCO in the VIKING-3 study).
Overall, the MAICs were determined to be inconclusive due to the limitations of the available evidence.
Other Relevant Evidence
There were no extensions and no other relevant studies in the population of interest identified for this review.
Conclusions
Evidence from 1 DB, randomized controlled trial (RCT) suggests that when combined with a failing ARV regimen, fostemsavir reduces viral load after 8 days of therapy, compared to placebo, establishing proof of concept. In the subsequent OL phase of this trial, improvements in viral load appeared to be maintained, and CD4+ counts increased, over a 240-week treatment period; however, analysis of these outcomes is potentially biased by attrition and other sources of missing data over such a long follow-up period. There were AIDS-related deaths and indicators of progression to AIDS in the OL phase; however, with the lack of a control group, there is a lack of context for these findings. In the brief, 8-day DB phase of the trial, there were no clear indications of safety or tolerability issues with fostemsavir compared with placebo. There were numerical increases in the number of patients experiencing specific AEs or overall SAEs from 96 weeks to 240 weeks of follow-up in the OL phase, and it is not clear whether this represents an increased risk of these AEs or whether this is simply a consequence of the increased duration of follow-up. Evidence from indirect comparisons of fostemsavir versus OBT were generally inconclusive, largely due to the small sample size after a majority of patients from the index trial was excluded and to the concerns of incomplete matching adjustment on effect modifiers.
Introduction
Disease Background
HIV consists of HIV-1 and HIV-2, with HIV-1 being the most common globally. HIV-1 is transmitted by bodily fluids, including blood, semen, genital secretions, and breast milk. Infection with HIV-1 selectively destroys CD4+ immune cells, which play a critical role in fighting infection. This gradual weakening of the immune system over time leaves the patient in an immunocompromised state, and therefore susceptible to opportunistic infections. HIV-1 can progress to AIDS, which is ultimately fatal if untreated.1
According to PHAC, in 2020, there were an estimated 62,790 patients living with HIV in Canada. Among those with HIV, it is estimated that 90% were diagnosed, and of those diagnosed, 87% were on treatment and 95% had a suppressed viral load.2 According to PHAC, 75% of patients living with HIV were male. According to PHAC, females may have a lower percentage of awareness of infection, as well as lower treatment and viral suppression rates. There are also specific populations that appear to be disproportionately impacted by HIV, such as Indigenous people and those who inject drugs.2
Standards of Therapy
HIV-1 is treated using combinations of antivirals; combination therapy is necessary to achieve sustained control of HIV-1 viremia because resistance occurs quickly when HIV-1 is exposed to insufficient treatment regimens, according to the clinical expert consulted by CADTH. The 4 main drug classes used in these combination regimens are NRTIs, NNRTIs, PIs, and INSTIs, according to the clinical expert consulted by CADTH on this review. There are typically 2 or 3 of these classes used in a given ARV regimen and, according to the clinical expert, infection control is achievable in most patients using combinations involving these classes; however, there are 2 additional classes, CCR5 inhibitors and fusion inhibitors, that can be used as rescue therapies in patients who experience issues with resistance to the conventional 4 classes. The goal of therapy, according to the clinical expert consulted by CADTH on this review, is to control viral replication and/or viremia, which in turn prevents HIV disease progression, prolongs life, prevents transmission, reduces the incidence of HIV-affected chronic diseases, and improves quality of life.
According to the clinical expert consulted by CADTH on this review, patients with HIV are defined as being HTE if they have 2 or fewer classes of fully active medications available (i.e., with expected ability to treat that patient). These classes tend to be administered in the second line, according to the clinical expert, because of their lower tolerability, higher burden of side effects, and because they present challenges with administration, all of which complicate the safety and stability of long-term therapy.
Drug
Fostemsavir is a first-in-class inhibitor of HIV-1 attachment. After being converted to its active form, temsavir, it inhibits the gp120 subunit within the gp160 envelope glycoprotein, preventing attachment and viral entry. It is indicated for adults with HIV-1 who are HTE and have multidrug-resistant HIV-1, and for whom it is otherwise not possible to construct a suppressive antiviral regimen due to resistance, intolerance, or safety considerations. Fostemsavir is administered orally, at a dose of 600 mg twice daily. The sponsor’s reimbursement request is identical to the indication. Fostemsavir underwent the priority review process through Health Canada.3
Table 3: Key Characteristics of Various ARV Regimens
Comparator regimensa | Brand name | Dosage strength | Indicationb | Key side effects and/or safety issues |
|---|---|---|---|---|
Single-tablet regimens | ||||
DTG/3TC | Dovato | DTG: 50 mg 3TC: 300 mg | A complete regimen for the treatment of HIV-1 infection in adults and adolescents aged ≥ 12 years and weighing ≥ 40 kg | DTG: insomnia, headache, depression; early benign increase in SCr 3TC: generally well tolerated |
DOR/TDF/3TC | Delstrigo | DOR: 100 mg TDF: 300 mg 3TC: 300 mg | A complete regimen for the treatment of HIV-1 infection in adults without past or present evidence of viral resistance to doravirine, lamivudine, or tenofovir | DOR: dizziness, abnormal dreams, insomnia, nightmares, headache, sleepiness, nausea, diarrhea, vomiting, feeling tired and weak, depression TDF: renal toxicity, decreased BMD, increased osteoporotic fractures; reports of lactic acidosis, hepatotoxicity 3TC: generally well tolerated |
BIC/TAF/FTC | Biktarvy | BIC: 50 mg FTC: 200 mg TAF: 25 mg | A complete regimen for the treatment of HIV-1 infection in adults with no known substitution associated with resistance to the individual components | BIC: diarrhea, nausea, headache, fatigue, abnormal dreams, dizziness, and insomnia FTC: discoloration of skin (hands and/or feet) TAF: similar to TDF, but may have less renal and bone toxicity |
DTG/ABC/3TC | Triumeq | DTG: 50 mg | Treatment of HIV-1 infection in adults and adolescents aged ≥ 12 years and weighing ≥ 40 kg | DTG: insomnia, headache, depression; early benign increase in SCr ABC: risk of severe hypersensitivity reaction in genetically susceptible patients; possible increased risk for MI 3TC: generally well tolerated |
ABC: 600 mg | ||||
3TC: 300 mg | ||||
EVG/c/TAF/FTC | Genvoyac | EVG: 150 mg | A complete regimen for the treatment of HIV-1 infection in adults and adolescents aged ≥ 12 years (and weighing ≥ 35 kg) and with no known RAMs to the individual components | EVG: nausea, diarrhea, insomnia, headache, depression; early benign increase in SCr c: can falsely increase SCr FTC: discoloration of skin (hands and/or feet) TAF: similar to TDF, but may have less renal and bone toxicity |
c: 150 mg | ||||
FTC: 200 mg | ||||
TAF: 10 mg | ||||
RPV/TAF/FTC | Odefseyc | RPV: 25 mg | A complete regimen for the treatment of adults infected with HIV-1 with no known RAMs to the NNRTI class, tenofovir or FTC, and with a VL ≤ 100,000 c/mL | RPV: depression, insomnia, rash, headache; early benign increase in SCr TAF: Similar to TDF, but may have less renal and bone toxicity FTC: discoloration of skin (hands and/or feet) |
TAF: 25 mg | ||||
FTC: 200 mg | ||||
DTG/RPV | Juluca | DTG: 50 mg RPV: 25 mg | A complete regimen to replace the current ARV regimen for the treatment of HIV-1 infection in adults who are virologically stable and suppressed (HIV-1 RNA < 50 c/mL) | DTG: insomnia, headache, depression; early benign increase in SCr RPV: depression, insomnia, rash, headache; early benign increase in SCr |
DRV/c/TDF/FTC | Symtuza | DRV: 800 mg c: 150 mg TAF: 10 mg FTC: 200 mg | a complete regimen for the treatment of HIV-1 infection in adults and adolescents (aged ≥ 12 years and weighing ≥ 40 kg) and with no known mutations associated with resistance to the individual components | DRV: diarrhea, nausea, headache, rash, hyperlipidemia; drug-induced hepatotoxicity in DRV/r (rare); all PIs: risk of ECG abnormalities (i.e., PR interval prolongation) c: can falsely increase SCr TAF: Similar to TDF, but may have less renal and bone toxicity FTC: discoloration of skin (hands and/or feet) |
EVG/c/TDF/FTC | Stribildc | EVG: 150 mg c: 150 mg FTC: 200 mg TDF: 300 mg | A complete regimen for the treatment of adults aged ≥ 18 years infected with HIV-1 with no known mutations to the INSTI class, tenofovir, or FTC | EVG: nausea, diarrhea, insomnia, headache, depression; early benign increase in SCr c: can falsely increase SCr FTC: discoloration of skin (hands and/or feet) TDF: renal toxicity; decreased BMD, increased osteoporotic fractures; reports of lactic acidosis, hepatotoxicity |
RPV/TDF/FTC | Complerac | RPV: 25 mg TDF: 300 mg FTC: 200 mg | A complete regimen for the treatment of adults infected with HIV-1 with no known RAMs to the NNRTI class, tenofovir, or FTC, and with a VL ≤ 100,000 c/mL | RPV: depression, insomnia, rash, headache; early benign increase in SCr TDF: renal toxicity; decreased BMD, increased osteoporotic fractures; reports of lactic acidosis, hepatotoxicity FTC: discoloration of skin (hands and/or feet) |
EFV/TDF/FTC | Atriplad | EFV: 600 mg TDF: 300 mg FTC: 200 mg | For use alone as a complete regimen or in combination with other ARVs for the treatment of HIV-1 infection in adults | EFV: insomnia, vivid dreams, depressed mood, dizziness, headache, rash; avoid in patients with a history of anxiety, depression, or psychosis; contraindicated in the first trimester of pregnancy TDF: renal toxicity; decreased BMD, increased osteoporotic fractures; reports of lactic acidosis, hepatotoxicity FTC: discoloration of skin (hands and/or feet) |
Additional relevant comparator regimens | ||||
DRV/c + TAF/FTC | Prezcobixc Descovy | DRV/c: 800 mg/150 mg TAF/FTC: 10 mg/200 mg 25 mg/200 mg | In combination with other ARVs for the treatment of HIV infection in treatment-naive and in treatment-experienced patients without DRV RAMs In combination with other ARVs (such as NNRTIs or PIs) for the treatment of HIV-1 infection in adults and adolescents aged ≥ 12 years (and weighing ≥ 35 kg) | DRV: diarrhea, nausea, headache, rash, hyperlipidemia; drug-induced hepatotoxicity in DRV/r (rare); all PIs: risk of ECG abnormalities (i.e., PR interval prolongation) c: can falsely increase SCr TAF: similar to TDF, but may have less renal and bone toxicity FTC: discoloration of skin (hands and/or feet) |
DTG + TAF/FTC | Tivicay Descovy | DTG: 50 mg TAF/FTC: 10 mg/200 mg 25 mg/200 mg | Treatment of HIV-1 infection in adults and in INSTI-naive children weighing ≥ 30 kg In combination with other ARVs (such as NNRTIs or PIs) for the treatment of HIV-1 infection in adults and adolescents aged ≥ 12 years (and weighing ≥ 35 kg) | DTG: insomnia, headache, depression; early benign increase in SCr TAF: similar to TDF, but may have less renal and bone toxicity FTC: discoloration of skin (hands and/or feet) |
DRV+ r + TDF/FTC | Prezistac | DRV: 800 mg | Coadministered with 100 mg ritonavir and with other ARVs for the treatment of HIV-1 infection | DRV: diarrhea, nausea, headache, rash, hyperlipidemia; drug-induced hepatotoxicity in DRV/r (rare); all PIs: risk of ECG abnormalities (i.e., PR interval prolongation) r: diarrhea, nausea, headache, paresthesias, rash, hyperlipidemia; drug-induced hepatotoxicity in DRV/r (rare); all PIs: risk of ECG abnormalities (i.e., PR interval prolongation) TDF: renal toxicity; decreased BMD, increased osteoporotic fractures; reports of lactic acidosis, hepatotoxicity FTC: discoloration of skin (hands and/or feet) |
Norvirc | r: 100 mg | In combination with other ARVs for the treatment of HIV infection when therapy is warranted | ||
Truvada, generics | TDF: 300 mg | In combination with other ARVs (such as NNRTIs or PIs) for the treatment of HIV-1 infection in adults | ||
FTC: 200 mg | ||||
DTG + TDF/FTC | Tivicay | DTG: 50 mg | Treatment of HIV-1 infection in adults and in INSTI-naive children weighing ≥ 30 kg | DTG: insomnia, headache, depression; early benign increase in SCr TDF: renal toxicity; decreased BMD, increased osteoporotic fractures; reports of lactic acidosis, hepatotoxicity FTC: discoloration of skin (hands and/or feet) |
Truvada, generics | TDF: 300 mg | In combination with other ARVs (such as NNRTIs or PIs) for the treatment of HIV-1 infection in adults | ||
FTC: 200 mg | ||||
3TC = lamivudine; ABC = abacavir; ARV = antiretroviral; BIC = bictegravir; BMD = bone mineral density; c = cobicistat; c/mL = copies per millilitre; DOR = doravirine; DRV = darunavir; DTG = dolutegravir; ECG = electrocardiogram; EFV = efavirenz; EVG = elvitegravir; FTC = emtricitabine; HIV-1 = HIV, type 1; INSTI = integrase strand transfer inhibitor; MI = myocardial infarction; NNRTI = nonnucleoside reverse transcriptase inhibitor; PI = protease inhibitor; PR interval = time from the beginning of the P wave, indicating atrial depolarization, to the beginning of the QRS complex; r = low-dose ritonavir; RAM = resistance-associated mutation; RPV = rilpivirine; SCr = serum creatinine; TAF = tenofovir alafenamide; TDF = tenofovir disoproxil fumarate; VL = viral load.
aAll regimens are administered orally once daily.10
bHealth Canada indication.
cMust be taken with food or a meal.10
dMust be taken on an empty stomach.10
Source: Cabotegravir and rilpivirine (Vocabria, Cabenuva) clinical guidance report (Table 2).11
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups.
One patient group submitted input: the CBRC. The CBRC is a nonprofit charitable organization based in Vancouver, British Columbia, that promotes the health of people of diverse sexualities and genders through research and intervention. The CBRC collected information via Sex Now, a community-based research initiative and Canada’s largest running survey of gay, bisexual, and queer men (cis and trans), and nonbinary and 2-spirit people’s health in 2021 (n = 325) and in 2022 (n = 144).
The group said the outcome of untreated HIV is disability and premature death. According to the input,, people living with HIV, the most stigmatized disease worldwide, are too often viewed by society, public health, governments, the legal system, and researchers as a vector of disease. As a result, the experience of living with HIV is reduced to whether someone can transmit HIV, rather than being viewed as a health condition that is part of lived experience with disease. Pill burden and medication adherence are challenges for many, and certain socioeconomic factors and/or social determinants of health (e.g., housing and food insecurity) make it more challenging. About a third of Sex Now 2021 survey respondents reported that, due to the U = U (undetectable = untransmittable) campaign, they experienced a reduction in stigma, shame, and rejection, and about a third experienced an improvement in mental, social, and sexual well-being. However, nearly 20% of respondents felt pressured to take medication or maintain an undetectable viral load due to the U = U campaign. Moreover, the Sex Now 2021 survey conducted online showed a positive correlation between a suppressed viral load and having a health care provider, and between viral load and the ease of taking medicine. According to the Sex Now 2022 survey conducted at Pride Festivals and other queer spaces, 19% of respondents said they prefer taking daily oral pills, whereas 47% of respondents said they prefer injectables. This result shows a strong desire among the 2-spirit, lesbian, gay, bisexual, transgender, queer or questioning, and additional sexual orientations and gender identities (2SLGBTQ+) community for innovation in HIV treatments (e.g., long-acting drugs) to reduce the burden of taking medication.
The input stated that for HTE people living with HIV, there are no other treatment options. The patient group noted that it would be highly unethical for this drug to be unavailable, because untreated HIV can lead to disability and premature death and can increase the likelihood of passing on HIV when sexually active or sharing injection supplies with others. In general, this population faces barriers in the social determinants of health. The input suggests that considerations need to be made as to how pharmaceutical companies are supporting medication adherence outside of the medical model (e.g., social supports, income supports, food security, housing security, mental health support).
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol; assisting in the critical appraisal of clinical evidence; interpreting the clinical relevance of the results; and providing guidance on the potential place in therapy). The following input was provided by a clinical specialist with expertise in the diagnosis and management of patients living with HIV.
Unmet Needs
According to the clinical expert consulted by CADTH on this review, not all patients respond to available treatments. According to the clinical expert, many patients with prior exposure to ARVs develop resistance to individual medications and whole classes of medications, depending on the type of resistance mutations. The clinical expert noted that patients with increasing ARV resistance experience increasingly significant negative outcomes related to HIV (including lower life expectancy, higher burden of opportunistic and other chronic diseases, and greater treatment-related complications), as do those with multiple classes of resistance.
According to the clinical expert consulted by CADTH, there continue to be patients for whom medication intolerance or adverse reactions (including lipodystrophy, neuropsychiatric consequences, weight gain, metabolic disease) are a consistent barrier to use; therefore, more treatments are needed to address these gaps.
The clinical expert noted that, by definition, there are limited treatment options for HTE patients because all or nearly all of the safe, effective, and easily administered regimens have lost any efficacy due to resistance. Therefore, the clinical expert noted that clinicians are forced to use therapies with lower viral efficacy, greater associated harms (in the form of AEs), and that are more difficult to administer (i.e., subcutaneous injections). All of these factors all make treatment adherence challenging to maintain in the long-term. In summary, the clinical expert noted that access to well-tolerated, effective, new antiviral drugs from novel classes are needed to improve care for this group.
Place in Therapy
According to the clinical expert consulted by CADTH on this review, fostemsavir would be used for HTE patients and for other patients for whom there are limited options for treatment as a result of underlying disease state or drug resistance. The clinical expert noted that fostemsavir would provide a new class of anti-HIV therapies for use in treatment-experienced patients with drug-resistant HIV infection, for whom outcomes are poor. The clinical expert went on to note that a well-tolerated oral treatment for this patient population would be used in cases of drug resistance. The clinical expert noted that currently, patients with resistance to 3 or more classes of ARV therapy typically require medications from 3 or 4 classes and the use of multiple modalities (combining oral and injectable) therapies, or they are dependent on access to clinical trials. Therefore, according to the clinical expert, new oral therapies are needed to improve virologic response, clinical outcomes, and adherence to treatment to manage HIV in this context.
Patient Population
According to the clinical expert consulted by CADTH on this review, patients who are treatment-experienced and patients who have multidrug-resistant HIV would respond to treatment with fostemsavir. These patients are in need of such interventions.
The clinical expert noted that HIV specialists would identify patients for whom fostemsavir would be appropriate, based on clinical history, treatment history (i.e., ARV exposure and outcomes), and resistance testing of the patient’s virus. According to the clinical expert, these patients are not difficult to identify in clinical practice, and the testing required to facilitate treatment is already routinely performed in their care and management. The clinical expert noted that the patients most likely to respond to fostemsavir can be identified with the previously mentioned assessments.
Assessing Response to Treatment
According to the clinical expert consulted by CADTH on this review, viral load is the most important test to determine response to treatment. Clinical response (e.g., resolution of disease-related symptoms, immune reconstitution, rate of opportunistic infections, survival) will add supplemental evidence of treatment response.
The clinical expert noted that a clinically meaningful response to treatment would be improvement in or suppression of viral load, recovery of immune function (predominantly measured by CD4+ count), alongside resolution or stability in HIV-related symptoms, if present, the presence and/or prevention of opportunistic infections, and improvement or stability in related chronic diseases (e.g., anemia, thrombocytopenia), if present. The treatment outcome is unlikely to vary by physician.
Discontinuing Treatment
The clinical expert consulted by CADTH on this review noted that the following factors would be considered when deciding to discontinue fostemsavir:
lack of response to treatment and/or evidence of resistance based on phenotypic or genotypic resistance testing
AEs (i.e., untreatable or irreversible side effects that render the medication intolerable to the patient, or cases in which continuation of the drug would be life-threatening or organ-threatening, such as hypersensitivity, liver disease, or unstable cardiac arrhythmia)
patient preference.
Prescribing Conditions
According to the clinical expert consulted by CADTH on this review, specialty clinics (e.g., infectious diseases, internal medicine), and in some cases community clinics with HIV expertise, are the most appropriate settings for the treatment and monitoring of HTE patients with HIV. The clinical expert noted that, in Canada, the majority of patients with HIV are managed in these settings.
Clinician Group Input
No clinician group input was received for this submission.
Drug Program Input
Drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical expert consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Considerations for initiation of therapy | |
The indication corresponds to the unstable and unsuppressed HTE population with 4-class resistance and 0 to 2 fully active classes of treatment remaining who are unable to construct a suppressive regimen due to resistance, intolerance, or safety considerations. 1. What would provide clarity or define acceptable intolerance and safety considerations? 2. What parameters are used to measure resistance? | 1. The clinical expert noted that although safety issues can be measured more objectively, the impact of tolerability is based on agreement between physician and patient and the ability to mitigate side effects. 2. The clinical expert noted that resistance is measured objectively, and resistance profile and genotyping are assessed based on laboratory assessments. There is also clinically defined resistance, in which a patient is not responding to the drug (e.g., viral load is not reduced) and, in these patients, the most common issue is nonadherence to therapy. |
Considerations for discontinuation of therapy | |
Virologic response (HIV-1 RNA < 40 c/mL) was assessed at each time point using the FDA snapshot algorithm, which considers only HIV-1 RNA level at the visit of interest. The indication is for combination use with other ARVs. End points in the study included virologic response, change in CD4+ cell count, and gp120 polymorphisms. 1. The clinical report mentions clinically meaningful CD4+ T-cell count. Could this be defined, and at what point would fostemsavir be discontinued? 2. Virologic response (HIV-1 RNA < 40 c/mL) was assessed at each time point using the FDA snapshot algorithm. Does this align with the way virologic response is assessed in Canada? 3. Would both the HIV-1 RNA and CD4+ T-cell count be used to determine discontinuation, and at what level would this be? For the gp120 polymorphism additional end point, what is the clinical significance in the use fostemsavir? 4. If the patient can no longer take their current ARV due to intolerance or safety considerations, is fostemsavir discontinued or can a patient continue on fostemsavir? 5. What would be a reasonable amount of time for a patient on fostemsavir to see a clinical meaning full response before discontinuing? | 1. The clinical expert consulted by CADTH on this review noted that CD4+ count is unlikely to be used to determine whether or not a patient should discontinue therapy. 2. The clinical expert noted that virologic response is typically assessed every 6 months in stable patients. In patients who are changing therapies, virologic response could be assessed every 1 to 3 months. When initiating a new treatment or when a patient is unstable, assessment of virologic response is typically limited by availability of the patient and what is permitted by jurisdictions; therefore, an assessment every 4 to 6 weeks would be sufficient. 3. The clinical expert noted that virologic response targeting < 40 c/mL or < 50 c/mL (depending on the assay used) would be used to determine response. The clinical expert went on to note that a higher viral load may be tolerated in this HTE population if a clinical response is observed (so a target of 250 to 1,000 c/mL). The clinical expert noted that gp120 is important for establishing HIV subtype and susceptibility, vis-a-vis resistance, to the drug. The clinical expert went on to note that there were no concerns with fostemsavir in this regard, based on the results from the BRIGHTE study. 4. The clinical expert noted that patients who need to discontinue their ARV due to intolerance and/or safety would not continue on fostemsavir as monotherapy. 5. The clinical expert noted that a trial of 3 to 6 months would be used to assess a clinically meaningful response, depending on the patient’s viral load. |
Based on the clinical trial, the sponsor claims that for the HTE multidrug-resistant patient population, fostemsavir undoubtedly provides substantial clinical and economic certainty for patients, clinicians, and payers for a minimum of 5 years, per the 240-week data. An ARV regimen is typically composed of 2 or 3 fully active ARV drugs from 2 different classes to suppress HIV-1 RNA to below assay quantification limits (< 20 to 50 c/mL). An undetectable viral load is clinically presented as HIV-1 RNA < 50 c/mL.
| The clinical expert consulted by CADTH on this review noted that treatment would indeed continue past 5 years if the patient demonstrated sustained virologic efficacy. |
Considerations for prescribing of therapy | |
Concerns related to accessing clinical specialists and/or special settings Is access to infection disease specialists a concern in all jurisdictions? Could a physician or nurse practitioner provide this medication? | Patients could be followed by a physician or nurse practitioner trained to manage the treatment of THE patients living with HIV. Jurisdictional issues are a concern, but most patients with HIV live near large centres with access to care. Always initiated by, or in conjunction with, an HIV specialist, but could be managed in a community care setting. |
ARV = antiretroviral therapy; c/mL = copies per millilitre; CD4+ = cluster of differentiation 4; gp = glycoprotein; HTE = heavily treatment-experienced.
Clinical Evidence
The clinical evidence included in the review of fostemsavir is presented in 3 sections. The first section, the Systematic Review, includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as studies that were selected according to an a priori protocol. The second section includes indirect evidence from the sponsor and indirect evidence selected from the literature that met the selection criteria specified in the review. The third section normally includes sponsor-submitted long-term extension studies and additional relevant studies considered to address important gaps in the evidence included in the Systematic Review; however, none were identified for this report.
Systematic Review (Pivotal and Protocol-Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of fostemsavir (extended-release tablets, 600 mg) in combination with other ARVs for the treatment of HIV-1 infection in HTE adults with multidrug-resistant HIV-1 infection.
Methods
Studies selected for inclusion in the Systematic Review include pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 5. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
Table 5: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Population | HTE adults who have multidrug-resistant HIV-1 infection, and for whom it is otherwise not possible to construct a suppressive antiviral regimen due to resistance, intolerance, or safety considerations Subgroups of interest
|
Intervention | Fostemsavir extended-release tablets, 600 mg, administered orally twice daily, plus standard of care |
Comparator | Any effective combination of antiviral therapies from susceptible classes:
|
Outcomes | Key outcomes:
Harms outcomes:
Notable harms (QT prolongation, immune reconstitution inflammatory syndrome, elevated liver enzymes [patients coinfected with hepatitis B virus or hepatitis C virus]) |
Study designs | Published and unpublished phase III and IV RCTs |
ARV = antiretroviral; c/mL = copies per millilitre; CCR5 = C-C chemokine receptor 5; CD4+ = cluster of differentiation 4; HIV-1 = HIV, type 1; HRQoL = health-related quality of life; HTE = heavily treatment-experienced; INSTI = integrase strand transfer inhibitor; NNRTI = nonnucleoside reverse transcriptase inhibitor; NRTI = nucleoside reverse transcriptase inhibitor; PI = protease inhibitor; RCT = randomized controlled trial.
The literature search was performed by an information specialist using a peer-reviewed search strategy. The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy, according to the PRESS Peer Review of Electronic Search Strategies checklist.1
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946–) via Ovid and Embase (1974–) via Ovid. All Ovid searches were run simultaneously as a multifile search. Duplicates were removed using Ovid deduplication for multifile searches, followed by manual deduplication in Endnote. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concept was Rukobia (fostemsavir). The following clinical trials registries were searched: the US National Institutes of Health’s clinicaltrials.gov, WHO’s International Clinical Trials Registry Platform (ICTRP) search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
No filters were applied to limit the retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. Refer to Appendix 1 for the detailed search strategies. The initial search was completed on November 22, 2022. Regular alerts updated the search until the meeting of the CADTH Canadian Drug Expert Committee (CDEC) on March 22, 2023.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool For Searching Health-Related Grey Literature checklist.2 Included in this search were the websites of regulatory agencies (FDA and European Medicines Agency). Google was used to search for additional internet-based materials. Refer to Appendix 1 for more information on the grey literature search strategy. These searches were supplemented by reviewing bibliographies of key papers and through contacts with appropriate experts. In addition, the sponsor of the drug was contacted for information regarding unpublished studies.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
A focused literature search for indirect treatment comparisons (ITCs) dealing with Rukobia (fostemsavir) and HIV-1 was run in MEDLINE All (1946–) on November 22, 2022. No limits were applied.
Findings From the Literature
A total of 1 study was identified from the literature for inclusion in the Systematic Review (Figure 1). The included study is summarized in Table 6.
Table 6: Details of the Included Study
Detail | BRIGHTE |
|---|---|
Designs and populations | |
Study design | DB RCT |
Locations | 108 centres, 22 countries in Africa, Asia-Pacific, Europe, North America, and South America |
Patient enrolment dates | February 23, 2015 (first visit) to August 11, 2016 (first dose in last patient) |
Randomized (N) | 272 |
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Fostemsavir 600 mg orally twice daily Plus, failed ARV regimen (DB phase) then OBT (OL phase) |
Comparators | Placebo (matched) 600 mg orally twice daily for 8 days (DB phase) Fostemsavir 600 mg orally twice daily thereafter (OL phase) Plus, failed ARV regimen (DB phase) then OBT (OL phase) |
Duration | |
Phase | |
Screening | 42 days |
DB | 8 days |
OL | At least 96 weeks, continuing until an additional option, a rollover study, or marketing approval is in place |
Follow-up | NR |
Outcomes | |
Primary end point | HIV-1 RNA at day 8 in the randomized cohort |
Secondary and exploratory end points | Percentage of patients with a decrease in the HIV-1 RNA level of > 0.5 log10 copies and of > 1.0 log10 c/mL at day 8 Virologic response (HIV-1 RNA level, < 40 c/mL) at weeks 24, 48, and 96 Resistance testing for all patients meeting the criteria for PDVF Mean change in CD4+ T-cell count through week 96 Events resulting in a diagnosis of AIDS, class C of the classification system of the Centers for Disease Control and Prevention Safety Adverse events Serious adverse events Adverse events leading to discontinuation |
Notes | |
Publications | Kozal et al. (2020)12 |
ALT = alanine transaminase; ARV = antiretroviral; AST = aspartate transaminase; ATV = atazanavir; c/mL = copies per millilitre; CD4+ = cluster of differentiation 4; CHF = congestive heart failure; DB = double-blind; HBV = hepatitis B virus; HIV-1 = HIV, type 1; HIV-2 = HIV, type 2; NR = not reported; OBT = optimized background therapy; OL = open-label; PDVF = protocol-defined virologic failure; RCT = randomized controlled trial; ULN = upper limit of normal.
Note: Five additional reports were included (CSRs for the BRIGHTE study, the sponsor’s submission, FDA clinical and statistical review).
Source: Clinical Study Report for BRIGHTE.6
Description of Studies
One pivotal, phase III, sponsor-funded, multinational (108 centres in 22 countries, including 3 sites in Canada) study was included in this review (Table 6). The BRIGHTE study consisted of an initial DB phase that lasted 8 days and a subsequent OL phase that will remain ongoing through 240 weeks. The primary objective was to compare the efficacy of fostemsavir relative to placebo, when given on the background of a failing regimen, by determining the mean change in log10 HIV-1 RNA from day 1 to day 8 in the randomized cohort. The secondary objectives were to assess the durability of patients’ responses to fostemsavir when given with OBT by determining the proportion of participants with a plasma HIV-1 RNA level below 40 copies/mL at weeks 24, 48 and 96 in the randomized cohort, safety and tolerability, disease progression (by measuring emergence of AIDS-defining events or death), the emergence of resistance in patients with PDVF, and the efficacy of fostemsavir plus OBT by examining changes from baseline in log10 HIV-1 RNA, CD4+ cell counts, and percent of CD4+ T-cells through weeks 24, 48 and 96.
In the DB phase, 272 patients with HIV-1 who were eligible to receive at least 1 fully active, approved ARV in 1 or 2 ARV classes at baseline were randomized, in a 3:1 ratio, to fostemsavir 600 mg twice daily or placebo, plus their baseline failing ARV regimen, for 8 days. Randomization was stratified by baseline HIV-1 RNA (up to 1,000 copies/mL or greater than 1,000 copies/mL). The primary analysis was conducted after 8 days and consisted of the primary outcome, the mean change from baseline to day 8 in HIV-1 RNA. In addition to this randomized cohort, there was a nonrandomized cohort that consisted of 99 patients who had no other options for fully active and approved ARV; these patients received fostemsavir plus OBT (determined based on resistance testing and treatment history) from the beginning of the study. In the randomized cohort, after day 8, patients entered an OL phase during which they all received fostemsavir plus OBT. The study began in August 2016, and was expected to last at least 96 weeks and continue until an additional option, a rollover study, or marketing approval was in place. The data for this review were primarily obtained from the 96-week CSR, dated May 9, 2019, and the 240-week CSR, dated March 10, 2022. The 96-week CSR was provided by the sponsor as part of their submission to CADTH, whereas the 240-week CSR is the most recent CSR available from this ongoing study and was provided to CADTH after a request to the sponsor. A schematic of study design is presented in Figure 2.
Figure 2: Study Design for BRIGHTE
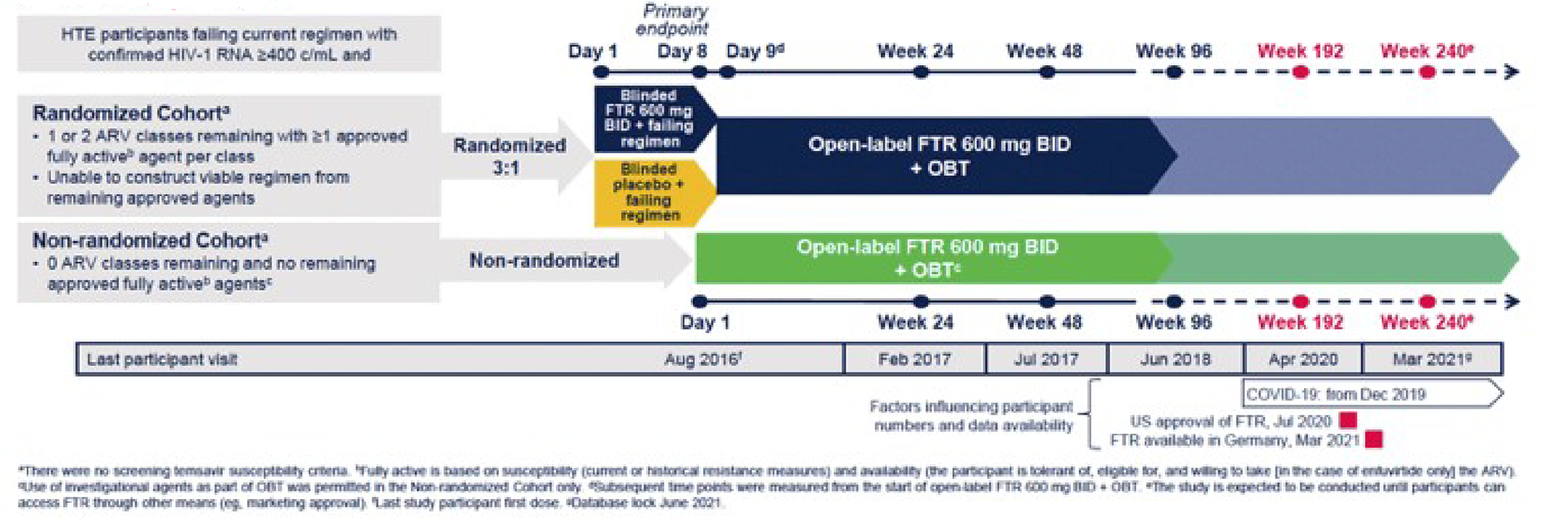
ARV = antiretroviral; BID = twice daily, c/mL = copies per millilitre; FTR = fostemsavir; HIV-1 = HIV, type 1; HTE = heavily treatment-experienced; OBT = optimized background therapy.
Source: Aberg et al. (2022).13
Populations
Inclusion and Exclusion Criteria
Adults with chronic HIV-1 who were ARV experienced with documented historical baseline resistance, intolerance, and/or contraindications to ARV in at least 3 classes were randomized in the BRIGHTE study. Patients also had to be failing their current ARV regimen, with a confirmed plasma HIV-1 RNA level of at least 400 copies/mL, and must have had at least 1 fully active drug available in 2 or fewer classes, based on current and/or documented historical resistance testing, taking into account tolerability and other safety concerns. Patients in the nonrandomized cohort had 0 ARV classes remaining and had no remaining approved fully active drugs.
Patients were excluded if they had chronic untreated hepatitis B virus, indicators of poor hepatic function, a history of decompensated cirrhosis or active decompensated cirrhosis, or a history of congestive heart failure or long QT syndrome.
Baseline Characteristics
Patients in the randomized cohort were approximately 44 years of age, and the majority were male (74% of patients) and white (68% of patients) (Table 7). Most patients (89%) had a baseline viral load of 1,000 copies/mL or higher, and more than half (57% of patients) had a baseline viral load of 30,000 copies/mL or more. Approximately one-quarter of patients had a CD4+ count of less than 20 cells/mm3, and a similar percentage had a baseline CD4+ count of 200 cells/mm3 or more. Approximately one-third of patients had been treated for HIV for more than 20 years, and 85% of patients overall had a positive AIDS history, meaning that they either had a nadir CD4+ count of less than 200 cells/mm3 or a response of yes to the question, “Does participant have AIDS?” on the disease history component of the CRF. Most patients (90% or more) had prior exposure to an NNRTI, NRTI, or PI, and 75% had prior exposure to an INSTI. Other ARV patients had prior exposure to included entry inhibitors (39%), CCR5 antagonists (26%), and other investigational ARV (11%). The most common ARV classes in the failing regimen were NRTIs (81%), PIs (67%), INSTIs (44%), and NNRTIs (28%); other classes included CCR5 antagonists (12%) and entry inhibitors (4%).
There were some numerical differences in baseline characteristics between the fostemsavir and placebo groups, including a lower percentage of males in the fostemsavir group than in the placebo group (70% versus 83%), a lower mean (± SD) CD4+ count in the fostemsavir group (146.6 ± 173.8 cells/mm3 versus 170.0 ± 204.8 cells/mm3), a lower percentage of patients on fostemsavir with 2 or more fully active and available ARV classes at screening (60% versus 70%) and in their initial OBT (39% versus 49%).
Table 7: Summary of Baseline Characteristics (ITT-E Population)
Characteristic | Randomized cohort | Nonrandomized cohort | |
|---|---|---|---|
Fostemsavir N = 203 | Placebo N = 69 | Fostemsavir N = 99 | |
Mean (SD) age | 45.2 (12.7) | 43.0 (11.0) | 48.1 (11.5) |
Male, n (%) | 143 (70) | 57 (83) | 89 (90) |
Race, n (%) | |||
White | 137 (67) | 48 (70) | 74 (75) |
Black or African American | 42 (21) | 18 (26) | 23 (23) |
Asian | 2 (< 1) | 0 | 0 |
American Indian or Alaska Native | 6 (3) | 1 (1) | 1 (1) |
Native Hawaiian or other Pacific Islander | 1 (< 1) | 0 | 0 |
Other | 15 (7) | 2 (3) | 1 (1) |
Baseline HIV-1 RNA, c/mL, n (%) | |||
< 400 | 14 (7) | 7 (10) | 5 (5) |
400 to < 1,000 | 7 (3) | 3 (4) | 4 (4) |
||||| || ||||||| | || |||| | || |||| | || |||| |
|||||| || ||||||| | || |||| | | |||| | || |||| |
|||||| || |||||||| | || |||| | || |||| | || |||| |
||||||| || |||||||| | || |||| | || |||| | || |||| |
|||||||| | || ||| | | |||| | | ||| |
Mean (SD) HIV-1 RNA, log10 c/mL | 4.44 (0.98) | 4.38 (1.18) | 4.20 (0.89) |
Baseline CD4+, cells/mm3, n (%) | |||
< 20 | 55 (27) | 17 (25) | 40 (40) |
20 to < 50 | 19 (9) | 6 (9) | 14 (14) |
|| || |||| | || |||| | || |||| | || |||| |
||| || |||| | || |||| | || |||| | || |||| |
||| || |||| | || |||| | || |||| | || |||| |
||| || |||| | || ||| | | ||| | | ||| |
≥ 500 | 11 (5) | 4 (6) | 2 (2) |
Mean (SD) CD4+ cells/mm3 | 146.6 (173.8) | 170.0 (204.8) | 99.4 (130.8) |
Number of years treated for HIV, n (%) | |||
|| | | ||| | | |||| | ||| |
|||| | || |||| | | |||| | | ||| |
||||| | || |||| | || |||| | || |||| |
||||| | || |||| | || |||| | || |||| |
||| | || |||| | || |||| | || |||| |
||||||| | | ||| | | ||| | | ||| |
AIDS history, n (%) | |||
Yes | 170 (84) | 61 (88) | 89 (90) |
No | 33 (16) | 8 (12) | 10 (10) |
Prior exposure, n (%) | |||
INSTI | 149 (73) | 55 (80) | 94 (95) |
NRTI | 202 (> 99) | 68 (99) | 97 (98) |
PI | 193 (95) | 64 (93) | 97 (98) |
NNRTI | 188 (93) | 60 (87) | 93 (94) |
Entry inhibitor | 81 (40) | 26 (38) | 67 (68) |
CCR5 antagonist | 52 (26) | 20 (29) | 40 (40) |
Other investigational ARV | 24 (12) | 7 (10) | 21 (21) |
||| ||||||| || ||||||| |||||||| | ||| | |||
|||| |||||||||| | || |||| | | |||| | || |
||||| ||||||||| | | ||| | | ||| | || |
||||| | || |||| | || |||| | || |
||||| | || |||| | || |||| | || |
|||| | ||| |||| | || |||| | || |
||| | ||| |||| | || |||| | || |
||||| |||||| ||| ||||||||| ||| ||||||| || |||||||||| | ||| | |||
||| ||||||| | | | | ||| | || | || |||| |
||| ||||||| | | | || |||| | || |||| | | ||| |
||| ||||||| | | | ||| |||| | || |||| | || |
||| ||||||| || | | ||| | | ||| | || |
||||| |||||| ||| ||||||||| ||| ||||||| || ||||||| |||| | ||| | |||
ARV classes = 0 | 15 (7) | 1 (1) | 80 (81) |
ARV classes = 1 | 108 (53) | 34 (49) | 19 (19) |
ARV classes = 2 | 80 (39) | 34 (49) | 0 |
||| ||||||| || | || | || | || |
ARV = antiretroviral; c/mL = copies per millilitre; CCR5 = C-C chemokine receptor 5; CD4+ = cluster of differentiation 4; HIV-1 = HIV, type 1; INSTI = integrase strand transfer inhibitor; ITT-E = intention-to-treat, exposed; NNRTI = nonnucleoside reverse transcriptase inhibitor; NRTI = nucleoside reverse transcriptase inhibitor; PI = protease inhibitor; SD = standard deviation.
Notes: AIDS history data are positive if a patient has nadir CD4+ count < 200 cells/mm3, or if the response to the question, “Does patient have AIDS?” on the disease history CRF is yes.
HIV subtype was determined from baseline entry and PhenoSense GT plus Integrase resistance testing performed at Monogram Biosciences. The determination of subtype prioritized envelope gene sequences, given the fostemsavir mechanism of action.
Initial OBT is the combination of ARVs that the patient is initiated on when they start participation in the OL phase of the study. Fostemsavir is not counted in the calculation of the number of fully active and available ARVs in the initial OBT.
Source: Clinical Study Report.6
Interventions
Fostemsavir was administered orally, at a dose of 600 mg twice daily, and during the 8-day DB phase, a matching placebo was used as a control. With respect to background therapy, during the 8-day DB phase in the randomized cohort, patients continued on their current failing ARV regimen, whereas during the OL phase, patients switched to OBT. Patients in the nonrandomized cohort began on fostemsavir plus OBT from the beginning of the study.
For patients enrolled in the study who were unable to construct a fully active regimen with existing ARV therapies, the selection of ARVs used as part of the OBT in combination with fostemsavir was based on remaining available active ARVs or other investigational drugs (investigational drugs were allowed, per protocol, only for the nonrandomized cohort) and was consistent with contemporaneous guidelines, which recommended the use of at least 2 but preferably 3 active drugs, based on analysis of current and/or prior drug resistance testing, to replace a previously failing regimen. A fully active drug is a drug that meets the following criteria: sensitive on the net assessment of the PhenoSense GT Plus Integrase; yes on the anticipated activity of the CCR5 coreceptor in HIV entry, per the Trofile coreceptor tropism assay; or susceptible on the PhenoSense Entry assay for enfuvirtide. Documented ARV or drug classes that patients are either intolerant of, ineligible for, or unwilling to take (due to route of administration, for example) did not apply toward the total number of active remaining ARVs.
The ARV drugs taken as the failing regimen or as OBT were administered based on local prescribing information. Dose modifications to the failing regimen used by patients in the randomized cohort during day 1 to day 8 were not allowed. During the conduct of the study, modifications to OBT (dosing or ARVs used) were permitted to be made by the investigator in both the randomized and the nonrandomized cohorts. Patients who switched OBT due to a lack of efficacy were counted as virologic failures in the reported data.
Outcomes
Progression to AIDS
The sponsor reported the occurrence of AIDS-defining events, or events that may result in a diagnosis of AIDS, over the course of the OL phase. The list of potential events included various infections (parasitic, fungal, bacterial, viral) and neoplasias (such as Kaposi’s sarcoma), as well as HIV dementia and HIV wasting syndrome. These appear consistent with the AIDS-defining conditions listed on the CDC website.14
Viral Load
The primary outcome of the BRIGHTE study was the change from day 1 to day 8 in HIV-1 RNA. Plasma HIV-1 RNA levels were quantified, using the Abbott RealTime HIV-1 assay, at screening, day 1 and day 8 during the DB phase, and at weeks 4, 8, 12, 16, 24, 36, 48 and every 12 weeks thereafter in the OL phase. The FDA snapshot algorithm was used in determining virologic response (achievement of HIV-1 RNA < 40 copies/mL). In the snapshot algorithm, patients without a value for HIV-1 RNA at the relevant time point or those who changed OBT due to lack of efficacy up to each time point were counted as treatment failures. A modified analysis was also performed, in which a change in OBT was not counted as treatment failure.
CD4+ Count
CD4+ counts were reported as a secondary outcome in both the DB and OL phases; however, no formal statistical significance tests were planned. CD4+ counts were performed using flow cytometry, and these assays were conducted at screening, day 1, day 8, and weeks 4, 8, 12, 16, 24, 36, 48 and every 12 weeks thereafter throughout the OL phase, or within 5 days of early termination.
Health-Related Quality of Life
The FAHI was reported as a secondary patient-reported outcome in the OL phase, with data collected at baseline and every 12 weeks thereafter. The recall period was not reported. No formal comparisons were planned, as there was no comparator in the OL phase. FAHI consists of 47 items, grouped into 5 subscales: physical well-being, global/functional well-being, emotional well-being/living with HIV, social well-being, and cognitive function. A scale of 0 (not at all) to 4 (very much) is used to score 44 of the items, and a total score is calculated from the subscales. Higher scores are associated with better quality of life.15 In treatment-experienced patients with HIV, based on EQ-5D index, the minimally important difference (MID) for total score ranged from 6.5 to 9.0, and based on ED-5D visual analogue scale (VAS), the MID for total score ranged from 3.2 to 5.8. Based on the distribution approach, the MID for total score ranged from 3.9 (standard error of mean) to 14.0 (0.5 × SD).
The 3-Level EQ-5D (EQ-5D-3L) was reported as a secondary outcome in the OL phase, with data collected at baseline and every 12 weeks thereafter. The recall period was not reported. The EQ-5D-3L consists of 5 questions covering the domains of mobility, pain, self-care, usual activities, and anxiety/depression, with 3 levels of response for each. Scores of these 5 dimensions can be converted to a single summary index utility score, with higher scores indicating improved HRQoL. In addition to the single index utility score, a 0 mm (worst health you can imagine) to 100 mm (best health you can imagine) VAS is also used as a simple measure of a patient’s self-rated health.16 An MID specific to patients living with HIV was not identified.
Resistance
The emergence of drug resistance or reduced susceptibility was assessed using phenotypic and genotypic resistance testing of isolates from participants identified as meeting the criteria for PDVF. Phenotypic results were expressed as the fold-change in IC50 for the test sample relative to a laboratory control HIV-1.12 The criteria for PDVF depended on time on the study. Prior to week 24, PDVF was defined as a confirmed measure of, or as the last available measure before discontinuation, HIV-1 RNA of at least 400 copies/mL at any time after before confirmed suppression to less than 400 copies/mL; or it was defined as confirmed, or as last available measure before discontinuation, of more than a 1 log10 copies/mL increase in HIV-1 RNA at any time above nadir level, where nadir is at least 40 copies/mL. PDVF at or beyond week 24 was defined as a confirmed measure, or as the last available measure before discontinuation, of HIV-1 RNA of at least 400 copies/mL. Genotype and phenotype testing was carried out by Monogram Biosciences using the PhenoSense GT plus Integrase, PhenoSense Entry, and Trofile assays.
Adherence
Adherence was evaluated at each treatment visit by staff, using dosing diaries, self-completed Modified-Medication Adherence Self-Report Inventory (M-MASRI) questionnaires, interviews with the patients, and examination of returned medication. The M-MASRI is a self-reported questionnaire that covers the percentage of times in the previous month that medication was taken and the percentage of doses taken within 2 hours of the correct time in the previous month.
A list of efficacy end points identified in the CADTH review protocol that were assessed in the clinical trials included in this review is provided in Table 8. These end points are summarized here. A detailed discussion and critical appraisal of the outcome measures is provided in Appendix 4.
Table 8: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure | BRIGHTE |
|---|---|
Mortality | Reported as harms |
Progression to AIDS | Reported as AIDS-related events in the DB phase and the OL phase |
Hospitalizations (overall and AIDS-related) | Not reported |
Viral load | Primary outcome (DB phase): change from baseline to day 8 in HIV-1 RNA |
Change in CD4+ | Secondary outcome (DB phase): change from baseline to day 8 in CD4+ |
HRQoL | Not reported for the DB phase change from baseline to week 96 reported for the OL phase |
Resistance | Not reported for the DB phase Reported for the OL phase |
Adherence | Not reported for the DB phase Reported for the OL phase, week 24 |
DB = double-blind; HIV-1 = HIV, type 1; HRQoL = health-related quality of life; OL = open-label.
Source: Clinical Study Report.6
Statistical Analysis
Primary Outcome of the Studies
Power Calculation
The sponsor planned to randomize at least 140 patients, in a 3:1 ratio, to either fostemsavir or placebo, respectively. The power of a single superiority comparison between fostemsavir and placebo was determined to be more than 95%, assuming the following: a 2-sided test; an alpha level of 0.05; a 0.5 log10 difference between the treatment groups; and a common SD of 0.6 log10. There was no target sample size for the nonrandomized cohort.
Statistical Test or Model
The primary end point of the efficacy of fostemsavir relative to placebo was assessed using the mean change in log10 HIV-1 RNA from day 1 to day 8, estimated by 1-way analysis of covariance (ANCOVA) in the randomized cohort, with a log10 HIV-1 RNA change from day 1 to day 8 as the dependent variable, treatment (fostemsavir or placebo) as an independent variable, and log10 HIV-1 RNA at day 1 as a continuous covariate. The mean was adjusted by day 1 log10 HIV-1 RNA. The hypothesis tested was that fostemsavir 600 mg would have antiviral efficacy superior to placebo in a randomized cohort of HTE patients infected with multidrug-resistant HIV-1 when given in combination with a failing background ARV regimen over a period of 8 days.
Data Imputation Methods
For the primary outcome, missing HIV-1 RNA values at day 8 were imputed using day 1 observation carried forward (i.e., imputing a change of 0 from day 1) for patients without a value during the DB phase, and last observation carried forward for patients with an early value during blinded treatment but before day 8.
The FDA snapshot analysis was used to assess dichotomous outcomes, such as virologic response. In this method, missing data are counted as failures.
Subgroup Analyses
No statistical testing of subgroups was performed.
Secondary Outcomes of the Studies
There were no statistical significance test analyses planned for secondary outcomes; rather, descriptive analyses were reported.
Analysis Populations
The safety population comprised all patients who received at least 1 dose of the study treatment. This population was based on the treatment the patient actually received. The intention-to-treat exposed (ITT-E) population included all randomized patients who received at least 1 dose of the study treatment. The randomized cohort is based on the treatment to which the patient was randomized (placebo or fostemsavir), regardless of the treatment actually received. Any patient who received a treatment randomization number was considered to have been randomized. The per-protocol population comprised all randomized patients in the ITT-E population who complied with the protocol.
Results
Patient Disposition
Disposition was not reported for the DB phase. For the OL phase, after 96 weeks, 22% of patients had withdrawn in the randomized cohort and 38% of patients had withdrawn in the nonrandomized cohort. The most common primary reasons for withdrawing in the randomized cohort were lack of efficacy (4%) and noncompliance with the study drug (4%), and the most common primary reason for withdrawing in the nonrandomized cohort was death (15%).
Disposition | Randomized cohort | Nonrandomized cohort | |
|---|---|---|---|
Fostemsavir | Placebo | Fostemsavir | |
Screened, N | 731 | ||
Enrolled and treated, n | 272 | 99 | |
Randomized, n | 203 | 69 | NA |
Week 96 | |||
Ongoing, n (%) | 159 (78) | 54 (78) | 61 (62) |
Completed | 0 | 0 | 0 |
Withdrawn, n (%) | 44 (22) | 15 (22) | 38 (38) |
Primary reason for study withdrawal, n (%) | |||
Lack of efficacy | 9 (4) | 3 (4) | 6 (6) |
Adverse event | 4 (2) | 3 (4) | 4 (4) |
Withdrawn by patient | 5 (2) | 0 | 1 (1) |
Death | 7 (3) | 2 (3) | 15 (15) |
Lost to follow-up | 4 (2) | 4 (6) | 1 (1) |
Noncompliance with study drug | 8 (4) | 3 (4) | 6 (6) |
Pregnancy | 1 (< 1) | 0 | 0 |
Patient no longer meets study criteria | 5 (2) | 0 | 4 (4) |
Other | 1 (< 1) | 0 | 1 (1) |
Randomized | 203 (100) | 69 (100) | NA |
ITT-E population | 203 (100) | 69 (100) | 99 (100) |
Week 240 | |||
Ongoing, n (%) | 104 (51) | 29 (42) | 23 (23) |
Completed | 39 (19) | 16 (23) | 25 (25) |
Withdrawn, n (%) | 60 (30) | 24 (35) | 51 (52) |
Primary reason for study withdrawal, n (%) | |||
Lack of efficacy | 11 (5) | 6 (9) | 10 (10) |
Adverse event | 4 (2) | 4 (6) | 5 (5) |
Withdrawn by patient | 10 (5) | 0 | 2 (2) |
Death | 8 (4) | 3 (4) | 17 (17) |
Lost to follow-up | 7 (3) | 5 (7) | 3 (3) |
Noncompliance with study drug | 11 (5) | 4 (6) | 6 (6) |
Pregnancy | 2 (< 1) | 0 | 0 |
Patient no longer meets study criteria | 6 (3) | 1 (1) | 6 (6) |
Other | 1 (< 1) | 1 (1) | 2 (2) |
ITT-E = intention-to-treat, exposed; NA = not applicable.
Source: Clinical Study Report.6
Exposure to Study Treatments
During the DB phase, the mean (SD) exposure in the fostemsavir group was 8.1 (1.1) days and in the placebo group was 8.2 (0.9) days. In the OL phase, after 96 weeks, the mean (SD) exposure was 722.0 (285.2) days in the cohort originally randomized to fostemsavir and 747.1 (265.6) in the cohort originally randomized to placebo. The ARV regimens that patients were on at screening and the ARV regimens that were part of their OBT are listed in Table 7 and described in the section on baseline characteristics. The percentage of patients who had their OBT switched during the DB and OL phases was not reported in the CSR; however, patients who were counted as virologic failures due to switching of their OBT was reported, and these data are reported in Table 11 as part of the virologic response analysis. After 96 weeks, 7% of patients in the randomized cohort had been originally randomized to fostemsavir and 10% had been originally randomized to placebo; in the nonrandomized cohort, 19% of patients had had their OBT switched, presumably due to lack of efficacy. After 240 weeks, 11% of patients in the randomized cohort had been originally randomized to fostemsavir and 13% had been originally randomized to placebo; in the nonrandomized cohort, 24% of patients had had their OBT switched.|Efficacy
Only efficacy outcomes and analyses of subgroups identified in the review protocol are reported here. Refer to Appendix 3 for detailed efficacy data.
Mortality
During the OL phase, after 96 weeks, 4% of patients had died in the randomized cohort, as had 17% of patients in the nonrandomized cohort. Overall, 7 deaths (2% of patients) were considered to be AIDS-related (Table 11). After 240 weeks, 6% of patients in the randomized cohort and 20% of patients in the nonrandomized cohort had died, and || |||||| ||| || ||||||||), overall, across both cohorts, were considered to be AIDS-related.
Progression to AIDS
In the DB phase, after 8 days there were 2 patients in the fostemsavir group who had an AIDS-related event (grade 3 SAE of recurrent pneumonia; grade 2 AEs of herpes simplex virus, gastrointestinal other than mouth, throat, perirectal) and 1 patient in the placebo group (grade 3 SAE of Candida esophagitis) (Table 10).
After 96 weeks in the OL phase, 9% of patients who were originally assigned to the fostemsavir group and 7% of patients who were originally assigned to the placebo group had an AIDS-related event. In the nonrandomized cohort, after 96 weeks, 15% had an AIDS-related event. ||||| ||| |||||| || of patients originally assigned to the fostemsavir group and || originally assigned to the placebo group had an AIDS-related event, whereas ||| of patients in the nonrandomized cohort had an AIDS-related event by |||| ||| (Table 11).
Hospitalizations
On-study hospitalizations were not reported.
Viral Load
The mean change from baseline to day 8 in plasma HIV-1 RNA log10 copies/mL was –0.791 (95% CI, –0.885 to –0.698) log10 copies/mL in the fostemsavir group and –0.166 (95% CI, –0.326 to –0.007) log10 copies/mL in the placebo group, for a difference between groups of –0.625 (95% CI, –0.810 to –0.441; P < 0.0001). There were 65% of fostemsavir patients and 19% of placebo patients who achieved a decrease in HIV-1 RNA of more than 0.5 log10 copies/mL by day 8 and 46% of fostemsavir patients and 10% of placebo patients who achieved a decrease in HIV-1 RNA of more than 1.0 log10 copies/mL (Table 10).
Figure 3: Percentage of Patients Achieving HIV-1 RNA < 40 Copies/mL Through Week 240
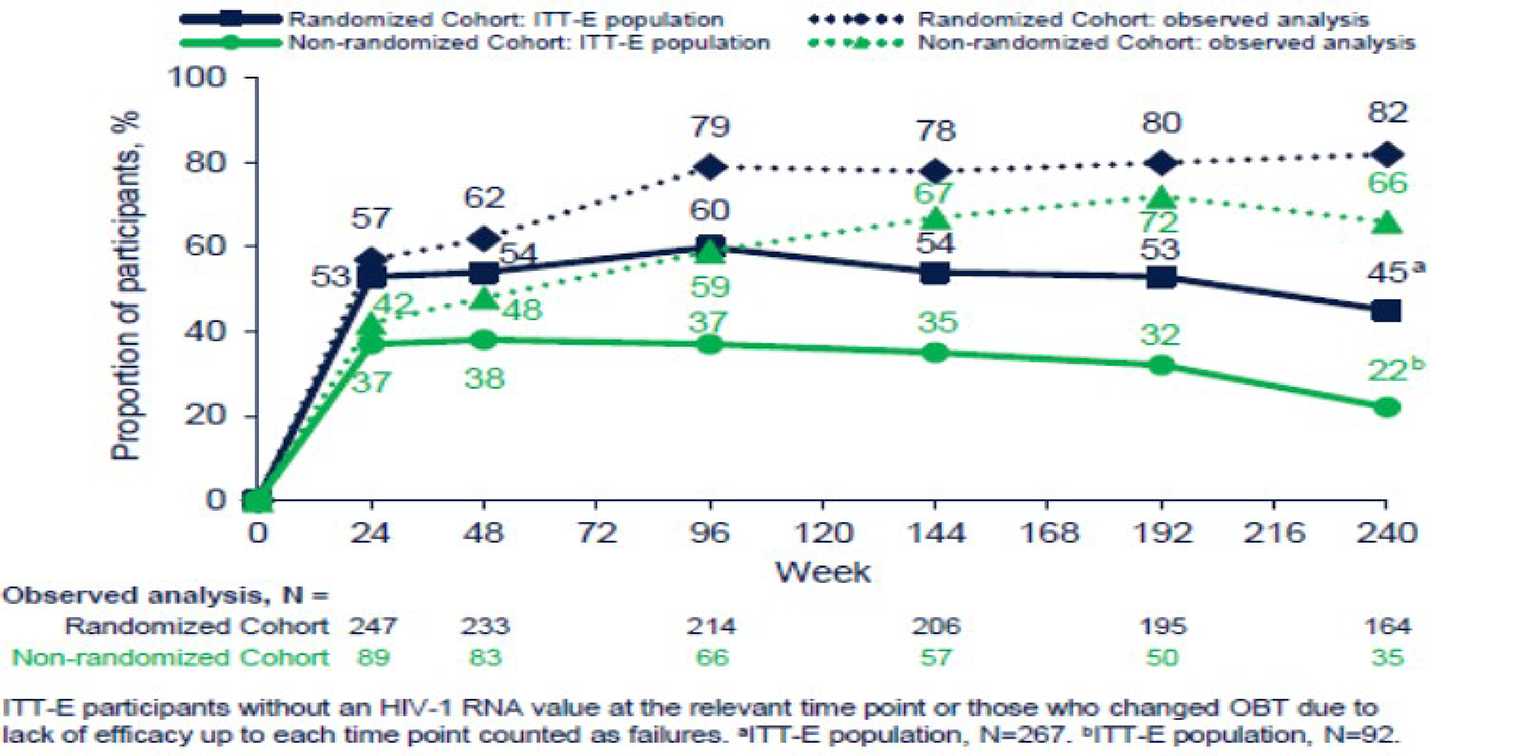
ITT-E = intention-to-treat, exposed.
Source: Aberg et al. (2022).13
Viral load was also reported during various time points in the OL phase as the percentage of patients who achieved an HIV-1 RNA level below 40 copies/mL. Figure 3 reports results for both the FDA snapshot analysis (ITT-E population) and the observed analysis. Note that in the figure, data for the randomized cohort combines the group that began on fostemsavir and the group that began on placebo in the DB phase.
Preplanned subgroup data were reported for the primary outcome for a number of subgroups of interest in our protocol, although no formal statistical significance test analyses were presented (Table 10). ||| |||||||| |||| | |||||||| |||||| ||||| ||| ||||| ||||||||| || |||||| ||||| ||| | |||| |||| ||||||||| |||| |||||||| || |||||| ||||| ||| ||||| ||||||||| || |||||| ||||||| |||| ||||||||||| ||| |||||| ||||||| ||||| ||||||||| |||| ||||||| ||| ||| |||||||| |||| |||||||| ||||| ||| || |||| |||| |||| ||||| ||||||||| ||||| ||| | |||| |||| ||||||||| || |||||| ||||| ||| || |||||| ||||||| ||||| ||||||||| |||| ||||||||||| ||| ||||| ||||||| ||||| ||||||||| |||| ||||||||
|||||||| |||| ||| ||||| |||| ||||| || |||||||| |||| ||||| ||| |||| ||||||||| |||||||| |||| | |||||||| |||| || ||| ||||||||| ||| | ||||||||| || |||||| ||||| ||| || |||||| |||| ||| |||||| || ||||||| ||||| ||||||||| |||| ||||||||||| ||| |||||| |||| ||| |||||| || |||||| ||||| ||||||||| |||| |||||||| ||||| |||||||| |||| |||||||| |||| |||| || || ||| ||||||||| ||| | ||||||||| || |||||| ||||| ||| || |||||| |||| ||| |||||| || ||||||| ||||| ||||||||| || ||| ||||||||||| ||||| ||| ||||| |||| ||| |||||| || |||||| ||||| ||||||||| || ||| ||||||| |||||| ||| |||||||| |||| |||||||| |||| || || || |||| ||||||||| ||| |||| |||||| |||| |||||||| || ||| | || |||||| ||||| ||| ||| |||||| |||| ||| |||||| || ||||||| ||||| ||||||||| |||| ||||||||||| ||| ||||| |||| ||| |||||| || |||||| ||||| ||||||||| |||| |||||||| ||| ||| |||||||| |||| | |||||||| |||| || ||| || |||| ||||||||| ||| |||||||| |||| |||||| |||| |||||||| || ||| | ||| |||||| |||| ||| |||||| || ||||||| ||||| ||||||||| |||| ||||||||||| ||| |||||| |||| ||| |||||| || |||||| ||||||||| |||| |||||||| |||||||| || |||||||| |||| | |||||||| |||| || ||| || |||| |||||||||| ||| |||||||| |||| |||||| |||| |||||||| || ||| | || ||||| ||| ||| |||||| |||| ||| |||||| || ||||||| ||||| ||||||||| |||| ||||||||||| ||| |||||| |||| ||| |||||| || |||||| ||||| ||||||||| || ||| ||||||| ||||||
CD4+ Counts
At day 8, the mean (SD) change from day 1 in CD4+ counts was |||| |||||| ||||||||| in the fostemsavir group (from a baseline of ||||| ||||||| ||||||||| ||| |||| |||||| ||||||||| in the placebo group (from a baseline || ||||| ||||||| ||||||||| (Table 10).
In the OL phase, the mean (SD) change from baseline to week 96 in CD4+ counts was 204.7 (191.3) cells/mm3 in the randomized cohort and 119.1 (201.76) cells/mm3 in the nonrandomized cohort (Table 11). After 240 weeks, the mean (SD) change from baseline in CD4+ counts was 296.4 (227.5) cells/mm3 in the randomized cohort and 240.0 (318.5) cells/mm3 in the nonrandomized cohort. Figure 4 illustrates changes in CD4+ counts over time.
Figure 4: Change From Baseline to Week 240 in CD4+ Count
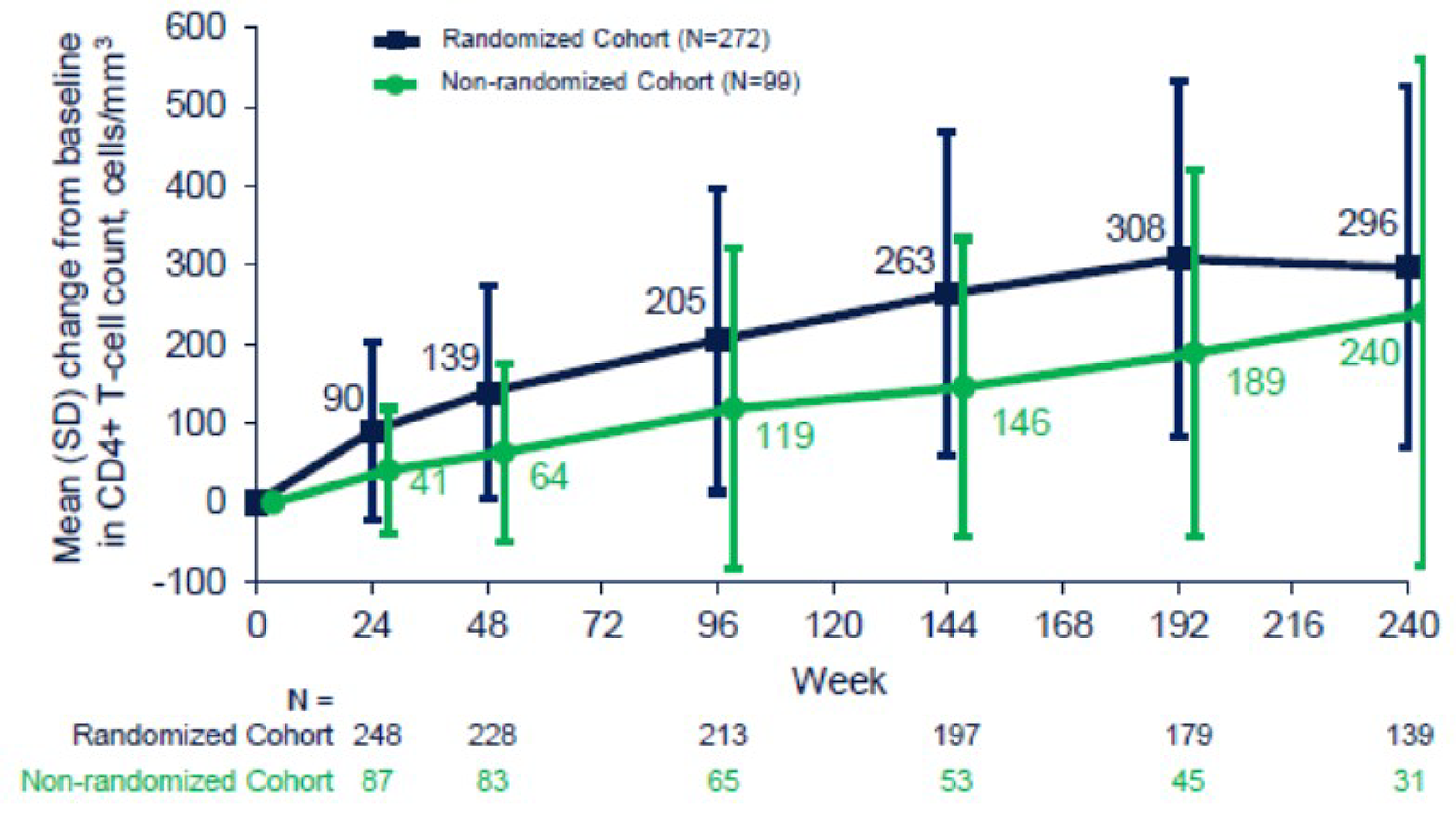
SD = standard deviation.
Source: Aberg et al. (2022).13
HRQoL
HRQoL was not assessed in the DB phase but was assessed in the OL phase up to week 96. The mean (SD) EQ VAS score increased (improved) from baseline to week 96 in both the randomized cohort, by 9.7 (20.6) points, and the nonrandomized cohort, by 5.3 (19.3) points, and US centric index scores increased (improved) by 0.302 (1.15137) points in the randomized cohort and by 0.0020 (0.20231) points in the nonrandomized cohort (Table 11).
The mean (SD) FAHI total score increased (improved) from baseline to week 96 in both cohorts, by 5.3 (24.0) points in the randomized cohort and by 4.9 (26.4) points in the nonrandomized cohort (Table 11).
Resistance
The FDA performed an analysis of virologic response (defined as > 0.5 log10 reduction in HIV-1 RNA from baseline to day 8) by resistance-associated polymorphism, using the as-treated population. According to that analysis, which was only performed for the fostemsavir group, the overall responder rate was 71%, the responder rate for those with no resistance-associated phenotypes was 81%, and the responder rate for those with changes at S375, M426, M434, or M475 was 64% (Table 10).17
The sponsor reported the occurrence of various resistance-associated mutations at week 96. For patients at week 96 who had an undetectable viral load (HIV-1 RNA < 40 copies/mL), the responses were similar in patients who had no predefined substitutions at positions of interest in the gp160 domain (62%) and in those who did have predefined substitutions at the gp160 domain (60%) (Table 11).
Adherence
At day 8 in the fostemsavir group, 97% of patients’ medications had been taken in the previous 7 days, whereas in the placebo group, adherence was 99%. At week 24, 95% of anti-HIV medications were taken in the previous 30 days in the randomized cohort, as were 96% in the nonrandomized cohort (Table 11).
Table 10: Efficacy, DB Phase (ITT-E Population)
Characteristic | Randomized cohort | |
|---|---|---|
Fostemsavir N = 203 | Placebo N = 69 | |
Viral load | ||
Plasma HIV-1 RNA log10 (c/mL) change from day 1 to day 8 | ||
N | 201 | 69 |
Adjusteda mean, log10 c/mL (95% CI) | –0.791 (–0.885 to –0.698) | –0.166 (–0.326 to –0.007) |
Difference between groups, log10 c/mL (95% CI) | –0.625 (–0.810 to –0.441) | Reference |
P value | < 0.0001b | Reference |
Subgroups based on primary outcome: change from day 1 to day 8 in viral load | ||
By baseline plasma HIV-1 RNA log10, c/mL | ||
> 1,000, n | 180 | 59 |
Mean (SD) | –0.861 (0.715) | –0.202 (0.597) |
≤ 1,000, n | 21 | 10 |
Mean (SD) | –0.219 (0.514) | 0.099 (0.718) |
|| |||||||| |||| |||||| ||||||||| | ||
|||| | | || | || |
|||||||| |||| |||| ||| | |||||| ||||||| || ||||||| | |||||| ||||||| || |||||| |
|||||||||| ||||||| |||||| |||| ||| | |||||| ||||||| || |||||| | ||||||||| |
|| || |||| | | || | || |
|||||||| |||| |||| ||| | |||||| ||||||| || ||||||| | ||||| ||||||| || |||||| |
|||||||||| ||||||| |||||| |||| ||| | |||||| ||||||| || ||||||| | ||||||||| |
|| || ||||| | | || | || |
|||||||| |||| |||| ||| | |||||| ||||||| || ||||||| | ||||| ||||||| || |||||| |
|||||||||| ||||||| |||||| |||| ||| | |||||| ||||||| || ||||||| | ||||||||| |
||| || ||||| | | || | || |
|||||||| |||| |||| ||| | |||||| ||||||| || ||||||| | |||||| ||||||| || |||||| |
|||||||||| ||||||| |||||| |||| ||| | |||||| ||||||| || ||||||| | ||||||||| |
||||| | | || | || |
|||||||| |||| |||| ||| | |||||| ||||||| || ||||||| | |||||| ||||||| || |||||| |
|||||||||| ||||||| |||||| |||| ||| | |||||| ||||||| || ||||||| | ||||||||| |
Virologic response | ||
HIV-1 RNA decrease > 0.5 log10 c/mL | ||
Achieved HIV-1 RNA outcome, n (%) | 131 (65) | 13 (19) |
Difference (%) between groups (95% CI) | 46 (32.95 to 55.45) | Reference |
HIV-1 RNA decrease > 1.0 log10 c/mL | ||
Achieved HIV-1 RNA outcome, n (%) | 93 (46) | 7 (10) |
Difference (%) between groups (95% CI) | 36 (24.16 to 44.25) | Reference |
Change in CD4+, cells/mm3 | ||
Day 1, n | 196 | 69 |
|||| |||| |||||||| ||||| | ||||| |||||||| | ||||| |||||||| |
|||||| ||||||| | ||||| ||| ||||| | ||||| ||| |||| |
||| || | | ||| | || |
|||| |||| |||||| |||| ||| | | |||| ||||||| | |||| ||||||| |
|||||| ||||||| | ||| |||||| |||| | ||| |||||| |||| |
||||||||||| || |||| | ||
||||||||||||| |||||||| ||||||||| | | ||| | ||| |
Resistance (FDA analysis, as-treated population) | ||
Patients with > 0.5 log10 decline in HIV-1 RNA by day 8, n/N (%) | ||
Overall | 107/151 (71) | NR |
Any change at S375, M426, M434, or M475 | 56/88 (64) | NR |
No EN RAP at the above sites | 51/63 (81) | NR |
Predefined EN RAP: S375I/M/N/T, M426L, M434I, M475I/V | 37/68 (54) | NR |
||||||||| | ||
||||||| || |||||||| ||||||||||| ||||| || ||| |||| | ||||| |||| |||| | |||| |||||| | |||| |||||| |
c/mL = copies per millilitre; CD4+ = cluster of differentiation 4; CI = confidence interval; EN = envelope; HIV-1 = HIV, type 1; ITT-E = intention-to-treat, exposed; NR = not reported; RAP = resistance-associated polymorphisms; SD = standard deviation.
aMean adjusted by day 1 log10 HIV-1 RNA.
bEstimated by 1-way ANCOVA in the randomized cohort, with log10 HIV-1 RNA change from day 1 to day 8 as the dependent variable, treatment (fostemsavir or placebo) as an independent variable, and log10 HIV-1 RNA at day 1 as a continuous covariate.
cAIDS-defining events were based on the CDC class C classification system, which includes events, typically infectious diseases, that tend to be characteristic of AIDS.
Source: Clinical Study Report.6
Table 11: Efficacy, OL Phase (ITT-E Population)
Characteristic | Randomized cohort | Nonrandomized cohort Fostemsavir N = 99 | |
|---|---|---|---|
Fostemsavir N = 203 | Placebo N = 69 | ||
Mortality | |||
Deaths by week 96, n (%) | 12 (4) | 17 (17) | |
AIDS-related deaths, n | 7 | ||
|||||| || |||| |||| | ||| | || ||| | || |||| | |
|||||||||||| ||||||| | | || | ||
||||||||||| || |||| | |||
||||||||||||| ||||||| || |||| ||| | ||| | || ||| | | ||| | || |||| |
||||||||||||| ||||||| || |||| |||| | ||| | || ||| | | ||| | || |||| |
Viral load | |||
HIV-1 RNA < 40 c/mL at week 96 using snapshot analysis, OBT change due to lack of efficacy as failure, n (%) | 124 (61) | 39 (57) | 37 (37) |
HIV-1 RNA ≥ 40 c/mL, n (%) | 57 (28) | 24 (35) | 43 (43) |
Data in window not below threshold | 24 (12) | 9 (13) | 15 (15) |
Discontinued for lack of efficacy | 8 (4) | 2 (3) | 3 (3) |
Discontinued for other reason while not below threshold | 11 (5) | 6 (9) | 6 (6) |
Change in background therapy | 14 (7) | 7 (10) | 19 (19) |
No virologic data, n (%) | 22 (11) | 6 (9) | 19 (19) |
Discontinued study due to adverse event or death | 10 (5) | 5 (7) | 14 (14) |
Discontinued study for other reasons | 5 (2) | 0 | 0 |
Missing data during window but on study | 4 (2) | 0 | 4 (4) |
||||| ||| ||| |||| || |||| ||| ||||| |||||||| ||||||||| ||| |||||| ||| || |||| || |||||||| || |||||||| ||| ||| | |||||| |||| | ||||| |||| | ||||| |||| |
HIV-1 RNA ≥ 40 c/mL, n/N (%) | 62/198 (31) | 27/69 (39) | 43/92 (47) |
Data in window not below threshold | 14 (7) | 6 (9) | 5 (5) |
Discontinued for lack of efficacy | 9 (5) | 5 (7) | 6 (7) |
Discontinued for other reason while not below threshold | 17 (9) | 7 (10) | 10 (11) |
Change in background therapy | 22 (11) | 9 (13) | 22 (24) |
No virologic data, n/N (%) | 45/198 (23) | 13/69 (19) | 29/92 (32) |
Discontinued study due to adverse event or death | 11 (6) | 6 (9) | 18 (20) |
Discontinued study for other reasons | 17 (9) | 2 (3) | 4 (4) |
Missing data during window but on study | 1 (< 1) | 2 (3) | 2 (2) |
Missing data during window but on study (due to pandemic) | 16 (8) | 3 (4) | 5 (5) |
CD4+ count, cells/mm3 | |||
Baseline, n | 272 | 99 | |
Mean (SD) | 152.5 (182.01) | 99.4 (130.81) | |
Mean (SD) change: | |||
From baseline to week 96, n | 213 | 65 | |
Mean change (SD) | 204.7 (191.28) | 119.1 (201.76) | |
From baseline to week 240, n | 139 | 31 | |
Mean change (SD) | 296.4 (227.52) | 240.0 (318.50) | |
HRQoL | |||
||||| |||| |||| ||||| ||||| |||||||| | ||||| ||||||||||||| | ||||| |||||||||||| | |
Mean (SD) change from baseline to week 96 | 5.3 (23.97) N = 206 | 4.9 (26.40) N = 63 | |
EQ VAS, mean (SD) baseline score | 75.0 (21.26) N = 263 | 70.6 (21.70) N = 97 | |
Mean (SD) change from baseline to week 96 | 9.7 (20.60) N = 207 | 5.3 (19.25) N = 64 | |
EQ-5D-3L US centric index scores mean (SD) baseline score | 0.8312 (0.16901) N = 263 | 0.7985 (0.19702) N = 97 | |
Mean (SD) change from baseline to week 96 | 0.0302 (0.15137) N = 208 | 0.0020 (0.20231) N = 64 | |
Resistance | |||
Patients with HIV-1 RNA < 40 c/mL at week 96, n/N (%): | 163/272 (60) | NR | |
All patients with HIV-1 RNA < 40 c/mL at week 96 who had the following at baseline, n/N (%): | Reference | ||
No predefined substitutions at positions of interest in gp160 domain | 87/141 (62) | NR | |
Predefined substitutions at positions of interest in gp160 domain | 73/122 (60) | NR | |
Predefined S375 (S375H/I/M/N/T) | 52/86 (60) | NR | |
Predefined M426 (M426L/P) | 18/32 (56) | NR | |
Predefined M434 (M434I/K) | 9/17 (53) | NR | |
Predefined M475 (M475I) | 3/3 (100) | NR | |
Adherence | |||
N | 165 | 61 | 84 |
Anti-HIV medication taken (past 30 days), %, mean (SD) at week 24 | 95.2 (9.78) | 96.1 (5.85) | 94.8 (10.24) |
Hospitalizations | |||
Data only gathered at baseline | NR | NR | NR |
c/mL = copies/millilitre; CD4+ = cluster of differentiation 4; EQ VAS = EQ visual analogue scale; EQ-5D-3L = 3-Level EQ-5D; gp = glycoprotein; HIV-1 = HIV, type-1; HRQoL = health-related quality of life; ITT-E = intention-to-treat, exposed; NR = not reported; OBT = optimized background therapy; OL = open-label; SD = standard deviation.
aAIDS-defining events were based on the CDC class C classification system, which includes events, typically infectious diseases, that tend to be characteristic of AIDS.
Source: Clinical Study Report.6
Harms
Only harms identified in the review protocol are reported here. Refer to Table 12 for detailed harms data.
Adverse Events
In the 8-day DB phase, 41% of patients in the fostemsavir group and 32% of patients in the placebo group experienced an AE. The most common AEs (fostemsavir versus placebo) were nausea (|| |||||| ||), diarrhea (|| || |||| |||||), and headache (|| |||||| ||) (Table 12).
|| ||| || |||||| ||||| || ||||| || ||| |||| ||||||||||| || ||| || |||||||| || ||| |||||||||| |||||| ||| || ||| || |||||||| || ||| |||||||||||||| ||||||| ||| ||||| ||| |||||| || ||| |||| ||||||||||| || ||| || |||||||| || ||| |||||||||| |||||| ||| ||| || |||||||| || ||| |||||||||||||| ||||||| ||| |||| |||||| || ||||||||| ||||| || |||||| |||||||||| ||| |||||||||||||| ||||||| |||| |||||||| |||| ||| ||||| |||||| |||| ||| ||||| ||| ||||| ||||||||||| ||||| ||||||||| |||| || |||| ||||||| ||||| ||| |||| |||||| || ||||| ||| ||||| |||| |||||||| |||| ||| ||||| |||||| |||| || |||| |||||||| ||| ||||| ||||||||||| ||||| ||||||||| |||| ||| ||||| |||||| ||||
Serious Adverse Events
In the 8-day DB phase, || of patients in the fostemsavir group and || of patients in the placebo group experienced an SAE. The only SAE that occurred in more than 1 patient in either group was |||||||||| ||||| |||||||| || | |||||||| ||||| || ||| ||||||||||| ||||| ||| || || |||||||| || ||| ||||||| ||||| |||||| |||| ||||||| ||| || |||||| ||||| || |||||| ||| || |||||||| || ||| |||||||||| |||||| ||| ||| || |||||||| || ||| |||||||||||||| |||||| ||||||||||| || |||| ||||| ||||| ||| |||||| ||| || |||||||| || ||| |||||||||| |||||| ||| ||| || |||||||| || ||| |||||||||||||| |||||| ||| ||||||||||| || |||| ||| |||| |||||| ||| ||| |||||||||| ||||||||| || || || |||||||| || ||| |||||||||| |||||| ||| || || |||||||| || ||| |||||||||||||| |||||| ||||| || |||||| ||| ||||| ||| |||||| || || |||||||| || ||| |||||||||| |||||| ||| || || |||||||| || ||| |||||||||||||| |||||| |||||| ||||
Withdrawals Due to Adverse Events
In the 8-day DB phase, || of patients in the fostemsavir group and ||| of patients in the placebo group who discontinued treatment due to an AE (Table 12).
|| ||| || |||||| ||||| || ||||| ||||| |||| || || |||||||| || ||| |||||||||| |||||| ||| ||| || |||||||| || ||| |||||||||||||| ||||||| ||| ||||| ||| ||||| ||||| |||| || || |||||||| || ||| |||||||||| |||||| ||| ||| || |||||||| || ||| |||||||||||||| |||||| ||| |||||||||||| ||||||||| ||| || || ||| ||| |||| |||||| |||||| ||| ||||||||||| ||| |||||||||||||| ||||||||| || || || |||||||| || ||| |||||||||| |||||| ||||| || ||| ||||| ||| ||||| ||| || || |||||||| ||||| || ||||| ||| || || |||||||| ||||| ||| ||||| || ||| |||||||||||||| |||||| |||||| ||||
Notable Harms
||||||| ||||| |||| |||||||||| |||||| ||| || |||||| |||| ||| ||||||||| |||||| ||||||||| || ||| || |||||||| ||||||| |||| |||||||||||| ||||| ||| ||||||||||||| ||| ||||||||| ||||| |||||||| ||||||||||| |||||| |||| |||||| ||| ||||| || ||| || |||||| |||| ||| |||||||| || || || |||||||| |||||| ||||
Detail | |||||||||| |||||| | |
|---|---|---|
||||||| |||| | |||| || |||| | |
|||||||| |||| | | ||||||| ||||| |||||| | ||| || ||||| | ||
| ||| | || |||| | || |||| |
|||| |||||| |||||||| | ||| | ||
|||||| | || ||| | | ||| |
|||||||| | | ||| | | ||| |
|||||||| | | ||| | | ||| |
|||||||| |||| | | ||| |||||| | ||| || ||||| | ||
| ||| | | ||| | | ||| |
|||| |||||| |||||||| |||| | ||
||||||||| | | |||| | || |
|||||||| ||| ||||||| ||||||||| ||| || ||||||| |||||| |||||| | ||| || ||||| | ||
| ||| | | ||| | | |||| |
|||||||| ||||||| | ||
|||||||||||||||| || | | |||| | || |
||||||||| | | | |||| | || |
|||||||| ||||||||| | | |||| | || |
|||| || |||||| || | | |||| | || |
||||| ||||| ||||| | || | || |
||||||| ||||| |||||| | ||| || ||||| | ||
||||| | ||| | | |||| | || |
||| ||||||||||||| | ||| | | |||| | || |
||||| |||||||| ||||||||||| |||||||||| | ||| | | |||| | || |
AE = adverse event; CAP = community-acquired pneumonia; FOST = fostemsavir; IRIS = immune reconstitution inflammatory syndrome; PLA = placebo.
aOccurring in 3% or more of patients in any group.
bOccurring in more than 1 patient in any group.
Source: Clinical Study Report.6
Detail | |||||||||| |||||| | |||||||||||||| |||||| | |||||||||| |||||| | |||||||||||||| |||||| |
|---|---|---|---|---|
||||||| |||| | |||||||||| | ||||||| |||| | |||||||||| | |
|| ||||| | ||||| || ||||| | ||||| ||| ||||| | ||
|||||||| |||| | | ||||||| ||||| | ||||
| ||| | ||| |||| | || |||| | ||| |||| | || |||| |
|||| |||||| ||||||| ||||||||| || ||| || |||| |||||||| || ||| |||||| | ||| | ||||
|||||||| | || |||| | || |||| | || |||| | || |||| |
|||||| | || |||| | || |||| | || |||| | || |||| |
||||| ||||||||||| ||||| ||||||||| | || |||| | || |||| | || |||| | || |||| |
|||||||| | || |||| | || |||| | || |||| | || |||| |
||||| | || |||| | || ||| | || |||| | || |||| |
||||||||||||||| | || |||| | || |||| | || |||| | || |||| |
||||||| | || |||| | || |||| | || |||| | || |||| |
||||||||| | || |||| | || |||| | || |||| | || |||| |
|||||||| | || |||| | | ||| | || |||| | || |||| |
|||||||||| | || |||| | | ||| | || |||| | | ||| |
||||||| | || ||| | || |||| | || ||| | || |||| |
|||||||||| | || ||| | | ||| | || |||| | || |||| |
|||| |||| | || ||| | || |||| | || |||| | || |||| |
||||||||| | || ||| | || |||| | || |||| | || |||| |
|||||||| | || ||| | || |||| | || ||| | || |||| |
|||| ||||||||||| | || ||| | || |||| | || ||| | || |||| |
||||||| ||||| ||||||||| | || ||| | || |||| | || ||| | || |||| |
|||||||| |||| | | ||| | ||||
| ||| | || |||| | || |||| | ||| |||| | || |||| |
|||| |||||| ||||||| | |||| ||||||||| || || ||||| || || |||||||| || ||| ||||| | ||||
||||||||| | || ||| | | ||| | || ||| | | ||| |
|||||||||| | | ||| | | ||| | | ||| | | ||| |
|||||||||||| ||||||||| ||||||||| | | |||| | | ||| | | |||| | | ||| |
|||||| | | |||| | | ||| | | ||| | | ||| |
||||||||||||||| ||||||| | || | | ||| | || | | ||| |
||||||||| ||||||| | | |||| | | ||| | | |||| | | ||| |
||||| |||||| |||||| | | |||| | | ||| | | ||| | | ||| |
||||| ||||||| | || | | ||| | | |||| | | ||| |
||||||| | | |||| | | ||| | | |||| | | ||| |
|||||||| | || | | ||| | || | || |
||||||| | | |||| | | ||| | | |||| | | ||| |
||||||| ||||||| | || | | ||| | || | | ||| |
|||| |||| |||||||||| | | |||| | | ||| | | |||| | | ||| |
|||||||| ||| ||||||| ||||||||| ||| || ||||||| |||||| | ||||
| ||| | || ||| | || |||| | || ||| | || |||| |
|||||||||| ||| |||||||||||| | | ||| | | ||| | | ||| | | ||| |
|||||||||||||||| ||||||||| | | |||| | | ||| | | |||| | | ||| |
||||||||||| ||||| |||| | | |||| | | ||| | | |||| | | ||| |
||||||||||| ||||| | || | | ||| | || | | ||| |
||| || ||||||||| | | |||| | | ||| | | |||| | | ||| |
||||||| |||||| ||||||||| | || | | ||| | || | | ||| |
||||||||| ||||||| ||||||||| ||| ||||||||||| ||||| ||||| ||| ||||||| | | |||| | | ||| | | |||| | | ||| |
||||||||||||| ||||||||| | | |||| | | ||| | | |||| | | ||| |
|||| | | |||| | || | | |||| | || |
||||||||||| | || | | ||| | || | | ||| |
|||| |||||||||| | || | | ||| | || | | ||| |
|||||||||||||| | | |||| | || | | |||| | || |
|||||||| | || | | ||| | || | | ||| |
||||| |||||| |||||| | || | | ||| | || | | ||| |
||||||| ||||| | ||||
||||| ||| ||| | ||||| ||| | ||||| ||| | ||
||| ||||||||||||| ||| ||| | |||||| ||| | || | || | |
|||||||| || |||||||| | | | ||| | || | || | |
||| || | ||| ||| ||||| ||||||||| || | |||| | ||| | | ||| | | ||| | ||
ALT = alanine aminotransferase; ECG = electrocardiogram; FOST = fostemsavir; IRIS = immune reconstitution inflammatory syndrome; NR = not reported; PLA = placebo; SD = standard deviation; ULN = upper limit of normal.
Source: Clinical Study Report.6
Critical Appraisal
Internal Validity
The BRIGHTE study appeared to be reasonably well conducted with respect to the steps taken to ensure adequate blinding during the 8-day DB phase and allocation concealment during the randomization process. Blinding was facilitated by use of a matching placebo, and Interactive Response Technology was used in the randomization process to maintain allocation concealment. A power calculation was performed, a priori, and the planned sample size was easily met.
Although the 8-day DB phase is not of sufficient duration to adequately assess CD4+ counts or any clinical outcomes, such as AIDS-related mortality or progression to AIDS, the clinical expert consulted by CADTH on this review believed that fostemsavir was able to elicit a reduction in viral load over a relatively short follow-up period in the DB phase, which was supported by its impact on outcomes such as CD4+ counts in the OL phase.
HRQoL was assessed in the OL phase of the BRIGHTE study; however, with a lack of control group and lack of blinding, the results for HRQoL are difficult to place into context. Patient-reported outcomes such as HRQoL are particularly prone to bias from lack of blinding, which limits any conclusions that can be drawn from the HRQoL data reported in the BRIGHTE study. Additionally, there was no MID found in the literature that was specific to HIV for the EQ-5D-3L or EQ VAS. The psychometric properties of the FAHI have been assessed in treatment-experienced patients with HIV; however, the data reported have the same limitations as data for the EQ-5D-3L, namely a lack of blinding and a lack of control group.
The sponsor used the FDA snapshot method to account for missing data when reporting data for dichotomous outcomes such as virologic response. In the FDA snapshot analysis, patients with missing data were counted as treatment failures. This is a conservative approach to account for missing data and is endorsed by the FDA. This approach could underestimate the effect of treatment if a group has a high rate of withdrawals or missed assessments; however, because the snapshot analysis only appeared to be applied to outcomes assessed in the OL phase of the study, concerns about biasing results for or against fostemsavir or placebo would be mitigated. That said, the snapshot analysis does complicate interpretation of the results, as attrition increases over the course of this relatively long-term trial. For example, when looking at virologic response, the percentage of patients experiencing virologic response appears to plateau || |||| ||| |||||||| || | ||||||| |||||||| ||||||| |||| ||| ||| |||| |||| ||||||||| ||| |||| |||| |||||||| ||| |||||| || |||||||| |||| ||| ||||||||| ||||| ||||||||| ||||||| ||||| || ||| ||| || |||| ||| |||||||||| |||||| ||||||||| |||| ||| || |||| ||| || ||| |||||||||||||| |||||| ||||||||| |||| ||| || ||||| ||||| ||||||| |||||||| ||| ||| |||| || ||||||||| |||||||||| |||| |||||||| || |||| |||||| |||| |||| |||| |||||. The sponsor also noted that this time frame corresponds to the pandemic, and at week 240, missing data due to the pandemic occurred in 7% of patients in the randomized cohort and 5% of patients in the nonrandomized cohort.
Another confounder of the interpretation of data from the BRIGHTE study is the different OBTs used in the OL phase. Each patient had their own tailored OBT; therefore, OBT was not standardized in either the randomized or nonrandomized cohorts. This assumes that all of those regimens were appropriate and are indeed the optimal choice for a given patient. For assessment of virologic response, patients whose OBT was switched during the OL phase were counted as nonresponders, which helps to account for patients whose OBT was not truly optimized. For example, change in background therapy was the reason 12% of patients in the randomized cohort and 24% of patients in the nonrandomized cohort were categorized as not having a virologic response.
Data for preplanned subgroups of interest to this review protocol, such as response by baseline viral load and by CD4+ count, were reported; however, due to the small sample size, no formal analyses of these data were planned. This limits any conclusions that can be drawn regarding response to fostemsavir in any of the subgroups in our protocol.
There were some numerical differences in baseline characteristics between fostemsavir and placebo. Most notably, patients on fostemsavir had a lower CD4+ count and fewer had 2 or more fully active and available ARV regimens at screening and in their initial OBT. These differences may have resulted in the fostemsavir group having slightly more advanced disease than those in the placebo group. However, the impact of such imbalances may be limited, as the DB phase focused on viral load, for which there was only a small difference between groups and which was not expected to be considered meaningful, with higher values in the fostemsavir group.
Patient disposition was not reported for the 8-day DB phase, so it is unknown whether there was a numerical difference in withdrawals between the fostemsavir and placebo groups for the only comparative phase of the trial. Although 1 would not expect a large number of dropouts during the 8-day DB phase, the only data available show that there were 2% of patients in the fostemsavir group and less than 1% in the placebo group who stopped treatment due to an AE. Despite the short duration of the DB phase, complete reporting of disposition would allow for an assessment of whether there were imbalances in withdrawals between treatment groups, which can potentially bias assessment of efficacy and harms.
External Validity
The clinical expert consulted by CADTH on this review noted that the population enrolled in the BRIGHTE study was generally consistent with the population that is expected to receive treatment with fostemsavir in Canadian clinical practice, with the exception of a relatively small number of female participants and the absence of pregnant women, who were excluded from the trial. The clinical expert noted that this is not uncommon in HIV trials, as women are less often recruited to clinical trials of ARVs and pregnant women are generally excluded from clinical trials. There were Canadian sites in the trial, which is relevant for the generalizability of the data to the Canadian population.
The primary outcome of the BRIGHTE study was the change from baseline to day 8 in HIV-1 RNA. The change in HIV-1 RNA is a very common primary outcome in the assessment of ARVs and is considered to be the main predictor of complications of HIV, including progression to AIDS, according to the clinical expert consulted by CADTH on this review. CD4+ count was assessed in the DB and OL phases of the BRIGHTE study and is also considered to be an important outcome in the assessment of efficacy of ARVs, according to the clinical expert.
The DB, controlled phase of the BRIGHTE study lasted only 8 days, which is not sufficient follow-up to adequately assess harms from fostemsavir. The sponsor explained that the short controlled phase was necessary because of the risk of emergence of resistance when using a failing background regimen with placebo, rather than a modified regimen; this appears to be consistent with FDA guidance, which recommends the use of placebo in a population such as this for only 7 to 14 days.17 The sponsor chose the lowest end of the treatment period due to concerns about the development of resistance. The OL phase, with follow-up as long as 240 weeks, continued to collect harms data on patients; however, with the lack of a control group and a lack of blinding, it is difficult to place these data into context.
Fostemsavir or placebo were initially combined with the patient’s failing ARV regimen for the DB phase of the BRIGHTE study, then patients transitioned to OBT during the OL phase of the trial. The drugs used in the failing regimen, as well as those used for OBT, appeared consistent with what would be used in Canada for these patients, and the decisions regarding what drugs to include in OBT were at the discretion of the physician and patient, allowing for the individualization of therapy, and the classes used were consistent with what would be expected for HTE patients in Canadian clinical practice, according to the clinical expert consulted by CADTH on this review. The eligibility criteria for the nonrandomized cohort indicated that only patients who did not have any remaining fully active approved ARVs would be included; however, 4% of patients were identified as having 1 fully active and available ARV at screening and 19% had 1 fully active and available ARV in their initial OBT. In their comments on the Clinical Review Report, the sponsor clarified that the 4% of patients with a remaining fully active regimen were protocol deviations, and 15% received an investigational drug as part of their regimen, which was permitted in the protocol.5 It is not clear how available those investigational drugs would be to all HTE patients, which could potentially present a generalizability issue.
Indirect Evidence
Objectives and Methods for the Summary of Indirect Evidence
The objective of this section is to summarize and appraise the indirect evidence for fostemsavir (extended-release tablets, 600 mg, administered orally twice daily in addition to standard of care) for the treatment of HTE adults with HIV-1 who have multidrug-resistant HIV-1 infection, and for whom it is otherwise not possible to construct a suppressive antiviral regimen due to resistance, intolerance, or safety considerations. The BRIGHTE trial included an 8-day randomized phase comparing fostemsavir plus OBT to placebo plus OBT, followed by a single-arm phase in which all patients received fostemsavir plus OBT. In the absence of direct comparative evidence, indirect comparisons were summarized to address the gap in the evidence comparing fostemsavir to other treatments used for HTE adults with HIV-1 who have multidrug-resistant HIV-1 infection. This included 1 MAIC submitted by the sponsor. In addition, a focused literature search for ITCs dealing with fostemsavir and HIV-1 was run in MEDLINE All (1946–) on November 22, 2022. No limits were applied. One relevant published ITC was found in the literature search.
Description of Indirect Comparisons
The sponsor submitted 1 MAIC to CADTH for review, which is summarized and appraised in this report and is hereafter referred to as the sponsor-submitted MAIC.
CADTH also retrieved 1 published ITC,8 which is summarized and appraised in this report and is hereafter referred to as the MAIC by Anderson et al.
Methods of Sponsor-Submitted MAIC
Objectives
The objective of the sponsor-submitted MAIC was to generate long-term comparative efficacy estimates for fostemsavir plus OBT versus OBT alone for the management of HTE patients with HIV using IPD from the BRIGHTE study.
Efficacy was quantified in terms of:
change (from baseline) in CD4+ cell count
rates of virologic suppression
rates of PDVF
rates of treatment discontinuation.
Secondary analyses assessed the relative safety profile of fostemsavir based on the rates of SAEs, discontinuation due to AEs, and death.
Study Selection Methods
The BRIGHTE study IPD were used to inform estimates of efficacy for fostemsavir. Two distinct cohorts were included in the study: a randomized cohort, which included patients with 1 or 2 remaining ARV classes, randomized in a 3:1 ratio to either blinded fostemsavir or placebo, plus their existing background regimen for 8 days before switching to fostemsavir plus OBT; and a nonrandomized cohort, which included patients with no remaining approved active ARVs who commenced OL fostemsavir plus OBT on day 1 and could take other investigational ARVs along with their OBT regimen.
A SLR was conducted to identify sources suitable for informing a comparator for the ITC. Studies published between January 1, 2003, and February 23, 2021, were included. As there are no relevant active comparators for this patient population, the sponsor sought to identify the most appropriate and contemporary comparator evidence source for OBT. As such, the sponsor identified trials from the SLR that comprised HTE cohorts sufficiently similar (as judged by the reviewer) to those recruited in the BRIGHTE study, based on the following HTE inclusion criteria:
ARV experienced with documented historical or baseline resistance, intolerability, and/or contraindications to ARVs in at least 3 classes, and
no more than 2 classes with at least 1 but no more than 2 fully active ARVs remaining that can be effectively combined to form a viable new regimen, based on current and/or documented historical resistance testing and tolerability and safety, and
able to receive at least 1 fully active approved ARV as part of the OBT.
Of the 52 studies included, 13 were considered to be closely aligned with the BRIGHTE study in terms of patient eligibility criteria regarding treatment history, resistance, and available treatments remaining. Studies were excluded from consideration for reasons such as expanded-access programs, no OBT comparator arm, no objective to suppress HIV replication, a focus on treatment interruption, insufficient information on the number of active drugs in the background regimen, and insufficient treatment experience in the patient population. Ultimately, 3 phase III studies were identified for potential use in the MAIC analyses: TMB-301 (ibalizumab + OBT; ClinicalTrials.gov identifier NCT02475629), BENCHMRK-1 and BENCHMRK-2 (OBT alone; NCT00293267 identifier NCT00293254), and VIKING-3 (OBT alone; ClinicalTrials.gov identifier NCT01328041).
Ultimately, the VIKING-39 single-arm study of dolutegravir-containing regimens was identified by the reviewers to be the most relevant contemporary study. The VIKING-3 study had the most comparable HTE population and patients were treated with ARV regimens that were the most closely reflective of those used in the BRIGHTE study and in Canadian clinical practice, primarily including dolutegravir, darunavir, and tenofovir disoproxil fumarate plus emtricitabine.9 The ARVs used in the VIKING-3 study most closely matched those used in the BRIGHTE study, in which 303 of 371 (82%) patients received dolutegravir as part of their OBT.8 In contrast, the TMB-301 study evaluated a treatment not currently approved or used in Canadian clinical practice (ibalizumab), and the BENCHMRK studies began in 2006 and do not reflect the ARV regimens used in the BRIGHTE study or in current Canadian practice, most notably because they lack dolutegravir.8 The VIKING-3 study included Canadian treatment centres (in addition to centres in Europe and the US) and, according to the OSS, the majority of patients had no more than 2 ARVs remaining and resistance to at least 3 classes. VIKING-4 was not used despite being more recent because of the small population size (N = 40).
ITC Analysis Methods
Analysis Populations
Data for the combined randomized and nonrandomized BRIGHTE study ITT-E population was employed to characterize outcomes for patients managed with fostemsavir plus OBT. The randomized cohort includes patients assigned to fostemsavir plus OBT and those assigned to placebo plus OBT during the initial 8-day DB phase. The combined cohort was used to maximize the sample size of the analysis population. Scenario analyses evaluated outcomes for fostemsavir plus OBT based on the randomized ITT-E population alone in the BRIGHTE study.
The BRIGHTE and VIKING-3 studies differed in their screening HIV-1 RNA inclusion criteria. BecauseHIV-1 RNA is prognostic of outcomes, the comparison of studies that differ in their HIV-1 RNA eligibility criteria may be biased in nature. Consequently, the BRIGHTE study ITT-E population was used to form analysis populations comparable to the VIKING-3 population. Patients with a screening HIV-1 RNA level of less than 500 copies/mL were excluded from the analysis populations; from the combined cohort, 13 patients were excluded, 8 of which were from the randomized cohort.
Outcomes for patients managed with OBT were characterized by the ITT-E population in the VIKING-3 study.
Matching Variables
Because the analyses were unanchored, matching variables were selected to include both prognostic factors and treatment-effect modifiers, which were determined by referencing literature and consulting with an international steering committee of consultant physicians who had a special interest in HIV. The matching variables were as follows:
baseline viral load (log10 copies/mL)
baseline CD4+ cell count (cells/μL)
baseline age (years)
sex (male or female)
history of AIDS
number of active ARVs in initial OBT (OSSnew profiles).
MAICs
Unanchored MAICs were conducted for the target outcomes based on the methodology described by the National Institute for Health and Care Excellence (NICE) Decision Support Unit. Because there was no common comparator between the studies and the long-term data are single-arm, anchored MAICs were not possible.
A logistic propensity score model was fitted, using the method of moments, to derive weights for the index trial to balance the summary statistics of the baseline characteristics between the index trial and comparator trial. Treatment effects were estimated using weighted regression with sandwich standard errors. Continuous outcome variables were estimated as mean differences and binary outcome variables were estimated as odds ratios. The effective sample size was calculated to represent the effective number of subjects remaining in the study after weighting.
Outcomes were assessed at week 24 and week 48 relative study time. Comparisons that examined subsequent assessment points were not considered appropriate because patients in the VIKING-3 study would withdraw after their week 48 visit when commercial dolutegravir became available at their study site.
Results of Sponsor-Submitted MAIC
Summary of Included Studies
Design
BRIGHTE is a phase III, 2-cohort study of fostemsavir use in HTE patients with HIV. Patients in the randomized cohort were randomized and blinded in a 3:1 ratio to fostemsavir plus current regimen or placebo plus current regimen for the 8-day randomized phase of the study. Thereafter, all patients received OL fostemsavir plus current regimen until the end of the study. The nonrandomized cohort (N = 371) received OL fostemsavir from study day 1. The BRIGHTE study included centres in Canada, as well as in the US, South America, UK, Europe, Asia, and Australia.
VIKING-3 was a single-arm, OL, phase III study (N = 183) of dolutegravir-containing regimens in HTE adults with HIV. Patients received dolutegravir 50 mg twice daily in an initial 7-day functional monotherapy phase, followed by the same dose in addition to an OBT. The primary and key secondary end points were assessed at day 8 and week 24 of treatment. The VIKING-3 study included treatment centres in Canada, Europe, and the US.
The key patient eligibility criteria for the BRIGHTE and VIKING-3 studies are summarized in Table 14.
Table 14: Key Patient Inclusion and Exclusion Criteria for the BRIGHTE and VIKING-3 Studies
Eligibility | BRIGHTE9 | VIKING-39 |
|---|---|---|
Population | Nonpregnant adults with HIV-1 infection | Nonpregnant adults with HIV-1 infection |
Screening plasma HIV-1 RNA | ≥ 400 c/mL | ≥ 500 c/mL |
Treatment history | Randomized cohort: ART-experienced patients with ≤ 2 classes and at least 1 but no more than 2 fully active ARVs remaining (randomized cohort) Nonrandomized cohort: patients with 0 fully active ARVs | ART-experienced, INI-experienced, DTG naive |
Virologic failure history | Failing on current regimen | Current or prior failure on RAL or ELV |
Resistance status | Historic or baseline resistance, intolerability, and/or contraindications to ARVs in ≥ 3 classes | ≥ 1 drug from each of 3 or more of all approved ART classes Resistance to RAL or ELV at screening or time of prior virologic failure |
Other | Randomized cohort: able to receive ≥ 1 fully active approved drug as part of OBT in the open-label phase (randomized cohort) Nonrandomized cohort: patients without fully active ARVs remaining | Able to receive ≥ 1 fully active drug as part of OBT from day 8 |
ART = antiretroviral therapy; ARV = antiretroviral; c/mL = copies per millilitre; DTG = dolutegravir; ELV = elvitegravir; HIV-1 = HIV, type 1; INI = integrase inhibitor; OBT = optimized background therapy; RAL = raltegravir.
Source: Sponsor-submitted MAIC report.7
Baseline Patient Characteristics
Demographic and baseline characteristics of the overall BRIGHTE cohort (including randomized and nonrandomized patients) and the VIKING-3 cohort are reported in Table 15.
In the BRIGHTE study, history of AIDS was higher than the corresponding value for AIDS at baseline reported in the VIKING-3 study. However, the definitions of this characteristic differed: in BRIGHTE, it was defined as a history of AIDS if a patient has a nadir CD4+ count below 200 cells/mm3, or if the response to, “Does subject have AIDS?” on disease history CRF is yes. In VIKING-3, it was defined as CDC classification C at baseline. However, in the CDC classification system, once a category C condition has occurred, the person will remain in category C, so these definitions are comparable. Assuming clinical comparability of the definition of this characteristic, the BRIGHTE population appears to be appreciably more severe than the VIKING-3 population with regard to AIDS history at baseline.
Table 15: Baseline Patient Characteristics in the BRIGHTE and VIKING-3 Studies
Characteristic | BRIGHTE | VIKING-3 |
|---|---|---|
N | 371a | 183 |
Mean (SD) viral load, log10 c/mL | 4.36 (1.00) | 4.26 (0.93) |
Mean (SD) CD4+ cell count, cells/μL | 138.34 (171.32) | 199.90 (192.43) |
Mean (SD) age, years | 45.58 (12.20) | 47.00 (9.26) |
Sex: male, n %) | 289 (77.90) | 141 (77.05) |
AIDS history, n (%)b,c | 320 (86.25) b | 102 (55.74) c |
GSS, n (%) | ||
0 drug classes available | 23 (6.20) | 8 (4.37) |
> 0 to 1 drug classes available | 95 (25.61) | 58 (31.69) |
> 1 to 2 drug classes available | 161 (43.40) | 87 (47.54) |
> 2 drug classes available | 82 (22.10) | 30 (16.39) |
Missing | 10 (2.70) | 0 |
PSS, n (%) | ||
0 drug classes available | 5 (1.35) | 8 (4.37) |
> 0 to 1 drug classes available | 52 (14.02) | 57 (31.15) |
> 1 to 2 drug classes available | 159 (48.86) | 83 (45.36) |
> 2 drug classes available | 144 (38.81) | 35 (19.13) |
Missing | 11 (2.96) | 0 |
OSS, n (%) | ||
0 drug classes available | 8 (2.16) | 9 (4.92) |
> 0 to 1 drug classes available | 63 (16.98) | 70 (38.25) |
> 1 to 2 drug classes available | 157 (42.32) | 78 (42.62) |
> 2 drug classes available | 125 (33.69) | 26 (14.21) |
Missing | 18 (4.85) | 0 |
OSSnew, n (%)d | ||
0 drug classes available | 90 (24.26) | 68 (37.16) |
> 0 to 1 drug classes available | 135 (36.39) | 78 (42.62) |
> 1 to 2 drug classes available | 113 (30.46) | 35 (19.13) |
> 2 drug classes available | 18 (4.85) | 2 (1.09) |
Missing | 15 (4.04) | 0 |
c/mL = copies per millilitre; CD4+ = cluster of differentiation 4; GSS = genotypic susceptibility score; OSS = overall susceptibility score; PSS = phenotypic susceptibility score; SD = standard deviation.
aCombined population (N = 371) includes randomized (n = 272) and nonrandomized cohorts (n = 99) based on BRIGHTE 96 week CSR.
bBRIGHTE: History of AIDS is yes if a patient has nadir CD4+ count < 200 cells/mm3, or if response to, “Does subject have AIDS?” on disease history CRF is yes.
cVIKING-3: AIDS at baseline defined as CDC classification C at baseline.
dOSSnew is the overall susceptibility score for new (not previously taken) drugs in the initial OBT. If a drug was taken as part of a prior regimen, then the susceptibility rating for that component is assumed to be 0 (resistant).
Source: Sponsor-submitted MAIC report,7 based on the BRIGHTE 96-week CSR and the VIKING-3 48-week CSR.
Based on the published baseline data, the percentage of patients with 0 available treatments according to OSSnew was lower in the BRIGHTE study than in the VIKING-3 study. However, comparing these side by side is complex due to between-trial differences in exposure to ARVs and differences in the calculation of this variable. OSSnew is the overall susceptibility score for new (not previously taken) drugs in the initial OBT. If an drug was taken as part of a prior regimen, then the susceptibility rating for that component is assumed to be 0 (resistant). The VIKING-3 and BRIGHTE studies differed in the approach used to derive overall susceptibility ratings (OSRs) and, subsequently, OSS. An OSR score of 0.5 in the BRIGHTE study indicated that net assessment is partial sensitivity, whereas the VIKING-3 study employed a binary scoring system with partial sensitivity considered to be not fully active and assigned a score of 0. The data in Table 15 reflects the published data; however, for the MAICs, to ensure consistency in the employed approach, BRIGHTE study OSR values were reclassified with partial (0.5) OSR scores assigned 0 values. The resulting OSSnew values for the BRIGHTE study are reported in Table 18.
Notably, OSSnew as reported in the VIKING-3 study at baseline also does not reflect the activity of dolutegravir, given that all treated patients in that study would have been exposed to dolutegravir, per the design of the study. For the purpose of this comparison, outcome data reflect the total regimen efficacy and are therefore inclusive of dolutegravir, so adjustments were employed in the MAIC to account for this. For the VIKING-3 baseline data used in the MAIC, dolutegravir was assumed to be fully active (i.e., attributed an OSRnew of 1) in the primary analyses, and sensitivity analyses were conducted with alternative assumptions of partial OSSnew adjustment (i.e., dolutegravir was assumed to be fully active only for the proportion of patients virologically suppressed at week 48 in theVIKING-3 study [63.39%]) and no OSSnew adjustment (i.e., assuming an OSRnew of 0). The distributions of OSSnew based on these assumptions are summarized in Table 21.
Other metrics of susceptibility include genotypic susceptibility score (GSS) and phenotypic susceptibility score (PSS). The distributions of GSS and PSS differed in the BRIGHTE and VIKING-3 studies; by GSS, there were more patients in the BRIGHTE study than in the VIKING-3 study with 0 drug classes available, but by PSS, there were fewer patients in the BRIGHTE study with 0 drug classes available. However, more patients in the BRIGHTE study than in the VIKING-3 study had more than 2 drug classes available, according to both GSS and PSS.
There were also more missing data in susceptibility scores for baseline characteristics in the BRIGHTE population than in the VIKING-3 population.
In the BRIGHTE study, most patients (90% or more) had prior exposure to NNRTIs, NRTIs, or PIs and 75% had prior exposure to an INSTI. Other ARVs that patients had prior exposure to included entry inhibitors (39%), CCR5 antagonists (26%), and other investigational ARVs (11%). The most common ARV classes in the failing regimen were NRTIs (81%), PIs (67%), INSTIs (44%), and NNRTIs (28%), whereas other classes included CCR5 antagonists (12%) and entry inhibitors (4%).
In the VIKING-3 study, 56% of patients had prior exposure to etravirine, 49% to enfuvirtide, and 73% to darunavir plus ritonavir. The median (interquartile range) number of prior ARV therapies was 14 (3 to 23). The most frequently used OBT regimens (≥ 25%) in combination with dolutegravir were darunavir plus ritonavir (65%), tenofovir plus emtricitabine (60%), etravirine (37%), enfuvirtide (32%), and maraviroc (25%). Other regimens used by at least 5% of patients included darunavir plus ritonavir and tenofovir plus emtricitabine (11%); darunavir plus ritonavir, tenofovir plus emtricitabine, and enfuvirtide (6%); darunavir plus ritonavir, tenofovir plus emtricitabine, enfuvirtide, and etravirine (5%); darunavir plus ritonavir, tenofovir plus emtricitabine, and etravirine (5%); darunavir plus ritonavir and maraviroc (5%); and darunavir plus ritonavir and etravirine (5%).
Four efficacy outcomes were analyzed at week 24 and week 48 relative study time: change from baseline in CD4+ cell count, percent of patients with virologic suppression, percent of patients with PDVF, and all-cause discontinuation. There were between-trial differences in the definitions of the outcomes of virologic suppression and PDVF, which are detailed in Table 16, along with an explanation of any adjustments applied for the MAIC, where applicable, or justification in cases where adjustments were not applied. In summary, a different HIV-1 RNA threshold was used in the BRIGHTE study (< 40 copies/mL) than in the VIKING-3 study (< 50 copies/mL), but for the MAIC with the BRIGHTE IPD, the threshold from the VIKING-3 study was used. The differences in PDVF definition were not possible to eliminate with adjustment of the BRIGHTE IPD; the sponsor’s clinical expert determined that this difference was unlikely to be of clinical significance.
Table 16: Outcome Definitions in the Included Studies and Adjustments Applied for MAIC
Outcome | BRIGHTE | VIKING-3 | Adjustment applied for MAIC or justification provided by sponsor if no adjustment applied |
|---|---|---|---|
Change (from baseline) in CD4+ cell count, cells/μL | Change (from baseline) in CD4+ cell count (cells/μL), evaluated on an observed case basis | NA | |
Virologic suppression | HIV-1 RNA < 40 c/mL evaluated according to the FDA snapshot algorithm | HIV-1 RNA < 50 c/mL evaluated according to the FDA snapshot algorithm | The threshold used in VIKING-3 was employed for BRIGHTE in the MAIC |
PDVF | Prior to Week 24 Confirmed, or last available before discontinuation, HIV-1 RNA ≥ 400 c/mL at any time after prior confirmed suppression to < 400 c/mL or confirmed, or last available before discontinuation, > 1 log10 c/mL increase in HIV-1 RNA at any time above nadir level, where nadir is ≥ 40 c/mL At or after week 24 Confirmed, or last available before discontinuation, HIV-1 RNA ≥ 400 c/mL | During the functional monotherapy treatment phase < 0.5 log10 c/mL decrease in plasma HIV-1 RNA at day 8, unless absolute value is < 400 c/mL During the optimized phase Virological nonresponse: A decrease in plasma HIV-1 RNA of less than 1 log10 c/mL by week 16, with subsequent confirmation, unless plasma HIV-1 RNA is < 400 c/mL, or confirmed plasma HIV-1 RNA levels ≥ 400 c/mL on or after week 24 Virological rebound: Confirmed rebound in plasma HIV-1 RNA levels to ≥ 400 c/mL after prior confirmed suppression to < 400 c/mL or confirmed plasma HIV-1 RNA levels > 1 log10 c/mL above the nadir value, where nadir is ≥ 400 c/mL | It was not possible to homogenize the outcome definitions, but the sponsor sought clinical expert input and determined that the differences were unlikely to be of clinical significance |
Discontinuation | Premature discontinuation was required for patients meeting at least 1 of the study withdrawal criteria | NR | |
c/mL = copies per millilitre; CD4+ = cluster of differentiation 4; HIV-1 = HIV, type 1; MAIC = matching-adjusted indirect comparison; NA = not applicable; NR = not reported; PDVF = protocol-defined virologic failure.
Source: Sponsor-submitted MAIC report.7
Safety data available for the VIKING-3 study reflected all events observed through the CSR DCO date, rather than only those observed within the week 24 and week 48 relative study time assessment points. The median (range) exposure times of 336 (14 to 509) days and 507 (14 to 757) days were reported for the week 24 and week 48 DCO dates for the VIKING-3 study. The sponsor therefore concluded that comparisons with BRIGHTE safety results observed only within 24-week or 48-week relative study times would be subject to significant bias, and instead compared safety outcomes based on all data available to the week 48 DCO of the respective studies (i.e., week 48 DCO of the VIKING-3 study) (median [range] exposure: 507 [14 to 757] days) versus week 48 DCO of BRIGHTE (median [range] exposure: 621 [1 to 1,039] days). A sensitivity analysis was also conducted comparing the VIKING-3 week 48 DCO to the BRIGHTE week 24 DCO (median [range] exposure: 421 [1 to 1,036] days).
A summary of efficacy and safety outcomes before matching is presented in Table 17 for the BRIGHTE (randomized, nonrandomized, and combined cohorts) and VIKING-3 studies. The efficacy outcomes reported reflect the 24-week or 48-week relative study time, and the safety outcomes provided reflect those observed as of the 24-week or 48-week DCO.
Table 17: Summary of Outcomes in the BRIGHTE and VIKING-3 Studies
Outcome | BRIGHTE | VIKING-3 N = 183 | ||
|---|---|---|---|---|
Randomizeda N = 272 | Nonrandomized N = 99 | Combined N = 371 | ||
Week 24 | ||||
Mean (SD) change from baseline in CD4+ cell count, cells/μL | 90.36 (112.10) | 41.03 (78.56) | 77.51 (106.52) | 81.00 (117.94) |
Percent virologic suppression (HIV-1 RNA < 50 c/mLb) | 52.94 | 37.37 | 48.79 | 68.85 |
Percent PDVFc | 11.40 | 28.28 | 15.90 | 19.67 |
Percent discontinued: all-cause | 6.99 | 4.04 | 6.20 | 17.49 |
Percent discontinued: virologic | 6.25 | 0 | 5.66 | NR |
Percent discontinued: nonvirologic | 0.74 | 0 | 0.54 | NR |
Week 48 | ||||
Mean (SD) change from baseline in CD4+ cell count, cells/μL | 138.86 (135.06) | 63.49 (112.60) | 118.74 (133.51) | 114.80 (130.69) |
Percent virologic suppression (HIV-1 RNA < 50 c/mL) | 53.68 | 38.38 | 49.60 | 63.39 |
Percent PDVF | 18.01 | 46.46 | 25.61 | 22.40 |
Percent discontinued: all-cause | 11.03 | 11.11 | 11.05 | 22.40 |
Percent discontinued: virologic | 9.93 | 9.09 | 9.70 | NR |
Percent discontinued: nonvirologic | 1.10 | 2.02 | 1.35 | NR |
c/mL = copies per millilitre; CD4+ = cluster of differentiation 4; HIV-1 = HIV, type 1; NR = not reported; PDVF = protocol-defined virologic failure; SD = standard deviation.
Note: Cause of discontinuation (virologic or nonvirologic) was determined by a patient’s viral load at the time of discontinuation; patients with a viral load of < 50 copies/mL at the time of discontinuation were considered to have discontinued for nonvirologic reasons, and those with a viral load of ≥ 50 copies/mL were considered to have discontinued for virologic reasons.
aThe randomized cohort includes participants randomized to placebo plus their existing background regimen for the first 8 days.
bThe BRIGHTE study defined this outcome as < 40 copies/mL, but for the MAIC, it was adjusted using the IPD to align with the VIKING-3 definition of < 50 copies/mL.
cThere were differences in the PDVF outcome definition that could not be adjusted for the MAIC.
Source: Sponsor-submitted MAIC report.7
Results
Matching
A summary of baseline characteristics before and after matching is presented in Table 18, along with the corresponding target values from the VIKING-3 study. Relative to the VIKING-3 study population, the unadjusted BRIGHTE analysis population had more poorly controlled disease at baseline, as indicated by the notably higher viral load, the notably lower CD4+ cell count, the substantially higher proportion of patients with a history of AIDS, as well as the OSSnew distributions.
Table 18: MAIC Matching Variable Summary Statistics
Parameter | BRIGHTEa fostemsavir plus OBT | VIKING-3 OBT alone | |
|---|---|---|---|
Unadjusted | Adjusted | ||
N | 358 | 78 | 183 |
Mean (SD) age at baseline, years | 45.54 (12.30) | 47.00 (9.26) | 47.00 (9.26) |
Sex: male (%) | 78.49 | 77.05 | 77.05 |
History of AIDS (%) | 86.87b | 55.74 b | 55.74c |
Mean (SD) viral load at baseline (log10 c/mL) | 4.413 (0.968) | 4.257 (0.934) | 4.257 (0.934) |
Mean (SD) CD4+ cell count at baseline, cells/μL | 131.53 (164.09) | 199.90 (192.43) | 199.90 (192.43) |
OSSnew, %d,e | |||
0 drug classes available | 31.50 | 0.00 | 0.00 |
1 drug class available | 35.26 | 37.16 | 37.16 |
2 drug classes available | 31.21 | 42.62 | 42.62 |
> 2 drug classes available | 2.02 | 20.22 | 20.22 |
c/mL = copies per millilitre; CD4+ = cluster of differentiation 4; MAIC = matching-adjusted indirect comparison; OBT = optimized background therapy; OSS = overall susceptibility score; SD = standard deviation.
aRestricted to patients with a screening HIV-1 RNA ≥ 500 copies/mL; OSSnew recalculated to reflect the way OSR was derived in the VIKING-3 study.
bHistory of AIDS is yes if a patient has a nadir CD4+ count < 200 cells/mm3, or if response to “Does subject have AIDS?” on disease history CRF is yes.
cDefined as CDC classification C at baseline.
dOSSnew is the overall susceptibility score for new (not previously taken) drugs in the initial OBT. If a drug was taken as part of a prior regimen, then the susceptibility rating for that component is assumed to be 0 (resistant).
eThe VIKING-3 OSSnew distribution has been recalculated to account for the activity of dolutegravir.
Source: Sponsor-submitted MAIC report.7
Notably, the OSSnew distributions were recalculated for the MAIC. The unadjusted baseline distributions (before recalculation) are reported in Table 15. Recalculated distributions for the primary analysis MAIC are reported in Table 18. For the MAIC, distribution in the BRIGHTE study was recalculated using the approach taken in the VIKING-3 study to derive OSR for partial susceptibility, as previously described. For the VIKING-3 study, the recalculation was performed to account for the activity of dolutegravir by assigning an OSR of 1 (refer to Table 21 for alternative assumptions used in the sensitivity analyses).
During matching adjustment, weights were rescaled such that a weight of 1 is equivalent to a single patient in the BRIGHTE analysis population (n = 358). Weights were predominantly clustered around 1, with the largest single count of patients having weight values of less than 1. The largest rescaled weight was 13.
After matching, the effective sample size of BRIGHTE was 78 in the primary MAIC, which is a 78% reduction from the original sample size.
||||||| || || ||||| | |||||||||| | |||||||| |||||| | |||||
|---|---|---|---|---|---|---|---|
||| ||||| |||||||| | ||| |||| ||| ||||||| | |||| |||||||||| |||||| | ||||||| | ||| |||| || |||||||||| | |||| |||||||||| || |||| ||||| | ||||||| | |
|||||| |||| | ||| | ||| | ||| | ||| | || | ||| | ||| |
|||| |||||| |||| |||||||| || |||| |||| ||||| ||||||||||||||| |||| ||| | ||||||||||||| | | |||||||||||||||||| | |||||||||||||| || | |||||| | ||||||||||||||||||| | |||||||||||||||||| | ||||| |
||||||| ||||||||| |||||||||||||||||| ||| ||| ||||||||||||||| ||| | | ||||||||||||| || | ||||||||||||| |||||| | ||||||||||| ||||| | |||||| | |||||||||||||||||| | ||||||||||| ||||| | ||||| |
||||||| |||| |||| ||| | ||||||||||||| || | ||||||||||||| |||||| | ||||||||||| ||||| | ||||| | ||||||||||| ||||| | ||||||||||| ||||| | |||||| |
||||||| ||||||||||||| ||||||||| |||| ||| | ||||||||||||| | | ||||||||||| ||||| | ||||||||||| ||||| | |||||| | ||||||||||| ||||| | ||||||||||| ||||| | ||||| |
||||||| ||||||||||||| ||||||||| |||| ||| | | || | ||||||||||| ||||| | || | || | ||||||||||| ||||| | || | || |
||||||| ||||||||||||| ||||||||||||| |||| ||| | | || | ||||||||||| ||||| | || | || | ||||||||||| ||||| | || | || |
CD4+ = cluster of differentiation 4; CI = confidence interval; FTR = fostemsavir; MAIC = matching-adjusted indirect comparison; NR = not reported; OBT = optimized background therapy; PDVF = protocol-defined virologic failure; SD = standard deviation.
Note: P values were assessed at the 5% (alpha = 0.05) significance level.
aRestricted to patients with a screening HIV-1 RNA ≥ 500 copies/mL.
bA threshold of < 50 copies/mL was employed for both studies.
cIn the BRIGHTE study, cause of discontinuation (virologic or nonvirologic) was determined by a patient’s viral load at the time of discontinuation; patients with a viral load of < 50 copies/mL at the time of discontinuation were considered to have discontinued for nonvirologic reasons, and those with a viral load of ≥ 50 copies/mL were considered to have discontinued for virologic reasons.
Source: Sponsor-submitted MAIC report.7
Efficacy Outcomes at the Week 24 Assessment Point
|||||||| || ||||||||| ||| ||| ||||| || |||||||| || ||||||||| ||||||||||| | ||| ||| |||||||||| |||| | |||||||||| |||||| |||| |||||| || |||| |||| |||||| ||| ||| |||||||||| ||| ||| ||||||||||||| ||||||||||| || ||| | | |||| |||||||||| ||||||| |||| ||||||||| ||||||||||| ||| ||||||||||||| ||||||||| || ||| |||||||||| |||||||| ||| | ||||| ||| || | |||| || ||||| | | ||||||| ||| ||| || ||| |||| ||| | ||||| ||| || | |||| || ||||| | | ||||||||||| ||| ||||||| || ||||| ||| |||||||||| |||||||| ||| ||| |||| | ||||||||||| ||||||||||| ||||| ||||||||||| ||| |||| || |||| |||| ||||| || ||| |||||||| ||||||| |||| ||||||||||| | ||| |||| ||| ||||| ||| | ||||| ||| || | |||| || ||||| ||| ||| |||||||||| ||| ||||||||||||| ||||||||||| || | |||||||
|||||||| || ||||||||||| | ||| ||| |||||||||| |||| ||||||||||||| ||||||||||| ||||||||| || ||||||||| ||||||||||||||| |||||||| || ||| ||||| || |||| ||| |||||||||| ||| |||||||| |||||||||
Efficacy Outcomes at the Week 48 Assessment Point
Results for the week 48 assessment ITCs were similar to those for the week 24 assessment.
In summary, the difference in mean change in CD4+ cell count from baseline was not statistically significant in any analyses. Fostemsavir plus OBT exposure was associated with a higher percent of virologic suppression in the unadjusted analyses, but the result was not statistically significant in the MAIC analyses. Fostemsavir plus OBT was not associated with a statistically significant reduction in PDVF in the unadjusted analyses, but after adjustment, the difference was statistically significant. Finally, fostemsavir plus OBT was associated with a significantly lower all-cause discontinuation in both the unadjusted and adjusted analyses.
Table 20: MAIC Primary Analyses — Efficacy Outcomes, Week 48
Outcome at 48 weeks | Unadjusted (naive) | Adjusted (MAIC) | |||||
|---|---|---|---|---|---|---|---|
OBT alone (VIKING-3) | ||| |||| ||| |||||||| | |||| |||||||||| || |||| ||||| | ||||||| | Fostemsavir plus OBT (BRIGHTE)a | Mean difference or odds ratio | P value | |
Sample size | 183 | ||| | ||| | ||| | 78 | NA | NA |
Mean change from baseline in CD4+ cell count, cells/μL, [SD] (95% CI) | 114.80 [||||||| |||||||| ||||||| | |||||| ||||||||| ||||||||| ||||||| | |||| ||||||||| |||||| | ||||| | 141.66 [||||||| ||||||||| ||||||) | 26.86 (–10.79 to 64.52) | 0.162 |
Percent virologic suppression (HIV-1 RNA < 50 c/mL) (95% CI)b | 63.39 ||||||| |||||| | ||||| |||||||| |||||| | |||| ||||||| ||||| | ||||| | 69.81 (|||||| |||||) | 1.34 (0.78 to 2.30) | 0.297 |
Percent PDVF (95% CI) | 22.40 ||||||| |||||| | ||||| |||||||| |||||| | |||| ||||||| ||||| | ||||| | 7.25 (||||| |||||) | 0.27 (0.15 to 0.48) | < 0.001 |
Percent discontinued: all-cause (95% CI) | 22.40 (|||||| |||||) | ||||| ||||||| |||||| | |||| ||||||| ||||| | |||||| | 6.19 (||||| ||||) | 0.23 (0.11 to 0.47) | < 0.001 |
Percent discontinued: virologic (95% CI)c | NR | |||| ||||||| |||||| | ||| | ||| | 5.41 (||||| ||||) | — | — |
Percent discontinued: nonvirologic (95% CI)c | NR | |||| ||||||| ||||| | ||| | ||| | 0.78 (||||| ||||) | — | — |
c/mL = copies per millilitre; CD4+ = cluster of differentiation 4; CI = confidence interval; MAIC = matching-adjusted indirect comparison; NA = not applicable; NR = not reported; OBT = optimized background therapy; PDVF = protocol-defined virologic failure; SD = standard deviation.
Note: P values were assessed at the 5% (alpha = 0.05) significance level.
aRestricted to patients with a screening HIV-1 RNA ≥ 500 copies/mL.
bA threshold of < 50 copies/mL was employed for both studies.
cIn the BRIGHTE study, the cause of discontinuation (virologic or nonvirologic) was determined by a patient’s viral load at the time of discontinuation; patients with a viral load of < 50 copies/mL at the time of discontinuation were considered to have discontinued for nonvirologic reasons, and those with a viral load of ≥ 50 copies/mL were considered to have discontinued for virologic reasons.
Source: Sponsor-submitted MAIC report.7
Sensitivity Analyses for Efficacy Outcomes: OSSnew Adjustment in the VIKING-3 Population
|| ||| ||||||| ||||||||| |||||||||||| ||| ||||||| || || ||||| |||||| |||||| |||||| | || || ||| |||||||| ||||||||||| ||| ||||||||||| |||||||| |||| ||||||||| ||| |||||||| |||||||| ||||||||| ||| ||||||||||| ||| |||||||||| || |||||| || |||||||| |||||| || ||||| || ||| |||||||||||| |||||||||||||||
||||||| |||||| ||||||||||| ||| |||||||||| || |||||||| ||| |||| |||||||||||| ||| ||||| |||||| ||| ||||||| || || |||||||||| || ||| |||||||||| || |||||||| ||||||||||||| |||||||||| || ||| |||| || |||||||||| ||||| || ||||||||| |||||| || |||||||| ||| | ||||| ||| |||||||| |||| |||| ||| || ||||||||| ||||||||||||||||| |||||||||| ||||||||| |||||||||||| ||| ||||||| || || ||| ||||| |||||| |||||| |||||||| || |||||| || || ||| ||| ||||||||||
Table 21: OSSnew in the VIKING-3 Primary Analysis vs. Sensitivity Analyses
OSSnew (%) | Fostemsavir plus OBT (BRIGHTE), unadjusted, combined cohorta | OBT alone (VIKING-3) | |||
|---|---|---|---|---|---|
Published baseline data | Primary analysis, recalculated | ||||||||||| ||||||||| ||||||| |||||||||| | ||||||||||| ||||||||| |||||||||| |||||||| | ||
0 drug classes available | 31.50 | 37.16 | 0.00 | ||||| | ||||| |
1 drug class available | 35.26 | 42.62 | 37.16 | ||||| | ||||| |
2 drug classes available | 31.21 | 19.13 | 42.62 | ||||| | ||||| |
> 2 drug classes available | 2.02 | 1.09 | 20.22 | ||||| | |||| |
OBT = optimized background therapy; OSSnew = overall susceptibility score (for new [not previously taken] drugs in the OBT).
aRestricted to patients with a screening HIV-1 RNA ≥ 500 copies/mL; OSSnew recalculated to reflect the way OSR was derived in the VIKING-3 study.
Source: Sponsor-submitted MAIC report.7
|| ||| ||||||||||| |||||||| || ||||||| |||||| |||||||||| || |||| ||| ||| ||||||||| |||||| |||| || ||||||| ||||| |||||||||| ||| |||| | ||| ||||||||| |||| ||| |||||||| |||||| ||||| || ||| ||||||||||| |||||||| || ||||||||| |||| |||||||||| || |||| ||| ||| ||||||||| |||||| |||| || ||||||| ||||| |||||||||| ||| |||| | ||| ||||||||| |||| ||| |||||||| |||||| |||||||||||||| || ||| ||||||| ||||||||| | ||||| || |||||||||||| |||||| ||| ||||| |||||| |||||||||| ||| |||||||| ||| ||| ||||||| |||||||||| ||| |||| |||| || ||| ||| ||||||||| || |||||| ||||||||||| ||||||| || ||| ||||||| ||||||||| ||||||||||| | ||| ||| |||||||||| |||| | ||||||||||||| ||||||||||| ||||||||| || ||||||||| ||||||||||||||| || |||| || ||| ||||||||||| |||||||| || |||| |||| || ||| || |||||||| || ||| |||||| ||| ||||||| ||| |||| |||||| ||| ||||||||| ||||||||||| |||| |||||||||||| ||| || ||| ||| |||| |||||||| ||| |||| |||||| |||||||| |||||| ||| ||||||| ||||||||| ||| |||| |||||| ||| |||| ||| ||| ||||||||||||| ||||||||||| || ||| ||||||||||| |||||||||
Sensitivity Analyses for Efficacy Outcomes: Randomized Population of the BRIGHTE Study Only
In the primary analysis, both the randomized and nonrandomized cohorts of the BRIGHTE study were included to maximize the available sample size. | ||||||||||| |||||||| ||| ||||||||| ||||| |||| ||| |||||||||| |||||| || |||||||| |||| |||| ||||||||||| |||||||||| |||||||| ||| |||||||||| || ||||| |||| | ||||||||| ||| || |||| |||||||||| ||| |||||||||| |||||| |||| ||| ||||||| ||| |||| ||| ||||| |||| ||| ||| ||| ||| | ||||||||| || |||||| ||||||||||||| ||||||||||| |||||||||| ||| |||||||| || ||||||| |||| || |||||| || ||||||||||| | ||| ||||| |||| || |||| || ||| ||| |||||||| |||| |||||||||| ||| ||| ||||||||||||| ||||||||||| || ||| |||||||||| ||||||| |||||||| || |||||| |||||||||| ||||||||||||| || ||| ||||||| ||||||||| ||||| ||| || ||||||||||||| ||||||||||| |||||||||| || ||| |||| |||||| || |||| |||| ||||| ||| || ||||||| ||||||||| |||||||||||| ||| | ||||||||||||| ||||||||||| ||||||||| || |||| || ||||||||| ||||||||||||||| ||| |||||||| || |||| || ||| || || |||| ||| |||||||||| ||| |||||||| |||||| |||||||||
Safety Outcomes
For the week 48 DCO analyses (Table 22), the proportion of patients who experienced any SAE was significantly higher for fostemsavir-exposed patients in the unadjusted comparison, but the difference was not statistically significant in the MAIC. Note that the median (range) exposure in the VIKING-3 study was 507 (14 to 757) days, whereas in the BRIGHTE study, it was 621 (1 to 1,039) days.
There were no statistically significant differences between any individual SAE, death, or discontinuation due to AEs. In general, a high degree of uncertainty was observed for the comparative treatment-effect estimates.
| ||||||||||| |||||||| ||| ||||||||| ||||||||| ||| ||||||| ||| |||| ||||||| ||||||| ||||||| ||||||||| ||| || || ||||| ||||| || ||| ||||||| ||| |||| |||||||| ||||||| ||||||| ||||||||| ||| ||| || |||| |||||| ||||| |||| || ||||||||||||| ||||||||||| ||||||||||| || ||| ||||||| || |||| ||||||||||| |||||||||
Table 22: MAIC Primary Analysis — SAEs, Week 48
Event | Unadjusted (naive) | Adjusted (MAIC) | |||||
|---|---|---|---|---|---|---|---|
OBT alone (VIKING-3), proportion (95% CI) | ||| |||| ||| ||||||||| ||||||||||||| |||| ||| | |||| ||||| |||| ||| | ||||||| | Fostemsavir plus OBT (BRIGHTE),a proportion (95% CI) | Odds ratio (95% CI) | P value | |
N | 183 | ||| | ||| | ||| | 78 | NA | NA |
Any SAE | 0.213 (0.156 to 0.280) | ||||| |||||||| |||||| | ||||| ||||||||||||| | ||||| | 0.289 (0.242 to 0.339) | 1.499 (0.833 to 2.698) | 0.177 |
Cellulitis | 0.000 (0.000 to 0.02) | ||||| |||||||| |||||| | ||| | ||| | 0.015 (0.005 to 0.034) | — | — |
Dehydration | 0.016 (0.003 to 0.047) | ||||| |||||||| |||||| | ||||| |||||||| |||| | ||||| | 0.000 (0.000 to 0.010) | — | — |
Pneumonia | 0.027 (0.009 to 0.063) | ||||| |||||||| |||||| | ||||| |||||||| |||| | ||||| | 0.015 (0.005 to 0.034) | 0.545 (0.177 to 1.677) | 0.290 |
Pyrexia | 0.016 (0.003 to 0.047) | ||||| |||||||| |||||| | ||||| |||||||| |||| | ||||| | 0.000 (0.000 to 0.010) | — | — |
Acute kidney injury | 0.006b (0.000 to 0.030) | ||||| |||||||| |||||| | ||||| |||||||||||| | ||||| | 0.003 (0.000 to 0.016) | 0.570 (0.053 to 6.186) | 0.644 |
Death | 0.011 (0.001 to 0.039) | ||||| |||||||| |||||| | ||||| |||||| ||||| | ||||| | 0.019 (0.007 to 0.039) | 1.720 (0.353 to 8.392) | 0.502 |
Discontinuation due to adverse events | 0.044 (0.019 to 0.084) | ||||| |||||||| |||||| | ||||| ||||||||||||| | ||||| | | 0.041 (0.023 to 0.067) | 0.930 (0.312 to 2.774) | 0.896 |
CI = confidence interval; MAIC = matching-adjusted indirect comparison; NA = not applicable; OBT = optimized background therapy; SAE = serious adverse event.
aRestricted to patients with a screening HIV-1 RNA ≥ 500 copies/mL.
bNot reported; incidence of acute renal failure assumed.
Source: Sponsor-submitted MAIC report.7
Critical Appraisal of Sponsor-Submitted MAIC
An SLR was conducted to identify potentially relevant studies for the sponsor-submitted MAIC. Three trials were identified as potentially relevant for the purposes of informing an OBT comparator arm in ITCs: VIKING-3, BENCHMRK, and TMB-301. The VIKING-3 study9 was selected as the trial most similar to the BRIGHTE study with regard to the patient population, particularly HIV-1 RNA and treatment history at baseline, and was also considered the most relevant to the Canadian clinical context in terms of available treatments and patient population. The TMB-301 study evaluated ibalizumab, which is not currently available or marketed for use in Canada. In the BENCHMRK-1 and BENCHMRK-2 studies, the ARV regimens used in the OBT-alone arm did not closely reflect the combination of regimens used in the BRIGHTE study and, in particular, lacked dolutegravir. Because 303 of 371 patients (82%) in the BRIGHTE study received dolutegravir as part of their OBT,8 it more closely matches the ARVs used in the VIKING-3 study.
VIKING-3 was a single-arm, open-label, phase III study of dolutegravir-containing regimens in which HTE adults with HIV received dolutegravir 50 mg twice daily in an initial 7-day monotherapy phase, followed by the same dose in addition to OBT. The clinical expert consulted by CADTH concurred that the VIKING-3 study was a reasonable representation of the Canadian treatment landscape and patient population for the purpose of informing the OBT arm in the sponsor-submitted MAIC.
There were some important differences between the population of the VIKING-3 study and that of the BRIGHTE study, including the screening RNA threshold for trial eligibility, differences in the proportion of patients with AIDS at baseline, and the distribution of OSSnew, GSS, and PSS. There were also between-study differences in the outcome definitions of virologic suppression and PDVF. For virologic suppression, the sponsor was able to use the threshold in the VIKING-3 study and apply it to the BRIGHTE IPD. However, the differences in PDVF could not be accounted for. The clinical expert consulted by CADTH noted that PDVF may be more strictly defined in the BRIGHTE study, but agreed with the sponsor’s clinical expert guidance that this residual difference was unlikely to be of clinical significance. Nonetheless, it complicates interpretation of the MAIC results.
In the unanchored MAICs, patients from the BRIGHTE study who would not have met the VIKING-3 screening RNA threshold were excluded. The MAICs adjusted for a list of prognostic factors and treatment-effect modifiers that were determined through examination of literature and consultation with an international steering committee of consultant physicians who had a special interest in HIV. The reason these factors were selected was not reported in detail, nor was justification presented for the exclusion of other potential factors, such as history of treatment failure, resistance status by class, or coinfection with hepatitis B or hepatitis C. Unanchored ITCs require strong assumptions about the data that are nearly impossible to meet, as unbiased estimates depend in part on adjustment for all known and unknown prognostic factors and treatment-effect modifiers. Although the sponsor followed a comprehensive and expert-guided process to identify prognostic factors and treatment-effect modifiers, it is unknown whether all relevant variables were captured. Additionally, prognostic factors and treatment-effect modifiers for outcomes such as discontinuation and SAEs may or may not be similar to those relevant to efficacy outcomes.
In the VIKING-3 study, patients could withdraw after week 48, once commercial dolutegravir was available, which was recorded as a failure. Therefore, comparisons were conducted only up to week 48.
Efficacy outcomes were assessed at 24week and 48-week relative study time and included the mean change in CD4+ count from baseline, the proportion of patients with virologic suppression (aligned with the VIKING-3 definition of < 50 HIV-1 RNA copies/mL), PDVF, and all-cause discontinuation. Although the populations appeared to be balanced after MAIC, based on the selected list of prognostic factors and treatment-effect modifiers, the balance in treatment history and other variables was not reported. Importantly, the effective sample size in the BRIGHTE study was reduced by nearly 80% in the primary MAIC analyses, reflecting poor overlap in the studies and potentially unstable MAIC results. |||||| |||| |||||||||| |||| |||| |||||| || ||| ||||||||||| |||||||| |||| ||||||||||| ||||||||||| |||||| |||||| || ||| |||||||| ||||||||||| ||| ||||| |||||||| ||||
Balance in OSSnew is difficult to interpret due to the number of assumptions required to facilitate MAIC. The difference in the derivation of OSRs between the BRIGHTE and VIKING-3 studies was resolved through alignment with the method of the VIKING-3 study; a net assessment of partial sensitivity to a drug was assigned an OSR of 0.5 in the BRIGHTE study and of 0 in the VIKING-3 study, so for the MAIC, the distribution of OSSnew was recalculated for the BRIGHTE study using the binary approach of the VIKING-3 study. Additionally, by design, all patients entering the VIKING-3 study were dolutegravir-naive, which is reflected in the baseline values contributing to the distribution of OSSnew. However, results from the VIKING-3 study would be inclusive of patient exposure to dolutegravir through the duration of the study. Three different assumptions were used to resolve this: in the primary analysis, it was assumed that dolutegravir was fully active (OSRnew = 1), ||||| || ||| ||||||| |||||||||| ||||||||||| ||||||||| |||||||||||| ||| ||||||| || || ||||| |||||| |||| || ||| |||||||||| || |||||||| ||| ||| ||||||||| ||||||||||| || |||| || || |||||||| |||||| |||||||| ||| |||||||| | ||||||||||| |||||||| ||| ||||||||| |||||||| |||||||||||| ||| ||||||| || || ||| ||||| |||||| |||||| |||||| | ||| ||| ||||||| || ||||| ||||||||||| |||||||| |||| ||||||||| ||||||| || ||| ||||||| ||||||||| |||||| |||| ||| |||||| ||| ||||||| |||| ||| || |||||| ||||||||||||| ||||||||||| || |||||| || ||| ||||||||||| ||||||||| |||||||||| |||| ||| ||||||| |||||||| ||| || |||||| || |||||| || ||||||||||| | ||| ||| |||| |||||||| ||| ||||||||| ||| ||||||||| || ||||||||| |||| |||||| || |||| ||||||||||| |||||||||| ||| ||| |||||||||||| || ||||||| |||||||||| |||| ||||||| || |||||| || |||||||||||| || ||||||||| || |||| |||||||||.
Because OSSnew was identified as a key prognostic factor and/or treatment-effect modifier for this patient population and because there were no other factors adjusted to account for between-trial heterogeneity related to prior treatment exposure or failure, uncertainty in this assumption complicates the interpretation of the MAICs.
Additionally, the BRIGHTE study allowed patients without fully active ARVs remaining (in the nonrandomzied cohort), whereas the VIKING-3 study required at least 1. |||||||| ||| ||||||||||| |||||||| ||||||||| |||| ||| |||||||||| |||||| || ||||||| ||| ||||||| ||||||| || ||| ||||||| |||||||||
Adjustment of the BRIGHTE IPD to reflect the population of the VIKING-3 study necessarily results in shifting toward a population with more treatment options remaining, as defined by the distribution of OSSnew. This may, therefore, be less representative of the target population than before adjustment, and may not be generalizable to the full population eligible for fostemsavir, particularly with regard to patients with the most highly resistant disease, who are in the greatest need of new treatment options.
The unadjusted and MAIC analyses were both presented. || ||| |||||||||| ||||||||| ||||||||||| |||||||| ||| |||||||||| |||| | ||||||||||| ||||||||| || ||| |||| || ||||||||| ||||||||||||||| ||| | ||||||||||| |||||||| || ||| |||| || |||||||||||| ||| |||| ||| ||||| |||||||| ||| |||||| ||||||| |||| |||||||||||| ||| || ||| |||| |||||||| ||| |||| |||||. In the primary MAIC analyses, the results were similar, except that fostemsavir was significantly associated with a reduced risk of PDVF, and the odds of experiencing any SAE became inconclusive because the 95% CI included the null value. The results for most safety outcomes were generally imprecise, and interpretation is confounded by substantial differences in study-drug exposure at the 48-week DCO of each trial, which may have resulted in bias against fostemsavir. | ||||||||||| |||||||| ||| ||||||||| ||||||||| ||| ||||||| ||| || ||||||| || ||| ||||||| ||| || ||||||||| ||| ||| ||||||||||| || |||||||| || ||||| |||| ||||| || |||||| || |||||| || |||||||||||. Ultimately, the safety results of both the unadjusted analyses and the MAICs were considered by CADTH to be inconclusive due to these limitations.
In general, results for important efficacy outcomes (CD4+ count and virologic suppression) and all safety outcomes were inconclusive in both the unadjusted analyses and MAICs due to a combination of confounding factors and uncertainty, and wide CIs that included the null value. The MAICs demonstrated that fostemsavir may have lower rates of PDVF and discontinuation, but this must be interpreted with caution in light of the limitations and strong assumptions of the unanchored MAICs.
Methods of MAIC by Anderson et al.
The SLR conducted by CADTH also identified a published MAIC (Anderson et al. [2022]8), which included comparisons between the BRIGHTE study and each of the TMB-301, BENCHMRK, and VIKING-3 studies. The MAIC between the BRIGHTE and VIKING-3 studies was the same analysis as the sponsor-submitted MAIC.
As discussed by the sponsor with regard to the sponsor-submitted MAIC, eligibility criteria in the VIKING-3 study were more similar to those in the BRIGHTE study than to those in the other 2 studies (TMB-301 and BENCHMRK) with regard to HIV-1 RNA and treatment history.
The methodology and results of the MAIC of the BRIGHTE and VIKING-3 studies were the same as previously described (i.e., the sponsor-submitted MAIC).
The MAIC methodology was the same for comparisons of the BRIGHTE and TMB-301 studies and the BRIGHTE and BENCHMRK studies. As these were considered less relevant comparisons, the results will not be considered in detail, but will be summarized briefly here (MAIC primary analyses).
Virologic suppression:
For the BRIGHTE versus TMB-301 (ibalizumab and OBT) studies, there was no statistically significant difference at week 24 (odds ratio [OR] = 1.44; 95% CI, 0.74 to 2.80; P = 0.284).
For the BRIGHTE versus BENCHMRK (OBT) studies, fostemsavir plus OBT was associated with significantly higher odds of virologic suppression at week 96 (OR = 3.26; 95% CI, 2.08 to 5.11; P < 0.001).
Change from baseline in CD4+ cell count:
For the BRIGHTE versus TMB-301 (ibalizumab and OBT) studies, there was no statistically significant difference at week 24 (mean difference = 7.05 cells/mm3; 95% CI, −60.88 to 74.98; P = 0.834).
For the BRIGHTE versus BENCHMRK (OBT) studies, fostemsavir plus OBT was associated with significant improvement in mean CD4+ cell count at week 96 (mean difference = 135.78 cells/mm3; 95% CI, 91.93 to 179.63; P < 0.001).
All-cause discontinuation:
For the BRIGHTE versus TMB-301 (ibalizumab and OBT) studies, there was no statistically significant difference at week 24 (OR = 0.38; 95% CI, 0.13 to 1.09; P = 0.073).
For the BRIGHTE versus BENCHMRK (OBT) studies, there was no statistically significant difference (OR = 1.14; 95% CI, 0.66 to 1.99; P = 0.634).
Discontinuation due to AEs:
For the BRIGHTE versus TMB-301 (ibalizumab and OBT) studies, fostemsavir plus OBT was associated with significantly lower odds of discontinuation due to AEs (OR = 0.26; 95% CI, 0.08 to 0.89, P not reported).
For the BRIGHTE versus BENCHMRK (OBT) studies, there was no statistically significant difference (OR = 0.66; 95% CI, 0.19 to 1.91; P not reported).
Development of any SAE:
For the BRIGHTE versus TMB-301 (ibalizumab and OBT) studies, there was no statistically significant difference (OR = 0.65; 95% CI, 0.30 to 1.56, P not reported).
For the BRIGHTE versus BENCHMRK (OBT) studies, there was no statistically significant difference (OR = 1.41; 95% CI, 0.86 to 2.30, P not reported).
Note that before adjustment, fostemsavir plus OBT was associated with significantly higher odds of experiencing any SAE at week 96 (OR = 1.79; 95% CI, 1.23 to 2.62, P not reported).
Mortality:
For the BRIGHTE versus TMB-301 (ibalizumab and OBT) studies, fostemsavir plus OBT was associated with significantly lower odds of mortality (OR = 0.11; 95% CI, 0.02 to 0.54).
For the BRIGHTE versus BENCHMRK (OBT) studies, there was no statistically significant difference (OR = 0.92; 95% CI, 0.21 to 3.23).
Note that before adjustment, fostemsavir plus OBT was associated with significantly higher odds of mortality at week 96 (OR = 2.33; 95% CI, 1.03 to 5.96).
Because the results of the BRIGHTE versus VIKING-3 comparison are the same as those previously discussed (i.e., the sponsor-submitted MAIC), refer to the critical appraisal of the sponsor-submitted MAIC.
The TMB-301 and BENCHMRK comparisons were considered to be less relevant, so a formal critical appraisal was not conducted by CADTH for the MAIC by Anderson et al. Limitations similar to those in the sponsor-submitted MAIC apply, with additional concerns related to generalizability, as follows:
The TMB-301 study did not include participants who were comparable to the nonrandomized cohort of the BRIGHTE study, in which patients had no fully active ARVs available.
The TMB-301 OSS did not count fostemsavir as a fully active ARV.
The TMB-301 study had limited follow-up time available (i.e., 24 weeks).
The TMB-301 study assessed a therapy not currently available in Canada (ibalizumab + OBT).
In the TMB-301 study, 43% of patients were taking fostemsavir as a part of their OBT, and in the BRIGHTE study, 4% of patients were taking ibalizumab as part of their OBT. It was not possible to remove the 43% of patients in the TMB-301 study because no subgroup data were available.
Although the BENCHMRK studies included an OBT-alone group in an HTE population, the trial began in 2006, and the ARVs used in the OBT-alone group (e.g., darunavir and tipranavir) do not reflect those used in the BRIGHTE study. In particular, the BENCHMRK studies lacked dolutegravir, which was a component of OBT in 82% of patients in the BRIGHTE study.
Other Relevant Evidence
No other relevant studies in the population of interest that addressed an important gap in the available evidence were identified for this review.
Discussion
Summary of Available Evidence
One sponsor-funded, multinational, phase III, DB, RCT was included in this review. The BRIGHTE study randomized 272 adults with HIV-1 who were ARV experienced and had documented historical or baseline resistance, intolerability, and/or contraindications to ARVs in at least 3 classes. Patients were randomized, in a 3:1 ratio, to fostemsavir 600 mg orally, or placebo, added to their failing ARV regimen for a DB period of 8 days. This DB phase was followed by an OL phase of at least 96 weeks, during which all patients were on fostemsavir plus OBT. The primary outcome, assessed during the DB phase, was the change from baseline to day 8 in HIV-1 RNA. Secondary outcomes, none of which were formally assessed, included the percentage of patients with a decrease in HIV-1 RNA of greater than 0.5 log10 copies/mL and greater than 1.0 log10 copies/mL at day 8, virologic response (HIV-1 RNA level of < 40 copies/mL at weeks 24, 48, and 96), resistance testing for patients experiencing virologic failure, mean change in CD4+ count through week 96, and events resulting in a diagnosis of AIDS, using the CDC classification system, Evidence was also available from a MAIC submitted by the sponsor, as well as a published MAIC. There were no extensions or other relevant studies identified for this review.
Patients in the randomized cohort were approximately 44 years of age, and the majority were male (74%) and white (68%). Most patients (89%) had a baseline viral load of 1,000 copies/mL or higher, and more than half (57%) had a baseline viral load of 30,000 copies/mL or more. Approximately one-quarter of patients had a CD4+ count of less than 20 cells/mm3, and a similar percentage had a baseline CD4+ count of 200 cells/mm3 or more. Approximately one-third of patients had been treated for HIV more than 20 years, and 85% of patients, overall, had a positive AIDS history, meaning that they either had a nadir CD4+ count of less than 200 cells/mm3 or a response of yes to the question, Does participant have AIDS? on the disease history component of the CRF. Most patients (90% or more) had prior exposure to a NNRTI, NRTI, or PI, whereas 75% had prior exposure to an INSTI. Other ARV that patients had prior exposure to included entry inhibitors (39%), CCR5 antagonists (26%), and other investigational ARVs (11%). The most common ARV classes in the failing regimen were NRTIs (81%), PIs (67%), INSTIs (44%), and NNRTIs (28%); other classes included CCR5 antagonists (12%) and entry inhibitors (4%).
Interpretation of Results
Efficacy
For the past 2 decades, the vast majority of patients with HIV-1 have maintained control of viremia using combinations of ARVs, initially primarily consisting of NRTIs, NNRTIs, and PIs, and joined more recently by INSTIs. However, there remains a small minority of patients whose HIV-1 remains uncontrolled and/or whose treatment options are very limited, due in large part to issues with resistance. According to the clinical expert consulted by CADTH on this review, these HTE patients are managed with whatever remaining drugs they are still sensitive to, many of which are suboptimal due to limitations with respect to safety or tolerability issues or route of administration. Fostemsavir is the first in a series of ARVs with a novel mechanism of action, intended to address the treatment gap seen in the HTE population. Because these novel ARVs that were designed to address the HTE population are just coming on to the market, there is a lack of direct comparative data between these new drugs, and instead patients in the BRIGHTE study were randomized to either fostemsavir or placebo, plus their current failing regimen, in the DB phase, and then switched to fostemsavir plus OBT in the OL phase of the study. The DB phase of the BRIGHTE study lasted only 8 days, and therefore the only outcome that was formally assessed was the mean change from baseline in HIV-1 RNA. The explanation provided by the sponsor for the short DB phase was that it did not want to continue using a failing ARV regimen without adding another active drug to the regimen, due to concerns over increased resistance. The sponsor also asserted that 8 days was enough to establish proof of concept or, in other words, to establish that fostemsavir, when added to a failing ARV regimen, could elicit a reduction in viral load. The 8-day DB phase also appears to be consistent with FDA guidance.17
Another issue with such a short comparative phase is that it does not produce a reliable estimate of the ability of fostemsavir to elicit an increase in CD4+ counts. CD4+ counts have traditionally been a key surrogate marker for assessing the efficacy of ARV, as there has traditionally been a well-established inverse relationship between viral load and CD4+ count, and CD4+ counts have, in turn, been used to predict the risk of developing various complications of HIV, including progression to AIDS and death. However, according to the clinical expert consulted by CADTH on this review, it can take several months, or more, for CD4+ counts to begin to increase in response to a reduction in HIV-1 RNA. Indeed, results from the OL phase of the BRIGHTE study demonstrate a gradual increase in CD4+ counts over time, continuing out to both the 96-week and 240-week follow-up time periods. This gradual increase in CD4+ counts needs to be interpreted with caution, however, as there is a gradual attrition of patients from the trial, many because of death due to AIDS, and presumably these patients would have much lower CD4+ counts. Ultimately, however, no conclusions can be drawn from the BRIGHTE study regarding the ability of fostemsavir to improve CD4+ counts compared to placebo. The clinical expert consulted by CADTH on this review noted that although CD4+ counts remain an important clinical prediction tool, the goal of therapy in HIV has always been, and continues to be, reduction in viral load. For example, in a study by Shoko and Chikobvu (2019),18 the authors used a Markov model and longitudinal data from an HIV clinic in South Africa to conclude that viral load is a superior predictor of HIV and/or AIDS progression than CD4+ counts. Ultimately, according to the clinical expert, the goal in assessing novel antivirals is, and should continue to be, their ability to reduce viral load.
The short DB phase also makes it difficult to assess the impact of fostemsavir on the risk of patients progressing to AIDS and AIDS-related death, outcomes that are clearly of concern for patients, according to the input provided to CADTH. According to the clinical expert consulted by CADTH on this review, the HTE population is at far higher risk of progressing to AIDS than typical patients living with HIV; therefore, these outcomes are important to track and assess in this population, and 8 days is not a sufficient duration to conduct a comparison with placebo. There were patients who died of AIDS in the fostemsavir group, and this number increased from the 96-week to the 240-week follow-up; these percentages were higher in the nonrandomized cohort than in the randomized cohort, which is consistent with expectations, as patients in the nonrandomized cohort no longer have any viable treatment options left and are likely, therefore, to have more advanced disease. However, aside from drawing inferences when comparing longer-term results in these 2 cohorts, whether fostemsavir reduces the risk of AIDS-related morbidity and mortality relative to currently available treatments (OBT) cannot be concluded based on results of the BRIGHTE trial. What is known and has become well-established dogma in HIV and AIDS is that reducing viral load to undetectable levels does indeed reduce the risk of complications from AIDS, including death.
HRQoL is clearly an important outcome for patients living with HIV, and likely particularly important for HTE patients, as these patients are often experiencing AIDS-related complications, such as opportunistic infections and neurologic complications. In addition, patient input to CADTH suggests that HTE patients are particularly concerned about the increased risk of disability and death associated with infection. Although an instrument that was modified to assess HRQoL in patients living with HIV, the FAHI, was used in the BRIGHTE study, the lack of a control group, and perhaps more important, the lack of blinding, make it challenging to draw any conclusions from these data. Although scores on the FAHI increased from baseline, suggesting an improvement in HRQoL, the lack of blinding means that it is not possible to know whether the improvement in HRQoL was due to a patient’s knowing that they were assigned a new ARV that held the promise of improving their prognosis, or whether this was due to an improvement in the symptoms and/or complications they were experiencing from HIV. This is further complicated by the lack of interpretable data for hard clinical outcomes, such as AIDS-related complications.
Resistance is clearly an issue in the HTE population; such patients exhibit multidrug resistance to ARV and are, therefore, very limited in their treatment options. Resistance data were reported for the DB and OL phases of the BRIGHTE study and, according to the clinical expert consulted by CADTH on this review, the evidence suggests that the efficacy of fostemsavir in patients expressing resistance-associated polymorphisms may not be an issue. There are some challenges associated with interpreting data for specific resistance-associated polymorphisms due to the small sample size of the study and the relatively rare occurrence of certain polymorphisms. For example, after 96 weeks in the OL phase, only 3 patients were identified as having an M475I mutation, and all had a virologic response; however, 1 cannot assume that in clinical practice, all patients who have this mutation will respond to fostemsavir. The clinical expert noted that resistance testing and resistance-associated polymorphisms are used to guide therapeutic decisions in clinical practice. Presumably, the tracking of resistance-associated polymorphisms associated with fostemsavir will continue as the drug goes into widespread use in clinical practice, providing a much larger sample to draw upon, which will allow for enhanced precision in therapeutic decision-making.
There were no direct comparisons of fostemsavir and other ARVs that can be used in this HTE population, leaving only indirect comparisons, the results of which were largely inconclusive.
Harms
There were no obvious concerns related to the harms of fostemsavir during the 8-day DB phase, although this is an inadequate duration of follow-up to provide an assessment of harms versus placebo, particularly for a novel, first-in-class drug. Notable harms such as IRIS and QT prolongation were reported infrequently. Assessment of safety and tolerability in the OL phase is limited by the lack of comparator. From the 96-week follow-up to the 240-week follow-up, there were numerical increases in the percentage of patients experiencing specific AEs, such as diarrhea, nausea, upper respiratory tract infection, and influenza, as well as overall SAEs. Numerical increases in these harms may be explained by the difference in length of follow-up; however, the possibility of an increasing risk of harms with longer-term therapy cannot be excluded.
Conclusions
Evidence from 1 DB RCT suggests that when combined with a failing ARV regimen, fostemsavir reduces viral load after 8 days of therapy, compared to placebo, establishing proof of concept. In the subsequent OL phase of the BRIGHTE trial, improvements in viral load appeared to be maintained and CD4+ counts increased over a 240-week treatment period; however, analysis of these outcomes is potentially biased by attrition and other sources of missing data over such a long follow-up period. There were AIDS-related deaths and indicators of progression to AIDS in the OL phase; however, without a control group, there is a lack of context for these findings. In the brief, 8-day DB phase of the trial, there were no clear indications of safety or tolerability issues compared to placebo. There were numerical increases in the number of patients experiencing specific AEs and overall SAEs from 96 weeks to 240 weeks of follow-up in the OL phase, but it is not clear whether this represents an increased risk of these AEs or is simply a consequence of the increased duration of follow-up. Evidence from indirect comparisons of fostemsavir versus OBT were generally inconclusive, largely due to a small sample size after a majority of patients from the index trial was excluded and concerns about incomplete matching adjustment on effect modifiers.
References
1.Centers for Disease Control and Prevention. About HIV. 2022; https://www.cdc.gov/hiv/basics/whatishiv.html. Accessed 2023 Jan 19.
2.Challacombe L. The epidemiology of HIV in Canada. CATIE: Canada's Source for HIV and hepatitis C information.2021: https://www.catie.ca/the-epidemiology-of-hiv-in-canada. Accessed 2023 Jan 19.
3.Rukobia (fostemsavir): 600 mg extended release tablets [product monograph]. Laval (QC): ViiV Healthcare ULC; 2021 Oct 1.
4.Drug Reimbursement Review sponsor submission: Rukobia (fostemsavir), 600 mg extended-release tablets [internal sponsor's package]. Montreal (QC): ViiV Healthcare ULC; 2022 Oct 21.
5.ViiV Healthcare ULC. CADTH reimbursement review sponsor comments template: rukobia (fostemsavir) 2023 Feb 14.
6.Clinical Study Report: 205888 [BRIGHTE Study]. Week 96 results, 205888 (AI438047): a multi-arm, phase 3, randomized, placebo-controlled, double-blind clinical trial to investigate the efficacy and safety of BMS-663068 (GSK3684934, fostemsavir) in heavily treatment-experienced subjects infected with multi-drug-resistant HIV-1 [internal sponsor's report]. Research Triangle Park (NC): ViiV Healthcare Company; 2019 May 9.
7.Comparative effectiveness of fostemsavir and Optimised Background Therapy (OBT) versus optimised regimens of existing agents in heavily treatment experienced HIV patients for use in health economic evaluation. (Vol.2) [VIKING-3 MAIC report]. [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Rukobia (fostemsavir), 600 mg extended-release tablets. Montreal (QC): ViiV Healthcare ULC; 2022 Oct 21.
8.Anderson SJ, van Doornewaard A, Turner M, et al. Comparative efficacy and safety of fostemsavir in heavily treatment-experienced people with HIV-1. Clin Ther. 2022;44(6):886-900. PubMed
9.Castagna A, Maggiolo F, Penco G, et al. Dolutegravir in antiretroviral-experienced patients with raltegravir- and/or elvitegravir-resistant HIV-1: 24-week results of the phase III VIKING-3 study. J Infect Dis. 2014;210(3):354-362. PubMed
10.Complera (emtricitabine/rilpivirine/tenofovir disoproxil fumarate) tablets: 200 mg emtricitabine, 25 mg rilpivirine, 300 mg tenofovir disoproxil fumarate [product monograph]. Mississauga (ON): Gilead Sciences Canada, Inc; 2018 Dec 3: https://pdf.hres.ca/dpd_pm/00048610.PDF. Accessed 2023 Jan 10.
11.Drug Reimbursement Review clinical review report: cabotegravir tablets, cabotegravir extended-release injectable suspension, and rilpivirine extended-release injectable suspension (Vocabria, Cabenuva) for HIV-1 infection. Ottawa (ON): CADTH; 2020 Sep: https://www.cadth.ca/sites/default/files/cdr/clinical/sr0628-vocabria%2Bcabenuva-clinical-review-report.pdf. Accessed 2023 Mar 5.
12.Kozal M, Aberg J, Pialoux G, et al. Fostemsavir in adults with multidrug-resistant HIV-1 infection. N Engl J Med. 2020;382(13):1232-1243. PubMed
13.Aberg J, Shepherd B, Wang M, et al. Efficacy and safety of fostemsavir plus optimized background therapy in heavily treatment-experienced adults with HIV-1: week 240 results of the phase 3 BRIGHTE study [internal sponsor's report] [poster]. In: Drug Reimbursement Review sponsor submission: Rukobia (fostemsavir), 600 mg extended-release tablets. Montreal (QC): ViiV Healthcare ULC; 2022 Oct 21.
14.Centers for Disease Control and Prevention. Recommendations and reports: appendix A: AIDS-defining conditions. MMWR CDC Surveill Summ. 2008;57(RR10):9.
15.Viala-Danten M, Dubois D, Gilet H, Martin S, Peeters K, Cella D. Psychometric evaluation of the functional assessment of HIV Infection (FAHI) questionnaire and its usefulness in clinical trials. Qual Life Res. 2010;19(8):1215-1227. PubMed
16.van Reenen M, Oppe M, Secnik Boye K, et al. EuroQol Research Foundation. EQ-5D-3L user guide. 2018; https://euroqol.org/publications/user-guides. Accessed 2022 Jan 13.
17.U.S. Food and Drug Administration. Center for Drug Evaluation and Research: integrated review [for fostemsavir]: application number: 212950Orig1s000 Silver Spring (MD): U.S. FDA; 2020: https://www.fda.gov/media/140641/download. Accessed 2023 Jan 19.
18.Shoko C, Chikobvu D. A superiority of viral load over CD4 cell count when predicting mortality in HIV patients on therapy. BMC Infect Dis. 2019;19(1):169. PubMed
19.Sinnott PL, Joyce VR, Barnett PG. Guidebook: preference measurement in economic analysis. HERC (Health Economics Resource Center). Menlo Park (CA): VA Palo Alto Healthcare System; 2007: https://www.herc.research.va.gov/files/BOOK_419.pdf. Accessed 2023 Jan 26.
20.FACIT.org. FAHI: Functional Assessment of HIV Infection. 2021; https://www.facit.org/measures/FAHI. Accessed 2023 Jan 13.
21.Peterman AH, Cella D, Mo F, McCain N. Psychometric validation of the revised Functional Assessment of Human Immunodeficiency Virus Infection (FAHI) quality of life instrument. Qual Life Res. 1997;6(6):572-584. PubMed
22.Cohen J. A power primer. Psychol Bull. 1992;112(1):155-159. PubMed
23.Reeve BB, Wyrwich KW, Wu AW, et al. ISOQOL recommends minimum standards for patient-reported outcome measures used in patient-centered outcomes and comparative effectiveness research. Qual Life Res. 2013;22(8):1889-1905. PubMed
Appendix 1: Literature Search Strategy
Note this appendix has not been copy-edited.
Databases: MEDLINE, Embase
Limits: none
Filters: none
Conference abstracts removed
Submission indication: in combination with other antiretroviral agents for the treatment of HIV type 1 (HIV-1) infection in HTE adults with multidrug-resistant (MDR) HIV-1 infection for whom it is otherwise not possible to construct a suppressive antiviral regimen due to resistance, intolerance, or safety considerations
Drug class: attachment inhibitor
MEDLINE
Rukobia (fostemsavir): (fostemsavir* OR Rukobia* OR bms 663068* OR bms663068* OR gsk 3684934* OR gsk3684934* OR 97IQ273H4L OR 2X513P36U0).ti,ab,kf,ot,hw,nm,rn
Embase
Rukobia (fostemsavir): *fostemsavir/ OR (fostemsavir* OR Rukobia* OR bms 663068* OR bms663068* OR gsk 3684934* OR gsk3684934*).ti,ab,kf,dq
NOT
(conference review OR conference abstract).pt
OVID search conducted on November 22, 2022
Database(s): Embase 1974 to 2022 November 21, Ovid MEDLINE(R) ALL 1946 to November 21, 2022
Searches | Results |
|---|---|
1. (fostemsavir* or Rukobia* or bms 663068* or bms663068* or gsk 3684934* or gsk3684934* or 97IQ273H4L or 2X513P36U0).ti,ab,kf,ot,hw,nm,rn. | 324 |
2. 1 use medal | 85 |
3. *fostemsavir/ or (fostemsavir* or Rukobia* or bms 663068* or bms663068* or gsk 3684934* or gsk3684934*).ti,ab,kf,dq. | 247 |
4. use oemezd | 168 |
5. 4 not (conference review or conference abstract).pt. | 110 |
6. 2 or 5 | 195 |
7. remove duplicates from 6 | 119 |
Appendix 2: Excluded Studies
There were no excluded studies for this report.
Appendix 3: Description and Appraisal of Outcome Measures
Note this appendix has not been copy-edited.
Aim
To describe the following outcome measures and review their measurement properties (validity, reliability, responsiveness to change, and MID):
EQ-5D-3L utility index and VAS
FAHI.
Findings
Table 24: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about Measurement Properties | MID |
|---|---|---|---|
EQ-5D-3L utility and VAS | A generic, preference-based HRQoL measure consisting of descriptive questions and a VAS. For the EQ-5D-3L, the descriptive questions cover 5 dimensions, while each dimension is divided into 3 levels of perceived problems (no, some, extreme problems labelled 1 to 3). Raw 5-digit score and weighted score based on population preference can be calculated. At individual level, higher descriptor, 5-digit score indicates worse quality of life. For example, 55555 (extreme problems in all of the dimensions) represents the worst health state and 11111 (no problems in all of the dimensions) represents the best state. At population level, higher utility index score represents better health (0 = death; 1 = perfect health; negative scores = worse than death). The VAS records the patient’s self-rated health on the day with end points 0 (the worst health you can imagine) to 100 (the best health you can imagine).16 | No studies assessing psychometric properties in HIV population have been identified from literature search. | Unknown in patients with HIV In general populations, MID for EQ-5D utility index score ranges from 0.033 to 0.074.19 |
FAHI | An HIV-specific adaptation of FACT-G which addresses concerns of patients with cancer. The questionnaire is self-administered, but an interview can be conducted when applicable. The recall period is based on the past 7 days.20 The most recent version (v.4) of FAHI contains 47 items in 5 subscales (PWB, EWB, FGWB, SWB, CF). Each item is a 5-point Likert-type scale with score ranges from 0 (not at all) to 4 (very much).15 Forty-four items (27 from the original core instrument, FACT-G and the remaining 17 items reflect HIV/AIDS-specific additional concerns) are scored yielding total scores that range from 0 to 176.21,15 For each subscale (items), score ranges are20:
For all subscales and total scores, higher scores indicate better HRQoL.15 | Psychometric properties have been assessed in treatment-experienced HIV patients (N = 565 and 1,096) enrolled in 2 clinical trials (POWER and DUET trials).15 Validity: Patients in earlier HIV stage (CDC categories) showed higher total score compared to patients in later HIV stage who showed lower total score(P < 0.0001 and 0.024). Similar trend has been observed in most domain scores, some of which have shown to be different depending on HIV clinical stages (CDC categories) (P < 0.001 to 0.616). Weak correlations22 were found between total score and CD4 cell count (Spearman r = 0.17 to 0.19) or viral load (r = −0.14 to −0.13).15 The previous version of FAHI showed total score could discriminate between groups differing in ECOG PSR (P = 0.0001) or CD4+ count (P = 0.0439).21 Reliability: For 5 domains, Cronbach alphas ranged from 0.72 to 0.92, which were acceptable (alpha > 0.7).23,15 In the previous version of FAHI, alpha for domain scores ranged from 0.73 to 0.90 and alpha for a total score was shown to be 0.91.21 Responsiveness to change: When measured in patients categorized based on EQ-5D VAS at baseline and Week 24, a positive change in total score in improved patients, a negative total score change in worsened patients, and no change in total score in stable patients (between group P < 0.001) have been observed.15 | In treatment-experienced HIV patients15: Based on EQ-5D index, MID for total score ranged from 6.5 to 9.0. Based on EQ-5D VAS, MID for total score ranged from 3.2 to 5.8. Based on distribution approach, MID for total score ranged from 3.9 (SEM) to 14.0 (0.5xSD). |
CF = cognitive functioning; EWB = emotional well-being/living with HIV; FACT-G = Functional Assessment of Cancer Therapy-General core questionnaire; FAHI = Functional Assessment of HIV Infection; FGWB = functional and global well-being; MID = minimal important difference; PWB = physical well-being; SWB = social well-being; VAS = visual analogue scale
Sources: Viala-Danten, et al. (2010),15 FACIT.org (2021),20 Sinnott et al. (2007),19 EQ-5D-3L User Guide version 6.0 (2018).16
Pharmacoeconomic Review
Abbreviations
ADE
AIDS-defining event
ARV
antiretroviral
BIA
budget impact analysis
CD4+
cluster of differentiation 4
HIV-1
HIV, type 1
HTE
highly treatment-experienced
ICER
incremental cost-effectiveness ratio
MAIC
matching-adjusted indirect comparison
MDR
multidrug-resistant
OBT
optimized background therapy
QALY
quality-adjusted life-year
ST
salvage therapy
WTP
willingness to pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Fostemsavir (Rukobia), 600 mg tablet |
Submitted price | Fostemsavir, 600 mg, tablet = $62.77 |
Indication | For adults with HIV-1 who are heavily treatment-experienced and have multidrug-resistant HIV-1, and for whom it is otherwise not possible to construct a suppressive antiviral regimen due to resistance, intolerance, or safety considerations |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | October 1, 2021 |
Reimbursement request | Per indication |
Sponsor | Viiv Health care ULC |
Submission history | Previously reviewed: no |
HIV-1 = HIV, type 1; NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov model |
Target population(s) | Adults with multidrug-resistant HIV for whom it is otherwise not possible to construct a suppressive antiviral regimen (per indication) |
Treatment | Fostemsavir |
Comparators | OBT, defined as average mix of most commonly used regimens, based on mix of treatments available in the BRIGHTE randomized trial cohort (including NRTIs, NNRTIs, FIs, PIs, and INSTIs) |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (53 years) |
Key data source |
|
Submitted results | ICER = $469,086 per QALY gained (incremental cost = $315,607; incremental QALYs = 0.673) |
Key limitations |
|
CADTH reanalysis results |
|
CD4+ = cluster of differentiation 4; FI = fusion inhibitor; ICER = incremental cost-effectiveness ratio; INSTI = integrase strand transfer inhibitor; LY = life-year; MAIC = matching-adjusted indirect comparison; NNRTI = nonnucleoside reverse transcriptase inhibitor; NRTI = nucleotide reverse transcriptase inhibitor; OBT = optimized background therapy; PI = protease inhibitor; QALY = quality-adjusted life-year; WTP = willingness to pay.
Conclusions
The CADTH clinical review of the BRIGHTE trial found that fostemsavir reduced viral load compared to placebo during the randomized period of the trial. Conclusions about the impact of fostemsavir on cluster of differentiation 4 (CD4) count, AIDS-related mortality, or progression to AIDS could not be drawn during this period; however, CADTH’s appraisal of the sponsor’s matching-adjusted indirect comparison (MAIC), in which the BRIGHTE trial population was matched with the VIKING-3 trial population, found wide uncertainty around the efficacy of fostemsavir plus OBT compared to OBT alone, such that definitive conclusions could not be made. The effectiveness of fostemsavir plus OBT compared to OBT alone is highly uncertain, both in the short-term and over the course of 48 weeks.
CADTH identified several key limitations in the economic analysis that could not be addressed due to lack of model flexibility and availability of information. As such, CADTH was unable to use the sponsor’s economic model to derive a more reliable estimate of the cost-effectiveness of fostemsavir. Using the sponsor’s base case, fostemsavir plus OBT is $315,607 more costly and produces 0.673 more QALYs than OBT alone, resulting in an ICER of $469,086 per QALY gained. The sponsor’s results suggest that a 94% reduction in the price of fostemsavir would be required for fostemsavir plus OBT to be considered cost-effective compared to OBT alone at a willingness-to-pay (WTP) threshold of $50,000 per QALY gained. CADTH noted several limitations that add uncertainty to these results. The analysis relies on the assumption that the long-term incremental effectiveness of fostemsavir (compared to OBT alone) is maintained over time, which was beyond the scope of the submitted evidence supporting the economic model. The incremental effectiveness of treatment may, therefore, be overestimated.
Interpretation of the estimates produced in the sponsor’s economic evaluation, including the incremental cost-effectiveness ratio (ICER) and price reduction, must consider the fact that the model likely does not fully capture the amount of uncertainty around the decision. In the probabilistic analysis, 36% of the estimated incremental quality-adjusted life-years (QALYs) associated with the addition of fostemsavir to OBT was less than 0. This figure may be best understood as the probability that the addition of fostemsavir to OBT will not produce any increase in effectiveness, and would, therefore, not be considered cost-effective at any WTP threshold.
Treatment with fostemsavir is expected to add $45,864 per year in drug costs. Based on information from the BRIGHTE trial, fostemsavir plus OBT reduces viral load, compared to placebo, by 0.625 copies/mL over 8 days, with other clinical effects being highly uncertain. The impact that this short-term reduction in viral load may have on patient health beyond the study period is unknown. As such, the clinical effectiveness and cost-effectiveness of fostemsavir plus OBT is unknown.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
CADTH received 1 patient group input submission from the Community-Based Research Centre, collected through an online survey of people living with HIV (n = 325) conducted in 2021. This information was supplemented by an additional venue-based survey that collected information from people living with HIV (n = 144) in 2022. Results from these surveys suggested that nearly all (> 99%) respondents were taking antiretroviral (ARV) therapies, and 94% had a suppressed viral load. The majority (72%) of patients responding to the surveys reported that they found it very easy to take their daily medication; however, it was noted that 26% of people without a suppressed viral load found it somewhat or very difficult to take daily medication. The surveys also suggested that only 19% of respondents would prefer a daily pill to an injectable medication taken every 2 months. The patient group input also highlighted that people in the indicated population (heavily treatment-experienced [HTE] with multidrug-resistant HIV) tend to face additional burdens related to the social determinants of health. Namely, they are more likely to face issues related to housing and food insecurity, which exacerbates problems related to treatment adherence. The patient input argued for consideration of nonmedical supports — such as social supports, income supplements, mental health support — to account for all the health needs facing this population. Suppression of viral load was identified as a goal of treatment, but several respondents to the 2022 survey noted that they experienced pressure and stigma related to having an undetectable viral load.
CADTH did not receive any input from clinician groups for this review.
Drug plan input did not raise any specific economic concerns. The input noted that fostemsavir is a first-in-class attachment inhibitor with a unique mechanism of action, and that it has no relevant comparators.
Although the sponsor’s economic model considered a societal perspective, it solely considered costs due to loss of productivity, which does not address the aspects of interest identified by patient groups (e.g., the relationship between treatment adherence and factors like route of administration, viral load, and socioeconomic barriers, and the role that nonmedical supports have on treatment adherence and patient well-being). As such, the economic analysis does not allow for the consideration of these aspects.
Economic Review
The current review is for fostemsavir for the treatment of HIV type 1 (HIV-1) infection in HTE adults with multidrug-resistant (MDR) HIV-1 infection for whom it is otherwise not possible to construct a suppressive antiviral regimen due to resistance, intolerance, or safety considerations.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis of fostemsavir in combination with optimized background therapy (OBT) compared with OBT alone. The model population comprised HTE adults with MDR HIV-1 infection and who cannot be treated with other ARV drugs due to resistance, intolerance, or safety considerations. The target population is aligned with the Health Canada–indicated population.
Fostemsavir is available as an extended-release tablet containing 600 mg of fostemsavir. The recommended dose of fostemsavir is 600 mg taken orally twice daily. At the submitted price of $62.77 per tablet, the annual cost of fostemsavir was estimated to be $45,822.47 per patient.
OBT was defined as a frequency-weighted basket of treatments intended to represent the typical mix of treatments available to patients in the indicated population treated in Canada. They included (as combinations and as single-drug therapies): abacavir, ||||||||||, ||||||||||||||||||||, dolutegravir, |||||||||||||, enfuvirtide, etravirine, ||||||||||, |||||||||, maraviroc, |||||||||||, |||||||||||, |||||||||, tenofovir disoproxil fumarate, ||||||||||, and ||||||||||. A full list of included therapies, as well as their frequency within the basket, is included in Table 11 in Appendix 3. The estimated annual cost of OBT was $26,499 per patient.
The sponsor also considered a subgroup of patients who have no approved treatment options available. In this subpopulation, fostemsavir plus salvage therapy (ST) was compared to ST alone. ST was defined as a frequency-weighted basket of treatments that differed in proportion to OBT. The therapies comprising ST are also described in Table 11 in Appendix 3. The estimated annual cost of ST was $29,341.
The sponsor’s economic evaluation considered QALYs and life-years over a time horizon of 53 years. The base-case and subgroup analyses were conducted from the perspective of the Canadian public health care system, with an annual discount rate of 1.5% applied to both costs and outcomes.
Model Structure
The sponsor submitted a Markov model containing a decision tree to simulate the clinical course of HIV disease progression. The model contained 5 states defined by CD4+count (< 50 cells/mm3, 50 to 199 cells/mm3, 200 to 349 cells/mm3, 350 to 500 cells/mm3, and > 500 cells/mm3). The model had a cycle length of 1 month. Patients begin the model in health states defined by viral load and CD4+level. Each cycle, the patients’ CD4+T-cell counts could improve, remain the same, or worsen, depending on their current CD4+T-cell count. In addition to CD4+T-cell count, health states were based on HIV-1 RNA viral loads (< 50 copies/mL or ≥ 50 copies/mL). Discontinuation of treatment for virologic or nonvirologic reasons was modelled separately to account for the differential impact of these reasons on the efficacy of subsequent lines of therapy. Discontinuation occurring while patients were in the low viral-load state were assumed to be nonvirologic discontinuations, whereas discontinuations in the high viral-load state were assumed to be virologic discontinuations. After discontinuation (either virologic or nonvirologic), patients could receive ST. Patients experiencing virologic discontinuation moved to the corresponding high-viral-load CD4-based health state.
While in any health state, patients could develop AIDS-defining events (ADE) (acute viral, bacterial, fungal, protozoan, or other opportunistic infection) based on their CD4+T-cell count and time on treatment. Treatment-associated adverse events were omitted from the model. Patients could move from any health state to the absorbing death state. A schematic of the model is presented in Figure 1 in Appendix 3.
Model Inputs
Patients were assumed to be 47 years old at the start of the model, and 23% were female, aligned with participants in the BRIGHTE trial.1,2 Patients begin the model in CD4+states based on the efficacy of the treatment they received (i.e., fostemsavir plus OBT or OBT alone). To estimate treatment efficacy in the base case, the sponsor performed a MAIC between patient populations in the BRIGHTE trial and the VIKING-3 trial. Transition between CD4+states was estimated with a cohort simulation process, in which the transition rate was simulated for patients using a multinomial distribution based on their CD4+value at different times of interest. The output of this simulation was an empirically derived cohort of patients with CD4+counts estimated over multiple time points. From these empirical cohorts, the sponsor estimated the transition matrix for patients receiving fostemsavir plus OBT from the MAIC, and for OBT alone from the VIKING-3 trial. Viral load was incorporated into the cohort simulation, informing the transitions between CD4+states. Consequently, CD4+transition probabilities in the model were assumed to be equal for patients with fewer than 50 copies/mL and at least 50 copies/mL.
The probability of patients discontinuing treatment was estimated from the sponsor’s MAIC (for fostemsavir plus OBT) and from the 48-week results from the VIKING-3 trial (for OBT alone). In the subgroup analysis, discontinuation probabilities were estimated from BRIGHTE patient data.1,2 Transition probabilities between CD4+states for patients receiving ST were derived from the OPTIMA trial, and were assumed to follow the natural history of HIV in patients not on treatment. The probability of developing an ADE was estimated from literature sources.3,4 The probability of mortality was derived from all-cause mortality life tables,5 by multiplying age-specific mortality rates by CD4-associated mortality risk ratios that were derived from the literature.6,7 AIDS-related mortality was estimated as the sum of CD4-associated mortality and AIDS-specific mortality, with the latter estimated from the Multicenter AIDS Cohort Study.3,8
Utility estimates were applied to each CD4+ state, based on the 36-Item Short Form Health Survey values published in the literature.9 Alternative utility estimates collected in the BRIGHTE trial using the 5-level EQ-5D questionnaire were available in the scenario analysis. Utility estimates were adjusted for age using Canadian gender-specific utility values.10 Utility decrements related to ADEs and end-of-life health status were applied in the appropriate states, and were derived from the literature.11,12
The daily cost of treatment with fostemsavir was estimated by multiplying the sponsor’s list price for a 600 mg tablet by the daily dose in the product monograph (two 600 mg tablets per day), which was then multiplied by a monthly cost to match the cycle length. The sponsor estimated the monthly cost of OBT with a similar process, based on the weighted average of treatments received by participants in the randomized BRIGHTE trial, adjusted for availability in Canada. The cost of ST was estimated using a similar treatment-weighting method, based on the mix of treatments used in the nonrandomized BRIGHTE trial. A summary of the treatment weights used to define OBT and ST are provided in Table 11 in Appendix 3. Costs associated with other health care system resource use (opportunistic infection prophylaxis; primary care visits; testing for CD4, HIV, and drug resistance; non-HIV medications; emergency department visits; and inpatient days) were estimated for each CD4+health state, with values derived from the literature and from the Ontario Drug Benefits Formulary.13,14 Costs for end-of-life care were estimated using values published by the Canadian Institute for Health Information.15 Costs associated with the management of ADEs were derived from the literature.16
Summary of Sponsor’s Economic Evaluation Results
The sponsor submitted a probabilistic analysis based on 2,500 model iterations. The sponsor also submitted deterministic analyses, where the results were notably different from the probabilistic analyses; estimated costs, life-years, and QALYs were higher for the fostemsavir arm in the probabilistic analysis than in the deterministic results.
Base-Case Results
In the sponsor’s base-case probabilistic analysis, treatment with fostemsavir plus OBT was associated with an ICER of $469,100 per QALY gained, compared to OBT alone. Of incremental QALYs 100% were gained beyond the BRIGHTE observation period of 8 days, and 97% (0.65 QALYs) were gained beyond the VIKING-3 trial period of 48 weeks, indicating that the vast majority of benefit was derived through extrapolation beyond the available clinical evidence. There was a 0% probability of fostemsavir being cost-effective at a WTP threshold of $50,000 per QALY gained. In addition, 36% of probabilistically sampled incremental QALYs were less than 0 and would not be considered cost-effective at any WTP threshold. A scatterplot of probabilistically sampled ICERs is presented in Figure 2 in Appendix 3.
The submitted analysis is based on the publicly available process of all treatments, including subsequent therapies.
Table 3: Summary of the Sponsor’s Probabilistic Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total LYs | Total QALYs | Incremental QALYs | ICER vs. OBT ($/QALY) |
|---|---|---|---|---|---|---|
Deterministic results | ||||||
Fostemsavir plus OBT | 716,745 | Reference | 8.52 | 6.39 | Reference | Reference |
OBT alone | 453,390 | 263,355 | 7.78 | 5.81 | 0.59 | 449,670 |
Probabilistic results | ||||||
Fostemsavir plus OBT | 791,486 | Reference | 9.35 | 7.02 | Reference | Reference |
OBT alone | 475,879 | 315,607 | 8.49 | 6.35 | 0.67 | 469,086 |
ICER = incremental cost-effectiveness ratio; LY = life-year; OBT = optimized background therapy; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.17
Sensitivity and Scenario Analyses Results
The key scenario analysis considered by the sponsor concerned a subgroup of HTE patients with no approved treatment options available and for whom a suppressive ARV regimen, therefore, cannot be constructed. In this population, because OBT cannot be used, fostemsavir was added to ST and was compared to ST alone. Comparative treatment efficacy was estimated from nonrandomized data from the BRIGHTE trial. Deterministic and probabilistic results are presented in Appendix 3.
The sponsor’s base-case and subgroup analyses were robust to changes in OBT and ST cost and utility values. Both analyses were highly sensitive to a shorter (2-year) time horizon, producing estimated probabilistic ICERs of $3,301,647 and $384,313 per QALY gained for the base case and subgroup, respectively. The sponsor also conducted a scenario analysis that considered lost productivity costs, and found probabilistic ICERs of $440,295 and $129,137 per QALY gained for the base case and subgroup, respectively.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis:
The comparative clinical effectiveness of fostemsavir is highly uncertain. The CADTH clinical review showed that the duration of the randomized comparative phase of the BRIGHTE trial (8 days) was too short to draw conclusions about the comparative efficacy of fostemsavir on CD4+counts, AIDS-related mortality, or progression to AIDS, compared to placebo. In the absence of long-term comparative clinical evidence from the pivotal trial, the sponsor’s method for modelling treatment efficacy in the economic model was derived primarily from a MAIC in which BRIGHTE trial participants were matched to VIKING-3 trial participants. The CADTH clinical review found that the results for clinically important efficacy outcomes (change in CD4+count, rate of virologic suppression) were inconclusive due to a high degree of uncertainty and the role of confounding factors, and that a high drop in sample size suggested poor overlap between the BRIGHTE and VIKING-3 trials. Given the central role that the BRIGHTE and VIKING-3 results play in the estimation of survival, the limitations in the trials and in the MAIC add considerable uncertainty to estimated QALYs for patients in the indicated population, irrespective of treatment.
CADTH was not able to address this limitation in its reanalysis.
The method for modelling of the natural history of CD4+progression lacked transparency and has uncertain validity. The sponsor estimated Markov transition probabilities between CD4+states using a statistical simulation approach. This approach allowed it to simulate the clinical trajectory of a population of patients over the course of 48 weeks. The resulting population was used to estimate monthly transition probabilities. This approach is not typical in pharmacoeconomic analyses, and was conducted external to the model file. The simulation was thus a nontransparent approach to estimating the primary driver of survival in both arms. In addition to the uncertainty contributed by the sponsor’s chosen method, the values of baseline CD4+and change in CD4+cell count that are used as inputs into the simulation are highly uncertain, with standard deviations that are comparable in size to the estimated mean CD4+values. Given the central role that changes in CD4+counts play in the estimation of survival, the cumulative uncertainty contributed by the nontransparent methodology and the high level of variance in the underlying data adds considerable uncertainty to the estimated QALYs for patients in the indicated population, irrespective of treatment.
CADTH was not able to address this limitation in its reanalysis.
The durability of long-term response is uncertain. In the sponsor’s base-case analysis, nearly 100% of incremental QALYs were estimated beyond the 48-week observation period of the VIKING-3 trial that informed the MAIC. The sponsor-submitted scenario analysis showed that the ICER for fostemsavir plus OBT compared to OBT alone was highly sensitive to the time horizon. Nearly all of the benefit of fostemsavir is estimated from extrapolation of uncertain evidence, through a process that lacks transparency. According to clinical expert input solicited by CADTH for this review, patients who achieve stable virologic response beyond 3 months are likely to maintain that response for as long as they continue treatment. However, it is important to recognize that there is no direct comparative evidence to support the long-term effectiveness of fostemsavir over OBT. The sponsor’s pharmacoeconomic model did not incorporate the possibility of treatment efficacy waning or the effect of changes in treatment adherence beyond 48 weeks. Given that the near entirety of incremental benefit is expected to occur after patients have been using fostemsavir (and accruing associated costs) for a year, the lack of long-term efficacy data adds considerable uncertainty to the estimated incremental QALYs associated with fostemsavir.
CADTH was not able to address this limitation in its reanalysis.
The relationship between CD4+count and viral load is uncertain. The sponsor’s calculated transition probabilities between CD4+states were assumed to be independent of viral load, such that patients with fewer than 50 HIV-1 RNA copies/mL had the same risk of moving between CD4-based health states as those with 50 copies/mL or more. The clinical expert consulted by CADTH for this review indicated that although this assumption of independence likely holds at the chosen categorical threshold, patients with higher viral load (above 500 to 1,000 copies/mL) are likely to see a more rapid decrease in their CD4+count compared to patients with a viral load below 500 copies/mL. The feedback also suggested that the 50-copy threshold may not be clinically meaningful to distinguish between patients with high and low viral loads. The clinical expert also suggested that it was unlikely that a CD4+count would be used to determine whether or not a patient would discontinue therapy, and that virologic response would be assessed every 6 months in stable patients. The disaggregated results of the sponsor’s analysis (refer to Table 9 in Appendix 3) show that the pharmacoeconomic model predicts that fewer patients receiving fostemsavir plus OBT will reach a CD4+count below 200 cells/mm3 than those receiving OBT alone; however, failure to consider the relationship between viral load and CD4+count suggests that there is additional uncertainty around the QALYs generated in these states that is not reflected in the results.
CADTH was not able to address this limitation in its reanalysis.
Parameter uncertainty around influential model values is poorly characterized. Several parameters in the model were assumed to have standard error values that were equal to 10% of the mean parameter estimate. These parameters included the risk of death relative to all-cause mortality among patients with CD4+counts below 200 cells/mm3; morality and disutility associated with AIDS; the probability of developing an ADE; and the costs associated with disease management, ADEs, and death. No justification was provided for this approach. It is implausible that all of these values would have an identical mathematical relationship between the mean and standard error. In fact, CADTH notes that for the relative risk of death by CD4+count, the parameter uncertainty for the higher CD4+count states (200 to < 350 cells/mm3, 350 to < 500 cells/mm3, ≥ 500 cells/mm3) was closer to 20%. The sponsor’s submitted analysis does not accurately reflect the uncertainty around parameters, and appears to underestimate it for at least 1 pair of highly influential survival parameters. This methodological limitation has unknown influence on the ICER. The sponsor’s base-case analysis estimated that 36% of estimated incremental QALY values were less than 0. If parameter uncertainty is underestimated, that proportion may be higher.
CADTH was not able to address this limitation in its reanalysis.
Parameter uncertainty in the model appears to introduce asymmetric bias in results. There is a wide discrepancy between the deterministic and probabilistic results produced by the sponsor’s pharmacoeconomic model. Compared to the deterministic results, fostemsavir plus OBT is $74,741 more costly and produces an additional 0.63 QALYs in the probabilistic analysis. Similarly, the probabilistic results of OBT alone were $22,489 more costly and were associated with 0.539 more QALYs than estimated in the deterministic results. These results suggest the presence of high levels of variance in 1 or more parameters that are distributed asymmetrically through the model. This asymmetric variance creates a directional survival bias that favours fostemsavir. The lack of transparency in the model’s natural history function and the use of arbitrary parameter uncertainty values precluded CADTH from identifying the source of this bias. CADTH notes, however, that this apparent asymmetry does not have a meaningful impact on the estimated ICER (deterministic = $449,670 per QALY gained; probabilistic = $469,086 per QALY gained).
CADTH was not able to address this limitation in its reanalysis
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
Health state utility values captured in the BRIGHTE trial were excluded in favour of values published in the literature.9 | Reasonable. The BRIGHTE utility values were evaluated in a scenario analysis and had minimal impact on the ICER. |
Patient virologic suppression status was assumed to remain constant beyond 48 weeks. | Reasonable. Clinical expert input suggested that if a patient is stable at 48 weeks, their virologic status should remain stable as long as they continue therapy. |
The discontinuation rate is assumed to be the average rate in the BRIGHTE MAIC, and equal for both the fostemsavir plus OBT and OBT arms in the model. | Reasonable. The clinical data did not suggest a reason that patients would disproportionately discontinue fostemsavir. |
Adverse events were assumed to be equal in both arms. | Acceptable. The sponsor asserted that AEs would be AIDS-related, and that patients who had achieved a stable response would not experience any non-AIDS-related AEs. Consequently, the sponsor asserted that AEs were captured in the AIDS-related disutilities used in the model. The clinical data did not suggest the existence of fostemsavir-related AEs, although firm conclusions could not be drawn from the submitted MAICs. |
AE = adverse event; ICER = incremental cost-effectiveness ratio; MAIC = matching-adjusted indirect comparison; OBT = optimized background therapy.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
Given the limitations CADTH identified in the sponsor’s economic submission, CADTH was unable to use the model to derive more reliable estimates of the cost-effectiveness of fostemsavir or to help quantify the impact of uncertainty.
When reviewing the sponsor’s base-case results, the probability that fostemsavir plus OBT is cost-effective at a WTP threshold of $50,000 per QALY gained was 0%. As such, even when uncertainty cannot be effectively included in the model, it is highly unlikely, based on the sponsor’s analysis, that fostemsavir would be cost-effective at this threshold. In the probabilistic analysis, 36% of sampled ICERs had incremental QALY values lower than 0, suggesting a probability that fostemsavir would not be cost-effective at any WTP threshold.
Scenario Analysis Results
CADTH undertook price reduction analyses based on the sponsor’s base case. These analyses demonstrated that a 94% reduction in the price of fostemsavir would be required for fostemsavir plus OBT to be considered cost-effective at a WTP threshold of $50,000 per QALY gained compared to OBT alone.
Table 5: CADTH Price Reduction Analyses (Probabilistic and Deterministic)
Analysis | ICERs for fostemsavir plus OBT vs. OBT alone ($/QALY gained) | |
|---|---|---|
Price reduction | Probabilistic | Deterministic |
No price reduction | 469,086 | 449,670 |
10% | 430,958 | 407,330 |
20% | 386,574 | 364,989 |
30% | 342,067 | 322,649 |
40% | 297,470 | 280,308 |
50% | 253,056 | 236,946 |
60% | 208,581 | 194,203 |
70% | 164,106 | 151,459 |
80% | 119,630 | 108,716 |
90% | 75,155 | 65,973 |
100% | 28,460 | 23,230 |
ICER = incremental cost-effectiveness ratio; OBT = optimized background therapy; QALY = quality-adjusted life-year.
Note: Probabilistic price reduction analyses were run with 2,500 probabilistic iterations.
The results of the sponsor’s model were robust to changes in the costs of OBT and ST, the use of alternative utility values, and the time point beyond which CD4+counts were allowed to change. To explore the impact of model parameters with arbitrarily chosen standard errors (risk of death relative to all-cause mortality among patients with CD4+counts < 200 cells/mm3; morality and disutility associated with AIDS; the probability of developing an ADE; and costs associated with disease management, ADEs, and death), CADTH performed a scenario analysis in which these standard errors were assumed to be 20% of the mean.
In scenario analysis of the subgroup identified in the nonrandomized BRIGHTE population (patients who have no approved treatment options available), a 90% reduction in the price of fostemsavir would be required for fostemsavir plus ST to be cost-effective, compared to ST alone, at a WTP threshold of $50,000 per QALY gained. CADTH notes that this estimate is subject to additional uncertainty beyond the limitations previously identified, owing to the nonrandomized nature of the patients in this population. Due to the presence of multiple forms of structural and parametric uncertainty, additional price reduction may be warranted.
Issues for Consideration
Input from the clinical expert consulted for the review was aligned with patient input that identified the socioeconomic vulnerability that disproportionately affects HTE patients, compared to other patients with HIV. These socioeconomic barriers (poverty, housing concerns, other structural barriers) affect treatment adherence and access to care. The clinical expert also indicated that new oral therapies are needed to improve virologic response, clinical outcomes, and adherence to treatment. Should fostemsavir be approved in this population, it is likely that there will be a desire to use it in a broader population, for which clinical and economic information is not available.
The sponsor submitted a scenario described as a societal perspective. In this analysis, the sponsor considered costs generated due to productivity loss. Productivity loss was estimated using average age- and sex-specific monthly wages and labour force participation. Patient input and clinical expert input note that patients eligible for fostemsavir are disproportionately likely to face financial barriers and have unstable housing and food security. Given the specific needs of this population, the human capital approach chosen by the sponsor likely does not represent the societal perspective, and does not include the most relevant patient-important outcomes for this population.
The analysis did not consider the method of delivery of the treatment (i.e., oral versus injectable medication, multiple drug regimens versus a single-drug regimen). Based on patient input, only 19% preferred a daily pill over an injectable. The clinical expert consulted by CADTH suggested that patients who are HTE may have issues with adherence to previously prescribed regimens. This difficulty could possibly be due to the complexity and pill burden that accompanies multidrug regimens or obstacles related to accessing injectable treatments. The impact of this on fostemsavir is unknown.
The economic analysis results rely on an assumption that adverse events will occur with equal frequency and severity if patients receive OBT with or without the addition of fostemsavir. Fostemsavir employs a novel mechanism of action. Accordingly, the long-term safety of fostemsavir alone and in combination with OBT is unknown beyond the 240-week period of the BRIGHTE trial.
Overall Conclusions
The CADTH clinical review of the BRIGHTE trial showed that fostemsavir reduced viral load compared to placebo during the randomized period of the trial. Conclusions about the impact of fostemsavir on CD4+count, AIDS-related mortality, or progression to AIDS could not be drawn during this period, however. CADTH’s appraisal of the sponsor’s MAIC in which the BRIGHTE trial population was matched with the VIKING-3 trial population showed wide uncertainty around the efficacy of fostemsavir plus OBT, compared to OBT alone, such that definitive conclusions could not be made. The effectiveness of fostemsavir plus OBT, compared to OBT alone, is highly uncertain, both in the short-term and over the course of 48 weeks.
CADTH identified additional limitations in the economic analysis: a nontransparent method for modelling the natural history of patients with HIV, the uncertain long-term durability of fostemsavir response, an uncertain relationship between viral load and CD4+count, an inappropriate incorporation of parameter uncertainty, and a modelling approach that produces a notable discrepancy between deterministic and probabilistic results. None of these major limitations could be addressed due to lack of model flexibility and availability of information, and CADTH, therefore, did not conduct a reanalysis.
Using the sponsor’s base case, fostemsavir plus OBT was $315,607 more costly and produced 0.67 more QALYs than OBT alone, resulting in an ICER of $469,086 per QALY gained. A 94% reduction in the price of fostemsavir would be required for fostemsavir plus OBT to be considered cost-effective compared to OBT alone at a WTP threshold of $50,000 per QALY gained. Given the limitations of the MAIC, the short duration of the trial, and the methodological concerns identified in the economic model, these estimates remain highly uncertain.
The cost-effectiveness results are driven by the estimated changes in CD4+count. The sponsor’s economic model used a nontransparent method to simulate the way that CD4+count may change over time. This simulation was based on 8 days of trial data and a MAIC that was designed to extend that data over the course of 48 weeks, and then over the lifetime of a cohort of patients. The inputs into the simulation process were themselves highly uncertain, with standard errors nearly as large as mean estimates (or larger in some cases). The output of these overlapping forms of uncertainty was a range of probabilistic estimates that varied from a gain of more than 8 QALYs to a loss of nearly 6 QALYs. In the probabilistic analysis, 36% of the estimated incremental QALYs associated with the addition of fostemsavir to OBT were less than 0. There was no evidence in the BRIGHTE trial or the sponsor’s submitted MAIC that suggested that patients would have worse outcomes with fostemsavir than if they received only OBT, and the model did not consider adverse events for fostemsavir that would potentially produce increased morbidity or mortality. The 36% figure may be best understood as the probability that the addition of fostemsavir to OBT will not produce any increase in effectiveness. Another way to interpret the value is that there is a 36% chance that fostemsavir will not be cost-effective at any WTP threshold.
Interpretation of the estimates produced in the economic evaluation, including the ICER and price reduction, must consider the fact that the model likely does not fully capture the amount of uncertainty around the decision. The various forms of structural, methodological, and parametric uncertainty identified by CADTH have an unknown effect on both the estimated mean ICER and the amount of statistical uncertainty around that mean estimate, which is already wide. Given that the sponsor’s base case estimated a 36% chance that fostemsavir would not be cost-effective at any WTP, the price reduction analysis performed using that base case is likely an underestimate.
References
1.Data on File: REF-152618. BRIGHTE IPD. 2019. Non-randomized cohort initial optimized background therapy regimen agents (N=99) – frequency and percentage of patients by agent. In: Drug Reimbursement Review sponsor submission: Rukobia (fostemsavir), 600 mg extended-release tablets [internal sponsor's package]. Montreal (QC): ViiV Healthcare ULC; 2022 Oct 21.
2.Data on File; REF-152539. BRIGHTE IPD. 2019. Randomized cohort initial optimized background therapy regimen agents (N=267) – frequency and percentage of patients by agent. In: Drug Reimbursement Review sponsor submission: Rukobia (fostemsavir), 600 mg extended-release tablets [internal sponsor's package]. Montreal (QC): ViiV Healthcare ULC; 2022 Oct 21.
3.OPTUMInsight. Cost-effectiveness model for dolutegravir (ARAMIS-DTG) in treatment-experienced HIV patients: report version 3.0. [2013]. In: Drug Reimbursement Review sponsor submission: Rukobia (fostemsavir), 600 mg extended-release tablets [internal sponsor's package]. Montreal (QC): ViiV Healthcare ULC; 2022 Oct 21.
4.d'Arminio Monforte A, Sabin CA, Phillips A, et al. The changing incidence of AIDS events in patients receiving highly active antiretroviral therapy. Arch Intern Med. 2005;165(4):416-423. PubMed
5.Statistics Canada. Table 13-10-0114-01: Life expectancy and other elements of the life table, Canada, all provinces except Prince Edward Island. 2018-2020. 2022; https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011401. Accessed 2022 Feb 9.
6.Lewden C, Chêne G, Morlat P, et al. HIV-infected adults with a CD4+cell count greater than 500 cells/mm3 on long-term combination antiretroviral therapy reach same mortality rates as the general population. J Acquir Immune Defic Syndr. 2007;46(1):72-77. PubMed
7.Ledergerber B, Lundgren JD, Walker AS, et al. Predictors of trend in CD4-positive T-cell count and mortality among HIV-1-infected individuals with virological failure to all three antiretroviral-drug classes. Lancet. 2004;364(9428):51-62. PubMed
8.Rydzak CE, Cotich KL, Sax PE, et al. Assessing the performance of a computer-based policy model of HIV and AIDS. PLoS One. 2010;5(9):e12647. PubMed
9.Kauf TL, Roskell N, Shearer A, et al. A predictive model of health state utilities for HIV patients in the modern era of highly active antiretroviral therapy. Value Health. 2008;11(7):1144-1153. PubMed
10.Guertin JR, Feeny D, Tarride JE. Age- and sex-specific Canadian utility norms, based on the 2013-2014 Canadian Community Health Survey. CMAJ. 2018;190(6):E155-E161. PubMed
11.Paltiel AD, Scharfstein JA, Seage GR, 3rd, et al. A Monte Carlo simulation of advanced HIV disease: application to prevention of CMV infection. Med Decis Making. 1998;18(2 Suppl):S93-105. PubMed
12.Anis AH, Nosyk B, Sun H, et al. Quality of life of patients with advanced HIV/AIDS: measuring the impact of both AIDS-defining events and non-AIDS serious adverse events. J Acquir Immune Defic Syndr. 2009;51(5):631-639. PubMed
13.Mauskopf J, Brogan AJ, Talbird SE, Martin S. Cost-effectiveness of combination therapy with etravirine in treatment-experienced adults with HIV-1 infection. AIDS. 2012;26(3):355-364. PubMed
14.Ontario Ministry of Health and Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2022; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2022 Nov 14.
15.Canadian Institute for Health Information. Health care use at the end of life in Saskatchewan. Ottawa (ON): CIHI; 2008: https://secure.cihi.ca/free_products/EOL_Report_Saskatchewan.pdf. Accessed 2023 Jan 19.
16.Anis AH, Guh D, Hogg RS, et al. The cost effectiveness of antiretroviral regimens for the treatment of HIV/AIDS. Pharmacoeconomics. 2000;18(4):393-404. PubMed
17.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Rukobia (fostemsavir), 600 mg extended-release tablets. Montreal (QC): ViiV Healthcare ULC; 2022 Oct 21.
18.Rukobia (fostemsavir): 600 mg extended release tablets [product monograph]. Laval (QC): ViiV Healthcare ULC; 2021 Oct 1.
19.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Rukobia (fostemsavir), extended release 600 mg tablet. Montreal (QC): ViiV Healthcare ULC.; 2022 Sept 19.
20.DeltaPA [database on the Internet]. Ottawa (ON): IQVIA; 2022: https://www.iqvia.com/. Accessed 2022 Nov 14.
21.Haddad N, Robert A, Weeks A, Popovic N, Siu W, Archibald C. HIV in Canada: surveillance report, 2018. Can Commun Dis Rep. 2019;45(12):304-312. PubMed
22.Public Health Agency of Canada. Estimates of HIV incidence, prevalence and Canada’s progress on meeting the 90-90-90 HIV targets, 2018. Ottawa (ON): PHAC; 2020: https://www.canada.ca/content/dam/hc-sc/documents/services/publications/diseases-conditions/summary-estimates-hiv-incidence-prevalence-canadas-progress-90-90-90/national-hiv-estimates-report-2018-en.pdf. Accessed 2021 Nov 1.
23.IQVIA. Pharmastat data [Data on file]. 2021; https://pharmastat.iqvia.com/. Accessed 2023 Jan 19.
Appendix 1: Cost Comparison Table
Note this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts and drug plan. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 6: CADTH Cost Comparison for Adults With HIV-1 Infection Who Are HTE and Are Unable to Construct a Suppressive ARV Regimen Due to MDR
Treatment | Strength | Form | Price ($) | Recommended Dosage | Daily Cost ($) | Annual Drug Cost ($) |
|---|---|---|---|---|---|---|
Fostemsavir (Rukobia) | 600 mg | Tablet | 62.7705a | 600 mg twice daily | 125.54 | 45,854 |
ARV = antiretroviral; HTE = heavily treatment-experienced; MDR = multidrug resistance; OBT = optimized background therapy.
Note: All prices are from the Ontario Drug Benefit Formulary14 (accessed December 2022), unless otherwise indicated, and do not include dispensing fees. Recommended dosages from the respective product monographs.18
aSponsor submitted price.19
Table 7: Cost Comparison of Antiretrovirals for Adults With HIV-1 Infection Who Are HTE With MDR
Treatment | Strength | Form | Price ($) | Recommended Dosage | Daily Cost ($) | Annual Drug Cost ($) |
|---|---|---|---|---|---|---|
NRTIs | ||||||
|||||||| ||||||||| | ||| || | |||||| | |||||| | ||| || |||| || |||||||| | |||| | ||||| |
|||||||||| ||||||||| | ||| || | |||||| | |||||| | ||| || ||||| | |||| | ||||| |
Tenofovir (Generic) | 300 mg | Tablet | 4.8884 | 300 mg daily | 4.89 | 1,784 |
|||||||||| ||||||||| | ||| || | |||||| | |||||| | ||| || ||||| | |||| | ||||| |
2 NRTIs | ||||||
||||||||| |||||||||| ||||||||| | ||| || | ||| || | |||||| | |||||| | ||| |||||| ||||| | |||| | ||||| |
||||||||||||| | ||||||||| |||||||||| ||||| | ||| || | ||| || | |||||| | |||||| | ||| |||||| ||||| | |||| | ||||| |
|||||||||| | |||||||||| ||||||||| | ||| || | ||| || | |||||| | |||||| | ||| |||||| ||||| ||||| | |||| | ||||| |
NNRTIs | ||||||
Efavirenz (Generic) | 600 mg | Tablet | 3.8030 | 600 mg daily | 3.80 | 1,389 |
Etravirine (Intelence) | 100 mg 200 mg | Tablet | 6.5710 12.6195 | 200 mg daily | 12.62 | 4,609 |
Doravirine (Pifeltro) | 100 mg | Tablet | 16.6500 | 100 mg once daily | 16.65 | 6,081 |
Nevirapine (Generic) | 200 mg | Tablet | 1.2346 | 200 mg twice daily | 2.47 | 902 |
Rilpivirine (Edurant) | 25 mg | Tablet | 16.2870 | 25 mg once daily | 16.29 | 5,949 |
PIs | ||||||
|||||||||| ||||||||| |||| ||||||||| ||||||| | ||| |||| |||||| |||||| || | |||||| | |||||||||||||||||||| | ||| || || ||||||||| ||||| ||||| |||| ||| || ||||||||| |||| |||| | ||||| | ||||| |
Tipranavir (Aptivus) | 250 mg | Capsule | 8.7203 | 500 mg twice daily | 34.88 | 12,740 |
Darunavir (Generic) with ritonavir (Generic) | 600 mg 800 mg 100 mg | Tablet | 4.2970 5.8295 1.1745 | 600 mg darunavir twice daily with 100 mg of ritonavir once daily 800 mg darunavir with 100 mg of ritonavir once daily | 7.00 | 2,558 |
|||||||||||||||||||| ||||||||||| | ||| || | ||| || | |||||| | ||||||| | ||| || ||||||||||| ||| || || ||||||| |||| ||||| | ||||| | ||||| |
Fosamprenavir (Telzir) with ritonavir (Generic) | 700 mg 50 mg/mL 100 mg | Tablet Oral suspension Tablet | 9.4389 0.6535 1.1745 | 700 mg fosamprenavir and 100 mg ritonavir twice daily | 21.23 | 7,753 |
||||||||||||||||||| ||||||||| | ||| || | || |||||| || | || ||||||||| ||||| | |||||||||||||| |||||||| | ||||||||||||||||||| | ||||||| |||||| ||||| || ||| |||||||||||| |||| |||||| |||| || ||||||||||||| ||||||| | ||||| | ||||| |
INSTIs | ||||||
Dolutegravir (Tivicay) | 50 mg | Tablet | 20.8317 | Treatment-experienced, INSTI-naive: once daily Treatment-experienced, INSTI-resistant: twice daily | 20.8317 41.6634 | 7,609 15,218 |
||||||||||| ||||||||||| | ||| || | |||||| | ||||||| | ||| || ||||| ||||| | ||||| | |||||| |
FIs | ||||||
Enfuvirtide (Fuzeon) | 108 mg/vial | Single-use Vial | 42.323020 | 90 mg twice daily injected SC | 84.65 | 15,459 |
CCR5 | ||||||
Maraviroc (Celsentri) | 150 mg 300 mg | Tablet | 19.1525 | 300 mg twice daily | 38.31 | 13,991 |
INSTI + 2 NRTIs | ||||||
||||||||||||||||||||| ||||||||||| ||||||||| | |||| | ||| || | ||| || | |||||| | ||||||| | ||| |||||| ||||| | ||||| | |||||| |
|||||||||||||||||| |||||||||||||||||||||| ||||||||||||||||| |||||||||| | ||| ||| || | ||| || ||| || | ||| | ||||||| | ||| |||||| ||||| | ||||| | |||||| |
||||||||||||||||||||| ||||||||| ||||||||||| |||||||||| | || || | ||| || | || || | ||| | ||||||| | ||| |||||| ||||| | ||||| | |||||| |
||||||||||||||||||||| ||||||||||||||||||||| ||||||||| ||||||||| | ||| ||| || | ||| || | || || | ||| | ||||||| | ||| |||||| ||||| | ||||| | |||||| |
NNRTI + 2 NRTIs | ||||||
Efavirenz/tenofovir /fosamprenavir (Generic) | 600 mg / 300 mg / 200 mg | Tablet | 11.3300 | One tablet daily | 11.33 | 4,138 |
|||||||||||||||||||||||||| ||||||||| |||||||||| |||||||| |||||||||| | |||||||||| || | ||| | ||||||| | ||| |||||| ||||| | ||||| | |||||| |
Regimen Costsa | ||||||
Optimized Background Therapy | 63.36 | 25,333a | ||||
Salvage Therapy | 74.20 | 27,101a | ||||
CCR5 = chemokine receptor antagonists; FI = fusion inhibitors; INSTI = integrase strand transfer inhibitors; NNRTI = nonnucleoside reverse transcriptase inhibitors; NRTI = nucleotide reverse transcriptase inhibitors; PI = protease inhibitors.
Note: All prices are from the Ontario Drug Benefit Formulary (accessed December 2022),14 unless otherwise indicated, and do not include dispensing fees. Recommended dosages from the respective product monographs.
aCosts derived using drug costs from Table 7 and prescription frequencies from sponsor submission highlighted in Table 11.
Appendix 2: Submission Quality
Note this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | Yes | No comment |
Model has been adequately programmed and has sufficient face validity | No | Model results are highly reliant on externally-calculated values whose uncertainty cannot be directly adjusted for within the model file. |
Model structure is adequate for decision problem | No | Model fails to directly reflect treatment effectiveness differences due to treatment adherence or viral load. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | Model’s probabilistic results are asymmetric, producing marked difference between probabilistic and deterministic analyses. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | Sponsor used arbitrary values to characterize statistical uncertainty around multiple parameters. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | The model and the technical report did not present discounted disaggregated LYs by state |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note this appendix has not been copy-edited.
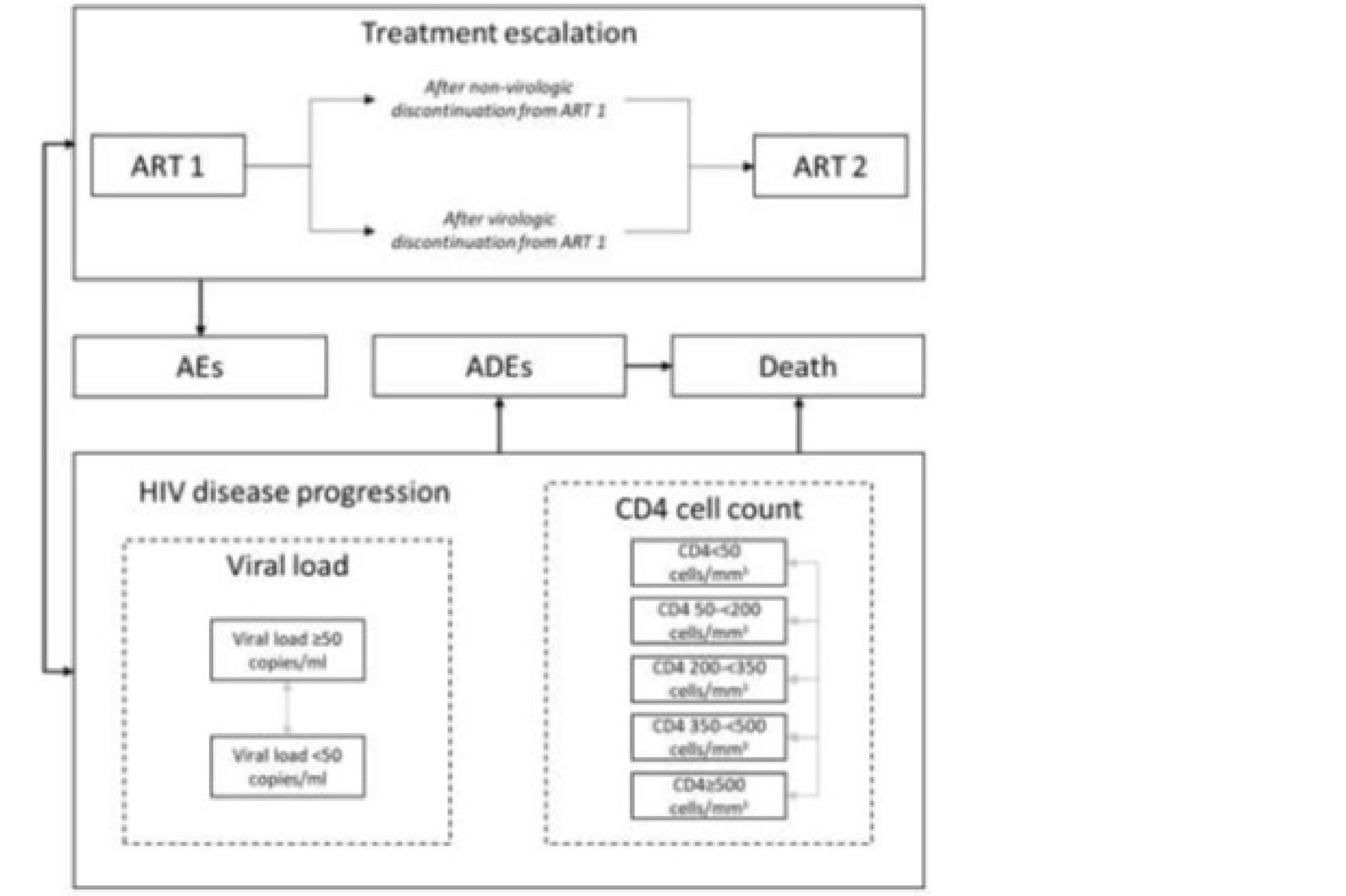
ART = antiretroviral; HIV = HIV; ADE = AIDS-defining event.
Source: Sponsor’s pharmacoeconomic submission.17
Figure 2: Probabilistic Results on the Cost-Effectiveness Plane
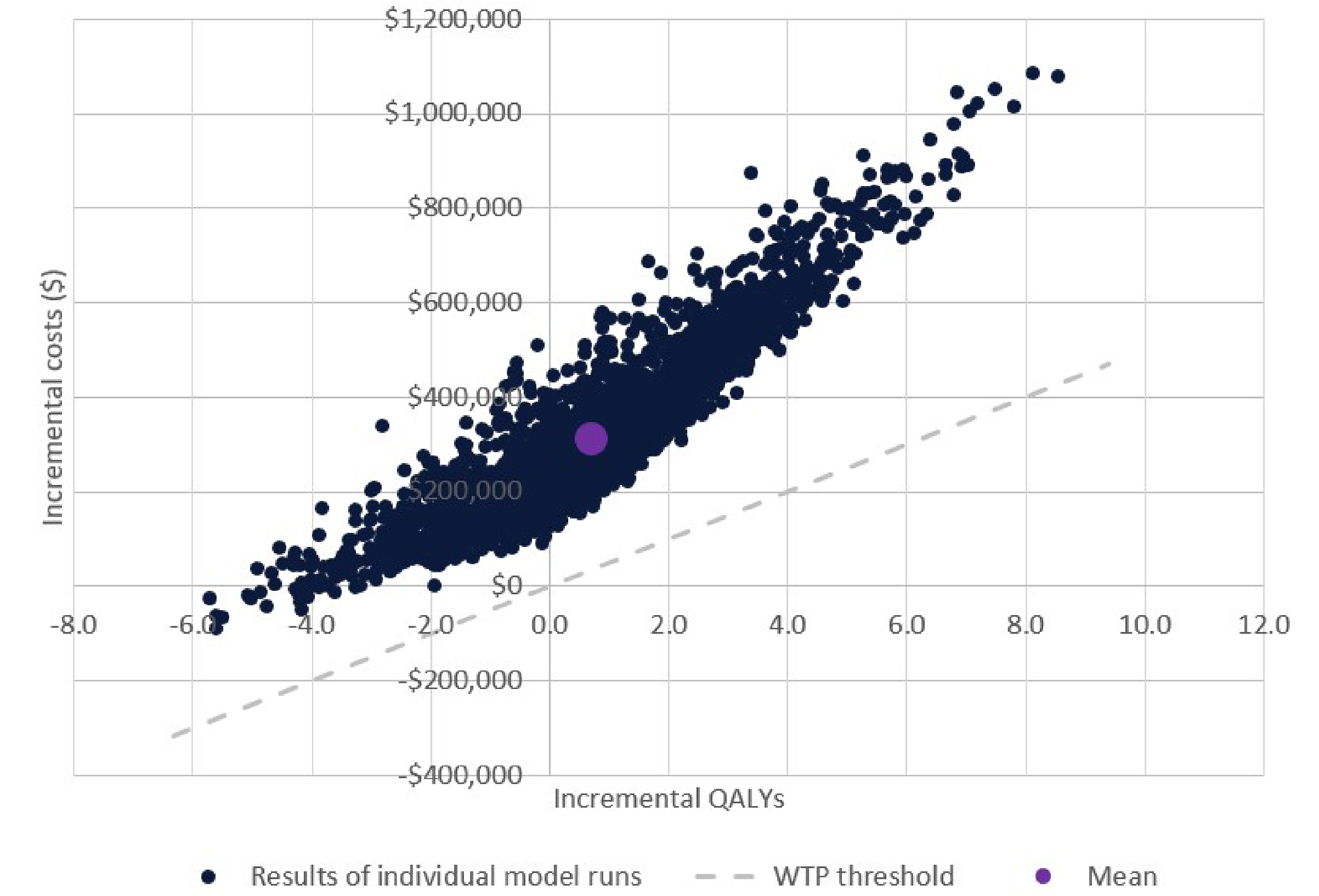
QALY = quality-adjusted life-year; WTP = willingness to pay.
Source: Sponsor’s pharmacoeconomic submission.17
Detailed Results of the Sponsor’s Base Case
Table 9: Disaggregated Summary of Sponsor’s Probabilistic Economic Evaluation Results
Parameter | Fostemsavir plus OBT | OBT Alone | Incremental | |
|---|---|---|---|---|
Discounted LYs | ||||
Total | 8.315 | 3.979 | 4.335 | |
Discounted QALYs | ||||
Total | 6.165 | 2.864 | 3.301 | |
By health state | ||||
CD4+ < 50 | 1.087 | 1.934 | −0.848 | |
CD4 + 50- < 200 | 1.739 | 0.847 | 0.892 | |
CD4 + 200- < 350 | 1.909 | 0.166 | 1.743 | |
CD4 + 350- < 500 | 1.233 | 0.019 | 1.214 | |
CD4+ ≥ 500 | 0.401 | 0.004 | 0.397 | |
QALY decrementsa | −0.202 | −0.105 | −0.099 | |
Discounted costs ($) | ||||
Total | 791,486 | 475,879 | 315,607 | |
Treatment | ||||
ART 1 | 467,930 | 143,592 | 324,338 | |
ART 2 | 84,479 | 90,224 | −5,745 | |
By health state | ||||
CD4+ < 50 | 99,897 | 112,160 | −12,263 | |
CD4 + 50- < 200 | 44,986 | 48,310 | −3,324 | |
CD4 + 200- < 350 | 28,738 | 26,714 | 2,024 | |
CD4 + 350- < 500 | 20,936 | 17.020 | 20,919 | |
CD4+ ≥ 500 | 23,008 | 15.910 | 22,992 | |
AIDS-defining events | 1,740 | 1,885 | −145 | |
Mortality | 19,773 | 20,063 | −290 | |
ICER ($/QALY) | 449,670 | |||
OBT = optimized background therapy; QALY = quality-adjusted life-year; LY = life-years; AIDS = AIDS; ICER = incremental cost-effectiveness ratio.
aQALY decrements include disutility associated with age, AIDS-defining events (opportunistic infection) and end-of-life events.
Table 10: Summary of the Sponsor’s Subgroup Scenario Analysis Results
Drug | Total costs ($) | Incremental costs ($) | Total LYs | Total QALYs | Incremental QALYs | ICER vs. OBT ($/QALY) |
|---|---|---|---|---|---|---|
Deterministic results | ||||||
Fostemsavir plus ST | 691,945 | Ref. | 7.48 | 5.53 | Ref. | Ref. |
ST alone | 338,720 | 353,225 | 3.94 | 2.84 | 2.69 | 129,814 |
Probabilistic results | ||||||
Fostemsavir plus ST | 773,678 | Ref. | 8.32 | 6.17 | Ref. | Ref. |
ST alone | 341,475 | 432,203 | 3.98 | 2.86 | 3.30 | 131,939 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; Ref. = reference; vs. = versus; ST = salvage therapy.
Source: Sponsor’s pharmacoeconomic submission.17
Table 11: Proportions Used to Define Antiretroviral Therapy
Treatment | Strength | Adjusteda proportion: OBT (%) | Adjusted proportion: ST (%) |
|---|---|---|---|
Nucleotide reverse transcriptase inhibitors (NRTIs) | |||
|||||||| ||||||||| | ||| || | |||| | |||| |
|||||||||| ||||||||| | ||| || | ||||| | |||| |
Tenofovir (Generic) | 300 mg | ||||| | ||||| |
|||||||||| ||||||||| | ||| || | |||| | |||| |
2 NRTIs | |||
||||||||| |||||||||| ||||||||| | ||| || | ||| || | |||| | |||| |
|||||||||||||| ||||||||| |||||||||| ||||||||| | ||| || | ||| || | |||| | |||| |
|||||||||| | |||||||||| ||||||||| | ||| || | ||| || | |||| | |||| |
Nonnucleoside reverse transcriptase inhibitors (NNRTIs) | |||
Efavirenz (Generic) | 600 mg | |||| | |||| |
Etravirine (Intelence) | 100 mg 200 mg | ||||| | ||||| |
Doravirine (Pifeltro) | 100 mg | |||| | |||| |
Nevirapine (Generic) | 200 mg | |||| | |||| |
Rilpivirine (Edurant) | 25 mg | |||| | |||| |
Protease Inhibitors (PIs) | |||
|||||||||| ||||||||| |||| ||||||||| ||||||||| | ||| |||||| |||||| |||||| || | |||| | |||| |
Tipranavir (Aptivus) | 250 mg | |||| | |||| |
Darunavir (Generic) with ritonavir (Generic) | 600 mg 800 mg 100 mg | ||||| | ||||| |
|||||||||||||||||||| ||||||||||| | ||| || | ||| || | |||| | |||| |
Fosamprenavir (Telzir) with ritonavir (Generic) | 700 mg 50 mg/mL 100 mg | |||| | |||| |
||||||||||||||||||| ||||||||| | ||| || | || |||||| || | || |||||||| ||||| | |||| | |||| |
Integrase strand transfer inhibitors (INSTIs) | |||
Dolutegravir (Tivicay) | 50 mg (BID) 50 mg (QD) | ||||||||||| | |||||||||| |
||||||||||| ||||||||||| | ||| || | |||| | |||| |
Fusion inhibitors (FIs) | |||
Enfuvirtide (Fuzeon) | 108 mg/vial | ||||| | ||||| |
Chemokine receptor antagonists (CCR5) | |||
Maraviroc (Celsentri) | 150 mg 300 mg | ||||| | |||| |
INSTI + 2 NRTIs | |||
||||||||||||||||||||| ||||||||||| ||||||||| | |||| | ||| || | ||| || | |||| | |||| |
|||||||||||||||||||||||| ||||||||||||||||||||||| |||||||||| |||||||| |||||||||| | ||| || | ||| || | ||| || | ||| || | |||| | |||| |
|||||||||||||||||||||||||| ||||||||| ||||||||||| |||||||||| | || || | ||| || | || || | |||| | |||| |
|||||||||||||||||||||||| ||||||||||||||||||||||| ||||||||||| ||||||||| | ||| || | ||| || | ||| || | || || | |||| | |||| |
NNRTI + 2 NRTIs | |||
Efavirenz/tenofovir /fosamprenavir (Generic) | 600 mg / 300 mg / 200 mg | |||| | |||| |
|||||||||||||||||||||||||| ||||||||| |||||||||| |||||||| |||||||||| | ||| || | || || | ||| || | |||| | |||| |
BID = twice daily; CCR5 = chemokine receptor antagonists; FI = fusion inhibitors; INSTI = integrase strand transfer inhibitors; NNRTI = nonnucleoside reverse transcriptase inhibitors; NRTI = nucleotide reverse transcriptase inhibitors; PI = protease inhibitors; QD = once daily.
aProportions were adjusted from their frequency in the BRIGHTE trial to match expected proportion used in Canadian practice.
Note: proportions do not add to 100%, as patients may receive multiple treatment regimens over their lifetime.
Appendix 4: Submitted BIA and CADTH Appraisal
Note this appendix has not been copy-edited.
Table 12: Summary of Key Takeaways
Key Takeaways of the BIA |
|---|
|
Summary of Sponsor’s BIA
The submitted budget impact analysis (BIA) assessed expected budgetary impact resulting from reimbursing fostemsavir to be used in combination with other antiretroviral (ARV) agents for the treatment of HIV-1 infection in heavily treatment-experienced (HTE) adults with multidrug-resistant (MDR) HIV-1 infection.19 In this population, it is otherwise not possible to construct a suppressive ARV regimen due to resistance, intolerance, or safety considerations. The BIA was conducted from the perspective of the Canadian public drug plans over a 3-year time horizon using an epidemiologic approach. The sponsor’s pan-Canadian estimates reflect the aggregated results from provincial budgets (excluding Quebec) and the Non-Insured Health Benefits (NIHB) program. The analysis was performed using jurisdiction-specific values by summing up individual provincial results to obtain consolidated results. Key inputs to the BIA are documented in Table 15.
The following key assumptions were made by the sponsor:
The sponsor did not include background therapy cost in the base-case analysis.
In the year that patients initiate treatment with fostemsavir, only a half-year cost is applied. The sponsor assumed that, on average, patients will start treatment halfway through a year.
The sponsor assumed 0.44% of the prevalent HIV-1 population will be HTE with MDR and unable to construct a suppressive antiviral regimen due to resistance, intolerance, or safety.
The sponsor assumed fostemsavir would capture 100% of the market if reimbursed.
Table 13: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as Year 1 / Year 2 / Year 3 if appropriate) |
|---|---|
Target population | |
Number of adults with HIV-1 | 53,347 / 53,995 / 54,64221 |
Proportion receiving ARV treatment | 85% / 85% / 85%22 |
Number of patients reimbursed through public drug plans | |||||| | |||||| | |||||||| |
Proportion considered HTE and eligible for fostemsavir | 0.44% / 0.44% / 0.44%a |
Proportion with zero classes of treatment remaining and zero fully active approved agents remaining | |
Proportion with 1 to 2 classes of treatment remaining and ≥ 1 fully active approved drug remaining | |
Number of patients eligible for drug under review Number with zero classes of treatment remaining and zero fully active approved agents remaining Number with 1 to 2 classes of treatment remaining and ≥ 1 fully active approved drug remaining | ||| | ||| | ||||||| | || | |||||||| | ||| | ||| |
Market uptake (3 years) | |
Uptake (reference scenario) | |
Target population with zero remaining classes of treatment and zero fully approved agents remaining: Salvage therapy | 100% / 100% / 100% |
Target population with 1 to 2 classes of treatment remaining and ≥ 1 fully active approved drug remaining: OBT | 100% / 100% / 100% |
Uptake (new drug scenario) | |
Target population with zero remaining classes of treatment and zero fully approved agents remaining: Fostemsavir + Salvage therapy Salvage therapy | 100% / 100% / 100% 0% / 0% / 0% |
Target population with 1 to 2 classes of treatment remaining and ≥ 1 fully active approved drug remaining: Fostemsavir plus OBT OBT | 100% / 100% / 100% 0% / 0% / 0% |
Cost of treatment (per patient) | |
Cost of treatment over one year Fostemsavir OBT Salvage therapy | $45,854 $26,561 $29,405 |
Abbreviations; ARV = antiretroviral; HIV = HIV; HTE = heavily treatment-experienced; OBT = optimized background therapy.
Note: the annual cost of fostemsavir is of the drug alone, not including background therapy costs.
aSponsor obtained clinical expert feedback.
Summary of the Sponsor’s BIA Results
The sponsor’s base case reported that the reimbursement of fostemsavir to be used in combination with other ARV agents for the treatment of HIV-1 infection in HTE adults with MDR HIV-1 infection would lead to an incremental budget impact of $3,877,855 in Year 1, $7,803,294 in Year 2, $7,898,369 in Year 3. The total 3-year incremental cost was $19,579,518. Sensitivity analyses were completed to (i) include background therapy costs, (ii) include markups, and (iii) vary the proportion of patients who are eligible for fostemsavir. These sensitivity analyses impacted the 3-year incremental budget impact from + 10% to −10% in terms of % change from the base case. These changes suggested the 3-year total incremental budget impact may vary from $17,621,567 to $21,537,470.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The market uptake of fostemsavir may be overestimated: The sponsor’s submitted budget impact analysis indicated that fostemsavir would result in a market uptake of 100% in all 3 years. However, according to the clinical experts consulted by CADTH for this review, the market uptake of 100% in Year 1 and Year 2 does not align with clinical expectations and indicate the sponsor likely overestimated fostemsavir uptake. The clinical experts consulted by CADTH for this review expect difficulties in transitioning HTE patients to a new therapy right away. The experts deemed the sponsor’s estimate in Year 3 to by reasonable, but they indicated that the fostemsavir market share would likely be 50% in Year 1 and gradually increase to 75% by Year 2.
To address this limitation, CADTH undertook a scenario analysis by revising market shares for fostemsavir in the new drug scenario to 50% in Year 1, 75% in Year 2 and 100% in Year 3. This revision was completed in both the salvage therapy and OBT groups.
Additional limitations were identified but were not considered to be key limitations. These limitations include the underestimation of select background therapy costs, further highlighted in Table 14.
CADTH Reanalyses of the BIA
Table 14: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
1. Comparator cost | Unit cost: Darunavir 600 mg: $8.5940 Dolutegravir (Tivicay) 50 mg: $20.8094 Elvitegravir/cobicistat/ emtricitabine/tenofovir disoproxil fumarate (Stribild): $45.5200 Fosamprenavir (Telzir) 700 mg: $9.1869 Maraviroc (Celsentri): $18.5790 Emtricitabine/rilpivirine/ tenofovir disoproxil fumarate (Complera): $41.9140 | Unit cost: Darunavir 600 mg: $4.2970 Dolutegravir (Tivicay) 50 mg: $20.8317 Elvitegravir/cobicistat/ emtricitabine/tenofovir disoproxil fumarate (Stribild): $48.0177 Fosamprenavir (Telzir) 700 mg: $9.4389 Maraviroc (Celsentri): $19.1525 Emtricitabine/rilpivirine/ tenofovir disoproxil fumarate (Complera): $44.8643 |
Changes to derive the CADTH base case | ||
CADTH base case | No changes | |
CADTH did not undertake a base-case reanalysis. Instead, CADTH explored the potential impact of several scenario analyses which included:
Incorporating background therapy costs with corrected costs using publicly available prices.
Assuming 50% of market uptake of fostemsavir in Year 1, 75% in Year 2, and 100% in Year 3, due to feedback obtained from clinical experts consulted by CADTH.
94% price reduction of fostemsavir.
Results are presented in Table 17. The reimbursement of fostemsavir was associated with a 3-year incremental budget impact of $19,579,518 in the base case. A price reduction of 94% significantly reduces the budget impact, for a 3-year incremental budget impact of $1,174,771. Similarly, the revised market uptake of fostemsavir using feedback obtained from clinical experts consulted by CADTH resulted in a 3-year budget impact of $13,738,944.
Table 15: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $0 | $0 | $0 | $0 | $0 |
New drug | $0 | $3,877,855 | $7,803,294 | $7,898,369 | $19,579,518 | |
Budget impact | $0 | $3,877,855 | $7,803,294 | $7,898,369 | $19,579,518 | |
CADTH scenario analysis: 94% price reduction | Reference | $0 | $0 | $0 | $0 | $0 |
New drug | $0 | $232,671 | $468,198 | $473,902 | $1,174,771 | |
Budget impact | $0 | $232,671 | $468,198 | $473,902 | $1,174,771 | |
CADTH scenario analysis: including background therapy costs | Reference | $4,308,549 | $4,362,297 | $4,415,859 | $4,469,317 | $13,247,472 |
New drug | $4,308,549 | $8,240,152 | $12,219,153 | $12,367,686 | $32,826,991 | |
Budget impact | $0 | $3,877,855 | $7,803,294 | $7,898,369 | $19,579,518 | |
CADTH scenario analysis: revised marked uptake | Reference | $0 | $0 | $0 | $0 | $0 |
New drug | $1,938,928 | $4,883,007 | $6,917,010 | $13,738,944 | $1,938,928 | |
Budget impact | $0 | $1,938,928 | $4,883,007 | $6,917,010 | $13,738,944 |
BIA = budget impact analysis
Stakeholder Input
Patient Input
Community-Based Research Centre
About Community-Based Research Centre
Community-Based Research Centre (CBRC) promotes the health of people of diverse sexualities and genders through research and intervention development.
CBRC's core pillars - community-led research, knowledge exchange, network building, and leadership development - position the organization as a thought leader, transforming ideas into actions that make a difference in our communities.
CBRC was incorporated in 1999 and is a non-profit charitable organization. Our main office is located in Vancouver, British Columbia, and we also have satellite offices located in Edmonton, Toronto, and Halifax.
Information Gathering
We are a non-profit that provides leadership to 2SLGBTQ+ community organizations and conduct community-led research with PI Dr. Nathan Lachowsky with research ethics provided through the University of Victoria. Research collected is from Sex Now, which is CBRC’s principal community-based research initiative and Canada’s largest and longest running survey of gay, bisexual, queer men (cis and trans), non-binary and Two-Spirit people’s health. Originating at Pride festivals across British Columbia in 2002, Sex Now has been administered both online and in-person at events across Canada in both official languages. Often referred to as “the gay census”, Sex Now has become an essential source of data on the health and well-being of GBT2Q in Canada, and is widely used by community, public health, research, and policy stakeholders.
Disease Experience
HIV is one of the most stigmatized diseases worldwide. The outcome of untreated HIV is disability and premature death. People living with HIV are no homogenous and come from all walks of life, however HIV disproportionately impacts gay, bi, queer and trans men, Indigenous people, African, Caribbean and Black people, and people who inject drugs. Pill burden and medication adherence is a challenge for many people, whether it comes down to socioeconomic factors or other social determinants of health (e.g. housing insecurity, food insecurity) that make adherence more challenging.
Experiences With Currently Available Treatments
From our recent surveys amongst gay, bisexual, queer men (cis and trans), non-binary and Two-Spirit people, there is evidence that there are barriers, challenges and dissatisfaction with current medications. This can lead to poor medication adherence can lead to people who have heavily treatment resistant HIV.
In our Sex Now 2021 survey conducted online, amongst people living with HIV (n=325), greater than 99% had a healthcare provider and were taking ARV, with 94% having a suppressed viral load. Yet, only 72% found taking daily medication very easy, with 5% identifying that it was somewhat or very difficult. For those without a suppressed viral load or who were unsure about their viral load there was a statistically significant difference in ease of medication adherence, only 42% found it very easy to take medication on a daily basis, with 26% identifying that it was somewhat or very difficult.
In our Sex Now 2022 survey conducted through venue-based recruitment at Pride Festivals and other queer spaces, amongst people living with HIV (n=144), only 19% preferred taking daily oral pills versus an injectable medication taken every 2 months, with 47% preferring the injectable. This shows a strong desire for innovation in the HIV sector is needed to reduce the burden of taking medication on our community (e.g. long-acting agents).
The social impacts of living with HIV cannot be ignored when considering treatment issues. Especially in the context of U=U, people who are not able to maintain an undetectable viral load are not able to benefit from social advances. From Sex Now 2021, people living with HIV (n=325), due to the U=U campaign, slightly more than one-third have experienced reductions in stigma, shame and rejection. As well, nearly one-third have experienced improvement in mental, social and sexual well-being. In fact, more than not benefiting, harm may be exacerbated as there is an increased pressure to take medication and maintain an undetectable viral load. While overall there have been many successes, some queer and trans people living with HIV have experienced unintended consequences from the U=U campaign, such as nearly 20% feeling increased pressure to take medication or maintain an undetectable viral load. This highlights a common issue that existed long before U=U. Because of the stigma associated with the virus, people living with HIV are too often viewed by society, public health, governments, the legal system and researchers as a vector of disease. The experience of living with HIV is thus reduced to whether someone is able to pass HIV to someone else, rather than a health condition being just one part of their lived experience.
Improved Outcomes
This medication is intended for use with people living with heavily treatment resistant HIV. There are no other treatment options for people this medication is used for. The consequence of not treating HIV is progression to AIDS, which includes disability and premature death. For our community there is an increased likelihood of those people passing on HIV if they are sexually active or sharing injection supplies with others. It would be highly unethical for this to not be available. We cannot state with stronger words, the importance and urgency to approve this medication.
Generally folks in this situation are also facing barriers in the social determinants of health. Outside of the medical model, considerations need to be made for how pharmaceutical companies are finding ways to support medication adherence (e.g. social supports, income supports, food security, housing security, mental health support).
Experience With Drug Under Review
N/A
Companion Diagnostic Test
N/A
Anything Else?
N/A
Conflict of Interest Declaration — Community-Based Research Centre Patient Group Conflict of Interest Declaration
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
ViiV Healthcare ULC provided CBRC with a PowerPoint presentation outlining information about Rukobia detailing unmet needs, clinical indication, efficacy/safety and explaining the CADTH process.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
No
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
Table 1: Financial Disclosures for Community-Based Research Centre
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Gilead Sciences Canada, Inc. | — | — | — | X |
Merck Canada Inc. | X | — | — | — |
ViiV Healthcare ULC | — | — | — | X |
Clinician Input
No clinician group input was received for this submission.
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines.
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.