CADTH Reimbursement Review
Difelikefalin (Korsuva)
Sponsor: Otsuka Canada Pharmaceutical Inc.
Therapeutic area: Chronic kidney disease
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
ANCOVA
analysis of covariance
CHW
Cui, Hung, and Wang
CI
confidence interval
CKD
chronic kidney disease
CKD-aP
chronic kidney disease–associated pruritus
EOT
end of treatment
ESRD
end-stage renal disease
ET
early termination
HRQoL
health-related quality of life
ITT
intention to treat
LS
least squares
MID
minimal important difference
MMRM
mixed effects model with repeated measures
OLE
open-label extension
PCIG
Patient Global Impression of Change
PP
per protocol
QoL
quality of life
RCT
randomized controlled trial
SAE
serious adverse events
SD
standard deviation
TEAE
treatment-emergent adverse event
WI-NRS
Worst Itching Intensity Numerical Rating Scale
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Difelikefalin (Korsuva), 50 mcg/mL, IV |
Indication | For the treatment of moderate-to-severe pruritus associated with chronic kidney disease in adult patients on hemodialysis |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | August 16, 2022 |
Sponsor | Otsuka Canada Pharmaceutical Inc. |
NOC = Notice of Compliance.
Source: Difelikefalin Product Monograph.1
Introduction
Chronic kidney disease (CKD) is a progressive disease characterized by the gradual loss of renal function and/or abnormalities of renal structure over 3 months. CKD constitutes a major health burden worldwide and is associated with high morbidity and mortality.2,3 CKD-associated pruritus (CKD-aP), also known as uremic pruritus, is a common, distressing, and underrecognized systemic itch CKD comorbidity that affects more than 60% of patients undergoing hemodialysis, with 20% to 40% of patients reporting moderate-to-severe pruritus.4-9 Intense and generalized systemic itching in these patients is associated with poor sleep quality, depression, reduced quality of life, increased risk of infection, and an increased risk of death.5,6,10-12 In Canada, the estimated overall prevalence of CKD-aP in adults undergoing hemodialysis is about 70%, according to the international, observational Dialysis Outcomes and Practice Patterns Study.8 More than one-third of patients with CKD who are undergoing hemodialysis experience moderate-to-severe itch.8,13,14 There is currently no standardized lab or diagnostic testing for the diagnosis of CKD-aP. A complaint of pruritus by someone with a diagnosis of CKD is presumed to be CKD-aP unless assessment determines a different diagnosis.6
There is no approved therapy for CKD-aP in Canada. The standard of care for moderate-to-severe pruritus associated with CKD includes the use of topical moisturizing treatments, steroids in combination with menthol and camphor, calcineurin inhibitors, and the systemic use of gabapentin or pregabalin, naltrexone, thalidomide, biologics (i.e., dupilumab or tralokinumab), kappa opioid receptor agonists (i.e., difelikefalin), and topical phototherapy to reduce itch intensity and improve sleep quality, per feedback provided by the clinical experts consulted by CADTH.
Difelikefalin is a selective kappa opioid receptor agonist that acts in the peripheral nervous system. The drug exerts antipruritic effects by means of activation of kappa opioid receptors on peripheral neurons and immune cells. The selective activity of difelikefalin at kappa opioid receptors mostly avoids mu opioid–associated side effects, such as respiratory depression, dependence, and euphoria. Difelikefalin is anticipated to have no meaningful abuse and dependence potential.1
The objective of this report was to perform a systematic review of the beneficial and harmful effects of difelikefalin 0.5 mcg/kg IV 3 times per week for the treatment of moderate-to-severe pruritus associated with CKD in adults undergoing hemodialysis.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient groups that responded to CADTH’s call for patient input and from clinical experts consulted by CADTH for the purpose of this review.
Patient Input
One patient advocacy group, The Kidney Foundation of Canada, provided input for the treatment of adults with CKD who are undergoing hemodialysis. Patient input was gathered from independent surveys of people living with CKD and their caregivers across Canada in September 2022. In total, 19 responses were gathered from the survey (10 fully completed and 9 partially completed).
More than 90% of patients who responded to the survey reported experiencing itchy skin as part of their kidney disease, with 50% of respondents experiencing itchiness every day, 40% experiencing itchiness several times per week, and 10% experiencing itchiness occasionally. Although 60% of respondents reported living with pruritus for 1 to 2 years, 20% reported living with it for 2 to 5 years and 20% reported living with it for more than 5 years. The itchiness was described as moderate to severe by 80% of respondents. When describing disease experience, several respondents reported developing scabs and/or sores because of their itchy skin. Many respondents also reported having trouble sleeping as a result of itchiness. One-third (33%) of patients who responded to this survey reported taking medication to treat their itchiness associated with kidney disease. Although 33% of respondents reported that their treatments were covered by their provincial drug plan, 67% reported paying out of pocket. Most respondents to this survey expressed satisfaction with their current medication and/or combination of treatments, whereas 33% were neither satisfied nor unsatisfied. On the other hand, more than 66% of respondents expressed uncertainty regarding the improvement of their skin appearance in response to currently available treatment.
When describing their expectations for CKD therapies in general, patients who responded to the survey mentioned improvement in their well-being or quality of life (QoL), and 90% mentioned hoping for increased energy. Other important considerations mentioned included fewer hospital visits, less medication overall, fewer side effects, and efficacy. None of the respondents had experience with the drug under review.
Clinician Input
Input From Clinical Experts Consulted by CADTH
According to the clinical experts consulted by CADTH, treatment for moderate-to-severe pruritus associated with CKD includes the use of topical moisturizing treatments, steroids in combination with menthol and camphor, calcineurin inhibitors, and the systemic use of gabapentin or pregabalin, naltrexone, thalidomide, biologics (i.e., dupilumab or tralokinumab), kappa opioid receptor agonists (i.e., difelikefalin), and topical phototherapy to reduce itch intensity and improve sleep quality. The clinical experts stated that the goal of treatment is to reduce of itch intensity, improve QoL and sleep quality, decrease itch-related depression, and, in some cases, suicidality. They also stated that currently used off-label treatments do not adequately address these issues in all patients.
The clinical experts indicated that difelikefalin is a selective kappa opioid receptor agonist that acts in the peripheral nervous system, and thus the drug has less neuromodulation effects than a nonselective kappa opioid receptor agonist. Difelikefalin is the first treatment approved in Canada for moderate-to-severe pruritus in patients undergoing hemodialysis, although the clinical experts consulted by CADTH were uncertain about whether the drug would address the underlying disease process that causes pruritus, as the etiology of CKD-aP is not yet fully understood. The clinical experts indicated that difelikefalin would be used in combination with other therapies in a later-line setting in which existing therapies are intolerable, have failed to control symptoms, or are contraindicated. The clinical experts stated that the prior lines of therapy include a menthol-containing topical steroid and/or a topical calcineurin inhibitor, a systemic drug (gabapentin or naltrexone), and phototherapy. The clinical experts did not expect that difelikefalin would replace any treatments or cause a shift in the current treatment paradigm but, instead, would be used after other more accessible topical and systemic treatment options have failed.
The clinical experts indicated that only patients with end-stage renal disease (ESRD) on hemodialysis with moderate-to-severe pruritus who are not responsive to existing therapies would be candidates for treatment with difelikefalin. According to the clinical experts, the drug is not suitable for patients on peritoneal dialysis, patients with nondialysis CKD, or patients who have received a kidney transplant.
The clinical experts stated that clinical evaluations, such as the itch numeric rating scale, can help identify pruritus severity at baseline and assess response to treatment, but they are not typically used in clinical practice. The clinical experts indicated that it is important to consider other causes of pruritus in patients, such as dermatologic conditions, CKD mineral bone disease, hepatobiliary disorders, diabetes, and hematologic conditions, as patients with these conditions may not respond to difelikefalin. Further, the clinical experts mentioned that it is possible that misdiagnosis of the cause of pruritus may occur in clinical practice due to the various potential causes of pruritus, as well as the lack of unique laboratory findings associated with CKD-aP.
The clinical experts consulted by CADTH indicated that in clinical practice, subjective patient-reported improvement in symptoms is the primary outcome used to determine whether a patient is responding to treatment. The clinical experts highlighted that reductions in the frequency and/or severity of symptoms would be considered when evaluating response to treatment, along with other improvements in sleep quality, depression, adherence to dialysis, depression and suicidality, and overall QoL. The clinical experts consulted by CADTH stated that a patient may discontinue treatment if there is a lack of response, and that this could be assessed at 12 weeks. The development of significant and persistent side effects, such as diarrhea, dizziness, and recalcitrant nausea and/or vomiting, may also be cause for discontinuing treatment.
According to the clinical experts consulted by CADTH, difelikefalin would most often be prescribed in dialysis units or clinics setting by nephrologists or other physicians.
Clinician Group Input
Clinician group input on the review of difelikefalin for the treatment of moderate-to-severe pruritus associated with CKD in adults on hemodialysis was received from 3 clinician groups: Saskatchewan Kidney Doctors; Hemodialysis Specialty Physician Group, Division of Nephrology, The Ottawa Hospital; and Division of Nephrology, Department of Medicine, Dalhousie University and Nova Scotia Health.
The clinician groups agreed that currently only off-label medications in Canada are available for the treatment of pruritus in patients with kidney disease. There have been some unmet needs as the current treatment options are unsatisfactory and not effective in reducing the symptom burden. The debilitating symptoms in most severe cases may lead to a deterioration in QoL. The clinician groups mentioned the need for a new treatment option that could help relieve symptoms with better tolerability, affordability, and ease of administration. One clinician group in particular indicated that the most important treatment goal would be to have a therapy that would reduce or maintain the severity of itch below the threshold of clinical importance, which is known to be the level above which itch negatively impacts QoL and outcomes important to patients.
The clinician groups mentioned that difelikefalin may cause a paradigm shift in the treatment of itching, with data from randomized controlled trials (RCTs) and a pooled analysis demonstrating objective effectiveness in reducing the severity of the symptoms of CKD-associated (uremic) pruritus. Input from clinician groups provided different opinions about the place in therapy for difelikefalin. Some groups suggested that it would likely be used as first-line therapy, whereas others recommended it as an add-on or second-line treatment. Given the route of administration of difelikefalin, the clinician groups pointed out that patients on hemodialysis with moderate-to-severe pruritus would benefit the most from this treatment. The clinician groups mentioned the possibility of underreporting or underdiagnosing CKD-aP, which was also pointed out by the clinical experts consulted by CADTH. Regarding the diagnosis of this indication, the clinician groups noted the lack of diagnostic tests. One group mentioned using clinical history and exclusion criteria to identify patients, whereas other groups pointed out that the identification of patients and severity of symptoms can be done through self-administered questionnaires and screening tools (e.g., the 5-D itch scale, the Uremic Pruritus in Dialysis Patient [UP-Dial] questionnaire, and the Skindex-10 scale). The clinician groups stated that the reduction in itch severity measured on a numeric rating scale is used to evaluate symptoms. According to the clinician groups, lack of a clinically meaningful response and intolerable side effects should be considered discontinuation criteria. The clinician groups added that a meaningful response to treatment for this disease would be symptom reduction leading to better sleep, improved QoL, improved mood, and return to activities of daily living.
Drug Program Input
Input was obtained from the drug programs that participate in the CADTH reimbursement review process. The following were identified as key factors that could potentially impact the implementation of a CADTH recommendation for difelikefalin:
considerations for relevant comparator
considerations for initiation of therapy
considerations for prescribing of therapy
considerations for care provision issues
considerations for system and economic issues.
Clinical Evidence
Pivotal Studies and Protocol-Selected Studies
Description of Studies
The KALM-1 and KALM-2 studies met the inclusion criteria for the CADTH systematic review. Both KALM trials were designed as multicentre, randomized, double-blind, phase III clinical trials to compare difelikefalin with placebo in patients being treated with hemodialysis who were experiencing moderate-to-severe pruritus. The primary objective of both KALM trials was to evaluate the efficacy of difelikefalin at a dose of 0.5 mcg/kg, compared with placebo, in reducing the intensity of itch in patients undergoing hemodialysis and experiencing moderate-to-severe pruritus. The shared key secondary objectives of the KALM-1 and KALM-2 trials were to evaluate the efficacy of difelikefalin at a dose of 0.5 mcg/kg, compared with placebo, in improving itch-related QoL and safety in patients undergoing hemodialysis and experiencing moderate-to-severe pruritus. A total of 378 patients in the KALM-1 trial and 473 patients in the KALM-2 trial were randomized, in a 1:1 ratio, to difelikefalin or placebo. The KALM-1 trial was limited to study centres in the US, and the KALM-2 trial was conducted globally, and included 5 sites in Canada. For both KALM trials, the randomization was stratified based on the use of concomitant medications to treat itch during the week before randomization (yes or no), specific medical conditions (such as a history of falls or fracture or fall-related fracture, confusional state or mental status change or altered mental status or disorientation, or gait disturbance or movement disorder [presence or absence]).
The primary efficacy end point in the KALM-1 and KALM-2 trials was the percentage of patients at week 12 who achieved an improvement of at least 3 points from baseline in the weekly mean score on the daily Worst Itch Intensity Numerical Rating Scale (WI-NRS). Key secondary efficacy end points for both trials were mean change from baseline at week 12 in itch-related QoL (as measured by the 5-D itch scale total score), mean change from baseline at week 12 in itch-related QoL (as measured by the Skindex-10 scale total score), and the percentage of patients at week 12 achieving an improvement of at least 4 points from baseline in the weekly mean score of the daily WI-NRS. In addition, the proportions of patients who achieved an improvement of at least 3 points or at least 4 points from baseline at week 4 and week 8 were considered as secondary end points in the KALM-2 trial.
The baseline patient characteristics were roughly balanced between treatment groups. The median age of all randomized patients was similar in the 2 studies, at 58.0 years (range, 22 to 88 years) in the KALM-1 trial and 60.0 years (range, 23 to 87 years) in the KALM-2 trial. Most patients in the KALM-1 and KALM-2 studies were male (61.0% and 58.2%, respectively). The predominant races in the KALM-1 and KALM-2 studies were white (48.8% and 70.3%, respectively) and Black or African American (41.6% and 19.3%, respectively). The median prescription dry body weight was 84.0 kg (range, 42.0 to 135.0 kg) in the KALM-1 trial and 78.0 kg (range, 42.0 to 135.0 kg) in the KALM-2 trial. The median baseline WI-NRS score was 7.14 (range, 4.1 to 10.0) in the KALM-1 trial and 7.13 (range, 4.5 to 10.0) in the KALM-2 trial. At baseline, 39.8% of patients in the KALM-1 trial and 36.5% in the KALM-2 trial were using antiitch medications, and 14.1% and 16.6%, respectively, reported at least 1 of the specified medical conditions (i.e., history of falls or fracture or fall-related fracture, confusional state or mental status change or altered mental status or disorientation, or gait disturbance or movement disorder). The median duration of CKD-aP for all patients was 2.5 years (range, 0.1 to 26.5 years) in the KALM-1 trial and 2.3 years (range, 0.0 to 58.4 years) in the KALM-2 trial. The median time interval since the diagnosis of CKD and of ESRD for all patients was 5.45 years (range, 0.3 to 42.9 years) and 3.92 years (range, 0.3 to 28.7 years), respectively, in the KALM-1 trial, and 7.53 years (range, 0.3 to 48.3 years) and 4.03 years (range, 0.3 to 30.2 years), respectively, in the KALM-2 trial.
Efficacy Results
A summary of key efficacy results is provided in Table 2. No evidence was identified in the KALM trials for mood, days of missed work, days of missed school, and days of missed dialysis, which were identified in the CADTH systematic review protocol and considered to be important outcomes of interest by patients and clinicians.
Pruritus Severity
Worst Itching Intensity Numerical Rating Scale
The severity of pruritus was assessed in both the KALM-1 and KALM-2 trials using the WI-NRS. The primary and key secondary end points in both trials were the proportion of patients with at least a 3-point improvement in the WI-NRS score from baseline to week 12 and the proportion of patients with at least a 4-point improvement in the WI-NRS score from baseline to week 12, respectively.
At week 12 in the KALM-1 trial, 52% and 31% of patients randomized to difelikefalin and placebo, respectively, had a WI-NRS score that improved by at least 3 points from baseline to week 12. This corresponded to an odds ratio of 2.72 (95% confidence interval [CI], 1.72 to 4.30; P < 0.001) in favour of difelikefalin. In the KALM-2 trial, 50% and 37% of patients randomized to difelikefalin and placebo, respectively, had a WI-NRS score that improved by at least 3 points from baseline to week 12. This corresponded to an odds ratio of 1.61 (95% CI, 1.08 to 2.41; P = 0.02) in favour of difelikefalin. A 3-point improvement on the WI-NRS has been validated as an appropriate threshold specifically for patients with CKD-aP in an assessment of the psychometric properties published by the sponsor that was based on data from a phase II study (CR845-CLIN2101, NCT02858726) and the KALM-1 and KALM-2 trials.15 However, the FDA report states that, “in several communications to the sponsor the Agency recommended the primary efficacy end point to be the proportion of patients achieving at least a 4-point improvement in WI-NRS score from baseline to week 12.”16 The clinical experts consulted by CADTH were aligned with the FDA guidance regarding a 4-point improvement, but input from clinician groups suggested a preference for a 3-point improvement. In summary, there is uncertainty regarding the most appropriate minimally important difference (MID) for the WI-NRS score; therefore, results for an assessment of both outcomes have been presented. With regard to the proportion of patients achieving at least a 4-point improvement from baseline to week 12 with respect to the weekly mean of the daily 24-hour WI-NRS, 41% and 21% of patients randomized to difelikefalin and placebo, respectively, in the KALM-1 trial, and 38% and 25% of patients randomized to difelikefalin and placebo respectively, in the KALM-2 trial had a WI-NRS score that improved by at least 4 points from baseline to week 12. This corresponded to an odds ratio of 2.89 (95% CI, 1.75 to 4.76; P < 0.001) in the KALM-1 trial and of 1.77 (95% CI, 1.14 to 2.74; P = 0.01) in the KALM-2 trial in favour of difelikefalin. Overall, difelikefalin demonstrated an improvement in the WI-NRS that was clinically meaningful, based on both of the reported assessments of 3- and 4-point improvements in the WI-NRS.
The supportive analyses performed for the primary end point in the KALM-1 and KALM-2 trials was conducted using the per-protocol (PP) population. The results were consistent with the primary analysis. Similarly, the sensitivity analyses for the primary end point conducted using the intention-to-treat (ITT) population showed efficacy with difelikefalin, and the results were consistent with the efficacy shown in the primary efficacy analysis.
The proportion of patients with at least a 3-point or 4-point improvement from baseline in the WI-NRS was also reported at week 4 and week 8 in both trials. In the KALM-1 trial, the proportion of patients with at least a 3-point or at least a 4-point improvement from baseline in the WI-NRS at week 8 and week 4 were considered exploratory, but in the KALM-2 trial, they were included as key secondary end points. In the KALM-2 trial, a benefit from treatment with difelikefalin, compared to placebo, was also observed at week 4 (35% versus 22%) and week 8 (45% versus 33%) for the proportion of patients with at least a 3-point improvement from baseline in the WI-NRS. Similar results were observed in the KALM-1 trial for difelikefalin versus placebo at week 4 (33% versus 18%) and at week 8 (43% versus 28%). Similarly, with regard to the proportion of patients with at least a 4-point improvement from baseline in the KALM-2 trial, a benefit from treatment with difelikefalin, compared to placebo, was observed at week 4 (20% versus 13%) and at week 8 (31% versus 20%). Similar results were observed in the KALM-1 trial at week 4 (18% versus 8%) and at week 8 (31% versus 20%). In the KALM trials, data for the primary and secondary or other outcomes were reported up to 12 weeks. According to the clinical experts consulted by CADTH, patients with CKD-aP would receive difelikefalin beyond 12 weeks for the desired treatment effect. A pooled analysis of the 2 open-label extension (OLE) studies —KALM-1 and KALM-2 — assessed the 5-D itch scale for 52 weeks after the 12-week pivotal trials, and reported that the proportion of patients achieving at least a 5-point improvement (reduction) was maintained up to 64 weeks.17 Due to the amount of missing data, the lack of reporting for other outcome measures (i.e., WI-NRS and Skindex-10), and the absence of a control group after week 12, it is difficult to draw conclusions about whether the efficacy of difelikefalin beyond 12 weeks would be consistent with the efficacy at week 12.
Patient Global Impression of Change
At week 12 in the KALM-1 trial, ||| and ||| of patients randomized to difelikefalin and placebo, respectively, were complete responders. This corresponded to an odds ratio of |||| |||| ||| |||| || ||||| | | |||||). In the KALM-2 trial, ||| and ||| of patients randomized to difelikefalin and placebo, respectively, were complete responders. This corresponded to an odds ratio of |||| |||| ||| |||| || ||||| | | ||||||. Of note, the P values were not adjusted for multiple testing.
Health-Related Quality of Life
Health-related quality of life (HRQoL) was assessed in both the KALM-1 and KALM-2 trials using the Skindex-10 scale score and 5-D itch scale score. The change from baseline to week 12 in the Skindex-10 scale total score and the 5-D Itch scale score were included as key secondary end points in both of the trials.
Skindex-10 Scale Total Score
Skindex-10 is a modified scale that contains 10 questions to evaluate CKD-aP and QoL and relevant subdomains for patients in the hemodialysis setting, with higher scores indicating more bothered and lower scores indicating less bothered.4,5 At the end of week 12, the least squares (LS) mean change (reduction) in the Skindex-10 scale total score was –17.2 versus –12.0 for difelikefalin versus placebo in the KALM-1 trial, –16.6 versus –14.8, respectively, in the KALM-2 trial. In the KALM-1 trial, the difference in LS mean change (reduction) from baseline to week 12 in the Skindex-10 scale total score between the difelikefalin and placebo groups was –5.1 points (95% CI, –8.0 to –2.3 points, P < 0.001). In the KALM-2 trial, the results for the change from baseline to week 12 in the Skindex-10 scale total score indicated no difference between treatment groups, with a LS mean difference of –1.8 points (95% CI, –4.3 to 0.8 points; P = 0.171). A 3-point to 12-point change on the Skindex-10 scale was identified as a clinically meaningful change in patients with moderate-to-severe CKD-aP on hemodialysis.5 Therefore, the reported within-group differences in Skindex-10 scores were clinically meaningful, as they met the MID identified by the literature search conducted by CADTH. With regard to Skindex-10 domain scores, the results were in favour of difelikefalin; however, there was no adjustment for multiplicity, so definitive conclusions could not be drawn with respect to the individual domains of the Skindex-10 scale: disease total, mood/emotional distress total, and social functioning total. No long-term data were reported in the KALM trials for the Skindex-10 scale.
5-D Itch Scale Total Score
The 5-D itch scale is a questionnaire that assesses CKD-aP and QoL and has relevant subdomains for patients in the hemodialysis setting, with higher scores indicating more bothered and lower scores indicating less bothered.18 At the end of week 12, the LS mean change in the 5-D itch scale total score was greater in the difelikefalin group than in the placebo group in the KALM-1 trial (–5.0 versus –3.7) and the KALM-2 trial (–4.9 versus –3.8), indicating that patients in the difelikefalin group experienced greater improvement (reduction) than those in the placebo group. Overall, the difference in LS mean change from baseline to week 12 in the 5-D itch scale total score between the difelikefalin and placebo groups was –1.3 points (95% CI, –2.0 to –0.5 points, P < 0.001) in the KALM-1 trial and –1.1 points (95% CI, –1.7 to –0.4 points; P = 0.002) in the KALM-2 trial. Of note, the analysis of the difference in LS means for the 5-D itch score at week 12 in the KALM-2 trial was at risk for a type I error, as the difference in LS means for Skindex-10 at week 12, which came first in the testing hierarchy, was not statistically significant. In addition, no published MID was identified for 5-D itch scores in patients with CKD-aP. Therefore, it is unclear whether the reported within-group differences are clinically meaningful. No long-term data were reported in the KALM trials for the 5-D itch scale.
Table 2: Summary of Key Efficacy Results From Pivotal and Protocol-Selected Studies
End point | KALM-1 | KALM-2 | |||
|---|---|---|---|---|---|
Difelikefalin 0.5 mcg/kg N = 189 | Placebo N = 189 | Difelikefalin 0.5 mcg/kg N = 237 | Placebo N = 236 | ||
Patients with ≥ 3-point WI-NRS improvement (ITT analysis set) | |||||
Week 12 | |||||
Proportion of patients with ≥ 3-point WI-NRS improvement at week 12,a n (%) | 82 (52.2) | 51 (30.9) | 95 (49.7) | 77 (37.2) | |
Difference, % (95% CI) | 22 (12 to 32) | 11 (1 to 20) | |||
LS means estimate of percent with improvementb (95% CI) | 51.0 (42.9 to 58.9) | 27.6 (20.2 to 36.6) | 54.0 (43.9 to 63.9) | 42.2 (32.5 to 52.5) | |
Odds ratioc (95% CI) | 2.72 (1.72 to 4.30) | 1.61 (1.08 to 2.41) | |||
P valued | < 0.001 | 0.020 | |||
Patients with ≥ 4-point WI-NRS improvement (ITT analysis set) | |||||
Week 12 | |||||
Proportion of patients with ≥ 3-point WI-NRS improvement at week 12,a n (%) | 64 (40.8) | 35 (21.2) | 72 (37.7) | 52 (25.1) | |
Difference, % (95% CI) | 19 (9 to 28) | 12 (3 to 20) | |||
LS means estimate of percent with improvementb (95% CI) | 38.9 (29.8 to 48.7) | 18.0 (12.1 to 26.0) | 41.2 (33.0 to 50.0) | 28.4 (21.3 to 36.7) | |
Odds ratioc (95% CI) | 2.89 (1.75 to 4.76) | 1.77 (1.14 to 2.74) | |||
P valued | < 0.001 | 0.010 | |||
Patient Global Impression of Change | |||||
Total, n | ||| | ||| | ||| | ||| | |
Responders,e n (%) | ||| |||||| | || |||||| | ||| |||||| | || |||||| | |
95% CI of percent | |||| || |||| | |||| || |||| | |||| || |||| | |||| || |||| | |
Odds ratio (95% CI) | |||| ||||| || ||||| | |||| ||||| || ||||| | |||
P valuef | ||||||| | ||||| | |||
Change from baseline in Skindex-10 scale total score at week 12 (ITT analysis set) | |||||
End of week 12 change from baseline, LS mean (95% CI) | –17.2 (–19.6 to –14.7) | –12.0 (–14.5 to –9.6) | –16.6 (–19.3 to –14.0) | –14.8 (–17.4 to –12.2) | |
Difference in LS meansf,h,i (95% CI) | –5.1 (–8.0 to –2.3) | –1.8 (–4.3 to 0.8) | |||
P value | < 0.001 | 0.171 | |||
Change from baseline in 5-D itch scale total score at week 12 (ITT analysis set) | |||||
End of week 12 change from baseline, LS mean (95% CI) | –5.0 (–5.7 to –4.4) | –3.7 (–4.4 to –3.1) | –4.9 (–5.6 to –4.2) | –3.8 (–4.5 to –3.1) | |
Difference in LS means (95% CI) | –1.3 (–2.0 to –0.5) | –1.1(–1.7 to –0.4) | |||
P value | < 0.001 | 0.002j | |||
CI = confidence interval; ITT = intention to treat; LS = least squares; WI-NRS = Worst Itching Intensity Numerical Rating Scale.
Note: Combined analysis for the primary efficacy end point used the separate interim and postinterim results to generate an adjusted overall estimate and P value using the Lawrence and Hung and/or Cui, Hung. and Wang (CHW) methodology.
aCounts and percentages are based on nonmissing data.
bEstimated percent, odds ratio, and P value used a logistic regression model with terms for treatment group, baseline WI-NRS score, region (for the KALM-2 trial only), use of antiitch medication during the week before randomization, and the presence of specific medical conditions. Missing values were imputed using multiple imputation under the missing-at-random missing data assumption for interim patients and postinterim patients separately.
cEstimated using the Lawrence and Hung approach.
dEstimated using the CHW approach.
ePatient Global Impression of Change responders were those with responses of very much improved or much improved.
eThe P value is calculated with the Cochran-Mantel-Haenszel exact test.
gThe P value was not adjusted for multiple testing (i.e., the type I error rate has not been controlled).
hUsed the placebo group as reference.
iLS means and 95% CIs were based on an ANCOVA, with fixed effects for treatment, with baseline score, region (for the KALM-2 trial only), and the randomization stratification variables as covariates for the KALM-2 trial. Missing values were imputed using multiple imputation under the missing-at-random missing data assumption.
jThe P value was not considered inferential and the null hypotheses was not rejected, according to the gate-keeping strategy, based on the hierarchical testing order of key secondary end points, as the test of the prior end point (change from baseline in Skindex-10 total score) in the sequence was not statistically significant.
Sources: KALM-1 Clinical Study Report;19 KALM-2 Clinical Study Report;20 FDA report.16
Harms Results
Selected harms outcomes from the KALM-1 and KALM-2 trials are summarized in Table 3.
The percentage of patients with any reported treatment-emergent adverse events (TEAE) was comparable in the difelikefalin and placebo groups in the KALM-1 trial (68.8% versus 62.2%) and the KALM-2 trial (68.1% versus 61.4%). In the KALM-1 trial, the most common TEAEs reported for patients randomized to difelikefalin and placebo, respectively, were diarrhea (9.5% versus 3.7%), dizziness (6.9% versus 1.1%), vomiting (5.3% versus 3.2%), and nasopharyngitis (3.2% versus 5.3%). Diarrhea, dizziness, and vomiting were reported more commonly in the difelikefalin treatment group than the placebo treatment group in the KALM-1 trial. In the KALM-2 trial, the most common TEAEs reported for patients randomized to difelikefalin and placebo, respectively, were diarrhea (8.1% versus 5.5%), vomiting (6.4% versus 5.9%), falls (6.8% versus 5.1%), dizziness (5.5% versus 5.1%), and nausea (6.4% versus 4.2%), all of which were reported more frequently in the difelikefalin treatment group than in the placebo group. Serious adverse events (SAEs) were reported in 25.9% of patients in the difelikefalin group and 21.8% of patients in the placebo group in the KALM-1 trial, and in 24.7% and 21.6%, respectively, in the KALM-2 trial. The clinical experts stated that the proportion of patients reporting an SAE was consistent with what would be expected, given the characteristics of the patients enrolled in these studies. Specific SAEs reported in the trials were hyperkalemia, sepsis, pneumonia, fluid overload, and chest pain, all of which were infrequently reported, occurring in less than 4% of patients in any treatment group.
The proportion of patients who discontinued treatment due to TEAEs was 7.9% for difelikefalin and 4.8% for placebo in the KALM-1 trial and 5.5% for difelikefalin and 3.4% for placebo in the KALM-2 trial. Dizziness was the most frequently reported TEAE that caused discontinuation for both KALM trials. Deaths were reported in 1.1% of patients in the difelikefalin group and 1.6% of patients in the placebo group in the KALM-1 trial, and in 0.9% for difelikefalin and 0.8% for placebo in the KALM-2 trial. In the KALM-1 trial, sepsis was the cause of death for 2 patients randomized to difelikefalin (but for 0 patients randomized to placebo and 0 patients in the KALM-2 trial), and septic shock was the cause of death for 2 patients randomized to placebo (but for 0 patients randomized to difelikefalin and 0 patients in the KALM-2 trial). All other reported causes of death (dyspnea and/or hypotension, cardiac arrest, unknown) were infrequently reported, with no more than 1 patient in any treatment group. No specific adverse event (AE) was identified to account for the majority of deaths in either group in the KALM-2 trial.
The following harms of particular interest were included in the CADTH systematic review protocol: diarrhea, nausea, vomiting, gait disturbance, falls, dizziness, headache, somnolence, seizures, syncope, mental status changes, mood changes, paresthesia (unusual feeling or sensation), hyperkalemia, back pain, tachycardia, and palpitation. The most common notable harms were diarrhea, dizziness, and vomiting in both KALM trials, as well as falls and nausea in the KALM-2 trial. Overall, during the 12-week treatment period of the KALM-1 and KALM-2 trials, patients who received difelikefalin reported notable harms at a similar or slightly higher frequency than patients who received placebo. There were imbalances in the proportion of patients (difelikefalin versus placebo) reporting diarrhea (9.5% versus 3.7%) and dizziness (6.9% versus 1.1%) as an AE in the KALM-1 trial.
Table 3: Summary of Key Safety Results From Pivotal and Protocol-Selected Studies
Outcome | KALM-1 | KALM-2 | ||
|---|---|---|---|---|
Difelikefalin 0.5 mcg/kg N = 189 | Placebo N = 188 | Difelikefalin 0.5 mcg/kg N = 235 | Placebo N = 236 | |
Harms, n (%) (double-blind safety analysis set) | ||||
Patients with ≥ 1 TEAEsa | 130 (68.8) | 117 (62.2) | 160 (68.1) | 145 (61.4) |
Patients with ≥ 1 serious TEAEsa | 49 (25.9) | 41 (21.8) | 58 (24.7) | 51 (21.6) |
Patients who discontinued treatment due to TEAEa | 15 (7.9) | 9 (4.8) | 13 (5.5) | 8 (3.4) |
Deaths | 2 (1.1) | 3 (1.6) | 2 (0.9) | 2 (0.8) |
Notable harms, n (%) | ||||
Diarrhea | 18 (9.5) | 7 (3.7) | 19 (8.1) | 13 (5.5) |
Dizzinessb | 13 (6.9) | 2 (1.1) | 13 (5.5) | 12 (5.1) |
Vomiting | 10 (5.3) | 6 (3.2) | 15 (6.4) | 14 (5.9) |
Hyperkalemia | 8 (4.2) | 5 (2.7) | 9 (3.8) | 6 (2.5) |
Headache | 7 (3.7) | 4 (2.1) | 10 (4.3) | 6 (2.5) |
Somnolenceb | 6 (3.2) | 4 (2.1) | 11 (4.7) | 5 (2.1) |
Nausea | 6 (3.2) | 9 (4.8) | 15 (6.4) | 10 (4.2) |
Back pain | 6 (3.2) | 1 (0.5) | 0 | 0 |
Fallsb | 5 (2.6) | 5 (2.7) | 16 (6.8) | 12 (5.1) |
Paresthesia (unusual feeling or sensation) | 5 (2.6) | 7 (3.7) | 11 (4.7) | 6 (2.5) |
Mental status changesb | 3 (1.6) | 3 (1.6) | 3 (1.3) | 1 (0.4) |
Tachycardiab,c | 2 (1.1) | 1 (0.5) | 1 (0.4) | 6 (2.5) |
Gait disturbanceb | 1 (0.5) | 2 (1.1) | 7 (3.0) | 2 (0.8) |
Seizureb | 1 (0.5) | 1 (0.5) | NR | NR |
Syncopeb | 1 (0.5) | 1 (0.5) | 4 (1.7) | 3 (1.3) |
Altered moodb | 1 (0.5) | 0 | 0 | 1 (0.4) |
Palpitationsb | 0 | 2 (1.1) | 3 (1.3) | 1 (0.4) |
TEAE = treatment-emergent adverse event.
aTEAEs relative to the double-blind treatment period are identified as any AE with an onset date after the first dose of the study drug up to the study end of treatment or early termination visit, the start of the discontinuation period, or 10 days after the last dose if no end of treatment or early termination visit was conducted, whichever is later.
bReported based on TEAEs of special interest identified by the sponsor.
cTachycardia included the following preferred terms: tachycardia, sinus tachycardia, and tachyarrhythmia.
Sources: KALM-1 Clinical Study Report;19 KALM-2 Clinical Study Report.20
Critical Appraisal
The overall study design of the KALM-1 and KALM-2 trials was appropriate for the objectives of the study. There was no particular concern with the methods of randomization or allocation concealment. According to the clinical experts consulted by CADTH, the frequency of hemodialysis (i.e., optimization of hemodialysis) is a potential effect modifier and prognostic factor that was not considered in either KALM trial. Patients with more frequent hemodialysis visits would have better control of the disease and more exposure to difelikefalin than patients who had less frequent hemodialysis visits. In addition, more frequent hemodialysis visits indicated better compliance with difelikefalin treatment. Therefore, the frequency of hemodialysis may have a potential impact on the validity of the study results for the treatment effect of difelikefalin; however, the magnitude of the impact is unknown, as there were no data with regard to the treatment effect in patients with different frequencies of hemodialysis in either KALM trial. The 3-point reduction in WI-NRS scores was adopted in both KALM studies as the cut-off point to define improvement and response of treatment in pruritus intensity based on the results of a phase II study of difelikefalin (CR845-CLIN2101, NCT02858726) and the 2 phase III trials (KALM-1 and KALM-2) conducted by the sponsor.15 The FDA recommended an improvement of at least 4 points as the cut-off for the primary efficacy end point (i.e., proportion of patients with a ≥ 4-point improvement in WI-NRS at week 12).16 Overall, although odds ratios at week 12 were similar with the 3-point and 4-point cut-offs, the proportion of patients in each treatment group that met the threshold was higher with the 3-point threshold, which was used for the primary end point. It is worth mentioning that odds ratios were used throughout the KALM trials for the primary and key secondary end points, and that odds ratios tend to give an inflated impression of the treatment effects, compared with relative risks.21 Therefore, the results of the treatment effect of difelikefalin compared to placebo estimated using odds ratios should be interpreted with caution. Although there was a large proportion of patients with at least 1 major protocol deviation (around 30%) in both studies, the sensitivity and supplemental analyses were consistent with the primary estimand. In addition, a notable response was observed in the placebo treatment groups in both KALM trials, albeit not as great as in the difelikefalin treatment groups. According to the clinical experts consulted by CADTH, the placebo response may be due to the optimized hemodialysis treatment associated with the trials, as patients enrolled in trials are more likely to attend hemodialysis visits more frequently and regularly than those in real-world settings, which could have provided a benefit to these patients. In addition, the CADTH review team considered the placebo effect to be a contributing factor to the response in the placebo group; however, the extent to which the placebo effect influenced the results is unclear.
In terms of the generalizability of the pivotal KALM studies, pruritus severity was measured using the WI-NRS and the Patient Global Impression of Change (PGIC), and HRQoL was measured using the Skindex-10 and 5-D itch scales. However, the experts consulted by CADTH stated that these outcome measures are not routinely used in clinical practice to assess itch intensity or HRQoL in patients. Therefore, there is uncertainty about how changes in pruritus severity measured with the WI-NRS and PGIC and changes in HRQoL measured with the Skindex-10 and 5-D itch scales translate to clinical practice. A limitation to note is that the studies included patients with better hemodialysis adherence and less pruritus severity than is seen in clinical practice in Canada, per feedback from the clinical experts consulted by CADTH. The clinical experts for this review also indicated that the patients in the KALM-1 and KALM-2 trials had better adherence to hemodialysis than in Canadian clinical practice. As such, the generalizability of efficacy outcomes may be overestimated and safety outcomes may be underestimated. In addition, the baseline median WI-NRS score was around 7 for both KALM trials (7.14 for the KALM-1 trial and 7.13 for the KALM-2 trial), and the clinical experts consulted by CADTH indicated the patients with CKD-aP would have a worse itch intensity (with a numeric rating scale score greater than 8) in dermatology clinical practice in Canada. Overall, the selection of patients with better hemodialysis adherence and less severe pruritus (based on the WI-NRS) may limit the applicability of the study results to the patient population in Canada and introduce selection bias, which may lead to uncertainty in the efficacy results. In the KALM trial, data for the primary and secondary or other outcomes were reported up to 12 weeks. According to the clinical expert, patients with CKD-aP would receive the difelikefalin treatment beyond 12 weeks for the desired treatment effect. Although a reduction in symptoms of CKD-aP may be observed during the first 12 weeks of treatment, it is uncertain whether the treatment effect or the safety of difelikefalin beyond 12 weeks would be consistent with the results at week 12.
Indirect Comparisons
No indirect evidence was identified for this review.
Other Relevant Evidence
Additional safety data for the open-label, phase III CR845-CLIN3101 study for up to 52 weeks was summarized in this report.
Description of the CR845-CLIN3101 Trial
One open-label, multicentre, phase III study (CR845-CLIN3101) that evaluated the long-term safety of difelikefalin at a dose of 0.5 mcg/kg administered for up to 52 weeks was included as other relevant evidence to address the gap in the long term safety of difelikefalin for this review.
The long-term safety study included patients who participated in the phase II studies for difelikefalin (CR845-CLIN2005 and CR845-CLIN2101). The long-term safety study also included de novo patients with moderate-to-severe CKD-aP undergoing hemodialysis who had not been previously exposed to difelikefalin and who had not participated in the phase II studies for difelikefalin. The open-label, phase III study consisted of a screening visit, a 52-week treatment period, an end of treatment (EOT) visit, and a follow-up visit 7 to10 days after the EOT visit. Patients received difelikefalin at a dose of 0.5 mcg/kg after each dialysis session, 3 times per week for up to 52 weeks, for a total of approximately 156 doses of the study drug. All scheduled study visits were conducted on dialysis days during the treatment period. The last dose was administered at the last dialysis visit on week 52, at or early termination (ET). The EOT visit was conducted at the dialysis visit following the last dose. A final safety follow-up was conducted 7 to 10 days after the EOT or ET visit. In the long term safety study, AEs, SAEs, withdrawal due to adverse events, and deaths were reported descriptively for each of the 3 treatment groups.
Efficacy Results
No efficacy results were evaluated in the open-label, phase III study.
Harms Results
In the open-label, phase III study, |||||| |||||, and ||||| patients experienced at least 1 AE in the placebo and difelikefalin groups in the phase II study and in the de novo group in the open-label study, respectively. The most common AEs were (frequency ≥ 5%) were nausea, diarrhea, falls, vomiting, hypotension, noncardiac chest pain, hyperkalemia, dizziness, abdominal pain, fluid overload, pneumonia, dyspnea, acute myocardial infarction, pain in extremity, arthralgia, and asthenia. The proportion of patients with at least 1 SAE was |||||| |||||, and ||||| in the placebo and difelikefalin groups in the phase II study and in the de novo group in the open-label study, respectively. The most common SAEs (frequency > 4%) among patients were acute myocardial infarction, angina pectoris, gastrointestinal hemorrhage, pneumonia, cellulitis, fluid overload, hyperkalemia, respiratory failure, pulmonary edema, and hypotension. A total of || patients discontinued the study drug due to AEs, with the proportions being 20.0%, 15.4%, and 10.2% in the placebo and difelikefalin groups in the phase II study and in the de novo group in the open-label study, respectively. A total of 16 deaths occurred during the study, with the proportions being ||||| ||||, and |||| in the placebo and difelikefalin groups in the phase II study and in the de novo group in the open-label study, respectively.
Critical Appraisal
The objective of the open-label, phase III study was to evaluate the long-term safety of difelikefalin, administered intravenously after each dialysis session for up to 52 weeks. Because results for the open-label, phase III trial were only reported descriptively, they should be interpreted with caution. Discontinuation rates were high in all 3 treatment groups. One of the issues with discontinuation from an open-label study, particularly when patients discontinue due to AEs, is that the summary of harms may underestimate the frequency of AEs, because those who remained in the study are more likely to have responded well to treatment. Overall, the high discontinuation rates, as well as the descriptive nature of analysis, introduce uncertainty into the long-term safety results. The lack of comparative evidence makes it difficult to interpret the safety results.
Although the patient population in the open-label study is different than the KALM-1 and KALM-2 populations, the external validity points related to demographic factors from the main report could be applicable to this study population. Although the 0.5 mcg/kg dose is consistent with the Health Canada–approved dose, the duration of the open-label, phase III trial (up to 52 weeks) is more than that in the pivotal KALM-1 and KALM-2 trials (12 weeks). Because it is expected that patients would receive this treatment beyond 12 weeks, the safety results of this open-label, phase III study may be generalizable to this time frame to some extent, but not completely, as the rates of AEs are expected to increase with longer treatment times.
Summary of Pooled Analysis of OLE of KALM-1 and KALM-2
A pooled analysis by Topf et al. (2022)17 of 2 OLE studies of the KALM-1 and KALM-2 trials assessed the 5-D itch scale for 52 weeks after the 12-week pivotal trials. The study reported the proportion of patients achieving at least a 5-point reduction in the 5-D itch scale was maintained up to 52 weeks in the open-label phase of the KALM-1 and KALM-2 trials. However, there was some uncertainty in the reported long-term treatment effect due to the amount of missing data, with 26.5% (189 of 712) of patients in the pooled analysis population contributing data at week 52 in the OLE phase. In addition, it is difficult to discern much about the long-term efficacy of difelikefalin due to a lack of reporting on other outcomes (i.e., WI-NRS and Skindex-10 scores) and the absence of a control group.
Conclusions
The CADTH systematic review identified 2 phase III, double-blind, placebo-controlled RCTs (KALM-1 and KALM-2) that compared the efficacy and safety of difelikefalin 0.5 mcg/kg with placebo over 12 weeks in patients on hemodialysis with moderate-to-severe CKD-aP. In both of the KALM trials, patients randomized to difelikefalin were more likely than patients randomized to placebo to report an improvement in WI-NRS score or a clinically meaningful reduction in the intensity of the worst itch, based on a 3-point (primary end point) or 4-point (key secondary end point) improvement in the score at week 12. This corresponded to 38% to 41% of patients randomized to difelikefalin and 21% to 25% of patients randomized to placebo with a 4-point improvement in WI-NRS score in the 2 trials. Other outcomes related to pruritus severity, including the change from baseline in WI-NRS and the PGIC scores, were consistent with the primary end point. In the analyses of other secondary end points that assessed HRQoL, measured by the Skindex-10 and 5-D itch scales, treatment with difelikefalin was associated with an improvement in QoL in the KALM-1 trial, but statistical superiority for the same outcomes were not demonstrated in the KALM-2 trial. Generally, AEs were reported at similar rates in the difelikefalin and placebo groups. Treatment with difelikefalin revealed no new safety issues in either KALM trial. No evidence was identified in the KALM trials for mood, days of missed work, days of missed school, or days of missed dialysis, which were considered to be important outcomes of interest by patients and clinicians. A pooled analysis of the OLE studies of KALM-1 and KALM-2 and the phase III, 52-week, open-label study (CLIN3101) provided evidence of long-term safety associated with difelikefalin, and no new safety signals were reported, but the study was subject to many limitations, including the lack of comparative evidence and high discontinuation rates. Overall, based on evidence from the KALM trials, difelikefalin may reduce the severity of itch and improve HRQoL, compared with placebo, but there is uncertainty regarding the magnitude of the treatment effect for patients with moderate-to-severe CKD-aP.
Introduction
Disease Background
CKD is a progressive disease characterized by a gradual loss of renal function and/or abnormalities of renal structure over 3 months. CKD constitutes a major health burden worldwide and is associated with high levels of morbidity and mortality.2,3
CKD-aP, also known as uremic pruritus, is a CKD comorbidity that is a common, distressing, and underrecognized systemic itch that affects more than 60% of patients undergoing hemodialysis, with 20% to 40% of patients reporting moderate-to-severe pruritus.4-9 It is marked by an unpleasant sensation and the desire to scratch. The intensity of the itch sensation can fluctuate over time, from barely noticeable to relentless and disturbing.13 The distribution of the itch sensation across the body is also inconsistent. Approximately 50% of patients with CKD-aP report generalized pruritus that is usually symmetrically distributed, whereas the remainder report that the itch is mainly localized to the back, face, and dialysis shunt arm.13,22 The itch can occur either intermittently (i.e., sporadic discomfort) or persistently (i.e., constant itching), and it may present before, during, and/or after dialysis.13,22 Intense and generalized systemic itching in these patients is associated with poor sleep quality, depression, reduced QoL, increased risk of infection, and an increased risk of death.5,6,10-12 In Canada, the estimated overall prevalence of CKD-aP in adult hemodialysis patients is about 70%, according to the international, observational Dialysis Outcomes and Practice Patterns Study.8 Among patients with CKD on hemodialysis, more than one-third experience moderate-to-severe itch.8,13,14
The pathophysiology of CKD-aP is not fully understood. a multitude of hypotheses have been proposed, including systemic inflammation and an imbalance in the endogenous opioid system (e.g., overexpression of mu opioid receptors and concomitant downregulation of kappa opioid receptors). Opioid receptors are known to modulate itch signals and inflammation, with kappa opioid receptor activation reducing itch and producing immunomodulatory effects.11,22-25
The treatment of patients with CKD-aP involves clinical specialists such as nephrologists and dermatologists. There are currently no standardized lab or diagnostic tests for the diagnosis of CKD-aP. A complaint of pruritus in someone with a diagnosis of CKD is presumed to be CKD-aP unless assessment determines a different diagnosis.6
Standards of Therapy
There is no approved therapy for CKD-aP in Canada. The clinical experts stated that the goal of treatment is to reduce itch intensity, improve QoL and sleep quality, and decrease itch-related depression and suicidality.
Currently, the treatment for moderate-to-severe pruritus associated with CKD is heterogeneous, as there are no existing guidelines and/or frameworks. The standard of care for CKD-aP includes the use of topical moisturizing treatments, steroids in combination with menthol and camphor, topical calcineurin inhibitors, and the systemic use of gabapentin or pregabalin, naltrexone, thalidomide, biologics (i.e., dupilumab or tralokinumab), kappa opioid receptor agonists (i.e., difelikefalin), and topical phototherapy to reduce itch intensity and improve sleep quality, per feedback provided by the clinical experts consulted by CADTH. Generally, the clinical experts indicated that they would typically start with topical treatments and then move on to systemic drugs and phototherapy. The clinical experts stated that phototherapy is a useful treatment option in clinical practice, but only a small number of patients can receive the treatment due to limited access.
Difelikefalin is the first treatment approved in Canada for moderate-to-severe pruritus in patients on hemodialysis. The clinical experts indicated that difelikefalin would be used in combination with other therapies in a later-line setting when existing therapies are intolerable or have failed to control symptoms or contraindicated. The clinical experts stated that the prior lines of therapy include menthol-containing topical steroids and/or topical calcineurin inhibitors, systemic drugs (gabapentin or naltrexone), and phototherapy.
Drug
Difelikefalin is a selective kappa opioid receptor agonist that acts in the peripheral nervous system. The drug exerts antipruritic effects by means of activation of kappa opioid receptors on peripheral neurons and immune cells. The selective activity of difelikefalin on kappa opioid receptors mostly avoids mu opioid–associated side effects, such as respiratory depression, dependence, and euphoria. Difelikefalin is anticipated to have no meaningful abuse or dependence potential.1
This is the first CADTH review for difelikefalin. Difelikefalin was granted a Health Canada Notice of Compliance on August 16, 2022, for the indication of treatment of moderate-to-severe pruritus associated with CKD in adults on hemodialysis. Per the Health Canada product monograph, the recommended dose of difelikefalin is 0.5 mcg/kg dry body weight (i.e., the target postdialysis weight).1
The sponsor is seeking reimbursement of difelikefalin per the indication.
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups. The full original patient inputs received by CADTH have been included in the stakeholder section at the end of this report.
One patient advocacy group, The Kidney Foundation of Canada, provided input for the treatment of adults with CKD on hemodialysis. Patient input was gathered from independent surveys of people living with CKD and their caregivers across Canada in September 2022. A total of 19 responses were gathered from the survey (10 fully completed and 9 partially completed).
More than 90% of patients who responded to the survey reported experiencing itchy skin as part of their kidney disease, with 50% of respondents experiencing itchiness every day, 40% experiencing it several times per week, and 10% experiencing it occasionally. Although 60% of respondents reported living with pruritus for 1 to 2 years, 20% reported living with it for 2 to 5 years, and 20% reported living with it for more than 5 years. The itchiness was described as moderate-to-severe by 80% of respondents. When describing their disease experience, several respondents reported developing scabs and/or sores because of their itchy skin. Many respondents also reported having trouble sleeping as a result of itchiness. One-third (33%) of patients who responded to the survey reported taking medication to treat itchiness associated with kidney disease. Although 33% of respondents reported treatments being covered by their provincial drug plan, 67% reported paying out of pocket. Most survey respondents expressed satisfaction with their current medication or combination of treatments, but 33% reported being neither satisfied nor unsatisfied. However, more than 66% of respondents expressed uncertainty regarding the improvement of their skin appearance related to currently available treatment.
When describing their expectations for CKD therapies in general, patients who responded to the survey mentioned improvement in their well-being or QoL, with 90% of respondents hoping for increased energy. In addition, fewer hospital visits, less medication overall, and side effects and efficacy were important considerations. None of the respondents had experience with the drug under review.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of moderate-to-severe pruritus associated with CKD.
Unmet Needs
According to the clinical experts consulted by CADTH, treatment for moderate-to-severe pruritus associated with CKD includes the use of topical moisturizing treatments, steroids in combination with menthol and camphor, calcineurin inhibitors, and the systemic use of gabapentin or pregabalin, naltrexone, thalidomide, biologics (i.e., dupilumab or tralokinumab), kappa opioid receptor agonists (i.e., difelikefalin), and topical phototherapy to reduce itch intensity and improve sleep quality. The clinical experts stated that the goal of treatment is to reduce itch intensity, improve QoL and sleep quality, and decrease itch-related depression and, in some cases, suicidality. They also stated indicated that currently used off-label treatments do not adequately address these issues in all patients.
Place in Therapy
The clinical experts indicated that difelikefalin is a selective kappa opioid receptor agonist that acts in the peripheral nervous system, and thus the drug has fewer neuromodulation effects than nonselective kappa opioid receptor agonists. Difelikefalin is the first treatment approved in Canada for moderate-to-severe pruritus in patients on hemodialysis, although the clinical experts consulted by CADTH were uncertain about whether the drug would address the underlying disease process that causes pruritus, as the etiology of CKD-aP is not yet fully understood. The clinical experts indicated that difelikefalin would be used in combination with other therapies in a later-line setting in which existing therapies are intolerable, have failed to control symptoms, or are contraindicated. The clinical experts stated that the prior lines of therapy include menthol-containing topical steroids and/or topical calcineurin inhibitors, systemic drugs (gabapentin or naltrexone), and phototherapy. The clinical experts did not expect that difelikefalin would replace any treatments or cause a shift in the current treatment paradigm, but instead would be used after other more accessible topical and systemic treatment options have failed.
Patient Population
The clinical experts indicated that only patients with ESRD on hemodialysis and with moderate-to-severe pruritus who are not responsive to existing therapies would be candidates for treatment with difelikefalin. According to the clinical experts, the drug is not suitable for patients on peritoneal dialysis, with nondialysis CKD, or who have received a kidney transplant.
Assessing Response to Treatment
The clinical experts stated that clinical evaluations, such as the itch numeric rating scale, can help identify pruritus severity at baseline and assess response to treatment, but they are not typically used in clinical practice. The clinical experts indicated that it is important to consider other causes of pruritus in patients, such as dermatologic conditions, CKD mineral bone disease, hepatobiliary disorders, diabetes, and hematologic conditions, as patients with these conditions may not respond to difelikefalin. The clinical experts mentioned that it is possible that underdiagnosis may occur in clinical practice due to the lack of unique laboratory findings associated with CKD-aP.
Discontinuing Treatment
The clinical experts consulted by CADTH indicated that in clinical practice, subjective patient-reported improvement in symptoms is the primary outcome used to determine whether a patient is responding to treatment. The clinical experts highlighted that reductions in the frequency and/or severity of symptoms would be considered when evaluating response to treatment, along with improvements in sleep quality, depression, adherence to dialysis, depression and suicidality, and overall QoL. The clinical experts consulted by CADTH stated that a patient may discontinue treatment if there is a lack of response, and that this could be assessed at 12 weeks. The development of significant and persistent side effects, such as diarrhea, dizziness, and recalcitrant nausea and/or vomiting may also be cause for discontinuing treatment.
Prescribing Conditions
According to the clinical experts consulted by CADTH, difelikefalin would mostly be prescribed in dialysis units or clinic settings by nephrologists or other physicians.
Clinician Group Input
This section was prepared by CADTH staff based on the input provided by clinician groups. The full original clinician group inputs received by CADTH have been included in the stakeholder section at the end of this report.
Clinician group input on the review of difelikefalin for the treatment of moderate-to-severe pruritus associated with CKD in adults on hemodialysis was received from 3 clinician groups: Saskatchewan Kidney Doctors; the Hemodialysis Specialty Physician Group, Division of Nephrology, The Ottawa Hospital; and Division of Nephrology, Department of Medicine, Dalhousie University and Nova Scotia Health.
The clinician groups agreed that, currently, only off-label medications in Canada are available for the treatment of pruritus in patients with kidney disease. There have been some unmet needs, as current treatment options are unsatisfactory and not effective in reducing the symptom burden. Debilitating symptoms in most severe cases may lead to deterioration in QoL. The clinician groups mentioned the need for a new treatment option that would help relieve symptoms, but with better tolerability, affordability, and ease of administration. One clinician group, in particular, indicated that the most important treatment goals would be to have a therapy that would reduce or maintain the severity of itch below the threshold of clinical importance that is known to be the level above which itch negatively impacts QoL and outcomes important to patients.
The clinician groups mentioned that difelikefalin may cause a paradigm shift in the treatment of itching, with data from RCTs and pooled analyses demonstrating objective effectiveness in reducing the severity of the symptoms of CKD-associated (uremic) pruritus. Input from clinician groups provided different opinions about the place in therapy for difelikefalin. Some groups suggested that it would likely be used as a first-line therapy, whereas others recommended it as an add-on or second-line treatment. Given the route of administration of difelikefalin, the clinician groups pointed out that patients on hemodialysis with moderate-to-severe pruritus would benefit the most from this treatment. The clinician groups mentioned the possibility of underreporting or underdiagnosing CKD-aP, which was also pointed out by the clinical experts consulted by CADTH. Regarding the diagnosis of this indication, the clinician groups noted the lack of diagnostic tests. Although 1 group mentioned using clinical history and exclusion criteria to identify patients, other groups pointed out that the identification of patients and the severity of their symptoms can be done with self-administered questionnaires and screening tools (e.g., the 5-D itch, UP-Dial, and Skindex-10 scales). The clinician groups stated that the reduction in itch severity measured on a numeric rating scale is used to evaluate symptoms. According to the clinician groups, lack of a clinical meaningful response, as well as intolerable side effects, should be considered as discontinuation criteria. The clinician groups added that a meaningful response to treatment for this disease would be symptom reduction leading to better sleep, improved QoL, improved mood, and return to activities of daily living.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparator | |
In Canada, physicians prescribe lifestyle changes, topical treatments, and systemic treatments for patients with CKD-aP. In more severe cases, for which lifestyle changes and topical treatments are insufficient, patients with CKD-aP in Canada are treated with a basket of off-label, interchangeable, BSC therapies, including antihistamines, gabapentinoids, phototherapy, and/or antidepressants. Consequently, in this submission, which focused on patients with CKD and moderate-to-severe pruritus on hemodialysis, the comparators considered are that basket of BSC products and difelikefalin (which includes BSC). At the basis of the Health Canada–approved indication, the KALM trials demonstrate that patients can be treated with difelikefalin regardless of whether they are receiving concomitant antiitch BSC. That is, prescribing difelikefalin has minimal impact on BSC use. Considering that difelikefalin is not expected to significantly displace any currently reimbursed treatments, no other treatments were included in this submission. | Comment from the drug programs will inform CDEC deliberations. |
Initiation of therapy | |
Difelikefalin may be given as an additional therapy, with or without other drugs considered BSC (antihistamines, gabapentin, antidepressants), all of which are off-label. Difelikefalin is not expected to displace other drugs. | Comment from the drug programs will inform CDEC deliberations. |
Prescribing of therapy | |
IV administration in hospital following hemodialysis treatment. | Comment from the drug programs will inform CDEC deliberations |
Care provision issues | |
Hospital administration in the setting of hemodialysis treatment. | Comment from the drug programs will inform CDEC deliberations. |
System and economic issues | |
In the probabilistic base case, difelikefalin was dominant compared to BSC, with an absolute decrease in costs (mean incremental decrease of $2,223) while generating higher mean total QALYs (mean incremental QALYs of 0.08). The probabilistic base case ICER (discounted) of difelikefalin vs. BSC is therefore-$27,195 per QALY gained. In 84% of simulations, at a willingness-to-pay threshold of $100,000/QALY, difelikefalin was found to be a cost-effective treatment strategy. Results remained robust and consistent across all scenario analyses. A budget impact analysis was developed to determine the total drug costs and budget impact to CADTH-participating drug plans associated with funding difelikefalin for the treatment of moderate-to-severe pruritus associated with CKD in adults undergoing hemodialysis. Following listing, the aggregated incremental budget impact across CADTH-participating plans was estimated to be $1.5 million, $2.5 million, and $3.2 million in years 1, 2, and 3, respectively. | Comment from the drug programs will inform CDEC deliberations. |
The administration of difelikefalin requires single-use vials. More than 1 vial may be needed, depending on dry weight. Wastage was not included in the sensitivity analysis. What is the impact of both factors on the budget impact analysis? | The clinical experts consulted by CADTH indicated that the impact of wastage on the budget impact analysis would be minimal. The clinical expert mentioned that routinely used injections in dialysis, such as erythropoietin, do not require single-use vials but require proper storage for reuse. |
This is a hospital-administered drug by the IV route. | Comment from the drug programs will inform CDEC deliberations. |
BSC = best supportive care; CDEC = CADTH Canadian Drug Expert Committee; CKD = chronic kidney disease; CKD-aP = chronic kidney disease–associated pruritus; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Clinical Evidence
The clinical evidence included in the review of difelikefalin is presented in 2 sections. The first section, the Systematic Review, includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as studies that were selected according to an a priori protocol. The second section includes sponsor-submitted long-term extension studies and additional relevant studies that were considered to address important gaps in the evidence included in the systematic review.
Systematic Review (Pivotal and Protocol-Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of difelikefalin 0.5 mcg/kg IV 3 times per week for the treatment of moderate-to-severe pruritus associated with CKD in adults undergoing hemodialysis.
Methods
Studies selected for inclusion in the systematic review included pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 6. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
Table 5: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Population | Adults with moderate-to-severe pruritus associated with CKD who are on hemodialysis Subgroups:
|
Intervention | Difelikefalin 0.5 mcg/kg IV 3 times per week |
Comparator | Standard of care |
Outcomes | Efficacy outcomes:
Harms outcomes:
|
Study designs | Published and unpublished phase III and phase IV RCTs |
AE = adverse event; CKD = chronic kidney disease; HRQoL = quality of life; RCT = randomized controlled trial; SAE = serious adverse event; WDAE = withdrawal due to adverse event.
aThis outcome was identified as being of particular importance to patients in the input received by CADTH from patient groups.
The literature search was performed by an information specialist using a peer-reviewed search strategy in accordance with the PRESS Peer Review of Electronic Search Strategies checklist.26
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946–) via Ovid and Embase (1974–) via Ovid. All Ovid searches were run simultaneously as a multifile search. Duplicates were removed using Ovid deduplication for multifile searches, followed by manual deduplication in Endnote. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concepts were difelikefalin, Korsuva, and Kapruvia. The following clinical trials registries were searched: the US National Institutes of Health’s clinicaltrials.gov, WHO’s International Clinical Trials Registry Platform (ICTRP) search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
No filters were applied to limit the retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. Refer to Appendix 1 for the detailed search strategies.
The initial search was completed on October 20, 2022. Regular alerts updated the search until the meeting of the CADTH Canadian Drug Expert Committee (CDEC) on February 22, 2023.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool For Searching Health-Related Grey Literature checklist.27 Included in this search were the websites of regulatory agencies (FDA and European Medicines Agency). Google was used to search for additional internet-based materials. Refer to Appendix 1 for more information on the grey literature search strategy.
These searches were supplemented with a review of bibliographies of key papers and through contacts with appropriate experts. In addition, the manufacturer of the drug was contacted for information regarding unpublished studies.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
Findings From the Literature
A total of 2 studies were identified from the literature for inclusion in the systematic review (Figure 1). The included studies are summarized in Table 6. A list of excluded studies is presented in Appendix 2.
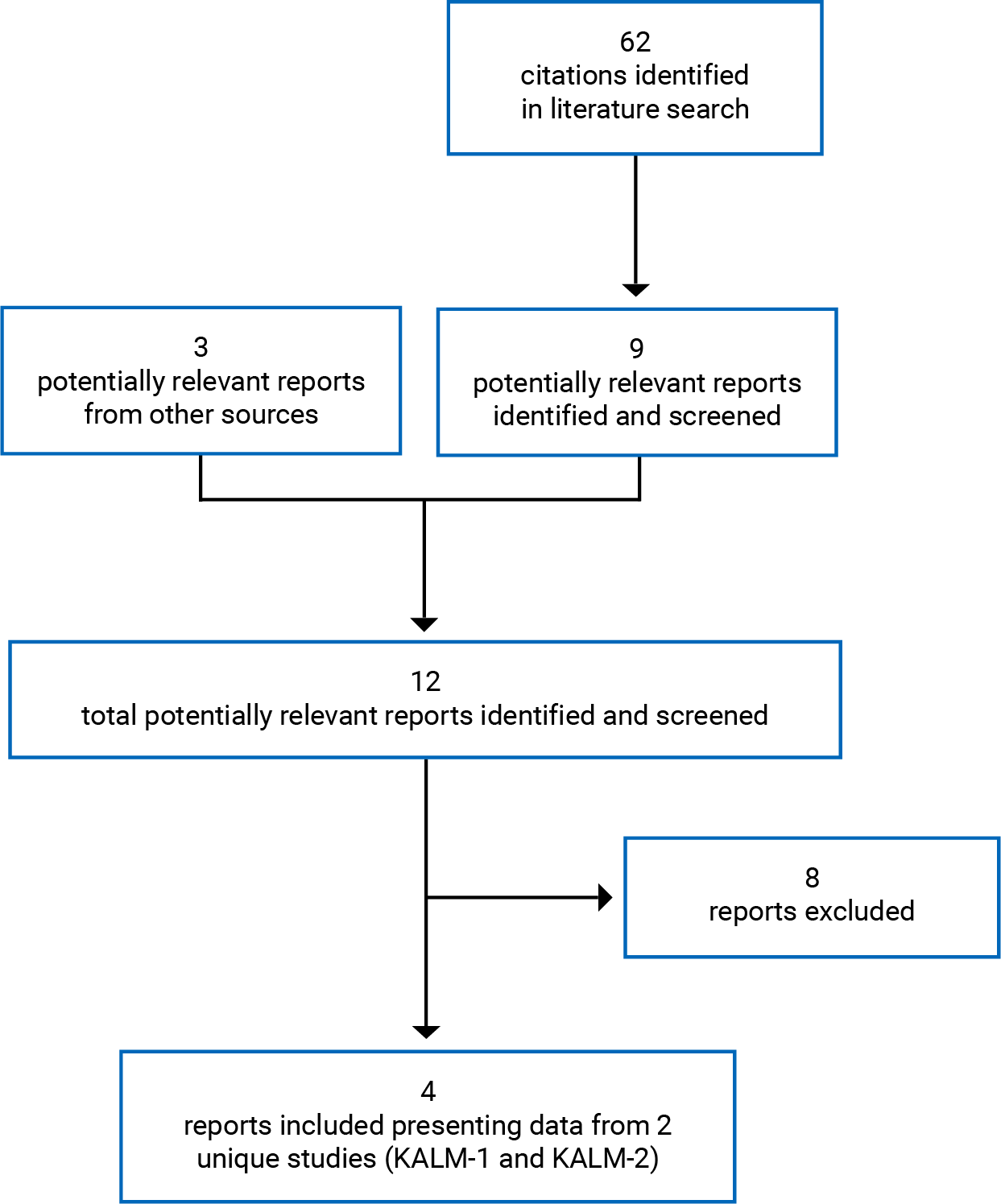
Table 6: Details of Included Studies
Details | KALM-1 | KALM-2 |
|---|---|---|
Designs and populations | ||
Study design | Phase III, multicentre, double-blind, randomized, placebo-controlled trial | |
Locations | US (80 sites) | Global (95 sites): Europe, US, Asia-Pacific, and Canada |
Study duration | Start date: February 20, 2018 Completion date: March 26, 2020 | Start date: July 17, 2018 Completion date: March 30, 2020 |
Randomized (N) | 378 | 471 |
Inclusion criteria |
| |
Has a mean baseline WI-NRS ≥ 4, which was indicative of moderate-to-severe uremic pruritus | Has a mean baseline WI-NRS ≥ 5, which was indicative of moderate-to-severe uremic pruritus | |
Exclusion criteria |
| |
Drugs | ||
Intervention | Difelikefalin 0.5 mcg/kg IV 3 times per week administered after each dialysis session | |
Comparator | Placebo IV 3 times per week administered after each dialysis session | |
Duration | ||
Phase | ||
Screening | 1 to 4 weeks | |
Run-in | 1 week | |
Double-blind | 12 weeks | |
Discontinuation | 2 weeks | NA |
Open-label extension | 52 weeks | |
Outcomes | ||
Primary end point | Proportion of patients with ≥ 3-point improvement from baseline in WI-NRS at week 12 of the double-blind treatment period | |
Secondary end points |
| |
NA | Proportion of patients with ≥ 3-point and ≥ 4-point improvement from baseline in WI-NRS at week 8 and week 4 of the double-blind treatment period | |
Exploratory end points | Itch-intensity end points:
Itch-related QoL end points:
| |
NA | Proportion of patients with ≥ 3 and ≥ 4-point improvement from baseline in WI-NRS at week 12 of the double-blind treatment period by region and dialysis type (each individually) | |
Notes | ||
Publications | Fishbane et al. (2020)28 | Wooldridge et al. (2020)29 |
ESRD = end-stage renal disease; Kt/V = dialyzer clearance of urea (K), dialysis time (t), volume of distribution of urea (V); NA = not applicable; PGIC = Patient Global Impression of Change; QoL = quality of life; WI-NRS = Worst Itching Intensity Numerical Rating Scale.
Sources: KALM-1 Clinical Study Report;19 KALM-2 Clinical Study Report;20 Fishbane et al. (2020);28 Wooldridge et al. (2020).29
Description of Studies
The primary objective for both KALM trials was to compare the efficacy of difelikefalin at a dose of 0.5 mcg/kg with placebo in reducing the intensity of itch in patients undergoing hemodialysis and experiencing moderate-to-severe pruritus. The shared key secondary objectives of the KALM-1 and KALM-2 trials were to compare the efficacy of difelikefalin at a dose of 0.5 mcg/kg with placebo in improving itch-related QoL and safety in patients on hemodialysis experiencing moderate-to-severe pruritus.
Both KALM trials were designed as multicentre, randomized, double-blind, phase III clinical trials to compare difelikefalin to placebo in patients being treated with hemodialysis who were experiencing moderate-to-severe pruritus. An overview of the study schematic for the KALM-1 and KALM-2 trials is presented in Figure 2 and Figure 3, respectively. The KALM-1 and KALM-2 studies included a double-blind phase and an OLE phase. The double-blind phase consisted of a screening visit (day –28 to day –7), a 7-day run-in period, and a 12-week double-blind treatment period. During the double-blind treatment period, difelikefalin was evaluated relative to placebo over 12 weeks. In the KALM-1 trial, there was a 2-week monitored discontinuation period after the double-blind treatment period during which patients did not receive any study drug. The KALM-2 trial did not have a discontinuation period. At the end of the discontinuation period in the KALM-1 trial and at the end of the double-blind treatment period in the KALM-2 trial, patients who had received at least 30 doses of the study drug during the 12-week double-blind treatment period had the option to receive open-label difelikefalin for up to an additional 52 weeks in the OLE phase. Results for the double-blind treatment period were presented and critically appraised in this report.
A total of 378 patients in the KALM-1 trial and 473 patients in the KALM-2 trial were randomized, in a 1:1 ratio, to treatment with difelikefalin or placebo. The KALM-1 trial was limited to study centres in the US and the KALM-2 trial was conducted globally and included 5 sites in Canada. The primary efficacy end point in the KALM-1 and KALM-2 trials was the percentage of patients at week 12 who achieved at least a 3-point improvement from baseline in the weekly mean score on the daily WI-NRS. Key secondary efficacy end points for both KALM trials were mean change from baseline at week 12 in itch-related QoL (measured with the 5-D itch scale total score and the Skindex-10 scale total score) and the percentage of patients at week 12 achieving at least a 4-point improvement from baseline in the weekly mean score of the daily WI-NRS. In the KALM-2 trial, the proportion of patients with at least a 3-point improvement and at least a 4-point improvement from baseline in WI-NRS at week 8 and week 4 were considered as key secondary outcomes.
For both KALM trials, randomization was based on the following 2 stratification variables:
the use or nonuse of concomitant medications to treat itch during the week before randomization
the presence or absence of specific medical conditions (i.e., history of falls or fracture or fall-related fracture, confusional state or mental status change or altered mental status or disorientation, or gait disturbance or movement disorder).
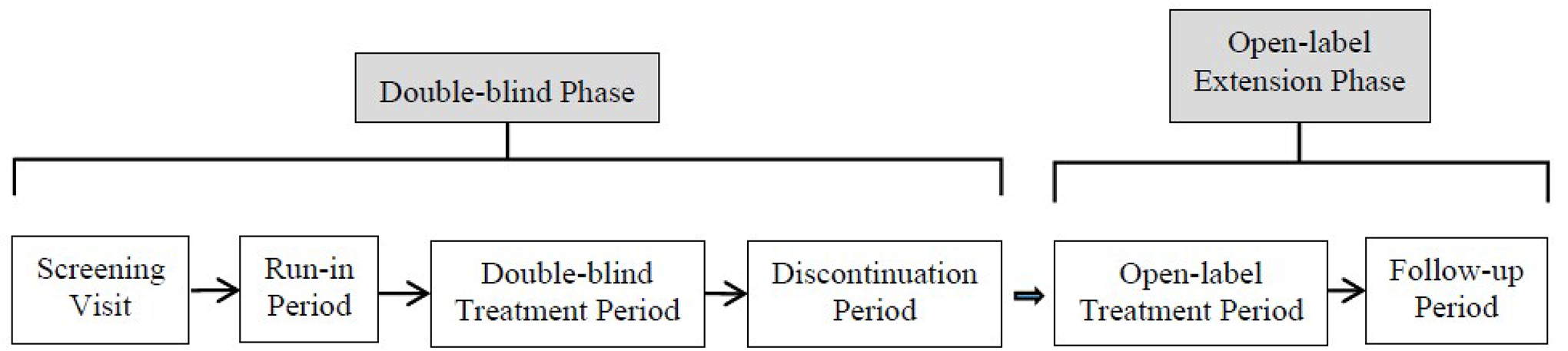
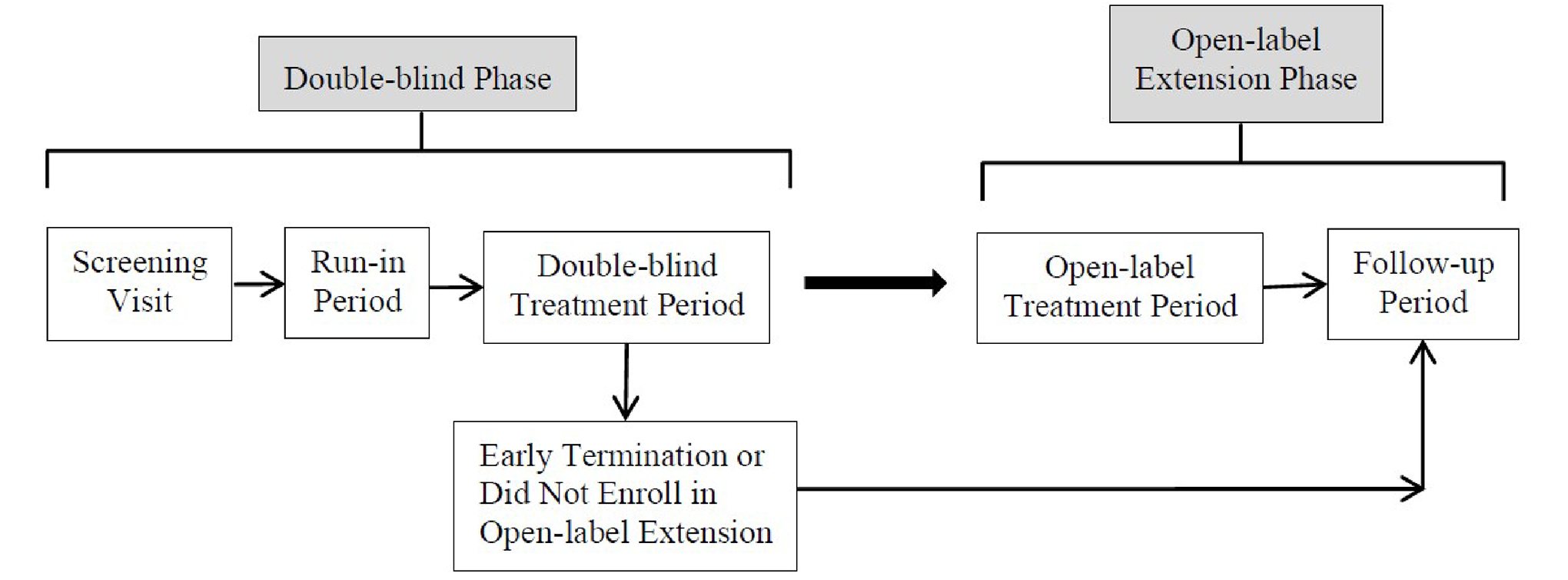
Populations
Inclusion and Exclusion Criteria
The key inclusion and exclusion criteria applied to the KALM-1 and KALM-2 trials are summarized in Table 6.
Eligible patients for both KALM trials had ESRD, had been on hemodialysis 3 times per week for at least 3 months before the start of screening, had moderate-to-severe pruritus, and had a body weight between 40.0 kg and 135.0 kg, inclusive. For the KALM-1 trial, eligible patients were 18 years and older, and for the KALM-2 trial, eligible patients were aged 18 to 85 years. The KALM-1 trial included patients with a mean baseline WI-NRS score of at least 4 and at least 4 of 8 worksheets completed in the run-in period, whereas the KALM-2 trial included patients with a mean baseline WI-NRS score of at least 5.
Patients were excluded from both KALM trials if they were with known to be noncompliant with dialysis treatments; scheduled to receive a kidney transplant during the study; started or changed treatment for itch, including antihistamines and corticosteroids (oral, IV, or topical) in the 14 days before screening; had received a new or changed prescription for opioids, gabapentin, or pregabalin in the 14 days before screening; received another investigational drug in the 30 days before the start of screening; or planned to participate in another clinical study while enrolled in this study. Patients who had pruritus attributed to a cause other than ESRD or its complications (e.g., patients with concomitant pruritic dermatological disease or cholestatic liver disease) were not enrolled. Patients who reported pruritus only during the dialysis session or who were receiving ongoing UV B therapy and anticipated receiving such treatment during the study were also excluded from both trials. Patients who participated in a previous clinical study with difelikefalin were excluded.
Baseline Characteristics
A summary of baseline characteristics for the KALM-1 and KALM-2 trials is presented in Table 7.
Baseline demographic characteristics were well balanced between the treatment arms in both KALM trials. The median age of all randomized patients was similar in the 2 studies, at 58.0 years (range, 22 to 88 years) in the KALM-1 trial and 60.0 years (range, 23 to 87 years) in the KALM-2 trial, and the majority of patients were older than 45 years (56.8% and 51.6%, respectively, were 45 to 64 years and 20.2% and 22.1%, respectively, were 65 to 74 years). Most of the patients in the KALM-1 and KALM-2 studies were male (61.0% and 58.2%, respectively), and the predominant races were white (48.8% and 70.3%, respectively) and Black or African American (41.6% and 19.3%, respectively), with Asian patients representing 3.4% and 6.8% of patients, respectively, and other races representing less than 3%. The median prescription dry body weight was 84.0 kg (range, 42.0 kg to 135.0 kg) in the KALM-1 trial and 78.0 kg (range, 42.0 kg to 135.0 kg) in the KALM-2 trial. With the exception that a greater percentage of patients was 75 years or older in the placebo group than in the difelikefalin group (10.6% versus 5.3%) in the KALM-1 trial, the treatment groups were comparable in terms of demographic characteristics in the 2 KALM trials.
In the KALM-1 and KALM-2 trials, the median baseline WI-NRS score was 7.14 (range, 4.1 to 10.0) and 7.13 (range, 4.5 to 10.0), respectively, and the vast majority of patients (91.8% and 99.6%, respectively) had a baseline WI-NRS score of 5 or greater. The mean baseline 5-D itch scale total score was 16.9 (standard deviation [SD] = 3.5) for difelikefalin and 17.9 (SD = 3.5) for placebo in the KALM-1 trial, and 16.7 (SD = 3.5) and 16.2 (SD = 3.3), respectively, in the KALM-2 trial. The mean baseline Skindex-10 scale total score was 36.2 (SD = 14.4) for difelikefalin and 38.3 (SD = 15.4) for placebo in the KALM-1 trial, and 35.5 (SD = 15.0) and 34.2 (SD = 14.7), respectively, in the KALM-2 trial. At baseline, 39.8% of patients in the KALM-1 trial and 36.5% in the KALM-2 trial were using antiitch medications, and 14.1% and 16.6%, respectively, reported at least 1 of the specified medical conditions (i.e., history of falls or fracture or fall-related fracture, confusional state or mental status change or altered mental status or disorientation, or gait disturbance or movement disorder). The median duration of CKD-aP for all patients was 2.5 years (range, 0.1 to 26.5 years) in the KALM-1 trial and 2.3 years (range, 0.0 to 58.4 years) in the KALM-2 trial. The median time interval since the diagnosis of CKD and ESRD for all patients was 5.45 years (range, 0.3 to 42.9 years) and 3.92 years (range, 0.3 to 28.7 years), respectively, in the KALM-1 trial, and 7.53 years (range, 0.3 to 48.3 years) and 4.03 years (range, 0.3 to 30.2 years) in the KALM-2 trial.
All patients in both KALM trials reported at least 1 prior medical condition. The most frequently reported medical conditions in the KALM-1 trial were hypertension (95.8%), anemia of CKD (73.7%), diabetes (62.9%), hyperphosphatemia (51.7%), secondary hyperparathyroidism (49.6%), gastroesophageal reflux disease (45.9%), hyperlipidemia (44.3%), nausea (41.6%), hypotension (40.8%), muscle spasms (39.0%), iron deficiency anemia (37.4%), constipation (35.3%), diarrhea (33.7%), and headache (31.0%). In the KALM-2 trial, the most frequently reported medical conditions were hypertension (95.8%), anemia of CKD (77.3%), hyperphosphatemia (59.0%), diabetes (55.6%), hyperparathyroidism secondary (54.8%), hyperlipidemia (38.0%), gastroesophageal reflux disease (34.4%), iron deficiency anemia (34.2%), and nausea (32.1%).
Table 7: Summary of Baseline Characteristics (Double-Blind Safety Analysis Set)
Characteristic | KALM-1 | KALM-2 | ||
|---|---|---|---|---|
Difelikefalin 0.5 mcg/kg N = 189 | Placebo N = 188 | Difelikefalin 0.5 mcg/kg N = 235 | Placebo N = 236 | |
Demographics | ||||
Age, years | ||||
Mean (SD) | 58.2 (11.16) | 56.8 (13.89) | 59.7 (13.11) | 59.6 (13.07) |
Median (range) | 59.0 (22.0 to 85.0) | 57.5 (24 to 88) | 61.0 (23 to 87) | 60.0 (24 to 85) |
Sex, n (%) | ||||
Female | 77 (40.7) | 70 (37.2) | 100 (42.6) | 97 (41.1) |
Male | 112 (59.3) | 118 (62.8) | 135 (57.4) | 139 (58.9) |
Race, n (%) | ||||
American Indian or Alaska Native | 6 (3.2) | 5 (2.7) | 1 (0.4) | 1 (0.4) |
Asian | 6 (3.2) | 7 (3.7) | 12 (5.1) | 20 (8.5) |
Black or African American | 82 (43.4) | 75 (39.9) | 53 (22.6) | 38 (16.1) |
Native Hawaiian or other Pacific Islander | 2 (1.1) | 4 (2.1) | 1 (0.4) | 3 (1.3) |
White | 91 (48.1) | 93 (49.5) | 162 (68.9) | 169 (71.6) |
Unknown | 1 (0.5) | 2 (1.1) | NR | NR |
Other | 1 (0.5) | 2 (1.1) | 6 (2.6) | 5 (2.1) |
Region, n (%) | ||||
US | 189 (100.0) | 188 (100.0) | 145 (61.7) | 133 (56.4) |
Asia | 0 | 0 | 8 (3.4) | 12 (5.1) |
Eastern Europe | 0 | 0 | 54 (23.0) | 60 (25.4) |
Western Europe and/or European origin | 0 | 0 | 28 (11.9) | 31 (13.1) |
Prescription dry body weight (kg) | ||||
Mean (SD) | 85.91 (20.26) | 84.98 (21.08) | 81.56 (19.73) | 79.95 (19.45) |
Median (range) | 84.00 (47.0 to 135.0) | 82.00 (42.0 to 135.0) | 79.50 (42.0 to 130.0) | 77.00 (44.0 to 135.0) |
Disease characteristics | ||||
Baseline WI-NRS | ||||
Mean (SD) | 7.06 (1.49) | 7.25 (1.61) | 7.27 (1.36) | 7.12 (1.36) |
Median (range) | 7.00 (4.2 to 10.0) | 7.44 (4.1 to 10.0) | 7.20 (4.5 to 10.0) | 7.00 (4.8 to 10.0) |
Patients with baseline WI-NRS score < 5 | || ||||| | || ||||| | | ||||| | | ||||| |
Patients with baseline WI-NRS score ≥ 5 | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| |
Baseline 5-D itch scale total score, mean (SD) | 16.9 (3.5) | 17.9 (3.5) | 16.7 (3.5) | 16.2 (3.3) |
Baseline Skindex-10 scale total score, mean (SD) | 36.2 (14.4) | 38.3 (15.4) | 35.5 (15.0) | 34.2 (14.7) |
Baseline antiitch medication use,a n (%) | ||||
Yes | 72 (38.1) | 78 (41.5) | 87 (37.0) | 85 (36.0) |
No | 117 (61.9) | 110 (58.5) | 148 (63.0) | 151 (64.0) |
Specific medical conditions,a n (%) | ||||
Yes | 25 (13.2) | 28 (14.9) | 41 (17.4) | 37 (15.7) |
No | 164 (86.8) | 160 (85.1) | 194 (82.6) | 199 (84.3) |
Duration of pruritus, years | ||||
Mean (SD) | 3.19 (3.24) | 3.45 (3.37) | 3.21 (4.57) | 3.20 (3.18) |
Median (range) | 2.20 (0.2 to 26.5) | 2.57 (0.1 to 24.3) | 2.03 (0.0 to 58.4) | 2.50 (0.0 to 23.2) |
Years since diagnosis of ESRD | ||||
Mean (SD) | 4.66 (3.90) | 5.66 (5.18) | 5.23 (4.68) | 5.46 (4.51) |
Median (range) | 3.67 (0.3 to 26.5) | 4.10 (0.3 to 28.7) | 3.97 (0.3 to 30.2) | 4.11 (0.3 to 27.9) |
Years since diagnosis of CKD | ||||
Mean (SD) | 6.92 (5.93) | 7.03 (5.74) | 9.28 (7.64) | 9.76 (7.01) |
Median (range) | 5.50 (0.5 to 42.9) | 5.45 (0.3 to 28.9) | 7.19 (0.3 to 46.3) | 7.85 (0.6 to 48.3) |
Years on chronic hemodialysis | ||||
Mean (SD) | 4.37 (3.98) | 4.73 (4.22) | 4.83 (4.59) | 5.09 (4.33) |
Median (range) | 3.27 (0.2 to 26.5) | 3.55 (0.0 to 22.9) | 3.68 (0.3 to 30.2) | 4.00 (0.3 to 27.9) |
Medical history (≥ 30% of patients), n (%) | ||||
Any medical history | ||| ||||||| | ||| ||||||| | |||||||| | |||||||| |
Hypertension | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| |
Anemia of CKD | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| |
Diabetes | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| |
Hyperphosphatemia | || |||||| | ||| |||||| | ||| |||||| | ||| |||||| |
Secondary hyperparathyroidism | || |||||| | || |||||| | ||| |||||| | ||| |||||| |
Gastroesophageal reflux disease | || |||||| | || |||||| | || |||||| | || |||||| |
Hyperlipidemia | || |||||| | || |||||| | || |||||| | || |||||| |
Nausea | || |||||| | || |||||| | || |||||| | || |||||| |
Hypotension | || |||||| | || |||||| | || |||||| | || |||||| |
Muscle spasms | || |||||| | || |||||| | || |||||| | || |||||| |
Iron deficiency anemia | || |||||| | || |||||| | || |||||| | || |||||| |
Constipation | || |||||| | || |||||| | || |||||| | || |||||| |
Diarrhea | || |||||| | || |||||| | || |||||| | || |||||| |
Headache | || |||||| | || |||||| | || |||||| | || |||||| |
CKD = chronic kidney disease; ESRD = end-stage renal disease; SD = standard deviation; WI-NRS = Worst Itching Intensity Numerical Rating Scale.
aIncluding history of falls or fracture (related to fall), confusional state or mental status change or altered mental status or disorientation, or gait disturbance or movement disorder. Observed stratum values.
Sources: KALM-1 Clinical Study Report;19 KALM-2 Clinical Study Report.20
Interventions
In each of the included studies, the intervention employed was difelikefalin 0.5 mcg/kg administered intravenously 3 times per week, after each hemodialysis session. The duration of treatment was up to 64 weeks, which included 12 weeks of double-blind treatment and 52 weeks of open-label treatment in both KALM trials. Patients were provided with difelikefalin as an IV bolus into the venous line of the hemodialysis circuit at the end of each hemodialysis session and could be given either during or after rinse back of the hemodialysis circuit. The dose of difelikefalin to be administered throughout the double-blind treatment period was calculated using the patient’s prescription dry body weight (i.e., the target postdialysis weight, determined by the patient’s nephrologist or at the dialysis unit during screening). If a patient received additional hemodialysis during a given week for any reason, an additional dose of difelikefalin or placebo was administered after hemodialysis. A maximum of 4 doses per week was allowed. No additional doses were given for patients receiving an additional unscheduled ultrafiltration treatment.
The KALM trials were double-blind, placebo-controlled, and used matching placebo vials for IV administration. The matching placebo (0.04 mol isotonic acetate buffer, pH = 4.5) was provided in 2 mL glass vials that contained a minimum extractable volume of 1.3 mL. The placebo buffer solution was composed of acetic acid, sodium acetate trihydrate, sodium chloride, hydrochloric acid, and water for injection. The difelikefalin buffer and placebo buffer were identical in appearance and were packaged, stored, and shipped in an identical manner.
Concomitant medications were defined as medications taken any time after the first dose of difelikefalin on day 1 of the double-blind treatment period through the end of the open-label treatment period or ET (i.e., EOT or ET visit). During either the double-blind phase or the OLE phase, no new medication to treat itch could be initiated.
A summary of concomitant medication and of concomitant antiitch medications use reported by patients in the included studies is presented in Table 8 and Table 9, respectively. In the KALM-1 trial, 99.7% and 100.0% patients randomized to difelikefalin and placebo, respectively, reported any concomitant medication use. The most frequently reported concomitant medications in the KALM-1 trial were beta-blocking drugs, antiparathyroid drugs, antithrombotic drugs, other analgesics and antipyretics, plain lipid-modifying drugs, drugs for peptic ulcer and gastroesophageal reflux disease, antihistamines, selective calcium channel blockers, vitamin A and D, and insulins and analogues. With regard to the use of concomitant antiitch medications, 45.0% and 50.0% of patients randomized to difelikefalin and placebo, respectively, reported using at least 1 concomitant antiitch medication. The most common antiitch medication used was diphenhydramine, which was used 33.3% of patients in the difelikefalin group and 37.8% in the placebo group. All other antiitch medications were used by less than 10.0% of patients and were comparable in the treatment groups.
In the KALM-2 trial, all patients reported concomitant medication use. The most common concomitant medications were antithrombotic drugs, beta-blocking drugs, other antianemic preparations, iron preparations, plain lipid-modifying drugs, drugs for peptic ulcer and gastroesophageal reflux disease, vitamin A and D, selective calcium channel blockers, antiparathyroid drugs, other analgesics and antipyretics, antihistamines, insulins and analogues, and high-ceiling diuretics. With regard to the use of concomitant antiitch medications, 39.1% and 38.6% of patients randomized to difelikefalin and placebo, respectively, reported using at least 1 concomitant antiitch medication. The most common antiitch medication, diphenhydramine, was used by a higher proportion of patients in the difelikefalin group (19.1%) than in the placebo group (11.0%). All other antiitch medications were used by less than 10.0% of patients and were comparable in the treatment groups.
Table 8: Common Concomitant Medications (in > 30% of Patients) (Double-Blind Safety Analysis Set)
Medication | KALM-1 | KALM-2 | ||
|---|---|---|---|---|
Difelikefalin 0.5 mcg/kg N = 189 | Placebo N = 188 | Difelikefalin 0.5 mcg/kg N = 235 | Placebo N = 236 | |
Any concomitant medications, n (%) | ||| |||||| | ||| ||||||| | ||| ||||||| | ||| ||||||| |
Beta-blocking drugs | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| |
Antiparathyroid drugs | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| |
Antithrombotic drugs | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| |
Other analgesics and antipyreticsa | ||| |||||| | ||| |||||| | || |||||| | || |||||| |
Lipid-modifying drugs, plain | || |||||| | || |||||| | ||| |||||| | ||| |||||| |
Selective calcium channel blockers with mainly vascular effects | || |||||| | || |||||| | ||| |||||| | ||| |||||| |
Antihistamines for systemic use | || |||||| | || |||||| | || |||||| | || |||||| |
Drugs for peptic ulcer and gastroesophageal reflux disease | || |||||| | ||| |||||| | ||| |||||| | ||| |||||| |
Insulins and analogues | || |||||| | || |||||| | || |||||| | || |||||| |
Vitamin A, vitamin D, or both | || |||||| | || |||||| | ||| |||||| | ||| |||||| |
Vitamin B complex, including combinations | || |||||| | || |||||| | || |||||| | || |||||| |
Antiemetics and antinauseants | || |||||| | || |||||| | || |||||| | || |||||| |
Opioids | || |||||| | || |||||| | || |||||| | || |||||| |
Other antianemic preparationsb | || | || | ||| |||||| | ||| |||||| |
Iron preparations | || |||||| | || |||||| | ||| |||||| | ||| |||||| |
High-ceiling diuretics | || |||||| | || |||||| | || |||||| | || |||||| |
NR = not reported.
aIncluding paracetamol.
bIncluding methoxy polyethylene glycol-epoetin beta and darbepoetin alfa.
Sources: KALM-1 Clinical Study Report;19 KALM-2 Clinical Study Report.20
Table 9: Concomitant Antiitch Medications (in > 2% of Patients) (Double-Blind Safety Analysis Set)
Drug class | KALM-1 | KALM-2 | ||||
|---|---|---|---|---|---|---|
Difelikefalin 0.5 mcg/kg N = 189 | Placebo N = 188 | Difelikefalin 0.5 mcg/kg N = 235 | Placebo N = 236 | |||
Any concomitant antiitch medications, n (%) | 85 (45.0) | 94 (50.0) | || |||||| | || |||||| | ||
Diphenhydramine | 63 (33.3) | 71 (37.8) | || |||||| | || |||||| | ||
Hydroxyzine | 18 (9.5) | 19 (10.1) | || ||||| | || |||||| | ||
Clemastine | NR | NR | | ||||| | || ||||| | ||
Hydrocortisone | 6 (3.2) | 8 (4.3) | | ||||| | | ||||| | ||
Cetirizine | NR | NR | | ||||| | | ||||| | ||
Triamcinolone | 5 (2.6) | 3 (1.6) | || | || | ||
Ammonium lactate | 2 (1.1) | 4 (2.1) | || | || | ||
Loratadine | NR | NR | | ||||| | | ||||| | ||
Chlorphenamine | NR | NR | | ||||| | | ||||| | ||
NR = not reported.
Sources: KALM-1 Clinical Study Report;19 KALM-2 Clinical Study Report.20
Outcomes
A list of efficacy end points identified in the CADTH review protocol that were assessed in the clinical trials included in this review is provided in Table 10 and subsequently summarized. A detailed discussion and critical appraisal of the outcome measures is provided in Appendix 4.
Table 10: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure | KALM-1 | KALM-2 |
|---|---|---|
Pruritus severity | ||
Proportion of patients with ≥ 3-point WI-NRS improvement at week 12 of the double-blind treatment period | Primary | Primary |
Proportion of patients with ≥ 4-point improvement from baseline in WI-NRS at week 12 of the double-blind treatment period | Secondary | Secondary |
Proportion of patients with ≥ 3-point and ≥ 4-point improvement from baseline in WI-NRS at week 8 and week 4 of the double-blind treatment period | Not specified | Secondary |
Proportion of patients reporting very much improved or much improved itch on PGIC at week 12 of the double-blind treatment period | Other | Other |
HRQoL | ||
Change from baseline in Skindex-10 scale total score at week 12 of the double-blind treatment period | Secondary | Secondary |
Change from baseline in each of the 3 Skindex-10 scale scores at week 12 of the double-blind treatment period | Other | Other |
Proportion of patients with ≥ 15-point improvement from baseline in Skindex-10 scale total score at week 12 of the double-blind treatment period | Other | Other |
Change from baseline in 5-D itch scale score at week 12 of the double-blind treatment period | Secondary | Secondary |
Change from baseline in the five 5-D itch scale domains at week 12 of the double-blind treatment period | Other | Other |
Proportion of patients with ≥ 5-point improvement from baseline in 5-D itch scale total score at week 12 of the double-blind treatment period | Other | Other |
HRQoL = health-related quality of life; PGIC = Patient Global Impression of Change; WI-NRS = Worst Itch Intensity Numerical Rating Scale.
Sources: KALM-1 Clinical Study Report;19 KALM-2 Clinical Study Report.20
Pruritus Severity
Worst Itching Intensity Numerical Rating Scale
Pruritus severity was measured using the WI-NRS on a worksheet that asked patients to indicate the intensity of the worst itching they experienced during the previous 24 hours by marking 1 of 11 numbers, from 0 (labelled with the anchor phrase, no itching) to 10 (labelled as worst itching imaginable), that best described it. Patients were provided with these worksheets to record their 24-hour worst itching assessment scores, both at the clinic on hemodialysis days and at home on nonhemodialysis days.19,20 The WI-NRS has been widely used for evaluation of chronic itch, including CKD-aP.5,10,30 The phase II CR845-CLIN2101 study showed that a 3-point difference in the WI-NRS score from baseline was a clinically meaningful improvement.31 This threshold was validated in other studies.15,30
Patient Global Impression of Change
The PGIC is a global patient-reported outcome measure that assesses the overall change in itch (no change, improvement, or worsening) relative to the start of the study.32 The scale has only 1 item; the patient is asked to mark the category that best describes the change in itch, ranging from very much improved to very much worse. The PGIC has been used to evaluate the threshold of meaningful change within pruritus populations for the WI-NRS, based on the FDA recommendation.30,33 No validity, reliability, or responsiveness outcome results in the CKD-aP population were found from the literature search. No MID results could be validated either.
Health-Related Quality of Life
Skindex-10 Scale
The Skindex-10 scale is an instrument for the measurement of QoL that correlates with itch intensity.5 Patients are asked to mark 1 of 7 boxes, numbered from 0 (labelled with the anchor phrase, “never bothered”) to 6 (labelled as “always bothered”) for each of the 10 questions asking how often they had been bothered by their itch and its impact over the previous week. The total score is the sum of the numeric value of each answered question. The total score is subdivided into 3 domain scores, which are sums of the scores for the following question topics: disease domain (questions 1 to 3), mood/emotional distress domain (questions 4 to 6), and social functioning domain (questions 7 to 10).19,20 The phase II CR845-CLIN2101 study conducted by the sponsor showed that a 15-point difference in the Skindex-10 total score from baseline represented a clinically meaningful improvement.19,20,31 In a hemodialysis setting, among 103 patients with moderate-to-severe CKD-aP, changes of at least 20% on a visual analogue scale that measured itching intensity was considered clinically meaningful, which is associated with a 3-point to 12-point change on the Skindex-10 scale in patients on hemodialysis.5
5-D Itch Scale
The 5-D itch scale was developed as a brief multidimensional questionnaire designed to be useful as an outcome measure in clinical studies. The scale has been validated in patients with chronic pruritus, including patients undergoing hemodialysis, and has been shown to be sensitive to changes in pruritus over time.18 The 5 dimensions of itch assessed by the scale are degree, duration, direction, disability, and distribution. Patients are asked to mark boxes that best describe the impact of their itch over the previous 2 weeks. The phase II CR845-CLIN2101 study conducted by the sponsor showed that a 5-point difference in the total 5-D itch score from baseline represented a clinically meaningful improvement.19,20 The threshold for MID was not found from the literature search conducted by CADTH for the CKD-aP population.
Safety
TEAEs included any AE that occurred after the start of the study drug administration and any AE that was present at baseline but worsened in severity after the start of the study drug administration. SAEs included any event that resulted in death, was life-threatening, required hospitalization or prolongation of existing hospitalization, resulted in persistent or significant disability, was a congenital anomaly, and was an important medical event that may have jeopardized the patient or required intervention to prevent any of those outcomes.
Statistical Analysis
A summary of the statistical analyses of efficacy outcomes in the KALM-1 and KALM-2 trials is shown in Table 11.
Sample Size and Power Calculation
The sample size calculations for the KALM-1 and KALM-2 trials are based on the results of a completed phase II, double-blind, placebo-controlled study of difelikefalin in patients with ESRD on hemodialysis who had moderate-to-severe pruritus.31 In that phase II study, 30% of patients randomized to the placebo group reported at least a 3-point improvement from baseline with respect to the 24-hour WI-NRS score at the EOT (week 8). The proportion of patients who received difelikefalin and reported a similar improvement in itch scores ranged from approximately 60% to 45% (i.e., a 30% to 15% difference from placebo), depending on the dose of the active study drug (0.5 mcg/kg, 1.0 mcg/kg, or 1.5 mcg/kg).31
Assuming a true response rate of 30% for the placebo group and a true response rate of 50% for the difelikefalin group (defining response as an improvement from baseline of ≥ 3 points with respect to the WI-NRS at week 12), the KALM-1 and KALM-2 trials required a sample size of 350 patients (175 per treatment group) to be randomized in a 1:1 ratio to difelikefalin or placebo to detect a treatment difference of 0.2 at the alpha of 0.05 with a power of 96% using a 2-sided continuity corrected Chi-square test. The power of this test statistic would be 84% or greater for differences from placebo as low as 0.16 in both trials.
Interim Analysis and Sample Size Re-estimation
There was 1 planned unblinded interim analysis conducted by an Independent Data Monitoring Committee for both KALM trials for sample size re-estimation. The sample size in the KALM-1 and KALM-2 trials could be increased to 500 patients (250 per treatment group) or keep the original sample size based on the results of the unblinded interim assessment. The interim analysis was conducted when 175 (50%) of the first 350 patients had been randomized and had either completed the 12-week treatment period or had discontinued treatment early. The re-estimation of the sample size was conducted using a conditional power approach, which used the primary end point (the proportion of patients achieving a ≥ 3-point improvement from baseline with respect to the weekly mean of daily 24-hour WI-NRS scores at week 12 of the double-blind treatment period) and 1 key secondary end point (the proportion of patients achieving a ≥ 4-point improvement from baseline with respect to the weekly mean of daily 24-hour WI-NRS scores at week 12 of the double-blind treatment period) as conditions.34 A weighted test statistic, the Cui, Hung. and Wang (CHW) approach, in which the z score is a weighted average of the z score at the interim and the z score observed for data collected after the interim was used to calculate the final P value.35 The Lawrence and Hung approach was used to calculate the point estimates and CIs.36 After the interim analysis, the Independent Data Monitoring Committee recommended that the original sample size be kept.
Multiplicity Adjustment
In both KALM trials, testing of the primary efficacy end point was 2-sided and conducted at the 5% error level. The study was considered positive if the null hypothesis of no treatment difference in the primary efficacy analysis of the primary end point was rejected (i.e., deemed statistically significant) in favour of the alternative, in which patients randomized to difelikefalin experienced significantly less itching than patients randomized to placebo.
To protect against type I errors, a sequential gate-keeping strategy was implemented in both KALM trials. Although the P values corresponding to the hypothesis testing of the secondary variables were reported, they were only considered inferential if the primary analysis was statistically significant. Testing of the secondary efficacy end points was performed sequentially at a 2-sided 5% error level. If the test of an end point in the sequence was not statistically significant, the P value for the tests corresponding to the remaining end points in the sequence would not be considered inferential and the null hypotheses for the subsequent tests would not be rejected.
The testing order for secondary efficacy end points in the KALM-1 trial is as follows:
Difference between patients assigned to difelikefalin and placebo with respect to the 5-D itch scale total score.
Difference between patients assigned to difelikefalin and placebo with respect to the Skindex-10 scale total score.
The proportion of patients achieving at least a 4-point improvement from baseline with respect to the weekly mean of daily 24-hour WI-NRS scores at week 12 of the double-blind treatment period.
The testing order for secondary efficacy end points in the KALM-2 trial is as follows:
The proportion of patients achieving at least a 4-point improvement from baseline with respect to the weekly mean of daily 24-hour WI-NRS scores at week 12 of the double-blind treatment period will be tested first.
The proportion of patients achieving at least a 3-point improvement from baseline with respect to the WI-NRS at week 8 will be tested next, followed by week 4 testing.
Testing will continue with the proportion of patients achieving at least a 4-point improvement at week 8, followed by week 4, in an identical manner.
Change from baseline in the Skindex-10 scale total score at week 12 of the double-blind treatment period.
Change from baseline in 5-D itch scale total scores at week 12 of the double-blind treatment period.
Primary Efficacy Analysis
In both KALM trials, the primary end points, the proportion of patients who have an improvement from baseline with respect to the weekly mean of daily 24-hour WI-NRS scores of 3 points or greater, were calculated using the ITT datasets. Differences between difelikefalin and placebo were compared using a logistic regression model that contained terms for treatment group, baseline WI-NRS score, use of antiitch medication during the week before randomization, and the presence of specific medical conditions. For the KALM-2 trial, region was also included in the logistic regression model for the primary efficacy analysis. A summary of the statistical analysis of primary efficacy end points is included in the description provided in Table 11.
Data Imputation
In both KALM trials, patients had to report at least 4 daily values for a week for the weekly value to be considered as nonmissing. Missing WI-NRS data for the primary efficacy analysis at the end of week 12 was imputed using a multiple imputation approach, under the assumption that patients who discontinue double-blind treatment early would have similar WI-NRS scores as other patients in their respective treatment arm who have complete data. The multiple imputation with the missing-at-random assumption was performed using logistic regression within treatment group, with covariates for baseline WI-NRS score, randomization stratification factors (i.e., the use or nonuse of concomitant antiitch medications and the presence or absence of a specific medical condition), and the nonmissing WI-NRS scores for each week. For the KALM-2 trial, region was also included in the multiple imputation. In addition, multiple imputation was performed separately for the interim analysis cohort and the postinterim analysis cohort. The missing data were imputed 20 times.
Sensitivity Analyses
Three sensitivity analyses of the primary efficacy end point were conducted to evaluate the robustness of the study results under different assumptions and imputation algorithms (described here) for both KALM trials. For each of the following sensitivity analyses, the final P value was calculated using the CWH procedure.
Sensitivity analysis under assumption 1: Early discontinuations as nonresponders. Patients who discontinue the study drug early were considered to be nonresponders (including patients who discontinued the study drug but continued to report WI-NRS scores). Patients who did not discontinue but who had missing week 12 data were imputed via multiple imputation, as was done in the primary analysis. The imputed data were analyzed using a logistic regression model similar to the 1 used in the primary analysis.
Sensitivity analysis under assumption 2: Multiple imputation with the missing-not-at-random assumption. This sensitivity analysis is an implementation of a pattern mixture model that draws from different populations based on the reason for withdrawal.
Intermittent missing WI-NRS scores were first imputed using the Markov chain Monte Carlo method.
For patients who discontinued due to AEs, WI-NRS scores missing after discontinuation were imputed using the distribution of the baseline value of all patients’ daily worst itching score, assuming a trimmed normal (from 4 to 10).
For patients who discontinued for reasons other than an AE, missing WI-NRS scores after patients discontinued were multiply imputed using data from patients in the same treatment group who had complete data at that time.
Sensitivity analysis under assumption 3: Tipping point analysis. Multiple imputation with mixed missing data mechanisms (missing not at random for difelikefalin and missing at random for placebo) were used to assess the robustness of the missing-at-random assumption. This sensitivity analysis investigates the departure from the missing-at-random assumption by progressively decreasing the treatment differences with respect to WI-NRS scores over the missing visits in the active treatment group until a conclusion from the primary analysis is overturned. This sensitivity analysis was applied to only week 12 values.
Markov chain Monte Carlo methodology was used to impute the intermittent missing data to a monotone missing pattern.
A standard missing-at-random–based multiple imputation approach was used to impute data from monotone missing data.
For patients in the active treatment group, a shift parameter was progressively applied to impute the missing data, until the P value > 0.05.
Secondary Efficacy Analyses
For both KALM trials, the secondary efficacy analyses were performed in the ITT and PP populations. The 5-D itch scale and the Skindex-10 scale scores were analyzed using an analysis of covariance (ANCOVA) and/or a mixed effects model with repeated measures (MMRM). The ANCOVA contains treatment as fixed effects, with baseline score and randomization stratification variables as covariates. The MMRM model contains treatment, week, and treatment-by-week interaction as fixed effects, and baseline score and randomization stratification variables as covariates. The baseline 5-D Itch scale score and the Skindex-10 scale total score were defined as the value collected on day 1, before randomization. Repeated measures included values assigned at the end of weeks 4, 8, 10, and 12 (EOT). A summary of the statistical analyses of secondary efficacy end points is included in the description provided in Table 11.
For both KALM trials, standard descriptive statistics were reported at each time point for the values and changes from baseline, along with the LS means, standard errors, 95% CIs, and differences between treatment groups reported with LS means, standard errors, and 95% CIs.
Missing data were handled implicitly in the MMRM or explicitly with multiple imputation, where intermittent missing WI-NRS scores were first imputed using the Markov chain Monte Carlo method and the monotone missing WI-NRS values were then imputed using the regression method for both KALM trials. For each domain in the 5-D itch scale and the Skindex-10 scale, missing values at week 12 were imputed using a multiple imputation approach, assuming that patients who discontinued double-blind treatment early would have 5-D itch and Skindex-10 scores similar to other patients in their respective treatment arm who have complete data. All available visits were included in the multiple imputation to better inform the week 12 imputed results. The multiple imputation approach was used in the ITT population.
Other Efficacy Analysis
In both pivotal trials, other efficacy analyses were tested in a nonhierarchical fashion without adjustments for multiplicity. Methods used and data reporting 5-D itch scale scores and Skindex-10 scale scores were identical to the secondary efficacy analyses previously mentioned. The treatment difference between difelikefalin and placebo was tested using the Cochran-Mantel-Haenszel exact test with respect to the PGIC, adjusted for the randomization stratification variables. The Mantel-Haenszel estimate of common odds ratio, the exact 95% CI for the common odds ratio, and the Cochran-Mantel-Haenszel exact test P value were reported for this treatment comparison. Additionally, the exact Clopper-Pearson 95% CIs for the proportion of patients who rated their itch condition as very much improved or much improved were reported.
Subgroup Analyses
The subgroup analyses for both KALM trials were prespecified and based on age (< 65 years and ≥ 65 years), sex, race (white, Black or African American, and other), prior antiitch medication use, specific medical conditions, and country (US and outside the US) for the primary end point (i.e., proportion of patients with a ≥ 3-point improvement in WI-NRS from baseline at week 12) and key secondary end point (i.e., proportion of patients with a ≥ 4-point improvement in WI-NRS from baseline at week 12). Subgroup analyses by use of concomitant medications (e.g., antihistamines, gabapentin, pregabalin) to treat pruritus, comorbid conditions, or pruritus severity were included in the CADTH systematic review protocol. None of the subgroups analyzed in the KALM-1 and KALM-2 trials were aligned with those of interest to this review.
Table 11: Statistical Analysis of Efficacy End Points
End point | Statistical model | Adjustment factors | Sensitivity analyses |
|---|---|---|---|
KALM-1 | |||
Proportion of patients with a ≥ 3-point improvement from baseline in WI-NRS at week 12 of the double-blind treatment period | Logistic regression | Treatment group, baseline WI-NRS score, use of antiitch medication during the week before randomization, and presence of specific medical conditions |
|
Proportion of patients with a ≥ 3-point improvement from baseline in WI-NRS at weeks 8 and 4 of the double-blind treatment period | Not specified | ||
Proportion of patients with a ≥ 4-point improvement from baseline in WI-NRS at weeks 12, 8, and 4 of the double-blind treatment period | |||
Change from baseline in total Skindex-10 scale score at week 12 of the double-blind treatment period | ANCOVA (for each domain in each questionnaire, missing values at week 12 were imputed using a multiple imputation approach, assuming that patients who discontinued double-blind treatment early would have 5-D itch and Skindex-10 scores similar to other patients in their respective treatment arm who have complete data) | Treatment as fixed effects, with baseline score, use of antiitch medication during the week before randomization, and presence of specific medical conditions variables as covariates | MMRM sensitivity analysis with no imputation |
Change from baseline in 5-D itch scale total score at week 12 of the double-blind treatment period | |||
Change from baseline in each of the 3 Skindex-10 scale domain scores at week 12 of the double-blind treatment period | |||
Change from baseline in the five 5-D itch scale domain scores at week 12 of the double-blind treatment period | |||
Proportion of patients with a ≥ 15-point improvement from baseline in Skindex-10 scale total score at week 12 of the double-blind treatment period | Not specified | ||
Proportion of patients with a ≥ 5-point improvement from baseline in 5-D itch scale total score at week 12 of the double-blind treatment period | |||
Proportion of patients very much improved or much improved on the PGIC scale at week 12 of the double-blind treatment period | CMH exact test | Use of antiitch medication during the week before randomization and presence of specific medical conditions | Not specified |
KALM-2 | |||
Proportion of patients with a ≥ 3-point improvement from baseline in WI-NRS at week 12 of the double-blind treatment period | Logistic regression | Treatment group, baseline WI-NRS score, region, use of antiitch medication during the week before randomization, and presence of specific medical conditions |
|
Proportion of patients with a ≥ 4-point improvement from baseline in WI-NRS at week 12 of the double-blind treatment period | |||
Proportion of patients with a ≥ 3 and ≥ 4-point improvement from baseline in WI-NRS at week 8 and week 4 of the double-blind treatment period | |||
Change from baseline in Skindex-10 scale total score at week 12 of the double-blind treatment period | ANCOVA (For each domain in each questionnaire, missing values at Week 12 will be imputed using a multiple imputation approach, assuming that patients who discontinue double-blind treatment early would have similar 5-D itch and Skindex-10 scores as other patients in their respective treatment arm that has complete data.) | Treatment as fixed effects, with baseline score, use of antiitch medication during the week before randomization, and presence of specific medical conditions |
|
Change from baseline in 5-D itch scale total score at week 12 of the double-blind treatment period | |||
Change from baseline in each of the 3 Skindex-10 scale domain scores at week 12 of the double-blind treatment period | MMRM sensitivity analysis with no missing data imputation | ||
Change from baseline in the five 5-D itch scale domain scores at week 12 of the double-blind treatment period | |||
Proportion of patients with a ≥ 15-point improvement from baseline in Skindex-10 scale total score at week 12 of the double-blind treatment period | Not specified | ||
Proportion of patients with a ≥ 5-point improvement from baseline in 5-D itch scale total score at week 12 of the double-blind treatment period | |||
Proportion of patients very much improved or much improved on the PGIC scale at week 12 of the double-blind treatment period | CMH exact test | Use of antiitch medication during the week before randomization and presence of specific medical conditions | Not specified |
ANCOVA = analysis of covariance; CMH = Cochran-Mantel-Haenszel; MMRM = mixed effects model with repeated measures; PGIC = Patient Global Impression of Change; WI-NRS = Worst Itching Intensity Numerical Rating Scale.
Analysis Populations
Six analysis sets were used for the KALM-1 study: the enrolled set, the ITT set, the double-blind safety set, the double-blind discontinuation set, the double-blind discontinuation safety set, and the PP set.
Four analysis sets were used for the KALM-2 study: the enrolled set, the ITT set, the double-blind safety set, and the PP set.
The definitions of each analysis set are as follows:
The enrolled set is defined as the group of patients who sign an informed consent form.
The ITT set is defined as the group of patients randomized to a treatment group. Following the ITT principle, patients in the ITT set were analyzed according to their randomized treatment, regardless of the actual treatment received. The ITT set was used to analyze all efficacy end points collected during the double-blind phase.
The double-blind safety set is defined as the group of randomized patients who received at least 1 dose of the double-blind study drug during the double-blind treatment period. Patients in the double-blind safety set were analyzed according to the actual treatment received. The double-blind safety set was used to analyze all safety end points collected during the double-blind phase.
The double-blind discontinuation safety set is defined as the subset of patients in the double-blind safety set who have at least 1 visit in the discontinuation period. The double-blind discontinuation safety set was used to analyze all end points collected during the discontinuation period. The double-blind discontinuation safety set was not reviewed in this report due to limited relevance.
The double-blind discontinuation set is defined as the subset of patients in the double-blind safety set who have completed 12 weeks of treatment, have received at least 6 doses in the 2 weeks before the start of the discontinuation period, and have at least 1 visit in the discontinuation period. The double-blind discontinuation set repeated analyses on the end points related to drug withdrawal that are also presented for the double-blind discontinuation safety population. The double-blind discontinuation set was not reviewed in this report due to limited relevance.
The PP set is defined as the subset of patients in the ITT population who do not have any major protocol deviations that could affect the efficacy analyses of the double-blind data.
Results
Patient Disposition
A summary of patient disposition for the KALM-1 and KALM-2 trials is shown Table 12.
Of the 503 screened patients in the KALM-1 trial, 378 patients were randomized, 189 to the difelikefalin group and 189 to the placebo group. The proportion of patients who completed the double-blind treatment period was 85.7% for the difelikefalin group and 90.4% for the placebo group. A greater proportion of patients discontinued early from the double-blind treatment period in the difelikefalin group than in the placebo group (14.3% versus 9.6%). The most common reasons for early discontinuation from the double-blind treatment period were AEs (6.1%) and patient withdrawal of consent (3.7%). The most frequently reported TEAEs leading to study drug discontinuation were septic shock (0.8%), dizziness (0.8%), and pneumonia (0.5%). With respect to treatment group differences in reasons for early discontinuation, only the proportion of patients who discontinued due to an AE was greater in the difelikefalin group than in the placebo group (7.4% versus 4.8%).
Of 620 screened patients in the KALM-2 trial, 473 patients were randomized, 237 to the difelikefalin group and 236 to the placebo group. The proportion of patients who completed the double-blind treatment period was 87.7% for the difelikefalin group and 94.5% for the placebo group. A greater proportion of patients discontinued early from the double-blind treatment period in the difelikefalin group than in the placebo group (12.3% versus 5.5%). The most common reason for early discontinuation from the double-blind treatment period was AEs (4.2%), followed by other (1.9%) and patient withdrawal of consent (1.3%). The most frequently reported TEAEs leading to study drug discontinuation were anxiety (0.9%) and insomnia (0.9%) in the difelikefalin group. A higher proportion of patients in the difelikefalin group than in the placebo group discontinued due to AEs (5.5% versus 3.0%), withdrew consent (2.1% versus 0.4%), or discontinued for other reasons (2.6% versus 1.3%).
Table 12: Patient Disposition (All Patients, Enrolled Analysis Set)
Patient disposition | KALM-1 | KALM-2 | ||
|---|---|---|---|---|
Difelikefalin 0.5 mcg/kg | Placebo | Difelikefalin 0.5 mcg/kg | Placebo | |
Screened, n | 503 | 620 | ||
Randomized, n (%) | 189 | 189 | 237 | 236 |
Treated in double-blind phase | 189 | 188 | 235 | 236 |
Completed the double-blind treatment period, n (%) | 162 (85.7) | 170 (90.4) | 206 (87.7) | 223 (94.5) |
Discontinued the double-blind treatment period, n (%) | 27 (14.3) | 18 (9.6) | 29 (12.3) | 13 (5.5) |
Reason for discontinuation, n (%) | ||||
Adverse events | 14 (7.4) | 9 (4.8) | 13 (5.5) | 7 (3.0) |
Lack of therapeutic efficacy | 0 | 0 | 1 (0.4) | 0 |
Lost to follow-up | 0 | 0 | 1 (0.4) | 0 |
Pregnancy | 0 | 0 | 0 | 0 |
Eligibility (inclusion and exclusion criteria) | 1 (0.5) | 2 (1.1) | 2 (0.9) | 0 |
Noncompliance | 1 (0.5) | 1 (0.5) | 1 (0.4) | 2 (0.8) |
Withdrew consent | 8 (4.2) | 6 (3.2) | 5 (2.1) | 1 (0.4) |
Administrative | 0 | 0 | 0 | 0 |
Other | 3 (1.6) | 0 | 6 (2.6) | 3 (1.3) |
ITT analysis set, n | 189 | 189 | 237 | 236 |
PP analysis set, n | 163 | 169 | 205 | 213 |
Double-blind safety analysis set, n | 189 | 188 | 235 | 236 |
ITT = intention to treat; PP = per protocol.
Sources: KALM-1 Clinical Study Report;19 KALM-2 Clinical Study Report.20
Protocol Deviation
The incidence of major and minor protocol deviations in the KALM-1 and KALM-2 trials is shown in Table 13. The proportion of patients who reported at least 1 major protocol deviation was ||||| for the difelikefalin group and ||||| for the placebo group in the KALM-1 trial, and ||||| for the difelikefalin group and ||||| for the placebo group in the KALM-2 trial. In the KALM-1 trial, the most frequently identified categories of major deviations were informed consent |||||||, investigational product accountability management |||||||, delegation of authority |||||||, more than 25% of WI-NRS scores missing |||||||, tests and/or assessments and/or procedure |||||||, no dosing in either week 11 or week 12 || ||||||, and receiving less than ||| of planned study doses |||||||. In the KALM-2 trial, the most frequently identified categories of major deviations were dosing noncompliance ||||||||, procedure not performed ||||||||, and procedure performed out of window |||||||. The proportion of patients who reported at least 1 minor protocol deviation was ||||| for the difelikefalin group and ||||| for the placebo group in the KALM-1 trial, and ||||| for the difelikefalin group and ||||| for the placebo group in the KALM-2 trial, which is comparable between the treatment groups in the 2 KALM trials.
Table 13: Protocol Deviations (Double-Blind Safety Analysis Set)
Protocol deviations | KALM-1 | KALM-2 | ||
|---|---|---|---|---|
Difelikefalin 0.5 mcg/kg N = 189 | Placebo N = 188 | Difelikefalin 0.5 mcg/kg N = 235 | Placebo N = 236 | |
Number of patients with any major protocol deviation, n (%) | || |||||| | || |||||| | || |||||| | || |||||| |
Dosing noncompliance | || | || | || |||||| | || |||||| |
Inclusion criteria | | ||||| | | ||||| | | ||||| | | ||||| |
Exclusion criteria | | ||||| | | ||||| | | ||||| | || |
Prohibited comedication | | ||||| | || | | ||||| | | ||||| |
Procedure not performed | || | || | || |||||| | || |||||| |
Procedure performed out of window | || | || | || ||||| | || ||||| |
Tests, assessments, procedure | | ||||| | | ||||| | || | || |
Visit not performed | || | || | | ||||| | | ||||| |
Visit performed out of window | || | || | | ||||| | | ||||| |
IP accountability | || ||||| | | ||||| | || | || |
AE, SAE reporting | || | | ||||| | || | || |
Informed Consent | | ||||| | || ||||| | || | || |
Delegation of authority | | ||||| | | ||||| | || | || |
IRB | || | | ||||| | || | || |
≥ 25% WI-NRS weeks missing | || ||||| | | ||||| | || | || |
Received < 80% of planned study doses | || ||||| | | ||||| | || | || |
Patients was not dosed in either week 11 or week 12 | | ||||| | || ||||| | || | || |
Insufficient values for baseline worst itching intensity calculation | | ||||| | || | || | || |
Others | | ||||| | || ||||| | || |||||| | || |||||| |
Missing | || | || | | ||||| | | ||||| |
AE = adverse event; IP = investigational product; IRB = Institutional Review Board; NR = not reported; SAE = serious adverse event; WI-NRS = Worst Itching Intensity Numerical Rating Scale.
Sources: KALM-1 Clinical Study Report;19 KALM-2 Clinical Study Report.20
Exposure to Study Treatments
Exposure to study treatments for the KALM-1 and KALM-2 trials is summarized in Table 14. The median duration of treatment during the double-blind treatment period was 85.0 days (range, 5 to 93 days) in the difelikefalin group and 85.0 days (range, 3 to 94 days) in the placebo group in the KALM-1 trial, and 85.0 days (range, 3 to 90 days) in the difelikefalin group and 85.0 days (range, 7 to 91 days) in the placebo group in the KALM-2 trial. The median average dose per administration was 0.5 mcg/kg in both groups for KALM-1 and KALM-2 trials, which is consistent with the dose in the product monograph. During the 12 weeks of the double-blind treatment period, the majority of patients in the safety population (60.2% in the KALM-1 trial and 69.4% in the KALM-2 trial) had 34 to 36 doses (times) administered. Most patients (54.6% in the KALM-1 trial and 41.6% in the KALM-2 trial) missed 1 to 3 doses during the 12 weeks of the double-blind treatment period. Overall, median dosing compliance was 94.44% (range, 66.7% to 100.0%) for patients in the difelikefalin group and 97.22% (range, 71.4% to 100.0%) for patients in the placebo group in the KALM-1 trial, and 97.22% (range, 51.4% to 100.0%) for patients in the difelikefalin group and 97.22% (range, 50.0% to 100.0%) for patients in the placebo group in the KALM-2 trial.
Efficacy
Only efficacy outcomes and analyses of subgroups identified in the review protocol are reported here. Refer to Appendix 3 for the hierarchical testing order of key secondary end points and detailed efficacy data.
Table 14: Study Drug Exposure (Double-Blind Safety Analysis Set)
Drug class | KALM-1 | KALM-2 | ||
|---|---|---|---|---|
Difelikefalin 0.5 mcg/kg N = 189 | Placebo N = 188 | Difelikefalin 0.5 mcg/kg N = 235 | Placebo N = 236 | |
Duration of double-blind treatment, days | ||||
Mean (SD) | 78.2 (18.37) | 80.4 (16.77) | 78.8 (18.33) | 82.5 (11.51) |
Median (range) | 85.0 (5 to 93) | 85.0 (3 to 94) | 85.0 (3 to 90) | 85.0 (7 to 91) |
Duration of double-blind period, daysa | ||||
Mean (SD) | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| |
Median (range) | |||| || || |||| | |||| || || |||| | |||| || || |||| | |||| || || |||| |
Average dose (mcg/kg) per administration | ||||
Mean (SD) | 0.5 (0.01) | NA | 0.50 (0.01) | NA |
Median (range) | 0.5 (0.5 to 0.5) | NA | 0.50 (0.5 to 0.6) | NA |
% complianceb | ||||
Mean (SD) | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| |
Median (range) | ||||| |||||| || |||||| | ||||| |||||| || |||||| | ||||| |||||| || |||||| | ||||| |||||| || |||||| |
NA = not applicable; SD = standard deviation.
aIncluding discontinuation period.
b% compliance = actual doses / planned doses.
Sources: KALM-1 Clinical Study Report;19 KALM-2 Clinical Study Report.20
Pruritus Severity
Worst Itching Intensity Numerical Rating Scale
The primary efficacy end point for both KALM trials was the proportion of patients achieving at least a 3-point improvement from baseline with respect to the weekly mean of daily 24-hour WI-NRS scores at week 12 of the double-blind treatment period. Both of the KALM trials met the primary end point. Table 15 summarizes these results for the ITT population, based on the combined imputed data from patients in the interim and postinterim analyses. The results were adjusted for the interim analysis using the CHW methodology. In the KALM-1 trial, 52% and 31% of patients randomized to difelikefalin and placebo, respectively, had a WI-NRS score that improved by at least 3 points from baseline to week 12. This corresponded to an odds ratio of 2.72 (95% CI, 1.72 to 4.30; P < 0.001) in favour of difelikefalin. In the KALM-2 trial, 50% and 37% of patients randomized to difelikefalin and placebo, respectively, had a WI-NRS score that improved by at least 3 points from baseline to week 12. This corresponded to an odds ratio of 1.61 (95% CI, 1.08 to 2.41; P = 0.02) in favour of difelikefalin.
The supportive analyses performed for the primary end point in the KALM-1 and KALM-2 trials were conducted using the PP population. The results were consistent with the primary analysis (P < 0.001 with CHW adjustment for the interim analysis and P ≤ 0.001 without CHW adjustment for the interim analysis in the KALM-1 trial, and P = 0.019 with CHW adjustment for the interim analysis and P = 0.027 without CHW adjustment for the interim analysis in the KALM-2 trial). Similarly, the sensitivity analyses conducted using the ITT population showed that efficacy results with difelikefalin were consistent with the efficacy shown in the primary efficacy analysis, with P values less than 0.01 across 3 sensitivity analyses (P < 0.001 for patients who discontinued the study drug early designated as nonresponders, P < 0.001 for multiple imputation followed a missing-not-at-random approach, and P = 0.009 for the tipping point analysis) in the KALM-1 trial. In the KALM-2 trial, the P value was 0.168 for the sensitively analysis with patients who discontinued the study drug early designated as nonresponders, P = 0.029 for the multiple imputation followed a missing-not-at-random approach, and P = 0.044 for the tipping point analysis.
The percentage of patients with at least a 3-point improvement from baseline in WI-NRS score at week 4 and at week 8 was considered to be part of other efficacy end points in the KALM-1 trial, but was considered to be a key secondary efficacy end point in the KALM-2 trial. At week 8, 43% and 28% of patients randomized to difelikefalin and placebo, respectively, in the KALM-1 trial, and 45% and 33% of patients randomized to difelikefalin and placebo, respectively in the KALM-2 trial had a WI-NRS score that improved by at least 3 points from baseline. This corresponded to an odds ratio of 2.33 (95% CI, 1.40 to 3.55; P < 0.001, with the P value not adjusted for multiple testing) in the KALM-1 trial and of 1.69 (95% CI, 1.13 to 2.53; P = 0.01) in the KALM-2 trial in favour of difelikefalin. At week 4, 33% and 18% of patients randomized to difelikefalin and placebo, respectively, in the KALM-1 trial and 35% and 22% of patients randomized to difelikefalin and placebo, respectively, in the KALM-2 trial had a WI-NRS score that improved by at least 3 points from baseline. This corresponded to an odds ratio of 2.5 (95% CI, 1.50 to 4.17; P < 0.001, with the P value not adjusted for multiple testing) in the KALM-1 trial and 1.99 (95% CI, 1.13 to 2.53; P = 0.02) in the KALM-2 trial in favour of difelikefalin.
Table 15: Proportion of Patients Achieving a ≥ 3-Point Improvement From Baseline in WI-NRS Score at Weeks 4, 8, and 12 (ITT Analysis Set)
End point | KALM-1 | KALM-2 | ||
|---|---|---|---|---|
Difelikefalin 0.5 mcg/kg N = 189 | Placebo N = 189 | Difelikefalin 0.5 mcg/kg N = 237 | Placebo N = 236 | |
Proportion of patients achieving a ≥ 3-point improvement from baseline in WI-NRS scorea,b | ||||
Week 12 | ||||
Patients with a ≥ 3-point WI-NRS improvement at week 12, n (%) | 82 (52.2) | 51 (30.9) | 95 (49.7) | 77 (37.2) |
Difference, % (95% CI) | 22 (12 to 32) | 11 (1 to 20) | ||
LS means estimate of percent with improvement (95% CI) | 51.0 (42.9 to 58.9) | 27.6 (20.2 to 36.6) | 54.0 (43.9 to 63.9) | 42.2 (32.5 to 52.5) |
Odds ratio (95% CI) | 2.72 (1.72 to 4.30) | 1.61 (1.08 to 2.41) | ||
P value | < 0.001 | 0.02 | ||
Patients with complete data, n (%) | 157(83.1) | 165(89.4) | 191 (80.6) | 207 (87.7) |
Week 8 | ||||
Patients with a ≥ 3-point WI-NRS improvement at week 8, n (%) | 71 (42.5) | 48 (27.7) | 93 (44.5) | 73 (33.0) |
Difference, % (95% CI) | 16 (6 to 26) | 11 (2 to 20) | ||
LS means estimate of percent with improvement (95% CI) | 42.7 (33.5 to 52.6) | 25.1 (18.0 to 33.7) | 49.0 (38.3 to 59.9) | 36.2 (27.3 to 46.2) |
Odds ratio (95% CI) | 2.23 (1.40 to 3.55) | 1.69 (1.13 to 2.53) | ||
P value | < 0.001c | 0.01 | ||
Patients with complete data, n (%) | 167 (88.4) | 173 (91.5) | 209 (88.2) | 221 (93.6) |
Week 4 | ||||
Patients with a ≥ 3-point WI-NRS improvement at week 4, n (%) | 58 (33.0) | 32 (18.0) | 75 (35.0) | 50 (22.2) |
Difference, % (95% CI) | 15 (6 to 24) | 14 (5 to 22) | ||
LS means estimate of percent with improvement (95% CI) | 33.0 (24.9 to 43.2) | 16.7 (11.1 to 24.4) | 38.3 (28.5 to 49.1) | 23.8 (16.6 to 32.8) |
Odds ratio (95% CI) | 2.50 (1.50 to 4.17) | 1.99 (1.29 to 3.06) | ||
P value | < 0.001c | 0.002 | ||
Patients with complete data, n (%) | 176 (93.1) | 178 (94.2) | 214 (90.3) | 225 (95.3) |
CI = confidence interval; ITT = intention to treat; LS = least squares; WI-NRS = Worst Itching Intensity Numerical Rating Scale.
Note: Combined analysis used the separate interim and postinterim results to generate an adjusted overall estimate and P value using the Lawrence and Hung and/or CHW methodology.
aEstimated %, odds ratio, and P value used a logistic regression model with terms for treatment group, baseline WI-NRS score, region (for the KALM-2 trial only), use of antiitch medication during the week before randomization, and the presence of specific medical conditions. Missing values were imputed using multiple imputation under the missing-at-random missing data assumption for interim patients and postinterim patients separately. Counts and percentages were based on nonmissing data.
bOdds ratio was estimated using the Lawrence and Hung approach. P value was estimated using the CHW approach.
cThe P value was not adjusted for multiple testing (i.e., the type I error rate has not been controlled).
Sources: KALM-1 Clinical Study Report;19 KALM-2 Clinical Study Report;20 FDA report.16
The percentage of patients with at least a 4-point improvement from baseline in WI-NRS score at week 12, week 8, and week 4 is summarized in Table 16. At week 12, 41% and 21% of patients randomized to difelikefalin and placebo, respectively, in the KALM-1 trial and 38% and 25% of patients randomized to difelikefalin and placebo respectively, in the KALM-2 trial had a WI-NRS score that improved by at least 4 points from baseline. This corresponded to an odds ratio of 2.89 (95% CI, 1.75 to 4.76; P < 0.001) in the KALM-1 trial and 1.77 (95% CI, 1.14 to 2.74; P = 0.01) in the KALM-2 trial in favour of difelikefalin.
The percentage of patients with at least a 4-point improvement from baseline in WI-NRS score at week 4 and week 8 was considered to be part of other efficacy end points in the KALM-1 trial, but was considered to be a key secondary efficacy end point in the KALM-2 trial. At week 8, 31% and 19.7% of patients randomized to difelikefalin and placebo, respectively, in the KALM-1 trial and 31% and 20.4% of patients randomized to difelikefalin and placebo, respectively, in the KALM-2 trial had a WI-NRS score that improved by at least 4 points from baseline. This corresponded to an odds ratio of 2.11 (95% CI, 1.26 to 3.53; P = 0.005, with the P value not adjusted for multiple testing) in the KALM-1 trial and 1.82 (95% CI, 1.16 to 2.86; P = 0.01) in the KALM-2 trial in favour of difelikefalin. At week 4, 18% and 8% of patients randomized to difelikefalin and placebo, respectively, in the KALM-1 trial and 20% and 13% of patients randomized to difelikefalin and placebo, respectively, in the KALM-2 trial had a WI-NRS score that improved by at least 4 points from baseline. This corresponded to an odds ratio of 2.75 (95% CI, 1.40 to 5.39; P = 0.003, with the P value not adjusted for multiple testing) in the KALM-1 trial and 1.76 (95% CI, 1.04 to 2.98; P = 0.036) in the KALM-2 trial in favour of difelikefalin.
Table 16: Proportion of Patients Achieving a ≥ 4-Point Improvement From Baseline in WI-NRS Score at Weeks 4, 8, and 12 (ITT Analysis Set)
WI-NRS score | KALM-1 | KALM-2 | ||
|---|---|---|---|---|
Difelikefalin 0.5 mcg/kg N = 189 | Placebo N = 189 | Difelikefalin 0.5 mcg/kg N = 237 | Placebo N = 236 | |
Proportion of patients achieving a ≥ 4-point improvement from baseline in WI-NRS scorea | ||||
Week 12 | ||||
Patients with a ≥ 4-point WI-NRS improvement at week 12,b,c n (%) | 64 (40.8) | 35 (21.2) | 72 (37.7) | 52 (25.1) |
Difference, % (95% CI) | 19 (9 to 28) | 12 (3 to 20) | ||
LS means estimate of percent with improvementd (95% CI) | 38.9 (29.8 to 48.7) | 18.0 (12.1 to 26.0) | 41.2 (33.0 to 50.0) | 28.4 (21.3 to 36.7) |
Odds ratioe (95% CI) | 2.89 (1.75 to 4.76) | 1.77 (1.14 to 2.74) | ||
P valuef | < 0.001 | 0.01 | ||
Patients with complete data, n (%) | 157 (83.1) | 165 (87.3) | 191 (80.6) | 207 (87.7) |
Week 8 | ||||
Patients with a ≥ 4-point WI-NRS improvement at week 8,b,g n (%) | 51 (30.5) | 34 (19.7) | 64 (30.6) | 45 (20.4) |
Difference, % (95% CI) | 12 (3 to 21) | 10 (2 to 19) | ||
LS means estimate of percent with improvementd (95% CI) | 26.9 (18.8 to 37.0) | 14.9 (9.6 to 22.4) | 36.1 (28.0 to 45.1) | 23.7 (17.2 to 31.8) |
Odds ratioe (95% CI) | 2.11 (1.26 to 3.53) | 1.82 (1.16 to 2.86) | ||
P valuef | 0.005h | 0.01 | ||
Patients with complete data, n (%) | 167 (88.4) | 173 (91.5) | 209 (88.2) | 221 (93.6) |
Week 4 | ||||
Patients with a ≥ 4-point WI-NRS improvement at week 4,b,g n (%) | 32 (18.2) | 15 (8.4) | 43 (20.1) | 30 (13.3) |
Difference, % (95% CI) | 10 (3 to 16) | 7 (0 to 14) | ||
LS means estimate of percent with improvementd (95% CI) | 16.4 (10.1 to 25.4) | 6.6 (3.5 to 12.3) | 26.1 (18.8 to 34.9) | 16.7 (11.4 to 23.9) |
Odds ratioe (95% CI) | 2.75 (1.40 to 5.39) | 1.76 (1.04 to 2.98) | ||
P valuef | 0.003h | 0.036 | ||
Patients with complete data, n (%) | 176 (93.1) | 178 (94.2) | 214 (90.3) | 225 (95.3) |
CI = confidence interval; ITT = intention to treat; LS = least squares; WI-NRS = Worst Itching Intensity Numerical Rating Scale.
Note: Combined analysis used the separate interim and postinterim results to generate an adjusted overall estimate and P value using the LH and/or CHW methodology.
aMultiple imputation with missing-at-random assumption.
bCounts and percentages were based on nonmissing data.
cSecondary end point for both the KALM-1 and KALM-2 trials.
dEstimated %, odds ratio, and P value used a logistic regression model with terms for treatment group, baseline WI-NRS score, use of antiitch medication during the week before randomization, region (KALM-2 trial only), and the presence of specific medical conditions. Missing values were imputed using multiple imputation under the missing-at-random missing data assumption for interim patients and postinterim patients separately.
eEstimated using the Lawrence and Hung approach.
fEstimated using the CHW approach.
gSecondary end points for the KALM-2 trial only.
hThe P value was not adjusted for multiple testing (i.e., the type I error rate has not been controlled).
Sources: KALM-1 Clinical Study Report;19 KALM-2 Clinical Study Report;20 FDA report.16
Results for the LS mean change from baseline in the weekly WI-NRS score at week 12, week 8, and week 4 in both KALM trials have been summarized in Table 17, Figure 4, and Figure 5. The LS mean change from baseline to week 12 was –3.2 points for difelikefalin versus –2.0 points for placebo in the KALM-1 trial and –3.1 points for difelikefalin versus –2.5 points for placebo in the KALM-2 trial. Similar results were observed at week 8, when the LS mean change from baseline was –2.7 points for difelikefalin versus –1.8 points for placebo in the KALM-1 trial and –2.8 points for difelikefalin versus –2.1 points for placebo in the KALM-2 trial. At week 4, the LS mean change from baseline was –2.2 points for difelikefalin versus –1.3 points for placebo in the KALM-1 trial and –2.3 points for difelikefalin versus –1.6 points for placebo in the KALM-2 trial. The difference in LS means between difelikefalin and placebo was –1.1 points (95% CI, –1.6 to –0.6 points; P < 0.001) at week 12, –0.9 points (95% CI, –1.4 to –0.6 points; P < 0.001) at week 8, and –0.9 points (95% CI, –1.3 to –0.5 points; P < 0.001) at week 4 in the KALM-1 trial, and –0.6 points (95% CI, –1.1 to –0.2 points; P = 0.008) at week 12, –0.7 points (95% CI, –1.1 to –0.3 points; P = 0.001) at week 8, and –0.7 points (95% CI, –1.1 to –0.3 points; P = 0.001) at week 4 in the KALM-2 trial. Of note, the P values for the difference in LS means between difelikefalin and placebo at week 12, week 8, and week 4 were not adjusted for multiplicity in either KALM trial.
Table 17: MMRM Analysis of Weekly WI-NRS Score Changes From Baseline (ITT Analysis Set)
WI-NRS score | KALM-1 | KALM-2 | ||
|---|---|---|---|---|
Difelikefalin 0.5 mcg/kg N = 189 | Placebo N = 189 | Difelikefalin 0.5 mcg/kg N = 237 | Placebo N = 236 | |
Change from baseline in weekly WI-NRS scorea | ||||
Week 12 | ||||
Change from baseline, LS mean (95% CI) | |||||||||| || ||||| | |||||||||| || ||||| | |||||||||| || ||||| | |||||||||| || ||||| |
Difference in LS means, %b (95% CI) | |||| ||||| || ||||| | |||| ||||| || ||||| | ||
P valuec | |||||| | ||||| | ||
Week 8 | ||||
Change from baseline, LS mean (95% CI) | |||||||||| || ||||| | |||||||||| || ||||| | |||||||||| || ||||| | |||||||||| || ||||| |
Difference in LS means, %b (95% CI) | |||| ||||| || ||||| | |||| ||||| || ||||| | ||
P valuec | |||||| | ||||| | ||
Week 4 | ||||
Change from baseline, LS mean (95% CI) | |||||||||| || ||||| | |||||||||| || ||||| | |||||||||| || ||||| | |||||||||| || ||||| |
Difference in LS means, %b (95% CI) | |||| ||||| || ||||| | |||| ||||| || ||||| | ||
P valuec | |||||| | |||||| | ||
CI = confidence interval; ITT = intention to treat; LS = least squares; MMRM = mixed effects model with repeated measures; WI-NRS = Worst Itching Intensity Numerical Rating Scale.
aLS means, P values, and CIs were based on an MMRM analysis with effects for treatment, visit, treatment-by-visit interaction, baseline score, use of antiitch medication during the week before randomization, region (the KALM-2 trial only), and the presence of specific medical conditions. The model was fit using an unstructured covariance structure. Missing values were imputed using multiple imputation under the missing-at-random missing data assumption.
bPlacebo group was used as reference.
cThe P value was not adjusted for multiple testing (i.e., the type I error rate has not been controlled).
Sources: KALM-1 Clinical Study Report;19 KALM-2 Clinical Study Report.20
Figure 4: KALM-1 Mean Change From Baseline in WI-NRS Score by Study Week (Primary Efficacy Imputation, ITT Analysis Set) — Redacted

Note: This figure has been redacted at the request of the sponsor. LS means, standard errors, and CIs were based on an MMRM analysis with effects for treatment, visit, treatment-by-visit interaction, baseline score, use of antiitch medication during the week before randomization, and the presence of specific medical conditions. The model was fit using an unstructured covariance structure. Missing values were imputed using multiple imputation under the missing-at-random missing data assumption.
Source: KALM-1 Clinical Study Report.19
Figure 5: KALM-2 Mean Change From Baseline in WI-NRS Score by Study Week (Primary Efficacy Imputation, ITT Analysis Set) — Redacted

CI = confidence interval; ITT = intention to treat; LS = least squares; WI-NRS = Worst Itching Intensity Numerical Rating Scale; SE = standard error.
Note: This figure has been redacted at the request of the sponsor. LS means, standard errors, CIs, and P values were based on an MMRM analysis with effects for treatment, visit, treatment-by-visit interaction, baseline score, region, use of antiitch medication during the week before randomization, and the presence of specific medical conditions. The model was fit using an unstructured covariance structure. Missing values were imputed using multiple imputation under the missing-at-random missing data assumption.
Source: KALM-2 Clinical Study Report.20
Patient Global Impression of Change
The proportion of responders on the PGIC scale at week 12 is summarized in Table 18. At week 12 in the KALM-1 trial, ||| and ||| of patients randomized to difelikefalin and placebo, respectively, were complete responders. This corresponded to an odds ratio of |||| |||| ||| |||| || ||||| | | |||||||. At week 12 in the KALM-2 trial, ||| and ||| of patients randomized to difelikefalin and placebo, respectively, were complete responders. This corresponded to an odds ratio of |||| |||| ||| |||| || ||||| | | ||||||.
Table 18: Summary of PGIC Responders (ITT Analysis Set)
PGIC scale | KALM-1 | KALM-2 | ||
|---|---|---|---|---|
Difelikefalin 0.5 mcg/kg N = 189 | Placebo N = 189 | Difelikefalin 0.5 mcg/kg N = 237 | Placebo N = 236 | |
Total, n | ||| | ||| | ||| | ||| |
Responders,a n (%) | ||| |||||| | || |||||| | ||| |||||| | || |||||| |
95% CI of % | |||| || |||| | |||| || |||| | |||| || |||| | |||| || |||| |
Odds ratio (95% CI) | |||| ||||| || ||||| | |||| ||||| || ||||| | ||
P valueb | |||||| | |||| | ||
CI = confidence interval; PGIC = Patient Global Impression of Change.
Note: The denominator for all percentages is the number of patients with available PGIC scores. Clopper-Pearson CIs for proportions are reported. Odds ratio and CI were based on the Mantel-Haenszel estimate, stratified by the use of antiitch medication during the week before randomization and the presence of specific medical conditions.
aPGIC responders were those with responses of very much improved or much improved.
bThe P value is calculated with the Cochran-Mantel-Haenszel exact test and was not adjusted for multiple testing (i.e., the type I error rate has not been controlled).
Sources: KALM-1 Clinical Study Report;19 KALM-2 Clinical Study Report.20
Symptom Reduction (e.g., Pruritus, Pain, Sleep Disturbance)
Evidence of outcomes related to an improvement in symptoms of pruritus were reported in the previous section (Pruritus Severity). Evidence of outcomes related to symptom reduction, such as pain and sleep disturbance, was not identified for this review.
Health-Related Quality of Life
Itch-Related Quality of Life (Skindex-10 Scale Score)
Summary of the Main Analyses (Total Score and Domain Scores)
The change from baseline in itch-related QoL, assessed by the Skindex-10 scale total and domain scores, at the end of week 12 was summarized in Table 19. At the end of week 12, the LS mean change in Skindex-10 scale total score was –17.2 for difelikefalin versus –12.0 for placebo in the KALM-1 trial –16.6 for difelikefalin versus –14.8 for versus placebo in the KALM-2 trial. Overall, the difference in LS mean change from baseline to week 12 on the Skindex-10 scale total score between the difelikefalin and placebo groups was –5.1 points (95% CI, –8.0 to –2.3 points, P < 0.001). In the KALM-2 trial, results for the change from baseline to week 12 in the Skindex-10 scale total score indicated no difference between treatment groups (LS mean difference = –1.8 points; 95% CI, –4.3 to 0.8 points; P = 0.171).
The findings on the Skindex-10 scale by domain scores (disease domain, mood/emotional distress domain, and social functioning domain) demonstrated trends of change similar to those in the preceding analysis of Skindex-10 scale total scores, in which the difelikefalin group showed a greater reduction in scores from baseline at week 12 than the placebo group in the KALM-1 trial. For the disease total, the LS mean difference was –1.8 points (95% CI, –2.8 to –0.9 points; P < 0.001); for the mood/emotional distress total, the LS mean difference was |||| |||||| |||| ||| |||| || ||||||||||| | ||||||); and for the social functioning total, the LS mean difference was –||| |||||| |||| ||| |||| || ||||||||||| | | |||||||.
In the KALM-2 trial, the LS mean difference for the disease domain was –0.9 points (95% CI, –1.7 to 0.0 points; P = 0.047); a difference between treatment groups was not observed in the other analyses of the Skindex-10 scale by the domains of mood/emotional distress total (LS mean difference = |||| ||||||| ||| ||| |||| || ||| ||||||| ||||||| in the KALM-2 trial) or social functioning total (LS mean difference = |||| ||||||| ||| ||| |||| || ||| ||||||| ||||||| in the KALM-2 trial).
Summary of Supportive Analyses
The findings for the supportive analysis of the Skindex-10 scale total score in the PP population were consistent with the analysis of the ITT population in the KALM-1 trial, but in the KALM-2 trial, the results were not statistically significant (LS mean difference = |||| ||||||| ||| ||| |||| || |||| ||||||; | |||||| in the KALM-1 trial and LS mean difference = |||| ||||||| ||| ||| |||| || ||| ||||||| | || |||| || ||||||).
Summary of Sensitivity Analyses
The results of the MMRM sensitivity analysis (no imputation) of the Skindex-10 scale total score at week 12 for the ITT population were similar to the preceding ANCOVA with multiple imputation for the KALM-1 trial, but for the KALM-2 trial, the results were not statistically significant. In the KALM-1 trial, the treatment group difference in LS means (difelikefalin minus placebo) was |||| |||| ||| |||| || ||||| | ||||||| in favour of difelikefalin. In the KALM-2 trial, the treatment group difference in LS means was |||| |||| ||| |||| || |||| | |||||||, which was not statistically significant.
In the KALM-2 trial, sensitivity analyses were also performed for Skindex-10 scale scores using ANCOVA with multiple imputation of missing data and control distribution and using ANCOVA with multiple imputation of missing data and baseline distribution. The treatment group difference in LS means was |||| |||| ||| |||| || |||| | ||||||| using ANCOVA with multiple imputation of missing data and control distribution and |||| |||| ||| |||| || |||| | | ||||| using ANCOVA with multiple imputation of missing data and baseline distribution).
Table 19: Change From Baseline in Skindex-10 Scale Score at Week 12 (ITT Analysis Set)
Skindex-10 scale score | KALM-1 | KALM-2 | |||
|---|---|---|---|---|---|
Difelikefalin 0.5 mcg/kg N = 189 | Placebo N = 189 | Difelikefalin 0.5 mcg/kg N = 237 | Placebo N = 236 | ||
Change from baseline in Skindex-10 scale scorea | |||||
Total Skindex-10 scale score | |||||
End of week 12 change from baseline LS mean (95% CI) | –17.2 (–19.6 to –14.7) | –12.0 (–14.5 to –9.6) | –16.6 (–19.3 to –14.0) | –14.8 (–17.4 to –12.2) | |
Difference in LS meansb (95% CI) | –5.1 (–8.0 to –2.3) | –1.8 (–4.3 to 0.8) | |||
P value | < 0.001 | 0.171 | |||
Disease domain, total | |||||
End of week 12 change from baseline LS mean (95% CI) | |||| |||||| || ||||| | |||| |||||| || ||||| | |||| |||||| || ||||| | |||| |||||| || ||||| | |
Difference in LS meansb (95% CI) | |||| ||||| || ||||| | |||| ||||| || |||| | |||
P valuec | |||||| | ||||| | |||
Mood/emotional distress total | |||||
End of week 12 change from baseline LS mean (95% CI) | |||| |||||| || ||||| | |||| |||||| || ||||| | |||| |||||| || ||||| | |||| |||||| || ||||| | |
Difference in LS meansb (95% CI) | |||| ||||| || ||||| | |||| ||||| || |||| | |||
P valuec | |||||| | ||||| | |||
Social functioning total | |||||
End of week 12 change from baseline LS mean (95% CI) | |||| |||||| || ||||| | |||| |||||| || ||||| | |||| |||||| || ||||| | |||| |||||| || ||||| | |
Difference in LS meansb (95% CI) | |||| ||||| || ||||| | |||| ||||| || |||| | |||
P valuec | ||||| | ||||| | |||
CI = confidence interval, ITT = intention to treat; LS = least squares.
aLS means and 95% CIs were based on ANCOVA with fixed effects for treatment, with baseline score, region (for the KALM-2 trial only), and the randomization stratification variables as covariates for the KALM-2 trial. Missing values were imputed using multiple imputation under the missing-at-random missing data assumption.
bPlacebo group was used as reference.
cThe P value was not adjusted for multiple testing (i.e., the type I error rate has not been controlled).
Sources: KALM-1 Clinical Study Report;19 KALM-2 Clinical Study Report.20
The proportion of patients with at least a 15-point improvement from baseline in Skindex-10 scale total score at week 12 is summarized in Table 20. At week 12, the LS mean percentage of patients with at least a 15-point improvement from baseline in the Skindex-10 scale total score was ||||| in the difelikefalin group and ||||| in the placebo group for the KALM-1 trial, and ||||| for difelikefalin and ||||| for placebo in the KALM-2 trial. The odds ratio for at least a 15-point improvement from baseline with difelikefalin versus placebo was |||| |||| ||| |||| || ||||| |||||||| in the KALM-1 trial and |||| |||| ||| |||| || ||||| |||||||| in the KALM-2 trial.
Table 20: Proportion of Patients With a ≥ 15-Point Improvement in Skindex-10 Scale Total Scores (ITT Analysis Set)
Skindex-10 scale score | KALM-1 | KALM-2 | ||
|---|---|---|---|---|
Difelikefalin 0.5 mcg/kg N = 189 | Placebo N = 189 | Difelikefalin 0.5 mcg/kg N = 237 | Placebo N = 236 | |
Proportion of patients with a ≥ 15-point improvement in Skindex-10 scale total score,a n (%) | || |||||| | || |||||| | ||| |||||| | || |||||| |
LS means estimate of percent with improvementb (95% CI) | |||| |||||| || ||||| | |||| |||||| || ||||| | |||| |||||| || ||||| | |||| |||||| || ||||| |
Odds ratio (95% CI) | |||| ||||| || ||||| | |||| ||||| || ||||| | ||
P valuec | 0.011 | 0.005 | ||
CI = confidence interval; ITT = intention to treat; LS = least squares.
aCounts and percents are based on nonmissing data.
bEstimated percent, odds ratio, and P value use a logistic regression model with terms for treatment group, baseline score, use of antiitch medication during the week before randomization, and the presence of specific medical conditions. No imputation was performed.
cThe P value was not adjusted for multiple testing (i.e., the type I error rate has not been controlled).
Sources: KALM-1 Clinical Study Report;19 KALM-2 Clinical Study Report.20
Itch-Related Quality of Life (5-D Itch Scale Score)
Summary of the Main Analyses (Total Score and Domain Scores)
The change from baseline in itch-related QoL, assessed by the 5-D itch scale total and domain scores at the end of week 12 is summarized in Table 21. At the end of week 12, the LS mean change in total 5-D itch scale score was greater in the difelikefalin group than in the placebo group for the KALM-1 and KALM-2 trials (–5.0 versus –3.7and –4.9 versus –3.8, respectively). Overall, the difference in LS mean change from baseline to week 12 for the 5-D itch scale total score between the difelikefalin and placebo groups was –1.3 points (95% CI, –2.0 to –0.5 points, P < 0.001) in the KALM-1 trial and –1.1 points (95% CI, –1.7 to –0.4 points; P = 0.002) in the KALM-2 trial. Of note, the P value for the difference in LS means on the 5-D itch scale at week 12 for the KALM-2 trial was not adjusted for multiple testing, as the preceding test in the testing hierarchy (i.e., difference in LS means for Skindex-10 score at week 12 in the KALM-2 trial) was not statistically significant.
In the KALM-1 trial, the findings of the 5-D itch domain scores were consistent with the analysis of the 5-D itch scale total score for duration (|| |||| |||||||||| | |||| ||||||| ||| ||| |||| || |||| ||||||| | | |||||), degree (|| |||| |||||||||| | |||| ||||||| ||| ||| |||| || |||| ||||||| | | |||||), direction (|| |||| |||||||||| | |||| ||||||| ||| ||| |||| || |||| ||||||| | ||||||), disability(|| |||| |||||||||| | |||| ||||||| ||| ||| |||| || ||| ||||||| |||||||), and distribution (|| |||| |||||||||| | |||| ||||||| ||| ||| |||| || |||| ||||||| | | |||||). In the KALM-2 trial, the findings of the 5-D itch scale domain scores did not show a difference in duration (|| |||| |||||||||| | |||| ||||||| ||| ||| |||| || ||| ||||||| |||||||) or disability (|| |||| |||||||||| | |||| ||||||| ||| ||| |||| || |||| ||||||| |||||||). The remainder of the analyses by domain were consistent with the analysis of the 5-D itch scale total score for degree (|| |||| |||||||||| | |||| ||||||| ||| ||| |||| || ||| ||||||| |||||||), direction (|| |||| |||||||||| | |||| ||||||| ||| ||| |||| || |||| ||||||| |||||||), and distribution (|| |||| |||||||||| | |||| ||||||| ||| ||| |||| || |||| ||||||| |||||||).
Summary of Supportive Analyses
The findings for the supportive analysis of the 5-D itch scale total score in the PP population were consistent with the analysis in the ITT population for both KALM trials (LS mean difference = –1.4 points; 95% CI, –2.2 to –0.6 points; P < 0.001 in the KALM-1 trial and LS mean difference = –1.3 points; 95% CI, –2.0 to –0.6 points; P < 0.001 in the KALM-2 trial).
Summary of Sensitivity Analyses
The results of the MMRM sensitivity analysis (no imputation) of the5-D itch scale total score at week 12 for the ITT population were similar to the preceding ANCOVA with multiple imputation for both KALM trials (LS mean difference = |||| ||||||| ||| ||| |||| || |||| ||||||| | | ||||| in the KALM-1 trial and LS mean difference = |||| ||||||| ||| ||| |||| || |||| ||||||| | ||||||| in the KALM-2 trial).
In the KALM-2 trial, sensitivity analyses were also performed for the 5-D itch scale total score using ANCOVA with multiple imputation of missing data and control distribution and using ANCOVA with multiple imputation of missing data and baseline distribution: the treatment group difference in LS means was –1.2 (95% CI, –1.9 to –0.5; P = 0.004) using ANCOVA with multiple imputation of missing data and control distribution and –0.7 (95% CI, –1.4 to 0.0; P = 0.046) using ANCOVA with multiple imputation of missing data and baseline distribution).
Table 21: Change From Baseline in 5-D Itch Scale Score at Week 12 (ITT Analysis Set)
5-D itch score | KALM-1 | KALM-2 | ||
|---|---|---|---|---|
Difelikefalin 0.5 mcg/kg N = 189 | Placebo N = 189 | Difelikefalin 0.5 mcg/kg N = 237 | Placebo N = 236 | |
5-D itch scale total scorea | ||||
End of week 12 change from baseline LS mean (95% CI) | –5.0 (–5.7 to –4.4) | –3.7 (–4.4 to –3.1) | –4.9 (–5.6 to –4.2) | –3.8 (–4.5 to –3.1) |
Difference in LS means (95% CI) | –1.3 (–2.0 to –0.5) | –1.1(–1.7 to –0.4) | ||
P value | < 0.001 | 0.002b | ||
Duration domain score | ||||
End of week 12 change from baseline LS mean (95% CI) | |||| |||||| || ||||| | |||| |||||| || ||||| | |||| |||||| || ||||| | |||| |||||| || ||||| |
Difference in LS means (95% CI) | |||| ||||| || ||||| | |||| ||||| || |||| | ||
P valuec | ||||| | ||||| | ||
Degree domain score | ||||
End of week 12 change from baseline LS mean (95% CI) | |||| |||||| || ||||| | |||| |||||| || ||||| | |||| |||||| || ||||| | |||| |||||| || ||||| |
Difference in LS means (95% CI) | |||| ||||| || ||||| | |||| ||||| || |||| | ||
P valuec | ||||| | ||||| | ||
Direction domain score | ||||
End of week 12 change from baseline LS mean (95% CI) | |||| |||||| || ||||| | |||| |||||| || ||||| | |||| |||||| || ||||| | |||| |||||| || ||||| |
Difference in LS means (95% CI) | |||| ||||| || ||||| | |||| ||||| || ||||| | ||
P valuec | |||||| | ||||| | ||
Disability domain score | ||||
End of week 12 change from baseline LS mean (95% CI) | |||| |||||| || ||||| | |||| |||||| || ||||| | |||||||||| || ||||| | |||| |||||| || ||||| |
Difference in LS means (95% CI) | |||| ||||| || |||| | ||||||||| || |||| | ||
P valuec | ||||| | ||||| | ||
Distribution domain score | ||||
End of week 12 change from baseline LS mean (95% CI) | |||| |||||| || ||||| | |||| |||||| || ||||| | |||| |||||| || ||||| | |||| |||||| || ||||| |
Difference in LS means (95% CI) | |||| ||||| || ||||| | |||| ||||| || ||||| | ||
P valuec | ||||| | ||||| | ||
CI = confidence interval; ITT = intention to treat; LS = least squares.
aLS means and 95% CIs were based on ANCOVA with fixed effects for treatment, with baseline score, region (for the KALM-2 trial only), and the randomization stratification variables as covariates for the KALM-2 trial. Missing values were imputed using multiple imputation the missing-at-random missing data assumption.
bThe P value was not considered inferential and the null hypothesis was not rejected, according to the gate-keeping strategy based on the hierarchical testing order of key secondary end points, as the test of the prior end point (change from baseline in Skindex-10 scale total score) in the sequence was not statistically significant.
cThe P value was not adjusted for multiple testing (i.e., the type I error rate has not been controlled).
Sources: KALM-1 Clinical Study Report;19 KALM-2 Clinical Study Report.20
The proportion of patients with at least a 5-point improvement from baseline in 5-D itch scale total score at week 12 of the double-blind treatment period in the ITT analysis set is summarized in Table 22. At week 12, the LS mean percentage of patients with at least a 5-point improvement from baseline in the 5-D itch scale total score was greater in the difelikefalin group than in the placebo group for the KALM-1 and KALM-2 trials (||||| ||| ||||| for difelikefalin versus placebo and ||||| ||| ||||| for difelikefalin versus placebo, respectively).The odds ratio for at least a 15-point improvement from baseline for difelikefalin versus placebo was |||| |||| ||| |||| || ||||| |||||||) in the KALM-1 trial and |||| |||| ||| |||| || ||||| |||||||) in the KALM-2 trial. Of note, the P values were not adjusted for multiple testing.
Table 22: Proportion of Patients With a ≥ 5-Point Improvement in 5-D Itch Scale Total Score at Week 12 (ITT Analysis Set)
5-D itch score | KALM-1 | KALM-2 | ||
|---|---|---|---|---|
Difelikefalin 0.5 mcg/kg N = 189 | Placebo N = 189 | Difelikefalin 0.5 mcg/kg N = 237 | Placebo N = 236 | |
Proportion of patients with a ≥ 5-point improvement in 5-D itch scale total score,a n (%) | || |||||| | || |||||| | ||| |||||| | || |||||| |
LS means estimate of percent with improvementb (95% CI) | |||| |||||| || ||||| | |||| |||||| || ||||| | |||| |||||| || ||||| | |||||||||| || ||||| |
Odds ratio (95% CI) | |||| ||||| || ||||| | |||| ||||| || ||||| | ||
P valuec | ||||| | ||||| | ||
CI = confidence interval; ITT = intention to treat; LS = least squares.
aCounts and percents are based on nonmissing data.
bEstimated percent, odds ratio, and P value use a logistic regression model with terms for treatment group, baseline score, use of antiitch medication during the week before randomization, and the presence of specific medical conditions for the KALM-1 trial. Estimated percent, odds ratio, and P value used a logistic regression model with terms for treatment group, baseline score, region, use of antiitch medication during the week before randomization, and the presence of specific medical conditions for the KALM-2 trial. For both trials, no imputation was performed.
cThe P value was not adjusted for multiple testing (i.e., the type I error rate has not been controlled).
Sources: KALM-1 Clinical Study Report;19 KALM-2 Clinical Study Report.20
Mood (e.g., Depression)
Evidence of outcomes related to mood, such as depression, was not identified for this review.
Days of Missed Work, Missed School, or Missed Dialysis
Evidence of outcomes related to days of missed work, missed school, or missed dialysis was not identified for this review.
Harms
Only harms identified in the review protocol are reported here. Refer to Table 23 or detailed harms data.
Adverse Events
The percentage of patients with any reported TEAE was similar in the difelikefalin and placebo groups (68.8% versus 62.2% in the KALM-1 trial and 68.1% versus 61.4% in the KALM-2 trial). The TEAEs more commonly reported in the difelikefalin group than in the placebo group were diarrhea (9.5% versus 3.7%), dizziness (6.9% versus 1.1%), and vomiting (5.3% versus 3.2%); the TEAE more commonly reported in the placebo group was nasopharyngitis (3.2% versus 5.3%) in the KALM-1 trial. In the KALM-2 trial, the TEAEs more commonly reported in the difelikefalin than in the placebo groups were diarrhea (8.1% versus 5.5%), vomiting (6.4% versus 5.9%), falls (6.8% versus 5.1%), dizziness (5.5% versus 5.1%), and nausea (6.4% versus 4.2%). Overall, the frequency of TEAEs was roughly balanced in the difelikefalin and placebo groups in both KALM trials, with the exception of dizziness (6.9% versus 1.1%) and diarrhea (9.5% versus 3.7%), which were notably higher in the difelikefalin group than in the placebo group in the KALM-1 trial.
Serious Adverse Events
Serious TEAEs were reported in 25.9% of patients in the difelikefalin group and 21.8% of patients in the placebo group in the KALM-1 trial, and in 24.7% and 21.6%, respectively, in the KALM-2 trial (Table 23). The most common SAEs (difelikefalin versus placebo) were hyperkalemia (2.1% versus 2.1% in the KALM-1 trial and 1.7% versus 1.3% in the KALM-2 trial), sepsis (1.6% versus 2.1% in the KALM-1 trial and 1.3% versus 1.3% in the KALM-2 trial), and pneumonia (1.6% versus 2.7% in the KALM-1 trial and 0.9% versus 0.0% in the KALM-2 trial), as well as fluid overload (1.1% versus 2.1% in the KALM-1 trial), and chest pain (3.4% versus 0.4% in the KALM-2 trial). Overall, the frequency of SAEs was roughly balanced between treatment groups in both KALM trials.
Withdrawals Due to Adverse Events
Withdrawals due to TEAEs were comparable in the 2 treatment groups in both KALM trials. The proportion of patients who discontinued treatment due to TEAEs was 7.9% for difelikefalin and 4.8% for placebo in the KALM-1 trial and 5.5% for difelikefalin and 3.4% for placebo in the KALM-2 trial. Dizziness was the TEAE that most frequently caused discontinuation (difelikefalin versus placebo) in both KALM trials (1.6% versus 0.% for in the KALM-1 trial and 0.4% versus 0.4% in the KALM-2 trial).
Mortality
Deaths were reported in 1.1% of patients in the difelikefalin group and 1.6% of patients in the placebo group in the KALM-1 trial, and in 0.9% for difelikefalin and 0.8% for placebo in the KALM-2 trial. In the KALM-1 trial, sepsis was the cause of death for 2 patients randomized to difelikefalin (and for 0 patients randomized to placebo and 0 patients in the KALM-2 trial), and septic shock was the cause of death for 2 patients randomized to placebo (and for 0 patients randomized to difelikefalin and 0 patients in the KALM-2 trial). All other causes of death (dyspnea and/or hypotension, cardiac arrest, unknown) were infrequently reported, with no more than 1 patient in any treatment group. No specific AE was identified to account for the majority of deaths in either group in the KALM-2 trial.
Notable Harms
The following notable harms were included in the CADTH systematic review protocol: diarrhea, nausea, vomiting, gait disturbance, falls, dizziness, headache, somnolence, seizures, syncope, mental status changes, mood changes, paresthesia (unusual feeling or sensation), hyperkalemia, back pain, tachycardia, and palpitation. The most common serious notable harms (difelikefalin versus placebo) were diarrhea (9.5% versus 3.7% in the KALM-1 trial and 8.1% versus 5.5% in the KALM-2 trial), dizziness (6.9% versus 2.1% in the KALM-1 trial and 5.5% versus 5.1% in the KALM-2 trial), and vomiting (5.3% versus 3.2% in the KALM-1 trial and 6.4% versus 5.9% in the KALM-2 trial), as well as falls (6.8% versus 5.1% in the KALM-2 trial) and nausea (6.4% versus 4.2% in the KALM-2 trial. Overall, during the 12-week treatment period of the KALM-1 and KALM-2 trials, patients who received difelikefalin reported notable harms at a similar or slightly higher frequency than patients who received placebo. There were imbalances in the proportion of patients (difelikefalin versus placebo) reporting diarrhea (9.5% versus 3.7%) and dizziness (6.9% versus 1.1%) as an AE in the KALM-1 trial.
Table 23: Summary of Harms (Double-Blind Safety Analysis Set)
Harms | KALM-1 | KALM-2 | ||
|---|---|---|---|---|
Difelikefalin 0.5 mcg/kg N = 189 | Placebo N = 188 | Difelikefalin 0.5 mcg/kg N = 235 | Placebo N = 236 | |
Patients with ≥ 1 TEAEa | ||||
Patients with any reported TEAE, n (%) | 130 (68.8) | 117 (62.2) | 160 (68.1) | 145 (61.4) |
Most common events,b n (%) | ||||
Diarrhea | 18 (9.5) | 7 (3.7) | 19 (8.1) | 13 (5.5) |
Dizziness | 13 (6.9) | 2 (1.1) | 13 (5.5) | 12 (5.1) |
Vomiting | 10 (5.3) | 6 (3.2) | 15 (6.4) | 14 (5.9) |
Nasopharyngitis | 6 (3.2) | 10 (5.3) | 3 (1.3) | 5 (2.1) |
Nausea | 6 (3.2) | 9 (4.8) | 15 (6.4) | 10 (4.2) |
Falls | 5 (2.6) | 5 (2.7) | 16 (6.8) | 12 (5.1) |
Patients with ≥ 1 serious TEAEs | ||||
Patients with any reported serious TEAE, n (%) | 49 (25.9) | 41 (21.8) | 58 (24.7) | 51 (21.6) |
Most common events,c n (%) | ||||
Hyperkalemia | 4 (2.1) | 4 (2.1) | 4 (1.7) | 3 (1.3) |
Sepsis | 3 (1.6) | 4 (2.1) | 3 (1.3) | 3 (1.3) |
Pneumonia | 3 (1.6) | 5 (2.7) | 2 (0.9) | 0 |
Fluid overload | 2 (1.1) | 4 (2.1) | NR | NR |
Chest pain | 1 (0.5) | 3 (1.6) | 8 (3.4) | 1 (0.4) |
Patients who stopped treatment due to TEAEs | ||||
Patients with any TEAE leading to study drug discontinuation, n (%) | 15 (7.9) | 9 (4.8) | 13 (5.5) | 8 (3.4) |
Most common events,d n (%) | ||||
Dizziness | 3 (1.6) | 0 | 1 (0.4) | 1 (0.4) |
Septic shock | 0 | 3 (1.6) | NR | NR |
Pneumonia | 0 | 2 (1.1) | NR | NR |
Death | ||||
n (%) | 2 (1.1) | 3 (1.6) | 2 (0.9) | 2 (0.8) |
Cause of death, n (%) | ||||
Septic shock | 0 | 2 (1.1) | 0 | 0 |
Sepsis | 2 (1.1) | 0 | 0 | 0 |
Dyspnea and/or hypotension | 0 | 0 | 1 (0.4) | 1 (0.4) |
Cardiac arrest | 0 | 0 | 1 (0.4) | 1 (0.4) |
Unknown | 0 | 1 (0.5) | 0 | 0 |
Notable harms | ||||
Diarrhea | 18 (9.5) | 7 (3.7) | 19 (8.1) | 13 (5.5) |
Dizzinesse | 13 (6.9) | 2 (1.1) | 13 (5.5) | 12 (5.1) |
Vomiting | 10 (5.3) | 6 (3.2) | 15 (6.4) | 14 (5.9) |
Hyperkalemia | 8 (4.2) | 5 (2.7) | 9 (3.8) | 6 (2.5) |
Headache | 7 (3.7) | 4 (2.1) | 10 (4.3) | 6 (2.5) |
Somnolencee | 6 (3.2) | 4 (2.1) | 11 (4.7) | 5 (2.1) |
Nausea | 6 (3.2) | 9 (4.8) | 15 (6.4) | 10 (4.2) |
Back pain | 6 (3.2) | 1 (0.5) | 0 | 0 |
Fallsd | 5 (2.6) | 5 (2.7) | 16 (6.8) | 12 (5.1) |
Paresthesia (unusual feeling or sensation) | 5 (2.6) | 7 (3.7) | 11 (4.7) | 6 (2.5) |
Mental status changese | 3 (1.6) | 3 (1.6) | 3 (1.3) | 1 (0.4) |
Tachycardiae,f | 2 (1.1) | 1 (0.5) | 1 (0.4) | 6 (2.5) |
Gait disturbanced | 1 (0.5) | 2 (1.1) | 7 (3.0) | 2 (0.8) |
Seizuree | 1 (0.5) | 1 (0.5) | NR | NR |
Syncopee | 1 (0.5) | 1 (0.5) | 4 (1.7) | 3 (1.3) |
Altered moode | 1 (0.5) | 0 | 0 | 1 (0.4) |
Palpitationse | 0 | 2 (1.1) | 3 (1.3) | 1 (0.4) |
NR = not reported; TEAE = treatment-emergent adverse event.
aTEAEs relative to the double-blind treatment period are identified as any AE with an onset date after the first dose of the study drug up to the EOT or ET visit or the start of the discontinuation period or 10 days after the last dose if no EOT or ET visit was conducted, whichever is later.
bFrequency > 5%.
cFrequency > 2%.
dFrequency > 1%.
eReported based on the TEAEs of special interest identified by the sponsor.
fTachycardia included the following preferred terms: tachycardia, sinus tachycardia, and tachyarrhythmia.
Sources: KALM-1 Clinical Study Report;19 KALM-2 Clinical Study Report.20
Critical Appraisal
Internal Validity
The KALM-1 and KALM-2 trials appear to have used accepted methods for blinding, allocation concealment, and randomization with stratification. Overall, study baseline demographic and disease characteristics were well balanced between treatment groups in both KALM trials. In the KALM-1 trial, there was an imbalance between the treatment groups in years since diagnosis of ESRD (median = 3.67 years [range, 0.3 to 26.5] versus 4.10 [range, 0.3 to 28.7) for difelikefalin versus placebo], indicating patients in the difelikefalin group had less disease burden and may have had a better response to treatment than those in the placebo group. This difference in patients may introduce a high risk of selection bias and lead to uncertainty in the results. In the KALM-2 trial, there was a difference in the proportion of patients who had diabetes (||||| versus ||||| for difelikefalin versus placebo), and the proportion of patients with hyperphosphatemia was higher in the difelikefalin treatment group than in the placebo group (||||| versus |||||). The clinical experts consulted by CADTH indicated that diabetes and mineral disorders (i.e., hyperphosphatemia) have direct associations with pruritus. The impact of the differences in disease history on efficacy results is uncertain due to limited data reported with respect to the duration of medical history and the severity of these issues. The frequency of hemodialysis (i.e., optimization of hemodialysis) is a potential effect modifier and prognostic factor that was not considered in either KALM trial. Patients with more frequent hemodialysis visits would have better control of the disease and more exposure to difelikefalin than patients with less frequent hemodialysis visits. In addition, more frequent hemodialysis visits indicated better compliance with difelikefalin treatment. Therefore, the frequency of hemodialysis may have a potential impact on the validity of the study results for the treatment effect of difelikefalin; however, the magnitude of the impact is unknown, as there were no data with regard to the treatment effect in patients with different frequencies of hemodialysis in either KALM trial.
Approximately one-third of patients in the studies had at least 1 major protocol deviation (||||| to ||||| across treatment arms at 12 weeks). Particularly in the KALM-2 trial, there were high rates of major protocol deviation due to noncompliance in both treatment groups (||||| versus ||||| for difelikefalin versus placebo) and procedure not performed (||||| versus ||||| for difelikefalin versus placebo). Although the number of major protocol deviations is a limitation, these events were roughly balanced between treatment arms in each study, and the results of the sensitivity and supplementary analyses (including PP analysis) were consistent with the primary estimand.
The primary end point in the KALM-1 and KALM-2 studies was the proportion of patients who achieved a 3-point reduction in WI-NRS scores at week 12, which was used to define improvement and response to treatment for pruritus intensity. The 3-point improvement in WI-NRS scores was considered the MID in patients with CKD-aP, based on a study that used data from a phase II study (CR845-CLIN2101, NCT02858726) and the KALM-1 and KALM-2 trials conducted by the sponsor.30 However, the FDA multidiscipline review for difelikefalin recommended an improvement of at least 4 points as the cut-off for the primary efficacy end point (i.e., proportion of patients with a ≥ 4-point improvement in WI-NRS at week 12) in the target population for both KALM trials.16 In both the KALM-1 and KALM-2 trials, although odds ratios at week 12 were similar based on the 3-point and 4-point cut-offs, the proportion of patients in each treatment group who met the threshold was higher with the 3-point threshold, which was used for the primary end point. The CADTH review team observed that the sponsor used odds ratios to estimate the treatment effect in the primary and key secondary outcomes. It is important to note that odds ratios tend to give an inflated impression of the treatment effects compared with relative risks.21 Therefore, the results of the treatment effect of difelikefalin, compared to placebo, estimated using odds ratios should be interpreted with caution.
There was an imbalance in the proportion of patients (difelikefalin versus placebo) who discontinued the double-blind treatment period (14.3% versus 9.6% in the KALM-1 trial and 12.3% versus 5.5% in the KALM-2 trial). The most common reason for discontinuation in both KALM trials was AEs. Discontinuation due to AEs was slightly higher in the difelikefalin group than in the placebo group in both KALM trials (7.4% versus 4.8% in the KALM-1 trial and 5.5% versus 3% in the KALM-2 trial). Although the clinical experts consulted by CADTH indicated that imbalances in discontinuation were not seen as having a differential effect on blinding or outcomes, as the majority of patients (88.1% in the KALM-1 trial and 91.1% in the KALM-2 trial) completed the study treatment in both KALM trials, there is a potential risk that patients may have been aware of their treatment assignment as a result of the differential rate of reported AEs. Moreover, the WI-NRS scale used in the KALM trials to collect and assess pruritus severity in patients with CKD-aP is a subjective patient-reported outcome measure, so the awareness of treatment may introduce performance bias in the reporting of the results.
In the KALM trials, between ||||| and ||||| of the data were missing for assessments of the primary end points at week 12. Of note, the rate of missing data was higher in the difelikefalin group than in the placebo group (||||| versus ||||| in the KALM-1 trial and ||||| versus ||||| in the KALM-2 trial) for the primary end point of both KALM trials. There were no data reported regarding the demographic and disease characteristics of the patients with missing WI-NRS scores. Given that AEs were the most common cause for discontinuation in both of the KALM trials, there is a high possibility that patients with missing WI-NRS scores dropped out due to AEs or lack of response. The sponsor performed 3 sensitivity analyses with different assumptions (i.e., early discontinuations as nonresponders, missing not at random, and mixed missing data mechanisms with missing not at random for difelikefalin and missing at random for placebo) to assess the robustness of the missing-at-random assumptions. The results of the sensitivity analyses were generally similar, irrespective of the method used to impute the missing data, indicating that the missing-at-random assumption for the multiple imputation in the ITT population was reasonable.
In addition, whether patients with missing WI-NRS scores would satisfy the multiple imputation assumption, which assumes that patients who discontinue double-blind treatment early would have WI-NRS scores similar to other patients in their respective treatment arm who have complete data, is uncertain, given the limited available data. Thus, there was a potential risk of attrition bias, as there was a disproportionally high rate of patients with missing WI-NRS scores in the treatment group, compared with the placebo groups, and it is uncertain whether those patients would systematically differ from those who remained in their respective study arms. The direction of the observed attrition bias is uncertain, as there were no data available with regard to the demographics and disease characteristics for patients with missing WI-NRS scores in either KALM trial.
The clinical experts consulted by CADTH indicated that the clinical relevance of the subgroup variables in the KALM-1 and KALM-2 trials were limited. Subgroups of particular relevance, such as the use of concomitant medications to treat pruritus (e.g., antihistamines, gabapentin, pregabalin), comorbid conditions (e.g., diabetes, neuropathies, liver disease, dermatologic conditions), and pruritus severity (moderate versus severe) were not assessed in the KALM trials. It is not clear whether the effect of difelikefalin would be consistent across these potentially relevant subgroups.
In the KALM trials, only the primary (i.e., proportion of patients with a ≥ 3-point improvement from baseline in WI-NRS score at week 12) and key secondary outcomes (i.e., proportion of patients with a ≥ 3-point improvement from baseline in WI-NRS score at week 4 and week 8, proportion of patients with a ≥ 4-point improvement from baseline in WI-NRS score at week 4, week 8, and week 12, and change from baseline in Skindex-10 scale total score and 5-D itch scale total score) were controlled for multiple statistical testing using a gate-keeping procedure. The lack of adjustments for multiplicity for the domain scores for the Skindex-10 and 5-D itch scales and the PGIC makes it challenging to draw conclusions surrounding any difference between treatment groups for these end points in the KALM trials. Moreover, neither the KALM-1 trial nor the KALM-2 trial were powered to detect a change in secondary end points. Therefore, these outcomes should be interpreted with caution.
The WI-NRS is a valid, reliable, and responsive questionnaire for assessing pruritus severity in patients with CKD-aP, and a 3-point improvement in Wi-NRS scores in both KALM trials was considered clinical meaningful, according to the reported MID threshold for symptom reduction (≥ 3 points). The PGIC used to evaluate improvement in pruritus severity relative to baseline status in the KALM trials is commonly used in clinical trials; however, the validity, reliability, and responsiveness of the instrument has not been studied in patients with CDK-aP. In addition, the Skindex-10 and 5-D itch scales used for HRQoL assessment are generally appropriate for patients with CKD-aP; however, no evidence of MIDs for these 2 measurements in patients with CKD-aP were identified in the literature search conducted by CADTH.
External Validity
The median age of all randomized patients in both KALM trials was around 60 years (58.0 years in the KALM-1 trial and 60.0 years in the KALM-2 trial), and majority of patients were aged 45 years to 64 years (56.8% in the KALM-1 trial and 51.6% in the KALM-2 trial). The clinical experts consulted by CADTH felt that patients on hemodialysis in Canada would be older, on average, with most patients 65 years and older. The KALM-1 trial included study sites in the US only, whereas the KALM-2 trial included 5 study sites in Canada; however, the clinical experts indicated that the demographics of the patient population in both trials was consistent with patients living with CKD in Canada. Thus, the differences between the study population and the patient population in Canada would not likely bias the generalizability of the study results to patients with CKD-aP in Canada.
In the KALM trials, difelikefalin was administered with an IV bolus injection into the venous line of the dialysis circuit at the end of each hemodialysis session. The majority of patients (||||| in the KALM-1 trial and ||||| in the KALM-2 trial) had 34 to 36 doses (times) administered, and ||||| of patients in the KALM-1 trial and ||||| in the KALM-2 trial missed 1 to 3 doses during the 12 weeks of the double-blind treatment period. The clinical experts for this review indicated that the patients in the KALM-1 and KALM-2 trials had better hemodialysis adherence than patients in Canadian clinical practice. The clinical experts stated that better hemodialysis adherence (i.e., hemodialysis optimization) may have contributed to the observed improvement in pruritus severity, and thus the observed treatment effect of difelikefalin may have been overestimated in both the treatment and placebo groups in the KALM trials. In addition, the baseline median WI-NRS score was around 7 in both KALM trials (7.14 in the KALM-1 trial and 7.13 in the KALM-2 trial). The clinical experts consulted by CADTH indicated that patients with CKD-aP would be expected to have a worse itch intensity (with a numeric rating scale greater than 8) in dermatology clinical practice in Canada. Overall, the selection of patients with better hemodialysis adherence and less pruritus severity may limit the applicability of the study results to the real-world patient population in Canada, and may have introduced selection bias, which may lead to uncertainty about the efficacy results.
In the KALM trials, data for the primary and secondary or other outcomes were reported up to 12 weeks. According to the clinical experts, patients with CKD-aP would receive the difelikefalin treatment beyond 12 weeks for the desired treatment effect. As there were no long-term data reported in the KALM trials, it is uncertain whether the treatment effect or the safety of difelikefalin beyond 12 weeks would be consistent with the results at week 12.
The comparator used for both KALM trials was placebo. The clinical experts consulted by CADTH indicated that other relevant treatment options, such as topical treatments, gabapentin, and phototherapy, are commonly used in clinical practice. The lack of active comparators for the KALM trials does not allow conclusions to be drawn about the efficacy of difelikefalin relative to commonly used antiitch therapies in clinical practice.
In the pivotal KALM studies, pruritus severity was measured using the WI-NRS and PGIC, and HRQoL was measured using the Skindex-10 and 5-D itch scales. However, the experts consulted by CADTH stated that these outcome measures are not routinely used in clinical practice to assess itch intensity or HRQoL in patients. Therefore, there is uncertainty about how the changes in pruritus severity measured with the WI-NRS and PGIC and HRQoL measured with the Skindex-10 and 5-D itch scales translate to clinical practice.
Indirect Evidence
No indirect evidence was identified for this review.
Other Relevant Evidence
One open-label, multicentre, phase III study (CR845-CLIN3101) that evaluated the long-term safety of difelikefalin at a dose of 0.5 mcg/kg administered for up to 52 weeks was included as other relevant evidence to address the gap in the long term safety of difelikefalin for this review.
Description of Studies
The long-term safety study included patients who participated in the phase II studies of difelikefalin (CR845-CLIN2005 and CR845-CLIN2101). The long-term safety study also included de novo patients with moderate-to-severe CKD-aP undergoing hemodialysis who had not been previously exposed to difelikefalin and had not participated in the phase II studies of difelikefalin. Figure 6 shows the study design and key features of the open-label, phase III study, which consisted of a screening visit, a 52-week treatment period, an EOT visit, and a follow-up visit 7 to 10 days after the EOT visit. To confirm eligibility, the screening visit was scheduled in the 14 days before administration of the first dose of the study drug. The eligibility criteria for de novo patients required moderate-to-severe pruritus, measured with the 24-hour WI-NRS. Scores for the WI-NRS were collected at each dialysis visit in the week before administration of the first dose (including the score on day 1 of dosing). At least 1 WI-NRS score greater than 4 was needed to be eligible for participation. Patients remained unaware of these eligibility criteria.
Patients received difelikefalin at a dose of 0.5 mcg/kg 3 times per week after a dialysis session for up to 52 weeks, for a total of approximately 156 doses of the study drug. All scheduled study visits were conducted on dialysis days during the treatment period. The final dose was administered at the final dialysis visit on week 52, or at the ET visit. The EOT visit was conducted at the dialysis visit after the final dose. A final safety follow-up was conducted 7 to 10 days after the EOT or ET visit.
Figure 6: Study Schematic for the Open-Label, Phase III Study
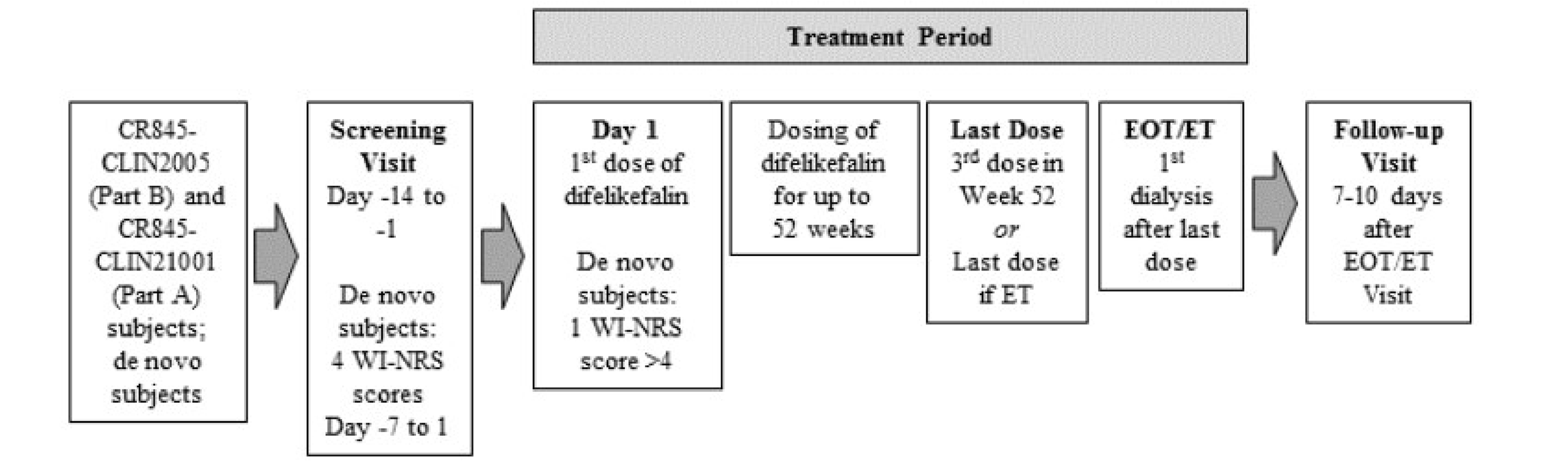
EOT = end of treatment; ET = early termination; WI-NRS = Worst Itching Intensity Numerical Rating Scale.
Source: Clinical Study Report for the open-label, phase III study.37
Populations
Inclusion and Exclusion Criteria
Patients meeting the following inclusion criteria were included in this study:
being 18 years or older and participating in either of the phase II studies or being a de novo patient
being currently on hemodialysis for ESRD and being categorized as experiencing moderate-to-severe CKD-aP as part of either of the phase II studies or as a de novo patient
having continued to experience CKD-aP since the previous phase II studies or, for de novo patients, having experienced CKD-aP at the time of screening
having a dry body weight of at least 40.0 kg at screening (prescription target dry body weight)
having adequacy of dialysis, defined as meeting any 1 of the following criteria during the 3 months before screening —
at least 2 single-pool measurements of (dialyzer clearance of urea × dialysis time) / (volume of distribution of urea) of ≥ 1.2 or
at least 2 urea reduction ratio measurements of at least 65% or
1 single-pool measurement of (dialyzer clearance of urea × dialysis time) / (volume of distribution of urea) of at least 1.2 and 1 urea reduction ratio measurement of at least 65%
for de novo patients, having recorded up to 4 WI-NRS scores at dialysis visits over the week before the first dose (including the score on day 1 of dosing) and having at least 1 of the WI-NRS scores be above 4.
Patients meeting the following criteria were excluded from the study:
having received an investigational drug (other than the study drug while participating in the phase II studies) in the 30 days before the first dose of the study drug, or planning to participate in another interventional clinical study while enrolled in the study
having a concomitant disease or any medical condition that could have posed undue risk to the patient, impeded completion of the study procedures, or compromised the validity of the study measurements
having abnormal liver function, defined as —
serum alanine aminotransferase or aspartate aminotransferase more than 2.5 times the reference upper limit of normal at screening and/or
total bilirubin more than 2 times the upper limit of normal at screening.
De novo patients meeting the following criteria were excluded from the study:
a known history of allergic reaction to opiates, such as hives or anaphylaxis
having pruritus attributed to a cause other than ESRD or its complications (e.g., patients with concomitant pruritic dermatological disease or cholestatic liver disease), determined by the investigator
having localized itch restricted to the palms of the hands.
Baseline Characteristics
Table 24 Summarizes the baseline characteristics of participants in the open-label, phase III trial.
Table 24: Summary of Baseline Characteristics in the Open-Label, Phase III Study (Safety Population)
Characteristic | Pbo/CR845 (N = 30) | CR845/CR845 (N = 52) | De novo (N = 206) |
|---|---|---|---|
Demographics | |||
Age, years | |||
Mean (SD) | |||| |||||||| | |||| |||||||| | |||| |||||||| |
Median (range) | |||| ||||| || |||||| | |||| ||| || |||| | |||| ||| || |||| |
Sex, n (%) | |||
Female | | ||||||| | || ||||||| | || |||||||| |
Male | || ||||||| | || ||||||| | ||| |||||||| |
Race, n (%) | |||
American Indian or Alaska Native | | |||||| | | ||||||||| | | ||||| |
Asian | | ||||||| | | ||||||| | | ||||| |
Black or African American | || |||||||| | || |||||||| | ||| |||||| |
Native Hawaiian or other Pacific Islander | ||| | ||| | | ||||||| |
White | | |||||||| | || |||||| | || |||||| |
Not reported | ||| | ||| | | ||||| |
Unknown | ||| | | ||||||| | | ||||| |
Other | ||| | | ||||||| | | ||||| |
Ethnicity, n (%) | |||
Hispanic or Latino | | ||||||| | || |||||| | || |||||| |
Not Hispanic or Latino | || ||||||| | || |||||| | ||| |||||| |
Target dry body weight (kg) | |||
Mean (SD) | ||||| |||||||| | ||||| |||||||| | ||||| |||||||| |
Median (range) | ||||||||||||| || ||||||| | |||||||||||| || ||||||| | ||||||||||| || ||||||||| |
Disease characteristics | |||
Baseline WI-NRS (de novo only) | |||
Mean (SD) | ||| | ||| | |||| ||||||| |
Median (range) | ||| | ||| | |||| |||| || |||||| |
Baseline maximum WI-NRS (de novo only) | |||
Mean (SD) | ||| | ||| | |||| ||||||| |
Median (range) | ||| | ||| | |||| |||| || |||||| |
Baseline antiitch medication use, n (%) | |||
Yes | | |||||| | || |||||| | || |||||| |
No | || |||||||| | || |||||| | ||| |||||| |
Duration of pruritus (years) | ||| | ||| | ||| |
Mean (SD) | |||| ||||||| | |||| ||||||| | |||| ||||||| |
Median (range) | |||| |||| || |||||| | |||| |||| || |||||| | |||| |||| || |||||| |
Years since diagnosis of ESRD | ||| | ||| | ||| |
Mean (SD) | |||| |||||| | |||| |||||| | |||| ||||||| |
Median (range) | |||| |||| || |||||| | |||| |||| || |||||| | |||| |||| || |||||| |
Years since diagnosis of CKD | ||| | ||| | ||| |
Mean (SD) | |||| |||||| | |||| |||||| | |||| |||||| |
Median (range) | |||| |||| || |||||| | |||| |||| || |||||| | |||| |||| || ||||||| |
Years on chronic hemodialysis | ||| | ||| | ||| |
Mean (SD) | |||| ||||||| | |||| ||||||| | |||| ||||||| |
Median (range) | |||| |||| || |||||| | |||| |||| || |||||| | |||| || ||| || |||||| |
Medical history (≥ 3% of patients), n (%) | |||
Hypertension | || |||||| | || |||||| | ||| |||||| |
Diabetes | || |||||| | || |||||| | ||| |||||| |
Large vessel disease | | ||||| | | ||||| | | ||||| |
Glomerulonephritis | | |||||| | | ||||| | || ||||| |
Interstitial nephritis | | ||||| | ||| | | ||||| |
Other | | |||||| | | |||||| | || ||||||| |
CKD = chronic kidney disease; ESRD = end-stage renal disease; SD = standard deviation; WI-NRS = Worst Itch Intensity Numerical Rating Scale.
Note: The Pbo/CR845 group consists of patients randomized to the placebo group in a previous phase II study; the CR845/CR845 group consists of patients randomized to the difelikefalin group in a previous phase II study; and the de novo group consists of patients who did not participate in a previous phase II study.
Source: Clinical Study Report for the open-label, phase III study.37
Results
Patient Disposition
A summary of patient disposition for the open-label, phase III study is summarized in Table 25.
A total of 315 patients were enrolled in the study, of which 288 (91.4%) received the study treatment. Among these 288 patients, 30 (10.4%) had been randomized to the placebo group, 52 (18.1%) had been randomized to the active treatment group in 1 of the phase II studies, and the remaining 206 (71.5%) were de novo patients.
A total of 133 (46.2%) patients out of 288 completed the study treatment. Overall, 57 (19.8%) patients discontinued the study treatment because the sponsor stopped the study for administrative reasons unrelated to safety concerns (0.0%, 1.9%, and 27.2% for the placebo and difelikefalin group in the previous studies and the de novo group in the open-label, phase III study, respectively), whereas 98 (34.0%) patients discontinued the study treatment for other reasons (40.0%, 32.7%, and 33.5%, respectively). For the group of patients who discontinued for other reasons, the most common reasons for discontinuation were AEs, withdrawal of consent, and other reasons.
Table 25: Patient Disposition (All Patients, Enrolled Analysis Set) for the Open-Label, Phase III Study
Patient disposition | Pbo/CR845 | CR845/CR845 | De novo |
|---|---|---|---|
Screened/enrolled, n | 315 | ||
Randomized, n (%) | || | || | ||| |
Completed study treatment, n (%) | || |||||| | || |||||| | || |||||| |
Discontinued study treatment (except due to the sponsor stopping the study), n (%) | || |||||| | || |||||| | || ||||||| |
Reason for discontinuation, n (%) | |||
Adverse events | | |||||| | | |||||| | || |||||| |
Lack of therapeutic efficacy | ||| | ||| | ||| |
Lost to follow-up | ||| | ||| | | ||||| |
Pregnancy | ||| | ||| | ||| |
Eligibility (inclusion and exclusion criteria) | ||| | ||| | | ||||| |
Noncompliance | | ||||| | ||| | | ||||| |
Withdrawal of consent | | ||||| | | ||||| | || ||||| |
Administrative | ||| | ||| | | ||||| |
Other | | |||||| | | |||||| | || |||||| |
Discontinued study treatment because the sponsor stopped the study, n (%) | ||| | | ||||| | || |||||| |
Completed follow-up, n (%) | || |||||| | || |||||| | || |||||| |
Safety population,a n (%) | || ||||||| | || ||||||| | ||| ||||||| |
Note: The Pbo/CR845 group consisted of patients randomized to the placebo group in a previous phase II study; the CR845/CR845 group consisted of patients randomized to the difelikefalin group in a previous phase II study; and the de novo group consisted of patients who did not participate in a previous study.
aThe safety population included any patient who received the study treatment.
Source: Clinical Study Report for the open-label, phase III study.37
Protocol Deviation
The incidence of major and minor protocol deviations in the open-label CR845-CLIN3101 study is presented in Table 27. The proportion of patients reporting at least 1 major protocol deviation was ||||| and at least 1 minor deviation was ||||||. The most frequently identified categories of major deviations (≥ 2% of all patients) were investigational product accountability management (|||||), good clinical practice noncompliance (||||), and informed consent (||||).
Table 26: Protocol Deviations (Safety Population) for the Open-Label, Phase III Study
Protocol deviations | Pbo/CR845 N = 30 | CR845/CR845 N = 52 | De novo N = 206 |
|---|---|---|---|
Number of patients with any major protocol deviation, n (%) | || |||||| | || |||||| | || |||||| |
Informed consent | | ||||| | | ||||| | | |||||| |
Eligibility criteria | | ||||| | | ||||| | | ||||| |
Tests, assessments, procedure | | ||||| | ||| | | |||||| |
IP accountability management | || |||||| | || |||||| | || |||||| |
AE and SAE reporting | | ||||| | | ||||| | | ||||| |
GCP noncompliance | ||| | | ||||| | || ||||| |
Others | | ||||||| | || ||||||| | || ||||||| |
Number of patients with any minor protocol deviation, n (%) | || |||||| | || |||||| | ||| |||||| |
Informed consent | | ||||| | | ||||| | | ||||| |
Eligibility criteria | | ||||| | | ||||| | || |
Prohibited concomitant medication | ||| | | |||||| | || |
Tests, assessments, procedure | || |||||| | || |||||| | ||| ||||||| |
IP accountability management | || |||||| | || |||||| | ||| |||||| |
AE and SAE reporting | | ||||| | ||| | | ||||| |
GCP noncompliance | ||| | | ||||| | || |
Other | | ||||| | ||| | | ||||| |
AE = adverse event; GCP = good clinical practice; IP = investigational product; SAE = serious adverse event.
Note: The Pbo/CR845 group consisted of patients randomized to the placebo group in a previous phase II study; the CR845/CR845 group consisted of patients randomized to the difelikefalin group in a previous phase II study; and the de novo group consisted of patients who did not participate in a previous phase II study.
Source: Clinical Study Report for the open-label, phase III study.37
Efficacy
No efficacy outcomes were assessed in the open-label, phase III study.
Harms
Table 28 summarizes the harms outcome for the safety population in the open-label, phase III study. The safety population was defined as all enrolled patients who had received at least 1 dose of the study drug during the open-label phase. The harms results were reported descriptively.
Adverse Events
The percentage of patients with any reported TEAEs in the open-label, phase III study was ||||| ||| ||||| in the placebo and difelikefalin groups in the previous studies, respectively, and ||||| in the de novo group. The most common TEAEs (> 10%) were nausea ||||||| |||||| ||| |||||||, diarrhea ||||||| |||||| ||| |||||||, falls ||||||| |||||| ||| |||||||, vomiting ||||||| |||||| ||| |||||||, hypotension ||||||| |||||| ||| |||||||, noncardiac chest pain |||||| |||||| ||| |||||||, hyperkalemia (||||| |||||| ||| ||||||), dizziness ||||||| ||||| ||| |||||, abdominal pain ||||||| ||||| ||| |||||, and acute myocardial infarction ||||||| |||||| ||| ||||| in the placebo and difelikefalin groups in the previous studies and in the de novo group in the open-label, phase III study, respectively. The percentage of patients with any TEAEs in the open-label, phase III study was higher than the percentages seen in the pivotal KALM-1 and KALM-2 studies.
Serious Adverse Events
Serious TEAEs (SAEs) were reported in ||||| and ||||| in the placebo and difelikefalin group in the previous studies, respectively, and in ||||| in the de novo group. The most common SAEs (> 4%) were acute myocardial infarction (|||||| ||||| ||| ||||||), angina pectoris |||||| ||||| ||| ||||||, gastrointestinal hemorrhage |||||| ||||| ||| ||||||, pneumonia ||||||| ||||| ||| ||||||, cellulitis |||||| ||||| ||| ||||||, fluid overload |||||| ||||| ||| ||||||, hyperkalemia |||||| ||||| ||| ||||||, respiratory failure ||||||| ||||| ||| ||||||, pulmonary edema |||||| ||||| ||| |||||, and hypotension |||||| ||||| ||| ||||| in the placebo and difelikefalin groups in the previous studies and in the de novo group in the open-label, phase III study, respectively. The percentage of patients with any SAEs in the open-label, phase III study was higher than the percentages seen in the pivotal KALM-1 and KALM-2 studies.
Withdrawals Due to Treatment-Emergent Adverse Events
The proportion of patients who discontinued treatment due to TEAEs was |||||| |||||| ||| ||||| in the placebo and difelikefalin groups in the previous studies and in the de novo group in the open-label, phase III study, respectively. Cardiac arrest was the most frequently reported TEAE (> 3%) to cause discontinuation in the difelikefalin group |||||||, whereas myocardial infarction, acute myocardial infarction, chills, fatigue, Clostridium difficile colitis, decreased blood sugar, visual hallucinations, and insomnia were the most frequently reported TEAEs to cause discontinuation in the placebo crossover group |||||||. The de novo group had the least reported TEAEs leading to study drug discontinuation compared with other 2 groups. The percentage of patients with any TEAE leading to study drug discontinuation in the open-label, phase III study was higher than the percentage seen in the pivotal KALM-1 and KALM-2 studies.
Mortality
Deaths were reported in |||| of patients in the placebo crossover group, |||| in the difelikefalin group, and |||| in the de novo group. No specific AE was identified to account for the majority of deaths in any group in the open-label, phase III study.
Notable Harms
The most common serious notable harms were diarrhea ||||||| |||||| ||| |||||||, nausea ||||||| |||||| ||| |||||||, falls ||||||| |||||| ||| |||||||, vomiting ||||||| |||||| ||| |||||||, dizziness ||||||| ||||| ||| ||||||, hyperkalemia |||||| |||||| ||| ||||||, mental status changes |||||| |||||| ||| ||||||, unusual feeling or sensation |||||| |||||| ||| ||||||, syncope |||||| ||||| ||| ||||||, and altered mood |||| ||||| ||| ||||| in the placebo crossover, difelikefalin, and de novo groups, respectively. The percentage of patients with the serious notable harms in the open-label, phase III study was higher than the percentage seen in the pivotal KALM-1 and KALM-2 studies.
Table 27: Summary of Harms (Safety Population) for the Open-Label, Phase III Study
Harms | Pbo/CR845 N = 30 | CR845/CR845 N = 52 | De novo N = 206 |
|---|---|---|---|
Patients with ≥ 1 TEAEs | |||
Patients with any reported TEAE, n (%) | || |||||| | || |||||| | ||| |||||| |
Most common events,a n (%) | |||
Nausea | | |||||| | || |||||| | || |||||| |
Diarrhea | | |||||| | || |||||| | || |||||| |
Falls | | |||||| | || |||||| | || |||||| |
Vomiting | | |||||| | || |||||| | || |||||| |
Hypotension | | |||||| | || |||||| | || |||||| |
Noncardiac chest pain | | ||||| | | |||||| | || |||||| |
Hyperkalemia | | ||||| | | |||||| | || ||||| |
Dizziness | | |||||| | | ||||| | || ||||| |
Abdominal pain | | |||||| | | ||||| | || ||||| |
Fluid overload | | ||||| | | ||||| | || ||||| |
Pneumonia | ||| | | ||||| | || ||||| |
Dyspnea | ||| | | ||||| | || ||||| |
Acute myocardial infarction | | |||||| | | |||||| | | ||||| |
Pain in extremity | | ||||| | | ||||| | || ||||| |
Arthralgia | | ||||| | | ||||| | | ||||| |
Asthenia | | ||||| | | ||||| | | ||||| |
Patients with ≥ 1 serious TEAEs | |||
Patients with any reported serious TEAE, n (%) | || |||||| | || |||||| | || |||||| |
Most common events,b n (%) | |||
Acute myocardial infarction | | |||||| | | ||||| | |||| ||||| |
Angina pectoris | | ||||| | | ||||| | | ||||| |
Gastrointestinal hemorrhage | | ||||| | | ||||| | | ||||| |
Pneumonia | ||| | | ||||| | | ||||| |
Cellulitis | | ||||| | ||| | | ||||| |
Fluid overload | | ||||| | | ||||| | || ||||| |
Hyperkalemia | | ||||| | | ||||| | | ||||| |
Respiratory failure | | |||||| | ||| | | ||||| |
Pulmonary edema | | ||||| | | ||||| | | ||||| |
Hypotension | | ||||| | | ||||| | | ||||| |
Patients who stopped treatment due to TEAEs | |||
Patients with any TEAE leading to study drug discontinuation, n (%) | | |||||| | | |||||| | |||| |||||| |
|||| |||||| |||||||| | |||| | |||
Cardiac arrest | ||| | | |||||| | | ||||| |
Myocardial infarction | | |||||| | ||| | | ||||||| |
Acute myocardial infarction | | |||||| | ||| | ||| |
Chills | | |||||| | ||| | ||| |
Fatigue | | |||||| | ||| | ||| |
Clostridium difficile colitis | | |||||| | ||| | ||| |
Decreased blood glucose | | |||||| | ||| | ||| |
Hallucination, visual | | |||||| | ||| | ||| |
Insomnia | | |||||| | ||| | ||| |
Deaths | |||
n (%) | | ||||| | | ||||| | || ||||| |
Cause of death, n (%) | |||
Cardiac arrest | ||| | | ||||| | | ||||| |
Sudden death | ||| | ||| | | ||||||| |
Congestive heart failure | ||| | ||| | | ||||| |
Intestinal ischemia | ||| | ||| | | |||||| |
Myocardial infarction | | ||||| | ||| | | |||||| |
Acute myocardial infarction | | ||||| | ||| | ||| |
Upper gastrointestinal hemorrhage and/or aortic stenosis | ||| | | |||||| | ||| |
Cardiac tamponade and/or pericardial effusion | ||| | | |||||| | ||| |
Gastric hemorrhage and/or myocardial infarction | ||| | ||| | | |||||| |
Unknown | ||| | | |||||| | | |||||| |
Notable harms | |||
Diarrhea | | |||||| | || |||||| | || |||||| |
Dizzinessd | | |||||| | | ||||| | || ||||| |
Vomiting | | |||||| | || |||||| | || |||||| |
Hyperkalemia | | ||||| | | |||||| | || ||||| |
Headache | | ||||||| | | ||||| | | ||||| |
Somnolenced | ||| | | ||||| | | ||||| |
Nausea | | |||||| | || |||||| | || |||||| |
Back pain | | ||||| | | ||||| | | ||||| |
Fallsd | | |||||| | || |||||| | || |||||| |
Unusual feeling or sensation | | ||||| | | |||||| | || ||||| |
Mental status changesd | | ||||| | | |||||| | || ||||| |
Tachycardiad,e | ||| | | ||||| | | ||||| |
Gait disturbanced | ||| | ||| | | |||||| |
Seizured | ||| | | ||||| | | ||||| |
Syncoped | | ||||| | | ||||| | | ||||| |
Altered moodd | ||| | | ||||| | | ||||| |
Palpitationsd | | ||||| | | ||||| | ||| |
TEAE = treatment-emergent adverse event.
Note: The Pbo/CR845 group consisted of patients randomized to the placebo group in a previous phase II study; the CR845/CR845 group consisted of patients randomized to the difelikefalin group in a previous phase II study; and the de novo group consisted of patients who did not participate in a previous phase II study.
aFrequency ≥ 5%.
bFrequency > 4%.
cFrequency > 3%.
dReported based on the TEAEs of special interest identified by the sponsor.
eTachycardia included the following preferred terms: tachycardia, sinus tachycardia, and tachyarrhythmia.
Source: Clinical Study Report for the open-label, phase III study.37
Critical Appraisal
Internal Validity
The objective of the open-label, phase III study was to evaluate the long-term safety of difelikefalin, administered intravenously after each dialysis session for up to 52 weeks. The difference in years since the diagnosis of ESRD across treatment groups may be associated with a higher disease burden, which may have affected the safety results (median [range]: |||| ||||| |||| || |||||| |||| ||||| |||| || |||||| ||| |||| ||||| |||| || ||||||) for the placebo and difelikefalin groups in the previous study and in the de novo group in the open-label, phase III study, respectively. Similar trends were seen for years since the diagnosis of CKD and for years on chronic hemodialysis. As mentioned previously, patients with more frequent hemodialysis visits would have more exposure to difelikefalin than patients with fewer hemodialysis visits. Therefore, the frequency of hemodialysis may have a potential impact on the validity of the study results for the treatment effect of difelikefalin; however, the direction and magnitude of the impact is unknown, as there were no data with regard to the treatment effect in patients with different frequencies of hemodialysis in the open-label study. The duration of pruritus was similar in the placebo and difelikefalin groups in the previous study (median [range]: |||| ||||| |||| || ||||||||| |||| ||||| |||| || |||||, respectively). However, for the de novo group, the duration of pruritus (median [range]) was |||| years (||| || ||||). This difference may be associated with a different level of disease burden, which may have affected the safety results as well. Because the results for the open-label, phase III trial were only reported descriptively, the interpretations should be made with caution. Discontinuation rates were high in all 3 treatment groups. One of the issues with discontinuation from an open-label study, particularly when patients discontinue due to AEs, is that the summary of harms may underestimate the frequency of AEs, because those who remained in the study are more likely to have responded well to treatment. Overall, the high discontinuation rates, as well as the descriptive nature of analysis, introduce uncertainty to the long-term safety results. The lack of comparative evidence makes it difficult to interpret the safety results.
External Validity
Although the patient population in the open-label study is different than the KALM-1 and KALM-2 populations, the external validity points related to demographics factors mentioned previously could be applicable to this study population. Patients were expected to receive treatment with difelikefalin at a dose of 0.5 mcg/kg for up to 52 weeks. Although the dose is consistent with the Health Canada–approved dose, the duration of the open-label, phase III trial is longer than the pivotal KALM-1 and KALM-2 trials (12 weeks). It was previously mentioned that that patients would likely receive this treatment beyond 12 weeks, so the safety results of this open-label, phase III study may be generalizable to this time frame to an extent, but not completely, as the rates of AEs are expected to increase with longer treatment times.
Pooled Analysis of OLE of the KALM-1 and KALM-2 Trials
The aim of this section was to summarize and appraise evidence from the study by Topf et al. (2022)17 that was used to inform the clinical report.
Topf et al. (2022)17 conducted a pooled analysis based on data from the KALM-1 and KALM-2 studies evaluating the efficacy of difelikefalin in adults with moderate-to-severe CKD-aP treated with hemodialysis. The aim was to obtain a combined estimate of the treatment effects of difelikefalin in this population. The primary end points assessed in this pooled analysis were itch intensity (evaluated by WI-NRS) and itch-related QoL (evaluated with the Skindex-10 and 5-D itch scales). Outcomes based on the WI-NRS and Skindex-10 scales were assessed up to week 12. The only outcome that was assessed beyond 12 weeks was the 5-D itch scale score, which will be the focus of this summary of the evidence of long-term efficacy. The long-term impacts of difelikefalin on itch intensity and itch-related QoL were evaluated with the 5-D itch scale for up to week 52 of the OLE.
For the statistical analysis, efficacy analyses were conducted in the randomized ITT population pooled from the KALM-1 and KALM-2 studies. A logistic regression model was used to assess differences between the placebo and difelikefalin groups, which included terms for the treatment group, baseline WI-NRS score, geographic region, use of an antiitch medication during the week before randomization, and presence of specific medical conditions. For itch-related QoL assessments, the proportion of patients achieving clinically meaningful improvements in the 5-D itch total score was analyzed without imputation for missing values. The proportion of participants achieving at least a 5-point improvement in 5-D itch total score was reported for the pooled population during both the placebo-controlled, double-blind period (12 weeks) and the OLE period (up to 52 weeks). Mean improvements from baseline in 5-D itch total score were reported for the pooled population during both the placebo-controlled, double-blind period and the OLE period.
Table 28 presents patient disposition for the pooled analysis up to week 12. Patient disposition up to week 52 was not reported.
Table 28: Patient Disposition in the Pooled KALM-1 and KALM-2 Studies
Patient disposition | Pooled KALM-1 and KALM-2 | |
|---|---|---|
Difelikefalin 0.5 mcg/kg | Placebo | |
Screened, n | 1,123 | |
Randomized, n | 426 | 425 |
Completed the double-blind treatment period, n (%) | 368 (86.4) | 393 (92.5) |
Discontinued the double-blind treatment period, n (%) | 56 (13.1) | 31 (7.3) |
Reason for discontinuation, n (%) | ||
Adverse events | 27 (6.3) | 16 (3.8) |
Lack of therapeutic efficacy | 1 (0.2) | 0 |
Lost to follow-up | 1 (0.2) | 0 |
Eligibility (inclusion and exclusion criteria) | 3 (0.7) | 2 (0.5) |
Noncompliance | 2 (0.5) | 3 (0.7) |
Withdrew consent | 13 (3.1) | 7 (1.6) |
Other | 9 (2.1) | 3 (0.7) |
Entered the open-label phase, n | 340 | 372 |
Source: Topf et al. (2022).17
Figure 7: Achievement of a ≥ 5-Point Improvement in 5-D Itch Scale Total Score in the Pooled KALM-1 and KALM-2 Studies
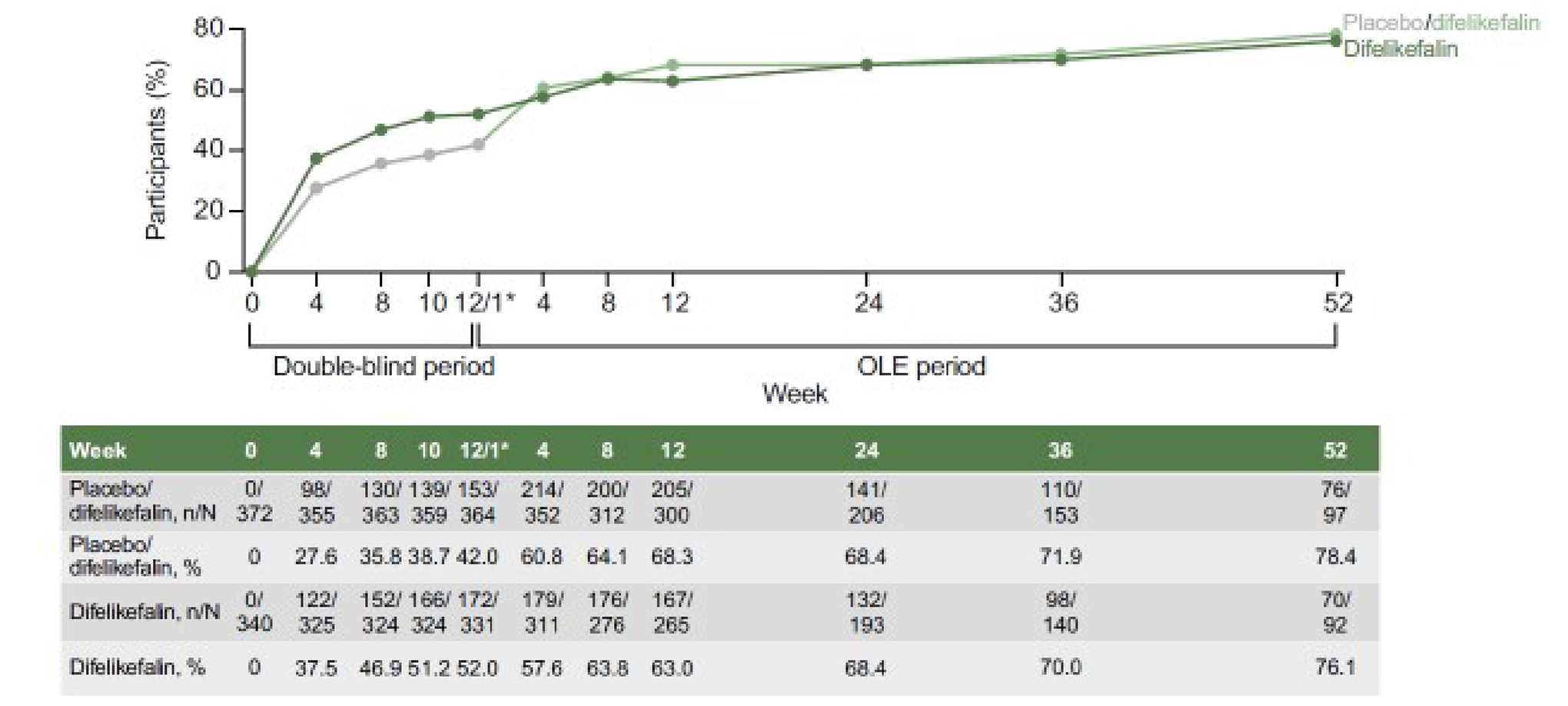
OLE = open-label extension.
Note: Data given as n/N indicate the number of participants who achieved a ≥ 5-point improvement in the 5-D itch scale total score / the total number of participants assessed at each time point, respectively. Data are as observed.
*Week 12 of the double-blind period and week 1 of the OLE period, during which participants taking placebo in the double-blind period switched to active treatment with difelikefalin. In the KALM-2 trial, in addition to the participants who discontinued during the OLE period, 313 of 399 (78.4%) participants could not complete the 52-week OLE period because of the sponsor’s decision to stop the study for reasons unrelated to safety or lack of drug effects. A 2-week discontinuation period after the end of the double-blind period of the KALM-1 trial is not depicted in the figure.
Source: Topf et al. (2022).17
© 2022 The Authors. Published by Elsevier Inc. on behalf of the National Kidney Foundation, Inc.
Reprinted in accordance with Creative Commons Attribution Licence CC BY-NC-ND 4.0.
Figure 7 presents the proportion of patients achieving a clinically meaningful 5-D itch response (≥ 5-point improvement) in the pooled KALM-1 and KALM-2 studies during the 12-week double-blind treatment period and the 52-week OLE period.
The pooled analysis evaluating the long-term efficacy of difelikefalin suggested that the proportion of patients achieving a clinically meaningful 5-D itch response was maintained after the double-blind phase and for up to 52 weeks of the long-term extension phase. The proportion of patients who entered the OLE from the difelikefalin and placebo groups were 97.4% and 97.8%, respectively, and the proportion of patients achieving a clinically meaningful 5-D itch response at week 12 and/or week 1 of the OLE in the difelikefalin and placebo crossover groups were 52.0% and 42.0%, respectively. At week 52 of the OLE, the proportion of patients in the difelikefalin and placebo crossover groups with data available were 27.1% and 26.1%, respectively, and the proportion of patients achieving a clinically meaningful 5-D itch response at week 52 in these 2 groups were 76.1% and 78.4%, respectively. Although the pooled results suggested the long-term efficacy of difelikefalin in patients with moderate-to-severe CKD-aP treated with hemodialysis, the substantial amount of missing data over time introduces uncertainty to the results. The sponsor indicated that a high proportion of missing data in the OLE of the KALM-2 trial was due to the sponsor’s decision to stop the OLE study early for administrative reasons. As a result of the missing data, it is difficult to determine the magnitude and direction of bias. In addition to the amount of missing data, methods to account for missing data were not implemented (i.e., no imputation for missing data). Moreover, the lack of comparative evidence during the OLE made it difficult to interpret the results and the durability of the efficacy of difelikefalin.
Some limitations of this pooled analysis were identified by the authors (such as the small number of subgroup samples and the WI-NRS, Skindex-10 scale, and 5-D itch scale not being used in the routine clinical care of dialysis patients), so the findings may not be appropriate in the real-world setting. In addition, some important efficacy outcomes (e.g., WI-NRS and Skindex-10 scores) were not measured for 52 weeks. Thus, the long-term efficacy outcomes of difelikefalin from this pooled analysis based on 1 outcome, a very small number of patients, and without any comparative evidence need to be interpreted with caution.
Discussion
Summary of Available Evidence
Two phase III, placebo-controlled, RCTs, KALM-1 and KALM-2, were included in the CADTH systematic review. The studies aimed to compare the efficacy of difelikefalin at a dose of 0.5 mcg/kg with placebo in reducing the intensity of itch in patients undergoing hemodialysis and experiencing moderate-to-severe pruritus. The KALM trials used a parallel design. In total, 378 to 473 patients were randomized in a 1:1 ratio to treatment with difelikefalin or matched placebo in the KALM trials for up to 12 weeks. For both KALM trials, the randomization was stratified based on prespecified stratification variables. The dose of difelikefalin was 0.5 mcg/kg IV 3 times per week after each hemodialysis section. In the KALM trials, the primary outcome was the proportion of patients with at least a 3-point improvement in WI-NRS score at week 12, and the secondary outcomes were the proportion of patients with at least a 4-point improvement in WI-NRS score at week 12, the proportion of patients who were PGIC responders at week 12, and the proportions of patients with improvements in HRQoL, as measured by Skindex-10 and 5-D itch scores at week 12.
The median age of all randomized patients was similar in the 2 studies, at 58.0 years (range, 22 to 88 years) in the KALM-1 trial and 60.0 years (range, 23 to 87 years) in the KALM-2 trial. Most of the patients in the 2 studies were male (61.0% and 58.2%). The predominant races were white (48.8% and 70.3%) and Black or African American (41.6% and 19.3% in the KALM-1 and KALM-2 trials, respectively). The median prescription dry body weight was 84.0 kg (range, 42.0 to 135.0 kg) in the KALM-1 trial and 78.0 kg (range, 42.0 to 135.0 kg) in the KALM-2 trial. The median baseline WI-NRS score was 7.14 (range, 4.1 to 10.0) in the KALM-1 trial and 7.13 (range, 4.5 to 10.0) in the KALM-2 trial. At baseline, 39.8% of patients in the KALM-1 trial and 36.5% in the KALM-2 trial were using antiitch medications, and 14.1% and 16.6%, respectively, reported at least 1 of the specific medical conditions (i.e., history of falls or fracture or fall-related fracture, confusional state or mental status change or altered mental status or disorientation, or gait disturbance or movement disorder). The median duration of CKD-aP for all patients was 2.5 years (range, 0.1 to 26.5 years) in the KALM-1 trial and 2.3 years (range, 0.0 to 58.4 years) in the KALM-2 trial. The median time since the diagnosis of CKD and ESRD for all patients was 5.45 years (range, 0.3 to 42.9 years) and 3.92 years (range, 0.3 to 28.7 years), respectively, in the KALM-1 trial and 7.53 years (range, 0.3 to 48.3 years) and 4.03 years (range, 0.3 to 30.2 years), respectively, in the KALM-2 trial.
No indirect comparison was identified for this review. The CR845-CLIN3101 study, a phase III, multicentre, open-label study, was also summarized and appraised as supporting evidence.
Interpretation of Results
Efficacy
The KALM-1 and KALM-2 trials met their primary end point of the proportion of patients with at least a 3-point WI-NRS improvement, indicating that treatment with difelikefalin 0.5 mcg/kg resulted in an improvement in pruritus severity (a reduction in itch intensity) after 12 weeks. Additionally, the supportive analysis of the primary end point that was performed in the PP population and the sensitivity analyses performed in the ITT population with 3 multiple imputation assumptions (i.e., patients who discontinued the study drug early designated as nonresponders, multiple imputation followed a missing-not-at-random approach, and tipping point analysis) for both KALM trials demonstrate benefit with difelikefalin compared to placebo, which is consistent with the primary analysis.
In KALM-1 and KALM-2 studies, the 3-point reduction in WI-NRS score was used as the clinically meaningful cut-off point to define improvement and response of treatment in pruritus intensity, based on the results of a phase II study of difelikefalin (CR845-CLIN2101, NCT02858726) and 2 phase III trials (KALM-1 and the KALM-2) conducted by the sponsor. However, the 4-point cut-off was recommended by the FDA as the primary end point cut-off.16 The clinical experts consulted by CADTH were aligned with the FDA guidance regarding a 4-point improvement, but input from clinician groups suggested preference for a 3-point improvement. In summary, there is uncertainty regarding the most appropriate MID for the WI-NRS score; therefore, results from an assessment of both outcomes have been presented. Although statistical significance at week 12 was reached for the primary end point in both of the KALM trials, only about half of the patients in the difelikefalin group reported at least a 3-point improvement in WI-NRS score (52.2% in the KALM-1 trial and 49.7% in the KALM-2 trial). With regard to the proportion of patients achieving at least a 4-point improvement in WI-NRS score at week 12, fewer than half of patients in the difelikefalin group reported an improvement (40.8% in the KALM-1 trial and 37.7% in the KALM-2 trial). Moreover, a notable response was observed in the placebo treatment groups in both KALM trials, albeit not as great as in the difelikefalin group. According to the clinical experts consulted by CADTH, the placebo response may be due to the optimized hemodialysis treatment associated with the trials, as patients who were enrolled in the trials tended to attend their hemodialysis more frequently and regularly, which would provide a benefit. The CADTH review team considered the placebo effect to be a contributing factor for the response in the placebo group; however, the extent to which the placebo effect influenced the results is unclear. With regard to the PGIC, there was a higher proportion of complete responders in the difelikefalin group than in the placebo group in both KALM trials; however, there were no adjustments for multiple testing. Thus, the results for the PGIC should be interpretated with caution. Overall, the effect size of difelikefalin, compared with placebo, in reducing itch intensity, as measured by the WI-NRS, is small in the KALM trials but showed meaningful clinical efficacy in patients with moderate-to-severe CKD-aP on hemodialysis.
According to the clinical experts consulted by CADTH, patients with CKD-aP would receive the difelikefalin treatment beyond 12 weeks for the desired treatment effect. A pooled analysis of 2 OLE studies of the KALM-1 and KALM-2 trials assessed the 5-D itch scale for 52 weeks after the 12-week pivotal trials.17 The study reported that the proportion of patients achieving at least a 5-point reduction in 5-D itch scale score was maintained up to 52 weeks in the open-label phase of the KALM-1 and KALM-2 trials. However, there was some uncertainty about the reported long-term treatment effect due to the amount of missing data, with 26.5% (189 of 712) of patients in the pooled analysis population contributing data at week 52 in the OLE phase. In addition, although there is evidence based on 1 validated outcome designed to measure the severity of itch (the 5-D itch scale), outcomes related to itch intensity and HRQoL, as measured by the WI-NRS, PGIC, and Skindex-10 scale in the protocol, were not available. Thus, it is difficult to discern much about the long-term efficacy of difelikefalin because of the lack of other reported outcomes and the absence of a control group.
Although difelikefalin is the first treatment approved by Health Canada specifically indicated for the treatment of CKD-aP, the generalizability of the study results is uncertain, considering that the treatments included in the comparator arm do not align with clinical practice in Canada. Of note, commonly prescribed antiitch treatments and medications, such as topical therapies, phototherapy, gabapentin, and pregabalin, identified by the clinical experts for this review were not included in the comparator arm. Although not indicated by Health Canada for this condition, gabapentin is often used to treat CKD-aP in Canada, according to the clinical experts consulted by CADTH. The lack of direct evidence comparing difelikefalin with active comparators represents an evidence gap in the treatment of patients with moderate-to-severe CKD-aP on hemodialysis.
HRQoL was included as a secondary end point in both KALM trials and was noted to be an important outcome of interest by patients and clinicians. The secondary efficacy analyses of HRQoL, as measured by the change from baseline at week 12 in the total score of the Skindex-10 and 5-D itch scales, for difelikefalin compared to placebo were controlled for type I error in both KALM trials. The results of the analysis of HRQoL, based on the Skindex-10 scale, indicated that patients treated with difelikefalin reported a greater improvement in QoL than patients who received placebo at week 12 (–17.2 versus –12.0 for difelikefalin versus placebo in the KALM-1 trial and –16.6 versus –14.8, respectively, in the KALM-2 trial). A 3-point to 12-point change on the Skindex-10 scale was identified as a clinically meaningful change in patients on hemodialysis with moderate-to-severe CKD-aP.5 Therefore, the reported within-group differences in Skindex-10 scores were clinically meaningful, as they met the MID identified in the literature search conducted by CADTH. Although a greater improvement in HRQoL, as measured by the 5-D itch scale, was reported in the difelikefalin group than in the placebo group for the KALM trials, it is worth mentioning that results for the analysis of the 5-D itch total score in the KALM-2 trial was not considered inferential, as the preceding analyses in the testing hierarchy (i.e., difference in LS means for Skindex-10 scores at week 12 in the KALM-2 trial) was not statistically significant. In addition, no published MID was identified for the 5-D itch scale in patients with CKD-aP. Therefore, it is unclear whether the reported within-group differences are clinically meaningful. With regard to the Skindex-10 domain scores, the results were in favour of difelikefalin; however, there was no adjustment for multiplicity, so definitive conclusions could not be drawn with respect to the individual domains of the Skindex-10 scale (disease total, mood/emotional distress total, and social functioning total). For the same reason, the results of 5-D-Itch domain scores, which suggested a change in 5-D itch total score from baseline at week 12 between treatment arms in favour of difelikefalin, should be considered exploratory. Also, considering that these instruments are not routinely administered in patients with CKD-aP in clinical practice, per the clinical experts, it is a challenge to draw a conclusion about the effect of difelikefalin on HRQoL based on available data.
No evidence was identified in the KALM trials for mood, days of missed work, days of missed school, or days of missed dialysis, which were identified in the CADTH systematic review protocol and considered to be important outcomes of interest by patients and clinicians.
The KALM trials assessed the efficacy of difelikefalin, compared with placebo, in reducing the intensity of itch and improving itch-related QoL and safety in patients undergoing hemodialysis and experiencing moderate-to-severe pruritus. According to the clinical experts consulted by CADTH, sleep quality, mood (e.g., depression), days of missed work, days of missed school, and days of missed dialysis are clinically relevant outcomes that were not assessed in the KALM trials. Although sleep quality was assessed in an open-label, phase III study (CR845-CLIN3105 study, NCT03998163) and the results suggest that difelikefalin improves sleep quality at week 12, the study was subject to limitations of the noncomparative study design.38 Overall, the comparative efficacy of difelikefalin in these outcomes is uncertain.
The dose for difelikefalin in both KALM trials was 0.5 mcg/kg, which is aligned with the Health Canada–approved dose. The clinical experts consulted for this review indicated that there is a likelihood of increasing the dose in real-world settings to achieve the desired treatment effect. This may increase the side-effect profile of difelikefalin in real-world clinical settings. In the KALM trials, the concomitant use of antiitch medications were allowed, which is supported by the clinical experts’ statement that difelikefalin would likely to be used in combination with other treatment options (e.g., topical therapies and gabapentinoids) rather than as monotherapy. In additional, the clinical experts mentioned that difelikefalin would likely be used intermittently to achieve a response. However, the comparative efficacy of the intermittent use of difelikefalin was not assessed in either KALM trial. Thus, it is uncertain how applicable the results of the KALM trials would be to patients who received a higher dose of difelikefalin or used it intermittently.
Harms
In both KALM trials, the proportion of patients with TEAEs, SAEs, TEAEs leading to study drug discontinuation, and death was similar in the 2 treatment groups. The most common TEAEs, occurring in at least 0.5% of patients, in both trials were diarrhea, dizziness, and vomiting. The clinical experts consulted by CADTH for this review indicated that the mechanism of action of difelikefalin can leads to dizziness because of kappa opioid receptor activation. The rates of SAEs were comparable in the difelikefalin and placebo groups in both trials (25.9% versus 21.8% in the KALM-1 trial and 24.7% versus 21.6% in the KALM-2 trial), with hyperkalemia and sepsis pneumonia being the most commonly reported. According to the clinical experts, the proportion of patients with SAEs is consistent with that observed in clinical practice for patients who are on hemodialysis. The clinical experts also indicated that hyperkalemia, sepsis, volume overload, and chest pains, which were commonly reported SAEs in the KALM trials, are commonly seen in clinical practice in this patient population. The most frequently reported TEAEs leading to treatment discontinuation was dizziness. Diarrhea, nausea, vomiting, gait disturbance, falls, dizziness, headache, somnolence, seizures, syncope, mental status changes, mood changes, paresthesia (unusual feeling or sensation), hyperkalemia, back pain, tachycardia, and palpitation were considered notable harms in this review. Overall, the notable harms appeared at a similar frequency in both treatment groups in the KALM trials, except for diarrhea and dizziness, which were reported at a notably higher rate in the difelikefalin group than in the placebo group in the KALM-1 trial. The clinical experts consulted by CADTH commented that diarrhea and dizziness are clinically concerning but manageable. Overall, treatment with difelikefalin generally revealed no new safety issues in either KALM trial and was consistent with its known safety profile in patients on hemodialysis with moderate-to-severe CKD-aP, per feedback from the clinical experts.
As noted in the discussion of efficacy results, direct evidence of difelikefalin was limited to placebo-controlled comparisons. There was a gap in the evidence assessing the safety profile of difelikefalin relative to other clinically relevant comparators that are commonly used in clinical practice.
Evidence of the long-term safety of difelikefalin up to 52 weeks in patients on hemodialysis with moderate-to-severe CKD-aP was assessed in an open-label, phase III study (CR845-CLIN3101) and summarized as other relevant evidence for this review. Generally, patients reported higher rates of TEAEs, SAEs, and notable harms in the open-label, phase III study than patients in the KALM trials. Although the demographic and disease characteristics of patients in the open-label, phase III study were similar to those enrolled in the KALM-1 and KALM-2 trials, the source of the open-label study population was not from the KALM trials. In addition, the disproportional withdrawal rates and discontinuation of the study drug due to AEs made it difficult to interpret the safety outcomes in the open-label study. In addition, a pooled analysis39 that included patients enrolled in the12-week double-blind period and the 52-week OLE in the KALM-1 and KALM-2 trials, the 52-week open-label CLIN3101 study previously described, and a 12-week open-label CLIN3105 study39 was also included in the sponsor’s submission. The pooled safety analysis included an assessment of safety in patients receiving difelikefalin for up to 64 weeks. This pooled safety analysis was not formally summarized or critically appraised, as it included studies that were excluded from the CADTH systematic review (CLIN3101 and CLIN3105). Further, analysis of the 52-week CLIN3101 study alone was summarized under Other Relevant Evidence, as previously described, and the CLIN3105 study did not assess treatment beyond 12 weeks. However, the results for frequent TEAEs with up to 64 weeks of treatment presented by Fishbane et al. (2022)39 suggest that there were no new safety signals, based on the similar rate of AEs reported in the KALM-1 and KALM-2 trials.
Conclusions
The CADTH systematic review identified 2 phase III, double-blind, placebo-controlled RCTs (KALM-1 and KALM-2) that assessed the efficacy and safety of difelikefalin 0.5 mcg/kg compared to placebo over 12 weeks in patients on hemodialysis with moderate-to-severe CKD-aP. In both of the KALM trials, patients randomized to difelikefalin were more likely than patients randomized to placebo to report an improvement in WI-NRS score or a clinically meaningful reduction in the intensity of the worst itch, based on a 3-point (primary end point) or 4-point (key secondary end point) improvement in the score at week 12. This corresponded to 38% to 41% of patients randomized to difelikefalin and 21% to 25% of patients randomized to placebo having a 4-point improvement in WI-NRS score in the 2 trials. Other outcomes related to pruritus severity, including changes from baseline in WI-NRS scores and PGIC scores, were consistent with the primary end point. In the analyses of other secondary end points that assessed HRQoL, measured with the Skindex-10 and 5-D itch scales, treatment with difelikefalin was associated with an improvement in QoL in the KALM-1 trial, but statistical superiority for the same outcomes were not demonstrated in the KALM-2 trial. Generally, AEs were reported at a similar rate in the difelikefalin and placebo groups. Treatment with difelikefalin revealed no new safety issues in either KALM trial. No evidence was identified in the KALM trials for mood, days of missed work, days of missed school, or days of missed dialysis, which were considered to be important outcomes of interest by patients and clinicians. A pooled analysis of the OLE studies of the KALM-1 and KALM-2 trials, as well as a phase III, 52-week, open-label study (CLIN3101), provided evidence of long-term safety associated with difelikefalin, and no new safety signals were reported, but the study was subject to many limitations, including the lack of comparative evidence and high discontinuation rates. Overall, based on evidence from the KALM trials, difelikefalin may reduce the severity of itch and improve HRQoL compared with placebo, but there is uncertainty regarding the magnitude of the treatment effect in patients with moderate-to-severe CKD-aP.
References
1.PrKorsuva® (difelikefalin): 50 micrograms/mL intravenous solution [product monograph]. Saint-Laurent (QC): Otsuka Canada Pharmaceutical; 2022 Aug 12.
2.Braun MM, Khayat M. Kidney disease: end-stage renal disease. FP Essent. 2021;509:26-32. PubMed
3.Crawford PW, Lerma EV. Treatment options for end stage renal disease. Prim Care. 2008;35(3):407-432, v. PubMed
4.Lopes MB, Karaboyas A, Sukul N, et al. Utility of a single itch-related question and the Skindex-10 questionnaire for assessing pruritus and predicting health-related quality of life in patients receiving hemodialysis. Kidney Med. 2022;4(6):100476. PubMed
5.Mathur VS, Lindberg J, Germain M, et al. A longitudinal study of uremic pruritus in hemodialysis patients. Clin J Am Soc Nephrol. 2010;5(8):1410-1419. PubMed
6.Shirazian S, Aina O, Park Y, et al. Chronic kidney disease-associated pruritus: impact on quality of life and current management challenges. Int J Nephrol Renovasc Dis. 2017;10:11-26. PubMed
7.Hayani K, Weiss M, Weisshaar E. Clinical findings and provision of care in haemodialysis patients with chronic itch: new results from the German Epidemiological Haemodialysis Itch Study. Acta Derm Venereol. 2016;96(3):361-366. PubMed
8.Rayner HC, Larkina M, Wang M, et al. International comparisons of prevalence, awareness, and treatment of pruritus in people on hemodialysis. Clin J Am Soc Nephrol. 2017;12(12):2000-2007. PubMed
9.Aresi G, Rayner HC, Hassan L, et al. Reasons for underreporting of uremic pruritus in people with chronic kidney disease: a qualitative study. J Pain Symptom Manage. 2019;58(4):578-586.e572. PubMed
10.Pisoni RL, Wikström B, Elder SJ, et al. Pruritus in haemodialysis patients: international results from the Dialysis Outcomes and Practice Patterns Study (DOPPS). Nephrol Dial Transplant. 2006;21(12):3495-3505. PubMed
11.Narita I, Alchi B, Omori K, et al. Etiology and prognostic significance of severe uremic pruritus in chronic hemodialysis patients. Kidney Int. 2006;69(9):1626-1632. PubMed
12.Kuypers DR. Skin problems in chronic kidney disease. Nat Clin Pract Nephrol. 2009;5(3):157-170. PubMed
13.Verduzco HA, Shirazian S. CKD-associated pruritus: new insights into diagnosis, pathogenesis, and management. Kidney Int Rep. 2020;5(9):1387-1402. PubMed
14.Sukul N, Karaboyas A, Csomor PA, et al. Self-reported pruritus and clinical, dialysis-related, and patient-reported outcomes in hemodialysis patients. Kidney Med. 2021;3(1):42-53.e41. PubMed
15.Vernon MK, Swett LL, Speck RM, et al. Psychometric validation and meaningful change thresholds of the Worst Itching Intensity Numerical Rating Scale for assessing itch in patients with chronic kidney disease-associated pruritus. J Patient Rep Outcomes. 2021;5(1):134. PubMed
16.Center for Drug Evaluation Research. Multidiscipline review: Korsuva (difelikefalin) 0.5mcg/kg by intravenous injection. Company: Cara Therapeutics, Inc. Application No.:214916. Approval date: 08/23/2021 (FDA approval package). Silver Spring (MD): U.S. Food and Drug Administration (FDA); 2021: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2021/214916Orig1s000MultidisciplineR.pdf. Accessed 2022 Dec 20,
17.Topf J, Wooldridge T, McCafferty K, et al. Efficacy of difelikefalin for the treatment of moderate to severe pruritus in hemodialysis patients: pooled analysis of KALM-1 and KALM-2 phase 3 studies. Kidney Med. 2022;4(8):100512. PubMed
18.Elman S, Hynan LS, Gabriel V, Mayo MJ. The 5-D itch scale: a new measure of pruritus. Br J Dermatol. 2010;162(3):587-593. PubMed
19.Clinical Study Report: KALM-1 CR845-CLIN3102. A multicenter, double-blind, randomized, placebo-controlled study to evaluate the safety and efficacy of intravenous CR845 in hemodialysis patients with moderate-to-severe pruritus, with a 52-week open-label extension [internal sponsor's report]. Stamford (CT): Cara Therapeutics; 2020.
20.Clinical Study Report: KALM-2 CR845-CLIN3103. A multicenter, double-blind, randomized, placebo-controlled study to evaluate the safety and efficacy of intravenous CR845 in hemodialysis patients with moderate-to-severe pruritus, with a 52-week open-label extension [internal sponsor's report]. Stamford (CT): Cara Therapeutics; 2020.
21.Davies HT, Crombie IK, Tavakoli M. When can odds ratios mislead? BMJ. 1998;316(7136):989-991. PubMed
22.Mettang T, Kremer AE. Uremic pruritus. Kidney Int. 2015;87(4):685-691. PubMed
23.Kimmel M, Alscher DM, Dunst R, et al. The role of micro-inflammation in the pathogenesis of uraemic pruritus in haemodialysis patients. Nephrol Dial Transplant. 2006;21(3):749-755. PubMed
24.Phan NQ, Lotts T, Antal A, Bernhard JD, Ständer S. Systemic kappa opioid receptor agonists in the treatment of chronic pruritus: a literature review. Acta Derm Venereol. 2012;92(5):555-560. PubMed
25.Ko MJ, Peng YS, Wu HY. Uremic pruritus: pathophysiology, clinical presentation, and treatments. Kidney Res Clin Pract. 2022;04:04.
26.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. J Clin Epidemiol. 2016;75:40-46. PubMed
27.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2022 Oct 7.
28.Fishbane S, Jamal A, Munera C, Wen W, Menzaghi F, Investigators K-T. A phase 3 trial of difelikefalin in hemodialysis patients with pruritus. N Engl J Med. 2020;382(3):222-232. PubMed
29.Wooldridge TD, McCafferty K, Schoernig M, et al. Efficacy and safety of difelikefalin for moderate-to-severe CKD–associated pruritus: a global phase 3 study in hemodialysis patients (KALM-2) [abstract]. Presented at: American Society of Nephrology Annual Meeting; Oct 22-25, 2020. https://www.asn-online.org/education/kidneyweek/2020/program-abstract.aspx?controlId=3444152#prettyPhoto. Accessed 2022 Dec 6.
30.Vernon M, Stander S, Munera C, Spencer RH, Menzaghi F. Clinically meaningful change in itch intensity scores: An evaluation in patients with chronic kidney disease-associated pruritus. J Am Acad Dermatol. 2021;84(4):1132-1134. PubMed
31.Menzaghi F. Clinically meaningful reduction of itch and improvement in multiple quality of life measures in hemodialysis patients with moderate-to-severe pruritus following treatment with difelikefalin [abstract]. Presented at: the 27th Annual Congress of the European Academy of Dermatology and Venereology; Paris, FR; Sep 12–16, 2018.
32.Dworkin RH, Turk DC, Farrar JT, et al. Core outcome measures for chronic pain clinical trials: IMMPACT recommendations. Pain. 2005;113(1-2):9-19. PubMed
33.Incorporating clinical outcome assessments into endpoints for regulatory decision-making. Patient-focused drug development guidance public workshop. Silver Spring (MD): U.S. Food & Drug Administration (FDA); 2019: https://www.fda.gov/media/132505/download. Accessed 2022 Dec 6.
34.Bovin MJ, Marx BP, Weathers FW, et al. Psychometric properties of the PTSD checklist for Diagnostic and Statistical Manual of Mental Disorders-fifth edition (PCL-5) in veterans. Psychol Assess. 2016;28(11):1379-1391. PubMed
35.Cui L, Hung HM, Wang SJ. Modification of sample size in group sequential clinical trials. Biometrics. 1999;55(3):853-857. PubMed
36.Lawrence J, Hung HMJ. Estimation and confidence intervals after adjusting the maximum information. Biom J. 2003;45(2):143-152.
37.Clinical Study Report: Study CLIN3101. An open-label, multicenter, extension study to evaluate the long-term safety of intravenous CR845 in hemodialysis patients with chronic kidney disease-associated pruritus [internal sponsor's report]. Stamford (CT): Cara Therapeutics; 2020.
38.Clinical Study Report: Study CLIN3105. An open-Label, multicenter, study to evaluate the safety and effectiveness of intravenous CR845 in hemodialysis patients with moderate-to-severe pruritus [internal sponsor's report]. Stamford (CT): Cara Therapeutics; 2020.
39.Fishbane S, Wen W, Munera C, et al. Safety and tolerability of difelikefalin for the treatment of moderate to severe pruritus in hemodialysis patients: pooled analysis from the phase 3 clinical trial program. Kidney Med. 2022;4(8):100513. PubMed
40.Cheung HN, Chan YS, Hsiung NH. Validation of the 5-D itch scale in three ethnic groups and exploring optimal cutoff values using the itch numerical rating scale. Biomed Res Int. 2021;2021:7640314. PubMed
Appendix 1: Literature Search Strategy
Clinical Literature Search
Overview
Interface: Ovid
Databases
MEDLINE All (1946-present)
Embase (1974-present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: October 19, 2022
Alerts: Bi-weekly search updates until project completion
Search filters applied: No filters were applied to limit the retrieval by study type.
Limits: Conference abstracts: excluded.
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
MeSH | Medical Subject Heading |
exp | Explode a subject heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Author keyword heading word (MEDLINE) |
.dq | Candidate term word (Embase) |
.pt | Publication type |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multidatabase Strategy
(Korsuva* or difelikefalin* or Kapruvia* or CKD943 or CKD 943 or CR845 or CR 845 or FE202845 or FE 202845 or MR13A9 or MR 13A9 or NA1U919MRO or 0P70AR5BYB).ti,ab,ot,kf,hw,nm,rn.
1 use medall
*difelikefalin/
(Korsuva* or difelikefalin* or Kapruvia* or CKD943 or CKD 943 or CR845 or CR 845 or FE202845 or FE 202845 or MR13A9 or MR 13A9).ti,ab,kf,dq.
3 or 4
5 use oemezd
6 not (conference abstract or conference review).pt.
2 or 7
remove duplicates from 8
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
[Search terms – difelikefalin, Korsuva, Kapruvia]
WHO ICTRP
International Clinical Trials Registry Platform, produced by the WHO. Targeted search used to capture registered clinical trials.
[Search terms – difelikefalin, Korsuva, Kapruvia]
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
[Search terms – difelikefalin, Korsuva, Kapruvia]
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
[Search terms – difelikefalin, Korsuva, Kapruvia]
Other Databases
Cochrane Central Register of Controlled Trials
Same MeSH, keywords, and limits used as per MEDLINE search, excluding study types and human restrictions. Syntax adjusted for Wiley platform. The search strategy is available on request.
Grey Literature
Search dates: October 7 to 17, 2022
Keywords: Korsuva, Kapruvia, difelikefalin, pruritus
Limits: none
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Appendix 2: Excluded Studies
Note this appendix has not been copy-edited.
Reference | Reason for Exclusion |
|---|---|
Fishbane S, Wen W, Munera C, et al. Safety and tolerability of difelikefalin for the treatment of moderate to severe pruritus in hemodialysis patients: pooled analysis from the phase 3 clinical trial program. Kidney Med. 2022;4(8):100513. | Pooled analysis |
Fishbane S, Wen W, Munera C, Menzaghi F, McCafferty K. Long-term safety and efficacy of difelikefalin in patients with chronic kidney disease-associated pruritus: analysis from KALM-1 and KALM-2. Am J Kidney Dis. 2021;77(4):593 to 594. | |
Topf J, Wen W, Munera C, Menzaghi F, Schomig M. Efficacy of difelikefalin in patients with moderate-to-severe chronic kidney disease-associated pruritus: pooled subgroup analysis of KALM-1 and KALM-2. Am J Kidney Dis. 2021;77(4):658. | |
Topf J, Wooldridge T, McCafferty K, et al. Efficacy of Difelikefalin for the treatment of moderate to severe pruritus in hemodialysis patients: pooled analysis of KALM-1 and KALM-2 phase 3 studies. Kidney Med. 2022;4(8):100512. | |
Narita I, Tsubakihara Y, Uchiyama T, et al. Efficacy and safety of difelikefalin in Japanese patients with moderate to severe pruritus receiving hemodialysis: a randomized clinical trial. JAMA Netw. 2022;5(5):e2210339. | Study design |
Weiner DE, Vervloet MG, Walpen S, et al. Safety and effectiveness of difelikefalin in patients with moderate-to-severe pruritus undergoing hemodialysis: an open-label, multicenter study. Kidney Med. 2022;4(10):100542. | |
Fishbane S, Mathur V, Germain MJ, et al. Randomized controlled trial of difelikefalin for chronic pruritus in hemodialysis patients. Kidney Int Rep. 2020;5(5):600 to 610. | |
Viscusi ER, Torjman MC, Munera CL, Stauffer JW, Setnik BS, Bagal SN. Effect of difelikefalin, a selective kappa opioid receptor agonist, on respiratory depression: a randomized, double-blind, placebo-controlled trial. Clin Transl Sci. 2021;14(5):1886 to 1893. | Study population |
Appendix 3: Detailed Outcome Data
Note this appendix has not been copy-edited.
Table 31: Hierarchical Testing Order of Key Secondary End Points
Key Secondary End points | P value | Significant |
|---|---|---|
KALM-1 | ||
Difference between patients assigned to difelikefalin arm and placebo arm in the 5-D itch total score at week 12 | P < 0.001 | Yes |
Difference between patients assigned to difelikefalin arm and placebo arm in the Skindex-10 total score at week 12 | P < 0.001 | Yes |
The proportion of patients achieving ≥ 4-point improvement from baseline in the weekly mean of the daily 24-hour WI-NRS at week 12 | P < 0.001 | Yes |
KALM-2 | ||
The proportion of patients achieving ≥ 4-point improvement from baseline in the weekly mean of the daily 24-hour WI-NRS at week 12 | P = 0.01 | Yes |
The proportion of patients achieving a ≥ 3-point improvement from baseline in the daily 24-hour WI-NRS at week 8 | P = 0.01 | Yes |
The proportion of patients achieving a ≥ 3-point improvement from baseline in the daily 24-hour WI-NRS at week 4 | P = 0.002 | Yes |
The proportion of patients achieving a ≥ 4-point improvement from baseline in the weekly mean of the daily 24-hour WI-NRS at week 8 | P = 0.002 | Yes |
The proportion of patients achieving a ≥ 4-point improvement from baseline in the weekly mean of the daily 24-hour WI-NRS at week 4 | P = 0.036 | Yes |
Change from baseline in the total Skindex-10 scale score at Week 12 | P = 0.171 | No |
Change from baseline in the total 5-D itch scale score itch-related QoL at week 12 | P = 0.002 | No |
WI-NRS = Worst itching intensity numerical rating scale
Sources: KALM-1 Clinical Study Report;19 KALM-2 Clinical Study Report.20
Table 32: Subgroup Analysis of Proportion of Patients With a ≥ 3-Point Improvement or a ≥ 4-Point Improvement in WI-NRS at Week 12 (ITT Analysis Set)
End points | KALM-1 | KALM-2 | ||||
|---|---|---|---|---|---|---|
Difelikefalin 0.5 mcg/kg N = 189 | Placebo N = 189 | Difference (95% CI) | Difelikefalin 0.5 mcg/kg N = 237 | Placebo N = 236 | Difference (95% CI) | |
Proportion of patients with a ≥ 3-point Improvement in WI-NRS at Week 12 | ||||||
Prior antiitch medication use, % | ||||||
Yes | n = 72 52 | n = 78 30 | 23 (7 to 39) | n = 87 41 | n = 85 26 | 15 (0 to 30) |
No | n = 117 52 | n = 111 30 | 20 (8 to 33) | n = 150 39 | n = 151 32 | 7 (–4 to 17) |
Proportion of patients with a ≥ 4-point Improvement in WI-NRS at Week 12 | ||||||
Prior antiitch medication use, % | ||||||
Yes | n = 72 40 | n = 78 21 | 19 (4 to 34) | n = 87 42 | n = 85 22 | 20 (6 to 34) |
No | n = 117 41 | n = 111 22 | 19 (7 to 30) | n = 150 35 | n = 151 29 | 6 (–5 to 17) |
CI = confidence interval; NA = not applicable; WI-NRS = worst itching intensity numerical rating scale.
Note: Combined analysis used the separate interim and postinterim results to generate an adjusted overall estimate using the Cui, Hung, and Wang (CHW) methodology.
Source: FDA report16
Appendix 4: Description and Appraisal of Outcome Measures
Note this appendix has not been copy-edited.
Aim
To describe the following outcome measures and review their measurement properties (validity, reliability, responsiveness to change, and MID):
Worst Itching Intensity Numerical Rating Scale (WI-NRS)
Skindex-10 Questionnaire
5-D itch scale
Patient Global Impression of Change (PGIC)
Findings
Table 33: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measuremnt properties | MID |
|---|---|---|---|
WI-NRS | The WI-NRS is a single-item patient-reported outcome measure in which patients indicate the intensity of the worst itching they experienced over the past 24 hour, by marking a number between ‘0’ (corresponding to no itch) and ‘10’ (the worst itching imaginable).15 | Validity: Content validity for the WI-NRS has been assessed through qualitative interviews with 23 hemodialysis patients with CKD-aP of any severity, with feedback from patients considering the scale as straightforward, comprehensive, and relevant to their experiences with CKD-aP.15 Construct validity of the WI-NRS was evaluated by examining both convergent and divergent validity. For convergent validity, moderate (r ≥ 0.3 to < 0.5) or large (r ≥ 0.5) correlations by Cohen standards were hypothesized for the PGI-S in a phase II trial and for items within the Skindex-10 and the 5-D itch that measure similar concepts to the WI-NRS. For divergent validity test, the MOS Sleep Scale domain scores were used to assess the extent to which less related concepts, e.g., sleep and itch, exhibit low correlations (r < 0.3) with one another. Analyses indicated a high correlation between the WI-NRS scores and other measures of itch, e.g., Skindex-10 and 5-D itch measures in both phase II and phase III datasets, particularly with the conceptually related Skindex-10 Disease domain (r = 0.7 to 0.8) and the 5-D itch Degree domain (r = 0.65 to 0.67) at the end of treatment. Correlations with the conceptually unrelated domains of the MOS Sleep measure (Sleep Problem Index I and II, and Sleep Disturbance) were small (r = 0.16 to 0.26) by Cohen standards, as hypothesized for phase II trial patients.15 Known groups validity was evaluated to assess the discriminant properties of the WI-NRS. Groups were created using the PROs collected from a phase II study (PGI-S, Patient Self-categorization of Pruritus Disease Severity, Skindex-10, 5-D itch, MOS Sleep Problem Index II) and 2 pooled phase III studies (Skindex-10, 5-D itch). Two-sample t tests and linear model ANOVA were used to compare differences in WI-NRS for known groups with 2 categories and for known groups with more than 2 categories, respectively. The baseline WI-NRS scores were significantly different between known groups of the conceptually related 5-D Itch total score and Skindex-10 measures both the phase II and phase III cohorts. Known group comparisons of WI-NRS against Patient Self-Categorization of Pruritus Disease Severity as well as PGI-S were also statistically significant. But the differences in WI-NRS scores at baseline were not significantly different when grouped by the quartiles of the conceptually unrelated MOS Problem Index II (P = 0.1049; phase II cohort only).15 Reliability: Test-retest reliability was evaluated by using intraclass correlation coefficients (ICCs) between weeks 1 and 2 and between weeks 2 and 4 for the phase II cohort, and by using the same time points with all evaluable patients included for the phase III cohort. Patients from the phase II trial that were stable on the PGI-S demonstrated good reproducibility on their weekly mean WI-NRS scores between week 1 and week 2 (ICC = 0.76) and between week 2 and week 4 (ICC = 0.81). WI-NRS scores for patients from the pooled phase III trials also demonstrated reproducibility, with ICC = 0.80 between week 1 and week 2 and ICC = 0.81 between week 3 and week 4, with ICC values greater than the acceptable threshold 0.70.15 | The threshold for meaningful reduction of WI-NRS was estimated using anchor- and distribution-based methods through a secondary analysis of pooled data from a phase II study in hemodialysis patients with moderate to severe pruritus. Analyses showed that a reduction of ≥ 3 points from baseline score on the WI-NRS represents an appropriate clinically meaningful change (within-patient) in pruritus in patients with CKD-aP.30 This clinically meaningful within-patient change score threshold was confirmed using same anchor-based approach in an analysis among a phase III trial patients. PGIC was used as an anchor, which specifically asked patients to indicate the improvement of their condition taking into consideration treatment effect and patient expectation. For the pooled phase III cohorts, the mean change in WI-NRS associated with a change from baseline to ‘minimally improved’ on the PGIC was −1.85 points (26% change), whereas the mean change in WI-NRS associated with a change to a much improved response on the PGIC was −3.54 points (51% change), based on the secondary anchor-based approach, representing larger changes.15 Mixed-method exit interviews to determine what constituted a meaningful change from patients’ perspectives. When reviewing actual WI-NRS change scores experienced in the exit interviews, all patients with a change ≥ 3 points considered the change meaningful, mentioning reduced intensity, frequency, and duration of itch and improvements in HRQoL. Meaningful changes were also reported by two-thirds of participants with score changes in the range 1 to 1.99-points, suggesting changes on the WI-NRS do not have to be large in this population. The exit interview result indicates individual differences in the magnitude of change considered meaningful by patients as well as the possibility that many patients with CKD-aP will experience meaningful improvements with changes below the ≥ 3-point change threshold.15 |
Skindex-10 Questionnaire | While the original Skindex questionnaires containing at least 16 questions is a reliable and valid instrument measuring the effects of skin disease on the quality of life and supplementing clinical judgments of the severity of the disease, Skindex-10 is a modified version containing 10 questions, designed specifically to evaluate CKD-aP with relevant subdomains for patients in the hemodialysis setting.5 The responses to each of the 10 questions (0 to 6 scale per question) pertaining to how often the patients were bothered by itchy skin in the past week, are summed to create a total summary score, ranging between 0 and 60, with 0 indicating not at all bothered, and 3 subdomain scores (e.g., itching and its impact on mood or emotions and social functioning).4,5 | Validity: No validity outcome results were found from the literature search in the CKD-aP population. Reliability: An analysis was conducted using data from hemodialysis 4,940 patients from 17 countries enrolled in phase 5 of the Dialysis Outcomes and Practice Patterns Study. The Spearman correlation coefficient was calculated between Skindex-10 (total score and scores for each of its 3 domains, i.e., social, emotional, and functional) and a single itch-related question KDQOL-36 to evaluate their relationship in predicting HRQoL. Internal consistency was evaluated using the Cronbach alpha value of the correlation of the patients’ responses to individual Skindex-10 questions with its overall score and each of its 3 domains. Unadjusted linear regression model was used to analyze the association between CKD-aP and HRQoL.4 The Skindex-10 questionnaire demonstrated a high internal consistency, with the Cronbach alpha values of > 0.83 for all 10 questions. The internal consistency with the overall Skindex-10 score was higher for the emotional domain (Cronbach alpha, 0.91 to 0.93), followed by that for the disease (Cronbach alpha, 0.90 to 0.91) and social domains (Cronbach alpha, 0.83 to 0.86). Within the domains, the Cronbach alpha values ranged from 0.92 to 0.97 for the 3 questions in the disease domain, 0.93 to 0.97 for the 3 questions in the emotional domain, and 0.92 to 0.94 for the 4 questions in the social domain.4 The Spearman correlation between the single itch-related question and the overall Skindex-10 score was 0.72, showing a strong correlation. The correlation between the single itch-related question and each of the domains was 0.72 for the disease, 0.62 for the social, and 0.70 for the emotional domains. The correlations between the single itch-related question and each of the 10 individual Skindex-10 questions ranged between 0.47 and 0.74. The correlations with the single itch-related question were the strongest for the 4 Skindex-10 questions pertaining to being bothered by itching, the persistence or recurrence of itching, and being frustrated and annoyed by itching during the past week.4 PCS and MCS scores were modelled to investigate the relative strength of the Skindex-10 in predicting HRQoL. A 10-point higher Skindex-10 score was associated with a 1.2-point lower PCS score (95% CI, −1.4 to −0.9) and a 1.5-point lower MCS score (95% CI, −1.7 to −1.3). The single itch-related question model outperformed Skindex-10 while predicting the PCS (R2, 0.065 vs 0.033, respectively). Similar results were found for predicting the MCS.4 Responsiveness: No responsiveness results were found from the literature search in the CKD-aP population. | A greater than 20% of change in a VAS measurement of itching intensity is considered clinically meaningful, which is associated with a 3- to 12-point change on the Skindex-10 in patients on hemodialysis.5 |
5-D itch scale | The 5-D itch questionnaire is specifically developed measure of itch. The questionnaire is brie, easy to complete and score (either manually at the bedside or electronically as part of a large clinical trial), sensitive to the multidimensional nature of pruritus and its effect on quality of life, applicable to multiple diseases, and capable of detecting change over time. The 5-D itch questionnaire is consisted of 5 domains - duration, degree, direction, disability, and distribution. The duration, degree and direction domains each has one item, while the disability domain has 4 items. All items of the first 4 domains are measured on a 5-point Likert scale. The distribution domain has 16 potential locations of itch, including 15 body part items and one point of contact with clothing or bandages.18 | Validity: The 5-D was administered to 234 individuals with chronic pruritus due to different diseases (e.g., liver, kidney, dermatological disorders, HIV/AIDS, burn injuries) to assess its validity and reliability. Convergent validity was evaluated using Spearman rank order correlations separately for each time point to measure the association between the total score of the 5-D itch scale with the VAS and the PBC-40. Spearman rank order correlations were used to assess the association between the individual domain scores on the 5-D and the VAS and PBC-40. Six-week change in each of the 5 domains and the 5-D itch score, was correlated with the VAS using a Spearman rank order correlation. The 5-D score demonstrated a strong correlation with the VAS score each time the measures were administered together. The Pearson's correlation coefficients were r = 0.727 at baseline (P < 0.0001), r = 0.868 at the 3-day repeat (P < 0.0001), and r = 0.892 at the 6-week follow-up (P < 0.0001). The individual 5-D domains that were hypothesized to correlate best with other measures based on similarity of concept had demonstrated expected correlation – the degree domain at 6 weeks with change in VAS (r = 0.55, P < 0.0001), the direction domain at 6 weeks with change in VAS (r = 0.70, P < 0.0001), and the disability domain with the quality of life assessment of pruritus from the PBC-40 at baseline (r = 0.69, P < 0.0001) and at follow-up (r = 0.87, P < 0.0001).18 A significantly high concurrent validity was also achieved in another validation study of the 5-D itch scale with the Itch-NRS and WI-NRS.40 Reliability: The intraclass correlation coefficient (ICC) was calculated to examine the agreement between repeated measures in a subgroup of 50 individuals who repeated the questionnaire within 2 to 3 days. Cronbach alpha was determined to assess the internal consistency of the measure, whereas a Bland-Altman plot was used to evaluate the association between repeated measures. No change was observed in mean 5-D score between day 1 and day 3 in the 50 patients repeating the questionnaire at days 1 and 3 for test-retest reliability (16.5 vs. 16.5, respectively). The ICC between the 5-D score obtained on day 1 and day 3 was 0.96 (95% CI, 0.92 to 0.98), showing highly significant correlation/ test-retest reliability (P < 0.0001). The responses on some individual items changed over the 3-day interval (paired differences, P < 0.0001), whereas the total score did not change, which indicated that patients could not always remember their previous answers. The Bland-Altman plot demonstrated an excellent Association/reliability between repeated measures. The internal consistency (Cronbach alpha) of the 5-D itch scale was 0.734. If one item of the 5-D scale were deleted, the average decrease in reliability was 0.067 (range 0.035 to 0.095).18 Responsiveness: The 5-D itch scale demonstrated a good responsiveness, by detecting significant changes in pruritus over the follow-up period. Each of the 5 individual domains of the scale was sensitive to change over time. The change in 5-D score demonstrated a strong correlation with the change in VAS in all subjects (r = 0.862, P < 0.0001).18 | The MID was not found from the literature search for the CKD-aP population. |
PGIC | The PGIC evaluates patient impression of change (improvement or worsening) in overall status relative to the baseline status. The measure is consisted of 7 categories, ranging from ‘1’ (Very Much Improved) to ‘7’ (Very Much Worse); with higher scores reflecting worse status. The PGIC has been recommended to be used as an anchor variable by the FDA to generate appropriate thresholds representing a meaningful within-patient change in the target patient population.33 For this reason, this measure has been used to evaluate the threshold meaningful change within populations for the WI-NRS measure with pruritus (described earlier). “Minimally improved,” “minimally and much improved,” and “much improved” anchor categories were used to represent minimal to larger improvements.30 | No validity, reliability, and responsiveness outcome results were found from the literature search in the CKD-aP population. | No MID results were found from the literature search in the CKD-aP population. |
CI = confidence interval; CKD-aP = chronic kidney disease–associated pruritus; HRQoL = health-related quality of life; ICC = intraclass correlation coefficient; KDQOL-36 = Kidney Disease Quality of Life 36-item survey; MCS = mental component summary; MID = minimal important difference; MOS = Medical Outcomes Study; NRS = Numerical Rating Scale; PBC = primary biliary cirrhosis; PCS = physical component summary; PGIC = Patient Global Impression of Change; PGI-S = Patient Global Impression of Worst Itch Severity; PRO = patient-reported outcomes; VAS = visual analogue scale; WI-NRS = Worst Itching Intensity Numerical Rating Scale.
Pharmacoeconomic Review
Abbreviations
AE
adverse event
BIA
budget impact analysis
BSC
best supportive care
CKD
chronic kidney disease
CKD-aP
chronic kidney disease–associated pruritus
CUA
cost-utility analysis
HRQoL
health-related quality of life
ICER
incremental cost-effectiveness ratio
QALY
quality-adjusted life-year
QoL
quality of life
WI-NRS
Worst Itching Intensity Numerical Rating Scale
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Difelikefalin (Korsuva), 50 mcg/1.0 mL solution for IV injection |
Submitted price | Difelikefalin, 50 mcg vial: $27.00 per single-use vial |
Indication | For the treatment of moderate-to-severe pruritus associated with chronic kidney disease in adult patients on hemodialysis |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | August 16, 2022 |
Reimbursement request | Per indication |
Sponsor | Otsuka Canada Pharmaceutical Inc. |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov model |
Target population | Moderate-to-severe pruritus associated with chronic kidney disease in adults on hemodialysis |
Treatment | Difelikefalin added to BSC |
Comparators | BSC alone, which includes topical therapies, antihistamines, gabapentinoids (i.e., gabapentin or pregabalin), antidepressants, and/or UV type B phototherapy |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs and LYs |
Time horizon | Lifetime (10 years) |
Key data source | KALM-1 and KALM-2 trials |
Submitted results | $286,717 per QALY (incremental costs = $23,413.77; incremental QALYs = 0.08) |
Key limitations |
|
CADTH reanalysis results |
|
BSC = best support care; HRQoL = health-related quality of life; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; QoL = quality of life; WI-NRS = Worst Itching Intensity Numerical Rating Scale.
Conclusions
The CADTH clinical review concluded that difelikefalin (Korsuva) demonstrated a statistically significant and clinically meaningful improvement in itch intensity, compared to placebo, in patients on hemodialysis with moderate-to-severe chronic kidney disease–associated pruritis (CKD-aP) based on the KALM-1 and KALM-2 trials, as well as a statistically significant improvement in disease severity, as measured by at least a 3-point improvement in WI-NRS scores. However, the clinical meaningfulness of a 3-point improvement in the WI-NRS score, which was the scale used to categorize health states in the sponsor’s submitted model, was uncertain. Furthermore, although analyses of secondary end points measuring health-related quality of life (HRQoL) showed results in favour of difelikefalin, no conclusions could be drawn due to a lack of statistical testing or control for multiplicity. No data using generic preference-based measures were collected in the trials. The economic model, therefore, attempted to capture these benefits by mapping the WI-NRS to EQ-5D through a de novo data collection study. However, this method may not have accounted for all confounders, and this introduced significant uncertainty into the measurement of HRQoL, which was a driver of quality-adjusted life-year (QALY) gains in the submitted economic model.
The CADTH reanalysis assumed no difference in mortality, hospitalization, or primary care visits by pruritus states and excluded phototherapy from the best supportive care (BSC) costs. The CADTH results were similar to the sponsor’s, in that difelikefalin was not cost-effective at the $50,000 per QALY gained threshold. The CADTH base case incremental cost-effectiveness ratio (ICER) for difelikefalin plus BSC compared to BSC alone was $582,515 per QALY gained; difelikefalin had a 0% probability of being cost-effective at a willingness-to-pay threshold of $50,000 per QALY gained. A price reduction of at least 92% would be required for difelikefalin to achieve a mean cost-effective estimate of $50,000 per QALY. Because of the small clinical benefits expected from difelikefalin, the ICER was found to be less stable. However, the conclusions remained the same across all CADTH reanalyses, which indicated a substantial price reduction would be necessary for difelikefalin to be considered cost-effective.
Although the CADTH reanalysis attempted to address some of the identified limitations, uncertainty remained in the CADTH base case due to uncertain but optimistic assumptions favouring difelikefalin. Specifically, if the true QoL benefit is less than the estimated values from mapping, the ICER is expected to be much greater. Further, unless strict stopping rules related to achievement of a specific pruritus severity (i.e., achievement of mild or no pruritus within 12 weeks) are enforced, the ICER is expected to be greater. In both of these plausible scenarios, the ICERs were found to be closer to $1 million per QALY gained, with a price reduction greater than 95% required to achieve an ICER of $50,000 per QALY.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
Patient input was provided by the Kidney Foundation of Canada. Survey respondents indicated that CKD-aP was interfering with their QoL, as they had developed an uncomfortable itch that hindered their ability to participate in day-to-day activities and dramatically reduced the quality of their sleep. Patients reported using antihistamines and topical creams to treat their CKD-aP, and most said they were satisfied with their current combination of treatments. However, many patients reported paying out of pocket for these treatments.
Clinician Input was received from 3 physician groups across Canada: the Dalhousie University Division of Nephrology, Department of Medicine, Dalhousie University and Nova Scotia Health, Hemodialysis Specialty Physician Group, Division of Nephrology, The Ottawa Hospital, and the Saskatchewan Kidney Doctors. Clinician groups noted that there was an unmet need addressed by this treatment, as there are currently no available treatment options that modify the underlying mechanism of CKD-aP. Current treatment options only function to reduce the burden of systemic side effects and have adverse effects that prevent their widespread use in the treatment of CKD-aP. They explained that their treatment goals would be to have a therapy that reduces or maintains the severity of the itch below a threshold of clinical importance that is known to negatively impact QoL and outcomes important to patients. These clinician groups also indicated that their ideal treatment should also be affordable, easy to administer, and have a tolerable side-effect profile. There are currently no approved therapies for the management of CKD-aP in Canada, as medications (mainly gabapentin and opioid receptor antagonists) are used off-label to help patients manage symptoms. The physician groups commented that difelikefalin would likely be used as a complement to these existing treatments or as a second-line option when other off-label treatments have failed; others noted that it should be a first-line treatment. Clinician input also noted that UV type B therapy has demonstrated some clinical efficacy in the treatment of CKD-aP, but it is not feasible for patients on hemodialysis who spend 12 to 15 hours a week getting dialysis to travel to centres to get this therapy.
Input received from participating drug plans noted concerns regarding the potential use of more than 1 vial of difelikefalin per treatment course, depending on the patient’s dry weight. This led to concerns that the budget impact may be underestimated under the assumption that a single vial would be used for each eligible patient. Plans also had concerns about vial waste and the impact this might have on budget impact analysis (BIA) results.
Several of these concerns were addressed in the sponsor’s model:
Health states were based on the severity of pruritus, with decreasing QoL accompanying increasing pruritus severity.
The comparator in the model was BSC to reflect the fact that no other indicated options are available. Off-label medications were considered as part of BSC.
The sponsor’s base case for both the economic evaluation and the BIA included drug waste.
CADTH addressed the following concerns raised from stakeholder input:
CADTH removed phototherapy from BSC.
CADTH conducted a scenario analysis in which a proportion of patients required 2 vials.
Economic Review
The current review is for difelikefalin for moderate-to-severe CKD-aP in adults on hemodialysis.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis (CUA) comparing difelikefalin plus BSC (hereafter referred to as difelikefalin) with BSC alone (topical therapies, antihistamines, gabapentinoids [i.e., gabapentin or pregabalin], antidepressants, and UV type B phototherapy) for patients with chronic kidney disease (CKD) on hemodialysis with moderate-to-severe pruritus. This population aligns with the approved Health Canada indication and reimbursement request.1
The drug-acquisition cost for difelikefalin is $27.00 per 2 mL vial (concentration of 50 mg/mL). It is administered 3 times per week by IV bolus injection into the venous line of the dialysis circuit at the end of the hemodialysis treatment during or after rinse-back. The recommended dose of difelikefalin is 0.5 mcg/kg dry body weight (i.e., the target postdialysis weight). Accounting for potential drug waste, no vial sharing, and a patient weight of |||| kg (per the KALM-1 trial),2 on average patients are expected to use 1 vial of difelikefalin per administration, resulting in a 4-week cycle cost of $324, or an annual cost of $4,212.
The clinical outcomes reported were QALYs and life-years. The analyses were conducted from the Canadian public payer perspective. The time horizon in the base case was lifetime (10 years due to the high mortality in patients with CKD-aP), with a 1.5% annual discount rate for costs and effects.
Model Structure
A 5-state Markov model structure (Figure 1 in Appendix 3) was used to assess the cost-effectiveness of difelikefalin relative to BSC. The Markov cycle length was 4 weeks. The cohort could transition through 5 health states defined by pruritus severity, measured with the Worst Itching Intensity Numerical Rating Scale (WI-NRS), including severe (WI-NRS score ≥ 7), moderate (WI-NRS score > 4 to < 7), mild (WI-NRS score > 0 to 4), none (WI-NRS score of 0), and death.
All patients in both treatment arms enter the model in the moderate or severe pruritus health state, per the stated indication. The number and proportion of patients with moderate or severe pruritus was informed by the baseline health state proportions in the KALM-1 trial,2 excluding any patients with mild or no pruritus. In each Markov cycle, patients may either remain in or transition to 1 of the 5 mutually exclusive health states.
Model Inputs
The characteristics of the simulated patient cohort in the model were informed by the KALM-1 trial.2 The starting age of the cohort was 58.0 years, and 62.8% of the cohort was assumed to be male and had a mean body weight of |||| kg. In the 2 treatment arms of KALM-1 trial, 162 patients were experiencing moderate pruritus at baseline and 215 were experiencing severe pruritusat baseline.2,3 The modelled population reflected this baseline severity, with ||% of patients entering the model in the moderate pruritus health state and ||% of patients entering the model in the severe health state.
Due to small patient numbers and a small amount of missing and/or unreported patient WI-NRS scores, treatment efficacy was informed by pooled patient-level data from both the KALM-1 and KALM-2 trials (efficacy of BSC alone in the model was informed by pooled patient-level data from the placebo arms). Transition probabilities were calculated based on a median frequency of 4-week WI-NRS data; the change in patients’ scores, categorized according to the WI-NRS, from the previous time point was used to generate transition probabilities. For example, if, on average, 30% of patients who experienced moderate pruritus transitioned to mild pruritus at the relevant time point, the mean transition probability between these 2 states would be 0.30. The proportion of patients in each health state at each time point was adjusted for half-cycle corrections. Patients’ pruritus severity was assumed to stabilize after 12 weeks, regardless of treatment arm. Adverse event (AE) data were sourced from the KALM-1 trial, and the sponsor’s model included all AEs that occurred during the 12-week double-blind treatment period in at least 2% of patients. Cost and QoL implications associated with AEs were applied at the first model cycle. The cohorts in the model could transition to death from any live health state. Patients with moderate or severe CKD-aP were assumed to have a higher mortality rate than patients with mild or no CKD-aP (5.7% versus 3.6%) based on analysis from the Dialysis Outcomes and Practice Patterns Study.4
To inform the health state utilities in the model, a mapping exercise that involved a de novo data collection study was completed using the WI-NRS data collected from the KALM trials. An adjusted limited dependent variable mixture model was implemented to map utility weightings from the EQ-5D to the WI-NRS, while accounting for age, sex, the presence of diabetes, and the number of years in dialysis. Weekly mean WI-NRS data from patients in the KALM-1 and KALM-2 trials were then converted to a 3-Level EQ-5D utility, ranging from |||||| for severe CKD-aP to |||||| for no CKD-aP (Table 11). Disutility associated with each AE included in the model was sourced from the relevant literature, and ranged from 0.01 (e.g., arthralgia) to 0.26 (diarrhea or nausea).
Drug-acquisition costs for difelikefalin were based on the sponsor’s submitted price. No additional administration cost was included, given that difelikefalin is administered by IV bolus injection into the venous line of the dialysis circuit at the end of each hemodialysis treatment. The model assumed potential drug waste, no vial sharing, and a patient weight of |||| kg, per the KALM-1 trial. Based on these assumptions, 1 vial of difelikefalin per administration was assumed in the model. Patients who did not respond (i.e., remained in the moderate or severe pruritus state) to difelikefalin after 12 weeks for the treatment of CKD-aP were assumed to discontinue difelikefalin.
Resource use frequency was sourced from consulted Canadian clinical experts to inform on the use of BSC, antisedatives, IV antibiotics, and iron, and on hospital admissions, outpatient visits, physician visits (dermatologist and general practitioner), and hemodialysis. All resource use frequencies, except hemodialysis, were greater for patients with moderate or severe CKD-aP than for those with less severe pruritus. Resource use costs were informed by the Ontario Drug Benefit Formulary,5 Canadian Institute for Health Information cost estimator,6 Ontario Schedule of Benefits,7 and British Columbia Medical Services Commission.
Summary of the Sponsor’s Economic Evaluation Results
The sponsor’s cost-effectiveness analysis was based on 5,000 probabilistic iterations; the findings are presented here. The results of the deterministic analysis were similar to the results of the probabilistic analysis.
Base-Case Results
In the sponsor’s base-case analysis, the ICER for difelikefalin plus BSC was $286,717 per QALY, compared to BSC alone. Specifically, difelikefalin was associated with 0.08 additional QALYs and $23,414 additional costs compared to BSC over a 10-year lifetime horizon (disaggregated results are presented in Table 10 and Table 11 in Appendix 3). The cost-effectiveness acceptability curves indicated that 0% of the results were cost-effective at a willingness-to-pay threshold of $50,000 per QALY.
Most of the incremental cost was attributable to the drug-acquisition costs associated with difelikefalin, with cost offsets due to the aversion of pruritus-related health state costs (pruritus treatment and health care use, including hospitalization) assigned to the moderate and severe pruritus states (Table 12). The key driver contributing to QALY gains for difelikefalin was a reduction in mortality because more patients were in the no CKD-aP or mild pruritus states (Table 13). Of the QALYs gained, 95% were accrued after the 12-week trial period.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. BSC ($/QALY) |
|---|---|---|---|---|---|
BSC | $639,740 | Reference | 7.31 | Reference | Reference |
Difelikefalin | $663,154 | $23,414 | 7.42 | 0.08 | 286,717 |
BSC = best support care; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.8
Sensitivity and Scenario Analyses Results
The sponsor conducted several scenario analyses to test the impact of alternative parameters and assumptions on the modelled results. The largest change from the base-case results was seen with alternative utility weightings (ICER of $143,737 per QALY) and a decreased time horizon of 5 years (ICER of $441,104 per QALY).
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations in the sponsor’s analysis that have notable implications on the economic analysis.
Assumption of mortality benefit with difelikefalin. The sponsor assumed increased mortality with increasing severity of pruritus, with patients in the moderate and severe pruritus states having a greater probability of mortality than patients in the mild or no pruritus states (3.6% versus. 5.7%), based on observational data from the Dialysis Outcomes and Practice Patterns Study. The modelled benefit of difelikefalin on pruritus subsequently led to a modelled reduction in mortality. Pruritus is not an accepted or validated surrogate for mortality, and the evidence provided in support of this assumption was observational in nature and did not account for all potentially relevant confounders. Therefore, the assumption that a reduction in pruritus from treatment with difelikefalin would result in a mortality benefit is not supported. The clinical expert input obtained by CADTH for this review confirmed that this is an unjustified assumption, as there is no evidence to support a causal link between pruritus severity and mortality. This assumption resulted in the majority of QALY (and all life-year) gains, and overestimated the benefits associated with difelikefalin.
In the CADTH reanalysis, mortality rates were assumed to be equal among all CKD-aP states.
Assumption of reduced all-cause hospitalization in patients treated with difelikefalin. The model assumed a greater rate of hospital admissions (0.01 to 0.05 per 4-week cycle) for patients with an elevated severity of pruritus, based on expert opinion obtained by the sponsor. As with the mortality benefit assumption, this resulted in a modelled reduction in hospitalizations with difelikefalin treatment. There is no evidence provided to support the theory that a reduction in pruritus severity with treatment would reduce all-cause hospitalizations, and removing this assumption was supported by the clinical expert input obtained by CADTH. This assumption overestimated the cost savings associated with difelikefalin.
In the CADTH reanalysis, hospitalization rates were the same among all CKD-aP states.
Assumption of reduced primary care visits in patients treated with difelikefalin. The model assumed more primary care visits (0.025 to 0.05 per 4-week cycle) for patients with an elevated severity of pruritus, based on expert opinion obtained by the sponsor. Patients on hemodialysis attend routine visits and receive care from nephrologists, which includes the identification and treatment of kidney disease and dialysis-related symptoms, including pruritus; non-CKD-related and nondialysis-related issues may be managed by a primary care physician. According to clinical expert input obtained by CADTH, no reduction in primary care physician visits is expected with a treatment-associated reduction in pruritus severity with difelikefalin, as this symptom is managed by nephrologists and treatment is administered in the dialysis unit where patients are seen regularly. This assumption overestimated the cost savings associated with difelikefalin.
In the CADTH reanalysis, the number of primary care and nephrologist visits were assumed to be the same, regardless of a patient’s pruritus severity health state.
Inclusion of phototherapy in BSC. The model assumed that phototherapy was part of the BSC costs. However, phototherapy was not used in the KALM trials. Furthermore, there are issues with access to phototherapy in clinical practice in Canada, according to clinical expert input obtained by CADTH, and the inclusion of differential phototherapy costs based on pruritus severity may overestimate BSC costs for patients on BSC alone, although the costs are relatively small.
In the CADTH reanalysis, the cost of phototherapy was removed.
Mapping of WI-NRS to EQ-5D. To inform the health state utilities in the model, a mapping exercise that involved a de novo data collection study was completed using the WI-NRS data collected from the KALM trials. The sponsor mapped values from the WI-NRS to the 3-Level EQ-5D. The sponsor provided limited details on the mapping exercise, and mapping is not recommended for the derivation of utilities in the CADTH Guidelines for the Economic Evaluation of Health Technologies in Canada.9 Mapping is unlikely to successfully capture the utility relationship between 2 measures due to the high variability in predictive values, depending on the instrument being mapped, the algorithm used, and the severity of the health states included. The mapping exercise accounted for only 4 parameters (age, sex, diabetes status, dialysis vintage), but there are numerous other potential confounders in the relationship of pruritus and preference-based HRQoL. These factors include those that may be on the causal pathway to both itchiness-related and nonitchiness-related QoL, including underdialysis, other comorbid conditions, and health behaviours (adherence). Those with itchiness may have a worse QoL due to itchiness and other factors that are not accounted for (e.g., nausea, anorexia, restless legs), which may confound mapping. This approach introduced uncertainty in the derivation of the utility values informing the sponsor’s model; it is plausible that it led to an overestimate of the QALY benefit with difelikefalin because relevant confounders were not included in the mapping.
In a scenario analysis, utility values were increased or decreased by 25% to examine the impact of CKD-aP severity and HRQoL on the results.
Discontinuation criteria with difelikefalin are uncertain. The model assumed that difelikefalin would be discontinued in patients with moderate and severe pruritus who did not enter the mild or normal pruritus health state after 12 weeks. This assumption is likely to underestimate ongoing difelikefalin use and costs, as, according to clinical expert opinion obtained by CADTH, patients who remain in the moderate or severe pruritus health state may still have a 3-point to 4-point reduction in WI-NRS score, a change deemed potentially clinically significant, and would likely remain on difelikefalin. Furthermore, as the use of the WI-NRS is not standard practice, it is plausible that many patients with a subjectively minor improvement may be deemed by clinicians to be responders and would be likely to continue treatment.
In a CADTH scenario analysis, only patients who started in the severe pruritus health state and remained in this state would cease difelikefalin; patients who moved from the severe to moderate pruritus health state or remained in the moderate health state would continue on difelikefalin (as would those who moved to the mild or no pruritus health states).
Severity stabilization. Pruritus severity was assumed to stabilize after 12 weeks, at which point treatment stopping assumptions were applied (as discussed previously). Although the clinical experts consulted by CADTH found it reasonable to assume pruritus stabilization at 12 weeks, given that the observed response to other pruritus treatments is likely to be known at 12 weeks, it is plausible that a trial of treatment may continue and that stabilization of pruritus severity related to treatment may continue to occur over a longer time period.
In the CADTH scenario analysis, pruritus severity was assumed to stabilize after 52 weeks, rather than at 12 weeks.
Identification of patients with moderate or severe pruritus. The WI-NRS was used to determine the pruritus severity and transition probabilities in the model; however, this scale is not used in routine care and, as such, patients with a wider spectrum of symptoms, including mild pruritus defined by the WI-NRS score, may be deemed by clinicians to have symptoms meriting treatment with difelikefalin. The clinical benefit in these patients is unknown, given the inclusion criteria in the KALM trials and, according to the clinical experts consulted by CADTH, these patients are likely to have a smaller incremental benefit with regard to the improvement of symptoms and QoL.
CADTH could not address this limitation in its reanalysis, and the impact of this limitation is uncertain; it is plausible that this will lead to a greater ICER and may lead to a larger proportion of patients treated with difelikefalin (with budget impact implications).
CADTH also identified the following minor limitations:
Inclusion of nonpruritus-related AEs. Some AEs reported in the KALM trials, and subsequently included in the sponsor’s submission, appear to be unrelated to pruritus or its treatment, such as fluid overload, sepsis, upper respiratory tract infection, and urinary tract infection (but are included in disutility and associated costs, including IV antibiotics). The inclusion of these AE costs might overestimate the costs associated with BSC alone, although the health and resource use implications of the additional AEs were relatively small.
In a CADTH scenario analysis, the costs and disutilities of the aforementioned AEs were removed.
Assumption on BSC use. The model assumed less use of BSC therapies for milder states (no pruritus or mild pruritus). However, in the trials, patients continued to use BSC drugs (reduction in use was not reported). According to the CADTH clinical experts, patients are likely to remain on their BSC treatments even after transitioning to milder health states, and the length of time they would remain on these drugs after a resolution of symptoms would be determined on a case-by-case basis by physicians.
In a CADTH scenario analysis, BSC costs (antihistamines, gabapentin or pregabalin, oral steroids, topical steroids, antidepressants) were set to the same amounts across all pruritus states.
Possibility of up-dosing or use of another vial due to patient weight in clinical practice. The model assumed 1 vial per administration, based on a patient weight of |||| kg. The clinical experts consulted by CADTH for this review noted that up-dosing is possible and that more than 1 vial might be used in practice.
In a CADTH scenario analysis, 25% of patients were assumed to be up-dosed and to require an additional vial per administration (i.e., vials per administration was changed from 1 to 1.25).
Additionally, the key assumptions outlined in Table 4 were made by the sponsor and have been appraised by CADTH.
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations in the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
Patients in the KALM studies are representative of patients who will be treated, if approved. | There is a reasonable likelihood of difelikefalin being used in patients with less severe pruritus. Therefore, generalizability is of concern. |
No additional health care provider time required to administer difelikefalin. | Although health care provider time is likely to be minimal, given the method of administration, it is another task in often busy hemodialysis units that may require some time to complete. The impact of this is uncertain, although it is likely to be minimal. |
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
The CADTH base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts. These changes, summarized in Table 5, involved the removal of a benefit in mortality, hospitalization, and primary care visits with decreasing pruritus severity, and also the removal of phototherapy.
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Changes to derive the CADTH base case | ||
1. Mortality and pruritus severity | Patients in the moderate and severe pruritus states have a greater probability of mortality than patients in the mild or no pruritus states (3.6% vs. 5.7% per cycle) | No mortality benefit by pruritus severity, with the same mortality probability applied for all pruritus states (3.6% per cycle) |
2. Hospitalizations and pruritus severity | More hospital admissions (0.01 to 0.05 per 4-week cycle) with a greater severity of pruritus | Hospital admissions assumed to be the same (0.01 per cycle) for all pruritus states |
3. Primary care visits and pruritus severity | More primary care visits (0.025 to 0.05 per 4-week cycle) with a greater severity of pruritus | Primary care visits assumed to be the same (0.025 per cycle) for all pruritus states |
4. Inclusion of phototherapy as part of BSC | The model included phototherapy as part of the BSC costs | Phototherapy was excluded as part of the BSC costs |
CADTH base case | 1 + 2 + 3 + 4 | |
BSC = best support care.
The CADTH base case demonstrated that, relative to BSC alone, difelikefalin plus BSC was more expensive by $16,500 and more effective by 0.03 QALYs, resulting in an ICER of $582,515 per QALY (Table 6). Deterministic 1-way analyses were also provided to show how each of the parameters altered the results as part of the CADTH base-case impact, while holding everything else constant. Due to the small QALY gain (0.03) in the denominator of the ICER calculation, the results of the deterministic analysis and the results of the probabilistic analysis were not similar. The change to the CADTH base case with the greatest impact on the results was setting mortality to be the same across all pruritus states (reanalysis 1), reducing the QALYs gained from 0.08 to 0.02. This supports the statement that the main driver contributing to QALY gains in the sponsor’s base-case analysis was mortality benefits associated with less severe health states, as more patients entered the less severe pruritus states with difelikefalin. In the CADTH reanalysis, after removal of the mortality benefit, there were no longer life-years gained with difelikefalin. As a result, the primary parameter that contributed to an incremental QALY benefit was the difference in QoL between pruritus severity health states. The total costs for both arms also increased under the assumption that there was no difference in mortality (as patients who survive longer incur more health care costs).
Table 6: Summary of the Stepped Analysis of the CADTH Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
Sponsor’s base case (probabilistic) | BSC | 639,740.48 | 3.77 | Reference |
Difelikefalin | 663,154.26 | 3.85 | 286,717 | |
Sponsor’s base case (deterministic) | BSC | 639,916.48 | 3.77 | Reference |
Difelikefalin | 660,402.86 | 3.85 | 254,401 | |
CADTH reanalysis 1 (deterministic) | BSC | 688,400.74 | 4.06 | Reference |
Difelikefalin | 699,852.86 | 4.08 | 407,449 | |
CADTH reanalysis 2 (deterministic) | BSC | 616,917.98 | 3.77 | Reference |
Difelikefalin | 639,044.20 | 3.85 | 274,765 | |
CADTH reanalysis 3 (deterministic) | BSC | 639,771.54 | 3.77 | Reference |
Difelikefalin | 660,285.50 | 3.85 | 254,743 | |
CADTH reanalysis 4 (deterministic) | BSC | 639,846.69 | 3.77 | Reference |
Difelikefalin | 660,346.35 | 3.85 | 254,566 | |
CADTH base case (deterministic) | BSC | 663,196.86 | 4.06 | Reference |
Difelikefalin | 676,715.40 | 4.08 | 480,969 | |
CADTH base case (probabilistic) | BSC | 663,205.52 | 4.06 | Reference |
Difelikefalin | 679,706.34 | 4.09 | 582,515 |
BSC = best support care; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
CADTH notes that the reanalysis focused on modifying the sponsor’s model assumptions and parameters to more accurately reflect clinical course, patient management, and treatment in Canada, where possible. However, it should be noted that significant uncertainty exists in this reanalysis due to limitations that could not be addressed or were only addressed as part of scenario analyses. This includes multiple optimistic model assumptions in favour of difelikefalin that remained part of the CADTH base case. The assumption that pruritus severity was assumed to stabilize after 12 weeks, regardless of treatment arm, even among patients who discontinued difelikefalin treatment, could not be tested due to model restrictions. Uncertainty of mapping the WI-NRS to EQ-5D, and assumptions on the stopping rules after 12 weeks in those who did not transition to the mild or no severity pruritus health state likely favour difelikefalin; further uncertainties are the inclusion of nonrelated AEs, the possibility of up-dosing, and severity stabilization.
Scenario Analysis Results
To address some of this uncertainty, CADTH performed probabilistic scenario analyses to determine the impact of varying the utility weights (± 25% for each pruritus health state), discontinuing difelikefalin only in patients remaining in the severe state after 12 weeks, and assuming that disease severity stabilization only occurs after 52 weeks rather than 12 weeks. The results of the analyses with the largest impact on the ICER results are presented in Table 7, with further scenario analyses provided in Table 16 in Appendix 4. In the scenario in which health state utilities were assumed to be less (smaller change in HRQoL with varying CKD-aP severity) (scenario 1b in Table 7), the ICER increased to $933,591 per QALY gained, whereas difelikefalin was associated with only a QALY gain of 0.02 compared to BSC. This highlights how sensitive the model is to the assumed utility difference between pruritus severity health states. Additionally, the model is sensitive to difelikefalin drug use and associated costs, which increased as fewer patients discontinued treatment based on pruritus severity (scenario 2 in Table 7) or when patients discontinued treatment after a longer period of time had elapsed (scenario 3 in Table 7).
CADTH undertook a price reduction analysis based on the sponsor’s base case and the CADTH base case to determine the price reduction of difelikefalin required to achieve an ICER under $50,000 per QALY (Table 8). These analyses demonstrated that a price reduction greater than 92% for difelikefalin would be required to reach this willingness-to-pay threshold in the CADTH base case.
Table 7: Additional Scenario Analysis Results
Analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
CADTH base case | BSC | 663,205.52 | 4.06 | Reference |
Difelikefalin | 679,706.34 | 4.09 | 582,515 | |
1.a. Increase health state utilities by 25% | BSC | 663,207.19 | 5.08 | Reference |
Difelikefalin | 679,808.77 | 5.12 | 408,527 | |
1.b. Decrease health state utilities by 25% | BSC | 663,207.46 | 3.03 | Reference |
Difelikefalin | 679,719.28 | 3.05 | 933,591 | |
2. Discontinue only severe patients after 12 weeks | BSC | 663,210.41 | 4.06 | Reference |
Difelikefalin | 694,076.96 | 4.09 | 1,022,496 | |
3. Severity stabilized after 52 weeks | BSC | 662,296.56 | 4.17 | Reference |
Difelikefalin | 693,134.83 | 4.19 | 1,629,408 |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Table 8: CADTH Price Reduction Analyses
Analysis Price reduction | ICERs for difelikefalin vs. BSC ($/QALY) | |
|---|---|---|
Sponsor’s base case | CADTH reanalysis | |
No price reduction | 286,717 | 582,515 |
10% | 273,763 | 496,000 |
20% | 254,850 | 441,967 |
30% | 235,938 | 387,933 |
40% | 217,025 | 333,900 |
50% | 198,113 | 279,867 |
60% | 179,200 | 225,833 |
70% | 160,288 | 171,800 |
80% | 141,375 | 117,767 |
90% | 122,463 | 63,733 |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-years.
Issues for Consideration
According to the clinical expert feedback obtained by CADTH, difelikefalin is not likely to be used in patients undergoing dialysis other than hemodialysis or in patients CKD not yet on dialysis because of its administration method. However, the cost-effectiveness of difelikefalin in such patients, should it be used, is unknown.
According to the clinical experts consulted by CADTH, difelikefalin would probably be an add-on treatment and would likely not lead to a reduction in the use of other BSC drugs. The length of time patients would remain on BSC after the resolution of symptoms would be determined on a case-by-case basis by physicians.
Overall Conclusions
The CADTH clinical review concluded that difelikefalin demonstrates a statistically significant and clinically meaningful improvement in itch intensity, compared to placebo, in patients on hemodialysis with moderate-to-severe CKD-aP, based on the KALM-1 and KALM-2 trials, as well as a statistically significant improvement in disease severity measured by at least a 3-point improvement in WI-NRS scores. However, the clinical meaningfulness of a 3-point improvement in WI-NRS score, which was the scale used to categorize health states in the sponsor’s submitted model, is uncertain. Furthermore, although analyses of the secondary end points that measure HRQoL showed results in favour of difelikefalin, no conclusions could be drawn due to a lack of statistical testing or control for multiplicity. No data using generic preference-based measures were collected in the trials. The economic model, therefore, attempted to capture these benefits by mapping the WI-NRS to EQ-5D through a de novo data collection study. However, this method may not have accounted for all confounders and may have introduced significant uncertainty into the measurement of HRQoL, which was a driver of QALY gains in the submitted economic model.
The sponsor’s base case included several assumptions that were not substantiated by the available evidence and were counter to CADTH clinical expert opinion on current practice in Canada. These include assumptions of a mortality benefit and a reduction in all-cause hospitalization mediated by improvement of CKD-aP. Further, change in primary care use was assumed. However, in Canada, CKD and its related symptoms (i.e., CKD-aP) would be managed by nephrologists rather than in the primary care setting. Finally, phototherapy was included as part of BSC. This informed costs but did not impact the efficacy of BSC, given that the efficacy inputs were based on the KALM trials, which excluded phototherapy as a possible concomitant treatment.
The CADTH reanalysis assumed no difference in mortality, hospitalization, or primary care visits by pruritus health state and excluded phototherapy from the BSC costs. The CADTH results were similar to those of the sponsor, in that difelikefalin was not cost-effective at the threshold of $50,000 per QALY gained. The CADTH base-case ICER for difelikefalin plus BSC, compared to BSC alone, was $582,515 per QALY gained; difelikefalin has a 0% probability of being cost-effective at a willingness-to-pay threshold of $50,000 per QALY gained. A price reduction of at least 92% would be required for difelikefalin to achieve a mean cost-effective estimate of $50,000 per QALY. Because of the small clinical benefits expected from difelikefalin, the ICER was found to be less stable. However, the conclusions remained the same across all CADTH reanalyses, which indicated that a substantial price reduction would be necessary for difelikefalin to be considered cost-effective.
Although the CADTH reanalysis attempted to address some of the identified limitations, uncertainty remained in the CADTH base case due to remaining uncertain but optimistic assumptions favouring difelikefalin. Specifically, if the true QoL benefit is less than the estimated values from mapping, the ICER is anticipated to be much greater. Further, unless strict stopping rules related to the achievement of a specific pruritus severity (i.e., achievement of mild or no pruritus within 12 weeks) are enforced, the ICER is expected to be greater. In both of these plausible scenarios, the ICERs were found to be closer to $1 million per QALY gained, with a price reduction greater than 95% required to achieve an ICER of $50,000 per QALY.
References
1.Drug Reimbursement Review sponsor submission: Korsuva® (difelikefalin), 50 micrograms/mL intravenous solution [internal sponsor's package]. Saint-Laurent (QC): Otsuka Canada Pharmaceutical; 2022 Sep 22.
2.Clinical Study Report: KALM-1 CR845-CLIN3102. A multicenter, double-blind, randomized, placebo-controlled study to evaluate the safety and efficacy of intravenous CR845 in hemodialysis patients with moderate-to-severe pruritus, with a 52-week open-label extension [internal sponsor's report]. Stamford (CT): Cara Therapeutics; 2020.
3.Clinical Study Report: KALM-2 CR845-CLIN3103. A multicenter, double-blind, randomized, placebo-controlled study to evaluate the safety and efficacy of intravenous CR845 in hemodialysis patients with moderate-to-severe pruritus, with a 52-week open-label extension [internal sponsor's report]. Stamford (CT): Cara Therapeutics; 2020.
4.Pisoni RL, Wikström B, Elder SJ, et al. Pruritus in haemodialysis patients: International results from the Dialysis Outcomes and Practice Patterns Study (DOPPS). Nephrol Dial Transplant. 2006;21(12):3495-3505. PubMed
5.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2022; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2022 Dec 15.
6.Patient cost estimator. 2022; https://www.cihi.ca/en/patient-cost-estimator. Accessed 2022 Dec 15.
7.Schedule of benefits for physician services under the Health Insurance Act: (January 25, 2022 (effective July 1, 2022)). Toronto (ON): Ontario Ministry of Health; 2022: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf. Accessed 2022 Dec 15.
8.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Korsuva® (difelikefalin), 50 micrograms/mL intravenous solution. Saint-Laurent (QC): Otsuka Canada Pharmaceutical; 2022 Sep 22.
9.Guidelines for the economic evaluation of health technologies: Canada. 4th ed. Ottawa (ON): CADTH; 2017: https://www.cadth.ca/guidelines-economic-evaluation-health-technologies-canada-4th-edition. Accessed 2022 Dec 15.
10.Table: 17-10-0005-01: population estimates on July 1st, by age and sex. Ottawa (ON): Statistics Canada; 2022 Dec 21: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710000501. Accessed 2022 Dec 15.
11.Organ replacement in Canada: CORR annual statistics. Ottawa (ON): Canadian Institue for Health Information (CIHI); 2021: https://www.cihi.ca/en/organ-replacement-in-canada-corr-annual-statistics. Accessed 2022 Dec 15.
12.Treatment of end-stage organ failure in Canada, Canadian Organ Replacement Register, 2011 to 2020: end-stage kidney disease and kidney transplants —data tables. Data table 16. Ottawa (ON): Canadian Institute for Health Information (CIHI); 2021.
13.Trends in end-stage kidney disease in Canada, 2020 - infographic. Ottawa (ON): Canadian Institute for Health Information (CIHI); 2021: https://www.cihi.ca/en/trends-in-end-stage-kidney-disease-in-canada-2020-infographic. Accessed 2022 Dec 15.
14.Sukul N, Karaboyas A, Csomor PA, et al. Self-reported pruritus and clinical, dialysis-related, and patient-reported outcomes in hemodialysis patients. Kidney Med. 2021;3(1):42-53 e41. PubMed
15.Sutherland G, Dinh T. Understanding the gap: a pan-Canadian analysis of prescription drug insurance coverage. Ottawa (ON): The Conference Board of Canada; 2017.
Appendix 1: Cost Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s) and drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 9: CADTH Cost Comparison for CKD-aP
Treatment | Strength / concentration | Form | Price | Recommended dosage | Daily cost | Annual cost |
|---|---|---|---|---|---|---|
Difelikefalin (Korsuva) | 50 mcg / 1 mL | Solution for IV Injection | $27.0000 | 0.5 mcg/kg dry body weight 3 times per week | $11.53 | $4,212 |
Note: The drug price is based on the sponsor submitted price. The annual cost was based on an assumed patient dry weight of |||| kg and the daily cost was achieved by dividing the annual cost by the number of days in a year (365.25).
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | Yes | No comment |
Model has been adequately programmed and has sufficient face validity | No | The original model was sent back to the sponsor because incorrect formula has been applied when discounting the costs in the severe health state for the BSC group. The sponsor provided CADTH with an updated model. |
Model structure is adequate for decision problem | Yes | No comment |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | No | The resource use costs table (Table 10) in the sponsor submitted report did not align with the values identified in the model. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
CKD-aP = Pruritus associated with chronic kidney disease
Source: Sponsor’s pharmacoeconomic submission8
Table 11: Health State Utilities Derived From the Mapping of WI-NRS to EQ-5D
Disease Severity | Weighting | Standard Deviation |
|---|---|---|
No CKD-aP | |||||| | |||||||| |
Mild CKD-aP | |||||| | |||||||| |
Moderate CKD-aP | |||||| | |||||||| |
Severe CKD-aP | |||||| | ||||||| |
CKD-aP = Pruritus associated with chronic kidney disease
Source: Sponsor’s pharmacoeconomic submission.8
Detailed Results of the Sponsor’s Base Case
Table 12: Discounted Disaggregated Mean Costs for the Sponsor’s Reference Case Analysis (Probabilistic)
Cost ($) | Difelikefalin | BSC | Incremental |
|---|---|---|---|
No CKD-aP | $14,929.40 | $5,279.99 | $9,649.41 |
Mild CKD-aP | $254,052.62 | $167,423.23 | $86,629.39 |
Moderate CKD-aP | $223,778.84 | $264,671.62 | -$40,892.78 |
Severe CKD-aP | $153,629.56 | $202,307.31 | -$48,677.75 |
Drug acquisition | $16,682.52 | $0.00 | $16,682.52 |
AEs | $81.33 | $58.33 | $23.00 |
Total costs | $663,154.26 | $639,740.48 | $23,413.79 |
AE = adverse event; BSC = best support care; CKD-aP = Pruritus associated with chronic kidney disease.
Source: Sponsor’s pharmacoeconomic submission.8
Table 13: Discounted Disaggregated Mean QALYs for the Sponsor’s Reference Case Analysis (Probabilistic)
Health State or Event | Difelikefalin | BSC | Incremental |
|---|---|---|---|
No CKD-aP | 0.10 | 0.04 | 0.06 |
Mild CKD-aP | 1.61 | 1.06 | 0.55 |
Moderate CKD-aP | 1.34 | 1.58 | −0.24 |
Severe CKD-aP | 0.87 | 1.14 | −0.27 |
AEs | −0.07 | −0.05 | −0.02 |
Total QALYs | 3.85 | 3.77 | 0.08 |
AE = adverse event; BSC = best support care; CKD-aP = Pruritus associated with chronic kidney disease. QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.8
Table 14: Discounted Disaggregated Mean LYs for the Sponsor’s Reference Case Analysis (Probabilistic)
Health State | Difelikefalin | BSC | Incremental |
|---|---|---|---|
No CKD-aP | 0.18 | 0.06 | 0.12 |
Mild CKD-aP | 2.95 | 1.95 | 1.00 |
Moderate CKD-aP | 2.55 | 3.02 | −0.47 |
Severe CKD-aP | 1.74 | 2.29 | −0.55 |
Total LYs | 7.42 | 7.32 | 0.10 |
BSC = best support care; CKD-aP = Pruritus associated with chronic kidney disease; LY = life-year.
Source: Sponsor’s pharmacoeconomic submission.8
Appendix 4: Additional Details on the CADTH Reanalyses and the Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Detailed Results of CADTH Base Case
Table 15: Disaggregated Summary of CADTH’s Economic Evaluation Results
Parameter | Difelikefalin | BSC | Incremental |
|---|---|---|---|
Discounted LYs | |||
No CKD-aP | 0.18 | 0.06 | 0.12 |
Mild CKD-aP | 2.95 | 1.95 | 1.00 |
Moderate CKD-aP | 2.82 | 3.33 | −0.51 |
Severe CKD-aP | 1.91 | 2.52 | −0.61 |
Total LYs | 7.87 | 7.87 | 0.00 |
Discounted QALYs | |||
No CKD-aP | 0.10 | 0.04 | 0.06 |
Mild CKD-aP | 1.61 | 1.07 | 0.54 |
Moderate CKD-aP | 1.48 | 1.74 | −0.26 |
Severe CKD-aP | 0.96 | 1.26 | −0.30 |
AEs | −0.07 | −0.05 | −0.02 |
Total QALYs | 4.09 | 4.06 | 0.03 |
Discounted costs ($) | |||
No CKD-aP | $14,900.42 | $5,299.33 | $9,601.09 |
Mild CKD-aP | $248,202.41 | $163,864.24 | $84,338.17 |
Moderate CKD-aP | $238,070.90 | $281,063.55 | -$42,992.65) |
Severe CKD-aP | $161,614.15 | $212,920.03 | -$51,305.88) |
Drug Acquisition | $16,837.14 | $0.00 | $16,837.14 |
AEs | $81.32 | $58.37 | $22.95 |
Total costs | $679,706.34 | $663,205.52 | $16,500.82 |
ICER ($/QALY) | 582,515 | ||
AE = adverse event; BSC = best support; CKD-aP = Pruritus associated with chronic kidney disease care; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year.
Scenario Analyses
Table 16: Additional Scenario Analysis Results
Analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
CADTH base case | BSC | $663,205.52 | 4.06 | Ref. |
Difelikefalin | $679,706.34 | 4.09 | 582,515 | |
Up-dose 25% | BSC | $663,207.71 | 4.06 | Ref. |
Difelikefalin | $680,086.75 | 4.09 | 584,423 | |
Exclude unrelated AEs and IV antibiotics | BSC | $662,712.83 | 4.06 | Ref. |
Difelikefalin | $679,419.22 | 4.09 | 567,418 | |
Same BSC costs across all pruritus states | BSC | $662,023.77 | 4.06 | Ref. |
Difelikefalin | $678,842.41 | 4.08 | 604,341 | |
Vial sharing with difelikefalin | BSC | — | 4.05 | Ref. |
Difelikefalin | $674,675.38 | 4.08 | 399,659 |
AE = adverse event; BSC = best support; CKD-aP = Pruritus associated with chronic kidney disease care; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year.
Appendix 5: Submitted BIA and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 17: Summary of Key Takeaways
Key Takeaways of the BIA |
|---|
|
Summary of Sponsor’s BIA
The submitted BIA assessed the introduction of difelikefalin as a novel treatment for moderate-to-severe CKA-aP in adult patients on hemodialysis. The analysis took the perspective of Canadian public drug plans. A time horizon of 3 years was taken. The base-case analysis considered BSC alone in the reference scenario as the only available treatment option and the new drug scenario considered the reimbursement of difelikefalin. Key inputs to the BIA, included the parameters used to determine the size of the population eligible for difelikefalin, are documented in Table 18.
Key assumptions to the sponsor’s BIA included:
Drug-acquisition costs included the use of 1 vial per patient per treatment administration based on an assumed dry body weight of |||| kg. This incorporated potential wastage.
The sponsor assumed no patients would discontinue difelikefalin based on treatment response.
Table 18: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as Year 1 / Year 2 / Year 3 if appropriate) |
|---|---|
Target population | |
Base Year Population of CADTH-Participating Drug Plans | 29,834,46310 |
% of Canadians with ESRD | 0.14%11 |
% of Canadians with ESRD on Dialysis | |
% of Dialysis Patients on HD | 79.40%13 |
% of Canadian HD patients with moderate-to-severe pruritus | 40.00%14 |
Public plan eligibility | 79.08%15 |
Number of patients eligible for drug under review | 6,170 / 6,314 / 6,461 |
Market Uptake (3 years) | |
Uptake (Reference scenario) | |
Best Supportive Care | 100% / 100% / 100% |
Uptake (New drug scenario) | |
Difelikefalin | ||% / ||% / ||% |
Best Supportive Care | ||% / ||% / ||% |
Cost of treatment (per patient per year) | |
Difelikefalin plus Best Supportive Care | $4,226.46 |
Best Supportive Care | $0 |
ESRD = End Stage Renal Disease; HD = hemodialysis.
Summary of the Sponsor’s BIA Results
The sponsor’s estimated budget impact of funding difelikefalin as a treatment for patients with moderate-to-severe CKD-aP was $1,468,123 in year 1, $2,545,492 in year 2, and $3,243,486 in year 3, for a 3-year total of $7,257,101.
The sponsor conducted several sensitive and scenario analyses testing alternative assumptions. The changes with the greatest impact on the results included a change in the assumption in the number of vials required based on the assumed patient dry body weight, as well as the assumed market share and public drug plan eligibility.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The anticipated market uptake of difelikefalin is underestimated: The sponsor assumed that difelikefalin would achieve a ||% market share over a 3-year time horizon but did not provide sufficient evidence to support this assumption. Clinical expert feedback obtained by CADTH indicated that physicians would be likely to prescribe difelikefalin to a far greater proportion of eligible patients due to the lack of therapies indicated for CKD-aP. The likelihood for increased uptake is also further supported by its ease of administration, according to the clinical experts consulted by CADTH. Clinical expert input obtained by CADTH indicated that difelikefalin would likely achieve a market share of 50%, and even up to 75%, in 3 years if available. The underestimation of the market uptake of difelikefalin led to an underestimation of the anticipated budget impact.
CADTH addressed this limitation by revising market share of difelikefalin to 10% in Year 1, 30% in Year 2, and 50% in Year 3 in the CADTH base case.
The estimated population eligible and assumed to have moderate-to-severe pruritus is uncertain: The sponsor assumed that 40% of Canadian HD patients would have moderate or severe pruritus and thus be eligible for treatment with difelikefalin. This value was taken from a 2021 study where HD patients self-reported pruritus and other dialysis-related clinical outcomes.14 This estimate was deemed plausible by the clinical experts consulted by CADTH; however, they noted that the classification of pruritus in clinical practice in Canada can be challenging. The reliance on subjective ranking introduces a high level of uncertainty into the estimation of the eligible patient population. Should the proportion of patients determined to have moderate-to-severe pruritus be greater than 40%, this could significantly underestimate the eligible population size and potential budget impact.
CADTH assumed the proportion of patients on HD with moderate-to-severe pruritus to be 60% in an exploratory scenario analysis.
There is uncertainty in the number of vials needed per patient per course: The sponsor assumed that 1 vial of difelikefalin would be needed to treat a single patient, assuming a mean patient dry body weight of |||| kg. It is unclear whether this assumption would be reflective of clinical practice as the proportion of CKD-pruritus patients who have a dry body weight above 104kg and, would require a second vial, is uncertain. Moreover, the clinical experts consulted by CADTH indicated that up-dosing with this class of medications is common in Canadian clinical practice; however, as difelikefalin is the first medication with this indication, it is unclear how frequently up-dosing will occur in clinical practice. The product monograph notes that 1 additional dose per week may be given, however the safety and efficacy of an extra dose has not been established,
CADTH conducted an exploratory scenario analysis with the assumption that 25% of patients would require a second vial due to up-dosing or a greater dry body weight.
Discontinuation criteria with difelikefalin are uncertain. The model assumed difelikefalin would not be discontinued. This is not aligned with the assumption from the submitted CUA. In the CUA, patients with moderate and severe pruritus who did not enter the “mild” or “normal” pruritus state after 12 weeks were assumed to discontinue. Assuming all patients remained on treatment may have overestimated the budget impact associated with difelikefalin, however the extent of this is uncertain given the discontinuation rules with difelikefalin remain unclear. It is possible patients would be trialled for a period longer than 12 weeks, as assumed in the CUA, and it is also possible patients who achieved some improvement, but did not achieve “mild” or “no” pruritus, would remain on treatment, which would significantly decrease the likelihood of discontinuation. As a result, the magnitude by which the sponsor’s BIA is overestimated is highly uncertain.
CADTH could not address this limitation in reanalyses.
CADTH Reanalyses of the BIA
CADTH revised the sponsor’s base case by updating market shares to align with the expected market uptake of difelikefalin in clinical practice, Table 19.
Table 19: CADTH Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | — | — |
Changes to derive the CADTH base case | ||
1. Market Uptake of difelikefalin over a 3-year period | ||% in Year 1, ||% in Year 2, and ||% in Year 3 | 10% in Year 1, 30% in Year 2, 50% in Year 3 |
CADTH base case | Reanalysis 1 | |
The results of the CADTH reanalysis are presented in summary format in Table 20 and a more detailed breakdown is presented in Table 21. Based on the CADTH base case, the expected budget impact of difelikefalin for the treatment of moderate-to-severe CKD-aP for patients receiving HD was $2,607,678 in Year 1, $8,004,690 in Year 2, $13,651,036 in Year 3, for a 3-year total of $24,263,405.
Scenario analyses were conducted to assess alternative assumptions, and these included changes related to the proportion of patients with moderate-to-severe pruritus (60% instead of the 40% assumed in the base case) and the proportion of patients who would be receiving more than 1 vial per treatment course (assumed 30% in scenario analysis). The results of these scenario analyses ranged from $52,437,971 to $60,505,351 over a 3-year period. The impact of discontinuation criteria could not be assessed.
Table 20: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total |
|---|---|
Submitted base case | $7,257,101 |
CADTH base case | $24,263,405 |
BIA = budget impact analysis
Table 21: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 0 | 0 | 0 | 0 | 0 |
New drug | 0 | $1,468,123 | $2,545,492 | $3,243,486 | $7,257,101 | |
Budget impact | 0 | $1,468,123 | $2,545,492 | $3,243,486 | $7,257,101 | |
CADTH base case | Reference | 0 | 0 | 0 | 0 | 0 |
New drug | 0 | $2,607,678 | $8,004,690 | $13,651,036 | $24,263,405 | |
Budget impact | 0 | $2,607,678 | $8,004,690 | $13,651,036 | $24,263,405 | |
CADTH sensitivity analysis: 60% of patients have moderate-to-severe pruritus | Reference | 0 | 0 | 0 | 0 | 0 |
New drug | 0 | $3,911,518 | $12,007,035 | $20,476,554 | $36,395,107 | |
Budget impact | 0 | $3,911,518 | $12,007,035 | $20,476,554 | $36,395,107 | |
CADTH sensitivity analysis: 25% of patients will require 2 vials of difelikefalin | Reference | 0 | 0 | 0 | 0 | 0 |
New drug | 0 | $3,259,598 | $10,005,863 | $17,063,795 | $30,329,256 | |
Budget impact | 0 | $3,259,598 | $10,005,863 | $17,063,795 | $30,329,256 | |
CADTH sensitivity analysis: 92% price reduction | Reference | 0 | 0 | 0 | 0 | 0 |
New Drug | 0 | $208,614 | $640,375 | $1,092,083 | $1,941,072 | |
Budget Impact | 0 | $208,614 | $640,375 | $1,092,083 | $1,941,072 | |
CADTH sensitivity analysis: Patients would be able to share vials | Reference | 0 | 0 | 0 | 0 | 0 |
New Drug | 0 | $2,346,911 | $7,204,221 | $12,285,932 | $21,837,064 | |
Budget Impact | 0 | $2,346,911 | $7,204,221 | $12,285,932 | $21,837,064 | |
CADTH sensitivity analysis: Uptake of difelikefalin up to a maximum of 75% in year 3 | Reference | 0 | 0 | 0 | 0 | 0 |
New drug | 0 | $6,519,196 | $13,341,151 | $20,476,554 | $40,336,901 | |
Budget impact | 0 | $6,519,196 | $13,341,151 | $20,476,554 | $40,336,901 |
BIA = budget impact analysis
Stakeholder Input
Patient Input
Kidney Foundation of Canada
About the Kidney Foundation of Canada
Over nearly six decades, the Kidney Foundation of Canada has been guided by the fundamental principles of innovation, leadership, and collaboration, and has been committed to excellent kidney health, optimal quality of life for those affected by kidney disease, and a cure.
The Kidney Foundation of Canada is the leading charity committed to eliminating the burden of kidney disease through:
Funding and stimulating innovative research for better prevention, treatments and a cure;
Providing education and support to prevent kidney disease in those at risk and empower those with kidney disease to optimize their health status;
Advocating for improved access to high quality health care;
Increasing public awareness and commitment to advancing kidney health and organ donation.
For more information, please visit kidney.ca.
Information Gathering
Patient input was collected via independent surveys in September 2022 by the Kidney Foundation of Canada. Each survey was a self-administered questionnaire directed at people living with chronic kidney disease, as well as their caregivers. The surveys inquired about respondents’ lived experience with pruritus and chronic kidney disease, including questions on medications and expectations for new drug therapies in Canada. Awareness about the surveys was generated through the Kidney Foundation’s website and social media channels (Twitter and Facebook).
A total of 19 people responded to the survey; 10 questionnaires were fully completed and 9 were partially completed. 10 respondents identified as being a person living with kidney disease, 1 is a kidney transplant recipient, and 1 is a caregiver.
Disease Experience
Chronic kidney disease (CKD) is the presence of kidney damage or a decreased level of kidney function for a period of three months or more. In the early stages of CKD, self-management strategies may slow or prevent further damage to the kidneys. These lifestyle strategies may include engaging in regular physical activity, maintaining a healthy body weight, stopping smoking, reducing sodium, and managing other medical conditions and medications.
There are usually no specific symptoms of kidney disease until the damage is severe. In some cases, chronic kidney disease can lead to kidney failure (also called end-stage kidney disease, or ESKD). When the kidneys fail, wastes accumulate in the body and dialysis treatments or a kidney transplant are needed to survive.
Canadians with kidney failure and their families face significant out-of-pocket costs regardless of the treatment they receive. This financial burden is further compounded by the loss of income that is often associated with starting dialysis, which is the most common treatment for kidney failure. Because poverty is an important determinant of health, patients and their families who live in poverty may not be able to achieve optimal management of their kidney disease.
One of the most common symptoms experienced by kidney patients is pruritus, or itchy skin. Studies show that pruritus is present in about 40% to 84% of patients with end-stage kidney disease1. Over 90% of the respondents to The Kidney Foundation of Canada’s survey reported that they have experienced itchy skin as part of their kidney disease. 50% said they experience itchiness every day, 40% said they experience it several times per week, and 10% said they experience itchiness occasionally.
In terms of the duration of time patients have been experiencing itchiness, 60% reported that they had been living with pruritus for 1-2 years, while 20% said they’d been living with it for 2-5 years, and 20% over 5 years. 80% of respondents described their itchiness as moderate or severe.
Several respondents reported that they develop scabs and/or sores as a result of their itchy skin:
“I often put of (sic) things like going out or getting a haircut as the itching is so bad I scratch until there are sores.”
“Scratching so often that I end up with scabs.”
“[…] I would scratch and bleed VERY easy (sic). I had scabs, I would bleed on clothes and bedsheets, and I had to use expensive creams (protopic) to control it. Quite ugly and often had to use multiple bandaids all over my legs.”
Many respondents also reported that they have trouble sleeping as a result of itchiness, with one person saying that they sometimes take Benadryl to help them sleep. Another said “I’m unable to sleep and be intimate with my partner.”
Experiences With Currently Available Treatments
33% of survey respondents reported taking medication to treat itchiness associated with kidney disease. The types of treatments used to control itchiness that were cited by survey respondents were:
Antihistamines (e.g., Benadryl, Claritin, Aerius, Atarax, etc.)
Corticosteroid creams or ointments (e.g., hydrocortisone, betamethasone)
Moisturizing creams or ointments (e.g., Uremol, Cetaphil, Vaseline, etc.)
67% of respondents said that they paid out of pocket for these treatments, while 33% had their treatments covered by their provincial drug plan. When asked how satisfied they are with their current medication/combination of treatments, most said they were satisfied, with 33% saying they were neither satisfied nor unsatisfied.
In terms of the challenges or difficulties experienced with existing treatments, one respondent had this to say:
“Cost. I don’t use it that often. But paying $25 for a small jar of cream for the pharmacy to make up and insurance does not cover.”
The current medication/combination of medications taken by respondents were largely reported to leave skin itchiness about the same. Over 66% of respondents said they didn’t know whether their skin appearance was improved. One person reported that their itchiness symptoms disappeared after transplant.
Improved Outcomes
When asked about their expectations for CKD therapies in general, respondents rated certain questions as important or very important, including: “Does it interfere with my sleep?”, “How much does it cost?”, and “Does it interfere with my other medications?”.
All survey respondents said that they hoped that new medications would increase their well-being or quality of life, and 90% said that they hoped for increased energy. Other expectations included fewer hospital visits and less medication overall.
In talking about what else is important when choosing medication for kidney disease, several respondents mentioned side effects and efficacy. One respondent said:
“Cost, making sure theee (sic) isn’t a whole lot of work to get something covered […]”
Experience With Drug Under Review
None of the those surveyed reported experience with difelikefalin.
Companion Diagnostic Test
Not applicable to this submission.
Anything Else?
Living with chronic kidney disease can involve not only health and quality of life challenges, but significant financial challenges as well. People may experience a decrease in income if they must limit their working hours due to their symptoms, and out-of-pocket costs increase as they change their diet, follow up more often with their health care team, and use recommended non-prescription treatments.
Those living with kidney disease also tend to be part of a low income and high cost population, and government coverage and financial support varies across jurisdictions, which can lead to inequities.
The burden of chronic kidney disease means that many would benefit from effective, affordable treatments that they can access equitably and in a timely manner. Itchy skin is a very common symptom for kidney patients, especially for those on dialysis, therefore difelikefalin should be available as an option for people living with kidney disease.
Clinician Input
Division of Nephrology, Department of Medicine, Dalhousie University/Nova Scotia Health
About the Division of Nephrology, Department of Medicine, Dalhousie University/Nova Scotia Health
We are a group of academic nephrologists working in a quaternary care institution in Halifax Nova Scotia. We provide comprehensive kidney care for a catchment area of 750,000 individuals in Nova Scotia and additional care (i.e. transplantation) to all 4 Atlantic provinces We provide clinical and research expertise in the area(s) of dialysis therapy and outcomes, health economic research and chronic kidney disease. As an academic division, we participate in and support national and international efforts to improve health outcomes for people living with kidney disease. Division members are clinicians, educators and clinical researchers.
https://medicine.dal.ca/departments/department-sites/medicine/divisions/nephrology/about.html
Information Gathering
The information gathered in this proposal was done through direct experience, clinical knowledge of chronic kidney disease associated pruritus (CKD-aP) and literature review. As nephrologists with expertise/experience in the area of CKD-aP and for myself (Dr. Tennankore) having previously conducted two systematic reviews in the area of CKD-aP and a local quality improvement initiative, I am well-versed in this area. Importantly, this was done independently with no involvement from members of industry.
References:
Jaiswal D, Uzans D, Hayden J, Kiberd BA, Tennankore KK. Targeting the opioid pathway for uremic pruritus: a systematic review and meta-analysis. Can J Kidney Health Dis., 2016
Bailey AMJ, Burns W, Cheng W, Collister EP, Westby EP, Purdy KS, Walsh M, Tennankore KK. Targeting the opioid pathway for the treatment of chronic kidney disease-associated pruritus: a systematic review and meta-analysis of randomized controlled trials. Br J Dermatol. 186(3):575-577, 2022.
Current Treatments and Treatment Goals
CKD-aP begins with recognition; dialysis patients or individuals with advanced chronic kidney disease are asked about and told to rate the severity of itch. Specifically, they are asked about the presence or absence of daily or near daily itch that is often generalized and not related to another chronic condition. CKD-aP is common; in an International study the prevalence of CKD-aP that was moderate to extremely bothering was >40% in Canadian in-center hemodialysis patients, which was higher than most countries. Patients were bothered by the appearance of their skin, depressed about the severity of their itch and noted that it made it hard to work and interact with others. Given the paucity of effective treatments, patients inflicted with CKD-aP have a relative risk of mortality that is 1.5-2X those without itch and a profound decrease in quality of life.
CKD-aP is typically treated using a stepwise approach that includes the use of 1. Ruling out other conditions that warrant other therapies. 2. Optimization of dialysis therapy including ensuring adequate dialyzer clearance and better treatment of elevated phosphate and parathyroid hormone levels. 3. Hydrous emollients, to ensure the skin is well moisturized. 4. Targeted topical treatments for those who continue to experience itch. 5. Oral agents aimed at modifying the neural pathways that lead to sensation of itch including (at our center) gabapentin and pregabalin. 6. UV-B light if it is provided at the center at which the dialysis population is being managed.
Of these treatments, gabapentin is routinely used in Canadian clinical practice, however, this therapy is not usually available through special access programs and is difficult to get public or private funding approved for the treatment of CKD-aP. Topical therapies largely do not have a Health Canada approval for the management of itch but are often used as first line treatment due to their reduced burden of systemic side effects. Unfortunately, current treatments do not modify the underlying disease mechanism, rather, they are focused on treating symptoms (itch), Furthermore, they all have adverse effects that prevent their widespread use in the management of CKD-aP. These are listed in the next section (treatment gaps).
The most important treatment goals would be to have a therapy that would reduce or maintain the severity of itch below a threshold of clinical importance that is known to be the level above which itch negatively impacts quality of life and outcomes important to patients. In addition, the treatment should be easy to administer, should be affordable and should have a tolerable side effect profile.
References:
Rayner et al. International comparisons of prevalence, awareness, and treatment of pruritus in people on hemodialysis. CJASN 12(12):2000-2007, 2017
Westby EP, Purdy KS, Tennankore KK. A review of the management of uremic pruritus: current perspectives and future directions. Itch, 1-7. 2020.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
As highlighted below in more detail, not all patients who are treated for CKD-aP receive benefit. This is evident in the literature review provided above as well as our own local data which suggests that only 25-30% of patients with CKD-aP are receiving a treatment that is actually managing their itch. This is very concerning, especially considering that our center likely has more awareness of CKD-aP than many others across Canada and yet, we are not able to treat patients effectively. Other treatment goals that are not being met include a lack of treatments that address other patient-important outcomes such as the impact of itch on function and quality of life. Most focus on resolution of symptoms as opposed to the effect on other health outcomes. As noted, treatments are needed that are better tolerated (given the side effect profiles of currently available oral and topical therapies) and given the polypharmacy faced by dialysis patients, treatments to improve compliance/adherence would also be valued in this patient population.
Specific considerations for each arm of a typical CKD-aP management pathway include the following:
There is no definitive data that escalation of a dialysis prescription or treatment of phosphate removes CKD-aP. Treating the former requires an increase in either dialysis frequency or duration which can be profoundly burdensome to patients who are already attending thrice-weekly dialysis. Treating phosphate requires additional medications that then come with their own burden of side effects, polypharmacy and drug interactions necessitating complex spacing of medication regimens.
Hydrous emollients are used to sooth and moisturize the skin, however CKD-aP does not always associate with xerosis (drying of skin) and can be present without xerosis. Furthermore, addressing dry skin doesn’t treat the underlying disease mechanism.
Topical treatments are primarily used as symptom relief but are hard to apply due to their texture (potentially impairing patient quality of life) and require pharmacy compounding in many cases. Furthermore, they require 2-4X/day application in most cases, limiting their effectiveness. While it is ideal to have patients that respond to topical therapy, in our center, only about 25% respond to/receive a topical therapy for itch that they are able to take and that works. Furthermore, the availability of topical therapies (i.e. menthol/camphor, capsaicin, tacrolimus) is limited to site-to-site availability or compounding through community pharmacy which can be expensive.
Although some units use sedatives (Benadryl/Atarax), these medications have no data and have side effects including dependency and somnolence. We do not typically use them in our algorithm for treating itch at our institute
Pregabalin and Gabapentin do have benefits in resolving itch in some patients. Our local experience has been poor. In a review using a locally developed treatment algorithm (Westby et al. ASN annual meeting 2018), we identified that of the 11/56 patients who entered our algorithm and were treated with gabapentin, only 3 had a complete response to gabapentin despite up titration. A recent systematic review (Lau et al. CJKHD 2016), demonstrated that while beneficial in treating itch, the studies evaluating the efficacy of gabapentin were generally of low quality. In addition, there were important safety outcomes including somnolence, dizziness, fatigue, nausea ultimately leading to treatment discontinuation in many cases.
UV light is a last line treatment for refractory pruritus; however, it is simply not available at many Canadian centers. Furthermore, it requires daily attendance to clinics to receive the treatment which patients are not keen on doing. In our local experience quality improvement initiative, some patients considered UV-light treatment, but ultimately discontinued it due to an inability to attend the appointments or lack of interest in committing to travel long distances to a UV-light treatment center in addition to the significant time burden of dialysis they were already facing.
References:
Tennankore et al. Management of uremic pruritus in hemodialysis: effectiveness of a quality improvement treatment algorithm. ASN Kidney Week Annual Meeting, 2018. https://www.asn-online.org/education/kidneyweek/archives/KW18Abstracts.pdf
Lau et al. Gabapentin for uremic pruritus in hemodialysis patients: a qualitative systematic review. Can J Kidney Health Dis., 2016.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
To date, there is no approved therapy for the management of CKD-aP in Canada. Medications that are being used are being done so off-label, and often the side effects of medications (i.e. sedatives) are being used to treat this condition. Difelikefalin is unique in the fact that it is administered intravenously making it easier to give to hemodialysis patients who suffer the most with CKD-aP relative to all other patients with kidney failure. The mechanism would certainly complement current treatments, i.e. those other options that are listed above do not affect kappa-opioid receptors. There is currently no therapy that truly affects the underlying disease process for CKD-aP. It is theorized that opioid receptors do play a role as a neurologic mediator of itch, potentially related to uremic toxins that accumulate with kidney failure. Thus, targeting the opioid receptor may impact CKD-aP and lead to benefit for patients. This medication would be a second line treatment after failure of or in complement to topical treatments. Given the side effect profile of gabapentin, it would replace gabapentin in the algorithm order for CKD-aP management. In this regard, topical treatments and optimization of dialysis would still be the first line course of action, acknowledging their limited roles. Once those factors are considered/approaches are optimized, Difelikefalin would be the next step. We believe this treatment will shift the current treatment paradigm. Given this is the first treatment that has shown clear benefit with a tolerable side effect profile, there will be great interest in screening for and measuring itch. Said differently, having a treatment of this quality will encourage physicians to focus more on capturing and measuring this symptom in chronic dialysis patients. Therefore, we think this will change the recognition of CKD-aP in addition to providing a treatment for it. As noted, we do feel that less systemic treatments (topicals, emollients), and optimizing dialysis where feasible should remain the first line therapy for CKD-aP. However, this treatment would be superior to gabapentin given the side effect profile and limited benefit of gabapentin in treating CKD-aP.
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
Chronic dialysis patients with CKD-aP who have failed topical therapies would be the most likely to respond to this treatment. I suspect given the route of administration, hemodialysis patients would benefit the most (they are coming to a facility or dialyzing at home which requires access to a blood vessel improving the feasibility of this treatment given its an IV preparation). Thankfully, hemodialysis patients are the most affected by CKD-aP. Whether or not a patient would benefit from this treatment would not be expected to differ based on patient characteristics, but it is acknowledged that other conditions should be ruled out as not all itch is related to CKD-aP. Identifying who is suitable for this drug would require a screening tool (many exist including a numerical or visual analogue scale) that rates the severity of pruritus on a scale of 1-10. It would be helpful but not necessary to also ensure patients have an assessment of the impact of CKD-aP on quality of life. In addition to this, patients should have a physical exam to rule-out other conditions causing itch and a review of blood work to ensure dialysis is optimized as best as can be. Related to diagnosis, it is important to note that some conditions cause itch but are not CKD-aP. These are typically fairly obvious and are accompanied by skin manifestations, a history that is compelling or an obvious bloodwork abnormality. It is likely that CKD-aP is severely underdiagnosed in our population. Administering routine screening tools for the presence of/severity of itch are not established practices and patients do not always volunteer symptoms, especially in the face of few therapies. Treatment response would best be assessed through an algorithmic approach whereby itch severity would be reassessed once treatment has been initiated to ensure patients are benefiting.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
The best outcomes would be to evaluate whether the itch severity is reduced on a numerical rating scale, standard practice for symptom assessment in clinical trials. The threshold for clinical significance varies, but a 3 point improvement in itch severity is what we typically use to ascertain benefit of topical/gabapentin. Ideally the NRS score should fall to <3 to emphasize that the itch is only in the mild range, but even a ≥3 point improvement despite the score remaining above 3 would be beneficial. Given CKD-aP is not well-recognized it is possible that some clinicians may expect complete resolution of itch or on the contrary, may view small changes with the same degree of importance. Using data from clinical trials, the best approach would be to standardize the goal of a ≥3 point improvement, thus ensuring that a lack of response leads to medication discontinuation and that other complementary therapies are considered if there is no response. Clinically meaningful benefits in addition to itch severity would be a reduction on the impact of itch-related outcomes that are important to patients including sleep, depression/mood, and the effects of itch on work, interactions and desire to be with people. These elements are covered in an evaluation tool known as the Skindex-10 questionnaire. These are secondary factors that should accompany the primary goal which is a reduction in NRS score as noted.
What factors should be considered when deciding to discontinue treatment with the drug under review?
Factors that should be considered include side effects that are intolerable to patients irrespective of the itch/itch severity or non-response (lack of improvement in NRS itch level).
What settings are appropriate for treatment with difelikefalin? Is a specialist required to diagnose, treat, and monitor patients who might receive difelikefalin?
This medication would be easiest to administer (given it is intravenous) in a dialysis unit (either outpatient or inpatient). Monitoring would be as per Canadian drug product monograph and is not anticipated to require special monitoring. It may be feasible to allow select patients to administer this at home whereby they are already accessing the intravenous route (i.e. home dialysis), or, medical day units. Ideally, this medication would be prescribed by nephrologists or internal medicine specialists delivering dialysis care given it has a key role in the dialysis population. Other groups that could potentially get involved in prescribing it would be dermatologists and palliative care specialists, but always in consultation with a nephrologist.
Additional Information
Conflict of Interest Declarations for Kidney Foundation of Canada
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
No outside help was received outside of a review from our kidney pharmacist working with our provincial kidney program given her expertise in medications and treatment protocols/pathways. She has been added onto this review as a co-author.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review
Declaration for Clinician 1
Name: Karthik Tennankore
Position: Nephrologist, Associate professor, MD SM FRCPC
Date: 12-SEP-2022
Table 1: COI Declaration for Division of Nephrology, Department of Medicine, Dalhousie University/Nova Scotia Health — Clinician 1
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Otsuka | X | — | — | — |
Vifor Pharmaceuticals | X | — | — | — |
Bayer | X | — | — | — |
Astra Zeneca | X | — | — | — |
GSK | X | — | — | — |
Declaration for Clinician 2
Name: Steven Soroka
Position: Professor of Medicine and Adult Nephrology Consultant, Dalhousie University and Nova Scotia Health, Senior Medical Director Nova Scotia Health Renal Program and Pharmacy Services
Date: 2022- 09-12
Table 2: COI Declaration for Division of Nephrology, Department of Medicine, Dalhousie University/Nova Scotia Health — Clinician 2
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Otsuka | X | — | — | — |
Astra Zeneca | — | X | — | — |
Bayer | X | — | — | — |
Declaration for Clinician 3
Name: Dr. Jo-Anne Wilson, BSc.Pharm, ACPR, M.Ed (student), PharmD
Position: Associate Professor of Pharmacy, Faculty of Health, Dalhousie University and Clinical Pharmacy Specialist, NS Health Renal
Date: 12-9-2022
Table 3: COI Declaration for Division of Nephrology, Department of Medicine, Dalhousie University/Nova Scotia Health — Clinician 3
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
None to disclose | — | — | — | — |
Declaration for Clinician 4
Name: Neil Finkle
Position: Division Head and Central Zone Renal Program Physician Lead
Date: 20-09-2022
Table 4: COI Declaration for Division of Nephrology, Department of Medicine, Dalhousie University/Nova Scotia Health — Clinician 4
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
None to Disclose | — | — | — | — |
Declaration for Clinician 5
Name: Dr. David Clark
Position: Consultant Nephrologist, Director-Central Zone Renal Program
Date: 20-09-2022
Table 5: COI Declaration for Division of Nephrology, Department of Medicine, Dalhousie University/Nova Scotia Health — Clinician 5
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Bayer | X | — | — | — |
Baxter | X | — | — | — |
Otsuka | X | — | — | — |
Hemodialysis Specialty Physician Group, Division of Nephrology, The Ottawa Hospital
About the Hemodialysis Specialty Physician Group, Division of Nephrology, The Ottawa Hospital
Our group is made up of 6 academic nephrologists affiliated with the University of Ottawa. All of us have a subspecialty practice focusing on hemodialysis. We provide longitudinal hemodialysis care to patients within followed the Division Nephrology at the University of Ottawa and at the Ottawa Hospital. We oversee the care of patients in various dialysis units at the 3 campuses of the Ottawa Hospital and various satellite dialysis units across Eastern Ontario including Hawkesbury & District General Hospital, Queensway Carleton Hospital, Cornwall Community Hospital, and at 2 independent dialysis health facilities, Ottawa Carleton dialysis clinic and Cornwall Dialysis Clinic. Altogether, our group provides care to more than 400 dialysis patients.
Information Gathering
As we work closely together, we often collaborate and review patient care. We therefore have met and discussed the problem of CKD associated pruritus as a group and are providing the information below based on our clinical experience with patients and our clinical expertise on the subject matter.
Current Treatments and Treatment Goals
Pruritus is a common and very bothersome symptom in patients with chronic kidney disease but especially in patient with end-stage kidney disease treated via hemodialysis. In one of the larger studies reporting on this problem, it has been shown to affect up to affect 1 in 2 dialysis patients [1]. The percentage of patients that report being extremely bothered by uremic pruritus is ~18%, [1]. Given its prevalence and the impact it can have on quality of life, uremic pruritus remains an important area of research priority for patients with CKD [2].
The exact etiology and pathophysiology of this phenomenon remains very poorly understood. Studies have not shown a clear association with sex, age, ethnicity, duration of dialysis, or actual underlying pathology that led to renal failure. It is therefore a very challenging problem to treat given the poorly understood mechanism of underlying it [3].
Current treatment options for this condition include some non-pharmacological options, as well as pharmacological options. We note that all pharmacological options for this condition are currently medications used "off label" (i.e. not used following Health Canada approved indications).
Xerosis is a potent potentiator of uremic pruritus symptoms and the first line of non-pharmacological treatment remains adequate skin care and aggressive use of non-scented skin emollients.
With regards to non-pharmacological options, ultraviolet phototherapy with UVB rays has been found to provide some effective relief of uremic pruritus symptoms [4]. However, this therapy is extremely burdensome for patients on hemodialysis as it requires almost daily appointments. Hemodialysis patients already spend more than 12 to 15 hours/week undergoing treatment for renal replacement at a minimum of 3 times per week, excluding transportation time. Practically speaking, it is almost impossible for patients to attend both UVB therapy and dialysis treatments routinely and maintain a meaningful quality of life. Adjustment of the hemodialysis prescription may help provide some relief [4]. High flux hemodialysis filters are helpful at improving pruritus symptoms; however, these filters are now in use broadly across Canada and as such patients who currently develop pruritus do so despite their use. Hemodiafiltration rather than hemodialysis alone has also been shown to lead to some improvement in pruritus, but this modality requires specific hemodialysis machines and water treatments which are not commonly available and remain rather rare across Canada. Thus hemodiafiltration cannot be offered to most hemodialysis patients and is unlikely to be available broadly anytime soon given the large capital investment sums required to convert hemodialysis centres to be able to provide type of therapy.
With regards to pharmacological interventions, gabapentin is the only medication, when used off label, with some evidence supporting a benefit in patients with CKD and end-stage kidney disease associated uremic pruritus. Several studies have shown a statistically significant benefit in favor of its use with respect to decreasing the severity of pruritus [5]. However, gabapentin’s adverse effects, including drowsiness, dizziness, somnolence and neurotoxicity, combined with its pharmacokinetics (predominantly renal excretion) are dose-limiting in the end-stage kidney disease population. This significantly impairs clinicians’ ability to titrate the medication to achieve control of symptoms.
Numerous other medications have been tried with various results. Agents with some evidence in the literature include montelukast, nalfurafine, ondensatron, naltrexone, primrose oil, cholestyramine and thalidomide. A systematic review of the literature published in 2017 concluded that "there remains considerable uncertainty about effective treatments for this important and burdensome symptom in patients with kidney failure. Despite a large trial literature examining multiple different interventions, the combination of flawed methodology, high risk of bias, small sample size, and study heterogeneity prevent the generation of robust treatment recommendations” [6].
References:
Pisoni RL, Wikström B, Elder SJ, et al. Pruritus in haemodialysis patients: International results from the Dialysis Outcomes and Practice Patterns Study (DOPPS). Nephrol Dial Transplant 2006;21(12):3495–505.
Manns B, Hemmelgarn B, Lillie E, et al. Setting research priorities for patients on or nearing dialysis. Clin J Am Soc Nephrol 2014;9(10):1813–21.
Manenti L, Tansinda P, Vaglio A. Uraemic pruritus: clinical characteristics, pathophysiology and treatment. Drugs 2009;69(3):251–63.
Gilchrest BA. Ultraviolet phototherapy of uremic pruritus. Int J Dermatol 1979;18(9):741–8.
Simonsen E, Komenda P, Lerner B, et al. Treatment of Uremic Pruritus: A Systematic Review. Am J Kidney Dis 2017;70(5):638–55.
Lau T, Leung S, Lau W. Gabapentin for uremic pruritus in hemodialysis patients: a qualitative systematic review. Can J Kidney Health Dis 2016;3:14.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
As mentioned above, there are at this time with no effective treatment approved for relief of CKD associated (uremic) pruritus. The current treatments are not very effective, and most patients require a multimodal approach to treat this disease. Treatments remain often modestly effective at best and fraught with adverse events.
In most severe cases, the symptoms can be overbearing and lead to significant adverse quality of life.
As there are currently no treatments that are available to reverse the course of the disease, difelikefalin would address an unmet need as the first available indicated therapy to target this significant complication. It will be easily and conveniently administered 3 times weekly intravenously at the end of each dialysis session. The medication is well-tolerated. Randomized control trials have demonstrated objective effectiveness in reducing the severity of the symptoms of CKD associated (uremic) pruritus [1-2]. This has also been confirmed in a pooled analysis [3]. These data show that difelikefalin provides effective relief of common symptom that is currently without an adequate therapy.
References:
Fishbane S, Jamal A, Munera C, Wen W, Menzaghi F, KALM-1 Trial Investigators. A Phase 3 Trial of Difelikefalin in Hemodialysis Patients with Pruritus. N Engl J Med 2020;382(3):222–32.
Fishbane S, Mathur V, Germain MJ, et al. Randomized Controlled Trial of Difelikefalin for Chronic Pruritus in Hemodialysis Patients. Kidney Int Rep 2020;5(5):600–10.
Topf J, Wooldridge T, McCafferty K, et al. Efficacy of Difelikefalin for the Treatment of Moderate to Severe Pruritus in Hemodialysis Patients: Pooled Analysis of KALM-1 and KALM-2 Phase 3 Studies. Kidney Med 2022;4(8):100512.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
As difelikefalin is the first Health Canada approved medication for treatment of CKD associated (uremic) pruritus, we believe it should be amongst the first line treatments prescribed to patients. Consistent with the current evidence, this medication would be indicated for moderate to severe symptoms. Patients with mild symptoms should be treated with non-pharmacological interventions +/- gabapentin if the side effects can be tolerated
Current data suggests that it is effective in patients with moderate to severe symptoms and therefore it should be limited to that population. After a trial of non-pharmacological interventions, patients who experience moderate to severe symptoms should be treated with difelikefalin. Patients with partial response might benefit from additional, off label therapies such as gabapentin in addition, although there exists no data at this time on whether this would have additive, synergistic or detrimental impact on symptom control.
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
Patients on hemodialysis with moderate to severe pruritus and no obvious reversible causes (e.g. other underlying skin disease, scabies, etc) would be best suited for treatment with difelikefalin.
Identification of these patients and the severity of the symptoms can only be achieved via self-reported questionnaire such as the 5-D itch scale [1], UP-Dial [2], Skindex-10 [3] or other validated scales. There are no diagnostic laboratory or imaging tests to confirm the diagnosis of CKD associated (uremic) pruritus. Rather, it is a diagnosis of “exclusion”, i.e. chronic pruritus in patients with advanced CKD and no obvious skin pathology in patients with advanced CKD imaging or laboratory based. Patients more likely to exhibit a response are those that suffer from moderate to severe pruritic symptoms.
References:
Lai J-W, Chen H-C, Chou C-Y, et al. Transformation of 5-D itch scale and numerical rating scale in chronic hemodialysis patients. BMC Nephrol 2017;18(1):56.
Nochaiwong S, Ruengorn C, Koyratkoson K, et al. Clinical interpretation of the Uremic Pruritus in Dialysis Patients (UP-Dial) scale: a novel instrument for the assessment of uremic pruritus. J Eur Acad Dermatol Venereol 2018;32(7):1188–94.
Lopes MB, Karaboyas A, Sukul N, et al. Utility of a Single Itch-Related Question and the Skindex-10 Questionnaire for Assessing Pruritus and Predicting Health-Related Quality of Life in Patients Receiving Hemodialysis. Kidney Med 2022;4(6):100476.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
An improvement which is documented objectively by a reduction in the severity of the symptoms, assessed by one or more of the pruritic symptom scales [1-3] would be the best way to determine clinical response. Patients should be assessed regularly, likely monthly initially and then surveyed every 3 to 6 months chronically.
References:
Lai J-W, Chen H-C, Chou C-Y, et al. Transformation of 5-D itch scale and numerical rating scale in chronic hemodialysis patients. BMC Nephrol 2017;18(1):56.
Nochaiwong S, Ruengorn C, Koyratkoson K, et al. Clinical interpretation of the Uremic Pruritus in Dialysis Patients (UP-Dial) scale: a novel instrument for the assessment of uremic pruritus. J Eur Acad Dermatol Venereol 2018;32(7):1188–94.
Lopes MB, Karaboyas A, Sukul N, et al. Utility of a Single Itch-Related Question and the Skindex-10 Questionnaire for Assessing Pruritus and Predicting Health-Related Quality of Life in Patients Receiving Hemodialysis. Kidney Med 2022;4(6):100476.
What factors should be considered when deciding to discontinue treatment with the drug under review?
Lack of any meaningful response/effectiveness to the medication would be an important reason to discontinue therapy. This could be evaluated by changes in an objective pruritus scale. Adverse events such as diarrhea, vertigo, vomiting or other known adverse events related to difelikefalin would also be considered as indications for stopping therapy.
What settings are appropriate for treatment with [drug under review]? Is a specialist required to diagnose, treat, and monitor patients who might receive [drug under review]?
The only appropriate setting is in hemodialysis units. Nephrology consultation and follow-up is required for the diagnosis, treatment, and monitoring of these patients. Patient should be followed by a nephrologist to receive this therapy.
Additional Information
N/A
Conflict of Interest Declarations for Hemodialysis Specialty Physician Group - Division of Nephrology, The Ottawa Hospital
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict-of-interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
No outside help was received.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No outside help was received.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
Declaration for Clinician 1
Name: Pierre Antoine Brown
Position: Medical Director, Hemodialysis Program, The Ottawa Hospital; Assistant Professor, Division of Nephrology, Department of Medicine, University of Ottawa
Date: 30-08-2022
Table 6: COI Declaration for Hemodialysis Specialty Physician Group, Division of Nephrology, The Ottawa Hospital — Clinician 1
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Otsuka Canada | — | X | — | — |
Astra Zeneca Canada | X | — | — | — |
Declaration for Clinician 2
Name: Brendan McCormick
Position: Medical Director, Home Dialysis Program, The Ottawa Hospital; Associate Professor, Division of Nephrology, Department of Medicine, University of Ottawa
Date: 30-08-2002
Table 7: COI Declaration for Hemodialysis Specialty Physician Group, Division of Nephrology, The Ottawa Hospital — Clinician 2
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 3
Name: Peter Magner
Position: Nephrologist, The Ottawa Hospital; Associate Professor, Division of Nephrology, Department of Medicine, University of Ottawa
Date: 30-08-2022
Table 8: COI Declaration for Hemodialysis Specialty Physician Group, Division of Nephrology, The Ottawa Hospital — Clinician 3
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Janssen Canada | X | — | — | — |
Declaration for Clinician 4
Name: Marcel Ruzicka
Position: Nephrologist, The Ottawa Hospital; Professor, Division of Nephrology, Department of Medicine, University of Ottawa
Date: 30-08-2022
Table 9: COI Declaration for Hemodialysis Specialty Physician Group, Division of Nephrology, The Ottawa Hospital — Clinician 4
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Medtronics Canada | X | — | — | — |
Declaration for Clinician 5
Name: Edward Clark
Position: Nephrologist, The Ottawa Hospital; Associate Professor, Division of Nephrology, Department of Medicine, University of Ottawa
Date: 30-08-2022
Table 10: COI Declaration for Hemodialysis Specialty Physician Group, Division of Nephrology, The Ottawa Hospital — Clinician 5
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 6
Name: Swapnil Hiremath
Position: Nephrologist, The Ottawa Hospital; Associate Professor, Division of Nephrology, Department of Medicine, University of Ottawa
Date: 30-08-2022
Table 11: COI Declaration for Hemodialysis Specialty Physician Group, Division of Nephrology, The Ottawa Hospital — Clinician 6
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Saskatchewan Kidney Doctors
About Saskatchewan Kidney Doctors
We are a group of 8 kidney doctors from Saskatchewan who care for persons living with end-stage kidney failure receiving dialysis therapy. Our group is interested in patient related outcome measures.
Information Gathering
This information was gathered after an exhaustive review of the existing literature,
Current Treatments and Treatment Goals
Dialysis Outcomes and Practice Patterns Study (DOPPS) which collects data from 20 countries including Canada reported that the majority (65%) of patients on hemodialysis (HD) experience pruritus, with 37% being moderately bothered and 7% being extremely bothered. Pruritus is often due to the uremic state of kidney failure. It can be debilitating and is inadequately controlled by dialysis therapy alone. It has been associated with increased recovery time post sessions, insomnia, reduced quality of life, depression, missed dialysis sessions, and withdrawal from therapy.
The list of currently available topical and non-pharmacological therapies is listed below.
A: Topical:
Emollients with low pH and /or high-water content have been found to be helpful as xerosis exists in 50–85% of patients and is an aggravating factor of pruritus. There are several over the counter options (OTC) for the clinicians.
i. Cream containing 2.2% gamma linolenic acid: In a double blind, placebo controlled, crossover study (n=17) with refractory pruritus, a cream containing 2.2% gamma linolenic acid (GLA), significantly improved pruritus compared to a placebo. GLA is an essential fatty acid that is metabolized to a prostaglandin precursor with known anti-inflammatory properties and is available OTC as evening primrose oil.
ii. Cream containing 1% pramoxine (topical anesthetic): A randomized, double-blind, controlled comparative trial with 28 patients using 1% pramoxine lotion vs placebo, there was 61% reduction in pruritus scores in comparison to 12% in the placebo group. The cream is available OTC in 0.5% -1.0% concentrations as CeraVe, Gold Bond and Aveeno anti Itch cream. Both topical GLA and pramoxine show promise and should be attempted initially in the management of pruritus. Allergic/hypersensitivity reactions to pramoxine can occur and prevent its use.
iii. Topical cannabinoids can be effective, but more data is needed: A single study of 21 patients showed that topical cannabinoids were effective in reducing xerosis and pruritus and was well tolerated. The baseline itch intensity measured by VAS was (6.24 ± 2.19) and had improved by day 21 (1.29 ±.41). The long-term effects of cannabis use, particularly in CKD, is yet unknown and there is no data on systemic absorption. In the absence of randomized trials, a recent Canadian review on the topic suggested caution regarding its use until further.
B. Nonpharmacological therapies:
i. Trail phototherapy (UVB radiation), if available at your institution: 18 HD patients with severe pruritus randomized to UVB (290- to 320-nm wavelength) or UVA therapy light showed a greater reduction (four week follow up) in the UVB therapy group (80% vs 20%. Gilchrest et al showed that side effects of narrow-band UVB-radiation were less frequent and just as effective as treatment using broadband UVB. Phototherapy appears to be the most promising nonpharmacological therapy available to clinicians but is often not available in many the jurisdictions.
ii. Acupuncture has been trialed, but more evidence is needed. A review of 3 RCTs and 3 uncontrolled observational trials found that acupuncture reduced itching in all trials. However, due to a high risk of bias in these studies, there is insufficient evidence to recommend acupuncture.
ii. While there is data on benefits of exercise in CKD patients with pruritus, there are no trials yet looking at Intradialytic exercise on itch reduction.
C: Low quality evidence exists for optimizing the dialysis prescription, changing patients from high flux to medium cut off dialyzers. There is data to support parathyroidectomy in patients with secondary hyperparathyroidism.
In a randomized controlled trial (RCT) of 22 HD patients with severe pruritus, higher small solute clearance (mean Kt/V, 1.28) achieved through increased dialysis prescription, led to a decrease in pruritus scores (12.6 ± 5.1 to 6.3 ± 3.2). In comparison when the dialysis prescription was left unchanged (mean Kt/V, 1.09) pruritus scores remained unchanged (12.3 ± 4.7 to 12.7 ± 6.4). Ko et al. also found a kT/V >1.5 and use of high flux dialyzers was associated with less itch. However, other studies have not shown a similar relationship between kT/V and pruritus implying that it’s middle molecules and not small molecules that play a role in pruritus.
Unfortunately, none of the trials comparing short daily or nocturnal hemodialysis with incentre HD evaluated pruritus as an outcome measure. We identified only one RCT comparing medium cut-off (MCO) dialyzer (n=24) with high-flux dialyzer (n=25). Patients with MCO dialyzers had better pruritus scores (1.29 ± 0.46) vs. (1.64 ± 0.64, p= 0.034) and sleep, reflecting a relationship between efficient elimination of middle molecules and symptom relief.
Secondary hyperparathyroidism is a complication of kidney failure and is associated with severe complications and symptoms such as pruritus. Parathyroidectomy for severe secondary/tertiary hyperparathyroidism has been found to be consistently associated with itch reduction. A prospective, uncontrolled study of 37 dialysis patients (22 had symptomatic pruritus) with mean parathyroid hormone level of 156 pmol/L, showed a significant decrease in visual analogue (VAS) score pre and post parathyroidectomy (5.4 +/- 3.2 to 1.8 +/- 1.5 (p < 0.001). A couple of other pre/post parathyroidectomy studies from the mid 1960’s has shown similar reduction in pruritus.
There are currently no approved medications in Canada for treatment in pruritus in patients with kidney disease.
i. Antihistamines are routinely prescribed in clinical practice, but majority of the studies have been unsuccessful in reducing pruritus. Medications such as hydroxyzine do not have supportive trial data and there is a likelihood for side effects in the elderly (over sedation). Mast cell stabilizers such as oral montelukast, cromolyn sodium, and zinc sulfate, all probably help pruritus, but additional high-quality evidence is required before we can start using the medications on a consistent basis.
ii. Gabapentin and Pregabalin: In a RCT involving 42 HD patients, 21 received gabapentin and 21 received pregabalin. Both medications produced a significant difference in itching intensity. Another study reported significant reduction in mean pruritus score pre and post administration of 300 mg of oral gabapentin post dialysis over four weeks in HD patients with pruritus (8.4 +/- 0.94 vs. 1.2 +/- 1.8) (P = 0.0001), with no demonstrable side effects at that dose. Both the drugs were initially approved to treat epilepsy and not pruritus. However, the use of gabapentin and pregabalin in patients with pruritus is supported by European Dermatology Guidelines and a recent Cochrane systematic review.
iii. μ-opioid receptors (MORs) antagonists (Naltrexone): A placebo-controlled, double-blind, crossover trial (n=15) showed administration with oral naltrexone 50 mg once a day for 1 week led to an almost complete resolution of itch with few side effects. However, another double-blind placebo-controlled crossover study of 23 patients over four weeks failed to show a statistically significant reduction in itch scores. Naltrexone is approved for use in Canada only for alcohol and opiate use disorder and used as a component of an alcohol counselling program.
iv. Selective κ-opioid receptor (KORs) agonists (Nalfurafine): A meta-analysis of two randomized, double blind, and placebo-controlled studies on 144 HD patients showed nalfurafine reduced worst itching (p= 0.02) and itching intensity (p= 0.04). In a study of 337 patients on HD with pruritus, participants were randomized 1:1:1 to 5.0 μg of nalfurafine, 2.5 μg of nalfurafine or placebo, and followed for 2 weeks. Both 5.0 and 2.5 μg of nalfurafine significantly improved pruritus intensity compared to placebo. This drug is approved for use in Japan for management of pruritus, but not in Canada.
v. Combination of κ-opioid receptor (KORs) agonists and μ-opioid receptors (MORs) antagonists (Nalbuphine): A multicenter, randomized, double-blind, placebo-controlled trial of 373 hemodialysis patients with moderate to severe pruritus demonstrated that the group receiving nalbuphine 120 mg twice daily for 8 weeks reported significantly decreased pruritus. Nalbuphine is approved for use in Canada for pain management and as a surgical anesthesia supplement.
None of the treatment options that are currently available in Canada modify disease mechanisms. The current options merely assist in symptom reduction with variable impact and insufficient relief.
An ideal treatment should treat the disease mechanism leading to pruritus. It is proposed that overstimulation of the central μ opioid receptors, antagonism of peripheral κ-opioid receptors, or a discrepancy of stimulation and antagonism of μ and κ-opioid receptors lead to itching in conjunction with immune system dysregulation. The current choices available in Canada do not address the above pathways. It is only by offering therapy that addresses these pathways, that we can effectively treat pruritus. Treating this important patient reported symptoms will improve patient care by improving their quality of life, improve mood, reduce dependence on care providers and hopefully, they can return to work and maintain employment.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
The current options are unsatisfactory to help reduce the symptom burden. The best option for treatment of pruritus is kidney transplantation. In a prospective study of 1298 dialysis patients and 521 transplant recipients, pruritus (of 11 symptoms measured) had the greatest improvement post-transplant. However, there is scarcity of organs and majority of patients with pruritus struggle with symptom relief and have very poor quality of life. Nocturnal or longer sessions of dialysis might be helpful, but we do not have any RCT data. The best nonpharmacological option is UV-B radiation, but the response is variable not every jurisdiction has access to phototherapy. The best pharmacological option is Gabapentin and Pregabalin but not everyone responds similarly, and their metabolites accumulate in kidney failure. Parathyroidectomy helps but only in patients with elevated PTH levels (>150 pmol/L). So, we need a new option that helps relieve symptoms and better tolerated.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Difelikefalin (CR845) is a peripherally restricted, selective kappa opioid receptor agonist that exerts antipruritic effects by means of activation of kappa opioid receptors on peripheral neurons and immune cells. The hydrophilic small-peptide structure restricts passive diffusion across membranes, thereby limiting access to kappa opioid receptors in the central nervous system. This treatment option will address the underlying disease process.
The medication will be used as an add on after attempting other options: emollient creams, optimizing phosphorus levels, improving PTH levels and trial of Gabapentin/Pregabalin. We do not believe that it should be the initial medication for treatment of pruritus.
Difelikefalin is certainly expected to cause a paradigm shift in the treatment of itching.
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
Patients whose itching is from causes that cannot be attributed to kidney disease
Pruritus is most intense in patients on renal replacement therapy (in comparison to earlier stages of kidney disease).
There are no diagnostic tools / lab tests. The decision is made on clinical history and exclusion of other causes. There are no companion tests. It’s under reported with 17% of symptomatic patients not reporting symptoms to health care providers, and it’s underappreciated by healthcare professionals in dialysis units.
The ones that need the intervention the most are the ones that have not responded to emollients, antihistamines, Gabapentin, and phototherapy (not available at all sites).
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
As the diagnosis is a based on symptoms- a meaningful response will be symptom reduction leading to better sleep, improved quality of life, improved mood, and return to activities of daily living. Symptom burden can be quantified and followed using tools such as the Edmonton Symptom Score, etc.
What factors should be considered when deciding to discontinue treatment with the drug under review?
Lack of a clinical meaningful response (persistence of symptoms, no impact on mood, no impact on quality of life and inability to return to activities of daily living) should be factors considered to discontinue treatment with the medication.
What settings are appropriate for treatment with [drug under review]? Is a specialist required to diagnose, treat, and monitor patients who might receive [drug under review]?
Dialysis centres in community or hospital setting.
This is a specialist diagnosis (Dermatology and Nephrology).
Additional Information
There is an urgent unmet need in the treatment of pruritus in patients with pruritus with chronic kidney disease. Its debilitating and as Physicians we have been witness to its impact on patients (increased recovery time post sessions, insomnia, reduced quality of life, depression, missed dialysis sessions, withdrawal from dialysis, and increased mortality).
Conflict of Interest Declarations for Saskatchewan Kidney Doctors
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
Yes, my colleagues helped me in creating this submission.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
There are no conflicts for any of the physicians.
Declaration for Clinician 1
Name: Dr Bhanu Prasad, FRPCP
Position: Professor of Medicine, University of Saskatchewan
Date: 15th September 2022
Table 12: COI Declaration for Saskatchewan Kidney Doctors — Clinician 1
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 2
Name: Dr. Sachin Shah, FRCPC
Position: Nephrologist, Saskatchewan Health Authority
Date: 15th September 2022
Table 13: COI Declaration for Saskatchewan Kidney Doctors — Clinician 2
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 3
Name: Dr Rodrick Stryker
Position: Division Head for Nephrology, North, Saskatchewan Health Authority
Date: 15th September 2022
Table 14: COI Declaration for Saskatchewan Kidney Doctors — Clinician 3
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 4
Name: Dr Siva Karunakaran, FRCPC
Position: Nephrologist, Saskatchewan Health Authority
Date: 15th September 2022
Table 15: COI Declaration for Saskatchewan Kidney Doctors — Clinician 4
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 5
Name: Dr Mohammed Abdulhadi, FRCPC
Position: Nephrologist, Saskatchewan Health Authority
Date: 15TH Sept 2022
Table 16: COI Declaration for Saskatchewan Kidney Doctors — Clinician 5
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 6
Name: Dr Olumide Ogundare, FRCPC
Position: Nephrologist, Division Head, Internal Medicine (South), Saskatchewan Health Authority
Date: 15TH Sept 2022
Table 17: COI Declaration for Saskatchewan Kidney Doctors — Clinician 6
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 7
Name: Dr Lookman Abdul, FRCPC
Position: Nephrologist, Saskatchewan Health Authority
Date: 15TH Sept 2022
Table 18: COI Declaration for Saskatchewan Kidney Doctors — Clinician 7
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 8
Name: Dr David Reid, FRCPC
Position: Nephrologist, Saskatchewan Health Authority
Date: 15TH Sept 2022
Table 19: COI Declaration for Saskatchewan Kidney Doctors — Clinician 8
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines.
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.