CADTH Reimbursement Review
Dupilumab (Dupixent)
Sponsor: Sanofi-Aventis Canada Inc.
Therapeutic area: Asthma
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
ACQ
Asthma Control Questionnaire
ACQ-5
5-item Asthma Control Questionnaire
ACQ-7
7-item Asthma Control Questionnaire
ACQ-5-IA
5-item Asthma Control Questionnaire–Interviewer Administered
ACQ-7-IA
7-item Asthma Control Questionnaire–Interviewer Administered
AE
adverse event
CI
confidence interval
ED
emergency department
EOS
eosinophils
EQ-5D-Y
EQ-5D-Youth
FeNO
fractional exhaled nitric oxide
FEV1
forced expiratory volume in 1 second
GINA
Global Initiative for Asthma
HRQoL
health-related quality of life
ICS
inhaled corticosteroid
IgE
immunoglobulin E
IL
interleukin
ITC
indirect treatment comparison
ITT
intention to treat
LABA
long-acting beta agonist
LAMA
long-acting muscarinic antagonist
LS
least squares
LTRA
leukotriene receptor antagonist
MID
minimal important difference
MMRM
mixed-effect model with repeated measures
OCS
oral corticosteroid
OR
odds ratio
PACQLQ
Paediatric Asthma Caregiver’s Quality of Life Questionnaire
PAQLQ(S)
Standardised Paediatric Asthma Quality of Life Questionnaire
PEF
peak expiratory flow
Ppb
parts per billion
PRQLQ
Pediatric Rhinoconjunctivitis Quality of Life Questionnaire
RCT
randomized controlled trial
RR
relative risk
SABA
short-acting beta agonist
SAE
serious adverse events
SCS
systemic corticosteroid
SD
standard deviation
SE
standard error
VAS
visual analogue scale
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Dupilumab (Dupixent), 200 mg or 300 mg, single-use syringe, solution for subcutaneous injection |
Indication | As an add-on maintenance treatment in patients aged ≥ 6 years with severe asthma with a type 2 or eosinophilic phenotype or oral corticosteroid-dependent asthma. |
Reimbursement request | As an add-on maintenance treatment in patients aged 6 to < 12 years with severe asthma with a type 2 or eosinophilic phenotype characterized by:
|
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | March 25, 2022 |
Sponsor | Sanofi-Aventis Canada Inc. |
EOS = eosinophils; FeNO = fractional exhaled nitric oxide; ICS = inhaled corticosteroid; NOC = Notice of Compliance; SCS = systemic corticosteroid.
Introduction
Asthma is a chronic respiratory disorder characterized by reversible airway obstruction.1 In Canada, it is estimated that 14% and 19% of children aged 5 years to 9 years and 10 years to 14 years suffer from asthma, respectively.2 According to the clinical experts consulted for this CADTH review, asthma has several diverse phenotypes, 1 of which is primarily driven by type 2 inflammation, presenting with an allergic or atopic profile and/or eosinophilic asthma.
The management of asthma is traditionally carried out using “reliever” medication for the acute relief of exacerbations, combined with controllers used on a regular or chronic basis, in an effort to prevent the onset of exacerbations.1 Treatment of patients in Canada follows an asthma management continuum, with inhaled corticosteroids (ICSs) as the backbone of maintenance anti-inflammatory therapy, and other medications added on as necessary.3 Monoclonal antibodies are the newest entrants into the asthma treatment paradigm, such as immunoglobulin E (IgE) inhibitors, interleukin (IL)-5 inhibitors, and IL-4 and IL-13 inhibitors. None of the monoclonal antibodies are intended to be used first line, and are reserved for those patients whose asthma is not well controlled despite optimized controller medications.
Dupilumab is an IL-4 and IL-13 inhibitor, indicated as add-on maintenance treatment in patients aged 6 years and older with severe asthma with a type 2 or eosinophilic phenotype or with oral corticosteroid (OCS)-dependent asthma. Dupilumab was previously reviewed by CADTH for the indication of severe asthma with a type 2/eosinophilic phenotype or OCS-dependent asthma in patients aged 12 years and older and received a positive recommendation in June 2021.4 Dupilumab is administered by subcutaneous injection in pediatric patients aged 6 years to 11 years at the dose of 100 mg every 2 weeks or 300 mg every 4 weeks for patients with a body weight from 15 kg to less than 30 kg, 200 mg every 2 weeks or 300 mg every 4 weeks for patients with a body weight from 30 kg to less than 60 kg, and 200 mg every 2 weeks for patients with a body weight of 60 kg or more. Dupilumab also received a Health Canada indication for atopic dermatitis and for chronic rhinosinusitis with nasal polyposis.5 It was also previously reviewed by CADTH for the atopic dermatitis indication in patients older than 12 years of age and received a positive recommendation in April 2020.6
The sponsor has requested that dupilumab be reimbursed for patients aged 6 years to younger than 12 years with severe asthma with the type 2 or eosinophilic phenotype characterized by the following: symptoms that are not controlled despite optimal treatment, defined by the daily use of a medium or high-dose ICS plus 1 controller medication or high-dose ICS alone; eosinophils (EOS) of 150 cells/μL or greater, fractional exhaled nitric oxide (FeNO) of 20 parts per billion (ppb) or greater, or allergy-driven asthma; uncontrolled asthma having at least 1 severe exacerbation, defined by having experienced 1 or more hospitalizations or emergency care visits or treatment with a systemic corticosteroid (SCS) (oral or parenteral) in the past year; and a baseline assessment of asthma symptom control using a validated asthma control questionnaire (ACQ) must be completed before initiation of dupilumab treatment.
The objective of this report was to perform a systematic review of the beneficial and harmful effects of dupilumab for the add-on maintenance treatment of severe asthma with a type 2 or eosinophilic phenotype or OCS-dependent asthma in patients aged 6 years to younger than 12 years.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient groups who responded to CADTH’s call for patient input and from the clinical experts consulted by CADTH for the purpose of this review.
Patient Input
Input from patients was provided by Asthma Canada, based on a survey conducted between February and March 2022, clinical practice guidelines, the product monograph, non-for-profit organization websites, and research papers. More than 100 patients (92%) and caregivers (8%) across all provinces responded to survey with 4 patients having had experience with dupilumab. In addition, the Lung Health Foundation submitted patients’ input based on a survey conducted between January 2021 and June 2022 from 27 patients with asthma and 2 caregivers, all living in Ontario.
Even with currently available treatments, 1 in 4 respondents to the Asthma Canada survey indicated that they have poor symptom control. Approximately 60% of respondents worry about or have a fear of exacerbations, 47% of respondents are concerned about potential hospital admissions, and 47% of survey participants are concerned with missing school or work. The survey findings highlighted challenges for children with asthma, including difficulties in inhaler use techniques, difficulties with making and keeping friends due to fatigue and less energy, activity limitations, inability to attend and concentrate at school, and sleep disturbances. Patients, parents, and caregivers noted several barriers to accessing health care providers (e.g., respirologists, specialized asthma clinics) including travel time and cost, missed school or work, and the financial burden of prescription refills. The Lung Health Foundation input from patients indicated common symptoms of asthma, such as shortness of breath (74.2%), fatigue (67.7%), cough (51.6%), as well as difficulties in activities of daily living such as climbing stairs (43.4%), housework (40.0%), and physical activities (40.0%). Some of the negative impacts of asthma that were highlighted by the patients included: night or early morning waking due to breathing problems (34.5%), emotional well-being (37.9%), and being short-tempered or impatient with others (31.0%).
Patients and caregivers identified the following expectations for new treatment for children with severe asthma: increasing lung function, making management of symptoms easier, reducing exacerbations, and reducing reliance on OCSs. Moreover, children with asthma and their parents expect to see improved day-to-day activities affecting quality of life (school attendance, sleep, energy, participation in activities), less health care visits including those to the emergency department (ED), less anxiety and panic for potential exacerbations, less time off work, and decreased financial hardships. Respondents indicated they would like to minimize side effects of medication; however, they are willing to tolerate certain side effects to improve management of asthma. Decreasing frequency and easing the administration of medication was an additional priority reported by the participants. Finally, it was noted that children on dupilumab cannot be vaccinated with live vaccines, which can pose challenges for children who are not fully immunized.
Clinician Input
Input From Clinical Experts Consulted by CADTH
According to the clinical experts consulted by CADTH for this review, the needs of the majority of patients with asthma are met with current standard therapies; however, a subset of patients remains poorly controlled despite maximized pharmacological treatment and nonpharmacological interventions such as inhaler education and improved medication adherence.
In Canada, pediatric patients with uncontrolled moderate-to-severe type 2 inflammatory asthma have access to treatment with biologics such as anti-IgE, anti-IL5, or anti-IL4/IL13 monoclonal antibodies. Clinical experts reported that the patients who would most likely benefit from dupilumab treatment include individuals with moderate-to-severe asthma who have not achieved optimal asthma control despite conventional therapy (i.e., high-dose ICS with add-on therapy [long-acting beta agonist {LABA} and/or leukotriene receptor antagonist {LTRA}] and requiring ongoing or multiple courses of SCSs) and presenting with a clear inflammatory phenotype, as assessed by peripheral blood eosinophil levels.
According to the clinical experts consulted by CADTH, relevant outcomes to assess treatment response in children include improvements in pulmonary function testing (improvement or stabilization of forced expiratory volume in 1 second [FEV1], elimination of airflow reversibility to bronchodilator), decreases in acute asthma exacerbations, improvements in symptom control, and improvements in health-related quality of life (HRQoL). The clinical experts believed that the primary factor in deciding whether to discontinue dupilumab treatment would be a lack of improvement in asthma control outcomes over many months. Moreover, treatment with dupilumab should be discontinued in case of serious adverse events (SAEs) (e.g., serious immune or allergic reactions, serious dermatological reactions, malignancy, and ophthalmologic adverse events [AEs]). Initiation of the drug should be limited to pediatric respirologists or allergy specialists with significant pediatric asthma experience.
Clinician Group Input
Input was received from 6 clinicians, on behalf of the Canadian Thoracic Society. There were no contrary views reported between the clinician group and the clinical experts consulted for this review. The clinician group indicated that children with severe asthma have limited treatment options compared to the adult population. In addition, there is a lack of effective add-on therapies in younger children with severe asthma with nontype 2 inflammation involving neutrophilic inflammation and recurrent exacerbations caused by viral respiratory infections. According to the clinician group, key outcomes in asthma management include prevention of asthma exacerbations, maximization of quality of life, symptom prevention, and maximization of exercise tolerance. Members of the Canadian Thoracic Society agreed that the use of dupilumab should be restricted to patients aged 6 years to 11 years with type 2 inflammation, moderate-to-severe asthma not adequately controlled on medium-dose ICS plus LABA (or other second controller) or high-dose ICS (or OCS), and who experienced a severe exacerbation in the past year. The Canadian Thoracic Society clinicians suggested that dupilumab should be discontinued if a lack of clinically meaningful positive outcomes over an expected time frame is observed, as well as in case of safety concerns. Assessment of pediatric patients’ eligibility for biologic asthma therapy should be limited to asthma specialists (e.g., respirologists, allergists, pediatricians with a focus on childhood asthma), according to the clinician group input.
Drug Program Input
Input was obtained from the drug programs that participate in the CADTH reimbursement review process. The following were identified as key factors that could potentially impact the implementation of a CADTH recommendation for dupilumab.
Relevant comparators
Considerations for initiation of therapy
Considerations for continuation or renewal of therapy
Considerations for discontinuation of therapy
Considerations for prescribing of therapy
System and economic issues
Clinical Evidence
Pivotal Studies and Protocol Selected Studies
Description of Studies
The VOYAGE study is a multinational, multicentre, randomized, double-blind, placebo-controlled study that compared dupilumab to placebo in patients aged 6 years to younger than 12 years with asthma who were already receiving standard of care. Four hundred and 8 patients with persistent asthma were randomized in 2:1 ratio to 1 dose of dupilumab (100 mg or 200 mg) every 2 weeks or placebo every 2 weeks, over a treatment course of 52 weeks. The primary outcome was the annualized rate of severe exacerbations, while the key secondary outcome was pulmonary function measurement (i.e., change from baseline in prebronchodilator percent predicted FEV1 at week 12). There were 2 main efficacy populations assessed in the trial: type 2 inflammatory asthma phenotype population, characterized by a baseline blood eosinophil count of 150 cells/µL or greater or baseline FeNO of 20 ppb or greater, and baseline blood eosinophils of 300 cells/µL or greater.
The median age of patients included in the VOYAGE trial was 9 years (range = 6 to 11). Across both efficacy populations, the majority of patients were male (range = 64.4% to 69%), White (range = 86.3% to 89.5%), and had a body weight of greater than 30 kg (range = ||||||||||||||||||||). Greater than 60% of patients in the VOYAGE study had experienced 1 or 2 severe asthma exacerbations in the past year. At baseline, FEV1 reversibility was slightly higher in the dupilumab versus placebo group, with a mean (standard deviation [SD]) of 21.5% (21.37) versus 15.81% (16.40) in the type 2 asthma population, and with a mean (SD) of 22.9% (23.23) versus 16.2 (15.8) in the population with baseline eosinophils of 300 cells/µL or greater. Regarding ICS dosing, greater than 40% of patients were on high dosing (dupilumab versus placebo: 43.2% versus 43.9% in the type 2 asthma population and 42.3% versus 48.8% in the baseline blood eosinophils ≥ 300 cells/µL population) and greater than 50% of patients were receiving medium ICS dosing (dupilumab versus placebo: 55.5% versus 56.1% in the type 2 population and 56.0% versus 51.2% in the baseline blood eosinophils ≥ 300 cells/µL population).
Efficacy Results
Mortality
In the VOYAGE trial, there were no deaths reported across the dupilumab and placebo groups.
Acute Asthma Exacerbation
The adjusted annualized rates of severe asthma exacerbations over 52 weeks in the type 2 inflammatory asthma population were 0.305 (95% CI, 0.223 to 0.416) with dupilumab and 0.748 (95% CI, 0.542 to 1.034) with placebo, for a relative risk (RR) of 0.407 (95% CI, 0.274 to 0.605; P < 0.0001) and a risk difference of ||||||||||||||||||||||||||||||. In the population with a baseline eosinophil count of at least 300 cells/µL at baseline, the adjusted rates of exacerbations were 0.235 (95% CI, 0.16 to 0.345) in the dupilumab and 0.665 (95% CI, 0.467 to 0.949) in the placebo group (RR = 0.353; 95% CI, 0.222 to 0.562; P < 0.0001; risk difference = |||||||||||||||||||||||||||||||||||||||| (Table 2).
RRs for the dupilumab versus placebo comparison of severe exacerbation events associated with ED visits or hospitalizations were |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| for the populations with type 2 asthma and baseline blood eosinophils of 300 cells/µL or greater, respectively. Rates associated with hospitalizations only were not estimable due to |||||||||||||||||||||||||||||| events experienced by the patients during the trial.
Asthma Symptoms
Symptoms were assessed using the 7-item ACQ (ACQ-7). At week 24, ACQ-7 scores decreased (improved) in both the dupilumab and placebo groups. The least squares (LS) mean (standard error [SE]) was –1.33 (0.05) in the dupilumab group and –1.00 (0.07) in the placebo group, for a LS mean difference versus placebo of –0.33 (95% CI, –0.50 to –0.16; P = 0.0001) in the type 2 population. In the population with a baseline blood eosinophil count of 300 cells/µL or greater, the LS mean (SE) change from baseline to week 24 was –1.34 (0.06) with dupilumab and –0.88 (0.09) with placebo, for a difference between groups of –0.46 (95% CI, –0.66 to –0.26; P < 0.0001).
Results were maintained during the trial period (to week 52) across both efficacy populations.
Reduction in Use of OCSs
The proportions of patients experiencing treatment with SCS during the trial was higher in the placebo compared to dupilumab arm (dupilumab versus placebo: 24.2% versus 40.4% within the type 2 inflammatory asthma phenotype and 22.3% versus 41.7% within the baseline blood eosinophils ≥ 300 cells/µL population). Adjusted RRs in annualized SCS courses, for the comparison of dupilumab to placebo, were 0.407 (95% CI, 0.272 to 0.609) and 0.340 (95% CI%, 0.212 to 0.545), within the type 2 inflammatory asthma phenotype and population with baseline eosinophils of 300 cells/µL or greater, respectively.
Pulmonary Function
The percent predicted prebronchodilator FEV1 at week 12 increased in both the dupilumab and placebo groups in the type 2 inflammatory asthma phenotype population, with an LS mean difference between groups of 5.21% (95% CI, 2.14 to 8.27%; ||||||||||). Similarly, the LS mean difference at week 12 between the dupilumab and placebo group of 5.32% (95% CI, 1.76 to 8.88%, ||||||||||), in the population with a baseline blood eosinophil count of 300 cells/µL or greater, was reported. In both primary efficacy populations, LS mean changes in the percent predicted prebronchodilator FEV1 were sustained through week 52.
Reduction in Dose of ICSs
The VOYAGE study protocol allowed a permanent increase in background medications after 2 or more severe asthma exacerbations. During the treatment period of the trial, ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||, across all efficacy populations assessed.
Health-Related Quality of Life–Paediatric Asthma Quality of Life Questionnaire
In type 2 inflammatory asthma phenotype population of the VOYAGE study, Standardised Paediatric Asthma Quality of Life Questionnaire PAQLQ(S) scores increased (improved) from baseline to week 52, with an LS mean difference between dupilumab and placebo groups of 0.34 (95% CI, 0.16 to 0.52, ||||||||||). In the population with at least 300 cells/µL baseline blood eosinophils values, similar differences at week 52 between groups were observed (LS mean = 0.33; 95% CI, 0.12 to 0.53; ||||||||||).
Reduction in Use of Rescue Medication
An overall decrease in number of puffs of reliever medications across the 24-hour period was observed in both treatment arms (LS mean differences between dupilumab and placebo groups at week 52 were |||||||||||||||||||||||||||||||||||||||| and |||||||||||||||||||||||||||||||||||||||| for the type 2 and baseline blood eosinophils ≥ 300 cells/µL populations, respectively).
Harms Results
In the VOYAGE trial, AEs occurred in 83% and 79.9% of patients in the dupilumab and placebo groups, respectively. The most common AEs in the dupilumab versus placebo groups were: nasopharyngitis (18.5% versus 21.6%), viral upper respiratory tract infection (12.2% versus 9.7%), pharyngitis (8.9% versus 10.4%), bronchitis (6.3% versus 10.4%), allergic rhinitis (5.9% versus 11.9%), injection site erythema (12.9% versus 9.7%), and injection site edema (10.3% versus 5.2%). SAEs were reported by 4.8% of patients receiving dupilumab and 4.5% of patients receiving placebo, the majority of which were asthma (dupilumab versus placebo: 1.5% versus 0%) and eosinophilia (dupilumab versus placebo: 0.7% versus 0%). Discontinuation due to an AE occurred in 1.8% versus 1.5% of patients of the VOYAGE study, in the dupilumab versus placebo groups, respectively.
Regarding notable harms, injection site reactions were the most commonly reported, by 17.7% and 13.4% of patients in the dupilumab and placebo groups, respectively. Hypersensitivity and anaphylactic reactions occurred in |||||||||||||||||||||||||||||| placebo patients and 0 dupilumab patients versus 1.5% placebo patients, respectively. In terms of infections, severe cases occurred in |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| of patients in the placebo group. Parasitic infections were reported only among patients in the dupilumab group (2.6%). Eosinophilia was more frequently occurring in dupilumab arm compared to placebo (6.6% versus 0.7%, respectively). More patients in the placebo group experienced conjunctivitis compared to dupilumab group (dupilumab versus placebo: 2.6% versus 6.7% for conjunctivitis [narrow] and 3.0% versus 7.5% for conjunctivitis [broad]).
Table 2: Summary of Key Results From Pivotal and Protocol Selected Study
Efficacy outcome | VOYAGE | |||
|---|---|---|---|---|
Type 2 inflammatory asthma phenotype population | Baseline blood eosinophils ≥ 300 cells/µL population | |||
Outcome | Dupilumab n = 236 | Placebo n = 114 | Dupilumab n = 175 | Placebo n = 84 |
Severe asthma exacerbations | ||||
Number of patients with ≥ 1 events | |||||||||| | |||||||||| | |||||||||| | |||||||||| |
Annualized rate, unadjusteda | |||||||||| | |||||||||| | |||||||||| | |||||||||| |
Annualized rate, adjusted (95% CI)a | 0.305 (0.223 to 0.416) | 0.748 (0.542 to 1.034) | 0.235 (0.160 to 0.345) | 0.665 (0.467 to 0.949) |
Relative risk (95% CI)a | 0.407 (0.274 to 0.605) | 0.353 (0.222 to 0.562) | ||
P valuea | < 0.0001 | < 0.0001 | ||
Risk difference (95% CI) b | –0.444 (–0.683 to –0.204) | –0.431 (–0.664 to 0.197) | ||
Severe asthma exacerbations associated with emergency department visit or hospitalization | ||||
Number of patients with ≥ 1 events | |||||||||| | |||||||||| | |||||||||| | |||||||||| |
Annualized rate, adjusted (95% CI)a | |||||||||| | |||||||||| | |||||||||| | |||||||||| |
Relative risk (95% CI)a | |||||||||| | |||||||||| | ||
P valuea | |||||||||| | |||||||||| | ||
Risk difference (95% CI)b | |||||||||| | |||||||||| | ||
Severe asthma exacerbations associated with hospitalization | ||||
Number of patients with ≥ 1 events | |||||||||| | |||||||||| | |||||||||| | |||||||||| |
Annualized rate, adjusted (95% CI)a | NE | NE | NE | NE |
Relative risk (95% CI)a | NE | NE | ||
P valuea | NE | NE | ||
Risk difference (95% CI)b | NE | NE | ||
ACQ-7, total score | ||||
Baseline, N | 236 | 114 | 175 | 84 |
Mean (SD) baseline | 2.15 (0.70) | 2.12 (0.76) | 2.16 (0.73) | 2.15 (0.77) |
Change from baseline, N | |||||||||| | |||||||||| | |||||||||| | |||||||||| |
CFB week 24, LS mean (SE)c | –1.33 (0.05) | –1.00 (0.07) | –1.34 (0.06) | –0.88 (0.09) |
Difference vs. placebo (95% CI) week 24c | –0.33 (–0.50 to –0.16) | –0.46 (–0.66 to –0.26) | ||
P valuec | 0.0001 | < 0.0001 | ||
CFB week 52, LS mean (SE)c | |||||||||| | |||||||||| placebo (95% CI) week 52c | |||||||||| | |
Harm | Dupilumab (n = 271) | Placebo (n = 134) | ||
Any AE | 225 (83.0) | 107 (79.9) | ||
Any SAE | 13 (4.8) | 6 (4.5) | ||
AE leading to treatment discontinuation | 5 (1.8) | 2 (1.5) | ||
Notable harms | ||||
Harm | Dupilumab (n = 271) | Placebo (n = 134) | ||
Anaphylactic reaction | 0 (0.0) | 2 (1.5) | ||
Hypersensitivity (medically reviewed) | |||||||||| | |||||||||| | ||
Severe infections | |||||||||| | |||||||||| | ||
Opportunistic infections | 0 (0.0) | 0 (0.0) | ||
Parasitic infections | 7 (2.6) | 0 (0.0) | ||
Serious injection site reactions | 2 (0.7) | 0 (0.0) | ||
Injection site reactions | 48 (17.7) | 18 (13.4) | ||
Conjunctivitis (narrow) | 7 (2.6) | 9 (6.7) | ||
Conjunctivitis (broad) | 8 (3.0) | 10 (7.5) | ||
Eosinophilia | 18 (6.6) | 1 (0.7) | ||
ACQ-7 = 7-item Asthma Control Questionnaire; AE = adverse event; CFB = change from baseline; CI = confidence interval; LS = least squares; NE = not estimable; PAQLQ(S)-IA = Standardised Paediatric Asthma Quality of Life Questionnaire–Interviewer Administered; SAE = serious adverse event; SD = standard deviation; SE = standard error; vs. = versus.
Notes: Values are n (%) unless otherwise indicated.
Type 2 inflammatory asthma phenotype population is defined as the randomized patients with baseline blood eosinophils of 0.15 Giga/L or greater, or baseline FeNO of 20 parts per billion or greater.
All severe exacerbation events that occurred during the 52-week treatment period are included, regardless of whether the patient is on treatment or not.
For the PAQLQ(S)-IA assessment, only patients aged 7 years or older at randomization are included in the analysis.
Definitions of harms: Conjunctivitis (narrow company medical query): preferred term in (conjunctivitis, conjunctivitis allergic, conjunctivitis bacterial, conjunctivitis viral, atopic keratoconjunctivitis). Conjunctivitis (broad company medical query): preferred term in (conjunctivitis, conjunctivitis allergic, conjunctivitis bacterial, conjunctivitis viral, atopic keratoconjunctivitis, blepharitis, dry eye, eye irritation, eye pruritus, lacrimation increased, eye discharge, foreign body sensation in eyes, photophobia, xerophthalmia, ocular hyperaemia, conjunctival hyperaemia).
aDerived using negative binomial model with the total number of events onset from randomization up to week 52 visit or last contact date (whichever comes earlier) as the response variable, with the treatment group, age, baseline weight group, region, baseline eosinophil level, baseline fractional exhaled nitric oxide level, baseline inhaled corticosteroid dose level, and number of severe exacerbation events within 1 year before the study as covariates, and log-transformed standardized observation duration as an offset variable.
bDerived using delta method.
cDerived from mixed-effect model with repeated measures with change from baseline in ACQ-7–Interviewer Administered up to week 24 as the response variable, and treatment, age, weight group, region, baseline eosinophil level, baseline fractional exhaled nitric oxide level, baseline inhaled corticosteroid dose level, visit, treatment by-visit interaction, baseline ACQ-7–Interviewer Administered and baseline-by-visit interaction as covariates.
Source: Clinical Study Report.7
Critical Appraisal
The VOYAGE trial is a multinational, multicentre, randomized, double-blind, placebo-controlled study. The study used a matching placebo-controlled design, and patients and investigators were blinded to the study treatment assignment, but not the dosing of the injections. Potential for unblinding might have also occurred because of higher frequencies of injection site reactions and eosinophilic reactions in the dupilumab arm compared to the placebo arm. Multiplicity adjustments were implemented adequately for the analysis of severe exacerbation events during the 52-week treatment period, change from baseline in prebronchodilator percent predicted FEV1 at week 12, and change in the ACQ-7–Interviewer Administered (ACQ-7-IA) at week 24. Baseline characteristics were largely balanced between the groups of the study, except for FEV1 reversibility, which was slightly higher in the dupilumab compared to the placebo group. Clinical experts consulted by CADTH regarded the selection of specific time points for outcome assessment and their inclusion in the hierarchy (i.e., FEV1 at week 12 and ACQ-7 at week 24) as not optimal, noting that 52-week assessments would have been more clinically relevant. Many important outcomes such as HRQoL, exposure to OCSs, and ICS dose adjustments were not controlled for multiple comparisons. Even though treatment withdrawals were higher in the dupilumab group compared to placebo, proportions of individuals discontinuing study treatment due to an AE were balanced across the 2 study arms. The number of study withdrawals was generally low (< 6%) and appropriate sensitivity analyses were implemented to handle missing data for the primary and key secondary outcomes, suggesting limited impact on the validity of observed findings.
Dupilumab is indicated as add-on maintenance treatment in patients aged 6 years and older with severe asthma with a type 2 or eosinophilic phenotype or with OCS-dependent asthma. The current review focuses on the patient population of 6 years to 11 years of age, as dupilumab was previously reviewed by CADTH and received a positive recommendation for patients aged 12 years and older. Patients who were OCS-dependent were not included in the VOYAGE trial. The type 2 population was 1 of the main efficacy populations in the trial, defined as having a baseline blood eosinophil count of 150 cells/µL or greater or baseline FeNO of 20 ppb or greater, but the clinicians noted that FeNO assessments are not routinely performed in Canadian clinical practice, which represents an implementation limitation. Most of the VOYAGE trial population was White; hence, generalizability of study findings to people living in Canada may be limited in this regard. Even though background medications administered in the trial were considered reflective of treatments used in Canadian practice by the clinician experts, it was not clear whether inhaler technique was checked throughout the trial. Despite this, adherence to background therapy was high across both treatment groups and placebo responses were robust for many outcomes, suggesting that patients may have benefited from the close attention and monitoring they received in a clinical trial setting per the clinical experts consulted by CADTH. The VOYAGE trial compared dupilumab to placebo (added on to standard of care), which represents a limitation as comparative effectiveness and safety of dupilumab to other biologics approved for management of asthma in pediatric population is limited to available indirect comparisons.
Indirect Comparisons
Description of Studies
The sponsor submitted 1 indirect treatment comparison (ITC). No published ITCs were identified after a systematic search of the literature performed by CADTH. The sponsor-submitted ITC aimed to compare dupilumab to other biologics for the treatment of pediatric patients aged 6 to younger than 12 years with uncontrolled, moderate-to-severe asthma with a type 2 inflammatory phenotype. After a systematic literature review and a feasibility assessment, a total of |||||||||| connected via placebo as a common comparator were identified as eligible. A series of pairwise Bucher ITCs were performed on various outcomes (severe exacerbations, deterioration of asthma [post hoc analysis], asthma symptoms, rescue medication use, and HRQoL), comparing dupilumab (100 mg to 200 mg every 2 weeks) with the IgE inhibitor omalizumab (75 mg to 375 mg once or twice a month). Subgroup data were generated from the dupilumab trial population to match the allergic phenotype and inclusion criteria of omalizumab trials.
Efficacy Results
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Harms Results
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Critical Appraisal
Several limitations of the sponsor-submitted ITC were noted. There was considerable heterogeneity in study characteristics, patient populations and outcomes assessed across the studies included in the network. Since the population of interest for the ITC was the type 2 inflammatory population, an assumption was made that the efficacy of the IgE inhibitor omalizumab would be maintained in these patients. Even though there is clinical overlap between severe allergic and eosinophilic asthma according to the clinical experts, the amount of population concordance and its impact on indirect estimates could not be determined as omalizumab trials were not designed to include an eosinophilic asthma population. In addition, it is unclear whether the placebo link for the ITC was sufficiently similar for making comparisons, since data from the VOYAGE trial suggested a robust placebo response on several outcomes assessed in the trial. In reference to the subgroup analysis, matching specific groups of patients with dupilumab to the omalizumab studies lead to considerable reductions in sample size. A limited number of studies as well as limited available data restricted the possibility to perform meta-regression and account for differences across trials. There were no direct comparisons between treatments; therefore, the assessment of consistency was not feasible. In summary, due to various methodological limitations, no robust conclusions can be drawn about the comparative clinical efficacy of dupilumab versus omalizumab in the treatment of patients aged 6 years to 11 years with uncontrolled moderate-to-severe asthma.
Other Relevant Evidence
Description of Studies
The LIBERTY ASTHMA EXCURSION (EXCURSION) study was an open-label, noncomparative longer-term extension study that enrolled patients who completed the VOYAGE trial. The primary objective of EXCURSION was to assess long-term safety and tolerability of dupilumab. All patients received open-label treatment with dupilumab during the period of 52 weeks. A total of 365 patients were enrolled in EXCURSION, of which 240 patients had been assigned to dupilumab treatment in the parent trial (dupilumab–dupilumab group) and 125 had been assigned to placebo treatment in the parent trial (placebo–dupilumab group). All patients in EXCURSION were receiving their background medication (ICS with or without a second controller) as well as reliever therapy, if necessary. As per the database lock of January 17, 2022, the median duration of study was ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| for the dupilumab–dupilumab and placebo–dupilumab groups, respectively.
Efficacy Results
Severe Asthma Exacerbations
As of January 17, 2022, a total of ||||||||||||||||| of patients with type 2 inflammatory asthma phenotype in the dupilumab–dupilumab and placebo–dupilumab groups, respectively, experienced a severe exacerbation event. When looking at the patients with eosinophils of 300 cells/µL or greater at baseline of the parent study, ||||||||||||||||| experienced an event in dupilumab–dupilumab and placebo–dupilumab groups, respectively. The unadjusted annualized rate of severe exacerbation was 0.118 and 0.124 for the dupilumab–dupilumab and placebo–dupilumab groups, respectively, in the type 2 inflammatory asthma phenotype population. Similarly, in the subgroup with baseline eosinophils of 300 cells/µL or greater, the unadjusted annualized severe exacerbation event rate was 0.120 and 0.119, for the dupilumab–dupilumab and placebo–dupilumab groups, respectively.
Pulmonary Function
At week 52, mean (SD) changes from baseline in percent predicted prebronchodilator FEV1 were |||||||||||||||||||||||||||||||||| for the dupilumab–dupilumab and placebo–dupilumab groups, respectively, in the type 2 inflammatory population, and ||||||||||||||||||||||||||||||||||||||||||||||||||| for the 2 groups, respectively, in the population with eosinophils of 300 cells/µL or greater.
Harms Results
Among patients who entered the EXCURSION study from the VOYAGE study, 61.3% of patients in the dupilumab–dupilumab and 68.0% of patients in the placebo–dupilumab groups reported at least 1 AE as per data cut-off of January 17, 2022. SAEs were experienced by 2.5% of patients in the dupilumab–dupilumab group and 0.8% in the placebo–dupilumab group. There were no deaths reported during the study period. AEs leading to discontinuation of the treatment were reported by 3 (1.3%) patients in the dupilumab–dupilumab group (pulmonary tuberculosis, ascariasis, and allergic conjunctivitis), and no patients in the placebo–dupilumab group.
In terms of notable harms, hypersensitivity was experienced |||||||||||||||||% of patients in the dupilumab–dupilumab and placebo–dupilumab groups, respectively, with ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| experiencing anaphylactic reaction. Other notable harms of interest reported during the long-term extension study period in the dupilumab–dupilumab versus placebo–dupilumab included: injection site reactions ||||||||||||||||||||||||||||||||||||||||||||||||||| conjunctivitis (4.2% versus 4.8%), eosinophilia (3.3% versus 8.0%), severe or serious infections (|||||||||||||||||), and parasitic infections (1.7% versus 1.6%).
Critical Appraisal
The EXCURSION trial provided additional data on the longer-term safety and efficacy of dupilumab relative to placebo. The validity of observed findings is limited due to the open-label and noncomparative study design. Statistical hypothesis testing was not part of the design. Furthermore, as the EXCURSION trial is a 1-year study, rare AEs might not be captured as of the data cut-off date. Given that the patients enrolled in the long term extension study were originally from the VOYAGE parent study, and the eligibility criteria remained the same, it is reasonable to expect that the same limitations to generalizability are relevant to EXCURSION.
Conclusions
One sponsor-submitted, multicentre, randomized, double-blind, phase III trial (VOYAGE), comparing add-on therapy with dupilumab to placebo in patients aged 6 years to younger than 12 years with persistent asthma demonstrated that dupilumab reduced the annualized rate of severe exacerbations and improved pulmonary function (FEV1) in patients whose asthma remains uncontrolled despite background therapy with medium to high doses of ICSs. There was supportive evidence on the overall treatment benefit on asthma-related symptoms, as measured by ACQ-7, but the differences between the dupilumab and placebo groups did not exceed the minimal important difference (MID). HRQoL analyses were not controlled for multiple comparisons in this randomized controlled trial (RCT); thus, it remains unclear what the effect of add-on dupilumab on patients’ HRQoL is compared to placebo. Likewise, observed reductions in OCS usage were not part of the statistical hierarchy, which precluded drawing conclusions about the effects of dupilumab on this outcome. With respect to harms, there were no obvious safety or tolerability issues associated with the use of dupilumab in children. A longer-term extension study did not identify any new safety issues. Findings from the sponsor-submitted ITC were inconclusive with respect to the efficacy and safety of dupilumab compared to omalizumab due to numerous methodological limitations.
Introduction
Disease Background
Asthma is a chronic respiratory disorder characterized by reversible airway obstruction. Hallmarks of asthma include inflammation, bronchoconstriction, and airway remodelling, as well as hyperresponsive airways and mucous production.1 Symptoms of asthma include wheezing, dyspnea, chest tightness, sputum production, and coughing, and these symptoms can be exacerbated by exogenous influences such as allergens, upper respiratory tract infections, or environmental factors such as smoke or cold air.1 It is estimated that 2.4 million people living in Canada aged 12 years or older have asthma, or 12% of all children and 8% of adults.8 Moreover, prevalence data from the population of people living in Canada in 2011 and 2012 reported asthma estimates of 14% and 19% in children aged 5 years to 9 years and 10 years to 14 years, respectively.2
According to the clinical experts consulted for this CADTH review, asthma has several diverse phenotypes, 1 of which is primarily driven by type 2 inflammation, presenting with allergic or atopic profile and/or eosinophilic asthma. Eosinophils, among other functions, promote airway inflammation and contribute to airway hyperresponsiveness and remodelling,9,10 and eosinophilic asthma is characterized by increased peripheral blood eosinophil counts. Severe type 2 or eosinophilic asthma tends to be asthma that is poorly controlled despite optimized ICSs, plus add-on therapy with a LABA or LTRA.11
Standards of Therapy
Traditionally, the management of asthma is carried out using: medications for the acute relief of exacerbations (colloquially, “asthma attacks”), often referred to as “relievers” or “rescue medications”; and controllers, or maintenance drugs, which are used on a regular or chronic basis in an effort to prevent the onset of exacerbations.1
The Global Initiative for Asthma (GINA) guidelines support a stepwise approach for asthma treatment in children aged 6 years to younger than 12 years. Of note, detailed GINA recommendations for the management of children aged 6 years to younger than 12 years with severe asthma are currently under development and only the stepwise diagram for treatment is available in the available GINA guidance documents. In step 1, patients begin using a low-dose ICS whenever a reliever medication (short-acting beta agonist [SABA]) is used. As symptoms persist, step 2 involves a low-dose ICS with as-needed SABA, with an alternative treatment option of a LTRA. From here, patients may need to escalate to step 3, which includes a combination of low-dose ICS and LABA with as-needed reliever (SABA), or medium-dose ICS with as-needed reliever (SABA), or maintenance and reliever therapy with a very low dose of ICS plus formoterol. Step 4 includes stepping up to a medium-dose ICS and LABA combination or maintenance and reliever therapy of a low dose of ICS plus formoterol, with an alternative addition of tiotropium or LTRA and recommendation for expert advice in case the asthma is not well controlled. Finally, step 5 involves referral to a phenotypic assessment and the use of a higher-dose ICS and LABA combination, with additional possibility of add-on treatment (i.e., anti-IgE, anti-IL5/5R, anti-IL4R) in case asthma control is not achieved.1 Similarly, The Canadian Asthma Consensus Guidelines approach to therapy proposes SABA, or budesonide plus formoterol as needed for symptom control, and outlines low-dose, medium-dose, and high-dose ICSs for anti-inflammatory maintenance therapy, with add-on LTRA, LABA, and or tiotropium as necessary.3 In case the high-dose ICS with add-on therapy does not achieve control, treatment with OCSs and/or biologic therapy can be considered. According to the clinical experts consulted for this CADTH review, a very limited proportion of pediatric patients is on daily OCS therapy, but many patients may experience frequent or repeated short courses of OCSs. Monoclonal antibodies are the newest entrants into the asthma treatment paradigm for children aged 6 years to 12 years of age, such as an IgE inhibitor (omalizumab), IL-5 inhibitor (mepolizumab), and most recently IL-4 and IL-13 inhibitors (dupilumab). Nonpharmacologic therapies include asthma education, improvement of inhaler technique, allergen avoidance, and a written asthma action plan.1 The clinical experts consulted by CADTH stated that patients aged 6 years to younger than 12 years who are not well controlled on an ICS and another controller (e.g., GINA step 4) would receive a high-dose ICS plus a LABA (or alternative controller) when considering adding a biologic. The experts indicated that the goal would be to gain asthma control in a timely way and to lower the ICS dose when control is achieved. Aligned with previously mentioned guidelines, a Canadian Thoracic Society statement from 2017 provides guidance on the management of severe asthma.11
With respect to harms associated with pharmacologic therapies, chronic ICS use in children can have a number of concerning adverse effects, including growth retardation and adrenal suppression, particularly at high doses.12 The use of OCSs heightens the risk of harms such as fractures and decreased bone density, and their chronic use should be limited in children, according to the clinical experts.
According to the clinical experts consulted by CADTH, the goals of asthma therapy in children aged 6 years to 12 years are to achieve asthma symptom control, decrease future risk of worsening, prevent persistent airflow limitation, and decrease the frequency of exacerbations, which will improve HRQoL. Additionally, limiting complications and long-term adverse effects of current therapy is another important goal of asthma management in children.
Drug
Dupilumab is an IL-4 and IL-13 inhibitor.5 Both IL-4 and IL-13 are thought to play a role in inflammation and in the pathophysiology of asthma, and dupilumab is a monoclonal antibody that targets both. Dupilumab is administered by subcutaneous injection in pediatric patients aged 6 years to 11 years at the dose of 100 mg every 2 weeks or 300 mg every 4 weeks for patients with a body weight from 15 kg to less than 30 kg, 200 mg every 2 weeks or 300 mg every 4 weeks for patients with a body weight from 30 kg to less than 60 kg, and 200 mg every 2 weeks for patients with a body weight of 60 kg or more. Dupilumab is indicated as add-on maintenance treatment in patients aged 6 years and older with severe asthma with a type 2 or eosinophilic phenotype or with OCS-dependent asthma. Dupilumab is also indicated for atopic dermatitis in patients aged 6 years and older and for chronic rhinosinusitis with nasal polyposis in adults.5
Dupilumab was previously reviewed by CADTH for the indication of severe asthma with a type 2 or eosinophilic phenotype or OCS-dependent asthma in patients aged 12 years and older (June 2021; recommendation to reimburse with clinical criteria and/or conditions), and for the atopic dermatitis indication in patients older than 12 years (April 2020; recommendation to reimburse with clinical criteria and/or conditions).4,6
The sponsor has requested that dupilumab be reimbursed for patients aged 6 years to younger than 12 years with severe asthma with type 2 or eosinophilic phenotype characterized by the following: symptoms that are not controlled despite optimal treatment, defined by the daily use of a medium or high-dose ICS plus 1 controller medication or high-dose ICS alone; EOS of 150 cells/μL or greater or FeNO of 20 ppb or greater, or allergy-driven asthma; uncontrolled asthma having at least 1 severe exacerbation, defined by having experienced 1 or more hospitalization/emergency care visit or treatment with an SCS (oral or parenteral) in the past year; and a baseline assessment of asthma symptom control using a validated ACQ must be completed before initiation of dupilumab treatment. These criteria are in addition to the Health Canada indication.
Key characteristics of dupilumab and other biologics used for severe asthma are summarized in Table 3.
Table 3: Key Characteristics of Dupilumab, Mepolizumab, and Omalizumab
Drug | IL-4 and IL-13 inhibitors (dupilumab) | IL-5 inhibitors (mepolizumab) | IgE inhibitors (omalizumab) |
|---|---|---|---|
Mechanism of action | A recombinant human IgG4 monoclonal antibody that inhibits IL-4 and IL-13 signalling and blocks proinflammatory cytokines, resulting in an anti-inflammatory effect. | Mepolizumab is a targeted IL-5 IgG1 kappa monoclonal antibody that reduces the production and survival of eosinophils, acting as an anti-inflammatory drug for asthma. | Omalizumab is an IgE inhibitor that reduces the amount of free IgE, which is available to trigger the allergic–inflammatory cascade in asthma. |
Indicationa | Add-on maintenance treatment in patients aged ≥ 6 years with severe asthma with a type 2 or eosinophilic phenotype or OCS-dependent asthma. | Add-on maintenance treatment for adults, adolescents, and children (aged ≥ 6 years) with severe eosinophilic asthma who: - are inadequately controlled with high-dose ICS (patients ≥ 18 years of age) or medium-to-high-dose ICS (patients 6 to 17 years of age) and an additional asthma controller (e.g., LABA); and - have a blood eosinophil count of ≥ 150 cells/μL at initiation of treatment with mepolizumab or ≥ 300 cells/μL in the past 12 months. | Treatment of adult and pediatric patients (≥ 6 years of age) with moderate-to-severe persistent asthma who have a positive skin test or in vitro reactivity to a perennial aeroallergen and whose symptoms are inadequately controlled on ICS. |
Route of administration | SC | SC | SC |
Recommended dose | Based on patient’s body weight:
| Children (aged 6 to 11 years): 40 mg q.4.w. | 75 mg to 375 mg q.2.w. or q.4.w. depending on body weight and serum IgE |
Serious adverse effects or safety issues | Anaphylaxis, injection site reactions, eosinophilia, helminth infections, eye disorders | Anaphylaxis, injection site reactions, infection | Anaphylaxis, injection site reactions, infection |
ICS = inhaled corticosteroid; IgE = immunoglobulin E; IgG = immunoglobulin G; IL = interleukin; LABA = long-acting beta agonist; OCS = oral corticosteroid; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; SC = subcutaneously.
aHealth Canada–approved indication.
Source: Product monographs for dupilumab,5 mepolizumab,13 and omalizumab.14
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups. The full original patient input(s) received by CADTH have been included in the stakeholder section at the end of this report.
Asthma Canada, the only national charity advocating for people with asthma and respiratory allergies, submitted patient group input based on a survey conducted between February and March, 2022, in addition to several other sources. More than 100 patients (92%) and caregivers (8%) across all provinces responded to the survey, including 4 patients having with experience with dupilumab. Another patient group, Lung Health Foundation, a registered charity that empowers people living with or caring for others with lung disease, submitted input based on a survey conducted between January 2021 and June 2022 from 27 patients with asthma and 2 caregivers, all living in Ontario.
One in 4 Asthma Canada survey participants indicated they have poor symptom control even with currently available treatments. Furthermore, many patients reported challenges in accessing the needed health providers, such as respirologists and specialized asthma clinics, to manage their health. When patients live in rural areas and must travel to get necessary care, children miss school and parents and caregivers miss work. The respondents said that managing care can require a significant amount of time and be a burden, which can be made worse with poor asthma control. Respondents emphasized that timely access to care becomes critical when patient must urgently travel to the ED for exacerbations that can be life-threatening and to restore airway function. Approximately 60% of respondents said that they worry about or have a fear of exacerbations; 47% respondents said they are concerned about potential hospital visits or admissions, which can be stressful; and 47% of survey participants said missed work and school days are concerning. In addition, some of the challenges that affect children with asthma identified by respondents were difficult techniques involved with inhaler use, other children not adequately understanding the impact of asthma on daily lives, making and keeping friends made difficult by fatigue and less energy, activity limitations that could be worsened by environmental triggers, inability to attend and concentrate at school, sleep disturbances, and significant time spent on educating friends, daycares, schools, and others about the seriousness of asthma. According to the patient group input, financial hardships associated with paying for asthma treatments are added stress. Lastly, the respondents to Asthma Canada survey said that drug shortages caused by limited supply have also impacted children living with asthma.
The Lung Health Foundation input highlighted that asthma symptoms such as shortness of breath (74.2%), fatigue (67.7%), and cough (51.6%), as well as difficulties in activities of daily living such as climbing stairs (43.4%), housework (40.0%), and physical activities (40.0%) are the challenging part of living with asthma. In addition, respondents to the Lung Health Foundation survey said that the negative impacts of asthma included waking up at night or in the early morning due to breathing problem (34.5%), emotional well-being (37.9%), and being short-tempered or inpatient with others (31.0%). They said they want treatments that improve symptom management, energy, and quality of life, as well as reduce exacerbations. In addition, the respondents indicated they wanted treatments with a reduced cost to the patient and caregiver.
Patients indicated that they want a new medication that can be another treatment option for children with severe asthma and their families to ease burden on patients, families, caregivers, and the health care system by increasing lung function (73%), making management of symptoms easier (61%), reducing exacerbations (56%) and reducing reliance on OCSs (56%). Also, children with asthma and their parents expect to see improved day-to-day activities affecting quality of life, such as attendance at school, sleep, energy, participation in activities, as well as less health care visits including those to the ED, less anxiety and panic (for potential exacerbations), less time off work, and less financial hardships. Respondents said they would like to minimize side effects of medication; however, they are willing to tolerate certain side effects to improve management of their asthma. One in 4 survey participants indicated that they need too many daily doses, therefore, a medication that can be taken less frequently and administered easily would be helpful for children with severe asthma.
Lastly, it was noted that children receiving dupilumab cannot be vaccinated with live vaccines (e.g., hepatitis, meningococcal, and HPV vaccines), which can be challenging for children who may not be fully immunized.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of asthma.
Unmet Needs
According to the clinical experts consulted by CADTH, the main goals of asthma therapy in children include achieving asthma symptom control, decreasing the risk and frequency of acute asthma exacerbations, and preventing sequelae of long-term uncontrolled asthma (i.e., fixed or only partially reversible airflow limitation). Additionally, limiting complications and long-term adverse effects of inhaled and OCSs among children was identified as important goal by the clinicians.
There are no treatments available that would cure asthma, but long-term control can be achieved for many patients, according to the experts. The clinical experts reported that the majority of patients achieve excellent control with existing medications and treatments, aligned with the Canadian Asthma Consensus Guidelines.3 The experts noted that increasing medication adherence, adopting self-management techniques and inhaler education are also important, yet challenging to adopt, factors in reaching asthma treatment goals. The clinical experts also stressed that a subset of patients will remain poorly controlled despite adoption of the best practices for asthma treatment.
Place in Therapy
The clinical experts reported that dupilumab would be utilized in treating children and youth with uncontrolled moderate-severe type 2 inflammatory asthma (i.e., individuals who remain uncontrolled on high-dose ICSs with add-on therapy [LABA and/or LTRA] and requiring ongoing or multiple courses of SCSs). Hence, they indicated that dupilumab would not be implemented as a first-line therapy, but would be a therapy used when conventional treatment has not achieved asthma control.
According to the clinical experts, biologic agents targeting IgE-mediated disease (e.g., the anti-IgE drug omalizumab) and decreasing eosinophilic inflammation (e.g., the anti-IL5 drug mepolizumab) have found limited use among pediatric patients in the Canadian setting so far. The clinicians noted that dupilumab therapy addresses a specific aspect of the inflammatory cascade (the IL-4 and IL-13 pathway) and highlighted its novelty in this regard.
Patient Population
The clinical experts reported that the target population for dupilumab would include individuals with moderate-to-severe asthma who have not achieved optimal asthma control despite conventional therapy and clearly present with an inflammatory phenotype. The type 2 inflammatory profile can be assessed by peripheral blood eosinophil levels in the Canadian context to determine initiation of therapy. The experts highlighted that the VOYAGE trial enrolled patients with type 2 inflammatory asthma (i.e., peripheral blood EOS > 150 cells/μL or FeNO > 20 ppb), and that the eosinophilic criteria might have implementation difficulties in certain laboratory facilities across Canada, which routinely measure rounded values of EOS (i.e., 100 cells/μL, 200 cells/μL, 300 cells/μL, and so forth). Furthermore, clinicians noted that FeNO assessments are not routinely performed in most Canadian centres.
Both clinical experts stated that misdiagnosis of asthma is common. Clinicians felt that 6 years of age represents a limit for reliable and reproducible pulmonary function measurement. Therefore, children older than 6 years can undergo pulmonary function and reversibility testing, which is required for confirming the diagnosis of asthma. According to the pediatric clinical experts, under-diagnosis of patients with moderate-to-severe asthma can sometimes be observed in the clinical practice.
Assessing Response to Treatment
The clinical experts reported that outcomes used clinically to assess response to treatment are typically targeting assessment of asthma control. Specifically, clinicians reported the following outcomes to be of relevance: improvements in pulmonary function testing, decreases in acute asthma exacerbations, improvements in symptom control, and improvements in quality of life scores. A validated measure such as the ACQ can be used to objectively assess improved control; however, 1 clinician reported that the instrument is mostly used for research purposes and is not used on a regular basis within the everyday clinical care. In addition, the experts reported that decreased health care resource utilization, improvement in ability to perform activities of daily living, and ability to participate in recreational activities can be considered supportive of a positive treatment effect of dupilumab. Regarding the frequency of disease assessments, the clinical experts reported that patients with moderate-to-severe asthma would be assessed in specialty asthma clinics approximately 2 to 4 times per year, depending on the asthma severity and clinic resources.
Discontinuing Treatment
According to the clinical experts, evidence of lack of impact on pulmonary function testing, asthma symptom control, acute exacerbations, and quality of life assessment need to be accounted for when considering discontinuation from therapy. Moreover, treatment with dupilumab should be discontinued the in case of SAEs (e.g., serious immune/allergic reactions, serious dermatological reactions, malignancy, and ophthalmologic AEs).
Prescribing Conditions
Clinical experts indicated that treatment with dupilumab should be initiated by a pediatric respirologist or allergy specialist with significant pediatric experience. For patients living in rural or remote areas, physician care could be provided in liaison with 1 of the previously mentioned specialists, while optimal access to care can be achieved though the implementation of telehealth practices.
Additional Considerations
One clinician reported that concomitant treatment of atopic dermatitis as an outcome should be considered, as atopic dermatitis may have significant adverse effects upon quality of life of patients.
Clinician Group Input
This section was prepared by CADTH staff based on the input provided by clinician groups. The full original clinician group input(s) received by CADTH have been included in the stakeholder section at the end of this report.
A total of 6 clinicians from the Canadian Thoracic Society (2 Canadian Thoracic Society Asthma Assembly Co-chairs and 4 pediatric respirologists from the Asthma Assembly Steering Committee) submitted clinician group input. The Canadian Thoracic Society is a national professional association for health care providers in respiratory care and research. The Canadian Thoracic Society promotes lung health and enhances the ability of health care providers through leadership, collaboration, research, learning, and advocacy, as well as providing the best respiratory practices in Canada. The Canadian Thoracic Society is also an accrediting body for specialist education and continuing professional development.
Unmet Needs
The clinician group stated that key goals of asthma control are to prevent asthma exacerbations, which can be life-threatening, maximize quality of life, prevent symptoms, and maximize exercise tolerance.
Secondary goals in asthma management include normalizing lung function, reducing airway inflammation, avoiding permanent airway remodelling, avoiding chronic use of OCSs, and using the lowest effective dose of ICSs to avoid growth suppression, adrenal suppression, and other significant side effects of corticosteroids in children.
The clinician group reported that in a relatively small subgroup of children with severe asthma, acceptable control requires high doses of ICSs and/or OCSs and add-on therapies, which puts them at a high risk of significant AEs. In some children, acceptable control cannot be achieved with these therapies and these patients experience frequent exacerbations that require OCSs. The clinician group indicated that chronic OCSs are not a viable treatment option given their side effect profile and highlighted that the recurrent use of OCSs is associated with a high risk of side effects.
According to the clinicians, children with severe asthma have limited treatment options compared to the adult population. Currently, the American Thoracic Society/European Respiratory Society,15 but not the Canadian Thoracic Society3 recommend tiotropium. However, tiotropium is not approved for those younger than 18 years in Canada and is not regularly used off-label in children aged 6 years to 11 years. The Canadian Thoracic Society and the American Thoracic Society/European Respiratory Society both recommend omalizumab for children aged 6 years to 11 years who meet the initiation criteria. However, the guidelines only include clinical trials published up to 2018. Lastly, mepolizumab is approved in Canada for children aged 6 years to 11 years with severe asthma but has not been widely used.
The clinician group identified other unmet needs. In younger children with severe asthma who have nontype 2 inflammation involving neutrophilic inflammation and recurrent severe exacerbations caused by viral acute respiratory tract infections, effective add-on therapies have not been identified when ICSs fail. Similarly, in a subgroup of older children with severe asthma who have neutrophil-triggered inflammation, effective add-on therapies are also needed.
Place in Therapy
The clinician group stated that dupilumab (an anti-IL4/IL-13 drug) targets different inflammatory pathways than omalizumab (an anti-IgE drug) and mepolizumab (an anti-IL5 drug); therefore, complements other available treatments. Following the Canadian Thoracic Society3 Severe Asthma Management Continuum, dupilumab would be considered as an add-on therapy in severe asthma when other biologics are indicated. Also, the group reported dupilumab could be helpful when asthma is not well controlled on a combination of a high-dose ICS, LABA, and LTRA, or when a child experiences significant side effects from these medications, or when these medications are contraindicated. The clinician group emphasized that since injectable medications can be less acceptable due to a higher burden on families and higher costs for the health care system, it is recommended to use biologics based on the child’s inflammatory profile and only after standard therapy has been adequately tried with good adherence.
Moreover, the clinician group suggested that when children with severe asthma concomitantly have atopic dermatitis, when children are allergic to or have had SAEs with mepolizumab, or when there is supply issue with mepolizumab, dupilumab can be a therapeutic option.
Patient Population
Based on input, patients aged 6 years to 11 years with type 2 inflammation, moderate-to-severe asthma not adequately controlled on a medium-dose ICS plus LABA (or other second controller) or high-dose ICS (or OCS) and who experienced a severe exacerbation in the past year are most likely to respond to dupilumab treatment. (Type 2 inflammation is defined as having a serum eosinophil count ≥ 150 cells/μL and FeNO ≥ 20 ppb or a serum eosinophil count ≥ 300 cells/μL.) The group added that those with an eosinophil count of greater than 300 cells/μL and a baseline FeNO of 25 ppb have shown to have the best response to dupilumab. However, the group said that there is still a good response with a lower-level eosinophil count as long as it is greater than 150 cells/μL.
According to the clinician group, the 2 markers of type 2 inflammation, namely an eosinophil count of greater than 150 cells/μL and FeNO elevation, are the only disease characteristics that would make differences in responses.
The clinician group indicated that the best-suited physicians to identify eligible patients are respirologists or allergists, who must confirm diagnosis based on symptoms and airway reversibility measured by spirometry (FEV1 change > 10%). If not under the care of a respirologist or allergist, the clinician group indicated that patients must be referred and assessed with spirometry, clinical exam, differential complete blood count (serum EOS), and FeNO, if available.
Assessing Response to Treatment
The clinician group said that clinically meaningful outcomes include reduced frequency and severity of symptoms, improved quality of life, and minimized side effects from existing maximal therapy (e.g., corticosteroids). Reducing daily medication burden and improving compliance are also noted as potential outcomes to measure response. Other outcomes, such as reduced inflammatory markers, improved lung function, and prevention of long-term airway remodelling are additional outcomes to determine response to treatment. The clinical measures, such as ACQs, FEV1, and exacerbation rates are routinely used in clinical practice and do not differ among specialists. However, according to the clinician group, the inflammatory markers may not be routinely used or accessible in clinical practice and may vary in their application.
Discontinuing Treatment
The clinician group said that a lack of clinically meaningful positive outcomes over an expected time frame, such as similar (or worse) rate of exacerbation, prebronchodilator FEV1, or patient symptom scores compared to pretreatment levels, is a reason to consider discontinuing treatment. Additionally, the group mentioned that safety concerns and patient’s choice could also play roles in making decision to discontinue treatment.
Prescribing Conditions
The clinician group cited the Canadian Thoracic Society Position Statement for the Recognition and Management of Severe Asthma11 for prescribing conditions. They said that “asthma specialists,” including but not limited to respirologists, allergists, and pediatricians with a focus on childhood asthma or who are specialists in asthma, general respirology, or allergy/immunology, and who have access to lung function tests and certified asthma or respiratory educators or nurse practitioners should assess a child for eligibility for biologic asthma therapy. According to the clinician group, an asthma specialist should diagnose, treat, and monitor for adherence and correct administration techniques.
The clinician group suggested the first 1 or more doses of dupilumab be administered by a health care practitioner in a hospital or medical setting. Following this, injection by a caregiver who has received proper training on correct administration can be undertaken in the community setting, such as in the home, if the health care practitioner determines it appropriate.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
There are a number of biologics used for severe asthma. Mepolizumab (Nucala) and omalizumab (Xolair) may have been better comparators than placebo in the trials as they are indicated for severe asthma in patients aged ≥ 6 years. | Comment from the drug plans to inform CDEC deliberations. |
There are other biologics indicated for severe asthma (omalizumab, mepolizumab, reslizumab, benralizumab); however, only omalizumab and mepolizumab have Health Canada approval for patients aged ≥ 6 years. Omalizumab and mepolizumab are not funded through government-sponsored drug plans for patients ≥ 6 years of age; therefore, there is limited access to biologic therapy for severe asthma for the pediatric population. Asthma is a heterogeneous condition and the biologics are indicated in a variety of phenotypes: omalizumab for allergic asthma and mepolizumab, reslizumab, and benralizumab for eosinophilic asthma. Dupilumab is indicate for type 2 or eosinophilic asthma. Question for CDEC: Given the different phenotypes and treatments for severe asthma, would mepolizumab or omalizumab have been more appropriate comparators than placebo in the clinical trials? | Even though the VOYAGE trial did not compare dupilumab to any of the other biologics approved for management of asthma, the clinical experts consulted by CADTH for this review reported that placebo represents an appropriate comparator as long as patients were not deprived of their background medication. According to the clinical experts, biologic agents targeting IgE-mediated disease (omalizumab) and decreasing eosinophilic inflammation (mepolizumab) have found limited use among pediatric patients in the Canadian setting so far. |
Considerations for initiation of therapy | |
Populations in the study included children with the type 2 inflammatory asthma phenotype: baseline eosinophil count ≥ 150 cells/μL or FeNO ≥ 20 parts per billion Question for CDEC: FeNO is not part of any other asthma criteria. As this was included in the primary efficacy population and requested to be included by the sponsor, would this be a consideration to include for initiation/renewal criteria? | According to the clinical experts, assessment of eligibility for dupilumab treatment should be based on peripheral blood eosinophil counts, as a surrogate for type 2 inflammation. The clinicians reported that FeNO assessments are not routinely performed in clinical practice in Canada. |
Question for CDEC: For dupilumab, will there be differences in the initiation criteria between the populations aged 6 years to 11 years and ≥ 12 years? | Question for CDEC to be addressed in the recommendation. The clinical experts indicated that the initiation criteria should be identical. |
Patients in the studies were on medium-dose ICS therapy with a second controller (LABA, LTRA, LAMA, or methylxanthines) high-dose ICS, or high-dose ICS with a second controller. The sponsor is requesting the initiation criteria be for patients:
Question for CDEC: Is the alignment in the following initiation criteria for the use of dupilumab for patients ≥ 12 years for severe asthma reviewed by CADTH appropriate?
If the criteria are not aligned, how will patients qualify for dupilumab when they age into the ≥ 12 years of age criteria? | Question for CDEC to be addressed in the recommendation. The clinical experts suggested aligning criteria; however, they also noted the lack of data among OCS-dependent children. The experts felt that this situation would occur rarely in the Canadian setting and proposed that the drug plans handle this situation as a special exception. Moreover, 1 clinical expert highlighted some implementation challenges for the initiation criteria and noted that asthma control questionnaires are not routinely used in asthma clinical care, but more as a research tool. |
Considerations for continuation or renewal of the therapy | |
Question for CDEC: Would alignment with the dupilumab criteria for patients aged ≥ 12 years and other biologics for severe asthma reviewed by CADTH be appropriate? | Question for CDEC to be addressed in the recommendation. |
Considerations for discontinuation of therapy | |
Question for CDEC: Should the discontinuation criteria align with dupilumab for 12 years and older and mepolizumab and benralizumab:
| Questions for CDEC to be addressed in the recommendation. According to the clinical experts, discontinuation criteria for the pediatric and adult populations should be aligned. The experts also noted 2 implementation considerations in younger children: younger children experience frequent upper respiratory tract infections during the school year, and it is harder to implement exacerbation frequency as a stopping rule. |
Considerations for prescribing of therapy | |
No evidence was identified to support combination use. Combination use in this space would significantly impact cost for jurisdictions. | Comment from the drug plans to inform CDEC deliberations. |
Question for CDEC: Is alignment with the following criteria appropriate?
| Clinical experts indicated that treatment with dupilumab should be managed by a pediatric respirologist or allergy specialist with significant pediatric experience. In specific circumstances (i.e., once patient is stable or for patients who live in remote areas), management of asthma with dupilumab could be performed by a family physician in conjunction with an asthma specialist (e.g., linking through telehealth services). |
System and economic issues | |
The decision regarding the use of dupilumab as an add-on maintenance treatment in patients aged ≥ 12 years with severe asthma with a type 2 or eosinophilic phenotype or OCS-dependent asthma concluded without an agreement at the pCPA meeting on June 28, 2022. Mepolizumab, benralizumab, and omalizumab have agreements in place with the pCPA. | Comment from the drug plans to inform CDEC deliberations. |
CDEC = Canadian Drug Expert Committee; EOS = eosinophils; ER = emergency room; FeNO = fractional exhaled nitric oxide; ICS = inhaled corticosteroid; IgE = immunoglobulin E; LABA = long-acting beta agonist; LAMA = long-acting muscarinic antagonist; LTRA = leukotriene receptor antagonist; OCS = oral corticosteroid; pCPA = pan-Canadian Pharmaceutical Alliance.
Clinical Evidence
The clinical evidence included in the review of dupilumab is presented in 3 sections. The first section, the systematic review, includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those studies that were selected according to an a priori protocol. The second section includes indirect evidence from the sponsor and indirect evidence selected from the literature that met the selection criteria specified in the review. The third section includes sponsor-submitted long-term extension studies and additional relevant studies that were considered to address important gaps in the evidence included in the systematic review.
Systematic Review (Pivotal and Protocol Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of dupilumab (200 mg/1.14 mL [prefilled syringe)] and 300 mg/2 mL [prefilled syringe]) for the add-on maintenance treatment of severe asthma with a type 2 or eosinophilic phenotype or OCS-dependent asthma in patients aged 6 years to younger than 12 years.
Methods
Studies selected for inclusion in the systematic review included pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 5. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
Table 5: Inclusion Criteria for the Systematic Review
Criteria | Description | |
|---|---|---|
Population | Patients aged 6 to < 12 years with severe asthma with a type 2 or eosinophilic phenotype or with OCS-dependent asthma. | |
Subgroups:
|
| |
Intervention | Dupilumab as add-on therapy.a Dosing is based on patients’ body weight (subcutaneous administration):
| |
Comparator | Maintenance therapy with ICSs in combination with long-acting beta agonists alone or leukotriene receptor antagonists in combination with ≥ 1 of the following:
Note: Rescue medications (SABAs or SAMAs) may be part of any regimen for asthma. | |
Outcomes | Efficacy outcomes:
Harms outcomes: AEs, SAEs, WDAEs Notable harms: hypersensitivity reactions, helminth infections, conjunctivitis and keratitis, eosinophilic pneumonia, vasculitis consistent with eosinophilic granulomatosis with polyangiitis, injection site reactions | |
Study designs | Published and unpublished phase III and IV RCTs. | |
ACQ = Asthma Control Questionnaire; AE = adverse event; ED = emergency department; FEV1 = forced expiratory volume in 1 second; HRQoL = health-related quality of life; ICS = inhaled corticosteroid; IgE = immunoglobulin E; IL = interleukin; OCS = oral corticosteroid; PEF = peak expiratory flow; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; RCT = randomized controlled trial; SABA = short-acting beta agonist; SAE = serious adverse event; SAMA = short-acting muscarinic antagonist; WDAE = withdrawal due to adverse event.
aFor children (6 years to 11 years) with asthma, no initial loading dose is recommended.5
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy according to the PRESS Peer Review of Electronic Search Strategies checklist.16
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946–) via Ovid and Embase (1974–) via Ovid. All Ovid searches were run simultaneously as a multifile search. Duplicates were removed using Ovid deduplication for multifile searches, followed by manual deduplication in Endnote. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concepts were Dupixent (dupilumab) and asthma. Clinical trials registries were searched: the US National Institutes of Health’s clinicaltrials.gov, WHO’s International Clinical Trials Registry Platform search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
CADTH-developed search filters were applied to limit retrieval to RCTs or controlled clinical trials. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. Refer to Appendix 1 for the detailed search strategies.
The initial search was completed on July 27, 2022. Regular alerts updated the search until the meeting of the CADTH Canadian Drug Expert Committee on November 23, 2022.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool For Searching Health-Related Grey Literature checklist.17 Included in this search were the websites of regulatory agencies (US FDA and European Medicines Agency). Google was used to search for additional internet-based materials. Refer to Appendix 1 for more information on the grey literature search strategy.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
Findings From the Literature
One study was identified from the literature for inclusion in the systematic review (Figure 1). The included study is summarized in Table 6. A list of excluded studies is presented in Appendix 2.
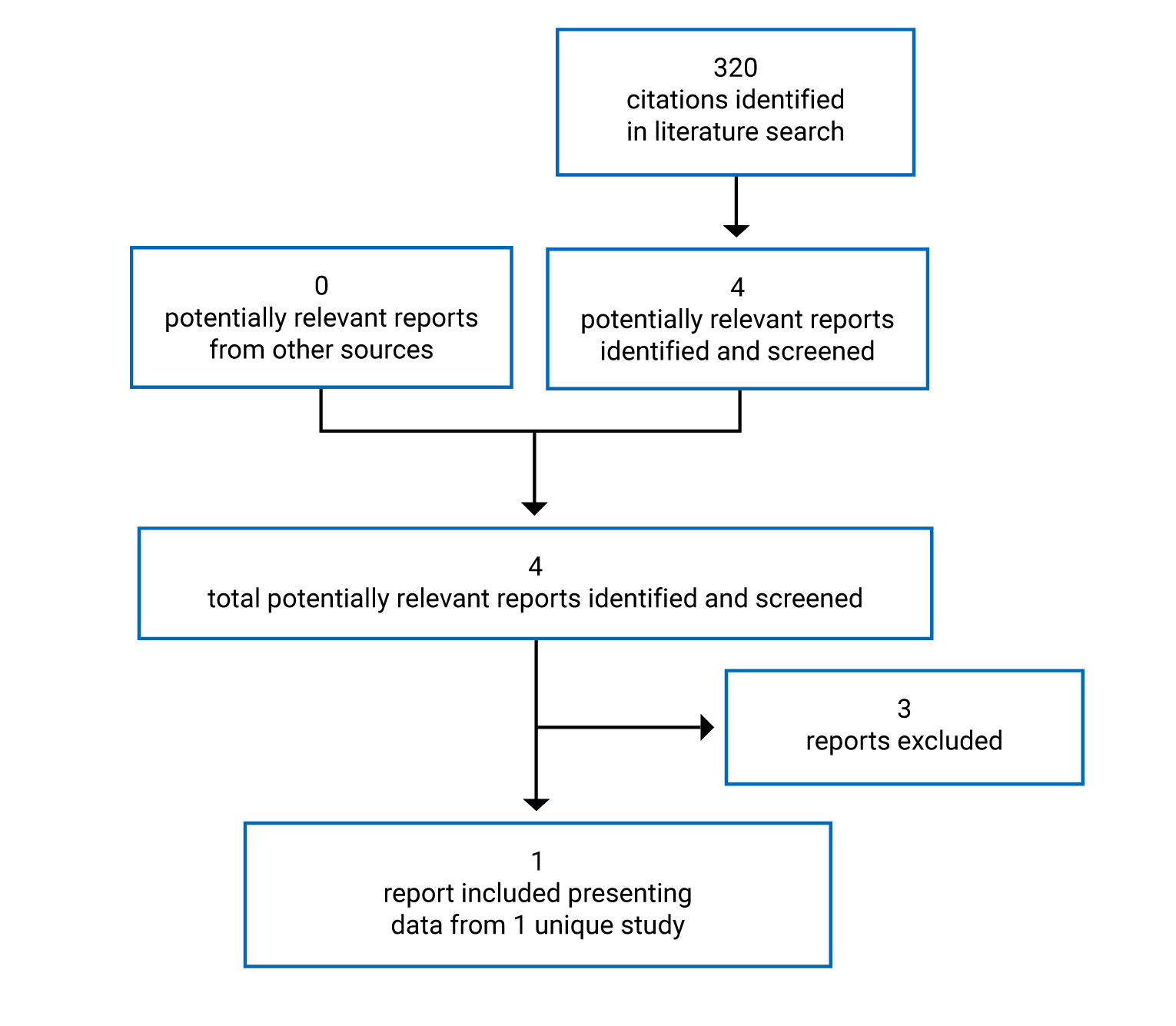
Table 6: Details of Included Studies
Trial details | VOYAGE |
|---|---|
Designs and populations | |
Study design | Multinational, multicentre, randomized, double-blind, phase III, placebo-controlled, parallel-group study |
Locations | 90 centres in 17 countries worldwide (Argentina, Australia, Brazil, Canada, Chile, Colombia, Hungary, Italy, Lithuania, Mexico, Poland, Russia, South Africa, Spain, Turkey, Ukraine, and the US). |
Patient enrolment dates | April 21, 2017, to August 26, 2020, (first patient randomized to last visit) |
Randomized (N) | 408 |
Inclusion criteria | Children aged 6 to < 12 years, with a physician diagnosis of persistent asthma for ≥ 12 months before screening, based on clinical history and examination, pulmonary function parameters according to GINA 2015 guidelines18 and the following criteria:
|
Exclusion criteria |
|
Drugs | |
Intervention | For patients weighing ≤ 30 kg at randomization: 100 mg in a 0.67 mL subcutaneous injection once q.2.w. For patients weighing > 30 kg at randomization: 200 mg in a 1.14 mL subcutaneous injection once q.2.w. |
Comparator(s) | For patients weighing ≤ 30 kg at randomization: 0.67 mL of matching placebo as a subcutaneous injection once q.2.w. For patients weighing > 30 kg at randomization: 1.14 mL of matching placebo as a subcutaneous injection once q.2.w. |
Duration | |
Phase | |
Screening period | 4 (± 1) weeks |
Treatment period | 52 weeks |
Posttreatment period | 12 weeks (for patients who choose not to participate in the 1-year long-term extension study) |
Outcomes | |
Primary end point | Annualized rate of severe exacerbation events during the 52-week placebo-controlled treatment period |
Secondary and exploratory end points | Key secondary end point: change from baseline in prebronchodilator percent predicted FEV1 at week 12 Other secondary end points:
Exploratory end points:
|
Notes | |
Publications | Bacharier et al. (2021)19 |
ACQ-5-IA = 5-item Asthma Control Questionnaire–Interviewer Administered; AE = adverse event; EQ-5D-Y = EQ-5D-Youth; FEF = forced expiratory flow; FeNO = fractional exhaled nitric oxide; FEV1 = forced expiratory volume in 1 second; FVC = forced vital capacity; GINA = Global Initiative for Asthma; ICS = inhaled corticosteroid; IgE = immunoglobulin E; IgG = immunoglobulin G; LABA = long-acting beta agonist; LAMA = long-acting muscarinic antagonist; LOAC = loss of asthma control; LTRA = leukotriene receptor antagonist; PACQLQ = Paediatric Asthma Caregiver's Quality of Life Questionnaire; PAQLQ(S)-IA = Standardised Paediatric Asthma Quality of Life Questionnaire–Interviewer Administered; PEF = peak expiratory flow; PRO = patient-reported outcome; PRQLQ-IA = Pediatric Rhinoconjunctivitis Quality of Life Questionnaire–Interviewer Administered; q.2.w. = every 2 weeks; SCS = systemic corticosteroid; TARC = thymus and activation regulated chemokine.
Source: Clinical Study Report.7
Description of Studies
The VOYAGE RCT was designed with a primary objective to assess the efficacy of add-on dupilumab compared to placebo in children aged 6 year to younger than 12 years with uncontrolled persistent asthma who were already receiving standard of care. Secondary goals of the VOYAGE trial were to investigate safety and tolerability of dupilumab, evaluate patient-reported outcomes and HRQoL, and assess systemic exposure and association between treatment and pediatric immune responses to vaccines.
There were 2 primary efficacy populations in the trial: the type 2 inflammatory population, defined as either a baseline blood eosinophil count of 150 cells/µL or greater or baseline FeNO of 20 ppb or greater; and a population with a baseline blood eosinophil count of 300 cells/µL or greater.
The study randomized patients in a 2:1 ratio to receive a subcutaneous injection of dupilumab or placebo. Patients were randomized from 90 centres in 17 countries, including Canada. Patients and investigators were blinded to the study treatment assignment, but not to the dose of the injections. Dupilumab and placebo were provided in identically matched prefilled syringes, labelled with a treatment kit number generated by the sponsor.
The randomized treatment kit number list was generated centrally using interactive voice response system or interactive web response system. Randomization was stratified by ICS dose level (medium or high) at screening, blood eosinophil count (< 300 cells/µL and ≥ 300 cells/µL) at screening, and region (Latin America, Eastern Europe, and Western countries).
The study consisted of 3 periods (Figure 2):
Screening (4 ± 1 weeks) to collect baseline data on asthma control and assure eligibility criteria
Randomized double-blind treatment (up to 52 weeks)
Posttreatment period (12 weeks)
Eligible patients who completed the randomized treatment period of the study were offered the opportunity to participate in the 1-year long-term extension study, EXCURISON.20 Patients who enrolled in the extension study were excluded from the posttreatment period of the trial.
The first patient was randomized on April 21, 2017, and the last patient completed the study on August 26, 2020. According to the VOYAGE trial protocol, the database lock was planned based on the time when all randomized patients completed the week 52 visit or discontinued from the study before week 52.
Figure 2: Study Design of the VOYAGE Trial
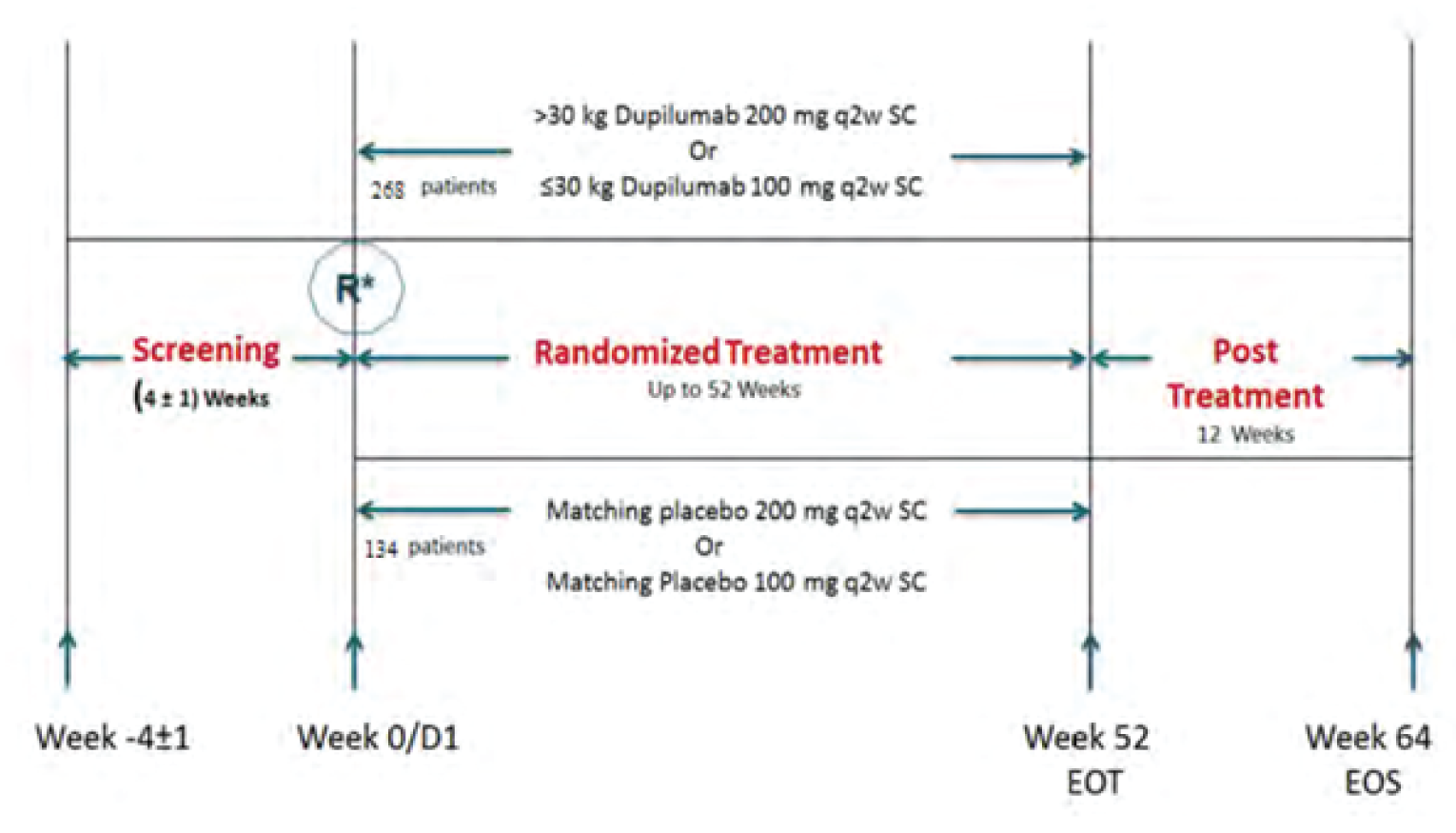
D = day; EOS = end of study; EOT = end of treatment; ICS = inhaled corticosteroids; q.2.w. = every 2 weeks; R = randomization; SC = subcutaneous.
Note: Background medications include medium-dose ICS plus second controller, or high-dose ICS alone, or high-dose ICS plus second controller.
Source: Clinical Study Report.7
Populations
Inclusion and Exclusion Criteria
VOYAGE included patients with asthma aged 6 years to younger than 12 years who were on existing background therapy of medium-dose ICSs in combination with a second controller (i.e., LABA, LTRA, long-acting muscarinic antagonist [LAMA], or methylxanthines) or high-dose ICSs alone or in combination with a second controller for at least 3 months and on a stable dose of at least 1 month before the study. Patients had to have a prebronchodilator FEV1 of 95% or less of predicted normal or postbronchodilator reversibility of at least 10% or greater, showed evidence of uncontrolled asthma during the screening period and had either been treated with an SCS or been hospitalized or visited an ED for worsening asthma within the past year.
Patients were excluded if their body weight was less than 16 kg, if they had other chronic lung diseases or history of life-threatening asthma (e.g., requiring intubation), and if they had a history of any malignancies or current comorbidities that may interfere with the drug under study. Moreover, nonadequate adherence with background therapy during the screening period (< 80% of total number of prescribed doses) was also an exclusion criterion in the trial.
Baseline Characteristics
In the VOYAGE trial, median age of patients was 9 years (range = 6 to 11) across type 2 inflammatory asthma phenotype and population with baseline blood EOS of 300 cells/µL or greater. Most patients were male (range = 64.4% to 69% across the study groups in the 2 efficacy populations), White (range = 86.3% to 89.5% across the study groups in the 2 efficacy populations) and weighed more than 30 kg (range = 66.7% to 68.4% across the study groups in the 2 efficacy populations). Within the type 2 inflammatory asthma phenotype population, a greater proportion of individuals had blood eosinophilic counts of 300 cells/µL or greater (74.2% in the dupilumab and 73.7% in the placebo group). Although slight imbalances were observed across the groups in terms of number severe exacerbations (1, 2, 3, or ≥ 4) in the year before the study enrolment, the average number of exacerbations (mean [SD]) reported were 2.61 (2.58) in the dupilumab and 2.18 (1.55) in the placebo groups for the type 2 asthma population, and 2.78 (2.90) in the dupilumab and 2.37 (1.71) in the placebo groups for the population with baseline blood EOS of 300 cells/µL or greater. FEV1 reversibility was larger in the dupilumab than placebo group, with a mean (SD) of 21.5% (21.37) versus 15.81 (16.4) in the type 2 asthma population, and with a mean (SD) of 22.9% (23.23) versus 16.2 (15.8) in the population with baseline blood EOS of 300 cells/µL or greater. Regarding the ICS dosage, approximately 43% of patients were on a high-dose ICS and 55% were on a medium-dose ICS within the type 2 asthma population. In the population with baseline blood EOS of 300 cells/µL or greater, approximately 45% of patients were using high doses of an ICS (dupilumab versus placebo: 42.3% versus 48.8%), and approximately 53% of patients were using medium doses of an ICS (dupilumab versus placebo: 56.0% versus 51.2%). Three patients in the dupilumab group were on a low-dose ICS, which was a protocol violation (Table 7).
Baseline characteristics of other efficacy populations (intention-to-treat [ITT] and baseline EOS ≥ 150 cells/µL populations) are presented in the Appendix 3.
Table 7: Summary of Baseline Characteristics
Trial, Population, and Characteristic | VOYAGE trial | |||
|---|---|---|---|---|
Type 2 inflammatory asthma phenotype population | Baseline blood eosinophils ≥ 300 cells/µL population | |||
Dupilumab n = 236 | Placebo n = 114 | Dupilumab n = 175 | Placebo n = 84 | |
Age, years, mean (SD) | 8.9 (1.6) | 9.0 (1.6) | 8.9 (1.6) | 9.0 (1.5) |
Median, range | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
Male | 152 (64.4) | 78 (68.4) | 116 (66.3) | 58 (69.0) |
Race | ||||
White | 208 (88.1) | 102 (89.5) | 151 (86.3) | 75 89.3) |
Black/African descent | 9 (3.8) | 5 (4.4) | 8 (4.6) | 5 (6.0) |
Asian | 2 (0.8) | 0 (0.0) | 2 (1.1) | 0 (0.0) |
American Indian or Alaska Native | 1 (0.4) | 0 (0.0) | 1 (0.6) | 0 (0.0) |
Other | 16 (6.8) | 7 (6.1) | 13 (7.4) | 4 (4.8) |
Weight, kg, mean (SD) | 35.60 (10.00) | 37.08 (11.61) | 35.45 (9.98) | 36.97 (11.70) |
Weight, range | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
Weight group, kg | ||||
≤ 30 kg | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
> 30 kg | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
Regiona | ||||
East Europe | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
Latin America | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
Western countries | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
Blood eosinophils, giga cells/L, mean (SD) | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
Range | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
Eosinophil group, cells/µL | ||||
< 150 | 13 (5.5) | 6 (5.3) | 0 (0.0) | 0 (0.0) |
150 to < 300 | 48 (20.3) | 24 (21.1) | 0 (0.0) | 0 (0.0) |
≥ 300 | 175 (74.2) | 84 (73.7) | 175 (100) | 84 (100) |
FeNO, parts per billion, n | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
Mean (SD) | 31.83 (24.85) | 28.38 (23.44) | 34.53 (25.83) | 31.33 (23.52) |
FeNO group, parts per billion | ||||
< 20 | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
≥ 20 < 35 | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
≥ 35 | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
Severe exacerbations, past 12 monthsb | ||||
Mean (SD) | 2.61 (2.58) | 2.18 (1.55) | 2.78 (2.90) | 2.37 (1.71) |
1 | 85 (36.0) | 47 (41.2) | 64 (36.6) | 32 (38.1) |
2 | 75 (31.8) | 32 (28.1) | 53 (30.3) | 20 (23.8) |
3 | 29 (12.3) | 21 (18.4) | 17 (9.7) | 19 (22.6) |
≥ 4 | 47 (19.9) | 14 (12.3) | 41 (23.4) | 13 (15.5) |
Total IgE, IU/mL, n | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
Mean (SD) | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
Ongoing atopic medical condition | 226 (95.8) | 103 (90.4) | 171 (97.7) | 79 (94.0) |
Ongoing atopic dermatitis | 95 (40.3) | 41 (36.0) | 80 (45.7) | 35 (41.7) |
Ongoing allergic conjunctivitis | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
Ongoing allergic rhinitis | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
Time since asthma diagnosis, years, mean (SD) | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
Range | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
Prebronchodilator FEV1, L, mean (SD) | 1.48 (0.39) | 1.53 (0.46) | 1.45 (0.39) | 1.52 (0.48) |
Prebronchodilator FEV1, percent predicted, mean (SD) | 77.66 (14.38) | 78.36 (14.51) | 76.37 (14.60) | 77.87 (15.19) |
Postbronchodilator FEV1, L, n | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
Mean (SD) | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
FEV1 reversibility, percent, mean (SD) | 21.47 (21.37) | 15.81 (16.38) | 22.94 (23.23) | 16.20 (15.76) |
Baseline a.m. PEF, L/min, mean (SD) | 196.35 (67.63) | 187.96 (57.49) | 192.32 (67.62) | 185.87 (59.12) |
Baseline p.m. PEF, L/min, mean (SD) | 203.27 (65.78) | 198.72 (57.53) | 199.97 (65.44) | 197.06 (59.70) |
ACQ-5-IA score, mean (SD) | 2.18 (0.79) | 2.15 (0.84) | 2.18 (0.83) | 2.17 (0.86) |
ACQ-7-IA score, mean (SD) | 2.15 (0.70) | 2.12 (0.76) | 2.16 (0.73) | 2.15 (0.77) |
ICS dose at baseline | ||||
High | 102 (43.2) | 50 (43.9) | 74 (42.3) | 41 (48.8) |
Medium | 131 (55.5) | 64 (56.1) | 98 (56.0) | 43 (51.2) |
Low | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
ACQ-5-IA = 5-item Asthma Control Questionnaire–Interviewer Administered; ACQ-7-IA = 7-item Asthma Control Questionnaire–Interviewer Administered; FeNO = fractional exhaled nitric oxide; FEV1 = forced expiratory volume in 1 second; ICS = inhaled corticosteroid; IgE = immunoglobulin E; PEF = peak expiratory flow; SD = standard deviation.
Notes: Values are n (%) unless otherwise indicated.
A patient is considered to have an ongoing atopic medical condition if they have any of the following ongoing conditions: atopic dermatitis, allergic conjunctivitis, allergic rhinitis, eosinophilic esophagitis, food allergy, or hives or has baseline total IgE of 100 IU/mL or greater and at least 1 aeroantigen-specific IgE is positive (≥ 0.35 IU/mL) at baseline.
aLatin America includes Argentina, Brazil, Colombia, Chile, and Mexico; Eastern Europe includes Poland, Hungary, Romania, Lithuania, Russia, Ukraine, and Turkey; Western Countries include Australia, Canada, Italy, South Africa, Spain, and US.
bSevere asthma exacerbation before the study is defined as any treatment with 1 systemic (oral or parenteral) steroid burst or more for worsening asthma or hospitalization or an emergency/urgent medical care visit for worsening asthma.
Source: Clinical Study Report.7
Interventions
In the VOYAGE trial, the dupilumab 100 mg dose was administered subcutaneously in a 0.67 mL syringe (for patients with a body weight ≤ 30 kg) and the 200 mg dose was administered in a 1.14 mL syringe (for patients with a body weight > 30 kg), once every 2 weeks. To maintain blinding, the 100 mg dose was matched to a 0.67 mL placebo and the 200 mg dose was matched to a 1.14 mL placebo delivered once every 2 weeks.
After week 12, parents, caregivers, and legal guardians of patients were allowed to perform home administration of dupilumab if they elected to do so and if they followed a training demonstration and completed administration under close supervision by the investigator or delegate at not less than 3 visits.
Temporary treatment discontinuation could be considered by the investigator because of AEs, infections or infestations that do not respond to medical treatment, and laboratory abnormalities. Re-initiation of treatment was conducted after close monitoring, once clinical consideration that the AEs occurrence was not related to the treatment under study was complete and if the eligibility criteria for the study were still met.
Permanent discontinuation from the treatment and the study may have occurred on patients’ or patients’ legal representative request or based upon clinical investigators’ decision. Moreover, patients’ study withdrawal occurred for the following additional reasons: specific request from the sponsor, in case of protocol deviation, pregnancy, occurrence of anaphylactic reactions or systemic allergic reactions related to the treatment under study, malignancy diagnosis, opportunistic infections, levels of serum alanine transaminase greater than 3 times the upper limit of normal, and total bilirubin greater than 2 times the upper limit of normal.
After permanent treatment discontinuation, patients were encouraged to complete the remaining study visits according to the visit schedule until the end of study or up to recovery or stabilization of any AEs.
Background medications, including ICSs and second controller and reliever medications were administered during the trial and their usage was recorded. Patients supplied their controller medication.
Prior to study entry, patients needed to be on a stable dose of a medium-dose ICS with a second controller medication, high-dose ICS alone, or high-dose ICS for at least 3 months with at least 1 month of stable dose before the first visit. Dosing levels considered as medium- or high-dose ICS in children aged 6 years to younger than 12 years were aligned with the GINA guidelines, 2015 version.18 Only 1 second controller medication (LABA, LTRA, LAMA, or methylxanthines) for combined use with medium- or high-dose ICS was permitted. Use of a reliever medication other than albuterol/salbutamol or levalbuterol/levosalbutamol was discouraged.
During the treatment period, patients were on their baseline dose regimen of the controller medication(s) used during screening. Patients who experienced a deterioration of asthma during the trial were allowed to have an up to 4-fold increase in their ICS dose temporarily, for a maximum of 10 days (recorded as a loss of asthma control event). At that point, treatment could have been changed to SCSs (severe exacerbation event) or reverted to the original ICS dose depending on the progression of symptoms. Permanent adjustments to the controller medication dosing (i.e., step up in medium- to high-dose ICS or addition of second controller for patients on high-dose ICS monotherapy) were allowed only if the patient experienced 2 or more severe exacerbations events at any time during the trial. SCSs were allowed at any time in case of clinical symptoms of severe asthma exacerbation event, per the judgment of the study investigator.
During the posttreatment period, patients not continuing with the long-term open-label extension study, were treated with the controller medication regimen and dose used during the randomized treatment period. Adjustments were possible based on patients’ status and clinical judgment of the investigator.
Outcomes
A list of efficacy end points identified in the CADTH review protocol that were assessed in the clinical trials included in this review is provided in Table 8. These end points are further summarized in the following. A detailed discussion and critical appraisal of the outcome measures is provided in Appendix 4.
Table 8: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure (CADTH protocol) | VOYAGE trial | Adjusted for multiplicity | |
|---|---|---|---|
Description and type of outcome (primary, secondary, or exploratory) | Population assessed | ||
Mortality | Reported under harms | Safety population | No |
Asthma exacerbations:
| Annualized rate of severe exacerbation events during the 52-week placebo-controlled treatment period (primary) | Type 2 inflammatory asthma phenotype Baseline blood eosinophils ≥ 300 cells/µL Baseline blood eosinophils ≥ 150 cells/µL Baseline FeNO ≥ 20 parts per billion Full ITT | Yes |
Time to first severe exacerbation event during 52-week treatment period (secondary) Severe exacerbation leading to hospitalization/ED visit (post hoc) | Type 2 inflammatory asthma phenotype Baseline blood eosinophils ≥ 300 cells/µL | No | |
Asthma symptoms | Change from baseline in ACQ-7 (secondary) | Type 2 inflammatory asthma phenotype Baseline blood eosinophils ≥ 300 cells/µL Baseline blood eosinophils ≥ 150 cells/µL Baseline FeNO ≥ 20 parts per billion Full ITT | Only at week 24 |
Change from baseline in ACQ-5 (secondary) | Type 2 inflammatory asthma phenotype Baseline blood eosinophils ≥ 300 cells/µL | No | |
Reduction in use of OCS | Total systemic (oral or parenteral) corticosteroid exposure (secondary) | Type 2 inflammatory asthma phenotype Baseline blood eosinophils ≥ 300 cells/µL Baseline blood eosinophils ≥ 150 cells/µL Baseline FeNO ≥ 20 parts per billion Full ITT | No |
Change in pulmonary function (e.g., PEF, FEV1) | Change from baseline in prebronchodilator percent predicted FEV1 at week 12 (key secondary) | Type 2 inflammatory asthma phenotype Baseline blood eosinophils ≥ 300 cells/µL Baseline blood eosinophils ≥ 150 cells/µL Baseline FeNO ≥ 20 parts per billion Full ITT | Yes |
Change from baseline in prebronchodilator percent predicted FEV1 at weeks 2, 4, 8, 24, 36, and 52 (secondary) | Type 2 inflammatory asthma phenotype Baseline blood eosinophils ≥ 300 cells/µL Baseline blood eosinophils ≥ 150 cells/µL Baseline FeNO ≥ 20 parts per billion Full ITT | No | |
Change from baseline in morning and evening PEF (secondary) | Type 2 inflammatory asthma phenotype Baseline blood eosinophils ≥ 300 cells/µL | No | |
ICS dose reduction | The proportion of patients requiring a permanent step up in background controller medication (step up in medium- to high-dose ICS or addition of second controller for patients on high-dose ICS monotherapy) after ≥ 2 severe asthma exacerbation events (exploratory) | Type 2 inflammatory asthma phenotype Baseline blood eosinophils ≥ 300 cells/µL Baseline blood eosinophils ≥ 150 cells/µL Baseline FeNO ≥ 20 parts per billion Full ITT | No |
HRQoL | Change from baseline in PAQLQ(S)-IA) at weeks 12, 24, 36, 52, and 64 (secondary) Change from baseline in PACQLQ (exploratory) Change from baseline in EQ-5D-Y (exploratory) | Type 2 inflammatory asthma phenotype Baseline blood eosinophils ≥ 300 cells/µL | No |
Reduction in use of rescue medication | Use of reliever medication (secondary) | Type 2 inflammatory asthma phenotype Baseline blood eosinophils ≥ 300 cells/µL | No |
Improvements in symptoms of atopic dermatitis and rhinosinusitis | Change from baseline in PRQLQ-IA (exploratory) | Type 2 inflammatory asthma phenotype Baseline blood eosinophils ≥ 300 cells/µL | No |
Harms (AEs, SAEs, WDAEs, notable harms) | AEs, SAEs, and AEs of special interest | Safety population | No |
ACQ-5 = 5-item Asthma Control Questionnaire; ACQ-7 = 7-item Asthma Control Questionnaire; AE = adverse event; ED = emergency department; EQ-5D-Y = EQ-5D-Youth; FeNO = fractional exhaled nitric oxide; FEV1 = forced expiratory volume in 1 second; HRQoL = health-related quality of life; ICS = inhaled corticosteroid; ITT = intention to treat; OCS = oral corticosteroid; PEF = a.m./p.m. peak expiratory flow; PACQLQ = Paediatric Asthma Caregiver's Quality of Life Questionnaire; PAQLQ(S)-IA = Standardised Paediatric Asthma Quality of Life Questionnaire–Interviewer Administered; PRQLQ-IA = Pediatric Rhinoconjunctivitis Quality of Life Questionnaire–Interviewer Administered; SAE = serious adverse event; WDAE = withdrawal due to adverse event.
Source: Clinical Study Report.7
Severe Exacerbation Events
The annualized rate of severe asthma exacerbations during the 52-week period was the primary outcome of the VOYAGE trial. Severe exacerbations were defined as a deterioration in asthma that required the use of SCSs for at least 3 days or resulted in hospitalizations or ED visits requiring SCSs. Two events were considered as different if the interval between their start dates was at least 28 days. The reasons behind any exacerbation event (e.g., infections including viral and bacterial, allergen exposure, exercise, and others) were recorded in the electronic case report form. Study investigators were responsible for maintaining accurate and timely electronic case report forms, which were initially designed by the sponsor.
Patients who permanently discontinued the study medication were asked and encouraged to continue with all the remaining study visits and their additional off-treatment severe exacerbation events up to week 52 were recorded. Patients who discontinued treatment were not eligible for the 1-year long-term extension study.
Pulmonary Function
Spirometry was performed following American Thoracic Society/European Respiratory Society 2005 guidelines.21 The key secondary outcome was change from baseline in prebronchodilator percent predicted FEV1 at week 12, while the measurements at other time points (weeks 2, 4, 8, 24, 36, and 52) were considered secondary end points. Spirometry was to be performed preferably during morning hours and at the same time every day, using the same spirometer and standardized techniques.
Measurements of morning and evening peak expiratory flow (PEF) were recorded through daily assessments and collection using an electronic diary and PEF metre by caregiver(s) of the patients. Both morning and evening PEF measurements were performed before intake of any reliever medications. Three values were collected and the highest 1 was used for evaluation.
Electronic diaries were also used to record the number of inhalations of relievers used for symptom relief in a day, number of inhalations of background medication, and record OCS use for an exacerbation event.
ACQ-7-IA and 5-item ACQ–Interviewer Administered
The ACQ is a questionnaire designed to assess the degree of asthma control with questions on symptoms (completed by the patient and/or their caregiver), including activity limitation, nocturnal waking, shortness of breath, wheezing, and symptoms on waking. Moreover, the average number of daily doses of rescue inhaler used, as well as spirometry measured as FEV1 (completed by clinic staff, including prebronchodilator use, percent and percent predicted use). The ACQ-7 has 7 items with each measured on a 7-point scale scored from 0 (totally controlled) to 6 (severely uncontrolled). Clinical staff score the percent predicted FEV1 on a 7-point scale based on the spirometry result.22-24 The ACQ-7 score is computed as the unweighted mean of the responses to the 7 questions. Changes of at least 0.5 are considered to be clinically meaningful.24,25 ACQ assessments were conducted every 2 weeks up to the week 12, and then every 4 weeks until end of study visit of the VOYAGE trial.
The 5-item ACQ–Interviewer Administered (ACQ-5-IA) scores were deduced from the responses to the first 5 questions of ACQ-7-IA and were applied to patients aged 6 years to younger than 12 years in the VOYAGE trial.
PAQLQ(S)–Interviewer Administered
The PAQLQ(S) disease-specific instrument comprises 23 items across 3 domains: symptoms (10 items), activity limitation (5 items), and emotional function (8 items), which are graded on a 7-point Likert scale (1 = maximum impairment, 7 = no impairment).A standardized format of the instrument was used in the trial, which included generic activity questions (physical activity, activities with animals, and activities with friends and family). It was applied to patients who were aged 7 years and older in the VOYAGE trial. A global score ranges from 1 to 7, with higher score representing better HRQoL.26-28 The MID of approximately 0.5 was identified for the overall quality of life score.27 Assessments during VOYAGE trial were conducted every 12 weeks until end of study visit.
Paediatric Asthma Caregiver's Quality of Life Questionnaire
The PACQLQ is a self-administered 13-item questionnaire capturing the impact of child’s asthma on quality of life of the parent(s), caregiver(s), and legal guardian(s). The items are organized in 2 domains (4 items concern activity limitations and 9 concern emotional function). Responses to the individual items are given on a 7-point Likert scale where 1 (all of the time/very very worried or concerned) represents severe impairment and 7 (none of the time/not worried or concerned) represents no impairment. In the VOYAGE trial, the instrument was administered among parent(s), caregiver(s), and legal guardian(s) of children aged 7 years to younger than 12 years of age. Both domain and overall scores range from 1 to 7, with higher score representing better HRQoL.27,29 The estimated MID identified through the CADTH literature search is 0.5 for the overall score.27 Assessments during VOYAGE trial were conducted every 12 weeks until end of study visit.
Pediatric Rhinoconjunctivitis Quality of Life Questionnaire–Interviewer Administered in Patients With Comorbid Allergic Rhinitis
Pediatric Rhinoconjunctivitis Quality of Life Questionnaire (PRQLQ)–Interviewer Administered is a disease-specific instrument developed to measure HRQoL in children aged 6 years to younger than 12 years diagnosed with seasonal allergic rhinoconjunctivitis or hay fever. Twenty-three items of the instrument are organized into 5 domains (nose symptoms, eye symptoms, practical problems, activity limitation, and other symptoms), which are assessed on a 7-point Likert scale (0 = no impairment, 6 = maximum impairment). Both domain and overall scores range from 0 to 6, with higher score representing worse HRQoL.30,31 The CADTH literature search could not identify studies reporting MID values for this instrument. Assessments during VOYAGE trial were conducted every 12 weeks until end of study visit.
EQ-5D-Youth
The EQ-5D-Youth (EQ-5D-Y) is a generic preference-based HRQoL measure that contains a descriptive system comprising of 5 dimensions using child-friendly wording (mobility, looking after myself, doing usual activities, having pain or discomfort, feeling worried, sad or unhappy), each of which has 3 levels (no problems, some problems, a lot of problems). The next component of the instrument consists of a visual analogue scale (VAS) on which the respondent rates their perceived health from 0 to 100, with respective anchors of “worst imaginable health state” and “best imaginable health state.”32,33 Patients enrolled in the VOYAGE trial, who could read, were encouraged to complete the questionnaire by themselves.
The instrument produces 3 types of data for each respondent, a profile indicating the extent of problems on each of the 5 dimensions represented by a 5-digit descriptor, a population preference-weighted health index score based on the descriptive system, and a self-reported assessment of health status based on the VAS. The index score is arrived at by applying a multiattribute utility function to the descriptive system. Scores of 0 represent the health state “dead” and 1 represents “perfect health.” Lower scores on 5-digit health status and higher scores on index and VAS represent better HRQoL.32,33 No MID specific to asthma in children was identified during the CADTH literature review. Assessments during VOYAGE trial were conducted on weeks 2, 24, and 52.
Harms
For each patient, SAEs and AEs of special interest were monitored and documented from the initial visit until the end of the study or rollover to the extension study. AEs that were ongoing at database lock were also captured. An independent data monitoring committee was reviewing the safety data on a periodic basis throughout the course of the trial.
Statistical Analysis
Primary Outcome of the Study
Power Calculation
The sample size of this study was based on the primary outcome (i.e., annualized rate of severe exacerbations over 52 weeks of treatment) for the following populations: baseline blood EOS of 300 cells/µL or greater, baseline blood EOS of 150 cells/µL or greater, and patients with type 2 inflammatory phenotype (baseline blood EOS ≥ 150 cells/µL or baseline FeNO ≥ 20 ppb).
The following assumptions were made: the number of severe exacerbations follows a negative binomial distribution, a randomization ratio of 2:1 was used, and a linear discontinuation rate of 20% at 1 year (i.e., average exposure duration for patients of 0.9 year). Moreover, the assumed RR reductions were based on the results in the phase III study EFC13579 (QUEST) for dupilumab in adolescent and adult patients with asthma.34
For patients with baseline EOS of 300 cells/µL or greater, assuming a placebo annualized severe exacerbation rate of 0.8 and a dispersion parameter of 1.5, with approximately 255 patients randomized (170 for dupilumab and 85 for matching placebo group), there was 96% power to detect a RR reduction of 60% (i.e., annualized rate of 0.32 for the dupilumab group) at a 2-sided significance of 5%.
For patients with baseline EOS of 150 cells/µL or greater, assuming a placebo annualized severe exacerbation rate of 0.7 and a dispersion parameter of 1.5, with approximately 327 patients randomized (218 for dupilumab and 109 for matching placebo group), there was 93% power to detect a RR reduction of 54% (i.e., annualized rate of 0.322 for the dupilumab group) at a 2-sided significance of 5%.
For patients with type 2 inflammatory phenotype, assuming a placebo annualized severe exacerbation rate of 0.7 and a dispersion parameter of 1.5, with approximately 345 patients randomized (230 for dupilumab and 115 for matching placebo group), there was 94% power to detect a RR reduction 54% (i.e., annualized rate of 0.322 for the dupilumab group) at a 2-sided significance of 5%.
According to the sponsor, approximately 402 patients in the overall population (268 for dupilumab and 134 for placebo) needed to be randomized to achieve target sample sizes, with an assumption that 86%, 81%, and 64% of patients have a type 2 inflammatory phenotype, baseline blood EOS of 150 cells/µL or greater, and baseline blood EOS of 300 cells/µL or greater, respectively.
Multiplicity Considerations
In VOYAGE, a hierarchical testing strategy was implemented to test for superiority of dupilumab over placebo in the primary (annualized rate of severe exacerbation events during the 52-week placebo-controlled treatment period), key secondary (change from baseline in prebronchodilator percent predicted FEV1 at week 12), and secondary outcomes (change in ACQ-7-IA at week 24, change from baseline in FeNO at week 12), while controlling the overall type I error rate at 0.05 (2-sided). The sponsor included 2 distinct sequential testing procedures, based on approved indication in adults and adolescents.
For the US and US reference countries: testing hierarchy started with the population with a baseline blood EOS of 300 cells/µL or greater (Appendix 3; Table 40)
For European Union and European Union reference countries: testing hierarchy started with the population with a type 2 inflammatory phenotype (Appendix 3; Table 41)
Statistical Test or Model
The analysis of the primary end point estimated annualized exacerbation rates over 52 weeks for treatment groups as well as the RR, 95% CIs, and P values between treatment and placebo arms, and was performed using a negative binomial model. Details of the statistical model, adjustments, and populations analyzed are provided in Table 9. In the primary analysis approach, off-treatment measurements of patients who prematurely discontinued treatment were included in the analysis since patients who permanently discontinued study medication were encouraged to complete remaining study visits. For patients discontinuing the study before week 52, analyses were censored at the time of study discontinuation, and all observed severe exacerbation events up to the last contact date were included in the analysis. No imputations were performed for the unobserved events after study discontinuation and up to week 52.
An assessment of time to first severe asthma exacerbation event was conducted via Cox regression model that yielded hazard ratio estimates (dupilumab versus placebo) along with its 95% CIs. Kaplan-Meier curves were used to derive the proportion of patients with a severe asthma exacerbation event at various time points in each treatment group (Table 9).
Preplanned sensitivity analyses were conducted on the primary outcome in the VOYAGE trial for the type 2 inflammatory phenotype and baseline blood EOS of 300 cells/µL or greater populations (Refer to Table 9).
Key Secondary Outcome
The change from baseline in prebronchodilator percent predicted FEV1 at week 12 was compared to placebo using a mixed-effect model with repeated measures (MMRM), and LS mean of each treatment group as well as difference of LS means between treatment and placebo arms, 95% CIs, and P values were reported. Details of the statistical model, adjustments, and populations analyzed is provided in Table 9.
The primary analytical approach included additional off-treatment percent predicted FEV1 values measured up to week 12 for patients discontinuing the treatment before week 12, who were encouraged to complete all remaining study visits. Data from patients who withdrew from the study before week 12 were considered as missing after study discontinuation and no imputation was performed in the primary analysis.
Preplanned sensitivity analyses were conducted on the key secondary outcome in the VOYAGE trial for the type 2 inflammatory phenotype and baseline blood EOS of 300 cells/µL or greater populations (Refer to Table 9).
Subgroup Analyses
Preplanned subgroup analyses were conducted on the primary and key secondary outcome in the VOYAGE trial for the following subgroups identified in the CADTH review protocol:
baseline eosinophil counts ( < 300 cells/µL, ≥ 300 cells/µL; < 150 cells/µL, ≥ 150 cells/µL; < 150 cells/µL, ≥ 150 to < 300 cells/µL, ≥ 300 to < 500 cells/µL, ≥ 500 cells/µL) (performed in the ITT population)
ICS dose at baseline (medium or high) (performed in the type 2 inflammatory asthma phenotype population and baseline blood EOS ≥ 0.3 Giga/L population)
Number of exacerbations in the past year (≤ 1, 2, > 2) (performed in the type 2 inflammatory asthma phenotype population and baseline blood EOS ≥ 0.3 Giga/L population)
Atopic medical condition (yes, no) and comorbid atopic dermatitis (yes, no) (performed in the type 2 inflammatory asthma phenotype population and baseline blood EOS ≥ 0.3 Giga/L population)
For the primary outcome, treatment by subgroup interaction and its P value was derived from a negative binomial model. Similarly, treatment by subgroup interaction at week 12 and its P value was derived from an MMRM for the key secondary outcome of interest. RRs and risk differences for the primary outcome, and LS mean differences for the key secondary outcome comparing dupilumab versus placebo for the subgroups were reported. In both cases, evidence of quantitative treatment by subgroup interaction with nominal P < 0.05 for any subgroup would follow-up with an assessment of qualitative interaction via Gail-Simon test.
In VOYAGE, the specific subgroup of patients with eosinophil counts of 150 cells/μL or greater, with eosinophil counts of 300 cells/μL or greater, and with high ICS dosing at baseline were included as part of the statistical hierarchy. All other subgroups were not adjusted for multiplicity.
Other Secondary Outcomes
Analyses of change from baseline in other secondary end points with a continuous nature was derived through a similar MMRM (Table 9).
In addition to the MMRM analyses, a responder analysis was performed for the ACQ-7, 5-item ACQ (ACQ-5), and Asthma Quality of Life Questionnaire total scores at weeks 12, 24, 36, and 52. Specifically, a logistic regression model was used to obtain the odds ratio (OR) of being a responder comparing dupilumab and placebo group along with the corresponding 95% CI and P value (Table 9).
Analysis Populations
In the VOYAGE trial, there were 2 primary efficacy populations, for which all efficacy outcomes were analyzed:
Type 2 inflammatory asthma phenotype (randomized patients with baseline blood eosinophil count ≥ 150 cells/µL or baseline FeNO ≥ 20 ppb)
Baseline blood EOS count of 300 cells/µL or greater (randomized patients with baseline blood EOS count ≥ 0.3 Giga/L)
There were additional efficacy populations, for which primary and selected secondary end points were analyzed in a multiplicity-controlled manner, including patients with baseline blood eosinophil count of 150 cells/µL or greater.
Safety analyses were conducted according to the treatment patients actually received and were based on the safety population (i.e., all patients receiving at least 1 dose or part of a dose of the trial treatment).
Table 9: Statistical Analysis of Efficacy End Points
Outcome | End point assessed | Statistical model | Adjustment factors | Sensitivity analyses | |
|---|---|---|---|---|---|
Population | Adjustment factors | ||||
VOYAGE | |||||
Severe exacerbation events | Annualized rate of severe exacerbations during the 52-week period | Negative binomial regression analysis (dependent variable: total number of events that occur during the observation period; independent variable: treatment arm; offset variable: log-transformed observation duration) | Type 2 inflammatory phenotype; baseline blood eosinophils ≥ 150 cells/µL; full ITT | Age, baseline weight (≤ 30 kg, > 30 kg), region, baseline eosinophil level (< 0.3 Giga/L, ≥ 0.3 Giga/L), baseline FeNO level (< 20 ppb, ≥ 20 ppb), baseline ICS dose level (medium or high), and number of severe exacerbation events within 1 year before the study |
|
Baseline blood eosinophils ≥ 300 cells/µL population | Age, baseline weight (≤ 30 kg, > 30 kg), region, baseline FeNO level (< 20 ppb, ≥ 20 ppb), baseline ICS dose level (medium or high), and number of severe exacerbation events within 1 year before the study | None | |||
Time to first severe exacerbation during the 52-week treatment period | Cox proportional hazards model (dependent variable: time to the first event; independent variable: treatment arm;). Kaplan-Meier method used to derive the probabilities that a patient would experience events up to week 12, 24, 36, and 52 per each treatment group | Type 2 inflammatory phenotype | age, baseline weight (≤ 30 kg, > 30 kg), region, baseline eosinophil level, baseline FeNO level, baseline ICS dose level, and number of severe exacerbation events within 1 year before the study | None | |
Baseline blood eosinophils ≥ 300 cells/µL population | age, baseline weight (≤ 30 kg, > 30 kg), region, baseline FeNO level, baseline ICS dose level, and number of severe exacerbation events within 1 year before the study | None | |||
Pulmonary function (FEV1) | Change from baseline in percent predicted prebronchodilator FEV1 at week 12 (and other time points up to week 52) | MMRM (dependent variable: change from baseline in percent predicted FEV1; independent variable: treatment arm) | Type 2 inflammatory asthma phenotype; baseline blood eosinophils ≥ 150 cells/µL; full ITT | Baseline weight, region, ethnicity, baseline eosinophil level, baseline FeNO level, baseline ICS dose level, visit, treatment by-visit interaction, baseline percent predicted FEV1 value, and baseline-by-visit interaction |
|
Baseline blood eosinophils ≥ 300 cells/µL | Baseline weight, region, ethnicity, baseline FeNO level, baseline ICS dose level, visit, treatment by-visit interaction, baseline percent predicted FEV1 value, and baseline-by-visit interaction | ||||
SCS usage | Annualized SCS duration (days) and courses during the treatment period | Negative binomial regression model (dependent variable: number or duration of SCS courses taken during the treatment period; independent variable: treatment arm) | Type 2 inflammatory asthma phenotype; baseline blood eosinophils ≥ 300 cells/µL | Same as the primary efficacy end point | None |
Time to first SCS usage | Cox proportional hazards model (dependent variable: time to the first event; independent variable: treatment arm) | Type 2 inflammatory asthma phenotype; baseline blood eosinophils ≥ 300 cells/µL | Same as the primary efficacy end point | ||
Asthma symptoms | Change from baseline in ACQ-7-IA and ACQ-5-IA | MMRM (dependent variable: change from baseline in ACQ; independent variable: treatment arm) | Type 2 inflammatory asthma phenotype; baseline blood eosinophils ≥ 150 cells/µL; full ITT | Age, baseline weight, region (pooled country), baseline eosinophil level, baseline FeNO level, baseline ICS dose level, visit, treatment by-visit interaction, baseline end point value, and baseline-by-visit interaction | None |
Baseline blood eosinophils ≥ 300 cells/µL | Age, baseline weight, region (pooled country), baseline FeNO level, baseline ICS dose level, visit, treatment by-visit interaction, baseline end point value, and baseline-by-visit interaction | ||||
Responder analysis at week 12, 24, 36, and 52 | Logistic regression to compare percentage of patients who reached MCID (responders) in dupilumab and placebo group at selected time points | Type 2 inflammatory asthma phenotype | Age, baseline weight, region (pooled country), baseline eosinophil level, baseline FeNO level, baseline ICS dose level, and baseline ACQ score | None | |
Baseline blood eosinophils ≥ 300 cells/µL | Age, baseline weight, region (pooled country), baseline FeNO level, baseline ICS dose level, and baseline ACQ score | ||||
Other continuous time points | Change from baseline at all time points (a.m./p.m. PEF, number of puffs of reliever medication per 24 hours over time, PAQLQ(S)-IA global score, EQ-5D-Y VAS for Children, PACQLQ, PRQLQ-IA) | MMRM (dependent variable: change from baseline in the outcome of interest; independent variable: treatment arm) | Type 2 inflammatory asthma phenotype | Age, baseline weight, region (pooled country), baseline eosinophil level, baseline FeNO level, baseline ICS dose level, visit, treatment by-visit interaction, baseline end point value and baseline-by-visit interaction, ethnicity (only for spirometry parameters) | None |
Baseline blood eosinophils ≥ 300 cells/µL | Age, baseline weight, region (pooled country), baseline FeNO level, baseline ICS dose level, visit, treatment by-visit interaction, baseline end point value and baseline-by-visit interaction, ethnicity (only for spirometry parameters) | ||||
Responder analysis at week 12, 24, 36, and 52 (for PAQLQ[S]-IA) | Logistic regression to compare percentage of patients who reached MCID (responders) in dupilumab and placebo group at selected time points | Type 2 inflammatory asthma phenotype | Age, baseline weight, region (pooled country), baseline eosinophil level, baseline FeNO level, baseline ICS dose level, and baseline PACQLQ score | None | |
NA | Baseline blood eosinophils ≥ 300 cells/µL | Age, baseline weight, region (pooled country), baseline FeNO level, baseline ICS dose level, and baseline PACQLQ score | |||
ACQ = Asthma Control Questionnaire; ACQ-5-IA = 5-item Asthma Control Questionnaire–Interviewer Administered; ACQ-7-IA = 7-item Asthma Control Questionnaire–Interviewer Administered; EQ-5D-Y = EQ-5D-Youth; FeNO = fractional exhaled nitric oxide; FEV1 = forced expiratory volume in 1 second; ICS = inhaled corticosteroid; ITT = intention to treat; MCID = minimal clinically important difference; MMRM = mixed-effect model with repeated measures; PACQLQ = Paediatric Asthma Caregiver's Quality of Life Questionnaire; PAQLQ(S)-IA = Standardised Paediatric Asthma Quality of Life Questionnaire–Interviewer Administered; PEF = peak expiratory flow; ppb = parts per billion; PRQLQ-IA = Pediatric Rhinoconjunctivitis Quality of Life Questionnaire–Interviewer Administered; SCS = systemic corticosteroid; VAS = visual analogue scale.
Source: Clinical Study Report.7
Protocol Deviations
The sponsor identified major protocol deviations that may have a potential adverse impact on data integrity, patients’ rights, or safety. These were reported, within the type 2 inflammatory asthma phenotype population, in 5 (2.1%) and 3 (2.6%) patients in the dupilumab and placebo group, respectively. Within the population with a baseline blood EOS count of 300 cells/µL or greater, these were reported in 4 (2.3%) and 2 (2.4%) patients in the dupilumab and placebo group, respectively. The types of protocol deviations included missing assessments of FeNO or blood EOS at baseline, low adherence to background and study treatments, and use of prohibited concomitant medications.
Randomization and Dosing Irregularities
In the 2 primary efficacy populations, randomization and drug allocation irregularities were reported in 19 (8.1%) in the dupilumab and 8 (7.0%) patients in the placebo group (type 2 inflammatory asthma phenotype population) and in 12 (6.9%) in the dupilumab and 6 (7.1%) patients in the placebo group (baseline blood EOS ≥ 300 cells/µL population). The majority of errors were related to the ICS dose level stratum classification reported by the investigators, which occurred in both directions (i.e., medium erroneously classified as high and vice versa). Specifically, misclassification according to the ICS dose levels occurred in 18 (7.6%) and 7 (6.1%) of type 2 population patients in the dupilumab and placebo group, respectively, and in 11 (6.3%) and 5 (6.0%) patients in the population with baseline EOS of 300 cells/µL or greater in the dupilumab and placebo group, respectively. The sponsor reported that the analyses factoring ICS dose levels were conducted according to the actual calculated total daily dose of all medications containing ICSs and not on the investigators’ reported classification.
Breaking of the Blind
In the VOYAGE trial, breaking of the blind for regulatory purposes occurred due to serious adverse reactions suspected to be related to treatment (2 patients in the dupilumab group: 1 reported pneumonia and 1 patient had eosinophilia, headache, and blurred vision; 1 patient had lymphadenitis viral in the placebo group)
Local breaking of the blind by the investigators occurred only in the dupilumab group, among 4 patients from Brazil (discontinuation due to yellow fever vaccination) and 1 patient from the US (reporting AE of eosinophilia, headache, and blurred vision).
The sponsor specified that patient withdrawal from treatment would occur only in the case of breaking of the blind occurring at a local level, not a study level.
Amendments to the Protocol
The original protocol, dated August 04, 2016, was modified per 3 global amendments (Amendment 1: March 10, 2017; Amendment 2: June 18, 2018; Amendment 3: October 1, 2019) and 1 local amendment for Brazil (February 2, 2018).
Notably, Amendment 3 included following modifications:
Changes to the study primary efficacy analysis population (from an overall uncontrolled persistent asthma population to the population with baseline eosinophil count ≥ 0.3 Giga/L or with the type 2 inflammatory asthma phenotype)
Changes to the sample size
Specification of different hierarchy orders used for US and US reference countries and European Union and European Union reference countries
Removal of the limit in enrolling patients according to background therapy with medium-dose ICS or blood eosinophil count level
Defining the planned database lock
Classification of FeNO as a secondary end point instead of an exploratory end point
Additional changes to the Study Analysis plan were made as per regulatory agency request on August 14, 2020, to provide a detailed elaboration of the primary estimate and to provide further analyses to evaluate whether baseline FeNO values can independently predict treatment effect. Moreover, addition of outcome measures (SCS exposure, loss of asthma control), COVID-19 relevant analysis, and adjustments to subgroup analyses (removal of elevated IgE [yes/no] and age subgroup analyses; addition of treatment exposure-adjusted analysis for key AE end points) were implemented.
Post hoc analyses (i.e., implemented after database lock) was added to evaluate effects of dupilumab on some end points of interest. These included annualized rate of the subtype of severe asthma exacerbation resulting in hospitalization or ED visits resulting in hospitalization (for the type 2 inflammatory asthma phenotype and with baseline blood EOS ≥ 300 cells/µL populations), responder analyses for ACQ-7-IA (based on minimal clinically important difference cut-off points of 0.75,1, and 1.5 as a responder definition), and biomarker analyses (interaction of biomarkers of type 2 inflammation, baseline EOS and FeNO levels) on the effect of dupilumab on the primary end point and key secondary end point based on the ITT population.
Results
Patient Disposition
A total of ||||||||| patients were screened for the study. The screening failure rate was ||||||||| and the major reason for screening out of the study was failure to meet inclusion criteria described in the Table 6 (|||||||||), consisting of lack of appropriate background therapy, exceeding the specified maximum prebronchodilator FEV1, reversibility in FEV1 that was lower than specified minimum, or lack of evidence of uncontrolled asthma.
Out of ||||||||| screened patients, 408 underwent randomizations (ITT population). In the type 2 inflammatory phenotype and population with baseline blood EOS of 300 cells/µL or greater, there were a total of 350 (236 in dupilumab and 114 in the placebo arm) and 259 patients (175 in dupilumab and 84 in the placebo arm), respectively (Table 10). Three patients in the dupilumab group were randomized, but not treated (due to erroneous randomization despite ineligibility of 2 patients and withdrawal from the study before treatment by 1 patient).
Treatment discontinuations were generally below ||||||||| in each group across both efficacy populations, and were higher in the dupilumab group, compared to placebo group (dupilumab versus placebo: |||||||||||||||||| in the type 2 population; |||||||||||||||||| in the baseline EOS ≥ 300 cells/µL population). The most common reason for discontinuation was “other,” occurring in ||||||||||||||||||||||||||| of dupilumab versus placebo patients in both efficacy populations, respectively. The sponsor reported that |||||||||||||||||||||||||||||||||||| due to “other” reasons was related to the safety issues. Specifically, in the dupilumab group (ITT population), discontinuation due to “other reasons” included: |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| Discontinuation for “other reasons” in the placebo group (ITT population) was due to |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. Percentage of patients who withdrew due to AEs was similar in dupilumab and placebo arms, across all efficacy populations. Study withdrawals were reported in ||||||||||||||||||||||||||| of type 2 patients of the dupilumab and placebo group, and in ||||||||||||||||||||||||||| of patients with baseline EOS of 300 cells/µL or greater in the dupilumab and placebo groups, respectively.
Overall, more patients in the placebo group, compared to dupilumab group, continued to the long-term extension study (dupilumab versus placebo: ||||||||||||||||||||||||||| in the type 2 population; ||||||||||||||||||||||||||| in the baseline EOS ≥ 300 cells/µL population).
Disposition of study patients in the ITT and baseline blood EOS of 150 cells/µL or greater populations is presented in the Appendix 3.
Table 10: Patient Disposition (VOYAGE Trial)
Trial population | VOYAGE | |||
|---|---|---|---|---|
Type 2 inflammatory asthma phenotype population | Baseline blood eosinophils ≥ 300 cells/µL population | |||
Dupilumab n = 236 | Placebo n = 114 | Dupilumab n = 175 | Placebo n = 84 | |
Screened, n | ||||||||||||||||||||||||||||||||||||||||||||| | |||
Randomized, n | 236 | 114 | 175 | 84 |
Treated | 233 (98.7) | 114 (100) | 172 (98.3) | 84 (100) |
Completed study treatment during the treatment period | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Discontinued study treatment | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Reason for treatment discontinuation | ||||
Adverse events | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Poor compliance to protocol | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Other reason | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Discontinued study | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Patients who continued into LTE study | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Patients who did not continue into LTE study | ||||
Completed the follow-up period | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Discontinued from the follow-up period | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Reason for discontinuation from the follow-up | ||||
Adverse events | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Poor compliance to protocol | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Other reason | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Analyses sets | ||||
ITT population, n (dupilumab vs. placebo) | ||||||||| | |||
Safety, n (dupilumab vs. placebo) | ||||||||| | |||
ITT = intention to treat; LTE = long-term extension.
Values are n (%) unless otherwise indicated.
Source: Clinical Study Report.7
Exposure to Study Treatments
Study duration for the dupilumab and placebo groups of the VOYAGE trial was similar, with mean values of ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| for dupilumab and placebo arm, respectively, in the type 2 inflammatory asthma population and mean values of |||||||||||||||||||||||||||||||||||||||||||||||||||||| for dupilumab and placebo arm, respectively, in the population with baseline blood EOS of 300 cells/µL or greater. Median duration of the study treatment was the same for both groups (365 days) in both efficacy populations.
Regarding the adherence to study treatment, an average of |||||||||||||||||||||||||||||||||||| of adherence to injections was observed among the patients in dupilumab and placebo groups in the type 2 inflammatory asthma population, respectively. For the population with baseline blood EOS of 300 cells/µL or greater, mean adherence to injections was ||||||||||||||||||||||||||||||||||||||||||||| in the dupilumab and placebo arms, respectively.
Mean percentage of days in which patients were compliant with all background controller medications was ||||||||||||||||||||||||||| in the dupilumab group and ||||||||||||||||||||||||||| in the placebo group (for the type 2 population) and ||||||||||||||||||||||||||| in the dupilumab group and ||||||||||||||||||||||||||| in the placebo group (baseline blood EOS ≥ 300 cells/µL population).
Efficacy
Only those efficacy outcomes and analyses of subgroups identified in the review protocol are reported in the following. Refer to Appendix 3 for detailed efficacy data.
Mortality
In the VOYAGE trial, there were no deaths reported across the dupilumab and placebo groups.
Asthma Exacerbations
The adjusted rate over 52 weeks in the type 2 inflammatory asthma population was 0.31 (95% CI, 0.22 to 0.42) with dupilumab and 0.75 (95% CI, 0.54 to 1.03) with placebo, for a RR of 0.41 (95% CI, 0.27 to 0.61; P < 0.0001) and a risk difference of –0.44 (95% CI, –0.68 to –0.20) (Table 11). In the patients with an EOS count of at least 300 cells/µL at baseline, the adjusted rate of severe asthma exacerbations across 52 weeks was 0.24 (95% CI, 0.16 to 0.35) in the dupilumab group and 0.67 (95% CI, 0.47 to 0.95) in the placebo group (RR = 0.35; 95% CI, 0.22 to 0.56; P < 0.0001; risk difference: –0.43; 95% CI, –0.66 to –0.20).
Table 11: Outcome: Severe Asthma Exacerbations During the 52-Week Randomized Treatment Period (Type 2 Inflammatory Asthma Phenotype Population; Population With Baseline Blood Eosinophils of 300 cells/µL or Greater)
Trial population efficacy outcome | VOYAGE | |||
|---|---|---|---|---|
Type 2 inflammatory asthma phenotype population | Population with baseline blood eosinophils ≥ 300 cells/µL | |||
Dupilumab n = 236 | Placebo n = 114 | Dupilumab n = 175 | Placebo n = 84 | |
Severe asthma exacerbationsa | ||||
Number of patients with ≥ 1 events, n (%) | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Total number of events, n | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Total patient-years followed | 229.6 | 112.7 | 168.9 | 82.6 |
Annualized rate, unadjustedb | 0.357 | 0.719 | 0.314 | 0.738 |
Annualized rate, adjusted (95% CI)c | 0.305 (0.223 to 0.416) | 0.748 (0.542 to 1.034) | 0.235 (0.160 to 0.345) | 0.665 (0.467 to 0.949) |
Relative risk (95% CI)c | 0.407 (0.274 to 0.605) | 0.353 (0.222 to 0.562) | ||
P valuec | < 0.0001 | < 0.0001 | ||
Risk difference (95% CI)d | ||||||||||||||||||||||||||| | ||||||||||||||||||||||||||| | ||
Time to first severe exacerbation | ||||
HR (95% CI) | 0.443 (0.293 to 0.670) | 0.382 (0.233 to 0.629) | ||
P value | 0.0001 | 0.0002 | ||
Severe asthma exacerbations associated with ED visit or hospitalization | ||||
Number of patients with ≥ 1 events, n (%) | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Total number of events | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Total patient-years followed | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Annualized rate, unadjustedb | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Annualized rate, adjusted (95% CI)c | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Relative risk (95% CI)c | ||||||||| | ||||||||| | ||
P value | ||||||||| | ||||||||| | ||
Risk difference (95% CI)d | ||||||||| | ||||||||| | ||
Severe asthma exacerbations associated with hospitalization | ||||
Number of patients with ≥ 1 events, n (%) | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Total number of events | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Total patient-years followed | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Annualized rate, unadjustedb | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Annualized rate, adjusted (95% CI)c | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Relative risk (95% CI)c | ||||||||| | ||||||||| | ||
P value | ||||||||| | ||||||||| | ||
Risk difference (95% CI)d | ||||||||| | ||||||||| | ||
CI = confidence interval; ED = emergency department; FeNO = fractional exhaled nitric oxide; HR = hazard ratio; ICS = inhaled corticosteroid; NE = not estimable.
Note: All severe exacerbation events occurred during the 52-week treatment period are included, regardless of whether the patient is on treatment or not.
Type 2 inflammatory asthma phenotype population is defined as the randomized patients with baseline blood eosinophils of 0.15 Giga/L or greater or baseline FeNO of 20 parts per billion or greater.
The time-to-event variable is defined as (date of the first event - randomization date + 1). For patients who have no event on or before week 52 or the last contact date, the time will be censored at the date of date of visit at week 52 or the last contact date, whichever happens earlier.
aOutcome was part of statistical hierarchy testing and adjusted for multiplicity.
bThe total number of event that occurred during the 52-week treatment period divided by the total number of patient-years followed in the 52-week treatment period.
cDerived using negative binomial model with the total number of events onset from randomization up to the week 52 visit or last contact date (whichever comes earlier) as the response variable, with the treatment group, age, baseline weight group, region, baseline eosinophil level, baseline FeNO level, baseline ICS dose level, and number of severe exacerbation events within 1 year before the study as covariates, and log-transformed standardized observation duration as an offset variable.
dDerived using the delta method.
eDerived using Cox regression model including the time to the first event as the dependent variable, and treatment groups, age, baseline weight group, region, baseline eosinophil level, baseline FeNO level, baseline ICS dose level, and number of severe exacerbation events within 1 year before the study as covariates.
Source: Clinical Study Report.7
Various sensitivity analyses were conducted to account for missing data, and results were consistent with that of the primary analysis, (Appendix 3).
RRs for the comparison of severe exacerbations associated with an ED visit or hospitalization between dupilumab and placebo were ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||, while the risk differences were ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| for the populations with type 2 asthma and baseline blood EOS of 300 cells/µL or greater, respectively. Adjusted rates of severe exacerbations associated with a hospitalization were not estimable due to ||||||||| events reported in the placebo group.
Subgroup analyses for the primary outcome were performed for several subgroups relevant to the CADTH protocol (baseline eosinophil levels, baseline ICS dose, number of previous asthma exacerbations, atopic medical history) (Table 12, Table 13, Table 14, and Table 15). In the subgroup of patients with baseline blood EOS of 150 cells/µL or greater, RRs between dupilumab and placebo were 0.390 (95% CI, 0.261 to 0.583 to |||||||||||||) and the risk difference ||||||||||||||||||||||||||||||||||||, with a P value for the interaction of ||||||||||||||||||. Other subgroup analyses did not yield statistically significant interaction.
Results of the primary end point analyses in the ITT and baseline blood EOS of 150 cells/µL or greater populations were aligned with those presented in the type 2 and baseline blood EOS of 300 cells/µL or greater populations (Table 12, Appendix 3).
Table 12: Subgroup Analysis Results From VOYAGE Trial for the Primary Outcome by Baseline Blood Eosinophil Count (Adjusted Annualized Rate of Severe Exacerbation Events During the 52-Week Treatment; ITT population)
Trial population subgroup | VOYAGE | |||
|---|---|---|---|---|
ITT population | ||||
Dupilumab n = 273 | Placebo n = 135 | Dupilumab n = 273 | Placebo n = 135 | |
Baseline blood eosinophil (group), cells/µL | < 300 | ≥ 300a | ||
N | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Total severe exacerbation events, n | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Total patient-years followed | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Relative risk vs. placebo (95% CI)b | ||||||||| | ||||||||| | ||
P valueb | ||||||||| | ||||||||| | ||
Risk difference c vs. placebo (95% CI)c | ||||||||| | ||||||||| | ||
Overall P value for interactiond | ||||||||| | |||
Baseline blood eosinophil (group), cells/µL | < 150 | ≥ 150a | ||
N | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Total severe exacerbation events, n | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Total patient-years followed | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
Relative risk vs. placebo (95% CI)b | ||||||||| | ||||||||| | ||
P valueb | ||||||||| | ||||||||| | ||
Risk difference vs. placebo (95% CI)c | ||||||||| | ||||||||| | ||
Overall P value for interactiond | ||||||||| | |||
CI = confidence interval; FeNO = fractional exhaled nitric oxide; ICS = inhaled corticosteroid; ITT = intention to treat.
aOutcome was part of statistical hierarchy testing and adjusted for multiplicity.
bDerived using negative binomial model with the total number of events onset from randomization up to week 52 visit or last contact date (whichever comes earlier) as the response variable, with the treatment group, age, baseline weight group, region, baseline eosinophil level, baseline FeNO level, baseline ICS dose level, and number of severe exacerbation events within 1 year before the study as covariates, and log-transformed standardized observation duration as an offset variable.
cDerived using the delta method.
dSimilar mode as mentioned in footnote “b”, except replacing the biomarker level with corresponding subgroup and additionally including treatment by subgroup interaction as the covariate.
Source: Clinical Study Report.7
||||||||| | ||||||||| | |||||||
|---|---|---|---|---|---|---|---|---|
||||||||| | ||||||||| | |||||||
||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | |
||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||
||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| |
||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||
||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||
||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||||||| | ||||
||||||||| | ||||||||| | ||||||||| | ||||||
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Source: Clinical Study Report.7
||||||||||||||||| | ||||||||||||||||| | |||||
|---|---|---|---|---|---|---|
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | |
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | |||
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | |||
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | |||
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | |||
||||||||||||||||| | ||||||||||||||||| | |||||
||||||||||||||||| | ||||||||||||||||| | |||||
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | |||
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | |||
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | |||
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | |||
||||||||||||||||| | ||||||||||||||||| | |||||
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Source: Clinical Study Report.7
||||||||||||||||| | ||||||||||||||||| | |||||||
|---|---|---|---|---|---|---|---|---|
||||||||||||||||| | ||||||||||||||||| | |||||||
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | |
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| | ||||||
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Source: Clinical Study Report.7
Asthma Symptoms
ACQ-7 scores decreased (improved) from baseline to week 24 in both the dupilumab and placebo groups, for an LS difference versus placebo of –0.33 points (95% CI, –0.50 to –0.16; |||||||||||| in the type 2 population and of –0.46 points (95% CI, –0.66 to –0.26; |||||||||||| in the baseline blood eosinophil count of 300 cells/µL or greater population. In the type 2 inflammatory asthma population, there were |||||||||||| of dupilumab patients and |||||||||||| of placebo patients who were responders at week 24, for an OR of ||||||||||||||||||||||||||||||||||||||||||||||||. In the baseline eosinophil count of 300 cells/µL or greater population, there were |||||||||||| responder patients in the dupilumab and |||||||||||| responder patients in the placebo arm at week 24, with an OR of |||||||||||||||||||||||||||||||||||||||||||||||| (Table 16).
Results were maintained during the trial period (to week 52) |||||||||||||||||||||||||||||||||||||||||||||||| (Table 16, Figure 3, and Figure 4). Appendix 3 contains findings from the ACQ-7 assessments conducted across additional efficacy populations of the VOYAGE trial (ITT and baseline blood EOS ≥ 150 cells/µL populations), as well as results obtained from the ACQ-5 assessments in the type 2 inflammatory asthma phenotype and baseline eosinophil count of 300 cells/µL or greater populations.
Table 16: Symptoms According to the ACQ-7 (Weeks 24 and 52)
Trial population efficacy outcome | VOYAGE | |||
|---|---|---|---|---|
Type 2 inflammatory asthma phenotype population | Baseline blood eosinophils ≥ 300 cells/µL population | |||
Dupilumab n = 236 | Placebo n = 114 | Dupilumab n = 175 | Placebo n = 84 | |
ACQ-7, total score | ||||
Baseline | ||||
N | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Mean (SD) baseline | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Change from baseline at week 24a | ||||
N | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Mean (SD) CFB, week 24 | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
LS mean CFB week 24 (SE)b | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Difference vs. placebo (95% CI) week 24b | –0.33 (–0.50 to –0.16) | –0.46 (–0.66 to –0.26) | ||
P valueb | |||||||||||| | |||||||||||| | ||
Responders (patients with ≥ 0.5 CFB), n (%)c | |||||||||||| | |||||||||||| | |||||||||||| | || |||||| |
OR (95% CI)d | |||||||||||| | |||||||||||| | ||
P value (nominal)d | |||||||||||| | |||||||||||| | ||
Change from baseline at week 52 | ||||
N | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Mean (SD) CFB, week 52 | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
LS mean CFB week 52 (SE)b | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Difference vs. placebo (95% CI) week 52b | |||||||||||| | |||||||||||| | ||
P value (nominal) | |||||||||||| | |||||||||||| | ||
Responders (patients with ≥ 0.5 CFB), n (%)c | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
OR (95% CI)d | |||||||||||| | |||||||||||| | ||
P value (nominal)d | |||||||||||| | |||||||||||| | ||
ACQ-7 = 7-item Asthma Control Questionnaire; CFB = change from baseline; CI = confidence interval; FeNO = fractional exhaled nitric oxide; ICS = inhaled corticosteroid; LS = least squares; OR = odds ratio; SD = standard deviation; SE = standard error.
Note: Type 2 inflammatory asthma phenotype population is defined as the randomized patients with baseline blood eosinophils of 0.15 Giga/L or greater or baseline FeNO of 20 parts per billion or greater.
aOutcome was part of statistical hierarchy testing and adjusted for multiplicity.
bDerived from mixed-effect model with repeated measures with change from baseline in ACQ-7–Interviewer Administered up to week 24 as the response variable, and treatment, age, weight group, region, baseline eosinophil level, baseline FeNO level, baseline ICS dose level, visit, treatment by-visit interaction, baseline ACQ-7–Interviewer Administered, and baseline-by-visit interaction as covariates.
cPatients who met the corresponding criterion are considered as responders. Patients who did not meet the criterion or had missing value are considered as nonresponders.
dDerived from logistic regression model with treatment, age, weight group, region, baseline eosinophil level, baseline FeNO level, baseline ICS dose level, and baseline ACQ-7–Interviewer Administered score as covariates.
Source: Clinical Study Report.7
Figure 3: LS Mean Change From Baseline in ACQ-7-IA Over Time (MMRM Including Measurements Up to Week 52): Type 2 Inflammatory Asthma Phenotype Population
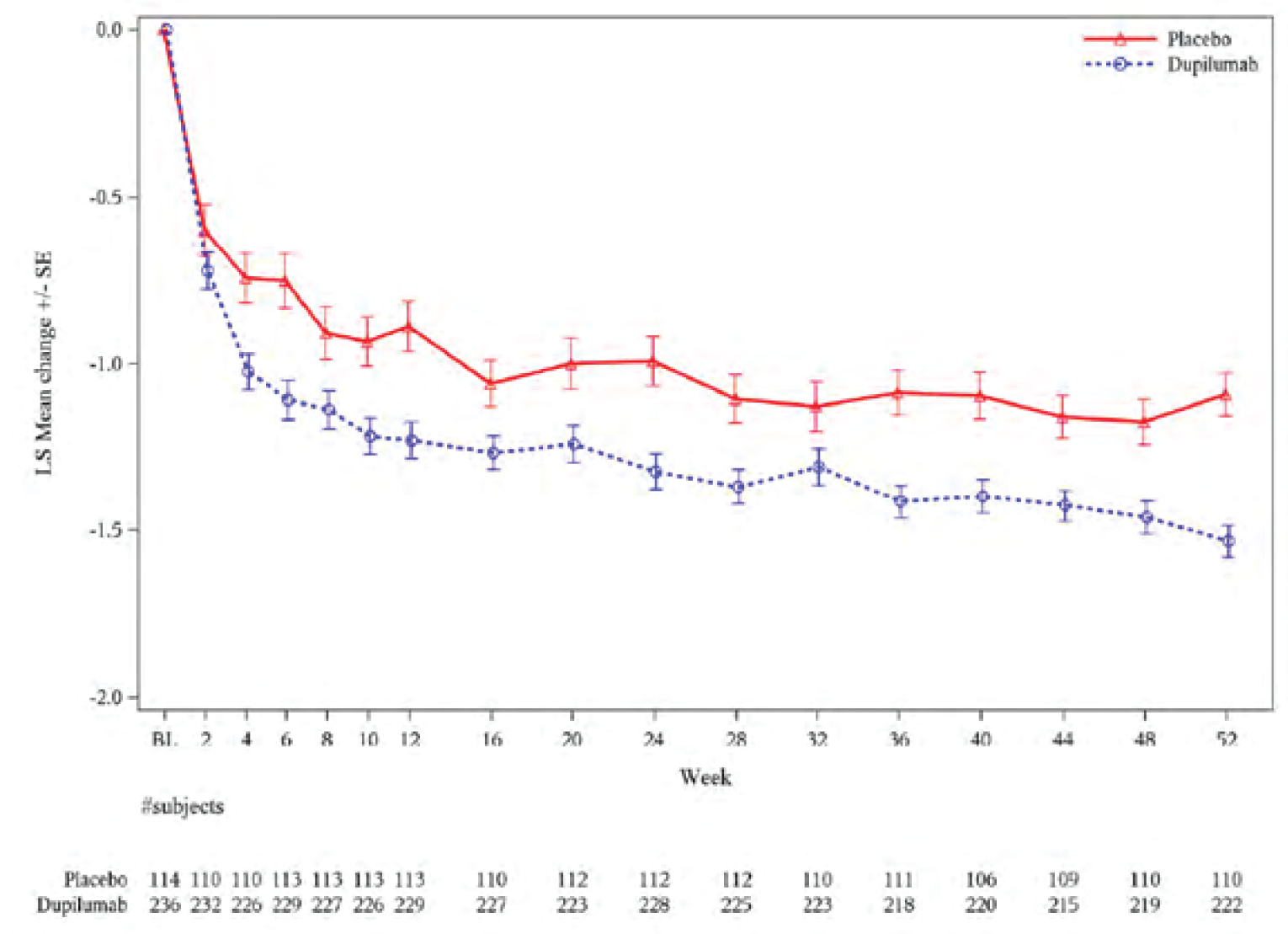
ACQ-7-IA = 7-item Asthma Control Questionnaire–Interviewer Administered; MMRM = mixed-effect model with repeated measures.
Source: Clinical Study Report.7
Reduction in Use of OCSs
Total SCSs usage during the treatment period was captured in the VOYAGE trial. Overall, more patients in the placebo arm (40.4% and 41.7%) compared to patients in the dupilumab arm (24.2% and 22.3%) received treatment with SCS during the trial within the type 2 inflammatory asthma phenotype and baseline blood EOS of 300 cells/µL or greater populations, respectively (Table 17).
The adjusted RR in annualized number of SCS courses, comparing dupilumab to placebo, was 0.407 (95% CI, 0.272 to 0.609) for the type 2 inflammatory asthma phenotype population and 0.340 (95% CI, 0.212 to 0.545) for the baseline blood EOS of 300 cells/µL or greater population.
Figure 4: Least Squares Mean Change From Baseline in ACQ-7-IA Over Time (MMRM Including Measurements Up to Week 52): Population With Baseline Blood Eosinophils of 300 cells/µL or Greater
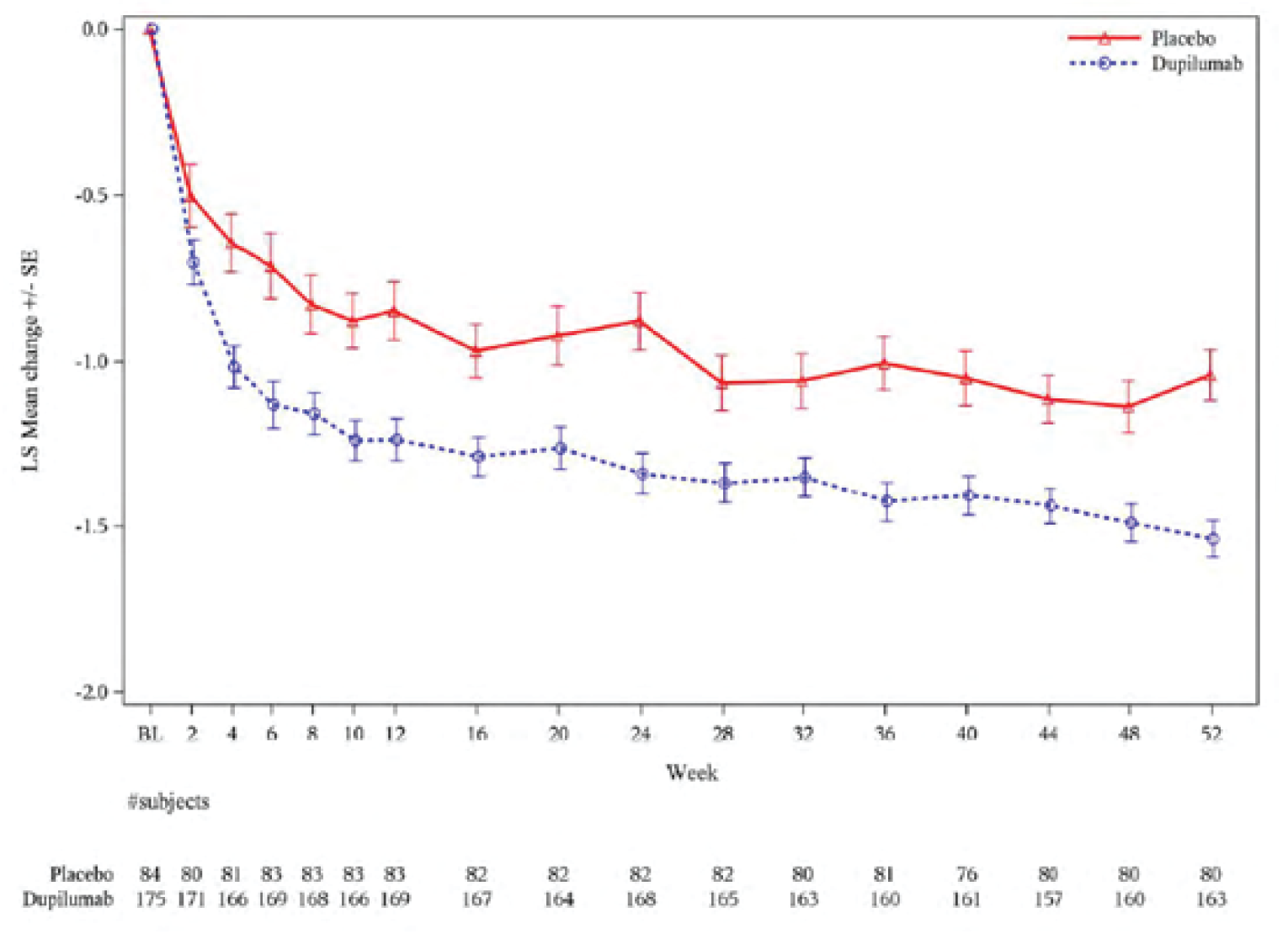
ACQ-7-IA = 7-item Asthma Control Questionnaire–Interviewer Administered; MMRM = mixed-effect model with repeated measures.
Source: Clinical Study Report.7
Table 17: Total SCS Usage During the Treatment Period
Trial population efficacy outcome | VOYAGE trial | |||
|---|---|---|---|---|
Type 2 inflammatory asthma phenotype population | Baseline blood eosinophils ≥ 300 cells/µL population | |||
Dupilumab n = 236 | Placebo n = 114 | Dupilumab n = 175 | Placebo n = 84 | |
SCS | ||||
Patients with SCS intake, n (%) | 57 (24.2) | 46 (40.4) | 39 (22.3) | 35 (41.7) |
Total SCS duration (days) in patients with SCS intake | ||||
n | 57 | 46 | 39 | 35 |
Mean (SD) | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Median (IQR) | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Individual annualized total SCS duration (days) | ||||
In patients with SCS intake, median (IQR)a | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
In all patients, median (IQR)a | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Total SCS courses in patients with SCS intakeb | ||||
n | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Mean (SD) | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Median (IQR) | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Total SCS courses in all patientsb | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Total patient-years followed | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Individual annualized total SCS courses | ||||
In patients with SCS intake, median (IQR)b,c | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
In all patients, median (IQR)b,c | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Adjusted annualized total SCS courses in all patients | ||||
Estimate (95% CI) | 0.350 (0.256 to 0.477) | 0.860 (0.616 to 1.200) | 0.274 (0.188 to 0.399) | 0.806 (0.563 to 1.154) |
Relative risk vs. placebo (95% CI)d | |||||||||||| | |||||||||||| | ||
P valued | |||||||||||| | |||||||||||| | ||
Time to first SCS usage for asthma | ||||
Patients with SCS usage, n (%) | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Patients censored, n (%) | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Median time to SCS usage, days (95% CI)e | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
HR (95% CI)f | |||||||||||| | |||||||||||| | ||
P valuef | |||||||||||| | |||||||||||| | ||
CI = confidence interval; HR = hazard ratio; IQR = interquartile range; SCS = systemic corticosteroid; SD = standard deviation.
aThe total dose per number of SCS days for each patient divided by the number of patient-years followed in the treatment period for that patient.
bA course of SCSs is considered continuous if treatment is separated by less than 7 days.
cThe number of SCS courses for each patient divided by the number of years followed in the treatment period for that patient.
dDerived using negative binomial model with the total number of courses of SCSs intake during the treatment period as the response variable, and the treatment groups, age, baseline weight group, region, baseline eosinophil level, baseline fractional exhaled nitric oxide level, baseline inhaled corticosteroid dose, and number of severe exacerbation events within 1 year before the study as covariates, and log-transformed standardized observation duration as an offset variable.
eDerived from Kaplan-Meier estimates.
fDerived using Cox regression model including the time to the first event as the dependent variable, and treatment groups, age, baseline weight group, region, baseline eosinophil level (not available for the population with baseline eosinophils of 0.3 Giga/L or greater), baseline fractional exhaled nitric oxide level, baseline inhaled corticosteroid dose, and number of severe exacerbation events within 1 year before the study as covariates.
Source: Clinical Study Report.7
Pulmonary Function
For the key secondary outcome (change from baseline in percent predicted prebronchodilator FEV1 at week 12) within the type 2 inflammatory asthma phenotype population, LS mean difference between the groups was 5.21% (95% CI, 2.14 to 8.27%; P = 0.0009). Similarly, in the baseline blood eosinophil count of 300 cells/µL or greater population, at week 12 the LS mean difference between groups was 5.32% (95% CI, 1.76 to 8.88%; P = 0.0036). In both primary efficacy populations, LS mean changes in the percent predicted prebronchodilator FEV1 were sustained through week 52 (Table 18).
Findings from other multiplicity-controlled analyses for the ITT and baseline blood EOS of 150 cells/µL or greater populations were similar to those observed in the 2 primary efficacy populations (Appendix 3).
A series of sensitivity analyses were conducted to account for missing data as well as the effects of SCS exposure, showing consistent and similar findings to the primary analysis (Appendix 3).
Subgroup analyses for change from baseline in percent predicted prebronchodilator FEV1 at week 12 in patients with baseline blood EOS of 300 cells/µL or greater and 150 cells/µL or greater were generally consistent with the primary analyses. Other subgroup analyses reported were not adjusted for multiplicity (Table 19, Table 20, Table 21, and Table 22).
In both the type 2 inflammatory asthma phenotype and baseline blood eosinophil count of 300 cells/µL or greater populations, there was an improvement in morning PEF scores at week 12 in both treatment groups, with the LS mean difference versus placebo of 14.02 L/min (95% CI, 4.92 to 23.12; P = 0.0026) and 14.48 L/min (95% CI, 3.49 to 25.47; P = 0.01) for the 2 populations, respectively (Table 18).
Changes at week 12 of evening PEF values were reported among the 2 populations, with LS mean differences versus placebo of 17.97 L/min (95% CI, 8.98 to 26.95; P = 0.0001) and 19.10 L/min (95% CI, 8.36 to 29.83; P = 0.0005) for the 2 populations (Table 18).
Table 18: Pulmonary Function Measurements (Percent Predicted Prebronchodilator FEV1, a.m. and p.m. PEF)
Trial population efficacy outcome | VOYAGE | |||
|---|---|---|---|---|
Type 2 inflammatory asthma phenotype population | Baseline blood eosinophils ≥ 300 cells/µL population | |||
Dupilumab n = 236 | Placebo n = 114 | Dupilumab n = 175 | Placebo n = 84 | |
Prebronchodilator percent predicted FEV1 | ||||
Baseline, N | 236 | 114 | 175 | 84 |
Mean (SD) baseline | 77.66 (14.38) | 78.36 (14.51) | 76.37 (14.60) | 77.87 (15.19) |
Change from baseline at week 12a | ||||
n | 229 | 110 | |||||||||||| | |||||||||||| |
Mean (SD) CFB, week 12 | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
LS mean CFB week 12 (SE)b | 10.53 (1.01) | 5.32 (1.36) | 10.15 (1.12) | 4.83 (1.54) |
Difference vs. placebo (95% CI) week 12b | 5.21 (2.14 to 8.27) | 5.32 (1.76 to 8.88) | ||
P valueb | 0.0009 | 0.0036 | ||
Change from baseline at week 52 | ||||
n | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Mean (SD) CFB, week 52 | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
LS mean CFB week 52 (SE)c | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Difference vs. placebo (95% CI) week 52c | |||||||||||| | |||||||||||| | ||
P valuec | |||||||||||| | |||||||||||| | ||
a.m. PEF (L/MIN) | ||||
Baseline, N | 236 | 114 | 175 | 84 |
Mean (SD) baseline | 196.35 (67.63) | 187.96 (57.49) | 192.32 (67.62) | 185.87 (59.12) |
Change from baseline at week 12 | ||||
N | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Mean (SD) CFB, week 12 | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
LS mean CFB week 12 (SE)c | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Difference vs. placebo (95% CI) week 12d | |||||||||||| | |||||||||||| | ||
P valued | |||||||||||| | |||||||||||| | ||
Change from baseline at week 52 | ||||
N | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Mean (SD) CFB, week 52 | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
LS mean CFB week 52 (SE)d | 31.45 (3.69) | 19.88 (5.16) | 32.80 (4.49) | 19.75 (6.33) |
Difference vs. placebo (95% CI) week 52d | 11.57 (–0.58 to 23.71) | 13.05 (–2.03 to 28.12) | ||
P valued | |||||||||||| | |||||||||||| | ||
p.m. PEF (L/MIN) | ||||
Baseline, N | 236 | 114 | 175 | 84 |
Mean (SD) baseline | 203.27 (65.78) | 198.72 (57.53) | 199.97 (65.44) | 197.06 (59.70) |
Change from baseline at week 12 | ||||
N | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Mean (SD) CFB, week 12 | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
LS mean CFB week 12 (SE)e | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Difference vs. placebo (95% CI) week 12e | |||||||||||| | |||||||||||| | ||
P valuee | |||||||||||| | |||||||||||| | ||
Change from baseline at week 52 | ||||
N | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Mean (SD) CFB, week 52 | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
LS mean CFB week 52 (SE)e | 28.20 (3.73) | 16.46 (5.20) | 26.60 (4.54) | 14.67 (6.39) |
Difference vs. placebo (95% CI) week 52e | 11.74 (–0.44 to 23.92) | 11.93 (–3.25 to 27.10) | ||
P valuee | |||||||||||| | |||||||||||| | ||
CFB = change from baseline; CI = confidence interval; FEV1 = forced expiratory volume in 1 second; LS = least squares; MMRM = mixed-effect model with repeated measures; PEF = peak expiratory flow; SD = standard deviation; SE = standard error.
Note: Type 2 inflammatory asthma phenotype population is defined as the randomized patients with baseline blood eosinophils of 0.15 Giga/L or greater or baseline fractional exhaled nitric oxide of 20 parts per billion or greater.
aOutcome was part of statistical hierarchy testing and adjusted for multiplicity.
bDerived from an MMRM with change from baseline in prebronchodilator percent predicted FEV1 values up to week 12 as the response variable, and treatment, baseline weight group, region, ethnicity, baseline eosinophil level, baseline fractional exhaled nitric oxide level, baseline inhaled corticosteroid dose, visit, treatment by-visit interaction, baseline percent predicted FEV1 value, and baseline-by-visit interaction as covariates.
cDerived from an MMRM with change from baseline in prebronchodilator percent predicted FEV1 values up to week 52 as the response variable, and treatment, baseline weight group, region, ethnicity, baseline eosinophil level, baseline fractional exhaled nitric oxide level, baseline inhaled corticosteroid dose, visit, treatment by-visit interaction, baseline percent predicted FEV1 value, and baseline-by-visit interaction as covariates.
dDerived from an MMRM with change from baseline in a.m. PEF (L/min) values up to week 52 as the response variable, and treatment, age, baseline weight group, region, baseline eosinophil level, baseline fractional exhaled nitric oxide level, baseline inhaled corticosteroid dose, visit, treatment by-visit interaction, baseline a.m. PEF (L/min) value, and baseline-by-visit interaction as covariates.
eDerived from an MMRM with change from baseline in p.m. PEF (L/min) values up to week 52 as the response variable, and treatment, age, baseline weight group, region, baseline eosinophil level, baseline fractional exhaled nitric oxide level, baseline inhaled corticosteroid dose, visit, treatment by-visit interaction, baseline p.m. PEF (L/min) value, and baseline-by-visit interaction as covariates.
Source: Clinical Study Report.7
Table 19: Subgroup Analysis Results From VOYAGE Trial for the Key Secondary Outcome by Baseline Blood Eosinophil Count (Change From Baseline in Prebronchodilator Percent Predicted FEV1 at Week 12; ITT population)
Trial population subgroup | VOYAGE | |||
|---|---|---|---|---|
ITT population | ||||
Dupilumab n = 273 | Placebo n = 135 | Dupilumab n = 273 | Placebo n = 135 | |
Baseline blood eosinophil (group), cells/µL | < 300 | ≥ 300a | ||
N | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Mean (SD) CFB, week 12 | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
n (patients in the model) | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
LS mean CFB week 12 (SE)b | |||||||||||| | |||||||||||| | 10.15 (1.12) | 4.83 (1.54) |
Difference vs. placebo (95% CI) week 24b | |||||||||||| | |||||||||||| | ||
P valueb | |||||||||||| | |||||||||||| | ||
Overall P value for interactionc | |||||||||||| | |||
Baseline blood eosinophil (group, cells/µL | < 150 | ≥ 150a | ||
N | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Mean (SD) CFB, week 12 | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
n (patients in the model) | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
LS mean CFB week 12 (SE)b | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Difference vs. placebo (95% CI) week 24b | |||||||||||| | 4.98 (1.83 to 8.13) | ||
P valueb | |||||||||||| | |||||||||||| | ||
Overall P value for interactionc | |||||||||||| | |||
CFB = change from baseline; CI = confidence interval; FEV1 = forced expiratory volume in 1 second; ITT = intention to treat; LS = least squares; MMRM = mixed-effect model with repeated measures; SD = standard deviation; SE = standard error.
aOutcome was part of statistical hierarchy testing and adjusted for multiplicity.
bDerived from an MMRM with change from baseline in prebronchodilator percent predicted FEV1 values up to week 12 as the response variable, and treatment, baseline weight group, region, ethnicity, baseline eosinophil level, baseline fractional exhaled nitric oxide level, baseline inhaled corticosteroid dose, visit, treatment by-visit interaction, baseline percent predicted FEV1 value, and baseline-by-visit interaction as covariates.
cSimilar an MMRM as mentioned in footnote “b”, except replacing baseline eosinophil level and baseline fractional exhaled nitric oxide level with corresponding subgroup level when performing relevant subgroup analysis, and additionally including treatment by subgroup interaction as covariate.
Source: Clinical Study Report.7
Table 20: Subgroup Analysis Results From VOYAGE Trial for the Key Secondary Outcome by Baseline ICS Dose (Change From Baseline in Prebronchodilator Percent Predicted FEV1 at Week 12)
Trial population subgroup | VOYAGE | |||||||
|---|---|---|---|---|---|---|---|---|
Type 2 inflammatory asthma phenotype population | Baseline blood eosinophils ≥ 300 cells/µL population | |||||||
Dupilumab n = 236 | Placebo n = 114 | Dupilumab n = 236 | Placebo n = 114 | Dupilumab n = 175 | Placebo n = 84 | Dupilumab n = 175 | Placebo n = 84 | |
Baseline ICS dose level | Medium | High | Medium | High | ||||
N | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Mean (SD) CFB, week 12 | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
n (patients in the model) | 131 | 61 | 98 | 49 | 98 | 40 | 70 | 40 |
LS mean CFB week 12 (SE)a | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Difference vs. placebo (95% CI) week ||||||||a | 7.19 (3.15 to 11.22) | 2.47 (–2.37 to 7.31) | 6.73 (1.92 to 11.53) | 3.02 (–2.54 to 8.58) | ||||
P valuea | 0.0006 | 0.3152 | 0.0064 | 0.2843 | ||||
Overall P value for interactionb | |||||||||||| | |||||||||||| | ||||||
CFB = change from baseline; CI = confidence interval; FEV1 = forced expiratory volume in 1 second; ICS = inhaled corticosteroid; LS = least squares; MMRM = mixed-effect model with repeated measures; SD = standard deviation; SE = standard error.
aDerived from an MMRM with change from baseline in prebronchodilator percent predicted FEV1 values up to week 12 as the response variable, and treatment, baseline weight group, region, ethnicity, baseline eosinophil level, baseline fractional exhaled nitric oxide level, visit, treatment by-visit interaction, baseline percent predicted FEV1 value, and baseline-by-visit interaction as covariates.
bSimilar model as that mentioned in footnote “a”, except additionally including the subgroup and subgroup-by-treatment interaction as covariate.
Source: Clinical Study Report.7
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||
|---|---|---|---|---|---|---|
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Source: Clinical Study Report.7
Reduction in Dose of ICSs
The VOYAGE study protocol allowed a permanent increase in background medications after 2 or more severe asthma exacerbations. During the treatment period of the trial, ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||, across all efficacy populations assessed.
Health-Related Quality of Life
PAQLQ(S)–Interviewer Administered was assessed in the VOYAGE trial among children aged 7 years to younger than 12 years at randomization. LS mean values of PAQLQ(S) scores showed increases (improvements) from baseline to week 24 in type 2 inflammatory asthma phenotype population, with a difference between groups of 0.19 points (95% CI, –0.03 to 0.40, |||||||||||||||) (Table 23). In the baseline blood EOS of 300 cells/µL or greater population, the LS mean difference between groups at week 24 was 0.30 points (95% CI, 0.06 to 0.54; ||||||||||).
PAQLQ(S) responders at week 24 were also reported, defined as a change from baseline of 0.5 or greater, and in type 2 inflammatory asthma population there were |||||||||| of dupilumab patients and |||||||||| of placebo patients who were responders (OR = |||||||||| |||||||||| |||||||||| ||||||||||. In the baseline blood EOS of 300 cells/µL or greater population, there were 72.8% of dupilumab responder patients and 63% placebo responder patients (OR = 1.84; 95% CI, 0.92 to 3.65).
Improvements in the PAQLQ(S) scores were maintained through the week 52 of the VOYAGE trial, across both efficacy populations (Figure 5 and Figure 6).
Findings from the PAQLQ(S) assessments conducted across additional efficacy populations of the VOYAGE trial (ITT and baseline blood EOS ≥ 150 cells/µL populations) are reported in the Appendix 3.
Trial population subgroup | VOYAGE | |||||||
|---|---|---|---|---|---|---|---|---|
Type 2 inflammatory asthma phenotype population | Baseline blood eosinophils ≥ 300 cells/µL population | |||||||
Dupilumab n = 236 | Placebo n = 114 | Dupilumab n = 236 | Placebo n = 114 | Dupilumab n = 175 | Placebo n = 84 | Dupilumab n = 175 | Placebo n = 84 | |
Atopic medical condition | Yes | No | Yes | No | ||||
N | 219 | 100 | 9 | 11 | 164 | 76 | 4 | 5 |
Mean (SD) CFB, week 12 | 10.58 (16.08) | 4.36 (13.11) | 2.44 (14.46) | 7.55 (13.62) | 11.24 (15.99) | 4.54 (13.87) | 3.75 (14.24) | 3.00 (12.59) |
n (patients in the model) | 220 | 99 | 9 | 11 | 164 | 75 | 4 | 5 |
LS mean CFB week 12 (SE)a | 11.08 (1.04) | 5.15 (1.44) | –8.46 (4.62) | 15.23 (3.74) | 10.62 (1.14) | 4.94 (1.59) | –13.54 (4.24) | 6.40 (3.73) |
Difference vs. placebo (95% CI) week 24a | 5.93 (2.73 to 9.12) | –23.68 (–33.54 to –13.83) | 5.67 (2.00 to 9.35) | –19.94 (–36.18 to –3.71) | ||||
P valuea | 0.0003 | 0.0002 | 0.0026 | 0.0270 | ||||
Overall P value for interactionb | 0.0244 | 0.2179 | ||||||
Comorbid atopic dermatitis | Yes | No | Yes | No | ||||
N | 103 | 48 | 125 | 63 | 86 | 41 | 82 | 40 |
Mean (SD) CFB, week 12 | 10.50 (14.35) | 2.79 (14.41) | 10.06 (17.42) | 6.11 (12.00) | 10.78 (13.78) | 2.10 (14.22) | 11.37 (18.03) | 6.85 (12.93) |
n (patients in the model) | 105 | 45 | 124 | 65 | 87 | 38 | 81 | 42 |
LS mean CFB week 12 (SE)a | 12.45 (1.50) | 6.14 (2.10) | 9.11 (1.54) | 4.96 (1.94) | 10.85 (1.36) | 4.24 (1.96) | 8.82 (1.96) | 4.65 (2.54) |
Difference vs. placebo (95% CI) week 24a | 6.31 (1.75 to 10.87) | 4.15 (–0.11 to 8.41) | 6.61 (1.99 to 11.23) | 4.17 (–1.45 to 9.80) | ||||
P valuea | 0.0070 | 0.0560 | 0.0054 | 0.1443 | ||||
Overall P value for interactionb | 0.5234 | 0.5200 | ||||||
CFB = change from baseline; CI = confidence interval; FEV1 = forced expiratory volume in 1 second; IgE = immunoglobulin E; LS = least squares; MMRM = mixed-effect model with repeated measures; SD = standard deviation; SE = standard error.
Note: The patient is considered to have an ongoing atopic medical condition if they have any of the following ongoing conditions: atopic dermatitis, allergic conjunctivitis, allergic rhinitis, eosinophilic esophagitis, food allergy, or hives; or has a baseline total IgE of 100 IU/mL or greater and at least 1 aeroantigen-specific IgE is positive (≥ 0.35 IU/mL) at baseline.
aDerived from an MMRM with change from baseline in prebronchodilator percent predicted FEV1 values up to week 12 as the response variable, and treatment, baseline weight group, region, ethnicity, baseline eosinophil level, baseline fractional exhaled nitric oxide level, baseline inhaled corticosteroid dose, visit, treatment by-visit interaction, baseline percent predicted FEV1 value, and baseline-by-visit interaction as covariates.
bSimilar model as that mentioned in the footnote “a”, except additionally including the subgroup (if different than the aforementioned covariates) and subgroup-by-treatment interaction as covariate.
Source: Clinical Study Report.7
The EQ-5D-Y VAS also showed improved HRQoL from baseline in both analysis populations in favour of dupilumab at week 24 (Table 24).
Impact of children’s (≥ 7 years and < 12 years old) asthma on the caregivers’ quality of life was assessed with PACQLQ. Increases from baseline to week 24 in the PACQLQ global score compared to placebo were observed in both the type 2 inflammatory asthma phenotype population (LS mean difference between groups = 0.25; 95% CI, 0.00 to 0.50; P = 0.0531) and baseline blood EOS of 300 cells/µL or greater population (LS mean difference between groups = 0.34; 95% CI, 0.01 to 0.67; P = 0.0445). At week 52, observed PACQLQ LS mean differences for comparison of dupilumab versus placebo were 0.47 (95% CI, 0.22 to 0.72; P = 0.0003) and 0.50 (95% CI, 0.21 to 0.79; P = 0.0007), in the 2 populations, respectively (Table 24).
Table 23: PAQLQ(S)-IA Global Score
Trial population efficacy outcome | VOYAGE | |||
|---|---|---|---|---|
Type 2 inflammatory asthma phenotype population | Baseline blood eosinophils ≥ 300 cells/µL population | |||
Dupilumab n = 211 | Placebo n = 107 | Dupilumab n = 158 | Placebo n = 81 | |
PAQLQ(S)-IA global score | ||||
Baseline | ||||
N | |||||||||| | |||||||||| | |||||||||| | |||||||||| |
Mean (SD) baseline | 4.95 (1.08) | 4.92 (1.13) | 4.96 (1.13) | 4.89 (1.09) |
Change from baseline at week 24 | ||||
N | |||||||||| | |||||||||| | |||||||||| | |||||||||| |
Mean (SD) CFB | |||||||||| | |||||||||| | |||||||||| | |||||||||| |
LS mean (SE), CFB week 24a | 1.30 (0.07) | 1.11 (0.09) | 1.36 (0.08) | 1.06 (0.10) |
Difference vs. placebo (95% CI)a | 0.19 (–0.03 to 0.40) | 0.30 (0.06 to 0.54) | ||
P value a | 0.0843 | 0.0141 | ||
Patients with ≥ 0.5 CFB, n (%)b | |||||||||| | |||||||||| | |||||||||| | |||||||||| |
OR (95% CI)c | |||||||||| | |||||||||| | ||
P valuec | |||||||||| | |||||||||| | ||
Change from baseline at week 52 | ||||
N | |||||||||| | |||||||||| | |||||||||| | |||||||||| |
Mean (SD) CFB | |||||||||| | |||||||||| | |||||||||| | |||||||||| |
LS mean (SE), CFB week 52 a | 1.53 (0.06) | 1.19 (0.08) | 1.56 (0.07) | 1.23 (0.09) |
Difference vs. placebo (95% CI)a | 0.34 (0.16 to 0.52) | 0.33 (0.12 to 0.53) | ||
P valuea | 0.0002 | 0.0020 | ||
Patients with ≥ 0.5 CFB, n (%)b | |||||||||| | |||||||||| | |||||||||| | |||||||||| |
OR (95% CI)c | |||||||||| | |||||||||| | ||
P valuec | |||||||||| | |||||||||| | ||
CFB = change from baseline; CI = confidence interval; LS = least squares; MMRM = mixed-effect model with repeated measures; OR = odds ratio; PAQLQ(S)-IA = Standardised Paediatric Asthma Quality of Life Questionnaire–Interviewer Administered; SD = standard deviation; SE = standard error.
Notes: Type 2 inflammatory asthma phenotype population is defined as the randomized patients with baseline blood eosinophils of 0.15 Giga/L or greater or baseline fractional exhaled nitric oxide of 20 parts per billion or greater.
Only patients of aged 7 years or older at randomization are included in the analysis.
aDerived from an MMRM with change from baseline in PAQLQ(S)-IA global score values up to week 52 as the response variable, and treatment, age, baseline weight group, region, baseline eosinophil level, baseline fractional exhaled nitric oxide level, baseline inhaled corticosteroid dose, visit, treatment by-visit interaction, baseline PAQLQ(S)-IA global score value, and baseline-by-visit interaction as covariates.
bA responder is defined as a patient with improvement from baseline in PAQLQ(S)-IA global score of 0.5 or greater. Patients with an improvement of less than 0.5 or with missing value are considered to be nonresponders.
cDerived from a logistic regression model with treatment, age, baseline weight group, region, baseline eosinophil level, baseline fractional exhaled nitric oxide level, baseline inhaled corticosteroid dose, and baseline PAQLQ(S)-IA global score as covariates.
Source: Clinical Study Reportt.7


||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||
|---|---|---|---|---|
|||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||
|||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||| | |||||||||| |||||||||| |||||||||| ||||| |
|||||||||| |||||||||| |||||||||| ||||| | ||||
|||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||| | |||||||||| |||||||||| |||||||||| ||||| |
|||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||| | |||||||||| |||||||||| |||||||||| ||||| |
|||||||||| |||||||||| |||||||||| ||||| | ||||
|||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||| | |||||||||| |||||||||| |||||||||| ||||| |
|||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||| | |||||||||| |||||||||| |||||||||| ||||| |
|||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||| | |||||||||| |||||||||| |||||||||| ||||| |
|||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | ||
|||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | ||
|||||||||| |||||||||| |||||||||| ||||| | ||||
|||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||| | |||||||||| |||||||||| |||||||||| ||||| |
|||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||| | |||||||||| |||||||||| |||||||||| ||||| |
|||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||| | |||||||||| |||||||||| |||||||||| ||||| |
|||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | ||
|||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | ||
|||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||| | |||||||||| |||||||||| |||||||||| ||||| |
|||||||||| |||||||||| |||||||||| ||||| | ||||
|||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||| | |||||||||| |||||||||| |||||||||| ||||| |
|||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||| | |||||||||| |||||||||| |||||||||| ||||| |
|||||||||| |||||||||| |||||||||| ||||| | ||||
|||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||| | |||||||||| |||||||||| |||||||||| ||||| |
|||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||| | |||||||||| |||||||||| |||||||||| ||||| |
|||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||| | |||||||||| |||||||||| |||||||||| ||||| |
|||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | ||
|||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | ||
|||||||||| |||||||||| |||||||||| ||||| | ||||
|||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||| | |||||||||| |||||||||| |||||||||| ||||| |
|||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||| | |||||||||| |||||||||| |||||||||| ||||| |
|||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||| | |||||||||| |||||||||| |||||||||| ||||| |
|||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | ||
|||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | ||
|||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||| | |||||||||| |||||||||| |||||||||| ||||| |
|||||||||| |||||||||| |||||||||| ||||| | ||||
|||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||| | |||||||||| |||||||||| |||||||||| ||||| |
|||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||| | |||||||||| |||||||||| |||||||||| ||||| |
|||||||||| |||||||||| |||||||||| ||||| | ||||
|||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||| | |||||||||| |||||||||| |||||||||| ||||| |
|||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||| | |||||||||| |||||||||| |||||||||| ||||| |
|||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||| | |||||||||| |||||||||| |||||||||| ||||| |
|||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | ||
|||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | ||
|||||||||| |||||||||| |||||||||| ||||| | ||||
|||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||| | |||||||||| |||||||||| |||||||||| ||||| |
|||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||| | |||||||||| |||||||||| |||||||||| ||||| |
|||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||| | |||||||||| |||||||||| |||||||||| ||||| |
|||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | ||
|||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | |||||||||| |||||||||| |||||||||| ||||| | ||
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Source: Clinical Study Report.7
Reduction in Use of Rescue Medication
Regarding the use of reliever medications in the VOYAGE trial, a decrease in number of puffs in 24-hour period was observed in both treatment arms. The LS mean difference versus placebo for the number of puffs of reliever medication in a 24-hour period at week 24 was |||||||||| |||||||||| ||||||||||||||| |||||||||| ||||| and |||||||||| |||||||||| ||||||||||||||| |||||||||| ||||| for the 2 populations, respectively. At week 52, |||||||||| |||||||||| ||||||||||||||| |||||||||| ||||||||||||||| |||||||||| ||||| (Table 25).
|||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| | |||
|---|---|---|---|---|
|||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| | |||
|||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| | |
|||||||||| |||||||||| ||||| | ||||
|||||||||| |||||||||| ||||| | ||||
|||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| |
|||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| |
|||||||||| |||||||||| ||||| | ||||
|||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| |
|||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| |
|||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| |
|||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| | ||
|||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| | ||
|||||||||| |||||||||| ||||| | ||||
|||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| |
|||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| |
|||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| |
|||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| | ||
|||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| | |||||||||| |||||||||| ||||| | ||
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Source: Clinical Study Report.7
Improvements in Symptoms of Atopic Dermatitis and Rhinosinusitis
In patients with comorbid allergic rhinitis, PRQLQ–Interviewer Administered global scores at weeks 24 were improved in favour of dupilumab (LS mean difference between groups = |||||||||| |||||||||| ||||||||||||||| |||||||||| |||||, in the type 2 phenotype population. Improvements in allergic rhinitis related quality of life were also observed in the baseline blood EOS of 300 cells/µL or greater population (LS mean difference between groups = |||||||||| |||||||||| ||||||||||||||| |||||||||| ||||||||||||||| |||||||||| |||||) (Table 24).
Harms
Only those harms identified in the review protocol are reported in the following. Refer to Table 26 for detailed harms data.
Adverse Events
AEs in the dupilumab versus placebo groups occurred in 83% versus 79.9% of patients in the safety population of the VOYAGE trial, respectively (Table 26). The most common AEs for dupilumab versus placebo were nasopharyngitis (18.5% versus 21.6%), viral upper respiratory tract infection (12.2% versus 9.7%), pharyngitis (8.9% versus 10.4%), bronchitis (6.3% versus 10.4%), allergic rhinitis (5.9% versus 11.9%), injection site erythema (12.9% versus 9.7%) and injection site edema (10.3% versus 5.2%).
Serious Adverse Events
SAEs occurred in 4.8% patients receiving dupilumab and 4.5% of patients receiving placebo, most common of which were asthma (dupilumab versus placebo: 1.5% versus 0%) and eosinophilia (dupilumab versus placebo: 0.7% versus 0%).
Withdrawals Due to AEs
AE resulting in discontinuation of study drug occurred in 1.8% versus 1.5% of patients of the VOYAGE study, in the dupilumab versus placebo groups, respectively.
Notable Harms
Injection site reactions were the most commonly reported notable harms, with 17.7% patients versus 13.4% of patients in the dupilumab versus placebo groups, respectively. Hypersensitivity and anaphylactic reactions occurred in 1.8% of dupilumab patients versus 3.7% of placebo patients and 0.0% of dupilumab patients versus 1.5% of placebo patients, respectively. Severe infections occurred in |||||||||| of patients in the dupilumab and |||||||||| of patients in the placebo group. Parasitic infections were reported among 7 patients (2.6%) in the dupilumab group, and no patients in the placebo arm. There were no opportunistic infections reported in the trial. Eosinophilia was more frequently occurring in dupilumab arm compared to placebo (6.6% versus 0.7%, respectively). Conjunctivitis, classified using a narrow custom Medical Dictionary for Regulatory Activities (MedDRA) Query search, occurred in 2.6% of the dupilumab group versus 6.7% of the placebo group. Similarly, conjunctivitis classified using a broad MedDRA search, occurred in 3.0% of the dupilumab group versus 7.5% of the placebo group.
Table 26: Summary of Harms (Safety Population)
Harms outcome | VOYAGE trial | |
|---|---|---|
Safety population | ||
Dupilumab (n = 271) | Placebo (n = 134) | |
AEs | ||
Any AE, n (%) | 225 (83.0) | 107 (79.9) |
Nasopharyngitis | 50 (18.5) | 29 (21.6) |
Viral upper respiratory tract infection | 33 (12.2) | 13 (9.7) |
Pharyngitis | 24 (8.9) | 14 (10.4) |
Influenza | 20 (7.4) | 12 (9.0) |
Bronchitis | 17 (6.3) | 14 (10.4) |
Sinusitis | 9 (3.3) | 7 (5.2) |
Eosinophilia | 16 (5.9) | 1 (0.7) |
Headache | 19 (7.0) | 10 (7.5) |
Rhinitis allergic | 16 (5.9) | 16 (11.9) |
Cough | 15 (5.5) | 9 (6.7) |
Injection site erythema | 35 (12.9) | 13 (9.7) |
Injection site edema | 28 (10.3) | 7 (5.2) |
Injection site nodule | 17 (6.3) | 3 (2.2) |
Accidental overdose | 3 (1.1) | 7 (5.2) |
SAEs | ||
Any SAE, n (%) | 13 (4.8) | 6 (4.5) |
Asthma | 4 (1.5) | 0 (0.0) |
Eosinophilia | 2 (0.7) | 0 (0.0) |
WDAE | ||
AE leading to treatment discontinuation, n (%) | 5 (1.8) | 2 (1.5) |
Deaths | ||
n | 0 (0.0) | 0 (0.0) |
Notable harms, n (%) | ||
Anaphylactic reaction | 0 (0.0) | 2 (1.5) |
Hypersensitivity (medically reviewed) | 5 (1.8) | 5 (3.7) |
Severe infections | |||||||||| | |||||||||| |
Opportunistic infections | 0 (0.0) | 0 (0.0) |
Parasitic infections | 7 (2.6) | 0 (0.0) |
Serious injection site reactions | 2 (0.7) | 0 (0.0) |
Injection site reactions | 48 (17.7) | 18 (13.4) |
Conjunctivitis (narrow) | 7 (2.6) | 9 (6.7) |
Conjunctivitis (broad) | 8 (3.0) | 10 (7.5) |
Eosinophilia | 18 (6.6) | 1 (0.7) |
AE = adverse event; SAE = serious adverse event; WDAE = withdrawal due to adverse event.
Note: Definitions of harms: Conjunctivitis (narrow company medical query): preferred term in (conjunctivitis, conjunctivitis allergic, conjunctivitis bacterial, conjunctivitis viral, atopic keratoconjunctivitis). Conjunctivitis (broad company medical query): preferred term in (conjunctivitis, conjunctivitis allergic, conjunctivitis bacterial, conjunctivitis viral, atopic keratoconjunctivitis, blepharitis, dry eye, eye irritation, eye pruritus, lacrimation increased, eye discharge, foreign body sensation in eyes, photophobia, xerophthalmia, ocular hyperaemia, conjunctival hyperaemia).
Source: Clinical Study Report.7
Critical Appraisal
Internal Validity
The VOYAGE trial is a multinational, multicentre, randomized, double-blind, placebo-controlled study. Treatment allocation was appropriately performed through a central interactive voice and web response systems. However, the randomization ratio of 2:1 was implemented in the trial, and no justification was provided from the sponsor. Furthermore, the study randomization was stratified by ICS dose level (medium or high) at screening, patients’ blood eosinophil count (< 300 cells/µL and ≥ 300 cells/µL populations) at screening, and region (Latin America, Eastern Europe, and Western countries).
The study was double-blind, and both the patients and investigators were blinded to the study treatment. Additional steps to maintain blinding were undertaken by using a matching placebo in the trial (i.e., 2 different volumes of injection solutions, corresponding to the 2 different doses of dupilumab were used; these were each matched to corresponding volumes of placebo injection). However, this meant that patients and investigators were aware of the dose or volume of the injections administered in the trial. Even though dosing awareness might have introduced the possibility of bias, the fact that both patients and investigators were blinded to the type of treatment would have mitigated this bias. Furthermore, patients in the dupilumab group experienced more injection site reactions and eosinophilic reactions compared to placebo, which might have created possibilities for study participants or investigators to anticipate the AEs of active treatment, therefore, introducing unblinding in the trial. The impact of such unblinding (i.e., patients believing they were assigned to the treatment group rather than placebo) might have introduced bias in assessing patient-reported and HRQoL outcomes of the trial. During the VOYAGE trial, breaking of the blind leading to study discontinuation occurred in a limited number of patients receiving dupilumab. These irregularities were reported at the local level among 4 patients from Brazil (discontinued due to yellow fever vaccination) and 1 patient from US (reported AE of eosinophilia, headache, and blurred vision, leading to study discontinuation).
The sponsor controlled for multiplicity using a hierarchical testing procedure, which was applied for the primary outcome (annualized rate of severe exacerbation events during the 52-week placebo-controlled treatment period), key secondary outcome (change from baseline in prebronchodilator percent predicted FEV1 at week 12) and secondary outcomes (change in ACQ-7-IA at week 24, change from baseline in FeNO at week 12). Each hypothesis was formally tested only if the preceding 1 was significant at the 2-sided 5% level. There were 2 distinct sequential testing procedures that incorporated the same end points, but in a different order, based on the approved indication in adults and adolescents in the US and European Union. For European Union and European Union reference countries, the hierarchy procedure started with the type 2 inflammatory asthma phenotype population, and for the US and US reference countries, the hierarchy procedure started with the population with baseline blood eosinophil count of 0.3 Giga/L or greater. Of note, statistical significance was reached across all multiplicity-controlled end points in both hierarchical testing procedures.
Sample size and power calculations were based on the primary outcome for the populations with a type 2 inflammatory phenotype, baseline blood EOS of 300 cell/μL or greater, and of 150 cells/μL or greater. When calculating sample size and power of the trial, the sponsor made assumptions regarding expected RR reductions in the pediatric population, based on data previously reported from trials on individuals aged older than 12 years.34 Considering that there is a paucity of data in pediatric populations, this approach was deemed reasonable for a priori estimation of the sample size.
Regarding baseline characteristics, there were slight imbalances across the groups in terms of severe exacerbations (1, 2, 3, or ≥ 4) in the year before the study enrolment, but the median number exacerbations for the dupilumab and placebo groups was the same (median = 2). Moreover, FEV1 reversibility was slightly higher in the dupilumab compared to placebo group, which might have introduced bias in the study findings either for or against the study drug. Bias for the active treatment group might have occurred due to the fact that the VOYAGE trial included patients with uncontrolled asthma, who were more likely to respond to a new therapy and show a difference versus placebo. On the other hand, clinical experts reported that higher baseline reversibility in the dupilumab group suggested that that this group may be more uncontrolled than the placebo, making it more difficult for the active treatment group to reach a beneficial effect, compared to placebo (bias against the drug). Nevertheless, other baseline and demographic characteristics were largely balanced within the 2 study arms, suggesting that randomization was successfully implemented. Screening failure was reported in 35.3% of the initial number of patients, most commonly occurring because patients failed to meet prespecified inclusion criteria of the trial. However, clinicians consulted during this CADTH review reported that higher levels of screening out are very common in asthma biologic trials, noting that virally triggered asthma presentation, commonly reported in pediatric population, might have accounted for these screening failures.
Withdrawals from the study were generally low in the VOYAGE study, but higher proportions were observed in the dupilumab group (dupilumab versus placebo for the type 2 population: ||||||||||||||||||||; dupilumab versus placebo for the baseline EOS ≥ 300 cells/µL population: ||||||||||||||||||||). Similarly, discontinuation from the treatment occurred in approximately 8% of individuals receiving dupilumab, compared to approximately 3.5% of patients receiving placebo across the 2 efficacy populations. According to the data presented in the study report, proportions of individuals who discontinued study treatment due to an AE were balanced across the 2 study arms (approximately 2% for both study arms in the type 2 population and approximately 1% for both study arms in the baseline EOS ≥ 300 cells/µL population), suggesting lower possibility of attrition bias. In an attempt to account for missing data for the primary and key secondary outcomes, the sponsor performed a variety of sensitivity analyses, which are appropriate for scenarios when data are not missing at random. Finding of the sensitivity analyses were aligned with the primary findings. Considering all the above, CADTH deemed that the impact of missing data on the estimated treatment effects was limited.
Annualized rate of severe exacerbation events during the 52-week placebo-controlled treatment period and change from baseline in prebronchodilator percent predicted FEV1 at week 12 were the primary and the key secondary outcomes included in the statistical hierarchy of the VOYAGE trial, respectively. Both outcomes were considered clinically relevant for patients with severe asthma, according to the clinician experts consulted and patient input provided. The definition of the primary outcome (i.e., deterioration in asthma requiring SCSs for at least 3 days or resulting in hospitalizations or ED visits requiring SCSs) as well as the interval between which 2 events were considered as separate (28 days) were regarded as appropriate, according to the clinical experts. Assessment and recording of severe exacerbations were conducted by a treating physician through the electronic case reports. Clinical experts reported that measuring percent predicted values of prebronchodilator FEV1, instead of absolute values, was appropriate for the pediatric population under study. However, the clinicians stated the 12-week assessment of FEV1 was not optimal for assessing the treatment effects of dupilumab versus placebo on pulmonary function because it would not be a sufficient duration to assess the seasonal and other factors that affect this outcome. The experts regarded the 52-week assessment of FEV1 as more clinically relevant.
In the primary analyses for the primary and key secondary outcomes, off-treatment measurements of patients prematurely discontinuing treatment were included in the models, while data of patients after study discontinuations were censored. A series of sensitivity analyses were conducted by the sponsor in an attempt to evaluate the robustness of the primary findings. Specifically, on-treatment analyses to assess treatment effects if patients adhere to the study treatment as directed and missing data imputations (PMM-MI, PMM placebo-based and tipping point analyses) were conducted for both outcomes, while censoring methods according to the SCS usage were performed only for the key secondary outcome. Considering that the multiple imputation methods applied in the VOYAGE trial are applicable to situations when data are not missing at random, CADTH evaluation deemed the methodology applied as appropriate. Findings from the sensitivity analyses were consistent with the primary analyses across the 2 main efficacy populations of the trial.
The VOYAGE trial prespecified a series of subgroup analyses for the primary and the key secondary outcome, aligned with the subgroups of interest identified in the CADTH protocol (baseline eosinophil levels, baseline ICS dose, number of previous asthma exacerbations, atopic medical history). However, only subgroups of patients with baseline blood EOS of 300 cells/µL or greater, baseline blood EOS greater than150 cells/µL and patients receiving high doses of ICS were adjusted for multiple statistical comparisons, as they were part of the hierarchical testing procedure. Statistical tests of interaction were performed to test whether treatment effects differed among subgroups. However, the analyses for other subgroups of interest were not adjusted for multiplicity, and results from these analyses were treated as supportive of the overall benefit of dupilumab but no decisive conclusion can be drawn.
Another secondary outcome included in the hierarchical testing of the VOYAGE trial covered asthma symptoms, measured by a well-established and validated questionnaire (ACQ-7). The MID of 0.5 was established, which is reviewed in detail in Appendix 4.24,25 Although not part of the statistical hierarchy, data regarding the responders’ analysis for the ACQ-7 was reported in the trial, with a response being defined using the 0.5 MID.
Of note, multiplicity adjustments were not conducted for other secondary and exploratory end points of interest, including annualized rates of severe asthma exacerbations resulting in hospitalization or ED visits and resulting in hospitalization only, HRQoL (PAQLQ, PACQLQ, EQ-5D-Y, PRQLQ), pulmonary function (PEF), SCS exposure, reliever medication use, and ICS dose adjustment; therefore, the results for these outcomes will be considered as supportive of the treatment effect but no definitive conclusions can be made.
The results reported in the Clinical Study Report of the VOYAGE trial were based on all the data collected up to the database lock on August 26, 2020. The study protocol underwent 3 global amendments and 1 local amendment for Brazil. Notable changes included the following: change to the study primary efficacy populations (from an overall uncontrolled persistent asthma population to the population with baseline eosinophil count ≥ 300 cells/µL or with the type 2 inflammatory asthma); changes to the sample size; specification of different testing hierarchies based on the US, European Union, and their reference countries; removal of the limitations for enrolling patients according to the background therapy, and EOS levels. These amendments were made before the database lock, suggesting that their ability to affect the end results or imply bias due to patient selection is limited.
Overall, no critical deviations from the protocol were reported during the VOYAGE trial. The sponsor identified a subset of major deviations, which had the potential adverse impact on integrity of the data, patients’ rights, or safety of the participants. These occurred in about 2% of patients across both the placebo and dupilumab arms in the 2 main efficacy populations and included: missing FeNO assessments at baseline, use of prohibited concomitant medications during the treatment period, less than 80% adherence with the treatment under study, and nonadherence with mandatory background therapy during the screening period. Considering that the number of patients with important deviations was low and balanced across the 2 study groups, CADTH reviewers deemed the risk of bias owing to protocol deviations to be low and to have negligible influence on comparative efficacy findings. Moreover, some randomization and dosing irregularities happened during the trial due to a difference in the ICS dose level as reported by the investigator at randomization in comparison to the actual calculated total daily dose of all medications containing ICSs, which was derived after converting to fluticasone dose equivalent according to GINA guidelines.1 The irregularities were fairly balanced between the study groups (7.6% and 6.1% of type 2 population patients in the dupilumab and placebo group, respectively; 6.3% and 6.0% of baseline EOS ≥ 300 cells/µL patients in the dupilumab and placebo group, respectively). To overcome this, the sponsor specified that the analyses factoring ICS dose levels were conducted according to the actual calculated total daily dose of all medications containing ICSs and not on the investigators’ reported classification.
External Validity
The VOYAGE study was a multinational, multicentre trial spanning across 90 sites in 17 countries, including Canada. The population requested for the reimbursement is narrower than the population included in the Health Canada–approved indication, as the OCS-dependant individuals were not taking part in the VOYAGE trial. According to the clinical experts, the number of pediatric patients on regular OCS therapy is limited, but many patients might experience frequent and repeated short course of OCS treatment to gain control of their symptoms or for exacerbations.
Overall, the clinical experts consulted by CADTH agreed that the inclusion and exclusion criteria of the trial were reflective for the severe asthma pediatric population. They also believed that the baseline and demographic characteristics of patients in the trial were largely consistent with the population that would be expected to use the drug in the real-world practice (i.e., poorly controlled individuals experiencing high FEV1 and high reversibility despite being on appropriate therapy). Of note, the majority of the study population included individuals who were White; hence, generalizability of study findings to people living in Canada may be limited in this regard. Moreover, 1 of the main efficacy populations in the VOYAGE trial required that the identification of eligible patients be performed based on FeNO levels. Clinical experts consulted for this review reported that FeNO assessments are not routinely performed in Canadian clinical practice, which limits the generalizability of the study population.
According to the experts, the dosing of dupilumab applied during the clinical trial was consistent with the dose that would be applied in the Canadian clinical practice per the Health Canada–approved indication. Background medications administered during the trial were considered appropriate by the clinical experts consulted and reflective of medications administered in the Canadian setting. The experts also noted that adherence to therapy is a major issue in the management of asthma. The study documentation did not report whether the inhaler technique was checked throughout the trial, which can be considered a limitation. However, adherence to background therapy was high in both groups across the 2 efficacy populations during the study, suggesting that patients might have benefited from the close attention and monitoring of the trial. This might have also explained the level of benefits observed in the placebo population, which was receiving only background medication.
The VOYAGE trial did not compare dupilumab to any of the other biologics approved for the management of asthma in pediatric population, such as IgE inhibitors or the IL-5 inhibitors. Comparison of dupilumab to placebo (added on to standard of care) may be appropriate for establishing efficacy; however, the lack of an active control represents a limitation when trying to assess the comparative effects of dupilumab, leaving indirect comparisons available to assess the relative efficacy and harms of dupilumab compared to other biologics.
Clinical experts consulted during this CADTH review highlighted that the primary and key secondary outcomes were relevant outcomes for assessing pediatric patients with asthma, notably exacerbation rate, pulmonary function (FEV1), and symptoms (ACQ-7). One of the clinical experts consulted for this CADTH review noted that ACQ is mostly applied for research purposes, but additionally commented that the tool can be regarded as appropriate and valid for assessing symptoms in the pediatric population.
Indirect Evidence
Objectives and Methods for the Summary of Indirect Evidence
Due to the lack of direct evidence comparing dupilumab with other existing therapies as an add-on maintenance treatment in patients aged 6 years to younger than 12 years with severe asthma with a type 2 or eosinophilic phenotype or with OCS-dependent asthma, the sponsor submitted 1 ITC analysis.35 In addition, CADTH’s focused literature search for ITCs dealing with asthma was run-in MEDLINE All (1946–) on July 26, 2022. No limits were applied to the search. No published ITCs were identified in the CADTH literature search. The objective of this section is to summarize and critically appraise the indirect evidence from the sponsor-submitted ITC. To align with the research protocol of this review, only information pertaining to the criteria outlined in Table 5 are presented in this section.
Description of Indirect Comparison
The sponsor submitted 1 ITC, which had the objective to identify, evaluate, and synthesize the empirical evidence on the clinical efficacy of dupilumab compared to other recommended biologics for the treatment of pediatric patients who were aged 6 years to younger than 12 years with uncontrolled, moderate-to-severe asthma with a type 2 inflammatory phenotype.
Methods of the Sponsor-Submitted ITC
Study Selection Methods
Systematic Literature Review
The systematic review was conducted in line with the PRISMA guidelines36,37 and Cochrane Handbook,38 and the search strategy was developed based on the PICOS-T criteria presented in Table 27 to identify relevant studies investigating the efficacy and safety of dupilumab against other existing treatments. The systematic literature search was conducted by screening electronic literature databases, spanning from 1998 (aligned with the earliest published omalizumab trial in pediatric asthma) to February 2021. The searches were limited to studies conducted in humans and published in English language. In addition, grey literature searches (January 2019 to February 2021) were conducted as well as snowball screening of the bibliographies of relevant systematic reviews identified across the electronic database. Methods for extracting data and performing quality assessment are presented in the Table 27.
The original searches identified 2,317 publications for further screening. Following screening and a feasibility assessment, a total of 3 trials were included in the ITCs (1 evaluating treatment with dupilumab7 and 2 trials evaluating treatment with omalizumab39,40).
Network Meta-Analysis Feasibility Assessment
To ensure that the assumptions inherent to ITCs were appropriate for any planned analyses, the clinical heterogeneity across all included studies was assessed. This was done by establishing a list of potential treatment effect modifiers (Table 28), which was developed by reviewing subgroup results across the included RCTs and consultations with medical experts to discuss selected subgroups as well as comparability of included trials and feasibility of the analyses.
Table 27: Study Selection Criteria and Methods for the Sponsor-Submitted ITC
Criteria | ITC |
|---|---|
Population | Pediatric patients of aged 6 to < 12 years with uncontrolled, moderate-to-severe asthmaa Subgroups of interest: Type 2 inflammation, EOS ≥ 300 cells/μL, EOS ≥ 150 cells/μL, FeNO ≥ 20 parts per billion |
Intervention | Biologics administered as add-on to standard of care such as dupilumab, mepolizumab, omalizumab, benralizumab, and reslizumab |
Comparator | Any intervention of interest or none for single-arm trials |
Outcome |
|
Study design | Phase II, III, and IV clinical trials, including: randomized controlled trials (including crossover designs), open-label trials (including long-term extensions), single-arm trials, and pooled analysis of eligible trials |
Time frame |
|
Publication characteristics | Only human studies and English-language articles included |
Exclusion criteria | Main exclusion:
|
Databases searched |
|
Selection process | Articles screened independently by 2 researchers; any discrepancies resolved by engaging a third researcher Pilot screening for testing the questions performed before formal screening |
Data extraction process | Data extraction performed by 1 researcher and quality-checked by an independent senior researcher. |
Quality assessment | Cochrane Risk of Bias Assessment Tool 1.0, as recommended by The National Institute for Health and Care Excellence (Section 2.5 of the User Guide for Single Technology Appraisal), conducted by 1 researcher and validated by a senior researcher.41 |
ACQ = Asthma Control Questionnaire; ACQ-5-IA = 5-item Asthma Control Questionnaire–Interviewer Administered; ACQ-7-IA = 7-item Asthma Control Questionnaire–Interviewer Administered; AE = adverse event; EOS = eosinophils; EQ-5D-Y = EQ-5D-Youth; FEF = forced expiratory flow; FeNO = fractional exhaled nitric oxide; FEV1 = forced expiratory volume in 1 second; FVC = forced vital capacity; ITC = indirect treatment comparison; PAQLQ = Paediatric Asthma Quality of Life Questionnaire–Interviewer Administered; PACQLQ = Paediatric Asthma Caregiver’s Quality of Life Questionnaire; PEF = peak expiratory flow; PRO = patient-reported outcome; PRQLQ-IA = Pediatric Rhinoconjunctivitis Quality of Life Questionnaire–Interviewer Administered; SAE = serious adverse event; TEAE = treatment-emergent adverse event.
aNote: Studies conducted in mixed age populations were included if subgroup data were reported for ages of interest, if 80% or more of the included patients were within the age group of interest, or if the mean/median age was less than 12 years.
Source: Sponsor-submitted ITC report.35
Table 28: Characteristics of Potential Treatment Effect Modifiers
Category | Potential effect modifiers |
|---|---|
Population | Age, gender, weight/BMI, race/region, biomarkers (EOSa, IgEa), FeNOa, FEV1/ppFEV1a, exacerbations history, symptoms (ACQ-7-IA), presence of comorbidities |
Treatment | Dosing, frequency of administration, treatment duration, prior use and dosing of ICSs, prior use and dosing of OCSs, allowance of treatment rescue |
Outcomes | Summary statistic (e.g., rate vs. relative risk), baseline risk, definition of severe exacerbations, time points of assessment, subgroup availability |
Study design | Phase II, III RCTs, open-label vs. blinded design, follow-up duration, run-in and posttreatment phase, enrolment period and seasonality, sample size, quality of included studies, study region/racea |
ACQ-7-IA = 7-item Asthma Control Questionnaire–Interviewer Administered; BMI = body mass index; EOS = eosinophils; FeNO = fraction of exhaled nitric oxide; FEV1 = forced expiratory volume in 1 second; ICS = inhaled corticosteroid; IgE = immunoglobulin E; OCS = oral corticosteroid; ppFEV1 = percent predicted forced expiratory volume in 1 second; RCT = randomized controlled trial.
aEvidence of potential effect modification observed across the included trials.
Source: Sponsor-submitted indirect treatment comparison report.35
ITC Analysis Methods
The ITC technical report noted that the clinical experts consulted for the feasibility assessment were split in their assessments of the clinical importance of the heterogeneity between the identified studies. Nonetheless, an “exploratory” Bucher ITC comparing dupilumab with omalizumab was conducted. The ITC analyses included a total of 3 trials connected with placebo as a common comparator (VOYAGE: dupilumab trial; IA05 and ICATA: omalizumab trials). Data from the VOYAGE and IA05 trials were used in the base case ITCs, while data from the ICATA study were included in separate analyses of certain outcomes, such as severe exacerbations, because of differences in key characteristics between the VOYAGE and ICATA trials that “would not allow for sufficient comparability” in the base case.35
The technical report specified that the type 2 inflammatory population from the VOYAGE study was selected as the primary population of interest for the ITCs, in line with the regulatory agencies’ approvals. Since the type 2 asthma population data were not available in the omalizumab trials, an assumption was made that the efficacy of omalizumab would be maintained in the population of interest.
ITC analyses were performed for the following outcomes: severe exacerbations (annualized rates); deterioration of asthma from the VOYAGE study (post hoc analysis) versus severe exacerbations from the IA05 study (annualized rates); morning asthma symptom score at 24 weeks; rescue medication use at 24 weeks; change in PAQLQ(S)-Interviewer Administer at 24 weeks; discontinuations due to AEs at 52 weeks.
Statistical Approach
Since most of the analyses included a single trial per comparison in the network, a fixed-effect model was applied to the Bucher ITCs. For the analysis of severe exacerbations data, which included additional data from the ICATA trial, random effects Bucher ITC was performed.
The P values of significance for the ITC effects were computed in 2-sided tests. The technical report states that due to the limited number of trials in the ITCs, indirectly estimated effects with a P value of 0.05 or less were classified as statistically favourable, a P value greater than 0.05 and 0.15 or greater, as numerically favourable and a P value greater than 0.15 as comparable.
Results of the Bucher ITCs were presented as a central estimate of the relative effect of interest (MID for a continuous outcome and OR or RR for a binary outcome) along with 95% CIs. Analyses were carried out using the metafor package in R 4.0.3.
In the Bucher ITC, statistical heterogeneity for a treatment effect between the same comparison (i.e., omalizumab versus placebo in the IA05 and ICATA trials when the latter study is included) was evaluated using the I2 statistic for the studies. A consistency assessment between direct and indirect sources of evidence was not applicable in the ITCs because there were no head-to-head comparisons between dupilumab and other comparators.
Subgroup Analysis
To account for differences in patient traits (i.e., omalizumab trials included patients with allergic phenotype, assessed via a positive skin prick test or a positive in vitro response to ≥ 1 perennial allergen) and biomarker or clinical characteristics (baseline EOS, percent predicted FEV1, and prior exacerbations), an additional set of analyses was conducted in the “omalizumab-eligible” subgroup with allergic phenotype and corresponding to the inclusion criteria of the IA05 study. The following criteria were used in a post hoc analysis of VOYAGE to better align type 2 inflammatory patients from the VOYAGE trial with those in the IA05 trial:
Baseline weight between 20 kg to 50 kg and serum IgE level of 30 IU/mL to 1,300 IU/mL and weight-IgE values combinations based on omalizumab dosing table
At least 1 positive perennial allergen-specific IgE (concentration ≥ 0.35 IU/mL) among the following allergens: Alternaria tenuis/alternata; Cladosporium herbarum/hormodendrum; Aspergillus Fumigatus; Cat Dander; D. Farinae; D. Pteronyssinus; Dog Dander; and German Cockroach
Of note, the VOYAGE trial did not include the performance of skin prick test, so the presence of 1 positive perennial allergen-specific IgE was used as a proxy for the allergic phenotype in the IA05 study.
Sensitivity Analyses
To account for differences in the definition of the outcome of interest (severe exacerbations) and increase the comparability between the 2 trials included in the ITC, an additional post hoc analyses of VOYAGE data were performed using an outcome labelled as deterioration of asthma (Table 29).
Table 29: Overview of Outcomes of Interest Assessed in the Eligible Trials and Included in the ITC
Outcome | Trial-specific definition and time points assessed | ||
|---|---|---|---|
VOYAGE | IA05 | ICATA | |
Severe exacerbations | Deterioration of asthma requiring any of the following:
Time point: 52 weeks | Worsening of asthma symptoms requiring any of the following:
|
Time point: 48 weeks (24 weeks end of fixed steroid dosing) |
Asthma deterioration | Deterioration of asthma resulting in any of the following:
Time point: 52 weeks | Worsening of asthma symptoms requiring any of the following:
Time point: 52 weeks (24 weeks end of fixed steroid dosing) | NA |
Morning asthma symptom score | Morning asthma symptom score (impact on sleep; score range of 0 to 4, with 4 indicating more severe symptoms) Time point: 52 weeks | Nocturnal asthma symptom score (scale of 0 to 4, where 0 = no symptoms and 4 = breathing problems resulting in nocturnal symptoms despite use of rescue medication) Time point: 52 weeks | NR |
Rescue medication use | Use of rescue medication as measured by patient-recorded number of puffs of short-acting bronchodilator used per day over 2 to 60 weeks Time point: 2 to 60 weeks (assessment every 2 weeks until 12 weeks, and every 4 weeks thereafter) | Daily puffs of rescue medication Time point: 24 weeks | NR |
PAQLQ(S)-IA | LS mean change from baseline in PAQLQ(S)-IA Time point: weeks 12, 24, and 52 | LS mean change from baseline in PAQLQ(S)-IA Time point: 24 weeks | NR |
Discontinuation due to AEs | Any TEAE leading to permanent treatment discontinuation Time point: 52 weeks | Discontinued because of AE Time point: 52 weeks | NR |
AE = adverse event; ICS = inhaled corticosteroid; ITC = indirect treatment comparison; LS = least squares; NA = not applicable; NR = not reported; PAQLQ(S)-IA = Standardised Paediatric Asthma Quality of Life Questionnaire–Interviewer Administered; TEAE = treatment-emergent adverse event.
Source: Sponsor-submitted ITC report.35
Results of ITC
Summary of Included Studies
The 3 double-blind, RCTs trials were VOYAGE (dupilumab 100 mg to 200 mg every 2 weeks), IA05 (omalizumab 75 to 375 mg every 1 month or every 2 months), and ICATA (omalizumab 150 mg to 375 mg every 2 weeks or every 4 weeks), connected via placebo as a common comparator. Characteristics of the study design and patient populations across the included studies are presented in Table 30 and Table 31.
Assessment of Risk of Bias of Included Trials
Methodological quality of all included studies was assessed using the Cochrane Risk of Bias Assessment Tool 1.0. The technical report for the ITC states that the assessed trials were rated as low risk of bias overall. Specifically, all studies showed no evidence of selection, performance, and attrition bias. Both omalizumab trials had unclear reporting, and the presence of detection bias was rated as unclear. Moreover, presence of reporting bias was also unclear in the IA05 study.
|||||||||||| | ||||||||||||||||||||||| | ||||||||||||||||||||||| | ||||||||||||||||||||||| | ||||||||||||||||||||||| | |||||||||||||||||||| | ||||||||||||||||||||||| | ||||||||||||||||||||||| |
|---|---|---|---|---|---|---|---|
|||||||||||||||||||||||||||||||||||||||||||||||||| | |||||||
|||||||||||| | ||||||||||||||||||||||| | ||||||||||||||||||||||| | ||||||||||||||||||||||| | ||||||||||||||||||||||| | |||||||||||||||||||| | ||||||||||||||||||||||| | ||||||||||||||||||||||| |
|||||||||||||||||||||||||||||||||||||||||||||||||| | |||||||
|||||||||||| | ||||||||||||||||||||||| | ||||||||||||||||||||||| | ||||||||||||||||||||||| | ||||||||||||||||||||||| | |||||||||||||||||||| | ||||||||||||||||||||||| | ||||||||||||||||||||||| |
|||||||||||| | ||||||||||||||||||||||| | ||||||||||||||||||||||| | ||||||||||||||||||||||| | ||||||||||||||||||||||| | |||||||||||||||||||| | ||||||||||||||||||||||| | ||||||||||||||||||||||| |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Source: Sponsor-submitted indirect treatment comparison report.35
|||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
|---|---|---|---|---|---|---|---|---|---|---|---|
|||||||||||||||||||||||||||||||||||||||||||||||| | |||||||||||
|||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
|||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |
|||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
|||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |
|||||||||||||||||||||||||||||||||||||||||||||||| | |||||||||||
|||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
|||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |
|||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
|||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| | |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Source: Sponsor-submitted indirect treatment comparison report.35
Results
The key findings of the ITC are presented in Table 32, both for the type 2 Inflammatory asthma phenotype as well as the omalizumab-eligible type 2 inflammatory asthma phenotype patient populations. A visual representation of the evidence network is provided in Figure 7.

Note: This figure has been redacted.
Source: Sponsor-submitted indirect treatment comparison report.35
Dupilumab Versus Omalizumab
For the type 2 population, there was an improvement in annualized rates of severe exacerbations (||||||||||||||||||||||||||||||||||||||||||||||||||), and an improvement in deterioration of asthma for the comparison of dupilumab versus omalizumab (||||||||||||||||||||||||||||||||||||||||) (Table 32). No statistically significant between-group differences were found in terms of changes from baseline in morning asthma score, rescue medication use, and PAQLQ(S)–Interviewer Administered as well as withdrawals due to AEs.
Subgroup Analyses
In the omalizumab-eligible type 2 inflammatory subgroup, no significant differences were observed for the comparison of dupilumab with omalizumab in any of the outcomes assessed.
Sensitivity Analyses
Overall, the results of the sensitivity analyses that included data from additional omalizumab trial (ICATA) were aligned with the primary analyses of reduction in severe exacerbation events.
|||||||||||||||||||||||||||||| | |||||||||||||||||||| | |||||||||||||||||||| | |||||||||||||||||||||||||||||| | |||
|---|---|---|---|---|---|---|
|||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||| | ||||
|||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||| | ||||||||||||||||||| | |||
||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | ||||||
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | ||||||
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | ||||||
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Source: Sponsor-submitted indirect treatment comparison report.35
Critical Appraisal of ITC
Regarding the systematic literature review, the technical report adopted standard methods for conducting and reporting of reviews, aligned with Cochrane and PRISMA guidelines.36,38 Methods of the systematic review included defining the research question according to the PICOS-T criteria, searching through multiple database sources with comprehensive literature searches, adequate screening processes (double reviewers), data extraction using single reviewers and quality control by an additional reviewer, and quality appraisal of included studies.
A feasibility assessment for the ITC was performed and the rationale for exclusion of initially eligible studies in the ITC was provided. The technical report identified several patient characteristics for which there was “strong evidence of effect modification” and the report also suggested that there were limitations regarding reporting of characteristics in trials thereby complicating the assessment of heterogeneity. Based on this and input from 2 clinical experts (who were reported as disagreeing on the degree of the clinical importance of the identified effect modifiers), the technical report states that the ITC was exploratory given the potential limitations related to comparability of the trials. It is not clear what an exploratory ITC is or the intended objective for informing a reimbursement review, especially when the drug of interest—dupilumab—was compared with only 1 relevant comparator for which the included trials indeed appear to be heterogeneous.
||||||||||||||||||| were incorporated in the ITC base case and 1 additional study included in the separate/sensitivity analysis of severe exacerbations. The population of interest was the type 2 inflammatory population; however, data on this population were not available in the omalizumab trials, and an assumption was made that the efficacy of omalizumab would be maintained in the population of interest. It has been acknowledged in previous CADTH reviews and with input from clinicians that severe allergic asthma may also have an eosinophilic component (i.e., there is an overlapping patient population of patients with severe asthma who would be eligible for omalizumab as well as IL inhibitors). Therefore, there is validity to the assumption used in the ITC, but it is unclear how well the subset of patients from the omalizumab studies included in the ITC reflect this overlap population given the omalizumab trials were not designed specifically to include and identify these patients. It could not be determined what impact this had on the indirect estimates between dupilumab and omalizumab.
To improve the similarities between the trials in the ITC, a post hoc subgroup of patients with type 2 inflammatory asthma from the VOYAGE trial who had characteristics that may make them eligible for omalizumab was identified. This subgroup was identified largely based on IgE levels, which is an important marker of allergic asthma. However, it could not be determined how well this subpopulation matched the omalizumab trials population in part because the latter did not report baseline IgE levels. As well, the use of subgroup data resulted in a reduced sample size when compared to the original trial population (i.e., it included approximately 50% of the VOYAGE patient population), limiting the certainty of the results due to smaller evidence base as well as the generalizability of findings relative to the original trial population.
The Bucher indirect method assumes similar relative effectiveness of treatments across all trials, requiring that the included studies involve sufficiently similar populations to be compared. As noted, in this ITC, there were variations related to the study design, patient populations, and outcomes assessed across the studies included. The omalizumab and dupilumab studies varied in study region, length of the run-in periods, and treatment duration. Moreover, the fixed steroid dosing requirement differed among the trials. In contrast to the dupilumab trial, omalizumab trials required fixed steroid dosing for a limited period of 24 weeks, which influenced the outcome assessment in the ITC analysis. Of note, patients included in the omalizumab trials had asthma with an allergic phenotype, while the dupilumab trial did not apply this limitation in the patient population. Moreover, there were differences observed between groups at baseline in the racial distribution, baseline ICS dosing, baseline EOS levels, number of previous exacerbations as well as percent predicated FEV1 values, all of which may be prognostic or affect modifying factors. Additionally, as noted from the VOYAGE trial, patients randomized to placebo also saw improvements from baseline on several outcomes, including asthma control and lung function (i.e., an observable placebo response likely owing to improved administration of standard of care treatments). Therefore, it is not clear whether the placebo link for the ITC was sufficiently similar for making comparisons. Thus, the available data suggest the presence of residual heterogeneity and that key assumption of homogeneity was violated for the ITC analysis. According to the clinical experts consulted by CADTH, the differences in effect modifiers were considered clinically important, impacting the validity of the presented ITC analysis.
The ITC covered many of the relevant clinical outcomes for patients with severe asthma, identified in the CADTH review protocol (asthma exacerbations, HRQoL, symptoms, rescue medication usage, and AEs). However, there were variations in terms of the outcome definitions and outcome estimation time points across trials. To account for differences in severe exacerbation definition, a new outcome, called deterioration of asthma, was identified via post hoc analyses. Clinical experts consulted by CADTH reported that deterioration of asthma as an outcome could be considered comparable across the trials, but also noted that deterioration is already included in the definition of exacerbation and ACQ. Of note, a validated definition for this outcome was not identified. Given the fixed steroid dosing requirement for the omalizumab trials and variations in time points assessed across the studies, the ITC analyses were limited to 24 weeks for the majority of outcomes. Harms data were assessed at 52 weeks, a period which included ICS dose reduction in the omalizumab trial, which limits the interpretability of these findings. Finally, the majority of the analyses included single trial available per comparison in the network, and fixed effects Bucher ITCs were adequately applied to these assessments (except for the sensitivity analysis of severe exacerbations that included 2 omalizumab trials). However, the approach for considering diverse P value cut-offs for ITC effects as comparable, numerically, or statistically favourable in the technical report were not supported by literature and were deemed inappropriate by CADTH reviewers. Also, limited data availability restricted the possibility to perform a meta-regression and account for differences across trials, such as variation in the time points or study and patient characteristics. There were no direct comparisons between treatments, therefore, the assessment of consistency was not feasible.
Due to the limitations described above, the findings of the ITCs can be regarded as highly uncertain, and no conclusions can be drawn on the comparative efficacy of dupilumab compared to omalizumab in patients with uncontrolled moderate-to-severe asthma.
Other Relevant Evidence
This section includes an additional study that could address important gaps in the evidence, such as a short follow-up in the pivotal study.
Long-Term Extension Studies
One sponsor-submitted long-term extension study, EXCURSION, has been summarized to provide evidence regarding dupilumab as add-on therapy to ICSs with or without another controller for the treatment of moderate-to-severe uncontrolled asthma in children aged 6 years to younger than 12 years.20 This report presents the results from the ongoing EXCURSION study as of the data cut-off date of August 18, 2020, and from a second interim analysis with a database lock date of January 17, 2022.42
Methods
The EXCURSION study (N = 365) is a multicentre (involving 17 countries including Canada), open-label, noncomparative study evaluating dupilumab given subcutaneously to pediatric patients aged 6 years to younger than 12 years with asthma who participated in the VOYAGE study. All patients enrolled in EXCURSION received open-label treatment with dupilumab for a period of 52 weeks. After completion of the 1-year treatment period, all patients were to continue into the 12-week posttreatment period. This study was designed to evaluate the long-term safety and tolerability of dupilumab, as well as its long-term efficacy.
Populations
To be eligible for enrolment into the EXCURSION study, pediatric patients with asthma must have completed the parent study, the VOYAGE trial. The placebo–dupilumab group is defined as the patients who were in the placebo group of the parent study and then exposed to dupilumab in the long term study. The dupilumab–dupilumab group is defined as the patients who were in the dupilumab group of the parent study and exposed to dupilumab in the long term study.
The baseline characteristics were reported from the time of enrolment in the parent study, VOYAGE. For a full list of baseline characteristics of the VOYAGE study, refer to Table 7. At week 0 of the EXCURSION study, age, weight, height, body mass index, ICS dose level, serum total IgE, blood eosinophil count, and FEV1 (pre- and postbronchodilator, percent predicted, and reversibility [%]) were re-assessed; however, these are not reported in this section.
Interventions
The dosing regimens in the EXCURSION study are the same as the ones evaluated in the VOYAGE trial.
Patients whose weight changed from 30 kg or less to greater than 30 kg during the study switched their dosing regimen from 100 mg every 2 weeks or 300 mg every 4 weeks to 200 mg every 2 weeks and maintained the same dosing regimen regardless of further weight changes.
All patients in EXCURSION were receiving ICSs with or without a second controller (e.g., LABA, LTRA, LAMA, or methylxanthines) as background maintenance therapy. Also, patients were allowed to use albuterol/salbutamol or levalbuterol/levosalbutamol as a reliever therapy as needed during the study.
All patients were required to remain on stable dosing regimens of their background medications. During deterioration, the ICS dose could be increased or switched to SCS (for a severe exacerbation) temporarily as indicated by the physician and/or the sponsor. When the exacerbation resolved, patients could change back to the original ICS dose or modified ICS dose depending on their control of asthma symptoms.
Outcomes
The primary objective of the EXCURSION study was to assess the long-term safety and tolerability of dupilumab. The efficacy outcome definitions in the EXCURSION study are the same as those in the VOYAGE study.
Statistical Analysis
The results from the EXCURSION study were presented as descriptive summary statistics with observed data only. No model-based imputation for missing data was performed. Also, statistical adjustment including multiplicity adjustment was not conducted. The results were presented by treatment categories according to the actual treatment group in the parent study VOYAGE (i.e., the dupilumab–dupilumab and the placebo–dupilumab groups).
The full analysis set contained all data observed in the study without excluding any data or censoring and was used for the planned analyses. For efficacy outcomes, results are presented by 2 main subgroups, namely patients with type 2 inflammatory asthma phenotype at baseline of the parent study (blood eosinophil ≥ 0.15 Giga/L or FeNO ≥ 20 ppb) and patients with blood eosinophil of 300 cells/µL or greater at baseline of the parent study.
The term “baseline” refers to the baseline of the parent study VOYAGE, while the term “week 0” refers to the beginning of the EXCURSION study (at enrolment, before the first dupilumab administration). The full analysis set includes data collected from all enrolled patients regardless of the dosing schedules they had been assigned to in the parent trial, VOYAGE.
Patient Disposition
Patient disposition in the EXCURSION study as of both interim analyses is summarized in Table 33. A total of 365 patients (ITT population from the VOYAGE study, ||||||||||||||||||| from dupilumab group, ||||||||||||||||||| from placebo group) were rolled over from the parent study and received open-label dupilumab for an additional 52 weeks in the EXCURSION study. Of |||||||||||||||||||||||||||||||||||||| previously received dupilumab and ||||||||||||||||||| previously received placebo in the VOYAGE study.
At the cut-off date of August 18, 2020, |||||||||||| of the dupilumab–dupilumab group and |||||||||||| of the placebo–dupilumab group completed the study treatment, respectively. It was noted that more patients in the dupilumab–dupilumab group discontinued treatment compared to the placebo–dupilumab group (||||||||||||) due to AEs, poor treatment adherence, voluntary withdrawal, and other reasons A total of |||||| additional patients (|||||| in dupilumab–dupilumab group and |||||| in placebo–dupilumab group) discontinued study after completion of treatment period due to AEs, poor treatment adherence, or other reasons (|||||| in dupilumab–dupilumab group versus |||||| in placebo–dupilumab group).
As of the database lock date of January 17, 2022, almost all patients either completed treatment (95% in the dupilumab–dupilumab group and 97.6% in the placebo–dupilumab group) or the study period (95% and 94.4%, respectively). Compared to the previous data cut-off date, 1 more patient discontinued treatment due to an AE in the dupilumab–dupilumab group and 1 more patient discontinued study for other reason in the placebo–dupilumab group. Overall, 12 (5.0%) and 3 (2.4%) patients prematurely discontinued treatment in dupilumab–dupilumab and placebo–dupilumab groups, respectively. The number (%) of patients who did not complete the end of study visit were 12 (5%) and 7 (5.6%) in the dupilumab–dupilumab and placebo–dupilumab arms, respectively (Table 32).
Table 33: Patient Disposition (EXCURSION)
Disposition | August 18, 2020, DCO | January 17, 2022, DCO | ||
|---|---|---|---|---|
Dupilumab–dupilumab n = 240 | Placebo–dupilumab n = 125 | Dupilumab–dupilumab n = 240 | Placebo–dupilumab n = 125 | |
Screened, N | |||||||||||||||||||||||| | |||||||||||||||||||||||| | ||
Enrolled and treated | 240 (100) | 125 (100) | 240 (100) | 125 (100) |
Ongoing treatment | |||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| |
Completed the study treatment period | |||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| |
Discontinued treatment | |||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| |
Adverse event | |||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| |
Poor compliance to protocol | |||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| |
Withdrawal by patient | |||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| |
Other | |||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| |
Ongoing in the study | |||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| |
Completed study period | |||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| |
Discontinued study | |||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| |
Adverse event | |||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| |
Poor compliance to protocol | |||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| |
Other | |||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| |
FAS | |||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| |
DCO = data cut-off; FAS = full analysis set; NR = not reported.
Note: Values are n (%) unless otherwise indicated.
Source: Clinical Study Report–EXCURSION (DCO: August 18, 2020,20 and January 17, 2022).42
Exposure to Study Treatments
Exposure to study treatments is summarized in ||||||||||||||||||||||||.
At the cut-off date of August 18, 2020, the median duration of the study was 372 days (range = 1 to 488) and 375 days (range = 2 to 518 days) for the dupilumab–dupilumab and placebo–dupilumab groups, respectively. The total cumulative study duration was 217 patient-years and 116 patient-years for the dupilumab–dupilumab and placebo–dupilumab groups, respectively, reflecting a 2:1 randomization scheme from the parent trial.
As of the January 17, 2022, data cut-off date, the median duration of study was |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| for the dupilumab–dupilumab and placebo–dupilumab groups, respectively. The cumulative study duration |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| for the dupilumab–dupilumab and placebo–dupilumab groups, respectively. All enrolled patients received ICSs with or without a second controller in addition to dupilumab. In general, all patients had high treatment adherence with nearly everyone having achieved equal or more than |||||||||||||||||||||||||||||||||||||||||||||||| in the dupilumab–dupilumab group and placebo–dupilumab groups, respectively, in the August 18, 2020, dataset; |||||||||||||||||||||||||||||||||||||||||||||||| respectively, in the January 17, 2022, dataset).
|||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| | ||
|---|---|---|---|---|
|||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| | |
|||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| |
|||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| |
|||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| |
|||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| |
|||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| | |||||||||||||||||||||||| |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Source: Clinical Study Report–EXCURSION (Data cut-off: August 18, 202220 and January 17, 2022).42
Efficacy
All efficacy end points were secondary end points in the EXCURSION study.
Severe Asthma Exacerbation
Exacerbation data are summarized in ||||||||||||||||||||||||.
When analyzed as a subgroup, patients with type 2 inflammatory asthma phenotype at baseline of the parent study |||||||||||||||||||||||| experienced an event in the dupilumab–dupilumab and placebo–dupilumab groups, respectively. When analyzed as a subgroup, patients with EOS of 300 cells/µL or greater at baseline of the parent study |||||||||||||||||||||||| experienced an event in the dupilumab–dupilumab and placebo–dupilumab groups, respectively. The unadjusted annualized rate of severe exacerbation was |||||||||||||||||||||||| for the dupilumab–dupilumab and placebo–dupilumab groups, respectively |||||||||||||||||||||||| in a type 2 inflammatory asthma phenotype subgroup; |||||||||||||||||||||||| in an eosinophil ≥ 300 cells/µL subgroup).
As of the January 17, 2022, data cut-off, for both the type 2 inflammatory asthma and EOS of 300 cells/µL or greater at baseline subgroups, there was |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| whereas the numbers |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| compared to the previous dataset. Compared to the previous dataset, in a type 2 inflammatory asthma phenotype subgroup, the unadjusted annualized severe exacerbation event rate decreased from ||||||||||||||| to 0.118 and from ||||||||||||| to 0.124 for the dupilumab–dupilumab and placebo–dupilumab groups, respectively. Similarly, in the subgroup with eosinophil of 300 cells/µL or greater, the unadjusted annualized severe exacerbation event rate decreased from |||||||||||| to 0.120 and from |||||||||||| to 0.119, for the dupilumab–dupilumab and placebo–dupilumab groups, respectively. There were |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| who experienced a severe exacerbation requiring hospitalization or ED visit during the treatment period.
||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||| | |||
|---|---|---|---|---|---|---|
||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | |
||||||||||||||||||||||||| | ||||||
||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| |
||||||||||||||||||||||||| | ||||||
||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| |
||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| |
||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| |
||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| |
||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| |
||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| |
||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| |
||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| |
||||||||||||||||||||||||| | ||||||
||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| |
||||||||||||||||||||||||| | ||||||
||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| |
||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| |
||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| |
||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| |
||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| |
||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| |
||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| |
||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Source: Clinical Study Report–EXCURSION (Data cut-off: August 18, 2020,20 and January 17, 2022).42
Change in Percent Predicted FEV1
Data for change in the percent predicted FEV1 are presented in Table 35.
As of the August 18, 2022, data cut-off, at week 52, improvement in percent predicted prebronchodilator FEV1 was |||||||||||||||||||||||||||||||||||||||||||||||||| for the dupilumab–dupilumab and placebo–dupilumab groups, respectively, in the type 2 inflammatory asthma subgroup. In the same dataset, at week 52, improvement in percent predicted prebronchodilator FEV1 was ||||||||||||||||||||||||| and ||||||||||||||||||||||||| for the dupilumab–dupilumab and placebo–dupilumab groups, respectively, in the baseline blood EOS of 300 cells/µL or greater subgroup. Of note, the total number of patients contributing to the analysis reduced to almost half (n = 111 versus n = 58 for the dupilumab–dupilumab and placebo–dupilumab groups, respectively) compared to the number of enrolled patients (n = 240 versus n = 125) at week 52 in data from the August 18, 2020, cut-off .
The second interim analysis based on the updated dataset (as of January 17, 2022) included more patients (n = ||||||||||||||||||||||||| for the dupilumab–dupilumab and placebo–dupilumab groups, respectively) than previous analysis. Additional data showed sustained lung function improvement in the type 2 inflammatory asthma subgroup as shown by the FEV1 increase from baseline by |||||||||||||||||||||||||||||||||||||||||||||||||| in the dupilumab–dupilumab and placebo–dupilumab groups, respectively, at week 52. Similarly, in subgroup with baseline blood EOS of 300 cells/µL or greater, improvement in FEV1 was sustained at week 52 as shown by increase of |||||||||||||||||||||||||||||||||||||||||||||||||| in the dupilumab–dupilumab and placebo–dupilumab groups, respectively.
Table 36: Change From Baseline in Percent Predicted Prebronchodilator FEV1 Over Time (EXCURSION)
Trial population | EXCURSION | ||||||
|---|---|---|---|---|---|---|---|
FAS | Type 2 inflammatory asthma phenotypea at baseline subgroup | Baseline blood eosinophils ≥ 300 cells/µL subgroup | |||||
Dupilumab–dupilumab n = 240 | Placebo–dupilumab n = 125 | Dupilumab–dupilumab n = 209 | Placebo–dupilumab n = 106 | Dupilumab–dupilumab n = 155 | Placebo–dupilumab n = 76 | ||
Baseline of the parent study | |||||||
N | 240 | 125 | 209 | 106 | 155 | 76 | |
Mean (SD) | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | |
Week 0 | |||||||
N | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | 209 | 106 | 155 | 76 | |
Mean (SD) | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | |
Change from baseline, mean (SD) | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | 12.50 (18.70) | 3.79 (14.40) | 13.34 (19.61) | 3.47 (15.56) | |
Week 52 (August 18, 2022, dataset) | |||||||
N | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | |
Mean (SD) | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | |
Change from baseline, mean (SD) | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | |
Week 52 (January 17, 2022, dataset) | |||||||
N | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | 184 | 98 | 133 | 69 | |
Mean (SD) | NR | NR | NR | NR | NR | NR | |
Change from baseline, mean (SD) | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | |
FAS = full analysis set; FEV1 = forced expiratory volume in 1 second; NR = not reported; SD = standard deviation.
aType 2 inflammation asthma phenotype is defined as blood eosinophils of 0.15 Giga/L or greater or fractional exhaled nitric oxide of 20 parts per billion or greater at baseline of parent the study.
Source: Clinical Study Report–EXCURSION (data cut-off: August 18, 2020,20 and January 17, 2022).42
Harms
Only those harms identified in the review protocol outlined in Table 5 are reported below. Refer to Table 36 for detailed harms data.
Adverse Events
In the initial analysis with data from the August 18, 2020, cut-off, the most frequently reported (≥ 5%) treatment-emergent AEs were nasopharyngitis (7.5% and 8.8% for the dupilumab–dupilumab and placebo–dupilumab groups, respectively), pharyngitis (5.0% and 8.8% for the dupilumab–dupilumab and placebo–dupilumab groups, respectively), and upper respiratory tract infection (6.7% and 3.2%, for the dupilumab–dupilumab and placebo–dupilumab groups, respectively). As of the January 17, 2022, data cut-off, the number of treatment-emergent AEs reported slightly increased for both groups (8.8% and 9.6% for nasopharyngitis, 6.3% and 9.6% for pharyngitis, and 7.9% and 4.0% for upper respiratory tract infections for the dupilumab–dupilumab and placebo–dupilumab groups, respectively).
Serious Adverse Events
As of the data cut-off date of August 18, 2020, there were 4 (1.7%) and 1 (0.8%) patient(s) from the dupilumab–dupilumab and placebo–dupilumab groups, respectively, who experienced SAEs during the EXCURSION study period. ||||||||||||||||||||||||||||||||||||||||||||||||||. As of the January 17, 2022, data cut-off, 2 more cases of SAEs were reported in the dupilumab–dupilumab group (for a total of 6 patients or 2.5%); whereas the number remained the same for the placebo–dupilumab group compared to the previous data cut-off.
Withdrawals Due to AEs
Based on data from the cut-off date of August 18, 2020, there were 2 (0.8%) patients in the dupilumab–dupilumab group who discontinued treatment due to AEs (pulmonary tuberculosis and allergic conjunctivitis). As of January 17, 2022, 1 more patient discontinued treatment due to ascariasis and a total of 3 (1.3%) patients discontinued treatment. No patient in the placebo–dupilumab group discontinued treatment due to an AE.
Mortality
No deaths were reported during the EXCURSION study period.
Notable Harms
As of the August 18, 2020, data cut-off, a total of |||||||||||||||||||||||||||||||||||||||||||||||||| patient(s) in the dupilumab–dupilumab and placebo–dupilumab groups, respectively, experienced hypersensitivity (medically reviewed) reactions with ||||||||||||||||||||||||| patients in the dupilumab–dupilumab group and none in the placebo–dupilumab group experiencing anaphylactic reaction. According to the study report, |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| in intensity and none led to treatment interruption or discontinuation.
As of the August 18, 2020, data cut-off, severe or serious infection was reported in |||||||||||| patients in the dupilumab–dupilumab group (complicated appendicitis, pulmonary tuberculosis, and upper respiratory tract infection) and |||||||||||| patient in the placebo–dupilumab group (pneumonia). One (0.4%) patient from the dupilumab–dupilumab group and none in the placebo–dupilumab group experienced an opportunistic infection (pulmonary tuberculosis). |||||||||||||||||||||||||||||||||||| patient(s) in the dupilumab–dupilumab and placebo–dupilumab groups, respectively, reported to have had parasitic infections during the study period.
As of the August 18, 2020, data cut-off, injection site reaction was reported more frequently in the placebo–dupilumab group than the dupilumab–dupilumab group (||||||||||||||||||||||||).
As of the August 18, 2020, data cut-off, conjunctivitis occurred in |||||||||||||||||||||||||||||||||||| of patients in the dupilumab–dupilumab and placebo–dupilumab groups, respectively. Based on the Clinical Study Report, ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Lastly, eosinophilia was reported in |||||||||||||||||||||||||||||||||||| patients in the dupilumab–dupilumab and placebo–dupilumab groups, respectively as of the August 18, 2020, data cut-off. According to the study report, all cases of eosinophilia were asymptomatic and a transient increase in blood eosinophil counts that showed a gradual decrease over time. For some eosinophilia episodes, corrective treatment was administered.
As of January 17, 2022, the number of patients who experienced hypersensitivity (medically reviewed), parasitic infections, injection site reaction, conjunctivitis (broad), and eosinophilia increased by 1 to 3 patients per category from those of the earlier cut-off date, August 18, 2020. For a detailed description, refer to Table 37.
Table 37: Summary of Harms–FAS (EXCURSION)
Harms | August 18, 2020, DCO | January 17, 2022, DCO | ||
|---|---|---|---|---|
Dupilumab–dupilumab n = 240 | Placebo–dupilumab n = 125 | Dupilumab–dupilumab n = 240 | Placebo–dupilumab n = 125 | |
Patients with ≥ 1 adverse event | ||||
Patients who experienced an adverse event | 122 (50.8) | 79 (63.2) | 147 (61.3) | 85 (68.0) |
Most common eventsa | ||||
Nasopharyngitis | 18 (7.5) | 11 (8.8) | 21 (8.8) | 12 (9.6) |
Pharyngitis | 12 (5.0) | 11 (8.8) | 15 (6.3) | 12 (9.6) |
Upper respiratory tract infection | 16 (6.7) | 4 (3.2) | 19 (7.9) | 5 (4.0) |
Influenza | 12 (5.0) | 6 (4.8) | 13 (5.4) | 7 (5.6) |
Rhinitis allergic | 6 (2.5) | 7 (5.6) | 7 (2.9) | 9 (7.2) |
Injection site reaction | 7 (2.9) | 9 (7.2) | 8 (3.3) | 9 (7.2) |
Patients with ≥ 1 SAE | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Complicated appendicitis | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Pneumonia | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Upper respiratory tract infection | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Pulmonary tuberculosis | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Atelectasis | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Constipation | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Radius fracture | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Patients who stopped treatment due to adverse events | 2 (0.8) | 0 (0.0) | 3 (1.3) | 0 (0.0) |
Conjunctivitis allergic | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Pulmonary tuberculosis | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Ascariasis | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Deaths | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Notable harms | ||||
Hypersensitivity (medically reviewed) | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Anaphylactic reaction | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Angioedema | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Urticaria cholinergic | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Rash | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Urticaria | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Severe or serious infection | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Complicated appendicitis | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Pneumonia | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Pulmonary tuberculosis | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Upper respiratory tract infection | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Opportunistic infection | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Pulmonary tuberculosis | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Parasitic infections | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Enterobiasis | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Ascariasis | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Injection site reaction | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Injection site reaction | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Injection site erythema | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Injection site nodule | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Injection site rash | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Injection site urticaria | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Injection site induration | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Injection site edema | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Injection site pain | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Conjunctivitis (broad) | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Conjunctivitis | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Conjunctivitis allergic | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Conjunctivitis bacterial | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Eosinophilia | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Eosinophilia | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Eosinophilia count increased | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Allergic eosinophilia | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
DCO = data cut-off; FAS = full analysis set; NR = not reported; SAE = serious adverse event.
Note: Values are n (%) unless otherwise indicated.
aNumber (%) of patients with treatment-emergent adverse event(s) that occurred with a frequency of 5% or greater in any treatment category by Primary System Organ Class and Preferred Term.
Source: Clinical Study Report–EXCURSION (data cut-off: August 18, 2020,20 and January 17, 2022).42
Critical Appraisal
Internal Validity
The EXCURSION study was an open-label, noncomparative, longer-term extension study that evaluated the safety of dupilumab as a primary objective. A lack of blinding could have had impact on patient report of AEs. However, most AEs reported in the EXCURSION study could be assessed by signs, thereby reducing the potential biases. Furthermore, as the EXCURSION study is a 1-year study, rare AEs could not be captured as of the data cut-off date. Lastly, as efficacy outcomes were secondary objectives and given this is a noncomparative trial, no definitive conclusion about long-term comparative efficacy of dupilumab as add-on therapy to standard of care can be drawn based on the data from the EXCURSION study. Of note, 300 mg dosing of dupilumab was not assessed in the parent study, VOYAGE, which represents an additional limitation for the assessment of safety and efficacy of treatment of this dosage regimen.
External Validity
As patients were rolled over from the VOYAGE study, the same generalizability issues and limitations hold for the EXCURSION study. In the EXCURSION study, although most dupilumab injections were administered by a health care provider, home administration of 300 mg every 4 week dosing following first administration onsite was allowed with a protocol amendment (n = 18). The home administration of dupilumab was not regarded as a generalizability concern according to the clinical experts consulted, as this approach may be implemented in the Canadian real-world practice setting.
Discussion
Summary of Available Evidence
Dupilumab has been resubmitted to CADTH to expand the reimbursement for severe asthma to include patients aged 6 years to younger than 12 years. The sponsor provided 1 multinational, multicentre, double-blind RCT, VOYAGE, that was included in the systematic review section. The VOYAGE trial was conducted in 408 patients aged 6 years to younger than 12 years with persistent uncontrolled asthma, despite standard treatment use. The VOYAGE study compared dupilumab to placebo (both added to standard of care) administered over 52 weeks. The primary outcome was annualized rate of severe exacerbations. There were 2 efficacy populations in the trial that informed the reimbursement request: the type 2 inflammatory asthma phenotype population, characterized by a baseline blood eosinophil count of 150 cells/µL or greater or baseline FeNO of 20 ppb or greater, and a population with baseline blood EOS of 300 cells/µL or greater.
Additional evidence was available from an ITC submitted by the sponsor, comparing the efficacy of dupilumab to another biologic treatment (the IgE inhibitor omalizumab), and a longer-term extension study, EXCURSION (N = 365), evaluating the safety and tolerability as well as efficacy of dupilumab for an additional 52 weeks of treatment after the VOYAGE trial.
Interpretation of Results
Efficacy
Dupilumab was previously reviewed by CADTH for the indication of severe asthma with a type 2 or eosinophilic phenotype or OCS-dependent asthma in patients aged 12 years and older and received a positive recommendation in June 2021. On March 25, 2022, the Health Canada indication was revised and now dupilumab has a Health Canada indication for add-on maintenance treatment in patients aged 6 years and older with severe asthma with a type 2 or eosinophilic phenotype or OCS-dependent asthma. It is important to consider that the VOYAGE trial did not include patients who were OCS-dependant, therefore the treatment effect in OCS-dependant individuals aged 6 years to younger than 12 years remains unclear. However, the clinical experts consulted by CADTH for this review noted that very few pediatric patients undergo daily or alternate daily OCS treatment, but many may experience frequent short courses of treatment with OCSs.
Efficacy in the VOYAGE trial was assessed in 2 main populations, defined as the type 2 inflammatory asthma phenotype population (baseline blood eosinophil count ≥ 150 cells/µL or baseline FeNO ≥ 20 ppb) and a population with a baseline blood eosinophil count of 300 cells/µL or greater, due to differences in approved label indications across the US and European Union reference countries. Additional efficacy populations that were multiplicity controlled were reported, including ITT, baseline blood eosinophil count of 150 cells/µL or greater, and baseline FeNO of 20 ppb or greater. Certain implementation issues were highlighted by the clinical experts, mainly the applicability of utilizing FeNO measurements to identify eligible patients in the Canadian settings. Even though this restriction was suggested by the sponsor in their reimbursement request, the experts reported that FeNO assessments are not routinely performed in clinical practice in Canada.
Both clinical experts and patient groups consulted during the CADTH review highlighted that reducing asthma exacerbations is considered an important treatment goal for patients with asthma. In the VOYAGE trial, the number of severe asthma exacerbations over 52 weeks was lower in the dupilumab group versus placebo. The observed between-group effects were both statistically significant and clinically meaningful, favouring patients randomized to the dupilumab group compared to placebo. Clinical experts noted that the effects of asthma treatments may be driven by external and environmental factors that can vary within 1 year and even between years, such as viral infection, air pollution, and allergen exposure. Hence, treatment effects in asthma studies with limited time intervals may be biased by these factors, and longer-term studies are needed for assessment of effectiveness. The VOYAGE trial assessed exacerbation rates at 52 weeks which is appropriate given the aforementioned variation in asthma over time; however, asthma is a chronic condition and longer follow-up periods are needed to be able to extrapolate the benefits in the longer-term. The sponsor provided additional evidence from an interim analysis of a longer-term extension study (EXCURSION), which followed the VOYAGE trial population for up to an additional year after the parent study. Even though the EXCURSION study showed continued low severe asthma exacerbation event rates, these findings are limited by the lack of formal hypothesis testing in this open-label, noncomparative trial.
The CADTH systematic review protocol prespecified acute asthma exacerbations leading to hospitalization and/or to ED visits as outcomes of interest for this review. There was a limited number of these events experienced by the patients during the VOYAGE trial, thus limiting the interpretability of the observed results. Moreover, these analyses were introduced after the database lock occurred in the trial, suggesting that no conclusions can be made on the efficacy of dupilumab in preventing hospitalizations and ED visits.
Change from baseline in prebronchodilator percent predicted FEV1 at week 12 was the key secondary outcome included in the statistical testing hierarchy of the VOYAGE trial. Despite being the key secondary efficacy outcome in the pivotal study, prebronchodilator percent predicted FEV1 was rated with a lower ranking of importance by the clinical experts, as listed in the CADTH protocol, and was labelled as a secondary goal of asthma management, according to the clinical group input. However, this outcome was regarded as important by the patient groups as more than two-thirds of respondents from the patient survey submitted to CADTH reported an increase in lung function to be an important expectation for new asthma medication. At week 12, the differences in mean change from baseline for the key secondary end point of percent predicted prebronchodilator FEV1 were statistically significant. The clinical experts reported that the magnitude of change from baseline between groups at week 12 in percent predicted prebronchodilator FEV1 was modest yet acceptable. Furthermore, the experts reported that the 12-week assessment of FEV1 was not optimal as this duration might be insufficient to evaluate seasonal and other factors affecting the outcome; hence, the experts placed more clinical relevance on the 52-week assessment. While no definitive conclusions can be drawn because multiplicity considerations were not accounted for, the results of pulmonary function analyses across the other time points are supportive of a treatment benefit with dupilumab versus placebo.
Subgroup analyses was performed for the primary and key secondary outcome of the VOYAGE trial, based on the following factors of interest for this CADTH review: baseline eosinophil levels, baseline ICS dose, number of previous asthma exacerbations, and atopic medical history. Subgroups of patients with baseline blood EOS of 300 cells/µL or greater, baseline blood EOS of greater than 150 cells/µL, and patients receiving high doses of ICSs were part of the hierarchical testing procedure and, thus, adjusted for multiplicity. The findings from the baseline eosinophil count subgroup analyses are indicative of greater treatment benefit, in terms of annualized exacerbation rates, among patients with higher eosinophil counts. This is aligned with the input received from clinical experts, who expected an almost linear relationship to be observed between baseline blood eosinophil count and exacerbation frequency. It is also supportive of the dupilumab-indicated patient population as well as suggested reimbursement criteria to use dupilumab in patients with baseline EOS of 150 cells/µL or greater, a requirement that is also consistent with the GINA definition of type 2 inflammatory asthma.1 Still, clinical experts expressed some implementation concerns and reported that different provinces across Canada may report lab values with diverse acuity (i.e., some labs only report round numbers of 100 cells/μL, 200 cells/μL, 300 cells/μL, and so forth). In terms of the analyses for other subgroups, findings were considered only supportive of the overall treatment effect with dupilumab by CADTH as there were no multiplicity adjustments performed for these analyses.
Reduction in asthma symptoms represents an important outcome to patients and clinicians. In the VOYAGE trial, asthma symptoms were captured through a validated questionnaire (ACQ-7), with an established MID of 0.5, and the analyses at the week 24 time point were incorporated in the hierarchical testing procedures. Mean changes from baseline in the ACQ-7 in both groups exceeded the MID of 0.5 points; however, differences in ACQ scores between dupilumab and placebo were less than 0.5 at week 24. Responder analyses showed that approximately 80% of patients receiving dupilumab patients were considered responders, exceeding the established MID, across both the type 2 and baseline eosinophil of 300 cells/µL or greater populations. However, it should be noted that slightly more than 60% of the patients in the placebo group were also considered responders. Despite the lack of multiplicity adjustments for the responder analyses, findings can be considered supportive of improved asthma control with dupilumab treatment compared to placebo. According to the clinical experts, robust placebo responses observed in the trial indicate that getting patients who have uncontrolled asthma to adhere to their background medication is very important when treating asthma. Indeed, adherence to background therapy was high in both groups during the VOYAGE study (approximately 80%), which suggests that patients might have benefited from the close attention and monitoring of the trial. As a result, clinicians stressed that patients who are well managed should not be eligible for dupilumab treatment and highlighted the importance of going through an asthma clinic or subspecialist or specialist care when initiating treatment with dupilumab.
Asthma has a substantial impact on HRQoL, according to the patient input received by CADTH. HRQoL, assessed through the validated, disease-specific PAQLQ instrument among children aged 7 years to younger than 12 years, was a secondary outcome of the VOYAGE trial. The within-group mean changes from baseline exceeded the MID of 0.5 regardless of treatment group or subpopulation; however, the differences in PAQLQ global scores between dupilumab and placebo were less than 0.5. Other HRQoL measures (PACQLQ, EQ-5D-Y, and PRQLQ) were prespecified as exploratory outcomes in the VOYAGE study. The clinical experts consulted agreed with the CADTH reviewers that the clinical significance of the comparative estimates of effect on HRQoL are uncertain. As well, the HRQoL analyses presented in the trial were without adjustments for multiplicity; thus, the results presented were considered supportive by CADTH, yet it remains unclear what the impact of add-on dupilumab on patients’ HRQoL is, compared to placebo.
Additional important treatment goals of asthma, according to the clinical experts and the patients, included reducing OCS usage and limiting complications of the existing maximal therapy. The VOYAGE study findings showed that more than 40% of patients in the placebo arm and more than 20% of patients in the dupilumab arm received at least 1 course of SCSs. The decreases in SCS burden observed between the 2 groups were regarded as clinically significant, with possible impact on linear growth in patients, according to the clinical experts. Regarding the ICS usage, permanent increases in dosing were captured during the trial only after 2 or more severe asthma exacerbations, as specified in the study protocol. Even though none of the patients had their background medications increased during trial, clinical experts reported assessment concerns and noted that there should have been earlier escalation of therapy or at least reassessment for need for escalation before the 2 events occurring. In addition to these concerns, it is important to note that OCS usage was reported under additional efficacy analyses and the ICS was considered an exploratory outcome in the pivotal trial. As such, they were absent from the statistical testing hierarchy. This resulted in an inability to draw conclusions about the efficacy of dupilumab to decrease the OCS use and reduce the burden of existing ICS therapy; thus, an evidence gap remains regarding treatment effect on these outcomes of importance in the clinical setting.
Currently, there are no head-to-head trials that have evaluated the efficacy of dupilumab with relevant biologics of interest for patients with severe uncontrolled asthma. The sponsor submitted a Bucher ITC analysis that compared the efficacy of dupilumab to omalizumab in patients who are aged 6 years to 11 years of age with uncontrolled moderate-to-severe asthma. Limitations of the ITC include the heterogeneity of included studies in terms of study characteristics, patient populations, and outcomes assessed. The exploration of between study differences and potential biases was further limited by the small number of trials included in the ITC. Considering that prognostic factors and effect modifiers are likely imbalanced between treatment groups, the results of the ITC are subject to an unknown amount and direction of bias. Thus, the findings of the ITC are highly uncertain. According to the clinical experts, presented data are limited and the results do not support superiority of dupilumab, compared to omalizumab, in patients aged 6 years to younger than 12 years with severe uncontrolled asthma. Evidence on comparative efficacy of dupilumab to the other Health Canada–approved biologic drug for patients 6 and older (mepolizumab) was not identified.
Harms
Dupilumab appeared to have an acceptable safety profile, based on the small number of patients who discontinued therapy due to an AE in the VOYAGE study. The proportions of patients experiencing SAEs during the VOYAGE trial were also balanced between the dupilumab and placebo arms. In terms of notable harms, there seemed to be no indication of an increased risk of hypersensitivity reactions with dupilumab versus placebo. Even though parasitic infections are common with monoclonal antibodies, due to the creation of an immunosuppressive environment that decreases the host’s resistance to infections,5 only 2.6% of dupilumab patients had a parasitic infection, compared to none experienced in the placebo group. The incidence of conjunctivitis was low across both studied groups, which was unexpected according to the clinical experts. Clinical experts reported that the higher incidence of injection site reactions and eosinophilia reported in the dupilumab compared to the placebo group are aligned with the data from the adult and adolescent population.
Longer-term follow-up data from the EXCURSION study did not reveal any new safety issues from those observed in the parent trial VOYAGE; however, these findings are limited considering the open-label and noncomparative design.
The sponsor-submitted ITC reported on safety outcomes, mainly withdrawals due to AEs. There were no significant differences observed between dupilumab and omalizumab at 52 weeks, but the interpretation of these findings is limited due to numerous methodological limitations as well as the fact that the period of assessment for harm outcomes included an ICS dose reduction period from the omalizumab trial. As a result, no definitive conclusions can be drawn regarding the comparative safety of dupilumab relative to omalizumab.
Conclusions
One sponsor-submitted, multicentre, randomized, double-blind, phase III trial (VOYAGE), comparing add-on therapy with dupilumab to placebo in patients aged 6 years to younger than 12 years with persistent asthma demonstrated that dupilumab reduced the annualized rate of severe exacerbations and improved pulmonary function (FEV1) in patients whose asthma remains uncontrolled despite background therapy with medium to high doses of ICSs. There was supportive evidence on the overall treatment benefit on asthma-related symptoms, as measured by ACQ-7, but the differences between the dupilumab and placebo groups did not exceed the MID. HRQoL analyses were not controlled for multiple comparisons in this RCT; thus, the impact of add-on dupilumab on patients’ HRQoL compared to placebo remains. Likewise, observed reductions in OCS usage were not part of the statistical hierarchy, which precluded drawing conclusions about the effects of dupilumab on this outcome. With respect to harms, there were no obvious safety or tolerability issues associated with the use of dupilumab in children. A longer-term extension study did not identify any new safety issues. Findings from the sponsor-submitted ITC were inconclusive with respect to the efficacy and safety of dupilumab compared to omalizumab due to numerous methodological limitations.
References
1.Global strategy for asthma management and prevention (2022 update). Fontana-on-Geneva Lake (WI): Global Initiative for Asthma; 2022: https://ginasthma.org/wp-content/uploads/2022/07/GINA-Main-Report-2022-FINAL-22-07-01-WMS.pdf. Accessed 2022 Sep 08.
2.Public Health Infobase. Asthma in Canada (2018-05-01). Data Blog. Ottawa (ON): Government of Canada; 2018: https://health-infobase.canada.ca/datalab/asthma-blog.html. Accessed 2022 Sep 08.
3.Yang CL, Hicks EA, Mitchell P, et al. Canadian Thoracic Society 2021 Guideline update: Diagnosis and management of asthma in preschoolers, children and adults. Can J Respir Crit Care Sleep Med. 2021;5(6):348-361.
4.CADTH Drug Reimbursement Expert Review Committee final recommendation: dupilumab (Dupixent - Sanofi Genzyme). Can J Health Technol. 2021;1(6). https://www.cadth.ca/sites/default/files/attachments/2021-06/CADTH_reimbursement_recommendation_dupilumab_%28dupixent%29_1.pdf. Accessed 2022 Sep 17.
5.Dupixent (dupilumab injection): solution for subcutaneous injection, 300 mg single-use syringe (300 mg/2 mL), 200 mg single-use syringe (200 mg/1.14 mL), and 100 mg single-use syringe (100 mg/0.67 mL); Dupixent (dupilumab injection): solution for subcutaneous injection, 300 mg single-use pen (300 mg/2 mL) and 200 mg single-use pen (200 mg/1.14 mL) [product monograph]. Laval (QC): Sanofi-aventis Canada Inc.; 2022 Mar 25.
6.CADTH Drug Reimbursement Expert Review Committee final recommendation: dupilumab (Dupixent — Sanofi-aventis Canada Inc.). Ottawa (ON): CADTH; 2020 Apr 24: https://www.cadth.ca/sites/default/files/cdr/complete/SR0636%20Dupixent%20-%20CDEC%20Final%20%20Recommendation%20April%2024%2C%202020%20for%20posting.pdf. Accessed 2022 Sep 17.
7.Clinical Study Report: SAR231893-EFC14153. VOYAGE: A randomized, double-blind, placebo-controlled, parallel group study to evaluate the efficacy and safety of dupilumab in children 6 to <12 years of age with uncontrolled persistent asthma [internal sponsor's report]. Paris (FR): Sanofi Group; 2020 Dec 07.
8.Asthma, 2014. Health Fact Sheets. Ottawa (ON): Statistics Canada; 2014: https://www150.statcan.gc.ca/n1/pub/82-625-x/2015001/article/14179-eng.htm. Accessed 2022 Sep 08.
9.Carr TF, Zeki AA, Kraft M. Eosinophilic and Noneosinophilic Asthma. Am J Respir Crit Care Med. 2018;197(1):22-37. PubMed
10.Wenzel S. Severe asthma phenotypes. In: Post TW, ed. UpToDate. Waltham (MA): UpToDate; 2021: http://www.uptodate.com. Accessed 2022 Jul 18.
11.FitzGerald JM, Lemiere C, Lougheed MD, et al. Recognition and management of severe asthma: A Canadian Thoracic Society position statement. Can J Respir Crit Care Sleep Med. 2017;1(4):199-221.
12.Sawicki G, Haver K. Asthma in children younger than 12 years: managementof persistent asthma with controller therapies. In: Post TW, ed. UpToDate. Waltham (MA): UpToDate; 2021: http://www.uptodate.com. Accessed 2022 Jul 18.
13.Nucala (mepolizumab for injection): 100 mg/mL lyophilized powder for subcutaneous injection in single-use vial; Nucala (mepolizumab injection): 100 mg/mL solution for subcutaneous injection in single-use pre-filled autoinjector or safety syringe [product monograph]. Mississauga (ON): GlaxoSmithKline Inc.; 2021 Nov 05: https://pdf.hres.ca/dpd_pm/00063467.PDF. Accessed 2022 Sep 08.
14.Xolair (omalizumab): 150 mg vial, sterile powder for reconstitution for subcutaneous injection; Xolair (omalizumab): 75 mg and 150mg solution for subcutaneous injection in pre-filled syringe [product monograph]. Dorval (QC): Novartis Pharmaceuticals Canada Inc.; 2022 Aug 11: https://pdf.hres.ca/dpd_pm/00066970.PDF. Accessed 2022 Sep 08.
15.Holguin F, Cardet JC, Chung KF, et al. Management of severe asthma: a European Respiratory Society/American Thoracic Society guideline. Eur Respir J. 2020;55(1):1900588. PubMed
16.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. J Clin Epidemiol. 2016;75:40-46. PubMed
17.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2022 Jul 14.
18.Pocket guide for asthma management and prevention (for adults and children older than 5 years): a pocket guide for physicians and nurses, updated 2015. Fontana-on-Geneva Lake (WI): Global Initiative for Asthma; 2015: https://ginasthma.org/wp-content/uploads/2016/01/GINA_Pocket_2015.pdf. Accessed 2022 Sep 08.
19.Bacharier LB, Maspero JF, Katelaris CH, et al. Dupilumab in Children with Uncontrolled Moderate-to-Severe Asthma. N Engl J Med. 2021;385(24):2230-2240. PubMed
20.Clinical Study Report: SAR231893 - LTS14424. EXCURSION: One-year study to evaluate the long-term safety and tolerability of dupilumab in pediatric patients with asthma who participated in a previous dupilumab asthma clinical study [internal sponsor's report]. Paris (FR): Sanofi Group; 2020 Dec 03.
21.Miller MR, Hankinson J, Brusasco V, et al. Standardisation of spirometry. Eur Respir J. 2005;26(2):319-338. PubMed
22.Barnes PJ, Casale TB, Dahl R, Pavord ID, Wechsler ME. The Asthma Control Questionnaire as a clinical trial endpoint: past experience and recommendations for future use. Allergy. 2014;69(9):1119-1140. PubMed
23.Sastre J, Olaguibel J, Vega JM, Del Pozo V, Picado C, Lopez Viña A. Cut-off points for defining asthma control in three versions of the Asthma Control Questionnaire. J Asthma. 2010;47(8):865-870. PubMed
24.Juniper EF, Gruffydd-Jones K, Ward S, Svensson K. Asthma Control Questionnaire in children: validation, measurement properties, interpretation. Eur Respir J. 2010;36(6):1410-1416. PubMed
25.Nguyen JM, Holbrook JT, Wei CY, et al. Validation and psychometric properties of the Asthma Control Questionnaire among children. J Allergy Clin Immunol. 2014;133(1):91-97.e91-96.
26.Qoltech, Measurement of Health-Related Quality of Life & Asthma Control. Standardised Paediatric Asthma Quality of Life Questionnaire (PAQLQ(S)). https://www.qoltech.co.uk/paqlq_s.html. Accessed 2022 Jul 28.
27.Juniper EF, Guyatt GH, Feeny DH, Ferrie PJ, Griffith LE, Townsend M. Measuring quality of life in children with asthma. Qual Life Res. 1996;5(1):35-46. PubMed
28.Wing A, Upton J, Svensson K, Weller P, Fletcher M, Walker S. The standardized and mini versions of the PAQLQ are valid, reliable, and responsive measurement tools. J Clin Epidemiol. 2012;65(6):643-650. PubMed
29.American Thoracic Society Quality of Life Resource. Pediatric Asthma Caregiver's Quality of Life Questionnaire (PACQLQ). 1999; https://qol.thoracic.org/sections/instruments/pt/pages/pacqlq.html. Accessed 2022 Aug 09.
30.Juniper EF, Howland WC, Roberts NB, Thompson AK, King DR. Measuring quality of life in children with rhinoconjunctivitis. J Allergy Clin Immunol. 1998;101(2 Pt 1):163-170. PubMed
31.Qoltech, Measurement of Health-Related Quality of Life and Asthma Control. Paediatric Rhinoconjunctivitis Quality of Life Questionnaire (PRQLQ). http://www.qoltech.co.uk/prqlq.html. Accessed 2022 Jul 27.
32.EQ-5D, EuroQol Research Foundation. EQ-5D-Y (Youth) | About. 2021; https://euroqol.org/eq-5d-instruments/eq-5d-y-about/. Accessed 2022 Aug 05.
33.EQ-5D-Y User Guide, version 2.0. Rotterdam (NL): EuroQol Research Foundation; 2020: https://euroqol.org/publications/user-guides/. Accessed 2022 Aug 05.
34.Busse WW, Maspero JF, Rabe KF, et al. Liberty Asthma QUEST: Phase 3 Randomized, Double-Blind, Placebo-Controlled, Parallel-Group Study to Evaluate Dupilumab Efficacy/Safety in Patients with Uncontrolled, Moderate-to-Severe Asthma. Adv Ther. 2018;35(5):737-748. PubMed
35.Efficacy and safety of dupilumab in uncontrolled moderate-to-severe asthma in 6 to <12-year-old children: systematic literature review and indirect treatment comparison technical report, version 9 [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Dupixent (dupilumab), 200 mg and 300 mg solution for subcutaneous injection. Mississauga (ON): Sanofi-aventis Canada Inc; 2022 Jul 14.
36.Hutton B, Salanti G, Caldwell DM, et al. The PRISMA extension statement for reporting of systematic reviews incorporating network meta-analyses of health care interventions: checklist and explanations. Ann Intern Med. 2015;162(11):777-784. PubMed
37.Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 2009;6(7):e1000097. PubMed
38.Cochrane handbook for systematic reviews of interventions, version 5.1.0 (updated March 2011). In: Higgins JPT, Green S, eds. London (UK): The Cochrane Collaboration; 2011: https://handbook-5-1.cochrane.org/. Accessed 2022 Sep 08.
39.Lanier B, Bridges T, Kulus M, Taylor AF, Berhane I, Vidaurre CF. Omalizumab for the treatment of exacerbations in children with inadequately controlled allergic (IgE-mediated) asthma. J Allergy Clin Immunol. 2009;124(6):1210-1216. PubMed
40.Szefler SJ, Casale TB, Haselkorn T, et al. Treatment Benefit with Omalizumab in Children by Indicators of Asthma Severity. J Allergy Clin Immunol Pract. 2020;8(8):2673-2680.e2673. PubMed
41.National Institute for Health and Care Excellence. Single technology appraisal and highly specialised technologies evaluation: user guide for company evidence submission template. (Process and methods PMG24) 2022; https://www.nice.org.uk/process/pmg24/chapter/clinical-effectiveness#quality-assessment-of-the-relevant-clinical-effectiveness-evidence. Accessed 2022 Sep 08.
42.Clinical Study Report: SAR231893 - LTS14424. Liberty Asthma Excursion: One year study to evaluate the long-term safety and tolerability of dupilumab in pediatric patients with asthma who participated in a previous dupilumab asthma clinical study [internal sponsor's report]. Chilly-Mazarin (FR): Sanofi-Aventis Research and Development; 2022 Apr 12.
43.Canonica GW, Bourdin A, Peters AT, et al. Dupilumab Demonstrates Rapid Onset of Response Across Three Type 2 Inflammatory Diseases. J Allergy Clin Immunol Pract. 2022;10(6):1515-1526. PubMed
44.Gallagher A, Edwards M, Nair P, et al. Anti-interleukin-13 and anti-interleukin-4 agents versus placebo, anti-interleukin-5 or anti-immunoglobulin-E agents, for people with asthma. Cochrane Database Syst Rev. 2021;10:CD012929. PubMed
45.Miller MA, Davis KL. Dupilumab Efficacy in Patients With Uncontrolled, Moderate-to-Severe Allergic Asthma. Pediatrics. 2020;146(Supplement_4):S375-S375.
46.Juniper EF, O'Byrne PM, Guyatt GH, Ferrie PJ, King DR. Development and validation of a questionnaire to measure asthma control. Eur Respir J. 1999;14(4):902-907. PubMed
47.Cohen J. A power primer. Psychol Bull. 1992;112(1):155-159. PubMed
48.Juniper EF, Guyatt GH, O'Byrne PM, Viveiros M. Aqueous beclomethasone diproprionate nasal spray: regular versus “as required” use in the treatment of seasonal allergic rhinitis. J Allergy Clin Immunol. 1990;86(3 Pt 1):380-386. PubMed
49.Mayoral K, Garin O, Lizano-Barrantes C, et al. Measurement properties of the EQ-5D-Y administered through a smartphone app in children with asthma: a longitudinal questionnaire study. Health Qual Life Outcomes. 2022;20(1):51. PubMed
50.Bergfors S, Astrom M, Burstrom K, Egmar AC. Measuring health-related quality of life with the EQ-5D-Y instrument in children and adolescents with asthma. Acta Paediatr. 2015;104(2):167-173. PubMed
51.Juniper EF, Chauhan A, Neville E, et al. Clinicians tend to overestimate improvements in asthma control: an unexpected observation. Prim Care Respir J. 2004;13(4):181-184. PubMed
52.Honkoop PJ, Loijmans RJ, Termeer EH, et al. Comparison between an online self-administered and an interviewer-administered version of the Asthma Control Questionnaire: a cross-sectional validation study. Prim Care Respir J. 2013;22(3):284-289. PubMed
53.Guyatt GH, Kirshner B, Jaeschke R. Measuring health status: what are the necessary measurement properties? J Clin Epidemiol. 1992;45(12):1341-1345. PubMed
54.Juniper EF, Wisniewski ME, Cox FM, Emmett AH, Nielsen KE, O'Byrne PM. Relationship between quality of life and clinical status in asthma: a factor analysis. Eur Respir J. 2004;23(2):287-291. PubMed
55.Reeve BB, Wyrwich KW, Wu AW, et al. ISOQOL recommends minimum standards for patient-reported outcome measures used in patient-centered outcomes and comparative effectiveness research. Qual Life Res. 2013;22(8):1889-1905. PubMed
56.Leidy NK, Wyrwich KW. Bridging the gap: using triangulation methodology to estimate minimal clinically important differences (MCIDs). COPD. 2005;2(1):157-165. PubMed
57.American Thoracic Society Quality of Life Resource. Pediatric Rhinoconjunctivitis Quality of Life Questionnaire (PRQLQ). https://qol.thoracic.org/sections/instruments/pt/pages/prqlq.html. Accessed 2022 Jul 27.
Appendix 1: Literature Search Strategy
Clinical Literature Search
Overview
Interface: Ovid
Databases:
MEDLINE All (1946-present)
Embase (1974-present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: July 27, 2022
Alerts: Bi-weekly search updates until project completion
Search filters applied: Randomized controlled trials or controlled clinical trials.
Limits:
Publication date limit: none
Language limit: none
Conference abstracts: excluded.
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
MeSH | Medical Subject Heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Author keyword heading word (MEDLINE) |
.dq | Candidate term word (Embase) |
.pt | Publication type |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multidatabase Strategy
(dupixent* or dupilumab* or SAR-231893 or SAR231893 or REGN-668 or REGN668 or 420K487FSG).ti,ab,kf,ot,hw,nm,rn.
exp Asthma/ or Bronchial Spasm/
(asthma* or antiasthma* or wheez*).ti,ab,kf.
(bronchospas* or bronchiospas* or (bronch* adj2 spas*)).ti,ab,kf.
(lung adj5 allerg*).ti,ab,kf.
or/2-5
1 and 6
7 use medall
*dupilumab/
(dupixent* or dupilumab* or SAR-231893 or SAR231893 or REGN-668 or REGN668).ti,ab,kf,dq.
or/9-10
exp Asthma/ or exp bronchospasm/
(asthma* or antiasthma* or wheez*).ti,ab,kf,dq.
(bronchospas* or bronchiospas* or (bronch* adj2 spas*)).ti,ab,kf,dq.
(lung adj5 allerg*).ti,ab,kf,dq.
or/12-15
11 and 16
17 use oemezd
18 not (conference abstract or conference review).pt.
8 or 19
(Randomized Controlled Trial or Controlled Clinical Trial or Pragmatic Clinical Trial or Equivalence Trial or Clinical Trial, Phase III).pt.
Randomized Controlled Trial/
exp Randomized Controlled Trials as Topic/
"Randomized Controlled Trial (topic)"/
Controlled Clinical Trial/
exp Controlled Clinical Trials as Topic/
"Controlled Clinical Trial (topic)"/
Randomization/
Random Allocation/
Double-Blind Method/
Double Blind Procedure/
Double-Blind Studies/
Single-Blind Method/
Single Blind Procedure/
Single-Blind Studies/
Placebos/
Placebo/
Control Groups/
Control Group/
(random* or sham or placebo*).ti,ab,hw,kf.
((singl* or doubl*) adj (blind* or dumm* or mask*)).ti,ab,hw,kf.
((tripl* or trebl*) adj (blind* or dumm* or mask*)).ti,ab,hw,kf.
(control* adj3 (study or studies or trial* or group*)).ti,ab,kf.
(Nonrandom* or non random* or non-random* or quasi-random* or quasirandom*).ti,ab,hw,kf.
allocated.ti,ab,hw.
((open label or open-label) adj5 (study or studies or trial*)).ti,ab,hw,kf.
((equivalence or superiority or non-inferiority or noninferiority) adj3 (study or studies or trial*)).ti,ab,hw,kf.
(pragmatic study or pragmatic studies).ti,ab,hw,kf.
((pragmatic or practical) adj3 trial*).ti,ab,hw,kf.
((quasiexperimental or quasi-experimental) adj3 (study or studies or trial*)).ti,ab,hw,kf.
(phase adj3 (III or "3") adj3 (study or studies or trial*)).ti,hw,kf.
or/21-51
20 and 52
remove duplicates from 53
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
[Search -- Studies with results | dupixent OR dupilumab OR SAR-231893 OR SAR231893 OR REGN-668 OR REGN668 | asthma]
WHO ICTRP
International Clinical Trials Registry Platform, produced by the World Health Organization. Targeted search used to capture registered clinical trials.
[Search terms -- (dupixent OR dupilumab OR SAR-231893 OR SAR231893 OR REGN-668 OR REGN668) AND asthma]
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
[Search terms -- (dupixent OR dupilumab) AND asthma]
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
[Search terms -- (dupixent OR dupilumab OR SAR-231893 OR SAR231893 OR REGN-668 OR REGN668) AND asthma]
Grey Literature
Search dates: July 14, 2022 – July 20, 2022
Keywords: [dupixent OR dupilumab OR SAR-231893 OR SAR231893 OR REGN-668 OR REGN668 | asthma]
Limits: Publication years: 2017-present for guidelines; none for other sections
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Internet Search.
Appendix 2: Excluded Studies
Note that this appendix of tables has not been copy-edited.
Reference | Reason for Exclusion |
|---|---|
Canonica, G. W., et al.43 | Study design (Post hoc analyses), Study population |
Gallagher, A., et al.44 | Study design, Study population |
Miller, M.A., et al.45 | Study design, Study population |
Appendix 3: Detailed Outcome Data
Note that this appendix of tables has not been copy-edited.
Table 40: Hierarchical Testing Order for US and US Reference Countries in the VOYAGE Trial
End points | Population | Test Order |
|---|---|---|
Annualized rate of severe exacerbation events during the 52-week placebo-controlled treatment period | Patients with baseline blood eosinophils ≥0.3 Giga/L | 1 |
Annualized rate of severe exacerbation events during the 52-week placebo-controlled treatment period | Patients with baseline blood eosinophils ≥0.15 Giga/L | 2 |
Annualized rate of severe exacerbation events during the 52-week placebo-controlled treatment period | Type 2 inflammatory asthma phenotype (Baseline blood eosinophils ≥0.15 Giga/L or Baseline FeNO ≥20 ppb) | 3 |
Change from baseline in prebronchodilator % predicted FEV1 at Week 12 | Patients with baseline blood eosinophils ≥0.3 Giga/L | 4 |
Change from baseline in prebronchodilator % predicted FEV1 at week 12 | Patients with baseline blood eosinophils ≥0.15 Giga/L | 5 |
Change from baseline in prebronchodilator % predicted FEV1 at week 12 | Type 2 inflammatory asthma phenotype (Baseline blood eosinophils ≥0.15 Giga/L or Baseline FeNO ≥20 ppb) | 6 |
Change in ACQ-7-IA at week 24 | Patients with baseline blood eosinophils ≥0.3 Giga/L | 7 |
Change in ACQ-7-IA at week 24 | Patients with baseline blood eosinophils ≥0.15 Giga/L | 8 |
Change in ACQ-7-IA at week 24 | Type 2 inflammatory asthma phenotype (Baseline blood eosinophils ≥0.15 Giga/L or Baseline FeNO ≥20 ppb) | 9 |
Annualized rate of severe exacerbation events during the 52-week placebo-controlled treatment period | Baseline FeNO ≥20 ppb | 10 |
Change from baseline in prebronchodilator % predicted FEV1 at week 12 | Baseline FeNO ≥20 ppb | 11 |
Change in ACQ-7-IA at week 24 | Baseline FeNO ≥20 ppb | 12 |
Annualized rate of severe exacerbation events during the 52-week placebo-controlled treatment period | Full ITT | 13 |
Change from baseline in prebronchodilator % predicted FEV1 at week 12 | Full ITT | 14 |
Annualized rate of severe exacerbation events during the 52-week placebo-controlled treatment period | Patients with baseline blood eosinophils ≥0.3 Giga/L and High ICS at baseline | 15 |
Annualized rate of severe exacerbation events during the 52-week placebo-controlled treatment period | Patients with baseline blood eosinophils ≥0.15 Giga/L and High ICS at baseline | 16 |
Annualized rate of severe exacerbation events during the 52-week placebo-controlled treatment period | Type 2 inflammatory asthma phenotype (Baseline blood eosinophils ≥0.15 Giga/L or Baseline FeNO ≥20 ppb) with High ICS at baseline | 17 |
Annualized rate of severe exacerbation events during the 52-week placebo-controlled treatment period | Baseline FeNO ≥20 ppb with High ICS at baseline | 18 |
Annualized rate of severe exacerbation events during the 52-week placebo-controlled treatment period | Full ITT with High ICS at baseline | 19 |
Change from baseline in FeNO at week 12 | Patients with baseline blood eosinophils ≥0.3 Giga/L | 20 |
Change from baseline in FeNO at week 12 | Patients with baseline blood eosinophils ≥0.15 Giga/L | 21 |
Change from baseline in FeNO at week 12 | Type 2 inflammatory asthma phenotype (Baseline blood eosinophils ≥0.15 Giga/L or Baseline FeNO ≥20 ppb) | 22 |
Change from baseline in FeNO at week 12 | Baseline FeNO ≥20 ppb | 23 |
Change from baseline in FeNO at week 12 | Full ITT | 24 |
ACQ-IA = Asthma Control Questionnaire–Interviewer Administered; FeNO = fractional exhaled nitric oxide; FEV1= forced expiratory volume in 1 second; ICS = inhaled corticosteroids; ITT = intention to treat.
Source: Clinical Study Report.7
Table 41: Hierarchical Testing Order for EU and EU Reference Countries in the VOYAGE Trial
End points | Population | Test Order |
|---|---|---|
Annualized rate of severe exacerbation events during the 52-week placebo-controlled treatment period | Type 2 inflammatory asthma phenotype (Baseline blood eosinophils ≥0.15 Giga/L or Baseline FeNO ≥20 ppb) | 1 |
Annualized rate of severe exacerbation events during the 52-week placebo-controlled treatment period | Patients with baseline blood eosinophils ≥0.15 Giga/L | 2 |
Annualized rate of severe exacerbation events during the 52-week placebo-controlled treatment period | Patients with baseline blood eosinophils ≥0.3 Giga/L | 3 |
Change from baseline in prebronchodilator % predicted FEV1 at week 12 | Type 2 inflammatory asthma phenotype (Baseline blood eosinophils ≥0.15 Giga/L or Baseline FeNO ≥20 ppb) | 4 |
Change from baseline in prebronchodilator % predicted FEV1 at week 12 | Patients with baseline blood eosinophils ≥0.15 Giga/L | 5 |
Change from baseline in prebronchodilator % predicted FEV1 at week 12 | Patients with baseline blood eosinophils ≥0.3 Giga/L | 6 |
Change in ACQ-7-IA at week 24 | Type 2 inflammatory asthma phenotype (Baseline blood eosinophils ≥0.15 Giga/L or Baseline FeNO ≥20 ppb) | 7 |
Change in ACQ-7-IA at week 24 | Patients with baseline blood eosinophils ≥0.15 Giga/L | 8 |
Change in ACQ-7-IA at week 24 | Patients with baseline blood eosinophils ≥0.3 Giga/L | 9 |
Annualized rate of severe exacerbation events during the 52-week placebo-controlled treatment period | Baseline FeNO ≥20 ppb | 10 |
Change from baseline in prebronchodilator % predicted FEV1 at week 12 | Baseline FeNO ≥20 ppb | 11 |
Change in ACQ-7-IA at week 24 | Baseline FeNO ≥20 ppb | 12 |
Annualized rate of severe exacerbation events during the 52-week placebo-controlled treatment period | Full ITT | 13 |
Change from baseline in prebronchodilator % predicted FEV1 at week 12 | Full ITT | 14 |
Annualized rate of severe exacerbation events during the 52-week placebo-controlled treatment period | Type 2 inflammatory asthma phenotype (Baseline blood eosinophils ≥0.15 Giga/L or Baseline FeNO ≥20 ppb) with High ICS at baseline | 15 |
Annualized rate of severe exacerbation events during the 52-week placebo-controlled treatment period | Patients with baseline blood eosinophils ≥0.15 Giga/L and High ICS at baseline | 16 |
Annualized rate of severe exacerbation events during the 52-week placebo-controlled treatment period | Patients with baseline blood eosinophils ≥0.3 Giga/L and High ICS at baseline | 17 |
Annualized rate of severe exacerbation events during the 52-week placebo-controlled treatment period | Baseline FeNO ≥20 ppb and High ICS at baseline | 18 |
Annualized rate of severe exacerbation events during the 52-week placebo-controlled treatment period | Full ITT and High ICS at baseline | 19 |
Change from baseline in FeNO at week 12 | Type 2 inflammatory asthma phenotype (Baseline blood eosinophils ≥0.15 Giga/L or Baseline FeNO ≥20 ppb) | 20 |
Change from baseline in FeNO at week 12 | Patients with baseline blood eosinophils ≥0.15 Giga/L | 21 |
Change from baseline in FeNO at week 12 | Patients with baseline blood eosinophils ≥0.3 Giga/L | 22 |
Change from baseline in FeNO at week 12 | Baseline FeNO ≥20 ppb | 23 |
Change from baseline in FeNO at week 12 | Full ITT | 24 |
ACQ-IA = Asthma Control Questionnaire–Interviewer Administered; FeNO = fractional exhaled nitric oxide; FEV1= forced expiratory volume in 1 second; ICS = inhaled corticosteroids; ITT = intention to treat.
Source: Clinical Study Report.7
Table 42: Patient Disposition (ITT and Baseline Blood Eosinophils ≥ 150 cells/µL Populations)
Trial population | Voyage trial | |||
|---|---|---|---|---|
ITT | Baseline blood eosinophils ≥150 cells/µL population | |||
Dupilumab (N=273) | Placebo (N=135) | Dupilumab (N=223) | Placebo (N=108) | |
Screened, N | |||||||||||| | |||
Randomized, N | 273 | 135 | |||||||||||| | |||||||||||| |
Treated, N (%) | 270 (98.9) | 135 (100) | |||||||||||| | |||||||||||| |
Completed study treatment during the treatment period, N (%) | 248 (90.8) | 130 (96.3) | |||||||||||| | |||||||||||| |
Discontinued study treatment, N (%) | 22 (8.1) | 5 (3.7) | |||||||||||| | |||||||||||| |
Reason for discontinuation, N (%) | |||||||||||| | |||||||||||| | ||
Adverse events | 5 (1.8) | 2 (1.5) | |||||||||||| | |||||||||||| |
Poor compliance to protocol | 2 (0.7) | 0 | |||||||||||| | |||||||||||| |
Other reason | 15 (5.5) | 3 (2.2) | |||||||||||| | |||||||||||| |
Discontinued from the study, N (%) | 22 (8.1) | 5 (3.7) | |||||||||||| | |||||||||||| |
Continued into LTE study | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Patients who did not continue into LTE study | ||||
Completed the follow-up period, N (%) | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Discontinued from the follow-up period, N (%) | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Reason for discontinuation from the follow-up, N (%) | ||||
Adverse events | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Poor compliance to protocol | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Other reason | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
ITT = intention to treat; LTE = long-term extension.
Source: Clinical Study Report.7
Table 43: Summary of Sensitivity Analyses for Adjusted Annualized Severe Exacerbation Event Rate (Type 2 Inflammatory Asthma Phenotype and Baseline Blood Eosinophils ≥ 300 cells/µL Populations)
Trial population efficacy outcome | VOYAGE | |||
|---|---|---|---|---|
Type 2 inflammatory asthma phenotype population | Baseline blood eosinophils ≥ 300 cells/µL population | |||
Dupilumab N=236 | Placebo N=114 | Dupilumab N=175 | Placebo N=84 | |
On-treatment analysisa | ||||
Number of patients | 233 | 114 | 172 | 84 |
Estimate (95% CI) | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Relative risk (95% CI) | 0.403 (0.269, 0.603) | 0.351 (0.219, 0.563) | ||
P value b | |||||||||||| | |||||||||||| | ||
Risk difference (95% CI) | |||||||||||| | |||||||||||| | ||
PMM-MIb | ||||
Number of patients | 236 | 114 | 175 | 84 |
Estimate (95% CI) | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Relative risk (95% CI) | 0.411 (0.277, 0.609) | 0.360 (0.227, 0.569) | ||
P value | |||||||||||| | |||||||||||| | ||
Risk difference (95% CI) | |||||||||||| | |||||||||||| | ||
Control-based PMM-MIc | ||||
Number of patients | 236 | 114 | 175 | 84 |
Estimate (95% CI) | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Relative risk (95% CI) | 0.440 (0.293, 0.661) | 0.393 (0.244, 0.632) | ||
P value | |||||||||||| | |||||||||||| | ||
Risk difference (95% CI) e | |||||||||||| | |||||||||||| | ||
PMM = Pattern mixture model; MI = multiple imputation.
aOn-treatment analysis including severe exacerbation events occurred during the treatment epoch only. Any exacerbation obtained after the first permanent stepping up of background asthma medication (following at least 2 severe exacerbations per protocol) will also be excluded from the analysis.
bPattern mixture model - Multiple imputation. Treatment group, age, baseline weight group, baseline eosinophils level, and number of severe exacerbation events within 1 year prior to the study are used to determine the pattern.
cPMM-MI with imputation based on the observed events in the placebo group, and in addition, age, baseline weight group, and number of severe exacerbation events within 1 year prior to the study are used to determine the pattern.
eDerived using delta method
Source: Clinical Study Report.7
Table 44: Severe Asthma Exacerbations (ITT and Baseline Blood Eosinophils ≥ 150 cells/µL Populations)
Trial population efficacy outcome | VOYAGE | |||
|---|---|---|---|---|
ITT | Baseline blood eosinophils ≥ 150 cells/µL population | |||
Dupilumab N=273 | Placebo N=135 | Dupilumab N=223 | Placebo N=108 | |
Severe asthma exacerbations | ||||
Number of patients with ≥ 1 events, n (%) | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Total number of events, n | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Total patient-years followed | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Annualized rate, unadjusted a | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Annualized rate, adjusted (95% CI) b | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Relative risk (95% CI) b | 0.458 (0.313 to 0.671) | 0.390 (0.261, 0.583) | ||
P value b | |||||||||||| | |||||||||||| | ||
Risk difference (95% CI) c | |||||||||||| | |||||||||||| | ||
CI = confidence interval.
aThe total number of event that occurred during the 52-week treatment period divided by the total number of patient-years followed in the 52-week treatment period.
bDerived using negative binomial model with the total number of events onset from randomization up to week 52 visit or last contact date (whichever comes earlier) as the response variable, with the treatment group, age, baseline weight group, region, baseline eosinophil level, baseline FeNO level, baseline ICS dose level and number of severe exacerbation events within 1 year prior to the study as covariates, and log-transformed standardized observation duration as an offset variable.
cDerived using delta method.
Note: All severe exacerbation events occurred during the 52-week treatment period are included, regardless of whether the patient is on treatment or not.
Type 2 inflammatory asthma phenotype population is defined as the randomized patients with baseline blood eosinophils >= 0.15 Giga/L or baseline FeNO >= 20ppb.
Source: Clinical Study Report.7
Table 45: Symptoms ACQ-7 (Weeks 24 and 52; ITT and Baseline Blood Eosinophils ≥ 150 cells/µL Populations)
Trial population efficacy outcome | VOYAGE | |||
|---|---|---|---|---|
ITT | Baseline blood eosinophils ≥ 150 cells/µL population | |||
Dupilumab N=273 | Placebo N=135 | Dupilumab N=223 | Placebo N=108 | |
ACQ-7, total score | ||||
Baseline | ||||
N | 273 | 135 | 223 | 108 |
Mean (SD) baseline | 2.14 (0.72) | 2.11 (0.75) | |||||||||||| | |||||||||||| |
Change from baseline at week 24 | ||||
N | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Mean (SD) CFB, week 24 | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
LS mean CFB week 24 (SE) a | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Diff vs placebo (95% CI) week 24 a | |||||||||||| | -0.36 (-0.53, -0.18) | ||
P value a | |||||||||||| | |||||||||||| | ||
Change from baseline at week 52 | ||||
N | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Mean (SD) CFB, week 52 | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
LS mean CFB week 52 (SE) a | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Diff vs placebo (95% CI) week 52 a | |||||||||||| | |||||||||||| | ||
P value (nominal) | |||||||||||| | |||||||||||| | ||
ACQ = Asthma Control Questionnaire; CFB = change from baseline; CI = confidence interval; LS = least square; OR = Odds ratio; SE = Standard error; SD = standard deviation.
aDerived from an MMRM with change from baseline in ACQ-7-IA up to week 24 as the response variable, and treatment, age, weight group, region, baseline eosinophil level, baseline FeNO level, baseline ICS dose level, visit, treatment by-visit interaction, baseline ACQ-7-IA and baseline-by-visit interaction as covariates.
bPatients who met the corresponding criterion are considered as responders. Patients who did not meet the criterion or had missing value are considered as nonresponders.
cDerived from logistic regression model with treatment, age, weight group, region, baseline eosinophil level, baseline FeNO level, baseline ICS dose level, and baseline ACQ-7-IA score as covariates.
Note: Type 2 inflammatory asthma phenotype population is defined as the randomized patients with baseline blood eosinophils >= 0.15 Giga/L or baseline FeNO >= 20 ppb
Source: Clinical Study Report.7
Table 46: Symptoms ACQ-5 (Type 2 Inflammatory Asthma Phenotype Population and Baseline Blood Eosinophils ≥ 300 cells/µL Populations)
Trial population efficacy outcome | VOYAGE | |||
|---|---|---|---|---|
Type 2 inflammatory asthma phenotype population | Baseline blood eosinophils ≥ 300 cells/µL population | |||
Dupilumab N=236 | Placebo N=114 | Dupilumab N=175 | Placebo N=84 | |
ACQ-5, total score | ||||
Baseline | ||||
N | 236 | 114 | 175 | 84 |
Mean (SD) baseline | 2.18 (0.79) | 2.15 (0.84) | |||||||||||| | |||||||||||| |
Change from baseline at week 24 | ||||
N | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Mean (SD) CFB, week 24 | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
LS mean CFB week 24 (SE) a | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Diff vs placebo (95% CI) a | |||||||||||| | |||||||||||| | ||
P value a | |||||||||||| | |||||||||||| | ||
Responders, Patients with ≥ 0.5 CFB, n (%) b | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
OR (95% CI) | |||||||||||| | |||||||||||| | ||
P value (nominal) | |||||||||||| | |||||||||||| | ||
Change from baseline at week 52 | ||||
N | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Mean (SD) CFB, week 52 | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
LS mean CFB week 52 (SE) a | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Diff vs placebo (95% CI) a | |||||||||||| | |||||||||||| | ||
P value a | |||||||||||| | |||||||||||| | ||
Responders, Patients with ≥ 0.5 CFB, n (%) b | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
OR (95% CI) | |||||||||||| | |||||||||||| | ||
P value (nominal) | |||||||||||| | |||||||||||| | ||
ACQ = Asthma Control Questionnaire; CFB = change from baseline; CI = confidence interval; LS=least square; OR = Odds ratio; SE = Standard error; SD = standard deviation.
aDerived from an MMRM with change from baseline in ACQ-5-IA values up to week 52 as the response variable, and treatment, age, baseline weight group, region, baseline eosinophil level, baseline FeNO level, baseline ICS dose level, visit, treatment by-visit interaction, baseline ACQ-5-IA value and baseline-by-visit interaction as covariates.
bA responder is defined as a patient with improvement from baseline in ACQ-5-IA >= 0.5. Patients with improvement < 0.5 or with missing value are considered as nonresponder.
Note: Type 2 inflammatory asthma phenotype population is defined as the randomized patients with baseline blood eosinophils >= 0.15 Giga/L or baseline FeNO >= 20 ppb.
Source: Clinical Study Report.7


Table 47: Pulmonary Function Measurements (Percent Predicted Prebronchodilator FEV1; ITT and Baseline Blood Eosinophils ≥ 150 cells/µL Populations)
Trial population efficacy outcome | VOYAGE | |||
|---|---|---|---|---|
ITT | Baseline blood eosinophils ≥ 150 cells/µL population | |||
Dupilumab N=273 | Placebo N=135 | Dupilumab N=223 | Placebo N=108 | |
Prebronchodilator % predicted FEV1 | ||||
Baseline | ||||
N | 273 | 135 | |||||||||||| | |||||||||||| |
Mean (SD) baseline | 77.63 (14.72) | 78.98 (14.74) | |||||||||||| | |||||||||||| |
Change from baseline at week 12 | ||||
N | |||||||||||| | |||||||||||| | 216 | 104 |
Mean (SD) CFB, week 12 | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
LS mean CFB week 12 (SE) a | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Diff vs placebo (95% CI) week 12 a | |||||||||||| | 4.98 (1.83, 8.13) | ||
P value a | |||||||||||| | |||||||||||| | ||
Change from baseline at week 52 | ||||
N | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Mean (SD) CFB, week 52 | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
LS mean CFB week 52 (SE) a | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Diff vs placebo (95% CI) week 52 a | |||||||||||| | |||||||||||| | ||
P value a | |||||||||||| | |||||||||||| | ||
CFB=change from baseline; CI = confidence interval; FEV1= forced expiratory volume in 1 second; LS=least square; SD = standard deviation; SE = standard error.
aDerived from an MMRM with change from baseline in prebronchodilator % predicted FEV1 values up to week 12 as the response variable, and treatment, baseline weight group, region, ethnicity, baseline eosinophil level, baseline FeNO level, baseline ICS dose level, visit, treatment by-visit interaction, baseline % predicted FEV1 value and baseline-by-visit interaction as covariates
Source: Clinical Study Report.7
Table 48: Summary of Sensitivity Analyses for Change From Baseline in Prebronchodilator % Predicted FEV1 at Week 12 (Type 2 Inflammatory Asthma Phenotype and Baseline Blood Eosinophils ≥ 300 cells/µL Populations)
Trial population efficacy outcome | VOYAGE | |||
|---|---|---|---|---|
Type 2 inflammatory asthma phenotype population | Baseline blood eosinophils ≥ 300 cells/µL population | |||
Dupilumab N=236 | Placebo N=114 | Dupilumab N=175 | Placebo N=84 | |
Excluding data within 14 days of SCS usea | ||||
Number of patients in the model | 229 | 110 | 168 | 80 |
LS mean CFB week 12 (SE) | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Diff vs placebo (95% CI) week 12 | 5.51 (2.48, 8.54) | 5.85 (2.32, 9.38) | ||
P value | |||||||||||| | |||||||||||| | ||
Excluding data after the start of SCS useb | ||||
Number of patients in the model | 218 | 105 | 161 | 75 |
LS mean CFB week 12 (SE) | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Diff vs placebo (95% CI) week 12 | 5.02 (1.91, 8.13) | 4.72 (0.98, 8.47) | ||
P value | |||||||||||| | |||||||||||| | ||
On-treatment analysisc | ||||
Number of patients in the model | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
LS mean CFB week 12 (SE) | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Diff vs placebo (95% CI) week 12 | |||||||||||| | |||||||||||| | ||
P value | |||||||||||| | |||||||||||| | ||
On-treatment analysis excluding data within 14 days of SCS use a,c | ||||
Number of patients in the model | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
LS mean CFB week 12 (SE) | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Diff vs placebo (95% CI) week 12 | |||||||||||| | |||||||||||| | ||
P value | |||||||||||| | |||||||||||| | ||
On-treatment analysis excluding data after the start of SCS use b,c | ||||
Number of patients in the model | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
LS mean CFB week 12 (SE) | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Diff vs placebo (95% CI) week 12 | |||||||||||| | |||||||||||| | ||
P value | |||||||||||| | |||||||||||| | ||
PMM-MI | ||||
Number of patients in the model | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
LS mean CFB week 12 (SE) | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Diff vs placebo (95% CI) week 12 | |||||||||||| | |||||||||||| | ||
P value | |||||||||||| | |||||||||||| | ||
Control-based PMM-MI d | ||||
Number of patients in the model | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
LS mean CFB week 12 (SE) | |||||||||||| | |||||||||||| | |||||||||||| | |||||||||||| |
Diff vs placebo (95% CI) week 12 | |||||||||||| | |||||||||||| | ||
P value | |||||||||||| | |||||||||||| | ||
CFB=change from baseline; CI = confidence interval; FEV1= forced expiratory volume in 1 second; LS=least square; MI= Multiple imputation; PMM = Pattern mixture model; SCS = systemic corticosteroid; SD = standard deviation; SE = standard error.
aAnalysis excluding prebronchodilator FEV1 measurements collected from systemic corticosteroid start date to systemic corticosteroid end date + 14 days.
bAnalysis excluding prebronchodilator FEV1 measurements collected on and after first day of systemic corticosteroid use.
cAnalysis including on-treatment prebronchodilator FEV1 up to week 12 and within last IMP date + 14 days.
dAssuming patients from the dupilumab groups would exhibit the same future evolution of prebronchodilator FEV1 as patients on the placebo.
Note: Type 2 inflammatory asthma phenotype population is defined as the randomized patients with baseline blood eosinophils >= 0.15 Giga/L or baseline FeNO >= 20 ppb.
Source: Clinical Study Report.7
|||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||| | |||
|---|---|---|---|---|
|||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||| | |||
|||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||||||||| | |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | ||||
|||||||||||||||||||||||||||||||||||| | ||||
|||||||||||||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| |
|||||||||||||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| |
|||||||||||||||||||||||||||||||||||| | ||||
|||||||||||||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| |
|||||||||||||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| |
|||||||||||||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| |
|||||||||||||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||
|||||||||||||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||
|||||||||||||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| |
|||||||||||||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||
|||||||||||||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||
|||||||||||||||||||||||||||||||||||| | ||||
|||||||||||||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| |
|||||||||||||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| |
|||||||||||||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| |
|||||||||||||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||
|||||||||||||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||
|||||||||||||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| |
|||||||||||||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||
|||||||||||||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Source: Clinical Study Report.7
Appendix 4: Description and Appraisal of Outcome Measures
Note that this appendix has not been copy-edited.
Aim
To describe the following patient-reported outcome measures and review their measurement properties (validity, reliability, responsiveness to change, and MID):
ACQ-IA (ACQ-7 and ACQ-5) for children aged 6 to less than 12 years
PAQLQ(S)-IA for patients older than 7 and younger than 12 years
PRQLQ-IA for children aged 6 to less than 12 years, with history of allergic rhinitis
PACQLQ for caregivers of patients older than 7 years
EQ-5D-Y
Findings
Table 50: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
ACQ-7 | ACQ is a patient-reported tool to assess asthma control in patients ≥ 6 years of age. It comprises the following 7 questions, of which the mean of the results is the overall score (0 indicates well-controlled asthma and 6 indicates extremely poorly controlled asthma):
| Validity: Content validity has been ensured during development phase by having incorporated input from pediatric asthma clinicians.46 Moderate to strong construct validity has been demonstrated with other instruments such as MiniPAQLQ, RCP, ACD, PACQLQ, ACT or cACT, ASUI, and PAQLQ (r ranges from 0.49 to 0.93).24,25 It has been shown that ACQ can differentiate between well-controlled patients and poorly controlled patients (P < 0.0001).25 Reliability: Test-retest reliability has been demonstrated (ICC = 0.22 to 0.95) in stable patients.24,25 Even though high concordance was found between ACQ-7 and -5 (ICC = 0.93), they are not interchangeable as differences in scores were statistically significant (P = 0.006 to 0.01).24 Internal consistency (Cronbach alpha = 0.74) was acceptable for group comparison (alpha > 0.7).25 Responsiveness Both ACQ-7 and -5 were able to detect changes in patients whose health status has either worsened or improved over a period of time.24,25 Also, changes in ACQ scores over time differed significantly across patients whose health status differed.25 | In pediatric patients with asthma: Anchor-based method ACQ-7:
ACQ-5: 0.65 based on Global Rating of Change24 Distribution-based method: Between 0.375 (0.5*SD) and 0.382 (SEM)25 Triangulation method: Estimated to be 0.4025 |
ACQ-5 | ACQ-5 is a shortened version of the original patient-reported ACQ-7 measure, which includes items relating exclusively to patient symptoms. Items relating to rescue bronchodilator use and lung function are excluded from the ACQ-5. All items are scored on a 7-point scale, which ranges from 0 (indicating good control) to 6 (poor control). The overall score is the mean of all questions, with a high score indicating poor control. | ||
PAQLQ(S) | A disease-specific, quality of life questionnaire available in both interviewer and self-administered formats, designed for children aged 7 to 17 years of age. There are 23 items grouped into 3 domains (symptoms, emotional aspects, activity limitation) that are graded on a 7-point Likert scale (1 = maximum impairment, 7 = no impairment). Both domain and overall scores range from 1 to 7 with higher score representing better HRQoL. Recall period is past 1 week. In standardized format, 3 patient-specific questions are replaced with generic activity questions (physical activity, activities with animals, activities with friends and family). | Validity: Criterion validity has been ensured by comparing with PAQLQ, which had been developed based on guidelines, input from patients, parents, and health professionals.28 All measurements were made in data from the interviewer-administered version.28 Construct validity was strong (Pearson r > 0.547) as measured by correlation with the ACQ, weak (r = 0.1 – 0.3) to moderate (r = 0.3 – 0.5) with PEF, moderate with HUI, and moderate to strong with ACD and PACQLQ.28 Reliability: Concordance with PAQLQ (ICC = 0.669 to 0.974) was high and internal consistency (Cronbach alpha = 0.90 – 0.96) was acceptable (alpha > 0.7), all exceeding individual monitoring level (alpha > 0.9).28 Test-retest reliability measured in stable patients 4 weeks apart (ICC = 0.81 – 0.92) also demonstrated to be acceptable.28 Responsiveness: PAQLQ(S) was able to detect changes in patients who either improved or worsened (score change > 0.5) over a 4-week period (P < 0.0001) and changes between stable and unstable patients (P < 0.0003).28 | In children with asthma: 0.42 ± 0.55 for overall quality of life score.27 |
PRQLQ | A disease-specific, interviewer-administered, health-related quality of life instrument developed in children 6 to 12 years of age diagnosed with SAR or hay fever. PRQLQ consists of 23 items grouped into 5 domains (nose symptoms, eye symptoms, practical problems, activity limitation and other symptoms) that are graded on a 7-point Likert scale (0 = no impairment, 6 = maximum impairment). Both domain and overall scores range from 0 to 6, with higher score representing worse HRQoL. Recall period is past 1 week. | Validity: Content validity has been ensured during development stage by incorporating input from pediatric patients and clinicians as well as referencing literature and other questionnaires.30 Construct validity has been demonstrated with diary symptom scores and global rating of change, with higher correlation between nose and eye specific domains of different questionnaires (Pearson r = ranges from 0.47 to 0.67) and poorer correlation found between nonrelated domains of different questionnaires (r near zero).30 Reliability: Test-retest reliability has been demonstrated in stable patients over a 3-week period (ICC = from 0.57 to 0.93).30 Responsiveness: PRQLQ was able to detect within-patient change over a 3-week period in patients whose health status changed as well as differentiate stable vs. unstable patients for the same period.30 | Not identified.48 |
PACQLQ | A self-administered questionnaire to measure impact of children’s asthma on their caregivers’ normal daily activities and emotional function. There are 13 questions grouped into 2 domains (activity limitation and emotional function) which are answered based on a 7-point Likert scale (1 = severe impairment, 7 = no impairment). Both domain and overall scores range from 1 to 7, with higher score representing better HRQoL. Recall period is previous 1 week. | Validity: Content validity has been ensured during development by pooling items generated by parents of children with asthma, a literature review, and input from health professionals.27 Construct validity has been demonstrated with caregiver burden of illness (as measured by generic questionnaire), child’s asthma severity, global ratings of change, and perception of change in child’s asthma survey, with higher correlations found between similar domains from different instruments (r > 0.5) and poorer correlations noted between nonrelated domains from different instruments (r < 0.3 or near zero).27 Poor correlations were found between child’s peak flow rates, asthma control, and FEV1.27 Reliability: Test-retest reliability was demonstrated (ICC = 0.84 to 0.85), with low within-patient variance (within-patient SD = 0.31 – 0.39).27 Responsiveness: PACQLQ was able to detect within-patient changes over a 4-week period (P < 0.001) and differentiate patients who stayed stable from those whose status has changed over the same period of time (P < 0.0003).27 | In pediatric patients with asthma and their caregivers: Based on anchor-based approach (global rating of change), estimated to be 0.5 for overall score, 0.64 for the emotional function domain, and 0.67 for activity limitation domain.27 |
EQ-5D-Y | A generic, preference-based HRQoL measure with child-friendly wording intended for younger population. Self-completion (ages 8-15 years) and proxy (ages 4-7 years) versions are available. Based on EQ-5D-3L, descriptive system comprises 5 dimensions (mobility, self-care, usual activities, pain or discomfort, anxiety/depression) with each dimension having 3 levels (no problems, some problems, a lot of problems). VAS remains the same as other EQ-5D. Lower scores on 5-digit health status, higher scores on index and VAS represent better HRQoL. | Validity: A moderate convergent validity of utility index has been demonstrated with PROMIS-PAIS in most questions (r = 0.38), a weak correlation was found with ACQ (r = 0.28). Correlations between VAS and both instruments were weak (r = 0.16 and 0.26, respectively).49 A weak (r = 0.015) to strong (r = 0.583) correlation has been shown with PACQLQ.50 Both utility index and VAS were able to differentiate patients defined by asthma control, reliever use, and second-hand smoke exposure most of the time.50 Reliability: Test-retest reliability was confirmed over a 6-month period in stable patients (defined by either VAS or ACQ) with high ICC values of utility index values (0.81 and 0.79, respectively). ICC for VAS when patients were classified by ACQ was also high, i.e., 0.70, proving reproducibility of EQ-5D-Y.49 Responsiveness: EQ-5D-Y utility index and VAS were able to detect health status changes over 10 months in patients who worsened during that time with greater than moderate effect sizes, albeit not statistically significantly.49 | Not identified |
ACD = Asthma Control Diary; ACQ = Asthma Control Questionnaire; ACT = Asthma Control Test; cACT = childhood Asthma Control Test; ASUI = Asthma Symptom Utility Index; EQ-5D-3L = EQ-5 Dimensions-3 Levels; EQ-5D-Y = EQ-5 Dimensions-Youth; MID = minimal important difference; MiniPAQLQ = Mini Pediatric Asthma Quality of Life Questionnaire; FEV1 = forced expiratory volume in the first second; FEV1% = FEV1/forced vital capacity (FVC) ratio; HRQoL = health-related quality of life; HUI = Health Utility Index; ICC = intraclass coefficient; PACQLQ = Paediatric Asthma Caregiver’s Quality of Life Questionnaire; PAQLQ = Pediatric Asthma Quality of Life Questionnaire; PAQLQ(S) = Pediatric Asthma Quality of Life Questionnaire with Standardised Activities; PEF = % predicted peak expiratory flow; PROMIS-PAIS = Patient-Reported Outcomes Measurement Information System-Pediatric Asthma Impact; PRQLQ = Pediatric Rhinoconjunctivitis Quality of Life Questionnaire; RCP = Royal College of Physicians’ “3 questions”; SABA = short-acting beta agonist; SAR = seasonal allergic rhinoconjunctivitis; VAS = visual analogue scale.
Asthma Control Questionnaire
The complete ACQ, also termed the ACQ-7, was developed to evaluate asthma control in adult patients with asthma.22 The ACQ is 1 of the most commonly used instruments measuring asthma control.22,23 The questionnaire comprises 7 questions, the responses of which are scored on a 7-point scale (0 = no impairment; 6 = maximum impairment).24 Questions regarding 6 aspects of the patient’s previous week’s experiences are answered by the patient and include questions on activity limitation, nocturnal waking, shortness of breath, wheezing, symptoms on waking, and the average number of daily doses of short-acting beta2-agonist used.23 In addition, the seventh item includes calculations performed by clinical staff with regard to prebronchodilator FEV1% predicted on a similar 7-point scale.22,23 The ACQ score is calculated as the mean of the 7 questions (as all questions are equally weighted), with scores ranging from 0 (well-controlled) to 6 (extremely poorly uncontrolled).22,23,51 The ACQ is used extensively in clinical trials to measure clinically meaningful change in asthma control.22
The ACQ also exists in abbreviated versions: the ACQ-5 focusing only on the symptoms (omitting bronchodilator use and FEV1) and the ACQ-6 scoring symptoms and bronchodilator use (only omitting FEV1).
During development of the ACQ, it was found that the self-administered version was easily and accurately understood by children aged ≥ 11 years and children < 6 years of age had difficulty understanding the concept of “during the last week.” Therefore, an interviewer-administered version of the ACQ was devised for children aged 6 – 10 years. ACQ should not be used in children < 6 years of age.24 In the IA version, there are instructions to the interviewer on how to ensure that children understand the 7-point scale and the concept of “during the last week.” For example, if the child does not understand the primary wording in the ACQ, such as ‘how limited were you in your activities,’ the interviewer uses the secondary wording, such as ‘how bothered were you in the things you do every day.’ Interviewers were asked to help only when children have difficulties understanding questions. It is recommended that in children < 10 years, the questionnaire should be administered by a trained health professional.24 The IA version of ACQ and an online self-administered version of ACQ are strongly correlated (Pearson r = 0.79), however, are not completely interchangeable. For example, a study showed that better control of asthma is detected when measured with ACQ-IA than by online self-assessment ACQ.52
Validity
Content validity of ACQ has been ensured by having involved pediatric asthma clinicians during development phase. They selected the items in ACQ that are considered the most important to determine asthma control.46
Juniper et al.24 tested measurement properties of the ACQ-7 in 35 children (aged 6 – 16 years) with a wide range of asthma severity and had current symptoms of asthma (ACQ score > 0.5) in England. The cross-sectional construct validity has been demonstrated by strong Pearson correlation coefficients (r > 0.547) between established measures (Mini Pediatric Asthma Quality of Life Questionnaire [MiniPAQLQ], Royal College of Physicians’ “3 questions” [RCP], Asthma Control Diary [ACD], PACQLQ) and ACQs (ACQ-7 = -0.83, 0.52, 0.77, -0.63; ACQ-5 = -0.84, 0.57, 0.71, -0.56, respectively). Longitudinal construct validity has also been shown by similarly strong Pearson correlation coefficients (r > 0.547) between established health status measures,53 (changes in MiniPAQLQ, RCP, ACD, PACQLQ) and changes in ACQ scores over 1-4 weeks (ACQ-7 = -0.89, 0.81, 0.83, -0.49; ACQ-5 = -0.93, 0.81, 0.79, -0.84, respectively). Multiple measures were used since there is no gold standard for measuring asthma control in children. These correlations were close to a priori predications about the degrees of correlations expected of the ACQ if it truly measures change in asthma control. Strong correlations demonstrated that established instruments and ACQ measure a similar concept. However, the author of the study noted that asthma control and quality of life are 2 distinct components of clinical asthma assessment, therefore, some degree of discrepancies are expected between ACQ and the HRQoL measurement instruments.54
The study by Juniper, et al.24 has several limitations in that it tested ACQ in 35 pediatric patients from a single centre with a UK version that had alternative wording to younger children and was administered by a trained interviewers. To overcome some of the limitations, Nguyen, et al.25 tested measurement properties of ACQ in 305 pediatric patients (aged 6 – 17 years old) with inadequately controlled asthma on ICS that were enrolled in a clinical trial using the North American English version of ACQ. They confirmed the moderate to strong construct validity of ACQ by measuring Pearson correlation coefficients with Asthma Control Test (ACT for ages 12 to 17; r = -0.73) or childhood ACT (cACT for ages 6 to 11; r = -0.57), the Asthma Symptom Utility Index (ASUI; for ages 6 to 11, r = -0.74; for ages 12 to 17, r = -0.74), and the PAQLQ (for ages 6 to 11, r = -0.71; for ages 12 to 17 r = -0.66). Furthermore, ACQ differentiated patients who had an episode of poor asthma control from those who did not (for ages 6 to 11, mean difference [SD] = 0.46 [0.01]; P < 0.0001; for ages 12 to 17, mean difference [SD] = 0.30 [0.02]; P < 0.0001) in all aspects such as decrease in PEF rate, increase in rescue medication use, urgent care (ED, hospital, clinic or doctor visit), and SCS use. However, the group did not find such correlation between exhaled NO (n = 146) or expired breath condensate pH (n = 239) and ACQ scores.25
Reliability
Juniper, et al.24 categorized 2 groups of children, e.g., a group of children who remained clinically stable and another group who experienced change in their asthma control between clinic visits (weeks 0-1 and 1-4) based on the Clinician’s Global Rating of Change score. Pearson correlation coefficients between ACQ changes and Global Rating of Change Questionnaire over a period of 1 – 4 weeks were considered to be strong (r > 0.5).47 Test-retest reliability was determined from the stable group by estimating intraclass correlation coefficient (ICC estimated as the within-patient SD and related to the total SD). The group made a priori predications about the level of correlation expected if the ACQ truly measures asthma control. In 19 children who remained stable, there was good evidence for reliability as ICC for the ACQ-7 was 0.79 and ACQ-5 was 0.67.
Even though the concordance between the ACQ-7 and ACQ-5 was high (ICC = 0.93), there was statistical difference between the ACQ-7 and ACQ-5 scores (mean ± SD = 0.12 ± 0.26; P = 0.010) with the greater change in the ACQ-5 than ACQ-7 in scores between baseline and 4 weeks (mean ± SD = 0.17 ± 0.34; P = 0.006).24 Therefore, the 2 versions of ACQ should not be used interchangeable.24
Another group25 found that Cronbach alpha for the ACQ to be 0.74 (n = 305), 0.75 for ages 6 to 11 group (n = 164) and 0.72 for ages 12 to 17 group (n = 141), all of which are considered acceptable for group-level internal consistency (alpha > 0.7).55 Of note, a value of alpha > 0.9 is considered necessary for an individual-level comparison. The ICC for ACQ scores between 2 consecutive visits (total 8 visits) among stable patients who reported no episode of poor asthma control for that period ranged between 0.42 to 0.82 for the overall group, 0.34 to 0.95 for the 6 to 11 age group and 0.22 to 0.74 for the 12 to 17 age group.25 The reliability for ACQ in the overall group and younger patient group was moderate to strong,47 whereas that for older children varied from weak to strong depending on a period of visit measured.
Responsiveness to Change
Responsiveness was measured by detecting within-patient change in the unstable group of children using a paired t-test and between-group changes (stable vs. unstable) using an unpaired t-test. The ACQ-7 and the ACQ-5 both were able to detect changes in asthma control in unstable patients between clinic visits 4 weeks apart (P = 0.026 and 0.007, respectively) and were able to distinguish between stable and unstable patients during the same period (P = 0.072 and 0.027, respectively), with ACQ-5 showing statistically better responsiveness than ACQ-7.24
Similarly, Nguyen, et al.25 demonstrated responsiveness of ACQ to changes in asthma control. Mean changes in ACQ scores between consecutive visits in continuously well-controlled (n = 196 over 547 days) and continuously poorly controlled patients (n = 148 over 337 days) were -0.02 (95% CI = -0.05, 0.01) and 0.06 (95% CI = 0.00, 0.12), respectively. For those who had worsening control (n = 155 over 177 days) and improved control (n = 165 over 200 days), mean changes in ACQ scores were 0.26 (95% CI = 0.11, 0.41) and -0.33 (95% CI = -0.44, -0.22), respectively. Also, mean changes in ACQ scores differed significantly across groups of patients categorized based on their health status for the overall group (P < 0.0001), 6 to 11 age group (P < 0.001), and 12 to 17 age group (P = 0.01).
MID
With the Global Rating of Change method (n = 11), Juniper, et al. estimated the MID for ACQ-7 to be 0.52 (SD = 0.45) and for ACQ-5 to be 0.65 (SD = not reported). The estimated MID was confirmed with the geometric mean regression method (n = 31), which gave a similar result with 0.50 (SEM = 0.05).24 The sponsor used an MID estimate of 0.5 based on the same reference as identified by CADTH literature search.24
Nguyen, et al.25 estimated MID to be 0.375 and 0.382 based on the distribution-based method using 0.5*SD and SEM for the overall group. For those aged 6 to 11 years, MID was estimated to be 0.40 and for those between the ages of 12 and 17 years old, estimated MID was 0.35 using both 0.5*SD and SEM. The group also estimated MIDs based on various anchors. Using the mean ACQ scores among patients experiencing a specified clinical event compared to those who did not as anchor, Nguyen, et al. estimated MID to be 0.4-0.5 for the combined age group, 0.5-0.6 for the 6 to 11 age group, and 0.3-0.4 for the 12 to 17 age group. Anchoring the ACQ against the ACT that uses MID of 3 points, an estimated MID for ACQ was 0.42 for the 12 to 17 age group, and against a 3-point change in the cACT, estimated MID was 0.33 for children ages 6 to 11. Lastly, using the triangulation method,56 the estimated MID was 0.40 for the overall group.
Standardized PAQLQ
The original PAQLQ was developed in McMaster University to measure the problems that children with asthma experience in their daily lives.27 The PAQLQ is the only disease-specific quality of life measurement tool to be specifically developed for use in children with asthma and referred to as the gold standard.28 The PAQLQ has 23 items grouped into 3 domains: symptoms (10 items), emotional aspects (8 items), and activity limitation (5 items) that ask about the most troublesome functional problems to children with asthma.26,28 In the activity domain, there are 3 questions that are patient specific, meaning the child is asked to choose 3 activities that they are most likely to undertake at the time they complete the questionnaire.26,28 These 3 activities remain constant at each follow-up visit. Although the individualized questionnaire enhances content validity by incorporating activities that are most relevant to children, the main disadvantage is that it is time-consuming to complete.28 Also, children tend to change their favourite activities.26 In the PAQLQ(S), the 3 patient-specific questions have been replaced by generic activity questions (physical activity, activities with animals, activities with friends and family) that are the same for all children. These 3 generic activity questions incorporate the activities that were most frequently chosen by children when the original PAQLQ was used. For some studies, such as in long-term clinical trials and patient monitoring, the generic activities become more appropriate since they are easier and more convenient to administer and compare across samples.28 PAQLQ(S) addresses this need, however, one or more of the generic activities may be irrelevant giving rise to a risk of a ceiling effect. The PAQLQ(S) is more appropriate for use in large-scale research projects, which aim to measure quality of life for children with asthma at a group level. The PAQLQ(S) is shorter, quicker to complete and more cost-effective for use in large cohort studies, such as clinical trials for long-term monitoring. The PAQLQ(S) provides standardized responses, which can be coded and analyzed relatively easily to provide broad comparisons across different populations.28
The PAQLQ(S) is a disease-specific, quality of life questionnaire available in both interviewer and self-administered formats, designed for children aged 7 to 17 years of age.26 Questions in each domain are scored on a Likert scale of 1 – 7 (1 = maximum impairment, 7 = no impairment) and are equally weighted to be combined to create mean scores for each domain as well as overall quality of life.28 Therefore, both the domain and overall scores range from 1 – 7, with higher score representing better HRQoL. The recall period is the past 1 week.27
Validity
Wing, et al.28 tested measurement properties in 42 children with asthma (mean age = 11.3 years of old, SD = 2.7; ranges 7 – 17) receiving standard asthma care in UK. All data from Wing, et al. study was generated from interviewer-led questionnaires and none from the self-administered versions. Criterion validity was ensured by comparing PAQLQ(S) overall and domain scores to those of the PAQLQ, which had been developed based on guidelines for a dozen validated disease-specific quality of life instruments and input from patients, parents and health professionals.27 There was no statistically significant difference in scores between the original PAQLQ and the PAQLQ(S), apart from the “activities” subscale (P < 0.03). Both cross-sectional (which demonstrates that the observed differences between children truly reflect differences in asthma-specific quality of life) and longitudinal construct validity (which demonstrates that observed changes in scores over a period of time correlate in a predictable manner with changes in other measures of health status) were evaluated by the Pearson’s correlation coefficients with the original PAQLQ. The cross-sectional construct validity measured at week 9 against ACQ, % predicted PEF, ACD, Health Utilities Index (HUI), and PACQLQ showed Pearson’s coefficients of -0.77 (P < 0.001), 0.25 (P < 0.05), -0.68 (P < 0.001), -0.40 (P < 0.01), and 0.51 (P < 0.001), respectively, for overall scores (data for domain scores are not shown). The longitudinal construct validity measured by Pearson coefficients between weeks 5 and 9 for ACQ, PEF, ACD, HUI, and PACQLQ were -0.79 (P < 0.001), 0.41 (P < 0.01), -0.45 (P < 0.001), -0.46 (P < 0.001), and 0.43 (P < 0.01), respectively for overall scores (data for domain scores are not shown).28
Reliability
The concordance between the original PAQLQ and PAQLQ(S) was measured by ICC (Pearson correlation coeifficient) with a paired Student’s independent t-test and was 0.974, 0.669, 0.955, and 0.863 for overall, activity, symptom, and emotion scores, respectively (P value < 0.001). Internal consistency estimated by Cronbach alpha values at baseline for overall, activity, symptom, and emotion scores were 0.96, 0.90, 0.90, and 0.94, respectively. Of note, reliability in activity domain was higher for PAQLQ(S) compared to PAQLQ probably due to generic questions included to standardize responses. These measures indicate that the correlation with PAQLQ is strong (r > 0.547) and PAQLQ(S) has an acceptable internal consistency (alpha > 0.755), all exceeding alpha = 0.90 set for individual monitoring. Test-retest reliability assessed in patients with stable asthma between weeks 1 – 5 and 5 – 9 (n = 16) was also strong (ICC [95% CI] = 0.92 [0.82, 0.97], 0.81 [0.63, 0.92], 0.89 [0.77, 0.96], and 0.92 [0.84, 0.97] for overall, activity, symptom, and emotion scores, respectively). These results exceed 0.70, which is commonly accepted minimal standard for reliability coefficients.28 However, Wing, et al. expected ICC for test-retest stability greater than 0.85 based on ICCs reported in previous studies.
Responsiveness to Change
The PAQLQ(S) was able to detect the changes in patients who either improved or deteriorated by a score > 0.5 between weeks 1 – 5 and 5 – 9 (paired Student’s t-test, all P < 0.0001) and differences between stable and unstable groups for each of the measures (unpaired Student’s t-test, overall differences all < 0.0003).28
MID
Juniper, et al.27 used anchor-based method to estimate an MID in children with asthma. Briefly, the group took the mean difference in PAQLQ score in patients who score -3, -2, +2, or +3 on the global rating of change as MIDs. According to this method, MID estimates for overall quality life, symptom, activity, and emotion domains were 0.42 ± 0.55 (n = 29), 0.54 ± 0.64 (n = 0.29), 0.70 ± 1.17 (n = 26), and 0.28 ± 0.67 (n = 17), respectively. The sponsor took 0.5 as an MID estimate for PAQLQ(S)-IA based on the same reference.27
Pediatric Rhinoconjunctivitis Quality of Life Questionnaire
The PRQLQ is a disease-specific, HRQoL instrument developed in children 6 to 12 years of age diagnosed with seasonal allergic rhinoconjunctivitis or hay fever who had experienced troublesome symptoms in the previous month.57 The PRQLQ was developed in Southern Ontario to measure the functional problems (physical, emotional, and social) that are most troublesome to children with SAR. The questionnaire should be administered by a trained interviewer. Parents should not be present during the interview31 The PRQLQ consists of 23 items grouped in 5 domains (nose symptoms, eye symptoms, practical problems, activity limitation and other symptoms). Children are asked to recall how they have been during the previous week and respond to each question on a 7-point Likert scale. Responses are given by children with two 7-point scales (a green card and a blue card), where 0 represents no impairment (not bothered/none of the time) and 6 represents maximum impairment (extremely bothered/all of the time). All items are weighted equally. Each domain score and overall score is the mean of the items in each domain and overall questionnaire, respectively. The overall score is estimated from the mean score of all items, thus ensuring that greater weight is given to the domains with the larger number of items. Both the domain and overall scores range from 0 to 6, with higher score representing worse HRQoL. All the words used in the PRQLQ are child-friendly language; however, the recall time (1 week) can be difficult for younger children to understand. Therefore, the PRQLQ should not be used in children under 6 years old.31
Validity
Content validity has been ensured by incorporating several measures during development period: 1) item pools from the Rhinoconjunctivitis Quality of Life Questionnaire, the Adolescent Rhinoconjunctivitis Quality of Life Questionnaire, and the PAQLQ; 2) discussion with children 6 to 12 years old with allergic rhinoconjunctivitis; 3) discussion with clinicians; and 4) a review of the pediatric HRQoL literature.30
Juniper, et al.30 conducted validation study in 75 children (a mean age of 9.8 years, SD = 1.9) who had troublesome allergic rhinoconjunctivitis that required additional medication during the fall pollen season in the US. They assessed longitudinal construct validity by correlating within-patient changes in quality of life scores with within-patient changes in diary symptom scores and global rating of change. The a priori prediction was that if the PRQLQ truly could measure rhinoconjunctivitis-specific quality of life, the correlations should be similar to those observed in adults with rhinoconjunctivitis. Actual and predicated correlations between change in the PRQLQ and diary symptom scores were close across all domains of the PRQLQ and the different symptoms of the diary. For example, stuffy nose (r = 0.58) and mean nose symptoms (r = 0.67) in symptom diary were highly correlated with nose symptom in the PRQLQ (r = 0.55 – 0.7 a priori prediction).
The group also assessed cross-sectional construct validity by correlating quality of life scores with daily symptom diary scores. Again, a priori prediction was that the correlations should be close to those observed in adults. Correlations between the PRQLQ and diary symptoms were very similar to those observed in adults and adolescents with rhinoconjunctivitis, with the highest correlations observed between the nasal domain of the PRQLQ and diary nasal symptoms (r = 0.47 actual vs. r = 0.45 – 0.6 for a priori prediction) and between the eye domain of the PRQLQ and eye symptom scores in the diary (r = 0.59 actual vs. r = 0.45 – 0.6 for a prior prediction). The poorest correlations were found between other symptom domain of the PRQLQ and the diary stuffy nose scores (r = 0.06 actual vs. r < 0.25 a priori prediction).30
Reliability
Juniper, et al.30 measured reliability in 13 children whose rhinoconjunctivitis remained stable between clinic visits at weeks 1 and 3 using ICC. The result shows that ICC was 0.93 for overall quality of life and 0.57, 0.91, 0.79, 0.88, and 0.87 for nose symptoms, eye symptoms, practical problems, other symptoms, and activity limitations, respectively. High ICCs indicate that within-patient variance was consistent in general, except for nose symptoms that showed lower ICC possibly as a result of a low between-patient variance.
Responsiveness to Change
Responsiveness was measured in 61 children who experienced a change in their rhinoconjunctivitis between weeks 1 and 3.30 Using a paired t-test, within-patient changes in children who changed (n = 61) between visits were detected (change in mean score ranges from 0.37 to 0.80). No such changes were found in children (n = 13) who had stable rhinoconjunctivitis (change in mean score ranges from -0.22 to 0.12). Using an unpaired t-test to examine the ability of the instrument to distinguish between patients whose quality of life changed from the beginning to end of the study period and those whose quality of life remained unchanged for the same period was tested. The PRQLQ was able to detect the difference on in overall quality of life (P = 0.005), not each domain (P value ranges from 0.061 to 0.087).
MID
An estimate for MID has not been identified through literature search. The sponsor has taken 0.5 to be an MID for the VOYAGE study based on their literature search.48 However, CADTH could not verify the MID estimate referenced by the sponsor.
Paediatric Asthma Caregiver’s Quality of Life Questionnaire
The PACQLQ is a self-administered questionnaire designed to measure the problems that are most troublesome to the parents (primary caregivers) of children with asthma.29 The questionnaire asks the ways in which child’s asthma has interfered with caregiver’s normal daily activities and how this has made them feel. The PACQLQ was developed and validated using the same methodology as other questionnaires such as PAQLQ(S) or PRQLQ.27
The problems experienced by the parents of younger children (< 7 years) may not be the same as those experienced by the parents of older children and adolescents. For example, by 7 years old, most children are at school and participating in activities without parental involvement and these children are often responsible for taking their own asthma medications. In contrast, children under 7 years may not understand their asthma and how to manage it (e.g., running until severe shortness of breath) and parents worry that they may not detect deterioration in symptoms. Due to these differences, the PACQLQ should only be used in the parents of children 7-17 years. It is likely to be inaccurate in assessing the impact of younger children’s asthma on their parents.29
There are 13 questions in 2 domains (4 items concern activity limitations and 9 concern emotional function). Parents recall the impact that their child’s asthma has had during the previous week.27 Responses to each item in the PACQLQ are given on a 7-point Likert scale where 1 (all of the time/very worried or concerned) represents severe impairment and 7 (none of the time/not worried or concerned) represents no impairment.27 Each item is weighted equally. Domain and overall quality of life scores are calculated as the mean scores; therefore, both range from 1 to 7, with higher score representing better HRQoL. The recall period is the previous 1 week.29
Validity
Content validity has been ensured during development of PACQLQ. Briefly, a pool of items was generated from interviews with parents of children with asthma, a literature review, and discussion with health professionals. The items that were identified most frequently and rated most bothersome by caregivers were selected for the final questionnaire.27
Juniper, et al.27 assessed measurement properties of PACQLQ in 52 children with current symptoms of asthma and their primary caregivers. The children were between 7 and 17 years and represented a wide range of asthma severity. The primary caregiver (age ranged from 30 to 63 years; mean 40.94, SD = 5.6 years) was usually a parent and lived with the child at least 75% of the time. The a priori predictions for both longitudinal and cross-sectional validity was that correlations would be observed if investigators can truly measure quality of life in caregivers. The longitudinal construct validity was measured by correlating within-patient changes in the caregiver’s quality of life scores over a 4-week period with within-patient changes in caregiver burden of illness (generic questionnaire), child’s asthma severity (FEV1% predicted, PEF, beta agonist use, and so forth), global ratings of change, and perception of change in child’s asthma survey. The highest correlations were observed between the PACQLQ emotional function and caregiver global ratings of change – emotions (r = 0.55 vs. r > 0.50 a priori prediction) or caregiver perception of change in child’s’ asthma survey (r = 0.52 vs. r = 0.20 – 0.35 a priori prediction). The poorest correlations were between the change in caregiver burden of illness – mastery and the PACQLQ emotional function (r = -0.0007 vs. r = 0.20 – 0.35 a priori prediction) or activity limitation (r = 0.02 vs. r = no a priori prediction made). The cross-sectional construct validity was measured by correlating caregiver quality of life scores at each clinic visit with the measures of the child’s asthma severity and with generic caregiver quality of life scores. The highest correlations were observed between the PACQLQ activity limitation and caregiver burden of illness – overall (r= -0.65), family/social (r = -0.62 vs. r = 0.20 – 0.35 a priori prediction), and personal strain (r = -0.70). The poorest correlations were observed between the caregiver burden of illness – mastery and the PACQLQ emotional function (r = 0.28 vs. r = 0.20 – 0.35 a priori prediction) and activity limitation (r = 0.26 vs. no a priori prediction). Similarly, such poor correlations were observed between child’s’ asthma severity – beta agonist use and the PACQLQ emotional function (r = -0.22 vs. r = 0.20 – 0.35 a priori prediction) or activity limitation (r = -0.28).27
As predicted, the correlation between the PACQLQ and change in child’s peak flow rates or asthma control was weak, with FEV1 even lower than predicted. In general, correlations between the PACQLQ and the global ratings of change or parental rating of change in the children’s severity of asthma were moderate to high as predicted, with the Impact on Family Scale substantially higher than predicted. These results suggest that problems associated with caring for a child with asthma is not entirely the same as general problems of looking after a sick child.27
Reliability
Reliability was measured in 44 caregivers who were stable between consecutive clinic visits.27 The ICCs for overall quality of life, emotional function, and activity limitation were 0.85, 0.80, and 0.84, respectively. The PACQLQ showed a high degree of reliability with a low within-patient variance (within-patient SD = 0.31 – 0.39) and ability to detect differences in quality of life between caregivers (between-patient SD = 0.71 – 0.88).
Responsiveness to Change
Responsiveness was measured using a paired t-test as ability to detect important within-patient changes when caregivers changed over 4 weeks.27 Caregivers who changed (n = 23) showed the change in mean score per item as 0.72, 0.71, and 1.80 for overall quality of life, emotional function, and activity limitation, respectively (P < 0.001 for all), whereas those who were stable showed -0.03, -0.02, and -0.01, respectively. Also, using an unpaired t-test of the differences between the beginning and end of each period, the ability of the instrument to distinguish between patients who remained stable and those who changed was measured. Changes in those caregivers who changed over 4 weeks were significantly different from the changes in score in the caregivers who reported staying stable (P < 0.0003 for all). The questionnaire proved highly responsive to within-patient changes over time.
MID
Based on global rating of change as anchor, the MID for overall quality of life was estimated to be 0.50 (SD = 0.51; n = 16) with similar values for the emotional function domain (0.64, SD = 0.56; n = 18) and activity limitation domain (0.67, SD = 0.63; n = 9).27
EQ-5D-Y
In 2009, EuroQol group introduced the EQ-5D-Y that is a child-friendly version of EQ-5D and a more comprehensible instrument suitable for children and adolescents. EQ-5D-Y is available in more than 100 languages and in various modes of administration. The EQ-5D-Y is based on the EQ-5D-3L and essentially consists of 2 pages: the EQ-5D descriptive system and the EQ VAS.32 EQ-5D-Y is designed for self-completion by children and adolescents aged 8-15 years. Between the ages of 4 and 7 years, a proxy-completed version should be used. Proxy versions, for completion by a caregiver or someone who knows the person well, are used when children or adolescents are mentally or physically incapable of reporting on their HRQoL, for instance because of severe intellectual disability or mental health problems. Four proxy versions are currently available.32 For adolescents between the ages of 12 and 15 years, both EQ-5D-Y and adult version of EQ-5D can be used depending on study design. All the questions in descriptive questionnaire and VAS ask to describe a health status on the day of administration or “today.”33
The EQ-5D-Y descriptive system comprises 5 dimensions: 1) mobility, 2) self-care, 3) usual activities, 4) pain or discomfort, 5) anxiety/depression. Each dimension has 3 levels: no problems, some problems and a lot of problems. The younger patient is asked to indicate their health state by ticking the box next to the most appropriate statement in each of the 5 dimensions. Even though the EQ-5D-Y descriptive system comprises the same 5 dimensions as the EQ-5D-3L, it uses more appropriate, child-friendly wording. The most relevant differences with the adult EQ-5D-3L are32:
The ‘Mobility’ dimension header includes ‘(walking about)’ to facilitate understanding.
The title of the second dimension was changed from ‘Self-Care’ to ‘Looking After Myself’.
The ‘Usual Activities’ dimension became more child relevant: the new title, ‘Doing Usual Activities,’ is followed by ‘(for example, going to school, hobbies, sports, playing, doing things with family or friends).’
For the fifth dimension, ‘Anxiety/Depression’ was replaced with ‘Feeling Worried, Sad or Unhappy.’
The wording of the items representing the highest level of severity were changed in all dimensions, from ‘confined to bed’ to ‘a lot of problems walking about,’ in the first dimension, and from ‘being unable to’ to ‘having a lot of problems’ (with washing or dressing myself, or doing usual activities) in the second and third dimensions. In the pain/discomfort dimension, the upper (worst) level was changed from ‘I have extreme pain or discomfort’ in the adult 3L version to ‘I have a lot of pain or discomfort’ in the Y version; in the final dimension, the upper level was changed from ‘I am extremely anxious or depressed’ to ‘I am very worried, sad or unhappy.’
The wording of the first response level in the Looking after Myself dimension was also changed from ‘I have no problems with self-care’ to ‘I have no problems washing or dressing myself.’
Each dimension results in a 1-digit number and the digits for the 5 dimensions can be combined to form a 5-digit score that describes the younger patient’s health state. Lower number in 5-digit score indicates better HRQoL. A summary index value can be obtained based on societal preference weights for the health state. The weights or ‘utilities’ are often used to compute QALYs for health economic analyses. Health state index scores generally range from < 0 (negative values represent a health state worse than dead; 0 = dead) to 1 (the value of full health), with higher scores indicating higher health utility. The health state preferences often represent national or regional values and can therefore differ between countries/regions. The second part of the questionnaire consists of a VAS on which the respondent rates their perceived health from 0 (the worst imaginable health) to 100 (the best imaginable health).33 The EQ VAS records the younger patient’s self-rated health and can be used as a quantitative measure of health outcome that reflects the younger patient’s own judgment.32 Higher scores on index and VAS represent better HRQoL.
So far, EQ-5D-Y utility index value sets for children have been published for Slovenian and Spanish population. Similar to EQ-5D-3L, ceiling effect (60.3%), but not floor effect (0%) has been reported with EQ-5D-Y.49
Validity
Mayoral et al.49 assessed measurement properties of EQ-5D-Y administered through a smartphone app in 119 children with asthma (81 self-responded and 38 through proxy response) in Spain between 2018 and 2020. Children aged 8 – 11 years completed self-response version. For those under the age of 8 years, their parents filled out the proxy response version of EQ-5D-Y.
For convergent validity, a hypothesis for a moderate correlation between the Patient-Reported Outcomes Measurement Information System-Pediatric Asthma Impact (PROMIS-PAIS) and the EQ-5D-Y utility index (except for certain questions where a weak correlation is expected) was made considering their asthma-specific and generic aspects, respectively. For divergent validity, a priori hypothesis was a weak correlation between the EQ-5D-Y utility index and the ACQ (except for certain questions that were expected to be moderately correlated), since they differ on the construct being measured (HRQoL and disease control, respectively). Multitrait–multimethod matrix method confirmed the associations for the EQ-5D-Y utility index with the PROMIS-PAIS to be moderate47 (Spearman r = 0.38) and with ACQ to be weak47 (r = 0.28). With the EQ VAS, the correlation coefficients with both PROIMS-PAS and ACQ were weak47 (r = 0.16 and 0.26, respectively).
Another research group tested convergent validity of EQ-5D-Y with PAQLQ in Swedish children (aged 8 – 16 years) with asthma (n = 53 – 90).50 This group hypothesized that most of the domains of 2 questionnaires would be correlated since they do not capture the same aspects of HRQoL as they are constructed differently. The absolute Spearman’s rank correlations between 5 domains of EQ-5D-Y and 3 subdomain scores of PAQLQ ranged from 0.015 (weak) to 0.583 (strong).47 The highest correlations were found in ‘doing usual activities’ and ‘having pain or discomfort’ domains, whereas the lowest correlation was found in ‘looking after myself’ domain of EQ-5D-Y. Also, in general, EQ VAS was moderately to strongly47 correlated with symptoms (r = 0.435), activity limitations (r = 0.567), emotional functions (r = 0.411), and total score (r = 0.517) of PAQLQ. Patients who reported no problems on the EQ-5D-Y consistently reported fewer problems on the PAQLQ.50
Based on the known-groups approach, a priori hypothesis was made by Mayoral, et al.49 that patients with worse control asthma (according to ACQ: well-controlled, immediate, and not well controlled), asthmatic exacerbations last 6 months, higher frequency of SABA inhaler use during the previous 4 weeks, and second-hand smoke exposure would have worse HRQoL. Statistically significant differences were found in the EQ-5D-Y utility index and EQ VAS between groups defined by asthma control, reliever inhalers use, and second-hand smoke exposure, but not with asthmatic exacerbations (which could be attributed to attrition bias effect). Of note, EQ-5D-Y utility index and second-hand smoke exposure were moderately correlated (effect size [ES] = -0.45 [95% CI = -1.01 to 0.1]), but not statistically significant (P = 0.35). The absolute ES coefficients (mean change over SD of change) ranged from 0.38 to 0.75 indicating moderate to strong relationship between the 3 known groups and EQ-5D-Y utility index and EQ VAS.
Reliability
In the same study,49 Mayoral et al. showed test-retest reproducibility of EQ-5D-Y in pediatric patients with asthma who remained stable (n = 19 – 30) and completed the questionnaire twice 6 months apart. When subsample of patients was classified to be stable based on EQ VAS, the ICC was 0.81 with EQ-5D-Y utility index. When the stable patients were classified according to the ACQ, the ICC was 0.79 with EQ-5D-Y utility index and 0.70 with EQ VAS. Based on high ICC values obtained, it can be concluded that EQ-5D-Y reliably reproduces results over time.
Responsiveness to Change
Regarding responsiveness, Mayoral, et al.49 hypothesized that the EQ-5D-Y would be able to detect change over time, though with a lower sensitivity than the asthma-specific instrument PROMIS-PAIS. At 10 months follow-up, responsiveness of EQ-5D-Y utility index for worsening subsamples (n = 6 – 17) presented a degree of change (ES = 0.68 based on EQ VAS and ES = 0.78 based on ACQ), though without statistical significance (P = 0.12 and 0.50, respectively). With respect to EQ VAS, the ES was 1.15 in worsening subsample as measured by ACQ, however, the statistical significance was not detected (P = 0.67), either. In contrast, the PROMIS-PAIS demonstrated better responsiveness in worsening subsample of patients over the same period when patients were evaluated by EQ VAS (ES = 1.08, P = 0.07) or ACQ (ES = 1.28, P = 0.82), though still not be able to detect statistically significant differences.
Minimal Important Difference
The MID for EQ-5D-Y in pediatric patients with asthma has not been estimated based the literature search results.
Pharmacoeconomic Review
Abbreviations
ACQ
Asthma Control Questionnaire
BIA
budget impact analysis
ED
emergency department
EQ-5D-Y
EQ-5D-Youth
FeNO
fractional exhaled nitric oxide
HRQoL
health-related quality of life
ICER
incremental cost-effectiveness ratio
ICS
inhaled corticosteroid
ITT
intention to treat
LABA
long-acting beta agonist
LAMA
long-acting muscarinic antagonist
LTRA
leukotriene receptor antagonist
OCS
oral corticosteroid
QALY
quality-adjusted life-year
SABA
short-acting beta agonist
SCS
systemic corticosteroid
WTP
willingness to pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Dupilumab (Dupixent), solution for subcutaneous injection (200 mg per 1.14 mL prefilled syringe [175 mg/mL]; 300 mg per 2 mL prefilled syringe [150 mg/mL]) |
Submitted price | Dupilumab 200 mg, 300 mg: $978.70 per prefilled syringe |
Indication | Add-on maintenance treatment in patients aged ≥ 6 years with severe asthma with a type 2 or eosinophilic phenotype or oral corticosteroid-dependent asthma |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | March 25, 2022 |
Reimbursement request | As an add-on maintenance treatment in patients aged 6 to < 12 years with severe asthma with a type 2 or eosinophilic phenotype characterized by:
|
Sponsor | Sanofi-Aventis Canada Inc. |
Submission history | Previously reviewed: Yes Indication: Asthma Recommendation date: June 8, 2021 Recommendation: Reimburse with clinical criteria and/or conditions Indication: Atopic dermatitis Recommendation date: April 22, 2020 Recommendation: Reimburse with clinical criteria and/or conditions |
EOS = eosinophils; FeNO = fractional exhaled nitric oxide; ICS = inhaled corticosteroid; NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov model |
Target population | Patients aged 6 to < 12 years with severe asthma with a type 2 or eosinophilic phenotype, characterized by symptoms that are not controlled despite optimal treatment, EOS ≥ 150 cells/µL or FeNO ≥ 20 parts per billion or allergy-driven asthma, and uncontrolled asthma having at least 1 severe exacerbation in the past 12 months |
Treatment | Dupilumab plus background therapy |
Comparator | Background therapy alone (consisting of ICS, ICS-LABA, LABA, LTRA, LAMA, theophylline, SABA) |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs, number of exacerbations |
Time horizon | Lifetime (up to patient age of 100 years) |
Key data source | VOYAGE trial, LIBERTY ASTHMA EXCURSION trial |
Submitted results | ICER = $407,473 per QALY (incremental costs: $231,319; incremental QALYs: 0.57) |
Key limitations |
|
CADTH reanalysis results |
|
ACQ-5 = 5-item Asthma Control Questionnaire; EOS = eosinophils; FeNO = fractional exhaled nitic oxide; GINA = Global Initiative for Asthma; ICER = incremental cost-effectiveness ratio; ICS = inhaled corticosteroid; LABA = long-acting beta agonist; LAMA = long-acting muscarinic antagonist; LTRA = leukotriene receptor antagonist; LY = life-year; QALY = quality-adjusted life-year, SABA = short-acting beta agonist.
Conclusions
Over the trial period of 52 weeks, dupilumab reduced severe asthma exacerbations (defined as a deterioration in asthma that required the use of systemic corticosteroids (SCSs) for at least 3 days or resulted in hospitalizations or emergency department (ED) visits requiring SCSs) compared to background therapy alone; the reduction in severe asthma exacerbations associated with dupilumab was found to be statistically significant and clinically meaningful, although there were few severe exacerbations associated with hospitalizations and small numerical differences between treatments for this outcome. Additionally, the effects of dupilumab on health-related quality of life (HRQoL) are uncertain. Furthermore, the durability of treatment effect is also associated with uncertainty given the lack of long-term efficacy data available to support sustained clinical benefit beyond the trial period of 52 weeks. The comparative effects of dupilumab relative to other biologic treatments for severe asthma are highly uncertain owing to a lack of direct comparative evidence and limitations within the sponsor’s indirect treatment comparisons. Notably, while other biologics are indicated for this age group, none are reimbursed by public drug plans in this population.
CADTH undertook reanalyses to address limitations in the sponsor’s economic submission. CADTH was unable to address the lack of head-to-head comparative clinical data and uncertainty regarding long-term clinical effectiveness.
The CADTH reanalysis findings are in line with the sponsor’s base case, in which dupilumab plus background therapy is not cost-effective at a willingness-to-pay (WTP) threshold of $50,000 per quality-adjusted life-year (QALY). In the CADTH base case, dupilumab plus background therapy was more effective and more costly than background therapy alone (incremental costs = $209,655; incremental QALYs = 0.07). Dupilumab is not cost-effective compared to background therapy at a WTP threshold of $50,000 (the incremental cost-effectiveness ratio [ICER] was $2,999,591 per QALY). The key driver of the ICER is the cost of dupilumab acquisition, and the amount of quality of life benefit attributed to the differences in severe exacerbations. A price reduction of at least 98% would be required for it to be considered the optimal treatment at a WTP threshold of $50,000 per QALY. This price reduction is likely conservative given the cost-effectiveness is reliant on patients maintaining long-term treatment benefit.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
Patient input from Asthma Canada and the Lung Health Foundation collected through patient surveys indicated that goals of asthma therapy included improved lung function, symptom management, improved energy, reduced exacerbations, reduced reliance on oral corticosteroids (OCSs), and improved quality of life. Patients who provided input had experience with short-acting beta agonists, ICSs, leukotriene receptor antagonists (LTRAs), ICS-long-acting beta agonist (LABA) combination inhalers, ICS-LABA-long-acting muscarinic antagonist (LAMA) combination inhalers, as well as OCSs. Current treatments were described as proving some relief from cough, shortness of breath, and reduced energy, as well as improved ability to exercise and participate in daily activities. The side effects reported from the medications were heart palpitations, poor sleep, voice hoarseness, and inability to fight infection. Side effects were reported to be particularly common with OCSs, including obesity, diabetes, osteoporosis, glaucoma, cataracts, hypertension, and adrenal suppression, as well as psychological side effects (e.g., depression, anxiety). The patient groups reported a desire for treatments that would reduce the number of exacerbations, as well as eliminate or reduce OCS use given their impact on a child’s growth. Patient feedback from those with dupilumab experience indicated that improvement was observed, although OCS use was not reduced in some patients.
Clinician input was received from the Canadian Thoracic Society. Clinician feedback indicated that children with severe asthma are those who require treatment with daily high-dose ICSs and a second controller medication, or who remain uncontrolled despite that treatment. Treatment options for children with severe asthma are limited to injectable biologic medications such as omalizumab, mepolizumab, and dupilumab. Clinicians indicated that treatment goals include well-controlled asthma, good activity tolerance, no-to-minimal severe exacerbations, normal lung function, improved quality of life, and minimal side effects. Notably, children are particularly susceptible to the side effects of high-dose ICS and OCS use, which is associated with irreversible growth suppression and life-threatening adrenal suppression. Clinicians stated that dupilumab is an important therapeutic option for children with severe asthma and atopic dermatitis, as well as for children who have had severe adverse reactions with mepolizumab.
Drug plan input received for this review noted that there is limited access to biologic therapy for severe asthma for the pediatric population, and other biologic treatments for use in this population under review (e.g., mepolizumab and omalizumab) are indicated for different asthma phenotypes. Drug plans noted that these other biologic treatments have not been reviewed by CADTH in the population in which dupilumab is being reviewed. Drug plans also expressed concern over the potential for differences in initiation and discontinuation criteria between patients aged 6 years to 11 years, and patients aged 12 and older. The plans also noted that negotiations for dupilumab for patients aged 12 years and older concluded without agreement at the pan-Canadian Pharmaceutical Alliance in June 2022. Other biologics (i.e., mepolizumab, benralizumab, and omalizumab) have had negotiations in the older asthma population concluded with agreement.
Several of these concerns were addressed in the sponsor’s model.
Clinical effectiveness was based on the rate of asthma exacerbations, with those who experienced a severe exacerbation assumed to have lower HRQoL for the duration of the exacerbation.
Adverse events were incorporated for OCSs.
CADTH was unable to address the following concerns raised from stakeholder input.
Lack of comparative evidence between dupilumab and other currently available biologic treatments for severe asthma in the pediatric population.
Improvements in lung function.
Adverse events related to dupilumab or background therapy.
Economic Review
The current review is for dupilumab (Dupixent) as add-on maintenance treatment in patients aged 6 years to younger than 12 years with severe asthma with a type 2 or eosinophilic phenotype.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis of dupilumab plus background therapy compared with background therapy alone. The model population comprised of patients aged 6 years to younger than 12 years with severe asthma with a type 2 or eosinophilic phenotype1 characterized by the following.
Symptoms that are not controlled despite optimal treatment, defined by the daily use of a medium or high-dose ICS plus 1 controller medication or high-dose ICS alone.
Eosinophils of 150 cells/µL or greater or fractional exhaled nitric oxide (FeNO) of 20 parts per billion or greater or allergy-driven asthma.
Uncontrolled asthma having at least 1 severe exacerbation, defined by having experienced 1 or more hospitalization or ED visit or treatment with an SCS (oral or parenteral) in the past 12 months.
The modelled population is consistent with the reimbursement request. The composition of background therapy reflected a basket of treatments including ICSs, LABAs, ICS-LABA combination inhalers, LAMAs, LTRAs, theophylline, and short-acting beta agonists. The cost-effectiveness of dupilumab relative to other monoclonal antibodies (mepolizumab and omalizumab) was assessed in scenario analyses.
Two strengths of dupilumab are available (200 mg/1.14 mL [175 mg/mL] and 300 mg/2 mL [150 mg/mL]) in prefilled syringes for self-administration.2 The recommended dosage for dupilumab is 200 mg every other week or 300 mg every 4 weeks for those weighing 30 kg to less than 60 kg and 200 mg every other week for those weighing 60 kg or more.2 The sponsor assumes that all patients weighing 15 kg to less than 30 kg will receive 300 mg every 4 weeks; 30% of those weighing 30 kg to less than 60 kg will receive 300 mg every 4 weeks and the remaining 70% will receive 200 mg every 2 weeks; and those weighing 60 kg or more will receive 200 mg every 2 weeks. The annual cost for those receiving dupilumab 200 mg every 2 weeks is $25,446 based on a unit cost of $978.70 per syringe. The annual cost for those receiving dupilumab 300 mg every 4 weeks is $12,723. The annual cost of background therapy was calculated by the sponsor to be $529 per patient.
The clinical outcomes were QALYs, life-years, and asthma exacerbations (moderate, severe). The sponsor adopted a lifetime horizon (defined by the sponsor as 100 years minus the starting age of the cohort) using 4-week cycles and undertook the analysis from the perspective of the publicly funded health care payer. Costs and clinical outcomes were discounted at a rate of 1.5% per year.
Model Structure
The sponsor submitted 2 Markov models (“5 substate model,” “4 substate model”) (Appendix 3).1 In the sponsor’s base case, the 5 substate model was used as it was considered more appropriate since it captures the impact of treatment on symptom control in addition to the impact of exacerbation reduction. The 5-substate model includes 2 asthma control-based states (“uncontrolled asthma,” “controlled asthma”), as well as 2 exacerbation states (“moderate exacerbations,” “severe exacerbations”). The asthma control health states were defined based on Asthma Control Questionnaire (ACQ) scores (“controlled asthma” = ACQ < 1.5 and no moderate or severe exacerbations; “uncontrolled asthma” = ACQ score3 1.5 and no moderate or severe exacerbation). The 4-substate model comprises health states related to “no exacerbations” (i.e., no moderate or severe exacerbations), “moderate exacerbations,” and “severe exacerbations” with no further breakdown by asthma control. Both models included a “death” state. In both models, patients in the severe exacerbation state were at risk of asthma-related mortality, while patients in all states were at risk of all-cause mortality.
Patients entered the model either “on add-on treatment plus background therapy” (dupilumab plus background therapy) or “on background therapy only.” Over time, patients who start on dupilumab would discontinue and move to background therapy alone. In the 5-substate model, patients start in the “uncontrolled asthma” substate. In the 4-substate model, they start in the “no exacerbation substate.” Movement between states was defined by a set of transition probabilities that varied depending on what treatment the patient was receiving and over time (< 12 weeks, 12 to 52 weeks, > 52 weeks).1
Model Inputs
The baseline patient characteristics were aligned with the VOYAGE trial (mean age = 9.0 years; 34.6% female; mean number of severe exacerbations in the past 12 months = 2.50).3 The VOYAGE trial was a phase III, multicentre, randomized, placebo-controlled trial that compared 2 doses of dupilumab (200 mg every 2 weeks or 100 mg every 2 weeks) to placebo that enrolled participants (6 to < 12 years) with severe asthma with a type 2 or eosinophilic phenotype.3
Clinical efficacy (i.e., probability of transition between health states) was based on the 52-week period of the VOYAGE trial. For patients that reach 12 years of age, clinical efficacy was then based on the 52-week period of the LIBERTY ASTHMA QUEST (QUEST) trial which has previously been reviewed by CADTH.4 Transition between health states was based on a count of patients in each health state in the VOYAGE study every 4 weeks, along with the frequency of transition to other health states in the model.
Patients were assumed to remain on treatment for the first 52 weeks in the model, at which time treatment response was assessed. Treatment response was defined as an improved exacerbation risk (reduced annualized rate of severe asthma exacerbation events > 50%). The proportion of responders was based on the VOYAGE trial. Treatment responders were assumed to continue to receive dupilumab, while nonresponders were assumed to continue on background therapy alone. Treatment effect was assumed to be maintained over the model time horizon. Long-term discontinuation was based on the VOYAGE trial and was assumed to occur at a constant rate regardless of health state.
Moderate exacerbations in the sponsor’s model were defined as per the VOYAGE and QUEST trials (at least 1 of the following: ≥ 6 additional reliever puffs of salbutamol/albuterol or levosalbutamol/levalbuterol in a 24-hour period on 2 consecutive days; ≥ 20% decrease in prebronchodilator forced expiratory volume in 1 second compared with baseline; increase in ICS dose ≥ 4 times than the dose at visit 2; decrease in a.m. or p.m. peak flow of ≥ 30% on 2 consecutive days).1 Severe exacerbations were defined as the use of SCSs for 3 days or more, admission to hospital, or an ED visit because of asthma, requiring SCSs. The proportion of exacerbations managed by office visits, ED visits, or admission to hospitalization varied by severe exacerbations (office visits = 56.38%; ED visits = 16.78%; hospitalization = 26.85%) or moderate exacerbations (office visits = 100%) based on a sponsor-submitted clinical practice research datalink study.1 The risk of asthma-related mortality among those with a severe exacerbation varied by age and whether the exacerbation resulted in an office visit, ED visit, or hospital admission.5-8 Annual mortality rates for other-cause death were based on general population life tables, after the exclusion of asthma-related deaths.9
Utility values were estimated for the “controlled asthma,” “uncontrolled asthma,” “moderate exacerbation,” and “severe exacerbation” health states in the 5-substate model. In the 4-substate model, the same utilities were applied to moderate and severe exacerbations, but a separate utility estimate was generated for the “no exacerbation” health state, which included all levels of asthma control. In the 4-substate model, a utility benefit was applied to patients receiving dupilumab to account for improvement in asthma control. Utilities for the asthma control health states were based on EQ-5D-Youth (EQ-5D-Y) data from the VOYAGE trial, mapped to 3-Level EQ-5D-3L.1 Disutilities for moderate and severe exacerbations were derived from EQ-5D data from Lloyd et al. and were assumed to be experienced for the duration of an exacerbation, which was dependent on the treatment received.10 Disutilities were applied for OCS-related adverse events only.
The model included drug costs, disease management costs, monitoring costs, and exacerbation-related costs to the health care system (i.e., office visits, ED visits, admission to hospital, intensive care unit stays, OCS use). Disease management costs included outpatient visits to a family physician or specialist and spirometry testing. The price of dupilumab was based on the sponsor’s submitted price,1 while the price of background therapy drugs was obtained from the Ontario Drug Benefit Formulary.11 For scenario analyses, the cost of mepolizumab and omalizumab was obtained from the Ontario Drug Benefit Exceptional Access Program.12 It was assumed that there would be no administration costs for background therapy, and dupilumab was assumed to be self-administered.1
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (5,000 iterations for the base-case and scenario analyses). The deterministic and probabilistic results differed notably. The probabilistic findings are presented below while the deterministic results are presented in Appendix 3.
Base-Case Results
The sponsor’s base-case results are shown in Table 3. The addition of dupilumab to background therapy was associated with incremental costs of $231,319 compared with background therapy alone for the lifetime horizon. The addition of dupilumab was associated with a gain of 0.57 QALYs over the same period, resulting in an ICER of $407,474 per QALY gained. Approximately 93% of the incremental QALYs in the sponsor’s base case were accrued beyond 52 weeks (per the VOYAGE trial duration). At a WTP threshold of $50,000 per QALY, dupilumab has a 0% probability of being cost-effective.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. reference ($/QALY) |
|---|---|---|---|---|---|
Background therapy alone | 275,602 | Reference | 36.51 | Reference | Reference |
Dupilumab plus background therapy | 506,921 | 231,319 | 37.08 | 0.57 | 407,474 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Note: The submitted analysis is based on the publicly available prices of the comparator treatments.
Source: Sponsor’s pharmacoeconomic submission.1
Additional results from the sponsor’s submitted economic evaluation base case are presented in Appendix 3.
Sensitivity and Scenario Analysis Results
The sponsor conducted several scenario analyses, most notably assessing exclusion of response assessment, alternate data sources (relative risk from in intention-to-treat [ITT] population for transition probabilities, post hoc analyses of the VOYAGE trial as the exacerbation setting, mortality based on general population life tables adjusted based on lung function), alternate utility values based on published study, EQ visual analogue scale to replace EQ-5D utility values, and the risk of experiencing exacerbations dependent on current health state and time since treatment initiation. Across these scenario analyses, the ICER for dupilumab compared to background therapy ranged from $270,547 to $548,147 per QALY.
The sponsor conducted several scenario analyses to explore the cost-effectiveness of dupilumab compared to other biologic treatments for severe asthma in subgroups aligned with the various reimbursement criteria; these analyses were informed by indirect treatment comparisons conducted by the sponsor. The cost-effectiveness of dupilumab relative to mepolizumab and omalizumab varied from being dominant (provide more QALYs at a lower cost) to being more costly and more effective based on the sponsor’s analysis. However, CADTH notes these results are highly uncertain as CADTH was unable to reproduce the sponsor’s results due to multiple errors within the submitted model. There is also substantial uncertainty in the available comparative clinical data.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis.
The sponsor-submitted 5-substate pharmacoeconomic model lacks face validity: The sponsor submitted 2 pharmacoeconomic models (5-substate and 4-substate, as described above), with the 5-substate model used to assess the cost-effectiveness of dupilumab among patients with a type 2 or eosinophilic phenotype. The “controlled” and “uncontrolled” asthma health states in the 5-substate model were defined based on ACQ score, with a threshold of 1.5 used to classify patients as controlled or uncontrolled (controlled: ACQ < 1.5; uncontrolled: ACQ score ≥ 1.5). This dichotomization lacks face validity, as it implies that a patient whose ACQ score improved by as little as 0.01 (i.e., from 1.50 to 1.49) would be considered to have controlled asthma and would receive the utility benefit for the controlled state (0.922) instead of that for the uncontrolled state (0.819). CADTH recognizes that an ACQ score of 1.5 is a commonly used threshold in clinical trials; however, the Global Initiative for Asthma guidelines13 consider an ACQ score of 0.75 to 1.5 to represent a “grey zone” between well-controlled and uncontrolled asthma. As noted in the clinical review, HRQoL (assessed via EQ-5D-Y) was improved from baseline in the VOYAGE trial; however, the minimum clinically important difference was not met, and the impact of dupilumab on patient HRQoL is unclear compared to placebo. Finally, a model structure based on exacerbations is more appropriate to address the decision problem, as according to clinical expert feedback, clinical benefit from dupilumab should come from reduction in exacerbations as opposed to “asthma control.”
CADTH reanalyses utilized the sponsor-provided 4-substate model, which comprised health states based on asthma exacerbations (“no exacerbations,” “moderate exacerbations,” and “severe exacerbations”) and does not include health states related to overall asthma control.
The number of hospitalizations predicted by the model is not aligned with clinical trial evidence as benefit due to reduced hospitalizations is overestimated: Both the 4-substate and 5-substate model overestimated the number of hospitalizations occurring following severe exacerbations during the clinical trial period relative to the data from the VOYAGE trial. For example, the sponsor uses external data to assume that 26.85% of severe exacerbations will result in hospitalizations in the child cohort, whereas the VOYAGE trial data demonstrated no hospitalizations occurred in those receiving background therapy alone and 5 hospitalizations (2.1%) occurred in those receiving dupilumab plus background therapy.3 Similarly in the adult cohort, the sponsor uses external data to assume that 20.31% of severe exacerbations will result in hospitalizations, which also does not align with the QUEST trial data. Over the lifetime time horizon, the sponsor’s model predicted that approximately 18 versus 21 hospitalizations would occur per patient receiving dupilumab versus background therapy alone, which was overpredicted and did not meet face validity according to clinical expert feedback. Based on the VOYAGE trial data and assuming that the estimated hospitalization rate was sustained over the patient lifetime, the predicted number of total hospitalizations would be less than 2 per patient. Furthermore, there is no evidence to suggest that dupilumab results in reduced hospitalizations based on the VOYAGE trial data. Therefore, the sponsor’s model results likely bias cost-effectiveness in favour of dupilumab.
CADTH removed the assumed benefits due to reduced hospitalizations occurring with dupilumab in the child cohort. In the adult cohort, a hospitalization benefit was maintained; however, CADTH applied ITT population data (3.66%) from the QUEST trial to more accurately capture the proportion of severe exacerbations that would result in hospitalization.
Assumption of increased mortality during severe asthma exacerbation is inappropriate: The sponsor assumed an increased risk of asthma-related death when patients had a severe exacerbation, and the risk varied by age group and treatment setting (i.e., hospital, ED, office visit). As dupilumab reduces the rate of severe exacerbations, this implies a survival benefit with dupilumab treatment, which has not been shown in clinical trials. Notably, in the VOYAGE clinical trial, no deaths were reported in the dupilumab or background therapy alone treatment arms.3 In the previous submission for adults, the CADTH clinical review noted few deaths across the included studies and no clear differences in mortality between groups within studies.4 This is despite a continued reduction in severe exacerbations in those randomized to dupilumab. No evidence is therefore reported from the clinical trials that suggest a reduction in mortality. Although dupilumab reduces exacerbations, there is no evidence that it reduces fatal exacerbations.
As noted in previous CADTH reviews and by clinical expert feedback, asthma-related mortality is rare and often linked to causes such as lack of adherence and incorrect management. This is extensively highlighted in the National Review of Asthma deaths in the UK, cited by the sponsor, which found 45% of asthma deaths occur without any medical intervention.6 Likewise, many individuals who died were misclassified as having mild or moderate asthma or were not seeing a specialist and therefore did not receive adequate care. A study by Suissa et al. using data from Saskatchewan found that asthma deaths were substantially reduced by regular use of low-dose ICSs.14 Clinical expert feedback also noted that deaths related to chronic obstructive pulmonary disease are contributors to the observed number of asthma deaths. All these factors that contribute to asthma deaths are therefore unlikely to be impacted by dupilumab use. Clinical expert feedback also indicated that the mortality rates applied in the sponsor’s model did not meet face validity; for example, the range of 2.05% to 4.54% for probability of death among adult patients being treated by an ED visit or hospitalization is not reflective of patients living in Canada. According to data from the Canadian Institute for Health Information, in 2019 to 2020 there were 60,492 ED visits related to asthma from participating facilities in Prince Edward Island, Nova Scotia, Quebec, Ontario, Manitoba, Saskatchewan, Alberta, British Columbia, and theYukon.15 Of those not admitted less than 5 people (< 0.009%) died before discharge.15 For the sponsor’s values to hold face validity, a substantial proportion of patients would have to die immediately postdischarge.
Finally, the model results lacked face validity when predicting expected deaths due to asthma. In the background therapy alone arm, the model predicted that 39% of patients would die from an exacerbation-related death, with the remainder occurring due to nonasthma-related causes. This ratio of asthma-related deaths to nonasthma-related deaths is not in line with what is seen from clinical trials or literature. According to a multinational cohort study of mortality in patients with severe asthma, deaths related to asthma accounted for 1.9% to 6.3% of deaths with the rest attributed to a nonasthma-related cause.16 Therefore, the sponsor’s model substantially overpredicts the number of asthma-related deaths occurring in patients, which does not align with what is observed in clinical practice, registry data, or the VOYAGE, LIBERTY ASTHMA EXCURSION (EXCURSION), or QUEST clinical trial data submitted by the sponsor. Therefore, if a survival benefit were to occur, it would likely be substantially smaller than what is predicted by the sponsor’s model.
The predicted survival benefit with dupilumab compared with background therapy is highly uncertain and is not supported by clinical trial data. This mortality benefit was removed in CADTH reanalysis, consistent with previous CADTH reviews.
The model structure does not adequately reflect the management of asthma in clinical practice: In the sponsor’s model, response to treatment is assessed at 52 weeks, with treatment response defined as an improved exacerbation risk (reduced annualized rate of severe asthma exacerbation events > 50%). The clinical expert consulted by CADTH for this review, as well as in prior CADTH reviews,17,18 indicated that response to biologic treatment is usually assessed in clinical practice at an earlier time point (i.e., 4 or 6 weeks initially) based on Canadian Asthma Consensus or Global Initiative for Asthma guidelines (i.e., practice in Canada). Treatment response is not typically assessed in terms of exacerbation risk, as exacerbations are a distinct clinical outcome that may be infrequent and influenced by factors other than asthma control (e.g., influenza, pneumonia). The sponsor assumed that patients with no treatment response at 52 weeks would receive background therapy alone for the remainder of the time horizon; however, it is likely that a proportion of patients who improve somewhat but not to the extent of the response criteria would likely continue to receive their current biologic treatment.
In CADTH reanalyses, treatment response assessment at 52 weeks was disabled, such that patients discontinued treatment based only on the constant long-term discontinuation rate as derived from the trial data.
Resource utilization was overestimated for moderate and severe exacerbations: The sponsor has overestimated costs associated with exacerbations leading to increased health care resource use. For moderate exacerbations, the sponsor incorporated costs associated with family physician and nurse visits, as well as spirometry and FeNO tests, estimated at approximately $54 and $725 per cycle for children and adults, respectively.1 However, clinical expert feedback indicated that moderate exacerbations generally reflect poor symptom control and would likely only require OCS use to resolve. Furthermore, the sponsor included costs for severe exacerbation related to office visits ($193 and $725 for childrenand adults, respectively), ED visits ($545 and $1,451 for children and adults, respectively), and hospitalizations ($3,764 and $5,902 for children and adults, respectively).1 These values were determined to be likely overestimated and did not meet face validity compared to clinical practice and provincial estimates. The sponsor also appeared to double count various costs across each exacerbation treatment setting. For example, costs for hospitalization included 2 spirometry and FeNO tests, 2 family physician visits, 2 nurse visits, and 1 specialist visit. Hospitalization costs are likely not expected to include these additional resource costs, as the costs for required care associated with hospitalizations are already incorporated. Overall, resource utilization costs were likely overestimated and did not meet face validity. CADTH adjusted these values in reanalysis using Ontario Physicians Schedule of Benefits and Ontario Case Costing Initiative data to better reflect clinical practice.19,20
CADTH removed costs associated with moderate exacerbations and adjusted costs associated with severe exacerbations to reflect Ontario Physicians Schedule of Benefits and Ontario Case Costing Initiative data.
Poor modelling practices were employed: The model was unnecessarily complex and lacked transparency. First, the sponsor used numerous IFERROR statements in their model. IFERROR statements lead to situations in which the parameter value is overwritten with an alternative value without alerting the user to the automatized overwriting. The systematic use of IFERROR statements makes thorough auditing of the sponsor’s model impossible, as it remains unclear whether the model is running inappropriately by overriding errors. Best programming practices are such that any errors alert the user to a specific error. Second, the model repeated the same information numerous times, making it unclear what values were being used to derive model estimates. As noted above, the results of the sponsor’s submitted model lacked face validity, which CADTH was unable to fully address owing to the complex coding to the model.
CADTH could not address limitations regarding the validity of the model and notes that the results presented should be treated with a degree of caution.
Uncertainty regarding long-term clinical effectiveness: In the sponsor’s pharmacoeconomic submission, the effects of dupilumab were considered to be consistent over the lifetime analysis horizon (up to patient lifetime of 100 years). The potential waning of treatment effect was not considered despite lack of long-term efficacy data. Furthermore, the sponsor applied an increased rate of severe exacerbations following the 52-week period of the VOYAGE clinical trial because a “lower severe exacerbation rate would be expected in the clinical trial setting as compared to the real world due to improved adherence and monitoring.”1 However, it remains unknown whether treatment effect would remain consistent. Additionally, patients who experience an increase in exacerbations would likely try different biologics once they become eligible (i.e., age out of the child cohort), meaning that differences captured at the end of trial would likely not be permanent for the rest of the patient’s life.
CADTH was unable to address this limitation owing to the structure of the sponsor’s economic model and a lack of long-term effectiveness data for dupilumab.
Comparative clinical efficacy versus other biologics is highly uncertain: There have been no head-to-head trials of dupilumab and other biologic treatments for asthma (i.e., omalizumab, mepolizumab) for the indicated population. The sponsor conducted indirect treatment comparisons to provide comparative clinical effectiveness data for scenario analyses. The CADTH clinical review raised several concerns regarding considerable heterogeneity across the included studies, lack of direct comparisons between treatments, and limited available data. As such, no robust conclusions can be drawn on the comparative clinical efficacy of dupilumab versus other currently available biologic treatments.
CADTH was unable to address this limitation owing to a lack of direct evidence and limitations with the sponsor’s indirect treatment comparison. The cost-effectiveness of dupilumab relative to other biologic treatments indicated for type 2 or eosinophilic asthma in the indicated population is unknown.
Additional limitations were identified, but were not considered to be key limitations. These limitations are outlined subsequently, and are also present in CADTH’s previous review of dupilumab for severe asthma with a type 2 or eosinophilic phenotype in the adult and adolescent population.
The cost-effectiveness of dupilumab among adolescents is uncertain: In the model, once patients are aged 12 years or older, clinical data used to model outcomes are derived from the QUEST trial. However, the QUEST trial enrolled relatively few participants aged 12 years to 17 years (5.6%).18 As noted by the clinical expert consulted by CADTH, health care utilization by those with severe asthma exacerbations may differ between adolescents and adults.
CADTH was unable to address this limitation owing to a lack of data on the effectiveness and health care resource utilization among adolescents. The cost-effectiveness of dupilumab among adolescents is thus unknown.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (Refer to Table 5).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
Health state utility values were mapped from EQ-5D-Y to EQ-5D-3L in the child cohort. | Uncertain. The sponsor mapped utility values from EQ-5D-Y (captured as part of the VOYAGE trial) to the EQ-5D-3L via a mapping function. Mapping utility values introduces uncertainty. |
Response data from the VOYAGE trial for those receiving dupilumab during weeks 0 to 12 were excluded from the derivation of transition probabilities. | Likely inappropriate. Reduction of exacerbations was greater in the data for weeks 12 to 52 from the VOYAGE trial, and the exclusion of 12 weeks of initial data may have biased results in favour of dupilumab. |
Adverse events related only to OCS treatment were included. | Reasonable. Adverse events other than those related to OCS were not considered in the sponsor’s model (no justification provided). Costs related to adverse events could have been included for completeness; however, this is unlikely to influence model results. |
The efficacy of the 300 mg q.4.w. regimen was assumed to be the same as the efficacy of the pooled 100 mg q.2.w. and 200 mg q.2.w. dupilumab arms from the VOYAGE trial. | Reasonable. The dosage used in the model (200 mg) is aligned with the application request and reimbursement request population. The 300 mg dosage is for patients with OCS-dependent asthma or for those with comorbid moderate to severe atopic dermatitis or severe chronic rhinosinusitis with nasal polyposis.2 |
EQ-5D-3L = 3-Level EQ-5D; EQ-5D-Y = EQ-5D-Youth; OCS = oral corticosteroid; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
Several limitations with the sponsor’s submission could not be adequately addressed (i.e., lack of head-to-head comparative clinical data, uncertainty regarding long-term clinical effectiveness). Further, CADTH could not fully validate the sponsor’s model owing to a lack of transparency and poor modelling practices employed. CADTH undertook a stepped reanalysis using the 4-substate model which involved: removing hospitalization benefit associated with dupilumab in the child cohort and applying QUEST hospitalization data for the adult cohort, assuming no mortality benefit associated with dupilumab, removing response assessment at 52 weeks, and adjusting resource utilization costs for moderate and severe exacerbations.
The CADTH base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts. Table 6 details each change made to derive the CADTH reanalysis, which was conducted in step-wise approach to the sponsor’s base case to highlight the impact of each change. The summary results of the CADTH reanalyses are presented in Table 7.
In the CADTH base case, dupilumab was associated with higher costs (incremental: $209,655) and higher QALYs (incremental: 0.07) compared to background therapy over a lifetime horizon (approximately 91 years). The ICER for dupilumab versus background therapy was $2,999,591 per QALY gained with a probability of being cost-effective at a WTP of $50,000 of 0%. Detailed information and disaggregated results are presented in Table 12 in Appendix 4.
Scenario Analysis Results
CADTH performed price reduction analyses based on the sponsor base-case and CADTH base-case reanalysis. Based on the CADTH base case, a price reduction of at least 98% would be required to achieve cost-effectiveness at a WTP threshold of $50,000 per QALY (Table 9).
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
1. Model choice | 5-substate model | 4-substate model |
Changes to derive the CADTH base case | ||
1. Reduction in hospitalizations | 26.85% of severe exacerbations will result in hospitalizations in the child cohort and 20.31% of severe exacerbations will result in hospitalizations in the adult cohort based on external data | Benefit due to reduction in hospitalizations attributed to dupilumab was removed in the child cohort. In the adult cohort, CADTH applied the sponsor-provided LIBERTY ASTHMA QUEST ITT population data to estimate that 3.66% of severe exacerbations would result in hospitalizations. |
2. Asthma-related mortality | Assumed a mortality benefit associated with dupilumab | Assumed no mortality benefit associated with dupilumab |
3. Response assessment | Treatment response assessed at 52 weeks | No response assessment at 52 weeks |
4. Resource utilization for moderate and severe exacerbations | The cost per cycle for moderate exacerbations was $53.69 (office visit) and the cost per cycle for severe exacerbations included $192.50 (office visit), $545.16 (ED visit), and $3,763.54 (hospitalization). For adults, the cost per cycle for moderate exacerbations was $725.38 (office visit) and for severe exacerbations was $1,451.41 (office visit), $3,184.07 (ED visit), and $5,902.14 (hospitalization). | CADTH removed the costs for moderate exacerbations for the child and adult cohort. For severe exacerbations, office visits costs were estimated to be $45.90 (OHIP SOB C006 or A006). ED visits were estimated to be $410.24 (OHIP SOB H055; Ontario Case Costing Diagnosis J4500, J4501, J4511, J4581, J4590, J4591). Hospitalization costs were estimated to be $4,453.49 (OHIP SOB A475; Ontario Case Costing; CMG Group 147). |
CADTH base case | Reanalysis 1 + 2 + 3 + 4 | |
CMG = case mix group; ED = emergency department; ITT = intention to treat; OHIP SOB = Ontario Health Insurance Plan Schedule of Benefits.
Table 6: Summary of the Stepped Analysis of the CADTH Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case (deterministic) | Background therapy | 307,315 | 33.43 | Reference |
Dupilumab plus background therapy | 486,456 | 33.92 | 366,710 | |
Sponsor’s corrected base case (4-substate model, deterministic) | Background therapy | 292,233 | 34.33 | Reference |
Dupilumab plus background therapy | 474,418 | 34.64 | 580,892 | |
CADTH reanalysis 1 (hospitalizations) | Background therapy | 243,087 | 35.60 | Reference |
Dupilumab plus background therapy | 434,717 | 35.86 | 755,947 | |
CADTH reanalysis 2 (mortality benefit) | Background therapy | 321,629 | 37.39 | Reference |
Dupilumab plus background therapy | 502,757 | 37.58 | 960,082 | |
CADTH reanalysis 3 (removal of response assessment) | Background therapy | 292,233 | 34.33 | Reference |
Dupilumab plus background therapy | 482,991 | 34.50 | 1,145,799 | |
CADTH reanalysis 4 (costs for moderate and severe exacerbations) | Background therapy | 139,422 | 34.33 | Reference |
Dupilumab plus background therapy | 338,412 | 34.64 | 634,474 | |
CADTH base case (reanalysis 1 + 2 + 3 + 4) probabilistic | Background therapy | 95,416 | 36.22 | Reference |
Dupilumab plus background therapy | 305,072 | 36.29 | 2,999,591 | |
CADTH base case (reanalysis 1 + 2 + 3 + 4) deterministic | Background therapy | 95,005 | 37.55 | Reference |
Dupilumab plus background therapy | 302,418 | 37.62 | 2,870,113 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: The CADTH reanalysis is based on publicly available prices of the comparator treatments. All presented analyses are deterministic, with the exception of the CADTH base case which is presented probabilistically.
Table 7: CADTH Price Reduction Analyses
Analysis | ICERs for dupilumab plus background therapy vs. background therapy ($/QALY) | |
|---|---|---|
Price reduction | Sponsor base case | CADTH reanalysis |
No price reduction | 366,710 | 2,870,113 |
10% | 324,037 | 2,583,115 |
20% | 281,364 | 2,296,117 |
30% | 238,691 | 2,009,118 |
40% | 196,018 | 1,722,120 |
50% | 153,345 | 1,435,122 |
60% | 110,672 | 1,148,123 |
70% | 67,999 | 861,125 |
80% | 25,326 | 574,127 |
90% | Dupilumab was dominanta | 287,128 |
98% | Dupilumab was dominanta | 57,530 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
aThis means that dupilumab in addition to background therapy was more effective and less costly than background therapy alone.
CADTH performed scenario analyses to determine the impact of alternative assumptions on the cost-effectiveness of dupilumab. CADTH assessed the cost-effectiveness of dupilumab in the child cohort subgroup of those aged 6 year to younger than 12 years; medium ICS with second controller or high ICS with second controller; and eosinophils of 300 cells/μL or greater with 2 or more prior exacerbations. The associated ICER was $1,282,219 per QALY gained. This shows that even in the most severely impacted subgroup of patients, dupilumab likely remains not cost-effective.
Issues for Consideration
Dupilumab was reviewed for add-on maintenance treatment in patients aged 12 years and older with severe asthma with a type 2 or eosinophilic phenotype or OCS-dependent asthma, for which it received recommendation to reimburse with conditions.18 However, no agreement was reached with the pan-Canadian Pharmaceutical Alliance negotiations on June 28, 2022.
There may be an indication creep with dupilumab used in pediatric patients with less severe asthma and who have comorbid atopic dermatitis. The clinical expert consulted by CADTH indicated that dupilumab may be considered in practice for patients with moderate uncontrolled asthma in the presence of these comorbidities.
The cost-effectiveness of 300 mg dupilumab is uncertain. Given the varying doses and indications of dupilumab, individual use of dupilumab may differ in clinical practice from what is modelled by the sponsor.2 The submitted costs of 200 mg and 300 mg dupilumab doses is the same; however, potential differences in effectiveness could result in a difference in cost-effectiveness.
Overall Conclusions
Over the trial period of 52 weeks, dupilumab reduced severe asthma exacerbations (defined as a deterioration in asthma that required the use of SCSs for at least 3 days or resulted in hospitalizations or ED visits requiring SCSs) compared to background therapy alone; the reduction in severe asthma exacerbations associated with dupilumab was found to be statistically significant and clinically meaningful, although there were few severe exacerbations associated with hospitalizations and small numerical differences between treatments for this outcome. Additionally, the effects of dupilumab on HRQoL are uncertain. Furthermore, the durability of treatment effect is also associated with uncertainty given the lack of long-term efficacy data available to support sustained clinical benefit beyond the trial period of 52 weeks. The comparative effects of dupilumab relative to other biologic treatments for severe asthma are highly uncertain owing to a lack of direct comparative evidence and limitations within the sponsor’s indirect treatment comparisons. Notably, while other biologics are indicated for this age group, none are reimbursed by public drug plans in this population.
CADTH undertook reanalyses to address limitations in the sponsor’s submission, including removing the hospitalization benefit associated with dupilumab in the child cohort and applying QUEST hospitalization data for the adult cohort, removing the risk of mortality with a severe exacerbation, removing response assessment at 52 weeks, and adjusting resource utilization costs for moderate and severe exacerbations. CADTH was unable to address the lack of head-to-head comparative clinical data and uncertainty regarding long-term clinical effectiveness.
The CADTH reanalysis findings are in line with the sponsor’s base case, in which dupilumab plus background therapy is not cost-effective at a WTP threshold of $50,000 per QALY. In the CADTH base case, dupilumab plus background therapy was more effective and more costly than background therapy alone (incremental costs: $209,655; incremental QALYs: 0.07). Dupilumab is not cost-effective compared to background therapy at a WTP threshold of $50,000 (ICER = $2,999,591 per QALY). A scenario analysis assessing the cost-effectiveness of dupilumab in the subgroup most impacted by severe asthma indicated that dupilumab would remain not cost-effective (ICER = $1,282,219). The key driver of the ICER is the cost of dupilumab acquisition, and the amount of quality of life benefit attributed to the differences in severe exacerbations. A price reduction of at least 98% would be required for it to be considered the optimal treatment at a WTP threshold of $50,000 per QALY. This price reduction is likely conservative given the cost-effectiveness is reliant on patients maintaining long-term treatment benefit.
Although the ICER for dupilumab compared with current standards of care for patients captured in the full Health Canada-indicated population is unknown, based on this analysis in patients aged 6 to 11 years, and the analysis in patients aged 12 years and older, dupilumab is unlikely to be cost-effective at a WTP threshold of $50,000 per QALY.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Dupixent (dupilumab), 200 mg and 300 mg solution for subcutaneous injection. Mississauga (ON): Sanofi-aventis Canada Inc; 2022 Jul 14.
2.Dupixent (dupilumab injection): solution for subcutaneous injection, 300 mg single-use syringe (300 mg/2 mL), 200 mg single-use syringe (200 mg/1.14 mL), and 100 mg single-use syringe (100 mg/0.67 mL); Dupixent (dupilumab injection): solution for subcutaneous injection, 300 mg single-use pen (300 mg/2 mL) and 200 mg single-use pen (200 mg/1.14 mL) [product monograph]. Laval (QC): Sanofi-aventis Canada Inc.; 2022 Mar 25.
3.Clinical Study Report: SAR231893-EFC14153. VOYAGE: A randomized, double-blind, placebo-controlled, parallel group study to evaluate the efficacy and safety of dupilumab in children 6 to <12 years of age with uncontrolled persistent asthma [internal sponsor's report]. Paris (FR): Sanofi Group; 2020 Dec 07.
4.Drug Reimbursement Review pharmacoeconomic report: dupilumab (Dupixent) for Type 2 or eosinophilic asthma. Ottawa (ON): CADTH; 2021: https://www.cadth.ca/sites/default/files/cdr/complete/SR0667-combined%20clinical%20and%20PE%20report.pdf. Accessed 2022 Sept 15.
5.National Institute for Health and Care Excellence. Benralizumab for treating severe eosinophilic asthma (Single technology appraisal ID1129) 2018; https://www.nice.org.uk/guidance/ta565/evidence/final-appraisal-determination-committee-papers-pdf-6715489647. Accessed 2022 Sept 15.
6.Levy ML. The national review of asthma deaths: what did we learn and what needs to change? Breathe (Sheff). 2015;11(1):14-24. PubMed
7.Watson L, Turk F, James P, Holgate ST. Factors associated with mortality after an asthma admission: a national United Kingdom database analysis. Respir Med. 2007;101(8):1659-1664. PubMed
8.Roberts NJ, Lewsey JD, Gillies M, et al. Time trends in 30 day case-fatality following hospitalisation for asthma in adults in Scotland: a retrospective cohort study from 1981 to 2009. Respir Med. 2013;107(8):1172-1177. PubMed
9.Table: 13-10-0114-01. Life expectancy and other elements of the life table, Canada, all provinces except Prince Edward Island. Ottawa (ON): Statistics Canada; 2022: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011401. 2022 Sept 15.
10.Lloyd A, Price D, Brown R. The impact of asthma exacerbations on health-related quality of life in moderate to severe asthma patients in the UK. Prim Care Respir J. 2007;16(1):22-27. PubMed
11.Ontario Ministry of H, Ontario Ministry of Long-Term C. Ontario drug benefit formulary/comparative drug index. 2022; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2022 Sept 15.
12.Exceptional Access Program (EAP). Toronto (ON): Ontario Ministry of Health; Ontario Ministry of Long-Term Care; 2022: https://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx. Accessed 2022 Sept 15.
13.Workman AD, Bleier BS. Biologic therapies versus surgical management for aspirin-exacerbated respiratory disease: A review of preliminary data, efficacy, and cost. World Journal of Otorhinolaryngology - Head and Neck Surgery. 2020;6(4):230-234. PubMed
14.Suissa S, Ernst P, Benayoun S, Baltzan M, Cai B. Low-dose inhaled corticosteroids and the prevention of death from asthma. N Engl J Med. 2000;343(5):332-336. PubMed
15.Canadian Institute for Health Information. Asthma emergency department visits: volume and median length of stay. 2021; https://www.cihi.ca/en/indicators/asthma-emergency-department-visits-volume-and-median-length-of-stay. Accessed 2022 Sept 15.
16.Engelkes M, de Ridder MA, Svensson E, et al. Multinational cohort study of mortality in patients with asthma and severe asthma. Respir Med. 2020;165:105919. PubMed
17.CADTH Common Drug Review pharmacoeconomic review report: benralizumab (Fasrena) for an add-on maintenance treatment of adult patients with severe eosinophilic asthma. Ottawa (ON): CADTH; 2018 Aug: https://cadth.ca/sites/default/files/cdr/pharmacoeconomic/SR0561_Fasenra_PE_Report.pdf. Accessed 2022 Sept 15.
18.CADTH reimbursement review: dupilumab (Dupixent) for type 2 eosinophilic asthma. Can J Health Technol. 2021;1(8). https://www.cadth.ca/sites/default/files/cdr/complete/SR0667-combined%20clinical%20and%20PE%20report.pdf. Accessed 2022 Sept 15.
19.Schedule of benefits for physician services under the Health Insurance Act: effective October 1, 2021. Toronto (ON): Ontario Ministry of Health; 2022: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf. Accessed 2022 Sept 15.
20.Ontario Case Costing Initiative (OCCI). Toronto (ON): Ontario Health and Long-Term Care; 2017: https://data.ontario.ca/dataset/ontario-case-costing-initiative-occi. Accessed 2022 Sept 15.
21.DeltaPA. [Ottawa (ON)]: IQVIA; 2022: https://www.iqvia.com/. Accessed 2022 Sept 15.
22.Saskatchewan Drug Plan: search formulary. 2022; https://formulary.drugplan.ehealthsask.ca/SearchFormulary. Accessed 2022 Sept 15.
23.Alberta Health Care Insurance Plan: medical price list as of 31 March 2020. Edmonton (AB): Government of Alberta; 2020: https://open.alberta.ca/dataset/30add047-29c2-4fc7-83b5-a8ab78605cdd/resource/48d0dd0b-bb33-478d-a85c-85a86d4555ad/download/health-somb-medical-price-list-2020-03.pdf. Accessed 2022 Sept 15.
24.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Dupixent (dupilumab), 200 mg and 300 mg solution for subcutaneous injection. Mississauga (ON): Sanofi-aventis Canada Inc; 2022 Jul 14.
25.Table: 17-10-0057-01 Projected population, by projection scenario, age and sex, as of July 1 (x 1,000). Ottawa (ON: Statistics Canada; 2022: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710005701. Accessed 2022 Sept 15.
26.Data Blog: Asthma in Canada. 2018; https://health-infobase.canada.ca/datalab/asthma-blog.html. Accessed 2022 September 15.
27.Annesi-Maesano I, Sterlin C, Caillaud D, et al. Factors related to under-diagnosis and under-treatment of childhood asthma in metropolitan France. Multidiscip Respir Med. 2012;7(1):24. PubMed
28.Europe TRf. Truven Claims Analyses. 2020.
29.Evaluation IoHMa. Global Disease Burden: SNY/REGN analyses. In: Group SGaRGEW, ed. Epidemiology (VERSION 02) DUPILUMAB INDICATIONS GLOBAL HEVA/HEOR20202020.
30.Statistics Canada. Table: 13-10-0096-01. Health characteristics, annual estimates. 2022; https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310009601. Accessed 2022 Sept 15.
31.Bosonea AM, Sharpe H, Wang T, et al. Developments in asthma incidence and prevalence in Alberta between 1995 and 2015. Allergy Asthma Clin Immunol. 2020;16:87. PubMed
32.Public Health Agency of Canada. Asthma and chronic obstructive pulmonary disease (COPD) in Canada, 2018. Report from the Canadian Chronic Disease Surveillance System 2018; https://www.canada.ca/en/public-health/services/publications/diseases-conditions/asthma-chronic-obstructive-pulmonary-disease-canada-2018.html#a1.2.2. Accessed 2022 Sept 15.
Appendix 1: Cost Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CADTH Cost Comparison Table of Biologics for Severe Eosinophilic Asthma
Treatment | Strength | Form | Price | Recommended dosage | Daily cost ($) | Annual cost ($) |
|---|---|---|---|---|---|---|
Dupilumab (Dupixent) | 200 mg | Prefilled syringe for SC injection | 978.7000a | ≥15 kg to < 30 kg 100 mg every other week or 300 mg every 4 weeks ≥30 kg to < 60 kg 200 mg every other week or 300 mg every 4 weeks ≥60 kg 200 mg every other week | 34.86 34.86 or 69.72 69.72 | 12,723 12,723 or 25,446 25,446 |
300 mg | ||||||
Biologics | ||||||
Mepolizumab (Nucala) | 100 mg/mL | Vial of powder for SC injection | 2,100.6100 | 40 mg every 4 weeks | 74.82 | 27,308 |
Omalizumab (Xolair) | 150 mg | Vial of powder for SC injection | 652.9800b | 75 to 375 mg every 2 or 4 weeksc | 23.26 to 139.54 | 8,489 to 50,932 |
75 mg | Prefilled syringe for SC injection | 281.2400 | 10.02 to 100.17 | 3,656 to 36,561 | ||
150 mg | 641.6000 | 22.85 to 137.11 | 8,341 to 50,045 | |||
SC = subcutaneous.
Note: All prices are from the Ontario Exceptional Access Program Formulary (accessed August 2022),12 unless otherwise indicated, and do not include dispensing fees. Drug wastage was included.
aBased on sponsor’s submission.1
bPrice obtained from Delta PA Database.21
cDosing is dependent upon body weight and baseline IgE and can range from 75 mg to 300 mg when dosed every 4 weeks, and 225 mg to 375 mg when dosed every 2 weeks.
Table 9: CADTH Cost Comparison Table of Other Medications for Asthma
Drug/ Comparator | Strength | Dosage Form | Price ($) | Recommended Dosage | Daily Drug Cost ($) | Annual Drug Cost ($) | |
|---|---|---|---|---|---|---|---|
Inhaled corticosteroids | |||||||
Beclomethasone dipropionate (QVAR) | 50 mcg 100 mcg | MDI (200 doses) | 37.1200 74.0200 | 50 to 400 mcg twice daily | 0.37 to 2.96 | 135 to 1,081 | |
Budesonide (Pulmicort Turbuhaler) | 100 mcg 200 mcg 400 mcg | MDPI (200 doses) | 33.9600 69.4600 101.3900 | 200 to 400 mcg twice daily | 0.69 to 1.01 | 254 to 370 | |
Ciclesonide (Alvesco) | 100 mcg 200 mcg | MDI (120 doses) | 47.8560 79.1880 | 100 to 800 mcg twice daily | 0.80 to 2.64 | 291 to 963 | |
Fluticasone furoate (Arnuity Ellipta) | 100 mcg 200 mcg | MDPI (30 doses) | 42.7700 85.5500 | 100 or 200 mcg once daily | 1.43 to 2.85 | 520 to 1,041 | |
Fluticasone propionate (Flovent Diskus) | 100 mcg 250 mcg 500 mcg | MDPI (60 doses) | 26.2900a 49.0200 76.2500 | 100 to 500 mcg twice daily | 0.88 to 2.54 | 320 to 927 | |
Fluticasone propionate (Flovent HFA) | 50 mcg 125 mcg 250 mcg | MDI (120 doses) | 28.4200 35.1150 45.0200 | 100 to 500 mcg twice daily | 0.95 to 1.50 | 346 to 548 | |
Mometasone furoate (Asmanex Twisthaler) | 100 mcg 200 mcg 400 mcg | MDPI (60 doses) | 76.1940b 40.4100 80.7900 | 200 or 400 mcg once daily | 0.67 to 1.35 | 246 to 491 | |
ICS/LABA Combinations | |||||||
Indacaterol acetate/mometasone furoate (Atectura Breezhaler) | 150/80 mcg | Inhalation powder hard capsules (30 doses) | 58.0800 | One capsule for inhalation daily | 1.94 | 707 | |
Budesonide/ formoterol fumarate dihydrate (Symbicort Turbuhaler) | 100/6 mcg 200/6 mcg | MDPI (120 dose pack) | 69.5400 90.3600 | Low | 100/6 mcg, 2 inhalations twice daily | 2.32 | 846 |
Med | 200/6 mcg, 2 to 4 inhalations daily | 1.51 to 3.01 | 550 to 1,099 | ||||
High | 200/6 mcg, > 4 inhalations dailyc | >3.01 | >1,099 | ||||
Fluticasone propionate/ salmeterol (Advair) | 125/25 mcg 250/25 mcg | MDI (120 pack) | 114.4100 162.4200 | Low | 125/25 mcg, 1 inhalation twice daily | 1.91 | 696 |
Med | 125/25 mcg, 2 inhalations twice daily | 3.81 | 1,392 | ||||
High | 250/25 mcg, 2 inhalations twice daily | 5.41 | 1,976 | ||||
Fluticasone propionate/ salmeterol (Advair Diskus, generic) | 100/50 mcg 250/50 mcg 500/50 mcg | MDPI (60 doses) | 42.4050 50.7600 72.0600 | Low | 100/50 mcg, 1 inhalation twice daily | 1.41 | 516 |
Med | 250/50 mcg, 1 inhalation twice daily | 1.69 | 618 | ||||
High | 500/50 mcg, 1 inhalation twice daily | 2.40 | 877 | ||||
Fluticasone furoate/vilanterol (Breo Ellipta) | 100/25 mcg 200/25 mcg | MDPI (30 doses) | 93.0500 144.7400 | Low | NA | NA | NA |
Med | 100/25 mcg, 1 inhalation once daily | 3.10 | 1,132 | ||||
High | 200/25 mcg, 1 inhalation once daily | 4.82 | 1,761 | ||||
Mometasone furoate/formoterol fumarate dihydrate (Zenhale) | 100/5 mcg 200/5 mcg | MDI (120 doses) | 107.6400 130.4300 | Low | NA | NA | NA |
Med | 100/5 mcg, 2 inhalations twice daily | 3.56 | 1,310 | ||||
High | 200/5 mcg, 2 inhalations twice daily | 4.35 | 1,587 | ||||
Long-Acting Beta2-Adrenergic Agonists (LABA) | |||||||
Salmeterol xinafoate (Serevent Diskhaler) | 50 mcg | Dry powder inhaler (60 doses) | 67.0000 | 50 mcg twice daily | 2.68 | 978 | |
Formoterol fumarate (Foradil) | 12 mcg | Dry powder capsules for inhalation (60 doses) | 55.7700 | 12 mcg twice daily | 1.86 | 679 | |
Formoterol fumarate dihydrate (Oxeze Turbuhaler) | 6 mcg 12 mcg | MDPI (60 doses) | 34.0200 45.2900 | 6 to 12 mcg twice daily | 1.13 to 1.51 | 414 to 551 | |
ICS/LABA/LAMA Combinations | |||||||
Indacaterol/ glycopyrronium/ mometasone furoate (Enerzair Breezhaler) | 150/50/160 mcg | Inhalation powder hard capsules (30 doses) | 102.83 | One capsule inhaled daily | 3.43 | 1,251 | |
Leukotriene receptor antagonists (LTRA) | |||||||
Montelukast (Singulair, generics) | 4 mg 5 mg 10 mg | Chew tablet Chew tablet Tablet | 0.2758 0.3082a 0.4231a | Age 6-14: 5 mg daily | 0.31 to 0.42 | 112 to 154 | |
Long-acting Muscarinic antagonists (LAMA) | |||||||
Tiotropium (Spiriva Respimat) | 2.5 mcg | Solution for inhalation (60 doses) | 54.8580 | 2 inhalations once daily | 1.83 | 667 | |
Oral corticosteroids | |||||||
Prednisone (generic) | 1 mg 5 mg 50 mg | Tablet | 0.1214a 0.0220 0.1735 | 5 to 60 mg daily | 0.02 to 0.17 | 8 to 85 | |
Note: All prices are from the Ontario Drug Benefit Formulary (accessed August 2022),11 unless otherwise indicated, and do not include dispensing fees. Drug wastage was included.
aPrice obtained from Saskatchewan Online Formulary Database.22
bPrice obtained from Alberta Online Formulary Database.23
cBased on clinical expert feedback.
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | Yes | No comment. |
Model has been adequately programmed and has sufficient face validity | No | Poor modelling practices were employed. Refer to the CADTH appraisal section for further details. |
Model structure is adequate for decision problem | No | The sponsor’s 4- and 5-substate models lacked face validity compared with the VOYAGE, EXCURSION, and QUEST clinical trial results. Owing to the poor modelling practices, CADTH was unable to fully validate the sponsor’s model. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | No | The model was unnecessarily complex, and the report lacked transparency. The utility mapping function was not well described. Inconsistencies were noted between the scenario analyses described in the submitted report and those seen in the model. Programming errors were observed in the scenario analyses sheets. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
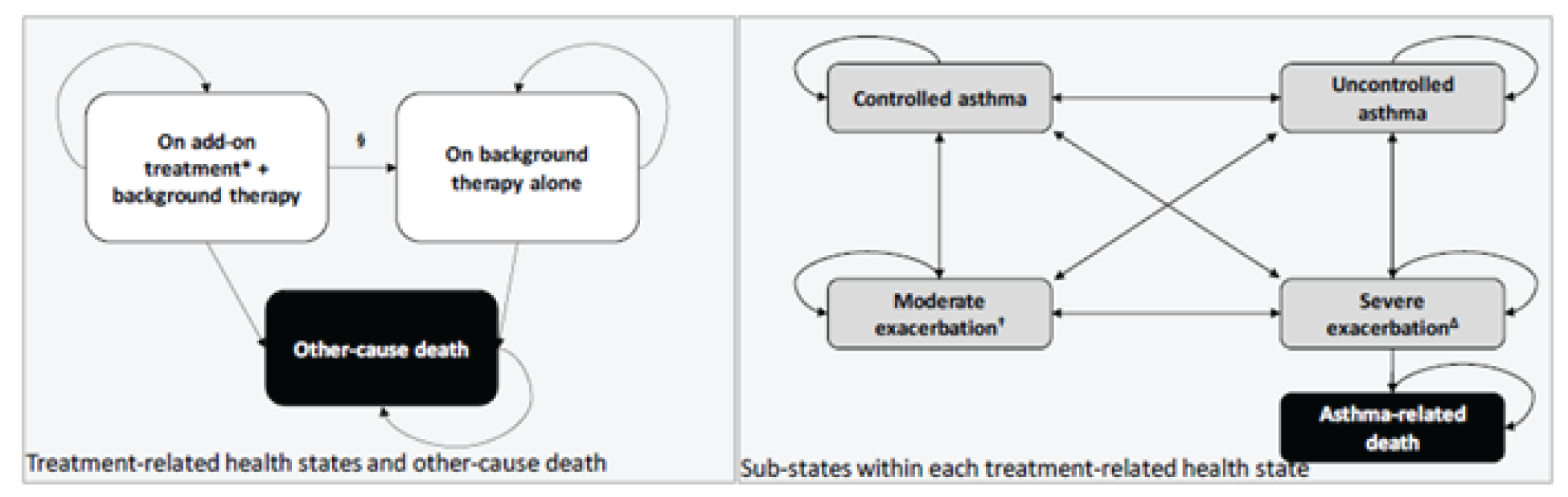
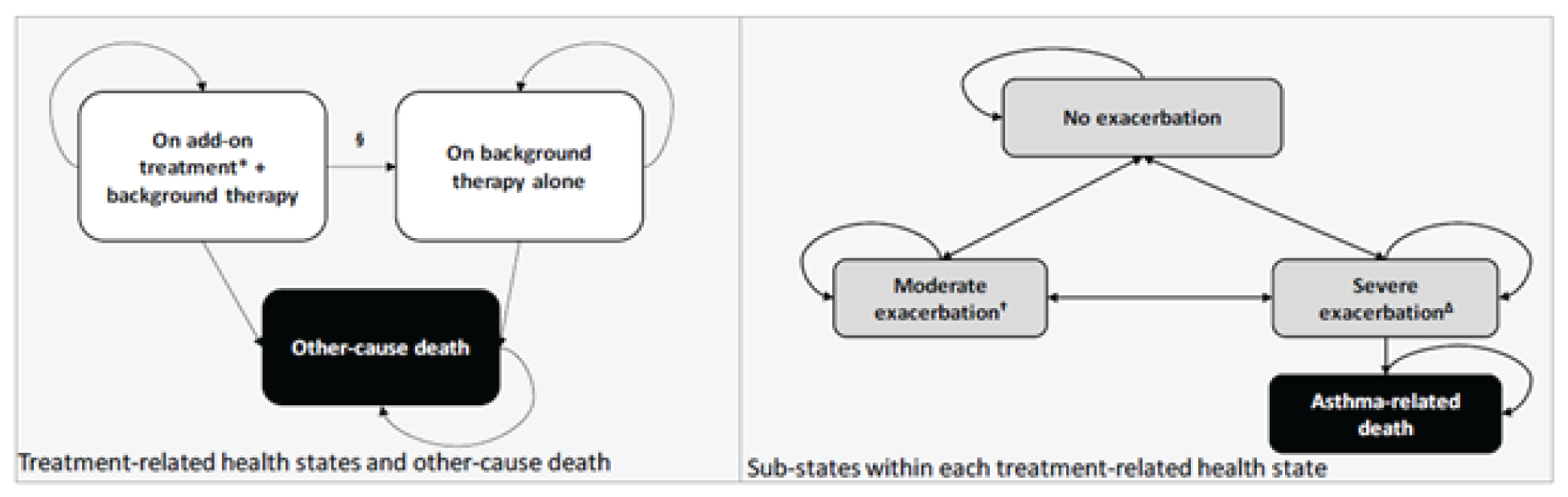
Detailed Results of the Sponsor’s Base Case
Table 11: Disaggregated Summary of Sponsor’s Economic Evaluation Results—Type 2 or Eosinophilic Phenotypic Asthma
Drug | Dupilumab | Background Therapy |
|---|---|---|
Discounted LYs | ||
Total | 42.11 | 42.02 |
Discounted QALYs | ||
Total | 37.08 | 36.51 |
Trial Period | 0.83 | 0.80 |
Extrapolation Period (Child) | 1.88 | 1.81 |
Extrapolation Period (Adult/Adolescent) | 34.36 | 33.90 |
Discounted Exacerbations | ||
Total | 219.97 | 248.41 |
Moderate | 130.37 | 141.65 |
Trial Period | 1.54 | 2.35 |
Extrapolation Period (Child) | 3.62 | 4.58 |
Extrapolation Period (Adult/Adolescent) | 125.21 | 134.73 |
Severe Exacerbations | 89.60 | 106.75 |
Trial Period | 0.35 | 0.67 |
Extrapolation Period (Child) | 0.77 | 2.57 |
Extrapolation Period (Adult/Adolescent) | 88.48 | 103.51 |
Discounted Costs ($) | ||
Total | 506,921 | 275,602 |
Drug Acquisition | 319,466 | 53,152 |
Dupilumab | 266,196 | 0 |
Background Treatment | 53,270 | 53,152 |
Monitoring Costs | 863 | 0 |
Disease Management | 27,818 | 28,715 |
Exacerbation Costs | 158,774 | 193,736 |
ICER ($/QALY) | 407,474 | |
ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.1
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Detailed Results of CADTH Base Case
Table 12: Disaggregated Summary of CADTH’s Economic Evaluation Results—Type 2 or Eosinophilic Phenotypic Asthma
Drug | Dupilumab plus background therapy | Background therapy |
|---|---|---|
Discounted LYs | ||
Total | 44.23 | 44.23 |
Discounted QALYs | ||
Total | 36.29 | 36.22 |
Trial Period | 0.81 | 0.80 |
Extrapolation Period (Child) | 1.75 | 1.72 |
Extrapolation Period (Adult/Adolescent) | 33.73 | 33.70 |
Discounted Exacerbations | ||
Total | 253.27 | 266.58 |
Moderate | 135.23 | 140.05 |
Trial Period | 1.53 | 2.22 |
Extrapolation Period (Child) | 3.63 | 5.03 |
Extrapolation Period (Adult/Adolescent) | 130.07 | 132.81 |
Severe Exacerbations | 118.04 | 126.53 |
Trial Period | 0.32 | 0.64 |
Extrapolation Period (Child) | 1.05 | 1.92 |
Extrapolation Period (Adult/Adolescent) | 116.67 | 123.97 |
Discounted Costs ($) | ||
Total | 305,072 | 95,416 |
Drug Acquisition | 266,085 | 56,417 |
Dupilumab | 209,667 | 0 |
Background Treatment | 56,417 | 56,417 |
Monitoring Costs | 795 | 0 |
Disease Management | 23,415 | 22,769 |
Exacerbation Costs | 14,777 | 16,229 |
ICER ($/QALY) | 2,999,591 | |
ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year.
Scenario Analyses
Table 13: Summary of CADTH’s Scenario Analyses Results
Drug | Total Costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|
Scenario 1: Subgroup of those aged ≥6 and under 12; medium ICS with second controller or high ICS with second controller; EOS ≥300 with ≥2 prior exacerbations | |||
Background therapy | 110,740 | 35.94 | Ref. |
Dupilumab + background Therapy | 319,359 | 36.10 | 1,282,219 |
ICER = incremental cost-effectiveness ratio; LY= life-year; QALY = quality-adjusted life-year; Ref. = reference.
Note: Reanalyses are based on publicly available prices of the comparator treatments.
Appendix 5: Submitted BIA and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 14: Summary of Key Take-Aways
Key Take-aways of the BIA |
|---|
|
Summary of Sponsor’s BIA
The submitted budget impact analysis (BIA) assessed the introduction of dupilumab as an add-on maintenance treatment in patients aged 6 years to <12 years with severe asthma with a type 2/eosinophilic phenotype or OCS-dependent asthma.24 The analysis took the perspective of CADTH-participating Canadian public drug plans using a top-down epidemiological approach and incorporated drug acquisition costs. A time horizon of 3 years was taken. The target population size was estimated using the prevalence of asthma in patients aged 6 to 11, the proportion of those treated, the proportion of those with moderate to severe asthma, the proportion of those uncontrolled, and the proportion of uncontrolled patients with severe asthma. Finally, the proportion of type 2 asthma and eligibility for biologics was used to determine the target population. The base-case analysis considers best supportive care (BSC) as the sole comparator in the model, with the option to assess add-on biologic treatments (omalizumab and mepolizumab) as a scenario analysis. It was assumed that all add-on therapies will be taken with background therapy (ICS, OCS, and rescue medications); background therapy was excluded from the analysis. The reference scenario included BSC and the new drug scenario considered the reimbursement of add-on treatment with dupilumab. Key inputs to the BIA and the sponsor’s methodology in calculating target population are documented in Table 15.
Table 15: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
Canadian population aged 6 to 1125 | 1,945,404 |
Asthma patients aged 6 to 1126 | 15.7% |
Proportion of treated asthma patients27 | 67.1% |
Proportion of moderate to severe asthma patients28 | 37.0% |
Proportion of moderate to severe patients that are uncontrolled27 | 32.2% |
Proportion of uncontrolled patients with severe asthma | 48.6% |
Proportion of severe asthma patients with type 2 asthma29 | 89.1% |
Proportion of those eligible for biologicsa | 100% |
Number of patients eligible for drug under review | 4,025 / 4,067 / 4,110 |
Market Uptake (3 years) | |
Uptake (reference scenario) BSC | 100% / 100% / 100% |
Uptake (new drug scenario) Dupilumab BSC | 19.5% / 20.0% / 20.5% 80.5% / 80.0% / 79.5% |
Cost of treatment (per patient) | |
Cost of treatment over one year Dupilumab BSC | $21,629 $0 |
BSC = best supportive care.
aParameter was informed by sponsor assumption.
Summary of the Sponsor’s BIA Results
The sponsor’s estimated budget of funding dupilumab as an add-on maintenance treatment in patients aged 6 years to <12 years with severe asthma with a type 2/eosinophilic phenotype or OCS-dependent asthma was $18,162,580 in Year 1, $18,647,960 in Year 2, and $19,314,858 in Year 3, for a 3-year total of $56,125,399.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Uncertainty regarding the number of patients eligible to receive dupilumab: In deriving target population, the sponsor estimated that 37.0% of treated asthma patients aged 6 to 11 have moderate to severe asthma. Clinical expert feedback indicated that this may be an overestimate, as the majority of patients are diagnosed with mild asthma. The number of patients receiving dupilumab may be lower than the sponsor’s analysis suggests if the proportion of those with moderate to severe asthma is overestimated.
Furthermore, clinical expert feedback indicated that clinicians may also consider dupilumab for patients with moderate asthma and comorbid atopic dermatitis, owing to dupilumab’s Health Canada-approved indication for atopic dermatitis for this patient population. The sponsor’s exclusion of potential use in this population may lead to an underestimated target population which would therefore underestimate the budget impact of reimbursing dupilumab.
CADTH assessed the impact of increasing market shares by 10% by Year 3 to account for potential indication creep in a scenario analysis.
Uncertainty regarding the sponsor’s epidemiological approach to calculate target population: The sponsor used prevalence of asthma to estimate target population and does not explicitly model incidence. It is assumed that the asthma population will change at the same rate as the province-specific population growth rate. Some evidence suggests that asthma prevalence, as a percentage of the population, may increase over time.30-32 Although the sponsor accounts for population growth they do not account for potential growth in asthma as well.
CADTH could not address the limitation regarding exclusion of incidence as this would require the entire structure of the model to be changed.
Uncertainty regarding market uptake of dupilumab: Given that there are no currently available biologic treatments for type 2/eosinophilic asthma for children aged 6 to 11, clinical expert feedback indicated that uptake would likely be higher than that estimated by the sponsor given the lack of currently available biologics for the indicated population.
CADTH increased the market shares of dupilumab to reflect clinical expert feedback to reach 25% by Year 3 in a scenario analysis.
Uncertainty surrounding the sponsor’s assumptions to calculate target population: Several of the sponsor’s sources used to estimate target population were derived from external countries which may not be applicable to the population of patients living in Canada. Furthermore, for example, the proportion of those with uncontrolled moderate to severe asthma was estimated using foreign claims data which could not be validated by CADTH and was not provided by the sponsor.28 The estimated target population is a key influence on the anticipated budget impact of dupilumab.
CADTH could not address this limitation in reanalysis.
CADTH Reanalyses of the BIA
Table 16: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Changes to derive the CADTH scenario analyses | ||
Scenario analysis 1: Increased dupilumab market shares due to anticipated indication creep | Dupilumab: 19.5% / 20.0% / 20.5% BSC: 80.5% / 80.0% / 79.5% | Dupilumab: 29.5% / 30.0% / 30.5% BSC: 70.5% / 70.0% / 69.5% |
Scenario analysis 2: Increased dupilumab market shares due to anticipated use | Dupilumab: 19.5% / 20.0% / 20.5% BSC: 80.5% / 80.0% / 79.5% | Dupilumab: 24.5% / 25.0% / 25.5% BSC: 75.5% / 75.0% / 74.5% |
The results of the CADTH scenario analyses are presented in Table 16. CADTH did not undertake a base-case reanalysis of the sponsor’s BIA, owing to the high degree of uncertainty around key model parameters, including the size of the eligible population and exclusion of incidence. Due to the uncertainty as well as the additional limitations described above, the impact of reimbursing dupilumab is highly uncertain.
The scenario analysis assessing the impact of increasing market uptake to reflect potential indication creep due to clinician’s anticipated use of dupilumab for those with comorbid atopic dermatitis and moderate asthma led to a 3-year budget impact of $84,185,405. A scenario analysis assessing the impact of increasing market uptake to reflect clinician’s preference for dupilumab given the current unavailability of biologic treatments for the indicated population led to a 3-year budget impact of $70,155,402.
Table 17: Detailed Breakdown of the CADTH Scenario Analyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $0 | $0 | $0 | $0 | $0 |
New drug | $0 | $18,162,580 | $18,647,960 | $19,314,858 | $56,125,399 | |
Budget impact | $0 | $18,162,580 | $18,647,960 | $19,314,858 | $56,125,399 | |
CADTH scenario analysis: Increased market shares (indication creep) | Reference | $0 | $0 | $0 | $0 | $0 |
New drug | $0 | $27,476,724 | $27,971,941 | $28,736,740 | $84,185,405 | |
Budget impact | $0 | $27,476,724 | $27,971,941 | $28,736,740 | $84,185,405 | |
CADTH scenario analysis: Increased market shares (anticipated use) | Reference | $0 | $0 | $0 | $0 | $0 |
New drug | $0 | $22,819,652 | $23,309,950 | $24,025,799 | $70,155,402 | |
Budget impact | $0 | $22,819,652 | $23,309,950 | $24,025,799 | $70,155,402 | |
CADTH scenario analysis: 98% price reduction | Reference | $0 | $0 | $0 | $0 | $0 |
New drug | $0 | $363,252 | $372,959 | $386,297 | $1,122,508 | |
Budget impact | $0 | $363,252 | $372,959 | $386,297 | $1,122,508 |
BIA = budget impact analysis.
Stakeholder Input
Patient Input
Asthma Canada
About Asthma Canada
Asthma Canada is the only national, patient-driven charity, solely devoted to enhancing the quality of life for people living with asthma and respiratory allergies. The Board of Directors has a strong patient and caregiver presence. For nearly 50 years, Asthma Canada has proudly served as the national voice for Canadians living with asthma. We empower patients with evidence-based information, education programs and support asthma research in Canada. For more information about our organization, visit our website at: www.asthma.ca
Information Gathering
A survey was independently developed and launched to seek the perspectives of people living with asthma, including caregivers. The survey was open from February 28th and closed on March 10th, 2022. Over 100 people responded to the survey and participants were from British Columbia (25%), Alberta (11%), Saskatchewan (2%), Manitoba (5%), Ontario (51%), Quebec (3%) and the Atlantic provinces (2%). Most participants lived with asthma (92%) while the rest were caregivers (8%). Four survey participants had experience taking Dupixent (dupilumab).
One of the authors (LP) of this submission lives with asthma that was diagnosed in childhood and also has a family history of severe asthma. Various reports and resources were referenced in developing this submission, such as clinical practice guidelines, the Dupixent (dupilumab) product monograph, websites of not-for-profit organizations, research papers, etc.
Disease Experience
Asthma is a chronic, inflammatory condition of the airways of the lungs that causes difficulty breathing, chest tightness, coughing and wheezing. Asthma affects people at any age, including children and can be intermittent to severe. When diagnosed in childhood, the condition can continue into adulthood and becomes a lifelong disease. The disease affects an estimated 850,000 children under the age of 14, making it the most common chronic disease among children in Canada, and is the leading cause of hospitalization and school absenteeism. More than 20,000 hospitalizations each year means that children with asthma spend more time in the hospital, and less time playing outside, socializing, or learning in school.
Parents and children aim to control asthma by identifying environmental triggers and finding the right combination of pharmacological treatment(s) to reduce or eliminate asthma exacerbations (known as “asthma attacks”) with the support of a respirologist, family physician and/or pediatrician. The pharmacological treatments used to treat asthma depend on the type of asthma, severity of the condition, and how the child responds to various treatment options. What works for one person with asthma changes over time and means that many treatment options are needed to manage their health successfully. One in 4 people who completed our survey indicated they have poor symptom control even with currently available treatments. Many people with asthma have challenges in accessing the needed health providers, like respirologists and specialized asthma clinics, to manage their health. There can be significant time and burden involved to manage care with health care providers made worse with poor asthma control. There can be travel involved for those living in rural areas to centres providing the health care need. This means children miss school and parents and caregivers may miss work. Some parents and caregivers may not have paid leave to attend these appointments or may need to use vacation leave, if available.
“Sometimes not being able to breathe is just too scary for a child to endure.”
Parents and children have significant daily responsibility to self-manage asthma symptoms with the goal of ensuring the child can fully participate in day-to-day activities, like school and play. Inhaler technique is important and can be particularly difficult to manage with children. When well-controlled, there can be periods of stability however exacerbations or asthma attacks can occur due to environmental triggers like pollution, smoking, and allergies. Asthma attacks can also be triggered by viral infection and uncontrolled disease. Often, children with asthma face several barriers as adults and other children don’t adequately understand the impact of asthma on their lives or how environmental triggers can trigger asthma attacks.
“(I wish) places like schools understood the seriousness of asthma. (The most frustrating thing is) the judgement from others who don’t understand what it’s like with a child with asthma”.
Parents and caregivers are often concerned with accessing adequate and necessary medical care within a short period of time, as exacerbations can lead to urgent trips to the Emergency Department (ED) to address and restore airway function. In severe asthma attacks, loss of consciousness or hypoxia can occur. Visits to the ED can be stressful as parents and caregivers navigate busy and overcrowded ED, particularly during the COVID-19 pandemic and still currently, as backlogs and staff shortages continue. Access to specialists with knowledge of asthma continues to be a challenge. People with asthma and caregivers that responded to our survey noted the worry and fear of an asthma attack was the most concerning (60%) followed by potential for hospital visits / admissions (47%) and missed work and school days (47%).
Asthma symptoms impact both the child and family’s quality of life. Children may experience fatigue and have less energy to play and exercise. Making and keeping friends can be made more difficult due to the symptoms of the disease, the presence of environmental triggers, or activity limitations. School is an important part of a child’s development however children may not be able to attend and concentrate at school due to disease symptoms, fatigue, and exacerbations. The sleep of children with asthma can be disturbed and parents and caregivers are often called on to support their child in the night (38% of survey participants noted sleep as a concern). Children and parents / caregivers are faced with barriers in understanding the seriousness of asthma. They spend a significant amount of time educating their children’s friends, daycares, schools, and others about the seriousness of asthma.
“Having an asthmatic child has changed a lot. I am nervous to let him sleep over at friends. Constantly reminding him when he walks out the door to make sure Ventolin is in his pocket.”
The impact of asthma on parents and caregivers means missing work to care for their child when they are suffering from an asthma attack or bringing them to medical appointments. Children, parents, and caregivers are affected due to stress and concerns of the current and future health of the child. Medications need to always be kept on hand in case of emergency. Managing an asthma attack can cause panic in the child and their parents due to the life-threatening nature asthma attacks. There can be added stress due to financial hardships with paying for current treatments which can be expensive and strain a family’s finances.
“I have had to decline full time employment as I couldn't guarantee my availability if she (my child) was sick or in hospital.”
The COVID-19 pandemic has impacted children with asthma and their families. Asthma attacks can be triggered by respiratory viruses, like COVID-19. Many parents and caregivers fear seeking needed health care at ED’s and other facilities due to the fear of contracting COVID-19. ED’s have been deeply affected by the pandemic and this has affected services provided to others needing emergency care, like the asthma community. Drug shortages have impacted children with asthma due to limited supply. During the COVID-19 pandemic, many pharmacies dispensed a limited 30-day supply of medications causing further stress and anxiety in the asthma community. Many families suffered financial stress during the pandemic, and these factors created additional strain for families dealing with asthma.
“The ER (Emergency Room) won't give oxygen treatments anymore because of covid.”
Experiences With Currently Available Treatments
The treatment of asthma involves both pharmacological and non-pharmacological treatments. Asthma self-management education is important especially since it is important to manage symptoms on a regular basis. It is important to identify triggers like respiratory viruses, indoor allergens, secondhand smoke, and exercise. However, access to self-management and appropriate specialist care can be challenging for many children with asthma.
“We worry about what this does to his body as he grows. He has been on and off since age 1 and he is 10 now.”
“Even with the inhalers, (I) still get out of breath when walking or climbing stairs.”
There are a range of pharmacological treatment options for children with asthma depending on the severity of the illness. There are “rescue medications’ like short-acting beta antagonists (SABA’s) that provide quick relief for asthma symptoms, like shortness of breath. There are “controller medications’ like inhaled corticosteroids, leukotriene modifiers, long-acting beta agonists (LABAs), Theophylline and combination inhalers. Varying doses of these medications are tried before finding the ideal dosage. Side effects of SABA’s include increased heart rate, nervousness, trembling, and sore throat. “Controller medications” can cause side effects, such as thrush, sore throat, and hoarseness. Many of the people who responded to our survey noted they took a variety of medications, such as inhaled corticosteroids (26%), rescue inhalers (61%), combination medications (75%), Leukotriene Receptor Antagonists (19%), and biologics (12%).
“(Managing asthma is) like a yo-yo. We’ve tried most biological, but none are long lasting.”
These medications are taken through a metered-dose inhalers or dry-powder inhaler and can be taken at home. It can be challenging to take these medications regularly given the frequency of administration. Children and parents are taught inhaler technique as it is crucial to maximizing the effectiveness of the medications. Often spacers are used to make administration easier but come at an additional cost and subject to varying degrees of insurance coverage. Nebulizers can be used at home and in a hospital to provide for easier administration however the child needs to cooperate and remain still. Nebulizer equipment comes at an additional cost to families.
“(I would like to) reduce usage of inhalers and antihistamine pills.”
For severe cases of asthma, children may take oral corticosteroids. Although helpful in the short-term, these medications have a long list of side effects if taken for longer periods of time and at higher doses. Side effects include weight gain, acne, excess facial hair, mood swings, high blood pressure, hyperactivity, high blood sugar, increased infection. In the long term, oral corticosteroids can cause osteopenia, osteoporosis, glaucoma, cataracts, and heart disease. The growth of the child can be affected by inhaled and oral corticosteroids and impact long-term self-image and confidence.
“Yes (I worry about) taking prednisone especially since he is so young.”
Immunomodulator medications, or biologics, are also available to treat severe asthma. Some biologics are for eosinophilic asthma or for those that are dependant on oral corticosteroids. Some biologics are available however only Omalizumab (Xolair), Mepolizumab (Nucala) and now Dupixent (dupilumab) are approved for use in children from age six to twelve. Many biologics are available in a self-injectable form (e.g., autoinjector pen). Side effects include allergic reactions, injection site reactions, and infections. Self-injection provides flexibility and reduces the time involved in taking medicine. However, parents can feel additional stress in learning how to give injections as proper training is needed before initiating therapy. Children may also have needle phobia which is common in children of any age. These stressors may affect the willingness to take the medication. It can also be difficult to travel as the self-injectors need to be constantly refrigerated.
Improved Outcomes
The expectations of the drug are to offer another treatment option for children with severe asthma and their families. New treatment options have the potential to ease the burden on patients’ families, caregivers, and the healthcare system. It also provides another option for children with severe asthma who have tried many other pharmacological and non-pharmacological treatments. The current risk-benefit profile of Dupixent (dupilumab) is similar to other biologics, e.g., allergic infection.
“I am always out of breath.”
Children and parents reported that current treatments can be difficult to take highlighting that the mode of administration and frequency of dosing is important. Medications taken less frequently can be especially useful in children as cooperation is important. About one in four survey participants indicated that they need too many daily doses.
“The family will have a better life when the child has a better health with this medicine.”
A variety of side effects of inhalers are difficult to manage such as elevated heart rate, anxiousness, and thrush. Minimizing these side effects are important outcomes that should be considered when evaluating new therapies. Survey participants noted that dry throat (50%), difficulty sleeping (42%), increased heart rate (38%), headaches (36%), hoarseness (35%) and weight gain (34%) were the most bothersome symptoms. Oral corticosteroids cause a long list of side effects and impact on a child’s growth and reducing or eliminating oral corticosteroids is also important to patients.
Survey participants indicated their expectations for a new medication and ranked these expectations in the following order:
Increase in lung function (73%)
Easier management of asthma symptoms (61%)
Reduction in asthma exacerbations or asthma attacks (56%)
Reduced reliance on oral corticosteroids (56%)
“The medications are expensive and without an extended health plan it would be challenging for me.”
Broadly speaking, children and parents expect to see improvements in a range of day-to-day activities affecting qualify of life, for example:
Improved attendance at school
Improved sleep
More energy
Less time off work for parents and caregivers
Participation in play, physical and social activities
Less health care visits, such as ED visits
Less anxiety and panic due to asthma attacks
Less financial hardships
“I can’t sleep very well and I often feel nauseated.”
“(The most difficult thing is) the exhaustion from actively working to breathe. It affects your whole life depending on your triggers.”
Over half of survey participants indicated that the benefits of the new treatment are worth tolerating the potential side effects to improve management of asthma.
Experience With Drug Under Review
Dupixent (dupilumab) is a new immunomodulator that can be prescribed as an add-on maintenance treatment in people living with severe type 2/eosinophilic phenotype or dependent on oral corticosteroids. We spoke with one person who is currently taking Dupixent (dupilumab) that was prescribed by a respirologist. They had tried several biologics, corticosteroids, rescue inhalers and controller inhalers before starting the medication. They indicated that managing severe asthma is “like a teeter totter” and they are regularly worried about viruses, like COVID-19, that could trigger an asthma attack that requires hospitalization. They also shared the following thoughts about living with asthma and taking Dupixent:
“For the first seven days after taking Dupixent, I feel pretty good. I feel in better control than without it.”
“I’ve noticed an improvement on this medication; however, I do need to go on dexamethasone about 10 days after taking Dupixent. I wish I could find a medication that addresses both my lower and upper airway. My respirologist feels I need medications that target both airways.”
“I feel agitated and depressed, but I can’t say if it comes from living with asthma, the medications, COVID-19, or what’s going on in the world. I am very unmotivated. I am tired...”
Many people with asthma will have tried and used many other treatments before using Dupixent. The addition of a new biologic for children with severe asthma provides another treatment option so they can tailor treatments to their needs. Patients and caregivers value a reduction in other medications, such as oral corticosteroids and inhalers and less medication side effects. The asthma community would also value improvements in quality of life, like participation in school. A variety of outcomes are also valued as described in Section 5 – Improved Outcomes.
Companion Diagnostic Test
Not applicable.
Anything Else?
Live vaccines cannot be given with Dupixent and this could be a challenge since children using this medication may not be fully immunized (e.g. Hepatitis, Meningococcal, HPV).
Conflict of Interest Declaration — Asthma Canada
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
|||||||||||||||||||, a patient who lives with asthma, diagnosed in childhood and also has a family history of severe asthma helped write and review the survey respondent’s data.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detai l the help and who provided it.
See above.
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
Table 1: Financial Disclosures for Asthma Canada
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AstraZeneca | — | — | — | X |
Sanofi Genzyme | — | — | — | X |
GSK | — | — | — | X |
Novartis | — | — | X | — |
Sanofi Pasteur | — | — | X | — |
Pfizer | — | — | X | — |
Teva | — | — | X | — |
Valeo | — | — | X | — |
Lung Health Foundation / The Ontario Lung Association
About the Lung Health Foundation / The Ontario Lung Association
The Ontario Lung Association (now named Lung Health Foundation) is registered with the CADTH and pCODR (www.lunghealth.ca). The Lung Health Foundation (Ontario Lung Association) is a registered charity that assists and empowers people living with or caring for others with lung disease. It is a recognized leader, voice and primary resource in the prevention and control of respiratory illness, tobacco cessation and prevention, and its effects on lung health. The Foundation provides programs and services to patients and health-care providers, invests in lung research and advocates for improved policies in lung health. It is run by a board of directors and has approximately 46 employees, supported by thousands of dedicated volunteers.
Information Gathering
The information provided from the Lung Health Foundation in this submission was obtained From a survey completed by 27 people living with Asthma and 2 caregivers to people living with Asthma. Input was received from Jan 2021 to June 2022. All respondents live in Ontario. Information on age and gender was not collected within this survey.
Disease Experience
The patient experience with asthma can be divided between symptoms and quality of life. The survey respondents describe symptoms such as fatigue (67.7%), cough (51.6%), shortness of breath (74.2%), excessive mucus (41.9%), wheezing (41.9%), and chest tightness (45.2%). Asthma affects their activities of daily living such as difficulty with housework (40.0%), difficulty with going to work (33.3%), difficulty climbing stairs (43.4%), difficulty with sports and physical activities (40.0%), and difficulty with leisure activities and hobbies (30.0%). The respondents’ comments regarding impacts of asthma on their quality of life included:
“I have challenges babysitting grandchildren”
“I can’t play soccer”
“When humid, I have to stay indoors”.
“It is horrible living with this. It is constantly on your mind and I feel like a burden to my family”
“I feel embarrassed from coughing and phlegm.”
Other negative impacts included waking at night or early morning because of breathing problems (34.5%), emotional well-being (37.9%), being short tempered or impatient with others (31.0%), managing symptoms (31%), being unable to do daily activities due to shortness of breath (55.2%), being unable to do daily activity due to fatigue (41.4%).
Experiences With Currently Available Treatments
The patients indicated that they were treated with Alvesco, Symbicort, Ventolin, Onbrez, Spiriva, Advair, Trelegy, Pulmicort, Breo, Flovent, Arnuity, Bricanyl, Breo, Incruse, Respimat, Fasenra, Singulair, Combivent and Xolair.
Patients reported that medications have helped to reduce cough (40.7%), reduce shortness of breath (74.1%), increase energy (29.6%), increase ability to exercise (44.4%), increase participation in daily activities (37.0%).
The side effects reported from the medications were heart palpitations, poor sleep, voice hoarseness and inability to fight infection.
Improved Outcomes
Key treatment outcomes for the patients interviewed include improving symptom management, improving energy, reducing cost, and improving quality of life. A survey respondent stated, “this condition affects negatively my emotional and social life. I have anxiety about nights, because quite often at night, I cannot breathe properly”. Another respondent stated, “each time meds are changed there is a fear that I will not be able to get the new drug. All have been via compassionate care program. Cost is prohibitive in most cases. Provincial and or federal coverage is a must.”
Experience With Drug Under Review
No patients within this evidence group submission had experience with the medication under review. Patients with uncontrolled asthma have a reduced quality of life due to exacerbations. There continues to be an unmet need in treatment options that reduce exacerbations.
Companion Diagnostic Test
Not applicable.
Anything Else?
Not applicable.
Conflict of Interest Declaration — Lung Health Foundation/The Ontario Lung Association
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
No.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
Table 2: Financial Disclosures for the Lung Health Foundation/The Ontario Lung Association
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Sanofi Genzymes | — | — | — | X |
Clinician Input
Canadian Thoracic Society
About the Canadian Thoracic Society
The Canadian Thoracic Society (CTS) is a national specialty society and membership-based professional association for health care providers (HCPs) working in respiratory care and research. Our mission is to promote lung health by enhancing the ability of HCPs through leadership, collaboration, research, learning and advocacy, and providing the best respiratory practices in Canada. CTS is recognized as an accrediting body of the Royal College of Physicians and Surgeons for specialist education and continuing professional development.
Website: https://cts-sct.ca/wp-content/uploads/2022/07/CTS-Sev-Asthma-Continuum.pdf
Information Gathering
The following document has been prepared as a submission to CADTH in response to its request for clinician input into the application by Sanofi Canada regarding its add-on maintenance treatment, DUPIXENT™. The submission represents the viewpoint of the Canadian Thoracic Society with respect to severe asthma therapy in individuals aged 6 to 11 years of age with severe asthma with a type 2/eosinophilic phenotype or oral corticosteroid-dependent asthma. The draft document was prepared by the Asthma Assembly Steering Committee Co-Chairs and Pediatric Respirologists. The final draft document was then submitted to the CTS Executive Committee for review, revision and approval prior to its submission.
Current Treatments and Treatment Goals
Children with severe asthma are those who require treatment with daily high dose inhaled corticosteroids and a second controller medication, or who remain uncontrolled despite that treatment. Unfortunately, non-pharmacologic treatments such as environmental modification are not effective enough in controlling asthma, and particularly severe asthma.
In Canada, children with severe asthma have limited treatment options in comparison to adults given that tiotropium is not approved for those under 18 years of age and is not regularly used off-label in children 6-11 years of age as there is no evidence for improvement in outcomes other than small benefits in lung function. The only other options include injectable biologic medications: omalizumab, mepolizumab and dupilumab. To date, omalizumab is the typical treatment recommended for children 6-11 years of age with severe asthma, evidence of aeroallergen sensitization and an IgE ranging from 30 IU/ml to 1300 IU/ml. Mepolizumab is approved for use in Canada for children 6-11 years of age with severe asthma, eosinophils >=150cells/uL at initiation of treatment or >=300cells/uL in the last 12 months. However, there has not been a large use of this medication in children 6-11 years of age given the limited longer term safety data (n=30 patients in 52 week open-label trial) and the paucity of efficacy data in this age group with no randomized-control data. Dupilumab was recently approved for use in children with severe asthma and there is safety data from trials of children 6-11 years of age with atopic dermatitis (>600 patients). It is currently being used in those 6 years of age and older with severe asthma (funded through extended health plans or compassionate release from the pharmaceutical company) and would be the first choice medication in those with severe asthma and concomitant eczema.
Clinical practice guidelines for severe asthma (CTS, ATS/ERS) only included clinical trials published up to 2018. For children 6-11 years of age, the guidelines recommend omalizumab (CTS, ATS/ERS) and the ATS/ERS but not the CTS recommends tiotropium.
An ideal asthma medication for children with severe asthma would allow them to have: well controlled asthma (less frequent and severe symptoms) including good activity tolerance, no to minimal severe exacerbations, normal lung function, improved quality of life, no to minimal side-effects from asthma medication, and be easy to administer. Compared to adults, it is particularly important for children to meet these asthma goals as their lungs are still growing and the consequences of poorly controlled asthma (e.g., decreased activity levels and missed school) have lifelong, irreversible detrimental effects. Children are also more susceptible to side effects from high dose inhaled corticosteroids compared to adults and can have irreversible growth suppression and life-threatening adrenal suppression. Chronic oral steroids are not considered a viable treatment option in children with severe asthma given the side-effect profile.
For those with severe asthma, asthma medications that have been shown to improve asthma control, decrease frequency of exacerbations and improve lung function in those with mild to moderate asthma are not as effective. In this age group, tiotropium can improve lung function and omalizumab improves lung function, asthma control and decreases exacerbations in those that meet the initiation criteria. There is no randomized control efficacy data for mepolizumab in this age group. To date, none of the asthma medication has been shown to cure asthma such that when medication is discontinued symptoms recur. Thus, we are in great need of additional asthma medications for this age group that can improve lung function and reduce exacerbations such as dupilumab.
Treatment Gaps (Unmet Needs)
Considering the treatment goals in Section 3, please describe goals (needs) that are not being met by currently available treatments.
In general, the key goals in asthma management include preventing asthma exacerbations (which are potentially life-threatening), maximizing quality of life, preventing symptoms, and maximizing exercise tolerance. Secondary goals include normalizing lung function, reducing airway inflammation, and avoiding permanent airway remodeling. In children, it is essential to achieve these goals while avoiding chronic use of oral corticosteroids and using the lowest effective dose of inhaled corticosteroids (ICS), to avoid growth suppression, adrenal suppression, and other important adverse events. In the majority of children, these goals can be achieved with low or moderate doses of ICS, with or without the addition of other controllers (long-acting beta-2 agonists or a leukotriene receptor antagonist). In the relatively small subgroup of children with severe asthma, achieving acceptable control typically requires high doses of ICS and/or oral steroids and add-on therapies, with a very high risk of important adverse events. In some children, acceptable control cannot be achieved even with these therapies. Children with severe asthma often have frequent severe exacerbations which are treated with oral steroids, so even if they’re not on chronic oral steroids, they can have recurrent exposures to oral steroids, with a high risk of side effects. Studies in adult Severe Asthma Clinics have demonstrated that many people with “severe asthma” have poor adherence, and evaluating adherence to existing therapies is challenging.
Biologic agents provide a means of improving asthma control in children with severe asthma with type II (eosinophilic or atopic) inflammation at significant risk of adverse events. In addition, if these agents are administered at a health care facility, adherence can be monitored. The first such agent, omalizumab, is limited to children with a serum IgE of 72 to 1680 mcg/L. Children with an IgE < 72 are generally not atopic and don’t require this type of product. However, many children with severe asthma and eczema have an IgE of 5,000 mcg/L or more, where omalizumab is not indicated. Biologics that target eosinophil survival and function require an elevated eosinophil count for efficacy but have no upper limit of eosinophil concentration, and don’t suffer from this limitation.
At present, mepolizumab is available for children with severe asthma ages 6-11 years with eosinophilic (type II inflammation). The indications for mepolizumab and dupilumab for the treatment of asthma overlap, and efficacy, while it has not directly compared, is probably fairly similar. According to their product monographs, mepolizumab is indicated for children with a blood eosinophil count of at least 150 cells/microliter at treatment initiation (or 300 in the past year) and dupilumab, in children with type 2/eosinophilic inflammation, although the pivotal trial (referenced in the monograph) also generally required an eosinophil count of 150 cells/microliter. Importantly, other indications vary between the products, and as children with severe asthma often have other, and often severe, atopic conditions, this is important. Mepolizumab is also indicated for individuals with chronic rhinosinusitis with nasal polyps and eosinophilic granulomatosis with polyangiitis, though both these conditions are uncommon in children. Dupilumab is also indicated for chronic rhinosinusitis with nasal polyps, but also for moderate to severe eczema, which is very common in children with asthma. Thus, for children with severe asthma and atopic dermatitis, dupilumab becomes an important therapeutic option. Other arguments for including dupilumab as a therapeutic option for this age group include children allergic to, or who have had severe adverse events with mepolizumab (although these circumstances are likely uncommon), and having an alternative therapy should supply issues render mepolizumab unavailable.
In terms of remaining unmet needs, younger children with severe asthma may have non-type 1 inflammation and recurrent severe exacerbations driven by viral acute respiratory tract infections. This involves neutrophilic inflammation, and effective add-on therapies for children with this phenotype who are incompletely controlled with inhaled corticosteroids have not been identified. Similarly, a subgroup of older children with severe asthma also have neutrophil-triggered inflammation, and effective add-on therapies are also needed in this population.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Dupilumab targets type II inflammation by blocking the alpha subunit of the interleukin (IL)-4 receptor and inhibits signaling by IL-4 and IL-13. This mechanism of action differs from current available therapies for children with severe asthma, which commonly include inhaled or oral corticosteroids (ICS, OCS), long-acting beta-agonists (LABA), leukotriene receptor antagonists (LTRA). Other biologics available for children 6-11 include omalizumab (anti-IgE) and mepolizumab (anti-IL5), although they target different pathways than the IL-4/IL-13 pathway. Thus, the mechanism of action of dupilumab is complementary to other available treatments Additionally, while dupilumab is not the first treatment approved to address eosinophilic type II inflammation in pediatric severe asthma. However, it is the first to specifically inhibit the IL-4/IL-13 pathway.
The use of dupilumab would follow the Canadian Thoracic Society Severe Asthma Management Continuum. Following the current treatment paradigm, dupilumab would be recommended as an add-on therapy in severe asthma and would be considered when other biologics are indicated. This occurs when a child is not well controlled on a combination of high-dose ICS, LABA, and LTRA, or when a child experiences significant side effects from these medications or when these medications are contraindicated. Furthermore, injectable medications such as biologics are generally associated with a higher burden for families (thus less acceptability) and incur higher costs for the health system. Thus, it is appropriate to recommend biologics based on the child’s inflammatory profile and only after standard therapy has been adequately tried with good adherence.
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
The patients most likely to respond to treatment are those aged 6-11 years old with type II inflammation, moderate to severe asthma not adequately controlled on medium dose ICS plus LABA or high dose ICS and with a severe exacerbation in the past year. Those with type 2 inflammation are defined as having a serum eosinophil count >=150 and FeNO >=20 ppm) or with a serum eosinophil count >= 300. Those patients with an eosinophil count > 300 and with a baseline FeNO of 25 ppb have been shown to have the best response to dupilumab.
The patients in most need of this intervention are those with frequent severe exacerbations with evidence of type 2 inflammation as defined above despite medium dose inhaled corticosteroids plus a second controller or those on a high dose ICS. Patients with frequent exacerbations and frequent doses of corticosteroids will benefit the most.
The only disease characteristics that would differ in patients most likely to respond are markers of type 2 inflammation (FeNO elevation and eosinophil count > 150). They must also have moderate to severe asthma and be on at least moderate to high dose ICS, oral steroids and/or a second controller medication and still have ongoing symptoms and exacerbations.
The patients best suited for treatment would be identified by their treating respirologist or allergist. They should have confirmation of asthma diagnosis based on symptoms and spirometry showing reversibility (>10% change in FEV1). If not under the care of a respirologist or allergist they should be referred before considering this treatment to ensure their disease management is otherwise optimized. They would have spirometry pre and post bronchodilator, a clinical assessment and CBC with differential count done and a FeNO if available.
If the appropriate pulmonary function testing is done along with a clinical assessment by a respirologist or allergist there should be little issues related to diagnosis of asthma.
The only diagnostics tests required are spirometry to confirm diagnosis and measures of type two inflammation of which serum eosinophil count is readily available. FeNO was also used in the studies but eosinophil count alone also predicted response and nothing beyond usual care is required. The only tool that would likely be helpful is broader access to exhaled nitric oxide (FeNO).
Misdiagnosis of asthma does occur, but it is our recommendation that use of this drug be restricted to respirologists and allergists who look after severe asthma and thus the appropriate diagnostic test will be conducted and the chance of any misdiagnoses and thus any misuse of the drug would be very small.
It is possible to identify those patients who are likely to exhibit a response to the drug as they are that patients described in detail in the studies. Those with both high eosinophil counts and high FeNO will respond best but there is also a good response with lower levels of eosinophil count as long as it is above 150. The patient should also have an exacerbations history as reduction in exacerbations was the main outcome in the clinical trials.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
The addition of biologic therapy such as dupilumab in severe asthma is done with specific treatment goals in mind. Clinically meaningful outcomes for patients would include lessening the frequency and severity of symptoms, improving quality of life (e.g. exercise tolerance, school/work attendance), reducing exacerbations and minimizing side effects from existing maximal therapy (e.g. steroid side effects). There is also the potential of reducing the burden of daily medication and improving compliance to treatment with injection therapy. Reduction of inflammatory markers, improvement of lung function and prevention of long-term airway remodeling, particularly relevant for the younger pediatric population aged 6-11, are also key outcomes.
In clinical trials, dupilumab was shown to significantly reduce the annualized rate of severe exacerbations in the pediatric population aged 6-11 years. It also resulted in higher asthma control scores (e.g. Asthma Control Questionnaire 7 -IA at week 24 where a 0.5 increase in score was considered clinically significant) and improved baseline FEV1 compared to existing therapy. Additional markers of airway (e.g. FeNO at week 12 from baseline) and circulatory (serum total IgE and serum thymus and activation-regulated chemokine or TARC) inflammation were also reduced with the drug under review compared to placebo. Dupilumab was shown to achieve these outcomes with a low incidence of severe adverse events in the pediatric group, similar to adolescents and adults, and importantly, a rate which did not differ from those on standard background therapy.
Clinical measures such as Asthma Control Questionnaires and assessment of FEV1 and exacerbation rates are routinely used in clinical practice and utilization would not differ amongst specialist physicians. However, secondary endpoints such as inflammatory markers may not be routinely used or accessible in practice and there may be variation in their application.
What factors should be considered when deciding to discontinue treatment with the drug under review?
Factors to consider for discontinuation of treatment with dupilumab would include lack of clinically meaningful positive outcomes over an expected timeframe, such as annualized rates of exacerbation similar to pre-treatment levels, similar or worse pre-bronchodilator FEV1 or patient symptom scores. Additionally, safety concerns around significant adverse local or systemic events, and patient choice e.g., regimen fatigue may play roles in the decision to discontinue this treatment.
Conversely, a positive response to treatment in these parameters may be relevant when considering renewal of treatment with dupilumab.
What settings are appropriate for treatment with [drug under review]? Is a specialist required to diagnose, treat, and monitor patients who might receive [drug under review]?
It is recommended that the first one or more doses of dupilumab should be administered by a healthcare professional in a hospital/medical setting to ensure correct technique for administration (e.g., identifying appropriate injection site, depth of injection) and tolerance by the patient, given their young age. Following this, injection by the caregiver can be undertaken in the community setting, at home, after the healthcare professional determines it is appropriate and the caregiver has received proper training on correct administration.
An asthma specialist is required to diagnose, treat and monitor patients who might receive the drug under review. This recommendation is made based on recommendations in the Canadian Thoracic Society Position Statement for the Recognition and Management of Severe Asthma (https://cts-sct.ca/wp-content/uploads/2018/01/Recognition-and-Management-of-Severe-Asthma.pdf) because this medication is indicated for children with moderate to severe asthma who are not responding to conventional therapies. Typically, the assessment of whether a child would qualify for a biologic asthma therapy (including determining adherence to typical therapies, correct administration technique and absence of an alternative diagnosis to explain suboptimal asthma control despite typical therapy) is best done by an asthma specialist. Asthma specialists can include (but may not be limited to): respirologists, allergists, pediatricians with a focus in childhood asthma. Per the Canadian Thoracic Society Position Statement for the Recognition and Management of Severe Asthma, “an operational definition of ‘asthma specialist’ would include specialists in asthma, general respirology, pediatrics, and/or allergy/immunology who have access to lung function, certified asthma/respiratory educators/nurse practitioners and FeNO C/¡ induced sputum analysis.”
Conflict of Interest Declarations — Canadian Thoracic Society
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
No outside help was sought or received.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No outside analysis was sought or received.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician who contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
Declaration for Clinician 1
Name: Dhenuka Radhakrishnan
Position: Co-Chair CTS Asthma Assembly - Pediatric Respirologist, Children’s Hospital of Eastern Ontario and Associate Professor, Department of Pediatrics, University of Ottawa
Date: 11-07-2022
Table 3: COI Declaration for Canadian Thoracic Society — Clinician 1
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Lung Health Foundation Ontario | X | — | — | — |
Declaration for Clinician 2
Name: Clare Ramsey
Position: Co-Chair CTS Asthma Assembly – Adult Respirologist, Health Sciences Centre Winnipeg, Associate Professor, Department of Respiratory Medicine, University of Manitoba
Date: 11-07-2022
Table 4: COI Declaration for Canadian Thoracic Society — Clinician 2
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Sanofi | X | — | — | — |
Astra Zeneca | X | — | — | — |
Valeo | X | — | — | — |
Declaration for Clinician 3
Name: Tom Kovesi
Position: Pediatric Respirologist, Children’s Hospital of Eastern Ontario, Professor, Department of Pediatrics, University of Ottawa
Date: 11-07-2022
Table 5: COI Declaration for Canadian Thoracic Society — Clinician 3
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
COVIS | X | — | — | — |
Declaration for Clinician 4
Name: Sze Man Tse
Position: Pneumologue pédiatrique, CHU Sainte-Justine, Professeure adjointe de clinique, Département de pédiatrie, Université de Montréal
Date: 11-07-2022
Table 6: COI Declaration for Canadian Thoracic Society — Clinician 4
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Fonds de recherche du Québec – Santé (FRQS) (salary award) | — | — | X | — |
Canadian Institute for Health Research (CIHR) (research operating funds) | — | — | — | X |
Quebec respiratory health research network (QRHN) (research operating funds) | — | — | X | — |
Institut TransMedTech (research operating funds) | — | — | X | — |
Declaration for Clinician 5
Name: Tania Samanta
Position: Pediatric Respirologist at North York General Hospital
Date: 11-07-2022
Table 7: COI Declaration for Canadian Thoracic Society — Clinician 5
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Valeo Pharma Inc | X | — | — | — |
Covis Pharma Canada Ltd | X | — | — | — |
Declaration for Clinician 6
Name: Connie Yang
Position: Past Co-Chair, CTS Asthma Assembly - Pediatric Respirologist, British Columbia Children's Hospital, Clinical Associate Professor, Division of Respiratory Medicine, Department of Pediatrics, University of BC
Date: 11-07-2022
Table 8: COI Declaration for Canadian Thoracic Society — Clinician 6
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Sanofi | X | — | — | — |
GSK | X | — | — | — |
Novartis | X | — | — | — |
Valeo | X | — | — | — |
Covis | X | — | — | — |
In last 5 years attended advisory board, chaired meetings sponsored by company, participated as an investigator in trials run by the company but did not receive any funding or sponsorship.
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.