CADTH Reimbursement Review
Deferiprone (Ferriprox)
Sponsor: Chiesi Canada Corp.
Therapeutic area: Transfusional iron overload
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
AE
adverse event
ANCOVA
analysis of covariance
CanHaem
Canadian Hemoglobinopathy Association
CHQ
Child Health Questionnaire
CHQ-CF87
Child Health Questionnaire Child Form 87 Questions
CHQ-PF50
Child Health Questionnaire Parent Form 50 Questions
CI
confidence interval
CIC
cardiac iron concentration
CrI
credible interval
DFO
deferoxamine
DFP
deferiprone
DFX
deferasirox
dw
dry weight
HRQoL
health-related quality of life
ITC
indirect treatment comparison
ITT
intention-to-treat
LIC
liver iron concentration
LOCF
last observation carried forward
LSM
least squares mean
MID
minimal important difference
ms
milliseconds
NMA
network meta-analysis
PP
per-protocol
RBC
red blood cell
RCT
randomized controlled trial
SAE
serious adverse event
SC
subcutaneous
SCD
sickle cell disease
SD
standard deviation
SF
serum ferritin
SF-36
Short Form (36) Health Survey
ULN
upper limit of normal
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Deferiprone (Ferriprox), 1,000 mg oral tablets and 100 mg/mL oral solution |
Indication | The treatment of patients with transfusional iron overload due to sickle cell disease or other anemias |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | October 13, 2021 |
NOC = Notice of Compliance.
Introduction
Ferriprox (deferiprone [DFP]) is an iron chelator that is approved for use in the treatment of patients with transfusional iron overload due to thalassemia syndromes when current chelation therapy is inadequate, or due to sickle cell disease (SCD) or other anemias. The indication under consideration for this review is the treatment of patients with transfusional iron overload due to SCD or other anemias.
SCD is an inherited blood disorder affecting an estimated 5,000 people in Canada.1 Patients with SCD usually have chronic anemia and may require blood transfusions. The approved indication for DFP does not identify what conditions are classified as “other anemias” for which patients require treatment for transfusional iron overload. These anemias are rare; the conditions studied in the pivotal trial for DFP included congenital dyserythropoietic anemia, pyruvate kinase deficiency, hereditary spherocytosis, hemoglobin s-beta thalassemia, dyserythropoietic anemia, autoimmune hemolytic anemia, other rare hemoglobinopathies, and chronic nonspherocytic hemolytic anemia.
DFP is available as 1,000 mg oral tablets and a 100 mg/mL oral solution. The recommended dose is 25 mg/kg to 33 mg/kg body weight per day taken orally 3 times a day for a total daily dosage of 75 mg/kg to 100 mg/kg body weight.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient and clinician groups that responded to CADTH’s call for patient input and from the clinical expert consulted by CADTH for the purpose of this review.
Patient Input
Two patient groups, the Sickle Cell Awareness Group of Ontario and the Thalassemia Foundation of Canada, submitted patient input for this review. The Thalassemia Foundation of Canada reiterated the input it submitted to CADTH in October 2015 for the initial review of DFP. That input gathered information from various sources, including a search of the medical literature, a collection of focus group reports, clinical practice guidelines, and data from other organizations representing the interests of patients with SCD.
The Thalassemia Foundation of Canada stated that, based on published literature, it believes the symptoms of excess iron are numerous, including endocrine disorders (growth retardation, failure of sexual maturation, diabetes mellitus, and insufficiency of the parathyroid, thyroid, pituitary and, less commonly, adrenal glands), dilated cardiomyopathy, arrhythmias, liver fibrosis, and cirrhosis. The Sickle Cell Awareness Group of Ontario noted that individuals with SCD face debilitating complications not limited to vaso-occlusive pain crisis and damage to their vital organs (including kidneys and liver). According to the input received, continuous blood transfusions in patients with SCD can lead to an excessive buildup of iron causing further organ damage and increased cancer risk. The patient input cited evidence in which patients and caregivers stated that this condition disrupted their ability to work or attend school as well as their physical and social interactions.
Respondents reported having experience with injectable treatments (e.g., deferoxamine [DFO]) and oral treatments (e.g., deferasirox [DFX]). They noted that DFO treatment has a demanding subcutaneous (SC) or IV administration schedule and can be associated with important side effects, such as local irritation, high-frequency hearing loss, deafness, retinal damage, impaired vision, growth retardation, and bone abnormalities. The patient groups also reported that oral treatments are associated with improvements in quality of life, treatment adherence, patient satisfaction, and reduced preventable organ damage from iron overload. The patient input stated that improved heart and endocrine function, reduced risk of premature death, and ease of oral administration (obtained by treating patients with DFP) are goals that will be meaningful to patients and their families. The patient groups concluded that expanding access to appropriate iron chelation therapies such as DFP is vital to effectively improve patient outcomes.
Clinician Input
Input From the Clinical Expert Consulted by CADTH
The clinical expert consulted by CADTH identified that the goal of treatments for patients with transfusional iron overload are to reduce hepatic iron to a safe level that will not lead to cirrhosis or hepatocellular carcinoma while minimizing or avoiding drug-related toxicity. Cardiac iron overload is uncommon in patients with SCD.
The clinical expert indicated that challenges with the existing treatments, DFX and DFO, include drug toxicity and nonadherence. DFX is contraindicated for patients with renal impairment. DFO via the IV route can be a suitable option for a select group of patients who are motivated and have an existing indwelling catheter; however, even under those circumstances, it can be challenging to maintain for a prolonged period. The risk of recurrent thrombosis or line-related infections is also present with IV DFO. Overnight SC infusion of DFO is even harder to maintain over the medium to long-term.
According to the clinical expert, the patients who are most suitable for treatment with DFP include those with sickle nephropathy, poor IV access, adverse events (AEs) with DFO or DFX, and normal liver enzymes. Patients for whom DFP may not be suitable or who may require closer monitoring due to the risk of mild or severe neutropenia include those with baseline neutropenia or concomitant therapy with hydroxyurea. Patients with very severe hepatic iron overload may also be less suitable or require dosing at the upper end of the dose range for efficacy.
In routine clinical practice, the clinical expert indicated that DFP may be administered until liver iron concentration (LIC) levels are at an acceptable threshold, not a fixed duration of 12 months. The goal of therapy is to reduce and then maintain liver iron at an acceptable range of 2 mg/g dry weight (dw) to 5 mg/g dw. DFP should be discontinued when: iron stores have reached the target range and there is no ongoing iron loading, during pregnancy and lactation, and in the presence of clinically significant toxicity that cannot be safely rechallenged or managed with a reduction in dose.
Clinician Group Input
This section was prepared by CADTH staff based on the input provided by clinician groups.
The input provided by 1 clinician group, the Canadian Hemoglobinopathy Association (CanHaem), generally aligned with the input provided by the clinical expert consulted by CADTH. The input from CanHaem noted there are currently 2 treatments available for transfusional iron overload: DFO (SC or IV infusion) or DFX (oral). The clinician group noted that not all patients will adequately respond to the currently available treatments and that many patients experience side effects with iron chelators; hence, additional treatment options would be beneficial for patients.
Renal dysfunction was highlighted as a common complication in patients with SCD and, consequently, some patients may experience intolerance to DFX. For others, the SC or IV administration of DFO may not be a feasible treatment option. The clinician group highlighted the unmet need for treatments that are better tolerated and that can improve compliance and patient convenience. In terms of place in therapy, CanHaem noted that DFP would be suitable for patients with SCD who demonstrate transfusional iron overload, particularly those who are unable to tolerate SC or IV drug infusion, those with liver or kidney dysfunction, and those who are not considered to be at risk of neutropenia. A clinically meaningful response to treatment was defined by the group as the maintenance or decrease of iron burden over time. CanHaem indicated that pediatric and adult patients receiving treatment with DFP should be under the care of a hematologist.
Drug Program Input
Input was obtained from the drug programs that participate in the CADTH reimbursement review process. The following were identified as key factors that could potentially impact the implementation of a CADTH recommendation for DFP:
relevant comparators
considerations for continuation or renewal of therapy
considerations for the prescribing of therapy
care provision issues
system and economic issues.
Clinical Evidence
Pivotal Studies and Protocol-Selected Studies
Description of Studies
Two studies were included in this review: FIRST (N = 213) and Calvaruso et al. (2014) (N = 60). The FIRST trial was a pivotal, late-phase (phase IV in the US, phase IIIb in other countries), multicentre, randomized, open-label, noninferiority study comparing the efficacy and safety of the iron chelator DFP versus DFO in patients with SCD or other transfusion-dependent anemias. Eligible patients were randomized (2:1) to receive either DFP or DFO for up to 12 months. Randomization was stratified by disease category (SCD versus other anemias) and burden of transfusional iron loading in the 3 months before baseline (≤ 0.3 mg/kg/day versus > 0.3 mg/kg/day). The mean age for patients was 16.9 years (standard deviation [SD] = 9.6). The primary end point of the study was change from baseline in LIC after 12 months. For noninferiority, the upper limit of the 96.01% confidence interval (CI) could be no more than 2 mg/g dw. Secondary outcomes included changes in cardiac iron concentration (CIC), serum ferritin (SF), and health-related quality of life (HRQoL). For CIC and SF, if the 96.01% CI contained zero (0), then no significant difference between the 2 treatment groups was assumed. The mean age was 16.9 years. Most patients were white (77.2%) and 46.5% had never received chelation therapy.
The second study was conducted by Calvaruso et al. (2014), and was a 5-year, multicentre, open-label, randomized controlled trial (RCT) to compare the safety and efficacy of DFP versus DFO in Italian patients. Eligible patients were randomized (1:1) to receive either DFP or DFO for up to 12 months. The primary outcome was a reduction in SF, and patients were considered responders if their SF values were less than 400 ng/mL. Patients were randomized consecutively after confirming eligibility; no stratification was conducted. Baseline characteristics for race and prior chelation therapy were not provided; the mean age ranged from 36.4 to 35.8 years (SD = 13.9 to 11.6).
Efficacy Results
Results for the key efficacy outcomes in the FIRST trial and Calvaruso et al. (2014) are summarized in Table 2.
Liver Iron Concentration
In the FIRST trial, in the analysis of covariance (ANCOVA) model, the mean change from baseline was similar between the 2 treatment groups █ █ █ █ ████ █ ██████ ███ and the upper limit of the 96.01% CI was ████, thereby supporting the noninferiority criterion. Subgroup analyses of SCD versus other anemias were generally supportive of the main analysis but sample sizes were too small for any definitive conclusions.
Cardiac Iron Concentration
In the FIRST trial, changes in CIC values were generally supportive of the primary end point. The least squares mean (LSM) of change in log-transformed cardiac MRI T2* difference in milliseconds (ms) between both groups was ███████ ███ ████████ █ ███ ███████ █ █ █ █ ██ █████ ████, thereby supporting the noninferiority of DFP to DFO.
Serum Ferritin
In the FIRST trial, changes in SF values were generally supportive of the primary end point. The LSM difference (DFP minus DFO) was ██ █ █ ████████████ █ █ ███████ █ █ ████████ ████, thereby supporting the noninferiority of DFP to DFO. In the Calvaruso et al. study, 36.6% of patients in the DFP group and 3.3% of patients in the DFO groups were responders. The changes over time in SF in patients receiving DFP versus DFO (as per a linear effects model) were statistically not significant; moreover, the study’s sample size was too small for any definitive conclusions.
Harms Results
In the FIRST trial, at least 1 AE was reported for 88.2% of patients in the DFP group and 88.2% of patients in the DFO group. The most frequently reported AE was pyrexia (28.3% of patients in the DFP group versus 32.9% of patients in the DFO group), followed by abdominal pain (25.0% of patients in the DFP group versus 13.2% of patients in the DFO group). Patients in the DFP group had higher rates of liver enzyme increases. The number of patients reporting agranulocytosis and neutropenia in both groups was low.
Table 2: Summary of Key Results From Pivotal and Protocol-Selected Studies
End points | FIRST | Calvaruso et al. (2014) | ||
|---|---|---|---|---|
DFP (N = 122) | DFO (N = 63) | DFP (N = 30) | DFO (N = 30) | |
LIC (mg/g dw), ITT set | ||||
Baseline, mean (SD) | █ █ █ █ █ ███████ | ██ █ ███████ | NA | |
Change from baseline, mean (SD) | ██ █ ███████ | ████████ | ||
P value for change in LIC (t-test)b | ███████ | ██████████ | ||
LS mean change (SE)a | █ █ █ █ █ ██████ | ████████ | ||
LSMD (96.01% CI) | █ █ ██ █ █ █████████ | █████████ | ||
CIC (ms), ITT set | ||||
Baseline, geometric mean ± CV (%) | ███ █ ██████ | ████████ | NA | |
Change from baseline, geometric mean ± CV (%) | █ █ ███ █ ██████ | █ █ ███ █ ██████ | ||
P valueb | ███████ | █████████ | ||
LS mean change (SE)a | ███████████████ | ███████████ | ||
LSMD (96.01% CI) | ████████████ | █████████ | ||
SF (mcg/L), ITT set | ||||
Baseline, mean (SD) | ███████████ | ██████████ | 1,440.13 (712.80) | 1,726.03 (694.01) |
LS means of change (SE) | ████████████ | ███████████ | NA | NA |
LSMD (96.01% CI), DFP minus DFX | ███████████████ | █████████ | NA | NA |
Responders (SF < 400 ng/mL), n (%) | ██ | ██ | 11 (36.6) | 1 (3.3) |
P valueb | ██ | ██ | 0.002 | Reference |
Harms, % (safety set) | ||||
At least 1 AE | █ ███████ | ████████ | NR | NR |
At least 1 SAE | ███████ | ████████ | NR | NR |
WDAE (from study treatment) | █████ | █ █ █████ | 16 | 13 |
Notable harms, N | ||||
Agranulocytosis | ██ | ██ | 0 | NR |
Neutropenia | ██ | ██ | NR | NR |
AE = adverse event; CI = confidence interval; CIC = cardiac iron concentration; CV = coefficient of variation; DFO = deferoxamine; DFP = deferiprone; DFX = deferasirox; dw = dry weight; ITT = intention-to-treat; LIC = liver iron concentration; LS = least squares; LSMD = least squares mean difference; ms = milliseconds; NA = not applicable; NR = not reported; SAE = serious adverse event; SD = standard deviation; SE = standard error; SF = serum ferritin; WDAE = withdrawal due to adverse event.
aTreatment was the main factor and average transfusional iron input, baseline LIC, and stratification factors (disease category and transfusional iron input in the 3 months before baseline) were covariates.
bP value has not been adjusted for multiple testing (i.e., the type I error rate has not been controlled).
Sources: FIRST Clinical Study Report2 and Calvaruso et al. (2014).3
Critical Appraisal
There were concerns in the FIRST trial regarding internal validity. Although the trial was open-label, most of the outcome measures were objective and relied on a central laboratory, thereby indicating a low risk of detection bias. On the other hand, nonobjective outcomes (e.g., HRQoL and subjective AEs) could be affected by the lack of blinding. Randomization and allocation were conducted by an electronic system, suggesting that the risk of bias due to the randomization process was probably low. Per the clinical expert consulted by CADTH, the noninferiority margin used in the FIRST trial was clinically appropriate. A high rate of protocol deviations and loss to follow-up occurred in the FIRST trial, which may create some uncertainty in the data, as it increases bias toward the null and falsely declaring noninferiority. The sensitivity analyses also did not support the noninferiority of DFP to DFO. Investigators used both an intention-to-treat (ITT) set and per-protocol (PP) set for efficacy analyses; however, a true ITT was not conducted.
There were concerns in the FIRST trial regarding external validity. The eligibility criteria and baseline characteristics were generally representative of the patient population in Canada, with a few notable exceptions (e.g., race). A baseline LIC of 7 mg/g dw was used in the inclusion criteria of the trial; however, in practice, patients with an LIC lower than 7 mg/g dw would be treated. Patients treated with hydroxyurea within the past 30 days were excluded; however, these patients would not be excluded in routine practice and often, hydroxyurea is a concomitant medication. A majority of the patients were white, and the rate of prior chelation therapy with DFO was higher than what would be expected in routine clinical practice. The efficacy and harms outcomes used in the FIRST trial were generally clinically meaningful and important to clinicians and patients. However, as noted by the clinical expert, few patients with SCD have cardiac iron overload in routine clinical practice, so this measure is not utilized as much as LIC. SF concentrations are a somewhat unreliable indicator in routine clinical practice because many factors affect these values outside of iron overload. Furthermore, there were concerns with the high discontinuation rates in the study.
In the Calvaruso et al. (2014)4 study, the investigators adequately used randomization and allocation concealment in the study and provided a rationale for why blinding was unfeasible. The use of SF as the only efficacy end point was a major limitation of the study, as this end point is not typically used in isolation to evaluate response to iron chelation in Canada. There were major concerns about generalizability to Canadian clinical practice in this study. With respect to the eligibility criteria, the study excluded patients with white blood cell counts of less than 3,000/μL, which may prevent Black patients and patients on hydroxyurea from accessing treatment (populations for which this drug would be used in routine clinical practice) because these populations typically have white blood cell counts that are lower. Per the clinical expert consulted by CADTH, the baseline transfusion burden appeared to be low in the trial, whereas patients in routine clinical practice with a higher transfusion burden would be treated. For the baseline SF values, the clinical expert indicated they are low compared with what would be expected in routine clinical practice, suggesting patients participating in the trial may be less iron overloaded than typically seen.
Indirect Comparisons
Description of Studies
In the absence of direct comparative evidence from trials, the aim of the network meta-analysis (NMA) conducted was to compare the relative efficacy of DFP versus DFX and DFO. The sponsor chose to restrict the NMA to the 2 RCTs. The FIRST trial was a late-phase (phase IIb and IV) open-label noninferiority trial comparing DFP with DFO in patients with SCD or anemias with transfusional iron overload, while the NCT00067080 trial was a phase II open-label trial comparing DFX with DFO in patients with SCD with transfusional iron overload. The mean duration of follow-up was 12 months in the FIRST trial and 13 months in the NCT00067080 trial. The inclusion and exclusion criteria were similar with some exceptions: the FIRST trial required a higher baseline LIC value compared with the NCT00067080 trial, but excluded patients with a baseline LIC exceeding 30 mg/g dw and patients who received treatment with hydroxyurea within 30 days of the study. The NMA used a Bayesian approach using random-effects models with a noninformative prior in all analyses. The clinical end points included change from baseline to 12 months in LIC and SF. The quality of the included studies was assessed by the sponsor according to the revised Cochrane risk-of-bias tool for randomized trials.
Efficacy Results
The results from the sponsor-submitted NMA suggested that in the overall population, no treatment was favoured when DFP was compared with DFX and DFO with respect to change from baseline to 12 months in LIC and SF. Compared with DFP, the mean difference for change at 12 months in LIC was –0.4 (95% credible interval [CrI], –1.7 to 0.9) for DFO, and –0.7 (95% CrI, –3.6 to 2.3) for DFX. Compared with DFP, the mean difference for change at 12 months in SF was –364.4 (95% CrI, –961.4 to 237.2) for DFO, and 11.2 (95% CrI, –688.2 to 712.5) for DFX.
Harms Results
No analysis of harms was included in the indirect comparisons.
Critical Appraisal
The sponsor-submitted NMA was based on a systematic literature review that identified studies according to prespecified inclusion criteria. Overall, based on the methods detailed in the report, the systematic literature review has an adequate search strategy, screening, and appraisal of the risk of bias of the included studies. The systematic review identified 11 primary studies for inclusion based on pre-identified study selection criteria, which were further refined on an ad hoc basis, potentially introducing selection bias. All titles, abstracts, and full texts of identified studies were screened by 2 independent reviewers and any discrepancies were resolved by a third reviewer. The main limitations of the NMA relate to data sparseness, network structure, and potential violation of the transitivity assumption. As the network was spare, fixed-effects models were used, and there was no opportunity to use meta-regression to adjust for variability in baseline characteristics and correct for potential bias. Furthermore, the evidence is imprecise in most of the effect estimates from the NMA, with wide CrIs that could include an appreciable threshold of benefit or lack of benefit. Additional sensitivity analyses were not performed due to limited data.
There were some important differences between the FIRST and NCT00067080 trials that increase the uncertainty of the NMA analyses. The FIRST trial required a higher baseline LIC (> 7 mg/g dw) than the NCT00067080 trial, which indicates a more severe iron overload. The clinical expert consulted highlighted that the exclusion of patients with an LIC exceeding 30 mg/g dw in the FIRST trial could result in the loss of a population that is nonadherent generally to iron chelators, which is likely to bias the study results in favour of DFP. The baseline patient characteristics differed between the 2 trials, with the patients enrolled in the FIRST trial appearing to have a more severe iron overload, as evidenced by the elevated SF and LIC values at baseline compared with the patients enrolled in the NCT00067080 trial, which could bias the results. Despite the described differences between the 2 studies, there does not appear to be evidence for a difference in treatment effects between DFP, DFX, and DFO with respect to change at 12 months in LIC and SF, aligning with the opinion of the clinical expert consulted. The aforementioned limitations must be considered when drawing conclusions based on the results of the NMA.
Other Relevant Evidence
Description of Studies
A total of 134 patients from the FIRST trial were enrolled in the FIRST-EXT extension study. The primary objective of the extension study was to assess the long-term safety and tolerability of DFP.
Efficacy Results
The mean change in LIC levels from baseline at year 1, year 2, and year 3 was supportive of the results from FIRST.
Harms Results
Harms were similar to the AEs reported in FIRST. A total of 104 patients (77.6%) reported AEs, with the most common being pyrexia (26.1%), bone pain (26.1%), abdominal pain (19.4%), and sickle cell crisis (18.7%), which were also reported in the pivotal trial. A total of 13 patients (9.7%) experienced serious AEs (SAEs) that were considered related to the study drug, including neutropenia (9.0%), agranulocytosis (1.5%), thrombocytopenia (0.7%), and generalized edema (0.7%). ██████████ █ ███ █ █ █ █████████ ██ ███████ █ ██████ █ █ █ █ ██████ █ █ █ █████ █ ███████████████████ █ ███ █ █ █████ █ ███ ██████████████ █ █████████████ █ ██████ █ █ ██ █ ██████████████ █ ██████████████████ █ █ █ ███████.
Critical Appraisal
Limitations of the extension study include the absence of an active comparator and the fact that potential confounders were not accounted for, which limits causal conclusions. Interpretation of some outcomes was also limited by the large amount of missing data due to attrition. Subgroup analyses were descriptive and often limited to few patients, reducing the chance of detecting a true effect. As the extension study consisted of patients who took part in the pivotal FIRST parent study, it is reasonable to expect that the same limitations related to generalizability apply to the extension study.
Conclusions
One 12-month, open-label, randomized, pivotal trial (FIRST; N = 230) demonstrated that orally administered DFP was noninferior to subcutaneously administered DFO for change from baseline in LIC, SF, and cardiac iron in patients with SCD and other anemias who require iron chelation therapy for transfusional iron overload. Despite limitations, the sponsor-submitted indirect treatment comparison (ITC) suggested that DFP also has similar efficacy for reducing LIC and SF at 12 months compared with orally administered DFX. The clinical expert consulted by CADTH indicated that evidence suggests DFP is an effective treatment option for the management of patients with SCD with transfusional iron overload. There is consensus across regulatory authorities, patient and clinician groups, and the clinical expert consulted by CADTH that DFP oral tablets and solution could help address an unmet need for patients with SCD. Cases for anticipated use include patients with renal impairment (i.e., those who cannot receive DFX), prior intolerance, or who have experienced an AE with DFX or DFO resulting in discontinuation or a dose reduction to a level that is subtherapeutic, and those who experience intolerance and/or adherence issues with IV or SC administration of DFO. Treatment with DFP may be associated with rare but serious AEs (i.e., severe neutropenia) as well as milder, more common side effects (e.g., transaminitis); typically, patients should be managed and monitored under the supervising care of health care teams with experience in the diagnosis and management of both SCD or rare anemias and transfusional iron overload. The long-term monitoring for AEs that is required for patients receiving DFP is not anticipated to be greater than current practice for patients receiving DFO or DFX.
Introduction
Disease Background
SCD is an inherited blood disorder affecting an estimated 5,000 people in Canada.1 Patients with SCD often have chronic anemia that worsens for reasons such as accelerated hemolysis, splenic sequestration, or transient red cell aplasia.5 Patients with SCD are at risk of serious vaso-occlusive events, including stroke. CanHaem’s Consensus Statement on the Care of Patients With Sickle Cell Disease in Canada recommends that patients with SCD receive red blood cell (RBC) transfusions to treat severe exacerbations of anemia and to treat and/or reduce the complications of SCD.6
In the absence of a natural mechanism to excrete excess iron in the body, patients with SCD who undergo transfusions may experience iron overload. The accumulation of excess iron may lead to free radicals and cause hepatic fibrosis, arrhythmias, congestive heart failure, renal tubular injury, and endocrinopathies.7-9 If left untreated, iron overload can lead to organ failure and/or death. Repeated transfusions lead to increasing iron concentrations and, eventually, iron overload,6 with a significant risk of overload observed in patients who receive more than 10 to 20 units of RBCs.10 In the majority of cases, liver iron overload is asymptomatic, so patients would not experience any symptom changes.
The approved indication for DFP does not identify what conditions are classified as “other anemias” for which patients require treatment for transfusional iron overload. These anemias are rare; the conditions studied in the pivotal trial for DFP included congenital dyserythropoietic anemia, pyruvate kinase deficiency, hereditary spherocytosis, hemoglobin s-beta thalassemia, dyserythropoietic anemia, autoimmune hemolytic anemia, other rare hemoglobinopathies, and chronic nonspherocytic hemolytic anemia.11
Iron burden is commonly measured through modalities such as liver MRI, cardiac MRI, and blood testing for SF levels. The clinical expert consulted by CADTH maintained that patients with SCD are unlikely to have cardiac iron deposition and indicated that SF levels alone are often an unreliable marker because factors beyond iron burden can affect SF values, for example, SCD flares, the presence of inflammation or infection, and vitamin C deficiency.12
Standards of Therapy
The use of blood transfusions can be episodic or chronic (80% and 20% of patients, respectively). With episodic transfusions, iron overload tends to begin after a number of events; ideally, it should be monitored and treated before any sequelae developing, which may mean a short period of chelation therapy that can then be discontinued once iron levels are acceptable. On the other hand, chronic transfusions consist of a prolonged period of monthly transfusions, including up to a patient’s entire lifetime.
Determination of the need, intensity, and duration of treatment for iron overload in patients with SCD is directly related to the transfusion burden. The clinical expert consulted by CADTH noted that the goal of therapy in patients with transfusional iron overload is to reduce and then maintain liver iron at an acceptable range (2 mg/g to 5 mg/g dw), essentially reducing hepatic iron to a safe level that will not lead to cirrhosis or hepatocellular carcinoma while minimizing or avoiding any drug toxicity. There are 2 approaches to addressing transfusional iron overload: switching the type of transfusion being undergone by the patient (i.e., switching from simple transfusion to exchange transfusion methods), and iron chelation therapy.
Prior to the approval of DFP for the treatment of transfusional iron overload in patients with SCD and other anemias, there were 2 iron chelation drugs marketed in Canada for use in these patients: DFO and DFX. DFO is administered as a slow infusion, either subcutaneously or intravenously. Both routes affect adherence; however, the IV route may be preferred by patients who already have an indwelling IV catheter to facilitate chronic transfusions. SCD is a prothrombotic state, so consideration should be given to anticoagulation for patients with IV catheters, adding to the overall burden of therapy. DFO IV may also be preferred by patients who struggle with daily pill compliance. Dose reduction for DFO is necessary for patients with end-stage renal disease but can be used safely in those on renal replacement therapy.
DFX is administered orally and is available in 2 dosage forms: dispersible tablets (Exjade) and film-coated tablets (Jadenu). The clinical expert consulted by CADTH noted that the primary issues with DFX are patient adherence (which can be a challenge, particularly with dispersible, but much less so with film-coated) and renal toxicity, which is 1 of the most common side effects of the medication. Given the toxicities caused by DFX, it is generally not prescribed in patients with end-stage renal disease. The clinical expert consulted by CADTH noted that patients with SCD are more likely to have renal impairment from an early stage of life compared with the patients with beta thalassemia.
Drug
Ferriprox (DFP) is an iron chelator that is approved by Health Canada for use in the treatment of patients with transfusional iron overload due to:
thalassemia syndromes when current chelation therapy is inadequate
SCD or other anemias.13
The current review is focused on the use of DFP for the treatment of patients with transfusional iron overload due to SCD or other anemias. The sponsor has requested that DFP be reimbursed in accordance with the Health Canada–approved indication.
Recommended Dosage
The recommended dose for DFP is 25 mg/kg to 33 mg/kg body weight administered orally 3 times a day for a total daily dosage of 75 mg/kg to 100 mg/kg body weight.13
Previous CADTH Reviews
DFP was previously reviewed by CADTH for the treatment of patients with transfusional iron overload due to thalassemia syndromes when current chelation therapy is inadequate. It received a recommendation in favour of reimbursement with a condition that it be reimbursed in a manner similar to DFX.14
Table 3: Key Characteristics of DFP, DFO, and DFX
Characteristic | DFP | DFO | DFX | |
|---|---|---|---|---|
Film-coated tablets | Dispersible tablets | |||
Mechanism of action | Iron-chelating drug | Iron-chelating drug | Iron-chelating drug | Iron-chelating drug |
Indicationa | Transfusional iron overload due to:
|
| Chronic iron overload:
| |
Route of administration | Oral | SC, IV, or IM | Oral | — |
Dosage form |
| 500 mg vial | Film-coated tablets: 90 mg, 180 mg, 360 mg | Dispersible tablets: 125 mg, 250 mg, and 500 mg |
Recommended dose | 25 mg/kg to 33 mg/kg body weight, orally, 3 times a day (total daily dosage of 75 mg/kg to 100 mg/kg body weight) |
|
|
|
Serious adverse effects or safety issues |
| Contraindicated in patients hypersensitive to DFO mesylate or a component of the container, except where desensitization is successful |
| |
DFO = deferoxamine; DFP = deferiprone; DFX = deferasirox; ESRD = end-stage renal disease; IM = intramuscular; RBC = red blood cell; SC = subcutaneous; SCD = sickle cell disease.
aHealth Canada–approved indication.
Sources: Product monographs.13,15-17
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups. The full original patient input received by CADTH has been included in the stakeholder section at the end of this report.
Two patient groups, the Sickle Cell Awareness Group of Ontario and the Thalassemia Foundation of Canada submitted patient input for this review. The Thalassemia Foundation of Canada reiterated the input it originally submitted to CADTH in October 2015 for the initial review of DFP. The input gathered information from various sources, including a search of the medical literature, a collection of focus group reports, clinical practice guidelines, and data from other organizations representing the interests of patients with SCD.
The Thalassemia Foundation of Canada stated that, based on published literature, it believes the symptoms of excess iron are numerous, including endocrine disorders (growth retardation, failure of sexual maturation, diabetes mellitus, and insufficiency of the parathyroid, thyroid, pituitary and, less commonly, adrenal glands), dilated cardiomyopathy, arrhythmias, liver fibrosis, and cirrhosis. The Sickle Cell Awareness Group of Ontario noted that individuals with SCD face debilitating complications not limited to vasoocclusive pain crisis and damage to their vital organs (including kidneys and liver). According to the input received, continuous blood transfusions in patients with SCD can lead to an excessive buildup of iron, causing further organ damage and increased cancer risk. The patient input cited evidence in which patients and caregivers stated that this condition disrupts their ability to work or attend school as well as their physical and social interactions.
Respondents reported having experience with injectable treatments (e.g., DFO) and oral treatments (e.g., DFX). They noted that DFO treatment has a demanding SC or IV administration schedule and can be associated with important side effects, such as local irritation, high-frequency hearing loss, deafness, retinal damage, impaired vision, growth retardation, and bone abnormalities. The patient groups also reported that oral treatments are associated with improvements in quality of life, treatment adherence, patient satisfaction, and reduction in preventable organ damage from iron overload. The patient input stated that improved heart and endocrine function, reduced risk of premature death, and ease of oral administration (obtained by treating patients with DFP) are goals that will be meaningful to patients and their families. The patient groups concluded that expanding access to appropriate iron chelation therapies such as DFP is vital to effectively improving patient outcomes.
Clinician Input
Input From the Clinical Expert Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 1 clinical specialist with expertise in the diagnosis and management of treatment of patients with transfusional iron overload due to SCD or other anemias.
Unmet Needs
The expert indicated that approved use of DFO and/or DFX IV are the 2 options available for chelation therapy in Canada (in addition to compassionate use of the drug under review). The expert noted that treatment goals are to reduce hepatic iron concentration to a safe level (i.e., where it will not lead to cirrhosis or hepatocellular carcinoma) while minimizing or avoiding any drug toxicity. The clinical expert consulted by CADTH noted there are 2 main reasons for treatment failure in hepatic iron overload: drug toxicity and nonadherence. Expanding the available options beyond the 2 existing drugs will provide the flexibility to switch therapies as circumstances change or to combine drugs when toxicity is seen at high doses of a single drug. The challenges with the 2 existing chelation therapies include difficulty in administering (i.e., those requiring an IV or SC route) or known renal toxicities. The clinical expert consulted by CADTH noted that an important exception is administering DFO intravenously to a select group of patients who are motivated and already have an existing indwelling catheter to facilitate their chronic blood transfusions. However, even under these circumstances, it can be challenging to maintain DFO IV for a prolonged period of time.
Place in Therapy
The clinical expert consulted by CADTH noted that DFP would serve as an additional chelation option for patients who are not suited to other available therapies, whether as a single drug or in combination with other therapies. For example, patients with renal dysfunction due to their SCD may have a relative contraindication to DFX.
Patient Population
The clinical expert consulted by CADTH indicated that DFP would be most suitable for patients with sickle nephropathy, poor IV access, AEs with DFX, and normal liver enzymes. Patients with the highest liver iron levels and those who have experienced high levels for the longest duration are most in need of treatment to reduce the risk of advancing fibrosis and cirrhosis. Most patients who would be candidates for chelation would be connected with a sickle cell clinic for expert management. For those patients not linked to a clinic (i.e., uninsured or undiagnosed with SCD), there is a risk of over- or under-diagnosis and a potential risk of chelator toxicity due to unfamiliarity in managing the condition in this population. The clinical expert consulted by CADTH noted that patients should not be treated empirically with iron chelators; rather, they should undergo a validated liver MRI with iron quantification when there is a history of significant transfusions and if the SF level is consistently greater than 1,000 ng/mL.
On the other hand, patients with baseline neutropenia or those receiving hydroxyurea may not be ideal candidates and may require closer monitoring due to the risk of mild and severe neutropenia with DFP. Patients with clinically significant transaminitis unrelated to iron overload may also warrant additional monitoring and care due to the known risk of transaminitis with DFP.
Monitoring efficacy with liver MRI iron quantification should be carried out approximately once a year while on therapy.
Assessing Response to Treatment
The clinical expert noted that outcomes in clinical practice are consistent across Canada’s SCD clinics and aligned with clinical trial data. The clinical expert consulted by CADTH noted that the most reliable efficacy outcome is liver iron quantification using a validated MRI technique. In rare cases, an MRI may not be possible, in which case a liver biopsy is required to quantify the iron levels. Trends in SF can provide a surrogate for a more frequent marker of success, although SF levels are prone to being influenced by SCD disease flares. The goal of therapy is to reduce and then maintain liver iron at an acceptable range (2 mg/g dw to 5 mg/g dw). Most patients will require an MRI every 6 to 24 months while on therapy, depending on their degree of iron overload and ongoing transfusion burden. In the vast majority of cases, liver iron overload is asymptomatic, so patients would not experience any symptom changes. As equally important as efficacy is lack of toxicity; therefore, patients should be monitored for signs of toxicity, including any new symptoms that might arise while on the drug.
Discontinuing Treatment
Per the clinical expert, DFP should be discontinued when: iron stores have reached the target range and there is no ongoing iron loading, during pregnancy and lactation, and in the presence of clinically significant toxicity that cannot be safely rechallenged. In addition, if a patient undergoes curative treatment, therapeutic phlebotomy could be introduced instead of DFP if the hemoglobin level permits. Similarly, a patient switching from chronic transfusions to automated exchange may de-iron without the need for chelation, though this is a highly variable and patient-specific phenomenon that is not well understood. Finally, the addition of other drugs that interact or have a toxicity profile that is similar to DFP should trigger a reassessment of the risk and benefit of continuing DFP (e.g., when starting treatment for a new cancer with a chemotherapy drug known to cause neutropenia).
Prescribing Conditions
The clinical expert consulted by CADTH noted that a pediatric or adult hematologist should supervise the treatment of transfusional iron overload either themselves or in collaboration with another provider type.
Additional Considerations
The largest group of patients within the target population for treatment with DFP are those with SCD. In Canada, SCD predominantly affects individuals who identify as Black. The Sickle Cell Awareness Group of Ontario and the clinicians who provided input to CADTH note that racialized communities experience health disparities and face significant barriers to health equity. The clinical expert consulted by CADTH noted that societal factors should be considered when evaluating the additional needs of those living with SCD and their caregivers to achieve optimal care outcomes and health.
Clinician Group Input
This section was prepared by CADTH staff based on the input provided by clinician groups. The full original clinician group input received by CADTH has been included in the stakeholder section at the end of this report.
Five clinicians from CanHaem provided input. The clinician group stated that transfusional iron overload is a clinically silent condition that requires appropriate long-term care to manage effectively. The group noted there are currently 2 treatments available for transfusional iron overload: DFO (SC or IV continuous) or DFX (oral). The clinician group suggested that not all patients respond to currently available treatments and many patients experience side effects with iron chelators and can only tolerate 1 type of medication. Renal dysfunction is a common complication in patients with SCD; consequently, some of these patients may not tolerate DFX, while the SC or IV administration of DFO may not be a feasible treatment option for others. The clinician group highlighted the need for more treatment options and for treatments that are better tolerated and improve compliance and convenience. In terms of place in therapy, the group noted that the treatment would be suitable for patients with SCD who are on transfusion therapy and demonstrating iron overload, particularly those who are unable to tolerate SC drug infusion, those with liver or kidney dysfunction, and those not at risk of neutropenia. A clinically meaningful response to treatment was defined as the maintenance of or decrease in iron burden over time. The submission indicated that the treatment should be managed by a hematologist (pediatric or adult, as appropriate).
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
Would DFX (Jadenu, Exjade, and generics), the other oral iron chelator therapy, have been a better comparator than the SC product DFO in FIRST and FIRST-EXT? | The clinical expert consulted by CADTH agreed that DFX would have been a better comparator than the DFO (SC) in the FIRST and FIRST-EXT studies. |
Considerations for continuation or renewal of therapy | |
Is there a response with SF, LIC, and/or CIC that would support continuation (or discontinuation) of therapy? | The clinical expert noted that appropriate reimbursement criteria to support renewal of DFP would be reduction in absolute or maintenance of low concentrations of LIC, CIC, or SF. |
Are there other considerations that could be used to evaluate whether therapy should be continued or renewed? | The clinical expert consulted by CADTH noted that other considerations used to evaluate whether therapy should be continued or renewed may include the presence of clinically significant AEs or SAEs and unanticipated gaps in medication use (e.g., due to illness or pregnancy). |
Considerations for the prescribing of therapy | |
Is adherence to DFP an issue due to its pill burden and/or frequency of administration? | The clinical expert consulted by CADTH noted adherence may be a challenge when the daily dosage is administered over 3 doses. |
Is there any experience for giving the total daily dosage of DFP in 2 doses rather than 3? If so, has this been successful in your practice? | The clinical expert consulted by CADTH noted there is clinical experience providing the total daily dosage of DFP as 2 doses per day rather than 3. The twice-daily regimen can be associated with greater convenience for patients and improved adherence to the treatment. |
Care provision issues | |
Are there any concerns with continuing to limit coverage of DFP to hematologists? Are there other specialties that should be able to obtain coverage (use) of DFP? | The clinical expert consulted by CADTH identified the concern of timely access to a pediatric or adult hematologist if coverage of DFP continues to be limited to hematologists. The clinical expert added that there is not usually an urgent need for chelation for SCD, and most SCD clinics would prioritize based on clinical need, though a patient’s uninsured status would impact this. The clinical expert indicated that other providers or specialists should not be directing care (i.e., the initial decision or request) but could act with support from hematologists. |
Is combination chelation therapy used in clinical practice? If so, how often and is it effective? | The clinical expert noted that combination chelation therapy is used in Canadian clinical practice. |
Is weekly blood work a significant burden for patients and clinicians with this life-long therapy? Are there any scenarios where the frequency of monitoring may be reduced? For example, if a patient was treated with DFP for a number of years without any AEs? | The clinical expert consulted by CADTH noted that weekly CBCs would be extremely burdensome for patients and caregivers and are unlikely to be performed in routine clinical practice for patients receiving longer-term treatment with DFP. The clinical expert added that patients with SCD who are receiving regular transfusions or are receiving hydroxyurea medication are also getting their CBCs monitored at each visit. The clinical expert indicated that in routine clinical practice, clinics may encourage weekly monitoring of CBC for the first 6 months of treatment; thereafter, it may be as often as their transfusions occur. The expert noted that patients and caregivers would be educated about the signs and symptoms of febrile neutropenia and instructed on when to seek medication attention and to ensure that the health care provider is aware they are receiving medication that poses a risk of neutropenia (e.g., by presenting the wallet card that is provided with DFP). |
How often are SF, LIC, and CIC monitored in routine clinical practice for patients with SCD? Are other labs or diagnostic tests required that were not mentioned by the sponsor (of note, zinc levels were also recommended by the sponsor and UpToDate)? | The clinical expert consulted by CADTH noted that LIC is measured yearly, with some variation depending on the degree of iron overload, either plus or minus 6 to 12 months. SF is measured every month if patients are being treated with DFO; otherwise, it is measured every 3 months. CIC may be checked at baseline and only repeated intermittently, as cardiac iron overload is uncommon with patients who have SCD. According to the expert, LIC, CIC, and SF concentration would be the only tests conducted to assess efficacy in this population. Furthermore, with respect to monitoring zinc levels, the expert indicated there is variation across routine clinical practice. |
AE = adverse event; CBC = complete blood count; CIC = cardiac iron concentration; DFO = deferoxamine; DFP = deferiprone; DFX = deferasirox; LIC = liver iron concentration; SAE = serious adverse event; SC = subcutaneous; SCD = sickle cell disease; SF = serum ferritin.
Clinical Evidence
The clinical evidence included in this review of DFP is presented in 3 sections. The first section, the systematic review, includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those studies that were selected according to an a priori protocol. The second section includes indirect evidence from the sponsor and indirect evidence selected from the literature that met the selection criteria specified in the review. The third section includes sponsor-submitted long-term extension studies and additional relevant studies that were considered to address important gaps in the evidence included in the systematic review.
Systematic Review (Pivotal and Protocol-Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of DFP (1,000 mg tablet and/or 100 mg/mL oral solution) for the treatment of patients with transfusional iron overload due to SCD or other anemias.
Methods
Studies selected for inclusion in the systematic review include pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 5. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
Table 5: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Patient population | Patients with transfusional iron overload due to SCD or other anemias. Subgroups:
|
Intervention | DFP (recommended dose of 25 mg/kg to 33 mg/kg body weight, orally, 3 times a day for a total daily dosage of 75 mg/kg to 100 mg/kg body weight) |
Comparators | Monotherapy or in combination:
|
Outcomes | Efficacy outcomes:
Harms outcomes:
|
Study design | Published and unpublished phase III and IV RCTs |
AE = adverse event; DFO = deferoxamine; DFP = deferiprone; DFX = deferasirox; HRQoL = health-related quality of life; RCT = randomized controlled trial; RBC = red blood cell; SAE = serious adverse event; SC = subcutaneous; SCD = sickle cell disease; WDAE = withdrawal due to adverse event.
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy according to the PRESS Peer Review of Electronic Search Strategies checklist.18
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946—) through Ovid and Embase (1974—) through Ovid. All Ovid searches were run simultaneously as a multifile search. Duplicates were removed using Ovid deduplication for multifile searches followed by manual deduplication in EndNote. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concept was Ferriprox (DFP). Clinical trial registries were searched: the US National Institutes of Health’s clinicaltrials.gov, WHO’s International Clinical Trials Registry Platform (ICTRP) search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
CADTH-developed search filters were applied to limit retrieval to RCTs or controlled clinical trials. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. Refer to Appendix 1 for the detailed search strategies.
The initial search was completed on July 5, 2022. Regular alerts updated the search until the meeting of the CADTH Canadian Drug Expert Committee (CDEC) on November 23, 2022.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool For Searching Health-Related Grey Literature checklist.19 Included in this search were the websites of regulatory agencies (FDA and European Medicines Agency). Google was used to search for additional internet-based materials. Refer to Appendix 1 for more information on the grey literature search strategy.
These searches were supplemented by reviewing bibliographies of key papers and through contact with appropriate experts. In addition, the sponsor of the drug was contacted for information regarding unpublished studies.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review and differences were resolved through discussion.
Findings From the Literature
A total of 2 studies were identified from the literature for inclusion in the systematic review (Figure 1). The included studies are summarized in Table 6. There were no excluded studies.
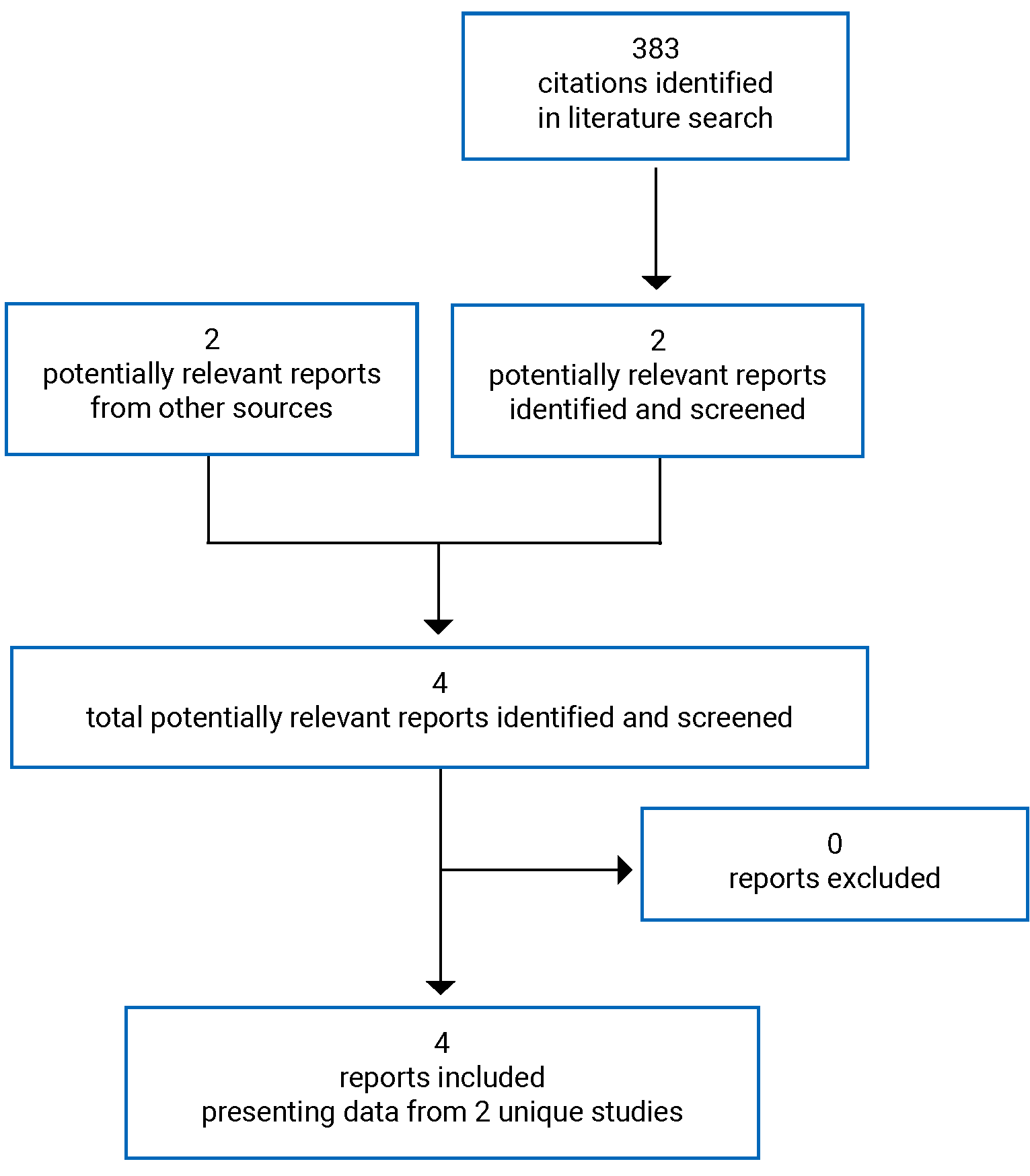
Table 6: Details of Included Studies
Characteristic | FIRST | Calvaruso et al. (2014)3 |
|---|---|---|
Designs and populations | ||
Study design | Late-phase (phase IV in the US, phase IIIb in other countries), multicentre, open-label, RCT | Multicentre, randomized, open-label study |
Locations | 27 sites in Egypt, US, Brazil, UK, Saudi Arabia, Tunisia, Canada, and Turkey | 9 sites in Italy |
Patient enrolment dates: | April 17, 2014, to November 30, 2019 | January 2001 and May 2011 |
Randomized (N) | 230; DFP = 152; DFO = 78 | 60; DFP = 30; DFO = 30 |
Inclusion criteria |
|
|
Exclusion criteria |
|
|
Drugs | ||
Intervention | DFP at a total daily dosage of 75 mg/kg to 99 mg/kg of body weight, divided t.i.d. 8 hours apart 7 days a week depending on iron load severity). Patients were titrated to the target dosage over 3 weeks | DFP at 75 mg/kg per day, divided into 3 oral daily doses, for 7 days per week |
Comparator | DFO SC infusion over 8 to 12 hours, 5 to 7 days a week as a total daily dosage of 20 mg/kg (children) or up to 40 mg/kg (adults) for less severe iron load; those with more severe iron load were prescribed a daily dosage of up to 40 mg/kg (children) or 50 mg/kg (adults) | DFO SC infusion (8 to 10 hours) at 50 mg/kg per day for 5 days per week |
Duration | ||
Phase | ||
Run-in | 7-day washout period | 1 week washout |
OL treatment | Up to 12 months | Up to 5 years |
Follow-up | 30 days or entry in long-term extension phase | Additional 5 years |
Outcomes | ||
Primary end point | Change from baseline to month 12 in LIC, as measured by MRI. | Change from baseline value in SF levels during the 5 years. Treatment failure was defined as an increase in SF levels to more than 1,000 ng/mL from baseline confirmed in at least 2 consecutive determinations. |
Secondary and exploratory end points |
| Safety and survival analysis at 5 years |
Notes | ||
Publications | Kwiatkowski et al. (2021)20 | Calvaruso et al. (2014)3 |
AE = adverse event; ALT = alanine aminotransferase; CHQ = Child Health Questionnaire; CIC = cardiac iron concentration; DFO = deferoxamine; DFP = deferiprone; dw = dry weight; ECG = echocardiogram; LIC = liver iron concentration; MDS = myelodysplastic syndrome; ms = milliseconds; OL = open-label; RBC = red blood cell; RCT = randomized controlled trial; SAE = serious adverse event; SC = subcutaneous; SCD = sickle cell disease; SF = serum ferritin; SF-36 = Short Form (36) Health Survey; t.i.d. = 3 times daily; ULN = upper limit of normal; WDAE = withdrawal due to adverse event.
Note: Additional reports included the Clinical Study Report for FIRST2 and the Health Canada Pharmaceutical Safety and Efficacy Assessment.11
Sources: FIRST Clinical Study Report2 and Calvaruso et al. (2014).3
Description of Studies
FIRST Trial
The FIRST trial was a late-phase (phase IV in the US; phase IIIb in other countries), multicentre, randomized, open-label study comparing the efficacy and safety of the iron chelator DFP versus DFO in patients with SCD or other transfusion-dependent anemias. Eligible patients were randomized in a 2:1 ratio to receive either DFP or DFO for up to 12 months. Randomization was stratified by disease category (SCD versus other anemias) and transfusional iron input in the 3 months before baseline (less than or equal to 0.3 mg/kg/day versus more than 0.3 mg/kg/day).
Patients visited the study sites monthly for evaluations of efficacy and/or safety and, additionally, underwent weekly or biweekly monitoring for hematology at a local laboratory. Safety assessments were performed at each site visit; SF was measured quarterly, and assessments of LIC, cardiac iron, and quality of life were carried out at baseline, month 6, and month 12. Patients who completed the 12 months of treatment were eligible to enrol in a 2-year extension study, FIRST-EXT, in which all participants received DFP.
Calvaruso et al. (2014)
Calvaruso et al. (2014) conducted a 5-year, multicentre, open-label RCT to compare the safety and efficacy of DFP versus DFO in Italian patients. Eligible patients were randomized in a 1:1 ratio to receive either DFP or DFO for up to 12 months.3
Populations
Inclusion and Exclusion Criteria
FIRST Trial
Patients eligible for enrolment in the FIRST trial included those at least 2 years of age with a diagnosis of SCD or a transfusion-dependent anemia with a baseline LIC exceeding 7 mg/g dw (measured by MRI) who had received at least 20 blood transfusions (at least 1 per year in the 2 years before screening) and who were expected to require blood transfusions through the planned duration of the trial. Patients were excluded if they had a diagnosis of a thalassemia syndrome, myelodysplastic syndrome, myelofibrosis, or Diamond-Blackfan anemia; primary bone marrow failure; treatment with hydroxyurea within 30 days of screening; a baseline LIC measurement exceeding 30 mg/g dw; and a baseline cardiac T2* MRI of less than 10 ms.
Calvaruso et al. (2014)
Patients eligible for enrolment in the Calvaruso et al. (2014) study included those who were aged 13 years or older with a diagnosis of SCD based on accepted clinical and molecular criteria (these were not defined in the publication) and an SF concentration of between 800 ng/mL and 3,000 ng/mL. Patients were excluded if they had a platelet count of less than 100,000/µL or a leucocyte count of less than 3,000/µL, Child-Pugh grade C classification of hepatic impairment, sepsis, or overt heart failure.3
Baseline Characteristics
FIRST Trial
The baseline and demographic characteristics for the FIRST trial are summarized in Table 7. Baseline characteristics were generally balanced across both treatment groups, with the exception of sex (DFP = 42.1% female; DFO = 52.2% female). The mean age was 16.2 years (SD = 9.5) in the DFP group and 16.5 years (SD = 8.0) in the DFO group. The majority of patients had a primary diagnosis of SCD (82.9% in both groups) and had not undergone prior chelation therapy (48.7% in the DFP group versus 42.1% in the DFO group).2
Table 7: Summary of Baseline Characteristics in FIRST Trial (ITT Set)
Characteristic | DFP | DFO |
|---|---|---|
Age (years), mean (SD) | 16.2 (9.5) | 16.5 (8.0) |
Sex, n (%) | ||
Female | 56 (42.1) | 36 (52.2) |
Male | 77 (57.9) | 33 (47.8) |
Ethnicity, n (%) | ||
Hispanic or Latino | 10 (7.5) | 5 (7.4) |
Other | 123 (92.5) | 64 (92.8) |
Race, n (%) | ||
Black | 22 (16.5) | 14 (20.3) |
Multiracial | 9 (6.8) | 5 (7.2) |
White | 102 (76.7) | 50 (72.5) |
Primary diagnosis,a n (%) | ||
Autoimmune hemolytic anemia | 1 (0.7) | 1 (1.3) |
Congenital anemia | 1 (0.7) | 1 (1.3) |
Congenital dyserythropoietic anemia | 4 (2.6) | 3 (3.9) |
Hemoglobin C disease | 2 (1.3) | 1 (1.3) |
Hemoglobinopathy | 1 (0.7) | 0 (0.0) |
Hemolytic anemia | 1 (0.7) | 0 (0.0) |
Pyruvate kinase deficiency anemia | 2 (1.3) | 1 (1.3) |
SCD | 126 (82.9) | 63 (82.9) |
Spherocytic anemia | 14 (9.2) | 6 (7.9) |
Prior chelation therapy 3 months before baseline,a n (%) | ||
DFP | 28 (18.4) | 19 (25.0) |
DFO | 25 (16.4) | 17 (22.4) |
DFX | 38 (25.0) | 17 (22.4) |
No chelation | 74 (48.7) | 32 (42.1) |
Transfusion iron input (mg/kg/day) 3 months before baseline,a n (%) | ||
N | 152 | 76 |
Mean (SD) | 0.18 (0.15) | 0.20 (0.18) |
DFO = deferoxamine; DFP = deferiprone; DFX = deferasirox; ITT = intention-to-treat; SCD = sickle cell disease; SD = standard deviation.
aSafety population; N = 152 for DFP and N = 76 for DFO.
Sources: FIRST Clinical Study Report2 and Kwiatkowski et al. (2021).20
Calvaruso et al. (2014)
The baseline and demographic characteristics for the Calvaruso et al. (2014) study are summarized in Figure 2. The groups were generally balanced, with the notable exception of the proportion of patients with splenectomy. Total blood transfusion values were lower in the DFP group compared with the DFO group (2,055.05 ± 1,282.01 versus 2,797.15 ± 2,018.08, respectively). In the DFP group, 45.4% of patients had undergone splenectomy versus 70.6% in the DFO group.3
Figure 2: Baseline Characteristics in Calvaruso et al. (2014) (N = 60)
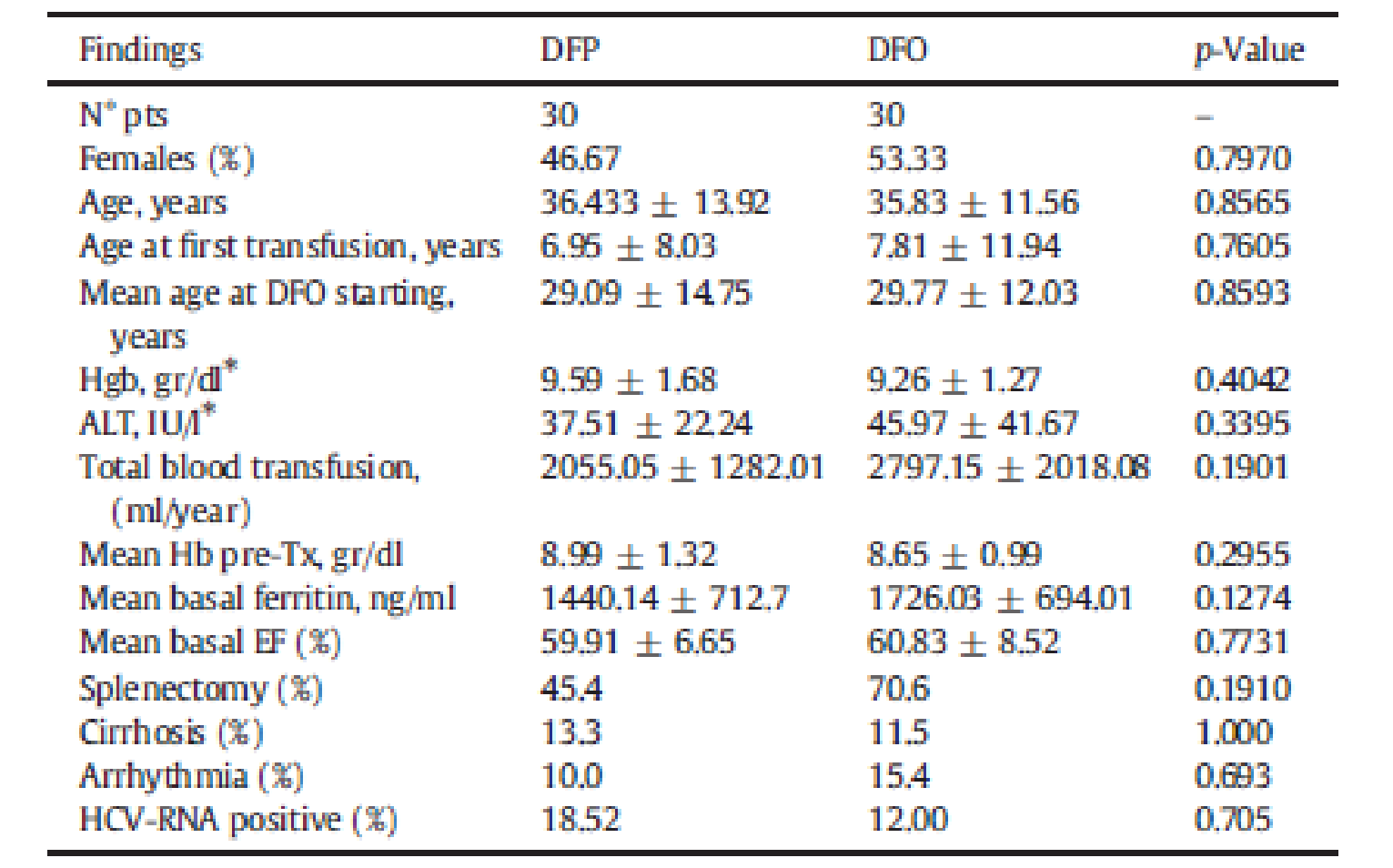
ALT = alanine aminotransferase; DFO = deferoxamine; DFP = deferiprone; EF = ejection fraction; Hb = hemoglobin; HCV = hepatitis C virus; Hgb = hemoglobin; RNA = ribonucleic acid.
Source: Calvaruso et al. (2014).3
Interventions
FIRST Trial
Patients were classified as having less severe iron overload at baseline if they met all of the following criteria at baseline: transfusional iron input of 0.3 mg/kg per day or greater, SF of less than 2,500 mcg/L, LIC of less than 15 mg/g dw, or cardiac T2* greater than 20 ms. Patients who failed to meet 1 or more of these criteria (i.e., they had a transfusional iron input of > 0.3 mg/kg per day and/or SF ≥ 2,500 mcg/L and/or an LIC of ≥ 15 mg/g dw and/or a cardiac T2* ≤ 20 ms) were deemed to have more severe iron overload.2
Deferiprone
DFP was administered for up to 12 months at a total daily dosage of 75 mg/kg to 99 mg/kg of body weight divided into 3 daily doses 8 hours apart 7 days a week depending on iron load severity. Patients could receive either of the following formulations depending on their preference and product availability: DFP immediate-release 500 mg tablets or DFP 80 mg/mL oral solution. Titration for all patients in the DFP group started at a dose of 15 mg/kg 3 times daily for week 1, increased to 20 mg/kg 3 times daily for week 2, and further increased to 25 mg/kg body weight 3 times daily in week 3. Patients remained at the 25 mg/kg 3 times daily dosage for the remainder of the 52-week treatment period unless they met the criteria for severe iron overload, in which case they received 33 mg/kg body weight 3 times daily starting in week 4.2
Deferoxamine
DFO was administered through SC infusion over 8 to 12 hours, 5 to 7 days a week, with a daily dose of 20 mg/kg (for children) for less severe iron load or 40 mg/kg (for adults) for less severe iron load. Those with a more severe iron load were prescribed it at a dosage of up to 40 mg/kg (children) or 50 mg/kg (adults).2
Calvaruso et al. (2014)
Patients randomized to DFP received 75 mg/kg per day divided into 3 oral doses for 7 days per week. Those randomized to DFO received 50 mg/kg per day for 5 days per week by SC infusion (8 to 10 hours). Treatment was administered for 5 years.
Outcomes
A list of efficacy end points identified in the CADTH review protocol that were assessed in the clinical trials included in this review is provided in Table 8. These end points are further summarized subsequently. A detailed discussion and critical appraisal of the outcome measures is provided in Appendix 3.
Table 8: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure | FIRST | Calvaruso et al. (2014) |
|---|---|---|
Change from baseline in LIC | Primary | NA |
Change from baseline in CIC | Secondary | NA |
Change from baseline in SF | Secondary | Primary |
Change from baseline in SF-36 | Secondary | NA |
Change from baseline in CHQ-PF50 and CHQ-CF87 | Secondary | NA |
Survival | NA | Secondary |
CHQ-CF87 = Child Health Questionnaire Child Form 87 Questions; CHQ-PF50 = Child Health Questionnaire Parent Form 50 Questions; CIC = cardiac iron concentration; HRQoL = health-related quality of life; LIC = liver iron concentration; NA = not applicable; SF = serum ferritin; SF-36 = Short Form (36) Health Survey.
Source: FIRST Clinical Study Report.2
Liver Iron Concentration
In the FIRST trial, MRI scans for the determination of LIC were performed at screening or baseline, month 6, and month 12 (or early termination), and were transmitted to a central laboratory for interpretation.
Cardiac Iron Concentration
In the FIRST trial, MRI scans for the assessment of cardiac MRI T2* were performed at screening or at baseline, month 6, and month 12 (or early termination), and the images were transmitted to a central laboratory for interpretation.
SF Concentration
In the FIRST trial, SF was assessed at baseline and at months 3, 6, 9, and 12 (or early termination). In the Calvaruso et al. (2014) study, SF was assessed yearly for 5 years.
Short Form (36) Health Survey
In the FIRST trial, HRQoL was measured using the Short Form (36) Health Survey (SF-36) and the Child Health Questionnaire (CHQ). The SF-36 is a 36-item questionnaire that yields an 8-scale profile of functional health and well-being scores as well as psychometrically based physical and mental health summary components and a preference-based health utility index. It is a generic measure. Higher scores represent better health and the minimal important difference (MID) for either the summary components of the SF-36 is typically between 2.5 points and 5 points; however, this estimate is not specific to the SCD population.21-23 The summary scales are scored using norm-based methods, with regression weights and constants derived from the general US population. Both the physical component and mental component summary scales are transformed to have a mean of 50 (SD = 10) in the general US population. Therefore, all scores above or below 50 are considered above or below average for the general US population.24 The SF-36 questionnaire was completed by patients and/or parents or guardians at baseline, month 6, and month 12 (or early termination).
Child Health Questionnaire (CHQ-PF50)
The CHQ is a group of generic quality-of-life instruments that have been designed and normed for children aged 17 years and older. It measures 14 unique physical and psychosocial concepts. The 14 domains covered in the CHQ include: physical function, role/social limitations due to physical problems, general health perceptions, bodily pain/discomfort, family activities, role/social limitations due to behavioural problems, role/social limitations due to emotional problems, impact on parent time, impact on parent emotions, self-esteem, mental health, general behaviour, family cohesion, and change in health.25 Each item has a 4- to 6-point response scale reported as levels of intensity or agreement.25 Scores for each domain can be transformed to a total score from 0 to 100, with higher scores indicating better HRQoL.26 Only the Child Health Questionnaire Parent Form 50 Questions (CHQ-PF50) also provides 2 summary scores for physical and psychosocial health.25 No MIDs were found in the SCD population. The versions used in this study were the CHQ-PF50, which contains 50 questions, and the Child Form 87 (CHQ-CF87), which contains 87 questions. When possible, both versions were completed: the CHQ-PF50 by parents of any patient younger than 18 years, and the CHQ-CF87 was completed by child patients aged 10 years or older. The CHQ was completed by patients and/or parents or guardians at baseline, month 6, and month 12 (or early termination).
Survival
Survival was a secondary end point in the study conducted by Calvaruso et al. (2014). The Kaplan-Meier method was used to estimate the survival probability for the 2 treated groups.3
Statistical Analysis
The following is a brief description of the statistical analyses conducted for each included study.
Power Calculation and Noninferiority Margin
For the FIRST trial, the investigators planned to enrol 300 patients (200 in the DFP group and 100 in the DFO group) and at least 80% had to have a diagnosis of SCD. For the primary end point, the investigators estimated that 300 patients would provide more than 95% power for showing noninferiority of DFP to DFO based on a noninferiority margin of 2 mg/g dw for the reduction of LIC and a 2-sided alpha of 0.05 (or a 1-sided alpha of 0.025). For reference, the investigators used patients with thalassemia receiving regular RBC transfusions, where such patients’ LIC could increase by 5 mg/g dw after 1 year without chelation therapy. Citing that the cause of iron overload is the same in thalassemia and SCD, the trial investigators claimed that the same increase in LIC may be expected if patients with SCD are transfused at the same rate and not treated with a chelator. The difference in reduction of LIC between DFO therapy and a placebo (effect size) was estimated to be 9 mg/g dw (i.e., 4 minus −5), where the noninferiority margin of 2 mg/g dw was about 20% of the effect size. Due to difficulties in recruitment, the number of patients actually enrolled was 228 (152 for DFP and 76 for DFO). The investigators conducted another power analysis with the same noninferiority margin for the reduction of LIC with a 2-sided alpha level of 0.0399 (Pocock alpha spending method), which indicated that this sample size would be sufficient to meet the primary end point.2
In Calvaruso et al. (2014), the investigators conducted a sample size estimation, based on Rochon et al. (1991) calculations for 2-group repeated-measures experiments, with the recommended number of patients ranging from 10 and 100.3,27 The minimum number of patients required in each treatment group was calculated assuming equal allocation under the hypothesis of equality at every point for the autoregressive correlation structure, for a 2-sided test at an alpha of 0.05, a beta of 0.80, and a delta of 0.41 (standardized effect; P = 0.60), and number of follow-up measurements (t = 5).3
Statistical Test
The primary outcome in the FIRST trial was change in LIC at 12 months from baseline, analyzed using an ANCOVA model using both the ITT and PP populations. In the ANCOVA model, treatment was the main factor, and overall average transfusion iron input during the study and the baseline iron load measure were stratification factors. It was prespecified that the noninferiority of DFP versus DFO was shown if the upper limit of the 96.01% CI of the difference between the 2 groups was 2 mg/g dw or greater. The 96.01% CI of the difference between the treatment groups in the change in LIC from baseline to month 12 was computed. The confidence level of 96.01% or 0.9601 was determined by 1 minus alpha, where an alpha of 0.0399 was based on the Pocock alpha spending function for the interim analysis. The safety analysis set was used to produce the differences in LSM change in LIC at month 6 and month 12 between the DFP and DFO groups and the corresponding 96.01% CIs. There was no adjustment for type I error because the investigators cited that all efficacy outcomes involved a comparison of only 2 treatment groups and each outcome assessed a different measure of iron load in the patients.2
In Calvaruso et al. (2014), statistical significance was declared when the P value was less than 0.05, with all tests being 2-sided. Baseline descriptive statistics were presented for each variable in the study. For categorical variables, the number and percentage were calculated for nonmissing data, and the difference in the treatment arms was compared using a Fisher exact test. The mean and SD values were derived for continuous variables, and the comparisons between the baseline mean difference in the 2 intervention groups were based on a t-test. For SF data, a linear mixed-effects model was used, wherein an autoregressive (AR) correlation of order 1, AR (1) structure was considered to model correlation within repeated observations.3
Data Imputation Methods
For all outcomes in the FIRST trial, the last observation carried forward (LOCF) method was used to fill in missing data for patients who dropped out of the study early. In the statistical analysis plan, for patients with early termination due to worsening of disease conditions or inadequate efficacy of the drug, as judged by assessing the AE log, the “worst value” method was expected to be used (i.e., the worst value of all patients from the corresponding treatment group was used to impute the missing data at that time point). Since there were no patients with early termination due to worsening of disease conditions or inadequate efficacy of the drug in this interim analysis dataset, this method was not applicable.2
In Calvaruso et al. (2014), missing scores for all outcomes were not included and observed case analyses were used.3
Subgroup Analyses
The only subgroup for efficacy analyses in FIRST was disease type (SCD versus other anemias). The majority of patients had a primary diagnosis of SCD (82.9%). There was no adjustment for type I error. For the subgroup analysis of only those patients with SCD, the investigators indicated that the power was 82% and there should be a 98% chance of the occurrence of at least 1 SAE with an anticipated incidence rate of 2%, or an 88% chance for an anticipated incidence rate of as low as 1%.2
In Calvaruso et al. (2014), subgroup analyses were conducted but were not consistent with the CADTH protocol and therefore not reported.3
Sensitivity Analyses
Two sensitivity analyses were performed on the primary efficacy outcome in the FIRST trial. The first analysis excluded any patients who had withdrawn before providing the month 6 data to assess the impact of early withdrawal on the results of the ITT analysis. The second analysis included all evaluable patients; however, whereas the ITT analysis used the LOCF method to impute missing data in both treatment groups, this analysis used a “worst-case” scenario, with LOCF being used only for dropouts in the DFO group, while the highest (i.e., worst) LIC value observed at the missing visit (month 6 or month 12) among the DFP patients was used for dropouts in that group, thereby producing the worst-case scenario for DFP recipients. For HRQoL data, sensitivity analyses were performed on datasets that included estimated scores using multiple imputation.2
In Calvaruso et al. (2014), information about sensitivity analyses was not reported.3
Secondary Outcomes of the Studies
The secondary outcomes in the FIRST trial were change from baseline to month 12 in cardiac MRI T2* and SF. The values for cardiac MRI T2* were log-transformed for normalization of the data. There was no adjustment for type I error.
The secondary outcome in Calvaruso et al. (2014) was survival. The Kaplan-Meier method was used from January 30, 2001, to January 30, 2006. The survival curves were compared using the log-rank test.3
Analysis Populations
In the FIRST trial, there were 3 analysis populations: ITT, PP, and safety. The ITT population was the primary analysis population for all efficacy end points and included all patients who had either completed or withdrawn from the study by December 17, 2018 (if withdrawn, they had to have undergone at least 1 postbaseline efficacy assessment). The PP population was the secondary analysis population for the primary end point and was used for assessments of the other efficacy end points, as well. It included all enrolled patients who had completed the study by December 17, 2018, had no major protocol violations, and had a measure of LIC at month 12. Major protocol violations were determined before database lock, and any patients with such violations were excluded from this population. The safety population included all enrolled patients who received at least 1 dose of the study drug. An interim analysis was conducted on the data of patients who were evaluable as of the cut-off date of December 17, 2018, which were predefined as the pivotal efficacy findings.
Protocol Amendments and Deviations
Major protocol deviations in the FIRST trial are summarized in Table 9. The most common deviation was noncompliance with treatment (DFP = 35.5%; DFO = 25.6%).
Table 9: Major Protocol Deviations in FIRST Trial
Deviation, n (%) | FIRST | ||
|---|---|---|---|
DFP | DFO | Overall | |
Noncompliance | ████████ | ████████ | █ █ ████████ |
Missing the end-of-study MRI | █ █ ████████ | █████ | ████████ |
Dosing error | ███████ | █ █ █████ | ███████ |
Violation of inclusion or exclusion criteria | █████ | █████ | █ █ █████ |
Problem with informed consent | █ █ █████ | █ █ █████ | █ █ █████ |
Extended drug exposure period | █ █ █████ | █ █ █████ | █████ |
Delayed end-of-study MRI | █ █ █████ | █████ | █ █ █████ |
Randomization error | █████ | █████ | █ █ █████ |
DFO = deferoxamine; DFP = deferiprone.
Source: FIRST Clinical Study Report.2
The Calvaruso et al. (2014) study did not report data on protocol amendments and deviations.3
Results
Patient Disposition
FIRST Trial
Patient disposition in the FIRST trial is summarized in Table 10. In total, 439 patients were screened; 209 failed screening.20 A total of 230 patients were randomized: 152 to the DFP group and 78 to the DFO group. The total number of patients, 230, does not reflect the total number of unique patients enrolled because 2 patients each registered twice at different study sites, with 1 randomized both times to the DFP arm and the other to DFP and DFO. Further, 2 patients who were assigned to the DFO group withdrew before receiving any treatment. Overall, the completion rates in the groups were similar (69.7% of DFP patients versus 74.4% of DFO patients). The most common reasons for discontinuation in both groups were patient request, protocol deviation, and AE. Ten ongoing patients were withdrawn by the sponsor when the decision was made to terminate the study. The number of patients in the ITT populations varied according to the outcome evaluated, as shown in Table 10.2
Table 10: Patient Disposition in FIRST Trial
Disposition, n (%) | FIRST | |
|---|---|---|
DFP | DFO | |
Screened, N | 439 | |
Randomized, N (%) | 152 | 78 |
Exposed, N (%) | 152 (100.0) | 76 (97.4) |
Discontinued from study, N (%) | 46 (30.3) | 18 (23.1) |
Reason for discontinuation, N (%) | ||
Patient request | █ █ ███████ | █ █ █████ |
Protocol deviation | ███████ | █ █ █████ |
Sponsor decision | █████ | █ █ █████ |
Investigator decision | █████ | █████ |
AEs | █████ | █ █ █████ |
Lost to follow-up | █████ | █████ |
Other | █ █ █████ | █████ |
ITT, N (%)a | 143 | 74 |
LIC | █ ███████ | ████████ |
Cardiac MRI T2* | █ ██ █ ██████ | ████████ |
SF | █ ██ █ ██████ | ████████ |
PP, N (%)a | 69 | 47 |
LIC | 69 (100.0) | 47 (100.0) |
Cardiac MRI T2* | 69 (100.0) | 47 (100.0) |
SF | 69 (100.0) | 47 (100.0) |
Safety, N | 152 | 76 |
AE = adverse event; DFO = deferoxamine; DFP = deferiprone; ITT = intention-to-treat; LIC = liver iron concentration; PP = per-protocol; SF = serum ferritin.
aPopulations noted were part of the final efficacy analysis, not limited to patients who were evaluable as of December 17, 2018 (i.e., the interim analysis).
Source: FIRST Clinical Study Report.2
Calvaruso et al. (2014)
Patient disposition in the Calvaruso et al. study is summarized in Figure 3. In total, 94 patients were assessed for eligibility. The majority of patients refused to participate (n = 30) or failed to meet the inclusion criteria (n = 4); the remaining 60 were randomized into the study. Patients were allocated 1:1 into the DFO (n = 30) and DFP groups (n = 30). No patients were lost to follow-up.3
Figure 3: Patient Disposition in Calvaruso et al. (2014)
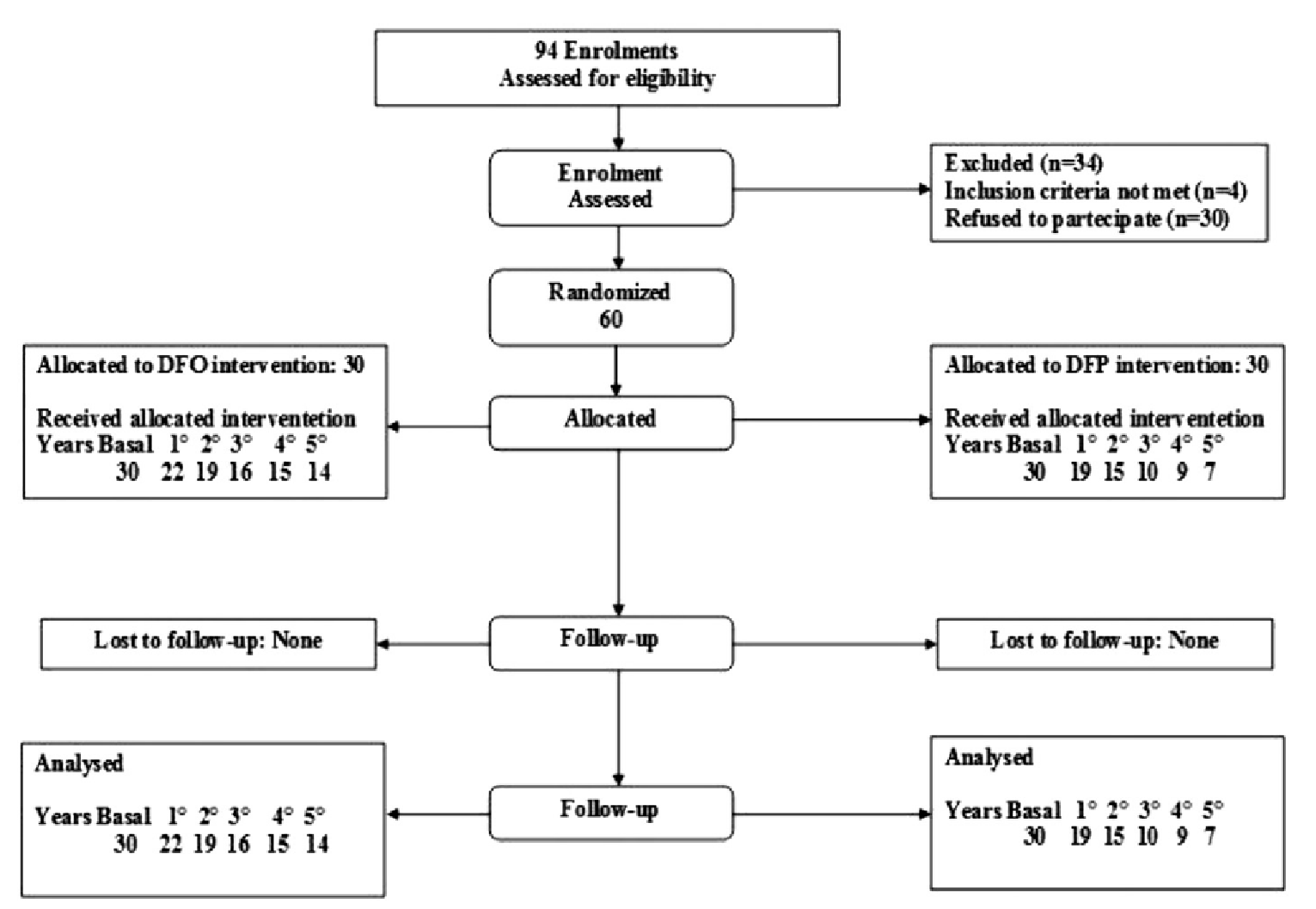
DFO = deferoxamine; DFP = deferiprone.
Source: Calvaruso et al. (2014).3
Exposure to Study Treatments
Study Treatments
FIRST Trial
As shown in Table 11, total exposure to the study treatments in the FIRST trial was 118.9 person-years in the DFP group (N = 152) and 62.5 person-years in the DFO group (N = 76). In both groups, the mean exposure was 0.8 years (SD = 0.3) with a range of 0.0 to 1.1. Treatment up to or beyond the point of month 12 (study completion) was received by 11.2% of patients in the DFP group and 10.5% of patients in the DFO group.2
Table 11: Treatment Exposure in FIRST Trial
Duration of exposure, n (%) | DFP (N = 152) | DFO (N = 76) |
|---|---|---|
Any exposure | 152 (100.0) | 76 (100.0) |
≥ 1 month | █ ███████ | ████████ |
≥ 3 months | █ ███████ | ████████ |
≥ 6 months | █ ███████ | ████████ |
≥ 12 months | ████████ | █ █ ██████ |
DFO = deferoxamine; DFP = deferiprone.
Source: FIRST Clinical Study Report.2
Calvaruso et al. (2014)
Detailed treatment exposure data were not reported.
Dose Interruptions and Reductions
Dose interruptions and reductions in the FIRST trial were not reported.
Adherence
FIRST Trial
In the FIRST trial, treatment adherence was evaluated at each monthly postbaseline visit. Patients were provided with a medication usage diary card and asked to record the number of DFP tablets, the volume of DFP oral solution taken, or the volume of DFO solution administered and the time of injection. For DFP, this was done by counting the number of tablets or the volume of oral solution returned by the patient. For DFO, adherence was evaluated by checking the record maintained by the infusion pump that tracked the number of infusions administered. Patients who took at least 80% and not more than 120% of the prescribed dosage were in adherence. Overall, 68.9% of patients in the DFP group and 78.9% in the DFO group were in adherence. All cases of nonadherence involved under-dosing, although no patients took more than 120% of the dosage.2
Calvaruso et al. (2014)
In Calvaruso et al. (2014), adherence to treatment was assessed by counting the pills in each returned bag of DFP and by assessing the number of infusions of DFO registered on the electronic pump. Adherence was reported as 89% in the DFP group versus 75% in the DFO group.3
Transfusional Iron Input
Figure 4 shows the mean transfusion iron input at baseline and at each study visit through the FIRST trial.
Figure 4: Mean Transfusional Iron Input Over Time
DFO = deferoxamine; DFP = deferiprone; EOS = end of study; MTH = month.
Source: FIRST Clinical Study Report.2
Efficacy
Only those efficacy outcomes and analyses of subgroups identified in the review protocol are reported subsequently. Refer to Appendix 3 for detailed efficacy data.
Liver Iron Concentration
FIRST Trial
Change in LIC values (mg/g dw) from the FIRST trial are summarized in Table 12 and Figure 5. A total of 185 patients (DFO = 122; DFO = 63) were included in the pivotal analysis. █ ███ █ ██ █ ██ █ ██ █ ██ █ ██ █ ██ █ ██ █ █ ███ █ ██ █ █ █ ██ █ ██ ██ █ █ █ █ █ █ ██ █ ██ █ ██ █ ██ █ ███ █ █ █ █ ██ █ ██ █ ███ █ ██ █ ██ █ ██ █ ██ █ █ █ ██ █ ██ █ ███████. The groups were similar at baseline and all postbaseline time points. In the ANCOVA model, the mean change from baseline to month 12 was similar between the 2 treatment groups (█ ██ █ █ █ █ █ █ ██ █ ██ █ ██ █ ██ █ ████). The LSM difference between the 2 treatment groups was 0.26 (96.01% CI, −0.97 to 1.48). The upper limit of the 96.01% CI was 1.48, thereby meeting the predefine noninferiority criterion of the upper limit of the 96.01% CI of the difference between treatment groups being 2 mg/g dw or less. Of the 185 patients evaluable for LIC, 155 (DFP = 104, DFO = 51) had SCD and 30 (DFP = 18, DFO = 12) had other anemias. The results of the subgroup analyses were supportive of the overall study effects.2
Figure 5: Mean Change in LIC (mg/g dw) at Month 12 in FIRST Trial (ITT Set) — Redacted

██████████ █ ██████████ █ █ █████ █ ███████████████████ █ ████████████
Confidential figure redacted at sponsor’s request.
Table 12: Summary of LIC (mg/g dw) in FIRST Trial (ITT Set)
Time point | Analysis | DFP █ █ █ ████ | DFO █ █ █ ███ |
|---|---|---|---|
Overall population (ITT) | |||
Baseline | N (%) | █ ███████ | █ ████████ |
Mean LIC (SD) | █ █ █ ███████ | █ █ █ ███████ | |
Month 6 | N (%) | █ ███████ | █ █ ████████ |
Mean LIC (SD) | ██ █ ██████ | █ █ █ ███████ | |
LS mean change (SE)a | ███████ | █ █ █ ██ █ ██████ | |
LSMD (96.01% CI)b | █ █ ██ █ ██████████ | █████████ | |
P value for change in LICc | ██████ | █████████ | |
Month 12 | N | █ ███████ | █ █ ████████ |
Mean LIC (SD) | █ █ █ ███████ | ███████ | |
LS mean change (SE)a | █ █ █ ███████ | ██ █ ███████ | |
LSMD (96.01% CI)b | █ █ ██ █ ██████████ | █████████ | |
P value for change in LICc | ██████ | █████████ | |
SCD (subgroup) | |||
Baseline | N | ███ | ██ |
Mean LIC (SD) | █ █ █ ███████ | ███████ | |
P value (t-test)c | ██████ | █████████ | |
Month 12 | N | ███ | ██ |
Mean LIC (SD) | ██ █ ██████ | █ █ █ ███████ | |
LS mean change (SE)a | █ █ █ ███████ | █ █ █ ██ █ ██████ | |
LSMD (96.01% CI)b | █ █ ██ █ ██ █ █ ███ █ ███████ | █████████ | |
P valuec | ██████ | █████████ | |
Other anemias (subgroup) | |||
Baseline | N | ██ | ██ |
Mean LIC (SD) | ██ █ ██████ | █ █ █ ███████ | |
P valuec | ██████ | █████████ | |
Month 12 | N | ██ | ██ |
Mean LIC (SD) | █ █ █ ██ █ ██████ | ███████ | |
LS mean change (SE)a | █ █ █ ███████ | ███████ | |
LSMD (96.01% CI)d | █ █ █ ██ █ █████████ | █████████ | |
P valuec | ██████ | █████████ | |
CI = confidence interval; DFO = deferoxamine; DFP = deferiprone; dw = dry weight; ITT = intention-to-treat; LIC = liver iron concentration; LS = least squares; LSMD = least squares mean difference; SCD = sickle cell disease; SD = standard deviation; SE = standard error.
aTreatment was the main factor and overall average transfusional iron input during the study, baseline LIC, and stratification factors (disease category and transfusional iron input in the 3 months before baseline) were covariates. The 96.01% CI of the difference between the treatment groups in the change in LIC from baseline to month 12 was computed. (The 96.01% was determined by 1 minus alpha, where alpha = 0.0399 based on the Pocock alpha spending function for the interim analysis.)
bFor the primary efficacy end point, it was predefined that noninferiority of DFP vs. DFO would be shown if the upper limit of the 96.01% CI of the difference between treatment groups was ≤ 2 mg/g dw. This met the noninferiority criterion.
cP value was derived using a t-test. Values have not been adjusted for multiple testing (i.e., the type I error rate has not been controlled).
dFor the primary efficacy end point, it was predefined that noninferiority of DFP vs. DFO would be shown if the upper limit of the 96.01% CI of the difference between treatment groups was ≤ 2 mg/g dw. As the upper limit of the 96.01% CI is 2.21, this did not meet the noninferiority criterion.
Source: FIRST Clinical Study Report.2
The sponsor conducted 2 sensitivity analyses on the primary end point. In the first, patients who had withdrawn before providing the month 6 LIC data were excluded from the ITT population. The other was a “worst-case scenario” analysis in which missing data from dropouts from the DFO group were imputed using LOCF while, for the DFP group, missing data were imputed using the worst (i.e., largest) value obtained among the DFP-treated patients at the missing visit (month 6 or month 12). For the first sensitivity analysis, the N was 179 patients (n = 119 for DFP; n = 60 for DFO). The results of these analyses using the ANCOVA model upheld the noninferiority in the first test (upper limit of the 96.01% CI = 1.53). For the worst-case scenario analysis, a smaller magnitude of change (i.e., a smaller reduction in LIC) from baseline was observed for the DFP group, resulting in a larger mean difference of plus or minus the standard error (SE) (2.37 ± 0.92) for change in LIC at month 12 between the treatment groups and a failure to meet the noninferiority criterion (upper limit of the 96.01% CI = 4.27).2
Cardiac Iron Concentration
FIRST Trial
The change in mean CIC values (ms) is summarized in Figure 6, Figure 7, and Table 13. At month 12, the change in log (cardiac MRI T2*) geometric mean (± the coefficient of variation %) was 1.01 ± 20.06 in the DFP group and 1.00 ± 21.00 in the DFO group (P = 0.87). For this outcome, support for noninferiority was prespecified to be demonstrated if the 96.01% CI contained 0. The results of the noninferiority analysis using the ANCOVA model show that the mean change from baseline at month 12 was similar between the groups (approximately −0.02 for both) and the 96.01% CI was ███ █ ███████ ███, supporting the noninferiority of DFP to DFO.2
Figure 6: Mean Change From Baseline in Log-Transformed Cardiac MRI T2* (ms) at Month 12 in FIRST Trial (ITT Set) — Redacted

Confidential figure redacted at sponsor’s request.
Figure 7: Geometric Mean Relative to Baseline Mean of Cardiac MRI T2* (ms) at Month 12 in FIRST Trial (ITT Set) — Redacted

██████ █ █ ███ █ █ █ █ ███████ █ █ ██ █ █ ████████████ █ █ █ █ ████ █ █ ██ █ █ █ █ ███████
Confidential figure redacted at sponsor’s request.
Table 13: Summary of Change in Cardiac Iron Concentrations (ms) in FIRST Trial (ITT Set)
Time point | Population | DFP N = 143 | DFO N = 74 |
|---|---|---|---|
Overall population (ITT) | |||
Baseline | N (%) | █ ██ █ ██████ | ████████ |
Geometric mean ± CV (%) | █ █ █ ███████ | ███ █ █████ | |
P valuea | ██████ | █████████ | |
Month 6 | N (%) | █ ███████ | ████████ |
Geometric mean ± CV (%) | █ █ █ ███ █ █████ | ███████ | |
P value (t-test) | ██████ | █████████ | |
Change in log (cardiac MRI T2*), geometric mean ± CV (%) | █ █ ███████ | █ █ ███████ | |
P valuea | ██████ | █████████ | |
Month 12 | N (%) | █ ███████ | ████████ |
Geometric mean ± CV (%) | ███████ | ███████ | |
P value (t-test) | ██████ | █████████ | |
Change in log (cardiac MRI T2*), geometric mean ± CV (%) | █ █ ███████ | █ █ ███████ | |
LS means of change in log-transformed cardiac MRI T2* (SE) | ███████████████ | ██████ █ ██████ | |
LSMD (96.01% CI)b | ███ █ █ █████ ███ | █████████ | |
P valuea | ██████ | █████████ | |
CI = confidence interval; CV = coefficient of variation; DFO = deferoxamine; DFP = deferiprone; ITT = intention-to-treat; LS = least squares; LSMD = least squares mean difference; ms = milliseconds; SE = standard error.
aP value was derived using a t-test. Values have not been adjusted for multiple testing (i.e., the type I error rate has not been controlled).
bFor this outcome, support for noninferiority would be demonstrated if the 96.01% CI contains zero (0), thereby achieving it.
Source: FIRST Clinical Study Report.2
SF Concentration
FIRST Trial
The mean SF (mcg/L) in the FIRST trial is shown in Figure 8. In the DFO group, the mean SF level went down at each time point except month 12. In the DFP group, it increased at month 3 and began to decrease after that (though still above baseline at month 6 ███████ █ █ ████████████████████ █ █████████████████ ████. The SF level continued to go down for both groups and no significant group differences were seen at months 9 and 12 (both P values > 0.05). In the DFP group at month 12, the mean change was slightly greater than 0 but was still not significantly different from that in the DFO group (Figure 9). For this outcome, noninferiority could be concluded if the 96.01% CI contains zero (0). The 96.01% CI at month 12 was ███████, supporting the noninferiority of DFP to DFO.2
Figure 8: Mean Serum Ferritin (mcg/L) Over Time in FIRST Trial
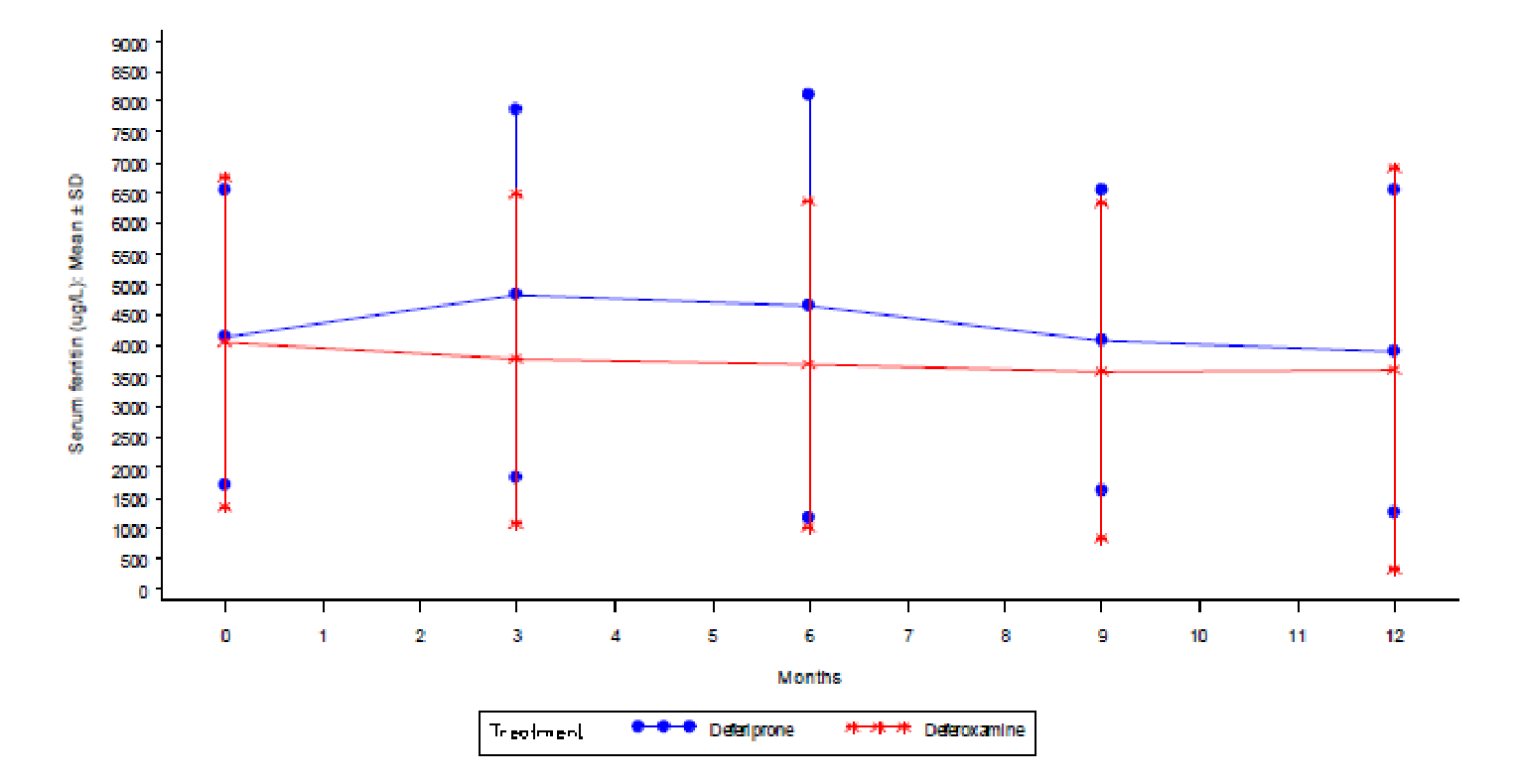
SD = standard deviation.
Source: FIRST Clinical Study Report.2
Figure 9: Mean Change in Serum Ferritin (mcg/L) at Month 12 in FIRST Trial (ITT Set) — Redacted

ITT = intention-to-treat.
Confidential figure redacted at sponsor’s request.
Table 14: Summary of Change in Mean Serum Ferritin in FIRST Trial (ITT Set)
Time point | Analysis | DFP (N = 133) | DFO (N = 67) |
|---|---|---|---|
Overall population (ITT) | |||
Month 3 | Means (SD) | ██ █ ████████ | ███ █ ████████ |
P valuea | ██████ | █████████ | |
Month 6 | Means (SD) | █ █ █ ██ █ ████████ | ██████████ |
P valuea | ██████ | █████████ | |
Month 9 | Means (SD) | █████████ | ██████████ |
P valuea | ██████ | █████████ | |
Month 12 | Means (SD) | █ █ ██ █ ████████ | ███ █ ████████ |
P valuea | ██████ | █████████ | |
LS mean (SE) | ███ █ █ ████████ | ███ █ █ ██ █ ████████ | |
LSMD (96.01% CI) | ██ █ █ ████ █ █ █ █ █ █████████ | █████████ | |
SCD (subgroup) | |||
Month 12 | N | ███ | ██ |
LS mean (SE) | ██████████ | ███████████ | |
LSMD (96.01% CI) | ███ █ ██████████ | █████████ | |
Other anemias (subgroup) | |||
Month 12 | N | ██ | ██ |
LS mean (SE) | ████ █ █ ██ █ ████████ | ███████████ | |
LSMD (96.01% CI) | ████ █ ████████████ | █████████ | |
CI = confidence interval; DFO = deferoxamine; DFP = deferiprone; ITT = intention-to-treat; LS = least squares; LSMD = least squares mean difference; SD = standard deviation; SE = standard error.
aThe P value was derived using a t-test. Values have not been adjusted for multiple comparisons.
Source: FIRST Clinical Study Report.2
Calvaruso et al. (2014)
In Calvaruso et al., the mean value from baseline to year 1 decreased for both the DFP and the DFO group (Figure 10). The incidence of patients with SF levels of less than 400 ng/mL was higher in the DFP group (11 patients, 36.6%) in comparison with the DFO group (1 patient, 3.3%) and was statistically significant (P = 0.002).3
Figure 10: Serum Ferritin Concentration Levels in Calvaruso et al. 2014 (N = 60)

DFO = deferoxamine; DFP = deferiprone; sd = standard deviation.
Source: Calvaruso et al. (2014).3
Survival
FIRST Trial
There were no deaths reported in the FIRST trial.
Calvaruso et al. (2014)
The 5-year probability of survival was similar between groups (P = 0.38).3
Figure 11: Kaplan-Meier Survival Probability Curves in Calvaruso et al. (2014)
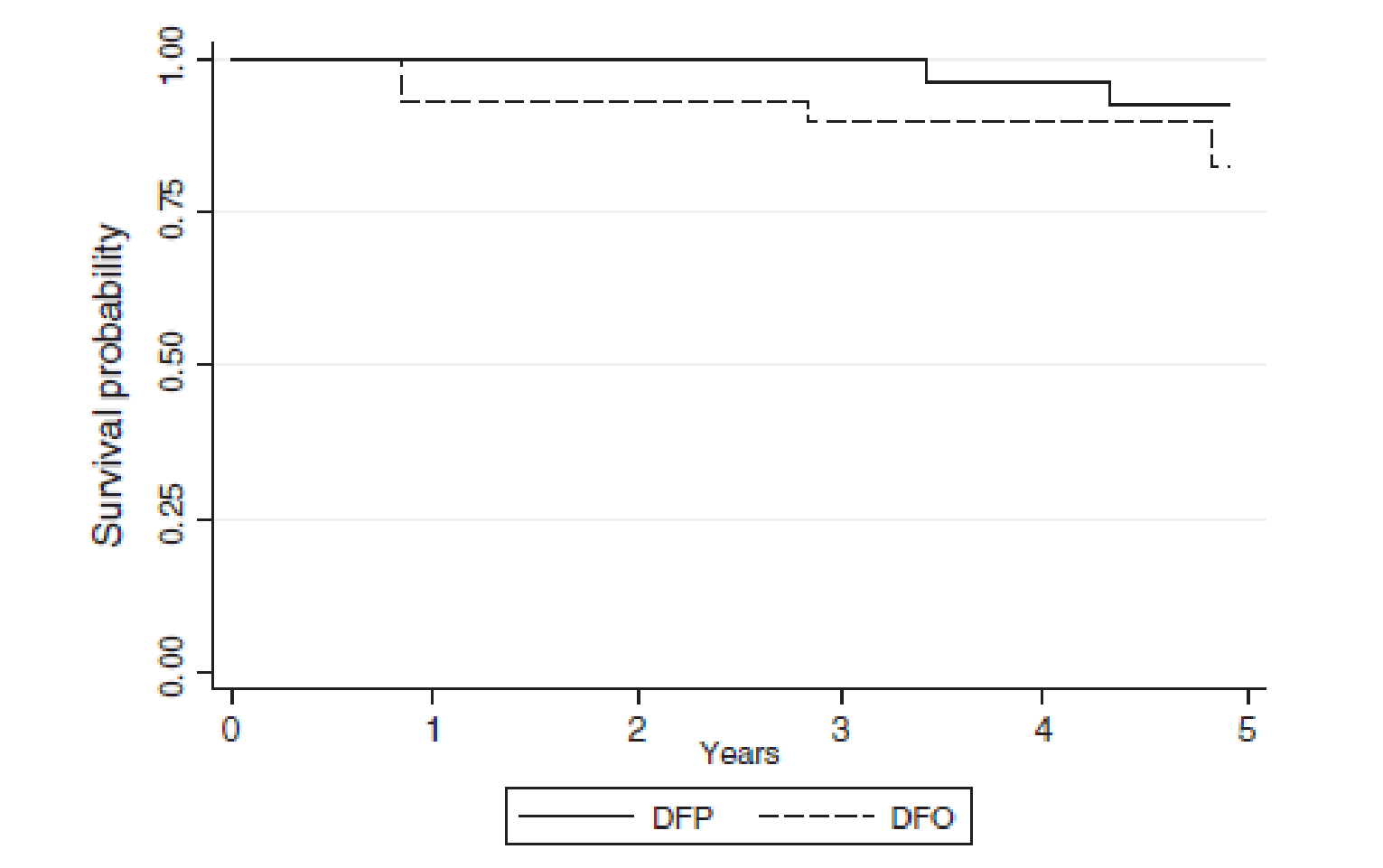
DFO = deferoxamine; DFP = deferiprone.
Source: Calvaruso et al. (2014).3
Figure 12: Summary of Deaths in Calvaruso et al. (2014)
DFO = deferoxamine; DFP = deferiprone.
Source: Calvaruso et al. (2014).3
Health-Related Quality of Life
The FIRST trial assessed HRQoL using the SF-36 and the Child Health Questionnaire (CHQ-PF50 and CHQ-CF87). Appendix 2 contains a description and appraisal of the HRQoL outcome measures. Approximately half of the adult patients in the ITT population and a large majority of the child patients (70% for DFP; 81% for DFO) were missing scores for at least 1 of 3 assessment visits, with the investigators citing administrative and other issues. Sensitivity analyses were done on datasets that included estimated scores using multiple imputation. For both the SF-36 and the CHQ-PF50 summary scores at month 12, there were no differences between the 2 treatment groups using the ANCOVA model and the repeated-measures mixed-effects models. There are no summary scores for the CHQ-CF. Table 16 includes the 8 individual measures of the SF-36; Table 17 includes the 14 individual measures of the CHQ-PF50.
Table 15: Comparison of HRQoL Mean Summary Scores at Month 12 in FIRST Trial (ITT Set)
Instrument | Measure | DFP | DFO | DFP vs. DFOa | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
N | Estimated mean | SE | N | Estimated mean | SE | MD | P value (original) | Hommel adjusted P value | Effect size (Cohen d) | ||
SF-36 | Physical summary | 35 | 43.1 | 1.8 | 19 | 43.0 | 2.0 | 0.2 | 0.9214 | 0.9520 | 0.03 |
Mental summary | 35 | 44.7 | 2.7 | 19 | 40.9 | 2.9 | 3.8 | 0.1174 | 0.8218 | 0.46 | |
CHQ-PF50 | Physical summary | 60 | 29.3 | 1.8 | 23 | 30.5 | 2.4 | −1.2 | 0.6488 | 0.9369 | 0.11 |
Psychosocial summary | 60 | 42.5 | 1.5 | 23 | 41.3 | 2.1 | 1.2 | 0.5915 | 0.9369 | 0.13 | |
ANOVA = analysis of variance; CHQ-PF50 = Child Health Questionnaire Parent Form 50 Questions; DFO = deferoxamine; DFP = deferiprone; HRQoL = health-related quality of life; ITT = intention-to-treat; MD = mean difference; SCD = sickle cell disease; SE = standard error; SF-36 = Short Form (36) Health Survey.
aUnivariate ANCOVA models with treatment arm as a between-patients fixed factor and with baseline values, disease category (SCD vs. non-SCD), baseline transfusional iron input category (≤ 0.3 mg/kg/day vs. > 0.3 mg/kg/day), and the overall mean transfusional iron input during study as covariates.
Source: FIRST Clinical Study Report.2
Table 16: Summary of SF-36 Mean Scores at Month 12 in FIRST Trial (ITT Set)
Measure | DFP | DFO | DFP vs. DFOa | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
N | Estimated mean | SE | N | Estimated mean | SE | MD | F | P value (original) | Hommel adjusted P value | Effect size, Cohen d | |
Physical functioning | ██ | ████ | ███ | ██ | ████ | ███ | ██ | ███ | ██████ | ██████ | ████ |
Role, physical | ██ | ████ | ███ | ██ | ████ | ███ | ██ | ███ | ██████ | ██████ | ████ |
Bodily pain | ██ | ████ | ███ | ██ | ████ | ███ | ██ | ███ | ██████ | ██████ | ████ |
General health | ██ | ████ | ███ | ██ | ████ | ███ | ██ | ███ | ██████ | ██████ | ████ |
Vitality | ██ | ████ | ███ | ██ | ████ | ███ | ██ | ███ | ██████ | ██████ | ████ |
Social functioning | ██ | ████ | ███ | ██ | ████ | ███ | ██ | ███ | ██████ | ██████ | ████ |
Role, emotional | ██ | ████ | ███ | ██ | ████ | ███ | ██ | ███ | ██████ | ██████ | ████ |
Mental health | ██ | ████ | ███ | ██ | ████ | ███ | ██ | ███ | ██████ | ██████ | ████ |
Physical summary | ██ | ████ | ███ | ██ | ████ | ███ | ██ | ███ | ██████ | ██████ | ████ |
Mental summary | ██ | ████ | ███ | ██ | ████ | ███ | ██ | ███ | ██████ | ██████ | ████ |
ANCOVA = analysis of covariance; DFO = deferoxamine; DFP = deferiprone; ITT = intention-to-treat; MD = mean difference; SCD = sickle cell disease; SE = standard error; SF-36 = Short Form (36) Health Survey.
aUnivariate ANCOVA models with treatment arm as a between-patients fixed factor and with baseline values, disease category (SCD vs. non-SCD), baseline transfusional iron input category (≤ 0.3 mg/kg/day vs. > 0.3 mg/kg/day), and the overall mean transfusional iron input during the study as covariates.
Source: FIRST Clinical Study Report.2
Table 17: Summary of CHQ-PF50 Month 12 Mean Scores (ITT Set)
Measure | DFP | DFO | DFP vs. DFOa | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
N | Estimated mean | SE | N | Estimated mean | SE | MD | F | P value (original) | Hommel adjusted P value | Effect size (Cohen d) | |
Physical functioning | ██ | ████ | ███ | ██ | ████ | ███ | ██ | ███ | ██████ | ██████ | ████ |
Role/social limitations, physical | ██ | ████ | ███ | ██ | ████ | ███ | ██ | ███ | ██████ | ██████ | ████ |
General health perceptions | ██ | ████ | ███ | ██ | ████ | ███ | ██ | ███ | ██████ | ██████ | ████ |
Bodily pain/discomfort | ██ | ████ | ███ | ██ | ████ | ███ | ██ | ███ | ██████ | ██████ | ████ |
Family activities | ██ | ████ | ███ | ██ | ████ | ███ | ██ | ███ | ██████ | ██████ | ████ |
Role/social limitations, emotional–behavioural | ██ | ████ | ███ | ██ | ████ | ███ | ██ | ███ | ██████ | ██████ | ████ |
Parental impact, time | ██ | ████ | ███ | ██ | ████ | ███ | ██ | ███ | ██████ | ██████ | ████ |
Parental impact, emotional | ██ | ████ | ███ | ██ | ████ | ███ | ██ | ███ | ██████ | ██████ | ████ |
Self-esteem | ██ | ████ | ███ | ██ | ████ | ███ | ██ | ███ | ██████ | ██████ | ████ |
Mental health | ██ | ████ | ███ | ██ | ████ | ███ | ██ | ███ | ██████ | ██████ | ████ |
Behaviour | ██ | ████ | ███ | ██ | ████ | ███ | ██ | ███ | ██████ | ██████ | ████ |
Family cohesion | ██ | ████ | ███ | ██ | ████ | ███ | ██ | ███ | ██████ | ██████ | ████ |
Global health | ██ | ████ | ███ | ██ | ████ | ███ | ██ | ███ | ██████ | ██████ | ████ |
Global behaviour | ██ | ████ | ███ | ██ | ████ | ███ | ██ | ███ | ██████ | ██████ | ████ |
Physical summary | ██ | ████ | ███ | ██ | ████ | ███ | ██ | ███ | ██████ | ██████ | ████ |
Psychosocial summary | ██ | ████ | ███ | ██ | ████ | ███ | ██ | ███ | ██████ | ██████ | ████ |
ANCOVA = analysis of covariance; CHQ-PF50 = Child Health Questionnaire Parent Form 50 Questions; DFO = deferoxamine; DFP = deferiprone; ITT = intention-to-treat; MD = mean difference; SCD = sickle cell disease; SE = standard error; SF-36 = Short Form (36) Health Survey.
aUnivariate ANCOVA models with treatment arm as a between-patients fixed factor and with baseline values, disease category (SCD vs. non-SCD), baseline transfusional iron input category (≤ 0.3 mg/kg/day vs. > 0.3 mg/kg/day), and the overall mean transfusional iron input during the study as covariates.
Source: FIRST Clinical Study Report.2
Harms
Only those harms identified in the review protocol are reported subsequently.
Adverse Events
Treatment-emergent AEs that occurred in the FIRST trial are summarized in Table 18. At least 1 AE was reported for 88.2% of patients in the DFP group and 88.2% of patients in the DFO group. The most frequent AEs were pyrexia, experienced by 28.3% of DFP patients and 32.9% of DFO patients, followed by abdominal pain in 25.0% and 13.2%, respectively. Alanine aminotransferase increase (11.8% versus 0.0%, respectively) and aspartate aminotransferase increase were higher in DFP patients than in DFO patients (11.2% versus 0.0%). Chromaturia, which is a renal and urinary disorder, was reported in 5.9% and 2.6% of patients in the DFP and DFO groups, respectively.
Table 18: Summary of AEs in FIRST Trial (Safety Set)
AEs, % (n)a | DFPb (N = 152) | DFOb (N = 76) |
|---|---|---|
Pyrexia | 28.3 (43) | 32.9 (25) |
Abdominal painc | 25.0 (38) | 13.2 (10) |
Bone pain | 25.0 (38) | 34.2 (26) |
Headache | 19.7 (30) | 13.2 (10) |
Vomiting | 19.1 (29) | 10.5 (8) |
Pain in extremity | 17.8 (27) | 14.5 (11) |
Sickle cell crisis | 17.1 (26) | 13.2 (10) |
Back pain | 13.2 (20) | 18.4 (14) |
ALT increased | 11.8 (18) | 0.0 (0) |
AST increased | 11.2 (17) | 0.0 (0) |
Oropharyngeal pain | 9.9 (15) | 14.5 (11) |
Nasopharyngitis | 9.2 (14) | 11.8 (9) |
Cough | 7.9 (12) | 14.5 (11) |
Arthralgia | 9.9 (15) | 7.9 (6) |
Neutrophil count decreased | 7.9 (12) | 3.9 (3) |
Nausea | 7.2 (11) | 9.2 (7) |
Chromaturia | 5.9 (9) | 2.6 (2) |
Pain | 5.3 (8) | 3.9 (3) |
Diarrhea | 4.6 (7) | 7.9 (6) |
Chest pain | 3.3 (5) | 5.3 (4) |
Influenza | 3.3 (5) | 6.6 (5) |
Toothache | 2.0 (3) | 6.6 (5) |
Injection site pain | 0.0 (0) | 6.6 (5) |
Injection site swelling | 0.0 (0) | 5.3 (4) |
AE = adverse event; ALT = alanine aminotransferase; AST = aspartate aminotransferase; DFO = deferoxamine; DFP = deferiprone.
aAEs reported in ≥ 5% of patients.
bDFP: Low dose = 75 mg/kg/day (25 mg/kg per dose); high dose = 99 mg/kg/day (33 mg/kg per dose). DFO: Low dose = 20 mg/kg/day (children) or 40 mg/kg/day (adults); high dose = up to 40 mg/kg/day (children) or 50 mg/kg/day (adults).
cIncludes the preferred terms of abdominal pain and abdominal pain upper.
Source: FIRST Clinical Study Report.2
AEs for the Calvaruso et al. trial are summarized in Figure 13. In the Calvaruso et al. study, patients in the DFP group most frequently reported AEs such as nausea, fever, or other infections and liver damage (13.3%, 10.0%, and 10.0%, respectively). The rate of SCD crisis was higher in the DFP group compared with the DFO group (6.7% versus 3.3%, respectively).3
Figure 13: Summary of AEs in Calvaruso et al. (2014)
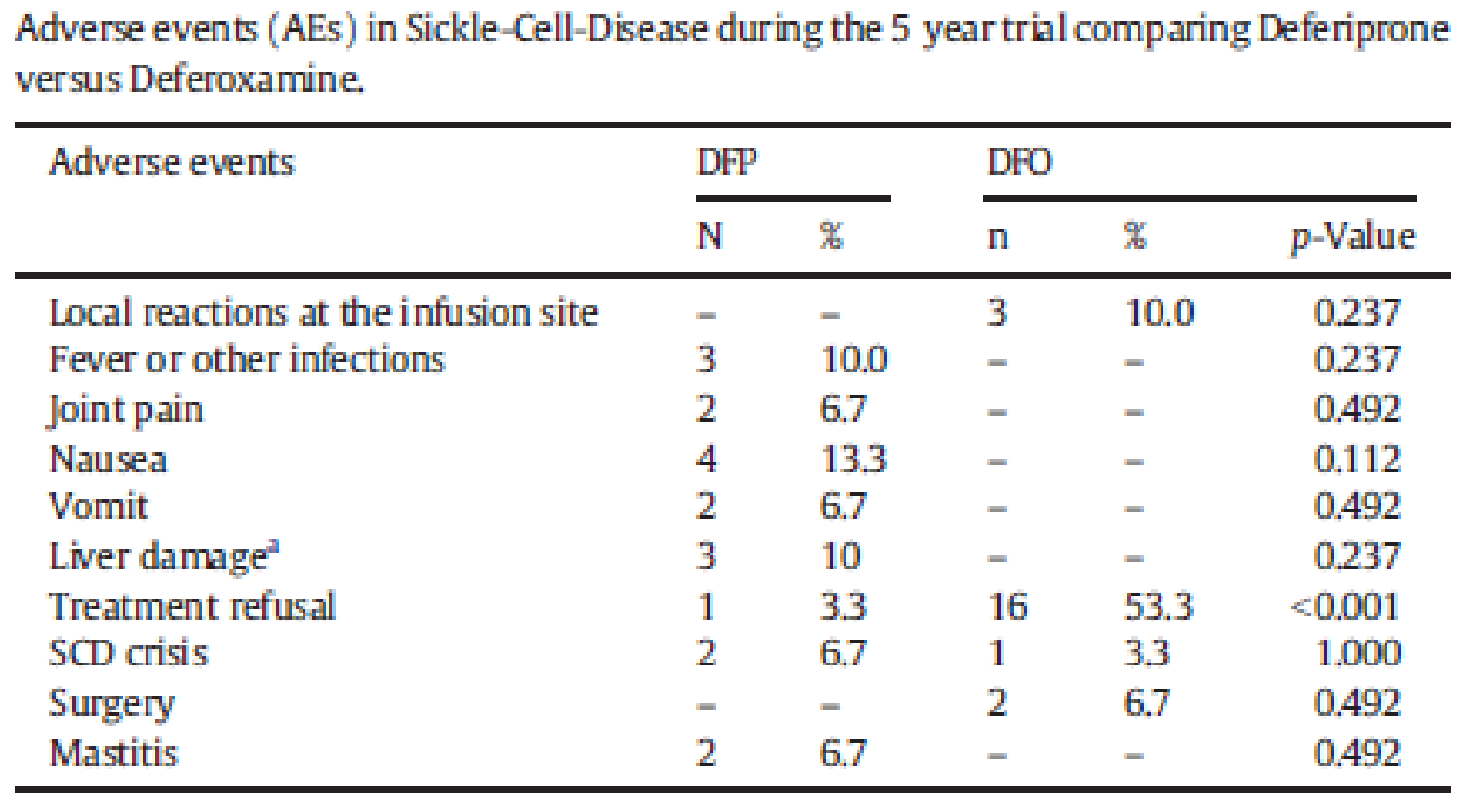
AE = adverse event; DFO = deferoxamine; DFP = deferiprone; SCD = sickle cell disease.
Source: Calvaruso et al. (2014).3
Serious Adverse Events
SAEs that occurred in the FIRST trial are summarized in Table 19. At least 1 SAE was reported for 26.3% of patients in the DFP group and 18.4% of patients in the DFO group. The most frequent SAE was sickle cell crisis, experienced by 10.5% of DFP patients and 5.3% of DFO patients, followed by pyrexia in 3.3% and 3.9%, respectively; neutropenia in 2.6% and 1.3%, respectively; abdominal pain in 2.0% and 1.3%, respectively; and acute chest syndrome in 2.0% and 0.0%, respectively. No other SAE was seen in more than 2 patients. With respect to dosage, the investigators reported that the rates of SAEs were numerically higher in high-dose recipients.
Table 19: Summary of SAEs in FIRST Trial (Safety Set)
SAEs, % (n)a | DFPb (N = 152) | DFOb (N = 76) |
|---|---|---|
Sickle cell anemia with crisis | 10.5 (16) | 5.3 (4) |
Pyrexia | 3.3 (5) | 3.9 (3) |
Neutropenia | 2.6 (4) | 1.3 (1) |
Abdominal painc | 2.0 (3) | 1.3 (1) |
Acute chest syndrome | 2.0 (3) | 0.0 (0) |
Parvovirus infection | █ ██ █ ███ | █ ████ |
Transaminases increased | █ ██ █ ███ | █ ████ |
Encephalopathy | █ ████ | █ ██ █ ███ |
Pneumonia | █ ██ █ ███ | █ ████ |
Sepsis | █ ████ | █ ████ |
Abortion spontaneous | █ ████ | █ ████ |
Deep vein thrombosis | █ ████ | █ ██ █ ███ |
Splenectomy | █ ████ | █ ████ |
Vomiting | █ ████ | █ ████ |
Back pain | █ ██ █ ███ | █ ██ █ ███ |
DFO = deferoxamine; DFP = deferiprone; SAE = serious adverse event.
aSAEs reported in ≥ 1 patient.
bDFP: Low dose = 75 mg/kg/day (25 mg/kg per dose); high dose = 99 mg/kg/day (33 mg/kg per dose)· DFO: Low dose = 20 mg/kg/day (children) or 40 mg/kg/day (adults); high dose = up to 40 mg/kg/day (children) or 50 mg/kg/day (adults).
cIncludes the preferred terms of abdominal pain and abdominal pain upper.
Source: FIRST Clinical Study Report.2
Withdrawals Due to Adverse Events
In the FIRST trial, 12 patients withdrew due to AEs, 5.9% versus 3.9% in the DFP and DFO groups, respectively. Seven of the 12 events, (5 in the DFP group and 2 in the DFO group) and the 2 fatalities were considered unrelated to the study treatment. Of the 5 cases considered to be at least possibly related, 4 were in the DFP group: 1 event of agranulocytosis, 1 of mild neutropenia that lasted beyond 14 days, and 2 instances of abdominal pain and vomiting (both moderate in 1 patient; both severe in the other). The 1 case in the DFO group was due to severe nausea.
Notable Harms
Agranulocytosis occurred in 1 patient in the DFP group compared with zero (0) patients in the DFO group. Neutropenia occurred in 4 patients in the DFP group compared with 1 patient in the DFO group. The instances of neutropenia were classified as less severe episodes, and all patients with agranulocytosis and neutropenia recovered (Table 20).
Table 20: Summary of Notable Harms in FIRST Trial (Safety Set)
Harms, n | DFP (N = 152) | DFO (N = 76) |
|---|---|---|
Agranulocytosis | 1 | 0 |
Neutropenia | 4 | 1 |
DFO = deferoxamine; DFP = deferiprone.
Source: FIRST Clinical Study Report.2
Critical Appraisal
Internal Validity
FIRST Trial
Randomization was performed using an appropriate methodology with adequate allocation concealment (i.e., interactive voice response system), and stratification was based on relevant prognostic factors (i.e., SCD versus other anemias and transfusional iron input in the 3 months before baseline). The treatment groups were well balanced at baseline, with minor differences noted for sex and prior chelation therapy. The clinical expert consulted by CADTH indicated there did not appear to be any clinically important differences across the treatment groups at baseline.
Approximately 30% and 23% of the patients in the DFP and DFO groups prematurely withdrew from the study. The Health Canada reviewers noted that this amount of missing data could affect the efficacy evaluation of the clinical trial. In response, the sponsor provided details about the patient characteristics for those who withdrew prematurely, which demonstrated balance between the 2 groups. Health Canada subsequently noted that the patients who withdrew prematurely would not have had an impact on the efficacy and safety data (citing the similar characteristics across the withdrawn patients and supportive subgroup analyses).11
The FIRST trial was an open-label study design, which can increase the risk of performance and detection bias, particularly for outcomes that are subjective in measurement and interpretation (e.g., symptoms, HRQoL, subjective AEs). However, the blinding of the study treatments would have been challenging, given the difference in the route of administration (e.g., oral tablet for DFP versus SC infusion for DFO) and the 12-month study duration. Given that the primary and secondary efficacy end points were objective measures relying on a central laboratory, the risk of detection bias for the objective outcomes is considered to be low. On the other hand, the open-label administration of the treatments may introduce bias for the nonobjective outcomes (i.e., symptoms, HRQoL, and subjective AEs), although it is unclear from the data if or how the bias could have affected these results.
Follow-up was completed, but the trial enrolment was stopped before the planned sample size (i.e., 300 patients) was reached, and an interim analysis was conducted on only those patients who had provided postbaseline efficacy data before a specified cut-off date (December 17, 2018, the pivotal analysis); some statistical analyses on all patients available when the study was terminated (April 26, 2019, the full analysis) were also provided. Although the sample size of 213 patients was smaller than the planned 300, it was adequate to estimate the primary end point of treatment response to detect a statistically significant difference at 89% power. The investigators analyzed patients according to the treatment they received and the groups to which they were assigned. Although the sponsor indicated an ITT analysis was conducted, not all patients were included for all outcomes, and a true ITT analysis was not completed. It is unclear how the exclusion of these patients from the ITT analysis would have affected the results.
The clinical expert consulted by CADTH noted that the primary end point, reduction in LIC from baseline to 12 months, was clinically meaningful and that a noninferiority margin of 2 mg/g dw for the reduction of LIC was appropriate.
The analysis populations used in the FIRST trial were appropriate for measuring the effect of assignment to the interventions. Both the PP and ITT sets were used for most analyses, and both supported the findings of noninferiority. Safety outcomes were assessed in all patients who were treated with a study drug. All analyses were prespecified.
As noted in the clinician input for this review and in the patient input in the previous CADTH review of DFP, patients often prefer orally administered treatments as opposed to IV or SC infusion. The open-label administration of the study drugs and the considerable amount of missing data for the HRQoL measures limit the ability to draw conclusions regarding patient preferences for DFP versus DFO.
A high rate of protocol deviations occurred in the FIRST trial and was mostly similar in both groups, which creates some uncertainty in the data. Key protocol deviations were related to study treatment compliance. Given that this was an inferiority study, it is important for participants to complete the study protocols and adhere as intended, or differences may be difficult to find. Approximately 30% of participants in each group had protocol violations and/or were nonadherent, which could introduce substantial bias into the claims of noninferiority. Sensitivity analyses were completed and were supportive of noninferiority, with the exception of the worst-case scenario for missing data, which suggested that noninferiority was not achieved. How the protocol violations, low adherence, and missing data affected the overall study results is uncertain. The Health Canada reviewers noted that both the ITT and PP analyses supported the conclusion of noninferiority. In addition, the clinical expert consulted by CADTH noted the protocol violations reported within the trial can be a more accurate reflection of routine clinical practice, where adherence to a treatment regimen can vary over the course of a 12-month period.
Calvaruso et al. (2014)
In the Calvaruso et al. (2014) study, the investigators adequately used randomization and allocation concealment in the study and provided a rationale for why blinding was not feasible. The use of SF as the only efficacy end point was a major limitation of the study. The clinical expert noted that SF values are not reliable on their own, as several factors beyond iron overload, including SCD flares, could impact values and provide inaccurate interpretations. In the DFP group, 45.4% of patients had had a splenectomy versus 70.6% in the DFO group.3 The clinical noted that this imbalance could affect the results, as the spleen is a sink for iron and macrophage activity that might be impacted by splenectomy and affect the SF numbers.
External Validity
FIRST Trial
The clinical expert consulted by CADTH indicated that the eligibility criteria used in the FIRST trial were appropriate and allowed enrolment of patients who were generally representative of the patient population in Canada, with a few notable exceptions. A baseline LIC of 7 mg/g dw was used in the inclusion criteria of the trial; however, in Canadian practice, patients with an LIC lower than 7 mg/g dw could be treated with iron chelation therapy (i.e., this is a higher threshold for initiating therapy for patients in Canada managed by a health care team experienced in the management of SCD). In the FIRST trial, patients treated with hydroxyurea within the past 30 days were excluded; this is not reflective of routine practice in Canada, where many patients would receive concomitant treatment with hydroxyurea, a drug that is recommended by the Guidelines for the Clinical Management of Patients With Sickle Cell Disease in Canada as a disease-modifying therapy for SCD. Although not reflective of routine care in Canada, the exclusion of these patients from the FIRST trial is not anticipated to limit the generalizability of the results for the efficacy end points that were studied in the trial. However, concomitant usage with hydroxyurea could pose additional risks for neutropenia, as hydroxyurea is associated with a risk of bone suppression and leukopenia, which could result in treatment interruption for patients.
The clinical expert indicated that the baseline demographic characteristics in the FIRST trial were generally representative of the patient population in Canada, with the exception of race (only 16.2% of patients were identified as Black) and prior chelation therapy with DFO. SCD is most common in individuals of African ancestry, and the proportion of patients who identified as Black in the FIRST trial is below what would be anticipated in Canadian practice and below what was reported in the clinical trial comparing DFX and DFO, which was used in the sponsor’s indirect comparison (i.e., approximately 90% of patients were Black). Prior chelation therapy with DFO in the FIRST trial (reported for 16.4% and 22.4% of those in the DFP and DFO groups, respectively) is higher than what would be expected in routine Canadian clinical practice. These differences were not anticipated to impact the generalizability of the study results to the target population in Canada.
The comparator used in the FIRST trial was identified by the clinical expert as appropriate, although the clinical expert consulted by CADTH stated that DFX would have been more appropriate. It is noted that the clinical trial was initiated before the approval of DFX for the first-line treatment of SCD; therefore, it was compared only with DFO.
The study treatments were administered in accordance with the dose ranges recommended in the Canadian product monographs. The clinical expert consulted by CADTH noted that DFP is often administered twice daily in routine clinical practice, as opposed to 3 times daily to improve patient adherence to the therapy; however, the same total daily dosage is administered. The definitions of “less severe” and “more severe” iron burden that were used to determine dose escalation in the FIRST trial may not be reflective of routine care in Canada. The clinical expert consulted by CADTH noted the severity of iron overload in Canada would likely be categorized as mild, moderate, or severe, and that the upper range for LIC of 15 mg dw that was used to differentiate between “less severe” and “more severe” patients is too high. In routine clinical practice, initial starting dosages may be higher and/or dose escalation could occur more frequently for patients with higher baseline LIC levels.
The efficacy and harms outcomes used in the FIRST trial were generally clinically meaningful and important to clinicians and patients. End points were evaluated in a manner consistent with Canadian practice (e.g., LIC evaluated using MRI). Baseline values for CIC suggest there were no significant cardiac iron depositions, which is generally reflective of patients with SCD in routine practice in Canada.
The clinical expert consulted by CADTH noted that the study discontinuation rates were relatively high for a trial with a 12-month duration; however, the breakdown of reasons for discontinuation in both arms appeared to be a reasonable reflection of the Canadian clinical context.
Calvaruso et al. (2014)
In the Calvaruso et al. (2014) study, there were major concerns about generalizability. With respect to the eligibility criteria, the study excluded patients with white blood cell counts of less than 3,000/μL.3 This may have excluded relevant patient populations, including those receiving treatment with hydroxyurea. The clinical expert consulted by CADTH noted that the baseline transfusion burden appeared to be low in the trial whereas, in routine clinical practice, patients with a higher transfusion burden would be treated. For the baseline SF values, the clinical expert indicated they are low compared with what would be expected in routine clinical practice, suggesting patients participating in the trial may be healthier.
Indirect Evidence
Objectives and Methods for the Summary of Indirect Evidence
The objective of this section is to summarize and critically appraise indirect evidence comparing DFP with other relevant treatments (identified in the protocol) in patients with SCD.
In the FIRST trial,2 DFP has been compared with DFO in patients with SCD and other anemias who underwent transfusional therapy. As no head-to-head evidence comparing DFP against other relevant comparators, specifically DFX, was identified, a focused literature search for ITCs was run in MEDLINE All (1946–) on July 5, 2022. No limits were applied. Titles, abstracts, and full-text articles were screened for inclusion by 1 reviewer based on the population, intervention, comparator, and outcome criteria outlined in accordance with the protocol for the CADTH review. No published ITCs were found from the CADTH literature search comparing DFP with the comparators of interest for this review.
Description of Indirect Comparison
A single sponsor-submitted NMA28 was provided as part of the submission and has been described and critically appraised in the following sections. It included a systematic review with an NMA of 2 studies comparing DFP with DFO2,20 and DFX with DFO.29
Methods of Sponsor-Submitted NMA
Objectives
The objective of the sponsor’s ITC was to conduct a systematic literature review and NMA to compare the relative efficacy of DFP versus DFO and DFX in patients with SCD with transfusional iron overload.
Study Selection Methods
The sponsor conducted a systematic review to select studies based on the criteria outlined in Table 21. The systematic review was restricted to phase II, III, and IV RCTs conducted in patients with an SCD and iron overload, with or without a control group with no restriction on blinding. The sponsor considered different comparators in the systematic review, as presented in Table 21. Database searches were done for MEDLINE and Embase (2001–), MEDLINE In-Process, the Cochrane Library Health Technology Assessments, and Database of Abstracts of Reviews of Effects (DARE). The literature search was conducted on January 12, 2022, and was limited to publication dates ranging from January 2001 to January 2022. Search terms consisted of SCD, anemia, hemoglobin S disease, iron overload, iron toxicity, iron poisoning, DFX, DFO, DFP, or iron chelating agent.
The following data were extracted from each trial: sample size, year of publication, change in LIC at 12 months, change in SF at 12 months, and change in cardiac MRI T2* at 12 months. All titles, abstracts, and full texts of identified studies were screened by 2 independent reviewers and any discrepancies were resolved by a third reviewer. The quality of the included studies was assessed using the revised Cochrane risk-of-bias tool for randomized trials. Randomized trials were categorized with respect to their perceived risk of bias (low risk of bias, unclear risk of bias, high risk of bias).
Table 21: Study Selection Criteria and Methods for ITCs
Characteristic | Sponsor-submitted ITC |
|---|---|
Population | Patients with SCD and transfusional iron overload |
Intervention (comparators) |
|
Outcome |
|
Study design |
|
Language | English |
Databases searched | Embase, MEDLINE, MEDLINE In-Process, Cochrane Library Health Technology Assessment, and DARE |
Selection process | NR |
Data-extraction process | NR |
Quality assessment | Assessed according to the revised Cochrane risk-of-bias tool for randomized trials |
DARE = Database of Abstracts of Reviews of Effects; DFO = deferoxamine; DFP = deferiprone; DFX = deferasirox; ITC = indirect treatment comparison; LIC = liver iron concentration; NR = not reported; RCT = randomized controlled trial; SCD = sickle cell disease; SF = serum ferritin.
Source: Sponsor ITC Report.28
ITC Analysis Methods
The base-case analysis utilized the ITT population of 2 RCTs: the FIRST trial, comparing DFP with DFO, and the NCT00067080 trial, comparing DFX with DFO. An NMA was conducted using a Bayesian framework with a noninformative prior. A fixed treatment-effect approach was used to accommodate simple networks, as a random-effects model was not appropriate due to the small sample size (small number of studies) included. Efficacy estimates were reported as a mean difference. DFP was considered as the reference treatment in this NMA analysis, as the study objective was to compare DFP with its comparators, including DFO and DFX. Effect estimates were summarized using the mean with 95% CrIs. Model convergence was assessed using trace plots and Gelman-Rubin-Brooks plots of the potential scale reduction factor, with a minimum cut-off below 1.05 by the final iteration. Two subgroup populations were examined in the NMA:
the SCD population only, as a difference in the proportion of patients with SCD was observed between the included trials
the subgroup of patients with a serum creatinine below the upper limit of normal (ULN), as the NCT00067080 trial excluded patients with a baseline serum creatinine above the ULN.
All analyses were performed using R Studio using the geMTC package with 5,000 burn-in iterations and 10,000 actual iterations.
Homogeneity in the evidence network was assessed by comparing baseline descriptive statistics between the trials. Due to the small number and sample size of the included studies, methods to account for heterogeneity in the populations, such as meta-regression or random-effects models, could not be used. The assessment of statistical consistency was not possible, as no closed loops were included in the NMA. The transitivity assumption was assessed by comparing trial definitions for common comparators within the network, including baseline characteristics imbalance, inclusion or exclusion criteria, and study design. As part of a feasibility analysis, an exploratory analysis was conducted based on the individual patient data from the FIRST trials, which identified no effect modifiers for changes at 12 months in SF and LIC. Additional sensitivity analyses were not performed due to limited data.
Study End Points
Treatments were compared with respect to 2 efficacy end points:
change from baseline to 12 months in LIC
change from baseline to 12 months in SF
Indirect mean differences with 95% CrIs were reported. Change in cardiac MRI T2* from baseline to 12 months was not analyzed because it was reported in the FIRST trial only.
Construction of the Networks
NMA results were provided for only 2 outcomes: change from baseline to 12 months in LIC and SF. There were 2 trials included in the analysis for both end points. The network diagram presented in Figure 14 summarizes the methodology by which the relative treatment effects for 2 drugs without head-to-head evidence were estimated. The common comparator arm of DFO in the FIRST and NCT00067080 trials was used to estimate the relative efficacy of DFP against DFX.
Figure 14: Schematic of the ITC Methodology
DFO = deferoxamine; DFP = deferiprone; DFX = deferasirox; ITC = indirect treatment comparison.
Source: Sponsor ITC Report.28
Results of the Sponsor-Submitted ITC
Summary of Included Studies
A total of 1,180 records were screened by title and abstract; of these, 77 records were screened in full text. Following a full-text review, a total of 14 records were included and extracted in the systematic review, including 11 primary studies and 3 substudies. From the 11 identified primary studies, the sponsor further refined the included post hoc studies for the purposes of the ITC. Only RCTs that reported at least 1 of the efficacy end points with SE or SD were included in the NMA. Based on the inclusion criteria, 2 RCTs were included in this NMA: FIRST20 and NCT00067080.29 The rest of the studies were excluded from the analysis because they:
were not RCTs (n = 5)
did not report efficacy end points at 12 months (n = 3)
did not report the SD or standard error of the efficacy end points (n = 1).
The quality of the included studies was assessed using the revised Cochrane risk-of-bias tool for randomized trials.
A summary of the included trials in the sponsor-submitted NMA is provided in Table 22. Both the FIRST and NCT00067080 trials were parallel-group open-label RCTs. The FIRST trial was a phase IIIb and phase IV noninferiority trial, while the NCT00067080 trial was a phase II superiority trial. The FIRST trial compared DFP against DFO and included the population with SCD or other transfusion-dependent anemia, while the NCT00067080 trial compared the efficacy of DFX against DFO and only included patients with SCD and a serum creatinine level below the ULN. The FIRST trial required a higher baseline LIC (> 7 mg/g dw) compared with the NCT00067080 trial, but excluded patients with a baseline LIC exceeding 30 mg/g dw and patients who received treatment with hydroxyurea within 30 days of the study.
Table 22: Summary of Included Trials in the Sponsor-Submitted ITC
Detail | FIRST | NCT00067080 |
|---|---|---|
Intervention | DFP at a total daily dosage of 75 mg/kg to 99 mg/kg, divided t.i.d. 8 hours apart 7 days a week, depending on iron load severity | DFX at a total daily dosage of 10 mg/kg to 30 mg/kg, according to baseline LIC |
Comparator | DFO SC infusion over 8 to 12 hours, 5 to 7 days a week for a total daily dosage of 20 mg/kg to 50 mg/kg, depending on iron load severity | DFO SC infusion over 8 to 12 hours, 5 to 7 days per week (i.e., 50 mg/kg administered 7 days per week would be reported as 70 mg/kg), depending on iron load severity |
Phase | Phase IIIb and phase IV | Phase II |
Study duration | Up to 12 months | Up to 13 months |
Method of blinding | Open label | Open label |
Population, N | 228 | 195 |
Population | Patients aged > 2 years with SCD or transfusion-dependent anemia | Patients with SCD aged > 2 years with transfusional iron overload |
Inclusion criteria |
|
|
Exclusion criteria |
|
|
ACE = angiotensin-converting enzyme; DFO = deferoxamine; DFP = deferiprone; DFX = deferasirox; dw = dry weight; ITC = indirect treatment comparison; LIC = liver iron concentration; RBC = red blood cell; SC = subcutaneous; SCD = sickle cell disease; SF = serum ferritin; t.i.d. = 3 times daily; ULN = upper limit of normal.
Source: Sponsor ITC Report.28
The baseline characteristics for the 2 included studies are summarized in Table 23. The included trials had differences in the baseline characteristics across groups with respect to sex, race, and baseline SF and LIC values. The FIRST trial included a higher proportion of males compared with the NCR00067080 trial. The majority of patients in the FIRST trial were white (77.2%), while most patients in the NCR00067080 trial were Black (90.8%). Most patients in the FIRST trial had a baseline LIC of greater than 7 mg/g dw, while almost half of the patients in the NCR00067080 trial had baseline LIC levels of less than 7 mg/g dw. In FIRST, the mean baseline SF was 4,114.5 mcg/L (SD = 2,385.7) in the DFP group and 4,136.9 mcg/L (SD = 2,649.1) in the DFX group while, in NCR00067080, the mean SF was 3,460.0 mcg/L (SD = 1,082.0) and 2,834.0 mcg/L (SD = 1,015.0) in the DFX and DFO groups, respectively.
Table 23: Summary of Patient Baseline Characteristics
Characteristic | FIRST (NCT02041299) | NCT00067080 | ||
|---|---|---|---|---|
DFP (N = 152) | DFO (N = 76) | DFX (N = 132) | DFO (N = 63) | |
Age, median (Q1 to Q3) | 15 (3 to 59) | 15 (4 to 40) | 15 (3 to 54) | 16 (3 to 51) |
Male, n (%) | 83 (54.6) | 38 (50.0) | 52 (39.4) | 28 (44.4) |
Race, n (%) | ||||
White | 120 (78.9) | 56 (73.7) | 8 (6.1) | 3 (4.8) |
Black | 23 (15.1) | 14 (18.4) | 118 (89.4) | 59 (93.7) |
Multiracial | 9 (5.9) | 6 (7.9) | — | — |
Other | — | — | 6 (4.5) | 1 (1.6) |
SCD, n (%) | 126 (82.9) | 63 (82.9) | 132 (100.0) | 63 (100.0) |
SF (mcg/L), mean (SD) | 4,114.5 (2,385.7) | 4,136.9 (2,649.1) | 3,460.0 (1,082.0) | 2,834.0 (1,015.0) |
LIC (mg/g dw), n (%) | ||||
≤ 3 | — | — | 4 (3.0) | 6 (9.5) |
> 3 to 7 | 1 (0.8) | — | 64 (48.5) | 21 (33.3) |
> 7 to 14 | 61 (45.9) | 37 (53.6) | 46 (34.8) | 20 (31.7) |
> 14 | 71 (53.4) | 32 (46.4) | 18 (13.6) | 16 (25.4) |
DFO = deferoxamine; DFP = deferiprone; DFX = deferasirox; dw = dry weight; ITC = indirect treatment comparison; LIC = liver iron concentration; Q1 = 25th percentile; Q3 = 75th percentile; SCD = sickle cell disease; SF = serum ferritin.
Source: Sponsor ITC Report.28
Table 24 shows an assessment of heterogeneity based on the study and patient characteristics.
Table 24: Assessment of Homogeneity for Sponsor-Submitted ITC
Detail | Description and handling of potential effect modifiers |
|---|---|
Disease severity | Patients in the FIRST trial had higher baseline SF and LIC values compared with the patients in the NCT00067080 trial, indicating more severe iron overload. Details of baseline transfusional iron input and transfusion category (simple, exchange, or simple and exchange) were not extracted in the report. |
Treatment history | Details of treatment history were not extracted in the report. |
Clinical trial eligibility criteria | The FIRST trial included a population with SCD or other transfusion-dependent anemia, while the NCT00067080 trial only included patients with SCD with a serum creatinine level below the upper limit of normal. As a result of this, 2 subgroup analyses were performed, 1 that included an SCD-only subpopulation and a second that included patients with a serum creatinine level below the upper limit of normal. The FIRST trial required a higher baseline LIC (> 7 mg/g dw) compared with NCT00067080 but excluded patients with a baseline LIC exceeding 30 mg/g dw and patients who received treatment with hydroxyurea within 30 days of the study. |
Dosing of comparators | A total daily dosage of DFP is 75 mg/kg to 99 mg/kg divided t.i.d. 8 hours apart and 7 days a week, while a daily dosage of DFX is 10 mg/kg to 30 mg/kg. Thus, the dosages for DFP and DFX in the FIRST and NCT00067080 trials cannot be considered equivalent, as the maximum daily dosage of DFP was 99 mg/kg, while the maximum for DFX was 40 mg/kg. The dosage of DFO used in both included studies ranged from 20 mg/kg per day to 50 mg/kg per day for 4 to 7 days per week. These variations reflected the flexibility of iron-chelator dosing, depending on the severity of iron overload in patients with SCD. |
Patient characteristics | The sponsor claimed that the results of the univariate treatment effect modifier assessments using the individual patient data of the FIRST trial indicated that none of the mutually reported patient baseline characteristics were identified as potential effect modifiers. However, the baseline mean SF and LIC values as well as sex and race were found to vary across the trials included in the NMA. |
Definitions of end points | Treatments in both trials were compared with respect to 2 efficacy end points: change from baseline to 12 months in LIC, and change from baseline to 12 months in SF. However, LIC was determined using MRI in the FIRST trial and SQUID biosusceptometry in the NCT00067080 trial. The change in LIC was adjusted in the NCT00067080 trial for transfusion category (simple, exchange, simple and exchange), while no adjustment was made in the FIRST trial. |
Timing of end point evaluation or trial duration | The median duration of follow-up was 12 months in FIRST and 13 months in NCT00067080. |
Withdrawal frequency | Withdrawal frequency was not reported in data extraction. |
Clinical trial setting | Details of the setting were not extracted in the report. |
Study design | Both included studies were parallel-group open-label RCTs. FIRST was a late-phase (IIb and IV) noninferiority study and NCT00067080 was a phase II superiority study. |
DFO = deferoxamine; DFP = deferiprone; DFX = deferasirox; dw = dry weight; ITC = indirect treatment comparison; LIC = liver iron concentration; NMA = network meta-analysis; RCT = randomized controlled trial; SCD = sickle cell disease; SF = serum ferritin; SQUID = superconducting quantum interference device; t.i.d. = 3 times daily.
Source: Sponsor ITC Report.28
Efficacy Results of the Sponsor-Submitted NMA
The mean difference from baseline to 12 months in SF and LIC from the individual studies, along with the relative efficacy estimates for DFO versus DFP, and DFX versus DFP from the NMA, are presented in Table 25. For change from baseline to 12 months in SF and LIC, the NMA results from fixed-effect models found there was no difference between DFP and DFO, or DFP and DFX. Compared with DFP, the mean difference for change at 12 months in LIC was –0.4 (95% CrI, –1.7 to 0.9) for DFO, and –0.7 (95% CrI, –3.6 to 2.3) for DFX. Compared with DFP, the mean difference for change at 12 months in SF was –364.4 (95% CrI, –961.4 to 237.2) for DFO and 11.2 (95% CrI, –688.2 to 712.5) for DFX. Subgroup analyses based on both the SCD-only subpopulation and the subpopulation with serum creatinine below the ULN were consistent with the primary analyses.
Table 25: Change in SF and LIC With DFP Relative to Comparators (ITT Population)
Characteristic | FIRST | NCT00067080 | Sponsor-submitted ITC | |
|---|---|---|---|---|
DFP vs. DFO | DFX vs. DFO | DFX vs. DFP | DFO vs. DFP | |
Change in LIC from baseline to 12 months | ||||
Mean difference (SE) | n = 202 0.4 (0.7) | n = 195 –0.2 (1.4) | NA | NA |
Mean difference (95% CrI) | N | NA | –0.7 (–3.6 to 2.3) | –0.4 (–1.7 to 0.9) |
Change in SF from baseline to 12 months | ||||
Mean difference (SE) | n = 217 367.70 (305.5) | n = 195 375.0 (187.1) | NA | NA |
Mean difference (95% CrI) | NA | NA | 11.2 (–688.2 to 712.5) | –364.4 (–961.4 to 237.2) |
CrI = credible interval; DFO = deferoxamine; DFP = deferiprone; DFX = deferasirox; ITC = indirect treatment comparison; ITT = intention-to-treat; LIC = liver iron concentration; NA = not applicable; SE = standard error; SF = serum ferritin.
Source: Sponsor ITC Report.28
Pairwise comparisons of interventions estimated from NMAs are presented in Table 26. They were also presented through forest plots that report mean differences with the 95% Crls.
Table 26: Change in SF and LIC With DFP Relative to Comparators — Pairwise Comparison Matrix (ITT Population)
Treatment | DFO Mean difference (95% CrI) | DFP Mean difference (95% CrI) | DFX Mean difference (95% CrI) |
|---|---|---|---|
Change in LIC from baseline to 12 months | |||
DFO | DFO | 0.4 (–0.9 to 1.7) | –0.3 (–2.9 to 2.4) |
DFP | –0.4 (–1.7 to 0.9) | DFP | –0.7 (–3.6 to 2.3) |
DFX | 0.3 (–2.4 to 2.9) | 0.7 (–2.3 to 3.6) | DFX |
Change in SF from baseline to 12 months | |||
DFO | DFO | 364.4 (–237.2 to 961.4) | 376.1 (5.29 to 739.1) |
DFP | –364 (–961.4 to 237.2) | DFP | 11.2 (–688.2 to 712.5) |
DFX | –376.1 (–739.1 to –5.3) | –11.2 (–712.5 to 688.2) | DFX |
CrI = credible interval; DFO = deferoxamine; DFP = deferiprone; DFX = deferasirox; ITC = indirect treatment comparison; ITT = intention-to-treat; LIC = liver iron concentration; SF = serum ferritin.
Source: Sponsor ITC Report.28
Critical Appraisal of Sponsor-Submitted NMA
The submitted NMA was conducted to assess the efficacy of DFP against relevant comparators among patients with SCD who underwent transfusional therapy. The sponsor-submitted NMA was based on a systematic literature review that identified studies according to prespecified inclusion criteria. Overall, based on the methods detailed in the report, the systematic literature review has an adequate search strategy, screening, and appraisal of the risk of bias of the included studies. The systematic review identified 11 primary studies for inclusion, based on preidentified study selection criteria. These identified studies were refined, post hoc, to include only RCTs reporting at least 1 efficacy end point with an SE or SD. Given that a large number of studies were identified but not included, it is not clear how the inclusion of these other studies may have affected the results of the study. All titles and abstracts and full texts of identified studies were screened by 2 independent reviewers, and any discrepancies were resolved by a third reviewer. The quality of the included studies was assessed using the revised Cochrane risk-of-bias tool for randomized trials; however, it was not used in any of the analyses.
Aside from the selection of studies, other limitations of the NMA relate to data sparseness, network structure, and potential violation of the transitivity assumption. The networks for analyses were small. Thus, the decision was made a priori to limit the analysis to fixed-effects models. The fixed-effects approach with noninformative priors was appropriate, given the sparsity of data. However, this approach entailed the assumption that between-study heterogeneity was zero and the true treatment effects in each study are the same, which was unlikely to be the case. Furthermore, the evidence is imprecise in the effect estimates from the NMA due to the sparseness of data, with wide CIs that could include an appreciable threshold of benefit or lack of benefit. Model fit was not evaluated, so it is not clear how well the model estimates treatment differences. The small number of studies also meant there was no opportunity to use meta-regression to adjust for variability in baseline characteristics and correct for potential bias. Thus, these limitations must be considered when drawing conclusions based on the results of the NMA.
There were some important differences between the FIRST and NCT00067080 trials that increase the uncertainty of the NMA analyses. The FIRST trial required a higher baseline LIC (> 7 mg/g dw) than the NCT00067080 trial, which indicated a more severe iron overload. According to the clinical expert consulted by CADTH for this review, treatment with iron chelators is typically initiated in patients receiving transfusion therapy with an LIC below 7 mg/g dw. The clinical expert consulted highlighted that the exclusion of patients with an LIC exceeding 30 mg/g dw in the FIRST trial could result in the loss of a population that is nonadherent generally to iron chelators, which is likely to bias the study results in favour of DFP. No potential effect modifiers were identified by the sponsor in a feasibility assessment using the individual patient data of the FIRST trial. However, the baseline patient characteristics differed between the 2 trials, with patients enrolled in the FIRST trial appearing to have a more severe iron overload, as evidenced by the elevated SF and LIC values at baseline compared with the patients enrolled in the NCT00067080 trial, which could likely bias the results. Details of baseline transfusional iron input and transfusion category (simple, exchange, or simple and exchange) were not extracted in the NMA report. The plausibility of the transitivity assumption is therefore uncertain.
Only 2 clinical efficacy outcomes were prespecified in the NMA. According to the clinical expert consulted by CADTH for this review, change from baseline to 12 months in LIC is an appropriate outcome, while change in SF is an unreliable and inaccurate marker for assessing and monitoring iron overload in patients with SCD. The LIC was determined using MRI scan in the FIRST trial, and superconducting quantum interference device (SQUID) biosusceptometry in the NCT00067080 trial. The clinical expert consulted noted that SQUID biosusceptometry is a very reliable method for determining LIC but is rarely used in clinical practice. The change at 12 months in LIC was adjusted in the NCT00067080 trial for transfusion category (simple, exchange, simple and exchange), while no adjustment was made in the FIRST trial. The results of subgroup analyses were consistent with the primary analyses. The results of the NMA comparing DFP with DFX and DFO in patients with SCD and iron overload showed no difference with regard to both of the assessed end points. Heterogeneity between the included studies would be expected to introduce bias into the study estimates observed between the comparators. Additional sensitivity analyses were not performed due to limited data. No results on patient-reported quality of life and safety were evaluated, which were outcomes considered for this review to be important to patients. Thus, these limitations must be considered when drawing conclusions on the results of the NMA.
Other Relevant Evidence
This section includes the long-term extension study included in the sponsor’s submission to CADTH that was considered to address important gaps in the evidence included in the systematic review.
FIRST-EXT Long-Term Extension Study
Methods
The FIRST-EXT trial was a 2-year, open-label, single-arm extension study conducted to evaluate the efficacy and safety of DFP consisting of 500 mg tablets or 80 mg/mL oral solution for the treatment of patients with transfusional iron overload due to SCD or other anemias.30 This oral solution was less than the 100 mg/mL oral solution approved by Health Canada; however, the total daily dosage used in the trial was consistent with the Health Canada–approved dosage of 75 mg/kg to 100 mg/kg per day.
A total of 164 patients completed the FIRST trial; of these, 134 were enrolled in the FIRST-EXT extension study, with the first patient enrolled in May 2015. A total of 89 patients received DFP and 34 received DFO in the pivotal study, all of whom either continued receiving or were switched to DFP in the extension study. An interim analysis found that a sufficient number of patients were enrolled to meet the pivotal trial’s primary end point; due to a lack of patient enrolment, the trial was terminated in April 2019. The extension study was also terminated, as existing methods of surveillance for the safety of DFP were equally as or more informative than what was being obtained from the extension study.30
The primary objective of the extension study was to assess the long-term safety and tolerability of DFP using the relevant safety outcomes assessed, including AEs, SAEs, and withdrawals due to AEs. The relevant efficacy outcomes assessed in the extension study as secondary end points included change in LIC as measured by MRI, cardiac iron load as measured by cardiac MRI T2*, and SF levels. In assessing these efficacy end points, baseline was defined as the start of DFP treatment. Therefore, for the patients who received DFP in the pivotal trial and were enrolled in the extension study (n = 89), year 1 of DFP treatment was at the completion of the pivotal trial, year 2 was at 12 months in the extension study, and year 3 was at the completion of the extension study. For patients who received DFO in the pivotal trial and continued into the extension to receive DFP (n = 45), year 1 of DFP treatment was defined as 12 months into the extension study and year 2 was at the completion of the extension study, with no year 3.30
Populations
A total of 134 patients were enrolled in the extension study from 14 sites in Egypt, the US, the UK, Saudi Arabia, and Canada. Of these patients, 115 (85.8%) had some form of SCD, while the remaining 19 (14.2%) had some other type of transfusion-dependent anemia. To enrol in the extension study, patients were required to have completed the pivotal FIRST study. The overall median age for all participants in the extension study was 14.0 years (range, 4 to 47), slightly more than half were male (60.4%), and most patients were white (85.1%).30 Overall, the baseline characteristics of the patients enrolled in the extension study were consistent with the baseline characteristics of the patients randomized in the pivotal trial. Refer to Table 27 for a summary of the baseline characteristics of the patients enrolled in the FIRST-EXT trial.
Table 27: Summary of Baseline Characteristics for FIRST-EXT Extension Study
Characteristic | FIRST-EXT N = 134 |
|---|---|
Age, years, median (range) | 14.0 (4 to 47) |
Males, n (%) | 81 (60.4) |
Race n (%) | |
White | 114 (85.1) |
Black | 20 (14.9) |
Ethnicity n (%) | |
Hispanic or Latino | 1 (0.7) |
Other | 133 (99.3) |
Primary diagnosisa n (%) | |
Sickle cell anemia | 73 (54.5) |
Hemoglobin s-beta-thalassemia | 40 (29.9) |
Sickle cell with hemoglobin C disease | 2 (1.5) |
Hereditary spherocytosis | 8 (6.0) |
Congenital dyserythropoietic anemia | 4 (3.0) |
Spherocytic anemia | 4 (3.0) |
Hemoglobinopathy | 1 (0.7) |
Hemolytic anemia | 1 (0.7) |
Autoimmune hemolytic anemia | 1 (0.7) |
LIC (mg/g dw) | |
n (%) | 133 (99.3) |
Mean (SD), range | 14.9 (7.6), 2.3 to 36.8 |
Cardiac MRI T2* (ms) | |
n (%) | 131 (97.8) |
Mean (SD), range | 32.69 (17.6), 20.70 to 48.2 |
Serum ferritin (mcg/L) | |
n (%) | 134 (100.0) |
Mean (SD), range | 3,894 (2,591), 134 to 12,397 |
Concomitant medications taken by ≥ 10% of patients, n (%) | |
Folic acid | █ ████████ |
Levocarnitine | █ ████████ |
Ibuprofen | █ ████████ |
Paracetamol | █ ████████ |
Decal B12 | █ ████████ |
Sodium chloride | █ ████████ |
Hydroxycarbamide | █ ████████ |
Ranitidine hydrochloride | █ ████████ |
Ketorolac tromethamine | █ ████████ |
Augmentin | █ ████████ |
Benzylpenicillin | █ ████████ |
Phenoxymethylpenicillin | █ █████████ |
dw = dry weight; LIC = liver iron concentration; ms = milliseconds; SCD = sickle cell disease; SD = standard deviation.
aThe diagnoses of sickle cell anemia, hemoglobin s-beta-thalassemia, and sickle cell with hemoglobin C disease are in the category of SCD; all remaining diagnoses are in the category of “other anemias.”
Source: Clinical Study Report for FIRST-EXT.30
Interventions
Upon inclusion into the extension study, patients who received DFP in the FIRST pivotal trial continued on the same dosing regimen. Patients who had been treated with DFO in the pivotal trial had the following doses of DFP 3 times daily in the extension study: 15 mg/kg in week 1, 20 mg/kg in week 2, followed by 25 mg/kg in week 3. The dosage could have been increased to 33 mg/kg 3 times daily if, in the 3 months prior, patients’ transfusional iron input was greater than 0.3 mg/kg body weight or if they had an SF of 2,500 mcg/L or greater, an LIC of 15 mg/g dw or greater, or a cardiac T2* of 20 ms or greater at visit 1 of the extension study.30
Any patient’s dose could be increased to the upper limit of 33 mg/kg at any time in the extension study if their transfusional iron input was greater than 0.3 mg/kg per day for 3 months or greater or if, in the previous 6 months, there had been an improvement of less than 10% in any of the measures indicative of iron overload. Doses could be decreased based on assessment of safety markers for adverse reactions that were possibly dose-dependent. Concomitant medications considered necessary for the patient’s well-being could be taken at the discretion of the investigator. Rescue medication (DFO or a combination of 2 iron chelators from among DFP, DFO, and DFX) for the treatment of iron overload was not permitted during the study.30
Outcomes
The primary objective of the extension study was to assess the long-term safety and tolerability of DFP using the relevant safety outcomes assessed, including AEs, SAEs, and withdrawals due to AEs. Secondary outcomes to evaluate the long-term efficacy of DFP were consistent with those assessed in the pivotal trials and included the change from baseline in LIC measured by MRI, cardiac MRI T2*, and SF levels. A responder analysis was also conducted, which was defined as the percentage of patients who showed a 20% or greater decline from baseline in LIC or SF, or a 20% or greater increase from baseline in cardiac MRI T2*. Subgroup analyses of these efficacy outcomes were conducted on patients with SCD compared with outcomes for other anemias.30
Statistical Analysis
All analyses were descriptive for the FIRST-EXT extension study. Efficacy analyses were conducted on the ITT population and safety analyses were conducted on the safety population, both of which consisted of enrolled patients who received 1 or more doses of the study medication (n = 134). A secondary efficacy analysis for the efficacy end points was conducted on the PP population that consisted of all enrolled patients who completed the study, had no major protocol violations, and had an efficacy assessment at the end of the study (n = 51). Analyses included mean change from baseline for efficacy outcomes, which was tested against no change using a 1-sample t-test. Cardiac MRI T2* data were log-transformed for normalization of the data, and the change in the geometric mean of the log-transformed data was expressed as a ratio (i.e., geometric mean at a time point divided by the geometric mean at baseline). No imputation was made for missing data.30
Patient Disposition
The patient disposition for the extension study is summarized in Table 28. Of the 164 patients who completed the pivotal trial, LA38 to 0411 (FIRST), 134 were enrolled in the extension study, FIRST-EXT. A total of 58 (43.3%) withdrew from the extension study, with 23.1% withdrawing involuntarily due to sponsor decision when the study was terminated. A total of 9 patients (6.7%) discontinued due to patient request and 9 (6.7%) discontinued due to protocol deviation (8 of the 9 patients missed 4 of the 6 weekly complete blood count tests). Other reasons for discontinuation included AEs (3.0%), investigator decision (2.2%), and loss to follow-up (1.5%).30
Table 28: Patient Disposition in FIRST-EXT Trial (ITT Population)
Disposition | FIRST-EXT N = 134 |
|---|---|
Completed LA38 to 0411, n | 164 |
Enrolled in FIRST-EXT, n | 134 |
Exposed, n | 134 |
Completed, n (%) | 76 (56.7) |
Discontinued, n (%) | 58 (43.3) |
Reason for discontinuation, n (%) | |
AE | 4 (3.0) |
Patient request | 9 (6.7) |
Lost to follow-up | 2 (1.5) |
Investigator decision | 3 (2.2) |
Protocol deviation | 9 (6.7) |
Sponsor decision | 31 (23.1) |
AE = adverse event; ITT = intention-to-treat.
Source: Clinical Study Report for FIRST-EXT.30
Exposure to Study Treatments
The median duration of treatment exposure to DFP throughout the pivotal trial and extension study was 285.5 person-years. The mean exposure was 2.1 years (SD = 0.8), with the duration ranging from 0.1 to 3.0 years. A total of 122 patients (91.0%) received DFP for at least 1 year, 69 (51.5%) received it for at least 2 years, and 56 (41.8%) received it for at least 2.5 years. Partly owing to the early termination of the study, only 3 patients received it for the maximum of 3 years.
Efficacy
Liver Iron Concentration
The mean change from baseline to year 1, year 2, and year 3 in LIC as measured by MRI scans was −2.64 (SD = 4.64), −3.91 (SD = 6.38), and −6.64 (SD = 7.72), respectively (Table 29). Subgroup analyses found that in patients with SCD, the mean change at year 1, year, 2, and year 3 from baseline was −2.33 (SD = 4.41), −3.41 (SD = 6.04), and −6.05 (SD = 7.71), respectively; in patients with other anemias, the mean change was −4.43 (SD = 5.62), −6.93 (SD = 7.68), and −9.96 (SD = 7.30) at year 1, year 2, and year 3, respectively. Similar trends were seen for the PP population.
Responders were defined as individuals who had at least a 20% decline in LIC since the start of DFP treatment. A total of 60 out of 129 patients (46.5%) were LIC responders at year 1, 64 out of 112 (57.1%) at year 2, and 39 out of 59 (66.1%) at year 3.
█████████ █ █ ███████ █ █ ████████ █ ██████ █ █ ███████████ ██ ███████████ █ █ █████ █ ███ █ █ ███████ █ █ █████ █ █ ████████████████████ █ █ ██ █ █████████████████ █ █ █ █ ██████████ █ █ █ ███ █ ██ █ █ ██ █ █ █ █████ █ █ ████████ █ █ ██████ █ █ ███████ █ █ ████████████████ █ █ ██ █ █ █ █ █████ █ ███████████ █ █ ██ █ █ █ ████ █ █ █████ █ ██████ █ ████ █ █ ████████ █ █ ██ █ ███ █ ██ █ █ █ ███ █ ███████████ █ █ ██ █ █████ █ █ ███ █ █ █ ███████████ █ █ █████████████████████ █ ███ █ █ ████████ █ █ ███ █ ██ █ █████ █ █ ██████████ █ ██████ █ ███ █ ██ █ █ █ █ ██ █ ████ █ █ ███ █ █ ██ ██ █ ██ █ █ █████████████ █ ██████ █ █ ███ █ ███ █ █ ████████████ █ ███ █ ████ █ █ ██ █ █████ █ ██ ████ █ ███ █ █ █████ █ ██
Serum Ferritin Levels
The mean change from baseline to year 1, year 2, and year 3 in SF levels was −1 mcg/L (SD = 1,986), −771 mcg/L (SD = 2,171), and −1,016 mcg/L (SD = 3,617), respectively. Subgroup analyses showed that in patients with SCD, the mean change was 130 mcg/L (SD = 2,086), −711 mcg/L (SD = 2,310), and −918 mcg/L (SD = 3,926) at year 1, year 2, and year 3, respectively. In patients with other anemias, the mean change was −733 mcg/L (SD = 1,066), −1,095 mcg/L (SD = 1,175), and −1,517 mcg/L (SD = 1,120) at year 1, year 2, and year 3, respectively. In the PP population, the mean SF values rose slightly from baseline at year 1, dropped at year 2, and then rose numerically at year 3, but remained below the baseline level.
Responders were defined as patients who had at least a 20% decline in SF (mcg/L) since the start of DFP treatment. A total of 44 out of 125 patients (35.2%) were deemed responders at year 1, 53 out of 96 (55.2%) at year 2, and 39 out of 55 (70.9%) at year 3.
Table 29: Efficacy Outcomes in FIRST-EXT Trial (ITT Population)
Efficacy outcomes | N | FIRST-EXT N = 134 | P valuea |
|---|---|---|---|
LIC (mg/g dw), mean (SD) | |||
Baselineb | ███ | ███████ | ███████ |
Year 1 | ███ | ███████ | |
Change from baseline to year 1 | ███ | █ █ █ ██ █ ██████ | |
Year 2 | ███ | ██ █ ██████ | ███████ |
Change from baseline to year 2 | ███ | ███████ | |
Year 3 | ██ | ███████ | ███████ |
Change from baseline to year 3 | ██ | ███████ | |
Responders for LIC,c n (%) | |||
Year 1 | ███ | █ █████████ | ██ |
Year 2 | ███ | █ █████████ | ██ |
Year 3 | ██ | █ █████████ | ██ |
Log cardiac MRI T2* (ms), geometric mean (% CV) | |||
Baseline | ███ | █ █ █ █ █ ████████ | ██████ |
Year 1 | ███ | █ █ █ █ █ ████████ | |
Change from baseline to year 1 | ███ | █ █ █ █ ████████ | |
Year 2 | ███ | █ █ █ █ █ ████████ | ██████ |
Change from baseline to year 2 | ███ | █ █ █ █ ████████ | |
Year 3 | ██ | █ █ ████████ | ██████ |
Change from baseline to year 3 | ██ | █ █ █ █ ████████ | |
Responders for cardiac MRI T2*,d n (%) | |||
Year 1 | ███ | █ █ █ ██ █ ████████ | ██ |
Year 2 | ███ | █ █ █ █████████ | ██ |
Year 3 | ██ | █ █ █ █████████ | ██ |
SF (mcg/L), mean (SD) | |||
Baseline | ███ | █ █ ██ █ ███████ | ██████ |
Year 1 | ███ | █ █ ██ █ ███████ | |
Change from baseline to year 1 | ███ | █ █ █ ██ █ █████████ | |
Year 2 | ██ | █ █ █ █ █ ████████ | ██████ |
Change from baseline to year 2 | ██ | █ █ █ █ ████████ | |
Year 3 | ██ | █ █ █ █ █ ████████ | ██████ |
Change from baseline to year 3 | ██ | ██ █ █ █ █ ██ █ ███████ | |
Responders for SF (mcg/L),e n (%) | |||
Year 1 | ██ | █ █ █ █████████ | ██ |
Year 2 | ██ | █ █ █ █████████ | ██ |
Year 3 | ██ | █ █ █ █████████ | ██ |
CV = coefficient of variation; DFO = deferoxamine; DFP = deferiprone; dw = dry weight; ITT = intention-to-treat; LIC = liver iron concentration; ms = milliseconds; NA = not applicable; SD = standard deviation; SF = serum ferritin.
aP values have not been adjusted for multiple testing (i.e., the type I error rate has not been controlled).
bFor patients who received DFP in LA38 to 0411, baseline was defined as the start of LA38 to 0411; for patients who received DFO in LA38 to 0411, baseline was defined as the start of FIRST-EXT.
cResponders were defined as patients who had a ≥ 20% decline in LIC since the start of DFP treatment.
dResponders were defined as patients who had a ≥ 20% increase in cardiac MRI T2* since the start of DFP treatment.
eResponders were defined as patients who had a ≥ 20% decline in SF since the start of DFP treatment.
Source: Clinical Study Report for FIRST-EXT.30
Table 30: Efficacy Outcomes in FIRST-EXT Trial by Subgroup — SCD and Other Anemias (ITT Population)
Efficacy outcomes | Patients with SCD | Patients with other anemias | ||||
|---|---|---|---|---|---|---|
N | FIRST-EXT N = 115 | P valuea | N | FIRST-EXT N = 19 | P valuea | |
LIC (mg/g dw), mean (SD) | ||||||
Baselineb | ███ | ███████ | ███████ | ██ | █ █ █ ██ █ ██████ | ██████ |
Year 1 | ███ | ██ █ ██████ | ██ | ███████ | ||
Change from baseline to year 1 | ███ | █ █ █ ███████ | ██ | ██ █ ██████ | ||
Year 2 | ██ | ███████ | ███████ | ██ | █ █ █ ███████ | ██████ |
Change from baseline to year 2 | ██ | ██ █ ██████ | ██ | ██ █ ██████ | ||
Year 3 | ██ | █ █ █ ███████ | ███████ | █ | █ █ █ ███████ | ██████ |
Change from baseline to year 3 | ██ | ██ █ ██████ | █ | ██ █ ██████ | ||
Responders for LIC,c n (%) | ||||||
Year 1 | ███ | █ ████████ | ██ | ██ | ████████ | ██ |
Year 2 | ██ | █ ████████ | ██ | ██ | █ █████████ | ██ |
Year 3 | ██ | █ ████████ | ██ | █ | █ ██ █ ██████ | ██ |
Log cardiac MRI T2* (ms), geometric mean (% CV) | ||||||
Baseline | ███ | █ █ █ ████████ | ██████ | ██ | ████████ | ██████ |
Year 1 | ███ | █ █ █ ████████ | ██ | █ █ █ ████████ | ||
Change from baseline to year 1 | ███ | █ █ ██ █ ███████ | ██ | █ █ ████████ | ||
Year 2 | ██ | ████████ | ██████ | ██ | █ █ █ ████████ | ██████ |
Change from baseline to year 2 | ██ | █ █ ████████ | ██ | █ █ ████████ | ||
Year 3 | ██ | █ █ █ ████████ | ██████ | █ | █ █ █ ████████ | ██████ |
Change from baseline to year 3 | ██ | █ █ ██ █ ███████ | █ | █ █ ██ █ ███████ | ||
Responders for cardiac MRI T2*,d n (%) | ||||||
Year 1 | ███ | █ ████████ | ██ | ██ | █ ██ █ ██████ | ██ |
Year 2 | ██ | █ ██ █ ████████ | ██ | ██ | █ ██ █ ██████ | ██ |
Year 3 | ██ | █ ██ █ ████████ | ██ | █ | █ ██ █ ██████ | ██ |
Serum ferritin (mcg/L), mean (SD) | ||||||
Baseline | ███ | ██ █ ███████ | ██████ | ██ | █ █ █ ████████ | ██████ |
Year 1 | ███ | █ █ █ ████████ | ██ | █ █ █ ████████ | ||
Change from baseline to year 1 | ███ | █ ██ █ ███████ | ██ | █ █ ████████ | ||
Year 2 | ██ | ████████ | ██████ | ██ | █ ██ █ █████ | ██████ |
Change from baseline to year 2 | ██ | █ █ ████████ | ██ | ██ █ █ ████████ | ||
Year 3 | ██ | ██ █ ███████ | ██████ | █ | ██████ | ██████ |
Change from baseline to year 3 | ██ | █ █ ██ █ ███████ | █ | ██ █ █ ████████ | ||
Responders for SF (mcg/L),e n (%) | ||||||
Year 1 | ███ | █ █████████ | ██ | ██ | █ █████████ | ██ |
Year 2 | ██ | █ █████████ | ██ | ██ | █ █████████ | ██ |
Year 3 | ██ | █ █████████ | ██ | █ | █ ███████ | ██ |
CV = coefficient of variation; DFO = deferoxamine; DFP = deferiprone; dw = dry weight; ITT = intention-to-treat; LIC = liver iron concentration; ms = milliseconds; NA = not applicable; SCD = sickle cell disease; SD = standard deviation; SF = serum ferritin.
aP values have not been adjusted for multiple testing (i.e., the type I error rate has not been controlled).
bFor patients who received DFP in LA38 to 0411, baseline was defined as the start of LA38 to 0411. For patients who received DFO in LA38 to 0411, baseline was defined as the start of FIRST-EXT.
cResponders were defined as patients who had a ≥ 20% decline in LIC since the start of DFP treatment.
dResponders were defined as patients who had a ≥ 20% increase in cardiac MRI T2* since the start of DFP treatment.
eResponders were defined as patients who had a ≥ 20% decline in SF since the start of DFP treatment.
Source: Clinical Study Report for FIRST-EXT.30
Harms
The summary of treatment-emergent AEs during the extension study is presented in Table 31. A total of 104 patients (77.6%) reported a total of 836 AEs. The most common AEs were pyrexia (26.1%), bone pain (26.1%), abdominal pain (19.4%), and sickle cell crisis (18.7%), which were also reported in the pivotal trial. The clinical expert noted that these events are commonly observed in patients living with SCD.30
A total of 35 patients (26.1%) reported SAEs during the extension study, with the most common being sickle cell crisis (14.2%) and neutropenia (9.0%). A total of 13 patients (9.7%) experienced SAEs that were considered related to the study drug, including neutropenia (9.0%), agranulocytosis (1.5%), thrombocytopenia (0.7%), and generalized edema (0.7%). One death was reported during the extension study in a patient hospitalized with generalized edema. The cause of death was not clarified, although hepatic encephalopathy was clinically suspected by the investigator. The investigator considered the generalized edema to be possibly related to DFP. Of the notable harms, 1 patient (0.70%) experienced severe neutropenia and 2 patients (1.5%) experienced agranulocytosis during the extension study.30
Table 31: Summary of Harms in FIRST-EXT Extension Study (Safety Population)
AE | FIRST-EXT N = 134 |
|---|---|
Patients with ≥ 1 AE, n (%) | █ ███████ |
Most common AEs,a n (%) | |
Pyrexia | █ ██ █ ████████ |
Bone pain | █ █████████ |
Abdominal painb | █ █████████ |
Sickle cell crisisc | █ █████████ |
Pain in extremity | █ █████████ |
Oropharyngeal pain | █ █████████ |
Nasopharyngitis | █ █████████ |
Back pain | █ ██ █ ████████ |
Neutrophil count decreasedd | █ ██ █ ████████ |
Neutropenia | █ ████████ |
Headache | █ ████████ |
Arthralgia | █ ████████ |
Vomiting | █ ████████ |
Pharyngitis | █ ██ █ ███████ |
Upper respiratory tract infection | █ ██ █ ███████ |
Musculoskeletal pain | █ ████████ |
Nausea | █ ██████ |
Cough | █ ██ █ █████ |
Patients with ≥ 1 SAE, n (%) | █ █████████ |
Most common SAEs, n (%)e | |
Sickle cell crisisc | █ ██ █ ████████ |
Neutropenia | █ ██ █ ███████ |
Pyrexia | █ ██████ |
Cholecystectomy | █ ██████ |
Agranulocytosis | █ ██ █ █████ |
Pneumonia | █ ██ █ █████ |
Arthralgia | █ ██████ |
Splenectomy | █ ██████ |
Hypotension | █ ██████ |
SAEs at least possibly related to study treatment, n (%) | █ ██ █ ███████ |
AEs leading to study withdrawal, n (%) | █ ██████ |
Deaths, n (%) | █ ███████ |
Notable harms, n (%) | |
Severe neutropenia | █ ██ █ █████ |
Agranulocytosis | █ ██████ |
AE = adverse event; SAE = serious adverse event.
aObserved in ≥ 5% of patients.
bIncludes the preferred terms of abdominal pain and abdominal pain upper.
cIn the source tables, this preferred term appears as “sickle cell anemia with crisis.”
dDefined as a single occurrence of an absolute neutrophil count of < 1.5 × 109/L with no confirmatory second value < 1.5 × 109/L within 3 days.
eSAEs affecting ≥ 2 patients.
Source: Clinical Study Report for FIRST-EXT.30
Critical Appraisal
Internal Validity
The FIRST-EXT extension study allowed for the investigation of the long-term efficacy and harms of DFP for up to 3 years.30 The limitations of the extension study include the absence of an active comparator and the fact that potential confounders were not accounted for, which limits causal conclusions. An additional limitation is the open-label study design and unblinding of the study drug in the extension trial, which can bias the reporting of harms outcomes, as they are subjective in measurement and interpretation. These harms could have been overestimated, since both patients and their treating clinicians were aware of the treatment received. The extension study only included patients who had successfully completed the pivotal FIRST trial, which may have introduced selection bias, as this could have resulted in the enrolment of patients into the extension study who were better able to tolerate DFP.
The interpretation of outcomes is also limited by the large amount of missing data due to attrition. The discontinuation rate of the extension study was high, with 20.1% of patients discontinuing before study termination and there was no imputation for missing data.30 This could have resulted in an increased risk of attrition bias in favour of the intervention, as patients not responding to treatment may be less likely to continue participation in the extension study and patients who are experiencing AEs may also be less likely to continue in the study. Subgroup analyses were descriptive and often limited to few patients, reducing the chance of detecting a true effect.
External Validity
As the extension study consisted of patients who took part in the pivotal FIRST parent study, it is reasonable to expect that the same limitations related to generalizability apply to the extension study. The characteristics of the patients enrolled in the trial were representative of the patient population in Canada, according to the clinical expert consulted. The clinical expert noted that concomitant use of levocarnitine is not common in Canadian clinical practice, which may limit the generalizability of the results. As is inherent in all studies of chronic disease, it is difficult to conclude with certainty if the safety and efficacy results can be extrapolated over the lifetime of a patient receiving DFP; however, the clinical expert consulted stated that the length of the extension study was appropriate to evaluate the long-term safety and efficacy of DFP. In any extension study, patients are likely selected in favour of being ideal study participants, which may overcall the long-term compliance and efficacy reflected in the real world.
Discussion
Summary of Available Evidence
The evidence for this review was derived from a systematic literature review of pivotal and phase III studies that was supplemented with additional studies to address important gaps in the RCT evidence. The systematic review included 2 RCTs: FIRST (N = 213) and Calvaruso et al. (2014) (N = 60).2,3 The FIRST trial was a pivotal, multicentre, randomized, open-label, noninferiority study comparing the efficacy and safety of DFP versus DFO in patients with SCD or other transfusion-dependent anemias.2 Calvaruso et al. (2014) conducted a 5-year, multicentre, open-label RCT to compare the safety and efficacy of DFP versus DFO in Italian patients.3
The FIRST trial compared the efficacy of DFP and DFO on end points that are important in the clinical management of transfusional iron overload, most notably LIC, over a 12-month period. The trial included HRQoL outcomes but was not designed to evaluate potential differences in the improvement and/or management of disease-related symptoms, physical functioning, or increased survival.11 The patients enrolled in the FIRST trial were considered by the clinical expert to be a reasonable representation of the target population in Canada. Calvaruso et al. (2014) conducted a 5-year, multicentre, open-label RCT to compare the safety and efficacy of DFP versus DFO in Italian patients.3 This was a small study that only evaluated change from baseline SF, an end point that is not typically used in Canada to evaluate longer-term treatment response. In addition, there were important limitations with the external validity of the study that limit the ability to draw any conclusions regarding the long-term comparative efficacy and safety of DFP versus DFO.
The evidence from the studies included in the systematic review was supplemented with 1 long-term extension-phase study (FIRST-EXT; N = 134) and 1 indirect comparison submitted by the sponsor. The FIRST-EXT trial was a 2-year, open-label, single-arm extension study conducted to evaluate the efficacy and safety of DFP. All patients from the FIRST trial who were enrolled in the FIRST-EXT extension study received DFP (i.e., either continued therapy from the pivotal trial or switched to DFP from DFO). The sponsor-submitted Bayesian NMA evaluated the relative efficacy of DFP versus DFO and DFX in patients with SCD and transfusional iron overload.
Interpretation of Results
Efficacy
CanHaem’s Consensus Statement on the Care of Patients With Sickle Cell Disease in Canada recommends that patients receiving iron chelation therapy have their LIC and CIC assessed every 6 to 12 months, depending on iron overload severity.6 The clinical expert consulted by CADTH noted that LIC is typically the most reliable clinical outcome for evaluating response to iron chelation therapy. In the FIRST trial, change from baseline in LIC was the primary end point and DFP was shown to be noninferior to DFO at 12 months. Results of the extension study supported the persistence of efficacy for those who remained on the treatment for up to 3 years, with LIC levels continuing to decline over the study period. Overall, the clinical expert consulted by CADTH concluded that the results suggest DFP is an effective treatment option for the management of transfusional iron overload for SCD and other anemias.
Similar to the approach used in the FIRST trial, LIC in patients in Canada is typically evaluated using a validated MRI technique. In rare cases where MRI is not possible (e.g., due to a contraindication), a liver biopsy may be required to quantify LIC. As liver biopsy is an invasive procedure that may be associated with risks for the patients, the decision to perform the biopsy would be made in consultation with a liver specialist and would take into account other indications for performing the biopsy (e.g., presence of cirrhosis). Alternatively, a combination of historical LIC and the corresponding ferritin level and a current downward trend or stability in SF would be a reasonable assessment of efficacy. However, a stably high or upward trend in SF would be an insufficient demonstration of efficacy, even though liver iron may be improving in some cases.
Change from baseline in cardiac iron was a secondary end point of the FIRST trial. Routine monitoring of cardiac iron is recommended by the CanHaem consensus statement on the management of SCD and currently occurs in Canadian practice.6 The clinical expert consulted by CADTH noted that cardiac iron deposition is not commonly observed in patients with SCD in Canada. This is reflective of the baseline characteristics in the FIRST trial, where patients in both treatment groups showed normal levels of cardiac iron at baseline, and no significant changes were seen at any time point throughout the study (with no differences between the groups).2 As such, monitoring changes in CIC would not be useful for evaluating response to treatment for the purposes of drug reimbursement, unless this is the primary indication for iron chelation in a particular patient.
Change from baseline in SF was a secondary end point of the FIRST trial and the primary end point of the Calvaruso et al. (2014) study.3 In the FIRST trial, DFP demonstrated noninferiority versus DFO for change from baseline in SF at 12 months. After initiating the study treatments, SF levels declined consistently in the DFO group but initially rose in the DFP group, resulting in significant treatment group differences at 3 and 6 months favouring DFO over DFP. The SF levels in the DFP group subsequently declined, with no significant group differences seen at 9 and 12 months.2 Health Canada reviewers noted that similar results were observed in the pivotal DFP study for the treatment of patients with transfusional iron overload due to thalassemia syndromes.11 The sponsor has indicated that the transient rise and subsequent decline in SF may be due to the mechanism of action of DFP, where it initially transfers intracellular labile iron to transferrin or other biologic iron acceptors, resulting in a transient rise in SF that is then followed by a decline as excess iron is cleared from the body.11
CanHaem and the clinical expert consulted by CADTH have noted that SF level is the most commonly used test to screen for patients with transfusional iron overload (as it is inexpensive and widely available). When observed in isolation, SF levels may not be an accurate marker of transfusional iron overload, as they may be increased in the presence of inflammation, liver disease, vitamin C deficiency, and in patients who are experiencing or have experienced a vasoocclusive episode. However, both the clinical expert consulted by CADTH and CanHaem note that SF can be a useful test for evaluating patients who are clinically stable when used in conjunction with monitoring transfusion volume and the routine monitoring of liver iron.6
Patients with transfusional iron overload with pre-existing renal impairment may have limited options for iron chelation therapy, particularly if there are challenges with tolerance and/or adherence with SC or IV administration. The clinical expert consulted by CADTH and the input from CanHaem indicated there is an unmet need and strong rationale for using DFP in this population, and Health Canada reviewers similarly noted that DFP may help fulfill an unmet need for patients with renal impairment.11
Subgroup analyses were conducted for patients with SCD (83% of the trial population) and those with other anemias (17% of the trial population). Due to the small sample size, there was considerable uncertainty in the estimates of effect for the subgroup analyses for patients with other anemias. Overall, the results were supportive of the primary analysis. Those with other anemias that were treated with DFP demonstrated statistically significant reductions from baseline in LIC throughout the FIRST trial and the FIRST extension-phase study. The sponsor and the clinical expert consulted by CADTH noted that the “other anemias” where patients could develop transfusional iron overload and require iron chelation therapy (i.e., those conditions other than SCD and thalassemia) are rare conditions and it would be challenging to design a clinical trial to specifically evaluate the comparative efficacy of therapeutic options. The sponsor’s perspective, that transfusional iron overload is a common pathophysiological process irrespective of the primary condition, was acceptable to regulatory authorities, with the exception of patients with myelodysplastic syndromes and Diamond-Blackfan anemia. As these patients were specifically excluded from the trial population, the Canadian product monograph states that the safety and effectiveness of DFP have not been established for the treatment of transfusional iron overload in patients with myelodysplastic syndrome or in patients with Diamond-Blackfan anemia.
The FIRST trial evaluated 2 HRQoL end points (SF-36 and CHQ scores) and there was no statistically significant difference between the DFP and DFO treatment groups,2 though important limitations such as open-label administration and missing data preclude drawing any conclusions regarding the impact of orally administered DFP versus SC-administered DFO. DFP is marketed in Canada as 500 mg and 1,000 mg oral tablets as well as a 100 mg/mL oral solution. No patient groups responded to the call for patient input for the current review of DFP; however, in a previous CADTH review of DFP for the treatment of transfusional iron overload in patients with thalassemia syndromes, the Thalassemia Foundation of Canada noted that patients would value additional oral treatment options. The clinical expert consulted by CADTH also noted that the availability of an oral solution could be a useful option for the management of pediatric patients who require chelation therapy.
The sponsor-submitted Bayesian NMA evaluated the relative efficacy of DFP with DFO and DFX in patients with SCD who underwent transfusional therapy. The NMA included 2 studies, 1 comparing DFP with DFO2,20 and the other comparing DFX with DFO.29 The NMA demonstrated no difference between DFP, DFO, and DFX for change from baseline in SF and LIC. A number of limitations were identified, including:
potential violation of the transitivity assumption
the dataset was sparse, leading to wide CrIs and potential failure to detect real differences
no data were reported on harms
no sensitivity analyses were performed due to a dearth of data.
Thus, these limitations must be considered when drawing conclusions based on the results of the NMA.
Harms
The most common AEs reported among the patients treated with DFP in the clinical trials included pyrexia, abdominal pain, bone pain, headache, vomiting, extremity pain, sickle cell anemia with crisis, back pain, increased alanine aminotransferase and aspartate transferase, arthralgia, oropharyngeal pain, nasopharyngitis, decreased neutrophil count, cough, and nausea.2 The clinical expert consulted by CADTH noted that these events are typically seen in patients with SCD.
Health Canada reviewers noted that agranulocytosis is the most serious risk with DFP, and the product monograph includes a black-box warning stating that the drug can cause agranulocytosis (severe neutropenia) that may lead to serious and life-threatening infections.11 The product monograph recommends that absolute neutrophil count be measured before starting therapy and monitored weekly during therapy. The clinical expert consulted by CADTH noted that the recommendation for weekly monitoring is unlikely to be followed in routine clinical practice for patients receiving long-term treatment with DFP, as it is a heavy burden on patients to undergo such frequent monitoring. This level of monitoring could be followed in the initial months after treatment initiation but would likely not continue in the longer term. Monitoring would typically occur when the patient receives a transfusion (if on a chronic transfusion protocol), as they would typically undergo a complete blood count test, which includes neutrophil count. In addition, patients and caregivers are educated about symptoms that may be indicative of febrile neutropenia and instructed on when to seek medical attention. Patients receive a wallet card and are instructed to present it to the health care professionals when seeking medical attention to alert them that they are receiving a drug that carries a risk for febrile neutropenia.
The clinical expert consulted by CADTH noted patients with SCD are at risk of developing renal impairment from an early stage of life. For such patients, chelation options are limited (e.g., DFX is contraindicated for patients with SCD who have renal impairment). The FIRST trial was not designed or powered to evaluate potential differences in renal toxicity associated with DFP compared with DFO. To support the claims used in the economic evaluation that DFP has a superior renal AE profile compared with DFO, the sponsor included an unpublished comparison of estimated glomerular filtration rate (eGFR) values in DFP-treated patients from the FIRST-EXT trial against DFO- and DFX-treated patients obtained from a real-world dataset.31 The sponsor reported that DFP-treated patients demonstrated a reduced rate of eGFR decline at 24 months compared with DFX and DFO, with an LS mean change for DFP of −0.3 mL/min/1.73 m2 (SD = 6.41) versus −7.3 mL/min/1.73 m2 (SD = 1.24) for DFX and −8.9 mL/min/1.73 m2 (SD = 3.25) for DFO.31 The clinical expert consulted by CADTH noted that both DFO and DFX can pose a risk for renal toxicity and it was plausible that DFP may pose a lower risk for renal toxicity; however, the available evidence is insufficient to draw any conclusions regarding the magnitude and clinical relevance of any potential differences across the treatments for patients with no known contraindications. Similarly, CADTH identified numerous limitations with the unpublished data that precluded any conclusions regarding the comparative safety of these drugs, including heterogeneity in treatment setting (e.g., within the setting of phase III clinical trial versus real-world data), generalizability concerns regarding registry data from the US with the management of patients with SCD in Canadian practice, and significant loss to follow-up. In addition, the clinical expert noted that eGFR values would not be used in isolation within Canadian practice to inform treatment decisions (e.g., proteinuria would also be evaluated).
Conclusions
One 12-month, open-label, randomized, pivotal trial (FIRST; N = 230) demonstrated that orally administered DFP was noninferior to SC-administered DFO for change from baseline in LIC, SF, and cardiac iron in patients with SCD and other anemias who require iron chelation therapy for transfusional iron overload. Despite limitations, the sponsor-submitted ITC suggested that DFP also has similar efficacy in comparison with orally administered DFX for reducing LIC and SF at 12 months. The clinical expert consulted by CADTH indicated the evidence suggests DFP is an effective treatment option for the management of SCD in patients with transfusional iron overload. There is consensus across regulatory authorities, patient and clinician groups, and the clinical expert consulted by CADTH that DFP oral tablets and solution could help address an unmet need for patients with SCD. Anticipated use cases include patients with renal impairment (i.e., those who cannot receive DFX), prior intolerance or AEs with DFX or DFO resulting in discontinuation or dose reduction to a level that is subtherapeutic, and those who experience intolerance and/or adherence issues with IV or SC administration of DFO. Treatment with DFP may be associated with rare but serious AEs (i.e., severe neutropenia) as well as milder, more common side effects (e.g., transaminitis), and patients should be typically managed and monitored under the supervising care of health care teams with experience in the diagnosis and management of SCD and rare anemias with transfusional iron overload. The amount of long-term monitoring for AEs that is required for patients receiving DFP is not anticipated to be greater than that in current practice for patients receiving DFO or DFX.
References
1.El-Haj N, Hoppe CC. Newborn screening for SCD in the USA and Canada. Int J Neonatal Screen. 2018;4(4):36. PubMed
2.Clinical Study Report: LA38-0411. The efficacy and safety of Ferriprox®for the treatment of transfusional iron overload in patients with sickle cell disease or other anemias [internal sponsor's report]. In: Toronto (ON): ApoPharma, a division of Apotex Inc; 2019 Sep 20.
3.Calvaruso G, Vitrano A, Di Maggio R, et al. Deferiprone versus deferoxamine in sickle cell disease: results from a 5-year long-term Italian multi-center randomized clinical trial. Blood Cells Mol Dis. 2014;53(4):265-271. PubMed
4.Calvaruso G, Vitrano A, Di Maggio R, et al. Deferiprone versus deferoxamine in thalassemia intermedia: results from a 5-year long-term Italian multicenter randomized clinical trial. Am J Hematol. 2015;90(7):634-638. PubMed
5.Chou ST, Alsawas M, Fasano RM, et al. American Society of Hematology 2020 guidelines for sickle cell disease: transfusion support. Blood Adv. 2020;4(2):327-355. PubMed
6.Consensus statement on the care of patients with sickle cell disease in Canada. Version 2.0. In: Ottawa (ON): Canadian Haemoglobinopathy Association; 2014: https://emergencymedicinecases.com/wp-content/uploads/filebase/pdf/Canadian%20Sickle%20Cell%20Guidelines%202014.pdf. Accessed 2022 Aug 8.
7.Hoffbrand AV, Taher A, Cappellini MD. How I treat transfusional iron overload. Blood. 2012;120(18):3657-3669. PubMed
8.Belmont A, Kwiatkowski JL. Deferiprone for the treatment of transfusional iron overload in thalassemia. Expert Rev Hematol. 2017;10(6):493-503. PubMed
9.Raghupathy R, Manwani D, Little JA. Iron overload in sickle cell disease. Adv Hematol. 2010;2010:272940. PubMed
10.Remacha A, Sanz C, Contreras E, et al. Guidelines on haemovigilance of post-transfusional iron overload. Blood Transfus. 2013;11(1):128-139. PubMed
11.Health Canada reviewer's report: Ferriprox (deferiprone) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Ferriprox (deferiprone), 1000 mg oral tablet and 100 mg/mL oral solution. In: Woodbridge (ON): Chiesi Canada Corp; 2022 Jun 15.
12.Ware HM, Kwiatkowski JL. Evaluation and treatment of transfusional iron overload in children. Pediatr Clin. 2013;60(6):1393-1406. PubMed
13.Ferriprox (deferiprone): 500 mg and 1000 mg tablets, Ph.Eur.; 100 mg/mL oral solution, Ph.Eur. [product monograph]. In: Woodbridge (ON): Chiesi Canada Corp; 2021 Oct 12.
14.CADTH Canadian Drug Expert Committee final recommendation: deferiprone (Ferriprox - ApoPharma Inc.). In: Ottawa (ON): CADTH; 2016 Mar 18: https://www.cadth.ca/sites/default/files/cdr/complete/SR0448_cdr_complete_Ferriprox_March-23-16_e.pdf. Accessed 2022 Sep 8.
15.Desferal (deferoxamine mesylate): 500 mg/vial lyophilized powder, for injection [product monograph]. In: Dorval (QC): Novartis Pharmaceuticals Canada Inc; 2019 Jul 3: https://pdf.hres.ca/dpd_pm/00052016.PDF. Accessed 2022 Aug 8.
16.Exjade (deferasirox): 125 mg, 250 mg, and 500 mg, oral dispersible tablets for suspension [product monograph]. In: Dorval (QC): Novartis Pharmaceuticals Canada Inc; 2022 Aug 16: https://pdf.hres.ca/dpd_pm/00067002.PDF. Accessed 2022 Aug 8.
17.Jadenu (deferasirox): 90 mg, 180 mg, 360 mg oral tablets [product monograph]. In: Dorval (QC): Novartis Pharmaceuticals Canada Inc; 2022 Aug 16: https://pdf.hres.ca/dpd_pm/00067003.PDF. Accessed 2022 Aug 8.
18.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. J Clin Epidemiol. 2016;75:40-46. PubMed
19.Grey matters: a practical tool for searching health-related grey literature. In: Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2022 Jun 23.
20.Kwiatkowski JL, Hamdy M, El-Beshlawy A, et al. Deferiprone vs deferoxamine for transfusional iron overload in SCD and other anemias: a randomized, open-label noninferiority study. Blood Adv. 2022;6(4):1243-1254. PubMed
21.Hays RD, Morales LS. The RAND-36 measure of health-related quality of life. Ann Med. 2001;33(5):350-357. PubMed
22.Strand V, Singh JA. Improved health-related quality of life with effective disease-modifying antirheumatic drugs: evidence from randomized controlled trials. Am J Manag Care. 2008;14(4). PubMed
23.Samsa G, Edelman D, Rothman ML, Williams GR, Lipscomb J, Matchar D. Determining clinically important differences in health status measures. Pharmacoeconomics. 1999;15(2):141-155. PubMed
24.Leung YY, Ho K, Zhu T, Tam L-S, Kun E, Li E. Testing scaling assumptions, reliability and validity of medical outcomes study short-form 36 health survey in psoriatic arthritis. Rheumatology (Oxford). 2010;49(8):1495-1501. PubMed
25.Panepinto JA, O'Mahar KM, DeBaun MR, Loberiza FR, Scott JP. Health-related quality of life in children with sickle cell disease: child and parent perception. Br J Haematol. 2005;130(3):437-444. PubMed
26.HealthActCHQ iNC. CHQ: Child Health Questionnaire™. In:2021: https://www.healthactchq.com/survey/chq. Accessed 2022 Jul 7.
27.Rochon J. Sample size calculations for two-group repeated-measures experiments. Biometrics. 1991;47(4):1383-1398.
28.Drug Reimbursement Review sponsor submission: Ferriprox (deferiprone), 1000 mg oral tablet and 100 mg/mL oral solution [internal sponsor's package]. In: Woodbridge (ON): Chiesi Canada Corp; 2022 Jun 15.
29.Vichinsky E, Onyekwere O, Porter J, et al. A randomised comparison of deferasirox versus deferoxamine for the treatment of transfusional iron overload in sickle cell disease. Br J Haematol. 2007;136(3):501-508. PubMed
30.Clinical Study Report: LA38-EXT. Long-term safety and efficacy study of Ferriprox® for the treatment of transfusional iron overload in patients with sickle cell disease or other anemias [internal sponsor's report]. In: Toronto (ON): ApoPharma, a division of Apotex Inc; 2020 May 26.
31.Gluhcheva Y, Pashkunova-Martic I, Schaier M, et al. Comparative effects of deferiprone and salinomycin on lead-induced disturbance in the homeostasis of intrarenal essential elements in mice. Int J Mol Sci. 2022;23(8):15. PubMed
32.Hays RD, Sherbourne CD, Mazel RM. The RAND 36‐Item Health Survey 1.0. Health Econ. 1993;2(3):217-227. PubMed
33.Sogutlu A, Levenson JL, McClish DK, Rosef SD, Smith WR. Somatic symptom burden in adults with sickle cell disease predicts pain, depression, anxiety, health care utilization, and quality of life: the PiSCES project. Psychosomatics. 2011;52(3):272-279. PubMed
34.Asnani MR, Lipps GE, Reid ME. Validation of the SF-36 in Jamaicans with sickle-cell disease. Psychol Health Med. 2009;14(5):606-618. PubMed
35.Asnani M, Lipps G, Reid M. Component structure of the SF-36 in Jamaicans with sickle cell disease. West Indian Med J. 2007;56(6):491-497. PubMed
36.Bjorner JB, Wallenstein GV, Martin MC, et al. Interpreting score differences in the SF-36 Vitality scale: using clinical conditions and functional outcomes to define the minimally important difference. Curr Med Res Opin. 2007;23(4):731-739. PubMed
37.Hullmann SE, Ryan JL, Ramsey RR, Chaney JM, Mullins LL. Measures of general pediatric quality of life: Child Health Questionnaire (CHQ), DISABKIDS Chronic Generic Measure (DCGM), KINDL‐R, Pediatric Quality of Life Inventory (PedsQL) 4.0 Generic Core Scales, and Quality of My Life Questionnaire (QoML). Arthritis Care Res (Hoboken). 2011;63(Suppl 11):S420-S430. PubMed
38.Palermo TM, Schwartz L, Drotar D, McGowan K. Parental report of health-related quality of life in children with sickle cell disease. J Behav Med. 2002;25(3):269-283. PubMed
39.Ware J, Kosinski M, Bjorner JB, Turner-Bowker DM, Gandek B, Maruish ME. Determining important differences in scores. In: User's manual for the SF-36v2 health survey. 2nd ed. Lincoln (RI): Quality Metric Inc; 2007.
Appendix 1: Literature Search Strategy
Note that this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases:
MEDLINE All (1946 to present)
Embase (1974 to present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: July 5, 2022
Alerts: Weekly search updates until project completion
Search filters applied: RCTs; controlled clinical trials
Limits:
Publication date limit: none
Language limit: none
Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
MeSH | Medical Subject Heading |
exp | Explode a subject heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
# | Truncation symbol for one character |
adj# | Requires terms to be adjacent to each other within # number of words (in any order) |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Keyword heading word |
.dq | Candidate term word (Embase) |
.pt | Publication type |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multidatabase Strategy
deferiprone/ or (deferiprone* or deferrum* or deferum* or kelfer* or ferriprox* or upkanz* or cruderan* or feripon* or neferi* or DMOHPO or HDMPP or apo-066 or apo066 or apo-66 or apo66 or cgp-37391 or cpg37391 or cp-020 or cp020 or cp-20 or cp20 or crmd-001 or crmd001 or BRN 1447108 or CCRIS 8318 or DN-180-01-AF or HSDB 8335 or NSC 758880 or 2bty8kh53l).ti,ab,kf,ot,hw,nm,rn.
1 use medall
*deferiprone/ or (deferiprone* or deferrum* or deferum* or kelfer* or ferriprox* or upkanz* or cruderan* or feripon* or neferi* or DMOHPO or HDMPP or apo-066 or apo066 or apo-66 or apo66 or cgp-37391 or cpg37391 or cp-020 or cp020 or cp-20 or cp20 or crmd-001 or crmd001 or BRN 1447108 or CCRIS 8318 or DN-180-01-AF or HSDB 8335 or NSC 758880).ti,ab,kf,dq.
3 use oemezd
4 not (conference review or conference abstract).pt.
2 or 5
(Randomized Controlled Trial or Controlled Clinical Trial or Pragmatic Clinical Trial or Equivalence Trial or Clinical Trial, Phase III).pt.
Randomized Controlled Trial/
exp Randomized Controlled Trials as Topic/
“Randomized Controlled Trial (topic)”/
Controlled Clinical Trial/
exp Controlled Clinical Trials as Topic/
“Controlled Clinical Trial (topic)”/
Randomization/
Random Allocation/
Double-Blind Method/
Double Blind Procedure/
Double-Blind Studies/
Single-Blind Method/
Single Blind Procedure/
Single-Blind Studies/
Placebos/
Placebo/
Control Groups/
Control Group/
(random* or sham or placebo*).ti,ab,hw,kf.
((singl* or doubl*) adj (blind* or dumm* or mask*)).ti,ab,hw,kf.
((tripl* or trebl*) adj (blind* or dumm* or mask*)).ti,ab,hw,kf.
(control* adj3 (study or studies or trial* or group*)).ti,ab,kf.
(Nonrandom* or non random* or non-random* or quasi-random* or quasirandom*).ti,ab,hw,kf.
allocated.ti,ab,hw.
((open label or open-label) adj5 (study or studies or trial*)).ti,ab,hw,kf.
((equivalence or superiority or non-inferiority or noninferiority) adj3 (study or studies or trial*)).ti,ab,hw,kf.
(pragmatic study or pragmatic studies).ti,ab,hw,kf.
((pragmatic or practical) adj3 trial*).ti,ab,hw,kf.
((quasiexperimental or quasi-experimental) adj3 (study or studies or trial*)).ti,ab,hw,kf.
(phase adj3 (III or “3”) adj3 (study or studies or trial*)).ti,hw,kf.
or/7-37
6 and 38
remove duplicates from 39
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
Search terms: Ferriprox (deferiprone); SCD/other anemias
WHO ICTRP
International Clinical Trials Registry Platform, produced by WHO. Targeted search used to capture registered clinical trials.
Search terms: Ferriprox (deferiprone); SCD/other anemias
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
Search terms: Ferriprox (deferiprone); SCD/other anemias
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
Search terms: Ferriprox (deferiprone); SCD/other anemias
Grey Literature
Search dates: June 23, 2022, to July 5, 2022
Keywords: Ferriprox (deferiprone); SCD/other anemias
Limits: None
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Internet Search
Open Access Journals
Appendix 2: Description and Appraisal of Outcome Measures
Note that this appendix has not been copy-edited.
Aim
To describe the following outcome measures and review their measurement properties (validity, reliability, responsiveness to change, and MID):
SF-36 version 2
Child Health Questionnaire: Parent Form 50 (CHQ-PF50) and Child Form 87 (CHQ-CF87)
Findings
Table 33: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
Short Form (36) Health Survey Version 2 | The SF-36 is a generic self-reported HRQoL measure consisting of 8 subdomains: physical functioning, physical role limitations, bodily pain, general health perceptions, energy/vitality, social functioning, emotional role limitations, and mental health. The 8 subdomains are each measured on a scale of 0 to 100, with an increase in score indicating improvement in health status. The SF-36 provides 2 component summaries, PCS and MCS.32 | Validity: Supported by strong to moderate correlations with various instruments in studies of adults with SCD.33,34 Reliability: Cronbach alpha values for SF-36 domains ≥ 0.85 for all subscales in a study of adults with SCD.35 Responsiveness: Not assessed in indicated population. | PCS or MCS: 2.5 to 5 points for various conditions.21-23 Vitality scale: 5 points as assessed in a study of patients with chronic conditions that cause fatigue.36 Not assessed in indicated population. |
Child Health Questionnaire: Parent Form 50 and Child Form 87 | Generic instrument assessing 14 domains.37 CHQ-PF50: completed by patient/caregiver of child aged 5 to 18. CHQ-CF87: self-administered by children ≥ 10 years old.37 CHQ-PF50 provides 2 summary scores for physical and psychosocial health.25 | Validity: Significant differences in mean scores between children with SCD and healthy controls in all scales for CHQ-PF50 except for family cohesion.38 Reliability: Strong correlation between CHQ-PF28 and CHQ-CF87 in domain of impact of bodily pain; moderate correlations in 5 domains; poor correlations for role/social physical, mental health, family activities, and family cohesion.25 Responsiveness: Not assessed in indicated population. | Not assessed in indicated population. |
CHQ-CF87 = Child Health Questionnaire: Child Form 87; CHQ-PF28 = Child Health Questionnaire: Parent Form 28; CHQ-PF50 = Child Health Questionnaire: Parent Form 50; HRQoL = health-related quality of life; MCS = mental component score; MID = minimal important difference; PCS = physical component score; PHQ-15 = 15-item Patient Health Questionnaire; SCD = sickle cell disease; SF-36 = Short Form (36) Survey.
Short Form (36) Health Survey Version 2
The SF-36 is a generic self-reported health assessment questionnaire that has been used in clinical trials to study the impact of chronic disease on HRQoL. The SF-36 consists of 8 domains: physical functioning, role physical, bodily pain, general health, vitality, social functioning, role emotional, and mental health. The SF-36 also provides 2 component summaries: the physical component summary (SF-36 PCS) and the mental component summary (SF-36 MCS), which are created by aggregating scores on the 8 domains. The SF-36 PCS, SF-36 MCS, and 8 domains are each measured on a scale of 0 to 100, with an increase in score indicating improvement in health status. In general use of the SF-36, a change of 2 to 4 points in each domain or 2 to 3 points in each component summary indicates a clinically meaningful improvement as determined by the patient.39 The summary scales are scored using norm-based methods, with regression weights and constants derived from the general US population. Both the PCS and MCS scales are transformed to have a mean of 50 and an SD of 10 in the general US population. Therefore, all scores above/below 50 are considered above/below average for the general US population.24
Validity and Reliability
The SF-36 was assessed in a study of 230 adults from the US with SCD by comparing it to an abridged version of the 15-item Patient Health Questionnaire (PHQ-15); an instrument used to measure somatic symptoms.33 Pearson’s correlation coefficient was used to assess the correlations between of SF-36 subscale scores and total PHQ-15 scores resulting in moderate negative correlations ranging from r = −0.34 to −0.47 across all items, providing evidence of convergent validity. It should be noted that is not clear which version of the SF-36 was assessed in the study.33
The SF-36 version 2 was assessed by Asnani et al., in a study of adult patients with SCD in Jamacia (n = 489).34 The study found the instrument to be strongly correlated with WHO’s Quality of Life-BREF (r = 0.69), moderately correlated with the Flanagan Quality-of-Life Scale (r = 0.48) and the UCLA Loneliness Scale (r = 0.39), supporting convergent validity. The study also found strong internal consistency reliability with Cronbach alpha ranging from 0.85 to 0.91 for all subscales including the PCS and MCS.34
Responsiveness
Responsiveness has not been formally assessed for the instrument in the indicated population.
MID
The MID for either the PCS or MCS of the SF-36 is typically between 2.5 points and 5 points; however, this estimate is not specific to the indicated patient population.21-23 An MID for the SF-36 vitality scale was estimated to be 5 points as assessed in a study of patients (n = 3,445) with chronic conditions that cause fatigue.36 No MID has been formally assessed for the SF-36 in the indicated population.
Child Health Questionnaire Parent Form 50 (CHQ-PF50) and Child Form 87 (CHQ-CF87)
The CHQ is a generic quality of life measure that assesses the physical, emotional, and social aspects of health status in children and adolescent aged 5 to 18 years of age over the last 4 weeks.26 There are both parent-reported and child-reported versions of the of the questionnaire with varying lengths. The pivotal trial LA38 to 0411 used both the 50-item Parent Form (CHQ-PF50) completed by parents or caretakers of children aged 5 to 18 years of age and the 87-item Child Form (CHQ-CF87) which can be self-administered to patients 10 years of age or older.2,25 The 14 domains covered in the CHQ include: physical function, role/social limitations due to physical problems, general health perceptions, bodily pain/discomfort, family activities, role/social limitations due to behavioural problems, role/social limitations due to emotional problems, impact on parent time, impact on parent emotions, self-esteem, mental health, general behaviour, family cohesion, and change in health.25 Each item has a 4 to 6 point response scale reported as levels of intensity or agreement.25 Scores for each domain can be transformed to a total score from 0 to 100, with higher scores indicating better HRQoL.26 Only the CHQ-PF50 also provides 2 summary scores for physical and psychosocial health.25
Validity
A US study examined the validity of the instrument by comparing mean scores on the CHQ-PF50 as responded by parents/caregivers of children with SCD (n = 58) versus healthy children (n = 120), aged 5 to 18 years.38 Results found that children with SCD had lower mean scores in all dimensions compared with the healthy controls, illustrating construct validity using the known-groups approach. The physical health summary score was significantly different (P = 0.000) between groups with a mean (SD) of 39.4 (6.4) and 54.9 (3.2) for each of the SCD and healthy control groups, respectively. The psychosocial summary score was also significantly different (P = 0.000) between groups with a mean (SD) of 45.8 (4.5) for children with SCD and 53.0 (4.4) for the healthy control group. Using univariate F tests, results found significant differences between the 2 groups for 13 of the individual subscales, except for family cohesion, which was comparable between the groups.38
Reliability
A cross-sectional sample of parents (n = 95) and their children with SCD (n = 53; aged 5 to 18 years) completed the CHQ-PF28 (a shorter version of the CHQ-PF50) and the CHQ-CF87, respectively.25 Strong correlations (> 0.50) were observed between parent and child assessments of impact of bodily pain on HRQL (r = 0.58) and moderate correlations (0.30 to 0.50) in physical functioning (0.44), behaviour (r = 0.45), general health (r = 0.44), self-esteem (r = 0.40), and changes in health (r = 0.33), indicating good interrater reliability for these items.25 Correlations were poor (< 0.30) for role/social physical, mental health, family activities, and family cohesion, indicating different perspectives between parents and their children, with parents reporting lower mean scores for almost all items.25
Responsiveness
Responsiveness has not been formally assessed for the instrument in the indicated population.
MID
No MID has been formally assessed for the instrument in the indicated population.
Pharmacoeconomic Review
Abbreviations
AE
adverse event
BIA
budget impact analysis
DFO
deferoxamine
DFP
deferiprone
DFX
deferasirox
eGFR
estimated glomerular filtration rate
ICER
incremental cost-effectiveness ratio
ICT
iron-chelating therapy
MMRM
mixed model for repeated measures
NMA
network meta-analysis
QALY
quality-adjusted life-year
SCD
sickle cell disease
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Deferiprone (Ferriprox), tablet and oral solution |
Submitted price | Deferiprone, tablet 1,000 mg: $33.47 Deferiprone, oral solution 100 mg/mL: $3.35 per mL |
Indication | Treatment of iron overload in patients with sickle cell disease or other anemias |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | October 13, 2021 |
Reimbursement request | As per indication |
Sponsor | Chiesi Canada Corp. |
Submission history | Previously reviewed: Yes Indication: Transfusional iron overload due to thalassemia syndromes Recommendation date: March 18, 2016 Recommendation: List with clinical criteria and/or conditions |
NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Semi-Markov structure |
Target populations | People with SCD or other anemias who have transfusional iron overload |
Treatment | DFP |
Comparators | DFO DFX |
Perspective | Canadian publicly funded health care payer |
Outcome(s) | QALYs |
Time horizon | Lifetime (62.5 years) |
Key data source | LA38 to 0411 (FIRST) trial and an indirect treatment comparison to support the assumption of equivalent efficacy of DFP vs. DFX vs. DFO on chelating iron Real-world US medical records (TriNetX): Changes in patients’ eGFR over time |
Submitted results | Compared with DFX, DFP is associated with more QALYs (5.20) and higher costs ($798,170), resulting in an ICER of $153,481 per QALY. DFO is dominated by DFX, as it is more costly and leads to fewer QALYs. Probability of DFP being cost-effective is 0% at a WTP threshold of $50,000 per QALY. |
Key limitations |
|
CADTH reanalysis results |
|
DFO = deferoxamine; DFP = deferiprone; DFX = deferasirox; eGFR = estimated glomerular filtration rate; ICER = incremental cost-effectiveness ratio; ICT = iron-chelating therapy; QALY = quality-adjusted life-year; SCD = sickle cell disease; WTP = willingness to pay.
Conclusions
Evidence from the FIRST trial demonstrated that orally administered deferiprone (DFP) was noninferior to subcutaneous deferoxamine (DFO) in terms of liver iron concentration, serum ferritin, and cardiac iron in patients with sickle cell disease (SCD) or other anemias receiving chronic transfusion therapy. A sponsor-submitted network meta-analysis (NMA) suggested that DFP has similar efficacy (in reducing liver iron concentration and serum ferritin) compared with orally administered deferasirox (DFX). Although this evidence is associated with uncertainty due to the limitations noted in the CADTH Clinical Report, the clinical expert feedback obtained by CADTH indicated that the assumption of comparable efficacy across iron-chelating therapy (ICT) options is plausible. The sponsor also suggested that DFP was associated with a renal protective effect compared with DFO or DFX, based on an unpublished comparison from real-world evidence. Clinical expert feedback noted that both DFO and DFX can pose a risk for renal toxicity and it was plausible that DFP may pose a lower risk for renal toxicity; however, the available evidence is insufficient to draw any conclusions regarding the magnitude and clinical relevance of any potential differences across the treatments for patients with no known contraindications. Furthermore, CADTH identified numerous methodological limitations with the unpublished data that precluded any conclusions regarding the comparative safety of the ICTs. Finally, and importantly, the clinical expert feedback noted that estimated glomerular filtration rate (eGFR) values would not be used in isolation within Canadian practice to inform treatment decisions (e.g., proteinuria would also be evaluated).
In addition to the concerns regarding the comparative clinical efficacy of the ICTs, CADTH identified several additional limitations, specifically, the inadequacy of a Markov model with only 3 health states for capturing the care pathway for patients receiving ICT, the omission of relevant comparators, and a questionable rate of ICT discontinuation due to nonrenal causes beyond 12 months. To address some of the identified limitations, in the CADTH base case, a smaller proportion of Exjade use (10%) and fewer eye tests (once per year) were assumed among patients receiving DFX. Additionally, CADTH assumed that changes in eGFR levels over time among patients receiving DFP or DFX were comparable to those of patients receiving DFO.
CADTH’s base case provided results consistent with the sponsor’s base case, indicating that DFP is associated with higher costs and improved quality-adjusted life-years (QALYs). However, CADTH’s base case resulted in a substantially higher incremental cost-effectiveness ratio (ICER) ($6,812,661 per QALY versus $164,364 per QALY). These results were driven by the higher drug cost of DFP relative to comparators and a very small incremental QALY benefit from a lower risk of iron overload when accounting for some of the sponsor’s assumptions. A price reduction of at least 79.5% would be required for DFP to be an optimal treatment option at a threshold of $50,000 per QALY. The cost-effectiveness of DFP was highly sensitive to the assumption of the clinical benefits of DFP on renal function and the baseline age of patients starting treatment in the model, but found to be robust in relation to DFP dosage regimen and the type of population (i.e., patients with SCD versus patients with other anemias). CADTH was unable to address several key limitations due to the quality of the available comparative data and constraints introduced by the submitted model structure. It is unclear how subsequent ICTs may affect the cost-effectiveness findings. Additionally, the cost-effectiveness of DFP compared with other relevant options for patients with iron overload, such as a combination ICT and exchange transfusion, is unknown, as they were not considered by the sponsor.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process (specifically, information that pertains to the economic submission).
No patient input was received for this review. To ensure the patient voice is reflected in the review process, CADTH reviewed patient group input that was previously provided for the review of DFP for the treatment of patients with transfusional iron overload due to thalassemia syndromes when current chelation therapy is inadequate. At that time, patients reported having experience with injectable treatments (e.g., DFO) and oral treatments (e.g., DFX). They noted that DFO has a demanding subcutaneous or IV administration schedule and can be associated with important side effects, such as local irritation, high-frequency hearing loss, deafness, retinal damage, impaired vision, growth retardation, and bone abnormalities. The patient groups also reported that oral treatments are associated with improvements in quality of life, treatment adherence, and patient satisfaction.
Clinician input was received from the Canadian Hemoglobinopathy Association (CanHaem), a not-for-profit organization dedicated to the care of individuals in Canada with hemoglobinopathies like SCD. The current pathway of care for patients with SCD with iron overload was described as subcutaneous or IV delivery of DFO for 12 to 18 hours or an oral delivery of DFX. The clinician input noted that the goal of treatment is to prolong life, reduce drug toxicity, and overcome the drug delivery challenges seen in current treatments. Clinicians noted that DFP would be used as the preferred therapy over other ICTs if the patient has liver or kidney dysfunction and is not at risk of neutropenia, or if the hemoglobinopathy specialist decides on this therapy due to multifaceted factors such as patient age, health status, family or lifestyle considerations (e.g., unable to tolerate subcutaneous or IV drug infusion), organ toxicity, and other potential considerations.
The drug plan input received for this review noted that patients would typically be prescribed 25 mg/kg to 33 mg/kg body weight, orally 3 times a day, for a total daily dosage of 75 mg/kg to 100 mg/kg body weight. The plans indicated that dose adjustments would be tailored to the patient’s response and therapeutic goals. The plans indicated that the initiation criteria for DFP should likely be aligned with DFX; however, it was questioned whether DFP would be listed in the same tier as DFX. It was also noted that patients taking DFP are at an increased risk of agranulocytosis (severe neutropenia), which may lead to serious and life-threatening infections. To account for this risk, the patient’s absolute neutrophil count should be measured before and while receiving therapy.
Several of these concerns were addressed in the sponsor’s model:
The sponsor included DFO and DFX as comparators in a cost-utility analysis.
Costs and health utility decrements due to adverse events (AEs) associated with each ICT were considered in the sponsor’s model.
Costs associated with routine monitoring of neutrophil counts were considered in the sponsor’s model.
In addition, CADTH addressed some of these concerns as follows:
The drug plans noted that dose adjustments for DFP would be tailored to the patient’s response and therapeutic goals. CADTH performed scenario analyses by allowing dose titration for DFP.
CADTH was unable to address the following concern raised from stakeholder input:
The drug plans’ concern about the initiation criteria for DFP.
Economic Review
The current review is for DFP (Ferriprox) for people with SCD or other anemias who have transfusional iron overload.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis comparing the cost-effectiveness of DFP with DFO or DFX for the treatment of iron overload in patients with SCD or other anemias. The modelled population was in line with the trial population.
DFP is available as a 1,000 mg tablet and 100 mg/mL multi-use oral solution (500 mL). Although the sponsor assumed that all patients on DFP receive the therapy in tablet form, since a unit cost (per mg) in the model is similar between forms of DFP, this was assumed to not impact the modelled results. According to the product monograph, the recommended dosage is 25 mg/kg to 33 mg/kg body weight, orally 3 times a day, for a total daily dosage of 75 mg/kg to 100 mg/kg body weight.1 The sponsor applied the distribution of patients receiving high and low doses of DFP and DFO based on the individual patient-level data from the LA38 to 0411 (FIRST) trial for the first 12 months, after which the proportion of patients receiving high and low doses were assumed to stay constant based on the data from the FIRST trial.2,3
The cost of DFP is $33.47 for a 1,000 mg tablet and $3.35 for a 100 mg/mL oral solution, equating to a monthly cost of $6,113 for the low dose (75 mg/kg daily) and $8,151 for the high dose (100 mg/kg daily). The monthly costs of the low (25 mg/kg) and high (35 mg/kg) doses of DFO were $1,927 and $2,827, respectively, while the monthly cost of DFX was assumed to be stable at $2,552, which was calculated as a weighted average of DFX 360 mg (Jadenu generic) and 500 mg (Exjade generic). The sponsor assumed drug wastage in the drug cost calculation.4
The clinical outcomes were QALYs and life-years. The sponsor did not appear to explicitly report patient age at the model entry, but the economic analysis was undertaken over a lifetime time horizon (assumed to be 62.5 years) from the perspective of a Canadian publicly funded health care system. Costs and QALYs were discounted at a rate of 1.5% per annum.
Model Structure
The sponsor submitted a semi-Markov model with a cycle length of 1 month (30.44 days) and the following mutually exclusive health states: on ICT, not on ICT, and dead (Appendix 3, Figure 1). All patients begin in the on-ICT health state, where they can either remain, transition to the not-on-ICT health state, or die. Patients in the not-on-ICT health state can remain in that state or die; they were not allowed to transition back to the on-ICT health state. All patients in the on-ICT state were assumed to receive ICT (either DFP, DFO, or DFX).
Model Inputs
The modelled population generally reflects the baseline characteristics of the enrolment population in the FIRST trial, a multicentre, 2-arm, randomized, open-label study that evaluates the efficacy of DFP versus DFO in the treatment of iron overload in patients with SCD or other anemias.2,3 However, the sponsor’s model assumed a mean weight of 75.5 kg, which accounted for the proportion of females reported by Statistics Canada. The average patient weight in the FIRST trial was 42.4 kg.2
The model assumed all ICTs have equivalent efficacy in chelating iron. This assumption was based on the findings of the LA38 to 0411 noninferiority trial and the NMA comparing the relative efficacy of DFP, DFO, and DFX for liver iron concentration, serum ferritin, and cardiac iron.2,5
Transitions from the on-ICT health state to the not-on-ICT health state were derived from treatment discontinuation data from the LA38 to 0411 trial (DFP, DFO) and Vichinsky et al. study (DFX).2,6 Reasons for treatment discontinuation were categorized as being due to either renal impairment or nonrenal impairment (patient request, AE, loss to follow-up, investigator decision, and other). Treatment discontinuation due to nonrenal impairment reasons was based on data observed in the 12-month follow-up period of the LA38 to 0411 trial. Beyond 12 months, treatment discontinuation from other causes was assumed to decrease to 0.5% per month. Treatment discontinuation due to renal impartment was derived from a mixed model for repeated measures (MMRM) analysis of real-world US medical records data from TriNetX, which converted patients’ creatinine measures over time into eGFR values.7 The analysis estimated eGFR at different time points for patients receiving each ICT while adjusting for baseline age, sex (proportion of the population that is male), race (proportion of the population that is Black), baseline eGFR (mL/min/1.73 m2), and baseline hemoglobin (g/dL). If the estimated eGFR fell below the eGFR thresholds for ICT discontinuation reported in the published literature (40 mL/min/1.73 m2 for DFX and 30 mL/min/1.73 m2 for DFO), patients were assumed to stop the associated ICT. The model assumed that none of the patients receiving DFP would stop the treatment due to renal impairment.
The sponsor derived the survival of patients with SCD by applying a standardized mortality ratio of 1.25 for people with SCD to the mortality rates for the general population.8 The model further assumed a higher risk of death (2.21% per month) for patients with an eGFR below 15 mL/min/1.73 m2, i.e., those with end-stage renal disease.9
Health state utility values were based on published studies. The sponsor assumed that a patient receiving ICT had the same health utility as patients with SCD (0.84). Although patients with and without ICT were assumed to have the same baseline utility value, the sponsor applied larger utility decrements due to iron overload complications for patients who did not receive ICT. The model further accounted for utility decrements due to AEs and chronic kidney disease, which were sourced from the published literature.10-12
Costs included drug-related (acquisition, administration, monitoring); AEs; health care utilization due to SCD, chronic kidney disease, or end-stage renal disease; iron overload complications; and death. Drug acquisition costs for DFP were obtained from the Ontario Exceptional Access Program, while DFO and DFX costs were based on the Ontario Drug Benefit Formulary and Alberta Drug Benefit Formulary, respectively.13-15 Administration costs for subcutaneous and IV ICTs were obtained from the Ontario Schedule of Benefits. Information on dosage regimens shown in the relevant product monographs was used to inform the drug acquisition cost of each ICT.
The sponsor assigned AE costs for each cycle. The proportion of severe AEs experienced by patients receiving DFP or DFO was obtained from the LA38 to 0411 trial, while the unit cost of each AE requiring hospital admission was obtained from the Ontario Case Costing Initiative. The AE cost for DFX was estimated as the mean AE costs for DFO and DFP. Frequencies for receiving monitoring tests were sourced from the Guidelines for the Clinical Care of Patients with Thalassemia in Canada and unit costs were based on the Ontario Schedule of Benefits for Physician Services. Health care costs associated with complications were obtained from US studies.16
Mortality costs of death were calculated by weighting the unit cost of death from sudden death, terminal illness, and organ failure reported by the Canadian Hospice Palliative Care Association and the percentage of occurrence of each cause of death shown in a Canadian study conducted by Fassbender et al.17
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically with 5,000 iterations. The deterministic and probabilistic results were comparable, and the probabilistic findings are presented subsequently.
Base-Case Results
In the sponsor’s base case, DFO was dominated by DFX, as it was more costly and generated fewer QALYs. Compared with DFX, DFP was associated with an ICER of $153,481 per QALY (Table 3). At a willingness-to-pay value of $50,000 per QALY, the probability of DFP being cost-effective was 0% compared with DFX.
All patients transitioned to dead at the end of the model time horizon (i.e., after 62.5 years). A breakdown of the sponsor-submitted results for the base-case population by trial duration and extrapolated period shows that 100% of the expected QALY gains come from the time beyond the trial period.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
Deferasirox | 2,669,889 | 12.54 | Reference |
Deferiprone | 3,373,441 | 17.74 | 153,481 |
Dominated treatment | |||
Deferoxamine | 2,575,271 | 11.46 | Dominated by deferasirox |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.18
Sensitivity and Scenario Analysis Results
The sponsor conducted a series of scenario analyses by varying a time horizon and discount rates, using alternative assumptions on costs associated with complications and acquisition costs, varying discontinuation rates post 12 months, assuming different rates of eGFR decline for each ICT, applying alternative health state–specific utility values, and assuming a different baseline age for the modelled population. Key drivers of the cost-effectiveness findings included the assumptions on eGFR decline, the time horizon, and treatment discontinuation rates after 12 months due to nonrenal impairment reasons. The sponsor’s subgroup analysis focusing on patients with SCD showed consistent cost-effectiveness results, suggesting that DFO was dominated by DFX, and the ICER for DFP was $142,448 per QALY compared with DFX.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis:
Clinical benefits of DFP compared with DFO or DFX are uncertain. The sponsor derived the comparative efficacy of DFP versus DFO versus DFX from a sponsor-commissioned NMA that suggested there were no statistically significant differences in liver iron concentration and serum ferritin between the 3 ICT options. Based on this indirect evidence, the sponsor assumed the 3 ICTs have equivalent efficacy in chelating iron. The CADTH Clinical Review appraisal of the sponsor-submitted NMA noted that due to incomplete reporting of NMA methods, small or sparse network size, and notable differences in the baseline characteristics of the patient population enrolled in the included studies, the evidence is imprecise in its effect estimates. Although the limitations identified with the NMA increase the uncertainty associated with the assumption that the 3 ICTs have equivalent efficacy, CADTH obtained clinical expert feedback that indicated that the assumption of equivalent effect is plausible.
Furthermore, the sponsor assumed that DFP has a renal protective effect compared with other ICT options and DFP patients would experience a slower decline in renal function over time compared with those on DFX or DFO, thereby allowing patients to remain on ICT and reducing iron overload complications. To support this value claim, the sponsor used the US medical records (TriNetX) and a published algorithm to predict eGFR values at different time points, while adjusting for baseline age, sex, race, baseline eGFR level, and baseline hemoglobin level. The predicted eGFR values were then used to determine probabilities of ICT discontinuation due to renal-related causes. An ICT-specific MMRM model was utilized to predict eGFR levels for each ICT; however, CADTH identified numerous limitations with the sponsor’s use of real-world evidence, These limitations included heterogeneity in treatment setting (e.g., within the setting of a phase III clinical trial versus real-world data), generalizability concerns regarding registry data from the US being applied to the management of patients with SCD in Canadian practice, and significant loss to follow-up. In addition, the clinical expert consulted by CADTH noted that both DFO and DFX can pose a risk for renal toxicity, and it was plausible that DFP may pose a lower risk for renal toxicity; however, the available evidence is insufficient to draw any conclusions regarding the comparative safety of these treatments, in relation to renal functions in particular, for patients with no known contraindications. Moreover, the sponsor did not describe how the ICT-specific model was developed and how the model’s goodness of fit was assessed in its submitted TriNetX data analysis report. The validity of these MMRM models is therefore questionable. Furthermore, the models adjusted for a limited number of confounding factors and did not account for the history of comorbid or concurrent conditions, such as diabetes and hypertension, which may confound the association between ICT options and eGFR values.
More importantly, CADTH obtained clinical expert feedback that noted that eGFR values would not be used in isolation to inform treatment decisions in Canadian practice, as eGFR values are typically used along with proteinuria results to inform whether a treatment should be discontinued.
CADTH was unable to address the relative efficacy of DFP compared with other ICTs due to the lack of robust data.
CADTH undertook reanalyses that applied the same MMRM model to predict eGFR levels for the 3 ICTs and varying eGFR thresholds of discontinuation for DFP in its scenario analyses.
For the CADTH base case, it was assumed that changes in eGFR levels for patients receiving DFP or DFX were the same as those receiving DFO. CADTH used DFO as a reference instead of DFX because it was a comparator in the FIRST trial and is not contraindicated in patients with renal impairment.
The submitted model structure is insufficient to capture the care pathways, outcomes, and costs associated with SCD or other anemias. The sponsor used a 3–health state semi-Markov model to simulate the lifetime costs and QALYs of each ICT option. The sponsor’s model included only on-ICT, not-on-ICT, and dead health states. In addition to this simplified 3-state model, the sponsor assumed that patients who discontinued an ICT would not receive a subsequent ICT. CADTH obtained clinical expert feedback indicating that this model structure did not align with clinical practice and oversimplified the clinical pathway for patients with SCD or other anemias. The limited number of health states might inaccurately estimate total costs and QALYs associated with each ICT. Furthermore, in Canadian practice, clinicians would consider switching to another ICT if the existing ICT is ineffective and ICT remains a requirement.
CADTH was unable to assess the uncertainty associated with the model structure due to data limitations and the lack of flexibility with the submitted model.
Relevant comparators were not considered. As noted in the CADTH Clinical Review, CADTH obtained clinical expert feedback that noted that 2 standard approaches are commonly used to manage transfusional iron overload in Canada: switching from a simple transfusion to exchange transfusion methods, and using ICT. The sponsor did not include exchange transfusion in its economic submission and no clinical evidence was identified comparing DFP and exchange transfusion. The clinical expert feedback also noted that combination therapy may be considered a relevant option; however, this was not included in the sponsor’s submitted economic analysis.
CADTH was unable to address the limitation regarding the omission of these relevant comparators due to the lack of direct and indirect comparative evidence on their relative safety and efficacy.
The assumption that ICT treatment would be discontinued after 12 months due to nonrenal causes was unjustified. The sponsor assumed that beyond 12 months, ICT discontinuation (due to causes other than renal impairment) decreased to a constant rate of 0.5% per month; this assumption was applied without any supporting evidence and considered inappropriate. According to the clinical expert feedback obtained by CADTH, the decision to stop ICT is dependent on iron burden, which can vary over time.
CADTH was able to partially assess the concerns identified within this limitation by undertaking scenario analyses using alternative ICT discontinuation rates after 12 months due to other causes.
Additional limitations were identified, but none were considered to be a key limitation:
The sponsor’s report indicated that 80% of patients on DFX received Jadenu and 20% received Exjade; however, these proportions were not applied to the generic versions of Jadenu and Exjade, which were used in the sponsor’s base case. Moreover, the sponsor assumed that patients on DFX required an eye test 4 times per year. The clinical expert consulted by CADTH advised that the sponsor’s assumptions on the proportions of Exjade and the frequency of an eye test did not align with clinical practice in Canada. The proportion of patients on DFX receiving Exjade in Canada is approximately 10%, and the frequency of an eye test for those receiving DFX is once a year.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (refer to Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
In the sponsor’s model, it appears the sponsor assumed the simulated cohorts start at an age of 33 years (in line with the mean age of patients included in the submitted real-world analysis). | This assumption is reasonable. CADTH obtained clinical expert feedback that the baseline characteristics of the patients included in the sponsor’s real-world data analysis reflected patients in Canada with SCD. CADTH performed a scenario analysis by applying an alternative starting age based on the FIRST trial. |
The decline in eGFR is irreversible and it is clinically impossible for renal function to improve. | This assumption is not entirely correct. A decline in eGFR could be reversed for patients experiencing an acute kidney injury. |
A health utility value for patients not on ICT was assumed to be equal to the health utility value for patients with SCD receiving oral ICT. However, the larger utility decrements due to iron overload complications were applied to patients who did not receive ICT. | This assumption was considered uncertain, given the lack of health utility data among patients not receiving any ICTs. This assumption would favour DFP under the sponsor’s base-case assumptions, given that more patients on DFP would remain on ICT. |
All patients on DFO therapy received the therapy via the subcutaneous route of administration. | The clinical expert consulted by CADTH found this assumption acceptable. This assumption is expected to have a minimal impact on the cost-effectiveness finding, as the sponsor’s model applied the same administration cost for IV and subcutaneous routes of administration. |
All patients on DFP receive the therapy in tablet form. | This assumption was deemed acceptable. As the unit cost of the solution form is slightly higher than the tablet form, the use of a solution formulation is likely to increase the ICER. CADTH performed a scenario analysis and assumed that 10% of patients on DFP receive a solution formulation. |
DFO = deferoxamine; DFP = deferiprone; eGFR = estimated glomerular filtration rate; ICER = incremental cost-effectiveness ratio; ICT = iron-chelating therapy; SCD = sickle cell disease.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
CADTH corrected the sponsor’s model by changing the proportion of patients on DFX receiving the generic version of Exjade to 20%, the proportion specified in the sponsor’s report. The CADTH’s base case was derived by changing the proportion of Exjade to 10% and assuming patients on DFX required an eye test once per year to align with Canadian clinical practice. The CADTH base case also assumed that changes in eGFR levels over time for patients on DFP and DFX were equal to those of patients receiving DFO. Results from CADTH’s reanalysis were consistent with the sponsor’s base case, suggesting DFO was dominated by DFX and that DFP was associated with higher costs ($600,356) and improved QALYs (0.09 QALYs), with an ICER of $6,812,661 per QALY compared with DFX. The estimated ICER was substantially higher than that reported in the sponsor’s base case due to CADTH’s assumption that the impact on renal function would be comparable across ICTs; this revised assumption removed the survival benefit of DFP. The probability that DFP is cost-effective was zero at a willingness-to-pay value of $50,000 per QALY. Table 5 details the changes made to derive the CADTH reanalysis; the summary results of that reanalysis are presented in Table 6. Additional results are shown in Appendix 4.
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to the sponsor’s base case | ||
The sponsor’s model did not use the same proportion of Exjade used in the sponsor report. | 0% | 20% |
Changes to derive the CADTH base case | ||
1. Proportion of patients on DFX receiving Exjade | 20% | 10% |
2. The frequency of an eye test for patients receiving DFX | 4 times per year | 1 time per year |
3. The changes in eGFR levels over time varied by type of ICT | A different eGFR regression model was used for each ICT | An eGFR regression model for DFO was applied to DFP and DFX. In other words, changes in eGFR levels among patients receiving DFP or DFX were assumed to be comparable to those receiving DFO. |
CADTH base case | 1 + 2 + 3 | |
DFO = deferoxamine; DFP = deferiprone; DFX = deferasirox; eGFR = estimated glomerular filtration rate; ICT = iron-chelating therapy.
Table 6: Summary of the CADTH Reanalysis Results
Drug | Total costs ($) | Total QALYs | ICER vs. reference ($ per QALY) | Sequential ICER ($ per QALY) |
|---|---|---|---|---|
Sponsor’s corrected base case | ||||
DFX | 2,511,727 | 12.53 | Reference | Reference |
DFP | 3,370,801 | 17.75 | 164,364 | 164,364 |
Dominated treatment | ||||
DFO | 2,671,285 | 11.49 | NA | Dominated |
CADTH base case | ||||
DFX | 2,591,562 | 12.78 | Reference | Reference |
DFP | 3,191,918 | 12.86 | 6,812,661 | 6,812,661 |
Dominated treatment | ||||
DFO | 2,683,545 | 11.48 | NA | Dominated |
DFO = deferoxamine; DFP = deferiprone; DFX = deferasirox; ICER = incremental cost-effectiveness ratio; NA = not applicable; QALY = quality-adjusted life-year.
Scenario Analysis Results
Based on CADTH’s base case, a series of scenario analyses was conducted. These analyses explored the impact of the following model parameters and assumptions: changes in eGFR levels by type of ICT, different model starting age, alternative assumptions on treatment discontinuation due to nonrenal impairment causes, different DFP dosing regimens, and an alternative source of patient weight data. CADTH also explored the impact of health utility value by removing health utility decrements due to iron overload complications from the model.
Results from scenario analyses (Table 12 in Appendix 4) demonstrated that the assumption regarding the impact of ICTs on renal function (through changes in eGFR levels) was the key driver of the cost-effectiveness findings, followed by the model starting age and ICT discontinuation after 12 months. The ICERs for DFP increased significantly if all ICT options were assumed to have a comparable effect on eGFR values (scenarios 1 and 2) because this assumption removed the survival benefits of DFP shown in the sponsor’s base case. DFP was dominated by DFX if the baseline age of patients enrolled in the FIRST trial was used as the model starting age (scenario 3). The ICERs were also influenced by the ICT discontinuation rate beyond 12 months. Cost-effectiveness findings were found to be robust to the DFP dosage regimen and the type of population (patients with SCD versus patients with SCD or other anemias).
CADTH undertook a price reduction analysis based on the sponsor’s base case and the CADTH reanalysis (Table 7). The results show that a price reduction of 79.5% is required for DFP to be considered cost-effective at a willingness-to-pay threshold of $50,000 per QALY.
Table 7: CADTH Price Reduction Analyses
Analysis | ICERs ($ per QALY) for deferiprone vs. deferasiroxa | |
|---|---|---|
Price reduction (%) | Sponsor’s corrected base case | CADTH’s base case |
No price reduction | 164,364 | 6,812,661 |
10 | 147,785 | 5,915,187 |
20 | 131,105 | 5,116,804 |
30 | 114,032 | 4,293,262 |
40 | 97,793 | 3,455,776 |
50 | 80,714 | 2,566,208 |
60 | 64,348 | 1,731,625 |
69 | 49,545 | 964,479 |
70 | — | 854,345 |
79.5 | — | 49,870 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
aDeferoxamine was dominated by deferasirox regardless of the price of deferiprone.
Issues for Consideration
The clinical expert consulted by CADTH noted that DFP could potentially be used as a combination therapy with DFX or DFO or as a subsequent ICT among patients whose condition does not respond to the first ICT. This expanded use of DFP has not been accounted for in the sponsor’s base case or in CADTH’s reanalysis due to the limitation of the sponsor’s model structure. The expanded use is expected to increase the budget impact of DFP. CADTH did not identify any relevant studies that examined using DFP in combination with other drugs used for iron chelation.
The definitions of “less severe” and “more severe” iron burden that were used to determine dose escalation in the FIRST trial may not be reflective of routine care in Canada. The clinical expert feedback obtained by CADTH noted that the upper range for liver iron concentration used to differentiate between patients with “less severe” versus “more severe” iron burden is too high. In routine clinical practice, initial starting dosages may be higher and/or dose escalation could occur more frequently for patients with higher baseline liver iron concentration levels. This discrepancy may result in differing treatment costs in the economic evaluation for DFO and DFP.
Overall Conclusions
Evidence from the FIRST trial demonstrated that orally administered DFP was noninferior to subcutaneous DFO in terms of liver iron concentration, serum ferritin, and cardiac iron in patients with SCD or other anemias receiving chronic transfusion therapy. The sponsor derived the comparative efficacy of DFP versus DFO and DFX from a sponsor-commissioned NMA that suggested there were no statistically significant differences in liver iron concentration and serum ferritin between the 3 ICT options. Although this evidence is associated with uncertainty due to the limitations noted in the CADTH Clinical Report — the limited number of included studies, incomplete reporting on indirect treatment comparison methods, and sparse networks and heterogeneity across the included studies — the clinical expert feedback obtained by CADTH indicated that the assumption of comparable efficacy across ICT options is plausible. The sponsor’s model asserted that DFP was associated with clinical benefits in terms of renal protective effect by assuming a slower decline in eGFR levels among patients receiving DFP compared with DFO or DFX, thereby allowing patients to remain on ICT and reducing iron overload complications. The sponsor used US medical records (TriNetX) and a published algorithm to predict eGFR values at different time points and to support this value claim. According to the clinical expert feedback, while it may be plausible that DFP is associated with less renal toxicity than DFO and DFX, the available evidence is insufficient to draw any conclusions regarding the magnitude and clinical relevance of any potential differences across the treatments for patients with no known contraindications, as eGFR values are typically used along with proteinuria results to inform treatment discontinuation decisions in Canadian practice. Furthermore, CADTH identified numerous methodological limitations with the unpublished data that precluded any conclusions regarding the comparative safety of the ICTs.
CADTH identified several limitations within the sponsor’s economic evaluation, specifically, the uncertainty associated with the clinical benefits of DFP on renal function, the inadequacy of a 3–health state Markov model to capture the care pathway for patients receiving ICT, the omission of relevant comparators, and a questionable rate of ICT discontinuation beyond 12 months due to nonrenal causes.
CADTH’s base case assumed a smaller proportion of Exjade use (10%) and fewer eye tests (once per year) among patients receiving DFX. Additionally, given the lack of robust evidence supporting the renal protective effect of DFP compared with other ICTs, CADTH assumed that changes in eGFR levels over time among patients receiving DFP or DFX were comparable to those receiving DFO in its base case. CADTH undertook scenario analyses to explore the impact of assumptions regarding the renal protective effect of DFP, starting age of the model cohort, rate of ICT discontinuation after 12 months due to causes other than renal impairment, alternative dosage regimens for DFP, and an alternative assumption for health utility decrements due to iron overload complications.
Although CADTH’s base case resulted in a substantially higher ICER than the sponsor’s base case ($6,812,661 per QALY versus $164,364 per QALY), both analyses provided consistent results, suggesting that DFO was dominated by DFX and that DFP was associated with higher costs and improved QALYs but was not cost-effective compared with DFX at the submitted price. Based on CADTH’s base case, a price reduction of at least 79.5% would be required to make DFP an optimal treatment option at a willingness-to-pay threshold of $50,000 per QALY. The cost-effectiveness of DFP was highly sensitive to the assumption on the clinical benefits of DFP on renal function and the model starting age, but found to be robust to DFP dosage regimen and the type of population (patients with SCD with anemias versus patients with SCD).
CADTH was unable to address several key concerns due to limited data and constraints introduced by the submitted model structure. The submitted model structure failed to account for subsequent ICTs, which would be reflective of real-world Canadian clinical practice; it is therefore questionable whether the QALY gains observed in the sponsor’s and CADTH’s models align with the expected benefit associated with DFP. The cost-effectiveness of DFP compared with other relevant options for patients with iron overload, such as a combination of ICT and exchange transfusion, is unknown, as these options were not considered by the sponsor.
References
1.Ferriprox (deferiprone): 500 mg and 1000 mg tablets, Ph.Eur.; 100 mg/mL oral solution, Ph.Eur. [product monograph]. Woodbridge (ON): Chiesi Canada Corp; 2021 Oct 12.
2.Clinical Study Report: . LA38-0411. The efficacy and safety of Ferriprox®for the treatment of transfusional iron overload in patients with sickle cell disease or other anemias [internal sponsor’s report]. Toronto (ON): ApoPharma, a division of Apotex Inc; 2019 Sep 20.
3.Kwiatkowski JL, Hamdy M, El-Beshlawy A, et al. Deferiprone vs deferoxamine for transfusional iron overload in SCD and other anemias: a randomized, open-label noninferiority study. Blood Adv. 2022;6(4):1243-1254. PubMed
4.Reimbursement Review sponsor submission: Ferriprox (deferiprone), 1000 mg oral tablet and 100 mg/mL oral solution [internal sponsor’s package]. Woodbridge (ON): Chiesi Canada Corp; 2022 Jun 15.
5.Indirect treatment comparisons of deferiprone and comparators for iron chelation in sickle cell disease [internal sponsor’s report]. In: Drug Reimbursement Review sponsor submission: Ferriprox (deferiprone), 1000 mg oral tablet and 100 mg/mL oral solution. Woodbridge (ON): Chiesi Canada Corp; 2022 Jun 15.
6.Vichinsky E, Onyekwere O, Porter J, et al. A randomised comparison of deferasirox versus deferoxamine for the treatment of transfusional iron overload in sickle cell disease. Br J Haematol. 2007;136(3):501-508. PubMed
7.Levey AS, Stevens LA, Schmid CH, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150(9):604-612. PubMed
8.Allali S, Taylor M, Brice J, de Montalembert M. Chronic organ injuries in children with sickle cell disease. Haematologica. 2021;106(6):1535. PubMed
9.McClellan AC, Luthi JC, Lynch JR, et al. High one year mortality in adults with sickle cell disease and end‐stage renal disease. Br J Haematol. 2012;159(3):360-367. PubMed
10.Kansal AR, Cowie MR, Kielhorn A, et al. Cost‐effectiveness of ivabradine for heart failure in the United States. J Am Heart Assoc. 2016;5(5):e003221. PubMed
11.Geale K, Saridogan E, Lehmann M, Arriagada P, Hultberg M, Henriksson M. Repeated intermittent ulipristal acetate in the treatment of uterine fibroids: a cost-effectiveness analysis. ClinicoEcon Outcomes Res. 2017;9:669. PubMed
12.Crossan C, Tsochatzis EA, Longworth L, et al. Cost-effectiveness of non-invasive methods for assessment and monitoring of liver fibrosis and cirrhosis in patients with chronic liver disease: systematic review and economic evaluation. Health Technol Assess. 2015;19(9):hta19090. PubMed
13.Exceptional Access Program (EAP). Toronto (ON): Ontario Ministry of Health; Ontario Ministry of Long-Term Care; 2022: https://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx. Accessed 2022 Jul 15.
14.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2022; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2022 Jul 15.
15.Government of Alberta. Interactive drug benefit list. 2022; https://idbl.ab.bluecross.ca/idbl/load.do. Accessed 2022 Jul 15.
16.Sayani F, Warner M, Wu J, Wong-Rieger D, Humphreys K, Odame I. Guidelines for the clinical care of patients with thalassaemia in Canada. Toronto (ON): Anemia Institute for Research & Education; 2009: https://www.thalassemia.ca/wp-content/uploads/Thalassemia-Guidelines_LR.pdf. Accessed 2022 Jul 15.
17.Fassbender K, Fainsinger RL, Carson M, Finegan BA. Cost trajectories at the end of life: the Canadian experience. J Pain Symptom Manage. 2009;38(1):75-80. PubMed
18.Pharmacoeconomic evaluation [internal sponsor’s report]. In: Drug Reimbursement Review sponsor submission: Ferriprox (deferiprone), 1000 mg oral tablet and 100 mg/mL oral solution [internal sponsor’s package]. Woodbridge (ON): Chiesi Canada Corp; 2022 Jun 15.
19.Saskatchewan Drug Plan: search formulary. 2022; https://formulary.drugplan.ehealthsask.ca/SearchFormulary. Accessed 2022 Jul 15.
20.Budget Impact Analysis [internal sponsor’s report]. In: Drug Reimbursement Review sponsor submission:Ferriprox (deferiprone), 1000 mg oral tablet and 100 mg/mL oral solution [internal sponsor’s package]. Woodbridge (ON): Chiesi Canada Corp; 2022 Jun 15.
21.Shields M, Gorber SC, Janssen I, Tremblay MS. Bias in self-reported estimates of obesity in Canadian health surveys: an update on correction equations for adults. Health Rep. 2011;22(3):35. PubMed
22.Consensus statement on the care of patients with sickle cell disease in Canada. Version 2.0. Ottawa (ON): Canadian Haemoglobinopathy Association; 2014: https://emergencymedicinecases.com/wp-content/uploads/filebase/pdf/Canadian%20Sickle%20Cell%20Guidelines%202014.pdf. Accessed 2022 Jul 15.
23.Simpson E, Klaassen RJ, Chakraborty P, et al. Increasing incidence and prevalence of pathologic hemoglobinopathies among children in Ontario, Canada from 1991-2013. Blood. 2018;132:4698.
24.El-Haj N, Hoppe CC. Newborn screening for SCD in the USA and Canada. Int J Neonatal Screen. 2018;4(4):36. PubMed
Appendix 1: Cost Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts and drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and, as such, the table may not represent the actual costs to public drug plans.
Table 8: CADTH Cost Comparison Table for Deferiprone for the Treatment of Transfusional Iron Overload Due to Sickle Cell Disease
Treatment | Strength | Form | Price ($) | Recommended dosage | Daily cost ($)a | Average annual cost ($) |
|---|---|---|---|---|---|---|
Deferiprone | 1,000 mg 100 mg/mL | Tabletb Oral solution | 33.4740 3.3495 | 25 mg/kg to 33 mg/kg daily, 3 times daily (total daily dosage: 75 mg/kg to 100 mg/kg)1 | 117.16 to 150.63b 113.88 to 150.73 | 42,763 to 54,981b 41,567 to 55,016 |
Iron-chelating drugs | ||||||
Deferasirox | 125 mg 250 mg 500 mg | Tablet for oral suspension | 5.2408 10.4820 20.9649 | 10 mg/kg, 20 mg/kg, or 30 mg/kg dailyc | 20.96 (10 mg) 36.69 (20 mg) 57.65 (30 mg) | 7,652 (10 mg) 13,390 (20 mg) 21,042 (30 mg) |
Deferasirox | 90 mg 180 mg 360 mg | Tablet | 2.6303 5.2610 10.5228 | 7 mg/kg, 14 mg/kg, or 21 mg/kg dailyc | 10.52 (7 mg) 18.41 (14 mg) 28.93 (21 mg) | 3,840 (7 mg) 6,720 (14 mg) 10,561 (21 mg) |
Deferoxamine mesylate | 500 mg/vial 2 g/vial | Lyophilized powder in vials for IV infusion, or SC injectiond | 14.6700e 28.3500e | SC/IV: 1 g to 4 g (20 mg/kg to 60 mg/kg) daily over a period of 12 hoursf Dosing is 4 to 7 times per week | 28.35 to 56.70g | 10,348 to 20,696g |
IM = intramuscular; SC = subcutaneous.
Note: All prices are from the Ontario Drug Benefit Formulary14 or Ontario Exceptional Access Program Formulary13 (accessed July 2022), unless otherwise indicated, and do not include dispensing fees. Prices represent the amount paid by the drug plans. Annual costs are based on 365 days per year. CADTH obtained clinical expert feedback which noted that dosing is dependent upon iron load.
aBased on a mean weight of 45 kg assumed by CADTH.
bTablets can be broken in half. Dosage used to calculate costs were scaled to the nearest 500 mg.
cRecommended dosages are from the respective products’ monograph. CADTH obtained clinical expert feedback that the dose range for deferasirox (Exjade) may increase to 40 mg/kg as needed.
dSingle-use vial.
eSaskatchewan Drug Benefit Formulary (accessed July 2022).19 Unit price (per vial).
fCADTH obtained clinical expert feedback that IM infusion is no longer typically used in practice.
gCADTH obtained clinical expert feedback that the dose range for deferoxamine was 20 mg/kg to 50 mg/kg, so this range is used to determine the daily and annual drug costs. Dosing was assumed as 7 days per week.
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/no | Commentsa |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | The modelled population is deemed relevant and generalizable to patients with SCD and other anemias in Canada. However, the sponsor’s model did not consider standard approaches for the management of iron overload, including switching a transfusion method and using a combination ICT. |
Model has been adequately programmed and has sufficient face validity | Yes | The model programming is acceptable. |
Model structure is adequate for decision problem | No | Refer to CADTH appraisal section. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | The sponsor provided insufficient descriptions regression models used to predict eGFR levels for each ICT. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | No | The sponsor provided insufficient descriptions of how the ICT-specific eGFR level was predicted. There was also no justification for the assumption on the use of a 0.5% monthly rate of post–12-month ICT discontinuation due to nonrenal causes. |
eGFR = estimated glomerular filtration rate; ICT = iron-chelating therapy; SCD = sickle cell disease.
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
Detailed Results of the Sponsor’s Base Case
Figure 2: Cost-Effectiveness Acceptability Curves (DFP Versus DFX Versus DFO)
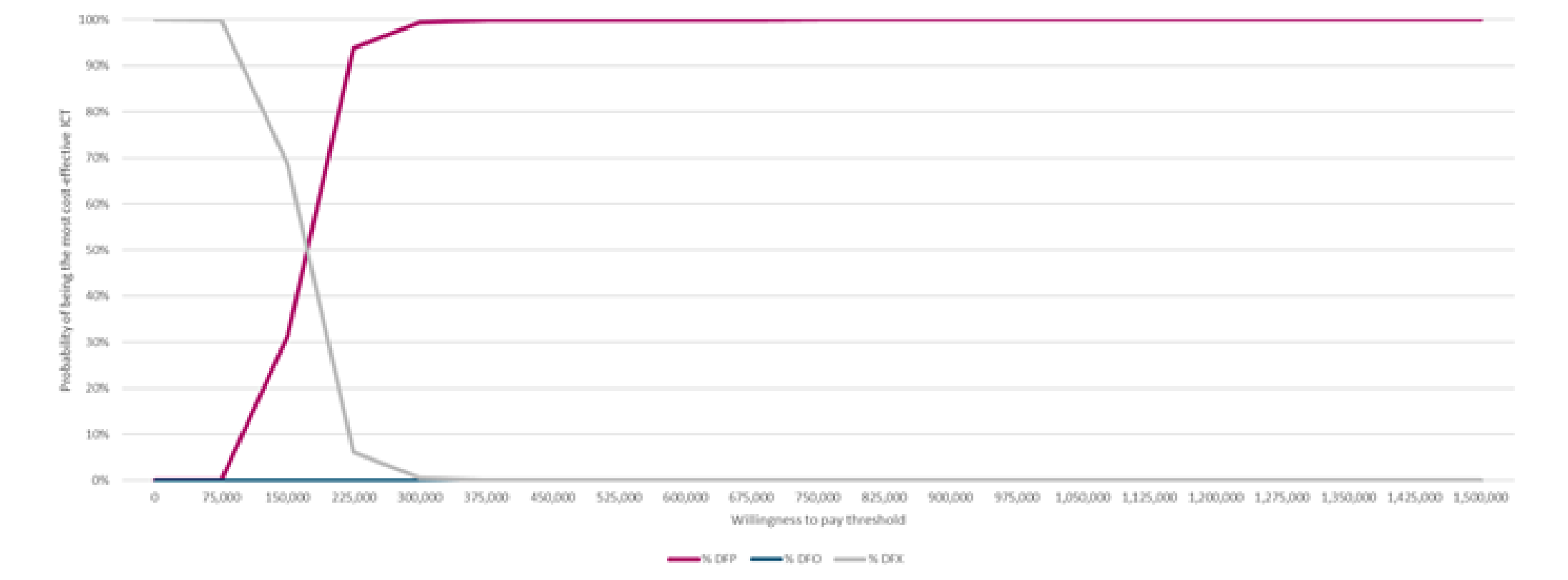
DFO = deferoxamine; DFP= deferiprone; DFX = deferasirox; ICT = iron chelation therapy.
Source: Sponsor’s pharmacoeconomic submission.18
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Table 10: Disaggregated Summary of CADTH’s Economic Evaluation Results
Treatment | Component | Value | Incremental (vs. DFX) | Incremental (sequential) |
|---|---|---|---|---|
Discounted LYs | ||||
DFX | On ICT | 7.37 | NA | NA |
Not on ICT | 8.27 | NA | NA | |
Total | 15.64 | NA | NA | |
DFO | On ICT | 7.30 | –0.07 | NA |
Not on ICT | 8.34 | 0.07 | NA | |
Total | 15.64 | 0.00 | NA | |
DFP | On ICT | 8.41 | 1.11 | 1.04 |
Not on ICT | 7.22 | –1.12 | –1.05 | |
Total | 15.63 | –0.01 | –0.01 | |
Discounted QALYs | ||||
DFX | On ICT | 6.19 | NA | NA |
Not on ICT | 6.59 | NA | NA | |
Total | 12.78 | NA | NA | |
DFO | On ICT | 4.82 | –1.37 | NA |
Not on ICT | 6.66 | 0.07 | NA | |
Total | 11.48 | –1.29 | NA | |
DFP | On ICT | 7.06 | 2.24 | 0.87 |
Not on ICT | 5.80 | –0.86 | –0.79 | |
Total | 12.86 | 1.38 | 0.09 | |
Discounted costs ($) | ||||
DFX | Drug | 140,410 | NA | NA |
Monitoring | 1,554 | NA | NA | |
AEs | 486 | NA | NA | |
Disease management | 2,401,184 | NA | NA | |
Mortality | 47,928 | NA | NA | |
Total | 2,591,562 | NA | NA | |
DFO | Drug | 219,110 | 78,700 | NA |
Monitoring | 1,455 | –99 | NA | |
AEs | 424 | –62 | NA | |
Disease management | 2,398,929 | –2,255 | NA | |
Mortality | 47,915 | –13 | NA | |
Total | 2,667,833 | 76,271 | NA | |
DFP | Drug | 749,553 | 530,443 | 609,143 |
Monitoring | 2,087 | 632 | 533 | |
AEs | 3,181 | 2,757 | 2,695 | |
Disease management | 2,389,276 | –9,653 | –11,908 | |
Mortality | 47,821 | –94 | –107 | |
Total | 3,191,918 | 524,085 | 600,356 | |
Treatment | ICER vs. DFX ($) | Sequential ICER ($) | ||
DFX | Reference | Reference | ||
DFO | Dominated by DFX | Dominated by DFX | ||
DFP | 6,812,661 | 6,812,661 vs. DFX | ||
DFO= deferoxamine; DFP= deferiprone; DFX = deferasirox; ICER = incremental cost-effectiveness ratio; ICT = iron-chelating therapy; LY = life-year; NA = not applicable; QALY = quality-adjusted life-year.
Detailed Results of CADTH Base Case
Table 11: Summary of the Stepped Analysis of the CADTH Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
Sponsor’s corrected base case | DFX | 2,511,727 | 12.53 | Reference |
DFP | 3,370,801 | 17.75 | 164,364 | |
Dominated treatment | ||||
DFO | 2,671,285 | 11.49 | NA | |
CADTH reanalysis 1: proportion of Exjade use | DFX | 2,502,705 | 12.54 | Reference |
DFP | 3,370,536 | 17.76 | 166,310 | |
Dominated treatment | ||||
DFO | 2,669,782 | 11.49 | NA | |
CADTH reanalysis 2: frequency of eye test for DFX | DFX | 2,510,414 | 12.54 | Reference |
DFP | 3,371,298 | 17.76 | 165,055 | |
Dominated treatment | ||||
DFO | 2,670,305 | 11.49 | NA | |
CADTH reanalysis 3: change in eGFR levels | DFX | 2,602,721 | 12.74 | Reference |
DFP | 3,192,429 | 12.83 | 6,612,929 | |
Dominated treatment | ||||
DFO | 2,669,339 | 11.45 | NA | |
CADTH base case (1+2+3) | DFX | 2,591,562 | 12.78 | Reference |
DFP | 3,191,918 | 12.86 | 6,812,661 | |
Dominated treatment | ||||
DFO | 2,667,833 | 11.48 | NA |
DFO= deferoxamine; DFP= deferiprone; DFX = deferasirox; eGFR = estimated glomerular filtration rate; ICER = incremental cost-effectiveness ratio; NA = not applicable; QALY = quality-adjusted life-year.
Scenario Analyses
Table 12: Summary of CADTH Scenario Analyses
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
Sponsor’s corrected base case | |||
DFX | 2,511,727 | 12.53 | Reference |
DFO | 2,671,285 | 11.49 | Dominated by DFX |
DFP | 3,370,801 | 17.75 | 164,364 |
CADTH’s base case | |||
DFX | 2,591,562 | 12.78 | Reference |
DFO | 2,683,545 | 11.48 | Dominated by DFX |
DFP | 3,191,918 | 12.86 | 6,812,661 |
CADTH’s scenario analysis 1: Applying changes in eGFR among patients on DFP or DFO to those receiving DFX | |||
DFX | 2,497,457 | 12.59 | Reference |
DFO | 2,572,755 | 11.28 | Dominated by DFX |
DFP | 3,093,782 | 12.67 | 6,988,363 |
CADTH’s scenario analysis 2: Applying changes in eGFR among patients on DFO or DFX to those receiving DFP | |||
DFX | 2,679,738 | 17.70 | Reference |
DFO | 2,768,458 | 16.03 | Dominated by DFX |
DFP | 3,362,501 | 17.75 | 13,742,764 |
CADTH’s scenario analysis 3: Using the starting age based on a mean age of the FIRST trial’s participants | |||
DFX | 2,682,176 | 16.33 | Reference |
DFO | 2,768,340 | 14.75 | Dominated by DFX |
DFP | 3,328,782 | 16.33 | Dominated by DFX |
CADTH’s scenario analysis 4: Assuming post 12-month ICT discontinuation rate due to nonrenal causes to be equal to 0% per month | |||
DFX | 2,614,075 | 12.78 | Reference |
DFO | 2,726,011 | 11.05 | Dominated by DFX |
DFP | 3,552,901 | 13.02 | 4,021,266 |
CADTH’s scenario analysis 5: Assuming post 12-month ICT discontinuation rate due to nonrenal causes to be equal to 25% per month | |||
DFX | 2,526,614 | 12.69 | Reference |
DFO | 2,536,032 | 12.49 | Dominated by DFX |
DFP | 2,600,093 | 12.69 | Dominated by DFX |
CADTH’s scenario analysis 6: Assuming DFP dosage as 2 times a day | |||
DFX | 2,603,126 | 12.78 | Reference |
DFO | 2,679,710 | 11.48 | Dominated by DFX |
DFP | 3,204,048 | 12.87 | 6,813,591 |
CADTH’s scenario analysis 7: Applying patient weight based on the FIRST trial | |||
DFX | 2,540,221 | 12.79 | Reference |
DFO | 2,585,502 | 11.51 | Dominated by DFX |
DFP | 2,927,540 | 12.88 | 4,205,654 |
CADTH’s scenario analysis 8: Assuming no health utility decrements due to iron overload complications | |||
DFX | 2,587,502 | 15.58 | Reference |
DFO | 2,663,869 | 15.58 | Dominated by DFX |
DFP | 3,187,149 | 15.57 | Dominated by DFX |
CADTH’s scenario analysis 9: Assuming a titrated dose for DFP | |||
DFX | 2,332,239 | 12.77 | Reference |
DFO | 2,678,843 | 11.47 | Dominated by DFX |
DFP | 3,200,262 | 12.86 | 6,845,465 |
CADTH’s scenario analysis 10: Focusing on patients with SCD | |||
DFX | 2,658,071 | 12.78 | Reference |
DFO | 2,671,422 | 11.48 | Dominated by DFX |
DFP | 3,193,506 | 12.86 | 6,153,024 |
DFO= deferoxamine; DFP= deferiprone; DFX = deferasirox; ICER = incremental cost-effectiveness ratio; LY = life-year; NA = not applicable; QALY = quality-adjusted life-year.
Appendix 5: Submitted Budget Impact Analysis and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 13: Summary of Key Take-Aways
Key take-aways of the BIA |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
The submitted budget impact analysis (BIA) assessed the expected budgetary impact resulting from reimbursing DFP for the treatment of transfusional iron overload due to SCD and other anemias in pediatric and adult patient groups.20 The BIA was conducted from the perspective of the Canadian public drug plans over a 3-year horizon using an epidemiologic approach. The analysis was performed using jurisdiction-specific values which informed individual provincial results. The sponsor’s pan-Canadian estimates reflect the aggregated results from provincial drug plans (excluding Quebec), as well as the Non-Insured Health Benefits Program.20 Key inputs to the BIA are documented in Table 15.
The sponsors’ submission considered a reference (i.e., current) scenario in which patients received DFX (Jadenu or Exjade) or DFO, and a new-drug scenario in which DFP was included in the treatment paradigm.
The following key assumptions were made by the sponsor:
The sponsor assumed a weight of 75.54 kg by taking the average of the reported body weights of males and females in Canada to determine the size of the dose to be administered, as each therapy is weight dependent.21
The sponsor assumed DFP will capture market share proportionally from each comparator treatment.
The sponsor assumed a median dose for all treatments to determine the 3-year budget impact. The median doses listed by the sponsor were as follows: DFO = 30 mg/kg, Jadenu = 14 mg/kg, Exjade = 20 mg/kg, DFP = 29 mg/kg.20 Alternate costs based on low and high doses of drug were assessed by the sponsor in sensitivity analyses.
The sponsor assumed there would be no discontinuation of treatment across all therapies.
Table 14: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
Pediatric | 15.268 per 100,000a |
Adults | 15.268 per 100,000a |
Proportion receiving ICT | |
Pediatric | 12.5%b |
Adults | 20%b |
Proportion of public drug program patients | |
Pediatric | 68%b |
Adults | 68%b |
Total number of patients eligible for drug under review, year 1 / year 2 / year 3 | 603 / 611 / 619b |
Market uptake (3 years), %, year 1 / year 2 / year 3b | |
Uptake (reference scenario) Deferoxamine mesylate Deferasirox (Jadenu) Deferasirox (Exjade) | 10.0% / 10.0% / 10.0% 80.0% / 80.0% / 80.0% 10.0% / 10.0% /10.0% |
Uptake (new-drug scenario) Deferiprone Deferoxamine mesylate Deferasirox (Jadenu) Deferasirox (Exjade) | 10.0% / 12.5% / 17.5% 9.0% / 8.8% / 8.3% 72.0% / 70.0% / 66.0% 9.0% / 8.8% / 8.3% |
Annual cost of treatment (per patient) | |
Deferiprone Deferoxamine mesylate Deferasirox (Jadenu) Deferasirox (Exjade) | $82,472 $26,771 $11,521 $22,955 |
ICT = iron chelation therapy; SCD = sickle cell disease.
aThe sponsor assumed there are 5,000 patients in Canada with SCD, averaged from a reported number between 3,000 to 7,000, and calculated a prevalence rate for the adult and pediatric population using the projected population in Canada over a 3-year horizon.22-24
bSponsor’s internal market research; data not provided.
Summary of the Sponsor’s BIA Results
The sponsor’s base case reported that the reimbursement DFP for the treatment of iron overload due to SCD and other anemia patients would lead to an incremental budget impact of $4,119,437 in year 1, $5,215,016 in year 2, and $7,393,032 in year 3. The total 3-year incremental cost of $16,727,485. Sensitivity analyses were completed by the sponsor to adjust prevalence rate of SCD and other anemias, proportion of pediatric and adult patients on ICT, low and high dosage for drug costs, DFP market share, patient weight, and DFP formulation. The lower and upper bounds were –25% and +25% of the base value. The sensitivity analysis demonstrated that the 3-year total incremental budget impact may vary from $12,545,614 up to $20,909,35.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Relevant comparators and treatment regimens excluded: The sponsor’s submitted budget impact detailed 3 competitors of DFP: DFO, and DFX (Jadenu and Exjade). Clinical expert feedback obtained by CADTH highlighted that in Canadian clinical practice, patients often cycle between ICTs, or, in a small number of patients, combination therapy of more than 1 ICT. In addition, automated exchange transfusion was identified as a nondrug therapy for patients to de-iron without the need for chelation. The sponsor did not include these alternate regimens or treatments in the BIA.
CADTH could not undertake reanalysis to address this limitation as the sponsor’s BIA model lacked flexibility to incorporate concomitant therapy and exchange transfusion therapy.
Average patient weight was overestimated leading to overestimated treatment costs: The average patient weight used to determine dosage was noted by the sponsor as 75.54 kg. This average weight was calculated by taking the weighted average of reported male and female weights in Canada (84.6 kg for males and 70.1 kg for females) and their weighted distributions (37.5% and 62.5%, respectively). This calculated weight may not be representative of the proportion of pediatric SCD and other anemias population with transfusional overload. Conversely, the average weight from the FIRST study was reported as 42.4 kg, where 46.9% of patients were female and 53.1% were male. As these distributions differed from the those used in the drug cost calculation, the treatment costs derived using average patient weight was overestimated.
To address this limitation, CADTH undertook a reanalysis with an average weight of 42.4 kg as part of the base case.
Market share and market capture is uncertain: Clinical expert feedback obtained by CADTH suggested that the sponsor overestimated the proportion of patients who would receive Exjade and underestimated the proportion of patients who would receive Jadenu. The clinical expert feedback indicated that of the total market share that both treatments capture, 90% of the market is captured by Jadenu and 10% of the market is captured by Exjade.
CADTH altered the market share distribution in a scenario analysis to increase the market uptake of Jadenu to 81% and reduced uptake of Exjade to 9%.
Duration of treatments were overestimated: In the economic evaluation and BIA, the sponsor assumed the duration of treatment would be ongoing across all identified therapies and that the patient would not switch treatments. The clinical expert indicated that this assumption was not likely to be appropriate as patients stop and/or switch treatment throughout due to a variety of reasons.
CADTH could not undertake reanalysis to address this limitation.
Additional limitations were identified but were not considered to be key limitations. These limitations include the overestimation of DFO drug costs, and different dose range of DFO. In the sponsor’s submission, drug cost of DFO was sourced from the Alberta Drug Benefit list.15 In CADTH’s reanalysis, DFO drug costs were sourced from the Saskatchewan Drug Benefit Formulary.19 The sponsor noted that the dose range of DFO was 25 mg/kg to 35 mg/kg daily, while CADTH identified a range of 20 mg/kg to 50 mg/kg daily. As a median dose of 30 mg/kg was used, the dose range does not impact the overall results.
CADTH Reanalyses of the BIA
Table 15: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
1. Treatment cost | Deferoxamine cost for 2 g strength was sourced from the Alberta Drug Benefit List at a cost of $58.68 per unit.15 | Deferoxamine cost for 2 g was sourced from the Saskatchewan Drug Benefit Formulary at a cost of $28.35 per unit.19 |
Changes to derive the CADTH base case | ||
1. Patient weight | 75.54 kg using Canadian population data. | 42.4 kg using data from the FIRST trial.2 |
CADTH base case | Reanalysis 1 | |
The results of the CADTH stepwise reanalysis are presented in summary format in Table 16 and a more detailed breakdown is presented in Table 17. Based on the CADTH base case, the budget impact associated with DFP’s reimbursement in the indicated target population is expected to be $2,246,254 in year 1, $2,843,654 in year 2, and $4,031,286 in year 3, with a 3-year total of $9,121,193.
Table 16: Summary of the CADTH Reanalyses of the Budget Impact Analysis
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case | 16,727,485 |
Corrected base case | 16,355,174 |
CADTH reanalysis 1: patient weight | 9,149,309 |
CADTH base case (reanalysis 1) | 9,149,309 |
Note: Submitted analysis is based on the publicly available prices of the comparator treatments.
CADTH conducted additional scenario analyses to address remaining uncertainty, using the CADTH base case. Results are provided in Table 17. The scenario analyses conducted included exploring the impact of updating DFX (Jadenu and Exjade) reference scenario market shares to align with clinical expert feedback and considering a 79.5% price reduction in the price of DFP, consistent with that required for the CADTH base case to be cost-effective at a willingness to pay of $50,000 per QALY.
Table 17: Detailed Breakdown of the CADTH Reanalyses of the Budget Impact Analysis
Stepped analysis | Scenario | Year 0 (current situation) ($) | Year 1 ($) | Year 2 ($) | Year 3 ($) | Three-year total ($) |
|---|---|---|---|---|---|---|
Submitted base case | Reference | 8,450,961 | 8,560,216 | 8,669,470 | 8,778,725 | 26,008,411 |
New drug | 8,450,961 | 12,679,652 | 13,884,487 | 16,171,757 | 42,735,896 | |
Budget impact | 0 | 4,119,437 | 5,215,016 | 7,393,032 | 16,727,485 | |
Corrected base case | Reference | 8,089,113 | 8,193,689 | 8,298,266 | 8,402,842 | 24,894,798 |
New drug | 8,089,113 | 12,221,438 | 13,397,209 | 15,631,324 | 41,249,971 | |
Budget impact | 0 | 4,027,748 | 5,098,943 | 7,228,482 | 16,355,174 | |
CADTH base case | Reference | 4,615,932 | 4,675,607 | 4,735,282 | 4,794,957 | 14,205,845 |
New drug | 4,615,932 | 6,928,784 | 7,587,701 | 8,838,669 | 23,355,153 | |
Budget impact | 0 | 2,253,178 | 2,852,419 | 4,043,712 | 9,149,309 | |
CADTH scenario analysis A: 79.5% price reduction | Reference | 4,615,932 | 4,675,607 | 4,735,282 | 4,794,957 | 14,205,845 |
New drug | 4,615,932 | 4,765,797 | 4,849,459 | 4,956,819 | 14,572,075 | |
Budget impact | 0 | 90,191 | 114,177 | 161,863 | 366,231 | |
CADTH scenario analysis B: market share and capture of deferasirox | Reference | 4,576,206 | 4,635,368 | 4,694,529 | 4,753,691 | 14,083,588 |
New drug | 4,576,206 | 6,892,570 | 7,552,042 | 8,804,624 | 23,249,236 | |
Budget impact | 0 | 2,257,202 | 2,857,513 | 4,050,934 | 9,165,648 |
Stakeholder Input
Patient Input
Sickle Cell Awareness Group of Ontario
October 21, 2022
To: CADTH Review Team and Expert Committee Members
RE: Recommendation Letter from the Sickle Cell Awareness Group of Ontario to CADTH Reimbursement Review of Ferriprox (Deferiprone) for Transfusional Iron Overload due to Sickle Cell Disease (SCD) and other Anemias
There are currently three iron chelators readily available for patients- deferiprone/DFP (branded as Ferriprox), deferasirox/DFX (branded as Exjade and Jadenu) and deferoxamine/DFO (branded as Desferal).1
According to a study by Pustika Amalia Wahidiyat et al, comparing Deferiprone to Deferasirox and Deferoxamine to Cardiac and Hepatic T2* MRI in Thalassemia Patients: Evidence-based Case Report; it was found that DFP is superior in controlling or reducing myocardial iron load (as proven by mT2* MRI) and DFO had better capabilities in controlling or reducing hepatic iron load (as proven by liver T2* MRI)1.
Ferriprox (deferiprone) is an oral iron-chelating agent that lowers iron levels by removing toxic iron from organ tissues and fluids. This prescription medicine is used to treat iron overload from blood transfusions in people with Thalassemia, Sickle cell disease and other anemias.2
People with sickle cell disease face debilitating complications not limited to vaso-occlusive pain crisis and damage to their vital organs (including kidney and liver), which may require them to receive blood transfusions. Unfortunately, continuous blood transfusions can lead to an excessive build up of iron in people with sickle cell disease who are unable to excrete excess iron from their body, thus causing further organ damage and increase cancer risk.
To reduce iron overload in organs (a preventable complication) in people with sickle cell disease, it becomes doubly important to ensure that patients have access to appropriate iron chelation therapies.
While DFP or Ferriprox was prior approved by Health Canada for thalassemia iron overload treatment, it is heart warming that Health Canada recently approved this therapy for sickle cell disease.
As a patient advocacy and support association, the Sickle Cell Awareness Group of Ontario (SCAGO) believes that Ferriprox will provide Ontarians living with sickle cell disease and who require treatment for chronic iron overload an option that will improve their adherence to physician prescribed treatment and reduced preventable organ damage from iron overload.
With the foregoing, the SCAGO is recommending where Ferriprox meets CADTH reimbursement criteria to be duly reimbursed in Canada.
In the long run, this will reduce preventable deaths and the burden on the Canadian health system budget.
We thank you in advance for your time and should you have any questions, we would be happy to respond.
Warm regards,
President/Chief Executive Officer, Sickle Cell Awareness Group of Ontario
Web: www.sicklecellanemia.ca | Email: sicklecellawarenessontario@gmail.com
References
1. Wahidiyat PA, Yosia M, Sari TT. Comparison of Deferiprone to Deferasirox and Deferoxamine to Cardiac and Hepatic T2* MRI in Thalassemia Patients: Evidence-based Case Report. Acta Med Indones. 2018 Apr;50(2):168-176. PMID: 29950538.
Thalassemia Foundation of Canada
Wednesday, October 26, 2022
Dear CADTH Reviewers and Expert Committee Members,
I am writing to you on behalf of the Thalassemia Community in Canada appealing for your kind consideration in favor of equitable and sustainable access to deferiprone (an oral iron chelation agent) for thalassemia and all other chronically transfused patients.
The Thalassemia Foundation of Canada (TFC) is a patient driven support organization serving the thalassemia community across Canada. The TFC started as a patient and parent peer support group back in the eighties and has grown to become a national patient organization leading activities and objectives that benefit all thalassemia patients. TFC has been a registered Canadian charity since 1988. The mission of TFC is to support and fund thalassemia scientific research, treatment, patient services, public awareness and education.
Thalassemia disorders are genetic blood disorders where the patient’s body does not produce sufficient red blood cells. Patients living with the severe forms of thalassemia have life dependency on regular red blood cell transfusions, which causes iron overload. If untreated results in progressive organ dysfunction and premature death. Because of iron overload, many thalassemia patients suffer from comorbidities such as heart and liver disease, pulmonary hypertension, diabetes, and other endocrinopathies that are known to reduce survival.
Fortunately, and thanks to medical advances, the use of iron chelation agents over the past several decades has been instrumental in preventing serious health complications and comorbidities that would add to an already disease burdened patient and family. However, not all iron chelation agents are equally affective for all thalassemia and other chronically transfused patients. Often chelation agents cause harmful undesired side effects such that alternative chelation agents must be prescribed. The most common side effects include renal failure, kidney complications, gastrointestinal discomforts, high-frequency hearing loss, deafness, retinal damage, impaired vision, growth retardation, and bone abnormalities. In addition, it is naturally understandable and evidently proven that better adherence to treatment is achievable through oral iron chelation than a cumbersome self-injections regimen alternative. It has been demonstrated and well known that patient outcomes could be improved through an equitable and sustainable access to a variety of available iron chelation options for thalassemia and other chronically transfused patients.
Deferiprone (Feriprox), while it has been licensed and accessible to thalassemia patients living in Europe and other parts of the world for over two decades was finally available for Canadian patients in 2015. This life saving medication which has been proven to be a very affective iron chelation agent is prescribed to patients who could not tolerate or benefit from using other available iron chelation options. Expanding an equitable and sustainable access to various chelation agents is what thalassemia and other chronically transfused patients deserve rather than limiting treatment options because of access denial that would burden patients and their families with the high costs of life saving therapies.
The Thalassemia Foundation of Canada provided a patient input submission to the Common Drug Review back in October 2015 for the initial review of Feriprox referencing evidence and facts gathered from credible sources which remains valid and true, and we strongly reaffirm our position on the points presented in the submission.
We ask your honorable committee members to grant recommendations in favor of an equitable and sustainable access to deferiprone (Feriprox) to thalassemia and other chronically transfused patients that will expand the choices for life saving oral chelation therapy options that will effectively improve patient outcomes.
We look forward to receiving the Committee’s recommendations and we greatly value the opportunity to express and share the views and position of the Thalassemia Community.
Clinician Group Input
Canadian Hemoglobinopathy Association
About the Canadian Hemoglobinopathy Association
The Canadian Hemoglobinopathy Association/ L’Association canadienne d’hémoglobinopathie (CanHaem) is a not-for-profit organization dedicated to the care of individuals across Canada with Hemoglobinopathies. Since its birth in 2013, CanHaem has had several important milestones: the sickle cell consensus statement (2014), the patient emergency sickle cell disease card (2014-2015), and the development of active subcommittees such as the Education subcommittee and the Nursing collaboration. CanHaem is dedicated to encouraging collaboration as well as fostering research, education and quality of care. CanHaem is actively participating in the development of a peer-review process, aimed at helping Canadian centres reach the highest standards of care. CanHaem is a group of subspecialists (Pediatric and adult hematologists) and associated allied health care professionals that care for patients with Sickle Cell Disease and Thalassemia. Home - Canhaem
Information Gathering
From members of our organization, working knowledge of treating sickle cell disease and iron overload, discussions. Current literature, ational and international guidelines, and standards of care.
Current Treatments and Treatment Goals
Sickle cell disease care can necessitate chronic red blood cell transfusions to decrease morbidity, mortality and for disease prophylaxis (i.e. decreasing the risk of stroke in a child with sickle cell disease – RJ, McKie VC, Hsu L, et al. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. N Engl J Med. 1998;339:5-11). Iron overload results from the transfusions and can lead to end organ complications and premature death if not managed. See CanHaem guidelines on when to use chronic transfusions in Sickle Cell Disease. Sickle-Cell-Consensus.pdf (canhaem.org)
Current treatments include: deferoxamine Subcutaneous (12-18 hours) or iv continuous or deferasirox (oral).
Treatment of iron overload in Canada for patients with Sickle Cell Disease is limited as the number of medications available are few. Many patients experience side effects with iron chelators and can only tolerate one type of medication (i.e., avoidance of deferasirox in renal dysfunction. Renal dysfunction is a common complication in sickle cell disease). Deferoxamine is often not feasible due to need for iv or sc delivery.
Iron chelators modify the risk associated with iron overload by removing iron from the body slowly. The iron chelators as a group also require monitoring to avoid drug toxicity. While the chelators share some overlap there is a need to expand the drug choices to provide options when a patient cannot tolerate the available choices.
An ideal iron chelator would prolong life, drug toxicity, and overcome drug delivery challenges.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
Not all patients respond to available treatments.
Treatments are needed that are better tolerated (some patients cannot take available medications due to organ toxicity).
Treatments are needed to improve compliance.
Formulations are needed to improve convenience.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
The hemolgobinopathy specialist would decide this based on the patient and a number of factors such as patient age, health status, family/lifestyle considerations, other organ toxicity and other potential considerations.
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
The population are those who have Sickle Cell Disease and on transfusion therapy demonstrating iron overload. The medication is critical in this population as many have kidney dysfunction and the other available oral chelator should be avoided with kidney dysfunction.
Identification of potential patients best suited for them medication include determination of social factors (unable to tolerate subcutaneous drug infusion, need for oral agent), those not at risk of neutropenia, those with liver or kidney dysfunction. Those known to have iron overload with a validated tool: MRI T2* liver, heart ferriscan.
Are there any issues related to diagnosis? Ideally the care will be managed by a hematologist. Sickle Cell Disease requires subspecialty expertise to manage.
Ferritin is also used to follow patients but the above-mentioned tests are required in Sickle Cell Disease.
Is it likely that misdiagnosis occurs in clinical practice (e.g., under diagnosis)? It is the recommendation that a hematologist with expertise in hemoglobinopathies manages patients (pediatric and adult) with sickle cell disease. Sickle Cell Disease is often not diagnosed if newborn screening the child is not born in a province with newborn screening or if the patient is new to Canada. Iron overload is silent without appropriate long-term care and can have long term consequences.
Is it possible to identify those patients who are most likely to exhibit a response to treatment with drug under review? It is possible to determine response to treatment with a medication trial (at least 6months to one year) but not those most likely to respond. Again, there are no other available medications suitable often.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
The outcomes used in clinical practice align with the outcomes used in clinical trials. These are usually MRI T2* heart and liver (tracking with ferritin to test between MRI scans). An alternative test is the ferriscan used in some clinical practices and clinical trials.
A clinically meaningful response to treatment would be a declining iron burden over time or maintenance of iron burden (no change rather than further accumulation) while still transfusion therapy based on MRI T2* heart/liver. All hematologists would have the same criteria.
What factors should be considered when deciding to discontinue treatment with the drug under review?
Not applicable.
What settings are appropriate for treatment with deferiprone? Is a specialist required to diagnose, treat, and monitor patients who might receive deferiprone?
Sickle Cell Disease with iron overload should be managed by a hematologist (pediatric or adult as appropriate).
Additional Information
Sickle Cell Disease is a complex disease involving every organ. The multitude and unique presentations of this condition require subspecialty care. Iron overload is clinically silent and deadly. Without appropriate medication options available, ramifications to patients are widespread. For example, if a patient has iron overload and kidney dysfunction currently the only option is burdensome to the patient (requiring an iv continuous infusion or subcutaneous infusion run on a pump). This burden leads to no adherence, skin breakdown and expensive equipment (pump).
Conflict of Interest Declarations
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please refer to the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
No.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician who contributed to the input.
Declaration for Clinician 1
Name: Aisha Bruce
Position: Chair, Canadian Hemoglobinopathy Association (Associate professor University of Alberta, Staff Physician Stollery Children’s Hospital, Pediatric Hematologist)
Date: 21-6-2022
Table 1: COI Declaration for Canadian Hemoglobinopathy Association — Clinician 1
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Bristol-Meyers-Squibb (quality of life analysis NOT sickle cell disease, chelation, or thalassemia relevant) | — | — | X | — |
Declaration for Clinician 2
Name: Catherine Corriveau-Bourque
Position: Vice-Chair Canadian Hemoglobinopathy Association (Associate professor University of Alberta, Staff Physician Stollery Children’s Hospital, Pediatric Hematologist)
Date: June 22, 2022
Table 2: COI Declaration for Canadian Hemoglobinopathy Association — Clinician 2
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
Declaration for Clinician 3
Name: Dr. Lauren D. Bolster
Position: Member, Canadian Hemoglobinopathy Association (assistant professor University of Alberta, Staff Physician University of Alberta Hospital, Edmonton
Date: June 22, 2022
Table 3: COI Declaration for Canadian Hemoglobinopathy Association — Clinician 3
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Bristol-Myers-Squibb | X | — | — | — |
Declaration for Clinician 4
Name: Suzan Williams
Position: Staff Physician, Division of Haematology/Oncology, Department of Paediatrics, Hospital for Sick Children
Date: 22-06-2022
Table 4: COI Declaration for Canadian Hemoglobinopathy Association — Clinician 4
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Cheisi | X | — | — | — |
Declaration for Clinician 5
Name: Ali Amid
Position: member, Canadian Hemoglobinopathy Association, Pediatric Hematologist Oncologist, Director of Hemoglobinopathy and Iron Overload Clinic, BC Children's Hospital, Vancouver, BC
Date: June 28, 2022
Table 5: COI Declaration for Canadian Hemoglobinopathy Association — Clinician 5
Company | $0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 |
|---|---|---|---|---|
No COI | — | — | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines.
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.