CADTH Reimbursement Review
Guselkumab (Tremfya)
Sponsor: Janssen Inc.
Therapeutic Area: Psoriatic arthritis
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
ACR
American College of Rheumatology
ACR 20
American College of Rheumatology 20% improvement
ACR 50
American College of Rheumatology 50% improvement
ACR 70
American College of Rheumatology 70% improvement
ANCOVA
analysis of covariance
ASQoL
Ankylosing Spondylitis Quality of Life
BASDAI
Bath Ankylosing Spondylitis Disease Activity Index
bDMARD
biologic disease-modifying antirheumatic drug
BSA
body surface area
CAPA
Canadian Arthritis Patient Alliance
CAPP
Canadian Association of Psoriasis Patients
CDEC
CADTH Canadian Drug Expert Committee
cDMARD
conventional disease-modifying antirheumatic drug
CI
confidence interval
CMH
Cochran-Mantel-Haenszel
CPN
Canadian Psoriasis Network
CrI
credible interval
CRP
C-reactive protein
CSR
Clinical Study Report
DAPSA
Disease Activity Index for Psoriatic Arthritis
DAS 28
Disease Activity Score 28
DAS 28 CRP
Disease Activity Score 28 using C-reactive protein
DLQI
Dermatology Life Quality Index
DMARD
disease-modifying antirheumatic drug
ESR
erythrocyte sedimentation rate
EULAR
European League Against Rheumatism
FACIT-Fatigue
Functional Assessment of Chronic Illness Therapy – Fatigue Scale
FAS
full analysis set
GRAPPA
Group for Research and Assessment of Psoriasis and Psoriatic Arthritis
HAQ-DI
Health Assessment Questionnaire Disability Index
HRQoL
health-related quality of life
ICC
intra-class correlation coefficient
IGA
Investigator’s Global Assessment of Psoriasis
IL
interleukin
ITC
indirect treatment comparison
JAK
Janus kinase
LEI
Leeds Enthesitis Index
LS
least squares
MCS
Mental Component Summary
MDA
minimal disease activity
MID
minimal important difference
MMRM
mixed-effects model for repeated measures
NMA
network meta-analysis
NSAID
nonsteroidal anti-inflammatory drug
PASI
Psoriasis Area and Severity Index
PASI 75
75% improvement from baseline in Psoriasis Area and Severity Index score
PASI 90
90% improvement from baseline in Psoriasis Area and Severity Index score
PASI 100
100% improvement from baseline in Psoriasis Area and Severity Index score
PCS
Physical Component Summary
PsA
psoriatic arthritis
PsAID-FC
Psoriatic Arthritis Impact of Disease Instrument Functional Capacity
RCT
randomized controlled trial
SC
subcutaneous
SD
standard deviation
SF-36
Short Form (36) Health Survey
SRM
standardized response mean
TNF
tumour necrosis factor
VAS
visual analogue scale
vdH-S
van der Heijde-Sharp
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Guselkumab (Tremfya) 100 mg/mL in a 1 mL pre-filled syringe or patient-controlled injector |
Indication | For the treatment of adult patients with active psoriatic arthritis. Guselkumab can be used alone or in combination with a conventional DMARD (e.g., methotrexate). |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | September 4, 2020 |
Sponsor | Janssen Inc. |
DMARD = disease-modifying antirheumatic drug; NOC = Notice of Compliance.
Introduction
Psoriatic arthritis (PsA) is an inflammatory musculoskeletal disease with a heterogenous presentation and disease course. While it is associated with psoriasis, PsA also presents with variable clinical features involving multiple domains, including peripheral arthritis, enthesitis (tenderness and swelling at the insertion of tendons and ligaments into bone), dactylitis (swelling of whole digits), and axial disease (inflammation of the joints of the back).1,2 Pain and stiffness of the affected joints are the most predominant presenting symptoms, with fatigue also occurring in many patients.1
The prevalence of PsA varies, depending on the case definition and geography, but it is estimated to be 1 to 2 cases per 1,000 in the general population.1 A population-based Canadian study estimated the age- and sex-standardized cumulative prevalence of PsA in Ontario to range from 0.09% in 2008 to 0.15% in 2015.3
Several drug classes are used in the pharmacologic treatment of PsA, including nonsteroidal anti-inflammatory drugs (NSAIDs), conventional disease-modifying antirheumatic drugs (cDMARDs) (i.e., methotrexate, sulfasalazine, leflunomide), biologic disease-modifying antirheumatic drugs (bDMARDs) (i.e., tumour necrosis factor [TNF] inhibitors, interleukin [IL]-23 inhibitors, IL-12/23 inhibitors, and IL-17 inhibitors), and targeted synthetic DMARDs (e.g., apremilast, upadacitinib, or tofacitinib).4
Guselkumab is a human immunoglobulin G1 lambda monoclonal antibody that binds to the IL-23 protein and inhibits its binding with the cell surface IL-23 receptor.5 Guselkumab is approved for the treatment of adult patients with active PsA.5 It may be used alone or in combination with a cDMARD (e.g., methotrexate) and is available as a 100 mg/mL solution for subcutaneous (SC) injection in either 1 mL pre-filled syringes or patient-controlled injector devices. The recommended dose for PsA is 100 mg SC at week 0, week 4, and every 8 weeks thereafter.5
The objective of this report was to perform a systematic review of the beneficial and harmful effects of guselkumab SC injection for the treatment of active PsA in adults.
Stakeholder Perspectives
The information in this section is a summary of the input provided by the patient groups that responded to CADTH’s call for patient input and from the clinical expert(s) consulted by CADTH for the purpose of this review.
Patient Input
Three patient group inputs were submitted for this review by 6 different patient groups: Arthritis Consumer Experts, the Canadian Association of Psoriasis Patients (CAPP) in partnership with the Canadian Psoriasis Network (CPN), and the Canadian Arthritis Patient Alliance (CAPA) in partnership with the Arthritis Society and CreakyJoints. Five patient organizations (CAPP, CPN, CAPA, the Arthritis Society, and CreakyJoints Canada) collaborated by collectively developing survey questions using SurveyMonkey for the inputs submitted. Each of the 3 patient group inputs used the same survey data. There were 71 respondents to the joint survey. In addition, Arthritis Consumer Experts gathered information from 1 respondent through email on May 4, 2022 and from 5 respondents through an online survey from December 18, 2020 to January 26, 2021.
Respondents reported a range of symptoms that are difficult to manage, including joint stiffness (79%), fatigue (75%), changes in fingernails and toes (63%), hip pain (61%), back pain (51%), anxiety (47%), and stress (33%). With regards to the most significant impacts of PsA on their daily quality of life, respondents expressed that PsA interfered with work (54%), social connections (52%), self-esteem (50%), mental health (50%), intimacy (50%), family life (38%), and friendships (24%). Other impacts included embarrassment and self-consciousness from the symptoms caused by PsA. Given that the disease would reduce their mobility and ability to participate in activities and affect their mental and social health, respondents indicated that there were additional tasks or chores for caregivers, such as cooking, cleaning, shopping, and helping patients get to and from medical appointments.
Survey respondents indicated that they had experience with several treatment approaches, including NSAIDs, corticosteroids, cDMARDs (such as methotrexate), and bDMARDs. Among respondents, 32% considered biologics as very effective, followed by oral steroids (23%) and other DMARDs (21%). Respondents expressed their ongoing unmet needs for symptom management and more tolerable side effects with current treatments.
Two respondents who had experience with guselkumab indicated that the drug was effective in terms of improving psoriasis and arthritis and slowing disease progression. Both respondents stated that they did not experience side effects.
Respondents expected new treatments to improve the following key outcomes: management of symptoms (e.g., reducing pain and fatigue, increasing mobility); tolerability of side effects; ease of drug administration; improved ability to work and carry out tasks and daily activities; and quality of life.
Clinician Input
Input From the Clinical Expert Consulted by CADTH
A substantial proportion of patients lose their response to therapy over time or do not achieve a minimal response with their first therapy. In addition, some treatments have more adverse effects than others. There is a need for medications with new mechanisms of action or a different safety profile to offer alternate treatment options for these patients who have an inadequate response or intolerance to therapy. According to the clinical expert, guselkumab may be used as first- or second-line biologic therapy. It may be a preferred first-line treatment for patients with moderate psoriasis in addition to musculoskeletal disease. Guselkumab may be used in combination with methotrexate or leflunomide; however, there is no evidence to support its use in combination with other biologics.
Response to therapy is based on a reduction in the number of inflamed joints, improvement in the skin, and improvements in patient-reported outcomes (i.e., based on assessments of pain, function, and fatigue). According to the clinical expert, a major improvement would constitute at least a 50% improvement, but may also include achievement of minimal disease activity (MDA) and remission as measured by specific instruments. An initial response may be expected within 3 months, with more significant improvement by 6 months after initiating therapy. The expert indicated that if a patient shows no change within 3 to 6 months, they would be considered a nonresponder and may be switched to another medication. The expert stated that ideally, guselkumab would be prescribed by specialists who are familiar with the drug and its uses, or at least in consultation with a dermatologist or rheumatologist.
Clinician Group Input
CADTH received 1 clinician group input submission from the Canadian Rheumatologist Psoriatic Arthritis Interest Group, based on responses from 6 clinicians who practise in academic and community settings. This clinician input largely agreed with the input received from the clinician consulted by CADTH. No major contrary views among the views provided by the clinical experts consulted by CADTH for this review were presented.
Drug Program Input
The drug programs identified the following issues that may affect these programs’ ability to implement a recommendation: relevant comparators, consideration for initiation of therapy, consideration for continuation or renewal of therapy, consideration for discontinuation of therapy, consideration for prescribing of therapy, and system and economic issues. Refer to Table 5.
Clinical Evidence
Pivotal Studies and Protocol Selected Studies
Description of Studies
Three double-blind, placebo-controlled, randomized controlled trials (RCTs) met the inclusion criteria for the systematic review. The pivotal trials (DISCOVER-1 and DISCOVER-2) included patients with active PsA who had experienced an inadequate response to cDMARDs, apremilast, and/or NSAIDs. The DISCOVER-1 study (N = 381) enrolled a mixed population that included patients with no prior biologic treatment experience; however, up to 30% of patients had previously received 1 or 2 TNF alpha inhibitors. In the DISCOVER-2 study, all enrolled patients were biologic-naive (N = 741). The COSMOS study enrolled patients with active PsA who were intolerant to or had experienced an inadequate response to 1 or 2 TNF alpha inhibitors (N = 285). The trials were conducted mainly in Europe, with some sites in Asia, the US, Australia, Israel, and Canada (DISCOVER-1 only).
Patients were randomized to receive placebo or guselkumab 100 mg SC at week 0, week 4, and every 8 weeks thereafter for 24 weeks. The DISCOVER trials included a third treatment group (guselkumab 100 mg every 4 weeks). However, the latter dosage is not consistent with the Health Canada–recommended dose; therefore, results for this treatment group are not included in this report. The total trial durations were 52 weeks (DISCOVER-1), 100 weeks (DISCOVER-2), and 48 weeks (COSMOS), with placebo patients switching to guselkumab starting at week 24. During the trials, patients could continue receiving methotrexate, leflunomide, hydroxychloroquine, sulfasalazine, oral corticosteroids, or NSAIDs if the doses were stable and did not exceed the protocol-specified maximum doses. Early escape therapy consisting of cDMARDs, corticosteroids, or NSAIDs — or a switch to guselkumab (COSMOS) — was available at week 16 for patients who had an improvement of less than 5% in tender and swollen joint counts.
The primary outcome in all trials was the proportion of patients who achieved an American College of Rheumatology (ACR) 20% improvement (ACR 20) at week 24. The ACR 20 was defined as a greater than or equal to 20% improvement from baseline in both swollen joint count (66 joints) and tender joint count (68 joints), and a greater than or equal to 20% improvement from baseline in 3 of the 5 assessments: patient’s assessment of pain, patient’s global assessment of disease activity, physician’s global assessment of disease activity, the Health Assessment Questionnaire Disability Index (HAQ-DI), and C-reactive protein (CRP). Other outcomes of interest included the change from baseline in the HAQ-DI and Short Form (36) Health Survey (SF-36) Physical Component Summary (PCS) and the impact on plaque psoriasis (as measured by the Investigator’s Global Assessment of Psoriasis [IGA] or Psoriasis Area and Severity Index [PASI] response).
The mean age of patients enrolled ranged from 44.9 years (standard deviation [SD] = 11.9 years) to 49.1 years (SD = 12.3 years) across treatment groups in the 3 trials. The proportion of female patients ranged from 46% to 55%, and most patients were White (89% to 98%; not reported for the COSMOS study). The mean number of swollen joints ranged from 9.0 (SD = 5.7) to 12.3 (SD = 6.9), and the mean number of tender joints ranged from 18.2 (SD = 10.7) to 21.6 (SD = 13.1). Approximately 2-thirds of patients had psoriatic involvement affecting at least 3% of their body surface area (BSA). Two-thirds of patients reported enthesitis, while approximately 40% had dactylitis at baseline. The majority of patients (54% to 63%) were receiving methotrexate at baseline, with a lower percentage (0% to 7%) receiving other permitted cDMARDs. In the DISCOVER trials, 14% to 20% of patients were receiving oral corticosteroids at baseline versus 4% to 5% of patients in the COSMOS study.
Efficacy Results
In the DISCOVER-1 study, 52.0% of patients in the group receiving guselkumab every 8 weeks achieved ACR 20 at 24 weeks, compared with 22.2% of patients in the placebo group. The absolute difference was 29.8% (95% confidence interval [CI], 18.6% to 41.1%; P < 0.001), favouring guselkumab versus placebo (Table 2). The proportion of patients who achieved at least an American College of Rheumatology 50% improvement (ACR 50) was 29.9% versus 8.7% (absolute difference = 21.4%; 95% CI, 12.1% to 30.7%) for guselkumab every 8 weeks versus placebo; the proportion of patients who achieved an American College of Rheumatology 70% improvement (ACR 70) was 11.8% versus 5.6% (absolute difference = 6.4%; 95% CI, −0.3% to 13.1%) for guselkumab every 8 weeks versus placebo. However, ACR 50 and ACR 70 were not controlled for multiple testing and should be interpreted considering the inflated risk of type I error rate.
Among patients who were biologic-naive (DISCOVER-2), 64.1% and 32.9% achieved ACR 20 at 24 weeks in the group receiving guselkumab every 8 weeks and the placebo group, respectively, with an absolute difference of 31.2% (95% CI, 22.9% to 39.5%; P < 0.001) (Table 2). The proportion of patients who achieved ACR 50 was 31.5% versus 14.2% (absolute difference = 17.2%; 95% CI, 10.0% to 24.4%), and the proportion of patients who achieved ACR 70 was 18.5% versus 4.1% (absolute difference = 14.5%; 95% CI, 9.1% to 19.9%). ACR 50 and ACR 70 were not controlled for multiple testing (i.e., the type I error rate has not been controlled).
For biologic-experienced patients who were enrolled in the COSMOS study, 44.4% and 19.8% achieved ACR 20 at week 24 in the group receiving guselkumab every 8 weeks and the placebo group, respectively. The absolute difference between groups favoured guselkumab: 24.6% (95% CI, 14.1% to 35.2%; P < 0.001) (Table 2). The difference also favoured guselkumab every 8 weeks versus placebo for the proportion who achieved ACR 50 (19.6% versus 5.2%; absolute difference = 14.3%; 95% CI, 7.2% to 21.4%; P < 0.001). ACR 70 was achieved by 7.9% versus 1.0% of patients in the group receiving guselkumab every 8 weeks versus the placebo group, with an absolute difference of 6.8% (95% CI, 2.6% to 11.1%). ACR 70 was not controlled for multiple testing.
In the DISCOVER trials, the odds ratios of ACR 20 response were generally consistent across subgroups based on prior TNF alpha inhibitor use, use of non-biologic DMARDs, oral corticosteroids, or NSAIDs at baseline; however, the trials may not have been powered to detect subgroup differences. The COSMOS study did not report data for any subgroups of interest to this review.
Disability was assessed using the HAQ-DI, a patient-reported, 20-question instrument that assesses the degree of difficulty a person has in accomplishing tasks in 8 functional areas (dressing, arising, eating, walking, hygiene, reaching, gripping, and activities of daily living). The overall score is the average of 8 domains ranging from 0 (no disability) to 3 (completely disabled). The change from baseline to week 24 in the HAQ-DI favoured guselkumab every 8 weeks versus placebo in all trials (Table 2). The least squares (LS) mean differences versus placebo reported were −0.25 (95% CI, −0.36 to −0.13; P < 0.001) in the DISCOVER-1 study; −0.24 (95% CI, −0.32 to −0.15; P < 0.001) in the DISCOVER-2 study; and −0.17 (95% CI, −0.28 to −0.06; P = 0.003) in the COSMOS study. Across the trials, the between-group and within-group differences did not exceed the 0.35 minimal important difference (MID) cited by the sponsor,6 with the exception of the change from baseline within the group receiving guselkumab every 8 weeks in the DISCOVER-2 study.
The change from baseline to week 24 in the SF-36 PCS favoured guselkumab every 8 weeks versus placebo in all 3 studies (Table 2). The LS mean differences were 4.1 (95% CI, 2.4 to 5.9; P < 0.001) in DISCOVER-1, 4.0 (95% CI, 2.7 to 5.2; P = 0.011) in DISCOVER-2, and 3.9 (95% CI, 2.5 to 5.4; P < 0.001) in the COSMOS study. The Clinical Study Report defined at least a 5-point increase as clinically meaningful, but a MID of 3.74 points has also been reported in the literature.7 No statistically significant differences were detected between guselkumab every 8 weeks and placebo in the change from baseline to week 24 in the SF-36 Mental Component Summary (MCS).
In all trials, psoriasis skin disease outcome measures were analyzed in the subgroup of patients who had psoriasis affecting greater than or equal to 3% BSA and an IGA score of greater than or equal to 2 at baseline (55% to 74% of patients per treatment group). Psoriasis severity was assessed using composite physician-reported assessments: IGA and PASI response. For the IGA, the severity of a patient’s psoriasis is scored as cleared (0), minimal (1), mild (2), moderate (3), or severe (4). IGA response was defined as a score of 0 or 1, and at least a 2-point decrease from baseline. The PASI evaluates the extent and severity of psoriasis and is scored from 0 to 72 points, with a PASI score greater than 10 considered to represent severe disease. Patients with 90% improvement in their PASI score (PASI 90) or 100% improvement in their PASI score (PASI 100) would meet PASI 90 or PASI 100 response criteria, respectively.
The proportion of patients who achieved an IGA response at week 24 was higher in the groups receiving guselkumab every 8 weeks than in the placebo groups in the DISCOVER-1 study (57.3% versus 15.4%; absolute difference = 42.0% [95% CI, 28.9% to 55.1%; P < 0.001]) and DISCOVER-2 study (70.5% versus 19.1%; absolute difference = 50.9% [95% CI, 42.2% to 59.7%; P < 0.001]) (Table 2). In the COSMOS study, 48.1% versus 9.4% of patients in the guselkumab-every-8-weeks versus placebo group achieved in IGA response, with an absolute difference of 38.8% (95% CI, 27.3% to 50.4%); however, the P value has not been adjusted to control for multiple testing and should be interpreted with caution because of the potential for inflated type I error rate. PASI 100 response at week 24 was a secondary outcome in the COSMOS study. In the guselkumab group, 30.8% of patients achieved a PASI 100 response compared with 3.8% of patients in the placebo group (absolute difference = 27.4% [95% CI, 17.9% to 36.8%; P < 0.001). In the DISCOVER trials, the proportion of patients who achieved a PASI 100 response was nominally higher for the guselkumab versus placebo groups; however, these outcomes were not controlled for multiple testing and should be interpreted considering the inflated risk of type I error rate.
For patients with enthesitis or dactylitis at baseline, the results of the DISCOVER-2 and COSMOS studies suggest an improvement in enthesitis or dactylitis end points, with guselkumab every 8 weeks relative to placebo, but no statistically significant difference was detected between groups in DISCOVER-1. Based on the preplanned pooled analysis of data from DISCOVER-1 and DISCOVER-2, 49.6% and 29.4% of patients in the guselkumab-every-8-weeks and placebo groups had resolution of enthesitis at week 24, with a between-group difference of 20.1% (95% CI, 11.8% to 28.5%; P = 0.03). The proportion of patients whose dactylitis resolved at week 24 was 59.4% versus 42.2% in the guselkumab-every-8-weeks versus placebo groups (between-group difference = 18.0%; (95% CI, 7.4% to 28.6%; P = 0.03). None of the trials detected a statistically significant difference between guselkumab every 8 weeks and placebo in the proportion of patients who reported a clinically important improvement in axial disease based on the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI). It is noteworthy that these outcomes were tested in subgroups of patients that may not have been balanced with respect to baseline demographic and disease characteristics between treatment groups, due to the lack of stratification at randomization.
Symptoms of fatigue were assessed using the Functional Assessment of Chronic Illness Therapy – Fatigue Scale (FACIT-Fatigue). The scores ranged from 0 to 52, with lower scores reflecting more severe fatigue. Estimates of the MID ranged from 3.1 points to 4 points.8,9 The DISCOVER-1 study reported an LS mean difference of 3.4 points (95% CI, 1.4 to 5.4); DISCOVER-2 reported an LS mean difference of 4.0 (95% CI, 2.5 to 5.5); and COSMOS reported an LS mean difference of 3.6 (95% CI, 1.7 to 5.4) for guselkumab every 8 weeks versus placebo. This outcome was not controlled for multiple testing (i.e., the type I error rate has not been controlled).
Radiographic progression was a major secondary outcome in the DISCOVER-2 study. Progression was assessed using the modified van der Heijde-Sharp (vdH-S) score, which ranges from 0 (best) to 528 (worst) and is the sum of the joint erosion score and the joint space narrowing score.10 At 24 weeks, the study failed to detect a statistically significant difference between guselkumab every 8 weeks and placebo in the change from baseline in vdH-S score. However, the duration of the trial may have been insufficient to detect a difference.
Harms Results
The frequency of adverse events was generally similar between groups in all trials, with 42% to 54% of patients in the groups receiving guselkumab every 8 weeks and 41% to 60% of patients in the placebo groups reporting 1 or more adverse events during the 24-week treatment period (Table 3). Nasopharyngitis (4% to 13%), upper respiratory tract infection (2% to 5%), and increased alanine aminotransferase (2% to 6%) were the most common adverse events in the guselkumab groups, with a comparable frequency of these events reported in the placebo groups. Generally, the frequency of infections was similar in the guselkumab groups (16% to 26%) and placebo groups (18% to 25%) across trials. Few serious infections were reported (0% to 0.5% in the guselkumab groups).
The frequency of serious adverse events ranged from 1% to 4% in the groups receiving guselkumab every 8 weeks and 3% to 4% in the placebo groups. No specific serious adverse events were reported in more than 1 patient per treatment group. Adverse events that resulted in treatment discontinuation were generally low and similar between groups (1% to 3%). In the DISCOVER-1 study, 1 patient in the placebo group died due to cardiac failure. No other deaths were reported in the first 24 weeks of the trials.
Table 2: Summary of Key Efficacy Results From Pivotal and Protocol Selected Studies
Outcome | DISCOVER-1 (FAS) | DISCOVER-2 (FAS) | COSMOS (FAS) | |||
|---|---|---|---|---|---|---|
GUSE q8w N = 127 | PBO N = 126 | GUSE q8w N = 248 | PBO N = 246 | GUSE q8w N = 189 | PBO N = 96 | |
ACR 20 at week 24a | ||||||
ACR 20 response at week 24, n (%) | 66 (52.0) | 28 (22.2) | 159 (64.1) | 81 (32.9) | 84 (44.4) | 19 (19.8) |
% difference vs. PBO (95% CI) | 29.8 (18.6 to 41.1) | Reference | 31.2 (22.9 to 39.5) | Reference | 24.6 (14.1 to 35.2) | Reference |
P value | < 0.001 | Reference | < 0.001 | Reference | < 0.001 | Reference |
Change from baseline to week 24 in HAQ-DI scoreb | ||||||
Number of patients contributing to the analysis | 127 | 126 | 248 | 246 | 189 | 96 |
Baseline, mean (SD) | 1.05 (0.57) | 1.11 (0.63) | 1.28 (0.63) | 1.29 (0.56) | 1.33 (0.60) | 1.22 (0.60) |
Change from baseline, LS mean (95% CI) | −0.32 (−0.41 to −0.24) | −0.07 (−0.16 to 0.01) | −0.37 (−0.43 to −0.31) | −0.13 (−0.19 to −0.07) | −0.18 (−0.27 to −0.09) | −0.001 (−0.12 to 0.10) |
LS mean difference (95% CI) vs. placebo | −0.25 (−0.36 to −0.13) | Reference | −0.24 (−0.32 to −0.15) | Reference | −0.17 (−0.28 to −0.06) | Reference |
P value | < 0.001 | Reference | < 0.001 | Reference | 0.003 | Reference |
Change from baseline to week 24 in SF-36 PCSb | ||||||
Number of patients contributing to the analysis | 127 | 126 | 248 | 246 | 188 | 96 |
Baseline, mean (SD) | 34.1 (7.6) | 33.8 (8.5) | 32.6 (7.9) | 32.4 (7.0) | 33.0 (7.0) | 33.9 (7.7) |
Change from baseline, LS mean (95% CI) | 6.1 (4.8 to 7.4) | 2.0 (0.7 to 3.2) | 7.4 (6.5 to 8.3) | 3.4 (2.5 to 4.3) | 3.5 (2.3 to 4.7) | −0.4 (−1.8 to 1.1) |
LS mean difference (95% CI) vs. placebo | 4.1 (2.4 to 5.9) | Reference | 4.0 (2.7 to 5.2) | Reference | 3.9 (2.5 to 5.4) | Reference |
P value | < 0.001 | Reference | 0.011c | Reference | < 0.001 | Reference |
IGA response at week 24a | ||||||
Number of patients contributing to analysis, N (%) | 82 (65) | 78 (62) | 176 (71) | 183 (74) | 133 (70) | 53 (55) |
IGA response at week 24, n (%) | 47 (57.3) | 12 (15.4) | 124 (70.5) | 35 (19.1) | 64 (48.1) | 5 (9.4) |
% difference vs. PBO (95% CI) | 42.0 (28.9 to 55.1) | Reference | 50.9 (42.2 to 59.7) | Reference | 38.8 (27.3 to 50.4) | Reference |
P value | < 0.001 | Reference | < 0.001 | Reference | < 0.001d | Reference |
ACR 20 = American College of Rheumatology 20% improvement; ANCOVA = analysis of covariance; CI = confidence interval; CMH = Cochran-Mantel-Haenszel; CRP = C-reactive protein; DMARD = disease-modifying antirheumatic drug; FAS = full analysis set; GUSE = guselkumab; HAQ-DI = Health Assessment Questionnaire Disability Index; IGA = Investigator’s Global Assessment of Psoriasis; LS = least squares; MMRM = mixed-effects model for repeated measures; PBO = placebo; q8w = every 8 weeks; SD = standard deviation; SF-36 PCS = Short Form (36) Health Survey Physical Component Summary; TNF = tumour necrosis factor.
aBased on composite estimand (either observed response data or nonresponse for patients who met the treatment failure criteria). Patients with missing data were imputed as nonresponders. P values were based on stratified CMH tests and 95% CIs were based on the Wald statistic. The DISCOVER-1 study CMH test stratified patients by baseline use of non-biologic DMARDs (yes, no) and prior exposure to TNF alpha inhibitors (yes, no). The DISCOVER-2 study CMH test stratified patients by baseline use of non-biologic DMARDs (yes, no) and CRP before randomization (< 2 mg/dL vs. ≥ 2 mg/dL). The COSMOS study CMH test stratified patients by baseline use of non-biologic DMARDs (yes, no) and number of prior TNF alpha inhibitors (1 or 2).
bChange from baseline was based on observed data or 0 (no improvement) if a patient met the treatment failure criteria before week 24 (composite estimand). The DISCOVER-1 and DISCOVER-2 studies used an ANCOVA model, with missing data imputed using multiple imputation, assuming data were missing at random. The COSMOS study used MMRM under the missing-at-random assumption.
cThere were 2 statistical testing procedures used to control the type I error rate. According to the US-based statistical testing procedure, this outcome was statistically significant, with a P value of 0.011. According to the global testing procedure, this outcome was not formally tested for statistical significance due to the failure of a prior outcome. The US-specific procedure was preferred by Health Canada.
dThe P value has not been adjusted for multiple testing (i.e., the type I error rate has not been controlled).
Source: Clinical Study Report for DISCOVER-1 (24 weeks);11 Clinical Study Report for DISCOVER-2 (24 weeks);12 Clinical Study Report for COSMOS (24 weeks);13 Coates et al. (2022).14
Table 3: Summary of Key Safety Results From Pivotal and Protocol Selected Studies
Adverse event | DISCOVER-1 GUSE q8w N = 127 | DISCOVER-1 PBO N = 126 | DISCOVER-2 GUSE q8w N = 248 | DISCOVER-2 PBO N = 246 | COSMOS GUSE q8w N = 189 | COSMOS PBO N = 96 |
|---|---|---|---|---|---|---|
Patients with ≥ 1 adverse event, n (%) (safety population) | ||||||
Any adverse event | 68 (54) | 75 (60) | 114 (46) | 100 (41) | 80 (42) | 46 (48) |
Infections | 33 (26) | 32 (25) | 40 (16) | 45 (18) | 40 (21) | 19 (20) |
Serious infections | 0 | 2 (1.6) | 1 (0.4) | 1 (0.4) | 1 (0.5) | 0 |
SAEs | 4 (3) | 5 (4) | 3 (1) | 7 (3) | 7 (4) | 3 (3) |
Stopped treatment due to adverse events | 3 (2) | 3 (2) | 2 (1) | 4 (2) | 5 (3) | 2 (2) |
Deaths | 0 | 1 (0.8) | 0 | 0 | 0 | 0 |
GUSE = guselkumab; PBO = placebo; q8w = every 8 weeks; SAE = serious adverse event.
Source: Clinical Study Report for DISCOVER-1 (24 weeks);11 Clinical Study Report for DISCOVER-2 (24 weeks);12 Clinical Study Report for COSMOS (24 weeks).13
Critical Appraisal
The risk of bias related to randomization and treatment allocation concealment was rated as low for all studies. In general, patient characteristics and co-interventions appeared to be balanced between groups at baseline. The trials were double blind and took steps to maintain blinding of patients and investigators. Joint assessments were conducted by an independent rater who was not otherwise involved in the trial. Therefore, the risk of bias in the measurement of the outcomes was low for all trials. The frequency of withdrawal in all trials was low and similar between groups; therefore, there is a low risk of bias due to missing outcome data. The full analysis set (FAS), which excluded only 1 randomized patient in DISCOVER-2, was used for all efficacy outcomes; therefore, the analyses were appropriate for estimating the effect of assignment to the intervention.
In all trials, the primary and other dichotomous end points were analyzed using a Cochran-Mantel-Haenszel (CMH) test that was stratified by randomization stratification factors, with missing data imputed as nonresponders. The DISCOVER trials used an adjusted analysis of covariance (ANCOVA) model, and the COSMOS study used an unadjusted mixed-effects model for repeated measures (MMRM) to analyze continuous outcomes. Missing data were imputed under the missing-at-random assumption. This assumption may not hold true, but was not thought to be a major source of bias. Efficacy analyses were based on the composite estimand, with any patients meeting treatment failure criteria considered nonresponders for binary end points, or as no change from baseline for continuous measures. Treatment failure criteria included early study withdrawal or discontinuation of the study drug, or initiation of new treatments for PsA. This estimand, which considers any treatment failure end point to be an unfavourable outcome, may be considered a more conservative estimate of treatment effects. Of note, the COSMOS study incorrectly assigned 20 patients to early escape despite these patients not having met the escape criteria. Although the sponsor conducted sensitivity analyses to explore the impact of this error, these analyses cannot fully address the potential bias. The type I error rate was controlled for the primary and selected secondary outcomes in all studies. However, several outcomes of interest to this review were not controlled for multiplicity; these data should be interpreted with caution, given the potential for inflated type I error rate. Randomization was not stratified by the presence of psoriasis, enthesitis, dactylitis, or axial disease; thus, interpretation of the results for these outcomes should consider the possibility of imbalances in baseline demographic and disease characteristics between treatment groups in these subpopulations. The primary outcome was ACR 20 response; however, according to the clinical expert, this represents the minimum level of improvement that may be relevant to patients. In practice, the goal of therapy is to achieve higher levels of response.
Although the trials were 48 weeks to 100 weeks in duration, the comparative period was limited to 24 weeks for this chronic condition. For outcomes such as radiologic changes, the duration of treatment may have been insufficient to detect the impact of guselkumab. Moreover, none of the trials included an active control group; thus, direct evidence comparing guselkumab to other DMARDs available in Canada is not available.
With regards to external validity, the clinical expert did not identify any substantial limits to generalizability based on the patient population enrolled. The guselkumab dosing regime used in the trials (i.e., once every 8 weeks) was consistent with the Health Canada–recommended dose, and the expert stated that concomitant utilization of cDMARDs was similar to what may be expected in clinical practice. However, the expert also noted that the use of oral corticosteroids in the DISCOVER trials was higher than would be expected in Canada (14% to 20%). The use of a placebo comparator as an add-on to cDMARDs and NSAIDs is not consistent with Canadian practice for patients who have demonstrated an inadequate response to cDMARDs or bDMARDs. The trials excluded patients who had previously been treated with biologics other than TNF inhibitors; thus, the efficacy in patients with intolerance or inadequate response to other biologics, such as Janus kinase (JAK) or other IL inhibitors, is not known.
Indirect Treatment Comparisons
Description of Studies
The sponsor conducted a network meta-analysis (NMA) of RCTs that assessed the comparative efficacy and safety of guselkumab and 13 other bDMARDs for the short-term treatment of acute PsA. The indirect treatment comparison (ITC) was based on a systematic literature review, and 34 RCTs provided data to inform the Bayesian NMA. Analyses were conducted for the overall PsA population, with subgroup analyses restricted to patients who were biologic-naive or biologic-experienced. Treatment durations were 12 weeks to 24 weeks.
One other NMA was identified by CADTH through a literature search. The NMA by McInnes et al. (2022)15 evaluated the efficacy and safety of licensed and unlicensed bDMARDs for patients with active PsA. A total of 46 RCTs, which were identified through a systematic review, were included in the Bayesian analyses. The MNA included 19 biologics with outcomes assessed at 12 weeks to 26 weeks.
Efficacy Results
For the overall PsA population in the sponsor-submitted NMA, the results suggest that patients who received guselkumab every 8 weeks ||| || |||| |||||| || ||||||| || ||| || |||||||| than those who received apremilast or abatacept, but were |||| |||||| || respond than infliximab, golimumab, and secukinumab 300 mg (34 RCTs; ordinal baseline risk-adjusted random-effects model). Comparisons between guselkumab and other biologics had 95% credible intervals (CrIs) that |||||||||| ||| |||| |||||. The NMA results for PASI 90 response |||||||| |||||||||| |||||| ||||||||||| ||| ||| ||| ||||| |||||||||| ||||||| |||||||||||| ||| ||||||||||| ||||||||| ||| ||||||||||. Other comparisons with biologics reported 95% CrIs that |||||||| ||| ||||. For the change in HAQ-DI, SF-36 PCS or MCS, or vdH-S scores, |||| || ||| ||||||||||| |||||||| guselkumab over other biologics, with analyses reporting 95% CrIs that |||||||||| ||| ||||. Across all networks, many comparisons showed imprecise 95% CrIs that included the possibility of appreciable benefit and/or worse outcomes, and || |||||||||| ||||||| |||||||||| ||| ||||| |||||||||, which limits the ability to draw conclusions from these data.
The results in the biologic-naive population were generally |||||||||| |||| ||| |||||||| || ||| ||||||| ||||||||||, although the networks were smaller (17 studies to 30 studies), not all comparators were available for all outcomes, and with |||| || ||| |||||||| ||||||| ||||||| |||||||||. The NMAs in biologic-experienced patients included fewer trials (8 studies to 16 studies) and the reported results were imprecise, limiting the ability to draw conclusions.
The results for ACR 20 and PASI 90 in the NMA by McInnes et al. (2022)15 were largely consistent with the findings of the sponsor-submitted NMA.
Harms Results
In the overall population, the analysis of serious adverse events in the sponsor-submitted NMA |||||| ||| ||| |||| |||||||||| ||| |||| ||| |||| |||||||||||| |||| |||||||| ||||| |||| ||| |||||||||| |||||| |||||||||| || |||||||||||| ||| || |||| ||| |||||||| |||||||| ||||||||||||| |||||| ||||||| ||||||. For all other comparisons, the 95% CrI was |||||||||| limiting the ability to draw a conclusion. In most cases, ||| ||||||| || ||| ||| || ||||||| |||||| |||||| ||||||| ||||||||.
||| ||| ||| ||||||| |||||| || ||||||| ||||||| |||||| || |||||||||||||| ||| ||||||| ||| |||||||||||||||||||| |||||||| || ||||||| |||||| ||| ||| |||| |||||||||| ||| ||||| |||| |||| ||||||||||| ||||||| |||| |||| |||||||| ||| ||||||| || |||| | ||||||||||. No comparative safety data for guselkumab were reported in the NMA by McInnes et al. (2022).15
Critical Appraisal
Although the sponsor-submitted ITC was based on a systematic review, 46 RCTs were excluded from the NMA, and the criteria for selecting trials or outcomes for analysis were not stated. Heterogeneity in patient and study characteristics was identified, and it is unclear if the transitivity assumption has been met. The authors of the NMA attempted to address potential variability in effect modifiers by using a baseline risk-adjusted model, but it is unclear if these effect modifiers have the same level of effect on the active arms. Given that it is unclear to what extent placebo response is an adequate proxy for specific characteristics or effect modifiers, uncertainty remains in these analyses. Subgroup analyses based on prior treatment exposure were conducted to create more homogenous patient populations, but some of these analyses included data from a limited number of trials, and often showed substantial uncertainty, with wide CrIs. There were no subgroup or sensitivity analyses conducted to explore the potential impact of differences in the timing of outcome assessment, duration of disease, background therapies, or year of study.
ACR and PASI percentage improvements were analyzed using an ordinal model, which assumed that the relative treatment effects were the same for each response level. Thus, although data were reported separately for each response level, the inferences for each comparison are the same across the ACR 20, ACR 50, and ACR 70 levels or PASI levels. It is not clear if this assumption of the model holds true (i.e., if relative treatment effects are consistent across response levels), given that data were pooled for different time points. No sensitivity analyses were run to examine the impacts of this assumption.
In the sponsor-submitted NMA, there was limited ability to assess the consistency between direct and indirect evidence because there were few closed loops (i.e., there were only 4 head-to-head studies), and the statistical tests for inconsistency are generally underpowered. Further, most of the contributing trials were judged to be at high or unclear risk of bias in at least 1 domain.
Issues with heterogeneity in patient and study characteristics, lack of ability to assess consistency, and potential bias in the included RCTs were also identified as limitations for the NMA by McInnes et al. (2022).15
The indirect evidence was limited to short-term efficacy and safety; thus, longer-term comparative effects are uncertain.
Other Relevant Evidence
Description of Studies
Efficacy and safety data were available for the uncontrolled extension phase of DISCOVER-1 (52 weeks), DISCOVER-2 (100 weeks), and COSMOS (48 weeks) trials. Descriptive results for patients who received guselkumab 100 mg every 8 weeks are summarized in this section, including results for patients from the placebo group in the COSMOS study who crossed over to guselkumab.
Efficacy Results
The extension phase data suggest that treatment effects may be maintained in patients who remain on guselkumab therapy every 8 weeks for 48 weeks to 100 weeks. In DISCOVER-1, 76 patients out of 112 patients (68%), and in COSMOS, 120 patients out of 172 patients (70%), achieved ACR 20 at week 48 or week 52. In the DISCOVER-2 study, 85 patients out of 234 patients (79%) achieved ACR 20 at week 52, and 183 patients out of 223 patients (82%) achieved ACR 20 at week 100. PASI 100 response was reported by 36 patients out of 75 patients (48%) in DISCOVER-1 (week 52), by 94 patients out of 169 patients (57%) in DISCOVER-2 (100 weeks), and by 80 patients out of 121 patients (66%) in COSMOS (48 weeks).
Harms Results
During the extension period, 31% to 72% of patients reported 1 or more adverse event; 3% to 9% reported a serious adverse event; and 1% to 3% stopped treatment due to adverse events. No deaths were reported. Infections were reported in 43% of patients in DISCOVER-1 and in 29% and 38% of patients at week 52 and week 100 of the DISCOVER-2 study. Over the 48-week treatment period of the COSMOS study, 22% of patients who received guselkumab every 8 weeks experienced an infection.
Critical Appraisal
Limitations of the extension study include selection bias and lack of a control group. Data were available only as descriptive statistics; and because there were no comparator groups, the interpretation of the results is limited. The outcomes were based on observed case data, with no imputation for missing data, and reflect treatment effects in patients who continue on therapy. As such, the results may overestimate the response in the broader population, given that patients who drop out are more likely to have unfavourable outcomes or poor tolerance to therapy.
Conclusions
Based on data from 3 double-blind RCTs, adults with active PsA who received guselkumab 100 mg every 8 weeks were more likely to show clinically relevant improvements in PsA disease activity and tender and swollen joint counts than patients who received placebo, based on the proportion who achieved an ACR 20 response at week 24. Favourable clinical responses in PsA activity and symptoms were observed among patients who were biologic-naive or had prior intolerance or inadequate response to TNF alpha inhibitors. This was also the case for a mixed population that included patients with and without prior TNF inhibitor exposure.
Patients on guselkumab also showed statistically significant improvements in disability as measured using the HAQ-DI, although the clinical relevance of the difference versus placebo is uncertain. Improvements in the physical component (but not the mental component) of the SF-36 were observed favouring guselkumab versus placebo. Outcomes related to psoriatic skin lesions demonstrated superiority of guselkumab every 8 weeks versus placebo at 24 weeks. Among patients with enthesitis or dactylitis at baseline, pooled data from the pivotal trials suggest that patients who receive guselkumab every 8 weeks may be more likely to have enthesitis or dactylitis resolved at 24 weeks than those receiving placebo. The impact of guselkumab on radiographic progression is unclear, given that no statistically significant differences were detected between guselkumab every 8 weeks and placebo for the change in the modified vdH-S score at 24 weeks among patients with active PsA who were biologic-naive.
No new safety signals were identified in the controlled or extension phases of the PsA trials. The frequency of infection was similar in the guselkumab and placebo groups up to 24 weeks.
There is no direct evidence comparing guselkumab to other bDMARDs available in Canada. The indirect evidence for ACR response rates, change in HAQ-DI scores, change in SF-36 PCS and MCS, and risk of adverse events or serious adverse events for guselkumab versus most biologic comparators showed imprecise results; this imprecision limits the ability to draw conclusions from these data. Based on the indirect evidence, short-term PASI response rates may favour guselkumab versus some other biologics. However, there is uncertainty in these findings, given that several sources of heterogeneity were identified across the trials included in the NMAs and that it is unclear whether the methods used to control for potential bias were adequate. In addition, many of the studies included in the NMAs were at an unclear or a high risk of bias in 1 or more study domains.
The direct comparative evidence versus placebo and indirect evidence versus other biologics was limited to short-term outcomes (i.e., up to 24 weeks). Although results from the extension phase of the trial suggest that treatment effects may be maintained up to 100 weeks, these data are difficult to interpret because of the lack of comparator group and bias due to attrition. Thus, the longer-term comparative efficacy and safety of guselkumab in patients with PsA is unclear.
Introduction
Disease Background
PsA is an inflammatory musculoskeletal disease with a heterogenous presentation and disease course. While it is associated with psoriasis, PsA also presents with variable clinical features involving multiple domains, including peripheral arthritis, enthesitis (tenderness and swelling at the insertion of tendons and ligaments into bone), dactylitis (swelling of the whole digit), and axial disease (inflammation of the joints of the back).1,2 Extra-articular manifestations include inflammation of the eye and inflammatory bowel disease. Diagnosis of PsA can be a challenge, given that there is no gold standard diagnostic test; it is typically diagnosed based on clinical findings and imaging features that evaluate specific patterns of joint inflammation or involvement of the different domains. Patients with PsA also present with psoriatic skin lesions and are usually seronegative for rheumatoid factor (95%).2,16 Pain and stiffness of the affected joints are the most predominant presenting symptoms, with fatigue also occurring in many patients.1 Patients with psoriasis and PsA are at risk for the development of comorbidities, including cardiovascular disease, diabetes, gout, metabolic syndrome, and depression.1
The prevalence of PsA varies, depending on the case definition and geography, and is estimated to be 1 to 2 cases per 1,000 in the general population.1 A population-based Canadian study estimated the age- and sex-standardized cumulative prevalence of PsA in Ontario to range from 0.09% in 2008 to 0.15% in 2015. The same study estimated the age- and sex-standardized incidence in 2015 to be 14 per 100,000.3
About 30% of patients with psoriasis develop PsA; skin disease usually precedes manifestations of PsA by several years (10 years on average); however, in some individuals, both can occur simultaneously, or PsA may occur before the onset of psoriasis.2 A Canadian prospective cohort study estimated the annual incidence of PsA to be 2.7 cases per 100 psoriasis patients.17 Over time, PsA can lead to deformities and joint damage.2 This can lead to significant functional impairment, which in turn can affect work productivity and reduce health-related quality of life (HRQoL).2,16
Standards of Therapy
Treatment goals for patients with PsA include: achieving the lowest possible level of disease activity in all domains of disease; optimizing functional status; improving quality of life and well‐being; preventing structural damage to the greatest extent possible; and avoiding or minimizing complications, both from untreated active disease and from therapy. This disease affects more than just patients’ joints; therefore, treatment is individualized based on various factors, including disease activity, structural damage, comorbid conditions, and previous therapies.18 Recent recommendations by the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA) have suggested the use of a domain-based approach (peripheral arthritic, axial disease, enthesitis, dactylitis, psoriasis, nail disease, inflammatory bowel disease, and uveitis) that considers patient preference and previous and/or concomitant therapies; the choice of therapy should address as many domains as possible (Figure 1).
Several drug classes are employed in the pharmacologic treatment of PsA, including NSAIDs, cDMARDs (i.e., methotrexate, sulfasalazine, or leflunomide), bDMARDs (i.e., TNF inhibitors, IL-23 inhibitors, IL-12/23 inhibitors, and IL-17 inhibitors), and targeted synthetic DMARDs (e.g., apremilast, upadacitinib, or tofacitinib). Key characteristics of select drugs used in the treatment of PsA that are relevant to this review are summarized in Table 4.
Figure 1: GRAPPA Treatment Schema — Redacted

This figure was removed by CADTH because permission to reproduce could not be obtained from the publisher. Please refer to Figure 2 in the source publication.
Source: Reproduced from Coates et al. (2022).19
Drug
Guselkumab is human immunoglobulin G1 lambda monoclonal antibody that binds to the IL-23 protein and inhibits its binding with cell surface IL-23 receptor.5 IL-23 is a cytokine that is involved in normal inflammatory and immune response. Guselkumab is approved for the treatment of adult patients with active PsA.5 It may be used alone or in combination with a cDMARD (e.g., methotrexate). Guselkumab is available as a 100 mg/mL solution for SC injection in either 1 mL pre-filled syringes or patient-controlled injector devices. The recommended dose is 100 mg SC at week 0, week 4, and every 8 weeks thereafter.5
Guselkumab underwent a standard Health Canada review and was approved by for PsA in September 2020. The sponsor has requested reimbursement as per the Health Canada indication.20
Guselkumab is also approved for the treatment of adult patients with moderate to severe plaque psoriasis who are candidates for systemic therapy or phototherapy.5 In 2018, the CADTH Canadian Drug Expert Committee (CDEC) recommended that guselkumab be reimbursed for adults with plaque psoriasis if the following clinical criteria and conditions were met:
Reimburse in a manner similar to other biologics for the treatment of moderate to severe plaque psoriasis.
Treatment should be discontinued if a response has not been demonstrated after 16 weeks.
The drug plan cost for guselkumab should not exceed the drug plan cost of treatment with the least costly biologic reimbursed for moderate to severe plaque psoriasis.21
Table 4: Key Characteristics of Select Drugs Used in the Treatment of PsA
Drug class or drug | Mechanism of action | Indicationa | Recommended dose | SAE |
|---|---|---|---|---|
IL-23 inhibitor | ||||
Guselkumab | Human IgG1 lambda monoclonal antibody that binds and inhibits pro-inflammatory cytokine IL-23 | Treatment of adult patients with active PsA; may be used alone or in combination with a cDMARD (e.g., MTX) | 100 mg SC at week 0, week 4, and every 8 weeks thereafter | Infections (TB and serious infection in particular), hypersensitivity reactions |
Risankizumab | Humanized IgG1 monoclonal antibody that binds and inhibits pro-inflammatory cytokine IL-23 | Treatment of adult patients with active PsA; can be used alone or in combination with a cDMARD (e.g., MTX) | 150 mg SC at week 0, week 4, and every 12 weeks thereafter | Infections (TB and serious infection in particular), hypersensitivity reactions |
IL-17 inhibitor | ||||
Ixekizumab | Humanized IgG4 monoclonal antibody that selectively binds and neutralizes the pro-inflammatory cytokine IL-17A | Treatment of adult patients with active PsA who have responded inadequately, or who are intolerant to 1 or more DMARDs; can be used alone or in combination with a cDMARD (e.g., MTX) | For PsA or PsA with coexistent, mild PP: 160 mg SC at week 0, followed by 80 mg q4w For PsA with coexistent, moderate to severe PP: 160 mg SC at week 0, followed by 80 mg at weeks 2, 4, 6, 8, 10, and 12, then 80 mg q4w | Infections (TB and serious infection, in particular), hypersensitivity reactions, and inflammatory bowel disease (exacerbations or new onset) |
Secukinumab | Fully human IgG1k monoclonal antibody that selectively binds and neutralizes the pro-inflammatory cytokine IL-17A | Treatment of adult patients with active PsA when response to previous DMARD therapy has been inadequate; can be used alone or in combination with MTX | 150 mg SC at weeks 0, 1, 2, 3, and 4 followed by monthly maintenance dosing; for PsA patients with coexistent, moderate to severe PP: 300 mg SC at weeks 0, 1, 2 3, and 4, followed by monthly maintenance dosing For patients with PsA who are inadequate responders to anti-TNF alpha or continue to have active PsA: a 300 mg SC dose should be considered | Same as for ixekizumab |
IL-12/23 inhibitor | ||||
Ustekinumab | Fully human IgG1k monoclonal antibody that inhibits the bioactivity of IL-12 and IL-23 | Treatment of adult patients with active PsA; can be used alone or in combination with MTX | 45 mg SC at weeks 0 and 4, then every 12 weeks thereafter 90 mg SC may be used in patients with a body weight > 100 kg | Infections and reactivation of latent infections (TB and serious infections), hypersensitivity reactions, malignancies, RPLS |
TNF alpha inhibitor | ||||
Adalimumab | TNF inhibitor; recombinant human IgG1κ monoclonal antibody | Reducing the signs and symptoms of active arthritis, inhibiting the progression of structural damage, and improving physical function in adult PsA patients; can be used in combination with MTX in patients who do not respond adequately to MTX alone | 40 mg SC every other week | Serious infections (bacterial, mycobacterial, invasive fungal, viral, parasitic, or other opportunistic); malignancies; hypersensitivity reactions (allergic reactions and injection-site reactions); neurologic events (e.g., demyelinating disease); congestive heart failure |
Certolizumab pegol | TNF inhibitor; recombinant, humanized antibody Fab’ fragment | Reducing signs and symptoms and inhibiting the progression of structural damage as assessed by X-ray in adult patients with moderately to severely active PsA who have failed one or more DMARDs; can be used alone or in combination with MTX | Loading dose of 400 mg SC initially (week 0) and at week 2 and week 4, followed by a maintenance dose of 200 mg q2w or 400 mg q4w | Same as for adalimumab |
Etanercept | TNF inhibitor; recombinant human TNF receptor: fusion protein | Reducing signs and symptoms, inhibiting the progression of structural damage of active arthritis, and improving physical function in adult patients with PsA; can be used in combination with MTX in adult patients who do not respond adequately to MTX alone | 50 mg SC once a week; can be given as 1 injection of 50 mg or 2 injections of 25 mg on the same day once weekly or 3 days or 4 days apart | Same as for adalimumab |
Golimumab | TNF inhibitor; recombinant human IgG1κ monoclonal antibody | Reducing signs and symptoms, inhibiting the progression of structural damage, and improving physical function in adult patients with moderately to severely active PsA; can be used in combination with MTX in patients who do not respond adequately to MTX alone | 50 mg SC once a month on the same date each month 2 mg/kg IV at week 0, week 4, and every 8 weeks thereafter | Same as for adalimumab |
Infliximab | TNF inhibitor; recombinant chimeric IgG1κ monoclonal antibody | Reduction of signs and symptoms, induction of major clinical response, inhibition of the progression of structural damage of active arthritis, and improvement in physical function in patients with PsA; can be used with or without MTX | 5 mg/kg given as an IV infusion followed with additional similar doses at 2 weeks and 6 weeks after the first infusion, then q8w thereafter | Same as for adalimumab |
JAK inhibitor | ||||
Upadacitinib | JAK inhibitor; greater inhibitory potency at JAK1 relative to JAK2, JAK3, and TYK2 | Treatment of adults with active PsA who have had an inadequate response or intolerance to MTX or other DMARDs; may be used as monotherapy or in combination with MTX | 15 mg oral once daily | Serious infections (TB, invasive fungal infections, opportunistic infections); malignancies; thrombosis; liver enzyme elevation |
Tofacitinib | JAK inhibitor; pan-JAK inhibitor | Reducing the signs and symptoms of PsA in adult patients with active PsA when the response to previous DMARD therapy has been inadequate; can be used in combination with MTX or another cDMARD | 5 mg oral twice daily | Same as for upadacitinib |
Other | ||||
Abatacept | Selective co-stimulation modulator; selectively modulates a key costimulatory signal required for full activation of T lymphocytes expressing CD28 | Treatment of adult patients with active PsA when the response to previous DMARD therapy has been inadequate; can be used with or without non-biologic DMARDs | 125 mg SC once weekly Weight-based IV dosing (500 mg to 1 g); administered at week 0, week 2, week 4, and every 4 weeks thereafter | Infection, COPD exacerbations |
Apremilast | Selective immunosuppressant; a small-molecule inhibitor of phosphodiesterase type 4; works intracellularly to modulate a network of pro-inflammatory and anti-inflammatory mediators | Alone or in combination with MTX; indicated for the treatment of active PsA in adult patients who have had an inadequate response, intolerance, or contraindication to a prior DMARD | 30 mg oral twice daily | Tachyarrhythmia, weight loss |
cDMARD = conventional disease-modifying antirheumatic drug; COPD = chronic obstructive pulmonary disease; DMARD = disease-modifying antirheumatic drug; Fab’ = fragment, antigen binding; IgG1κ = immunoglobin G1 kappa; IL = interleukin; JAK = Janus kinase; MTX = methotrexate; PP = plaque psoriasis; PsA = psoriatic arthritis; q2w = every 2 weeks; q4w = every 4 weeks; q8w = every 8 weeks; RPLS = reversible posterior leukoencephalopathy syndrome; SAE = serious adverse event; SC = subcutaneous; TB = tuberculosis; TNF = tumour necrosis factor.
Note: only bDMARDs and targeted synthetic DMARDs identified as relevant comparators in the CADTH systematic review are included in this table. Comparators are drugs used in the treatment of PsA that are publicly funded. Although tofacitinib, upadacitinib, abatacept, and apremilast are not currently reimbursed by public drug programs in Canada, these were deemed to be relevant comparators to guselkumab by the clinical expert consulted for this review.
aHealth Canada–approved indication for the condition under review, according to product monographs.
Source: Product monographs.5,22-34
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups. The full original patient inputs received by CADTH are included in the stakeholder section at the end of this report.
Three inputs were submitted for this review from 6 different patient groups: Arthritis Consumer Experts, CAPP in partnership with CPN, and CAPA in partnership with the Arthritis Society and CreakyJoints.
Arthritis Consumer Experts is Canada’s largest, longest-running national arthritis patient organization. It is headquartered in Vancouver, British Columbia, with 50,000 members from coast to coast. It provides free, science-based information and education programs to people with arthritis and those who care for and support them.
CAPP is a national, not-for-profit organization formed to better serve the needs of psoriasis patients across the country. Its mission is to be a resource and advocate for psoriatic patients and their families to improve patient care and quality of life.
CPN is a national, not-for-profit organization dedicated to improving the quality of life of people in Canada who live with psoriasis and PsA. It does this by providing current information on research and treatment options and by working with others to build awareness and advocacy about the complexities of these conditions.
CAPA is a grassroots, patient-driven and managed, independent, national education and advocacy organization with members and supporters across Canada. It creates links between people in Canada with arthritis, assists them to become more effective advocates, and seeks to improve the quality of life of all people living with the disease.
The Arthritis Society is dedicated to a vision of a world where people are free from the devastating effects of arthritis. It is Canada’s principal health charity, providing education, programs, and support to the more than 6 million people in Canada living with arthritis.
CreakyJoints Canada has served for more than 2 decades as a digital community for millions of arthritis patients and caregivers worldwide who seek education, support, advocacy, and patient-centred research. All of its programming and services are provided free of charge. CreakyJoints is part of the non-profit Global Healthy Living Foundation, whose mission is to improve the quality of life for people living with chronic illnesses.
Five patient organizations (CAPP, CPN, CAPA, the Arthritis Society, and CreakyJoints Canada) collaborated by collectively developing survey questions using SurveyMonkey for the inputs submitted. Each organization shared the surveys with its respective memberships or patient communities through email, social media, and organization websites. CAPA, CreakyJoints Canada, and the Arthritis Society submitted 1 collaborative patients’ input; the CAPP and CPN analyzed data and prepared 1 submission collectively, while the Arthritis Consumer Experts made its own submission based on survey response data and its unique community perspectives. Survey data were collected from April 20, 2022 to May 16, 2022. In addition, Arthritis Consumer Experts gathered information from 1 respondent through email on May 4, 2022 and from 5 respondents through an online survey from December 18, 2020 to January 26, 2021.
There were 71 respondents to the joint survey: 12 respondents were from British Columbia (17%), 7 were from Alberta (10%), 3 were from Manitoba (4%), 34 were from Ontario (49%), 4 were from Quebec (6%), 4 were from New Brunswick (6%), 4 were from Nova Scotia (6%), and 3 were from Newfoundland and Labrador (4%). Two survey participants had experience taking guselkumab.
Respondents reported a range of symptoms that were difficult to manage, including joint stiffness (79%), fatigue (75%), changes in fingernails and toes (63%), hip pain (61%), back pain (51%), anxiety (47%), and stress (33%). With regards to the most significant impacts of PsA on their daily quality of life, respondents reported that PsA interfered with their work (54%), social connections (52%), self-esteem (50%), mental health (50%), intimacy (50%), family life (38%), and friendships (24%). Other impacts included embarrassment and self-consciousness from symptoms caused by PsA. Because the disease would reduce their mobility and ability to participate in activities and affect their mental and social health, respondents indicated that caregivers had to take on additional tasks or chores, such as cooking, cleaning, shopping, and helping patients get to and from medical appointments.
Survey respondents indicated that they had experience with several treatment approaches, including NSAIDs, corticosteroids, cDMARDs (such as methotrexate), and bDMARDs. Among responders, 32% considered biologics to be highly effective, followed by oral steroids (23%) and other DMARDs (21%). Respondents expressed their ongoing unmet need to manage symptoms and side effects with current treatments.
Two respondents who had experience with guselkumab indicated that the drug was effective in terms of improving psoriasis and arthritis and slowing disease progression. Both stated that they did not experience side effects.
Respondents expected new treatments to improve the following key outcomes: management of symptoms (i.e., reducing pain and fatigue, increasing mobility); tolerability of side effects; ease of drug administration; improved ability to work and carry out tasks and daily activities; and quality of life.
Clinician Input
Input From the Clinical Expert Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol; assisting in the critical appraisal of clinical evidence; interpreting the clinical relevance of the results; and providing guidance on the potential place in therapy). The following input was provided by 1 clinical specialist with expertise in the diagnosis and management of PsA.
Unmet Needs
The goals of therapy are to control the symptoms of PsA (i.e., pain stiffness, swelling, fatigue), reduce impacts on patients’ quality of life and function, and prevent the progression of joint damage. A substantial proportion of patients do not achieve a minimal response with their first therapy or lose their response over time. In addition, some treatments have more adverse effects than others. Thus, there is a need for medications with new mechanisms of action or a different safety profile to offer alternate treatment options. There also needs to be a choice for patients in terms of preference with regards to the frequency of injections, injection versus infusion, and oral administration versus injection.
Place in Therapy
According to the clinical expert, guselkumab may be used as first- or second-line biologic therapy. It may be a preferred first-line treatment for patients with moderate psoriasis in addition to musculoskeletal disease. Guselkumab may be used in combination with methotrexate or leflunomide. There is no evidence to support its use in combination with other biologics. The expert stated that it would not be appropriate to recommend that patients try other biologics before receiving guselkumab.
Patient Population
There are currently no biomarkers that may be used to identify the patients who will be most suited for a particular drug. Clinicians make these decisions based on patient history, physical examination, imaging, and discussion.
There is currently no diagnostic test for PsA. Rheumatologists can usually diagnose PsA after evaluating the patient and may seek confirmation of the psoriasis diagnosis from a dermatologist. The expert stated that 30% to 80% of psoriasis patients had a missed PsA diagnosis.
Assessing Response to Treatment
Response to therapy is based on a reduction in the number of inflamed joints, improvement in the skin, and patient-reported outcomes. According to the clinical expert, a major improvement would be considered an improvement of at least 50%, but may also include achievement of MDA and remission as measured by specific instruments. Assessment of response requires the physician to perform a physical examination, including a joint count and skin evaluation, and also includes patient-reported outcomes (i.e., assessment of pain, function, fatigue). Less frequently, structural damage may be assessed using X-rays.
An initial response may be expected within 3 months, with more significant improvement by 6 months after initiating therapy. The expert indicated that if a patient shows no change within 3 months to 6 months, they would be considered a nonresponder and may be switched to another medication.
Discontinuing Treatment
Patients may be switched to another medication if no improvement is observed within 3 months to 6 months or if the patient experiences intolerable or serious adverse effects. If a patient has responded in some but not all manifestations, another medication may be added to address the area that has not responded.
Prescribing Conditions
Ideally, guselkumab would be prescribed by specialists who are familiar with the drug and its uses, or at least in consultation with a dermatologist or rheumatologist. The administration of guselkumab may take place in any setting, including self-administration by the patient in their home.
Clinician Group Input
This section was prepared by CADTH staff based on the input provided by clinician groups. The full original clinician group input(s) received by CADTH have been included in the stakeholder section at the end of this report.
CADTH received 1 submission from Canadian Rheumatologist Psoriatic Arthritis Interest Group, based on responses from 6 clinicians practising in academic and community settings. The Canadian Rheumatologist Psoriatic Arthritis Interest Group consists of a group of clinical rheumatologists across Canada who have extensive experience managing inflammatory arthritis, including PsA. No major views contrary to those provided by the clinical experts consulted by CADTH for this review were presented.
The clinician group stated that PsA is a complex disease with varied manifestations. The current treatment options include nonpharmacological treatments and pharmaceutical interventions, such as cDMARDs and biologic therapies (i.e., TNF alpha inhibitors and IL-17A inhibitors) as first-line therapies and IL-12/23 inhibitors and JAK inhibitors as second-line therapies. The clinician group highlighted that not all patients achieve a good response (defined as ACR 20 or MDA) to current treatments. In addition, the adverse effects of TNF inhibitors (lupus-like syndromes or multiple sclerosis) and IL-17A (inflammatory bowel disease) limit the use of the 2 most common classes of biologics used to manage PsA. Moreover, biologic therapies are associated with increased risk of serious infections. Thus, the clinician group identified significant unmet need in the management of PsA.
According to the clinician group, the goals of treatment are to improve quality of life and physical function by controlling symptoms (i.e., joint pain and stiffness, concomitant psoriasis, and extra-articular manifestations) and to limit structural damage and minimize long-term complications arising from inadequately controlled inflammation related to PsA. The clinician group expressed that guselkumab may be used alone or in combination with cDMARDs as a first-line biologic therapy or as a second-line treatment after failure to respond to a TNF inhibitor. The clinician group stated that rheumatologists can identify patients who are suitable for treatment with guselkumab based on inflammatory arthritis in the presence of psoriasis; the clinicians did not identify any potential challenges associated with the diagnosis.
According to the clinician group, patients with early disease and moderate to severe skin and joint involvement are most likely to respond to guselkumab. Those who are refractory to current therapeutic drugs or unable to take current classes of biologic drugs have the greatest need for an intervention such as guselkumab. The clinicians noted that improvement in tender and swollen joint counts, psoriasis, enthesitis, patient global impression, and ACR 20 are commonly used to determine whether a patient is responding to guselkumab. They also noted that composite measures — including MDA, Disease Activity Index for PsA, and PsA Disease Activity Score — would be considered as well. With regards to discontinuation, the clinician group noted several factors that would be considered when deciding to stop guselkumab treatment, such as inflammatory pain and stiffness, swollen or tender joint count, worsening psoriasis, poor function, intolerability due to adverse effects, or the development of severe extra-articular manifestations. The clinician group indicated that rheumatologists or experienced nurse practitioners with extensive rheumatology experience are required to diagnose and manage PsA. In addition, the clinician group mentioned that guselkumab has shown effectiveness in the treatment of joints in both biologic naive patients and patients with an inadequate response to TNF, and has been shown to improve pain and fatigue in clinical trials.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 5.
Table 5: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparator | |
DISCOVER-1 (a phase III, multi-centre, double-blind RCT) did not compare the effect of guselkumab to another biologic comparator. However, it included patients who had failed standard therapies (i.e., apremilast,a non-biologic DMARDs, NSAIDs, TNF inhibitors). DISCOVER-2 (a phase III, multi-centre, double-blind RCT) assessed patients who had failed standard non-biologic therapies (apremilast,a non-biologic DMARDs, and NSAIDs). COSMOS (an RCT) included patients who had stopped < 2 TNF inhibitors due to lack of efficacy or intolerance. There were no head-to-head, phase III RCTs comparing guselkumab to other biologics, such as TNF alpha inhibitors (certolizumab, etanercept, infliximab, adalimumab, golimumab) or interleukin inhibitors (secukinumab, ixekizumab, ustekinumab). The sponsor included patients who had failed TNF inhibitors, but randomized patients to the trial drug or placebo. There is no evidence comparing the noninferiority or superiority of guselkumab to current therapeutic options that are available on government-sponsored drug plans in Canadian jurisdictions. | For CDEC consideration |
Adalimumab, etanercept, and infliximab biosimilars are available for this condition and offer substantial discounts to jurisdictions. Ustekinumab’s patent has expired, with future biosimilars for the treatment of PsA in phase III trials (i.e., Amgen Inc.’s ABP 564). Head-to-head trials in this space are essential for public payers to consider listing drugs on government-sponsored drug plans. | For CDEC consideration |
Considerations for initiation of therapy | |
In DISCOVER-1, only 30% of the study population had been treated with up to 2 anti-TNF drugs; however, the COSMOS study did require all patients to have failed 1 or 2 TNF inhibitors to be enrolled. | For CDEC consideration |
DISCOVER-1 and DISCOVER-2 patients had to have experienced an inadequate response to a non-biologic DMARD, apremilast, or NSAIDs. DISCOVER-1 also included some patients who had received TNF inhibitors. | For CDEC consideration |
Alignment is preferred with the medications currently listed among jurisdictions. This includes anti-TNF drugs and IL inhibitors. Alignment with JAK inhibitors that have a reimbursement recommendation from CADTH for PsA would be helpful to drug plans as well. 1. Should the initiation criteria for PsA biologics and JAK inhibitors be applied to guselkumab? | The clinical expert’s view is that because guselkumab is a biologic drug, its reimbursement recommendations should be similar to those for other biologics. The clinical expert noted that guselkumab should be made available as either the first or subsequent medication. |
Considerations for continuation or renewal of therapy | |
Alignment with other biologic drugs and JAK inhibitors in this therapeutic space would be desirable for the drug plans. 2. Should the continuation or renewal criteria for PsA biologics and JAK inhibitors be applied to guselkumab? | The clinical expert explained that as a biologic drug, guselkumab should have criteria similar to those for other biologics. |
Considerations for discontinuation of therapy | |
Alignment with other biologic drugs and JAK inhibitors would be preferred by the drug plans. 3. Should the discontinuation criteria for PsA biologics and JAK inhibitors be applied to guselkumab? | The clinical expert reiterated that as a biologic drug, guselkumab should have criteria similar to those for other biologics. |
Considerations for prescribing of therapy | |
In general, patients are restricted to 1 biologic at a time and permitted to switch from 1 biologic drug to another following an adequate trial of the first biologic drug if unresponsive to therapy, or due to serious adverse effects or contraindications. Patients are not permitted to switch back to a previously trialled biologic drug if they were deemed unresponsive to therapy. No restrictions are in place regarding the combination with cDMARDs. 4. Should the same combination criteria for PsA biologics and JAK inhibitors be applied to guselkumab? | According to the clinical expert, patients should be permitted to switch back to a biologic they had previously tried. The clinical expert explained that sometimes there is response, but not complete remission, and a drug switch occurs to try to achieve remission. However, the patient may feel that although they are not in remission, they did better on the original drug, and they may prefer to resume that drug. It should be available for them. The clinical expert believes this approach should apply to guselkumab. |
Alignment with other biologic drugs and JAK inhibitors would be preferred. 5. Should similar prescribing criteria to those used for PsA biologics and JAK inhibitors be applied to guselkumab? | The clinical expert’s view is that the prescribing criteria should be similar, taking into account the individual drug formulation. |
System and economic issues | |
Many jurisdictions have biosimilar initiatives and policies in place. These initiatives involve removing originator biologics and listing only the biosimilar molecule in this therapeutic space. When the pan-Canadian budget impact assessment is adjusted to model all jurisdictions that have a biosimilar initiative in place for adalimumab, etanercept, and infliximab, the addition of guselkumab increases the incremental cost from $2 million (3-year total with no biosimilar policy) to $10 million (3-year total with biosimilar policies in place). As a result, the addition of guselkumab to public drug plans results in a higher incremental cost, especially in jurisdictions with biosimilar initiatives in place. Negotiated values will require a price reduction comparable to the confidential price of the least costly biosimilar to ensure the sustainability of drug plans. | For CDEC consideration |
A significant number of biologics have been negotiated by the pCPA for the treatment of PsA and listed on government-sponsored drug plans. 6. Is there evidence to support drug plans paying a price premium for guselkumab vs. the lowest-cost biosimilar TNF inhibitor? | The clinical expert expressed that from a clinician’s perspective, having access to treatment that is deemed appropriate for patients is of greater importance than treatment cost and that as a result, drugs should be available without a price premium. If a particular drug is most appropriate for a patient, its price should not be compared to that of another drug (unless within the same mechanism of action, possibly). |
CDEC = CADTH Canadian Drug Expert Committee; cDMARD = conventional disease-modifying antirheumatic drug; DMARD = disease-modifying antirheumatic drug; IL = interleukin; JAK = Janus kinase; NSAID = nonsteroidal anti-inflammatory drug; pCPA = pan-Canadian Pharmaceutical Alliance; PsA = psoriatic arthritis; RCT = randomized controlled trial; TNF = tumour necrosis factor.
aApremilast is not covered for PsA in any Canadian jurisdiction.
Clinical Evidence
The clinical evidence included in the review of guselkumab is presented in 3 sections. The first section, the Systematic Review, includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those studies that were selected according to an a priori protocol. The second section includes indirect evidence from the sponsor and indirect evidence selected from the literature that met the selection criteria specified in the review. The third section includes sponsor-submitted long-term extension studies and additional relevant studies that were considered to address important gaps in the evidence included in the systematic review.
Systematic Review (Pivotal and Protocol Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of guselkumab 100 mg/mL administered through a 1 mL pre-filled syringe or patient-controlled injector for SC injection for the treatment of active PsA in adults.
Methods
Studies selected for inclusion in the systematic review included pivotal studies provided in the sponsor’s submission to CADTH and Health Canada as well as those meeting the selection criteria presented in Table 6. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
Table 6: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Patient population | Adults with active psoriatic arthritis Subgroups:
|
Intervention | Guselkumab 100 mg SC injection at week 0 and week 4, followed by maintenance dosing every 8 weeks thereafter (alone or in combination with a cDMARD [e.g., methotrexate]) |
Comparators |
|
Outcomes | Efficacy outcomes:
Harms outcomes: AEs, SAEs, WDAEs, mortality, and notable harms (serious infections, hypersensitivity reactions, elevated hepatic enzymes, hepatic disorders, injection-site reactions) |
Study design | Published and unpublished phase III and IV RCTs |
ACR 20 = American College of Rheumatology 20% improvement; ACR 50 = American College of Rheumatology 50% improvement; ACR 70 = American College of Rheumatology 70% improvement; AE = adverse event; bDMARD = biologic disease-modifying antirheumatic drug; cDMARD = conventional disease-modifying antirheumatic drug; DAS 28 = Disease Activity Score 28; DMARD = disease-modifying antirheumatic drug; HAQ-DI = Health Assessment Questionnaire Disability Index; IGA = Investigator’s Global Assessment of Psoriasis; IL = interleukin; JAK = Janus kinase; MDA = minimal disease activity; PASI 75 = 75% improvement from baseline in Psoriasis Area and Severity Index score; PASI 90 = 90% improvement from baseline in Psoriasis Area and Severity Index score; PASI 100 = 100% improvement from baseline in Psoriasis Area and Severity Index score; PsA = psoriatic arthritis; RCT = randomized controlled trial; SAE = serious adverse event; SC = subcutaneous; TNF = tumour necrosis factor; WDAE = withdrawal due to adverse event.
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy according to the PRESS Peer Review of Electronic Search Strategies checklist.35
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946—) through Ovid and Embase (1974—) through Ovid. All Ovid searches were run simultaneously as a multi-file search. Duplicates were removed using Ovid deduplication for multi-file searches, followed by manual deduplication in Endnote. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concept was Tremfya (guselkumab). Clinical trial registries were searched: the US National Institutes of Health’s clinicaltrials.gov, WHO’s International Clinical Trials Registry Platform search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
No filters were applied to limit retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. Refer to Appendix 1 for the detailed search strategies.
The initial search was completed on May 31, 2022. Regular alerts updated the search until the meeting of the CADTH CDEC on September 28, 2022.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the CADTH Grey Matters: A Practical Tool For Searching Health-Related Grey Literature checklist.36 Included in this search were the websites of regulatory agencies (FDA and the European Medicines Agency). Google was used to search for additional internet-based materials. Refer to Appendix 1 for more information on the grey literature search strategy.
In addition to the literature search, the manufacturer of the drug was contacted for information regarding unpublished studies.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
Findings From the Literature
A total of 3 studies were identified from the literature for inclusion in the systematic review (Figure 2). The included studies are summarized in Table 7. A list of excluded studies is presented in Appendix 2.
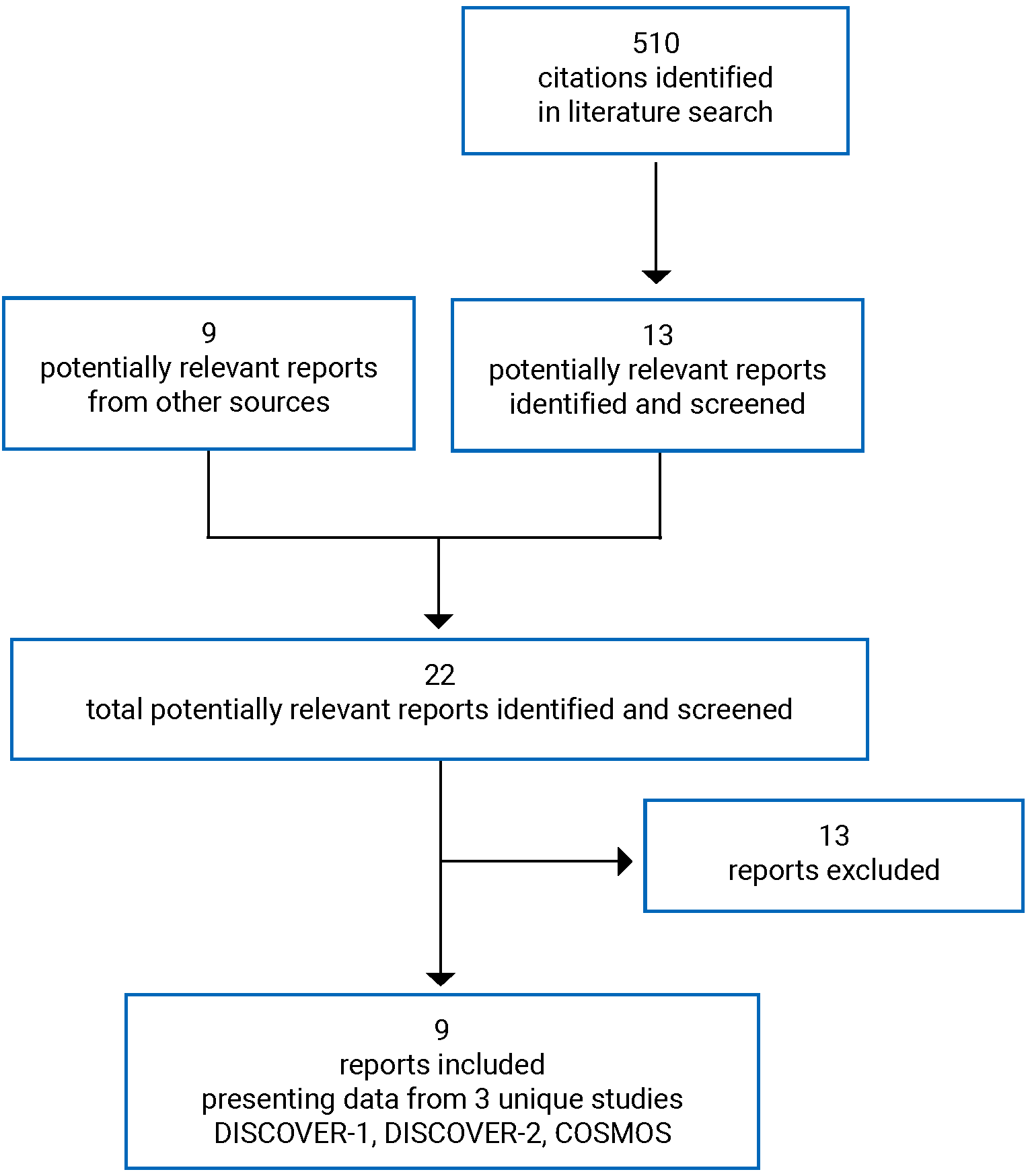
Table 7: Details of Included Studies
Detail | DISCOVER-1 | DISCOVER-2 | COSMOS |
|---|---|---|---|
Designs and populations | |||
Study design | DB RCT | DB RCT | DB RCT |
Locations | Canada (6 sites), US, Europe, Asia, Australia | Europe, Asia, US | Europe, Israel |
Patient enrolment dates | August 28, 2017 to March 14, 2019 (interim 24-week data cut-off) | July 13, 2017 to March 6, 2019 (interim 24-week data cut-off) | March 22, 2019 to August 3, 2020 (interim 24-week data cut-off) |
Randomized (N) | 381 | 741 | 285 |
Inclusion criteria | Adults ≥ 18 years of age with active PsA (≥ 3 tender and swollen joints and CRP ≥ 0.3 mg/dL) and inadequate response to standard therapiesa PsA diagnosis at least 6 months prior and met CASPAR at screening Active plaque psoriasis (≥ 1 lesion of ≥ 2 cm in diameter or nail changes of psoriasis or documented history of plaque psoriasis) 30% of patients may have been previously exposed to 1 or 2 TNF alpha inhibitors | Adults ≥ 18 years of age with active PsA (≥ 5 tender and swollen joints and CRP ≥ 0.6 mg/dL) and inadequate response to standard therapiesa PsA diagnosis at least 6 months prior and met CASPAR at screening Active plaque psoriasis (≥ 1 lesion of ≥ 2 cm in diameter or nail changes of psoriasis or documented history of plaque psoriasis) Biologic-naive | Adults ≥ 18 years of age with moderate to severe active PsA (≥ 3 tender and swollen joints) Inadequate response or intolerance to 1 or 2 TNF inhibitorsb PsA diagnosis at least 6 months prior and met CASPAR at screening Active plaque psoriasis (≥ 1 lesion of ≥ 2 cm in diameter or nail changes of psoriasis or documented history of plaque psoriasis) |
Exclusion criteria | Other inflammatory diseases (e.g., RA, axial spondyloarthritis, systemic lupus erythematosus, or Lyme disease), or a form of nonplaque or current drug-induced psoriasis Prior exposure to JAK inhibitors High suicide risk Recent serious infections, herpes zoster, latent or active TB, HIV, hepatitis B, or hepatitis C History of malignancy (within 5 years), lymphoproliferative disease, unstable cardiovascular disease, or other severe, progressive, or uncontrolled conditions | Received biologic for PsA (e.g., TNF inhibitor or IL-12, IL-17, or IL-23 inhibitors) Other criteria were the same as for DISCOVER-1. | Other inflammatory diseases (e.g., RA, axial spondyloarthritis, systemic lupus erythematosus, or Lyme disease) Prior treatment with > 2 TNF alpha inhibitors, guselkumab, any other biologic, or JAK inhibitor Received prohibited medication or psoriasis therapy within the preceding 4 weeks (refer to the Interventions section for details) Unstable suicide ideation or behaviour in the preceding 6 months History of other severe, progressive, or uncontrolled conditions |
Drugs | |||
Interventionsc | Guselkumab 100 mg SC at week 0, week 4, and every 8 weeks thereafterd Guselkumab 100 mg SC every 4 weeksd,e | Guselkumab 100 mg SC at week 0, week 4, and every 8 weeks thereafterd Guselkumab 100 mg SC every 4 weeksd,e | Guselkumab 100 mg SC at week 0, week 4, and every 8 weeks thereafterf |
Comparatorsc | Placebo SC every 4 weeks until week 20,d then switch to guselkumab 100 mg SC every 4 weeks from week 24 to 48 | Placebo SC every 4 weeks until week 20,d then switch to guselkumab 100 mg SC every 4 weeks from week 24 to week 100 | Placebo SC at week 0 and week 4, then every 8 weeks until week 20;f then switch to guselkumab 100 mg SC at week 24, week 28, and every 8 weeks thereafter |
Durations | |||
Phases | |||
Screening | 6 weeks | 6 weeks | 6 weeks |
DB | 52 weeks | 100 weeks | 48 weeks |
Safety follow-up | 8 weeks | 12 weeks | 8 weeks |
Outcomes | |||
Primary end points | Proportion of patients who achieve ACR 20 response at week 24 | Proportion of patients who achieve ACR 20 response at week 24 | Proportion of patients who achieve ACR 20 response at week 24 |
Secondary and exploratory end points | Major secondary:
Other secondary:
| Major secondary:
Other secondary:
| Secondary:
Exploratory:
|
Notes | |||
Publications | Deodhar et al. (2020)37 Rahman et al. (2021)38 | Mease et al. (2020)39 Rahman et al. (2021)38 | Coates et al. (2021)14 |
ACR 20 = American College of Rheumatology 20% improvement; ACR 50 = American College of Rheumatology 50% improvement; ACR 70 = American College of Rheumatology 70% improvement; ACR 90 = American College of Rheumatology 90% improvement; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; CASPAR = Classification Criteria for Psoriatic Arthritis; CRP = C-reactive protein; DAPSA = Disease Activity Index for Psoriatic Arthritis; DAS 28 = Disease Activity Score 28; DB = double blind; DLQI = Dermatology Life Quality Index; DMARD = disease-modifying antirheumatic drug; FACIT-Fatigue = Functional Assessment of Chronic Illness Therapy – Fatigue Scale; GRACE = GRAPPA composite score; HAQ-DI = Health Assessment Questionnaire Disability Index; IGA = Investigator’s Global Assessment of Psoriasis; IL = interleukin; JAK = Janus kinase; MCS = Mental Component Summary; MDA = minimal disease activity; NSAID = nonsteroidal anti-inflammatory drug; PASDAS = Psoriatic Arthritis Disease Activity Score; PASI 75 = Psoriasis Area and Severity Index 75% improvement; PASI 90 = Psoriasis Area and Severity Index 90% improvement; PASI 100 = Psoriasis Area and Severity Index 100% improvement; PCS = Physical Component Summary; PsA = psoriatic arthritis; PsARC = Psoriatic Arthritis Response Criteria; RA = rheumatoid arthritis; RCT = randomized controlled trial; SC = subcutaneous; SF-36 = Short Form (36) Health Survey; TB = tuberculosis; TNF = tumour necrosis factor; VLDA = very low disease activity; vdH-S = van der Heijde-Sharp; WPAI = Work Productivity and Activity Impairment.
Note: Six additional reports were included (Health Canada reviewer’s report;40 EPAR;41 Deodhar et al. [2020];37 Mease et al. [2020];39 Rahman et al. [2021];38 and Coates et al. [2021]14).
aNon-biologic DMARD (≥ 3 months), apremilast (≥ 4 months), and/or NSAID therapy (≥ 4 weeks).
bPatients had shown either a lack of benefit after at least 12 weeks of etanercept, adalimumab, golimumab or certolizumab pegol (or biosimilars) and/or at least 14 weeks of infliximab (or biosimilar), or intolerance to 1 of the TNF alpha inhibitors. A washout period for TNF inhibitors was required before study entry (8 weeks for infliximab, 6 weeks for golimumab, adalimumab or certolizumab pegol, and 4 weeks for etanercept).
cStable doses of NSAIDs or other analgesics, oral corticosteroids (≤ 10 mg of prednisone per day or equivalent), and select cDMARDs (i.e., 1 of the following: methotrexate [≤ 25 mg/week], sulfasalazine [≤ 3 g/day], hydroxychloroquine [≤ 400 mg/day], or leflunomide [≤ 20 mg/day]) were allowed during the study.
dAt week 16, patients with less than 5% improvement from baseline in both tender and swollen joint counts were considered to have met the early escape criteria and were allowed to start therapy or increase the dose of 1 of the permitted concomitant psoriatic medications (methotrexate, sulfasalazine, hydroxychloroquine, leflunomide, oral corticosteroids, or NSAIDs) up to the maximum dose specified in the protocol.
eNot a Health Canada–recommended dosage regimen.
fAt week 16, patients with less than 5% improvement from baseline in both tender and swollen joint counts were considered to have met the early escape criteria and were allowed to start therapy or increase the dose of 1 of the permitted concomitant psoriatic medications (up to the maximum dose specified in the protocol). Patients in the placebo group who qualified for early escape received guselkumab at week 16, week 20, and every 8 weeks thereafter.
Source: Clinical Study Report for DISCOVER-1 (24 weeks);11 Clinical Study Report for DISCOVER-2 (24 weeks);12 Clinical Study Report for COSMOS (24 weeks).13
Description of Studies
Three double-blind, randomized, placebo-controlled trials met the inclusion criteria for the systematic review.
The objective of the DISCOVER-1 study was to evaluate the efficacy of guselkumab in patients with active PsA who had inadequate response to standard therapies (e.g., non-biologic DMARDs, apremilast, and/or NSAIDs). Up to 30% of patients enrolled had received prior treatment with 1 or 2 TNF alpha inhibitors. Patients were randomized (1:1:1) to guselkumab 100 mg SC every 4 weeks for 48 weeks; guselkumab 100 mg SC at week 0, week 4, and every 8 weeks thereafter (with placebo injections at other time points); or placebo every 4 weeks until week 20, crossing over to guselkumab 100 mg SC at week 24 and then every 4 weeks thereafter until week 48 (Figure 3). Patients were randomized centrally through an interactive web response system using permuted block randomization, stratified by non-biologic DMARD use at baseline (yes, no) and by prior exposure to TNF alpha inhibitors (yes, no) (N = 381). The trial was conducted at a total of 6 sites in Asia, Europe, the US, and Canada.
The objective of the DISCOVER-2 study was to evaluate the efficacy of guselkumab in patients with active PsA who were biologic-naive and had an inadequate response to standard therapies (e.g., non-biologic DMARDs, apremilast, and/or NSAIDs). Patients were randomized (1:1:1) to guselkumab 100 mg SC every 4 weeks for 100 weeks; guselkumab 100 mg SC at week 0, week 4, and every 8 weeks thereafter (with placebo injections at other time points); or placebo every 4 weeks until week 20, crossing over at week 24 to guselkumab 100 mg SC every 4 weeks to week 100 (similar to the design shown in Figure 3). Permuted block randomization was used in the trial, with randomization stratified by non-biologic DMARD use at baseline (yes, no) and most recent CRP value (< 2.0 mg/dL versus ≥ 2.0 mg/dL) (N = 741). The trial was conducted in Europe, Asia, and the US.
The objective of the COSMOS study was to evaluate the efficacy of guselkumab in patients with active PsA who had an inadequate response or intolerance to 1 or 2 prior TNF alpha inhibitors. Patients were randomized (1:1) to guselkumab 100 mg SC at week 0, week 4, and every 8 weeks thereafter (with placebo injections at other time points) or to placebo until week 20 before crossing over to guselkumab 100 mg SC at weeks 24, 28, 36, and 44 (Figure 4). Patients were randomized through an interactive web response system (N = 285). Central permuted block randomization was used, with randomization stratified by non-biologic DMARD use at baseline (yes, no) and number of prior TNF alpha inhibitors (1 or 2). The trial was conducted in Europe and Israel.
Data for the placebo-controlled portion of the 3 studies (i.e., first 24 weeks) have been summarized as part of the systematic review, including data from the interim database lock on March 2019 for the DISCOVER-1 and DISCOVER-2 studies and August 2020 for the COSMOS study. The study results after placebo patients crossed over to guselkumab have been summarized in the Other Relevant Evidence section, given that these uncontrolled follow-up periods did not meet the study design criteria for the systematic review.
Data from the groups receiving guselkumab every 4 weeks in the DISCOVER-1 and DISCOVER-2 studies have not been summarized in this report because this dose is not consistent with the Health Canada–recommended dose and dose adjustment.5
Figure 3: DISCOVER-1 Study Schematic

CO = placebo crossover; DBL = database lock; EE = early escape; GUS = guselkumab; PE = primary end point; q4w = every 4 weeks; q8w = every 8 weeks; SC = subcutaneous; W0 = week 0; W8 = week 8; W20 = week 20; W24 = week 24; W48 = week 48.
Source: Clinical Study Report for DISCOVER-1.11
Figure 4: COSMOS Study Schematic — Redacted

Figure redacted at the request of the sponsor.
Source: Clinical Study Report for COSMOS study (24 weeks).13
Populations
Inclusion and Exclusion Criteria
All 3 included studies enrolled adults greater than or equal to 18 years of age with active PsA diagnosed at least 6 months prior who met the Classification Criteria for Psoriatic Arthritis (Table 7). In the DISCOVER-1 and COSMOS studies, patients were required to have least 3 tender and swollen joints — and in DISCOVER-2, at least 5 tender and swollen joints — to qualify. Key inclusion criteria differences were as follows:
In the DISCOVER studies, patients had to have documented evidence of inadequate response or intolerance to standard PsA therapies, including non-biologic DMARDs (≥ 3 months), apremilast (≥ 4 months), and/or NSAID therapy (≥ 4 weeks). In DISCOVER-1, up to 30% of patients enrolled in had previously received 1 or 2 TNF alpha inhibitors. DISCOVER-2 enrolled patients with no prior exposure to bDMARDs.
COSMOS enrolled patients with an inadequate response or intolerance to 1 or 2 TNF alpha inhibitors.
In all trials, the patients enrolled were required to have at least 1 PsA subset: distal interphalangeal joint involvement, polyarticular arthritis with absence of rheumatoid nodules, arthritis mutilans, asymmetric peripheral arthritis, or spondylitis with peripheral arthritis. Patients were also required to have active plaque psoriasis with 1 or more lesions at least 2 cm in diameter, nail changes of psoriasis, or documented history of plaque psoriasis.
Key exclusion criteria were the presence of other inflammatory diseases, a nonplaque form of psoriasis or drug-induced psoriasis, significant risk of suicide, recent serious infection, or herpes zoster, tuberculosis, HIV, hepatitis B, or hepatitis C. Those with prior treatment with JAK inhibitors or recent exposure to a prohibited medication without an adequate washout period were also excluded.
Baseline Characteristics
The demographics of patients were generally similar within and across trials (Table 8). The mean age of patients enrolled ranged from 44.9 years (SD = 11.9 years) to 49.1 years (SD = 12.3 years) across treatment groups. The proportion of female patients ranged from 46% to 55%, and most patients were White (89% to 98% in the DISCOVER studies; not reported for the COSMOS study). The mean number of swollen joints ranged from 9.0 (SD = 5.7) to 12.3 (SD = 6.9), and the mean number of tender joints ranged from 18.2 (SD = 10.7) to 21.6 (SD = 13.1). Approximately 2-thirds of patients had psoriatic involvement affecting at least 3% of their BSA. Two-thirds of patients reported enthesitis, while approximately 40% had dactylitis at baseline. Some potential imbalances were noted in the COSMOS study in the proportion of female patients (55% versus 46%) and psoriasis at baseline (70% versus 55%), and in the DISCOVER-2 study in the proportion with enthesitis at baseline (64% versus 73%) in the groups receiving guselkumab every 8 weeks versus placebo, respectively.
Across the studies, 89% to 95% of patients per treatment group had received a prior non-biologic DMARD, of which methotrexate was the most commonly received drug (80% to 92%). Prior use of any immunosuppressive or apremilast was less common, and was reported by 2% to 6% of patients. In the DISCOVER-1 and DISCOVER-2 studies, 39% to 48% of patients had previously received systemic corticosteroids, while 87% to 92% had received NSAIDs (data not reported for COSMOS).
In the DISCOVER-1 study, 41 patients (32%) and 39 patients (31%) in the guselkumab and placebo groups, respectively, had received prior TNF alpha inhibitors, including 7 and 4 patients, respectively, who had received 2 prior drugs from this class. The reasons for stopping TNF alpha inhibitor therapy in the guselkumab and placebo groups, respectively, were financial (42% and 41%), inadequate response (37% and 31%), adverse events (2% and 8%), contraindications (5% and 3%), or unspecified (22% and 23%). In the COSMOS studies, all patients had received TNF alpha inhibitors. Among them, 88% had received 1 prior drug and 12% had received 2 prior TNF inhibitors. As per the inclusion criteria, no patients in DISCOVER-2 had prior exposure to TNF inhibitors.
Table 8: Summary of Baseline Characteristics
Characteristics | DISCOVER-1 (FAS) | DISCOVER-2 (FAS) | COSMOS (FAS) | |||
|---|---|---|---|---|---|---|
GUSE q8w N = 127 | PBO N = 126 | GUSE q8w N = 248 | PBO N = 246 | GUSE q8w N = 189 | PBO N = 96 | |
Mean age, years (SD) | 48.9 (11.5) | 49.0 (11.1) | 44.9 (11.9) | 46.3 (11.7) | 49.1 (12.3) | 49.1 (12.1) |
Female, n (%) | 59 (47) | 65 (52) | 119 (48). | 129 (52) | 103 (55) | 44 (46) |
Race, n (%) | NR | NR | ||||
Asian | 10 (8) | 12 (10) | 8 (3) | 4 (2) | NR | NR |
American Indian or Pacific Islander | 1 (1) | 0 | 0 | 0 | NR | NR |
White | 116 (91) | 112 (89) | 240 (97) | 242 (98) | NR | NR |
Other or not reported | 0 | 2 (2) | 0 | 0 | NR | NR |
BMI, kg/m2, mean (SD) | 29.9 (6.4) | 29.6 (5.7) | 28.7 (6.3) | 29.0 (6.4) | |||| ||||| | |||| ||||| |
Number of swollen joints, mean (SD) | 10.9 (9.3) | 10.1 (7.1) | 11.7 (6.8) | 12.3 (6.9) | 10.2 (6.8) | 9.0 (5.7) |
Number of tender joints, mean (SD) | 20.2 (14.5) | 19.8 (14.4) | 19.8 (11.9) | 21.6 (13.1) | 21.0 (13.2) | 18.2 (10.7) |
Patient’s assessment of pain (VAS, 0 cm to 10 cm), mean (SD) | 6.0 (2.1) | 5.8 (2.2) | 6.3 (2.0) | 6.3 (1.8) | 6.5 (1.9) | 6.0 (1.8) |
Duration of PsA, years, mean (SD) | 6.4 (5.9) | 7.2 (7.6) | 5.11(5.5) | 5.8 (5.6) | 8.3 (7.8) | 8.7 (7.2) |
Duration of psoriasis, years, mean (SD) | |||| |||||| | |||| |||||| | |||| |||||| | |||| |||||| | |||| |||||| | |||| |||||| |
PASI score (0 to 72), mean (SD) | 8.4 (9.8) | 7.7 (8.9) | 9.7 (11.7) | 9.3 (9.8) | 11.7 (11.9) | 9.2 (9.4) |
CRP mg/dL, mean (SD) | 1.56 (2.37) | 1.44 (1.89) | 2.04 (2.35) | 2.11 (2.67) | 1.23 (1.96) | 1.15 (2.54) |
Patients with psoriatic Involvement ≥ 3% BSA and an IGA score of ≥ 2 at baseline, n (%) | 82 (65) | 78 (62) | 176 (71) | 183 (74) | 133 (70) | 53 (55) |
Enthesitis at baseline, n (%) | 72 (57) | 77 (61) | 158 (64) | 178 (73) | 126 (67) | 64 (67) |
Dactylitis at baseline, n (%) | 49 (39) | 55 (44) | 111 (45) | 99 (40) | 67 (35) | 36 (38) |
Prior treatments for PsA, n (%) | ||||||
Any non-biologic DMARDs, immunosuppressive, or apremilast | 118 (93) | 115 (91) | 221 (89) | 229 (93) | 177 (94) | 91 (95) |
Any non-biologic DMARDs | 116 (91) | 113 (90) | 221 (89) | 229 (93) | ||| |||| | || |||| |
Methotrexate | 102 (80) | 101 (80) | 199 (80) | 215 (87) | 171 (91) | 88 (92) |
Any immunosuppressivea | | ||| | | ||| | || ||| | || ||| | | ||| | | ||| |
Apremilast | 6 (5) | 4 (3) | 4 (2) | 4 (2) | | ||| | | ||| |
Any TNF alpha inhibitor | 41 (32) | 39 (31) | NA | NA | 189 (100) | 96 (100) |
Systemic corticosteroids | || |||| | || |||| | ||| |||| | ||| |||| | NR | NR |
NSAIDs | 111 (87) | 109 (87) | 226 (91) | 227 (92) | NR | NR |
BMI = body mass index; BSA = body surface area; CRP = C-reactive protein; DMARD = disease-modifying antirheumatic drug; FAS = full analysis set; GUSE = guselkumab; IGA = Investigator’s Global Assessment of Psoriasis; NA = not applicable; NR = not reported; NSAID = nonsteroidal anti-inflammatory drug; PASI = Psoriasis Area and Severity Index; PBO = placebo; PsA = psoriatic arthritis; q8w = every 8 weeks; SD = standard deviation; TNF = tumour necrosis factor; VAS = visual analogue scale.
aImmunosuppressives included cyclosporine, azathioprine, tacrolimus, mycophenolate mofetil, and others.
Source: Clinical Study Report for DISCOVER-1 (24 weeks);11 Clinical Study Report for DISCOVER-2 (24 weeks);12 Clinical Study Report for COSMOS (24 weeks);13 Coates et al. (2022).14 Additional data provided by the sponsor.42
Interventions
In all trials, the study drug was supplied as 1 mL pre-filled syringes that contained either placebo solution or guselkumab 100 mg. In the DISCOVER-1 and DISCOVER-2 trials, the study drugs were described as identical in appearance and packaging. Patients and site investigators remained blinded to treatment allocation until the ends of the studies (i.e., 60 weeks for DISCOVER-1, 112 weeks for DISCOVER-2, and 56 weeks for COSMOS). An interim database lock was planned for 24 weeks in all studies, and the sponsor’s personnel were unblinded to patient-level data at that time to conduct the primary data analysis. In the trials, the study drug was administered by health care workers at week 0 and week 4, but patients had the option to self-administer at later visits under the supervision of a health care professional at the study site.
In DISCOVER-1 and DISCOVER-2, patients were randomized (1:1:1) to guselkumab 100 mg SC every 4 weeks; guselkumab 100 mg SC at week 0, week 4, and every 8 weeks thereafter (with placebo injections at other time points); or placebo every 4 weeks until week 20, crossing over at week 24 to guselkumab 100 mg SC and continuing every 4 weeks until the end of the study. The double-blind treatment durations were 48 weeks for DISCOVER-1 and 100 weeks for DISCOVER-2. At week 16, patients were assessed to determine if they met the early escape criteria. Patients with less than 5% improvement from baseline in tender and swollen joint counts were considered to have met the early escape criteria and were allowed to start therapy or increase the dose of 1 of the permitted concomitant psoriatic medications (up to the daily maximum dose outlined in the study’s protocol). Titration to a stable dose was to be completed by week 24.
In the DISCOVER-1 and DISCOVER-2 studies, stable use of NSAIDs or other analgesics, oral corticosteroids (≤ 10 mg of prednisone per day or equivalent), and 1 of either methotrexate [≤ 25 mg/week], sulfasalazine [≤ 3 g/day], hydroxychloroquine [≤ 400 mg/day], or leflunomide [≤ 20 mg/day]) was allowed during the trial. A washout period was required for patients who had recently stopped treatment with 1 of these medications (2 weeks for NSAIDs or corticosteroids, 12 weeks for leflunomide, and 4 weeks for the other 3 DMARDs). Other psoriasis or PsA treatments had to be stopped 2 weeks to 4 weeks before the start of the study drug. Medications prohibited during the trial included other biologic or non-biologic DMARDs, systemic immunosuppressants, apremilast, JAK inhibitors, and cytotoxic drugs. Injectable systemic corticosteroids or topical psoriasis treatments (except salicylic acid or coal tar shampoo) were prohibited until after week 24. Low- and mid-potency topical or intralesional corticosteroids were allowed after week 24. In DISCOVER-2, high-potency topical corticosteroids, phototherapy, and systemic treatments for psoriasis were prohibited until week 24, whereas DISCOVER-1 prohibited these therapies for the entire study duration. In both trials, patients were allowed to have up to 2 intra-articular, tendon sheath, or bursal corticosteroid injections in no more than 2 sites within any 24-week period of the study.
In the COSMOS study, patients were randomized 2:1 to guselkumab 100 mg SC at week 0, week 4, and every 8 weeks thereafter (with placebo injections at other time points), or to placebo at week 0, week 4, week 12, and week 20 before crossing over to guselkumab 100 mg SC at week 24 and continuing with it at week 28, week 36, and week 44. The double-blind treatment duration was 48 weeks.
The same early escape criteria used in the DISCOVER trials were applied in the COSMOS study. Patients who met the early escape criteria could initiate or increase the dose of permitted concomitant medications, as described previously. In addition, patients randomized to placebo who met the escape criteria received guselkumab 100 mg SC at week 16, week 20, and every 8 weeks thereafter.
Stable doses of NSAIDs, oral corticosteroids, and select cDMARDs (i.e., 1 of methotrexate [≤ 25 mg/week], sulfasalazine [≤ 3 g/day], hydroxychloroquine [≤ 400 mg/day], or leflunomide [≤ 20 mg/day]) were allowed during the study. A washout period was required for patients who had recently stopped treatment with 1 of these medications (2 weeks for NSAIDs or corticosteroids, 12 weeks for leflunomide, and 4 weeks for the other 3 DMARDs).
Medications prohibited during the COSMOS study included other biologic or non-biologic DMARDs, systemic immunosuppressants, apremilast, JAK inhibitors, and lithium. Phototherapy, injectable systemic corticosteroids, and topical psoriasis treatments that could affect psoriasis assessments were also prohibited.
Outcomes
A list of efficacy end points identified in the CADTH review protocol that were assessed in the clinical trials included in this review is provided in Table 9. These end points are further summarized here. A detailed discussion and critical appraisal of the outcome measures is provided in Appendix 4.
After reviewing the outcomes reported, some exploratory or other end points were not summarized in this report if there were data for a similar end point that was listed as a secondary outcome or was part of the statistical testing hierarchy. Examples include the EQ-5D and some composite measures related to PsA symptoms (i.e., the Psoriatic Arthritis Disease Activity Score, GRAPPA Composite Score, and Disease Activity Index for Psoriatic Arthritis [DAPSA]).
Table 9: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure | DISCOVER-1 | DISCOVER-2 | COSMOS |
|---|---|---|---|
Proportion of patients with ACR 20 response at week 24 | Primary | Primary | Primary |
Change from baseline in HAQ-DI at week 24 | Major secondary | Major secondary | Secondary |
Change from baseline in SF-36 PCS at week 24 | Major secondary | Major secondary | Secondary |
Change from baseline in SF-36 MCS at week 24 | Major secondary | Major secondary | Exploratory |
Change from baseline in DAS 28 CRP at week 24 | Major secondary | Major secondary | Exploratory |
Proportion of patients with ACR 50 response at week 24 | Major secondary | Major secondary | Secondary |
Proportion of patients with ACR 20 response at week 16 | Major secondary | Major secondary | Exploratory |
Proportion of patients with ACR 70 response at week 24 | Major secondary | Major secondary | Exploratory |
Proportion of patients with ACR 50 response at week 16 | Major secondary | Major secondary | Exploratory |
Proportion of patients with psoriasis IGA response (0 out of 1 and ≥ 2 grade reduction from baseline) among patients with ≥ 3% BSA and ≥ 2 IGA at baseline | Major secondary | Major secondary | Exploratory |
Proportion of patients with PASI 100 response at week 24 in patients with ≥ 3% BSA and ≥ 2 IGA at baseline | Other secondary | Other secondary | Secondary |
Proportion of patients with resolution of enthesitis at week 24 (among patients with enthesitis at baseline) | Major secondary | Major secondary | Exploratory |
Proportion of patients with resolution of dactylitis at week 24 (among patients with dactylitis at baseline) | Major secondary | Major secondary | Exploratory |
Change from baseline in enthesitis score at week 24 (based on LEI) | Major secondary | Major secondary | Exploratory |
Change from baseline in dactylitis score at week 24 | Major secondary | Major secondary | Exploratory |
Proportion of patients with ≥ 50% improvement in BASDAI score at week 24 | Other secondary | Other secondary | Exploratory |
Proportion of patients achieving MDA at week 24 | Other secondary | Other secondary | Exploratory |
Proportion of patients with PASI 90 or PASI 75 response at week 24 in patients with ≥ 3% BSA and ≥ 2 IGA at baseline | Other secondary | Other secondary | Exploratory |
Change from baseline in FACIT-Fatigue score at week 24 | Other secondary | Other secondary | Exploratory |
Change from baseline in modified vdH-S score at week 24 | NR | Major secondary | NR |
ACR 20 = American College of Rheumatology 20% improvement; ACR 50 = American College of Rheumatology 50% improvement; ACR 70 = American College of Rheumatology 70% improvement; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; BSA = body surface area; CRP = C-reactive protein; DAS 28 = Disease Activity Score 28; DAS 28 CRP = Disease Activity Score 28 using C-reactive protein; FACIT-Fatigue = Functional Assessment of Chronic Illness Therapy – Fatigue Scale; HAQ-DI = Health Assessment Questionnaire Disability Index; IGA = Investigator’s Global Assessment of Psoriasis; LEI = Leeds Enthesitis Index; MDA = minimal disease activity; MCS = Mental Component Summary; NR = not reported; PASI 75 = Psoriasis Area and Severity Index 75% improvement; PASI 90 = Psoriasis Area and Severity Index 90% improvement; PASI 100 = Psoriasis Area and Severity Index 100% improvement; PCS = Physical Component Summary; SF-36 = Short Form (36) Health Survey; vdH-S = van der Heijde-Sharp.
Source: Clinical Study Report for DISCOVER-1 (24 weeks);11 Clinical Study Report for DISCOVER-2 (24 weeks);12 Clinical Study Report for COSMOS (24 weeks).13
Clinical Response in PsA Symptoms
In all studies, a trained, independent joint assessor who was not otherwise involved in the conduct of the trial evaluated each of 68 joints for tenderness and each of 66 joints for swelling (hips were excluded for swelling). This assessor also conducted enthesitis and dactylitis assessments.
ACR 20, ACR 50, and ACR 70 are measurements of improvement in multiple disease assessment criteria. The ACR 20 is defined as a greater than or equal to 20% improvement from baseline in both swollen joint count (66 joints) and tender joint count (68 joints), and a greater than or equal to 20% improvement from baseline in 3 of the following 5 assessments: the patient’s assessment of pain (visual analogue scale [VAS]), the patient’s global assessment of disease activity (arthritis, VAS), the physician’s global assessment of disease activity (VAS), the HAQ-DI, and CRP. ACR 50 and ACR 70 response criteria require changes from baseline of 50% and 70%, respectively. The ACR 20 is generally accepted as the MID, indicating a response to treatment, while the ACR 50 and ACR 70 more likely reflect truly important change for the long-term management of arthropathy.43,44
The MDA is a composite measure that encompasses different aspects of PsA disease.45 To meet the MDA criteria, patients had to achieve at least 5 out of the following 7 items: tender joint count less than or equal to 1, swollen joint count less than or equal to 1, PASI score less than or equal to 1, patient’s assessment of pain less than or equal to 15 (VAS), patient’s global assessment of disease activity less than or equal to 20 (arthritis and psoriasis, VAS), HAQ-DI score less than or equal to 0.5, and tender entheseal points less than or equal to 1.45
The Disease Activity Score 28 (DAS 28) using CRP (DAS 28 CRP) is composite measure of disease activity developed for patients with rheumatoid arthritis. It combines tender joints (28 joints) and swollen joints (28 joints) of the arms, shoulders, and knees with the CRP and the patient’s global assessment of disease activity.46 The range of the DAS 28 is 0 to 9.4, with higher scores indicating more active disease.47 No evidence was found to support the validity, reliability, or responsiveness in patients with PsA. According to the clinical expert consulted by CADTH, the relevance of this outcome measure in patients with PsA has been questioned because the DAS 28 does not include an assessment of inflammation in the feet, which is more common in patients with PsA than in those with rheumatoid arthritis. Assessment of 66 and 68 swollen and tender joints over the 28-joint count in patients with PsA has been endorsed by GRAPPA and the Outcome Measures in Rheumatology working group.48
Functioning or Disability
The HAQ-DI is a patient-reported, 20-question instrument that assesses the degree of difficulty a patient has in accomplishing tasks in 8 functional areas (dressing, arising, eating, walking, hygiene, reaching, gripping, and activities of daily living). Each functional area is scored from 0 (indicating no difficulty) to 3 (indicating inability to perform a task in that area). The overall score is the average of 8 domains, ranging from 0 (no disability) to 3 (completely disabled). The Clinical Study Report (CSR) states that in PsA, a decrease in score of 0.35 has been determined to indicate a clinically meaningful improvement.6 Other sources have reported MIDs of 0.131 and 0.16 in patients with PsA.49,50
Health-Related Quality of Life
The SF-36 is a 36-item general health status instrument that consists of 8 health domains: physical functioning, role physical, bodily pain, general health, vitality, social functioning, role emotional, and mental health. The SF-36 also provides 2 component summaries, the PCS and the MCS, which are derived by aggregating the 8 domains according to a scoring algorithm. All domain and component scores are based on a scale of 0 to 100, with higher scores indicating higher health status. Scores were standardized with a mean of 50 points and an SD of 10 points in the general US population. The CSR defined a change of greater than or equal to 5 points in the PCS and MCS as a clinically meaningful improvement; however, this threshold was not specific to patients with PsA.11,51 MIDs of 3.74 points for the PCS and 1.77 points for the MCS have been reported for patients with PsA.7,52
Psoriasis-Specific Measures
Psoriasis-specific end points were evaluated in the subgroup of patients with psoriatic involvement affecting at least 3% of their BSA and an IGA score of 2 (mild) or higher at baseline.
The IGA is a composite score of physician assessment of the overall severity of the patient’s psoriatic lesions. The static version of the IGA was used in all included studies, and is a measurement of disease severity at a given time point. The investigator assessed the overall severity of induration, erythema, and scaling of lesions based on a 5-point scale scored as 0 (no evidence), 1 (minimal), 2 (mild), 3 (moderate), and 4 (severe). The IGA score was the average of the induration, erythema, and scaling scores. The patient’s psoriasis was assessed as cleared (0), minimal (1), mild (2), moderate (3), or severe (4).53 IGA response was defined as a score of 0 or 1 and at least a 2-point decrease from baseline.
The PASI is a widely used instrument in psoriasis trials that grades the extent and severity of psoriatic lesions. It combines an assessment of the BSA affected in 4 anatomic regions (head, trunk, arms, and legs) and the severity of desquamation, erythema, and plaque induration or infiltration (thickness) in each region. Scores range from 0 points to 72 points, with a PASI score greater than 10 representing more severe disease.54 A PASI 75, PASI 90, or PASI 100 response is defined, respectively, as a greater than or equal to 75%, greater than or equal to 90%, or greater than or equal to 100% improvement in PASI score from baseline. No MID for the PASI has been estimated among patients with PsA.
Other Musculoskeletal Outcomes
Dactylitis was assessed in both hands and feet using a scoring system from 0 to 3 for each digit (where 0 = no dactylitis, 1 = mild dactylitis, 2 = moderate dactylitis, and 3 = severe dactylitis). The results were summed to produce a final score ranging from 0 to 60. Enthesitis was assessed using the Leeds Enthesitis Index (LEI), which counts the number of painful entheses among the left and right lateral epicondyle humerus, left and right medial femoral condyle, and left and right Achilles tendon insertion. The LEI index ranges from 0 to 6.55 No MID was identified for the LEI and dactylitis score. In all trials, only patients with dactylitis or enthesitis at baseline were included in the analysis of these end points.
The BASDAI is a patient-reported assessment for ankylosing spondylitis. It consists of 6 questions relating to the 5 major symptoms of ankylosing spondylitis (fatigue, spinal pain, peripheral joint pain, localized tenderness, and morning stiffness). Each question is scored using a 10-unit VAS to indicate the degree of symptoms over the past week. The total score ranges from 0 to 10, with higher scores indicate greater disease severity.56,57 According to the CSR, a 50% decrease in the score is clinically meaningful.11,58 The MID in patients with PsA is unclear. In the clinical trials, only patients with spondylitis with peripheral arthritis as the primary arthritic presentation of PsA completed the BASDAI.
Symptoms
FACIT-Fatigue consists of 13 questions that assess a patient’s level of fatigue and tiredness over the last 7 days. Each question was graded on a 5-point scale (where 0 = not at all, 1 = a little bit, 2 = somewhat, 3 = quite a bit, and 4 = very much). The score ranges from 0 to 52, with lower scores reflecting more severe fatigue.8 The CSR states that in rheumatology, a change of 4 points is considered meaningful and has been used in the PsA population.8 An MID of 3.1 points has also been reported for patients with PsA.9
Radiologic End Points
For the assessment of radiographic changes, single radiographs of the hands and feet were evaluated by central independent readers and scored using the modified vdH-S score. The modified vdH-S score, which ranges from 0 (best) to 528 (worst), is the sum of the joint erosion score and the joint space narrowing score.10 In addition to reporting the change from baseline in the modified vdH-S score, the DISCOVER-2 study also reported the number of patients with radiographic progression based on the smallest detectable change. The smallest detectable change is defined as the amount of change from baseline for which any smaller change cannot be reliably distinguished from random error in the measurement. The MID in patients with PsA is not known.
Safety
Treatment-emergent adverse events included any adverse events that occurred after the start of study drug administration and any adverse events that were present at baseline but worsened in severity after the start of study drug administration. Serious adverse events included any event that resulted in death, was life-threatening, required hospitalization or prolongation of existing hospitalization, resulted in persistent or significant disability, was a congenital anomaly, was the suspected transmission of an infectious drug through a medicinal product, or was medically important (e.g., required intervention to prevent 1 of the other outcomes listed).
Statistical Analysis
Primary Analysis Methods
In all of the trials, the primary analysis of efficacy outcomes was based on the composite estimand, which assessed the treatment effects based on the outcome variable as well as events that met the treatment failure criteria. Patients who met any of the treatment failure criteria up to week 24 were considered nonresponders for the binary (responder) outcomes, or were considered as no change from baseline for the continuous outcomes. Treatment failure was defined as 1 of the following: discontinued study drug injections due to any reason; terminated study participation due to any reason; initiated or increased the dose of non-biologic DMARDs (methotrexate, sulfasalazine, hydroxychloroquine, leflunomide) or oral corticosteroids over baseline for PsA; initiated protocol-prohibited medications or therapies for PsA; or met early escape criteria (COSMOS only). An alternate estimand, the treatment policy strategy, was the primary estimand for radiologic outcomes in DISCOVER-2 (refer to the Sensitivity and Subgroup Analyses section for details).
The statistical methods used in the included studies have been summarized in Table 10. The primary outcome (i.e., ACR 20 at week 24) and other binary outcomes were tested using a CMH test (or Fisher’s exact test, if CMH was not appropriate) stratified by baseline factors specific to each trial. In DISCOVER-1, the stratification factors were the use of a non-biologic DMARD at baseline (yes, no) and prior exposure to TNF alpha inhibitors (yes, no). DISCOVER-2 patients were stratified by baseline use of non-biologic DMARDs (yes, no) and most recent CRP value (< 2.0 mg/dL versus ≥ 2.0 mg/dL). In the COSMOS study, the stratification factors were non-biologic DMARD use at baseline (yes, no) and number of prior TNF alpha inhibitors (1 or 2). The differences in response rates and 95% CI between the guselkumab and placebo groups were calculated based on the Wald statistic. Patients who met any of the treatment failure criteria were considered nonresponders at week 24, regardless of their ACR 20 response status. In addition, any patients with missing data were imputed as not achieving the response.
In the DISCOVER trials, major secondary continuous outcomes were analyzed using an ANCOVA model based on multiple imputation data, with missing data imputed under the missing-at-random assumption (Table 10). The model included covariates for baseline score and each studies’ stratification factors. Other continuous outcomes (e.g., FACIT-Fatigue, BASDAI) were analyzed using an MMRM with no imputation for missing data in the DISCOVER trials. In the COSMOS study, continuous outcomes were analyzed using an MMRM with no imputation for missing data. Any patients who met the treatment failure criteria were imputed as no change (i.e., no improvement) from baseline in the model.
Sensitivity and Subgroup Analyses
Several sensitivity analyses were planned for the primary outcome in the DISCOVER-1 and DISCOVER-2 studies. These included the following: a tipping point analysis where all combinations of missing data, imputed as responders and nonresponders, were analyzed; a treatment policy analysis, which was based on observed ACR 20 response data for all patients regardless of whether treatment failure criteria were met before week 24 and using multiple imputation methods for missing data (missing-at-random assumption); a tipping point analysis based on multiple imputation methods for missing data for the treatment policy estimand; an alternate composite analysis where patients who stopped treatment for reasons other than lack of efficacy were not considered as nonresponders; and a per-protocol analysis that was based on the last non-missing data before stopping therapy or meeting treatment failure criteria in patients who did not have any major protocol violations.
In the DISCOVER trials, subgroup analyses based on treatment history and concomitant DMARD use were of interest to this review. These preplanned analyses for the ACR 20 end point were based on a logistic regression model and reported as odds ratios and 95% CIs. P values for the treatment by subgroup interaction terms were reported.
In the COSMOS study, a supplementary analysis was conducted based on the treatment policy estimand, which evaluated treatment effects using all observed data, regardless of the intercurrent events that met the treatment failure criteria, with imputation for missing data. In this trial, this estimand ignored the week 16 crossover of placebo patients who met the early escape criteria and received guselkumab. The sponsor stated that this analysis biases the results against guselkumab because it does not consider any benefit that patients in the placebo group may have received from switching to active treatment. A second supplementary analysis was conducted where discontinuation of the study drug due to reasons other than lack of efficacy was not considered as treatment failure.
A total of 20 patients in the COSMOS study (8 patients [8%] in the placebo group and 12 patients [6%] in the guselkumab group) were incorrectly assigned to early escape. Patients in the placebo group were incorrectly switched to guselkumab, and there was no change to the study drug received by the patients who had been incorrectly assigned to early escape in the guselkumab group. Two sensitivity analyses were conducted for these patients. The first ignored the early escape criterion, which meant that the 12 patients on guselkumab were not considered to have experienced treatment failure (i.e., no change in therapy); however, the 8 placebo patients were analyzed as a treatment failure because they had received a prohibited medication. The second analysis set the week 16 to week 24 end point values as missing. These were imputed using multiple imputation methods for binary outcomes or MMRM for continuous end points.
The preplanned subgroup analyses based on patient demographics were not of interest to this review and have not been summarized in this report.
Statistical Power
The DISCOVER-1 study was estimated to have greater than 90% power at a 2-sided alpha level of 0.05 for the primary outcome of ACR 20 response. This estimate was based on enrolling a total of 360 patients and assuming a 40% ACR 20 response rate in the guselkumab group and a 20% ACR 20 response in the placebo group at week 24.
For the DISCOVER-2 study, assuming ACR 20 response rates of 45% and 20% in the guselkumab and placebo groups, respectively, a sample size of 228 patients per group would have 99% power to detect a significant treatment effect at a 2-sided significance level of 0.05. With a sample size of 228 patients per group, the trial would have 90% power to detect a difference in the mean change from baseline in the vdH-S score, assuming a mean change of 0.9 in the placebo group and 0.3 in the guselkumab group, with an SD of 2.5. No references were listed to support the assumptions used in the calculations.
The COSMOS study was planned to have 90% power to detect a difference in the primary outcome (i.e., ACR 20) at a 2-sided significance level of 0.05 based on a sample size of 163 patients in the guselkumab group and 82 patients in the placebo group, assuming a response rate of 41% versus 20% for guselkumab and placebo, respectively. The estimated ACR 20 response rates were based on data from an ustekinumab trial in patients previously treated with TNF alpha inhibitors.59
Table 10: Statistical Analysis of Efficacy End Points
End point | Statistical model | Adjustment factors | Sensitivity analyses |
|---|---|---|---|
DISCOVER-1 | |||
Proportion of patients with ACR 20 response at week 24; other binary outcome measuresa | Stratified Cochran-Mantel-Haenszel test (p value), Wald statistic for difference in response rates (95% CI) (Missing data imputed as nonresponders) Composite estimand |
|
|
Change from baseline in HAQ-DI score, DAS 28 CRP, SF-36 PCS and MCS, enthesitis score, and dactylitis score at week 24 | ANCOVA (multiple imputation methods for missing data assuming MAR) Composite estimand |
|
|
Other continuous outcomes | MMRM (no imputation for missing data) Composite estimand | None | NR |
DISCOVER-2 | |||
Proportion of patients with ACR 20 response at week 24; other binary outcome measuresa | Stratified Cochran-Mantel-Haenszel test (p value), Wald statistic for difference in response rates (95% CI) (Missing data imputed as nonresponders) Composite estimand |
|
|
Change from baseline in HAQ-DI score, DAS 28 CRP, SF-36 PCS and MCS, enthesitis score, and dactylitis score at week 24a | ANCOVA (multiple imputation methods for missing data, assuming MAR) Composite estimand |
|
|
Change from baseline in modified vdH-S score at 24 weeks | ANCOVA (multiple imputation methods for missing data, assuming MAR) Treatment estimand |
|
|
Other continuous outcomes (e.g., BASDAI, FACIT-Fatigue) | MMRM (no imputation for missing data) Composite estimand | None | NR |
COSMOS | |||
Proportion of patients with ACR 20 response at week 24; other binary outcome measuresb | Stratified Cochran-Mantel-Haenszel test (p value), Wald statistic for difference in response rates (95% CI) (Missing data imputed as nonresponder) Composite estimand |
|
|
Continuous outcomesb | MMRM (no imputation for missing data; missing-at-random assumption) Composite estimand | None |
|
ACR 20 = American College of Rheumatology 20% improvement; ANCOVA = analysis of covariance; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; CI = confidence interval; CRP = C-reactive protein; DAS 28 = Disease Activity Score 28; DAS 28 CRP = Disease Activity Score 28 using C-reactive protein; DMARD = disease-modifying antirheumatic drug; FACIT-Fatigue = Functional Assessment of Chronic Illness Therapy – Fatigue Scale; HAQ-DI = Health Assessment Questionnaire Disability Index; MAR = missing at random; MCS = Mental Component Summary; MMRM = mixed-effects model for repeated measures; NR = not reported; PCS = Physical Component Summary; SF-36 = Short Form (36) Health Survey; TNF = tumour necrosis factor; vdH-S = van der Heijde-Sharp.
aThe sensitivity analyses for major secondary outcomes included a tipping point analysis based on the treatment policy estimand and multiple imputation for missing data.
bThe sensitivity analyses for major secondary outcomes included analyses based on treatment policy estimand and 2 analyses that corrected for early escape errors for 20 patients.
Source: Clinical Study Report for DISCOVER-1 (24 weeks);11 Clinical Study Report for DISCOVER-2 (24 weeks);12 Clinical Study Report for COSMOS (24 weeks).13
Control of Type I Error Rate
In the DISCOVER trials, the overall type I error was controlled at a significance level of less than or equal to 0.05 for the primary outcome (i.e., ACR 20) and select major secondary outcomes. Due to regulatory requirements, there were 2 pre-specified multiplicity adjustment procedures used: 1 for the US and 1 for countries except the US (global). In both procedures, the primary outcome was tested first for the group receiving guselkumab every 4 weeks versus the placebo group, and then for the group receiving guselkumab every 8 weeks versus the placebo group. Testing of select major secondary outcomes proceeded if the primary outcome was statistically significant. In the DISCOVER-1 study, the global testing procedure included 8 major secondary outcomes that were tested for each guselkumab dose group independently in the order specified in Table 11. In the US testing procedure, the 2 dosage groups were tested in parallel, as shown in Figure 5. The overall significance level of less than or equal to 0.05 was controlled for the 24-week IGA response and the HAQ-DI and SF-36 PCS outcomes. Other secondary outcomes were not controlled for type I error rate. The testing procedures stated that ACR 20 at week 16, ACR 50 at week 16, ACR 50 at week 24, ACR 70 at week 24, and change from baseline DAS 28 at week 24 were tested (with a nominal P value) only if the primary outcome for the corresponding guselkumab dosage group was statistically significant.
In the DISCOVER-2 study, the US-based testing procedure controlled the type I error at an overall significance level of less than or equal to 0.05 for the primary outcome (i.e., ACR 20) and for 3 major secondary end points that were not highly correlated with each other (IGA response, HAQ-DI, and SF-36 PCS at week 24). Figure 6 shows the testing procedure for these 4 outcomes in the 2 dosage groups. The global testing procedure controlled the type I error rate for 12 outcomes, as shown in Figure 7, and included a pooled analysis of the resolution of enthesitis or dactylitis based on data from DISCOVER-1 and DISCOVER-2. Five other outcomes highly correlated with ACR 20 were tested (with nominal P values) only if statistical significance was achieved for ACR 20 response at week 24 for the corresponding guselkumab dose group. These outcomes included ACR 20 and ACR 50 response at week 16, ACR 50 and ACR 70 response at week 24, and change from baseline in DAS 28 at week 24. Other secondary outcomes were not formally tested, and nominal P values were reported. Of note: the Health Canada reviewer’s report stated that the US analyses were deemed the preferred method.40
For the COSMOS study, the type I error rate was controlled for the primary outcome and 4 major secondary outcomes. These were tested in a fixed sequence (week 24 HAQ-DI, ACR 50, SF-36 PCS, PASI 100 response). Statistical testing was performed sequentially only if the previous outcome reported a P value of less than 0.05. The P values for the other outcomes reported were not controlled for multiple testing, and the sponsor stated that these were descriptive and not to be used to determine statistical significance.
Table 11: Hierarchical Sequence for Testing Secondary Outcomes
Study | Global testing hierarchy | US testing hierarchya |
|---|---|---|
DISCOVER-1 | 1. Proportion of patients with IGA response at week 24 among patients who had ≥ 3% BSA and IGA score ≥ 2 at baseline 2. Change from baseline DAS 28 CRP at week 24 3. Change from baseline HAQ-DI at week 24 4. Change from baseline SF-36 PCS at week 24 5. Proportion of patients with ACR 20 response at week 16 6. Proportion of patients with ACR 50 response at week 24 7. A split of:
| 1. Proportion of patients with IGA response at week 24 among patients who had ≥ 3% BSA and IGA score ≥ 2 at baseline 2. Change from baseline HAQ-DI at week 24 3. Change from baseline SF-36 PCS at week 24 |
DISCOVER-2 | 1. Proportion of patients with IGA response at week 24 among patients who had ≥ 3% BSA and IGA score ≥ 2 at baseline 2. Change from baseline DAS 28 CRP at week 24 3. Change from baseline HAQ-DI at week 24 4. Change from baseline in modified vdH-S score at week 24 5. Change from baseline SF-36 PCS at week 24 6. Proportion of patients with ACR 20 response at week 16 7. Proportion of patients with ACR 50 response at week 24 8. A split of:
9. Dactylitis resolution (pooled DISCOVER-1 and DISCOVER-2 data) 10. Enthesitis resolution (pooled DISCOVER-1 and DISCOVER-2 data) 11. Change from baseline in SF-36 MCS at week 24 | 1. Proportion of patients with IGA response at week 24 among patients who had ≥ 3% BSA and IGA score ≥ 2 at baseline 2. Change from baseline HAQ-DI at week 24 3. Change from baseline in modified vdH-S score at week 24 4. Change from baseline SF-36 PCS at week 24 5. Dactylitis resolution (pooled DISCOVER-1 and DISCOVER-2 data) 6. Enthesitis resolution (pooled DISCOVER-1 and DISCOVER-2 data) 7. Change from baseline in SF-36 MCS at week 24 |
ACR 20 = American College of Rheumatology 20% improvement; ACR 50 = American College of Rheumatology 50% improvement; ACR 70 = American College of Rheumatology 70% improvement; CRP = C-reactive protein; DAS 28 = Disease Activity Score 28; DAS 28 CRP = Disease Activity Score 28 using CRP; HAQ-DI = Health Assessment Questionnaire Disability Index; IGA = Investigator’s Global Assessment of Psoriasis; MCS = Mental Component Summary; PCS = Physical Component Summary; SF-36 = Short Form (36) Health Survey; vdH-S = van der Heijde-Sharp.
aThe US-based testing procedure was the preferred method, according to the Health Canada reviewer’s report.
Source: Clinical Study Report for DISCOVER-1 (24 weeks);11 Clinical Study Report for DISCOVER-2 (24 weeks).12
Figure 5: US-Specific Multiplicity Testing Procedure — DISCOVER-1 — Redacted

ACR 20 = American College of Rheumatology 20% improvement; HAQ-DI = Health Assessment Questionnaire Disability Index; IGA = Investigator’s Global Assessment of Psoriasis; PCS = Physical Component Summary; q4w = every 4 weeks; q8w = every 8 weeks.
Figure redacted at the request of the sponsor.
Source: Clinical Study Report for DISCOVER-1.11
Figure 6: US-Specific Multiplicity Testing Procedure — DISCOVER-2 — Redacted

ACR 20 = American College of Rheumatology 20% improvement; ACR 50 = American College of Rheumatology 50% improvement; ACR 70 = American College of Rheumatology 70% improvement; DAS 28 = Disease Activity Score 28; HAQ-DI = Health Assessment Questionnaire Disability Index; IGA = Investigator’s Global Assessment of Psoriasis; MCS = Mental Component Summary; PCS = Physical Component Summary; q4w = every 4 weeks; q8w = every 8 weeks; vdH-S = van der Heijde-Sharp.
Figure redacted at the request of the sponsor.
Source: Clinical Study Report for DISCOVER-2 (24 weeks).12
Figure 7: Global Multiplicity Testing Procedure — DISCOVER-2 — Redacted

ACR 20 = American College of Rheumatology 20% improvement; ACR 50 = American College of Rheumatology 50% improvement; ACR 70 = American College of Rheumatology 70% improvement; DAS 28 = Disease Activity Score 28; HAQ-DI = Health Assessment Questionnaire Disability Index; IGA = Investigator’s Global Assessment of Psoriasis; PCS = Physical Component Summary; vdH-S = van der Heijde-Sharp.
Figure redacted at the request of the sponsor
Source: Clinical Study Report for DISCOVER-2 (24 weeks).12
Analysis Populations
In all studies, most efficacy outcomes were analyzed based on the FAS, which included all randomized patients who received at least 1 dose of the study drug according to their randomized group. Psoriasis-specific outcomes (i.e., IGA or PASI response) were analyzed in specific FAS subpopulations that had greater than or equal to 3% BSA psoriatic involvement and an IGA score of greater than or equal to 2 at baseline. Patients included in the enthesitis analyses had to have at least 1 tender enthesis among the 6 sites included in the LEI at baseline. The dactylitis outcomes were analyzed in patients with dactylitis at baseline, and the BASDAI was analyzed in the subset of patients with spondylitis and peripheral joint PsA as their primary presentation. DISCOVER-2 also included a preplanned, pooled analysis of enthesitis and dactylitis outcomes that were based on relevant subgroup data from the DISCOVER-1 and DISCOVER-2 trials.
The safety population included all randomized patients who received at least 1 dose of the study drug and were analyzed according to the actual treatment received.
Results
Patient Disposition
In the DISCOVER-1 study, 624 patients were screened, and 381 patients (61%) were randomized to 1 of 3 treatment groups. By week 24, 7% versus 2% of patients in the placebo group and the group receiving guselkumab every 8 weeks, respectively, had discontinued from the study, and 10% versus 3% stopped treatment early. Table 12 describes the reasons for discontinuation. More patients met the treatment failure criteria (17% versus 6%) and qualified for early escape (19% and 3%) in the placebo group than in the guselkumab group.
Of the 1,153 patients screened in the DISCOVER-2 study, 741 patients were randomized, and 739 patients were treated (64%). The proportion of patients who discontinued the study was low (1% and 2%), as were the proportions who discontinued the study drug (2% and 3% in both the placebo and guselkumab groups, respectively). The treatment failure criteria were met by 7% and 5% of patients — and the early escape criteria were met by 15% and 5% of patients — in the placebo group and the group receiving guselkumab every 8 weeks, respectively.
In the COSMOS study, 328 patients were screened, of whom 285 patients (87%) were randomized. By week 24, 2% of patients in each group had discontinued the study, and 10% and 8% in the placebo and guselkumab groups, respectively, had discontinued the study drug. Approximately half of the patients in the placebo group met the treatment failure criteria or early escape criteria (54% and 47%, respectively), compared with 27% and 21% of patients in the guselkumab group. The CSR reported that a total of 20 patients were incorrectly assigned to early escape without having met the escape criteria. This affected 8 patients in the placebo group (8%) and 12 patients (6%) in the guselkumab group (Table 12).
Detail Week 0 to 24 | DISCOVER-1 | DISCOVER-2 | COSMOS | |||
|---|---|---|---|---|---|---|
GUSE q8w | PBO | GUSE q8w | PBO | GUSE q8w | PBO | |
Screened, N | 624 | 1,153 | 328 | |||
Randomized and treated, total N (%) | 381 (61)a | 739 (64)b | 285 (87)c | |||
Randomized and treated, N (%) | 127 | 126 | 248 | 246d | 189 | 96 |
Discontinued from study by week 24, n (%) | 3 (2) | 9 (7) | 6 (2) | 2 (1) | 3 (2) | 2 (2) |
Reason for discontinuation, n (%) | ||||||
Lost to follow-up | 1 (1) | 1 (1) | 1 (0.4) | 0 | 1 (1) | 0 |
Withdrawal by patient | 1 (1) | 5 (4) | 1 (0.4) | 0 | 2 (1) | 1 (1) |
Death | 0 | 1 (1) | 0 | 0 | 0 | 0 |
Other | 0 | 0 | 0 | 0 | 0 | 1 (1) |
Not specified (discontinued study, but completed protocol-required follow-up) | 1 (1) | 2 (2) | 4 (2) | 2 (1) | NR | NR |
Discontinued study drug by week 24, N (%) | 4 (3) | 12 (10) | 8 (3) | 6 (2) | 15 (8) | 10 (10) |
Reason for discontinuation, n (%) | ||||||
Adverse events | 3 (2) | 2 (2) | 2 (1) | 4 (2) | 4 (2) | 3 (3) |
Lost to follow-up | 0 | 1 (1) | 1 (0.4) | 0 | 1 (1) | 0 |
Withdrawal of consent | 0 | 3 (2) | 1 (0.4) | 1 (0.4) | 5 (3) | 1 (1) |
Death | 0 | 1 (1) | 0 | 0 | 0 | 0 |
Other | 1 (1) | 0 | 1 (0.4) | 1 (0.4) | 0 | 2 (2) |
Lack of efficacy | 0 | 4 (3) | 3 (1) | 0 | 4 (2) | 2 (2) |
Initiation of prohibited medication | 0 | 1 (1) | 0 | 0 | 1 (1) | 2 (2) |
Met treatment failure criteria through week 24, n (%) | 7 (6) | 21 (17) | 12 (5) | 17 (7) | 51 (27)e | 52 (54)e |
Qualified for early escape at week 16, n (%) | 4 (3) | 24 (19) | 13 (5) | 38 (15) | 39 (21)e | 45 (47)e |
FAS, N (%) | 127 | 126 | 248 | 246 | 189 | 96 |
Safety, N (%) | 127 | 126 | 248 | 246 | 189 | 96 |
FAS = full analysis set; GUSE = guselkumab; NR = not reported; PBO = placebo; q8w = every 8 weeks.
aIn the DISCOVER-1 study, 624 patients were screened and 381 were randomized (including 128 patients who were assigned to the guselkumab-every-4-weeks group whose data have not been summarized in this table).
bIn the DISCOVER-2 study, out of the 1,153 patients screened, 741 patients were randomized and 739 patients were treated (including 245 patients who were randomized to the guselkumab-every-4-weeks group whose data have not been summarized in this table).
cIn the COSMOS study, 43 patients failed screening due to exclusionary lab criteria (9 patients), TB criteria (7 patients), underlying conditions (7 patients), unwillingness to adhere to protocols (6 patients), or other reasons (16 patients).
dOne additional patient was randomized to placebo, but was not treated.
eIncludes 12 patients (6.3%) in the guselkumab group and 8 patients (8.3%) in the placebo group who were incorrectly assigned to early escape therapy (without having met the early escape criteria).
Source: Clinical Study Report for DISCOVER-1 (24 weeks);11 Clinical Study Report for DISCOVER-2 (24 weeks);12 Clinical Study Report for COSMOS (24 weeks);13 Coates et al. (2022).14
Exposure to Study Treatments
The durations of study follow-up were similar across the studies (mean = 23.7 weeks [SD = 2.4 weeks] to 24.0 weeks [SD = 0.5 weeks]). In the DISCOVER studies, the mean number of study drug administrations ranged from 5.8 weeks (SD = 0.8 weeks) to 5.9 weeks (SD = 0.5 weeks). In the COSMOS study, these numbers were 4.1 weeks (SD = 0.6 weeks) in the group receiving guselkumab every 8 weeks and 4.3 weeks (SD = 0.9 weeks) in the placebo group (Table 13).
In general, the proportions of patients receiving permitted concomitant therapies for PsA were similar between groups in the trials. In the DISCOVER trials, at baseline, 54% to 63% of patients per group were receiving methotrexate, || || || were receiving sulfasalazine, || || || were receiving leflunomide, and 14% to 20% were receiving oral corticosteroids. Mean daily or weekly doses of permitted concomitant therapies are listed in Table 13. In the COSMOS study, 58% and 61% of patients were receiving methotrexate at baseline.
In the DISCOVER-1 study, 3% of patients in the group receiving guselkumab every 8 weeks met the early escape criteria compared with 19% of patients in the placebo group. Similarly, 5% and 15% in the guselkumab and placebo groups of the DISCOVER-2 study met the early escape criteria. In the COSMOS study, 39 patients (21%) in the group receiving guselkumab every 8 weeks versus 45 patients (47%) in the placebo group were assigned to early escape. However, 20 patients were erroneously assigned to receive early escape therapy without having met the criteria. This error affected 12 patients (6.3%) in the guselkumab and 8 patients (8.3%) in the placebo group.
Table 13: Exposure to Study Treatments and Permitted Concomitant Therapies
Treatment and time point Week 24 | DISCOVER-1 (FAS) | DISCOVER-2 (safety set) | COSMOS (safety set) | |||
|---|---|---|---|---|---|---|
GUSE q8w N = 127 | PBO N = 126 | GUSE q8w N = 248 | PBO N = 246 | GUSE q8w N = 189 | PBO N = 96 | |
Duration of study follow-up, weeks, mean (SD) | 23.9 (0.8) | 23.7 (2.4) | 23.9 (1.3) | 24.0 (0.5) | 23.9 (1.5) | 23.8 (2.1) |
Number of study drug administrations, mean (SD) | 5.9 (0.5) | 5.8 (0.8) | 5.9 (0.5) | 5.9 (0.3) | 4.1 (0.6) | 4.3 (0.9) |
Total dose of guselkumab, mg, mean (SD) | 395.3 (30.5) | 0 | 394.8 (30.1) | 0 | 389.4 (43.7) | N = 45 197.8 (14.9) |
Permitted non-biologic DMARDs at baseline, n (%) | 83 (65) | 82 (65) | 170 (69) | 172 (70) | NR | NR |
Methotrexate | 68 (54) | 71 (56) | 141 (57) | 156 (63) | 105 (61) | 51 (58) |
Sulfasalazine | | ||| | | ||| | || ||| | | ||| | | ||| | | ||| |
Leflunomide | | ||| | | ||| | || ||| | | ||| | || ||| | | ||| |
Hydroxychloroquine | | ||| | || ||| | | ||| | || ||| | | ||||| | || ||| |
Permitted non-biologic DMARDs at baseline, mean dose (SD) | ||||||
Methotrexate, mg/week | |||| ||||| | |||| ||||| | |||| ||||| | |||| ||||| | NR | NR |
Sulfasalazine, g/day | ||| ||||| | ||| ||||| | ||| ||||| | ||| ||||| | NR | NR |
Leflunomide, mg/day | |||| ||||| | |||| ||||| | |||| ||||| | |||| ||||| | NR | NR |
Hydroxychloroquine, mg/day | || |||| | ||| ||| | ||| ||| | ||| ||| | NR | NR |
Oral corticosteroids for PsA at baseline, n (%) | 18 (14) | 20 (16) | 50 (20) | 49 (20) | || ||| | | ||| |
Oral corticosteroids, prednisone equivalent dose in mg/day, mean (SD) | 6.0 (1.9) | 6.4 (2.4) | 6.8 (2.5) | 7.8 (2.5) | NR | NR |
NSAIDs for PsA at baseline, n (%) | 71 (56) | 77 (61) | 165 (67) | 168 (68) | NR | NR |
Met early escape criteria at 16 weeks, n (%) | 4 (3) | 24 (19) | 13 (5) | 38 (15) | 39 (21)a | 45 (47)a |
Escape therapy: initiated or increased dose of permitted concomitant medications,b n (%) | | ||| | || |||| | | ||| | || ||| | | ||| | | ||| |
Patients with any joint procedure up to week 24,c n (%) | |||| | |||| | |||| | | ||||| | | ||| | | ||| |
Patients with any joint injection up to week 24, n (%) | | ||| | | ||| | |||| | | ||| | | ||| | | ||| |
DMARD = disease-modifying antirheumatic drug; FAS = full analysis set; GUSE = guselkumab; NR = not reported; NSAID = nonsteroidal anti-inflammatory drug; PBO = placebo; PsA = psoriatic arthritis; q8w = every 8 weeks; SD = standard deviation.
aIncludes 12 patients (6.3%) in the guselkumab group and 8 patients (8.3%) in the placebo group who were incorrectly assigned to early escape therapy without having met the early escape criteria.
bPermitted concomitant medications for PsA included oral corticosteroids (≤ 10 mg of prednisone per day or equivalent) and 1 of either methotrexate (≤ 25 mg/week), sulfasalazine (≤ 3 g/day), hydroxychloroquine (≤ 400 mg/day), or leflunomide (≤ 20 mg/day), or NSAIDs.
cIncluded joint replacement, arthroscopy (surgical or diagnostic), amputation, arthrocentesis, synovectomy, excision or resection, and radio synovectomy.
Source: Clinical Study Report for DISCOVER-1 (24 weeks);11 Clinical Study Report for DISCOVER-2 (24 weeks);12 Clinical Study Report for COSMOS (24 weeks).13
Efficacy
Only those efficacy outcomes and analyses of subgroups identified in the review protocol are reported. Refer to Appendix 3 for detailed efficacy data.
The systematic review protocol identified pain as an outcome of interest. None of the trials included pain as a secondary or exploratory outcome; however, patient-reported pain severity was included as part of the ACR and MDA response criteria and the SF-36.
Clinical Response in PsA Symptoms
In the DISCOVER-1 study, which enrolled both biologic-naive and biologic-experienced patients, 52.0% of patients in the group receiving guselkumab every 8 weeks achieved ACR 20 at 24 weeks compared with 22.2% of patients in the placebo group. The absolute difference was 29.8% (95% CI, 18.6% to 41.1%; P < 0.001) (Table 14) favouring guselkumab versus placebo. At week 24, 29.9% of patients versus 8.7% of patients in the guselkumab versus placebo groups, respectively, achieved ACR 50 (absolute difference = 21.4%; 95% CI, 12.1% to 30.7%; P < 0.001), and 11.8% versus 5.6% achieved ACR 70 (6.4%; 95% CI, −0.3% to 13.1%; P = 0.086). ACR 50 and ACR 70 outcomes were controlled for multiple testing based on the global testing procedure rather than the US-specific testing procedure. The US-specific procedure was preferred by Health Canada.
Among patients who were biologic-naive (in DISCOVER-2), 64.1% and 32.9% of patients achieved ACR 20 in the group receiving guselkumab every 8 weeks and the placebo group, respectively, with an absolute difference of 31.2% (95% CI, 22.9% to 39.5%; P < 0.001) (Table 14). The difference between guselkumab and placebo in the proportion of patients who achieved ACR 50 was 17.2% (95% CI, 10.0% to 24.4%), and the proportion of patients who achieved ACR 70 was 14.5% (95% CI, 9.1% to 19.9%). However, neither outcome was statistically tested according to the global testing procedure due to failure of a prior outcome in the hierarchy. ACR 50 and ACR 70 were not controlled for multiple testing in the US-specific procedure.
Among biologic-experienced patients in the COSMOS study, 44.4% and 19.8% achieved ACR 20 at week 24 in the guselkumab and placebo groups, respectively. The absolute difference between groups (24.6%) favoured guselkumab (95% CI, 14.1% to 35.2%; P < 0.001) (Table 14). The difference for guselkumab versus placebo for the proportion who achieved ACR 50 was 19.6% versus 5.2%; the absolute difference was 14.3% (95% CI, 7.2% to 21.4%; P < 0.001). ACR 70 response was achieved by 7.9% versus 1.0% of patients in the guselkumab versus placebo groups, with an absolute difference of 6.8% (95% CI, 2.6% to 11.1%). ACR 70 was not controlled for multiple testing and should be interpreted considering the inflated risk of type I error rate.
In the DISCOVER-1 and 2 studies, the results of the sensitivity analyses for ACR 20 were generally consistent with the results of the primary analysis. In the COSMOS study, the analysis that was based on observed case data regardless of meeting treatment failure criteria reported an absolute difference of 10.9% (95% CI, −1.3% to 23.2%), which the investigators stated was due to the doubled ACR 20 response rate in the placebo group, noting that almost half of patients in the placebo group received early escape therapies starting at week 16. Other sensitivity analyses (per-protocol population and analyses adjusting for errors in early escape assignment) showed results that were consistent with the primary analysis.
The proportion of patients who achieved MDA at week 24 was higher in the group receiving guselkumab every 8 weeks versus the placebo group in all 3 studies. However, this outcome was not controlled for multiple testing and should be interpreted with caution because of the potential for inflated type I error rate. The absolute differences between guselkumab and placebo were 11.9% (95% CI, 2.9% to 20.9%) for DISCOVER-1, 18.9% (95% CI, 12.8% to 25.0%) for DISCOVER-2, and 11.7% (95% CI, 5.6% to 17.7%) for the COSMOS study (Table 14).
Subgroup analyses of interest to this review for the DISCOVER studies are shown in figures 11 to 14 in Appendix 3. The odds ratios for ACR 20 response were generally consistent across subgroups based on prior use of TNF alpha inhibitors, non-biologic DMARDs, oral corticosteroids, or NSAIDs at baseline. The COSMOS study did not report data for any subgroups of interest.
Table 14: ACR and MDA Response at Week 24
Outcome | DISCOVER-1 (FAS) | DISCOVER-2 (FAS) | COSMOS (FAS) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
GUSE q8w N = 127 | PBO N = 126 | GUSE q8w N = 248 | PBO N = 246 | GUSE q8w N = 189 | PBO N = 96 | |||||
Number of patients contributing to the analysis | 127 | 126 | 248 | 246 | 189 | 96 | ||||
ACR 20 at week 24a | ||||||||||
ACR 20 response at week 24, n (%) | 66 (52.0) | 28 (22.2) | 159 (64.1) | 81 (32.9) | 84 (44.4) | 19 (19.8) | ||||
% difference vs. PBO (95% CI) | 29.8 (18.6 to 41.1) | Reference | 31.2 (22.9 to 39.5) | Reference | 24.6 (14.1 to 35.2) | Reference | ||||
P value | < 0.001 | Reference | < 0.001 | Reference | < 0.001 | Reference | ||||
ACR 50 at week 24a | ||||||||||
ACR 50 response at week 24, n (%) | 38 (29.9) | 11 (8.7) | 78 (31.5) | 35 (14.2) | 37 (19.6) | 5 (5.2) | ||||
% difference vs. PBO (95% CI) | 21.4 (12.1 to 30.7) | Reference | 17.2 (10.0 to 24.4) | Reference | 14.3 (7.2 to 21.4) | Reference | ||||
P value | < 0.001b | Reference | Not testedc | Reference | 0.001 | Reference | ||||
ACR 70 at week 24a | ||||||||||
ACR 70 response at week 24, n (%) | 15 (11.8) | 7 (5.6) | 46 (18.5) | 10 (4.1) | 15 (7.9) | 1 (1.0) | ||||
% difference vs. PBO (95% CI) | 6.4 (−0.3 to 13.1) | Reference | 14.5 (9.1 to 19.9) | Reference | 6.8 (2.6 to 11.1) | Reference | ||||
P value | 0.086b | Reference | Not testedc | Reference | 0.018d | Reference | ||||
MDA at week 24ae | ||||||||||
Patients with MDA at week 24 | 29 (22.8) | 14 (11.1) | 62 (25.0) | 15 (6.1) | 28 (14.8) | 3 (3.1) | ||||
% difference vs. PBO (95% CI) | 11.9 (2.9 to 20.9) | Reference | 18.9 (12.8 to 25.0) | Reference | 11.7 (5.6 to 17.7) | Reference | ||||
P value | 0.012d | Reference | < 0.001d | Reference | 0.003 d | Reference | ||||
ACR = American College of Rheumatology; ACR 20 = American College of Rheumatology 20% improvement; ACR 50 = American College of Rheumatology 50% improvement; ACR 70 = American College of Rheumatology 70% improvement; CI = confidence interval; FAS = full analysis set; GUSE = guselkumab; HAQ-DI = Health Assessment Questionnaire Disability Index; MDA = minimal disease activity; PBO = placebo; q8w = every 8 weeks; VAS = visual analogue scale.
aBased on composite estimand (either observed response data or nonresponse for patients who met the treatment failure criteria). Patients with missing data were imputed as nonresponders. P values were based on stratified CMH tests and 95% CIs were based on Wald statistic. The DISCOVER-1 study CMH test stratified patients by baseline use of non-biologic DMARDs (yes, no) and prior exposure to TNF alpha inhibitors (yes, no). The DISCOVER-2 study CMH test stratified patients by baseline use of non-biologic DMARDs (yes, no) and CRP before randomization (< 2 mg/dL vs. ≥ 2 mg/dL). The COSMOS study CMH test stratified patients by baseline use of non-biologic DMARDs (yes, no) and number of prior TNF alpha inhibitors (1 or 2).
bControlled for multiple testing based on the global testing procedure, but not the US-specific testing procedure. The US-specific procedure was preferred by Health Canada.
cOutcome was not formally tested for statistical significance due to failure of a prior outcome, according to the global testing procedure. This outcome was not controlled for multiple testing, according to the US-based testing procedure.
dThe P value has not been adjusted for multiple testing (i.e., the type I error rate has not been controlled).
eMDA is achieved if at least 5 of 7 criteria are met (tender joint count ≤ 1, swollen joint count ≤ 1, Psoriasis Activity and Severity Index ≤ 1, patient’s assessment of pain VAS ≤ 15, patient’s global assessment of disease activity VAS ≤ 20 [arthritis and psoriasis], HAQ-DI score ≤ 0.5, and tender entheseal points ≤ 1).
Source: Clinical Study Report for DISCOVER-1 (24 weeks);11 Clinical Study Report for DISCOVER-2 (24 weeks);12 Clinical Study Report for COSMOS (24 weeks).13
Additional data on the ACR 20 and ACR 50 response rates at week 16 are summarized in Appendix 3, Table 42. These findings were generally consistent with the ACR 20 results at week 24; however, the ability to draw statistical inferences from the data may be limited, whether because no statistical testing was performed (DISCOVER-2), there was no control for multiple testing (COSMOS), or control of the type I error rate was based on global rather than US-specific testing procedures (DISCOVER-1). For ACR 50, the DISCOVER-1 study failed to detect a statistically significant difference between guselkumab every 8 weeks and placebo at week 16 (based on global testing procedure). No statistical testing was performed in DISCOVER-2 (due to failure of a prior outcome). The response rates were nominally higher for the guselkumab versus placebo groups in the COSMOS study, but there was no control of the type I error rate.
Data on the changes from baseline in DAS 28 CRP scores are summarized in Appendix 3, Table 43. The point estimates for the LS mean differences ranged from −0.61 to −0.73, suggesting an improvement with guselkumab every 8 weeks versus placebo in all trials; however, in the COSMOS study, this outcome was not controlled for multiple testing, and in the DISCOVER studies, it was controlled for multiple testing based on the global testing procedure rather than the US-specific procedure.
Function or Disability
Changes from baseline to week 24 in the HAQ-DI favoured guselkumab every 8 weeks versus placebo in all trials. The LS mean differences reported were −0.25 (95% CI, −0.36 to −0.13; P < 0.001) in the DISCOVER-1 study, −0.24 (95% CI, −0.32 to −0.15; P < 0.001) in the DISCOVER-2 study, and −0.17 (95% CI, −0.28 to −0.06; P = 0.003) in the COSMOS study. Across the trials, only the group receiving guselkumab every 8 weeks in the DISCOVER-2 study achieved a change from baseline that exceeded the 0.35 MID cited in the study’s protocol.6,7,51
The proportion of HAQ-DI responders was reported as another secondary outcome in the DISCOVER trials (i.e., no control for type I error rate). Response was defined achieving a point improvement of greater than or equal to a 0.35 in the HAQ-DI score from baseline (among the subset of patients who had a HAQ-DI score ≥ 0.35 points at baseline). At 24 weeks in the DISCOVER-1 study, 50.9% of patients (57 of 112) in the guselkumab group and 29.1% of patients (32 of 110) in the placebo group achieved HAQ-DI responses (absolute difference = 21.8%; 95% CI, 9.3% to 34.2%). In DISCOVER-2, 50.0% of patients (114 of 228) and 31.4% of patients (74 of 236) in the guselkumab versus placebo groups, respectively, achieved HAQ-DI responses (absolute difference = 18.7%; 95% CI, 10.0% to 27.3%).
Table 15: Change From Baseline in HAQ-DI to Week 24
Outcome | DISCOVER-1 (FAS) | DISCOVER-2 (FAS) | COSMOS (FAS) | ||||
|---|---|---|---|---|---|---|---|
GUSE q8w N = 127 | PBO N = 126 | GUSE q8w N = 248 | PBO N = 246 | GUSE q8w N = 189 | PBO N = 96 | ||
Change from baseline to week 24 in HAQ-DI scorea | |||||||
Number of patients contributing to the analysis | 127 | 126 | 248 | 246 | 189 | 96 | |
Baseline, mean (SD) | 1.05 (0.57) | 1.11 (0.63) | 1.28 (0.63) | 1.29 (0.56) | 1.33 (0.60) | 1.22 (0.60) | |
Change from baseline, LS mean (95% CI) | −0.32 (−0.41 to −0.24) | −0.07 (−0.16 to 0.01) | −0.37 (−0.43 to −0.31) | −0.13 (−0.19 to −0.07) | −0.18 (−0.27 to −0.09) | −0.001 (−0.12 to 0.10) | |
LS mean difference (95% CI) vs. placebo | −0.25 (−0.36 to −0.13) | Reference | −0.24 (−0.32 to −0.15) | Reference | −0.17 (−0.28 to −0.06) | Reference | |
P value | < 0.001 | Reference | < 0.001 | Reference | 0.003 | Reference | |
ANCOVA = analysis of covariance; CI = confidence interval; FAS = full analysis set; GUSE = guselkumab; HAQ-DI = Health Assessment Questionnaire Disability Index; LS = least squares; MMRM = mixed-effects model for repeated measures; PBO = placebo; q8w = every 8 weeks; SD = standard deviation.
aChange from baseline was based on observed data or 0 (no improvement) if a patient met the treatment failure criteria before week 24 (composite estimand). The DISCOVER-1 and DISCOVER-2 analyses used an ANCOVA model with missing data imputed using multiple imputation, assuming data were missing at random. The COSMOS analysis used an MMRM under the missing-at-random assumption.
Source: Clinical Study Report for DISCOVER-1 (24 weeks);11 Clinical Study Report for DISCOVER-2 (24 weeks);12 Clinical Study Report for COSMOS (2 weeks).13
Health-Related Quality of Life
The change from baseline in the SF-36 PCS was a secondary outcome in all studies. Most treatment groups reported an improvement at week 24, with a difference that favoured guselkumab every 8 weeks versus placebo in all studies (Table 16). The LS mean difference was 4.1 (95% CI, 2.4 to 5.9; P < 0.001) in the DISCOVER-1 study, 4.0 (95% CI, 2.7 to 5.2; P = 0.011) in the DISCOVER-2 study, and 3.9 (95% CI, 2.5 to 5.4; P < 0.001) in the COSMOS study. The CSR defined an increase of at least 5 points as clinically meaningful, but a MID of 3.74 has also been reported in the literature.7,51
All trials failed to detect a statistically significant difference between guselkumab every 8 weeks and placebo in the change from baseline to week 24 in the SF-36 MCS (Table 16).
Table 16: Change From Baseline in the SF-36 PCS and MCS to Week 24
Outcome | DISCOVER-1 (FAS) | DISCOVER-2 (FAS) | COSMOS (FAS) | |||
|---|---|---|---|---|---|---|
GUSE q8w N = 127 | PBO N = 126 | GUSE q8w N = 248 | PBO N = 246 | GUSE q8w N = 189 | PBO N = 96 | |
Change from baseline to week 24 in SF-36 PCSa | ||||||
Number of patients contributing to the analysis | 127 | 126 | 248 | 246 | 188 | 96 |
Baseline, mean (SD) | 34.1 (7.6) | 33.8 (8.5) | 32.6 (7.9) | 32.4 (7.0) | 33.0 (7.0) | 33.9 (7.7) |
Change from baseline, LS mean (95% CI) | 6.1 (4.8 to 7.4) | 2.0 (0.7 to 3.2) | 7.4 (6.5 to 8.3) | 3.4 (2.5 to 4.3) | 3.5 (2.3 to 4.7) | −0.4 (−1.8 to 1.1) |
LS mean difference (95% CI) vs. placebo | 4.1 (2.4 to 5.9) | Reference | 4.0 (2.7 to 5.2) | Reference | 3.9 (2.5 to 5.4) | Reference |
P value | < 0.001 | Reference | 0.011b | Reference | < 0.001 | Reference |
Change from baseline to week 24 in SF-36 MCSa | ||||||
Number of patients contributing to the analysis | 127 | 126 | 248 | 246 | 188 | 96 |
Baseline, mean (SD) | 47.0 (11.1) | 48.7 (9.6) | 47.4 (10.8) | 47.2 (12.0) | 47.1 (12.1) | 46.1 (11.5) |
Change from baseline, LS mean (95% CI) | 3.2 (1.8 to 4.6) | 2.4 (0.9 to 3.8) | 4.2 (3.1 to 5.2) | 2.1 (1.1 to 3.2) | 2.1 (0.5 to 3.7) | 0.4 (−1.5 to 2.3) |
LS mean difference (95% CI) vs. placebo | 0.8 (−1.1 to 2.8) | Reference | 2.0 (0.6 to 3.5) | Reference | 1.7 (−0.1 to 3.6) | Reference |
P value | 0.398c | Reference | 0.072d | Reference | 0.07c | Reference |
ANCOVA = analysis of covariance; CI = confidence interval; FAS = full analysis set; GUSE = guselkumab; LS = least squares; MCS = Mental Component Summary; MMRM = mixed-effects model for repeated measures; PBO = placebo; PCS = Physical Component Summary; q8w = every 8 weeks; SD = standard deviation; SF-36 = Short Form (36) Health Survey.
aChange from baseline was based on observed data or 0 (no improvement) if a patient met the treatment failure criteria before week 24 (composite estimand). The DISCOVER-1 and DISCOVER-2 analyses used an ANCOVA model with missing data imputed using multiple imputation, assuming data were missing at random. The COSMOS analysis used an MMRM under the missing-at-random assumption.
bAccording to the US-based testing procedure, this outcome was statistically significant, with a P value of 0.011. According to the global testing procedure, this outcome was not formally tested for statistical significance due to failure of a prior outcome.
cP value has not been adjusted for multiple testing (i.e., the type I error rate has not been controlled).
dThis outcome was not formally tested for statistical significance due to failure of a prior outcome, according to the global testing procedure. According to the US-based testing procedure, this outcome was not statistically significant (P = 0.072).
Source: Clinical Study Report for DISCOVER-1 (24 weeks);11 Clinical Study Report for DISCOVER-2 (24 weeks);12 Clinical Study Report for COSMOS (24 weeks);13 Coates et al. (2022).14
Psoriasis Skin Disease
In all trials, psoriasis skin disease outcome measures were analyzed in the subgroup of patients who had psoriasis affecting greater than or equal to 3% of their BSA and an IGA score of greater than or equal to 2 at baseline (55% to 74% of patients per treatment group). Randomization was not stratified by the presence of skin disease at baseline.
The proportion of patients who achieved an IGA response at week 24 was higher in the group receiving guselkumab every 8 weeks than in the placebo group in the DISCOVER-1 study (57.3% versus 15.4%; absolute difference = 42.0% [95% CI, 28.9% to 55.1%; P < 0.001]) and the DISCOVER-2 study (70.5% versus 19.1%; absolute difference = 50.9% [95% CI, 42.2% to 59.7%; P < 0.001]). In the COSMOS study, 48.1% versus 9.4% of patients achieved an IGA response in the guselkumab and placebo groups; the absolute difference was 38.8% (95% CI, 27.3% to 50.4%). However, the P value was not adjusted to control for multiple testing; it should be interpreted with caution because of the potential for inflated type I error rate.
PASI 100 response at week 24 was a secondary outcome in the COSMOS study. In the group receiving guselkumab every 8 weeks, 30.8% of patients achieved a PASI 100 response compared with 3.8% of patients in the placebo group (absolute difference = 27.4%; 95% CI, 17.9% to 36.8%; P < 0.001]). The PASI 90 and PASI 75 results in the COSMOS study were nominally higher in the guselkumab versus placebo groups, but the type I error rate was not controlled for these outcomes. The PASI 100, PASI 90, and PASI 75 response rates in the DISCOVER trials were nominally higher for guselkumab versus placebo, but these outcomes were not part of the statistical testing procedure and should be interpreted with caution, considering the potential for inflated type I error rate (Table 17).
Table 17: IGA and PASI Response at Week 24
Outcome | DISCOVER-1 (FAS) | DISCOVER-2 (FAS) | COSMOS (FAS) | |||
|---|---|---|---|---|---|---|
GUSE q8w N = 127 | PBO N = 126 | GUSE q8w N = 248 | PBO N = 246 | GUSE q8w N = 189 | PBO N = 96 | |
Number of patients contributing to analysis, N (%) | 82 (65) | 78 (62) | 176 (71) | 183 (74) | 133 (70) | 53 (55) |
IGA response at week 24a | ||||||
IGA response at week 24, n (%) | 47 (57.3) | 12 (15.4) | 124 (70.5) | 35 (19.1) | 64 (48.1) | 5 (9.4) |
% difference vs. PBO (95% CI) | 42.0 (28.9 to 55.1) | Reference | 50.9 (42.2 to 59.7) | Reference | 38.8 (27.3 to 50.4) | Reference |
P value | < 0.001 | Reference | < 0.001 | Reference | < 0.001b | Reference |
PASI 100 response at week 24a | ||||||
PASI 100 response at week 24, n (%) | 21 (25.6) | 5 (6.4) | 80 (45.5) | 5 (2.7) | 41 (30.8) | 2 (3.8) |
% difference vs. PBO (95% CI) | 19.9 (9.6 to 30.2) | Reference | 42.4 (34.8 to 50.1) | Reference | 27.4 (17.9 to 36.8) | Reference |
P value | < 0.001b | Reference | < 0.001b | Reference | < 0.001 | Reference |
PASI 90 response at week 24a | ||||||
PASI 90 response at week 24, n (%) | 41 (50.0) | 9 (11.5) | 121 (68.8) | 18 (9.8) | 68 (51.1) | 4 (7.5) |
% difference vs. PBO (95% CI) | 38.6 (25.8 to 51.4) | Reference | 58.6 (50.6 to 66.6) | Reference | 43.7 (32.7 to 54.7) | Reference |
P value | < 0.001b | Reference | < 0.001b | Reference | < 0.001b | Reference |
PASI 75 response at week 24a | ||||||
PASI 75 response at week 24, n (%) | 62 (75.8) | 11 (14.1) | 139 (79.0) | 42 (23.0) | 79 (59.4) | 5 (9.4) |
% difference vs. PBO (95% CI) | 61.7 (49.8 to 73.7) | Reference | 55.7 (47.2 to 64.2) | Reference | 49.6 (38.3 to 60.9) | Reference |
P value | < 0.001b | Reference | < 0.001b | Reference | < 0.001b | Reference |
CI = confidence interval; CMH = Cochran-Mantel-Haenszel; DMARD = disease-modifying antirheumatic drug; FAS = full analysis set; GUSE = guselkumab; IGA = Investigator’s Global Assessment of Psoriasis; PASI = Psoriasis Area and Severity Index; PASI 75 = 75% improvement from baseline in Psoriasis Area and Severity Index score; PASI 90 = 90% improvement from baseline in Psoriasis Area and Severity Index score; PASI 100 = 100% improvement from baseline in Psoriasis Area and Severity Index score; PBO = placebo; q8w = every 8 weeks; TNF = tumour necrosis factor.
aBased on composite estimand (either observed response data or nonresponse for patients who met the treatment failure criteria). Patients with missing data were imputed as nonresponders. P values were based on stratified CMH tests and 95% CIs were based on the Wald statistic. The DISCOVER-1 study CMH test stratified patients by baseline use of non-biologic DMARDs (yes, no) and prior exposure to TNF alpha inhibitors (yes, no). The DISCOVER-2 study CMH test stratified patients by baseline use of non-biologic DMARDs (yes, no) and CRP before randomization (< 2 mg/dL vs. ≥ 2 mg/dL). The COSMOS study CMH test stratified patients by baseline use of non-biologic DMARDs (yes, no) and number of prior TNF alpha inhibitors (1 or 2).
bThe P value has not been adjusted for multiple testing (i.e., the type I error rate has not been controlled).
Source: Clinical Study Report for DISCOVER-1 (24 weeks);11 Clinical Study Report for DISCOVER-2 (24 weeks);12 Clinical Study Report for COSMOS (24 weeks).13
Other Musculoskeletal Disease
Outcomes related to enthesitis and dactylitis were analyzed for patients reporting these symptoms at baseline. Approximately 2-thirds of patients reported enthesitis at baseline (57% to 72%), whereas 35% to 45% of patients reported dactylitis across treatment groups in the 3 trials. Randomization was not stratified by the presence of enthesitis or dactylitis at baseline. These outcomes were not controlled for multiple testing; thus, P values of less than 0.05 should be interpreted with caution, considering the potential for inflated type I error rate. The results for these outcomes in each trial are shown in Table 18.
The DISCOVER-2 study planned a prior pooled analysis of the resolution of enthesitis and dactylitis end points using combined data from the DISCOVER-1 and DISCOVER-2 trials. Based on the pooled analysis, 49.6% and 29.4% of patients in the group receiving guselkumab every 8 weeks and the placebo group had resolution of enthesitis at week 24, with a between-group difference of 20.1% (95% CI, 11.8% to 28.5%; P = 0.03 [based on the US-specific testing procedure]). The proportion of patients whose dactylitis resolved at week 24 was 59.4% versus 42.2% in the guselkumab versus placebo group; the between-group difference was 18.0% (95% CI, 7.4% to 28.6%; P = 0.03 [based on the US-specific testing procedure]). In accordance with the global testing procedure, these outcomes were not formally tested due to the failure of a prior outcome in the testing hierarchy.
Table 18: Enthesitis and Dactylitis Outcomes at 24 Weeks
Outcome | DISCOVER-1 (FAS) | DISCOVER-2 (FAS) | COSMOS (FAS) | |||
|---|---|---|---|---|---|---|
GUSE q8w N = 127 | PBO N = 126 | GUSE q8w N = 248 | PBO N = 246 | GUSE q8w N = 189 | PBO N = 96 | |
Resolution of enthesitis at week 24a | ||||||
Number of patients contributing to the analysis, N (%) | 72 (57) | 77 (61) | 158 (64) | 178 (72) | 126 (67) | 64 (67) |
Resolution of enthesitis, n (%) | 29 (40.3) | 21 (27.3) | 85 (53.8) | 54 (30.3) | 50 (39.7) | 12 (18.8) |
% difference vs. PBO (95% CI) | 13.0 (−1.6 to 27.5) | Reference | 23.3 (13.1 to 33.5) | Reference | 21.6 (8.8 to 34.4) | Reference |
P value | 0.094b | Reference | < 0.001b | Reference | 0.003b | Reference |
Resolution of dactylitis at week 24a | ||||||
Number of patients contributing to the analysis, N (%) | 49 (39) | 55 (44) | 111 (45) | 99 (40) | 67 (35) | 36 (38) |
Resolution of dactylitis, n (%) | 32 (65.3) | 27 (49.1) | 63 (56.8) | 38 (38.4) | 30 (44.8) | 9 (25.0) |
% difference vs. PBO (95% CI) | 16.6 (−1.5 to 34.8) | Reference | 18.7 (5.7 to 31.7) | Reference | 19.9 (2.7 to 37.1) | Reference |
P value | 0.088b | Reference | 0.007b | Reference | 0.04b | Reference |
Change from baseline to week 24 in enthesitis score (based on LEI)c | ||||||
Number of patients contributing to the analysis, N (%) | 72 (57%) | 77 (61) | 158 (64) | 178 (72) | 126 (67) | 64 (67) |
Baseline, mean (SD) | 2.7 (1.6) | 2.8 (1.6) | 2.6 (1.5) | 2.8 (1.6) | 2.9 (1.5) | 2.7 (1.5) |
Change from baseline, LS mean (95% CI) | −1.4 (−1.7 to −1.0) | −1.0 (−1.4 to −0.7) | −1.6 (−1.8 to −1.4) | −1.0 (−1.3 to −0.8) | −1.4 (−1.7 to −1.0) | −0.7 (−1.1 to −0.2) |
LS mean difference (95% CI) vs. placebo | −0.3 (−0.8 to 0.2) | Reference | −0.6 (−0.9 to −0.3) | Reference | −0.7 (−1.1 to −0.2) | Reference |
P value | 0.185b | Reference | < 0.001b | Reference | 0.003b | Reference |
Change from baseline to week 24 in dactylitis scorec | ||||||
Number of patients contributing to the analysis, N (%) | 49 (39) | 55 (44) | 111 (45) | 99 (40) | 67 (35) | 36 (38) |
Baseline, mean (SD) | 8.2 (10.1) | 6.6 (7.4) | 8.0 (9.6) | 8.4 (9.3) | 6.7 (6.5) | 7.4 (8.3) |
Change from baseline, LS mean (95% CI) | −6.1 (−7.8 to −4.4) | −4.3 (−6.0 to −2.6) | −6.0 (−6.8 to −5.1) | −4.0 (−5.0 to −3.1) | −2.5 (−3.7 to −1.3) | −1.3 (−2.7 to 0.1) |
LS mean difference (95% CI) vs. placebo | −1.8 (−4.1 to 0.5) | Reference | −1.9 (−3.2 to −0.7) | Reference | −1.2 (−2.6 to 0.2) | Reference |
P value | 0.12 b | Reference | 0.002 b | Reference | 0.09 b | Reference |
ANCOVA = analysis of covariance; CI = confidence interval; CMH = Cochran-Mantel-Haenszel; CRP = C-reactive protein; DMARD = disease-modifying antirheumatic drug; FAS = full analysis set; GUSE = guselkumab; LEI = Leeds Enthesitis Index; LS = least squares; MMRM = mixed-effects model for repeated measures; PBO = placebo; q8w = every 8 weeks; SD = standard deviation; TNF = tumour necrosis factor.
Note: LEI is the total score based on 6 enthesitis sites with 1 point per digit (range = 0 to 6). Dactylitis was scored on a scale of 0 to 60, with a maximum of 3 points per digit. For both scales, a negative change indicates improvement. Resolution of enthesitis or dactylitis was defined as an LEI score or dactylitis score of 0 at 24 weeks among patients with scores greater than or equal to 0 at baseline.
aBased on composite estimand (either observed response data or nonresponse for patients who met the treatment failure criteria). Patients with missing data were imputed as nonresponders. P values were based on stratified CMH tests and 95% CIs were based on the Wald statistic. The DISCOVER-1 study CMH test stratified patients by baseline use of non-biologic DMARDs (yes, no) and prior exposure to TNF alpha inhibitors (yes, no). The DISCOVER-2 study CMH test stratified patients by baseline use of non-biologic DMARDs (yes, no) and CRP before randomization (< 2 mg/dL vs. ≥ 2 mg/dL). The COSMOS study CMH test stratified patients by baseline use of non-biologic DMARDs (yes, no) and number of prior TNF alpha inhibitors (1 or 2).
bThe P value has not been adjusted for multiple testing (i.e., the type I error rate has not been controlled).
cChange from baseline was based on observed data or 0 (no improvement) if a patient met the treatment failure criteria before week 24 (composite estimand). The DISCOVER-1 and DISCOVER-2 analyses used an ANCOVA model with missing data imputed using multiple imputation, assuming data were missing at random. The COSMOS study used an MMRM with a missing-at-random assumption.
Source: Clinical Study Report for DISCOVER-1 (24 weeks);11 Clinical Study Report for DISCOVER-2 (24 weeks);12 Clinical Study Report for COSMOS (24 weeks).13
The change in the BASDAI score was analyzed for the subgroup of patients with spondylitis and peripheral joint PsA as their primary presentation at baseline (18% to 37% of patients per treatment group across the studies). Randomization was not stratified by the presence of axial disease at baseline. The sponsor stated that a 50% improvement in the BASDAI represents a clinically important change.58
Among biologic-naive patients (DISCOVER-2), 39% and 22% in the guselkumab and placebo groups, respectively, achieved at least a 50% improvement in the BASDAI score at week 24 (absolute difference = 15%) (95% CI, 0.4% to 30%; not controlled for type I error rate) (Table 19). The proportion of patients with an improvement of greater than or equal to 50% was the same in both treatment groups (19%) among patients who were biologic-experienced in the COSMOS study. In the mixed population enrolled in the DISCOVER-1 study, 42% and 13% of patients in the guselkumab and placebo groups achieved at least a 50% improvement, respectively, with an absolute difference of 24% (95% CI, −1% to 49%).
Table 19: Improvement in BASDAI Score at Week 24
Outcome | DISCOVER-1 (FAS) | DISCOVER-2 (FAS) | COSMOS (FAS) | |||
|---|---|---|---|---|---|---|
GUSE q8w N = 127 | PBO N = 126 | GUSE q8w N = 248 | PBO N = 246 | GUSE q8w N = 189 | PBO N = 96 | |
Proportion of patients with ≥ 50% improvement in BASDAI score at week 24a | ||||||
Number of patients contributing to the analysis, N (%) | 24 (19) | 23 (18) | 67 (27) | 92 (37) | 47 (25) | 26 (27) |
≥ 50% response at week 24, n (%) | 10 (41.7) | 3 (13.0) | 26 (38.8) | 20 (21.7) | 9 (19.1) | 5 (19.2) |
% difference vs. PBO (95% CI) | 24.1 (−1.1 to 49.2) | Reference | 15.0 (0.4 to 29.6) | Reference | NE | Reference |
P value | 0.082b | Reference | 0.048b | Reference | 1.00 | Reference |
BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; CI = confidence interval; CMH = Cochran-Mantel-Haenszel; DMARD = disease-modifying antirheumatic drug; FAS = full analysis set; GUSE = guselkumab; NE = not estimable; PBO = placebo; q8w = every 8 weeks; TNF = tumour necrosis factor.
aBased on composite estimand (either observed response data or nonresponse for patients who met the treatment failure criteria). Patients with missing data were imputed as nonresponders. P values were based on stratified CMH tests and 95% CIs were based on Wald statistic. The DISCOVER-1 study CMH test stratified patients by baseline use of non-biologic DMARDs (yes, no) and prior exposure to TNF alpha inhibitors (yes, no). The DISCOVER-2 study CMH test stratified patients by baseline use of non-biologic DMARDs (yes, no) and CRP before randomization (< 2 mg/dL vs. ≥ 2 mg/dL). The COSMOS study CMH test stratified patients by baseline use of non-biologic DMARDs (yes, no) and number of prior TNF alpha inhibitors (1 or 2).
bThe P value has not been adjusted for multiple testing (i.e., the type I error rate has not been controlled).
Source: Clinical Study Report for DISCOVER-1 (24 weeks);11 Clinical Study Report for DISCOVER-2 (24 weeks);12 Clinical Study Report for COSMOS (24 weeks).13
PsA Symptoms
FACIT-Fatigue scores range from 0 to 52, with higher scores indicating less fatigue. This outcome was not controlled for multiple testing; thus, P values of less than 0.05 should be interpreted with caution, considering the potential for inflated type I error rate. MIDs of 3.1 to 4 points have been reported in the literature.8,9
The FACIT-Fatigue scores suggest improvement in fatigue, with point estimates for the within-group change from baseline to week 24 that exceeded the MID for all groups receiving guselkumab every 8 weeks and for the placebo group in the DISCOVER-2 study (Table 20). The DISCOVER-1 study reported an LS mean difference of 3.4 points (95% CI, 1.4 to 5.4); the DISCOVER-2 study reported an LS mean difference of 4.0 (95% CI, 2.5 to 5.5); and the COSMOS study reported an LS mean difference of 3.6 (95% CI, 1.7 to 5.4) for guselkumab every 8 weeks versus placebo. In all studies, the between-group difference exceeded the MID of 3.1, while only the DISCOVER-2 study met the 4-point threshold.
Radiologic Outcomes
The change from baseline to week 24 in the modified vdH-S score was a major secondary outcome in the DISCOVER-2 study. At 24 weeks, the LS mean difference observed for guselkumab versus placebo was −0.43 (95% CI, −0.90 to 0.03) (P = 0.068) (Table 21).
The proportions of patients with no progression (defined as a change from baseline that was less than or equal to the smallest detectable change for the modified vdH-S total score) were 89.0% in the group receiving guselkumab every 8 weeks and 84.0% in the placebo group (absolute difference = 5.0%; 95% CI, –1.0% to 10.9%).
Table 20: Change From Baseline in FACIT-Fatigue Score to Week 24
Outcome | DISCOVER-1 (FAS) | DISCOVER-2 (FAS) | COSMOS (FAS) | |||
|---|---|---|---|---|---|---|
GUSE q8w N = 127 | PBO N = 126 | GUSE q8w N = 248 | PBO N = 246 | GUSE q8w N = 189 | PBO N = 96 | |
Change from baseline to week 24 in FACIT-Fatigue scorea | ||||||
Number of patients contributing to the analysis | 127 | 126 | 248 | 246 | 189 | 96 |
Baseline, mean (SD) | 29.5 (11.3) | 30.2 (9.9) | 29.3 (9.9) | 29.1 (9.5) | 29.2 (11.3) | 29.2 (10.6) |
Change from baseline, LS mean (95% CI) | 5.6 (4.2 to 7.0) | 2.2 (0.8 to 3.6) | 7.6 (6.5 to 8.6) | 3.6 (2.5 to 4.6) | 4.6 (3.1 to 6.1) | 1.1 (−0.8 to 2.9) |
LS mean difference (95% CI) vs. placebo | 3.4 (1.4 to 5.4) | Reference | 4.0 (2.5 to 5.5) | Reference | 3.6 (1.7 to 5.4) | Reference |
P value | < 0.001b | Reference | < 0.001b | Reference | < 0.001b | Reference |
CI = confidence interval; FACIT-Fatigue = Functional Assessment of Chronic Illness Therapy – Fatigue Scale; FAS = full analysis set; GUSE = guselkumab; LS = least squares; MMRM = mixed-effects model for repeated measures; PBO = placebo; q8w = every 8 weeks; SD = standard deviation.
aChange from baseline based on observed data or 0 (no improvement) if a patient met the treatment failure criteria before week 24 (composite estimand). Analyzed using an MMRM under a missing-at-random assumption.
bThe P value has not been adjusted for multiple testing (i.e., the type I error rate has not been controlled).
Source: Clinical Study Report for DISCOVER-1 (24 weeks);11 Clinical Study Report for DISCOVER-2 (24 weeks);12 Clinical Study Report for COSMOS (24 weeks);13 Coates et al. (2022).14
Table 21: Change From Baseline in Modified vdH-S of PsA to 24 Weeks
Outcome | DISCOVER-2 (FAS) | |
|---|---|---|
GUSE q8w N = 248 | PBO N = 246 | |
Change from baseline to week 24 in modified vdH-S scorea | ||
Number of patients contributing to the analysis | 248 | 246 |
Baseline, mean (SD) | 23.04 (37.75) | 23.75 (37.79) |
Change from baseline, LS mean (95% CI) | 0.52 (0.18 to 0.86) | 0.95 (0.61 to 1.29) |
LS mean difference (95% CI) vs. placebo | −0.43 (−0.90 to 0.03) | Reference |
P value | 0.068 | Reference |
ANCOVA = analysis of covariance; CI = confidence interval; FAS = full analysis set; GUSE = guselkumab; LS = least squares; PBO = placebo; q8w = every 8 weeks SD = standard deviation; vdH-S = van der Heijde-Sharp.
aChange from baseline is based on observed data regardless of meeting the treatment failure criteria before week 24 (treatment policy estimand). Analyzed using ANCOVA model with missing data imputed using multiple imputation and assuming data were missing at random.
Source: Clinical Study Report for DISCOVER-2 (24 weeks).12
Harms
Only those harms identified in the review protocol are reported in this section. Refer to Table 22 for detailed harms data.
Adverse Events
The frequency of adverse events was generally similar between groups, with 42% to 54% of patients in the groups receiving guselkumab every 8 weeks and 41% to 60% of patients in the placebo groups reporting 1 or more adverse events during the 24-week treatment period (Table 22). Nasopharyngitis (4% to 13%), upper respiratory tract infection (2% to 5%), and increased alanine aminotransferase (2% to 6%) were the most commonly reported adverse events in the guselkumab groups. The corresponding proportions of patients experiencing these adverse events in the placebo groups were 4% to 6% for nasopharyngitis, 3% to 7% for upper respiratory tract infections, and 3% to 6% for increased alanine aminotransferase.
In the COSMOS study, adverse event data were reported separately for 45 patients who switched from placebo to guselkumab every 8 weeks before week 24. Six patients (13%) experienced an adverse event, including 1 patient with a serious adverse event (a buttock injury) and 2 patients with infections (4%).
Serious Adverse Events
The frequency of serious adverse events ranged from 1% to 4% in the groups receiving guselkumab every 8 weeks and from 3% to 4% in the placebo groups (Table 22). No specific serious adverse events were reported in more than 1 patient per treatment group (Table 23). In the DISCOVER-1 study, only 1 of the serious adverse events (plasma cell myeloma in the guselkumab group) resulted in treatment discontinuation. One patient in the guselkumab group of the COSMOS study experienced increased alanine aminotransferase that was categorized as a serious adverse event. This patient was subsequently diagnosed with autoimmune hepatitis and the study drug was stopped.
Treatment Discontinuation Due to Adverse Events
Adverse events that resulted in treatment discontinuation were generally low (1% to 3%) and similar between groups (Table 22). Other than 2 patients in the placebo group of the DISCOVER-1 study, who stopped treatment due to psoriasis, all other events were reported in 1 patient (Table 23).
Mortality
In the DISCOVER-1 study, 1 patient in the placebo group died due to cardiac failure. No other deaths were reported in the guselkumab groups or placebo groups during the first 24 weeks of the 3 studies.
Notable Harms
Generally, the frequency of infections was similar in the groups receiving guselkumab every 8 weeks (16% to 26%) and the placebo groups (18% to 25%) across trials (Table 22). In the DISCOVER-1 study, no patients in the guselkumab group reported a serious infection. Two patients in the placebo group experienced a serious infection, which included a serious upper respiratory tract infection and an abscessed limb. In the DISCOVER-2 study, 1 patient in each group reported a serious infection, including pyrexia in the guselkumab group and post-procedural fistula in the placebo group. In the COSMOS study, 1 patient in the guselkumab group reported a serious community-acquired pneumonia that required hospitalization. No serious infections were reported among patients who received placebo. No cases of tuberculosis or opportunistic infections were reported in the first 24 weeks of all 3 trials.
The other notable harms identified in the systematic review protocol were infrequently reported (i.e., injection-site reactions, anaphylactic adverse events, and liver-related adverse events).
Table 22: Summary of Harms Up to 24 Weeks (Safety Set)
Adverse event | DISCOVER-1 GUSE q8w N = 127 | DISCOVER-1 PBO N = 126 | DISCOVER-2 GUSE q8w N = 248 | DISCOVER-2 PBO N = 246 | COSMOS GUSE q8w N = 189 | COSMOS PBO N = 96 |
|---|---|---|---|---|---|---|
Patients with ≥ 1 adverse event | ||||||
n (%) | 68 (54) | 75 (60) | 114 (46) | 100 (41) | 80 (42) | 46 (48) |
Most common events,a n (%) | ||||||
Nasopharyngitis | 16 (13) | 8 (6) | 10 (4) | 9 (4) | 9 (5) | 5 (6) |
URTI | 7 (5) | 9 (7) | 6 (2) | 8 (3) | 6 (4) | 3 (3) |
Enthesopathy | 6 (5) | 6 (5) | 5 (2) | 7 (3) | NR | NR |
ALT increased | 3 (2) | 8 (6) | 15 (6) | 11 (5) | 4 (2) | 3 (3) |
AST increased | 3 (2) | 9 (7) | 14 (6) | 6 (2) | 1 (0.6) | 2 (2) |
Patients with ≥ 1 SAE | ||||||
n (%) | 4 (3) | 5 (4) | 3 (1) | 7 (3) | 7 (4) | 3 (3) |
Patients who stopped treatment due to adverse events | ||||||
n (%) | 3 (2) | 3 (2) | 2 (1) | 4 (2) | 5 (3) | 2 (2) |
Deaths | ||||||
n (%) | 0 | 1 (0.8) | 0 | 0 | 0 | 0 |
Notable harms | ||||||
Infections, n (%) | 33 (26) | 32 (25) | 40 (16) | 45 (18) | 40 (21) | 19 (20) |
Serious infections, n (%) | 0 | 2 (1.6) | 1 (0.4) | 1 (0.4) | 1 (0.5) | 0 |
Opportunistic infections | 0 | 0 | 0 | 0 | 0 | 0 |
Injection-site reactions, n (%) | 2 (2) | 0 | 3 (1) | 1 (0.4) | 4 (2) | 1 (1) |
Anaphylactic adverse events, n (%) | 0 | 0 | 0 | 0 | 0 | 0 |
Increase in ALT, grade 3 or 4, n (%) | 0 | 1 (1) | 2 (1) | 3 (1) | NR | NR |
Increase in AST, grade 3 or 4, n (%) | 1 (1) | 2 (2) | 1 (0.4) | 2 (1) | NR | NR |
Hepatobiliary disorders SOC, n (%) | 1 (1) | 1 (1) | 2 (1) | 3 (1) | 0 | 1 (1) |
ALT = alanine aminotransferase; AST = aspartate aminotransferase; GUSE = guselkumab; NR = not reported; PBO = placebo; q8w = every 8 weeks; SAE = serious adverse event; SOC = system organ class; URTI = upper respiratory tract infection.
Note: The data reported for the placebo group include patients who received only placebo. For any patients on placebo who received guselkumab by mistake (DISCOVER-1) or who received escape therapy with guselkumab (COSMOS), the data excluded any events that occurred after the patients received the active drug.
aFrequency greater than 5%.
Source: Clinical Study Report for DISCOVER-1 (24 weeks);11 Clinical Study Report for DISCOVER-2 (24 weeks);12 Clinical Study Report for COSMOS (24 weeks).13
Table 23: Description of SAEs or Adverse Events Leading to Treatment Discontinuation Up to 24 Weeks (Safety Set)
Adverse event type | DISCOVER-1 GUSE q8w N = 127 | DISCOVER-1 PBO N = 126 | DISCOVER-2 GUSE q8w N = 248 | DISCOVER-2 PBO N = 246 | COSMOS GUSE q8w N = 189 | COSMOS PBO N = 96 |
|---|---|---|---|---|---|---|
SAEsa | Arrhythmia, ileus, plasma cell myeloma, cervical dysplasia | Cardiac failure, pain, abscessed limb, URTI, COPD | Ankle fracture, coronary artery disease, pyrexia | Fistula, UA, IBD, drug-induced liver injury, obesity, clear cell renal cell carcinoma, tubulointerstitial nephritis | Pneumonia, ALT increase, depression, conversion disorder, prostate cancer, lumbosacral radiculopathy, intervertebral disc protrusion | Intervertebral disc protrusion, umbilical hernia, vomiting |
Adverse events leading to treatment discontinuationa | Bronchitis, psoriatic arthropathy, plasma cell myeloma | Cardiac failure, psoriasis (2 patients) | Rash, malignant melanoma | Drug-induced liver injury, clear cell renal cell carcinoma, IBD, tubulointerstitial nephritis | Psoriasis, urticaria, influenza-like illness, ALT increase, psoriatic arthropathy | Arthralgia, vomiting |
ALT = alanine aminotransferase; COPD = chronic obstructive pulmonary disease; GUSE = guselkumab; IBD = inflammatory bowel disease; PBO = placebo; q8w = every 8 weeks; SAE = serious adverse event; UA = unstable angina; URTI = upper respiratory tract infection.
aUnless otherwise stated, each event described was reported by a single patient.
Source: Clinical Study Report for DISCOVER-1 (24 weeks);11 Clinical Study Report for DISCOVER-2 (24 weeks);12 Clinical Study Report for COSMOS (24 weeks).13
Critical Appraisal
Internal Validity
The risk of bias related to randomization and treatment allocation concealment was rated as low for all studies, and, in general, the patient characteristics and co-interventions appeared to be balanced between groups at baseline. However, some differences were noted in the proportion of female patients and patients with psoriasis at baseline in the COSMOS study and in the proportion of patients with enthesitis at baseline in the DISCOVER-2 study. No issues were identified in the methods used to randomize patients and conceal allocation. The trials were double blind and took steps to maintain blinding of patients and investigators. In the DISCOVER studies, where the frequency of dosing varied across treatment groups (i.e., every 4 weeks or every 8 weeks), patients in the group receiving the study drug every 8 weeks also received placebo injections at alternate visits to match the every-4-weeks dosing schedule of the other guselkumab group. In all studies, an independent joint assessor not otherwise involved in the conduct of the trial evaluated patients’ joints for tenderness and swelling and the presence of enthesitis and dactylitis. Central raters assessed radiographs in DISCOVER-2. Therefore, the risk of bias in the measurement of the outcomes was low for all trials. In general, the type and frequency of adverse events were similar across groups, so there was low risk that adverse events led to substantial unblinding.
In all trials, the primary and other dichotomous end points were analyzed using a CMH test that was stratified by randomization stratification factors, with missing data imputed as nonresponders. The DISCOVER trials used an adjusted ANCOVA model, while the COSMOS study used an unadjusted MMRM model to analyze continuous outcomes. Missing data were imputed under the missing-at-random assumption, which may not hold true; however, this was not thought to be a major source of bias, given the low frequency of withdrawals and the use of the composite estimand to conduct the analyses. The efficacy analyses were based on the FAS (randomized and treated), which is not a true intention-to-treat population; however, only 1 patient in the DISCOVER-2 study was excluded. As a result, the analyses were appropriate for estimating the effect of assignment to the intervention.
All efficacy analyses were based on the composite estimand, where any patients who met the treatment failure criteria were considered nonresponders for binary end points, or to have experienced no change from baseline for continuous measures (e.g., HAQ-DI). Based on the composite estimand, patients who discontinued the study or study drug, initiated or increased the dose of cDMARDs or corticosteroids, initiated prohibited medication, or, in the COSMOS study, met early escape criteria, were considered treatment failures. This estimand, which considers any treatment failure end points to be unfavourable outcomes, may be considered a more conservative estimate of treatment effects, and was the estimand preferred by Health Canada. Of note, the COSMOS study incorrectly assigned 20 patients to early escape even though they had not met the escape criteria. As a result, 8 patients in the placebo group (8.3%) were switched to guselkumab treatment at week 16, and 12 patients (6.3%) in the guselkumab group were analyzed as nonresponders in the primary analysis. Although the sponsor conducted sensitivity analyses to explore the impact of this error, these analyses cannot fully address the potential bias.
The type I error rate was controlled for the primary and selected secondary outcomes in all studies. Two different testing strategies were used in the DISCOVER trials to meet US and other regulators’ requirements. The Health Canada reviewer’s report indicated that the US-based strategy was preferred.40 Several outcomes of interest to this review were tested, and nominal P values reported (e.g., MDA, ACR 70, PASI response, resolution of enthesitis and dactylitis, FACIT-Fatigue); however, any results with a P of less than 0.05 should be interpreted with caution, considering the potentially inflated type I error rate. Although the subgroup analyses were pre-specified, there is no evidence that the studies were powered to detect subgroup differences, and the 95% CIs for some subgroups were imprecise.
Of note, skin-related end points — as well as enthesitis, dactylitis, and axial disease outcome measures — were tested in subgroups of patients affected with these conditions at baseline. Randomization was not stratified for the presence of these conditions; thus, the interpretation of the results for these outcomes should consider the possibility of imbalances in baseline demographic and disease characteristics between treatment groups in these subpopulations. According to the expert consulted by CADTH, the DAS 28 is not relevant for PsA because it assesses only 28 joints and excludes the lower extremities (which may be important in PsA).48 Moreover, CADTH found limited data on the validity — and no data on the reliability and responsiveness — of the DAS 28 in patients with PsA. Thus, although this end point was a major secondary outcome in the DISCOVER trials, it should be interpreted with caution. Based on CADTH’s review, there was also limited evidence to support the validity or reliability of the BASDAI and the enthesitis and dactylitis scales used in the clinical trials. In addition, the MID for the change in vdH-S scores was unknown.
Although the trials were 48 weeks to 100 weeks in duration, the comparative period was limited to 24 weeks for this chronic condition. For outcomes such as radiologic changes, this duration of treatment may have been insufficient to detect the impact of guselkumab every 8 weeks. Moreover, none of the trials included an active control group (other than the group receiving guselkumab every 4 weeks, which is not a Health Canada–approved dosage); thus, direct evidence comparing guselkumab every 8 weeks to other biologics available in Canada is not available.
The primary outcome was ACR 20 response, but according to the clinical expert, this represents the minimum level of improvement that may be relevant to patients. In practice, the goal of therapy is to achieve higher levels of response, such as MDA or low disease activity as measured by the DAPSA. The COSMOS study included ACR 50 in its statistical testing procedures to control the type I error rate, but according to the US-based procedure in the DISCOVER trials, ACR 50 and ACR 70 were not controlled for multiple testing. The MDA end point was not controlled for multiple testing in any of the trials, which limits the ability to draw statistical inference from these data.
External Validity
The trials enrolled adults who, on average, were in their mid to late forties and predominantly White. There were a roughly equal number of male and female patients enrolled. Although patients with only 3 or 5 tender or swollen joints could enrol, the mean number of tender joints was about 20 and the mean number of swollen joints was about 11. The mean PASI score was generally less than 10, which the expert stated was consistent with scores seen in rheumatology clinics, given that most patients had received DMARDs (which may help to control psoriasis). Approximately 8% of patients in the trials had not previously received a cDMARD before being enrolled. Of the patients screened, 13%, 36%, and 39% were excluded. However, based on reporting in the CSRs, there was limited information about the characteristics of these patients and the reasons for their screening failures. Most of the trials were conducted in Europe, with some sites in Asia, the US, Australia, and Israel. Only the DISCOVER-1 study included sites in Canada, which enrolled 15 patients (4%). Based on the inclusion and exclusion criteria and the characteristics of patients, the clinical expert did not identify any substantial limits to the generalizability of the results of the trials.
The guselkumab-every-8-weeks dosing regime used in the trials was consistent with the Health Canada–recommended dose, and the clinical expert stated that concomitant utilization of cDMARDs was similar to what may be expected in practice. However, the clinical expert also noted that the use of oral corticosteroids in the DISCOVER trials (14% to 20%) was higher than would be expected in Canada, where chronic corticosteroid use is generally avoided. The trials excluded patients who had previously been treated with biologics other than TNF inhibitors; thus, the efficacy in patients with intolerance or inadequate response to other biologics is not known.
The trials assessed several of the outcomes that patients report as important. However, not all were part of the statistical testing strategy; as a result, the ability to draw conclusions from these data may be limited. The 24-week duration of the comparative part of the trials may be considered short, given that PsA is a chronic condition. Moreover, the comparator used in the trials (i.e., placebo plus cDMARD) may not reflect current practice, given that most patients who show an inadequate response cDMARDs or bDMARDs DMARDs would be offered biologic therapy.
Indirect Evidence
Objectives and Methods for the Summary of Indirect Evidence
A review of indirect evidence was required because none of the clinical trials included in the systematic review included an active control group (other than guselkumab every 4 weeks, which is not a Health Canada–approved dosage). In addition, an appraisal of the ITC submitted by the sponsor was necessary, given that these data were used to inform the pharmacoeconomic model.
A focused literature search for ITCs dealing with PsA was run in MEDLINE All (1946–) on May 30, 2022. No limits were applied to the search. The results were reviewed by 1 researcher to select any ITCs that met the patient, intervention, comparator, and outcome criteria listed in the review protocol (Table 6).
Nine potentially relevant ITCs15,60-67 were identified in the literature search. However, 4 of these trials did not include guselkumab, and were excluded.60,63,64,67
Another 4 ITCs were not summarized in this report. In 1 ITC, the only guselkumab data available was from a phase II trial; thus, it was missing key evidence to inform the indirect comparison.61 Another ITC was an earlier version of the sponsor’s ITC and was considered out of date,62 and a third ITC had a limited scope (comparing guselkumab to secukinumab only) and was missing many comparators that were relevant to the Canadian context.65 The ITC by Torres et al. (2020)66 included guselkumab PsA trials in the systematic review, but these studies were excluded from the NMA. Thus, Torres et al. (2020) was excluded because it did not provide any additional data to support the reimbursement decision.
This section provides a critical appraisal of the sponsor’s ITC20,68 as well as a brief summary of the published ITC by McInnes et al. (2022)15 that was identified in the literature search conducted by CADTH.
Description of ITC
The sponsor conducted an NMA of RCTs that assessed the comparative efficacy and safety of guselkumab and other bDMARDs for the short-term treatment of acute PsA. The NMA by McInnes et al. (2022)15 evaluated the efficacy and safety of licensed and unlicensed bDMARDs for patients with active PsA.
Methods of the Sponsor-Submitted ITC
Objectives
The objective of the sponsor-submitted ITC was to assess the relative efficacy and safety of guselkumab compared with other biologics after 12 weeks to 24 weeks of therapy in patients with active PsA.
Study Selection Methods
The ITC was based on a systematic literature review of RCTs that evaluated biologic and non-biologic DMARDs in adults with active PsA. The review was based on a protocol that was drafted a priori and published to PROSPERO in April 2020. The systematic review was first conducted in 2018, was updated in 2020 and 2021, and was published in 2021.62,68 Table 24 outlines the patients, interventions, comparators, outcomes, and study designs included in the current version of the sponsor’s systematic review. The review included phase II and III RCTs in adults with active PsA who were treated with 1 or more DMARD or placebo for at least 12 weeks. The review included a broad scope of treatments for PsA; however, the authors made no statement with regards to doses of treatments that would be accepted in the systematic review. No limits were placed on the outcomes of interest to the review.
The systematic review was based on a peer-reviewed literature search of the Embase, MEDLINE, and Cochrane Central databases and a search of ClinicalTrials.gov. Studies were selected based on the criteria listed in Table 24, with screening conducted by 2 reviewers independently. Data were extracted by 1 reviewer and verified by a second. Trials were assessed for study quality by a single reviewer using the National Institute for Health and Care Excellence clinical effectiveness quality assessment checklist. Although not explicitly stated, it appears that no trials were excluded based on study quality.
Additional criteria were applied to determine which studies were included in the quantitative analysis; however, no rationale was provided for these criteria, and it is unclear if these were determined a priori. Based on the description of the excluded studies, the ITC was limited to phase III RCTs of a biologic (including small-molecule inhibitors apremilast, tofacitinib, and upadacitinib) at dosages approved by the FDA or European Medicines Agency. It is not clear how outcomes were selected for analysis in the ITC.
Table 24: Study Selection Criteria and Methods for Sponsor-Submitted ITC
Category | Criteria |
|---|---|
Population | Active psoriatic arthritis, ≥ 18 years of age |
Intervention or comparator |
|
Outcome | No restriction on outcomes |
Study design |
|
Publication characteristics | English |
Exclusion criteria | Open-label extension phases of RCTs; phase IV studies |
Search strategy |
|
Selection process | Articles screened independently by 2 reviewers, with disagreement settled by discussion or consultation with a third reviewer |
Data extraction process |
|
Quality assessment | 1 reviewer evaluated RCTs using the NICE clinical effectiveness quality assessment checklist |
CSR = Clinical Study Report; CTLA-4 = cytotoxic T‐lymphocyte‐associated protein-4; DMARD = disease-modifying antirheumatic drug; EMA = European Medicines Agency; IL = interleukin; ITC = indirect treatment comparison; JAK = Janus kinase; NICE = National Institute for Health and Care Excellence; PDE-4 = phosphodiesterase type 4; RCT = randomized controlled trial; TNF = tumour necrosis factor.
Source: Sponsor’s submission for Tremfya.20
ITC Analysis Methods
The authors conducted an NMA using Bayesian methods (Table 25). The authors stated that a random-effects model was selected a priori as the primary analysis method due to the known clinical heterogeneity in PsA trials. Unadjusted fixed-effects models would also be run, but would be accepted only if there were compelling reasons, such as the between-trial heterogeneity prior not being updated by the data, leading to conclusions (based on overlap of the 95% CrIs) that are contrary to primary trial results. Further, a baseline risk-adjusted model (adjusting for placebo response, described by the authors as a proxy for measured and unmeasured patient characteristics) was planned to adjust for known heterogeneity in patient and study characteristics, as has been done in other NMAs for PsA.69 Model fit was assessed based on 4 factors: estimated regression coefficient (beta) and a 95% CrI that excludes the null for a meta-regression covariate; lower between-study SD compared with the unadjusted model; lower deviance information criterion (by ≥ 5 points) compared with the unadjusted model; and posterior mean of the residual deviance approximately equal to the number of treatment arms. The first 2 factors were given more weight when determining if the baseline risk-adjusted model fit best.
An inconsistency model was used to identify loops with inconsistency, although the authors stated that this method had low power to detect a difference. Of note, the network had few closed loops; only 5 studies had an active comparator. The authors stated that their interpretation of the reliability of estimates was based on their quality assessment of the transitivity assumption and evaluation of the estimated between-trial heterogeneity.
The NMA included trials with outcomes assessed at 12 weeks to 24 weeks. Although not explicitly stated, it appears that each dosage regimen was a separate node. Outcomes assessed include ACR response, PASI response, change from baseline in HAQ-DI, Psoriatic Arthritis Response Criteria response, SF-36 PCS and MCS, change from baseline in vdH-S, adverse events, and serious adverse events. No rationale was provided for the selection of these outcome measures.
The primary analysis included a mixed population of biologic-naive and biologic-experienced patients. Subgroup analyses were run, restricted to patients who were biologic-naive and biologic-experienced. For the overall and biologic-experienced populations, sensitivity analyses were run to test the impact of the early escape errors in the COSMOS study. The primary analysis used the main findings from COSMOS, with a secondary analysis that accounted for errors in early escape.
Table 25: ITC Analysis Methods
Detail | Sponsor-submitted ITC |
|---|---|
ITC methods | Bayesian baseline risk-adjusted random-effects models as well as unadjusted fixed-effects and random-effects models (where feasible) for the overall PsA population (biologic-naive and biologic-experienced) ACR and PASI response: ordinal model with probit link for each category of response (i.e., ACR 0, ACR 20, ACR 50, and ACR 70 or PASI 0, PASI 50, PASI 75, PASI 90, and PASI 100); relative probit effects converted to RR. Dichotomous outcomes: logit scale with OR transformed to RR using the unweighted average of trial placebo responses estimated by the model. Treatments with 0 events dropped from networks. Continuous outcomes: mean differences scale Software: R and JAGS; based on code from the NICE Evidence Synthesis Decision Support Unit Technical Support Document series |
Priors | Vague priors for all parameters |
Assessment of model fit | Assessed based on 4 factors: estimated regression coefficient (beta) and its 95% CrI; lower between-study SD compared with the unadjusted model; lower DIC (by ≥ 5 points) compared with the unadjusted model; posterior mean of the residual deviance approximately equal to the number of treatment arms. |
Assessment of consistency | Unrelated mean-effects model (inconsistency model). The posterior mean deviance of the individual data points in the inconsistency model were plotted against their posterior mean deviance in the consistency model to help identify loops in which there was inconsistency. |
Assessment of convergence | Analyses based on 4 chains, with a burn-in and sampling duration of 20,000 iterations or more. The model was considered converged if the Gelman-Rubin diagnostic (Rhat) was ≤ 1.05. After convergence was reached, the effective sample size and Monte Carlo standard error estimate were used to ensure sufficient post-convergence samples were taken to support inference. Model thinning was applied so that 10,000 iterations of each parameter would be saved (i.e., every eighth iteration). The authors stated that thinning was applied to facilitate probabilistic cost-effectiveness analysis, which requires a consistent number of samples for each outcome. |
Outcomes | ACR response,a PASI response,a PsARC response Change from baseline in HAQ-DI,a SF-36 MCS, SF-36 PCS, vdH-S Adverse events and SAEa |
Follow-up time points | Efficacy: based on primary time point of the trial (i.e., 12 weeks to 24 weeks) Safety: based on longest placebo-controlled time point up to 24 weeks |
Construction of nodes | Although not explicitly stated, it appears that each dose was a separate node. |
Sensitivity analyses |
|
Subgroup analysis |
|
Methods for pairwise meta-analysis | Not reported |
ACR = American College of Rheumatology; ACR 0 = American College of Rheumatology 0% improvement; ACR 20 = American College of Rheumatology 20% improvement; ACR 50 = American College of Rheumatology 50% improvement; ACR 70 = American College of Rheumatology 70% improvement; DIC = deviance information criterion; HAQ-DI = Health Assessment Questionnaire Disability Index; ITC = indirect treatment comparison; MCS = Mental Component Summary; NICE = National Institute for Health and Care Excellence; OR = odds ratio; PASI = Psoriasis Area and Severity Index; PASI 0 = % improvement from baseline in Psoriasis Area and Severity Index score;;PASI 50 = 50% improvement from baseline in Psoriasis Area and Severity Index score;;PASI 75 = 75% improvement from baseline in Psoriasis Area and Severity Index score; PASI 90 = 90% improvement from baseline in Psoriasis Area and Severity Index score; PASI 100 = 100% improvement from baseline in Psoriasis Area and Severity Index score; PCS = Physical Component Summary; PsARC = Psoriatic Arthritis Response Criteria; RR = relative risk; SAE = serious adverse events; SD = standard deviation; SF-36 = Short Form (36) Health Survey; vdH-S = van der Heijde-Sharp.
aUsed to inform the pharmacoeconomic model.
Source: Sponsor’s submission for Tremfya.20
Results of Sponsor-Submitted ITC
Summary of Included Studies
The systematic review included 169 citations of 80 RCTs. Of these, 46 RCTs were excluded from the ITC because the trials included a treatment or dose that was not approved by the European Medicines Agency or FDA (17 studies), included only 1 relevant treatment (1 study), did not include a biologic (21 studies), or were a phase II study (8 studies). A total of 34 RCTs published between 2004 and 2021 were included in the ITCs.
Fourteen bDMARDs were included in the NMA: abatacept, adalimumab, apremilast; certolizumab pegol, etanercept, golimumab, guselkumab, infliximab, ixekizumab, risankizumab, secukinumab, tofacitinib, upadacitinib, and ustekinumab. Most studies included a placebo group (32 trials). Of the 16 studies that compared active treatments, most included 2 dosing regimens of the same drug. Four trials compared 2 different active treatments. The sample size of the studies ranged from 100 patients to 1,281 patients.
The authors of the ITC acknowledged that there was heterogeneity across studies and provided data tables describing the patient and study characteristics of the included trials. The enrolment criteria of all studies specified adults (≥ 18 years) with active PsA; however, the definition of active PsA varied across trials. Most trials required patients to have at least 3 tender or swollen joints and a history or presence of plaque psoriasis at enrolment. The mean age of patients enrolled ranged from 45.7 years to 53.6 years. In terms of gender and race, 41.9% to 61% of patients enrolled were male, and 76.5% to 99.7% were White (not reported in 2 studies). The mean duration of PsA ranged from 3.1 years to 13.4 years across the studies. At baseline, the mean number of swollen and tender joints ranged from 6.1 to 18.3 and 14 to 27.3, respectively. The proportion of patients with psoriasis affecting more than 3% of their BSA ranged from 42% to 100% (not available in 6 studies). At baseline, the mean PASI score ranged from 5.9 to 12.2 (not reported in 9 studies).
Eighteen trials involved patients who were biologic-naive, 4 trials involved patients who were biologic-experienced, and 12 trials involved a mixed population. In the mixed-population studies, 14% to 61% of patients had prior experience with a biologic treatment. Where reported, 21% to 100% of patients had previously received cDMARDs (not reported in 16 studies). In most studies, patients were receiving methotrexate at baseline (38% to 83% of those enrolled). No information was provided about other co-interventions received during the trials.
The primary time point in 9 trials was 12 weeks. Ten trials had time points of 14 weeks to 16 weeks, and 15 trials lasted 24 weeks. In general, the trials of TNF inhibitors, JAK inhibitors, and apremilast were 16 weeks or shorter, whereas the studies of abatacept and the IL-17, IL-12 to 23, and IL-23 inhibitors were 24 weeks in duration. The exceptions were adalimumab and secukinumab — for which there were data from both shorter- and longer-duration trials — and etanercept, for which there were data from only a 24-week trial. Twelve of the 16 24-week studies (75%) had an early escape route available before the primary efficacy end point, which is a potential source of confounding. There were no details provided on how patients who received escape therapy were handled in each study.
To explore clinical heterogeneity within and across the studies, bar plots of the distribution of placebo responses and scatterplots of the relationships between placebo response and the treatment effect from each study were examined by the authors of the ITC (data not available). The authors stated that the placebo response varied across trials and tended to be lower for stricter ACR categories. Across the trials, the ACR 20 response rate in the placebo groups ranged from 9% to 33% (median = 20%; N = 32). The authors also noted that the trials evaluating guselkumab tended to have a higher-than-average placebo response (i.e., ACR 20 = 20% to 33%). The authors stated that a strong, negative association between treatment effect and placebo response was evident for all ACR response levels, with studies with larger placebo responses appearing to have smaller treatment effects. The authors of the ITC stated that failure to adjust for baseline risk could result in biased estimates of comparative efficacy. A summary of the CADTH assessment of clinical heterogeneity is provided in Table 26.
The authors of the ITC rated most of the included studies as having a low overall risk of bias; however, 53% were rated as having an unclear risk of bias, and 18% were rated as having a high risk of bias in at least 1 domain. Eight of 34 studies (24%) had unclear risk of bias related to randomization and/or allocation concealment. Blinding of patients, care providers, and/or outcome assessors was unclear in 6 studies (18%); and study participants were unblinded in 1 study (3%). The authors of the ITC assessed that the groups were balanced at the start of all studies, and that only 1 study had an unclear risk of bias related to imbalances in dropouts. Seventeen studies (50%) had an unclear risk of bias and 1 study (3%) had a high risk of bias related to the methods used to account for missing data. Three studies (9%) had a high risk of reporting bias.
Table 26: Assessment of Homogeneity for Sponsor-Submitted ITC
Detail | Description and handling of potential effect modifiers |
|---|---|
Disease severity | Not specifically assessed. There was variation across trials in the duration of PsA and tender and swollen joint counts. |
Treatment history | The overall PsA population included trials of only biologic-naive or only biologic-experienced patients as well as trials that included both types (i.e., mixed populations). The proportion of patients who had previously used non-biologic DMARDs also varied, as did the proportion who were receiving methotrexate at baseline. Subgroup analyses limited to biologic-naive and biologic-experienced patients were conducted. |
Clinical trial eligibility criteria | All trials included patients with PsA, but the trials varied in terms of the minimum number of tender or swollen joints and other criteria (e.g., presence of skin disease, enthesitis, dactylitis). |
Dosing of comparators | Each dosage regimen was analyzed as a separate node. The model included some dosages that have not been approved for use in Canada. |
Placebo response | The ACR 20 response rate ranged from 9% to 33% (median = 20%; N = 32). |
Definitions of end points | The ACR and PASI response and HAQ-DI are standardized end points. |
Timing of end point evaluation or trial duration | Variable; ranged from 12 weeks to 24 weeks for efficacy outcomes. |
Withdrawal frequency | Not evaluated |
Clinical trial setting | Not evaluated |
Study design | All were English-language, phase III RCTs. |
Time frame | Trials published between 2004 and 2021; potential source of heterogeneity, given that newer biologics have come to market and replaced TNF alpha inhibitors. |
ACR = American College of Rheumatology; ACR 20 = American College of Rheumatology 20% improvement; DMARD = disease-modifying antirheumatic drug; HAQ-DI = Health Assessment Questionnaire Disability Index; ITC = indirect treatment comparison; PASI = Psoriasis Area and Severity Index; PsA = psoriatic arthritis; RCT = randomized controlled trial; TNF = tumour necrosis factor.
Source: Sponsor’s submission for Tremfya.20
Results
ACR Response
The evidence network for ACR response in the overall PsA population (biologic-naive or experienced) is shown in Figure 8. All 34 studies reported data for ACR 20; 33 studies reported data for ACR 50; and 31 studies reported data for ACR 70 (number of patients not reported). Based on the multinomial model used in this analysis, the conclusions for ACR 50 and ACR 70 are the same as for ACR 20.
For the overall PsA population, the results of the baseline risk-adjusted random-effects model suggest that patients who received guselkumab every 8 weeks may || |||| |||||| || ||||||| || ||| || |||||||| |||| ||||| ||| |||||||| apremilast, abatacept, or placebo, given that the 95% CrI for the pairwise comparisons excluded the null (Table 27). The ACR 20 results |||||||| ||||||||||| ||||||||| ||| ||||||||||| ||| || |||||| |||||||||| |||. Comparisons between |||||||||| ||| ||| ||||| ||||||||| ||| ||| ||| |||| |||||||||| ||| |||| |||||| || |||| |||||| ||| ||| ||| ||| ||||||||||| |||||| |||||||| ||| ||||||||||| || ||||||||||| ||||||| |||||| ||||| ||||||||| ||| || |||||||||||| |||||||| ||| ||||||| || |||| | ||||||||||.
In the analysis of biologic-naive patients (30 studies, Figure 9), the baseline risk-adjusted random-effects model suggested patients who received guselkumab were |||| |||||| || ||||||| ||| || |||||||| |||| |||||||||| || ||||||| (Table 28). For other comparisons, the 95% CrI |||||||| ||| |||| |||||. In some cases, the 95% CrI was |||||||||| |||||||| ||| ||||||| || |||| | ||||||||||. The results in biologic-experienced patients (16 studies, Figure 10) |||||||| |||||||||| |||||| ||||||| ||||| || || |||||||||| |||||| ||||||| |||||| ||| ||| ||| ||| ||||| ||||||||||| |||||||||| ||| |||| ||||| ||| || ||| ||||| ||| ||| ||| ||| |||||||||| |||||||| ||| ||||||| || |||| | |||||||||| (Table 29).
Change in HAQ-DI
The NMA of the change in HAQ-DI in the overall, biologic-naive, and biologic-experienced populations were informed by data from 32 RCTs (unadjusted random-effects model), 21 RCTs (unadjusted random-effects model), and 8 RCTs (unadjusted fixed-effects model), respectively. In all 3 populations, ||| |||||| || |||||| |||||||| |||||||||| |||||| |||||||| ||| ||||||| |||||||| ||| ||||| |||| ||||| ||||||||||| |||||||| ||| ||||| ||||||||| |||| | |||| ||| |||| |||||||| ||| ||||||| || |||| | |||||||||| (Table 27, Table 28, Table 29). ||| ||||||| |||||||| ||||||||| || |||||| |||||||||| || ||| ||||||| ||| |||||||||||||| |||||||||||| ||| |||||||| |||||||||| || || ||||| |||||| |||||| |||||||||| || ||| ||||||| ||||||||||.
PASI Response
Thirty RCTs provided PASI response data to inform the ordinal baseline-adjusted random-effects Bayesian NMA. Based on this model, the results of the PASI 90 response are the same for other PASI thresholds. ||| ||||||| |||||||| |||||||||| |||||| ||||||||||| ||| ||| ||| ||||| |||||||||| ||||||| |||||||||||| ||| ||||||||||| |||||||||| |||||||||| ||| ||||||| (Table 27). ||| ||||||| |||||||| ||| ||||| ||||||||||| |||||||| ||| ||||| ||| ||| ||| ||| ||| ||||||||| |||||||||| |||||||| ||| ||||||| || |||| | ||||||||||| ||| ||| |||||||| |||| ||||||| ||| ||| |||||||||||||| |||||||| || | || |||||||| (Table 28).
|| ||| |||||||| || ||||||| || |||||||||||||||||||| |||||||| || | || ||||||||| ||| ||||||| |||||||| |||||||||| |||||| |||||||||||| ||||||||| ||| ||||||| |||||| |||| ||| ||||||| ||| ||| ||||| ||||||||||| |||| |||||||||| |||||||| ||| ||||||| || |||| | |||||||||||
Changes in SF-36 PCS and MCS
For the overall PsA population, ||| ||||||| |||||||| |||||||||| ||| ||||||||| || |||||| |||||||||| ||| ||| |||||| || ||||| ||| ||| |||||||| ||| |||||||| |||||||||| ||| |||||||||| |||||| |||||||||| ||| ||| |||||| || ||||| ||| ||| ||||||||| |||| ||| ||||| |||||||| ||||||||||| ||||||||| ||| |||| |||| |||||||| ||| |||| |||||| || |||||||||| |||||| ||||||| |||||||. The findings were similar for the biologic-naive subgroup (MCS = 13 studies and PCS = 18 studies; unadjusted random-effects models). The NMAs in the biologic-experienced population were conducted using unadjusted fixed-effects models that included 6 trials for the MCS and 7 trials for the PCS. ||| |||| ||||||||| ||| ||||||||||| ||||||| |||||||||| ||| ||||| ||||||||| |||||||| ||| |||| |||| |||||||| ||| ||||| |||| || ||||| |||| |||||||||.
Change in VdH-S Score
|| ||| ||||||| ||||||||||| ||| |||||||||| ||||| |||||| ||| ||| |||||||| |||||||| |||||||||| ||| ||||||||| || |||||| |||||||||| ||| ||| |||||| |||| |||||||| || ||| ||||| |||||| ||| ||||| ||||||||||| |||||||| ||| ||| |||| |||||||| ||| ||||| ||| |||||||| |||| ||||||| ||| ||| |||||||||||||| ||||||||| |||| ||||||| ||| ||| |||||||| || ||| |||||||||||||||||||| |||||||||||||.
Serious Adverse Events
The NMA of serious adverse events in the overall population included data from 32 RCTs and was analyzed using a baseline risk-adjusted, random-effects model. ||| ||||||| ||||||| |||||||||| ||| |||| | ||||| |||| || ||||||| ||||||| |||||| |||| |||||||||| ||| |||||||||||| ||||| |||| ||| ||||| ||||||||||| ||||||| |||||||||| ||| ||||||| || ||||||||| |||||||| ||||||||| ||| |||| |||| |||||||||| ||| ||||| |||||||| ||| ||||||| || |||| ||||||||||| |||||| |||| ||| ||||||||||| ||||||| |||||||||| ||| ||||||| || ||||||||| || ||| ||| || |||||||| ||| |||| |||||||||||||| || | || |||||||| ||||| ||| || |||||||||||||||||||| || | | |||||||| ||||| ||| |||||| ||| |||| |||| |||| |||||||||| ||||| |||||| ||| ||||||| || |||| | |||||||||| |||| ||||| ||||.
Adverse Events
The NMA of adverse events was conducted using a baseline risk-adjusted, random-effects model for the overall population (32 studies) and biologic-naive populations (17 studies). ||| ||||||| ||||||| |||| |||||||||| ||| |||| | ||||| |||| || |||||||||| ||||||| |||||| |||| ||||||||||| |||||||||| || ||||||||||| ||| ||||| ||||||||||| |||||| ||| |||| |||| |||||||| ||| ||||| ||| || |||| ||||| |||| |||||||||| || ||| |||||||||||||| ||||||||||| ||| ||||||||||| ||||||| |||||||||| ||| ||||||||| |||||||| ||| ||| |||| |||||||| ||| |||| ||| |||| |||||||||| ||| ||| || ||| |||||||||||||||||||| |||||||||| || |||||||| |||||||||| ||||| |||||| |||||| ||||||||| |||| ||||||||||| ||| |||| | ||||| |||| || ||||||| |||||| |||| |||||||||| ||| ||| |||||||||||| ||||| ||||||||||| |||||||| ||| |||| |||| |||||||| ||| ||||| |||| |||| ||||||| ||||||||| |||||||.
Figure 8: Evidence Network for ACR Response in the Overall PsA Population
ABA = abatacept; ADA = adalimumab; APR = apremilast; CERT = certolizumab; ETA = etanercept; GOL = golimumab; GUS = guselkumab; IFX = infliximab; IXE = ixekizumab; LD = loading dose; PBO = placebo; Q2W = every 2 weeks; Q4W = every 4 weeks; Q8W = every 8 weeks; RIS = risankizumab; SEC = secukinumab; TOF = tofacitinib; UPA = upadacitinib; UST = ustekinumab; WT = weight based.
Source: Sponsor’s submission for Tremfya.20
Table 27: Key NMA Results for the Overall PsA Population — Guselkumab Every 8 Weeks Versus Biologics
Comparator | ACR 20 | Change in HAQ-DI | PASI 90 | SAE |
|---|---|---|---|---|
Median RR (95% CrI) | MD (95% CrI) | Median RR (95% CrI) | Median RR (95% CrI) | |
Model; number of studies | |||||||| |||||||||||||| |||||| ||||||| |||||| || ||||||| | |||||||||| |||||| ||||||| |||||| || ||||||| | |||||||| |||||||||||||| |||||| ||||||| |||||| || ||||||| | |||||||| |||||||||||||| |||||| ||||||| |||||| || ||||||| |
Placebo | |||| ||||| || ||||| | ||||| |||||| || |||||| | ||||| |||| || |||||| | |||| ||||| || |||| |
IL-17 inhibitor | ||||
IXE 80 mg q4w | |||| ||||| || ||||| | |||| |||||| || |||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
IXE 80 mg q2w | |||| ||||| || ||||| | |||| |||||| || |||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
IXE 80 q4w/q2w | |||| ||||| || ||||| | |||||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
SEC 150 mg no LD | |||| ||||| || ||||| | ||||| |||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
SEC 150 mg | |||| ||||| || ||||| | |||| |||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
SEC 150/300 mg | |||| ||||| || ||||| | |||| |||||| || ||||| | |||||||| | |||||||| |
SEC 300 mg | |||| ||||| || ||||| | |||| |||||| || |||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
IL-12/23 or IL-23 inhibitor | ||||
RIS 150 mg | |||| ||||| || ||||| | ||||| |||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
UST 90 mg | |||| ||||| || ||||| | |||| |||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
UST 45 mg | |||| ||||| || ||||| | ||||| |||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
TNF inhibitor | ||||
ADA 40 mg | |||| ||||| || ||||| | ||||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
CERT 200 mg | |||| ||||| || ||||| | |||| |||||| || |||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
CERT 400 mg | |||| ||||| || ||||| | | |||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
ETA 25 mg BIW | |||| ||||| || ||||| | ||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
GOL 2 mg/kg | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
GOL 50 mg | |||| ||||| || ||||| | |||| |||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || |||||| |
IFX 5 mg/kg | |||| ||||| || ||||| | |||| |||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
JAK inhibitor | ||||
TOF 5 mg | |||| ||||| || ||||| | ||||| |||||| || |||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
UPA 15 mg | |||| ||||| || ||||| | |||| |||||| || ||||| | |||| ||||| || ||||| | |||| |||| || ||||| |
Other | ||||
ABA125 mg | |||| ||||| || ||||| | ||| |||||| || ||||| | |||| ||||| || |||||| | |||| ||||| || ||||| |
APR 30 mg | |||| ||||| || ||||| | ||||| |||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
ABA = abatacept; ADA = adalimumab; ACR 20 = American College of Rheumatology 20% improvement; APR = apremilast; BIW = twice weekly; CERT = certolizumab; Crl = credible interval; ETA = etanercept; GOL = golimumab; HAQ-DI = Health Assessment Questionnaire Disability Index; IFX = infliximab; IL = interleukin; IXE = ixekizumab; JAK = Janus kinase; LD = loading dose; MD = mean difference; NA = not applicable; NMA = network meta-analysis; PASI 75 = 75% improvement from baseline in Psoriasis Area and Severity Index score; PASI 90 = 90% improvement from baseline in Psoriasis Area and Severity Index score; PsA = psoriatic arthritis; q2w = every 2 weeks; q4w = every 4 weeks; RIS = risankizumab; RR = relative risk; SAE = serious adverse event; SEC = secukinumab; TNF = tumour necrosis factor; TOF = tofacitinib; UPA = upadacitinib; UST = ustekinumab.
Note: Data reported for guselkumab every 8 weeks vs. comparator. An RR is greater than 1 favours guselkumab for ACR 20 and PASI 75. For the change in HAQ-DI, negative MD values favour guselkumab. For SAE, an RR less than 1 favours guselkumab. Results shown in bold had 95% CrIs that excluded the null value.
Source: Sponsor’s submission for Tremfya.20
Figure 9: Evidence Network for ACR Response in Biologic-Naive PsA Population
ABA = abatacept; ACR = American College of Rheumatology; ADA = adalimumab; APR = apremilast; CERT = certolizumab; ETA = etanercept; GOL = golimumab; GUS = guselkumab; IFX = infliximab; IXE = ixekizumab; LD = loading dose; PBO = placebo; PsA = psoriatic arthritis; Q2W = every 2 weeks; Q4W = every 4 weeks; Q8W = every 8 weeks; RIS = risankizumab; SEC = secukinumab; TOF = tofacitinib; UPA = upadacitinib; UST = ustekinumab; WT = weight based.
Source: Sponsor’s submission for Tremfya.20
Table 28: Key NMA Results for the Biologic-Naive Population — Guselkumab Every 8 Weeks Versus Biologics
Comparator | ACR 20 | Change in HAQ-DI | PASI 90 | SAE |
|---|---|---|---|---|
Median RR (95% CrI) | MD (95% CrI) | Median RR (95% CrI) | Median RR (95% CrI) | |
Model; number of studies | |||||||| |||||||||||||| |||||| ||||||| |||||| || ||||||| | |||||||||| |||||| ||||||| |||||| || ||||||| | |||||||| |||||||||||||| |||||| ||||||| |||||| || ||||||| | ||||||||||| |||||| ||||||| |||||| || ||||||| |
Placebo | |||| ||||| || ||||| | ||||| |||||| || |||||| | ||||| |||||| || |||||| | |||| ||||| || ||||| |
IL-17 inhibitor | ||||
IXE 80 mg q4w | |||| ||||| || ||||| | |||| |||||| || |||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
IXE 80 mg q2w | |||| ||||| || ||||| | ||| |||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || |||| |
IXE 80 q4w/q2w | |||| ||||| || ||||| | |||||| | |||| ||||| || ||||| | |||| ||||| || |||||| |
SEC 150 mg no LD | |||| ||||| || ||||| | |||||| | |||||| | |||||| |
SEC 150 mg | |||| ||||| || ||||| | ||||| |||||| || ||||| | |||| ||||| || ||||| | |||| |||| || |||| |
SEC 150/300 mg | |||| ||||| || ||||| | |||| |||||| || ||||| | |||||| | |||||| |
SEC 300 mg | |||| ||||| || ||||| | ||| |||||| || ||||| | |||| ||||| || ||||| | ||| ||||| || ||||| |
IL-12/23 or IL-23 inhibitor | ||||
RIS 150 mg | |||| ||||| || ||||| | ||||| |||||| || |||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
UST 90 mg | |||| ||||| || ||||| | |||| |||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || |||||| |
UST 45 mg | |||| ||||| || ||||| | ||||| |||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
TNF inhibitor | ||||
ADA 40 mg | |||| ||||| || ||||| | ||||| |||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || |||| |
CERT 200/400 mg | |||| ||||| || ||||| | |||||| | |||| ||||| || ||||| | |||||| |
ETA 25 mg BIW | |||| ||||| || ||||| | |||| |||||| || |||| | |||| ||||| || ||||| | |||| ||||| || |||| |
GOL 2 mg/kg | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
GOL 50 mg | |||| ||||| || ||||| | |||| |||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||| |
IFX 5 mg/kg | |||| ||||| || ||||| | |||| |||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
JAK inhibitor | ||||
TOF 5 mg | |||| ||||| || ||||| | ||||| |||||| || |||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
UPA 15 mg | |||| ||||| || ||||| | |||| |||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
Other | ||||
ABA 125 mg | |||| ||||| || ||||| | ||||| |||||| || ||||| | |||| ||||| || |||||| | |||||| |
APR 30 mg | |||| ||||| || ||||| | ||||| |||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || |||||| |
ABA = abatacept; ACR 20 = American College of Rheumatology 20% improvement; ADA = adalimumab; APR = apremilast; BIW = twice weekly; CERT = certolizumab; Crl = credible interval; ETA = etanercept; GOL = golimumab; HAQ-DI = Health Assessment Questionnaire Disability Index; IFX = infliximab; IL = interleukin; IXE = ixekizumab; JAK = Janus kinase; LD = loading dose; MD = mean difference; NA = not applicable; NMA = network meta-analysis; PASI 75 = 75% improvement from baseline in Psoriasis Area and Severity Index score; PASI 90 = 90% improvement from baseline in Psoriasis Area and Severity Index score; PsA = psoriatic arthritis; q2w = every 2 weeks; q4w = every 4 weeks; RIS = risankizumab; RR = relative risk; SAE = serious adverse event; SEC = secukinumab; TNF = tumour necrosis factor; TOF = tofacitinib; UPA = upadacitinib; UST = ustekinumab.
Note: Data reported guselkumab every 8 weeks vs. comparator. RR greater than 1 favours guselkumab for ACR 20 and PASI 75. For the change in HAQ-DI, negative MD values favour guselkumab. For SAE, RR less than 1 favours guselkumab. Results shown in bold had 95% CrIs that excluded the null value.
Source: Sponsor’s submission for Tremfya.20
Figure 10: Evidence Network for ACR Response in Biologic-Experienced PsA Population
ABA = abatacept; American College of Rheumatology; APR = apremilast; CERT = certolizumab; GUS = guselkumab; IXE = ixekizumab; LD = loading dose; PBO = placebo; PsA = psoriatic arthritis; Q2W = every 2 weeks; Q4W = every 4 weeks; Q8W = every 8 weeks; RIS = risankizumab; SEC = secukinumab; TOF = tofacitinib; UPA = upadacitinib; UST = ustekinumab.
Source: Sponsor’s submission for Tremfya.20
Table 29: Key NMA Results for the Biologic-Experienced Population — Guselkumab Every 8 Weeks Versus Biologics
Comparator | ACR 20 | Change in HAQ-DI | PASI 90 | SAE |
|---|---|---|---|---|
Median RR (95% CrI) | MD (95% CrI) | Median RR (95% CrI) | Median RR (95% CrI) | |
Model; number of studies | ||||||||||| |||||| ||||||| |||||| || ||||||| | |||||||||| ||||| ||||||| |||||| | ||||||| | |||||||||| ||||| ||||||| |||||| | ||||||| | ||||||||||| ||||| ||||||| |||||| | ||||||| |
Placebo | |||| ||||| || ||||| | ||||| |||||| || |||||| | |||| ||||| || |||||| | |||| ||||| || ||||| |
IL-17 inhibitor | ||||
IXE 80 mg q4w | |||| |||||| || ||||| | |||| |||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || |||||| |
IXE 80 mg q2w | |||| ||||| || ||||| | ||||| |||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
SEC 150 mg no LD | |||| ||||| || ||||| | |||||| | |||||| | |||||| |
SEC 150 mg | |||| ||||| || ||||| | ||||| ||||| || ||||| | |||| ||||| || ||||| | |||||| |
SEC 300 mg | |||| ||||| || ||||| | |||| |||||| || ||||| | |||| ||||| || ||||| | |||||| |
IL-12/23 or IL-23 inhibitor | ||||
RIS 150 mg | |||| ||||| || ||||| | |||||| | |||| ||||| || ||||| | |||||| |
UST 90 mg | |||| ||||| || ||||| | ||||| |||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || |||||| |
UST 45 mg | |||| ||||| || ||||| | ||||| |||||| || ||||| | |||| ||||| || ||||| | |||||| |
TNF inhibitor | ||||
CERT 200/400 mg | |||| ||||| || ||||| | |||||| | |||| ||||| || ||||| | |||||| |
JAK inhibitor | ||||
TOF 5 mg | |||| ||||| || ||||| | |||| |||||| || ||||| | |||| ||||| || |||||| | |||| ||||| || |||| |
UPA 15 mg | |||| ||||| || ||||| | ||||| |||||| || ||||| | |||| ||||| || ||||| | |||| ||||| || ||||| |
Other | ||||
ABA125 mg | |||| ||||| || ||||| | ||||| |||||| || ||||| | |||| ||||| || |||||| | |||||| |
APR 30 mg | |||| ||||| || ||||| | |||||| | |||||| | |||||| |
ABA = abatacept; ACR 20 = American College of Rheumatology 20% improvement; APR = apremilast; CERT = certolizumab; Crl = credible interval; HAQ-DI = Health Assessment Questionnaire Disability Index; IL = interleukin; IXE = ixekizumab; JAK = Janus kinase; LD = loading dose; MD = mean difference; NA = not applicable; NMA = network meta-analysis; PASI 75 = 75% improvement from baseline in Psoriasis Area and Severity Index score; PASI 90 = 90% improvement from baseline in Psoriasis Area and Severity Index score; q2w = every 2 weeks; q4w = every 4 weeks; RIS = risankizumab; RR = relative risk; SAE = serious adverse event; SEC = secukinumab; TNF = tumour necrosis factor; TOF = tofacitinib; UPA = upadacitinib; UST = ustekinumab.
Note: Data reported guselkumab every 8 weeks vs. comparator. RR greater than 1 favours guselkumab for ACR 20 and PASI 75. For the change in HAQ-DI, negative MD values favour guselkumab. For SAE, RR less than 1 favours guselkumab. Results shown in bold had 95% CrIs that excluded the null value.
Source: Sponsor’s submission for Tremfya.20
Critical Appraisal of Sponsor-Submitted ITC
The ITC was based on a systematic literature review that used standard methods to conduct the literature search, screen articles, and extract data. The protocol for the review was registered, and the inclusion criteria were clearly defined. However, the risk of bias was assessed by a single reviewer and not assessed by outcome — an approach that is not aligned with the highest standards for evidence synthesis. Thus, there is uncertainty regarding the methodological rigour of the trials. It is noteworthy that 46 studies that met the criteria for inclusion in the systematic review were excluded from the NMA, which had a narrower scope than the systematic review. The authors did not provide a rationale for the exclusion criteria applied to the NMA, and it was unclear if the criteria were determined a priori. Further, there was some uncertainty regarding which dosages of drugs were eligible for inclusion, and it appears that some dosage regimens that are relevant to the Canadian context were excluded from the NMA (e.g., etanercept 50 mg weekly). According to the ITC authors’ ratings, the quality of the included studies was generally good; however, it is unclear how the overall risk of bias was assessed, given that 53% the studies were at unclear risk of bias and 18% were at high risk of bias in at least 1 domain. Specifically, 24% of the studies had an unclear risk of bias related to randomization or allocation concealment, 18% had an risk of bias was categorized as unclear and 3% had a high risk of bias related to blinding. Also of note, the report provided little information on the extent of withdrawals and or on the imputation methods for missing data.
There is some uncertainty as to whether the transitivity assumption has been met for the NMAs. There was heterogeneity across trials in the distribution of sexes, the durations of PsA, the number of tender and swollen joints, the proportions of patients with psoriasis affecting more than 3% of their BSA, and patients’ baseline PASI scores. For each study, there was limited information about the co-interventions patients received, the extent of withdrawals, or how patients with missing data were handled in the analyses.
The NMA pooled data from different time points ranging from 12 weeks to 24 weeks. According to the sponsor, this was done to align with the decision problem and Canadian practice. McInnes et al. (2022)15 notes that this is a significant source of heterogeneity, particularly for outcomes that require a higher degree of improvement (e.g., PASI 100 versus PASI 75). For the higher-response end points, the relative efficacy has been shown to increase over time, and McInnes et al. (2022) state that pooling data reported at different time points biases the results against drugs that are studied over a shorter period.15 Moreover, many of the longer-term studies allowed for early escape for nonresponders. This may be another source of heterogeneity, given that it is unclear how these patients were analyzed and whether the approach was consistent across trials.
The authors of the ITC used different methods in an attempt to control for heterogeneity in the NMA. The authors used a baseline risk-adjusted, random-effects model as the preferred model in most NMAs. Adjusting for the variation in placebo response rates across trials is a practice that has been endorsed by the National Institute for Health and Care Excellence and others, given that the placebo rate and the relative effect versus placebo may be related in psoriasis.71,72 This method assumes that study and patient characteristics that are effect modifiers of the relative treatment effect are also prognostic factors of the outcome with placebo.73,74 The use of a placebo response rate is an attempt to account for potential variability in effect modifiers, but it is unclear if these effect modifiers have the same level of effect on the active arms. Given that it is unclear to what extent placebo response is an adequate proxy for specific characteristics or effect modifiers, uncertainty remains in these analyses.
To minimize heterogeneity related to prior treatments, subgroup analyses were conducted in patients who were biologic-naive and biologic-experienced. Prior exposure to biologics has been identified as an important effect modifier for psoriasis, and limiting networks to more homogeneous subgroups is a strategy that has been used by others to reduce heterogeneity.75 In this ITC, the subgroup analyses in the biologic-naive population were generally consistent with the overall population in terms of direction of effect compared with guselkumab, but the networks included a smaller number of trials, and data were not available for all relevant comparators for each population. The networks in the biologic-experienced population were sparse, with only 6 of 16 RCTs reporting data on serious adverse events. The lack of a robust evidence network may contribute to uncertainty in the findings in this population. Among the potential sources of heterogeneity, the ITC did not conduct any subgroup or specific sensitivity analyses to explore the potential impact of differences in the timing of outcome assessment, duration of disease, background therapies, or year of study. Of these, the clinical expert consulted for this review identified disease duration and prior biologic therapy as important potential effect modifiers.
There was limited information available to assess whether the consistency assumption was met, given that there were only 4 head-to-head studies and few closed loops within the networks (no closed loops with guselkumab). Although the authors of the NMA took steps to assess consistency, these methods had low power to detect differences. Based on the model fit data presented and the authors’ explanation of the criteria used to assess fit, it appears the models selected for the primary analyses were appropriate.
ACR and PASI responses were analyzed using an ordinal model, which assumed that the relative treatment effects were the same for each response level. Thus, although data were reported separately for each response level, the inferences for each comparison are the same across the ACR 20, ACR 50, and ACR 70 levels. It is not clear if this assumption of the model holds true (i.e., relative treatment effects are consistent across response levels), given that data were pooled for different time points. No sensitivity analyses were run to examine the impacts of this assumption. There was greater uncertainty in the PASI response results, with some comparisons showing wide 95% CrIs. In most of the PsA clinical trials, assessments of PASI end points were based on a subgroup of patients with skin lesions at baseline. It is not known if randomization was stratified by this factor or if groups were balanced for potential confounders.
Regarding the NMA of the change in HAQ-DI, McInnes et al. (2022)15 stated that the HAQ-DI may be affected by the presence of comorbidities and the duration of PsA. Given the variability in the duration of PsA across the trials, this could affect the findings of the ITC.
Across all networks, many comparisons showed imprecise 95% CrIs that included the possibility of appreciable benefit and/or worse outcomes, and no difference between guselkumab and other biologics.70 Imprecision was an issue for all outcomes for the biologic-experienced population in particular, and for serious adverse events in all populations; this imprecision limits the ability to draw conclusions from these data. With regards to external validity, the comparators included in the model were relevant to the Canadian context, although not all comparators were available in all NMAs. The trials enrolled predominantly White patients; thus, the trials may not reflect the racial distribution of the population in Canada with PsA. Due to the availability of data, the NMAs assessed short-term treatment effects; thus, the comparative evidence on longer-term safety and efficacy is unclear. Although the NMA addressed outcomes of interest to patients, in many cases the uncertainty in the results limits the ability to draw conclusions on the comparative safety and efficacy of guselkumab.
Summary of Published ITC
The objective of the ITC by McInnes et al. (2022)15 was to evaluate the safety and efficacy of bDMARDs licensed for psoriasis or PsA. This report was funded by Leo Pharma A/S. The ITC was based on a systematic literature review (last search August 2020) of English-language RCTs that included licensed and unlicensed DMARDs for the treatment of active PsA (abatacept, apremilast, adalimumab, bimekizumab, brodalumab, certolizumab pegol, etanercept, filgotinib, golimumab, guselkumab, infliximab, ixekizumab, netakimab, risankizumab, secukinumab, tildrakizumab, tofacitinib, upadacitinib, and ustekinumab). The authors conducted a Bayesian NMA of ACR and PASI response rates, resolution of enthesitis and dactylitis (measured at 12 weeks to 16 weeks or up to 26 weeks, if an early time point was not available), and discontinuation due to adverse events (at study end point). ACR and PASI response were analyzed using a multinomial probit model, and other outcomes were based on a binomial likelihood with logit link. Both random and fixed effects, as well as placebo-adjusted models, were analyzed for the overall population with PsA. Subgroup analyses for biologic-naive and biologic-experienced patients were run for ACR response only. Inconsistency between direct and indirect estimates was assessed for closed loops in the network using the 2-stage Bucher method.
A total of 64 studies were included in the systematic review, and 46 studies were included in the NMAs. The authors noted differences across trials in the duration of PsA, percentage of female patients, and prior exposure to biologic therapies or cDMARDs. Also, there was variation in the timing of the end points, particularly PASI response, with approximately 40% of studies reporting data at 24 weeks and 60% of studies reporting at week 12 to week 16. Discontinuations due to adverse events were not reported consistently across studies, with some reporting discontinuation of the study drug and others reporting discontinuation from the trial.
Overall, the results of the ITC by McInnes et al.15 were generally consistent with the sponsor-submitted ITC. The odds ratios of ACR 20 response for guselkumab every 8 weeks versus most comparators at week 12 to week 16 showed 95% CrIs that overlapped the null and were imprecise, limiting the ability to draw a conclusion. The ACR 20 results favoured guselkumab versus ustekinumab 45 mg and apremilast 30 mg, but favoured infliximab versus guselkumab. The results for the NMA of PASI 90 response favoured guselkumab versus several comparators (ixekizumab, secukinumab, tildrakizumab, ustekinumab, adalimumab, certolizumab pegol, etanercept, golimumab, abatacept, apremilast, and tocilizumab). Guselkumab showed favourable resolution of enthesitis or dactylitis versus apremilast, but not versus 9 other active comparators with available data. No comparative results for guselkumab were reported for discontinuation due to adverse events; however, the results versus placebo showed wide CrIs (median odds ratio = 0.77; 95% CrI, 0.25 to 2.31).
The key limitation of the ITC was the heterogeneity in patient and study characteristics across trials. The authors noted several differences in patient demographics, disease, or treatment history. Also noted were differences in outcome definitions (discontinuations) and in the timing of PASI response and the resolution of enthesitis and dactylitis end points, which could not be addressed in the analysis. Although a placebo-adjusted model was run for some outcomes, it is unclear if these methods can adequately control for heterogeneity. Also noteworthy was the uncertainty in the NMA results for ACR 20, PASI response, and discontinuations — as shown in some comparisons with wide 95% CrIs — which limits the ability to draw a conclusion. In this NMA, there was limited ability to assess the consistency between direct and indirect evidence, given that there were few closed loops (only 6 head-to-head studies), and the statistical tests for inconsistency were generally underpowered. Further, most of the contributing trials were judged to be at high or unclear risk of bias in at least 1 domain, including 65% and 33% of trials with unclear risk of bias related to allocation concealment and random sequence generation, respectively. With respect to external validity, the NMA did not address outcomes of interest to patients, such as HRQoL, disability, or safety.
Other Relevant Evidence
This section includes the uncontrolled extension period of the phase III RCTs DISCOVER-1, DISCOVER-2, and COSMOS, which are summarized and appraised. These provide an overview of the long-term (> 24 weeks) efficacy and safety of guselkumab every 8 weeks among patients with active PsA.
Extension Phase of the Sponsor-Submitted Phase III Studies
Description of Studies
The extension period for DISCOVER-1 was from week 24 to week 52, with a safety follow-up phase of 8 weeks after week 52. For DISCOVER-2, the active treatment phase was from week 24 to week 100, with follow-up for efficacy at week 52 and week 100 and a safety follow-up phase of 12 weeks after week 100. For the COSMOS study, the extension period was from week 24 to week 48, with a safety follow-up phase of 8 weeks.
In all 3 trials, during the extension period, groups of patients who had been randomized to receive guselkumab 100 mg every 8 weeks or every 4 weeks continued on the same regimen, whereas those randomized to placebo switched to guselkumab 100 mg every 8 weeks or every 4 weeks. Data from the guselkumab every-4-weeks groups in the DISCOVER-1 and DISCOVER-2 studies (including patients who crossed over from placebo to guselkumab every 4 weeks at 24 weeks) have not been summarized in this section because this dose is not consistent with the Health Canada–recommended dose and dose adjustment, as mentioned earlier in the report. Results for the patients in the placebo group of the COSMOS trial who crossed over to guselkumab 100 mg every 8 weeks at 24 weeks have been provided as supporting information (hereafter referred to as the placebo crossover group).
Statistical Analysis
For the extension periods in the DISCOVER-1, DISCOVER-2, and COSMOS studies, no hypothesis testing of treatment differences was conducted. Descriptive statistics were used to summarize most efficacy data. Analyses of end points were conducted according to randomized treatment groups and based on observed data.
Results
Patient Disposition
Table 30 describes the patient disposition for the extension period. For DISCOVER-1 (week 52), 13 patients (10%) discontinued from the study early, whereas 13 patients (5%) and 27 patients (11%) discontinued the study early in DISCOVER-2 (week 52) and DISCOVER-2 (week 100), respectively. Moreover, in DISCOVER-1 (week 52), DISCOVER-2 (week 52), and DISCOVER-2 (week 100), 11 patients (9%), 14 patients (6%) and 25 patients (10%) discontinued the study drug before end of the study, respectively. The rates of discontinuation of the study drug were higher for DISCOVER-1 and for DISCOVER-2 during the active treatment period compared to the randomized phase.
In the COSMOS study, the rates of discontinuation of the study drug were 12% in the guselkumab-every-8-weeks group and 14% in the placebo group before week 44. However, the rates for early discontinuation of the study drugs were lower during the randomized phase, with 15 patients (8%) and 10 patients (10%) for the guselkumab-every-8-weeks and placebo groups, respectively.
Table 30: Patient Disposition for the Extension Period
Detail | DISCOVER-1 (Week 52) | DISCOVER-2 (Week 52) | DISCOVER-2 (Week 100) | COSMOS (Week 48) | |
|---|---|---|---|---|---|
GUSE q8w | GUSE q8w | GUSE q8w | GUSE q8w | PBO crossover | |
Randomized | 127 | 248 | 248 | 189 | 96 |
Continued at week 24, N (%) | 123 (97) | 240 (97) | NA | NR | NR |
Continued at week 48, N (%) | 116 (91) | NA | NA | NR | NR |
Continued at week 52, N (%) | NA | 235 (95) | NA | NA | NA |
Continued at week 60, N (%) | 114 (90) | NA | NA | NA | NA |
Continued at week 100, N (%) | NA | NA | 221 (89) | NA | NA |
Discontinued from study early, n (%) | 13 (10) | 13 (5) | 27 (11) | NR | NR |
Reason for discontinuation, n (%) | |||||
Lost to follow-up | 1 (1) | 1 (0.4) | 1 (0.4) | NR | NR |
Withdrawal by patient | 4 (3) | 3 (1) | 8 (3) | NR | NR |
Death | 0 | 0 | 0 | NR | NR |
Discontinued study, but completed protocol-required follow-up | 7 (6) | 9 (4) | 15 (6) | NR | NR |
Other | 1 (1) | 0 | 3 (1) | NR | NR |
Discontinued study drug early, N (%) | 11 (9) | 14 (6) | 25 (10) | 22 (12) | 13 (14) |
Reason for discontinuation from study drug, n (%) | |||||
Adverse event | 5 (4) | 2 (1) | 8 (3) | 7 (4) | 3 (3) |
Lost to follow-up | 0 | 1 (0.4) | 1 (0.4) | 1 (0.5) | 1 (1) |
Lack of efficacy | 3 (2) | 6 (2) | 7 (3) | 5 (3) | 3 (3) |
Withdrawal of consent | 2 (2) | 3 (1) | 6 (2) | 5 (3) | 3 (3) |
Death | 0 | 0 | 0 | 0 | 0 |
Other | 1 (1) | 2 (1) | 3 (1) | 4 (2) | 3 (3) |
GUSE = guselkumab; NA = not applicable; NR = not reported; PBO = placebo; q8w = every 8 weeks.
Source: Clinical Study Report for DISCOVER-1 (52 weeks);76 Clinical Study Report for DISCOVER-2 (52 weeks);77 Clinical Study Report for DISCOVER-2 (100 weeks);78 Clinical Study Report for COSMOS (48 weeks).79
Efficacy
Clinical Response in PsA Symptoms
Table 31 describes ACR and MDA responses for the active treatment period. In the DISCOVER-1 study, 67.9%, 43.4%, and 28.9% of patients achieved an ACR 20, ACR 50, and ACR 70 responses, respectively, at week 52. In the DISCOVER-2 study, 79.1%, 51.3%, and 29.5% of patients achieved ACR 20, ACR 50, and ACR 70 responses, respectively, at week 52. At week 100, the corresponding values were 82.1%, 60.7%, and 39.3%.
For COSMOS, 69.8%, 47.1%, and 27.9% of patients achieved ACR 20, ACR 50, and ACR 70 responses, respectively, at week 48.
Although differences were not tested statistically, in all cases, the rate of response appeared to increase numerically with longer follow-ups. The proportions of patients who maintained ACR 20, ACR 50, and ACR 70 responses at week 52 were 88.5%, 83.8%, and 80.0%, respectively, in DISCOVER-1, and were 91.7%, 87.2%, and 82.6%, respectively, in DISCOVER-2. The proportions of patients who maintained ACR 20, ACR 50, and ACR 70 responses at week 100 were 90.4%, 81.9%, and 80.9% in DISCOVER-2. The proportions of patients who maintained ACR 20, ACR 50, and ACR 70 responses at week 48 were 85.6%, 85.4%, and 81.3% in COSMOS.
The proportions of patients with an MDA response at week 24 were 23.6% in DISCOVER-1, 26.5% in DISCOVER-2, and 17.4% in COSMOS. At 52 weeks, the proportions of patients with MDA responses were 33.9% in DISCOVER-1 and 32.9% in DISCOVER-2. At 100 weeks, 44.6% of patients had an MDA response in DISCOVER-2. At 48 weeks, 32.5% of patients had an MDA response in COSMOS. Although the differences were not tested statistically, in all cases, the rate of response appeared to increase numerically with longer follow-up.
For the COSMOS study, the proportions of patients with MDA response were 32.5% in the guselkumab-every-8-weeks group and 29.8% in the placebo group at week 48. The proportions of patients who maintained MDA response at week 52 were 75.9% and 82.5% in DISCOVER-1 and DISCOVER-2, respectively. The proportions of patients who maintained MDA response at week 100 were 78.3% (based on patients achieving MDA response at week 24) and 85.3% (based on patients achieving MDA response at week 52) for DISCOVER-2.
Table 31: ACR and MDA Response for the Extension Period (FAS)
Outcome | DISCOVER-1 (Week 52) | DISCOVER-2 (Week 52) | DISCOVER-2 (Week 100) | COSMOS (Week 48) | |
|---|---|---|---|---|---|
GUSE q8w N = 123 | GUSE q8w N = 240 | GUSE q8w N = 232 | GUSE q8w N = 189 | PBO crossover N = 96 | |
ACR 20 response | |||||
Number of patients contributing to the analysis at week 24, N | 123 | 238 | 238 | 184 | 93 |
ACR 20 response at week 24, n (%) | 67 (54.5) | 159 (66.8) | 159 (66.8) | 101 (54.9) | 41 (44.1) |
Number of patients contributing to the analysis for longest follow-up, N | 112 | 234 | 223 | 172 | 85 |
ACR 20 response for longest follow-up, n (%) | 76 (67.9) | 185 (79.1) | 183 (82.1) | 120 (69.8) | 52 (61.2) |
ACR 50 response | |||||
Number of patients contributing to the analysis at week 24, N | 123 | 238 | 238 | 184 | 93 |
ACR 50 response at week 24, n (%) | 38 (30.9) | 78 (32.8) | 78 (32.8) | 44 (23.9) | 9 (9.7) |
Number of patients contributing to the analysis for longest follow-up, N | 113 | 234 | 224 | 172 | 85 |
ACR 50 response for longest follow-up, n (%) | 49 (43.4) | 120 (51.3) | 136 (60.7) | 81 (47.1) | 27 (31.8) |
ACR 70 response | |||||
Number of patients contributing to the analysis at week 24, N | 123 | 238 | 238 | 184 | 93 |
ACR 70 response at week 24, n (%) | 15 (12.2) | 46 (19.3) | 46 (19.3) | 16 (8.7) | 2 (2.2) |
Number of patients contributing to the analysis for longest follow-up, N | 114 | 234 | 224 | 172 | 85 |
ACR 70 response for longest follow-up, n (%) | 33 (28.9) | 69 (29.5) | 88 (39.3) | 48 (27.9) | 18 (21.2) |
MDA response | |||||
Number of patients contributing to the analysis at week 24, N | 123 | 238 | 238 | 184 | 93 |
Patients with MDA at week 24, n (%) | 29 (23.6) | 63 (26.5) | 63 (26.5) | 32 (17.4) | 5 (5.4) |
Number of patients contributing to the analysis for longest follow-up, N | 112 | 234 | 224 | 169 | 84 |
Patients with MDA for longest follow-up, n (%) | 38 (33.9) | 77 (32.9) | 100 (44.6) | 55 (32.5) | 25 (29.8) |
ACR = American College of Rheumatology; ACR 20 = American College of Rheumatology 20% improvement; ACR 50 = American College of Rheumatology 50% improvement; ACR 70 = American College of Rheumatology 70% improvement; FAS = full analysis set; GUSE = guselkumab; MDA = minimal disease activity; PBO = placebo; q8w = every 8 weeks.
Source: Clinical Study Report for DISCOVER-1 (52 weeks);76 Clinical Study Report for DISCOVER-2 (52 weeks);77 Clinical Study Report for DISCOVER-2 (100 weeks);78 Clinical Study Report for COSMOS (48 weeks).79
Function or disability
Table 32 describes the changes from baseline in HAQ-DI scores for the active treatment period. The mean changes in HAQ-DI at week 24 were –0.34 (SD = 0.57), –0.40 (SD = 0.54), and –0.29 (SD = 0.55) for DISCOVER–1, DISCOVER–2, and COSMOS, respectively. The mean changes from baseline at 52 weeks were –0.44 (SD = 0.56) and –0.48 (SD = 0.56) for DISCOVER–1 and DISCOVER–2, respectively. The mean changes from baseline at week 48 and week 100 were –0.47 (SD = 0.60) and –0.59 (SD = 0.58) for COSMOS and DISCOVER-2, respectively.
The proportions of patients who achieved a clinically meaningful HAQ-DI response (defined as a score greater than or equal to 0.35 improvement from baseline) were 53.2% at week 24 and 57.4% at week 52 for DISCOVER-1 and 52.5% at week 24, 60.9% at week 52, and 70.7% at week 100 for DISCOVER-2. For COSMOS, the proportions of patients achieving an improvement of greater than or equal to 0.35 from baseline were 47.4% at week 24 and 62.3% at week 48 for the guselkumab-every-8-weeks group, and 39.3% at week 24 and 48.7% at week 48 for the placebo crossover group.
The proportions of patients who maintained an improvement of greater than or equal to 0.35 from baseline in HAQ-DI score at week 52 were 84.9% and 92.0% in DISCOVER-1 and DISCOVER-2, respectively. The proportions of patients who maintained an improvement of greater than or equal to 0.35 from baseline in HAQ-DI score at week 100 were 88.7% (based on patients with HAQ-DI response at week 24) and 90.4% (based on patients with HAQ-DI response at week 52) in DISCOVER-2.
Table 32: Change From Baseline in HAQ-DI Scores for the Extension Period (FAS)
Outcome | DISCOVER-1 (Week 52) | DISCOVER-2 (Week 52) | DISCOVER-2 (Week 100) | COSMOS (Week 48) | |
|---|---|---|---|---|---|
GUSE q8w N = 123 | GUSE q8w N = 240 | GUSE q8w N = 232 | GUSE q8w N = 189 | PBO crossover N = 96 | |
Change from baseline in HAQ-DI score | |||||
Number of patients contributing to the analysis, N | 114 | 234 | 224 | 171 | 85 |
At week 24, mean (SD) | –0.34 (0.57) | –0.40 (0.54) | –0.40 (0.54) | –0.29 (0.55) | –0.14 (0.51) |
At longest follow-up, mean (SD) | –0.44 (0.56) | –0.48 (0.56) | –0.59 (0.58) | –0.47 (0.60) | –0.35 (0.55) |
FAS = full analysis set; GUSE = guselkumab; HAQ-DI = Health Assessment Questionnaire Disability Index; PBO = placebo; q8w = every 8 weeks; SD = standard deviation.
Source: Clinical Study Report for DISCOVER-1 (52 weeks);76 Clinical Study Report for DISCOVER-2 (52 weeks);77 Clinical Study Report for DISCOVER-2 (100 weeks);78 Clinical Study Report for COSMOS (48 weeks).79
Health-Related Quality of Life
The mean changes from baseline in SF-36 PCS and MCS scores from week 24 through the long-term follow-up period are provided in Table 33. The mean changes from baseline in the SF-36 PCS for week 24 were 6.51 (SD = 7.71), 7.84 (SD = 8.03), and 5.83 (SD = 7.17) for DISCOVER-1, DISCOVER-2, and COSMOS, respectively. The mean changes from baseline at 52 weeks were 7.28 (SD = 8.07) and 9.51 (SD = 8.32) for DISCOVER-1 and DISCOVER-2, respectively. The mean changes from baseline at week 48 and week 100 were 8.44 (SD = 7.27) and 11.28 (SD = 9.28) for COSMOS and DISCOVER-2, respectively.
The proportions of patients who achieved a clinically meaningful SF-36 PCS score (≥ 5-point improvement from baseline) were 53.7% at week 24 and 53.5% at week 52 for DISCOVER-1; for DISCOVER-2, they were 63.4% at week 24, 67.1% at week 52, and 70.1% at week 100. For COSMOS, the proportions of patients achieving a greater than or equal to 5-point improvement from baseline in SF-36 PCS were 46.0% at week 24 and 55.6% at week 48 for the group receiving guselkumab every 8 weeks, and 22.9% and 26.0% at week 24 and week 48, respectively, for the placebo crossover group.
The proportions of patients who maintained a greater than or equal to 5-point improvement in SF-36 PCS scores at week 52 were 74.1% and 81.8% in DISCOVER-1 and DISCOVER-2, respectively. The proportions of patients who maintained a greater than or equal to 5-point improvement from baseline in SF-36 PCS scores at week 100 were 82.0% (based on SF-36 responders at week 24) and 85.4% (based on responders at week 52).
The mean changes from baseline in SF-36 MCS for week 24 were 3.03 (SD = 10.62), 4.45 (SD = 9.96), and 2.59 (SD = 10.18) for DISCOVER-1, DISCOVER-2, and COSMOS, respectively. The mean changes from baseline at 52 weeks were 5.14 (SD = 9.17) and 4.47 (SD = 9.78) for DISCOVER-1 and DISCOVER-2, respectively. The mean changes from baseline at week 48 and week 100 were 4.98 (SD = 9.94) and 3.64 (SD = 10.95) for COSMOS and DISCOVER-2, respectively. The change from baseline in the SF-36 MCS was either maintained or increased numerically (i.e., was not tested statistically) at the various time points evaluated in the 3 studies.
The proportions of patients who achieved a clinically meaningful (≥ 5-point improvement from baseline) SF-36 MCS score were 39.0% at week 24 and 46.5% at week 52 for DISCOVER-1, and 40.3% at week 24, 44.9% at week 52, and 46.4% at week 100 for DISCOVER-2. For COSMOS, the proportions of patients achieving a greater than or equal to 5-point improvement from baseline in SF-36 MCS were 36.6% at week 24 and 42.7% at week 48 for the group receiving guselkumab every 8 weeks, and 30.1% and 45.9%, respectively, at week 24 and week 48 for the placebo crossover group.
The proportions of patients who maintained a greater than or equal to 5-point improvement in SF-36 MCS scores at week 52 were 82.2% and 82.3% in DISCOVER-1 and DISCOVER-2, respectively. The proportions of patients who maintained a greater than or equal to 5-point improvement from baseline in SF-36 MCS scores at week 100 were 79.1% (based on SF-36 responders at week 24) and 77.2% (based on responders at week 52).
Table 33: Change From Baseline in SF-36 PCS and MCS for the Extension Period (FAS)
Outcome | DISCOVER-1 (Week 52) | DISCOVER-2 (Week 52) | DISCOVER-2 (Week 100) | COSMOS (Week 48) | |
|---|---|---|---|---|---|
GUSE q8w N = 123 | GUSE q8w N = 240 | GUSE q8w N = 232 | GUSE q8w N = 189 | PBO crossover N = 96 | |
Change from baseline in SF-36 PCS | |||||
Number of patients contributing to the analysis, N | 114 | 234 | 224 | 172 | 85 |
At week 24, mean (SD) | 6.51 (7.71) | 7.84 (8.03) | 7.84 (8.03) | 5.83 (7.17) | 3.15 (6.94) |
At longest follow-up, mean (SD) | 7.28 (8.07) | 9.51 (8.32) | 11.28 (9.28) | 8.44 (7.27) | 6.99 (8.92) |
Change from baseline in SF-36 MCS | |||||
Number of patients contributing to the analysis, N | 114 | 234 | 224 | 172 | 85 |
At week 24, mean (SD) | 3.03 (10.62) | 4.45 (9.96) | 4.45 (9.96) | 2.59 (10.18) | 2.07 (8.84) |
At longest follow-up, mean (SD) | 5.14 (9.17) | 4.47 (9.78) | 4.72 (9.90) | 3.64 (10.95) | 4.98 (9.94) |
FAS = full analysis set; GUSE = guselkumab; MCS = Mental Component Summary; PBO = placebo; PCS = Physical Component Summary; q8w = every 8 weeks; SD = standard deviation; SF-36 = Short Form (36) Health Survey.
Source: Clinical Study Report for DISCOVER-1 (52 weeks);76 Clinical Study Report for DISCOVER-2 (52 weeks);77 Clinical Study Report for DISCOVER-2 (100 weeks);78 Clinical Study Report for COSMOS (48 weeks).79
Psoriasis Skin Disease
Table 34 shows patients’ IGA and PASI responses for the active treatment period. In all trials, psoriasis skin disease outcome measures were analyzed in the subgroup of patients who had psoriasis affecting greater than or equal to 3% of their BSA and an IGA score of greater than or equal to 2 at baseline for the randomized phase. Although the difference was not tested statistically, the proportions of patients who achieved an IGA response at week 52 were numerically higher than at week 24 in the DISCOVER-1 and DISCOVER-2 studies (58.0% versus 69.3%, and 72.1% versus 77.1%, respectively). For the COSMOS study, the proportions of patients who achieved an IGA response at week 48 were 81.0% for the group receiving guselkumab every 8 weeks and 78.4% for the placebo crossover group.
The proportions of patients who maintained their IGA responses at week 52 were 81.8% and 85.4% in the DISCOVER-1 and DISCOVER-2 studies; at week 100 in the DISCOVER-2 study, these proportions were 88.2% and 89.1% at week 24 and week 52, respectively.
The proportions of patients who achieved PASI 100, PASI 90, and PASI 75 responses at week 24 were 25.9%, 50.6%, and 76.5% in DISCOVER-1; 46.5%, 70.3%, and 80.8% in DISCOVER-2; and 41.1%, 65.1%, and 76.0% for COSMOS, respectively. For DISCOVER-1 and DISCOVER-2, the proportions of patients who achieved PASI 100, PASI 90, and PASI 75 responses at week 52 were 48%, 66.7%, and 80.0%; and 54.7%, 77.1%, and 88.8%, respectively. For DISCOVER-2, the proportions at week 100 were 57.3%, 75.0%, and 87.8% for PASI 100, PASI 90, and PASI 75, respectively. For COSMOS, the proportions achieving PASI 100, PASI 90, and PASI 75 responses at week 48 were 66.1%, 84.3%, and 93.4%, respectively, for the group receiving guselkumab every 8 weeks, and 49.0%, 66.7%, and 88.2%, respectively, for the placebo crossover group.
The proportions of patients who maintained PASI 100, PASI 90, and PASI 75 responses at week 52 were 80.0%, 84.6%, and 86.2% in DISCOVER-1; and 75.9%, 88.3%, and 97.1% in DISCOVER-2, respectively. For DISCOVER-2, the proportions of patients who maintained PASI 100, PASI 90, and PASI 75 responses at week 100 were 76.3%, 86.2%, and 93.9% at week 24 and 86.7%, 88.2%, and 94.5% at week 52.
Table 34: IGA and PASI Response for the Extension Period (FAS)
Outcome | DISCOVER-1 (Week 52) | DISCOVER-2 (Week 52) | DISCOVER-2 (Week 100) | COSMOS (Week 48) | |
|---|---|---|---|---|---|
GUSE q8w N = 123 | GUSE q8w N = 240 | GUSE q8w N = 232 | GUSE q8w N = 189 | PBO crossover N = 96 | |
IGA response | |||||
Number of patients contributing to analysis, N | 75 | 170 | 165 | 121 | 51 |
IGA response at week 24, n (%) | 47 (58.0) | 124 (72.1) | 124 (72.1) | 81 (62.8) | 19 (35.8) |
IGA response at longest follow-up, n (%) | 52 (69.3) | 131 (77.1) | 126 (76.4) | 98 (81.0) | 40 (78.4) |
PASI 100 response | |||||
Number of patients contributing to analysis, N | 75 | 170 | 169 | 121 | 51 |
PASI 100 response at week 24, n (%) | 21 (25.9) | 80 (46.5) | 80 (46.5) | 53 (41.1) | 8 (15.1) |
PASI 100 response at longest follow-up, n (%) | 36 (48.0) | 93 (54.7) | 94 (57.3) | 80 (66.1) | 25 (49.0) |
PASI 90 response | |||||
Number of patients contributing to analysis, N | 75 | 170 | 169 | 121 | 51 |
PASI 90 response at week 24, n (%) | 41 (50.6) | 121 (70.3) | 121 (70.3) | 84 (65.1) | 12 (22.6) |
PASI 90 response at longest follow-up, n (%) | 50 (66.7) | 131 (77.1) | 123 (75.0) | 102 (84.3) | 34 (66.7) |
PASI 75 response | |||||
Number of patients contributing to analysis, N | 75 | 170 | 169 | 121 | 51 |
PASI 75 response at week 24, n (%) | 62 (76.5) | 139 (80.8) | 139 (80.8) | 98 (76.0) | 18 (34.0) |
PASI 75 response at longest follow-up, n (%) | 60 (80.0) | 151 (88.8) | 144 (87.8) | 113 (93.4) | 45 (88.2) |
FAS = full analysis set; GUSE = guselkumab; IGA = Investigator’s Global Assessment of Psoriasis; PASI = Psoriasis Area and Severity Index; PASI 75 = 75% improvement from baseline in Psoriasis Area and Severity Index score; PASI 90 = 90% improvement from baseline in Psoriasis Area and Severity Index score; PASI 100 = 100% improvement from baseline in Psoriasis Area and Severity Index score; PBO = placebo; q8w = every 8 weeks..
Source: Clinical Study Report for DISCOVER-1 (52 weeks);76 Clinical Study Report for DISCOVER-2 (52 weeks);77 Clinical Study Report for DISCOVER-2 (100 weeks);78 Clinical Study Report for COSMOS (48 weeks).79
Other Musculoskeletal Disease
Table 35 presents enthesitis and dactylitis outcomes for the extension period. The proportions of patients with resolution of enthesitis and dactylitis at week 52 were 56.3% and 79.5% for DISCOVER-1; these were 65.5% and 81.9% for DISCOVER-2, respectively. The proportions of patients with resolution of enthesitis and dactylitis at week 100 were 77.5% and 91.1% for DISCOVER-2, respectively. For COSMOS, the proportions of patients with resolution of enthesitis and dactylitis at week 48 were 67.5% and 80.0%, respectively.
The mean reductions from baseline in enthesitis scores (based on the LEI) and dactylitis scores (based on observed data) at week 52 were –1.8 (SD = 1.66) and –7.8 (SD = 10.55) for DISCOVER-1, and –1.9 (SD = 1.65) and –7.3 (SD = 9.74) for DISCOVER-2, respectively; at week 100, these scores were –2.1 (SD = 1.65) and –7.9 (SD = 10.12) for DISCOVER-2, respectively. For COSMOS, the mean reductions from baseline in enthesitis scores and dactylitis scores were –2.0 (SD = 1.92) and –6.1 (SD = 6.41) for the group receiving guselkumab every 8 weeks, and –1.8 (SD = 1.70) and –7.3 (SD = 7.04) for the placebo crossover group, respectively.
The proportions of patients who maintained resolution of enthesitis (based on the LEI) and dactylitis at week 52 were 74.1% and 96.4% in DISCOVER-1, and 88.4% and 92.2% in DISCOVER-2, respectively. For DISCOVER-2, the proportions of patients who maintained resolution of enthesitis (based on the LEI) and dactylitis at week 100 were 94.0% and 95.7%, respectively, at week 24, and 96.8% and 96.4%, respectively, at week 52.
Table 35: Enthesitis and Dactylitis Outcomes for the Extension Period (FAS)
Outcome | DISCOVER-1 (Week 52) | DISCOVER-2 (Week 52) | DISCOVER-2 (Week 100) | COSMOS (Week 48) | |
|---|---|---|---|---|---|
GUSE q8w N = 123 | GUSE q8w N = 240 | GUSE q8w N = 232 | GUSE q8w N = 189 | PBO crossover N = 96 | |
Resolution of enthesitis | |||||
Number of patients contributing to analysis, N | 64 | 148 | 142 | 114 | 53 |
At week 24, n (%) | 29 (40.8) | 87 (57.6) | 87 (57.6) | 64 (52.0) | 26 (42.6) |
At longest follow-up, n (%) | 36 (56.3) | 97 (65.5) | 110 (77.5) | 77 (67.5) | 33 (62.3) |
Resolution of dactylitis | |||||
Number of patients contributing to analysis, N | 44 | 105 | 101 | 60 | 33 |
At week 24, n (%) | 33 (67.3) | 65 (60.7) | 65 (60.7) | 40 (61.5) | 23 (63.9) |
At longest follow-up, n (%) | 35 (79.5) | 86 (81.9) | 92 (91.1) | 48 (80.0) | 29 (87.9) |
Change from baseline in enthesitis score (based on LEI) | |||||
Number of patients contributing to analysis, N | 64 | 147 | 141 | 114 | 53 |
At week 24, mean (SD) | –1.2 (1.95) | –1.6 (1.75) | –1.6 (1.75) | –1.7 (1.88) | –1.3 (1.74) |
At longest follow-up, mean (SD) | –1.8 (1.66) | –1.9 (1.65) | –2.1 (1.65) | –2.0 (1.92) | –1.8 (1.70) |
Change from baseline dactylitis score (based on observed data) | |||||
Number of patients contributing to analysis, N | 44 | 105 | 101 | 60 | 33 |
At week 24, mean (SD) | –6.2 (10.31) | –6.1 (7.83) | –6.1 (7.83) | –4.8 (6.09) | –5.7 (6.16) |
At longest follow-up, mean (SD) | –7.8 (10.55) | –7.3 (9.74) | –7.9 (10.12) | –6.1 (6.41) | –7.3 (7.04) |
FAS = full analysis set; GUSE = guselkumab; LEI = Leeds Enthesitis Index; PBO = placebo; q8w = every 8 weeks; SD = standard deviation.
Source: Clinical Study Report for DISCOVER-1 (52 weeks);76 Clinical Study Report for DISCOVER-2 (52 weeks);77 Clinical Study Report for DISCOVER-2 (100 weeks);78 Clinical Study Report for COSMOS (48 weeks).79
Table 36 shows the improvement in BASDAI scores for the active treatment period. The changes in BASDAI scores were analyzed for the subgroup of patients who had spondylitis and peripheral joint PsA with BASDAI scores greater than 0 as their primary presentation at baseline. The proportions of patients with a greater than or equal to 50% improvement at week 24 were |||||| ||||| ||| ||||| in DISCOVER-1, DISCOVER-2, and COSMOS, respectively. The proportions at week 52 were ||||| ||| ||||| in DISCOVER-1 and DISCOVER-2; the proportions at week 100 were ||||| in DISCOVER-2; and at week 48, the proportions were ||||| ||| ||||| for the group receiving guselkumab every 8 weeks and the placebo crossover group in the COSMOS study, respectively.
Table 36: Improvement in BASDAI Scores for the Extension Period (FAS)
Outcome | DISCOVER-1 (Week 52) | DISCOVER-2 (Week 52) | DISCOVER-2 (Week 100) | COSMOS (Week 48) | |
|---|---|---|---|---|---|
GUSE q8w N = 123 | GUSE q8w N = 240 | GUSE q8w N = 232 | GUSE q8w N = 189 | PBO crossover N = 96 | |
Proportion of patients with ≥ 50% improvement in BASDAI score | |||||
Number of patients contributing to analysis, N | |||| | |||| | |||| | |||| | |||| |
> 50% response at week 24 | || |||||| | || |||||| | || |||||| | || |||||| | | |||||| |
≥ 50% response at longest follow-up, n (%) | || |||||| | || ||||||| | || |||||| | || |||||| | || |||||| |
BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; FAS = full analysis set; GUSE = guselkumab; PBO = placebo; q8w = every 8 weeks.
Source: Clinical Study Report for DISCOVER-1 (52 weeks);76 Clinical Study Report for DISCOVER-2 (52 weeks);77 Clinical Study Report for DISCOVER-2 (100 weeks);78 Clinical Study Report for COSMOS (48 weeks).79
PsA Symptoms
Table 37 shows the changes in FACIT-Fatigue scores for the active treatment period. The mean improvements from baseline in FACIT-Fatigue scores at week 24 were 5.86 (SD = 10.39), 8.03 (SD = 9.89), and 5.9 (SD = 9.56) in DISCOVER-1, DISCOVER-2, and COSMOS, respectively. The mean improvements from baseline in FACIT-Fatigue scores at week 52 were 7.48 (SD = 9.63) and 8.93 (SD = 9.47) in DISCOVER-1 and DISCOVER-2; at week 100, these were ||||| ||| | |||||| in DISCOVER-2; and at week 48, these were ||| ||| | |||||| ||| ||| ||| | |||||| for the group receiving guselkumab every 8 weeks and the placebo crossover group in the COSMOS study, respectively.
Table 37: Change From Baseline in FACIT-Fatigue Score for the Extension Period (FAS)
Outcome | DISCOVER-1 (Week 52) | DISCOVER-2 (Week 52) | DISCOVER-2 (Week 100) | COSMOS (Week 48) | |
|---|---|---|---|---|---|
GUSE q8w N = 123 | GUSE q8w N = 240 | GUSE q8w N = 232 | GUSE q8w N = 189 | PBO crossover N = 96 | |
Change from baseline in FACIT-Fatigue score | |||||
Number of patients contributing to analysis, N | 114 | 234 | |||||| | |||||| | ||||| |
At week 24, mean (SD) | 5.86 (10.39) | 8.03 (9.89) | |||| |||||| | |||| |||||| | |||| ||||||| |
At longest follow-up, mean (SD) | 7.48 (9.63) | 8.93 (9.47) | ||||| ||||||| | ||| ||||||| | ||| ||||||| |
FACIT-Fatigue = Functional Assessment of Chronic Illness Therapy – Fatigue Scale; FAS = full analysis set; GUSE = guselkumab; PBO = placebo; q8w = every 8 weeks; SD = standard deviation..
Source: Clinical Study Report for DISCOVER-1 (52 weeks);76 Clinical Study Report for DISCOVER-2 (52 weeks);77 Clinical Study Report for DISCOVER-2 (100 weeks);78 Clinical Study Report for COSMOS (48 weeks).79
Radiologic Outcomes
Table 38 shows the modified vdH-S scores for PsA for the DISCOVER-2 study at week 52 and week 100, based on the smallest detectable change.
The proportion of patients with no progression (defined as a change from baseline that was less than or equal to the smallest detectable change for the modified vdH-S total score) was ||||| from week 24 to week 52, and ||||| from week 52 to week 100 during the extension period. These results were consistent with the results for the group receiving guselkumab every 8 weeks ||||||| during the randomized phase.
Table 38: Modified vdH-S Scores of PsA (Based on the Smallest Detectable Change) for the Extension Period (FAS)
Outcome | DISCOVER-2 (Week 52) | DISCOVER-2 (Week 100) |
|---|---|---|
GUSE q8w N = 240 | GUSE q8w N = 232 | |
Patients without radiographic progression in the modified vdH-S score | ||
Number of patients contributing to analysis through the end of study, N | |||||| | |||||| |
From week 24 to week 52, n (%) | |||| |||||| | ||||| |
From week 52 to week 100, n (%) | ||||| | ||| |||||| |
FAS = full analysis set; GUSE = guselkumab; NA = not applicable; PsA = psoriatic arthritis; q8w = every 8 weeks; vdH-S = van der Heijde-Sharp..
Source: Clinical Study Report for DISCOVER-2 (52 weeks);77 Clinical Study Report for DISCOVER-2 (100 weeks).78
Harms
The summary of harms is presented in Table 39. More than half of patients had at least 1 adverse event through the end of the studies in DISCOVER-1 (69%) and DISCOVER-2 (63% after 52 weeks and 72% after 100 weeks). In the COSMOS study, the proportions of patients with at least 1 adverse event (cumulative) in the group receiving guselkumab every 8 weeks and the placebo crossover group were 50% and 46%, respectively. The most common events were nasopharyngitis, upper respiratory tract infection, bronchitis, enthesopathy, arthralgia, increase of alanine aminotransferase and aspartate aminotransferase, neutropenia, hypertension, and headache. No deaths were reported during the active treatment period. The rates of patients with at least 1 adverse or serious adverse event increased numerically through the end of study versus week 24. No deaths were reported through the end of the study. There were few serious adverse events in all trials through the end of the extension period.
The rate of infections was highest (43%) in the DISCOVER-1 study (week 52), following 38% and 29% in the DISCOVER-2 study (week 100 and week 52, respectively). In the COSMOS study, the rates of infection (cumulative) were 22% and 14% for the group receiving guselkumab every 8 weeks and the placebo crossover group, respectively. All other notable harms occurred only rarely during the long-term follow-up period. Some may have increased numerically, but the frequency was still quite low.
Table 39: Summary of Harms (Safety Set) for the Extension Period
Adverse events | DISCOVER-1 (Week 52) GUSE q8w N = 127 | DISCOVER-2 (Week 52) GUSE q8w N = 248 | DISCOVER-2 (Week 100) GUSE q8w N = 248 | COSMOS (Week 24 to Week 56) GUSE q8w N = 174 | COSMOS (Cumulative) GUSE q8w N = 279 | COSMOS (Week 56) PBO crossover N = 90 |
|---|---|---|---|---|---|---|
Patients with ≥ 1 adverse event | ||||||
n (%) | 87 (69) | 155 (63) | 178 (72) | 53 (31) | 139 (50) | 41 (46) |
Most common events,a n (%) | ||||||
Nasopharyngitis | 21 (17) | 20 (8) | 25 (10) | 5 (3) | 16 (6) | 2 (2) |
URTI | 10 (8) | 17 (7) | 24 (10) | 2 (1) | 10 (4) | 2 (2) |
Bronchitis | 10 (8) | 6 (2) | 10 (4) | 1 (1) | 4 (1) | 2 (2) |
Enthesopathy | 7 (6) | 7 (3) | 8 (3) | NR | NR | NR |
Arthralgia | 6 (5) | 4 (2) | 5 (2) | 1 (1) | 6 (2) | 1 (1) |
ALT increased | 9 (7) | 23 (9) | 29 (12) | 3 (2) | 11 (4) | 4 (4) |
AST increased | 11 (9) | 18 (7) | 23 (9) | 3 (2) | 6 (2) | 2 (2) |
Neutropenia | 0 | 12 (5) | 12 (5) | 0 | 3 (1) | 0 |
Hypertension | 6 (5) | 6 (2) | 12 (5) | 3 (2) | 4 (1) | 0 |
Headache | 3 (2) | 10 (4) | 16 (7) | 1 (1) | 6 (2) | 2 (2) |
Patients with ≥ 1 SAE | ||||||
n (%) | 8 (6) | 10 (4) | 22 (9) | 5 (3) | 15 (5) | 4 (4) |
Patients who stopped treatment due to adverse events | ||||||
n (%) | 2 (2) | 3 (1) | 8 (3) | 3 (2) | 7 (3) | 0 |
Deaths | ||||||
n (%) | 0 | 0 | 0 | 0 | 0 | 0 |
Notable harms | ||||||
Infections, n (%) | 54 (43) | 71 (29) | 94 (38) | 16 (9) | 61 (22) | 13 (14) |
Serious infection, n (%) | 2 (2) | 3 (1) | 8 (3) | 0 | 2 (1) | 1 (1) |
Opportunistic infections, n (%) | 0 | 0 | 2 (1) | 0 | 0 | 0 |
Injection-site reactions, n (%) | 2 (2) | 4 (2) | 8 (3) | 0 | 5 (2) | 1 (1) |
Anaphylactic adverse events, n (%) | 0 | 0 | 0 | 0 | 0 | 0 |
Increase in ALT, grade 3 or 4, n (%) | 1 (1) | 3 (1) | 4 (2) | 0 | NR | 3 (3) |
Increase in AST, grade 3 or 4, n (%) | 1 (1) | 0 | 3 (1) | 0 | NR | 3 (3) |
Hepatobiliary disorders, SOC, n (%) | 4 (3) | 6 (2) | 12 (5) | 2 (1) | NR | 1 (1) |
ALT = alanine aminotransferase; AST = aspartate aminotransferase; GUSE = guselkumab; NR = not reported; PBO = placebo; q8w = every 8 weeks; SAE = serious adverse event; SOC = system organ class; URTI = upper respiratory tract infection.
aFrequency greater than 5%.
Source: Clinical Study Report for DISCOVER-1 (52 weeks);76 Clinical Study Report for DISCOVER-2 (52 weeks);77 Clinical Study Report for DISCOVER-2 (100 weeks);78 Clinical Study Report for COSMOS (48 weeks).79
Critical Appraisal
Internal Validity
Although the treatment responses appeared to improve or be maintained, for the most part, there was no statistical testing; therefore, definitive conclusions could not be drawn from the extension period. Because this portion of the trial remained blinded, there is a low risk of bias in the measurement of the outcomes. Rates of early discontinuation from study were higher through the end of the DISCOVER-1 and DISCOVER-2 trials. Given that the reason for discontinuation was mainly unspecified (because the highest number of participants fell under the discontinued-from-study category, but completed the protocol-required follow-up), it is difficult to fully address any potential attrition bias. The same validity, reliability, and responsiveness concerns for the tools used to measure the outcomes described for the randomized phase also apply to the extension period. As with the randomized phase, there is no evidence for the validity or reliability of the IGA, PASI, or BASDAI in patients with PsA.
Because no hypothesis testing of treatment differences was conducted for the active treatment period, it is not possible to draw causal conclusions based on the descriptive results. The lack of a comparator group also poses a risk of confounding. Without a randomized comparator, it is not possible to draw causal conclusions (i.e., changes following the randomized phase cannot be attributed to the treatment with any certainty).
External Validity
The external validity of the extension phase is equivalent to that reported for the randomized phase. As with the randomized phase, per the clinical expert, there are no major concerns regarding the generalizability of the findings to Canadian clinical practice.
Discussion
Summary of Available Evidence
Three double-blind, placebo-controlled RCTs met the inclusion criteria for the systematic review. Two trials included patients with active PsA who were either biologic-naive (DISCOVER-2, N = 741) or were intolerant to or had experienced an inadequate response to TNF alpha inhibitors (COSMOS, N = 285). One trial included a mixed population of biologic-nave or TNF inhibitor-experienced patients (DISCOVER-1, N = 381). Patients were randomized to receive placebo or guselkumab 100 mg SC at week 0, week 4, and every 8 weeks thereafter for 24 weeks. The DISCOVER trials also included a third treatment group of patients who received guselkumab 100 mg every 4 weeks. This dosage was not consistent with the Health Canada–recommended dose; therefore, it has not been included in this report. The total trial durations were 52 weeks (DISCOVER-1), 100 weeks (DISCOVER-2), and 48 weeks (COSMOS), with patients on placebo switching to guselkumab at week 24. During the trials, patients could continue stable doses of methotrexate, leflunomide, hydroxychloroquine, sulfasalazine, oral corticosteroids, or NSAIDs up to the protocol-specified maximum doses. The primary outcome in all trials was the proportion of patients who achieved ACR 20 at week 24. Other outcomes of interest included changes from baseline in HAQ-DI and SF-36 PCS scores and the impact on plaque psoriasis (as measured by IGA or PASI response).
The mean age of patients enrolled ranged from 44.9 years (SD = 11.9) to 49.1 years (SD = 12.3) across treatment groups in the 3 trials. The proportion of female patients ranged from 46% to 55%, and most patients were White (89% to 98% in the DISCOVER trials; not reported for COSMOS). The mean number of swollen joints ranged from 9.0 (SD = 5.7) to 12.3 (SD = 6.9) and the mean number of tender joints ranged from 18.2 (SD = 10.7) to 21.6 (SD = 13.1). Approximately 2-thirds of patients had psoriatic involvement affecting at least 3% of their BSA. Two-thirds of patients reported enthesitis, while approximately 40% had dactylitis at baseline. The majority of patients in the studies were receiving methotrexate at baseline (54% to 63%). In the DISCOVER trials, 14% to 20% of patients were receiving oral corticosteroids at baseline, compared with 4% to 5% of patients in the COSMOS study.
Supportive data on longer-term efficacy and safety were available from the uncontrolled extension phase of the 3 trials that included data from approximately 530 patients who received guselkumab for 48 weeks to 100 weeks.
In addition, the sponsor submitted an ITC that examined the comparative efficacy and safety of guselkumab versus other bDMARDs available in Canada. Another published ITC of patients with PsA is also summarized in this report.
Interpretation of Results
Efficacy
Among patients who were biologic-naive, intolerant to, or had an inadequate response to TNF alpha inhibitors, and among a mixed population that included both biologic-naive and TNF inhibitor-experienced patients with active PsA, guselkumab every 8 weeks showed clinically important differences in the proportion of patients who achieved ACR 20 at 24 weeks. Secondary and exploratory measures of PsA activity (ACR 50, ACR 70, and MDA) also favoured guselkumab every 8 weeks versus placebo, except for ACR 70 in the DISCOVER-1 trial. However, most of the end points were not controlled for multiple testing; thus, these should be interpreted with consideration for the potential for inflated type I error rate. While an ACR 20 threshold may be the minimum change that may be considered important, the expert noted that higher thresholds, such as ACR 50 or MDA, are goals of therapy.
Other outcomes, such as the change from baseline in HAQ-DI, also showed improvement favouring guselkumab every 8 weeks versus placebo; however, the within-group changes for only 1 of the guselkumab groups, and none of the between-group differences versus placebo exceeded the clinically important difference of 0.35 points reported by the sponsor. Thus, the clinical relevance of the change in disability is uncertain. The change from baseline in the SF-36 PCS showed improvements that favoured guselkumab versus placebo in all studies, with both within- and between-group differences that exceeded some estimates of the MID that, in the DISCOVER trials, may be considered clinically relevant. No differences between groups were detected from the change from baseline in the SF-36 MCS; however, it should be noted that the baseline scores (46.1 points to 48.7 points) were near the US population average (50 points). Thus, there may be limited room for improvement. The changes from baseline in the FACIT-Fatigue scores suggest a reduction in fatigue relative to placebo, but this outcome was not controlled for type I error rate.
Among patients with psoriasis affecting greater than or equal to 3% of their BSA at baseline, the IGA and PASI response end points favoured guselkumab versus placebo, with 20% to 42% more patients achieving the PASI 100 threshold at week 24 across trials. However, these data should be interpreted with consideration for the potential for inflated type I error rate, given that only the IGA response in the DISCOVER trials and the PASI 100 response in the COSMOS study were controlled for multiple testing.
The DISCOVER-2 study failed to detect a difference between guselkumab every 8 weeks and placebo in radiographic progression. Although the study was powered for this end point, the 24-week duration may have limited the trial’s ability to detect a change. The results of the enthesitis and dactylitis end points in the DISCOVER-1 study failed to detect a difference between guselkumab and placebo, while the DISCOVER-2 and COSMOS study data suggest a favourable effect with guselkumab; however, none of these end points were controlled for multiple testing. To increase statistical power, a preplanned pooled analysis of the DISCOVER trials was conducted for the proportion of patients with resolution of enthesitis and dactylitis. The pooled analyses suggest that more patients who receive guselkumab every 8 weeks may have their enthesitis or dactylitis resolved by 24 weeks versus patients who receive placebo. No differences were detected between groups in the proportions of patients who achieved at least a 50% improvement in BASDAI scores. As with the skin-related end points, these analyses were conducted in subgroups of the overall population, which were not stratified at baseline. Thus, there is potential that the groups differed in their baseline demographic and disease characteristics.
The trials compared guselkumab plus background therapy (cDMARDs, corticosteroids, or NSAIDs) with placebo plus background therapy. Thus, direct evidence for guselkumab versus other DMARDs used to treat PsA is not available. Indirect evidence was available from 1 published and 1 unpublished NMA in adults with PsA. The sponsor submitted an NMA that evaluated the short-term (12-week to 24-week) efficacy and safety of guselkumab in the treatment of PsA compared with other biologic treatments. The NMA was based on a systematic review of the literature; data from 34 RCTs was used to inform the Bayesian models. The NMA by McInnes et al. (2022)15 evaluated the short-term safety and efficacy of bDMARDs licensed for psoriasis or PsA and used Bayesian methods to pool data from 46 RCTs. These reports suggest that PASI response may favour guselkumab versus some comparators. However, generally — due to imprecision in the results comparing guselkumab to other biologics — the ability to draw conclusions about ACR response rates and changes in HAQ-DI scores, SF-36 PCS and MCS scores, and vdH-S scores was limited. The findings for the subgroup of patients who were biologic-naive were generally similar to those for the overall population in the sponsor-submitted NMA. The NMAs in the biologic-experienced population were sparse and showed imprecision in their results. Both NMAs were limited by the heterogeneity across trials, which included differences in prior exposure to biologic or non-biologic therapies, duration of disease, study years, timing of outcome assessments, and placebo response rates. Due to this heterogeneity, the NMAs used a baseline risk-adjusted model for some analyses; however, it is uncertain whether this approach is adequate to control for differences in patient characteristics that may bias results. Other limitations include the inability to assess consistency (due to the lack of closed loops in the network) and the unclear or high risk of bias of the RCTs used to inform the networks. Given these limitations and the imprecision of the results, it is difficult to draw conclusions from the NMAs about the comparative efficacy of guselkumab.
The direct comparative evidence versus placebo and indirect evidence versus other biologics was limited to short-term outcomes. Although results from the extension phase of the trials suggest that treatment effects may be maintained up to 100 weeks, these data are difficult to interpret due to the lack of a comparator group and bias due to attrition. The treatment effects observed may be inflated relative to the broader population with PsA.
With regards to external validity, the clinical expert did not identify any major issues that may limit the generalizability of the trials. However, the clinical expert did note that the use of oral corticosteroids in the DISCOVER trials was higher than would be expected in Canada (14% to 20%), where chronic corticosteroid use is generally avoided. The trials excluded patients who had previously been treated with biologics other than TNF inhibitors; thus, the efficacy in patients with intolerance or inadequate response to other biologics, such as JAK inhibitors or other IL inhibitors, is not known.
Harms
Guselkumab was first approved in Canada in 2017. Thus, its safety profile is familiar to clinicians. No new safety signals were identified in the controlled and extension phases of the clinical trials in patients with PsA. Generally, the frequency of adverse events was similar in the guselkumab and placebo groups, including the risk of infection. The proportion of patients who stopped treatment due to adverse events, or who reported serious adverse events, was generally low. However, due to the sample sizes and follow-up durations, the studies may not detect rare events or those that take a longer time to develop. Of note, most patients in the active and placebo groups were receiving background therapies; thus, some adverse events reported may be attributable to these other drugs. No conclusions could be drawn regarding the comparative safety of guselkumab based on the indirect evidence, which reported imprecise results and was limited by the clinical heterogeneity across trials.
Conclusions
Based on data from 3 double-blind RCTs, adults with active PsA who received guselkumab 100 mg every 8 weeks were more likely to show clinically relevant improvements in PsA disease activity and tender and swollen joint counts than patients who received placebo, based on the proportion who achieved an ACR 20 response at week 24. Favourable clinical responses in PsA activity and symptoms were observed among patients who were biologic-naive or had experienced prior intolerance or inadequate response to TNF alpha inhibitors and among a mixed population of patients with and without exposure to a prior TNF inhibitor.
Guselkumab also showed statistically significant improvements in disability as measured using the HAQ-DI; however, the clinical relevance of the difference versus placebo is uncertain. Improvements in the PCS but not the MCS of the SF-36 were observed, favouring guselkumab versus placebo. Outcomes related to psoriatic skin lesions demonstrated superiority of guselkumab every 8 weeks versus placebo at 24 weeks. Among patients with enthesitis or dactylitis at baseline, pooled data from the pivotal trials suggest that patients who receive guselkumab every 8 weeks may be more likely to have enthesitis or dactylitis resolved at 24 weeks than those receiving placebo. The impact of guselkumab on radiographic progression is unclear because no statistically significant differences were detected between guselkumab every 8 weeks and placebo for the change in the modified vdH-S score at 24 weeks among patients with active PsA who were biologic-naive.
No new safety signals were identified in the controlled and extension phases of the PsA trials. The frequency of infection was similar in the guselkumab and placebo groups up to 24 weeks.
There is no direct evidence comparing guselkumab to other bDMARDs available in Canada. The indirect evidence for ACR response rates, change in HAQ-DI scores, change in SF-36 PCS and MCS, and risk of adverse events or serious adverse events for guselkumab versus most biologic comparators showed imprecise results, which limits the ability to draw conclusions from these data. Based on the indirect evidence, short-term PASI response rates may favour guselkumab versus some other biologics. However, there is uncertainty in these findings, given that several sources of heterogeneity were identified across the trials included in the NMAs and because it is unclear whether the methods used to control for potential bias were adequate. In addition, many of the studies included in the NMAs were at an unclear or high risk of bias in 1 or more study domains.
The direct comparative evidence versus placebo and indirect evidence versus other biologics was limited to short-term outcomes (up to 24 weeks). Although results from the extension phase of the trials suggest that treatment effects may be maintained up to 100 weeks, these data are difficult to interpret due to the lack of comparator group and bias due to attrition. Thus, the longer-term comparative efficacy and safety of guselkumab in patients with PsA are unclear.
References
1.Gladman D, Ritchlin C. Clinical manifestations and diagnosis of psoriatic arthritis. In: Post TW, ed. UpToDate. Waltham (MA): UpToDate; 2022: www.uptodate.com. Accessed 2022 May 18.
2.Ritchlin CT, Colbert RA, Gladman DD. Psoriatic Arthritis. N Engl J Med. 2017;376(10):957-970. PubMed
3.Eder L, Widdifield J, Rosen CF, et al. Trends in the Prevalence and Incidence of Psoriasis and Psoriatic Arthritis in Ontario, Canada: A Population-Based Study. Arthritis Care Res (Hoboken). 2019;71(8):1084-1091. PubMed
4.Gossec L, Baraliakos X, Kerschbaumer A, et al. EULAR recommendations for the management of psoriatic arthritis with pharmacological therapies: 2019 update. Ann Rheum Dis. 2020;79(6):700-712. PubMed
5.Tremfya (guselkumab): 100 mg/1 mL sterile solution for injection in pre-filled syringe or Tremfya One-pass (guselkumab): 100 mg/1 mL sterile solution for injection in a patient-controlled [product monograph]. Toronto (ON): Janssen Inc.; 2022 Apr 13.
6.Mease PJ, Woolley JM, Bitman B, Wang BC, Globe DR, Singh A. Minimally important difference of Health Assessment Questionnaire in psoriatic arthritis: relating thresholds of improvement in functional ability to patient-rated importance and satisfaction. J Rheumatol. 2011;38(11):2461-2465. PubMed
7.Leung YY, Zhu TY, Tam LS, Kun EW, Li EK. Minimal important difference and responsiveness to change of the SF-36 in patients with psoriatic arthritis receiving tumor necrosis factor-alpha blockers. J Rheumatol. 2011;38(9):2077-2079. PubMed
8.Cella D, Yount S, Sorensen M, Chartash E, Sengupta N, Grober J. Validation of the Functional Assessment of Chronic Illness Therapy Fatigue Scale relative to other instrumentation in patients with rheumatoid arthritis. J Rheumatol. 2005;32(5):811-819. PubMed
9.Højgaard P, Klokker L, Orbai AM, et al. A systematic review of measurement properties of patient reported outcome measures in psoriatic arthritis: A GRAPPA-OMERACT initiative. Semin Arthritis Rheum. 2018;47(5):654-665. PubMed
10.Salaffi F, Carotti M, Di Donato E, et al. Preliminary validation of the Simplified Psoriatic Arthritis Radiographic Score (SPARS). Skeletal Radiol. 2019;48(7):1033-1041. PubMed
11.Clinical Study Report: CNTO1959PSA3001. DISCOVER-1 24 week: a phase 3, multicenter, randomized, double-blind, placebo-controlled study evaluating the efficacy and safety of guselkumab administered subcutaneously in subjects with active psoriatic arthritis including those previously treated with biologic anti-TNFα agent(s) [internal sponsor's report]. Raritan (NJ): Janssen Research & Development, LLC; 2019 Aug 28.
12.Clinical Study Report: CNTO1959PSA3002. DISCOVER-2 24 week: a phase 3, multicenter, randomized, double-blind, placebo-controlled study evaluating the efficacy and safety of guselkumab administered subcutaneously in subjects with active psoriatic arthritis [internal sponsor's report]. Raritan (NJ): Janssen Research & Development, LLC; 2019 Aug 28.
13.Clinical Study Report: CNTO1959PSA3003. COSMOS week 24: a phase 3b, multicenter, randomized, double-blind, placebo-controlled study to evaluate the efficacy and safety of guselkumab administered subcutaneously in participants with active psoriatic arthritis and an inadequate response to anti-tumor necrosis factor COSMOS [internal sponsor's report]. High Wycombe (UK): Janssen-Cilag Ltd.; 2020 Sep 23.
14.Coates LC, Gossec L, Theander E, et al. Efficacy and safety of guselkumab in patients with active psoriatic arthritis who are inadequate responders to tumour necrosis factor inhibitors: results through one year of a phase IIIb, randomised, controlled study (COSMOS). Ann Rheum Dis. 2022;81(3):359-369. PubMed
15.McInnes IB, Sawyer LM, Markus K, LeReun C, Sabry-Grant C, Helliwell PS. Targeted systemic therapies for psoriatic arthritis: a systematic review and comparative synthesis of short-term articular, dermatological, enthesitis and dactylitis outcomes. RMD Open. 2022;8(1):03.
16.Van den Bosch F, Coates L. Clinical management of psoriatic arthritis. Lancet. 2018;391(10136):2285-2294. PubMed
17.Eder L, Haddad A, Rosen CF, et al. The Incidence and Risk Factors for Psoriatic Arthritis in Patients With Psoriasis: A Prospective Cohort Study. Arthritis Rheumatol. 2016;68(4):915-923. PubMed
18.Coates LC, Kavanaugh A, Mease PJ, et al. Group for Research and Assessment of Psoriasis and Psoriatic Arthritis 2015 Treatment Recommendations for Psoriatic Arthritis. Arthritis Rheumatol. 2016;68(5):1060-1071. PubMed
19.Coates LC, Soriano ER, Corp N, et al. Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA): updated treatment recommendations for psoriatic arthritis 2021. Nat Rev Rheumatol. 2022;18(8):465-479. PubMed
20.Drug Reimbursement Review sponsor submission: Tremfya (guselkumab), 100 mg/mL solution for injection [internal sponsor's package]. Toronto (ON): Janssen Inc.; 2022 May 17.
21.CADTH Canadian Drug Expert Committee (CDEC) final recommendation: guselkumab (Tremfya — Janssen Inc.) for moderate-to-severe plaque psoriasis. Ottawa (ON): CADTH; 2018 Feb 21: https://www.cadth.ca/sites/default/files/cdr/complete/SR0530_Tremfya_complete_Feb-23-18.pdf. Accessed 2022 Jun 09.
22.Humira (adalimumab injection): 50 mg/mL or 100 mg/mL of sterile solution for subcutaneous injection [product monograph]. St-Laurent (QC): AbbVie Corporation; 2021 Apr 21: https://pdf.hres.ca/dpd_pm/00061690.PDF. Accessed 2022 Jun 09.
23.Taltz (ixekizumab): 80 mg /1.0 mL solution for injection [product monograph]. Toronto (ON): Eli Lilly Canada Inc.; 2021 Mar 29: https://pdf.hres.ca/dpd_pm/00060574.PDF. Accessed 2022 Jun 09.
24.Stelara (ustekinumab): 45 mg/0.5 mL or 90 mg/1.0 mL solution for subcutaneous injection or Stelara I.V. (ustekinumab): 130 mg/26 mL (5 mg/mL) solution for intravenous infusion [product monograph]. Toronto (ON): Janssen Inc.; 2021 Sep 9: https://pdf.hres.ca/dpd_pm/00062938.PDF. Accessed 2022 Jun 09.
25.Cosentyx (secukinumab): 75 mg/0.5 mL or 150 mg/1 mL solution for injection or 150 mg powder for solution for injection [product monograph]. Dorval (QC): Novartis Pharmaceuticals Canada Inc.; 2022 Apr 08: https://pdf.hres.ca/dpd_pm/00065374.PDF. Accessed 2022 Jun 09.
26.Skyrizi (risankizumab): 90 mg/mL or 150 mg/mL, sterile solution subcutaneous injection [product monograph]. St-Laurent (QC): AbbVie Corporation; 2022 Mar 16: https://pdf.hres.ca/dpd_pm/00065082.PDF. Accessed 2022 May 25.
27.Xeljanz (as tofacitinib citrate): 5 mg or 10 mg, oral tablets or Xeljanz XR (as tofacitinib citrate): 11 mg, extended-release oral tablets [product monograph]. Kirkland (QC): Pfizer Canada ULC; 2022 May 09: https://pdf.hres.ca/dpd_pm/00065725.PDF. Accessed 2022 May 25.
28.Cimzia (certolizumab pegol): 200 mg/mL solution for injection [product monograph]. Oakville (ON): UCB Canada Inc; 2019 Nov 13: https://pdf.hres.ca/dpd_pm/00053920.PDF. Accessed 2022 Jun 28.
29.Enbrel (etanercept): 50 mg/mL solution in a prefilled syringe or Enbrel (etanercept): 25 mg/vial of lyophilized powder in a vial for reconstitution for subcutaneous injection [product monograph]. Mississauga (ON): Amgen Canada Inc.; 2021 Mar 19: https://pdf.hres.ca/dpd_pm/00060454.PDF. Accessed 2022 Jun 28.
30.Simponi (golimumab injection): 50 mg/0.5 mL or 100 mg/1.0 mL solution for subcutaneous injection or Simponi (golimumab for injection): 50 mg/4.0 mL solution for intravenous infusion [product monograph]. Toronto (ON): Janssen Inc.; 2019 Jun 19: https://pdf.hres.ca/dpd_pm/00052895.PDF. Accessed 2022 Jun 28.
31.Remicade (infliximab): 100 mg/vial sterile lyophilized powder for solution, for intravenous infusion [product monograph]. Toronto (ON): Janssen Inc.; 2021 Oct 15: https://pdf.hres.ca/dpd_pm/00063218.PDF. Accessed 2022 Jun 28.
32.Rinvoq (upadacitinib): 15 mg or 30 mg, oral extended-release tablets [product monograph]. St-Laurent (QC): AbbVie Corporation; 2022 Jan 27: https://pdf.hres.ca/dpd_pm/00064540.PDF. Accessed 2022 Jun 28.
33.Orencia (abatacept): 250 mg /vial for intravenous infusion or Orencia (abatacept): 125 mg /mL solution for subcutaneous injection [product monograph]. Montreal (QC): Bristol-Myers Squibb Canada; 2019 Sep 09: https://pdf.hres.ca/dpd_pm/00052970.PDF. Accessed 2022 Jun 28.
34.Otezla (apremilast): 10 mg, 20 mg, and 30 mg oral tablets [product monograph]. Mississauga (ON): Amgen Canada Inc.; 2020 Aug 05: https://pdf.hres.ca/dpd_pm/00057499.PDF. Accessed 2022 Jun 28.
35.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 Guideline Statement. J Clin Epidemiol. 2016;75:40-46. PubMed
36.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2022 May 13.
37.Deodhar A, Helliwell PS, Boehncke WH, et al. Guselkumab in patients with active psoriatic arthritis who were biologic-naive or had previously received TNFalpha inhibitor treatment (DISCOVER-1): a double-blind, randomised, placebo-controlled phase 3 trial. Lancet. 2020;395(10230):1115-1125. PubMed
38.Rahman P, Mease PJ, Helliwell PS, et al. Guselkumab demonstrated an independent treatment effect in reducing fatigue after adjustment for clinical response-results from two phase 3 clinical trials of 1120 patients with active psoriatic arthritis. Arthritis Res Ther. 2021;23(1):190. PubMed
39.Mease PJ, Rahman P, Gottlieb AB, et al. Guselkumab in biologic-naive patients with active psoriatic arthritis (DISCOVER-2): a double-blind, randomised, placebo-controlled phase 3 trial. Lancet. 2020;395(10230):1126-1136. PubMed
40.Health Canada reviewer's report: Tremfya (guselkumab) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Tremfya (guselkumab), 100 mg/mL solution for injection. Toronto (ON): Janssen Inc.; 2022 May 17.
41.Committee for Medicinal Products for Human Use. Extension of indication variation assessment report: Tremfya (guselkumab). Amsterdam (NL): European Medicines Agency; 2020 Oct 15: https://www.ema.europa.eu/en/documents/variation-report/tremfya-h-c-4271-ii-17-epar-assessment-report-variation_en.pdf. Accessed 2022 Jun 09.
42.Janssen Inc. response to July 11, 2022 DRR request for additional information regarding Tremfya (guselkumab) DRR review [internal additional sponsor's information]. Toronto (ON): Janssen Inc.; 2022 Jul 18.
43.Fransen J, Antoni C, Mease PJ, et al. Performance of response criteria for assessing peripheral arthritis in patients with psoriatic arthritis: analysis of data from randomised controlled trials of two tumour necrosis factor inhibitors. Ann Rheum Dis. 2006;65(10):1373-1378. PubMed
44.Albert DA, Huang G, Dubrow G, Brensinger CM, Berlin JA, Williams HJ. Criteria for improvement in rheumatoid arthritis: alternatives to the American College of Rheumatology 20. J Rheumatol. 2004;31(5):856-866. PubMed
45.Coates LC, Fransen J, Helliwell PS. Defining minimal disease activity in psoriatic arthritis: a proposed objective target for treatment. Ann Rheum Dis. 2010;69(1):48-53. PubMed
46.van Riel PL. The development of the disease activity score (DAS) and the disease activity score using 28 joint counts (DAS28). Clin Exp Rheumatol. 2014;32(5 Suppl 85):S-65-74. PubMed
47.Wells G, Becker JC, Teng J, et al. Validation of the 28-joint Disease Activity Score (DAS28) and European League Against Rheumatism response criteria based on C-reactive protein against disease progression in patients with rheumatoid arthritis, and comparison with the DAS28 based on erythrocyte sedimentation rate. Ann Rheum Dis. 2009;68(6):954-960. PubMed
48.Duarte-García A, Leung YY, Coates LC, et al. Endorsement of the 66/68 Joint Count for the Measurement of Musculoskeletal Disease Activity: OMERACT 2018 Psoriatic Arthritis Workshop Report. J Rheumatol. 2019;46(8):996-1005. PubMed
49.Kwok T, Pope JE. Minimally important difference for patient-reported outcomes in psoriatic arthritis: Health Assessment Questionnaire and pain, fatigue, and global visual analog scales. J Rheumatol. 2010;37(5):1024-1028. PubMed
50.Leung YY, Holland R, Mathew AJ, et al. Clinical trial discrimination of physical function instruments for psoriatic arthritis: A systematic review. Semin Arthritis Rheum. 2020;50(5):1158-1181. PubMed
51.Ware JE, Jr. SF-36 health survey update. Spine (Phila Pa 1976). 2000;25(24):3130-3139. PubMed
52.Leung YY, Ho KW, Zhu TY, Tam LS, Kun EW, Li EK. Testing scaling assumptions, reliability and validity of medical outcomes study short-form 36 health survey in psoriatic arthritis. Rheumatology (Oxford). 2010;49(8):1495-1501. PubMed
53.Langley RG, Feldman SR, Nyirady J, van de Kerkhof P, Papavassilis C. The 5-point Investigator's Global Assessment (IGA) Scale: A modified tool for evaluating plaque psoriasis severity in clinical trials. J Dermatolog Treat. 2015;26(1):23-31. PubMed
54.Mrowietz U, Kragballe K, Reich K, et al. Definition of treatment goals for moderate to severe psoriasis: a European consensus. Arch Dermatol Res. 2011;303(1):1-10. PubMed
55.Healy PJ, Helliwell PS. Measuring clinical enthesitis in psoriatic arthritis: Assessment of existing measures and development of an instrument specific to psoriatic arthritis. Arthritis Care Res (Hoboken). 2008;59(5):686-691. PubMed
56.Calin A, Nakache JP, Gueguen A, Zeidler H, Mielants H, Dougados M. Defining disease activity in ankylosing spondylitis: is a combination of variables (Bath Ankylosing Spondylitis Disease Activity Index) an appropriate instrument? Rheumatology (Oxford). 1999;38(9):878-882. PubMed
57.Garrett S, Jenkinson T, Kennedy LG, Whitelock H, Gaisford P, Calin A. A new approach to defining disease status in ankylosing spondylitis: the Bath Ankylosing Spondylitis Disease Activity Index. J Rheumatol. 1994;21(12):2286-2291. PubMed
58.Zochling J, van der Heijde D, Burgos-Vargas R, et al. ASAS/EULAR recommendations for the management of ankylosing spondylitis. Ann Rheum Dis. 2006;65(4):442-452. PubMed
59.Janssen Research & Development, LLC. NCT01077362: A Study of the Safety and Efficacy of Ustekinumab in Patients With Psoriatic Arthritis With and Without Prior Exposure to Anti-TNF Agents. ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2014: https://www.clinicaltrials.gov/ct2/show/results/NCT01077362. Accessed 2022 Aug 08.
60.Gladman DD, Orbai AM, Gomez-Reino J, et al. Network Meta-Analysis of Tofacitinib, Biologic Disease-Modifying Antirheumatic Drugs, and Apremilast for the Treatment of Psoriatic Arthritis. Curr Ther Res Clin Exp. 2020;93:100601. PubMed
61.Lu C, Wallace BI, Waljee AK, Fu W, Zhang Q, Liu Y. Comparative efficacy and safety of targeted DMARDs for active psoriatic arthritis during induction therapy: A systematic review and network meta-analysis. Semin Arthritis Rheum. 2019;49(3):381-388. PubMed
62.Mease PJ, McInnes IB, Tam LS, et al. Comparative effectiveness of guselkumab in psoriatic arthritis: results from systematic literature review and network meta-analysis. Rheumatology (Oxford). 2021;60(5):2109-2121. PubMed
63.Qiu M, Xu Z, Gao W, et al. Fourteen small molecule and biological agents for psoriatic arthritis: A network meta-analysis of randomized controlled trials. Medicine. 2020;99(31):e21447. PubMed
64.Ruyssen-Witrand A, Perry R, Watkins C, et al. Efficacy and safety of biologics in psoriatic arthritis: a systematic literature review and network meta-analysis. RMD Open. 2020;6(1):02.
65.Song GG, Lee YH. Relative efficacy and safety of apremilast, secukinumab, and ustekinumab for the treatment of psoriatic arthritis. Z Rheumatol. 2018;77(7):613-620. PubMed
66.Torres T, Barcelos A, Filipe P, Fonseca JE. A Systematic Review With Network Meta-Analysis of the Available Biologic Therapies for Psoriatic Disease Domains. Front Med (Lausanne). 2020;7:618163. PubMed
67.Wu D, Yue J, Tam LS. Efficacy and safety of biologics targeting interleukin-6, −12/23 and −17 pathways for peripheral psoriatic arthritis: a network meta-analysis. Rheumatology (Oxford). 2018;57(3):563-571. PubMed
68.Guselkumab (Tremfya®) for psoriatic arthritis: systematic review and network metaanalyses. Global NMA technical report [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Tremfya (guselkumab), 100 mg/mL solution for injection. Toronto (ON): Janssen Inc.; 2022.
69.CADTH Reimbursement Review clinical guidance and pharmacoeconomic reports: upadacitinib (Rinvoq) for psoriatic arthritis. Can J Health Technol. 2021;1(10). https://www.cadth.ca/sites/default/files/DRR/2021/SR0658-combined-report.pdf. Accessed 2022 Aug 02.
70.Guyatt GH, Oxman AD, Kunz R, et al. GRADE guidelines 6. Rating the quality of evidence--imprecision. J Clin Epidemiol. 2011;64(12):1283-1293. PubMed
71.Wade R, Grosso A, South E, et al. Brodalumab for the treatment of moderate-to-severe plaque psoriasis: an evidence review group evaluation of a NICE single technology appraisal. Pharmacoeconomics. 2019;37(2):131-139. PubMed
72.Cameron C, Hutton B, Druchok C, et al. Importance of assessing and adjusting for cross-study heterogeneity in network meta-analysis: a case study of psoriasis. J Comp Eff Res. 2018;7(11):1037-1051. PubMed
73.Jansen JP, Trikalinos T, Cappelleri JC, et al. Indirect treatment comparison/network meta-analysis study questionnaire to assess relevance and credibility to inform health care decision making: an ISPOR-AMCP-NPC Good Practice Task Force report. Value Health. 2014;17(2):157-173. PubMed
74.Jansen JP, Schmid CH, Salanti G. Directed acyclic graphs can help understand bias in indirect and mixed treatment comparisons. J Clin Epidemiol. 2012;65(7):798-807. PubMed
75.Wade R, Sharif-Hurst S, Dias S. Patient characteristics as effect modifiers for psoriasis biologic treatment response: an assessment using network meta-analysis subgroups. Syst Rev. 2020;9(1):132. PubMed
76.Clinical Study Report: CNTO1959PSA3001. DISCOVER-1 60 week: a phase 3, multicenter, randomized, double-blind, placebo-controlled study evaluating the efficacy and safety of guselkumab administered subcutaneously in subjects with active psoriatic arthritis including those previously treated with biologic anti-TNFα agent(s) [internal sponsor's report]. Raritan (NJ): Janssen Research & Development, LLC; 2020 Apr 09.
77.Clinical Study Report: CNTO1959PSA3002. DISCOVER-2 52 week: a phase 3, multicenter, randomized, double-blind, placebo-controlled study evaluating the efficacy and safety of guselkumab administered subcutaneously in subjects with active psoriatic arthritis [internal sponsor's report]. Raritan (NJ): Janssen Research & Development, LLC; 2020 Mar 27.
78.Clinical Study Report: CNTO1959PSA3002. DISCOVER-2 112 week: a phase 3, multicenter, randomized, double-blind, placebo-controlled study evaluating the efficacy and safety of guselkumab administered subcutaneously in subjects with active psoriatic arthritis [internal sponsor's report]. Raritan (NJ): Janssen Research & Development, LLC; 2021 Jul 12.
79.Clinical Study Report: CNTO1959PSA3003. COSMOS week 56: phase 3b, multicenter, randomized, double-blind, placebo-controlled study to evaluate the efficacy and safety of guselkumab administered subcutaneously in participants with active psoriatic arthritis and an inadequate response to anti-tumor necrosis factor alpha (Anti-TNFα) therapy COSMOS [internal sponsor's report]. High Wycombe (UK): Janssen-Cilag Ltd.; 2021 Jul 12.
80.McInnes IB, Rahman P, Gottlieb AB, et al. Efficacy and Safety of Guselkumab, an Interleukin-23p19-Specific Monoclonal Antibody, Through One Year in Biologic-Naive Patients With Psoriatic Arthritis. Arthritis Rheumatol. 2021;73(4):604-616. PubMed
81.McInnes IB, Rahman P, Gottlieb AB, et al. Long-Term Efficacy and Safety of Guselkumab, a Monoclonal Antibody Specific to the p19 Subunit of Interleukin-23, Through Two Years: Results From a Phase III, Randomized, Double-Blind, Placebo-Controlled Study Conducted in Biologic-Naive Patients With Active Psoriatic Arthritis. Arthritis Rheumatol. 2022;74(3):475-485. PubMed
82.Mease PJ, Helliwell PS, Gladman DD, et al. Efficacy of guselkumab on axial involvement in patients with active psoriatic arthritis and sacroiliitis: a post-hoc analysis of the phase 3 DISCOVER-1 and DISCOVER-2 studies. Lancet Rheumatol. 2021;3(10):e715-e723.
83.Ritchlin CT, Mease PJ, Boehncke WH, et al. Sustained and improved guselkumab response in patients with active psoriatic arthritis regardless of baseline demographic and disease characteristics: pooled results through week 52 of two phase III, randomised, placebo-controlled studies. RMD Open. 2022;8(1). PubMed
84.Ritchlin CT, Helliwell PS, Boehncke WH, et al. Guselkumab, an inhibitor of the IL-23p19 subunit, provides sustained improvement in signs and symptoms of active psoriatic arthritis: 1 year results of a phase III randomised study of patients who were biologic-naive or TNFalpha inhibitor-experienced. RMD Open. 2021;7(1):02.
85.Deodhar A, Gottlieb AB, Boehncke WH, et al. Efficacy and safety of guselkumab in patients with active psoriatic arthritis: a randomised, double-blind, placebo-controlled, phase 2 study. Lancet. 2018;391(10136):2213-2224. PubMed
86.Helliwell PS, Deodhar A, Gottlieb AB, et al. Composite Measures of Disease Activity in Psoriatic Arthritis: Comparative Instrument Performance Based on the Efficacy of Guselkumab in an Interventional Phase II Trial. Arthritis Care Res (Hoboken). 2020;72(11):1579-1588. PubMed
87.Mease PJ, Gladman DD, Deodhar A, et al. Impact of guselkumab, an interleukin-23 p19 subunit inhibitor, on enthesitis and dactylitis in patients with moderate to severe psoriatic arthritis: results from a randomised, placebo-controlled, phase II study. RMD Open. 2020;6(2):07.
88.van Gestel AM, Haagsma CJ, van Riel PL. Validation of rheumatoid arthritis improvement criteria that include simplified joint counts. Arthritis Rheum. 1998;41(10):1845-1850. PubMed
89.Felson DT, Anderson JJ, Boers M, et al. The American College of Rheumatology preliminary core set of disease activity measures for rheumatoid arthritis clinical trials. The Committee on Outcome Measures in Rheumatoid Arthritis Clinical Trials. Arthritis Rheum. 1993;36(6):729-740. PubMed
90.Gladman D, Cook R, Schentag C, et al. The clinical assessment of patients with psoriatic arthritis (PsA): Results of a validation study of the Spondyloarthritis Research Consortium of Canada. J Rheumatol. 2004;31(6):1126-1131. PubMed
91.Felson DT, Anderson JJ, Boers M, et al. American College of Rheumatology. Preliminary definition of improvement in rheumatoid arthritis. Arthritis Rheum. 1995;38(6):727-735. PubMed
92.Braun J, Davis J, Dougados M, et al. First update of the international ASAS consensus statement for the use of anti-TNF agents in patients with ankylosing spondylitis. Ann Rheum Dis. 2006;65(3):316-320. PubMed
93.Haywood KL, A MG, Jordan K, Dziedzic K, Dawes PT. Disease-specific, patient-assessed measures of health outcome in ankylosing spondylitis: reliability, validity and responsiveness. Rheumatology (Oxford). 2002;41(11):1295-1302. PubMed
94.Reilly MC, Gooch KL, Wong RL, Kupper H, van der Heijde D. Validity, reliability and responsiveness of the Work Productivity and Activity Impairment Questionnaire in ankylosing spondylitis. Rheumatology (Oxford). 2010;49(4):812-819. PubMed
95.Healy PJ, Helliwell PS. Psoriatic arthritis quality of life instrument: an assessment of sensitivity and response to change. J Rheumatol. 2008;35(7):1359-1361. PubMed
96.Cella D, Wilson H, Shalhoub H, et al. Content validity and psychometric evaluation of Functional Assessment of Chronic Illness Therapy-Fatigue in patients with psoriatic arthritis. J Patient Rep Outcomes. 2019;3(1):30. PubMed
97.Chandran V, Bhella S, Schentag C, Gladman DD. Functional assessment of chronic illness therapy-fatigue scale is valid in patients with psoriatic arthritis. Ann Rheum Dis. 2007;66(7):936-939. PubMed
98.Fries JF, Spitz P, Kraines RG, Holman HR. Measurement of patient outcome in arthritis. Arthritis Rheum. 1980;23(2):137-145. PubMed
99.Leung YY, Orbai AM, de Wit M, et al. Comparing the Patient-Reported Physical Function Outcome Measures in a Real-Life International Cohort of Patients With Psoriatic Arthritis. Arthritis Care Res (Hoboken). 2021;73(4):593-602. PubMed
100.Leung YY, Tillett W, Hojgaard P, et al. Test-retest Reliability for HAQ-DI and SF-36 PF for the Measurement of Physical Function in Psoriatic Arthritis. J Rheumatol. 2021;15:15. PubMed
101.Callis Duffin K, Bushmakin AG, Cappelleri JC, Mallbris L, Mamolo C. A multi-item Physician Global Assessment scale to assess psoriasis disease severity: validation based on four phase III tofacitinib studies. BMC Dermatol. 2019;19(1):8. PubMed
102.Cappelleri JC, Bushmakin AG, Harness J, Mamolo C. Psychometric validation of the physician global assessment scale for assessing severity of psoriasis disease activity. Qual Life Res. 2013;22(9):2489-2499. PubMed
103.Robinson A, Kardos M, Kimball AB. Physician Global Assessment (PGA) and Psoriasis Area and Severity Index (PASI): why do both? A systematic analysis of randomized controlled trials of biologic agents for moderate to severe plaque psoriasis. J Am Acad Dermatol. 2012;66(3):369-375. PubMed
104.Coates LC, Cook R, Lee K-A, Chandran V, Gladman DD. Frequency, predictors, and prognosis of sustained minimal disease activity in an observational psoriatic arthritis cohort. Arthritis Care Res (Hoboken). 2010;62(7):970-976. PubMed
105.Coates LC, Helliwell PS. Validation of minimal disease activity criteria for psoriatic arthritis using interventional trial data. Arthritis Care Res (Hoboken). 2010;62(7):965-969. PubMed
106.Coates LC, Strand V, Wilson H, et al. Measurement properties of the minimal disease activity criteria for psoriatic arthritis. RMD Open. 2019;5(2):e001002. PubMed
107.Coates LC, Helliwell PS. Defining Low Disease Activity States in Psoriatic Arthritis using Novel Composite Disease Instruments. J Rheumatol. 2016;43(2):371-375. PubMed
108.Simpson MJ, Chow C, Morgenstern H, Luger TA, Ellis CN. Comparison of three methods for measuring psoriasis severity in clinical studies (Part 2 of 2): use of quality of life to assess construct validity of the Lattice System Physician's Global Assessment, Psoriasis Area and Severity Index and Static Physician's Global Assessment. J Eur Acad Dermatol Venereol. 2015;29(7):1415-1420. PubMed
109.Berth-Jones J, Grotzinger K, Rainville C, et al. A Study examining inter- and intrarater reliability of three scales for measuring severity of psoriasis: Psoriasis Area and Severity Index, Physician's Global Assessment and Lattice System Physician's Global Assessment. Br J Dermatol. 2006;155(4):707-713. PubMed
110.Bozek A, Reich A. The reliability of three psoriasis assessment tools: psoriasis area and severity index, body surface area and physician global assessment. Adv Clin Exp Med. 2017;26(5):851-856. PubMed
111.Langley RG, Ellis CN. Evaluating psoriasis with Psoriasis Area and Severity Index, Psoriasis Global Assessment, and Lattice System Physician's Global Assessment. J Am Acad Dermatol. 2004;51(4):563-569. PubMed
112.Fink C, Alt C, Uhlmann L, Klose C, Enk A, Haenssle HA. Intra- and interobserver variability of image-based PASI assessments in 120 patients suffering from plaque-type psoriasis. J Eur Acad Dermatol Venereol. 2018;32(8):1314-1319. PubMed
113.Puzenat E, Bronsard V, Prey S, et al. What are the best outcome measures for assessing plaque psoriasis severity? A systematic review of the literature. J Eur Acad Dermatol Venereol. 2010;24 (Suppl 2):10-16. PubMed
114.Mattei PL, Corey KC, Kimball AB. Psoriasis Area Severity Index (PASI) and the Dermatology Life Quality Index (DLQI): the correlation between disease severity and psychological burden in patients treated with biological therapies. J Eur Acad Dermatol Venereol. 2014;28(3):333-337. PubMed
115.Ware JE, Jr., Sherbourne CD. The MOS 36-item short-form health survey (SF-36). I. Conceptual framework and item selection. Med Care. 1992;30(6):473-483. PubMed
116.Frendl DM, Ware JE, Jr. Patient-reported functional health and well-being outcomes with drug therapy: a systematic review of randomized trials using the SF-36 health survey. Med Care. 2014;52(5):439-445. PubMed
117.Tillett W, Jadon D, Shaddick G, et al. Feasibility, reliability, and sensitivity to change of four radiographic scoring methods in patients with psoriatic arthritis. Arthritis Care Res (Hoboken). 2014;66(2):311-317. PubMed
118.Gladman DD, Mease PJ, Healy P, et al. Outcome measures in psoriatic arthritis. J Rheumatol. 2007;34(5):1159-1166. PubMed
119.Bruce B, Fries JF. The Health Assessment Questionnaire (HAQ). Clin Exp Rheumatol. 2005;23(5 Suppl 39):S14-18. PubMed
120.Anderson J, Caplan L, Yazdany J, et al. Rheumatoid arthritis disease activity measures: American College of Rheumatology recommendations for use in clinical practice. Arthritis Care Res (Hoboken). 2012;64(5):640-647. PubMed
121.Bruce B, Fries JF. The Stanford Health Assessment Questionnaire: dimensions and practical applications. Health Qual Life Outcomes. 2003;1:20. PubMed
122.Bruce B, Fries JF. The Stanford Health Assessment Questionnaire: a review of its history, issues, progress, and documentation. J Rheumatol. 2003;30(1):167-178. PubMed
123.Mease P, Strand V, Gladman D. Functional impairment measurement in psoriatic arthritis: Importance and challenges. Semin Arthritis Rheum. 2018;48(3):436-448. PubMed
124.Blackmore MG, Gladman DD, Husted J, Long JA, Farewell VT. Measuring health status in psoriatic arthritis: the Health Assessment Questionnaire and its modification. J Rheumatol. 1995;22(5):886-893. PubMed
125.Husted JA, Gladman DD, Long JA, Farewell VT. A modified version of the Health Assessment Questionnaire (HAQ) for psoriatic arthritis. Clin Exp Rheumatol. 1995;13(4):439-443. PubMed
126.Feldman SR, Krueger GG. Psoriasis assessment tools in clinical trials. Ann Rheum Dis. 2005;64 (Suppl 2):ii65-68. PubMed
127.Perez-Chada LM, Salame NF, Ford AR, et al. Investigator and Patient Global Assessment Measures for Psoriasis Clinical Trials: A Systematic Review on Measurement Properties from the International Dermatology Outcome Measures (IDEOM) Initiative. Am J Clin Dermatol. 2020;21(3):323-338. PubMed
128.Gourraud PA, Le Gall C, Puzenat E, Aubin F, Ortonne JP, Paul CF. Why statistics matter: limited inter-rater agreement prevents using the psoriasis area and severity index as a unique determinant of therapeutic decision in psoriasis. J Invest Dermatol. 2012;139(9):2171-2175. PubMed
129.Queiro R, Cañete JD, Montilla C, et al. Minimal disease activity and impact of disease in psoriatic arthritis: a Spanish cross-sectional multicenter study. Arthritis Res Ther. 2017;19(1):72. PubMed
130.Lubrano E, Spadaro A. Pharmacoeconomic burden in the treatment of psoriatic arthritis: from systematic reviews to real clinical practice studies. BMC Musculoskelet Disord. 2014;15:25. PubMed
131.Feldman SR, Menter A, Koo JY. Improved health-related quality of life following a randomized controlled trial of alefacept treatment in patients with chronic plaque psoriasis. Br J Dermatol. 2004;150(2):317-326. PubMed
132.Ashcroft DM, Wan Po AL, Williams HC, Griffiths CE. Clinical measures of disease severity and outcome in psoriasis: a critical appraisal of their quality. Br J Dermatol. 1999;141(2):185-191. PubMed
133.Weisman S, Pollack CR, Gottschalk RW. Psoriasis disease severity measures: comparing efficacy of treatments for severe psoriasis. J Dermatolog Treat. 2003;14(3):158-165. PubMed
134.Carlin CS, Feldman SR, Krueger JG, Menter A, Krueger GG. A 50% reduction in the Psoriasis Area and Severity Index (PASI 50) is a clinically significant endpoint in the assessment of psoriasis. J Am Acad Dermatol. 2004;50(6):859-866. PubMed
135.Belinchon Romero I, Dauden E, Ferrandiz Foraster C, Gonzalez-Cantero A, Carrascosa Carrillo JM, Psoriasis Group of the Spanish Academy of Dermatology and Venereology. PASI 100 response rates in moderate to severe psoriasis: a systematic literature review and analysis of clinical practice guidelines. J Dermatolog Treat. 2022;33(3):1661-1669. PubMed
136.Spuls PI, Lecluse LL, Poulsen ML, Bos JD, Stern RS, Nijsten T. How good are clinical severity and outcome measures for psoriasis?: quantitative evaluation in a systematic review. J Invest Dermatol. 2010;130(4):933-943. PubMed
137.Maruish ME. Determining important differences in scores. User's manual for the SF-36v2 health survey. 3rd ed. Lincoln (RI): Quality Metric Incorporated; 2011:169-177.
138.McDowell I. Measuring health: a guide to rating scales and questionnaires. New York (NY): Oxford University Press; 2006.
139.Hays RD, Morales LS. The RAND-36 measure of health-related quality of life. Ann Med. 2001;33(5):350-357. PubMed
140.Samsa GP, Edelman D, Rothman M, Williams G, Lipscomb J, Matchar D. Determining Clinically Important Differences in Health Status Measures: A General Approach With Illustration to the Health Utilities Index Mark II. Pharmacoeconomics. 1999;15:141-155. PubMed
141.Strand V, Khanna D, Singh JA, Forsythe A, Edwards NL. Improved health-related quality of life and physical function in patients with refractory chronic gout following treatment with pegloticase: evidence from phase III randomized controlled trials. J Rheumatol. 2012;39(7):1450-1457. PubMed
Appendix 1: Literature Search Strategy
Note that this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases:
MEDLINE All (1946-present)
Embase (1974-present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: May 31, 2022
Alerts: Bi-weekly search updates until project completion
Search filters applied: No filters were applied to limit the retrieval by study type.
Limits:
Publication date limit: none
Language limit: none
Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
MeSH | Medical Subject Heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Author keyword heading word (MEDLINE) |
.dq | Candidate term word (Embase) |
.pt | Publication type |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multi-Database Strategy
(tremfya* or guselkumab* or cnto 1959 or cnto1959 or 089658A12D).ti,ab,kf,ot,hw,rn,nm.
1 use medall
*guselkumab/
(tremfya* or guselkumab* or cnto 1959 or cnto1959).ti,ab,kf,dq.
or/3-4
5 use oemezd
6 not (conference abstract or conference review).pt.
2 or 7
remove duplicates from 8
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
[Search – Studies with results | tremfya OR guselkumab OR cnto-1959 OR cnto1959 | Psoriatic Arthritis]
WHO ICTRP
International Clinical Trials Registry Platform, produced by the WHO. Targeted search used to capture registered clinical trials.
[Search terms – (tremfya OR guselkumab OR cnto-1959 OR cnto1959) AND psoriatic arthritis]
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
[Search terms – tremfya OR guselkumab]
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
[Search terms – guselkumab AND psoriatic arthritis]
Grey Literature
Search dates: May 18, 2022 – May 31, 2022
Keywords: [tremfya OR guselkumab OR cnto-1959 OR cnto1959 OR psoriatic arthritis OR psoriasis OR arthritis]
Limits: Publication years: none
Updated: Search updated before the meeting of the CADTH CDEC
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Internet Search.
Appendix 2: Excluded Studies
Note that this appendix has not been copy-edited.
Reference | Reason for exclusion |
|---|---|
Rahman P, Mease PJ, Helliwell PS, et al. Guselkumab demonstrated an independent treatment effect in reducing fatigue after adjustment for clinical response-results from two phase 3 clinical trials of 1120 patients with active psoriatic arthritis. Arthritis Res Ther. 2021;23(1):190. McInnes IB, Rahman P, Gottlieb AB, et al. Efficacy and Safety of Guselkumab, an Interleukin-23p19-Specific Monoclonal Antibody, Through One Year in Biologic-Naive Patients With Psoriatic Arthritis. Arthritis rheumatol. 2021;73(4):604 to 616. McInnes IB, Rahman P, Gottlieb AB, et al. Long-Term Efficacy and Safety of Guselkumab, a Monoclonal Antibody Specific to the p19 Subunit of Interleukin-23, Through Two Years: Results From a phase III, Randomized, Double-Blind, Placebo-Controlled Study Conducted in Biologic-Naive Patients With Active Psoriatic Arthritis. Arthritis rheumatol. 2022;74(3):475 to 485. Mease PJ, Helliwell PS, Gladman DD, et al. Efficacy of guselkumab on axial involvement in patients with active psoriatic arthritis and sacroiliitis: a post-hoc analysis of the phase 3 DISCOVER-1 and DISCOVER-2 studies. The Lancet Rheumatology. 2021;3(10):e715-e723. Ritchlin CT, Mease PJ, Boehncke WH, et al. Sustained and improved guselkumab response in patients with active psoriatic arthritis regardless of baseline demographic and disease characteristics: pooled results through week 52 of two phase III, randomized, placebo-controlled studies. RMD Open. 2022;8(1). Ritchlin CT, Helliwell PS, Boehncke WH, et al. Guselkumab, an inhibitor of the IL-23p19 subunit, provides sustained improvement in signs and symptoms of active psoriatic arthritis: 1 year results of a phase III randomized study of patients who were biologic-naive or TNF alpha inhibitor-experienced. RMD Open. 2021;7(1):02. Clinical Study Report: CNTO1959PSA3001. DISCOVER-1 60 week: A phase 3, multicenter, randomized, double-blind, placebo-controlled study evaluating the efficacy and safety of guselkumab administered subcutaneously in subjects with active psoriatic arthritis including those previously treated with biologic anti-TNFAlpha agent(s) [internal sponsor's report]. Raritan (NJ): Janssen Research and Development, LLC; 2020 Apr 09. Clinical Study Report: CNTO1959PSA3002. DISCOVER-2 52 week: a phase 3, multicenter, randomized, double-blind, placebo-controlled study evaluating the efficacy and safety of guselkumab administered subcutaneously in subjects with active psoriatic arthritis [internal sponsor's report]. Raritan (NJ): Janssen Research and Development, LLC; 2020 Mar 27. Clinical Study Report: CNTO1959PSA3002. DISCOVER-2 112 week: a phase 3, multicenter, randomized, double-blind, placebo-controlled study evaluating the efficacy and safety of guselkumab administered subcutaneously in subjects with active psoriatic arthritis [internal sponsor's report]. Raritan (NJ): Janssen Research and Development, LLC; 2021 Jul 12. Clinical Study Report: CNTO1959PSA3003. COSMOS week 56: phase 3b, multicenter, randomized, double-blind, placebo-controlled study to evaluate the efficacy and safety of guselkumab administered subcutaneously in participants with active psoriatic arthritis and an inadequate response to anti-tumour necrosis factor alpha (Anti-TNFAlpha) therapy COSMOS [internal sponsor's report]. High Wycombe (UK): Janssen-Cilag Ltd.; 2021 Jul 12. | |
Deodhar A, Gottlieb AB, Boehncke WH, et al. Efficacy and safety of guselkumab in patients with active psoriatic arthritis: a randomized, double-blind, placebo-controlled, phase 2 study. Lancet. 2018;391(10136):2213 to 2224. Helliwell PS, Deodhar A, Gottlieb AB, et al. Composite Measures of Disease Activity in Psoriatic Arthritis: Comparative Instrument Performance Based on the Efficacy of Guselkumab in an Interventional phase II Trial. Arthritis Care Res (Hoboken). 2020;72(11):1579 to 1588. Mease PJ, Gladman DD, Deodhar A, et al. Impact of guselkumab, an interleukin-23 p19 subunit inhibitor, on enthesitis and dactylitis in patients with moderate to severe psoriatic arthritis: results from a randomized, placebo-controlled, phase II study. RMD Open. 2020;6(2):07. |
Appendix 3: Detailed Outcome Data
Note this appendix has not been copy-edited.
Table 42: ACR Response at Week 16
Outcome | DISCOVER-1 (FAS) | DISCOVER-2 (FAS) | COSMOS (FAS) | |||
|---|---|---|---|---|---|---|
GUSE q8w N = 127 | PBO N = 126 | GUSE q8w N = 248 | PBO N = 246 | GUSE q8w N = 189 | PBO N = 96 | |
ACR 20 at week 16a | ||||||
ACR 20 response at week 16, n (%) | 66 (52.0) | 32 (25.4) | 137 (55.2) | 83 (33.7) | 78 (41.3) | 16 (16.7) |
% difference vs. PBO (95% CI) | 26.7 (15.3 to 38.1) | reference | 21.5 (13.1 to 30.0) | reference | 24.6 (14.5 to 34.7) | reference |
P value | < 0.001b | reference | Not testedc | reference | < 0.001d | reference |
ACR 50 at week 16a | ||||||
ACR 50 response at week 16, n (%) | 29 (22.8) | 16 (12.7) | 71 (28.6) | 23 (9.3) | 27 (14.3) | 5 (5.2) |
% difference vs. PBO (95% CI) | 10.2 (1.0 to 19.3) | reference | 19.3 (12.6 to 25.9) | reference | 9.0 (2.4, 15.6) | reference |
P value | 0.086 b | reference | Not testedc | reference | < 0.001d | reference |
ACR 20 = American College of Rheumatology 20% improvement; ACR 50 = American Collect of Rheumatology 50% improvement; CI = confidence interval; FAS = full analysis set; GUSE = guselkumab; PBO = placebo; q8w = every 8 weeks.
aBased on composite estimand (either observed response data or nonresponse for patients who met treatment failure criteria). P valued based on stratified CMH test 95% CI based on the Wald statistic. DISCOVER-1 study CMH stratified by baseline use of non-biologic DMARDs (yes, no) and prior exposure to TNF alpha inhibitors (yes, no). DISCOVER-2 study CMH stratified by baseline use of non-biologic DMARDs (yes, no) and CRP before randomization (< 2 mg/dL vs. ≥ 2 mg/dL).COSMOS study stratified by baseline use of non-biologic DMARDs (yes, no) and number of prior TNF alpha inhibitors (1 or 2). Patients with missing data were imputed as nonresponders.
bControlled for multiple testing based on the global-specific testing procedure but not US-specific testing procedure. US-specific procedure was preferred by Health Canada.
cOutcome was not formally tested for statistical significance due to failure of a prior outcome according to the global testing procedure. This outcome was not controlled for multiple testing according to the US-based testing procedure.
dP value has not been adjusted for multiple testing (i.e., the type I error rate has not been controlled).
Source: Clinical Study Report for DISCOVER-1 (24-weeks),11 Clinical Study Report for DISCOVER-2 (24-weeks),12 Clinical Study Report for COSMOS (24-week).13
Table 43: Change From Baseline in DAS 28 CRP Scores at Week 24
Outcome | DISCOVER-1 (FAS) | DISCOVER-2 | COSMOS (FAS) | |||
|---|---|---|---|---|---|---|
GUSE q8w N = 127 | PBO N = 126 | GUSE q8w N = 248 | PBO N = 246 | GUSE q8w N = 189 | PBO N = 96 | |
Change from baseline to week 24 in DAS 28 CRP a | ||||||
Number of patients contributing to the analysis | 127 | 126 | 248 | 246 | 187 | 96 |
Baseline, mean (SD) | 4.92 (1.08) | 4.94 (0.99) | 5.14 (0.99) | 5.26 (0.91) | 4.9 (1.0) | 4.6 (0.8) |
Change from baseline, LS mean (95% CI) | −1.43 (−1.61 to −1.24) | −0.70 (−0.89 to −0.51) | −1.59 (−1.72 to −1.45) | −0.97 (−1.11 to −0.84) | −0.95 (−1.15 to −0.75) | −0.30 (−0.54 to −0.05) |
LS mean difference (95% CI) vs. placebo | −0.73 (−0.98 to −0.48) | reference | −0.61 (−0.80 to −0.43) | reference | −0.66 (−0.90 to −0.41) | reference |
P value | < 0.001b | reference | < 0.001 b | reference | < 0.001c | reference |
CI = confidence interval; DAS 28 CRP = Disease Activity Score 28-Joint Count with C-reactive protein; FAS = full analysis set; GUSE = guselkumab; LS = least squares; PBO = placebo; q8w = every 8 weeks, SD = standard deviation.
aChange from baseline based on observed data or 0 (no improvement) if a patient met the treatment failure criteria before week 24 (composite estimand). DISCOVER-1 analyzed using ANCOVA model with missing data imputed using multiple imputation assuming data were missing at random. COSMOS analyzed using MMRM under missing at random assumption.
bControlled for multiple testing based on the global testing procedure but not US-specific testing procedure. US-specific procedure was preferred by Health Canada.
cP value has not been adjusted for multiple testing (i.e., the type I error rate has not been controlled).
Source: Clinical Study Report for DISCOVER-1 (24-weeks),11 Clinical Study Report for DISCOVER-2 (24-weeks),12 Clinical Study Report for COSMOS (24-week).13
Figure 11: ACR 20 Response for Subgroups Based on Prior Treatment History — DISCOVER-1 (FAS) — Redacted

ACR 20 = American College of Rheumatology 20% improvement; CI = confidence interval; DMARD = disease-modifying antirheumatic drug; eCRF = electronic case report form; FAS = full analysis set; HAQ-DI = Health Assessment Questionnaire Disability Index; IR = inadequate response; OR = odds ratio; q8w = every 8 weeks; TNF = tumour necrosis factor.
a Patients either have an observed ACR 20 response status or met a treatment failure criterion.
b Odds ratio, CI for odds ratio, and interaction p values are based on logistic regression.
c Patients with missing data are assumed to be nonresponders.
d ACR 20 response is defined as ≥ 20% improvement from baseline in both tender joint count (68 joints) and swollen joint count (66 joints), and ≥ 20% improvement from baseline in at least 3 of the 5 assessments: patient’s assessment of pain, patient’s global assessment of disease activity, physician’s global assessment of disease activity, HAQ-DI, and CRP.
Figure redacted at the request of the sponsor.
Source: Clinical Study Report for DISCOVER-1 (24-weeks).11
Figure 12: ACR 20 Response for Subgroups Based on Baseline Treatments — DISCOVER-1 (FAS) — Redacted

ACR 20 = American College of Rheumatology 20% improvement; CI = confidence interval; CRP = C-reactive protein; DMARD = disease-modifying antirheumatic drug; eCRF = electronic case report form; FAS = full analysis set; HAQ-DI = Health Assessment Questionnaire Disability Index; IR = inadequate response; MTX = methotrexate; NSAID = nonsteroidal anti-inflammatory drug; OR = odds ratio; q8w = every 8 weeks; TNF = tumour necrosis factor.
a Patients either have an observed ACR 20 response status or met a treatment failure criterion.
b Odds ratio, CI for odds ratio, and interaction p values are based on logistic regression.
c Patients with missing data are assumed to be nonresponders.
d ACR 20 response is defined as ≥ 20% improvement from baseline in both tender joint count (68 joints) and swollen joint count (66 joints), and ≥ 20% improvement from baseline in at least 3 of the 5 assessments: patient’s assessment of pain, patient’s global assessment of disease activity, physician’s global assessment of disease activity, HAQ-DI, and CRP.
Figure redacted at the request of the sponsor.
Source: Clinical Study Report for DISCOVER-1 (24-weeks).11
Figure 13: ACR 20 Response for Subgroups Based on Prior Treatment History — DISCOVER-2 (FAS) — Redacted

ACR 20 = American College of Rheumatology 20% improvement; CI = confidence interval; CRP = C-reactive protein; DMARD = disease-modifying antirheumatic drug; eCRF = electronic case report form; FAS = full analysis set; HAQ-DI = Health Assessment Questionnaire Disability Index; IR = inadequate response; MTC = methotrexate; OR = odds ratio; q8w = every 8 weeks.
a Patients either have an observed ACR 20 response status or met a treatment failure criterion.
b Odds ratio, CI for odds ratio, and interaction p values are based on logistic regression.
c Patients with missing data are assumed to be nonresponders.
d ACR 20 response is defined as ≥ 20% improvement from baseline in both tender joint count (68 joints) and swollen joint count (66 joints), and ≥ 20% improvement from baseline in at least 3 of the 5 assessments: patient’s assessment of pain, patient’s global assessment of disease activity, physician’s global assessment of disease activity, HAQ-DI, and CRP.
Figure redacted at the request of the sponsor.
Source: Clinical Study Report for DISCOVER-2 (24-weeks).12
Figure 14: ACR 20 Response for Subgroups Based on Baseline Treatments — DISCOVER-2 (FAS) — Redacted

ACR 20 = American College of Rheumatology 20% improvement; CI = confidence interval; CRP = C-reactive protein; DMARD = disease-modifying antirheumatic drug; eCRF = electronic case report form; FAS = full analysis set; HAQ-DI = Health Assessment Questionnaire Disability Index; IR = inadequate response; OR = odds ratio; q8w = every 8 weeks.
a Patients either have an observed ACR 20 response status or met a treatment failure criterion.
b Odds ratio, CI for odds ratio, and interaction p values are based on logistic regression.
c Patients with missing data are assumed to be nonresponders.
d ACR 20 response is defined as ≥ 20% improvement from baseline in both tender joint count (68 joints) and swollen joint count (66 joints), and ≥ 20% improvement from baseline in at least 3 of the 5 assessments: patient’s assessment of pain, patient’s global assessment of disease activity, physician’s global assessment of disease activity, HAQ-DI, and CRP.
Figure redacted at the request of the sponsor.
Source: Clinical Study Report for DISCOVER-2 (24-weeks).12
Appendix 4: Description and Appraisal of Outcome Measures
Note this appendix has not been copy-edited.
Aim
To describe the following 13 outcome measures and review their measurement properties (validity, reliability, responsiveness to change, and MID):
ACR 20, ACR 50, ACR 70
BASDAI
DAS 28 CRP
FACIT-Fatigue
HAQ-DI
IGA
LEI
MDA
PASI 75, PASI 90, PASI 100)
Resolution of Dactylitis – Dactylitis score
vdH-S score
SF-36 MCS and PCS.
Findings
Table 44: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
ACR 20, 50, 70 | ACR 20, ACR 50, and ACR 70 responses represent at least a 20%, 50%, and 70% improvement, respectively, in tender and swollen joint counts and in 3 of the 5 additional criteria: Patient global assessment of disease activity; Physician Global Assessment of disease activity; patient assessment of pain; health assessment questionnaire; CRP or ESR.88 | Validity Content validity: The ACR tender and swollen joint counts have been deemed as appropriate for disease activity for patients with rheumatoid arthritis by the ACR expert committee.89 Construct validity was assessed using the known-group method in a study of patients with PsA and peripheral arthritis using data from 2 RCTs (n = 164) of TNF inhibitors. the number of patients who achieved ACR 20, 50, 70 in both active drug groups was significantly higher than the placebo groups.43 Reliability Inter-observer reliability: Generally, Gladman et al. (2004) reported poor to substantial inter-observer reliability (ICC ranged from 0.10 to 0.80) in a study of 10 patients with PsA each assessed by 10 observers.90 Responsiveness In patients with PsA and peripheral arthritis from 2 RCTs of TNF inhibitors (n = 164), the SRM ranged from –1.98 to –0.61 and ES ranged from –2.84 to –0.57 for the active drug groups while the SRM ranged from –0.40 to 0.02 and ES ranged from –0.37 to 0.15 in the placebo groups.43 | ACR 20 is generally used to define improvement, indicating a response to treatment.43,44,91 The MID for ACR has not been estimated in patients with PsA. |
BASDAI | The BASDAI is a self-administered disease-specific questionnaire, a composite index containing 6 questions related to 5 major symptoms of ankylosing spondylitis (fatigue, spinal pain, peripheral joint pain and swelling, localized tenderness, and morning stiffness). Scores range from 0 to 10 with higher scores indicating greater disease activity.56,57,92 | Validity The construct validity was assessed using the known-group approach. In 1 study (n = 349), patients with AS who were able to work had significantly higher BASDAI scores compared with those unable to work due to ill health.93 Convergent validity: A strong correlation was observed between the BASDAI and ASQoL (Pearson’s correlation coefficient, r = 0.79) in 1 study (n = 349) of patients with AS.93 Reliability The BASDAI demonstrated strong internal consistency (Cronbach alpha = 0.87) in 1 study (n = 349) of patients with AS.93 The BASDAI demonstrated a test-retest reliability with a 2-week interval (ICC 0.87, 95% CI, 0.83 to 0.91) in 1 study (n = 349) of patients with AS.93 Responsiveness In 1 study (n = 349), the BASDAI produced high levels of responsiveness for groups of patients whose AS-specific (modified SRM ranged from –0.47 to 0.60) or general health (modified SRM ranged from –1.02 to 0.75) had improved or deteriorated according to transition question responses.93 The validity, reliability, and responsiveness of the BASDAI instrument have not been evaluated in patients with PsA. | In 1 RCT of adalimumab (n = 205), a change of −1.96 on the BASDAI scale was considered meaningful in patients with AS.94 The MID for the BASDAI has not been estimated in patients with PsA. |
DAS 28 | The DAS 28 is a measure of disease activity in RA based on a count of 28 swollen and tender joints plus the ESR or CRP, and a general health assessment using VAS.46 The range of the DAS 28 is 0 to 9.4 with higher scores indicating more active disease.47 | Validity Criterion validity: In an analysis of data from 2 RCTs of abatacept (n = 391), there was good agreement in classifying patients with RA as none, moderate and good EULAR responders using DAS 28 CRP and DAS 28 (ESR), with a k of 0.80 (95% CI, 0.76 to 0.83).47 Convergent validly: Moderate to weak correlations were observed between DAS 28 and the swollen and tender joint counts, patient and physician global VAS, CRP level, HAQ, and LEI (r ranged from –0.157 to 0.462) in 1 study of patients with PsA.95 Reliability No evidence of reliability was identified for DAS 28 in patients with PsA. Responsiveness In an analysis of data from 2 RCTs of abatacept (n = 246), responsiveness was assessed for the DAS 28 CRP using EULAR response criteria as anchors. The SRM was –0.31 in patients with RA.47 The reliability and responsiveness of the DAS 28 instrument have not been evaluated in patients with PsA. | In 1 RCT methotrexate, sulfasalazine, or both (n = 105), a value of ≤ 3.2 was defined as the DAS 28 threshold for a low disease activity state and < 2.6 as the threshold for remission in patients with early RA.88 The MID for the DAS 28 has not been estimated in patients with PsA. |
FACIT-Fatigue | The FACIT-Fatigue is a 13-item patient-reported measure of fatigue symptoms. It is scored from 0 to 52 with lower scores indicating higher levels of fatigue.8 | Validity Content validity: The FACIT-Fatigue demonstrated good content validity in 1 study of 12 patients with PsA. Patients considered the FACIT-Fatigue items relevant to their disease experience and understood the item content and response options well.8,96 Convergent validity: Strong correlations with SF-36 domains, generally exceeding 0.60 (all were > 0.50; P < 0.0001) and SF-36 Vitality domain (r > 0.80) in patients with PsA from 2 RCTs (n = 817).96 A strong correlation was observed between the FACIT-Fatigue and modified Fatigue Severity (r = −0.79, 95% CI −0.85 to −0.72) in 1 study of 135 patients with PsA.97 Construct validity was assessed using the known-group approach, FACIT-Fatigue scores were lower in patients with overwhelming fatigue and fibromyalgia than in those without (P < 0.001).97 Reliability In 3 studies, FACIT-Fatigue demonstrated good internal consistency (Cronbach coefficient alpha ≥ 0.90) and test-retest reliability (ICC ≥ 0.95) patients with PsA.9,96,97 Responsiveness No evidence of responsiveness was identified for FACIT-Fatigue in patients with PsA. | A suggested MID for the FACIT-Fatigue in patients with RA is between 3 and 4 points using the Multidimensional Assessment of Fatigue and SF-36 Vitality as anchors.8 Using disease activity measured by Patient’s Global Assessment of Psoriasis and Arthritis and SF-36 Vitality domain as anchors, a MID for the FACIT-Fatigue total score was estimated as 3.1 points using data from patients with PsA from 2 RCTs (n = 817).96 |
HAQ-DI | The HAQ-DI is the disability assessment component of the HAQ, a self-reported assessment of functional status.98 The overall HAQ-DI score ranges from zero (no disability) to 3 (completely disabled).98 | Validity Convergent validity: In a systematic review of 31 RCTs, strong to weak correlations were observed between HAQ-DI and PCS12, PsAID-FC, patient global assessments for arthritis, pain, tender and swollen joint counts, DAPSA, and patient global assessment for skin (ρ ranged from 0.19 to 78).50,99 Reliability Data from a longitudinal study in 14 countries of 414 patients with PsA assessed internal consistency, Cronbach alpha for HAQ-DI was 0.92.50,99 HAQ-DI demonstrated almost perfect test-retest reliability in patients with PsA (ICC ranged from 0.90 to 0.94).100 Responsiveness In a systematic review of 31 RCTs of patients with PsA, moderate effect sizes (Cohen d) were seen in HAQ-DI, PCS12 and PsAID-FC in the measurement of worsening (HAQ-DI 0.46, 95% CI 0.12 to 0.78; PCS12 − 0.57, 95% CI −0.92 to −0.24; PsAID-FC 0.51, 95% CI 0.16 to 0.87). The SRM for worsening for HAQ-DI, PCS12, and PsAID-FC were 0.37 (95% CI 0.10 to 0.61), −0.45 (95% CI 0. −0.74 to −0.19), and 0.38 (95% CI 0.12 to 0.66) respectively.50 | In 1 systematic review and 3 unique studies, MID estimates for HAQ-DI ranged from −0.35 to −0.13 for improvement, and 0.13 to 0.30 for worsening in patients with PsA.6,49,50,99 The MIDs were estimated using the anchor-based approach. |
IGA | The IGA is a 5-point scale used to measure the severity of disease at a single point in time (static IGA). IGA scores range from 0 (clear) to 4 (severe).53 | Validity Data from 5 RCTs of tofacitinib (n = 3,838) showed a relatively high correlation with both the PASI and PtGA (Pearson correlation coefficient r > 0.5),101,102 moderate correlation with DLQI (r ranged from 0.44 to 0.57),79 and a low correlation with the OCI (r < 0.2),102 supporting convergent and divergent validity in patients with psoriasis. Construct validity was assessed using a known-group approach, using the PASI score as a categorical anchor, the differences in the IGA scores between the ‘clear’ group (PASI score of 0) and the other groups (PASI score > 0) were statistically significant, and increased as psoriasis became more severe (i.e., with larger PASI scores), indicating that the IGA could discriminate between different degrees of disease severity.101 Reliability Data from 4 RCTs of tofacitinib (n = 3,641) showed acceptable test-retest reliability (ICC 0.8).101 Data from RCTs of tofacitinib showed high internal consistency reliability in IGA scores, Cronbach coefficient alpha > 0.80 at week 2 and onwards (1 RCT, n = 197)102 and ≥ 0.90 (4 RCTs, n = 3,641) in patients with psoriasis.101 Responsiveness No evidence of reliability was identified for IGA in patients with PsA. The validity, reliability, and responsiveness of the IGA instrument have not been evaluated in patients with PsA. | A score of 0 or 1 is generally accepted as clinically meaningful.53 Some trials of patients with moderate to severe psoriasis define efficacy as a 2-point reduction in the total score.103 In patients with psoriasis, clinically important difference estimates included point changes of about half of a PGA category; 0.52 and 0.55 points in 2 studies, respectively.101,102 The MID for IGA has not been estimated in patients with PsA. |
LEI | The LEI is an enthesitis index designed for use in PsA RCTs assessing lateral humeral epicondyles (elbows), medial femoral epicondyles (knees), and Achilles tendons (heels) bilaterally and scored as 0 (no pain) and 1 (painful) with an overall score range of 0 to 6.95 | Validity Convergent validity: Moderate to weak correlations were observed between LEI and the swollen and tender joint counts, patient and physician global VAS, CRP level, HAQ, and DAS 28 (r ranged from –0.157 to 0.462) in a study of 28 patients with PsA.55 Reliability No evidence of reliability was identified in patents with PsA. Responsiveness The LEI index showed a large effect size of 0.82 at 6 months and significant response to change indicating adequate responsiveness in a study of 28 patients with PsA.55,95 | The MID for LEI has not been estimated in patients with PsA. |
MDA | The MDA is a composite outcome measure developed as a target of treatment for patients with PsA that encompasses the different aspects of disease domains.45 | Validity Construct validity was assessed using the known-group approach and data from observational study (n = 344). Patients with PsA who achieved sustained MDA had a reduction in joint damage progression compared with those in the control group (P < 0.001).104,105 Convergent validity: Based on a review of 20 studies, the κ coefficient between MDA and patients’ rating of whether they were in a minimal disease state: PASDAS, Composite Psoriatic Disease Activity Index-4 and Composite Psoriatic Disease Activity Index-3 was ranged from 0.30 to 0.75 in patients with PsA.106 Reliability Agreement was also strong for the MDA skin domain (MDA-joints) (κ = 0.86) indicating adequate internal consistency in patents with PsA.105 Responsiveness No evidence of responsiveness was identified in patents with PsA. | The MDA-5, defined as meeting 5 of 7 of the cut-offs for the MDA domains, is generally accepted as the threshold for achieving MDA.107 |
PASI 75/90/100 | The PASI is a disease-specific composite severity index based on an average score of erythema, scaling, and thickness of the lesions, weighted by the area of involvement. PASI scores range from 0 to 72, with higher scores indicating greater severity.54 | Validity Construct validity was demonstrated through correlation of the PASI and DLQI scores (0.36 ≤ r ≤ 0.54) using data from an RCT (n = 445) of calcineuron inhibitors in patients with psoriasis.108 Correlation between the PASI and the LS-IGA and IGA (Spearman’s rank correlation 0.92 and 0.73) in 1 study of 16 patients with plaque psoriasis.109 A study of 9 adult patients with plaque-type psoriasis assessed the correlations of PASI with other commonly used instruments in psoriasis, including the BSA and the IGA, and reported a strong correlation with both measures (Pearson correlation coefficient > 0.78 and > 0.61, respectively).110 Content validity was assessed using relative impact of the individual components of the measures on HRQoL: BSA was most consistently associated with DLQI scores, followed by plaque induration and erythema while the scaling score was found to be minimally and inconsistently associated with DLQI scores.108 Reliability Excellent intra-rater and inter-rater reliability for the PASI have been observed in studies of patients with plaque psoriasis or psoriasis (ICC ranged from 0.72 to 0.97).109-112 Responsiveness Based on data from a systematic review, the PASI score was found to have a moderate sensitivity to change in psoriasis patients (ICC ranged from 0.5 to 0.8).113 The validity, reliability, and responsiveness of the PASI instrument have not been evaluated in patients with PsA. | Base on a systematic review of 13 RCTs evaluating biologics in psoriasis, a ≥ 75% reduction in the PASI score translates to clinically-significant HRQoL improvement in patients assessed using the DLQI.114 The MID for PASI has not been estimated in patients with PsA. |
Resolution of Dactylitis- Dactylitis score | The Dactylitis score is a total score of presence and severity of dactylitis in each digit using a scoring system from 0 (no dactylitis) to 3 (severe dactylitis). The final dactylitis score ranges from 0 to 60.13 | Validity No evidence of validity was identified in patents with PsA. Reliability No evidence of reliability was identified in patents with PsA. Responsiveness No evidence of responsiveness was identified in patents with PsA. | The MID for the dactylitis score has not been estimated in patients with PsA. |
SF-36 V2 MCS and PCS | The SF-36 is a 36-item general health status instrument. It consists of 8 domains. A PCS and a MCS can be computed. All domain and component scores are based on a scale of 0 to 100, with higher scores indicating higher health status.115,116 | Validity In 1 study of 168 patients with PsA, construct validity was assessed using the known-group approach, patients with PsA were grouped under severe disease based on HAQ > 1.0, BASDAI > 50 and DAS 28 > 5.1. All 8 scales and summary scores in the severe groups were significantly worse than the less severe groups.7,52 Reliability Internal consistency: Substantial to moderate levels of agreement were observed for most of scales of SF-36 (Cronbach a ranged from 0.33 to 0.70) in patients with PsA.7,52 SF-36 physical function domain demonstrated almost perfect test-retest reliability in patients with PsA (ICC 0.96, 95% CI 0.92 to 0.98).100 Responsiveness In a systematic review of 31 RCTs, the median ES were 0.77 (95% CI 0.60 to 0.93) and 0.23 (95% CI 0.09 to 0.36) for intervention and control groups in patients with PsA, respectively.50 | In 1 study of 20 patients with PsA, the MIDs were estimated as 3.74 and 1.77 for the PCS and MCS subsections, respectively.7 |
Modified van der Heijde- Sharp (vdH-S) | The vdH-S for PsA evaluates erosions, JSN, subluxation, ankylosis, gross osteolysis, and pencil-in-cup lesions. Erosions are assessed in 20 joints of the hands and wrists and 12 joints of the feet. The modified vdH-S score, which ranges from 0 (best) to 528 (worst), is the sum of the joint erosion score and the joint space narrowing score. | Validity In a study of 105 patients with PsA (mean age: 50.2, SD: 12.1), convergent validity was assessed through correlation with SPARS (r = 0.926, P < 0.0001).10 Reliability In 1 study of 10 patients with PsA, the inter- and intra-rater reliability were almost perfect agreement: ICC = 0.95 (95% CI, 0.83 to 0.99) at baseline and 0.99 (95% CI, 0.96 to 1.00) at follow-up (mean: 26 months, SD: 9.6 months) for inter-rater reliability; Rater 1 ICC = 0.97 (95%, CI 0.90 to 0.99 and Rater 2 0.99 (95% CI 0.98 to 0.99) for intra-rater reliability.117 Responsiveness In 1 study of 10 patients with PsA, vdH-S had the ability to detect change at 1.2%. vdH-S had the ability to detect change at a level of 0.79.117 | The MID for the vdH-S score has not been estimated in patients with PsA. |
AS = ankylosing spondylitis; ASQoL = ankylosing spondylitis quality of life questionnaire; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; BSA = body surface area; CRP = C-reactive protein; DAPSA = Disease Activity Index for Psoriatic Arthritis; DAS 28 = Disease Activity Score in 28 Joints; DAS 28 CRP = Disease Activity Score 28 using C-reactive protein; DLQI = Dermatology Life Quality Index; ES = effect size; ESR = erythrocyte sedimentation rate; EULAR = European League Against Rheumatism; FACIT-Fatigue = Functional Assessment of Chronic Illness Therapy – Fatigue; HAQ = Health Assessment Questionnaire; HAQ-DI = Health Assessment Questionnaire Disability Index; ICC = intra-class correlation coefficient; IGA = Investigator’s Global Assessment; JSN = joint space narrowing; LEI = Leeds Enthesitis Index; LS-IGA = Lattice System Investigator Global Assessment; MCS = Mental Component Score; MDA = minimal disease activity; MID = minimal important difference; OCI = Ocular Comfort Index; PASI = Psoriasis Area Severity Index; PCS = Physical Component Score; PCS12 = Physical Component Summary score of the 12-Item Short Form Survey; PGA = Physician Global Assessment; PsA = psoriasis arthritis; PsAID-FC = Psoriatic Arthritis Impact of Disease instrument functional capacity score; PtGA = Patient Global Self-Assessment; RA = rheumatoid arthritis; RCT = randomized controlled trial; SD = standard deviation; SF-36 = Short Form-36 Health Survey; vdH-S = van der Heijde/ Sharp); SRM = standardized response mean; VAS = visual analogue scale.
American College of Rheumatology 20, 50, 70
The ACR criteria for assessing joint status was originally developed for patients with rheumatoid arthritis, and provides a composite measure of ≥ 20%, ≥ 50%, or ≥ 70% improvement in both swollen and tender joint counts and at least 3 of 5 additional disease criteria including: patient’s global assessment of disease activity using a 10-cm VAS, physician’s global assessment of disease activity on VAS, HAQ-DI, patient assessment of pain intensity, and an acute-phase reactant value (CRP or erythrocyte sedimentation rate [ESR]).88 The ACR joint count assesses 68 joints for tenderness and 66 joints for swelling. Assessment of the proximal interphalangeal and distal interphalangeal joints of the hands and feet (i.e., 78 joints for tenderness and 76 for swelling) is not typically included for PsA because of difficulty distinguishing proximal and distal interphalangeal joint inflammation in the toes.118 The ACR is a general measure of clinical response of peripheral joint disease and does not include assessment of enthesitis, dactylitis, the spine, or the skin. Consequently, it represents only part of the clinical features of PsA; therefore, the use of additional assessment instruments to assess other clinical features is necessary.88,118
Assessment of Validity, Reliability, and Responsiveness
The ACR tender and swollen joint counts have been deemed as appropriate for disease activity in patients with rheumatoid arthritis by the ACR expert committee and shown to be a valid outcome measure in RCTs in terms of content and construct validity, inter-rater reliability, and responiiveness.43,44 The construct validity was also assessed using the known-group approach in a study of 164 patients with PsA and peripheral arthritis from 2 randomized placebo-controlled trials of TNF inhibitors (IMPACT trial, N = 104; etanercept trial, N = 60).43 The number of patients who achieved ACR 20, 50, 70 in both active drug groups was significantly higher than the placebo groups.43 The ACR criteria has demonstrated to have poor to substantial inter-observer reliability in PsA. Gladman and colleagues90 conducted study of 10 patients with PsA who represented a broad range of joint inflammation, joint damage, and spinal involvement. The study was based on a Latin Square design. Each patient was examined by each of 10 rheumatologists who were members of the Spondyloarthritis Research Consortium of Canada. Generally, the study reported that the inter-observer reliability was poor to substantial (intra-class correlation coefficients [ICC] ranged from 0.10 to 0.80). However, right grip strength, number of damaged joints, left lateral flexion, and cervical flexion demonstrated almost perfect agreement (ICC ranged from 0.81 to 0.92).90
The responsiveness of the ACR criteria was in the same study that assessed construct validity.43 The study compared responsiveness and discriminative capacity of the Psoriatic Arthritis Response Criteria, ACR, European League Against Rheumatism (EULAR) response criteria, the DAS, and core-set measures. The ACR improvement criteria with 20%, 50% and 70% (ACR 20, 50, 70) cut-off points, EULAR response criteria, and the Psoriatic Arthritis Response Criteria were used as criteria for improvement. Standardized response mean (SRM) and effect size were calculated as statistics for responsiveness. In both active drug groups, the SRM ranged from –1.98 to –0.61 and effect size ranged from –2.84 to –0.57 while the SRM ranged from –0.40 to 0.02 and effect size ranged from –0.37 to 0.15 in the placebo groups.43
Minimal Important Difference
The MID for ACR has not been estimated in patients with PsA, however, the ACR 20 is generally accepted as the MID indicating a response to treatment, while the ACR 50 and 70 more likely reflect truly important change for the long-term management of arthropathy.43,44,91
Bath Ankylosing Spondylitis Disease Activity Index
The most common and widely used validated measure of inflammatory activity of ankylosing spondylitis is the BASDAI.57 This instrument for disease activity is a self-administered patient questionnaire. The BASDAI is a composite index that records patients’ responses to major symptoms of ankylosing spondylitis. It includes 6 questions addressing 5 major symptoms: fatigue, axial (spinal) and peripheral joint pain, localized tenderness and morning stiffness (both degree of stiffness and length of time for which stiffness persists).56,57 Patients’ responses for each question are recorded on a 10 cm VAS. The final BASDAI score has a range from 0 to 10. The higher the score, the greater the degree of disease activity. A reduction in the BASDAI score is considered improvement. The definition of treatment response includes a change in the BASDAI value defined as 2 units (on a scale of 0 to 10) of the BASDAI.92 The recall period for BASDAI is “past week.”
Assessment of Validity, Reliability, and Responsiveness
No studies were found that validated BASDAI in PsA patients. In previous research, the BASDAI has been shown to have good validity, test-retest reliability, and responsiveness in patients with ankylosing spondylitis.93 Haywood and colleagues conducted study of patients with a confirmed diagnosis of ankylosing spondylitis (N = 349) in England and Scotland assessing the validity, reliability, and responsiveness of the BASDAI.93 Convergent validity of the BASDAI was assessed by testing a priori hypotheses about the strength of correlation with the Ankylosing Spondylitis Quality of Life Questionnaire (ASQoL). A high level of correlation (Pearson’s correlation coefficient r > 0.70) was hypothesized between the BASDAI and ASQoL. A strong correlation was observed between the BASDAI and ASQoL (r = 0.79).93 The construct validity was assessed using the known-group approach. Patients who were able to work had significantly higher BASDAI scores compared with those unable to work due to ill health.93 The BASDAI demonstrated strong internal consistency (Cronbach alpha = 0.87) and 2-week test-retest reliability (ICC 0.87, 95% 0.83 to 0.91).93 The BASDAI were compared for responsiveness to change over the 6-month period by calculating the modified SRM were calculated for patients reporting an improvement or deterioration on health transition (general or ankylosing spondylitis-specific). The BASDAI produced high levels of responsiveness for groups of patients whose ankylosing spondylitis-specific (modified SRM ranged from –0.47 to 0.60) or general health (modified SRM ranged from –1.02 to 0.75) had improved or deteriorated according to transition question responses.93
MID
The MID for the BASDAI has been determined based on data from 1 RCT (n = 205) as a change of −1.96 on the 10-point BASDAI scale in patients with ankylosing spondylitis.94 The MID for BASDAI has not been estimated in patients with PsA.
DAS in 28 Joints Calculated Using CRP
The DAS 28 is a measure of disease activity in rheumatoid arthritis based on a count of 28 swollen and tender joints.46 The set of 28 joint count is based on evaluation of the shoulder, elbow, wrist, metacarpophalangeal joints 1 to 5, proximal interphalangeal joints 1 to 5 of both the upper right extremity and the upper left extremity as well as the knee joints of lower right and lower left extremities.13 The DAS 28 is a modified version of DAS, which includes 2 comprehensive joint counts, the Ritchie Articular Index, and a 44-swollen-joint count, plus the ESR or CRP, and a general health assessment using VAS.46 The features of the original DAS generally apply to the DAS 28, due to the same developmental procedure. However, the DAS and DAS 28 values are not directly interchangeable.46 The DAS 28 CRP was developed as an alternative of DAS 28 based on ESR.47 The range of the DAS 28 is 0 to 9.4 and higher scores indicating more active disease.47
Assessment of Validity, Reliability, and Responsiveness
No studies were found that validated DAS 28 in PsA patients. The validity and responsiveness of the DAS 28 was assessed in a study of 391 patients with active rheumatoid arthritis from 2 RCTs of abatacept (ATTAIN trial, N = 258; AIM trial, N = 133).47 The k coefficients with quadratic weights were calculated as statistic for criterion validity. There was general agreement in classifying patients as none, moderate and good EULAR responders using DAS 28 CRP and ESR, with a k of 0.80 (95% CI 0.76 to 0.83) indicating good agreement. Healy and colleagues conducted a study of 28 patients with PsA underwent clinical assessment over a period of 6 months. Convergent validity was assessed using Spearman’s correlation was used to determine the relationship between the entheseal indices and disease activity as measured by the swollen and tender joint counts, patient and physician global VAS, CRP level, HAQ, and LEI. Strong to moderate correlations were observed between DAS 28 and the previously mentioned measures (r ranged from 0.344 to 0.651).95
Responsiveness was assessed for the DAS 28 CRP using EULAR response criteria as anchors, the treatment difference was –18.83; the percentage improvement was –14.42%, the SRM was –0.31, and the relative efficiency was 1.93.47
MID
No evidence of MID for DAS 28 was identified in patients with PsA. A study of 105 patients with early rheumatoid arthritis from a RCT assessed the thresholds for DAS 28 using an absolute level of disease activity, the study reported a value of ≤ 3.2 defined as the threshold for a low disease activity state and < 2.6 as the threshold for remission.88
Functional Assessment of Chronic Illness Therapy – Fatigue (FACIT-Fatigue)
The FACIT-Fatigue scale was originally developed for use in patients with cancer. It is 1 of a series of symptom subscales in the Functional Assessment of Chronic Illness Therapy measurement system and has since been validated for use in patients with rheumatoid and PsA.8 FACIT-Fatigue is a patient self-report measure consisting of 13 statements. Patients are asked to indicate to what extent the statement applies to them over the course of the previous 7 days.8 Each statement has 5 possible levels of response, scored on a scale of 0 to 4 (0 representing “not at all” and 4 representing “very much”), resulting in scores ranging from 0 to 52. Lower scores indicate higher levels of fatigue.8
Assessment of Validity, Reliability, and Responsiveness
The FACIT-Fatigue demonstrated good content validity. Cella et al. (2005) completed a qualitative study with 12 patients; 2 (17%) had mild, 8 (67%) had moderate, and 2 (17%) had severe PsA disease activity; 7 (58%) attributed fatigue to PsA, and 7 (58%) rated fatigue as important or extremely important. Most patients considered the FACIT-Fatigue items relevant to their PsA experience and understood the item content and response options well.8,96 A Toronto PsA cohort study of 135 patients with PsA assessed the validity and reliability of FACIT-Fatigue.97 The study reported high levels of internal consistency (Cronbach alpha = 0.96) and 1-week test-retest reliability (ICC = 0.95).97 A strong correlation was observed between the FACIT-Fatigue and modified Fatigue Severity (r = −0.79, 95% CI −0.85 to −0.72) indicating good convergent validity.97 Construct validity was assessed using the known-group approach, FACIT-Fatigue scores were lower in patients with overwhelming fatigue and fibromyalgia than in those without (P < 0.001).97 FACIT-Fatigue demonstrated good internal consistency (Cronbach coefficient alpha ≥ 0.90) and test-retest reliability (ICC = 0.95).96 Apart from the Health Transition Item (which has a recall period of 1 year), correlations between FACIT-Fatigue and SF-36 domains generally exceeded 0.60 (all were > 0.50; P < 0.0001). There was a strong correlation with SF-36 Vitality (ρ > 0.80).96
MID
A suggested MID for the FACIT-Fatigue in patients with rheumatoid arthritis is between 3 and 4 points using the Multidimensional Assessment of Fatigue and SF-36 Vitality as anchors.8 This MID was found in a sample of 271 patients (77% female, 81% White) with a median age of 56 years (range = 28 years to 84 years), a median tender joint count of 26 (range = 9 to 68), and a median swollen joint count of 15 (range = 2 to 43).8 Using disease activity measured by Patient’s Global Assessment of Psoriasis and Arthritis and SF-36 Vitality domain as anchors, a minimal clinically important difference for the FACIT-Fatigue total score was estimated as 3.1 points in patients with PsA.96
Health Assessment Questionnaire Disability Index
The HAQ was originally developed in 1978 at Stanford University.98 It was 1 of the first self-reported functional status (disability) measures and has become the dominant instrument in many disease areas, including arthritis.119 The full HAQ collects data on 5 generic patient-centred health dimensions: 1) to avoid disability, 2) to be free of pain and discomfort, 3) to avoid adverse treatment effects, 4) to keep dollar costs of treatment low, and 5) to postpone death.120
The HAQ-DI is the disability assessment component of the HAQ. It assesses a patient’s level of functional ability. There are 20 questions in 8 categories to assess a patient’s physical functional status: dressing, arising, eating, walking, hygiene, reach, grip, and common activities.121,122 For each of these categories, patients report the amount of difficulty they have in performing specific activities, and their responses are made on a scale from 0 (no difficulty) to 3 (unable to do). The 8 category scores are averaged into an overall HAQ-DI score on a scale from 0 (no disability) to 3 (completely disabled). The HAQ-DI was developed to assess physical disability and pain in rheumatoid arthritis98 and has been used extensively in RCTs in arthritis, including for PsA.99
Assessment of Validity, Reliability, and Responsiveness
The HAQ-DI, PCS score of the 12-Item Short Form Survey (PCS 12) and the Psoriatic Arthritis Impact of Disease instrument functional capacity score (PsAID-FC) were assessed for construct validity. The 3 patient-reported outcome measures correlated strongly with each other (ρ > 0.7); and moderately to strongly with patient global assessments for arthritis (ρ = 0.61 to 0.78), pain (ρ = 0.61 to 0.77); moderately with tender joint count (P = 0.39 to 0.51) and DAPSA (ρ = 0.55 to 0.72); weakly with swollen joint count (ρ = 0.19 to 0.32); and very weakly with patient global assessment for skin (ρ = 0.24 to 0.36).50 Leung et al. (2021) analyzed data available from a longitudinal study in 14 countries of consecutive adults with definite PsA of at least 2 years in duration.50 A total of 414 patients (52% male) were analyzed. They reported an internal consistency measured by Cronbach alpha of 0.92 for HAQ-DI. Ceiling effects were noted in a third of patients.99 A study collected data from 2 studies conducted in UK and Singapore, respectively. A total of 69 patients with PsA (UK study, N = 31; Singapore study, N = 38) were assessed for test-retest reliability.100 The study reported almost perfect test-retest reliability in the UK study (ICC 0.90, 95% CI 0.79 to 0.95) in a 1-week interval and Singapore study (ICC 0.94, 95% CI 0.89 to 0.97) in a 2-week interval.100 All 3 patient-reported outcome measures for physical function were more sensitive for worsening than improvement. Moderate effect sizes (Cohen d) were seen in all 3 patient-reported outcome measures in measurement of worsening (HAQ-DI 0.46,95% CI 0.12 to 0.78; PCS12 − 0.57, 95% CI −0.92 to −0.24; PsAID-FC 0.51, 95% CI 0.16 to 0.87). The SRM for worsening for HAQ-DI, PCS12, and PsAID-FC were 0.37(95% CI 0.10 to 0.61), −0.45(95% CI 0. −0.74 to −0.19), and 0.38(95% CI 0.12 to 0.66) respectively.50
MID
Mease et al. (2011)6 have estimated that the MID for the HAQ-DI in PsA patients using anchor-based methods is 0.35 (unlike 0.22 for rheumatoid arthritis), while the MID has been estimated to be 0.131 in PsA patients using an anchor-based approach (equal bidirectional magnitudes for improvement and worsening) by Kwok and Pope (2010).49 Discrepancies in the MID estimates may partly be explained by differences in the HAQ-DI score of the patients studied at baseline.123 In the study by Mease et al. (2011),6 patients had a mean HAQ-DI score at baseline of 1.16, corresponding to moderate functional impairment. In contrast, patients in the study by Kwok and Pope (2010) had less functional impairment at baseline, with a mean HAQ-DI score of 0.732.50 Blackmore and colleagues (1995) have shown the HAQ-DI adequately captures clinically important changes in functional status and pain in patients with PsA.124 However, the HAQ-DI focuses on physical disability and may not adequately capture disability in patients with predominantly skin disease.124 Modified versions of the HAQ to include spinal domains or skin disease assessment have not proven to be significantly better in assessment of health status in PsA than the original HAQ-DI.124,125 Leung et al. (2020), reported the MID for improvement and worsening for HAQ-DI were –0.16 (SD: 0.87) for improvement, and 0.30 (SD: 0.81) for worsening using patient-rated “improved” or “worse” disease status as anchors in patients with PsA.99
Investigator’s Global Assessment
The IGA, also known as the Physician Global Assessment, is a simple measurement of the clinical signs of psoriasis, frequently used as a co-primary end point with the PASI score in psoriasis clinical trials.126 Various IGAs have been used in psoriasis with different descriptions and scores, with the most common IGA versions using 5- to 6-point scales.111,126 The 5-point scale is reported to have a more strict criteria for the score of 1 (“almost clear”) than the 6-point scale score of 1 (“minimal”).53 There are 2 types of IGAs, a static form which measures the physician’s measurement of the disease at a given time point, and a dynamic form in which the physician evaluates the level of improvement or deterioration from a baseline.103,126 The static form of the IGA is preferred as it does not rely on the investigator’s recall of the patient’s disease severity observed at baseline or a previous visit.
Validity
The most recent study assessing the validity of the IGA evaluated data from 4 phase III clinical studies of tofacitinib in patients with psoriasis (N = 3,641).101 Construct validity was assessed using a known-group approach, measuring the relationship between IGA and PASI through a repeated measures model. Using the PASI score as a categorical anchor, the differences in the IGA scores between the ‘clear’ group (PASI score of 0) and the other groups (PASI score > 0) were statistically significant, and increased as psoriasis became more severe (i.e., with larger PASI scores), indicating that the IGA could discriminate between different degrees of disease severity.101
Convergent validity was assessed in the aforementioned study by comparing the IGA with 3 additional outcome measures: the PASI, patient global assessment, and Dermatology Life Quality Index (DLQI).101 Pearson correlation coefficients between IGA and the 3 scales ranged from 0.4 to 0.79 at primary assessment time points in the 4 clinical studies. Results showed a strong correlation between the IGA and the PASI (r ranged from 0.77 to 0.79) even though the IGA does not consider the amount of BSA affected by psoriasis. A moderately strong correlation was found between the IGA and DLQI (0.44 to 0.57). None of the correlations were large (> 0.8), indicating that the IGA considers some different information than the PASI and DLQI. Baseline correlations were smaller likely due to the limited range of responses from relatively homogeneous patients. An earlier study by Cappelleri et al. (2013)102 conducted a psychometric validation of the IGA using data from a phase II study of tofacitinib among 197 patients. Results found a relatively high correlation between the IGA and Patient Global Assessment as well as the IGA and PASI with correlation coefficient r > 0.5, except for at baseline. The study also examined correlations between the IGA and the Ocular Comfort Index (OCI) and the pain/discomfort assessment with all correlation coefficient r < 0.2, indicating relatively high divergent validity. These findings were consistent with several other studies examining the 6-point IGA.109-111
Reliability
A recent systematic review noted that the 5-point IGA appears to be based on a formative model where a change in the severity of the disease does not necessarily mean a change in all 3 components of erythema, induration, and scaling; therefore, an assessment of internal consistency would not be applicable.127 However, Callis Duffin et al. (2019)101 assessed the internal consistency reliability, demonstrating that the scoring items (erythema, induration, and scaling) were highly consistent with each other (Cronbach coefficient alpha ≥ 0.90) at the primary assessment points in all 4 trials. The internal consistency reliability was less convincing (Cronbach coefficient alpha 0.50 to 0.63) for the values observed at baseline, likely a result of the specific inclusion criteria of the trials.101 This study also evaluated the consistency of IGA measurements between screening and baseline visits, when no change in terms of disease severity was expected. The ICC for the pooled data was 0.70, suggesting an acceptable test-retest reliability over a stable period.
Cappellleri et al. (2013)102 also examined the test-retest reliability of IGA scores at baseline and week 2 among patients with little to no change in their PASI scores, resulting in a high ICC of 0.80. The study also examined internal consistency reliability of the IGA, with Cronbach coefficient alpha > 0.80 at all time points from week 2 onwards, indicating high correlations after baseline. Langley et al. (2013)111 conducted an analysis of variance to examine the variability in the IGA and PASI. The intra-rater variation in the 6-point IGA was lower than the PASI with SDs of 0.2 and 2.5, respectively.111 The systematic review by Puzenat et al. also reported low intra-observer variability but moderate inter-observer variability for the IGA.128
Responsiveness
No evidence regarding the responsiveness of the IGA was identified from the literature at this time.
The validity, reliability, and responsiveness of the IGA instrument have not been evaluated in patients with PsA.
MID
The MID for IGA has not been estimated in patients with PsA. It is generally accepted that a clinically meaningful score in the IGA is a score of 0 (“clear”) or 1 (“almost clear” or “minimal”).53 Furthermore, some trials of patients with moderate to severe psoriasis define efficacy as a 2-point reduction in the total IGA score.103 Both Cappellleri et al. (2013)102 and Callis Duffin et al. (2019)101 assessed the clinically important difference for the IGA. Both studies used the patient global assessment score as a continuous anchor and determined a clinically important difference score of 0.52 (95% CI, 0.42 to 0.56) and 0.55 (95% CI, 0.546 to 0.563) in each study, respectively. It should be noted that the clinically important difference does not necessarily imply a minimum clinically important difference and although it is not possible to measure less than 1 category difference for an individual on the IGA scale, the clinically important difference was deemed appropriate to determine a group difference.102
Leeds Enthesitis Index
Enthesitis, the inflammation at the bone insertion of a tendon or ligament, is common in PsA. The LEI is an enthesitis index designed for use in PsA and has been adopted for use in randomized controlled studies involving patients with PsA.55 Enthesitis was assessed by examining 6 sites, i.e., the lateral humeral epicondyles (elbows), medial femoral epicondyles (knees), and Achilles tendons (heels) bilaterally and scored as 0 (no pain) and 1 (painful), with an overall range of 0 to 6.55
Assessment of Validity, Reliability, and Responsiveness
Healy and colleagues conducted a study of 28 patients with PsA underwent clinical assessment over a period of 6 months. Convergent validity was assessed using Spearman’s correlation was used to determine the relationship between the entheseal indices and disease activity as measured by the swollen and tender joint counts, patient and physician global VAS, CRP level, HAQ, and DAS 28. Moderate to weak correlations were observed between LEI and the previously mentioned measures (r ranged from –0.157 to 0.462).55 The LEI index showed a large effect size of 0.82 at 6 months and significant response to change indicating adequate responsiveness.55 No evidence was identified to support the reliability of the LEI for patients with PsA.
MID
The MID for LEI has not been estimated in patients with PsA.
Minimal Disease Activity
MDA is a composite outcome measure that was developed as a target of treatment for patients with PsA that encompasses the different aspects of disease domains.45 Patients are considered as achieving MDA if they fulfilled the following 5 of 7 outcome measures: ≤ 1 tender joint count, ≤ 1 swollen joint count, PASI ≤ 1 or BSA ≤ 3%, patient pain VAS ≤ 15, patient global VAS ≤ 20, HAQ-DI ≤ 0.5, tender entheseal points ≤ 1.105 These criteria for MDA were validated in patients with active PsA using interventional trial data.50
Assessment of Validity, Reliability, and Responsiveness
Construct validity was assessed using the know-group approach in an observational PsA cohort study of 344 patients, it was found that patients who achieved sustained MDA (sustained MDA was defined as achieving MDA on consecutive visits for a minimum duration of 12 months) had a reduction in joint damage progression, where 69% of patients who achieved sustained MDA showed no progression of joint damage, compared with 51% in the control group, in addition the mean change in damaged joint counts was 0.931 in the sustained MDA group and 2.245 in the controls (P < 0.001).104,105 In addition, Queiro et al. (2017) reported the relationship between MDA and presence of radiographic erosions in the hands and feet in a cross-sectional study. Patients in MDA were less likely to have evidence of hand erosions compared with those who were not (P < 0.05); however, there were no significant differences among patients when evaluating presence of erosions in the feet.106,129 Coates et al.(2019) conducted a review of 20 studies evaluated the relationship between MDA and disease activity reported by the patient (measured as a patient-reported overall indicator of disease activity). The κ coefficient between MDA and patients’ rating of whether they were in a minimal disease state was 0.30 indicating weak convergent validity.106 Coates and colleagues also reported moderate agreement (κ ranged from 0.73 to 0.75) with 3 alternative definitions of treatment responses: PASDAS, Composite Psoriatic Disease Activity Index-4 and Composite Psoriatic Disease Activity Index-3 to assess convergent validity in the same study assessed the construct validity of MDA.130 Agreement was also strong for the MDA skin domain (MDA-joints) (κ = 0.86) indicating adequate internal consistency.106
Threshold for MDA
The MDA-5, defined as meeting 5 of 7 of the cut-offs for the previously mentioned MDA domains, is generally accepted as the threshold for achieving MDA.105,107
Psoriasis Area and Severity Index
The PASI is the most used instrument for the assessment of psoriasis severity.107 It is a single estimate of disease severity based on lesion characteristics weighted by area of body involvement. Psoriatic lesion characteristics are assessed separately for erythema, induration, and scaling in the 4 major body areas: head, upper extremities, trunk, and lower extremities. Severity of each item is graded on a scale of 0 to 4 (0 = clear, 1 = mild, 2 = slight, 3 = moderate, 4 = severe), which is then summed by body region and weighted by the percentage of BSA involvement converted on a scale of 0 to 6 (0 = no involvement, 1 = 1‒9%, 2 = 10‒29%, 3 = 30‒49%, 4 = 50‒69%, 5 = 70‒89%, 6 = 90‒100%). The individual body region scores are then multiplied by weighting factors representing their respective proportion of the total BSA (0.1 for head, 0.2 for upper extremities, 0.3 for trunk and 0.4 for lower extremities), as in the following formula131,132:
The generated PASI score is a numeric score ranging from 0 to 72, with a score greater than 10 representing more severe disease.131 In clinical trials, PASI is often reported as an overall mean percentage improvement with treatment, and is used most commonly for responder analyses.133 A 75% reduction in the PASI score, i.e., PASI 75, is used as a benchmark in clinical trials in psoriasis.134 While the PASI 75 is still used for legacy drugs, the treatment goal in clinical practice for newer treatment should be the achievement of PASI 90, according to the clinical expert consulted for this review. A recent systematic review noted that achieving a PASI 100 indicates total clearing of the skin and is commonly used in clinical trials, although more real-world evidence is required to determine its role in daily practice.135
Validity
Simpson et al. (2015)108 studied data from a phase III clinical trial (N = 445) in patients with plaque psoriasis to validate 3 systems of physician-scoring psoriasis severity, which included the PASI, static IGA, and Lattice System IGA measures. Construct validity of PASI was assessed by evaluating the correlation between the PASI score and the DLQI score, a skin-related HRQoL measure in grading psoriasis severity. The PASI correlated moderately with both the DLQI overall score as well as a single item of DLQI related to psoriasis symptoms (0.36 ≤ r ≤ 0.54), demonstrating that psoriasis severity is correlated with the DLQI score. The same study also investigated the content validity of the 3 measures by assessing the relative impact of the individual components of the measures on HRQoL using multiple linear regression analysis; BSA was most consistently associated with DLQI scores, followed by plaque induration and erythema. The scaling score was found to be minimally and inconsistently associated with DLQI scores which may be in part due to the static measurement of scaling which does not encompass the flaking of the skin over time which can be very distressing to patients.108 The authors therefore concluded that weighing erythema, induration and scaling equally would not accurately capture the varying degrees to which these factors affect the patient’s rating of quality of life. Lastly, the construct and content validity of the PASI were found to be stronger during active treatment compared to pre-therapy.108
Another study of 10 trained dermatologists evaluating 9 adult patients with plaque-type psoriasis assessed the correlations of PASI with other commonly used instruments in psoriasis, including the BSA and the IGA.108 The authors reported a strong correlation with both measures (Pearson correlation coefficient > 0.78 and > 0.61, respectively).110 Similarly, Berth-Jones et al. (2016) (14 trained dermatologists, 16 patients with chronic plaque psoriasis) reported a strong correlation between PASI and the Lattice System IGA (Spearman’s rank correlation, r = 0.92), and a moderate correlation with the IGA (r = 0.73).109 Berth-Jones et al. (2016) also found that the PASI and IGA were in good agreement for the clearance state (kappa = 0.64) but poor agreement for the severe state (kappa = 0.18).109
Reliability
The reliability of the PASI measure has been assessed in several studies.109-112 Bożek et al. (2017) reported the ICCs for all components of the PASI to be > 0.75, indicating very good intra-rater reliability, except for scaling (ICC = 0.72) in 9 patients with plaque-type psoriasis. The highest ICC was observed for the area score (ICC = 0.97). The coefficient of variation for the PASI was 36.9 overall, indicating moderate inter-rater reliability. The highest variability was observed for the head and neck (coefficients of variation = 117.8) and the lowest variability was for the area score (coefficients of variation = 26.8).109-112 Langley et al. (2004) (17 physicians, 25 patients with psoriasis) reported similar results, with higher variability observed in the PASI scores derived by inexperienced physicians compared with experienced investigators (σ = 3.2 versus 1.2).111 Berth-Jones et al. (2016) found excellent intra-rater and inter-rater reliability for the PASI score (ICCs > 0.81) in.109 Fink et al. (2018) validated the methodology of ‘image-based’ versus commonly used ‘live’ PASI measurements in a pilot study, followed by validating in an observational cohort study.112 They investigated the precision and reproducibility of automated, computer- guided PASI measurements in comparison with 3 trained physicians. PASI scores of 120 patients affected by plaque psoriasis of various severities were prospectively evaluated by 3 formally trained physicians by means of total body images. Each observer independently performed 2 rounds of image-based PASI calculations in all patients at 2 different time points.112 Overall, 720 image-based PASI scores were calculated with a mean PASI of 8.8 (range 0.7 to 34.8). An inter-rater variability with an ICC of 0.895 and mean absolute difference of 3.3 PASI points were observed. Intra-rater variability showed a mean ICC of 0.877 and a mean absolute difference of 2.2 points.112
Responsiveness
The PASI score was found to have a moderate sensitivity to change in psoriasis patients (ICC ranged from 0.5 to 0.8).113 In a review by Spuls et al. (2010)136 the authors commented on the responsiveness of PASI being weak when less than 10% of the BSA is affected given that the PASI score would be entirely dependent on the plaque severity scores, and therefore may underestimate the general degree of improvement.
Minimal Important Difference
The MID for PASI has not been estimated in patients with PsA. A systematic review by Mattei et al. (2013)114 including 13 RCTs evaluating biologics in psoriasis, reported that a ≥ 75% reduction in the PASI score translates to clinically-significant HRQoL improvement in patients assessed using the DLQI. This is based on the several studies that have demonstrated that a reduction in PASI scores can predict a reduction in DLQI scores, particularly when the patients were achieving a PASI 75 or higher (PASI 75 versus PASI 50 to 75 versus mean difference of 3.24).114
Resolution of Dactylitis – Dactylitis Score
Dactylitis score is a total score of presence and severity of dactylitis in each digit using a scoring system from 0 (no dactylitis) to 3 (severe dactylitis). The final dactylitis score ranges from 0 to 60. A negative change from baseline indicates improvement. Dactylitis resolution at a visit is established when a participant with a baseline non-zero dactylitis score has a score of 0 at the analysis visit.13
Assessment of Validity, Reliability, and Responsiveness
No evidence of validity, reliability, and responsiveness is identified for the dactylitis score in patients with PsA.
MID
The MID for the dactylitis score has not been estimated in patients with PsA.
Short Form-36 Health Survey Mental Component and Physical Component Scores (SF-36 MCS/PCS)
The SF-36 is a 36-item, general health status instrument that has been used extensively in clinical trials in many disease areas.115,116 The SF-36 consists of 8 health domains: physical functioning, role physical, bodily pain, general health, vitality, social functioning, role emotional, and mental health. For each of the 8 domains, a subscale score can be calculated. The SF-36 also provides 2 component summaries, the PCS and the MCS, derived from aggregating the 8 domains according to a scoring algorithm. All scores are based on a scale of 0 to 100, with higher scores indicating higher HRQoL. The scores can also be standardized to the general US population, where an average score is 50, with a SD of 10 (t score). Version 2 of the SF-36 was made available to researchers in 1996137 and was developed to address the shortcomings of the version 1 of the survey; including changing response options for role function scales from dichotomous to 5-point scale responses and altering the wording of certain items to make them easier to understand.138
Assessment of Validity, Reliability, and Responsiveness
Validity and reliability were assessed in a study of 168 patients with PsA.52 Construct validity was assessed using the known-group approach, patients with PsA were grouped under severe disease based on HAQ > 1.0, BASDAI > 50 and DAS 28 > 5.1. All 8 scales and summary scores in the severe groups were significantly worse than the less severe groups.52 Inter-scale coefficients (Cronbach alpha) were calculated to assess internal reliability, substantial to moderate levels of agreement were observed for most of scales of SF-36 (Cronbach alpha ranged from 0.33 to 0.70).52 Leung and colleagues (2021) analyzed data of 31 patients with PsA in a UK observational study. The study reported that SF-36 physical function domain demonstrated almost perfect test-retest reliability in patients with PsA (ICC 0.96, 95% CI 0.92 to 0.98).100 The responsiveness has been assessed for the SF-36 in RCTs in patients with PsA. The median effect sizes were 0.77 (95% CI 0.60 to 0.93) and 0.23 (95% CI 0.09 to 0.36) for intervention and control groups, respectively.139-141
Minimal Important Difference
The MID for either the PCS or MCS of the SF-36 is typically between 2.5 and 5 points.52 Leung and colleagues (2010) reported MIDs of 3.74 and 1.77 for the PCS and MCS subsections, respectively, in PsA patients treated with anti-TNF alpha drugs using an anchor-based approach.7,52
Modified vdH-S Score
The modified vdH-S score is based on the Sharp–van der Heijde method. The original scoring system evaluates erosions and joint space narrowing of joints of hands and feet in rheumatoid arthritis. The proposed method for PsA evaluates erosions, joint space narrowing, subluxation, ankylosis, gross osteolysis, and pencil-in-cup lesions. Erosions are assessed in 20 joints of hands and wrists: 10 distal interphalangeal joints and interphalangeal joints of the thumbs, 10 metacarpophalangeal joints, 2 first metacarpal bones, 2 radial and ulnar bones, 2 multangular units (trapezium and trapezoid combined) and in 12 joints of the feet (10 metatarsophalangeal joints and 2 interphalangeal joints of the big toes, joint space narrowing, subluxation, ankylosis, gross osteolysis and pencil in cup are assessed in the hands in 10 distal interphalangeal joints and interphalangeal joints of the thumbs, 10 metacarpophalangeal joints, second, third, fourth, and fifth carpometacarpal joints, 2 multangular units, 2 capitate-navicular-lunate joints, 2 radiocarpal joints, 10 metatarsophalangeal joints, and 2 interphalangeal joints of the big toes. The maximum score for erosions is 5 in the joints of the hands and 10 in the joints of the feet. Scores for erosions are as follows: 0 = no erosions; 1 = discrete erosions; 2 = large erosions not passing the midline; 3 = large erosions passing the midline. A combination of these scores leads to a maximum of 5 for a whole joint in the hands, and 5 at each site of the joint (for the entire joint a maximum of 10) in the feet. The joint space narrowing scoring is: 0 = normal; 1 = asymmetric or minimal narrowing up to a maximum of 25%; 2 = definite narrowing with loss of up to 50% of the normal space; 3 = definite narrowing with loss of 50% to 99% of the normal space or subluxation; 4 = absence of a joint space, presumptive evidence of ankylosis, or complete luxation. Gross osteolysis and pencil in cup are scored separately. If present, these lesions are scored with the maximum score for both erosions and joint space narrowing. The maximum possible score for erosions is 200 for the hands and 120 for the feet; the maximum possible score for joint space narrowing is 160 for the hands and 48 for the feet. Finally, the maximum possible score is 528.10
Assessment of Validity, Reliability, and Responsiveness
In a study of 105 patients with PsA (mean age = 50.2, SD = 12.1), the vdH-S method showed strong convergent validity, when correlated with Simplified Psoriatic Arthritis Radiographic Score (SPARS) (r = 0.926, P < 0.0001).117
In 1 study, hand and feet radiographs from 50 patients with PsA were scored at 2 time points by 2 assessors for the modified vdH-S score. The radiographs of 10 patients were scored by both the readers using all 4 techniques in random order to assess inter-rater reliability and then scored 1 month later to estimate intra-rater reliability using ICCs. Sensitivity to change was estimated using a SRM and smallest detectable change. The inter- and intra-rater reliability were almost perfect agreement: ICC = 0.95 (95% CI 0.83 to 0.99) at baseline and 0.99 (95% CI 0.96 to 1.00) at follow-up (mean: 26 months, SD: 9.6 months) for inter-rater reliability; Rater 1 ICC = 0.97 (95% CI 0.90 to 0.99) and Rater 2 0.99 (95% CI 0.98 to 0.99) for intra-rater reliability. The modified vdH-S score has the ability to detect change at 1.2%. The sensitivity to change of the methods using the SRM demonstrated the modified vdH-S as having the greatest ability to detect change at a level of 0.79. The feasibility of the modified vdH-S method was estimated based on the mean time taken to score each film. The modified vdH-S method took 14.4 minutes to score.117
Minimal Important Difference
No reported MID was found for PsA patients.
Pharmacoeconomic Review
Abbreviations
ACR
American College of Rheumatology
ACR 20
American College of Rheumatology 20% improvement
bDMARD
biologic disease-modifying antirheumatic drug
BSC
best supportive care
cDMARD
conventional disease-modifying antirheumatic drug
DMARD
disease-modifying antirheumatic drug
HAQ-DI
Health Assessment Questionnaire Disability Index
ICER
incremental cost-effectiveness ratio
IL
interleukin
JAK
Janus kinase
NMA
network meta-analysis
NSAID
nonsteroidal anti-inflammatory drug
ODB
Ontario Drug Benefit
PASI
Psoriasis Area and Severity Index
PASI 75
75% improvement from baseline in Psoriasis Area and Severity Index score
PsA
psoriatic arthritis
QALY
quality-adjusted life-year
SAE
serious adverse event
SEB
subsequent entry biologic
TNF
tumour necrosis factor
WTP
willingness to pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Guselkumab (Tremfya), solution for injection |
Submitted price | $3,059.74 per 100 mg/mL pre-filled syringe or patient-controlled injector |
Indication | For the treatment of adult patients with active psoriatic arthritis. Guselkumab may be used alone or in combination with a conventional disease-modifying antirheumatic drug. |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | April 9, 2020 |
Reimbursement request | As per indication |
Sponsor | Janssen Inc. |
Submission history | Previously reviewed: Yes Indication: Moderate to severe plaque psoriasis Recommendation date: February 21, 2018 Recommendation: Recommended for reimbursement in a manner similar to other biologics. Treatment should be discontinued if response has not been demonstrated after 16 weeks. Drug plan cost should not exceed that of the least costly reimbursed biologic for the treatment of moderate to severe plaque psoriasis. |
NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Semi-Markov model |
Target population |
The sponsor also presented subgroup analyses for:
|
Treatment | Guselkumab, alone or in combination with a cDMARD |
Comparators |
|
Perspective | Canadian publicly funded health care payer |
Outcome | QALYs |
Time horizon | Lifetime (53 years) |
Key data sources | The DISCOVER-1, DISCOVER-2, and COSMOS trials informed the efficacy of guselkumab. Sponsor-submitted NMAs for each population informed comparator efficacy. |
Submitted results |
|
Key limitations |
|
CADTH reanalysis results |
|
BSC = best supportive care; cDMARD = conventional disease-modifying antirheumatic drug; CEF = cost-effectiveness frontier; ICER = incremental cost-effectiveness ratio; NMA = network meta-analysis; NSAID = nonsteroidal anti-inflammatory drug; PsA = psoriatic arthritis; QALY = quality-adjusted life-year; TNF = tumour necrosis factor.
Conclusions
Based on the CADTH clinical review of the placebo-controlled trials of guselkumab (DISCOVER-1, DISCOVER-2, and COSMOS), compared to placebo, adults with active psoriatic arthritis (PsA) who received guselkumab were more likely to show improvement as measured by the proportion of patients who achieved an American College of Rheumatology (ACR) response, the Health Assessment Questionnaire Disability Index (HAQ-DI), and the Psoriasis Area and Severity Index (PASI). These responses were observed among patients who were biologic-naive or had prior intolerance or inadequate response to tumour necrosis factor (TNF) alpha inhibitors. The indirect evidence comparing guselkumab to other biologic disease-modifying antirheumatic drug (DMARDs) available in Canada in the full population showed imprecise results for ACR response rates, change in HAQ-DI scores, and risk of serious adverse events (SAEs). This imprecision limits the ability to draw conclusions from these data. Based on this indirect evidence, short-term PASI response rates may favour guselkumab versus some biologics. However, there is uncertainty in these findings, given that several sources of heterogeneity were identified across the trials included in the sponsor-submitted indirect treatment comparison and that is unclear whether the methods used to control for potential bias were adequate. In addition, there was limited ability to assess consistency between direct and indirect evidence. The biologic-naive findings were consistent with the full population analysis. Due to the sparse network for the biologic-experienced population, findings on all outcomes (including PASI) are uncertain.
CADTH could not address all of the identified key limitations in the sponsor’s economic submission. CADTH undertook reanalyses that equalized discontinuation rates among biologic comparators; removed double-counted resource use; removed apremilast as a comparator; and did not consider the mixed-population analysis. These reanalyses indicated that guselkumab is not cost-effective at the submitted price because it is dominated by (i.e., it is more costly and less effective than) other available treatments in both the biologic-naive and biologic-experienced populations of adult patients with active PsA. A price reduction of 20% (in the biologic-experienced population) to 42% (in the biologic-naive population) is required for guselkumab to enter the cost-effectiveness frontier, based on CADTH reanalyses.
Due to the lack of available evidence, CADTH could not assess the cost-effectiveness of guselkumab in the full population indicated by Health Canada. CADTH analyses assumed differences between treatments in clinical benefit; if no differences in clinical benefit are applied, different price reductions may be required for guselkumab. The price reductions are based on publicly available drug prices for the comparators. Further reduction may be required if there are lower confidential prices for comparator products.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
Input was received from 6 patient groups: Arthritis Consumer Experts, the Canadian Association of Psoriasis Patients in partnership with the Canadian Psoriasis Network, and the Canadian Arthritis Patient Alliance in partnership with the Arthritis Society and CreakyJoints. Five of these organizations (all except Arthritis Consumer Experts) collaborated by developing survey questions using SurveyMonkey, and each organization shared the surveys with its respective communities. There were 71 respondents to the joint survey from across Canada, with almost half (34 respondents out of 71 respondents) being from Ontario. Additionally, Arthritis Consumer Experts gathered information from 1 patient by email and from 5 patients through an online survey. Patients reported nausea, vomiting, and general malaise with use of conventional disease-modifying antirheumatic drugs (cDMARDs), as well as concerns about toxicity, weight gain, and increased infection risk, all of which decreased their willingness to use them. Biologic therapies were associated with better efficacy (11 out of 20 responding patients considered them to be “very effective” versus 9 out of 30 patients who said the same about cDMARDs); however, some patients reported side effects that were difficult to tolerate, loss of efficacy between administrations or over time, and concerns about increased risk of malignancy. One-third of respondents noted that they had experienced an inadequate response to currently available treatments, whether conventional or biologic. Patients responding to the survey desired new treatments to improve the management of their symptoms (pain, mobility, fatigue, psoriasis), make side effects more tolerable, improve their quality of life, provide improved routes of administration (i.e., infusion versus self-injection versus oral pills), improve their ability to be productive and carry out activities of daily living, and improve affordability. Some patients reported that the cost of medications was a significant barrier to accessing appropriate treatment, particularly given the lifetime maximums of some private plans and the loss of private coverage when the disease was advanced enough to interfere with full-time employment. Others reported difficulties accessing a rheumatologist and the distance they were required to travel to visit 1. Two respondents to the survey reported experience with guselkumab; 1 felt that it treated their psoriasis better than it treated their arthritis, while the other felt that their skin was not as clear as with other biologics, but that it helped their arthritis. Both stated that they did not experience side effects. One respondent reported that they had tried guselkumab and needed steroid injections during treatment with it, stating “it wasn’t the best for me.” One respondent emphasized that the subcutaneous administration of guselkumab and the ability to inject it at home made it more convenient than therapies requiring doctor appointments and time off work.
Clinical input was received from a group of 6 rheumatologists in Canada with an interest in PsA. The group noted that current biologic therapies result in only 60% of patients (approximately) achieving an ACR improvement of 20% (ACR 20), and that side effect profiles and comorbidities limit the use of the 2 most common classes of biologics, the TNF alpha and interleukin-17A (IL-17A) inhibitors. The group emphasized that the heterogenous nature of psoriatic disease requires nuanced treatment considerations, and that guselkumab, as an IL-23 inhibitor, may be a good option for some patients as a first-line treatment with or without a traditional DMARD, while in other patients, it may be a second-line option following intolerance to or inefficacy of other therapies.
CADTH-participating drug plans noted the lack of head-to-head trials comparing guselkumab to currently funded therapeutic options and emphasized that with the availability of biosimilars for adalimumab, etanercept, and infliximab — and the patent expiration and current testing of biosimilars for ustekinumab — newer drugs in this space would need to prove more effective than available therapies to justify a price premium. The drug plans indicated that prescribing criteria that were aligned with those of other biologic drugs and Janus kinase (JAK) inhibitors would be preferred. Concerns were raised regarding the budget impact of reimbursing guselkumab when jurisdictional biosimilar initiatives are considered (i.e., removing originator brands from funding where biosimilars are available).
Several of these concerns were addressed in the sponsor’s model:
Quality of life was incorporated.
Biologic-naive and biologic-experienced patients were considered separately.
The pharmacoeconomic analyses considered adalimumab, etanercept, and infliximab at the same cost as their biosimilar versions.
In addition, CADTH addressed some of these concerns as follows:
100% of adalimumab, etanercept, and infliximab use was considered to be biosimilar in the budget impact reanalysis.
CADTH was unable to address the following concerns raised from stakeholder input:
uncertainties in relative effectiveness compared to other biologics
the cost-effectiveness of guselkumab as a first-line therapy before or concurrent with cDMARDs, as mentioned by the clinician group.
Economic Review
The current review is for guselkumab (Tremfya) for adults with PsA in line with the DISCOVER-1, DISCOVER-2, and COSMOS trials. These trials predominantly included patients who had experienced an inadequate response to standard therapies (e.g., non-biologic DMARDs [non-bDMARDs], apremilast, or nonsteroidal anti-inflammatory drugs [NSAIDs]).
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
Guselkumab is indicated for adults with PsA and can be used alone or in combination with a cDMARD.1 The sponsor’s reimbursement request is aligned with the indication.2 The sponsor submitted 1 model that incorporated 3 cost-utility analyses assessing the cost-effectiveness of guselkumab in 3 populations.
One analysis assessed the cost-effectiveness of guselkumab in all adult patients with PsA from the trials who had not responded to standard therapies compared to 10 active treatments: TNF inhibitors (adalimumab, certolizumab pegol, etanercept, infliximab, and golimumab), IL-17 inhibitors (ixekizumab, secukinumab), IL-12/23 inhibitors (ustekinumab), phosphodiesterase type 4 inhibitors (apremilast), JAK inhibitors (upadacitinib), and best supportive care (BSC). This population differs slightly from the Health Canada–approved indication; however, it aligns with those in the guselkumab trials, in which patients had to have failed standard therapy. As such, no cost-utility analysis was provided on the use of guselkumab in adults with active PsA who had no prior therapies.
Another analysis assessed the cost-effectiveness of guselkumab in a subpopulation of adult patients with PsA who had not previously been treated with a bDMARD (i.e., were biologic-naive) compared to 9 active treatments: TNF inhibitors (with the exception of certolizumab, which was excluded due to a lack of data in this subpopulation), IL-17 inhibitors, IL-12/23 inhibitors, phosphodiesterase type 4 inhibitors, JAK inhibitors, and BSC.
The other analysis assessed the cost-effectiveness of guselkumab in a subpopulation of adult patients with PsA who had previously experienced an inadequate response or intolerance to at least 1 bDMARD (i.e., were biologic-experienced) compared to 5 treatments: IL-17 inhibitors, IL-12/23 inhibitors, JAK inhibitors, and BSC.3 The modelled populations in the sponsor’s analyses were based on pooled data from the DISCOVER-1, DISCOVER-2, and COSMOS clinical trials.4-6 Subsequent lines of active therapies were not considered in these main analyses. BSC was assumed to include a mix of cDMARDs and supportive and/or palliative care.
Guselkumab is available as a 100 mg/mL solution for injection in either a pre-filled syringe or a patient-controlled injector, with a recommended dose of 100 mg at week 0 and week 4 and every 8 weeks thereafter.1 The submitted price of guselkumab is $3,059.74 per syringe or injector, which corresponds to a cost of $21,418 per patient in the first year (52 weeks), and $19,888 per patient per year thereafter (365 days).3 The annual maintenance cost of the comparator treatments in the sponsor’s model ranged from $12,253 for adalimumab subsequent entry biologics (SEBs) to $21,716 for ixekizumab. Wastage of excess medication in vials was considered, where applicable (i.e., for infliximab). BSC costs were captured as disease-related costs rather than as drug-acquisition costs, and were based on disease severity, as defined in the model inputs section that follows.
The clinical outcome of interest was quality-adjusted life-years (QALYs). The sponsor adopted a lifetime time horizon (53 years), with the analysis conducted from the perspective of a publicly funded health care payer. Future costs and benefits were discounted at a rate of 1.5% per year, and the model cycle length was 28 days.
Model Structure
The sponsor’s model took the form of a semi-Markov model consisting of a trial period (i.e., an initial efficacy assessment period) and a maintenance period (refer to Figure 1 in Appendix 3). The trial period consisted of a series of tunnel states dependent on the time point at which each therapy was assessed for response in its relevant clinical trials (i.e., at 12 weeks for certolizumab, golimumab, infliximab, and upadacitinib; at 16 weeks for apremilast; and at 24 weeks for guselkumab, adalimumab, etanercept, ixekizumab, secukinumab, and ustekinumab). Patients who achieved ACR 20 after the trial period entered the maintenance phase and continued therapy until loss of response or an adverse event, then discontinued. Patients who did not achieve ACR 20 discontinued their current therapy at the end of the trial period and began treatment with the next line of therapy. In the main analyses, this was a direct move to BSC as the final line of therapy. The sponsor assumed that treatment type and status had no effect on patient mortality. Risk of death was based on age- and gender-matched all-cause mortality rates of the Canadian population,7 adjusted for excess mortality due to having PsA.8
Model Inputs
The baseline population characteristics used to inform the model were based on pooled data from the DISCOVER-1, DISCOVER-2, and COSMOS trials,4-6 which included adults older than 18 years of age with active PsA and inadequate response to standard therapies, defined as non-bDMARDs for more than 3 months, and/or apremilast for more than 4 months, and/or NSAID therapy for more than 4 weeks. Baseline characteristics included starting age, percentage of patients who were female, mean body weight, proportion of patients weighing more than 100 kg, and baseline HAQ-DI and PASI scores. These characteristics varied slightly among the populations.
The sponsor submitted network meta-analyses (NMAs) that were informed by the DISCOVER-1, DISCOVER-2, and COSMOS trials and relevant comparator trials found in a systematic literature review for the population of all included patients — as well as for biologic-naive and biologic-experienced subpopulations — to assess the relative efficacy and safety of guselkumab compared to other biologics for the short-term treatment of active PsA in adults.9 NMAs were conducted for the outcomes of ACR response, PASI response, change in HAQ-DI score, and SAEs. The best-fitting model (i.e., a baseline risk-adjusted fixed-effect, baseline risk-adjusted random-effect, unadjusted fixed-effect, or unadjusted random-effect model) was chosen for each analyzed population (i.e., biologic-naive, biologic-experienced, and mixed) and outcome (i.e., ACR 20 response, HAQ-DI score, PASI response, SAEs), with different models selected across the populations and outcomes.
Response in the model was defined as achieving ACR 20 after the trial period (12 weeks, 16 weeks, or 24 weeks) for each comparator; ACR 20 responders entered the maintenance phase of treatment, while non-responders discontinued the drug and began BSC. Additional NMAs for change in HAQ-DI score and PASI response were performed to estimate changes for both responders and non-responders. Prior to response assessment (i.e., before the end of the 12-week to 24-week trial period), all patients were assumed to have baseline HAQ-DI and PASI scores. After responders entered the maintenance phase, the NMA-derived mean HAQ-DI score change from baseline for responders was applied, as was a weighted average improvement in PASI score based on the distribution of patients in PASI improvement categories (e.g., 75% improvement from baseline in Psoriasis Area and Severity Index score [PASI 75], 90% improvement from baseline in PASI). This treatment effect was then assumed to be maintained for the duration of therapy; loss of response was not explicitly modelled, but was assumed to be captured through all-cause discontinuation. When patients moved to BSC, HAQ-DI scores reverted to baseline and continued to worsen at a rate equivalent to natural history progression (i.e., 0.072 per year) until plateauing at the maximum value of 3.
Constant, annual, and treatment-specific discontinuation rates were applied to each active treatment in the maintenance period. These were based on weighted average 1-year discontinuation rates from the clinical trials for each treatment.3
Health state utilities were based on the York algorithm10 (i.e., utility = 0.897 to 0.298 × HAQ-DI score – 0.004 × PASI score). Because HAQ-DI and PASI scores were assumed equivalent to baseline for the duration of the trial period for each comparator, utility values were also the same as at baseline for the duration of these trial periods (i.e., until response assessment). Responders then entered the maintenance phase with utility weights consistent with HAQ-DI and PASI scores calculated from mean change from baseline scores for responders for each comparator, as determined in the NMA.
SAE rates were based on probabilities reported by the sponsor’s NMA for each comparator. SAEs were associated with a disutility of –0.195, using serious infection to represent any SAE,11 and applied for 12 days (the average duration of serious infections from the DISCOVER 1 and 2 trials).
Costs included those for drug acquisition, treatment administration, monitoring, arthritis- and psoriasis-related disease management, and adverse events. Drug-acquisition costs for guselkumab were based on the sponsor’s submitted price, while the costs of comparators were based on Ontario, Saskatchewan, or Quebec formulary list prices.12-15 Administration costs were applied to IV therapies (i.e., infliximab),16 but not to oral or subcutaneous therapies. Resource usage, including rheumatology visits and regular monitoring, was based on the National Institute for Health and Care Excellence review for upadacitinib,17 with costs from Ontario benefit schedules.18,19 The model did not directly include drug costs for cDMARDs, given that these were assumed to be captured in the annual medical costs, based on algorithms that estimated health care costs from absolute PASI and HAQ-DI scores, which were treatment-specific. The equation used to determine costs related to the HAQ-DI were derived from a study that reported costs of treating rheumatoid arthritis patients in the UK and Sweden.10,20,21 Costs associated with psoriasis were based on a Canadian study of the economic burden of psoriasis22 and differed by both severity and whether the patient achieved a PASI 75 response.10 Costs associated with SAEs were based on the weighted average cost of SAEs occurring in the DISCOVER-1, DISCOVER-2, and COSMOS trials.4-6
Summary of Sponsor’s Economic Evaluation Results
The sponsor submitted 3 probabilistic analyses intended to represent the full Health Canada population (i.e., adults with active PsA), the biologic-naive population (i.e., adults with active PsA who had not been treated previously with a bDMARD), and the biologic-experienced population (i.e., adults with active PsA who had been treated previously with a TNF inhibitor). The sponsor’s probabilistic analyses were based on 2,000 iterations; the findings are presented here. The results of the deterministic analyses were similar to those of the probabilistic analyses except where indicated. The submitted analyses are based on publicly available list prices for the comparators. Additional results from the sponsor’s economic evaluation are presented in Appendix 3.
Base-Case Results
Among the full population of adult patients with active PsA included in the trials (i.e., the mixed-population analysis), the sponsor’s model predicted that there are 3 treatments on the cost-effectiveness frontier: BSC, etanercept, and golimumab. Guselkumab was dominated by golimumab; i.e., guselkumab was more costly and less effective than golimumab (Table 3). This means that at the submitted price, guselkumab would not be chosen as the optimal treatment strategy, regardless of a decision-maker’s willingness-to-pay (WTP) threshold.
The sponsor’s model predicted that guselkumab treatment of adults with active PsA was associated with 7.82 QALYs and a cost of $304,881 over a lifetime time horizon; 97% of these QALYs were accrued after the trial assessment point (24 weeks) on the basis of extrapolated long-term data. At a WTP threshold of $50,000 per QALY, guselkumab had a 4% chance of being the most cost-effective therapy.
In the sponsor’s deterministic results, guselkumab was not dominated; rather, it was associated with an incremental cost-effectiveness ratio (ICER) of $278,488 per QALY (i.e., 0.039 incremental QALYs and $10,826 in incremental costs) relative to golimumab.
Table 3: Summary of the Sponsor’s Economic Evaluation Results for Mixed-Population Analysis
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
BSC | 226,012 | 5.912 | Reference |
Etanercept | 260,594 | 7.361 | 23,877 vs. BSC |
Golimumab | 295,732 | 7.850 | 71,781 vs. etanercept |
Dominated treatments | |||
Guselkumab | 304,881 | 7.820 | Dominated by golimumab |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; vs. = versus.
Note: The table presents the results of the sponsor’s probabilistic analysis. Only the drug under review and treatments that are on the efficiency frontier are reported in the main report body. Full results are in Appendix 3.
Source: Sponsor’s pharmacoeconomic submission.3
Among the biologic-naive population, 3 treatments are on the cost-effectiveness frontier: BSC, etanercept, and guselkumab. Guselkumab was associated with an ICER of $62,265 (incremental costs = $49,587; incremental QALYs = 0.796) compared to etanercept (Table 4). At a WTP threshold of $50,000 per QALY, the sponsor’s model predicted a 16% probability of guselkumab being the most cost-effective therapy.
Table 4: Summary of the Sponsor’s Economic Evaluation Results for Biologic-Naive Adults With PsA
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
BSC | 225,884 | 6.914 | Reference |
Etanercept | 263,358 | 7.549 | 27,657 vs. BSC |
Guselkumab | 312,945 | 8.346 | 62,265 vs. etanercept |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; PsA = psoriatic arthritis; QALY = quality-adjusted life-year; vs. = versus.
Note: The table presents the results of the sponsor’s probabilistic analysis. Only the drug under review and treatments that are on the cost-effectiveness frontier are reported in the main report body. Full results are in Appendix 3.
Source: Sponsor’s pharmacoeconomic submission.3
Among the biologic-experienced population of patients who had previously failed 1 or 2 TNF inhibitors, 4 treatments are on the cost-effectiveness frontier: BSC, secukinumab, guselkumab, and ixekizumab. Guselkumab was associated with an ICER of $71,801 (incremental costs = $19,086; incremental QALYs = 0.266) compared to secukinumab (Table 5). At a WTP threshold of $50,000 per QALY, the sponsor’s model predicted a 21% probability of guselkumab being the most cost-effective therapy. If a payer’s WTP threshold is $267,048 per QALY, then ixekizumab becomes the optimal therapy.
In the sponsor’s deterministic analysis of this population, the results were similar, with the exception that ixekizumab was dominated by guselkumab: it remained more costly while accruing 6.451 QALYs (compared to the 6.456 QALYs accrued by guselkumab).
Table 5: Summary of the Sponsor’s Economic Evaluation Results for Biologic-Experienced Adults With PsA
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
BSC | 218,852 | 5.143 | Reference |
Secukinumab | 265,715 | 6.399 | 37,313 vs. BSC |
Guselkumab | 284,801 | 6.664 | 71,801 vs. secukinumab |
Ixekizumab | 290,250 | 6.685 | 267,048 vs. guselkumab |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; PsA = psoriatic arthritis; QALY = quality-adjusted life-year; vs. = versus.
Note: The table presents the results of the sponsor’s probabilistic analysis. Only the drug under review and treatments that are on the cost-effectiveness frontier are reported in the main report body. Full results are in Appendix 3.
Source: Sponsor’s pharmacoeconomic submission.3
Sensitivity and Scenario Analysis Results
The sponsor also conducted scenario analyses. These included assuming a Psoriatic Arthritis Response Criteria–based responder definition, assuming a different utility algorithm, assuming an average second-line (subsequent) therapy, considering lost productivity costs, using a different source for long-term discontinuation, varying discount rates, and using an NMA that incorporated COSMOS data using an analysis correcting for early escape error (ACEEE) by ignoring the early escape treatment failure criterion. Of these scenario analyses, those with the greatest impact on the sequential ICER associated with guselkumab included changing the response definition to a Psoriatic Arthritis Response Criteria response (which increased the ICER for biologic-naive patients and decreased it for biologic-experienced patients); including productivity costs (which decreased the ICER for the biologic-naive population and caused guselkumab to fall off the cost-effectiveness frontier for biologic-experienced patients); and incorporating real-world psoriasis discontinuation data (which decreased the ICER for both subpopulations).
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis.
Relative treatment effects are uncertain: The sponsor’s mixed population represents a combination of multiple patient populations studied in clinical trials (e.g., trials focusing on biologic-experienced patients, such as COSMOS; trials focusing on biologic-naive patients, such as DISCOVER-2; and trials that included a mixture of both types of patients, such as DISCOVER-1) that have been assessed in a single NMA. Combining these heterogenous patient populations into a single analysis leads to substantial uncertainty in the interpretation of the output used to inform the mixed-population analysis results. For example, prior exposure to biologics has been identified as an important effect modifier in psoriasis23; thus, the inclusion of trials with biologic-naive, biologic-experienced, and mixed-experience patients in potentially different proportions between comparators increases uncertainty in results. As stated in CADTH’s Guidelines for the Economic Evaluation of Health Technologies,24 “when a stratified analysis is conducted […] the appropriate estimate of the overall result is determined by weighting the estimates for each subgroup by their respective prevalence.” The sponsor’s mixed-population analysis does not represent the cost-effectiveness of guselkumab in the full population of adults with active PsA in Canada because no such weighting of the 2 subgroups by prevalence was conducted.
CADTH was unable to adjust for the uncertainty resulting from heterogeneity in the mixed population in its reanalyses; a prevalence-weighted combined analysis was not possible due to the different comparators included in the 2 subgroup analyses. CADTH reanalyses instead focused on the subgroup analyses for the biologic-naive and biologic-experienced populations of adults with active PsA. The cost-effectiveness of guselkumab in its full indicated population of adults with active PsA is unknown.
Relative treatment effects in the model are derived from 4 sponsor-conducted NMAs comparing guselkumab to BSC and other biologics for the outcomes of ACR 20 response, HAQ-DI improvement, PASI improvement, and SAEs. The outputs of these NMAs suggests that guselkumab was ||||||||||||| |||||| |||| ||||||| ||| ACR 20 response, change in HAQ-DI score, and change in PASI score ||| ||| ||| ||||||||||||| ||||||||| || ||||||| ||| total SAEs. The NMAs also suggest guselkumab may |||| || ||||||||| || ||||| || ||||||||| |||||||| ||||| |||||| |||||||| || ||| ||| ||| ||| ||||||||||, while indicating that ||||| |||||||||| |||||| || |||||| |||||| |||||||| || |||||| |||||||||||. However, pairwise comparison between guselkumab and other active comparators for these outcomes as well as for SAEs |||||||||| ||| |||| |||||| |||||||| ||| ||||||||||| || ||||||||||| ||||||| || ||||| ||||||| || || |||||||||||. The NMAs were associated with a number of limitations, including a lack of closed loops allowing assessment of consistency among comparisons as well as heterogeneity in the patient population for gender proportion, duration of PsA, number of affected joints, proportion of patients with psoriasis affecting greater than 3% of their body surface area, and baseline PASI score. Only limited information was available on the co-interventions received, patient withdrawals, and imputation of missing data. In particular, the NMAs pooled data from time points ranging from 12 weeks to 24 weeks. This range may bias outcomes, especially those requiring a higher degree of improvement (e.g., 100% improvement from baseline in PASI score versus PASI 75) in favour of comparators that were studied for longer time periods (e.g., guselkumab). The PASI outcome is also associated with a greater degree of uncertainty due to wide credible intervals for some comparators, the assumption of similar relative treatment effects across each response level, and the lack of information on whether randomization was stratified by the presence of skin lesions at baseline (i.e., the subgroup of patients analyzed for the outcome) and whether groups were balanced for confounders. The indirect evidence was limited to short-term outcomes and showed imprecise results for ACR response rates, change in HAQ-DI, and risk of SAEs, limiting the ability to draw a conclusion from the data. PASI response may favour guselkumab versus some biologics, but it included the null for other comparisons, sometimes with wide 95% credible intervals, and was associated with uncertainty. The findings in the subgroup of patients who were biologic-naive were generally similar to those for the overall population in the sponsor-submitted NMA. The NMAs in the biologic-experienced population were sparse and showed greater imprecision in their results.
CADTH was unable to adequately address the uncertainty associated with the point estimates derived from the sponsor’s NMA.
Although the sponsor considered cDMARDs to be captured within BSC, there is no analysis comparing the clinical efficacy of guselkumab to that of cDMARDs. The clinical trials of guselkumab allowed for the use of cDMARDs concomitantly with either treatment. Thus, the sponsor has incorporated cDMARD costs within BSC without fully assessing the relative effects of guselkumab compared with cDMARDs.
CADTH was unable to assess the relative effectiveness of guselkumab compared with cDMARDs.
Long-term treatment efficacy is uncertain: The sponsor’s model is based on an NMA comparing treatment efficacy over the short-term (12 weeks to 24 weeks). It assumes that patients who respond to and continue on their biologic therapy will continue receiving the full benefit of that treatment for as long as they remain on therapy. This implicitly assumes that treatment waning does not occur for the modelled outcomes (i.e., HAQ-DI and PASI). The sponsor assumed no attenuation of efficacy while on treatment.
CADTH could not address this limitation in its reanalyses. The impact of this limitation on the results is unclear.
Long-term discontinuation is uncertain and based on a naive comparison: The sponsor’s model used treatment-specific long-term discontinuation rates (i.e., discontinuation after ACR 20 response has been established) for all comparators except BSC, based on the rates observed in the clinical trials for each comparator.3 By using discontinuation rates directly from each comparator’s clinical trials, the model naively compares trial results without consideration for between-trial heterogeneity in patient populations, trial design, concurrent therapies, subsequent therapies, or time point of response assessment. There is insufficient evidence to conclude that differential discontinuation rates would exist in the same population of patients in Canadian clinical practice. Furthermore, modelled patients experienced only 1 line of therapy before transitioning to BSC; therefore, they would experience worsening of their disease (and the associated QALY loss and cost increase thereafter) to a degree that would not occur in Canadian clinical practice; meanwhile, patients who discontinued would transition to another biologic and gain the associated QALY benefit and avoid greater disease-related costs. Therefore, the sponsor’s included differences in discontinuation rates have an implausibly large impact on the results and are the largest driver of the model.
Disease-related costs are uncertain and may double count some resource use: Disease-related costs in the model were based on algorithms from the literature correlating costs to absolute HAQ-DI and PASI scores.10,20-22 This method assumes that the cost of treating a patient with PsA, excluding biologic therapies, is the same as the cost of treating a patient with rheumatoid arthritis plus the cost of treating a patient with psoriasis; however, this is unlikely to be the case because some aspects of patient care would overlap for patients with PsA, such as pain control, hospitalizations, and community care. Additionally, some aspects of care included in the algorithms are costed separately within the model, such as health care provider visits and laboratory tests.20,22 Also, given the availability of both cDMARDs and bDMARDs in recent years (representing better treatment), it is unlikely that resource use is as closely correlated to disease severity as it was in previous years. It is also unlikely that European resource use in the 1980s and 1990s adequately approximates current Canadian practice. Finally, the assumption that modelled patients discontinue their initial bDMARD and continue on BSC for their remaining years further inflates disease-related costs beyond that which would actually be seen in practice.
CADTH reanalyses removed the separate resource-use costs in the model on the basis that these costs are already included in the disease-related cost algorithms. CADTH explored the uncertainty in the disease-related cost algorithms by halving the HAQ-DI–based coefficient and the difference in costs between mild to moderate psoriasis and moderate to severe psoriasis in a scenario analysis.
Treatments modelled were not reflective of current Canadian clinical practice: Apremilast received a conditional positive recommendation from CADTH for the treatment of PsA27; as a result, it was included in the sponsor’s main analyses. However, because negotiations have twice concluded without an agreement with the pan-Canadian Pharmaceutical Alliance, apremilast is not currently listed for reimbursement by public drug programs.28,29 Additionally, because the guselkumab trials included patients who had failed at least 1 standard therapy that could include apremilast, it may not be appropriate to consider apremilast in patients’ next line of therapy.3-5 CADTH also noted that the limited list of comparators in the biologic-experienced population did not reflect the treatments used in clinical practice. The sponsor stated that this was due to a lack of trial data for other available treatments in this line of therapy.3
CADTH also noted that, based on the Health Canada indication for guselkumab,1 patients are not required to have failed any previous therapies. However, the included clinical trials required patients to have previously failed at least 1 prior therapy, which could include a cDMARD, apremilast, and/or an NSAID. Therefore, the relative clinical and cost-effectiveness of guselkumab in patients who have received no previous therapies is unknown. Additionally, bDMARDs in Canada are typically reimbursed only if a patient has previously failed at least 2 cDMARDs (e.g., Ontario,30 Saskatchewan31). Given that all comparators in the sponsor’s submitted analysis are bDMARDs (except for BSC), CADTH considered BSC to be the only relevant comparator for patients who have failed only 1 prior cDMARD. This consideration aligns with clinical practice and current reimbursement criteria. However, data informing the efficacy of guselkumab and BSC were not available for this subgroup.
CADTH removed apremilast as a comparator from the biologic-naive population analysis. Due to a lack of data, CADTH was unable to explore the cost-effectiveness of guselkumab in patients who had not failed at least 1 standard therapy, nor in patients who had failed only a single cDMARD.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (refer to Table 6).
Table 6: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
The 53-year time horizon represents a lifetime. | Acceptable. The mean starting age is approximately 47 years. |
Patients with moderate to severe psoriasis received ixekizumab every 2 weeks during the induction period. | Acceptable. Thirty-three percent of the mixed population, 31% of biologic-naive patients, and 36% of biologic-experienced patients received ixekizumab every 2 weeks rather than every 4 weeks, consistent with the proportions of patients with moderate to severe psoriasis. |
Sixty-one percent of secukinumab patients received the 300 mg dose. | Acceptable. |
Adalimumab, etanercept, and infliximab are assumed to be 100% biosimilar. | Acceptable. |
Administration costs are included only for IV comparators (infliximab). | Acceptable. |
Mortality is unrelated to treatment arm and is based on the general population with a modifier for having PsA. | Uncertain. The standardized mortality ratio used by the sponsor (i.e., 1.058 relative to the general population) has been accepted in previous CADTH25 and NICE26 reviews for PsA. However, more recent data (2016) from Ontario suggest that the SMR associated with PsA may be higher, at 1.34.32 However, given that the SMR affects all comparators equally, it has minimal impact on the ICER. Thus, it is not considered a key limitation of the analyses. There is no evidence to suggest that choice of biologic therapy will have a differential impact on mortality. As a result, the assumption that mortality is equivalent across treatment groups is acceptable. |
Patients who discontinue their bDMARD continue on BSC for the remainder of their lives. | This is unlikely to reflect clinical practice, but is reasonable as a simplifying assumption. In clinical practice, a patient who has an inadequate response to, loses response to, or cannot tolerate their current bDMARD is likely to switch to other bDMARDs until a suitable therapy is found. However, due to an absence of data to inform the efficacy and safety of treatment sequences (including second, third, and later lines of biologic therapy), the sponsor used the simplifying assumption of no additional lines of therapy except for BSC. |
Mean HAQ-DI scores worsen over time until these reach the maximum score for patients using BSC. | Uncertain. The clinical expert consulted by CADTH indicated that a HAQ-DI score of 3 was very severe and that it was unlikely that the average patient treated with cDMARDs would ever reach it. However, previous PsA models have used the same natural history assumptions as the sponsor.10,25 It is unclear what impact this assumption would have on results. |
The biologic-experienced population can be represented by a population of patients with inadequate response or intolerance to anti-TNFs. | Uncertain. The guselkumab clinical trials4-6 excluded patients who had previously been treated with biologics other than TNF inhibitors; thus, the clinical and cost-effectiveness of guselkumab in patients with an inadequate response or intolerance to other biologics, such as JAK inhibitors or other IL inhibitors, is unknown. |
bDMARD = biologic disease-modifying antirheumatic drug; BSC = best supportive care; cDMARD = conventional disease-modifying antirheumatic drug; HAQ-DI = Health Assessment Questionnaire Disability Index; IL = interleukin; JAK = Janus kinase; NICE = National Institute for Health and Care Excellence; PsA = psoriatic arthritis; SMR = standardized mortality ratio; TNF = tumour necrosis factor.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
The CADTH main reanalyses were derived by making changes to model parameter values and assumptions in consultation with clinical experts. CADTH did not consider the sponsor’s mixed-population analysis due to limitations with the sponsor’s approach. Thus, CADTH focused on the biologic-naive and biologic-experienced populations individually.
The CADTH reanalyses addressed some key limitations of the submitted economic model by equalizing long-term discontinuation among comparators, removing the double-counted resource-use costs, and removing apremilast from the biologic-naive analysis (Table 7). Additionally, CADTH removed the sponsor’s randomization seed for all reanalyses.
CADTH’s main reanalyses are presented in Table 8 (biologic-naive population) and Table 9 (biologic-experienced population). Additional reanalyses and disaggregated results are presented in Appendix 4.
Among patients who are biologic-naive, 3 treatments were on the cost-effectiveness frontier (BSC, etanercept, and infliximab); however, guselkumab was dominated by numerous biologic therapies because it was more costly and less effective (refer to Table 8). Among patients who are biologic-experienced, 3 treatments were also on the cost-effectiveness frontier (in this case, these were BSC, secukinumab, and ixekizumab); however, guselkumab was dominated by secukinumab and upadacitinib (refer to Table 9).
Table 7: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s main analyses | ||
Randomization seeding | Seeded | Not seeded |
Changes to derive the CADTH main analyses | ||
1. Discontinuation rates after response | Rates range from 9% to 25%, depending on biologic, based on a naive comparison of discontinuation rates from trials specific to each biologic. | All biologics have 16.5% as the annual discontinuation rate. |
2. Resource-use costs | Costs include rheumatologist visits, lab tests, and X-rays. | These costs were assumed to already be included in disease-related costs; therefore, they were removed. |
3. Apremilast | This drug was included in the sponsor’s base-case and biologic-naive analyses and excluded in the biologic-experienced analysis. | Excluded in all analyses. |
CADTH base case | — | 1 + 2 + 3 |
Table 8: Summary of the CADTH Reanalysis Results for Biologic-Naive Adults With PsA
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
Sponsor’s corrected analysis | |||
BSC | 224,391 | 6.190 | Reference |
Etanercept | 261,574 | 7.529 | 27,778 vs. BSC |
Guselkumab | 310,965 | 8.325 | 62,038 vs. etanercept |
CADTH analysis | |||
BSC | 218,981 | 6.199 | Reference |
Etanercept | 256,957 | 7.576 | 27,584 vs. BSC |
Infliximab | 280,869 | 7.707 | 182,188 vs. etanercept |
Dominated treatment | |||
Guselkumab | 277,946 | 7.317 | Dominated by adalimumab, etanercept, upadacitinib, secukinumab, and golimumab |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; PsA = psoriatic arthritis; QALY = quality-adjusted life-year; vs. = versus.
Note: The table presents the results of CADTH’s probabilistic analysis. Only the drug under review and treatments that are on the cost-effectiveness frontier are reported in the main body of the report. Full results are in Appendix 4.
Table 9: Summary of the CADTH Reanalysis Results for Biologic-Experienced Adults With PsA
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
Sponsor’s corrected analysis | |||
BSC | 219,238 | 5.092 | Reference |
Secukinumab | 265,945 | 6.359 | 36,858 vs. BSC |
Guselkumab | 284,377 | 6.599 | 62,038 vs. secukinumab |
Ixekizumab | 289,766 | 6.623 | 227,573 vs. guselkumab |
CADTH analysis | |||
BSC | 210,013 | 5.153 | Reference |
Secukinumab | 250,541 | 6.142 | 40,942 vs. BSC |
Ixekizumab | 269,605 | 6.344 | 94,793 vs. secukinumab |
Dominated treatment | |||
Guselkumab | 254,021 | 5.985 | Dominated by upadacitinib, secukinumab |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; PsA = psoriatic arthritis; QALY = quality-adjusted life-year; vs. = versus.
Note: The table presents the results of CADTH’s probabilistic analysis. Only the drug under review and treatments that are on the cost-effectiveness frontier are reported in the main body of the report. Full results are in Appendix 4.
Scenario Analysis Results
Scenario analyses were conducted using the main CADTH reanalyses for both the biologic-naive and biologic-experienced populations to investigate the impact of halving the disease-related cost differences associated with worsening HAQ-DI scores and psoriasis severity and increasing the standardized mortality rate of 1.34 for patients with PsA relative to the general population. Guselkumab remained dominated in both populations. Details and results of these scenarios are presented in Table 21 and Table 22, respectively.
CADTH also undertook price reduction analyses based on the sponsor-submitted analyses for the biologic-naive and biologic-experienced populations as well as for the CADTH main reanalyses of the same populations. For patients with active PsA who are naive to biologics, a 42% price reduction would be required for guselkumab to be considered the most cost-effective option at a WTP threshold of $50,000 per QALY (Table 23), while a 20% price reduction would be required when considering patients with active PsA who are biologic-experienced (Table 24).
Issues for Consideration
Availability of risankizumab for this population is unclear: Risankizumab (Skyrizi), an IL-23 inhibitor, has a Health Canada indication for PsA that is identical to that of Tremfya.33 Risankizumab is reimbursed by public plans for plaque psoriasis13,14,34; however, it has not been reviewed by CADTH for the treatment of PsA. At the list price of the 150 mg dose on the Saskatchewan formulary, the annual maintenance cost of risankizumab for PsA would be $21,458 per patient per year. This is more expensive than most other bDMARDs available for the treatment of PsA, including guselkumab at its submitted price.
The analysis is based on publicly available prices for comparators: The modelled prices of biologic therapies are based on publicly accessible list prices and do not reflect existing confidential pricing that has been negotiated by public plans. When existing confidential discounts on biologic therapies are considered, greater price reductions than those estimated in this report would be required to achieve cost-effectiveness.
Guselkumab was not successfully negotiated for reimbursement for the treatment of plaque psoriasis: In 2018, the CADTH Canadian Drug Expert Committee recommended that guselkumab be funded for the treatment of moderate to severe plaque psoriasis in a similar manner to other biologic therapies, with the condition that the drug plan cost for guselkumab should not exceed the drug plan cost of treatment with the least costly biologic reimbursed for moderate to severe plaque psoriasis.35 However, in 2019, price negotiations between the sponsor and the pan-Canadian Pharmaceutical Alliance concluded without an agreement. As a result, guselkumab is not currently listed for the treatment of plaque psoriasis by public drug plans in Canada.36
Patents for some comparators have recently expired: The patents for ustekinumab and certolizumab expired in June 2021 and August 2021, respectively.37,38 As such, it is possible that biosimilar versions of 1 or both products could become available within the next few years. Should this happen, the incremental cost of guselkumab compared to ustekinumab and/or certolizumab would increase.
The dosing regimen of guselkumab may be preferred by some patients: The recommended maintenance dose schedule for guselkumab is every 8 weeks,1 which is a longer interval than most of the comparator bDMARDs (the exceptions are ustekinumab, which is dosed every 12 weeks in the maintenance phase, and infliximab, which involves an infusion every 8 weeks; refer to Appendix 1). It is likely that some patients will prefer less frequent injections. These patients may prefer options such as guselkumab.
Overall Conclusions
Based on the CADTH clinical review of the DISCOVER-1, DISCOVER-2, and COSMOS clinical trials, compared to placebo, adults with active PsA who received guselkumab were more likely to show improvements in terms of ACR response, HAQ-DI improvement, and — in patients with psoriasis affecting 3% or more of their body surface area at baseline — PASI response. Favourable clinical responses were observed among patients who were biologic-naive, among patients who had experienced prior intolerance or inadequate response to TNF alpha inhibitors, and among a mixed population that included patients with and without prior TNF inhibitor exposure. There is no direct evidence comparing guselkumab to other biologic DMARDs available in Canada. The indirect evidence comparing guselkumab to other biologic DMARDs available in Canada in the full population showed imprecise results for ACR response rates, change in HAQ-DI scores, and risk of SAEs. This imprecision limits the ability to draw conclusions from these data. Based on this indirect evidence, short-term PASI response rates may favour guselkumab versus some biologics. However, there is uncertainty in these findings, given that several sources of heterogeneity were identified across the trials included in the sponsor-submitted indirect treatment comparison and that it is unclear whether the methods used to control for potential bias were adequate. In addition, there was limited ability to assess consistency between the direct and indirect evidence. The findings for the biologic-naive population were consistent with those for the full population in the analyses. Due to the sparse network for the biologic-experienced population, findings on all outcomes (including PASI) are uncertain.
CADTH identified several key limitations with the sponsor’s economic submission. First, the relative clinical effectiveness of guselkumab compared to other biologics is uncertain because the sponsor’s mixed-population analysis combined the heterogenous subpopulations of biologic-naive and biologic-experienced patients into 1 analysis without consideration for the prevalence of the 2 subpopulations in Canadian practice or the proportions of each subpopulation in the included trials of different comparators. Additionally, the relative clinical effectiveness of guselkumab compared to other biologic comparators is uncertain as the sponsor’s NMA is associated with limitations and must be interpreted with caution, given the imprecision in the point estimates. Second, the trial evidence is short-term (12 weeks to 24 weeks); long-term outcomes have not been assessed in the clinical trials. Third, the sponsor naively compared discontinuation rates from individual trials for each comparator without regard to heterogeneity in patient populations and trial design. When combined with the assumption that patients would discontinue directly to BSC, this approach amplified differences in the QALYs gained and costs derived from these discontinuation differences. Fourth, disease-related resource use was uncertain; therefore, it was likely double counted. Finally, the modelled treatments were not reflective of current Canadian practice; apremilast is not currently reimbursed by public dug plans, and the comparators considered in the biologic-experienced population may be too limited.
CADTH undertook reanalyses that equalized the discontinuation rates between biologic comparators, removed some double-counted resource use, removed apremilast, and did not consider the mixed-population analysis. These reanalyses indicated that guselkumab is not cost-effective at the submitted price because it is dominated by (i.e., it is more costly and less effective than) other available treatments in both the biologic-naive and biologic-experienced populations of adult patients with active PsA. A price reduction of 20% (in the biologic-experienced population) to 42% (in the biologic-naive population) is required for guselkumab to move onto the cost-effectiveness frontier, based on CADTH reanalyses. However, CADTH was unable to address all limitations, including uncertainty in the relative short- and long-term clinical effectiveness of guselkumab; the unknown cost-effectiveness of guselkumab against excluded comparators; and the lack of cost-effectiveness information for the full population of patients indicated by Health Canada. The CADTH reanalysis assumed differences between treatments in clinical benefit based on the point estimates from the NMAs; if no differences in clinical benefit are applied, then different price reductions may be required for guselkumab. The price reductions are based on publicly available drug prices for the comparators. Further reduction may be required if confidential discounts exist for comparator products.
References
1.Tremfya (guselkumab): 100 mg/1 mL sterile solution for injection in pre-filled syringe or Tremfya One-pass (guselkumab): 100 mg/1 mL sterile solution for injection in a patient-controlled injector [product monograph]. Toronto (ON): Janssen Inc.; 2022 Apr 13.
2.Drug Reimbursement Review sponsor submission: Tremfya (guselkumab), 100 mg/mL solution for injection [internal sponsor's package]. Toronto (ON): Janssen Inc.; 2022 May 17.
3.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Tremfya (guselkumab), 100 mg/mL solution for injection. Toronto (ON): Janssen Inc.; 2022 May 17.
4.Clinical Study Report: CNTO1959PSA3001. DISCOVER-1 24 week: a phase 3, multicenter, randomized, double-blind, placebo-controlled study evaluating the efficacy and safety of guselkumab administered subcutaneously in subjects with active psoriatic arthritis including those previously treated with biologic anti-TNFα agent(s) [internal sponsor's report]. Raritan (NJ): Janssen Research & Development, LLC; 2019 Aug 28.
5.Clinical Study Report: CNTO1959PSA3002. DISCOVER-2 24 week: a phase 3, multicenter, randomized, double-blind, placebo-controlled study evaluating the efficacy and safety of guselkumab administered subcutaneously in subjects with active psoriatic arthritis [internal sponsor's report]. Raritan (NJ): Janssen Research & Development, LLC; 2019 Aug 28.
6.Clinical Study Report: EDMS-RIM-142397:1.0. COSMOS: a phase 3b, multicenter, randomized, double-blind, placebo-controlled study to evaluate the efficacy and safety of guselkumab administered subcutaneously in participants with active psoriatic arthritis and an inadequate response to anti-tumor necrosis factor COSMOS [internal sponsor's report]. High Wycombe (UK): Janssen-Cilag Ltd.; 2020 Sep 23.
7.Table: 13-10-0114-01. Life expectancy and other elements of the life table, Canada, all provinces except Prince Edward Island. Ottawa (ON): Statistics Canada; 2022: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011401. Accessed 2022 Jun 03.
8.Ali Y, Tom BD, Schentag CT, Farewell VT, Gladman DD. Improved survival in psoriatic arthritis with calendar time. Arthritis Rheum. 2007;56(8):2708-2714. PubMed
9.Guselkumab (Tremfya®) for psoriatic arthritis: systematic review and network metaanalyses. Global NMA technical report [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Tremfya (guselkumab), 100 mg/mL solution for injection. Toronto (ON): Janssen Inc.; 2022.
10.Rodgers M, Epstein D, Bojke L, et al. Etanercept, infliximab and adalimumab for the treatment of psoriatic arthritis: a systematic review and economic evaluation. Health Technol Assess. 2011;15(10):i-xxi, 1-329. PubMed
11.Tolley K, Goad C, Yi Y, Maroudas P, Haiderali A, Thompson G. Utility elicitation study in the UK general public for late-stage chronic lymphocytic leukaemia. Eur J Health Econ. 2013;14(5):749-759. PubMed
12.Exceptional Access Program (EAP). Toronto (ON): Ontario Ministry of Health; Ontario Ministry of Long-Term Care; 2022: https://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx. Accessed 2022 Jun 06.
13.Saskatchewan Drug Plan: search formulary. 2022; https://formulary.drugplan.ehealthsask.ca/SearchFormulary. Accessed 2022 Jun 13.
14.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2022; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2022 Jun 06.
15.Régie de l'assurance maladie du Québec. List of medications, May 26, 2022. Quebec (QC): Government of Quebec; 2022: https://www.ramq.gouv.qc.ca/sites/default/files/documents/liste_med_2022-05-26_en.pdf. Accessed 2022 Jun 13.
16.Tam VC, Ko YJ, Mittmann N, et al. Cost-effectiveness of systemic therapies for metastatic pancreatic cancer. Curr Oncol. 2013;20(2):e90-e106. PubMed
17.National Institute for Health and Care Excellence. Upadacitinib for treating active psoriatic arthritis after inadequate response to DMARDs. (Technology appraisal guidance TA768) 2022; https://www.nice.org.uk/guidance/ta768. Accessed 2022 Jun 13.
18.Schedule of benefits for physician services under the Health Insurance Act: effective October 1, 2021. Toronto (ON): Ontario Ministry of Health; 2022: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf. Accessed 2022 Jun 13.
19.Schedule of benefits for laboratory services: effective July 1, 2020. Toronto (ON): Ontario Ministry of Health; 2020: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/lab/lab_mn2020.pdf. Accessed 2022 Jun 13.
20.Kobelt G, Jonsson L, Lindgren P, Young A, Eberhardt K. Modeling the progression of rheumatoid arthritis: a two-country model to estimate costs and consequences of rheumatoid arthritis. Arthritis Rheum. 2002;46(9):2310-2319. PubMed
21.Bansback NJ, Ara R, Barkham N, et al. Estimating the cost and health status consequences of treatment with TNF antagonists in patients with psoriatic arthritis. Rheumatology (Oxford). 2006;45(8):1029-1038. PubMed
22.Levy AR, Davie AM, Brazier NC, et al. Economic burden of moderate to severe plaque psoriasis in Canada. Int J Dermatol. 2012;51(12):1432-1440. PubMed
23.Wade R, Sharif-Hurst S, Dias S. Patient characteristics as effect modifiers for psoriasis biologic treatment response: an assessment using network meta-analysis subgroups. Syst Rev. 2020;9(1):132. PubMed
24.Guidelines for the economic evaluation of health technologies: Canada. 4th ed. Ottawa (ON): CADTH; 2017: https://www.cadth.ca/guidelines-economic-evaluation-health-technologies-canada-4th-edition. Accessed 2022 Jul 21.
25.Drug Reimbursement Review clinical guidance and pharmacoeconomic reports: upadacitinib (Rinvoq) for psoriatic arthritis. Can J Health Technol. 2021;1(10). https://www.cadth.ca/sites/default/files/DRR/2021/SR0658-combined-report.pdf. Accessed 2022 Jun 13.
26.National Institute for Health and Care Excellence. Ixekizumab for treating active psoriatic arthritis after inadequate response to DMARDs. (Technology appraisal guidance TA537) 2018; https://www.nice.org.uk/guidance/ta537. Accessed 2022 Jul 21.
27.CADTH Canadian Drug Expert Committee (CDEC) final recommendation: apremilast (Otezla — Celgene) for psoriatic arthritis. Ottawa (ON): CADTH; 2015 Dec 17: https://www.cadth.ca/sites/default/files/cdr/complete/SR0437_complete_Otezla_PsA-Dec-21-15_e.pdf. Accessed 2022 Jul 06.
28.pan-Canadian Pharmaceutical Alliance. Otezla (apremilast). 2021; https://www.pcpacanada.ca/negotiation/21210. Accessed 2022 Jul 06.
29.pan-Canadian Pharmaceutical Alliance. Otezla (apremilast). 2017; https://www.pcpacanada.ca/negotiation/20909. Accessed 2022 Jul 06.
30.Exceptional Access Program Reimbursement Criteria for Frequently Requested Drugs. Toronto (ON): Ontario Ministry of Health; 2022: https://www.health.gov.on.ca/en/pro/programs/drugs/docs/frequently_requested_drugs.pdf. Accessed 2022 Jul 06.
31.Saskatchewan Drug Plan. Appendix A: Exception Drug Status Program. Regina (SK): Government of Sakatchewan; 2022: https://formulary.drugplan.ehealthsask.ca/PDFs/APPENDIXA.pdf. Accessed 2022 Jul 06.
32.Colaco K, Widdifield J, Luo J, et al. Trends in mortality and cause-specific mortality among patients with psoriasis and psoriatic arthritis in Ontario, Canada. J Am Acad Dermatol. 2021;84(5):1302-1309. PubMed
33.Skyrizi (risankizumab): 150 mg/mL or 90 mg/mL sterile solution for subcutaneous injection [product monograph]. St-Laurent (QC): AbbVie Corporation; 2022 Jun 24: https://pdf.hres.ca/dpd_pm/00066510.PDF. Accessed 2022 Jul 23.
34.Government of Alberta. Interactive drug benefit list. 2022; https://idbl.ab.bluecross.ca/idbl/load.do. Accessed 2022 Jul 23.
35.CADTH Canadian Drug Expert Committee (CDEC) final recommendation: guselkumab (Tremfya — Janssen Inc.) for moderate-to-severe plaque psoriasis. Ottawa (ON): CADTH; 2018 Feb 21: https://www.cadth.ca/sites/default/files/cdr/complete/SR0530_Tremfya_complete_Feb-23-18.pdf. Accessed 2022 Jul 04.
36.pan-Canadian Pharmaceutical Alliance. Tremfya (guselkumab). 2019; https://www.pcpacanada.ca/negotiation/21013. Accessed 2022 Jul 04.
37.Health Canada. Patent Register - Form IV summaries - Ustekinumab. 2022; https://pr-rdb.hc-sc.gc.ca/pr-rdb/index-eng.jsp. Accessed 2022 Jul 06.
38.Health Canada. Patent Register - Form IV summaries - Certolizumab pegol. 2022; https://pr-rdb.hc-sc.gc.ca/pr-rdb/index-eng.jsp. Accessed 2022 Jul 06.
39.Table: 17-10-0057-01 Projected population, by projection scenario, age and sex, as of July 1 (x 1,000). Ottawa (ON): Statistics Canada; 2022: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710005701. Accessed 2022 Jun 16.
40.Chart 1: Population growth rate, 1999/2000 to 2019/2020, Canada. Canada's population estimates: Age and sex, July 1, 2020. Ottawa (ON): Statistics Canada; 2020: https://www150.statcan.gc.ca/n1/daily-quotidien/200929/cg-b001-eng.htm. Accessed 2022 Jun 16.
41.Eder L, Widdifield J, Rosen CF, et al. Trends in the Prevalence and Incidence of Psoriasis and Psoriatic Arthritis in Ontario, Canada: A Population-Based Study. Arthritis Care Res (Hoboken). 2019;71(8):1084-1091. PubMed
42.Kavanaugh A, Singh R, Karki C, et al. Disease activity and biologic use in patients with psoriatic arthritis or rheumatoid arthritis. Clin Rheumatol. 2018;37(8):2275-2280. PubMed
43.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Tremfya (guselkumab), 100 mg/mL solution for injection. Toronto (ON): Janssen Inc.; 2022 May 17.
44.Non-Insured Health Benefits program: First Nations and Inuit Health Branch: Annual report 2020 to 2021. Ottawa (ON): Indigenous Services Canada; 2021: https://www.sac-isc.gc.ca/eng/1645718409378/1645718500555. Accessed 2022 Jul 23.
Appendix 1: Cost Comparison Table
Note this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s). Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 10: CADTH Cost Comparison Table for PsA
Treatment | Strength/ concentration | Form | Price ($) | Recommended dosage | Daily cost ($) | Annual cost ($) |
|---|---|---|---|---|---|---|
Guselkumab | 100 mg/mL | Pre-filled syringe or patient-controlled injector | 3,059.7400a | 100 mg at weeks 0 and 4 then every 8 weeks thereafter | First year: 56.73 Subsequent: 54.64 | First year: 20,722 Subsequent: 19,957 |
TNF inhibitors | ||||||
Adalimumab | 40 mg/0.8 mL | Pre-filled syringe or pen | 794.1000 | 40 mg every 2 weeks | 56.72 | 20,718 |
SEB Adalimumab (biosimilars) | 20 mg/0.4 mL 40 mg/0.8 mL | Pre-filled syringe or pen | 235.6350 471.2700 | 40 mg every 2 weeks | 33.66 | 12,295 |
Certolizumab pegol (Cimzia) | 200 mg/mL | Single-use pre-filled syringe | 664.5100 | 400 mg SC injection at weeks 0, 2, and 4, then 200 mg every 2 weeks or 400 mg every 4 weeks | First year: 52.92 Subsequent: 47.47 | First year: 19,330 Subsequent: 17,337 |
Etanercept | 25 mg/vial | Vial | 202.9300 | 50 mg weekly (one 50 mg injection or two 25 mg injections on the same day or 3 or 4 days apart) | 57.98 | 21,177 |
50 mg/mL | Pre-filled syringe or auto-injector | 405.9850 | 58.00 | 21,184 | ||
SEB Etanercept | 25 mg/0.5 mL | Vial | 120.5000 | 34.43 | 12,566 | |
50 mg/mL | Pre-filled syringe or auto-injector | 241.0000 | ||||
Golimumab SC | 50 mg/0.5 mL 100 mg/mL | Pre-filled syringe or Auto-injector | 1,555.1700 | 50 mg monthly | 51.09 | 18,662 |
Infliximab | 100 mg/vial | Vial | 987.5600 | 5 mg/kg initial dose followed with additional similar doses at 2 and 6 weeks after the first infusion, then every 8 weeks thereafter | First year: 105.07 Subsequent: 88.18 | First year: 38,378 Subsequent: 32,206 |
SEB Infliximab | 100 mg/vial | Vial | 525.0000 | First year: 55.86 Subsequent: 46.88 | First year: 20,402 Subsequent: 17,121 | |
SEB Infliximab | 100 mg/vial | Vial | 493.0000 | First year: 52.45 Subsequent: 44.02 | First year: 19,159 Subsequent: 16,078 | |
IL-17A inhibitors | ||||||
Secukinumab | 150 mg/mL | Pre-filled syringe, pen, or vial | 840.0000 | 150 mg SC at weeks 0,1,2,3, and 4 followed by monthly thereafter 300 mg with coexistent moderate to severe plaque psoriasis | First year: 34.15 to 68.30 Subsequent: 27.60 to 55.20 | First year: 12,474 to 24,948 Subsequent: 10,080 to 20,160 |
Ixekizumab | 80 mg/mL | Pre-filled syringe or pen | 1,670.4400 | 160 mg SC at week 0, followed by 80 mg every 4 weeksb 160 mg SC at week 0, followed by 80 mg weeks 2,4,6,8,10, and 12, then 80 mg every 4 weeksc | First year: 64.23 to 77.95 Subsequent: 59.66 | First year: 23,461 to 28,472 Subsequent: 21,790 |
IL-12/23 inhibitor | ||||||
Ustekinumab | 45 mg/0.5 mL 90 mg/mL | Pre-filled syringe or vial | 4,593.1400 4,593.1400 | Patients < 100 kg: 45 mg at weeks 0 and 4, then every 12 weeks thereafter Patients > 100 kg: 90 mg at weeks 0 and 4, then every 12 weeks thereafter | First year: 63.06 Subsequent: 54.68 | First year: 23,034 Subsequent: 19,972 |
Janus kinase inhibitor | ||||||
Upadacitinib | 15 mg | Tablet | 48.6800d | 15 mg daily | 48.68 | 17,768 |
Phosphodiesterase type 4 inhibitor | ||||||
Apremilast | 10 mg 20 mg 30 mg | Tablet | 18.9041e | 30 mg twice daily, following titration | 37.81 | 13,809 |
Conventional synthetic disease-modifying antirheumatic drugs | ||||||
Methotrexate | 2.5 mg 10 mg | Tablet | 0.5027 2.7067f | 7.5 to 25 mg per week until response achieved. Dose adjusted to optimal clinical response; 30 mg/week not ordinarily be exceeded | 0.22 to 0.92 | 79 to 335 |
20 mg/2 mL 50 mg/2 mL | Injection | 12.5000 8.9200 | 10 to 25 mg per week until response achieved. Dose adjusted to optimal clinical response; 25 mg/week not ordinarily be exceeded | 127 | 465 | |
Leflunomide | 10 mg 20 mg | Tablet | 2.0000 | Loading: 100 mg daily for 3 days Maintenance: 20 mg daily | First year: 2.07 Subsequent: 2.00 | First year: 755 Subsequent: 731 |
Sulfasalazine | 500 mg | Tablet | 0.2533 | Titration: Week 1: 500 mg/day Week 2: 1,000 mg/day Week 3: 1,500 mg/day Maintenance: 2000 mg/day | First year: 0.98 Subsequent: 1.01 | First year: 359 Subsequent: 370 |
500 mg | EC tab | 0.3630 | First year: 1.41 Subsequent: 1.45 | First year: 515 Subsequent: 530 | ||
EC = enteric coated; SC = subcutaneous; SEB = subsequent entry biologic.
Note: All prices are from the Ontario Drug Benefit formulary (accessed June 2022),12,14 unless otherwise indicated, and do not include dispensing fees. Annual cost is based on a 365.25-day year. All weight-based doses assume an average patient weight of 85 kg and wastage of excess medication in vials.
aSponsor-submitted price.3
bDosing regimen for adult patients with PsA or patients with PsA with coexistent mild plaque psoriasis.
cDosing regimen for adult patients with PsA with coexistent moderate to severe plaque psoriasis.
dCADTH Rinvoq review for psoriatic arthritis.25
eRégie de l’assurance maladie du Québec formulary (accessed June 2022).15
fSaskatchewan formulary (accessed June 2022).13
Appendix 2: Submission Quality
Note this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | The mixed population is not representative of full population of patients with PsA in Canada, as weighting of the biologic-naive and biologic-experienced subgroups was not conducted |
Model has been adequately programmed and has sufficient face validity | Yes | No comment |
Model structure is adequate for decision problem | Yes | No comment |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note this appendix has not been copy-edited.
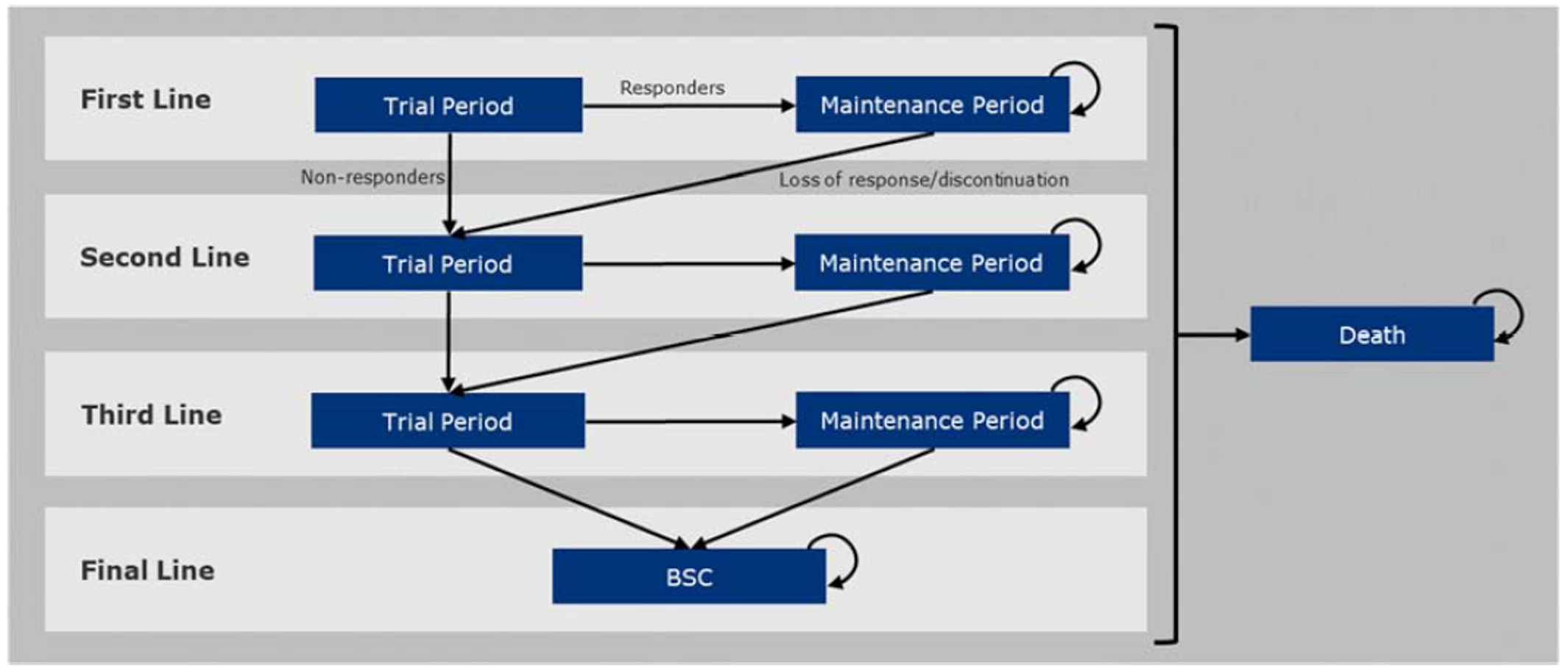
BSC = best supportive care.
Note: In the sponsor’s base-case analyses, second and third line therapies are excluded; patients move directly from first-line therapy to BSC.
Source: Sponsor’s pharmacoeconomic submission.3
Detailed Results of the Sponsor’s Base Case
Table 12: Summary of the Sponsor’s Economic Evaluation Results for the Mixed-Population Analysis (Adults With Active PsA in the Included Trials)
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
BSC | 226,012 | 5.912 | Reference |
Etanercept | 260,594 | 7.361 | 23,877 vs. BSC |
Golimumab | 295,732 | 7.850 | 71,781 vs. etanercept |
Dominated therapiesa | |||
Apremilast | 243,268 | 6.025 | Extendedly dominated through BSC and any of adalimumab, etanercept, golimumab, upadacitinib, infliximab, ustekinumab, certolizumab, secukinumab, guselkumab, or ixekizumab |
Adalimumab | 253,072 | 6.803 | Extendedly dominated through BSC and etanercept |
Upadacitinib | 271,111 | 6.983 | Dominated by etanercept |
Infliximab | 273,388 | 6.901 | Dominated by etanercept and upadacitinib |
Ustekinumab | 279,517 | 6.852 | Dominated by etanercept |
Certolizumab | 280,603 | 7.225 | Dominated by etanercept |
Secukinumab | 280,745 | 7.368 | Dominated by BSC and golimumab |
Guselkumab | 304,881 | 7.820 | Dominated by golimumab |
Ixekizumab | 304,977 | 7.596 | Dominated by golimumab and guselkumab |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; PsA = psoriatic arthritis; QALY = quality-adjusted life-year; Ref. = reference; vs. = versus.
Note: Table presents the results of the sponsor’s probabilistic analyses.
aExtended dominance reported as determined by CADTH.
Source: Sponsor’s pharmacoeconomic submission.3
Table 13: Summary of the Sponsor’s Economic Evaluation Results for the Biologic-Naive Population (Adults With Active PsA Without Prior bDMARD Experience)
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
BSC | 225,884 | 6.194 | Reference |
Etanercept | 263,358 | 7.549 | 27,657 vs. BSC |
Guselkumab | 312,945 | 8.346 | 62,265 vs. etanercept |
Dominated therapiesa | |||
Apremilast | 246,162 | 6.303 | Extendedly dominated through BSC and any of adalimumab, etanercept, golimumab, upadacitinib, infliximab, ustekinumab, certolizumab, secukinumab, guselkumab, or ixekizumab |
Adalimumab | 256,670 | 7.112 | Extendedly dominated through BSC and etanercept |
Infliximab | 273,492 | 7.075 | Dominated by adalimumab, etanercept |
Upadacitinib | 276,131 | 7.374 | Dominated by etanercept |
Secukinumab | 286,020 | 7.728 | Extendedly dominated through BSC and golimumab or BSC and guselkumab |
Ustekinumab | 287,262 | 7.219 | Dominated by etanercept, upadacitinib, secukinumab |
Golimumab | 296,137 | 7.999 | Extendedly dominated through BSC and guselkumab |
Ixekizumab | 311,270 | 7.924 | Dominated by golimumab |
bDMARD = biologic disease-modifying antirheumatic drugs; BSC = best supportive care; ICER = incremental cost-effectiveness ratio; PsA = psoriatic arthritis; QALY = quality-adjusted life-year; Ref. = reference; vs = versus.
aExtended dominance reported as determined by CADTH.
Note: Table presents the results of the sponsor’s probabilistic analyses.
Source: Sponsor’s pharmacoeconomic submission.3
Table 14: Summary of the Sponsor’s Economic Evaluation Results for the Biologic-Experienced Population (Adults With Active PsA With Prior Anti-TNF Experience)
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
BSC | 218,852 | 5.143 | Reference |
Secukinumab | 265,715 | 6.399 | 37,313 vs. BSC |
Guselkumab | 284,801 | 6.664 | 71,801 vs. secukinumab |
Ixekizumab | 290,250 | 6.685 | 267,048 vs. guselkumab |
Dominated therapiesa | |||
Upadacitinib | 258,683 | 5.986 | Extendedly dominated through BSC and any of secukinumab, guselkumab, or ixekizumab |
Ustekinumab | 265,655 | 5.974 | Dominated by upadacitinib |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; PsA = psoriatic arthritis; QALY = quality-adjusted life-year; Ref. = reference; TNF = tumour necrosis factor; vs = versus.
aExtended dominance reported as determined by CADTH.
Note: Table presents the results of the sponsor’s probabilistic analyses.
Source: Sponsor’s pharmacoeconomic submission.3
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note this appendix has not been copy-edited.
Detailed Results of CADTH Base Case
Table 15: Summary of the Stepped Analysis of the CADTH Reanalysis Results, Biologic-Naive Population
Stepped analysis | Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case, deterministic | BSC | 234,388 | 5.799 | Reference |
Etanercept | 270,881 | 7.249 | 25,178 | |
Guselkumab | 321,602 | 8.138 | 57,001 | |
Sponsor’s corrected base case, probabilistic | BSC | 224,391 | 6.190 | Reference |
Etanercept | 261,574 | 7.529 | 27,778 | |
Guselkumab | 310,965 | 8.325 | 62,038 | |
CADTH reanalysis 1: Equal discontinuation after response, deterministic | BSC | 234,388 | 5.799 | Reference |
Etanercept | 270,881 | 7.249 | 25,178 | |
Infliximab | 298,321 | 7.386 | 200,301 | |
Guselkumab | 291,709 | 6.976 | Dominated by adalimumab, etanercept, upadacitinib, secukinumab, golimumab | |
CADTH reanalysis 2: extra resource costs removed, deterministic | BSC | 225,921 | 5.799 | Reference |
Etanercept | 262,415 | 7.249 | 25,178 | |
Guselkumab | 313,136 | 8.138 | 57,001 | |
CADTH reanalysis 3: Apremilast removed, deterministic | BSC | 234,388 | 5.799 | Reference |
Etanercept | 270,881 | 7.249 | 25,178 | |
Guselkumab | 321,602 | 8.138 | 57,001 | |
CADTH base case, deterministic | BSC | 225,921 | 5.799 | Reference |
Etanercept | 262,415 | 7.249 | 25,178 | |
Infliximab | 289,788 | 7.386 | 199,813 | |
Guselkumab | 283,242 | 6.976 | Dominated by adalimumab, etanercept, upadacitinib, secukinumab, golimumab | |
CADTH base case, probabilistic | BSC | 218,981 | 6.199 | Reference |
Etanercept | 256,957 | 7.576 | 27,584 | |
Infliximab | 280,869 | 7.707 | 182,188 | |
Guselkumab | 277,946 | 7.317 | Dominated by adalimumab, etanercept, upadacitinib, secukinumab, golimumab |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: Only treatments that are on the cost-effectiveness frontier as well as the drug under review (guselkumab) are reported.
Table 16: Summary of the Stepped Analysis of the CADTH Reanalysis Results, Biologic-Experienced Population
Stepped analysis | Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|---|
Sponsor’s base case, deterministic | BSC | 225,558 | 4.814 | Reference |
Secukinumab | 272,387 | 6.187 | 34,103 | |
Guselkumab | 291,495 | 6.456 | 71,141 | |
Sponsor’s corrected base case, probabilistic | BSC | 219,238 | 5.092 | Reference |
Secukinumab | 265,945 | 6.359 | 36,858 | |
Guselkumab | 284,377 | 6.599 | 76,675 | |
Ixekizumab | 289,766 | 6.623 | 227,573 | |
CADTH reanalysis 1: Equal discontinuation after response, deterministic | Best supportive care | 225,558 | 4.814 | Reference |
Secukinumab | 265,834 | 5.896 | 37,225 | |
Ixekizumab | 284,979 | 6.100 | 93,995 | |
Guselkumab | 269,679 | 5.727 | Dominated by secukinumab | |
CADTH reanalysis 2: extra resource costs removed, deterministic | BSC | 217,574 | 4.814 | Reference |
Secukinumab | 264,402 | 6.187 | 34,103 | |
Guselkumab | 283,511 | 6.456 | 71,141 | |
CADTH base case, deterministic | Best supportive care | 217,574 | 4.814 | Reference |
Secukinumab | 257,849 | 5.896 | 37,225 | |
Ixekizumab | 276,995 | 6.100 | 93,995 | |
Guselkumab | 261,695 | 5.727 | Dominated by secukinumab | |
CADTH base case, probabilistic | Best supportive care | 210,013 | 5.123 | Reference |
Secukinumab | 250,541 | 6.142 | 40,942 | |
Ixekizumab | 269,605 | 6.344 | 94,793 | |
Guselkumab | 254,021 | 5.985 | Dominated by upadacitinib, secukinumab |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: Only treatments that are on the cost-effectiveness frontier as well as the drug under review are reported.
Table 17: Summary of CADTH Base-Case Economic Evaluation Results for the Biologic-Naive Population (Adults With Active PsA Without Prior bDMARD Experience)
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
BSC | 218,981 | 6.199 | Reference |
Etanercept | 256,957 | 7.576 | 27,584 vs. BSC |
Infliximab | 280,869 | 7.707 | 182,188 vs. etanercept |
Dominated therapies | |||
Adalimumab | 253,422 | 7.346 | Extendedly Dominated through BSC and Etanercept |
Upadacitinib | 270,223 | 7.422 | Dominated by Etanercept |
Secukinumab | 271,584 | 7.396 | Dominated by Etanercept, Upadacitinib |
Golimumab | 274,877 | 7.462 | Dominated by Etanercept |
Ustekinumab | 276,115 | 7.070 | Dominated by Adalimumab, Etanercept, Upadacitinib, Secukinumab, Golimumab SC |
Guselkumab | 277,946 | 7.317 | Dominated by Adalimumab, Etanercept, Upadacitinib, Secukinumab, Golimumab SC |
Ixekizumab | 292,242 | 7.525 | Dominated by Etanercept, Infliximab |
bDMARD = biologic disease-modifying antirheumatic drugs; BSC = best supportive care; ICER = incremental cost-effectiveness ratio; PsA = psoriatic arthritis; QALY = quality-adjusted life-year; Ref. = reference; vs = versus.
Note: Table presents the results of the CADTH’s probabilistic analyses.
Table 18: Summary of CADTH Base-Case Economic Evaluation Results for the Biologic-Experienced Population (Adults With Active PsA With Prior Anti-TNF Experience)
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
BSC | 210,013 | 5.153 | Reference |
Secukinumab | 250,541 | 6.142 | 40,942 vs. BSC |
Ixekizumab | 269,605 | 6.344 | 94,793 vs. secukinumab |
Dominated therapies | |||
Upadacitinib | 249,466 | 5.990 | Extendedly Dominated through BSC and Secukinumab |
Ustekinumab | 253,073 | 5.872 | Dominated by Upadacitinib, Secukinumab |
Guselkumab | 254,021 | 5.985 | Dominated by Upadacitinib, Secukinumab |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; PsA = psoriatic arthritis; QALY = quality-adjusted life-year; Ref. = reference; TNF = tumour necrosis factor; vs = versus.
Table 19: Disaggregated Costs for CADTH’s Probabilistic Base-Case Analyses
Drug | Drug costs ($) | Disease-related costs ($) | Administration costs ($) | Resource-use costs ($) | Adverse Event costs ($) | Total costs ($) |
|---|---|---|---|---|---|---|
Biologic-naive population | ||||||
BSC | 0 | 202,516 | 0 | 0 | 16,465 | 218,981 |
Adalimumab | 41,939 | 194,028 | 0 | 0 | 17,455 | 253,422 |
Etanercept | 43,339 | 195,072 | 0 | 0 | 15,925 | 256,422 |
Upadacitinib | 59,158 | 193,480 | 0 | 0 | 17,585 | 270,223 |
Secukinumab | 61,396 | 192,860 | 0 | 0 | 17,329 | 271,584 |
Golimumab | 64,560 | 194,392 | 0 | 0 | 15,925 | 274,877 |
Ustekinumab | 62,809 | 195,209 | 0 | 0 | 18,098 | 276,115 |
Guselkumab | 68,944 | 192,876 | 0 | 0 | 16,127 | 277,946 |
Infliximab | 58,132 | 191,809 | 10,188 | 0 | 20,740 | 280,869 |
Ixekizumab | 80,847 | 191,866 | 0 | 0 | 19,529 | 292,242 |
Biologic-Experienced Population | ||||||
BSC | 0 | 194,128 | 0 | 0 | 15,885 | 210,013 |
Upadacitinib | 42,197 | 187,561 | 0 | 0 | 19,709 | 249,466 |
Secukinumab | 48,451 | 186,383 | 0 | 0 | 15,706 | 250,541 |
Ustekinumab | 49,161 | 187,084 | 0 | 0 | 16,828 | 253,073 |
Guselkumab | 50,675 | 187,126 | 0 | 0 | 16,220 | 254,021 |
Ixekizumab | 67,340 | 185,613 | 0 | 0 | 16,652 | 269,605 |
BSC = best supportive care.
Table 20: Disaggregated QALYs for CADTH’s Probabilistic Base-Case Analyses
Drug | First line | Final line (BSC) | AE disutilities | Total QALYs |
|---|---|---|---|---|
Biologic-naive population | ||||
BSC | 0 | 6.211 | −0.012 | 6.199 |
Ustekinumab | 2.023 | 5.060 | −0.013 | 7.070 |
Guselkumab | 2.321 | 5.007 | -0.011 | 7.317 |
Adalimumab | 2.367 | 4.992 | −0.012 | 7.346 |
Secukinumab | 2.430 | 4.979 | −0.012 | 7.396 |
Upadacitinib | 2.431 | 5.004 | −0.012 | 7.422 |
Golimumab | 2.485 | 4.989 | −0.011 | 7.462 |
Ixekizumab | 2.565 | 4.973 | −0.014 | 7.525 |
Etanercept | 2.601 | 4.988 | −0.013 | 7.576 |
Infliximab | 2.758 | 4.964 | −0.015 | 7.707 |
Biologic-Experienced Population | ||||
BSC | 0 | 5.164 | −0.011 | 5.153 |
Ustekinumab | 1.383 | 4.501 | −0.012 | 5.872 |
Guselkumab | 1.515 | 4.482 | -0.011 | 5.985 |
Upadacitinib | 1.524 | 4.480 | −0.014 | 5.990 |
Secukinumab | 1.710 | 4.444 | −0.011 | 6.142 |
Ixekizumab | 1.943 | 4.412 | −0.012 | 6.344 |
AE = adverse event; BSC = best supportive care; QALY = quality-adjusted life-years.
Scenario Analyses
Table 21: CADTH Scenario Analyses
Detail | CADTH base case | CADTH scenario |
|---|---|---|
Scenario analyses | ||
A. Disease-related cost differences halved | Annual HAQ-DI costs: $3,085 + ($934 x HAQ-DI score) Annual uncontrolled PsO costs: Mild to moderate PsO = $1,907, moderate to severe = $5,434 | Annual HAQ-DI costs: $3,085 + ($467 x HAQ-DI score) Annual uncontrolled PsO costs: Mild to moderate PsO = $1,907, moderate to severe = $3,670 |
B. SMR | 1.058 | 1.3432 |
HAQ-DI = Health Assessment Questionnaire Disability Index; PsO = psoriasis; SMR = standardized mortality ratio.
Table 22: Summary of CADTH Scenario Analyses
Stepped analysis | Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALYs) |
|---|---|---|---|---|
Biologic-naive population | ||||
CADTH base case | BSC | 218,981 | 6.199 | Reference |
Etanercept | 256,957 | 7.576 | 27,584 vs. BSC | |
Infliximab | 280,869 | 7.707 | 182,188 vs. etanercept | |
Guselkumab | 277,946 | 7.317 | Dominated by adalimumab, etanercept, upadacitinib, secukinumab, golimumab | |
CADTH Scenario A: disease-related cost differences halveda | BSC | 178,648 | 6.163 | Reference |
Etanercept | 218,805 | 7.531 | 29,369 vs. BSC | |
Infliximab | 244,000 | 7.668 | 183,398 vs. etanercept | |
Guselkumab | 240,275 | 7.280 | Dominated by adalimumab, etanercept, upadacitinib, secukinumab, golimumab | |
CADTH Scenario B: SMR 1.34 | BSC | 206,149 | 6.143 | Reference |
Etanercept | 243,513 | 7.468 | 28,196 vs. BSC | |
Infliximab | 268,109 | 7.624 | 157,232 vs. etanercept | |
Guselkumab | 264,358 | 7.222 | Dominated by adalimumab, etanercept, upadacitinib, secukinumab, golimumab | |
Biologic-experience population | ||||
CADTH base case | BSC | 210,013 | 5.123 | Reference |
Secukinumab | 250,541 | 6.142 | 40,942 vs. BSC | |
Ixekizumab | 269,605 | 6.344 | 94,793 vs. secukinumab | |
Guselkumab | 254,021 | 5.985 | Dominated by upadacitinib, secukinumab | |
CADTH Scenario A: disease-related cost differences halveda | BSC | 170,697 | 5.149 | Reference |
Secukinumab | 213,482 | 6.138 | 43,283 vs. BSC | |
Ixekizumab | 232,916 | 6.340 | 96,300 vs. secukinumab | |
Guselkumab | 216,861 | 5.983 | Dominated by upadacitinib, secukinumab | |
CADTH Scenario B: SMR 1.34 | BSC | 200,703 | 5.100 | Reference |
Secukinumab | 241,064 | 6.072 | 41,544 vs. BSC | |
Ixekizumab | 260,072 | 6.266 | 97,797 vs. secukinumab | |
Guselkumab | 244,487 | 5.912 | Dominated by secukinumab | |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; Ref. = reference; SMR = standardized mortality ratio; vs. = versus.
aScenario A halves the coefficient associated with HAQ-DI score as well as the difference in cost between mild to moderate psoriasis and moderate to severe psoriasis.
Table 23: CADTH Deterministic Price Reduction Analyses — Biologic-Naive Population
Analysis Price reduction | ICERs for guselkumab vs. comparators | |
|---|---|---|
Sponsor base case | CADTH reanalysis | |
Guselkumab at submitted price | λ < $25,178: BSC $25,178 < λ < $57,001: Etanercept λ ≥ $57,001: Guselkumab | λ < $25,178: BSC $25,178 < λ < $199,813: Etanercept λ ≥ $199,813: Infliximab |
6% reduction | λ < $25,178: BSC $25,178 < λ < $49,838: Etanercept λ ≥ $49,838: Guselkumab | |
10% reduction | ||
20% reduction | ||
30% reduction | λ < $23,660: BSC λ ≥ $23,660: Guselkumab | |
40% reduction | ||
42% reduction | λ < $24,619: BSC $24,619 < λ < $27,596: Guselkumab $27,596 < λ < $199,813: Etanercept λ ≥ $199,813: Infliximab | |
50% reduction | ||
60% reduction | ||
70% reduction | ||
80% reduction | ||
83% reduction | Guselkumab dominant over all comparators including BSC | |
85% reduction | λ < $134,192: Guselkumab $134,192 < λ $199,813: Etanercept λ ≥ $199,813: Infliximab | |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; vs. = versus.
Price reduction scenarios are based on deterministic analyses.
Table 24: CADTH Deterministic Price Reduction Analyses — Biologic-Experienced Population
Analysis Price reduction | ICERs for guselkumab vs. comparators | |
|---|---|---|
Sponsor base case | CADTH reanalysis | |
Guselkumab at submitted price | λ < $34,103: BSC $34,103 < λ < $71,141: Secukinumab λ ≥ $71,141: Guselkumab | λ < $37,225: BSC $37,225 < λ < 93,995: Secukinumab λ ≥ $93,995 Ixekizumab |
8% reduction | λ < $34,103: BSC $34,103 < λ < $47,890: Secukinumab λ ≥ $47,890: Guselkumab | |
10% reduction | ||
13% reduction | λ < $33,981: BSC λ ≥ $33,981: Guselkumab | |
20% reduction | λ < $37,104: BSC $37,104 < λ < $37,879: Guselkumab $37,879 < λ < $93,995: Secukinumab λ ≥ $93,995: Ixekizumab | |
30% reduction | ||
40% reduction | λ < $25,886: BSC $25,886 < λ < $96,048: Guselkumab λ ≥ $96,048: Ixekizumab | |
50% reduction | ||
60% reduction | ||
70% reduction | ||
80% reduction | ||
85% reduction | Guselkumab dominant over all comparators including BSC | |
86% reduction | λ < $160,652: Guselkumab λ ≥ $160,652: Ixekizumab | |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; vs. = versus.
Price reduction scenarios are based on deterministic analyses.
Appendix 5: Submitted Budget Impact Analysis and CADTH Appraisal
Note this appendix has not been copy-edited.
Table 25: Summary of Key Take-aways
Key take-aways of the budget impact analysis |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
In the submitted budget impact analysis (BIA), the sponsor assessed the introduction of guselkumab for patients with active PsA who require a biologic or advanced therapy. The BIA was undertaken from the perspective of a Canadian public payer over a 3-year time horizon (2023 to 2025) using an epidemiological approach. The sponsor’s analysis included drug-acquisition costs; dispensing fees and markups were not included in the base case. Data from the model were obtained from various sources including Statistics Canada,39,40 the published literature,41,42 Ontario Drug Benefit (ODB) formulary list prices,12,14 Saskatchewan formulary list prices,13 RAMQ formulary list prices,15 and the sponsor’s internal data.43 Key inputs to the BIA are documented in Table 26. Key assumptions included:
Growth in the population of patients with PsA in addition to overall population growth.
Patients will only receive guselkumab if they would have otherwise received an alternate bDMARD. The sponsor does not consider differences in potential populations reimbursed.
Guselkumab will take 31% of its share from adalimumab (Humira), 5% from etanercept (Embrel), 15% from etanercept (biosimilar), 17% from secukinumab (Cosentyx), 16% from ixekizumab (Taltz), 3% from ustekinumab (Stelara), and 13% from apremilast (Otezla).
All patients in year 1 are in the induction year of their current therapy.
Table 26: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) | |
|---|---|---|
Target Population | ||
Population of Canada in base year, excluding Quebec and the territories | 24,255,72139 | |
Annual population growth rate | 1.41%40 | |
Prevalence of psoriatic PsA | 0.15%41 | |
Annual PsA growth rate | 7.20%41 | |
Percentage of patients with PsA requiring biologic/advanced therapy | 62.00%42 | |
Proportion of patients with PsA on biologic/advanced therapy accessing through a public plan | 52.31%a | |
Eligible patients | 12,742 / 13,852 / 15,059 | |
Market Uptake | Reference scenariob | New drug scenariob |
Guselkumab (Tremfya) | 0% / 0% / 0% | 1.6% / 2.0% / 2.9% |
Etanercept (Enbrel) | 9.1% / 8.6% / 8.2% | 9.1% / 8.5% / 8.1% |
Etanercept biosimilar | 4.0% / 4.0% / 4.2% | 3.8% / 3.8% / 3.8% |
Infliximab (Remicade) | 4.5% / 4.5% / 4.5% | 4.5% / 4.5% / 4.5% |
Infliximab biosimilar | 2.3% / 2.3% / 2.3% | 2.3% / 2.3% / 2.3% |
Certolizumab pegol (Cimzia) | 5.3% / 4.8% / 4.3% | 5.3% / 4.8% / 4.3% |
Secukinumab (Cosentyx) | 16.1% / 15.8% / 15.6% | 15.8% / 15.5% / 15.1% |
Adalimumab (Humira) | 23.1% / 22.8% / 22.7% | 22.6% / 22.2% / 21.8% |
Adalimumab biosimilar | 3.0% / 3.1% / 3.1% | 3.0% / 3.1% / 3.1% |
Ustekinumab (Stelara) | 3.1% / 3.1% / 3.1% | 3.0% / 3.0% / 3.0% |
Ixekizumab (Taltz) | 5.5% / 5.1% / 4.9% | 5.3% / 4.8% / 4.4% |
Golimumab (Simponi) | 19.9% / 19.3% / 18.7% | 19.9% / 19.3% / 18.7% |
Apremilast (Otezla) | 2.5% / 2.5% / 2.6% | 2.3% / 2.3% / 2.3% |
Upadacitinib (Rinvoq) | 1.6% / 4.0% / 5.8% | 1.6% / 4.0% / 5.8% |
Cost of treatment per patient (induction year / maintenance year) | ||
Guselkumab (Tremfya) | $21,418 / $19,888 | |
Etanercept (Enbrel) | $21,105 | |
Etanercept biosimilar | $12,532 | |
Infliximab (Remicade) | $39,080 / $31,753 | |
Infliximab biosimilar | $19,720 / $16,023 | |
Certolizumab pegol (Cimzia) | $19,271 / $17,277 | |
Secukinumab (Cosentyx) | $20,286 / $16,173 | |
Adalimumab (Humira) | $20,422 | |
Adalimumab biosimilar | $12,253 | |
Ustekinumab (Stelara) | $22,966 / $19,904 | |
Ixekizumab (Taltz) | $25,028 / $21,716 | |
Golimumab (Simponi) | $18,672 / $18,608 | |
Apremilast (Otezla) | $13,743 / $13,762 | |
Upadacitinib (Rinvoq) | $17,720 | |
PsA = psoriatic arthritis.
aSponsor’s internal market research, data not provided.43
bCited as based on the upadacitinib submission to CADTH, with years 2 and 3 reduced by half as guselkumab would not benefit from the patient preference for new oral therapies which affected upadacitinib uptake projections.43
Summary of the Sponsor’s BIA Results
Results of the sponsor’s analysis suggest that the reimbursement of guselkumab for adults with PsA will be associated with an incremental cost of $444,327 in year 1, $587,636 in year 2, and $927,602 in year 3, for a 3-year total incremental budgetary impact of $1,959,564.
In a scenario where all adalimumab, etanercept, and infliximab use is assumed to be biosimilars, the reimbursement of guselkumab would be associated with an incremental cost of $1,040,621 in year 1, $1,413,112 in year 2, and $2,228,814 in year 3, for a 3-year total incremental budget impact of $4,682,546.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Analysis assumes guselkumab will not grow the biologic market: The sponsor’s analysis assumes that patients will only receive guselkumab if they would otherwise have received a different biologic therapy. The sponsor is therefore implicitly assuming that guselkumab will not be reimbursed for its full indication of adults with active PsA, but instead will be reimbursed in a manner similar to other biologic drugs currently reimbursed by public drug plans. Should guselkumab instead be reimbursed under criteria which are less strict than existing biologic therapies, the budget impact of such reimbursement would be substantially larger than estimated.
CADTH was unable to estimate the budget impact of reimbursing guselkumab for its full indication due to a lack of data on how much the market would expand and hard-coding of market shares within the model making adjustments impractical. Under such an assumption, the budget impact of reimbursing guselkumab would be substantially larger than that estimated by either the sponsor’s or CADTH’s base case.
Proportion of patients using biologics is uncertain: The sponsor estimated that the proportion of patients with PsA who have active disease and would use a biologic therapy was 62%, based on 2012/2013 data from a retrospective cohort study of US registry data.42 It is unlikely that overall US biologic use can be generalized to current Canadian practice, nor to the proportion of publicly reimbursed patients who will receive a biologic within the next 3 years. The expert consulted by CADTH estimated that biologic use by publicly reimbursed patients with PsA is likely lower than estimated by the sponsor due to reimbursement criteria implemented by public plans but was unable to provide a precise estimate.
CADTH could not adjust for this limitation in its base-case analysis. A scenario analysis was conducted exploring the impact of assuming only 50% of publicly reimbursed patients with PsA in Canada are using a biologic.
Market uptake and displacement uncertain: The sponsor based the proportion of the market guselkumab would take as well as the proportions of other comparators displaced on those submitted to and estimated by CADTH for the review of upadacitinib for PsA, with uptake in years 2 and 3 halved from that of upadacitinib, as the latter drug was expected to benefit from a patient preference for oral therapies which would not apply to guselkumab. The clinical expert consulted by CADTH considered the uptake estimates to be reasonable, but noted that the perceived benefit of guselkumab for psoriasis symptoms and the lack of association with irritable bowel disease symptoms that may be experienced with some comparators (e.g., secukinumab, ixekizumab), may lead to increased uptake of guselkumab relative to other biologic options. However, the sponsor’s model as programmed was not capable of estimating increased market share for guselkumab while also assuming 100% biosimilar use.
CADTH was unable to formally explore different guselkumab uptake scenarios around its base case due to limitations in the way the sponsor’s model was programmed. However, if uptake for guselkumab is assumed to be double that predicted, the estimated budget impact of reimbursing guselkumab would also be doubled.
Biosimilar use is underestimated: The sponsor’s base case assumes that the majority of patients on adalimumab, etanercept, and infliximab are using the originator products, citing internally conducted market forecasts. However, public drug plans have instituted or are planning to institute policies limiting reimbursement of biologic products to less expensive SEBs where available. As such, it is expected that publicly funded SEB use will increase and therefore become the most relevant comparator for new entries into affected therapeutic spaces. Additionally, the CUA submitted by the sponsor assumes 100% biosimilar use for adalimumab, etanercept, and infliximab, therefore the BIA should also do so to ensure consistency between the analyses. Finally, the patents for ustekinumab and certolizumab recently expired, making it possible for biosimilar versions to become available within the 3-year time frame of the BIA. However, assuming the sponsor’s estimates of the market share and expected displacement of ustekinumab and certolizumab, this is not expected to have a large effect on the projected budget impact of guselkumab.
CADTH reanalyses assumed that all use of adalimumab, etanercept, and infliximab is SEB products.
The adult non-insured health benefits (NIHB) population was inaccurately estimated: The sponsor’s BIA for the NIHB population did not limit the population to clients older than 18 years of age, and therefore did not reflect the Health Canada indication for guselkumab which is limited to adults with active PsA. Additionally, adults residing within the borders of Ontario who are older than 65 years of age are reimbursed under the ODB program first, rather than NIHB, and therefore NIHB clients in Ontario who are older than 65 should be considered part of the Ontario population for the purpose of the BIA. Finally, the sponsor’s pan-Canadian analysis considers the full population of Canada and subtracts the populations of Quebec and the territories, however this fails to account for the NIHB population residing within Quebec and the territories who, as clients of a CDR-participating plan, should be counted within the pan-Canadian population.
CADTH considered only the adult NIHB population in reanalyses and moved patients older than 65 years of age who reside within the borders of Ontario from the NIHB population to the ODB population. The pan-Canadian population was calculated by adding the eligible non-NIHB adult provincial populations39 except Quebec to the adult NIHB population44 rather than subtracting the populations of Quebec and the territories from the total population of Canada.
Induction year assumption is inappropriate: The sponsor’s analysis assumes that every patient initiates treatment with their assigned biologic in year 1, continuing into the maintenance phase of treatment in years 2 and 3. However, as the model uses prevalence data to calculate population size, it should be assumed that patients with PsA existing in the base year would already be using a biologic, and only those who initiated biologic therapy (i.e., new patients) or those who switched therapies would be in an induction phase in any year of the model. The CADTH CUA reanalyses assume that 16.5% of patients will discontinue their biologic therapy per year, therefore it follows that 16.5% of patients within the BIA who were on biologic therapy the previous year would be in an induction year, while 100% of new patients would be. As guselkumab is not available in the base year of the analysis, all guselkumab patients in year 1 would be in an induction year.
CADTH reanalyses assumed that 100% of incident patients (patients not in the model the previous year) would be in an induction year, regardless of biologic therapy, while 16.5% of patients continuing from a previous year (prevalent patients) would be in an induction year to represent those in the process of switching therapies. 100% of guselkumab patents in year 1 were assumed to be in an induction year, dropping to 16.5% of pre-existing patients in years 2 and 3.
CADTH Reanalyses of the BIA
CADTH revised the sponsor’s submitted analysis by assuming 100% SEB use for products where they were available, correcting the NIHB population estimates, and assuming that 100% of incident (new) patients and 16.5% of prevalent patients (pre-existing) were in an induction phase of their biologic during each year of the model. The changes made to derive the CADTH base case are described in Table 27.
Table 27: CADTH Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Changes to derive the CADTH base case | ||
1. 100% SEB use (reference Y1/Y2/Y3)a | Market shares as in Table 26. | Adalimumab SEB: 26.1% / 25.9% / 25.8% Etanercept SEB: 13.1% / 12.7% / 12.4% Infliximab SEB: 6.8% / 6.8% / 6.7% No Humira, Enbrel, or Remicade share. |
2. NIHB population correction, 2021 numbers | NIHB population: 873,312 ODB population: 12,076,175 Pan-Canadian population:b 23,918,471 | NIHB population: 631,801 ODB population: 11,917,765 Pan-Canadian population:b 24,008,615 |
3. Induction year assumptions | All patients assumed to be in an induction year in year 1, 100% of new patients are in an induction year in years 2 and 3, 0% of returning patients are in an induction year in years 2 and 3 | 16.5% of pre-existing patients are assumed to be in an induction year in year 1 for non-guselkumab patients, 100% of guselkumab patients are in an induction year in year 1. In years 2 and 3, 16.5% of returning patients are assumed to be in an induction year for all comparators, and 100% of new patients. |
CADTH base case | Reanalysis 1 + 2 + 3 | |
NIHB = non-insured health benefits; ODB = Ontario Drug Benefit; SEB = subsequent entry biologic.
aEquivalent to the sponsor’s 100% SEB scenario.
bPan-Canadian population includes populations eligible under CDR-participating public plans. The sponsor’s estimate subtracts the populations of Quebec and the territories from the total Canadian population, while the CADTH estimate adds the 10 provincial populations, excluding Quebec and NIHB-eligible clients within the provinces, to the total NIHB population, including NIHB clients in Quebec and the 3 territories.
The results of the CADTH step-wise reanalysis are presented in summary format in Table 28 and a more detailed breakdown is presented in Table 29. CADTH reanalyses suggest that the reimbursement of guselkumab for adult patients with active PsA who would otherwise have received a different biologic will be associated with at 3-year incremental budgetary cost of $4,711,697.
Table 28: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | 3-year total |
|---|---|
Submitted base case | $1,959,564 |
CADTH reanalysis 1: 100% SEB use | $4,682,546 |
CADTH reanalysis 2: NIHB population correction | $1,966,950 |
CADTH reanalysis 3: Induction year assumptions | $2,019,031 |
CADTH base case | $4,711,697 |
BIA = budget impact analysis; NIHB = Non-Insured Health Benefits; SEB = subsequent entry biologic.
CADTH conducted additional scenario analyses (Table 29) to highlight the uncertainty associated with the potential budget impact. Scenarios were conducted assuming: a lower proportion of patients with PsA are assumed to require a biologic/advanced therapy, a price reduction of 20% consistent with that estimated by the CADTH CUA reanalysis for the biologic-naive population, and a 42% price reduction consistent with that estimated by the CADTH CUA reanalysis for the biologic-experienced population. While CADTH was unable to formally explore a scenario with increased guselkumab uptake due to limitations in the programming of the sponsor’s model, if guselkumab uptake was assumed to be double that predicted, the resulting budgetary impact would also be doubled.
All analyses assume that guselkumab will be used only in patients who would otherwise receive an alternate biologic. Should guselkumab be reimbursed with broader reimbursement criteria than that of other reimbursed biologics, the associated budget impact would be substantially higher.
Table 29: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | 3-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | NR | $260,952,294 | $263,882,559 | $286,552,501 | $811,387,355 |
New drug | NR | $260,507,968 | $263,294,923 | $285,624,899 | $809,427,790 | |
Budget impact | $0 | $444,327 | $587,636 | $927,602 | $1,959,564 | |
CADTH base case | Reference | NR | $203,394,999 | $220,794,481 | $240,082,626 | $664,272,107 |
New drug | NR | $204,439,542 | $222,217,468 | $242,326,794 | $668,983,804 | |
Budget impact | $0 | $1,044,542 | $1,422,986 | $2,244,168 | $4,711,697 | |
CADTH scenario A: 50% of patients with PsA require a biologica | Reference | NR | $164,028,225 | $178,039,368 | $193,596,858 | $535,664,451 |
New drug | NR | $164,870,598 | $179,175,819 | $195,393,964 | $539,440,381 | |
Budget impact | $0 | $842,373 | $1,136,451 | $1,797,106 | $3,775,930 | |
CADTH scenario analysis: 20% price reduction | Reference | NR | $203,394,999 | $220,768,816 | $240,060,104 | $664,223,920 |
New drug | NR | $203,579,023 | $221,038,086 | $240,481,772 | $665,098,880 | |
Budget impact | $0 | $184,024 | $269,270 | $421,667 | $874,960 | |
CADTH scenario: 42% price reduction | Reference | NR | $203,394,999 | $220,768,816 | $240,060,104 | $664,223,920 |
New drug | NR | $202,632,452 | $219,784,163 | $238,494,354 | $660,910,968 | |
Budget impact | $0 | -$762,547 | -$984,653 | -$1,565,751 | -$3,312,951 |
BIA = budget impact analysis; PsA = psoriatic arthritis.
aas opposed to 62.5% assumed by the sponsor
Stakeholder Input
Patient Input
Canadian Association of Psoriasis Patients and the Canadian Psoriasis Network
About the Canadian Association of Psoriasis Patients (CAPP) and the Canadian Psoriasis Network (CPN)
The Canadian Association of Psoriasis Patients (CAPP) is partnering with the Canadian Psoriasis Network (CPN) to develop this submission.
CAPP (www.canadianpsoriasis.ca) is a national, not-for-profit organization formed to better serve the needs of psoriasis patients across the country. CAPP’s mission is to be a resource and advocate for psoriatic patients and their families to improve patient care and quality of life.
CPN (www.canadianpsoriasisnetwork.com) is a national, not-for-profit organization dedicated to improving the quality of life of people in Canada who live with psoriasis and psoriatic arthritis. CPN does this by providing current information on research and treatment options and by working with others to build awareness and advocacy about the complexities of these conditions.
Information Gathering
CAPP and CPN collaborated with the Arthritis Society (arthritis.ca), the Canadian Arthritis Patient Alliance (arthritispatient.ca), and Creaky Joints Canada (creakyjoints.ca) to develop a survey in English using Survey Monkey that asked patients and caregivers about their experiences with psoriatic arthritis (PsA) and the treatment under review (guselkumab or Tremfya). Each organization participated in the development of the survey and shared the surveys with their respective memberships or patient communities via their communication channels (e.g., websites, social media channels, newsletters, etc.). CAPP and CPN analyzed the data and prepared this submission.
The survey was available from April 20, 2022 to May 16, 2022. Overall, there were 71 responses to the survey: 12 respondents were from British Columbia (17.1%), 7 from Alberta (10.0%), 3 from Manitoba (4.3%), 34 from Ontario (48.6%), 4 from Quebec (5.7%), 4 from New Brunswick (5.7%), 4 from Nova Scotia (5.7%), 3 from Newfoundland & Labrador (4.3%); no responses were received from Saskatchewan, Prince Edward Island, Yukon, the Northwest Territories or Nunavut.
In addition to PsA, 68.0% of 25 respondents (n=17) indicated they lived with psoriasis, 24.0% (n=6) live with another inflammatory condition, 48.0% (n=12) also live with another type of arthritis, and 32.0% (n=8) live with at least one other condition, including osteoporosis, hypertension, metabolic syndrome, fibromyalgia, type 1 diabetes, herniated discs, rosacea, kidney problems, inflammatory bowel disease and Celiac disease.
Twenty-nine respondents provided information about their gender: 79.3% (n=23) were female, 20.7% (n=6) were male. As well, 29 respondents provided information about their age, which is grouped here for ease of reference: under 26 years old (n=0), 26-34 years old (n=0), 35-44 years old (n=1), 45-54 years old (n=5), 55-64 years old (n=12), 65-74 years old (n=8), and 75 years or older (n=3). When asked how else they might describe their experience, 6 respondents (20.7%) identified as people with a disability, and 67.0% of respondents identified as White.
Household income levels of the 29 respondents who provided information represented a wide range.
Of 28 respondents, 57.1% (n=16) had private insurance, 21.4% (n=6) had coverage through a public plan, not including unemployment or disability supports, and 7.1% (n=2) had coverage through a disability support program. One respondent noted that they had reached the lifetime maximum on their private plan and were now covered through Nova Scotia’s provincial coverage.
There were two responses from people who have experience with Tremfya (guselkumab) for PsA.
Disease Experience
Psoriatic arthritis (PsA) is a form of arthritis linked to psoriasis and that is chronic and progressive. This inflammatory disease causes swelling and pain in multiple joints and can sometimes result in permanent and debilitating joint damage.
Fifty respondents identified aspects of their day-to-day lives and quality of life that are affected by PsA:
Table 1: Aspects of Day-to-Day Lives and Quality of Life Affected by PsA
Aspect | Percentage | Number of respondents | Aspect | Percentage | Number of respondents |
|---|---|---|---|---|---|
Ability to work | 16.0% | 8 | Participation in school | 2.0 | 1 |
Social connections | 52.5 | 26 | Friendships | 24.0 | 12 |
Parenting | 8.0% | 4 | Family life | 38.0% | 19 |
Intimacy | 50.0% | 25 | Self-esteem | 50.0% | 25 |
Mental health | 50.0% | 25 | None of the above | 16.0% | 8 |
PsA can curtail or diminish participation in the workforce and work opportunities. Among those who miss work or school because of symptoms or side effects of PsA, 28.3% (n=13 out of 46 respondents) miss 1-3 days on average each month, 17.4% (n=8) miss 4-6 days, 2.2% (n=1) miss 7-10 days, 2.2% (n=1) miss 16-20 days and 6.5% (n=3) cannot work or attend school due to PsA. Several respondents shared how their work lives had changed because of their PsA:
“no longer able to work full time…just part time due to fatigue, lack of grip strength in my hands, etc.”
“I take things day by day. I miss out on lots of things due to pain and fatigue. I work shift work usually 4 hrs per shift any more, even an hour, keeps me awake at night with agonizing foot and toe pain especially.”
“I push through but exhausted by the end of the working day.”
“No longer able to work full time as a Nurse due to my PsA.”
“I always worked FTE & now down to 3d/wk as a nurse practitioner, I have daily fatigue, painful sore joints, my fingers with tensosynovitis is now causing my keyboard work to become more difficult with lagging fingers - extra letters etc. My fatigue is exhausting & barely make it though a full day of work & need to sleep 11-12h /d for recovery. I awaken multiple times a night with pain or numbness to digits causing difficulty falling back to sleep. Losing contact with friends as cannot do evening out. Most frustration is being told by rheumatologist that my condition falls through the cracks in terms of financial support OHIP for TNFi which I need to stop the progression of my enthesitis. I am currently seeking a second opinion in [a large city].”
“Cannot do much when hands and feet are flaring”
“I only work online with a Modified work schedule. I’m not able to do housework, exercise, socialize or travel like I used to.”
For many respondents, their PsA reduced their mobility and ability to participate in activities and impacted their mental and social health:
“Can’t walk for long periods”
“Had to stop running/walking because I need to stop or reduce the diclofenac. Gained weight. Harder on the joints. A longer time to work out the stiffness in the morning. Aches in the evening. Lethargy.”
“I live very independently. The only difficulty I have is I get tired more often and very occasionally have issues with my knees and feet/ankles. Pain and swelling. Stairs can sometimes present an issue if it's multiple stories. This happens after prolonged walking, standing or excessive exercise.”
“Walking any distance. Lifting grandchildren. Anything that requires hand strength.”
“PsA causes significant pain, fatigue, and the dexterity I once had in my hands is severely deteriorated.”
“Joint stiffness impedes activities and being tired most of the time makes one lose desire to participate in activities.”
“Yes, it has affected what activities That I can still do. For example because of my range of motion in my knee I can not play hockey any More[. N] o support needed.”
“No motivation, mental health.”
When it comes to managing and completing everyday tasks, many respondents told us that the pain and joint symptoms have reduced their ability to complete activities of daily living on their own:
“It has in the past – my hands are very week with two joint replacements and a bit of pain - needing someone to help dress, cook, carry things, everyday living tasks.”
“Challenges of doing everyday tasks like housekeeping, shopping and travelling. Had to sell my home and move to apartment sooner than necessary for my friends of similar age group. Challenges with sleep and being able to visit certain areas or homes. Always balancing every day to stay healthy and active. Travel is limited.”
Several respondents receive support from their spouse, family and friends.
“Due to me being a Nurse, I am able to pace myself to complete my ADLs independently. I also have lots of support from my husband.”
“None at the moment per se but spouse drives me to grocery store and helps pack bags, does washing of dishes after I cook and bake. I do not qualify for other supports from my community at the moment.”
“Husband has taken on many chores and he and my family all lend emotional support. I am very lucky.”
“Assistance with making meals in particular because opening jars, peeling vegetables very difficult.”
“Spouse helps with errands, finances, cooking, cleaning. Sister and father understand and provide emotional support. Friends do not care.”
Others pay for services around the home, including housework and yard work: “I hire a cleaning lady twice a month.”
“I have a cleaning lady and several very good friends who are on call to help with lifting groceries, retrieving heavy items at low or high heights, packing for a trip, being on watch when with them so as not to fall. Assist with meal prep and clean up. My family are not here.”
Experiences With Currently Available Treatments
Respondents were asked about their experiences with different PsA symptoms, and how effective they were at treating their PsA symptoms:
Table 2: Experiences with Different PsA Treatments
PsA treatment | Very ineffective | Mildly ineffective | No difference | Mildly effective | Very effective | N/A |
|---|---|---|---|---|---|---|
Non-steroidal anti- inflammatory drugs (NSAIDs) | 22.2% n=10 | 8.9% N=4 | 4.4% N=2 | 37.8% N=17 | 11.1% N=5 | 15.6% N=7 |
Disease-modifying antirheumatic drugs (DMARDs) | 16.7% N=7 | 4.8% N=2 | 7.1% N=3 | 21.4% N=9 | 21.4% N=9 | 23.1% N=9 |
Leflunomide | 11.7% N=4 | 2.9% N=1 | 5.9% N=2 | 5.9% N=2 | 2.9% N=1 | 70.6% N=24 |
Apremilast | 10.0% N=3 | 0 | 0 | 3.3% N=1 | 6.7% N=2 | 80.0% N=24 |
Tofacitinib | 0 | 0 | 0 | 0 | 0 | 100% |
Hydroxychloroquine | 10.0% N=3 | 3.3% N=1 | 6.7% N=2 | 6.7% N=2 | 6.7% N=2 | 66.7% N=20 |
Biologics | 5.9% N=2 | 2.9% N=1 | 0 | 17.7% N=6 | 32.4% N=11 | 41.2% N=14 |
Steroid injections | 16.7% N=6 | 5.6% N=2 | 2.8% N=1 | 13.9% N=5 | 22.2% N=8 | 38.9% N=14 |
Oral steroids | 3.3% N=1 | 0 | 0 | 6.7% N=2 | 23.3% N=7 | 66.7% N=20 |
Medical cannabis | 11.8% N=4 | 0 | 5.9% N=2 | 17.7% N=6 | 8.8% N=2 | 57.6% N=19 |
The survey asked how well their PsA was managed by treatments they had previously tried. Some respondents told us that the drugs they were currently taking were managing their symptoms well. Others shared their experiences with the side effects (that made it difficult to continue with those drugs), and their ongoing unmet needs with current treatments.
“Mostly side effects stopped me from using certain medicines.”
“Side effects have been worse to manage than disease symptoms in some cases. None of my need (pain control, swelling, quality of life) have been managed with treatment to date.”
“Feeling sick to my stomach tired constant pain.”
“Took naproxen for 10 years but no longer take it as it ruined my stomach.”
“Stomach issues from ibuprofen, PsA effects my eyes and the drops I use can exacerbate my symptoms if used too much.”
“All Dmards made me 'sick', severe drug hangover, tingling in arms, nauseated. Celebrex caused GERD. Biologic is the best, but after 3 years, it's not working as well now.”
“Over the counter Tylenol arthritis - Not well managed; Diclofenac (daily)- very well managed steroidal meds didn do anything. Biweekly injections [golimumab] do a relatively good job when combined with others. Trying to get off the diclofenac because it’s so hard on my stomach. But methotrexate didn’t help. So I’m trying a combo of other items. But the diclofenac does it best for me. When taking 2 per day, I was able to run 5-10 Km per day. Without it, I ache. Daily.”
“Hydrochloroquine caused ear pustules & ++ ichiness NSAIDs causes GERD & IBS I have enthesitis as predominant feature with tenosynovitis to fingers & nail psoriasis”
“Methotrexate controls the disease well but I spend one day a week recovering from the injections. Mostly on the couch.”
“My rheumatologist won’t even talk about anything else and doesn’t seem to grasp that living only 6 days a week is too much side effects.”
“Have to plan ahead to make sure I’m not scheduled for anything on recovery day. I have to rest every afternoon or face the consequences.”
“Side effects of Cosentyx are hard to tolerate and since monthly injection at max amount it is only effective for 10 days after injections, then back in flare.”
“Enbrel has worked wonders for me, sore to inject but worth the relief of pain. All other medication tried did not work and lots of side effects.”
“Side effects (Liver and skin) are monitored with blood work, taking other meds to counter the adverse effects ..have a strong health team including a Doctor of Chinese medicine and acupuncture.”
Risks of malignancies are elevated for people with psoriatic disease:
“I have tried 3 different biological injections. My present is Cymzia and is working well. I have concerns tho regarding possible side effects ie cancers”
One responded who was undergoing treatment for cancer told us about their experiences:
“My PsA was doing well on Cosentyx until I was diagnosed with Ovarian Cancer then had to stop Biologics, & Methotrexate, now back on Methotrexate.”
“Side effects steroids weight gain and cancer. Side effects methotrexate weak and sick. Nothing has helped much in 45 years. No quality of life.”
“Chemo finishes the end of May 2022 so treatment is less now and will only go to Cancer Clinic once a month for blood work and IV Bone treatment; kind of a wait and see for at least three months; no consideration of my other autoimmune issues or support offered; still work with kidney specialist but now on a three month basis with blood and urine requisition and a consult by telephone or in-office. So my biggest concerns are kidney support, eye support & possible cataract eye surgery; concern about becoming a diabetic due to medications taken; working on a plan of care as I move forward.”
Despite the availability of several treatment options, many of which work well for people, there remain significant unmet needs for people living today with PsA:
“Overall health mental and physical takes a back seat to just joint issues.”
“My inflammation markers continue to increase; my blood sugars and blood pressure are rising, I have eye issues now, cataracts, due to chemo medications and other autoimmune related symptoms. My body is very sensitive to change, certain food sensitivities like histamines and oxalates. stress, sleep issues, share symptoms of lupus and chronic inflammatory response syndrome; I have various skin rashes and family history of various autoimmune conditions. My MDs work in silos and do NOT consult each other & I have to do my own medical research to figure out my issues and possible solutions.”
“I still have pain in some joints all the time when I exercise.”
“Not met, fatigue, swelling in hand joints and feet; poor sleep. No side effects from Apremelist (otezla).”
“Not managed very well at all, and my doc has been pushing me to start methotrexate however I am reluctant to start it.”
“I was told given enthesitis as my predominant feature I do not qualify for biologics. My disease is worsening from my feet, hands, arms, shoulder & now neck. i am seeking a second opinion from another rheumatologist as our local one said there is nothing left he can offer Tx.”
“Medications don't all work, I've spent hundreds of dollars.”
One respondent noted a change in the effectiveness of their medication around the time they received COVID-19 immunizations:
“Biologic was very effective until the 3rd [Pfizer] vaccine shot.”
The costs of medications borne by patients remain a significant barrier to accessing appropriate treatments that manage the symptoms and guard against irreversible joint damage.
“My present regime was covered by my insurance plan to the cost of $4,000 every 3 months but since I’ve had to reduce my hours of work I’m no longer covered. I’m a front line Nurse. I’ve had to use the Trillium Plan to get my medication covered. I have to travel over 100 km to see my Rheumatologist.”
“Yes, high cost of biologics. No drug benefit coverage, under age 65.”
“Between Apremilast manufacturer and private health plan the cost of the drug is free. If not for this coverage I would be on another medication.”
“Prescribing particular treatment options: Expiration (life time limit) of drug plans and provincial coverage do not cover the cost of some drugs. Takes a great deal of time, dollars and energy to stay well and be able to move.”
For others, travel and access to care present important barriers to optimal treatment of their PsA.
“I live 2.5 hours away from my Rheumatologist. This is a problem since I do not drive. We have been able to manage my appointments with phone appointments every 3 months. This has worked well. If I was having problems with my disease or medications, etc., it would be problematic to have in-person appointments. I live in a rural environment and there is very limited access to specialists.”
“Covid has Dr shortages in my rural area has made it difficult to get any treatment.”
Improved Outcomes
The survey asked respondents what they were looking for in this treatment option or in a new treatment option. One person commented that they wanted a treatment that would stop the disease process: “Stop the process of the disease, pain, easy access to support, medication. Being able to function daily. Some side effects are tolerable.”
Other respondents emphasized that they wanted better management of the symptoms that impact their lives, including reducing side effects from the medications currently available, while being cautious about needing to deal with new side effects:
“Fewer side effects.”
“Alleviate fatigue.”
“Looking to get back to a regular exercise routine. That’s my ultimate goal. Not as many stomach issues. Perhaps an easier time in the mornings.”
“I really an not interested in changing my present medication of Humira. I am to afraid of the pain I will feel changing. I have had a flare up that even my jaw was in pain along with my whole body. I do not want to go through that again although I may.”
“Hopeful I would qualify to as after 10 days on current treatment ( monthly injections) I go back to hardly being able to do daily tasks or walk but I continuously push through. Would love to have that quality of life and able to function and would absolutely accept side effects if less pain and flares could happen. At this moment even a 15% improvement would be amazing! This would help with less er visits, and in constant pain.”
“Waiting for treatment to begin hoping for a big improvement of my quality of life in every aspect. I am willing to try this treatment even if I get enough relief to exercise again.”
“I have never taken this drug nor have I heard of it. I rely on my Rheumatologist to present medication options to me that she feels would be helpful. My medication is costly but I have good insurance. If I had to pay for it myself I would likely look for a less expensive option. If I live relatively pain free I consider that my medication is working. If my blood work indicates elevated inflammation then it's discussed and treated. I have researched my disease thoroughly but I rely on my doctor's expertise. The only personal change I made was to my diet once I got diagnosed. I became a vegetarian and i believe it helped me. I would not accept medication with too many side effects, especially if they were long term. To me a medication is working if it reduces my symptoms. Quality of life is everything to me. No one wants to simply exist. Living pain free is always the goal.”
“What are the serious side effects? That sounds intimidating. I want to be pain free though and would try anything to accomplish that.”
“Not willing, at the moment, to experience serious side effects, More agility would be acceptable.”
“Right now I'll trial anything to stop the progression to improve my QoL as the last 2y have been shocking how fast things have progressed. I do not hav sick time but am willing to give anything a trial if means using holidays without pay right now. I just want to regain some basic exercise, get this excessive weight off , improve my energy again & feel alive . Currently my future looks pretty bleak & I'm still in shock with my deteriorating physical health over the last 2y.”
“Being able to have a life balance with not so much pain.”
“I was on Tremfy for a while, and during that time I needed steroid shots in my shoulders. It wasn't the best for me.”
“I would like to use a drug that does more for me.”
“Enhanced - better sleep, less swelling in joints. Unmet need - see above; side effects - possible if it enhances my lifestyle; improvement -less pain and swilling less fatigue, better sleep. No other benefits come to mind.”
“I am not willing to accept serious side effects with any medication. Or it would have to guarantee putting my PsA into remission for me to consider taking it.”
“Enhanced quality of life is very important to me, management is very important to me and would consider treatment if recommended by my rheumatologist who knows me well from the past 20 plus years. Reduced costs and the ability to not have to go to biosimilars would be wonderful. It is difficult to measure the amount of improvement but less pain and having more energy would be very helpful.”
Experience With Drug Under Review
Two survey participants indicated that they have used Tremfya (guselkumab). When asked what positive and/or negative effects they experienced with this drug, the participants stated:
“80% clearer skin, much improved mobility, and range of motion. No negative effects.”
“Nothing negative. Took longer for clearer skin.”
Demonstrating the heterogeneity of responses to treatment, one participant thought that the drug treated their psoriasis better than their arthritis. The other said that their skin was not as clear as on another biologic, but that it helped the arthritis in their shoulders, though they are not certain yet if they are experiencing PsA or osteoarthritis.
Both participants stated that they did not experience side effects.
When asked if this drug was easier to use than other therapies they have tried, the two participants provided the following insights:
“Easier than some. Would be more convenient to inject at home rather than make a doctor appointment and have to travel and take time off work.”
“Less injections. I get severe hangover after injecting any biologic. Headache, feel off.”
Both participants expect that this drug will change their long-term health and well-being. Specifically, they shared that:
“I expect to live longer because life isn't as miserable as it once was.”
“If it slows disease progression, that is good.”
They shared examples of how Tremfya (guselkumab) is already helping them achieve these outcomes. One participant reflected that there is “much less aggravation for myself and my family because I feel better” and the other shared that it has “kept PsA from getting worse.”
Companion Diagnostic Test
N/A
Anything Else?
Like psoriasis, PsA is complicated, frustrating and can be debilitating without access to appropriate treatments. Patients are very different in how they react to changes in lifestyle, topical treatments, oral treatments and biologics. What works for one patient may not work for the other, even if their symptoms are very similar.
For many, PsA is a disease that often “falls through the cracks.” Some patients are seen by a dermatologist while others are seen by rheumatologists. Joint pain is not always discussed with a dermatologist and plaques on the skin are not always discussed with rheumatologists. These challenges often lead to delays in diagnosis of PsA and consequently severe and irreversible damage to the joints.
It is still not clear how many people who have psoriasis will get PsA. It is estimated that up to 30% of people with psoriasis will develop PsA (estimates range from 4-30%, depending on what part of the world you live in). Both diseases are caused by the immune system being inappropriately activated. Most of the time (80%), psoriasis comes first but it remains difficult to predict whether a person living with psoriasis will later develop PsA, despite research advances that have identified several biomarkers associated with PsA and advances in developing predictive screening tools. (Eder, L. et al., The Prediction of Psoriatic Arthritis Tool (PRESTO) Study – Interim Report, American College of Rheumatology Convergence 2020 conference, online: https://acrabstracts.org/abstract/the-prediction-of-psoriatic-arthritis-tool-presto-study-interim-report/.)
All patients are looking for a treatment that will control all their symptoms but, ultimately, they would like a cure to this debilitating disease. Earlier treatment of PsA can result in better outcomes and reduce the risk of permanent and debilitating joint damage. PsA is also linked with an increased risk of cardiovascular disease, specifically atherosclerotic disease (low grade inflammation in blood vessels). Controlling inflammation can reduce the risk of PsA patients later developing heart disease. (Eder, L. et al. Cardiovascular Diseases in Psoriasis and Psoriatic Arthritis, The Journal of Rheumatology Supplement June 2019, 95 20-27; DOI: https://doi.org/10.3899/jrheum.190114)
Patient Group Conflict of Interest Declaration
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
CAPP and CPN collaborated with the Canadian Arthritis Patient Alliance, The Arthritis Society, and Creaky Joints (Global Healthy Living Foundation) to develop and distribute a survey of patients’ experiences with PsA and with Tremfya (guselkumab) to treat PsA.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
CAPP and CPN collaborated to analyze the data and prepare this submission, without any assistance outside our organizations.
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
Table 3: Financial Disclosures for the Canadian Association of Psoriasis Patients
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
AbbVie Canada | — | — | — | X |
Amgen Canada | — | — | X | — |
Bausch Health Canada | — | X | — | — |
Janssen Canada | — | — | X | — |
Novartis Canada | — | X | — | — |
Pfizer Canada | — | — | X | — |
Sun Pharma | — | — | X | — |
UCB Canada | — | — | X | — |
Table 4: Financial Disclosures for the Canadian Psoriasis Network
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
AbbVie Canada | — | — | — | X |
Amgen Canada | — | — | X | — |
Bausch Health | — | — | X | — |
Bristol Myers Squibb | — | — | X | — |
Boehringer Ingelheim International | — | X | — | — |
Boehringer Ingelheim Canada | — | — | X | — |
Janssen Canada | — | — | X | — |
LEO Pharma Canada | — | — | X | — |
Novartis Canada | — | — | X | — |
Sun Pharma | — | — | X | — |
UCB Canada | — | — | X | — |
Canadian Arthritis Patient Alliance, Arthritis Society, and CreakyJoints Canada
About Canadian Arthritis Patient Alliance, Arthritis Society, and CreakyJoints Canada
The Canadian Arthritis Patient Alliance (CAPA) is a grassroots, patient-driven and managed, independent, national education and advocacy organization with members and supporters across Canada. CAPA creates links between Canadians with arthritis, assists them to become more effective advocates and seeks to improve the quality of life of all people living with the disease.
CAPA believes the first expert on arthritis is the individual who has the disease, as theirs is a unique perspective. We assist members to become advocates not only for themselves but all people with arthritis. CAPA welcomes all Canadians with arthritis and those who support CAPA's goals to become members. Our website is updated regularly and can be viewed at:
The Arthritis Society (AS) has been setting lives in motion for over 70 years. Dedicated to a vision of living in a world where people are free from the devastating affects that arthritis has on the lives of Canadians, the Arthritis Society is Canada’s principal health charity providing education, programs, and support to the over 6 million Canadians living with arthritis. Since its founding in 1948, the Arthritis Society has been the largest non‐government funder of arthritis research in Canada, investing more than $200 million in projects that have led to breakthroughs in the diagnosis, treatment, and care of people with arthritis. The Arthritis Society is accredited under Imagine Canada’s Standards Program. The website www.arthritis.ca provides more detailed information.
For more than two decades, CreakyJoints has served as a digital community for millions of arthritis patients and caregivers worldwide who seek education, support, advocacy, and patient-centered research. All of our programming and services are always provided free of charge. CreakyJoints is part of the non-profit Global Healthy Living Foundation, whose mission is to improve the quality of life for people living with chronic illnesses. In keeping with our work at CreakyJoints USA, CreakyJoints Canada inspires, empowers, and supports arthritis patients – and patients living with other chronic conditions – and their caregivers to put themselves at the center of their care by providing evidence-based education and tools that help people make informed decisions about the daily and long-term management of arthritis and other chronic conditions. At the heart of CreakyJoints Canada is collaboration. We will continue and strengthen our work with Canadian arthritis organizations and patient advocates that you know, love, and respect. We are all stronger together.
Information Gathering
We developed a survey to hear directly from people living with psoriatic arthritis (PsA) about their experiences with PsA and experiences taking guselkumab (Tremfya). The survey was collaboratively developed by the Canadian Arthritis Patient Alliance (CAPA), the Arthritis Society, Canadian Spondylitis Association (CSA), Canadian Association of Psoriasis Patients (CAPP), CreakyJoints Canada, and Canadian Psoriasis Network (CPN). The design was informed by the lived experiences of the organizations’ members, many of whom live with various forms of arthritis. The survey was shared via e-mail, social media, and organization websites from all five organizations, through our respective Canadian networks and communities. The survey was open from April 20, 2022, to May 16, 2022.
CAPA, CreakyJoints Canada, and the Arthritis Society are making this collaborative submission, while the three remaining organizations who collaborated on the survey design will each be making their own submissions based on survey response data and their unique community perspectives.
There were 70 people that completed the survey with most participants living in Ontario (49%) followed by British Columbia (17%), Alberta (10%), Quebec (6%), New Brunswick (6%), Nova Scotia (6%), Newfoundland and Labrador (4%) and Nova Scotia (4%). Under 15% of survey participants were under the fifty years of age while the age range of the remaining survey participants ranged from fifty to eighty-five. Two survey participants have experience taking Tremfya. Four out of 5 people with psoriatic arthritis identified as a woman and one out of 5 identified as men. Over 20% of participants identified as living with a disability and about 70% of participants identified as white. A low number (4%) of survey participants sleep in government or group housing, 7% of survey participants rent housing while the remaining amount (89%) own their current housing.
Disease Experience
Psoriatic arthritis (PsA) is a chronic, inflammatory, systemic disease of the skin and joints. Symptoms include musculoskeletal pain, stiffness, fatigue, and limited range of motion in the joints as well as psoriasis plaques that are itchy, sensitive, red, flaky and cause pain. PsA is also a systemic disease meaning that other parts of the body in addition to joints can be affected, including the eyes and heart, metabolic disease, and related musculoskeletal diseases like osteoarthritis.
“I live with hypertension, depression, metabolic syndrome, fibromyalgia, and the awful side effects of that, and Inflammatory bowel disease.” Person living with Psoriatic arthritis
PsA can vary in severity from mild to very severe.
Survey participants rated their disease severity as an average of 57 out of 100. A person may experience active periods of disease (commonly known as flares or flare-ups) and times where there is decreased activity or even inactivity (remission). While people who have PsA live with several symptoms, how they experience those symptoms and the severity of PsA can be different from person to person – PsA is unique to each person who lives with it. There is currently no cure for PsA. Survey participants indicated a range of symptoms that are difficult to manage including joint stiffness (79%), fatigue (75%), changes in fingernails and toes (63%), hip pain (61%), back pain (51%), anxiety (47%), and stress (33%).
“I have to plan ahead to make sure I’m not scheduled for anything on recovery day. I have to rest every afternoon or face the consequences.” Person living with psoriatic arthritis
“(I am) not well (and) in constant pain.” Person living with psoriatic arthritis
People living with the disease are also at risk of co-morbidities, such as depression and mental health issues, diabetes, and cardiovascular disease.
Periods of very active disease are called a ‘flare’ and for some people, flares can be incapacitating. Flares are not predictable in terms of how severe they will be or how long they will last. They may last for a few hours, days, weeks or even months. Because of their unpredictability, flares must be dealt with reactively by people. The unpredictable nature of PsA also often makes it feel like a person is not in control of their disease and can impact their ability to carry out day to day activities and life roles, such as contributing to the workplace.
“I live very independently. The only difficulty I have is I get tired more often and very occasionally have issues with my knees and feet/ankles. Pain and swelling. Stairs can sometimes present an issue if it's multiple stories. This happens after prolonged walking, standing or excessive exercise.” Person living with psoriatic arthritis
The disease impacts all aspects of a person’s life including a variety of activities that people without PsA take for granted such as walking, sleeping, holding a phone, standing, and taking care of everyday tasks, such as shopping, running errands, and cooking. Given the limitations in activities of daily living, PsA impacts all aspects of a person’s life including workplace participation and productivity, carrying out parenting and other social roles, and relationships with spouses, friends, and family members. When asked about the most significant impacts of PsA on their daily quality of life, respondents expressed that PsA interfered with work (54%), social connections (52%), self-esteem (50%), mental health (50%), intimacy (50%), family life (38%) and friendships (24%). Other impacts included embarrassment and self-consciousness from symptoms caused by PsA.
“(I don’t receive any support) at the moment per se but spouse drives me to grocery store and helps pack bags, does washing of dishes after I cook and bake. I do not qualify for other supports from my community at the moment. Person living with psoriatic arthritis
People indicated difficulties in contributing and participating at school or work due to the fatigue, pain, and other symptoms of the psoriatic arthritis.
People with psoriatic arthritis that completed the survey indicated that they missed work due to their disease, including 28% missing 1-3 workdays per month, 17% missing 4-6 days per month, and 7% indicating they do not work due to PsA. PsA impacts lives in many ways: daily tasks that many well individuals take for granted may become too difficult or exhausting to complete; participating in leisure activities or hobbies can be challenging; while caring for or spending time with family members, including children, spouses/partners, and other loved ones, also becomes difficult.
“I only work online with a modified work schedule. I’m not able to do housework, exercise, socialize or travel like I used to.” Person living with psoriatic arthritis
“I take things day by day. I miss out on lots of things due to pain and fatigue. I work shift work, usually 4 hrs per shift anymore, even an hour, keeps me awake at night with agonizing foot and toe pain especially.” Person living with psoriatic arthritis
PsA’s impacts also extend to others within a person’s support circle, including caregivers such as spouses/partners and children who provide valuable emotional and direct support in complete activities of daily living. Often, these people take on additional chores or tasks such as cooking, cleaning, shopping, etc. to support the person living with PsA, and family roles change as spouses / partners take on more tasks, such as supporting their spouses / partners in getting to and from medical appointments. They may provide emotional support to help the person living with psoriatic arthritis in navigating the health care system and the anxiety and stress of living with PsA.
Experiences With Currently Available Treatments
Clinical practice guidelines emphasize early aggressive treatment of PsA, which provides the best long-term outcomes for people living with the disease. A number of treatment approaches are used to manage PsA including non-steroidal anti-inflammatory drugs (NSAIDS), corticosteroids and conventional synthetic Disease Modifying Anti-Rheumatic drugs (csDMARDS) such as Methotrexate, as well as biologic Disease-Modifying Anti-Rheumatic Drugs (bDMARDs), such as Etanercept and Infliximab. Effective treatments mean that people with PsA do not need to live with the permanent damage, high medical costs (e.g., surgery, mobility aids, accessible housing) and disability. Early intervention is critical to allow people with Psoriatic Arthritis the opportunity to fully participate in all aspects of life.
“Spouse helps with errands, finances, cooking, cleaning. Sister and father understand and provide emotional support. Friends do not care.” Person living with psoriatic arthritis
“(I tried) Chloroquine for my enthesis was ineffective - I have not been offered other DMARDs as apparently, I don't qualify.” Person living with psoriatic arthritis
Notwithstanding the fact that numerous medication options exist, patients’ responses to medication can vary significantly. Some medications are effective for some people with inflammatory arthritis while not effective for others. Survey participants indicated that they had experience with many medications, such as Methotrexate (61%), Hydroxychloroquine (29%), Sulfasalazine (32%), and Leflunomide (22%).
“My rheumatologist wasn’t seeing much improvement. Nor was I. So, after a month trial of some we changed them.” Person living with psoriatic arthritis
Some treatments will only manage the disease for a short period of time before the patient’s immune system adapts to a drug’s presence (i.e., becomes non-responsive to it) and they will have to switch to another medication. In some cases, patients with PsA may not adequately respond to any of the DMARD’s (conventional and biological) currently available. A third of survey participants noted that they had an inadequate response to currently available treatments. As a result, patients need a number of medication options in order to effectively manage their disease throughout their lives. There are also no specific tests that identify which medication will be effective for a person living with PsA. This means that a person with the disease will need to go on one or more medications on a trial-and-error basis in order to find a medication that is effective. Often, the treating physician determines which medication is most appropriate based on a number of factors such as patient preferences, mode of administration, anticipated side effects, etc. It is also an anxious and stressful experience if medications are not effective and cost thousands of dollars out of pocket. Oftentimes, people with PsA need to make difficult financial choices in order to pay for their medications.
It is also important to note that conventional synthetic DMARDs (csDMARDS) are difficult to take for people living with PsA. Nausea, vomiting and a general malaise can persist for days after treatment with csDMARDS. Due to these experiences, many patients may not wish to take the medicine in question because the medication(s) is too difficult to take. This impacts adherence to treatment, increases health care costs (e.g., more visits to the doctor) and makes it difficult for people living with PsA to work, carry out social roles and participate in other activities of daily living. Toxicity issues (e.g., liver) can also be of concern for people taking csDMARDS, such as Methotrexate, Immuran, and Leflunomide.
“I had terrible side effects. I just did not feel right.” Person living with psoriatic arthritis
“I discontinued methotrexate as I felt it was of no benefit to me and has very harsh side effects.” Person living with psoriatic arthritis
Patients may also pursue medical cannabis and/or non-pharmacological approaches to manage PsA symptoms, such as physiotherapy, occupational therapy, massage therapy, counselling, or acupuncture. These approaches can often help to address the symptoms of the disease, such as pain and fatigue. However, there are significant unmet patient needs in terms of accessing non- pharmacological treatments, often because they are not reimbursed through provincial health care systems, the treatment options are simply not offered, or there are lengthy waits.
Patients identified many challenges in accessing treatment options. Expense, travel, and time required for treatment were all cited as being prohibitive. Some patients identified a difficulty in access to treatment relating to lack of access to specialists and general practitioner, and/or the COVID-19 pandemic restrictions. Survey participants accessed reimbursement of medication costs in multiple ways, such as through private drug plans (57%), provincial or federal government drug programs (21%) or disability support programs (7%).
Improved Outcomes
PsA patients have identified several outcomes that are important to them and that should be considered when evaluating new therapies, including:
route of drug administration (pills vs infusion vs self-injections)
a reduction in pain and fatigue
effective for psoriasis symptoms as well as psoriatic arthritis symptoms.
increased mobility
ability to work and be productive at work
ability to carry out activities of daily living
ability to effectively carry out parenting tasks and other important social roles
reduced infection rates
affordability of the medication
increased quality of life
improved sleep
“I would hope it works to alleviate fatigue.” Person living with psoriatic arthritis
“Looking to get back to a regular exercise routine. That’s my ultimate goal. Not as many stomach issues. Perhaps an easier time in the mornings.” Person living with psoriatic arthritis
“I have scar on lung. (I) don’t want infections either.” Person living with psoriatic arthritis
Current medications for the treatment of PsA also have a number of negative side effects, such as fatigue which often persists beyond 24 hours (Methotrexate), nausea (Methotrexate, Arava, Immuran), increased infection risk (most DMARDs), liver toxicity and weight gain (Prednisone).
“Hopeful I would qualify to as after 10 days on current treatment (monthly injections) I go back to hardly being able to do daily tasks or walk but I continuously push through. Would love to have that quality of life and able to function and would absolutely accept side effects if less pain and flares could happen. At this moment even a 15% improvement would be amazing! This would help with less er visits, and in constant pain.” Person living with psoriatic arthritis
Experience with Drug Under Review
From those surveyed, two people identified having experience with taking guselkumab (Tremfya). The respondents shared positive and negative effects of taking guselkumab:
“I had 80% clearer skin, much improved mobility, and range of motion. No negative effects.” “Nothing negative. Took longer for clearer skin.”
“(It was) better at psoriasis than arthritis.”
“Overall, skin wasn't as clear as on another biologic, and it's not ruled out if my shoulders have PsA or OA, so I cannot say that it did not help for PsA in shoulders.”
“(It was) easier to take than some. (It) would be more convenient to inject at home rather than make a doctor’s appointment and have to travel and take time off work.”
“(There were) less injections. I get severe hangover after injecting any biologic. Headache, feel off.”
In terms of long-term benefits of taking the medication, the two people noted the following:
“I expect to live longer because life isn't as miserable as it once was.” “If it slows disease progression, that is good.”
“There was much less aggravation for myself and my family because I feel better.” “(The medication) kept PsA from getting worse.”
Companion Diagnostic Test
Not applicable
Anything Else?
Not applicable
Patient Group Conflict of Interest Declaration
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
No, we did not receive any outside help.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
No, we did not receive any outside help.
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
Table 5: Financial Disclosures for the Canadian Arthritis Patient Alliance
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Abbvie Corporation | — | — | X | |
ACE Planning and Consulting | — | — | — | — |
Canadian Rheumatology Association | X | — | — | — |
CAPDM | X | — | — | — |
Janssen | X | — | — | — |
CORECOM | X | — | — | — |
Government of Canada | X | — | — | — |
Brooks Group | X | — | — | — |
UCB Canada | — | X | — | — |
CADTH | X | — | — | — |
SmithSolve LLC | X | — | — | — |
The University of British Columbia | X | — | — | — |
Arthritis Society | X | — | — | — |
Table 6: Financial Disclosures for the Arthritis Society
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Abbvie | — | — | X | — |
Alcon | — | — | — | — |
Amgen | — | — | X | — |
Boehringer Ingelheim | — | — | X | — |
BMS | — | — | X | — |
Celgene | — | — | — | — |
Eli Lilly | X | — | — | — |
Eupraxia Pharmaceuticals | — | — | — | — |
Gilead | — | — | — | — |
Innovative Medicines Canada | — | X | — | — |
J+J Shared Services | — | — | — | X |
Janssen | — | X | — | — |
Merck | — | — | — | X |
Novartis | — | — | X | — |
Pfizer | — | — | — | X |
Sanofi | — | — | — | — |
UCB | — | X | — | — |
Table 7: Financial Disclosures for CreakyJoints Canada
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
AbbVie Corporation | — | — | X | — |
Arthritis Consumer Experts
About Arthritis Consumer Experts
Arthritis Consumer Experts (ACE) is Canada’s largest, longest running national arthritis patient organization headquartered in Vancouver, BC, with 50,000 members from coast-to-coast. ACE and its team members acknowledge that they gather and work on the traditional, ancestral and unceded territory of the Coast Salish peoples - xwməθkwəỳəm (Musqueam), Skwxwú7mesh (Squamish), and SəÌílwətaʔ/Selilwitulh (Tsleil-Waututh) Nations.
ACE provides free, science-based information and education programs in English and French to people with arthritis and those who care for and support them. ACE serves people living with all forms of arthritis by helping them take control of their disease and improve their quality of life through education and (em)powerment. Founded and led by people with arthritis, ACE also advocates on arthritis health policy and provides research-based education through ACE’s JointHealth™ family of programs, directly to consumers/patients, media and government. ACE operates as a non-profit in a fully transparent manner and is guided by a strict set of guiding principles, set out by an advisory board comprised of leading scientists, medical professionals and informed arthritis consumers. Ultimately, we are guided by the needs of our members, who are people living with arthritis, and their caregivers.
Link to website: www.jointhealth.org
Information Gathering
The information was gathered from a patient who submitted their response via email on May 4, 2022 (Patient A) and patients who completed ACE's patient input survey through SurveyMonkey from December 18, 2020 to January 26, 2021 (Patients B to F).
Disease Experience
How does the disease impact the patients’ day-to-day life and quality of life?
Psoriatic arthritis (PsA) has an unique and significant effect on the lives of people living with it and they constantly consider the state of their disease and decide what they can, or more likely, cannot, cope with or achieve, how they can go about their daily lives, and how much help they may need along the way. Because of the visible symptoms of the disease, such as a scaly rash on the skin, patients also experience mental stress.
Patient A: “I am in pain most of the time - day and night. It is not severe and not always constant, but staying too long in one position or certain moves trigger it. The pain is in my hands, mostly the wrists, one hip at the sacroiliac and sometimes the lower back. Due to the pain, I cannot enjoy long walks or many other activities which I used to do. It also affects my mood."
Patient B: Living with PsA for 47 years and also has fibromyalgia and is living with obesity. “My psoriatic arthritis is under control because of the medication I take and my quality of life is very good as a result.”
Patient C: Living with PsA for 22 years. They have “restricted ability to walk distances, difficulty opening door knobs and lids on jars, and using cutlery for meals”. This patient experiences joint pain on a daily basis.
Patient D: Living with PsA for 11 years. With medication therapies, they are able to control their PsA and maintain an active and busy life.
Patient E: Living with PsA for 6 months. As a result of their PsA, they experience pain and reduced mobility function.
Patient F was “diagnosed with PsA in 2015, but started to show symptoms in mid-eighties.” They experience pain in many joints and require 45 minutes warm-up exercises every morning before their day starts. They also experience big toes problems and have “psoriasis in scalp and on face” that requires lotions.
How does the disease impact the caregivers’ day-to-day life and quality of life?
Patient A: “My mood affects my family. Also, I will soon have a granddaughter and want to be able to take care of her and hold her without fear that my wrists will give out.”
Patient B: “Since I am not a caregiver, I cannot say what these may be.”
Patient C, D, E: N/A
Patient F: “None in my case.”
Are there any aspects of the illness that are more important to control than others?
Patient A: “I would like the pain to disappear permanently if possible.”
Patient B: “Joint damage from inflammation.”
Patient C: “Walking, using hands.”
Patient D: “Maintaining treatment schedule. Missing treatments, particularly my weekly methotrexate shot catches up with me…aches, stiffness, inflammation.”
Patient E: “Lack of mobility.”
Patient F: “Toes and psoriasis.”
Experiences with Currently Available Treatments
How well are patients managing their disease/condition with currently available treatments?
Patient A: “I have just started Methotrexate. It has only been a month. I notice no positive effects yet, but have some side effects which so far are not too bad. Though it seems to me the pain has increased since I started taking it. The side effects are headache, some bouts of nausea and I find I am not able to sleep as well. Folic tablets help with the nausea. I would have liked to have been put on biosimilars as apparently, they can lead to permanent remission in many cases if they are used early enough. Due to cost, I was put on Methotrexate, at least for the beginning. No problem in taking the pills. My lifestyle is not impacted."
Patient B is currently taking Humira and methotrexate. “This is very effective in controlling my psoriatic arthritis. I have so far been lucky in that I haven’t experienced any adverse effects.” This patient has no hardships accessing Humira and methotrexate.
Patient C is currently taking Erelzi, methotrexate, Plaquenil, Tylenol and ibuprofen and is not aware of any adverse effects. “All have moderate success.” This patient has no hardships accessing current therapies.
Patient D is taking a combination of a Remicade infusion every 7 weeks and a weekly dose of methotrexate. They experience feeling of nausea for a couple days after methotrexate and higher level of fatigue all the time. When asked if there are any needs that are not met by current therapy, this patient stated: “Not for me to say – but many friends and relatives have asked about my treatments and their inability to access them or their acceptance of arthritis/pain as part of getting old.” This patient has no hardships in accessing current therapies; they added: “But I have private coverage for Remicade, which is roughly $3K every 7 weeks – roughly $21-25,000.”
Patient E is controlling their psoriatic arthritis with medication and exercise; they state that both are not very useful and have no adverse effects. This patient would like warm water therapy to be available but this has stopped due to the pandemic.
Patient F: “Exercise. Scalp lotion. Soak toes in yellow Listerine daily. Ointment on big toes twice a day. Skin cream on face once a day. Tylenol before heavy exercise. No therapy adverse effects.” When asked if there are any needs that are not met by current therapy, this patient stated: “Access to gyms and recreation centres. COVID-19 has closed gyms – tough for exercise issue.” This patient has trouble finding the time to exercise.
Improved Outcomes
This section asked patients what they would consider when choosing a therapy.
Patient A: “I would like the pain to disappear permanently if possible. When on anti- inflammatory for 2 weeks at one point, I felt like a different person. My mood improved tremendously. I was happy. This is not the case when I have pain every day. Of course, my mood affects my family. Also, I will soon have a granddaughter and want to be able to take care of her and hold her without fear that my wrists will give out.
All arthritis medications seem to have serious adverse effects. Depending on how much they help with the disease, we are all willing to make trade offs. So far with the Methotrexate, I do not see any improvement so I am less willing to accept more side effects. If it improves the pain, then of course that would make me more willing to tolerate the downsides."
Patient B: “Potential side effects. In my case, how small an amount of these powerful drugs can be taken while still being effective (in order to minimize side effects).”
Patient C: Cost
Patient D: Side effects.
Patient E: Investment of time when not really working.
Patient F: Avoid drugs.
Experience with Drug Under Review
Patient A does not have any experience with guselkumab to treat their psoriatic arthritis, but have provided an additional comment for this section. Patients B to F completed a survey on SurveyMonkey for the call for patient input on upadacitinib. As the survey was conducted anonymously, ACE could not confirm if patients B to F have had any experience with guselkumab.
Patient A: None. It has an interesting name.
Companion Diagnostic Test
Not applicable to this submission.
Anything Else?
Arthritis Consumer Experts is providing this patient input submission based on a patient who submitted their response via email on May 4, 2022 (Patient A) and patients living with psoriatic arthritis who completed ACE’s patient input survey on SurveyMonkey between December 18, 2020 and January 26, 2021 (Patients B to F).
ACE made minor grammatical corrections to input where needed but in no way altered the meaning or intent of the input.
Appendix: Conflict of Interest Declaration
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
This submission was summarized and written solely by the staff of Arthritis Consumer Experts, free from consultation, advice, influence, or financial support from any outside individual, group or company.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
A full list of private and public sector organizations providing Arthritis Consumer Experts grants-in-aid over the past 12 months can be found here: https://jointhealth.org/about-principles.cfm?locale=en-CA
Specific to this input, Arthritis Consumer Experts has not received any financial payments (grants-in-aid) from Janssen Biotech Inc.
Table 8: Conflict of Interest Declaration for Arthritis Consumer Experts
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
N/A | — | — | — | — |
Clinician Input
Canadian Rheumatologist Psoriatic Arthritis Interest Group
About the Canadian Rheumatologist Psoriatic Arthritis Interest Group
We are a group of clinical rheumatologists across Canada who have extensive experience managing inflammatory arthritis, including psoriatic arthritis. We are practicing in academic and community settings. Dr. Proton Rahman has had a long-standing interest in the pathogenesis, prognosis, and management of psoriatic disease over the last 20 years and published widely on this topic. Dr. Rahman is also involved in Phase III Guselkumab studies in PsA and multiple secondary sub-analyses of guselkumab in PsA. Dr. Chan is an Assistant Clinical Professor at UBC and also has a clinical and research interest in psoriatic arthritis and axial spondyloarthritis. Dr. Haaland is a clinician and clinical researcher involved in assessing and treating patients with psoriatic disease. He is involved in several clinical trials and observational cohorts, including phase 3 trials of guselkumab in psoriasis and psoriatic arthritis and several collaborative real-world cohort initiatives in psoriasis. Dr. Jean-Pierre Raynauld, Is an Assistant Professor at Université de Montréal and a Rheumatologist at the Montreal Rheumatology Institute and CHUM since 1993. His main research interests focuses on the evaluation of new investigation and treatment techniques in inflammatory arthritis. Dr. Chandran is an associate professor at the University of Toronto and has a long standing interest in psoriatic arthritis, with a research interest in genomics of psoriatic disease. He has participated in the design and execution of multiple PsA clinical trials. Dr. Gladman is Professor of Medicine, University of Toronto and Senior Scientist Schroeder Arthritis Institute, Krembil Research Institute, Toronto Western Hospital. She has been investigating psoriatic arthritis for over 40 years including clinical features, prognosis, genetic and other biomarkers for disease susceptibility and progression as well as clinical trails. She is founding member of the Groups for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA) International Psoriasis and Arthritis Research Team (IPART, both international groups, as well as the Spondyloarthritis Research Consortium Canada (SPARCC), a Canadian group investigating spondyloarthritis and psoriatic arthritis. Drs. Chandran, Chan and Rahman are also members of SPARCC, GRAPPA, and have been involved in formulating treatment guidelines in Canada and internationally.
Information Gathering
Information was gathered through our clinical experience of current products, knowledge of the existing literature, including results from the published clinical trials, real-world studies, network meta-analysis and presentation at the recent congress of guselkumab and other therapeutic agents in PsA.
Current Treatments and Treatment Goals
PsA arthritis is a complex disease with varied manifestations. Broadly, therapy goals are to improve quality of life and improve physical function by controlling joint pain and stiffness, clearing concomitant psoriasis and extra-articular manifestations. Furthermore, our goal is to limit structural damage and minimize long term complications arising from inadequately controlled inflammation related to PsA.
In the Canadian context, nonpharmacological treatment is used as an adjunct rather than the sole form of therapy. In our experience, less than fifteen percent of PsA patients are solely managed by nonpharmacological therapies. These therapies include graded exercise programs, structured physical and occupational therapy, diet, weight loss, and smoking cessation.
Regarding pharmaceutical intervention, for mild articular symptoms, nonsteroidal anti-inflammatory drugs and local glucocorticoid injections are implemented, and very infrequently, systemic steroids for a limited period. For patients with persistent pain, stiffness, and swelling despite NSAIDs, conventional disease-modifying anti-rheumatic drugs (DMARDs) are utilized, usually starting with either oral or subcutaneous weekly methotrexate. Additional DMARDs used in combination or sequentially include sulfasalazine and leflunomide. Hydroxychloroquine is used by Canadian rheumatologist given its safety profile when used in combination with methotrexate or leflunomide. Apremilast is usually reserved for traditional DMARD failures.
If two or more DMARDs are not effective after at least three to six months at the optimal dose, then biologic agents or JAK inhibitors are considered.
The usual first-line biologic therapies include either TNF inhibitors (etanercept, infliximab, adalimumab, golimumab, certolizumab pegol) or IL-17A inhibitors (secukinumab, ixekizumab). IL-12/23 inhibitor (ustekinumab) or JAK inhibitor (tofacitinib, upadacitinib) are usually relegated to second-line therapies without contraindications to TNF inhibitors or IL-17A inhibitors. All classes of biologics agents can be used after failure to initial biologic treatment. If there is a primary failure to a particular mechanism of action, strong consideration is given to a drug-using an alternate mechanism of action. Prsence of specific extra-articualr manifestations need to be taken into account when considering which medication to use.
All approved treatments improve symptoms and can help reduce concomitant steroid use. Therapies convincingly demonstrating disease modification include TNF inhibitors, IL-17A inhibitors, tofacitinib and upadacitinib.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments
Response: There is still a significant unmet need in the management of PsA. Limitations to current therapies include:
Current biologics only result in approximately 60% of patients achieving a good response (achieving ACR 20 response*)
Minimal disease activity (MDA*- a measure of a state of low disease activity) is achieved in approximately one in three to one in four PsA patients with biologic therapy
Inability to sustain the reduction in joint inflammation results in either dose creep and/or secondary failure
Persistence of active psoriasis despite improvement in MSK symptoms results in a sub-optimal response / poor quality of life for PsA patients.
Side effect profiles such as lupus-like syndromes or multiple sclerosis (with TNFi) and inflammatory bowel disease (IL-17A) limits the use of the two most common classes of biologics used to manage PsA (Humira monograph; Cosentyx monograph)
Current biologics are associated with a small but significant increased risk of serious infections (Jin Y et al, 2021, Li X et al, 2020)
(*ACR 20 and MDA are defined in the Appendix)
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Response: Guselkumab is a fully human monoclonal antibody that selectively inhibits the p19 subunit of IL‐ 23. This is of direct relevance to psoriatic disease as the IL-23 cytokine pathway plays a central role in the pathogenesis of psoriasis and PsA. Furthermore, IL-23 is the key driver of Th17 cells and the downstream release of pro-inflammatory cytokines, including IL-17A, IL-17F, IL-21, IL-22, and TNF. Furthermore, the entheses contain a special resident T-cell population that reacts to localised microdamage through IL-23 receptor to create a pro-inflammatory loop that results in arthritis.
The heterogenous nature of psoriatic disease necessitates nuanced treatment considerations; precision medicine demands granular thinking and should take into account the multiple facets of disease, disease state, comorbidities and concomitant medications. Thus, consideration of these factors that leads to the best treatment decision of advanced therapy in appropriate patients. Thus in some patients IL-23 inhibition with guselkumab will be the best option as a first line treatment. In other patients it may be a second line option following intolerance to or inefficacy of other approaches.
Thus, based on the current clinical evidence, guselkumab can be used as first-line biologic therapy (with or without traditional DMARDs) and after TNF inhibitor failure (with or without conventional DMARDs). It effectively treats all articular domains of PsA, and psoriasis and improves fatigue and physical function, which are significantly compromised in the PsA population.
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
Which patients are most likely to respond to treatment with drug under review?
Response: A broad range of patients with PsA will improve with guselkumab treatment, including those with articular inflammation, enthesitis, psoriasis, and axial disease. In particular, psoriatic arthritis patients with moderate to severe arthritis and psoriasis are the most likely to exhibit the best response, given the superior response to cutaneous inflammation with guselkumab (Ritchlin C et al, 2021; McInnes I et al, 2022; McGonagle D et al, 2021; Mease P et al, 2021)
Which patients are most in need of an intervention?
Response: Patients are refractory to current therapeutic agents (TNFi, IL-17A, and JAKi) or those unable to take current classes of biologic agents due to relative contraindications are most in keed for an intervention. Additionally, patients with severe psoriasis, psoriatic arthritis, and inflammatory bowel disease would be good candidates, as well as patients prone to infections as the serious infection rate with guselkumab in the clinical trials was extremely low , with a serious infection rate of 1.2 to 1.7/100 PYs (Rahman P et al, 2021a). Our clinician group felt that guselkumab is a very safe medication, and in fact, advances medicine and clinical immunology beyond frankly immunosuppressive agents, which have largely dominated the landscape, into the realm of more immunomodulatory approaches, which optimizes disease control while minimizing adverse events.
Would this differ based on any disease characteristics (e.g., presence or absence of certain symptoms, stage of disease)?
Response: Guselkumab has demonstrated efficacy compared to placebo regardless of sex, disease duration, various disease characteristics, or disease severity (Ritchlin C et al, 2022)
How would patients best suited for treatment with drug under review be identified (e.g., clinician examination/judgement, laboratory tests (specify), diagnostic tools (specify)
Response: Patients suited for guselkumab can be identified by rheumatologists due to inflammatory arthritis in the presence of psoriasis. Articular symptoms can be measured and followed by tender and swollen joint counts, active enthesitis, dactylitis or composite scores such as psoriatic arthritis disease activity score (PASDAS), disease activity index for psoriatic arthritis (DAPSA) or minimal disease activity (MDA).
Are there any issues related to diagnosis?
Response: Diagnosis of PsA is relatively straightforward when assessed by a rheumatologist.
Is a companion diagnostic test required?
Response: No companion diagnostic test that is clinically actionable exists, and it is not required.
Is it likely that misdiagnosis occurs in clinical practice (e.g., underdiagnosis)?
Response: Rheumatoid arthritis, ankylosing spondylitis, and erosive osteoarthritis can mimic PsA; however, this should not be a problem for a rheumatologist under most circumstances.
Is it possible to identify those patients who are most likely to exhibit a response to treatment with drug under review?
Patients with early disease and moderate to severe skin and joint involvement are most likely to respond to guselkumab. That being said, guselkumab can improve multiple domains of PsA and can be used broadly to manage PsA.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
Response: Improvement in tender and swollen joint counts, psoriasis, enthesitis, patient global, and ACR 20 are the most common outcome measures used. Other composite measures include MDA, DAPSA and PSDAS.
What factors should be considered when deciding to discontinue treatment with the drug under review?
Response: Ongoing inflammation characterized by inflammatory pain/stiffness, (swollen or tender joint count), worsening psoriasis, poor function, or intolerability due to side effects. Also, the development of severe extra-articular manifestations that are not treated by this medication, such as uveitis, may prompt a clinician to change to another biologic that can treat pertinent manifestations.
What settings are appropriate for treatment with [drug under review]? Is a specialist required to diagnose, treat, and monitor patients who might receive [drug under review]?
Response: A rheumatologist or experienced nurse practitioner with extensive rheumatology experience is needed to diagnose and manage PsA confidently. This can be done in the community, outpatient clinic, or specialty clinic.
Additional Information
Psoriatic arthritis can involve multiple domains (joints, enthesis, spine, skin, nails, and extra-articular manifestations such as uveitis and inflammatory bowel disease) (Coates L et al., 2021). Over two-thirds of PsA patients have simultaneous involvement of multiple domains (Ogdie A et al., 2021). Currently, available treatments may either not treat specific manifestations or only be mild/moderately effective (Coates L et al., 2021). Guselkumab has been shown to be effective for the treatment of joints in both naive and TNF inadequate responsive patients (McInnes et al., 2022; Coates LC et al., 2022). It has also been shown to be highly effective for cutaneous psoriasis and inflammatory bowel disease (Sandborn et al., 2022). Finally, it does not seem to have an overlap with the same contraindications to therapy that currently available biologics have (ie: drug induced lupus/multiple sclerosis with TNF inhibitors (Humira Monograph) , IBD with IL-17 inhibitors (Costentyx Monograph), shingles/CVD/malignancy risk with JAK inhibitors (Xeljanz Monograph) and also has a very low risk of serious infections (lower than placebo in both guselkumab and risankizumab trials (Guselkumab Monograph).
The highest priority for patients is pain relief (Gudu T et al 2018). Patients treated with guselkumab reported twice the improvement in patient pain, spinal pain, joint pain and bodily pain intensity by week 24 compared with the control group. These improvements were maintained or further increased by week 52 and week 100 (Nash P et al, 2021). .
Up to 50% of patients with PsA report high levels of fatigue and those patients place fatigue as the second or third domain of health in terms of impact on their life and in terms of priority for improvement (Reygaerts et al, 2018). When the effect of guselkumab on fatigue was evaluated, it was demonstrated that patients with active PsA, guselkumab 100 mg q4w or q8w led to clinically meaningful and sustained improvements in fatigue through 1 year (Rahman P et al, 2021b). A substantial portion of the improvement in FACIT-Fatigue scores induced by guselkumab was independent of effects on the achievement of other select outcomes.
Conflict of Interest Declarations
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
No
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician who contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
Declaration for Clinician 1
Name: Proton Rahman
Position: Professor of Medicine, Memorial University
Date: 27-May-2022
Table 9: Conflict of Interest Declaration for Canadian Rheumatologist Psoriatic Arthritis Interest Group Clinician 1
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Janssen | — | — | X | — |
Abbvie | X | — | — | — |
Novartis | — | X | — | — |
Eli Lilly | — | X | — | — |
UCB | X | — | — | — |
Pfizer | X | — | — | — |
Declaration for Clinician 2
Name: Jonathan Chan
Position: Assistant Clinical Professor UBC
Date: May 26/2022
Table 10: Conflict of Interest Declaration for Canadian Rheumatologist Psoriatic Arthritis Interest Group Clinician 2
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Janssen | — | X | — | — |
Abbvie | — | X | — | — |
Novartis | — | X | — | — |
Eli Lilly | X | — | — | — |
UCB | X | — | — | — |
Pfizer | X | — | — | — |
Viatris | X | — | — | — |
Fresenius Kabi | X | — | — | — |
Declaration for Clinician 3
Name: Dr. Derek Haaland, M.D., M.Sc., F.R.C.P.C.
Position: Medical Director, The Waterside Clinic, Barrie, ON; Associate Clinical Professor, McMaster University, Hamilton, ON; Assistant Professor, Northern Ontario School of Medicine, Laurentian University Campus, Sudbury, ON
Date: 27-05-2022
Table 11: Conflict of Interest Declaration for Canadian Rheumatologist Psoriatic Arthritis Interest Group Clinician 3
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
AbbVie | — | — | X | — |
Amgen | — | — | X | — |
AstraZeneca | — | — | X | — |
Bristol-Myers Squibb | — | — | X | — |
Eli-Lily | — | — | X | — |
GlaxoSmithKline | — | — | X | — |
Janssen | — | — | X | — |
Merck | — | — | X | — |
Novartis | — | — | X | — |
Pfizer | — | — | X | — |
Roche | — | — | X | — |
Sanofi Genzyme | — | — | X | — |
Takeda | — | — | X | — |
UCB | — | — | X | — |
Declaration for Clinician 4
Name: Jean-Pierre Raynauld
Position: Assistant Professor of Medicine, University of Montreal
Date: May 27th, 2022
Table 12: Conflict of Interest Declaration for Canadian Rheumatologist Psoriatic Arthritis Interest Group Clinician 4
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Janssen | — | — | X | — |
Pfizer | — | — | X | — |
AbbVie | — | — | X | — |
Novartis | — | X | — | — |
Sanofi | — | X | — | — |
Sandoz | X | — | — | — |
Organon | X | — | — | — |
Declaration for Clinician 5
Name: Vinod Chandran
Position: Associate Professor,University of Toronto
Date: 28-05-2022
Table 13: Conflict of Interest Declaration for Canadian Rheumatologist Psoriatic Arthritis Interest Group Clinician 5
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
AbbVie | — | X | — | — |
Eli-Lilly | — | X | — | — |
Novartis | — | X | — | — |
Janssen | — | X | — | — |
Declaration for Clinician 6
Name: Dafna Glaldman
Position: Professor, University of Toronto
Date: 28-05-2022
Table 14: Conflict of Interest Declaration for Canadian Rheumatologist Psoriatic Arthritis Interest Group Clinician 6
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Abbive | — | X | — | — |
Amgen | X | — | — | — |
BMS | X | — | — | — |
Eli Lilly | X | — | — | — |
Janssen | X | — | — | — |
Novartis | X | — | — | — |
Pfizer | X | — | — | — |
Novartis | X | — | — | — |
References
Coates LC, Gossec L, Theander E, et al. Efficacy and safety of guselkumab in patients with active psoriatic arthritis who are inadequate responders to tumour necrosis factor inhibitors: results through one year of a phase IIIb, randomised, controlled study (COSMOS) Annals of the Rheumatic Diseases 2022;81:359-369.
Coates, L.C., Soriano, E., Corp, N., Bertheussen, H., Callis-Duffin, K., Barbosa Campanholo, C., … Kavanaugh, A. (2021). OP0229 THE GROUP FOR RESEARCH AND ASSESSMENT OF PSORIASIS AND PSORIATIC ARTHRITIS (GRAPPA) TREATMENT RECOMMENDATIONS 2021. Annals of the Rheumatic Diseases, 80(Suppl 1), 139–140. https://doi.org/10.1136/annrheumdis-2021-eular.4091
COSENTYX® (secukinumab) Product Monograph; Updated April 8, 2022. https://pdf.hres.ca/dpd_pm/00065374.PDF
Danese S, Panaccione R, Rubin DT, Sands BE, Reinisch W, D’Haens G, Panés J, Gonzalez S, Weisel K, Sahoo A, Frustaci ME, Yang Z, Sandborn WJ, Afzali A, Hisamatsu T, Andrews JM, Feagan B, on behalf of the GALAXI-1 investigators, OP24 Clinical efficacy and safety of guselkumab maintenance therapy in patients with moderately to severely active Crohn’s Disease: Week 48 analyses from the phase 2 GALAXI 1 study, Journal of Crohn's and Colitis, Volume 16, Issue Supplement_1, January 2022, Pages i026–i027, https://doi.org/10.1093/ecco-jcc/jjab232.023
Dignass A, Rubin D, Bressler B, Huang KH, Shipitofsky N, Germinaro M, Zhang H, Johanns J, Feagan B, Sandborn W, Sands B, Hisamatsu T, Lichtenstein G, Panes J, Allegretti J, QUASAR Investigators, OP23 The efficacy and safety of guselkumab induction therapy in patients with moderately to severely active Ulcerative Colitis: Phase 2b QUASAR Study results through week 12, Journal of Crohn's and Colitis, Volume 16, Issue Supplement_1, January 2022, Pages i025–i026, https://doi.org/10.1093/ecco-jcc/jjab232.022
Gudu T, Gossec L. Quality of life in psoriatic arthritis. Expert Rev Clin Immunol. 2018 May;14(5):405-417.
HUMIRA® (adalimumab) Product Monograph; Updted April 21, 2021. https://www.abbvie.ca/content/dam/abbvie-dotcom/ca/en/documents/products/HUMIRA_PM_EN.pdf
Jin Y, Lee H, Lee MP, Landon JE, Merola JF, Desai RJ, Kim SC. Risk of Hospitalized Serious Infection After Initiating Ustekinumab or Other Biologics for Psoriasis or Psoriatic Arthritis. Arthritis Care Res (Hoboken). 2021 May 10. doi: 10.1002/acr.24630. Epub ahead of print. PMID: 33973371.
Li X, Andersen KM, Chang HY, Curtis JR, Alexander GC. Comparative risk of serious infections among real-world users of biologics for psoriasis or psoriatic arthritis. Ann Rheum Dis. 2020 Feb;79(2):285-291. doi: 10.1136/annrheumdis-2019-216102. Epub 2019 Oct 31. PMID: 31672774; PMCID: PMC6989349.
McGonagle D, McInnes IB, Deodhar A, Schett G, Shawi M, Kafka S, Karyekar CS, Kollmeier AP, Hsia EC, Xu XL, Sheng S, Agarwal P, Zhou B, Ritchlin CT, Rahman P, Mease PJ. Resolution of enthesitis by guselkumab and relationships to disease burden: 1-year results of two phase 3 psoriatic arthritis studies. Rheumatology (Oxford). 2021 Nov 3;60(11):5337-5350. doi: 10.1093/rheumatology/keab285. PMID: 33822898; PMCID: PMC8566200.
McInnes IB, Rahman P, Gottlieb AB, Hsia EC, Kollmeier AP, Xu XL, Jiang Y, Sheng S, Shawi M, Chakravarty SD, van der Heijde D, Mease PJ. Long-Term Efficacy and Safety of Guselkumab, a Monoclonal Antibody Specific to the p19 Subunit of Interleukin-23, Through Two Years: Results From a Phase III, Randomized, Double-Blind, Placebo-Controlled Study Conducted in Biologic-Naive Patients With Active Psoriatic Arthritis. Arthritis Rheumatol. 2022 Mar;74(3):475-485. doi: 10.1002/art.42010. Epub 2022 Feb 7. PMID: 34719872.
Mease, PJ, Helliwell, PS, Gladman, DD et al. Efficacy of guselkumab on axial involvement in patients with active psoriatic arthritis and sacroiliitis: a post-hoc analysis of the phase 3 DISCOVER-1 and DISCOVER-2 studies. The Lancet Rheumatology, 2021; 3 (10).
Nash P, Tam L, Tsai W, Leung Y, Furtner D, Sheng S, Wang Y, Shawi M, Kollmeier A, Sherlock J, Cua D. Guselkumab (TREMFYA®) Provides Consistent and Durable Pain Improvement in Patients with Active Psoriatic Arthritis: Results of 2 Phase 3, Randomized, Controlled Clinical Trials [abstract]. Arthritis Rheumatol. 2021; 73 (suppl 10).
Ogdie A, Hur P, Liu M, Rebello S, McLean RR, Dube B, Glynn M, Mease PJ. Effect of Multidomain Disease Presentations on Patients With Psoriatic Arthritis in the Corrona Psoriatic Arthritis/Spondyloarthritis Registry. J Rheumatol. 2021 May;48(5):698-706. doi: 10.3899/jrheum.200371. Epub 2020 Oct 1. PMID: 33004532.
Rahman P, Ritchlin CT, Helliwell PS, Boehncke WH, Mease PJ, Gottlieb AB, Kafka S, Kollmeier AP, Hsia EC, Xu XL, Shawi M, Sheng S, Agarwal P, Zhou B, Ramachandran P, Zhuang Y, McInnes IB. Pooled Safety Results Through 1 Year of 2 Phase III Trials of Guselkumab in Patients With Psoriatic Arthritis. J Rheumatol. 2021a Dec;48(12):1815-1823. doi: 10.3899/jrheum.201532. Epub 2021 May 1. PMID: 33934076.
Rahman P, Mease PJ, Helliwell PS, Deodhar A, Gossec L, Kavanaugh A, Kollmeier AP, Hsia EC, Zhou B, Lin X, Shawi M, Karyekar CS, Han C. Guselkumab demonstrated an independent treatment effect in reducing fatigue after adjustment for clinical response-results from two phase 3 clinical trials of 1120 patients with active psoriatic arthritis. Arthritis Res Ther. 2021b Jul 14;23(1):
Reygaerts T, Mitrovic S, Fautrel B, Gossec L. Effect of biologics on fatigue in psoriatic arthritis: A systematic literature review with meta-analysis. Joint Bone Spine. 2018 Jul;85(4):405-410.
Ritchlin CT, Helliwell PS, Boehncke WH, Soriano ER, Hsia EC, Kollmeier AP, Chakravarty SD, Zazzetti F, Subramanian RA, Xu XL, Zuraw QC, Sheng S, Jiang Y, Agarwal P, Zhou B, Zhuang Y, Shawi M, Karyekar CS, Deodhar A. Guselkumab, an inhibitor of the IL-23p19 subunit, provides sustained improvement in signs and symptoms of active psoriatic arthritis: 1 year results of a phase III randomised study of patients who were biologic-naïve or TNFα inhibitor-experienced. RMD Open. 2021 Feb;7(1):e001457.
Ritchlin CT, Mease PJ, Boehncke W, et al. Sustained and improved guselkumab response in patients with active psoriatic arthritis regardless of baseline demographic and disease characteristics: pooled results through week 52 of two phase III, randomised, placebo-controlled studies. RMD Open 2022;8:e002195. doi: 10.1136/rmdopen-2022-002195
Sandborn WJ, D'Haens GR, Reinisch W, Panés J, Chan D, Gonzalez S, Weisel K, Germinaro M, Frustaci ME, Yang Z, Adedokun OJ, Han C, Panaccione R, Hisamatsu T, Danese S, Rubin DT, Sands BE, Afzali A, Andrews JM, Feagan BG; GALAXI-1 Investigators. Guselkumab for the Treatment of Crohn's Disease: Induction Results From the Phase 2 GALAXI-1 Study. Gastroenterology. 2022 May;162(6):1650-1664.e8. doi: 10.1053/j.gastro.2022.01.047. Epub 2022 Feb 5. PMID: 35134323.
Tremfya® (guselkumab) Product Monograph; Updated April 13, 2022. https://pdf.hres.ca/dpd_pm/00065455.PDF
XELJANZ® (tofacitinib) Product Monograph; Updated May 9, 2022. https://pdf.hres.ca/dpd_pm/00065725.PDF
Appendix
ACR criteria: Improvement is defined by at least 20% improvement in TJC and in SJC, and at least 20% improvement in 3 of the 5 measures: ESR or CRP PhGA of disease activity PtGA of disease activity Patient assessment of pain Disability:
Fransen J, Antoni C, Mease PJ, et al. Performance of response criteria for assessing peripheral arthritis in patients with psoriatic arthritis: analysis of data from randomised controlled trials of two tumour necrosis factor inhibitors. Ann Rheum Dis. 2006;65(10):1373-1378. doi:10.1136/ard.2006.051706
MDA: MDA was developed based on the PsA core set of outcomes, a patient achieves MDA when 5 of the following 7 criteria are met: tender joint count ≤ 1; swollen joint count ≤ 1; Psoriasis Area and Severity Index ≤ 1 or body surface area ≤ 3%; patient pain visual analog score (VAS) ≤ 15; patient global disease activity VAS ≤ 20; Health Assessment Questionnaire (HAQ) Disability Index ≤ 0.5; tender entheseal points ≤ 1.
Coates LC, Fransen J, Helliwell PS. Defining minimal disease activity in psoriatic arthritis: a proposed objective target for treatment. Ann Rheum Dis 2010;69:48–53
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines.
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.