CADTH Reimbursement Review
Tezepelumab (Tezspire)
Sponsor: AstraZeneca Canada Inc.
Therapeutic area: Asthma
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
AAER
annualized asthma exacerbation rate
ACQ
Asthma Control Questionnaire
ACQ-6
6-item Asthma Control Questionnaire
AE
adverse event
AQLQ(S)12+
Asthma Quality of Life Questionnaire (Standardized) for patients 12 years of age and older
CI
confidence interval
CIC
Clinical Investigator Collaborative
CrI
credible interval
DSU
Decision Support Unit
ED
emergency department
EQ-5D-5L
5-Level EQ-5D
EQ VAS
EQ-5D visual analogue scale
FAS
full analysis set
FeNO
fractional exhaled nitric oxide
GLM
generalized linear model
FEV1
forced expiratory volume in the first second
HRQoL
health-related quality of life
ICS
inhaled corticosteroids
IgE
immunoglobulin E
IL
interleukin
IPD
individual patient-level data
ITC
indirect treatment comparison
ITT
intention-to-treat
LABA
long-acting beta2-agonist
LAMA
long-acting muscarinic antagonist
LSM
least squares mean
LTE
long-term extension
LTRA
leukotriene receptor antagonist
MAIC
matching adjusted indirect comparison
MAR
missing at random
MCMC
Markov chain Monte Carlo
NICE
National Institute for Health and Care Excellence
MID
minimal important difference
NMA
network meta-analysis
NR
not reported
OCS
oral corticosteroids
OR
odds ratio
PEF
peak expiratory flow
RCT
randomized controlled trial
RR
relative risk
SABA
short-acting beta2-agonist
SAE
serious adverse event
SD
standard deviation
SE
standard error
STC
simulated treatment comparison
T2
type 2
Th2
T-helper 2
TSLP
thymic stromal lymphopoietin
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Tezepelumab (Tezspire), 210 mg, 110 mg/mL solution for subcutaneous injection |
Indication | Proposed: indicated as an add-on maintenance treatment in adults and adolescents 12 years and older with severe asthma |
Reimbursement request | As per indication |
Health Canada approval status | Under review (pre-NOC) |
Health Canada review pathway | Standard review |
NOC date | To be determined |
Sponsor | AstraZeneca Canada Inc. |
NOC = Notice of Compliance.
Introduction
Asthma is a chronic respiratory disorder characterized by reversible airway obstruction. Hallmarks of asthma include inflammation, bronchoconstriction, and airway remodelling, as well as hyper-responsive airways and mucous production.1 Symptoms of asthma include wheezing, dyspnea, chest tightness, sputum production, and coughing, all of which can be exacerbated by exogenous influences such as allergens, upper respiratory tract infections, or environmental factors such as smoke or cold air.1 Eosinophils are believed to be a major contributor to the inflammatory processes that are characteristic of the disease, according to the clinical expert consulted by CADTH for this review. An estimated 2.4 million Canadians 12 years or older suffer from asthma, representing 12% of all children and 8% of adults.2
The management of mild asthma is carried out using “relievers” such as short-acting beta2-agonists (SABAs) or rapid-acting beta2-agonists such as formoterol, combined with controllers such as inhaled corticosteroids (ICS) on an as-needed basis.1 Alternatively, regular, daily treatment with low-dose ICS is used.1 If regular low-dose ICS do not achieve good asthma control then treatment is typically escalated to the use of long-acting bronchodilators, most commonly long-acting beta2-agonists (LABAs), always in combination with ICS.1 Oral corticosteroids (OCS) are used for acute exacerbations on a short-term basis in “bursts,” although some patients’ asthma can be severe enough to require OCS on an ongoing basis, according to the clinical expert consulted by CADTH for this review. According to the expert, the approach to managing asthma has evolved, such that patients are now routinely grouped into those who have type 2 inflammation and those who do not. Type 2 inflammation is mediated, in part, by cytokines such as interleukin (IL)-4, IL-5, and IL-13, and this explains why this phenotype may be more responsive to the biologics that target these cytokines, according to the clinical expert. Monoclonal antibodies are the newest entrants into the asthma treatment paradigm, beginning with an immunoglobin E (IgE) inhibitor (omalizumab) and, more recently, IL-5 inhibitors (mepolizumab, reslizumab, and benralizumab), an inhibitor of IL-4 and IL-13 (dupilumab), and now a thymic stromal lymphopoietin (TSLP) inhibitor, tezepelumab. According to the clinical expert consulted by CADTH for this review, none of the monoclonal antibodies are intended to be used in the first line but are reserved instead for those patients whose asthma is not well controlled with high doses of ICS plus a LABA.
As TSLP is a cytokine found near the beginning of the inflammatory cascade, it is therefore thought that blockading TSLP may have a broad effect on many mediators that play a role in the pathophysiology of various asthma phenotypes, including eosinophils, IgE, and a variety of ILs.3 Tezepelumab is administered by subcutaneous injection at a dose of 210 mg every 4 weeks. Its proposed indication is as an add-on maintenance treatment in adults and adolescents 12 years and older with severe asthma, and the sponsor’s reimbursement request is the same as the proposed indication. At the time of writing this report, tezepelumab was being reviewed by Health Canada under the regular review process, and it has been approved by the FDA.
The objective of this report was to perform a systematic review of the beneficial and harmful effects of tezepelumab injections as an add-on maintenance treatment in adults and adolescents 12 years and older with severe asthma.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient groups who responded to CADTH’s call for patient input and from a clinical expert consulted by CADTH for the purpose of this review.
Patient Input
Patient input for this review was received from Asthma Canada and the Lung Health Foundation (formally known as the Ontario Lung Association). Patients reported that living with asthma negatively affected their psychological and social well-being and results in poor quality of life. The patients indicated that their asthma affects their ability to complete daily activities, attend school or work, and participate in outdoor and/or physical activity, and interferes with social interactions. Patients also reported loss of productivity at school or work due to their asthma, leading to a decrease in performance or quality of work and/or schoolwork. Parents and caregivers expressed concern about accessing adequate and necessary medical care as severe exacerbations can cause loss of consciousness or hypoxia, in addition to urgent emergency department (ED) care to restore airway functions. Patients, parents, and caregivers noted that there was an unmet need for treatment options for severe asthma. Even with currently available treatment, 1 in 4 respondents indicated that they have poor symptom control. Patients, parents, and caregivers noted several barriers to accessing health care providers (e.g., respirologists and specialized asthma clinics) including travel time and cost, missed school or work, and the financial burden of prescription refills. Patients reported that the long-term use of OCS, while providing some degree of inflammation control after failing other options, is associated with notable side effects, which were reportedly a considerable source of distress. Patients and caregivers identified the following treatment priorities: the ability to control their day-to-day symptoms, the ability to control exacerbation, reduction of cost or coverage for current and upcoming treatments, and reduction in medication-associated side effects. Other key treatment outcomes highlighted by patients included improvement in quality of life, reduction in the number of medications required to take to maintain asthma control, and treatments with minimal side effects.
Clinician Input
Input From the Clinical Expert Consulted by CADTH
According to the clinical expert consulted by CADTH for this review, half of all patients with type 2 asthma remain poorly controlled with existing nonbiologic interventions. Although various measures, including improved adherence to therapy, can improve control in these patients, a subset of approximately 5% of patients remain poorly controlled no matter what the intervention. The minority of patients who have non–type 2 asthma are unlikely to benefit from current biologics.
According to the clinical expert, tezepelumab can treat patients with type 2 and non–type 2 asthma, and no other biologics are available that can reduce exacerbation frequency in patients with non–type 2 disease. The expert stated that tezepelumab could be used in the first line in patients with type 2 asthma or in patients who failed to improve on other biologics, as airway inflammation in type 2 asthma may be driven by factors other than those targeted by current biologics.
As the definition of severe asthma can vary, an indication for use in severe asthma is imprecise, according to the clinical expert, although there is no evidence of efficacy in patients who are OCS-dependent. The subset of patients who are OCS-dependent likely only represents a relatively small segment of patients with asthma in Canada.
According to the clinical expert consulted by CADTH, relevant outcomes to assess treatment response include improved health-related quality of life (HRQoL), decreased frequency of exacerbations, and improved asthma control, which would include improvement in or stabilization of forced expiratory volume in the first second (FEV1), elimination of airflow reversibility to a bronchodilator, and reduction in symptoms. Once a patient meets the criteria for initiation, clear stopping rules will be difficult to develop, as asthma control can be affected by environmental factors. The decision to initiate administration of the drug should be limited to respirologists or allergists with experience using biologics, and, once started, tezepelumab could be maintained by a generalist.
Clinician Group Input
Input was received from 6 clinicians on behalf of the AllerGen Clinical Investigator Collaborative (CIC). The clinician group indicated that, while some treatments are effective for patients with severe eosinophilic type 2 (T2)-high asthma, no effective treatment options are available for those patients who have severe asthma that is not persistently T2-high asthma. Members of the CIC agreed that the use of tezepelumab in asthma should be restricted to patients with severe asthma, regardless of their eosinophilic asthma status. According to the CIC, severe exacerbation risk remains the single most important outcome to improve in severe asthma. The CIC suggested that tezepelumab should be discontinued if patients continue to experience severe exacerbations while on treatment. The only other reason cited for discontinuation by the CIC was side effects.
Drug Program Input
In response to a question from the drug plans regarding whether tezepelumab should be restricted based on phenotyping and/or biomarkers, the clinical expert consulted by CADTH stated that this is likely unnecessary as tezepelumab appears to have efficacy across all phenotypes. The drug plans asked about alignment with initiation criteria for other biologics, and the clinical expert confirmed that: 1) patients should have a documented diagnosis of asthma; 2) the patient should be inadequately controlled with high-dose ICS and 1 or more additional controllers; 3) the patient should have experienced 2 or more clinically significant asthma exacerbations in the past year; and 4) the patient should have completed a baseline assessment of asthma symptom control using a validated instrument. The clinical expert also confirmed that, in their opinion, the renewal criteria tezepelumab should be aligned with the renewal criteria for other biologics for severe asthma, and that there is no evidence to suggest tezepelumab should be combined with any other biologics. The clinical expert also noted that, with respect to prescribing criteria, tezepelumab should be restricted to respirologists and allergists for initiation of therapy; however, family physicians should be able to maintain treatment once initiated.
Clinical Evidence
Pivotal Studies and Protocol-Selected Studies
Description of Studies
Three multinational, sponsor-funded, double-blind randomized controlled trials (RCTs) were included in this systematic review. The NAVIGATOR study randomized 1,061 patients who were on medium- or high-dose ICS and who had 2 or more exacerbations in the past year at a 1:1 ratio to either tezepelumab or placebo over a treatment course of 52 weeks.4 The primary outcome was the annualized asthma exacerbation rate (AAER) and key secondary outcomes included the AAER in patients with baseline eosinophil counts of less than 300 cells/µL, change from baseline in pre-bronchodilator FEV1, the Asthma Quality of Life Questionnaire Standardized for patients 12 years of age and older (AQLQ[S]12+), and the 6-item Asthma Control Questionnaire (ACQ-6). The SOURCE study randomized 150 patients with OCS-dependent asthma, 1:1, to either tezepelumab or placebo over a treatment course of 48 weeks.5 The primary outcome was the percent reduction in OCS dose while not losing asthma control, and key secondary outcomes included the AAER, time to first asthma exacerbation, rate of asthma exacerbation associated with ED visits, urgent-care visits or hospitalization, and patients who did not experience an asthma exacerbation over 48 weeks. The PATHWAY study was a phase II double-blind RCT that randomized 550 patients on medium- to high-dose ICS and at least 2 exacerbations (or 1 severe asthma exacerbation) in the past year, 1:1:1:1, to 3 different doses of tezepelumab, including the proposed dose in the draft product monograph, or placebo, over a treatment course of 52 weeks.6 Results are reported for the tezepelumab treatment group in the PATHWAY study that received the dose recommended in the draft product monograph (i.e., 210 mg subcutaneously every 4 weeks) only; results from the other tezepelumab arms are not reported in this review. The primary outcome was the AAER, and secondary outcomes included subgroups based on the primary outcome, change from baseline in FEV1, and ACQ-6 score.
Across studies, the mean age of patients was between 49 and 53.5 years, and the majority were female, ranging between 59% and 68% of patients across studies. In the NAVIGATOR study, 62% of patients were White and 28% were Asian, while 84% of patients in the SOURCE study and 91% of patients in the PATHWAY study were White. In the NAVIGATOR study, 60% of patients had 2 exacerbations in the past 12 months and the remainder had more than 2, while in the PATHWAY study 78% of patients had 1 or 2 exacerbations and the remainder had 3 or more. In the SOURCE study, the protocol for which did not require more than 1 exacerbation in the past 12 months, 43% of patients had 1 exacerbation, 35% had 2, and 23% had more than 2 exacerbations. In the NAVIGATOR study, 75% of patients were on high-dose ICS and the remaining were on medium-dose ICS, while in the SOURCE study, all but 1 patient were on high-dose ICS. All patients in the SOURCE study were on OCS at baseline, while 9% were on OCS in the NAVIGATOR study and 8% were on OCS in the PATHWAY study.
Table 2: Summary of Key Results From Pivotal and Protocol-Selected Studies
Outcome | NAVIGATOR | SOURCE | PATHWAY | |||
|---|---|---|---|---|---|---|
Tez N = 529 | Placebo N = 532 | Tez N = 74 | Placebo N = 76 | Tez 210 mg N = 137 | Placebo N = 138 | |
Asthma exacerbations, full analysis set population | ||||||
AAER | ||||||
Number of events, n | 425 | 878 | 78 | 116 | NR | NR |
AAER, 52 weeks (95% CI) | 0.93 (0.80 to 1.07) | 2.10 (1.84 to 2.39) | NR | NR | 0.20 (0.13 to 0.30) | 0.72 (0.59 to 0.88) |
AAER, 48 weeks (95% CI) | NR | NR | 1.38 (0.98 to 1.95) | 2.00 (1.46 to 2.74) | NR | NR |
Rate ratio (95% CI) | 0.44 (0.37 to 0.53)a | 0.69 (0.44 to 1.09)b | 0.29 (0.16 to 0.51)c | |||
P value | < 0.001 | 0.111d | < 0.001d | |||
AAER associated with emergency department visit or hospitalization | ||||||
Number of events, n | 30 | 115 | 8 | 19 | NR | NR |
Rate (95% CI) | 0.06 (0.04 to 0.09) | 0.28 (0.20 to 0.39) | 0.16 (0.06 to 0.44) | 0.28 (0.13 to 0.58) | NR | NR |
Rate ratio (95% CI) | 0.21 (0.12 to 0.37)a | 0.59 (0.19 to 1.82)b | NR | |||
P value | < 0.001d | 0.361d | NR | |||
AAER associated with hospitalization | ||||||
Number of events, n | 14 | 78 | NR | NR | NR | NR |
Rate (95% CI) | 0.03 (0.01 to 0.06) | 0.19 (0.12 to 0.30) | NR | NR | 0.02 (0.00 to 0.07) | 0.14 (0.08 to 0.22) |
Rate ratio (95% CI) | 0.15 (0.07 to 0.33)a | NR | 0.14 (0.03 to 0.71)a | |||
P value | < 0.001d | NR | 0.017d | |||
Pulmonary function, full analysis set population | ||||||
Pre-BD FEV1, L | ||||||
Baseline, N | 528 | 531 | 74 | 76 | 137 | 138 |
Mean (SD) | 1.83 (0.72) | 1.85 (0.71) | 1.56 (0.50) | 1.59 (0.64) | 1.83 (0.58) | 1.82 (0.59) |
Change from baseline, N | 471 | 453 | 65 | 64 | 121 | 131 |
LSM (SE) CFB | 0.23 (0.018) | 0.10 (0.018) | 0.21 (0.046) | −0.04 (0.046) | 0.08 (NR) | 0.10 (NR) |
LSM difference (95% CI) | 0.13 (0.08 to 0.18)e | 0.26 (0.13 to 0.39)e | 0.13 (0.03 to 0.23)e | |||
P value | < 0.001 | < 0.001d | 0.009d | |||
Reduction in OCS, full analysis set population | ||||||
Patients achieving reduction in OCS dose, n (%) | ||||||
≥ 90% to ≤ 100% | NR | NR | 40 (54) | 35 (46) | NR | NR |
≥ 75% to < 90% | NR | NR | 5 (7) | 4 (5) | NR | NR |
≥ 50% to < 75% reduction | NR | NR | 10 (14) | 14 (18) | NR | NR |
> 0% to < 50% | NR | NR | 5 (7) | 9 (12) | NR | NR |
No change or increase | NR | NR | 14 (19) | 14 (18) | NR | NR |
Cumulative OR (95% CI) | NR | 1.28 (0.69 to 2.35)f | NR | |||
P value | NR | 0.434 | NR | |||
Reduction in rescue medication use, full analysis set population | ||||||
Daily rescue medication use, puffs/day | ||||||
Baseline, N | 528 | 531 | 74 | 76 | NR | NR |
Mean (SD) baseline | 4.36 (5.20) | 4.35 (5.09) | 3.11 (3.30) | 3.16 (3.03) | NR | NR |
Change from baseline, N | 439 | 428 | 60 | 70 | NR | NR |
Mean (SD) CFB, week 52 | −2.44 (4.21) | −2.49 (4.02) | −0.92 (2.83) | −0.43 (2.99) | NR | NR |
LSM CFB (SE) | −2.53 (0.137) | −2.36 (0.137) | −0.85 (0.280) | −0.37 (0.268) | NR | NR |
LSM difference (95% CI) | −0.17 (−0.55 to 0.21)g | −0.47 (−1.24 to 0.29)g | NR | |||
P value | 0.382d | 0.22d | NR | |||
Health-related quality of life, full analysis set population | ||||||
AQLQ(S)12+ total | ||||||
Baseline, n | 527 | 529 | 74 | 74 | 123 | 121 |
Mean (SD) baseline | 3.87 (1.02) | 3.90 (1.00) | 4.14 (1.18) | 4.11 (1.02) | 4.20 (0.91) | 4.09 (0.87) |
Change from baseline, n | 480 | 467 | 66 | 67 | 41j | 47j |
Mean (SD) CFB | 1.48 (1.26) | 1.16 (1.17) | 0.96 (1.17) | 0.59 (1.01) | NR | NR |
LS mean (SE), CFB | NR | NR | 0.94 (0.124) | 0.58 (0.123) | 1.17 | 0.97 |
Difference vs. placebo (95% CI) | 0.33 (0.20 to 0.47)h | 0.36 (0.01 to 0.70)h | 0.20 (−0.09 to 0.48)h | |||
P value | 0.001 | 0.042d | 0.185d | |||
Symptoms | ||||||
ACQ-6, total score | ||||||
Baseline, n | 528 | 531 | 74 | 76 | 137 | 138 |
Mean (SD) baseline | 2.82 (0.81) | 2.79 (0.82) | 2.48 (1.07) | 2.46 (1.03) | 2.70 (0.80) | 2.66 (0.69) |
Change from baseline | ||||||
n | 486 | 472 | 66 | 68 | 44j | 53j |
Mean (SD) CFB, week 52 | −1.55 (1.15) | −1.24 (1.10) | −0.93 (1.25) | −0.52 (1.02) | NR | NR |
LSM CFB week 52 | −1.53 (0.045) | −1.20 (0.046) | NR | NR | −1.20 | −0.91 |
LSM CFB week 48 | NR | NR | −0.87 (0.125) | −0.51 (0.123) | NR | NR |
Difference vs placebo (95% CI) | −0.33 (−0.46 to −0.20)i | −0.37 (−0.71 to −0.02)i | −0.29 (−0.56 to −0.01)i | |||
P value | < 0.001 | 0.038d | 0.039d | |||
HARMS, safety analysis set | ||||||
AEs, patients, n (%) | 407 (77) | 422 (80) | 53 (72) | 65 (86) | 90 (66) | 91 (66) |
SAEs, patients, n (%) | 46 (9) | 70 (13) | 11 (15) | 16 (21) | 13 (10) | 18 (13) |
Treatment WDAEs | 11 (2) | 19 (4) | 2 (3) | 2 (3) | 2 (1) | 1 (1) |
AAER = annualized asthma exacerbation rate; ACQ-6 = 6-item Asthma Control Questionnaire; AE = adverse event; AQLQ(S)12+ = Asthma Quality of Life Questionnaire (Standardized) for patients 12 years of age and older; BD = bronchodilator; CFB = change from baseline; FEV1 = forced expiratory volume in the first second; ICS = inhaled corticosteroids; LSM = least squares mean; NR = not reported; OCS = oral corticosteroids; OR = odds ratio; SAE = serious adverse events; SD = standard deviation; SE = standard error; Tez = tezepelumab; WDAE = withdrawal due to adverse event; vs. = versus.
aModel: a negative binomial regression analysis with treatment, region, age group, and history of exacerbations as covariates. The logarithm of the time at risk is used as an offset variable. Annual exacerbation rates displayed are estimated marginal rates from the model. Absolute difference is the difference between the marginal rates. Confidence intervals for annual exacerbation rates and absolute differences are estimated via the delta method.
bModel: a negative binomial regression analysis with treatment, region, and history of exacerbations as covariates. The logarithm of the time at risk is used as an offset variable. Annual exacerbation rates displayed are estimated marginal rates from the model. Confidence intervals for annual exacerbation rates and absolute differences are estimated via the delta method.
cRate ratio, and 95% CI for the rate ratio were estimated from negative binomial regression with treatment group, and the stratification factors- baseline blood eosinophil count (≥ or < 250 cells/μL) and baseline ICS dose level (medium or high) as the covariates.
dP values are not controlled for multiplicity or failed in the hierarchy and should be considered supportive in nature.
eEstimate of the mean change from baseline at each week in tezepelumab is compared to placebo using a repeated measures analysis. Estimates are LSMs. The model with unstructured covariance structure is: change from baseline in FEV1 = treatment group + region + age + baseline FEV1 + visit + treatment × visit.
fThe estimate of the cumulative odds ratio is obtained using a proportional odds model with treatment, region, and daily OCS dose at baseline as covariates
gEstimate of the mean change from baseline at each week in tezepelumab is compared to the placebo using a repeated measures analysis. Estimates are least squares means. The model with Unstructured covariance structure is: Change from baseline in Rescue medication use weekly means = Treatment group + region + age + baseline rescue medication use + week + treatment * week.
hEstimate of the mean change from baseline at each week in tezepelumab is compared to placebo using a repeated measures analysis. Estimates are LSMs. The model with unstructured covariance structure is: change from baseline in AQLQ(S) + 12 = treatment group + region + baseline AQLQ(S) + 12 + visit + treatment × visit .
iEstimate of the mean change from baseline at each week in tezepelumab is compared to placebo using a repeated measures analysis. Estimates are LSMs. The model with unstructured covariance structure is: change from baseline in ACQ-6 = treatment group + region + baseline ACQ-6 + visit + treatment × visit.
jSmall sample sizes for these outcomes in the PATHWAY study were due to an error in collecting electronic patient-reported outcome data.
Source: Clinical Study Report for NAVIGATOR,4 SOURCE,5 and PATHWAY.6
Efficacy Results
Mortality
Across all studies, only 1 death in the tezepelumab group and 2 deaths in the placebo group were reported. The 2 deaths in the placebo group were due to an unknown cause and heart failure, both in the NAVIGATOR study, and 1 patient in the tezepelumab group in the SOURCE study died due to cardiac arrest.
Acute Asthma Exacerbation
The AAER was the primary outcome of both the NAVIGATOR and PATHWAY studies. All results reported for the PATHWAY study included only the proposed Health Canada dosing. In the NAVIGATOR study the AAER over 52 weeks was 0.93 (95% confidence interval [CI], 0.80 to 1.07) with tezepelumab and 2.10 (95% CI, 1.84 to 2.39) with placebo, for a rate ratio of 0.44 (95% CI, 0.37 to 0.53; P < 0.001). In the PATHWAY study, the AAER over 52 weeks was 0.20 (95% CI, 0.13 to 0.30) with tezepelumab and 0.72 (95% CI, 0.59 to 0.88) with placebo, for a rate ratio of 0.29 (95% CI, 0.16 to 0.51; P < 0.001). In the SOURCE study, the rate ratio for AAER over 48 weeks was 0.69 (95% CI, 0.44 to 1.09).
The AAER associated with an ED visit or hospitalization was 0.06 (95% CI, 0.04 to 0.09) with tezepelumab and 0.28 (95% CI, 0.20 to 0.39) with placebo, for a rate ratio of 0.21 (95% CI, 0.12 to 0.37) in the NAVIGATOR study, and 0.16 (95% CI, 0.06 to 0.44) with tezepelumab and 0.28 (95% CI, 0.13 to 0.58) with placebo in the SOURCE study, for a rate ratio of 0.59 (95% CI, 0.19 to 1.82). The AAER associated with hospitalization was 0.03 (95% CI, 0.01 to 0.06) with tezepelumab and 0.19 (95% CI, 0.12 to 0.30) with placebo, for a rate ratio of 0.15 (95% CI, 0.07 to 0.33; P < 0.001) in the NAVIGATOR study, and 0.02 (95% CI, 0.00 to 0.07) with tezepelumab and 0.14 (95% CI, 0.08 to 0.22) with placebo, for a rate ratio of 0.14 (95% CI, 0.03 to 0.71) in the PATHWAY study.
Change in Pulmonary Function
The pre-bronchodilator FEV1 increased in both the tezepelumab and placebo groups in the NAVIGATOR study, with a least squares mean (LSM) change from baseline to 52 weeks of 0.23 L (standard error [SE] = 0.018) with tezepelumab and 0.10 L (SE = 0.018) with placebo and a LSM difference between groups of 0.13 L (95% CI, 0.08 to 0.18; P < 0.001). In the SOURCE study, the change from baseline to week 48 was 0.21 L (SE = 0.046) with tezepelumab and −0.04 L (SE = 0.046) with placebo, for a LSM difference between groups of 0.26 litres (95% CI, 0.13 to 0.39), and in the PATHWAY study, the LSM change from baseline to week 52 was 0.076 L with tezepelumab and −0.056 L with placebo, for a LSM difference between groups of 0.132 (95% CI, 0.033 to 0.231).
Reduction in Oral Corticosteroids
Reduction in OCS use was the primary outcome of SOURCE. The cumulative odds ratio (OR) for patients having a reduction in OCS dose was 1.28 (95% CI, 0.69 to 2.35; P = −0.434). Tezepelumab therefore failed to demonstrate superiority over placebo for the primary outcome of this study.
Reduction in Use of Rescue Medication
Reduction in daily rescue medication was observed in both the tezepelumab and placebo groups in the NAVIGATOR study, an LSM change from baseline of −2.53 puffs (SE = 0.137) with tezepelumab and −2.36 puffs (0.137) with placebo, for an LSM difference between groups of −0.17 (95% CI, −0.55 to 0.21). Rescue medication use also declined in both groups in the SOURCE study, with an LSM change from baseline to 48 weeks of −0.85 puffs (SE = 0.280) with tezepelumab and −0.37 puffs (SE = 0.268) with placebo, for an LSM difference between groups of −0.47 (95% CI, −1.24 to 0.29).
Health-Related Quality of Life
Mean AQLQ(S)12+ scores increased (improved) from baseline to 52 weeks in the NAVIGATOR study, both in the tezepelumab group, at 1.48 (SD = 1.26) and the placebo group, at 1.16 (SD = 1.17), with a difference between groups of 0.33 (95% CI, 0.20 to 0.47; P = 0.001). In the SOURCE study, the LSM change from baseline to week 48 was 0.94 (SD = 0.124) with tezepelumab and 0.58 (SD = 0.123) with placebo, for a difference between groups of 0.36 (95% CI, 0.01 to 0.70). In the PATHWAY study, the LSM change from baseline to week 52 was 1.17 (SE = not reported [NR]) with tezepelumab and 0.97 (SE = NR) with placebo for a difference between groups of 0.20 (95% CI, −0.09 to 0.48). Responders to the AQLQ(S)12+ were also reported, defined as those with a change from baseline of 0.5 or greater. In the NAVIGATOR study, 78% of tezepelumab patients and 72% of placebo patients were responders, for an OR of 1.36 (95% CI, 1.02 to 1.82). In the SOURCE study, 62% of patients in the tezepelumab group and 52% of the placebo group were responders, for an OR of 1.66 (95% CI, 0.81 to 3.43). In the PATHWAY study, 73% of patients treated with tezepelumab and 62% of those given a placebo were responders.
Symptoms
Symptoms were assessed using the ACQ-6. In the NAVIGATOR study, ACQ-6 scores decreased (improved) from baseline to week 52 in both the tezepelumab and placebo groups, for a difference versus placebo of 0.33 (95% CI, 0.20 to 0.47; P < 0.001). Responders to the ACQ-6 were also reported in the NAVIGATOR study, defined as those with a change from baseline of 0.5 or greater. In the NAVIGATOR study, 86% of tezepelumab patients and 77% of placebo patients were responders, for an OR of 1.99 (95% CI, 1.43 to 2.76). In the SOURCE study, the LSM change from baseline to 48 weeks for tezepelumab was −0.87 (SE = 0.125) and −0.51 (SE = 0.123) with placebo, for a difference between groups of −0.37 (95% CI, −0.71 to −0.02). In the PATHWAY study, the LSM change from baseline to week 52 was −1.20 (SE = NR) with tezepelumab and −0.91 (SE = NR) with placebo, for a difference between groups of −0.29 (95% CI, −0.56 to −0.01).
Harms Results
Adverse events (AEs) in the tezepelumab versus placebo groups occurred in 77% versus 80% of patients, respectively, in NAVIGATOR; 72% versus 86%, respectively, in the SOURCE study; and 66% of patients in each group in the PATHWAY study.
The most common AE was nasopharyngitis, occurring in 21% versus 21% of patients in the NAVIGATOR study, 15% versus 25% of patients in the SOURCE study, and 14% versus 12% of patients in the PATHWAY study in the tezepelumab versus placebo groups, respectively. Other common events (occurring in 10% or more of patients in any group of any study) were upper respiratory tract infections, headaches, and asthma.
Serious adverse events (SAEs) in the NAVIGATOR study for the tezepelumab versus placebo groups occurred in 9% versus 13% of patients respectively; in the SOURCE study they occurred in 15% versus 21% of patients, respectively, and in the PATHWAY study they occurred in 10% versus 13% of patients, respectively. The most common SAE was asthma.
Adverse events resulting in discontinuation of the study drug occurred in 2% versus 4% of patients in the NAVIGATOR study, 3% in each group in the SOURCE study, and 2% versus 1% of patients in the PATHWAY study in the tezepelumab versus placebo groups, respectively.
Notable harms in the CADTH systematic review protocol included infections. Severe infections occurred in the tezepelumab versus placebo groups in 9% versus 8% of patients in the NAVIGATOR study, respectively, and 5% versus 9% of patients in the SOURCE study, respectively. In the PATHWAY study, infections were reported as SAEs rather than severe infections, and these occurred in 1% versus 3% of patients in the tezepelumab versus placebo groups, respectively. No opportunistic infections and no helminth infections were reported across the studies. Injection-site reactions occurred infrequently across the studies. In the NAVIGATOR study, injection-site reactions occurred in 1.5% versus 0.9% of patients in the tezepelumab versus placebo groups, respectively; in the SOURCE study, none occurred with tezepelumab, and 1.3% of patients in the placebo group experienced these events. In the PATHWAY study, these events were reported by injection volume; at the 1 mL volume they occurred in 1.5% versus 2.9% of patients and at the 1.5 mL volume they occurred in 1.5% versus 1.4% of patients in the tezepelumab versus placebo groups, respectively. Hypersensitivity reactions reported as SAEs were infrequent; just 1 patient in each of the tezepelumab and placebo groups in the NAVIGATOR study and none in the other studies reported such a reaction.
Critical Appraisal
Although the NAVIGATOR and SOURCE trials accounted for multiplicity, early failure of the hierarchy in the SOURCE study meant that all of the P values for the key secondary outcomes should only be considered supportive and not suitable for drawing conclusions. Although the number of study withdrawals was generally low (less than 5%) across studies, additional data appeared to be missing for many of the continuous outcomes, including the patient-reported outcomes such as ACQ-6, AQLQ(S)12+, and EQ-5D 5-Levels questionnaire (EQ-5D-5L). Because the missing data also exceed the reported number of treatment withdrawals, it is unclear why the data were missing. In the SOURCE study, fewer patients at baseline had more than 2 asthma exacerbations in the past year in the tezepelumab group compared to the placebo group, and this may have biased the results in favour of tezepelumab if patients in the placebo group were more prone to having an asthma exacerbation.
With respect to external validity, the clinical expert consulted by CADTH for this review noted that 25% of patients in the NAVIGATOR study were on medium-dose ICS, suggesting that these patients may have been undertreated rather than having severe asthma. The clinical expert noted that they would not start a patient on a biologic for asthma until trying high-dose ICS. The lack of an active control, particularly another biologic, in any of the included trials is a limitation, as only indirect comparisons were available to assess the relative efficacy and harms of tezepelumab compared to other biologics.
Indirect Comparisons
Description of Studies
No head-to-head trials comparing the efficacy of tezepelumab with other biologics used to treat patients with severe uncontrolled asthma are currently available. The sponsor submitted 2 indirect treatment comparisons (ITCs), a network meta-analysis (NMA) and a matching adjusted indirect comparison (MAIC)–simulated treatment comparison (STC). Three additional ITCs7-9 were identified in a systematic search of the literature performed by CADTH. Of the sponsor-submitted ITCs, both the NMA and MAIC-STC compared tezepelumab with dupilumab, mepolizumab, benralizumab, omalizumab, and reslizumab for uncontrolled moderate-to-severe asthma in adults and adolescents.10,11 The 3 published ITCs identified by CADTH indirectly compared tezepelumab with dupilumab, benralizumab, mepolizumab, reslizumab, and omalizumab. Data on reslizumab are not reported in this review because reslizumab was not considered a relevant comparator in the CADTH systematic review protocol.
Efficacy Results
In the sponsor-submitted NMA, no differences were identified in terms of reduction of AAER, reduction of hospitalization due to AAER, FEV1 improvement, symptom reduction (change of ACQ-6 score), and an OCS reduction of 50% or greater when comparing tezepelumab with dupilumab, mepolizumab, benralizumab, and omalizumab. The results of the sponsor-submitted MAIC-STC were aligned with that reported in the sponsor’s NMA. Findings from the 3 published ITCs were also aligned with the results reported in the sponsor’s NMA.
Harms Results
Safety outcomes (i.e., any AEs) were assessed in a published ITC by Ando et al. (2022)7 that compared tezepelumab with mepolizumab, benralizumab, and dupilumab. No difference in the risk of AEs was found in this ITC.
Critical Appraisal
Due to the considerable methodological limitations of the ITCs, such as heterogeneity across the included studies and the significantly reduced effective sample size after the match adjustment in the MAIC-STC, as well as the lack of subgroup analysis for the pure severe uncontrolled asthma group, the ITC results are subject to uncertainty. No definitive conclusion can be drawn on the comparative effectiveness and safety profile between tezepelumab and other relevant biologics as an add-on maintenance treatment of adults and adolescents 12 years and older with severe asthma.
Other Relevant Evidence
Description of Studies
The DESTINATION study12 is a phase III, multi-centre, double-blind, randomized, placebo-control, parallel-group, long-term extension (LTE) study for patients who completed the NAVIGATOR4 or SOURCE5 trials. The DESTINATION study was designed to provide evidence of the long-term safety and tolerability of tezepelumab 210 mg administered every 4 weeks subcutaneously in adults and adolescents with severe, uncontrolled asthma for up to 2 continuous years, including 1 year of treatment in the predecessor NAVIGATOR and SOURCE parent studies. Adults (18 to 80 years old) and adolescents (12 to 17 years old) who had continued to receive the investigational product and attended the end-of-treatment visit in 1 of the parent studies were eligible for enrolment. A total of 951 patients were enrolled and randomized to the DESTINATION trial: 827 patients from the NAVIGATOR study and 124 from the SOURCE study. Patients previously randomized to 210 mg tezepelumab in either parent study were assigned to and remained on 210 mg tezepelumab administered every 4 weeks subcutaneously in the DESTINATION study (tezepelumab plus tezepelumab group). Patients previously randomized to the placebo arm in the parent studies were re-randomized in a 1:1 ratio to either 210 tezepelumab (placebo plus tezepelumab group) or matching placebo (placebo plus placebo group) administered every 4 weeks subcutaneously. Patients recruited from the SOURCE study were followed post-treatment for 12 weeks. Patients who enrolled from the NAVIGATOR trial who completed 100 weeks of tezepelumab treatment were eligible for either 12 weeks of follow-up or a 36-week extended follow-up. The primary outcome for the DESTINATION trial was to evaluate the long-term safety of tezepelumab in patients with severe asthma. The secondary outcome was to assess the effect of tezepelumab on the AAER over 104 weeks. This review of the DESTINATION study focused on the results from the tezepelumab plus tezepelumab and placebo plus placebo groups.
Efficacy Results
Asthma Exacerbations
Among patients enrolled in the LTE of the NAVIGATOR study, administration of tezepelumab plus tezepelumab resulted in a reduction in the rate of asthma exacerbation compared to placebo plus placebo (AAER = 0.50; 95% CI, 0.40 to 0.63). Similarly, treatment with tezepelumab plus tezepelumab reduced the rate of asthma exacerbations associated with hospitalization or ED visits compared with placebo plus placebo (AAER = 0.39; 95% CI, 0.22 to 0.69).
In patients enrolled in the LTE from the SOURCE study, the AAER for asthma exacerbations between tezepelumab plus tezepelumab and placebo plus placebo was 0.66 (95% CI, 0.37 to 1.19). For asthma exacerbations associated with hospitalization or ED visits, the AAER for tezepelumab plus tezepelumab versus placebo plus placebo was 0.27 (95% CI, 0.05 to 1.63).
Asthma Control
Improvement from baseline ACQ-6 scores over the LTE study period was observed in the tezepelumab plus tezepelumab group compared to the placebo plus placebo group in patients who were originally enrolled in the NAVIGATOR study (LSM difference = 0.31; 95% CI, −0.47 to −0.14). Similar trends in ACQ-6 scores were observed in patients originally enrolled in the SOURCE study, with the tezepelumab plus tezepelumab group seeing an improvement in ACQ-6 scores over the LTE study period compared to placebo plus placebo group (LSM difference = −0.74; 95% CI; −1.12 to −0.25).
Harms Results
Among patients who entered the DESTINATION study from the NAVIGATOR trial, 66.7% and 71.4% of patients in the tezepelumab plus tezepelumab and the placebo plus placebo groups reported at least 1 AE during the LTE study period, respectively. Among patients who remained on tezepelumab during the LTE period, AEs leading to discontinuation of the investigational product were reported by 4 patients (1%) and AEs leading to death were reported by 7 patients (1.7%). Among those who continued to received placebo in the LTE period, AEs leading to discontinuation of the investigational product were reported by 2 patients (1%) and AEs leading to death were reported by 1 patient (0.5%). Finally, SAEs during the LTE study period were reported in 35 (8.4%) and 22 (10.7%) of patients in the tezepelumab plus tezepelumab and placebo plus placebo groups, respectively. Notable harms of interest reported during the LTE study period included hypersensitivity (0.5% in both the tezepelumab plus tezepelumab group and placebo plus placebo group) and injection-site reactions (0.5% and 1.5% in the tezepelumab plus tezepelumab group and placebo plus placebo group, respectively).
Among patients who entered the DESTINATION study from the SOURCE trial, 71.1% and 68.8% of patients in the tezepelumab plus tezepelumab and placebo plus placebo groups reported at least 1 AE during the LTE study period, respectively. Among the tezepelumab plus tezepelumab group during the LTE period, no AEs led to discontinuation of the investigational product and 1 case (1.7%) involved an AE leading to death. In the placebo plus placebo group in the LTE period, there were no reported AEs leading to discontinuation of the investigational product or death. Finally, SAEs during the LTE study period were reported in 7 patients (11.7%) and 4 patients (12.5%) in the tezepelumab plus tezepelumab and the placebo plus placebo groups, respectively. No notable harms of interest were reported among patients enrolled in the DESTINATION study during the LTE study period.
Critical Appraisal
The DESTINATION trial provided additional data on the long-term efficacy of tezepelumab relative to placebo. Statistical hypothesis-testing was not part of the design. Blinding may have been compromised by accidental publishing of individual test results on the investigator’s portal (November 23, 2021) by the laboratory vendor before the primary database lock, which may have led to unblinding for investigators who may have viewed the data. There were several imbalances between treatment groups among those who enrolled from the SOURCE study. First, fewer patients in the placebo plus placebo group completed the treatment protocol. Second, a greater proportion of patients in the placebo plus placebo group reported use of additional controller medications at baseline. Although the direction of any bias is unclear, it is possible that the differential dropout rate between the 2 treatment groups may have introduced attrition bias in favour of the tezepelumab plus tezepelumab group. Likewise, while the direction of any bias is unclear, it is possible that the differential use of controller medication may have been a surrogate of disease severity and biased the results in favour of the tezepelumab plus tezepelumab group. Overall, the DESTINATION study population represented the population of patients with severe, uncontrolled asthma and severe, OCS-dependent asthma as derived from the parent NAVIGATOR and SOURCE studies, respectively. More than 90% of patient from the parent studies were enrolled in the DESTINATION trial. At LTE baseline, patient characteristics were similar to parent studies’ baseline. Completion of the LTE exceeded 96% across all treatment groups from the NAVIGATOR study. While completion of the LTE was lower among patients who entered the LTE from the SOURCE trial, completion of the LTE remained above 80%. Given that the patients enrolled in the LTE study were originally from the NAVIGATOR and SOURCE parent studies, and the eligibility criteria remained the same, it is reasonable to expect that the same limitations to generalizability are relevant to DESTINATION.
Conclusions
There is evidence that tezepelumab reduces the rate of asthma exacerbations in patients whose asthma remains uncontrolled despite the administration of medium- to high-dose ICS. This reduction in exacerbation risk appears to occur regardless of whether patients have type 2 or non–type 2 asthma. Additionally, tezepelumab appears to improve pulmonary function, as well as HRQoL and symptoms of asthma as measured by the AQLQ(S)12+ and ACQ-6. Data from an LTE suggest these benefits of tezepelumab on exacerbations and symptoms may continue through 2 years of treatment; however, these findings need to be confirmed in a study that formally compares tezepelumab to placebo over this time frame. There is no evidence that tezepelumab facilitates the reduction of OCS doses in patients with OCS-dependent asthma, or reduces exacerbations in these patients. With respect to harms, there are no obvious safety or tolerability issues associated with tezepelumab, and this conclusion takes into account data from an extension with at least 2 years of follow-up. Indirect evidence suggests that the efficacy and harms of tezepelumab are similar to those of other biologics used to treat asthma, although the degree of heterogeneity between the studies included in the indirect comparisons precludes drawing concrete conclusions on the comparative results.
Introduction
Disease Background
Asthma is a chronic respiratory disorder characterized by reversible airway obstruction. Hallmarks of asthma include inflammation, bronchoconstriction, and airway remodelling, as well as hyper-responsive airways and mucous production.1 Symptoms of asthma include wheezing, dyspnea, chest tightness, sputum production, and coughing, and these symptoms can be exacerbated by exogenous influences such as allergens, upper respiratory tract infections, or environmental factors such as smoke or cold air.1 An estimated 2.4 million Canadians 12 years or older, or 12% of all children and 8% of adults, suffer from asthma.2
According to the clinical expert consulted by CADTH for this review, there are several asthma phenotypes, 1 of which is associated with eosinophilic airway inflammation and an increased peripheral blood eosinophil count, and this may persist despite treatment with moderate- to high-dose ICS. Eosinophils promote airway inflammation and contribute to airway hyper-responsiveness and remodelling, among other functions, according to the clinical expert. The clinical expert noted that tissue eosinophilia is present in 40% to 60% of patients with asthma and the use of ICS typically reduces eosinophilic inflammation in these patients; however, a subset of patients (5% to 10% overall, and 50% of patients with severe asthma) continue to experience exacerbations despite treatment with high-dose ICS.
Standards of Therapy
Traditionally, the management of asthma is carried out using medications for the acute relief of exacerbations (colloquially known as “asthma attacks” and often referred to as “relievers” or “rescue medications”) and controllers, or maintenance drugs, which are used on a regular or chronic basis in an effort to prevent the onset of exacerbations.1 According to the clinical expert consulted by CADTH for this review and based on the updated Global Initiative for Asthma guidelines, the pharmacologic management of asthma in Canada has recently evolved.1 In step 1, patients begin using a low-dose ICS whenever a reliever medication is used. As symptoms persist, step 2 involves daily low-dose ICS or ICS plus formoterol on an as-needed basis. From there, patients may need to escalate to regular use of low-dose (step 3) or medium-dose (step 4) ICS plus a LABA.1 Finally, step 5 involves the use of daily high-dose ICS plus a LABA, and if control of asthma is not achieved at that point, additional treatments are considered, such as low-dose OCS, inhaled tiotropium, and/or biologics.1 According to the clinical expert consulted by CADTH for this review, other drugs that may be considered as add-on therapy include leukotriene receptor antagonists (LTRAs) and long-term therapy with macrolides, with the latter considered off-label. Nonpharmacologic therapies include asthma education, improvement of inhaler technique, allergen avoidance, and a written asthma action plan.1 According to the clinical expert consulted by CADTH on this review, the treatment of comorbidities such as tobacco dependence, depression, and obstructive sleep apnea are also important in the management of asthma. With respect to harms associated with pharmacologic therapies, ICSs have short-term side effects such as oral candidiasis (“thrush”) and dysphonia; however, a number of concerning adverse effects, including osteoporosis, are associated with their long-term use, particularly at high doses.13 The use of systemic corticosteroids heightens the risk of harms, and their chronic use is to be avoided, according to the clinical expert.
According to the clinical expert, the approach to managing asthma has also evolved, such that patients are now routinely grouped into those who have type 2 inflammation and those who do not. According to the clinical expert, type 2 inflammation is mediated, in part, by cytokines such as IL-4, IL-5, and IL-13, and this explains why the phenotype may be more responsive to biologics that target this cytokine. Monoclonal antibodies are the newest entrants into the asthma treatment paradigm, with an IgE inhibitor (omalizumab14) being the first drug approved, and, more recently, IL-5 inhibitors (mepolizumab,15 reslizumab,16 and benralizumab17), an IL-4 and IL-13 inhibitor (dupilumab18), and now a TSLP inhibitor, tezepelumab. According to the clinical expert, none of the monoclonal antibodies are intended to be used in the first line but are reserved instead for those patients whose asthma is not well controlled with high doses of ICS plus a LABA.
According to the clinical expert consulted by CADTH, the goals of asthma therapy are to maintain control of asthma, indicated by an absence of exacerbations, stable lung function, and improved symptoms. Improving these symptoms should improve HRQoL. The longer-term goal is to prevent airway remodelling, preventing future risk from severe exacerbations and, ultimately, reducing the risk of death. Reducing the risk of harms from pharmacologic therapies is also an important goal.
Drug
Tezepelumab is a TSLP inhibitor. As TSLP is a cytokine found at the beginning of the inflammatory cascade following epithelial signalling, blockading TSLP may have a broad effect on many mediators that play a role in the pathophysiology of various asthma phenotypes, including eosinophils, IgE, and a variety of interleukins.3 Tezepelumab is administered by subcutaneous injection at a dosage of 210 mg every 4 weeks.3 Its proposed indication is as an add-on maintenance treatment in adults and adolescents 12 years and older with severe asthma,3 and the sponsor’s reimbursement request is the same as the indication.19 It is currently being reviewed at Health Canada under the regular review process, and it has been approved by the FDA.20
Table 3: Key Characteristics of Tezepelumab, Dupilumab, IL-5 Inhibitors, and Omalizumab
Characteristic | Tezepelumab | Dupilumab | IL-5 inhibitors | Omalizumab |
|---|---|---|---|---|
Mechanism of action | Inhibits TSLP, an upstream regulator of various cytokines and mediators that play a role in asthma, including eosinophils, IgE, and various interleukins | Blocking IL-4R alpha, which inhibits IL-4 and IL-13 signalling; these ILs promote the release of a variety of pro-inflammatory cytokines; therefore, dupilumab blocks the actions of these cytokines, resulting in an anti-inflammatory effect | IL-5 inhibition results in destruction of eosinophils, which are thought to participate in the inflammatory component of asthma; IL-5 inhibitors therefore act as anti-inflammatories in asthma | IgE facilitates degranulation of mast cells, which leads to release of numerous mediators of the allergic component of asthma; IgE inhibitors therefore prevent mast cell degranulation and inhibit the allergic component of asthma |
Indicationa | Proposed: indicated as an add-on maintenance treatment in adults and adolescents 12 years and older with severe asthma | Add-on maintenance treatment in patients 12 years and older with severe asthma with a type 2 or eosinophilic phenotype or with OCS-dependent asthma | Add-on maintenance treatment for adult patients with severe eosinophilic asthma; the following criteria are added for mepolizumab and reslizumab:
For mepolizumab:
For reslizumab:
| Treatment of adults and adolescents with moderate-to-severe persistent asthma who have a positive skin test or in vitro reactivity to a perennial aeroallergen and whose symptoms are inadequately controlled with ICS |
Route of administration | Subcutaneous | Subcutaneous | Benralizumab: subcutaneous Reslizumab: mepolizumab IV infusion | Subcutaneous |
Recommended dosage | 210 mg subcutaneous every 4 weeks | Patients with severe asthma with a type 2 or eosinophilic phenotype:
Patients with OCS-dependent asthma or with comorbid moderate-to-severe AD or adults with comorbid severe chronic rhinosinusitis with nasal polyposis for which dupilumab is indicated:
| Benralizumab: 30 mg once every 4 weeks for the first 3 doses, then once every 8 weeks thereafter Mepolizumab: 100 mg every 4 weeks Reslizumab: 3 mg/kg every 4 weeks | 150 mg to 375 mg every 2 or 4 weeks depending on body weight and serum IgE |
Serious adverse effects or safety issues | Infections, particularly helminth Hypersensitivity reactions | Anaphylaxis, injection-site reactions, eosinophilia, helminth infections, eye disorders | Anaphylaxis, injection-site reactions, infection | Anaphylaxis, injection-site reactions, infection |
AD = atopic dermatitis; ICS = inhaled corticosteroid; IgE = immunoglobulin E; IL = interleukin; LABA = long-acting beta2-agonist; OCS = oral corticosteroids; TSLP = thymic stromal lymphopoietin.
aHealth Canada–approved indication.
Sources: Product monographs for tezepelumab,3 dupilumab,18 mepolizumab,15 benralizumab,17 reslizumab,16 and omalizumab.14
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups.
Patient input for this review was received from Asthma Canada and the Lung Health Foundation (formally known as the Ontario Lung Association), both of which are registered charities. The information gathered from Asthma Canada was obtained from approximately 600 people living with asthma or caring for someone living with asthma via multiple online surveys and in-depth individual interviews. The information from Lung Health Foundation was obtained during phone interviews conducted in April 2021 with 3 female patients living with asthma. None of the patients, parents, or caregiver respondents had experience with tezepelumab.
Patients reported that living with asthma negatively affects their psychological and social well-being, results in poor quality of life that affects their ability to complete daily activities, attend school or work, and participate in outdoor and/or physical activity, and interferes with social interactions. Patients also reported loss of productivity at school or work due to symptoms, fatigue, and exacerbations, leading to a decrease in performance or quality of work or schoolwork. Parents and caregivers expressed concerns about accessing adequate and necessary medical care as severe exacerbations can cause loss of consciousness or hypoxia, in addition to urgent ED care to restore airway functions. The need for urgent medical attention was noted to be stressful when parents and caregivers try to navigate busy and overcrowded EDs.
Patients, parents, and caregivers noted that there was an unmet need for treatment options for severe asthma. Even with currently available treatment, 1 in 4 respondents indicated that they have poor symptom control. Patients, parents, and caregivers identified several barriers to accessing health care providers (e.g., respirologists and specialized asthma clinics), including travel and missed school or work. Moreover, a third of patients and caregivers reported skipping prescription refills for asthma medications due to the financial burden as many patients with severe asthma have a low income or are unable to work due to living with asthma or caring for someone with asthma. The patients, parents, and caregivers reported that the long-term use of OCS provided some degree of inflammation control after failing other options, but is associated with notable side effects, including weight gain, acne, excess facial hair, mood swings, high blood pressure, hyperactivity, high blood sugar, and increased infections, as well as osteopenia, osteoporosis, glaucoma, cataracts, and heart disease. The source of distress associated with side effects from current medications taken to manage asthma was further emphasized by the patients who were interviewed. Patients, parents, and caregivers reported that current treatments can be difficult to take due to the mode of administration and frequency of dosing.
Patients and caregivers identified the following treatment priorities: the ability to control their day-to-day symptoms; the ability to control an exacerbation; reduction of cost or coverage for current and upcoming treatments; and reduction in medication-associated side effects. Other key treatment outcomes highlighted by patients included improvement in quality of life, reduction in the number of medications required to take to maintain asthma control, and treatments with minimal side effects.
Clinician Input
Input From the Clinical Expert Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 1 clinical specialist with expertise in the diagnosis and management of asthma.
Unmet Needs
According to the clinical expert consulted by CADTH, patients with asthma can be subdivided into those predominantly driven by type 2 inflammation (T-helper 2 [Th2] cells and type 2 innate lymphoid cell cellular pathways), who present with allergic and/or eosinophilic asthma, and those who are non–type 2, and it is the latter group of patients who are difficult to control. Non–type 2 includes patients with obesity-associated asthma, pauci-granulocytic asthma, very late onset disease, and smooth-muscle hypertrophy–associated disease. Currently these patients are managed with ICS-LABA treatments and often require additional treatments such as chronic macrolide administration (not Health Canada–approved) and/or addition of a long-acting muscarinic antagonist (LAMA), according to the clinical expert. The expert indicated that patients who smoke often require smoking cessation interventions to help improve asthma control. The subset of patients with primary airway smooth-muscle hypertrophy may respond to bronchial thermoplasty. The clinical expert reported that these non–type 2 patients typically do not respond to current biologic therapies.
The clinical expert reported that corticosteroids play an important role in managing non–type 2 asthma and tend to target airway inflammation in a nonspecific manner, and their systemic side effects increase with increasing dose.
According to the clinical expert, approximately half of patients with type 2 asthma remain poorly controlled with existing nonbiologics. Although nonpharmacologic treatments such as environmental control, medication adherence, inhaler education, and self-management techniques can improve asthma control, approximately 5% of patients will remain poorly controlled despite otherwise good treatment, according to the clinical expert. The clinical expert indicated that these poorly controlled patients are at greater risk for severe asthma exacerbations and worsened quality of life. Although OCS can reduce the frequency of exacerbations, they carry a risk of severe long-term side effects. A recent position statement from Asthma Canada21 and a peer-reviewed retrospective cohort analysis recommend limiting regular OCS use and the frequency of short-term OCS “bursts.”22 Treatment of comorbidities of asthma is an integral part of therapy, as highlighted in a recent Canadian Thoracic Society position statement on the treatment of severe asthma.23
Place in Therapy
The clinical expert indicated that tezepelumab can treat patients with type 2 asthma as well as those with non–type 2 disease. No other biologics are available that can effectively reduce exacerbation frequency in patients with non–type 2 disease, according to the clinical expert. The clinical expert noted that tezepelumab could be used either as a first-line biologic in patients with type 2 asthma or in patients who fail to improve on other biologics, as airway inflammation in type 2 asthma may be driven by factors that fall outside of those targeted by current biologics.
The clinical expert indicated that tezepelumab could drive a shift in how biologic medications are currently prescribed. Because it is effective in patients with type 2 or non–type 2 disease, the clinical expert anticipated that it could become the preferred biologic. Although good asthma care should include careful phenotyping, tezepelumab could simplify the assessment of patients for initiation of biologics as well as the assessment of maintenance, according to the clinical expert. The clinical expert added that, although there is no evidence that tezepelumab is effective in patients who use OCS on a chronic basis, other biologics with evidence of efficacy in this population are available, and regular OCS use is not common in patients with asthma in Canada.
Patient Population
The clinical expert indicated that most patients with asthma respond to therapy regardless of the underlying severity of the disease; however, the proposed Health Canada indication for tezepelumab is for add-on maintenance therapy in adults and adolescents 12 years and older with severe asthma. The clinical expert noted that this is not a precise indication as the definition of severe asthma can vary. In general, patients who remain poorly controlled on high-dose ICS-LABA or whose control worsens when they try to decrease the dose of ICS-LABA could be treated with tezepelumab, according to the clinical expert, who also noted that patients with type 2 or non–type 2 asthma would be eligible for treatment.
The clinical expert reported that patients with a clinical presentation suggestive of asthma require confirmation with spirometry or a peak expiratory flow (PEF) measurement showing reversibility of airflow obstruction. Alternatively, airway hyper-responsiveness, reactivity to cold air, exercise, or methacholine can also be used to confirm a diagnosis of asthma. The clinical expert reported that, when physicians rely on clinical judgment rather than these objective measures, there is evidence that asthma is over-diagnosed in about one-third of patients.24 The clinical expert also noted that there is evidence that Canadian physicians under-diagnose asthma,25 and more importantly, may under-diagnose the presence of severe asthma. Over-diagnosis of asthma can be prevented by restricting prescribing to a clearly defined group of physicians (respirologists and allergists) who then use standard practices to diagnose asthma and to assess asthma severity, according to the clinical expert.
The clinical expert highlighted that non–type 2 asthma is not a uniform disease. It is not clear which subtypes were chosen for the clinical trials but, for the time being, there is no clear method to define who is most likely to benefit. The default would be that all non–type 2 patients should be eligible if a treatment is funded.
Assessing Response to Treatment
The clinical expert indicated that outcomes of relevance to tezepelumab include improved HRQoL, decreased exacerbation frequency, improved asthma control, and decreased ED utilization. Biomarkers such as fractional exhaled nitric oxide (FeNO) as an indicator of airway inflammation or induced eosinophil counts are not used clinically in most Canadian sites due to access and lack of funding, according to the clinical expert, who reported that lung function can be measured in most Canadian sites and can be used to assess some airway responses (e.g., increased FEV1 and/or stabilization with loss of reversibility). Airway hyper-responsiveness, as measured by methacholine challenge testing, is also expected to improve with tezepelumab, unlike the lack of effect seen with most other biologic medications. Airway hyper-responsiveness is not a typical outcome measured clinically but such services are available in many lung-function laboratories in Canada. The clinical expert noted that the most relevant clinical outcome is asthma control, including improvement or stabilization of FEV1, elimination of airflow reversibility to bronchodilator, and reduction of night-time and daytime symptoms. A validated measure such as the ACQ-6 can be used to objectively assess improved control.
Discontinuing Treatment
The clinical expert indicated that, once the patient meets the criteria for initiation of tezepelumab, it is difficult to develop clear stopping rules other than drug intolerance. The clinical expert reported that the frequency of exacerbations is determined not just by medication use but also by environmental factors. For example, in Western Canada, the wildfire season could be expected to increase exacerbation frequency in a susceptible population, while in Central Canada, the pollen season could have a similar effect for type 2 asthma. Asthma control would have comparable environmental factors, according to the clinical expert, who noted that asthma control over a longer time frame than months would be needed to properly evaluate drug efficacy, and therefore any stopping rule would need exceptions for environmental factors.
Prescribing Conditions
The clinical expert indicated that tezepelumab should be started by either an allergist or a respirologist with experience using biologics. Ideally this should be restricted to asthma clinics but there is no accreditation process to support this designation, according to the clinical expert. The clinical expert noted that, once started, tezepelumab could be maintained by generalists. The expert noted that, in an era of virtual clinics, regional disparities in access to specialist care can be improved and should not prevent controlled access to drug initiation.
Clinician Group Input
This section was prepared by CADTH staff based on the input provided by clinician groups.
Input was received by 6 clinicians on behalf of the AllerGen CIC, which is a group of clinical investigator sites that investigate potential new therapies for the management of asthma. Six sites from 5 provinces in Canada are involved in the CIC.
Unmet Needs
The clinician group reported that, while some treatments are effective for patients with T2-high asthma, there are no effective treatment options for those patients who have severe asthma that is not persistently T2-high asthma. Currently, the only treatment options for those patients who do not have T2-high asthma are conventional asthma therapies, according to the CIC. However, the CIC indicated that these patients are less responsive to ICS or OCS. The CIC reported that, although various treatments have been developed for these patients, limited impact on clinical outcomes has been reported.
Place in Therapy and Patient Population
Members of the CIC agreed that the use of tezepelumab in asthma should be restricted to patients with severe asthma. However, in contrast to other biologic therapies, the CIC reported that tezepelumab may be used in patients with both T2-high asthma and T2-low asthma. The CIC suggested that tezepelumab should be discontinued if patients continue to experience severe exacerbations while on treatment. The only other indication cited for discontinuation by the CIC was side effects.
Assessing Response to Treatment
According to the CIC, severe exacerbation risk remains the most important outcome to improve in severe asthma. The CIC reported that severe asthma exacerbation events are potentially life-threatening and have significant effects on both patients’ and their families’ lives and functioning. The CIC indicated that other outcomes that may be used to assess response to treatment include improved lung function, other measurements considered important in asthma control, and reduction in biomarkers associated with severe asthma, including blood eosinophil count, sputum eosinophil count, and exhaled nitric oxide levels.
Discontinuing Treatment
Drug intolerance would be the main reason to discontinue the drug; otherwise, once a patient has been initiated on tezepelumab, it is difficult to develop clear stopping rules. Response to a drug with respect to exacerbation frequency can be affected by external factors, such as seasonal wildfires and pollen levels, which vary in intensity across the country. To adequately assess response and account for these external factors, one would need to try the drug for more than simply months, and any stopping rules would need exceptions for those factors.
Prescribing Conditions
The CIC noted that, in general, severe asthma is managed in specialty practices in Canada. The CIC indicated that treatment with tezepelumab would be expected to be initiated by an expert in managing severe asthma. Likewise, the decision to continue or discontinue treatment with tezepelumab would be made by an expert in managing severe asthma.
Additional Considerations
The clinical expert noted that non–type 2 asthma is not a homogeneous disease. Because it is not clear what subtypes were chosen in the clinical trials there is no clear method for defining who among patients with type 2 asthma would benefit most from this drug or whether all patients with non–type 2 asthma should be eligible for it. The expert also wondered whether post-marketing follow-up could be requested to determine the optimal characteristics for a non–type 2 responder.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical expert consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Implementation issues | |
The comparator in the submitted trials was placebo, whereas other biologics indicated for severe asthma are potentially relevant comparators. | For consideration by CDEC. |
Other biologics indicated for severe asthma include omalizumab, mepolizumab, reslizumab, benralizumab, and dupilumab. Health Canada indications for these agents generally relate to specific phenotypes — allergic (omalizumab) and eosinophilic (mepolizumab, reslizumab, benralizumab). The indication for dupilumab is broader and includes “severe asthma with a type 2/eosinophilic phenotype or oral corticosteroid-dependent asthma.” Omalizumab is reimbursed in Alberta and Ontario for patients with allergic asthma refractory to optimized standard therapy and a history of exacerbations. Mepolizumab and benralizumab are reimbursed by most public drug plans for patients with eosinophilic asthma refractory to optimized standard therapy and a history of exacerbations or dependence on OCS. At the time the drug plans provided input for this review, dupilumab was undergoing pCPA negotiations for severe asthma with type 2 or eosinophilic phenotype or OCS-dependent asthma. The pCPA negotiations for dupilumab concluded without agreement on June 28, 2022. Reslizumab is not currently funded by any of the jurisdictions, as pCPA negotiations concluded without agreement in 2019. | For consideration by CDEC. |
Considerations for initiation of therapy | |
The sponsor is positioning tezepelumab as the preferred first-line biologic across all patients with severe asthma, noting that it has clinical benefit across all asthma phenotypes, irrespective of biomarker status. Should initiation criteria for tezepelumab include any restrictions related to diagnostic phenotype or biomarker status? | Phenotyping will likely not matter with this drug as it appears to have efficacy across all phenotypes. |
Is alignment with the following aspects of initiation criteria for other biologics for severe asthma reviewed by CADTH appropriate?
| Alignment with each of the following criteria would be appropriate:
|
Considerations for continuation or renewal of therapy | |
Is alignment with renewal criteria for other biologics for severe asthma reviewed by CADTH (e.g., mepolizumab, benralizumab, or dupilumab) appropriate? | The renewal criteria for tezepelumab should be aligned with the renewal criteria for other biologics. |
Considerations for prescribing of therapy | |
There appears to be no evidence to support use of tezepelumab in combination with other biologics indicated for severe asthma, and combination use would significantly increase costs. | There is no evidence to support combinations of biologics. Another alarmin was combined with dupilumab and showed no benefit. |
Is alignment with the below-noted prescribing criteria for other biologics for severe asthma reviewed by CADTH (e.g., mepolizumab, benralizumab, or dupilumab) appropriate?
| Tezepelumab should be restricted to respirologists and allergists for initiation. Family physicians would be able to maintain a patient once initiated by a specialist. |
System and economic Issues | |
Mepolizumab and benralizumab have successfully completed pCPA price negotiations. There could be confidential prices for omalizumab in some jurisdictions. At the time the drug plans provided input on this review, dupilumab for asthma was under active negotiations through pCPA. Negotiations with pCPA concluded without agreement on June 28, 2022. | For consideration by CDEC. |
ACQ-6 = 6-item Asthma Control Questionnaire; OCS = oral corticosteroids; pCPA = pan-Canadian Pharmaceutical Alliance.
Clinical Evidence
The clinical evidence included in the review of tezepelumab is presented in 3 sections. The first section, the systematic review, includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those studies that were selected according to an a priori protocol. The second section includes indirect evidence from the sponsor and indirect evidence selected from the literature that met the selection criteria specified in the review. The third section includes sponsor-submitted LTE studies and additional relevant studies that were considered to address important gaps in the evidence included in the systematic review.
Systematic Review (Pivotal and Protocol-Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of tezepelumab injection as an add-on maintenance treatment in adults and adolescents 12 years and older with severe asthma.
Methods
Studies selected for inclusion in the systematic review included pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 5. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
The literature search was performed by an information specialist using a peer-reviewed search strategy.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
Table 5: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Patient population | Adults and adolescents 12 years and older with severe asthma Subgroups:
|
Intervention | Tezepelumab 210 mg subcutaneous every 4 weeks, as add-on therapy |
Comparators | Maintenance therapy with ICS in combination with long-acting beta2-agonists alone or in combination with 1 or more of the following:
Rescue therapy with SABA (or SAMA) is also assumed to be part of any regimen for asthma |
Outcomes | Key efficacy outcomes:
Harms: AE, SAE, withdrawal due to AE, and notable harms, including hypersensitivity reactions, helminth and other infections, injection-site reactions |
Study design | Published and unpublished phase III and IV randomized controlled trials |
ACQ-6 = 6-item Asthma Control Questionnaire; AE = adverse event; AQLQ(S) + 12 = Asthma Quality of Life Questionnaire for patients 12 years of age and older; ED = emergency department; FEV1 = forced expiratory volume in the first second; HRQoL = health-related quality of life; ICS = inhaled corticosteroids; IgE = immunoglobulin E; IL-5 = interleukin-5; OCS = oral corticosteroids; PEF = peak expiratory flow; SABA = short-acting beta2-agonists; SAE = serious adverse event; SAMA = short-acting muscarinic antagonists; vs. = versus.
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy according to the PRESS Peer Review of Electronic Search Strategies reference.26
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946–) via Ovid and Embase (1974–) via Ovid. All Ovid searches were run simultaneously as a multi-file search. Duplicates were removed using Ovid deduplication for multi-file searches, followed by manual deduplication in EndNote. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concept was tezepelumab. Clinical trials registries were searched: the US National Institutes of Health’s clinicaltrials.gov, WHO’s International Clinical Trials Registry Platform (ICTRP) search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
No filters were applied to limit the retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. Appendix 1 provides detailed search strategies.
The initial search was completed on May 6, 2022. Regular alerts updated the search until the meeting of the CADTH Canadian Drug Expert Committee on September 28, 2022.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool For Searching Health-Related Grey Literature reference.27 Included in this search were the websites of regulatory agencies (FDA and European Medicines Agency). Google was used to search for additional internet-based materials. Appendix 1 provides more information on the grey literature search strategy.
Findings From the Literature
Three studies were identified from the literature for inclusion in the systematic review (Figure 1). The included studies are summarized in Table 6. A list of excluded studies is presented in Appendix 2.
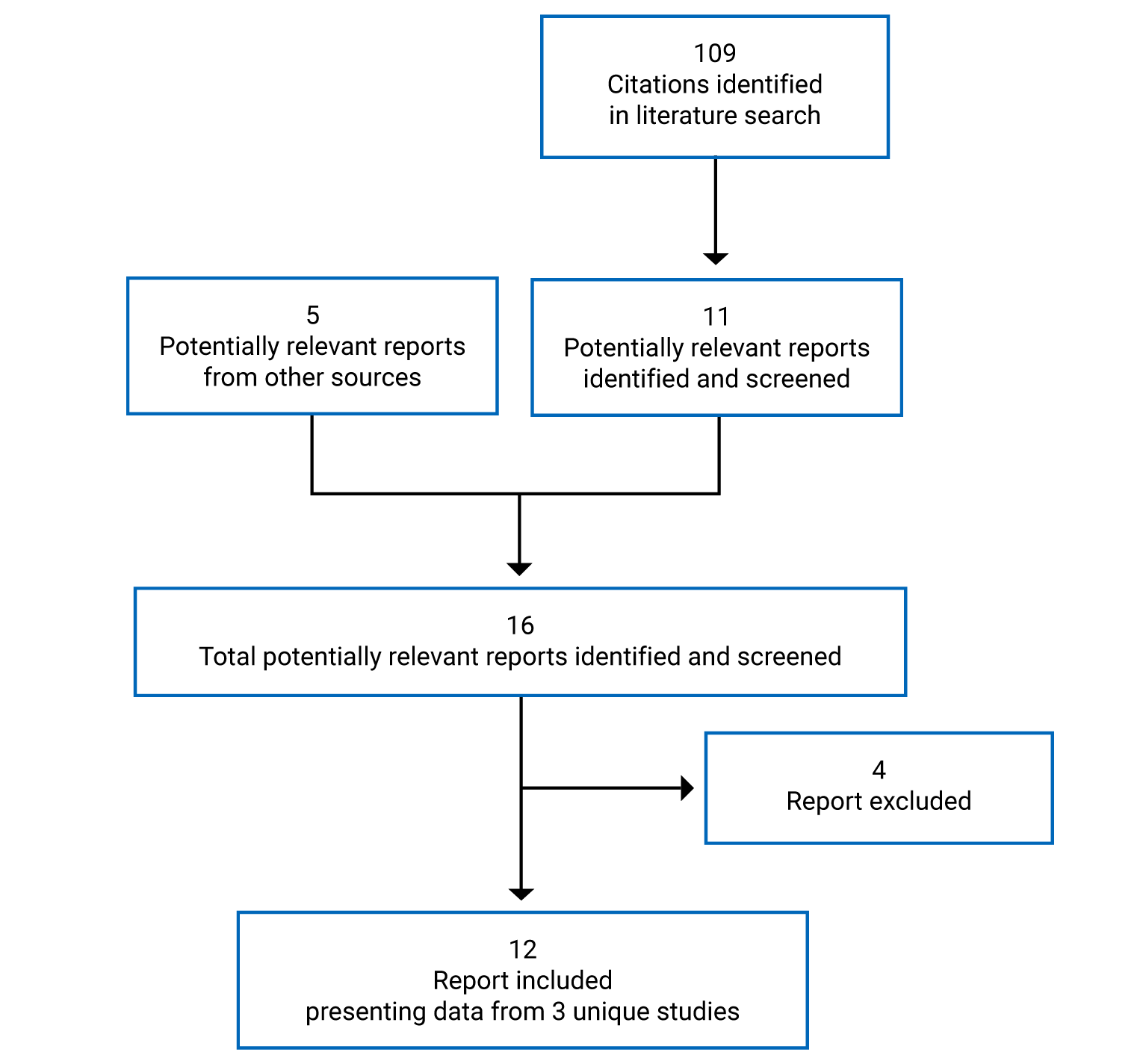
Table 6: Details of Included Studies
Characteristic | NAVIGATOR | SOURCE | PATHWAY |
|---|---|---|---|
Designs and populations | |||
Study design | Double-blind RCT, phase III | Double-blind RCT, phase III | Double-blind RCT, phase II |
Locations | 297 centres, 18 countries (Canada, US, South America, Australia, Europe, Israel, Japan, South Korea, South Africa, Taiwan, Vietnam, Saudi Arabia) | 60 centres, 7 countries (US, South Korea, Argentina, Germany, Ukraine, Poland, Turkey) | 98 centres, 12 countries (US, Europe, Israel, Japan, South Africa) |
Patient enrolment dates | November 23, 2017, to September 8, 2020 (October 29, 2020, database lock) | March 5, 2018, to September 25, 2020 (November 18, 2020, database lock) | December 19, 2013, to March 1, 2017 (first patient signing informed consent to last visit) |
Randomized (N) | 1,061 | 150 | 550 |
Inclusion criteria | Inclusion for screening/run-in:
Inclusion at randomization:
| Inclusion for screening/run-in:
Inclusion at randomization:
| Inclusion for screening/run-in:
|
Exclusion criteria |
|
|
|
Drugs | |||
Intervention | Tezepelumab 210 mg SC every 4 weeks | Tezepelumab 210 mg SC every 4 weeks | Tezepelumab 70 mg SC every 4 weeks Tezepelumab 210 mg SC every 4 weeks Tezepelumab 280 mg SC every 2 weeks |
Comparator(s) | Placebo SC every 4 weeks | Placebo SC every 4 weeks | Placebo SC every 2 weeks |
Duration | |||
Phase | |||
Run-in | 5 to 6 weeks | Run-in: 2 weeks OCS optimization: 8 weeks | Up to 5 weeks |
Double-blind | 52 weeks | 48 weeks | 52 weeks |
Follow-up | 12 weeks | 12 weeks | 12 weeks |
Outcomes | |||
Primary end point | AAER | Categorized percent reduction from baseline in the daily OCS dose at week 48 while not losing asthma control The categories for percent change from baseline in daily OCS dose are defined as: 1) ≥ 90% to ≤ 100% reduction 2) ≥ 75% to < 90% reduction 3) ≥ 50% to < 75% reduction 4) > 0% to < 50% reduction 5) no change or any increase | AAER |
Secondary and exploratory end points | Key secondary:
Other:
Safety:
| Key secondary:
Secondary:
Safety:
| Secondary: Subgroup analyses for:
Safety:
|
Notes | |||
Publications | Menzies-Gow (2020)28 Menzies-Gow (2021)29 Menzies-Gow NEJM (2021)30 | Wechsler (2020)31 | Corren (2017)32 Corren (2020)33 Corren, AAAI (2021)34 |
AAER = annualized acute exacerbation rate; ACQ-6 = 6-item Asthma Control Questionnaire; AE = adverse event; AQLQ(S)12+ = Asthma Quality of Life Questionnaire Standardized for patients 12 years of age and older; ASD = asthma symptom diary; BD = bronchodilator; BMI = body mass index; CGI-C = Clinical Global Impression of Change; COPD = chronic obstructive pulmonary disease; DPI = dry powder inhaler; ECG = electrocardiogram; ED = emergency department; ePRO = electronic patient-reported outcome; EQ-5D-5L = EQ-5D 5-Levels questionnaire; FeNO = fraction of exhaled nitric oxide; FEV1 = forced expiratory volume in the first second; FTP = fluticasone propionate; GINA = Global Initiative for Asthma; ICS = inhaled corticosteroids; IgE = immunoglobulin E; LABA = long-acting beta2-agonist; MDI = metered-dose inhaler; OCS = oral corticosteroids; PEF = peak expiratory flow; PGI-C = Patient Global Impression of Change; PGI-S = Patient Global Impression of Severity; PN = predicted normal; RCT = randomized controlled trial; SABA = short-acting beta2-agonist; SAE = serious adverse event; SC = subcutaneous; SCS = systemic corticosteroids; TB = tuberculosis.
Note: Three additional reports were included (FDA Clinical and Statistical Review, sponsor’s submission).
Sources: Clinical Study Reports for NAVIGATOR,4 SOURCE,5 and PATHWAY.6
Description of Studies
NAVIGATOR
NAVIGATOR was a phase III, multinational, sponsor-funded double-blind RCT that compared tezepelumab to placebo in adults and adolescents with severe, uncontrolled asthma. The primary objective was to assess the effect of 210 mg tezepelumab subcutaneous every 4 weeks on asthma exacerbations in adult and adolescent patients with severe, uncontrolled asthma compared with placebo. The key secondary objectives were to assess the effect of 210 mg tezepelumab subcutaneous every 4 weeks on pulmonary function compared with placebo, on health status and health-related quality of life, asthma control, and asthma symptoms.
A total of 1,061 patients were randomized 1:1 to either tezepelumab 210 mg every 4 weeks or matched placebo. Patients were randomized from 297 centres in 18 countries, including Canada. Randomization was stratified by region and age (adult or adolescent).
The screening and run-in period for the NAVIGATOR study was 5 to 6 weeks. During this time patients were to undergo a variety of assessments, including pulmonary function; fill out and receive training on symptom diaries; and undergo a number of screening assessments, including a history and physical, electrocardiogram, and a review of concomitant medication. During the 7 days before randomization, patients had to meet at least 1 indicator of continued asthma symptoms, indicated by at least 2 days with a daytime or night-time asthma symptom score of 1 or more, or use of a SABA as a reliever on more than 2 days, or at least 1 night-time awakening due to asthma, and at baseline patients had to have an ACQ-6 score of 1.5 or greater to be randomized.
Figure 2: Study Design for NAVIGATOR
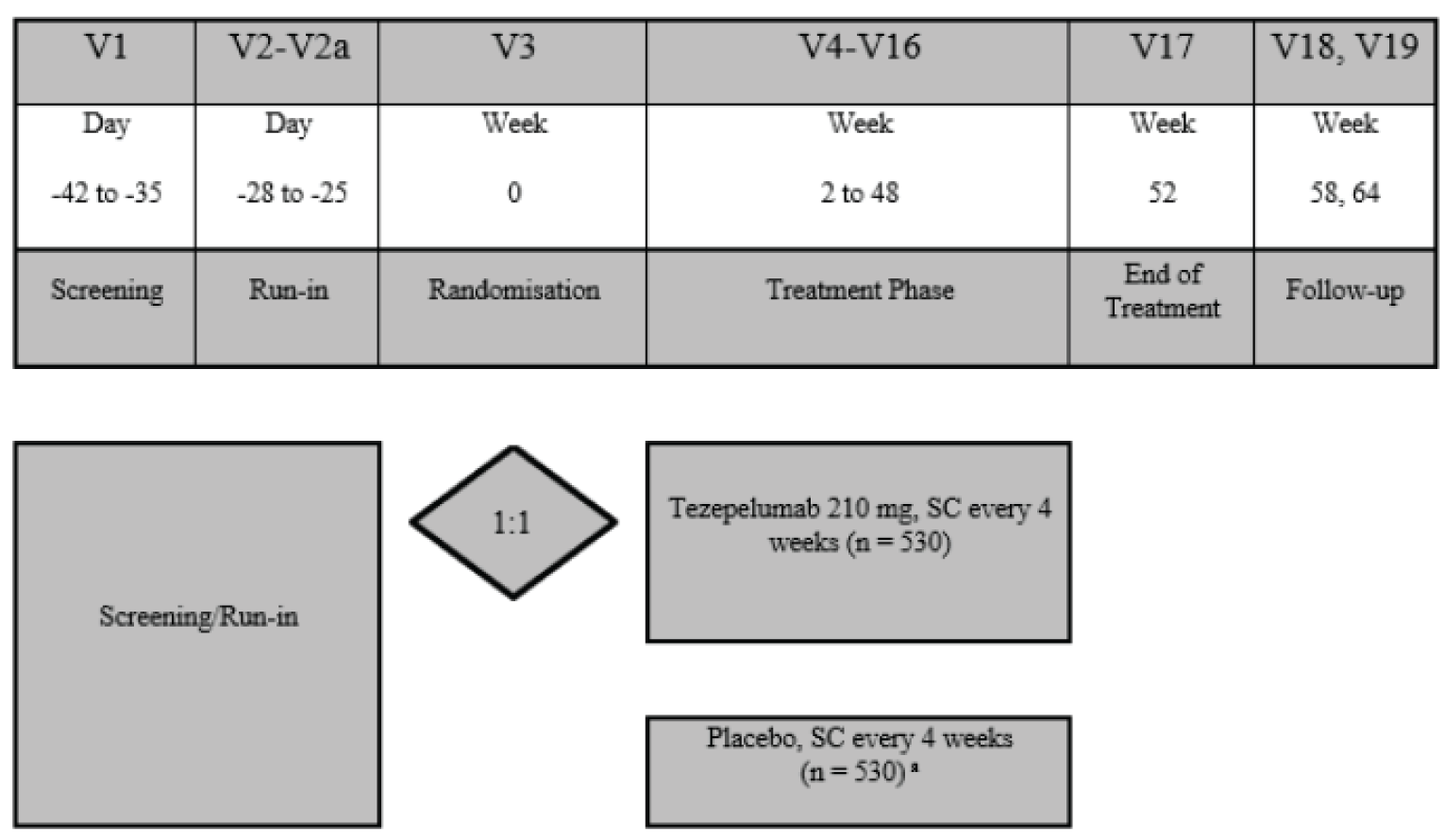
SC = subcutaneous; V = visit.
Source: Clinical Study Report for NAVIGATOR.4
SOURCE
The SOURCE study was a phase III, multinational, sponsor-funded, double-blind RCT that compared tezepelumab to placebo in patients with severe, refractory asthma who were on maintenance OCS and ICS and/or LABA, with or without additional asthma controllers. The primary objective of the study was to evaluate the effect of tezepelumab compared with placebo in reducing the prescribed OCS maintenance dose in patients with asthma requiring chronic treatment with maintenance OCS in addition to high-dose ICS plus a LABA. The key secondary objective was to evaluate the effect of tezepelumab compared with placebo on asthma exacerbations. A total of 150 patients were randomized 1:1 to either tezepelumab 210 mg subcutaneous every 4 weeks or matched placebo over a treatment course of 48 weeks. Patients were enrolled from 60 sites in 7 countries, and there does not appear to have been any Canadian sites. The run-in period in the SOURCE study was longer than in the NAVIGATOR and PATHWAY studies, as it included an 8-week OCS dose-optimization phase, in which the minimum dose at which patients were able to maintain asthma control was determined. The details of the dose-optimization phase are described in the Interventions section.
Figure 3: Study Design for SOURCE
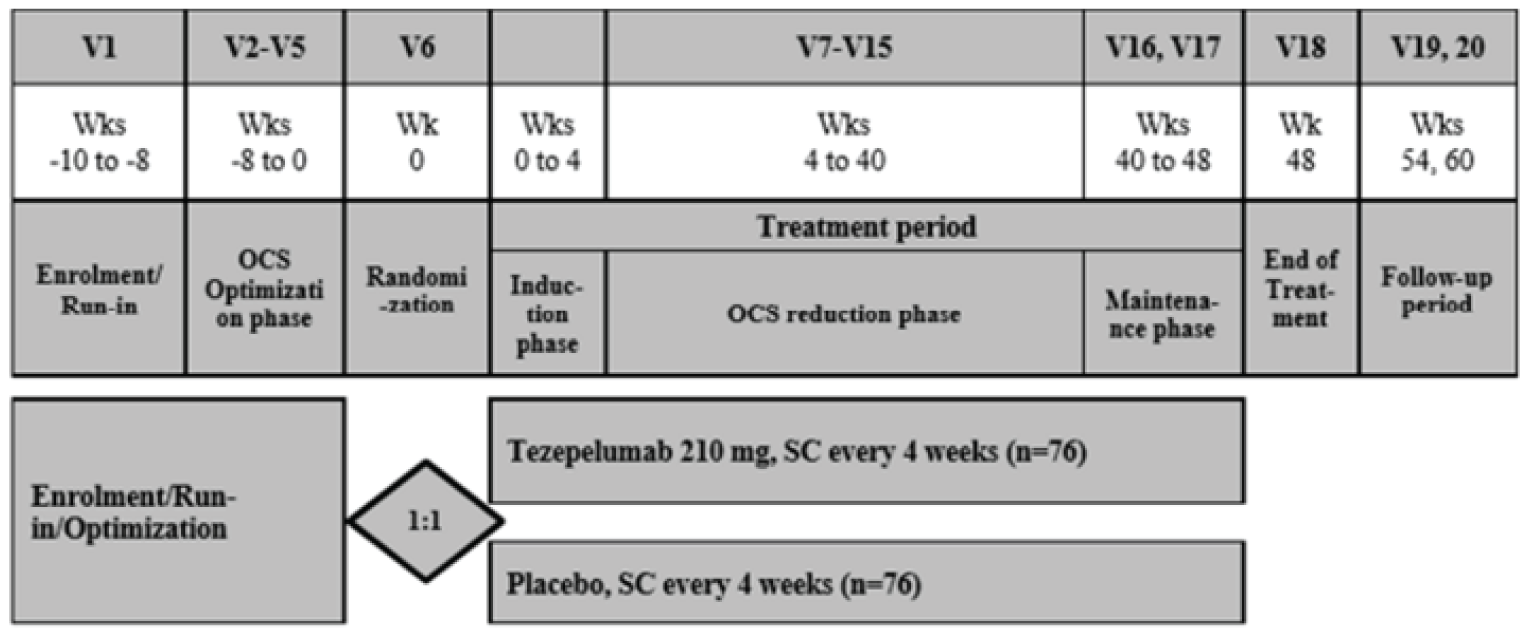
SC = subcutaneous; V = visit; Wk(s) = week(s).
Source: Clinical Study Report for SOURCE.5
PATHWAY
The PATHWAY study was a phase II, multinational, sponsor-funded, double-blind RCT that compared 3 different dose levels tezepelumab to placebo in adults with inadequately controlled, severe asthma. The primary objective was to evaluate the effect of 3 dose levels of tezepelumab on asthma exacerbations in adult patients with inadequately controlled severe asthma. There were a number of secondary objectives, including to evaluate the effect of tezepelumab on asthma exacerbations, lung function, and asthma symptoms in the pre-specified subpopulations of asthma; the effect of tezepelumab on lung function, asthma symptoms, and other metrics related to asthma control and on other parameters of asthma exacerbations; the effect on tezepelumab on HRQoL; and assessment of the safety and tolerability of tezepelumab. A total of 552 patients were randomized 1:1:1:1 to tezepelumab 70 mg, tezepelumab 210 mg, tezepelumab 280 mg, or placebo subcutaneous every 4 weeks. This report will focus on the results for the tezepelumab 210 mg group, which is the recommended dose in the draft product monograph, compared to placebo. Results will not be presented for tezepelumab 70 mg and 280 mg because these doses are not recommended by Health Canada. Randomization was stratified by study site (Japanese or non-Japanese), eosinophil count, and ICS dose (250 cells/µL or greater and medium-dose ICS; 250 cells/µL or greater and high-dose ICS; less than 250 cells/µL and medium-dose ICS; and less than 250 cells/µL and high-dose ICS). Patients were randomized at 98 sites across 12 countries, and there does not appear to have been any Canadian sites.
The screening and run-in period for PATHWAY was up to 5 weeks and had a purpose similar to that of NAVIGATOR, with similar assessments performed.
Figure 4: Study Design for PATHWAY
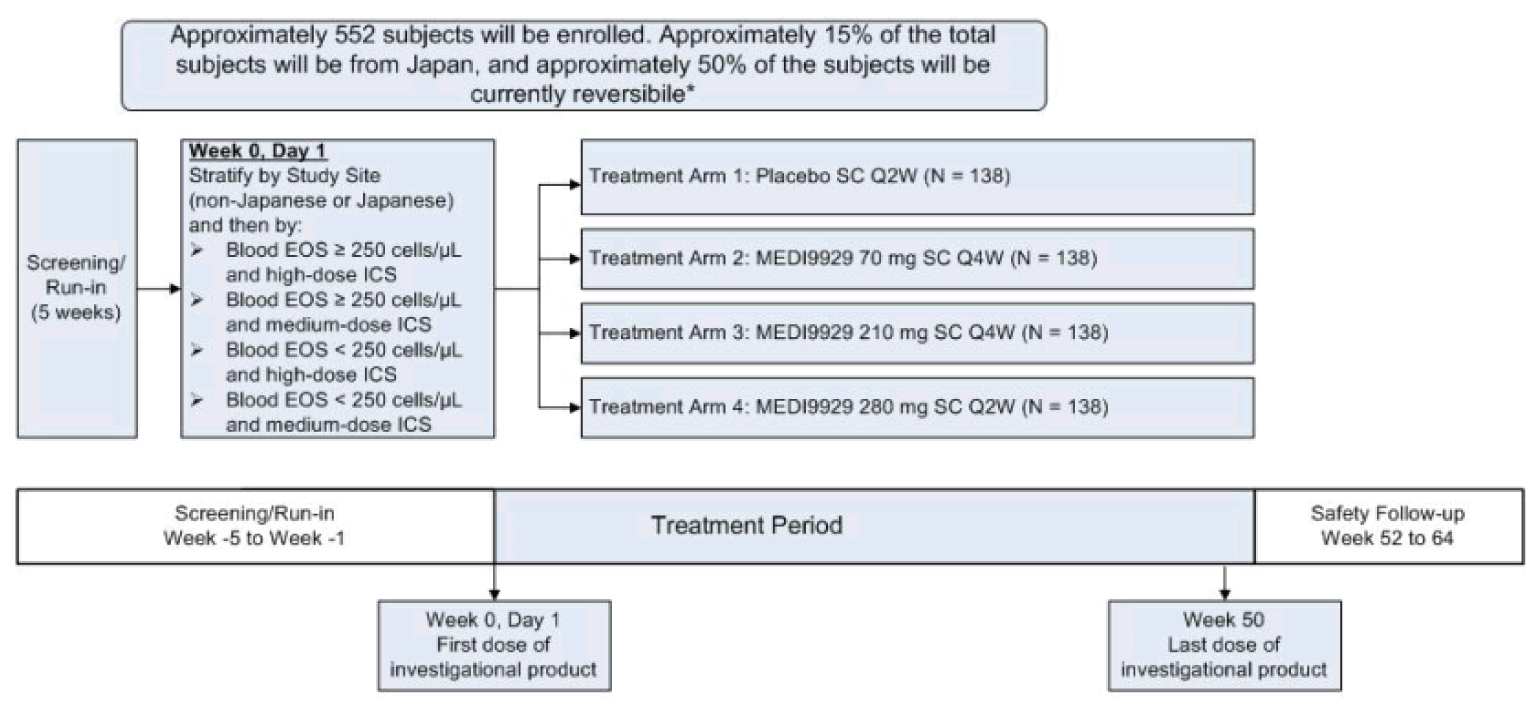
EOS = eosinophils; ICS = inhaled corticosteroids; Q2W = every 2 weeks; Q4W = every 4 weeks; SC = subcutaneous.
Source: Clinical Study Report for PATHWAY.6
Populations
Inclusion and Exclusion Criteria
In the NAVIGATOR and SOURCE studies, enrolled patients were 12 to 80 years of age, with a documented diagnosis of asthma for at least 12 months before the study. The NAVIGATOR and SOURCE studies specified that patients had to have a morning pre-bronchodilator FEV1 of less than 80% of predicted normal (< 90% for patients 12 to 17 years of age), and evidence of asthma, such as reversibility of 12% or greater or 200 mL or greater, either historically or during screening. In the NAVIGATOR study and PATHWAY, patients had to have 2 or more asthma exacerbations in the 12 months before screening (or in the PATHWAY study patients could have had 1 severe exacerbation resulting in hospitalization), while in the SOURCE study patients had to have 1 or more. An asthma exacerbation was defined by a worsening of asthma that required treatment with systemic corticosteroids for at least 3 consecutive days, an ED or urgent-care visit, or a hospitalization. For patients who were on OCS at baseline, a temporary increase for 3 or more days qualified as an exacerbation. In the NAVIGATOR and PATHWAY studies, patients had to have an ACQ-6 score of 1.5 or greater at screening. In the NAVIGATOR and PATHWAY study, patients had to have their asthma controlled by either medium- or high-dose ICS. In the SOURCE study, all patients had to have been receiving high-dose ICS and a LABA for at 3 months before the study, and had to have been using OCS for asthma for at least 6 months prior and received a stable dose of between 7.5 mg and 30 mg prednisone or prednisone equivalent, inclusive, for at least 1 month before the study.
Patients who had any other clinically relevant pulmonary disease, including chronic obstructive pulmonary disease, were excluded for all studies, as were patients who were current smokers, or those with a 10 pack-year history of smoking, and patients using vaping products.
Baseline Characteristics
Across studies, the median age of patients was between 49 and 53.5 years of age, and the majority were female, ranging between 59% and 68% of patients across studies. In the NAVIGATOR study, 62% of patients were White and 28% were Asian, while 84% of patients in the SOURCE study and 91% of patients in the PATHWAY study were White. In the NAVIGATOR study, 60% of patients had 2 exacerbations in the past 12 months, while the remainder had more than 2, while in the PATHWAY study 78% of patients had 1 or 2 exacerbations while the remainder had 3 or more. In the SOURCE study, which did not require more than 1 exacerbation in the past 12 months, 43% of patients had 1 exacerbation, 35% had 2, and 23% had more than 2 exacerbations. Mean eosinophil counts were higher in the NAVIGATOR study (340 cells/µL) and the PATHWAY study (372 cells/µL) than in the SOURCE study (242 cells/µL). The mean total IgE was also higher in the NAVIGATOR study than in the SOURCE study, at 565 IU/mL versus 299 IU/mL, respectively, and this was not reported in the PATHWAY study. In the NAVIGATOR study, 75% of patients were on high-dose ICS and the remaining were on medium-dose ICS, while in the SOURCE study, all but 1 patient were on high-dose ICS. According to the protocol, all patients in the SOURCE study were on OCS, while 9% in the NAVIGATOR study and 8% in the PATHWAY study were on OCS.
There were imbalances between the tezepelumab versus placebo groups within studies for mean baseline eosinophils in the NAVIGATOR (327 [SD = 293] versus 353 [SD = 488]), SOURCE (253 [SD = 203] versus 232 [[SD = 154]), and PATHWAY (365 [SD = 351] versus 380 [SD = 328]) studies. In the SOURCE study more patients in the tezepelumab group had 1 exacerbation in the past 12 months (46% versus 40%, respectively) and fewer in the placebo group had more than 2 (18% versus 28%, respectively). In the NAVIGATOR study, mean baseline IgE levels were lower with tezepelumab versus placebo at 516 (SD = 960) versus 614 (SD = 1,159). Reversibility of FEV1 was larger with tezepelumab than with placebo, with a mean of 16.5% (SD = 14.9) versus 13.9 (SD = 14.6) and fewer tezepelumab patients had an FEV1 reversibility of less than 12% (41% versus 54%, respectively).
Table 7: Summary of Baseline Characteristics
Characteristic | NAVIGATOR | SOURCE | PATHWAY | |||
|---|---|---|---|---|---|---|
Tez N = 528 | Placebo N = 531 | Tez N = 74 | Placebo N = 76 | Tez 210 mg N = 137 | Placebo N = 138 | |
Age, years, mean (SD) | 49.9 (16.3) | 49.0 (15.9) | 53.5 (12.1) | 53.4 (11.9) | 52.7 (12.7) | 52.3 (11.7) |
Range | 12 to 80 | 12 to 80 | 24 to 75 | 22 to 76 | (21.0 to 75.0) | (20.0 to 74.0) |
Female, n (%) | 335 (63) | 337 (64) | 49 (66) | 45 (59) | 87 (64) | 94 (68) |
Race, n (%) | ||||||
White | 332 (63) | 327 (62) | 62 (84) | 64 (84) | 128 (93) | 123 (89) |
Black and/or African-American | 30 (6) | 31 (6) | 1 (1) | 0 | 3 (2) | 6 (4) |
Asian | 146 (28) | 149 (28) | 11 (15) | 11 (15) | 5 (4) | 6 (4) |
Native Hawaiian or other Pacific Islander | 1 (0.2) | 0 (0.0) | 0 | 0 | 0 | 0 |
American Indian or Alaska Native | 0 (0.0) | 1 (0.2) | 0 | 0 | 0 | 0 |
Other | 19 (4) | 23 (4) | 0 | 1 (1) | 0 | 2 (1) |
Multiple | 0 | 0 | 0 | 0 | 1 (1) | 1 (1) |
BMI in kg/m2, mean (SD) | 28.69 (7.09) | 28.30 (6.89) | 29.29 (6.67) | 29.44 (7.44) | 28.50 (4.91) | 28.45 (5.55) |
Range | 17.1 to 62.9 | 16.2 to 63.1 | 18.5 to 48.3 | 19.5 to 55.9 | 19.8 to 39.5 | 18.0 to 44.4 |
Eosinophils, mean cells/µL (SD) | 326.7 (293.3) | 353.4 (488.4) | 253.2 (203.1) | 231.8 (153.8) | 365.0 (350.9) | 380.4 (328.0) |
Range | 0.0 to 3650.0 | 0.0 to 8170.0 | 20.0 to 1160.0 | 30.0 to 700.0 | NR | NR |
Eosinophils group, cells/µL (%) | ||||||
< 150 | 138 (26) | 138 (26) | 27 (37) | 24 (32) | NR | NR |
150 to < 300 | 171 (32) | 171 (32) | 19 (26) | 28 (37) | NR | NR |
300 to < 450 | 99 (19) | 95 (18) | 20 (27) | 16 (21) | NR | NR |
≥ 450 | 120 (23) | 127 (24) | 8 (11) | 8 (11) | NR | NR |
≥ 250 | NR | NR | NR | NR | 76 (56) | 78 (57) |
< 250 | NR | NR | NR | NR | 61 (45) | 60 (44) |
FeNO, ppb | 522 | 527 | 68 | 69 | 137 | 138 |
Mean (SD) | 41.4 (36.3) | 46.3 (44.7) | 38.7 (40.8) | 42.4 (37.4) | 31.5 (29.8) | 37.8 (39.7) |
Range | 5.0 to 235.0 | 5.0 to 265.0 | 9.0 to 279.0 | 6.0 to 159.0 | NR | NR |
FeNO group in ppb, n (%) | ||||||
< 25 | 213 (41) | 220 (42) | 32 (47) | 26 (38) | NR | NR |
25 to < 50 | 158 (30) | 151 (29) | 20 (29) | 27 (39) | NR | NR |
> 50 | 151 (29) | 156 (30) | 16 (24) | 16 (23) | NR | NR |
≥ 24 | NR | NR | NR | NR | 60 (44) | 65 (47) |
< 24 | NR | NR | NR | NR | 75 (56) | 72 (53) |
Exacerbations in past 12 months, n (%) | ||||||
1 | 0 (0) | 1 (0.2) | 34 (46) | 30 (40) | 105 (77) | 110 (80) |
2 | 310 (59) | 324 (61) | 27 (37) | 25 (33) | ||
> 2 | 218 (41) | 206 (39) | 13 (18) | 21 (28) | 32 (23.4) | 28 (20.3) |
Total IgE, mean IU/mL (SD) | 515.7 (959.8) | 614.1 (1,159.5) | 298.7 (576.3) | 300.9 (521.4) | NR | NR |
Range | 1.5 to 12,823.2 | 1.5 to 9,740.9 | 1.5 to 2,866.6 | 1.5 to 3,295.0 | NR | NR |
Allergic Asthma Status n (%) | ||||||
Allergic | NR | NR | NR | NR | 77 (61) | 80 (62) |
Perennial aeroallergen-specific IgE status (FEIA) n (%) | ||||||
Any perennial FEIA positive | 339 (64) | 341 (64) | 25 (34) | 34 (45) | NR | NR |
All perennial FEIA negative | 184 (35) | 177 (33) | 44 (60) | 39 (51) | NR | NR |
Unknown perennial FEIA | 5 (1) | 13 (2) | 5 (7) | 3 (4) | NR | NR |
Time since asthma diagnosis in years, mean (SD) | 22.3 (16.5) | 22.4 (15.8) | 23.5 (16.2) | 22.5 (13.9) | NR | NR |
Range | 1.0 to 69.0 | 1.0 to 65.0 | 1.4 to 66.0 | 2.0 to 55.0 | NR | NR |
FEV1 pre-BD, mean % PN (SD) | 62.8 (18.0) | 62.7 (18.0) | 54.3 (18.1) | 53.3 (18.4) | 59.0 (12.5) | 60.0 (13.5) |
Range | 18 to 106 | 5 to 127 | 20 to 104 | 19 to 91 | NR | NR |
FEV1 reversibility, mean % (SD) | 15.0 (15.6) | 15.1 (15.2) | 16.5 (14.9) | 13.9 (14.6) | 20.9 (18.7) | 22.5 (19.1) |
Range | −17.05 to 126.98 | −22.15 to 88.64 | −8.68 to 87.30 | −11.95 to 73.20 | NR | NR |
Reversibility in FEV1 (%) n(%) | ||||||
< 12% | 267 (51) | 276 (52) | 30 (41) | 41 (54) | NR | NR |
> 12% and < 15% | 50 (10) | 49 (9) | 11 (15) | 6 (8) | NR | NR |
≥ 15% | 211 (40) | 206 (39) | 33 (45) | 29 (38) | NR | NR |
Patients on high-dose ICS, n (%) | 397 (75) | 398 (75) | 73 (99) | 76 (100) | NR | NR |
Medium-dose ICS, n (%) | 131 (25) | 132 (25) | 1 (1) | 0 | NR | NR |
ICS dose in mcg/day, mean (SD) | NR | NR | NR | NR | 665.8 (362.0) | 680.2 (360.3) |
OCS use, n (%) | 49 (9) | 51 (10) | 74 (100) | 76 (100) | 9 (7) | 14 (10) |
OCS dose > 10 mg daily, n (%) | NR | NR | 17 (23) | 20 (26) | NR | NR |
BD = bronchodilator; BMI = body mass index; DPI = dry power inhaler; FEIA = fluorescence enzyme immunoassay; FeNO = fractional exhaled nitric oxide; FEV1 = forced expiratory volume in the first second; FTP = fluticasone propionate; ICS = inhaled corticosteroid; IgE = immunoglobulin E; NR = not reported; OCS = oral corticosteroids; PN = predicted normal; SD = standard deviation; Tez = tezepelumab.
Note: Medium-dose ICS defined as 500 mcq or greater FTP DPI-equivalent; high-dose ICS defined as greater than 500 mcg FTP DPI-equivalent.
Sources: Clinical Study Reports for NAVIGATOR,4 SOURCE,5 and PATHWAY.6
Interventions
Tezepelumab 210 mg or matching placebo was administered by subcutaneous injection every 4 weeks in all studies. The PATHWAY study was a dose-ranging study, and 2 other doses of tezepelumab were tested in the study: 70 mg every 4 weeks and 280 mg every 2 weeks, neither of which were of interest for this review as both were outside the recommended dose in the draft product monograph. In all studies, the study drug was administered by a qualified health care professional (a pharmacist or study nurse) at the site. After March 2020, due to the COVID-19 pandemic, the study drug could be administered in a patient’s home. The sponsor provided specific instructions as to drug administration, storage, and preparation. No changes were allowed to background medications except during the management of an asthma exacerbation. A SABA was to be held for at least 6 hours before scheduled spirometry, FeNO, or electrocardiogram, with the exception of any unscheduled visits due to asthma worsening. Both LABAs and LAMAs were also to be held, at least 12 hours for twice-daily dosing and 24 hours for once-daily dosing, and LTRAs were to be held for 24 hours before the assessment.
In the SOURCE study, patients had to be on OCS for at least 6 months before the study. There were a number of phases related to OCS use during the study, including an 8-week optimization phase before randomization, an induction phase that lasted the first 4 weeks after randomization, a reduction phase that lasted from weeks 4 to 40, and a maintenance phase that lasted from weeks 40 to 48. Details about each are provided in the following section.
During the OCS optimization phase of SOURCE, the minimum OCS dose while maintaining asthma control (optimized dose) was reached for all patients. The optimized OCS dose was kept stable for 2 weeks before randomization and was considered the baseline OCS dose. The baseline OCS dose was to be maintained at the same level from 2 weeks before randomization to the end of the induction phase. A reduction in the OCS dose commenced at this point, and continued at 4-week intervals until visit 15, in accordance with the OCS dose-titration schedule (reduction phase). During the reduction phase, a minimum stable OCS dose, or complete elimination of requirement for OCS, while maintaining asthma control, was reached for each patient. No adjustments should have been made to the OCS dose during the maintenance period until the end of treatment; however, exceptions may have occurred, such as in cases in which a patient had an exacerbation or for safety reasons.
In the SOURCE study, the magnitude of the OCS dose reduction depended on the dose the patient was taking. For patients on more than 10 mg to 30 mg of prednisone and/or prednisolone daily, their dose could be reduced by 5 mg per day, and for patients taking 7.5 mg to 10 mg daily, their dose could be reduced by 2.5 mg daily. Patients who were not considered to be candidates for OCS dose reduction in the optimization phase, or who were noncompliant with electronic diary completion and OCS, ICS-LABA, or other asthma-controller use, meaning they were unable to reduce their OCS dose, were considered screen failures. If a patient achieved asthma control at an OCS dose of less than 7.5 mg during the optimization phase, or asthma control was maintained after 3 consecutive OCS dose reductions, they were considered screen failures.
A set of criteria determined whether a patient was eligible to have an OCS dose reduced in the SOURCE study, and all criteria had to be met. These included pulmonary function (morning PEF of 80% or greater of mean morning measures as compared with the baseline mean), symptoms (no more than 2 nights with asthma-related awakenings [requiring rescue medications] over a 7-day period compared with baseline), rescue medication use (no more than 4 puffs per day above the baseline mean and fewer than 12 puffs per day on all days in the prior 14 days), exacerbations (none requiring increased systemic corticosteroids or hospitalization since the previous visit), investigator judgment (asthma control is sufficient to allow OCS dose reduction and no signs of adrenal insufficiency [at OCS dose reductions below 5 mg]). Whether to continue with OCS dose reduction was at the discretion of investigator even if the criteria had not been met, as long as the decision was documented and justified.
Patients across studies were allowed to take medications for conditions other than asthma, as deemed medically necessary. Some medications were prohibited within 4 to 12 weeks of the first study visit and for 4 weeks after the last dose of the study drug, largely because they had immunosuppressive or immunomodulatory effects. Exceptions were made for medical need, as judged by the investigator. Other monoclonal antibodies for asthma were not allowed within 4 months or 5 half-lives (whichever is longer) before randomization and for 4 weeks after the last dose of the study drug.
Outcomes
A list of efficacy end points identified in the CADTH review protocol that were assessed in the clinical trials included in this review is provided in Table 8. These end points are summarized in the following section. A detailed discussion and critical appraisal of the outcome measures are provided in Appendix 4.
Table 8: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure | NAVIGATOR | SOURCE | PATHWAY |
|---|---|---|---|
Mortality | Reported under harms | Reported under harms | Reported under harms |
Asthma exacerbations | AAER (primary) [1] AAER in patients with eosinophils < 300 cells/µL [2] | AAER (key secondary) [2] | AAER (primary) |
Time to first asthma exacerbation | Other secondary | Supportive | Secondary |
Patients with > 1 asthma exacerbation | Other secondary | Supportive | Secondary |
Hospitalization due to an exacerbation | Other secondary | Supportive | Secondary |
ED visit due to an exacerbation | Not reported | ||
Primary care visit due to an asthma exacerbation | Not reported | Not reported | Not reported |
Acute OCS “burst” | Not reported | Not reported | Not reported |
Pulmonary function | CFB in pre-BD FEV1 (key secondary) [3] | CFB in pre-BD FEV1 (other secondary) | CFB in pre-BD FEV1 (secondary) |
Use of OCS | Not reported | % reduction in daily OCS dose while not losing asthma control (primary) [1] | Not reported |
Patients with 100% reduction in daily OCS dose | Not reported | Other secondary | Not reported |
Patients with daily OCS dose ≤ 5mg | Not reported | Other secondary | Not reported |
Patients with ≥ 50% reduction in daily OCS dose | Not reported | Other secondary | Not reported |
Use of rescue medication | Weekly mean rescue med use (other secondary) | Weekly mean rescue med use (other secondary) | Not reported |
Health-related quality of life | EQ-5D-5L (other secondary) | EQ-5D-5L (other secondary) | EQ-5D-5L (secondary) |
CFB in AQLQ(S) + 12 total (key secondary) [4] | CFB in AQLQ(S) + 12 total (other secondary) | CFB in AQLQ(S) + 12 total (secondary) | |
Symptoms ACQ-6 | CFB in ACQ-6 (key secondary) [5] | CFB in ACQ-6 | CFB in ACQ-6 (secondary) |
ICS dose reduction | Not reported | Not reported | Not reported |
AAER = annualized acute exacerbation rate; ACQ-6 = 6-item Asthma Control Questionnaire; AQLQ12+ = Asthma Quality of Life Questionnaire (Standardized) for patients 12 years of age and older; BD = bronchodilator; CFB = change from baseline; ED = emergency department; EQ-5D-5L = EQ-5D 5-Levels questionnaire; FEV1 = forced expiratory volume in the first second; ICS = inhaled corticosteroids; OCS = oral corticosteroids.
Note: Numbers in square brackets, [ ], indicate ranking in the multiple testing hierarchy.
Sources: Clinical Study Reports for NAVIGATOR,4 SOURCE,5 and PATHWAY.6
Asthma Exacerbations
In the included studies, an asthma exacerbation (as recorded on the exacerbation electronic case report form page) was defined as a worsening of asthma that leads to any of the following:
a temporary bolus or burst of systemic corticosteroids (or a temporary increase in stable OCS background dose) for at least 3 consecutive days to treat symptoms of asthma worsening; a single depot-injectable dose of corticosteroids will be considered equivalent to a 3-day bolus or burst of systemic corticosteroids
an ED or urgent-care visit (defined as evaluation and treatment for less than 24 hours in an ED or urgent-care centre due to asthma that required systemic corticosteroids, as described previously)
an inpatient hospitalization (defined as admission to an inpatient facility and/or evaluation and treatment in a health care facility for ≥ 24 hours) due to asthma.
The start of an exacerbation was defined as the start date of systemic corticosteroids, ED or urgent-care visits requiring systemic corticosteroids, or hospital admissions due to asthma, whichever occurs earlier. The end date was defined as the last day of systemic corticosteroids or ED, urgent-care facility, or hospital discharge, whichever occurs later.
Two or more exacerbations with the same start date and end date were counted as 1 exacerbation for the purposes of calculating the number and duration of exacerbations for a patient. In cases in which 1 or more exacerbations were recorded as starting or ending during another exacerbation, these were counted as 1 exacerbation, using the earliest exacerbation start date and the latest exacerbation stop date to calculate duration.
Additional systemic corticosteroid treatments, ED or urgent-care visits requiring use of systemic corticosteroids, or inpatient hospitalization due to asthma occurring during an exacerbation were not regarded as a new exacerbation. To be counted as a new exacerbation it must have been preceded by at least 7 days during which neither criterion is fulfilled. If the end date of the first exacerbation and the start date of the second exacerbation were less than 7 days apart, then these were counted as 1 exacerbation.
Asthma Quality of Life Questionnaire (Standardized) for Patients 12 Years of Age and Older
In the AQLQ(S) + 12 the patients are asked to recall their experiences during the previous 2 weeks and to score each of the 32 questions on a 7-point scale ranging from 7 (no impairment) to 1 (severe impairment).35 The total score is calculated as the mean response to all questions. The 4 individual domain scores (assessing symptoms, activity limitations, emotional function, and environmental stimuli) are the means of the responses to the questions in each of the domains. No specific minimal important difference (MID) has been established for the AQLQ(S) + 12; however, due to similarities with the Asthma Quality of Life Questionnaire (Standardized), which has an MID of 0.5, a cut point of 0.5 points is considered to be clinically meaningful.36-38 The AQLQ(S)12+ was assessed at weeks 4, 12, 24, 36, and 52 in the NAVIGATOR study, and weeks 4, 40, 44, and 48 in the SOURCE study. In PATHWAY study, the sponsor noted in the schedule of assessments that compliance with patient-reported outcome assessments were checked every 2 weeks.
Six-Item Asthma Control Questionnaire
The ACQ-6 questionnaire39 includes questions on awakening at night by symptoms, limitations of normal daily activities, waking in the morning with symptoms, dyspnea, wheezing, and daily rescue medication. The questions of the ACQ-6 are measured on a 7-point scale scored from 0 (totally controlled) to 6 (severely uncontrolled). The ACQ-6 score computed as the unweighted mean of the responses to the 6 questions. If a response to any of the questions is missing, the ACQ-6 will be missing. A mean score of 0.75 or lower indicates well-controlled asthma, scores between 0.75 and up to 1.5 indicate partly controlled asthma, and a score higher than 1.5 indicates poorly controlled asthma.40 Changes of at least 0.5 are considered clinically meaningful.1,40-43 The ACQ-6 was to be completed at the beginning of each site visit by use of an electronic diary. This corresponded to week 2, 4, and then every 4 weeks until week 52 in the NAVIGATOR study, and every 4 weeks in the SOURCE study. In the PATHWAY study, the sponsor noted in the schedule of assessments that compliance with patient-reported outcome assessments was checked every 2 weeks.
5-Level EQ-5D Questionnaire
The EQ-5D-5L44-46 is a generic HRQoL instrument. The first 2 components of the EQ-5D-5L assess 5 domains: mobility, self-care, usual activities, pain and/or discomfort, and anxiety and/or depression, and each domain has 5 levels ranging from no problem to extreme problems. The next component of the EQ-5D-5L consists of a 20 cm Visual Analogue Scale (EQ VAS) that has end points labelled 0 and 100, with respective anchors of “worst imaginable health state” and “best imaginable health state.” Respondents are asked to draw a line from an anchor box to the point on the EQ VAS that best represents their health on that day. The EQ-5D-5L produces 3 types of data for each respondent, a profile indicating the extent of problems on each of the 5 dimensions represented by a 5-digit descriptor, a population-preference weighted health index score based on the descriptive system, and a self-reported assessment of health status based on the EQ VAS. The index score is calculated by applying a multi-attribute utility function to the descriptive system. Scores of 0 represent the health state “dead” and 1 represents “perfect health.” No MID was identified for the EQ-5D-5L that was specific to asthma; however, in the general population an MID of 0.056 has been established for index scores.47 The EQ-5D-5L was administered every 2 weeks via electronic diaries in the NAVIGATOR and SOURCE studies.
Pulmonary Function
Spirometry was performed by the investigator or authorized designate, and followed guidelines set by the American Thoracic Society and European Respiratory Society. Patients were advised to avoid strenuous exertion for at least 30 minutes or large meals for the 2 hours before the assessment. Patients were to withhold SABAs at least 6 hours before their scheduled assessment and LABA-LAMAs for 12 hours prior (for twice-daily formulations) or 24 hours (for once-daily formulations). Administration of LTRAs was to be held for 24 hours, twice-daily theophyllines for at least 12 hours, and once-daily theophyllines at least 24 hours before scheduled assessment. In the NAVIGATOR study, spirometry was carried out at weeks 2 and 4 and then every 4 weeks until week 52. In the SOURCE study, spirometry was conducted at weeks 4, 12, 24, 40, and 48.
Statistical Analysis
The statistical analysis methods used in the NAVIGATOR, SOURCE, and PATHWAY trials are summarized in Table 9.
Primary Outcome(s) of the Studies
Power Calculation
In the NAVIGATOR study, the planned number of 530 patients were enrolled, with an overall type I error control at an alpha of 0.05 and type I error control for the primary outcome at an alpha of 0.01, resulting in an estimated 90% power for key secondary outcomes. For the primary outcome, with a placebo rate of 0.9, a shape parameter of 2.4, and a dropout rate of 10%, there was greater than 99% power to detect a rate reduction of 50% at a 2-sided significance level of 1%. For the primary outcome in patients with baseline eosinophils below 300 cells/µL, with a placebo rate of 0.6 and assuming half the patients will be in this group, there was 94% power to detect a rate reduction of 50% at a 2-sided significance of 5%. For the key secondary outcome, the nominal power was 95% or higher, assuming SDs of 400 mL for FEV1 and 1.3 for all questionnaires.
For the SOURCE study, with 152 patients assigned to 2 treatment groups, it was estimated that a 2-sided 5% significance level would produce at least 90% power to reject the null hypothesis for the primary outcome, assuming an OR of 2.75 and the proportional odds assumption. Proportions assumed for placebo were 10% of patients for each of category 1 (90% to 100% reduction in OCS dose) and 2 (75% to < 90% reduction), 15% of patients for each of category 3 (50% to < 75% reduction) and category 4 (> 0% to < 50% reduction), and 50% for category 5 (no reduction or an increase). The minimum detectable OR that was still significant with these assumptions was 1.86. For the key secondary outcome, it was estimated that, with 76 patients per group, the study had greater than 80% power to reject the null hypothesis for rate ratios up to 0.39, using a 2-sided 5% significance level, and under the following assumptions: a placebo rate of 1.3 exacerbations per year in this study population, a conservative assumption on the dispersion parameter (2.4), and a uniform dropout rate of 10%.
For the PATHWAY study, the primary analysis was based on the intention-to-treat (ITT) population. A total of 124 patients per group were required to detect a 40% reduction in the AAER for each tezepelumab dose group compared to placebo. Assuming an AAER of 0.7 in the placebo group, a 2-sided significance of 0.1 was applied to the hypothesis of comparing a single tezepelumab dose group to placebo. An AAER of 0.7 in the placebo group was estimated based on internal and external studies with a similar patient population (before considering dropouts). The dispersion parameter was selected from a mepolizumab study in patients with severe eosinophilic asthma. The sample size was increased to accommodate an estimated 10% dropouts. The minimal detectable difference was approximately a 28% reduction in AAER. The minimum acceptable reduction in AAER for the study population was based upon the expected reduction of approximately 50% in a Th2-driven population, and a more modest reduction in a non-Th2 asthma population for which there were few data and no current competitors.
Statistical Test or Model
In the NAVIGATOR study, a hierarchical testing strategy was implemented to test for superiority of tezepelumab over placebo in each of the primary and key secondary end points, while controlling the overall type I error rate at 0.05 (2-sided). The primary end point in all patients was tested at a 2-sided significance level of 0.01. The primary analysis of the primary end point compared the AAER over 52 weeks between treatment groups using a negative binomial model. The response variable was the number of asthma exacerbations experienced by the patient over the study period. Treatment, region, age, and history of exacerbations were included as factors in the model. The logarithm of the time at risk for exacerbation in the study was used as an offset variable.
In the SOURCE study, a hierarchical testing strategy was also employed, and the order of outcomes in the hierarchy can be found in Table 8. The null hypothesis for the primary outcome was tested first, at a 2-sided 5% level of significance. If this was rejected, the key secondary outcome was tested at a 2-sided 5% significance level, and so on.
For the 5 ordered categories of the primary outcome, the cumulative OR of the percentage reduction category from baseline in daily OCS dose at 48 weeks while not losing asthma control, there are 4 possible cumulative odds for each treatment group, corresponding to the 4 different possible binary splits. These binary splits were defined as:
category 1 versus categories (2, 3, 4, and 5)
categories (1 and 2) versus categories (3, 4, and 5)
categories (1, 2, and 3) versus categories (4 and 5)
categories (1, 2, 3, and 4) versus category 5.
The ordered categories for OCS daily dose reduction were in turn defined as:
category 1 = 90% to 100% reduction
category 2 = 75% to lower than 90% reduction
category 3 = 50% to lower than 75% reduction
category 4 = 0% to lower than 50% reduction
category 5 = no reduction or an increase.
Key secondary outcomes in the SOURCE study were tested in a similar manner to that used for the NAVIGATOR study.
In the PATHWAY study, there does not appear to have been a strategy for controlling for multiplicity. The primary and secondary outcomes were tested in a similar manner to those in the NAVIGATOR study. Analysis of the primary outcome, AAER, was conducted using a negative binomial regression model with treatment group, baseline eosinophils, and baseline ICS dose as covariates and the response variable in the model was the number of exacerbations experienced by a patient over the 52-week study period. The follow-up time was adjusted by the offset option in the model to adjust for different exposure times among patients during the events that occurred as the logarithm of number of days in the study.
Subgroup Analyses
Pre-planned subgroup analyses were conducted on the primary and key secondary outcomes in each of the included studies for the following subgroups identified in our review protocol: baseline eosinophil counts (various cut points), ICS dose (medium and/or high), exacerbations in the past year (various cut points), OCS use at baseline and allergy status (allergic or not allergic). In the NAVIGATOR study, the specific subgroup of patients with eosinophil counts lower than 300 cells/µL was included as part of the statistical hierarchy; however, no other subgroups across all the studies were adjusted for multiplicity, and no P values were reported for any analyses presented.
Sensitivity Analyses
In the NAVIGATOR study, sensitivity analyses were performed on the primary and key secondary end points, including analyses to explore the impact of missing data and early discontinuation from investigational product. Further analyses were also performed to explore the consistency of treatment effects across demographic and baseline subgroups. Tipping-point analyses assessed pre-specified degrees of improvement in the placebo group and degrees of worsening in the tezepelumab group after study withdrawal.
In the SOURCE study, sensitivity analyses were to be performed if the amount of missing data was sufficient to potentially affect interpretation, using multiple imputation-by-pattern mixture models, where imputation of missing OCS doses at each visit up to and including week 48 was performed in 2 steps. The non-monotone (intermediate) missing values were to be imputed first, assuming they were missing at random (MAR). A Markov chain Monte Carlo (MCMC) method was used to partially impute the data. The remaining missing values at each visit were to be imputed using a sequential regression method. At each iteration, missing values were to be imputed sequentially, 1 time point at a time. Specifically, the model for each visit focused on the OCS dose at the visit as the response variable and included the following predictors: the covariates incorporated in the primary statistical model, including treatment, region, and baseline OCS dose, and the OCS dose from baseline at all the prior visits. A total of 100 imputations were to be carried out with the seed of 518,009. These imputation processes were to be carried out twice depending on the assumed mechanism of missingness, MAR, or dropout reason–based multiple imputation, which assumes that the trajectories for patients in the tezepelumab group who drop out for a treatment-related reason will follow the pattern of placebo observations based on an assumption of missing not at random, whereas remaining patients who dropped out were to be imputed assuming they were MAR.
A single imputation method, average dose, was also used; where a patient who withdrew at any point after their baseline assessment, the final OCS dose was to be imputed to the average daily dose that a patient was taking in the 14 days before their discontinuation from study drug.
Additionally, the actual percentage reduction in daily OCS dose was compared between groups using a van Elteren test. Patients who received systemic corticosteroids for other (non-asthma) reasons were summarized. All sensitivity analyses were also repeated using on-treatment data only. The final OCS dose for those patients discontinuing the study drug was to be the actual dose received from 1 dose level higher when asthma stability was verified, at the time of discontinuation of study drug.
In the PATHWAY study, sensitivity analyses were to be carried out using a Poisson regression model to assess robustness of the distributional assumptions. Correction for potential over-dispersion was to be made by the Pearson chi-square method.
Secondary Outcomes of the Studies
In the NAVIGATOR study, the main analysis of key secondary outcomes analyzed patients according to the treatment they were randomized to and included results from patients who received other controller or rescue therapies, as well as those who discontinued therapy early. Missing data from study discontinuations were modelled based on what was observed during the study using direct likelihood approaches, which are valid under the assumption that data are MAR. The change from baseline was compared to placebo using a mixed method for repeated measures model. The response variable in the model was change from baseline at each scheduled post-randomization visit, and treatment, visit, region, age (adolescent or adult) and treatment-by-visit interaction will be included as factors in the model. Baseline value of the corresponding outcome will also be included as a continuous linear covariate. Key secondary outcomes in the SOURCE and PATHWAY studies were tested in a similar manner to those in the NAVIGATOR study, although no testing hierarchy was planned for the PATHWAY study.
Table 9: Statistical Analysis of Efficacy End Points
End point | Statistical model | Adjustment factors | Sensitivity analyses |
|---|---|---|---|
NAVIGATOR | |||
AAER | Negative binomial regression analysis |
| Multiple imputation |
CFB in FEV1 CFB in ACQ-6 CFB in AQLQ(S)12+ | Repeated measures analysis |
| Controlled sequential multiple imputation based on pattern mixture models |
ACQ-6 responders AQLQ(S)12+ responders | Generalized linear model for repeated measures using a logit link function |
| NA |
SOURCE | |||
Reduction in OCS dose | Proportional odds model | Treatment, region, and daily OCS dose at baseline as covariates |
|
AAER | Negative binomial model |
| Multiple imputation |
CFB in FEV1 CFB in rescue med use CFB in ACQ-6 CFB in AQLQ(S)12+ CFB in EQ-5D-5L | MMRM model |
| NA |
ACQ-6 responders AQLQ(S)12+ responders | Generalized linear model for repeated measures using a logit link function | Treatment, region, visit, treatment-by-visit interaction, and baseline of the corresponding outcome as continuous linear covariates | NA |
PATHWAY | |||
AAER | Negative binomial regression |
| Poisson regression |
CFB in ACQ-6 CFB in AQLQ(S)12+ | Generalized linear mixed model using a linear contrast test | Treatment group, visit, treatment-by-visit interaction, baseline blood eosinophil level (≥ or < 250 cells/µL), baseline ICS dose level (medium or high), and baseline overall ACQ-6/AQLQ(S) + 12 score as fixed effects | NA |
AAER = annual asthma exacerbation rate; ACQ-6 = 6-item Asthma Control Questionnaire; AQLQ(S)12+ = Asthma Quality of Life Questionnaire (Standardized) for patients 12 years of age and older; CFB = change from baseline; EQ-5D-5L = EQ-5D 5-Levels questionnaire; FEV1 = forced expiratory volume in the first second; ICS = inhaled corticosteroids; MCMC = Markov chain Monte Carlo; MMRM = mixed model for repeated measures; NA = not applicable.
Sources: Clinical Study Report for NAVIGATOR,4 SOURCE,5 and PATHWAY.6
Analysis Populations
In the NAVIGATOR and SOURCE studies, efficacy analyses were performed on the full analysis set (FAS), which consisted of all randomized patients who received any study drug. The safety analysis set included all patients who received at least 1 dose of the study drug.
In the PATHWAY study, the ITT population was defined as patients who were randomized and received any study drug, and patients were analyzed according to their randomized treatment group. The as-treated population included any patients who received the study drug, and patients were analyzed according to the treatment they actually received. There was also a per-protocol population, which included patients who did not have significant protocol violations and received at least 80% of the intended doses of investigational product.
Results
Patient Disposition
Study withdrawals were generally below 10% in each group in each study, with the exception of the PATHWAY study, from which 11% of tezepelumab patients and 6% of placebo patients withdrew. This difference in study withdrawals between groups in the PATHWAY study was also the largest difference between groups across all the studies. The most common reason for withdrawal was “withdrawal by patient” and “other,” which occurred in 5% and 3% of tezepelumab versus placebo patients, respectively.
Exposure to Study Treatments
In the NAVIGATOR study, the mean duration of exposure with tezepelumab was 352.3 days (SD = 59.1) and in the placebo group mean duration of exposure was 342.2 (SD = 74.9) days. In the SOURCE study, the mean duration of exposure was 318.7 days (SD = 59.8) in the tezepelumab group and 325.8 days (SD = 51.7) in the placebo group. In the PATHWAY study, the mean duration of exposure was not reported; however, 70% of patients in each of the tezepelumab and placebo groups received all 26 planned doses.
Adherence to the study drug was monitored at clinic visits, at which patients had drugs administered and adherence to background therapy was monitored by electronic diaries. Days with missing diary entries were assumed to indicate nonadherence. Adherence to study drug was 99% across both groups in the NAVIGATOR study, and adherence to background medications for asthma was 80% in the tezepelumab group and 78% in the placebo group. In the SOURCE study, adherence to the study drug was 97% across both groups, and adherence to OCS during the optimization phase was 97% and during the down-titration period was 90% with tezepelumab and 93% with placebo. Adherence to background asthma therapy was 92% with tezepelumab and 93% in the placebo group. Adherence was not reported in the PATHWAY study, although it was monitored, according to the sponsor.
Characteristic | NAVIGATOR | SOURCE | PATHWAY | |||
|---|---|---|---|---|---|---|
Tez | Placebo | Tez | Placebo | Tez 210 mg | Placebo | |
Enrolled | 2,420 | 243 | 873 | |||
Screen failure | 909 | 63 | 323 | |||
Death | 1 | 0 | NR | |||
Withdrawal by patient | 17 | 7 | NR | |||
Other | 432 | 23 | NR | |||
Randomized | 529 | 532 | 74 | 76 | 412 | 138 |
Treated | 528 (99.8) | 531 (99.8) | 74 (100) | 76 (100) | 412 (100) | 138 (100) |
Completed treatment | 492 (93.0) | 474 (89.1) | 66 (89.2) | 71 (93.4) | 121 (88.3) | 129 (93.5) |
Discontinued treatment | 36 (6.8) | 57 (10.7) | 8 (10.8) | 5 (6.6) | 16 (11.7) | 9 (6.5) |
Withdrawal by patient | 14 (2.6) | 26 (4.9) | 4 (5.4) | 2 (2.6) | 7 (5.1) | 5 (3.6) |
Adverse event | 7 (1.3) | 14 (2.6) | 1 (1.4) | 2 (2.6) | 2 (1.5) | 1 (0.7) |
Protocol deviation | 2 (0.4) | 1 (0.2) | 0 | 0 | 0 | 0 |
Development of study-specific withdrawal criteria | 4 (0.8) | 5 (0.9) | 1a (1.4) | 0 | 0 | 0 |
Lost to follow-up | 5 (0.9) | 0 (0.0) | 0 | 1 (1.3) | 1 (0.7) | 0 |
Otherb | 4 (0.8) | 11 (2.1) | 2 (2.7) | 0 | 6 (4.4) | 3 (2.2) |
Discontinued treatment but completed study assessment | 22 (4.2) | 38 (7.1) | 4 (5.4) | 2 (2.6) | NR | NR |
Completed study | 509 (96.2) | 505 (94.9) | 68 (91.9) | 73 (96.1) | 122 (89.1) | 130 (94.2) |
Withdrawn from study | 16 (3.0) | 23 (4.3) | 6 (8.1) | 3 (3.9) | 15 (10.9) | 8 (5.8) |
Death | 0 | 2 (0.4) | 1 (1.4) | 0 | 0 | 0 |
Lost to follow-up | 5 (0.9) | 2 (0.4) | 0 | 1 (1.3) | 1 (0.7) | 0 |
Withdrawal by patient | 8 (1.5) | 15 (2.8) | 5 (6.8) | 2 (2.6) | 7 (5.1) | 4 (2.9) |
Otherb | 3 (0.6) | 4 (0.8) | 0 | 0 | 7 (5.1)b | 4 (2.9)b |
Completed treatment and study | 487 (92.1) | 467 (87.8) | 64 (86.5) | 71 (93.4) | NR | NR |
Analysis sets | ||||||
Safety set and/or as-treated | 528 | 531 | 74 | 76 | 137 | 138 |
Full analysis set and/or ITT | 528 | 531 | 74 | 76 | 137 | 138 |
ITT = intention-to-treat; NR = not reported; Tez = tezepelumab.
aThis patient withdrew from the study as they were unable to continue due to an adverse event (preferred term: invasive breast carcinoma).
bOther reasons included patient’s decision, AE, failure to meet eligibility criteria, visit out of window, sponsor decision, lack of available investigational product, pregnancy, use of prohibited medications, and missed dose.
Sources: Clinical Study Report for NAVIGATOR,4 SOURCE,5 and PATHWAY.6
Efficacy
Only those efficacy outcomes and analyses of subgroups identified in the review protocol are reported in the following section. Appendix 3 provides detailed efficacy data.
Mortality
Across all studies, there was only 1 death in the tezepelumab group and 2 deaths in the placebo group. The 2 deaths in the placebo group were due to an unknown cause and heart failure, both in the NAVIGATOR study, and 1 patient in the tezepelumab group died in the SOURCE study, due to cardiac arrest.
Asthma Exacerbations
The AAER was the primary outcome of both the NAVIGATOR and PATHWAY studies. In the NAVIGATOR study, the AAER over 52 weeks was 0.93 (95% CI, 0.80 to 1.07) with tezepelumab and 2.10 (95% CI, 1.84 to 2.39) with placebo, for a rate ratio of 0.44 (95% CI, 0.37 to 0.53; P < 0.001). Various sensitivity analyses were conducted to account for missing data, and results were consistent with that of the primary analysis, according to the sponsor. In the PATHWAY study, the AAER over 52 weeks was 0.20 (95% CI, 0.13 to 0.30) with tezepelumab and 0.72 (95% CI, 0.59 to 0.88) with placebo, for a rate ratio of 0.29 (95% CI, 0.16 to 0.51). In the SOURCE study, the AAER over 48 weeks was 0.69 (95% CI, 0.44 to 1.09) (Table 11).
The AAER associated with an ED visit or hospitalization was 0.06 (95% CI, 0.04 to 0.09) with tezepelumab and 0.28 (95% CI, 0.20 to 0.39) with placebo, for a rate ratio of 0.21 (95% CI, 0.12 to 0.37) in the NAVIGATOR study, and was 0.16 (95% CI, 0.06 to 0.44) with tezepelumab and 0.28 (95% CI, 0.13 to 0.58) with placebo in the SOURCE study, for a rate ratio of 0.59 (95% CI, 0.19 to 1.82) (Table 11).
The AAER associated with a hospitalization was 0.03 (95% CI, 0.01 to 0.06) with tezepelumab and 0.19 (95% CI, 0.12 to 0.30) with placebo, for a rate ratio of 0.15 (95% CI, 0.07 to 0.33; P < 0.001) in the NAVIGATOR study, and 0.02 (95% CI, 0.00 to 0.07) with tezepelumab and 0.14 (95% CI, 0.08 to 0.22) with placebo in the PATHWAY study, for a rate ratio of 0.14 (95% CI, 0.03 to 0.71) (Table 11).
Subgroup analyses were performed for a number of subgroups relevant to our protocol and those reported for the primary outcome of each study are reported in Table 25. In the NAVIGATOR study, the AAER in patients with baseline eosinophil counts below 300 cells/µL was part of the statistical hierarchy and was therefore controlled for multiplicity, with an AAER for tezepelumab versus placebo of 1.02 versus 1.73, and a rate ratio of 0.59 (95% CI, 0.46 to 0.75; P < 0.001). None of the other subgroup analyses reported were adjusted for multiplicity and no P values were reported.
Pulmonary Function
The pre-bronchodilator FEV1 increased in both the tezepelumab and placebo groups in the NAVIGATOR study, with a LSM change from baseline to 52 weeks of 0.23 L (SE = 0.018) with tezepelumab and 0.10 L (0.018) with placebo for an LSM difference between groups of 0.13 L (95% CI, 0.08 to 0.18; P < 0.001). In the SOURCE study, the change from baseline to week 48 was 0.21 L (SE = 0.046) with tezepelumab and −0.04 L (SE = 0.046) with placebo, for an LSM difference between groups of 0.26 L (95% CI, 0.13, 0.39), and in the PATHWAY study, the LSM change from baseline to week 52 was 0.076 L (SE = NR) with tezepelumab and −0.056 L (NR) with placebo, for an LSM difference between groups of 0.132 L (95% CI, 0.033 to 0.231) (Table 11).
Reduction in Oral Corticosteroid Use
Reduction in OCS use while still maintaining asthma control was the primary outcome of SOURCE. The cumulative OR for patients having a reduction in OCS dose was 1.28 (95% CI, 0.69 to 2.35; P = 0.434) (Table 11). Tezepelumab therefore failed to demonstrate superiority over placebo for the primary outcome of this study. There were 54% of patients in the tezepelumab group and 46% of placebo patients who achieved a dose reduction of 90% or more. Other thresholds for dose reduction for tezepelumab versus placebo included 75% or greater to less than 90% (7% versus 5%, respectively), 50% or greater to less than 75% (14% versus 18% respectively), and greater than 0 to less than 50% (19% versus 18%, respectively).
This outcome was not assessed in the NAVIGATOR or PATHWAY study.
Reduction in Rescue Medication
Reduction in daily rescue medication was observed in both the tezepelumab and placebo groups in the NAVIGATOR study, with an LSM change from baseline of −2.53 puffs (SE = 0.137) with tezepelumab and −2.36 puffs (0.137) with placebo, for an LSM difference between groups of −0.17 (95% CI, −0.55 to 0.21) (Table 11). Rescue medication use also declined in both groups in the SOURCE study, with an LSM change from baseline to 48 weeks of −0.85 puffs (SE = 0.280) with tezepelumab and −0.37 puffs (SE = 0.268) with placebo, for a LSM difference between groups of −0.47 (95% CI, −1.24 to 0.29).
This outcome was not assessed in the PATHWAY study.
Health-Related Quality of Life
Health-related quality of life was assessed using the AQLQ(S)12+ and the EQ-5D-5L instruments. Mean AQLQ(S)12+ scores increased (improved) from baseline in the NAVIGATOR study, both in the tezepelumab 1.48 (SD = 1.26) and placebo 1.17 (SD = 1.16) groups, with a difference between groups of 0.33 (95% CI, 0.20 to 0.47; P = 0.001) (Table 11). In the SOURCE study, the LSM change from baseline was 0.94 (SE = 0.124) with tezepelumab and 0.58 (SE = 0.123) with placebo for a difference between groups of 0.36 (95% CI, 0.01 to 0.70). In the PATHWAY study, the LSM change from baseline to week 52 was 1.17 (SE = NR) with tezepelumab and 0.97 (SE = NR) with placebo for a difference between groups of 0.20 (−0.09 to 0.48). Responders to the AQLQ(S)12+ were also reported, defined as those with a change from baseline of 0.5 or greater. In the NAVIGATOR study 78% of tezepelumab patients and 72% of placebo patients were responders, for an OR of 1.36 (95% CI, 1.02 to 1.82). In the SOURCE study, 62% of tezepelumab patients and 52% placebo were responders, for an OR of 1.66 (95% CI, 0.81 to 3.43). In the PATHWAY study, 73% of tezepelumab patients and 62% of placebo patients were responders.
In the NAVIGATOR study, on the EQ VAS, the LSM change from baseline was 14.64 (SE = 0.708) and 11.86 (SE = 0.712) with placebo, for a LSM difference between groups of 2.78 (95% CI, 0.81, 4.75) (Table 11). In the SOURCE study, the LSM change from baseline to week 48 was 9.21 (SE = 2.209) with tezepelumab and 2.00 (SE = 2.226) with placebo, for an LSM difference between groups of 7.21 (95% CI, 1.01 to 13.41). In the PATHWAY study, the LSM change from baseline was 11.7 (SE = 19.1) with tezepelumab and 10.5 (SE = 17.4) with placebo. No between-group differences were reported.
The EQ-5D-5L health index scores were also reported. In the NAVIGATOR study, the LSM change from baseline for tezepelumab was 0.11 (SE = 0.008) and 0.07 (SE = 0.009) for placebo, for a LSM difference between groups of 0.03 (95% CI, 0.01 to 0.06). In the SOURCE study, the LSM change from baseline with tezepelumab was 0.07 (SE = 0.026) and 0.0 (SE = 0.027) for placebo, for a LSM difference between groups of 0.07 (95% CI, −0.01 to 0.14). In the PATHWAY study, the LSM change from baseline to week 52 was 0.0858 (SE = 0.1994) with tezepelumab and 0.0719 (SE = 0.1824) with placebo.
Asthma Symptoms
Symptoms were assessed as a key secondary outcome using the ACQ-6. In the NAVIGATOR study, ACQ-6 scores decreased (improved) from baseline in both the tezepelumab and placebo groups, for a difference versus placebo of 0.33 (95% CI, 0.20 to 0.47; P < 0.001) (Table 11). Responders to the ACQ-6 were also reported, defined as those with a change from baseline of 0.5 or greater; 86% of tezepelumab patients and 77% of placebo patients were responders, for an OR of 1.99 (95% CI, 1.43 to 2.76). In the SOURCE study, the LSM change from baseline to 48 weeks was −0.87 (SE = 0.125) for tezepelumab and −0.51 (SE = 0.123) with placebo, for a difference between groups of −0.37 (95% CI, −0.71 to −0.02). In the PATHWAY study, the LSM change from baseline to week 52 was −1.20 with tezepelumab and −0.91 with placebo, for a difference between groups of −0.29 (95% CI, −0.56 to −0.01).
Reduction in ICS Dose
This outcome was not investigated.
Table 11: Efficacy, FAS Population (NAVIGATOR, SOURCE) and ITT Population (PATHWAY)
Efficacy outcome | NAVIGATOR | SOURCE | PATHWAY | |||
|---|---|---|---|---|---|---|
Tez N = 529 | Placebo N = 532 | Tez N = 74 | Placebo N = 76 | Tez 210 mg N = 137 | Placebo N = 138 | |
Asthma exacerbations | ||||||
AAER | ||||||
Number of events, n | 425 | 878 | 78 | 116 | NR | NR |
AAER, 52 weeks (95% CI) | 0.93 (0.80 to 1.07) | 2.10 (1.84 to 2.39) | NR | NR | 0.20 (0.13 to 0.30) | 0.72 (0.59 to 0.88) |
AAER, 48 weeks (95% CI) | NR | NR | 1.38 (0.98 to 1.95) | 2.00 (1.46 to 2.74) | NR | NR |
Absolute difference vs. placebo (95% CI) | −1.17 (−1.47 to −0.88) | −0.62 (−1.40 to 0.15) | NR | |||
Rate ratio (95% CI) | 0.44 (0.37 to 0.53)a | 0.69 (0.44 to 1.09)b | 0.29 (0.16 to 0.51)c | |||
P value | < 0.001 | 0.111d | < 0.001d | |||
Time to first exacerbation | ||||||
HR (95% CI) | 0.59 (0.50 to 0.70) | 0.74 (0.48 to 1.15) | 0.45 (0.26 to 0.75) | |||
P value | NR | 0.181d | 0.002d | |||
AAER associated with ED visit or hospitalization | ||||||
Number of events, n | 30 | 115 | 8 | 19 | NR | NR |
Rate (95% CI) | 0.06 (0.04 to 0.09) | 0.28 (0.20 to 0.39) | 0.16 (0.06 to 0.44) | 0.28 (0.13, 0.58) | NR | NR |
Absolute difference vs. placebo (95% CI) | −0.22 (−0.31 to −0.12) | −0.11 (−0.35 to 0.12) | NR | NR | ||
Rate ratio (95% CI) | 0.21 (0.12 to 0.37) | 0.59 (0.19 to 1.82) | NR | |||
P value | < 0.001 | 0.361d | NR | |||
AAER associated with hospitalization | ||||||
Number of events, n | 14 | 78 | NR | NR | NR | NR |
Rate (95% CI) | 0.03 (0.01 to 0.06) | 0.19 (0.12 to 0.30) | NR | NR | 0.02 (0.00 to 0.07) | 0.14 (0.08 to 0.22) |
Absolute difference vs. placebo (95% CI) | −0.16 (−0.25 to −0.07) | NR | NR | |||
Rate ratio (95% CI) | 0.15 (0.07 to 0.33) | NR | 0.14 (0.03 to 0.71)b | |||
P value | < 0.001d | NR | 0.017d | |||
Pulmonary function | ||||||
Pre-BD FEV1, L | ||||||
Baseline, n | 528 | 531 | 74 | 76 | 137 | 138 |
Mean (SD) | 1.830 (0.718) | 1.851 (0.706) | 1.556 (0.504) | 1.593 (0.637) | 1.828 (0.582) | 1.823 (0.587) |
Change from baseline, n | 471 | 453 | 65 | 64 | 121 | 131 |
LSM (SE) CFB | 0.23 (0.018) | 0.10 (0.018) | 0.21 (0.046) | −0.04 (0.046) | 0.076 (NR) | 0.097 (NR) |
LSM MD (95% CI) | 0.13 (0.08 to 0.18)e | 0.26 (0.13 to 0.39)e | 0.132 (0.033 to 0.231)e | |||
P value | < 0.001 | < 0.001d | 0.009d | |||
Reduction in OCS | ||||||
Patients achieving reduction in OCS dose | ||||||
≥ 90% to ≤ 100% | NR | NR | 40 (54) | 35 (46) | NR | NR |
≥ 75% to < 90% | NR | NR | 5 (7) | 4 (5) | NR | NR |
≥ 50% to < 75% | NR | NR | 10 (14) | 14 (18) | NR | NR |
> 0% to < 50% | NR | NR | 5 (7) | 9 (12) | NR | NR |
No change or increase | NR | NR | 14 (19) | 14 (18) | NR | NR |
Cumulative OR (95% CI) | NR | NR | 1.28 (0.69 to 2.35)f | NR | ||
P value | NR | NR | 0.434 | NR | ||
Reduction in rescue | ||||||
Daily rescue med use | ||||||
Baseline, N | 528 | 531 | 74 | 76 | NR | NR |
Mean (SD) baseline | 4.36 (5.20) | 4.35 (5.09) | 3.11 (3.30) | 3.16 (3.03) | NR | NR |
Change from baseline, N | 439 | 428 | 60 | 70 | NR | NR |
Mean (SD) CFB, week 52 | −2.44 (4.21) | −2.49 (4.02) | −0.92 (2.83) | −0.43 (2.99) | NR | NR |
LSM CFB (SE) | −2.53 (0.137) | −2.36 (0.137) | −0.85 (0.280) | −0.37 (0.268) | NR | NR |
LSM MD (95% CI) | −0.17 (−0.55 to 0.21)g | −0.47 (−1.24 to 0.29)g | NR | |||
P value | 0.382d | 0.22d | NR | |||
Health-related quality of life | ||||||
AQLQ(S)12+ total | ||||||
Baseline, N | 527 | 529 | 74 | 76 | 123 | 121 |
Mean (SD) baseline | 3.87 (1.02) | 3.90 (1.00) | 4.14 (1.18) | 4.11 (1.02) | 4.20 (0.91) | 4.09 (0.87) |
Change from baseline, N | 480 | 467 | 66 | 67 | 41f | 47f |
Mean (SD) CFB | 1.48 (1.26) | 1.16 (1.17) | 0.96 (1.17) | 0.59 (1.01) | NR | NR |
LS mean (SE), CFB | NR | NR | 0.94 (0.124) | 0.58 (0.123) | 1.17 | 0.97 |
Difference vs. placebo (95% CI) | 0.33 (0.20 to 0.47)h | 0.36 (0.01 to 0.70)h | 0.20 (−0.09 to 0.48)h | |||
P value | 0.001 | 0.042d | 0.185d | |||
Patients with ≥ 0.5 CFB, n (%) | 372 (78) | 335(72) | 41 (62) | 35 (52) | 87 (73) | 74 (62) |
OR (95% CI) | 1.36 (1.02 to 1.82) | 1.66 (0.81 to 3.43) | NR | |||
P value | 0.036d | 0.169d | NR | |||
EQ-5D-5L Visual Analogue Scale | ||||||
Baseline | ||||||
N | 527 | 527 | 74 | 73 | 123 | 120 |
Mean (SD) | 62.1 (17.8) | 62.5 (17.2) | 62.5 (18.4) | 61.5 (17.4) | 60.6 (16.5) | 60.5 (15.0) |
Change from baseline, N | 448 | 435 | 62 | 58 | 121 | 120 |
Mean (SD) CFB | 15.2 (20.0) | 12.2 (18.8) | 9.5 (18.8) | 2.4 (22.0) | 11.7 (19.1) | 10.5 (17.4) |
LSM CFB (SE) | 14.64 (0.708) | 11.86 (0.712) | 9.21 (2.209) | 2.00 (2.226) | NR | NR |
LSM difference (95% CI) | 2.78 (0.81 to 4.75)i | 7.21 (1.01 to 13.41)i | NR | |||
P value | 0.006d | 0.023d | NR | |||
EQ-5D-5L health-state valuation index score | ||||||
Baseline, N | 527 | 527 | 74 | 73 | 123 | 120 |
Mean (SD) baseline | 0.686 (0.219) | 0.691 (0.203) | 0.674 (0.239) | 0.649 (0.216) | 0.687 (0.179) | 0.677 (0.182) |
Change from baseline, N | 448 | 435 | 62 | 58 | 121 | 120 |
Mean (SD) CFB | NR | NR | NR | NR | 0.086 (0.199) | 0.072 (0.182) |
LSM (SE) CFB | 0.11 (0.01) | 0.07 (0.01) | 0.07 (0.03) | 0.0 (0.03) | NR | NR |
LSM MD (95% CI) | 0.03 (0.01 to 0.06)i | 0.07 (−0.01 to 0.14)i | NR | |||
P value | 0.006d | 0.084d | NR | |||
Symptoms | ||||||
ACQ-6, total score | ||||||
Baseline, N | 528 | 531 | 74 | 76 | 137 | 138 |
Mean (SD) baseline | 2.82 (0.81) | 2.79 (0.82) | 2.48 (1.07) | 2.46 (1.03) | 2.70 (0.80) | 2.66 (0.69) |
Change from baseline, N | 486 | 472 | 66 | 68 | 44j | 53j |
Mean (SD) CFB, week 52 | −1.55 (1.15) | −1.24 (1.10) | −0.93 (1.25) | −0.52 (1.02) | NR | NR |
LSM CFB week 52 | −1.53 (0.05) | −1.20 (0.05) | NR | NR | −1.20 | −0.91 |
LSM CFB week 48 | NR | NR | −0.87 (0.13) | −0.51 (0.12) | NR | NR |
Difference vs. placebo (95% CI) | −0.33 (−0.46 to −0.20)h | −0.37 (−0.71 to −0.02)h | −0.29 (−0.56 to −0.01)h | |||
P value | < 0.001 | 0.038d | 0.039d | |||
Responders, patients with ≥ 0.5 CFB, n (%) | 418 (86) | 361 (77) | 43 of 66 (65) | 31 of 68 (46) | 103 (76) | 83 (63) |
OR (95% CI) | 1.99 (1.43 to 2.76)k | 2.30 (1.10 to 4.81)k | NR | |||
P value | < 0.001d | 0.028d | 0.002d | |||
ICS dose reduction | ||||||
NR | NR | NR | NR | NR | NR | NR |
AAER = annualized asthma exacerbation rate; ACQ-6 = 6-item Asthma Control Questionnaire; AQLQ12+ = Asthma Quality of Life Questionnaire (Standardized) for patients 12 years of age and older; BD = bronchodilator; CFB = change from baseline; CI = confidence interval; ED = emergency department; EQ-5D-5L = EQ-5D 5-Levels questionnaire; FEV1 = forced expiratory volume in the first second; ICS = inhaled corticosteroid; LSM = least squares mean; NR = not reported; OCS = oral corticosteroids; OR = odds ratio; SD = standard deviation; SE = standard error; Tez = Tezepelumab; vs. = versus.
aModel: a negative binomial regression analysis with treatment, region, and history of exacerbations as covariates. The logarithm of the time at risk is used as an offset variable. Annual exacerbation rates displayed are estimated marginal rates from the model. All CIs for annual exacerbation rates and absolute differences are estimated via the delta method.
bThe rate ratio, and 95% CI for the rate ratio were estimated from negative binomial regression with treatment group, with stratification factors of baseline blood eosinophil count (≥ or < 250 cells/μL) and baseline ICS dose level (medium or high) as the covariates.
cModel: a negative binomial regression analysis with treatment, region, age group, and history of exacerbations as covariates. The logarithm of the time at risk is used as an offset variable. Annual exacerbation rates displayed are estimated marginal rates from the model. Absolute difference is the difference between the marginal rates. All CIs for annual exacerbation rates and absolute differences are estimated via the delta method.
dP values are not controlled for multiplicity or failed in the hierarchy and should be considered supportive in nature.
eEstimate of the mean change from baseline at each week in tezepelumab is compared to placebo using a repeated measures analysis. Estimates are least squares means. The model with unstructured covariance structure is: change from baseline in FEV1 = treatment group + region + age + baseline FEV1 + visit + treatment × visit.
fThe estimate of the cumulative odds ratio is obtained using a proportional odds model with treatment, region, and daily OCS dose at baseline as covariates.
gEstimate of the mean change from baseline at each week in Tezepelumab is compared to the Placebo using a repeated measures analysis. Estimates are least squares means. The model with Unstructured covariance structure is: Change from baseline in Rescue medication use weekly means = Treatment group + region + age + baseline rescue medication use + week + treatment * week.
hEstimate of the mean change from baseline at each week in tezepelumab is compared to placebo using a repeated measures analysis. Estimates are least squares means. The model with unstructured covariance structure is: change from baseline in AQLQ(S)12+/ACQ-6 = treatment group + region + baseline AQLQ(S)12+/ACQ-6 + visit + treatment × visit.
iEstimate of the mean change from baseline at each week in Tezepelumab is compared to the Placebo using a repeated measures analysis. Estimates are least squares means. The model with Unstructured covariance structure is: Change from baseline in VAS/index score = Treatment group + region + age + baseline VAS/index score + visit + treatment * visit.
jSample sizes for these outcomes in the PATHWAY study were low due to an error in collection of ePRO data.
kThe estimate of the odds ratio is obtained using a generalized estimating equation model for repeated measures binary data with an unstructured covariance structure and treatment, region, age, visit, visit × treatment, and baseline ACQ-6 and/or AQLQ(S)12+ score as covariates.
Sources: Clinical Study Report for NAVIGATOR,4 SOURCE,5 and PATHWAY.6
Harms
Only those harms identified in the review protocol are reported here. Table 12 provides detailed harms data.
Adverse Events
Adverse events in the tezepelumab versus placebo groups occurred in 77% versus 80% of patients, respectively, in the NAVIGATOR study; 72% versus 86%, respectively, in the SOURCE study; and 66% of patients in each group in the PATHWAY study (Table 12).
The most common AE was nasopharyngitis, occurring in 21% versus 21% of patients in the NAVIGATOR study, 15% versus 25% of patients in the SOURCE study, and 14% versus 12% of patients in the PATHWAY study in the tezepelumab versus placebo groups, respectively. Other common events (occurring in 10% or more of patients in any group of any study) were upper respiratory tract infections, headaches, and asthma.
Serious Adverse Events
Serious adverse events in the NAVIGATOR study, tezepelumab versus placebo, occurred in 9% versus 13% of patients. In the SOURCE study they occurred in 15% versus 21% of patients, respectively, and in the PATHWAY study they occurred in 10% versus 13% of patients, respectively (Table 12). The most common SAE was asthma.
Table 12: Summary of Harms, Safety Analysis Set
Harms outcome | NAVIGATOR | SOURCE | PATHWAY | |||
|---|---|---|---|---|---|---|
Tez N = 528 | Placebo N = 531 | Tez N = 74 | Placebo N = 76 | Tez 210 mg N = 137 | Placebo N = 138 | |
AE | ||||||
Any AE, n (%) | 407 (77) | 422 (80) | 53 (72) | 65 (86) | 90 (66) | 91 (66) |
Specific AE ≥ 10% in any group, n (%) | ||||||
Nasopharyngitis | 112 (21) | 113 (21) | 11 (15) | 19 (25) | 19 (14) | 16 (12) |
URTI | 58 (11) | 84 (16) | 9 (12) | 7 (9) | NR | NR |
Asthma | 25 (5) | 56 (11) | 7 (10) | 13 (17) | 27 (20) | 50 (36) |
Headache | 43 (8) | 44 (8) | 3 (4) | 8 (11) | 11 (8) | 6 (4) |
SAE | ||||||
Any SAE, n (%) | 46 (9) | 70 (13) | 11 (15) | 16 (21) | 13 (10) | 18 (13) |
Most common (asthma) | 12 (2) | 40 (7) | 2 (3) | 8 (11) | 4 (3) | 10 (7) |
Withdrawal due to AE | ||||||
AE leading to discontinuation of investigational product, n (%) | 11 (2) | 19 (4) | 2 (3) | 2 (3) | 2 (2) | 1 (1) |
Deaths | ||||||
n | 0 | 2 | 1 | 0 | 0 | 0 |
Notable harms, n (%) | ||||||
Severe infections | 46 (9) | 44 (8) | 4 (5) | 7 (9) | NA | NA |
Opportunistic infections | 0 | 0 | 0 | 0 | 0 | 0 |
Helminth infections | 0 | 0 | 0 | 0 | 0 | 0 |
Infections (SAEs) | NA | NA | NA | NA | 1 (1) | 4 (3) |
Injection-site reactions | 8 (1.5) | 5 (0.9) | 0 | 1 (1.3) | NA | NA |
1 mL volume | NA | NA | NA | NA | 2 (1.5) | 4 (2.9) |
1.5 mL volume | NA | NA | NA | NA | 2 (1.5) | 2 (1.4) |
Hypersensitivity reactions (SAEs) | 1 | 1 | 0 | 0 | 0 | 0 |
AE = adverse event; NA = not assessed; SAE = serious adverse event; Tez = tezepelumab; URTI = upper respiratory tract infection.
Sources: Clinical Study Report for NAVIGATOR,4 SOURCE,5 and PATHWAY.6
Withdrawals due to Adverse Events
Adverse events resulting in discontinuation of the study drug occurred in 2% versus 4% of patients in the tezepelumab versus placebo groups, respectively, in the NAVIGATOR study, 3% in each group in the SOURCE study, and 2% versus 1% of patients, respectively, in the PATHWAY study (Table 12).
Notable Harms
Notable harms in the CADTH systematic review protocol included infections. Severe infections occurred in the tezepelumab versus placebo groups in 9% versus 8% of patients in the NAVIGATOR study, respectively, and 5% versus 9% of patients in the SOURCE study, respectively (Table 12). In the PATHWAY study, infections were reported as SAEs rather than severe infections, and these occurred in 1% versus 3% of patients in the tezepelumab versus placebo groups, respectively. No opportunistic infections and no helminth infections were reported across the studies.
Injection-site reactions occurred infrequently across the studies. In the NAVIGATOR study, injection-site reactions occurred in 1.5% versus 0.9% of patients in the tezepelumab versus placebo groups, respectively; in the SOURCE study, there were none with tezepelumab group, and 1.3% of patients in the placebo group experienced these events. In the PATHWAY study, these events were reported by injection volume, and at the 1 mL volume they occurred in 1.5% versus 2.9% of patients in the tezepelumab versus placebo groups, respectively, and at the 1.5 mL volume they occurred in 1.5% versus 1.4% of patients, respectively. Hypersensitivity reactions reported as SAEs were infrequent, at 1 patient in each of the tezepelumab and placebo groups in the NAVIGATOR study, and none in the other studies.
Critical Appraisal
Internal Validity
All 3 studies appeared to take appropriate steps for the randomization to maintain allocation concealment (use of an interactive voice- or web-response system), and to maintain blinding (matching placebo injections). The phase III studies (NAVIGATOR and SOURCE) used a hierarchical testing procedure to control for multiplicity. The lack of multiplicity control in the PATHWAY study means that all reported P values should be considered supportive and not used to draw conclusions. The phase II dose-ranging design is an additional limitation when interpreting data from the PATHWAY study, as this design was not appropriate for determining superiority. Given the statistical testing could not reject the null hypothesis for the primary outcome in the SOURCE study, failure of the statistical hierarchy means that any P values less than the pre-specified 2-sided 5% significance level reported for outcomes after the primary outcome should also be considered supportive of efficacy. Failure of statistical comparison on the primary outcome and the impact on the hierarchy for the secondary outcome limits any conclusions that can be drawn from the SOURCE trial. The FDA review of tezepelumab focused on the results of the NAVIGATOR study because of these limitations with the other studies.
Withdrawals from the study were generally below 10% across the studies, and the only study for which this was not the case, PATHWAY, had numerically more withdrawals in the tezepelumab group compared to the placebo group (11% versus 6%, respectively). This difference in withdrawals could potentially bias results in favour of tezepelumab for outcomes such as exacerbations, as patients no longer in the study are no longer at risk of an exacerbation. Additionally, as the number of withdrawals increases there is an increasing risk that baseline characteristics will no longer be balanced between groups, and that these imbalances may bias results either for or against the study drug. The sponsor performed a variety of sensitivity analyses in an attempt to account for missing data. Despite steps to account for missing data, more than 10% of data were missing from end-of-study assessments for continuous outcomes such as FEV1, ACQ-6, AQLQ(s)12+, and EQ-5D-5L. For example, in the NAVIGATOR study, for the week-52 assessment of FEV1, the reported sample was 471 patients (of 528 at baseline) with tezepelumab and 453 of 531 patients in the placebo group, which were 11% and 14% less, respectively, than the baseline FAS population. Similar but slightly smaller amounts of missing data were seen for the ACQ-6 (8% tezepelumab and 11% placebo) and the AQLQ(S)12+ (10% and 12%, respectively). Larger amounts of data were missing for the EQ-5D-5L (15% and 17% in the tezepelumab and placebo groups, respectively, in the NAVIGATOR study). All of these numbers are higher than the reported number of patients who withdrew from study treatment (7% tezepelumab and 11% placebo), and higher than the number of patients reported to have withdrawn from the study (3% and 4%, respectively). It is unclear why data were missing for patients who were not only in the study but remaining on treatment, and this should be considered a limitation of the findings for these outcomes. Similar amounts of missing data were seen for these outcomes in the SOURCE study. In the PATHWAY study, there was a larger amount of missing data for the week-52 assessments for the ACQ-6 and AQLQ(S)12+, and this was attributed to an error in the administration of the electronic patient-reported outcome instruments. This resulted in 60% to 70% of the FAS missing from the week-52 analysis, meaning that these data are unlikely to be reliable. The sponsor reported data for these outcomes for the last reliable time point, week 48, and those results were consistent with data reported at week 52.
There were pre-specified subgroups in each of the included studies; however, only 1 of these in the NAVIGATOR study (AAER in patients with eosinophil counts < 300 cells/µL) were adjusted for multiple statistical comparisons, as it was part of the hierarchical testing procedure. No P values were therefore reported for the remaining subgroups across all the included studies, and no P values were reported for interactions. As a result, data for pre-specified subgroups were reported, and only limited conclusions can be drawn from these data. Additionally, for this subgroup in the NAVIGATOR trial (AAER in patients with eosinophils < 300 cells/µL at baseline) it was not clear from the Clinical Study Report whether this outcome was a co-primary outcome, and the FDA rejected it as a co-primary outcome in their review.
The primary and key secondary outcomes assessed in the included trials are all well-established, well-validated outcomes used for asthma. Minimal important differences have been established for the ACQ-6 and AQLQ(S)12+, and these are reviewed in detail in Appendix 4. Although not part of the statistical hierarchy, data were reported for ACQ-6 and AQLQ(S)12+ “responders,” and a response was defined using the MID for each of these instruments.
Noteworthy differences between groups in baseline characteristics were observed in the SOURCE study, in which fewer patients in the tezepelumab than in the placebo group had more than 2 exacerbations in the past year (18% versus 28% of patients, respectively). Given the importance of asthma exacerbations as a marker of asthma control, and the fact that AAER was a key secondary outcome in the SOURCE study, this difference may be important. If patients in the tezepelumab group were less prone to having multiple exacerbations the results may have been biased in favour of tezepelumab for the AAER outcome. Although a lower exacerbation rate with tezepelumab may also indicate their asthma was better controlled than in the placebo population, baseline ACQ-6 scores, another measure of asthma control, were similar between groups.
External Validity
The primary and key secondary outcomes were all relevant outcomes for assessing patients with asthma, notably exacerbation rate, pulmonary function (FEV1), HRQoL (AQLQ and EQ-5D-5L) and symptoms (ACQ-6). Reduction in OCS dose without losing asthma control was the primary outcome of the SOURCE study, and this was an appropriate outcome given the primary objective of the study. However, failure to demonstrate superiority of tezepelumab over placebo at this early stage of the hierarchy meant that all testing of all subsequent outcomes yielded P values that were “supportive.”
Although the clinical expert consulted by CADTH for this review noted that they would not consider putting a patient on tezepelumab, or any biologic, until they trialled them on high-dose ICS, 25% of patients in the NAVIGATOR study were on a medium-dose ICS at baseline. This, and the relatively large number of patients who demonstrated a high degree of airway reversibility suggests that this population may have been undertreated upon entering the trial, and this may affect the generalizability of the findings. The clinical expert also noted that the proposed indication for tezepelumab is for patients with severe asthma, and the expert would not consider patients who are taking a medium dose of ICS to have severe asthma.
None of the included trials compared tezepelumab to any of the other biologics approved for management of asthma, the IgE inhibitor omalizumab, the IL-5 inhibitors, or the IL4 and IL-13 inhibitor (dupilumab). As omalizumab and the IL-5 inhibitors have been approved for several years in patients with eosinophilic asthma or those with allergic asthma, it is plausible that a trial involving these patient populations that included 1 or more of these drugs as comparators could have been designed. These direct comparisons would have been valuable given the unique mechanism of tezepelumab. The lack of an active-controlled trial is a limitation of this review.
The clinical expert consulted by CADTH for this review noted that adherence to therapy is a major issue in the management of asthma. It is unknown what patient adherence to therapy was like before entering the NAVIGATOR or PATHWAY study. For example, all that was known is that patients were poorly controlled on their current asthma regimens, and it is possible that poor adherence was a major contributor to this poor asthma control. It is also possible that patients, when observed more closely in a clinical trial, may have increased their adherence to their background therapy. It is not known whether this may have biased results in favour of tezepelumab or placebo, or would have affected both groups equally.
The clinical expert consulted by CADTH noted that excluding current smokers from the included trials is a generalizability issue, although it is not an uncommon practice in asthma trials. Otherwise, the demographics of the patients included in the clinical trials generally reflected those patients that would be expected to be treated with tezepelumab, in the clinical expert’s opinion.
Indirect Evidence
Objectives and Methods for the Summary of Indirect Evidence
Due to the lack of direct evidence comparing tezepelumab with other existing therapies as add-on maintenance treatment in adults and adolescents 12 years and older with severe asthma, the sponsor submitted 2 ITCs.10,11 In addition, CADTH conducted an independent literature search for published ITCs that compared tezepelumab with other relevant comparators for the treatment of patients with in adults and adolescents 12 years and older with severe asthma. A focused literature search for ITCs dealing with tezepelumab and asthma was run in MEDLINE All (1946–) on May 9, 2022. No limits were applied. Three additional ITCs7-9 comparing these treatments were identified. The objective of this section is to summarize and critically appraise the indirect evidence from the 2 sponsor-submitted ITCs and the 3 ITCs identified in the CADTH literature search. To align with the CADTH systematic review protocol, only information pertaining to the criteria outlined in Table 5 are presented in this section.
Description of Sponsor-Submitted Indirect Comparisons
The sponsor submitted 2 ITC reports.10,11 One was an NMA10 and the other was an MAIC-STC.11 The objectives of the ITCs were to identify, evaluate, and synthesize the evidence on the clinical efficacy and safety of tezepelumab compared with other biologics in patients with moderate-to-severe uncontrolled asthma. The MAIC-STC analyses were performed to account for key areas of heterogeneity between trials that could not be addressed by NMA methods.
Systematic Literature Review of the Indirect Treatment Comparisons
A systematic literature search strategy was developed based on the population, intervention, comparator, outcome, and study design (PICOS) criteria presented in Table 13 to identify relevant studies investigating the efficacy and safety of tezepelumab with other existing treatments. Supplemental grey literature searches were also performed for key clinical conferences in January 2021. A targeted grey literature search of the US National Institutes of Health ClinicalTrials.gov and searches of health technology assessment websites were also completed to identify additional studies of interest. Hand-searches of the bibliographies of relevant systematic reviewers, meta-analysis, and NMAs (published within the past 3 years) identified from the database search were performed to identify additional relevant studies.
Following screening and a feasibility assessment, a total of 36 studies4,5,32,49-80 were included in the sponsor-submitted ITCs.
Network Meta-Analysis Feasibility Assessment
An NMA feasibility assessment was conducted by assessing heterogeneity across all included studies to ensure they were clinically and methodologically similar for comparison. The available study characteristics, inclusion and exclusion criteria, and potential treatment-effect modifiers were assessed. A list of potential treatment-effect modifiers is presented in Table 14. It was determined that a NMA was feasible, but MAIC and STC methods would be used to better adjust for differences between studies.
Methods of the Sponsor-Submitted Network Meta-Analysis
Based on the findings from the feasibility assessment, a full network Bayesian NMA10 was performed. Various subgroup analysis and sensitivity analyses were conducted. Methods for the NMA analysis are briefly summarized in Table 15.
Table 13: Study Selection Criteria and Methods for Indirect Treatment Comparison
PICOS and process | NMA, MAIC, or STC |
|---|---|
Population | Patients at least 12 years of age who have asthma that remains uncontrolled despite adherence with maximal optimized GINA step 4 or 5 treatment, including medium- to high-dose ICS and LABAa |
Intervention | Tezepelumab (210 mg every 4 weeks) |
Comparator | Approved dosages of the following based on prescribing information from FDA, EMA, and/or Japan:b Anti-IgE:
Anti–IL-5 pathways:
Anti–IL-4 and anti–IL-13:
|
Outcome | Efficacy, safety, and patient-reported outcomes including (but not limited to):
|
Study design |
|
Study language | Englishd |
Exclusion criteria |
|
Databases searched | No restriction: from the inception of the databases to date of search; additionally, regular alerts will be established |
Selection process | Articles screened independently by 2 researchers |
Data extraction process | Data extraction was performed by a single reviewer and independently assessed for accuracy and completeness by a second reviewer; a third independent reviewer resolved disagreements |
Quality assessment | Using the National Institute for Health and Care Excellence quality appraisal checklist of quantitative intervention studies48 |
AQLQ = Asthma Quality of Life Questionnaire; EMA = European Medicines Agency; ED = emergency department; FeNO = fractional exhaled nitric oxide; GINA = Global Initiative for Asthma; ICS = inhaled corticosteroids; IgE = immunoglobulin E; IL = interleukin; LABA = long-acting beta2-agonist; PICOS = population, intervention, comparator, outcome, and study design; RCT = randomized controlled trial; SGRQ = St. George's Respiratory Questionnaire.
aThis criterion was relaxed after initial screening to permit inclusion of studies in which at least 75% of patients reported LABA use (plus at least medium-dose ICS), despite not requiring use of a LABA or other controllers as a part of their inclusion criteria.
bLatest version of Japan prescribing information (as of March 2020).
cSystematic reviews, meta-analyses, network meta-analyses, and bibliographies of these records were reviewed and cross-referenced with the included study lists to ensure that no primary studies were missed.
dSearch captured all languages, but non-English citations were excluded during full-text screening.
Sources: Sponsor-submitted indirect treatment comparisons.10,11
Table 14: Categorized List of Potential Treatment-Effect Modifiers
Category | Potential treatment-effect modifier |
|---|---|
Asthma control–related characteristics | 1. Number of exacerbations in past 12 months |
Biomarkers | 2. Blood eosinophil count |
3. Fractional exhaled nitric oxide | |
4. Total immunoglobin E | |
Demographics | 5. Age |
6. Sex | |
Lung function | 7. FEV1 (% predicted) |
Other clinical characteristics | 8. Body mass index |
9. Disease duration | |
10. ACQ score | |
11. Nasal polyps | |
Treatment-related characteristics | 12. OCS users |
13. OCS dose at entry | |
14. ICS dose at entry |
ACQ = Asthma Control Questionnaire; FEV1 = forced expiratory volume in the first second; ICS = inhaled corticosteroid; OCS = oral corticosteroids.
Sources: Sponsor-submitted indirect treatment comparisons.10,11
Table 15: Indirect Treatment Comparison Analysis Methods
Components | Methods |
|---|---|
Indirect treatment comparison methods | Network meta-analysis |
Priors | Vague or flat priors, such as N (0 to 1,002), were assigned for basic parameters |
Assessment of model fit | RE or/and FE models Both FE and RE models were considered for NMAs of each outcome; vague or flat priors, such as N (0 to 1,002), were assigned for basic parameters |
Assessment of consistency | Not applicable |
Assessment of convergence | Yes, assessed |
Outcomes | AAER, AAER leading to hospitalization, ACQ, FEV1 and OCS reduction |
Follow-up time points | Up to 64 weeks as reported in the included studies |
Construction of nodes | Yes |
Sensitivity analyses | Yes |
Subgroup analysis | Yes |
Methods for pairwise meta-analysis | Bayesian |
AAER = annualized asthma exacerbation rate; ACQ = Asthma Control Questionnaire; FE = fixed-effect; FEV1 = forced expiratory volume in the first second; OCS = oral corticosteroids; RE = random-effect.
Source: Sponsor’s network meta-analysis.10
Statistical Approach
Bayesian NMAs were performed for each of the 5 outcomes using ITC methodology based on the National Institute for Health and Care Excellence (NICE) Decision Support Unit (DSU) Technical Support Document.81 Model selection was based on model fit statistics, total number of studies, and studies per connection, and plausibility of the results. The model fit statistics assessed were the deviance information criterion, total residual deviance, and the random-effects standard deviation. When model fit was unclear, both fixed-effect and random-effect model results were presented.
The NMAs were performed using R statistical software. The MCMC sampling process used to construct the posterior distribution for all model parameters involved discarding a burn-in of 40,000 or more iterations before accepting a subsequent 40,000 or more iterations for NMA estimation. Three MCMC chains were run in parallel. Monte Carlo errors, trace plots, and Gelman–Rubin diagnostic criteria (ideally close to 1, with values > 1.05 indicating nonconvergence) were generated and reviewed to assess model convergence.
Primary Analyses and Summary Measures of Treatment Effect by Outcome
All NMAs were conducted using the ITT populations of relevant RCTs. Changes in the AAER and changes in the AAER leading to hospitalization were modelled using a Poisson likelihood and a log link, with corresponding rate ratios and 95% credible intervals (CrIs) calculated. For changes from baseline in the Asthma Control Questionnaire (ACQ) score and pre-bronchodilator FEV1, normal likelihood functions were used. The relative treatment effect was defined as the mean difference (95% CrI) in change from baseline between treatments. For change from baseline in OCS dose by predefined reduction categories, treatment effects were modelled using an ordinal model (probit scale) considering each category reported in the included RCTs (no reduction or increase in use, 1% to 49% reduction, 50% to 74% reduction, 75% to 89% reduction, or 90% to 100% reduction). The proportion of patients in each of the predefined reduction categories for each treatment were calculated. Additionally, analyses were conducted for each predefined reduction category, where risk ratios and 95% CrIs were generated. All RCTs included in this analysis assessed OCS reduction between week 20 and week 28. However, OCS reduction in the SOURCE study5 was assessed at week 48.
Assessment of Heterogeneity and Inconsistency
The available study characteristics, inclusion and exclusion criteria, potential treatment-effect modifiers, and outcome definitions were assessed to ensure similarity. Additionally, clinical experts were consulted to further investigate the potential impact of heterogeneity on effect estimates. Because no direct evidence connects any of the relevant biologics, consistency could not be assessed.
Subgroup and Sensitivity analysis
Clinically relevant subpopulation and/or subgroup analysis (e.g., baseline eosinophil count, number of exacerbations at baseline) were performed.
Sensitivity NMA analyses were also conducted by removing some studies with different characteristics (such as those studies that used best standard care as a comparator and not a true placebo group).
Results of Sponsor-Submitted Network Meta-Analysis
Summary of Included Studies
A total of 36 trials4,5,32,49-80 were included in the NMA. An overview of the included trials is presented in Table 26. Overall, 3 trials for tezepelumab, 6 for benralizumab, 3 for dupilumab, 3 for mepolizumab, 16 for omalizumab, and 5 for reslizumab were included. For the purpose of this review, NMA results related to reslizumab are not summarized because reslizumab was not identified as a relevant comparator in the protocol for the CADTH systematic review.
Assessment of Risk of Bias of Included Trials
It was reported that the methodological quality of all included studies was assessed using the NICE quality appraisal checklist of quantitative intervention studies.48 As the technical report for the NMA states that the assessed trials were considered to have adequate internal validity and generalizability to the target population, sensitivity analyses based on the results of the quality assessment of individual studies were not conducted.82
Network Meta-Analysis Results
The key findings of the sponsor-submitted NMA are presented in Table 16 and Table 17.
Reduction in Annualized Asthma Exacerbation Rate
Primary Analyses
A visual representation of the evidence network for reduction in the AAER is provided Figure 5. The common comparator to all trials is placebo, with a single multi-dose study creating 1 closed loop.
Figure 5: Evidence Network for Annualized Asthma Exacerbation Rate
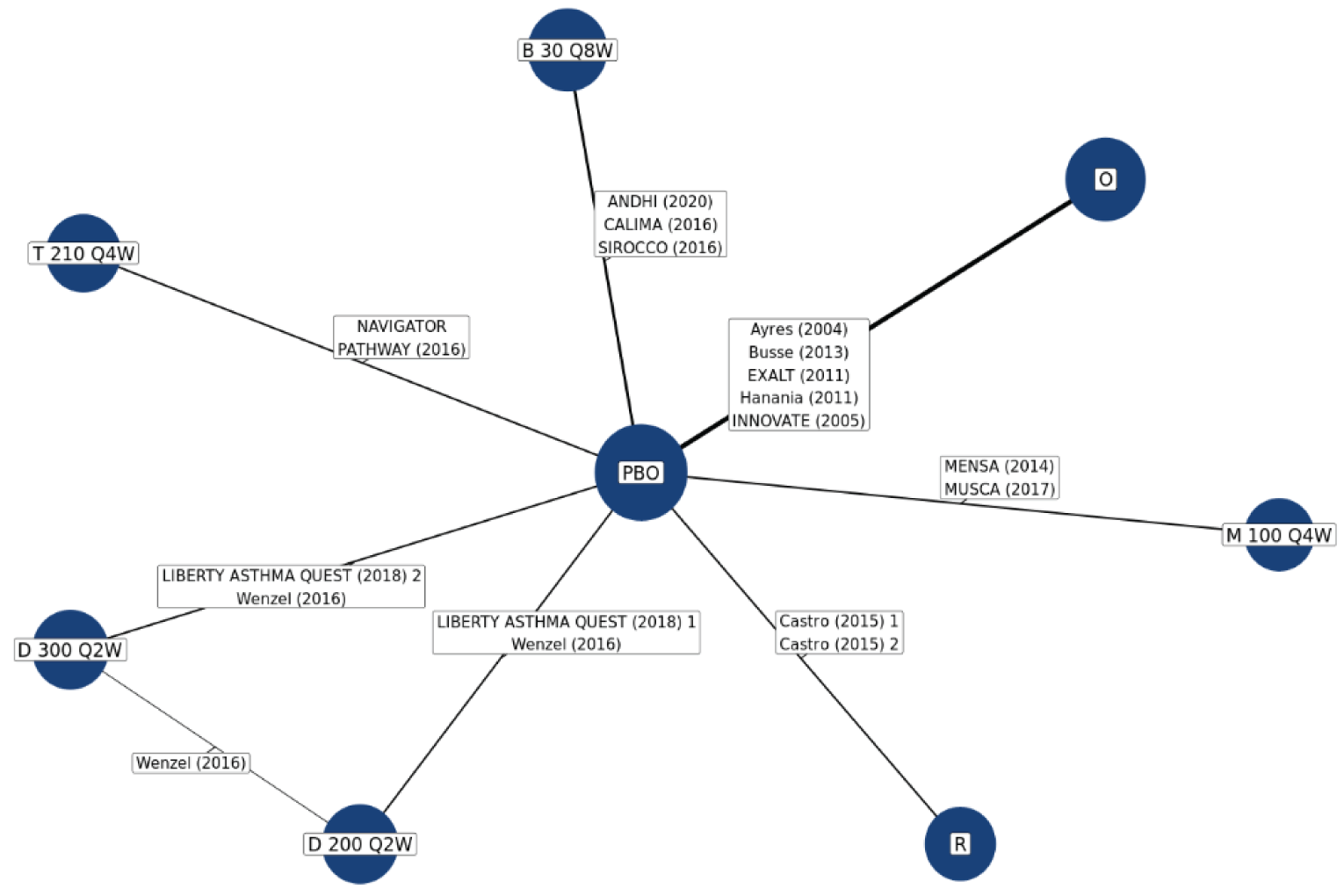
AAER = annualized asthma exacerbation rate; B = benralizumab; BSC = best standard care; D = dupilumab; M = mepolizumab; O = omalizumab; OAT = optimized asthma therapy; PBO = placebo; Q2W = every 2 weeks; Q4W = every 4 weeks; R = reslizumab; t = tezepelumab.
Source: Sponsor-submitted Indirect treatment comparison.10
Sixteen relevant studies reporting AAER were included, resulting in a network of 10,092 patients. Based on the number of studies included and the model fit statistics, the random-effects model was considered the best fit. In terms of the AAER, tezepelumab was not favoured over other biologics (Table 16).
Subgroup Analyses
For patients with an eosinophil count of 150 cells/μL or higher, tezepelumab showed a lower AAER compared with omalizumab (relative risk [RR] = 0.63; 95% Crl, 0.43 to 0.94) and benralizumab (RR = 0.63; 95%Crl, 0.49 to 0.82). There appeared to be no differences between tezepelumab when compared with mepolizumab and dupilumab.
For patients with an eosinophil count of 300 cells/μL or higher and lower than 300 cells/µL, 3 or more exacerbations in past 12 months, and allergic asthma, tezepelumab was not favoured over the other biologics.
For eosinophil counts below 150 cells/μL, tezepelumab had a lower AAER compared with dupilumab 300 mg (RR = 0.48; 95% Crl, 0.28 to 0.84).
Sensitivity Analyses
Overall, the results of the sensitivity analyses were aligned with the primary analyses of reduction in AAER.
Reduction in AAER Leading to Hospitalizations
Primary Analyses
A visual representation of the evidence network for reduction of AAER leading to hospitalizations is provided in Figure 6.
Figure 6: Evidence Network for AAERs Leading to Hospitalizations
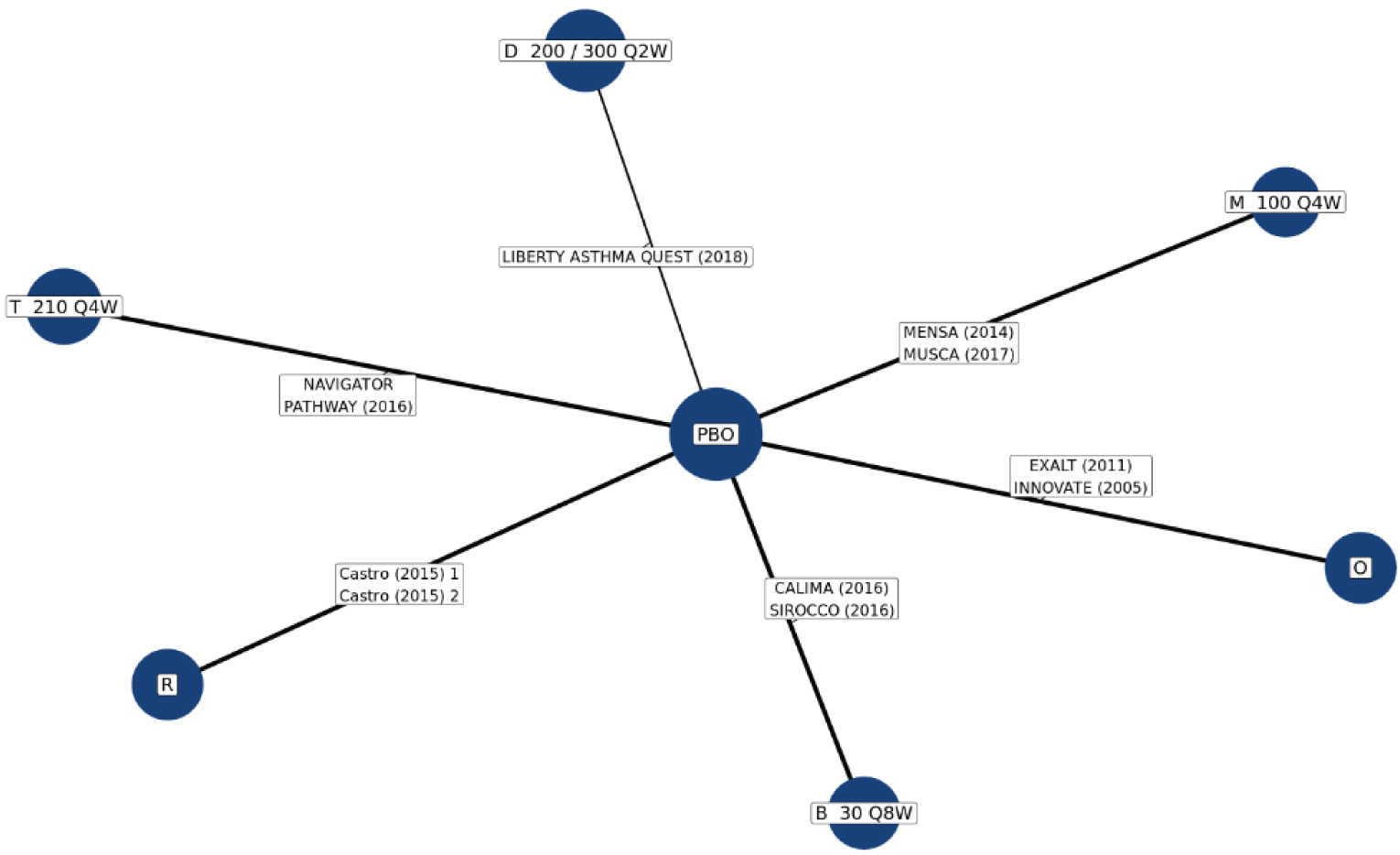
AAER = annualized asthma exacerbation rate; B = benralizumab; BSC = best standard care; D = dupilumab; M = mepolizumab; O = omalizumab; OAT = optimized asthma therapy; PBO = placebo; Q2W = every 2 weeks; Q4W = every 4 weeks; R = reslizumab; t = tezepelumab.
Source: Sponsor-submitted indirect treatment comparison.10
Eleven relevant studies reporting AAER leading to hospitalizations were included, which resulted in a network of 6,965 patients. Unlike AAER, only pooled data were available for the 2 dosages (200 mg and 300 mg every 2 weeks) of dupilumab. All trials except for 1, the EXALT (2011) study for omalizumab, reported the rate of exacerbations resulting in hospitalization or emergency room/department visit. The EXALT (2011) study reported the rate of exacerbations resulting in hospitalization. Based on the number of studies included and the adequate model fit statistics, the random-effects model was considered the best fit. The NMA found that tezepelumab was not favoured over other biologics for AAER leading to hospitalizations (Table 16).
Subgroup Analysis
No subgroup analyses were conducted for AAER leading to hospitalizations due to the lack of available data.
Sensitivity Analysis
The results of the sensitivity analysis for AAER leading to hospitalizations were consistent with the primary analysis.
Change From Baseline in Asthma Control Questionnaire Score
Primary Analysis
A visual representation of the evidence network for change in baseline ACQ score is provided in Figure 7.
Figure 7: Evidence Network for Change From Baseline in ACQ Score
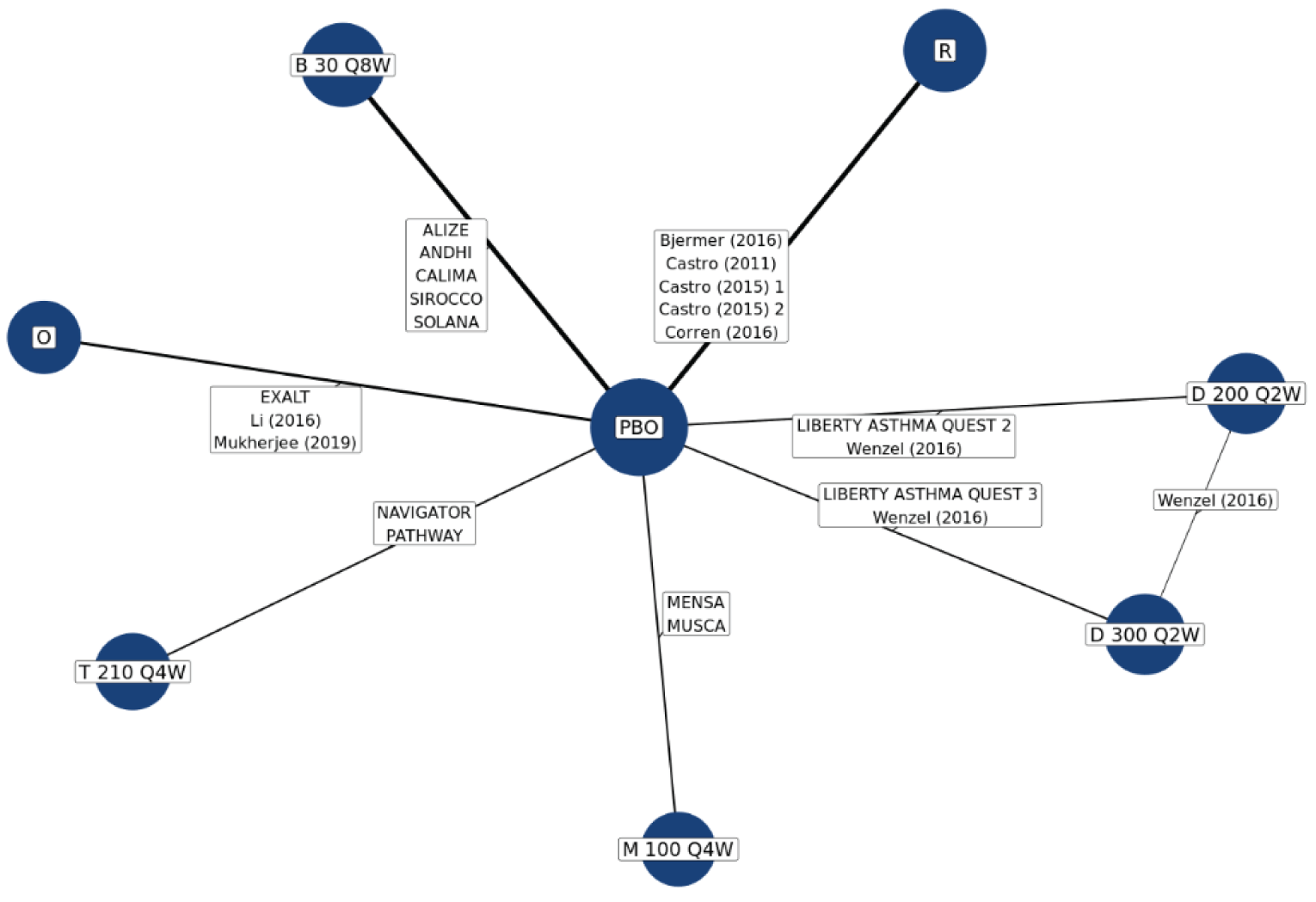
ACQ = Asthma Control Questionnaire; B = benralizumab; BSC = best standard care; D = dupilumab; M = mepolizumab; O = omalizumab; PBO = placebo; Q2W = every 2 weeks; Q4W = every 4 weeks; R = reslizumab; t = tezepelumab.
Source: Sponsor-submitted Indirect treatment comparison.10
A total of 19 relevant studies reporting ACQ score were included, resulting in a network of 8,791 patients. Based on the number of studies included and the adequate model fit statistics, the random-effects model was considered the best fit. The NMA indicated that tezepelumab was not favoured over other biologics for the change in ACQ (Table 17).
Subgroup Analysis
For patients with an eosinophil count of 150 cells/μL of higher, compared with benralizumab, tezepelumab demonstrated increased improvement in ACQ score (mean difference = −0.23; 95% CI, −0.4 to −0.04). Tezepelumab was not favoured over other biologics in any other subgroup analysis.
Sensitivity Analysis
The results of the sensitivity analysis for ACQ score were consistent with the primary analysis.
Change From Baseline in Pre-Bronchodilator FEV1
Primary Analysis
A visual representation of the evidence network for change from baseline in pre-bronchodilator FEV1 is provided in Figure 8.
Figure 8: Evidence Network for Change From Baseline in Pre-Bronchodilator FEV1
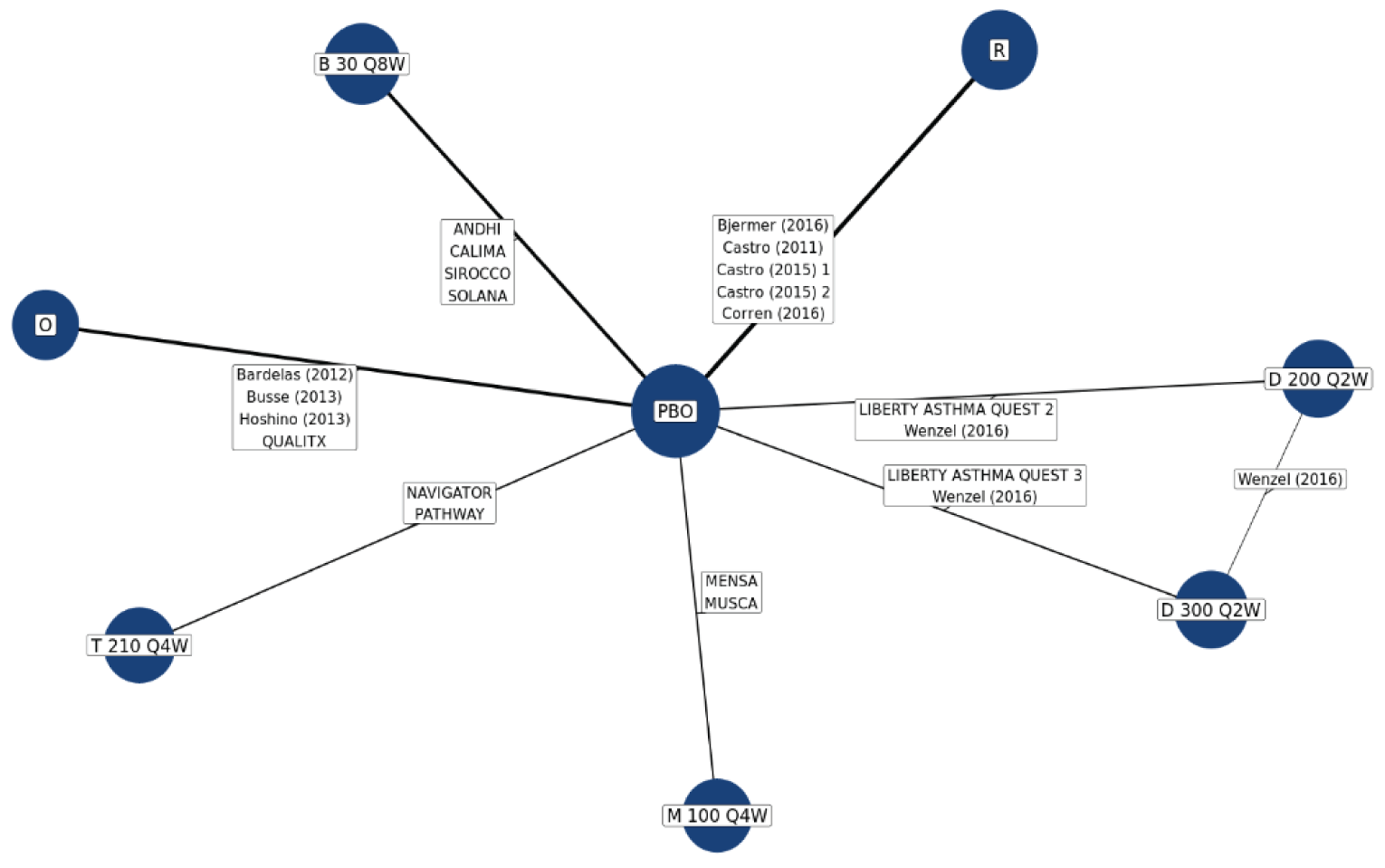
B = benralizumab; D = dupilumab; FEV1 = forced expiratory volume in 1 second; M = mepolizumab; O = omalizumab; PBO = placebo; Q2W = every 2 weeks; Q4W = every 4 weeks; Q8W = every 8 weeks; R = reslizumab; t = tezepelumab.
Source: Sponsor-submitted indirect treatment comparison.10
A total of 19 relevant studies reporting pre-bronchodilator FEV1 were included, which resulted in a network consisting of 9,046 patients. Based on the number of studies included and the adequate model fit statistics, the random-effects model was considered the best fit. The NMA indicated that, in terms of FEV1, tezepelumab was not favoured over other biologics studied (Table 17).
Subgroup Analysis
In terms of FEV1, the results of all subgroup analyses were consistent with the primary analysis, meaning tezepelumab was not favoured over other biologics.
Sensitivity Analysis
In terms of FEV1, the results of all sensitivity analyses were consistent with the primary analysis, meaning tezepelumab was not favoured over other biologics.
Change From Baseline in the OCS Dose Reduction of 50% or Greater
Primary Analysis
A visual representation of the evidence network for change from baseline in the OCS dose by predefined reduction categories is provided in Figure 9.
Figure 9: Evidence Network for Change From Baseline in the OCS Dose by Predefined Reduction Categories
B = benralizumab; D = dupilumab; M = mepolizumab; PBO = placebo; Q2W = every 2 weeks; Q4W = every 4 weeks; Q8W = every 8 weeks; t = tezepelumab.
Source: Sponsor-submitted indirect treatment comparison.10
Oral Corticosteroid Dose Reduction of 50% or Greater
Based on the number of single study connections in the network and the model fit statistics, the fixed-effects model was considered the best fit. The NMA indicated that tezepelumab was favoured to dupilumab 300 mg (OR = 0.36; 95% Crl, 0.14 to 0.93) for the outcome of OCS dose reduction of 50% or greater. No differences between tezepelumab and other biologics were identified (Table 17).
Subgroup Analysis
For patients with an eosinophil count of 300 cells/μL or greater, compared with mepolizumab, tezepelumab was favoured for the outcome of an OCS reduction of 50% or greater (OR = 11.07; 95% Crl, 1.17 to 312.70). Tezepelumab was not favoured over other biologics in this subgroup. Tezepelumab was not favoured over other biologics for the remaining subgroup analyses.
Sensitivity Analysis
Data of the sensitivity analysis for an OCS reduction of 50% or greater were consistent with that observed in primary analysis.82
Description of MAIC-STC
The MAIC-STC approach used individual patient-level data (IPD) from the tezepelumab trials, and weighted the trial population to match average baseline characteristics reported for the comparator trials. The IPD from the NAVIGATOR4 and SOURCE5 studies was used for tezepelumab for all MAIC and STC analyses. For comparators, the most relevant study or studies were selected based on a comparison of the study characteristics (e.g., follow-up duration), inclusion and exclusion criteria, availability of key potential treatment-effect modifiers, and reporting of outcomes. For some comparators, multiple studies were pooled as no clear rationale was identified for selecting 1 study over the other. Potential treatment-effect modifiers were identified based on use in previous published ITCs, clinical relevance, and input from the external experts.
Methods of Sponsor-Submitted MAIC-STC
MAIC-STC Analysis Methods
Anchored MAIC and STC analyses were performed using methods outlined by the NICE DSU Technical Support Document.81 The IPD for tezepelumab from the NAVIGATOR and SOURCE studies were used in the analyses, and aggregate data from relevant published trials were used for the comparators.
MAIC Analysis
Matching patients (i.e., IPD to match the inclusion and exclusion criteria of the comparator trial): First, the inclusion and exclusion criteria were aligned between the 2 studies being compared. Patients from the NAVIGATOR4 and SOURCE5 studies were removed from the IPD if they did not satisfy the eligibility criteria used in the relevant comparator RCT. The estimated treatment effect therefore represents that for tezepelumab in the populations studied in the comparator trials. Bucher ITCs, matched but unadjusted for any differences in potential treatment effect modifiers, were conducted and presented alongside MAIC results.
Adjustment for differences in treatment effect modifiers: After completing the matching phase of the anchored MAIC, patients remaining in the NAVIGATOR study IPD were re-weighted using the ranked treatment-effect modifiers (Table 14 provides a pre-specified list) that were available from RCTs of both treatments so that both their means and SDs matched those reported in the comparator trial. Inverse propensity score weighting, in which patients in the tezepelumab trial were weighted by the inverse odds of being in that trial versus the comparator trial, was used. The propensity score model was estimated using the generalized method of moments based on the aggregate data and IPD. Matching and adjusting were successful if the relevant treatment effect modifiers were similar between the treatment arms of the studies. Treatment outcomes were then compared between balanced trial populations.
Estimating indirect treatment effects: After matching patients from relevant comparator studies, a weighted estimate of the outcome was derived using anchored MAIC adjustment weights. The anchored MAIC was first performed using all of the ranked treatment-effect modifiers, data permitting. That is, the effect sample size decreases as the number of matching variables increases. Statistical significance testing was defined using a 2-tailed P value of less than 0.05, and all comparisons between groups were reported with point estimates (e.g., ratio ratios) and 95% CIs.
Primary analyses and summary measures of treatment effect by outcome: Exacerbation rates were modelled using a negative binomial generalized linear model (GLM) with anchored MAIC adjustment weights, using the number of person-years at risk. Rate ratios and 95% CIs were calculated. For changes from baseline in ACQ score and pre-bronchodilator FEV1, estimates were derived by fitting a GLM with anchored MAIC adjustment weights. The treatment effect between tezepelumab and the comparator was then derived as the difference between the predicted mean from the adjusted NAVIGATOR trial data and the estimated mean (based on summary-level data) from the comparator study. For the ACQ and FEV1, the relative treatment effect was defined as the mean difference in change from baseline between treatments. For change from baseline in OCS dose, a binomial GLM with anchored MAIC adjustment weights was used, and ORs with 95% CIs were calculated.
Performance assessment: The performance and suitability of each anchored MAIC model were assessed based on the following criteria: the effective sample size, distribution, and characteristics (i.e., zero or extreme values) of patient weights, and balance between patient populations on potential treatment-effect modifiers. All MAIC scenarios were conducted first with all potential treatment-effect modifiers, and then modifiers were subsequently dropped 1-by-one, starting with those ranked last, until a single modifier remained.
STC Analysis
Primary analyses and summary measures of treatment-effect by outcome: The AAER and AAER leading to hospitalizations were modelled using negative binomial regression, where the number of person-years at risk was used. Rate ratios and 95% CIs were calculated. For change from baseline in ACQ score and pre-bronchodilator FEV1, linear regression was used. The relative treatment effect was defined as the mean difference in change from baseline between treatments. For reduction in the OCS dose by 50% or greater, logistic regression was used. Odds ratios and 95% CIs were calculated.
General Model Selection Considerations
Regression models were fit using IPD from the NAVIGATOR and SOURCE studies, incorporating interaction terms between treatment and clinically relevant treatment-effect modifiers. The statistical performance of each regression model was assessed based on the criteria of model convergence and model fit statistics (e.g., Akaike information criterion). When models failed to converge, variables were either recategorized (i.e., by collapsing categorical variables into broader categories) or removed in order of least importance.
Results from MAIC-STC
Eleven studies4,32,49-51,55-60,67,68,77,78 were included in the MAICs and STCs.
A summary of MAIC and STC results from the primary analyses is presented in Table 16 and Table 17 (results are shown for the scenario that adjusted for the maximum number of variables, scenario A). Overall, MAIC-STC results for all of the outcomes evaluated did not demonstrate that tezepelumab was favoured against comparator treatments.
Critical Appraisal of the Sponsor-Submitted Network Meta-Analysis and MAIC-STC
Overall, the ITCs were conducted according to accepted methodological guidance. A systematic literature review was used as the basis for each ITC method. A pre-specified analysis plan for conducting NMAs and MAICs-STCs was used to guide the analyses. This included identification of potential sources of heterogeneity between trials and key clinical subpopulations that were then analyzed to further validate findings from the primary results. The ITCs were performed according to well-established methods outlined by the NICE DSU technical support documents.81,83 Geometry of the evidence networks was provided for each outcome analysis. Distribution and characteristics (i.e., zero or extreme values) of patient weights were assessed in each MAIC-STC.
Table 16: ITCs Results of AAER and Hospitalization due to AAER
Comparators | Outcome (tezepelumab vs. comparators) | |||||
|---|---|---|---|---|---|---|
AAER ratio (95% CrI for NMAs, 95% CI for MAICs and STCs) | Hospital AAER ratio (95% CrI for NMAs, 95% CI for MAICs and STCs) | |||||
NMA | MAICa | STCa | NMA | MAICa | STCa | |
Benralizumab | 0.63 (0.35 to 1.09) | 0.52 (0.18 to 1.45) | 0.69 (0.25 to 1.91) | 0.35 (0.08 to 1.16) | 0.08 (0.01 to 0.70) | 0.21 (0.03 to 1.22) |
Dupilumab 200 mg | 0.84 (0.45 to 1.56) | 0.82b (0.40 to 1.68) | 0.96 (0.47 to 1.96) | 0.36 (0.07 to 1.59) | 0.38b (0.13 to 1.13) | 0.38 (0.12 to 1.22) |
Dupilumab 300 mg | 0.84 (0.45 to 1.56) | 0.79b (0.39 to 1.59) | 0.92 (0.46 to 1.86) | |||
Omalizumab | 0.60 (0.35 to 1.01) | 1.23b (0.53 to 2.87) | 0.80 (0.41 to 1.58) | 0.40 (0.10 to 1.55) | 1.15b (0.19 to 6.94) | 0.88 (0.19 to 4.04) |
Mepolizumab | 0.82 (0.43 to 1.49) | 0.74 (0.33 to 1.67) | 0.92 (0.42 to 2.02) | 0.54 (0.13 to 2.00) | 0.80 (0.19 to 3.30) | 0.73 (0.19 to 2.81) |
AAER = annualized asthma exacerbation rate; CI = confidence interval; CrI = credible interval; MAIC = matching adjusted indirect comparison; NMA = network meta-analysis; STC = simulated treatment comparison.
aScenario A, which adjusts for the maximum number of variables, is presented.
bResults may not be robust due to removal of variables or a significant drop in effect sample size.
Sources: Sponsor-submitted indirect treatment comparisons.10,11
Table 17: Indirect Treatment Comparison results of ACQ1, FEV1, and OCS Reduction of 50% or Greater
Comparators | Outcome (Tez vs. comparators) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
ACQ mean difference (95% CrI for NMAs and CIs for MAICs and STCs) | FEV1 mean differences (95% CrI for NMAs and CIs for MAICs and STCs) | OCS ≥ 50% reduction, odds ratio (95% CrI for NMAs and CIs for MAICs and STCs) | |||||||
NMA | MAICa | STCa | NMA | MAICa | STCa | NMA | MAICa | STCa | |
Benralizumab | −0.01 (−0.30 to 0.28) | −0.17 (−0.48 to 0.14) | −0.08 (−0.62 to 0.46) | 0.02 (−0.07 to 0.11) | 0.14 (0.02 to 0.26) | 0.08 (−0.25 to 0.42) | 0.38 (0.14 to 1.07) | 5.25b (0.19 to 141.30) | 24.93 (1.24 to 502.97) |
Dupilumab 200 mg | 0.04 (−0.29 to 0.36) | 0.14b (−0.06 to 0.34) | 0.14 (−0.41 to 0.68) | −0.01 (−0.10 to 0.08) | 0.02b (−0.06 to 0.09) | −0.01 (−0.34 to 0.33) | NA | NA | NA |
Dupilumab 300 mg | −0.06 (−0.38 to 0.27) | −0.05b (−0.25 to 0.15) | −0.05 (−0.59 to 0.50) | −0.00 (−0.09 to 0.09) | 0.03b (−0.04 to 0.11) | 0.01 (−0.33 to 0.34) | 0.36 (0.14 to 0.93) | 0.63 (0.22 to 1.83) | 2.85 (0.41 to 19.81) |
Omalizumab | 0.16 (−0.19 to 0.51) | 0.61b (0.31 to 0.91) | 0.48 (−0.10 to 1.07) | 0.08 (−0.01 to 0.18) | 0.12 (−0.00 to 0.16) | 0.05 (−0.29 to 0.40) | NA | NA | NA |
Mepolizumab | 0.10 (−0.24 to 0.45) | 0.29b (−0.54 to 1.12) | 0.11 (−0.50 to 0.72) | 0.02 (−0.07 to 0.12) | 0.15 (0.04 to 0.27) | 0.07 (−0.27 to 0.42) | 0.54 (0.20 to 1.47) | 1.24b (0.04 to 35.09) | 10.12 (1.00 to 102.48) |
ACQ = Asthma Control Questionnaire; CI = confidence interval; CrI = credible interval; FEV1 = forced expiratory volume in the first second; MAIC = matching adjusted indirect comparison; NA = not applicable; NMA = network meta-analysis; STC = simulated treatment comparison; Tez = tezepelumab.
aScenario A, which adjusts for the maximum number of variables, is presented.
bResults may not be robust due to removal of variables or a significant drop in effect sample size.
Sources: Sponsor-submitted indirect treatment comparisons.10,11
The key limitation of the NMA was the heterogeneity across the included studies in terms of the eligibility criteria, study designs, patient characteristics, comparators, and outcome assessment. Several trials, including the tezepelumab trials, did not select patients based on peripheral eosinophil levels. As a result, patients were included in the analyses with wide ranges of baseline eosinophil counts, and the analyses likely included patients who would not actually be eligible for mepolizumab or benralizumab, based on their respective trial inclusion criteria and approved indications. Some trials included adult patients only (≥ 18 years) and some trials included those aged ≥ 12 years, although patients younger than 18 years accounted for a small percentage of the populations. As well, some of the included studies enrolled patients with moderate-to-severe uncontrolled asthma. The proposed tezepelumab indication and requested reimbursement criteria are for adults and adolescents 12 years and older with severe asthma. No subgroup analysis was conducted for severe asthma only. Doses of ICE also varied. For example, mepolizumab studies used higher ICS doses, whereas ICS doses for the benralizumab studies were medium or high. There were some variations in terms of the outcome evaluation time points across trials and differences in the study design (e.g., randomization versus partial randomization, blind versus open label, and parallel versus cross over). Differences were also observed for potential treatment-effect modifiers, exacerbations in the past 12 months, and number of OCS users in the included studies. The differences in these potential treatment-effect modifiers were described as clinically important by the clinical expert consulted by CADTH and may affect the validity of NMA results. Indeed, the technical report for the NMA pre-specified numerous potential sources of heterogeneity and effect modifiers, and acknowledged that key assumptions, including homogeneity, may have been violated given the differences between the included studies. No meta-regression analyses to adjust for factors that may bias comparisons were conducted, but random-effects models were used where possible and the impact of the various sources of heterogeneity was explored using subgroup and sensitivity analyses where data were available.
Appropriate MAIC and STC methods were used to try to account for the heterogeneity. All analyses were anchored through placebo, except for some outcomes comparing tezepelumab to omalizumab, in which an assumption was made that the best supportive care arm in the EXALT trial of omalizumab and the placebo arm of the NAVIGATOR trial could be considered a common comparator. It is difficult to determine to what extent this may have affected comparative estimates, particularly when considering that many (perhaps all) patients in other trials who received placebo would have been receiving standard-of-care treatment (i.e., ICS with or without a LABA) in the background.
The ITC technical report outlined the pre-specified steps involved in identifying and clinically validating the important effect modifiers and matching factors for the ITCs. The clinical expert consulted by CADTH agreed that the list of potential effect modifiers included most of the key factors to consider. Given the availability of only aggregate data for the comparator trials, limited or a lack of reporting on certain covariates, and narrow criteria used in certain trials, many of the identified effect modifiers could not be included in the models, likely leading to residual heterogeneity between populations. The MAIC method reduces sample sizes when the matching process includes many variables, as was the case in the provided MAICs, which further compromised the already constrained precision of estimates on comparative efficacy. Reductions in effective sample size (i.e., > 90%) were observed in some MAIC analyses. In many instances randomization was not preserved and could result in biased estimates of the treatment effect. Differences in trial design, such as timing of outcome evaluation and duration of follow-up, cannot be accounted for using either method. These factors likely led to the observed similarity of the results between the NMA and MAIC or STC, unrealistic treatment-effect differences for some comparisons and outcomes, and the generally wide CIs that indicated limited precision and lack of confidence in the validity of the results. Other limitations included the exclusion of non-English publications. As there were no direct comparisons between treatments, the assessment of inconsistence was not applicable. Furthermore, safety outcomes and quality of life outcomes (such as the AQLQ[S]12+ questionnaire) were not assessed in the sponsor-submitted ITCs.
Description of Indirect Treatment Comparisons identified by a CADTH Literature Search
A total of 116 citations were identified in the literature search. Following screening of titles and abstracts, 113 citations were excluded and 3 potentially relevant reports from the electronic search were retrieved for full-text review. All of the 3 ITCs7-9 were considered relevant to this review.
All 3 published ITCs used Bayesian NMA methods to compare the available biologics, including tezepelumab, for moderate-to-severe asthma. The results of NMAs were similar to the sponsor-provided NMA, indicating that tezepelumab was not clearly favoured over the other biologics for any of the outcomes evaluated. The NMAs had the same limitations as the sponsor-provided analysis, primarily the high degree of heterogeneity across the studies.
Other Relevant Evidence
This section includes submitted LTE studies and additional relevant studies included in the sponsor’s submission to CADTH that were considered to address important gaps in the evidence included in the systematic review.
Long-Term Extension Studies
One LTE study, DESTINATION,12 provided evidence of the long-term safety and tolerability of tezepelumab 210 mg administered every 4 weeks subcutaneously in adults and adolescents with severe, uncontrolled asthma for up to 2 continuous years, including 1 year of treatment in the predecessor NAVIGATOR4 and SOURCE84 parent studies. Data presented are based on a clinical database lock date of December 9, 2021.
Methods
The DESTINATION trial is a phase III, multi-centre, double-blind, randomized, placebo-controlled, parallel-group LTE study for patients who completed the NAVIGATOR4 or SOURCE study.84 The primary objective of the DESTINATION trial was to evaluate the long-term safety and tolerability of tezepelumab over 140 weeks, inclusive of the treatment period of either parent study. Adults (18 to 80 years old) and adolescents (12 to 17 years old) who had continued to receive the investigational product and attended the end-of-treatment visit in 1 of the parent studies were eligible for enrolment. A total of 951 patients were enrolled and randomized at 182 centres in 18 countries into the DESTINATION study: 827 patients from the NAVIGATOR study and 124 from the SOURCE study. The study design and sequence of treatment periods are illustrated in Figure 10.
Patients previously randomized to 210 mg tezepelumab in either parent study were assigned to remain on 210 mg tezepelumab administered every 4 weeks subcutaneously in the DESTINATION study (tezepelumab plus tezepelumab group; n = 415 from NAVIGATOR, n = 60 from SOURCE). Patients previously randomized to the placebo arm in the parent studies were re-randomized at a 1:1 ratio to either tezepelumab 210 mg (placebo plus tezepelumab group; n = 206 from NAVIGATOR, n = 32 from SOURCE) or placebo (placebo plus placebo group; n = 205 from NAVIGATOR; n = 32 from SOURCE) administered every 4 weeks subcutaneously. Randomization was stratified by parent study. Patients recruited from the SOURCE study were followed up post-treatment for 12 weeks. Patients who were enrolled from the NAVIGATOR study who completed 100 weeks of tezepelumab treatment were eligible for either 12 weeks of follow-up or a 36-week extended follow-up. This review of the DESTINATION study focused on the results from the tezepelumab plus tezepelumab and placebo plus placebo groups.
The study design of DESTINATION is depicted in Figure 10.
Figure 10: DESTINATION Study Design
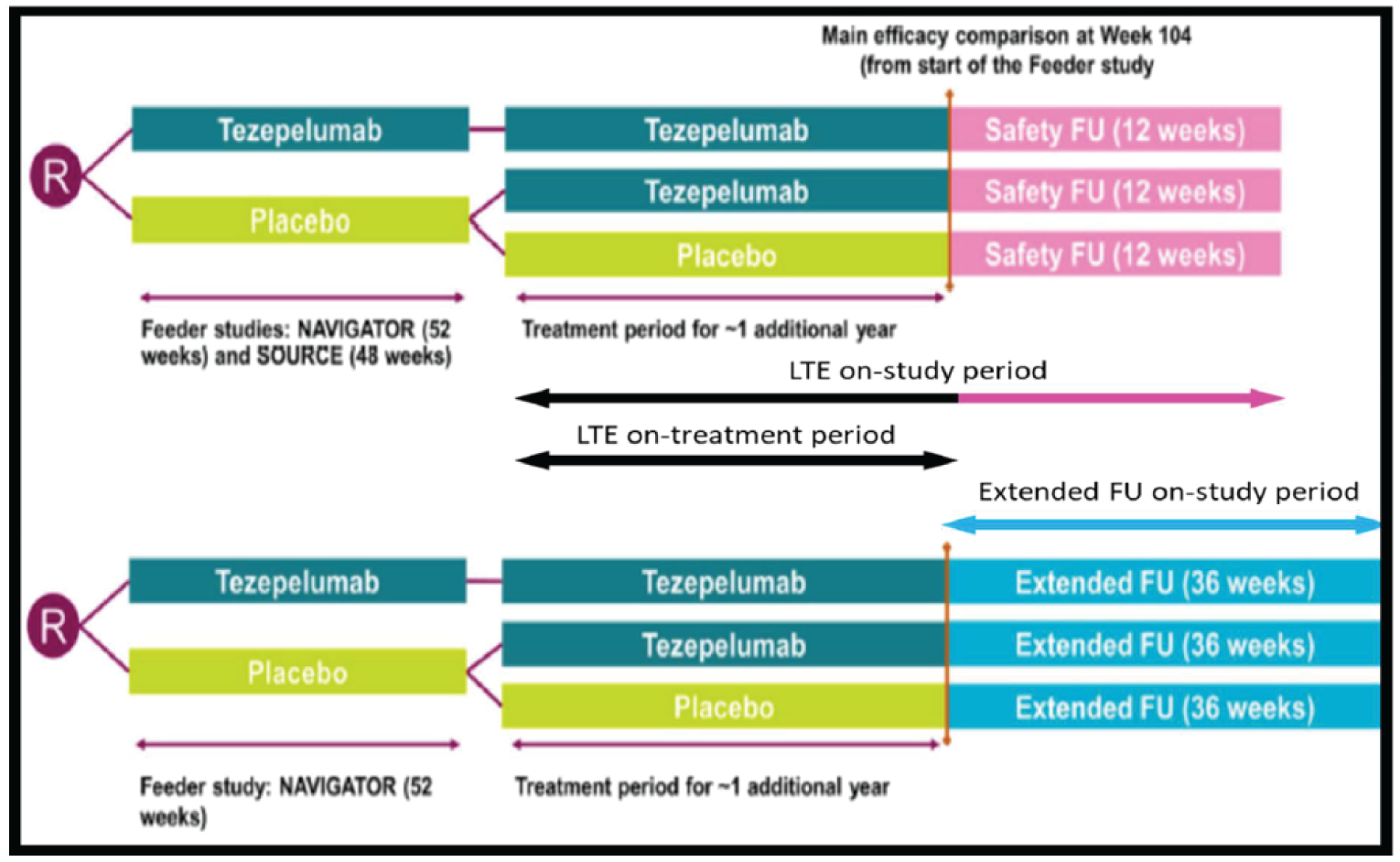
EOT = end of treatment; FU = follow-up; LTE = long-term extension; R = randomization; V = visit.
Source: Clinical Study Report for DESTINATION.12
The screening and/or randomization visit of the DESTINATION study was the same as the end-of-treatment visit from the parent NAVIGATOR (week 52) or SOURCE (week 48) studies.
Populations
Baseline patient and disease characteristics were as determined at baseline at time of enrolment in the parent study. Among patients who entered the DESTINATION study from the NAVIGATOR study, the mean age was 49 years (range = 12 to 80 years), and the majority were White (67%) and female (63%), and had baseline eosinophil counts of fewer than 300 cells/μL (58.4%) and FeNO levels below 25 ppb. Of patients who entered the LTE from the SOURCE study, the average age was 53.4 years (range = 22 to 76), and the majority were White (84%) and female (62.7%), with baseline eosinophil counts below 300 cells/μL (65.3%) and FeNO levels below 25 ppb. Overall, patients’ baseline characteristics were similar between the parent and LTE studies. The initial imbalances between groups in mean baseline eosinophil counts within the parent studies were no longer notable at LTE baseline.
Interventions
Investigational Products
The treatment protocol employed in the DESTINATION trial was similar to those described in the parent studies
The first dose of the investigational product was administered the same day as the end-of-treatment visit at week 52 and week 48 from the parent NAVIGATOR and SOURCE studies, respectively. The treatment-period duration was 52 weeks for patients who previously completed the NAVIGATOR study and 56 weeks for those who previously completed the SOURCE study. The last dose of the investigational product was administered at week 100 and the end-of-treatment visit was conducted at week 104. No investigational products were administered at week 104 or during the 12-week safety follow-up period.
Background Asthma Medication
In DESTINATION, tezepelumab was administered as an add-on maintenance therapy to ICS treatment plus at least 1 additional asthma-controller mediation with or without OCS, as described in the NAVIGATOR and SOURCE studies. Patients who were maintained on OCS, LABA, LAMA, theophylline, or LTRA in addition to ICS were allowed to continue treatment with these medications through the LTE. Starting from visit 1, investigators were also encouraged to consider stepping down the background medications when asthma symptoms were well controlled with stable lung function for at least 3 months in line with Global Initiative for Asthma guidelines.1
Concomitant Therapy
Investigators were permitted to prescribe concomitant medications or treatment to provide adequate supportive care as necessary, except for those medications identified as excluded in the prohibited medication list previously described for the NAVIGATOR and SOURCE studies.
Patients were permitted to use inhaled short-acting bronchodilators as needed as reliever or rescue medication due to worsening asthma. Any medications or vaccines, including over-the-counter or prescription medicines, vitamins and/or herbal supplements that the patients received at the time of enrolment or during the study were documented, along with the reason for use and dates of administration.
Outcomes
Primary Outcome
The primary outcome in DESTINATION was the exposure-adjusted incidence rates of AEs and SAEs over 104 weeks. For each treatment group, exposure-adjusted incidence rates were defined as the number of patients reporting AEs divided by the total exposure duration for that treatment group. For individual patients, exposure duration (days) was determined using the start and end dates of the applicable analysis period.
Secondary Outcome
The secondary objective of the trial was to assess the long-term effects of tezepelumab on asthma exacerbations which was measured as the AAER over 104 weeks where baseline was week 0 in the parent study.
Exploratory Outcomes
Exploratory outcomes included the long-term effects of tezepelumab on pulmonary function, asthma control, health status of patients with airway obstructive disease, lung function, other end points associated with asthma exacerbations; maintaining asthma control; and maintaining OCS dose at 5 mg or less.
Harms
All AEs and SAEs (new and ongoing from the parent study) were collected from the time patients signed the informed consent to the end of the treatment and follow-up periods. In addition, the following AEs of special interest were documented: anaphylactic reactions, immune complex disease (type III hypersensitivity reactions), hypersensitivity reactions, malignancy, helminth infections, severe infections, injection-site reactions, opportunistic infections, Guillain-Barré syndrome, and adrenal crisis.
Statistical Analysis
Sample-Size Determination
The sample size for the DESTINATION study was not based on statistical consideration but determined by the number of patients who completed the double-blind treatment periods in either of the parent studies and met all eligibility to enter the LTE study.
Statistical Methods
No formal hypothesis-testing was conducted in the DESTINATION study. Analyses of the primary (safety) and secondary (efficacy) end points and the exploratory end points were performed on data from the primary database lock (December 9, 2021), which was conducted after the last patient completed week 104 (October 26, 2021). Presentation of all results were stratified by parent study. Data were not aggregated across the 2 parent studies.
Analysis Population
Safety analyses were performed using the safety analysis set consisting of all patients who were randomized and received at least 1 dose of the investigational product in either of the parent studies, regardless of protocol adherence and continued participation in either the studies, and regardless of enrolment in the LTE study.
Efficacy analyses were performed using the FAS consisting of all patients who were randomized and received at least 1 dose of the investigational product in either parent study, irrespective of their protocol adherence and continued participation in either the studies or their enrolment in the LTE study. Efficacy analyses were also performed using the FAS-LTE set consisting of patients who were randomized and received at least 1 dose of the investigational product in the DESTINATION study. Patients were assigned to their randomized treatment according to ITT principles.
Patient Disposition
Patient disposition is summarized in Table 18.
Patient rollover into the DESTINATION study was 94.6% and 90.5% of patients who completed the NAVIGATOR and SOURCE studies, respectively. Of the 951 adults and adolescents from the parent studies enrolled in the DESTINATION study, 827 patients (including 72 adolescents) were from the NAVIGATOR study and 124 were from the SOURCE study. A total of 950 patients received treatment in the DESTINATION study. Overall, patient disposition was similar across treatment groups among patients with NAVIGATOR as the parent study. For patients with SOURCE as the parent study, the overall patient disposition in the LTE was similar across the tezepelumab plus tezepelumab, and placebo plus tezepelumab groups with slightly fewer patients who completed both treatment and study for the placebo plus placebo groups.
Exposure to Study Treatments
Extent of Exposure
Durations of exposure to tezepelumab by parent study are summarized in Table 19. The overall total exposure to tezepelumab was 1,283.27 patient-years, consisting of 575.62 patient-years from treatment of 602 patients in the parent study period and 708.94 patient-years from treatment of 712 patients in the LTE study period.
Table 18: Patient Disposition for DESTINATION (LTE Analysis Set)
Disposition | Parent study | |||||
|---|---|---|---|---|---|---|
NAVIGATOR | SOURCE | |||||
Tez + Tez | Pbo + Pbo | Pbo + Tez | Tez + Tez | Pbo + Pbo | Pbo + Tez | |
N | 827 | 124 | ||||
Randomized | 415 | 206 | 206 | 60 | 32 | 32 |
Received treatment | 415 (100) | 206 (100) | 205 (99.5) | 60 (100) | 32 (100) | 32 (100) |
Did not receive treatment | 0 (0) | 0 (0) | 1 (0.5) | 0 (0) | 0 (0) | 0 (0) |
Death | 0 (0) | 0 (0) | 1 (0.5) | 0 (0) | 0 (0) | 0 (0) |
Completed treatment | 390 (94.0) | 192 (93.2) | 193 (93.7) | 58 (96.7) | 27 (84.4) | 32 (100) |
Discontinued treatment | 25 (6.0) | 14 (6.8) | 12 (5.8) | 2 (3.3) | 5 (15.6) | 0 (0) |
Patient withdrawal | 11 (2.7) | 7 (3.4) | 8 (3.9) | 1 (1.7) | 3 (9.4) | 0 (0) |
AE | 1 (0.2) | 2 (1.0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
Study-specific withdrawal criteria | 3 (0.7) | 1 (0.5) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
Lost to follow-up | 0 (0) | 1 (0.5) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
Due to COVID-19 pandemic | 1 (0.2) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
Other | 9 (2.2) | 3 (1.5) | 4 (1.9) | 1 (1.7) | 3 (9.4) | 0 (0) |
Discontinued treatment but completed study assessment | 14 (3.4) | 10 (4.9) | 7 (3.4) | 1 (1.7) | 3 (9.4) | 0 (0) |
Completed studya | 400 (96.4) | 200 (97.1) | 198 (96.1) | 54 (90.0) | 26 (81.3) | 29 (90.6) |
Withdrawn from study | 15 (3.6) | 6 (2.9) | 8 (2.9) | 2 (3.3) | 4 (12.5) | 1 (3.1) |
Death | 8 (1.9) | 2 (1.0) | 2 (1.0) | 1 (1.7) | 3 (9.4) | 0 (0) |
Lost to follow-up | 2 (0.5) | 1 (0.5) | 1 (0.5) | 0 (0) | 1 (3.1) | 0 (0) |
Withdrawal by patient | 3 (0.7) | 3 (1.5) | 3 (1.5) | 1 (1.7) | 3 (9.4) | 0 (0) |
Due to COVID-19 pandemic | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (3.1) |
Other | 2 (0.5) | 0 (0) | 2 (1.0) | 0 (0) | 0 (0) | 0 (0) |
Completed treatment and study | 386 (93.0) | 190 (92.2) | 191 (92.7) | 53 (88.3) | 23 (71.9) | 29 (90.6) |
Safety analysis set LTE | 415 | 206 | 205 | 60 | 32 | 32 |
Full analysis set LTE | 415 | 206 | 205 | 60 | 32 | 32 |
LTE = long-term extension; Pbo + Pbo = placebo parent plus placebo LTE; Pbo + Tez = placebo parent plus tezepelumab LTE; Tez + Tez tezepelumab parent plus tezepelumab LTE.
aIncluded patients who completed treatment and the LTE period, and patients who discontinued treatment but completed study assessments during LTE period.
Source: Clinical Study Report for DESTINATION.12
Table 19: Extent of Exposure for DESTINATION (LTE Analysis Set)
Exposure | Parent studies | |||
|---|---|---|---|---|
NAVIGATOR (N = 827) | SOURCE (N = 124) | |||
Tez + Tez (n = 415) | Pbo + Pbo (n = 206) | Tez + Tez (n = 60) | Pbo + Pbo (n = 32) | |
Overall | ||||
Mean (SD) | 725.4 (50.8) | 721 (60.1) | 732.1 (25.9) | 704.8 (71.2) |
Total patient-year exposure | 824.22 | 406.62 | 120.26 | 61.75 |
Parent period, days | ||||
Mean (SD) | 367.8 (8.1) | 366.9 (6.5) | 339,3 (7.3) | 338.4 (3.4) |
Total patient-year exposure | 417.91 | 206.95 | 55.73 | 29.65 |
LTE period, days | ||||
Mean (SD) | 358.6 (50.6) | 355 (60.8) | 393.8 (24.7) | 367.4 (71.0) |
Total patient-years exposure | 407.44 | 200.23 | 64.69 | 32.19 |
LTE = long-term extension; Pbo + Pbo = placebo parent plus placebo LTE; Pbo + Tez = placebo parent plus tezepelumab LTE; SD = standard deviation; Tez + Tez = tezepelumab parent plus tezepelumab LTE
Source: Clinical Study Report for DESTINATION.12
Treatment Adherence
Treatment adherence to the investigational product was consistently high (> 97%) during the parent and LTE study periods.
Efficacy
Asthma Exacerbations
Asthma exacerbations over 104 weeks are summarized in Table 20.
Among patients enrolled in the LTE from the NAVIGATOR study, tezepelumab plus tezepelumab resulted in a reduction in the rate of asthma exacerbation compared to placebo plus placebo (AAER = 0.50; 95% CI, 0.40 to 0.63). Similarly, treatment with tezepelumab plus tezepelumab reduced the rate of asthma exacerbations associated with hospitalization or ED visits compared with placebo plus placebo (AAER = 0.39; 95% CI, 0.22 to 0.69).
In patients enrolled in the LTE from the SOURCE study, the AAER for asthma exacerbations between tezepelumab plus tezepelumab and placebo plus placebo was 0.66 (95% CI, 0.37 to 1.19). For asthma exacerbations associated with hospitalization or ED visits, the AAER for tezepelumab plus tezepelumab versus placebo plus placebo was 0.27 (95% CI, 0.05 to 1.63).
Asthma Control
The ACQ-6 change scores are summarized in Table 21.
Table 20: Asthma Exacerbation for DESTINATION (FAS-LTE)
Asthma exacerbation | Parent study | |||
|---|---|---|---|---|
NAVIGATOR (N = 827) | SOURCE (N = 124) | |||
Tez + Tez (n = 415) | Pbo + Pbo (n = 206) | Tez + Tez (n = 60) | Pbo + Pbo (n = 32) | |
Annualized asthma exacerbation rate ratio over 104 weeks | ||||
Patients with at least 1 exacerbation, n (%) | 138 (33.3) | 88 (42.7) | 19 (31.7) | 14 (43.8) |
Number of events, n | 542 | 515 | 93 | 70 |
Total time at risk (years) | 804.8 | 389.6 | 115.6 | 59.1 |
Crude rate | 0.66 | 1.31 | 0.80 | 1.18 |
Annual exacerbation rate (95% CI) | 0.71 (0.62 to 0.83) | 1.43 (1.18 to 1.72) | 0.88 (0. 61 to 1.27) | 1.34 (0.84 to 2.15) |
Absolute difference from placebo | −0.71 (−1.00 to −0.43) | −0.46 (−1.16 to 0.25) | ||
Rate ratio (95% CI) | 0.50 (95% CI, 0.40 to 0.63) | 0.66 (0.37 to 1.19) | ||
P valuea | < 0.001 | 0.65 | ||
Annualized asthma exacerbation associated with hospitalization or emergency department visit | ||||
Patients with at least 1 exacerbation, n (%) | 12 (2.9) | 11 (5.3) | 1 (1.7) | 2 (6.3) |
Number of events, n | 37 | 44 | 6 | 9 |
Total time at risk (years) | 825.5 | 409.3 | 119.6 | 61.8 |
Crude rate | 0.04 | 0.11 | 0.05 | 0.15 |
Annual exacerbation rate (95% CI) | 0.04 (0.03 to 0.07) | 0.11 (0.07 to 0.18) | 0.06 (0.01 to 0.21) | 0.20 (0.04 to 0.93) |
Absolute difference from placebo | −0.07 (−0.12 to −0.02) | −0.15 (0.45 to 0.15) | ||
Rate ratio (95% CI) | 0.39 (0.22 to 0.69) | 0.27 (0.05 to 1.63) | ||
P valuea | 0.001 | 0.154 | ||
CI = confidence interval; FAS = full analysis set; LTE = long-term extension; Pbo + Pbo = placebo parent plus placebo LTE; Pbo + Tez = placebo parent plus tezepelumab LTE; Tez + Tez = tezepelumab parent plus tezepelumab LTE.
Note: The model used a negative binomial regression analysis with treatment, region, age group, history of exacerbations as covariates. The logarithm of the time at risk is used as an offset variable. Annual exacerbation rates displayed are estimated marginal rates from the model. Absolute difference is the difference between the marginal rates. The CIs for annual exacerbation rates and absolute differences are estimated via the delta method.
aNo formal hypothesis-testing was pre-specified.
Source: Clinical Study Report for DESTINATION.12
Table 21: Asthma Control Questionnaire-6, Time-Point Summary, and Change From Baseline for DESTINATION (FAS-LTE)
AC6 to 6 | Parent study | |||
|---|---|---|---|---|
NAVIGATOR (N = 826) | SOURCE (N = 124) | |||
Tez + Tez (n = 415) | Pbo + Pbo (n = 206) | Tez + Tez (n = 60) | Pbo + Pbo (n = 32) | |
Baselinea (week 0), parent period | ||||
n | 415 | 206 | 60 | 32 |
Mean (SD) | 2.84 (0.78) | 2.81 (0.83) | 2.42 (1.06) | 2.45 (1.24) |
Week 104, LTE period | ||||
n | 309 | 196 | 58 | 27 |
Mean (SD) | 1.18 (1.02) | 1.44 (1.08) | 1.26 (1.05) | 1.90 (1.45) |
Mean change from baseline | −1.65 (1.05) | −1.37 (1.14) | −1.15 (1.23) | −0.53 (1.26) |
LSM (SE) | −1.65 (0.05) | −1.34 (0.07) | −1.10 (0.14) | −0.36 (0.20) |
LSM difference (95% CI) comparison with placebo | −0.31 (−0.47 to −0.14) | −0.74 (−1.23 to −0.25) | ||
P value | < 0.001 | 0.004 | ||
CI = confidence interval; FAS = full analysis set; LSM = least squares mean; LTE = long-term extension; Pbo + Pbo = placebo parent plus placebo LTE; Pbo + Tez = placebo parent plus tezepelumab LTE; SE = standard error; Tez + Tez = tezepelumab parent plus tezepelumab LTE.
aBaseline is defined as the last non-missing measurement recorded before randomization in the parent study.
bNo formal hypothesis-testing was pre-specified.
Source: Clinical Study Report for DESTINATION.12
Improvement from baseline ACQ-6 score over the LTE study period was observed in the tezepelumab plus tezepelumab group compared to the placebo plus placebo group in patients who were originally enrolled in the NAVIGATOR study (LSM difference = 0.31; 95% CI, −0.47 to −0.14). Similar trends in ACQ-6 were observed in patients originally enrolled in the SOURCE study, in which the tezepelumab plus tezepelumab group saw an improvement from ACQ-6 over the LTE study period compared to placebo plus placebo (LSM difference = −0.74; 95% CI; −1.12 to −0.25).
Harms
The long-term safety of tezepelumab in patients with severe asthma was the primary end point of the DESTINATION study. Most patients enrolled in the DESTINATION study (> 66%) reported experiencing at least 1 AE. Reported harms for patients in the tezepelumab plus tezepelumab group and for those in the placebo plus placebo group during the LTE study period are summarized in Table 22.
Among patients who entered DESTINATION from NAVIGATOR, 66.7% and 71.4% of patients in the tezepelumab plus tezepelumab and placebo plus placebo reported at least 1 AE during the LTE study period, respectively. Among patients who remained on tezepelumab during the LTE period, AEs leading to discontinuation of the investigational product were reported by 4 patients (1%) and AEs leading to death were reported by 7 patients (1.7%). Among those who continued to received placebo in the LTE period, AEs leading to discontinuation of the investigational product were reported by 2 patients (1%) and AEs leading to death were reported by 1 patient (0.5%). Finally, SAEs during the LTE study period were reported in 35 patients (8.4%) and 22 patients (10.7%) in the tezepelumab plus tezepelumab and placebo plus placebo groups, respectively. Notable harms of interest reported during the LTE study period included hypersensitivity (0.5% in both the tezepelumab plus tezepelumab arm and placebo plus placebo arm) and injection-site reactions (0.5% and 1.5% in the tezepelumab plus tezepelumab group and the placebo plus placebo group, respectively).
Among patients who entered the DESTINATION study from the SOURCE trial, 71.1% and 68.8% of patients in the tezepelumab plus tezepelumab and placebo plus placebo groups reported at least 1 AEs during the LTE study period, respectively. Among the tezepelumab plus tezepelumab group during the LTE period, there were no AEs leading to discontinuation of the investigational product and 1 patient (1.7%) reported an AE leading to death. Among the placebo plus placebo in the LTE period, there were no reported AEs leading to discontinuation of the investigational product or death. Finally, SAEs during the LTE study period were reported in 7 patients (11.7%) and 4 patients (12.5%) in the tezepelumab plus tezepelumab and the placebo plus placebo group, respectively. No notable harms of interest were reported among patients who enrolled in the DESTINATION study during the LTE study period.
Table 22: Summary of Harms — DESTINATION, Safety Analysis Set
Harms | Parent studies | |||
|---|---|---|---|---|
NAVIGATOR | SOURCE | |||
Tez + Tez (n = 415) | Pbo + Pbo (n = 206) | Tez + Tez (n = 60) | Pbo + Pbo (n = 32) | |
Any AEs | ||||
Number of patients (%)b | 277 (66.7) | 147 (71.4) | 43 (71.1) | 22 (68.8) |
Any AEs leading to treatment discontinuation | ||||
Number of patients (%)b | 4 (1.0) | 2 (1.0) | 0 (0) | 0 (0) |
Any AEs with death outcome | ||||
Number of patients (%)b | 7 (1.7) | 1 (0.5) | 1 (1.7) | 0 (0) |
Any SAE, including events death outcome | ||||
Number of patients (%)b | 35 (8.4) | 22 (10.7) | 7 (11.7) | 4 (12.5) |
Notable harms, n (%) | ||||
Hypersensitivity reactions | 2 (0.5) | 1 (0.5) | 0 (0) | 0 (0) |
Helminth | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
Opportunistic infections | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
Injection-site reactions | 2 (0.5) | 3 (1.5) | 0 (0) | 0 (0) |
AE = adverse event; LTE = long-term extension; Pbo + Pbo = placebo parent plus placebo LTE; Pbo + Tez = placebo parent plus tezepelumab LTE; SAE = serious adverse event; Tez + Tez = tezepelumab parent plus tezepelumab LTE.
aNumber of subjects with AEs/SAEs divided by total time at risk across all subjects in given treatment group, multiplied by 100.
bSubjects with multiple events in the same category are counted only once in that category. Subjects with events in more than 1 category are counted once in each of those categories.
Source: Clinical Study Report for DESTINATION.12
Critical Appraisal
Internal Validity
The DESTINATION trial provided additional data on the long-term efficacy of tezepelumab relative to placebo. Statistical hypothesis-testing was not part of the design. A notable attribute of the DESTINATION LTE study was it was designed to maintain blinding from the parent studies and allow for longer-term tezepelumab versus placebo comparisons. However, blinding may have been compromised via accidental publishing of individual test results on the investigator’s portal (November 23, 2021) by the laboratory vendor before the primary database lock, which may have led to unblinding for investigators who may have viewed the data. Investigation confirmed that the results of 264 patients were viewed by investigators, of whom 214 had already completed the study, 47 were in extended follow-up period, and 3 were in the 12-week follow-up period. No new asthma exacerbations were reported during the period of November 23, 2021, to the primary database lock (December 9, 2021). The incident therefore appeared to have minimal impact on the integrity of the data of the study. It was reported that corrective and preventive actions were implemented thereafter.
Disposition and baseline characteristics were well balanced between treatment groups among patients who enrolled in the LTE period from the NAVIGATOR study. However, there were several imbalances between treatment groups among those who enrolled from the SOURCE study. First, fewer patients in the placebo plus placebo group completed the treatment protocol. Second, a greater proportion of patients in the placebo plus placebo group reported the use of additional controller medications at baseline. Although the direction of any bias is unclear, it is possible that the differential dropout rate between the 2 treatment groups may have introduced attrition bias in favour of the tezepelumab plus tezepelumab group. Likewise, while the direction of any bias is unclear, it is possible that the differential use of controller medication may have been a surrogate of disease severity and biased the results in favour of the tezepelumab plus tezepelumab group.
External Validity
Overall, the DESTINATION study population represented the population of patients with severe, uncontrolled asthma and severe, OCS-dependent asthma as derived from the NAVIGATOR and SOURCE parent studies, respectively. Patient enrolment from the parent studies into the DESTINATION trial was high (> 90%). At LTE baseline, patient characteristics were similar to parent studies’ baselines. Completion of the LTE study was consistently high (> 96%) across all treatment groups from the NAVIGATOR study. While completion of the LTE was lower among patients who entered the LTE study from the SOURCE study, completion of the LTE study remained greater than 80%. Given that the patients enrolled in the LTE study were originally from the NAVIGATOR and SOURCE parent studies, and the eligibility criteria remained the same, it is reasonable to expect that the same limitations to generalizability are relevant to the DESTINATION study.
Discussion
Summary of Available Evidence
Three multinational, sponsor-funded double-blind RCTs were included in this review. The NAVIGATOR study randomized 1,061 patients who were on medium- or high-dose ICS and who had 2 or more exacerbations in the past year, 1:1, to either tezepelumab or placebo over a treatment course of 52 weeks. The primary outcome was the AAER and key secondary outcomes included the AAER in patients with baseline eosinophil counts of less that 300 cells/µL, change from baseline in pre-bronchodilator FEV1, AQLQ(S)12+, and ACQ-6. The SOURCE study randomized 150 patients with OCS-dependent asthma, 1:1, to either tezepelumab or placebo over a treatment course of 48 weeks. The primary outcome was the percent reduction in OCS dose while not losing asthma control, and key secondary outcomes included the AAER, time to first asthma exacerbation, rate of asthma exacerbation associated with ED visits, urgent-care visits or hospitalization, and patients who did not experience an asthma exacerbation over 48 weeks. The PATHWAY study was a phase II, double-blind RCT that randomized 550 patients on medium- to high-dose ICS and at least 2 exacerbations (or 1 severe asthma exacerbation) in the past year, 1:1:1:1, to 3 different doses of tezepelumab, including the approved dose, or placebo, over a treatment course of 52 weeks. The primary outcome was the AAER and secondary outcomes included subgroups based on the primary outcome, change from baseline in FEV1, and the ACQ-6. Additional evidence was available from 2 ITCs submitted by the sponsor, an NMA and a MAIC-STC, and 3 published ITCs. There was also evidence from a LTE extension, the DESTINATION study, which followed patients from the NAVIGATOR and SOURCE trials for an additional year of treatment.
Across studies, the mean age of patients was between 49 and 53.5 years of age, and the majority were female, ranging between 59% and 68% of patients across studies. In the NAVIGATOR study, 62% of patients were White and 28% Asian, while 84% of patients in the SOURCE study and 91% of patients in the PATHWAY study were White. In the NAVIGATOR study, 60% of patients had 2 exacerbations in the past 12 months, while the remainder had more than 2, while in the PATHWAY study 78% of patients had 1 or 2 exacerbations while the remainder had 3 or more. In the SOURCE study, which did not require more than 1 exacerbation in the past 12 months, 43% of patients had 1 exacerbation, 35% had 2 exacerbations, and 23% had more than 2 exacerbations. In the NAVIGATOR study, 75% of patients were on high-dose ICS and the remaining were on medium-dose ICS, while in the SOURCE study, all but 1 patient were on high-dose ICS. According to the protocol, all patients in the SOURCE study were on OCS, while in the NAVIGATOR study 9% were on OCS and 8% were in the PATHWAY study.
Interpretation of Results
Efficacy
Evidence from the NAVIGATOR study, the largest of the included trials, suggests that tezepelumab reduces the AAER and improves symptoms and HRQoL in a population of patients whose asthma is poorly controlled on medium- to high-dose ICS. Tezepelumab has a proposed indication for use in severe asthma; however, the treatment effect in patients with OCS-dependent asthma is unclear. The SOURCE study featured a population with OCS-dependent asthma and failed to demonstrate superiority for tezepelumab over placebo for the primary outcome of OCS dose reduction and there was also no statistically significant difference in exacerbations between tezepelumab and placebo. The clinical expert consulted by CADTH for this review noted that it is possible that many of the patients in the SOURCE study were not OCS-dependent, as the placebo response was much higher than would be expected. Nevertheless, based on the reported findings from SOURCE, the clinical expert stated that there is currently no evidence that patients who are OCS-dependent will experience a clinically important benefit from tezepelumab treatment, especially for outcomes such as reduction in OCS use and the AAER. The clinical expert indicated that patients who are OCS-dependent are the most challenging to treat, and the chronic use of OCS can have a significant negative impact on patient health, given the litany of serious harms associated with this class of drugs. In their input to CADTH, patients identified OCS side effects as a major concern.
As an upstream inhibitor of TSLP, tezepelumab is designed to inhibit various mediators of asthma, including those currently targeted by biologics (IL-4 and IL-13, IL-5, and IgE), and others under investigation for asthma. A key potential advantage to this approach is that it may address an unmet need in asthma management by targeting pathways that mediate the pathophysiology of non–type 2 asthma, in addition to targeting type 2 asthma. Subgroup analyses from the NAVIGATOR study suggest that tezepelumab appears to be effective across these subtypes of asthma in reducing asthma exacerbations in patients with high eosinophil counts (450 cells/µL or greater; rate ratio = 0.23; 95% CI, 0.15 to 0.34) or low eosinophil counts (150 cells/µL or lower; rate ratio = 0.61; 95% CI, 0.42 to 0.88) and patients with allergic induced asthma (rate ratio = 0.42; 95% CI, 0.33 to 0.53) or non-allergen induced asthma (rate ratio = 0.49; 95% CI, 0.36 to 0.67). According to the clinical expert consulted by CADTH for this review, if tezepelumab has consistent efficacy across these subgroups, this would fill an important gap in the management of asthma.
There is evidence from a LTE study (DESTINATION), which included the populations of the NAVIGATOR and SOURCE studies, that the improvement in the AAER and symptoms seen in the NAVIGATOR study may continue through 2 years of treatment, although these findings are limited by the lack of formal hypothesis-testing in this study. Longer-term studies are important when assessing drugs for asthma, particularly for outcomes such as exacerbations, as, according to the clinical expert consulted by CADTH for this review, asthma is often influenced by environmental factors that can vary in intensity within the year and even between years. For example, wildfire season in parts of Canada can result in a worsening of asthma control, while pollen season may have the same effect in other parts of the country, and each can vary in intensity from year to year. A treatment period that lasts more than 1 year would therefore help compensate for these variations.
Currently, there are no head-to-head trials that have compared the efficacy of tezepelumab with relevant biologics of interest for patients with severe uncontrolled asthma. The sponsor submitted an ITC (NMA, MAIC-STC). In addition, 3 published ITCs were identified by CADTH. In terms of reduction of the AAER, reduction of hospitalization due to AAER, FEV1 improvement, symptom reduction (change of ACQ-6 score) and OCS reduction of 50% or greater, no clinical meaningful difference between tezepelumab and other biologics for the treatment of severe asthma were identified. Heterogeneity between the included studies was a major limitation of the NMA. The MAIC and STC approaches that were designed to adjust for the differences did not appear to improve confidence in the findings. In the opinion of the clinical expert consulted by CADTH, the data do not support the superiority of tezepelumab in patients with type 2 asthma. The clinical expert indicated that it is reasonable to expect noninferiority compared with other drugs for type 2 asthma.
Harms
Tezepelumab appears to be a relatively well-tolerated drug, based on the number of patients who discontinued therapy due to AEs, which ranged between 2% and 3% of patients on tezepelumab, and the number of patients were similar to that seen in the placebo group. There were no serious safety issues (i.e., black-box warnings) in the draft product monograph for tezepelumab, and the notable harms identified for this review were those that are seen, in general, with monoclonal antibodies, such as injection-site reactions, hypersensitivity reactions, and infections. Although infections are notable harms associated with all monoclonal antibodies, from a mechanistic standpoint there is potentially a greater concern with tezepelumab compared to other monoclonal antibodies used to treat severe asthma, given its inhibition of TSLP. Tezepelumab works high up in the inflammatory-immune response, and inhibiting TSLP is expected to inhibit a large variety of cytokines and other factors that are important in the immune response. However, there was no indication from the included studies that there is a higher risk of infections or serious or severe infections in patients who are taking tezepelumab.
Conclusions
There is evidence that tezepelumab reduces the rate of asthma exacerbations in patients whose asthma remains uncontrolled despite administration of medium- to high-dose ICS. This reduction in exacerbation risk appears to occur regardless of whether patients have type 2 or non–type 2 asthma. Additionally, tezepelumab appears to improve pulmonary function, as well as HRQoL and symptoms of asthma as measured by the AQLQ(S)12+ and ACQ-6. Data from an LTE suggest these benefits of tezepelumab on exacerbations and symptoms may continue through 2 years of treatment; however, these findings need to be confirmed in a study that formally compares tezepelumab to placebo over this time frame. There is no evidence that tezepelumab facilitates the reduction of OCS dose in patients with OCS-dependent asthma, or reduces exacerbations in these patients. With respect to harms, no obvious safety or tolerability issues are associated with tezepelumab, and this includes data from an extension with at least 2 years of follow-up. Indirect evidence suggests that the efficacy and harms of tezepelumab are comparable to those of other biologics used in the treatment of asthma, although the degree of heterogeneity between the studies included in the indirect comparisons precludes drawing concrete conclusions on the comparative results.
References
1.Global Initiative for Asthma. Global Initiative for Asthma. 2022; https://ginasthma.org/. Accessed 2022 Jun 7.
2.Statistics Canada. Table: 12-10-0096-08. Asthma, by age group. 2022; https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310009608, 2022 Jun 7.
3.Tezspire (tezepelumab): 110 mg/mL solution for injection, subcutaneous use; 210 mg single-use, pre-filled syringe; 210 mg single-use, pre-filled pen [product monograph]. Mississauga (ON): AstraZeneca Canada Inc; 2022 Jul 28.
4.Clinical Study Report: D5180C00007 [NAVIGATOR]. A multicentre, randomised, double-blind, placebo controlled, parallel group, phase 3 study to evaluate the efficacy and safety of tezepelumab in adults and adolescents with severe uncontrolled asthma (NAVIGATOR) [internal sponsor's report]. Gaithersburg (MD): AstraZeneca; 2021 Feb 26.
5.Clinical Study Report: D5180C00009 [SOURCE]. A multicentre, randomized, double-blind, placebo controlled, phase 3 study to evaluate the efficacy and safety of tezepelumab in reducing oral corticosteroid use in adults with oral corticosteroid dependent asthma (SOURCE) [internal sponsor's report]. Gaithersburg (MD): AstraZeneca; 2021 Mar 22.
6.Clinical Study Report: CD-RI-MEDI9929-1146 [PATHWAY]. A phase 2 randomized, double-blind, placebo-controlled study to evaluate the efficacy and safety of MEDI9929 in adult subjects with inadequately controlled, severe asthma [internal sponsor's report]. Cambridge (GB): MedImmune, LLC, a wholly owned subsidiary of AstraZeneca; 2018 Apr 5.
7.Ando K, Fukuda Y, Tanaka A, Sagara H. Comparative efficacy and safety of tezepelumab and other biologics in patients with inadequately controlled asthma according to thresholds of type 2 inflammatory biomarkers: a systematic review and network meta-analysis. Cells. 2022;11(5):26. PubMed
8.Edris A, De Feyter S, Maes T, Joos G, Lahousse L. Monoclonal antibodies in type 2 asthma: a systematic review and network meta-analysis. Respir Res. 2019;20(1):179. PubMed
9.Edris A, Lahousse L. Monoclonal antibodies in type 2 asthma: an updated network meta-analysis. Minerva Medica. 2021;112(5):573-581. PubMed
10.Network meta-analyses comparing tezepelumab with biologics in patients with moderate-to-severe uncontrolled asthma [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: TBC (tezepelumab), 210 mg, 110 mg/mL solution for subcutaneous injection. Mississauga (ON): AstraZeneca Canada Inc; 2022 Apr 1.
11.MAIC and STC analyses comparing tezepelumab with biologics among patients with moderate-to-severe uncontrolled athma [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: TBC (tezepelumab), 210 mg, 110 mg/mL solution for subcutaneous injection. Mississauga (ON): AstraZeneca Canada Inc; 2022 Apr 1.
12.Clinical Study Report: D5180C00018 [DESTINATION]. A multicentre, double-blind randomized, placebo controlled parallel group, phase 3 safety extension study to evaluate the safety and tolerability of tezepleumab in adults and adolescents with severe uncontrolled asthma (DESTINATION) [internal sponsor's report]. Gaithersburg (MD): AstraZeneca; 2022 Apr 8.
13.Flovent HFA (fluticasone propionate inhalation aerosol): 50, 125, and 250 mcg/metered dose; Flovent Diskus (fluticasone propionate powder for inhalation): 100, 250, and 500 mcg/blister [product monograph]. Mississauga (ON): GlaxoSmithKline Inc; 2021 Mar 1: https://pdf.hres.ca/dpd_pm/00060119.PDF. Accessed 2022 Jun 7.
14.Xolair (omalizumab): sterile powder for reconstitution, 150 mg vial; solution for injection, 75 mg and 150 mg pre-filled syringe [product monograph]. Dorval (QC): Novartis Pharmaceuticals Canada Inc; 2021 Sep 8: https://pdf.hres.ca/dpd_pm/00062845.PDF. Accessed 2022 Jun 7.
15.Nucala (mepolizumab for injection): 100 mg/mL lyophilized powder for subcutaneous injection; 100 mg/mL solution for subcutaneous injection [product monograph]. Mississauga (ON): GlaxoSmithKline Inc; 2021 Nov 5: https://pdf.hres.ca/dpd_pm/00063467.PDF. Accessed 2022 Jun 7.
16.Cinqair (reslizumab): concentrate for solution for intravenous infusion 10 mg/mL vial [product monograph]. Toronto (ON): Teva Canada Limited; 2017 Mar 16: https://pdf.hres.ca/dpd_pm/00038484.PDF. Accessed 2022 Jun 7.
17.Fasenra (benralizumab injection): 30 mg/mL solution for subcutaneous injection; Fasenra Pen, 30 mg/mL solution for subcutaneous injection [product monograph]. Mississauga (ON): AstraZeneca Canada Inc; 2020 Feb 10: https://pdf.hres.ca/dpd_pm/00054955.PDF. Accessed 2022 Jun 7.
18.Dupixent (dupilumab injection): solution for subcutaneous injection, 300 mg single-use syringe (300 mg/2 mL), 300 mg single-use pen (300 mg/2 mL), 200 mg singl-use syringe (200 mg/1.14 mL), 200 mg single-use pen (200 mg/1.14 mL), 100 mg single-use syringe (100 mg/0.67 mL) [product monograph]. Laval (QC): Sanofi-aventis Canada Inc; 2022 Mar 25: https://pdf.hres.ca/dpd_pm/00065186.PDF. Accessed 2022 Jun 7.
19.Drug Reimbursement Review sponsor submission: Tezspire (tezepelumab), 210 mg, 110 mg/mL solution for subcutaneous injection [internal sponsor's package]. Mississauga (ON): AstraZeneca Canada Inc; 2022 Apr 1.
20.AstraZeneca. Prescribing information: Tezspire (tezepelumab-ekko) for injection, for subcutaneous use Silver Spring (MD): U.S. Food and Drug Administration; 2021: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761224s000lbl.pdf. Accessed 2022 Jun 7.
21.Position statement: appropriate use of oral corticosteroids in asthma. Toronto (ON): Asthma Canada; 2021: https://asthma.ca/wp-content/uploads/2021/12/OCS-Position-Statement-2021-EN.pdf. Accessed 2022 Jun 7.
22.Sullivan PW, Ghushchyan VH, Globe G, Schatz M. Oral corticosteroid exposure and adverse effects in asthmatic patients. J Allergy Clin Immunol. 2018;141(1):110-116.e117. PubMed
23.FitzGerald JM, Lemiere C, Lougheed MD, et al. Recognition and management of severe asthma: a Canadian Thoracic Society position statement. Can J Respir Crit Care Sleep Med. 2017;1(4):199-221.
24.Aaron SD, Vandemheen KL, FitzGerald JM, et al. Reevaluation of diagnosis in adults with physician-diagnosed asthma. JAMA. 2017;317(3):269-279. PubMed
25.Tsuyuki RT, Midodzi W, Villa-Roel C, et al. Diagnostic practices for patients with shortness of breath and presumed obstructive airway disorders: a cross-sectional analysis. CMAJ Open. 2020;8(3):E605-E612. PubMed
26.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. J Clin Epidemiol. 2016;75:40-46. PubMed
27.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2022 May 3.
28.Menzies-Gow A, Colice G, Griffiths JM, et al. NAVIGATOR: a phase 3 multicentre, randomized, double-blind, placebo-controlled, parallel-group trial to evaluate the efficacy and safety of tezepelumab in adults and adolescents with severe, uncontrolled asthma. Respir Res. 2020;21(1):266. PubMed
29.Menzies-Gow A, Corren J, Bourdin A, et al. Efficacy and safety of tezepelumab in adults and adolescents with severe, uncontrolled asthma: results from the Phase 3 NAVIGATOR study. J Allergy Clin Immunol. 2021;147(2):AB249.
30.Menzies-Gow A, Corren J, Bourdin A, et al. Tezepelumab in adults and adolescents with severe, uncontrolled asthma. N Engl J Med. 2021;384(19):1800-1809. PubMed
31.Wechsler ME, Colice G, Griffiths JM, et al. SOURCE: a phase 3, multicentre, randomized, double-blind, placebo-controlled, parallel group trial to evaluate the efficacy and safety of tezepelumab in reducing oral corticosteroid use in adults with oral corticosteroid dependent asthma. Respir Res. 2020;21(1):264. PubMed
32.Corren J, Parnes JR, Wang L, et al. Tezepelumab in adults with uncontrolled asthma. N Engl J Med 2017;377(10):936-946. PubMed
33.Corren J, Chen S, Callan L, Gil EG. The effect of tezepelumab on hospitalizations and emergency department visits in patients with severe asthma. Ann Allergy Asthma Immunol. 2020;125(2):211-214. PubMed
34.Corren J, Garcia Gil E, Griffiths JM, et al. Tezepelumab improves patient-reported outcomes in patients with severe, uncontrolled asthma in the PATHWAY study. Ann Allergy Asthma Immunol. 2021;126(2):187-193. PubMed
35.Bateman ED, Esser D, Chirila C, et al. Magnitude of effect of asthma treatments on Asthma Quality of Life Questionnaire and Asthma Control Questionnaire scores: systematic review and network meta-analysis. J Allergy Clin Immunol. 2015;136(4):914-922. PubMed
36.Wyrwich KW, Khan SA, Navaratnam P, Nolte H, Gates DF, Jr. Validation and agreement across four versions of the asthma control questionnaire in patients with persistent asthma. Respir Med. 2011;105(5):698-712. PubMed
37.Wyrwich KW, Ireland AM, Navaratnam P, Nolte H, Gates DF, Jr. Validation of the AQLQ12+ among adolescents and adults with persistent asthma. Qual Life Res. 2011;20(6):903-912. PubMed
38.Juniper EF, Guyatt GH, Epstein RS, Ferrie PJ, Jaeschke R, Hiller TK. Evaluation of impairment of health related quality of life in asthma: development of a questionnaire for use in clinical trials. Thorax. 1992;47(2):76-83. PubMed
39.Sastre J, Olaguibel J, Vega JM, Del Pozo V, Picado C, Lopez Viña A. Cut-off points for defining asthma control in three versions of the Asthma Control Questionnaire. J Asthma. 2010;47(8):865-870. PubMed
40.Juniper EF, Bousquet J, Abetz L, Bateman ED. Identifying 'well-controlled' and 'not well-controlled' asthma using the Asthma Control Questionnaire. Respir Med. 2006;100(4):616-621. PubMed
41.Juniper EF, Svensson K, Mörk AC, Ståhl E. Measurement properties and interpretation of three shortened versions of the asthma control questionnaire. Respir Med. 2005;99(5):553-558. PubMed
42.Juniper EF, O'Byrne PM, Guyatt GH, Ferrie PJ, King DR. Development and validation of a questionnaire to measure asthma control. Eur Respir J. 1999;14(4):902-907. PubMed
43.Barnes PJ, Casale TB, Dahl R, Pavord ID, Wechsler ME. The Asthma Control Questionnaire as a clinical trial endpoint: past experience and recommendations for future use. Allergy. 2014;69(9):1119-1140. PubMed
44.Buchholz I, Janssen MF, Kohlmann T, Feng YS. A systematic review of studies comparing the measurement properties of the three-level and five-level versions of the EQ-5D. Pharmacoeconomics. 2018;36(6):645-661. PubMed
45.Feng YS, Kohlmann T, Janssen MF, Buchholz I. Psychometric properties of the EQ-5D-5L: a systematic review of the literature. Qual Life Res. 2021;30(3):647-673. PubMed
46.van Reenen M, Janssen B. EQ-5D-5L user guide: basic information on how to use the EQ-5D-5L instrument. Rotterdam (NL): EuroQol Research Foundation; 2015: https://apersu.ca/wp-content/uploads/2020/10/EQ-5D-5L_User-Guide.pdf. Accessed 2022 May 15.
47.McClure NS, Sayah FA, Xie F, Luo N, Johnson JA. Instrument-defined estimates of the minimally important difference for EQ-5D-5L index scores. Value Health. 2017;20(4):644-650. PubMed
48.National Institute for Health and Care Excellence. Methods for development of NICE public health guidance. Appendix F: quality appraisal checklist – quantitative intervention studies. London (BG): National Institute for Health and Care Excellence; 2012: https://www.nice.org.uk/process/pmg4/chapter/appendix-f-quality-appraisal-checklist-quantitative-intervention-studies. Accessed 2022 Jun 7.
49.Bleecker ER, FitzGerald JM, Chanez P, et al. Efficacy and safety of benralizumab for patients with severe asthma uncontrolled with high-dosage inhaled corticosteroids and long-acting beta2-agonists (SIROCCO): a randomised, multicentre, placebo-controlled phase 3 trial. Lancet. 2016;388(10056):2115-2127. PubMed
50.FitzGerald JM, Bleecker ER, Nair P, et al. Benralizumab, an anti-interleukin-5 receptor alpha monoclonal antibody, as add-on treatment for patients with severe, uncontrolled, eosinophilic asthma (CALIMA): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2016;388(10056):2128-2141. PubMed
51.Nair P, Wenzel S, Rabe KF, et al. Oral glucocorticoid-sparing effect of benralizumab in severe asthma. N Engl J Med. 2017;376(25):2448-2458. PubMed
52.Zeitlin PL, Leong M, Cole J, et al. Benralizumab does not impair antibody response to seasonal influenza vaccination in adolescent and young adult patients with moderate to severe asthma: results from the phase iiib ALIZE trial. J Asthma Allergy. 2018;11:181-192. PubMed
53.Harrison TW, Chanez P, Menzella F, et al. Onset of effect and impact on health-related quality of life, exacerbation rate, lung function, and nasal polyposis symptoms for patients with severe eosinophilic asthma treated with benralizumab (ANDHI): a randomised, controlled, phase 3b trial. Lancet Respir Med. 2021;9(3):260-274. PubMed
54.Panettieri RA, Welte T, Shenoy KV, et al. Onset of effect, changes in airflow obstruction and lung volume, and health-related quality of life improvements with benralizumab for patients with severe eosinophilic asthma: phase iiib randomized, controlled trial (SOLANA). J Asthma Allergy. 2020;13:115-126. PubMed
55.Wenzel S, Castro M, Corren J, et al. Dupilumab efficacy and safety in adults with uncontrolled persistent asthma despite use of medium-to-high-dose inhaled corticosteroids plus a long-acting beta2 agonist: a randomised double-blind placebo-controlled pivotal phase 2b dose-ranging trial. Lancet. 2016;388(10039):31-44. PubMed
56.Busse WW, Maspero JF, Rabe KF, et al. Liberty asthma QUEST: phase 3 randomized, double-blind, placebo-controlled, parallel-group study to evaluate dupilumab efficacy/safety in patients with uncontrolled, moderate-to-severe asthma. Adv Ther. 2018;35(5):737-748. PubMed
57.Rabe KF, Nair P, Brusselle G, et al. Efficacy and safety of dupilumab in glucocorticoid-dependent severe asthma. N Engl J Med. 2018;378(26):2475-2485. PubMed
58.Ortega HG, Liu MC, Pavord ID, et al. Mepolizumab treatment in patients with severe eosinophilic asthma. N Engl J Med. 2014;371(13):1198-1207. PubMed
59.Chupp GL, Bradford ES, Albers FC, et al. Efficacy of mepolizumab add-on therapy on health-related quality of life and markers of asthma control in severe eosinophilic asthma (MUSCA): a randomised, double-blind, placebo-controlled, parallel-group, multicentre, phase 3b trial. Lancet Respir Med. 2017;5(5):390-400. PubMed
60.Bel EH, Wenzel SE, Thompson PJ, et al. Oral glucocorticoid-sparing effect of mepolizumab in eosinophilic asthma. N Engl J Med. 2014;371(13):1189-1197. PubMed
61.Ayres JG, Higgins B, Chilvers ER, Ayre G, Blogg M, Fox H. Efficacy and tolerability of anti-immunoglobulin E therapy with omalizumab in patients with poorly controlled (moderate-to-severe) allergic asthma. Allergy. 2004;59(7):701-708. PubMed
62.Holgate ST, Chuchalin AG, Hebert J, et al. Efficacy and safety of a recombinant anti-immunoglobulin E antibody (omalizumab) in severe allergic asthma. Clinical and Experimental Allergy 34 (4): 632. Clin Exp Allergy. 2004;34(4):632-638. PubMed
63.Humbert M, Beasley R, Ayres J, et al. Benefits of omalizumab as add-on therapy in patients with severe persistent asthma who are inadequately controlled despite best available therapy (GINA 2002 step 4 treatment): INNOVATE. Allergy. 2015;60(3):309-316. PubMed
64.Novartis. NCT00567476: Omalizumab use and asthma-related quality of life in patients with severe persistent allergic asthma. ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2011: https://clinicaltrials.gov/ct2/show/NCT00567476. Accessed 2022 Jun 7.
65.Ohta K, Miyamoto T, Amagasaki T, Yamamoto M, 1304 Study Group. Efficacy and safety of omalizumab in an Asian population with moderate-to-severe persistent asthma. Respirology. 2009;14(8):1156-1165. PubMed
66.Chanez P, Contin-Bordes C, Garcia G, et al. Omalizumab-induced decrease of FcξRI expression in patients with severe allergic asthma. Respir Med. 2010;104(11):1608-1617. PubMed
67.Bousquet J, Siergiejko Z, Swiebocka E, et al. Persistency of response to omalizumab therapy in severe allergic (IgE-mediated) asthma. Allergy. 2011;66(5):671-678. PubMed
68.Hanania NA, Alpan O, Hamilos DL, et al. Omalizumab in severe allergic asthma inadequately controlled with standard therapy: a randomized trial. Ann Intern Med. 2011;154(9):573-582. PubMed
69.Bardelas J, Figliomeni M, Kianifard F, Meng X. A 26-week, randomized, double-blind, placebo-controlled, multicenter study to evaluate the effect of omalizumab on asthma control in patients with persistent allergic asthma. J Asthma. 2012;49(2):144-152. PubMed
70.Hoshino M OJ. Effects of adding omalizumab, an anti-immunoglobulin E antibody, on airway wall thickening in asthma. Respiration. 2012;83(6):520-528. PubMed
71.Rubin AS, Souza-Machado A, Andradre-Lima M, et al. Effect of omalizumab as add-on therapy on asthma-related quality of life in severe allergic asthma: a Brazilian study (QUALITX). J Asthma. 2012;49(3):288-293. PubMed
72.Busse W, Spector S, Rosen K, Wang Y, Alpan O. High eosinophil count: a potential biomarker for assessing successful omalizumab treatment effects. J Allergy Clin Immunol. 2013;132(2):485-486.e411. PubMed
73.Garcia G, Magnan A, Chiron R, et al. A proof-of-concept, randomized, controlled trial of omalizumab in patients with severe, difficult-to-control, nonatopic asthma. Chest. 2013;144(2):411-419. PubMed
74.Pasha MA, Jourd'Heuil D, Jourd'Heuil F, et al. The effect of omalizumab on small airway inflammation as measured by exhaled nitric oxide in moderate-to-severe asthmatic patients. Allergy Asthma Proc. 2014;35(3):241-249. PubMed
75.Li J, Kang J, Wang C, et al. Omalizumab improves quality of life and asthma control in Chinese patients with moderate to severe asthma: a randomized phase III study. Allergy Asthma Immunol Res. 2016;8(4):319-328. PubMed
76.Mukherjee M, Kjarsgaard M, Radford K, et al. Omalizumab in patients with severe asthma and persistent sputum eosinophilia. Allergy Asthma Clin Immunol. 2019;15:21. PubMed
77.Castro M, Mathur S, Hargreave F, et al. Reslizumab for poorly controlled, eosinophilic asthma: a randomized, placebo-controlled study. Am J Respir Crit Care Med. 2011;184(10):1125-1132. PubMed
78.Castro M, Zangrilli J, Wechsler ME, et al. Reslizumab for inadequately controlled asthma with elevated blood eosinophil counts: results from two multicentre, parallel, double-blind, randomised, placebo-controlled, phase 3 trials. Lancet Respir Med. 2015;3(5):355-366. PubMed
79.Bjermer L, Lemiere C, Maspero J, Weiss S, Zangrilli J, Germinaro M. Reslizumab for inadequately controlled asthma with elevated blood eosinophil levels: a randomized phase 3 study. Chest. 2016;150(4):789-798. PubMed
80.Corren J, Weinstein S, Janka L, Zangrilli J, M G. Phase 3 study of reslizumab in patients with poorly controlled asthma: effects across a broad range of eosinophil counts. Chest. 2016;150(4):799-810. PubMed
81.Dias S, Welton N, Sutton A, A A. A generalised linear modelling framework for pairwise and network meta-analysis of randomised controlled trials. (NICE DSU Technical Support Document 2). Sheffield (GB): Decision Support Unit, ScHARR, University of Sheffield; 2014: https://www.ncbi.nlm.nih.gov/books/NBK310366/pdf/Bookshelf_NBK310366.pdf. Accessed 2022 Jun 7.
82.AstraZeneca Canada Inc response to May 30, 2022 request for additional information regarding Tezspire (tezepelumab): requested addition tables and figure related to the long-term extension study DESTINATION [internal additional sponsor's information]. Mississauga (ON): AstraZeneca Canada Inc; 2022 Jun 3.
83.Phillippo DM, Ades AE, Dias S, Palmer S, Abrams KR, Welton NJ. Methods for population-adjusted indirect comparisons in submissions to NICE. (NICE DSU Technical Support Document 18). Sheffield (GB): Decision Support Unit, ScHARR, University of Sheffield; 2016: https://www.sheffield.ac.uk/media/34216/download?attachment. Accessed 2022 Jun 7.
84.Chierrito de Oliveira D, Guerrero de Sousa P, Borges Dos Reis C, et al. Safety of treatments for ADHD in adults: pairwise and network meta-analyses. J Atten Disord. 2019;23(2):111-120. PubMed
85.Diver S, Khalfaoui L, Emson C, et al. Effect of tezepelumab on airway inflammatory cells, remodelling, and hyperresponsiveness in patients with moderate-to-severe uncontrolled asthma (CASCADE): a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Respir Med. 2021;9(11):1299-1312. PubMed
86.Corren J, Ambrose CS, Salapa K, et al. Efficacy of tezepelumab in patients with severe, uncontrolled asthma and perennial allergy. J Allergy Clin Immunol Pract. 2021;9(12):4334-4342.e4336. PubMed
87.Corren J, Karpefors M, Hellqvist A, Parnes JR, Colice G. Tezepelumab reduces exacerbations across all seasons in patients with severe, uncontrolled asthma: a post hoc analysis of the PATHWAY phase 2b study. J Asthma Allergy. 2021;14:1-11. PubMed
88.Juniper EF, Chauhan A, Neville E, et al. Clinicians tend to overestimate improvements in asthma control: an unexpected observation. Prim Care Respir J. 2004;13(4):181-184. PubMed
89.Juniper EF, Guyatt GH, Ferrie PJ, Griffith LE. Measuring quality of life in asthma. Am Rev Respir Dis. 1993;147(4):832-838. PubMed
90.Hernandez G, Garin O, Dima AL, et al. EuroQol (EQ-5D-5L) validity in assessing the quality of life in adults with asthma: cross-sectional study. J Med Internet Res. 2019;21(1):e10178. PubMed
91.Crossman-Barnes CJ, Sach T, Wilson A, Barton G. The construct validity and responsiveness of the EQ-5D-5L, AQL-5D and a bespoke TTO in acute asthmatics. Qual Life Res. 2020;29(3):619-627. PubMed
92.van Dalen C, Harding E, Parkin J, Cheng S, Pearce N, Douwes J. Suitability of forced expiratory volume in 1 second/forced vital capacity vs percentage of predicted forced expiratory volume in 1 second for the classification of asthma severity in adolescents. Arch Pediatr Adolesc Med. 2008;162(12):1169-1174. PubMed
93.Juniper EF, Svensson K, Mork AC, Stahl E. Modification of the asthma quality of life questionnaire (standardised) for patients 12 years and older. Health Qual Life Outcomes. 2005;3:58. PubMed
94.Brooks R. The EuroQol Group after 25 years. New York (NY): Springer; 2013.
95.Sinnott PL, Joyce VR, Barnett PG. Guidebook: preference measurement in economic analysis. Menlo Park (CA): Health Economics Research Center; 2007: https://www.herc.research.va.gov/files/BOOK_419.pdf. Accessed 2022 Jun 7.
96.Hernandez HG, Pont À, Gari O, et al. AB022. Validity of the five-level new version of the EQ-5D in asthma patients. J Thorac Dis. 2016;8(S5):AB022-AB022.
97.Whalley D, Globe G, Crawford R, et al. Is the EQ-5D fit for purpose in asthma? Acceptability and content validity from the patient perspective. Health Qual Life Outcomes. 2018;16(1):160. PubMed
Appendix 1: Literature Search Strategy
Note that this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases:
MEDLINE All (1946–)
Embase (1974–)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: May 6, 2022
Alerts: Weekly search updates until project completion
Search filters applied: No filters were applied to limit the retrieval by study type.
Limits:
Publication date limit: none
Language limit: none
Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
MeSH | Medical Subject Heading |
exp | Explode a subject heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
# | Truncation symbol for one character |
adj# | Requires terms to be adjacent to each other within # number of words (in any order) |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Keyword heading word |
.dq | Candidate term word (Embase) |
.pt | Publication type |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multi-Database Strategy
(tezepelumab* or Tezspire* or amg-157 or amg157 or medi-9929 or medi9929 or medi-19929 or medi19929 or GTPL-8933 or GTPL8933 or RJ1IW3B4QX).ti,ab,kf,ot,hw,nm,rn.
1 use ezep
*tezepelumab/ or (tezepelumab* or Tezspire* or amg-157 or amg157 or medi-9929 or medi9929 or medi-19929 or medi19929 or GTPL-8933 or GTPL8933).ti,ab,kf,dq.
3 use oemezd
4 not (conference review or conference abstract).pt.
2 or 5
remove duplicates from 6
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
Search terms: Tezspire (tezepelumab); asthma
WHO ICTRP
International Clinical Trials Registry Platform, produced by the World Health Organization. Targeted search used to capture registered clinical trials.
Search terms: Tezspire (tezepelumab); asthma
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
Search terms: Tezspire (tezepelumab); asthma
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
Search terms: Tezspire (tezepelumab); asthma
Grey Literature
Search dates: May 3-9, 2022
Keywords: tezepelumab, asthma
Limits: none
Updated: Search updated prior to the completion of stakeholder feedback period
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Internet Search
Open Access Journals
Appendix 2: Excluded Studies
Note that this appendix has not been copy-edited.
Reference | Reason for exclusion |
|---|---|
Diver et al. (2021)85 | Comparator (placebo) |
Corren et al. (2021)86 | Post hoc analysis |
Corren et al. (2021)87 | Post hoc analysis |
Corren et al. (2021)34 | Outcome |
Appendix 3: Detailed Outcome Data
Note that this appendix has not been copy-edited.
Table 25: Subgroup Analysis Results From NAVIGATOR, SOURCE, and PATHWAY for the Primary Outcomes
Subgroup | NAVIGATOR | SOURCE | PATHWAY | |||
|---|---|---|---|---|---|---|
Tez N = 529 | Placebo N = 532 | Tez N = 74 | Placebo N = 76 | Tez N = 137 | Placebo N = 138 | |
AAER | Reduction in OCS dose | AAER | ||||
Eosinophil counts | ||||||
<150 | 1.04 N = 138 | 1.70 N = 138 | — | — | — | — |
Rate ratio (95% CI) | 0.61 (0.42, 0.88) | — | — | — | — | |
Cumulative OR (95% CI) | — | 0.40 (0.14, 1.13) | — | — | ||
≥150 | 0.89 N = 390 | 2.24 N = 393 | — | — | — | — |
Rate ratio (95% CI) | 0.39 (0.32, 0.49) | — | — | — | — | |
Cumulative OR (95% CI) | — | 2.58 (1.16, 5.75) | — | — | ||
<300 | 1.02 | 1.73 | — | — | 0.26 N = 75 | 0.80 N = 67 |
Rate ratio (95% CI) | 0.59 (0.46, 0.75) P < 0.001a | — | — | — | — | |
Cumulative OR (95% CI) | — | 0.70 (0.33, 1.47) | — | — | ||
≥300 | 0.79 | 2.66 | — | — | 0.21 N = 62 | 0.65 N = 71 |
Rate ratio (95% CI) | 0.30 (0.22, 0.40) | 3.49 (1.16, 10.49) | — | — | ||
Allergy status | ||||||
Any perennial FEIA positive | 0.85 N = 339 | 2.03 N = 341 | — | — | 0.23 N = 71 | 0.75 N = 80 |
Rate ratio (95% CI) | 0.42 (0.33, 0.53) | — | — | — | — | |
— | 2.21 (0.78, 6.28) | — | — | |||
All perennial FEIA negative | 1.09 N = 184 | 2.21 N = 177 | — | — | 0.22 N = 58 | 0.65 N = 50 |
Rate ratio (95% CI) | 0.49 (0.36, 0.67) | 0.89 (0.39, 2.02) | — | — | ||
ICS dose at study entry | ||||||
Medium | 0.85 N = 131 | 1.33 N = 132 | — | — | 0.20 N = 71 | 0.38 N = 73 |
Rate ratio (95% CI) | 0.64 (0.43, 0.95) | - | — | — | - | |
High | 0.87 N = 397 | 2.04 N = 398 | — | — | 0.27 N = 66 | 1.12 N = 65 |
Rate ratio (95% CI) | 0.40 (0.32, 0.49) | — | — | — | — | |
Exacerbations in the year before study | — | — | — | — | ||
2 or less | 0.70 N = 310 | — | — | — | — | - |
Rate ratio (95% CI) | 0.51 (0.40, 0.66) | — | — | — | — | |
>2 | 1.22 N = 218 | — | — | — | — | - |
Rate ratio (95% CI) | 0.37 (0.28, 0.49) | — | — | — | — | |
1 or 2 | — | — | — | — | 0.19 N = 117 | 0.37 N = 110 |
3 or more | — | — | — | — | 0.48 N = 20 | 2.13 N = 28 |
OCS at baseline | ||||||
Present | 2.12 N = 49 | 2.94 N = 51 | — | — | NR | NR |
Rate ratio (95% CI) | 0.72 (0.41, 1.26) | — | — | — | NR | |
Absent | 0.82 N = 479 | — | — | — | NR | NR |
Rate ratio (95% CI) | 0.41 (0.33, 0.50) | — | — | — | — | |
OCS ≤10mg Cumulative OR (95% CI) | — | — | 1.91 (0.92, 3.94) | — | — | |
OCS >10mg Cumulative OR (95% CI) | — | — | 0.56 (0.17, 1.87) | — | — | |
CI = confidence interval; FEIA = food and exercise-induced anaphylaxis; ICS = inhaled corticosteroids; OCS = oral corticosteroid; OR = odds ratio.
aThis outcome was part of the statistical hierarchy for NAVIGATOR.
Source: Clinical Study Report for NAVIGATOR,4 Clinical Study Report for SOURCE,5 and Clinical Study Report for PATHWAY6
Table 26: Summary of Study Populations in the Included RCTs for the ITCs
Study (year) | Population | Age | Weight | Blood EOS (cells/μL) | Asthma duration (years) | Other treatment | ACQ score |
|---|---|---|---|---|---|---|---|
Tezepelumab | |||||||
PATHWAY (2017)32 | Uncontrolled asthma | 18-75 | ≥40 kg | NR | NR | Medium-high ICS+LABA | ≥1.5 |
NAVIGATOR (2020)4 | Severe uncontrolled asthma | 12-80 | NR | NR | NR | (i)Medium-high ICS+LABA for 12 m (ii) > 500 μg fluticasone equivalent daily for ≥3 m. (iii) ≥ 1 additional controller for ≥3 m | ≥1.5 |
SOURCE (2020)5 | OCS-dependent asthma | 18-80 | ≥40 kg | NR | ≥1 | (i)Medium or high-dose ICS for ≥ 12 months. (ii) LABA + high-dose ICS (>500μg fluticasone equivalent) for ≥ 3 months. (iii) Additional maintenance controllers are allowed according to standard practice of care. The use of these medications must be documented for at least 3 m. (iv) OCS for ≥ 6 m prior to screening and on a stable dose of between ≥ 7.5 to ≤ 30mg (prednisolone equivalent) daily for ≥ 1 m | NR |
Benralizumab | |||||||
SIROCCO(2016)49 | Severe, uncontrolled eosinophilic asthma | 12-75 | ≥40 kg | NR | ≥1 | (i)>250μg fluticasone equivalent + LABA for ≥ 12 m before enrolment. (ii) ≥ 500 μg fluticasone equivalents/day + LABA for ≥ 3 m before enrolment | ≥1.5 |
CALIMA(2016)50 | Severe, uncontrolled eosinophilic asthma | 12-75 | ≥40 kg | NR | ≥1 | (i)>250μg fluticasone equivalent + LABA for ≥ 12 m before enrolment. (ii) >500 μg fluticasone equivalents/day + LABA for ≥ 3 m before enrolment | ≥1.5 |
ZONDA(2017)51 | OCS-dependent asthma | 18-80 | ≥40 kg | ≥150 | ≥1 | (i) >500 μg fluticasone and LABA for ≥ 6 months prior to enrolment. (ii) Chronic OCS for ≥6 continuous m directly preceding enrolment | NR |
ALIZE (2018)52 | Patients aged 12–21 years receiving medium-to high-dosage ICS/LABA | 12-21 | ≥40 kg | NR | NR | All patients had current treatment with ICS and LABA (ICS dosage ≥500 μg/day fluticasone propionate dry powder formulation or equivalent/day) | ≥1.5 |
ANDHI (2020)53 | Patients with uncontrolled, severe eosinophilic asthma | 18-75 | ≥40 kg | ≥300 OR ≥150 to <300 | ≥1 | Documented current treatment with high daily dosages of ICS plus ≥ 1 other asthma controller for at least 3 months prior to visit 1; other acceptable asthma controllers included a long-acting bronchodilator (LABA or LAMA), a leukotriene inhibitor, theophylline preparations or maintenance OCS (daily or every other day OCS requirement to maintain asthma control; maximum total daily dosage 20 mg prednisone or equivalent) | ≥1.5 |
SOLANA (2020)54 | Patients with severe eosinophilic asthma | 18-75 | ≥40 kg | ≥300 | NR | Severe asthma required treatment with ICS/LABA for ≥ 30 days before enrolment; patients needed to have ≥ 2 documented asthma exacerbations requiring systemic corticosteroid therapy or a temporary increase in maintenance OCS dosage within 12 months before enrolment | ≥1.5 |
Dupilumab | |||||||
Wenzel (2016)55 | Uncontrolled persistent asthma | ≥18 | NR | NR | ≥1 | (i) >250 μg fluticasone equivalent + LABA for ≥ 1 m | ≥1.5 |
LIBERTY ASTHMAQUEST (2018)56 | Uncontrolled asthma | ≥12 | ≥30 kg | NR | ≥1 | (i) Fluticasone ≥500 μg equivalent + up to two additional controllers | ≥1.5 |
LIBERTY ASTHMAVENTURE (2018)57 | OCS-dependent asthma | ≥12 | ≥30 kg | NR | ≥1 | (i) Maintenance OCS in the 6 months prior to visit 1 (stable dose for 4 weeks). (ii) 5 to 35 mg/day of prednisone/prednisolone or an equivalent at visit 1 and at the randomization visit. (iii) ≥500 μg daily dose of fluticasone equivalent + a second controller (i.e., LABA or leukotriene receptor antagonist for ≥3 m, with a stable dose of ICS for ≥1 month | NR |
Mepolizumab | |||||||
MENSA(2014)58 | Severe eosinophilic asthma | 12-82 | ≥45 kg | ≥150 at screening or ≥300 during the 1 year before screening | NR | (i) ≥ 880 μg/day fluticasone equivalent for ≥ 12 m. And (ii) current additional controller medication for ≥ 3 m, or having used and failed it for ≥ 3 successive m | NR |
MUSCA (2017)59 | Severe eosinophilic asthma | ≥12 | NR | ≥150 at screening or ≥300 during the 1 year before screening | NR | (i) ≥ 880 μg of fluticasone in the prior 12 m + controller for ≥ 3 m. For ICS-LABA: the local highest dose will meet criterion. For 12-17 y/o: ≥ 440 μg/day. For ICS-LABA: the local mid-strength dose will meet criterion | NR |
SIRIUS(2014)60 | OCS-dependent asthma | ≥12 | NR | ≥150 during optimization phase or ≥300 during the 1 year before screening | NR | Previous 6 months: (i) 5 to 35 mg/day of prednisone equivalent (stable dose for ≥ 4 w) and (ii) ≥ 880 μg/day fluticasone equivalent (≥ 440 for 12-17 y/o). (iii) Current additional controller medication for ≥ 3 m, or having used and failed it for ≥ 3 successive m in the prior 12 m | NR |
Omalizumab | |||||||
Ayres (2004)61 | Poorly controlled (moderate-to-severe) allergic asthma | 12-75 | NR | NR | ≥2 | ≥ 800 μg/day beclomethasone equivalent (400 for adolescents) | NR |
Holgate (2004)62 | Patients with severe allergic asthma requiring ≥ 1,000 mg/day fluticasone for symptom control | 12-75 | NR | NR | NR | All patients required ≥ 1,000 mg/day fluticasone for symptom control. Short-acting beta2-agonists were allowed as needed, along with continued use of LABA | NR |
INNOVATE (2005)63 | Severe persistent asthma who are inadequately controlled despite best available therapy | 12-75 | NR | NR | (i) > 1,000 μg/day of beclomethasone equivalent and LABA. (ii) Maximum 20 mg/day of OCS permitted, as long as ≥ 1 exacerbations occurred while on this therapy | NR | Positive to ≥ 1 perennial allergen 30 IU/mL to 700 IU/mL |
NCT00567476 (2007)64 | Severe persistent allergic asthma | 12-75 | 20-150 kg | NR | NR | Subject taking >500 μg/day of fluticasone or equivalent associated to a LABA | NR |
Ohta (2009)65 | Moderate-to-severe asthma | 20-75 | NR | NR | NR | Treatment with beclomethasone dipropionate chlorofluorocarbon-containing metered-dose inhaler at ≥800 μg/day (or equivalent), and ≥1 of the following additional controller medications recommended as Step 3 and Step 4 treatments (LABA, sustained-release theophylline, LTRA, OCS) | NR |
Chanez (2010)66 | Patients with severe persistent allergic asthma | ≥18 | According to omalizumab dosing tables | NR | NR | High dose ICS > 1,000 μg BDP or equivalent and an inhaled LABA | NR |
EXALT (2011)67 | Severe allergic (IgE-mediated) asthma | 12-75 | 20-150 kg | NR | NR | ≥ 800 μg/day beclomethasone equivalent + LABA during the 3 years prior to screening | NR |
Hanania (2011)68 | Severe, inadequately controlled, allergic asthma | 12-75 | 30-150 kg | NR | ≥1 | High-dose ICS-LABA | NR |
Bardelas (2012)69 | Patients with inadequately controlled, persistent, allergic asthma treated with Step 4 or higher asthma maintenance therapy according to the 2007 NHLBI guidelines. | ≥12 | ≤150 kg | NR | ≥1 | Prescription for at least a medium-dose ICS plus LABA (fluticasone 250μg/ salmeterol 50 μg one inhalation or budesonide 160 μg/formoterol 4.5 μg 2 inhalations twice daily); a medium-dose ICS plus either a LTRA, theophylline, or zileuton for ≥ 3 m | NR |
Hoshino (2012)70 | Severe allergic asthma | 20-75 | NR | NR | NR | Treatment with ≥400μg fluticasone propionate or its equivalent ICS and LABA for 8 weeks; other medications were also permitted | NR |
QUALITX (2012)71 | Severe persistent uncontrolled asthma | 12-75 | 20-150 kg | NR | NR | At least, ICS (≥500 μg/day of fluticasone equivalent) + LABA | NR |
Busse (2013)72 | Atopic asthma who remained symptomatic and uncontrolled on ICS with or without other controller medications despite having normal lung function | 12-75 | NR | NR | NR | NR | NR |
NATAIR (2013)73 | Severe, persistent, non-atopic uncontrolled asthma | 18-70 | NR | NR | NR | Daily high-dose ICS treatment (> 1,000 μg beclomethasone dipropionate or equivalent per day) plus a LABA with or without maintenance OCS | NR |
Pasha (2014)74 | Moderate-to-severe asthma | ≥12 | NR | NR | NR | ICS with or without LABA; ≥3 months of stable immunotherapy | NR |
Li (2016)75 | Chinese patients with moderate-to-severe allergic asthma | 18-75 | >20 and ≤150 | NR | ≥1 | Inadequately controlled symptoms despite medium-to-high-dose ICS+LABA; received medium- to high-dose ICS > 500 μg BDP or equivalent plus regularly inhaled LABA, either separately or in combination, for at least 8 weeks prior to screening | NR |
Mukherjee (2019)76 | Patients with confirmed asthma, atopy, who were symptomatic, with evidence of sputum eosinophils, despite high-dose maintenance corticosteroid therapy | 18-75 | NR | NR | NR | ICS (≥ 1,500 μg fluticasone propionate or equivalent) with or without additional prednisone | ≥1.5 |
ACQ = Asthma Control Questionnaire; BDP = beclomethasone dipropionate; DPI = dry powder inhaler; EOS = eosinophils; ICS = inhaled corticosteroid; IgE = immunoglobulin; IU = international units; kg = kilogram; LABA = long-acting β-agonists; LAMA = long-acting muscarinic antagonists; LTRA = leukotriene receptor antagonist; m = months; mg = milligram; NR = not reported; RAST = radioallergosorbent; SABA = short-acting beta2-agonist; μg = microgram; μL = microliter.
aIndicated 1 of the 2 studies, the study 1, presented in Castro (2015)78
bIndicated 1 of the 2 studies, the study 2, presented in Castro (2015)78
Source: Sponsor-submitted indirect treatment comparisons.10,11
Appendix 4: Description and Appraisal of Outcome Measures
Note that this appendix has not been copy-edited.
Aim
To describe the following outcome measures and review their measurement properties (validity, reliability, responsiveness to change, and MID):
Asthma Control Questionnaire-6 (ACQ-6)
Standardized Asthma Quality of Life Questionnaire for 12 years and older (AQLQ[S]+12)
EQ-5D-5L
Findings
Table 27: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions About Measurement Properties | MID |
|---|---|---|---|
ACQ-6 | A 7-point Likert type patient-reported instrument to assess the adequacy of asthma control and change in asthma control which occurs either spontaneously or as a result of treatment. The ACQ-6 comprises of the following questions:
The ACQ-6 scores are calculated as a mean of the 6 items, with scores of 0 meaning the patients has asthma that is well controlled and those with 6 indicating the patients has asthma that is extremely poorly controlled. Mean scores of ≤ 0.75 indicated adequately controlled asthma, scores between 0.75 and < 1.5 indicated partly controlled asthma, and a score of ≥ 1.5 indicated asthma that was not well controlled. | Validity: Demonstrated good internal consistency with a Cronbach alpha of 0.82 and 0.87 for the ACQ-6 excluding daily SABA use and the ACQ-6 excluding FEV1 respectively. The internal consistency was good with a Cronbach alpha of 0.84 and 0.88 for the ACQ-6 excluding daily SABA and the ACQ-6 excluding FEV1, respectively.36 Reliability: The test-retest reliability showed good agreement with ICC scores ranging from 0.84 for the ACQ-6 excluding daily SABA) to 0.88 for the ACQ-6 excluding FEV1.36 Responsiveness: Strongly for both baseline and change scores on the AQLA(S)+12 (|r| > 0.74),36 and demonstrated similar responsiveness to change as the original ACQ-7.36,88,89 | In adult patients with asthma, a MID of 0.49 on the ACQ-6 (all symptoms plus rescue SABA) is considered meanginful.41 The MID for adolescents was not identified in the literature. |
AQLQ(S)+12 | A 7-point Likert-type patient-reported instrument to assess symptoms, limitations to activity, emotional function, and environmental stimuli as a result of asthma in adults and children aged 12 and older. The overall score of the AQLQ(S)+12 is derived as a mean of the 32 questions, ranging from 1 to 7. Higher scores indicate less severe impairment. Furthermore, domain specific mean scores may be reported. | Validity: Demonstrated weak to moderate construct validity with other measures of clinical status, including % predicted FEV1, PEF, symptoms, night waking, and amount of rescue medication.37 When correlated with baseline ACQ score and daytime and night-time symptoms, the AQLA(S)+12 demonstrated moderate to strong construct and known-group validity.36 Reliability: Demonstrated high internal consistency (Cronbach alpha 0.96 and 0.97) and good test-retest reliability (ICC of 0.86 and 0.83).36 Responsiveness: Responsiveness to change correlated with change in ACQ.36 | No established MID for the AQLQ(S)+12 was identified; however, given the significant overlap between the AQLQ+S and the AQLQ, a cut point of 0.5 points is considered clinically meaningful.36,37 |
EQ-5D-5L | A generic preference based HRQoL instrument consisting of a VAS and a composite index score of 5 dimensions: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression. Scores less than 0 represent health states that are valued by society as being worse than dead, while scores of 0 and 1.00 are assigned to the health states “dead” and “perfect health,” respectively. | Validity: Demonstrated moderate to strong convergent validity with the Asthma Quality of Life Utility Index.90 Known-group validity demonstrated when the ACQ-5 was used to classify asthma severity,91 but not demonstrated when severity was classified using PEF values.47 Reliability: No evidence of reliability in patients with asthma was identified. Responsiveness: Effectively discriminates between patient-reported improvement or deterioration in asthma.47 | No established MID for patients with asthma was identified. A MID of 0.056 is established for the general use in the Canadian population.92 |
ACQ-5; 5-Item Asthma Control Questionnaire; ACQ-6 = 6-Item Asthma Control Questionnaire; AQLQ(S)+12 = Standardized Asthma Quality of Life Questionnaire for 12 years and older (AQLA[S]+12); EQ VAS = EuroQol Visual Analogue Scale; FEV1 = forced expiatory volume in the first second; ICC = intraclass coefficient; MID = minimal important difference; PEF = pea expiratory flow; SABA = short-acting beta agonist
6-Item Asthma Control Questionnaire (ACQ-6)
The ACQ-6 is a shortened version of the ACQ, which was developed as a simple assessment of adequacy of- and change in asthma control that can be completed in the clinic setting without daily recordings of symptom, medication use and airway calibre.42 The ACQ is used extensively in clinical trials to measure clinically meaningful change in asthma control.43 The original ACQ is comprised of 7 questions, the responses to which are scored on a 7-point Likert type scale. The questionnaire consists of 6 patient-reported items concerning their previous week’s experience regarding activity limitation, nocturnal waking, shortness of breath, wheezing, symptoms on waking, and use of a SABA.39 The seventh item includes a clinic-administered pre-bronchodilator FEV1 or PEF.39,43 Since the development of the 7-item ACQ (ACQ-7), shortened version of the instrument have been constructed: a 5-item ACQ (ACQ-5) and 2 versions a 6-item ACQ (ACQ-6). The ACQ-5 includes only the symptoms items of the of the ACQ-7. The first ACQ-6 excludes the average daily SABA use from the original 7 items; and while the second ACQ-6 excludes the FEV1% predicted item from the original 7 items. The ACQ-6 excluding FEV1% predicted was used in the studies included in this report.
Scoring
All items in the ACQ are equally weighted. The ACQ-6 scores are calculated as a mean of the 6 items, with scores of 0 meaning the patients has asthma that is well controlled and those with 6 indicating the patients has asthma that is extremely poorly controlled.39,43,88 Mean scores of ≤ 0.75 indicated adequately controlled asthma, scores between 0.75 and < 1.5 indicated partly controlled asthma, and a score of ≥ 1.5 indicated asthma that was not well controlled.40
Psychometric Properties
The original ACQ-7 has demonstrated strong construct validity to the domains of the AQLQ(S)+12 (Pearson r = |0.77|),36 high test-retest reliability (ICC = 0.90),88,89 good internal consistency (Cronbach alpha > 0.70) in patients with stable and persistent asthma,36 and responsive to change.36,88,89 The measurement properties of all 3 shortened versions of the ACQ were found to be similar to the original ACQ-7.41 Based on the Global Initiative of Asthma and National Institute of Health guidelines and using data collected from 1,323 patients from clinical trials diaries and clinic records, a crossover score of 1.00 was acknowledged as the best single cut point to differentiate between “well-controlled” and “not well-controlled” asthma for the ACQ-7, ACQ-6 excluding FEV1 and ACQ-5 versions.40
The psychometric properties of 3 shortened version of the ACQ and their comparative performance with the ACQ-7 was assessed in post hoc analysis of 2 (n, low dose = 740; n, medium dose = 778) phase III, randomized, double-blinded, placebo-controlled efficacy studies of mometasone furoate/formoterol fumarate (MF/F) combination compared with monotherapy inpatients with persistent asthma.36 In the low-dose study, all 4 versions of the ACQ demonstrated good internal consistency with a Cronbach alpha of 0.83, 0.82, 0.87 for the ACQ-7, ACQ-6 excluding daily SABA use and the ACQ-6 excluding FEV1 and ACQ-5, respectively. The internal consistency of the ACQ-7, ACQ-6 excluding daily SABA se and the ACQ-6 excluding FEV1 was higher in the medium study with a Cronbach alpha of 0.85, 0.84 and 0.88, respectively. The test-retest reliability showed good agreement with ICC scores ranging from 0.75 (ACQ-7) to 0.80 (0.80) in the low-dose study and 0.84 (ACQ-6 excluding daily SABA) to 0.88 (ACQ-6 excluding FEV1 and ACQ-5) in the medium-dose study.36 Moreover, all 4 versions of the ACQ were strongly correlated with each other (r > 0.97) and with the overall score from the AQLA(S)+12 for both baseline and change scores (|r| > 0.74).36
Minimal Important Difference
Individual changes of at least 0.5 on the ACQ-6 are considered clinically meaningful in adult patients with asthma.1,40-43 The MID for adolescents was not identified in the literature.
Standardized Asthma Quality of Life Questionnaire for 12 Years and Older (AQLQ[S]+12)
The AQLQ(S)+12, a patient-reported, disease-specific HRQoL measure, is a modified version of the validated AQLQ(S)38 to accommodate those patients with asthma who are aged 12 years and older versus adult patients only.93 The original AQLQ(S) was altered to change the questions regarding “work-related limitations” to “work-/school-related limitation” to create the AQLQ(S)+12.93 The AQLQ(S)+12 consist of 32 questions grouped into 4 domains: (1) symptoms; (2) activity limitations; (3) emotion function; and (4) environmental stimuli.
Scoring
Patients score each item using a 7-point Likert type scale based on their recall of experiences during the previous 2 weeks. The Likert scale to each question is anchored by 7 (no impairment) and 1(severe impairment). The overall score of the AQLQ(S)+12 is derived as a mean of the 32 questions, ranging from 1 to 7. Higher scores indicate less severe impairment. Furthermore, domain specific mean scores may be reported.35
Psychometric Properties
The initial validation study of the AQLQ(S)+12 was conducted in a cohort of 2,433 patients with asthma between the ages of 12 to 75 years of age using database from 2 clinical trials.93 The AQLQ(S)+12 demonstrated excellent overall and domain specific internal consistency in patients aged 12 to 17 years (Cronbach alpha 0.77 to 0.97) and in those aged 18 years and older (Cronbach alpha 0.82 to 0,97).93 Moreover, the correlation between each domain of the AQLQ(S)12+ and other measures of asthma clinical status were also consistent in the 2 age groups.93
The psychometric properties of the AQLQ(S)+12 was further assessed in post hoc analysis of 2 (n trial 1 = 740 and n trial 2 = 778) Phase III, randomized, double-blinded, placebo-controlled efficacy studies of MF/F combination compared with monotherapy inpatients with persistent asthma.37 The study found that the AQLQ(S)+12 demonstrated moderate to strong construct validity with other measures of asthma health at baseline and over time Moreover the AQLQ(S)+12 demonstrated excellent reproducibility (ICC range 0.76 to 0.85).37
Minimal Important Difference
Currently, there is no established MID for the AQLQ(S)+12. However, given the significant overlap between the AQLQ(S)+12 and the original AQLQ(S), a cut point of 0.5 points is considered clinically meaningful.36,37,93
EQ-5D-5L
The EQ-5D-5L is a generic self-reported HRQoL outcome measure that may be applied to a variety of health conditions and treatments.94 The EQ-5D-5L tool have been applied to a wide range of health conditions and treatments, including patients with asthma or chronic obstructive pulmonary disease.44-46 The first 2 components of the EQ-5D-5L assesses 5 domains: mobility, self-care, usual activities, pain/discomfort and anxiety/depression.94 Each domain has 5 levels: no problem; slight problems; moderate problems; severe problems; and extreme problems. A descriptive system that classifies respondents (aged ≥ 12 years) based on the following 5 dimensions: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression. The EQ-5D-5L has 5 possible levels for each domain and respondents are asked to choose the level that reflects their health state for each of the 5 domains resulting in 3,125 possible health states.94 The second component of the EQ-5D-5L part is the 20 cm Visual Analogue Scale (EQ VAS) that has end points labelled 0 and 100, with respective anchors of “worst imaginable health state” and “best imaginable health state.” Respondents are asked to rate their health by drawing a line from an anchor box to the point on the EQ VAS which best represents their health on that day. Thus, the EQ-5D-5L produces 3 types of data for each respondent:
A profile indicating the extent of problems on each of the 5 dimensions represented by a 5-digit descriptor, e.g., 15121, 33211;
A population-preference weighted health index score based on the descriptive system; and
A self-reported assessment of health status based on the EQ VAS.
Scoring
The EQ-5D-5L index score is generated by applying a multi-attribute utility function to the descriptive system.95 Different utility functions are available that reflect the preferences of specific populations (e.g., US or UK). Scores less than 0 represent health states that are valued by society as being worse than dead, while scores of 0 and 1.00 are assigned to the health states ‘dead’ and “perfect health,’ respectively.
Psychometric Properties
The validity of the EQ-5D-5L in patients with asthma was first conducted in a 316 adolescents and adults with asthma included in the ASTRO-LAB cohort.96 In this study, the mean EQ-5D-5L index with well-controlled, intermediate-controlled, and poorly controlled asthma were 0.91 (95% CI, 0.89 to 0.93), 0.84 (95% CI 0.81 to 0.87) and 0. 73 (95% CI, 0.69 to .78). Relevant differences across known asthma control groups indicated that the EQ-5D-5L demonstrated good construct validity for assessing HRQoL in patients with asthma.96
In a follow-up validation study in 312 patients with asthma from a primary care setting in France and England, the EQ-5D-5L demonstrated acceptable floor and ceiling effect (0% and 26.5%, and respectively) and good construct validity based on the discriminant ability to distinguishing among health related known-groups (Cronbach alpha, 0.69).90 The mean EQ-5D-5L decreased significantly with an increase in the number of chronic condition from 0.91 to 0.82 with the English value set and from 0.86 to 0.75 in the French value set.90
The validity and responsiveness of the EQ-5D-5L in patients who were experienced an asthma exacerbation requiring emergency department care was most recently conducted using data from a prospective cohort study of 121 patients with asthma living in the UK.91 In this study, the EQ-5D-5L demonstrated good convergent validity with the Asthma Quality of Life Utility Index (Spearman’s r = 0.626) with no evidence of know-groups validity with patients were grouped by asthma severity as defined by PEF.91 However, the EQ-5D-5L demonstrated responsiveness to the question, “Compared to your asthma state when you were in the hospital 4 weeks ago, how would you rate your asthma now?” with a standardized response mean of 0.75, 0.95, 0.30 and -1.03 for the very good, good, moderate, and poor response groups.
In contrast, a qualitative study of 40 adults with asthma between the aged of 20 to 57 years old living in the UK found that the content of the EQ-5D-5L was poorly aligned with patient-perceived impact of asthma.97 The mobility and self-care dimension prompted strong negative reactions from most participants. Variations in interpretation of the mobility dimension and difficulties with multiple concepts in the pain/discomfort and anxiety/depression dimension were also noted. Moreover, the study found that the EQ-5D-5L failed to meet basic standards for acceptability and content validity as a measure to assess the impact of asthma from the patient perspective.97 Concepts reported missing by patients included environmental triggers, asthma symptoms, emotions, and sleep. Additionally, patients indicated shortness of breath and impact on activities as especially salient issues.97
Minimal Important Difference
A literature search was conducted to identify MID of the EQ-5D-5L in patients with asthma and none were identified. Below is a summary of MID of the EQ-5D-5L in patients within the general population.
A simulation-based approach using an instrumental-defined single-level transitions was used to estimate the MID of the EQ-5D-5L in the general population for each country-specific scoring algorithm. An estimated MID between 0.037 and 0.069 was determined for 6 countries (Canada, China, Spain, Japan, England, and Uruguay).47 The country-specific scoring algorithm were as follows Canada, 0.056 ± 0.011; China, 0.069 ± 0.007; Spain, 0.061 ± 0.008; Japan, 0.048 ± 0.004; England, 0.063 ± 0.013; and Uruguay, 0.063 ± 0.019. Differences in MID estimates reflect differences in population preferences, in valuation techniques used, as well as in modelling strategies. After excluding the maximum-valued scoring parameters, the MID estimates (mean ± SD) were as follows: Canada, 0.037 ± 0.001; China, 0.058 ± 0.005; Spain, 0.045 ± 0.009; Japan, 0.044 ± 0.004; England, 0.037 ± 0.008; and Uruguay, 0.040 ± 0.010.47
Pharmacoeconomic Review
Abbreviations
ACQ-6
6-item Asthma Control Questionnaire
AE
adverse event
BIA
budget impact analysis
HRQoL
health-related quality of life
ICER
incremental cost-effectiveness ratio
ICS
inhaled corticosteroids
LABA
long-acting beta2-agonist
OCS
oral corticosteroids
QALY
quality-adjusted life-years
SOC
standard of care
T2
severe eosinophilic type 2
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Tezepelumab (Tezspire), solution for subcutaneous injection (210 mg per 1.91 mL prefilled syringe or pen [110 mg/mL]) |
Submitted price | Tezepelumab, 210 mg: $1938.46 per prefilled syringe or pen |
Indication | Proposed: Add-on maintenance treatment in adults and adolescents 12 years and older with severe asthma |
Health Canada approval status | Under review (pre-NOC) |
Health Canada review pathway | Standard review |
NOC date | Anticipated: July 31, 2022 |
Reimbursement request | As per indication |
Sponsor | AstraZeneca Canada Inc. |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov model |
Target population | Add-on maintenance treatment in adults and adolescents 12 years and older with severe asthma |
Treatment | Tezepelumab plus SOC |
Comparator | SOC alone (high-dose ICS-LABA, OCS for OCS-dependent patients) |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, life-years |
Time horizon | Lifetime (50 years) |
Key data sources | NAVIGATOR trial, SOURCE trial |
Submitted results | ICER = $184,075 per QALY (incremental costs: $207,101; incremental QALYs: 1.13) |
Key limitations |
|
CADTH reanalysis results |
|
ACQ-6 = 6-item Asthma Control Questionnaire; FEV1 = forced expiratory volume in the first second; ICER = incremental cost-effectiveness ratio; LABA = long-acting beta2-agonist; OCS = oral corticosteroids; QALY = quality-adjusted life-year; SOC = standard of care.
Conclusions
Evidence indicates that tezepelumab reduces the rate of asthma exacerbations compared to standard of care (SOC) alone in patients whose asthma remains uncontrolled despite medium- to high-dose inhaled corticosteroids (ICS). This reduction in exacerbations appears to occur regardless of whether patients have type 2 or non–type 2 asthma. Additionally, tezepelumab appears to improve pulmonary function, as well as health-related quality of life (HRQoL) and symptoms of asthma as measured by the Asthma Quality of Life Questionnaire (Standardized) for patients 12 years of age and older and the 6-item Asthma Control Questionnaire (ACQ-6). There is no evidence that tezepelumab leads to a reduction in doses of oral corticosteroids (OCS) in patients with OCS-dependent asthma or reduces exacerbations in these patients. Finally, the comparative effects of tezepelumab relative to other biologic treatments for severe asthma are limited due to a lack of direct comparative evidence and heterogeneity within the sponsor’s indirect treatment comparisons.
CADTH undertook reanalyses to address limitations in the sponsor’s submission by assuming no mortality benefit associated with tezepelumab, using utility values associated with health states only, and removing response assessment at 26 weeks. CADTH was unable to address the lack of data providing a head-to-head comparison with other biologics and concerns remained with transparency over how transition probabilities were derived.
In the CADTH base case, tezepelumab plus SOC was more effective and more costly than SOC alone, with incremental costs of $228,312 and 0.171 incremental quality-adjusted life-years (QALYs), resulting in an incremental cost-effectiveness ratio (ICER) of $1,334,178 per QALY gained. Relative to SOC alone, treatment with tezepelumab led to an additional $232,952 in drug costs but was associated with $4,643 in cost savings from reduced hospitalizations and outpatient, emergency department, and community care visits arising from fewer severe exacerbations and improved asthma control. The additional 0.171 QALYs from tezepelumab were associated with improved asthma control and a reduction in exacerbations. A scenario analysis was conducted that explored a potential optimistic survival benefit associated with tezepelumab. This increased the gain in QALYs slightly to 0.23, resulting in an improved ICER of $972,451 per QALY. In the CADTH base case, the probability of tezepelumab being cost-effective at a willingness-to-pay threshold of $50,000 per QALY was 0%. A price reduction of approximately 95% is necessary to achieve cost-effectiveness at this threshold.
CADTH noted a small number of incremental QALYs in the CADTH base case are derived from benefits to patients who are OCS-dependent. However, the CADTH clinical review found no evidence that tezepelumab reduces OCS use and exacerbations in these patients. The incremental benefit as calculated in the CADTH base case is therefore likely overestimated. In a population consisting of non-chronic users of OCS, the ICER increases to $1,403,821 per QALY.
Given the lack of head-to-head comparative data and limitations associated with the indirect comparison there is no clinical evidence to support a price premium for tezepelumab above other biologics. The cost-effectiveness of tezepelumab in comparison to other biologics available in Canada is therefore unknown.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
CADTH received patient input from Asthma Canada and the Lung Health Foundation (formerly the Ontario Lung Foundation), organizations dedicating to supporting patients in Canada living with asthma and lung cancer, respectively. Patient input indicated that the goals of asthma therapy are day-to-day symptom relief, controlling exacerbations, reducing hospitalizations, improving quality of life, and increasing lung function. Patients who provided input had experience with bronchodilators and long-term controller medications such as ICS and OCS. However, 1 in 4 patients with severe asthma on current treatments reported poor symptom control that affected quality of life and highlighted issues with potential systemic dependence of OCS. Side effects related to current treatments were reported to be dry throats, elevated heart rates, difficulty sleeping, headaches, hoarseness, and weight gain. Side effects related to OCS noted to be particularly problematic included weight gain, acne, high blood pressure, high blood sugar in the short term, and osteopenia, osteoporosis, glaucoma, cataracts, and heart disease in the long-term. Patient input did not capture any patients with experience with tezepelumab. Notably, patient input indicated that the high cost of medications was a barrier to receiving proper asthma treatment.
Clinician input was received from the AllerGen Clinical Investigator Collaborative. Severe asthma was described as affecting between 5% and 8% of all asthmatic patients, who were considered to be patients already on ICS plus a long-acting beta2-agonist (LABA) and/or OCS but with uncontrolled asthma and at risk of severe exacerbations. Clinicians indicated approximately 40% to 50% of patients with severe asthma do not possess biomarkers for persisting severe eosinophilic type 2 (T2)-high asthma, therefore precluding them from treatment with currently available biologic therapies. Input from the clinician group stated that tezepelumab would be restricted to patients with severe asthma, but it would be used in patients with T2-low asthma who are ineligible for current biologics. Goals of treatment for this patient population include the reduction of severe asthma exacerbation, improved asthma control, and improved lung function.
Drug plan input noted implementation issues regarding other biologics indicated for severe asthma that are generally related to specific phenotypes. The reimbursement status of each of these comparator biologics varies by criteria and province. Furthermore, drug plans highlighted uncertainty regarding the positioning of tezepelumab as first-line therapy for all asthma phenotypes irrespective of biomarker status. As such, it was noted that initiation criteria may require restrictions related to diagnostic phenotype or biomarker status. Drug plans commented on the lack of evidence to support the use of tezepelumab in combination with other biologics indicated for severe asthma, and pointed out that combination use would considerably increase costs.
Two of these concerns were addressed in the sponsor’s model:
Clinical effectiveness was based on the rate of asthma exacerbations, with those who experienced a severe exacerbation assumed to have lower HRQoL for the duration of the exacerbation.
Adverse events (AEs) were incorporated for OCS.
In addition, CADTH addressed 1 additional concern by exploring the impact of tezepelumab uptake in patients with T2-low asthma who are ineligible for current biologic treatments as a scenario analysis in the budget impact analysis.
CADTH was unable to address the concerns raised in stakeholder input regarding the lack of comparative evidence between tezepelumab and other currently available biologic treatments for severe asthma.
Economic Review
The current review is for tezepelumab (Tezspire) as add-on maintenance treatment in patients aged 12 years and older with severe asthma.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis of tezepelumab plus SOC compared with SOC alone in patients 12 years and older with severe asthma.1 The modelled population is consistent with the reimbursement request. The SOC comprised a basket of ICS-and-LABA combination inhalers, and OCS. The cost-effectiveness of tezepelumab relative to other biologic treatments (omalizumab, mepolizumab, benralizumab, and dupilumab) was assessed in scenario analyses.
Tezepelumab is available in 210 mg (110 mg/mL) prefilled syringes or prefilled pens for self-administered subcutaneous injection. The recommended dosage for tezepelumab is 210 mg every 4 weeks and the annual cost of treatment is $25,200 based on a unit cost of $1,938.46 per syringe or pen.2 The weighted average annual cost of SOC per patient was calculated to be $1,478 for ICS-LABA and $40 for chronic OCS, as calculated by the sponsor.
Outcomes modelled included QALYs and life-years over a lifetime time horizon of 50 years and a cycle length of 28 days. The base-case analysis was conducted from the Canadian public health care system, with costs and outcomes discounted at 1.5% annually.
Model Structure
The sponsor submitted a 5-state Markov model with 2 asthma control–based states (“uncontrolled asthma” and “controlled asthma”), as well as 2 exacerbation states (“previously controlled asthma with exacerbation” and “previously uncontrolled asthma with exacerbation”).1 The asthma-control health states were defined based on 6-item Asthma Control Questionnaire (ACQ-6) scores (controlled asthma: ACQ-6 score < 1.5 and no history of exacerbation in previous 4-week period; uncontrolled asthma: ACQ-6 score ≥ 1.5 and no history of exacerbation in the previous 4-week period). The exacerbation health states were defined as a worsening of asthma symptoms that causes 1 of 3 events: a burst of OCS for at least 3 consecutive days, an emergency department visit, or hospitalization. The submitted model also included a “death” state. Canadian general population mortality was used to estimate mortality risk in non-exacerbation health states; in the exacerbation state patients were at elevated risk of dying. A figure of the submitted model is provided in Appendix 3.
Patients entered the model either on treatment with tezepelumab plus SOC or SOC alone, with the proportion of patients receiving chronic OCS based on characteristics of the NAVIGATOR trial. All patients enter the model with uncontrolled asthma and those who start treatment on tezepelumab can transition to SOC alone either due to discontinuation (i.e., AEs or nonefficacy-related reasons) or no response after assessment. Patients also can transition from treatment with OCS to without OCS following OCS sparing and discontinuation. Movement between health states was defined by transition probabilities that varied by treatment and over time (4-week observation windows).
Model Inputs
The baseline patient characteristics in the sponsor’s model were aligned with the NAVIGATOR and SOURCE trials (mean age of 50 years; 37% male; 9.4% OCS users with a baseline dose of 11.29 mg/day).
Clinical efficacy (i.e., the probability of a transition between health states) was based on the 52-week period of the NAVIGATOR trial or the 48-week period of the SOURCE trial. The NAVIGATOR trial compared tezepelumab to placebo in patients with severe asthma, with the primary end point being the annualized asthma exacerbation rate. The SOURCE trial compared tezepelumab to placebo in patients with severe asthma who required chronic OCS, with a primary end point of the categorized percentage reduction from baseline in daily OCS use at week 48. Definition of response in the model was assumed to be any reduction in rate of exacerbation or chronic OCS dose from baseline, assessed at 26 weeks. Patients classified as responders were assumed to continue treatment with tezepelumab, whereas nonresponders were assumed to discontinue tezepelumab and switch to treatment with SOC alone. Time spent in health states was defined by transition probabilities calculated from patient-level count data taken every 4 weeks during the pivotal trials. Four transition matrices (i.e., efficacy profiles) were calculated based on pre- and post-assessment OCS use and non-use for patients receiving tezepelumab; 2 efficacy profiles based on OCS use and non-use were calculated for patients receiving SOC alone.1 Post-assessment transition probabilities were applied following response assessment up until treatment discontinuation or the end of the model time horizon, meaning that treatment effect was assumed to be maintained for the remainder of the time horizon.
Chronic OCS use was defined by overall dose reduction or discontinuation, which was captured in the model by applying reductions in the proportion of OCS users (baseline of 9.4%) and mean OCS dose (baseline of 11.29 mg/day) at an OCS-sparing week. It was assumed that patients with a 90% to 100% reduction in chronic OCS will discontinue OCS.
The risk of exacerbation-related mortality varied by type of exacerbation (i.e., OCS burst, emergency department visit, or hospitalization) and age. Data from the National Review of Asthma Deaths, Roberts et al. (2013), and Watson et al. (2007) were adjusted to reflect exacerbation types aligned with the NAVIGATOR and SOURCE trials.3-5 Asthma-related deaths were estimated to range from 0.048% to 0.31% for OCS bursts; 0.49% to 3.2% for emergency department visits; and 0.15% to 4.5% for hospitalization.1 Annual mortality rates for all-cause mortality were based on general population life tables and applied to non-exacerbation states.6
Treatment-specific utility values were estimated using a mixed regression analysis pooled from NAVIGATOR and SOURCE data and a Canadian-specific value set from Xie et al. (2015).7 The estimated utility of a patient receiving current SOC without an exacerbation and with controlled asthma was |||||||||. Various utility decrements associated with uncontrolled asthma (-0.05), OCS burst (|||||||||), emergency department visit (|||||||||), and hospitalization (|||||||||) were also applied. Utility loss associated with exacerbation was experienced for the duration of an exacerbation, based on the pivotal clinical trials. Finally, the mixed regression model estimated an additional utility gain of ||||||||| with tezepelumab regardless of level of control and whether an exacerbation was occurring. Alternative health state–specific utility values were provided based on EQ-5D 5-Levels questionnaire data from the NAVIGATOR and SOURCE trials. A fixed utility value was applied for OCS-related AEs only.
Costs included those associated with drug acquisition for tezepelumab and SOC, treatment administration for some biologics, disease management, and AEs. Relevant costs were inflated to 2022 Canadian dollars. Drug acquisition costs for tezepelumab were sourced from the sponsor while costs for SOC were sourced from the Ontario Drug Benefit formulary.8 In the scenario analysis, the costs of comparator biologics were sourced from the Ontario Drug Benefit Exceptional Access Program, while the cost of reslizumab was obtained from the sponsor’s CADTH Common Drug Review submission.9,10 The drug acquisition cost of SOC was calculated as a weighted average based on estimated relative percentage of patient use per SOC drug for ICS-LABA and OCS, separately. Treatment administration costs for tezepelumab were included for 45% of patients assumed to receive treatment from a health care provider. Similar assumptions were applied to all comparator biologics in the scenario analysis, with the exception of omalizumab (which requires a longer duration of administration).11 No administration costs were included for SOC, which was assumed to be self-administered. Resource-use costs for disease management were dependent on health state occupancy and differed by the 3 exacerbation states (OCS burst, emergency department visit, and hospitalization) and sourced from the Ontario Schedule of Benefits, Ontario Case Costing Initiative, Government of Canada Job Bank, and literature.11-14 Varying disease-management costs included those related to general practitioner visits, nurse visits, specialist nurse visits, spirometry, flu vaccines, respirologist and emergency department visits, hospitalizations, and IV chair time. Costs of OCS-related AEs were included based on a study of a UK cohort reporting health care utilization use to estimate the average annual cost based on OCS treatment dose.11,12,15
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (1,000 iterations for the base-case and scenario analyses). The deterministic and probabilistic results were similar. The probabilistic findings are presented below.
Base-Case Results
Tezepelumab was associated with incremental costs of $207,101 and 1.13 incremental QALYs in comparison to SOC alone, resulting in an ICER of $184,075 per QALY gained (Table 3).
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. SOC alone ($ per QALY) |
|---|---|---|---|---|---|
SOC alone | 59,445 | Ref. | 18.82 | Reference | Reference |
Tezepelumab | 266,546 | 207,101 | 19.95 | 1.13 | 184,075 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SOC = standard of care.
Note: The submitted analysis is based on the publicly available prices of the comparator treatments.
Source: Sponsor’s pharmacoeconomic submission.1
Additional results from the sponsor’s submitted economic evaluation base case are presented in Appendix 3.
Sensitivity and Scenario Analysis Results
In a key sensitivity analysis, tezepelumab was compared to other comparator biologics using relative efficacy estimates obtained from a simulated treatment comparison conducted by the sponsor. Probabilistic results demonstrated that tezepelumab was associated with more QALYs (incremental QALYs: 0.061 to 0.404) and lower costs (incremental costs: −$1,974 to −$6,878). The sponsor’s analyses demonstrated that tezepelumab dominated other biologics (i.e., it was more effective and less costly). These analyses relied on indirect treatment comparisons, which were associated with limitations.
The sponsor conducted further scenario analyses involving an alternative response assessment week (52 weeks), a longer exacerbation duration of 4 weeks, exclusion of treatment administration, increased proportion of patients receiving chronic OCS to 15%, low and high values for percent reduction in OCS dose, exclusion of OCS-related AEs, and alternative sources for resource use and utilities. The ICER was most sensitive to a shorter time horizon of 10 years, resulting in an ICER of $303,478 per QALY gained. Application of a lower percentage reduction in OCS dose increased the ICER to $260,682, and the exclusion of OCS-related AEs resulted in an increased ICER of $260,659.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
Submitted model lacks face validity: The sponsor’s submitted model dichotomizes asthma control into “controlled” and “uncontrolled” based on ACQ score (controlled: < 1.5; uncontrolled: ≥ 1.5). Based on these thresholds, a change in ACQ-6 score of 0.01 (from 1.50 to 1.49) would result in a patient being classified as having controlled asthma. For example, while CADTH acknowledges that an ACQ-6 score of 1.5 is a commonly used threshold in clinical trials, the Global Initiative for Asthma guidelines consider an ACQ-6 score of 0.75 to 1.5 to represent a partially controlled health state in between controlled and well-controlled asthma.16 The degree of improvement in asthma control from baseline may be more indicative of patient utility change as opposed to whether they moved beyond the ACQ-6 cut-off for control. The clinical expert consulted by CADTH noted that an ACQ-6 score is a continuous variable and as such, differences as small as 0.01 would not affect patient quality of life.
Second, the sponsor differentiated exacerbations that occur in patients with controlled asthma versus those that occur in patients with uncontrolled asthma. This means a patient who is hospitalized has a better utility if their asthma was previously controlled before hospitalization. The expert concluded that this lacked face validity as asthma control before the asthma exacerbation would not influence quality of life during the exacerbation episode.
Due to the structure of the submitted model, CADTH could not address the limitation pertaining to asthma-control dichotomization in reanalysis.
A scenario analysis was conducted in which the utility experienced during an asthma exacerbation was not contingent on prior asthma control. This analysis assumed the utility experienced during an exacerbation for those who were previously uncontrolled is the same as the utility of a patient experiencing an exacerbation for those previously controlled.
Assumption of increased mortality during severe asthma exacerbation: The sponsor assumes an increased risk of death when patients have a severe exacerbation, with the risk varying by age group and type of exacerbation (i.e., OCS burst, emergency department visit, or hospitalization). As tezepelumab reduces the rate of severe exacerbations, this implies a guaranteed survival benefit with tezepelumab, which has not been shown in clinical trials. Across the SOURCE and NAVIGATOR trials, 2 deaths were reported in the placebo group and 1 was reported in the tezepelumab group; none were noted as being related to an exacerbation. In the long term extension trial (DESTINATION), 11 patients (incidence rate = 0.80 per 100 patient-years; 95% CI, 0.40 to 1.43) randomized to tezepelumab died versus 5 patients (incidence rate = 0.58 per 100 patient -years; 95% CI, 0.19 to 1.34) randomized to placebo.17 This is despite a continued reduction in severe exacerbations in those randomized to tezepelumab. No evidence suggesting a reduction in mortality was therefore reported from the clinical trials. Although tezepelumab reduces exacerbations there is no evidence that it reduces fatal exacerbations.
As noted in previous CADTH reviews and by the clinical expert consulted for this review, asthma-related mortality is rare and often linked to causes such as lack of adherence and incorrect management. For example, this is extensively highlighted in the National Review of Asthma Deaths in the UK cited by the sponsor, which found 45% of asthma deaths occur without any medical intervention.18 Likewise, many individuals who died were misclassified as having mild or moderate asthma or were not seeing a specialist and therefore did not receive adequate care. A study by Suissa et al. (2000) using data from Saskatchewan found that asthma deaths were substantially reduced by regular use of low-dose ICS.19 The clinical expert consulted by CADTH also noted that deaths related to chronic obstructive pulmonary disease are contributors to the observed number of asthma deaths. All these factors that contribute to asthma deaths are therefore unlikely to be affected by tezepelumab use. The clinical expert consulted by CADTH also noted that the mortality rates applied in the sponsor’s model did not meet face validity; for example, the 3.2% probability of death among patients aged 45 years or older with a severe exacerbation who visit an emergency department but are not admitted is not reflective of patients in Canada. According to data from the Canadian Institute for Health Information, between 2019 and 2020 there were 60,492 emergency department visits related to asthma from participating facilities in Prince Edward Island, Nova Scotia, Quebec, Ontario, Manitoba, Saskatchewan, Alberta, British Columbia, and Yukon.20 Of those not admitted, fewer than 5 people (< 0.009%) died before discharge.20 For the sponsor’s values to hold face validity, a substantial proportion of patients would have to die immediately post-discharge.
Finally, the model results lacked face validity when predicting expected deaths due to asthma. After 1 year in the model, asthma exacerbations are responsible for 77% of deaths that occur (approximately 0.7% die from an asthma exacerbation and 0.2% die from all-cause mortality). After 5 years in the model, asthma exacerbations are responsible for 74% of deaths that occur (3.4% die from an asthma exacerbation and 1.2% die from all-cause mortality). This ratio of asthma-related deaths to non–asthma-related deaths is not in line with what is seen from the clinical trial or the literature. According to a multinational cohort study of mortality in patients with severe asthma, deaths related to asthma accounted for 1.9% to 6.3% of deaths, with the rest attributed to a non–asthma-related cause.21 The sponsor’s model therefore substantially overpredicts the number of asthma-related deaths occurring in patients, which does not align with what is observed in clinical practice, registry data, or the NAVIGATOR or SOURCE clinical trial data submitted by the sponsor. If a survival benefit were to occur it would therefore likely be substantially smaller than what is predicted by the sponsor’s model. The sponsor does not appear to account for all-cause mortality in the exacerbation state, meaning that the probability of dying after an exacerbation is underestimated by the sponsor’s own calculations.
The predicted survival benefit with tezepelumab compared with background therapy is highly uncertain and is not supported by clinical trial data. This mortality benefit was removed in CADTH reanalysis, consistent with previous CADTH reviews.
In a scenario analysis CADTH applied a hazard ratio of 2 to all-cause mortality in the exacerbation state in line with evidence from Engelkes et al. (2020).21 Overall this assumes 9% of deaths in those on SOC are attributable to an asthma exacerbation, which was considered high in the Canadian context. Likewise, this analysis assumes tezepelumab has a direct impact on fatal exacerbations and should therefore be considered exploratory as this evidence was not presented from the trial or based on Canadian data.
The number of exacerbations predicted by the model is not aligned with clinical trial evidence: The sponsor’s model does not appear to accurately predict the number of severe exacerbations in either the tezepelumab or SOC patient groups during the clinical trial period (52 weeks) relative to the clinical data from the SOURCE and NAVIGATOR trials. For example, the model predicts 0.73 and 1.53 severe exacerbations per patient would occur, on average, during the first 52 weeks for tezepelumab and SOC alone, respectively. In the NAVIGATOR trial, the annualized rate of severe exacerbations was 0.93 and 2.1 for tezepelumab and placebo, respectively. In the SOURCE trial the rates were 1.37 and 2.0 for tezepelumab and placebo, respectively. The model therefore appears to routinely underestimate the number of exacerbations expected to occur. The rate ratio predicted by the model of approximately 0.47 is generally aligned with that of the clinical trials (0.44 in the NAVIGATOR trial) but the results presented should be interpreted with caution due to lack of validity in the absolute number of predicted outcomes. In addition, because the number of hospitalizations is dependent on how many exacerbations occur, the rate of hospitalization also appears to be underestimated in the analysis. Transition probabilities are hard coded into the sponsor’s model, and it is therefore unclear how they are estimated. Transition probabilities are calculated by pooling across trials and across different subgroups, and this makes validation difficult because the model predictions cannot be compared against the output from a single trial.
CADTH was unable to fully validate the estimation of transition probabilities in the sponsor’s model due to a lack of transparency. The sponsor also declined to provide an additional subgroup analysis using data from only the NAVIGATOR trial, which further limited CADTH’s ability to validate the submitted model.
CADTH conducted a scenario analysis that increased the probability of an exacerbation occurring in those on SOC alone such that the number of exacerbations prevented from tezepelumab matched that of the NAVIGATOR trial. This analysis was conducted to explore the potential impact of underestimating exacerbations avoided due to treatment with tezepelumab.
Inappropriate derivation of utilities: The sponsor derived utilities for each health state using a mixed regression model, which also included a treatment-specific utility. Treatment-specific utilities are inappropriate and health state–specific utilities are preferred, according to the CADTH Guidelines for the Economic Evaluation of Health Technologies.22
The sponsor’s utility estimates indicate that treatment with tezepelumab results in improved quality of life independent of whether it improves asthma control or prevents asthma exacerbations. The clinical expert consulted by CADTH noted that this assumption did not meet face validity, as any perceived improvement in quality of life would be due to improved asthma control or reduced exacerbations. The values presented by the sponsor also lack face validity. A patient receiving tezepelumab experiences a utility gain of |||||||||, regardless of asthma control, while a severe exacerbation requiring an OCS burst results in a utility loss of |||||||||. This means that the sponsor assumes patients treated with tezepelumab experiencing an exacerbation will have a higher utility than those not on tezepelumab with controlled asthma and not experiencing an exacerbation. The CADTH clinical expert stated that this was not clinically valid.
Finally, evidence from the trial as measured using the 5-Level EQ-5D questionnaire indicates that the expected utility benefit from receiving tezepelumab is 0.02 at 52 weeks. Using estimates from the sponsor’s regression, the model predicts that the utility for patients on tezepelumab at 52 weeks is approximately ||||||||| higher than that of patients on SOC.
In reanalysis, CADTH replaced treatment-specific utilities with sponsor-provided health state–specific utilities.
The model structure does not adequately reflect the management of asthma in clinical practice: In the sponsor’s model, response to treatment is assessed at 26 weeks, with treatment response defined as any reduction in the rate of exacerbation or chronic OCS use from baseline. The clinical expert consulted by CADTH for this review indicated that response to biologic treatment is usually assessed in clinical practice using ACQ-6 scores and lung function (i.e., forced expiratory volume in the first second) from a baseline assessment. This was also stated in prior CADTH reviews.23,24 Treatment response is not typically assessed in terms of exacerbation risk, as exacerbations may be infrequent and may be influenced by factors other than asthma control (e.g., influenza and pneumonia).
The sponsor assumed that patients with no treatment response at 26 weeks would receive background therapy alone for the remainder of the time horizon; however, the clinical expert consulted by CADTH indicated that patients with an inadequate treatment response would be switched to an alternative biologic, not moved to background therapy alone. Further, it is likely that a proportion of patients who improve, but not to the extent of the response criteria, would likely continue to receive their current biologic treatment.
In CADTH reanalyses, treatment response assessment at 26 weeks was disabled, such that patients discontinued treatment based only on the constant long-term discontinuation rate, as derived from the trial data.
Uncertainty regarding long-term clinical effectiveness: In the sponsor’s pharmacoeconomic submission, the effects of tezepelumab were consistent over the lifetime model horizon (approximately 50 years) after response assessment at 26 weeks. The potential waning of treatment effect over time was not explored in the sponsor’s model. Although the clinical expert consulted by CADTH noted that it may be feasible for treatment benefit to be sustained, there is limited clinical evidence to support the assumption that it would persist for approximately 50 years.
CADTH was unable to address this limitation due to the structure of the sponsor’s economic model and a lack of long-term effectiveness data for tezepelumab.
Highly uncertain comparative clinical efficacy versus biologics: There is an absence of head-to-head clinical evidence comparing tezepelumab to other biologic treatments for asthma (i.e., dupilumab, omalizumab, mepolizumab, reslizumab, and benralizumab). The sponsor conducted indirect treatment comparisons to provide comparative clinical effectiveness data for scenario analyses. The CADTH clinical report appraised the submitted indirect treatment comparisons and identified no differences when comparing tezepelumab to other biologics. The CADTH clinical report stated that no definite conclusions can be drawn on the comparative effectiveness and safety profile between tezepelumab and other relevant biologics in the treatment of adults and adolescents 12 years and older with severe asthma due to heterogeneity and small sample size. As such, the cost-effectiveness of tezepelumab compared to other biologic treatments is unknown.
CADTH was unable to address this limitation due to a lack of direct evidence and limitations with the sponsor’s indirect treatment comparisons. The cost-effectiveness of tezepelumab relative to other biologic treatments is unknown.
Uncertainty surrounding clinical evidence for reductions in OCS use: The CADTH clinical report concluded that there is no evidence to support a conclusion that tezepelumab facilitates the reduction of OCS doses in patients with OCS-dependent asthma, or reduces exacerbations in these patients. When a subgroup analysis looked only at patients who are not on chronic OCS, the ICER increased. The model is therefore generating an additional benefit for tezepelumab that is unsupported by clinical evidence. Given the small number of patients who require chronic OCS use in the model, the impact of this is slight.
A scenario analysis was conducted that looked at the cost-effectiveness of tezepelumab in those not on chronic OCS.
Additionally, the following key assumptions made by the sponsor were appraised by CADTH (Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
Adverse events related only to OCS treatment were included. | Reasonable. Adverse events other than those related to OCS were not modelled by the sponsor. This was unlikely to influence model results; however, costs related to adverse events could have been included for completeness. |
Rates of OCS discontinuation were informed by data on > 90% reduction of OCS dose. | Uncertain. It was noted that the relative probability of discontinuing chronic OCS could not be estimated in the stimulated treatment comparison, although this was unlikely to influence model results. |
OCS = oral corticosteroids.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
Several limitations with the sponsor’s submission could not be adequately addressed (i.e., the lack of head-to-head comparative clinical data, uncertainty regarding long-term clinical effectiveness, and lack of face validity regarding the model structure). Furthermore, CADTH could not fully validate the sponsor’s model due to a lack of transparency and the poor modelling practices employed. The CADTH base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts. CADTH undertook a stepped reanalysis that assumed no mortality benefit associated with tezepelumab, applied health state–specific utility values, and removed response assessment at 26 weeks. Details for each stepwise change to derive the CADTH reanalysis are presented in Table 5. The summary results of the CADTH reanalyses are presented in Table 6 (disaggregated results are presented in Appendix 4).
In the CADTH base case, tezepelumab was associated with a total cost of $295,314 and 22.04 QALYs compared to $67,003 and 21.87 QALYs for patients receiving SOC. The ICER for tezepelumab compared to SOC was $1,334,178 per QALY gained with a 0% probability of being cost-effective at a willingness-to-pay threshold of $50,000. Detailed information and disaggregated results are presented in Table 12 in Appendix 4.
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption | |
|---|---|---|---|
Corrections to sponsor’s base case | |||
None. | — | — | |
Changes to derive the CADTH base case | |||
1. Survival benefit | Assumed a mortality benefit associated with tezepelumab | Assumed no mortality benefit associated with tezepelumab | |
2. Treatment-specific utility values | Used treatment-specific utility values | Used health state–specific utility values | |
3. Assessment of response based on exacerbation and OCS use | Treatment response assessed at 26 weeks | No response assessment at 26 weeks. | |
CADTH base case | — | Reanalysis 1 + 2 + 3 | |
OCS = oral corticosteroids.
Table 6: Summary of the Stepped Analysis of the CADTH Reanalysis Results
Stepped analysis | Treatment | Total costs ($) | Total QALYs | ICER ($ per QALY) |
|---|---|---|---|---|
Sponsor’s base case (deterministic) | SOC | 59,384 | 18.94 | Reference |
Tezepelumab | 266,189 | 20.01 | 192,546 | |
CADTH reanalysis 1 (survival benefit) | SOC | 67,089 | 21.36 | Reference |
Tezepelumab | 275,094 | 21.75 | 533,936 | |
CADTH reanalysis 2 (substitution with health state utilities) | SOC | 59,384 | 19.39 | Reference |
Tezepelumab | 266,189 | 20.25 | 240,125 | |
CADTH reanalysis 3 (removal of response assessment) | SOC | 59,384 | 18.94 | Reference |
Tezepelumab | 285,535 | 20.11 | 192,600 | |
CADTH base case (reanalysis 1 + 2 + 3) Probabilistic | SOC | 67,003 | 21.87 | Reference |
Tezepelumab | 295,314 | 22.04 | 1,334,178 | |
Reanalysis 1 + 2 + 3 | SOC | 67,089 | 21.87 | Reference |
Tezepelumab | 294,571 | 22.04 | 1,329,802 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SOC = standard of care.
Note: The CADTH reanalysis is based on publicly available prices of the comparator treatments. All presented analyses are deterministic, with the exception of the CADTH base case, which is presented probabilistically.
Scenario Analysis Results
CADTH performed price-reduction analyses based on the sponsor’s base case and CADTH’s base-case reanalysis. Based on the CADTH base case, a price reduction of approximately 95% would be required to achieve cost-effectiveness at a willingness-to-pay threshold of $50,000 per QALY (Table 7).
Table 7: CADTH Price-Reduction Analyses
Analysis | ICERs for tezepelumab vs. standard of care ($ per QALY) | |
|---|---|---|
Price reduction | Sponsor base case | CADTH reanalysis |
No price reduction | 184,075 | 1,334,178 |
10% | 165,518 | 1,198,386 |
20% | 146,962 | 1,062,594 |
30% | 128,405 | 926,803 |
40% | 109,848 | 791,011 |
50% | 91,291 | 655,219 |
60% | 72,734 | 519,427 |
70% | 54,178 | 383,635 |
80% | 35,621 | 247,844 |
90% | 17,064 | 112,052 |
95% | 7,786 | 44,156 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
CADTH performed scenario analyses to determine the impact of alternative assumptions on the cost-effectiveness of tezepelumab. CADTH assessed the impact of including a mortality benefit and a response assessment at 26 weeks and removing the difference in uncontrolled and controlled exacerbation health state utility values.
Inclusion of a mortality benefit resulted in a decreased ICER of $972,451 per QALY. This shows that even if a mortality benefit were to occur, the impact on cost-effectiveness would be small given the rarity of exacerbation-related deaths. This analysis assumes a direct impact on mortality reduction not demonstrated from the trial. Inclusion of response assessment at 26 weeks under the assumption that reductions in exacerbations or OCS use are adequate proxies for asthma control resulted in a minimally decreased ICER of $1,333,639 per QALY. This is because patients who remain on tezepelumab in the model continue to receive benefit; although costs are higher, QALYs are higher as well. Removing the difference between uncontrolled and controlled exacerbation health state utility values resulted in an increased ICER of $1,928,386 per QALY. This is because the utility reduction from having an exacerbation is reduced for those who are uncontrolled. This represents the upper limit of the ICER as a pooled estimate of exacerbation disutility was not provided by the sponsor and would likely result in a greater disutility associated with an exacerbation than what is used in this scenario analysis.
Issues for Consideration
CADTH notes that benralizumab and mepolizumab have concluded letter-of-intent agreements with the pan-Canadian Pharmaceutical Alliance.
Overall Conclusions
Evidence indicates that tezepelumab reduces the rate of asthma exacerbations compared to SOC alone in patients whose asthma remains uncontrolled despite being treated with medium- to high-dose ICS. This reduction in exacerbations appears to occur regardless of whether patients have type 2 or non–type 2 asthma. Additionally, tezepelumab appears to improve pulmonary function, as well as HRQoL and symptoms of asthma as measured by the Asthma Quality of Life Questionnaire (Standardized) for patients 12 years of age and older and the ACQ-6. There is no evidence that tezepelumab leads to a reduction of OCS doses in patients with OCS-dependent asthma or reduces exacerbations in these patients. Finally, the comparative effects of tezepelumab relative to other biologic treatments for severe asthma are limited due to a lack of direct comparative evidence and heterogeneity within the sponsor’s indirect treatment comparisons.
CADTH undertook reanalyses to address limitations in the sponsor’s submission, including assuming no mortality benefit associated with tezepelumab, using utility values associated with health states only, and removing response assessment at 26 weeks. CADTH was unable to address the lack of head-to-head comparative clinical data to other biologics and concerns with transparency over how transition probabilities were derived.
In the CADTH base case, tezepelumab plus SOC was more effective and more costly than SOC alone (incremental costs = $228,312; incremental QALYs = 0.171) resulting in an ICER of $1,334,178 per QALY gained. Relative to SOC alone, treatment with tezepelumab led to an additional $232,952 in drug costs but was associated with $4,643 in cost savings from reduced hospitalizations, outpatient, emergency department, and community care visits arising from fewer severe exacerbations and improved asthma control. The additional 0.171 QALYs from tezepelumab are associated with improved asthma control and a reduction in exacerbations. A scenario analysis was conducted that explored a potential optimistic survival benefit associated with tezepelumab. This slightly increased QALY gains to 0.23, resulting in an improved ICER of $972,451 per QALY. In the CADTH base case, the probability of tezepelumab being cost-effective at a willingness-to-pay threshold of $50,000 per QALY was 0%. A price reduction of approximately 95% is necessary to achieve cost-effectiveness at this threshold.
A small number of incremental QALYs in the CADTH base case are derived from benefits to patients who are OCS-dependent. However, based on the CADTH clinical review, there is no evidence that tezepelumab reduces OCS use and exacerbations in these patients. The incremental benefit as calculated in the CADTH base case is therefore likely overestimated. In a population consisting of non-chronic OCS users only, the ICER increases to $1,403,821 per QALY.
Given the lack of head-to-head comparative data and limitations associated with the indirect comparisons, there is no clinical evidence to support a price premium for tezepelumab above other biologics. The cost-effectiveness of tezepelumab in comparison to other biologics available in Canada is therefore unknown.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Tezspire (tezepelumab), 210 mg, 110 mg/mL solution for subcutaneous injection [internal sponsor's package]. Mississauga (ON): AstraZeneca Canada Inc; 2022 Apr 1.
2.Tezspire (tezepelumab): 110 mg/mL solution for injection, subcutaneous use; 210 mg single-use, pre-filled syringe; 210 mg single-use, pre-filled pen [product monograph]. Mississauga (ON): AstraZeneca Canada Inc; 2021 Jul 28. Accessed 2022 Jul 6.
3.Roberts NJ, Lewsey JD, Gillies M, et al. Time trends in 30 day case-fatality following hospitalisation for asthma in adults in Scotland: a retrospective cohort study from 1981 to 2009. Respir Med. 2013;107(8):1172-1177. PubMed
4.Royal College of Physicians. Why asthma still kills. The National Review of Asthma Deaths (NRAD). Confidential inquiry report. London (GB): Royal College of Physicians; 2014: https://www.hqip.org.uk/wp-content/uploads/2018/04/NRAD-Annual-Report-2014.pdf. Accessed 2022 Jul 6.
5.Watson L, Turk F, James P, Holgate ST. Factors associated with mortality after an asthma admission: a national United Kingdom database analysis. Respir Med. 2007;101(8):1659-1664. PubMed
6.Statistics Canada. Table: 13-10-0114-01. Life expectancy and other elements of the complete life table, three-year estimates, Canada, all provinces except Prince Edward Island. 2022; https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011401. Accessed 2022 Jul 6.
7.Xie F, Pullenayegum E, Gaebel K, et al. A time trade-off-derived value set of the EQ-5D-5L for Canada. Med Care. 2016;54(1):98-105. PubMed
8.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2022; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2022 Jul 6.
9.Exceptional Access Program (EAP). Toronto (ON): Ontario Ministry of Health; Ontario Ministry of Long-Term Care; 2022: https://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx. Accessed 2021 Sep 1.
10.CADTH Canadian Drug Expert Committee (CDEC) final recommendation: reslizumab (Cinqair - Teva Canada Innovation). Ottawa (ON): CADTH; 2017 Mar 22: https://www.cadth.ca/sites/default/files/cdr/complete/SR0495_complete_Cinqair-Mar-24-17.pdf. Accessed 2022 Jun 15.
11.Ontario Case Costing Initiative (OCCI). Toronto (ON): Ontario Health and Long-Term Care; 2017: https://data.ontario.ca/dataset/ontario-case-costing-initiative-occi. Accessed 2021 Dec 15.
12.Schedule of benefits for physician services under the Health Insurance Act: effective October 1, 2021. Toronto (ON): Ontario Ministry of Health; 2022: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf. Accessed 2021 Sep 15.
13.Government of Canada. Job Bank. Wages: registered nurse (N.R.) in Canada. 2022; https://www.jobbank.gc.ca/marketreport/wages-occupation/993/ca. Accessed 2022 Jul 11.
14.Tam VC, Ko YJ, Mittmann N, et al. Cost-effectiveness of systemic therapies for metastatic pancreatic cancer. Curr Oncol. 2013;20(2):e90-e106. PubMed
15.Clinical Practice Research Datalink (CPRD). An observational retrospective cohort study to evaluate chronic disease onset associated with long-term oral corticosteroid use and the related cost impact on patients in the OPCRD / CPRD databases. 2017; https://cprd.com/protocol/observational-retrospective-cohort-study-evaluate-chronic-disease-onset-associated-long. Accessed 2021 Nov 15.
16.Global strategy for asthma management and prevention. Fontana (WI): Global Initiative for Asthma; 2020: https://ginasthma.org/wp-content/uploads/2020/06/GINA-2020-report_20_06_04-1-wms.pdf. Accessed 2022 Jul 6.
17.Menzies-Gow A, Ponnarambil S, Downie J, Bowen K, Hellqvist A, Colice G. DESTINATION: a phase 3, multicentre, randomized, double-blind, placebo-controlled, parallel-group trial to evaluate the long-term safety and tolerability of tezepelumab in adults and adolescents with severe, uncontrolled asthma. Respir Res. 2020;21(1):279. PubMed
18.Levy ML. The national review of asthma deaths: what did we learn and what needs to change? Breathe (Sheff). 2015;11(1):14-24. PubMed
19.Suissa S, Ernst P, Benayoun S, Baltzan M, Cai B. Low-dose inhaled corticosteroids and the prevention of death from asthma. N Engl J Med. 2000;343(5):332-336. PubMed
20.Canadian Institute for Health Information. Asthma emergency department visits: volume and median length of stay. 2021; https://www.cihi.ca/en/indicators/asthma-emergency-department-visits-volume-and-median-length-of-stay. Accessed 2022 Jul 6.
21.Engelkes M, de Ridder MA, Svensson E, et al. Multinational cohort study of mortality in patients with asthma and severe asthma. Respir Med. 2020;165:105919. PubMed
22.Guidelines for the economic evaluation of health technologies: Canada. 4th ed. Ottawa (ON): CADTH; 2017: https://www.cadth.ca/guidelines-economic-evaluation-health-technologies-canada-4th-edition. Accessed 2022 Jul 6.
23.CADTH Common Drug Review pharmacoeconomic review report: benralizumab (Fasrena) for an add-on maintenance treatment of adult patients with severe eosinophilic asthma. Ottawa (ON): CADTH; 2018 Aug: https://cadth.ca/sites/default/files/cdr/pharmacoeconomic/SR0561_Fasenra_PE_Report.pdf. Accessed 2022 Jul 6.
24.CADTH reimbursement review: dupilumab (Dupixent) for type 2 eosinophilic asthma. Can J Health Technol. 2021;1(8). https://www.cadth.ca/sites/default/files/cdr/complete/SR0667-combined%20clinical%20and%20PE%20report.pdf. Accessed 2022 Jul 6.
25.DeltaPA. Ottawa (ON): IQVIA; 2022: https://www.iqvia.com/. Accessed 2022 Jul 6.
26.Saskatchewan Drug Plan: search formulary. 2022; https://formulary.drugplan.ehealthsask.ca/SearchFormulary. Accessed 2022 Jul 6.
27.Alberta Health Care Insurance Plan: medical price list as of 31 March 2020. Edmonton (AB): Government of Alberta; 2020: https://open.alberta.ca/dataset/30add047-29c2-4fc7-83b5-a8ab78605cdd/resource/48d0dd0b-bb33-478d-a85c-85a86d4555ad/download/health-somb-medical-price-list-2020-03.pdf. Accessed 2021 Feb 23.
28.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Tezspire (tezepelumab), 210 mg, 110 mg/mL solution for subcutaneous injection [internal sponsor's package]. Mississauga (ON): AstraZeneca Canada Inc; 2022 Apr 1.
29.CompassRx, 6th edition: annual public drug plan expenditure report, 2018/2019. Ottawa (ON): Patented Medicine Prices Review Board; 2020: https://www.canada.ca/content/dam/pmprb-cepmb/documents/npduis/analytical-studies/compassrx-6th-edition/NPDUIS-CompassRx6th-2018-2019_en.pdf. Accessed 2021 Nov 15.
30.Dinh T, Sutherland G. Understanding the gap: a pan-Canadian analysis of prescription drug insurance coverage. Ottawa (ON): The Conference Board of Canada; 2017: https://www.conferenceboard.ca/e-library/abstract.aspx?did=9326. Accessed 2022 Jul 6.
31.Statistics Canada. Table: 17-10-0057-01. Projected population, by projection scenario, age and sex, as of July 1 (x 1,000). 2022: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710005701. Accessed 2022 Jul 6.
32.Statistics Canada. Table: 13-10-0096-01. Health characteristics, annual estimates. 2022; https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310009601. Accessed 2022 Jul 11.
33.Noorduyn SG, Johnston K, Osenenko K, Sriskandarajah N, Gendron A, Mbuagbaw L. Discontinuation of benralizumab in Canadian patients with severe eosinophilic asthma. ERJ Open Res. 2021;7(4). PubMed
34.Severe asthma - the Canadian patient journey. Toronto (ON): Asthma Society Canada; 2014: https://asthma.ca/wp-content/uploads/2017/06/SAstudy.pdf. Accessed 2022 Feb 15.
35.Chapman K, Remtulla A, Gendron A, Xu S, Nan C. Regional variation in asthma prevalence and oral corticosteroid use for Canadian patients: heat map analysis. A48 Environmental Asthma. 2020:A1835. https://www.atsjournals.org/doi/epdf/10.1164/ajrccm-conference.2020.201.1_MeetingAbstracts.A1835. Accessed 2022 Jul 6.
36.Bosonea AM, Sharpe H, Wang T, et al. Developments in asthma incidence and prevalence in Alberta between 1995 and 2015. Allergy Asthma Clin Immunol. 2020;16:87. PubMed
37.Public Health Agency of Canada. Asthma and chronic obstructive pulmonary disease (COPD) in Canada, 2018. Report from the Canadian Chronic Disease Surveillance System 2018; https://www.canada.ca/en/public-health/services/publications/diseases-conditions/asthma-chronic-obstructive-pulmonary-disease-canada-2018.html#a1.2.2. Accessed 2022 Jul 10.
38.Husereau D, Goodfield J, Leigh R, Borrelli R, Cloutier M, Gendron A. Severe, eosinophilic asthma in primary care in Canada: a longitudinal study of the clinical burden and economic impact based on linked electronic medical record data. Allergy Asthma Clin Immunol. 2018;14:15. PubMed
Appendix 1: Cost Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CADTH Cost Comparison Table of Biologics for Severe Asthma
Treatment | Strength | Form | Price | Recommended dosage | Daily cost ($) | Annual cost ($) |
|---|---|---|---|---|---|---|
Tezepelumab (TBC) | 110 mg/mL | Prefilled syringe for SC injection; prefilled pen for SC injection | 1,938.4600a | 210 mg every 4 weeks | 69.04 | 25,200 |
Biologics | ||||||
Benralizumab (Fasenra) | 30 mg/mL | Prefilled syringe for SC injection | 3,919.5700 | 30 mg every 4 weeks for first 3 doses, then once every 8 weeks | Year 1: 85.91 Year 2+: 69.8 | Year 1: 31,357 Year 2+: 25,477 |
Dupilumab (Dupixent) | 200 mg | Prefilled syringe for SC injection | 978.7000 | Initial dose of 400 mg or 600 mg, followed by 200 or 300 mg every 2 weeks | Year 1: 72.40 Year 2+ 69.72 | Year 1: 26,425 Year 2+: 25,446 |
Mepolizumab (Nucala) | 100 mg/mL | Vial of powder for SC injection | 2,100.6100 | 100 mg every 4 weeks | 74.82 | 27,308 |
Prefilled syringe for SC injection | ||||||
Prefilled autoinjector for SC injection | ||||||
Omalizumab (Xolair) | 150 mg | Vial of powder for SC injection | 652.9800b | 150 to 375 mg every 2 or 4 weeksc | 23.26 to 139.54 | 8,489 to 50,932 |
75 mg | Prefilled syringe for SC injection | 281.2400 | 20.03 to 100.17 | 7,312 to 36,561 | ||
150 mg | 641.6000 | 22.85 to 137.11 | 8,341 to 50,045 | |||
Reslizumab (Cinqair) | 10 mg/mL | Vial of solution for IV infusion | 640.0000d | 3 mg/kg every 4 weeks | 22.80 to 91.18e | 8,320 to 33,280e |
IV = intravenous; SC = subcutaneous.
Note: All prices are from the Ontario Exceptional Access Program Formulary9 (accessed May 2022), unless otherwise indicated, and do not include dispensing fees. Drug wastage was included.
aBased on sponsor’s submission.1
bPrice obtained from Delta PA Database.25
cDosing is dependent upon body weight and baseline immunoglobin E and can range from 150 mg to 300 mg when dosed every 4 weeks, and 225 mg to 375 mg when dosed every 2 weeks.
dPrice obtained from the CADTH Canadian Drug Expert Committee Recommendation for reslizumab.10
eAssumed weight range 30 kg to 120kg.
Table 9: CADTH Cost Comparison Table of Other Medications for Asthma
Drug/comparator | Strength | Dosage form | Price ($) | Recommended dosage | Daily drug cost ($) | Annual drug cost ($) | |
|---|---|---|---|---|---|---|---|
Inhaled corticosteroids | |||||||
Beclomethasone dipropionate (QVAR) | 50 mcg 100 mcg | MDI (200 doses) | 37.1200 74.0200 | 50 to 400 mcg twice daily | 0.37 to 2.96 | 135 to 1,081 | |
Budesonide (Pulmicort Turbuhaler) | 100 mcg 200 mcg 400 mcg | MDPI (200 doses) | 33.9600 69.4600 101.3900 | 200 to 400 mcg twice daily | 0.69 to 1.01 | 254 to 370 | |
Ciclesonide (Alvesco) | 100 mcg 200 mcg | MDI (120 doses) | 47.8560 79.1880 | 100 to 800 mcg twice daily | 0.80 to 2.64 | 291 to 963 | |
Fluticasone furoate (Arnuity Ellipta) | 100 mcg 200 mcg | MDPI (30 doses) | 42.7700 85.5500 | 100 or 200 mcg once daily | 1.43 to 2.85 | 520 to 1,041 | |
Fluticasone propionate (Flovent Diskus) | 100 mcg 250 mcg 500 mcg | MDPI (60 doses) | 26.2900a 49.0200 76.2500 | 100 to 500 mcg twice daily | 0.88 to 2.54 | 320 to 927 | |
Fluticasone propionate (Flovent HFA) | 50 mcg 125 mcg 250 mcg | MDI (120 doses) | 28.4200 49.0200 45.0200 | 100 to 500 mcg twice daily | 0.95 to 1.50 | 346 to 548 | |
Mometasone furoate (Asmanex Twisthaler) | 100 mcg 200 mcg 400 mcg | MDPI (60 doses) | 76.1940b 40.4100 80.7900 | 200 or 400 mcg once daily | 0.67 to 1.35 | 246 to 491 | |
ICS/LABA Combinations | |||||||
Indacaterol acetate/mometasone furoate (Atectura Breezhaler) | 150/80 mcg 150/160 mcg 150/320 mcg | Inhalation pwd hard capsules (30 doses) | 58.0800 | One capsule for inhalation daily | 1.9360 | 707 | |
Budesonide/ formoterol fumarate dihydrate (Symbicort Turbuhaler) | 100/6 mcg 200/6 mcg | MDPI (120 dose pack) | 69.5400 90.3600 | Low | 100/6 mcg, 2 inhalations twice daily | 2.32 | 846 |
Med | 200/6 mcg, 2 to 4 inhalations daily | 1.51 to 3.01 | 550 to 1,099 | ||||
High | 200/6 mcg, > 4 inhalations dailyc | >3.01 | >1,099 | ||||
Fluticasone propionate/ salmeterol (Advair) | 125/25 mcg 250/25 mcg | MDI (120 pack) | 114.4100 162.4200 | Low | 125/25 mcg, 1 inhalation twice daily | 1.91 | 696 |
Med | 125/25 mcg, 2 inhalations twice daily | 3.81 | 1,392 | ||||
High | 250/25 mcg, 2 inhalations twice daily | 5.41 | 1,976 | ||||
Fluticasone propionate/ salmeterol (Advair Diskus, generic) | 100/50 mcg 250/50 mcg 500/50 mcg | MDPI (60 doses) | 42.4050 50.7600 72.0600 | Low | 100/50 mcg, 1 inhalation twice daily | 1.41 | 516 |
Med | 250/50 mcg, 1 inhalation twice daily | 1.69 | 618 | ||||
High | 500/50 mcg, 1 inhalation twice daily | 2.40 | 877 | ||||
Fluticasone furoate/vilanterol (Breo Ellipta) | 100/25 mcg 200/25 mcg | MDPI (30 doses) | 93.0500 144.7400 | Low | NA | NA | NA |
Med | 100/25 mcg, 1 inhalation once daily | 3.10 | 1,132 | ||||
High | 200/25 mcg, 1 inhalation once daily | 4.82 | 1,761 | ||||
Mometasone furoate/formoterol fumarate dihydrate (Zenhale) | 100/5 mcg 200/5 mcg | MDI (120 doses) | 107.6400 130.4300 | Low | NA | NA | NA |
Med | 100/5 mcg, 2 inhalations twice daily | 3.56 | 1,310 | ||||
High | 200/5 mcg, 2 inhalations twice daily | 4.35 | 1,587 | ||||
Long-Acting Beta2-Adrenergic Agonists (LABA) | |||||||
Salmeterol xinafoate (Serevent Diskhaler) | 50 mcg | Dry powder inhaler (60 doses) | 67.0000 | 50 mcg twice daily | 2.68 | 978 | |
Formoterol fumarate (Foradil) | 12 mcg | Dry powder capsules for inhalation (60 doses) | 55.7700 | 12 mcg twice daily | 1.86 | 679 | |
Formoterol fumarate dihydrate (Oxeze Turbuhaler) | 6 mcg 12 mcg | MDPI (60 doses) | 34.0200 45.2900 | 6 to 12 mcg twice daily | 1.13 to 1.51 | 414 to 551 | |
ICS/LABA/LAMA Combinations | |||||||
Indacaterol/ | 150/50/160 mcg | Inhalation pwd hard capsules (30 doses) | 102.82 | One capsule inhaled daily | 3.43 | 1,251 | |
Leukotriene receptor antagonists (LTRA) | |||||||
Montelukast (Singulair, generics) | 4 mg 5 mg 10 mg | Chew tab Chew tab Tablet | 0.2758 0.3082a 0.4231a | Age 6-14: 5 mg daily | 0.31 to 0.42 | 112 to 154 | |
Long-acting muscarinic antagonists (LAMA) | |||||||
Tiotropium (Spiriva Respimat) | 2.5 mcg | Solution for inhalation (60 doses) | 54.8580 | 2 inhalations once daily | 1.83 | 667 | |
Oral corticosteroids | |||||||
Prednisone (generic) | 1 mg 5 mg 50 mg | Tab | 0.1166a 0.0220 0.1735 | 5 to 60 mg daily | 0.02 to 0.17 | 8 to 85 | |
Note: All prices are from the Ontario Drug Benefit Formulary (accessed May 2022), unless otherwise indicated, and do not include dispensing fees.8 Drug wastage was included.
aPrice obtained from Saskatchewan Online Formulary Database.26
bPrice obtained from Alberta Online Formulary Database.27
cBased on clinical expert feedback.
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | Cost-effectiveness of tezepelumab was assessed relative to SOC; cost-effectiveness relative to other biologic treatments available in Canada is unknown |
Model has been adequately programmed and has sufficient face validity | No | Poor modelling practices were employed (see CADTH critical appraisal section) |
Model structure is adequate for decision problem | No | The sponsor’s submitted model lacked face validity in consistency and ability to predict 52-week results from the NAVIGATOR and SOURCE trials; due to poor modelling practices, CADTH could not fully validate the sponsor’s submitted model |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | No | The model lacked transparency and technical documentation (see CADTH critical appraisal section) for several parameter calculations (i.e., health state–specific utilities, transition probabilities, exacerbations) which limited CADTH’s ability to validate the model |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
CA = controlled asthma; ER = emergency visit; OCS = oral corticosteroids; UA = uncontrolled asthma.
Source: Sponsor’s pharmacoeconomic submission.1
Detailed Results of the Sponsor’s Base Case
Table 11: Disaggregated Summary of the Sponsor’s Economic Evaluation Results
Parameter | Tezepelumab | SOC | Incremental |
|---|---|---|---|
Discounted life-years | |||
Total | 24.13 | 23.29 | 0.85 |
Discounted QALYs | |||
Total | 19.95 | 18.82 | 1.13 |
Controlled | 11.37 | 9.75 | 1.62 |
Uncontrolled | 7.01 | 7.12 | -0.11 |
OCS Burst | 1.40 | 1.74 | -0.33 |
ER Visit | 0.06 | 0.08 | -0.02 |
Hospitalization | 0.10 | 0.14 | -0.04 |
Discounted costs ($) | |||
Total | 266,546 | 59,445 | 207,101 |
Acquisition | 244,964 | 34,587 | 210,377 |
Biologic | 209,124 | 0 | 209,124 |
SOC | 35,823 | 34,567 | 1,256 |
OCS | 17 | 20 | -2 |
Controlled | 3,102 | 2,716 | 386 |
Uncontrolled | 9,479 | 9,786 | -307 |
OCS Burst | 524 | 653 | -129 |
ER Visit | 468 | 601 | -133 |
Hospitalization | 7,926 | 11,007 | -3,081 |
Adverse Events | 84 | 96 | -12 |
ICER ($/QALY) | 184,075 | ||
ER = emergency room; ICER = incremental cost-effectiveness ratio; LY= life-year; OCS = oral corticosteroid; QALY = quality-adjusted life-year; SOC = standard of care.
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Detailed Results of CADTH Base Case
Table 12: Disaggregated Summary of CADTH’s Economic Evaluation Results
Parameter | Tezepelumab | SOC | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total | 26.25 | 26.25 | 0.00 |
Discounted QALYs | |||
Total | 22.04 | 21.87 | 0.17 |
Controlled | 13.02 | 11.78 | 1.23 |
Uncontrolled | 7.30 | 7.83 | -0.53 |
OCS Burst | 1.54 | 2.00 | -0.47 |
ER Visit | 0.07 | 0.10 | -0.02 |
Hospitalization | 0.11 | 0.15 | -0.05 |
Discounted costs ($) | |||
Total | 295,314 | 67,003 | 228,312 |
Acquisition | 271,939 | 38,987 | 232,952 |
Biologic | 232,955 | 0 | 232,955 |
SOC | 38,964 | 38,964 | 0 |
OCS | 19 | 23 | -3 |
Controlled | 3,372 | 3,052 | 320 |
Uncontrolled | 10,218 | 10,953 | -735 |
OCS Burst | 563 | 732 | -170 |
ER Visit | 511 | 686 | -175 |
Hospitalization | 8,617 | 12,482 | -3,865 |
Adverse Events | 95 | 111 | -15 |
ICER ($/QALY) | 1,334,178 | ||
ER = emergency room; ICER = incremental cost-effectiveness ratio; LY= life-year; OCS = oral corticosteroid; QALY = quality-adjusted life-year; SOC = standard of care.
Scenario Analyses
Table 13: Summary of CADTH’s Scenario Analyses Results
Drug | Total Costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|
Scenario 1: Mortality Benefit Included | |||
SOC | 65,640 | 21.42 | Ref. |
Tezepelumab | 294,020 | 21.65 | 972,451 |
Scenario 2: Include Response Assessment at 26 Weeks | |||
SOC | 67,116 | 21.87 | Ref. |
Tezepelumab | 276,122 | 22.03 | 1,333,639 |
Scenario 3: Removal of Utility Difference in Uncontrolled and Controlled Exacerbation Health States | |||
SOC | 67,093 | 22.09 | Ref. |
Tezepelumab | 295,079 | 22.21 | 1,928,386 |
Scenario 4: Cost-effectiveness in those not on chronic OCS | |||
SOC | $66,517 | 21.90 | Ref. |
Tezepelumab | $299,006 | 22.07 | 1,403,821 |
Scenario 5: Increase the probability of a severe exacerbation in those on SOC alone | |||
SOC | $69,720 | 21.85 | Ref. |
Tezepelumab | $296,269 | 22.03 | 1,283,926 |
ICER = incremental cost-effectiveness ratio; LY= life-year; OCS = oral corticosteroid; QALY = quality-adjusted life-year; Ref. = reference.
Note: Reanalyses are based on publicly available prices of the comparator treatments.
Appendix 5: Submitted Budget Impact Analysis and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 14: Summary of Key Take-Aways
Key Take-aways of the budget impact analysis |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
The submitted budget impact analysis assessed the introduction of tezepelumab as an add-on maintenance treatment in patients aged 12 years and older with severe asthma.28 The analysis took the perspective of CADTH-participating Canadian public drug plans using a top-down epidemiological approach and incorporated drug acquisition costs. A time horizon of 3 years was taken. The target population size was estimated using number of beneficiaries 12 years and older, prevalence of asthma in Canada (8.7%), the proportion of asthmatic patients with severe asthma (8.0%), and lastly proportion of those eligible for biologics in Canada (5.5%). The base-case analysis considers SOC alone (ICS/LABA with or without OCS) in addition to currently reimbursed add-on biologic treatments as comparators in the model. The reference scenario included SOC alone, omalizumab, mepolizumab, benralizumab, and dupilumab, and the new drug scenario considered the reimbursement of tezepelumab. Key inputs to the BIA and the sponsor’s methodology in calculating target population are documented in Table 16.
Table 15: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as Year 1 / Year 2 / Year 3 if appropriate) |
|---|---|
Target population | |
8,606,653 | |
Asthma prevalence32 | 8.7% |
8.0% | |
Proportion of patients with severe asthma eligible for biologics35 | 5.5% |
Number of patients eligible for drug under review | 3,335 / 3,376 / 3,417 |
Market uptake (3 years) | |
Uptake (reference scenario) SOC Omalizumab Mepolizumab Benralizumab Dupilumab | ||||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||||||||||||||||||||||||||||||||| |
Uptake (new drug scenario) Tezepelumab SOC Omalizumab Mepolizumab Benralizumab Dupilumab | ||||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||||||||||||||||||||||||||||||||| ||||||||||||||||||||||||||||||||||||||||||||| |
Cost of treatment (per patient) | |
Cost of treatment over one year Tezepelumab SOC (plus OCS) Omalizumab Mepolizumab Benralizumab Dupilumab | $25,287 $1,484 ($1,511) $25,554 $26,172 $26,159 $25,680 |
OCS = oral corticosteroid; SOC = standard of care.
Summary of the Sponsor’s Budget Impact Analysis Results
The sponsor’s estimated budget impact of funding tezepelumab as an add-on maintenance treatment in patients aged 12 years and older with severe asthma was $84,456 in Year 1, $28,552 in Year 2, and $101,069 in Year 3, for a 3-year total of $214,077.
In the sponsor’s scenario analyses, increasing several parameters (i.e., prevalence of asthma, proportion of patients with severe asthma, proportion of patients with severe asthma eligible for biologic treatment) resulted in increased costs to the drug plans over 3 years by 20%.
CADTH Appraisal of the Sponsor’s Budget Impact Analysis
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Uncertainty regarding the sponsor’s epidemiological approach to calculate target population: The sponsor used prevalence of asthma to estimate target population and does not explicitly model incidence. It is assumed that the asthma population will change at the same rate as the province specific population growth rate. In some provinces, such as Newfoundland, the sponsor predicts asthma cases to fall over time in line with an assumed decreasing population size. Some evidence suggests that asthma prevalence, as a percentage of the population, may increase over time.32,36,37 Although the sponsor accounts for population growth they do not account for potential growth in asthma as well.
Secondly, uptake of tezepelumab is likely to be different in the incident population versus the prevalent population. For those well controlled on a current biologic they are unlikely going to switch to tezepelumab. For those who require a biologic for the first time the clinical expert noted that uptake in tezepelumab may be higher. It can only be assumed that the market uptake estimates calculated by the sponsor take this into account, but it is not explicitly modelled which would provide a more transparent and accurate calculation.
CADTH could not address the limitation regarding exclusion of incidence as this would require the entire structure of the model to be changed.
Estimating the percentage of patients eligible for biologics: The clinical expert consulted by CADTH noted that patient eligibility for biologics is potentially underestimated at 5.5%. This value considers all patients with severe asthma who have had more than 2 OCS prescriptions lasting more than 10 days in the past year. The Health Canada indication for tezepelumab does not specify that patients treated with tezepelumab are required to have uncontrolled asthma with previous exacerbations. Furthermore, estimates from literature indicate that approximately 20% of patients with severe asthma are uncontrolled in Canada.38 Although not all uncontrolled patients will receive biologic treatment, current treatment eligibility dictated by eosinophil count may not be applicable to tezepelumab given its broader indication. Lastly, the clinical expert stated that there may also be indication creep with patients with moderate asthma also being treated with tezepelumab given its efficacy and infrequent administration.
In a scenario analysis, CADTH increased the proportion of patients eligible for biologics from 5.5% to 20%.
Uncertainty regarding sponsor’s use of beneficiaries to calculate eligible patients for treatment: The sponsor used data across various years (2018 through 2020) and various data sources to estimate the number of beneficiaries eligible for tezepelumab age 12 or older. However, using the number of beneficiaries may underestimate the number of eligible patients for tezepelumab, as this assumes that tezepelumab will only be used in patients who have previously made claims. There may be patients who have not made a claim but will do so upon receiving a biologic. It also assumes that asthma prevalence is the same in the general population as it is for those currently making a claim. As asthma requires chronic medication the likelihood of a claimant having asthma may be different than that of the general population. For example, the estimated eligible patients using BC PharmaCare beneficiary data is 787,839 patients whereas enrolment in BC public drug coverage calculated using the Understanding the Gap report led to CADTH estimating that this number may be closer to 3,690,661 enrolled patients.30 Therefore, the budget impact of tezepelumab may be impacted by the underestimation of estimated eligible patients using beneficiary information.
CADTH recalculated the target population using enrolment in public plans as per the Understanding the Gap report and 2021 Canadian population estimates.30
Uncertainty regarding uptake of tezepelumab: The CADTH clinical expert indicated that patients with non–type 2 asthma who are currently ineligible for biologics would be eligible for tezepelumab given the broader indication. In the sponsor’s analysis they assume |||||||||% of patients are currently not receiving a biologic despite having uncontrolled severe asthma. It is assumed this accounts for patients who do not meet the eligibility criteria for biologics plus those who are not seeing a specialist. The sponsor assumes minimal uptake of tezepelumab in this group (roughly |||||||||% of patients move from SOC to tezepelumab). This means the sponsor assumes minimal uptake in those who have severe uncontrolled asthma but are currently ineligible for a currently funded biologic given the type of asthma they have. The clinical expert felt that of those currently receiving SOC many will continue to not receive a biologic as they are not seeing a specialist however there is considerable uncertainty as to what uptake will be in this group.
CADTH performed a scenario analysis where reimbursement of tezepelumab resulted in capture of 10% of SOC market shares; representing potential uptake in patients who are ineligible for current biologics due to their asthma type.
Uncertainty regarding market shares of tezepelumab and displacement of existing biologic treatments: The market uptake of tezepelumab was assumed to be |||||||||% in year 1, |||||||||% in year 2, and |||||||||% in year 3 based on the sponsor’s internal assumptions. The accuracy of the sponsor’s internal market shares could not be validated by CADTH. Comparator market shares were estimated from “AstraZeneca market research data” Based on discussion with clinical experts, it was noted that uptake of tezepelumab would likely capture market shares from benralizumab.
CADTH could not address this limitation due to inability to validate the sponsor’s market shares and uncertainty surrounding market shares of tezepelumab and biologic comparators.
CADTH Reanalyses of the Budget Impact Analysis
Table 16: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Changes to derive the CADTH scenario analyses | ||
Scenario analysis 1: Target population recalculated using proportion of patients covered by public drug plans30 | British Columbia: 792,485 Alberta: 726,674 Saskatchewan: 632,789 Manitoba: 121,838 Ontario: 5,176,474 New Brunswick: 122,08 Nova Scotia:141,749 Newfoundland and Labrador: 93,837 Prince Edward Island: 45,081 NIHB: 753,646 pan-Canadian: 8,606,653 | British Columbia: 3,394,263 Alberta: 843,984 Saskatchewan: 448,207 Manitoba: 672,359 Ontario: 3,734,781 New Brunswick: 124,378 Nova Scotia: 199,854 Newfoundland and Labrador: 296,542 Prince Edward Island: 49,140 NIHB: 812,445 pan-Canadian: 10,575,954 |
Scenario analysis 2: Increased proportion of those eligible for biologic | Eligibility for biologic: 5.5% (calculated by assuming 5.5% of patients with severe asthma are uncontrolled) | Eligibility for biologic: 20% (calculated by assuming 20% of patients with severe asthma are uncontrolled) |
Scenario analysis 3: Increased tezepelumab market shares through uptake of SOC | SOC: ||||||||||||||||||||||||||||||||||||||||||||| Tezepelumab: ||||||||||||||||||||||||||||||||||||| | SOC: 24.35% / 20.81% / 17.08% Tezepelumab: 5.44% / 19.12% / 26.74% |
NIHB; non-insured health benefits; SOC = standard of care.
The results of the CADTH scenario analyses are presented in Table 17. CADTH did not undertake reanalysis of the sponsor’s BIA, owing to the high degree of uncertainty around key model parameters, including the size of the eligible population and exclusion of incidence. Due to the uncertainty as well as the additional limitations described above, the impact of reimbursing tezepelumab is highly uncertain. CADTH did undertake several scenario analyses to assess the impact of changing key parameters within the sponsor BIA as outlined in Table 16.
The scenario analysis assessing a target population comprised of those enrolled and covered by public plans led to a 3-year budget impact of $348,107 in cost savings. This was due to an increase in the population covered by BC public funding where the sponsor predicts cost savings to occur; this caused the pan-Canadian estimate to become cost saving. A scenario analysis assessing the budget impact of 20% of patients with severe asthma being uncontrolled led to a 3-year budget impact of $778,461. An additional scenario analysis assessing the budget impact if tezepelumab were to capture 10% of SOC market shares led to a 3-year budget impact of $17,356,108. CADTH notes that although the scenario analyses lead to small changes in the budget impact the change in spend on tezepelumab varied substantially. Varying from $28 million in the sponsor’s base case to $103 million when considering a higher proportion of uncontrolled asthma in the severe population.
Table 17: Detailed Breakdown of the CADTH Reanalyses of the Budget Impact Analysis
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total | |
|---|---|---|---|---|---|---|---|
Submitted base case | Reference | Tezepelumab | $0 | $0 | $0 | $0 | $0 |
Comparatorsa | $66,462,396 | $67,722,062 | $68,506,880 | $69,528,910 | $205,757,853 | ||
Total | $66,462,396 | $67,722,062 | $68,506,880 | $69,528,910 | $205,757,853 | ||
New drug | Tezepelumab | $0 | $1,885,694 | $11,241,832 | $15,306,068 | $28,433,594 | |
Comparatorsa | $66,462,396 | $65,920,825 | $57,293,600 | $54,323,911 | $177,538,336 | ||
Total | $66,462,396 | $67,806,518 | $68,535,432 | $69,629,979 | $205,971,929 | ||
Budget impact | $0 | $84,456 | $28,552 | $101,069 | $214,077 | ||
CADTH scenario analysis: recalculated target population (enrolled) | Reference | Tezepelumab | $0 | $0 | $0 | $0 | $0 |
Comparatorsa | $82,705,692 | $84,206,449 | $84,971,926 | $86,152,302 | $255,330,677 | ||
Total | $82,705,692 | $84,206,449 | $84,971,926 | $86,152,302 | $255,330,677 | ||
New drug | Tezepelumab | $0 | $4,010,713 | $15,025,472 | $19,756,112 | $38,792,298 | |
Comparatorsa | $82,705,692 | $80,152,743 | $69,770,948 | $66,266,581 | $216,190,272 | ||
Total | $82,705,692 | $84,163,456 | $84,796,420 | $86,022,694 | $254,982,570 | ||
Budget impact | $0 | -$42,993 | -$175,506 | -$129,608 | -$348,107 | ||
CADTH scenario analysis: increased % eligible for biologic) | Reference | Tezepelumab | $0 | $0 | $0 | $0 | $0 |
Comparatorsa | $241,681,440 | $246,262,045 | $249,115,927 | $252,832,401 | $748,210,373 | ||
Total | $241,681,440 | $246,262,045 | $249,115,927 | $252,832,401 | $748,210,373 | ||
New drug | Tezepelumab | $0 | $6,857,068 | $40,879,389 | $55,658,429 | $103,394,886 | |
Comparatorsa | $241,681,440 | $239,712,090 | $208,340,364 | $197,541,495 | $645,593,948 | ||
Total | $241,681,440 | $246,569,158 | $249,219,752 | $253,199,924 | $748,988,834 | ||
Budget impact | $0 | $307,113 | $103,825 | $367,523 | $778,461 | ||
CADTH scenario analysis: 10% increased tezepelumab uptake of SOC | Reference | Tezepelumab | $0 | $0 | $0 | $0 | $0 |
Comparatorsa | $66,462,396 | $67,722,062 | $68,506,880 | $69,528,910 | $205,757,853 | ||
Total | $66,462,396 | $67,722,062 | $68,506,880 | $69,528,910 | $205,757,853 | ||
New drug | Tezepelumab | $0 | $4,860,506 | $17,263,812 | $24,448,022 | $46,572,340 | |
Comparatorsa | $66,462,395.95 | $65,757,366.14 | $56,962,698.23 | $53,821,556.51 | $176,541,621 | ||
Total | $66,462,396 | $70,617,872 | $74,226,510 | $78,269,579 | $223,113,961 | ||
Budget impact | $0 | $2,895,810 | $5,719,630 | $8,740,669 | $17,356,108 | ||
BIA = budget impact analysis; SOC = standard of care.
aComparators includes costs associated with standard of care (consisting of ICS/LABA with or without OCS); biologics (omalizumab, mepolizumab, benralizumab, dupilumab).
Stakeholder Input
Patient Input
Lung Health Foundation / Ontario Lung Association
About the Lung Health Foundation / Ontario Lung Association
The Ontario Lung Association (now named Lung Health Foundation) is registered with the CADTH and pCODR (www.lunghealth.ca). The Lung Health Foundation (Ontario Lung
Association) is a registered charity that assists and empowers people living with or caring for others with lung disease. It is a recognized leader, voice and primary resource in the prevention and control of respiratory illness, tobacco cessation and prevention, and its effects on lung health. The Foundation provides programs and services to patients and health-care providers, invests in lung research and advocates for improved policies in lung health. It is run by a board of directors and has approximately 46 employees, supported by thousands of dedicated volunteers.
Information Gathering
CADTH is interested in hearing from a wide range of patients and caregivers in this patient input submission. Describe how you gathered the perspectives: for example, by interviews, focus groups, or survey; personal experience; or a combination of these. Where possible, include when the data were gathered; if data were gathered in Canada or elsewhere; demographics of the respondents; and how many patients, caregivers, and individuals with experience with the drug in review contributed insights. We will use this background to better understand the context of the perspectives shared.
The information provided from the Lung Health Foundation in this submission was obtained from three phone interviews that were conducted in April 2021. The interviews were with three female patients living with Asthma. All the patients interviewed were over the age of 50 and are based in Ontario. Input from a certified respiratory educator was also obtained for this submission. The individual reviewed sections related to disease experience, experiences with available treatments and outcomes.
Disease Experience
CADTH involves clinical experts in every review to explain disease progression and treatment goals. Here we are interested in understanding the illness from a patient’s perspective. Describe how the disease impacts patients’ and caregivers’ day-to-day life and quality of life. Are there any aspects of the illness that are more important to control than others?
The three patients interviewed were diagnosed with asthma in childhood. Patient A was diagnosed at 3 years old. She reported that having asthma has been very limiting. In childhood, she was not able to attend gym class or socialize well with others because she had to pay attention to her surroundings and environmental triggers. Because of the prevalence of indoor smoking at that time and her sensitivity to it, she was frequently hospitalized due to exacerbations. She was also prone to respiratory infections, gastric issues and frequent anaphylaxis. The effects of asthma and side effects of some medications impacted her ability to pursue a career or have a family.
Patient B reports that asthma has been limiting throughout her life. In childhood she frequently missed classes due to frequent exacerbations. The farm she grew up on was a triggering environment for her due to the animals and she had to relocate to the city with her family. She reported that she has not able to travel long distances or by air because she worries she will have a flare up in transit. In her adult life, she still feels the impacts of asthma. She cannot make spontaneous plans to visit friends or family without assessing the environment for triggers ahead of time.
Patient C discussed the limitations caused by requiring oxygen supplementation. Her ability to mobilize has been greatly impacted. The has negatively affected her quality of life.
Experiences With Currently Available Treatments
The patients interviewed were treated with prednisone, fluticasone, salmeterol, Xolair, montelukast, budesonide, tiotropium bromide, bilastine, breo ellipta and Alvesco.
All the patients interviewed reported that medications have helped to control the number of exacerbations and hospitalizations related to asthma. They have benefitted greatly from the addition of biologics to their asthma treatment. They still require oral corticosteroids intermittently to manage flare-ups.
The side effects reported from the medications were heart palpitations, poor sleep, oral thrush, adrenal insufficiency, adrenal crisis, systemic fungal infection, hair loss, skin changes, osteoporosis, weight loss, urticaria and jaundice.
Most of the medications used were easily accessible through the pharmacy. Challenges were experienced while accessing specialty drugs through the Exceptional Access Program.
Patient B reported that she was not well informed about the reimbursement process and was not able to facilitate or follow up on drug coverage issues which led to some interruptions in treatment. The patients were also required to go to an infusion clinic twice a month which was concerning during the pandemic. The risk of exposure was a stressor especially since asthma increased their risk of COVID- 19 related illness.
Improved Outcomes
Key treatment outcomes for the patients interviewed include reducing exacerbations, reducing hospitalizations and improving quality of life. Patients are interested in medications that are effective in treating asthma so that they don’t need to take several medications for the same condition. Unmet patient needs persist in severe asthma.
The reimbursement process and criteria is another concern for patients. Patients want medications to be easily accessible so their healthcare providers have more flexibility when prescribing, allowing patients to have more options.
Side effects are a great source of distress for patients. Patient C reported that as a result of oral corticosteroid use, she has suffered from adrenal insufficiency, systemic fungal infections and liver damage. Patients would like treatments with minimal side effects that don’t impact their health and improve quality of life.
Experience With Drug Under Review
No patients within this evidence group submission had experience with the medication under review.
Companion Diagnostic Test
Not applicable.
Anything Else?
Is there anything else specifically related to this drug review that CADTH reviewers or the expert committee should know? Not applicable
Conflict of Interest Declaration — Lung Health Foundation / Ontario Lung Association
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
No.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
Table 1: Conflict of Interest Declaration for the Lung Health Foundation / Ontario Lung Association
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
AstraZeneca | — | — | — | X |
Asthma Canada
About Asthma Canada
Asthma Canada is the only national health charity solely dedicated to providing evidence-based, education, management tools and support programs for Canadians living with asthma. We also advocate to improve the quality of life for people living with asthma and invest and support strategic research to ultimately find a cure. For nearly 50 years, Asthma Canada has proudly served as the national voice for Canadians living with asthma. We empower patients with evidence-based information, education programs and support asthma research in Canada.
Asthma Canada is a registered charitable organization (BIN 89853-7048-RR0001). Our vision is a world without asthma, and our mission is to help Canadians with asthma lead healthy lives through education, advocacy and research.
Based in Toronto, Ontario, we operate under the direction of a volunteer Board of Directors and provide programs and services to people living with asthma and their caregivers through our website, e-newsletters, social media channels, and the Asthma & Allergy HelpLine.
Asthma Canada provides our services freely for all Canadians - coast to coast, via phone, email, social media, print resources and online. In addition, the Asthma Canada Member Alliance (ACMA) is the community arm and voice of Asthma Canada made up of people living with asthma, parents/caregivers, healthcare professionals, and anyone who has been affected by asthma. ACMA has more than 8,000 people living with asthma and allergies, caregivers, healthcare providers, and other interested participants from all regions of Canada.
Information Gathering
The majority of the patient perspective in this submission were pulled from:
An online survey, independently developed and launched in 2022 to seek the perspectives of people living with asthma, including caregivers. The survey was open from February 28th and closed on March 10th, 2022. Over 100 people responded to the survey and participants were from British Columbia (25%), Alberta (11%), Saskatchewan (2%), Manitoba (5%), Ontario (51%), Quebec (3%) and the Atlantic provinces (2%).
A Snapshot of Asthma in Canada: 2021 Annual Asthma Survey Report. The 2021 survey was launched online and shared with our Asthma Canada Member Alliance (ACMA) on May 3rd, 2021. Data collection period lasted for nine weeks, closing on July 4, 2021. We received a total of 256 responses with a 68% (174) completion rate. Criteria for participation was to be living in Canada and have asthma, or be a parent or caregiver to someone with asthma. Nearly half (48%) of the respondents indicated that they had moderate asthma. The percentage of respondents indicating mild and Severe Asthma was 25% and 27% respectively.
Lastly, a study conducted by Asthma Canada (formerly the Asthma Society of Canada) in 2014, entitled “Severe Asthma: The Canadian Patient Journey”. This study included Canadian adults 18 years or older who live with Severe Asthma as defined by their symptoms, their level of asthma control, and a review of their clinical profiles by a team of expert advisors. A total of 24 patients participated in in-depth personal interviews about their condition and its impact on their personal, social, medical, and economic circumstances. A complimentary online quantitative survey of 200 individuals with Severe Asthma accompanied the data from the interviews to validate and quantitate the in-person findings. Further details of the study population, investigators, and process are available on pages 28 to 31 of the full report.
Disease Experience
Asthma is a chronic lung disease which restricts the airflow into the lungs, making it difficult for nearly 3.8 million Canadians to breathe. It is the third most common chronic disease in Canada. Asthma symptoms are triggered by many environmental factors that cause symptoms such as shortness of breath, chest tightness, wheezing and coughing which constrict the airways (bronchial tubes). Asthma affects people at any age and can be intermittent to severe. When diagnosed in childhood, the condition can continue into adulthood and becomes a lifelong disease. Patients aim to control asthma by identifying environmental triggers and finding the right combination of pharmacological treatment(s) to reduce or eliminate asthma exacerbations (known as “asthma attacks”) with the support of a family physician or specialist, such as a respirologist, immunologist or allergist. The pharmacological treatments used to treat asthma depend on the type of asthma, severity of the condition, and how the patient responds to various treatment options. What works for one person with asthma changes over time and means that many treatment options are needed to manage their health successfully. It is estimated that nearly 300 Canadians are diagnosed with asthma every day, and roughly 250 Canadians die from an asthma attack each year.
“Because people don't "see" it as a debilitating disease they just assume when you are having an Asthma attack that you have a cold or something. There’s such a lack of knowledge on how serious Asthma is.”
The most recent international guidelines from the Global Initiative for Asthma (GINA) recommend classifying patients’ asthma as severe if still uncontrolled despite optimal use of long-term controller and short-acting reliever medications (Holguin, 2020). In addition, the recommendations for asthma management were updated by the Canadian Thoracic Society (CTS) in 2021, as asthma being uncontrolled if the patient ‘yes’ to any of the following:
Do you have daytime symptoms (cough, wheeze, shortness of breath and/or chest tightness) more than 2 days per week? Do you require your reliever medication more than 2 times per week? Do you have mild nighttime symptoms more than once a week? Do you have any physical activity limitation? Do you miss any work or school due to your asthma?
"Some of the simplest tasks are difficult, playing outside with the kids and running around. Well not so much running. Stairs."
Severe Asthma has many different effects and consequences that can impair patients’ quality of life. Asthma can negatively impact the psychological and social-well-being of those living with asthma, resulting in avoidance and lack of participation in physical activity. In A Snapshot of Asthma in Canada: 2021 Annual Asthma Survey Report, we found:
The majority (79%) of people living with asthma reported having some form of health-related anxiety and 74% reported feeling emotionally stressed due to their asthma.
More than half (62%) of respondents felt that living with asthma interfered with the quality of their social interactions.
The majority (71%) of respondents indicated that in the past 12 months their asthma symptoms prevented them from participating in outdoor and/or physical activities. This was seen in 81% of respondents with Severe Asthma, 76% with moderate asthma and 49% with mild asthma.
"I am not me anymore. I need so many meds."
It has been shown that people with Severe Asthma can have poor quality of life that impacts their ability to complete daily activities and participate in outdoor and/or physical activities. They may also experience a loss in productivity at work or school and can be limited in their leisure and lifestyle. One major limitation is not being able to participate in sports and vigorous activities and is more common in individuals with uncontrolled asthma.
“The symptoms are always there. Never goes away completely. It has a negative impact on my life.”
Asthma is the leading cause of absenteeism (time taken off/missed) from school and one of the leading causes of work loss through both absenteeism and presenteeism (loss in productivity). Absenteeism from school is more prevalent among low-income and racial/ethnic minorities. Differences associated with asthma control, such as access and quality of health care, adherence to medication use, and social factors such as psychosocial stressors also contribute to absenteeism.
"Employers do not want to hire me when I reveal my condition. Very upsetting. Hard to find work and live a productive life."
In the past 12-months, (43%) of respondents missed work or school because of their asthma symptoms, of which 67% had Severe Asthma. The loss of productivity at school or work due to being sick or feeling unwell in individuals with asthma, can lead to a decrease in performance or quality of work or schoolwork. Presenteeism is often overlooked and it has been reported that productivity loss in work and school can be avoided by achieving and maintaining control of asthma, which can sometimes be difficult in individuals with Severe Asthma.
"My partner is in many ways my caregiver - and their entire life has changed. They have so many more stresses on them than before. We were together before I got asthma, and their role has entirely changed. It's incredibly stressful for them."
Patients and caregivers have significant daily responsibilities to self-manage asthma symptoms with the goal of ensuring they can fully participate in day-to-day activities, like work and activities. When well-controlled, there can be periods of stability however exacerbations or asthma attacks can occur due to environmental triggers like pollution, smoking, and allergies. Asthma attacks can also be triggered by viral infection and uncontrolled disease.
Patients and caregivers are often concerned with accessing adequate and necessary medical care within a short period of time, as exacerbations can lead to urgent trips to the Emergency Department (ED’s) to address and restore airway function. In severe asthma attacks, loss of consciousness or hypoxia can occur. Visits to the Emergency Department can be stressful as parents and caregivers navigate busy and overcrowded Emergency Departments, particularly during the COVID-19 pandemic. Access to specialists with knowledge of asthma continues to be a challenge. People with asthma and caregivers that responded to our survey noted the worry and fear of an asthma attack was the most concerning (60%) followed by potential for hospital visits / admissions (47%) and missed work and school days (47%).
"My partner's asthma makes it impossible for her to go out, which is detrimental to her mental health. We cannot live in the same place because of our pets, and we cannot share a sleeping space due to nocturnal asthma making her a very light sleeper. I want her to be able to enjoy a higher quality of life and participate in activities and relationships that make her fulfilled."
Asthma symptoms impact both the patient and family’s quality of life. Patients may experience fatigue and have less energy to work and exercise. Making and keeping friends and colleagues can be made more difficult due to the symptoms of the disease, the presence of environmental triggers, or activity limitations. School and work are important parts of everyone’s live however patients may not be able to attend and concentrate due to disease symptoms, fatigue, and exacerbations. Sleep can be disturbed and patients and caregivers are often called on to deal with symptoms in the night (38% of survey participants noted sleep as a concern). Patients and caregivers are faced with barriers in understanding the seriousness of asthma. They spend a significant amount of time educating their friends, schools, workplaces, and others about the seriousness of asthma.
"It has impacted every single part of our lives as we have had to factor in her asthma into every situation. It’s also hard because most people don't understand that severe asthma is so different. I constantly hear "oh my kid had asthma too, but they outgrew it" - they don’t understand that it’s an entirely different situation for us."
The impact of asthma on patients and caregivers means missing work when suffering from an asthma attack or having to attend to medical appointments. Patients and caregivers are affected due to stress and concerns of current and future health perspectives and anxiety often is connected with asthma as a co-morbidity. Medications need to always be kept on hand in case of emergency. Managing an asthma attack can cause panic in the patient and caregivers due to the life-threatening nature of asthma attacks. There can be added stress due to financial hardships with paying for current treatments which can be expensive and strain a family’s finances.
Experiences With Currently Available Treatments
In current Canadian practice, the cornerstones of asthma management are:
Identification and avoidance of triggers that worsen symptoms or cause exacerbations;
Long-term controller medication(s) taken on an ongoing basis to reduce inflammation and reactivity in the airways. The most common controllers are inhaled corticosteroids (ICS) delivered through a puffer. Potential additional medications include leukotriene receptor antagonists and long-acting bronchodilators. For patients requiring a higher corticosteroid dose than an inhaler can provide, oral corticosteroids (OCS) are availed of by patients in healthcare settings; this is particularly common in patients with severe asthma;
A short-acting reliever (bronchodilator) taken through a puffer to provide rapid relief of exacerbations or severe symptoms.
While these measures are adequate to control asthma symptoms and exacerbations in people with mild asthma, a person whose asthma is severe may still experience symptoms that can drastically reduce their quality of life and lead to systemic dependence of oral corticosteroids (OCS). The definition of Severe Asthma, therefore, carries within it an unmet need for treatment options that go beyond the existing standard of care.
"Despite the Xolair, Breo Ellipta, Singulair, Nasonex, Alvesco, OTC allergy meds, etc. she [wife] is still needing frequent rounds of prednisone and/or Ventolin and Atrovent. She needs multiple daily doses of hydrocortisone due to a lifetime of heavy steroid use."
1 in 4 people who completed our 2022 online survey indicated they have poor symptom control even with currently available treatments. Many people with asthma have challenges in accessing the needed health providers, like respirologists and specialized asthma clinics, to manage their health. There can be significant time and burden involved to manage care with health care providers made worse with poor asthma control. There can be travel involved for those living in rural areas to centres providing the health care need. This means patents and caregivers may miss school and/or work. Some patients and caregivers may not have paid leave to attend these appointments or may need to use vacation leave, if available.
"It is extremely expensive and is preventing my husband from being able to switch jobs because we cannot afford to pay out of pocket if we lose his insurance. So he is stuck in a job that overworks their employees because of me."
Financial considerations are another critical barrier to optimal asthma medication use. In Asthma Canada’s 2021 survey, about one- third of patients reported that they had skipped filling a prescription for an asthma medication because they were unable to afford it. Many private insurers do not provide complete coverage for asthma medications, placing a significant portion of the burden on patients. Since many patients with Severe Asthma have lower incomes (more than one-third of survey respondents had household incomes under $50,000) or are unable to work because of their asthma, even having to pay a small percentage of the drug cost can be a significant financial concern (Severe Asthma, 2014).
"It has been suggested to me that I might do better on a biologic, but my extended health care plan and income would not cover the cost. My current drugs I can afford; although they’re expensive, I have an extended health care plan."
The use of oral corticosteroids in patients who fail to achieve adequate asthma control with inhaled corticosteroids (ICS) deserves special mention due to the short- and long-term side effects of the systemic use of oral corticosteroids. This issue is of particular concern to the population of patients with Severe Asthma, where many patients depend on long-term oral corticosteroids to provide some degree of inflammation control after other options prove to be inadequate. Although potentially helpful in the short-term, these medications have a long list of side effects if taken for longer periods of time and at higher doses. Side effects include weight gain, acne, excess facial hair, mood swings, high blood pressure, hyperactivity, high blood sugar, increased infection. In the long term, oral corticosteroids can cause osteopenia, osteoporosis, glaucoma, cataracts, and heart disease. (Refer to: Appropriate Use of Oral Corticosteroids in Asthma).
"I was on a prednisone treatment once as my asthma and a cold developed into a serious chest infection. Prednisone is harsh. I became extremely moody and angry when taking it. I hope ever to need it again. It's not a drug that I can tolerate."
Immunomodulator medications, or biologics, are also available to treat severe asthma. Some biologics are for eosinophilic asthma or for those that are dependent on oral corticosteroids. Some are now available in a self-injectable form (e.g., autoinjector pen). Side effects include allergic reactions, injection site reactions, and infections. Self-injection provides flexibility and reduces the time involved in taking medicine. However, patients and caregivers can feel additional stress in learning how to give injections as proper training is needed before initiating therapy.
"People with an autoimmune disease generally have more than one autoimmune disease. I have many. And I need many medications and Symbicort is VERY EXPENSIVE. I cannot afford my medications and have not been able to pay other bills like hydro in order to pay for my medications. Thankfully I was recently approved for ODSP so they pay for my prescription meds, but I have a lot of non-prescription meds that are not covered and I still can’t afford them but I need them to stay alive."
Improved Outcomes
Three major themes emerged from the data collected from our patient evidence submission survey in 2022.
First, the primary concern for people living with asthma and their caregivers was the ability to control their day-to-day symptoms. More than half of people with Severe Asthma do not regularly take their controller medication, leading to the possibility of an unnecessary increase in healthcare system usage. Parents and caregivers reported that current treatments can be difficult to take highlighting that the mode of administration and frequency of dosing is important. About one in four survey participants indicated that they need too many daily doses. Patients must be able to more easily control their Severe Asthma to live healthy and active lives.
"I would like to be more active with my kids. Right now, I have asthma symptoms from laughing. Not very much fun!"
The second most crucial factor for patients with Severe Asthma was the ability to control exacerbations. Asthma exacerbations put patients at risk of airway distress and hospitalization; therefore, the proper combination of appropriate medications is critical to managing Severe Asthma correctly.
“Not knowing what each day will bring. Not being able to perform my day to day activities. I am not able to walk my dog if it is too cold, too hot, too windy, or too humid. And with climate change, there are more bad asthma days than good ones. I am finding it more and more difficult to go outside, and I am an outdoor person by nature. I am not able to do the things that I enjoy; gardening, nature walks, picnics, dog park, etc. I feel trapped in my own home where I can control the temperature and humidity.”
Third, people living with asthma cited the cost of medications as a vital factor in receiving proper asthma treatment. Without coverage for current and upcoming treatments, people living with asthma and their caregivers may have to reduce their medication usage or stop taking them altogether. This lack of coverage may add to the staggering statistics of more than 1.6 million Canadians unable to fill a prescription due to cost. In addition to the cost of more widely-available medications, specialized and novel medication therapies (such as the ones under review for this patient evidence submission) play a critical role in reducing the cost of healthcare utilization and help improve the lives of over 250,00 Canadians living with Severe Asthma.
“Getting older I find it harder on a day to day to do most things. And still not being able to afford the treatment I need. I’ve had 37 years of asthma and there hasn’t been 1 in the 37 years that I haven’t had to stress or worry about breathing and affording medications. To have that stress lifted would be great.”
Survey participants indicated their expectations for a new medication and ranked these expectations in the following order:
Increase in lung function (73%)
Easier management of asthma symptoms (61%)
Reduction in asthma exacerbations or asthma attacks (56%)
Reduced reliance on oral corticosteroids (56%)
Broadly speaking, parents and caregivers expect to see improvements in a range of day-to-day activities affecting qualify of life, for example:
Improved attendance at school and/or work
Improved sleep
More energy
Less time off work for patients and caregivers
Participation in play, physical and social activities
Less health care visits, such as ED visits
Less anxiety and panic due to asthma attacks
Less financial hardships
A variety of side effects of inhalers are difficult to manage such as elevated heart rate, anxiousness, and thrush. Minimizing these side effects are important outcomes that should be considered when evaluating new therapies. Survey participants noted that dry throat (50%), difficulty sleeping (42%), increased heart rate (38%), headaches (36%), hoarseness (35%) and weight gain (34%) were the most bothersome symptoms. Oral corticosteroids cause a long list of side effects and impact on a child’s growth and reducing or eliminating oral corticosteroids is also important to patients.
Over half of survey participants indicated that the benefits of the new treatment are worth tolerating the potential side effects to improve management of asthma.
"All I wanna do is be able to breathe. I am so frustrated that I can’t do things like take long walks, I can’t run, or go outside when it’s cold. It will bring on an asthma attack."
Experience With Drug Under Review
Our survey showed no one with experience with the drug under review at this time. Though 65% of respondents in our survey said they'd tolerate the potential side effects of tezepelumab treatment to see an improvement in the management of their asthma. Most patients will have tried and used many other treatments before using tezepelumab. The addition of a new biologic for Severe Asthma provides more options so they can tailor treatments to their needs. Patients and caregivers value a reduction in other medications, such as oral corticosteroids and inhalers and less medication side effects. The asthma community would also value improvements in quality of life, like participation in work and school. A variety of outcomes are also valued as described in Section 5 – Improved Outcomes.
"If it worked for me, it would offer peace of mind. I’m 66 and know I have damaged lungs following years of pneumonia and bronchitis."
"It will depend on the cost of the treatment. Xolair already adds such a huge cost that we are held hostage by needing to keep her [daughter] on extended medical even though it doesn't align with her chosen career path."
Companion Diagnostic Test
Asthma Canada is not aware of any current companion diagnostic test for the drug under review beyond standard asthma diagnostics. The diagnosis of asthma happens through spirometry or the methacholine test.
Anything Else?
While asthma cannot be cured, it can be managed by using appropriate medications. Using prescribed medications reduces exacerbations, prevents hospital admissions and deaths. It allows people to work, attend school and live productive, symptom-free lives. The ability for those living with asthma to access and afford new and innovative drugs in Canada is essential to our community’s wellbeing. It can be the difference between living an active, productive life and not being able to function or even breathe. Close to one- third (30%) of respondents indicated that their current drug coverage is not sufficient to help them keep their asthma symptoms under control. The addition of a new biologic for severe asthma provides another treatment option so they can tailor treatments to their needs. Patients and caregivers value a reduction in other medications, such as oral corticosteroids and inhalers and less medication side effects.
“[My daughter’s] current maintenance medication list includes 4 prescription meds for asthma and allergies: Xolair, Breo Ellipta, Singulair and Nasonex, another 2 for side-effects, Ciprolex and Losec, as well as 2 OTC medications for allergies and weight control.
All of them are for daily use except for the Xolair. This doesn’t even include her Ventolin, Atrovent and Prednisone for treating exacerbations. The cost of these medications is close to $20,000 a year. We are extremely fortunate that my husband’s work includes excellent coverage for prescription medications, but we are aware that there is a time limit on this coverage. Her life choices are limited to staying in school to continue this coverage, and we know she will need to move immediately into a job with a good extended health plan when she’s done.”
Conflict of Interest Declaration — Lung Health Foundation / Ontario Lung Association
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
No.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
A a patient who lives with asthma, diagnosed in childhood and also has a family history of severe asthma helped review the survey respondent’s data.
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
Table 2: Financial Disclosures for Asthma Canada
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AstraZeneca | — | — | — | X |
Sanofi Genzyme | — | — | — | X |
GSK | — | — | — | X |
Novartis | — | — | X | — |
Sanofi Pasteur | — | — | X | — |
Pfizer | — | — | — | X |
Clinician Input
AllerGen Clinical Investigator Collaborative
About AllerGen Clinical Investigator Collaborative
The AllerGen Clinical Investigator Collaborative (CIC) is a group of clinical investigator sites, initially funded by the AllerGen Network of Centres of Excellence to investigate potential new therapies for the management of asthma.
The six sites involved in the collaborative are McMaster University (Dr. Gail Gauvreau, Dr. Paul O’Byrne); University de Laval (Dr. Louis Philippe Boulet); University of Saskatchewan (Dr. Donald Cockcroft, Dr. Beth Davis); University of Calgary (Dr. Richard Leigh); University of Alberta (Dr. Irv Mayers) and University of British Columbia (Dr. Celine Bergeron). This consortium has existed for more than 15 years and has completed more than 40 studies of new molecules being considered for the management of asthma. The consortium utilizes well developed methodologies that are robust and reliable, and which have been widely accepted as being predictive for potentially new therapies for asthma. There is no other consortium of this sort to investigate new therapies for asthma worldwide. It was this group that originally identified the potential for blocking thymic stromal lymphopoietin (TSLP) as a potential therapy for asthma.
The clinical model utilized by the Allergen CIC enrolls mild asthmatic patients sensitized to environmental allergens who are subsequently challenged with these allergens by inhalation and who have been shown to develop allergen- induced bronchoconstriction and airway inflammation. The initial study of a drug which blocks TSLP was with a monoclonal antibody (AMG157), which demonstrated attenuation all the allergen-induced responses in mild asthmatic subjects. In addition, and quite unexpectedly, blocking TSLP with this antibody also improved all the inflammatory biomarkers even prior to the allergen challenge. This strongly suggested that this molecule would be effective in patients with asthma, and it was subsequently extensively studied in patients with severe asthma as a therapy now known as Tezepelumab.
Information Gathering
The information provided in this letter has been agreed upon by all the investigators involved in the AllerGen CIC.
Current Treatments and Treatment Goals
Severe asthma is confined to between 5 and 8% of the asthmatic population and is considered to be those patients already on optimal treatments, including high doses of inhaled combination therapy (inhaled corticosteroids and long- acting β2 agonists), but asthma remains uncontrolled, and patients are still at risk of having severe asthma exacerbations. Many of these patients require intermittent or even maintenance oral corticosteroids to attempt to manage their asthma. While the mechanisms of severe asthma remain poorly understood, it has been now accepted that there are two reasonably distinct phenotypes of patients with severe asthma. These are patients who have severe eosinophilic asthma with a number of biomarkers increased which would suggest important engagement of T helper 2 (Th2) cells and innate lymphoid cells type 2 (ILC2). These patients usually have elevated blood and airway eosinophil counts and these are patients for whom there are already a number of biologic therapies available which will be discussed below. The other approximately 40 to 50% of patients do not have a consistent elevation in blood or sputum eosinophil counts, often have increased airway neutrophils and these as a group are less responsive to inhaled and/or oral corticosteroids. There are no approved treatment options other than conventional asthma therapies for this patient population in Canada.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
As mentioned above, there are treatments which are effective for severe asthma patients identified to be severe eosinophilic asthma (T2 high) and these are monoclonal antibodies directed at inhibiting interleukin 5 (IL5) either the ligand (Mepolizumab or Reslizumab) or the IL5 receptor alpha (Benralizumab) or inhibiting the IL4 receptor alpha (Dupilumab). In addition, there is an antibody which binds IgE (Omalizumab) which has been approved for treatment of severe allergic asthma many of whom have concurrent severe eosinophilic asthma. All these therapies have been approved in Canada, predominantly on their ability to reduce severe asthma exacerbation risk with an average of approximately 50% risk reduction for these treatments. There have been less consistent findings in improving asthma control or lung function in large clinical trials with these treatment approaches.
The main treatment gap exists for those patients who have severe asthma but cannot be demonstrated to have persisting T2 high asthma. Various therapeutic interventions have been attempted in this patient population without success and indeed the only treatment that has been shown in clinical trials to reduce asthma exacerbation risk is Azithromycin administered three times weekly. This approach is not approved by Health Canada.
Place in Therapy
The use of Tezepelumab in asthma will be restricted to patients with severe asthma. However, in contrast to the other available biologic therapies, this medication can be used in those with both T2 high (severe eosinophilic asthma) or T2 low asthma. The risk reduction for severe exacerbations in T2 high patients is at least as good or better than clinical trials with other therapies (although no direct comparison has been made between biologics and asthma).
Tezepelumab has also been consistently shown to improve both lung function and asthma control. In T2 low patients, the risk reduction for severe exacerbations is in the range of 45%, where other biologics have not shown any significant effect in this population. This is the first therapeutic approach to be considered for approval for this specific patient population.
The main outcome variable that would be monitored in patients being treated with Tezepelumab is severe asthma exacerbation risk. Severe exacerbation risk remains the single most important outcome to improve in severe asthma. These events are potentially life threatening and have massive impact on the patient’s life, functioning as well as on their family. Tezepelumab will, in addition, improve lung function to a clinically significant degree and also improve all of the other measurements considered to be important in asthma control. Tezepelumab has also been shown to reduce all of the biomarkers that are regularly measured in severe asthma, including blood eosinophil counts, sputum eosinophil counts, and exhaled nitric oxide levels.
Tezepelumab should be considered for discontinuation if the patient continues to have severe exacerbations while being treated. Other indications for discontinuation would be side effects. None were identified to be increased above placebo during the clinical trial phase of drug development, but clearly any type of allergic reaction to the administration of Tezepelumab would be one obvious reason for immediate discontinuation.
Severe asthma is generally managed in specialty practice in Canada and the expectation would be that in general the initiation of this biological therapy and decision to continue or discontinue its treatment would be made by an expert in managing severe asthma.
In summary, we believe that Tezepelumab is a treatment advance for the management of patients with severe asthma. It is very effective in all types of severe asthma, including demonstrated efficacy in a patient population with T2 low asthma for which there has not been an available treatment option in Canada. The biologic appears to be safe in the clinical trials to date and we are looking forward to its approval and availability for use in the severe asthma patient population.
Conflict of Interest Declarations — AllerGen Clinical Investigator Collaborative
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
No outside assistance was received
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No outside assistance was received.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician who contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
Declaration for Clinician 1
Name: Paul M O’Byrne
Position: Dean and Vice President, Faculty of Health Sciences, McMaster University
Date: April 20, 2022
Table 3: Conflict of Interest Declaration for AllerGen Clinical Investigator Collaborative — Clinician 1
Company | Check appropriate dollar range | |||
|---|---|---|---|---|
$0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 | |
Astra Zeneca | — | — | X (personal) | X (grant in aid) |
Amgen | — | X (personal) | — | — |
Biohaven | — | — | — | X (grant in aid) |
Covis | X (personal) | — | — | — |
Chiesi | X (personal) | — | — | — |
Cipla | X (personal) | — | — | — |
GSK | — | X (personal) | — | X (grant in aid) |
Merck | — | — | — | X (grant in aid) |
Teva | X (personal) | — | — | — |
Declaration for Clinician 2
Name: Richard Leigh
Position: Senior Associate Dean – Faculty Affairs, Cumming School of Medicine, University of Calgary
Date: April 20, 2022
Table 4: Conflict of Interest Declaration for AllerGen Clinical Investigator Collaborative — Clinician 2
Company | Check appropriate dollar range | |||
|---|---|---|---|---|
$0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 | |
Astra Zeneca | — | X (personal) | — | X (grant in aid) |
Biohaven | — | — | — | X (grant in aid) |
GSK | — | X (personal) | — | X (grant in aid) |
Novartis | — | — | — | X (grant in aid) |
Oncovir | — | — | — | X (grant in aid) |
Sanofi | — | X (personal) | — | X (grant in aid) |
Valeo | X (personal) | — | — | — |
Declaration for Clinician 3
Name: Donald W. Cockcroft
Position: Professor University of Saskatchewan
Date: April 20, 2022
Table 5: Conflict of Interest Declaration for AllerGen Clinical Investigator Collaborative — Clinician 3
Company | Check appropriate dollar range | |||
|---|---|---|---|---|
$0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 | |
Astra Zeneca | — | — | — | X (grant in aid) |
Biohaven | — | — | X (grant in aid) | — |
Declaration for Clinician 4
Name: Irv Mayers
Position: Professor, University of Alberta
Date: April 22, 2022
Table 6: Conflict of Interest Declaration for AllerGen Clinical Investigator Collaborative — Clinician 4
Company | Check appropriate dollar range | |||
|---|---|---|---|---|
$0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 | |
AstraZeneca | — | X (personal) | — | X (grant in aid) |
Boehringer | — | X (personal) | — | — |
Sanofi | — | X (personal) | — | — |
Declaration for Clinician 5
Name: Louis-Phillippe Boulet
Position: Respirologist, IUCPQ, Quebec and Professor of Medicine, Laval University, Quebec
Date: April 19, 2022
Table 7: Conflict of Interest Declaration for AllerGen Clinical Investigator Collaborative — Clinician 5
Company | Check appropriate dollar range | |||
|---|---|---|---|---|
$0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 | |
AstraZeneca | — | X (personal) | — | X (grant in aid) |
GSK | X (personal) | — | — | X (grant in aid) |
Merck | — | X (personal) | — | X (grant in aid) |
Novartis | X (personal) | — | — | — |
Sanofi Regeneron | X (personal) | — | — | X (grant in aid) |
BioHaven | — | — | — | X (grant in aid) |
Covis | X (personal) | — | — | — |
Teva | X (personal) | — | — | — |
Cipla | X (personal) | — | — | — |
Declaration for Clinician 6
Name: Celine Bergeron
Position: Respiratory Physician, Vancouver General Hospital and Clinical Associate Professor, University of British Columbia
Date: April 19, 2022
Table 8: Conflict of Interest Declaration for AllerGen Clinical Investigator Collaborative — Clinician 6
Company | Check appropriate dollar range | |||
|---|---|---|---|---|
$0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In Excess of $50,000 | |
Grifols | X (personal) | — | — | — |
Astra-Zenaca | X (personal) | — | — | — |
Sanofi | X (personal) | — | — | — |
Valeo | X (personal) | — | — | — |
GSK | X (personal) | — | — | — |
Takeda | X (personal) | — | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.