CADTH Reimbursement Review
Upadacitinib (Rinvoq)
Sponsor: AbbVie
Therapeutic area: Ulcerative colitis
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
AE
adverse event
5-ASA
5-aminosalicylic acid
bio-IR
inadequate response to biologic therapy
CI
confidence interval
CrI
credible interval
CRP
C-reactive protein
EMS2
endoscopic Mayo subscore of at least 2
EQ-5D-5L
5-level EQ-5D
EQ VAS
EuroQoL Visual Analogue Scale
FACIT-F
Functional Assessment of Chronic Illness Therapy–Fatigue
FE
fixed-effects unadjusted
FEA
fixed-effects adjusted
FMS6/12
full Mayo score of 6 to 12
HRQoL
health-related quality of life
IBD
inflammatory bowel disease
IBDQ
Inflammatory Bowel Disease Questionnaire
IL
interleukin
ITT
intention to treat
ITT1
intention-to-treat population for part 1
ITT2
intention-to-treat population for part 2
ITC
indirect treatment comparison
JAK
Janus kinase
LOCF
last observation carried forward
nonbio-IR
inadequate response to conventional therapy
NMA
network meta-analysis
NRI
nonresponder imputation
NRI-C
NRI incorporating multiple imputation to handle missing data related to COVID-19
NRI-NC
NRI with no special data handling for missing due to COVID-19
OR
odds ratio
PGA
Physician’s Global Assessment
RBS
rectal bleeding subscore
RCT
randomized controlled trial
RE
random-effects unadjusted
REA
random-effects adjusted
RR
re-randomized (study design)
S1P
sphingosine 1-phosphate
SAE
serious adverse event
SD
standard deviation
SF-36
Short Form (36) Health Survey
SFS
stool frequency subscore
TNF
tumour necrosis factor
TT
treat-through (study design)
UC
ulcerative colitis
UC-SQ
Ulcerative Colitis Symptoms Questionnaire
VAS
visual analogue scale
VTE
venous thromboembolic event
WDAE
withdrawal due to adverse event
WPAI
Work Productivity and Activity Impairment
WPAI-UC
Work Productivity and Activity Impairment–Ulcerative Colitis
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Upadacitinib (Rinvoq) 15 mg, 30 mg, and 45 mg extended-released tablets, oral administration |
Indication | For the treatment of adult patients with moderately to severely active ulcerative colitis whose condition has not responded to treatment, i.e., have had an inadequate response, loss of response, or intolerance to at least 1 of the conventional and/or biologic therapies |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | July 21, 2023 |
Sponsor | AbbVie Corporation |
NOC = Notice of Compliance.
Introduction
Ulcerative colitis (UC) is the most common form of inflammatory bowel disease (IBD). Depending on the extent and severity of the disease, patients with UC may present with diarrhea with or without blood and mucus, urgency or tenesmus, incontinence, constipation, colicky abdominal pain, fever, malaise, and weight loss.1,2 Regardless of severity, UC is also associated with high rates of fatigue and sleep difficulties.3 The disease has negative physical, emotional, and social impacts on patients. Aggressive disease course is experienced in 10% to 15% of patients. It was estimated that more than 120,000 people in Canada lived with UC in 2018.4
The selection of treatment regimens for UC is guided by disease severity and extent.5 While different drug classes are available for the long-term management of moderately to severely active UC, biologic therapies are the mainstay of treatment for patients with moderate to severe UC and are used for induction and maintenance when other treatments have been unsuccessful, or in those who cannot tolerate other treatments. At present, biologics include tumour necrosis factor (TNF) alpha antagonists (adalimumab, infliximab, and golimumab), anti-integrin drugs (vedolizumab), and interleukin 12 (IL-12) and interleukin 23 (IL-23) antagonists (ustekinumab). Small-molecule drugs, which include Janus kinase (JAK) inhibitors (tofacitinib) and the sphingosine 1-phosphate (S1P) receptor agonist ozanimod, are also used in patients with moderate to severe UC. Despite access to a variety of treatment options, not all patients respond to the available treatments and their condition may become refractory to the current treatment regimens.
Upadacitinib is a selective JAK1 inhibitor approved by Health Canada for the treatment of adult patients with moderately to severely active UC who have demonstrated treatment failure, i.e., an inadequate response, loss of response, or intolerance to at least 1 conventional and/or biologic therapy. Upadacitinib is available as 15 mg, 30 mg, and 45 mg oral extended-release tablets. The recommended oral induction dose of upadacitinib is 45 mg once daily for 8 weeks. The recommended oral dose of upadacitinib for maintenance treatment is 15 mg once daily; 30 mg once daily may be appropriate for some patients, such as those with refractory, severe, or extensive disease. For patients 65 years and older, the only recommended maintenance dose is 15 mg once daily.
The objective of this report is to perform a systematic review of the beneficial and harmful effects of upadacitinib (15 mg, 30 mg, and 45 mg) for the treatment of adult patients with moderately to severely active UC who have demonstrated treatment failure, i.e., an inadequate response, loss of response, or intolerance to at least 1 conventional and/or biologic therapy.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient groups that responded to CADTH’s call for patient input and from the clinical expert consulted by CADTH for the purpose of this review.
Patient Input
Two patient groups submitted input: the Gastrointestinal (GI) Society and Crohn and Colitis Canada. The GI Society is a national charity committed to research, advocacy, and educational activities for people with gastrointestinal and liver conditions that works closely with health care professionals and governments at all levels to improve care and treatment. Crohn and Colitis Canada is a national, volunteer-based health charity committed to finding cures for IBD and improving the lives of children and adults affected by these diseases through research, patient programs, fundraising, spreading information, advocacy, and awareness activities. The information provided in the GI Society submission was gathered through various questionnaires distributed among patients with IBD in 2015 (n = 423), 2018 (n = 432), 2020 (n = 724), and 2022 (ongoing), as well as one-to-one conversations with patients; a patient roundtable; recent phone, email, and social media interactions; and stories submitted by patients over time. Crohn and Colitis Canada compiled data from 2 online surveys (respondents included 354 patients with moderate to severe UC and 2 participants in the Rinvoq clinical trial) conducted earlier this year and 1 phone interview with a patient who participated in the Rinvoq clinical trial.
Patients with UC commonly experience symptoms such as diarrhea (fecal urgency and poor control of bowel function), rectal bleeding, and abdominal pain. Patients commonly described flares, which occur at unpredictable times, as causing extreme pain and fatigue with a need to always be near a bathroom. Symptoms may be present even during periods of remission. UC has a profound effect on patients’ physical, emotional, and social lives at their home, school, or workplace, and is particularly difficult for children and young adults, since it affects their sense of self. Based on the Crohn and Colitis Canada survey, the most frequently reported UC-related complications among patients were mental stress (65%), joint inflammation and arthritis (51%), fissure and hemorrhoids (40%), anemia (33%), skin condition (approximately 30%), and malnutrition and weight loss (approximately 30%); other complications include bowel obstruction, intestinal fistulas, abscesses, stricture, liver conditions, and cancer. Patients said their social lives (including romantic relationships with partners) had been negatively affected by their UC diagnosis and that they felt isolated due to others’ misunderstanding of their condition. About 72% of respondents said they had to constantly adjust their lifestyle and expectations due to UC; 2 in 5 patients said they changed their travel plans and 1 in 5 patients said they changed their career aspirations.
According to the GI Society submission, patients consider sustained remission and treatment response more important than relieving any 1 symptom. Despite the available treatment options, patients have difficulty obtaining remission or symptom relief and there is a need for more diversity in effective treatments that achieve mucosal healing, reduce symptoms, and allow patients to live full and productive lives. Patients want adequate access to medications that work to reduce preventable suffering, unnecessary use of health care resources, and financial burden on the government and taxpayers. Finally, the GI Society stated that having a treatment with oral administration rather than infusion or injection would be helpful for many patients.
Clinician Input
Input From Clinical Experts Consulted by CADTH
According to the clinical expert, even though various treatment options are available for patients with moderately to severely active UC, not all patients respond to them, and they may become refractory to current treatments. In addition, some of the current treatments are associated with many safety concerns. Some treatments have lower patient adherence due to the inconvenient route of administration.
The expert indicated that patients with moderately to severely active UC, either biologic-naive or biologic-exposed, are suitable for treatment with upadacitinib. The expert also stated that if patients could access upadacitinib without the need for their disease to have previously failed to respond to conventional therapies, immunomodulators, or previously available biologics, then access to upadacitinib would potentially cause a shift in the current treatment paradigm.
The expert noted that in clinical practice, clinical response and remission are assessed using the partial Mayo score or components of the Mayo score, along with certain biomarkers. Clinicians usually schedule a colonoscopy 6 to 9 months after starting treatment with biologics or small-molecule drugs to examine endoscopic healing.
The expert also stated that treatment with upadacitinib should be discontinued if there is a lack of clinical response to induction therapy or if there is disease progression.
Clinician Group Input
Two clinician groups submitted input on upadacitinib: 3 clinicians on behalf of the IBD Centre of BC, and 12 gastroenterologists and a nurse practitioner on behalf of the Atlantic Specialist Group, who submitted jointly with members from the University of Calgary IBD Unit.
The clinician group input was consistent with that of the clinical expert consulted by CADTH in terms of unmet needs, place in therapy, patient population, assessing response to treatment, discontinuing treatment, and prescribing conditions. The clinician groups emphasized that upadacitinib use should be restricted among patients with a history of thrombosis or coronary artery disease. In terms of the place in therapy of upadacitinib in clinical practice, both clinician groups agreed that upadacitinib would be used in various circumstances for patients, including as first-line therapy.
Drug Program Input
Input was obtained from the drug programs that participate in the CADTH reimbursement review process. The following were identified as key factors that could potentially impact the implementation of a CADTH recommendation for upadacitinib:
considerations for discontinuation of therapy
care provision issues
system and economic issues.
The clinical expert consulted by CADTH provided advice on the potential implementation issues raised by the drug programs.
Clinical Evidence
Pivotal Studies and Protocol Selected Studies
Description of Studies
Three phase III randomized controlled trials (RCTs) submitted by the sponsor (U-ACHIEVE Induction, N = 474; U-ACCOMPLISH, N = 522; U-ACHIEVE Maintenance, N = 1,046) were included in this systematic review. The objective of all 3 studies was to evaluate the efficacy and safety of upadacitinib in patients with moderately to severely active UC. The studies enrolled adult patients with a diagnosis of moderate to severe UC who had an inadequate response, loss of response, or were intolerant to either conventional therapy or biologic drugs. In the induction trials (U-ACHIEVE Induction and U-ACCOMPLISH), eligible patients were randomized to receive oral upadacitinib 45 mg once daily or matching placebo for 8 weeks in a double-blind manner. At the end of the 8 weeks, those who were deemed clinical responders were eligible to enter the maintenance study (U-ACHIEVE Maintenance), while nonresponders were given open-label upadacitinib for an additional 8 weeks. Clinical response was defined as a decrease from baseline in the adapted Mayo score of 2 points or greater and a decrease of 30% or greater from baseline, plus a decrease in rectal bleeding subscore (RBS) of 1 point or greater or an absolute RBS score of 1 or less. Patients who entered the maintenance study were re-randomized and treated with oral upadacitinib 15 mg or 30 mg once daily or matching placebo for up to 52 weeks. The primary efficacy outcome of these 3 studies was the proportion of patients achieving or maintaining clinical remission according to the adapted Mayo score (defined as a stool frequency subscore [SFS] of ≤ 1, RBS of 0, and endoscopic subscore of ≤ 1).
In the 2 induction trials, about 60% of patients were male and 65% to 71% were White. The mean age of patients enrolled in the induction trials was 42 to 44 years. At baseline, 50% to 53% of patients had an inadequate response, loss of response, or intolerance to biologic therapy, and 47% to 50% of the patients had an inadequate response, loss of response, or intolerance to conventional therapy. The majority of the patients had a mean adapted Mayo score of 7 or less. Corticosteroids were the most commonly prescribed prior UC medications. During the maintenance therapy, patients’ baseline characteristics were generally comparable to those in the induction period.
Efficacy Results
During the induction period of U-ACHIEVE, clinical remission based on the adapted Mayo score at week 8 was achieved in 26.1% of patients in the upadacitinib group and 4.8% of patients in the placebo group; the between-group difference was 21.6% (95% confidence interval [CI], 15.8% to 27.4%). In U-ACCOMPLISH, clinical remission per adapted Mayo score was achieved in 33.5% of patients in the upadacitinib group and 4.1% of patients in the placebo group; the between-group difference was 29.0% (95% CI, 23.2% to 34.7%). At the end of the maintenance period of U-ACHIEVE at week 52, clinical remission was maintained in 42.3% of patients in the upadacitinib 15 mg group, 51.7% of patients in the upadacitinib 30 mg group, and 12.1% of patients in the placebo group; the between-group differences were 30.7% (95% CI, 21.7% to 39.8%) for upadacitinib 15 mg versus placebo and 39.0% (95% CI, 29.7% to 48.2%) for upadacitinib 30 mg versus placebo (Table 2). The proportion of patients achieving clinical remission at week 8 or maintaining clinical remission at week 52 was the primary efficacy outcome in all 3 studies.
Similarly, the results for the proportion of patients achieving clinical response, endoscopic improvement or remission, histologic improvement, and mucosal healing favoured patients who were treated with upadacitinib compared with those treated with placebo for both the induction and maintenance periods. For maintenance therapy, the treatment effect for upadacitinib 15 mg versus placebo was smaller than for upadacitinib 30 mg versus placebo. The clinical expert consulted by CADTH indicated that all of the between-group differences were clinically meaningful. The results of the subgroup analyses based on patients’ baseline characteristics were consistent with those in the overall population. The results for other efficacy outcomes suggested that treatment with upadacitinib was associated with better symptom relief and improved health-related quality of life (HRQoL) compared with placebo during both the induction and maintenance periods. The changes in HRQoL measured with the Inflammatory Bowel Disease Questionnaire (IBDQ) and Functional Assessment of Chronic Illness Therapy–Fatigue (FACIT-F) favoured the upadacitinib therapy. The impact of UC on work was evaluated between the upadacitinib group and the placebo group; however, this outcome was not adjusted for multiplicity and the results should be interpreted with caution. Treatment with upadacitinib may be associated with lower rates of hospitalization due to UC for both induction and maintenance periods.
Harms Results
The proportion of patients experiencing at least 1 adverse event (AE) during induction was different between the 2 induction trials. In U-ACHIEVE Induction, at least 1 AE was reported by 56.4% and 61.9% of patients in the upadacitinib group and placebo group, respectively. In U-ACCOMPLISH, at least 1 AE was reported by 52.9% and 39.5% of patients in the upadacitinib group and placebo group, respectively. UC was more often reported in the placebo groups and was a major driver when the risk of AEs, serious adverse events (SAEs), or withdrawal due to adverse events (WDAEs) was high in the placebo group compared with the upadacitinib group. This may be explained by the fact that the AE “ulcerative colitis” was used to classify the exacerbation of a patient’s existing UC. Patients who were treated with placebo may be more likely to experience this exacerbation due to the lack of efficacy from the treatment of placebo. During the maintenance period, AEs were reported in 75.2%, 75.3%, and 73.5% of the patients in the upadacitinib 15 mg, upadacitinib 30 mg, and placebo groups, respectively.
In the induction period, there were no AEs of active tuberculosis, malignancy, adjudicated venous thromboembolic events (VTEs), or gastrointestinal perforation reported in the upadacitinib groups. The incidence of opportunistic infection, excluding tuberculosis and herpes zoster, herpes zoster, lymphopenia, and neutropenia, was higher in the upadacitinib groups. At the end of the maintenance period, patients treated with up to 1 year of upadacitinib reported cases of herpes zoster, neutropenia, malignancy, hepatic disorder, lymphopenia, and VTEs. The numbers of events were low for malignancy and VTE at this time point. Longer-term data are needed to fully understand the long-term safety profile of upadacitinib in patients with UC.
Table 2: Summary of Key Efficacy Results From Pivotal and Protocol Selected Studies
Outcomes | U-ACHIEVE Induction (ITT1 population, week 8) | U-ACCOMPLISH (ITT1 population, week 8) | U-ACHIEVE Maintenance (ITT_A population, week 52) | ||||
|---|---|---|---|---|---|---|---|
UPA 45 mg (N = 319) | Placebo (N = 154) | UPA 45 mg (N = 341) | Placebo (N = 174) | UPA 15 mg (N = 148) | UPA 30 mg (N = 154) | Placebo (N = 149) | |
Clinical remission per adapted Mayo score | |||||||
N | 319 | 154 | 341 | 174 | 148 | 154 | 149 |
n (%) | 83 (26.1) | 7 (4.8) | 114 (33.5) | 7 (4.1) | 63 (42.3) | 80 (51.7) | 18 (12.1) |
Adjusted between-group difference, % (95% CI) | 21.6 (15.8 to 27.4) | Reference | 29.0 (23.2 to 34.7) | Reference | 30.7 (21.7 to 39.8) | 39.0 (29.7 to 48.2) | Reference |
P value | < 0.001 | Reference | < 0.001 | Reference | < 0.001 | < 0.001 | Reference |
Clinical response per adapted Mayo score | |||||||
N | 319 | 154 | 341 | 174 | 135 | 144 | 134 |
n (%) | 232 (72.6) | 42 (27.3) | 254 (74.5) | 44 (25.4) | 85 (63.0) | 110 (76.6) | 25 (18.8) |
Adjusted between-group difference, % (95% CI) | 46.3 (38.4 to 54.2) | Reference | 49.4 (41.7 to 57.1) | Reference | 44.6 (34.5 to 54.7) | 56.6 (47.2 to 66.0) | Reference |
P value | < 0.001 | Reference | < 0.001 | Reference | < 0.001 | < 0.001 | Reference |
Endoscopic remission | |||||||
N | 319 | 154 | 341 | 174 | 148 | 154 | 149 |
n (%) | 44 (13.7) | 2 (1.3) | 62 (18.2) | 3 (1.7) | 36 (24.2) | 40 (25.9) | 8 (5.6) |
Adjusted between-group difference, % (95% CI) | 12.7 (8.4 to 17.0) | Reference | 15.9 (11.4 to 20.3) | Reference | 18.7 (11.0 to 26.4) | 19.4 (11.7 to 27.2) | Reference |
P value | < 0.001 | Reference | < 0.001 | Reference | < 0.001 | < 0.001 | Reference |
Histologic improvement | |||||||
N | 319 | 154 | 341 | 174 | 148 | 154 | 149 |
n (%) | 175 (55.0) | 35 (22.5) | 212 (62.2) | 43 (24.5) | 63 (42.8) | 88 (56.9) | 31 (20.6) |
Adjusted between-group difference, % (95% CI) | 32.2 (23.8 to 40.7) | Reference | 37.9 (29.8 to 46.1) | Reference | 23.0 (12.9 to 33.1) | 36.0 (25.8 to 46.2) | Reference |
P value | < 0.001 | Reference | < 0.001 | Reference | < 0.001 | < 0.001 | Reference |
Mucosal healing | |||||||
N | 319 | 154 | 341 | 174 | 148 | 154 | 149 |
n (%) | 34 (10.7) | 2 (1.3) | 46 (13.5) | 3 (1.7) | 26 (17.6) | 29 (19.0) | 7 (4.7) |
Adjusted between-group difference, % (95% CI) | 9.7 (5.7 to 13.7) | Reference | 11.3 (7.2 to 15.3) | Reference | 13.0 (6.0 to 20.0) | 13.6 (6.6 to 20.6) | Reference |
P value | < 0.001 | Reference | < 0.001 | Reference | < 0.001 | < 0.001 | Reference |
bio-IR = inadequate response to biologic therapy; CI = confidence interval; ITT = intention to treat; ITT1 = intention-to-treat population in part 1; non-bio-IR = inadequate response to conventional therapy; UPA = upadacitinib;.
Note: The ITT_A population was a subset of the ITT population in U-ACHIEVE Maintenance comprising the first 451 (actual) responders who had been randomized to upadacitinib 45 mg once daily 8-week induction treatment and who were enrolled under the protocol for 52-week maintenance treatment period in cohort 1 of this maintenance study.
For clinical remission, clinical response, endoscopic remission, histologic improvement, and mucosal healing, the following statistical model was used: 95% CI for adjusted difference and P value were calculated according to the Cochran-Mantel-Haenszel test adjusted for strata (corticosteroid use at week 0 [yes or no], clinical remission status at week 0 [yes or no], bio-IR status at baseline [bio-IR or non-bio-IR]) for the comparison of 2 treatment groups. Within each stratum, the 95% CI for difference was calculated based on the normal approximation to the binomial distribution and P value was calculated using Chi-square test. The calculations were based on nonresponder imputation incorporating multiple imputation to handle missing data due to COVID-19 or nonresponder imputation if there were no missing data due to COVID-19.
Sources: Clinical Study Reports for U-ACHIEVE Induction,6 U-ACCOMPLISH,7 and U-ACHIEVE Maintenance.8
Table 3: Summary of Key Safety Results From Pivotal and Protocol Selected Studies
Outcomes | U-ACHIEVE Induction (SA1 population) | U-ACCOMPLISH (SA1 population) | U-ACHIEVE Maintenance (SA_C population) | ||||
|---|---|---|---|---|---|---|---|
UPA 45 mg (N = 319) | Placebo (N = 155) | UPA 45 mg (N = 344) | Placebo (N = 177) | UPA 15 mg (N = 250) | UPA 30 mg (N = 251) | Placebo (N = 245) | |
Patients with ≥ 1 AE, n (%) | 180 (56.4) | 96 (61.9) | 182 (52.9) | 70 (39.5) | 188 (75.2) | 189 (75.3) | 180 (73.5) |
Patients with ≥ 1 SAE, n (%) | 8 (2.5) | 9 (5.8) | 11 (3.2) | 8 (4.5) | 20 (8.0) | 20 (8.0) | 23 (9.4) |
Patients with ≥ 1 WDAE, n (%) | 6 (1.9) | 14 (9.0) | 6 (1.7) | 9 (5.1) | 10 (4.0) | 12 (4.8) | 25 (10.2) |
Notable harms, n (%) | |||||||
Any serious infections | 5 (1.6) | 2 (1.3) | 2 (0.6) | 1 (0.6) | 8 (3.2) | 6 (2.4) | 8 (3.3) |
Opportunistic infection, excluding tuberculosis and herpes zoster | 1 (0.3) | 0 (0.0) | 6 (1.9) | 1 (0.6) | 2 (0.8) | 1 (0.4) | 2 (0.8) |
Active tuberculosis | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 | 0 | 0 |
Herpes zoster | 1 (0.3) | 0 (0.0) | 2 (0.6) | 0 (0.0) | 11 (4.4) | 10 (4.0) | 0 |
Neutropenia | 16 (5.0) | 1 (0.6) | 15 (4.4) | 0 (0.0) | 7 (2.8) | 15 (6.0) | 5 (2.0) |
Malignancy | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (0.4) | 5 (2.0) | 1 (0.4) |
Malignancy, excluding NMSC | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (0.4) | 2 (0.8) | 1 (0.4) |
NMSC | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 | 3 (1.2) | 0 |
Hepatic disorder | 9 (2.8) | 7 (4.5) | 10 (2.9) | 1 (0.6) | 17 (6.8) | 12 (4.8) | 5 (2.0) |
Anemia | 10 (3.1) | 14 (9.0) | 15 (4.4) | 4 (2.3) | 10 (4.0) | 7 (2.8) | 15 (6.1) |
Lymphopenia | 10 (3.1) | 1 (0.6) | 6 (1.7) | 1 (0.6) | 7 (2.8) | 5 (2.0) | 4 (1.6) |
Adjudicated GI perforations | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (0.6) | 0 | 0 | 1 (0.4) |
Adjudicated VTE | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (0.6) | 2 (0.8) | 2 (0.8) | 0 |
AE = adverse event; GI = gastrointestinal; NMSC = nonmelanoma skin cancer; SA1 = safety population for part 1; SAE = serious adverse event; UPA = upadacitinib; VTE = venous thrombosis event; WDAE = withdrawal due to adverse event.
The SA1 population included all randomized patients who received at least 1 dose of the study drug in part 1 of U-ACHIEVE Induction and U-ACCOMPLISH. The SA_C population included all upadacitinib 45 mg once daily 8-week induction responders who were enrolled under the protocol for the 44- or 52-week maintenance treatment period in cohort 1.
Sources: Clinical Study Reports for U-ACHIEVE Induction,6 U-ACCOMPLISH,7 and U-ACHIEVE Maintenance.8
Critical Appraisal
Internal Validity
In the maintenance period, the discontinuation rates were high and imbalanced across treatment arms. In cohort 1, 30.4%, 18.8%, and 63.8% of patients in the upadacitinib 15 mg, upadacitinib 30 mg, and placebo arms, respectively, discontinued the study. “Other” was the main reason for study discontinuation, and the majority of patients in this category were labelled “lack of efficacy” or “loss of response.” These patients would have been considered nonresponders in the efficacy analyses; a bias is less likely to be introduced in that circumstance.
Prespecified subgroup analyses were performed to examine the consistency of the treatment effect observed for the primary efficacy end points. However, proper interpretation of all subgroups was not possible due to the lack of sample size considerations for these subgroups. The subgroups were underpowered to detect significant effect modification by subgroups of interest, such as inadequate response to previous biologics.
External Validity
According to the clinical expert consulted by CADTH, the population included in the pivotal studies was generally consistent with clinical practice. Based on the patient’s baseline characteristics, the study populations reflect a typical population in Canada that would receive upadacitinib in practice.
U-ACHIEVE Induction and U-ACCOMPLISH were 8 weeks of induction therapy. The clinical expert consulted for this review indicated this was a sufficient time frame to determine short-term treatment effects with upadacitinib. U-ACHIEVE Maintenance was a 52-week study. The expert noted that 52 weeks would not be considered sufficient to observe the long-term safety of this drug for rare events, such as malignancy.
The patient population in the maintenance period was likely enriched due to the study design. Approximately 72% of patients responded to the treatment after 8 weeks of induction therapy and, it should be noted that the interpretation of the maintenance period results differs between a re-randomized (RR) study design and a treat-through (TT) design.
Indirect Comparisons
The sponsor-submitted indirect treatment comparison (ITC) provided indirect evidence on the efficacy and safety of upadacitinib relative to other active treatments for moderately to severely active UC. The active comparators for upadacitinib included other JAK inhibitors (tofacitinib and filgotinib), TNF alpha antagonists (adalimumab, golimumab, and infliximab), anti-integrin drugs (vedolizumab), IL-12 and IL-23 antagonists (ustekinumab), and an S1P receptor agonist (ozanimod). Relevant RCTs were identified through a systematic literature search. Twenty-three RCTs were included in the network meta-analysis (NMA). Outcomes of clinical remission, clinical response, and endoscopic improvement were evaluated in both bio-naive and bio-exposed patients. Harm outcomes were evaluated in the overall population. A Bayesian NMA approach was taken for data synthesis.
In addition, 3 published ITCs were identified from the CADTH literature search. Limitations in these studies included: concerns of substantial heterogeneity from different sources and insufficient description on the methods used to address and adjust these heterogeneities, the underlying transitivity assumption of the NMA not being upheld, and wide CIs or credible intervals (CrIs) of the effect estimates, meaning that the magnitude of the effects is uncertain. The authors’ conclusions are provided in this report; however, due to the aforementioned limitations, the results of these ITCs are not described in detail.
Efficacy Results
Based on the results of the sponsor-submitted ITC, for the induction phase, treatment with upadacitinib 45 mg may be associated with higher rates of clinical remission, clinical response, and endoscopic improvement compared with some of the active comparators. The estimates are associated with considerable uncertainty due to the lack of direct evidence, the sparsity of the network, and the potential for the transitivity assumption to have been violated. The analysis of the findings for the maintenance phase required adjustment for differences in study designs and there were fundamental differences in the placebo arms across the studies. The statistical techniques adopted in the sponsor’s ITC are possible strategies to address cross-study heterogeneity, lessen the impact of potential clinical heterogeneity on the estimated treatment effect of upadacitinib, and make NMAs feasible; however, they cannot adequately remove uncertainty. Therefore, firm conclusions could not be established for the efficacy of upadacitinib compared with other relevant active treatments in achieving clinical response, clinical remission, and endoscopic improvement.
Harms Results
Due to the limitations in the sponsor-submitted ITC, a conclusion regarding the relative safety of upadacitinib versus other active treatments cannot be drawn.
Critical Appraisal
In the sponsor-submitted ITC, sources of heterogeneities and potential treatment effect modifiers (such as study design, e.g., inclusion and exclusion criteria, outcome definitions, and notable heterogeneity in a number of patients’ baseline characteristics, e.g., previous UC medications or differences across the placebo arms) in the included studies were identified and some of them were addressed in data analyses. However, in several studies, data for potential effect modifiers were unavailable. The maintenance phase in particular is problematic. Some of the placebo arms were considered fundamentally different from 1 another. Given all these concerns, the transitivity assumption in an NMA may not be upheld. Despite various statistical techniques being employed to lessen the impact of potential clinical heterogeneity, such as baseline risks on the estimated treatment effect of upadacitinib, there is still uncertainty in the ITC results. The approaches used to adjust the differences in study design (TT versus RR) are potential solutions to adjust the cross-study heterogeneity in UC trials; however, it is uncertain whether the adjustment is adequate. In addition, the network is sparse. Coherence could not be assessed due to the lack of relevant closed loops when comparing with other active treatments. All evidence is indirect, which reduces our certainty in the study findings. Wide CrIs are observed for many efficacy and safety outcomes, especially in the maintenance phase. This implies considerable uncertainty in the magnitude of the treatment effects of upadacitinib.
Safety data were sparse and only available in the overall population. These data are likely confounded by efficacy, since UC is commonly reported as an AE, SAE, and WDAE in clinical trials of UC.
Other Relevant Evidence
Data from a long-term clinical trial were not available at the time of this review. No other relevant evidence was identified for this review.
Conclusions
Based on the 2 induction trials and 1 maintenance trial, oral upadacitinib 15 mg, 30 mg, and 45 mg once daily was superior to placebo in achieving induction and maintenance of clinical remission, clinical response, endoscopic improvement, and mucosal healing in patients with moderately to severely active UC. Moreover, upadacitinib was also found to be effective in alleviating UC-related symptoms and improving HRQoL. Long-term data are needed to fully evaluate the safety profile of upadacitinib. Serious limitations in the available ITCs mean that it remains uncertain how upadacitinib compares with other active treatments in the efficacy and safety for moderately to severely active UC.
Introduction
Disease Background
UC is the most common form of IBD. It is characterized by recurring episodes of inflammation to the mucosal layer of the colon. Usually, the disease affects the rectum and may spread to other parts of the colon.1,2,5 Depending on the extent and severity of the disease, patients with UC may present with diarrhea with or without blood and mucus, urgency or tenesmus, incontinence, constipation, colicky abdominal pain, fever, malaise, and weight loss.1,2 Regardless of severity, UC is also associated with high rates of fatigue and sleep difficulties.3 Approximately half of all patients require UC-related hospitalization at some point during the disease course. Patients with UC are at increased risk of developing colon cancer. Previous research found that the risk of colon cancer was 2% in the first 10 years of UC, 8% during the first 20 years, and 18% during the first 30 years.9 Moreover, approximately 1.5% of patients with UC are diagnosed with colorectal cancer, typically after prolonged active inflammation. While UC is not associated with increased risk of all-cause mortality after the first year after diagnosis,10 gastrointestinal-specific mortality may be increased.3,10 Poor prognostic factors for UC include young age of disease onset, extensive colitis, and deep ulcerations.5 The disease has negative physical, emotional, and social impacts on patients.
The diagnosis of UC is based on a combination of symptoms, endoscopic findings, histology, and the absence of an alternative diagnosis. Typical endoscopic findings in UC include erythema, loss of normal vascular pattern, granularity, erosions, friability, bleeding, and ulcerations. While endoscopy with biopsies is the only way to confirm the diagnosis, other laboratory tests of blood and stool are also required to eliminate the possibility that symptoms are being caused by enteric infections from bacteria, a virus or parasite, or by other forms of IBD, such as Crohn disease. The levels of markers of inflammation, such as C-reactive protein (CRP), can assist in determining the severity of disease.5
The majority of patients living with UC have a mild to moderate disease course, generally with active disease at diagnosis followed by alternating exacerbations and longer periods of remission.3 However, aggressive disease course is experienced in 10% to 15% of patients, with a cumulative risk of relapse between 70% to 80% at 10 years postdiagnosis.3 Determining the severity and extent of UC is essential for selecting appropriate treatment and predicting long-term outcomes.1 In general, multiple factors are taken into account when assessing the disease severity: disease effect on patient symptoms (e.g., frequency and severity of diarrhea), HRQoL, and disability; measurable inflammatory burden using objective markers of disease activity (e.g., CRP) and extent; and disease course, including structural damage, number of flares, and extraintestinal manifestations.1,5 In clinical practice, patients with moderate UC may have frequent (4 to 6 per day) loose bloody stools, mild anemia not requiring blood transfusions, and abdominal pain, while patients with severe UC typically have frequent loose bloody stools (more than 6 per day) with severe cramps and evidence of systemic toxicity such as fever, tachycardia, anemia, and weight loss.11,12
UC has a worldwide annual incidence rate of 1.2 to 20.3 cases per 100,000 people and a prevalence of 7.6 to 246.0 cases per 100,000 people.2 Canada has among the highest reported incidence and prevalence of UC in the world. Estimated annual incidence rates for UC in Canada ranged from a low of 8.4 per 100,000 people in Alberta to a high of 21.4 per 100,000 people in Nova Scotia.4 It was estimated that more than 120,000 people in Canada were living with UC in 2018.4
Standards of Therapy
The selection of treatment regimens for UC is guided by disease severity and extent.5 Treatment goals in UC are stepwise. According to the clinical expert consulted by CADTH, the short-term goals of UC treatment are to improve patient symptoms (cessation of rectal bleeding and reduction in stool frequency), prevent disease progression that requires hospitalization, prevent the need for colectomy, and improve the patient’s HRQoL. The intermediate goals are to achieve steroid-free remission and endoscopic improvement and healing. The long-term goals in this patient population include maintaining steroid-free clinical remission, endoscopic healing, and mucosal remission. These goals are consistent with the published clinical practice guidelines.11 When selecting induction therapy for patients with moderate to severe UC, patient preferences, patient characteristics, risk of AEs, other medication use, prior therapy for UC, accessibility to an infusion centre, patient compliance, and coverage of medication costs by payers need to be considered.12
For mild to moderate disease, the first-line therapy is 5-aminosalicylic acid (5-ASA) drugs, or corticosteroids if the patients do not respond to or achieve remission on 5-ASA drugs. Due to their associated side effects, corticosteroids should be reserved for induction therapy and not considered for long-term maintenance therapy.5,11
Different drug classes are available for long-term management of moderately to severely active UC; many are immunomodulators (e.g., azathioprine, 6-mercaptopurine, or methotrexate) or biologics. Biologic therapies are the mainstay of treatment for patients with moderate to severe UC and are used for induction and maintenance when other treatments have been unsuccessful, or in those who cannot tolerate other treatments. At present, biologics include TNF alpha antagonists (adalimumab, infliximab, and golimumab), anti-integrin drugs (vedolizumab), and IL-12 and IL-23 antagonists (ustekinumab). Small-molecule drugs, which include JAK inhibitors (tofacitinib) and S1P receptors (ozanimod), are also used in patients with moderate to severe UC. The clinical practice guidelines developed by the American Gastroenterological Association suggest early use of biologics with or without immunomodulator therapy rather than gradual step-up after failure of 5-ASA.11 Systemic oral glucocorticoids can be used for inducing remission in patients with moderately to severely active UC, or given to provide more immediate symptom relief for patients who are started on a biologic drug for induction therapy. After clinical remission has been achieved, corticosteroids need to be tapered to avoid the adverse effects related to long-term use.12
Surgery is indicated in uncontrolled hemorrhage, perforation, and colorectal carcinoma or dysplastic lesions that are not amenable to endoscopic removal. In addition, surgery is indicated in refractory acute severe UC or medically refractory disease. Approximately 20% to 35% of patients with UC may require surgery.13 Risks associated with surgery may include anastomotic leak, pelvic sepsis, pouchitis, and bowel obstructions and strictures.5
Despite access to a variety of treatment options, not all patients respond to the available treatments and their disease may become refractory to the current treatment regimens. The results of previous studies showed that in patients with IBD who were treated with biologics, primary nonresponse was observed in 20% to 30% of these patients and another 30% became refractory due to secondary loss of response.14
Drug
JAKs are intracellular enzymes that transduce signals from cell-surface receptors for cytokines or growth factors involved in a broad range of cellular processes, including inflammatory responses, hematopoiesis, and immune surveillance. JAK inhibitors are a family of small molecules that block 1 or more of the intracellular tyrosine kinases: JAK1, JAK2, JAK3, and tyrosine kinase 2 (TYK2). Upadacitinib is a selective JAK1 inhibitor. It modulates the signalling pathway at the point of JAKs and has greater inhibitory potency at JAK1 and JAK1/JAK3 than at JAK2/JAK2.15,16
Upadacitinib is approved by Health Canada for the treatment of adult patients with moderately to severely active UC who have demonstrated treatment failure, i.e., an inadequate response, loss of response, or intolerance to at least 1 conventional and/or biologic therapy. The reimbursement criteria for upadacitinib requested by the sponsor are the same as the indication approved by Health Canada. Upadacitinib has been previously approved by Health Canada for the treatment of patients with rheumatoid arthritis, psoriatic arthritis, and atopic dermatitis.
Upadacitinib is available as 15 mg, 30 mg, and 45 mg oral extended-release tablets. The recommended oral induction dose of upadacitinib is 45 mg once daily for 8 weeks. The recommended oral dose of upadacitinib for maintenance treatment is a dose of 15 mg once daily; 30 mg once daily may be appropriate for some patients, such as those with refractory, severe, or extensive disease. For patients 65 years and older, the only recommended maintenance dose is 15 mg once daily. In patients who have responded to treatment with upadacitinib, corticosteroids may be reduced and/or discontinued in accordance with standard of care.
Key characteristics of commonly used medical treatments for UC are presented in Table 4.
Table 4: Key Characteristics of Upadacitinib and Other Drugs for Moderate to Severe Ulcerative Colitis
Detail | Upadacitinib | Adalimumab | Golimumab | Infliximab | Tofacitinib | Ustekinumab | Vedolizumab |
|---|---|---|---|---|---|---|---|
Mechanism of action | Selective JAK inhibitor. Blocks several cytokine pathways and has greater inhibitory potency at JAK1 and JAK1/JAK3. | Anti-TNF. Human IgG1 monoclonal antibody. Binds and blocks TNF alpha and its interactions with p55 and p75 cell-surface TNF receptors. | Anti-TNF. Human monoclonal antibody that binds with p55 or p75 human TNF receptors. | Anti-TNF. IgG1 kappa monoclonal antibody that neutralizes the biological activity of TNF alpha by specifically binding to its receptors. | Selective JAK inhibitor. Blocks several cytokine pathways and lymphocyte activation. | Human IgG1 monoclonal antibody. Neutralizes cellular responses mediated by IL-12 and IL-23. | IgG1 monoclonal antibody. Binds to the human alpha-4 beta-7 integrin, acting as a gut-selective anti-inflammatory biologic. |
Indicationa | For the treatment of adult patients with moderately to severely active UC who have had an inadequate response, loss of response, or were intolerant to either conventional therapy or a biologic drug (i.e., TNF alpha antagonists, integrin receptor antagonists or IL-12 or IL-23 inhibitors). | For the treatment of adult patients with moderately to severely active UC who have had an inadequate response to conventional therapy, including corticosteroids and/or azathioprine or 6-MP, or who are intolerant to such therapies. | Induction and maintenance of clinical response in adults with moderately to severely active UC who have had an inadequate response to or have medical contraindications for conventional therapy, including corticosteroids, aminosalicylates, azathioprine, or 6-MP. | Induction and maintenance of clinical remission and mucosal healing, and reduction or elimination of corticosteroid use in adult patients with moderately to severe active UC who have had an inadequate response to conventional therapy. | For the treatment of adult patients with moderately to severely active UC with an inadequate response, loss of response, or intolerance to either conventional UC therapy or a TNF alpha inhibitor. | Treatment of adult patients with moderately to severely active UC who have failed or were intolerant to treatment with immunomodulators or corticosteroids but never failed treatment with a biologic, or have failed or were intolerant to treatment with a biologic. | Treatment of adult patients with moderately to severely active UC who have had an inadequate response to, loos of response to, or were intolerant to either conventional therapy or infliximab, a TNF alpha antagonist. |
Route of administration | Oral | SC | SC | IV | Oral | IV induction followed by SC injection for maintenance | IV induction followed by SC injection for maintenance |
Recommended dose | Induction: 45 mg once daily for 8 weeks. Maintenance: 15 mg or 30 mg once daily for adults aged 18 to 64 years and 15 mg once daily for adults 65 years and older; 30 mg once daily may be appropriate for some patients, such as those with refractory, severe, or extensive disease. The lowest effective dose needed to maintain response should be used. | 160 mg at week 0 followed by 80 mg at week 2 administered by SC injection. | 200 mg initially administered by SC injection at week 0 followed by 100 mg at week 2 and then 50 mg every 4 weeks thereafter. | Induction dose of 5 mg/kg at 0, 2, and 6 weeks followed by 5 mg/kg every 8 weeks thereafter. | 10 mg (as tofacitinib citrate) twice daily. | Induction: IV infusion, single-use, weight-based dose (approximately 6 mg/kg):
Maintenance: 90 mg SC infections every 8 weeks. | 30 mg administered by IV infusion at 0, 2, and 6 weeks and then every 8 weeks thereafter. The SC maintenance dose is 108 mg every 8 weeks. |
Serious adverse effects or safety issues | Serious warnings for serious infections, malignancies, and thrombosis (DVT, pulmonary embolism, and arterial thrombosis). | Serious warnings for infections, hepatosplenic T-cell lymphoma, and pediatric malignancy. Contraindicated in patients with severe infections and patients with moderate or severe congestive heart failure. Serious infections (pneumonia), malignancies, and neurologic events have been reported more frequently in patients taking adalimumab. | Serious warnings for infections and malignancy. Contraindicated in patients with severe infections and patients with moderate or severe congestive heart failure. Upper respiratory infections and reactions at the site injection, but no clinically significant differences compared with placebo. | Serious warnings for infections, hepatosplenic T-cell lymphoma, and pediatric malignancy. Contraindicated in patients with severe infections and patients with moderate or severe congestive heart failure. | Serious warnings for serious infections, malignancies, thrombosis (DVT, pulmonary embolism, and arterial thrombosis), and major adverse cardiovascular events. | Immunomodulating drugs have the potential to increase the risk of infections and malignancy. No clinically significant differences have been found in terms of malignancies. Contraindicated in patients with severe infections. | Infections and malignancies have been reported in patients taking vedolizumab, but no clinically significant differences have been found. Contraindicated in patients with active severe infections or opportunistic infections. |
6-MP = 6-mercaptopurine; CNS = central nervous system; DVT = deep venous thrombosis; IgG1 = immunoglobulin G1; IL = interleukin; JAK = Janus kinase; SC = subcutaneous; TNF = tumour necrosis factor; UC = ulcerative colitis.
aHealth Canada–approved indication.
Source: Product monographs for upadacitinib (Rinvoq),15 adalimumab (Humira),17 golimumab (Simponi),18 infliximab (Remicade),19 tofacitinib (Xeljanz),20 ustekinumab (Stelara),21 and vedolizumab (Entyvio).22
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups.
Two patient groups submitted input: the Gastrointestinal (GI) Society and Crohn and Colitis Canada. The GI Society is a national charity committed to research, advocacy, and educational activities for people with gastrointestinal and liver conditions that works closely with health care professionals and governments at all levels to improve care and treatment. Crohn and Colitis Canada is a national, volunteer-based health charity committed to finding cures for IBD and improving the lives of children and adults affected by these diseases through research, patient programs, fundraising, spreading information, advocacy, and awareness activities. The information provided in the GI Society submission was gathered through various questionnaires distributed among patients with IBD in 2015 (n = 423), 2018 (n = 432), 2020 (n = 724), and 2022 (ongoing) as well as one-to-one conversations with patients; a patient roundtable; recent phone, email, and social media interactions; and stories submitted by patients over time. Crohn and Colitis Canada compiled data from 2 online surveys (respondents included 354 patients with moderate to severe UC and 2 participants in the Rinvoq clinical trial) conducted earlier this year and 1 phone interview with a patient who participated in the Rinvoq clinical trial.
Patients with UC commonly experience symptoms such as diarrhea (fecal urgency and poor control of bowel function), rectal bleeding, and abdominal pain. Patients commonly described flares, which occur at unpredictable times, as causing extreme pain and fatigue with a need to always be near a bathroom. Symptoms may be present even during periods of remission. UC has a profound effect on patients’ physical, emotional, and social lives at their home, school, or workplace, and is particularly difficult for children and young adults, since it affects their sense of self. Based on the Crohn and Colitis Canada survey, the most frequently reported UC-related complications among patients were mental stress (65%), joint inflammation and arthritis (51%), fissure and hemorrhoids (40%), anemia (33%), skin condition (approximately 30%), and malnutrition and weight loss (approximately 30%); other complications include bowel obstruction, intestinal fistulas, abscesses, stricture, liver conditions, and cancer. Patients said their social lives (including romantic relationships with partners) had been negatively affected by their UC diagnosis and that they felt isolated due to others’ misunderstanding of their condition. About 72% of respondents said they had to constantly adjust their lifestyle and expectations due to UC; 2 in 5 patients said they changed their travel plans and 1 in 5 patients said they changed their career aspirations.
According to the GI Society submission, patients consider sustained remission and treatment response more important than relieving any 1 symptom. Despite the available treatment options, patients have difficulty obtaining remission or symptom relief and there is a need for more diversity in effective treatments that achieve mucosal healing and reduce symptoms and allow patients to live full and productive lives. Patients want adequate access to medications that work to reduce preventable suffering, unnecessary use of health care resources, and financial burden on the government and taxpayers. Finally, the GI Society stated that having a treatment with oral administration rather than infusion or injection would be helpful for many patients.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 1 clinical specialist with expertise in the diagnosis and management of UC.
Unmet Needs
The expert noted several unmet needs related to the treatment of UC. Even though various treatment options are available for patients with moderately to severely active UC, not all patients respond to them, and they may become refractory to current treatments. Currently, there is no medication available to reverse the structural damage of the colon. However, if UC is treated and controlled early in the disease course, the progression of disease may be stopped or delayed, and the risk of dysplasia or colon cancer could be reduced. In addition, some of the current treatments are associated with many safety concerns; for example, patients treated with tofacitinib have an increased risk of herpes zoster and thrombosis. Treatments that are better tolerated and have safe profiles are needed. Some treatments have lower patient adherence due to the inconvenient route of administration. There is also a lack of evidence regarding the efficacy and safety of various medications among special populations, such as pregnant patients, older adults, patients with cardiovascular risk factors, or patients with previous or current malignancies.
Place in Therapy
The expert indicated that upadacitinib is a selective JAK1 inhibitor and would occupy a place in therapy similar to biologics and other targeted small-molecule drugs. Upadacitinib can be given after 5-ASA and used instead of immunomodulators. Due to its mechanism of action, it is thought that the safety profile of upadacitinib may be favourable compared with tofacitinib, which is a less selective JAK inhibitor.
The expert suggested that upadacitinib be offered to biologic-naive patients with UC as well as biologic-exposed patients. The expert stated that it may not be appropriate to have the patients try and fail prednisone and immunomodulators before initiating upadacitinib, considering the side effects and risks associated with treatment with prednisone and immunomodulators. For patients whose condition has already failed to respond to biologics or targeted small-molecule drugs, the expert was of the opinion that upadacitinib would likely be recommended before other medications that have known side effects, such as tofacitinib.
The expert also stated that if patients gain access to upadacitinib without the need for their disease to fail to respond to immunomodulators, access to upadacitinib would potentially cause a shift in the current treatment paradigm based on the current funding criteria; otherwise, upadacitinib would be at a level similar to biologics and the other JAK inhibitors.
Patient Population
The expert indicated that patients with moderately to severely active UC, either biologic-naive or biologic-exposed, are suitable for treatment with upadacitinib. These patients can be identified by clinician examination and judgment based on disease severity, age, sex, family planning stage, comorbidities, certain laboratory tests, and colonoscopy. The expert noted that misdiagnosis is less likely to occur in clinical practice. The expert indicated it is not possible to identify those patients whose condition is most likely to exhibit a response to treatment with upadacitinib. According to the expert, patients who are pregnant, or have active infections, active malignancy, or severe hepatic impairment would not be suitable for treatment with upadacitinib.
Assessing Response to Treatment
The expert noted that in clinical practice, clinical response and remission are assessed using the partial Mayo score or components of the Mayo score. Biomarkers such as CRP and fecal calprotectin are also measured. In terms of endoscopic response, clinicians usually schedule a colonoscopy 6 to 9 months after starting treatment to examine endoscopic healing. If a follow-up colonoscopy 8 weeks after treatment initiation is not feasible, postinduction response can be evaluated using the level of fecal calprotectin or a flexible sigmoidoscopy.
By the end of the induction period, patients who report no further rectal bleeding, no rectal incontinence, more solid stools, or whose symptoms have reduced to no more rectal urgency, a normal frequency of bowel movements, or no more abdominal pain, are considered to have had a clinically meaningful response to treatment. In general, clinical improvement may occur within 4 weeks of treatment, and clinical remission is expected within 8 weeks of treatment. However, depending on the severity of disease at the beginning of treatment and previous medication use, patients may experience a slower response or delay to remission; therefore, an additional 8 weeks of treatment may be required for induction therapy.
Discontinuing Treatment
Treatment with upadacitinib should be discontinued if there is a lack of clinical response to induction therapy, or disease progression occurs. Treatment should also be stopped if the patient is experiencing significant adverse effects, such as hematologic abnormalities, new-onset malignancy, or recurrent infections. Patients who are trying to conceive or are pregnant or breastfeeding should discontinue the treatment, as well.
Prescribing Conditions
Upadacitinib can be prescribed by a gastroenterologist who can perform colonoscopies and diagnose UC. This oral drug can be administered at home.
Clinician Group Input
This section was prepared by CADTH staff based on the input provided by clinician groups. The full original clinician group inputs received by CADTH have been included in the stakeholder section at the end of this report.
Two clinician groups submitted input on upadacitinib: 3 clinicians on behalf of the IBD Centre of BC, and a group of 12 gastroenterologists and a nurse practitioner on behalf of the Atlantic Specialist Group, who submitted jointly with members from the University of Calgary IBD Unit.
The clinician group input was consistent with that from the clinical expert consulted by CADTH in terms of unmet needs, place in therapy, patient population, assessing response to treatment, discontinuing treatment, and prescribing conditions. The clinician groups emphasized that upadacitinib should be restricted from patients with a history of thrombosis or coronary artery disease. In terms of the place in therapy of upadacitinib in clinical practice, both clinician groups agreed that upadacitinib would be used in various circumstances for these patients, including as first-line therapy.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 5.
Table 5: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Considerations for discontinuation of therapy | |
The use of tofacitinib for patients with moderately to severely active UC has been reviewed by CADTH. The CDEC recommendation for tofacitinib for UC is to discontinue initial treatment if clinical response is not achieved after 8 weeks. Clinical response can be based on total or partial Mayo score or the clinical judgment of the prescribing gastroenterologists.
| The clinical expert disagreed that upadacitinib therapy should be stopped if clinical response is not achieved after 8 weeks of therapy. In the clinical trials, if the patients do not have an adequate response to the first 8-week treatment, they are allowed an additional 8-week treatment with upadacitinib. In clinical practice, it is common for clinicians to prescribe an additional 8 weeks of treatment to patients whose condition does not respond well in the first 8 weeks. Some patients may benefit from this extended therapy. |
Care provision issues | |
The sponsor provided the following statement in the submission: “Studies suggest that inhibition of JAK1 may be largely responsible for the efficacy of JAK inhibition in immune-mediated diseases whereas differences in safety of JAK inhibitors may be due to selectivity for specific JAK isoforms.” Both tofacitinib and upadacitinib have Health Canada black box warnings for infection, malignancy, and thrombosis. Health Canada specifically states that the thrombosis warning is because these events have occurred in patients taking upadacitinib.
| The clinical expert responded that the study duration (up to 1 year) of the pivotal studies was short. Therefore, the studies were not adequately designed to assess the long-term safety of upadacitinib. Upadacitinib is a selective JAK inhibitor. The clinical expert indicated that given its unique mechanism of action, in theory, upadacitinib has a better safety profile compared with the pan-JAK inhibitors (i.e., tofacitinib). |
System and economic issues | |
There are currently 5 tofacitinib generics under review by Health Canada, which means that when the generics are available, their price will drop significantly for the typical UC maintenance dose (5 mg p.o. b.i.d.). The price of the lowest maintenance dose for upadacitinib (15 mg p.o. q.d.) is $18,000 per year for the treatment of UC.
In addition to the significantly reduced price for tofacitinib generics, there are negotiated confidential prices for the biosimilars of adalimumab and infliximab, which places their prices in the ballpark of tofacitinib generics. There is also a negotiated price for vedolizumab.
| The clinical expert indicated that if clinical evidence supports improved safety with upadacitinib over tofacitinib generics, then it is beneficial for the drug plans to pay a price premium for improved safety. Even though a drug with a better safety profile might be more expensive, it would save more health care resources in the long-term (e.g., the expense of hospitalizations from treatment-related complications). The clinical expert also noted that clinical trial data have suggested that treatment with JAK inhibitors improves patient outcomes faster compared with biologics. If patients can be steroid-free faster with upadacitinib vs. biologics, it may be worth paying a premium. However, there is a lack of head-to-head trials to directly compare upadacitinib with biologics and provide compelling evidence on its superiority. |
b.i.d. = twice daily; CDEC = CADTH Canadian Drug Expert Committee; JAK = Janus kinase; p.o. = oral; q.d. = once daily; TNF = tumour necrosis factor; UC = ulcerative colitis.
Clinical Evidence
The clinical evidence included in the review of upadacitinib is presented in 3 sections. The first section, the systematic review, includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those studies that were selected according to an a priori protocol. The second section includes indirect evidence from the sponsor and indirect evidence selected from the literature that met the selection criteria specified in the review. The third section includes sponsor-submitted long-term extension studies and additional relevant studies that were considered to address important gaps in the evidence included in the systematic review. No studies were included in this section.
Systematic Review (Pivotal and Protocol Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of oral upadacitinib 15 mg, 30 mg, and 45 mg per day for the treatment of adult patients with moderately to severely active UC who have had an inadequate response to, loss of response to, or were intolerant to either conventional therapy or a biologic drug (i.e., TNF alpha antagonists, integrin receptor antagonists, or IL-12 or IL-23 inhibitors).
Methods
The studies selected for inclusion in the systematic review included pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 6. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans. Of note, the systematic review protocol presented in Table 6 was established before the granting of a Notice of Compliance from Health Canada.
Table 6: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Population | Adult patients with moderately to severely active ulcerative colitis who have had an inadequate response, loss of response, or were intolerant to either conventional therapy or a biologic drug (i.e., TNF alpha antagonists, integrin receptor antagonists, or interleukin 12 or interleukin 23 inhibitors). Subgroups:
|
Intervention | Upadacitinib, oral tablets.
|
Comparator |
|
Outcomes | Efficacy outcomes:
Harms outcomes:
|
Study designs | Published and unpublished phase III and IV RCTs |
AE = adverse event; RCT = randomized controlled trial; SAE = serious adverse event; TNF = tumour necrosis factor; WDAE = withdrawal due to adverse event.
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy according to the PRESS Peer Review of Electronic Search Strategies checklist.23
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946–) through Ovid and Embase (1974–) through Ovid. All Ovid searches were run simultaneously as a multifile search. Duplicates were removed using Ovid deduplication for multifile searches, followed by manual deduplication in Endnote. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concepts were Rinvoq, upadacitinib, and ulcerative colitis. Clinical trials registries were searched: the US National Institutes of Health’s clinicaltrials.gov, the WHO’s International Clinical Trials Registry Platform (ICTRP) search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
No filters were applied to limit the retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. Refer to Appendix 1 for the detailed search strategies.
The initial search was completed on May 16, 2022. Regular alerts updated the search until the meeting of the CADTH Canadian Drug Expert Committee on September 28, 2022.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool For Searching Health-Related Grey Literature checklist.24 Included in this search were the websites of regulatory agencies (the FDA and European Medicines Agency). Google was used to search for additional internet-based materials. Refer to Appendix 1 for more information on the grey literature search strategy.
These searches were supplemented by reviewing bibliographies of key papers. In addition, the sponsor of the drug was contacted for information regarding unpublished studies.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
Findings From the Literature
A total of 46 studies were identified from the literature for inclusion in the systematic review (Figure 1). A total of 5 reports were included that presented data from 3 unique studies. The included studies are summarized in Table 7.
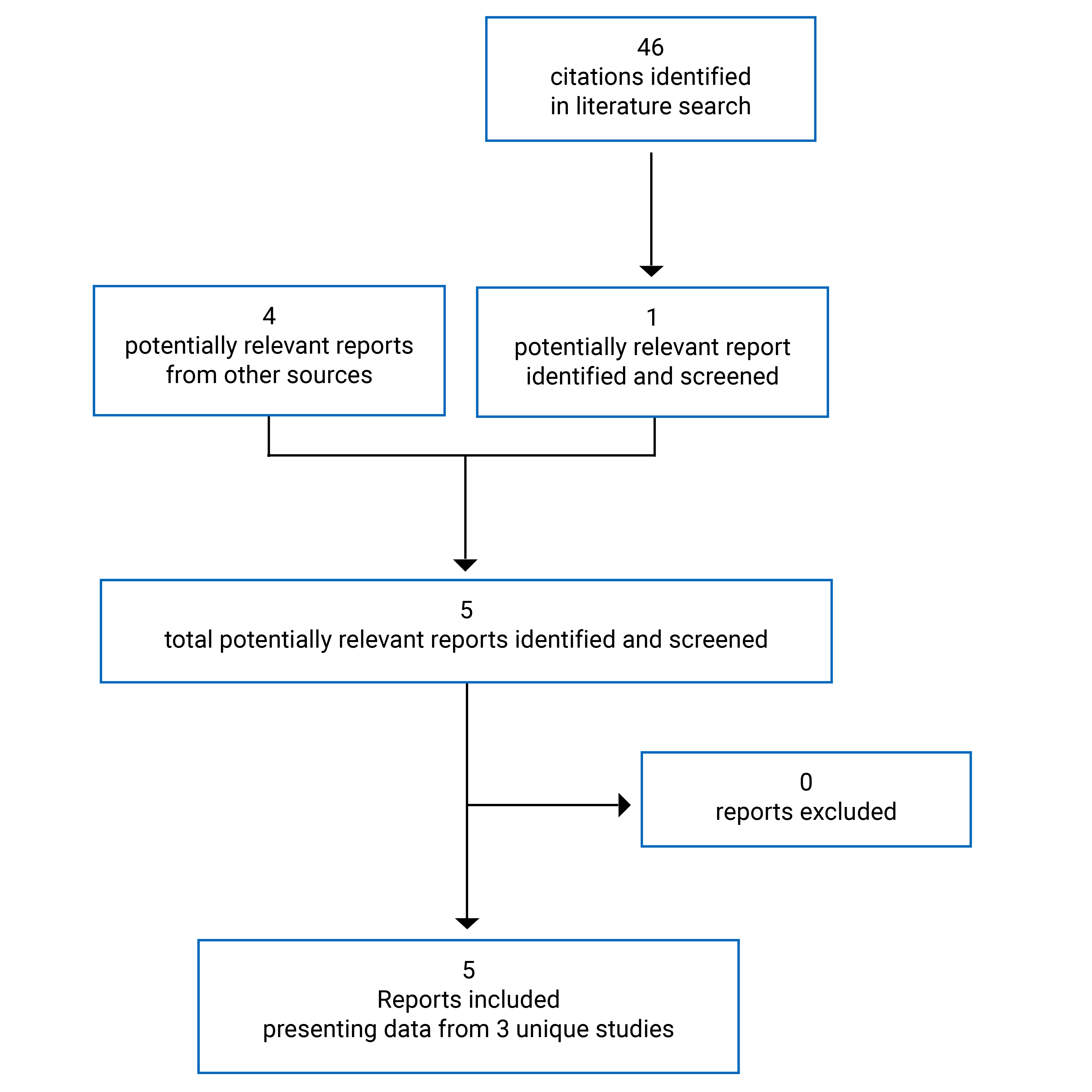
Table 7: Details of Included Studies
Detail | U-ACHIEVE Induction (substudy 2) | U-ACCOMPLISH | U-ACHIEVE Maintenance (substudy 3) |
|---|---|---|---|
Designs and populations | |||
Study design | Phase III, DB RCT, induction phase | Phase III, DB RCT, induction phase | Phase III trial, maintenance phase of U-ACHIEVE |
Locations | 199 sites in Europe, Australia, Asia, Africa, South America, and North America, including Canada | 204 sites in Europe, Australia, Asia, Africa, South America, and North America, including Canada | 302 sites in Australia, Europe, Asia, Africa, South America, and North America, including Canada |
Patient enrolment dates | October 3, 2018, to September 7, 2020 | December 6, 2018, to January 14, 2021 | December 14, 2016, to April 30, 2021 (data cut-off) |
Randomized (N) | 474 | 522 | 1,046 enrolled |
Inclusion criteria |
| ||
Inadequate response, loss of response, or intolerance to oral aminosalicylates, corticosteroids, immunosuppressants, and/or biologic therapies | Inadequate response, loss of response, or intolerance to oral aminosalicylates, corticosteroids, immunosuppressants, and/or biologic therapies |
| |
Exclusion criteria |
| ||
|
|
| |
Drugs | |||
Intervention |
| Upadacitinib 15 mg or 30 mg, oral, daily for 52 weeks | |
Comparator(s) | Placebo, oral, daily for 8 weeks | Placebo, oral, daily for 8 weeks | Placebo, oral, daily for 52 weeks |
Duration | |||
Phase | |||
Screening | Up to 5 weeks | No screening | |
Double blind | 8 weeks; nonresponders at week 8 received OL treatment of upadacitinib for an additional 8 weeks | Up to 52 weeks; treatment allocation depended on the previous treatment received in the induction studies | |
Follow-up | 30 days | ||
Outcomes | |||
Primary end point | Clinical remission at week 8 (per adapted Mayo score) | Clinical remission at week 44 or 52 per adapted Mayo score | |
Secondary and exploratory end points |
|
|
|
Notes | |||
Publications | Danese et al., 202225 | ||
ADA = adalimumab; CD = Crohn disease; CER = certolizumab; DB = double blind; GI = gastrointestinal; GOL = golimumab; HBV = hepatitis B virus; HCV = hepatitis C virus; HRQoL = health-related quality of life; IBDQ = Inflammatory Bowel Disease Questionnaire; INF = infliximab; JAK = Janus kinase; NAT = natalizumab; NR = not reported; NSAID = nonsteroidal anti-inflammatory drug; OL = open label; RBS = rectal bleeding subscore; RCT = randomized controlled trial; SFS = stool frequency subscore; TB = tuberculosis; TPN = total parenteral nutrition; UC = ulcerative colitis; UCEIS = Ulcerative Colitis Endoscopic Index of Severity; UST = ustekinumab; VED = vedolizumab.
Note: 1 additional report was included (submission26).
Source: Clinical Study Reports for U-ACHIEVE Induction,6 U-ACCOMPLISH,7 and U-ACHIEVE Maintenance.8
Description of Studies
Studies U-ACHIEVE (substudies 2 and 3) and U-ACCOMPLISH are pivotal trials submitted by the sponsor.
U-ACHIEVE comprises 3 substudies that included patients with moderately to severely active UC. Substudy 1 is a phase IIb dose-ranging induction trial and is not included in the CADTH systematic review. Substudies 2 and 3 are phase III trials evaluating the efficacy and safety of upadacitinib in the induction and maintenance phases, respectively. The results of substudies 2 (U-ACHIEVE Induction) and 3 (U-ACHIEVE Maintenance) are included in the current report. U-ACCOMPLISH is a phase III induction trial in the study population.
Induction
The primary objective of U-ACHIEVE Induction and U-ACCOMPLISH was to evaluate the efficacy and safety of upadacitinib 45 mg once daily compared with placebo in inducing clinical remission in patients with moderately to severely active UC who have had an inadequate response, loss of response, or intolerance to aminosalicylates, immunosuppressants, corticosteroids, or biologic therapies.
U-ACHIEVE Induction (N = 474) included 2 parts: part 1 was a randomized, double-blind placebo-controlled 8-week induction period; part 2 was an open-label 8-week extended-treatment period for clinical nonresponders from part 1 of this study (Figure 2). Clinical response was defined as a decrease from baseline in the adapted Mayo score of 2 points or greater and a reduction of 30% or greater from baseline, plus a decrease in RBS of 1 or greater or an absolute RBS of 1 or less. In part 1, eligible patients were randomized in a 2:1 ratio to upadacitinib 45 mg or matching placebo for 8 weeks. The randomization was stratified by response to previous treatment (insufficient response to biologic therapy [bio-IR] or insufficient response to conventional therapy [non-bio-IR]), corticosteroid use (yes or no), and adapted Mayo score (≤ 7 or > 7) at baseline. Within the bio-IR group, the randomization was further stratified by the number of prior biologic therapies (≤ 1 or > 1). Within the non-bio-IR group, the randomization was further stratified by previous biologic use (yes or no). In part 2, patients who did not achieve clinical response based on adapted Mayo score at week 8 in part 1 (regardless of initial randomized treatment) continued the treatment with oral upadacitinib 45 mg once daily. Clinical responders in part 1 and part 2 could enter substudy 3 of U-ACHIEVE (U-ACHIEVE Maintenance), while clinical nonresponders discontinued.
Similar to U-ACHIEVE Induction, U-ACCOMPLISH (N = 522) included 2 parts (Figure 3). Part 1 was a randomized, double-blind placebo-controlled 8-week induction period, and part 2 was an open-label 8-week extended-treatment period for clinical nonresponders from part 1. Eligible patients were randomized in a 2:1 ratio to 1 of the 2 treatment groups (oral upadacitinib 45 mg once daily or matching placebo) for 8 weeks. The randomization was stratified using the same categories as for U-ACHIEVE Induction. Clinical responders in part 1 and part 2 could enter substudy 3 of U-ACHIEVE (U-ACHIEVE Maintenance).
Maintenance
The primary objective of U-ACHIEVE Maintenance was to evaluate the efficacy and safety of oral upadacitinib 15 mg or 30 mg once daily compared with placebo in achieving clinical remission in patients with moderately to severely active UC who achieved clinical response after completion of induction treatment or the extended-treatment period from substudy 1 and substudy 2 of U-ACHIEVE Induction or U-ACCOMPLISH. This study included 4 cohorts (Figure 4). Treatment allocation in this maintenance study depended on the treatment received in U-ACHIEVE substudies 1 and 2 or U-ACCOMPLISH.
During this study, patients who met the criteria for loss of response after at least 4 weeks of follow-up had the option to enrol into an ongoing extension study (M14 to 533) and receive open-label upadacitinib. Loss of response was defined as follows: patient presents with both an SFS and an RBS that is at least 1 point greater than their value at the end of induction (week 8 of substudies 1 or 2 and U-ACCOMPLISH) on 2 consecutive visits at least 14 days apart.
The primary outcome of the 3 studies was the proportion of patients achieving clinical remission at week 8 or maintaining clinical remission at week 52; clinical remission was measured using the adapted Mayo score. The full Mayo score is composed of 4 components: rectal bleeding, stool frequency, Physician’s Global Assessment (PGA), and endoscopy findings, while the adapted Mayo score contains 3 components: rectal bleeding, stool frequency, and endoscopy findings.
Figure 2: Study Design of U-ACHIEVE Induction (Substudy 2)
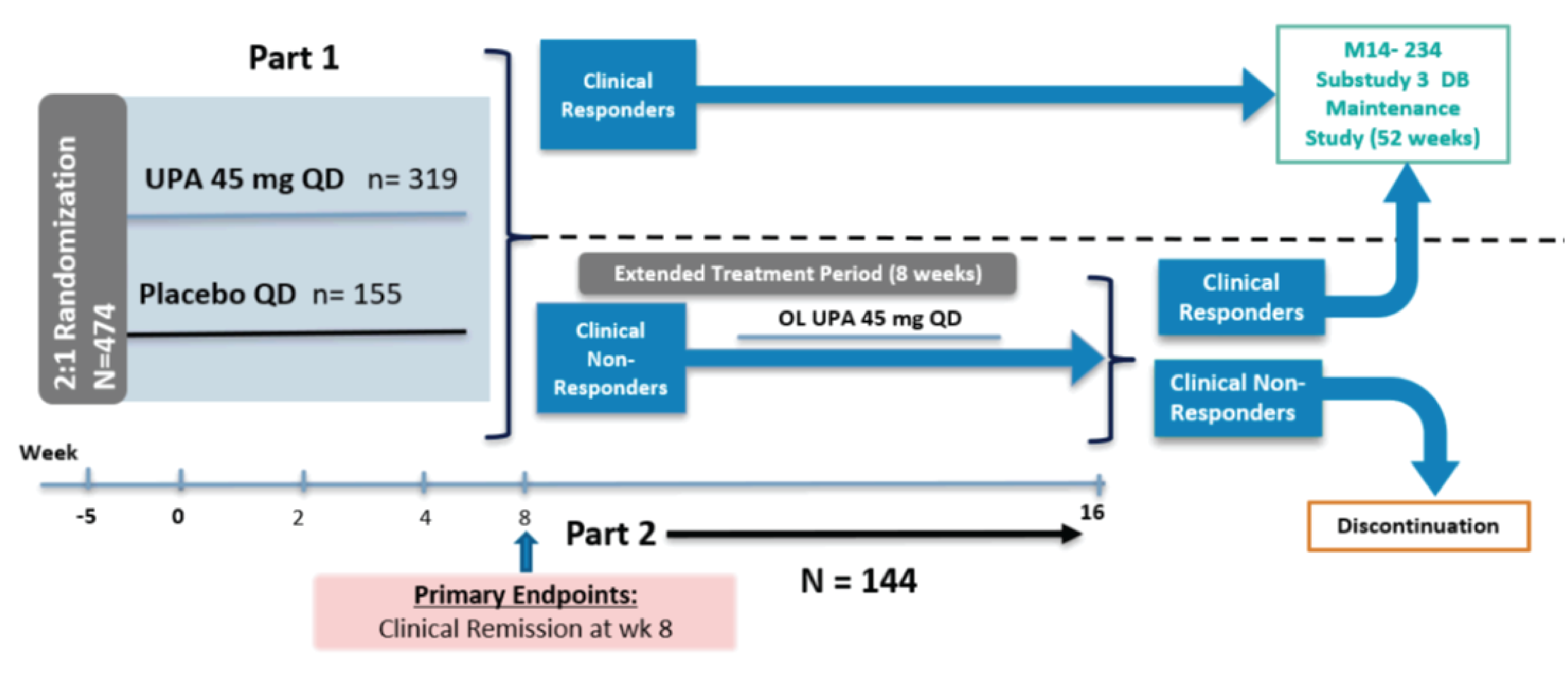
DB = double blind; OL = open label; QD = once daily; UPA = upadacitinib; wk = week.
Source: Clinical Study Report for U-ACHIEVE Induction.6
Figure 3: Study Design of U-ACCOMPLISH
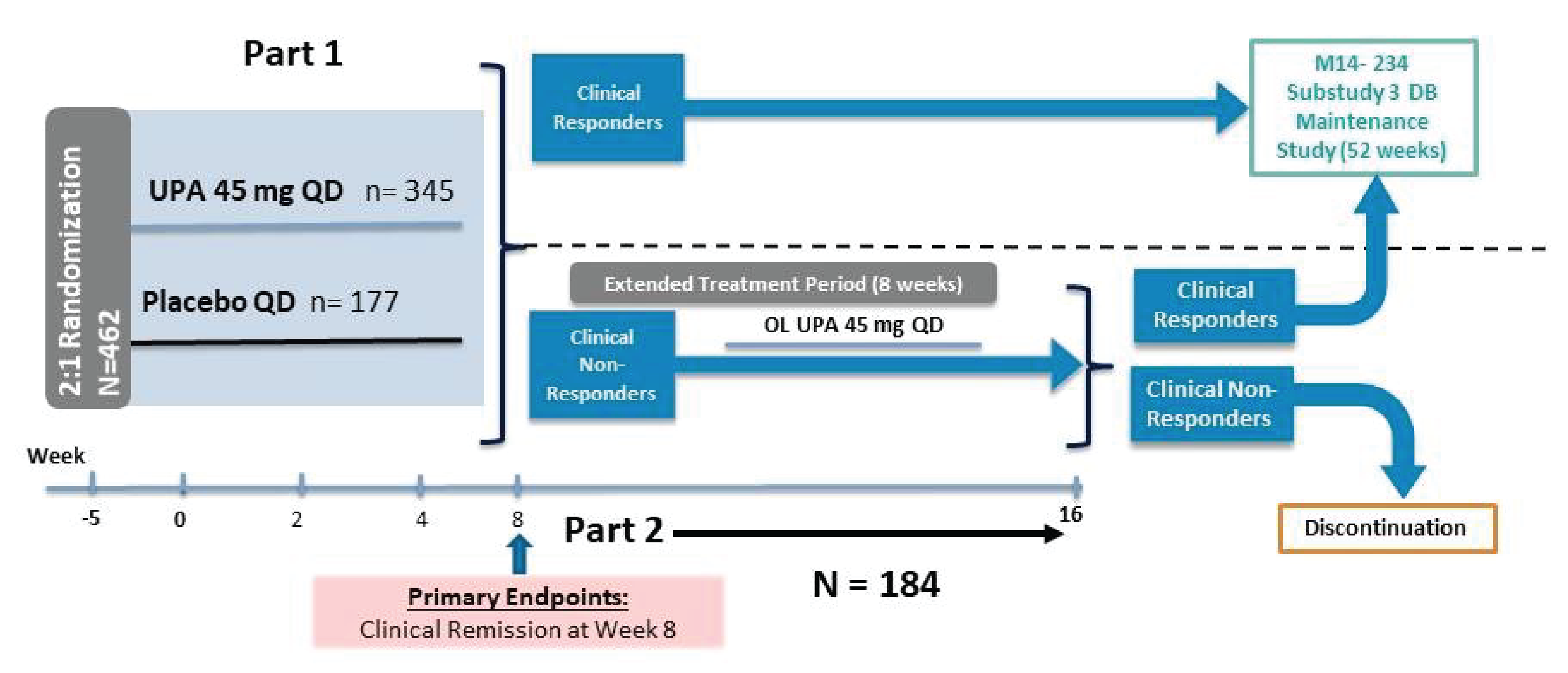
DB = double blind; OL = open label; QD = once daily; UPA = upadacitinib.
Source: Clinical Study Report for U-ACCOMPLISH.7
Figure 4: Study Design of U-ACHIEVE Maintenance (Substudy 3)
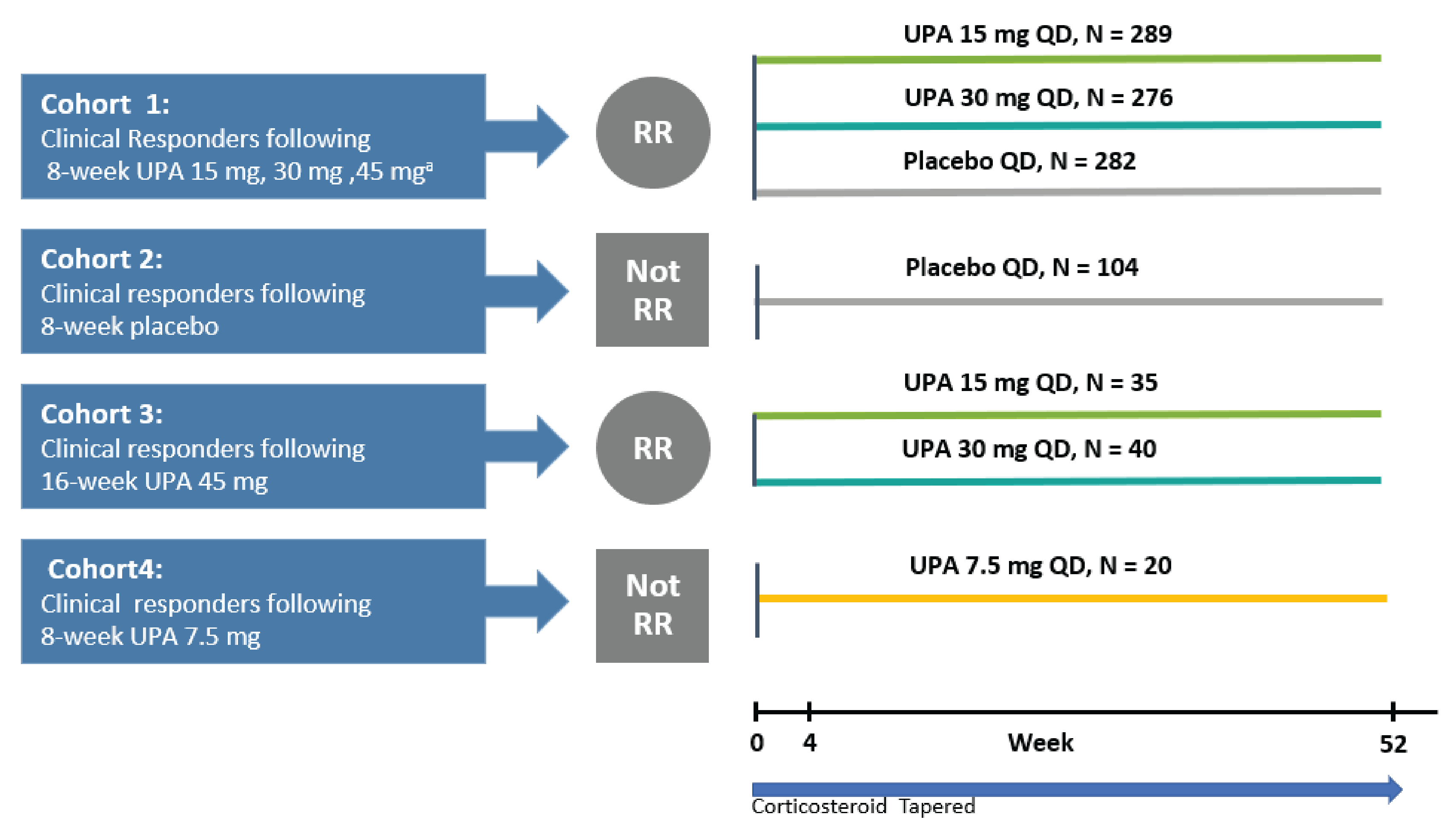
QD = once daily; RR = re-randomized; UPA = upadacitinib.
Source: Clinical Study Report for U-ACHIEVE Maintenance.8
Populations
Inclusion and Exclusion Criteria
Induction
Patients eligible for U-ACHIEVE Induction and U-ACCOMPLISH were between 18 and 75 years old with an inadequate response, loss of response, or intolerance to oral aminosalicylates, immunosuppressants, corticosteroids, and/or biologic therapies. The patients enrolled had to have received a diagnosis of UC at least 90 days before baseline, confirmed by colonoscopy during the screening period; patients with a current infection, colonic dysplasia, and/or malignancy were excluded. Eligible patients had to have an adapted Mayo score of 5 to 9 points and an endoscopy subscore of 2 to 3. The study allowed for up to 30% of enrolled bio-IR patients to have disease that had failed to respond to 3 or more biologics. Among non-bio-IR patients, patients who used a biologic for up to 1 year and discontinued for reasons other than inadequate response, loss of response, or intolerance (e.g., change of insurance or reimbursement, well-controlled disease) could be enrolled if other criteria for inadequate response, loss of response, or intolerance to aminosalicylates, corticosteroids, or immunosuppressants, as defined in the protocol, were met. The study allowed for enrolment of up to 20% of the enrolled non-bio-IR patients who had previously used biologic therapy but discontinued based on reasons other than inadequate response, loss of response, or intolerance.
Maintenance
Patients who achieved clinical response after completion of induction treatment or an extended-treatment period in the U-ACHIEVE Induction or U-ACCOMPLISH studies were eligible to enter U-ACHIEVE Maintenance. Clinical response was defined as a decrease from baseline in the adapted Mayo score of 2 points or greater and a decrease of at least 30% from baseline, plus a decrease in RBS of 1 point or greater or an absolute RBS score of 1 or less at week 8 or 16 of the U-ACHIEVE Induction study or in U-ACCOMPLISH, and who had not met any study discontinuation criteria.
Baseline Characteristics
The baseline characteristics of the patients who were enrolled in the induction period and the characteristics of the patients who entered into the maintenance period of U-ACHIEVE are summarized in Table 8 and Table 9. Baseline characteristics in the intention-to-treat (ITT) population for part 1 (ITT1) (i.e., all randomized patients who received at least 1 dose of the double-blinded study drug) are presented for both induction trials.
Most (62% to 63%) patients were male. The mean age of patients enrolled in the induction trials was 42 to 44 years. The majority (65% to 71%) of the patients were White. At baseline, 50% to 53% of patients had an inadequate response, loss of response, or intolerance to biologic therapy; the remaining 47% to 50% of the patients had inadequate response, loss of response, or intolerance to conventional therapy. The majority of the patients had a mean adapted Mayo score of 7 or less. Finally, almost all patients were previously treated with UC medications, although prior UC treatment was 1 of the inclusion criteria in the 2 induction trials. Corticosteroids were the most common previously prescribed UC medications (84% to 87%), followed by ASA, immunomodulators, and biologics. Overall, patients’ baseline characteristics in the 2 induction trials were comparable between the upadacitinib group and the placebo group.
During the maintenance therapy, patients’ baseline characteristics were generally comparable with those in the induction period. ITT_A is a subset of patients in the ITT population in U-ACHIEVE Maintenance who:
received at least 1 dose of the study drug in the maintenance phase
comprised the first 451 responders who had been randomized to upadacitinib 45 mg once daily 8-week induction treatment
were enrolled under the protocol for the 52-week maintenance treatment period in cohort 1.
The characteristics of the ITT_A population are presented in Table 9. It is unclear if the ITT_A population was similar to the overall ITT population in this study, although this was the largest subset of the overall ITT population and was also the primary analysis set for efficacy outcome assessment.
Table 8: Summary of Baseline Characteristics — Induction Trials (ITT1 Population)
Characteristics | U-ACHIEVE Induction | U-ACCOMPLISH | ||
|---|---|---|---|---|
UPA 45 mg (N = 319) | Placebo (N = 154) | UPA 45 mg (N = 341) | Placebo (N = 174) | |
Age, years, mean (SD) | 43.6 (14.0) | 44.4 (14.6) | 42.1 (14.7) | 42.2 (14.4) |
Sex, n (%) | ||||
Female | 121 (37.9) | 57 (37.0) | 127 (37.2) | 67 (38.5) |
Male | 198 (62.1) | 97 (63.0) | 214 (62.8) | 107 (61.5) |
Race, n (%) | ||||
White | 206 (64.6) | 100 (64.9) | 234 (68.6) | 124 (71.3) |
Black or African American | 12 (3.8) | 4 (2.6) | 11 (3.2) | 6 (3.4) |
Asian | 95 (29.8) | 46 (29.9) | 94 (27.6) | 41 (23.6) |
American Indian or Alaska Native | 0 (0.0) | 2 (1.3) | 0 (0.0) | 1 (0.6) |
Native Hawaiian or other Pacific Islander | 1 (0.3) | 0 (0.0) | 0 (0.0) | 1 (0.6) |
Multiple | 5 (1.6) | 2 (1.3) | 2 (0.6) | 1 (0.6) |
Disease duration, years, mean (SD) | 8.6 (7.2) | 9.1 (8.8) | 7.3 (6.4) | 7.4 (7.2) |
Bio-IR status, n (%) | ||||
Bio-IR | 168 (52.7) | 78 (50.6) | 172 (50.4) | 89 (51.1) |
Non-bio-IR | 151 (47.3) | 76 (49.4) | 169 (49.6) | 85 (48.9) |
Baseline corticosteroid use, n (%) | ||||
Yes | 124 (38.9) | 61 (39.6) | 120 (35.2) | 72 (41.4) |
No | 195 (61.1) | 93 (60.4) | 221 (64.8) | 102 (58.6) |
Baseline immunosuppressant use, n (%) | ||||
Yes | 2 (0.6) | 3 (1.9) | 1 (0.3) | 3 (1.7) |
No | 317 (99.4) | 151 (98.1) | 340 (99.7) | 171 (98.3) |
Baseline aminosalicylates use, n (%) | ||||
Yes | 220 (69.0) | 103 (66.9) | 233 (68.3) | 120 (69.0) |
No | 99 (31.0) | 51 (33.1) | 108 (31.7) | 54 (31.0) |
Baseline adapted Mayo score, n (%) | ||||
≤ 7 | 195 (61.3) | 94 (61.0) | 205 (60.3) | 103 (59.2) |
> 7 | 123 (38.7) | 60 (39.0) | 135 (39.7) | 71 (40.8) |
Missing | 1 | 0 | 1 | 0 |
Baseline full Mayo score, n (%) | ||||
≤ 9 | 162 (50.9) | 79 (51.3) | 160 (47.1) | 86 (49.4) |
> 9 | 156 (49.1) | 75 (48.7) | 180 (52.9) | 88 (50.6) |
Missing | 1 | 0 | 1 | 0 |
High-sensitivity CRP, mg/L, mean (SD) | 9.4 (15.3) | 12.2 (21.2) | 9.3 (15.3) | 10.8 (19.9) |
Fecal calprotectin, mg/kg, mean (SD) | 3,910.1 (5,698.7) | 3,135.5 (3,986.8) | 3,130.5 (4,719.8) | 3,126.7 (4,742.1) |
IBDQ total score, mean (SD) | 122.2 (36.5) | 121.5 (31.0) | 122.8 (34.5) | 122.7 (37.7) |
FACIT-F score, mean (SD) | 30.5 (11.7) | 31.6 (10.9) | 29.8 (11.8) | 31.4 (12.6) |
Previous UC-related medications, n (%) | 317 (99.4) | 154 (100.0) | 336 (98.5) | 170 (97.7) |
ASA | 259 (81.2) | 125 (81.2) | 267 (78.3) | 120 (69.0) |
Antibiotics | NR | 44 (12.9) | 27 (15.5) | |
Biologics | 174 (54.5) | 82 (53.2) | 173 (50.7) | 93 (53.4) |
ADA | 76 (23.8) | 42 (27.3) | 81 (23.8) | 48 (27.6) |
GOL | 32 (10.0) | 13 (8.4) | NR | |
INF | 124 (38.9) | 51 (33.1) | 114 (33.4) | 47 (27.0) |
VED | 91 (28.5) | 47 (30.5) | 91 (26.7) | 44 (25.3) |
Corticosteroids | 278 (87.1) | 131 (85.1) | 286 (83.9) | 147 (84.5) |
Immunosuppressants | 185 (58.0) | 87 (56.5) | 170 (49.9) | 95 (54.6) |
Other IBD medications | 40 (12.5) | 38 (24.7) | 50 (14.7) | 24 (13.8) |
ADA = adalimumab; bio-IR = patients with inadequate response, loss of response, or intolerance to biologic therapy; CRP = C-reactive protein; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; GOL = golimumab; IBD = inflammatory bowel disease; IBDQ = Inflammatory Bowel Disease Questionnaire; INF = infliximab; nonbio-IR = patients with inadequate response, loss of response, or intolerance to conventional therapy; NR = not reported; SD = standard deviation; UC = ulcerative colitis; UPA = upadacitinib; VED = vedolizumab.
Note: ITT1 population was the ITT population in part 1 of the 2 induction trials.
Source: Clinical Study Reports for U-ACHIEVE Induction6 and U-ACCOMPLISH.7
Table 9: Summary of Baseline Characteristics — Maintenance Trial (ITT_A Population)
Characteristics | U-ACHIEVE Maintenance | ||
|---|---|---|---|
UPA 15 mg (N = 148) | UPA 30 mg (N = 154) | Placebo (N = 149) | |
Age, years, mean (SD) | 42.6 (14.1) | 42.6 (14.8) | 43.3 (14.4) |
Sex, n (%) | |||
Female | 53 (35.8) | 68 (44.2) | 64 (43.0) |
Male | 95 (64.2) | 86 (55.8) | 85 (57.0) |
Race, n (%) | |||
White | 97 (65.5) | 101 (65.6) | 93 (62.4) |
Black or African American | 7 (4.7) | 3 (1.9) | 6 (4.0) |
Asian | 44 (29.7) | 48 (31.2) | 42 (28.2) |
American Indian or Alaska Native | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Native Hawaiian or other Pacific Islander | 0 (0.0) | 1 (0.6) | 1 (0.7) |
Multiple | 0 (0.0) | 1 (0.6) | 7 (4.7) |
Disease duration, years, mean (SD) | 8.9 (8.1) | 8.2 (7.6) | 8.7 (8.0) |
Disease extent, n (%) | |||
Rectosigmoid | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Left-sided | 66 (44.6) | 68 (44.2) | 79 (53.0) |
Extensive or pancolitis | 82 (55.4) | 86 (55.8) | 70 (47.0) |
Bio-IR status, n (%) | |||
Bio-IR | 71 (48.0) | 73 (47.4) | 81 (54.4) |
Nonbio-IR | 77 (52.0) | 81 (52.6) | 68 (45.6) |
Baseline corticosteroid use, n (%) | |||
Yes | 55 (37.2) | 57 (37.0) | 60 (40.3) |
No | 93 (62.8) | 97 (63.0) | 89 (59.7) |
Baseline immunosuppressant use, n (%) | |||
Yes | 1 (0.7) | 1 (0.6) | 0 (0.0) |
No | 147 (99.3) | 153 (99.4) | 149 (100.0) |
Baseline aminosalicylates use, n (%) | |||
Yes | 99 (66.9) | 106 (68.8) | 99 (66.4) |
No | 49 (33.1) | 48 (31.2) | 50 (33.6) |
Baseline adapted Mayo score, mean (SD) | 7.1 (1.3) | 7.0 (1.2) | 7.0 (1.2) |
Baseline adapted Mayo score, n (%) | |||
≤ 7 | 89 (60.1) | 88 (57.9) | 87 (58.4) |
> 7 | 59 (39.9) | 64 (42.1) | 62 (41.6) |
Missing | 0 | 2 | 0 |
Baseline full Mayo score, mean (SD) | 9.3 (1.3) | 9.4 (1.5) | 9.3 (1.4) |
High-sensitivity CRP, mg/L, mean (SD) | 8.4 (12.4) | 8.6 (14.7) | 9.8 (15.9) |
Fecal calprotectin, mg/kg, mean (SD) | 3,141.7 (4,694.0) | 2,737.3 (4,326.7) | 3,620.3 (5,222.3) |
IBDQ total score, mean (SD) | 125.8 (35.9) | 121.3 (35.0) | 122.6 (33.4) |
FACIT-F score, mean (SD) | 31.4 (11.5) | 29.9 (11.8) | 30.2 (11.1) |
Previous UC-related medications, n (%) | 148 (100.0) | 154 (100.0) | 149 (100.0) |
ASA | 141 (95.3) | 148 (96.1) | 140 (94.0) |
Antibiotics | 20 (13.5) | 25 (16.2) | 21 (14.1) |
Biologics | 73 (49.3) | 77 (50.0) | 84 (56.4) |
ADA | 31 (20.9) | 30 (19.5) | 41 (27.5) |
GOL | 16 (10.8) | 12 (7.8) | 16 (10.7) |
INF | 44 (29.7) | 53 (34.4) | 51 (34.2) |
VED | 33 (22.3) | 37 (24.0) | 42 (28.2) |
Corticosteroids | 139 (93.9) | 140 (90.9) | 137 (91.9) |
Immunosuppressants | 75 (50.7) | 86 (55.8) | 82 (55.0) |
Other IBD medications | 34 (23.0) | 40 (26.0) | 46 (30.9) |
ADA = adalimumab; bio-IR = patients with inadequate response, loss of response, or intolerance to biologic therapy; CRP = C-reactive protein; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; GOL = golimumab; IBD = inflammatory bowel disease; IBDQ = Inflammatory Bowel Disease Questionnaire; INF = infliximab; non-bio-IR = patients with inadequate response, loss of response, or intolerance to conventional therapy; NR = not reported; SD = standard deviation; UPA = upadacitinib; VED = vedolizumab.
Note: The ITT_A population was the subset of the ITT population comprising the first 451 responders who had been randomized to upadacitinib 45 mg once daily 8-week induction treatment and who were enrolled under the protocol for 52-week maintenance treatment period in cohort 1. The ITT_A population was the primary analysis population in cohort 1 for efficacy end points.
Source: Clinical Study Report for U-ACHIEVE Maintenance.8
Interventions
Induction
In both induction trials, patients were randomized to oral upadacitinib 45 mg once daily or matching placebo in a 2:1 ratio for 8 weeks in a double-blind manner. At the end of the 8-week induction period, patients who did not achieve clinical response were offered open-label upadacitinib 45 mg once daily for an additional 8 weeks until week 16. This is also known as the extended-treatment period.
During the induction period, concomitant UC-related medications (i.e., oral corticosteroids, antibiotics, aminosalicylates, and/or methotrexate) were allowed, and oral ASA and/or methotrexate were required to remain at a stable dose for the entire study period. For oral corticosteroids, a change in the dose during the induction period was not allowed, while the use of inhaled or topical dermatologic corticosteroids was not restricted. Rescue therapy with any allowed UC medications could be provided in the form of initiation or increased dosage at the investigator’s discretion to treat new or worsening UC symptoms. The allowed UC-related medications are locally acting, oral, or IV corticosteroids; aminosalicylates; methotrexate; or UC-related antibiotics.
Maintenance
U-ACHIEVE Maintenance included 4 cohorts (Figure 4). Treatment during the maintenance period continued for up to 52 weeks, and corticosteroid therapy was tapered during this period according to a prespecified schedule. Patients taking corticosteroids who had worsening of disease after steroid tapering could have their corticosteroid dose increased at the investigator’s discretion.
The treatment assignment in U-ACHIEVE Maintenance depended on the treatment received in substudies 1 and 2 in U-ACHIEVE Induction and U-ACCOMPLISH, as follows:
Placebo: Continue placebo
Upadacitinib 7.5 mg once daily: Continue upadacitinib 7.5 mg once daily
Upadacitinib 15 mg once daily: Randomized 1:1 to receive either upadacitinib 15 mg once daily or matching placebo
Upadacitinib 30 mg or 45 mg once daily: Randomized 1:1:1 to receive either upadacitinib 15 mg once daily, upadacitinib 30 mg once daily, or matching placebo
The dose of upadacitinib 7.5 mg once daily was not relevant to our review; therefore, the results of this dose are not reported.
During the maintenance period, concomitant UC-related medications were allowed, and oral aminosalicylates and/or methotrexate were required to remain at a stable dose for the entire study period. Doses could be decreased or terminated in the event of moderate to severe treatment-related toxicities. In addition, all patients receiving UC-related antibiotics could discontinue treatment starting at week 0 of this study at the discretion of the investigator. Use of inhaled or topical dermatologic corticosteroids was not restricted.
Outcomes
A list of efficacy end points identified in the CADTH review protocol that were assessed in the clinical trials included in this review is provided in Table 10. These end points are further summarized subsequently. A detailed discussion and critical appraisal of these outcome measures is provided in Appendix 2.
Table 10: Summary of Efficacy Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure | U-ACHIEVE Induction | U-ACCOMPLISH | U-ACHIEVE Maintenance |
|---|---|---|---|
Clinical remission | Primary | Primary | Primary |
Corticosteroid-free remission | NR | NR | Key secondary |
Clinical response | Key secondary | Key secondary | Key secondary |
Endoscopic remission | Key secondary | Key secondary | Key secondary |
Endoscopic improvement | Key secondary | Key secondary | Key secondary |
Histologic remission | NR | NR | Key secondary |
Histologic improvement | Key secondary | Key secondary | Other efficacy |
Mucosal healing | Key secondary | Key secondary | Key secondary |
Symptom relief (e.g., abdominal pain, rectal bleeding, bowel urgency) FACIT-F | Key secondary Key secondary | Key secondary Key secondary | Key secondary Key secondary |
HRQoL:
| Other efficacy Other efficacy Key secondary | Other efficacy Other efficacy Key secondary | Other efficacy Other efficacy Key secondary |
Need for colectomy | NR | ||
Extraintestinal manifestations | NR | ||
Emergency department visits or hospitalization | Other efficacy | ||
Work productivity | Other efficacy | Other efficacy | Other efficacy |
UC-related hospitalization | Other efficacy | Other efficacy | Other efficacy |
EQ-5D-5L = 5-level EQ-5D; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; HRQoL = health-related quality of life; IBDQ = Inflammatory Bowel Disease Questionnaire; NR = not reported; SF-36 = Short Form (36) Health Survey; UC = ulcerative colitis; WPAI-UC = Work Productivity and Activity Impairment–Ulcerative Colitis.
Note: The Mayo score, Geboes score, IBDQ, FACIT-F, SF-36, EQ-5D-5L, and WPAI-UC for work productivity are discussed in detail in Appendix 2.
Source: Clinical Study Reports for U-ACHIEVE Induction,6 U-ACCOMPLISH,7 and U-ACHIEVE Maintenance.8
Clinical Remission
Clinical remission was the primary efficacy end point in all 3 pivotal trials. It was expressed as the proportion of patients who were in clinical remission. Clinical remission was defined according to the adapted, full, or partial Mayo score.
The Mayo score is a disease-specific, physician-measured instrument that assesses disease severity and response to treatment in patients with UC.27,28 The Mayo scoring system is a combined endoscopic and clinical scale used to assess the severity of UC. In its complete form, the Mayo score is composed of 4 components: rectal bleeding, stool frequency, PGA (this was left out for the adapted Mayo score), and endoscopy findings. Each component is rated from 0 to 3 and may be summed to yield a total score of 0 to 12. A higher Mayo score indicates more severe disease: a higher SFS indicates more stools than normal and a higher RBS indicates more severe bleeding with stool or blood alone passed.
Clinical remission was measured at the conclusion of the induction period at week 8 or 16 and at the conclusion of the maintenance period at week 52. The definitions of clinical remission were as follows:
Clinical remission based on the adapted Mayo score: Defined as an SFS of 1 or less, an RBS of 0, and an endoscopic subscore of 1 or less. The adapted Mayo score was used to define the primary efficacy outcome in the 3 studies.
Clinical remission based on the full Mayo score: Defined as a total score of 2 or less, with no individual subscore greater than 1. The full Mayo score was used to define secondary efficacy outcomes in the 3 studies.
Clinical remission based on the partial Mayo score: A partial Mayo score is an adapted Mayo score minus the endoscopic finding.
Corticosteroid-free clinical remission: Patients who had been taking steroids at baseline in the induction trials but who discontinued corticosteroid use, remained corticosteroid-free for longer than 90 days immediately before week 52, and achieved clinical remission per adapted Mayo score.
Endoscopy recordings were centrally read by a gastroenterologist blinded to treatment assignment to determine the endoscopy subscore for the Mayo score.
Clinical Response
Clinical response was a key secondary efficacy end point of the 3 pivotal trials and was expressed as the proportion of patients who had clinical response. Clinical response was also defined according to the adapted or partial Mayo score. Clinical response was assessed at the conclusion of the induction period at week 8 and at the conclusion of the maintenance period at week 52. The definitions of clinical response were as follows:
Clinical response based on the adapted Mayo score was defined as a decrease from baseline in the adapted Mayo score of 2 points or greater and a reduction of 30% or greater, an RBS of 1 point or greater, or an absolute RBS of 1 point or less.
Clinical response based on the partial Mayo score was defined as a decrease from baseline of 1 point or greater and at least a 30% reduction, and a decrease in RBS of 1 point or greater or an absolute RBS of 1 or less.
Patients were considered nonresponders and were censored for efficacy assessments if they experienced worsening of disease during corticosteroid tapering (and may have had their corticosteroid dose increased up to or beyond the dose used at baseline or the maximum steroid dose exceeded the dose used at baseline). In addition, patients who were not taking UC-related conventional medications (oral aminosalicylates, systemic corticosteroids, and methotrexate) at baseline, but who initiated treatment with them during the study or who had their dosages of these medications increased to greater than the dosages they were taking at baseline, were also considered nonresponders.
Endoscopic Remission or Improvement
Endoscopic remission was a key secondary outcome in the 3 pivotal trials and was expressed as the proportion of patients with endoscopic remission. Endoscopic remission was defined as an endoscopic subscore of 0, while endoscopic improvement was defined as an endoscopic subscore of 1 or less.
Endoscopic improvement or remission was assessed at the conclusion of the induction period at week 8 and at the conclusion of the maintenance period at week 52.
Histologic Remission or Improvement
Histologic improvement was a key secondary efficacy end point in the induction trials. Histologic remission or improvement was assessed using the Geboes index score and was expressed as the proportion of patients with histologic remission or improvement.29,30 The Geboes score is a 6-item instrument that classifies histological changes into 1 of 6 grades (grade 0 to grade 5). Each grade is assessed on a 4-point scale and given equal weight, as follows: “no abnormality,” “mild abnormality,” “mild/moderate diffuse or multifocal abnormalities,” and “severe diffuse or multifocal abnormalities.” The Geboes score may also be converted into a continuous scale with each subgrade being assigned an ordinal value, yielding values between 0 and 22. Higher scores correspond to greater inflammation.
Histologic remission was defined as a Geboes index score of less than 2, and histologic improvement was defined as a decrease from baseline in Geboes score.
Histologic remission or improvement was assessed at the conclusion of the induction period at week 8 and at the conclusion of the maintenance period at week 52.
Mucosal Healing
Mucosal healing was a key secondary outcome in the 3 pivotal trials and expressed as the proportion of patients with mucosal healing. Mucosal healing was assessed using the endoscopy subscore of the Mayo score and the Geboes index score.
Mucosal healing was defined as an endoscopy subscore of 0 and a Geboes index score of less than 2. Mucosal healing was assessed during the induction period at week 8 and the maintenance period at week 52.
Symptoms Relief
Relief of symptoms such as abdominal pain, rectal bleeding, and bowel urgency was a key secondary efficacy end point in the included studies. The proportions of patients with no symptoms of abdominal pain, rectal bleeding, and bowel urgency were measured. Symptom relief was assessed during the induction period at week 8 and the maintenance period at week 52.
Fatigue was measured using FACIT-F. This is a questionnaire completed by the patients to assess fatigue during the past 7 days. It consists of 13 statements, each rated on a 4-point Likert scale, with higher FACIT-F scores representing less fatigue. A cut-off score of 30 or less is used to define fatigue in patients with IBD.
Health-Related Quality of Life
HRQoL in the 3 pivotal trials was generally classified as an “other efficacy end point” and was assessed using generic or disease-specific instruments: the 36-item Short Form Health Survey (SF-36), the 5-level EQ-5D (EQ-5D-5L), and/or the IBDQ total score. HRQoL outcomes were expressed as the change from baseline to week 8, and from baseline to week 52.
Hospitalization
UC-related hospitalization was measured in all 3 studies.
Work Productivity
Work productivity was classified as an “other efficacy end point” in the included studies and was assessed using the Work Productivity and Activity Impairment–Ulcerative Colitis (WPAI-UC) questionnaire. Work productivity was measured at week 8 of the induction studies and week 52 of the maintenance study.
The WPAI-UC questionnaire, version 2, is an instrument used to measure the impact of a disease on work and daily activities during the previous 7 days. The WPAI-UC consists of 6 questions: employment status (employed or not employed), hours at work missed because of UC, hours at work missed because of other reasons, hours actually worked, overall impairment in productivity while working (a visual analogue scale [VAS] from 0 to 10), and overall impairment in regular activities (VAS from 0 to 10) due to UC. Patients who are employed answer all questions, while those who are not employed answer only the first and last. Four scores are derived from the questionnaire. Scores are expressed as a percentage of impairment or productivity loss and range from 0 to 100%, with higher scores indicating greater impairment, for 4 domains:
absenteeism (work time missed)
presenteeism (percent of impairment while working)
percent of overall work impairment due to UC
impairment of regular activities due to UC.
Safety
The safety outcomes assessed in the included studies were:
treatment-emergent AEs, defined as any untoward medical occurrence in a patient administered a pharmaceutical product, that do not necessarily have a causal relationship with this treatment
SAEs, defined as death, life-threatening events, hospitalization or prolongation of hospitalization, congenital anomaly, persistent or significant disability or incapacity, or important medical event requiring medical or surgical intervention to prevent a serious outcome
AEs leading to discontinuation of the study drug or from the study
AEs of special interest, including serious or opportunistic infection, malignancy, thrombosis, hypersensitivity, hepatotoxicity, anemia, lymphopenia, neutropenia, gastrointestinal perforation, and hyperlipidemia.
All AEs that were reported from the time of the administration of the study drug until 30 days after the discontinuation of the study drug were collected, whether solicited or spontaneously reported by the patient.
Statistical Analysis
Primary Outcomes of the Studies
The primary outcome of the included studies was the proportion of patients who achieved clinical remission per adapted Mayo score at week 8 in the induction trial and at week 52 in the maintenance trial. The primary analysis of the proportion of patients in clinical remission at week 8 and week 52 and the 95% CI was carried out in the ITT population (including all randomized patients who received at least 1 dose of the double-blinded study drug) using the Cochran-Mantel-Haenszel test. The ITT1 (ITT population in part 1) and ITT2 (ITT population in part 2) populations were used for the primary efficacy analyses in the 2 induction trials, and the ITT-A population (defined as the subset of ITT population comprising the first 451 [actual] responders who had been randomized to upadacitinib 45 mg once daily 8-week induction treatment and who were enrolled under the protocol for 52-week maintenance treatment period in cohort 1) was used for the primary efficacy analyses in the maintenance trial. In the induction trials, the stratification factors included bio-IR status, baseline corticosteroid use (yes versus no), and baseline adapted Mayo score (≤ 7 versus > 7) while, in the maintenance trial, the stratification factors were bio-IR status at the baseline of the induction study, clinical remission status at week 0 (yes or no), and corticosteroid use at week 0 (yes or no). The nonresponder imputation (NRI) method was used to handle missing data while incorporating multiple imputation to handle missing data related to COVID-19 (NRI-C). Patients with missing data at a scheduled assessment visit for any reason (except COVID-19) were considered “not achieved” for clinical remission, which was used for the primary analysis. At or after the occurrence of UC-related corticosteroid intercurrent events (i.e., events occurring after treatment initiation), patients were also considered to be nonresponders. In addition, sensitivity analyses using various imputation methods (e.g., a hybrid multiple imputation, NRI with no special handling of data missing due to COVID-19 [NRI-NC], multiple imputation, and as observed) were conducted to address issues related to missing data. An analysis of observed cases that excluded the patients with missing data was performed as well, to determine the impact from missing data. Furthermore, prespecified subgroup analyses based on various patient demographic and disease characteristics were conducted for the primary outcome to explore the treatment effect of the study drug in different subgroups: sex, age, race, bio-IR status, baseline corticosteroid use, baseline adapted Mayo score, baseline full Mayo score, prior exposure to anti-TNF drugs for non-bio-IR, baseline weight, presence of pancolitis at baseline, disease duration at baseline, baseline high-sensitivity CRP, and region.
Power Calculation
The sample size calculation in U-ACHIEVE Induction and U-ACCOMPLISH was based on the expected proportion of patients who would achieve clinical remission per adapted Mayo score at week 8. Based on the results from the phase IIb upadacitinib study, U-ACHIEVE (substudy 1), the proportions of patients achieving clinical remission per adapted Mayo score in the upadacitinib 45 mg once daily group and placebo group were 19.6% and 0%, respectively. Considering the small sample size in the phase IIb study and the more stringent definition of primary end point used for phase III studies, the clinical remission rate was assumed to be 5% in the placebo group and 18% in the upadacitinib 45 mg once daily treatment group. Based on these assumptions, a sample size of 154 patients in the placebo group and 308 patients in the upadacitinib group would have greater than 95% power to detect the 13% treatment difference in the primary end point between the upadacitinib 45 mg once daily group and placebo group using a 2-sided Fisher’s exact test at a 0.05 significance level.
Sample size calculation in the maintenance trial was based on the expected proportion of patients who would achieve clinical remission per adapted Mayo score at week 52. Assuming a clinical remission rate of 12% in the placebo arm and 40% in 1 of the upadacitinib treatment arms at week 52, a sample size of 150 patients in the placebo group and 150 patients in each of the upadacitinib 15 mg and 30 mg treatment groups would have 95% power to detect the 28% treatment difference in the primary end point between an upadacitinib dose and placebo using a 2-sided Fisher exact test at a 0.025 significance level with multiplicity adjustment. Under the assumption that the average response rate to upadacitinib doses at the end of substudies 1 and 2 was 50%, a total of approximately 450 patients would be re-randomized.
Secondary and Other Efficacy Analysis
Secondary efficacy variables were divided into 2 groups. The first group included ranked secondary end points, which were ranked by clinical importance. The primary and ranked key secondary efficacy end points were tested with a fixed-sequence multiple testing procedure as well as a Holm procedure, to control for overall type I error rate at an alpha significance level of 0.05 (2-sided). Statistical significance was assessed at the prespecified alpha level (2-sided) in ranked end point order until the significance level exceeded the prespecified alpha level. No additional statistically significant treatment differences could be declared if the preceding ranked end point failed to achieve the prespecified alpha level. In the maintenance trial, multiplicity adjustment for the multiple-dose comparison (upadacitinib 15 mg versus placebo and upadacitinib 30 mg versus placebo) in the primary efficacy analysis was conducted using a 2-sided Fisher exact test at a significant level of 0.025. The second group included all other additional secondary variables. All analyses of secondary end points were performed using the ITT analysis set of each study: ITT1 and ITT2 for the induction trials, and the ITT-A population for the maintenance trial. In general, continuous secondary variables with repeated measurements were analyzed using a mixed-model for repeated measures at all visits, except that measurements at or after the occurrence of UC-related corticosteroid intercurrent events were excluded. The mixed model included the categorical fixed effects of treatment, visit and treatment-by-visit interaction, randomization stratification factors (bio-IR status [bio-IR versus non-bio-IR], baseline corticosteroid use [yes versus no], and baseline adapted Mayo score [≤ 7 versus > 7]), and the continuous fixed covariates of baseline measurements. An unstructured variance covariance matrix was used. If the model could not converge, an autoregressive covariance structure matrix was used. The parameter estimations were based on the method of restrictive maximum likelihood. The fixed effects were used to report model-based means at corresponding visits. These variables were also analyzed using an analysis of covariance model that included factors for treatment group and stratification variables. Both last observation carried forward (LOCF) and observed-case analyses were used for continuous end points. Binary secondary efficacy variables were analyzed using the Cochran-Mantel-Haenszel test, controlling for stratification variables. The handling of missing data for secondary efficacy end points was the same as for the primary end point: the NRI-C was the primary approach in the analyses of binary secondary efficacy end points. Sensitivity analyses for the secondary end points were performed using NRI-NC, hybrid multiple imputation, and as observed.
The ranked secondary efficacy variables for phase III induction and maintenance trials were as follows.
Induction Period (U-ACHIEVE Induction)
Proportion of patients with endoscopic improvement at week 8.
Proportion of patients with endoscopic remission at week 8.
Proportion of patients achieving clinical response per adapted Mayo score at week 8.
Proportion of patients achieving clinical response per Partial adapted Mayo score at week 2.
Proportion of patients achieving histologic-endoscopic mucosal improvement at week 8.
Proportion of patients who reported no bowel urgency at week 8.
Proportion of patients who reported no abdominal pain at week 8.
Proportion of patients who achieved histologic improvement at week 8.
Change from baseline in IBDQ total score at week 8.
Proportion of patients with mucosal healing at week 8.
Change from baseline in FACIT-F score at week 8.
Induction Period (U-ACCOMPLISH)
Proportion of patients with endoscopic improvement (defined as an endoscopic subscore ≤ 1) at week 8.
Proportion of patients with endoscopic remission (defined as an endoscopic subscore of 0) at week 8.
Proportion of patients achieving clinical response per adapted Mayo score (defined as a decrease from baseline in the adapted Mayo score ≥ 2 points and ≥ 30% from baseline, plus a decrease in RBS ≥ 1 or an absolute RBS ≤ 1) at week 8.
Proportion of patients achieving clinical response per adapted Mayo score (defined as a decrease from baseline ≥ 1 point and ≥ 30% from baseline, plus a decrease in RBS ≥ 1 or an absolute RBS ≤ 1) at week 2.
Proportion of patients achieving histologic-endoscopic mucosal improvement (endoscopic subscore ≤ 1 and Geboes score ≤ 3.1) at week 8.
Proportion of patients who reported no bowel urgency at week 8.
Proportion of patients who reported no abdominal pain at week 8.
Proportion of patients who achieved histologic improvement at week 8.
Change from baseline in IBDQ total score at week 8.
Proportion of patients with mucosal healing (endoscopic subscore = 0 and Geboes score < 2) at week 8.
Change from baseline in FACIT-F score at week 8.
Maintenance Period (U-ACHIEVE Maintenance)
Proportion of patients with endoscopic improvement.
Proportion of patients who maintain clinical remission per adapted Mayo score among patients who achieved clinical remission per adapted Mayo score in U-ACHIEVE Induction (substudy 1 or 2) or U-ACCOMPLISH.
Proportion of patients who achieved clinical remission at week 52 per adapted Mayo score and were corticosteroid-free for at least 90 days among patients in clinical remission in the end of the induction treatment in U-ACHIEVE Induction (substudies 1 or 2) or U-ACCOMPLISH.
Proportion of patients with endoscopic improvement among patients with endoscopic improvement in U-ACHIEVE Induction (substudy 1 or 2) or U-ACCOMPLISH.
Proportion of patients with endoscopic remission.
Proportion of patients who maintain clinical response per adapted Mayo score.
Proportion of patients with histologic-endoscopic mucosal improvement.
Change from baseline in the maintenance period in IBDQ total score.
Proportion of patients with mucosal healing.
Proportion of patients who reported no bowel urgency.
Proportion of patients who reported no abdominal pain.
Change from baseline in FACIT-F score.
Safety Analysis
The safety analysis in U-ACHIEVE Induction, U-ACCOMPLISH, and U-ACHIEVE Maintenance were carried out using the corresponding safety analysis set. Please refer to the following analysis populations section for descriptions of the safety analysis sets used in safety analyses.
In general, in the induction trials, all safety comparisons were to be performed between the treatment groups and placebo group using the ITT_A and safety analysis sets. The safety variables were summarized by treatment according to the treatment a patient actually received. The differences in safety parameters between treatment groups and placebo were evaluated using 2-sided tests at a significance level of 0.05. The treatment group differences in continuous safety variables were assessed using an analysis of variance model with the term of treatment; the treatment group differences in categorical safety variables were evaluated using a Fisher exact test. Missing safety data were not imputed.
In the maintenance trial, the standard safety analyses included reporting of AEs, AEs of special interest, and laboratory and vital sign measurements.
Analysis Populations
Induction
In U-ACHIEVE Induction (substudy 2) and U-ACCOMPLISH, the ITT population for the 8-week double-blind induction period (part 1) was denoted as ITT1 and included all randomized patients who received at least 1 dose of the double-blinded study drug in part 1. The ITT1 population was used for all efficacy and baseline analyses for part 1. These populations are similar to a true ITT population.
The ITT population for the 8-week open-label extended-treatment period (part 2) (ITT2) included all patients who received at least 1 dose of upadacitinib 45 mg in part 2.
The safety population for part 1 included all randomized patients who received at least 1 dose of upadacitinib 45 mg or placebo in part 1.
The safety population for part 2 included all patients who received at least 1 dose of upadacitinib 45 mg in part 2.
Maintenance
In U-ACHIEVE Maintenance, the ITT population included all patients who received at least 1 dose of the study drug in the maintenance study, and the patients were included in the analysis according to the treatment groups that they were randomized to. The following populations were subsets of this ITT population:
ITT_A population: This was the subset of the ITT population comprising the first 451 (actual) responders who had been randomized to upadacitinib 45 mg once daily 8-week induction treatment and who were enrolled under the protocol for 52-week maintenance treatment period in cohort 1. The planned number of patients in the ITT_A population was 450; however, the actual number of patients was 451 because patients 450 and 451 had the same enrolment date. The ITT_A population was the primary analysis population in cohort 1 for efficacy end points.
ITT_B population: This was the subset of the ITT population in cohort 3 who were responders to the upadacitinib 45 mg once daily 16-week induction.
In this maintenance study, the safety population included all patients who received at least 1 dose of the study drug in the maintenance study. The following safety populations were subsets of this safety population (note that the primary population for safety data presentations was the SA_C population, which included the SA_A population):
SA_C population, N = 746: A subset of the safety population comprising patients who were responders to upadacitinib 45 mg once daily 8-week induction and who were enrolled under the protocol for the 44- or 52-week maintenance treatment period in cohort 1 (i.e., all patients who responded to 8-week induction treatment with upadacitinib 45 mg and received maintenance therapy with upadacitinib 15 mg, 30 mg, or placebo).
SA_A population, N = 451: The planned subset of the safety population was to comprise the first 450 responders to upadacitinib 45 mg once daily 8-week induction treatment who were enrolled under the protocol for 52-week maintenance treatment period in cohort 1.
Results
Patient Disposition
Induction
In U-ACHIEVE Induction, 474 patients were randomized, with 319 randomized to the upadacitinib group and 155 randomized to the placebo group. In part 1 of this study, 13 patients (4.1%) in the upadacitinib group and 20 patients (12.9%) in the placebo group withdrew prematurely from the study. Patients treated with placebo (5.8%) were more likely to discontinue the study because of AEs compared with those treated with upadacitinib (1.9%).
In U-ACCOMPLISH, 522 patients were randomized, with 345 randomized to the upadacitinib group and 177 randomized to the placebo group. In part 1 of this study, 11 patients (3.2%) in the upadacitinib group and 13 patients (7.5%) in the placebo group withdrew prematurely from the study. Patients treated with placebo (7.5%) were more likely to discontinue the study because of AEs compared with those treated with upadacitinib (3.2%).
In both studies, UC symptoms were considered AEs.
Details of patient disposition in the induction trials are presented in Table 11.
Table 11: Patient Disposition — Induction Trials
Disposition | U-ACHIEVE Induction | U-ACCOMPLISH | ||
|---|---|---|---|---|
UPA 45 mg | Placebo | UPA 45 mg | Placebo | |
Screened, N | NR | NR | ||
Randomized, N | 319 | 155 | 345 | 177 |
Discontinued from study, N (%) | Week 8: 13 (4.1) | Week 8: 20 (12.9) | Week 8: 12 (3.2) | Week 8: 13 (7.5) |
Reason for discontinuation, N (%) | ||||
Adverse events | 6 (1.9) | 9 (5.8) | 5 (1.5) | 6 (3.4) |
Withdrew consent | 2 (0.6) | 3 (1.9) | 6 (1.8) | 4 (2.3) |
Lost to follow-up | 1 (0.3) | 1 (0.6) | 0 (0.0) | 0 (0.0) |
COVID-19 infection | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
COVID-19 logistical restrictions | 1 (0.3) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Other | 3 (0.9) | 7 (4.5) | 0 (0.0) | 3 (1.7) |
ITT, N | 319 | 154 | 341 | 174 |
ITT1 | 319 | 154 | 341 | 174 |
ITT2 | 59 | 84 | 66 | 113 |
Safety, N | 319 | 155 | 344 | 177 |
SA1 | 344 | 177 | 319 | 155 |
SA2 | 59 | 85 | 68 | 116 |
ITT1 = intention-to-treat population in part 1; ITT2 = intention-to-treat population in part 2; NR = not reported; SA1 = safety population in part 1; SA2 = safety population in part 2.
Source: Clinical Study Reports for U-ACHIEVE Induction6 and U-ACCOMPLISH.7
Maintenance
A total of 1,046 patients who achieved clinical response per adapted Mayo score after completion of induction treatment or an extended-treatment period in U-ACHIEVE substudy 1, substudy 2, or U-ACCOMPLISH entered U-ACHIEVE Maintenance, and 1,044 were treated with a blinded treatment assignment for up to 52 weeks. This study included 4 cohorts. In cohort 1, 847 patients who had achieved clinical response in U-ACHIEVE substudies 1 and 2 and U-ACCOMPLISH at either week 8 or week 16 were re-randomized in a 1:1:1 ratio to upadacitinib 15 mg once daily, upadacitinib 30 mg once daily, or placebo daily. At week 52 in cohort 1, 87 patients (30.4%) in the upadacitinib 15 mg group, 52 patients (18.8%) in the upadacitinib 30 mg group, and 178 patients (63.8%) in the placebo group withdrew prematurely from the study. The main reasons for early withdrawal were AEs and “other.”
According to the additional information provided by the sponsor, the majority of patients with reasons labelled as “other” withdrew due to lack of response or loss of efficacy. The ITT_A population was a subset of patients from cohort 1 who were the first 451 responders randomized to upadacitinib 45 mg induction treatment and who were enrolled under the protocol for 52-week maintenance treatment period in cohort 1. The ITT_A population was the primary analysis population in cohort 1 for efficacy end points. The SA_C population was a subset of the patients in the safety population who had responded to upadacitinib 45 mg once daily 8-week induction treatment and who were enrolled under the protocol for a 44- or 52-week maintenance treatment period in cohort 1. The safety results of this population are presented in this review.
Cohort 2 included patients who received double-blind placebo daily for 8 weeks during U-ACHIEVE Induction substudy 1, substudy 2 part 1, or U-ACCOMPLISH, and achieved a clinical response at week 8; these patients continued to receive blinded placebo daily in substudy 3. Treatment for this cohort was not randomly assigned to the patients.
Cohort 3 included 75 patients who received upadacitinib 45 mg once daily in the induction phase and did not achieve clinical response and received upadacitinib 45 mg as extended treatment in U-ACHIEVE Induction substudy 2 part 2, or U-ACCOMPLISH part 2, and achieved clinical response at week 16; these patients were re-randomized 1:1 and received blinded upadacitinib 30 mg once daily or upadacitinib 15 mg once daily in U-ACHIEVE Maintenance (substudy 3).
Details of patient disposition in the maintenance trials are presented in Table 12.
Table 12: Patient Disposition — Maintenance Trial
Detail | U-ACHIEVE Maintenance (ITT population) | |||||
|---|---|---|---|---|---|---|
Cohort 1 | Cohort 2 | Cohort 3 | ||||
UPA 15 mg | UPA 30 mg | Placebo | Placebo | UPA 15 mg | UPA 30 mg | |
Enrolled, N (%) | 289 | 276 | 282 | 104 | 35 | 40 |
Discontinued from study, N (%) | 87 (30.4) | 52 (18.8) | 178 (63.8) | 58 (55.8) | 14 (41.2) | 8 (20.5) |
Reason for discontinuation, N (%) | ||||||
Adverse events | 13 (4.5) | 16 (5.8) | 29 (10.4) | 11 (10.6) | 1 (2.9) | 1 (2.6) |
Withdrew consent | 3 (1.0) | 7 (2.5) | 11 (3.9) | 3 (2.9) | 0 (0.0) | 3 (7.7) |
Lost to follow-up | 1 (0.3) | 1 (0.4) | 1 (0.4) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
COVID-19 infection | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
COVID-19 logistical restrictions | 0 (0.0) | 1 (0.4) | 1 (0.4) | 2 (1.9) | 0 (0.0) | 1 (2.6) |
Othera | 74 (25.9) | 32 (11.6) | 149 (53.4) | 46 (44.2) | 13 (38.2) | 5 (12.8) |
ITT, N | 286 | 276 | 279 | 104 | 34 | 39 |
ITT_A | 148 | 154 | 149 | Not shown, since results from these cohorts are not presented in this review | ||
Safety, N | Not shown since results from these cohorts are not presented in this review. | |||||
ITT = intention to treat; NR = not reported; PP = per protocol; UPA = upadacitinib.
Note: Cohort 4 received UPA 7.5 mg. As this is not a recommended dosage for the study population, the results for this cohort are not included in the current report.
The ITT population included all patients who received at least 1 dose of the study drug. The ITT_A population was a subset of the ITT population in U-ACHIEVE Maintenance comprising the first 451 (actual) responders who had been randomized to upadacitinib 45 mg once daily 8-week induction treatment and who were enrolled under the protocol for 52-week maintenance treatment period in cohort 1 of this maintenance study.
The SA_C population comprised responders to UPA 45 mg 8-week induction who were enrolled for 44- or 52-week maintenance.
aThe majority of “other” results were labelled as lack of response or loss of efficacy, as per the additional information provided by the sponsor.
Source: Clinical Study Report for U-ACHIEVE Maintenance.8
Exposure to Study Treatments
In the induction period, the mean duration of exposure to upadacitinib 45 mg and placebo in U-ACHIEVE Induction was 56 days and 53 days, respectively, for part 1, and 54 days and 54 days, respectively, for part 2. In U-ACCOMPLISH, the mean duration of exposure to upadacitinib 45 mg and placebo was 56 days and 54 days, respectively, for part 1, and 56 days and 55 days, respectively, for part 2.
In the maintenance period, the mean duration of exposure to the study drug was 295 to 320 days in patients re-randomized to upadacitinib and 214 days to patients re-randomized to placebo.
Details of extent of exposure to the study drug in the induction and maintenance trials are presented in Table 13.
Table 13: Extent of Exposure — Induction and Maintenance Trials
Detail | U-ACHIEVE Induction (SA1 and SA2 population) | U-ACCOMPLISH (SA1 and SA2 population) | U-ACHIEVE Maintenance (SA_C population) | ||||
|---|---|---|---|---|---|---|---|
UPA 45 mg (n = 319) | Placebo (n = 155) | UPA 45 mg (n = 344) | Placebo (n = 177) | UPA 15 mg (n = 250) | UPA 30 mg (n = 251) | Placebo (n = 245) | |
Days on treatment, mean (SD) | Part 1: 56.1 (7.5) Part 2: 54.0 (10.6) | Part 1: 53.1 (12.0) Part 2: 53.8 (9.5) | Part 1: 55.8 (7.0) Part 2: 55.8 (7.23) | Part 1: 54.3 (11.1) Part 2: 55.4 (7.56) | 191.0 (123.4) | 289.2 (101.4) | 191 (123.5) |
SA = safety population; SA1 = safety population in part 1; SA2 = safety population in part 2; SD = standard deviation; UPA = upadacitinib.
Note: The SA_C population comprised responders to UPA 45 mg 8-week induction who were enrolled for 44- or 52-week maintenance.
Source: Clinical Study Reports for U-ACHIEVE Induction,6 U-ACCOMPLISH,7 and U-ACHIEVE Maintenance.8
Efficacy
Only those efficacy outcomes and analyses of subgroups identified in the review protocol are reported subsequently.
Clinical Remission
In the double-blind treatment period of U-ACHIEVE Induction, clinical remission per adapted Mayo score was achieved in 26.1% of patients in the upadacitinib group and 4.8% of patients in the placebo group; the between-group difference was 21.6% (95% CI, 15.8% to 27.4%; P < 0.001). In U-ACCOMPLISH, clinical remission per adapted Mayo score was achieved in 33.5% of patients in the upadacitinib group and 4.1% of patients in the placebo group; the between-group difference was 29.0% (95% CI, 23.2% to 34.7%; P < 0.001) (Table 14).
The results of the sensitivity analyses were consistent with those in the primary analysis for clinical remission.
The results of clinical remission based on the full Mayo score were consistent with those based on the adapted Mayo score. The proportion of patients who achieved clinical remission in the induction phase was higher in the upadacitinib-treated group compared with the placebo-treated group in both the bio-IR and non-bio-IR subgroups. The efficacy results for the primary end point across all other subgroups were also consistent with the overall population (data not shown). Note that these subgroup analyses are not controlled for multiplicity.
In U-ACHIEVE Induction, among 59 patients who received upadacitinib 45 mg treatment in part 1 and did not respond, 2 (3.4%) achieved clinical remission per adapted Mayo score at week 16 with additional 8-week treatment with upadacitinib 45 mg once daily. In U-ACCOMPLISH, among 66 patients who received upadacitinib 45 mg treatment in part 1 and did not respond, 5 (7.6%) achieved clinical remission per adapted Mayo score at week 16 with additional 8-week treatment with upadacitinib 45 mg once daily.
Table 14: Efficacy Outcomes, Clinical Remission — Induction Trials, ITT1 Population
Characteristics | U-ACHIEVE Induction | U-ACCOMPLISH | ||
|---|---|---|---|---|
UPA 45 mg (N = 319) | Placebo (N = 154) | UPA 45 mg (N = 341) | Placebo (N = 174) | |
Clinical remission per adapted Mayo score at week 8 | ||||
N | 319 | 154 | 341 | 174 |
n (%) | 83 (26.1) | 7 (4.8) | 114 (33.5) | 7 (4.1) |
Adjusted between-group difference, % 95% CI | 21.6 15.8 to 27.4 | Reference | 29.0 23.2 to 34.7 | Reference |
P value | < 0.001 | Reference | < 0.001 | Reference |
Subgroups | ||||
Bio-IR | ||||
N | 51 | 19 | 46 | 29 |
n (%) | 11 (21.8) | 0 (0) | 14 (30.4) | 0 (0) |
Between-group difference, % 95% CI | 21.8 10.4 to 33.2 | Reference | 30.4 17.1 to 43.7 | Reference |
P value | 0.025a | Reference | < 0.001a | Reference |
Non-bio-IR | ||||
N | 26 | 17 | 26 | 16 |
n (%) | 11 (42.3) | 2 (11.8) | 12 (46.2) | 1 (6.3) |
Between-group difference, % 95% CI | 30.5 6.1 to 54.9 | Reference | 39.9 17.4 to 62.4 | Reference |
P value | 0.033a | Reference | 0.007a | Reference |
Clinical remission per full Mayo score at week 8 | ||||
N | 319 | 154 | 341 | 174 |
n (%) | 73 (23.0) | 4 (2.8) | 93 (27.3) | 4 (2.4) |
Adjusted between-group difference, % 95% CI | 19.9 14.7 to 25.2 | Reference | 24.4 19.2 to 29.6 | Reference |
P value | < 0.001 | Reference | < 0.001 | Reference |
bio-IR = patients with inadequate response, loss of response, or intolerance to biologic therapy; CI = confidence interval; ITT1 = intention-to-treat population in part 1; non-bio-IR = patients with inadequate response, loss of response, or intolerance to conventional therapy; UPA = upadacitinib.
Note: Adapted Mayo score = stool frequency subscore + rectal bleeding subscore + endoscopic subscore. Clinical remission per adapted Mayo score is defined as an adapted Mayo score ≤ 2, with a stool frequency subscore ≤ 1 and not greater than baseline, rectal bleeding subscore of 0, and endoscopic subscore ≤ 1.
The 95% CI for adjusted difference and P value were calculated according to the Cochran-Mantel-Haenszel test adjusted for strata (baseline corticosteroid use [yes or no], baseline adapted Mayo score [≤ 7 or > 7], bio-IR status [bio-IR or non-bio-IR]) for the comparison of 2 treatment groups. If zero frequency occurred, the zero count was replaced by 0.1 to prevent dividing by zero. Within each stratum, the 95% CI for difference was calculated based on the normal approximation to the binomial distribution and P value was calculated using a Chi-square test. The calculations were based on nonresponder imputation incorporating multiple imputation to handle missing data due to COVID-19 or nonresponder imputation if there were no missing data due to COVID-19.
aP value not controlled for multiplicity.
Source: Clinical Study Reports for U-ACHIEVE Induction6 and U-ACCOMPLISH.7
In the maintenance trial (U-ACHIEVE Maintenance), the primary and multiplicity-controlled secondary efficacy end points were analyzed based on the ITT_A population, which was defined as the subset of the ITT population comprising the first 451 (actual) responders who had been randomized to upadacitinib 45 mg once daily 8-week induction treatment and who were enrolled under the protocol for 52-week maintenance treatment period in cohort 1.
At week 52, clinical remission per adapted Mayo score was maintained in 42.3% of patients in the upadacitinib 15 mg group, 51.7% of patients in the upadacitinib 30 mg group, and 12.1% of patients in the placebo group in the ITT_A population. The between-group differences were as follows: upadacitinib 15 mg versus placebo, 30.7% (95% CI. 21.7% to 39.8%; P < 0.0001), upadacitinib 30 mg versus placebo, 39.0% (95% CI, 29.7% to 48.2%; P < 0.0001). In the ITT_B population (the subset of the ITT population in cohort 3 who were responders to upadacitinib 45 mg once daily 16-week induction treatment), clinical remission was maintained in 24 patients (33.3%) treated with upadacitinib 30 mg and 21 patients (19.0%) treated with upadacitinib 15 mg at week 52.
At week 52, in patients who were corticosteroid-free for more than 3 months, clinical remission was maintained in 57.1% of patients in the upadacitinib 15 mg group, 68.0% of patients in the upadacitinib 30 mg group, and 22.2% of patients in the placebo group. The between-group differences were as follows: upadacitinib 15 mg versus placebo, 35.4% (95% CI, 18.2% to 52.7%), upadacitinib 30 mg versus placebo, 45.1% (95% CI, 28.7% to 61.6%) (Table 15).
The results of the sensitivity analyses were consistent with those in the primary analysis for clinical remission.
The efficacy results for the primary end point across all subgroups were consistent with the overall population (data not shown aside from the bio-IR and non-bio-IR subgroups in Table 15).
Table 15: Efficacy Outcomes, Clinical Remission — Maintenance Trial, ITT_A Population
Characteristic | U-ACHIEVE Maintenance | ||
|---|---|---|---|
UPA 15 mg (N = 148) | UPA 30 mg (N = 154) | Placebo (N = 149) | |
Clinical remission per adapted Mayo score at week 52 | |||
N | 148 | 154 | 149 |
n (%) | 63 (42.3) | 80 (51.7) | 18 (12.1) |
Adjusted between-group difference, % 95% CI | 30.7 21.7 to 39.8 | 39.0 29.7 to 48.2 | Reference |
P value | < 0.001 | < 0.001 | Reference |
Subgroups | |||
Bio-IR | |||
N | 71 | 73 | 81 |
n (%) | 29 (40.5) | 36 (49.1) | 6 (7.5) |
Between-group difference, % 95% CI | 33.0 20.1 to 45.9 | 41.6 28.6 to 54.7 | Reference |
Non-bio-IR | |||
N | 77 | 81 | 68 |
n (%) | 34 (43.9) | 44 (54.0) | 12 (17.6) |
Between-group difference, % 95% CI | 26.3 11.9 to 40.6 | 36.3 22.1 to 50.6 | Reference |
Corticosteroid-free clinical remission per adapted Mayo score at week 52 among patients who achieved clinical remission at end of the induction treatment in the induction study | |||
N | 47 | 58 | 54 |
n (%) | 27 (57.1) | 39 (68.0) | 12 (22.2) |
Adjusted between-group difference, % 95% CI | 35.4 18.2 to 52.7 | 45.1 28.7 to 61.6 | Reference |
P value | < 0.001 | < 0.001 | Reference |
bio-IR = inadequate response to treatment with biologics; CI = confidence interval; ITT = intention to treat; non-bio-IR = inadequate response to conventional therapy; UPA = upadacitinib.
Note: Adapted Mayo score = stool frequency subscore + rectal bleeding subscore + endoscopic subscore. Clinical remission per adapted Mayo score is defined as an adapted Mayo score ≤ 2, with stool frequency subscore ≤ 1 and not greater than baseline, rectal bleeding subscore of 0, and endoscopic subscore ≤ 1.
The ITT_A population is defined as the first 450 randomized patients administered 8 weeks of upadacitinib 45 mg once daily induction treatment and who were enrolled under the protocol for 52-week maintenance treatment period in cohort 1.
The 95% CI for adjusted difference and P value were calculated according to the Cochran-Mantel-Haenszel test adjusted for strata (corticosteroid use at week 0 [yes or no], clinical remission status at week 0 [yes or no], bio-IR status at baseline [bio-IR or non-bio-IR]) for the comparison of 2 treatment groups. If zero frequency occurred, the zero count was replaced by 0.1 to prevent dividing by zero. Within each stratum, the 95% CI for difference was calculated based on the normal approximation to the binomial distribution and P value was calculated using a Chi-square test. The calculations were based on nonresponder imputation incorporating multiple imputation to handle missing data due to COVID-19, or nonresponder imputation if there were no missing data due to COVID-19.
Source: Clinical Study Report for U-ACHIEVE Maintenance.8
Clinical Response
Details of the results for clinical response in the induction period are shown in Table 16. In the double-blind treatment period of U-ACHIEVE Induction, clinical response per adapted Mayo score was achieved in 72.6% of patients in the upadacitinib group and 27.3% of patients in the placebo group; the between-group difference was 46.3% (95% CI, 38.4% to 54.2%; P < 0.001). In U-ACCOMPLISH, clinical response per adapted Mayo score was achieved in 74.5% of patients in the upadacitinib group and 25.4% of patients in the placebo group; the between-group difference was 49.4% (95% CI, 41.7% to 57.1%; P < 0.001).
The results of clinical response based on the partial Mayo score were consistent with that based on the adapted Mayo score.
In U-ACHIEVE Induction, among 59 patients who received upadacitinib 45 mg treatment in part 1 and did not respond, 30 (50.8%) achieved clinical response per adapted Mayo score at week 16 with additional 8-week treatment with upadacitinib 45 mg once daily. In U-ACCOMPLISH, among 66 patients who received upadacitinib 45 mg treatment in part 1 and did not respond, 30 (45.5%) achieved clinical response per adapted Mayo score at week 16 with additional 8-week treatment with upadacitinib 45 mg once daily.
Table 16: Efficacy Outcomes, Clinical Response — Induction Trials, ITT1 Population
Characteristic | U-ACHIEVE Induction | U-ACCOMPLISH | ||
|---|---|---|---|---|
UPA 45 mg (N = 319) | Placebo (N = 154) | UPA 45 mg (N = 341) | Placebo (N = 174) | |
Clinical response per adapted Mayo score at week 8 | ||||
N | 319 | 154 | 341 | 174 |
n (%) | 232 (72.6) | 42 (27.3) | 254 (74.5) | 44 (25.4) |
Adjusted between-group difference, % 95% CI | 46.3 38.4 to 54.2 | Reference | 49.4 41.7 to 57.1 | Reference |
P value | < 0.001 | Reference | < 0.001 | Reference |
Clinical response per partial Mayo score at week 8 | ||||
N | 319 | 154 | 341 | 174 |
n (%) | 192 (60.1) | 42 (27.3) | 216 (63.3) | 45 (25.9) |
Adjusted between-group difference, % 95% CI | 33.3 24.8 to 41.8 | Reference | 37.0 28.8 to 45.1 | Reference |
P value | < 0.001 | Reference | < 0.001 | Reference |
CI = confidence interval; ITT1 = intention-to-treat population in part 1; UPA = upadacitinib.
Note: The 95% CI for adjusted difference and P value were calculated according to the Cochran-Mantel-Haenszel test adjusted for strata (baseline corticosteroid use [yes or no], baseline adapted Mayo score [≤ 7 or > 7], bio-IR status [bio-IR or non-bio-IR]) for the comparison of 2 treatment groups. If zero frequency occurred, the zero count was replaced by 0.1 to prevent dividing by zero. Within each stratum, the 95% CI for difference was calculated based on the normal approximation to the binomial distribution and P value was calculated using a Chi-square test. The calculations were based on nonresponder imputation incorporating multiple imputation to handle missing data due to COVID-19, or nonresponder imputation if there were no missing data due to COVID-19.
Sources: Clinical Study Reports for U-ACHIEVE Induction6 and U-ACCOMPLISH.7
At week 52 of U-ACHIEVE Maintenance, clinical response per adapted Mayo score was maintained in 63.0% of patients in the upadacitinib 15 mg group, 76.6% of patients in the upadacitinib 30 mg group, and 18.8% of patients in the placebo group. The between-group differences were as follows: upadacitinib 15 mg versus placebo, 44.6% (95% CI, 34.5% to 54.7%; P < 0.001), upadacitinib 30 mg versus placebo, 56.6% (95% CI, 47.2% to 66.0%; P < 0.001) (Table 17).
Table 17: Efficacy Outcomes, Clinical Response — Maintenance Trial, ITT_A Population
Characteristic | U-ACHIEVE Maintenance | ||
|---|---|---|---|
UPA 15 mg (N = 148) | UPA 30 mg (N = 154) | Placebo (N = 149) | |
Clinical response per adapted Mayo score at week 52 among patients who achieved clinical response at the end of the induction treatment in the induction study | |||
N | 135 | 144 | 134 |
n (%) | 85 (63.0) | 110 (76.6) | 25 (18.8) |
Adjusted between-group difference, % 95% CI | 44.6 34.5 to 54.7 | 56.6 47.2 to 66.0 | Reference |
P value | < 0.001 | < 0.001 | Reference |
bio-IR = inadequate response to biologic therapy; CI = confidence interval; ITT = intention to treat; non-bio-IR = inadequate response to conventional therapy; UPA = upadacitinib.
Note: The 95% CI for adjusted difference and P value were calculated according to the Cochran-Mantel-Haenszel test adjusted for strata (corticosteroid use at week 0 [yes or no], clinical remission status at week 0 [yes or no], bio-IR status at baseline [bio-IR or non-bio-IR]) for the comparison of 2 treatment groups. If zero frequency occurred, the zero count was replaced by 0.1 to prevent dividing by zero. Within each stratum, the 95% CI for difference was calculated based on the normal approximation to the binomial distribution and P value was calculated using a Chi-square test. The calculations were based on nonresponder imputation incorporating multiple imputation to handle missing data due to COVID-19, or nonresponder imputation if there were no missing data due to COVID-19.
The ITT_A population was a subset of the ITT population in U-ACHIEVE Maintenance comprising the first 451 (actual) responders who had been randomized to upadacitinib 45 mg once daily 8-week induction treatment and who were enrolled under the protocol for 52-week maintenance treatment period in cohort 1 of this maintenance study.
Source: Clinical Study Report for U-ACHIEVE Maintenance.8
Endoscopic Improvement and Remission
In the double-blind treatment period of U-ACHIEVE Induction, endoscopic improvement (defined as an endoscopic subscore of 0 or 1) was achieved in 36.3% of patients in the upadacitinib group and 7.4% of patients in the placebo group; the between-group difference was 29.3% (95% CI, 22.6% to 35.9%; P < 0.001). In U-ACCOMPLISH, endoscopic improvement was achieved in 44.0% of patients in the upadacitinib group and 8.3% of patients in the placebo group; the between-group difference was 35.1% (95% CI, 28.6% to 41.6%; P < 0.001).
In U-ACHIEVE Induction, endoscopic remission (defined as an endoscopic subscore of 0) was achieved in 13.7% of patients in the upadacitinib group and 1.3% of patients in the placebo group; the between-group difference was 12.7% (95% CI, 8.4% to 17.0%; P < 0.001). In U-ACCOMPLISH, endoscopic remission was achieved in 18.2% of patients in the upadacitinib group and 1.7% of patients in the placebo group; the between-group difference was 15.9% (95% CI, 11.4% to 20.3%; P < 0.001) (Table 18).
Table 18: Efficacy Outcomes, Endoscopic Remission — Induction Trials, ITT1 Population
Characteristic | U-ACHIEVE Induction | U-ACCOMPLISH | ||
|---|---|---|---|---|
UPA 45 mg (N = 319) | Placebo (N = 154) | UPA 45 mg (N = 341) | Placebo (N = 174) | |
Endoscopic improvement at week 8 | ||||
N | 319 | 154 | 341 | 174 |
n (%) | 116 (36.3) | 11 (7.4) | 150 (44.0) | 14 (8.3) |
Adjusted between-group difference, % 95% CI | 29.3 22.6 to 35.9 | Reference | 35.1 28.6 to 41.6 | Reference |
P value | < 0.001 | Reference | < 0.001 | Reference |
Endoscopic remission at week 8 | ||||
N | 319 | 154 | 341 | 174 |
n (%) 95% CI | 44 (13.7) 9.9 to 17.6 | 2 (1.3) 0.0 to 3.1 | 62 (18.2) 14.1 to 22.3 | 3 (1.7) 0.0 to 3.7 |
Adjusted between-group difference, % 95% CI | 12.7 8.4 to 17.0 | Reference | 15.9 11.4 to 20.3 | Reference |
P value | < 0.001 | Reference | < 0.001 | Reference |
CI = confidence interval; ITT1 = intention-to-treat population in part 1; UPA = upadacitinib.
Note: The 95% CI for adjusted difference and P value were calculated according to the Cochran-Mantel-Haenszel test adjusted for strata (baseline corticosteroid use [yes or no], baseline adapted Mayo score [≤ 7 or > 7], bio-IR status [bio-IR or non-bio-IR]) for the comparison of 2 treatment groups. If zero frequency occurred, the zero count was replaced by 0.1 to prevent dividing by zero. Within each stratum, the 95% CI for difference was calculated based on the normal approximation to the binomial distribution and P value was calculated using a Chi-square test. The calculations were based on nonresponder imputation incorporating multiple imputation to handle missing data due to COVID-19, or nonresponder imputation if there were no missing data due to COVID-19.
Source: Clinical Study Reports for U-ACHIEVE Induction6 and U-ACCOMPLISH.7
At week 52 of U-ACHIEVE Maintenance, endoscopic improvement was achieved in 48.7% of patients in the upadacitinib 15 mg group, 61.6% of patients in the upadacitinib 30 mg group, and 14.5% of patients in the placebo group; the between-group differences were as follows: upadacitinib 15 mg versus placebo, 34.4% (95% CI, 25.1% to 43.7%; P < 0.001), upadacitinib 30 mg versus placebo, 46.3% (95% CI, 36.7% to 55.8%; P < 0.001).
At week 52, endoscopic remission was achieved in 24.2% of patients in the upadacitinib 15 mg group, 25.9% of patients in the upadacitinib 30 mg group, and 5.6% of patients in the placebo group; the between-group differences were as follows: upadacitinib 15 mg versus placebo, 18.7% (95% CI, 11.0% to 26.4%; P < 0.001), upadacitinib 30 mg versus placebo, 19.4% (95% CI, 11.7% to 27.2%; P < 0.001) (Table 19).
Table 19: Efficacy Outcomes, Endoscopic Improvement, and Remission — Maintenance Trial, ITT_A Population
Characteristic | U-ACHIEVE Maintenance | ||
|---|---|---|---|
UPA 15 mg (N = 148) | UPA 30 mg (N = 154) | Placebo (N = 149) | |
Endoscopic improvement at week 52 | |||
N | 148 | 154 | 149 |
n (%) | 72 (48.7) | 95 (61.6) | 22 (14.5) |
Adjusted between-group difference, % 95% CI | 34.4 25.1 to 43.7 | 46.3 36.7 to 55.8 | Reference |
P value | < 0.001 | < 0.001 | Reference |
Endoscopic remission at week 52 | |||
N | 148 | 154 | 149 |
n (%) | 36 (24.2) | 40 (25.9) | 8 (5.6) |
Adjusted between-group difference, % 95% CI | 18.7 11.0 to 26.4 | 19.4 11.7 to 27.2 | Reference |
P value | < 0.001 | < 0.001 | Reference |
bio-IR = inadequate response to biologic therapy; CI = confidence interval; ITT = intention to treat; non-bio-IR = inadequate response to conventional therapy; UPA = upadacitinib.
Note: The 95% CI for adjusted difference and P value were calculated according to the Cochran-Mantel-Haenszel test adjusted for strata (corticosteroid use at week 0 [yes or no], clinical remission status at week 0 [yes or no], bio-IR status at baseline [bio-IR or non-bio-IR]) for the comparison of 2 treatment groups. If zero frequency occurred, the zero count was replaced by 0.1 to prevent dividing by zero. Within each stratum, the 95% CI for difference was calculated based on the normal approximation to the binomial distribution and P value was calculated using a Chi-square test. The calculations were based on nonresponder imputation incorporating multiple imputation to handle missing data due to COVID-19, or nonresponder imputation if there were no missing data due to COVID-19.
The ITT_A population was a subset of the ITT population in U-ACHIEVE Maintenance comprising the first 451 (actual) responders who had been randomized to upadacitinib 45 mg once daily 8-week induction treatment and who were enrolled under the protocol for 52-week maintenance treatment period in cohort 1 of this maintenance study.
Source: Clinical Study Report for U-ACHIEVE Maintenance.8
Histologic Remission or Improvement
In the double-blind treatment period of U-ACHIEVE Induction, histologic improvement (defined as a decrease from baseline in Geboes score) was achieved in 55.0% of patients in the upadacitinib group and 22.5% of patients in the placebo group; the between-group difference was 32.2% (95% CI, 23.8% to 40.7%; P < 0.001). In U-ACCOMPLISH, histologic improvement was achieved in 62.2% of patients in the upadacitinib group and 24.5% of patients in the placebo group; the between-group difference was 37.9% (95% CI, 29.8% to 46.1%; P < 0.001) (Table 20).
Table 20: Efficacy Outcomes, Histologic Improvement — Induction Trials, ITT1 Population
Characteristic | U-ACHIEVE Induction | U-ACCOMPLISH | ||
|---|---|---|---|---|
UPA 45 mg (N = 319) | Placebo (N = 154) | UPA 45 mg (N = 341) | Placebo (N = 174) | |
Histologic improvement at week 8 | ||||
N | 319 | 154 | 341 | 174 |
n (%) | 175 (55.0) | 35 (22.5) | 212 (62.2) | 43 (24.5) |
Adjusted between-group difference, % 95% CI | 32.2 23.8 to 40.7 | Reference | 37.9 29.8 to 46.1 | Reference |
P value | < 0.001 | Reference | < 0.001 | Reference |
CI = confidence interval; ITT1 = intention-to-treat population in part 1; UPA = upadacitinib.
Note: The 95% CI for adjusted difference and P value were calculated according to the Cochran-Mantel-Haenszel test adjusted for strata (baseline corticosteroid use [yes or no], baseline adapted Mayo score [≤ 7 or > 7], bio-IR status [bio-IR or non-bio-IR]) for the comparison of 2 treatment groups. If zero frequency occurred, the zero count was replaced by 0.1 to prevent dividing by zero. Within each stratum, the 95% CI for difference was calculated based on the normal approximation to the binomial distribution and P value was calculated using a Chi-square test. The calculations were based on nonresponder imputation incorporating multiple imputation to handle missing data due to COVID-19, or nonresponder imputation if there were no missing data due to COVID-19.
Source: Clinical Study Reports for U-ACHIEVE Induction6 and U-ACCOMPLISH.7
At week 52 of U-ACHIEVE Maintenance, histologic improvement was achieved in 42.8% of patients in the upadacitinib 15 mg group, 56.9% of patients in the upadacitinib 30 mg group, and 20.6% of patients in the placebo group; the between-group differences were as follows: upadacitinib 15 mg versus placebo, 23.0% (95% CI, 12.9% to 33.1%), upadacitinib 30 mg versus placebo, 36.0% (95% CI, 25.8% to 46.2%) (Table 21).
Table 21: Efficacy Outcomes, Histologic Improvement — Maintenance Trial, ITT_A Population
Characteristic | U-ACHIEVE Maintenance | ||
|---|---|---|---|
UPA 15 mg (N = 148) | UPA 30 mg (N = 154) | Placebo (N = 149) | |
Histologic improvement at week 52 | |||
N | 148 | 154 | 149 |
n (%) | 63 (42.8) | 88 (56.9) | 31 (20.6) |
Adjusted between-group difference, % 95% CI | 23.0 12.9 to 33.1 | 36.0 25.8 to 46.2 | Reference |
P value | < 0.001 | < 0.001 | Reference |
bio-IR = inadequate response to biologic therapy; CI = confidence interval; ITT = intention-to-treat; non-bio-IR = inadequate response to conventional therapy; UPA = upadacitinib.
Note: The 95% CI for adjusted difference and P value were calculated according to the Cochran-Mantel-Haenszel test adjusted for strata (corticosteroid use at week 0 [yes or no], clinical remission status at week 0 [yes or no], bio-IR status at baseline [bio-IR or non-bio-IR]) for the comparison of 2 treatment groups. If zero frequency occurred, the zero count was replaced by 0.1 to prevent dividing by zero. Within each stratum, the 95% CI for difference was calculated based on the normal approximation to the binomial distribution and P value was calculated using a Chi-square test. The calculations were based on nonresponder imputation incorporating multiple imputation to handle missing data due to COVID-19, or nonresponder imputation if there were no missing data due to COVID-19.
The ITT_A population was a subset of the ITT population in U-ACHIEVE Maintenance comprising the first 451 (actual) responders who had been randomized to upadacitinib 45 mg once daily 8-week induction treatment and who were enrolled under the protocol for 52-week maintenance treatment period in cohort 1 of this maintenance study.
This outcome was outside of the statistical hierarchy for multiplicity adjustment.
Source: Clinical Study Report for U-ACHIEVE Maintenance.8
Mucosal Healing
In U-ACHIEVE Induction, mucosal healing (defined as an endoscopic subscore of 0 and Geboes score < 2) was achieved in 10.7% of patients in the upadacitinib group and 1.3% of patients in the placebo group; the between-group difference was 9.7% (95% CI, 5.7% to 13.7%). In U-ACCOMPLISH, mucosal healing was achieved in 13.5% of patients in the upadacitinib group and 1.7% of patients in the placebo group; the between-group difference was 11.3% (95% CI, 7.2% to 15.3%; P < 0.001) (Table 22).
At week 52, mucosal healing was achieved in 17.6% of patients in the upadacitinib 15 mg group, 19.0% of patients in the upadacitinib 30 mg group, and 4.7% of patients in the placebo group; the between-group differences were as follows: upadacitinib 15 mg versus placebo, 13.0% (95% CI, 6.0% to 20.0%; P < 0.001), upadacitinib 30 mg versus placebo, 13.6% (95% CI, 6.6% to 20.6%; P < 0.001) (Table 23).
Symptoms Relief
At the end of the induction period of U-ACHIEVE, the proportion of patients who reported no bowel urgency was higher in the upadacitinib group (48.4%) compared with those in the placebo group (21.4%); the between-group difference was 27.4% (95% CI, 19.2% to 35.6%; P < 0.001). In U-ACCOMPLISH, the proportion of patients who reported no bowel urgency was higher in the upadacitinib group (53.7%) compared with those in the placebo group (25.9%); the between-group difference was 27.1% (95% CI, 19.0 to 35.3%; P < 0.001).
Table 22: Efficacy Outcomes, Mucosal Healing — Induction Trials, ITT1 Population
Characteristic | U-ACHIEVE Induction | U-ACCOMPLISH | ||
|---|---|---|---|---|
UPA 45 mg (N = 319) | Placebo (N = 154) | UPA 45 mg (N = 341) | Placebo (N = 174) | |
Mucosal healinga at week 8 | ||||
N | 319 | 154 | 341 | 174 |
n (%) | 34 (10.7) | 2 (1.3) | 46 (13.5) | 3 (1.7) |
Adjusted between-group difference, % 95% CI | 9.7 5.7 to 13.7 | Reference | 11.3 7.2 to 15.3 | Reference |
P value | < 0.001 | Reference | < 0.001 | Reference |
CI = confidence interval; ITT1 = intention-to-treat population in part 1; UPA = upadacitinib.
Note: The 95% CI for adjusted difference and P value were calculated according to the Cochran-Mantel-Haenszel test adjusted for strata (baseline corticosteroid use [yes or no], baseline adapted Mayo score [≤ 7 or > 7], bio-IR status [bio-IR or non-bio-IR]) for the comparison of 2 treatment groups. If zero frequency occurred, the zero count was replaced by 0.1 to prevent dividing by zero. Within each stratum, the 95% CI for difference was calculated based on the normal approximation to the binomial distribution and P value was calculated using a Chi-square test. The calculations were based on nonresponder imputation incorporating multiple imputation to handle missing data due to COVID-19, or nonresponder imputation if there were no missing data due to COVID-19.
aMucosal healing was defined as endoscopic subscore of 0 and Geboes score < 2.
Source: Clinical Study Reports for U-ACHIEVE Induction6 and U-ACCOMPLISH.7
Table 23: Efficacy Outcomes, Mucosal Healing — Maintenance Trial, ITT_A Population
Characteristic | U-ACHIEVE Maintenance | ||
|---|---|---|---|
UPA 15 mg (N = 148) | UPA 30 mg (N = 154) | Placebo (N = 149) | |
Mucosal healing at week 52 | |||
N | 148 | 154 | 149 |
n (%) | 26 (17.6) | 29 (19.0) | 7 (4.7) |
Adjusted between-group difference, % 95% CI | 13.0 6.0 to 20.0 | 13.6 6.6 to 20.6 | Reference |
P value | < 0.001 | < 0.001 | Reference |
CI = confidence interval; ITT = intention to treat; UPA = upadacitinib.
Note: The 95% CI for adjusted difference and P value were calculated according to the Cochran-Mantel-Haenszel test adjusted for strata (baseline corticosteroid use [yes or no], baseline adapted Mayo score [≤ 7 or > 7], bio-IR status [bio-IR or non-bio-IR]) for the comparison of 2 treatment groups. If zero frequency occurred, the zero count was replaced by 0.1 to prevent dividing by zero. Within each stratum, the 95% CI for difference was calculated based on the normal approximation to the binomial distribution and P value was calculated using a Chi-square test. The calculations were based on nonresponder imputation incorporating multiple imputation to handle missing data due to COVID-19, or nonresponder imputation if there were no missing data due to COVID-19.
The ITT_A population was a subset of the ITT population in U-ACHIEVE Maintenance comprising the first 451 (actual) responders who had been randomized to upadacitinib 45 mg once daily 8-week induction treatment and who were enrolled under the protocol for 52-week maintenance treatment period in cohort 1 of this maintenance study.
Source: Clinical Study Report for U-ACHIEVE Maintenance.8
At the end of the induction period of U-ACHIEVE, the proportion of patients who reported no abdominal pain was higher in the upadacitinib group (46.6%) compared with those in the placebo group (23.4%); the between-group difference was 23.6% (95% CI, 15.1% to 32.1%; P < 0.001). In U-ACCOMPLISH, the proportion of patients who reported no abdominal pain was higher in the upadacitinib group (53.7%) compared with those in the placebo group (24.1%); the between-group difference was 29.1% (95% CI, 20.9% to 37.4%; P < 0.001).
In the 2 induction trials, patients treated with upadacitinib 45 mg reported fewer stools per day than those treated with placebo at week 8 compared with baseline. More patients in the upadacitinib 45 mg group reported no rectal bleeding at week 8 compared with those in the placebo group. Patients in the upadacitinib group reported higher FACIT-F scores than those in the placebo group, which indicate less fatigue (Table 24).
At week 52 of U-ACHIEVE Maintenance, the proportion of patients who reported no bowel urgency was 56.1% in the upadacitinib 15 mg group, 63.6% in the upadacitinib 30 mg group, and 17.4% in the placebo group; the between-group differences were as follows: upadacitinib 15 mg versus placebo, 38.7% (95% CI, 28.9% to 48.5%; P < 0.001), upadacitinib 30 mg versus placebo, 45.1% (95% CI, 35.5% to 54.8%; P < 0.001).
At week 52, the proportion of patients who reported no abdominal pain was 45.9% in the upadacitinib 15 mg group, 55.3% in the upadacitinib 30 mg group, and 20.8% in the placebo group; the between-group differences were as follows: upadacitinib 15 mg versus placebo, 24.3% (95% CI, 14.2 to 34.5%; P < 0.001), upadacitinib 30 mg versus placebo, 33.7% (95% CI, 23.6% to 43.9%; P < 0.001).
In the maintenance trial, patients treated with upadacitinib 15 mg or 30 mg reported fewer stools per day than those treated with placebo at week 52 compared with baseline. More patients in the upadacitinib groups reported no rectal bleeding at week 52 compared with those in the placebo group. Patients in the upadacitinib groups reported higher FACIT-F scores than those in the placebo group at week 52, which indicate less fatigue (Table 25).
Table 24: Efficacy Outcomes, Symptom Relief — Induction Trials, ITT1 Population
Characteristic | U-ACHIEVE Induction | U-ACCOMPLISH | ||
|---|---|---|---|---|
UPA 45 mg (N = 319) | Placebo (N = 154) | UPA 45 mg (N = 341) | Placebo (N = 174) | |
No reported bowel urgency at week 8 | ||||
N | 319 | 154 | 341 | 174 |
n (%) | 155 (48.4) | 33 (21.4) | 183 (53.7) | 45 (25.9) |
Adjusted between-group difference, % 95% CI | 27.4 19.2 to 35.6 | Reference | 27.1 19.0 to 35.3 | Reference |
P value | < 0.001 | Reference | < 0.001 | Reference |
No reported abdominal pain at week 8 | ||||
N | 319 | 154 | 341 | 174 |
n (%) | 149 (46.6) | 36 (23.4) | 183 (53.7) | 42 (24.1) |
Adjusted between-group difference, % 95% CI | 23.6 15.1 to 32.1 | Reference | 29.1 20.9 to 37.4 | Reference |
P value | < 0.001 | Reference | < 0.001 | Reference |
Patient-reported stool frequency (absolute values) at week 8 | ||||
N | 293 | 128 | 329 | 160 |
Baseline | 7.96 | 7.39 | 7.67 | 7.90 |
Week 8 | 3.29 | 5.52 | 3.32 | 6.17 |
Change from baseline | −4.63 | −1.93 | −4.32 | −1.60 |
Adjusted between-group difference, % 95% CI | −2.71 −3.21 to −2.20 | Reference | −2.72,−3.22 to −2.22 | Reference |
P value | < 0.001 | Reference | < 0.001 | Reference |
% of patients with RBS 0 at week 8 | ||||
N | 319 | 154 | 341 | 174 |
n (%) | 234 (73.4) | 43 (27.9) | 235 (68.9) | 57 (32.8) |
Adjusted between-group difference, % 95% CI | 45.6 37.3 to 53.9 | Reference | 36.1 27.7 to 44.4 | Reference |
P value | < 0.001 | Reference | < 0.001 | Reference |
Change from baseline in FACIT-F score at week 8 | ||||
N | 291 | 125 | 312 | 155 |
Baseline | 30.8 | 31.4 | 29.8 | 32.1 |
Week 8 | 9.5 | 2.8 | 9.4 | 3.5 |
Adjusted between-group difference, LS mean 95% CI | 6.7 4.8 to 8.6 | Reference | 6.0 4.19 to 7.73 | Reference |
P value | < 0.001 | Reference | < 0.001 | Reference |
bio-IR = inadequate response to biologic therapy; CI = confidence interval; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; ITT = intention to treat; ITT1 = intention-to-treat population in part 1; LS = least squares; MMRM = mixed-model for repeated measures; non-bio-IR = inadequate response to conventional therapy; RBS = rectal bleeding subscore; UPA = upadacitinib.
Note: For bowel urgency, abdominal pain, and rectal bleeding, the 95% CI for adjusted difference and P value were calculated according to the Cochran-Mantel-Haenszel test adjusted for strata (baseline corticosteroid use [yes or no], baseline adapted Mayo score [≤ 7 or > 7], bio-IR status [bio-IR or non-bio-IR]) for the comparison of 2 treatment groups. If zero frequency occurred, the zero count was replaced by 0.1 to prevent dividing by zero. Within each stratum, the 95% CI for difference was calculated based on the normal approximation to the binomial distribution and P value was calculated using a Chi-square test. The calculations were based on nonresponder imputation incorporating multiple imputation to handle missing data due to COVID-19, or nonresponder imputation if there were no missing data due to COVID-19. Note that “rectal bleeding” was outside of the statistical hierarchy for multiplicity adjustment.
Change in stool frequency and FACIT-F score: MMRM was the mixed-effect model repeat measurement with baseline, treatment, visit, treatment-by-visit interaction, and strata (baseline corticosteroid use [yes or no], baseline adapted Mayo score [≤ 7 or > 7], bio-IR status [bio-IR or non-bio-IR]) in the model. Baseline was defined as the last nonmissing value before the first dose of study drug. Patients with only nonmissing change from baseline values were included in analysis. This outcome was outside of the statistical hierarchy for multiplicity adjustment. Note that this outcome was outside of the statistical hierarchy for multiplicity adjustment.
Sources: Clinical Study Reports for U-ACHIEVE Induction6 and U-ACCOMPLISH.7
Table 25: Efficacy Outcomes, Symptom Relief — Maintenance Trial, ITT_A Population
Characteristic | U-ACHIEVE Maintenance | ||
|---|---|---|---|
UPA 15 mg (N = 148) | UPA 30 mg (N = 154) | Placebo (N = 149) | |
No reported bowel urgency at week 52 | |||
N | 148 | 154 | 149 |
n (%) | 83 (56.1) | 98 (63.6) | 26 (17.4) |
Adjusted between-group difference, % 95% CI | 38.7 28.9 to 48.5 | 45.1 35.5 to 54.8 | Reference |
P value | < 0.001 | < 0.001 | Reference |
No reported abdominal pain at week 52 | |||
N | 148 | 154 | 149 |
n (%) | 68 (45.9) | 85 (55.3) | 31 (20.8) |
Adjusted between-group difference, % 95% CI | 24.3 14.2 to 34.5 | 33.7 23.6 to 43.9 | Reference |
P value | < 0.001 | < 0.001 | Reference |
Patient-reported stool frequency (absolute values) at week 52 | |||
N | 148 | 154 | 149 |
Baseline | 7.39 | 7.55 | 7.85 |
Week 52 | 3.30 | 2.43 | 5.92 |
Change from baseline | −4.02 | −5.07 | −1.34 |
Adjusted between-group difference, % 95% CI | −2.68 −3.58 to −1.78 | −3.73 −4.76 to −2.70 | Reference |
P value | < 0.001 | < 0.001 | Reference |
% of patients with RBS 0 at week 52 | |||
N | 148 | 154 | 149 |
n (%) | 95 (64.2) | 113 (73.2) | 32 (21.7) |
Adjusted between-group difference, % 95% CI | 42.4 32.5 to 52.4 | 50.6 41.1 to 60.2 | Reference |
P value | < 0.001 | < 0.001 | Reference |
Change from baseline in FACIT-F score at week 52 | |||
N | 148 | 154 | 149 |
Baseline | 31.3 | 29.9 | 30.2 |
Week 52 | 8.7 | 9.5 | 3.7 |
Adjusted between-group difference, LS mean 95% CI | 5.1 2.67 to 7.52 | 5.9 3.44 to 8.27 | Reference |
P value | < 0.001 | < 0.001 | Reference |
bio-IR = inadequate response to biologic therapy; CI = confidence interval; FACIT-F = Functional Assessment of Chronic Illness Therapy–Fatigue; ITT = intention to treat; LS = least squares; non-bio-IR = inadequate response to conventional therapy; RBS = rectal bleeding subscore; UC = ulcerative colitis; UPA = upadacitinib.
Note: For bowel urgency, abdominal pain, and rectal bleeding, the 95% CI for adjusted difference and P value were calculated according to the Cochran-Mantel-Haenszel test adjusted for strata (baseline corticosteroid use [yes or no], baseline adapted Mayo score [≤ 7 or > 7], bio-IR status [bio-IR or non-bio-IR]) for the comparison of 2 treatment groups. If zero frequency occurred, the zero count was replaced by 0.1 to prevent dividing by zero. Within each stratum, the 95% CI for difference was calculated based on the normal approximation to the binomial distribution and P value was calculated using a Chi-square test. The calculations were based on nonresponder imputation incorporating multiple imputation to handle missing data due to COVID-19, or nonresponder imputation if there were no missing data due to COVID-19.
The LS mean, 95% CI, and standard error were the synthetic result based on mixed-model for repeated measures or analysis of covariance with baseline, week 0, treatment, and strata (corticosteroid use at week 0 [yes or no], clinical remission status at week 0 [yes or no], bio-IR status at baseline [bio-IR or nonbio-IR]), and visit and treatment-by-visit interaction in the model from PROC MIANALYZE procedure. Baseline was defined as the last nonmissing value before the first dose in the phase IIb induction or in the induction studies. Patients with only nonmissing change from baseline values were included in the analysis.
The ITT_A population was a subset of the ITT population in U-ACHIEVE Maintenance comprising the first 451 (actual) responders who had been randomized to upadacitinib 45 mg once daily 8-week induction treatment and who were enrolled under the protocol for 52-week maintenance treatment period in cohort 1 of this maintenance study.
Source: Clinical Study Report for U-ACHIEVE Maintenance.8
Health-Related Quality of Life
At week 8, the between-group findings for change from baseline for the IBDQ total score favoured upadacitinib in both U-ACHIEVE Induction and U-ACCOMPLISH. In U-ACHIEVE Induction, the IBDQ total score was 33.7 points higher in the upadacitinib 45 mg group compared with the placebo group. In U-ACCOMPLISH, this score was 31.2 points higher in the upadacitinib group compared with placebo.
Results of the EQ-5D-5L index score, SF-36 Physical Component Summary and Mental Component Summary scores, and the Ulcerative Colitis Symptoms Questionnaire (UC-SQ) score showed more changes from baseline in the upadacitinib group compared with the placebo group at week 8 in the 2 induction trials. However, the outcomes of the EQ-5D-5L index score, SF-36 Physical Component Summary and Mental Component Summary scores, and the UC-SQ score were unadjusted for multiple comparisons.
Details of the results for each HRQoL instrument are provided in Table 26.
Table 26: Efficacy Outcomes, HRQoL — Induction Trials, ITT1 Population
Characteristic | U-ACHIEVE Induction | U-ACCOMPLISH | ||
|---|---|---|---|---|
UPA 45 mg (N = 319) | Placebo (N = 154) | UPA 45 mg (N = 341) | Placebo (N = 174) | |
Change from baseline in IBDQ total score at week 8 | ||||
N | 292 | 125 | 315 | 156 |
Week 8 | 55.3 | 21.7 | 52.2 | 21.1 |
Adjusted between-group difference, LS mean 95% CI | 33.7 27.0 to 40.4 | Reference | 31.2 24.98 to 37.36 | Reference |
P value | < 0.001 | Reference | < 0.001 | Reference |
Change from baseline in EQ-5D-5La index score at week 8 | ||||
N | 291 | 125 | 314 | 157 |
Baseline score | 0.693 | 0.658 | 0.677 | 0.687 |
Week 8 | 0,829 | 0.716 | 0.779 | 0.698 |
Change from baseline to week 8 | 0.142 | 0.045 | 0.097 | 0.017 |
Adjusted between-group difference, LS mean 95% CI | 0.097 0.0638 to 0.1306 | Reference | 0.080 0.0510 to 0.1092 | Reference |
P value | < 0.001 | Reference | < 0.001 | Reference |
Change from baseline in SF-36 PCSa at week 8 | ||||
N | 291 | 125 | 313 | 156 |
Baseline score | 43.666 | 42.793 | 42.374 | 43.935 |
Score at week 8 | 50.948 | 45.339 | 50.243 | 46.469 |
Change from baseline to week 8 | 7.374 | 2.283 | 7.594 | 2.882 |
Adjusted between-group difference, LS mean 95% CI | 5.091 3.8456 to 6.3369 | Reference | 4.713 3.5280 to 5.8974 | Reference |
P value | < 0.001 | Reference | < 0.001 | Reference |
Change from baseline in SF-36 MCSa at week 8 | ||||
N | 291 | 125 | 313 | 156 |
Baseline score | 40.395 | 40.901 | 40.905 | 40.690 |
Score at week 8 | 48.579 | 43.528 | 48.383 | 43.705 |
Change from baseline to week 8 | 8.331 | 2.397 | 7.302 | 2.745 |
Adjusted between-group difference, LS mean 95% CI | 5.934 4.1709 to 7.6969 | Reference | 4.557 2.9440 to 6.1707 | Reference |
P value | < 0.001 | Reference | < 0.001 | Reference |
Change from baseline in WPAIa overall work impairment score at week 8 | ||||
N | 180 | 70 | 166 | 89 |
Baseline score | 51.03 | 53.41 | 55.16 | 49.02 |
Score at week 8 | 23.72 | 46.50 | 25.42 | 36.50 |
Change from baseline to week 8 | −26.74 | −4.54 | −27.34 | −14.10 |
Adjusted between-group difference, LS mean 95% CI | −22.19 −28.954 to −15.434 | Reference | −13.24 −19.974 to −6.504 | Reference |
P value | < 0.001 | Reference | < 0.001 | Reference |
Change from baseline in UC-SQa score at week 8 | ||||
N | 287 | 124 | 312 | 155 |
Baseline score | 31.6 | 30.3 | 32.3 | 30.9 |
Score at week 8 | 13.1 | 23.1 | 13.5 | 23.5 |
Change from baseline to week 8 | −18.5 | −7.5 | −18.5 | −7.7 |
Adjusted between-group difference, LS mean 95% CI | −10.9 −13.06 to −8.84 | Reference | −10.7 −12.64 to −8.82 | Reference |
P value | < 0.001 | Reference | < 0.001 | Reference |
bio-IR = inadequate response to biologic therapy; CI = confidence interval; EQ-5D-5L = 5-level EQ-5D; HRQoL = health-related quality of life; IBDQ = Inflammatory Bowel Disease Questionnaire; ITT1 = intention-to-treat population for part 1; LS = least squares; MCS = Mental Component Summary; MMRM = mixed-model for repeated measures; non-bio-IR = inadequate response to conventional therapy; PCS = Physical Component Summary; SF-36 = Short Form (36) Health Survey; UC-SQ = Ulcerative Colitis Symptoms Questionnaire; UPA = upadacitinib; WPAI = Work Performance and Activity Impairment.
Note: IBDQ total score, EQ-5D index score, SF-36 PCS and MCS scores, WPAI scores, and UC-SQ score — MMRM is the mixed-effect model repeat measurement with baseline, treatment, visit, treatment-by-visit interaction, and strata (baseline corticosteroid use [yes or no], baseline adapted Mayo score [≤ 7 or > 7], bio-IR status [bio-IR or non-bio-IR]) in the model. Baseline was defined as the last nonmissing value before the first dose of the study drug. Patients with only nonmissing change from baseline values were included in the analysis.
aThese outcomes were unadjusted for multiple comparisons.
Sources: Clinical Study Reports for U-ACHIEVE Induction6 and U-ACCOMPLISH.7
HRQoL analyses were performed in the ITT_A population in the maintenance trial. At week 52, the between-group findings for change from baseline for the IBDQ total score favoured upadacitinib (Table 27).
The results of the UC-SQ score showed more changes from baseline in the upadacitinib groups compared with the placebo group at week 52; however, the UC-SQ score was unadjusted for multiple comparisons.
Table 27: Efficacy Outcomes, HRQoL — Maintenance Trial, ITT_A Population
Characteristic | U-ACHIEVE Maintenance | ||
|---|---|---|---|
UPA 15 mg (N = 148) | UPA 30 mg (N = 154) | Placebo (N = 149) | |
Change from baseline in IBDQ total score at week 52 | |||
N | 148 | 154 | 149 |
Mean | 49.2 | 58.9 | 17.9 |
Adjusted between-group difference, LS mean 95% CI | 31.3 21.98 to 40.70 | 41.0 31.39 to 50.55 | Reference |
P value | < 0.001 | < 0.001 | Reference |
Change in WPAI overall work impairment scorea from baseline at week 52 | |||
N | 148 | 154 | 149 |
Baseline | 50.25 | 55.50 | 53.53 |
Week 52 | 25.85 | 24.50 | 40.00 |
Change from baseline to week 52, LS mean 95% CI | −25.31 −30.472 to −20.155 | −26.26 −31.158 to −21.369 | −12.63 −18.456 to −6.805 |
Adjusted between-group difference, LS mean 95% CI | −12.68 −20.126 to −5.242 | −13.63 −20.966 to −6.300 | Reference |
P value | < 0.001 | < 0.001 | Reference |
Change in UC-SQ scorea from baseline at week 52 | |||
N | 148 | 154 | 149 |
Baseline | 31.3 | 32.0 | 32.6 |
Week 52 | 15.0 | 12.0 | 25.6 |
Change from baseline to week 52, LS mean 95% CI | −15.8 −18.02 to −13.54 | −19.0 −21.10 to −16.84 | −6.0 −8.43 to −3.63 |
Adjusted between-group difference, LS mean 95% CI | −9.7 −12.89 to −6.60 | −12.9 −16.02 to −9.86 | Reference |
P value | < 0.001 | < 0.001 | Reference |
bio-IR = inadequate response to biologic therapy; CI = confidence interval; HRQoL = health-related quality of life; IBDQ = Inflammatory Bowel Disease Questionnaire; LS = least squares; non-bio-IR = inadequate response to conventional therapy; SE = standard error; UC-SQ = Ulcerative Colitis Symptoms Questionnaire; UPA = upadacitinib; WPAI = Work Performance and Activity Impairment.
Note: IBDQ total score, WPAI score, and UC-SQ score The LS mean, 95% CI, and SE were the synthetic result based on analysis of covariance with baseline, week 0, treatment, and strata (corticosteroid use at week 0 [yes or no], clinical remission status at week 0 [yes or no], bio-IR status at baseline [bio-IR or non-bio-IR]) in the model from PROC MIANALYZE procedure. Baseline was defined as the last nonmissing value before the first dose in phase IIb Induction or Induction Studies. Patients with only nonmissing change from baseline values were included in the analysis.
The ITT_A population is defined as the first 450 patients randomized and administered 8-week upadacitinib 45 mg once daily induction treatment who were enrolled under the protocol for 52-week maintenance treatment period in cohort 1.
aThese outcomes were unadjusted for multiple comparisons.
Source: Clinical Study Report for U-ACHIEVE Maintenance.8
Need for Colectomy
Not assessed in the included studies.
Extraintestinal Manifestations
Not assessed in the included studies.
Emergency Department Visits or Hospitalization
At the end of the induction period of U-ACHIEVE, the proportion of patients with UC-related hospitalization was lower in the upadacitinib group (0.6%) compared with those in the placebo group (3.9%); the between-group difference was −3.5% (95% CI, −6.8% to −0.2%; P = 0.037). In U-ACCOMPLISH, the proportion of patients with UC-related hospitalization was similar in the upadacitinib group (2.1%) compared with those in the placebo group (2.3%); the between-group difference was −0.3% (95% CI, −3.2% to 2.6%, P > 0.05) (Table 28).
Table 28: Efficacy Outcomes, UC-Related Hospitalization — Induction Trials, ITT1 Population
Characteristic | U-ACHIEVE Induction | U-ACCOMPLISH | ||
|---|---|---|---|---|
UPA 45 mg (N = 319) | Placebo (N = 154) | UPA 45 mg (N = 341) | Placebo (N = 174) | |
Proportion of patients with UC-related hospitalizations through week 8 | ||||
N | 319 | 154 | 341 | 174 |
n (%) | 2 (0.6) | 6 (3.9) | 7 (2.1) | 4 (2.3) |
Adjusted between-group difference, % 95% CI | −3.5 −6.8 to −0.2 | Reference | −0.3 −3.2 to 2.6 | Reference |
P value | 0.037 | Reference | 0.817 | Reference |
bio-IR = inadequate response to biologic therapy; CI = confidence interval; ITT1 = intention-to-treat population in part 1; non-bio-IR = inadequate response to conventional therapy; UC = ulcerative colitis; UPA = upadacitinib.
Note: This outcome was not adjusted for multiplicity. The 95% CI for response rate was based on the normal approximation to the binomial distribution. The 95% CI for adjusted difference and P value were calculated according to the Cochran-Mantel-Haenszel test adjusted for strata (baseline corticosteroid use [yes or no], baseline adapted Mayo score [≤ 7 or > 7], bio-IR status [bio-IR or non-bio-IR]) for the comparison of 2 treatment groups. If zero frequency occurred, the zero count was replaced by 0.1 to prevent dividing by zero.
Source: Clinical Study Reports for U-ACHIEVE Induction6 and U-ACCOMPLISH.7
At week 52 of U-ACHIEVE Maintenance, the incidence rate of UC-related hospitalization was 0.76 in the upadacitinib 15 mg group, 1.36 in the upadacitinib 30 mg group, and 7.13 in the placebo group; the between-group differences were as follows: upadacitinib 15 mg versus placebo, 38.7% (95% CI, 28.9% to 48.5%; P < 0.001), upadacitinib 30 mg versus placebo, 45.1% (95% CI, 35.5% to 54.8%; P < 0.001) (Table 29).
The outcome of UC-related hospitalization was not included in the statistical hierarchy for multiplicity adjustment in these 3 studies.
Table 29: Efficacy Outcomes, UC-Related Hospitalization — Maintenance Trial, ITT_A Population
Characteristic | U-ACHIEVE Maintenance | ||
|---|---|---|---|
UPA 15 mg (N = 148) | UPA 30 mg (N = 154) | Placebo (N = 149) | |
Incidence rate UC-related hospitalizations through week 52 | |||
N | 148 | 154 | 149 |
N (incidence rate) | 1 (0.76) | 2 (1.36) | 7 (7.13) |
Adjusted between-group difference, % 95% CI | −6.36 −11.85 to −0.88 | −5.77 −11.37 to −0.16 | Reference |
P value | 0.023 | 0.044 | Reference |
CI = confidence interval; ITT = intention to treat; UC = ulcerative colitis; UPA = upadacitinib.
Note: The 95% CI for incidence rate difference and P value were based on the normal approximation to poisson distribution.
The ITT_A population was a subset of the ITT population in U-ACHIEVE Maintenance comprising the first 451 (actual) responders who had been randomized to upadacitinib 45 mg once daily 8-week induction treatment and who were enrolled under the protocol for 52-week maintenance treatment period in cohort 1 of this maintenance study.
Source: Clinical Study Report for U-ACHIEVE Maintenance.8
Work Productivity
Work productivity was assessed using the WPAI-UC.
In U-ACHIEVE Induction, patients in the upadacitinib group showed a greater reduction in the overall work impairment score compared with the placebo group (upadacitinib = −26.7; placebo = −4.5) from baseline at week 8. In U-ACCOMPLISH, patients in the upadacitinib group showed a greater reduction in the overall work impairment score (upadacitinib = −27.3; placebo = −14.1) compared with the placebo group.
In U-ACHIEVE Maintenance, at week 52, patients in the 2 upadacitinib groups reported a greater reduction from baseline in the overall work impairment score compared with the placebo group (upadacitinib 15 mg = −25.3; upadacitinib 30 mg = −26.26; placebo = −12.63).
The WPAI overall work impairment score was unadjusted for multiple comparisons.
Results of change in work productivity are shown in Table 26 and Table 27.
Harms
Only those harms identified in the review protocol are reported subsequently. See Table 30 and Table 31 for detailed harms data.
Adverse Events
In U-ACHIEVE Induction, at least 1 treatment-emergent AE was reported by 56.4% and 61.9% of patients in the upadacitinib group and the placebo group, respectively. In U-ACCOMPLISH, at least 1 treatment-emergent AE was reported by 52.9% and 39.5% of patients in the upadacitinib group and the placebo group, respectively.
In these 2 studies, the most commonly reported AEs in the upadacitinib groups were blood creatine phosphokinase increased (4.7% to 5% in part 1; 10.2% to 10.3% in part 2), acne (4.7% to 7.0% in part 1; 3.4% to 8.8% in part 2), nasopharyngitis (3.8% to 4.7% in part 1; 2.9% to 8.5% in part 2), headache (2.3% to 4.1% in part 1; 5.1% to 5.9% in part 2), and anemia (2.5% to 4.1% in part 1; 5.1% to 13.2% in part 2). The most commonly reported AEs in the placebo groups were nasopharyngitis (2.3% to 3.9% in part 1), anemia (2.3% to 5.8% in part 1), and UC (4.5% to 13.5% in part 1).
During the maintenance period, AEs were reported in 75.2%, 75.3%, and 73.5% of the patients in the upadacitinib 15 mg, upadacitinib 30 mg, and placebo groups, respectively. Commonly reported AEs were nasopharyngitis (8.2% to 10.4%), blood creatine phosphokinase increased (2.0% to 7.6%), UC (7.2% to 29.8%), arthralgia (2.8% to 9.8%), and herpes zoster (0% to 4.0%).
Serious Adverse Events
In U-ACHIEVE Induction, at least 1 SAE was reported by 2.5% and 5.8% of patients in the upadacitinib group and the placebo group, respectively. In U-ACCOMPLISH, at least 1 SAE was reported by 3.2% and 4.5% of patients in the upadacitinib group and the placebo group, respectively. In the upadacitinib group, isolated cases of SAEs were reported. UC was the most commonly reported SAE in the placebo group (1.7% to 3.2% in part 1).
During the maintenance period, at least 1 SAE was reported in 8.0%, 8.0%, and 9.4% of the patients in the upadacitinib 15 mg group, upadacitinib 30 mg group, and placebo group, respectively. Isolated cases of SAEs were reported in this period. Most of the SAEs occurred in less than 1% of the study population.
Withdrawal Due to Adverse Events
In U-ACHIEVE Induction, WDAEs were reported by 1.9% and 9.0% of patients in the upadacitinib group and the placebo group, respectively. In U-ACCOMPLISH, WDAEs were reported by 1.7% and 5.1% of patients in the upadacitinib group and the placebo group, respectively. UC was the most commonly reported reason for a WDAE in the placebo group (2.8% to 7.1% in part 1; 0 in part 2).
During the maintenance period, WDAEs were reported in 4.0%, 4.8%, and 10.2% of the patients in the upadacitinib 15 mg group, upadacitinib 30 mg group, and placebo group, respectively.
Mortality
No deaths were reported during the induction and maintenance periods of the included studies.
Notable Harms
During the induction period in U-ACHIEVE Induction and U-ACCOMPLISH, there were no AEs of active tuberculosis, malignancy, malignancy excluding nonmelanoma skin cancer, nonmelanoma skin cancer, adjudicated VTE, and gastrointestinal perforation reported in the upadacitinib groups. The incidence of opportunistic infection, excluding tuberculosis and herpes zoster, herpes zoster, lymphopenia, and neutropenia, was higher in the upadacitinib groups.
During the maintenance period, patients treated with upadacitinib were more likely to report herpes zoster, neutropenia, malignancy, nonmelanoma skin cancer, hepatic disorder, lymphopenia, and VTE. Patients in the placebo group reported more cases of anemia.
In general, the number of patients who reported AEs of particular interest was low.
Table 30: Harm Outcomes — Induction Trials, Safety Population in Part 1 and Safety Population in Part 2
Characteristic | U-ACHIEVE Induction (Part 1) | U-ACHIEVE (Part 2) | U-ACCOMPLISH (Part 1) | U-ACCOMPLISH (Part 2) | ||
|---|---|---|---|---|---|---|
UPA 45 mg (N = 319) | Placebo (N = 155) | Extended UPA 45 mg (N = 59) | UPA 45 mg (N = 344) | Placebo (N = 177) | Extended UPA 45 mg (N = 68) | |
Patients with ≥ 1 AE | ||||||
n (%) | 180 (56.4) | 96 (61.9) | 46 (78.0) | 182 (52.9) | 70 (39.5) | 44 (64.7) |
Most common events,a n (%) | ||||||
Blood CPK increased | 16 (5.0) | 3 (1.9) | 6 (10.2) | 16 (4.7) | 2 (1.1) | 7 (10.3) |
Acne | 15 (4.7) | 1 (0.6) | 2 (3.4) | 24 (7.0) | 3 (1.7) | 6 (8.8) |
Nasopharyngitis | 15 (4.7) | 6 (3.9) | 5 (8.5) | 13 (3.8) | 4 (2.3) | 2 (2.9) |
Headache | 13 (4.1) | 4 (2.6) | 3 (5.1) | 8 (2.3) | 9 (5.1) | 4 (5.9) |
Pyrexia | 9 (2.8) | 2 (1.3) | 5 (8.5) | 8 (2.3) | 3 (1.7) | 5 (7.4) |
Anemia | 8 (2.5) | 9 (5.8) | 3 (5.1) | 14 (4.1) | 4 (2.3) | 9 (13.2) |
Ulcerative colitis | 3 (0.9) | 21 (13.5) | 2 (3.4) | 6 (1.7) | 8 (4.5) | 5 (7.4) |
Insomnia | 14 (4.4) | 14 (9.1) | 3 (5.1) | 1 (0.3) | 2 (1.1) | 0 |
Patients with ≥ 1 SAE | ||||||
n (%) | 8 (2.5) | 9 (5.8) | 3 (5.1) | 11 (3.2) | 8 (4.5) | 4 (5.9) |
Events, n (%) | ||||||
Ulcerative colitis | 2 (0.6) | 5 (3.2) | 1 (1.7) | 4 (1.2) | 3 (1.7) | 1 (1.5) |
Colitis | 0 | 1 (0.6) | 1 (1.7) | 0 | 1 (0.6) | 0 |
Appendicitis | 2 (0.6) | 0 | 0 | NR | ||
Cellulitis | 0 | 1 (0.6) | 0 | NR | ||
Muscle abscess | 0 | 1 (0.6) | 0 | NR | ||
Hypoalbuminemia | 0 | 1 (0.6) | 0 | NR | ||
Anemia | NR | 2 (3.4) | 1 (0.3) | 1 (0.6) | 0 | |
Patients with ≥ 1 WDAE | ||||||
n (%) | 6 (1.9) | 14 (9.0) | 0 | 6 (1.7) | 9 (5.1) | 2 (2.9) |
Most common events,a n (%) | ||||||
Ulcerative colitis | 2 (0.6) | 11 (7.1) | 0 | 3 (0.9) | 5 (2.8) | 0 |
Deaths | ||||||
n (%) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Notable harms, n (%) | ||||||
Serious infections | 5 (1.6) | 2 (1.3) | 0 (0.0) | 2 (0.6) | 1 (0.6) | 0 (0.0) |
Opportunistic infection, excluding TB and herpes zoster | 1 (0.3) | 0 (0.0) | 0 (0.0) | 6 (1.9) | 1 (0.6) | 0 (0.0) |
Active TB | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Herpes zoster | 1 (0.3) | 0 (0.0) | 3 (5.1) | 2 (0.6) | 0 (0.0) | 1 (1.5) |
Malignancy | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
VTE | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (0.6) | 0 (0.0) |
Hepatic disorder | 9 (2.8) | 7 (4.5) | 1 (1.7) | 10 (2.9) | 1 (0.6) | 2 (2.9) |
Anemia | 10 (3.1) | 14 (9.0) | 2 (3.4) | 15 (4.4) | 4 (2.3) | 4 (5.9) |
Lymphopenia | 10 (3.1) | 1 (0.6) | 1 (1.7) | 6 (1.7) | 1 (0.6) | 1 (1.5) |
Neutropenia | 16 (5.0) | 1 (0.6) | 4 (6.8) | 15 (4.4) | 0 (0.0) | 1 (1.5) |
GI perforation | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (0.6) | 0 (0.0) |
Hyperlipidemia | NR | NR | ||||
Hypersensitivity | NR | NR | ||||
AE = adverse event; CPK = creatine phosphokinase; GI = gastrointestinal; NR = not reported; SAE = serious adverse event; TB = tuberculosis; UPA = upadacitinib; VTE = venous thromboembolic event; WDAE = withdrawal due to adverse event.
aFrequency > 5%.
Source: Clinical Study Reports for U-ACHIEVE Induction6 and U-ACCOMPLISH.7
Table 31: Harm Outcomes — Maintenance Trial
Characteristic | U-ACHIEVE Maintenance SA_C population | ||
|---|---|---|---|
UPA 15 mg (N = 250) | UPA 30 mg (N = 251) | Placebo (N = 245) | |
Patients with ≥ 1 AE | |||
n (%) | 188 (75.2) | 189 (75.3) | 180 (73.5) |
Most comment events,a (n, %) | |||
Nasopharyngitis | 22 (8.8) | 26 (10.4) | 20 (8.2) |
Ulcerative colitis | 28 (11.2) | 18 (7.2) | 73 (29.8) |
Blood CPK increased | 14 (5.6) | 19 (7.6) | 5 (2.0) |
Upper respiratory tract infection | 12 (4.8) | 11 (4.4) | 8 (3.3) |
Arthralgia | 14 (5.6) | 7 (2.8) | 24 (9.8) |
Rash | 7 (2.8) | 11 (4.4) | 8 (3.3) |
Herpes zoster | 10 (4.0) | 10 (4.0) | 0 (0.0) |
Acne | 8 (3.2) | 8 (3.2) | 8 (3.3) |
Headache | 7 (2.8) | 8 (3.2) | 11 (4.5) |
Anemia | 8 (3.2) | 5 (2.0) | 10 (4.1) |
Influenza | 7 (2.8) | 8 (3.2) | 3 (1.2) |
Pyrexia | 6 (2.4) | 8 (3.2) | 6 (2.4) |
Anemia | 8 (3.2) | 5 (2.0) | 10 (4.1) |
AST increased | 9 (3.6) | 4 (1.6) | 2 (0.8) |
Folliculitis | 4 (1.6) | 9 (3.6) | 4 (1.6) |
Neutrophil count decreased | 5 (2.0) | 8 (3.2) | 2 (0.8) |
COVID-19 | 4 (1.6) | 7 (2.8) | 9 (3.7) |
Back pain | 7 (2.8) | 1 (0.4) | 10 (4.1) |
Patients with ≥ 1 SAE | |||
n (%) | 20 (8.0) | 20 (8.0) | 23 (9.4) |
Events, n (%) | |||
Ulcerative colitis | 1 (0.4) | 2 (0.8) | 5 (2.0) |
Pneumonia | 0 (0.0) | 0 (0.0) | 3 (1.2) |
COVID-19 pneumonia | 0 (0.0) | 2 (0.8) | 1 (0.4) |
Invasive breast carcinoma | 1 (0.4) | 0 (0.0) | 1 (0.4) |
Pulmonary embolism | 2 (0.8) | 0 (0.0) | 0 (0.0) |
Cervical dysplasia | 0 (0.0) | 2 (0.8) | 0 (0.0) |
Abortion induced | 2 (0.8) | 0 (0.0) | 0 (0.0) |
Patients with ≥ 1 WDAE | |||
n (%) | 10 (4.0) | 12 (4.8) | 25 (10.2) |
Events, n (%) | |||
Ulcerative colitis | 5 (2.0) | 3 (1.2) | 13 (5.3) |
Invasive breast carcinoma | 1 (0.4) | 0 (0.0) | 1 (0.4) |
Deaths | |||
n (%) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Notable harms, n (%) | |||
Any serious infections | 8 (3.2) | 6 (2.4) | 8 (3.3) |
Opportunistic infection, excluding tuberculosis and herpes zoster | 2 (0.8) | 1 (0.4) | 2 (0.8) |
Active TB | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Herpes zoster | 11 (4.4) | 10 (4.0) | 0 (0.0) |
Neutropenia | 7 (2.8) | 15 (6.0) | 5 (2.0) |
Malignancy | 1 (0.4) | 5 (2.0) | 1 (0.4) |
Malignancy, excluding NMSC | 1 (0.4) | 2 (0.8) | 1 (0.4) |
NMSC | 0 (0.0) | 3 (1.2) | 0 (0.0) |
Hepatic disorder | 17 (6.8) | 12 (4.8) | 5 (2.0) |
Anemia | 10 (4.0) | 7 (2.8) | 15 (6.1) |
Lymphopenia | 7 (2.8) | 5 (2.0) | 4 (1.6) |
Adjudicated GI perforations | 0 (0.0) | 0 (0.0) | 1 (0.4) |
Adjudicated VTE | 2 (0.8) | 2 (0.8) | 0 (0.0) |
AE = adverse event; AST = aspartate aminotransferase; CPK = creatine phosphokinase; GI = gastrointestinal; NMSC = nonmelanoma skin cancer; SAE = serious adverse event; TB = tuberculosis; UPA = upadacitinib; VTE = venous thromboembolic event; WDAE = withdrawal due to adverse event.
Note: The SA_C population comprised responders to UPA 45 mg 8-week induction enrolled for 44- or 52-week maintenance.
aFrequency > 4%.
Source: Clinical Study Report for U-ACHIEVE Maintenance.8
Critical Appraisal
Internal Validity
U-ACHIEVE and U-ACCOMPLISH were both randomized, double-blind, placebo-controlled, parallel-group, multicentre trials. The trials used acceptable methods of randomization and allocation concealment; therefore, the risk of bias arising from the randomization process is low. In both studies, patients were randomized in a 2:1 ratio using a stratified randomization scheme, with allocation being accomplished using a web-based interactive response system. Overall, the baseline characteristics of patients appear to be balanced between trial arms within studies, indicating that the randomization was successful. In U-ACHIEVE Maintenance, patients were recruited from different studies. Some of the patients were re-randomized to the study drug, while the others continued their previous treatment. The efficacy analyses for this study were based on a re-randomized group, which is appropriate.
During the induction period, the discontinuation rates in U-ACHIEVE Induction were 4.1% in the upadacitinib group and 12.9% in the placebo group; in U-ACCOMPLISH, the discontinuation rates were 3.2% in the upadacitinib group and 7.5% in the placebo group. Patients in the placebo arms were more likely to discontinue the study compared with those in the upadacitinib arms, and AEs were the main reason for study discontinuation. Note that in these 3 included trials, UC was categorized as an AE. The incidence of UC was generally high in the placebo group compared with the upadacitinib group across the trials. In the maintenance period, the discontinuation rates increased substantially. In cohort 1, 30.4%, 18.8%, and 63.8% of patients in the upadacitinib 15 mg group, upadacitinib 30 mg group, and placebo group, respectively, discontinued the study. “Other” was the main reason for study discontinuation, and the majority of reasons for the patients in this category were labelled “lack of efficacy” or “loss of response.” These patients would have been considered nonresponders in the efficacy analyses. A bias is less likely to be introduced with this approach to imputation.
Regarding the statistical analysis, the trials were powered to assess the outcome of clinical remission. All analyses were performed using the ITT analysis set. The ITT analysis set included patients who received at least 1 dose of the study drug, which is not a true ITT population; however, this analysis set was deemed to be the same as a true ITT. Both trials used a fixed-sequence multiple testing procedure to control for overall type I error for the primary and key secondary outcomes. The trials also assessed HRQoL and work productivity, which were important to patients; however, these outcomes were outside the statistical testing procedure, which limited the ability to draw conclusions from these data.
Missing data pertaining to the primary and secondary outcomes was addressed using a variety of approaches. In U-ACCOMPLISH, NRI, while incorporating NRI-C, was the primary approach for the binary end points. For continuous end points, missing data were handled using a mixed-model for repeated measures. The use of the NRI approach is considered reasonable in this scenario. Sensitivity analyses were conducted using different imputation methods and observed cases with no imputation. The results of these analyses were comparable to the main analysis. In U-ACHIEVE Induction, the LOCF approach was used to impute missing continuous efficacy data. The NRI approach was used for binary efficacy end points. Sensitivity analyses were conducted using the hybrid multiple imputation method for the primary end point of clinical remission. A mixed-model for repeated measures was used in the analyses of continuous secondary efficacy end points. The observed-case analysis with no imputation was also performed. Results of the sensitivity analyses were consistent with the primary analysis.
Prespecified subgroup analyses were performed to examine the consistency of the treatment effect observed for the primary efficacy end points. However, proper interpretation of all subgroups was not possible due to lack of sample size considerations for these subgroups and the lack of testing for differences between subgroups.
External Validity
According to the clinical expert consulted by CADTH, the population included in the pivotal studies was generally consistent with clinical practice. Based on the patient’s baseline characteristics, the study populations reflect a typical population in Canada that would receive upadacitinib in practice.
The studies included outcomes that were important to patients. The primary outcome assessed in all trials was clinical remission based on the adapted Mayo score, where remission was defined as an SFS of 1 or less, an RBS of 0, and an endoscopic subscore of 1 or less. The adapted Mayo is considered the gold standard and is a reliable indicator of disease activity in clinical practice. The 3 components (RBS, SFS, and endoscopic subscore) in the adapted Mayo score (excluding PGA) have been recommended by the FDA as appropriate end point measures for clinical trials of UC.31 For this outcome, SFS and RBS data were collected from patient diaries; therefore, they were not obtained objectively. Endoscopies, however, were centrally read by clinicians blinded to the patient’s treatment assignment, which is ideal. In addition, patient-reported outcomes, such as symptoms or the results of some HRQoL instruments, would probably be at low risk of bias, unless unblinding occurred at any point; unblinding would occur more often in the placebo group if patients experiencing a lack of efficacy assumed they were in the placebo group. There appears to be a low risk of bias due to the selection of the reported results, and the results presented followed the prespecified analysis plan.
U-ACHIEVE Induction and U-ACCOMPLISH included 8 weeks of induction therapy. The clinical expert consulted for this review indicated this was a sufficient time frame to determine short-term treatment effects with upadacitinib. U-ACHIEVE Maintenance was a 52-week study. The expert noted that 52 weeks would be adequate to observe long-term efficacy; however, 52 weeks may not be considered sufficient to fully understand the long-term safety for rare events and those that take longer to develop, such as malignancy. In addition, a larger sample size may be needed to explore the risk of malignancy in the study population.
The patient population in the maintenance period was likely enriched due to the study design. Approximately 72% of these patients responded to the treatment after 8 weeks of induction therapy, and it is important to note that the interpretation of the maintenance period results differs between an RR study design and a TT study design.
Indirect Evidence
Objectives and Methods for the Summary of Indirect Evidence
As there was no direct evidence comparing upadacitinib with other active therapies for the treatment of moderate to severe UC in adult patients, a review of indirect evidence was undertaken.
The sponsor submitted an ITC in patients with moderate to severe UC.32 CADTH also conducted a literature search to identify potentially relevant ITCs in this patient population. A focused literature search for ITCs dealing with UC was run in MEDLINE All (1946–) on May 16, 2022. No limits were applied to the search. Titles, abstracts, and full-text articles were screened for inclusion by 1 reviewer based on the population, intervention, comparator, and outcome criteria outlined in Table 6. Three studies (Lasa et al., 2022;33 Bur et al., 2021;34 and Li et al., 202235) were identified describing NMAs of the efficacy and safety of biologics and small-molecule drugs, including upadacitinib, for patients with moderate to severe UC.
The objective of this section is to summarize and critically appraise the indirect evidence available in the sponsor-submitted NMA and the published NMAs.
Description of Sponsor-Submitted ITC
The sponsor’s ITC included a systematic review of the literature to identify trials investigating upadacitinib and comparator interventions in patients with moderate to severe UC, and a corresponding NMA that compared upadacitinib with other active treatments.
In the sponsor-submitted ITC, upadacitinib was compared with other JAK inhibitors (tofacitinib and filgotinib), TNF alpha antagonists (adalimumab, golimumab, and infliximab), anti-integrin drugs (vedolizumab), IL-12 and IL-23 antagonists (ustekinumab), S1P receptor modulators (ozanimod), and placebo. Phase III and higher RCTs were included. Clinical remission, clinical response, and endoscopic improvement were evaluated based on subgroups of patients who were biologic-naive or biologic-exposed and in the induction or maintenance phases of drug administration. Safety outcomes were also examined in this ITC.
The patient population, intervention and comparators, and outcome measures for study selection in this ITC are presented in Table 32.
Table 32: Study Selection Criteria and Methods for ITC
Criteria | Sponsor’s ITC |
|---|---|
Population | Moderately to severely active UC |
Intervention | Upadacitinib |
Comparators |
|
Outcomes | The following were measured after 6 to 10 weeks of induction treatment and 40 to 54 weeks of maintenance treatment:
Safety: All AEs, discontinuation due to AEs, SAEs, and serious infections |
Study design | Published phase III+ RCTs |
Publication characteristics | English only |
Exclusion criteria |
|
Databases searched |
|
Selection process | The SLR was conducted by searching multiple databases. It is not clear if a prior research protocol had been developed. Study selection and data extraction were conducted using a systematic review approach. The quality of the included studies was assessed using a validated tool. |
Data extraction process | |
Risk-of-bias assessment | Cochrane risk-of-bias tool on a per-study basis |
AE = adverse event; CDSR = Cochrane Database of Systematic Reviews; EMS = endoscopic Mayo subscore; FMS = full Mayo score; ITC = indirect treatment comparison; NHS = National Health Service (UK); PK = pharmacokinetics; RBS = rectal bleeding subscore; RCT = randomized controlled trial; SAE = serious adverse event; SLR = systematic literature review; UC = ulcerative colitis.
Source: Sponsor-submitted ITC.32
Methods of Sponsor-Submitted ITC
Objectives
The objective of this ITC was to compare the treatment efficacy and safety of upadacitinib relative to currently existing medications for the treatment of moderate to severe UC.
Systematic Review Methods
Phase III and higher RCTs that were used to inform the ITC were identified through a systematic literature search conducted by the ITC authors. Multiple databases were searched to identify clinical trials (published between the inception of the databases and January 2022) that evaluated the efficacy of drug therapies for moderately to severely active UC. The risk of bias of the included studies was assessed using the Cochrane risk-of-bias assessment tool (version 2) at the study level.
ITC Analysis Methods
For each feasible network, NMAs were conducted in a generalized linear model framework using Bayesian Markov Chain Monte Carlo simulations and 3 chains with 100,000 runs each, with a burn-in that was half of the convergence sequence (set size of 10,000).
Models were built using the Bayesian NMA package in the R statistical software, designed to fit NMAs in a Bayesian framework. Model selections were made after comparing the fit statistics, leverage plots, and density plots of posterior standard deviations (SDs) for each set of 4 possible models: fixed-effects unadjusted (FE) model, fixed-effects adjusted (FEA) model, random-effects unadjusted (RE) model, and random-effects adjusted (REA) model. Baseline risks were adjusted using these models.
Model fit statistics determined the selection between FE and RE models; all else equal, the RE model was selected over the FE model. The baseline risk–adjusted version was then selected if its beta coefficient was significant, and the SD decreased (for RE models).
In the NMA, relative treatment effects were modelled as log odds for binary outcomes. From the log odds, odds ratios (ORs) were derived. All posterior distributions, including those for ORs, were summarized by their medians and 95% CrIs.
Different doses for a treatment were treated as separate treatment nodes in the NMA networks.
Sources of potential heterogeneity across the relevant RCTs in UC were identified in the sponsor’s ITC:
Outcome definition: For example, full Mayo score versus adapted Mayo score–defined outcomes, duration of outcome assessment, central versus local endoscopy reading.
Maintenance study design (TT versus RR): Furthermore, in the maintenance phase, placebo arms were fundamentally different because they received and responded to different induction treatments, which potentially results in different levels of persistence after treatment is ended.
Prior UC medications: There were differences in the definition of “biologic experience” across the trials.
Demographic characteristics: Some trials were conducted primarily in Asian populations.
UC severity: Defined differently across the studies.
Differences between induction trials: Some heterogeneity in disease duration, extent of disease, CRP, and concurrent corticosteroids, and notable heterogeneity in the use of immunomodulators.
Differences between maintenance trials: Some heterogeneity in weight, disease duration, extent of disease, and CRP, and notable heterogeneity in use of immunomodulators.
Not all data reported: For all characteristics, there were studies where the data were not reported.
Table 33: Model Selections in Sponsor-Submitted NMA
Outcome | Biologic exposure | Selected logit model | Baseline assumption | Prior distribution for SD | Baseline model distribution | Placebo risk significanta | Baseline risk adjustment |
|---|---|---|---|---|---|---|---|
Induction phase | |||||||
Clinical remission | Naive | RE | Independent | Uniform | Predictive | No | Not significant (REA and FEA) |
Exposed | RE | Exchangeable | Half-normal | Posterior | No | Did not run (REA) or converge (FEA not significant) | |
Clinical response | Naive | REA | Independent | Uniform | Predictive | Yesb | Significant (REA and FEA) |
Exposed | RE | Independent | Half-normal | Predictive | Nob | Did not converge (REA and FEA not significant) | |
Endoscopic improvement | Naive | RE | Independent | Uniform | Posterior | Yes | Not significant (REA and FEA) |
Exposed | RE | Independent | Half-normal | Posterior | Yesc | Did not run (REA) or converge (FEA not significant) | |
All AEs | Overall | REA | Independent | Half-normal | Posterior | Yes | Significant (REA and FEA) |
Discontinuation due to AEs | Overall | RE | Independent | Half-normal | Posterior | Yesc | Did not converge (REA and FEA not significant) |
Serious AEs | Overall | RE | Independent | Half-normal | Predictive | No | Not significant (REA) or did not converge (FEA not significant) |
Serious infections | Overall | RE | Exchangeable | Half-normal | Predictive | No | Did not run (REA and FEA) |
Maintenance phase | |||||||
Clinical remission | Naive | RE | Independent | Half-normal | Predictive | Yesc | Did not converge (REA significant) or significant (FEA) |
Exposed | RE | Independent | Half-normal | Posterior | Nob | Did not run (REA) or not significant (FEA) | |
Clinical response | Naive | RE | Independent | Half-normal | Predictive | Yesc | Did not run (REA) or converge (FEA not significant) |
Exposed | RE | Independent | Half-normal | Posterior | Yesc | Did not run (REA and FEA) | |
Endoscopic improvement | Naive | RE | Independent | Half-normal | Predictive | Yesc | Did not converge (REA and FEA not significant) |
Exposed | RE | Independent | Half-normal | Posterior | Nocb | Did not run (REA and FEA) | |
All AEs | Overall | RE | Independent | Half-normal | Posterior | Yesc | Did not converge (REA and FEA not significant) |
Discontinuation due to AEs | Overall | RE | Independent | Half-normal | Posterior | Yes | Not significant (REA) or did not converge (FEA not significant) |
Serious AEs | Overall | RE | Independent | Half-normal | Posterior | Yes | Not significant (REA) or did not converge (FEA significant) |
Serious infections | Overall | RE | Independent | Half-normal | Posterior | No | Did not run (REA and FEA) |
AE = adverse event; FE = fixed-effects unadjusted; FEA = fixed-effects adjusted; ITT = intention to treat; PBO = placebo; RCT = randomized controlled trial; RD = risk difference; RE = random-effects unadjusted; REA = random-effects adjusted; SD = between-study standard deviation.
Note: By default, RCT-specific baselines (μi), shown in the following equation as representing the log odds of the outcome in the “control” treatment (i.e., the placebo arm), were modelled as independent, such that an unrelated model parameter was specified for each one: logit(pik) = μi+δi,1k. However, in networks with 1 or more placebo arm(s) having a value of zero (i.e., no events), an exchangeable baseline assumption with a half-normal (0 to 0.322) prior for heterogeneity was used to aid parameter estimation and numerical stability or convergence.
By default, the predictive distribution of the baseline probability was used to predict the absolute PBO probabilities. But if the resulting 95% CrI of a predicted PBO probability was too wide (i.e., differed from its median by greater than 2 factors), then the posterior distribution of the baseline probability, which is associated with less uncertainty in the natural history model, was used instead.
aStatistical significance with Wald test P value < 0.05; “yes” indicates that placebo rates were significantly heterogeneous.
bFE RD models tested to replicate the treat-through ITT rates in the sensitivity analysis.
cFE RD models were tested for networks with significant placebo rate heterogeneity, but baseline-risk adjustment was inconclusive (i.e., FEA and/or REA models did not converge or failed to run).
Source: Sponsor-submitted indirect treatment comparison.32
Two maintenance study designs, TT and RR, were employed across the UC RCTs. In TT studies, patients were randomized to treatment or placebo at baseline and outcomes were measured at the end of an induction phase and again at the end of a maintenance phase. In RR studies, patients were randomized to induction treatment or placebo at baseline, with outcomes measured at the end of the induction phase; induction responders were then re-randomized to maintenance treatment or placebo, with outcomes measured at the end of the maintenance phase strictly among induction responders. The use of both study designs across the included maintenance RCTs renders a standard NMA for maintenance outcomes not valid, and in prior National Institute for Health and Care Excellence (NICE) submissions, imputations were performed to make the outcomes more comparable. Two alternative approaches to the imputation were taken:
recalculate data from the TT RCTs to mimic an RR design and then perform NMAs, or
recalculate data from the RR RCTs to mimic a TT design and then perform NMAs.
Among the included maintenance RCTs, 3 followed a TT design: ACT-1 for infliximab, M10 to 447, and ULTRA-2 for adalimumab. Outcomes from these 3 RCTs were imputed to match more closely with those of the remaining RR trials. Briefly, the observed data from the TT RCTs were adjusted based on the assumption that the number of responders at the end of induction is a proxy for the total number of patients entering maintenance. So, to the extent they were reported, clinical outcomes for the induction treatment responder subset were used. Additional assumptions were made to impute clinical remission from clinical response.
The following baseline characteristics were identified a priori by the sponsor from clinical opinion to be potential treatment effect modifiers in UC: age (years), sex (% male), weight (kg), duration of disease (years), extent of disease (percentage of extensive colitis or pancolitis), baseline full Mayo score, baseline CRP (some may be high sensitivity), and concurrent medications for UC (percentage of corticosteroids, percentage of immunomodulators). A stepwise approach was taken to account for this expected heterogeneity, relying primarily on selecting the RE over the FE model when all else was equal, secondarily on adjusting for baseline (placebo) risk as a single proxy for characteristics that are thought to modify the treatment effect, and tertiarily on testing FE models with the risk difference link (instead of logit) that minimized the impact of placebo heterogeneity.
In the included induction trials, heterogeneity was observed for weights at baseline, disease duration, CRP levels, extent of disease, and previous or concomitant medications. The NRI method of handling missing binary outcomes was consistently employed across the included induction RCTs with minor exceptions, such as the use of LOCF in GEMINI 1 for vedolizumab and NRI-C uniquely to handle missing data due to COVID-19 in the upadacitinib RCTs.
For the maintenance trials, heterogeneity was observed in baseline weights, disease duration, CRP levels at baseline, extent of disease, notable heterogeneity in use of immunomodulators, concomitant medications, and UC severity definitions. Similar to induction, the NRI method of handling missing binary outcomes was consistently employed across the included maintenance RCTs, with minor exceptions, such as the use of LOCF in GEMINI 1 for vedolizumab and PURSUIT- J for golimumab, and NRI-C in U-ACHIEVE study 3 for upadacitinib.
Detailed statistical methods of ITC are provided in Table 34.
Table 34: ITC Analysis Methods
Detail | Sponsor’s NMA |
|---|---|
ITC methods | A Bayesian NMA approach was taken. |
Priors | Vague or flat prior distributions were given to the parameters to be estimated by default. For parameters assumed to be specified on a continuous scale, namely the relative treatment effects (d), RCT-specific baselines (μ), and baseline adjustment regression term B (for models with baseline-risk adjustment), a normal (0 to 100) prior distribution was used.2 For the between-study SD (for RE models), a uniform (0 to 5) prior distribution was used. |
Assessment of model fit | The models’ global fits were assessed and compared using their overall posterior mean residual deviance, effective number of parameters, DIC, leverage plots, and the posterior distribution of the between-study SD (σ or SD) associated with the RE model. |
Assessment of consistency | Even though the authors of this ITC tried to assess the consistency in their report, consistency could not be assessed, because the only closed loops were of different doses within individual trials; therefore, only indirect comparisons were available. |
Assessment of convergence | Assessed with the Brooks-Gelman-Rubin method using the PSRF. The PRSF should gradually shrink to 1 with increasing numbers of iterations; a value of < 1.05 was used to indicate convergence. |
Outcomes | Clinical remission, clinical response, endoscopic improvement, and safety outcomes. |
Follow-up time points | Induction phase: 6 to 10 weeks (taking the time point closest to 8 weeks). Maintenance phase: 40 to 54 weeks (taking the time point closest to 52 weeks). |
Construction of nodes | Each treatment and dose were treated as separate treatment nodes in the NMA networks. |
Sensitivity analyses | Not specified. |
Subgroup analysis | Subgroups: Biologic-naive vs. biologic-exposed. |
Methods for pairwise meta-analysis | NR |
DIC = deviance information criteria; ITC = indirect treatment comparison; NMA = network meta-analysis; NR = not reported; PRSF = potential scale reduction factor; RCT = randomized controlled trial; RE = random effects; SD = standard deviation.
Source: Sponsor’s ITC.32
Results of Sponsor-Submitted ITC
Summary of Included Studies
In total, 48 unique RCTs were identified in the systematic literature review, and 23 RCTs (of which 18 were used in the induction NMA and 14 in the maintenance NMA) were included in the NMA. All 18 induction RCTs randomized patients to relevant induction treatment(s) versus placebo for 6 to 10 weeks in a double-blinded manner, and all 14 maintenance RCTs randomized patients to relevant maintenance treatment(s) versus placebo (except for SERENE-C, which compared different induction regimens for adalimumab) for 40 to 54 weeks in a double-blinded manner.
Outcome Definitions
In the induction trials, for clinical remission, 3 RCTs (OCTAVE 1 and OCTAVE 2 for tofacitinib and TRUE NORTH for ozanimod) reported outcomes that deviated from the base-case definition of “full Mayo score ≤ 2 with no subscore > 1”. For clinical response, 1 RCT (TRUE NORTH for ozanimod) reported outcomes that deviated from the base-case definition of “decrease in [full Mayo] score ≥ 3 points and ≥ 30% from baseline, and a decrease in RBS ≥ 1 or an absolute RBS ≤ 1”.
The upadacitinib RCTs reported adapted Mayo score–defined clinical remission and response in their Clinical Study Reports, but outcomes using the base-case definitions were derived from upadacitinib patient-level data in ad hoc analyses. For endoscopic improvement, there was no deviation from the base-case definition of “[endoscopic Mayo subscore] ≤ 1.”
In the maintenance trials, for clinical remission, 2 RCTs (OCTAVE Sustain for tofacitinib and TRUE NORTH for ozanimod) reported outcomes that deviated from the base-case FM1 definition; for clinical response, only TRUE NORTH for ozanimod reported outcomes that deviated from the base-case definition.
Similar to induction, the upadacitinib RCTs reported adjusted Mayo score–defined clinical remission and response but outcomes using the FM1 definitions were derived from upadacitinib patient-level data in ad hoc analyses. Finally, for endoscopic improvement, there was no deviation from the base-case definition of “[endoscopic Mayo] subscore ≤ 1.”
Disease Severity
Induction trials: The UC disease severity criteria of each included induction RCT were relatively consistent, with all but 3 RCTs requiring patients to have a full Mayo score of 6 to 12 (FM6/12) and endoscopic Mayo subscore of at least 2 (EMS2) before enrolment. Three RCTs deviated from this: TRUE NORTH for ozanimod did not require EMS2 and the upadacitinib RCTs required an adjusted Mayo score of 5 to 9 instead of FM6/12. To the extent that PGA subscore affects UC severity, this could be a source of heterogeneity.
Maintenance trials: The UC disease severity criteria of each included maintenance RCT were relatively consistent, with all but 2 RCTs requiring FM6/12 and EMS2 before enrolment. The 2 RCTs that deviated from this were TRUE NORTH for ozanimod, which did not require EMS2, and U-ACHIEVE for upadacitinib, which required an adjusted Mayo score of 5 to 9 instead of FM6/12.
Table 35: Trial Characteristics of Studies Included in NMA
Study | UC severity | Prior use of biologics | Induction phase | Maintenance phase | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
Duration (weeks) | Total N | Treatment regimens (+ PBO) | RCT design | Induction treatment | Duration (weeks) | Total N | Treatment regimens (+ PBO) | |||
ACT-1 |
| Naive | 8 | 364 |
| TT |
| 46 | 364 |
|
ACT-2 |
| Naive | 8 | 364 |
| Excluded: Duration < 40 weeks | ||||
GEMINI 1 |
| Mixed | 6 | 374 | VED 300 mg | RR | VED 300 mg | 46 | 373 |
|
Japic CTI-060298 |
| Naive | 8 | 208 | INF 5 mg/kg | Excluded: Duration < 40 weeks | ||||
Jiang 2015 |
| Naive | 8 | 123 | INF 5 mg/kg (INF 3.5 mg/kg excluded) | Excluded: Duration < 40 weeks | ||||
M10 to 447 |
| Naive | 8 | 274 | ADA160/80 (ADA80/40 excluded) | TT |
| 44 | 274 | ADA 40 mg q.2.w. (ADA160/80 and ADA80/40 combined) |
NCT 01551290 |
| naïve | 8 | 99 | INF 5 mg/kg | Excluded: Duration < 40 weeks | ||||
NCT 02039505 |
| Mixed | 10 | 246 | VED 300 mg | RR | VED 300 mg | 50 | 83 | VED 300 mg q.8.w. |
OCTAVE 1 |
| Mixed | 8 | 598 | TOF 10 mg | Maintenance in OCTAVE Sustain | ||||
OCTAVE 2 |
| Mixed | 8 | 541 | TOF 10 mg | Maintenance in OCTAVE Sustain | ||||
OCTAVE Sustain |
| Mixed | Induction in OCTAVE 1 and OCTAVE 2 | RR |
| 52 | 593 |
| ||
PURSUIT-J |
| Mixed | Excluded: Open label | RR | GOL200/100 | 54 | 63 | GOL 100 mg | ||
PURSUIT-M |
| Mixed | Induction in PURSUIT-SC | RR |
| 54 | 464 |
| ||
PURSUIT-SC |
| Mixed | 6 | 774 | GOL200/100 (GOL400/200 excluded) | Maintenance in PURSUIT-M | ||||
SELECTION |
| Mixed | 10 | 1,348 |
| RR |
| 47 | 571 |
|
SERENE-UC |
| Mixed | Excluded: Intervention ADA HIR | RR |
| 44 | 371 |
| ||
TRUE NORTH |
| Mixed | 10 | 645 | OZA 0.92 mg | RR | OZA 0.92 mg | 42 | 457 | OZA 0.92 mg |
U-ACCOMPLISH |
| Mixed | 8 | 522 | UPA 45 mg | Induction in U-ACHIEVE study 3 | ||||
U-ACHIEVE Study 2 and 3 |
| Mixed | 8 | 474 | UPA 45 mg | RR | UPA 45 mg | 52 | 451 |
|
ULTRA-1 |
| Naive | 8 | 390 | ADA160/80 (ADA80/40 excluded) | No maintenance | ||||
ULTRA-2 |
| Mixed | 8 | 518 | ADA160/80 | TT |
| 44 | 518 | ADA 40 mg q.2.w. |
UNIFI |
| Mixed | 8 | 961 | UST 6 mg/kg (UST 130 mg excluded) | RR |
| 44 | 523 |
|
VISIBLE 1 |
| Mixed | Excluded: Open-label | RR | VED 300 mg | 46 | 216 | VED 300 mg q.8.w. (VED 108 mg q.2.w. SC excluded) | ||
ADA = adalimumab; ADA80/40 = adalimumab 40 mg every week or 80 mg every other week; ADA160/80 = adalimumab 160 mg at week 0 followed by 80 mg at week 2; AM = adapted Mayo score; AM5/9 = adapted Mayo score of 5 to 9; EMS2 = EMS of at least 2; FIL = filgotinib; FM = full Mayo score; FM6/12 = a full Mayo score of 6 to 12; GOL = golimumab; GOL200/100 = GOL 200 mg at week 0 and 100 mg at week 2; HIR = higher induction regimen; INF = infliximab; NMA = network meta-analysis; OZA = ozanimod; PBO = placebo; PGA2 = Physician’s Global Assessment score of 2; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; q.8.w. = every 8 weeks; q.12.w. = every 12 weeks; q.w. = once a week; RBS1 = rectal bleeding subscore of 1; RR = re-randomized; SC = subcutaneous; SFS1 = stool frequency subscore of 1; TDM = therapeutic drug monitoring; TOF = tofacitinib; TT = treat-through; UPA = upadacitinib; UST = ustekinumab; VED = vedolizumab.
Source: Sponsor-submitted ITC.32
Figure 5 and Figure 6 present the networks of evidence during induction or maintenance in the overall patient population. For the induction evidence network, the network is star-shaped, and all treatments are anchored on placebo as the common comparator. For the maintenance evidence network, the network is star-shaped, and all maintenance treatments are anchored on placebo as the common comparator. The only closed loops are formed by different doses used in single studies.
Figure 5: Network Diagram — Induction Phase
FIL = filgotinib; GOL = golimumab; INF = infliximab; PBO = placebo; OZA = ozanimod; TOF = tofacitinib; UPA = upadacitinib; UST = ustekinumab; VED = vedolizumab.
Source: Sponsor-submitted indirect treatment comparison.32
Figure 6: Network Diagram — Maintenance Phase
ADA40 = adalimumab 40 mg; FIL100 = filgotinib 100 mg; FIL200 = filgotinib 200 mg; GOL50 = golimumab 50 mg; GOL100 = golimumab 100 mg; OZA0.92 = ozanimod 0.92 mg; PBO = placebo; Q4W = every 4 weeks; Q8W = every 8 weeks; Q12W = every 12 weeks; TOF5 = tofacitinib 5 mg; TOF10 = tofacitinib 10 mg; UPA15 = upadacitinib 15 mg; UPA30 = upadacitinib 30 mg; UST90 = ustekinumab 90 mg; VED300 = vedolizumab 300 mg.
Source: Sponsor-submitted indirect treatment comparison.32
Results
Detailed efficacy and safety results are presented in Table 36 to Table 41. Only the results for upadacitinib versus relevant comparators are summarized in this section.
Table 36: Summary of NMA Results for Efficacy Outcomes at Induction, UPA 45 mg Versus Comparators, Random-Effects Models in the Biologic-Naive Population
Treatment | Clinical remission | Clinical response | Endoscopic improvement |
|---|---|---|---|
12 interventions, 18 studies | 12 interventions, 18 studies | 12 interventions, 18 studies | |
ADA160/80, median OR (95% CrI) | 5.45 (1.94 to 17.35) | 3.19 (1.88 to 5.56) | 4.36 (2.21 to 8.80) |
GOL200/100, median OR (95% CrI) | 2.98 (0.84 to 11.40) | 3.56 (1.83 to 6.87) | 3.81 (1.78 to 8.39) |
INF 10 mg/kg, median OR (95% CrI) | 3.01 (1.02 to 9.55) | 2.04 (1.16 to 3.62) | 2.26 (1.11 to 4.70) |
INF 5 mg/kg, median OR (95% CrI) | 2.46 (0.91 to 7.41) | 2.03 (1.22 to 3.33) | 2.27 (1.18 to 4.56) |
OZA 0.92 mg, median OR (95% CrI) | 2.35 (0.60 to 9.24) | 3.33 (1.69 to 6.49) | 1.93 (0.81 to 4.60) |
PLO, median OR (95% CrI) | 9.59 (4.02 to 25.83) | 6.90 (4.59 to 10.54) | 6.93 (3.92 to 12.81) |
TOF 10 mg, median OR (95% CrI) | 4.18 (1.25 to 14.65) | 2.21 (1.24 to 4.04) | 3.33 (1.49 to 7.43) |
UST 6 mg/kg, median OR (95% CrI) | 4.71 (1.28 to 18.33) | 1.91 (0.97 to 3.79) | 3.70 (1.58 to 8.75) |
VED 300 mg, median OR (95% CrI) | 2.94 (0.85 to 10.59) | 3.27 (1.75 to 6.06) | 2.74 (1.24 to 6.16) |
ADA = adalimumab; ADA160/80 = adalimumab 160 mg at week 0 followed by 80 mg at week 2; CrI = credible interval; GOL = golimumab; GOL200/100 = GOL 200 mg at week 0 and 100 mg at week 2; INF = infliximab; NMA = network meta-analysis; OR = odds ratio; OZA = ozanimod; PLO = placebo; TOF = tofacitinib; UPA = upadacitinib; UST = ustekinumab; VED = vedolizumab.
Note: Clinical response in bio-naive patients in induction trials was evaluated using an adjusted random-effects model.
OR > 1 indicates results favouring upadacitinib.
Bold values indicate statistical significance.
Source: Sponsor-submitted indirect treatment comparison.32
Clinical Remission Analyses
The bio-naive induction evidence base for clinical remission includes 12 interventions, 18 studies (none with a zero event), and 5,080 patients. An unadjusted version of the RE model was used in analysis.
The bio-exposed induction evidence base for clinical remission includes 9 interventions, 10 studies (2 with at least 1 zero event), and 2,839 patients. The unadjusted RE model is selected to account for expected heterogeneity.
The bio-naive maintenance evidence base for clinical remission includes 17 interventions, 12 studies (none with a zero event), and 2,648 patients. An unadjusted version of the RE model was used in the analysis.
The bio-exposed maintenance evidence base for clinical remission includes 13 interventions, 9 studies (none with a zero event), and 1,405 patients. The unadjusted RE model is selected to account for expected heterogeneity.
Clinical Response Analyses
The bio-naive induction evidence base for clinical response includes 12 interventions, 18 studies (none with a zero event), and 5,080 patients. The adjusted version of the RE model was selected in the analysis.
The bio-exposed induction evidence base for clinical response includes 9 interventions, 10 studies (2 with at least 1 zero event), and 2,823 patients. The unadjusted version of the RE model was selected in the analysis.
The bio-naive maintenance evidence base for clinical response includes 17 interventions, 11 studies (none with a zero event), and 2,579 patients. The unadjusted RE model is selected to account for expected heterogeneity.
The bio-exposed maintenance evidence base for clinical response includes 13 interventions, 8 studies (none with a zero event), and 1,348 patients. The unadjusted version of the RE model was selected in the analysis.
Endoscopic Improvement Analyses
The bio-naive induction evidence base for endoscopic improvement includes 12 interventions, 18 studies (none with a zero event), and 5,080 patients. The unadjusted version of the RE model was selected.
The bio-exposed induction evidence base for endoscopic improvement includes 9 interventions, 10 studies (none with a zero event), and 2,823 patients. The unadjusted version of the RE model was selected.
The bio-naive maintenance evidence base for endoscopic improvement includes 14 interventions, 9 studies (none with a zero event), and 2,230 patients. An unadjusted version of RE model was used in the analysis.
The bio-exposed maintenance evidence base for endoscopic improvement includes 12 interventions, 7 studies (none with a zero event), and 1,283 patients. An unadjusted version of RE model was used in the analysis.
Efficacy in the Induction Phase
All efficacy comparisons either favoured upadacitinib or showed no evidence for a difference between upadacitinib and the active comparators (i.e., the null was not excluded by the 95% CrIs).
Clinical Remission
For the induction phase, the results from the unadjusted RE models found that in the biologic-naive population, upadacitinib 45 mg was favoured over adalimumab (OR = 5.45; 95% CrI, 1.94 to 17.35), ustekinumab (OR = 4.71; 95% CrI, 1.28 to 18.33), tofacitinib (OR = 4.18; 95% CrI, 1.25 to 14.65), and infliximab 10 mg (OR = 3.01; 95% CrI, 1.02 to 9.55). In the comparisons between upadacitinib and other active treatments, 95% CrIs of clinical remission from RE models did not exclude the null. Among biologic-exposed patients, results showed that upadacitinib 45 mg was favoured over adalimumab (OR = 3.61; 95% CrI, 1.26 to 17.53), vedolizumab (OR = 3.03; 95% CrI, 1.32 to 11.21), and ozanimod (OR = 2.79; 95% CrI, 1.07 to 11.13). In the comparisons between upadacitinib and other active treatments, the 95% CrI of clinical remission did not exclude the null (Table 37).
During the maintenance phase, results from unadjusted RE models suggested that in the biologic-naive population, no treatment was favoured for clinical remission when upadacitinib was compared with other active treatments. Among biologic-exposed patients, results showed that upadacitinib 15 mg was favoured over tofacitinib (OR = 5.93; 95% CrI, 1.21 to 34.38), ustekinumab 90 mg every 12 weeks (OR = 8.12; 95% CrI, 1.66 to 45.69), and ustekinumab 90 mg every 8 weeks (OR = 4.89; 95% CrI, 1.04 to 27.09). Upadacitinib 30 mg was favoured over tofacitinib (OR = 7.50; 95% CrI, 1.53 to 43.12), ustekinumab 90 mg every 12 weeks (OR = 10.26; 95% CrI, 2.11 to 55.45), and ustekinumab 90 mg every 8 weeks (OR = 6.16; 95% CrI, 1.31 to 33.22). In the comparisons between upadacitinib 15 mg or 30 mg and other active treatments, the 95% CrI of clinical remission did not exclude the null.
Table 37: Summary of NMA Results for Efficacy Outcomes at Induction, UPA 45 mg Versus Comparators, Random-Effects Models in the Biologic-Experienced Population
Treatment | Clinical remission | Clinical response | Endoscopic improvement |
|---|---|---|---|
9 interventions, 10 studies | 9 interventions, 10 studies | 12 interventions, 7 studies | |
ADA160/80, median OR (95% CrI) | 3.61 (1.26 to 17.53) | 9.40 (3.26 to 27.55) | 13.80 (4.20 to 53.53) |
OZA 0.92, median OR (95% CrI) | 2.79 (1.07 to 11.13) | 5.18 (1.63 to 16.00) | 9.81 (2.33 to 42.79) |
PLO, median OR (95% CrI) | 9.80 (5.23 to 24.98) | 13.56 (7.24 to 26.41) | 15.08 (6.29 to 43.88) |
TOF 10 mg, median OR (95% CrI) | 1.89 (0.88 to 4.56) | 3.54 (1.45 to 8.69) | 3.13 (0.89 to 12.06) |
UST 6 mg/kg, median OR (95% CrI) | 1.67 (0.63 to 4.55) | 3.77 (1.41 to 10.50) | 4.11 (1.14 to 16.17) |
VED 300 mg, median OR (95% CrI) | 3.03 (1.32 to 11.21) | 8.62 (3.37 to 22.71) | 12.34 (3.94 to 43.49) |
ADA = adalimumab; ADA160/80 = adalimumab 160 mg at week 0 followed by 80 mg at week 2; CrI = credible interval; GOL = golimumab; GOL200/100 = GOL 200 mg at week 0 and 100 mg at week 2; INF = infliximab; NMA = network meta-analysis; OR = odds ratio; OZA = ozanimod; PLO = placebo; TOF = tofacitinib; UPA = upadacitinib; UST = ustekinumab; VED = vedolizumab.
Note: Clinical response in bio-naive patients in induction trials was evaluated using an adjusted random-effects model.
OR > 1 indicates results favouring upadacitinib.
Bold values indicate statistical significance.
Source: Sponsor-submitted indirect treatment comparison.32
Clinical Response
For the induction phase, results from unadjusted RE models found that in the biologic-naive population, upadacitinib 45 mg was favoured over adalimumab (OR = 3.19; 95% CrI, 1.88 to 5.56), golimumab (OR = 3.56; 95% CrI, 1.83 to 6.87), infliximab 5 mg (OR = 2.03; 95% CrI 1.22 to 3.33), infliximab 10 mg (OR = 2.04; 95% CrI, 1.16 to 3.62), tofacitinib (OR = 2.21; 95% CrI, 1.24 to 4.04), vedolizumab (OR = 3.27; 95% CrI 1.75 to 6.06) and ozanimod (OR = 3.33; 95% CrI 1.69 to 6.49). In the comparisons between upadacitinib and other active treatments, the 95% CrIs of clinical response from RE models did not exclude the null. Among biologic-exposed patients, results showed that upadacitinib 45 mg was favoured over adalimumab (OR = 9.40; 95% CrI, 3.26 to 27.55), vedolizumab (OR = 8.62; 95% CrI, 3.37 to 22.71), ozanimod (OR = 5.18; 95% CrI, 1.63 to 16.00), tofacitinib (OR = 3.54; 95% CrI, 1.45 to 8.69), and ustekinumab (OR = 3.77; 95% CrI, 1.41 to 10.50). In the comparisons between upadacitinib and other active treatments, the 95% CrIs of clinical response did not exclude the null.
Endoscopic Improvement
For the induction phase, results from unadjusted RE models found that in the biologic-naive population, upadacitinib 45 mg was favoured over adalimumab (OR = 4.36; 95% CrI, 2.21 to 8.80), golimumab (OR = 3.81; 95% CrI, 1.78 to 8.39), infliximab 5 (OR = 2.27; 95% CrI, 1.18 to 4.56), infliximab 10 (OR = 2.26; 95% CrI, 1.11 to 4.70), tofacitinib (OR = 3.33; 95% CrI, 1.49 to 7.43), vedolizumab (OR = 2.74; 95% CrI, 1.24 to 6.16) and ustekinumab (OR = 3.70; 95% CrI, 1.58 to 8.75). In the comparison between upadacitinib and ozanimod, the 95% CrIs of endoscopic improvement from the RE model did not exclude the null. Among biologic-exposed patients, results showed that upadacitinib 45 mg was favoured over adalimumab (OR = 13.80; 95% CrI, 4.20 to 53.53), ozanimod (OR = 9.81; 95% CrI, 2.33 to 42.79), vedolizumab (OR = 12.34; 95% CrI, 3.94 to 43.49), and ustekinumab (OR = 4.11; 95% CrI, 1.14 to 16.17). In the comparison between upadacitinib and tofacitinib, the 95% CrIs of endoscopic improvement did not exclude the null.
Efficacy in the Maintenance Phase
All efficacy comparisons either favoured upadacitinib or showed no evidence for a difference between upadacitinib and the active comparators (i.e., the null was not excluded by the 95% CrIs).
Clinical Remission
During the maintenance phase, results from the unadjusted RE models suggested that in the biologic-naive population, no treatment was favoured for clinical remission when upadacitinib was compared with other active treatments. Among biologic-exposed patients, results showed that upadacitinib 15 mg was favoured over tofacitinib (OR = 5.93; 95% CrI, 1.21 to 34.38), ustekinumab 90 mg every 12 weeks (OR = 8.12; 95% CrI, 1.66 to 45.69), and ustekinumab 90 mg every 8 weeks (OR = 4.89; 95% CrI, 1.04 to 27.09). Upadacitinib 30 mg was favoured over tofacitinib (OR = 7.50; 95% CrI, 1.53 to 43.12), ustekinumab 90 mg every 12 weeks (OR = 10.26; 95% CrI, 2.11 to 55.45), and ustekinumab 90 mg every 8 weeks (OR = 6.16; 95% CrI, 1.31 to 33.22). In the comparisons between upadacitinib 15 mg or 30 mg and other active treatments, the 95% CrI of clinical remission did not exclude the null.
Table 38: Summary of NMA Results for Efficacy Outcomes at Maintenance (UPA 15 mg or UPA 30 mg Versus Comparators), Random-Effects Models in the Biologic-Naïve Population
Treatment | Clinical remission | Clinical response | Endoscopic improvement |
|---|---|---|---|
17 interventions, 12 studies | 17 interventions, 11 studies | 14 interventions, 9 studies | |
ADA 40 mg q.2.w., median OR (95% CrI) | 2.61 (0.53 to 12.99) 3.65 (0.74 to 17.92) | 3.57 (0.96 to 13.56) 7.99 (2.07 to 30.59) | NA |
GOL 100 mg, median OR (95% CrI) | 1.04 (0.22 to 3.82) 1.44 (0.30 to 5.24) | 1.72 (0.52 to 5.24) 3.86 (1.12 to 12.15) | 1.26 (0.30 to 4.20) 2.43 (0.58 to 8.26) |
GOL 50 mg, median OR (95% CrI) | 1.33 (0.28 to 5.53) 1.86 (0.39 to 7.69) | 2.11 (0.63 to 7.06) 4.72 (1.35 to 16.05) | 1.55 (0.37 to 5.80) 2.98 (0.70 to 11.35) |
INF 10 mg/kg, median OR (95% CrI) | 2.03 (0.39 to 10.64) 2.84 (0.55 to 14.66) | 1.87 (0.48 to 17.26) 4.17 (1.04 to 16.73) | NA |
INF 5 mg/kg, median OR (95% CrI) | 2.14 (0.42 to 11.05) 2.97 (0.58 to 15.36) | 2.22 (0.58 to 8.52) 4.94 (1.25 to 19.68) | NA |
OZA 0.92 mg, median OR (95% CrI) | 1.22 (0.27 to 5.56) 1.71 (0.38 to 7.76) | 2.59 (0.78 to 8.86) 5.79 (1.68 to 20.63) | 1.61 (0.40 to 6.51) 3.11 (0.77 to 12.78) |
PLO, median OR (95% CrI) | 3.01 (0.98 to 9.62) 4.20 (1.37 to 13.19) | 4.64 (1.85 to 12.05) 10.40 (3.91 to 28.33) | 3.71 (1.29 to 10.81) 7.17 (2.46 to 21.26) |
TOF 10 mg, median OR (95% CrI) | 0.46 (0.09 to 2.22) 0.64 (0.13 to 3.08) | 0.83 (0.24 to 3.01) 1.85 (0.51 to 7.00) | 0.56 (0.13 to 2.43) 1.07 (0.24 to 4.72) |
TOF 5 mg, median OR (95% CrI) | 0.51 (0.10 to 2.47) 0.72 (0.15 to 3.41) | 1.16 (0.33 to 4.13) 2.59 (0.71 to 9.70) | 0.78 (0.18 to 3.41) 1.50 (0.34 to 6.62) |
UPA 15 mg, median OR (95% CrI) | NA 1.40 (0.48 to 4.02) | NA 2.22 (0.85 to 5.90) | NA 1.93 (0.70 to 5.37) |
UPA 30 mg, median OR (95% CrI) | 0.72 (0.25 to 2.07) NA | 0.45 (0.17 to 1.17) NA | 1.93 (0.70 to 5.37) NA |
UST 90 mg q.8.w., median OR (95% CrI) | 1.38 (0.29 to 6.57) 1.93 (0.41 to 9.10) | 1.48 (0.40 to 5.55) 3.30 (0.85 to 12.77) | 1.47 (0.34 to 6.30) 2.84 (0.65 to 12.33) |
UST 90 mg q.12.w., median OR (95% CrI) | 1.58 (0.34 to 7.45) 2.20 (0.47 to 10.28) | 1.52 (0.42 to 5.58) 3.41 (0.89 to 12.93) | 1.70 (0.40 to 7.27) 3.29 (0.76 to 14.08) |
VED 300 mg q.4.w., median OR (95% CrI) | 0.77 (0.17 to 3.56) 1.08 (0.24 to 4.92) | 1.37 (0.38 to 5.04) 3.06 (0.82 to 11.66) | 0.81 (0.20 to 3.51) 1.57 (0.37 to 6.83) |
VED 300 mg q.8.w., median OR (95% CrI) | 0.86 (0.23 to 3.37) 1.19 (0.32 to 4.66) | 0.98 (0.30 to 3.41) 2.19 (0.63 to 7.78) | 0.89 (0.24 to 3.51) 1.73 (0.45 to 6.87) |
ADA = adalimumab; CrI = credible interval; GOL = golimumab; INF = infliximab; NA = not available; NMA = network meta-analysis; OR = odds ratio; OZA = ozanimod; PLO = placebo; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; q.8.w. = every 8 weeks; q.12.w. = every 12 weeks; TOF = tofacitinib; UPA = upadacitinib; UST = ustekinumab; VED = vedolizumab.
Note: Bold values indicate statistical significance. Upper values are for UPA 15 mg, and lower values are for UPA 30 mg.
OR > 1 indicates results favouring upadacitinib.
Source: Sponsor-submitted indirect treatment comparison.32
Clinical Response
The results from the unadjusted RE models suggested that in the biologic-naive population, no treatment was favoured for clinical response when upadacitinib 15 mg was compared with other active treatments. Upadacitinib 30 mg was favoured over adalimumab (OR = 7.99; 95% CrI, 2.07 to 30.59), golimumab 50 mg (OR = 4.72; 95% CrI, 1.35 to 16.05), golimumab 100 mg (OR = 3.86; 95% CrI, 1.12 to 12.15), infliximab 5 mg (OR = 4.94; 95% CrI, 1.25 to 19.68), infliximab 10 mg (OR = 4.17; 95% CrI, 1.04 to 16.73), and ozanimod (OR = 5.79; 95% CrI, 1.68 to 20.63). Among biologic-exposed patients, results showed that no treatment was favoured when upadacitinib 15 mg was compared with other active treatments. Upadacitinib 30 mg was favoured over ustekinumab 90 mg every 8 weeks (OR = 4.09; 95% CrI, 1.11 to 15.24) and ustekinumab 90 mg every 12 weeks (OR = 6.02; 95% CrI, 1.59 to 22.93). In the comparisons between upadacitinib 30 mg and other active treatments, the 95% CrIs of clinical response did not exclude the null.
Endoscopic Improvement
Results from the unadjusted RE models suggested that in the biologic-naive population, no treatment was favoured for endoscopic improvement when upadacitinib 15 mg or 30 mg was compared with other active treatments. Among biologic-exposed patients, results showed that upadacitinib 15 mg was favoured over ustekinumab 90 mg every 12 weeks (OR = 8.06; 95% CrI, 1.93 to 36.68). Upadacitinib 30 mg was favoured over tofacitinib (OR = 4.71; 95% CrI, 1.06 to 21.49), ustekinumab 90 mg every 8 weeks (OR = 5.20; 95% CrI, 1.29 to 22.33) and ustekinumab 90 mg every 12 weeks (OR = 12.44; 95% CrI, 3.00 to 56.08). In the comparisons between upadacitinib 30 mg and other active treatments, the 95% CrIs of endoscopic improvement did not exclude the null (Table 39).
Table 39: Summary of NMA Results for Efficacy Outcomes at Maintenance (UPA 15 mg or UPA 30 mg Versus Comparators), Random-Effects Models in the Biologic-Exposed Population
Treatment | Clinical remission | Clinical response | Endoscopic improvement |
|---|---|---|---|
13 interventions, 9 studies | 13 interventions, 8 studies | 12 interventions, 7 studies | |
ADA 40 mg q.2.w., median OR (95% CrI) | 5.82 (0.71 to 43.74) 7.38 (0.90 to 55.92) | 2.58 (0.50 to 12.72) 4.24 (0.82 to 21.11) | NA |
OZA 0.92 mg, median OR (95% CrI) | 4.06 (0.73 to 24.05) 5.13 (0.92 to 30.01) | 1.91 (0.50 to 7.37) 3.15 (0.81 to 12.34) | 3.34 (0.77 to 15.35) |
PLO, median OR (95% CrI) | 15.37 (4.62 to 64.31) 19.37 (5.82 to 80.30) | 7.35 (2.84 to 19.58) 12.11 (4.63 to 32.92) | 9.52 (3.25 to 31.08) 14.59 (5.09 to 47.46) |
TOF 10 mg, median OR (95% CrI) | 3.26 (0.68 to 18.34) 4.09 (0.85 to 22.72) | 0.84 (0.22 to 3.22) 1.40 (0.35 to 5.42) | 1.99 (0.45 to 9.02) 3.05 (0.70 to 13.78) |
TOF 5 mg, median OR (95% CrI) | 5.93 (1.21 to 34.38) 7.50 (1.53 to 43.12) | 1.52 (0.39 to 5.94) 2.50 (0.63 to 9.93) | 3.07 (0.69 to 14.02) 4.71 (1.06 to 21.49) |
UPA 15 mg, median OR (95% CrI) | NA 1.26 (0.52 to 3.10) | NA 1.65 (0.66 to 4.08) | NA 0.65 (0.27 to 1.60) |
UPA 30 mg, median OR (95% CrI) | 0.80 (0.32 to 1.93) NA | 0.61 (0.25 to 1.51) NA | 1.54 (0.63 3.76) NA |
UST 90 mg q.12.w., median OR (95% CrI) | 8.12 (1.66 to 45.69) 10.26 (2.11 to 55.45) | 3.66 (0.99 to 13.63) 6.02 (1.59 to 22.93) | 8.06 (1.93 to 36.68) 12.44 (3.00 to 56.08) |
UST 90 mg q.8.w., median OR (95% CrI) | 4.89 (1.04 to 27.09) 6.16 (1.31 to 33.22) | 2.48 (0.69 to 9.12) 4.09 (1.11 to 15.24) | 3.37 (0.83 to 14.56) 5.20 (1.29 to 22.33) |
VED 300 mg q.4.w., median OR (95% CrI) | 1.92 (0.30 to 13.03) 2.41 (0.37 to 16.21) | 1.92 (0.43 to 8.68) 3.16 (0.70 to 14.51) | 1.00 (0.17 to 5.45) 1.54 (0.27 to 8.41) |
VED 300 mg q.8.w., median OR (95% CrI) | 1.89 (0.34 to 10.62) 2.38 (0.43 to 13.27) | 1.71 (0.43 to 6.70) 2.80 (0.70 to 11.11) | 1.35 (0.28 to 6.44) 2.06 (0.43 to 9.91) |
ADA = adalimumab; CrI = credible interval; GOL = golimumab; INF = infliximab; NA = not available; NMA = network meta-analysis; OR = odds ratio; OZA = ozanimod; PLO = placebo; q.2.w. = every 2 weeks; q.4.w. = every 4 weeks; q.8.w. = every 8 weeks; q.12.w. = every 12 weeks; TOF = tofacitinib; UPA = upadacitinib; UST = ustekinumab; VED = vedolizumab.
Note: Bold values indicate statistical significance. Upper values are for UPA 15 mg, and lower values are for UPA 30 mg.
OR > 1 indicates results favouring upadacitinib.
Source: Sponsor-submitted indirect treatment comparison.32
For the outcomes of clinical remission, clinical response, and endoscopic improvement, even though the associated 95% CrIs of the point estimate excluded the null, wide CrIs were reported in general, implying that the magnitude of these potential differences is uncertain. Conclusions on the potential clinical benefits related to the treatment with upadacitinib cannot be drawn based on the imprecise estimates of these outcomes.
Harms
Safety outcomes were assessed in the overall population (bio-naive and/or bio-exposed) (Table 40 and Table 41).
The results of the adjusted RE models showed there was no evidence for a difference between upadacitinib and other active comparators in terms of the incidence of all AEs in either the induction or maintenance phase.
For discontinuations due to AEs, upadacitinib 45 mg was favoured over tofacitinib (OR = 0.25; 95% CrI, 0.06 to 0.89) and adalimumab (OR = 0.29; 95% CrI, 0.09 to 0.88) in the overall population during induction therapy. Upadacitinib 15 mg was favoured over adalimumab (OR = 0.24; 95% CrI, 0.05 to 0.97) during maintenance therapy. There was no evidence for a difference between upadacitinib and other active comparators for discontinuation due to AEs.
Table 40: Summary of NMA Results for Safety Outcomes at Induction, UPA 45 mg Versus Comparators, Overall Population, RE Models
Treatment | All AEs | Discontinuation due to AEs | SAEs | Serious infections |
|---|---|---|---|---|
Median OR (95% Crl) | ||||
ADA 160/80 | 1.20 (0.83 to 1.81) | 0.29 (0.09 to 0.88) | 0.91 (0.35 to 2.42) | 0.98 (0.24 to 3.99) |
GOL 200/100 | 1.70 (0.99 to 2.81) | 0.96 (0.07 to 27.80) | 1.29 (0.38 to 4.61) | 5.22 (0.57 to 177.77) |
INF 5 mg/kg | 0.49 (0.19 to 1.42) | 0.42 (0.08 to 2.31) | 0.83 (0.23 to 3.12) | 1.57 (0.17 to 48.72) |
OZA 0.92 mg | 1.52 (0.84 to 2.74) | 0.23 (0.05 to 1.08) | 0.43 (0.11 to 1.56) | 1.08 (0.20 to 6.07) |
PLO | 1.13 (0.85 to 1.51) | 0.24 (0.10 to 0.61) | 0.54 (0.25 to 1.19) | 0.86 (0.28 to 2.33) |
TOF 10 mg | 1.08 (0.73 to 1.67) | 0.25 (0.06 to 0.89) | 0.86 (0.29 to 2.49) | 1.37 (0.32 to 5.76) |
UST 6 mg/kg | 1.16 (0.71 to 1.85) | NA | 1.15 (0.36 to 3.91) | 4.81 (0.54 to 167.40) |
VED 300 mg | 1.51 (0.98 to 2.27) | 0.36 (0.08 to 1.84) | 0.90 (0.29 to 2.84) | 2.55 (0.44 to 22.34) |
ADA160/80 = adalimumab 160 mg at week 0 followed by 80 mg at week 2; AE = adverse event; CrI = credible interval; GOL200/100 = GOL 200 mg at week 0 and 100 mg at week 2; INF = infliximab; NA = not available; NMA = network meta-analysis; OR = odds ratio; OZA = ozanimod; PLO = placebo; RE = random effects unadjusted model; REA = random-effects adjusted model; SAE = serious adverse event; TOF = tofacitinib; UPA = upadacitinib; UST = ustekinumab; VED = vedolizumab.
Note: All AEs in the overall population in the induction trials were evaluated using REAs. SAEs, discontinuation due to AEs, and serious infections in induction trials were evaluated using REs.
OR < 1 indicates results favouring upadacitinib.
Bold values indicate statistical significance.
Source: Sponsor-submitted indirect treatment comparison.32
Table 41: Summary of NMA Results for Safety Outcomes at Maintenance, UPA 15 mg or UPA 30 mg Versus Comparators, Overall Population, RE Models
Treatment | All AEs | Discontinuation due to AEs | SAEs | Serious infections |
|---|---|---|---|---|
Median OR (95% Crl) | ||||
ADA 40 mg q.2.w. | 0.79 (0.30 to 2.06) 0.83 (0.31 to 2.17) | 0.24 (0.05 to 0.97) 0.40 (0.09 to 1.50) | 0.28 (0.07 to 1.01) 0.24 (0.06 to 0.87) | 2.27 (0.09 to 113.62) 1.69 (0.07 to 86.28) |
GOL 100 mg | 0.58 (0.18 to 1.52) 0.62 (0.18 to 1.60) | 0.21 (0.04 to 1.04) 0.36 (0.08 to 1.69) | 0.34 (0.09 to 1.50) 0.29 (0.08 to 1.30) | 0.45 (0.05 to 3.87) 0.34 (0.04 to 2.93) |
GOL 50 mg | 0.70 (0.22 to 2.00) 0.74 (0.23 to 2.10) | 0.40 (0.07 to 2.16) 0.68 (0.13 to 3.45) | 0.53 (0.13 to 2.41) 0.46 (0.11 to 2.11) | 0.46 (0.05 to 3.77) 0.35 (0.04 to 2.99) |
INF 10 mg/kg | 0.62 (0.16 to 2.24) 0.65 (0.17 to 2.32) | 0.32 (0.06 to 1.62) 0.54 (0.11 to 2.58) | 0.54 (0.13 to 2.14) 0.46 (0.11 to 1.81) | 0.48 (0.07 to 3.14) 0.35 (0.05 to 2.48) |
INF 5 mg/kg | 0.89 (0.26 to 3.13) 0.93 (0.27 to 3.27) | 0.35 (0.06 to 1.83) 0.60 (0.12 to 2.88) | 0.62 (0.15 to 2.43) 0.53 (0.13 to 2.10) | 1.42 (0.16 to 13.38) 1.07 (0.12 to 10.52) |
OZA 0.92 mg | 0.66 (0.23 to 1.93) 0.70 (0.24 to 2.01) | 0.69 (0.09 to 5.55) 1.17 (0.17 to 9.12) | 0.76 (0.18 to 3.27) 0.65 (0.15 to 2.83) | 1.84 (0.17 to 25.79) 1.40 (0.13 to 20.74) |
PLO | 1.11 (0.50 to 2.49) 1.16 (0.53 to 2.60) | 0.32 (0.09 to 1.01) 0.53 (0.18 to 1.57) | 0.48 (0.17 to 1.36) 0.41 (0.14 to 1.19) | 0.82 (0.19 to 3.30) 0.62 (0.13 to 2.66) |
TOF 10 mg | 0.86 (0.29 to 2.64) 0.91 (0.30 to 2.75) | 0.68 (0.15 to 3.02) 1.16 (0.28 to 4.80) | 0.58 (0.13 to 2.56) 0.49 (0.10 to 2.22) | 1.98 (0.10 to 80.82) 1.47 (0.07 to 60.14) |
TOF 5 mg | 1.29 (0.44 to 3.90) 1.36 (0.46 to 4.09) | 0.74 (0.15 to 3.26) 1.25 (0.30 to 5.20) | 0.63 (0.14 to 2.92) 0.54 (0.12 to 2.53) | 0.84 (0.05 to 11.93) 0.63 (0.04 to 9.40) |
UPA 15 mg | NA 1.05 (0.47 to 2.36) | NA 1.69 (0.48 to 6.36) | NA 0.86 (0.26 to 2.71) | NA 0.76 (0.15 to 3.54) |
UPA 30 mg | 0.95 (0.42 to 2.13) NA | 0.59 (0.16 to 2.07) NA | 1.17 (0.37 to 3.81) NA | 1.31 (0.28 to 6.50) NA |
UST 90 q.12.w. | 1.84 (0.61 to 5.65) 1.94 (0.64 to 5.95) | 1.49 (0.26 to 8.70) 2.52 (0.50 to 13.73) | 0.65 (0.15 to 2.78) 0.55 (0.12 to 2.43) | 0.52 (0.06 to 3.83) 0.39 (0.04 to 2.94) |
UST 90 q.8.w. | 1.21 (0.40 to 7.75) 1.27 (0.42 to 3.94) | 1.49 (0.26 to 8.70) 2.52 (0.50 to 13.73) | 0.56 (0.13 to 2.40) 0.48 (0.11 to 2.05) | 1.19 (0.13 to 12.02) 0.91 (0.09 to 9.10) |
VED 300 q.4.w. | 1.27 (0.40 to 3.92) 1.33 (0.42 to 4.13) | 0.93 (0.17 to 5.25) 1.57 (0.33 to 8.21) | 0.79 (0.18 to 3.30) 0.67 (0.15 to 2.86) | 1.73 (0.17 to 21.89) 1.32 (0.12 to 17.05) |
VED 300 q.8.w. | 1.06 (0.38 to 2.82) 1.12 (0.40 to 3.01) | 0.82 (0.18 to 3.65) 1.37 (0.35 to 5.67) | 0.65 (0.17 to 2.22) 0.55 (0.12 to 2.43) | 1.03 (0.13 to 8.20) 0.77 (0.09 to 6.41) |
ADA = adalimumab; AE = adverse event; CrI = credible interval; GOL = golimumab; INF = infliximab; NA = not available or not applicable; OR = odds ratio; OZA = ozanimod; PLO = placebo; RE = random-effects unadjusted model; SAE = serious adverse event; TOF = tofacitinib; UPA = upadacitinib; UST = ustekinumab; VED = vedolizumab.
Note: RE models were used for all AEs, SAEs, discontinuation due to AEs, and serious infections in maintenance trials.
Upper values are for UPA 15 mg, and lower values are for UPA 30 mg.
OR < 1 indicates results favouring upadacitinib.
Bold values indicate statistical significance.
Source: Sponsor-submitted indirect treatment comparison.32
For SAEs, the results of the unadjusted RE models showed no evidence for a difference between upadacitinib and other active treatments during induction therapy, but upadacitinib 30 mg was favoured over adalimumab (OR = 0.24; 95% CrI, 0.06 to 0.87) during the maintenance therapy.
For serious infections, there was no evidence for a difference between upadacitinib and other active treatments in either the induction or maintenance phase.
Wide CrIs were noted in the comparisons between upadacitinib and other active treatments for the safety outcomes, particularly discontinuation due to AEs, SAEs, and serious infections; therefore, these results are generally untrustworthy.
Critical Appraisal of the Sponsor-Submitted ITC
In the sponsor-submitted ITC, studies were identified by searching multiple databases based on prespecified inclusion and exclusion criteria. The reviewers used appropriate methods for study selection and data extraction. The quality of the included studies was assessed using the Cochrane risk-of-bias tool (version 2). The authors of this ITC claimed that all of the included studies had a low risk of bias. Notably, in 1 of the published ITCs that included similar studies,34 9 out of 28 studies were considered to have a low risk of bias. Therefore, it is reasonable to assume that some of the included studies had more than a low risk of bias. In addition, the assessment of the risk of bias in these studies was not conducted by outcome, which is a flaw. It also appears that, as some trials are available only as trial registrations, it is unclear how the risk of bias would have been properly assessed.
Sources of heterogeneities and potential treatment effect modifiers in the included studies (such as study design [e.g., inclusion and exclusion criteria and outcome definitions] and notable heterogeneity in a number of patients’ baseline characteristics [e.g., previous UC medications or differences across the placebo arms]) were identified a priori and some of them were addressed in data analyses. However, in several studies, data for potential effect modifiers were unavailable. The maintenance phase, in particular, is problematic. In this phase, 1 of the key issues is that some of the placebo arms were considered fundamentally different from 1 another; patients received and responded to different induction treatments with potentially different persistence of treatment effect after treatment ended. Given all these concerns, the transitivity assumption in an NMA may not be upheld. Despite various statistical techniques being employed to lessen the impact of potential clinical heterogeneity, such as baseline risks on the estimated treatment effect of upadacitinib, there is still uncertainty in the ITC results. The approaches used to adjust the differences in study design (TT versus RR) are potential solutions to adjust the cross-study heterogeneity in UC trials; however, it is uncertain whether the adjustment is adequate. For example, when the placebo rates were heterogeneous, it could not be adequately addressed with an REA model.
In addition, the network is sparse, including, in general, a single trial for each treatment. Coherence could not be assessed due to the lack of relevant closed loops when comparing with other active treatments. As a result, all evidence is indirect, which reduces our certainty in the study findings. Furthermore, there was a lack of description on the sensitivity analyses that were conducted to adjust for the baseline risk. Despite the use of various strategies, such as RE models in addition to FE models or vague prior distributions on key parameters, wide CrIs are observed for many efficacy and safety outcomes, especially in the maintenance period. This implies considerable uncertainty in the magnitude of the treatment effects of upadacitinib versus the comparators.
Safety data were sparse and only available in the overall population. These data are likely confounded by treatment efficacy, since UC is commonly reported as an AE, SAE, and WDAE in trials for UC therapies.
In addition, some important patient outcomes were not included in the analyses, such as HRQoL, change in symptoms, and change in work productivity.
Description of Published ITCs
Three studies (Lasa et al., 2022;33 Bur et al., 2021;34 and Li et al., 202235) describing NMAs of the efficacy and safety of biologics and small-molecule drugs, including upadacitinib, for patients with moderately to severely active UC were identified. Two of them assessed the various biologic drugs and small-molecule drugs for UC: TNF alpha antagonists, anti-integrin drugs, IL-12 and IL-23 antagonists, S1P receptors, and JAK inhibitors. The analysis by Li et al. focused on JAK inhibitors only. Details on study design (patient population, intervention and comparators, outcome measures) and analysis methods are provided in Table 42.
Table 42: Summary of Published ITCs
Detail | Lasa, 2022 | Bur, 2021 | Li, 2022 |
|---|---|---|---|
Population | Adult patients (≥ 18 years) with moderate to severe UC who were either biologic-naive or biologic-exposed | Adult patients (≥ 18 years) with UC | |
Intervention or comparator |
|
| JAK inhibitors only:
|
Outcomes |
|
|
|
Study design | Phase III RCTs; 29 included | Phase III RCTs; 28 included | RCTs; 7 included |
ITC analysis methods | The NMA was conducted using the multivariate frequentist approach. Random-effects models were adopted. Results were reported for both the bio-naive and bio-experienced patient population at the end of the induction and maintenance phases. The quality of the included studies was assessed using the Cochrane risk-of-bias tool, version 2. Publication bias was examined. | The NMA was conducted using frequentist models (random-effects models were selected). The relative risk of failure to achieve each of the end points of interest was measured, instead of relative risk of improvement or OR, for some analyses. A priori subgroup analyses based on prior exposure to TNF alpha antagonists were performed. The quality of the included studies was assessed using the Cochrane risk-of-bias tool. Publication bias was examined. | A Bayesian NMA was performed. Fixed-effects models were used (the authors justified this by noting that because the network of evidence was sparse, there was insufficient evidence to effectively estimate heterogeneity between studies using a random-effects model). The quality of the included studies was assessed using the Cochrane risk-of-bias tool. |
Key limitations | Substantial heterogeneity was found across the included trials, for example, the follow-up durations of 6 to 14 weeks for induction therapy and 26 to 66 weeks for maintenance therapy, varied outcome measure definitions, and study design (some trials recruited primarily an Asian population; approximately half of the maintenance trials used an RR design and half used a TT design). In the direct pairwise meta-analysis, the I2 was 81% and 80% in the analyses of induction of clinical remission and induction of endoscopic improvement, respectively. There were insufficient details provided on whether the authors attempted to minimize the bias from the imbalance distribution of treatment effect modifiers across the different studies, except that for the maintenance trials with an TT or RR design, separate pairwise meta-analyses and NMAs were conducted for these different trial designs. All included studies were industry-sponsored. | Substantial heterogeneity existed in the included trials. Some trials were old; therefore, they differed in many ways, such as previous or concomitant UC medication use, patients’ demographic characteristics, time points assessed, and outcome measure definitions. Nine out of the 28 trials were considered to have a low risk of bias. Not much information was available regarding treatment effect modifiers and how the imbalance in these effect modifiers among the included studies were addressed. All included trials were industry-sponsored. | This study was not specific for moderate to severe UC, and some of the comparators have not been approved in Canada, (e.g., peficitinib and filgotinib). Not all relevant comparators were included; therefore, the network is incomplete. A Bayesian NMA was performed, but no additional regression adjustments, such as for baseline risk or adjustments for specific baseline characteristics, were made. Heterogeneity across the included trials was not discussed in detail. In addition, due to the limited number of trials that were included, subgroup analyses or sensitivity analyses could not be performed. Publication bias was not examined due to the small number of included trials. |
Authors’ key findings and conclusions | “Upadacitinib was significantly superior to all other interventions for the induction of clinical remission (infliximab [OR 2.70; 95% CI, 1.18–6.20], adalimumab [4.64, 2.47–8.71], golimumab [3.00, 1.32–6.82], vedolizumab [3.56, 1.84–6.91], ustekinumab [2.92, 1.31–6.51], etrolizumab [4.91, 2.59–9.31], tofacitinib [2.84, 1.28–6.31], filgotinib 100 mg [6.15, 2.98–12.72], filgotinib 200 mg [4.49, 2.18–9.24], and ozanimod [2.70, 1.18–6.20]). No differences between active interventions were observed when assessing adverse events and serious adverse events.” | “In a network meta-analysis, upadacitinib 45mg o.d. ranked first for clinical remission in all patients, patients naive to anti-TNF alpha drugs, and patients previously exposed. Upadacitinib was more likely to lead to adverse events, but serious adverse events were no more frequent, and withdrawals due to adverse events were significantly lower than with placebo.” | “Based on indirect comparisons, peficitinib 75 mg/75 mg BID/150 mg, tofacitinib 3 mg and filgotinib 100 mg were the most efficacious JAK inhibitor interventions in patients with UC” (upadacitinib was not mentioned in the conclusions). |
AE = adverse event; BID = twice daily; ITC = indirect treatment comparison; JAK = Janus kinase; NMA = network meta-analysis; OR = odds ratio; RCT = randomized controlled trial; RR = re-randomized study design; SAE = serious adverse event; TNF = tumour necrosis factor; TT = treat-through study design; UC = ulcerative colitis; WDAE = withdrawal due to adverse event.
aTD-1473 is an oral pan-JAK inhibitor indicated for inflammatory bowel disease.
Sources: Lasa, 2022;33 Bur, 2021;34 and Li, 2022.35
Similar to the sponsor-submitted ITC, the quality of the 3 published ITCs was compromised, and the effect estimates are uncertain, mostly due to the concerns of substantial heterogeneity from different sources and insufficient description on the methods used to address and adjust these heterogeneities. It is likely that the underlying transitivity assumption of the NMA was not upheld, and the lack of relevant closed loops meant that consistency could not be assessed. All evidence is therefore indirect, and wide CrIs or CIs of the effect estimates are observed, which means the magnitude of the effects is uncertain.
Other Relevant Evidence
The results of an ongoing long-term extension study, M14 to 533, were not available at the time of this review. (The study was designed to evaluate the long-term safety and efficacy of upadacitinib in patients with UC who had not responded at the end of the induction period in U-ACHIEVE substudy 1, who had a loss of response during the maintenance period of U-ACHIEVE substudy 3, or who had successfully completed U-ACHIEVE substudy 3). No other relevant evidence was identified.
Discussion
Summary of Available Evidence
Three phase III RCTs (U-ACHIEVE Induction, N = 474; U-ACCOMPLISH, N = 522; U-ACHIEVE Maintenance, N = 1,046) submitted by the sponsor were included in this systematic review. The objectives of all 3 studies were to evaluate the efficacy and safety of upadacitinib in patients with moderately to severely active UC. The studies enrolled adult patients with a diagnosis of moderate to severe UC who had an inadequate response, loss of response, or were intolerant to either conventional therapy or biologic drugs. In the induction trials (U-ACHIEVE Induction and U-ACCOMPLISH), eligible patients were randomized to receive oral upadacitinib 45 mg once daily or matching placebo for 8 weeks in a double-blind manner. At the end of the 8 weeks, those who were deemed clinical responders were eligible to enter the maintenance study (U-ACHIEVE Maintenance), while nonresponders were given open-label upadacitinib for an additional 8 weeks. Patients who entered the maintenance study were treated with oral upadacitinib 15 mg or 30 mg once daily, or placebo for up to 52 weeks. The primary efficacy outcome of these 3 studies was the proportion of patients achieving or maintaining clinical remission according to the 3-component Mayo score. Key secondary efficacy end points were clinical response, endoscopic improvement, mucosal healing, change in UC symptoms, and maintenance of remission. Other efficacy end points included rectal bleeding, histologic remission, HRQoL, and work productivity.
In the 2 induction trials, about 60% of patients were male and 65% to 71% were White. The mean age of patients enrolled in the induction trials was 42 to 44 years. At baseline, 50% to 53% of patients had inadequate response, loss of response, or intolerance to previous biologic therapy, and 47% to 50% of the patients had inadequate response, loss of response, or intolerance to previous conventional therapy. The majority of patients had a mean adapted Mayo score of 7 or less. Corticosteroids were the most commonly prescribed UC medications before study randomization. During maintenance therapy, patients’ baseline characteristics were generally comparable to those in the induction period.
The key limitations of these studies were the disproportionate discontinuation rates across the treatment arms, especially in the maintenance study. However, the AE of “ulcerative colitis” accounted for much of the imbalance in the induction studies and “lack of efficacy” and “loss of response” accounted for most of the discontinuations in the maintenance study. Since these patients were imputed as nonresponders, a bias is less likely to be introduced for the binary outcomes. Although subgroup analyses were performed to examine the consistency of the treatment effect observed for the primary efficacy end points, proper interpretation of all subgroups was not possible due to lack of sample size considerations for these subgroups and lack of comparisons between subgroups. In addition, these trials were not able to address long-term efficacy and safety, given the 1-year study duration.
Indirect evidence in 1 sponsor-submitted ITC was summarized in this review. In this study, upadacitinib was compared with adalimumab, golimumab, infliximab, tofacitinib, ustekinumab, and vedolizumab to evaluate the relative efficacy and safety of active treatments for patients with moderately to severely active UC. Clinical remission, clinical response, endoscopic improvement, and safety were evaluated. Twenty-three RCTs were included in the NMA. The key limitations of this study are sparse networks, heterogeneity in patients’ baseline characteristics and trial characteristics, and inadequate adjustments for the clinical heterogeneity. In addition, 3 published ITCs were identified from CADTH’s literature search. They were associated with substantial limitations and the results are not reported in this review.
Interpretation of Results
Efficacy
During the induction period of U-ACHIEVE, clinical remission based on the adapted Mayo score at week 8 was achieved in 26.1% of patients in the upadacitinib group and 4.8% of patients in the placebo group; the between-group difference was 21.6% (95% CI, 15.8% to 27.4%). In U-ACCOMPLISH, clinical remission per adapted Mayo score was achieved in 33.5% of patients in the upadacitinib group and 4.1% of patients in the placebo group; the between-group difference was 29.0% (95% CI, 23.2% to 34.7%). At the end of the maintenance period of U-ACHIEVE at week 52, clinical remission was maintained in 42.3% of patients in the upadacitinib 15 mg group, 51.7% of patients in the upadacitinib 30 mg group, and 12.1% of patients in the placebo group; the between-group differences were 30.7% (95% CI, 21.7% to 39.8%) for upadacitinib 15 mg versus placebo and 39.0% (95% CI, 29.7% to 48.2%) for upadacitinib 30 mg versus placebo.
Similarly, the results for the proportion of patients achieving clinical response, endoscopic improvement or remission, histologic improvement, and mucosal healing favoured patients who were treated with upadacitinib compared with those treated with placebo for both the induction and maintenance periods. For maintenance therapy, the treatment effect for upadacitinib 15 mg versus placebo was smaller than for upadacitinib 30 mg versus placebo. The clinical expert consulted by CADTH indicated that all of the between-group differences were clinically meaningful. The results of the subgroup analyses based on patients’ baseline characteristics were consistent with those in the overall population. The results for other efficacy outcomes suggested that treatment with upadacitinib was associated with better symptom relief and improved HRQoL compared with placebo during both the induction and maintenance periods. The changes in HRQoL measured with IBDQ and FACIT-F favoured upadacitinib therapy. The impact of UC on work was evaluated between the upadacitinib group and the placebo group; however, this outcome was not adjusted for multiplicity and the results should be interpreted with caution. Treatment with upadacitinib may be associated with lower rates of hospitalization due to UC for both the induction and maintenance periods.
The main efficacy outcome (clinical remission) and key secondary outcome that selected which patients moved into the maintenance period (clinical response) was based on the Mayo score. In the studies included in this review, the adapted version of the Mayo score was used to evaluate the treatment effect of the study drug. According to an FDA guidance document,31 the full Mayo score is subject to bias due to the PGA component, and that this component was poorly correlated to disease activity. Accordingly, the definitions for clinical remission and clinical response in the included trials for the current review conform to the definitions laid out by the FDA. Mucosal healing has emerged as a major therapeutic goal, since UC is a disease that is usually limited to the mucosal layer of the colon. It is important for this outcome to be evaluated in patients with UC, as this would help in assessing disease activity and in better understanding the treatment effect, and help improve patient prognosis.5,36
Based on the results of the sponsor-submitted ITC, for the induction phase, treatment with upadacitinib 45 mg may be associated with higher rates of clinical remission, clinical response, and endoscopic improvement compared with some of the active comparators. The estimates are associated with considerable uncertainty due to the lack of any direct evidence, the sparsity of the network, and the potential for the transitivity assumption to have been violated. The analysis of findings for the maintenance phase required adjustment for differences in study designs and there were fundamental differences in the placebo arms across the studies. The statistical techniques adopted in the sponsor’s ITC are possible strategies to address cross-study heterogeneity, lessen the impact of potential clinical heterogeneity on the estimated treatment effect of upadacitinib, and make NMAs feasible; however, they cannot adequately remove uncertainty. Therefore, firm conclusions could not be established for the efficacy of upadacitinib compared with other relevant active treatments in achieving clinical response, clinical remission, and endoscopic improvement.
Harms
The proportion of patients experiencing at least 1 AE during induction was different between the 2 induction trials. In U-ACHIEVE Induction, at least 1 AE was reported by 56.4% and 61.9% of patients in the upadacitinib group and placebo group, respectively. In U-ACCOMPLISH, at least 1 AE was reported by 52.9% and 39.5% of patients in the upadacitinib group and placebo group, respectively. UC was more often reported as an AE in the placebo groups and was a major driver when the risk of AEs, SAEs, or WDAEs was high in the placebo group compared with the upadacitinib group. Patients who were treated with placebo may be more likely to experience the AE of UC, due to the lack of efficacy of treatment with placebo. During the maintenance period, AEs were reported in 75.2%, 75.3%, and 73.5% of the patients in the upadacitinib 15 mg, upadacitinib 30 mg, and placebo groups, respectively.
In the induction period, there were no AEs of active tuberculosis, malignancy, adjudicated VTE, or gastrointestinal perforation reported in the upadacitinib groups. The incidence of opportunistic infection, excluding tuberculosis and herpes zoster, herpes zoster, lymphopenia, and neutropenia, was higher in the upadacitinib groups. At the end of the maintenance period, patients treated with up to 1 year of upadacitinib reported cases of herpes zoster, neutropenia, malignancy, hepatic disorder, lymphopenia, and VTEs. The numbers of events were low for malignancy and VTE at this time point. Longer-term data are needed to fully understand the long-term safety profile of upadacitinib in patients with UC.
Due to the limitations in the sponsor-submitted ITC, credible conclusions regarding the relative safety of upadacitinib versus other active treatments cannot be drawn. While there may be expectations that upadacitinib has a favourable safety profile compared with tofacitinib based on its mechanism of action as a selective JAK inhibitor, currently, there is insufficient evidence to determine the safety of upadacitinib versus tofacitinib in patients with moderately to severely active UC.
Conclusions
Based on the 2 induction trials and 1 maintenance trial, oral upadacitinib 15 mg, 30 mg, and 45 mg once daily was superior to placebo in achieving induction and maintenance of clinical remission, clinical response, endoscopic improvement, and mucosal healing in patients with moderately to severely active UC. Moreover, upadacitinib was also found to be effective in alleviating UC-related symptoms and improving HRQoL. Long-term data are needed to fully evaluate the safety profile of upadacitinib. Serious limitations in the available ITCs mean that it remains uncertain how upadacitinib compares with other active treatments in the efficacy and safety for moderately to severely active UC.
References
1.Peppercorn MA, Kane SV. Clinical manifestations, diagnosis, and prognosis of ulcerative colitis inadults. In: Lamont JT, Robson KM, eds. UpToDate. Waltham (MA): UpToDate; 2022: http://www.uptodate.com. Accessed 2022 Sep 7.
2.Danese S, Fiocchi C. Ulcerative colitis. N Engl J Med. 2011;365(18):1713-1725. PubMed
3.Fumery M, Singh S, Dulai PS, Gower-Rousseau C, Peyrin-Biroulet L, Sandborn WJ. Natural history of adult ulcerative colitis in population-based cohorts: a systematic review. Clin Gastroenterol Hepatol. 2018;16(3):343-356.e343. PubMed
4.Impact of inflammatory bowel disease in Canada. Toronto: Crohn's and Colitis Canada; 2018: https://crohnsandcolitis.ca/Crohns_and_Colitis/documents/reports/2018-Impact-Report-LR.pdf. Accessed 2022 Jul 2.
5.Ungaro R, Mehandru S, Allen PB, Peyrin-Biroulet L, Colombel JF. Ulcerative colitis. Lancet. 2017;389(10080):1756-1770. PubMed
6.Clinical study report: M14-234 (substudies 1 and 2, U-ACHIEVE induction). A multicenter, randomized, double-blind, placebo-controlled study to evaluate the safety and efficacy of upadacitinib (ABT-494) for induction and maintenance therapy in subjects with moderately to severely active ulcerative colitis [internal sponsor's report] North Chicago (IL): AbbVie; 2021 Aug.
7.Clinical study report: M14-675, U-ACCOMPLISH. A multicenter, randomized, double-blind, placebo-controlled induction study to evaluate the efficacy and safety of upadacitinib (ABT-494) in subjects with moderately to severely active ulcerative colitis [internal sponsor's report]. North Chicago (IL): AbbVie; 2021 Jun.
8.Clinical study report: M14-234 (substudy 3, U-ACHIEVE maintenance). A multicenter, randomized, double-blind, placebo-controlled study to evaluate the safety and efficacy of upadacitinib (ABT-494) for induction and maintenance therapy in subjects with moderately to severely active ulcerative colitis [internal sponsor's report]. North Chicago (IL): AbbVie; 2021 Aug.
9.Langan RC, Gotsch PB, Krafczyk MA, Skillinge DD. Ulcerative colitis: diagnosis and treatment. Am Fam Physician. 2007;76(9):1323-1330. https://www.aafp.org/dam/brand/aafp/pubs/afp/issues/2007/1101/p1323.pdf. Accessed 2022 Jul 22. PubMed
10.Bernstein CN, Nugent Z, Targownik LE, Singh H, Lix LM. Predictors and risks for death in a population-based study of persons with IBD in Manitoba. Gut. 2015;64(9):1403-1411. PubMed
11.Feuerstein JD, Isaacs KL, Schneider Y, Siddique SM, Falck-Ytter Y, Singh S. AGA clinical practice guidelines on the management of moderate to severe ulcerative colitis. Gastroenterology. 2020;158(5):1450-1461. PubMed
12.Cohen RD, Stein AC. Management of moderate to severe ulcerative colitis in adults. In: Kane SV, Robson KM, eds. UpToDate. Waltham (MA): UpToDate; 2021: http://www.uptodate.com. Accessed 2022 Sep 7.
13.Kuehn F, Hodin RA. Impact of modern drug therapy on surgery: ulcerative colitis. Visceral medicine. 2018;34(6):426-431. PubMed
14.Sabino J, Verstockt B, Vermeire S, Ferrante M. New biologics and small molecules in inflammatory bowel disease: an update. Therap Adv Gastroenterol. 2019;12:1756284819853208. PubMed
15.Rinvoq (upadacitinib): 15 mg, 30 mg, 45 mg extended-release oral tablets [product monograph]. St-Laurent (QC): AbbVie; 2021 Sep 30.
16.Olivera PA, Lasa JS, Bonovas S, Danese S, Peyrin-Biroulet L. Safety of Janus kinase inhibitors in patients with inflammatory bowel diseases or other immune-mediated diseases: a systematic review and meta-analysis. Gastroenterology. 2020;158(6):1554-1573.e1512.
17.Humira (adalimumab injection) [product monograph]. St-Laurent (QC): AbbVie Corporation; 2021 Apr 21: https://www.abbvie.ca/content/dam/abbvie-dotcom/ca/en/documents/products/HUMIRA_PM_EN.pdf. Accessed 2022 Sep 7.
18.Simponi (golimumab) solution for injection: 50 mg/0.5 mL, 100 mg/1.0 mL; and Simponi I.V. (golimumab) solution for infusion: 50 mg/4.0 mL [product monograph]. Toronto: Janssen Inc; 2018 Jan 15: https://pdf.hres.ca/dpd_pm/00043422.PDF. Accessed 2022 Sep 7.
19.Remicade (infliximab) powder for solution, sterile, lyophilized, 100 mg/vial [product monograph]. Toronto: Janssen Inc; 2017 Aug 4: https://crohnsandcolitis.ca/Crohns_and_Colitis/images/living-with-crohns-colitis/REMICADE-MONOGRAPH.PDF. Accessed 2022 Sep 7.
20.Xeljanz (tofacitinib) tablets, oral: 5 mg (as tofacitinib citrate), 10 mg (as tofacitinib citrate); and Xeljanz XR (tofacitinib extended-release) tablets, oral: 11 mg (as tofacitinib citrate) [product monograph]. Kirkland (QC): Pfizer Canada; 2022 May 9: https://www.pfizer.ca/sites/default/files/202206/Xeljanz_PM_EN_258173_09-May-2022.pdf. Accessed 2022 Sep 7.
21.Stelara (ustekinumab) solution for subcutaneous injection: 45 mg/0.5 mL, 90 mg/1.0 mL; and Stelara I.V. (ustekinumab) solution for intravenous infusion: 130 mg/26 mL (5 mg/mL) sterile solution [product monograph]. Toronto: Janssen Inc; 2017 Aug 18: https://crohnsandcolitis.ca/Crohns_and_Colitis/images/living-with-crohns-colitis/STELARA_MONOGRAPH.PDF. Accessed 2022 Sep 7.
22.Entyvio (vedolizumab) powder for concentrate for solution for infusion, 300 mg/vial [product monograph]. Oakville (ON): Takeda Canada; 2016 Mar 22: https://pdf.hres.ca/dpd_pm/00034239.PDF. Accessed 2022 Sep 7.
23.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. J Clin Epidemiol. 2016;75:40-46. PubMed
24.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2022 May 6.
25.Danese S, Vermeire S, Zhou W, et al. Upadacitinib as induction and maintenance therapy for moderately to severely active ulcerative colitis: results from three phase 3, multicentre, double-blind, randomised trials. Lancet. 2022;399(10341):2113-2128. PubMed
26.Drug Reimbursement Review sponsor submission: Rinvoq (upadacitinib) 15 mg, 30 mg, 45 mg extended-release oral tablets [internal sponsor's package]. Pointe-Claire (QC): AbbVie; 2022 Apr 18.
27.Lewis JD, Chuai S, Nessel L, Lichtenstein GR, Aberra FN, Ellenberg JH. Use of the noninvasive components of the Mayo score to assess clinical response in ulcerative colitis. Inflamm Bowel Dis. 2008;14(12):1660-1666. PubMed
28.Schroeder KW, Tremaine WJ, Ilstrup DM. Coated oral 5-aminosalicylic acid therapy for mildly to moderately active ulcerative colitis. A randomized study. N Engl J Med. 1987;317(26):1625-1629. PubMed
29.Geboes K, Riddell R, Ost A, Jensfelt B, Persson T, Löfberg R. A reproducible grading scale for histological assessment of inflammation in ulcerative colitis. Gut. 2000;47(3):404-409. PubMed
30.Bryant RV, Winer S, Travis SP, Riddell RH. Systematic review: histological remission in inflammatory bowel disease. Is 'complete' remission the new treatment paradigm? An IOIBD initiative. J Crohns Colitis. 2014;8(12):1582-1597. PubMed
31.Ulcerative colitis: clinical trial endpoints guidance for industry [draft guidance]. Rockville (MD): U.S. Food and Drug Administration (FDA); 2016: https://www.fda.gov/files/drugs/published/Ulcerative-Colitis--Clinical-Trial-Endpoints-Guidance-for-Industry.pdf. Accessed 2022 July 21.
32.The relative efficacy and safety of upadacitinib (UPA) versus relevant comparators for treating patients with moderately-to-severely active ulcerative colitis (UC): a mixed treatment comparison (MTC) using Bayesian network meta-analysis (NMA) [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Rinvoq (upadacitinib) 15 mg, 30 mg, 45 mg extended-release oral tablets [internal sponsor's package]. Pointe-Claire (QC): AbbVie; 2022.
33.Lasa JS, Olivera PA, Danese S, Peyrin-Biroulet L. Efficacy and safety of biologics and small molecule drugs for patients with moderate-to-severe ulcerative colitis: a systematic review and network meta-analysis. Lancet Gastroenterol Hepatol. 2022;7(2):161-170. PubMed
34.Burr NE, Gracie DJ, Black CJ, Ford AC. Efficacy of biological therapies and small molecules in moderate to severe ulcerative colitis: systematic review and network meta-analysis. Gut. 2021;22:22. PubMed
35.Li Y, Yao C, Xiong Q, et al. Network meta-analysis on efficacy and safety of different Janus kinase inhibitors for ulcerative colitis. J Clin Pharm Ther. 2022;06:06.
36.Chateau T, Feakins R, Marchal-Bressenot A, Magro F, Danese S, Peyrin-Biroulet L. Histological remission in ulcerative colitis: under the microscope is the cure. Am J Gastroenterol. 2020;115(2):179-189. PubMed
37.Dhanda AD, Creed TJ, Greenwood R, Sands BE, Probert CS. Can endoscopy be avoided in the assessment of ulcerative colitis in clinical trials? Inflamm Bowel Dis. 2012;18(11):2056-2062. PubMed
38.Mohammed Vashist N, Samaan M, Mosli MH, et al. Endoscopic scoring indices for evaluation of disease activity in ulcerative colitis. Cochrane Database Syst Rev. 2018;1(1):Cd011450. PubMed
39.Levesque B PS, King D, et al. Responsiveness of Central Endoscopic Assessment of Disease Activity Using the Modified Mayo Clinic Score in Ulcerative Colitis. Gastroenterology. 2013;144.
40.Walsh AJ, Ghosh A, Brain AO, et al. Comparing disease activity indices in ulcerative colitis. J Crohns Colitis. 2014;8(4):318-325. PubMed
41.Higgins PD, Schwartz M, Mapili J, Zimmermann EM. Is endoscopy necessary for the measurement of disease activity in ulcerative colitis? Am J Gastroenterol. 2005;100(2):355-361. PubMed
42.Mosli MH, Feagan BG, Zou G, et al. Reproducibility of histological assessments of disease activity in UC. Gut. 2015;64(11):1765-1773. PubMed
43.Lemmens B, Arijs I, Van Assche G, et al. Correlation between the endoscopic and histologic score in assessing the activity of ulcerative colitis. Inflamm Bowel Dis. 2013;19(6):1194-1201. PubMed
44.Kim DB, Lee K-M, Lee JM, et al. Correlation between histological activity and endoscopic, clinical, and serologic activities in patients with ulcerative colitis. Gastroenterol Res Pract. 2016;2016:5832051. PubMed
45.Mosli MH, Feagan BG, Zou G, et al. Development and validation of a histological index for UC. Gut. 2017;66(1):50-58. PubMed
46.Chen XL, Zhong LH, Wen Y, et al. Inflammatory bowel disease-specific health-related quality of life instruments: a systematic review of measurement properties. Health Qual Life Outcomes. 2017;15(1):177. PubMed
47.Alrubaiy L, Rikaby I, Dodds P, Hutchings HA, Williams JG. Systematic review of health-related quality of life measures for inflammatory bowel disease. J Crohns Colitis. 2015;9(3):284-292. PubMed
48.Irvine EJ. Quality of life of patients with ulcerative colitis: past, present, and future. Inflamm Bowel Dis. 2008;14(4):554-565. PubMed
49.Cohen BL, Zoega H, Shah SA, et al. Fatigue is highly associated with poor health-related quality of life, disability and depression in newly-diagnosed patients with inflammatory bowel disease, independent of disease activity. Aliment Pharmacol Ther. 2014;39(8):811-822. PubMed
50.Yarlas A, Bayliss M, Cappelleri JC, et al. Psychometric validation of the SF-36(®) Health Survey in ulcerative colitis: results from a systematic literature review. Qual Life Res. 2018;27(2):273-290. PubMed
51.Gibson PR, Vaizey C, Black CM, et al. Relationship between disease severity and quality of life and assessment of health care utilization and cost for ulcerative colitis in Australia: a cross-sectional, observational study. J Crohns Colitis. 2014;8(7):598-606. PubMed
52.Panés J, Domènech E, Aguas Peris M, et al. Association between disease activity and quality of life in ulcerative colitis: Results from the CRONICA-UC study. J Gastroenterol Hepatol. 2017;32(11):1818-1824. PubMed
53.Stark RG, Reitmeir P, Leidl R, König HH. Validity, reliability, and responsiveness of the EQ-5D in inflammatory bowel disease in Germany. Inflamm Bowel Dis. 2010;16(1):42-51. PubMed
54.Yarlas A, Maher SM, Bayliss MS, Lovley A, Cappelleri JC, DiBonaventura MD. Psychometric validation of the work productivity and activity impairment questionnaire in ulcerative colitis: results from a systematic literature review. J Patient Rep Outcomes. 2018;2(1):62-62. PubMed
55.Reilly MC, Gerlier L, Brabant Y, Brown M. Validity, reliability, and responsiveness of the work productivity and activity impairment questionnaire in Crohn's disease. Clin Ther. 2008;30(2):393-404. PubMed
56.Best WR, Becktel JM, Singleton JW, Kern F, Jr. Development of a Crohn's disease activity index. National Cooperative Crohn's Disease Study. Gastroenterology. 1976;70(3):439-444. PubMed
57.Sandborn W, Reilly M, Brown M, Brabant Y, Tan S, Gerlier L. P-046: Determination of the minimally important difference in WPAI: CD score that indicates a relevant impact on work productivity. Inflamm Bowel Dis. 2008;14(suppl_1):S24-S24.
58.Rubin D, Keyashian K, Bunnag A, et al. Correlation between clinical, endoscopic, and histologic disease activity in ulcerative colitis: 1712. [Abstracts: inflammatory bowel disease]. Am J Gastroenterol. 2012;107:S694.
59.Iacucci M, Cannatelli R, Gui X, et al. Assessment of endoscopic healing by using advanced technologies reflects histological healing in ulcerative colitis. J Crohns Colitis. 2020;14(9):1282-1289. PubMed
60.Samaan MA, Mosli MH, Sandborn WJ, et al. A systematic review of the measurement of endoscopic healing in ulcerative colitis clinical trials: recommendations and implications for future research. Inflamm Bowel Dis. 2014;20(8):1465-1471. PubMed
61.Travis SP, Stange EF, Lémann M, et al. European evidence-based Consensus on the management of ulcerative colitis: Current management. J Crohns Colitis. 2008;2(1):24-62. PubMed
62.U.S. Food and Drug Administration (FDA). Ulcerative colitis: developing drugs for treatment. (Draft guidance document, level 1) 2022; https://www.fda.gov/regulatory-information/search-fda-guidance-documents/ulcerative-colitis-developing-drugs-treatment. Accessed 2022 Sep 7.
63.Cooney RM, Warren BF, Altman DG, Abreu MT, Travis SP. Outcome measurement in clinical trials for ulcerative colitis: towards standardisation. Trials. 2007;8:17. PubMed
64.Magro F, Lopes J, Borralho P, et al. Comparison of the Nancy Index With Continuous Geboes Score: Histological Remission and Response in Ulcerative Colitis. J Crohns Colitis. 2020;14(7):1021-1025. PubMed
65.Lobatón T, Bessissow T, Ruiz-Cerulla A, et al. Prognostic value of histological activity in patients with ulcerative colitis in deep remission: A prospective multicenter study. United European Gastroenterol J. 2018;6(5):765-772. PubMed
66.Guyatt G, Mitchell A, Irvine EJ, et al. A new measure of health status for clinical trials in inflammatory bowel disease. Gastroenterology. 1989;96(3):804-810. PubMed
67.Irvine EJ. Development and subsequent refinement of the inflammatory bowel disease questionnaire: a quality-of-life instrument for adult patients with inflammatory bowel disease. J Pediatr Gastroenterol Nutr. 1999;28(4):S23-27. PubMed
68.Pallis AG, Mouzas IA, Vlachonikolis IG. The Inflammatory Bowel Disease Questionnaire: a review of its national validation studies. Inflamm Bowel Dis. 2004;10(3):261-269. PubMed
69.LeBlanc K, Mosli MH, Parker CE, MacDonald JK. The impact of biological interventions for ulcerative colitis on health-related quality of life. Cochrane Database Syst Rev. 2015(9):Cd008655. PubMed
70.Yellen SB, Cella DF, Webster K, Blendowski C, Kaplan E. Measuring fatigue and other anemia-related symptoms with the Functional Assessment of Cancer Therapy (FACT) measurement system. J Pain Symptom Manage. 1997;13(2):63-74. PubMed
71.Bonomi AE, Cella DF, Hahn EA, et al. Multilingual Translation of the Functional Assessment of Cancer Therapy (FACT) Quality of Life Measurement System. Qual Life Res. 1996;5(3):309-320. PubMed
72.Eremenco S, Cella D, Arnold BJ. A comprehensive method for the translation and cross-cultural validation of health status questionnaires. Eval Health Prof. 2005;28:212-232. PubMed
73.Tinsley A, Macklin EA, Korzenik JR, Sands BE. Validation of the functional assessment of chronic illness therapy-fatigue (FACIT-F) in patients with inflammatory bowel disease. Aliment Pharmacol Ther. 2011;34(11-12):1328-1336. PubMed
74.Reeve BB, Wyrwich KW, Wu AW, et al. ISOQOL recommends minimum standards for patient-reported outcome measures used in patient-centered outcomes and comparative effectiveness research. Qual Life Res. 2013;22(8):1889-1905. PubMed
75.Cohen J. A power primer. Psychol Bull. 1992;112(1):155-159. PubMed
76.Cella D, Yount SE, Sorensen M, Chartash E, Sengupta N, Grober JS. Validation of the Functional Assessment of Chronic Illness Therapy Fatigue Scale relative to other instrumentation in patients with rheumatoid arthritis. J Rheumatol. 2005;32(5):811-819. PubMed
77.Cella D, Eton DT, Lai JS, Peterman AH, Merkel DE. Combining anchor and distribution-based methods to derive minimal clinically important differences on the Functional Assessment of Cancer Therapy (FACT) anemia and fatigue scales. J Pain Symptom Manage. 2002;24(6):547-561. PubMed
78.Bernklev T, Jahnsen J, Lygren I, Henriksen M, Vatn M, Moum B. Health-related quality of life in patients with inflammatory bowel disease measured with the short form-36: psychometric assessments and a comparison with general population norms. Inflamm Bowel Dis. 2005;11(10):909-918. PubMed
79.Brooks R. The EuroQol Group after 25 years. eBook ed. Heidelberg (Germany): Springer Dordrecht; 2013.
80.Feng YS, Kohlmann T, Janssen MF, Buchholz I. Psychometric properties of the EQ-5D-5L: a systematic review of the literature. Qual Life Res. 2021;30(3):647-673. PubMed
81.Buchholz I, Janssen MF, Kohlmann T, Feng YS. A systematic review of studies comparing the measurement properties of the three-level and five-level versions of the EQ-5D. Pharmacoeconomics. 2018;36(6):645-661. PubMed
82.Sinnott PL, Joyce VR, Barnett P. Guidebook: preference measurement in economic analysis. Menlo Park (CA): Health Economics Resource Center (HERC); 2007: https://www.herc.research.va.gov/files/BOOK_419.pdf. Accessed 2022 Sep 7.
83.Herdman M, Gudex C, Lloyd A, et al. Development and preliminary testing of the new five-level version of EQ-5D (EQ-5D-5L). Qual Life Res. 2011;20(10):1727-1736. PubMed
84.Reilly MC, Bracco A, Ricci JF, Santoro J, Stevens T. The validity and accuracy of the Work Productivity and Activity Impairment questionnaire--irritable bowel syndrome version (WPAI:IBS). Aliment Pharmacol Ther. 2004;20(4):459-467. PubMed
Appendix 1: Literature Search Strategy
Note that this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases:
MEDLINE All (1946–present)
Embase (1974–present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: May 16, 2022
Alerts: Biweekly search updates until project completion
Search filters applied: No filters were applied to limit the retrieval by study type.
Limits:
Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
exp | Explode a subject heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
adj# | Requires terms to be adjacent to each other within # number of words (in any order) |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Author keyword heading word (MEDLINE) |
.dq | Candidate term word (Embase) |
.pt | Publication type |
.mp | Mapped term |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multidatabase Strategy
(Rinvoq* or upadacitinib* or abt494 or abt 494 or NEW4DV02U5).ti,ab,ot,kf,hw,nm,rn.
Colitis, ulcerative/
(Colitis or proctocolitis or colorectitis).ti,ab,kf.
(colon* adj3 ulcer*).ti,ab,kf.
2 or 3 or 4
1 and 5
6 use medall
*upadacitinib/
(Rinvoq* or upadacitinib* or abt494 or abt 494).ti,ab,kf,dq.
8 or 9
Ulcerative colitis/
(colitis or proctocolitis or colorectitis).ti,ab,kf,dq.
(colon* adj3 ulcer*).ti,ab,kf,dq.
11 or 12 or 13
10 and 14
15 use oemezd
16 not (conference review or conference abstract).pt.
7 or 17
remove duplicates from 18
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
Search — upadacitinib | “Colitis, Ulcerative”
WHO ICTRP
International Clinical Trials Registry Platform, produced by the WHO. Targeted search used to capture registered clinical trials.
Search terms — upadacitinib, colitis
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
Search terms — upadacitinib, colitis
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
Search terms — upadacitinib, colitis
Grey Literature
Search dates: May 5 to 10, 2022
Keywords: Rinvoq, upadacitinib, ulcerative colitis
Limits: none
Updated: Search updated before the completion of stakeholder feedback period
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free).
Appendix 2: Description and Appraisal of Outcome Measures
Note that this appendix has not been copy-edited.
Aim
To describe the following outcome measures and review their measurement properties (validity, reliability, responsiveness to change, and minimal important difference [MID]):
Primary efficacy end point: adapted Mayo score
Secondary efficacy end points: endoscopic subscore (ESS), Full Mayo score, Partial Mayo score, Geboes score, IBDQ, FACIT-F
Exploratory variables: WPAI-UC, EQ-5D-5L, SF-36, UC-SQ
Findings
Table 44: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
Mayo score | A disease-specific physician-measured and patient-reported score that includes the following components: rectal bleeding (0 to 3), stool frequency (0 to 3), PGA (0 to 3), and endoscopy findings (0 to 3). A total score ranges from 0 to 12 for Full Mayo score with higher score indicating more severe disease. An adapted Mayo score is calculated by subtracting PGA from the full Mayo score. A partial Mayo score is a noninvasive 9-point system consists of rectal bleeding, stool frequency, PGA only without endoscopy component. | Validity: Construct validity of the full Mayo score was demonstrated by a strong correlation with the patient’s assessment of disease activity (rho = 0.71 at week 12).27 A strong correlation was found between the partial and total Mayo scores (rho = 0.97 at weeks 4 and 8).37 Construct validity of the Mayo endoscopic subscore was supported by a strong correlation with the total Mayo score (Spearman rho = 0.97), the Riley histologic score (r = 0.55) and the Rubin histologic score (r = 0.60).38 Reliability and responsiveness: Endoscopic subscore was found to have moderate-to-substantial inter-rater agreement (r, 0.45 to 0.75). It was also found to be responsive to change over time with treatment.27,38-40 | Clinical response: ≥ 3 points decrease in the Mayo score or the partial Mayo score.27 Clinical remission: Clinical remission is indicated by a total Mayo score of ≤ 2 points, with or without an individual subscore of < 1.27,41 |
Geboes score | The Geboes score is a histologic activity index used to assess UC.29,30 A 6-grade classification system for inflammation (acute activity and chronicity), which could also be fine-tuned within each grade. The grades are: 0, structural change only; 1, chronic inflammation; 2, lamina propria neutrophils; 3, neutrophils in epithelium; 4, crypt destruction; and 5, erosions or ulcers.29 Higher the scores on major grades and/or subgrades, more progressive and increasing severity or activity of disease.29 | Validity: Criterion validity of the Geboes score was supported by a strong correlation between the Geboes score and a global disease activity evaluation assessed using VAS (r = 0.66; 95% CI, 0.57 to 0.72).42 Construct validity was supported by strong correlations between the Geboes score and the Mayo endoscopic subscore, endoscopic activity index, and clinical activity index (Spearman rank correlation range, 0.54 to 0.80).43,44 Reliability: The Geboes score was found to have substantial to almost perfect intrarater agreement (ICC range, 0.77 to 0.84) and moderate inter-rater agreement (ICC range, 0.51 to 0.60).42 The Geboes score was found to be responsive to treatment-related changes (SES = 1.87; 95% CI, 1.54 to 2.20).45 | Historical healing Histological healing was empirically defined in specimens of endoscopically uninflamed tissue as the average Geboes score below 2.29 |
IBDQ | Disease-specific, Likert-based health-related quality of life questionnaire consisting of 32 items classified into 4 dimensions: bowel symptoms (10 items), systemic symptoms (5 items), emotional function (12 items), and social function (5 items). It can be interviewer- or self-administered. Possible total IBDQ score ranges from 32 to 224 with higher scores representing better quality of life. | Validity: Content validity has been ensured by development process that involved patient interviews. Criterion validity was proven, as there was similar correlation with changes in IBDQ and other measures (P < 0.05).46,47 Reliability and responsiveness: The IBDQ was shown to be highly reliable through evaluation of internal consistency (Cronbach alpha 0.7) and test-retest assessment (ICC 0.9 to 0.99 or Pearson’s r ≥ 0.8). The IBDQ was also shown to be responsive to change in patients with IBD (P < 0.05).46,47 | Absolute score change of ≥ 30 points, or ≥ 15 points above the placebo score among patients with IBD.48 |
FACIT-F | Questionnaire completed by patients to assess fatigue during the past 7 days; consists of 13 statements, each rated on a 4-point Likert scale. | Validity: Scores were lower by 8.5 points (95% CI, 5.5 to 11.4; P < 0.001) in patients with active symptoms of UC, demonstrating construct validity. Scores were found to be correlated with ESR (−0.76; 95% CI, −0.89 to −0.50), CRP (−0.72; 95% CI, −0.88 to −0.43), and HCT (0.53; 95% CI, 0.22 to 0.74) in patients with UC. Reliability: Test-retest reliability within 180 days without change in disease state has been established (ICC = 0.81). Internal consistency also has been established (alpha = 0.94). | Unknown. Cut-off score ≤ 30 to define fatigue in patients with IBD has been proposed.49 |
SF-36 | A generic self-reported health-related quality of life questionnaire consisting of 8 domains: physical functioning, role physical, bodily pain, general health, vitality, social functioning, role emotional, and mental health. Scores for each individual domain, PCS, and MCS can range from 0 to 100 with higher scores representing improved HRQoL. | Validity: Construct validity was demonstrated through moderate to strong correlations (r > 0.4) between the 8 subscales of the SF-36 and corresponding domains of 5 patient-reported clinical constructs. The scale showed evidence of discriminant validity (against disease activity/symptom status).50 Reliability and responsiveness: The SF-36 was found to have good internal consistency for all 8 subscales (Cronbach alpha > 0.7) and good test-retest reliability for 6 of the 8 subscales (ICC > 0.7). The scale and its subscores were found to be responsive to treatment-related changes.50 | An absolute score increase of 3 to 5 points for PCS, MCS, and individual subscores for various conditions, including colitis.48 |
EQ-5D-5L | A generic preference-based HRQoL instrument consisting of a VAS and a composite index score of 5 dimensions: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression. Responses to items can be converted into a single measure of health utility using preference-based (typically country-specific) weights to generate health utility index score, where 0 and 1.0 are assigned to the health states “dead” and “perfect health,” respectively, with negative numbers denoting health state “worse than dead.” VAS is anchored by 0 (worst imaginable health) and 100 (best imaginable health). | Validity: Construct validity of index score and VAS was supported by a moderate to strong correlation of the EQ-5D-5L with the IBDQ (r = 0.69), physician-completed SCCAI (r = −0.53), and patient-completed SCCAI (r = −0.49). Also, EQ-5D-5L index scores could differentiate in patients in stable condition, worsening disease, or improving disease state.51,52 Reliability: A moderate agreement was observed for all domains of patient-completed and physician-completes SCCAIs (kappa, 0.41 to 0.58), except for the “anxiety/depression” domain (kappa = 0.28).52 | An EQ-5D-3L index score of 0.05 and VAS of 10.9 were estimated for improved health; VAS of 14.4 and the EQ-5D-3L index of 0.067 for deteriorated health in patients with UC.53 |
WPAI-UC | Self-rated, disease-specific questionnaire, consists of 6 items divided into 4 domains: absenteeism, presenteeism, percent overall work impairment, and regular activities impairment due to UC. | NRa | |
UC-SQ | A Likert-type, UC-specific instrument composed of 17 items to assess UC-related gastrointestinal symptoms (e.g., frequent bowel movements, abdominal pain, cramping) and nongastrointestinal symptoms (e.g., joint pain and sleep difficulties). Overall symptom scores are calculated by combining ratings of the individual items, ranging from 17 to 85; higher scores indicate greater symptom burden and/or greater severity. | NRb | Unknown |
CRP = C-reactive protein; ESR = erythrocyte sedimentation rate; FACIT- F = Functional Assessment of Chronic Illness Therapy–Fatigue; HCT = hematocrit; IBDQ = Inflammatory Bowel Disease Questionnaire; ICC = intraclass correlation; MID = minimal important difference; NR = not reported; PGA = physician’s global assessment; SCCAI = Simple Clinical Colitis Activity Index; SES = standardized effect size; SF-36 = Short Form (36) Health Survey; UC = ulcerative colitis; UC-SQ = Ulcerative Colitis – Symptoms Questionnaire; WPAI-UC = Work Productivity and Activity Impairment – Ulcerative Colitis
aPsychometric properties have been identified in literature search but have not been included in the report, as the scale is considered a lower priority end point in the Clinical Study Report.
bNo literature has been found regarding its development and/or validation.
Mayo Score
The Mayo scoring system is a combined endoscopic and clinical scale used to assess the severity of UC. It was first developed by Dr. Schroeder in 1987 and is now 1 of the most commonly used disease activity indices in UC.27,28 In its complete form, the Mayo score is composed of 4 components: rectal bleeding, stool frequency, PGA, and endoscopy findings. Each component is rated from 0 to 3, yielding a total score of 0 to 12. A score of 3 to 5 points indicates mildly active disease, while a score of 6 to 10 points indicates moderately active disease, and a score of 11 to 12 points indicates severe disease. Two abridged versions of the Mayo score have been developed and validated: 1) the noninvasive 9-point partial Mayo score that excludes the endoscopy subscore, resulting in a composite of the rectal bleeding, stool frequency, and PGA; and 2) the noninvasive 6-point score comprising only the bleeding and stool frequency subscores.27,38 Mucosal healing has been defined as a Mayo endoscopic subscore of 0 or 1 in major trials of biological therapies in UC. The grading of each component is defined in Table 45. In the pivotal trials for upadacitinib, the sponsor used 4 types of Mayo scores as follows:26
Full Mayo score: A composite score of UC disease activity based on the SFS 0 to 3, RBS 0 to 3, PGA 0 to 3, and ESS 0 to 3. Ranges from 0 to 12 points with higher scores representing severe disease
Partial Mayo score: Full Mayo score minus the ESS
Adapted Mayo score: Full Mayo score minus PGA subscore
Partial adapted Mayo score: Adapted Mayo score minus ESS
Table 45: The Mayo Score Disease Activity Index for Ulcerative Colitis
Descriptor | Level |
|---|---|
Stool frequency (SFS) based on past 3 days Normal no. stools for this patient 1 to 2 stools more than normal 3 to 4 stools more than normal 5 or more stools more than normal | 0 1 2 3 |
Rectal bleeding (RBS) based on past 3 days No blood seen Streaks of blood with stool less than half the time Obvious blood with stool most of the time Blood alone passed | 0 1 2 3 |
Findings of flexible proctosigmoidoscopy (ESS or MES) Normal or inactive disease Mild disease Moderate disease Severe disease | 0 1 2 3 |
Physician’s Global Assessment Normal Mild disease Moderate disease Severe disease | 0 1 2 3 |
Total score | (Range 0 to 12) |
ESS = endoscopic subscore; MES = Mayo endoscopic subscore; RBS = rectal bleeding subscore; SFS = stool frequency subscore.
Source: Clinical Study Report clinical summary.26
Psychometric Properties
A recent Cochrane systematic review, including 20 primary studies, assessed the validity, reliability, and responsiveness of endoscopic-scoring indices for evaluation of disease activity in UC.38 Content validity was not assessed in any of the included studies.38 The review identified 2 studies that assessed construct validity of the Mayo endoscopic subscore which found a strong correlation between the Mayo endoscopic subscore and 2 histologic indices, including the Riley index score (r = 0.55) and Rubin histologic index score (r = 0.60).37,58 However, the endoscopic subscore failed to discriminate between patients who achieved remission and response compared with those who did not.38 In terms of intra- and inter-rater reliability, the systematic review reported a moderate-to-substantial agreement in the inter-rater reliability estimates (r = 0.45 to 0.75) and a substantial agreement in the intrarater reliability estimates (r = 0.75) for the endoscopic subscore.38 A Canadian study consisting of 82 patients with UC (mean age = 49.9; SD = 14.8 years) demonstrated that the threshold of the Mayo endoscopic subscore for predicting histological healing was equal to 0, with sensitivity of 81.4% (95% CI, 25.4 to 90.9), specificity of 95.7% (95% CI, 67.0 to 100%), and accuracy of 85.4% (95% CI, 77.0 to 86.6%).59 Another study consisting of 149 patients with moderate to severe UC demonstrated a strong correlation between the partial and total Mayo scores (Spearman rho = 0.97 at weeks 4 and 8).37
An evaluation of the construct validity of the total and partial Mayo scores was conducted in 75 patients with UC.27 Both the total and partial Mayo scores were strongly correlated with patient assessment of disease activity (rho = 0.71 and rho = 0.70, respectively).27 Moreover, the Mayo score was found to correlated with patient assessment of change in UC activity,27 and with improvement in quality-of-life measures.60 A study evaluating the comparative inter-rater variation for 3 UC disease activity indices (n = 100) found that the inter-rater agreement for the total Mayo score was high (kappa = 0.72); however, the agreement was lower for the relatively subjective PGA and endoscopic subscores with kappa scores of 0.56 and 0.38, respectively.40 An evaluation of the reliability and responsiveness of the Mayo endoscopic subscore was assessed in a placebo-controlled trial evaluating change in UC disease activity after treatment with mesalamine.39 The authors reported both excellent inter- and intra-observer reliability with intraclass correlation (ICC) of 0.79 and 0.89, respectively. In addition, the Mayo endoscopic subscore was found to be responsive to change over time with treatment.48 Rubin et al. also reported a strong correlation between the Mayo Clinic Endoscopic subscore and the Simple Clinical Colitis Activity Index (SCCAI) (r = 0.53; P < 0.001).58
Minimal Important Difference
In a study of 105 patients with UC, the optimal cut point of change in the total Mayo score to identify a clinical improvement or response was 2.5 with sensitivity of 88%, specificity of 80%, using patient’s rating of the improvement as an anchor.27 What is considered the optimal cut point for clinical remission, however, varies. While Lewis et al. reported a cut point of change of 4.5 with sensitivity of 88% and specificity of 78%, cut points determined from other clinical trials ranged from a Mayo score of 0.6 to 2.27,41,61 As with remission, different definitions of response have been used, most commonly a reduction of the baseline total Mayo score of either 2 or 3 points.41 The FDA, on the other hand, defines clinical remission as a Mayo score of less than or equal to 2 with no individual subscore greater than 1 (SFS of 0 or 1, endoscopy subscore of 0 or 1, and RBS of 0).62 Also, the FDA defines clinical response as a reduction in the total Mayo score of 30% or more from baseline with a decrease in RBS greater than or equal to 1 point or absolute RBS of less than or equal to 1.62
Limitations
Cooney et al. argued that the PGA and the endoscopy subscore components of the Mayo score are subjective and, consequently, introduce variability and lack of precision into the index. However, in the trial, this concern was mitigated by centrally reading the scores. The PGA also includes a sigmoidoscopy score, which introduces double counts of some elements.63 Additionally, a single general item in the PGA is not sensitive to adequately capture benefits in all or some of the important signs and symptoms of UC. As a result, the FDA does not recommend using the PGA subscore or the full Mayo score to support a marketing decision; however, it does recommend the endoscopy, stool frequency, and rectal bleeding subscores as outcome measures for clinical trials until a well-defined and reliable instrument if available.62
Geboes Score
The Geboes score is a histologic index in UC for assessing disease severity and/or activity.29,30 It is a classification system consisting of 6 grades, with 4 to 5 subgrades each, that are meant to be progressive. Grading is performed on hematoxylin-eosin stained sections from biopsies obtained in the colonic mucosa. The grades and subgrades are defined as follows:29
Grade 0 (structural / architectural change): Subgrades — no abnormality (0.0), mild abnormality (0.1), mild or moderate diffuse or multifocal abnormalities (0.2), severe diffuse or multifocal abnormalities (0.3)
Grade 1 (chronic inflammatory increase): Subgrades — no increase (1.0), mild but unequivocal increase (1.1), moderate increase (1.2), marked increase (1.3)
Grade 2 (lamina propria neutrophils and eosinophils):
2A Eosinophils: No increase (2A.0), mild but unequivocal increase (2A.1), moderate increase (2A.2), marked increase (2A.3)
2B Neutrophilsa: None (2B.0), mild but unequivocal increase (2B.1), moderate increase (2B.2), marked increase (2B.3)
a A total of 7 different combinations for locations of neutrophils within the epithelium (surface epithelium, crypt epithelium, crypt abscesses) are possible.
Grade 3 (neutrophils in epithelium): None (3.0), < 5% crypts involved (3.1), < 50% crypts involved (3.2), > 50% crypts involved (3.3)
Grade 4 (crypt destruction): None (4.0), probable – local excess of neutrophils in part of crypt (4.1), probable – marked attenuation (4.2), unequivocal crypt destruction (4.3)
Grade 5 (erosions or ulcers): No erosion, ulceration, or granulation tissue (5.0), recovering epithelium + adjacent inflammation (5.1), probable erosion – focally stripped (5.2), unequivocal erosion (5.3), ulcer or granulation tissue (5.4)
Subgrades are assessed based on the worst area of the biopsy. The higher grade or subgrade indicates the greater degree of inflammation. The Geboes score may also be converted into a continuous scale with each subgrade being assigned an ordinal value, yielding values between 0 and 22.29
Psychometric Properties
An evaluation of the construct validity of the Geboes score in a cohort of 442 patients with UC previously enrolled in other studies found that the score was strongly correlated with the Nancy index score (r = 0.88; P < 0.001).64 Another study that evaluated the construct validity of the Geboes score in 131 patients with UC found that it was strongly correlated with the Mayo endoscopic subscore (r = 0.54, R < 0.001).43 Finally, in a study of 82 patients with UC (mean age = 47.5 years; SD = 15.9 years), the Geboes score was found to be strongly correlated with the endoscopic activity index (r = 0.77; P < 0.001) and weakly correlated with the clinical activity index (r = 0.40; P < 0.001) and CRP level (r = 0.42; P < 0.001).44 In a study of 49 patients with UC (mean age = 40.2 years; SD = 2.9 years), the criterion validity of the Geboes score was evaluated against a 100 mm global disease activity VAS (the most severe activity was scored as 1 and no disease activity was scored as 0).42 The Geboes scale, when used as a continuous scale, was found to be strongly correlated with the VAS (r = 0.66; 95% CI, 0.57 to 0.72). The Geboes and the VAS were moderately correlated when the Geboes score was used as a 6-grade ordinal scale (r = 0.61; 95% CI, 0.50 to 0.67), and weakly correlated when used as a categorical scale (inactive – grade 0 or 1, mildly active – grade 2 or 3, and severely active – grade 4 or 5) (r = 0.58; 95% CI, 0.48 to 0.64).42
Mosli et al. also evaluated intrarater and inter-rater reliability of the Geboes score by having 5 pathologists independently review 50 digital slide images 3 times, approximately 2 weeks apart.42 When used as a 6-grade ordinal scale, the Geboes score was found to have almost perfect intrarater agreement (ICC: 0.82; 95% CI, 0.73 to 0.88), and moderate inter-rater agreement (ICC: 0.56; 95% CI, 0.39 to 0.67). When used as a continuous scale, the Geboes score demonstrated almost perfect intrarater agreement (ICC = 0.84; 95% CI, 0.80 to 0.89) and moderate inter-rater agreement (ICC = 0.60; 95% CI, 0.46 to 0.71).42 Intrarater reliability of the individual items of the Geboes found strong agreement for the detection of erosions and ulcerations (ICC = 0.81; 95% CI, 0.72 to 0.86), substantial agreement for the detection of neutrophils in the epithelium (ICC = 0.71, 95% CI, 0.64 to 0.78), and erosion or ulceration (ICC = 0.78; 95% CI, 0.71 to 0.84), and moderate agreement for the detection of crypt destruction (ICC = 0.61; 95% CI, 0.53 to 0.68) and lamina propria eosinophils (ICC = 0.59; 95% CI, 0.50 to 0.67).42 Inter-rater reliability of the individual items of the Geboes scale ranged from weak (detection of lamina propria eosinophils: ICC = 0.26; 95% CI, 0.15 to 0.45; and detection of neutrophils in the epithelium: ICC = 0.48, 95% CI, 0.37 to 0.58) to moderate (erosions or ulcerations (ICC = 0.56; 95% CI, 0.43 to 0.67); and detection of chronic inflammatory infiltrate: ICC = 0.64; 95% CI, 0.50 to 0.74).42
In a later study by Mosli et al. consisting of 155 patients with UC (mean age, 41.7, SD = 14.1 years), the Geboes scoring system was found to have almost perfect intrarater agreement (ICC = 0.88; 95% CI, 0.79 to 0.93) and substantial inter-rater agreement (ICC = 0.79; 95% CI, 0.63 to 0.87).45 In the same study, the responsiveness of the Geboes scoring system was evaluated using an analysis of standardized effect size (SES) and the Guyatt responsiveness statistics (GRS).45 The responsiveness to change was moderate to large based on SES and GRS of 1.87 (95% CI, 1.54 to 2.20) and 1.23 (95% CI, 0.97 to 1.50), respectively, for the Geboes score based on treatment assignment, and 1.05 (95% CI, 0.78 to 1.31) and 0.84 (95% CI, 0.59 to 1.09), respectively, based on the Mayo clinical subscore of at least 2 points.45 Histological activity, defined as Geboes score ≥ 3.1, was found to be an independent risk factor for clinical relapse in patients with UC (OR = 4.31; 95% CI, 1.52 to 12.21; P = 0.006).65
Minimal Important Difference
Histological healing was empirically defined in specimens of endoscopically uninflamed tissue as the average Geboes score below 2.29
Inflammatory Bowel Disease Questionnaire
Developed by Guyatt et al., the IBDQ is an interviewer- or self-administered questionnaire to assess HRQoL in patients with IBD.66,67 It is a 32-item Likert-based questionnaire divided into 4 dimensions: bowel symptoms (10 items), systemic symptoms (5 items), emotional function (12 items), and social function (5 items). Patients are asked to recall symptoms and quality of life from the last 2 weeks with response graded on a 7-point Likert scale (1 being the worst situation, 7 being the best) with the total IBDQ score ranging from 32 to 224 (i.e., higher scores representing better quality of life). A total IBDQ score of at least 170 points or higher is considered clinical remission. This questionnaire has been validated in a variety of settings, countries, and languages, and is available in a 9-, 10-, and 36-item form.68
Validity
Two systematic reviews published in the past 3 years reported the measurement properties and methodological quality of a number of IBD-specific HRQoL instruments, including the IBDQ.46,47 Overall, the IBDQ was proven to be valid, reliable, and responsive scale; however, the methodological quality of these studies was poor to fair for some of these measurement properties. The IBDQ demonstrated content validity, as it was developed through patient interviews and covered the most frequent and important items. Results from factor analysis showed the items/domains of the scale explained at least 50% of the variance. In addition, criterion validity was proven, as there was similar correlation with changes in IBDQ and other measures.46,47 The scale showed lower discriminant validity in patients who required surgery.46,47
Reliability and Responsiveness
The reliability parameters showed high internal consistency (Cronbach alpha 0.7), test-retest reliability (ICC, 0.9 to 0.99 or Pearson’s r ≥ 0.8), and low measurement error (i.e., the SDs of the score changes were of similar magnitude and the smallest detectable change was less than the minimal important difference [MID]). Responsiveness was satisfactory, as the scale was sensitive to change corresponding to clinical improvement or deterioration. Floor and ceiling effects were not found, as less than 15% of the respondents achieved the highest or lowest possible score.46,47
Minimal Important Difference
Irvine et al. reported that a change of 30 or more points in actual score or an improvement of 15 or more points above the placebo score is associated with clinical benefits in patients with IBD.48 Several other studies have reported an increase of 15 to 32 points from baseline as clinically meaningful improvement.69
FACIT-F
The FACIT- F is a subscale of the general questionnaire, the FACT-General. It was developed to assess fatigue associated with anemia with item content established by combined expert and patient input.70 The FACIT-F is completed by patients (or interviewer when applicable) to assess fatigue. The current version is v.4. Patients are presented with a list of 13 statements and asked to rate each on a 5-point Likert scale (0 = not at all, 1 = a little bit, 2 = somewhat, 3 = quite a bit, 4 = very much) to indicate how true the statement was during the past 7 days. Examples of statements are “I feel fatigued” and “I feel weak all over.” In the scoring, the numbers are reversed so that higher scores denote better quality of life (i.e., 4 = not at all, 3 = a little bit, 2 = somewhat, 1 = quite a bit, and 0 = very much). For statements 7 (“I have energy”) and 8 (“I am able to do my usual activities”), the scores are not reversed. The total score ranges between 0 and 52 with a lower score representing a higher level of fatigue. FACIT-F questionnaire has been translated into 48 languages permitting cross-cultural comparisons of fatigue in patients of diverse backgrounds.71,72
Validity and Reliability
A validation study has been conducted with FACIT-F in adult US population with IBD. A total of 209 patients with IBD (77 patients with UC) completed the 13 items of the FACIT-F, alongside laboratory testing and disease activity assessment.73 The internal consistency of the 13 items of the FACIT-F questionnaire measured by Cronbach alpha was 0.94 for UC, which is acceptable (i.e., > 0.7).74 The test-retest reliability as represented by intraclass correlation coefficient (ICC) when FACIT-F assessments were repeated within 180 days in patients with stable health was 0.87 for UC (n = 19), which is considered strongly reliable.75 In UC, it was found that there was a strong correlation (−0.59 [−0.72 to −0.41, P ≤ 0.001]) between the FACIT-F and the SCCAI. Furthermore, in UC, FACIT-F scores strongly correlated with erythrocyte sedimentation rate at −0.77 (95% CI, −0.89 to −0.50; P ≤ 0.001), CRP at −0.73 (95% CI, −0.88 to −0.43, P ≤ 0.001) and hematocrit at 0.53 (95% CI, 0.22 to 0.74; P = 0.001). However, an important aspect of the validation process — assessing criterion validity — is severely limited by the lack of an accepted gold standard measure of fatigue. Lastly, FACIT-F scores were lower in patients with active UC (−8.5 points, 95% CI, −11.4 to −5.5, P ≤ 0.001) and physician’s assessments of change in patients’ health corresponded closely to changes in FACIT-F scores (adjusted mean [95% CI], P value: Much better = −11.8 [−15.9 to −7.8] P ≤ 0001, slightly better = −2.6 [−4.5 to −0.7] P = 0.007, same = 0.7 [−0.3 to 1.6] P = 0.17, slightly worse = + 2.4 [0.9 to 3.9] P = 0.002, much worse = + 5.2 [3.2 to 7.1] P ≤ 0001).
Even though some aspects of validity have been assessed for the FACIT-F in IBD, future studies designed to administer the FACIT-F before and after initiation of various treatments, with fatigue scores as the primary outcome are needed. Also, the unidimensional nature of the tool is another limitation. Multidimensional scales that may offer additional insights into the various underlying components of IBD-associated fatigue are needed.73
Minimal Important Difference
A difference of 3 to 4 units is considered a minimal important difference in patients with rheumatoid arthritis and/or cancer.76,77 However, no MID has been estimated in patients with UC. A cut-off score ≤ 30 to define fatigue in patients with IBD has been proposed and used in some studies.49
Short Form (36) Health Survey
The SF-36 is a generic self-reported health assessment questionnaire that has been used in clinical trials to study the impact of chronic disease on HRQoL. The original version (SF-36 version 1) was released in 1992; however, a revised version (SF-36 version 2), released in 1996, is used more commonly. The SF-36 consists of 8 domains: physical functioning, role limitations due to physical health problems, bodily pain, general health, vitality, social functioning, role limitations due to emotional health problems, and mental health. The SF-36 also provides 2 component summaries: the Physical Component Summary (PCS) and the Mental Component Summary (MCS), which are scores created by aggregating the 8 domains. The SF-36 PCS and MCS and individual domains are each measured on a scale of 0 to 100, with an increase in score indicating improvement in health status.78
Psychometric Properties
The construct validity, reliability, and responsiveness of the SF-36v2 among patients with UC was recently assessed in a systematic review that included 43 studies.50 Construct validity of the SF-36 subscales was supported by a moderate-to-high correlation with the corresponding domains of 5 patient-reported tools, including the IBD Quality of Life Questionnaire, Brief Pain Inventory, Short Health Scale, and Rating Form of IBD Patient Concerns (r ≥ 0.4).50 The SF-36 was found to be discriminative between subgroups of patients classified by disease activity, symptom status, and comorbidity status. In terms of reliability and responsiveness, 1 included study found that the SF-36 had high internal consistency of all 8 subscales (Cronbach alpha > 0.7) and high test-retest reliability for 6 of the 8 subscales (ICC > 0.7); the 2 subscales that had lower test-retest reliability were the role physical and role emotional subscales with ICCs of 0.64 and 0.63, respectively. The possibility of high floor and ceiling effect may explain the lower test-retest reliability for the role physical and role emotional subscale.50 Finally, the systematic review found that the SF-36 scale, and its subscores, were responsive to treatment-related changes following effective treatment in noncomparative trials or among treated patients relative to controls in RCTs.50
Minimal Important Difference
An absolute score increase of 3 to 5 points for both the PCS and MCS, as well as the individual scores in the SF-36, was shown to capture the MID across various conditions, including colitis.48
Five-Level EQ-5D
The EQ-5D-5L is a generic self-reported HRQoL outcome measure that may be applied to a variety of health conditions and treatments.79 The first 2 components of the EQ-5D-5L assess 5 domains: mobility, self-care, usual activities, pain/discomfort and anxiety/depression.79 Each domain has 5 levels: no problem; slight problems; moderate problems; severe problems; and extreme problems. A descriptive system that classifies respondents (aged ≥ 12 years) based on the following 5 dimensions: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression.
The EQ-5D-5L has 5 possible levels for each domain and respondents are asked to choose the level that reflects their health state for each of the 5 domains resulting in 3,125 possible health states.79 The EQ-5D-5L tool has been applied to a wide range of health conditions and treatments, including IBD.80,81 The EQ-5D-5L index score is generated by applying a multiattribute utility function to the descriptive system.82 Different utility functions are available that reflect the preferences of specific populations (e.g., US or UK). Scores less than 0 represent health states that are valued by society as being worse than dead, while scores of 0 and 1.00 are assigned to the health states “dead” and “perfect health,” respectively. The second component of the EQ-5D-5L is the 10 cm EuroQoL Visual Analogue Scale (EQ VAS). Its end points labelled 0 and 100, with respective anchors of “worst imaginable health state” and “best imaginable health state.” Respondents are asked to rate their health by drawing a line from an anchor box to the point on the EQ VAS which best represents their health on that day. Thus, the EQ-5D-5L produces 3 types of data for each respondent:
a profile indicating the extent of problems on each of the 5 dimensions represented by a 5-digit descriptor, e.g., 15121, 33211
a population preference-weighted health index score based on the descriptive system
a self-reported assessment of health status based on the EQ VAS
Psychometric Properties
The face and content validity of the EQ-5D-5L index score was investigated by Herdman et al. using focus groups.83 An Australian study of 175 patients with UC (mean age, 42, SD = 15 years) that examined the construct validity of the EQ-5D-5L found that it was strongly correlated with the disease-specific IBDQ (r = 0.69; P < 0.001).51 Mean EQ-5D-5L scores were found to be significantly greater for patients with UC in remission (mean = 0.81, SD = 0.18) than for patients who had active disease (mean = 0.72, SD = 0.19). Likewise, among patients with active UC, lower scores were observed in patients with mild disease (mean = 0.78, SD = 0.18) than in those with moderate to severe disease (mean = 0.68, SD = 0.19).51 A similar pattern was observed for EQ VAS scores.
A prospective, noninterventional study conducted at 37 hospital centres in Spain found a consistent and linear relationship between the EQ-5D-5L and the SCCAI among a group of 199 patients with UC (mean age = 39; SD = 11 years).52 In this study, patients with UC completed both the EQ-5D-5L and SCCAI at 3 and 6 months. The SCCAI was also completed by treating gastroenterologists who were blinded to patient responses. The construct validity of the EQ-5D-5L was then evaluated by mapping its index scores to those of patient- and physician-completed SCCAIs. The study found a moderate correlation between EQ-5D-5L index scores and patient-completed SCCAI (r = –0.49; P < 0.001), and a strong correlation between EQ-5D-5L index scores and physician-completed SCCAI (r = –0.53; P < 0.001).52 In particular, a moderate to strong correlation was observed between the “general well-being” item on the patient-completed and physician-completed SCCAIs to the “pain/discomfort” (r = 0.52 to 0.54) and “usual activities” items (r = 0.38 to 0.40) on the EQ-5D-5L scores at month 3; and for “general well-being” and “pain/discomfort” (r = 0.64 to 0.66) and “usual activities” items (r = 0.57 to 0.61) at month 6.52 In addition, decline in HRQoL was observed during disease flare. Indeed, the difference in EQ-5D-5L index scores from 3 to 6 months was lower in patients who experienced worsening disease (mean, –0.069 ± 0.07) compared with patients in stable condition (mean, 0.022 ± 0.11) or improving disease state (mean, 0.035 ± 0.13).52 In terms of reliability, a moderate agreement was observed between EQ-5D-5L index and all domains of patient-completed and physician-completes SCCAIs (kappa range, 0.41 to 0.58), except for fair agreement between the “anxiety/depression” domain and patient-completed SCCAI (kappa = 0.28).52 Finally, agreement between the EQ-5D-5L and patient-completed and physician-completed SCCAIs index scores was 74.2% and 68.8%, respectively.52 To date, there is no literature evaluating the responsiveness of the EQ-5D-5L among patients with UC over time.
Minimal Important Difference
A literature search was conducted to identify the MID of the EQ-5D-5L in patients with UC and no relevant studies were identified. However, Stark et al. estimated a disease-specific MID of the EQ-5D-3L using a regression model; the MIDs for improved health were reported to be 10.9 for the VAS, and 0.050 (European Union) and 0.076 (UK) for the EQ-5D-3L index score.53 This is within the range of other reported MIDs for the EQ-5D-3L index score of 0.033 to 0.074.82
Work Productivity and Activity Impairment Index–Ulcerative Colitis
The WPAI-UC questionnaire, version 2, is an instrument used to measure the impact of a disease on work and daily activities during the previous 7 days.84 The WPAI-UC consists of 6 questions: employment status (employed or not employed); hours at work missed because of UC, hours at work missed because of other reasons; hours actually worked; overall impairment in productivity while working (VAS from 0 to 10) and overall impairment in regular activities (VAS from 0 to 10) due to UC. Patients who are employed answer all questions, while those who are not employed answer the first and last. Four measures are derived from the questionnaire. All 4 domain scores are expressed as a percentage of impairment/productivity loss, and range from 0 to 100%, with higher scores indicating greater impairment.84
Absenteeism (work time missed)
Presenteeism (percent impairment while working)
Percent overall work impairment due to UC, and
Regular activities impairment due to UC
Minimal Important Difference
No reported MID was found for patients with UC. Yarlas, et al.54 who conducted a systematic review of literature from 13 sources (8 articles, 5 posters) on WPAI-UC in patients with ulcerative colitis, suggested a change of 7% be taken as an MID. The rationale was that several studies found a change of 7% in each WPAI–Crohn Disease domain corresponds to a clinically meaningful change in patients with CD activity and symptoms of the 2 conditions are similar. Also, previous research on the WAPI-UC has adopted this threshold of 7% change.55-57
Ulcerative Colitis Symptoms Questionnaire
The UC-SQ is a UC-specific instrument composed of 17 items. UC-SQ was developed to assess UC-related gastrointestinal symptoms (e.g., frequent bowel movements, abdominal pain, cramping) and nongastrointestinal symptoms (e.g., joint pain and sleep difficulties). Each symptom item from 1 to 9 is rated on a 5-point Likert scale: Not at all = 0 points; A little bit = 1 point; Somewhat = 2 points; Quite a bit = 3 points; Very much = 4 points. Each symptom item from 10 to 17 is rated on a Likert scale: Never: 0 points; Rarely: 1 point; Sometimes: 2 points; Often: 3 points; Always: 4 points. Overall symptom scores are calculated by combining ratings of the individual items, ranging from 17 to 85; higher scores indicate greater symptom burden and/or greater severity.26 The range for the overall score is 17 to 85, with higher scores corresponding to greater symptom burden.
No literature has been found to support the validity, reliability, responsiveness to change, nor an MID for the UC-SQ.
Pharmacoeconomic Review
Abbreviations
5-ASA
5-aminosalicylates
AE
adverse event
CEF
cost-effectiveness frontier
ICER
incremental cost-effectiveness ratio
JAK
Janus kinase
NMA
network meta-analysis
QALY
quality-adjusted life-year
TNF
tumour necrosis factor
UC
ulcerative colitis
WTP
willingness to pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Upadacitinib (Rinvoq), 15 mg, 30 mg, and 45 mg tablets |
Submitted price | Upadacitinib, 15 mg: $49.22 per tablet Upadacitinib, 30 mg: $74.00 per tablet Upadacitinib, 45 mg: $101.81 per tablet |
Indication | For the treatment of adult patients with moderately to severely active ulcerative colitis who have demonstrated prior treatment failure, i.e., an inadequate response to, loss of response to, or intolerance to at least one of conventional, and/or biologic therapy. |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | July 21, 2023 |
Reimbursement request | As per indication |
Sponsor | AbbVie |
Submission history | Previously reviewed: Yes Indication: Atopic dermatitis
Previously reviewed: Yes Indication: Psoriatic arthritis
Indication: Rheumatoid arthritis
|
NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Decision tree followed by a Markov cohort model |
Target population | Adult patients (≥ 18 years of age) with moderately to severely active UC with or without prior exposure to biologica drugs (i.e., biologic-experienced or biologic-naive) |
Treatment | Upadacitinib |
Comparators |
|
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (57 years) |
Key data source | A phase III clinical program comprising 3 pivotal multicentre, double-blind, placebo-controlled studies, of which 2 are replicate induction studies (U-ACHIEVE Induction, U-ACCOMPLISH), and 1 is a maintenance study (U-ACHIEVE Maintenance). These informed the efficacy and safety of upadacitinib, while a sponsor-commissioned NMA informed the efficacy and safety of comparators, including CT. |
Submitted results | For both the biologic-naive and biologic-experienced populations, the cost-effectiveness frontier was represented by CT and upadacitinib. All other treatments were either strictly or extendedly dominated.
|
Key limitations |
|
CADTH reanalysis results |
|
AE = adverse event; CT = conventional therapy; ICER = incremental cost-effectiveness ratio; IL = interleukin; JAK = Janus kinase; LY = life-year; NMA = network meta-analysis; QALY = quality-adjusted life-year; TNF = tumour necrosis factor; UC = ulcerative colitis; WTP = willingness to pay.
aBiologic refers to TNF alpha antagonists, integrin receptor antagonists, and interleukin 12 and interleukin 23 inhibitors.
Conclusions
Based on the appraisal of 2 induction trials and 1 maintenance trial, CADTH clinical reviewers found that upadacitinib was efficacious in inducing and maintaining clinical remission and clinical response when compared with placebo in patients with moderately and severely active ulcerative colitis (UC). Since there are no trials comparing upadacitinib with the advanced therapies of interest (i.e., biologics and small-molecule drugs), comparisons among treatments were based on the sponsor-commissioned network meta-analysis (NMA). CADTH identified considerable uncertainty in the results from the NMA due to sparse networks, heterogeneity in patient characteristics and trial designs, and inadequate adjustments for the substantial clinical heterogeneity. As such, the CADTH clinical appraisal concluded there is a paucity of evidence to determine how upadacitinib compares with other advanced treatments for efficacy and safety in patients with moderately to severely active UC.
CADTH undertook reanalyses to address limitations in the sponsor’s submission, including applying the probabilities of response without remission at the end of induction in the biologic-naive population, as derived from the NMA’s random-effects unadjusted model, and assuming the probabilities of clinical response and remission as well as the probabilities of serious infection of all advanced therapies to be equal to upadacitinib for all doses. In both the biologic-naive and biologic-exposed populations, upadacitinib was strictly dominated (i.e., higher cost, equally effective) by adalimumab.
CADTH was unable to address limitations relating to: the omission of relevant adverse events (AEs) and the implementation of AEs that were considered in the model; the assumption that treatment effect, and the corresponding loss of response, is constant throughout the maintenance phase and over the lifetime time horizon of the model; the absence of concomitant conventional therapy use while on primary advanced therapy; and the assumption of equivalent disease management and monitoring resource use across advanced therapies. Given the effect that many of these limitations have on incremental cost, a greater price reduction is likely necessary to achieve cost-effectiveness. There was insufficient economic evidence to justify a higher drug cost for upadacitinib compared with other available advanced therapies. When only considering drug acquisition costs, a price reduction of between 32% and 55% is necessary for upadacitinib to be no more costly than the least costly advanced therapy, depending on the dose of upadacitinib.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
Two patient groups provided input for the upadacitinib submission1 for UC: Crohn and Colitis Canada and the Gastrointestinal Society. The input was based on patient surveys, published literature, and interviews. The most important outcome for patients with moderate to severe UC is sustained remission and treatment response. Currently available first-line treatments include anti-inflammatory drugs together with corticosteroids, as well as second-line treatments typically consisting of immunomodulators or immunosuppressants and biologics, which tend to be prescribed concomitantly with corticosteroids. While patients with mild to moderate levels of UC may experience improvements in their overall condition with initial treatments, patients with moderately to severely active UC often experience loss of response and/or remission under various treatment options. Because of this, continual treatment switching is required to achieve an adequate response until all therapeutic options are exhausted. Overall, patients want treatments to be safe, improve quality of life, allow them to perform daily activities with ease, increase the duration of remission, and improve symptoms.
Registered clinician input was received from 2 groups: the Inflammatory Bowel Disease Centre of BC and the Atlantic Specialist Group, the latter of which was submitted jointly with the University of Calgary Inflammatory Bowel Disease Unit. According to clinician input, patients with moderate to severe UC need selective and efficacious treatments that could target histologic and endoscopic remission with rapid onset, achieve symptomatic remission, reduce extraintestinal manifestations, and be safe and convenient to administer. Clinicians noted that upadacitinib could be used as the first-line therapy for patients with moderate to severe UC who are not responding or are intolerant to 5-aminosalicylates (5-ASA), as well as patients who are refractory to, dependent on, or intolerant to corticosteroids. In addition, clinicians indicated that upadacitinib could replace corticosteroid induction, be offered to patients regardless of history of prior advanced therapy use, and eliminate the need for immunomodulators completely. Thus, clinicians noted that upadacitinib may shift the current treatment paradigm by providing an additional option for patients, and they expect the drug under review to become the most widely used therapy for moderately to severely active UC.
CADTH-participating drug plans provided feedback centred on the safety of upadacitinib relative to tofacitinib, the only other publicly reimbursed Janus kinase (JAK) inhibitor whose safety concerns warrant a black box warning. The drug plan input highlighted the need to understand how upadacitinib’s safety profile, particularly regarding risk of serious infections, malignancy, and thrombosis, compared with currently available advanced therapies used in the management of UC. Moreover, the participating drug plans inquired whether the discontinuation rules for upadacitinib should be aligned with those of tofacitinib in the way of a foreseeable recommendation to discontinue initial treatment if clinical response is not achieved within 8 weeks. Finally, the drug plans noted that generic tofacitinib would soon be available, leading to a reduction in costs of the only other JAK-inhibiting comparator in the moderate to severe UC space.
Several of these concerns were addressed in the sponsor’s model:
The most important outcome for patients with moderately to severely active UC is sustained clinical remission and/or response, which are the primary health states in the maintenance phase of the sponsor’s model.
Multiple lines of therapy were considered in the sponsor’s model.
CADTH was unable to address the following concerns raised from stakeholder input:
The patient and clinician input confirmed that the disease management journey for the population with moderate to severe UC is characterized by continual treatment switching until all therapeutic options are exhausted. However, CADTH was not able to consider multiple lines of treatment, as this would require currently unavailable efficacy data from populations who have failed 2 biologics.
Economic Review
The current review is for upadacitinib (Rinvoq) for adult patients (≥ 18 years of age) with moderately to severely active UC who have demonstrated prior treatment failure, i.e., an inadequate response to, loss of response to, or intolerance to at least 1 of conventional, and/or biologic therapy.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis (CUA) of upadacitinib compared with other advanced therapies and conventional therapy.2 Aligned with Health Canada’s indicated population, the modelled population comprised Canadian adults between the ages of 18 and 75 years with moderately to severely active UC (defined as a Mayo score of 6 to 12) who had an inadequate response, loss of response, or were intolerant to either conventional therapy or biologic agents. The CUA was conducted separately for the biologic-naive and -experienced populations.
Upadacitinib is a once-daily, orally administered and highly selective JAK1 inhibitor.1 Treatment with upadacitinib is initiated with an 8-week induction period where patients receive a dose of 45 mg once daily. The recommended dose for maintenance treatment is 15 mg once daily. For some patients, such as those with refractory, severe, or extensive disease, a maintenance dose of 30 mg once daily may be appropriate.3 The upadacitinib regimen captured in the economic model reflected the Health Canada dosing regimen. Assuming the lowest maintenance dose, at the sponsor’s reported price of $49.22, $74.00, and $101.81 per 15 mg, 30 mg, and 45 mg tablet, respectively, the annual cost of upadacitinib is $20,861 for year 1, and $17,965 thereafter.
The comparators for this analysis include tumour necrosis factor (TNF) alpha inhibitors (i.e., adalimumab, adalimumab biosimilar, infliximab, infliximab biosimilar, golimumab), JAK inhibitor (i.e., tofacitinib), 1 cell-adhesion molecule inhibitor (i.e., vedolizumab IV), and conventional therapy (i.e., mix of 5-ASA, corticosteroids, and immunomodulators). Due to lack of maintenance efficacy data for golimumab and infliximab among biologic-experienced patients, these advanced therapies were omitted from the experienced population analysis. The recommended dosing regimen of comparators was sourced from product monographs and their costs from the Ontario Drug Benefit Formulary. These are summarized in Table 11 (Appendix 1). The annual maintenance costs for the comparators ranged from $12,253 for adalimumab (Hulio) to $27,356 for infliximab (Remicade) based on the recommended doses.
The economic evaluation was conducted over a lifetime time horizon (approximately 57 years), from the perspective of the Canadian public health care payer. Costs and clinical outcomes (life-years and quality-adjusted life-years [QALYs]) are discounted at 1.5% per annum.
Model Structure
The sponsor submitted a hybrid model structure that considers a short-term induction phase (decision tree) and a longer-term maintenance phase (Markov model) to evaluate clinical outcomes and costs.1 The decision tree modelled initial response to treatment with either a primary (8-week) or extended (16-week) induction period, while the Markov model cycles were 4 weeks in duration. The same model structure was used for both biologic-naive and -experienced patient populations. Patients entered the model in the induction phase with active UC and initiated treatment. Patients who entered the model on advanced therapies could experience 1 of the following outcomes: remission, response without remission (hereafter termed response), or fail to achieve response. Patients who achieved remission or response at the end of the induction phase, entered the maintenance phase of the Markov model in their corresponding health states, while nonresponders received an additional line of non-conventional therapy. For patients who entered the model in the induction phase on conventional therapy, as well as for those who re-entered the induction phase on non-conventional therapy following failure on the first advanced therapy (i.e., the biologic-experienced population), it was possible to either achieve remission, response, or fail to achieve response. Those who responded to conventional therapy or second-line advanced therapy entered the maintenance phase in their corresponding health states, while those who failed entered the maintenance phase in “active UC.” If available for a drug, patients on maintenance treatment could be on a low- or high-dose formulation of the drug.
The maintenance phase was composed of 9 Markov health states: “remission,” “response without remission,” “active UC,” “first surgery,” “post first surgery remission,” “post first surgery complications,” “second surgery,” “post second surgery remission,” and “death.” During the maintenance phase, patients in the “remission” and “response” health states received treatment until they experienced loss of response, upon which patients who entered the model on upadacitinib or advanced therapies or conventional therapy transitioned to “active UC,” where they received conventional therapy. In the reference case, an additional line of advanced therapy was allowed after advanced therapy failure while on maintenance and before patients transitioned to relapse or loss of response. The sponsor assumed that all patients who failed advanced therapy would sequence to vedolizumab, unless they experienced loss of response while on vedolizumab, whereupon it was assumed they would sequence to tofacitinib. Transition from conventional therapy to advanced therapy was not possible. The model also included a proportion of patients in the active UC health state having surgery. Following the first colectomy, patients discontinued treatment, including conventional therapy and biologics, for the remainder of their lifetime. Post surgery, patients could experience complications or achieve remission. The former could transition to “post first surgery complications” or remain in remission, whereas the latter could remain in “post first surgery complications” or transition to the “second surgery” health state, upon which patients entered “post second surgery remission” for the remainder of the model’s time horizon. Finally, patients could transition to death from any of the maintenance model health states at any time.
Figure 1 and Figure 2 depict the model’s induction and maintenance phases, respectively.
Model Inputs
Baseline patient characteristics were derived from the U-ACHIEVE and U-ACCOMPLISH clinical trials and informed the drug dosage regimens, the age- and sex-specific distribution of the general mortality risk, and the length of the lifetime horizon. The average patient in the biologic-naive cohort was 43.0 years old, weighed 73.1 kg, and was more likely to be male (66.8%). Likewise, in the biologic-experienced cohort, the average patient was 42.7 years old, weighted 72.3 kg, and tended to be male (58.5%). The sponsor submitted an NMA in the absence of head-to-head trial data comparing upadacitinib with its advanced therapy comparators. Bayesian NMAs were performed using random- or fixed-effect models, with a focus on the clinical response and clinical remission outcomes in primary analyses. The model allowed the choice between the unadjusted efficacy data or efficacy data adjusted for baseline characteristics. For the latter, separate NMAs for induction and maintenance were used to allocate patients in the induction and maintenance phases of the model, respectively. These were conducted for 2 patient populations: biologic-naive and biologic-experienced. The efficacy of conventional therapy was represented by the weighted average response rates estimated from the placebo arm of the clinical trials included in the NMA.
In the model, the efficacy response criteria informed by the full Mayo score defined which patients continued treatment, switched to a second advanced therapy, or discontinued treatment and moved to conventional therapy i.e., patients achieving remission (Mayo score of 0 to 2), response (decrease from baseline in the total Mayo score of at least 3 points), and patients with active moderate to severe UC (Mayo score of 6 to 12). The mean absolute probabilities of achieving remission, response, or neither response nor remission that was used in the induction, extended induction, and maintenance phases of the model were derived from the random-effects NMA’s induction- and maintenance-specific phases of the clinical trials (Table 15, Table 16, Table 17, and Table 18). In the maintenance phase, the probability that patients stayed in remission and response was dependent on whether a low or high dose of the advanced therapy was prescribed. Due to the lack of long-term efficacy data for UC treatments beyond the typical trial duration of 1 year, a key assumption in the sponsor’s approach was that of constant treatment effect and corresponding loss of response over the lifetime time horizon.
Probabilities for progression to surgery and surgery with complications were derived from the literature. The annual probability of first and second surgery (0.47%) was derived from the estimate of the risk of colectomy by Misra et al. (2016) based on data from the UK’s Hospital Episode Statistics database, which contains all hospital episodes of colectomy between April 1997 and March 2012.4 The proportion of surgeries that resulted in postsurgery complications (33.5%) was taken from the UK-based clinical audit. The rate informing the proportion of patients who experienced delayed chronic complications post surgery was estimated as a weighted average (5.64%) across 4 studies that had at least 1 year of follow-up.5-8 AE data were gathered from clinical trials, a summary of product characteristics, and previous UC submissions to CADTH.9,10 The sponsor only modelled and costed AEs related to serious infections (i.e., sepsis, pneumonia, urinary tract infection, respiratory infection, and bronchitis) as a composite one-off AE applied during the induction phase. As such, the model applied a utility decrement (0.156) to patients experiencing serious infections once in the induction phase of the model to partially account for the impact that treatment-emergent AEs could have on quality of life.11 The proportion of patients who experienced an AE was obtained from evidence synthesized from a sponsor-commissioned NMA (Table 19). All-cause mortality was applied in the model based on the 2018 to 2020 life expectancy tables from Statistics Canada, without adjustment for UC-specific mortality.
Patients accrued health state–specific utilities, as well as treatment-related and health state–specific costs as they transitioned through changes in disease activity. Utility values for nonsurgical health states were sourced from Woehl et al. (2008),12 who used the EQ-5D questionnaire to collect utility scores from 180 patients with active UC in the UK and reported utility scores for patients in remission, with mild disease, and with moderate to severe disease, and those who were post colectomy without complications. These were used to inform the utility values for remission, response, and active UC, as well as first- and second-surgery remission. Values for surgical states were obtained from Arseneau et al. (2006).13 Utility weights reported in this study were obtained from 48 patients with UC using time trade-off and visual analogue rating scale methods. These were applied to all patients alive in each health state using an age- and sex-adjusted utility approach.14
Unit dose and dosing frequency during the model’s induction and maintenance phase, for both low and high doses, were derived from the respective product monographs for upadacitinib and advanced therapies. In the reference case, if available for a drug, patients on maintenance treatment could be on a low or high dose formulation of the drug. The sponsor derived the percentage of patients on high dose maintenance treatment for both the biologic-naive and biologic-experienced populations from market research as follows: upadacitinib (44%), infliximab (71%), and tofacitinib (44%). The proportions of patients with high doses treated with adalimumab, golimumab, and vedolizumab were set to zero in the sponsor’s model, providing that dose escalation had not been evaluated in the respective clinical trials.
Drug acquisition costs for upadacitinib were based on the sponsor’s submitted price, while the unit costs for every other biologic and nonbiologic comparator were obtained from the Intercontinental Medical Statistics Health Quintiles (IQVIA) DeltaPA database as of February 1, 2022. No administration costs were assumed for upadacitinib, tofacitinib, or conventional therapy due to their oral route of administration. For treatments with subcutaneous administration (i.e., adalimumab and golimumab), 10% of patients were assumed to require nurse-assisted administration, whereas 90% of patients were assumed to self-administer the therapy. For treatments requiring IV administration (i.e., vedolizumab), the cost of administration was estimated by multiplying the cost of IV material from Tam et al. (2013)15 by the time required to prepare the IV from Millar et al. (2008).16 Wastage costs for IV drugs with weight-based dosing were considered by assuming one-time vial usage.
Resource use by health state was informed from a sponsor-commissioned survey of 6 key opinion leaders in the management of UC in Canada. The estimated number of physician visits, tests, and diagnostic procedures gathered from the survey are presented in Table 20. Unit costs for disease management resource use, as well as costs associated with AE management, were obtained from a sponsor-commissioned costing report using the Ontario Ministry of Health and Long-Term Care’s Schedule of Benefits for Professional Services, as well as its Schedule of Benefits for Laboratory Services and the Ontario Case Costing Initiative for hospitalizations. Two phases of surgery were modelled, each lasting for 6 months to allow for staged procedures. Costs for first and second surgeries were allocated as single-event costs, whereas postsurgery remission and postsurgery complications were both inputted as annual costs applied per 4-week cycle for as long as patients remained in these health states after surgery. Resource use for postsurgery periods was informed by a study from Tsai et al. (2008).17
Summary of Sponsor’s Economic Evaluation Results
The sponsor conducted the reference case for the biologic-naive and biologic-experienced moderate to severe UC patient population through a probabilistic sensitivity analysis with 5,000 simulations.1 The deterministic and probabilistic results were similar. The probabilistic findings are presented subsequently.
Base-Case Results
The sequential multiple-comparison cost-utility findings for each population are presented in Table 3 and Table 4. For both the biologic-naive and biologic-experienced populations, the cost-effectiveness frontier (CEF) was represented by conventional therapy and upadacitinib. All other treatments were either strictly or extendedly dominated. In the biologic-naive population, the upadacitinib sequence was associated with a QALY gain of 1.53 at an additional cost of $91,338, resulting in an incremental cost-effectiveness ratio (ICER) of $59,609 per QALY, relative to conventional therapy. Of note, both in the biologic-naive and biologic-experienced populations, the majority of the QALY benefit, 82.5% and 77.1% respectively, was derived from the period beyond which there is observed data, which assumed continued efficacy beyond 52 weeks.
Table 3: Summary of the Sponsor’s Economic Evaluation Results, Biologic-Naive
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
Conventional therapy | 116,390 | 11.39 | Reference |
Upadacitinib | 207,728 | 12.92 | 59,609 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.2
In the biologic-experienced population, upadacitinib was associated with a QALY gain of 1.39 at an additional cost of $79,007, resulting in an ICER of $56,795 per QALY, relative to conventional therapy. Upadacitinib was cost-effective at a willingness-to-pay (WTP) threshold of $50,000 per QALY in 4% and 11% of the iterations in the biologic-naive and biologic-experienced populations, respectively. Table 13 and Table 14 present the results from the sponsor’s sequential analysis, which include dominated treatments, for the biologic-naive and biologic-experienced populations, respectively (Appendix 3).
Table 4: Summary of the Sponsor’s Economic Evaluation Results, Biologic-Experienced
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
Conventional therapy | 117,575 | 11.40 | Reference |
Upadacitinib | 196,582 | 12.79 | 56,795 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.2
Sensitivity and Scenario Analysis Results
The sponsor conducted sensitivity and scenario analyses. Pairwise one-way sensitivity analyses were conducted using the deterministic model to assess the impact of specific parameters on the ICER, incremental QALYs, and incremental costs, for the biologic-naive and biologic-experienced populations. For each one-way sensitivity analysis, the upadacitinib sequence was compared with conventional therapy for both patient populations. The parameters that had the largest impact on the model’s findings were the clinical remission rates and the utility values assigned to clinical remission and active UC, as well as the proportion of patients receiving escalated doses while on maintenance treatment.
The sponsor’s economic submission considered 16 alternative scenarios for further analysis, based on mean costs and QALYs from 1,000 simulations. These were pairwise comparisons that evaluated each comparator treatment relative to upadacitinib; hence, they do not provide relevant information when assessing the cost-effectiveness of upadacitinib in the current setting. The sponsor explored a scenario where conventional therapy was excluded as a comparator. In this scenario, the CEF was represented by adalimumab and upadacitinib in both the biologic-naive and biologic-experienced populations, with every other advanced therapy being either strictly or extendedly dominated.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
High degree of uncertainty in the comparative efficacy and safety of upadacitinib and comparators: Since only 1 published head-to-head trial exists to date concerning advanced therapies in the treatment of moderate to severe UC (i.e., adalimumab versus vedolizumab), comparisons between treatments were driven almost exclusively by the NMA. The evidence synthesized through the sponsor-commissioned NMA showed that upadacitinib had superior clinical efficacy relative to some of the other advanced therapy comparators. However, wider credible intervals of the point estimates were observed and implied considerable uncertainty in the magnitude of the treatment effects of upadacitinib, especially in the maintenance phase. The CADTH clinical review identified considerable uncertainty in the results from the NMA due to sparse networks, heterogeneity in patient characteristics and trial characteristics, and inadequate adjustment for the clinical heterogeneity, including baseline disease severity and treatment exposure. As such, CADTH concluded there is insufficient evidence to confirm how upadacitinib compares relative to other advanced treatments in patients with moderately to severely active UC, regardless of the dose received. Finally, the CADTH clinical review noted that due to limitations with the NMA, conclusions around the comparative safety of upadacitinib versus advanced therapies could not be drawn.
Moreover, the sponsor’s base case used unadjusted efficacy data from a random-effects model for all induction and maintenance clinical parameters, including remission and response. The proportion of patients achieving remission were subtracted from the proportion achieving response to derive the probability of achieving response without remission. However, there were incongruencies in the sponsor’s parametrization of the response without remission probabilities for the biologic-naive population. These were the only probabilities derived incorrectly by subtracting the results of unadjusted remission from the adjusted response. The values used to determine the response without remission should be derived from the same analysis set. The sponsor’s approach led to an overestimation of the proportion of patients achieving response without remission in comparison with using the values from the same analysis set. Finally, the NMA findings showed there was no evidence for a difference between upadacitinib and other relevant active treatments for the incidence of any AEs and serious infections for both induction and maintenance.
CADTH performed a reanalysis by assuming the clinical efficacy and safety of all advanced treatments to be equal to upadacitinib (with the lower maintenance dose [15 mg] used to inform maintenance efficacy for all doses), based on the sponsor’s NMA. Efficacy with conventional therapy was assumed to remain the same as estimated in the sponsor’s base case; however, this is noted to be uncertain, given it is derived from the sponsor’s NMA. Alternative efficacy estimates specific to the sponsor’s pivotal trials comparing upadacitinib and conventional therapy and compatible with the sponsor’s submission were unavailable. In addition, CADTH applied the probabilities of response without remission at the end of induction in the biologic-naive population as derived from the NMA’s random-effects unadjusted model.
AEs considered in the model and their implementation: The sponsor only modelled and costed AEs related to serious infections (i.e., sepsis, pneumonia, urinary tract infection, respiratory infection, and bronchitis) as a composite one-off AE applied during the induction phase. As such, the model applied a utility decrement (0.156) to the proportion of patients experiencing serious infections once in the induction phase of the model to partially account for the impact that treatment-emergent AEs could have on quality of life.11 First, this approach is incomplete because it does not consider important AEs reported in the pivotal trials of upadacitinib (i.e., malignancy and thrombosis) which, along with the incidence of serious infections, have warranted Health Canada–issued serious warnings and precautions on the product. Moreover, the sponsor’s approach to the incorporation of serious infections is inappropriate. In the first model cycle, 0.97% of patients receiving upadacitinib experienced a serious infection in the cohort, which is less than the overall proportion of patients who experienced treatment-emergent serious infections in the U-ACHIEVE Maintenance trial (2.7%), yet AEs were only applied in the first cycle of the model. Patients in the relevant trials of advanced therapies for UC, including upadacitinib, discontinued treatment due to AEs well beyond the first month of therapy. Moreover, the clinical experts consulted by CADTH noted the incidence of herpes zoster infection among patients receiving JAK inhibitors as nonnegligible. Indeed, 4.2% of patients experienced treatment-emergent herpes zoster in the 52-week U-ACHIEVE Maintenance trial. Thus, it would have been appropriate for a more comprehensive inventory of treatment-emergent AEs (including serious infections, malignancy, thrombosis, and herpes zoster) to be applied to the cohort receiving upadacitinib or advanced therapy as a constant rate in each cycle.
CADTH was unable to address this limitation, as the sponsor’s model lacked flexibility to consider AEs other than serious infections. Given the conclusions of the CADTH critical appraisal of the sponsor’s NMA, the issues related to the implementation of serious infections are unlikely to have a significant impact on model results. However, the omission of relevant AEs beyond serious infections likely biases the cost-effectiveness findings in favour of upadacitinib.
Treatment effect is assumed to be constant: In the absence of direct comparative evidence, the sponsor’s model is underpinned by a key efficacy assumption that treatment effect, and the corresponding loss of response, is constant throughout the maintenance phase and over the lifetime time horizon. The clinical experts consulted by CADTH noted that, given the potential for biologic-exposed patients with moderate to severe UC to develop anti-drug antibodies while receiving some advanced therapies, it would be reasonable to assume an increasing risk in the first year, followed by a relatively constant loss of response and remission after that. With regard to small-molecule advanced therapies like upadacitinib, the clinical experts consulted by CADTH noted that although patients are unlikely to develop anti-drug antibodies while on these treatments, loss of response occurs nonetheless, as the pathophysiology of UC may lead to the activation of different inflammation pathways. However, it should be noted that data regarding loss of response is sparse, and current clinical practice suggests that the risk may well be different across advanced therapies, as well as between patients with varying degrees of disease severity. This introduces uncertainty in the estimates of cost-effectiveness derived from the sponsor’s submitted model.
CADTH was unable to address this limitation, as the sponsor’s model lacked flexibility to consider a nonconstant risk of loss of response whereby one-time reductions in treatment efficacy could be applied.
Use of concomitant conventional therapy while on primary advanced therapy is not considered: The sponsor’s model did not include use of concomitant conventional therapy while on primary advanced therapy in the model. The clinical experts consulted by CADTH noted that while the objective is to taper the use of corticosteroids during induction as patients start to respond to the advanced therapies prescribed, it is common for patients to continue receiving immunomodulators during maintenance while on their primary biologic. In fact, current Canadian clinical guidelines18 emphasize the use of concomitant immunomodulators, as they may help optimize biologic pharmacokinetics, minimize immunogenicity, and improve outcomes. Of note, the clinical experts remarked that the practice of concomitant therapy varies widely according to the biologic drug; while corticosteroids may be prescribed for a lengthier period alongside therapies with slower onset of symptom relief, 5-ASAs tend to be more frequently used among patients on anti-TNFs to mitigate the development of antidrug antibodies. Given that the pivotal trials suggest that use of upadacitinib may result in fast-acting symptomatic relief and that, as a small-molecule therapy, it is unlikely that upadacitinib would be prescribed alongside 5-ASAs, the exclusion of concomitant conventional therapy from the model likely biases results in favour of the anti-TNFs (i.e., adalimumab, golimumab, and infliximab) and cell-adhesion molecule inhibitors (i.e., vedolizumab). Nonetheless, CADTH considers that in the moderate to severe UC therapeutic space, the inclusion of concomitant conventional therapy constitutes good modelling practice, as it better reflects current Canadian clinical practice.
CADTH was unable to address this limitation, as the sponsor’s model lacked flexibility to consider the differential use and costs associated with the practice of concomitant conventional therapy across advanced therapies.
Resource utilization not reflective of clinical practice: Resource use by health state was informed from a sponsor-commissioned survey of 6 key opinion leaders in the management of UC in Canada. The estimated number of physician visits, tests, and diagnostic procedures gathered from the survey are presented in Table 20. The clinical experts consulted by CADTH validated the sponsor’s estimates. However, the expert advised that the annual amount of disease management resource use relevant to Canadian practice could not be assumed to be equal across advanced therapies. Indeed, the clinical experts noted that given the uncertainty regarding the long-term safety profiles of the emerging advanced therapies prescribed to treat moderate to severe UC, disease management resource use, particularly as it pertains to JAK inhibitors, would tend to be higher. Specifically, the clinical experts remarked that considering their safety profile, patients receiving JAK inhibitors are more likely to require full blood panels every 2 weeks during induction and every 2 months during maintenance, whereby the frequency of resources used would be double that required for advanced therapies. The clinical experts also highlighted that patients receiving JAK inhibitors would require a greater number of liver function tests per annum, relative to those on advanced therapies. Thus, in alignment with Health Canada’s warning regarding AEs associated with the use of JAK inhibitors, the clinical experts consulted by CADTH confirmed that patients prescribed upadacitinib would require additional surveillance and disease monitoring relative to patients receiving advanced therapies. Thus, it is likely that the omission of resource utilization items especially relevant to JAK inhibitors underestimates the total costs of upadacitinib and biases the cost-effectiveness findings in its favour.
Though the sponsor’s model lacked flexibility to consider the differential disease management and monitoring resource utilization across advanced therapies, CADTH conducted scenario analyses that explored the impact that a 10% increase in the frequency of full blood panels and liver function tests assigned to treatment with upadacitinib would have on the total costs incurred by the drug under review and, consequently, its cost-effectiveness.
Relevant comparators not included in biologic-experienced analysis: The sponsor omitted golimumab and infliximab from the analysis of the biologic-experienced population due to lack of maintenance efficacy data. While the issues with data limitations are plausible, these comparators are relevant to this population. Their exclusion introduces substantial uncertainty into the sponsor’s submitted analysis.
CADTH was unable to address this limitation in reanalyses.
Model transparency: In its assessment of model behaviour, CADTH found the sponsor’s model did not produce identical life-years and QALYs among advanced therapies when implementing the CADTH base-case assumptions of equivalent efficacy when conducted probabilistically. These results did not meet face validity and did not align with the deterministic results, which did produce identical QALYs and life-years among advanced therapies. This introduces uncertainty in the results produced by the sponsor’s model.
CADTH was unable to address this limitation and conducted all reanalyses deterministically.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (Table 5).
Table 5: Key Assumptions of the Submitted Economic Evaluation
Sponsor’s key assumption | CADTH comment |
|---|---|
Patients undergoing revision surgery were assumed to achieve remission after the surgery and to have no surgery-related complications. | Acceptable as a simplifying assumption. However, the relapsing-remitting nature of the disease is not accurately captured post revision surgery. |
Four-week cycle length. | Acceptable. By accommodating varying regimens of UC treatment and allowing the inclusion of induction periods of different lengths, the 4-week cycle length may capture treatment-related costs more accurately. |
Resource use for disease management, treatment monitoring, and surgery-related inputs were assumed to be similar across the biologic-naive and biologic-experienced populations included in the model. | This is uncertain, although acceptable as a simplifying assumption. |
Patients were assumed to remain on any specified escalated dose regimens for the entire duration of the treatment. | According to the clinical experts consulted by CADTH, the proportion of patients with moderately to severely active UC who are prescribed dose de-escalation during the maintenance phase is marginal and thus deemed unlikely to significantly impact expected costs in the model. |
The sponsor assumed a proportion of patients would be on high or low maintenance doses of relevant advanced therapies, including upadacitinib. | Appropriate. Some patients are expected to receive a higher dose of an advanced therapy, such as upadacitinib or tofacitinib, based on their clinical presentation following induction. However, the comparative efficacy of these higher doses is highly uncertain based on the CADTH critical appraisal of the sponsor’s NMA. |
NMA = network meta-analysis; UC = ulcerative colitis.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
CADTH’s reanalysis addressed key limitations within the economic model. The CADTH base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts. The following changes were applied: using the probabilities of response without remission at the end of induction in the biologic-naive population as derived from the NMA’s random-effects unadjusted model, and assuming the probabilities of clinical response and remission, as well as the probabilities of serious infection of all advanced therapies, to be equal to upadacitinib. Table 6 details each change made to derive the CADTH revised base case, which was conducted in a stepwise approach to highlight the impact of each change. The summary of results from the stepped reanalysis are presented in Table 21 and Table 22. All CADTH analyses were conducted deterministically due to issues with the outputs of the sponsor’s probabilistic analyses when applying CADTH base-case changes.
Table 6: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | — | — |
Changes to derive the CADTH base case | ||
1. Estimates for response and remission derived from same analysis | Probabilities of response without remission at the end of induction in the biologic-naive population derived from the random-effects adjusted model. | Probabilities of response without remission at the end of induction in the biologic-naive population derived from the random-effects unadjusted model, in alignment with the probability of remission and the rest of the sponsor’s efficacy inputs. |
2. Comparative efficacy | Probabilities of clinical response and remission derived from the sponsor-commissioned NMA that indicated numerical effect differences between advanced therapies (Table 15, Table 16, Table 17, Table 18). | Clinical efficacy of all advanced treatments assumed to be equal to upadacitinib. |
3. Comparative safety | Probability of serious infection was obtained from evidence synthesized from the sponsor-commissioned NMA (Table 19). | Probability of serious infection for all advanced therapies assumed to be equal to upadacitinib. |
CADTH base case, biologic-naive | Combined revisions 1 + 2 + 3 | |
CADTH base case, biologic-experienced | Combined revisions 1 + 2 + 3 | |
NMA = network meta-analysis.
Regarding the biologic-naive population, conventional therapy and adalimumab were on the CEF. Conventional therapy was found to be the most cost-effective therapy for WTP threshold values below $39,631 per QALY gained, and adalimumab the most cost-effective advanced therapy for WTP threshold values above $39,631 per QALY gained (Table 7). Regarding the biologic-experienced population, conventional therapy and adalimumab were on the CEF. Conventional therapy was found to be the most cost-effective therapy for WTP threshold values below $38,919 per QALY gained, and adalimumab the most cost-effective advanced therapy for values above this threshold (Table 8). All other advanced therapies, including upadacitinib, were strictly dominated.
A detailed breakdown of the disaggregated results is available in Table 23, Table 24, Table 25, and Table 26.
Table 7: Summary of the CADTH Reanalysis Results, Biologic-Naive
Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|
Conventional therapy | 116,339 | 11.29 | Reference |
Adalimumab | 184,326 | 13.00 | 39,631 |
Upadacitinib | 209,692 | 13.00 | Strictly dominated by adalimumab |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: Only nondominated treatments are presented. Strictly dominated upadacitinib is presented as the drug under review.
Table 8: Summary of the CADTH Reanalysis Results, Biologic-Experienced
Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|
Conventional therapy | 117,027 | 11.31 | Reference |
Adalimumab | 181,343 | 12.97 | 38,919 |
Upadacitinib | 203,028 | 12.97 | Strictly dominated by adalimumab |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: Only nondominated treatments are presented. Strictly dominated upadacitinib is presented as the drug under review.
Scenario Analysis Results
CADTH undertook price reduction analyses based on the sponsor’s and CADTH’s base case. Based on the CADTH base case of the sponsor-submitted model, a price reduction of 45% would be necessary for upadacitinib to achieve cost-effectiveness in the biologic-naive population (Table 9) and in the biologic-experienced population (Table 10).
As the CADTH base case assumes equal comparative efficacy and safety across advanced therapies, CADTH also considered price reductions based on the submitted price for upadacitinib and the publicly accessible list prices of all other advanced therapies (Table 11), which indicated that a price reduction of 32% would be required for upadacitinib to be no more costly than a adalimumab biosimilar, which is the least costly advanced therapy for moderately to severely active UC. While the aforementioned price reduction would be required when assuming a low dose maintenance regimen (15 mg) for the drug under review, a price reduction of 50% during the first year, and 55% thereafter, would be required when assuming a high dose maintenance regimen (30 mg).
Table 9: CADTH Price Reduction Analyses, Biologic-Naive
Price reduction | Sponsor base-case ICER ($/QALY) | CADTH base-case ICER ($/QALY) |
|---|---|---|
Upadacitinib submitted price | λ < $57,390: CT λ ≥ $57,390: Upadacitinib | λ < $39,631: CT λ ≥ $39,631: Adalimumab |
10% | λ < $52,027: CT λ ≥ $52,027: Upadacitinib | λ < $39,631: CT λ ≥ $39,631: Adalimumab |
14% | λ < $49,881: CT λ ≥ $49,881: Upadacitinib | λ < $39,631: CT λ ≥ $39,631: Adalimumab |
20% | λ < $46,663: CT λ ≥ $46,663: Upadacitinib | λ < $39,631: CT λ ≥ $39,631: Adalimumab |
30% | λ < $41,300: CT λ ≥ $41,300: Upadacitinib | λ < $39,631: CT λ ≥ $39,631: Adalimumab |
40% | λ < $35,936: CT λ ≥ $35,936: Upadacitinib | λ < $39,631: CT λ ≥ $39,631: Adalimumab |
45% | λ < $33,255: CT λ ≥ $33,255: Upadacitinib | λ < $39,479: CT λ ≥ $39,479: Upadacitinib |
λ = willingness-to-pay threshold; CT = conventional therapy; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: Only nondominated strategies are presented.
Table 10: CADTH Price Reduction Analyses, Biologic-Experienced
Price reduction | Sponsor base-case ICER ($/QALY) | CADTH base-case ICER ($/QALY) |
|---|---|---|
Upadacitinib submitted price | λ < $56,747: CT λ ≥ $56,747: Upadacitinib | λ < $38,919: CT λ ≥ $38,919: Adalimumab |
10% | λ < $51,861: CT λ ≥ $51,861: Upadacitinib | λ < $38,919: CT λ ≥ $38,919: Adalimumab |
14% | λ < $49,907: CT λ ≥ $49,907: Upadacitinib | λ < $38,919: CT λ ≥ $38,919: Adalimumab |
20% | λ < $46,975: CT λ ≥ $46,975: Upadacitinib | λ < $38,919: CT λ ≥ $38,919: Adalimumab |
30% | λ < $42,090: CT λ ≥ $42,090: Upadacitinib | λ < $38,919: CT λ ≥ $38,919: Adalimumab |
40% | λ < $37,204: CT λ ≥ $37,204: Upadacitinib | λ < $38,919: CT λ ≥ $38,919: Adalimumab |
45% | λ < $34,761: CT λ ≥ $34,761: Upadacitinib | λ < $38,817: CT λ ≥ $38,817: Upadacitinib |
λ = willingness-to-pay threshold; CT = conventional therapy; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Note: Only nondominated strategies are presented.
In addition, CADTH conducted a series of exploratory analyses to determine the impact of alternative assumptions on the cost-effectiveness of upadacitinib, which are outlined as follows:
10% increase in the frequency of full blood panels and liver function tests assigned to treatment with upadacitinib.
Assuming all patients on upadacitinib are prescribed low maintenance dosage.
In the first scenario, the total cost of upadacitinib increased to $210,045 and $203,383 in the biologic-naive and the biologic-experienced populations, respectively. In the second scenario, the total cost of upadacitinib decreased to $200,401 and $195,257 in the biologic-naive and biologic-experienced populations, respectively. In both scenarios and across populations, upadacitinib remained strictly dominated by adalimumab.
Issues for Consideration
The drug plan representatives consulted by CADTH have indicated there are currently 5 tofacitinib generics under review by Health Canada, thereby suggesting that when these become available, the price of tofacitinib would be greatly reduced in comparison with the current list price for branded tofacitinib. In addition to the significantly reduced price for tofacitinib generics, there is negotiated confidential pricing for adalimumab biosimilar and infliximab biosimilar. Noting these important considerations with regard to the pricing landscape where advanced therapies like upadacitinib are anticipated to be introduced, the drug plans have highlighted the need to ascertain whether the efficacy and safety superiority of upadacitinib relative to currently available advanced therapies may be demonstrated. As per CADTH’s clinical appraisal, at the time of this review, comparative evidence to confirm the superiority of upadacitinib over other advanced therapies in the treatment of moderate to severe active UC is lacking.
Upadacitinib may be self-administered and would be the third oral, small-molecule advanced treatment to be introduced to the current moderately to severely active UC therapeutic space. This ease of administration was noted as an important outcome for patients and clinicians in their respective inputs.
The modelled price of advanced therapies is based on publicly accessible list prices and does not reflect the existing confidential pricing that has been negotiated by public plans. When further existing confidential discounts on advanced therapies are considered, greater price reductions than those referenced in this report would be required to achieve cost-effectiveness.
Ozanimod is currently under review at CADTH for the indication of UC. The cost-effectiveness of upadacitinib in comparison with ozanimod is unknown.
Overall Conclusions
Based on the appraisal of 2 induction trials and 1 maintenance trial, CADTH clinical reviewers found that upadacitinib was efficacious in inducing and maintaining clinical remission and clinical response when compared with placebo in patients with moderately and severely active UC. Since there are no trials comparing upadacitinib with the advanced therapies of interest (i.e., biologics and small-molecule drugs), comparisons among treatments were based on the sponsor-commissioned NMA. CADTH identified considerable uncertainty in the results from the NMA due to a sparse network, substantial heterogeneity in patient characteristics and trial designs, and inadequate adjustments for the clinical heterogeneity. As such, the CADTH clinical appraisal concluded there is a paucity of evidence to determine how upadacitinib compares with other advanced treatments for efficacy and safety in patients with moderately to severely active UC.
CADTH undertook reanalyses to address limitations in the sponsor’s submission, including applying the probabilities of response without remission at the end of induction in the biologic-naive population as derived from the NMA’s random-effects unadjusted model, and assuming the probabilities of clinical response and remission, as well as the probabilities of serious infection of all advanced therapies, to be equal to upadacitinib for all doses. In both the biologic-naive and biologic-exposed populations, upadacitinib was strictly dominated (i.e., higher cost, equally effective) by adalimumab.
CADTH was unable to address limitations relating to the following:
omission of relevant AEs and the implementation of AEs that were considered in the model
assumption that treatment effect, and the corresponding loss of response, is constant throughout the maintenance phase and over the lifetime time horizon of the model
absence of concomitant conventional therapy use while on primary advanced therapy
assumption of equivalent use of disease management and monitoring resources across advanced therapies.
Given the effect that many of these limitations have on incremental cost, a greater price reduction is likely necessary to achieve cost-effectiveness. There was insufficient economic evidence to justify a higher drug cost for upadacitinib compared with other available advanced therapies. When only considering drug acquisition costs, a price reduction of between 32% and 55% is necessary for upadacitinib to be no more costly than the least costly advanced therapy, depending on the dose of upadacitinib.
References
1.AbbVie. Drug Reimbursement Review sponsor submission: Rinvoq (upadacitinib) 15 mg, 30 mg, 45 mg extended-release oral tablets [internal sponsor's package]. Pointe-Claire (QC): AbbVie; 2022 Apr 18.
2.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Rinvoq (upadacitinib) 15 mg, 30 mg, 45 mg extended-release oral tablets. Pointe-Claire (QC): AbbVie; 2022 Apr 18.
3.AbbVie. PrRinvoq® (upadacitinib): 15 mg, 30 mg, 45 mg extended-release oral tablets [product monograph]. St-Laurent (QC): AbbVie; 2021 Sep 30.
4.Misra R, Askari A, Faiz O, Arebi N. Colectomy rates for ulcerative colitis differ between ethnic groups: results from a 15-year nationwide cohort study. Can J Gastroenterol Hepatol. 2016;2016:8723949. PubMed
5.Suzuki H, Hitoshi O, Chikashi S, et al. The long-term clinical course of pouchitis after total proctocolectomy and IPAA for ulcerative colitis. Dis Colon Rectum. 2012;55(3):330-336. PubMed
6.Ferrante M, Sarah Declerck, Gert De Hertogh, et al. Outcome after proctocolectomy with ileal pouch-anal anastomosis for ulcerative colitis. Inflamm Bowel Dis. 2008;14(1):20-28. PubMed
7.Hata K, Ishihara S, Nozawa H, et al. Pouchitis after ileal pouch-anal anastomosis in ulcerative colitis: Diagnosis, management, risk factors, and incidence. Dig Endosc. 2017;29(1):26-34. PubMed
8.Segal JP, McLaughlin SD, Faiz OD, Hart AL, Clark SK. Incidence and long-term implications of prepouch ileitis: an observational study. Dis Colon Rectum. 2018;61(472-5). PubMed
9.CADTH Common Drug Review pharmacoeconomic report: tofacitinib (Xeljanz) for ulcerative colitis. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/sites/default/files/cdr/pharmacoeconomic/sr0572-xeljanz-pharmacoeconomic-report.pdf. Accessed 2022 Sep 2.
10.CADTH Common Drug Review pharmacoeconomic report: vedolizumab (Entyvio SC) for ulcerative colitis. Ottawa (ON): CADTH; 2020: https://www.cadth.ca/sites/default/files/cdr/pharmacoeconomic/sr0635-entyvio-pharmacoeconomic-review-report.pdf. Accessed 2022 Sep 2.
11.Stevenson M, Archer R, Tosh J, et al. Adalimumab, etanercept, infliximab, certolizumab pegol, golimumab, tocilizumab and abatacept for the treatment of rheumatoid arthritis not previously treated with disease-modifying antirheumatic drugs and after the failure of conventional disease-modifying antirheumatic drugs only: systematic review and economic evaluation. Health Technol Assess. 2016;20(35):611-614. PubMed
12.Woehl A, Hawthorne AB, Morgan CL, Punekar Y, McEwan P. PG113: The epidemiology and health care resource use in patients with Crohn's disease: a population based UK study. Value Health. 2007;10(6).
13.Arseneau KO, Sultan S, Provenzale DT, et al. Do patient preferences influence decisions on treatment for patients with steroid-refractory ulcerative colitis? Clin Gastroenterol Hepatol. 2006;4(9):1135-1142. PubMed
14.Ara R, Brazier J. Populating an economic model with health state utility values: Moving toward better practice. Value Health. 2010;13(5). PubMed
15.Tam VC, Ko Y, Mittmann N, et al. Cost-effectiveness of systemic therapies for metastatic pancreatic cancer. J Clin Oncol. 2011;Meeting Abstract, 2011 ASCO Annual Meeting.
16.Millar D, Corrie P, Pulfer A. PCN74: A service evaluation to compare secondary care resource use between XELOX and FOLFOX-6 regimens in the treatment of metastatic colorectal cancer (MCRC) from a UK National Health Service (NHS) perspective. Value Health. 2008;11(6).
17.Tsai HH, Punekar YS, Morris J, Fortun P. A model of the long-term cost effectiveness of scheduled maintenance treatment with infliximab for moderate-to-severe ulcerative colitis. Aliment Pharmacol Ther. 2008;28(10):1230-1239. PubMed
18.Bressler B, Marshall JK, Bernstein CN, et al. Clinical practice guidelines for the medical management of nonhospitalized ulcerative colitis: the Toronto consensus. Gastroenterology. 2015;148(5):1035-1058 e1033. PubMed
Appendix 1: Cost Comparison Table
Table 11: CADTH Cost Comparison Table for Severely to Moderately Active Ulcerative Colitis
Drug/ comparator | Strength | Dosage form | Price ($) | Recommended dosage | Average cost per month ($) | Average cost per year ($) |
|---|---|---|---|---|---|---|
Upadacitinib (Rinvoq) | 15 mg 30 mg 45 mg | Tab | $49.2200a $74.0000a $101.8100a | 45 mg once daily for 8 weeks, then 15 mg or 30 mg once daily thereafterb | Low dose maintenance Year 1: $1,738.43 Thereafter: $1,497.11 | Low dose maintenance Year 1: $20,861 Thereafter: $17,965 |
High dose maintenance Year 1: $2,374.45 Thereafter: $2,250.83 | High dose maintenance Year 1: $28,493 Thereafter: $27,010 | |||||
Comparators: Biologics | ||||||
Adalimumab (Humira) | 40 mg/0.8 mL | Prefilled syringe or autoinjector for SC injection | $794.1000 | 160 mg at week 0, 80 mg at week 2, then 40 mg every other week thereafterd | Year 1: $1,985.25 Thereafter: $1,720.55 | Year 1: $23,823 Thereafter: $20,647 |
Adalimumab (Hulio) | 40 mg/0.8 mL | Prefilled syringe or autoinjector for SC injection | $471.2700e | 160 mg at week 0, 80 mg at week 2, then 40 mg every other week thereafterf | Year 1: $1,178.18 Thereafter: $1,021.09 | Year 1: $14,138 Thereafter: $12,253 |
Golimumab (Simponi) | 50 mg/0.5 mL 100 mg/1 mL | Prefilled syringe or autoinjector for SC injection | $1,555.1700g $1,557.0000g | 200 mg at week 0, 100 mg at week 2, then 50 mg every 4 weeks thereafterh | Year 1: $1944.42 Thereafter: $1,684.77 | Year 1: $23,333 Thereafter: $20,217 |
Infliximab (Inflectra) | 100 mg | Vial for IV infusion | $525.0000 | 5 mg/kg at week 0, 2, and 6, then every 8 weeks thereafteri | Year 1: $1,400.00 Thereafter: $1,225.00 | Year 1: $16,800 Thereafter: $14,700 |
Infliximab (Remicade) | 100 mg | Vial for IV infusion | $977.0000g | 5 mg/kg at week 0, 2, and 6, then every 8 weeks thereafterj | Year 1: $2,605.33 Thereafter: $2,279.67 | Year 1: $31,264 Thereafter: $27,356 |
Infliximab (Renflexis) | 100 mg | Vial for IV infusion | $493.0000 | 5 mg/kg at week 0, 2, and 6, then every 8 weeks thereafterk | Year 1: $1,314.67 Thereafter: $1,150.33 | Year 1: $15,776 Thereafter: $13,804 |
Ozanimod* (Zeposia) | 0.25 mg 0.5 mg 1 mg | Cap | $68.4932 | 0.25 mg daily on days 1 to 4, 0.5 daily on days 5 to 7, then 1 mg daily thereafterc | Year 1: $2,083.34 Thereafter: $2,083.34 | Year 1: $25,000 Thereafter: $25,000 |
Tofacitinib (Xeljanz) | 5 mg 10 mg | Tab | $23.9589 $42.3436 | 10 mg twice daily for at least 8 weeks, then 5 mg twice daily thereafterm | Year 1: $1,625.10 Thereafter: $1,453.51 | Year 1: $19,501 Thereafter: $17,442 |
Ustekinumab (Stelara) | 130 mg/26.0 mL 90 mg/1.0 mL | Vial for IV infusion Prefilled Syringe for SC injection | $2,079.8400n $4,593.1400n | 6 mg/kg IV at week 0, then 90 mg SC every 8 weeks thereaftern | Year 1: $2,816.53 Thereafter: $2,679.33 | Year 1: $33,798 Thereafter: $32,152 |
Vedolizumab (Entyvio) (IV) | 300 mg | Vial for IV infusion | $3,291.0000g | 300 mg at week 0, 2, 6, then every 8 weeks thereaftero | Year 1: $2,194.00 Thereafter: $1,919.75 | Year 1: $26,328 Thereafter: $23,037 |
Vedolizumab (Entyvio) (SC) | 108 mg/0.68 mL | Prefilled syringe or pen for SC injection | $822.5000p | Following 300 mg IV infusions at weeks 0 and 2, 108 mg SC injection is administered every 2 weeks as maintenance only (from week 4 onwards)o | Year 1: $2,193.50 Thereafter: $1,782.08 | Year 1: $26,322 Thereafter: $21,385 |
Comparators: Aminosalicylates | ||||||
5-ASA (Asacol, Asacol 800) | 400 mg 800 mg | Tab | $0.5597 $1.1358 | Active: 2 to 8 tabs daily in divided doses Maint: 4 tabs daily in divided dosesq | $34.05 to $136.19 | $409 to $1,634 |
5-ASA (Mesasal) | 500 mg | Ent. Tab | $0.6559 | Active: 1.5 to 3 g tabs daily in divided doses Maint: 1.5 g daily in divided dosesr | $59.85 to $119.70 | $718 to $1,436 |
5-ASA (Mezavant) | 1.2 g | Delayed ER-Tab | $1.7284 | Active: 2 to 4 tabs once daily Maint: 2 tabs dailys | $105.14 to $210.29 | $1,262 to $2,523 |
5-ASA (Pentasa) | 500 mg 1,000 mg | ER-Tab | $0.5881 $1.1761 | 0.5 to 1 g 4 times daily (2 g daily dose)t | $71.55 to $143.09 | $859 to $1,717 |
1g | Supp | $1.9962 | 1 g dailyt | $60.72 | $729 | |
1g/100mL 4g/100mL | Enema Enema | $4.4790 $6.0400 | 1 to 4 g daily | $136.24 to $183.72 | $1,635 to $2,205 | |
5-ASA (Salofalk) | 500 mg | Ent.Tab | $0.6445 | Active: 3 g to 4 g daily in divided dosesu Maint: 1.5 to 3 g per day in divided dosesu | $117.62 to $156.83 | $1,411 to $1,882 |
500 mg 1,000 mg | Supp Supp | $1.5314 $2.2495 | 1 to 1.5 g/dayd | $68.42 to $115.00 | $821 to $1,380 | |
4 g/60 g | Rect Susp | $8.1360 | Active: 4 g nightly Maint: 2 g nightly or 4 g every 2 nights | $247.47 $123.74 | $2,970 $1,485 | |
Olsalazine (Dipentum) | 250 mg | Cap | $0.5330 | Active: 1 g to 3 g daily in divided dosesl Maint: 1 g daily in divided dosesl | Year 1: 64.85 to 194.55 Thereafter: $64.85 | Year 1: $778 to $2,335 Thereafter: $778 |
Sulfasalazine (Salazopyrin, generics) | 500 mg | Tab | $0.1804 | Active: 1 g to 2 g 3 to 4 times dailyu Maint: 1 g 2 to 3 times dailyu | Year 1: $32.92 to $65.85 Thereafter: $21.95 to $32.92 | Year 1: $395 to $790 Thereafter: $263 to $395 |
Comparators: Corticosteroids | ||||||
Betamethasone enema (Betnesol) | 5mg/100mL | Enema | $11.8214 | 5 mg nightlyl | $359.57 | $4,315 |
Budesonide (Entocort) | 3 mg | Cap | $1.8653g | 3 mg 3 times per day up to 8 weeks, followed by 6 mg daily for up to 3 monthsl | $54.48 | $654 |
Hydrocortisone enema (Cortenema) (Cortifoam) | 100 mg/60 mL | Enema | $8.2541 | 60 mL nightly or every other night | $125.53 to $251.06 | $1,506 to $3,013 |
15 g/pack (14 doses) | Rect. Aerosol | $117.8800 | One dose nightly or every other nightl | $117.88 to $235.80 | $1,415 to $2,830 | |
Hydrocortisone (Solu-cortef) | 100 mg 250 mg | Vial | $4.1500g $7.2000g | 100 mg to 500 mg IV daily to induce remission; then switch to other agentl | $126.25 to $438.00 | $1,515 to $5,256 |
Prednisone (generic) | 1 mg 5 mg 50 mg | Tab | $0.1095g $0.0220 $0.1735 | 40 mg to 60 mg daily to induce remission; then lower dosel | $5.42 to $8.08 | $64 to $79, or lower |
Comparators: Immunomodulators | ||||||
Azathioprine (generic) | 50 mg | Tab | $0.2405 | up to 2.5 mg/kg dailyl | $29.26 | $351 |
Azathioprine (Imuran) | 50 mg | Tab | $1.0927 | $132.95 | $1,595 | |
Mercaptopurine (Purinethol and generic) | 50 mg | Tab | $2.8610 | 1.5 to 2.5mg/kg dailyl | $261.07 to $348.09 | $3,133 to $4,177 |
Methotrexate (generic) | 2.5 mg 10 mg | Tab | $0.6325 $2.7000g | 10 to 25mg weeklyl | $11.70 to $28.88 | $140 to $347 |
cap = capsule; ent = enteric; er = extended release; maint = maintenance; mg = milligram; sol inj = solution for injection; supp = suppository; tab = tablet
Note: All prices are from the Ontario Drug Benefit Formulary (effective August 2019), unless otherwise indicated, and do not include dispensing fees. Annual period assumes 52 weeks, 365 days.
Note that this table has not been copy-edited.
The comparators presented in this table have been deemed to be appropriate by clinical experts. Comparators may be recommended (appropriate) practice, vs. actual practice. Comparators are not restricted to drugs but may be devices or procedures. Costs are sponsor list prices, unless otherwise specified. Existing Product Listing Agreements are not reflected in the table and as such may not represent the actual costs to public drug plans.
*Ozanimod is currently under review at CADTH for the indication of ulcerative colitis. Price based on submitted price from prior review of ozanimod for multiple sclerosis.
aBased on price submitted by sponsor.
bBased on the product monograph the recommended maintenance dose is 15 mg or 30 mg depending upon patient presentation. For patients aged 65 years or older, the recommended maintenance dose is 15 mg once daily.
cReports dose of ozanimod hydrochloride (HCl); a 0.25 mg, 0.5 mg, and 1 mg of ozanimod HCl equivalents to 0.23 mg, 0,46, and 0.92 mg of ozanimod, respectively.
dHealth Canada Drug Database
ePrice obtained from Ontario Drug Benefit Formulary
fProduct Monograph Adalimumab (HULIO)
gPrice obtained from Saskatchewan Drug Benefit (August 2019)
hProduct Monograph Simponi golimumab injection
IProduct Monograph Infliximab (INFLECTRA).
jProduct Monograph Infliximab (REMICADE).
kProduct Monograph Infliximab (RENFLEXIS).
lXeljanz CADTH CDR PE Report.9
mProduct Monograph Tofacitinib (XELJANZ).
nUstekinumab’s sponsor submission.
oProduct Monograph Vedolizumab (ENTYVIO).
pPrice obtained from Ontario Exceptional Access Program.
q5-ASA Asacol.
r5-ASA Mesasal.
s5-ASA Mezavant.
t5-ASA Pentasa.
uRxTx.
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | Due to lack of maintenance efficacy data for golimumab and infliximab among biologic-experienced patients in the sponsor’s NMA, these advanced therapies were omitted from the biologic-experienced population analysis. |
Model has been adequately programmed and has sufficient face validity | Yes | No comment. |
Model structure is adequate for decision problem | Yes | The model structure is acceptable. However, the relapsing-remitting nature of the disease is not accurately captured post revision surgery. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | The reporting in the pharmacoeconomic and budget impact submissions is clear and consistent with the respective Excel models. Technical documentation regarding the sponsor-commissioned NMA reported the comparative efficacy findings in detail. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
Figure 1: Model Structure, Induction Phase
w/o = without; UC = ulcerative colitis
Source: Sponsor’s pharmacoeconomic submission.2
Figure 2: Model Structure, Maintenance Phase
w/o = without; UC = ulcerative colitis
Source: Sponsor’s pharmacoeconomic submission.2
Detailed Results of the Sponsor’s Base Case
Table 13: Summary of the Sponsor’s Economic Evaluation Results, Biologic-Naive
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
Conventional therapy | 116,390 | 11.39 | Reference |
Upadacitinib | 207,728 | 12.92 | 59,609 |
Dominated treatments | |||
Adalimumab | 145,543 | 11.82 | Extendedly dominated |
Golimumab | 168,711 | 12.21 | Extendedly dominated |
Infliximab | 169,844 | 12.03 | Strictly dominated |
Vedolizumab | 181,030 | 12.35 | Extendedly dominated |
Tofacitinib | 188,411 | 12.52 | Extendedly dominated |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission2
Table 14: Summary of the Sponsor’s Economic Evaluation Results, Biologic-Experienced
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
Conventional therapy | 117,575 | 11.40 | Reference |
Upadacitinib | 196,582 | 12.79 | 56,795 |
Dominated treatments | |||
Adalimumab | 144,035 | 11.76 | Extendedly dominated |
Tofacitinib | 166,469 | 12.06 | Extendedly dominated |
Vedolizumab | 166,582 | 12.06 | Strictly dominated |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.2
Sponsor’s Base Case Model Inputs
Table 15: Probability of Remission and Response by End of Induction, Biologic-Naive
Drug | Induction (8 weeks) | Extended induction (16 weeks) | ||
|---|---|---|---|---|
Remission (%) | Response (%) | Remission (%) | Response (%) | |
Upadacitinib | 50.26 | 41.02 | 5.60 | 42.40 |
Adalimumab | 15.60 | 38.30 | NA | NA |
Golimumab | 25.31 | 31.28 | 15.50 | 12.60 |
Infliximab | 29.03 | 34.15 | 15.50 | 12.60 |
Tofacitinib | 19.44 | 41.30 | 12.50 | 27.90 |
Vedolizumab | 25.63 | 28.50 | 16.00 | 20.00 |
Conventional therapy | 9.48 | 26.19 | NA | NA |
NA = not applicable.
Source: Sponsor’s pharmacoeconomic submission.2
Table 16: Probability of Remission and Response by End of Induction, Biologic-Experienced
Drug | Induction (8 weeks) | Extended induction (16 weeks) | ||
|---|---|---|---|---|
Remission (%) | Response (%) | Remission (%) | Response (%) | |
Upadacitinib | 18.21 | 60.41 | 5.60 | 42.40 |
Adalimumab | 5.60 | 22.48 | NA | NA |
Golimumab | NA | NA | NA | NA |
Infliximab | NA | NA | NA | NA |
Tofacitinib | 10.48 | 40.44 | 5.90 | 31.80 |
Vedolizumab | 6.59 | 23.29 | 6.70 | 19.70 |
Conventional therapy | 2.17 | 19.10 | NA | NA |
NA = not applicable.
Source: Sponsor’s pharmacoeconomic submission.2
Table 17: Probability of Remission and Response by End of Maintenance, Biologic-Naive
Drug | Low dose | High dose | ||
|---|---|---|---|---|
Remission (%) | Response (%) | Remission (%) | Response (%) | |
Upadacitinib | 43.40 | 28.72 | 51.68 | 33.59 |
Adalimumab | 22.69 | 19.37 | 22.69 | 19.37 |
Golimumab | 36.78 | 18.32 | 42.95 | 17.27 |
Infliximab | 26.44 | 27.53 | 27.35 | 30.90 |
Tofacitinib | 60.08 | 8.97 | 62.59 | 13.14 |
Vedolizumab | 47.24 | 25.29 | 49.80 | 15.68 |
Conventional therapy | 20.28 | 15.47 | 20.28 | 15.47 |
Source: Sponsor’s pharmacoeconomic submission2
Table 18: Probability of Remission and Response by End of Maintenance, Biologic-Experienced
Drug | Low dose | High dose | ||
|---|---|---|---|---|
Remission (%) | Response (%) | Remission (%) | Response (%) | |
Upadacitinib | 60.94 | 6.75 | 66.33 | 11.21 |
Adalimumab | 21.17 | 23.68 | 21.17 | 23.68 |
Golimumab | NA | NA | NA | NA |
Infliximab | NA | NA | NA | NA |
Tofacitinib | 20.75 | 37.25 | 32.34 | 38.90 |
Vedolizumab | 45.47 | 9.70 | 44.92 | 7.14 |
Conventional therapy | 9.19 | 12.98 | 9.19 | 12.98 |
NA = not applicable.
Source: Sponsor’s pharmacoeconomic submission.2
Table 19: Probability of Serious Infections During Induction Phase
Drug | Percentage of composite infection rate (%) |
|---|---|
Upadacitinib | 0.97 |
Adalimumab | 0.97 |
Golimumab | 0.19 |
Infliximab | 0.64 |
Tofacitinib | 0.71 |
Vedolizumab | 0.38 |
Conventional therapy | 1.15 |
Source: Sponsor’s pharmacoeconomic submission.2
Table 20: Annual Direct Medical Care Resource Use by Health State
Resource | Response | Remission | Active UC | Postsurgical remission | Postsurgical complications |
|---|---|---|---|---|---|
Gastroenterologist visit | 3.1 | 1.2 | 5.3 | 1.4 | 6.0 |
Other specialist | 0.3 | 0.3 | 0.5 | 0.6 | 1.1 |
Family physician visit | 2.0 | 0.6 | 2.8 | 0.8 | 3.0 |
Upper gastrointestinal endoscopy | 0.1 | 0.1 | 0.1 | 0.1 | 0.2 |
Colonoscopy | 1.3 | 0.4 | 1.5 | 0.4 | 1.5 |
Flexible sigmoidoscopy | 0.5 | 0.0 | 1.0 | 0.1 | 1.1 |
X-ray abdominal | 0.3 | 0.0 | 0.7 | 0.0 | 0.8 |
CT scan colon | 0.0 | 0.0 | 0.2 | 0.0 | 0.2 |
CT scan abdominal | 0.2 | 0.0 | 0.2 | 0.0 | 0.4 |
MRI colon | 0.2 | 0.0 | 0.0 | 0.0 | 0.0 |
Full blood count/blood panel | 3.0 | 1.6 | 5.1 | 1.8 | 5.7 |
Urea and electrolytes | 1.3 | 0.5 | 3.5 | 0.7 | 4.2 |
Stool culture | 0.8 | 0.0 | 1.8 | 0.0 | 1.9 |
FCP testing | 2.5 | 1.6 | 3.8 | 1.6 | 3.9 |
Liver function test | 1.9 | 1.4 | 3.6 | 1.6 | 4.0 |
Lipid profile | 0.0 | 0.0 | 0.2 | 0.0 | 0.2 |
Dietician/Nutritionist | 0.5 | 0.2 | 0.7 | 0.2 | 0.8 |
Surgical consult | 0.1 | 0.0 | 0.8 | 0.1 | 1.1 |
Source: Sponsor’s pharmacoeconomic submission.2
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Detailed Results of CADTH Base Case
Table 21: Summary of the Stepped Analysis of the CADTH Reanalysis Results, Biologic-Naive
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
Sponsor’s base case | Conventional therapy | 116,390 | 11.39 | Ref. |
Upadacitinib | 207,728 | 12.92 | 59,609 | |
CADTH reanalysis 1: Unadjusted estimates | Conventional therapy | 116,339 | 11.29 | Ref. |
Upadacitinib | 195,057 | 12.62 | 58,832 | |
CADTH reanalysis 2: Comparative efficacy | Conventional therapy | 116,339 | 11.29 | Ref. |
Adalimumab | 187,124 | 13.1336 | 38,310 | |
Golimumab | 210,221 | 13.1337 | 275,832,044 | |
CADTH reanalysis 3: Comparative safety | Conventional therapy | 116,339 | 11.29 | Ref. |
Upadacitinib | 204,157 | 12.82 | 57,256 | |
CADTH base case: 1 + 2 + 3 | Conventional therapy | 116,339 | 11.29 | Ref. |
Adalimumab | 184,326 | 13.00 | 39,631 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; Ref. = reference.
Table 22: Summary of the Stepped Analysis of the CADTH Reanalysis Results, Biologic-Experienced
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
Sponsor’s base case | Conventional therapy | 117,575 | 11.4 | Ref. |
Upadacitinib | 196,582 | 12.79 | 56,795 | |
CADTH reanalysis 1: Unadjusted estimates | Conventional therapy | 117,027 | 11.31 | Ref. |
Upadacitinib | 182,228 | 12.46 | 56,747 | |
CADTH reanalysis 2: Comparative efficacy | Conventional therapy | 117,027 | 11.31 | Ref. |
Adalimumab | 181,252 | 12.96516 | 38,861 | |
Tofacitinib | 204,782 | 12.96518 | 850,111,435 | |
CADTH reanalysis 3: Comparative safety | Conventional therapy | 117,027 | 11.31 | Ref. |
Upadacitinib | 183,191 | 12.48 | 56,574 | |
CADTH base case: 1 + 2 + 3 | Conventional therapy | 117,027 | 11.31 | Ref. |
Adalimumab | 181,343 | 12.97 | 38,919 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; Ref. = reference.
Table 23: Disaggregated Costs in the CADTH’s Reanalysis, Biologic-Naive
Drug | Induction | Maintenance | |||||||
|---|---|---|---|---|---|---|---|---|---|
Rem. | Resp. | Active UC | First surgery | Second surgery | Surgery rem. | Surgery comp. | Total costs | ||
Upadacitinib | 16,601 | 67,289 | 26,987 | 92,118 | 3,309 | 153 | 460 | 2,776 | 209,693 |
Adalimumab | 13,722 | 53,734 | 18,055 | 92,118 | 3,309 | 153 | 460 | 2,776 | 184,326 |
Golimumab | 15,553 | 64,955 | 25,449 | 92,118 | 3,309 | 153 | 460 | 2,776 | 204,773 |
Vedolizumab | 21,365 | 66,622 | 26,547 | 92,118 | 3,309 | 153 | 460 | 2,776 | 213,350 |
Tofacitinib | 15,640 | 69,331 | 28,332 | 92,118 | 3,309 | 153 | 460 | 2,776 | 212,119 |
Infliximab | 17,413 | 71,817 | 29,970 | 92,118 | 3,309 | 153 | 460 | 2,776 | 218,016 |
CT | 730 | 144 | 110 | 107,162 | 3,853 | 197 | 567 | 3,576 | 116,339 |
Rem. = remission; Resp. = response; Comp. = complications.
Table 24: Disaggregated QALYs Gained in the CADTH Reanalysis, Biologic-Naive
Drug | Induction | Maintenance | |||||||
|---|---|---|---|---|---|---|---|---|---|
Rem. | Resp. | Active UC | First surgery | Second surgery | Surgery rem. | Surgery comp. | Total QALYs | ||
Upadacitinib | 0.12 | 2.61 | 0.88 | 8.57 | 0.03 | 0.00 | 0.46 | 0.33 | 13.00 |
Adalimumab | 0.12 | 2.61 | 0.88 | 8.57 | 0.03 | 0.00 | 0.46 | 0.33 | 13.00 |
Golimumab | 0.12 | 2.61 | 0.88 | 8.57 | 0.03 | 0.00 | 0.46 | 0.33 | 13.00 |
Vedolizumab | 0.12 | 2.61 | 0.88 | 8.57 | 0.03 | 0.00 | 0.46 | 0.33 | 13.00 |
Tofacitinib | 0.12 | 2.61 | 0.88 | 8.57 | 0.03 | 0.00 | 0.46 | 0.33 | 13.00 |
Infliximab | 0.12 | 2.61 | 0.88 | 8.57 | 0.03 | 0.00 | 0.46 | 0.33 | 13.00 |
CT | 0.06 | 0.05 | 0.03 | 10.10 | 0.03 | 0.00 | 0.58 | 0.43 | 11.29 |
Rem. = remission; Resp. = response; Comp. = complications.
Table 25: Disaggregated Costs in the CADTH’s Reanalysis, Biologic-Experienced
Drug | Induction | Maintenance | |||||||
|---|---|---|---|---|---|---|---|---|---|
Rem. | Resp. | Active UC | First surgery | Second surgery | Surgery rem. | Surgery comp. | Total costs | ||
Upadacitinib | 16,675 | 74,139 | 11,351 | 93,979 | 3,376 | 158 | 474 | 2,876 | 203,028 |
Adalimumab | 13,796 | 57,758 | 8,928 | 93,979 | 3,376 | 158 | 474 | 2,876 | 181,343 |
Vedolizumab | 21,439 | 73,333 | 11,232 | 93,979 | 3,376 | 158 | 474 | 2,876 | 206,867 |
Tofacitinib | 15,714 | 76,606 | 11,716 | 93,979 | 3,376 | 158 | 474 | 2,876 | 204,899 |
CT | 730 | 33 | 60 | 107,922 | 3,880 | 200 | 573 | 3,630 | 117,027 |
Rem. = remission; Resp. = response; Comp. = complications.
Table 26: Disaggregated QALYs Gained in the CADTH Reanalysis, Biologic-Experienced
Drug | Induction | Maintenance | |||||||
|---|---|---|---|---|---|---|---|---|---|
Rem. | Resp. | Active UC | First surgery | Second surgery | Surgery rem. | Surgery comp. | Total QALYs | ||
Upadacitinib | 0.12 | 2.88 | 0.37 | 8.74 | 0.03 | 0.00 | 0.48 | 0.34 | 12.97 |
Adalimumab | 0.12 | 2.88 | 0.37 | 8.74 | 0.03 | 0.00 | 0.48 | 0.34 | 12.97 |
Vedolizumab | 0.12 | 2.88 | 0.37 | 8.74 | 0.03 | 0.00 | 0.48 | 0.34 | 12.97 |
Tofacitinib | 0.12 | 2.88 | 0.37 | 8.74 | 0.03 | 0.00 | 0.48 | 0.34 | 12.97 |
CT | 0.06 | 0.01 | 0.01 | 10.17 | 0.03 | 0.00 | 0.59 | 0.43 | 11.31 |
Rem. = remission; Resp. = response; Comp. = complications.
Appendix 5: Submitted BIA and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 27: Summary of Key Takeaways
Key Takeaways of the BIA |
|---|
|
Summary of Sponsor’s BIA
The sponsor sought to determine the incremental budget impact of reimbursing upadacitinib in patients with moderately to severely active UC from the perspective of Canadian public drug plans, which included all participating public drug plans (except for Quebec), as well as the Non-Insured Benefits Program (NIHB). The sponsor estimated the budget-impact analysis (BIA) via an incremental comparison of 2 scenarios: 1 that considers drug acquisition costs (including dispensing fees, markups, and copayments) associated with currently available therapies used to treat patients with moderate to severe UC (i.e., reference scenario), and a second 1 that considers costs in a world where upadacitinib is reimbursed for the same population (i.e., new scenario). The costs associated with the cohort of eligible patients were forecasted over a 3-year time horizon for both scenarios.
The size of the eligible population of treated patients with moderate to severe UC covered by public drug programs in the baseline “year zero” of the model (i.e., 2022) was estimated using a claims-based approach. The number of patients using advanced therapies for UC was retrieved from IQVIA GPM patient data covering the total Canadian UC market from February 2020 to January 2022. However, this dataset includes information from public and private programs. Thus, the proportion of claims of advanced therapies for UC by public drug plan source for the period between Q1 to 2020 and Q4 to 2021 was retrieved from IQVIA Pharmastat data to estimate the Canadian public coverage of UC patients. Since these datasets do not contain information regarding the NIHB population, the sponsor used a weighted average based on national IQVIA data using the latest estimation of the NIHB and Canadian population. Patient projections were made by forecasting each treatment based on a simple exponential smoothing forecast applied to the estimated number of patients with UC covered by public drug plans from February 2020 to January 2022. The market penetration estimates for upadacitinib over the 3 years of the BIA (i.e., 2023, 2024, 2025) were based on the sponsor’s internal projections. When projecting market shares across advanced therapies, the sponsor considered the implementation of a nonmedical switch (NMS), whereby patients receiving brand products are automatically switched to biosimilars. The number of patients expected to receive each therapy in each year of the BIA model derived from the sponsor’s available data on annual market shares based on the IQVIA GPM and Pharmastat datasets. Key inputs to the BIA are documented in Table 28.
Key model assumptions:
The model assumed that the reimbursement of upadacitinib would not result in additional growth in the advanced therapy-treated patient population.
The model assumed 100% patient compliance to drug treatment.
Treatment discontinuation was assumed to be equal across therapies and hence, is not modelled.
The model assumed that provinces that have not yet announced the NMS policy will start implementation as early as June 2022, with a transition period of 12 months.
The sponsor assumed that upadacitinib market shares would be aligned to the other small-molecule competitors in the UC market (i.e., tofacitinib). However, it was projected that, as the second oral therapy, upadacitinib adoption would be slower.
The sponsor assumed that upadacitinib market shares would come proportionally from currently reimbursed therapies via new patients or switches.
Table 28: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
Methodology used to estimate population size | Claims-based approach |
Number of patients eligible for drug under review | 11,463 / 13,067 / 14,710 |
Market uptake (3 years) | |
Uptake (reference scenario) Humira (adalimumab) Adalimumab biosimilar Remicade (infliximab) Infliximab biosimilar Simponi (golimumab) Xeljanz (tofacitinib) Entyvio (vedolizumab) Conventional therapy | 0.73% 0.00% 0.00% 11.27% 11.63% 11.22% 2.24% 0.00% 0.00% 32.32% 32.72% 31.94% 2.33% 2.17% 2.07% 8.27% 8.49% 8.66% 42.86% 44.99% 46.12% 0.00% 0.00% 0.00% |
Uptake (new-drug scenario) Rinvoq (upadacitinib) Humira (adalimumab) Adalimumab biosimilar Remicade (infliximab) Infliximab biosimilar Simponi (golimumab) Xeljanz (tofacitinib) Entyvio (vedolizumab) Conventional therapy | 3.66% 8.26% 12.08% 0.70% 0.00% 0.00% 10.86% 10.67% 9.86% 2.16% 0.00% 0.00% 31.13% 30.02% 28.07% 2.24% 1.99% 1.81% 7.96% 7.79% 7.62% 41.29% 41.27% 40.56% 0.00% 0.00% 0.00% |
Cost of treatment (per patient) | |
Annual cost of treatment over (year 1) Rinvoq (upadacitinib) Humira (Adalimumab) Adalimumab biosimilar Remicade (infliximab) Infliximab biosimilar Simponi (golimumab) Xeljanz (tofacitinib) Entyvio (vedolizumab) Conventional therapy Annual cost of treatment over (year 2+) Rinvoq (upadacitinib) Humira (adalimumab) Adalimumab biosimilar Remicade (infliximab) Infliximab biosimilar Simponi (golimumab) Xeljanz (tofacitinib) Entyvio (vedolizumab) Conventional therapy | $24,860 $23,564 $14,138 $47,403 $23,664 $23,328 $24,484 $27,965 $1,922 $21,945 $20,422 $12,253 $41,478 $20,706 $20,217 $23,331 $21,385 $1,922 |
Summary of the Sponsor’s BIA Results
The sponsor’s BIA estimated that 3,276 patients with moderate to severe UC would be treated with upadacitinib in the first 3 years of public reimbursement. The incremental expenditures associated with upadacitinib reimbursement in this patient population were estimated to result in savings of -$22,489 in year 1, and incremental costs of $807,980 in year 2, and $1,881,522 in year 3, for a combined 3-year budget impact of $2,667,014.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Uncertainty in the projected market share of upadacitinib: The sponsor assumed that upadacitinib market uptake would be aligned with tofacitinib’s, the only other small-molecule competitor in the moderate to severe UC market. However, it was projected that, as the second oral therapy, upadacitinib adoption would be slower (i.e., 3.66%, 8.26%, and 12.08%, for years 1, 2, and 3, respectively). The clinical experts consulted by CADTH highlighted that although the sponsor’s market projections appear reasonable, there is a notable degree of uncertainty regarding upadacitinib safety profile which may impact its market shares. In this regard, should data become available in the near future demonstrating that upadacitinib has a more advantageous safety profile relative to other advanced therapies, it is likely that upadacitinib would take a greater share of the market than currently anticipated. Likewise, the opposite would be true should the upadacitinib safety profile be less advantageous than known to date.
In light of uncertainty, CADTH conducted exploratory analyses that assumed upadacitinib market shares to be 25% higher and lower than anticipated by the sponsor’s BIA model.
Uncertainty in the projected capture rates of upadacitinib: The sponsor assumed that upadacitinib market shares would come proportionally from currently reimbursed therapies via new patients or switches. The clinical experts consulted by CADTH emphasized that the expectation would be for upadacitinib to capture market shares disproportionally from currently available advanced therapies. Indeed, the clinical experts indicated that should upadacitinib safety profile be more advantageous than tofacitinib, it would displace tofacitinib more intensely. It is also probable that upadacitinib would displace non–small molecule advanced therapies administered via IV considering the comparative ease of administration of an oral therapy.
CADTH could not undertake a reanalysis to address this limitation as the sponsor’s BIA model used a fixed market penetration across advanced therapies and lacked flexibility to alter the capture rates disproportionately.
Exclusion of costs associated with concomitant conventional therapy: The sponsor excluded costs relevant to the practice of concomitant conventional therapy for advanced therapies from the BIA model. The use of concomitant conventional therapy for the treatment of moderate to severe UC when co-administered with a primary biologic drug is widely acknowledged in Canadian clinical guidelines and practice. Moreover, the proportion of patients who are prescribed concomitant therapy is not negligible and, in fact, relevant to include in the BIA considering that the types of therapies and distribution of concomitant conventional therapy varies substantially across advanced therapies.
CADTH could not undertake a reanalysis to address this limitation as the sponsor’s BIA model lacked flexibility to incorporate concomitant conventional therapy costs for advanced therapies.
Inclusion of copayments: The sponsor considered copayments where applicable in the BIA model. Currently, CADTH does not require that patient copayments (i.e., proportion of drug prices paid by the patient) be incorporated into the BIA due to the variability between types of programs/provinces and type of co-payment system implemented (e.g., income based, fixed ceiling, variable stepped rate). Further, copayments are typically inclusive of all treatments received by the patient and may already reach the maximum amount to be paid before starting the drug under review.
CADTH conducted a reanalysis by removing copayments as part of the base case.
CADTH Reanalyses of the BIA
Table 29: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Changes to derive the CADTH base case | ||
1. Copayments | Copayments included. | Copayments excluded. |
CADTH base case | Revision 1 | |
CT = conventional therapy; BIA = budget impact analysis.
The results of the CADTH reanalysis are presented in summary format in Table 30 and a more detailed breakdown is presented in Assuming a 45% price reduction. Based on the CADTH base case, the budget impact associated with upadacitinib’s reimbursement in the indicated target population is expected to be $32,172 in year 1, $796,095 in year 2, and $1,873,060 in year 3, with a 3-year total of $2,636,982.
Table 30: Summary of the CADTH Reanalyses of the BIA, Mixed Population
Stepped analysis | Three-year total |
|---|---|
Submitted base case | $2,667,014 |
CADTH base case | $2,636,982 |
BIA = budget impact analysis. Submitted analysis is based on the publicly available prices of the comparator treatments.
CADTH conducted additional scenario analyses to address remaining uncertainty, using the CADTH base case. Results for the mixed population (biologic-naive and biologic-experienced) are provided in Table 31.
Assuming upadacitinib market shares to be 25% higher.
Assuming upadacitinib market shares to be 25% lower.
Excluding dispensing fees and markups.
Assuming a 55% price reduction.
Assuming a 45% price reduction.
Assuming a 32% price reduction.
Table 31: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $256,129,771 | $259,620,129 | $284,477,956 | $318,737,570 | $862,835,655 |
New drug | $256,129,771 | $259,597,641 | $285,285,936 | $320,619,091 | $865,502,669 | |
Budget impact | $0 | -$22,489 | $807,980 | $1,881,522 | $2,667,014 | |
CADTH base case | Reference | $260,742,633 | $265,082,883 | $290,747,748 | $325,817,121 | $881,647,752 |
New drug | $260,742,633 | $265,050,710 | $291,543,843 | $327,690,181 | $884,284,735 | |
Budget impact | $0 | -$32,172 | $796,095 | $1,873,060 | $2,636,982 | |
CADTH scenario analysis 1: Assuming upadacitinib market shares to be 25% higher | Reference | $260,742,633 | $265,082,883 | $290,747,748 | $325,817,121 | $881,647,752 |
New drug | $260,742,633 | $265,043,190 | $291,749,188 | $328,205,059 | $884,997,437 | |
Budget impact | $0 | -$39,692 | $1,001,440 | $2,387,937 | $3,349,685 | |
CADTH scenario analysis 2: Assuming upadacitinib market shares to be 25% lower | Reference | $260,742,633 | $265,082,883 | $290,747,748 | $325,817,121 | $881,647,752 |
New drug | $260,742,633 | $265,058,230 | $291,341,764 | $327,188,871 | $883,588,865 | |
Budget impact | $0 | -$24,653 | $594,015 | $1,371,750 | $1,941,113 | |
CADTH scenario analysis 3: Excluding dispensing fees and markups | Reference | $246,440,821 | $249,249,668 | $273,157,862 | $306,119,405 | $828,526,936 |
New drug | $246,440,821 | $249,269,385 | $274,045,409 | $308,105,408 | $831,420,201 | |
Budget impact | $0 | $19,716 | $887,546 | $1,986,003 | $2,893,265 | |
CADTH scenario analysis 4: Assuming a 55% price reduction | Reference | $260,742,633 | $265,082,883 | $290,747,748 | $325,817,121 | $881,647,752 |
New drug | $260,742,633 | $259,098,568 | $276,928,718 | $304,282,671 | $840,309,958 | |
Budget impact | $0 | -$5,984,315 | -$13,819,030 | -$21,534,450 | -$41,337,794 | |
CADTH scenario analysis 4: Assuming a 45% price reduction | Reference | $260,742,633 | $265,082,883 | $290,747,748 | $325,817,121 | $881,647,752 |
New drug | $260,742,633 | $260,170,981 | $279,560,144 | $308,495,994 | $848,227,120 | |
Budget impact | $0 | -$4,911,901 | -$11,187,604 | -$17,321,128 | -$33,420,633 | |
CADTH scenario analysis 4: Assuming a 32% price reduction | Reference | $260,742,633 | $265,082,883 | $290,747,748 | $325,817,121 | $881,647,752 |
New drug | $260,742,633 | $261,558,272 | $282,967,236 | $313,954,146 | $858,479,654 | |
Budget impact | $0 | -$3,524,611 | -$7,780,512 | -$11,862,975 | -$23,168,098 |
BIA = budget impact analysis.
Stakeholder Input
Patient Input
Gastrointestinal Society
About Gastrointestinal Society
As the Canadian leader in providing trusted, evidence-based information on all areas of the gastrointestinal tract, the GI (Gastrointestinal) Society is committed to improving the lives of people with GI and liver conditions, supporting research, advocating for appropriate patient access to healthcare, and promoting gastrointestinal and liver health.
Canadian healthcare professionals request more than 600,000 of our BadGut® Basics patient information pamphlets each year, and tens of thousands of Canadians benefit from our important quarterly publication, the Inside Tract® | Du coeur au ventreMD newsletter. GI Society support group meetings offer a wealth of information for those newly diagnosed with a gastrointestinal disorder, as well as those who have lived with a condition for years.
The GI Society is a national charity formed in 2008 on the groundwork of its partner organization, the Canadian Society of Intestinal Research (CSIR), which was founded in Vancouver in 1976. We receive national and international attention, simply because we have earned the respect of both the gastrointestinal medical community and Canadians who battle GI and liver issues daily. Our English (www.badgut.org) and French (www.mauxdeventre.org) websites received 7,839,520 pageviews by 5,753,826 unique visitors in 2021. This is increasing year over year.
All our programs and services focus on providing Canadians with trusted, commercial-free, medically-sound information on gut and liver diseases and disorders in both official languages. Our BadGut® lectures (currently on hiatus due to the pandemic), quarterly Inside Tract® newsletter, pamphlets, and educational videos arm Canadians with the information they require to better understand and manage their specific needs. We also work closely with healthcare professionals and governments at all levels toward system-wide improvements in care and treatment.
Information Gathering
The information we used to complete this questionnaire was obtained primarily through questionnaires:
2015 survey on biologics and biosimilars (then called subsequent entry biologics) completed by 423 Canadians (English: 317 and French: 106) with inflammatory bowel disease (IBD), including Crohn’s disease and ulcerative colitis,
2018 survey on the unmet need in IBD completed by 432 Canadians with IBD,
2020 survey completed by 579 respondents regarding the unmet needs of IBD,
2020 survey on biosimilars with 145 respondents, most of whom had IBD, and a
2022 still-open survey about the IBD patient journey.
We also had contact with patients affected by IBD through one-to-one conversations at our BadGut® Lectures; a patient roundtable; recent phone/email/social media interactions with individuals who have IBD; and stories submitted over time from patients.
Disease Experience
Ulcerative colitis can arise at any age, commonly occurring in young people. There is an increased risk for those who have a family member with the condition. Currently, Canada has among the highest prevalence and incidence yet reported in the world, with approximately 120,000 diagnosed individuals.
Diarrhea, rectal bleeding, and abdominal pain are common symptoms. Inflammation decreases the intestine’s absorptive surfaces, triggering watery stools that can lead to fecal urgency and poor control of bowel function. Low red blood cell count (anemia) can result from blood loss due to ulcerations in the intestine and from general malnutrition due to the debilitating effects of the disease.
Some patients have extra-intestinal manifestations, including fever, inflammation of the eyes or joints (arthritis), ulcers of the mouth or skin, tender and inflamed nodules on the shins, and numerous other conditions. Anxiety and stress are major factors.
Ulcerative colitis often has a profound effect on an individual’s life – physically, emotionally, and socially, both at home and at school or in the workplace. It is particularly difficult for children and young adults since it often affects a person’s sense of self.
More than anything, patients have told us that sustained remission/treatment response is more important than relieving any one symptom. As a chronic disease, it is never just one flare that dominates the impact of the disease, but the constant concern that there will be future flares, possibly worse than the last, and at unpredictable times, which can disastrously disrupt patients’ lives.
The following quotes are from individuals describing what it feels like during a flare of IBD (including ulcerative colitis), and what their biggest concern is, in their own words:
“Your gut aches and burns and there is often blood in the toilet. You lose your appetite and weight, unhealthily! My biggest concern is I'm going to run out of meds to help!”
“It’s like I can’t control anything, I feel weak and can barely get up. My biggest concern is usually when I see blood and determining at what point to go to the ER.”
“The pain is worse than childbirth...and I have 3 kids.1 labour without drugs.”
“Worst flu symptoms, fatigue, lethargy, like swallowing glass and chili and then having constipation and diarrhea at the same time. Gut cramps and hunger cramps at the same time. Want to die. Biggest concern is needing a toilet at all times with zero minutes waiting time.”
“It feels like my guts are in a vise. The nausea can be so bad I can't move or even vomit and the diarrhea is so painful I'll be literally screaming in the bathroom.”
“The worst part is fear of irreversible permanent damage that will affect your day to day life forever.”
“It is so exhausting and feels like it will never end. You start to question if you can still live the life you planned. And no-one gives you a break.”
“A flare can come out of nowhere and completely disrupt your life. Pain can sometimes be so bad that it keeps you in bed. You mostly spend life either asleep or on the toilet. My biggest concern during a flare is being able to keep up with my responsibilities (work, school, social, etc.).”
“It feels like your body is betraying you. You can’t plan anything in advance because you don’t know how your body will feel on a day-to-day basis.”
It’s one thing to read a list of common symptoms or data on how this disease affects patients, but it is the individual stories of these patients, as summarized above, which astound us and motivate us to support patients’ need for more diversity in effective treatments. In addition, treatments should improve quality of life, not cause more symptoms, pain, frustration, or hardship.
Experiences With Currently Available Treatments
The treatment of ulcerative colitis is multi-faceted; it includes managing the symptoms and consequences of the disease along with therapies targeted to reduce the underlying inflammation. Typically, a patient starts on one type of treatment and, if there is inadequate response, then switches to another type.
5-ASA helps to settle acute inflammation and, for some patients, keeps the inflammation inactive when taken on a long-term basis (maintenance). To reduce inflammation in moderate to severe cases, corticosteroids can help. For topical relief in the colon, corticosteroids are available in rectal formulations. These are inconvenient therapies that make it difficult for patients to keep a normal routine, even though they offer relief for those with mild to moderate disease. Also, if a patient has significant diarrhea, then the rectal medications may be difficult to hold in place for sufficient time to be effective. Immunosuppressive agents reduce dependence on steroids and help patients who have steroid-resistant disease, but it could take up to six months or more of therapy to see results.
Rinvoq® is a Janus kinase (JAK) inhibitor, a newer medication type that typically works faster than the other immunosuppressive medications and is in oral form, which many patients prefer. However, we are aware of many recent health risks have arisen with the first product of this type introduced to the market, and that Rinvoq® works in a more targeted way, which might mitigate the side effects.
Biologics treat ulcerative colitis when initially used medications fail to relieve symptoms. There are a variety of mechanisms through which they work. However, these also do not work for all patients, and sometimes an individual will experience remission upon beginning biologic therapy but might find that it stops working after some time.
While there are a few options available, patients still have a lot of difficulty obtaining remission or adequate symptom relief. In one of our recent surveys, we asked patients if the currently available medications are adequate to control their disease. Only 24% of those with IBD thought that the available medications are adequate. Conversely, 56% found them to be only somewhat adequate and 20% not adequate. Patients are still suffering, and they need new and effective options to achieve mucosal healing and reduce the debilitating symptoms of ulcerative colitis.
Improved Outcomes
Patients affected by ulcerative colitis need access to medications that work. Inadequate access to medication results in preventable patient suffering (e.g., continual, debilitating disease symptoms; secondary illnesses such as depression and anxiety disorders; and loss of family/social interactions). It also leads to unnecessary usage of healthcare resources (e.g., hospital stays, surgeries, diagnostic procedures, other medications) and a ripple effect of financial burden on the government and taxpayers (e.g., through inability to work, long-term disability claims, biologic-related debt, and even bankruptcy).
When the patient receives the right medication at the right time and for the right duration – as determined between physician and patient – these individuals can live full, rewarding lives as productive, valuable citizens who participate in the workforce and community. However, since patients respond differently to various medications, and in some cases stop responding to medications after using them for some time, it is important to have a variety of options available.
Experience With Drug Under Review
We haven’t spoken with contacts who have used this medication to treat ulcerative colitis. However, we know that patients want more options, particularly those in pill form, such as Rinvoq®. While biologic medications are very effective, the ongoing injections or infusions required for a person with a chronic disease are a lot of work and effort. Therefore, having more options to try before being prescribed a biologic is helpful for many patients.
Conflict of Interest Declaration — Gastrointestinal Society
To maintain the objectivity and credibility of the CADTH CDR and pCODR programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission?
No.
Did you receive help from outside your patient group to collect or analyze data used in this submission?
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
Table 1: Financial Disclosures for Gastrointestinal Society
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie in 2022 | — | — | X | — |
AbbVie in 2021 | — | — | — | X |
AbbVie in 2020 | — | — | — | X |
Crohn’s and Colitis Canada
About Crohn’s and Colitis Canada
Crohn’s and Colitis Canada is the only national, volunteer-based health charity focused on finding the cures for Crohn’s disease and ulcerative colitis, the two main forms of inflammatory bowel disease (UC) and improving the lives of children and adults affected by these diseases.
Crohn’s and Colitis Canada is one of the top health charity funders of Crohn’s and colitis research in the world, investing over $140 million in research since our founding in 1974. The organization also delivers on its promise through patient programs, advocacy and awareness. We help improve the quality of lives today by:
Sharing accurate and reliable information on treatments, research and issues related to life with Crohn’s and colitis through website, print materials, webinars and live events;
Increasing public washroom access through the GoHere program;
Raising awareness about these Canadian diseases with bilingual public communication;
Offering kids with Crohn’s or colitis camp experience;
Providing a peer support program to newly diagnosed people; and
Advocating on behalf of the patients and caregivers on priority concerns and needs.
Crohn’s and Colitis Canada is comprised of approximately 65,000 supporters including volunteers, donors or individuals interested in engaging with the organization. There is no paid membership. Crohn’s and Colitis Canada is governed by a national volunteer Board of Directors. The organization has a network of volunteer-led Chapters in 46 communities across the country, offering information, events, fundraising opportunities and encouragement. There are thousands of volunteers from coast-to-coast supporting Crohn’s and Colitis Canada’s mission. Crohn’s and Colitis Canada website (https://crohnsandcolitis.ca)/
Information Gathering
Information summarized in this submission was compiled from two online surveys and one phone interview with a patient who participated in the Rinvoq clinical trial.
Survey 1: Our first survey was deployed to our community to better understand unmet needs and priority concerns. The survey included responses from 1706 Canadians, of which 354 had moderate to severe ulcerative colitis.
Survey 2: The goal of our second survey, also deployed earlier this year, was to capture the experience of ulcerative colitis patients who participated in the Rinvoq clinical trial. We received a total of four responses, of which two participated in the Rinvoq clinical trial.
Our answers below are based on the responses from the respondents with moderate to severe ulcerative colitis.
Disease Experience
The results from the patient survey provide a window into how moderate to severe ulcerative colitis (UC) patients live and manage their symptoms. 78% of the respondents were female, 21% male and 1% non-binary.
When asked what UC related complications they are experiencing currently or within the past year, most frequently reported were mental health and stress (65%), followed by joint inflammation & arthritis (51%), anal fissures and hemorrhoids (40%), anemia (33%), and skin conditions and malnutrition and weight loss both at ~ 30%. Other complications include strictures, adhesions (scar tissue), bowel obstruction, eye inflammation, perianal or anal fistulas and abscesses, internal (or intra-abdominal) fistulas or abscesses, stricture, ankylosing spondylitis (arthritis of the spine), liver conditions, and cancer. 13% of the respondents were currently experiencing at least one complication of UC.
Thinking back to when they were first diagnosed, patients noted that they hid aspects of their diagnosis from friends, coworkers and classmates. There is a general misunderstanding of what UC is, which could impact how patients navigate social situations. Nine-in-ten agree that most people don’t know what UC is. This is further compounded by the fact that almost two thirds (63%) of patients agree that their family and friends don’t understand what they are going through. In spite of their medications, two thirds of the patients continue to experience at least one symptom of UC, the most frequent of which are bloating and urgent and frequent need to use the washroom. Over half (56%) believed that different treatment options could make them feel better. At least half of patients felt they could not be open about their UC, felt isolated due to their UC, and believe that their UC has had a negative impact on their romantic relationships with their spouse or partner.
A significant proportion of patients have adjusted their lifestyle and expectations. 72% agreed that they have changed the expectations they had of themselves or that they are always adapting their lifestyle to account for their UC. Two in five patients reported that they changed their travel plans and one in five changed their career aspirations.
Experiences With Currently Available Treatments
Disease management is incredibly important to ensuring patients can live a life of normalcy. Many patients have used a combination of medications to manage their disease, with systemic steroids and biologics being the most common ways (85% and 76% have taken these medications at least once). One third are currently on sulfasalazine & 5-aminosalicylates. Well over half are currently taking a biologic/biosimilar to help manage their UC, though it’s far more likely to be a biologic than a biosimilar. More than one in five are currently taking steroids (30% within last year). Roughly one third of the UC patients have also tried medical cannabis, anti- anxiety medications, and antidepressants to manage their symptoms.
Steroid use is also an important aspect in symptom management and patients aren’t particularly supportive of this treatment option. Almost all patients surveyed agree that they only take systemic steroids if absolutely necessary (93%) with four in five in agreement that they wish they could eliminate systemic steroids from the list of medications they use. Half of respondents say that systemic steroids is/was a burden in their UC management. This is particularly true among those with moderate to severe forms of UC, and among women. Those under the age of 55 are more likely to agree that they have had side effects from systemic steroids. Those with a severe state of UC indicate that they have also experienced side effects from systemic steroid use (90%).
Among those who are using steroids 84% have been on systemic steroids for less than 12 months; with 42% less than 3 months. 13% of the respondents have been on steroids for over 1 year. Two thirds of the respondents feel that systemic steroids are a burden to their UC treatment, with 71% indicating that they have experienced side effects of the steroids.
Among patients who say managing medication use is important, having enough of their treatment options, understanding side effects, and minimizing steroid use were most important. Women are more likely than men to find it important to ensure they have enough treatment options, understand the side effects of long-term use, and minimize the use of steroids.
Improved Outcomes
Patients seek any treatments that can mitigate these symptoms to protect a patient’s ability to work productively, attend school and social events, and even basic daily necessities like leaving the house to run errands or have the energy to maintain a household or raise children. Quality of life could be greatly improved in UC patients if their flares are brought into remission. Based on our survey results, the majority of patients with moderate to severe UC continue to experience symptoms with current treatment options.
Experience With Drug Under Review
Of the two survey responses, both were male, one between the ages of 26-35 and the other 56-65. The telephone interviewee was a female. The responses below are based on both the survey and the telephone interview.
When asked how their UC impacted their daily lives prior to the clinical trial, all three indicated that other therapies were not working well for them, and their UC had been significantly impacting their daily activities and quality of life. The phone interviewee elaborated on her life cycling between periods of remission where symptoms diminished but not completely and periods of active flares. During remission, three out of five of her workdays were “bad” because of bleeding, fatigue and abdominal pain. When asked which aspect of UC was more important to control, all three indicated frequency and urgency of needing to use the toilet, bleeding, pain and anxiety. The phone interviewee also mentioned chronic fatigue due to significant blood loss. Diminished quality of life was also mentioned.
All three trial participants indicated how Rinvoq significantly improved their quality of life and symptoms. Nonexperienced side effects nor had difficulty accessing the treatment.
The phone interviewee summed up her experience with:
“Before Rinvoq, my life was a roller coaster ride…When I got the drug, it was life changing…the bleeding is gone…I have never been better.”
Companion Diagnostic Test
Since all respondents were on a clinical trial, none had difficulty with companion diagnostics.
Anything Else?
No.
Conflict of Interest Declaration — Crohn’s and Colitis Canada
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission?
No.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
Yes. The initial analysis of the survey was conducted by Leger.
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
Table 2: Financial Disclosures for Crohn’s and Colitis Canada
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie (2020-2021 Support) | — | — | — | X |
Clinician Input
IBD Centre of British Columbia
About IBD Centre of British Columbia
The IBD Centre of BC is a charitable organization that is dedicated to providing multidisciplinary care to patients in BC living with IBD. Its secondary goals are to provide education and excel in research in IBD. https://www.ibdcentrebc.ca/
Information Gathering
Clinical experience.
Current Treatments and Treatment Goals
The current treatment paradigm in Canada for the management of ulcerative colitis for the most part focuses on capturing control of inflammation and then using medications to keep the immune system in line to avoid subsequent bouts of inflammation. The treatment options for moderative to severely active UC has evolved over the past 20 years. For the most part patients with UC qualify for advanced therapies when they are either steroid refractory or dependent. The majority of these medications are biologics with the exception of Xeljanz. The majority of these medications are covered by provincial formularies, and all are covered by private insurance companies. Limited free goods are available by drug companies for some of these medications when all other reasonable options have been tried and failed.
The most important goal to achieve in our UC management is enough control of the inflammatory burden in the colon to prevent symptomatic flares, progression of the disease which could either require a colectomy or colon cancer. Approximately 14% of patients with UC will require a colectomy in Canada. Although the target that defines optimal control of inflammation is evolving, the currently accepted target for UC treatment is to achieve mucosal healing defined as the lack of evidence of significant inflammation by the endoscopic appearance of the colon. All of the approved therapies to manage UC can achieve this target however clinical trials and real-world studies suggest that the majority of UC patients cannot achieve a sustainable optimal control.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
Appropriate treatment of any chronic disease should be effective such that the potential long-term consequences of the disease are not reached, and the patients can live their life without the psychological stress of worrying about when their current treatment will fail. Unfortuantely very few patients with UC ever receive this ideal treatment. The reason behind this failure rests on the point that our current choices for treatment fail to achieve mucosal healing in a large proportion of patients. Furthermore, about 15% of patients every year will lose the response they achieved with their current advanced therapy. This means by 3 years almost half of patients started on advanced therapies for UC will need a change of treatment. With that fact in mind it is not surprising that so many patients with UC worry about their disease and because of this their quality of life tremendously suffers. A few potential reasons of this problem centres around the type of medications that are so often being used. Most patients needing advanced therapies require biologics. The problem with biologics is in part related to mode of administration. They all require either an infusion or injection. These modes of administration are a problem for many patients which impacts the adherence to therapy. Patients are at risk of developing antibodies to biologics. Once antibodies have been formed usually the biologic will no longer work or would cause a serious adverse event for that patient.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Rinvoq would have an immediate, significant advantage compared to all other possible treatment options for moderate to severe UC. Rinvoq is an oral medication which deals so clearly with many of the limitation’s biologics have. The network meta-analysis comparing the available medications clearly demonstrate a meaningful efficacy (endoscopic and symptomatic remission) advantage for Rinvoq. Because of the profound improvement in efficacy compared to the biologics it would be reasonable to expect Rinvoq to be appropriate to use in many circumstances for patients including first line.
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
The majority of patients with UC who are steroid dependent of refractory are best suited for Rinvoq. Due to the adverse event profile, there are some patients with UC where other medications would be a better choice. Older patients (>60 years old), patients with multiple cardiac risk factors, and patients with a previous history of skin cancer are those patients.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
The outcomes and their timing to assess response are:
Short term (1-3 months) – symptomatic response
Intermediate term (3-9 months) – biomarkers (fecal calprotectin, CRP)
Long term (9-12 months) – endoscopic healing
What factors should be considered when deciding to discontinue treatment with the drug under review?
Rinvoq (and all other options) should be discontinued if the outcomes used to determine whether a patient is responding to treatment are not reached or the patient develops an intolerance or adverse event caused by the drug.
What settings are appropriate for treatment with [drug under review]? Is a specialist required to diagnose, treat, and monitor patients who might receive [drug under review]?
Rinvoq should only be prescribed by Gastroenterologists.
Conflict of Interest Declarations — IBD Centre of British Columbia
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please refer to the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission?
No.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission?
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
Declaration for Clinician 1
Name: Brian Bressler
Position: Staff Gastroenterologist at St Paul’s Hospital
Date: 28-04-2022
Table 3: COI Declaration for IBD Centre of British Columbia — Clinician 1
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Celltrion, Sandoz | X | — | — | — |
Organon, BMS, Gilead, Viatris | — | X | — | — |
Takeda, Abbvie, Janssen, Pfizer | — | — | — | X |
Declaration for Clinician 2
Name: Yvette Leung
Position: Staff Gastroenterologist at St Paul’s Hospital
Date: 05-05-2022
Table 4: COI Declaration for IBD Centre of British Columbia — Clinician 2
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Abbvie, Takeda, Janssen, Pfizer | — | — | — | X |
Merck, Viatris, BMS, Sandoz | — | — | X | — |
Celltrion, Sandoz | X | — | — | — |
Declaration for Clinician 3
Name: Greg Rosenfeld
Position: Clinical Associate Professor of Medicine, University of BC, Division of Gastroenterology
Date: 05-05-2022
Table 5: COI Declaration for IBD Centre of British Columbia — Clinician 3
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Abbvie, Takeda, Janssen, Pfizer | — | — | — | X |
Merck, Viatris, BMS, Sandoz | — | — | X | — |
Atlantic Specialist Group Managing Ulcerative Colitis & University of Calgary Inflammatory Bowel Disease Unit
About Atlantic Specialist Group Managing Ulcerative Colitis & University of Calgary Inflammatory Bowel Disease Unit
The Atlantic Specialist Group is a group of gastroenterologists and specialists in Inflammatory Bowel Disease (IBD) that treats patients with ulcerative colitis. We are located in various clinical settings across Atlantic Canada.
The University of Calgary Inflammatory Bowel Disease (IBD) Unit comprises a group of nine gastroenterologists, all of whom have fellowship training in inflammatory bowel disease. It comprises national and international experts in IBD ranging from clinical trials, epidemiology, diagnostics, and women’s health. The unit is one of the top three units in the world with respect to patient volume and research output. The group follows over 6000 patients from Western Canada and from around Canada. The mission is to further the care of those that suffer with Crohn’s disease and ulcerative colitis through excellence in patient care, research, and patient and health-care provider education. Dr. Panaccione, the Director of the Unit, holds the Crohn’s Colitis Canada Endowed Research Chair at the University of Calgary.
Information Gathering
A group of ulcerative colitis (UC) specialists from Atlantic Canada convened (12 gastroenterologists and a nurse practitioner) to consult on filling unmet needs in UC and broadening access to efficient treatment in UC (specifically newer biologics coming to market).
Over the course of the meeting, participants discussed Canadian regulatory processes, UC treatment options and recent updates, standards of care in UC, treatment goals in UC, UC patient burden and journey from different perspectives (e.g., patient, family members, and health system/society), the ideal UC care pathway, and gaps needed to address in this care pathway. Following the meeting, we, a subset of the attendees, used the key discussion points to build this submission.
Separately, members of the University of Calgary IBD group reviewed all the literature incorporating the unmet needs and therapeutic gaps in the management of ulcerative colitis.
Current Treatments and Treatment Goals
The most recent Canadian recommendations for the management of ulcerative colitis are the Toronto Consensus clinical practice guidelines for the medical management of non-hospitalized ulcerative colitis. (Bressler B, et al. Gastroenterology. 2015;148:1035–1058) Since this time, the development of new biologics and small molecule drugs, as well as new diagnostic tools, have increased the complexity of ulcerative colitis management. More recent guidelines, such as those from the Selecting Therapeutic Targets in Inflammatory Bowel Disease (STRIDE) Initiative of the International Organization for the Study of IBD (IOIBD) and the European Crohn’s and Colitis Organization (ECCO), reflect updated recommendations for treatment goals, treatment sequencing, and treatment administration. While these are not Canadian-specific guidelines, they are heavily referred to by Canadian physicians, and both publications do have physicians that practise in Canada on the author list. (Turner D, et al. Gastroenterology. 2021;160:1570–1583; Raine T, et al. J Crohns Colitis. 2022;16(1):2-17) As such, these guidelines incorporate the latest thinking in the management of moderate to severe ulcerative colitis in Canada and will be referred to moving forward in this document.
Patients with ulcerative colitis may suffer from mild, moderate, or severe activity that can impact on patients’ overall quality of life. In addition, the uncontrolled inflammation that accompanies symptoms leads to progressive bowel that can lead to mucosal fibrosis, functional bowel issues, stricture formation, and an increased risk of colorectal cancer.
Patients with mildly to moderately active or moderately to severely active ulcerative colitis are primarily treated with pharmacologic therapy. (Kobayashi T, et al. Nat Rev Dis Primers. 2020 Sep 10;6(1):74) The management of patients with acute severe ulcerative colitis requiring hospitalization will not be discussed here because this patient population is beyond the scope of this submission.
In patients with mildly to moderately active UC, ECCO recommends 5-aminosalicylates (5-ASA) (both systemic and topical) to induce remission. (Raine T, et al. J Crohns Colitis. 2022;16(1):2-17) In Canada, 5-ASA is approved for this indication and marketed under several brand names. (e.g., Mezavant®, Mezera®, OctasaTM, Pentasa®, Salofalk®, and Teva-5 ASA; See Product Monographs) ECCO believes topical or colonic-release corticosteroids (budesonide) may be appropriate for some patients who do not respond well to 5-ASA. (Raine T, et al. J Crohns Colitis. 2022;16(1):2-17) In Canada, these are approved and marketed as Entocort® and Cortiment®, respectively. (See Product Monographs) ECCO does not recommend the immunomodulator thiopurine for induction; they only recommend its use for maintenance in mildly to moderately active UC for patients who are steroid-dependent or 5-ASA intolerant. (Raine T, et al. J Crohns Colitis. 2022;16(1):2-17) In fact, in Canada, the most commonly used thiopurine (azathioprine [Imuran®]) is not approved for any UC indications. (See Product Monograph)
Induction of remission with systemic corticosteroids — prednisolone, methylprednisolone, or prednisone — marketed and approved in Canada under multiple brand names (e.g., Medrol, Depo-Medrol®, Solu-Medrol®, Apo-Prednisone, Teva-Prednisone, and Winpred®) is recommended for more moderately to severely active UC. (Raine T, et al. J Crohns Colitis. 2022;16(1):2-17; see Canadian Product Monographs) Induction of remission with advanced targeted agents is recommended if patients are intolerant or have an inadequate response to conventional therapies (e.g., 5-ASA, corticosteroids, and immunomodulators), or even if a patient responds well to corticosteroid induction, because corticosteroid use is not recommended for the long-term due to safety concerns. These advanced targeted agents include anti-tumour necrosis factor (TNF) agents (infliximab [Remicade® or biosimilars], adalimumab [Humira® or biosimilars], and golimumab [Simponi®]), the 𝛼4β7 integrin inhibitor vedolizumab (Entyvio®), the Janus kinase (JAK) inhibitor tofacitinib (Xeljanz®), or the interleukin (IL)-12/23 inhibitor ustekinumab (Stelara®) (Raine T, et al. J Crohns Colitis. 2022;16(1):2-17; see Canadian Product Monographs) Since the aforementioned advanced targeted agents have a proven ability to maintain corticosteroid-free remission, corticosteroid use can and should be limited, with tapering recommended after three months of use. Therefore, ECCO recommends that maintenance therapy should consist of these advanced targeted agents if the patient responds well to their use in induction. (Raine T, et al. J Crohns Colitis. 2022;16(1):2-17)
Only one head-to-head trial of these approved advanced targeted agents exists in ulcerative colitis (VARSITY; adalimumab vs. vedolizumab; Sands B, et al. N Engl J Med. 2019;381:1215-1226) ECCO recommends using vedolizumab rather than adalimumab for the induction and maintenance of remission in moderately to severely active UC based on the findings of this trial, which demonstrated significantly higher percentage of patients achieving clinical response, clinical remission, and endoscopic remission with vedolizumab versus adalimumab. In terms of the other targeted agents, ECCO endorses the use of ustekinumab based on a single randomised trial. Due to safety concerns, ECCO recommends the use of tofacitinib be accompanied by an assessment of the risk and benefits for a particular patient. (Raine T, et al. J Crohns Colitis. 2022;16(1):2-17.) These recommendations are reflected by the prescribing practices of our Canadian group, who prefer vedolizumab and anti-TNFs over ustekinumab and tofacitinib, due to limited efficacy data and safety events, respectively.
Current therapies target the inflammatory response that forms the basis of UC pathology. The conventional treatments described herein (5-ASA, corticosteroids, and thiopurines) act non-specifically, affecting multiple immune pathways. (de Barros Cardoso, C. R. et al. In: Biological Therapy for Inflammatory Bowel Disease [Internet]. London: IntechOpen; 2019) Advanced targeted therapies like vedolizumab, anti-TNF agents, tofacitinib, and ustekinumab inhibit specific cellular pathways. (Kobayashi T, et al. Nat Rev Dis Primers. 2020 Sep 10;6(1):74) All of these therapies target and improve symptoms.
We believe that clinical practice guidelines for ulcerative colitis focus on Health Canada approved medical therapies, but do not always capture the breadth of treatment options discussed with patients, such as combining therapies or fecal microbiota transplantation. As such, our response has focused on medical treatments. Nonmedical treatments, such as nutritional and psychological support, play an important role in the multidisciplinary approach to managing UC. As discussed earlier in this response, pharmacological treatment forms the basis of the treatment plan for patients with moderately to severely active UC. Despite the availability of advanced targeted agents, up to 25% of patients with UC will eventually require surgery, underscoring the need for new medical treatments. (Kobayashi T, et al. Nat Rev Dis Primers. 2020 Sep 10;6(1):74)
Due to the complexity of UC, there are multiple treatment goals to consider when developing a management plan, leading to the need to identify which are the most important to address. Many physicians rely on the STRIDE consensus recommendations and clinical algorithms of the STRIDE group to incorporate clinical targets and facilitate treatment of UC. The most recent initiative, STRIDE-II, proposed an updated, simple algorithm for using selected short-, intermediate-, and long-term treatment targets, with the timing of reaching the goals depending on the specific treatment. (Turner D, et al. Gastroenterology. 2021;160:1570–1583)
According to STRIDE-II, the most important long-term treatment targets are clinical remission, endoscopic healing, restoration of quality of life (QoL), and absence of disability. A particularly important immediate goal is symptomatic relief, a target that is rated highest by patients in studies. (Turner D, et al. Gastroenterology. 2021;160:1570–1583) Patients wish for quick relief to improve their quality of life that is significantly reduced by unpredictable relapses of gastrointestinal symptoms, such as diarrhea, abdominal pain, rectal bleeding, and bowel urgency, as well as systemic symptoms, such as fatigue. (Loftus EV, et al. UEG Journal. 2021. 9(S8): 37-38; Kobayashi T, et al. Nat Rev Dis Primers. 2020 Sep 10;6(1):74.)
STRIDE-II endorses using serum and fecal biomarkers as intermediate medium-term feasible treatment targets, allowing facilitation of UC management in a clinical setting by providing physicians a basis for changing treatment. (Turner D, et al. Gastroenterology. 2021;160:1570–1583) We concur with the STRIDE-II recommendations, but believe histologic remission in conjunction with endoscopic remission is increasingly becoming important for new drugs to achieve, because these measures are associated with a decreased likelihood of clinical relapse. (Yoon H, et al. Gastroenterology. 2020 October; 159(4): 1262–1275.e7.) We acknowledge that incorporating histologic healing in the clinic may be difficult, but achievement of this endpoint in clinical trials can be used to inform clinical practice. In addition, we believe that rapid onset of symptom relief, durability of treatment, and safety of treatment are important treatment goals.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
Overall, we believe the unmet need in patients with moderately to severely active UC is a highly effective therapy that works rapidly and responds in the majority of patients (75% in induction trials). We also believe there is an unmet need for a therapy that meets current treatment goals, which includes not only symptomatic remission, but also steroid sparing and the ability to achieve disease control by meeting important endoscopic and histologic endpoints. We also note the need for therapies that address symptoms important to patients such as bowel urgency, abdominal pain, and fatigue.
We believe that for moderately to severely active UC, no current therapy meets all of these objectives. Further, conventional treatments for UC (5-ASA, corticosteroids, and thiopurines) are not associated with sustained efficacy, have long-term safety concerns, and can sometimes be cumbersome to use.
While advanced targeted agents are available with varying mechanisms of action and have improved efficacy, up to 25% of patients with UC will eventually require surgery, underscoring the need for new medical treatments. (Kobayashi T, et al. Nat Rev Dis Primers. 2020 Sep 10;6(1):74) Further, they each have unique efficacy and safety concerns; for example:
Patients receiving vedolizumab may experience infusion-related reactions. (See Entyvio Product Monograph) In addition, vedolizumab may not be efficacious against the extra-intestinal manifestations (EIMs) often seen with UC (Chateau T, et al. J Crohns Colitis. 2019 Dec 10;13(12):1569-1577.) As discussed in earlier responses, patients rate symptomatic relief as their highest treatment goal; the slower onset of action of vedolizumab compared to some other treatments may therefore pose a problem for maintaining an optimal patient-centred approach to UC management (Vasudevan A, et al. World J Gastroenterol. 2017 Sep 21; 23(35): 6385–6402.)
In our experience, there are logistical concerns with anti-TNF agents; with the abundance of options, accessing support becomes difficult. Further, the practice of therapeutic drug monitoring (TDM) that best optimizes anti-TNF use can be difficult to coordinate if a clinic is not adequately staffed. We also have some concerns regarding the adverse effects of anti-TNF agents, which include infection risk as well as impact on vaccine efficacy, in particular that against Coronavirus disease 2019 (COVID-19).
In addition, patients and physicians all value therapy that is durable. Anti-TNF agents are associated with immunogenicity that often erodes their effects. (Atiqi S, et al. Front Immunol. 2020; 11: 312)
The JAK inhibitor tofacitinib has a Serious Warnings and Precautions Box for serious infections, malignancies, thrombosis, and major adverse cardiac events (MACE). An important property of tofacitinib to note is the fact that it is an inhibitor of JAK1, JAK2, JAK3, and Tyk2. (See Xeljanz Product Monograph) This limited selectivity within the JAK family may be responsible for some of the concerning adverse effects. (Parmentier JM, et al. BMC Rheumatol. 2018;2:23.)
There is limited data of ustekinumab in UC. (Turner D, et al. Gastroenterology. 2021;160:1570–1583) We believe this limits claims that can be made regarding its efficacy.
Taken together, there remains an unmet need for a selective, targeted treatment that meets current treatment targets, particularly histologic and endoscopic remission, reduces extraintestinal manifestations, is safe, and is convenient to administer.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Like already available advanced targeted agents (anti-TNF agents, vedolizumab, ustekinumab, and tofacitinib), upadacitinib targets cellular pathways involved in the inflammation underlying UC, by acting as a selective JAK1 inhibitor. JAKs are important in transducing signals in multiple cellular processes, including inflammation. Therefore, upadacitinib provides an additional mechanism to the UC armamentarium that physicians can choose from to treat their patients with UC.
Although these previously approved advanced targeted agents address some of the needs of some patients with moderately to severely active UC, a large unmet need still exists in this population. As discussed in earlier responses, despite the availability of advanced targeted agents, up to 25% of patients with UC will eventually require surgery, underscoring the need for new medical treatments. (Kobayashi T, et al. Nat Rev Dis Primers. 2020 Sep 10;6(1):74) Further, as discussed in earlier responses, each of these agents has unmet needs relating to efficacy, safety, and/or convenience of administration. In particular, while upadacitinib would not be the first JAK inhibitor approved in UC (tofacitinib), it is the first JAK1 selective inhibitor.
In addition to its JAK1 selectivity, we believe that upadacitinib should be the first choice to treat any patient with moderately to severely active UC who is not responding or intolerant to 5-ASA, or refractory, dependent, or intolerant to corticosteroids because upadacitinib addresses multiple unmet needs:
Safety: Through its JAK1 selectivity, upadacitinib may avoid some of the adverse events of other less selective oral JAK inhibitors, while maintaining the efficacy. The U-ACCOMPLISH and U-ACHIEVE phase 3 trials of 45 mg upadacitinib daily met all primary and ranked secondary endpoints. Eight-week upadacitinib 45 mg QD induction treatment led to statistically significant improvements in clinical, endoscopic, and histologic endpoints in patients with moderately to severely active UC. Upadacitinib was well tolerated; the thrombosis and MACE observed with the less-selective JAK inhibitor tofacitinib were not seen here with this JAK1 selective inhibitor (Danese S, et al. J Crohns Colitis. 2021;15(S1):S022-S024; Vermeire S, et al. J Crohns Colitis. 2021;15(S1):S021-S022.) Furthermore, experience with upadacitinib in other indications (e.g., rheumatoid arthritis, psoriatic arthritis, and atopic dermatitis) further confirms the absence of these safety signals. For example, in patients with rheumatoid arthritis with up to 4.5 years of upadacitinib exposure (over 10,000 patient-years), rates of thrombosis and MACE were equivalent with patients receiving adalimumab or methotrexate. (Cohen SB, et al. Ann Rheum Dis. 2021;80:328-329)
Fast-acting: The control of symptoms was observed as early as day 1; further, the rapid onset of symptom relief aligned with a reduction of inflammatory biomarkers as early as week 2; these findings are promising because they both address the concern of patients for symptomatic relief but also suggest a reduction in treatment targets deemed important by physicians. (Loftus EV, et al. UEG Journal. 2021. 9(S8): 37-38; Vermeire S, et al. J Crohns Colitis. 2022; 16(S1):S087–S088)
Long-term efficacy, including mucosal healing: The efficacy (clinical, endoscopic, and histologic) and safety findings with upadacitinib in patients with moderately to severely active UC were upheld in the phase 3 maintenance study. (Panaccione R, et al. UEG Week. 2021) When looking at maintenance histologic outcomes with and without endoscopy, mucosal healing was also shown to be significantly greater with upadacitinib maintenance treatment versus placebo. (Peyrin-Biroulet L, et al. J Crohns Colitis. 2022; 16(S1):S477–S478) Upadacitinib has been the only advanced treatment so far to achieve such positive findings for mucosal healing. Further, an oral small molecule such as upadacitinib is attractive as a once-a-day therapy that is not associated with immunogenicity that often erodes the effects of our other advanced therapies, diminishing their long-term efficacy.
Treatment of Immune-mediated inflammatory diseases (IMIDs)/Resolution of extraintestinal manifestations (EIMs): Upadacitinib is approved to treat multiple IMIDs (rheumatoid arthritis, atopic dermatitis, and psoriatic arthritis)(RINVOQ Product Monograph). Further, EIM symptom resolution was improved versus placebo following induction treatment with upadacitinib 45 mg and after maintenance treatment with upadacitinib 15 or 30 mg (Colombel J.F., et al. J Crohns Colitis. 2022; 16(S1): i036–i037) Both IMIDs and EIMs are common in patients with UC and impair their quality of life. As discussed in earlier responses, vedolizumab may not be efficacious against these conditions (Chateau T, et al. J Crohns Colitis. 2019 Dec 10;13(12):1569-1577.) Ustekinumab does not cover the same breadth of IMIDs as upadacitinib (STELARA Product Monograph). Further, more data is needed to determine if ustekinumab can resolve EIMs (Guillo L, et al. J Crohns Colitis. 2021 Jul 5;15(7):1236-1243; Guillo L, et al. United European Gastroenterol J. 2020 Nov;8(9):1013-1030) Therefore, upadacitinib may address the current unmet need for an advanced targeted agent that is effective against both UC as well as IMIDs and/or EIMs.
Oral treatment: In our experience, compliance to UC medications can be a major issue. Since upadacitinib is a once-daily oral medication, administration may be more convenient, and may be more likely to adhere to their UC treatment regimen. In fact, a recent survey of IBD patients showed a strong acceptance (91%) of tablets compared to granules (64%), infusions (33%), and subcutaneous injections (34%). (Denesh D, et al. Expert Rev Gastroenterol Hepatol. 2021 Sep;15(9):1091-1096.) Further, oral administration of upadacitinib allows patients to avoid some adverse events seen with medications like vedolizumab, such as infusion-related reactions. There are also patients who require oral therapy, such as those who travel, have needle phobia, or who have poor venous access.
Patients with limited treatment options: As discussed, we recommend patients should be the first choice to treat any patient with moderately to severely active UC who are not responding or intolerant to 5-ASA, or refractory, dependent, or intolerant to steroids.. While awaiting long-term safety data, we believe that upadacitinib should only be restricted from a small subset of patients: those with history of thrombosis or coronary artery disease, or those who could become pregnant.
In addition to 5-ASA and corticosteroids, upadacitinib is effective in those who have failed multiple prior treatments; e.g., those with inadequate response, loss of response, or intolerance to immunosuppressants, and/or biologics. (Danese S, et al. J Crohns Colitis. 2021;15(S1):S022-S024; Vermeire S, et al. J Crohns Colitis. 2021;15(S1):S021-S022.)
We also believe that upadacitinib could also be administered after or in place of corticosteroids even if they are effective at controlling UC. Corticosteroid use can and should be limited due to serious side effects, with tapering recommended after three months of use. (Raine T, et al. J Crohn’s Colitis. 2022;16(1):2-17) Therefore, at minimum, maintenance therapy should consist of upadacitinib if the patient responds well to its use in induction, and physicians can also consider replacing corticosteroid induction entirely with upadacitinib.
We do not believe upadacitinib needs to be administered after other biologic therapies. As discussed earlier in this response, upadacitinib was effective and safe in patients who were either biologic-naïve or experienced. (Danese S, et al. J Crohns Colitis. 2021;15(S1):S022-S024; Vermeire S, et al. J Crohns Colitis. 2021;15(S1):S021-S022.) Therefore, we believe that administering upadacitinib after conventional treatments for UC (e.g., 5-ASA or corticosteroids) would avoid unnecessary and costly use of other biologics prior to treatment with upadacitinib.
Further, we do not believe that patients should be required to have failed an immunomodulator. This aligns with the ECCO recommendations discussed in earlier responses that states immunomodulators should only be used as maintenance treatment in patients who are steroid-dependent or intolerant to 5-ASA. ECCO stresses that immunomodulator maintenance therapy needs to be paired with an effective induction regimen. (Raine T, et al. J Crohns Colitis. 2022;16(1):2-17) Since upadacitinib is a safe and effective option for both induction and long-term maintenance therapy, we believe that it can eliminate the need for immunomodulators completely.
Therefore, upadacitinib may further shift the current treatment paradigm by providing an additional option for patients with moderately to severely active UC whose disease is not adequately controlled with conventional medical treatments. If the strong safety profile continues to be validated in long-term studies, we believe upadacitinib could become the most widely used therapy for moderately to severely active UC.
Which patients would be best suited for treatment with the drug under review? Which patients would be least suitable for treatment with the drug under review?
In Phase 3 trials, patients were selected based on eligibility criteria including:
Diagnosis of UC prior to baseline, confirmed by colonoscopy
Appropriate documentation of biopsy results consistent with the diagnosis of UC in the assessment of the investigator
Active UC with an Adapted Mayo score of 5 to 9 points and endoscopic subscore of 2 to 3
Demonstrated an inadequate response, loss of response, or intolerance to at least one of the following treatments including oral aminosalicylates, corticosteroids, immunosuppressants, and/or biologic therapies.
(U-ACCOMPLISH, https://clinicaltrials.gov/ct2/show/NCT03653026; U-ACHIEVE, https://clinicaltrials.gov/ct2/show/NCT02819635)
Regarding the last bullet, we believe that upadacitinib should be the first choice to treat any patient with moderately to severely active UC who is not responding or intolerant to 5-ASA, or refractory, dependent, or intolerant to corticosteroids. However, several patients will also fail to respond, lose response, or be intolerant to other advanced therapies (anti-TNF agents, vedolizumab, ustekinumab, and tofacitinib). In these situations, upadacitinib would also be the treatment of choice. Recent network meta-analyses showed indirectly that upadacitinib is most efficacious out of the available advanced therapies. (Lasa JS, et al. Lancet Gastroenterol Hepatol. 2022 Feb;7(2):161-170; Burr NE, et al. Gut. 2021 Dec 22;gutjnl-2021-326390.)
These patients are in the most need of intervention as they lack long-term treatment options and are at high risk of disease progression. Once UC has progressed, patients are at higher risk of disabling digestive symptoms, emergency room visits, hospitalizations, and surgery.
Following an in-office consultation, patients require an endoscopy for a definitive UC diagnosis. In Canada, a lack of access to endoscopy delays diagnosis and increases wait time for adequate treatment. Physicians also may use other modalities to complete their diagnosis, including but not limited to tests for complete blood count (CBC), albumin, thiopurine methyltransferase (TPMT), C. difficile, hepatis B virus, and tuberculosis, as well as examination of stool, biologic work-up, intestinal ultrasonography, and abdominal x-ray.
Finally, UC patients who have mildly to moderately active disease, or those with previous mildly to moderately active disease and whose remission can be adequately maintained with conventional treatments are least suitable for treatment with upadacitinib.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice? How often should treatment response be assessed?
In clinical trials, outcomes are measured by the following endpoints:
Clinical remission/response (based on stool frequency, rectal bleeding subscore, and endoscopic subscore)
Endoscopic improvement/remission
Histologic improvement/Histologic-endoscopic mucosal improvement/mucosal healing
Inflammatory bowel disease questionnaire (IBDQ) to assess quality of life
Patient-reported outcomes including bowel urgency and abdominal pain
Biomarkers such as fecal calprotectin and C-reactive protein
Work Productivity and Activity Impairment (WPAI) Questionnaire to assess work productivity losses and impairment in daily activity
(U-ACCOMPLISH, https://clinicaltrials.gov/ct2/show/NCT03653026; U-ACHIEVE, https://clinicaltrials.gov/ct2/show/NCT02819635; Danese S, et al. J Crohns Colitis. 2021;15(S1):S022-S024; Vermeire S, et al. J Crohns Colitis. 2021;15(S1):S021-S022)
While not frequently employed in clinical practice, as discussed in earlier responses, histologic remission in conjunction with endoscopic remission is increasingly becoming important for new drugs to achieve, because these measures are associated with a decreased likelihood of clinical relapse. (Yoon H, et al. Gastroenterology. 2020 October;159(4):1262–1275.e7.) We acknowledge that incorporating histologic healing in the clinic may be difficult, but achievement of this endpoint in clinical trials can be used to inform clinical practice, which would then allow more patients to receive an effective treatment, reducing the need for hospitalizations and surgery.
In our opinion, the minimally clinical important difference that upadacitinib could achieve and be unlikely to vary across physicians would be a clinical response of 30% decrease from baseline in Mayo score.
However, it is important to note that upadacitinib has also demonstrated efficacy in multiple other scores, including:
Clinical remission: adapted Mayo score ≤2 (stool frequency subscore ≤1 and not higher than baseline, rectal bleeding subscore of 0, and endoscopic subscore ≤1)
Clinical response: decrease in partial adapted Mayo score ≥1 and ≥30% from baseline, plus a decrease in rectal bleeding subscore ≥1 or an absolute rectal bleeding subscore ≤1
Endoscopic improvement: endoscopic subscore of 0 or 1
Endoscopic remission: endoscopic subscore of 0
Histologic improvement defined by Geboes score (any decrease)
Histologic-endoscopic mucosal improvement: endoscopic subscore of 0 or 1 and Geboes score ≤3.1
Mucosal healing: endoscopic subscore of 0 and Geboes score <2.0
Inflammatory bowel disease questionnaire (IBDQ): positive change in score, based on questions regarding bowel and systemic symptoms, as well as emotional and social functions
Meaningful improvements in the Work Productivity and Activity Impairment (WPAI) Questionnaire
Meaningful and rapid reductions in patient-reported outcomes including bowel urgency and abdominal pain. These outcomes cannot be overlooked as they are most important to patients
Meaningful and rapid reductions in biomarkers such as fecal calprotectin and C-reactive protein
(U-ACCOMPLISH, https://clinicaltrials.gov/ct2/show/NCT03653026; U-ACHIEVE, https://clinicaltrials.gov/ct2/show/NCT02819635)
Importantly, a patient should not experience any severe side effects, including over sustained time periods, in order for the response to upadacitinib to be clinically meaningful.
The duration of treatment is highly dependent on the therapy and on the patient’s disease activity and personal preferences. Further, as discussed in earlier responses, some treatments have more rapid onset of action than others and this should be considered before deciding a treatment has failed. In general, we believe that a treatment should be used for two to three months, at which point a determination of treatment response can be made.
What factors should be considered when deciding to discontinue treatment with the drug under review?
We believe that ongoing clinical symptoms and need for continued systemic corticosteroid use or steroid intolerance would indicate the patient has failed that treatment.
Treatment should also be discontinued if the patient experiences adverse reactions or intolerance to the medication that are deemed to be unacceptable by the patient-physician team.
Treatment with upadacitinib should be interrupted if a patient develops a serious infection, until the infection is controlled. Treatment should also be interrupted to address abnormal laboratory results (ALC less than 500 cells/mm3, ANC less than 1000 cells/mm3, Hb less than 8 g/dL, or if drug-induced liver injury is suspected [based on hepatic transaminases]) and may be resumed once levels return to normal. (RINVOQ Product Monograph. 2022. Canada. AbbVie Inc)
What settings are appropriate for treatment with [drug under review]? Is a specialist required to diagnose, treat, and monitor patients who might receive [drug under review]?
Patients with UC receiving upadacitinib would ideally be managed in any non-emergent setting that they have access to, and that has a gastroenterologist well-versed in managing moderately to severely active UC, who would be responsible for the diagnosis and staging, as well as prescribing and managing treatment with upadacitinib for moderately to severely active UC.
Referring family physicians, nurse practitioners, or other health care providers should be counselled on the appropriate referral process and information needed for a complete UC patient work-up.
Additional Information
Upadacitinib presents a breakthrough in moderately to severely active UC management as reflected by its convenient oral mode of administration, selective mechanism of action, efficacy, rapid onset of impact on patients’ QoL, and acceptable safety profile for long term use. Early control of UC and achievement of outcomes such as endoscopic and histologic remission, and mucosal healing, could reduce the risk of clinical relapse, disease progression, and the associated costs of emergency room (ER) visits, hospitalizations, and surgeries.
Our clinical opinion has been validated indirectly in recent network metanalyses, which showed that upadacitinib is most efficacious out of the available advanced therapies. (Lasa JS, et al. Lancet Gastroenterol Hepatol. 2022 Feb;7(2):161-170; Burr NE, et al. Gut. 2021 Dec 22;gutjnl-2021-326390.)
Conflict of Interest Declarations — Atlantic Specialist Group Managing Ulcerative Colitis & University of Calgary Inflammatory Bowel Disease Unit
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please refer to the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission?
No.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission?
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
Declaration for Clinician 1
Name: Mark Borgaonkar
Position: Gastroenterologist, Eastern Health, NL
Date: 16-03-2022
Table 6: COI Declaration for Atlantic Specialist Group Managing Ulcerative Colitis & University of Calgary Inflammatory Bowel Disease Unit — Clinician 1
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Abbvie | — | — | X | X |
Pfizer | — | X | — | — |
Janssen | — | — | X | — |
Takeda | — | — | X | — |
Declaration for Clinician 2
Name: John Igoe
Position: Gastroenterologist, NB
Date: 17-03-2022
Table 7: COI Declaration for Atlantic Specialist Group Managing Ulcerative Colitis & University of Calgary Inflammatory Bowel Disease Unit — Clinician 2
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Abbvie | — | — | X | — |
Janssen | X | — | — | — |
Takeda | — | X | — | — |
Pfizer | X | — | — | — |
Declaration for Clinician 3
Name: Cathy Lu
Position: Gastroenterologist, University of Calgary, AB
Date: 08-05-2022
Table 8: COI Declaration for Atlantic Specialist Group Managing Ulcerative Colitis & University of Calgary Inflammatory Bowel Disease Unit — Clinician 3
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie | X | — | — | — |
Janssen | X | — | — | — |
Fresenius Kabi | X | — | — | — |
Declaration for Clinician 4
Name: Chris Ma
Position: Gastroenterologist, Univerity of Calgary, AB
Date: 09-05-2022
Table 9: COI Declaration for Atlantic Specialist Group Managing Ulcerative Colitis & University of Calgary Inflammatory Bowel Disease Unit — Clinician 4
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie | — | — | X | — |
Alimentiv | — | — | — | X |
Amgen | — | X | — | — |
AVIR Pharma Inc | — | X | — | — |
BioJAMP | X | — | — | — |
BMS | — | X | — | — |
Celltrion | X | — | — | — |
Ferring | — | — | X | — |
Fresenius Kabi | — | — | X | — |
Janssen | — | — | X | — |
McKesson | X | — | — | — |
Mylan | X | — | — | — |
Takeda | — | — | X | — |
Pendopharm | — | X | — | — |
Pfizer | — | — | X | — |
Roche | — | X | — | — |
Declaration for Clinician 5
Name: Mark MacMillan
Position: Gastroenterologist, MD, FRCPC, Assistant Professor Dalhousie University, Memorial University, President New Brunswick Association of Gastroenterology, NB
Date: 16-03-2022
Table 10: COI Declaration for Atlantic Specialist Group Managing Ulcerative Colitis & University of Calgary Inflammatory Bowel Disease Unit — Clinician 5
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie | X | — | — | — |
Pfizer | X | — | — | — |
Janssen | X | — | — | — |
Takeda | X | — | — | — |
Declaration for Clinician 6
Name: Bruce Musgrave
Position: Gastroenterologist, Valley Regional Hospital, Kentville, NS
Date: 25-03-2022
Table 11: COI Declaration for Atlantic Specialist Group Managing Ulcerative Colitis & University of Calgary Inflammatory Bowel Disease Unit — Clinician 6
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie | X | — | — | — |
Janssen | X | — | — | — |
Declaration for Clinician 7
Name: Kerri Novak
Position: Gastroenterologist, University of Calgary, AB
Date: 11-05-2022
Table 12: COI Declaration for Atlantic Specialist Group Managing Ulcerative Colitis & University of Calgary Inflammatory Bowel Disease Unit — Clinician 7
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie | — | X | — | — |
Amgen | X | — | — | — |
Fresenius Kabi | X | — | — | — |
Janssen | — | X | — | — |
Pfizer | X | — | — | — |
Sandoz | X | — | — | — |
Takeda | — | X | — | — |
Declaration for Clinician 8
Name: Remo Panaccione
Position: Director, Gastrointestinal Research, University of Calgary, AB
Date: 07-05-2022
Table 13: COI Declaration for Atlantic Specialist Group Managing Ulcerative Colitis & University of Calgary Inflammatory Bowel Disease Unit — Clinician 8
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie | — | — | X | — |
Alimentiv (formerly Robarts) | X | — | — | — |
Amgen | X | — | — | — |
Arena Pharmaceuticals | X | — | — | — |
AstraZeneca | X | — | — | — |
Bristol-Myers Squibb | X | — | — | — |
Boehringer Ingelheim | X | — | — | — |
Eli Lilly | X | — | — | — |
Ferring | X | — | — | — |
Fresenius Kabi | X | — | — | — |
Galapagos | X | — | — | — |
Gilead Sciences | X | — | — | — |
Glaxo-Smith Kline | X | — | — | — |
JAMP biomed | X | — | — | — |
Janssen | — | — | X | — |
Merck | X | — | — | — |
Mylan | X | — | — | — |
Oppilan | X | — | — | — |
Organon | X | — | — | — |
Pandion Pharma | X | — | — | — |
Pfizer | — | — | X | — |
Progenity | X | — | — | — |
Protagonist Therapeutics | X | — | — | — |
Roche | X | — | — | — |
Satisfai Health | X | — | — | — |
Sandoz | X | — | — | — |
Sublimity Therapeutics | X | — | — | — |
Takeda Pharmaceuticals | — | — | X | — |
Viatris | X | — | — | — |
Declaration for Clinician 9
Name: Cynthia Seow
Position: Gastroenterologist, Univerity of Calgary, AB
Date: 09-05-2022
Table 14: COI Declaration for Atlantic Specialist Group Managing Ulcerative Colitis & University of Calgary Inflammatory Bowel Disease Unit — Clinician 9
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie | — | X | — | — |
Amgen | X | — | — | — |
Fresenius Kabi | X | — | — | — |
Janssen | — | X | — | — |
Pfizer | X | — | — | — |
Sandoz | X | — | — | — |
Takeda | — | X | — | — |
Declaration for Clinician 10
Name: Tushar Shukla
Position: Gastroenterologist, South Health Campus, AB
Date: 09-05-2022
Table 15: COI Declaration for Atlantic Specialist Group Managing Ulcerative Colitis & University of Calgary Inflammatory Bowel Disease Unit — Clinician 10
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie | X | — | — | — |
Janssen | X | — | — | — |
Pfizer | X | — | — | — |
Takeda | X | — | — | — |
Declaration for Clinician 11
Name: Michael Stewart
Position: Gastroenterologist, Halifax, Nova Scotia
Date: 12-05-2022
Table 16: COI Declaration for Atlantic Specialist Group Managing Ulcerative Colitis & University of Calgary Inflammatory Bowel Disease Unit — Clinician 11
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
AbbVie | — | — | X | — |
Amgen | X | — | — | — |
Janssen | — | X | — | — |
Pfizer | X | — | — | — |
Takeda | X | — | — | — |
Sandoz | X | — | — | — |
Bristol-Myers-Squibb | X | — | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.