CADTH Reimbursement Review
Dalbavancin (Xydalba)
Sponsor: Paladin Labs Inc.
Therapeutic area: Acute bacterial skin and skin structure infections
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
ABSSSI
acute bacterial skin and skin structure infection
AE
adverse event
BMI
body mass index
CE
clinically evaluable
CI
confidence interval
CrCl
creatinine clearance
CrI
credible interval
cSSSI
complicated skin and skin structure infection
cSSTI
complicated skin and soft tissue infection
DB
double-blind
EOT
end of treatment
ER
emergency room
HRQoL
health-related quality of life
ITC
indirect treatment comparison
ITT
intention-to-treat
IVRS
interactive voice-activated randomization system
LFU
long-term follow-up
LOS
length of stay
microITT
microbiological intention-to-treat
MRSA
methicillin-resistant Staphylococcus aureus
MSSA
methicillin-susceptible Staphylococcus aureus
NMA
network meta-analysis
OR
odds ratio
PICC
peripherally inserted central catheter
RCT
randomized controlled trial
SAE
serious adverse event
SAP
statistical analysis plan
SFU
short-term follow-up
SIRS
systemic inflammatory response syndrome
SSSI
skin and skin structure infection
SSTI
skin and soft tissue infection
TEAE
treatment-emergent adverse event
TOC
test of cure
WBC
white blood cell
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Dalbavancin (Xydalba) for injection, 500 mg (as dalbavancin hydrochloride) per vial, lyophilized powder for solution, IV infusion |
Indication | Treatment of adults with ABSSSI caused by susceptible isolates of the following gram-positive micro-organisms: Staphylococcus aureus (including methicillin-susceptible and methicillin-resistant strains), Streptococcus pyogenes, Streptococcus agalactiae, Streptococcus dysgalactiae, Streptococcus anginosus group (including Streptococcus anginosus, Streptococcus intermedius, Streptococcus constellatus), and Enterococcus faecalis (vancomycin-susceptible strains) |
Reimbursement request | Per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Priority |
NOC date | May 5, 2021 |
Sponsor | Paladin Labs Inc. |
ABSSSI = acute bacterial skin and skin structure infection; NOC = Notice of Compliance.
Introduction
Acute bacterial skin and skin structure infections (ABSSSIs) are bacterial infections of skin and associated tissues. They are caused by the penetration of bacterial skin flora into soft tissues. ABSSSIs include cellulitis, erysipelas, wound infections, and major cutaneous abscesses.1 ABSSSIs may be acquired in either the community or hospital setting and are a frequent cause of morbidity.2-4 Worldwide, the most common cause of ABSSSIs is Staphylococcus aureus, including methicillin-resistant Staphylococcus aureus (MRSA).2,5 In Canada, the estimated prevalence of ABSSSIs requiring IV antibiotics is 0.39%,6,7 and First Nations communities are disproportionately affected.7 Severity ranges from nonsevere infections to severe, life-threatening infections that affect deeper tissue layers.2,8,9 In North America, the mortality rate among patients hospitalized for skin and soft tissue infections (SSTIs) is approximately 0.5%.10 Compared with non-MRSA infections, ABSSSIs caused by MRSA are associated with increased risk of mortality, longer hospital stays, and higher health care costs.11
The microbiologic diagnosis of ABSSSIs can be difficult, especially in cases of closed infection, where a microbiological specimen cannot be obtained without invasive procedures, such as biopsy. Hence, in many cases, the microbiological cause of ABSSSIs is unknown. Among outpatients, lack of compliance may contribute to failure of initial therapy, which may result in hospitalization.12-14 ABSSSIs can lead to missed work or school, the inconvenience of inpatient treatment, emotional distress, and embarrassment, all of which can affect a patient’s quality of life.15
The current standard of therapy for ABSSSIs in Canada is a cephalosporin for methicillin-sensitive Staphylococcus aureus (MSSA) infections (i.e., IV cefazolin, IV ceftriaxone, or oral cephalexin), and IV vancomycin, IV daptomycin, or oral or IV linezolid for MRSA infections (daptomycin and linezolid are rarely used). For severe ABSSSI, treatment includes IV piperacillin plus tazobactam or IV ertapenem. According to the clinical expert consulted by CADTH, an oral route of administration is well suited for the treatment of ABSSSI, as oral antibiotics are inexpensive, easy to use, and can be administered at home. However, the potential lack of adherence to oral antibiotics is a concern. The goals of therapy are prevention of mortality, improvements in accessibility and compliance, and reducing the risk of IV-related complications, hospital visits, and health care costs.
Dalbavancin is a semisynthetic bactericidal lipoglycopeptide active against susceptible strains of gram-positive bacteria.16
Dalbavancin is indicated for the treatment of adults with ABSSSI caused by susceptible isolates of the following gram-positive micro-organisms: Staphylococcus aureus (including methicillin-susceptible and methicillin-resistant strains), Streptococcus pyogenes, Streptococcus agalactiae, Streptococcus dysgalactiae, Streptococcus anginosus group (including Streptococcus anginosus, Streptococcus intermedius, and Streptococcus constellatus), and Enterococcus faecalis (vancomycin-susceptible strains).16 The Health Canada recommended dosage is 1,500 mg administered either as a single dose or as 1,000 mg followed 1 week later by a 500 mg IV infusion administered over 30 minutes. The sponsor’s requested reimbursement criteria for dalbavancin aligns with the Health Canada indication. Dalbavancin underwent a priority review process by Health Canada and received a Notice of Compliance on May 5, 2021. Dalbavancin has no other Health Canada–approved indication and has not previously been reviewed by CADTH.
Clinician Input
Input From Clinical Experts Consulted by CADTH
The clinical expert noted many gaps in the treatment of ABSSSIs, including the fact that current IV treatment is expensive and complicated (potentially requiring nursing effort, a peripherally inserted central catheter [PICC] line, repeated visits, and automated IV pumps), there is a risk of IV-line complications, patient adherence to treatment can be poor, obtaining a microbiological can be difficult, and the current standard of care can have toxic effects (e.g., IV vancomycin, which requires therapeutic monitoring). Sometimes, given the difficulty in obtaining a swab to identify the pathogen, the diagnosis may be incorrect and delay the treatment process. In addition, patients may be misdiagnosed with ABSSSIs and given unnecessary treatment. In terms of access, people in remote areas or Indigenous communities may have poor or limited access to IV treatment and may face geographic barriers accessing treatment after discharge.
According to the clinical expert, dalbavancin could change the current treatment paradigm, given its unique pharmacokinetics and its ease of use compared to other treatments. Dalbavancin would be considered a first-line treatment and would replace current IV antibiotic treatments for ABSSSIs caused by gram-positive bacteria (unless oral antibiotic treatment was available). In the opinion of the clinical expert, there would be no reason to try other treatments before dalbavancin. Given that dalbavancin does not require repeat IV infusion, PICC line placement, or therapeutic drug monitoring, it is much easier to access than other standards of therapy and may lead to better patient satisfaction. In addition, dalbavancin could prevent repeat visits, hospital admission, and poor adherence, which is observed with other therapies. According to the clinical expert, patients best suited for treatment with dalbavancin include those with MRSA infection, those who inject drugs, those identified by physician assessment at presentation, and those who are clinically stable and candidates for home IV, who have mild-to-moderate infection, are competent, and have access to a nearby hospital. Patients least suited for dalbavancin include those with infections that require surgical debridement and those with polymicrobial infections. According to the clinical expert, response in a trial setting is best assessed using FDA outcome criteria at 48 to 72 hours: alive, a 20% reduction in the affected area, and no rescue treatment. In clinical practice, there are typically daily assessments, which may not be necessary; also, assessment may be highly variable between physicians (e.g., determining time to resolution of redness). Patients can be re-treated, typically around day 8, if there is no response. However, physicians should investigate the reasons for nonresponse and adjust the treatment plan accordingly.
The clinical expert indicated that dalbavancin could be administered in any setting in which there is IV access (e.g., emergency room [ER], long-term care facility, hospital, clinic, or at a patient’s home). Dalbavancin does not require a specialist for prescription, although its use should be regulated carefully by an antimicrobial stewardship committee. Treatment should not be used off-label without trial evidence. The clinical expert added that trials should be done in patients with osteomyelitis, endocarditis, abscess, prosthetic joint infection, pneumonia, and meningitis.
The clinical expert also noted that dalbavancin has some potential harms. If an allergic reaction occurred, there would be no way to withdraw the drug. Also, the convenience of dalbavancin dosing could increase the unnecessary treatment of wrong diagnoses, lead to failure to change treatment after culture results are received, lead to increased IV treatment when oral treatment would be adequate, or increase unnecessary prophylaxis. According to the clinical expert, there may be concerns related to antimicrobial resistance with the use of dalbavancin, if used inappropriately; with dalbavancin, there is prolonged duration of selection toward antimicrobial resistance in human gut flora. Antimicrobial resistance is associated with enormous cost to the Canadian health system and mortality among Canadians; therefore, antimicrobial use must be minimized, both in the spectrum of action and the duration of treatment, to reduce antimicrobial resistance.
Drug Program Input
Input was obtained from the drug programs that participate in the CADTH reimbursement review process. The following were identified as key factors that could potentially affect the implementation of a CADTH recommendation for dalbavancin: considerations related to the initiation of therapy, discontinuation of therapy, prescribing, generalizability, and care provisions. The clinical expert consulted by CADTH provided advice on the potential implementation issues raised by the drug programs.
Clinical Evidence
Pivotal Studies and Protocol-Selected Studies
Description of Studies
Four randomized controlled trials (RCTs) were included in this review: the DISCOVER 1, DISCOVER 2, DUR001-303, and VER001-9 trials. The first 3 RCTs were considered pivotal trials by Health Canada. The DISCOVER 1, DISCOVER 2, and DUR001-303 studies were sponsored by Durata Therapeutics, and the VER001-9 study was sponsored by Vicuron Pharmaceuticals Inc.
The DISCOVER 1 (N = 573) and DISCOVER 2 (N = 739) studies were phase III, multicentre, 1:1 randomized, double-blind (DB), noninferiority studies comparing the efficacy and safety of dalbavancin to vancomycin (with a possible switch to oral linezolid) in patients with a known or suspected gram-positive ABSSSI. The primary objective in both trials was to compare clinical efficacy 48 to 72 hours after study-drug initiation between dalbavancin and a vancomycin and linezolid regimen. Clinical response was defined as no increase in lesion size, absence of pyrexia at 48 to 72 hours in the intention-to-treat (ITT) population, and no new systemic antibacterial treatment for gram-positive ABSSSIs. In both trials, the key secondary objectives included the following: clinical response 48 to 72 hours after study-drug initiation, based on measurements of ABSSSI lesion size (≥ 20% reduction in lesion area); clinical efficacy at day 14 or 15 after study-drug initiation (end of treatment [EOT] visit), based on lesion size, local signs, temperature, and receipt of nonstudy antibiotics; and clinical efficacy at the day 28 short-term follow-up (SFU) visit, based on lesion size, local signs, temperature, and receipt of nonstudy antibiotics. Patients assigned to dalbavancin received a 1,000 mg dose on day 1 followed by a 500 mg dose on day 8 with a possible switch to oral placebo (if switching criteria were met), for a total duration of 14 days. Patients assigned to IV vancomycin received at least 3 days of therapy with the option to switch to oral linezolid to complete 10 to 14 days of therapy. For both treatment arms, the total course of therapy (IV and oral) was 14 days. Treatment was initiated on day 1 and efficacy and safety assessments took place on days 2, 3, 4, and 8, as well as at the EOT visit (day 14). Following the EOT visit, patients were to return for SFU at day 28 and long-term follow-up (LFU) at day 70 (2 months after the EOT visit).
The DUR001-303 study (N = 698) was a phase III, multicentre, 1:1 randomized, DB, noninferiority study designed to compare single-dose versus 2-dose IV dalbavancin regimens in patients with known or suspected gram-positive ABSSSIs. The primary objective of this study was to compare the efficacy of treatment with a single dose of dalbavancin 1,500 mg to treatment with a 2-dose regimen of dalbavancin (1,000 mg on day 1 followed by 500 mg on day 8) 48 to 72 hours after the initiation of treatment. Clinical response was defined as the patient being alive, not receiving rescue therapy for ABSSSI, and having at least a 20% decrease in lesion area. The secondary objectives of this study were clinical status at day 14 or 15 (EOT visit) and day 28 (± 2 days) after study-drug initiation, and safety. Other objectives looked at health care resource use, including length of stay (LOS) in the hospital. Patients in the single-dose group received a single dose of IV dalbavancin on day 1 and a dalbavancin-matching placebo on day 8. Patients randomly assigned to the 2-dose dalbavancin group received the first dose of dalbavancin on day 1 and the second dose of dalbavancin on day 8. Treatment was initiated on day 1, and efficacy and safety assessments took place on day 3 to 4, day 8, day 14 or 15 (EOT visit), and day 28 (final visit).
The VER001-9 study (N = 854) was a phase III, multicentre, 2:1 randomized, DB, noninferiority study that aimed to determine whether dalbavancin is noninferior to IV linezolid (with a possible switch to oral linezolid) in adults with complicated skin and skin structure infections (cSSSIs) due to gram-positive pathogens, based on clinical response, defined as survival status, temperature, and no rescue therapy. The primary objective of the VER001-9 study was to compare the clinical efficacy and safety of dalbavancin (2-dose regimen) with that of a linezolid regimen in the treatment of adults with cSSSIs due to gram-positive pathogens. A clinical response of success was defined as sufficient resolution of the local and systemic signs and symptoms of skin and skin structure infections (SSSIs) such that the patient did not receive new systemic antibacterial treatment for SSSI. Additional objectives included hospital use and LOS. Treatment was initiated on day 1, and efficacy and safety assessments took place on day 4, day 8, in the 3 days following treatment completion on day 14 (EOT visit), day 28 (test of cure [TOC] visit), and on day 39 (LFU).
In the DISCOVER 1, DISCOVER 2, and DUR001-303 studies, patients were 18 to 85 years of age with a known or suspected ABSSSI (major cutaneous abscess, surgical-site or traumatic-wound infection, or cellulitis), accompanied by at least 75 cm2 of erythema, at least 2 signs of ABSSSI (purulent drainage or discharge, fluctuance, heat or localized warmth, tenderness on palpation, and swelling or induration), at least 1 systemic sign of infection, and infection severity requiring a minimum of 3 days of IV therapy. The most common infection type was cellulitis (47.3% to 54.2% of patients), followed by major cutaneous abscess (25.0% to 30.2%) and wound infection (18.2% to 26.4%). Of patients with a known pathogen, 16.7% to 28.8% had a MRSA infection and 41.8% to 58.0% had an MSSA infection.
In the VER001-9 study, inclusion and exclusion criteria were similar to the other 3 trials, although no threshold was set for the size of erythema and the threshold for increased white blood cells (WBCs) was > 10,000/mm3. Major cutaneous abscess was the most common infection type (30.4% to 33.3%), followed by cellulitis (27.5% to 29.7%) and wound infection (19.2% to 21.2%), and 62.5% to 64.3% of patients were hospitalized at study entry. Of patients with a known pathogen, 88.8% to 90.6% had a Staphylococcus aureus infection and 51% had a MRSA infection.
Efficacy Results
The key outcomes from the RCTs are summarized in Table 2. Overall, the 2-dose regimen of dalbavancin was considered noninferior to the comparator regimens, and single-dose dalbavancin was noninferior to the 2-dose regimen in terms of clinical response at 48 to 72 hours across the pivotal trials and at day 28 in the VER001-9 study.
Clinical Response or Success
In the DISCOVER 1 study, the clinical response rate at 48 to 72 hours was 83.3% in the dalbavancin group and 81.8% in the vancomycin group (treatment difference = 1.5%; 95% confidence interval [CI], –4.6% to 7.9%). The lower limit of the 95% CI for the treatment difference was higher than the pre-specified noninferiority margin of –10%. Therefore, the noninferiority of dalbavancin to vancomycin was concluded. The proportion of patients in the ITT population with clinical success at the EOT visit (day 14) was similar between groups, with 81.9% in the dalbavancin group and 86.7% in the comparator group, for a between-group difference of –4.8% (95% CI, –10.7% to 1.3%). The proportion of patients with clinical success at day 28 was 83.7% for dalbavancin and 88.1% for the comparator (treatment difference = –4.4%; 95% CI, –10.1% to 1.4%).
In the DISCOVER 2 study, clinical response rate at 48 to 72 hours was 76.8% in the dalbavancin group versus 78.3% in the vancomycin group (treatment difference = –1.5%; 95% CI, –7.4% to 4.6%). The lower limit of the 95% CI for the treatment difference was higher than the pre-specified noninferiority margin of –10%. Therefore, the noninferiority of dalbavancin to vancomycin was concluded. In addition, clinical response at the EOT visit (day 14 or 15) in the clinically evaluable (CE) population was consistent with the ITT population, with a treatment difference of 2.8% (95% CI, –6.7% to 0.7%) in favour of the comparator. At the EOT visit (day 14), 88.7% in the dalbavancin group and 85.6% in the comparator group had achieved clinical success, for a between-group difference of 3.1% (95% CI, –1.8% to 8.0%). At day 28, 88.1% in the dalbavancin group and 84.5% in the comparator group had achieved clinical success, for a between-group difference of 3.6% (95% CI, –1.1% to 8.9%).
Sensitivity analyses conducted in both the DISCOVER 1 and DISCOVER 2 studies were consistent with the results of the primary analysis.
In the DUR001-303 study, the clinical response rate at 48 to 72 hours was 84.2% in the 2-dose arm of dalbavancin and 81.4% in the single-dose arm, with a between-group difference of 2.9% (95% CI, –8.5% to 2.8%). The lower limit of the 95% CI for the treatment difference was higher than the pre-specified noninferiority margin of –10%. Therefore, the noninferiority of 1-dose dalbavancin to 2-dose dalbavancin was concluded. At the EOT visit (day 14) and at day 28, the treatment difference for the single-dose versus the 2-dose regimens was –0.9% (95% CI, –6.3% to 4.6%) and 0.6% (95% CI, –6.0% to 4.8%), respectively.
Clinical response at 48 to 72 hours across subgroups (MRSA versus MSSA; infection type) showed similar clinical response rates across all trials and groups.
In the VER001-9 study, clinical response was assessed at the EOT (day 14) and TOC (day 28) visits in the ITT population of the study. The clinical response rate at the TOC visit, the primary end point, was 76.5% in the dalbavancin arm and 82.7% in the linezolid arm. The treatment difference was –6.2% (95% CI, –12.03% to –0.27%). The lower limit of the 95% CI for the treatment difference of –6.5% remained above the pre-specified noninferiority margin of –12.5%. Therefore, the noninferiority of dalbavancin to linezolid was claimed. In terms of clinical response at the EOT visit, results were consistent with those observed at 48 to 72 hours, with 80.6% of patients treated with dalbavancin and 86.9% of patients treated with linezolid deemed to have achieved clinical success in the ITT analysis. Clinical response was also evaluated in the CE population at the TOC visit (day 28), and results were consistent with that of the ITT population (treatment difference = –2.21%; 95% CI, –7.28% to 2.86%).
Clinical Failure at EOT
In the DISCOVER 1 study, 38 patients (13.2%) in the dalbavancin group and 29 (10.2%) patients in the vancomycin group in the ITT population were clinical failures at the EOT visit (day 14). The most commonly reported reasons for clinical failure were that local signs of fluctuance and localized heat or warmth had not resolved (84.2% of patients on dalbavancin and 79.3% of patients on vancomycin), local signs of tenderness on palpation and swelling or induration were worse than mild (23.7% and 34.5%, respectively), and the patient received a new nonstudy systemic antibacterial treatment (34.2% and 13.8%, respectively).
In the DISCOVER 2 study, 32 (8.6%) patients in the dalbavancin group and 33 (9.0%) patients in the vancomycin group in the ITT population were clinical failures at the EOT visit. The most commonly reported reasons for clinical failure at the EOT visit in both treatment groups were the same as in the DISCOVER 1 study: local signs of fluctuance and localized heat or warmth had not resolved (53.1% of those on dalbavancin and 60.6% of those on vancomycin); local signs of tenderness on palpation and swelling or induration were worse than mild (34.4% and 48.5%, respectively); and the patient received a new nonstudy systemic antibacterial treatment (28.1% and 42.4%, respectively).
In the DUR001-303 study, 42 (12.0%) patients on single-dose dalbavancin and 36 (10.3%) patients on 2-dose dalbavancin in the ITT population were clinical failures at the EOT visit. The most common reasons for clinical failure were lesion size that did not decrease from baseline (73.8% on single-dose dalbavancin and 66.7% on 2-dose dalbavancin), local signs of tenderness on palpation and swelling or induration that were worse than mild (21.4% and 19.4%, respectively), and the patient received a new nonstudy systemic antibacterial treatment (21.4% and 16.7%, respectively).
Harms Results
A summary of the key harms reported in the RCTs are summarized in Table 2.
In the DISCOVER 1 and DISCOVER 2 studies, the incidence of treatment-emergent adverse effects (TEAEs) was lower in the dalbavancin group than in the vancomycin group (34.9% versus 39.4% in the DISCOVER 1 study and 31.3% versus 36.8% in the DISCOVER 2 study). Across the pivotal trials, the number and type of TEAEs were similar between groups, with the most commonly reported TEAEs being headache, nausea, hypertension, and rash. The TEAEs in the DISCOVER 2 study were similar to those in the DISCOVER 1 study. In the DUR001-303 study, the incidence of TEAEs was similar with the single-dose and 2-dose dalbavancin regimens (20.1% versus 19.9%). The most common (≥ 2%) TEAEs in the single-dose and 2-dose treatment groups, respectively, were nausea (3.4% versus 2.0%), headache (1.7% versus 1.2%), and vomiting (1.7% versus 0.9%). In the VER001-9 study, the most commonly (≥ 2%) reported TEAEs throughout the entire study period for the dalbavancin and linezolid groups, respectively, were nausea (3.2% versus 5.3%), diarrhea (2.5% versus 5.7%), and headache (1.9% versus 1.8%).
In the DISCOVER 1 study, treatment-emergent serious adverse events (SAEs) were less commonly reported in the dalbavancin group than in the comparator group (1.8% versus 4.2%, respectively). In the DISCOVER 2 study, the number of treatment-emergent SAEs was similar in the dalbavancin and comparator groups (3.3% versus 3.8%, respectively). In the DUR001-303 study, the percentage of patients with SAEs was similar in the single-dose and 2-dose treatment groups (2.0% versus 1.4%). In the VER001-9 study, the rate of SAEs was similar in the dalbavancin and linezolid groups (7.5% versus 8.4%).
The proportion of patients who discontinued treatment due to adverse events (AEs) was similar in the 2 treatment groups across the RCTs (1.8% or less in all groups).
The number of deaths was similar between groups in all trials (0.7% or less of each group), except in the DISCOVER 1 study, where there were deaths in 1.8% of the vancomycin group and none in the dalbavancin group. The most common notable harms were infusion-related reactions (1.8% or less of each group), rash or hypersensitivity reactions (0.8% to 2.1% of each group in the pivotal trials), and hepatic AEs (0.8% to 2.1% in the DISCOVER 1 and DISCOVER 2 studies).
Table 2: Summary of Key Results From Pivotal and Protocol-Selected Studies
Outcome | DISCOVER 1 | DISCOVER 2 | DUR001-303 | VER001-9 | ||||
|---|---|---|---|---|---|---|---|---|
DAL, N = 288 | VAN/LZD, N = 285 | DAL, N = 371 | VAN/LZD, N = 368 | 1-dose DAL, N = 349 | 2 dose DAL, N = 349 | DAL, N = 571 | LZD, N = 283 | |
Clinical response at 48 to 72 hours, ITT population | ||||||||
Patients with clinical response, n (%) | 240 (83.3) | 233 (81.8) | 285 (76.8) | 288 (78.3) | 284 (81.4) | 294 (84.2) | NR | NR |
Difference, % (95% CI) | 1.5 (–4.6 to 7.9)a | –1.5 (–7.4 to 4.6)a | –2.9 (–8.5 to 2.8)a | NR | ||||
OR (95% CI) | 1.1 (0.7 to 1.7) | 0.9 (0.7 to 1.3) | NR | NR | ||||
Clinical success at EOT [Day 14], ITT population | ||||||||
|||||||| |||| |||||||| |||||||| | ||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | 293 (84.0) | 296 (84.8) | ||| |||||| | ||| |||||| |
||||||||||| | |||| ||| | |||| |||||| || |||| | ||| ||||| || |||| | –0.9 (–6.3 to 4.6) | ||||| ||||||| || |||||| | ||||
Clinical success at day 28, ITT population | ||||||||
|||||||| |||| |||||||| |||||||| | ||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | 295 (84.5) | 297 (85.1) | ||| |||||| | ||| |||||| |
||||||||||| | |||| ||| | |||| |||||| || |||| | ||| ||||| || |||| | –0.6 (–6.0 to 4.8) | ||||| ||||||| || ||||||| | ||||
Harms, n (%), safety population | ||||||||
Patients with ≥ 1 AE | 113 of 284 (39.8) | 117 of 284 (41.2) | 122 of 368 (33.2) | 144 of 367 (39.2) | 70 of 349 (20.1) | 69 of 346 (19.9) | 318 of 571 (55.7) | 174 of 283 (61.5) |
Patients with ≥ 1 SAE | 5 of 284 (1.8) | 12 of 284 (4.2) | 12 of 368 (3.3) | 14 of 367 (3.8) | 7 of 349 (2.0) | 5 of 346 (1.4) | 43 of 571 (7.5) | 24 of 283 (8.5) |
WDAEs (from study treatment) | 5 of 284 (1.8) | 6 of 284 (2.1) | 9 of 368 (2.4) | 7 of 367 (1.9) | 6 of 349 (1.7) | 5 of 346 (1.4) | 22 of 571 (3.9) | 9 of 283 (3.2) |
Deaths | 0 | 5 of 284 (1.8) | 1 of 368 (0.3) | 2 of 367 (0.5) | 1 of 349 (0.3) | 1 of 346 (0.3) | 2 of 571 (0.4) | 2 of 283 (0.7) |
Notable harms, n (%), safety population | ||||||||
Antibiotic resistance | NR | NR | NR | NR | NR | NR | NR | NR |
|||||||| ||||||| ||||||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | |||||| | | ||||| |
Rash | 6 (2.1) | 6 (2.1) | 3 (0.8) | 6 (1.6) | 4 (1.1) | 4 (1.2) | 0 | 0 |
|||||||||||||||| ||||||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| |
||||||||||||| |||||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| |
||||||| || | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| |
||||||||||| ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| |
||||| ||||||| ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| |
AE = adverse event; CI = confidence interval; DAL = dalbavancin; EOT = end of treatment; ITT = intention-to-treat; LZD = linezolid; NR = not reported OR = odds ratio; SAE = serious adverse event; VAN = vancomycin; WDAE = withdrawal due to adverse event.
aThis was the primary end point. The 95% CI was calculated using the Miettinen and Nurminen method, without adjustment in the DUR001-303 study and with adjustment for the randomization stratification factor of presence or absence of fever at baseline in the DISCOVER 1 and DISCOVER 2 studies.
bPrimary end point.
Note: Linezolid in refers to oral linezolid in the pivotal trials, and linezolid in the VER001-9 study refers to IV linezolid (with a possible switch to oral linezolid). The noninferiority margin was –10% for the pivotal trials and –12.5% for the VER001-9 trial, and there was no adjustment for multiple outcomes. In the DISCOVER 1 and DISCOVER 2 studies, the OR was stratified for the presence or absence of fever at baseline, and the 95% CI for the OR was calculated using the Cochran-Mantel-Haenszel method.
Source: Clinical Study Reports.7-20
Critical Appraisal
Internal Validity
The noninferiority design was adequately powered, and the threshold used (–10% to –12.5%) was justified. According to the clinical expert consulted by CADTH, the 10% noninferiority margin is somewhat wide, but a loss of approximately 5% in clinical benefit would not be too concerning for the treatment of ABSSSI. Also, the expert indicated that although vancomycin is expected to be efficacious in this population (especially in those with MRSA), linezolid is a bacteriostatic rather than bactericidal drug and may not be as efficacious as other available options. Although the dosing of vancomycin aligned with the recommended dose for this population, it may not have been the most appropriate comparator, given that majority of patients with Staphylococcus aureus infections were MSSA and not MRSA. According to the clinical expert, more appropriate drugs for patients with MSSA include cephalosporins, such as IV ceftriaxone or oral cephalexin.
The primary outcome in the DUR001-303 study corresponds to the primary efficacy end point recommended in the FDA draft guidance for the development of drugs for ABSSSIs,1 and the primary outcomes in the DISCOVER 1 and DISCOVER 2 studies were also similar. The primary outcomes for the DISCOVER 1 and DISCOVER 2 studies also considered resolution of fever and did not require a decrease in lesion size for clinical response, which is more aligned with the way patients are assessed in clinical practice, as the requirement for a 20% reduction in lesion area (part of the FDA-recommended outcome) is arbitrary, and fever, WBC count, and pain are typically assessed. The clinical expert noted that the outcomes for the VER001-9 study were less well defined. Overall, the expert considered the efficacy outcomes and follow-up in the 4 studies to be appropriate for guidance on the use of dalbavancin in patients with ABSSSIs.
Treatment compliance was higher with dalbavancin than with other IV therapies and with the single-dose than with the 2-dose regimen. The use of concomitant antibiotic therapy was similar among groups across all trials; hence, bias toward treatment outcome is low. Subgroup analyses did not include statistical testing and were limited in number, so results should be considered exploratory. Overall, the subgroup results across all trials were similar to those in the primary analysis.
External Validity
The clinical expert consulted by CADTH agreed that the baseline patient characteristics in the pivotal trials and the VER001-9 study were reflective of the patients with ABSSSIs they encounter in Canadian clinical practice. Although the majority of patients in each study were enrolled at trial sites in the US and Europe, the population enrolled in the trial was consistent with the population expected to be treated in Canadian clinical practice. In general, lesion size was quite large across all trials, (e.g., approximately 300 cm2 up to approximately 750 cm2), which is indicative of more severe ABSSSI; this may limit generalizability to populations with smaller lesion sizes. Furthermore, the subgroup analyses (e.g., bacteremia, infection type, MRSA status) underwent no statistical comparisons and had smaller sample sizes, which limit the generalizability to a broader population. According to the clinical expert, the concomitant medications used in the trial were reflective of those encountered in Canadian clinical practice.
Indirect Comparisons
Description of Studies
The published indirect treatment comparison (ITC) included in the sponsor’s submission included a systematic review, and the authors used a Bayesian network meta-analysis (NMA) to evaluate the relative clinical and safety efficacy of IV antibiotics for the treatment of ABSSSI in adults. The efficacy outcomes of interest were clinical treatment success and microbiological success, and the safety outcomes were discontinuation due to AEs or SAEs, any AE or any SAE experienced by the patients, and all-cause mortality.
Efficacy Results
The NMA shows no evidence of a difference in clinical success between dalbavancin and vancomycin (odds ratio [OR] = 0.99; 95% credible interval [CrI], 0.68 to 1.51), linezolid (OR = 0.69; 95% CrI, 0.41 to 1.00), or daptomycin (OR = 1.05; 95% CrI, 0.61 to 2.10).
Safety Results
The ITC reported that dalbavancin is associated with a lower likelihood of AEs than linezolid, a lower likelihood of SAEs than vancomycin and daptomycin, and a lower risk of all-cause mortality than vancomycin, linezolid, and tigecycline. However, the CrIs were wide, particularly for SAEs and mortality.
Critical Appraisal
The ITC had several limitations, including high clinical heterogeneity in study design (e.g., patient selection criteria, definitions of response, and timing of assessment) and in patient characteristics. High statistical heterogeneity was also present in the pairwise meta-analyses. The structure of the network was star-shaped with 1 closed loop, with some contrasts represented by 1 or 2 trials. Because of the small number of trials included in the NMA, the ability to estimate between-trial variance was limited. As a result, analyses with uncommon events, such discontinuations due to AEs or SAEs, would likely produce imprecise estimates. Furthermore, there were limitations related to the lack of reporting of certain items that would have better informed the certainty of the indirect evidence; the authors did not report risk of bias when pooling the studies, and did not adequately report sensitivity and subgroup analyses to investigate the root of heterogeneity or conduct a meta-regression that would adjust for effect modifiers that could influence the results. Overall, there was substantial uncertainty around the ITC because of imprecision and risk of bias.
Other Relevant Evidence
Description of Studies
The ENHANCE21 and ADVANCE22 pre-post pragmatic studies provided further evidence on the efficacy (hospital LOS and hospital admission rate) and safety of dalbavancin in patients with ABSSSI.
The ENHANCE study21 was conducted to estimate the difference in infection-related total hospital admission days during initial care (the period between enrolment and 10 to 14 days after treatment initiation) and follow-up (the period between the end of initial care to 30 days after treatment initiation) between patients with ABSSSIs who received care before (pre-period) and after (post-period) the implementation of a critical pathway that was developed for the management of patients with ABSSSIs who were admitted to the hospital. The intervention was the critical pathway, which comprised the identification of patients who met criteria that were developed based on guidelines on the management of ABSSSIs in the hospital setting and outpatient parenteral antibiotic therapy; and the administration of dalbavancin to patients who met the criteria and were subsequently discharged to an outpatient setting at the discretion of the treating physician. During the pre-period, only the first component of the critical pathway was implemented; patients received usual care for ABSSSIs, which was defined as the antibiotic with coverage for the known or suspected gram-positive infection selected by the treating physician or site. During the post-period, both components of the critical pathway were implemented, with the initiation of 1,500 mg dalbavancin as a single IV dose over 30 minutes in a new set of patients enrolled based on the same guideline-based criteria used in the pre-period. For both the pre-period and the post-period, patients were assessed at baseline (date of enrolment), 48 to 72 hours after the date of enrolment or discharge, 10 to 14 days after the date of enrolment, and 44 to 51 days after date of enrolment.
The ADVANCE study22 was conducted to estimate the difference in hospital admission rates at initial care between patients with ABSSSIs who received care before (pre-period) and after (post-period) implementation of the critical pathway. The design of the ADVANCE study was similar to that of the ENHANCE study,21 with a few differences. First, the ENHANCE study recruited patients with ABSSSIs who were admitted to the ER, whereas the ADVANCE study recruited patients with ABSSSIs who presented to the ER. Therefore, patients enrolled in the ADVANCE study received dalbavancin at the point of care in the ER and were subsequently sent home at the discretion of the treating physician. Finally, patients enrolled in the ADVANCE study had an additional assessment 24 hours after enrolment in the post-period, whereas patients in the pre-period were not assessed 48 to 72 hours after enrolment.
Efficacy Results
The mean difference between the pre-period and post-period in total infection-related LOS during the entire ENHANCE study was 1.6 days (95% CI, 0.6 to 2.6 days; P = 0.003), in favour of the post-period. The results of the secondary analysis, after adjustment for age and immunocompromised status, were generally consistent with those of the primary analysis. The results from the analysis that included time spent by patients in prolonged (greater than 1 day) observation and patients who completed the study were generally consistent with the observed difference in the primary analysis for inpatients only for the entire duration of the study.
The ABSSSI-related hospital admission rate at initial care in the ADVANCE study was 38.5% and 17.6% in the pre-period and post-period groups, respectively. The difference between the pre-period and post-period groups in the ABSSSI-related hospital admission rate at initial care was in favour of the post-period group (P < 0.001). The results of the secondary analysis, after adjustment for age, race, insurance type, prior resource use, and systemic inflammatory response syndrome (SIRS) score, were generally consistent with the primary analysis.
All secondary outcomes were exploratory, as the studies were not powered for secondary outcomes, and no adjustments for multiple comparisons were made. In the ADVANCE study, the 12-Item Short Form Survey mental and physical health component scores at day 14, relative to baseline, were used to assess health-related quality of life (HRQoL). The trial found no difference between the 2 assessment groups.
Harms Results
In the ENHANCE study, 3 (6.2%) and 20 (47.6%) patients in the pre-period and post-period groups, respectively, reported at least 1 AE. The most common (> 5%) AE was pyrexia, which occurred in 3 patients (7.1%) in the post-period group. No patients discontinued the study due to any AE, and no deaths were reported in the study. A total of 1 (2.1%) and 3 (7.1%) patients in the pre-period and post-period groups, respectively, reported at least 1 SAE.
In the ADVANCE study, 22 patients (14.1%) and 68 (44.4%) patients in the pre-period and post-period groups, respectively, reported at least 1 AE. The most common (> 5%) AEs were cellulitis and diarrhea, both of which occurred in 8 (5.2%) patients in the post-period group. In the post-period group, no patients discontinued the study due to an AE, and no deaths were reported; in the pre-period group, 2 deaths were reported, 1 due to congestive cardiac failure and 1 due to a road traffic accident. A total of 11 (7.0%) and 16 (10.5%) patients in the pre-period and post-period groups, respectively, reported at least 1 SAE; most common SAEs were not reported.
Critical Appraisal
Internal Validity
The interpretation of the efficacy and safety results from both the ENHANCE and ADVANCE studies may be limited due to the pre-post pragmatic (nonrandomized and open-label) study design. Each site enrolled patients consecutively into both the pre-period and post-period groups and, as a result, the study lacked a concurrent control and patients were not randomized to a treatment. The study design may have also introduced the risk for time-related confounders, such as changes between the pre-period and the post-period in local antimicrobial resistance patterns and in the site-specific and physician-specific approach to usual care for the treatment of ABSSSI. In comparison to the post-period, in which site staff were trained on the critical pathway and the post-period protocol, staff were only trained on the pre-period protocol for the pre-period. As a result, it is uncertain how much of the treatment effect observed in the post-period group was due to the efficacy of dalbavancin, rather than the effectiveness of the entire critical pathway.
Of note, treating physicians were not trained on the protocol (e.g., study objectives, exclusion and inclusion criteria, intervention, and comparator treatment, and outcomes of interest) in the pre-period to limit performance bias when selecting usual care for the treatment of ABSSSIs. This was particularly important because patients were discharged or sent home from the ER at the discretion of the treating physician during the pre-period and the post-period, which may have had an impact on the LOS and hospital admission rate.
The interpretation of the results may be further limited by missing data. Because the analyses were conducted using the observed-cases approach and because of a relatively high and unbalanced study withdrawal rate due to patients lost to follow-up — 4 (8.3%) patients in the pre-period and 9 (20.9%) patients in the post-period in the ENHANCE study — the direction of attrition bias is uncertain. The interpretation of the results in the ADVANCE study may be further limited by the 104 (68.0%) patients in the post-period group having received concomitant therapy with other antibiotics at initial care, thereby removing the difference in the treatment received between the pre-period and the post-period.
External Validity
To optimize the selection of patients with ABSSSIs that best represented real-world outpatients, both the ENHANCE and ADVANCE studies used minimal inclusion and exclusion criteria. Although this would support generalizability to clinical practice, there may still be potential differences between study sites and Canadian practice in the approach to usual care for the treatment of ABSSSI, according to guidelines on the management of patients with ABSSSIs, in the acute-care setting and between the recommended treatment options or regimens and local antimicrobial resistance patterns. Further, the generalizability of the results to the population of patients with ABSSSIs in Canada may be limited, given that all patients were sourced from 1 hospital in the ENHANCE study.
Conclusions
In patients with ABSSSIs, dalbavancin demonstrated noninferiority to vancomycin (with a possible switch to oral linezolid) and IV linezolid (also with a possible switch to oral linezolid), with similar rates of clinical response and clinical success. The dalbavancin single-dose treatment regimen was also shown to be noninferior to the 2-dose treatment regimen. A key limitation from these trials was that the comparators did not fully reflect the standard of care typically used in Canadian practice for patients with ABSSSIs, as identified by the clinical expert, as there were no comparators specific to the treatment of MSSA infections, which may reduce the generalizability of the findings. The ITC compared dalbavancin with vancomycin, linezolid, and daptomycin, but the efficacy and safety results were uncertain because of the heterogeneity across trials and the sparse safety data. The pragmatic studies showed an improvement in total infection-related LOS (the ENHANCE study) and the hospital admission rate at initial care (the ADVANCE study) after the introduction of a dalbavancin care pathway; however, the pre-post study design and use of a pathway intervention limit interpretation. The AEs reported in the RCTs did not give rise to any safety concerns (in either the single-dose or 2-dose regimens), and the safety profile of dalbavancin was similar and possibly better than that of vancomycin and linezolid for patients with ABSSSIs.
Introduction
Disease Background
ABSSSIs are bacterial infections of skin and associated tissues, also referred to as SSTIs. They are caused by the penetration of bacterial skin flora into soft tissues. ABSSSIs include cellulitis, erysipelas, wound infection, and major cutaneous abscesses.1 ABSSSIs are defined by lesion size23 (at least 75 cm2), as measured by the area of redness, edema, or induration.2 ABSSSIs may be acquired in either the community or hospital setting, and are a frequent cause of morbidity.2-4 Staphylococcus aureus and streptococci are responsible for most simple community-acquired infections. Worldwide, the most common cause of ABSSSIs is Staphylococcus aureus, including MRSA.2,5 ABSSSIs are a frequent cause of morbidity in both community and hospital settings.3,4 In Canada, the estimated prevalence of ABSSSIs requiring IV antibiotics is 0.39%,6,7 and First Nations communities are disproportionately affected.7 Severity ranges from nonsevere infections to severe, life-threatening infections that affect deeper tissue layers.2,8,9 In North America, the mortality rate among patients hospitalized for SSTIs is approximately 0.5%.10 ABSSSIs caused by MRSA are associated with increased risk of mortality, longer hospital stays, and higher health care costs than non-MRSA infections.11
ABSSSIs may be closed (cellulitis) or open (wound infection). The microbiologic diagnosis can be difficult, especially in cases of closed infection, where a microbiological finding cannot be obtained without invasive procedures, such as biopsy. Hence, in many cases, the microbiological cause of ABSSSIs is unknown. Microbiologic diagnosis is associated with delay because of culture growth and antimicrobial susceptibility testing; in many cases, treatment is completed without microbiologic diagnosis, and appropriateness of treatment is unknown. Among outpatients, lack of compliance with antibiotic treatment also contributes to the failure of initial therapy.14 ABSSSIs affect patient quality of life due to missed work or school, the inconvenience of inpatient treatment, emotional distress, and embarrassment.15 In Canada, ABSSSIs represent a major burden to patients; for example, it is estimated that patients hospitalized for cellulitis have a mean hospital stay of 7.1 days.11,24
Standards of Therapy
According to the clinical expert consulted by CADTH, the current standard of therapy for ABSSSIs in Canada is a cephalosporin for MSSA infections (i.e., IV cefazolin, IV ceftriaxone, or oral cephalexin) and IV vancomycin, IV daptomycin, or oral or IV linezolid for MRSA infections (daptomycin and linezolid are rarely used). For severe ABSSSIs, treatment would include IV piperacillin plus tazobactam or IV ertapenem. According to the clinical expert consulted by CADTH, an oral route of administration is well suited for the treatment of ABSSSIs, as oral antibiotics are inexpensive, easy to use, and can be administered at home. However, the potential lack of adherence to oral antibiotics is still a concern. Treatment begins in a step-wise manner, beginning with microbiologic diagnosis (if possible), surgery (if needed), admission to hospital (if needed), IV or oral antibiotics, and follow-up with inpatient or outpatient care to monitor response to treatment. The physician will decide on the duration of treatment and will monitor for toxicities and AEs. The microbiologic diagnosis of ABSSSIs guides the course of treatment (e.g., treatment of MRSA is different than treatment for MSSA). However, treatment of ABSSSIs is difficult because the pathogen type is not always known. This may also contribute to antimicrobial resistance, given that treatment is often initiated before microbiologic diagnosis. Antimicrobial resistance affects the prognosis and duration of treatment. Therefore, a goal of therapy is to minimize antimicrobial resistance, both in spectrum of action and duration of treatment. The prognosis of ABSSSIs is generally cure, but some patients may have recurrent ABSSSIs, which is more likely in patients with lymphedema or other skin diseases. According to the clinical expert, the goals of therapy are prevention of mortality, improvement in accessibility and compliance, and reduction in the risk of IV-related complications, hospital visits, and health care costs.
According to the clinical expert, treatment is generally initiated in ERs, and patients are referred to outpatient infusion clinics where available. However, some patients receive care in the ER only. Given the lack of randomized trials, many factors are unknown about the treatment of ABSSSIs, including when to switch from an IV to oral antibiotic, the ideal duration of treatment, and the efficacy of various treatments, including penicillin G.
The wider impacts of antimicrobial resistance are outlined in a report by the Council of Canadian Academies Expert Panel on the Potential Socio-Economic Impacts of Antimicrobial Resistance in Canada.25 The panel estimated that in 2018, there were 5,400 deaths directly attributable to antimicrobial resistance, an additional $1.6 billion in health care system costs from expenses related to antimicrobial resistance, and a reduction in gross national product by $2.8 billion related to the impact on productivity.25
Drug
Dalbavancin is a semisynthetic bactericidal lipoglycopeptide antibiotic used against susceptible strains of gram-positive bacteria.16 Its mechanism of action is the interruption of cell-wall synthesis; the drug binds to the terminal D-alanyl-D-alanine of the stem peptide in nascent cell-wall peptidoglycan, which prevents cross-linking of disaccharide subunits, resulting in bacterial cell death.16
Dalbavancin is indicated for the treatment of adults with ABSSSI caused by susceptible isolates of the following gram-positive micro-organisms: Staphylococcus aureus (including methicillin-susceptible and methicillin-resistant strains), Streptococcus pyogenes, Streptococcus agalactiae, Streptococcus dysgalactiae, the Streptococcus anginosus group (including Streptococcus anginosus, Streptococcus intermedius, and Streptococcus constellatus), and E. faecalis (vancomycin-susceptible strains).16 The Health Canada recommended dosage is 1,500 mg, administered either as a single dose or as a 1,000 mg dose followed 1 week later by a 500 mg dose, all administered by IV infusion over 30 minutes. The sponsor’s requested reimbursement criteria for dalbavancin aligns with the Health Canada indication. Dalbavancin underwent a priority review by Health Canada and received a Notice of Compliance on May 5, 2021. Dalbavancin has no other Health Canada–approved indication and has not previously been reviewed by CADTH.
Key characteristics of treatments used for ABSSSIs are presented in Table 3.
Table 3: Key Characteristics of Dalbavancin, Vancomycin, Linezolid, and Other Antibiotics
Characteristic | Dalbavancin | Vancomycin | Linezolid | Cephalosporins | Cloxacillin (penicillin family) | SMX-TMP | |
|---|---|---|---|---|---|---|---|
Cephalexin | Cefazolin | ||||||
Mechanism of action | Bactericidal antibiotic that inhibits cell-wall synthesis | Bactericidal antibiotic that inhibits cell-wall synthesis and selectively inhibits RNA | Synthetic antibacterial of the oxazolidinone class | Bactericidal antibiotic that inhibits cell-wall synthesis | Bactericidal cephalosporin antibiotic that inhibits bacterial cell-wall synthesis | Semisynthetic antibiotic from the penicillin family | Antibacterial drug |
Indicationa | Treatment of adults with ABSSSI caused by susceptible isolates of the following gram-positive micro-organisms: Staphylococcus aureus (including methicillin-susceptible and methicillin-resistant strains), Streptococcus pyogenes, Streptococcus agalactiae, Streptococcus dysgalactiae, Streptococcus anginosus group (including Streptococcus anginosus, Streptococcus intermedius, Streptococcus constellatus), and E. faecalis (vancomycin-susceptible strains) | Therapy for severe or life-threatening staphylococcal infections in patients who cannot receive or have failed to respond to penicillins or cephalosporins, or who have infections with staphylococci resistant to other antibiotics, including methicillin | Treatment of adults with the following infections, when caused by susceptible strains of the designated aerobic gram-positive micro-organisms: VREF infections; complicated SSSIs, including nonlimb-threatening diabetic foot infections without concomitant osteomyelitis; uncomplicated SSSIs caused by Staphylococcus aureus (methicillin-susceptible strains only) or Streptococcus pyogenes | Treatment of bacterial infections of the respiratory tract, including otitis media, genitourinary tract, bones, and joints, skin, and soft tissue when the infection is caused by susceptible organisms; culture and susceptibility studies should be performed | Treatment of the following infections when caused by susceptible strains of the listed organisms: SSTIs caused by Staphylococcus aureus (penicillin-sensitive and penicillin-resistant); group A beta-hemolytic streptococci and other strains of streptococci | Treatment of infections caused by streptococci when associated with sensitive penicillinase-producing staphylococci, and treatment of all staphylococcal infections, whether sensitive or resistant to penicillin G | Treatment of infections associated with the following gram-positive organisms:
|
Route of administration | IV | IV | IV or oral | Oral | IV | Oral | Oral |
Recommended dose | 1,500 mg, administered either as a single dose or as 1,000 mg followed 1 week later by 500 mg, administered by IV infusion over 30 minutes | IV: 500 mg every 6 hours or 1 g every 12 hours; dosage varies for patients with obesity or impaired renal function | Dosage for uncomplicated SSSI is 400 mg orally every 12 hours for 10 to 14 days Dosage for vancomycin-resistant Enterococcus faecium infection is 600 mg, IV or oral, every 12 hours | Adults: 1 g to 4 g daily in divided doses (typically 1 g every 6 hours); larger doses may be needed for more severe infections or for those caused by less susceptible organisms | Adults: dosages of 6 g per day in serious infections, such as endocarditis; treatment should be continued for at least 10 days in beta-hemolytic streptococcal infections to minimize possible complications associated with the disease | Adults: 250 mg to 500 mg every 6 hours | Adults: 2 tablets or 1 tablet twice daily every 12 hours |
SAEs and safety issues | Infusion-related reactions (e.g., flushing, urticaria, pruritus, and/or rash); mixed infections | Warnings: Exaggerated hypotension, including shock and, rarely, cardiac arrest may result; risk of toxic serum levels; tinnitus and potential deafness; and the following SAEs
| May promote overgrowth of nonsusceptible organisms leading to risk of lactic acidosis; mortality imbalance; serotonin syndrome; carcinogenesis and mutagenesis; endocrine and metabolism disorders; hematologic myelosuppression; peripheral neuropathy; ophthalmologic, renal disorders; sensitivity or resistance | Warnings:
SAEs
| Warnings:
AEs
| Warnings:
AEs
| Risk of fatalities due to severe reactions, including Stevens-Johnson syndrome, toxic epidermal necrolysis (Lyell’s syndrome), fulminant hepatic necrosis, agranulocytosis, aplastic anemia, other blood dyscrasias, and hypersensitivity of the respiratory tract Rare life-threatening and fatal cases of immune thrombocytopenia Discontinue at the first sign of a skin rash or any sign of adverse reaction Susceptibility to resistance |
Other | None | Development of drug-resistant bacteria; contraindicated in patients with known hypersensitivity to the antibiotics | None | Contraindicated in patients with known allergy to the cephalosporin group of antibiotics | Use with caution in patients with penicillin hypersensitivity | None | Contraindicated in patients with a known hypersensitivity to the antibiotic class and in patients with documented megaloblastic anemia due to folate deficiency, evidence of marked liver parenchymal damage, or blood dyscrasias |
ABSSSI = acute bacterial skin and skin structure infection; AE = adverse event; SAE = serious adverse event; SGOT = serum glutamic oxaloacetic transaminase; SMX-TMP = sulfamethoxazole and trimethoprim; SSSI = skin and skin structure infection; SSTI = skin and soft tissue infection; VREF = vancomycin-resistant Enterococcus faecium.
aHealth Canada–approved indication.
Stakeholder Perspectives
Patient Group Input
No patient group input was received for this review.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 1 clinical specialist with expertise in the diagnosis and management of ABSSSIs.
Unmet Needs
According to the clinical expert, current IV treatment is expensive and complicated, requiring nursing effort and PICC line insertion, and increasing the risk of IV-line complications, repeated visits, automated IV pumps, and/or home IV visits. IV vancomycin is toxic and requires therapeutic drug monitoring. Sometimes, given the difficulty in obtaining a culture specimen to identify the pathogen, the diagnosis may be incorrect and delay the treatment process. There is no accurate diagnostic biomarker or imaging study to assist in diagnosis. In addition, many patients have red skin on the legs caused by other diseases, so ABSSSI treatment is often administered unnecessarily. Response to treatment is often slow, which leads to prescribers often treating for durations longer than necessary. The longer treatment duration is further perpetuated in the ER settings, where a different doctor assesses the skin each day without comparison to previous appearances. Compliance is also an issue with oral antibiotics, especially in people who inject drugs who may fail to return or fail to comply with oral antibiotics; patients who inject drugs are at higher risk of ABSSSI due to injection. In terms of access, people in remote areas or Indigenous communities may have poor or limited access to IV treatment and may face geographic barriers accessing treatment after discharge.
Place in Therapy
According to the clinical expert, dalbavancin could change the current treatment paradigm, given its unique pharmacokinetics and ease of use compared to other treatments. Dalbavancin would be considered a first-line treatment (unless oral antibiotics were available and suitable for use by the patient) and would replace current IV antibiotic treatments for ABSSSIs caused by gram-positive bacteria. In the opinion of the clinical expert, there would be no reason to try other IV treatments before dalbavancin. Given that dalbavancin does not require repeat IV infusion, PICC line placement, or therapeutic drug monitoring, it is much easier to access than other treatments and may lead to higher patient satisfaction. In addition, dalbavancin could prevent the repeat visits, hospital admission, and poor compliance observed with other therapies.
Patient Population
According to the clinical expert, patients best suited for treatment with dalbavancin include those with MRSA infection, those who inject drugs, those identified by physician assessment at presentation, and candidates for home IV who are clinically stable, have mild-to-moderate infection, are competent, and have access to a nearby hospital if they worsen after dalbavancin injection. Patients least suited for dalbavancin include those with infections that require surgical debridement.
Assessing Response to Treatment
According to the clinical expert, response in a trial setting is best assessed using the FDA outcome criteria at 48 to 72 hours: alive, a 20% reduction in affected area, and no rescue treatment. In clinical practice, there are typically daily assessments, which may not be necessary; also, assessment may be highly variable between physicians (e.g., determining time to resolution of redness). Patients can be re-treated, typically around day 8, if there is no response. However, physicians should investigate reasons for nonresponse and adjust the treatment plan accordingly.
Discontinuing Treatment
Dalbavancin is a single-dose or 2-dose regimen, so criteria for discontinuation are not applicable in most cases, according to the expert.
Prescribing Conditions
Dalbavancin could be given in any setting in which there is IV access (e.g., ER, long-term care, hospital, clinic, or home visit). Dalbavancin does not require a specialist for prescription, although it may be beneficial to have antimicrobial stewardship involved to ensure appropriate use. Treatment should not be used off-label without trial evidence. The clinical expert added that trials should be conducted in patients with osteomyelitis, endocarditis, abscess, prosthetic joint infection, pneumonia, and meningitis.
Additional Considerations
Antimicrobial treatment is associated with selection toward antimicrobial resistance in human flora. This antimicrobial resistance is transmitted among humans, animals, and the environment, and is associated with enormous cost to the Canadian health system and mortality. Therefore, antimicrobial use must be minimized, both in spectrum of action and duration of treatment, to reduce antimicrobial resistance. Furthermore, there has been little new drug development in antimicrobials.
Dalbavancin has potential harms, including an increased risk of antimicrobial resistance due to prolonged duration of selection toward antimicrobial resistance among gut flora (with fecal excretion for up to 70 days) and the inability to withdraw the drug if allergic reaction occurs. Because of the associated convenience, the availability of dalbavancin could increase unnecessary treatment by increasing treatment of the wrong diagnosis, increasing IV treatment when oral treatment would be adequate, and increasing unnecessary antimicrobial prophylaxis. Discontinuation of close patient follow-up due to prolonged dosing intervals could lead to a lack of recognition of worsening; patients could be discharged on dalbavancin pending culture results, which the provider might fail to review because the patient is discharged. If the culture reveals gram-negative infection, dalbavancin would not be a suitable treatment.
Clinician Group Input
No clinician group input was received for this review.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
Comparator drugs are listed in various ways in many jurisdictions, ranging from non-benefit to restricted access to full benefit. | For CDEC consideration. |
Considerations for initiation of therapy | |
Can the drug be given again to patients who may not have had full response or the infection returns? Can treatment be repeated? If so, what would be the appropriate timing of re-treatment? | According to the clinical expert, infection can recur and patients can be re-treated with dalbavancin at the time of recurrence. If the patient fails to clear the infection after a single dose of dalbavancin, a repeat dose could be given. |
Considerations for discontinuation of therapy | |
Is there a time period required before repeating treatment, if necessary? | The clinical expert indicated that clinicians will wait for the drug to clear from the patient’s body, which is typically 5 to 7 days after administration. If there is a treatment failure, the treating physician would reflect on the reasons the treatment did not work. |
Considerations for prescribing of therapy | |
Access to a hospital or infusion clinic may be necessary or the ability to provide infusion services as an outpatient. | The clinical expert stated that most patients would likely use the ER or outpatient infusion services. The proportion of patients accessing treatment through each route would depend on how care is delivered in different locations. |
Generalizability | |
Pediatric population may request usage for infections. currently under review in US for this population. | For CDEC consideration. |
Could this drug be used off-label for infections other than ABSSSIs, but with susceptible gram-positive organisms (osteomyelitis, endocarditis etc.)? | The clinical expert indicated that there would be great demand for dalbavancin to be used off-label for other infections because of the convenience of its use. The expert was hopeful that trials of dalbavancin would be conducted in patients with any serious gram-positive infection (e.g., diabetic foot infection, endocarditis, prosthetic joint infection). |
Care provision issues | |
Drug may need to be infused in hospital or infusion clinic or have ability to infuse in outpatient settings. | As described above, the clinical expert stated that most patients would likely use the ER or outpatient infusion services. |
System and economic issues | |
Outpatient infusion services may be associated with extra costs. Being a 1-time dose, where will dalbavancin fit in the health care system? Will it be funded by the health authorities or drug plans? | As described above by the clinical expert, the proportion of patients accessing the drug through a hospital or outpatient setting would depend on how care is delivered in different locations. |
This therapy may require resources and/or facilities that may not be available in all locations. Will drug plans be required to cover travel expenses for eligible patients? | For CDEC consideration. |
Relevant comparators have been around for some time and are much less expensive. | For CDEC consideration. |
Only 1 or 2 infusions are needed, therefore there is less time in hospital and less associated costs if successful in treating the infection. The cost of the drug itself ($957.1679 per vial) is significantly higher than the IV comparators that are used for 5 to 10 days. It has a better safety profile, less SAEs, less demand for monitoring, and less infusions needed. The cost-effectiveness model submitted by the sponsor showed dalbavancin to be dominant to all comparators in severe ABSSSI treatment. In non-severe infection, it was dominant to IV vancomycin, IV linezolid, and IV daptomycin. In kidney dysfunction patients it was dominant to all IV treatments except ceftriaxone. | For CDEC consideration. |
ABSSSI = acute bacterial skin and skin structure infection; CDEC = CADTH Canadian Drug Expert Committee; ER = emergency room; SAE = serious adverse event.
Clinical Evidence
The clinical evidence included in the review of dalbavancin is presented in 3 sections. The first section, the systematic review, includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as studies that were selected according to an a priori protocol. The second section includes indirect evidence from the sponsor and indirect evidence selected from the literature that met the selection criteria specified in the review. The third section includes sponsor-submitted additional relevant studies that were considered to address important gaps in the evidence included in the systematic review.
Systematic Review (Pivotal and Protocol-Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of dalbavancin injection (1,500 mg, administered as a single dose or as 1 dose of 1,000 mg followed by a second dose of 500 mg 1 week later) for the treatment of adults with ABSSSI caused by susceptible isolates of the following gram-positive micro-organisms: Staphylococcus aureus (including methicillin-susceptible and methicillin-resistant strains), Streptococcus pyogenes, Streptococcus agalactiae, Streptococcus dysgalactiae, Streptococcus anginosus group (including Streptococcus anginosus, Streptococcus intermedius, Streptococcus constellatus), and E. faecalis (vancomycin-susceptible strains).
Methods
Studies selected for inclusion in the systematic review included pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 5. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
Table 5: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Population | Adults with ABSSSI caused by susceptible isolates of the following gram-positive micro-organisms: Staphylococcus aureus (including methicillin-susceptible and methicillin-resistant strains), Streptococcus pyogenes, Streptococcus agalactiae, Streptococcus dysgalactiae, Streptococcus anginosus group (including Streptococcus anginosus, Streptococcus intermedius, Streptococcus constellatus), and E. faecalis (vancomycin-susceptible strains). Subgroups
|
Intervention | Dalbavancin for injection, 1,500 mg, administered as a single dose or as 1 dose of 1,000 mg followed by 500 mg 1 week later |
Comparator | Includes the following treatments as monotherapy or in combination Oral antibiotics:
IV antibiotics:
|
Outcomes | Efficacy outcomes:
Harms outcomes: AEs, SAEs, WDAEs, notable harms (e.g., development of antibiotic resistance, infusion-related reactions, hypersensitivity reactions, C. difficile infection, renal toxicity, hepatic toxicity) |
Study design | Published and unpublished phase III and IV RCTs |
ABSSSI = acute bacterial skin and skin structure infection; AE = adverse event; HRQoL = health-related quality of life; MRSA = methicillin-resistant Staphylococcus aureus; MSSA = methicillin-susceptible Staphylococcus aureus; RCT = randomized controlled trial; SAE = serious adverse event; SIRS = systemic inflammatory response system; vs. = versus; WDAE = withdrawal due to adverse event.
aSIRS criteria include tachycardia (heart rate > 90 beats/min), tachypnea (respiratory rate > 20 breaths/min), fever or hypothermia (temperature > 38°C or < 36°C), and leukocytosis, leukopenia, or bandemia (WBCs > 1,200/mm3, < 4,000/mm3, or bands ≥ 10%).
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy according to the Peer Review of Electronic Search Strategies (PRESS) checklist.31
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946—) via Ovid and Embase (1974—) via Ovid. All Ovid searches were run simultaneously as a multifile search. Duplicates were removed using Ovid deduplication for multifile searches, followed by manual deduplication in Endnote. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concepts were Xydalba (dalbavancin) and ABSSSIs. The following clinical trials registries were searched: the US National Institutes of Health’s clinicaltrials.gov, WHO’s International Clinical Trials Registry Platform search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
No filters were applied to limit retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. Refer to Appendix 1 for the detailed search strategies.
The initial search was completed on May 13, 2022. Regular alerts updated the search until the meeting of the CADTH Canadian Drug Expert Committee on September 28, 2022.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool For Searching Health-Related Grey Literature checklist.32 Included in this search were the websites of regulatory agencies (FDA and European Medicines Agency). Google was used to search for additional internet-based materials. Refer to Appendix 1 for more information on the grey literature search strategy.
These searches were supplemented by reviewing bibliographies of key papers and through contacts with appropriate experts. In addition, the sponsor was contacted for information regarding unpublished studies.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
A focused literature search for ITCs dealing with ABSSSIs was run in MEDLINE All (1946–) on May 12, 2022. No limits were applied to the search.
Findings From the Literature
A total of 4 studies were identified from the literature for inclusion in the systematic review (Figure 1). The included studies are summarized in Table 6. A list of excluded studies is presented in Appendix 2.
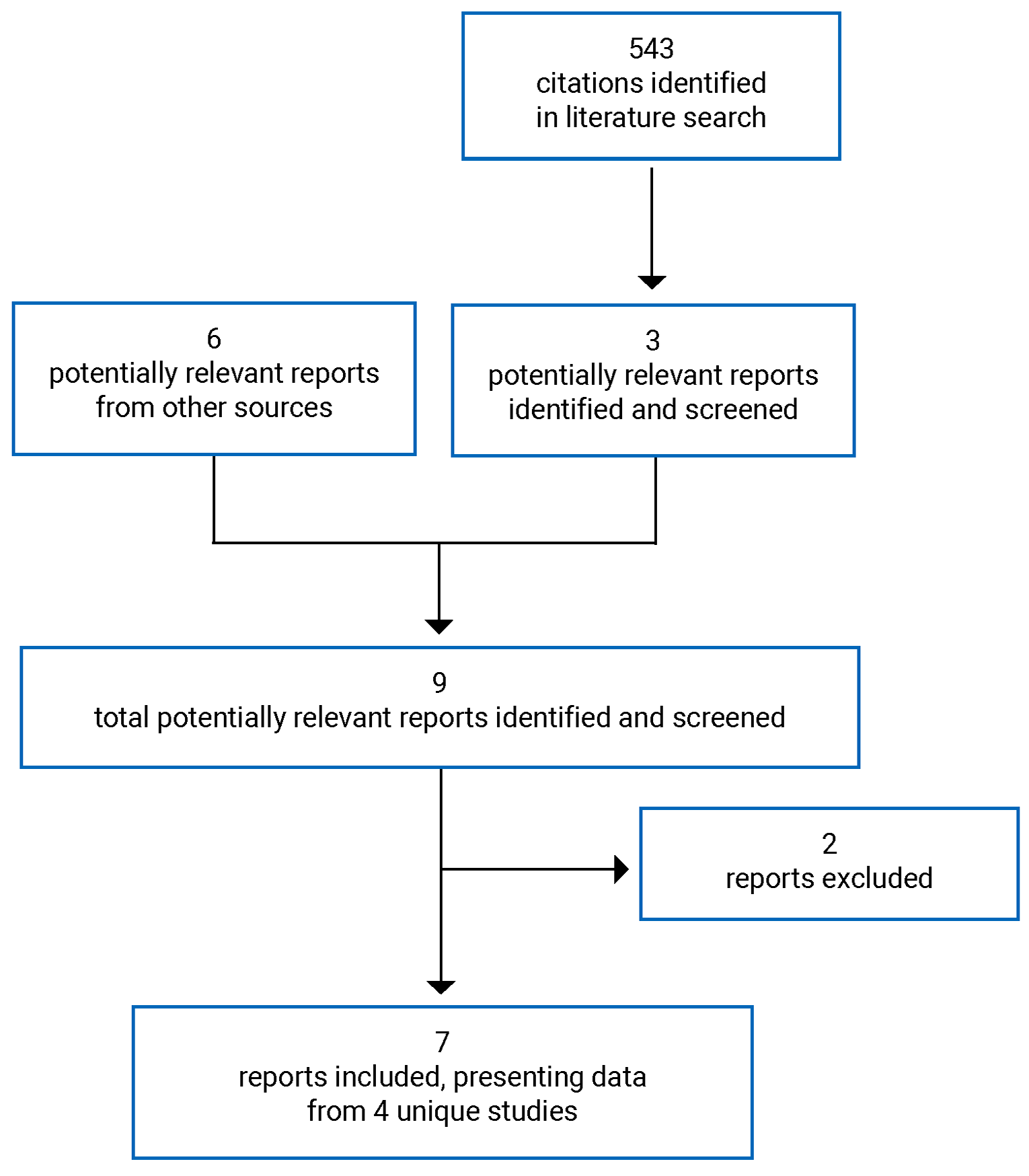
Table 6: Details of Included Studies
Detail | DISCOVER 1 | DISCOVER 2 | DUR001-303 | VER001-9 |
|---|---|---|---|---|
Designs and populations | ||||
Study design | DB RCT | DB RCT | DB RCT | DB RCT |
Locations | 7 countries (US, Canada, Croatia, Georgia, Russia, Serbia, Ukraine) | 14 countries (US, South Africa, Bulgaria, Estonia, Hungary, Israel, South Korea, Romania, Russia, Ukraine, Slovakia, Latvia, Lithuania, Taiwan) | US, South Africa, Bulgaria, Croatia, Estonia, Georgia, Hungary, Latvia, Romania, Russia, Ukraine, Serbia | US, Latvia, Lithuania, Canada, UK, Estonia, and Germany |
Patient enrolment dates | April 16, 2011, to November 7, 2012 | September 26, 2011, to December 27, 2012 | April 18, 2014, to March 11, 2015 | January 3, 2003, to May 21, 2004 |
Randomized, N | 573 | 739 | 698 | 854 |
Inclusion criteria for DISCOVER 1, DISCOVER 2, and DUR001-303 trials |
| |||
Inclusion criteria for VER001-9 trial |
| |||
Exclusion criteria for DISCOVER 1, DISCOVER 2, and DUR001-303 trials |
| |||
Exclusion criteria for VER001-9 trial |
| |||
Drugs | ||||
Intervention | IV dalbavancin 1,000 mg on day 1 and 500 mg on day 8, with placebo matched to vancomycin and oral linezolid | IV dalbavancin 1,000 mg on day 1 and 500 mg on day 8, with placebo matched to vancomycin and oral linezolid | Single-dose of dalbavancin 1,500 mg IV infusion over 30 minutes on day 1 followed by dalbavancin-matching placebo IV infusion over 30 minutes on day 8 | Dalbavancin 1,000 mg on day 1 followed by a 500 mg on day 8, with placebo matched to IV linezolid and oral linezolid |
Comparator(s) | IV vancomycin (1,000 mg every 12 hours or 15 mg/kg every 12 hours) with optional switch to oral linezolid (600 mg every 12 hours), plus placebo matched to dalbavancin; total duration of therapy is 10 to 14 days | IV vancomycin (1,000 mg every 12 hours or 15 mg/kg every 12 hours) with optional switch to oral linezolid (600 mg every 12 hours), plus placebo matched to dalbavancin; total duration of therapy is 10 to 14 days | Two doses of dalbavancin: 1,000 mg IV infusion over 30 minutes on day 1 followed by 500 mg IV infusion over 30 minutes on day 8 | IV linezolid (600 mg every 12 hours) with a possible switch to orally administered linezolid (600 mg every 12 hours) after at least 24 hours of IV therapy |
Duration | ||||
Phase (first dose given on day 1) | ||||
DB | 2 weeks | 2 weeks | 2 weeks | 2 weeks |
Follow-up | SFU: day 28 LFU: 2 months after EOT | SFU: day 28 LFU: 2 months after EOT | 28 days | TOC: day 28 (14 days ± 2 days after EOT) LFU: day 39 (17 days after EOT [only for patients with clinical success at TOC]) |
Outcomes | ||||
Primary end point | Clinical response 48 to 72 hours after study-drug initiation, based on measurements of ABSSSI lesion size and temperature | Clinical response 48 to 72 hours after study-drug initiation, based on measurements of lesion size and temperature | Clinical response 48 to 72 hours after study-drug initiation | Clinical response at the TOC visit (day 28) |
Secondary and exploratory end points | Secondary end points Clinical response 48 to 72 hours after study-drug initiation, based on measurements of ABSSSI lesion size (≥ 20% reduction in lesion area) Clinical efficacy at EOT visit (day 14 or 15) based on lesion size, local signs, temperature, and receipt of nonstudy antibiotics Clinical efficacy at the day 28 SFU visit based on lesion size, local signs, temperature, and receipt of nonstudy antibiotics | Secondary end points Clinical response 48 to 72 hours after study-drug initiation, based on measurements of ABSSSI lesion size (≥ 20% reduction in lesion area) Clinical efficacy at EOT visit (day 14 or 15) based on lesion size, local signs, temperature, and receipt of other therapy Clinical efficacy at the day 28 SFU visit based on lesion size, local signs, temperature, and receipt of other therapy | Secondary end points Clinical status at EOT (day 14 or 15) and final visit (28 ± 2 days after the initiation of study drug) Other outcome measures Clinical status based on localized fluctuance and heat or warmth at EOT Investigator assessment of clinical outcome; a successful outcome was based on resolution or improvement of all signs and symptoms of the infection to such an extent that no further antibacterial treatment was given (an unsuccessful outcome was the opposite) Clinical outcome of success based on key target pathogen at baseline by day 3 or 4 and by EOT Complete resolution of local signs of infection at day 3 or 4, day 8, EOT, and day 28 Change in patient’s assessment of pain from baseline to day 3 or 4, day 8, EOT, and final visit, based on Brief Pain Inventory Resource use categoriesa by day 28 Overall satisfaction response to Skin and Soft Tissue Infection-Convenience Questionnaire | Secondary end points Clinical response at EOT (day 14) and TOC (day 28) in the ITT, microITT, and ME populations Clinical response at EOT in the CE population, by-pathogen and by-patient microbiological response at EOT and TOC in the microITT and ME populations Overall response at EOT and TOC in the microITT and ME populations Additional end points Clinical response at LFU based on information obtained from a telephone contact post-TOC on day 39 (± 3 days) was examined for the ITT and CE populations in patients who were clinical successes at TOC to determine relapse of SSSI, defined as the need for additional antibiotics for SSSI post-TOC Health care resource use Safety Safety was evaluated after collection and analysis of data on AEs, clinical laboratory tests, physical examinations, vital signs, and concomitant medications |
Notes | ||||
Publications | Boucher et al. (2014)33 | Boucher et al. (2014)33 | Dunne et al. (2016)34 | Jauregui et al. (2005)35 |
ABSSSI = acute bacterial skin and skin structure infection; AE = adverse event; CD4 = cluster of differentiation 4; DB = double-blind; EOT = end of treatment; FV = first visit; ITT = intention-to-treat; LFU = long-term follow-up; ME = microbiologically evaluable; microITT = microbiological intention-to-treat; MRSA = methicillin-resistant Staphylococcus aureus; MSSA = methicillin-sensitive Staphylococcus aureus; RCT = randomized controlled trial; SFU = short-term follow-up; SSSI = skin and skin structure infection; TOC = test of cure; WBC = white blood cell.
Note: Day 14 after the initial dose of treatment was considered the EOT visit in all trials. Day 28 is the SFU for the DISCOVER 1 and DISCOVER 2 studies, the final visit for the DUR001-303 study, and the TOC visit for the VER001-9 study.
aResource use categories included any additional visits (including urgent care), any additional procedures, any additional tests, any home visits or nursing care, and any ER visits. The percentage of patients in each category is reported.
Source: Clinical Study Reports.17-20
Description of Studies
Four sponsor-submitted trials were included in this review: the DISCOVER 1, DISCOVER 2, DUR001-303, and VER001-9 trials. The DISCOVER 1, DISCOVER 2, and DUR001-303 studies were sponsored by Durata Therapeutics, and the VER001-9 study was sponsored by Vicuron Pharmaceuticals Inc.
The DISCOVER 1 (N = 573) and DISCOVER 2 (N = 739) studies were phase III, multicentre, 1:1 randomized, DB, noninferiority studies comparing the efficacy and safety of dalbavancin to a vancomycin and linezolid regimen in patients with known or suspected gram-positive ABSSSI. The DISCOVER 1 study had 54 sites in 7 countries (including 1 in Canada), and the DISCOVER 2 study had 86 sites in 14 countries (with no sites in Canada). The primary objective in both trials was to compare clinical efficacy 48 to 72 hours after study-drug initiation between the dalbavancin and vancomycin regimens. Clinical response was defined as no increase in lesion size and absence of pyrexia at 48 to 72 hours in the ITT population. In both trials, the key secondary objectives included the following: clinical response 48 to 72 hours after study-drug initiation, based on measurements of ABSSSI lesion size (≥ 20% reduction in lesion area); clinical efficacy day 14 or 15 after study-drug initiation (EOT visit) based on lesion size, local signs, temperature, and receipt of nonstudy antibiotics; and clinical efficacy at the day 28 SFU visit based on lesion size, local signs, temperature, and receipt of nonstudy antibiotics. Patients assigned to dalbavancin received a 1,000 mg dose on day 1 followed by a 500 mg dose on day 8, with a possible switch to oral placebo (if switching criteria were met), for a total duration of 14 days. Patients assigned to IV vancomycin received at least 3 days of therapy with the option to switch to oral linezolid to complete 10 to 14 days of therapy. Patients were stratified by the presence or absence of fever at baseline (a minimum of 25% of patients had to have fever at baseline), geographic region, and infection type (cellulitis, major abscess [to a maximum of 30% of the total study population], and traumatic-wound or surgical-site infection), using block randomization (block size of 4) with an interactive voice-activated randomization system (IVRS). For both treatment arms, the total course of therapy (IV and oral) was 14 days. Treatment was initiated on day 1, and efficacy and safety assessments took place on days 2, 3, 4, and 8 as well as at the EOT visit (day 14). Following the EOT visit, patients were to return for SFU at day 28 and LFU at day 70 (2 months after the EOT visit). Baseline assessments were performed in the 24 hours before the first dose.
The DUR001-303 study (N = 698) was a phase III, multicentre, 1:1 randomized, DB, noninferiority study designed to compare single-dose and 2-dose IV dalbavancin regimens in patients with known or suspected gram-positive ABSSSI. The DUR001-303 study had 60 sites in 11 countries, with no sites in Canada. The primary objective of this study was to compare the efficacy of a single dose of dalbavancin 1,500 mg to a 2-dose regimen of dalbavancin (1,000 mg on day 1 followed by 500 mg on day 8) 48 to 72 hours after the initiation of treatment. The secondary objectives of this study were clinical status at day 14 or 15 (EOT visit) and day 28 (± 2 days) after study-drug initiation and safety. Other objectives looked at health care resource use, including hospital LOS. Patients in the single-dose group received a single dose of IV dalbavancin on day 1, and a dalbavancin-matching placebo on day 8. Patients randomly assigned to the 2-dose dalbavancin group received the first dose on day 1 and the second dose on day 8. Treatment was initiated on day 1, and efficacy and safety assessments took place on day 3 or 4, day 8, day 14 or 15 (EOT visit), and day 28 (final visit). Baseline assessments were performed in the 24 hours before the first dose.
The VER001-9 study (N = 854) was a phase III, multicentre, 2:1 randomized, DB, noninferiority study that aimed to determine whether dalbavancin is noninferior to IV or oral linezolid treatment in adults with cSSSIs due to gram-positive pathogens, based on clinical response, defined by survival status, temperature, and use of rescue therapy. The VER001-9 study included 65 sites, including 21 in Canada, in 7 countries. Eligible patients in the VER001-9 study were randomized in a 2:1 ratio, with an IVRS, to receive treatment with IV dalbavancin or IV linezolid (possibly followed by oral linezolid). Patients were stratified by geographic location and site. The primary objective of the VER001-9 study was to compare the clinical efficacy and safety of dalbavancin with that of a linezolid regimen for the treatment of adults with cSSSIs due to gram-positive pathogens. The secondary objective was to compare the microbiological efficacy between treatment arms and to obtain dalbavancin pharmacokinetic and pharmacokinetic and/or pharmacodynamic data in patients with this disease entity. Additional objectives included hospital use and LOS. Treatment was initiated on day 1 and efficacy and safety assessments took place on day 4, day 8, in the 3 days after treatment completion (the day 14 [EOT], day 28 [TOC], and day 39 [LFU] visits). Baseline assessments were performed in the 24 hours before the first dose.
Populations
Inclusion and Exclusion Criteria
In the DISCOVER 1, DISCOVER 2, and DUR001-303 studies, patients were 18 to 85 years of age with a known or suspected ABSSSI (major cutaneous abscess, surgical-site or traumatic-wound infection, or cellulitis) accompanied by at least 75 cm2 of erythema, at least 2 signs of ABSSSI (purulent drainage or discharge, fluctuance, heat or localized warmth, tenderness on palpation, and swelling or induration), at least 1 systemic sign of infection (a body temperature of 38°C or greater; WBC count above 12,000 cells/mm3; a manually performed WBC differential count with at least 10% band forms, regardless of peripheral WBC count); and infection severity requiring a minimum of 3 days of IV therapy.
In the VER001-9 study, the inclusion and exclusion criteria were similar to those in the other 3 trials, although no threshold was set for the size of erythema and the threshold for an elevated WBC count was greater than 10,000/mm3.
Baseline Characteristics
Baseline characteristics of all included studies are presented in Table 7. Overall, baseline characteristics were well balanced in the groups and studies. In the DISCOVER 1 study, most patients in the dalbavancin and vancomycin treatment groups were male (59.0% versus 60.7%) and white (91.7% versus 90.9%), and median age was 50 years in both treatment groups. Cause, type, and presentation of ABSSSIs were similar in the dalbavancin and vancomycin treatment groups; the most common ABSSSI was cellulitis (48.6% versus 49.1%). Median body mass index (BMI) was elevated in both treatment groups (27.80 kg/m2 and 27.60 kg/m2, respectively). The median (minimum to maximum) area of ABSSSI erythema at baseline was 333.00 cm2 (25.6 to 3,400.0 cm2) and 367.75 cm2 (77.6 to 3,675.0 cm2) in the dalbavancin and vancomycin treatment groups, respectively, which was larger than the minimum (75 cm2) required. SIRS criteria were met by 61.6% of patients in each treatment group. More than 80% of patients in the dalbavancin and vancomycin treatment groups had fever at baseline (81.9% versus 82.2%). Approximately 1-third of patients in each treatment group had a temperature of at least 38°C and either a WBC count above 12,000 cells/mm3 or bands of at least 10%. Staphylococcus aureus was the most common organism isolated in the dalbavancin and vancomycin treatment groups (79.7% versus 82.6%); fewer than 30% of Staphylococcus aureus isolates were MRSA (28.8% versus 25.2%) and approximately half were MSSA (51.0% versus 56.8%).
In the DISCOVER 2 study, the majority of patients in the dalbavancin and vancomycin treatment groups were male (60.1% versus 54.6%) and white (88.4% versus 87.0%), and the median age was approximately 50 years (49.0 versus 51.0 years). Median BMI was elevated in both treatment groups (27.40 kg/m2 versus 27.60 kg/m2). Cause, type, and presentation of ABSSSIs were similar in the dalbavancin and vancomycin treatment groups, with cellulitis being the most common infection, accounting for a mean of 54.1% of infections in the 2 groups (53.4% versus 54.9%), followed by major abscesses, accounting for a mean of 24.0% of infections in the 2 groups (24.3% versus 23.6%), and traumatic-wound or surgical-site infections, accounting for a mean of 21.8% of infections in the 2 groups (22.1% versus 21.5%). The planned minimum area of ABSSSI erythema required for a patient to participate in the study was 50 cm2 on the face or 75 cm2 on other anatomic sites; however, actual ABSSSI erythema area measurements recorded in the studies were generally much larger than the minimum required. The median (minimum to maximum) area of ABSSSI erythema at baseline was 313.50 cm2 (85.1 to 5,100.0 cm2) and 362.40 cm2 (72.0 to 3,922.0 cm2) in the dalbavancin and vancomycin treatment groups, respectively. SIRS criteria were met by 42.7% of patients in the dalbavancin group. More than 80% of patients in the dalbavancin and vancomycin groups had fever at baseline (81.9% versus 82.2%). Approximately 1-third of patients in each treatment group had a temperature of at least 38°C and either a WBC count above 12,000 cells/mm3 or bands of at least 10%. Staphylococcus aureus was the most common organism isolated in the dalbavancin and vancomycin groups (73.4% versus 73.6%); no more than 25% of Staphylococcus aureus isolates were MRSA (25.0% versus 16.1%) and approximately half were MSSA (48.4% versus 58.0%).
In the DUR001-303 study, the single-dose and 2-dose dalbavancin groups were well matched with respect to demographic and baseline characteristics. The majority of patients in the 1-dose and 2-dose treatment groups were male (58.5% versus 58.2%) and White (89.4% versus 89.1%). Median age was 49 years in the 1-dose group and 50 years in the 2-dose group. Cause, type, and presentation of ABSSSI were similar in the 2 treatment groups. Cellulitis constituted a mean of 47.4% of infections in the 1-dose and 2-dose treatment groups (47.3% versus 47.6%), and median BMI was elevated in both groups (26.90 kg/m2 versus 27.80 kg/m2). The actual ABSSSI erythema area measurements recorded in this study were much larger than the minimum required. The median (minimum to maximum) area of ABSSSI erythema at baseline was 296.05 cm2 (56.0 to 4,325.0 cm2) and 293.25 cm2 (76.5 to 2,668.0 cm2) in the 1-dose and 2-dose dalbavancin groups, respectively. The largest lesions were observed in patients with cellulitis. SIRS criteria were met by 42.4% of patients in the single-dose group and by 44.4% of the 2-dose group. The majority of patients in the ITT population had fever at baseline (83.1% of patients in the 1-dose group and 81.6% of patients in the 2-dose group). The majority of patients in the 1-dose and 2-dose treatment groups in the microbiological intention-to-treat (microITT) population had monomicrobial infections (72.4% versus 73.6%). Staphylococcus aureus was the most common organism isolated in the 1-dose and 2-dose groups (65.2% versus 65.9%); approximately 1-third of the Staphylococcus aureus isolates were MRSA and approximately 2-thirds were MSSA.
In the VER001-9 study, groups were well matched with respect to demographic and baseline characteristics. The majority of patients were male (61.5% of the study population) and white (68.4%), while 18.0% of the patients identified as Hispanic or Latino and 10.7% were Black. Mean age was similar in the dalbavancin and linezolid treatment arms (47.1 years versus 46.0 years). Study-specific medical history was also similar in the 2 treatment arms, except a significantly higher percentage of patients in the dalbavancin arm than in the linezolid arm had vascular disease at study entry. Centres in North America enrolled and dosed 86.5% of the patient population. Cause, type, and presentation of SSSIs were similar in the treatment arms. Infections included predominantly major abscesses (32.3%), cellulitis (28.2%), and other deep soft tissue infections (16.6%). Hallmark signs and symptoms of SSSI reported in more than 95% of patients at baseline included erythema, heat or localized warmth, pain or tenderness on palpation, and swelling or induration. Drainage or discharge (69.9% of patients) and fluctuance (57.3% of patients) were also common.
Table 7: Summary of Baseline Characteristics (ITT Population)
Characteristic | DISCOVER 1 | DISCOVER 2 | DUR001-303 | VER001-9 | ||||
|---|---|---|---|---|---|---|---|---|
DAL, N = 288 | VAN/LZD, N = 285 | DAL, N = 371 | VAN/LZD, N = 368 | 1-dose DAL, N = 349 | 2-dose DAL, N = 349 | DAL, N = 571 | LZD, N = 283 | |
Age, years | ||||||||
|||||| | |||| | |||| | |||| | |||| | |||| | |||| | |||| | |||| |
Minimum to maximum | 18 to 84 | 18 to 84 | 18 to 85 | 18 to 84 | 18 to 85 | 19 to 84 | 18 to 93 | 18 to 92 |
Sex, n (%) | ||||||||
Male | 170 (59.0) | 173 (60.7) | 223 (60.1) | 201 (54.6) | 204 (58.5) | 203 (58.2) | 353 (61.8) | 172 (60.8) |
Female | 118 (41.0) | 112 (39.3) | 148 (39.9) | 167 (45.4) | 145 (41.5) | 146 (41.8) | 218 (38.2) | 111 (39.2) |
Race, n (%) | ||||||||
White | 264 (91.7) | 259 (90.9) | 328 (88.4) | 320 (87.0) | 312 (89.4) | 311 (89.1) | 390 (68.3) | 194 (68.6) |
Black or African American | 16 (5.6) | 19 (6.7) | 13 (3.5) | 17 (4.6) | 28 (8.0) | 31 (8.9) | 59 (10.3) | 32 (11.3) |
||||| | | ||||| | | ||||| | || ||||| | || ||||| | | ||||| | | ||||| | | ||||| | | ||||| |
|||||||| |||||| || |||||| |||||| | | ||||| | | ||||| | | ||||| | ||||| | | ||||| | | ||||| | ||||| | ||||| |
|||||| |||||||| || ||||| ||||||| |||||||| | | ||||| | ||||| | ||||| | | ||||| | | ||||| | ||||| | ||||| | ||||| |
Other | 2 (0.7) | 1 (0.4) | 2 (0.5) | 0 | 1 (0.3) | 5 (1.4) | 8 (1.4) | 3 (1.1) |
|||||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
||| ||||||| | ||||||||
||||| | ||| | ||| | ||| | ||| | ||| | ||| | |||||| | |||||| |
|||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | |||||| | |||||| |
|||| ||| | ||||| |||| | ||||| |||| | ||||| |||| | ||||| |||| | ||||| |||| | ||||| |||| | |||||| | |||||| |
|||||||||| ||||||||| ||||| | ||||||||
||| |||||| | || ||||| | | ||||| | | ||||| | | ||||| | ||||| ||||| | ||||| ||||| | |||||| | |||||| |
||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||||||| |||||| | ||||||| |||||| | |||||| | |||||| |
|||| |||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| | ||||| ||||||| | ||||| |||||| |
|||||||||||| || ||||| |||||| | ||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| | ||| |||||| | ||| |||||| |
|||| ||||| ||||||| | ||| | ||||||||
||||| || |||||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | |||||| | |||||| | |||||| | |||||| |
|||||||| || |||||||| | || |||||| | || |||||| | || |||||| | || |||||| | |||||| | |||||| | |||||| | |||||| |
|||||| || |||||||| |||| || |||||||| |||||||||||||||| ||||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | |||||| | |||||| | |||||| | |||||| |
|||||| |||||| | || |||||| | || |||||| | ||| |||||| | ||| |||||| | |||||| | |||||| | || |||||| | || |||||| |
Diabetes mellitus | 43 (14.9) | 30 (10.5) | 35 (9.4) | 62 (16.8) | 1 (0.3) | 0 | 139 (24.3) | 60 (21.2) |
|||||||| |||||| | || |||||| | || |||||| | || ||||| | || |||||| | |||||| | |||||| | |||||| | |||||| |
Current or recent IV drug abuse | 36 (12.5) | 51 (17.9) | 58 (15.6) | 56 (15.2) | NR | NR | NR | NR |
|||||||||| |||||| | || ||||| | || ||||| | || ||||| | || ||||| | |||||| | |||||| | |||||| | |||||| |
|||||||| |||||||| ||||||||| | | ||||| | || ||||| | || ||||| | || ||||| | |||||| | |||||| | || |||||| | || |||||| |
||||| ||||||||||||| | | ||||| | | ||||| | | ||||| | | ||||| | |||||| | |||||| | |||||| | |||||| |
|||||| |||||| | | ||||| | | ||||| | | ||||| | | ||||| | |||||| | |||||| | |||||| | |||||| |
||||||| ||||| | | ||||| | || ||||| | | ||||| | | ||||| | |||||| | |||||| | |||||| | |||||| |
|||||||||| |||||||| ||||||| | | ||||| | | ||||| | | ||||| | | ||||| | |||||| | |||||| | |||||| | |||||| |
||||||| ||||||||||||| | | ||||| | | ||||| | | ||||| | | ||||| | |||||| | |||||| | |||||| | |||||| |
||||||||| ||||| |||| |||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| | || ||||| | || ||||| |
|||||||||| ||||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| | || ||||| | || ||||| |
|||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| | | ||||| | | ||||| |
|||||||||| | |||||| | |||||| | |||||| | |||||||||| | |||||| | |||||| | ||||| | ||||| |
|||||||| ||||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| | || |||||| | || ||||| |
Type of infection | ||||||||
Cellulitis | 156 (54.2) | 147 (51.6) | 198 (53.4) | 202 (54.9) | 165 (47.3) | 166 (47.6) | 157 (27.5) | 84 (29.7) |
Major cutaneous abscess | 72 (25.0) | 86 (30.2) | 90 (24.3) | 87 (23.6) | 88 (25.2) | 91 (26.1) | 190 (33.3) | 86 (30.4) |
Wound infection | 60 (20.8) | 52 (18.2) | 82 (22.1) | 79 (21.5) | 96 (27.5) | 92 (26.4) | 110 (19.2) | 60 (21.2) |
|||||||| |||| | ||||| | | ||||| | || ||||| | || ||||| | |||||| | |||||| | || ||||| | || ||||| |
||||||||| | || |||||| | || |||||| | || |||||| | || |||||| | |||||| | |||||| | || |||||| | || |||||| |
Measurement of infection lesion size | ||||||||
All ABSSSI area, cm2, na | 284 | 284 | NR | NR | 349 | 346 | NR | NR |
Median | 333.00 | 367.75 | NR | NR | 296.05 | 293.25 | NR | NR |
Minimum to maximum | 25.6 to 3,400.0 | 77.6 to 3,675.0 | NR | NR | 56.0 to 4,325.0 | 76.5 to 2,668.0 | NR | NR |
|||||||||| |||| |||||| | | | ||| | ||| | ||| | ||| | ||| | ||| | |||||| | |||||| |
|||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
|||| ||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||| | ||||| |||| | |||||| | |||||| |
||||| |||||||| |||| |||||| | | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
|||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
|||| ||| | ||||| |||||| | ||||| |||||| | |||||| |||||| | ||||| |||||| | ||||| |||| | |||||| ||||| | |||||| | |||||| |
||||| |||||||||| |||| |||||| | | | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
|||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
|||| ||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||| | ||||| |||| | |||||| | |||||| |
||||| ||||| || ||||||||| | ||||||||
|||||||| ||||||||||||||||||| | | ||||||| | ||||||| | ||||||| | ||||||| | ||||||| | ||||||| | |||||| | |||||| |
|||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | |||||| | |||||| |
|||| | || |||||| | || ||||| | || |||||| | || |||||| | || |||||| | || |||||| | |||||| | |||||| |
|||||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | |||||| | |||||| |
|||||| | || |||||| | || |||||| | || |||||| | || ||||| | || |||||| | || |||||| | |||||| | |||||| |
||||||||||| |||||||||||||||||| |||||||| ||||||||||||| | | ||||||| | ||||||| | ||||||| | ||||||| | |||||| | |||||| | |||||| | |||||| |
|||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | |||||| | |||||| | |||||| | |||||| |
|||| | || |||||| | || |||||| | || |||||| | || |||||| | |||||| | |||||| | |||||| | |||||| |
|||||||| | || |||||| | || |||||| | || ||||| | || |||||| | |||||| | |||||| | |||||| | |||||| |
|||||| | | ||||| | || ||||| | || ||||| | | ||||| | |||||| | |||||| | |||||| | |||||| |
||||||||| | | ||||||| | ||||||| | ||||||| | ||||||| | ||||||| | ||||||| | |||||| | ||||||| |
|||| | | ||||| | | ||||| | || ||||| | | ||||| | | ||||| | || ||||| | |||||| | |||||| |
|||||||| | || |||||| | || |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | |||||| | |||||| |
|||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | |||||| | |||||| |
||||||||||| | | ||||||| | ||||||| | ||||||| | ||||||| | ||||||| | ||||||| | |||||| | |||||| |
|||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | |||||| | |||||| |
|||| | || |||||| | || ||||| | || |||||| | || |||||| | || |||||| | || |||||| | |||||| | |||||| |
|||||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | || |||||| | |||||| | |||||| |
|||||| | || |||||| | || ||||| | || ||||| | || ||||| | || |||||| | || |||||| | |||||| | |||||| |
|||||||||||||| ||||||| | | ||||||| | ||||||| | ||||||| | ||||||| | ||||||| | ||||||| | |||||| | |||||| |
|||||| | ||||| | | ||||| | | ||||| | ||||| | ||||| | | ||||| | |||||| | |||||| |
|||| | || ||||| | | ||||| | || ||||| | || ||||| | || ||||| | || ||||| | |||||| | |||||| |
|||||||| | ||| |||||| | || |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | |||||| | |||||| |
|||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | |||||| | |||||| |
|||||||||| || |||||||||| | | ||||||| | ||||||| | ||||||| | ||||||| | ||||||| | ||||||| | |||||| | |||||| |
|||||| | | ||||| | | ||||| | ||||| | | ||||| | | ||||| | | ||||| | |||||| | |||||| |
|||| | || ||||| | | ||||| | || ||||| | || ||||| | || ||||| | | ||||| | |||||| | |||||| |
|||||||| | || |||||| | || |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | |||||| | |||||| |
|||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | |||||| | |||||| |
|||||||||||||||||||| | | ||||||| | ||||||| | ||||||| | ||||||| | ||||||| | ||||||| | |||||| | |||||| |
|||||| | ||||| | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | |||||| | |||||| |
|||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | |||||| | |||||| |
|||||||| | || |||||| | || |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | |||||| | |||||| |
|||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | |||||| | |||||| |
|||||||| ||||| || ||||||||| | ||||||||
|||||||| |||| || ||||| ||| |||||||||||| ||||||||||||| |||| || |||||||| ||||||||||| | | | || | |||||| | |||||| | |||||| | |||||| | |||||| | ||| |||||| | ||| |||||| |
Temperature ≥ 38°C, n of N (%) | 243 of 284 (85.6) | 242 of 284 (85.2) | 306 of 365 (83.8) | 310 of 365 (84.9) | 290 of 349 (83.1) | 283 of 346 (81.6) | 184 of 571 (32.2) | 101 of 283 (35.7) |
WBC > 12,000 cells/mm3, n of N (%) | 98 of 259 (37.8) | 104 of 254 (40.9) | 149 of 368 (40.5) | 146 of 367 (39.8) | 132 of 348 (37.9) | 126 of 342 (36.8) | NR | NR |
||||||||| ||| |||||||||||||| ||| ||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| | ||||||| |||||| | ||||||| |||||| |
Bands ≥ 10%, n of N (%) | 63 of 238 (26.5) | 66 of 244 (27.0) | 48 of 241 (19.9) | 42 of 234 (17.9) | 56 of 263 (21.3) | 46 of 268 (17.2) | 18 of 571 (3.2) | 15 of 283 (5.3) |
||||| |||||||||||| ||||||| ||| ||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| | ||||||| |||||| | |||||| |||||| |
|||||||| |||||| ||||||| ||| |||| | ||||||| |||||| | ||||||| |||||| | ||||||| |||||| | ||||||| |||||| | |||||| | |||||| | |||||| | |||||| |
|||||||| || |||||||| |||||||||||||| ||| |||| | |||||| |||||| | |||||| |||||| | |||||| |||||| | |||||| |||||| | |||||| | |||||| | |||||| | |||||| |
Patients who meet SIRS criteria, n of N (%)c | 175 of 284 (61.6) | 175 of 284 (61.6) | 157 of 368 (42.7) | 161 of 368 (43.8) | 148 of 349 (42.4) | 154 of 347 (44.4) | NR | NR |
||||||||||||||| |||||||||| | ||||||||
|||||| || |||||||| |||| || |||||| |||| |||||||| ||||||||| ||| |||| | ||||||| |||||| | ||||||| |||||| | ||||||| |||||| | ||||||| |||||| | ||||||| |||||| | ||||||| |||||| | ||||||| |||||| | ||| |||||| |
||||| |||||| || |||||| ||||||||| ||||||||| | | ||||||| | ||||||| | ||||||| | ||||||| | ||||||| | ||||||| | ||||||| | ||||||| |
|||||| || ||||||||| ||||||||| | ||| | ||||||||
|||||| |||||||| | ||||||| |||||| | ||||||| |||||| | ||||||| |||||| | ||||||| |||||| | ||||||| |||||| | ||||||| |||||| | |||||| | |||||| |
|||||| | |||||| |||||| | |||||| |||||| | |||||| |||||| | |||||| |||||| | |||||| |||||| | |||||| |||||| | |||||| | |||||| |
|||||||| |||| | |||||| |||||| | |||||| |||||| | |||||| |||||| | |||||| |||||| | |||||| |||||| | ||||||| |||||| | |||||| | |||||| |
||||| | |||||| |||||| | |||||| |||||| | |||||| |||||| | |||||| |||||| | |||||| ||||| | |||||| ||||| | |||||| | |||||| |
||||| |||||||||| ||||||| |||||||| | ||| | ||||||||
|| ||||||| ||||||||| | ||||| ||||| | ||||| ||||| | |||||| ||||| | |||||| ||||| | |||||| | |||||| | |||||| | |||||| |
|||||||| ||| |||||||| | ||||||| |||||| | ||||||| |||||| | ||||||| |||||| | ||||||| |||||| | ||||||| |||||| | ||||||| |||||| | |||||| | |||||| |
|| |||||||||||||||||| | |||||| |||||| | |||||| |||||| | |||||| |||||| | |||||| |||||| | |||||| | |||||| | |||||| | |||||| |
||||||| |||||||||| ||||||| |||||||| | ||| | ||||||||
|||||||| ||| |||||||| | ||||||| |||||| | ||||||| |||||| | ||||||| |||||| | ||||||| |||||| | ||||||| |||||| | ||||||| |||||| | |||||| | |||||| |
|| |||||||||||||||||| | ||||| ||||| | ||||| ||||| | |||||| ||||| | ||||| ||||| | |||||| | |||||| | |||||| | |||||| |
|||||| || |||||||| |||| || |||||| |||||||| |||| ||| |||||| |||| || ||||| || |||||||| | ||||||| | ||||||| | ||||||| | ||||||| | ||||||| | ||||||| | ||||||| | ||||||| |
|||||||| |||| || ||||||||||||| |||||||| |||||||||| | ||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | |||||| | |||||| |
Staphylococcus aureus | 122 (79.7) | 128 (82.6) | 135 (73.4) | 128 (73.6) | 137 (65.2) | 145 (65.9) | 318 (88.8) | 174 (90.6) |
MRSA | 44 (28.8) | 39 (25.2) | 46 (25.0) | 28 (16.1) | 35 (16.7) | 54 (24.5) | 181 (50.6) | 97 (50.5) |
MSSA | 78 (51.0) | 88 (56.8) | 89 (48.4) | 101 (58.0) | 102 (48.6) | 92 (41.8) | NR | NR |
|||||| || |||||||| |||| || ||||||||||||| |||||||| ||||||||| | || |||||| | || |||||| | || |||||| | || |||||| | || ||||| | || |||||| | |||||| | |||||| |
ABSSSI = acute bacterial skin and skin structure infection; BMI = body mass index; DAL = dalbavancin; hs-CRP = high sensitivity C-reactive protein; ITT = intent-to-treat; IVRS = Interactive Voice Randomization System; LZD = linezolid; N = number of patients in the ITT population; MRSA = methicillin-resistant Staphylococcus aureus; MSSA = methicillin-sensitive Staphylococcus aureus; NR = not reported; SD = standard deviation; SIRS = systemic inflammatory response syndrome; SSTI = skin structure and tissue infection; VAN = vancomycin; WBC = white blood cell count.
Note: The percentages were based on nonmissing data, unless otherwise stated. Meeting SIRS criteria was defined as having 2 or more of the following: temperature < 36°C or > 38°C, heart rate > 90 beats per minute, respiratory rate > 20 breaths per minute, or WBC count < 4,000 cells/mm3 or > 12,000 cells/mm3 or > 10% bands. The values reported by the local laboratory were used to determine SIRS; wound infections include both surgical-site infection and traumatic-wound infection.
aArea was defined as the longest length × the widest perpendicular width.
bPatients can have more than 1 complicating factor and/or signs of systemic infection.
cDenominator n1 is the number of patients with nonmissing values.
dThe percentages were calculated as shown in the row labels. Specimen types identified as other include purulent fluid taken by aseptic aspiration; specimens taken during curettage, debridement, or incision and drainage; scrapings from the wound base; superficial wound smears; and various types of swabs.
Source: Clinical Study Reports.17-20
Interventions
In the DISCOVER 1 and DISCOVER 2 studies, patients were randomly assigned to 1 of 2 treatment groups on day 1 no more than 4 hours before their first dose of the study drug. Patients assigned to dalbavancin received a 1,000 mg dose on day 1 followed by a 500 mg dose on day 8, with a possible switch to oral placebo if criteria were met, for a total duration of 14 days. Reduced dalbavancin doses of 750 mg on day 1 and 375 mg on day 8 were given to patients with creatinine clearance (CrCl) values of less than 30 mL/min who were not receiving regular hemodialysis or peritoneal dialysis. An IV placebo infusion was given every 12 hours for 3 to 14 days to correspond to the vancomycin dosage regimen, and the patient was switched to an oral placebo regimen to match oral linezolid if they met the criteria for a switch. Vancomycin-matched placebo was prepared using the same diluent as the active vancomycin for IV infusion at each study site. All patients received a single IV dose of either dalbavancin (dalbavancin treatment arm) or its placebo (vancomycin arm) on day 8.
Patients assigned to the vancomycin treatment group received an IV infusion of vancomycin every 12 hours for 3 to 14 days. Patients with normal renal function received vancomycin doses of 1,000 mg or 15 mg/kg. Patients with impaired renal function had their dosages and intervals of vancomycin treatment adjusted by an unblinded pharmacist, as necessary, based on local standard of care, renal function, and vancomycin levels. An IV placebo infusion was also given on days 1 and 8 to match the dalbavancin dosing regimen. Dalbavancin-matched placebo was prepared using 250 mL of 5% dextrose. All unit doses were prepared and administered at the study site by an unblinded pharmacist or by a designated member of the clinical pharmacy staff. A total of 20 to 28 doses of vancomycin or linezolid (or matching placebo) were to be administered.
In the DISCOVER 1 and DISCOVER 2 studies, after a minimum of 72 hours, if the blinded investigator determined that a patient met the predefined criteria for a switch to oral therapy, the IV placebo or IV vancomycin treatment was stopped and the patient was given oral therapy (e.g., oral linezolid 600 mg every 12 hours for patients in the vancomycin treatment group or matching placebo for patients in the dalbavancin treatment group). A switch to oral therapy occurred if both of the following conditions were met:
In the previous 24 hours, the patient had 4 temperature measurements, each separated by approximately 6 hours, in which all 4 measurements were 37.6°C or lower.
There was unequivocal improvement in some or all of the clinical signs of the ABSSSI under study; if some signs had not improved, none should have worsened (i.e., stability for that sign was observed).
In the DUR001-303 study, patients in the single-dose dalbavancin group received a single dose of dalbavancin on day 1 and a dalbavancin-matched IV placebo on day 8. The dalbavancin dose on day 1 was 1,500 mg for patients with CrCl of 30 mL/min or greater and for patients with CrCl of less than 30 mL/min who were receiving regular hemodialysis or peritoneal dialysis. In patients with CrCl of less than 30 mL/min who were not receiving regular hemodialysis or peritoneal dialysis, the dalbavancin dose on day 1 was 1,000 mg. Patients randomly assigned to the 2-dose regimen of dalbavancin followed the same dosing schedule as that described for the 2 DISCOVER studies. Dalbavancin-matched IV placebo was prepared using 250 mL of 5% dextrose. All unit doses were prepared and administered at the study site by an unblinded pharmacist or by a designated member of the clinical pharmacy staff. Patients in the DUR001-303 study received the first dose of the study drug no more than 4 hours after randomization.
In the VER001-9 study, patients were randomly assigned in a 2:1 ratio to treatment with dalbavancin or linezolid. Patients in the IV dalbavancin group received a 1,000 mg dose on day 1 followed by a 500 mg dose on day 8. In addition, IV placebo was administered every 12 hours afters the initial dalbavancin dose, with a possible switch to oral placebo every 12 hours. IV placebo was supplied by the sites. IV linezolid was administered in a dose of 600 mg every 12 hours, with possible switch to oral linezolid if switching criteria were met (i.e., improvement in fever or clinical improvement at the SSSI site after at least 24 hours of parenteral therapy). IV linezolid and IV placebo were supplied by the sites.
Concomitant Medications
Across the pivotal trials (DISCOVER 1, DISCOVER 2, and DUR001-303), any medication taken by the patient other than the study drug was considered concomitant medication. Nonantibacterial medications were limited to those essential to the patient. Concomitant and adjunctive systemic and topical antibacterials were prohibited during the study, up to day 14, with the following exceptions:
Oral vancomycin in doses of 125 mg or 250 mg every 6 hours could be used in both treatment groups for the treatment of C. difficile infections and could be continued as required for the duration of the study. The sponsor did not provide oral vancomycin.
Metronidazole, IV or oral, in a dose of 500 mg every 8 hours could be used in both treatment groups for the treatment of C. difficile infections and could be continued as required for the duration of the study. The sponsor did not provide metronidazole. As an adjunctive therapy, it could be used in both treatment groups for suspected anaerobic pathogens and could be continued as required for the duration of the study.
Other antibacterials that did not achieve therapeutic levels in the serum (e.g., nitrofurantoin, fosfomycin, norfloxacin) or at the site of the ABSSSI could be considered.
Aztreonam could be used for the treatment of the ABSSSI caused by gram-negative bacteria, but only systemic aztreonam could be administered empirically at randomization for a presumed gram-negative contribution to the ABSSSI. Empirical use of aztreonam after randomization was not permitted. Use of aztreonam to treat a culture-confirmed infection at any time during the study was acceptable.
The use of any other investigational drug was prohibited, and patients could not participate in any other studies that involved marketed products during the study. Nonsteroidal anti-inflammatory drugs, such as acetaminophen, were administered if patients had a temperature of 38°C or higher. Patients in this study who were given the oral study drug were prohibited from the following medications: inhibitors of monoamine oxidase A or B (e.g., phenelzine, isocarboxazid, methylene blue), direct-acting and indirect-acting sympathomimetic drugs (e.g., pseudoephedrine), serotonin 5-HT1-receptor agonists (triptans), meperidine, and buspirone. Drugs such as dopamine and epinephrine were to be reduced and titrated to achieve the desired response. Patients were not to be switched to an oral study drug if they had received any of the following medications: any serotonergic psychiatric medication, any other psychiatric medication, or any related medication in the previous 2 weeks; fluoxetine in the previous 5 weeks; or both. In addition, patients treated with an oral study drug were not to receive any of these contraindicated medications until at least 24 hours after their final dose of the oral study drug.
In the VER001-9 study, all medications required by the patient to manage underlying illnesses were permitted. Systemic antimicrobials were prohibited during the study, up to the TOC visit, with the following exceptions:
metronidazole for the treatment of gram-negative anaerobes
fluconazole for the treatment of fungal infections
aztreonam for the treatment of gram-negative bacteria.
Topical antibacterial therapies (e.g., mupirocin), although permitted before study entry, were prohibited during the study. Nonabsorbable oral antimicrobial therapies for digestive tract decontamination (e.g., oral vancomycin or neomycin) were permitted.
The following nondrug adjunctive therapies were permitted for the treatment of ABSSSIs in all trials: debridement at the bedside; topical solutions, including antiseptic drugs such as iodopovidone (Betadine); and local bedside wound care per hospital protocol.
Outcomes
A list of efficacy end points identified in the CADTH review protocol that were assessed in the clinical trials included in this review is provided in Table 8. These end points are summarized below.
Table 8: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure | DISCOVER 1 | DISCOVER 2 | DUR001-303 | VER001-9 |
|---|---|---|---|---|
Clinical response (e.g., percent achieving, TOC, mortality) | Primary and secondary | Primary and secondary | Primary and secondary | Primary and secondary |
Systemic signs and symptoms of infection (e.g., heart rate, body temperature, blood cell count) | Secondary | Secondary | Secondary | Secondary |
Substitution with another antibiotic | NR | NR | NR | NR |
HRQoL | NR | NR | NR | NR |
Hospitalization rate | NR | NR | Other | Additional end point |
Hospital LOS | NR | NR | NR | NR |
HRQoL = health-related quality of life; LOS = length of stay; NR = not reported. TOC = test of cure.
Analysis Populations
Five analysis sets were defined: the ITT population, the safety population, the microITT population, the CE population, and the microbiologically evaluable population. For the purpose of this review, only the ITT, safety, CE, and microITT populations will be discussed.
ITT Population
For all pivotal trials (DISCOVER 1, DISCOVER 2, and DUR001-303), the ITT population consisted of all randomly assigned patients, regardless of whether or not they received the study drug. A patient was considered randomly assigned when the pharmacist or pharmacist’s designee received the treatment assignment from the IVRS.
Safety or Modified ITT Population
In the DISCOVER 1 and DISCOVER 2 studies, all patients in the ITT population who received at least 1 dose of the dalbavancin or vancomycin (active) study drug were included in the safety population. In the DUR001-303 study, all patients in the ITT population who received at least 1 (active) dose of dalbavancin were included in the microITT population, which also served as the safety population. In the VER001-9 study, all patients who received any dose of the study medication were included in the safety analyses (ITT population). Descriptive statistics were compiled for AEs, clinical laboratory test results, and vital signs.
CE Population
All trials involved a CE population. In the pivotal trials (DISCOVER 1, DISCOVER 2, and DUR001-303), 2 different CE populations were defined, based on the timing of the outcome assessment evaluated: CE-EOT and CE-final visit (the term CE population refers collectively to both of these populations). Any patient who met all of the following criteria was included in the CE population for the respective visit:
fulfilled patient selection criteria
received dalbavancin as randomized, specifically in the DUR001-303 study, which means 1 dose for patients in the single-dose treatment group and 2 doses for patients in the 2-dose treatment group
received a new, nonstudy systemic antibacterial drug (other than aztreonam or metronidazole) for the treatment of ABSSSI or received more than 1 dose of a concomitant systemic antibacterial therapy (except systemic aztreonam and oral or IV metronidazole) for a non-ABSSSI indication; these patients were considered to be evaluable failures and therefore included in the CE population for the respective visit
received appropriate adjunctive antibacterial coverage if the patient had a culture-documented mixed ABSSSI (1 or more gram-positive pathogens with 1 or more gram-negative aerobic or anaerobic organisms)
had an outcome assessment from which clinical status could be evaluated in the CE populations
for the CE-EOT population, that included all patients with an assessment in the time window from study day 12 to day 18, plus those who had assessments in the 3 days before premature discontinuation from the study drug and those who were considered clinical failures and received a concomitant antibiotic for worsening of the primary ABSSSI lesion before study day 12
for the CE-final visit population, that included all patients with an assessment in the time window from study day 26 through study day 30, plus those who were considered clinical failures at the EOT visit and received a concomitant antibiotic for worsening of the primary ABSSSI lesion.
In the VER001-9 study, patients in the CE population met all of the following conditions:
fulfilled inclusion and exclusion criteria such that a clinical response could be evaluated, unless an exemption was granted before randomization
received at least 3 days worth of study medication based on start and stop dates (i.e., stop date minus start date, plus 1 day)
had not received more than 24 hours of another systemic or topical antibacterial therapy with documented activity against the causative organism between the 7 days before the first dose of the study medication and the TOC assessment, unless the indication for the new antibiotic was lack of efficacy, in which case the outcome was considered a failure and the patient remained evaluable
did not have a clinical response considered indeterminate at the time point being analyzed
received adjunctive antibacterial coverage with aztreonam or metronidazole if the patient had a culture-documented mixed SSSI (i.e., concomitant gram-negative aerobic and/or anaerobic pathogens), unless the organism was not considered by the investigator to be causative
had not received unanticipated surgical intervention during the study, unless the response was considered a clinical failure.
Patients who were CE at the EOT visit (day 14) and did not return for a TOC visit (day 28) and/or violated the protocol in a manner that affected evaluability before the TOC visit were considered CE at the EOT visit, but not at the TOC visit. Similarly, patients who did not attend the EOT visit (and thus had a clinical response considered indeterminate for EOT and were not CE at the EOT visit) were CE for TOC if they returned for the TOC visit and had not otherwise violated the protocol before the TOC visit. Therefore, there were 2 groups in the CE population: CE at the EOT visit (day 14), and CE at the TOC visit (day 28).
Primary End Points
Clinical Response
All pivotal trials (DISCOVER 1, DISCOVER 2, and DUR001-303) used clinical response at 48 to 72 hours as the primary end point. In the VER001-9 study, the primary end point was clinical response at day 28.
In the DISCOVER 1 and DISCOVER 2 studies, clinical response was based on lesion size and temperature. A patient was defined as a clinical responder if the following 2 criteria were met:
The patient had a temperature no higher than 37.6°C (regardless of method of measurement) 48 to 72 hours after the first dose of the study therapy followed by 2 additional temperature measurements no higher than 37.6°C (regardless of method of measurement) separated by at least 3 hours and no more than 9 hours, with no intervening temperature above 37.6°C.
The patient had no increase in lesion area 48 to 72 hours after the first dose of study-drug therapy from the baseline measurement. Lesion area was defined as length × width, and lesion size was defined as length or width. The baseline lesion measurement was defined as the measurement taken closest to but before the first dose of the study drug. If multiple lesion measurements were taken in the 48 to 72 hours after the first dose of the study drug, the most recent lesion measurement was used.
Patients was met at least 1 of the following criteria were defined as a clinical nonresponder:
The patient had an increase over baseline in lesion size 48 to 72 hours after initiation of the study drug.
The patient did not have a temperature of 37.6°C or lower (regardless of method of measurement) 48 to 72 hours after the first dose of the study therapy followed by 2 additional temperature measurements below 37.6°C, each separated by approximately 6 hours, or had an intervening temperature above 37.6°C.
The patient died from any cause in the 72 hours after the first dose of the study drug.
The patient initiated a new, systemic antibacterial with gram-positive activity for the ABSSSI under study in the 72 hours after the first dose of the study drug.
The patient had missing data at 48 to 72 hours for lesion size or temperature, such that a clinical outcome could not be defined.
In the DISCOVER 1 and DISCOVER 2 studies, IV treatment was initiated and the patient’s temperature was recorded on day 1. Efficacy assessments were made on days 2, 3, 4, 8, and 14 or 15 of the treatment period. An EOT assessment took place on days 14 or 15, or in the 3 days after premature discontinuation of treatment. A SFU was planned for day 28 and a final LFU at day 70.
In the DUR001-303 study, patients were defined as clinical responders if they met the following 3 criteria:
alive
received no rescue therapy for ABSSSI before the infection-site assessment at 48 to 72 hours
examination of the ABSSSI lesion demonstrated a decrease of at least 20% in lesion area (calculated as the longest length multiplied by the longest perpendicular width) relative to the baseline measurement. If an antibiotic was given for a reason other than rescue therapy, the patient was not considered a nonresponder.
Nonresponders were those who did not meet the aforementioned criteria. Baseline assessments were performed in the 24 hours before the first dose of the study drug. On day 2, patients being treated on an outpatient basis were contacted by the investigator to check for worsening of the presenting ABSSSI lesion. Efficacy assessments were performed on day 3 to 4; on day 8, at which time patients were administered the second dose of the study drug; on day 14 or 15, which was defined as the EOT visit; and on day 28, which was defined as the final visit.
In all pivotal trials, patients were considered to have an indeterminate outcome if any data needed to determine whether the outcome was a success or failure were missing. For example, if the assessment of local signs was not completed at the EOT visit, the patient was considered to have an indeterminate response for the analysis at the EOT visit. Patients with an indeterminate response were included in the denominator for analyses of the ITT population and were considered treatment failures.
In the VER001-9 study, the primary efficacy end point was the clinical response rate in the CE population at day 28, which was defined as the TOC visit. Clinical response (success, indeterminate, failure) was assessed by the investigator, as follows:
Success meant sufficient resolution of the local and systemic signs and symptoms of SSSI, such that the patient did not receive new, systemic antibacterial treatment for SSSI.
Failure meant persistence of 1 or more local or systemic signs and symptoms of the SSSI, such that new, systemic antibacterial treatment was given for the SSSI, or the patient died during the study period and the SSSI was the sole cause or was thought to be contributory toward death. All EOT failures were carried forward as failures to the TOC visit.
Indeterminate meant there were no post-baseline local or systemic signs or symptoms data available to assess clinical response (e.g., patient lost to follow-up); the patient received the study medication for fewer than 72 hours; or another reason (must be specified). Indeterminate responses were grouped with failures for the ITT analysis.
Clinical status was assessed by the investigator at baseline; during treatment (day 4 + 1 day), day 8 (± 1 day), day 14 (defined as EOT); in the 3 days after completion of therapy; and at the TOC visit (14 ± 2 days after completion of therapy). The investigator assigned a clinical response at the EOT and TOC visits. Patients with a successful clinical response at the TOC visit were contacted for LFU assessment.
Secondary End Points
Clinical Response
In the VER001-9 study, clinical response was additionally evaluated in the CE population at day 14 (EOT) and, as a secondary end point, in the ITT population at the EOT and day 28 (TOC) visits. Clinical response and failure were defined as they were for the primary efficacy variable.
Clinical Status of Success
For all pivotal trials (DISCOVER 1, DISCOVER 2, and DUR001-303), a secondary outcome measure was clinical status at the EOT (day 14 or 15) and day 28 visits. To accommodate a specific request from the European Medicines Agency directed to the sponsor as part of scientific advice, the clinical status at day 14 or 15 in the CE population was selected as a clinical end point of special interest in the DISCOVER 1 and DISCOVER 2 studies.
In the DISCOVER 1 and DISCOVER 2 studies, a response was considered a clinical success if it met following 5 criteria:
The patient’s lesion size, as defined by erythema, was decreased from baseline.
The patient’s temperature was no higher than 37.6°C (by any measurement method).
Local signs of fluctuance and localized heat or warmth were absent.
Local signs of tenderness on palpation and swelling or induration were no worse than mild.
For patients with a wound infection, the severity of purulent drainage was improved and no worse than mild, relative to baseline.
In the DUR001-303 study, a response was defined as a clinical success if it met the following 5 criteria:
lesion area must be decreased by at least 80% from baseline and, at the final visit, must be decreased by at least 90% from baseline
temperature is no higher than 37.6°C
local signs of tenderness on palpation and swelling or induration are no worse than mild
at the EOT visit, local signs of fluctuance and localized heat or warmth must be improved from baseline and no worse than mild, and at the final visit, local signs of fluctuance and localized heat or warmth must be absent; for patients with a wound infection, the severity of purulent drainage must be improved and no worse than mild relative to baseline
for patients with a wound infection the severity of purulent drainage is improved and no worse than mild relative to baseline.
For all trials, a patient was defined as having indeterminate clinical status if any of the data needed to determine clinical success or clinical failure, as defined above, were missing.
|||||||| ||| ||||||| | |||||||| |||| | | ||| ||||||||| |||| ||||||||| |||| | | |||||||| |||| ||||||||| |||| | |
|---|---|---|---|
|||||| |||| ||| ||||||| || ||||||||| | ||||||||| |||| |||||||| | ||||||||| |||| |||||||| | |||||||||| || ||||||||||| | |
||||||||||| ||||| ||| |||||||| |||| ||||| || ||||||||| | ||||||||| ||||||||| ||||| ||| |||||||||| |||||||| || ||||| |||| | |||||||||| ||||||||||| |||||||||||| ||||||| ||||||||||| || |||||| || |||||||||||| ||||| | || ||||| |||||| ||| ||||| |||||||||| || | |||||||||| |||||||||||| ||||||| ||| ||||| | ||||||| || ||||||||| ||||| ||| |||||||||| |||||||| || ||||| |||| | |||||||||| ||||||||||| |||||||||||| ||||||| ||||||||||| || |||||| || |||||||||||| ||||| | || ||||| |||||| ||| ||||| |||||||||| || | |||||||||| |||||||||||| ||||||| ||| ||||| | |||||||||| || ||||||||||| | |
||||| ||||| || |||||||||| ||| ||||||||| ||||||||||| | |||||| | || ||||||| || ||||||||| |||| || |||||||| |||| |||||||| | |||||||||| || ||||||||||| | |
||||| ||||| || |||||||||| || ||||||||| ||| ||||||||||||||||||| | ||| ||||| |||| |||| | ||| ||||| |||| |||| | |||||||||| || ||||||||||| | |
|||||||| |||||||| | |||||||| |||||||| ||| || ||||| |||| |||| |||||||| || |||||||| ||||| ||| |||||||| |||| ||||| |||||||||| | |||||||| |||||||| ||| || ||||| |||| |||| |||||||| || |||||||| ||||| ||| |||||||| |||| ||||| |||||||||| | ||||||||||| || ||||||||||| | |
|||||||||| |||||||||| ||| ||||||| ||||||||| | |||| | |||| | |||| |
|||||||| |||||||||||| ||| |||||| | || ||||||||| ||||||||| | || ||||| ||||| ||||||||| |||||||||| | || ||||||||| ||||||||| | || ||||| ||||| ||||||||| |||||||||| | || ||||||||| ||||||||| | || ||||| ||||| ||||||||| |||||||||| |
|||||||| | ||||| | ||||| | ||||| |
|||||| | ||||| ||||||||| |||| ||| |||| ||||||||| ||||||||||||| ||| |||||||||||| |||||||||| |||||||| |||| ||||||||||| |||| |||| || ||||||| ||||||||||||| ||||||||| ||| ||||| || |||||||| ||||||| |||||| ||| |||||||| ||||||| ||||||||| |||||||||||||| |||||||| ||||| ||||||||||||
Clinical Status of Failure
In the DISCOVER 1 and DISCOVER 2 studies, a response was defined as a clinical failure if at least 1 of the following criteria was met:
the patient’s lesion size, defined by erythema, was not decreased from baseline
local signs of fluctuance and localized heat or warmth had not resolved
local signs of tenderness on palpation and swelling or induration were worse than mild
for patients with a wound infection, the severity of the purulent drainage was the same as it was at baseline, or worse, or was worse than mild
the patient had a temperature higher than 37.6°C (by any measurement method) at the visit
the patient received a new, nonstudy, systemic antibacterial treatment for the ABSSSI at any time from the first dose of the study drug through the visit
the patient died before the visit
unless preplanned as part of the nondrug therapy for the ABSSSI, the patient required surgical intervention more than 72 hours after the start of therapy for the treatment of the ABSSSI under study
the patient received study therapy for the ABSSSI under study beyond the protocol treatment period as a result of the investigator’s assessment that additional drug therapy was needed for treatment of the underlying skin infection.
In the DUR001-303 study, clinical failure was defined as the opposite of success, death before the visit, or the administration of the study therapy for ABSSSIs beyond the protocol treatment period.
Systemic Signs and Symptoms
In the DISCOVER 1 and DISCOVER 2 studies, time to resolution of fever was based on the patient’s first temperature measurement that indicated the patient was afebrile, although resolution required 3 sequential temperatures no higher than 37.6°C. Patients who were lost to follow-up while on the study drug and continued to have fevers were censored at their last visit. Patients who continued to have fevers were censored on the date they last received the study drug.
In the DUR001-303 study, time to resolution of fever was based on the patient’s first temperature measurement of ≤ 37.6°C, regardless of method of measurement. Patients who continued to have fever were censored on the last date they received the study drug or on the last visit date, whichever was later.
Need for Substitution With Another Antibiotic
The need for substitution with another antibiotic was considered a reason for failure in the assessments of clinical response and clinical status (EOT).
Health-Related Quality of Life
HRQoL was not assessed in the trials.
Hospitalization Rate and Hospital LOS
In the DUR001-303 and VER001-9 studies, an efficacy end point was resource use. In the DUR001-303 study, resource use was based on days in the hospital, outpatient visits, and ER visits. Data were collected on day 14 or 15 and on day 28 (± 2 days).
Safety
Safety assessments were made at every visit. For all trials, the safety parameters included AEs, clinical laboratory evaluations, vital signs, and physical examination assessments. AEs were coded using the Medical Dictionary of Regulatory Activities (MedDRA), version 14.0 (DISCOVER 1 and DISCOVER 2 studies) or version 17.0 (DUR001-303 study) or higher of the System Organ Class and Preferred Term levels. In the VER001-9 study, AEs included any adverse experience, whether or not it was considered drug-related, that occurred during a patient’s study participation. This included any side effect, injury, toxicity, sensitivity reaction, intercurrent illness, or sudden death. A pre-existing condition was 1 that was present on physical examination at the start of the study and was reported as part of the subject’s medical history. Any change from the previous physical examination during study treatment (e.g., worsening in frequency, intensity, or character of the pre-existing condition) was considered an AE.
Safety evaluations included TEAEs, which were defined as AEs that appeared, increased in frequency, or worsened in severity during the period that began with the first dose of the study drug (including events that occurred during study-drug administration) and ended at the final visit.
Statistical Analysis
Primary Outcome of the Studies
Sample Size Determination
In the DISCOVER 1 and DISCOVER 2 studies, the point estimate used for sample size determination was based on a retrospective analysis of the VER001-9 study, and included 428 patients with cellulitis, major abscess, or surgical- or traumatic-wound infection with a baseline lesion area of at least 75 cm2. In the VER001-9 study, clinical response was 70% (95% CI, 65% to 74%) for the outcome measures of cessation of spread and absence of fever approximately 48 to 72 hours after the first dose. The sponsor noted that there are study design and analysis issues (e.g., measurement error, missing data, different data-collection times) that could have biased the point estimate. Thus, because the VER001-9 study may have provides a reduced estimate of the true clinical response rate, a point estimate of 85%, which corresponds to the upper limit of the CI for the analysis, excluding missing data, was used to determine the sample size. In addition to a 1-sided alpha of 0.025, a noninferiority margin of 10% and a 90% power were assumed. Based on the method of Farrington and Manning,36 the sample size for the DISCOVER 1 and DISCOVER 2 studies was 556 randomized patients (ITT population). There is also sufficient power for the secondary outcome of clinical status at the EOT visit, assuming a 10% noninferiority margin. Data from the VER001-9 study indicate an 81% clinical success rate at the EOT visit (in the ITT population). The outcome rate for the programmatic determination of clinical status at the EOT visit was expected to be similar to that of the investigator’s assessment in the VER001-9 study. Assuming an outcome rate of 80% in the ITT population and 90% in the CE population (and an 85% evaluability rate), there is an 83% and 94% power, respectively, for the outcome of clinical status at the EOT visit.
In the DUR001-303 study, the proposed sample size was 205 patients per arm, based on the method of Farrington and Manning.36 This assumed a noninferiority margin of 10%, a power of 90%, a 1-sided alpha level of 0.025, and a 90% treatment response, which is consistent with FDA guidance. The expected treatment response was estimated from the DISCOVER 1 and DISCOVER 2 trials, given the similar patient population. In those studies, the response rate for the primary outcome measure was 89.9% in the DISCOVER 1 study and 87.6% in the DISCOVER 2 study. Therefore, the overall response rate was expected to be approximately 90% in the DUR001-303 study. The aggregate blinded response rate was assessed when approximately 60% of patients were enrolled. If the aggregate response rate was less than 90%, a sample size adjustment could be made. To ensure that the point estimate of greater than a 20% reduction used in the estimation of sample size was valid for this study, an interim analysis for sample size re-estimation was performed when early clinical response data, at 48 to 72 hours, were available for 60% of the patients. The interim analysis involved a sample size re-estimation to either confirm that the initial sample size estimate was adequate or to increase the sample size (number of randomly assigned patients) to ensure that the study had adequate power to determine whether a single 1,500 mg dose of dalbavancin is noninferior to the same total dose given as 1,000 mg on day 1 and 500 mg on day 8 for the primary outcome measure. The sample size re-estimation was based on the blinded overall (not by treatment group) clinical response rate and was conducted by an independent, blinded statistician.
In the VER001-9 study, the sample size calculation assumed that if the 2 treatment groups (dalbavancin and linezolid) were equally effective and the clinical success rate was 85% at the day 28 visit, then 258 patients in the dalbavancin group and 129 patients in the linezolid group (CE population) would be required to ensure, with 90% power, that the lower bound of a 1-sided 97.5% CI for the true difference in efficacy between the groups would not exceed –12.5%. Assuming an evaluability rate of at least 70%, approximately 555 patients will need to be enrolled to obtain 386 CE patients. An additional 150 patients were enrolled in the study in a 2:1 ratio (dalbavancin:linezolid), resulting in a total patient enrolment of approximately 705 to augment the safety database.
Statistical Test
All trials used a noninferiority analysis for the primary efficacy end point.
In the DISCOVER 1 and DISCOVER 2 studies, the primary efficacy analysis was performed in the ITT population. The noninferiority test was a 1-sided hypothesis test performed at the 2.5% level of significance and was based on the lower limit of the 2-sided 95% CI. The primary efficacy analysis was adjusted for the randomization stratification factor of the presence or absence of fever at baseline. The number and percentage of patients in each treatment group defined as clinical responders and nonresponders were tabulated. To test the null hypothesis, a 2-sided 95% CI for the observed difference in primary outcome rates (dalbavancin treatment group minus vancomycin treatment group) was calculated. If the lower limit of the 95% CI for the treatment difference in the ITT population exceeded –10%, then the null hypothesis was rejected and the noninferiority of dalbavancin to vancomycin was concluded. The 2-sided 95% CI for noninferiority testing based on the difference in clinical response rates at 48 to 72 hours was computed using the method proposed with stratification by Miettinen and Nurminen (1985).37
In the DUR001-303 study, the primary efficacy analysis used the same noninferiority test as the DISCOVER 1 and DISCOVER 2 studies for the observed difference in the primary outcome measure (single-dose dalbavancin group minus 2-dose dalbavancin group). If the lower limit of the 95% CI for the difference in ITT population exceeds –10%, then the null hypothesis will be rejected and the noninferiority of 1-dose dalbavancin to 2-dose dalbavancin will be declared. In the primary efficacy analysis, the 95% CI was computed using Cochran-Mantel-Haenszel stratum weights, without adjustments for the stratification factors employed in randomization (i.e., geographic region, infection type, and prior use of antibiotics).
In the VER001-9 study, the primary analysis was a 2-step comparison of the clinical success rates (success versus failure) between treatment arms (Hwang 1999). A 1-sided 97.5% CI was calculated for the true difference in efficacy (dalbavancin minus linezolid) using the normal approximation method for CIs. In the first step, noninferiority was concluded if the lower limit of the 1-sided CI did not exceed –12.5%. In the second step, if the lower limit of the 1-sided CI was greater than 0, dalbavancin was considered strict-sense superior to linezolid. In practice, the 2-sided 95% CI of the treatment difference is displayed. The lower bound of this interval is identical to the lower bound of the planned 1-sided 97.5% CI. The primary end point (clinical response rate at the TOC visit in the CE population) was the sole end point for confirmatory statistical testing. All other end points, populations, and time points were considered secondary and supportive. Therefore, no adjustments for multiple end points were considered necessary by the sponsor.
Data Imputation Methods
For the primary outcome measure (clinical response at 48 to 72 hours), patients were considered to have missing data if there was no lesion measurement at baseline and/or in the 48- to 72-hour time period (after the first dose of the study drug). Patients were considered to have missing data if there were 3 missing temperature measurements in the 48- to 72-hour time period taken 6 hours (± 3 hours) apart. Patients with missing data were defined as a nonresponders for the primary analysis (ITT analysis).
The DUR001-303 study handled missing data as described for the DISCOVER 1 and DISCOVER 2 studies. In addition, the DUR001-303 study used multiple imputation, with a Markov chain Monte Carlo method, for the secondary outcomes. Two models were run; the first used the type of infection as a predictive variable and the second used the clinical response at 48 to 72 hours as a predictive variable. For both analyses, 50 imputation datasets were created with imputed data for the missing values and existing values for complete cases.
For all pivotal trials (DISCOVER 1, DISCOVER 2, and DUR001-303), patient responses were considered indeterminate if data were not available for the evaluation of efficacy for any reason. By definition, patients with an indeterminate response were included in the denominator for analyses in the ITT and microITT populations and are considered failures.
In the VER001-9 study, there were no plans to impute missing data in the study. Responses for patients with missing data related to the clinical response were considered indeterminate.
Subgroup Analyses
In the pivotal trials and the VER001-9 study, the subgroup analyses were planned a priori in the statistical analysis plan (SAP) for specific groups of patients. For each subgroup, a forest plot and the respective outcome’s 95% CI was provided. In the DISCOVER 1 and DISCOVER 2 studies, subgroup analyses were conducted for the primary outcome, clinical response at 48 to 72 hours, and clinical status at the EOT and day 28 visits, based on the following groups:
infection type
sex (female versus male)
geographic region (North America versus the rest of the world)
prior surgical intervention
presence or absence of fever
presence or absence of bacteremia
nonsteroidal anti-inflammatory drug use before administration of the study drug
antipyretic use before administration of the study drug, excluding patients with an infection-related major abscess
SIRS criteria met.
In the DUR001-303 study, subgroup analyses were conducted for the primary outcome, clinical response at 48 to 72 hours, and clinical status at the EOT and day 28 visits, based on the following groups:
age (< 65 or ≥ 65 years)
sex
race
ethnicity
randomization before or after interim analysis
geographic region
infection type
prior use of antibiotics
presence or absence of fever
presence or absence of bacteremia
SIRS criteria met
inpatient or outpatient treatment.
In the VER001-9 study, subgroup analyses were conducted for the primary outcome, clinical response at the TOC visit (day 28) in the CE population, and clinical response at the TOC visit (day 28) in the ITT population, based on the following categories: age, sex, ethnicity, presence of diabetes, presence of vascular disease, infection category, geographic area, and surgical intervention.
The following subgroup, planned a priori in the statistical analyses plan, aligned with the subgroup pre-specified in the protocol for this CADTH review: presence or absence of bacteremia, MRSA versus MSSA infection, and infection type (major abscess, cellulitis, and wound). Only the subgroup identified in the CADTH review protocol is reported in the efficacy section.
Sensitivity Analyses
In the DISCOVER 1 and DISCOVER 2 studies, 13 separate pre-specified sensitivity analyses of the primary efficacy analysis were performed to determine how missing data and different measurement criteria would affect the results.
In the DUR001 to 3 study, sensitivity analysis of the primary efficacy analysis was conducted using an expanded window of 36 to 75 hours. An additional analysis of clinical status at the EOT visit (day 14 or 15) and on day 28 was conducted in which the definition of clinical success was switched from “local signs of fluctuance and localized heat/warmth are absent” to “local signs of fluctuance and localized heat/warmth are improved from baseline and no worse than mild.” In addition, the primary efficacy outcome was adjusted for the randomization stratification factors of prior use of antibiotics, infection type, and geographic region as captured in the IVRS.
In the VER001-9 study, a post hoc sensitivity analysis was conducted that excluded patients with an indeterminate clinical response in both treatment arms for clinical response at the TOC visit.
Secondary Outcomes of the Studies
Secondary efficacy end points were for descriptive purposes and no conclusions of noninferiority were made.
In the DISCOVER 1 and DISCOVER 2 studies, the observed difference in percentage of patients with a clinical success (dalbavancin group minus the vancomycin group) was determined, and a 2-sided 95% CI for the observed difference was computed using the method of Miettinen and Nurminen (1985),37 with stratification for the presence or absence of fever at baseline. Cochran-Mantel-Haenszel weights were used for the stratum weights in the calculation of the CI. The reasons (a patient may have more than 1 reason for clinical failure) for clinical failure were summarized by treatment group, by the presence or absence of fever at baseline, and by infection type. The reasons for an indeterminate response were also summarized by treatment group. Two-sided 95% CIs for the observed differences in clinical success rates were calculated for descriptive purposes. Exploratory subgroup analyses of clinical status at the EOT visit were also conducted.
In the DUR001-303 study, noninferiority of the secondary end points was not formally tested; however, from previous phase III studies, it was expected that with a 1-sided hypothesis test performed at the 2.5% level of significance, the lower limit of the 95% CI for the difference in response rates in the ITT population would be greater than –15%.
In the VER001-9 study, clinical response end points were analyzed using the same method as for the primary end point. For the additional end points of time from IV to oral switch and discontinuation of IV access following IV to oral switch, summary statistics were produced.
Sensitivity Analyses
In the DISCOVER 1 and DISCOVER 2 studies, a sensitivity analysis of the secondary outcome measure of clinical status was conducted, which emphasized improvement, rather than absence, in fluctuance and warmth of the lesion. In this analysis, the criteria for success, “local signs of fluctuance and localized heat/warmth are absent,” were replaced with “if present at baseline, local signs of fluctuance and localized heat/warmth must be improved from baseline.” The number and percentage of patients with a clinical status of success and failure at the EOT and SFU visits, based on the revised definition, is determined in each treatment group in the ITT and CE populations. The observed difference in the percentage of patients with a clinical success (dalbavancin group minus the vancomycin group) was determined, and a 2-sided 95% CI for the observed difference was computed using the method of Miettinen and Nurminen (1985),37 with stratification for the presence or absence of fever at baseline. Cochran-Mantel-Haenszel weights are used for the stratum weights in the calculation of the CI. Multiple imputation methods, using a Markov chain Monte Carlo full-data imputation, will be used to define missing data (i.e., patients with an indeterminate outcome). Exploratory subgroup analyses (patients with bacteremia) of clinical status at the EOT visit were also conducted.
In the DUR001-303 study, sensitivity and additional analyses for the secondary efficacy end point were performed in the same manner as described for the primary efficacy analysis, except that the patient’s lesion size had to be decreased by at least 80% from baseline at the EOT visit and at least 90% from baseline at the final visit. A further sensitivity analysis was conducted in which 95% CIs were calculated in which clinical failures at the EOT visit carried forward to the final visit and clinical status was adjusted by stratification factors.
The VER001-9 study included post hoc sensitivity analyses that will not be considered by CADTH.
|||||||| ||||||||||||| |||||||| || ||||||||| | || ||| ||||| |||||||| ||||||| ||||| ||||||||| || ||| |||||||| ||||||||| ||||| ||||||| |||||||| |||||||| ||| ||||||||| |||||||| || ||||||| |||||||| |||| |||||||||| |||||||| ||||||||| | ||| ||| ||||| |||||||| ||||||| ||||| ||||||||| || ||||||||| | ||||||||| ||||||| || ||| ||||||||| |||||||| || ||||||| |||||||| |||| ||||| || ||||||||| ||| ||||||||||| || |||| | |||| || ||||||| |||| |||||||| ||||||||||||||||| ||| ||||||| ||| |||||| ||| |||||||||| || || || ||||||| || ||||||| |||| || ||||| |||| ||| |||||||||| |||||||| || ||| ||||| |||||||| ||||| ||| || |||||||||| || || ||| ||||||||| | ||| ||||| ||||| |||||||| || |||||| || ||| ||||||||||||| |||||||| || ||||| || |||| |||| ||| || |||||||| |||||||| |||||||| || |||| | ||||| ||||||| || ||||| |||||||||| ||||||||| ||||||| ||||||||||| ||||| ||| |||||||| || ||||||||| | || ||| ||||| |||||||| ||||||| ||||| ||||||||| || ||| |||||||| ||||||||| ||| |||| ||||||| || ||||||||| ||| ||||||||| |||| |||| || |||||||| || || ||||||||| ||| |||||| |||| ||||||||||||| ||| ||||||| || |||||||| ||| ||||||||| || |||||| |||| |||| ||| || ||||||| ||||||||||||| |||||||| |||| ||||||| || ||| ||||| |||||| || |||||||| || | ||||||||| || |||||||||||||| ||| ||||||| || ||||| ||| || |||||||| |||||||| |||||||| || |||| ||||| || ||||||||| ||||||||| | ||| ||||| |||||| |||||||| ||||||| || ||| ||||||||||||| |||||||| || ||||| || |||| |||| ||| || |||||||| |||||||| |||||||| || |||| | ||||| ||||||| || ||||| |||||||||| ||||||||| ||||||| ||||||||||| ||||| ||||||||| | ||| |||||| ||||| |||||||| ||||||||||| ||||||| || ||| ||||||| |||||| |||| ||||| || ||| ||||||| ||||||| ||||||||| |||||||||| ||| |||||| || |||||||| ||||||| |||| ||| || ||||||| ||||||||||| | |||||||||| |||| |||| || ||| ||||||||| || ||||||||| | ||||||||| ||| ||||| ||| |||||||| |||||| |||| ||| ||||||| |||| ||| |||||||| || ||| ||||| || ||| |||||||| |||||||| |||| || ||| ||||||| ||||||||| ||||||| || ||| ||||||| |||||||| || |||||||||| |||||||| || ||| |||| ||||||| ||| |||||||||| |||| ||||| ||||||||| ||||||||| || ||| |||||||||| || | |||| || ||| || |||| ||||| ||||||||| || |||||||| || |||||||| ||||||||| |||||||||| || ||||| |||||||| ||||||||| ||||| |||| | |||||||||| || ||| ||||||||| ||||||||| | || ||| ||||| |||||||| |||||||||| || |||||||||| |||||||| || ||||||| ||| |||||| ||||||||| ||| ||| |||||||||| || ||||||||||||| |||||||| |||||||| || ||||||| || ||||||| |||||||| ||| |||||||| | || ||||| || ||||| ||||||||||| ||| |||| ||||||| || ||| | |||||||| |||| ||||||||| ||| || ||||| |||||||||| || |||||||||| |||||||| || || | ||||| || |||| || ||||||||| |||||||| ||| ||| || |||||||||| |||| ||||||||| || |||||||| |||| |||||||| ||| |||||||| ||| ||||| || ||||| ||||||||||| ||||||||| |||||||| ||||| || ||||||||| |||||||||||| || ||||||||| ||||
Results
Patient Disposition
A summary of patient disposition in the pivotal trials (DISCOVER 1, DISCOVER 2, and DUR001-303) and the VER001-9 study is available in Table 10. The most common reasons for failing screening (≥ 10% of patients) were receipt of a systemic antibiotic with gram-positive activity for more than 24 hours before screening, the potential presence of osteomyelitis or septic arthritis, and refusal to provide informed consent. The rate of study discontinuation was similar between treatment groups for all trials. In all trials, the major reason for discontinuation was loss to follow-up (4.0% to 5.2% in the pivotal trials and 7.7% to 7.8% in the VER001-9 study).
Disposition | DISCOVER 1 | DISCOVER 2 | DUR001-303 | VER001-9 | ||||
|---|---|---|---|---|---|---|---|---|
DAL | VAN/LZD | DAL | VAN/LZD | 1-dose DAL | 2-dose DAL | DAL | LZD | |
Screened, N | 659 | 836 | NR | 3,417 | ||||
Randomized, N | 288 | 285 | 371 | 368 | 349 | 349 | 583 | 290 |
Discontinued from study, N (%) | 27 (9.4) | 28 (9.8) | 39 (10.5) | 35 (9.5) | 26 (7.4) | 27 (7.7) | 75 (12.9) | 30 (10.3) |
Reason for discontinuation, N (%) | ||||||||
Lost to follow-up | 15 (5.2) | 14 (4.9) | 23 (6.2) | 15 (4.1) | 14 (4.0) | 14 (4.0) | 44 (7.5) | 22 (7.6) |
Withdrew consent and refused further contact | 6 (2.1) | 2 (0.7) | 9 (2.4) | 13 (3.5) | 5 (1.4) | 3 (0.9) | 20 (3.4) | 5 (1.7) |
Adverse event | 0 | 0 | 0 | 0 | 2 (0.6) | 1 (0.3) | 0 | 0 |
Other | 6 (2.1) | 7 (2.5) | 6 (1.6) | 4 (1.1) | 4 (1.1) | 7 (2.0) | 9 (1.5) | 2 (0.7) |
Death | 0 | 5 (1.8) | 1 (0.3) | 3 (0.8) | 1 (0.3) | 1 (0.3) | 2 (0.3) | 1 (0.3) |
Pregnancy | 0 | 0 | 0 | 0 | 0 | 1 (0.3) | 0 | 0 |
ITT, N | 288 | 285 | 371 | 368 | 349 | 349 | 571 | 283 |
CE-EOT, N | ||| | ||| | ||| | ||| | ||| | ||| | ||| | ||| |
MicroITT, N (%) | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| | ||| |
Safety, N | 284 | 284 | 368 | 367 | 349 | 346 | 571 | 283 |
CE-EOT = clinically evaluable population at end of treatment; DAL = dalbavancin; ITT = intention-to-treat; LZD = linezolid; MicroITT = microbiological intention-to-treat; VAN = vancomycin.
Source: Clinical Study Reports.17-20
Exposure to Study Treatments
DISCOVER 1 and DISCOVER 2 Studies
In the DISCOVER 1 and DISCOVER 2 studies, at least 98% of patients in the ITT population had a compliance of at least 80% in each treatment group for both placebo and the active study drug, and the majority of patients in both treatment groups had 100% compliance with treatment.
DUR001-303 Study
In the ITT population, 93.0% of patients in both treatment groups completed treatment with both doses of the randomly assigned study drug, per-protocol (active and placebo considered together); more patients were compliant with the first dose (99.6% of patients) than with both doses (93.0% of patients).
VER001-9 Study
All patients received at least 1 dose of IV study medication in this study. Each IV dose was considered to represent 7 days of therapy because of the long half-life. Therefore, patients in the dalbavancin arm were exposed to treatment for either 7 or 14 days, depending on whether the day 8 dalbavancin infusion was administered. Exposure for patients who received linezolid depended on the number of IV and oral doses received. Oral linezolid was taken by 265 (93.6%) patients. Most patients in both treatment arms were exposed to treatment for at least 14 days (87.2% in the dalbavancin group versus 85.2% in the linezolid group). Average exposure was 3.8 days (range, 1 to 16 days) for IV linezolid and 11.4 days (range, 1 to 20 days) for oral linezolid.
|||||||| |||||||||| || |||| || ||||||||| |||||||| |||||||||| ||| |||||||| |||||||||| ||| ||||||||| |||||| |||||| ||| ||||||||| || ||||| || ||| ||||| ||| ||||||||| |||||||| |||||||||| ||| |||||||| ||||||||||| |||| ||||| ||||||||| ||| |||||||| || |||||| ||| ||||||| |||||||| |||||||| |||||||||| |||| ||||||| ||||||| |||||| |||||| |||| ||||||
|||||||||||||| | |||||||| | | |||||||| | | ||
|---|---|---|---|---|
||| |||||||| | ||||||||||||||| | ||||||||||| | |||||||| ||||||| | |
|||||||| ||| |||||||| | || |||| || ||||||||| | ||||
||||||| ||||| |||| ||||||||||| | ||||
|||| | ||| | ||| | ||| | ||| |
|||||| | ||||| | ||||| | ||||| | ||||| |
|||| ||| | ||| ||| | ||| ||| | ||| ||| | ||| ||| |
|||||||||| |||| | |||| | |||| | | ||||| | | ||||| |
|||||||||| |||| | ||| ||||||| | ||| ||||||| | ||| |||||| | ||| |||||| |
|||||||||| ||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| |
||||||| ||||||||||| | ||||
||| | ||| | ||| | ||| | ||| |
|||||| | ||||| | ||||| | ||||| | ||||| |
|||| ||| | ||| ||| | ||| ||| | ||| ||| | ||| ||| |
|||||||||| |||| | |||| | | ||||| | | ||||| | | ||||| |
|||||||||| |||| | ||| ||||||| | ||| ||||||| | ||| |||||| | ||| |||||| |
|||||||||| ||||| | ||| | ||| | ||| |||||| | ||| |||||| |
|||||| ||||| |||| ||||||||||| | ||||
|||| | ||| | ||| | ||| | ||| |
|||||| | ||| | ||| | ||||| | ||||| |
|||| ||| | |||| ||| | ||| ||| | ||| ||| | ||| ||| |
|||||||||| |||| | |||| | | ||||| | | ||||| | | ||||| |
|||||||||| |||| | ||| ||||||| | ||| |||||| | ||| |||||| | ||| |||||| |
|||||||||| ||||| | ||| ||||||| | ||| |||||| | ||| |||||| | ||| |||||| |
||| | |||||||||||| ||| | |||||||||||||||| ||| | |||||||||| ||| | ||||||| |||||| ||| | ||||||| |||||| | | |||||| || |||||||| |||| || |||||||||||| || | ||| ||||||||| || | |||||||| |||||||||| ||| | ||||||||||||| |||| |||||||||| ||| ||||||| || ||| |||||| || ||||| |||||||| ||||||| || ||| |||||| || |||||||| |||||| ||||| |||||||| ||||| ||| ||||| || ||| |||||| || |||| |||| ||||||| ||||| ||||||| ||| ||||||||| ||||| |||| || ||||| |||| ||| |||||||| || ||| ||||||| || |||||||| ||| ||||||| ||| || ||||| ||||| ||||||| |||||||||| |||||||| |||||||||||| ||||||||||| |||||||||| ||| ||| ||||||||||||| |||||||||||||||| |||||||| ||||| ||||||||||||
||||||||| ||||| | ||| | |||||||||| | |||||||||| | |||||||||| | |||
|---|---|---|---|---|---|---|
|||||| | |||| | |||||||||| | |||| | ||| |||||||| | ||||||||||||||| | ||| | |||||||||||| | ||| | ||||||||||||| | |
|||||||| |||| || ||||| ||| ||||||||| |||||||||| |||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| |
|||||||||| |||||||||| ||| |||| | || |||||| | || |||||| | || |||||| | || |||||| | |||||| | |||||| |
|||||||| |||||||||| ||||| | | ||||| | | ||||| | || ||||| | || ||||| | |||||| | |||||| |
|||||||||| |||||| ||||||||| | | ||||| | | ||||| | |||||| | | ||||| | |||||| | |||||| |
||||||||| | ||||||||| |||||| | || ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | |||| |
|||||||| ||||||| |||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| |
|| |||||| | || |||||| | || |||||| | || |||||| | || |||||| | |||||| | |||||| |
|||||||||| |||||| | || |||||| | || |||||| | || |||||| | || |||||| | |||||| | |||||| |
||||| | || |||||| | || |||||| | || ||||| | || ||||| | | ||||| | | ||||| |
||| || ||||| |||||| | || |||||| | || |||||| | || |||||| | || |||||| | |||||| | |||||| |
||||||||||||| ||||||||||| | || |||||| | || |||||| | || |||||| | || |||||| | | ||||| | |||||| |
||||||||||||| ||||| | |||||| | | ||||| | | ||||| | |||||| | | ||||| | | ||||| |
||||| |||| |||||||||||||| | |||||| | |||||| | |||||| | |||||| | | ||||| | | ||||| |
||||| ||| |||||||||| | |||||| | |||||| | || |||||| | || |||||| | || |||||| | || |||||| |
||||| |||||||| | |||||| | |||||| | || |||||| | || |||||| | || |||||| | || |||||| |
|||||||||| ||| | |||||| | |||||| | | ||||| | |||||| | ||||||| | |||||| |
||| | |||||||||||| || | ||||||||||||||| |||||||| ||| | |||||||||||||||| ||| | |||||||||| | | |||||| || |||||||| |||| || |||||||||||| | | |||||| || ||| |||||||| || ||| ||| ||||||||||| || | ||| ||||||||| ||| | ||||||| ||||||| |||||| ||| | ||||||||||||||||| ||| ||||||||||| |||| |||||||||| || ||||||||| || ||| ||| ||||||| | ||||||| ||| |||| |||| |||| | |||| || |||||||||| |||| ||||| |||||||||| ||| ||| |||||||| || ||| ||||||||
||||||||| ||||| | ||| | |||||||||| | |
|---|---|---|
||||||||||| | ||||||||||| | |
|||||||| |||| || ||||| ||| |||||||| |||||||||| | ||| | ||| |||||| | ||| |||||| |
||| ||| |||| ||||||||||||| ||||||||||| |||||||| |||| ||| |||| |||| |||||| ||||| || |||||||| | ||| |||||| | || |||||| |
||| |||||||||| ||||||||| | ||| |||||| | || |||||| |
||| | |||||||| |||||||| || ||||||||||||| || ||| | || |||||| | || |||||| |
||| | |||||||| |||||||| || ||||||||||||| || ||| | || |||||| | || ||||| |
||||||||||| ||||| || ||||||| |||||||| |||||| ||||||| ||||||||||||| ||||||| |||| |||||||||| |||||||| ||||||| ||| ||||||||| |||||||| |||||| | |||| ||||| || ||| ||||| |||| || ||||| | || |||||| | || |||||| |
||| | ||||||||||||| ||||||||||||||||| |||||||||| ||| ||| ||| ||||||| ||||||||||| ||||||||| |||| ||||||||| || ||||||||||||| | || |||||| | || |||||| |
||| |||||| ||| ||||| ||| |||||| ||||||||| |||||| | || ||||| | || ||||| |
|||||||| || |||| || ||||| ||||||||||| ||||||||| ||||||| | || ||||| | | ||||| |
||||||||| ||||||||||||| ||||||||||||||| |||||||| |||||||||||||||||||||||| |||||| ||| ||||| ||| ||| | |||||||| ||||||| | || ||||| | | ||||| |
||||||||| | ||||||||| |||||| | | ||||| | | ||||| |
||| | |||||||||||| ||| | |||||||||||||||| ||| | |||||||||||||||| ||| ||||||||||| |||| |||||||||| || ||||||||| || ||| ||| ||||||| | ||||||| ||| |||| |||| |||| | |||| || |||||||||| |||| ||||| |||||||||| ||| ||| |||||||| || ||| ||||||||
Concomitant Medication
Overall, in the DISCOVER 1 and DISCOVER 2 studies, the numbers of patients who used antibacterial and/or nonantibacterial medications either before the first dose of study drug or concomitantly during the study were similar across treatment groups. In the ITT population of the DISCOVER 1 study, the number of patients who took any systemic or topical antibacterial medications in addition to the study drugs from the start of the study drug through to the EOT visit was similar in the dalbavancin and vancomycin treatment groups (26 [9.0%] versus 25 [8.8%]); all medications were systemic. The medication most commonly taken in both treatment groups was aztreonam. The most common nonantibacterial medications were anesthetics, antihistamines, antiseptics and disinfectants, and antithrombotic medications. Medications taken in the dalbavancin treatment group were metronidazole and aztreonam and in the vancomycin treatment group were amoxicillin plus clavulanic acid, penicillin, and aztreonam. Medications taken in both treatment groups were most commonly in the Anatomic Therapeutic Chemical classes, including anti-inflammatory and antirheumatic products, other analgesics and antipyretics, and opioids. In the ITT population, approximately 1-fifth of patients had incision and drainage of their ABSSSI before the start of the study drug (18.8% of patients and 20.7% patients in the dalbavancin and vancomycin treatment groups, respectively). Other commonly performed nondrug interventions included aspiration, application or change of surface dressings, and wound packing.
In the DUR001-303 study, overall, the number of patients who used antibacterial and/or nonantibacterial medications either before the first dose of the study drug or concomitantly during the study was similar across treatment groups. For antibacterial medications, patients who used any systemically administered antibiotic with a gram-positive spectrum that achieves therapeutic concentrations in the serum or at the ABSSSI site were excluded from the study. However, an exception to this exclusion criterion was allowed for patients who received, during the 14 days before the first dose of the study drug, a single dose of an antibacterial drug with a half-life no longer than 12 hours. Twenty-two patients (6.3%) in the single-dose group and 19 patients (5.4%) in the 2-dose group received a systemic antibacterial drug in the 14 days before study-drug assignment. The number of patients who used any systemic antibacterial medication during the period from the first dose of the study drug through to the EOT visit was similar across treatment groups: 39 patients (11.2%) in the single-dose group and 54 patients (15.5%) in the 2-dose group. Per protocol, any patient who used a systemic antibacterial medication for ABSSSIs during this period was to be considered a treatment failure. The number of patients who used any systemic antibacterial medication during the period from the first dose of the study drug through to the final visit (day 28) was also similar between groups.
In the VER001-9 study, the majority of patients in each treatment arm received at least 1 concomitant medication. Analgesics were the most common class of concomitant medications, taken by 72.2% of patients overall, and by a similar percentage of patients in each treatment arm. Vicodin and paracetamol were the most prescribed analgesics. Systemic antibacterials other than study medication, taken by 52.5% of patients overall, were the second most common class of concomitant medication. Twelve percent of patients in each treatment arm received antibacterial therapy in violation of the protocol. In addition to protocol violators, the overall use of concomitant antibiotics included protocol-defined use of systemic antibacterials, such as antibiotics administered to patients who failed treatment, aztreonam and/or metronidazole given for gram-negative aerobe and/or anaerobe coverage in patients with mixed infection, no more than 24 hours of antibacterial therapy (either before or concomitant), and antibiotics with a documented lack of in vitro activity against baseline gram-positive pathogens. Aztreonam and metronidazole were administered concomitantly to a similar proportion of patients in the 2 treatment arms (for aztreonam, 15.6% and 14.8% of patients in the dalbavancin and linezolid groups, respectively, and for metronidazole, 12.6% and 11.7% of patients, respectively).
Efficacy
Only efficacy outcomes and analyses of subgroups identified in the review protocol are reported here.
Clinical Response
Clinical response results for all trials are presented in Table 14. In the DISCOVER 1 study, clinical response at 48 to 72 hours was reported in 83.3% of the dalbavancin group and 81.8% of the vancomycin group, with a between-group difference of 1.5% (95% CI, –4.6% to 7.9%). The lower limit of the 95% CI for the treatment difference (1.5%) in the ITT population was –4.6%, which was higher than the pre-specified lower limit of –10%. Therefore, noninferiority of dalbavancin to vancomycin was concluded. The results of clinical response at the EOT visit by investigator assessment also showed similar results between treatment groups (90.3% with dalbavancin and 91.9% with comparator), with a 1.6% treatment difference (95% CI, –6.4% to 3.1%).
In the DISCOVER 2 study, clinical response at 48 to 72 hours was also similar between groups, with a greater response in the comparator arm (78.3%) than in the dalbavancin (76.8%), and a –1.5% treatment difference (95% CI, –7.4% to 4.6%). The lower limit of the 95% CI for the treatment difference of –1.5% in the ITT population was –7.4, which was higher than the pre-specified lower limit of –10%. Therefore, noninferiority of dalbavancin to vancomycin was concluded. In addition, clinical response at the EOT visit in the CE population was consistent with the ITT population, with a treatment difference of 2.8% (95% CI, –6.7% to 0.7%).
In the DISCOVER 1 and DISCOVER 2 studies, multiple sensitivity analyses were conducted for clinical response at 48 to 72 hours in the ITT population (refer to Appendix 3). Results of all of the sensitivity analyses were consistent with results of the primary efficacy analysis, with the treatment difference ranging from 0.2% to 3.3% in the DISCOVER 1 study and from 1.7% to 2.8% in the DISCOVER 2 study; the lower limit of the 95% CI for the treatment difference was always higher than –10%. This included the sensitivity analysis that excluded temperature from the primary outcome measure and defined success as at least a 20% decrease from baseline in lesion area.
In the DUR001-303 study, clinical response at 48 to 72 hours was observed in 84.2% of patients in the 2-dose dalbavancin arm and in 81.4% of the single-dose dalbavancin arm, with a between-group difference of –2.9% (95% CI, –8.5% to 2.8%). The lower limit of the 95% CI for the treatment difference of –2.9% in the ITT population was –8.5%, which was higher than the pre-specified lower limit of –10%. Therefore, noninferiority of 1-dose to 2-dose dalbavancin was concluded. In addition, clinical response at the EOT visit was almost identical between groups (treatment difference = –0.20%; 95% CI, –4.2% to 3.8%).
In the VER001-9 study, clinical response was assessed at day 14 (EOT) and day 28 (TOC) in the ITT population. The percentage of patients with clinical response at the TOC visit was 82.7% in the linezolid arm and 76.5% in the dalbavancin arm. The treatment difference was –6.15% (95% CI, –12.03% to –0.27%). The lower limit of the 95% CI for the treatment difference of –12.03% remained above the pre-specified lower limit of –12.5%. Therefore, noninferiority of dalbavancin to linezolid was claimed. However, because the upper bound of the 95% CI was just slightly below zero (–0.27%), the study authors felt that although dalbavancin may be considered statistically inferior to linezolid, the difference was not clinically meaningful. In terms of clinical response at the EOT visit, results were consistent with those observed at 48 to 72 hours, with 80.6% of patients in the dalbavancin arm and 86.9% of patients in the linezolid arm deemed clinical successes in the ITT analysis. Clinical response was also evaluated in the CE population at the TOC visit, and results were consistent with those of the ITT population (treatment difference = –2.21% in favour of linezolid; 95% CI, –7.28% to 2.86%).
Table 14: Clinical Response at 48 to 72 Hours Across Pivotal Trials and at the TOC Visit in the VER001-9 Trial (ITT Population)
Outcome | DISCOVER 1 | DISCOVER 2 | DUR001-303 | VER001-9 | ||||
|---|---|---|---|---|---|---|---|---|
DAL, N = 288 | VAN/LZD, N = 285 | DAL, N = 371 | VAN/LZD, N = 368 | 1-dose DAL, N = 349 | 2-dose DAL N = 349 | DAL, N = 571 | LZD, N = 283 | |
Clinical response at 48 to 72 hours (day 28 for VER001-9) | ||||||||
Clinical responder, n (%) | 240 (83.3) | 233 (81.8) | 285 (76.8) | 288 (78.3) | 284 (81.4) | 294 (84.2) | 437 (76.5)b | 234 (82.7) |
Clinical nonresponder n (%) | 48 (16.7) | 52 (18.2) | 86 (23.2) | 80 (21.7) | 65 (18.6) | 55 (15.8) | 134 (23.5) | 49 (17.3) |
Difference (95% CI) | 1.5 (–4.6 to 7.9) | –1.5 (–7.4 to 4.6) | –2.9 (–8.5 to 2.8)a | –6.15 (–12.03 to –0.27) | ||||
OR (95% CI) | 1.1 (0.7 to 1.7) | 0.9 (0.7 to 1.3) | NR | NR | ||||
Clinical response at day 14 | ||||||||
Clinical responder, n (%) | 260 (90.3)c | 262 (91.9)c | NR | NR | 321 of 347 (92.5)c | 319 of 344 (92.7)c | 460 (80.6) | 246 (86.9) |
Clinical nonresponder n (%) | 16 (5.6) | 9 (3.2) | NR | NR | 10 of 347 (2.9) | 5 of 344 (1.5) | 111 (19.4) | 37 (13.1) |
Difference (95% CI) | –1.6 (–6.4 to 3.1) | NR | –0.2 (–4.2 to 3.8) | –6.37 (–11.73 to –1.01) | ||||
CI = confidence interval; DAL = dalbavancin; ITT = intention-to-treat; LZD = linezolid; NR = not reported; OR = odds ratio; VAN = vancomycin.
Note: In the DISCOVER 1 and DISCOVER 2 studies, the OR of clinical response in the dalbavancin treatment group relative to the vancomycin treatment group stratified for the presence or absence of fever at baseline and the 95% CI for the OR were calculated using the Cochran-Mantel-Haenszel method.
a95% CI was calculated using the Miettinen and Nurminen method without adjustment in the DUR001-303 study and with adjustment for the randomization stratification factor of presence or absence of fever at baseline in the DISCOVER 1 and DISCOVER 2 studies.
bThe time point for assessment in the VER001-9 study is TOC (e.g., day 28).
cBased on investigator assessment.
Source: Clinical Study Reports.17-20
Reasons for clinical nonresponse are presented in Table 15. In the DISCOVER 1 and DISCOVER 2 studies, reasons for clinical nonresponse at 48 to 72 hours were similar in the 2 treatment arms. In the DISCOVER 1 study, the most common reasons for clinical nonresponse were fever only (approximately 29% of patients in each group), increase in lesion size only (27.1% in the dalbavancin group and 25.0% in the vancomycin group), and no evidence of fever but temperature criteria not met, as described in the SAP (25.0% of patients in each group). Most patients in both treatment groups were nonresponders, either because of fever or not meeting the criteria for temperature measurement (54.2% in the dalbavancin group versus 53.8% in the vancomycin group). In the DISCOVER 1 study, missing data were the reason for nonresponse in approximately 20% of patients with nonresponse status (22.9% of patients in the dalbavancin group and 19.2% in the vancomycin group).
In the DISCOVER 2 study, the most common reasons for nonresponse in the dalbavancin and vancomycin groups were the same as in the DISCOVER 1 study: fever only (31.4% versus 35.0%), increase in lesion size only (29.1% versus 22.5%), and no evidence of fever but temperature criteria not met, as described in SAP (24.4% versus 22.5%). The majority of patients in both treatment groups were nonresponders, either because of fever or not meeting the criteria for temperature measurement (55.8% in the dalbavancin group and 57.5% in the vancomycin group). In the DISCOVER 2 study, missing data accounted for a relatively small proportion of clinical nonresponse in each treatment group.
In the DUR001-303 study, the proportion of patients determined to be nonresponders for each of these reasons was similar across treatment groups in the ITT population. The most common reason for clinical nonresponse was lesion area not decreased by at least 20% relative to baseline (63.1% of patients in the single-dose group and 61.8% in the 2-dose group). Other reasons for clinical nonresponse included missing lesion data at any point (33.8% of patients in the single-dose group versus 32.7% in the 2-dose group), and lesion area collected outside of the 48- to 72-hour window (23.1% in the single-dose group versus 20.0% in the 2-dose group). A sensitivity analysis was conducted using a 36- to 75-hour time window instead of a 48- to 72-hour window, and 9 of the nonresponder patients in the single-dose group and 4 in the 2-dose group were considered responders. Deaths and use of rescue medications accounted for only a small proportion of the patients in each treatment group who were nonresponders (≤ 6.2% in the single-dose group and ≤ 7.3% in the 2-dose group).
In the VER001-9 study, there were 48 (8.4%) and 20 (7.1%) clinical failures in the dalbavancin and linezolid groups, respectively, in the CE population at the TOC visit (day 28). The most common reasons for clinical nonresponse were not reported.
Table 15: Reasons for Clinical Nonresponse at 48 to 72 Hours and on Day 28 Across Pivotal Trials (ITT Population)
Reason for clinical nonresponse | DISCOVER 1 | DISCOVER 2 | DUR001-303 | |||
|---|---|---|---|---|---|---|
DAL, N = 288 | VAN/LZD, N = 285 | DAL, N = 371 | VAN/LZD, N = 368 | 1-dose DAL, N = 349 | 2-dose DAL, N = 349 | |
Clinical nonresponse, n (%) | 48 (16.7) | 52 (18.2) | 86 (23.2) | 80 (21.7) | 65 (18.6) | 55 (15.8) |
Only fever | 14 (29.2) | 15 (28.8) | 27 (31.4) | 28 (35.0) | 0 | 0 |
Only increase in lesion size | 13 (27.1) | 13 (25.0) | 25 (29.1) | 18 (22.5) | 0 | 0 |
No evidence of fever, but temperature criteria not meta | 12 (25.0) | 13 (25.0) | 21 (24.4) | 18 (22.5) | 0 | 0 |
Missing data at 48 to 72 hours for both lesion measurement and determination of fever | 6 (12.5) | 5 (9.6) | 7 (8.1) | 9 (11.3) | 0 | 0 |
Missing data at baseline for lesion measurement | 4 (8.3) | 1 (1.9) | 3 (3.5) | 0 | 0 | 0 |
Both increase in lesion size and fever | 3 (6.3) | 3 (5.8) | 5 (5.8) | 4 (5.0) | 0 | 0 |
Both increase in lesion size and temperature criteria not met | 3 (6.3) | 1 (1.9) | 0 | 1 (1.3) | 0 | 0 |
Initiation of a new, systemic antibacterial in the first 72 hours or receipt of rescue therapy for ABSSSIb | 2 (4.2) | 2 (3.8) | 4 (4.7) | 5 (6.3) | 4 (6.2) | 4 (7.3) |
Missing data at 48 to 72 hours for lesion measurement only | 1 (2.1) | 3 (5.8) | 1 (1.2) | 1 (1.3) | NA | NA |
Missing data at 48 to 72 hours for determination of fever onlyc | 0 | 1 (1.9) | 0 | 1 (1.3) | NA | NA |
Died in the first 72 hours | 0 | 1 (1.9) | 0 | 0 | 0 | 1 (1.8) |
ABSSSI lesion size increased or decreased < 20% relative to baseline | NA | NA | NA | NA | 41 (63.1) | 34 (61.8) |
Missing lesion data at any point | NA | NA | NA | NA | 22 (33.8) | 18 (32.7) |
Lesion area collected outside of the 48- to 72-hour window | NA | NA | NA | NA | 15 (23.1) | 11 (20.0) |
ABSSSI = acute bacterial skin and skin structure infection; CE = clinically evaluable; DAL = dalbavancin; ITT = intention-to-treat; LZD = linezolid; NA = not applicable; TOC = test of cure; VAN = vancomycin.
Note: Patients could have had more than 1 reason for clinical failure. Lesion size was defined as lesion area only.
aThe patient had at least 1 temperature measurement, but did not have 3 temperature measurements 3 to 9 hours apart in the 48- to 72-hour window and had no temperature > 37.6°C after the 48-hour time point.
bThe patient initiated a new, systemic antibacterial with gram-positive activity for the ABSSSI under study in the first 72 hours.
cAll temperature data in the 48- to 72-hour window are missing for the patient.
Source: Clinical Study Reports.17-19
Clinical Status at EOT (Day 14) and Day 28
Summary results of clinical status at the EOT and day 28 visits in the ITT population and CE population are presented in Table 16. Clinical status of success or failure was not evaluated as defined in the pivotal trials in the VER001-9 study, and hence is not reported.
Clinical Status at EOT (Day 14)
In the DISCOVER 1 study, after adjustment for baseline infection type or fever status, the proportion of patients in the ITT population who achieved clinical success at the EOT visit (day 14) was similar in the comparator and dalbavancin groups (86.7 versus 81.9%), with a between-group difference of –4.8% (95% CI, –10.7% to 1.3%). Results of clinical success at the EOT visit were similar in the CE and ITT populations. In the DISCOVER 2 study, results were similar in the dalbavancin and comparator groups in the ITT population (88.7% versus 85.6%), with a between-group difference of 3.1% (95% CI, –1.8% to 8.0%). In the DISCOVER 2 study, clinical success at the EOT visit was higher in the CE population than in the ITT population for both treatment groups, and there was little difference between treatment arms (treatment difference = 0.8%; 95% CI, –3.3% to 5.0%). In the DUR001-303 study, there was little to no difference in the proportion of patients achieving clinical success at day 28 between the single-dose and 2-dose regimens of dalbavancin in the ITT population (treatment difference = –0.8%; 95% CI, –6.3% to 4.6%). This result was consistent with that in the CE population.
Clinical Status at Day 28
In all trials, the proportion of patients achieving clinical success were similar with dalbavancin and its comparator. In the DISCOVER 1 study, after adjustment for baseline infection type or fever status, the proportion of patients classified as a clinical success at day 28 was similar in treatment groups in the ITT populations (83.7% of patients in the dalbavancin group versus 88.1% in the comparator group; treatment difference = –4.4; 95% CI, –10.1% to 1.4%). In the DISCOVER 2 study, the proportion of patients achieving clinical success at SFU was similar in the 2 treatment groups in the ITT population (88.1% versus 84.5%; treatment difference = 3.6%; 95% CI, –1.1% to 8.9%). In the DUR001-303 study, there was little to no difference in the proportion of patients achieving clinical success at day 28 in the single-dose and 2-dose dalbavancin groups in the ITT population (treatment difference = –0.6%; 95% CI, –6.0% to 4.8%). Results for clinical success in the CE population at day 28 were consistent with those for the ITT population for all trials.
||||||| | |||||||| | | |||||||| | | |||||||||| | |||
|---|---|---|---|---|---|---|
|||||| | |||| | |||||||||| | |||| | |||||| | |||| | |||||||||| | |||| | ||| | ||||||| | |||| | ||| | |||||||| | |||| | |
|||||||| |||||| || ||| || | ||||||
||| ||||||||||| | | ||| | ||| | ||| | ||| | ||| | ||| |
|||||||| ||||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| |
|||||||| ||||||| | || |||||| | || |||||| | || ||||| | || ||||| | || |||||| | || |||||| |
||||||||||||| | || ||||| | | ||||| | || ||||| | || ||||| | || ||||| | || ||||| |
|||||||||| |||| ||| |||||| | |||| |||||| || |||| | ||| ||||| || |||| | |||| ||||| || |||| | |||
|||||| ||||||||||| | | ||| | ||| | ||| | ||| | ||| | ||| |
|||||||| ||||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| |
|||||||||| |||| ||| |||||| | |||| |||||| || |||| | ||| ||||| || |||| | |||| ||||| || |||| | |||
|||||||| |||||| || ||| || | ||||||
||| ||||||||||| | | ||| | ||| | ||| | ||| | ||| | ||| |
|||||||| ||||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| |
|||||||||| |||| ||| |||||| | |||| |||||| || |||| | |||| ||||| || |||| | |||| ||||| || |||| | |||
|| ||||||||||| | | ||| | ||| | ||| | ||| | ||| | ||| |
|||||||| ||||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| |
|||||||||| |||| ||| |||||| | ||||| ||||| || |||| | |||| ||||| || |||| | |||| ||||| || |||| | |||
|||||| | |||||||||| ||||||||| || ||| |||||||||||||||| |||||| || | |||||||||| ||||||||| ||| | |||||||||||| ||| | ||| || |||||||||| ||| | |||||||||||||||| ||| | |||||||||| ||| | |||||||||||| || ||||| || ||| ||||||||||| |||||||| ||||| ||| |||||||||| || ||||||||||| |||||| |||||||||| ||||||||| |||| |||||||||| ||||| ||||||||| ||| |||||||| |||||| |||| ||||||||||| || |||||| ||| | ||||| ||||||||| ||| ||| ||||| |||||| ||||||| ||||||| ||| || |||||||| | ||| || ||||| ||||| || ||||||||||| ||| ||| || |||||||| || ||| ||||| ||||||||| ||| ||| || ||| ||||| |||||||| |||||||| ||||| ||||||||||||
The proportion of clinical failures was similar between treatment groups across all pivotal trials.
In the DISCOVER 1 study, 38 patients (13.2%) in the dalbavancin treatment group and 29 (10.2%) patients in the vancomycin treatment group in the ITT population were clinical failures at the EOT visit (day 14). The most commonly reported reasons for clinical failure at the EOT visit in the dalbavancin and vancomycin treatment groups were that local signs of fluctuance and localized heat or warmth had not resolved (84.2% versus 79.3%), local signs of tenderness on palpation and swelling or induration were worse than mild (23.7% versus 34.5%), and the patient received a new, nonstudy systemic antibacterial treatment (34.2% versus 13.8%).
In the DISCOVER 2 study, 32 (8.6%) patients in the dalbavancin treatment group and 33 (9.0%) patients in the vancomycin treatment group in the ITT population were clinical failures at the EOT visit. The most commonly reported reasons for clinical failure at the EOT visit in the dalbavancin and vancomycin treatment groups were the same as in the DISCOVER 1 study: local signs of fluctuance and localized heat or warmth had not resolved (53.1% versus 60.6%), local signs of tenderness on palpation and swelling or induration were worse than mild (34.4% versus 48.5%), and the patient received a new, nonstudy systemic antibacterial treatment (28.1% versus 42.4%).
In the DUR001-303 study, 42 (12.0%) patients on single-dose dalbavancin and 36 (10.3%) patients on 2-dose dalbavancin in the ITT population were clinical failures at the EOT visit. The most common reasons for clinical failure in the 1-dose and 2-doses dalbavancin groups were that the lesion size did not decrease from baseline (73.8% versus 66.7%), local signs of tenderness on palpation and swelling or induration were worse than mild (21.4% versus 19.4%), and the patient received a new, nonstudy systemic antibacterial treatment (21.4% versus 16.7%).
Table 17: Reasons for Clinical Failure on Day 14 (ITT Population)
Reason for failure | DISCOVER 1 | DISCOVER 2 | DUR001-303 | |||
|---|---|---|---|---|---|---|
DAL, N = 288 | VAN/LZD, N = 285 | DAL, N = 371 | VAN/LZD, N = 368 | 1-dose DAL, N = 349 | 2-dose DAL, N = 349 | |
Clinical failures, n (%) | 38 (13.2) | 29 (10.2) | 32 (8.6) | 33 (9.0) | 42 (12.0) | 36 (10.3) |
|||||| ||| |||||||| |||||||| | || || |||||||| ||||||||| | ||||||
||||| ||||| |||||| ||||||||||||||||||||||| |||| ||| |||||||| | || |||||| | || |||||| | || |||||| | || |||||| | | ||||| | | |||||| |
|||||||| | ||| ||||||||| |||||||| ||||||||||||| |||||||||| | || |||||| | | |||||| | | |||||| | || |||||| | | |||||| | | |||||| |
||||| ||||| || |||||||||| || ||||||||| ||| ||||||||||||||||||| ||| ||||| |||| |||| | | |||||| | || |||||| | || |||||| | || |||||| | | |||||| | | |||||| |
|||||| |||| ||| ||| |||||||| |||| |||||||| | | |||||| | | |||||| | | |||||| | | ||||| | || |||||| | || |||||| |
|||||||| ||||||||||||| | | |||||| | |||| | | |||||| | | |||||| | |||| | |||| |
||| ||||| |||||||||| ||| |||||||| || ||| |||||||| |||||||| || ||| |||| || |||||||| |||| |||||||| || || ||||| |||| |||| | | ||||| | |||| | | ||||| | | ||||| | | ||||| | | ||||| |
||||||||||| |||||| || ||| ||| ||||| | | ||||| | | |||||| | | ||||| | | ||||| | | ||||| | | ||||| |
|||| |||||| ||| ||||| |||||| || || ||| ||| ||||| | |||| | | ||||| | |||| | |||| | | ||||| | | ||||| |
|||||||| ||||| ||||||| |||||| ||| |||||||| ||||||||| |||||| | | |||| | ||| | |||| | |||| | |||| | |||| |
DAL = dalbavancin; EOT = end-of-treatment visit; ITT = intention-to-treat; LZD = linezolid; VAN = vancomycin.
| ||| ||||||| |||||||| | ||| ||||||||| |||||||| ||||||||||||| ||||||||| ||| ||| |||||| ||||| ||||| || ||| |||| |||| ||| ||||| |||| || ||||| |||| ||||||| ||| |||||| || |||||| |||||||||| || |||| || |||||||| ||||||| ||| ||| ||||||| ||| ||||||| |||||||| |||||||| |||||||||||| |||| |||| || ||||| ||||| ||| ||||| || ||||||| ||| ||||||||| || ||| |||||| ||||| |||||| || |||||||| ||| ||||||| ||||| |||| ||||||| |||||| ||| |||||||| ||||||||| |||||| |||||| ||| |||||| || | |||||| || ||| |||||||||||||| |||||||||| |||| |||||||||| |||| ||||||| || |||||| ||| ||||||||| || ||| |||||| ||||| |||||| |||||| |||||||| ||||| |||| ||| |||| |||| ||| |||||| ||| |||||||| |||||||| |||||| |||| ||| ||||||| || |||||| |||| ||||||
Source: Clinical Study Reports.17-19
Subgroup Analyses
Presence or Absence of Bacteremia
The results for clinical response at 48 to 72 hours and clinical success at the EOT visit by presence or absence of bacteremia are presented in Table 18 and Table 19, respectively. Across the pivotal trials, there were no notable differences between the 2 treatments groups in the rate of clinical responders or the clinical status of patients with bacteremia at baseline. The only exception was clinical success at the EOT visit in patients with bacteremia at baseline, which was 50% higher in the linezolid arm than in the dalbavancin arm (100% versus 50%). However, the numbers in some of the subgroups were very small, limiting the conclusions that could be drawn. Results were generally similar to those for the primary efficacy analysis in all pivotal trials. In the VER001-9 study, clinical success was generally similar in the 2 groups for patients with bacteremia; 58.3% of patients with bacteremia at baseline were clinical successes at the EOT and day 28 visits in the dalbavancin group, whereas 50% of patients were clinical successes at the EOT and day 28 visits in the linezolid arm.
||||||||| | |||||||| |||||||| || || || || ||||| | |||||||||||||| | |
|---|---|---|---|
|||||||| | | ||| | ||||||| | |||||||||| |||| ||| |
||| ||||||||||| ||| ||| | ||| | ||| | ||| |
||| |||||||||| | ||| |||||| | ||| |||||| | |||| |||| |
|| |||||||||| | ||||||| |||||| | ||||||| |||||| | ||| ||||| || |||| |
|||||||| | | ||| | ||||||| | |||||||||| |||| ||| |
||| ||||||||||| ||| ||| | ||| | ||| | ||| |
||| |||||||||| | ||||| |||||| | ||||| |||||| | |||| ||||||| |||||| |
|| |||||||||| | ||||||| |||||| | ||||||| |||||| | |||| |||||| |||| |
|||||||||| | ||| || |||| | ||| || ||||| | |||||||||| |||| ||| |
|||||||| ||||||||||| ||| ||| | ||| | ||| | ||| |
||| |||||||||| | ||||| |||||| | || ||| ||||||| | ||||| |||||| || ||||| |
|| |||||||||| | ||| |||| |||||| | ||| |||| |||||| | ||| ||||| || |||| |
||| | |||||||||||| ||| | |||||||||| |||||||| | |||||||||||||| |||||| || ||||| ||||||||||| | | |||||||| |||||||| | | |||||| || |||||||| ||| ||| |||||||| |||||| ||| | |||||||||||||| ||| |||||| || |||||||| |||||| ||| ||||||||| ||| |||||||||||||||| || ||||| ||||||| ||||||||||| || |||| |||| ||| |||| |||||||||||| |||||| || |||| |||||||| |||||||| ||||| ||||||||||||
||||||||| | |||||||| ||||||| || ||| | |||||||||||||| | |
|---|---|---|---|
|||||||| | | ||| | ||||||| | |||||||||| |||| ||| |
|||||| ||||||||||| | | ||| | ||| | ||| |
||| |||||||||| | ||| |||||| | ||| ||||||| | ||| |||| |
|| |||||||||| | ||||||| |||||| | ||||||| |||||| | |||| ||||| || |||| |
|||||||| | | ||| | ||||||| | |||||||||| |||| ||| |
|||||| ||||||||||| | | ||| | ||| | ||| |
||| |||||||||| | ||||| |||||| | |||| |||||| | |||| |||||| || |||||| |
|| |||||||||| | ||||||| |||||| | ||||||| |||||| | ||| ||||| || |||| |
|||||||||| | ||| || |||| | ||| || ||||| | |||||||||| |||| ||| |
|||||||| |||||||||| | |||
||| |||||||||| | || ||| |||||||| | | ||| ||||||| | |||| |||||| || ||||| |
|| |||||||||| | ||| |||| ||||||| | ||| |||| ||||||| | |||| |||||| || |||| |
||| | |||||||||||| ||| | |||||||||| |||||||| | |||||||||||||| |||||| || ||||| |||||||||| | | |||||||| |||||||| | | |||||| || |||||||| ||| ||| |||||||| |||||| ||| | |||||||||||||| ||| |||||| || |||||||| |||||| ||| ||||||||| ||| |||||||||||||||| || ||||| ||||||| ||||||||||| || |||| |||| ||| |||| |||||||||||| |||||| || |||| |||||||| |||||||| ||||| ||||||||||||
MRSA Versus MSSA
The results for clinical response at 48 to 72 hours and clinical success at the EOT visit by MRSA versus MSSA are presented in Table 20. In the DISCOVER 1 study, clinical response and clinical success at the EOT visit were similar in the dalbavancin and vancomycin groups for both MRSA (82.9% versus 80.0%) and MSSA (86.6% versus 90.1%). However, the difference between groups was more pronounced at the EOT visit, when a greater proportion of patients who achieved clinical success was observed in the comparator group than in the dalbavancin group for both MRSA (92.3% versus 79.5%) and MSSA (87.5% versus 75.6%). Clinical success was similar in the 2 groups at day 28 for patients with MRSA; however, results at day 28 for patients with MSSA were similar to those observed at the EOT visit, with lower rates of success with dalbavancin than with the comparator (79.5% versus 94.3%). Similarly, in the DISCOVER 2 study, patients with Staphylococcus aureus who were treated with dalbavancin had lower response rates and lower clinical success at day 28 than those treated with vancomycin for MRSA (76.1% versus 85.7%) and MSSA (83.2% on comparator versus 76.4% on dalbavancin). Clinical status at the EOT visit and day 28 was similar in the 2 treatment groups; however, clinical success was slightly higher with dalbavancin than with vancomycin for patients with MRSA (95.7% versus 85.7%). In the DUR001-303 study, clinical response and clinical success were similar in the 2 treatment groups for both MRSA and MSSA. In the VER001-9 study, clinical responses at the EOT and TOC visits were similar in the 2 treatment groups for patients with MRSA and MSSA.
Outcome | |||||||| | | |||||||| | | DUR001-303 | |||||||| | ||||
|---|---|---|---|---|---|---|---|---|
||| | ||||||| | ||| | ||||||| | 1-dose DAL | 2-dose DAL | ||| | ||| | |
|||||||| |||||||| || || || || ||||| |||||||| ||||||| ||| || ||| || |||||||||| | ||||||||
|||||||||||||| |||||| |||||||| |||||||| |||| ||| | |||||| ||||||| | |||||| |||||| | ||||||| |||||| | ||||||| |||||| | ||||||| |||||| | ||||||| |||||| | ||||||| |||||| | ||||||| |||||| |
||||| |||| ||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||||| |||||| | ||||| |||||| |
||||| |||| ||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | |||||| |||||| | |||||| |||||| | ||||| |||||| | |||||||| | |||||||| |
|||||||| |||||||| || ||| || |||||||||| | ||||||||
|||||||||||||| |||||| |||||||| |||||||| |||| ||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| | ||||||| |||||| | ||||||| |||||| |
||||| |||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| | ||||||| |||||| | ||||| |||||| |
||||| |||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
|||||||| ||||||| || ||| || | ||||||||
|||||||||||||| |||||| |||||||| |||||||| |||| ||| | |||||| |||||| | ||||||| |||||| | ||||||| |||||| | ||||||| |||||| | ||||||| |||||| | ||||||| |||||| | |||||| | |||||| |
||||| |||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | |||||| | |||||| |
||||| |||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | |||||| |||||| | |||||| |||||| | ||||| |||||| | |||||| | |||||| |
|||||||| ||||||| || ||| || | ||||||||
|||||||||||||| |||||| |||||||| |||||||| |||| ||| | |||||| |||||| | ||||||| |||||| | ||||||| |||||| | ||||||| |||||| | ||||||| |||||| | ||||||| |||||| | |||||| | |||||| |
||||| |||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | |||||| | |||||| |
||||| |||| | ||||| |||||| | ||||| |||||| | ||||| |||||| | |||||| |||||| | |||||| |||||| | ||||| |||||| | |||||| | |||||| |
||| | |||||||||||| ||| | |||||||||||||||| |||||| ||| | |||||||||| ||| | |||||||||| ||||||||||| |||| | ||||||||||||||||||||| |||||||||||||| ||||||| |||| | ||||||||||||||||||||| |||||||||||||| ||||||| | | |||||| || |||||||| |||| ||||||| |||||||| |||| |||||||| |||||||| | | |||||| || ||| |||||||| || ||| |||||||| ||||||||||| || | |||||| || |||||||| || ||| |||||||| |||||||||| |||| ||| ||||| ||||||||| || | ||| ||||||||||| || | ||| ||||||||| ||| | |||||||||||| |||||||| |||||||| || |||||||| |||| ||||||||||||| |||||||||| || |||||||| |||||||| |||||||| ||||| ||||||||||||
Infection type
In the pivotal trials, when response rates for the primary end point were assessed by type of infection, the results were consistent with those from the overall ITT population. In the DISCOVER 1 study, 85.3% of dalbavancin-treated patients and 78.9% vancomycin-treated patients (treatment difference = 6.4%; 95% CI, –2.3% to 15.1%) with cellulitis were clinical responders at 48 to 72 hours. Responder rates for major abscess were 80.6% in dalbavancin-treated patients and 84.9% in vancomycin-treated patients (treatment difference = –4.3; 95% CI, –16.8% to 7.5%). Response rates for wound infection were 81.7% in dalbavancin-treated patients and 84.6% in vancomycin-treated patients (treatment difference = –2.9; 95% CI, –17.0% to 11.7%).
In the DISCOVER 2 study, clinical response rates at 48 to 72 hours were similar in the 2 treatment groups for patients with cellulitis, major abscess, and wound infection. In general, clinical response rates were higher for patients with major abscess than for those with either cellulitis or traumatic-wound infections. In the DISCOVER 2 study, responder rates for patients with cellulitis were 74.7% in the dalbavancin group and 75.7% in the vancomycin group, rates for major abscess were 83.3% and 87.4%, respectively, and rates for wound infection were 75.6% and 74.7%, respectively.
In the DUR001-303 study, responder rates for patients with cellulitis were 72.7% in the single-dose group and 78.3% in the 2-dose group, rates for patients with major abscess were 85.2% and 90.1%, respectively, and rates for patients with wound infection were 92.7% and 89.1%, respectively.
For all trials, the 95% CIs were wide and, especially in the case of surgical-site infections, patient numbers were low.
Systemic Signs and Symptoms of Infection (SIRS Criteria)
SIRS criteria that were documented as a secondary outcome included temperature and WBC count.
In the DISCOVER 1 and DISCOVER 2 studies, systemic signs of infection at each visit for the ITT population were similar in the 2 treatment groups. In the ITT population of the DISCOVER 1 study, 17 (6.1%) patients in each of the 2 treatment groups had a fever on day 3. At the EOT visit, 0 patients in the dalbavancin treatment group and 2 (0.8%) patients in the vancomycin treatment group had fever. In the ITT population, 24 (10.1%) patients in the dalbavancin treatment group and 29 (11.9%) patients in the vancomycin treatment group had a WBC count greater than 12,000 cells/mm3 on day 3. Median WBC count was 7.20 × 1,000 cells/mm3 in the dalbavancin treatment group and 7.40 × 1,000 cells/mm3 in the vancomycin treatment group. At the EOT visit, 10 (4.2%) patients in the dalbavancin treatment group and 6 (2.5%) patients in the vancomycin treatment group had a WBC count greater than 12,000 cells/mm3.
In the DISCOVER 2 study, for the ITT population, the proportion of patients with complete resolution of all local signs and symptoms and an absence of fever at the EOT visit was similar in the 2 treatment groups (253 [68.2%] patients and 250 [67.9%] patients in the dalbavancin and vancomycin treatment groups, respectively). A similar proportion of patients in each of the 2 treatment groups had a WBC count greater than 12,000 cells/mm3 on day 3 (33 [9.9%] patients in the dalbavancin treatment group and 28 [8.6%] patients in the vancomycin treatment group, respectively). Median (minimum to maximum) WBC count was 7,400 cells/mm3 (2,600 to 25,500 cells/mm3) in the dalbavancin treatment group and 7,100 cells/mm3 (2,000 to 26,700 cells/mm3) in the vancomycin treatment group. At the EOT visit, the proportion of patients with a WBC count greater than 12,000 cells/mm3 was higher in the dalbavancin treatment group (17 [5.3%] patients) than in the vancomycin treatment group (7 [2.2%] patients). Median (minimum to maximum) WBC count was 6,970 cells/mm3 (1,700 to 17,000 cells/mm3) in the dalbavancin treatment group and 6,350 cells/mm3 (1,200 to 28,500 cells/mm3) in the vancomycin treatment group.
In the DUR001-303 study, the majority of patients in the 2 treatment groups achieved a complete resolution of all local signs of ABSSSI by day 28. The percentage of patients who had a complete resolution of each of the various local signs of infection (defined as absence of purulence or drainage, erythema, heat or localized warmth, pain or tenderness on palpation, fluctuance, and swelling or induration) was similar across treatment groups at each study visit.
Time to Event Analysis
In the DISCOVER 1 and DISCOVER 2 studies, the Kaplan–Meier 75th percentile for time to resolution of fever was reached at 37.2 hours in the dalbavancin group and at 38.3 hours in the vancomycin group. In the DISCOVER 2 study, the Kaplan–Meier 75th percentile for time to resolution of fever was reached at 32.7 hours in both treatment groups.
Health-Related Quality of Life
HRQoL was not assessed in any of the included studies.
Hospitalization Rate and LOS
In the DUR001-303 study, more than 97% of patients in either treatment group had no additional clinic visits by the EOT visit and day 28 visit in the ITT population. Moreover, low per-patient use rates (< 3% each) were observed at both the EOT visit and the day 28 visit for ER visits and for additional unplanned visits, including urgent care visits.
In the VER001-9 study, the median length of hospitalization was 5 days in the dalbavancin arm and the linezolid arm. In the ITT population, 10 (1.8%) patients in the dalbavancin group and 6 (2.1%) patients in the linezolid group required hospital readmission for ABSSSI.
Harms
Only harms identified in the review protocol are reported here. Refer to Table 21 for detailed harms data.
Adverse Events
The incidence of TEAEs was lower in the dalbavancin treatment group than in the vancomycin treatment group in the DISCOVER 1 study (34.9% versus 39.4%) and the DISCOVER 2 study (31.3% versus 36.8%). Across the 2 trials, the number and type of TEAEs were similar in the 2 treatment groups. The most commonly reported TEAEs in the DISCOVER 1 study in the dalbavancin treatment group were headache, nausea, hypertension, and rash. For the vancomycin group, the common TEAES were headache, nausea, diarrhea, pruritus, hypertension, asthenia, and contact dermatitis. In the DISCOVER 2 study, the TEAEs reported were similar to those in the DISCOVER 1 study.
In the DUR001-303 study, the incidence of TEAEs was similar in the single-dose and 2-dose dalbavancin groups (20.1% versus 19.9%). The most common (≥ 2%) TEAEs in the single-dose and 2-dose treatment groups were nausea (3.4% versus 2.0%), headache (1.7% versus 1.2%), and vomiting (1.7% versus 0.9%). Most TEAEs were mild or moderate; severe TEAEs were observed for 8 patients (2.3%) in the single-dose group and 7 patients (2.0%) in the 2-dose group.
In the VER001-9 study, the incidence of TEAEs was not reported; however, the number of AEs were reported. The incidence of AEs in the dalbavancin and linezolid groups was similar. In the VER001-9 study, the most commonly (≥ 2%) reported AEs throughout the entire study period in both the dalbavancin and linezolid groups were nausea (3.2% versus 5.3%), diarrhea (2.5% versus 5.7%), and headache (1.9% versus 1.8%).
Serious Adverse Events
In the DISCOVER 1 study, treatment-emergent SAEs were less commonly reported in the dalbavancin treatment group than in the vancomycin treatment group (1.8% versus 4.2%).
In the DISCOVER 2 study, the number of treatment-emergent SAEs was similar in the dalbavancin and vancomycin treatment groups (3.3% versus 3.8%). The number of patients who experienced SAEs leading to death was very low (1 patient in the dalbavancin treatment group and 2 patients in the vancomycin treatment group).
In the DUR001-303 study, the percentage of patients with SAEs was low and similar in the single-dose and 2-dose treatment groups (2.0% versus 1.4%).
In the VER001-9 study, 7.5% and 8.4% of patients in the dalbavancin and vancomycin groups, respectively, experienced an SAE.
Withdrawals Due to Adverse Events
In the DISCOVER 1 study, the proportion of patients who discontinued treatment due to AEs was similar in the dalbavancin and vancomycin treatment groups (1.8% versus 2.1%).
In the DISCOVER 2 study, the proportion of patients who discontinued treatment due to AEs was similar in the dalbavancin and vancomycin treatment groups (2.4% versus 1.7%).
In the DUR001-303 study, the proportion of patients who discontinued treatment due to AEs was similar in the single-dose and 2-dose dalbavancin groups (1.7% versus 1.4%).
In the VER001-9 study, the proportion of patients who discontinued treatment due to AEs was 3.9% in the dalbavancin group and 3.2% in the linezolid group.
Mortality
In the DISCOVER 1 study, there were 0 deaths in the dalbavancin group and 5 deaths (1.8%) in the vancomycin group (caused by congestive heart failure, systemic lupus erythematosus, acute cardiac failure, pulmonary embolism, and hypovolemia, cardiopulmonary failure).
In the DISCOVER 2 study, rates of mortality were similar in the 2 groups, with 1 death (0.3%) in the dalbavancin group (caused by sepsis) and 2 deaths (0.5%)in the vancomycin group (caused by cardiorespiratory insufficiency and sudden death).
During the DUR001-303 study, 2 patients died. For the 1 patient in the single-dose group, cause of death was reported as toxicity to various drugs (i.e., combined heroin and methamphetamine intoxication), and for the 1 patient in the 2-dose group, cause of death was reported as pulmonary embolism (autopsy report terms: thromboembolism of main pulmonary artery, thrombosis of deep veins of lower extremities [including right tibia near cellulitis] and ischemic heart disease).
In the VER001-9 study, there were 2 deaths in each group (0.4% versus 0.7%).
Notable Harms
In the DISCOVER 1 study, notable harms in the dalbavancin and vancomycin groups included infusion-related reactions (0% versus 0.4%), rash (2.1% versus 2.1%), hepatic AE (1.7% versus 2.1%), acute renal failure (0.0% versus 0.4%), and nephropathy toxic (0.0% versus 0.4%).
In the DISCOVER 2 study, notable harms in the dalbavancin and vancomycin groups included infusion-related reactions (0.8% versus 0%), rash (0.8% versus 1.6%), hypersensitivity reactions (0.8% versus 0.3%), anaphylactoid reaction (0.3% versus 0.0%), hepatic AE (1.0% versus 0.8%), and acute renal failure (0.0% versus 0.5%).
In the DUR001-303 study, notable harms included rash (1.2% in both the 1-dose and 2-dose groups), hypersensitivity reaction (0.6% in both groups), and acute renal failure in the 2-dose group (0.3%).
In the VER001-9 study, notable harms in the dalbavancin and linezolid groups included infusion-related reactions (0.4% versus 0.0%), hypersensitivity reactions (0.2%% versus 0.0%), hepatic AE (0.4% versus 0.4%), and acute renal failure (0.0% versus 0.7%).
Table 21: Summary of Harms (Safety and ITT Population)
Harms | DISCOVER 1 | DISCOVER 2 | DUR001-303 | VER001-9 | ||||
|---|---|---|---|---|---|---|---|---|
DAL, N = 284 | VAN/LZD, N = 284 | DAL, N = 368 | VAN/LZD, N = 367 | 1-dose DAL, N = 349 | 2-dose DAL, N = 346 | DAL, N = 571 | LZD, N = 283 | |
Patients with ≥ 1 AE | ||||||||
TEAEs, n (%) | 99 (34.9) | 112 (39.4) | 115 (31.3) | 135 (36.8) | 70 (20.1) | 69 (19.9) | 318 (55.7) | 174 (61.5) |
Most common events,a n (%) | ||||||||
Headache | 14 (4.9) | 14 (4.9) | 11 (3.0) | 9 (2.5) | 6 (1.7) | 4 (1.2) | 33 (5.8) | 27 (9.5) |
Nausea | 12 (4.2) | 13 (4.6) | 15 (4.1) | 15 (4.1) | 12 (3.4) | 7 (2.0) | 34 (6.0) | 33 (11.7) |
Hypertension | 7 (2.5) | 7 (2.5) | 5 (1.4) | 5 (1.4) | 1 (0.3) | 1 (0.3) | 5 (0.9) | 2 (0.7) |
Rash | 6 (2.1) | 6 (2.1) | 4 (1.1) | 3 (0.8) | 2 (0.6) | 2 (0.6) | 15 (2.6) | 10 (3.5) |
Diarrhea | 4 (1.4) | 11 (3.9) | 4 (1.1) | 8 (2.2) | 4 (1.1) | 2 (0.6) | 30 (5.3) | 24 (8.5) |
Vomiting | 3 (1.1) | 6 (2.1) | 8 (2.2) | 4 (1.1) | 6 (1.7) | 3 (0.9) | 21 (3.7) | 12 (4.2) |
|||||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| |
|||||||||| ||||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| |
|||||||| | | ||||| | || ||||| | | ||||| | | ||||| | | ||||| | | ||||| | || ||||| | | ||||| |
|||||||||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | || ||||| | | ||||| |
||||| |||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | || ||||| |
|||||||||||||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| |
||||||| ||||| ||||||||| ||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | || ||||| | | ||||| |
|||||| ||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | || ||||| | | ||||| |
|||||||||||||| ||||||||||| ||||||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | || ||||| | | ||||| |
||||| ||||||| |||||||||||||| ||||||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | || ||||| | | ||||| |
||||| |||||||| |||| ||||||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | || ||||| | | ||||| |
|||||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | || ||||| | || ||||| |
||||||||||||| ||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | || ||||| | | ||||| |
||||||||||||||||| |||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| |
|||||||| |||| | | ||| | ||||||||
n (%) | 5 (1.8) | 12 (4.2) | 12 (3.3) | 14 (3.8) | 7 (2.0) | 5 (1.4) | 43 (7.5) | 24 (8.5) |
|||| |||||| |||||||| | |||| | ||||||||
||||||||| ||| |||||||||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| |
||||||||| ||||||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| |
|||||||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| |
||||||| ||||||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| |
||||||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | || ||||| | | ||||| | | ||||| |
||||||| | | ||||| | | ||||| | || ||||| | | ||||| | || ||||| | || ||||| | | ||||| | | ||||| |
|||||||||| | | ||||| | | ||||| | | ||||| | | |||||| | || ||||| | || ||||| | | ||||| | | ||||| |
|||||||| |||| ||||||||| | | ||||| | | ||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| |
|||||||| | | ||||| | || ||||| | || ||||| | | ||||| | || ||||| | || ||||| | || ||||| | || ||||| |
|||||| ||||||| | | ||||| | | ||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| |
||||||||||| ||||||||| | || ||||| | | |||||| | | ||||| | || ||||| | || ||||| | | ||||| | || ||||| | || ||||| |
|||||| | || ||||| | || ||||| | | ||||| | | ||||| | | ||||| | || ||||| | || ||||| | || ||||| |
|||||||||||| | || ||||| | || ||||| | || ||||| | | ||||| | || ||||| | || ||||| | || ||||| | || ||||| |
||||||||||| ||||||||| |||||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | | ||||| | || ||||| | || ||||| |
|||| ||||||||| |||||||||| | || ||||| | || ||||| | || ||||| | || ||||| | | ||||| | || ||||| | || ||||| | || ||||| |
||||||||||||| ||| | | ||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | | ||||| | | ||||| |
||||||||| ||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | | ||||| | || ||||| |
|||| |||| |||||||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | | ||||| | | ||||| |
|||||||||| ||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | | ||||| | || ||||| |
|||||||| ||||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | | ||||| | || ||||| |
||||||| |||| | || ||||| | | ||||| | || ||||| | || ||||| | || ||||| | || ||||| | | ||||| | || ||||| |
|||||||||| |||||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | | ||||| | || ||||| |
Patients who stopped treatment due to AEs | ||||||||
n (%) | 5 of 284 (1.8) | 6 of 284 (2.1) | 9 of 368 (2.4) | 7 of 367 (1.9) | 6 of 349 (1.7) | 5 of 346 (1.4) | 22 of 571 (3.9) | 9 of 283 (3.2) |
||||||||||||| | || ||||| | ||| ||||| | | ||||| | || ||||| | || ||||| | || ||||| | | ||||| | || ||||| |
||||||||||| |||| | || ||||| | || ||||| | || ||||| | || ||||| | | |||||| | | ||||| | | ||||| | || ||||| |
|||||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | | ||||| | || ||||| |
||||||||| ||| | || ||||| | || ||||| | | |||||| | || ||||| | || ||||| | || ||||| | | ||||| | || ||||| |
|||||||||| | | ||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | | ||||| | || ||||| |
|||||||||| ||||||||||||| | | ||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| |
|||||||||||||||| ||||| | | ||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| |
|||||||||| | | ||||| | || ||||| | | ||||| | || ||||| | || ||||| | || ||||| | | ||||| | || ||||| |
|||||| ||||||| | || ||||| | | ||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| |
|||||||||||||||| |||||||| | || ||||| | | ||||| | || ||||| | | ||||| | | ||||| | | ||||| | || ||||| | || ||||| |
||||||||| || |||||| | || ||||| | | ||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| |
||||||||||||| | || ||||| | | ||||| | || ||||| | || ||||| | || ||||| | | ||||| | || ||||| | || ||||| |
|||||| | || ||||| | | ||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| |
||||| ||||| ||||| ||||||| | || ||||| | | ||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| |
||||||||| ||| ||| ||| | || ||||| | || ||||| | | ||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| |
||||||||| ||||||||| | || ||||| | || ||||| | | ||||| | || ||||| | || ||||| | || ||||| | | ||||| | || ||||| |
|||| |||||||| | || ||||| | || ||||| | | ||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| |
||||||||||| ||||||||| | || ||||| | || ||||| | | ||||| | || ||||| | || ||||| | | ||||| | || ||||| | || ||||| |
||||||||||| | || ||||| | || ||||| | | ||||| | || ||||| | || ||||| | || ||||| | || ||||| | || ||||| |
|||||||||||||||||| | || ||||| | || ||||| | || ||||| | | ||||| | || ||||| | || ||||| | | ||||| | || ||||| |
||||| ||||||| ||||| | || ||||| | || ||||| | | ||||| | | ||||| | || ||||| | || ||||| | || ||||| | | ||||| |
|||||||| ||| |||||||| ||||||||| |||||||||| |||||||||||| ||||||||| | || ||||| | || ||||| | || ||||| | | ||||| | || ||||| | || ||||| | || ||||| | || ||||| |
||||| |||||||||||||||||||||| ||||||| | || ||||| | || ||||| | | |||||| | | ||||| | || ||||| | || ||||| | || ||||| | || ||||| |
|||||||||||| | || ||||| | || ||||| | || ||||| | | ||||| | || ||||| | || ||||| | || ||||| | || ||||| |
Deaths | ||||||||
n (%) | 0 | 5 (1.8) | 1 (0.3) | 2 (0.5) | 1 (0.3) | 1 (0.3) | 2 (0.4) | 2 (0.7) |
|||||| | |||| | |||| | | ||||| | |||| | ||||| | ||||| | ||||| | ||||| |
|||||||||| ||||||| |||||||| | ||||| | | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
|||||||| ||||| ||||||||||||| | ||||| | | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
||||| ||||||| ||||||| | ||||| | | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
||||||||| |||||||| | ||||| | | ||||| | ||||| | ||||| | ||||| | | ||||| | ||||| | ||||| |
||||||||||| | ||||| | | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
||||||||||||||| ||||||| | ||||| | | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
|||||||||||||||||| ||||||||||||| | ||||| | ||||| | ||||| | | ||||| | ||||| | ||||| | ||||| | ||||| |
|||||| ||||| | ||||| | ||||| | ||||| | | ||||| | ||||| | ||||| | ||||| | ||||| |
|||||||||| |||||||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | | ||||| | ||||| |
||||||| ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | | ||||| | ||||| |
||||||||||||||| |||||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | | ||||| |
||||||||| ||||| ||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | | ||||| |
|||||||| || ||||||| |||||| |||||||| | |||||||||||||||| | ||||| | ||||| | ||||| | ||||| | | ||||| | ||||| | ||||| | ||||| |
Notable harms, n (%) | ||||||||
|||||||||| |||||||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| | |||||| |
|||||||| ||||||| ||||||||| | ||||| | | ||||| | | ||||| | ||||| | ||||| | ||||| | | |||||| | ||||| |
Rash | 6 (2.1) | 6 (2.1) | 3 (0.8) | 6 (1.6) | 4 (1.1) | 4 (1.2) | 0 | 0 |
|||||||||||||||| ||||||||| | ||||| | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | | ||||| | ||||| |
||||||||||||| |||||||| | ||||| | ||||| | | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
||||||| || | | ||||| | | ||||| | | ||||| | | ||||| | ||||| | ||||| | | ||||| | | ||||| |
||||||||||| ||||| | ||||| | | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
||||| ||||||| ||||| | ||||| | | ||||| | ||||| | | ||||| | ||||| | | ||||| | ||||| | | ||||| |
||||||||||| ||||||||| ||||||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| | ||||| |
AE = adverse event; DAL = dalbavancin; ITT = intention-to-treat; LZD = linezolid; TEAE = treatment-emergent adverse event; VAN = vancomycin.
Note: If the same AE (based on MedDRA Preferred Term) was reported for the same patient more than once, the AE was counted only once for that Preferred Term. In addition, the ITT population was used for the VER001-9 study.
|||||||||| ||||||| ||| | |||||||||||| ||| | |||||||||| ||| | ||| ||||||||| |||||||||| ||| | ||||||| ||||||| ||||||| |||| | ||||||||| |||||||| ||||||| |||||| ||| | |||||||||||||||||||||| | ||| |
bFrequency ≥ 2% across treatment groups.
Source: Clinical Study Reports.17-20
Critical Appraisal
Internal Validity
In general, the noninferiority design, outcome assessments, and study populations in the pivotal trials and the VER001-9 study were appropriate for the purpose of establishing the clinical efficacy of dalbavancin in the treatment of ABSSSI. The noninferiority design was adequately powered, and the thresholds used (–10% and –12.5%) were justified. According to the clinical expert consulted by CADTH, the 10% noninferiority margin is somewhat wide, but a loss of approximately 5% in clinical benefit (corresponding to the reported between-group differences in the pivotal trials) would not be too concerning for the treatment of ABSSSI. Also, the expert indicated that although vancomycin is expected to be efficacious in this population (especially in those with MRSA),26 linezolid is a bacteriostatic, rather than bactericidal, drug and may not be as efficacious as other available options.27 Although the dosing of vancomycin aligned with the recommended dose for this population, it may not have been the most appropriate comparator, given that majority of patients with Staphylococcus aureus had known MSSA infection, not MRSA infection. According to the clinical expert, more appropriate drugs for patients with MSSA include cephalosporins, such as IV ceftriaxone and oral cephalexin. The primary outcome in the DUR001-303 study corresponds to the primary efficacy end point recommended in the FDA draft guidance for the development of drugs for ABSSSIs,1 and the primary outcome was similar in the DISCOVER 1 and DISCOVER 2 studies. The primary outcome for the DISCOVER 1 and DISCOVER 2 studies also considered resolution of fever and did not require a decrease in lesion size for clinical response, which is more aligned with the way patients are assessed in clinical practice, as the requirement for a 20% reduction in lesion area (part of the FDA-recommended outcome) is arbitrary, and fever, WBC count, and pain are typically assessed. The clinical expert noted that the outcomes in the VER001-9 study were less well defined. Overall, the expert considered the efficacy outcomes and follow-up in the 4 studies to be appropriate for advising on the use of dalbavancin in patients with ABSSSI. The time point of 48 to 72 hours seemed to be an appropriate surrogate for eventual cure, as there was consistency with the findings at the EOT and day 28 time points. Treatment compliance was higher with dalbavancin than with other IV therapies. The single-dose regimen also had higher compliance than the 2-dose regimen. The use of concomitant antibiotic therapy was similar between groups across all trials; hence, bias toward treatment outcome is low.
Patients were randomized with IVRS or other statistical software; hence, the risk of bias from inadequate allocation concealment and confounding was low. Patients and investigators were blinded to treatments, and the likelihood of unblinding was low, given the double-dummy design. Overall, baseline characteristics were generally similar and balanced between groups across all trials. There tended to be a greater percentage of MRSA infections in the dalbavancin group than in the vancomycin group, which may have biased the results against the intervention, as vancomycin is used specifically to treat MRSA and would have a treatment advantage. The pathogens identified at baseline were similar and balanced across all trials, with majority being MSSA. The clinical expert consulted for this review indicated that the differences between the dalbavancin and vancomycin or linezolid groups would not be expected to have an impact on the observed treatment effect. In all trials, discontinuation from the study was low (approximately 10%) and was similar between groups. The most common reason for discontinuation in all trials was loss to follow-up, which occurred in similar percentages in the treatment groups. Protocol deviations were high, especially in the VER001-9 study; however, these occurred at a similar frequency in the 2 treatment groups in each study. Hence, bias related to protocol deviations is of little concern.
Sensitivity analyses confirmed that results presented in the primary analysis across all pivotal trials and results of the CE population were similar to those of the ITT population. Data imputation methods were the same across pivotal trials. However, the VER001-9 study did not adjust for missing values. The statistical methods used were appropriate and pre-specified before database lock, and the number of randomized patients in each treatment group were sufficient, according to sample size calculations. An interim analysis was conducted by an independent unblinded statistician for sample size re-estimation, but this did not introduce bias into the primary outcome
Subgroup analyses did not include statistical testing, were limited in number, and did not preserve randomization; hence, results should be considered descriptive. Overall, there were no notable differences between subgroups, and results were similar to those in the primary analysis.
External Validity
The clinical expert consulted by CADTH agreed that the baseline patient characteristics of the pivotal trials and the VER001-9 study were reflective of patients encountered in Canadian clinical practice for the ABSSSI indication. Although the majority of patients in each study were enrolled at trial sites in the US and Europe, the population enrolled in the trial was consistent with the population expected to be treated in Canadian clinical practice. In general, lesion size was quite large across all trials, (e.g., approximately 300 cm2 to approximately 750 cm2), which is indicative of more severe ABSSSI; this may limit generalizability to populations with smaller lesion sizes or less severe disease. Furthermore, the subgroup analyses (e.g., bacteremia, infection type, and MRSA status) had no statistical comparisons and had small sample sizes in some cases, which limits the generalizability to a broader population. The rate of microbiologic diagnosis was higher in the trials than in clinical practice, and the selection of the comparator did not reflect treatment used for patients with ABSSSI caused by non-MRSA pathogens. This limited the generalizability and interpretation of the results. According to the clinical expert, the concomitant medications used in the trial were reflective of those encountered in Canadian clinical practice.
Indirect Evidence
Objectives and Methods for the Summary of Indirect Evidence
Evidence from head-to-head comparisons of dalbavancin and treatments other than vancomycin and linezolid for adults with ABSSSI caused by susceptible isolates of gram-positive micro-organisms was not available for this review. To fill this gap in evidence, the sponsor submitted a published ITC for CADTH to review. CADTH also conducted a literature search to identify other potentially relevant ITCs in adults with ABSSSI. The Ovid MEDLINE database was searched using a combination of MeSH and keywords. The main search concept was adults with ABSSSI. A filter was applied to limit study types to NMAs. Retrieval was not limited by publication date or by language. Titles, abstracts, and full-text articles were screened for inclusion by 1 reviewer, based on the population, intervention, comparator, and outcome criteria outlined in the CADTH systematic review protocol (Table 5).
One sponsor-submitted published ITC38 was summarized and critically appraised. Two other published ITCs were identified in a literature search: Agarwal et al.(2018)39 and Vlachaki et al. (2021).40 The Agarwal et al. (2018) study was excluded from this review because of a lack of relevant comparators. The Vlachaki et al. (2021) study was excluded because of the very limited evidence of dalbavancin within the evidence network of the NMA and incomplete reporting of the NMA results, such that there were no useful comparisons involving dalbavancin. The sponsor-submitted ITC was used to inform the pharmacoeconomic model.
Description of Indirect Comparison
The sponsor-submitted ITC, which is an NMA, aimed to evaluate the clinical efficacy of dalbavancin relative to daptomycin, linezolid, tigecycline, and vancomycin in adults with complicated ABSSSI caused by gram-positive bacteria. The sponsor performed a systematic review to identify relevant studies for inclusion in the ITC. The outcomes that were analyzed were clinical cure, microbiological cure, AEs, and mortality. The populations, interventions, comparators, outcomes, and designs of the studies included in the sponsor’s ITC are provided in Table 22.
Table 22: Study Selection Criteria and Methods for ITCs
Criteria | Sponsor-submitted ITC |
|---|---|
Population | Adults and children of any with complicated ABSSSIs (suspected or confirmed to be caused by gram-positive bacteria) |
Intervention | Dalbavancin, daptomycin, linezolid, tigecycline, and vancomycin |
Comparator | The same as the interventions |
Outcome | Clinical cure, microbiological cure, AEs, and mortality |
Study design | RCT of any size and duration |
Publication characteristics | Language of publication was not restricted |
Exclusion criteria | Studies were excluded if they:
|
Databases searched | PubMed, Cochrane, ClinicalTrials.gov |
Selection process | Articles were screened independently by 2 researchers |
Data extraction process | Data extraction was performed by pairs of reviewers and compared for discrepancies |
Quality assessment | CRD and ISPOR guidelines |
ABSSSI = acute bacterial skin and skin structure infection; CRD = Centre for Research and Dissemination; ISPOR = International Society for Pharmacoeconomics and Outcomes Research; ITC = indirect treatment comparison; RCT = randomized controlled trial; SSTIs = skin and soft tissue infections.
Source: Guest et al. (2017).38
Methods of ITC
Objectives
The primary objective of this ITC was to compare the efficacy and safety of IV antibiotics used in the current standard of care for the management of patients 18 years and older with ABSSSIs.
Study Selection Methods
As part of the sponsor’s ITC, a systematic literature review was conducted to identify current available evidence for the clinical efficacy and safety of antibiotics used to treat ABSSSI in adults. In the absence of direct head-to-head comparisons of treatments of interest, a NMA of dalbavancin, daptomycin, linezolid, tigecycline, and vancomycin was performed. The systematic review and NMA were conducted in adherence to the principles set out in the Centre for Research and Dissemination and International Society for Pharmacoeconomics and Outcomes Research guidelines.
The search strategy was based on the criteria outlined in Table 22. The study population was defined as adults with complicated ABSSSI (suspected or confirmed to be caused by gram-positive bacteria).
Only published RCTs (of any size and duration and with any blinding status) were eligible for inclusion in the analysis. Studies were excluded from the analysis if they were nonrandomized, observational studies or review articles; lacked relevant outcomes; included pooled analyses or subgroup analyses of other studies; or lacked the interventions of interest.
PubMed and the Cochrane database were searched without any language or time restrictions. The searches were conducted in November 2014. In addition, the ClinicalTrials.gov database was searched in January 2015. Websites for the European Society of Clinical Microbiology and Infectious Disease, Interscience Conference on Antimicrobial Agents and Chemotherapy, IDWeek, and International Society for Pharmacoeconomics and Outcomes Research were also searched. Additional methods to identify ongoing and recently completed research included searching the websites of Health Technology Assessment and related agencies, professional associations, and key conferences to identify conference abstracts from the past 3 years.
The selected outcomes of the analysis were chosen because they are frequently measured and reported in trials that involve patients with ABSSSI. The outcomes of interest were clinical treatment success, microbiological success, discontinuation due to AEs, the occurrence of any AE, the occurrence of any SAE, and all-cause mortality.
ITC Analysis Methods
The authors of the submitted ITC used a Bayesian NMA approach. A fixed effects model and a random-effects model were developed for each outcome and compared using the deviance information criterion. The choice of whether to use a fixed- or random-effects model was based on evaluation of the deviance information criterion statistic and assessment of statistical heterogeneity. Prior distributions used in the analyses were not reported. Statistical heterogeneity was assessed for each end point with a random-effects model.
A pairwise meta-analysis that used the frequentist approach was undertaken for each pairwise comparison that was informed by more than 1 trial. The authors used statistical measures to quantify the amount of heterogeneity between the trials included in the NMA. The authors computed the I2 statistic, representing the proportion of variation across trials caused by systematic differences rather than randomness, the τ2 value, the estimated between-study variance in a random-effects model, and the P value of the heterogeneity statistic Q, which represents the weighted total of the square of the differences between individual trial effects and the pooled effect across trials. In terms of the degree of heterogeneity, I2 statistics of 0% to 40% were classified as not important, of 30% to 60% were classified as moderate, of 50% to 90% were classified as substantial, and of 75% to 100% were classified as considerable.
In addition to statistical heterogeneity, an exploratory analysis was carried out to identify all the potential sources of heterogeneity. The potential heterogeneity sources were categorized into 2 distinct classes: clinical heterogeneity and methodological heterogeneity. Clinical heterogeneity refers to potential sources that stem from differences in patient characteristics that could influence study-specific effect estimates. Such differences can be related to variations in inclusion and exclusion criteria between the RCTs or to specific characteristics of the sampled individuals. Methodological heterogeneity includes potential causes of bias arising from the study design and variation in the definitions of the outcomes of interest. Inconsistency was assessed with the Bücher method.
The authors of the submitted ITC conducted a similarity assessment of the trials eligible for inclusion in the NMA and an assessment of risk of bias for each trial, but the details of these assessments were not available. The trials included in the NMA varied in design and quality, and the authors chose to include all trials in the NMA, regardless of their risk of bias.
For a particular treatment comparison, with estimates based on NMA results, the statistical significance (2-sided P value) of the difference between the direct and indirect evidence was tested.
All end points included in the NMA were binary and were summarized as an OR. The efficacy-related end points were clinical treatment success at the TOC visit and microbiological success at the TOC visit. The safety-related end points were the number of discontinuations due to AEs and/or SAEs, the occurrence of AEs, the occurrence of SAEs, and all-cause mortality.
Table 23: ITC Analysis Methods
Variable | ITC |
|---|---|
ITC methods | Multivariate fixed and random-effects NMA under a Bayesian framework |
Priors | Not reported |
Assessment of model fit | DIC |
Assessment of consistency | The Bücher method |
Assessment of convergence | NR |
Outcomes | Clinical treatment success, microbiological success, number of discontinuations due to AEs or SAEs, occurrence of AEs, occurrence of SAEs, all-cause mortality |
Follow-up time points | From 7 to 28 days after last dose |
Construction of nodes | NR |
Sensitivity analyses | Not conducted |
Subgroup analysis | Adults and mixed (adult and children) |
Methods for pairwise meta-analysis | Frequentist pairwise meta-analysis |
AE = adverse event; DIC = deviance information criterion; ITC = indirect treatment comparison; NR = not reported; SAEs = severe adverse event.
Source: Guest et al. (2017).38
Results of ITC
Summary of Included Studies
The systematic literature review identified 3,616 articles, of which 1,828 were screened, yielding 48 articles from 46 studies that provided clinical and safety-related data. Of these, 17 studies (19 articles) met the inclusion criteria for the NMA. One study that compared teicoplanin with linezolid was excluded from this review because results from the adult disease population were not provided. The network connecting all the articles included in the different analyses is illustrated in Figure 2.
Figure 2: Network of Trials Included in the NMA
DAL = dalbavancin; DAP = daptomycin; LZD = linezolid; NMA = network meta-analysis; TEI = teicoplanin; TIG = tigecycline VAN = vancomycin.
* Includes studies in which the comparator was vancomycin, vancomycin plus aztreonam, or vancomycin plus conventional treatment. Each node represents a treatment, and the connecting lines indicate pairs of treatments that have been directly compared in 1 RCT. The numbers in the connecting lines indicate the number of relevant study articles for each treatment comparison.
Source: Guest et al. Copyright 2017. Licensed under CC BY 4.0: https://creativecommons.org/licenses/by/4.0/
Population Characteristics
Although all trials contained in the NMA included patients with cSSSIs, the definition of cSSSI was not consistent across trials. In 2 trials, patients recruited had an ABSSSI, defined as an infection with a minimum lesion size of 75 cm2, involving deeper soft tissue or needing surgical intervention. Major cutaneous abscesses, surgical-site or traumatic-wound infection, and cellulitis were the infections included. On top of the presence of erythema, all patients needed to have a minimum of 2 of the following: purulent drainage or discharge, fluctuance, heat or localized warmth, tenderness on palpation, or swelling or induration. Moreover, the infection severity was required to be such that a minimum of 3 days of IV therapy was deemed as appropriate to treat the infection.
The trial population was defined as adults with cSSSI in 6 of the trials. In another 4 trials, the population comprised adults with cSSSI, but MRSA had to be separated from specimens before patients were recruited. In another 3 trials, the trial indication included pneumonia and complicated skin and soft tissue infections (cSSTI) induced by gram-positive bacteria (including MRSA), and in 2 other studies, the trial population had pneumonia, bacteremia, and cSSSI. But in 1 trial, only patients with MRSA-related serious infections were included. Another trial included patients infected with vancomycin-resistant E faecium or E. faecalis or MRSA, either isolated alone or as part of a polymicrobial infection. Furthermore, in another trial, patients with cSSTIs with or without bacteremia were included. In contrast, bacteremia was an exclusion criterion in 2 other trials. The trial population in 1 trial differed from all the other trials, in that patients with a central venous, pulmonary artery, or arterial catheter in place for 13 days and a suspected catheter-related infection were eligible for inclusion, as were patients with Staphylococcus aureus bacteremia who were to undergo echocardiography to rule out endocarditis. But the primary analysis population was restricted to patients with catheter-related blood infections and cSSSI. Therefore, the primary disease differed across trials. However, the clinical, microbiological, and safety-related outcomes analyzed in the NMA related only to 1 disease: cSSSI.
The trial population of the different NMA trials also differed in terms of patient age. The majority of trials recruited patients 18 years and older. However, 1 trial recruited patients 20 years and older. Another trial recruited patients 65 years and older, and 3 trials recruited patients 13 years and older.
Interventions
The interventions included in the NMA were linezolid (8 trials), daptomycin (5 trials), tigecycline (4 trials), and dalbavancin (3 trials). The dosage of an intervention tended to be the same across trials. However, dosage and treatment duration differed, depending on the nature of the infection.
In all trials in which daptomycin was the intervention, daptomycin was administered at a dose of 4 mg/kg IV once-daily if patients did not have bacteremia, and at 6 mg/kg 4 times a day for 14 to 42 days if bacteremia was detected. In 2 trials, the treatment duration was 5 to 14 days and 7 to 14 days if no Staphylococcus aureus bacteremia was detected, and for 10 to 28 days and 14 to 42 days, respectively, if bacteremia was detected. In all 3 studies in which dalbavancin was the intervention, the dose of dalbavancin was the same. It was administered as 1 1,000 mg IV dose on day 1 and a subsequent 500 mg IV dose on day 8. Likewise, the dosage of tigecycline was the same in all trials, and was administered as an initial IV dose of 100 mg followed by 50 mg twice a day. Linezolid was administered as 600 mg IV every 12 hours in all trials in which it was the intervention. But, in 4 studies, IV linezolid could be switched to the oral formulation at the investigator’s discretion. The minimum treatment duration differed by infection type. In general, the minimum duration was 7 days for patients with cSSTI or urinary tract infection, 14 days for patients with bacteremia, and 10 days for patients with pneumonia.
Comparators
Vancomycin was the comparator in all trials included in the NMA, except in 2 trials in which the comparator was linezolid and teicoplanin. In 5 trials, IV vancomycin 1 g was administered every 12 hours. In another trial, 1 g was administered if the patient was younger than 60 years, but the dose was reduced to 0.75 g in patents older than 60 years. The same dose was administered in all the trials irrespective of infection type. The minimum treatment duration was 7 days for patients with cSSTI or urinary tract infection, 14 days for patients with bacteremia, and 10 days for patients with pneumonia.
The comparator was either vancomycin (if the patient had MRSA or a penicillin allergy) or a penicillinase-resistant penicillin in 4 trials. The dosage of vancomycin was 1 g every 12 hours in all the trials. In 1 trial, the dose of penicillin differed for patients with bacteremia (2 g every 4 hours) and without bacteremia (2 g every 6 hours). The treatment duration differed between patients with bacteremia (10 to 28 days) and those without bacteremia (5 to 14 days) in this trial. The comparator was IV vancomycin if patients had MRSA infection but, in another trial, linezolid if patients had a vancomycin-resistant enterococci infection.
In 2 trials, the comparator was IV vancomycin, but patients could switch to oral linezolid after 3 days of therapy if:
in the previous 24 hours, a patient had 4 temperature measurements, each roughly 6 hours apart, in which all 4 measurements were less than 37.6°C (99.7°F)
unequivocal improvement was achieved in some or all of the clinical signs of the ABSSSI; if some signs had not improved, none should have deteriorated.
In most trials, aztreonam and other drugs with gram-negative coverage were permitted as adjunctive therapies. However, it was used in combination with vancomycin in 3 trials. In these trials, vancomycin was administered as 1 g IV over 60 minutes, followed by 2 g aztreonam administered over 60 minutes, twice a day. In another trial, aztreonam could be discontinued after 48 hours, according to the investigator’s judgment.
Linezolid was the comparator in 1 trial. In this trial, patients were randomized to receive IV linezolid 600 mg or IV or oral linezolid every 12 hours for 14 days. In another trial, patients received IV or intramuscular teicoplanin as the comparator for up to 28 days.
Trial Characteristics
All trials were randomized and had an active control. However, several differences in trial design were identified. Eight trials were double-blinded, 2 were single-blinded, and the other studies were all open-label. All trials were phase III, except for 1, which was phase IV. The phase of another trial was not stated. Five trials were conducted in a single country: 1 in China, 2 in the US, and 2 in Japan. The other trials were conducted in several countries and several continents. The timing of the trials also differed, and ranged from 1998 to 2012 for trials that reported information on the timing of the trial. The timing is not mentioned in 3 trials. In all trials, the outcomes were based on clinical and microbiologic assessments. The end points assessed aligned with those suggested by the FDA and included rates of clinical and microbiological success. However, the primary end point differed across trials.
Primary End Point
The primary end point was clinical success at the TOC visit in 3 trials. However, the TOC visit occurred from 12 to 16 days after completion of treatment at day 14 in 1 of trials, and from 7 to 14 days after treatment in another. The definition of clinical success was the same in all 3 trials. Improvement in signs and symptoms of SSSI such that no additional antibacterial therapy was necessary was defined as clinical success.
The assessment of homogeneity between trials is presented in Table 24. In 1 trial, the primary end point was defined as the clinical and microbiological response at the TOC visit, which occurred 7 to 14 days after the last dose of trial medication. Clinical and microbiological responses were affirmed by the efficacy adjudication committee. In another trial, the primary end point was clinical and microbiological success, but at the EOT visit and at the follow-up evaluation 5 to 16 days after treatment. In another trial, the primary end point was microbiologic success at the TOC visit, which took place 7 to 14 days after treatment, and clinical success at the TOC visit was a secondary end point.
In all the other trials, the primary end point was defined as the clinical outcome, success, or response. The timing of assessment of the clinical outcome was at the TOC visit in most trials, but the timing of the TOC visit differed across trials. The TOC visit occurred at different times:
between 12 and 92 days after the last dose in 3 trials
between 12 and 37 days after administration of the last dose of the study drug in 1 trial
between 7 and 14 days after administration of the last dose of the study drug in 1 trial
between 7 and 28 days after administration of the last dose of the study drug in 1 trial
between 15 and 21 days after the end of therapy in 2 trials
ten days after the EOT visit in 1 trial
seven days after the EOT visit in 1 trial.
In 2 trials, the primary end point included clinical outcome at the EOT visit, which occurred in the 72 hours after the last dose of the study medication. In 2 trials, the primary outcome was clinical response, defined by measurement of ABSSSI lesion size and temperature, 48 to 72 hours after study-drug initiation. In another trial, the primary end point was not specified, but both clinical and microbiological responses were assessed at the TOC visit, which occurred 10 days after the EOT visit.
Table 24: Assessment of Homogeneity for ITC
Variable | Assessment of between-trial heterogeneity |
|---|---|
Disease severity | Definition of cSSSI differed across studies; a random effect model was used |
Treatment history | NR |
Clinical trial eligibility criteria | Age varied across studies; a random-effects model was used |
Dosing of comparators | Varied across studies; a random-effects model was used |
Placebo response | NR |
Definitions of end points | Varied across studies; a random-effects model was used |
Timing of end point evaluation or trial duration | Varied between 7 and 92 days after administration of the last dose; a random-effects model was used |
Withdrawal frequency | NR |
Clinical trial setting | Multinational and single country; a random-effects model was used |
Study design | Varied across studies and included double-blinded, single-blinded, and open-label; a random-effects model was used |
cSSSI = complicated skin and skin structure infection; ITC = indirect treatment comparison; NR = not reported.
Source: Guest et al. (2017).38
Results
Not all of the trials reported all of the end points. Nineteen trials reported clinical success, 13 reported microbiological success, 11 reported treatment discontinuation due to AEs or SAEs, 11 reported the number of AEs, 10 reported the number of SAEs, and 12 reported all-cause mortality.
The NMA shows no evidence of a difference in clinical success between dalbavancin and vancomycin, linezolid, or daptomycin. In terms of safety, there was no evidence of differences between dalbavancin and any of the comparators for the rate of discontinuation due to AEs or SAEs. In contrast, dalbavancin was associated with a lower occurrence of AEs than linezolid, a lower occurrence of SAEs than vancomycin and daptomycin, and a lower risk of all-cause mortality than vancomycin and linezolid. The results are shown in Table 25.
Drug | OR (95% CrI) for clinical treatment success, RE model | OR (95% CrI) for discontinuation due to AEs or SAEs, FE model | OR (95% CrI) for occurrence of AEs, FE model | OR (95% CrI) for occurrence of SAEs, FE model | OR (95% CrI) for all-cause mortality, FE model |
|---|---|---|---|---|---|
Vancomycin | 0.99 (0.68 to 1.51) | 1.08 (0.59 to 1.98) | 0.85 (0.70 to 1.03) | 0.54 (0.30 to 0.96) | 0.26 (0.05 to 0.93) |
Linezolid | 0.69 (0.41 to 1.00) | 1.24 (0.68 to 2.30) | 0.78 (0.62 to 0.98) | 0.99 (0.61 to 1.66) | 0.20 (0.04 to 0.77) |
Daptomycin | 1.05 (0.61 to 2.10) | 1.28 (0.53 to 3.09) | 1.05 (0.76 to 1.46) | 0.48 (0.24 to 0.95) | 0.34 (0.05 to 1.71) |
AE = adverse event; CrI = credible interval; FE = fixed-effects; ITC = indirect treatment comparison; OR = odds ratio; RE = random-effects; SAE = severe adverse event.
Source: Guest et al. (2017).38
Critical Appraisal of Sponsor-Submitted ITC
The sponsor’s rationale for conducting the ITC (i.e., an absence of head-to-head studies comparing dalbavancin with relevant treatments for adults with ABSSSI) and the objectives of the ITC (to determine the efficacy and safety of dalbavancin compared with other treatments appropriate for adults with ABSSSI) were clearly reported. A comprehensive systematic review was performed. The language of publication was not restricted, which minimized publication bias. The efficacy outcome of interest to this review was clinical treatment success, and the safety outcomes were discontinuation due to AEs or SAEs, the occurrence of any AE, the occurrence of any SAE, and all-cause mortality.
Although no limits were placed on date or language during the publication search that was part of the systematic review, the impact of potential publication bias was not explored in the review. Although a full assessment of risk for bias for each RCT was conducted, methods and results were not described, and all trials in the NMA were included regardless of their risk of bias.
Clinical and statistical heterogeneity were present in the analysis because of variations in study duration, blinding, dosage, disease definition, publication date, patient demographic and clinical characteristics, and clinical-effect modifiers. Such heterogeneity likely resulted in bias and undermined the validity of the NMA results.
In the NMA, each of the comparators involved few trials, with some comparators informed by only 1 study. Furthermore, most of the active treatments were compared directly to vancomycin but not to each other, contributing to the large amount of uncertainty around the estimates, which is reflected in the wide CrIs for treatment differences.
The degree of statistical heterogeneity differed in the pairwise meta-analyses. The I2 statistic ranged from 17.3% to 85.6% in comparisons that reported I2. The τ2 value ranged from 0.047 to 0.719 in comparisons that reported τ2. All 23 pairwise comparisons reported the P value for the heterogeneity statistic Q, and 4 of them were than 0.05.
Because of the small number of trials included in the NMA, the ability to estimate between-trial variance was limited. As a result, analyses with uncommon events, such discontinuations due to AEs and SAEs, would likely produce imprecise estimates.
Given the differences in patient characteristics, such as age, disease, treatment indication, and statistical heterogeneity, exchangeability of patients in the trials included of the NMA was not guaranteed.
Inconsistency of the network was not reported, likely because of the limited ability to do so, given that the network involved mostly trials comparing against 1 common comparator.
Given these limitations, the results from the sponsor-submitted ITC are imprecise and at risk of bias. However, given the magnitude of the difference in the results between dalbavancin and the comparators for the efficacy outcomes and the wide CrIs, there is much uncertainty surrounding treatment effect estimates and firm conclusions cannot be drawn. For the safety outcomes, given the magnitude of the difference in results and the limitations of these results, reported improvements with dalbavancin, relative to certain comparators, are subject to bias and uncertainty and, therefore, are not reliable.
Other Relevant Evidence
This section includes 2 pre-post pragmatic studies — the ENHANCE study21 and the ADVANCE study22 — that were considered to provide additional information on the effect of dalbavancin on the outcomes of hospital LOS and hospitalization rate in patients with ABSSSI.
Description of Studies
ENHANCE and ADVANCE Studies
The ENHANCE21 pre-post pragmatic study was conducted to estimate the difference in infection-related total hospital admission days during initial care (the period from date of enrolment to 10 to 14 days later) and follow-up (the period from the end of initial care to 30 days later) between patients with ABSSSIs who received care before (pre-period) and after (post-period) the implementation of a critical pathway that was developed for the management of patients with ABSSSIs who were admitted to the hospital. The intervention was the critical pathway, which comprised the identification of patients using criteria that were developed from guidelines on the management of ABSSSIs in the hospital setting and outpatient parenteral antibiotic therapy and the administration of dalbavancin to patients who met the criteria and were subsequently discharged to an outpatient setting at the discretion of the treating physician. Training on the protocol for the pre-period, with an emphasis on patient identification strategies, was provided to the study team on-site, including investigators, the study coordinator, and data entry personnel. Training on the protocol for the post-period, with an emphasis on the critical pathway, was provided to the team on-site and treating physicians and pharmacists. During the pre-period, only the first component of the critical pathway was implemented, and usual care was initiated for patients with ABSSSI, which was defined as the antibiotic with coverage for the known or suspected gram-positive infection selected by the treating physician or site. Of note, treating physicians were not trained on the protocol and did not take part in the study enrolment decision-making in the pre-period. During the post-period, both components of the critical pathway were implemented, and 1,500 mg of dalbavancin was administered as a single IV dose over 30 minutes in a new set of patients enrolled according to the same guideline-based criteria used in the pre-period. Therefore, the criteria used for patient selection remained the same throughout the study (first step of the critical pathway was implemented in both the pre-period and the post-period), but the treatment received was different (patients in the pre-period received usual care, whereas patients in the post-period received dalbavancin, in accordance with the second step of the critical pathway). For both the pre-period and post-period, patients were assessed at baseline (date of enrolment), 48 to 72 hours after the date of enrolment or discharge, 10 to 14 days after the date of enrolment, and 44 to 51 days after the date of enrolment. The final assessment date could have been earlier if a patient prematurely discontinued from the study.
The ADVANCE study22 was conducted to estimate the difference in hospital admission rates at initial care between patients with ABSSSIs who received care before (pre-period) and after (post-period) implementation of the critical pathway. The study design of the ADVANCE study was similar to that of the ENHANCE study,21 with a few differences of note. First, the ENHANCE study recruited patients with ABSSSIs who were admitted to the hospital and required treatment for a known or suspected gram-positive infection, whereas the ADVANCE study recruited patients with ABSSSIs who presented to the ER and required treatment for a known or suspected gram-positive infection. Therefore, patients enrolled in the ADVANCE study received dalbavancin at the point of care in the ER and were subsequently sent home at the discretion of the treating physician. Further, compared with the ENHANCE study, the ADVANCE study had additional criteria in the newly developed critical pathway to ensure that patients who likely required parenteral antibiotic treatment were identified. Finally, patients enrolled in the ADVANCE study had an additional assessment 24 hours after enrolment in the post-period, whereas patients in the pre-period were not assessed 48 to 72 hours after enrolment.
Secondary outcomes of interest were total hospital admission days during initial care and follow-up, infection-related hospitalizations during initial care and follow-up (and resulting in admission to an intensive care unit), all-cause hospitalizations in the 30 days after discharge (or after release from the ER in the ADVANCE study22), patient-reported HRQoL, SAEs during initial care and follow-up, response to treatment at the EOT visit (the ENHANCE study21 only), and infection-related total hospital admission days during initial care and follow-up (the ADVANCE study only).
||||| || ||||| || ||| |||| ||||||| || ||| |||||||| | ||||||||||| || ||| ||||||||||| ||||||| || |||||||| || |||||||| | ||| |||||||| | ||| ||||||| ||| |||||||| ||||||||||||| ||||||||| | ||||| |||||||| ||||||||||||||| |||||| |||| ||||||| ||||||||||| |||||| |||| ||| |||||||||| ||| |||||| ||| || |||||||| |||||| ||| ||||||||||| ||| |||||||| |||| ||||||||| ||| || |||||||| |||| ||||||||| | ||||| || || |||||||| ||||||| ||||||||| ||| ||||| ||| | |||||||| ||||||| ||| |||||||||| | |||||||| |||||| |||| |||| || |||||||||| ||| |||| |||||||| |||| ||||| |||||||| |||||||| || ||| |||||||| ||| ||| |||||||| || ||||| | |||| || || ||||||||||| ||||||||| || || ||||||||| |||||| ||| |||||||||||| ||| |||||||| |||| ||||||||| ||| || |||||||| |||| ||||||||| | ||||| || || |||||||| ||||||| ||||||||| ||| ||||| ||| || |||||||| ||||||| ||| |||||||||| | |||||||| ||||||| |||| |||| || |||||||||| ||| |||| |||||||| ||| |||||||| || ||||||||| ||| |||| ||| ||| |||| ||||| ||||||||| ||||||||| |||| | |||||| ||| |||| ||||| ||| | |||||| || ||| |||| ||| |||||||||||| ||||||||||||| | ||||| || || |||||||| ||||||| ||| || |||||||| ||||||| |||| |||| || ||| |||| ||| |||||||||||| ||||||||||||| ||| |||| ||| ||| |||| ||||| ||| | ||||| ||| |||| ||||| ||| | |||||| || ||| |||| ||| |||||||||||| ||||||||||||| ||| |||||||| |||||| ||| |||||| || ||| |||||||||| |||| ||||||||||||||||||||| || ||| |||| ||| |||||||||||| ||||||||||||| ||| |||| |||||| || |||| ||||||| |||||||||| ||||||||| |||| || ||| |||||||||| |||| |||||||||| |||||||| |||||||||| |||||||| ||| ||||||||||| |||||||| ||| |||||||| |||||||| || ||| ||||||||||| |||||||| |||||||||||| || ||||| | |||||||| |||||||| ||||| |||| ||||||||||| ||||||||||||| |||| |||||||||||| ||||| |||| |||| |||||||||| || ||||||||||||||| ||||||| ||| |||| ||| ||||||||||||| || ||||||||||| ||||| ||| ||||| || |||||||| || ||| |||| ||| ||||||||||||| ||||||||||||| ||| |||||||||| ||||||||| ||||||| ||| ||||| |||||||||||||||| | ||||| |||||||| ||||||||||||||| |||||| |||| ||||||| ||||||||||| |||||| |||| ||| |||||||||| ||| |||||| ||| || |||||||| |||||| ||| ||||||||||| ||||| |||||||| |||| ||||||||| ||| ||| |||||||| |||| ||||||||| | ||||| || ||| |||||||| ||||||| ||||||||| ||| ||||| ||| || |||||||| ||||||| ||| |||||||||| || |||||||| ||||||| |||| |||| || |||||||||| ||| |||| |||||||| |||| ||||| |||||||| |||||||| || ||| |||||||| ||| ||| |||||||| || ||||| | |||| || || ||||||||||| ||||||||| || ||| ||||||||| |||||| ||| |||||||||||| ||||| |||||||| |||| ||||||||| ||| ||| |||||||| |||| ||||||||| | ||||| || ||| |||||||| ||||||| ||||||||| ||| ||||| ||| || |||||||| ||||||| ||| |||||||||| || |||||||| ||||||| |||| |||| || |||||||||| ||| |||| |||||||| ||| |||||||| ||| ||||||||| ||| |||| ||| ||| |||| ||||| ||| | |||||| ||| |||| ||||| ||| | |||||| || ||| |||| ||| |||||||||||| ||||||||||||| | ||||| || ||| |||||||| ||||||| ||| || |||||||| ||||||| |||| |||| || ||| |||| ||| |||||||||||| ||||||||||||| ||| |||| ||| ||| |||| ||||| ||| | ||||| ||| |||| ||||| ||| | |||||| || ||| |||| ||| |||||||||||| ||||||||||||| ||| |||||||| |||||| ||| |||||| || ||| |||||||||| |||| ||||||||||||||||||||| || ||| |||| ||| |||||||||||| ||||||||||||| ||| |||| |||||| || |||| ||||||| |||||||||| ||||||||| |||| || ||| |||||||||| |||| |||||||||| |||||||| ||||||||| |||||||| ||||||||||| |||||||| |||||||||||||||| |||||||||||| |||||||| ||| ||||||||| |||||||| | ||||| || ||| |||||||| ||||||| || ||| ||||||||||| |||||||| |||||||||||| | ||||||| ||| |||||||| || |||| |||||||| ||||||||||| | ||||| ||||| ||||||||||| || ||||| | |||||| || |||||||| || ||| ||||||||||| |||||||| ||||| ||||||||||| ||||||||||||| |||| |||||||||||||
Table 26: Details of the ENHANCE and ADVANCE Studies
Detail | ENHANCE | ADVANCE | |
|---|---|---|---|
Design and population | |||
Study design | Pre-post pragmatic study | Pre-post pragmatic study | |
Location | US (1 site) | US (11 sites) | |
||||||| ||||||||| ||||| | || |||| |||| || | |||||||| |||| ||||||||||||||| |||||||| |||| || || ||||||| |||| ||||||||||||| | | |||||||| |||| || || ||||||| |||| ||||||||||||||| ||||| |||| || || ||||| |||| ||||||||||||| | |
Enrolled, n | 48 (pre-period) 43 (post-period) | 160 (pre-period) 153 (post-period) | |
Inclusion criteria |
|
| |
||||||||| |||||||| |
|
| |
Drug | |||
Intervention (post-period only) |
or
|
or
| |
Comparators (pre-period only) | Usual care (the site or treating physician’s selection of antibiotic therapy for ABSSSI with coverage for a gram-positive infection; dosing of each drug should follow recommended guidelines) | Usual care (the site or treating physician’s selection of antibiotic therapy for ABSSSI with coverage for a gram-positive infection; dosing of each drug should follow recommended guidelines) | |
Duration | |||
Phase | |||
Enrolment | 3 months (pre-period); 9 months (post-period) | 11 months (pre-period); 12 months (post-period) | |
Initial care | 10 to 14 days | 10 to 14 days | |
Follow-up | 30 days | 30 days | |
Outcomes | |||
Primary end point | Difference in infection-related total hospital admission days during initial care (date of enrolment to 10 to 14 days) and follow-up (30 days after initial care) between patients with ABSSSI receiving care before and after implementation of the critical pathway | Difference in hospital admission rates at initial care between patients with ABSSSI receiving care before and after implementation of the critical pathway | |
Secondary and exploratory end points |
|
| |
Notes | |||
Publications | McCarthy et al. (2020)41 | Talan et al. (2021)42 | |
ABSSSI = acute bacterial skin and skin structure infection; CrCl = creatinine clearance; ER = emergency room; HRQoL = health-related quality of life; ICU = intensive care unit; MRSA = methicillin-resistant Staphylococcus aureus; MSSA = methicillin-susceptible Staphylococcus aureus; SAE = serious adverse event; SF-12 = 12-Item Short Form Survey.
aIn the ADVANCE study, 1 patient with a CrCl of 21 mL/min at baseline and not on dialysis received 1,125 mg of dalbavancin. Two other patients with a CrCl < 30 mL/min and not on dialysis were recorded as having received 500 mg of dalbavancin.
Source: Clinical Study Report for ENHANCE21 and ADVANCE.22
Results
ENHANCE
The full details of the difference between the pre-period and post-period in the total infection-related hospital admission days are shown in Table 32 in Appendix 3. The mean difference between the pre-period and post-period in the total infection-related LOS during the entire study was 1.6 days (95% CI, 0.6 to 2.6 days; P = 0.003) in favour of the post-period. The results of the secondary analysis, after adjustment for age and immunocompromised status, were generally consistent with the primary analysis. Analysis that included time spent in prolonged (> 1 day) observation and patients who completed the study were generally consistent with the observed difference in the primary analysis with inpatients only for the duration of the study.
Secondary Outcomes
All secondary outcomes were exploratory, as the study was not powered for secondary outcomes and no adjustments for multiple comparisons were made.
|||||||| || ||||| || ||||| ||| |||| ||||||| || ||| |||||||||| ||||||| |||| ||| ||||||||||| || ||| ||||| |||||||| |||||||| ||||| ||| |||| |||||||||| ||||||| |||| ||| ||||||||||| || ||| ||||| |||||| || |||| |||||| ||| |||||| ||||| ||| ||| |||| |||| ||| ||| || ||| |||||| || |||||| || ||| |||||||||||| |||||||| ||||||||| |||| ||||| || ||||||||| ||||||||||| ||| || |||||||| ||| ||||||||| ||| ||||| |||| ||||||||| |||||||||| |||| ||| ||||||| |||||||| |||| |||||||||| |||| ||| ||| |||||| |||||||| || ||| ||||||||||||||| || ||||| || ||||| ||| |||| ||||||| || ||||| ||||||||||||||||| ||||||||||||||||| ||||||||||||||||| |||||||||||||||| ||||||| || ||||||||| || || |||| ||| ||||||||| |||||||||||||||| || ||| || |||| |||||||||||||| |||||||| || ||| ||||||| |||||||||||||||| |||||| ||| ||||||| ||||| ||| |||||||| |||||||| || |||| ||| |||| ||| |||||||||||| ||| ||||||||||||||||| ||||||||||||||||| ||| |||||| ||| ||||||||| ||||||| || |||||||| ||| | |||||||| |||||| ||| ||||||||||||||||| |||||||||||||||| || ||| |||| ||| |||||||||||| ||||||||||||| || |||||||| ||||||||||| ||||||||||||||||| |||||||||||||||| |||| ||| || ||| |||||||||| |||||| ||| ||||||| ||||| | ||||||| |||| || ||| |||| ||| ||||| |||||| ||||| ||| ||||| ||||||||||||| |||| |||||||| || |||| ||| || ||||||||| ||||||||||||||| || ||| || |||| ||||||||| ||||||||| |||| ||| ||||||| |||||||||||||||| |||||| ||| ||||||||| ||||||| || |||||||| ||| | |||||||| |||||| || ||| |||| ||| |||||||||||| ||||||||||||| |||||||| ||||||||| ||||||||||||||| || ||| || |||| ||||||||| ||||||||| |||| ||| ||||||| ||||||||||||||||||||||||| || ||||| || ||||| ||| |||| ||||||| || |||||||| || ||||||||| || ||| ||| || ||||||||| |||||| || ||| ||||||||||| || |||||||| ||||||| ||| |||||||| ||||||||| || |||||||| ||| ||||||| ||||||||| || |||||||| ||||||| ||||||||||| ||||||| || |||||||||| ||| || |||||||| ||||||| ||| ||||||| ||||| || ||| |||||||||||| || |||||||| ||||||| ||| |||||||| ||||||||| || |||||||| ||| ||||||| ||||||||| | |||||||| ||||||| ||||||||||| ||||||| || |||||||||| ||| | |||||||| ||||||| ||| ||||||| |||||||||||||| || ||||| || ||||| ||| |||| ||||||| || ||| |||||| || ||||| |||||| ||| |||||||| |||||| ||||||||| |||||| || ||| || |||||||| || ||||||||| || |||| ||| |||| ||| ||||||||||||| |||||||| |||||||||||| ||||||||||| || ||| |||||| |||||| ||| ||||||||||| ||| |||| |||||| |||||| ||||||||| ||||| || |||||||| ||| ||| || ||| |||| ||| | |||||| ||| |||| ||| | ||||||| ||||||||||||| |||||| ||| |||||||||||| ||| |||| |||||| |||||| ||||||||| ||||| || |||||||| ||| ||| || ||| |||| ||| | |||||| ||| |||| ||| | ||||||| ||||||||||||| || ||| ||||||||||| ||| |||| |||||||| |||||| ||||||||| ||||| || |||||||| ||| ||| || ||| |||| ||| | ||||| ||| |||| ||| | ||||||| ||||||||||||| || ||| |||||||||||| ||| |||| |||||||| |||||| ||||||||| ||||| || |||||||| ||| ||| || ||| |||| ||| | |||||| ||| |||| ||| | ||||||| |||||||||||||||||||||| || ||||| || ||||| ||| |||| ||||||| || ||||| |||||||| || ||| |||| ||| |||||||||||| | ||||| || | |||||| ||| || ||||||| |||||||| || ||| |||| ||| ||||||||||| ||||||| ||||||||||||| |||||||| || ||||| ||| ||||||| |||||| ||| |||| |||||| || ||| ||||||| ||||| ||| ||||||| || | |||||||| |||||| || ||| |||||||||||| || |||||||| |||||||||||| ||||| ||| || ||| ||||||| ||||| ||| || |||||| |||| |||||||| || ||| |||||| | ||||| || | |||||| ||| | |||||| |||||||| || ||| |||| ||| ||||||||||| ||||||| ||||||||||||| |||||||| || ||||| ||| ||||||| ||||||| |||||| ||||||||||||||||| | ||||| || ||||| ||| |||| ||||||| || ||| |||||||| ||||||||| |||| ||||||| || |||||| || ||||||| |||| || ||| |||| ||| |||||||||||| ||| |||||||||||||| |||||||| ||||||||| |||| || ||||||| |||| ||| ||||| ||| ||||| || ||| |||| ||| ||||||||||| |||||| ||||||||||||| ||| |||||||||| ||||||| |||| ||| ||||||||||| || ||| |||||||||||||| |||||||| ||||||||| |||| || ||||||| |||| ||| || |||||| || ||| ||||||||||| ||||| || | ||||||| ||| ||||||| || ||| ||||||||| |||||||| ||||| ||||||||| ||| |||| ||||| ||||||||| ||||| ||||| |||||||| |||| ||| |||| ||||| |||| ||||||||| |||||||||| |||| ||| ||||||| ||||||||||||||||||| |||||||||||| ||||||||| |||||||| |||| ||||||||||| || ||| ||||| ||| ||| ||||||| ||| ||||||||| |||||||| ||| || ||||||||||| ||| |||||||| ||||||||||| |||| |||||
|||||||| || ||||| || ||||| ||| |||| ||||||| || ||| ||||| |||||||| |||||||| |||| ||| ||| |||||| || ||| |||| ||| ||||||||||||| ||| |||| ||||| |||||| || |||| || ||||||| |||| |||||| |||| ||| ||||||||||| ||| ||| |||| ||| | ||||| ||| ||| |||| ||| | |||||| ||||||||||||| ||| |||| ||||| |||||| || |||| || ||||||||| |||||| |||| ||| ||||||||||| ||| ||| ||| ||| | ||||| ||| ||| ||| ||| | |||||| ||||||||||||| |||||||| ||||||||| |||| ||||| || ||||||||| ||||||||||| ||| || |||||||| ||| ||||||||| ||| ||||| |||| ||||||||| |||||||||| |||| ||| ||||||| ||||||||| ||| |||| ||||| |||||| || |||| ||| |||||||| |||||||| || ||| |||||||| || ||||||| |||| |||||| |||| ||| ||||||||||| ||| ||| |||| ||| | ||||| ||| ||| |||| ||| | |||||| ||||||||||||| ||||||| |||| ||||||||| |||||||||| || |||||||| ||| ||||||||| ||| |||||| ||| |||| ||||| |||||| || |||| ||| |||||||| |||||||| || ||| |||||||| || ||||||||| |||||| |||| ||| ||||||||||| ||| ||| ||| ||| | ||||| ||| ||| ||| ||| | |||||| ||||||||||||||||||||| || ||||| || ||||| ||| |||| ||||||| || ||| ||||| ||||||||||||||||| |||||||| |||||||| |||| || ||| |||| ||| ||||||||||||| ||| |||| ||||| ||||||||||||||||| |||||| || |||| || ||||||| |||| |||||| |||| ||| ||||||||||| ||| ||| |||| ||| | ||||| ||| ||| ||| ||| | |||||| ||||||||||||| ||| |||| ||||| ||||||||||||||||| |||||| || |||| || ||||||||| |||||| |||| ||| ||||||||||| ||| ||| ||| ||| | ||||| ||| ||| ||| ||| | |||||| ||||||||||||| |||||||| ||||||||| |||| ||||| || ||||||||| ||||||||||| ||| || |||||||| ||| ||||||||| ||| ||||| |||| ||||||||| |||||||||| |||| ||| |||||||| |||||||||| || ||| ||||||| |||||||||||||||||| || ||||| || ||||| ||| |||| ||||||| || ||||| ||||||||||||||||| ||||||||||||||||| ||||||||||||||||| |||||||||||||||| ||||||| || ||||||||| || || |||| ||| ||||||||| |||||||||||||||| || ||| || |||| |||||||||||||| || ||||||| |||| ||| ||| |||||| ||| ||||||| ||||| || |||||||| ||||||| ||| || |||||||| ||||||| ||| ||||||||||||||||| |||||||||||||||| || ||| |||| ||| |||||||||||| ||||||||||||| ||| |||||| ||| ||||||||| ||||||| | |||||||| |||||| ||| | |||||||| |||||| ||| ||||||||||||||||| |||||||||||||||| || ||| |||| ||| |||||||||||| ||||||||||||| | ||||| || | |||||| ||| | ||||||| |||||| ||| || ||||||||||||||||| ||||||||||||||| |||| ||| || || ||| ||||||||| |||||| ||| |||||||||| || ||| ||||||| |||| ||| ||||||||| ||||||| ||||||||||||| || |||||||| || ||| ||||||||||| ||||| ||||||||||| || ||||||||||||||||| ||||||||||||||| |||| ||| || || ||| |||||||||| ||| ||| |||||| ||||| ||||||||| | |||||||| |||||| || ||| |||||||||| ||| || |||||||| |||||| || ||| ||||||||||| |||||||| || ||||||||| ||||||||||||||| || ||| || |||| |||||||||||||| |||| ||| ||||||| ||||||||||||||| || ||||||| |||| ||| ||||||| || ||||||||||||||| || ||||| || ||||| ||| |||| ||||||| || ||| |||||| || ||||| |||||| ||| |||||||| |||||| ||||||||| |||||| || ||| || |||||||| || ||||||||| || |||| ||| |||| ||| ||||||||||||| |||||||| |||||||||||| ||||||||||| || ||| |||||| |||||| ||| ||||||||||| ||| |||| |||||| |||||| ||||||||| ||||| || |||||||| ||| ||| || ||| |||| ||| | |||||| ||| |||| ||| | ||||||| ||||||||||||| |||||| ||| |||||||||||| ||| |||| |||||| |||||| ||||||||| ||||| || |||||||| ||| ||| || ||| |||| ||| | |||||| ||| |||| ||||||| ||||||||||||| || ||| ||||||||||| ||| |||| |||||||| |||||| ||||||||| ||||| || |||||||| ||| ||| || ||| |||| ||| | |||||| ||| |||| ||| | ||||||| ||||||||||||| || ||| |||||||||||| ||| |||| |||||||| |||||| ||||||||| ||||| || |||||||| ||| ||| || ||| |||| ||| | |||||| ||| |||| ||||||| |||||||||||||||||||||| || ||||| || ||||| ||| |||| ||||||| || ||||| |||||||| || ||| |||| ||| |||||||||||| | ||||| || || |||||||| ||||||| ||| || ||||||| |||||||| || ||| |||| ||| ||||||||||| ||||||| ||||||||||||| |||||||| || ||||| ||| ||||||| |||||| ||| |||| |||||| || ||| ||||||| ||||| ||| |||||||||| || | |||||||| |||||| ||| |||||||| || | |||||||| |||||| || ||| |||||||||||| || |||||||| |||||||||||| ||||| ||| || ||| ||||||| ||||| ||| || |||||| |||| |||||||| || ||| ||||||||||| |||||| | |||||| |||| |||||||| || ||| |||||||||| |||||| | |||| ||| || |||||||||| ||||||| ||||||| ||| |||| ||||||| ||||||||| | ||||| || || |||||| ||| || ||||||| |||||||| || ||| |||| ||| ||||||||||| ||||||| ||||||||||||| |||||||| || ||||| ||| ||||||| ||||||| |||||| |||| |||||| ||||||| ||||||| |||||| |||| ||| ||||||||.
Critical Appraisal
Internal Validity
The interpretation of the efficacy and safety results from both the ENHANCE21 and ADVANCE22 studies may be limited by the pre-post pragmatic (nonrandomized and open-label) study design. To limit any potential confounding variables associated with the setting, each site enrolled patients consecutively into both the pre-period and the post-period and, as a result, the study lacked a concurrent control and patients were not randomized to each arm. In the ENHANCE study, a notably greater proportion of patients in the pre-period had experienced antibiotic treatment failure for the index ABSSSI in the 3 months before study enrolment, and it is possible that this could have biased the results in favour of the post-period. Although both studies attempted to adjust for differences in baseline characteristics with a multivariable model, antibiotic failure was not adjusted for.
The study design may have also introduced the risk for time-related confounders, such as changes between the pre-period and post-period in local antimicrobial resistance patterns, as well as in site- and physician-specific approaches to usual care for the treatment of ABSSSIs. Compared with the post-period, in which site staff were trained on the critical pathway and the post-period protocol, for the pre-period, staff were only trained on the pre-period protocol. As a result, it is uncertain how much of the treatment effect observed in the post-period group was due to the efficacy of dalbavancin and how much was due to the effectiveness of the entire critical pathway.
Of note, treating physicians were not trained on the protocol (e.g., study objectives, exclusion and inclusion criteria, intervention and comparator treatment, and outcomes of interest) for the pre-period to limit performance bias when selecting usual care for the treatment of ABSSSIs. This was particularly important because patients were discharged or sent home from the ER at the discretion of the treating physician during the pre-period and post-period, which may have had an impact on LOS and the hospital admission rate.
Interpretation of the results may be further limited because of missing data. Although the pre-specified outcome results of the analysis for patients who had completed the study were generally consistent with those of the primary analysis, the primary analyses were based on observed cases only. The analyses were conducted using the observed-cases approach and there was a relatively high and unbalanced rate of study withdrawal due to loss to follow-up (8.3% in the pre-period and 20.9% in the post-period) in the ENHANCE study, and it is possible there was bias in favour of the post-period. Interpretation of the results in the ADVANCE study may be further limited because the 104 (68.0%) patients in the post-period group received concomitant therapy with other antibiotics at initial care, thereby removing differences in the treatment received between the pre-period and post-period groups. Finally, it should be noted that all secondary outcomes were exploratory, as the studies21,22 were not powered for secondary outcomes and no adjustments for multiple comparisons were made.
External Validity
To optimize the selection of patients with ABSSSIs that best represented the real world (e.g., patients who are usually admitted to the hospital but are ideal candidates to receive treatment in the outpatient setting), both the ENHANCE21 and ADVANCE22 studies used minimal inclusion and exclusion criteria. Although this would support generalizability to clinical practice, there may be potential differences between the study sites and Canadian practice in the approach to usual care for the treatment of ABSSSI based on guidelines on the management of patients with ABSSSIs in the acute-care setting and in the recommended treatment options or regimens and local antimicrobial resistance patterns. Further, the generalizability of the results to the population of patients with ABSSSI in Canada may be limited, given that all patients were sourced from 1 hospital in the ENHANCE study.
Discussion
Summary of Available Evidence
Results from 4 multinational, phase III, DB, noninferiority RCTs for dalbavancin in patients with ABSSSIs due to known or suspected gram-positive pathogens were included in this review. The primary end point was clinical response 48 to 72 hours after treatment initiation for the 3 pivotal trials (DISCOVER 1, DISCOVER 2, and DUR001-303) and clinical response at day 28 in the VER001-9 study. The DISCOVER 1 (N = 573) and DISCOVER 2 (N = 739) studies compared the efficacy and safety of dalbavancin (given as a 2-dose regimen) to IV vancomycin, with a possible switch to oral linezolid. The DUR001-303 study (N = 698) was designed to determine if a single dose of dalbavancin 1,500 mg was noninferior to a 2-dose regimen of dalbavancin (1,000 mg on day 1 followed by 500 mg on day 8). The VER001-9 study (N = 854) compared the safety and efficacy of the 2-dose regimen of dalbavancin to IV or oral linezolid. The primary objective of the VER001-9 study was to compare the clinical efficacy and safety of dalbavancin with that of IV linezolid (with a possible switch to oral linezolid) in the treatment of adults with cSSSI, which is synonymous with ABSSSI.
One relevant published ITC38 included a systematic review and Bayesian NMA to evaluate the relative clinical efficacy and safety of dalbavancin to other comparators (including daptomycin, linezolid, and vancomycin) for the treatment of ABSSSIs in adults. The efficacy outcomes of interest were clinical treatment success and microbiological success, and the safety outcomes were discontinuation due to AEs, the occurrence of any AEs, the occurrence of any SAEs, and all-cause mortality. In addition, 2 pre-post pragmatic studies included in the sponsor’s submission to CADTH were reviewed, as they were considered to provide more information on the effect of dalbavancin on the outcomes of hospitalization rate and hospital LOS in patients with ABSSSIs.
Interpretation of Results
Efficacy
Overall results in the DISCOVER 1 and DISCOVER 2 studies showed that dalbavancin was noninferior in clinical response 2 to 3 days after treatment initiation to the comparator treatment of vancomycin with a possible switch to oral linezolid. In the VER001-9 study, dalbavancin was noninferior to IV linezolid, with a possible switch to oral linezolid, in clinical response at day 28. In addition, the DUR001-303 study showed that the single-dose dalbavancin regimen was noninferior to the 2-dose dalbavancin regimen (which was used in the DISCOVER 1, DISCOVER 2, and VER001-9 studies) in terms of clinical response 2 to 3 days after treatment initiation. There were no major concerns with internal validity in the studies that would limit the interpretation of the primary outcome results. Although there was no control for multiple outcomes in any of the studies, the results for the other relevant efficacy outcomes were generally supportive of the primary outcome results.
There were some external validity issues in the RCTs that limit the generalizability of the results to clinical practice. According to the clinical expert, the comparators of vancomycin and linezolid did not seem to reflect the standard of care used in Canada. Linezolid is considered a weaker antibiotic (bacteriostatic) and is not commonly used in clinical practice for patients with ABSSSIs. Further, although the proportion of patients with MRSA and MSSA was generally balanced between groups, there seemed to be a slightly higher proportion of MRSA in the dalbavancin group in the DISCOVER 1 and DISCOVER 2 studies, which may have put it at a disadvantage, compared to vancomycin, which is used specifically for MRSA. The clinical expert felt that the difference between dalbavancin and the comparator might be smaller if a more appropriate mix of comparator drugs was used in the control arm, based on presence of MSSA or MRSA (e.g., a cephalosporin for MSSA infections). In addition, vancomycin is typically only used in cases of MRSA and would not be needed in cases where MRSA is not present. Furthermore, the majority of patients (> 80%) were able to obtain a microbiologic diagnosis for their ABSSSI, which, according to the clinical expert, is not reflective of the rate of diagnosis in the real world. Given the invasive nature of needle aspiration and biopsy of closed infections (which represent majority of ABSSSIs), often patients are treated without a microbiologic diagnosis and treatments are adjusted on a dose-response basis.
The clinical expert emphasized that the convenience of dalbavancin, with its 1- or 2-dose regimen, makes it an attractive and potentially more accessible treatment option for ABSSSIs than other IV antibiotic treatments (but not oral antibiotics). This could translate into improved treatment adherence, the elimination of multiple reassessments, a reduction in the need to travel to the ER or outpatient clinic (or home visits), and a reduction in demand for outpatient resources. The expert also suggested that the single-dose regimen would be the preferred regimen. However, the less frequent dosing of dalbavancin also makes it less suitable under some circumstances, as it could increase unnecessary treatment in patients who receive an MSSA diagnosis after treatment initiation and increase IV treatment when oral antibiotics would be adequate.
The sponsor-submitted ITC reported that there was no evidence of a difference in clinical treatment success between dalbavancin and vancomycin, daptomycin, and linezolid. However, estimates showed wide CrIs. Given the limitations (e.g., heterogeneity due to variations in study duration, blinding, dosage, disease definition, and patient demographics and, for some contrasts and outcomes, sparse data), CADTH’s conclusions are that the results from the sponsor-submitted ITC are uncertain and at risk of bias.
Results from the pre-post pragmatic trials showed that dalbavancin was associated with a shorter LOS and lower hospital admission rate at initial care, compared to standard of care. However, several limitations of these studies were inherent in the nonrandomized, open-label study design. The study design was prone to risk of selection and performance bias, given that the patients were discharged at the discretion of the treating physician during the pre-period and post-period. Other limitations included a lack of a concurrent comparator, lack of randomization, time-related confounders, missing data, and attrition bias. Generalizability is limited because of the small sample size and limited number of sites. The risk of bias may have had an impact on the LOS and hospital admission rate in favour of IV dalbavancin; however, the magnitude of this bias could not be inferred. Hence, the results should be interpreted with caution.
Harms
Overall, dalbavancin was safe and well tolerated across the pivotal trials. In the DISCOVER 1 and DISCOVER 2 studies, there were fewer TEAEs in the dalbavancin treatment group than in the vancomycin treatment group, and similar numbers of SAEs and deaths (1 and 2, respectively) were reported. According to the clinical expert, given the renal toxicity posed by vancomycin, dalbavancin had a better safety profile in comparison. Overall, the clinical expert felt that dalbavancin had a safety profile similar to standard therapies encountered in Canadian practice. A similar safety profile was observed between the single-dose and 2-dose dalbavancin regimens. In the VER001-9 study, there were no notable differences between dalbavancin and linezolid. One concern raised by the clinical expert was that because dalbavancin is long-acting, any allergic reactions cannot be mitigated by withdrawing treatment. In the dalbavancin group of the DISCOVER 1 study, 0.4% of patients reported infusion-related reactions, and in the dalbavancin group of the DISCOVER 1, DISCOVER 2, and DUR001-303 studies, 0.3% to 2.1% reported rash or hypersensitivity reactions.
The sponsor-submitted ITC reported that dalbavancin is associated with a lower likelihood of AEs than linezolid, a lower likelihood of SAEs than vancomycin and daptomycin, and a lower risk of all-cause mortality than vancomycin and linezolid. For the safety outcomes, given the magnitude of the difference in results and the limitations of these results, reported significant improvements with dalbavancin compared with certain comparators are subject to bias and uncertainty and, therefore, are not reliable.
Conclusions
In patients with ABSSSIs, dalbavancin demonstrated noninferiority to vancomycin (with a possible switch to oral linezolid) and IV linezolid (also with a possible switch to oral linezolid), with similar rates of clinical response and clinical success. The dalbavancin single-dose treatment regimen was shown to be noninferior to the 2-dose treatment regimen. A key limitation of these trials was that the comparators did not fully reflect the standard of care typically used in Canadian practice for patients with ABSSSIs, as identified by the clinical expert, as there were no comparators specific to the treatment of MSSA infections, which may reduce the generalizability of the findings. The ITC compared dalbavancin with vancomycin, linezolid, and daptomycin, but the efficacy and safety results were uncertain because of heterogeneity across trials and sparse safety data. The pragmatic trials showed an improvement in total infection-related LOS (the ENHANCE study) and hospital admission rate at initial care (the ADVANCE study) after the introduction of a dalbavancin care pathway; however, the pre-post study design and use of a pathway intervention limits interpretation. The AEs reported in the RCTs did not give rise to any safety concerns (for the single-dose or 2-dose regimen) and the safety profile of dalbavancin was similar to and possibly better than that of vancomycin and linezolid for the treatment of ABSSSIs.
References
1.Guidance for industry. Acute bacterial skin and skin structure infections: developing drugs for treatment. Silver Spring (MD): U.S. Food and Drug Administration (FDA); 2013: https://www.fda.gov/files/drugs/published/Acute-Bacterial-Skin-and-Skin-Structure-Infections---Developing-Drugs-for-Treatment.pdf Accessed 2022 Jul 28.
2.Pulido-Cejudo A, Guzman-Gutierrez M, Jalife-Montano A, et al. Management of acute bacterial skin and skin structure infections with a focus on patients at high risk of treatment failure. Ther Adv Infect Dis. 2017;4(5):143-161. PubMed
3.Ramakrishnan K, Salinas RC, Agudelo Higuita NI. Skin and soft tissue infections. Am Fam Physician. 2015;92(6):474-483. PubMed
4.Soriano A, Rossolini GM, Pea F. The role of dalbavancin in the treatment of acute bacterial skin and skin structure infections (ABSSSIs). Expert Rev Anti Infect Ther. 2020;18(5):415-422. PubMed
5.Sofia Antao H, Guimaraes JP, Froes F, Almeida J, Marques S. Dalbavancin: a new pathway for the treatment of acute bacterial skin and skin structure infections in Portugal. Health Econ Outcome Res. 2017;03(03).
6.Bridgman AC, Fitzmaurice C, Dellavalle RP, et al. Canadian burden of skin disease from 1990 to 2017: results from the global burden of disease 2017 study [formula: refer to text]. J Cutan Med Surg. 2020;24(2):161-173. PubMed
7.Jeong D, Nguyen HNT, Tyndall M, Schreiber YS. Antibiotic use among twelve Canadian First Nations communities: a retrospective chart review of skin and soft tissue infections. BMC Infect Dis. 2020;20(1):118. PubMed
8.Stevens DL, Bisno AL, Chambers HF, et al. Practice guidelines for the diagnosis and management of skin and soft tissue infections: 2014 update by the Infectious Diseases Society of America. Clin Infect Dis. 2014;59(2):e10-52. PubMed
9.Scott LJ. Dalbavancin: a review in acute bacterial skin and skin structure infections. Drugs. 2015;75(11):1281-1291. PubMed
10.Kaye KS, Petty LA, Shorr AF, Zilberberg MD. Current epidemiology, etiology, and burden of acute skin infections in the United States. Clin Infect Dis. 2019;68(Suppl 3):S193-S199. PubMed
11.Potashman MH, Stokes M, Liu J, Lawrence R, Harris L. Examination of hospital length of stay in Canada among patients with acute bacterial skin and skin structure infection caused by methicillin-resistant Staphylococcus aureus. Infect Drug Resist. 2016;9:19-33. PubMed
12.Schrock JW, Laskey S, Cydulka RK. Predicting observation unit treatment failures in patients with skin and soft tissue infections. Int J Emerg Med. 2008;1(2):85-90. PubMed
13.Walsh TL, Chan L, Konopka CI, et al. Appropriateness of antibiotic management of uncomplicated skin and soft tissue infections in hospitalized adult patients. BMC Infect Dis. 2016;16(1):721. PubMed
14.Eells SJ, Nguyen M, Jung J, Macias-Gil R, May L, Miller LG. Relationship between adherence to oral antibiotics and postdischarge clinical outcomes among patients hospitalized with Staphylococcus aureus Skin Infections. Antimicrob Agents Chemother. 2016;60(5):2941-2948. PubMed
15.Mascitti KB, Gerber JS, Zaoutis TE, Barton TD, Lautenbach E. Preferred treatment and prevention strategies for recurrent community-associated methicillin-resistant Staphylococcus aureus skin and soft-tissue infections: a survey of adult and pediatric providers. Am J Infect Control. 2010;38(4):324-328. PubMed
16.Xydalba (dalbavancin for Iinjection): 500 mg dalbavancin (as dalbavancin hydrochloride)/ vial lyophilized powder for solution, intravenous antibacterial agent [product monograph]. St-Laurent (QC): Paladin Labs Inc; 2021 Apr 30.
17.Clinical Study Report: DUR001-301. A phase 3, randomized, double-blind, double-dummy study to compare the efficacy and safety of dalbavancin to a comparator regimen (vancomycin and linezolid) for the treatment of acute bacterial skin and skin structure infections [internal sponsor's report]. Chicago (IL): Durata Therapeutics, Inc; 2013 Sep 4.
18.Clinical Study Report: DUR001-302. A phase 3, randomized, double-blind, double-dummy study to compare the efficacy and safety of dalbavancin to a comparator regimen (vancomycin and linezolid) for the treatment of acute bacterial skin and skin structure infections [internal sponsor's report]. Chicago (IL): Durata Therapeutics, Inc; 2013 Sep 4.
19.Clinical Study Report: DUR001-303. A phase 3b, double-blind, multicenter, randomized study to compare the efficacy and safety of single dose dalbavancin to a two dose regimen of dalbavancin for the treatment of acute bacterial skin and skin structure infections [internal sponsor's report]. Amsterdam (NL): Durata Therapeutics International B.V; 2015 Jun 26.
20.Clinical Study Report: VER001-9. Phase 3, randomized, double-blind, multi-center study to evaluate the safety and efficacy of dalbavancin versus linezolid in the treatment of complicated skin and soft tissue infections with suspected or confirmed gram-positive bacterial pathogens[internal sponsor's report]. King of Prussia (PA): Vicuron Pharmaceuticals Inc; 2004 Sep 30.
21.Clinical Study Report: Protocol CMO-US-ID 0528. Development of a new critical pathway for treatment of acute bacterial skin and skin structure infections - ENHANCE. V2.0 (amendment) [internal sponsor's report]. Irvine (CA): Allergan; 2019 Dec 4.
22.Clinical Study Report: Protocol CMO-US-ID-0476. A pragmatic trial designed to evaluate a new critical pathway for treatment of patients with acute bacterial skin and skin structure infections - ADVANCE. V2.0 (amendment) [internal sponsor's report]. Irvine (CA): Allergan; 2020 Apr 28.
23.Esposito S, Leone S, Petta E, Noviello S, Ianniello F. Treatment options for skin and soft tissue infections caused by meticillin-resistant Staphylococcus aureus: oral vs. parenteral; home vs. hospital. Int J Antimicrob Agents. 2009;34:S30-S35. PubMed
24.Baibergenova A, Drucker AM, Shear NH. Hospitalizations for cellulitis in Canada: a database study. J Cutan Med Surg. 2014;18(1):33-37. PubMed
25.When antibiotics fail. The Expert panel on the potential socio-economic impacts of antimicrobial resistance in Canada Ottawa (ON): Council of Canadian Academies; 2019: https://cca-reports.ca/wp-content/uploads/2018/10/When-Antibiotics-Fail-1.pdf. Accessed 2022 Jul 25.
26.Vancomycin Hydrochloride for injection (vancomycin hydrochloride):500 mg, 1 g, 5 g and 10 g of vancomycin (as vancomycin hydrochloride) per vial, sterile lyophilized powder for solution [product monograph]. Kirkland (QC): Pfizer Canada ULC; 2021 Jul 21: https://www.pfizer.ca/sites/default/files/202108/Vancomycin_Hydrochloride_PM_EN_249989_21-July-2021.pdf. Accessed 2022 Jul 7.
27.Zyvoxam (linezolid): solution, 2 mg/mL, for intravenous infusion, powder for suspension, 100 mg/5mL when reconstituted, for oral use [product monograph]. Kirkland (QC): Pfizer Canada ULC; 2022 Jan 27: https://www.pfizer.ca/sites/default/files/202203/ZYVOXAM_PM_EN_256427_27-Jan-2022.pdf. Accessed 2022 Jul 8.
28.Keflex (cephalexin): tablets and oral suspensions, 250 mg, 500 mg, 125 mg/5 mL and 250 mg/5 mL [product monograph]. Montreal (QC): Pendopharm, Division of Pharmascience Inc; 2018 May 1: https://pdf.hres.ca/dpd_pm/00045523.PDF. Accessed 2022 Jul 8.
29.Cefazolin for injection USP (cefazolin as cefazolin sodium): 500 mg, 1 g & 10 g per vials [product monograph]. Boucherville (QC): Sandoz Canada Inc; 2010 Nov 15: https://www.sandoz.ca/sites/www.sandoz.ca/files/Cefazolin%20Monograph.pdf. Accessed 2022 Jul 8.
30.Teva-Cloxacillin (cloxacillin sodium): 250 and 500 mg capsules, 125 mg/5 ml granules for oral solution [product monograph]. Toronto (ON): Teva Canada Limited; 2018 Jun 21: https://pdf.hres.ca/dpd_pm/00046029.PDF. Accessed 2022 Jul 8.
31.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. J Clin Epidemiol. 2016;75:40-46. PubMed
32.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2022 May 2.
33.Boucher HW, Wilcox M, Talbot GH, Puttagunta S, Das AF, Dunne MW. Once-weekly dalbavancin versus daily conventional therapy for skin infection. N Engl J Med. 2014;370(23):2169-2179. PubMed
34.Dunne MW, Puttagunta S, Giordano P, Krievins D, Zelasky M, Baldassarre J. A randomized clinical trial of single-dose versus weekly dalbavancin for treatment of acute bacterial skin and skin structure infection. Clin Infect Dis. 2016;62(5):545-551. PubMed
35.Jauregui LE, Babazadeh S, Seltzer E, et al. Randomized, double-blind comparison of once-weekly dalbavancin versus twice-daily linezolid therapy for the treatment of complicated skin and skin structure infections. Clin Infect Dis. 2005;41(10):1407-1415. PubMed
36.Farrington CP, Manning G. Test statistics and sample size formulae for comparative binomial trials with null hypothesis of non-zero risk difference or non-unity relative risk. Stat Med. 1990;9(12):1447-1454. PubMed
37.Miettinen O, Nurminen M. Comparative analysis of two rates. Stat Med. 1985;4(2):213-226. PubMed
38.Guest JF, Esteban J, Manganelli AG, Novelli A, Rizzardini G, Serra M. Comparative efficacy and safety of antibiotics used to treat acute bacterial skin and skin structure infections: results of a network meta-analysis. PLoS One. 2017;12(11):e0187792. PubMed
39.Agarwal R, Bartsch SM, Kelly BJ, et al. Newer glycopeptide antibiotics for treatment of complicated skin and soft tissue infections: systematic review, network meta-analysis and cost analysis. Clin Microbiol Infect. 2018;24(4):361-368. PubMed
40.Vlachaki I, Vacchelli M, Zinzi D, et al. Comparative efficacy of delafloxacin for complicated and acute bacterial skin and skin structure infections: results from a network meta-analysis. BMC Infect Dis. 2021;21(1):1036. PubMed
41.McCarthy MW, Keyloun KR, Gillard P, et al. Dalbavancin reduces hospital stay and improves productivity for patients with acute bacterial skin and skin structure infections: the ENHANCE trial. Infect. 2020;9(1):53-67. PubMed
42.Talan DA, Mower WR, Lovecchio FA, et al. Pathway with single-dose long-acting intravenous antibiotic reduces emergency department hospitalizations of patients with skin infections. Acad Emerg Med. 2021;28(10):1108-1117. PubMed
Appendix 1: Literature Search Strategy
Note that this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases:
MEDLINE All (1946 to present)
Embase (1974 to present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: May 13, 2022
Alerts: Bi-weekly search updates until project completion
Search filters applied: No filters were applied to limit the retrieval by study type.
Limits:
No date or language limits were used.
Conference abstracts: excluded.
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
MeSH | Medical Subject Heading |
.fs | Floating subheading |
exp | Explode a subject heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Author keyword heading word (MEDLINE) |
.dq | Candidate term word (Embase) |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multidatabase Strategy
(Dalbavancin* or Xydalba* or Dalvance* or Exulett* or bi 397 or bi397 B 397 or B397 or Bi 387 or Bi387 or MDL-63397 or MDL63397 or MDL-63,397 or MDL63,397 or MDL-64397 or MDL64397 or MDL-64,397 or MDL64,397 or VER-001 or VER001 or A-A-1 or Zeven or 808UI9MS5K or B0U42518WL or 33WDQ7T81E).ti,ab,kf,ot,hw,rn,nm.
exp Skin Diseases, Bacterial/ or exp Staphylococcus/ or exp Methicillin-Resistant Staphylococcus aureus/ or exp Staphylococcal skin Infections/ or soft tissue infections/
((acute bacterial adj34 skin) or (complicated adj4 skin adj4 infection*)).ti,ab,kf.
(ABSSSI* or cSSSI* or cSSTI* or cellulitis* or erysipelas* or wound infection* or cutaneous abscess* or Staphylococcus* or staphylococcal* or MRSA*).ti,ab,kf.
or/2-4
1 and 5
6 use medall
* dalbavancin/
(Dalbavancin* or Xydalba* or Dalvance* or Exulett* or bi 397 or bi397 or B 397 or B397 or Bi 387 or Bi387 or MDL-63397 or MDL63397 or MDL-63,397 or MDL63,397 or MDL-64397 or MDL64397 or MDL-64,397 or MDL64,397 or VER-001 or VER001 or A-A-1 or Zeven).ti,ab,kf,dq.
8 or 9
exp bacterial skin disease/ or exp Staphylococcus/ or exp methicillin-resistant Staphylococcus aureus/ or soft tissue infection/ or exp staphylococcal skin infection/
((acute bacterial adj34 skin) or (complicated adj4 skin adj4 infection*)).ti,ab,kf.
(ABSSSI* or cSSSI* or cSSTI* or cellulitis* or erysipelas* or wound infection* or cutaneous abscess* or Staphylococcus* or staphylococcal* or MRSA*).ti,ab,kf.
or/11-13
10 and 14
15 use oemezd
16 not (conference review or conference abstract).pt.
7 or 17
remove duplicates from 18
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
[Search -- Studies with results | Xydalba or dalbavancin or Dalvance]
WHO ICTRP
International Clinical Trials Registry Platform, produced by the WHO. Targeted search used to capture registered clinical trials.
[Search terms -- Xydalba or dalbavancin or Dalvance]
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
[Search terms -- Xydalba or dalbavancin or Dalvance]
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
[Search terms -- Xydalba or dalbavancin or Dalvance]
Grey Literature
Search dates: May 2, 2022, to May 6, 2022
Keywords: Xydalba, dalbavancin, Dalvance, acute bacterial skin and skin structure infections
Limits: None
Updated: Search updated before the completion of stakeholder feedback period
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Health Statistics
Internet Search
Appendix 2: Excluded Studies
Note that this appendix has not been copy-edited.
Reference | Reason for exclusion |
|---|---|
McCarthy MW, Keyloun KR, Gillard P, et al. Dalbavancin Reduces Hospital Stay and Improves Productivity for Patients with Acute Bacterial Skin and Skin Structure Infections: The ENHANCE Trial. Infect Dis Ther. 2020;9(1):53 to 67. | Study design |
Talan DA, Mower WR, Lovecchio FA, et al. Pathway with single-dose long-acting IV antibiotic reduces emergency department hospitalizations of patients with skin infections. Acad Emerg Med. 2021;28(10):1108 to 1117. | Study design |
Appendix 3: Details of the ENHANCE Study
Note that this appendix has not been copy-edited.
Table 29: Summary of Baseline Characteristics in the ENHANCE Study (FAS Population)
Characteristics | ENHANCE | |
|---|---|---|
Pre-period (N = 48) | Post-period (N = 42) | |
Demographics | ||
Age (years) | ||
Mean (SD) | 59.0 (21.79) | 53.4 (21.52) |
Sex, n (%) | ||
Male | 24 (50.0) | 22 (52.4) |
Female | 24 (50.0) | 20 (47.6) |
Race, n (%) | ||
White | 36 (75.0) | 28 (66.7) |
Black or African American | 1 (2.1) | 7 (16.7) |
Asian | 1 (2.1) | 1 (2.4) |
Native Hawaiian or Other Pacific Islander | 0 | 1 (2.4) |
American Indian or Alaska Native | 0 | 0 |
Other | 0 | 0 |
Patient did not disclose | 10 (20.8) | 5 (11.9) |
BMI (kg/m2) | ||
n | 47 | 38 |
Mean (SD) | 30.2 (8.48) | 31.8 (12.38) |
|||||||||| ||||||||| |||||||| | ||
||| | |||| | |||| |
|||| |||| | ||||| ||||||| | ||||| ||||||| |
Relevant comorbid conditions and medical history, n (%)b | ||
Solid tumour | 13 (27.1) | 7 (16.7) |
Diabetes mellitus | 5 (10.4) | 6 (14.3) |
Moderate to severe chronic kidney disease | 10 (20.8) | 4 (9.5) |
Drug abuse (non-alcohol)a | 5 (10.4) | 3 (7.1) |
Illicit needle use | 4 (80.0) | 2 (66.7) |
Lymphedema/chronic venous stasis | 3 (6.3) | 3 (7.1) |
Connective tissue disease | 1 (2.1) | 2 (4.8) |
Peripheral vascular disease | 2 (4.2) | 2 (4.8) |
Alcohol abuse | 0 | 1 (2.4) |
AIDS (not just HIV positive) | 0 | 0 |
Leukemia | 1 (2.1) | 0 |
Liver disease | 4 (8.3) | 0 |
Malignant lymphoma | 0 | 0 |
Immunocompromising conditions | ||
Receiving any immune modulating medication including biologics other than TNF | 4 (8.3) | 5 (11.9) |
HIV (excluding AIDS) | 1 (2.1) | 2 (4.8) |
||||||||| || || |||||||||| ||||| ||| ||||||||||| |||||||||| |||||||| | | ||||| | | ||||| |
Organ transplant recipient | 2 (4.2) | 1 (2.4) |
Receiving chemotherapy | 2 (4.2) | 1 (2.4) |
Receiving TNF inhibitors | 0 | 0 |
|||||||| |||||||||| |||||||| ||||||||||| || ||| |||||||| | ||||||| | ||| | ||
||||||||||| ||||| |||||| | |||||| |||||| ||||| |||||||||| | | || |||||| | || ||||||| |
|||||||||| ||| ||||||||| || ||||| |||||| | || |||||| | || |||||| |
|||||||||| ||| ||||||||| || ||||||| |||||| | ||| | ||| |
|||||||||| ||| ||||||||| || ||||| ||||||||| || |||||||| ||||||||| | | ||||| | | ||||| |
|||||||||| ||||||||| ||||||| |||||| | |||||| |||||| ||||| |||||||||| || | | || |||||| | || |||||| |
|||||||||| ||| ||||||||| || ||||| |||||| | || |||||| | || ||||||| |
|||||||||| ||| ||||||||| || ||||||| |||||| | ||| | ||| |
|||||||||| ||| ||||||||| || ||||| ||||||||| || |||||||| ||||||||| | | ||||| | ||| |
||||| |||| ||| |||||||||| |||||||||| | ||| | || |||||| | | |||||| |
Infection and clinical characteristics | ||
Infection type, n (%)b | ||
Cellulitis/Erysipelas | 41 (85.4) | 37 (88.1) |
Wound Infection | 8 (16.7) | 4 (9.5) |
Abscess | 1 (2.1) | 3 (7.1) |
Purulent drainage from primary lesion, n (%) | ||
Yes | 13 (27.1) | 9 (21.4) |
No | 35 (72.9) | 33 (78.6) |
Primary lesion size (cm2) | ||
Mean (SD) | 533.5 (647.94) | 685.0 (703.53) |
Median | 255.0 | 458.0 |
Min, Max | 12.0 to 3,600.0 | 30.0 to 2,625.0 |
Fever, n (%)d | 4 (8.3) | 1 (2.4) |
SIRS criteria met, n (%)e | 11 (22.9) | 8 (19.0) |
Recurrent/ABSSSI infection in prior 6 months, n (%) | 1 (2.1) | 2 (4.8) |
|||||||||| |||||| | ||
||||| |||||||||| ||||| |||||||| ||||| ||| ||||| | ||| | ||
|||||||| | ||| | ||| |
|||||||| | ||| | | ||||| |
||| |||| | || ||||||| | || |||||| |
ABSSSI = acute bacterial skin and skin structure infection; BMI = body mass index; FAS = full analysis set; MRSA = methicillin-resistant Staphylococcus aureus; SD = standard deviation; SIRS = systemic inflammatory response syndrome.
| |||| || |||| ||| ||| |||| || ||| ||||||||||| ||| ||| |||||||||| |||||||| || ||| |||||||||| ||||||||||||||
bPatients can contribute to multiple categories; hence, percentage may not sum to 100.
| |||||||||| ||||||||| ||||||| ||| ||||||| || ||||||||||||||| ||| || ||||||||| || ||||||||| |||||| || ||| || || ||||||| ||||| ||||||| || ||| |||||||||| |||||||| || ||| |||| |||||| |||||
dFever was defined as a reported temperature of greater than 38°C.
eMeeting SIRS criteria was defined as having 2 or more of the following: temperature < 36°C or > 38°C, heart rate > 90 beats per minute, respiratory rate > 20 breaths per minute or arterial carbon dioxide tension < 32 mm hg, or white blood cell count < 4,000 cells/µL or > 12,000 cells/µL or > 10% bands.
Source: Clinical Study Report for ENHANCE.21
Table 30: Summary of Patient Disposition in the ENHANCE Study
Patient disposition | ENHANCE | |
|---|---|---|
Pre-period | Post-period | |
Screened, n | 112 | 131 |
Enrolled, n | 48 | 43a |
Completed, n (%) | 42 (87.5) | 33 (76.7) |
Withdrawn, n (%) | 6 (12.5) | 10 (23.3) |
Lost to follow-up | 4 (8.3) | 9 (20.9) |
Withdrawal of consent by patient | 2 (4.2) | 1 (2.3) |
FAS population, Nb | 48 | 42 |
FAS = full analysis set.
aOne patient was excluded because treatment with dalbavancin was not recorded after enrolment.
bFull analysis set included patients in the enrolled set who received at least 1 dose of an antibiotic in the pre-period and at least one dose of dalbavancin in the post-period.
Source: Clinical Study Report for ENHANCE.21
|||||||| || | ||||||| | |
|---|---|---|
||||||||||||| | ||| | |||||||||||||| | ||| | |
||||||| |||||||||| || |||||||| |||| || |||||||||| ||||||| || ||| |||| ||| ||||||||||| ||| |||||||| |||||||||||| ||||| | ||
|||| | ||| | ||| | | || |||||| |
||| | ||| | | || || | || |||||| |
||||||| |||||||| |||||||||| ||||||||||||||||||||| ||| || |||||| | ||| | | || |||||| |
||||||| |||||||| |||||||||| ||||||||||||||||||||| ||| || ||||| ||||||| | ||| | | | |||||| |
|||||||||| |||||||| ||| ||||||||| || ||||||||| ||||||||| | ||| | | | ||||| |
||||| ||||| ||| ||||||||| || |||||||||| ||||||||| | ||| | | || | |
||||| | | ||| | | | ||||| |
|||||| || |||||||||| |||||||| |||||| |||||| | ||| | ||
|| | | ||| | | ||| | |
| || | || |||||| | || |||||| |
|| | | || |||||| | | ||||| |
|| | | || |||||| | | ||||| |
| || | | | |||||| | | ||||| |
||||| || ||||||||||||||| | ||| | ||
||||||||||| | || |||||| | || ||||||| |
|| ||||| | || |||||| | | ||||| |
||||| |||| |||||||| |||||||||||||| ||||||||||||| ||||||| | | ||||| | || | |
||||||| |||||||||| |||||||||| | ||| | | ||
||||||||||| | || | | || ||||||| | |
|||||||||| | || |||||| | || | |
|||||||||| | || |||||| | | ||||| |
||||||||||| | || |||||| | | ||||| |
||||||| || | | |||||| | || | |
||||||||| | | |||||| | || | |
||||||| | | |||||| | || | |
||||||||||| | | |||||| | || | |
|||||| | | |||||| | || | |
|||||||||||| | | |||||| | || | |
||||| | | |||||| | || | |
||||||||||| ||||||||||| | | ||||| | | ||||| |
|||||||||||| |||||||||| | || | | | ||||| |
|||||| ||| ||||||| |||||||||| |||||||||||||||| | ||| | | ||
||||||||| ||||||| |||||| || ||||||| ||| |||||| | || |||||| | || ||||||| |
|| ||||||| ||||| ||||||| || ||| |||||||||| | | ||||| | | |||||| | |
||||||||| || ||||||||| |||||| | | |||||| | | ||| |
||||||| |||||||| | | ||||| | | ||| |
||||| | | ||||| | | ||| |
|||||||||| |||||||| ||||| ||||||||||||||| || ||||||| ||||||||||| | ||| | | ||
||| || | || | | | ||||| |
|| || | || ||||||| | || |||||| |
|||||| | ||||| ||||||||| |||| ||| |||| ||||||||| |||||||||| ||| | |||| |||||||| |||| || | ||| |||||||||||| |||| || |||| ||| ||| |||| || ||| ||||||||||| ||| ||| |||||||||| |||||||| || ||| |||||||||| |||||||||||||||| ||| ||||||| |||||| ||||||||| ||| || |||||||| | ||||| ||||| ||||||||| |||||||||||| || ||| || |||| |||||||| |||| ||||||||||| ||||| || ||| ||||||||||| ||| ||||||||| |||||||||||||| ||||| |||||||| |||||||| |||| ||||||||||| ||||||||||||| |||| |||||||||||| ||||||||||| ||| ||||||||||| ||| |||||||||||| |||||||||| || | ||| |||||||||| || | ||| ||| ||||||||||| || | ||| || |||||||| ||| |||||||||| || |||| |||| | ||||||||| ||| ||||||| |||||||||||| ||||||||||| ||| || |||||||| ||||||||| ||||| ||||||||| | |||| || |||| || |||||||||||| || |||||||| ||| |||| |||||||| |||||||| |||||||||| ||||||||| ||||||| |||||||||||||||||| |||||||| ||||| |||||| ||| |||||||||
||||||||||| |||||||| || |||||||||||| ||||||||| |||||| |||| || || |||||||| ||| ||| | ||||||||| |||||||||| |||| |||| | |||||||||||| ||||| || |||| |||| |||||||| ||||| |||| || |||||||| |||||| ||||||| ||| |||| ||| |||||||||||| |||| ||| |||||| ||| ||||||||||| |||| || ||||||| |||||||| ||| || | |||| |||| | || || | ||||| ||| | ||||||||| || ||| ||||||| |||||| || | ||||| ||||| || ||||| |||||||||||| || ||| ||||||||| |||| || |||||||| ||||| || |||||||| || |||| ||| || ||||||||||| | |||| ||||||||| || ||| || | ||||| ||||| ||||||||| ||| ||||||||| ||| |||||||||| || ||||||| ||||||||| ||| ||||| ||||||||| ||| || |||||||| |||| || |||||||| || |||| ||||||| |||| |||| |||||||| ||| ||||||||| || ||| |||||||| |||||||| ||| |||||||| || ||||| | |||| || || |||||||||| || ||| |||||||||| ||| || ||||| | |||| || ||||||||||| || ||| |||||||||||| ||||||||||| |||||||| |||| ||||||||| || ||||||||| |||||||||| ||| ||||||||||| |||||||||| ||||||||||| |||| |||| ||||||| ||| |||| ||| ||||||||||| |||||| || ||| ||||||||||| ||| ||| |||||||||| ||||||||||| ||||||||| || |||||||| ||||||||||||||| ||||| |||||||||| || |||||||| ||||| |||||| ||||||||||| ||| ||| ||||||||||||| ||| || |||| |||||||| ||||| ||||||||||| ||||||||||| |||| |||| ||||||| ||| |||| ||| ||||||||||| |||||| || ||| ||||||||||| |||||||||| ||| ||||||||||| ||| |||||||||| |||||||||| ||||||||| ||||| ||||||||| ||||||||| ||||||| || |||||||||||| | |||||| ||||||||||| ||| ||| ||||||||||||| ||| || |||| |||||||| ||||| ||||||||||| | |||||||||||| ||||| || |||| ||| |||| ||| ||| ||||||||||| |||||||| || ||||| ||| ||||||||| |||||||| |||| ||||||||||| || ||| ||||| ||| ||| ||||||| ||| ||||||||| |||||||| ||| || ||||||||||| ||| |||||||| ||||||||||| |||| ||||| |||||||| |||||||| |||| ||||||||| ||||| || ||| |||||||| |||| ||||||||| | ||||||||| |||||||| ||||| | ||||||||||||| |||||||||| |||||||| |||||||||||| |||||| ||||| |||||| ||| ||||||||| ||| ||| ||||||| |||||||| | |||||||| ||||||||| |||| |||||||||| ||||||||| ||| | ||||||||||| |||||||||||| ||||| || |||| |||| |||| || ||||||||| ||| |||||||||| |||||||| |||||||| ||||||||||||||| ||| ||| |||||||| |||||||||
Table 32: Infection-Related Total LOS in the ENHANCE Study (FAS Population)
Infection-related total LOS | ENHANCE | |||||
|---|---|---|---|---|---|---|
Initial Care (1 to 14 days) | Follow-up (15 to 44 days) | Entire Study Duration (44 days) | ||||
Pre-period (N = 48) | Post-period (N = 42) | Pre-period (N = 48) | Post-period (N = 42) | Pre-period (N = 48) | Post-period (N = 42) | |
Total infection-related LOS (inpatient only), days | ||||||
Mean (SD) | 4.8 (2.55) | 2.8 (1.38) | 0.0 (0.00) | 0.4 (1.78)a | 4.8 (2.55) | 3.2 (2.48) |
Mean difference (95% CI) | — | — | — | — | 1.6 (0.6 to 2.6) | |
Unadjusted P-value | — | — | — | — | 0.003 | |
Adjusted P-value | — | — | — | — | 0.002 | |
||||| ||||||||||||||||| ||| |||||||||| |||| ||||| || ||||||||| ||||||||||| |||||| ||| |||| | ||||||
|||| |||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | | ||| |||||| | ||| |||||| |
|||| |||||||||| |||| ||| | ||| | ||| | |||| | |||| | ||| |||| || |||| | |
|||||||||| ||||||| | | ||| | |||| | |||| | |||| | ||||| | |
CI = confidence interval; FAS = full analysis set; LOS = length of stay; SD = standard deviation; SE = standard error.
aDuring follow-up, 2 patients in the post-period contributed to the infection-related total length of stay; hospitalized for 10 days due to worsening of index ABSSSI in which the patient presented with severe refractory cellulitis (n = 1) and hospitalized for 6 days due to worsening of index ABSSSI in which the patient presented with moderate cellulitis (n = 1).
| |||||||| |||| |||||||| ||||| ||||||||||| |||||| || ||| |||| ||| ||||||| |||| | |||| || | |||||| ||| ||||||||||| |||| || ||||| ||| || ||||||| ||| |||| | ||||| |||||
Analysis: Unadjusted P values were from a 2-sided, 2 sample, equal variance t-test.
Multivariable analysis: Adjusted P values were from the generalized Linear Mixed Model with treatment cohort, age, and immunocompromised status as fixed effects and patient as a random effect. Immunocompromised at baseline was defined as: connective tissue disease, diabetes mellitus, leukemia, or malignant lymphoma.
Note: Infection-related hospital length of stay was a subset of all-cause hospitalization time for infection-related hospitalization stays. For all-cause hospitalizations, length of stay was the sum of all hospitalizations, calculated as the difference between hospitalization end date and start date, plus 1. If hospitalization was ongoing at the end of study or discontinuation date, the study completion date or discontinuation date was used as the end date.
Source: Clinical Study Report for ENHANCE.21
Total LOS | ||||||| | |||||
|---|---|---|---|---|---|---|
||||||| ||||||| || || ||||| | ||||||||| |||| || || ||||| | |||||| ||||| |||||||||||| ||||| | ||||
||||||||||||| | ||| | |||||||||||||| | ||| | ||||||||||||| | ||| | |||||||||||||| | ||| | ||||||||||||| | ||| | |||||||||||||| | ||| | |
||||| ||| |||||||||| |||||| |||| | ||||||
|||| |||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| |
|||| |||||||||| |||| ||| | |||| | |||| | ||||| | ||||| | ||| |||| || |||| | |
|||||||||| ||||||| | |||| | ||||| | ||||| | ||||| | ||||| | |
||||| ||| |||||||||| |||| ||||| || ||||||||| ||||||||||| |||||| ||| |||| | ||||||
|||| |||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| |
|||| |||||||||| |||| ||| | |||| | ||||| | ||||| | ||||| | ||| |||| || |||| | |
|||||||||| ||||||| | |||| | ||||| | ||||| | ||||| | ||||| | |
||| |||||||||| ||||||||| ||| | |||| |||||||| |||| ||| | |||||| || ||||| || | |||||||| |||||||||||| |||||||| |||| |||||||| ||||| ||||||||||| |||||| || ||| |||| ||| ||||||| |||| | |||| |||||||||| |||||||| ||| |||||||||| ||||||||| ||| |||| ||||||||| |||||| ||| |||||||||||| | |||| || ||||||||| |||||||||| |||| ||| ||||||||||| | |||||| ||| ||||||||||| |||| || ||||| ||| || ||||||| ||| |||| | ||||| ||||||||||||| |||||||| ||||| |||||| ||| |||||||||
Table 34: Hospitalizations in the ENHANCE Study (FAS Population)
Hospitalizations | ENHANCE | |||||
|---|---|---|---|---|---|---|
Initial care (1 to 14 days) | Follow-up (15 to 44 days) | Entire study duration (44 days) | ||||
Pre-period (N = 48) | Post-period (N = 42) | Pre-period (N = 48) | Post-period (N = 42) | Pre-period (N = 48) | Post-period (N = 42) | |
||||||||||||||||| ||||||||||||||||| | ||| | ||||||
|||| | ||| | |||| | || ||||||| | || |||||| | | ||||| | | ||||| |
||| | || ||||||| | || ||||||| | |||| | | ||||| | | || ||||||| | || ||||||| |
Infection-related hospitalizations that resulted in admission to ICU, n (%) | ||||||
None | |||| | |||| | |||| | |||| | 0 | 0 |
Any | |||| | |||| | |||| | |||| | 0 | 0 |
All-cause hospitalizations in the 30 days following discharge from the initial hospitalization, n (%) | ||||||
None | || |||||| | || |||||| | || ||||||| | || |||||| | || |||||| | 39 (92.9) |
Any | | ||||| | | ||||| | |||| | | ||||| | 1 (2.1) | 3 (7.1) |
FAS = full analysis set; ICU = intensive care unit.
Source: Clinical Study Report for ENHANCE.21
Table 35: Response to Treatment at End of Treatment Visit in the ENHANCE Study (FAS Population)
Response to treatment | ENHANCE | |
|---|---|---|
Entire study duration (44 days) | ||
Pre-period (N = 48) | Post-period (N = 42) | |
Complete response, n (%) | 24 (50.0) | 24 (57.1) |
Unadjusted P value for between-group mean difference in response to treatment | 0.58 | |
Partial Response, n (%) | 0 | 0 |
Failure, n (%) | 12 (25.0) | 9 (21.4) |
Missing, n (%) | 12 (25.0) | 9 (21.4) |
FAS = full analysis set.
Analysis: P values for categorical variables are from a Chi-square test (or Fisher’s exact test if distribution assumption is not met).
Note: Complete response was defined as a reduction in pain or no pain, tenderness, or fever, and the primary lesion area at the day 10 to 14. Partial response was defined as a reduction in pain or no pain, tenderness, or fever but the primary lesion remained the same size at day 10 to 14. Failure was defined as if none of the aforementioned criteria were met.
NOTE: P values are descriptive only as there was no control for type I error rate.
Source: Clinical Study Report for ENHANCE.21
Change in SF-12 | ||||||| | |||
|---|---|---|---|---|
||||||||||||| | ||| | |||||||||||||| | ||| | |||
|||||||| | ||| || | |||||||| | ||| || | |
|||||| |||||| ||||||||| ||||| | ||||
||| | ||| | ||| | ||| | ||| |
|||| |||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| |
|||| |||||||||| |||| ||| | ||| |||||| |||| | ||| |||||| |||| | ||
|||||||||| ||||||| | | |||| | |||| | ||
||||||| ||| ||||||||||||| |||||||||| || |||||| |||| |||||||| | | |||| | |||
|||||||| |||||| ||||||||| ||||| | ||||
||| | ||| | ||| | ||| | ||| |
|||| |||| | |||| |||||| | |||| ||||||| | |||| ||||||| | |||| ||||||| |
|||| |||||||||| |||| ||| | ||| |||||| |||| | ||| ||||| ||||| | ||
|||||||||| ||||||| | | ||||| | | ||||| | ||
||||||| ||| ||||||||||||| |||||||||| || |||||| |||| |||||||| | | ||||| | |||
||| |||||||||| ||||||||| ||| | |||| |||||||| |||| || | |||||||| |||||||||| ||||| | ||||||| |||||||| ||||| ||||| |||||||||| |||||||| |||| |||| | |||||| |||||| ||| |||||| |||| |||||||| || ||| ||| | |||||| ||| ||||||||||| |||| || ||||| ||| || ||||||| ||| |||| | ||||| |||||| |||||||| |||| |||| | ||||||||| |||||| ||| |||||||||||| | |||| || ||||||||| |||||||||| ||| ||| ||||| | |||||| ||| ||||||||||| |||| || ||||| ||| || ||||||| ||| |||| | ||||| ||||||||||| ||||| || | ||||||| |||||| |||| || |||||| |||||| ||| |||||| |||||| ||| | ||||| ||||| |||||||| ||| ||||||||| |||||||| |||||||||||| |||| |||||||||| ||| || |||||||| |||||| ||||||||| |||||| ||||| ||||||| ||||||| |||||||| ||||||||||||||||| |||||| |||||||||||| |||| ||||||||||| ||| || ||||||||| ||||||||| ||| |||||| ||||||| |||| |||| || ||||| || | ||||||| |||||| ||||| || ||||||| |||||| |||||| ||||| |||||| | |||||||| ||| |||||| |||||| ||||||||| ||||||| ||||| |||| |||||| |||||| |||||||||| |||||| |||||||||||||| |||||||| ||||| |||||| ||| |||||||||
Harms | ||||||| | |
|---|---|---|
||||||||||||| | ||| | |||||||||||||| | ||| | |
|||||||| |||| | | ||||||| ||||| | ||
|| ||| | | ||||| | || |||||| |
|||| |||||| ||||||| | ||| | | ||
||||||| | |||| | | ||||| |
|||||||||| | |||| | | ||||| |
|||| | |||| | | ||||| |
|||||||| | |||| | | ||||| |
|||||||| ||| |||||||||||| ||||| ||| || ||| ||||||| ||||| | ||
|| ||| | ||| | ||| |
||||| ||| || ||| ||||||| ||||| | ||
|| ||| | ||| | ||| |
|||||||| |||| | | ||| | ||
| ||| | | ||||| | | ||||| |
|||||||||| | ||| | | ||||| |
|||||||| | ||| | | ||||| |
|||||||||||| | | ||||| | ||| |
||||||| |||||| | ||| | ||
|||||||||| |||||||||| | |||| | |||| |
||||||||||| || |||||||||| |||||||||| | |||| | |||| |
|||||||| ||||||| |||||||| | |||| | |||| |
|||||||||||||||| ||||||||| | |||| | |||| |
|||| | ||| | | ||||| |
||||||||| | ||| | | ||||| |
||||||||||| ||||||||| ||||||||| | |||| | |||| |
||||| |||||||| | ||
||||| |||||| |||||| | | ||||| | ||| |
||||||| |||||||| | |||| | |||| |
||||| | |||| |||||||| |||| || | ||| ||||||||| ||| | ||||||| ||||||| ||||||| ||||||||| | || |||||||| ||| ||||| || ||| ||||||||||| |||||||||||| ||||||| |||||| |||| ||||| |||| ||||| ||||||| ||||||||||||| |||||||| ||||| |||||| ||| |||||||||
Appendix 4: Details of the ADVANCE Study
Note that this appendix has not been copy-edited.
Table 38: Summary of Baseline Characteristics in the ADVANCE Study (FAS Population)
Characteristics | ADVANCE | |
|---|---|---|
Pre-period (N = 156) | Post-period (N = 153) | |
Demographics | ||
Age (years) | ||
Mean (SD) | 46.5 (16.02) | 47.2 (15.58) |
Sex, n (%) | ||
Male | 100 (64.1) | 99 (64.7) |
Female | 56 (35.9) | 54 (35.3) |
Race, n (%) | ||
White | 89 (57.0) | 91 (59.5) |
Black or African American | 30 (19.2) | 39 (25.5) |
American Indian or Alaska Native | 6 (3.8) | 2 (1.3) |
Asian | 1 (0.6) | 2 (1.3) |
Native Hawaiian or Other Pacific Islander | 0 | 0 |
Other | 17 (10.9) | 16 (10.5) |
Patient did not disclose | 13 (8.3) | 3 (2.0) |
BMI (kg/m2) | ||
n | 137 | 146 |
Mean (SD) | 33.1 (9.87) | 32.7 (10.62) |
|||||||||| ||||||||| |||||||| | ||
||| | ||| | ||| |
|||| |||| | |||| ||||||| | |||| ||||||| |
Relevant comorbid conditions and medical history, n (%) | ||
Solid tumour | 6 (3.8) | 3 (2.0) |
Diabetes mellitus | 27 (17.3) | 31 (20.3) |
Moderate to severe chronic kidney disease | 4 (2.6) | 5 (3.3) |
|||| ||||| ||||||||||||| | | || |||||| | || |||||| |
Illicit needle use | 13 (59.1) | 19 (55.9) |
Lymphedema / chronic venous stasis | 6 (3.8) | 2 (1.3) |
Connective tissue disease | 1 (0.6) | 2 (1.3) |
Peripheral vascular disease | 5 (3.2) | 4 (2.6) |
||||||| ||||| | | ||||| | | ||||| |
AIDS (not just HIV positive) | 0 | 1 (0.7) |
Leukemia | 1 (0.6) | 0 |
Liver disease | 9 (5.8) | 9 (5.9) |
Malignant lymphoma | 3 (1.9) | 1 (0.7) |
Immunocompromising conditions | ||
Receiving any immune modulating medication including biologics other than TNF | 1 (0.6) | 0 |
HIV (excluding AIDS) | 3 (1.9) | 3 (2.0) |
||||||||| || || |||||||||| ||||| ||| ||||||||||| |||||||||| |||||||| | |||| | |||| |
Organ transplant recipient | 1 (0.6) | 1 (0.7) |
Receiving chemotherapy | 1 (0.6) | 1 (0.7) |
Receiving TNF inhibitors | 1 (0.6) | 0 |
|||||||| |||||||||| |||||||| ||||||||||| || ||| |||||||| | ||||||| | ||| | ||
||||||||||| ||||| |||||| | |||||| |||||| ||||| |||||||||| | | || |||||| | || |||||| |
|||||||||| ||| ||||||||| || ||||| |||||| | || |||||| | || |||||| |
|||||||||| ||| ||||||||| || ||||||| |||||| | | |||||| | | |||||| |
|||||||||| ||| ||||||||| || ||||| ||||||||| || |||||||| ||||||||| | || |||||| | | ||||| |
|||||||||| ||||||||| ||||||| |||||| | |||||| |||||| ||||| |||||||||| || | | || |||||| | || |||||| |
|||||||||| ||| ||||||||| || ||||| |||||| | || ||||||| | || |||||| |
|||||||||| ||| ||||||||| || ||||||| |||||| | |||| | | ||||| |
|||||||||| ||| ||||||||| || ||||| ||||||||| || |||||||| ||||||||| | |||| | |||| |
||||| |||| ||| |||||||||| |||||||||| | ||| | |||| | || |||||| |
Infection and clinical characteristics | ||
Infection type, n (%)b | ||
Cellulitis/Erysipelas | 127 (81.4) | 126 (82.3) |
Abscess | 51 (32.7) | 55 (35.9) |
Wound Infection | 15 (9.6) | 9 (5.9) |
Purulent drainage from primary lesion, n (%) | ||
Yes | 38 (24.4) | 34 (22.2) |
No | 118 (75.6) | 119 (77.8) |
Primary lesion size (cm2) | ||
|||| | ||| | ||| |
|||| |||| | ||||| |||||||| | ||||| ||||||||| |
Median | 255.0 | 289.0 |
Min, Max | 75.0 to 3,196.0 | 77.0 to 24,242.0 |
Fever, n (%)d | 7 (4.5) | 3 (2.0) |
SIRS criteria met, n (%)e | 31 (19.8) | 27 (17.6) |
|||||||||||||||| ||||||||| || ||||| | ||||||| | ||| | || |||||| | || |||||| |
|||||||||| |||||| | ||
||||| |||||||||| ||||| |||||||| ||||| ||| ||||| | ||| | ||
|||||||| | |||| | | ||||| |
|||||||| | | ||||| | |||| |
||| |||| | ||| |||||| | ||| |||||| |
ABSSSI = acute bacterial skin and skin structure infection; BMI = body mass index; FAS = full analysis set; MRSA = methicillin-resistant Staphylococcus aureus; SD = standard deviation; SIRS = systemic inflammatory response syndrome.
| |||| || |||| ||| ||| |||| || ||| ||||||||||| ||| ||| |||||||||| |||||||| || ||| |||||||||| ||||||||||||||
bPatients can contribute to multiple categories; hence, percentage may not sum to 100.
| |||||||||| ||||||||| ||||||| ||| ||||||| || ||||||||||||||| ||| || ||||||||| || ||||||||| |||||| || ||| || || ||||||| ||||| ||||||| || ||| |||||||||| |||||||| || ||| |||| |||||| |||||
dFever was defined as a reported temperature of greater than 38°C.
eMeeting SIRS criteria was defined as having 2 or more of the following: temperature < 36°C or > 38°C, heart rate > 90 beats per minute, respiratory rate > 20 breaths per minute or arterial carbon dioxide tension < 32 mm hg, or white blood cell count < 4,000 cells/µL or > 12,000 cells/µL or > 10% bands.
Source: Clinical Study Report for ADVANCE.22
Table 39: Summary of Patient Disposition in the ADVANCE Study
Patient disposition | ADVANCE | |
|---|---|---|
Pre-period | Post-period | |
Screened, n | 3,104 | 3,293 |
Enrolled, n | 160 | 153 |
Completed, n (%) | 121 (75.6) | 128 (83.7) |
|||||||||| | ||| | || |||||| | || |||||| |
|||| || ||||||||| | || |||||| | || |||||| |
|||||||| ||||||||||||||||||| | | |||| | | ||||| |
|||||||||| || ||||||| || ||||||| | | ||||| | | ||||| |
||||| | | | ||||| | | ||||| |
Enrolled Set population, N | 160c | 153 |
FAS population, N | 156 | 153 |
FAS = full analysis set.
| ||| ||||||| ||| | |||||||| ||||||||| ||| || || ||||||||| |||| |||||||| |||||||| || ||||||||||| || ||| ||||||||| ||||||| ||| || |||||| || ||||| |||||||||||||| || ||||| |||| || |||||||||| ||||||||
cOf the 160 patients enrolled, 4 patients had missing information on antibiotic use.
Source: Clinical Study Report for ADVANCE.22
Table 40: Summary of Antibiotic Use in the ADVANCE Study (FAS Population)
Antibiotic use | ADVANCE | |
|---|---|---|
Pre-period (N = 156) | Post-period (N = 153) | |
||||||| |||||||||| || |||||||| |||| || |||||||||| ||||||| || ||| |||| ||| ||||||||||| ||| |||||||| |||||||||||| ||||| | ||
|||| | ||| | ||| | ||| |||||| |
||| | ||| | | ||| | || |||||| |
||||||| |||||||| |||||||||| ||||||||||||||||||||| ||| || |||||| | ||| | || |||||| |
||||||| |||||||| |||||||||| ||||||||||||||||||||| ||| || ||||| ||||||| | ||| | | |||||| |
|||||||||| |||||||| ||| ||||||||| || ||||||||| ||||||||| | ||| | |||| |
||||| ||||| ||| ||||||||| || |||||||||| ||||||||| | ||| | |||| |
||||| | | ||| | | ||||| |
|||||| || |||||||||| |||||||| |||||| |||||| | ||| | ||
|||| | ||| | ||| |
|||| | || |||||| | || |||||| |
|||| | || |||||| | || |||||| |
|||| | || |||||| | || |||||| |
| || | || |||||| | || |||||| |
||||| || ||||||||||||||| | ||| | ||
||||||||||| | || |||||| | ||| |||||| |
|| ||||| | ||| |||||| | || |||||| |
||||||| |||||||||| |||||||||| | ||| | | ||
||||||||||| | | ||||| | ||| |||||| | |
Vancomycin | 66 (42.3) | 21 (13.7) |
||||||||| | || |||||| | || |||||| |
||||||||||| | || |||||| | | ||||| |
|||||||||||||||| |||||||||||| | || |||||| | || |||||| |
Cefazolin | 33 (21.2) | 7 (4.6) |
Ceftriaxone | 16 (10.3) | 7 (4.6) |
||||||||||| | || ||||| | | ||||| |
|||||||||||| |||||| |||||||||| |||||| | || ||||| | | ||||| |
|||||||| | | ||||| | | ||||| |
||||| | || |||||| | || ||||| |
|||||| ||| ||||||| |||||||||| |||||||||||||||| | ||| | | ||
||||||||| ||||||| |||||| || ||||||| ||| |||||| | 135 (86.5) | 153 (100.0) |
||||||| |||||||| | 6 (3.8) | 3 (2.0) |
||||||||| || ||||||||| |||||| | 10 (6.4) | 3 (2.0) |
|| ||||||| ||||| ||||||| || ||| |||||||||| | 3 (1.9) | 1 (0.7) |
||||| | 22 (14.1) | 18 (11.8) |
|||||||||| |||||||| ||||| ||||||||||||||| || ||||||| ||||||||||| | ||| | ||
||| | 19 (12.2) | 12 (7.8) |
||| | 137 (87.8) | 141 (92.2) |
ABSSSI = acute bacterial skin and skin structure infection; FAS = full analysis set; NA = not applicable.
| |||| || |||| ||| ||| |||| || ||| ||||||||||| ||| ||| |||||||||| |||||||| || ||| |||||||||| ||||||||||||||
| ||| ||||||| ||| ||||||||| |||||||| || ||||||| |||||||||| || ||||||||||| ||| ||||||| |||||||| || |||||||||||| || ||| || |||| |||||||| |||| ||||||||||| ||||| || ||| ||||||||||||| ||| ||||||| ||| |||||||| || |||||| |||||||| ||||||||||| | ||||| ||||| ||||||||||||| |||||||| ||| |||| ||||||||||| || |||| |||| | |||||||
Source: Clinical Study Report for ADVANCE.22
||||||||||| |||||||| || ||||||||||||||| || ||| ||||||| ||||||||| || ||| |||||||||| |||| || ||||| || |||||||| || ||||||| | ||||||||||||| ||||||||||| ||||||||| || ||| ||||| ||| || |||||||| |||||| || ||| ||||||||||| ||||| |||| ||| |||||||| ||||||||| |||||| |||| || ||| |||||||| ||| ||| |||||||||| ||||||| || ||| |||||||| ||||||| ||||||| |||||| |||||| || |||| ||||| || ||| ||||||||| || ||| |||||||| ||||||||||| |||| ||| |||| |||||||||| || ||||||||| ||| |||| || |||||| ||| ||||||| ||||||| || |||||||| ||||||||| |||| || ||| ||||||| ||||||| || ||||| || ||||||||| |||||| |||| || ||| |||||||| ||||||||| | ||||||||| |||||||||| |||| |||| | |||||||||||| ||||| || |||| ||| ||| |||||| ||| ||| ||||||||||| |||| | |||||||| ||||||||| |||| || ||| ||| ||| || ||| |||| ||| ||||||||||||| ||||||||||||| ||||| ||||||||| ||| |||||||||| ||| ||||| ||||||||| ||| ||| ||||||||| || ||||| ||| |||||||| ||||||||| |||| ||| ||| ||||| ||| ||| |||||||| || |||||||||| ||| |||||| ||||||||| |||| |||||||| ||| ||||||||| || ||| |||||||| |||||||| ||| |||||||| || ||||| | |||| || || |||||||||| || ||| |||||||||| ||| || ||||| | |||| || ||||||||||| || ||| |||||||||||| ||||||||||| |||||||| |||| ||||||||| || ||||||||| |||||||||| ||| ||||||||||| |||||||||| ||||||||||| |||| |||| ||||||| ||| |||| ||| ||||||||||| |||||| || ||| ||||||||||| ||| ||| |||||||||| ||||||||||| ||||||||| || |||||||| ||||||||||||||| ||||| |||||||||| || |||||||| ||||| |||||| ||||||||||| ||| ||| ||||||||||||| ||| |||||||||| |||||||| |||| |||| |||||||| ||||| ||||||||||| ||||||||||| |||| |||| ||||||| ||| |||| ||| ||||||||||| |||||| || ||| ||||||||||| |||||||||| ||| ||||||||||| ||| |||||||||| |||||||||| ||||||||| ||||| ||||||||| ||||||||| ||||||| || |||||||||||| | |||||| ||||||||||| ||| ||| ||||||||||||| ||| || |||| |||||||| ||||| ||||||||||| | |||||||||||| ||||| || |||| ||| |||| ||| ||| ||||||||| ||||||||||| |||||||| |||| ||| ||||||||| || ||| ||||||| |||||||| || ||||| ||| ||||||||| |||||||| |||| ||||||||||| || ||| ||||| ||| ||| ||||||| ||| ||||||||| |||||||| ||| ||||||||||| ||| |||||||| ||||||||||| |||| ||| ||||| |||||||| |||||||| |||| ||||||||| ||||| || ||| |||||||| |||| |||||||| ||| ||||||| |||||| |||| ||||||| ||||||||| || ||| |||||||| ||||||| ||||||| ||| ||||||||| |||||||||||| | ||||||||| |||||||| ||||| | ||||||||||||| |||||||| |||||||||| ||||| |||||||| ||| ||||||||| ||| ||| ||||||| ||||||||
Table 41: ABSSSI-Related Hospital Admission Rate at Initial Care in the ADVANCE Study (FAS Population)
ABSSSI-related hospital admission | ADVANCE | |
|---|---|---|
Initial care (1 to 14 days) | ||
Pre-period (N = 156) | Post-period (N = 153) | |
ABSSSI-related hospital admission at initial care, n (%) | ||
Yes | 60 (38.5) | 27 (17.6) |
No | 96 (61.5) | 126 (82.3) |
Unadjusted P-value | < 0.001 | |
Adjusted P-valuea | < 0.001 | |
ABSSSI = acute bacterial skin and skin structure infection; CI = confidence interval; FAS = full analysis set; LOS = length of stay; SD = standard deviation.
aAdjusted for age, race, insurance type, prior resource use (yes/no), and SIRS score (< 2 / ≥ 2).
Analysis: P-value was from Fisher's Exact test.
Source: Clinical Study Report for ADVANCE.22
Total LOS | ||||||| | |||||
|---|---|---|---|---|---|---|
||||||| ||||||| || || ||||| | ||||||||| |||| || || ||||| | |||||| ||||| |||||||||||| ||||| | ||||
||||||||||||| | |||| | |||||||||||||| | |||| | ||||||||||||| | |||| | |||||||||||||| | |||| | ||||||||||||| | |||| | |||||||||||||| | |||| | |
||||| ||| |||||||||| |||||| |||| | ||||||
|| | ||| | ||| | ||| | ||| | ||| | ||| |
|||| |||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| |
|||||||||| ||||||| | | ||||| | |||| | ||||| | |||
||||| ||| ||| |||||||| |||||||| || ||| |||||||| |||||||||| |||||| |||| | ||||||
|| | || | || | || | || | || | || |
|||| |||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| |
|||||||||| ||||||| | | |||| | |||| | |||| | |||
||||| ||| |||||||||| |||| ||||| || ||||||||| ||||||||||| |||||| ||| |||| | ||||||
|| | ||| | ||| | ||| | ||| | ||| | ||| |
|||| |||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| |
|||||||||| ||||||| | | ||||| | |||| | ||||| | |||
||| |||||||||| ||||||||| ||| | |||| |||||||| |||| ||| | |||||| || ||||| || | |||||||| |||||||||||| ||||||| ||| |||| |||||| ||||||||| ||||||||| |||||||||| |||||| |||||||||||||| |||||||| |||| |||||||| ||||| ||||||||||| |||||| || || ||| ||||||| |||| | |||||||||| | |||||| ||| ||||||||||| |||| || ||||| ||| || ||||||| ||| |||| | ||||| ||||||||||| ||||||||||||||||| |||||||| |||||| || |||| ||| | |||||| || ||||||||| ||||||||||||||| |||| ||| ||||||||||||||||| ||||||||||||||| |||||| ||| ||||||||| ||||||||||||||||| |||||| || |||| ||| ||| ||| || ||| ||||||||||||||||| |||||||||| || ||| |||||||||| ||||||| ||||||||||||||| ||| |||| ||| ||||| ||||| |||| || || ||||||||||||||| ||| ||||||| || ||| ||| || ||||| || ||||||||||||||| ||||| ||| ||||| |||||||||| |||| || ||||||||||||||| |||| ||| |||| || ||| ||| ||||||||||||| |||||||| ||||| |||||| ||| |||||||||
Infection-related LOS | ||||||| | |||||
|---|---|---|---|---|---|---|
||||||| ||||||| || || ||||| | ||||||||| |||| || || ||||| | |||||| ||||| |||||||||||| ||||| | ||||
||||||||||||| | |||| | |||||||||||||| | |||| | ||||||||||||| | |||| | |||||||||||||| | |||| | ||||||||||||| | |||| | |||||||||||||| | |||| | |
||||||||||||||||| |||| |||| | ||||||
|| | ||| | ||| | ||| | ||| | ||| | ||| |
|||| |||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| |
|||||||||| ||||||| | | ||||| | |||| | ||||| | |||
||||||||||||||||| ||| |||||||||| |||| ||||| || ||||||||| ||||||||||| |||||| ||| |||| | ||||||
|| | ||| | ||| | ||| | ||| | ||| | ||| |
|||| |||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| | ||| |||||| |
|||||||||| ||||||| | | | ||||| | |||| | ||||| | |||
||| |||||||||| ||||||||| ||| | |||| |||||||| |||| ||| | |||||| || ||||| || | |||||||| |||||||||||| ||||||| ||| |||| ||||||||| |||||| |||||||||||| |||||||| |||| |||||||| ||||| ||||||||||| |||||| || || ||| ||||||| |||| | |||||||||| | |||||| ||| ||||||||||| |||| || ||||| ||| || ||||||| ||| |||| | ||||| ||||||||||||| |||||||| ||||| |||||| ||| |||||||||
Hospitalizations | ||||||| | |||||
|---|---|---|---|---|---|---|
||||||| ||||||| || || ||||| | ||||||||| |||| || || ||||| | |||||| ||||| |||||||||||| ||||| | ||||
||||||||||||| | |||| | |||||||||||||| | |||| | ||||||||||||| | |||| | |||||||||||||| | |||| | ||||||||||||| | |||| | |||||||||||||| | |||| | |
||||||||||||||||| ||||||||||||||||| | ||| | ||||||
|||| | || |||||| | ||| |||||| | ||| |||||| | ||| |||||| | || |||||| | ||| |||||| |
||| | || |||||| | || |||||| | | ||||| | | ||||| | || |||||| | || |||||| |
||||||||||||||||| |||||||||||||||| |||| |||||||| || ||||||||| || |||| | ||| | ||||||
|||| | ||| |||||| | ||| ||||||| | ||| |||||| | ||| ||||||| | ||| |||||| | ||| ||||||| |
||| | | ||||| | |||| | | ||||| | |||| | | ||||| | |||| |
||||||||| |||||||||||||||| || ||| || |||| |||||||||||||| |||| ||||||| ||||||||||||||| || ||||||| |||| ||||||| || |||||| | |||| | ||||||
|||| | ||| | ||| | ||| | ||| | ||| |||||| | ||| |||||| |
||| | ||| | ||| | ||| | ||| | | ||||| | || ||||| |
||| | |||| |||||||| |||| ||| | ||||||||| |||| ||||| || | ||| ||||||||||||||| ||| ||| |||||||| ||| ||||||| |||| |||||||| |||| ||||||||||| ||||||||||||| |||||||| ||||| |||||| ||| |||||||||
Change in SF-12 | ||||||| | |||
|---|---|---|---|---|
||||||||||||| | |||| | |||||||||||||| | |||| | |||
|||||||| | ||| || | |||||||| | ||| || | |
|||||| |||||| ||||||||| ||||| | ||||
|||| | ||| | ||| | ||| | ||| |
|||| |||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| |||||| |
|||| |||||||||| |||| ||| | ||| |||| || |||| | ||| |||| || |||| | ||
|||||||||| ||||||| | | ||||| | ||||| | ||
||||||| ||| ||||||||||||| |||||||||| || |||||| |||| |||||||| | | |||| | |||
|||||||| |||||| ||||||||| ||||| | ||||
|||| | ||| | ||| | ||| | ||| |
|||| |||| | |||| ||||||| | |||| ||||||| | |||| ||||||| | |||| |||||| |
|||| |||||||||| |||| ||| | ||| |||| || |||| | ||| |||| || |||| | ||
|||||||||| ||||||| | | | ||||| | | ||||| | ||
||||||| ||| ||||||||||||| |||||||||| || |||||| |||| |||||||| | | || ||||| | |||
||| |||||||||| ||||||||| ||| | |||| |||||||| |||| || | |||||||| |||||||||| ||||| | ||||||| |||||||| ||||| ||||| |||||||||| |||||||| |||| |||| | |||||| |||||| ||| |||||| |||| |||||||| || ||| ||||| |||||||| |||| |||| | ||||||||| |||||| ||| |||||||||||| | |||| || ||||||||| |||||||||| || ||| ||||||||||| | |||||| ||| ||||||||||| |||| || ||||| ||| || ||||||| ||| |||| | ||||| ||||||||||| ||||| || | ||||||| |||||| |||| || |||||| |||||| ||| |||||| |||||| ||| | ||||| ||||| |||||||| ||| ||||||||| |||||||| |||||||||||| |||| |||||||||| ||| || |||||||| |||||| ||||||||| |||||| ||||| ||||||| ||||||| |||||||| ||||||||||||||||| |||||| |||||||||||| |||| ||||||||||| ||| || ||||||||| ||||||||| ||| |||||| ||||||| |||| |||| || ||||| || | ||||||| |||||| ||||| || ||||||| |||||| |||||| ||||| |||||| | |||||||| ||| |||||| |||||| ||||||||| ||||||| ||||| |||| |||||| |||||| |||||||||| |||||| |||||||||||||| |||||||| ||||| |||||| ||| |||||||||
Harms | ||||||| | |
|---|---|---|
||||||||||||| | |||| | |||||||||||||| | |||| | |
|||||||| |||| | | ||||||| ||||| | ||
|| ||| | || |||||| | || |||||| |
|||| |||||| ||||||| | ||| | | ||
|||||||||| | | ||||| | | ||||| |
|||||||| | | ||||| | | ||||| |
|||||| | | ||||| | | ||||| |
|||||||| ||| |||||||||||| ||||| ||| || ||| ||||||| ||||| | ||
|| ||| | ||| | ||| |
||||| ||| || ||| ||||||| ||||| | ||
|| ||| | | ||||| | ||| |
|||||||||| ||||||| ||||||| | | ||||| | ||| |
|||| ||||||| |||||||| | | ||||| | ||| |
|||||||| |||| | | ||| | ||
| ||| | || ||||| | || |||||| |
||||||| |||||| | ||| | ||
||||||||||| || |||||||||| |||||||||| | |||| | |||| |
||||||||| ||||||| | | ||||| | ||| |
|||||||| ||||||| |||||||| | | ||||| | | ||||| |
|||||||||||||||| ||||||||| | ||
|||||||||||| |||||||| | | ||||| | ||| |
|||| |||||||||||||||| | | ||||| | ||| |
||||||||||| ||||||||| ||||||||| | ||
||||||||||| ||||||||| ||||||| | ||| | | ||||| |
||||| |||||||| | |||| | |||| |
||||||| |||||||| | |||| | |||| |
||| | |||| |||||||| |||| || | ||| ||||||||| ||| | ||||||| ||||||| ||||||| ||||||||| | || |||||||| ||| ||||||||| || ||| |||||||||||||||||| ||||||| |||||| |||| ||||| |||| |||||| ||||||| ||||||||||||| |||||||| ||||| |||||| |||||||||
Pharmacoeconomic Review
Abbreviations
ABSSSI
acute bacterial skin and skin structure infection
BIA
budget impact analysis
CUA
cost-utility analysis
ICER
incremental cost-effectiveness ratio
QALY
quality-adjusted life-year
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Dalbavancin (Xydalba), 500 mg lyophilized powder in vial for IV infusion |
Submitted price | Dalbavancin, 500 mg, lyophilized powder in vial = $957.1679 |
Indication | For the treatment of adults with acute bacterial skin and skin structure infections (ABSSSIs) caused by susceptible isolates of gram-positive micro-organisms |
Health Canada approval status | NOC |
Health Canada review pathway | Priority review |
NOC date | May 5, 2021 |
Reimbursement request | Per indication |
Sponsor | Paladin Labs Inc. |
Submission history | Previously reviewed: No |
ABSSSI = acute bacterial skin and skin structure infection; NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Decision tree |
Target population | Adults with nonsevere and severe ABSSSIs |
Treatment | Dalbavancin |
Comparators |
|
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | 6 months (197 days) |
Key data source | Clinical efficacy and safety: DISCOVER 1 trial, DISCOVER 2 trial, and sponsor-submitted ITC Hospital discharge rates: clinical expert opinion and published literature |
Submitted results | Total population (patients with nonsevere and severe ABSSSIs): dalbavancin dominates (i.e., less costly, more effective) vancomycin IV, linezolid IV, cefazolin IV, and daptomycin IV. Dalbavancin IV was associated with an ICER of $401,168 per QALY compared to ceftriaxone IV (incremental costs = $239; incremental QALYs = 0.0006). Nonsevere ABSSSIs: ceftriaxone IV dominates (i.e., less costly, more effective) vancomycin IV, linezolid IV, cefazolin IV, and daptomycin IV. Dalbavancin IV was associated with an ICER of $793,404 per QALY compared to ceftriaxone IV (incremental costs = $434; incremental QALYs = 0.0005). Severe ABSSSIs: dalbavancin IV dominates (i.e., less costly, more effective) all active comparators (vancomycin IV, linezolid IV, cefazolin IV, ceftriaxone IV, and daptomycin IV). |
Key limitations | The assumption of equivalent efficacy between dalbavancin IV and active comparators was associated with a high degree of uncertainty. Based on the CADTH clinical review, the evidence of comparative efficacy from the DISCOVER trials for vancomycin IV, with a possible switch to oral linezolid, was uncertain due a lack of direct or indirect comparative evidence for ceftriaxone IV and cefazolin IV, relative to dalbavancin IV, and concerns about generalizability to standard treatment. There was no direct or indirect comparative evidence to support early discharge rates associated with dalbavancin IV. The sponsor modelled reduced hospitalization rates for nonsevere patients treated with dalbavancin IV, relative to active comparators, using the Talan et al. (2021) study, which performed a naive comparison of hospitalization rates before and after dalbavancin IV was administered in a hospital setting. The study design was prone to risk of selection and performance bias. Further, there was a lack of a concurrent comparator, lack of randomization, time-related confounders, missing data, and attrition bias, which may have introduced a bias in the length of hospital stay and admission rate in favour of dalbavancin IV. As such, this assumption was highly uncertain. There was insufficient evidence to support an association between early discharge rates and dalbavancin IV, which was a key driver of the cost savings reported in the sponsor’s economic evaluation. The sponsor did not include any oral therapies that clinical experts consulted by CADTH deemed to be relevant in clinical practice in Canada. The cost-effectiveness of dalbavancin IV compared to these omitted comparators is unknown. The sponsor assumed that patients switched to an oral antibiotic after hospital discharge, and adopted a higher rate of disease recurrence with oral treatments than with IV treatments. This increased the subsequent treatment costs for comparators, relative to dalbavancin IV. CADTH was unable to validate the sponsor’s estimated increase in recurrence rate with the sponsor’s reference. |
CADTH reanalysis results | Given the key limitations of the available clinical evidence, the clinical effects of dalbavancin IV compared to active comparators for ABSSSIs were highly uncertain. The CADTH reanalysis assumed that there would be no difference in treatment effects (i.e., no difference in total QALYs), and a cost comparison between dalbavancin IV and its comparators was conducted to highlight the differences in drug costs. The treatment cost of dalbavancin IV ($2,872 per patient) was higher than all active comparators, which range from $97 to $1,775 for IV treatments. There was insufficient clinical evidence to justify a price premium for dalbavancin IV above all other comparator treatments. The submitted price of dalbavancin IV would need to be reduced by at least 38% to 97% to be equivalent to the lowest-priced generic IV treatment. |
ABSSSI = acute bacterial skin and skin structure infection; ICER = incremental cost-effectiveness ratio; ITC = indirect treatment comparison; LY = life-year; QALY = quality-adjusted life-year.
Conclusions
The CADTH clinical review concluded that dalbavancin (Xydalba) IV demonstrated noninferiority to vancomycin IV (with an optional switch to oral linezolid) and linezolid IV. However, the evidence was uncertain because of concerns regarding the validity of outcomes and the selection of comparators. The evidence was not available for all comparators, and the indirect treatment comparison was limited due to heterogeneity and sparse data for safety end points. These factors hindered the generalizability and interpretation of the results. There was also no significant difference in length of hospital stay, but the study design of the pragmatic trials and use of a pathway intervention limited the interpretation of results.
Because of limitations with the available comparative evidence, CADTH reanalyses assumed no difference in treatment effects (i.e., equal quality-adjusted life-years [QALYs]) for dalbavancin IV and all active comparators for the treatment of acute bacterial skin and skin structure infections (ABSSSIs), and a cost comparison was conducted to assess annual drug costs. The treatment course of dalbavancin IV costs $2,872 per patient, which is more costly than all IV and oral treatments for ABSSSIs reported on publicly available list prices. The submitted price of dalbavancin IV would need to be reduced by at least 38% to 97% to be equivalent to the lowest-priced generic IV treatment.
Based on the CADTH clinical and economic reviews, there is insufficient evidence to support a price premium for dalbavancin IV over other oral and IV antibiotics for the treatment of ABSSSIs. CADTH notes that the incremental benefit with dalbavancin IV is highly uncertain, with wide credible intervals that include the possibility of negative QALYs, given the lack of justification to support an assumption of equal efficacy between dalbavancin IV and active comparators. Dalbavancin IV could be dominated (i.e., be more costly and less effective) by active comparators, based on the submitted evidence. In this case, a price reduction of even 100% would not make dalbavancin IV cost-effective. CADTH could not fully explore this uncertainty because of a lack of available evidence and, therefore, the possibility that dalbavancin IV generates fewer QALYs at a higher cost than active comparators at any price reduction should be considered.
CADTH further notes that the economic evaluation does not include oral comparators, which are relevant in the nonsevere ABSSSIs population and are far less costly than dalbavancin. The comparative effectiveness and cost-effectiveness of dalbavancin IV remains unknown for these comparators.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process, and specifically provides information that pertains to the economic submission.
No patient or clinician input was received for this review.
Drug plan input received for this review noted that the cost of dalbavancin is significantly higher than comparators, many of which have negotiated confidential prices. Dalbavancin IV has a slightly improved safety profile and less demand for monitoring and drug administrations. However, access to infusion services as an outpatient may be associated with extra costs to the drug plans. The plans also noted that public drug plan coverage of comparators varies in jurisdictions, and may range from no coverage to restricted access to full benefit. The plans also raised concerns about eligibility for re-treatment, and noted that dalbavancin may be used in the pediatric population for infections upon request.
Several of these concerns were addressed in the sponsor’s model:
The sponsor’s model included monitoring and drug administration costs for dalbavancin IV and comparators.
In addition, CADTH addressed some of these concerns, as follows:
CADTH conducted a cost comparison between dalbavancin IV and its comparators for the treatment of ABSSSIs, using the lowest publicly available price listed in public formularies.
CADTH was unable to address the following concern raised from stakeholder input:
Confidential prices of comparators receiving public drug plan coverage are unknown.
Economic Review
The current review is for dalbavancin for the treatment of adults with ABSSSIs caused by susceptible isolates of the following gram-positive micro-organisms: Staphylococcus aureus (including methicillin-susceptible and methicillin-resistant strains), Streptococcus pyogenes, Streptococcus agalactiae, Streptococcus dysgalactiae, Streptococcus anginosus group (including Streptococcus anginosus, Streptococcus intermedius, and Streptococcus constellatus), and Enterococcus faecalis (vancomycin-susceptible strains).
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis (CUA) of dalbavancin for the treatment of patients with ABSSSIs, assessed separately for severe and nonsevere disease.1 The modelled population is aligned with the Health Canada indication and reimbursement request. The sponsor only considered comparators delivered by IV infusion, which included vancomycin, linezolid, cefazolin, ceftriaxone, and daptomycin.
Dalbavancin IV is available as a solution for infusion (500 mg lyophilized powder in a vial). The recommended dose is 1,500 mg taken once, or 1,000 mg followed by 500 mg 1 week later. At the submitted price of $957 per vial, the treatment cost of dalbavancin was $2,872 per patient. The daily cost of comparators ranges from $9 to $177, depending on the comparator and its dosage.
The submitted model reported both QALYs and life-years over a time horizon of approximately 6 months (197 days) in the modelled population. The base-case analysis was conducted from the perspective of the Canadian public health care payer. Given the time horizon of less than 1 year, no discounting was applied to costs or health outcomes.
Model Structure
The sponsor submitted a decision-tree model to capture the efficacy and safety of ABSSSIs and associated treatments. All patients who entered the model with severe ABSSSIs received the empiric IV treatment on day 1 of a 3-day hospital stay. Patients were assessed for treatment success on day 4, and were either discharged or continued to receive treatment in the outpatient or hospital setting until the day 9 assessment. Patients who were not discharged until day 9 remained in the hospital for 5 days after the day 4 assessment to complete the treatment course. Patients who experienced treatment failure were switched to linezolid IV and were treated in the hospital for an additional 14 days. If the initial treatment was linezolid IV, patients were switched to dalbavancin IV.
Patients who entered the model with nonsevere ABSSSIs received the empiric IV treatment in the emergency department or on day 1 of a 3-day hospital stay. On day 4, if the treatment was a success, patients were prescribed an oral antibiotic and treated in the outpatient setting for 11 days. If treatment was a failure, patients were switched to another antibiotic and remained in the hospital for another 13 days to complete their treatment course.
Patients who had a recurrence of infection in the 6 months after treatment were readmitted to the hospital to complete the full treatment course (14 days), regardless of disease severity. The sponsor’s submitted model structure can be found in Appendix 3.
Model Inputs
Treatment efficacy and safety were assumed to be the same for all treatments, based on 2 randomized noninferiority trials — the DISCOVER 1 and DISCOVER 2 trials — and on published meta-analyses.2-4 Comparative efficacy data were not available for ceftriaxone IV or cefazolin IV. The sponsor assumed equivalence of efficacy between dalbavancin IV and these comparators. Based on the equivalent-efficacy assumption, the sponsor estimated a treatment failure rate of 8.6% for all comparators.2 The failure rate for patients starting treatment with vancomycin IV, cefazolin IV, or daptomycin IV was adjusted for kidney dysfunction. The sponsor assumed a clinical failure rate of 16.8% for an estimated 16.0% of patients with kidney dysfunction, based on feedback from sponsor-consulted clinical experts and the published literature, respectively.5 The rate of recurrent infection was higher with oral antibiotics (20.3%) than IV antibiotics (16.3%). It was assumed that 62.0% of patients switched to oral antibiotics and that 7.0% of patients switched to IV antibiotics administered daily, based on feedback from sponsor-consulted clinical experts. Mortality was not considered because of the short time horizon of the model.
The choice of treatment affected hospital discharge rates. For patients with severe ABSSSIs, the model adopted a 37.0% increase in early and mid-treatment discharge rates associated for dalbavancin IV compared to all other IV comparators.6 CADTH notes that these rates were estimated through expert opinion rather than clinical data. Patients with nonsevere ABSSSIs treated with dalbavancin IV were hospitalized at a lower rate (17.6%) than those treated with any other IV treatment (37.5%).7
Patients treated in the hospital were assumed to have a lower utility than patients discharged and treated in outpatient settings. At the end of the treatment course, patients were assumed to be cured and accrued the utility of an average patient 50 to 59 years of age. Utilities were obtained from published literature that measured health utilities with the 5-Level EQ-5D instrument.5,8
Costs included drug acquisition, subsequent treatment, hospitalization, drug administration (infusion in outpatient clinics), and other medical resources, such as physician visits and use of a catheter or peripherally inserted central catheter line. Drug costs for dalbavancin IV were based on the sponsor’s submitted price, whereas costs for comparators were based on the listed prices in public formularies.1,9,10 For comparators with variable dosing, the sponsor calculated drug costs using the average dose in the product monograph’s recommended range. For example, although the recommended dose of vancomycin IV ranges from 500 mg every 6 hours to 1,000 mg every 12 hours, the sponsor adopted the maximum recommended dose when estimating treatment costs. The recommended dose of ceftriaxone in the product monograph is 1,000 mg to 2,000 mg every 24 hours, and the sponsor adopted a dose of 1,500 mg once daily. Drug acquisition costs and dosing were consistent with those reported above. The cost of subsequent treatments was incurred by patients who switched treatments, and was calculated using the average daily cost of each treatment weighed by the market share of each treatment, based on clinical expert opinion consulted by the sponsor. The cost of hospitalization was calculated by multiplying the length of hospital stay with an estimate of the daily cost of hospitalization. The standardized daily cost of hospitalization was derived from the Patient Cost Estimator tool, developed by the Canadian Institute for Health Information, which allows the user to estimate the average cost of various services provided in hospitals.11
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (1,000 iterations). The deterministic results did not fully align with the probabilistic results because of small differences in incremental costs and QALYs. The deterministic incremental cost-effectiveness ratio (ICER) was $368,241 per QALY gained for dalbavancin IV compared to ceftriaxone IV (incremental cost = $222; incremental QALYs = 0.0006), whereas the probabilistic results estimated an ICER of $401,168 (incremental cost = $239; incremental QALYs = 0.0006).
Base-Case Results
In a sequential analysis conducted by the sponsor, ceftriaxone IV was associated with a lowest expected cost of $15,198. All comparators were dominated (i.e., were more costly and less effective) by ceftriaxone IV except dalbavancin IV. The undominated comparator, dalbavancin IV, was associated with incremental costs of $239 and incremental QALYs of 0.0006 compared to ceftriaxone IV, for an ICER of $401,168 per QALY. Results from the sponsor’s probabilistic analysis revealed that there was 66.6% chance that dalbavancin IV would be cost-effective at the willingness-to-pay threshold of $50,000 per QALY gained.
In a subgroup of patients with severe ABSSSIs, all comparators were dominated by (i.e., were more costly and less effective than) dalbavancin IV. In a subgroup of patients with nonsevere ABSSSIs, dalbavancin IV was associated with incremental costs of $446 and incremental 0.0005 QALYs, compared to ceftriaxone IV, resulting in an ICER of $832,245 per QALY gained (Table 3).
Table 3: Summary of the Sponsor’s Economic Evaluation Results — Total Population
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
Ceftriaxone IV | 15,198 | 0.4429 | Reference |
Dalbavancin IV | 15,437 | 0.4435 | 401,168 |
Cefazolin IV | 15,564 | 0.4428 | Dominateda |
Daptomycin IV | 16,092 | 0.4428 | Dominateda |
Vancomycin IV | 16,139 | 0.4428 | Dominateda |
Linezolid IV | 16,539 | 0.4429 | Dominateda |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
aDominated refers to a treatment having higher total costs and lower total QALYs compared to the previous less costly treatment.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analyses Results
The sponsor provided several scenario analyses. The scenario conducted on a subpopulation of patients with kidney dysfunction did not show an important impact on the ICER. In another scenario conducted on a subpopulation of people who are unhoused and people who misuse IV drugs, dalbavancin IV dominated (i.e., was less costly and more effective than) all other comparators. The sponsor reported that dalbavancin IV was associated with a cost savings of $21,793 and an incremental QALY gain of 0.0060 compared with vancomycin IV in the latter subpopulation.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis:
There is uncertainty in the assumption of equivalent efficacy between dalbavancin IV and comparators: The sponsor assumed equal clinical efficacy between dalbavancin IV and vancomycin IV, followed by oral linezolid, based on 2 double-blind, double-dummy, randomized, noninferiority trials: DISCOVER 1 and DISCOVER 2. There were concerns about the external validity of the trial comparators used in the randomized controlled trials, as they are not reflective of Canadian clinical practice, according to clinical expert feedback received by CADTH. In addition, the rate of microbiological diagnosis was higher in the trials than in clinical practice, and the selection of the comparator did not reflect treatment used to treat ABSSSIs caused by a non–methicillin-resistant Staphylococcus aureus pathogen. This limited the generalizability of the trial results.
In the absence of direct comparative evidence, the sponsor assumed equal clinical efficacy for dalbavancin IV, linezolid IV, and daptomycin IV, based on a sponsor-submitted indirect treatment comparison. However, the CADTH clinical review noted several methodological limitations that made the evidence uncertain, including inadequate adjustment for heterogeneity in patient and study characteristics, such as study duration, blinding, dosage, disease definition, and the sparse data for some contrasts and outcomes. Further, there was no direct or indirect comparative evidence to support the equal clinical efficacy of dalbavancin IV, ceftriaxone IV, and cefazolin IV. As such, the assumption of clinical efficacy between dalbavancin IV and these comparators is highly uncertain.
The probabilistic sensitivity analysis of the sponsor’s base case demonstrated that there was no meaningful difference in total QALYs accrued between dalbavancin IV and active comparators. There was a high degree of overlap in the confidence intervals (refer to Table 12 in Appendix 3), indicating the possibility that QALYs accrued with dalbavancin IV may be lower than QALYs accrued with active comparators.
In reanalyses, CADTH assumed that there would be no difference in total QALYs accrued among treatments.
There is insufficient evidence to support an early discharge rate associated with dalbavancin IV: The sponsor obtained early discharge rates associated with dalbavancin IV from sponsor-consulted clinical experts. There was no direct or indirect comparative evidence to support early discharge rates with dalbavancin IV, relative to comparators, in patients with severe ABSSSIs. The evidence supporting lower hospitalization rates for patients with nonsevere ABSSSIs treated with dalbavancin IV was obtained using the pre-post pragmatic study by Talan et al. (2021),7 which was a naive comparison of hospitalization rates before and after dalbavancin IV was administered in a hospital setting. The study design was prone to risk of selection and performance bias, given the patients were discharged at the discretion of the treating physician during the pre-treatment and post-treatment periods. The risk of bias may have affected the length of hospital stay and the admission rate in favour of dalbavancin IV; however, the magnitude of this bias could not be inferred.
In reanalysis, CADTH assumed no difference in discharge rates between dalbavancin and the comparators.
Incremental costs are highly uncertain: Under the assumption of equal clinical efficacy and safety between dalbavancin IV and comparators, the cost savings derived from early discharge rates were a key driver of the sponsor’s results. However, there was insufficient evidence to support early discharge rates with dalbavancin IV. The sponsor explored the uncertainty in hospitalization costs by assuming a standard deviation of 10% of the mean. This method lacks face validity and likely does not reflect the true uncertainty in cost, as uncertainty in cost would not be expected to be identical for all comparators, and the 10% value is arbitrary. The probabilistic sensitivity analysis of the sponsor’s base case demonstrated that there was no meaningful difference in total costs accrued with dalbavancin IV, relative to comparators, given that there was overlap in the confidence intervals associated with treatment costs (refer to Table 13 in Appendix 3). In reanalysis, CADTH assumed no difference in total costs between dalbavancin IV and active comparators.
Relevant comparators are excluded: The sponsor restricted the analysis to IV comparators. The clinical experts consulted by CADTH for this review noted that oral antibiotics, which are reimbursed by several of the public formularies, may be prescribed for the treatment of ABSSSIs in nonsevere cases. As these comparators were not included in the analysis, their cost-effectiveness compared to dalbavancin IV is unknown; however, notably, their list price in public formularies is less costly than dalbavancin IV (Appendix 1). The exclusion of oral comparators from the sponsor’s submission adds uncertainty to both costs and outcomes of dalbavancin.
CADTH was not able to address the cost-effectiveness of dalbavancin compared to oral treatments. The cost-effectiveness of dalbavancin in the nonsevere ABSSSI population is unknown.
The impact of treatment recurrence on costs and outcomes is uncertain: The sponsor assumed that patients on comparator treatments were switched to oral antibiotics at hospital discharge and experienced a 25% increase in recurrence rate, compared to dalbavancin IV. The sponsor’s assumption lacks face validity. Based on clinical expert feedback solicited by CADTH for this review, the risk of reinfection after cure is not solely dependent on the treatment used, but also depends on patient health and environmental factors. It was also unclear how the sponsor derived the estimated 25% increase in the risk of recurrence for oral antibiotics compared to IV antibiotics from an observational study that is noncomparative in design.12 As such, there is insufficient direct and indirect comparative evidence to support the sponsor’s assumption. The sponsor’s assumption results in subsequent treatment costs that are lower for dalbavancin IV than for comparators, which inappropriately biases the results in favour of dalbavancin IV.
In reanalysis, CADTH assumed no differences in recurrence rates between IV antibiotics and oral antibiotics.
The key assumptions made by the sponsor and appraised by CADTH are outlined in Table 4.
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
Treatment paradigm | The submitted model structure lacked logic and face validity. The clinical expert consulted by CADTH noted that treatment with comparators is adjusted, if needed, based on clinical response and results of micro-organism diagnosis. The sponsor assumed that patients who fail dalbavancin IV treatment switch to another antibiotic on day 4 after the initiation of treatment. According to the clinical expert, there is no opportunity to switch or withdraw treatment once the long-acting dose has been administered until approximately day 8. |
Impact of kidney dysfunction | Uncertain. The sponsor assumed no change in the rate of clinical failure for patients with kidney dysfunction who were treated with dalbavancin IV. In contrast, the rate of treatment failure was increased for patients with reduced renal function who were treated with vancomycin IV, daptomycin IV, or cefazolin IV. There is insufficient evidence to support the rate of clinical failure for patients with kidney dysfunction, which was estimated using feedback from clinical experts consulted by the sponsor. The sponsor’s assumption biases the results in favour of dalbavancin IV because a higher proportion of patients treated with comparators than with dalbavancin IV experienced treatment failure. |
Proportion of patients with nonsevere vs. severe ABSSSIs | Uncertain. The sponsor assumed that 72.3% of ABSSSI cases are nonsevere. The clinical expert consulted for this review by CADTH noted that there is no standardized severity classification available for ABSSSIs. Severity definition is subjective in clinical practice and may depend on a range of factors, including surface area affected, presence of fever, white blood cell count, and organ failure. |
Treatment duration | Uncertain. The sponsor assumed equal treatment duration for all antibiotics. However, the length of treatment varies. The clinical expert consulted for this review noted that treatment duration is higher individualized and subjective, depending on patient status at reassessment and physician’s discretion. |
Health state utilities were obtained from published literature | Uncertain. To inform the pharmacoeconomic model, the sponsor adopted health state utility values from a prospective observational study of 1,033 patients with complicated skin and soft tissue infection in the US. It is uncertain whether preferences of patients hospitalized in the US reflect preferences of patients hospitalized in Canada. |
ABSSSI = acute bacterial skin and skin structure infection.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
Key limitations were identified in the available clinical efficacy data that informed the treatment benefit of dalbavancin IV. The CADTH critical appraisal of the clinical evidence concluded that dalbavancin IV demonstrated noninferiority to vancomycin IV, followed by oral linezolid, but the evidence was uncertain because the comparator was not generalizable to Canadian clinical practice. The indirect comparative evidence for treatments such as linezolid IV and daptomycin IV was uncertain because of methodological limitations, which included concerns about heterogeneity in study duration, blinding, dosage, disease definition, and patient demographics. There was also no efficacy or safety data that compared dalbavancin IV to cefazolin IV or ceftriaxone IV. Additionally, CADTH identified key limitations of the evidence used to support early discharge rates with dalbavancin IV, missing comparators from the analysis, uncertain incremental costs, and imprecise use of disease recurrence rate. Given the uncertainty in the noninferiority assumption and in the absence of available evidence in support of a treatment benefit with dalbavancin IV relative to standard-of-care treatments, CADTH assumed no difference in treatment effects (i.e., no difference in total QALYs) and compared treatment-course costs of dalbavancin IV with active comparators in its reanalysis. CADTH does not present results for the severe and nonsevere ABSSSIs separately.
In the CADTH reanalysis, the cost of IV comparators (cefazolin, ceftriaxone, daptomycin, linezolid, and vancomycin) ranged from $97 to $1,775 per patient. All active comparators were less costly than dalbavancin IV. The treatment cost of each comparator, along with the difference in treatment costs between dalbavancin IV and comparators, can be found in Table 5.
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Price reduction scenarios | Treatment cost of dalbavancin IV ($) | Reduction needed (%) | Reduced treatment cost of dalbavancin IV ($) | Savings in treatment cost ($) |
|---|---|---|---|---|
IV drug | ||||
Cefazolin 250 mg to 500 mg every 8 hours, or 500 mg to 1,000 mg every 6 hours | 2,872 | 95 to 97 | 97 to 129 | 2,742 to 2,775 |
Ceftriaxone 1,000 mg to 2,000 mg every 24 hours | 2,872 | 92 to 96 | 125 to 241 | 2,630 to 2,747 |
Daptomycin 4 mg/kg every 24 hours | 2,872 | 44 | 1,610 | 1,262 |
Linezolid 600 mg IV every 12 hours | 2,872 | 38 | 1,775 | 1,097 |
Vancomycin 500 mg every 6 hours, or 1,000 mg every 12 hours | 2,872 | 86 to 87 | 376 to 395 | 2,477 to 2,496 |
Note: The range of prices was calculated using the dose recommended in the product monographs. Drug wastage was included in the estimated costs. A treatment duration of 10 days was adopted using the Practice Guidelines for the Diagnosis and Management of Skin and Soft Tissue Infections (2014).13 CADTH’s reanalysis is based on publicly available prices of the comparator treatments.
Price Reduction Analyses
In the absence of clinical information to justify a price premium for dalbavancin IV, and given that the treatment cost of dalbavancin IV is higher than all other treatments assessed, price reduction analyses were conducted to understand the percentage reductions required for the price of dalbavancin IV to be similar to that of active comparators. The submitted price of dalbavancin IV would need to be reduced by 38% to 97% for the treatment cost to be equivalent to that of the IV comparators.
Table 6: CADTH Price Reduction Analyses
Scenario | Sponsor’s submitted price ($) | Reduction needed (%) | Reduced price ($) | Savings in treatment cost ($) |
|---|---|---|---|---|
IV treatment | ||||
Cefazolin 250 mg to 500 mg every 8 hours, or 500 mg to 1,000 mg every 6 hours | 957 | 95 to 97 | 32 to 43 | 2,742 to 2,775 |
Ceftriaxone 1,000 mg to 2,000 mg every 24 hours | 957 | 92 to 96 | 42 to 80 | 2,630 to 2,747 |
Daptomycin 4 mg/kg every 24 hours | 957 | 44 | 537 | 1,262 |
Linezolid 600 mg IV every 12 hours | 957 | 38 | 592 | 1,097 |
Vancomycin 500 mg every 6 hours, or 1,000 mg every 12 hours | 957 | 86 to 87 | 125 to 132 | 2,477 to 2,496 |
Note: CADTH’s reanalysis is based on publicly available prices of the comparator treatments. Drug wastage was included in the estimated costs.
Issues for Consideration
Given that dalbavancin IV is a single-dose, long-acting drug, immediate treatment withdrawal or switch in cases of allergic reaction or treatment failure is difficult to implement. The sponsor assumed no difference in the time to a treatment switch or withdraw, which is inappropriate, given that treatment may be switched or withdrawn earlier with comparators than with dalbavancin IV. The effectiveness and cost-effectiveness of dalbavancin IV in patients who need to withdraw or switch treatment is unknown.
CADTH’s estimate of the 3-year budget impact are subject to a high degree of uncertainty. The sponsor’s estimate of market share was not transparent and clinical expert feedback received by CADTH suggested that it is underestimated to a degree that is unknown and could not be estimated. Further, there is uncertainty about whether the cost of dalbavancin IV would be covered by hospitals or the drug plan budget. These sources of uncertainty could not be explored by CADTH. The true budget impact is unknown but is likely to be higher than the sponsor’s estimate.
Overall Conclusions
The CADTH clinical review concluded that dalbavancin IV demonstrated noninferiority to vancomycin IV (with an optional switch to oral linezolid) and linezolid IV. However, the evidence was uncertain because of concerns regarding the validity of outcomes and the selection of comparators. The evidence was not available for all comparators, and the indirect treatment comparison was limited because of heterogeneity and sparse data for safety end points. These factors hindered the generalizability and interpretation of the results. There was also no significant difference in the length of hospital stay between treatments, but the design of the pragmatic trials and use of a pathway intervention limited the interpretation of results.
Because of the limitations of the available comparative evidence, the CADTH reanalysis assumed no difference in effectiveness (i.e., equal QALYs) between dalbavancin IV and all active comparators for the treatment of ABSSSIs, and a cost comparison to assess annual drug costs was conducted. The cost comparison adopted a treatment duration of 10 days. The treatment-course cost of dalbavancin IV is $2,872 per patient, which is more than the publicly available list prices of all IV and oral treatments for ABSSSIs. The submitted price of dalbavancin IV would need to be reduced by at least 38% to 97% to be equivalent to the lowest-priced generic IV treatment. When considering the most and least costly oral comparator, the submitted price would need to be reduced by 74% and 100%, respectively, for treatment to be cost-neutral.
Based on the CADTH clinical and economic reviews, there is insufficient evidence to support a price premium for dalbavancin IV over other oral and IV antibiotics for the treatment of ABSSSIs. CADTH notes that although the noninferiority of dalbavancin IV to vancomycin IV and linezolid IV has been demonstrated, the equivalence in effectiveness for other comparators was based on an assumption. The wide credible intervals around the estimated comparative effectiveness of dalbavancin suggest the possibility that dalbavancin is less effective than comparator treatments. Dalbavancin IV could be dominated (i.e., be more costly and less effective) by active comparators. CADTH could not fully explore this uncertainty because of the lack of available evidence and, therefore, the possibility that dalbavancin IV generates fewer QALYs at a higher cost than active comparators at any price reduction should be considered.
CADTH notes that clinical expert feedback suggested that although nonsevere ABSSSI does not have a consistent clinical definition, less-severe cases may be managed with oral antibiotics. Because of a lack of evidence to inform the way treatment adherence and recurrence affects both comparative efficacy and cost between dalbavancin IV and oral comparators, the cost-effectiveness of dalbavancin IV in nonsevere cases is unknown. The cost of oral treatments is, however, even lower than the comparators included in the sponsor’s analysis and further price reduction may be warranted.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Xydalba (dalbavancin for IV injection), 500 mg for injection. Montreal (QC): Paladin Labs Inc; 2022 Apr 11.
2.Boucher HW, Wilcox M, Talbot GH, Puttagunta S, Das AF, Dunne MW. Once-weekly dalbavancin versus daily conventional therapy for skin infection. N Engl J Med. 2014;5(370):2169-2179. PubMed
3.Guest JF, Esteban J, Manganelli AG, Novelli A, Rizzardin IG, Serra M. Comparative efficacy and safety of antibiotics used to treat acute bacterial skin and skin structure infections: results of a network meta-analysis. PLoS One. 2017;12(11):e0187792. PubMed
4.Vardakas KZ, Mavros MN, Roussos N, Falagas ME. Meta-analysis of randomized controlled trials of vancomycin for the treatment of patients with gram-positive infections: focus on the study design. Mayo Clin Proc. 2012;87(4):349-363. PubMed
5.Lipsky BA, Moran GJ, Napolitano LM, Vo L, Nicholson S, Kim M. A prospective, multicenter, observational study of complicated skin and soft tissue infections in hospitalized patients: clinical characteristics, medical treatment, and outcomes. BMC Infect Dis. 2012;12(227). PubMed
6.Marcellusi A, Viti R, Sciattella P, et al. Economic evaluation of the treatment of acute bacterial skin and skin structure infections (ABSSSIs) from the national payer perspective: introduction of a new treatment to the patient journey. A simulation of three European countries. Expert Rev Pharmacoecon Outcomes Res. 2019;19(5):581-599. PubMed
7.Talan DA, Mower WR, Lovecchio FA, et al. Pathway with single-dose long-acting intravenous antibiotic reduces emergency department hospitalizations of patients with skin infections. Acad Emerg Med. 2021;28(10):1108-1117. PubMed
8.EQ-5D-5L index norms for Alberta population. Calgary (AB): Health Quality Council of Alberta; 2016.
9.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2022; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2021 Nov 5.
10.Government of Alberta. Interactive drug benefit list. 2022.
11.Patient cost estimator. 2022; https://www.cihi.ca/en/patient-cost-estimator. Accessed 2021 Jul 18.
12.Eells SJ, Nguyen M, Jung J, Macias-Gil R, May L, Miller LG. Relationship between adherence to oral antibiotics and postdischarge clinical outcomes among patients hospitalized with staphylococcus aureus skin infections. Antimicrob Agents Chemother. 2016;60(5):2941-2948. PubMed
13.Stevens DL, Bisno AL, Chambers HF, et al. Practice guidelines for the diagnosis and management of skin and soft tissue infections: 2014 update by the Infectious Diseases Society of America. Clin Infect Dis. 2014;59(2):147-159. PubMed
14.Zyvoxam (linezolid): solution, 2 mg/mL, for intravenous infusion, powder for suspension, 100 mg/5mL when reconstituted, for oral use [product monograph]. Kirkland (QC): Pfizer Canada ULC; 2022 Jan 27: https://www.pfizer.ca/sites/default/files/202203/ZYVOXAM_PM_EN_256427_27-Jan-2022.pdf. Accessed 2022 Jul 7.
15.Vancomycin Hydrochloride for injection (vancomycin hydrochloride):500 mg, 1 g, 5 g and 10 g of vancomycin (as vancomycin hydrochloride) per vial, sterile lyophilized powder for solution [product monograph]. Kirkland (QC): Pfizer Canada ULC 2021 Jul 21: https://www.pfizer.ca/sites/default/files/202108/Vancomycin_Hydrochloride_PM_EN_249989_21-July-2021.pdf. Accessed 2022 Jul 7.
16.Amoxicillin Sodium and Potassium Clavulanate for injection: powder for solution for intravenous injection 500 mg amoxicillin (as amoxicillin sodium) and 100 mg clavulanic acid (as clavulanate potassium), 1000 mg amoxicillin (as amoxicillin sodium) and 200 mg clavulanic acid (as clavulanate potassium), 2000 mg amoxicillin (as amoxicillin sodium) and 200 mg clavulanic acid (as clavulanate potassium) [product monograph]. Boucherville (QC): Sandoz Canada Inc; 2020 Jan 31: https://www.sandoz.ca/sites/www.sandoz.ca/files/131-AmoxClav%20Inj%20PMe%2020200131.pdf?CID=mvbrief_cshp. Accessed 2022 Jul 7.
17.Cubicin/Cubicin (daptomycin for injection): lyophilized powder for solution, for intravenous use only 10 mL vial, 500 mg/vial [product monograph]. Lucerne (CH): Cubist Pharmaceuticals LLC; 2020 May 15.
18.Sulfatrim (sulfamethoxazole and trimethoprim tablets USP) 400/80 mg; Sulfatrim DS (sulfamethoxazole and trimethoprim tablets USP) 800/160 mg; Sulfatrim Pediatric (sulfamethoxazole and trimethoprim tablets USP) 100/20 mg [product monograph]. Vaughan (ON): AA Pharma Inc; 2019 Jan 17: https://pdf.hres.ca/dpd_pm/00049327.PDF. Accessed 2022 Jul 7.
19.Teva-Cloxacillin (cloxacillin sodium): 250 and 500 mg capsules, 125 mg/5 ml granules for oral solution [product monograph]. Toronto (ON): Teva Canada Limited; 2018 Jun 21: https://pdf.hres.ca/dpd_pm/00046029.PDF. Accessed 2022 Jul 7.
20.Keflex (cephalexin): tablets and oral suspensions, 250 mg, 500 mg, 125 mg/5 mL and 250 mg/5 mL [product monograph]. Montreal (QC): Pendopharm, Division of Pharmascience Inc; 2018 May 1: https://pdf.hres.ca/dpd_pm/00045523.PDF. Accessed 2022 Jul 7.
21.Vibramycin capsules (doxycycline hyclate capsules USP, doxycycline): 100 mg; Vibra-Tabs film coated tablets (doxycycline hyclate tablets USP, doxycycline): 100 mg [product monograph]. Kirkland (QC): Pfizer Canada Inc; 2015 Dec 22: https://pdf.hres.ca/dpd_pm/00033153.PDF. Accessed 2022 Jul 7.
22.Apo-Amoxi Clav (amoxicillin and clavulanic acid tablets USP): 250 mg amoxicillin and 125 mg clavulanic acid / tablet, 500 mg amoxicillin and 125 mg clavulanic acid / tablet, 875 mg amoxicillin and 125 mg clavulanic acid / tablet (as amoxicillin trihydrate and clavulanate potassium); Amoxicillin and Clavulanic acid powder for oral suspension USP: 125 mg amoxicillin and 31.25 mg clavulanic acid / 5 ml, 250 mg amoxicillin and 62.5 mg clavulanic acid / 5 ml, 400 mg amoxicillin and 57 mg clavulanic acid / 5 ml (as amoxicillin trihydrate and clavulanate potassium) [product monograph]. Toronto (ON): Apotex Inc; 2014 May 8: https://pdf.hres.ca/dpd_pm/00025149.PDF. Accessed 2022 Jul 7.
23.Cloxacillin for injection, Cloxacillin powder for solution (as cloxacillin sodium): 500 mg powder/vial, 1g powder/vial, 2g powder/vial, 10g powder/vial [product monograph]. Mississauga (ON): SteriMax Inc; 2013 Jan 29: https://pdf.hres.ca/dpd_pm/00019026.PDF. Accessed 2022 Jul 7.
24.Ceftriaxone Sodium for injection BP (ceftriaxone as ceftriaxone sodium BP): 0.25 gm, 1 gm, 2 gms, 10 gm per vial [product monograph]. Boucherville (QC): Sandoz Canada Inc; 2010 Nov 15: https://www.sandoz.ca/sites/www.sandoz.ca/files/Ceftriaxone%20Product%20Monograph.pdf. Accessed 2022 Jul 7.
25.Cefazolin for injection USP (cefazolin as cefazolin sodium): 500 mg, 1 g & 10 g per vials [product monograph]. Boucherville (QC): Sandoz Canada Inc; 2010 Nov 15: https://www.sandoz.ca/sites/www.sandoz.ca/files/Cefazolin%20Monograph.pdf. Accessed 2022 Jul 7.
26.Saskatchewan Drug Plan: search formulary. 2022; https://formulary.drugplan.ehealthsask.ca/SearchFormulary. Accessed 2022 Jul 31.
27.B. C. Government. BC PharmaCare formulary search. 2022; https://pharmacareformularysearch.gov.bc.ca. Accessed 2022 Jul 31.
28.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Xydalba (dalbavancin), 500 mg for injection [internal sponsor's package]. Montreal (QC): Paladin Labs Inc; 2022 Apr 11.
29.Institut national d'excellence en santé et en services sociaux. Zyvoxammc – Infections à staphylocoques résistants à la méthicilline. Montreal (QC): INESSS; 2013.
30.Kaye KS, Petty LA, Shorr AF, Zilberberg MD. Current epidemiology, etiology, and burden of acute skin infections in the United States. Clin Infect Dis. 2019;68(Suppl 3):S193-S199. PubMed
Appendix 1: Cost Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from the clinical expert(s). Comparators may be recommended (appropriate) practice or actual practice. Existing product listing agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 7: CADTH Cost Comparison for ABSSSIs
Treatment | Strength / concentration | Form | Price ($) | Recommended dosage | Daily cost ($) | Course cost ($) |
|---|---|---|---|---|---|---|
Dalbavancin | 500 mg | Vial IV infusion | 957.1679a | A single dose of 1,500 mg Alternative dosing: 1,000 mg followed by 500 mg 1 week later | 2,871.50 | 2,872 |
IV infusion drugs | ||||||
Cefazolin | 500 mg 1,000 mg 10,000 mg | Vial IV infusion | 4.0000 3.2308b 30.1500b | Mild infections caused by susceptible gram-positive cocci: 250 mg to 500 mg every 8 hoursf Moderate to severe infection: 500 mg to 1,000 mg every 6 to 8 hours | 9.69 Moderate to severe infection: 9.69 to 12.92 | 97 Moderate to severe infection: 97 to 129 |
Ceftriaxone | 250 mg 500 mg 1,000 mg 2,000 mg 10,000 mg | Vial IV infusion | 3.9501 NA 12.4950 24.1395 214.2000 | 1,000 mg to 2,000 mg every 24 hoursg | 12.50 to 24.14 | 175 to 338 |
Cloxacillin | 125 mg/5 mL | Vial IV infusion | 0.0606 | Mild to moderate infection: 250 mg to 500 mg every 6 hoursh Severe infection: up to 6,000 mg every 24 hours | Mild to moderate infection: 0.48 to 0.97 Severe infection: 2.91 | 2 to 5 Severe infection: 15 |
Daptomycin | 500 mg | Vial IV infusion | 161.0000b,c | 4 mg/kg should be administered intravenously once every 24 hoursg | 161.00 | 2,254 |
Linezolid | 2 mg/mL | Vial (300 mL) IV infusion | 88.7400 | 600 mg every 12 hoursg | 177.48 | 2,485 |
Vancomycin | 500 mg 1,000 mg 5,000 mg 10,000 mg | Vial IV infusion | 9.8669d 18.7810d 316.1160e 589.9000d | 500 mg every 6 hours or 1,000 mg every 12 hoursi | 37.56 to 39.47 | 376 to 395 |
Oral drugs | ||||||
Amoxicillin/ clavulanic acid | 250 mg / 125 mg | Tablet | 0.2467 | Tablet: 500 mg / 125 mg to 875 mg / 125 mg every 12 hours or 500 mg / 125 mg every 8 hours | 0.76 to 2.27 | 8 to 23 |
500 mg / 125 mg | 0.3778 | |||||
875 mg / 125 mg | 0.5551 | |||||
125 / 31.25 mg | Powder for oral suspension (in 5 mL vial for IV infusion) | 0.1009d | ||||
250 mg / 62.5 mg | 0.2173d | |||||
200 mg / 28.5 mg | 0.1537d | |||||
400 mg / 57 mg | 0.3039d | |||||
Cephalexin | 250 mg | Capsule | 0.3703 | 1,000 mg to 4,000 mg dailyf | 1.45 to 5.80 | 15 to 58 |
500 mg | 0.7252 | |||||
250 mg | Tablet | 0.0866 | 0.35 to 1.38 | 3 to 14 | ||
500 mg | 0.1731 | |||||
125 mg/5 mL | Oral suspension | 0.2193 | 1.47 to 5.88 | 15 to 59 | ||
250 mg/5 mL | 0.3675 | |||||
Cloxacillin | 250 mg 500 mg | Capsule | 0.2141d 0.4045d | Mild to moderate infection: 250 mg to 500 mg every 6 hoursh | 0.48 to 0.97 | 2 to 5 |
125 mg /5 mL | Granules for oral solution | 0.0606 | Severe infection: up to 6,000 mg every 24 hours | 4.85 | 24 | |
Doxycycline | 100 mg 100 mg | Capsule Tablet | 0.5860 0.5860 | Loading dose: 200 mg on Day 1i Maintenance dose: 100 mg to 200 mg once daily | Loading dose: 1.17 Maintenance dose: 0.59 | Loading dose: 1.17 Maintenance dose: 23 |
Sulfamethoxazole and trimethoprim | 400 / 80 mg 800 / 160 mg | Tablet | 0.0482d 0.2074 | Two 400 / 80 mg tablets or one 800 / 160 mg tablet twice dailyh Severe: three 400 / 80 mg tablets or one and a half 800 / 160 mg tablets twice daily | 0.19 to 0.41 0.29 to 0.62 | 1 to 2 1 to 3 |
Linezolid | 400 mg | Tablet | NA | 600 mg every 12 hoursg | 74.10 | 1,037 |
600 mg | 37.0500b,c | |||||
100 mg/5 mL | Powder for oral suspension | 2.6232 | 31.48 | 441 | ||
NA = not available.
Note: All prices are from the Ontario Drug Benefit Formulary (accessed July 2022), unless otherwise indicated, and do not include dispensing fees. Drug wastage was included. Recommended drug dose was obtained from respective product monographs.14-25 Cloxacillin injection is not included in the cost table because the product monograph noted that injection is not effective against the methicillin-resistant strains of Staphylococcus.23
aPrice obtained from sponsor’s submission.1
bAlberta Interactive Drug Benefit List,10 accessed July 2022.
cCoverage available by requesting special authorization.10
dSaskatchewan Drug Plan,26 accessed July 2022.
eBritish Columbia PharmaCare Formulary,27 accessed July 2022.
fThe recommended treatment duration of 10 days was adopted.16,20,22,25
gRecommended treatment duration of 14 days was adopted in estimating treatment-course cost.14,17,24
hRecommended treatment duration of 5 days was adopted in estimating treatment-course cost.18,19
iA treatment duration of 10 days was assumed based guidelines for management of skin and soft tissue infections.13
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/no | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | Refer to CADTH appraisal section. |
Model has been adequately programmed and has sufficient face validity | No | Refer to CADTH appraisal section. |
Model structure is adequate for decision problem | Yes | No comment. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | Refer to CADTH appraisal section. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | No | Refer to CADTH appraisal section. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | No | CADTH identified limitations with the submission related to transparency, including difficulty with tracing model parameters and locating source of evidence used in the model. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
Figure 1: Model Structure of the Decision Tree for Patients With Severe ABSSSIs
Source: Sponsor’s pharmacoeconomic submission.1
Figure 2: Model Structure of the Decision Tree for Patients With Nonsevere ABSSSIs
Source: Sponsor’s pharmacoeconomic submission.1
Detailed Results of the Sponsor’s Base Case
Table 9: Disaggregated Summary of the Sponsor’s Economic Evaluation Results — Total Population With ABSSSIs
Parameter | Dalbavancin IV | Vancomycin IV | Linezolid IV | Cefazolin IV | Ceftriaxone IV | Daptomycin IV |
|---|---|---|---|---|---|---|
LYs | ||||||
Total LYs | 0.54 | 0.54 | 0.54 | 0.54 | 0.54 | 0.54 |
Hospital days saved | ||||||
Number of hospital days | 5.7 | 6.9 | 6.7 | 6.9 | 6.7 | 6.9 |
QALYs | ||||||
Total QALYs | 0.4443 | 0.4436 | 0.4437 | 0.4436 | 0.4437 | 0.4436 |
QALYs generated within trial period | 0.0294 | 0.0290 | 0.0290 | 0.0290 | 0.0290 | 0.0290 |
QALYs generated after trial period | 0.4149 | 0.4146 | 0.4147 | 0.4146 | 0.4147 | 0.4146 |
Costs | ||||||
Total costs ($) | 15,488 | 16,207 | 16,609 | 15,630 | 15,266 | 16,159 |
Drug ($) | 2,872 | 286 | 1,166 | 179 | 173 | 663 |
Subsequent treatments ($) | 290 | 334 | 335 | 334 | 302 | 334 |
Hospitalization ($) | 12,188 | 14,803 | 14,445 | 14,803 | 14,445 | 14,803 |
Drug administration (infusion) ($) | 39 | 697 | 575 | 225 | 258 | 272 |
Other medical resources ($) | 100 | 87 | 88 | 87 | 88 | 87 |
Productivity ($) | 0 | 0 | 0 | 0 | 0 | 0 |
LY = life-year; QALY = quality-adjusted life-year.
Table 10: Disaggregated Summary of the Sponsor’s Economic Evaluation Results — Population With Severe ABSSSIs
Parameter | Dalbavancin IV | Vancomycin IV | Linezolid IV | Cefazolin IV | Ceftriaxone IV | Daptomycin IV |
|---|---|---|---|---|---|---|
LYs | ||||||
Total LYs | 0.54 | 0.54 | 0.54 | 0.54 | 0.54 | 0.54 |
Hospital days saved | ||||||
Number of hospital days | 9.9 | 11.4 | 11.3 | 11.4 | 11.3 | 11.4 |
QALYs | ||||||
Total QALYs | 0.4420 | 0.4412 | 0.4412 | 0.4412 | 0.4412 | 0.4412 |
QALYs generated within trial period | 0.0270 | 0.0265 | 0.0265 | 0.0265 | 0.0265 | 0.0265 |
QALYs generated after trial period | 0.4149 | 0.4146 | 0.4147 | 0.4146 | 0.4147 | 0.4146 |
Costs | ||||||
Total costs ($) | 24,653 | 25,574 | 26,145 | 25,267 | 24,983 | 25,770 |
Drug ($) | 2,872 | 286 | 1,166 | 179 | 173 | 663 |
Subsequent treatments ($) | 290 | 334 | 335 | 334 | 302 | 334 |
Hospitalization ($) | 21,423 | 24,525 | 24,266 | 24,525 | 24,266 | 24,525 |
Drug administration (infusion) ($) | 39 | 404 | 354 | 204 | 218 | 224 |
Other medical resources ($) | 30 | 24 | 24 | 24 | 24 | 24 |
Productivity ($) | 0 | 0 | 0 | 0 | 0 | 0 |
LY = life-year; QALY = quality-adjusted life-year.
Table 11: Disaggregated Summary of the Sponsor’s Economic Evaluation Results — Nonsevere ABSSSI Population
Parameter | Dalbavancin IV | Vancomycin IV | Linezolid IV | Cefazolin IV | Ceftriaxone IV | Daptomycin IV |
|---|---|---|---|---|---|---|
LYs | ||||||
Total LYs | 0.54 | 0.54 | 0.54 | 0.54 | 0.54 | 0.54 |
Hospital days saved | ||||||
Number of hospital days | 4.0 | 5.1 | 5.0 | 5.1 | 5.0 | 5.1 |
QALYs | ||||||
Total QALYs | 0.4452 | 0.4446 | 0.4447 | 0.4446 | 0.4447 | 0.4446 |
QALYs generated within trial period | 0.0303 | 0.0299 | 0.0300 | 0.0299 | 0.0300 | 0.0299 |
QALYs generated after trial period | 0.4149 | 0.4146 | 0.4147 | 0.4146 | 0.4147 | 0.4146 |
Costs | ||||||
Total costs ($) | 11,979 | 12,620 | 12,957 | 11,939 | 11,545 | 12,479 |
Drug ($) | 2,872 | 286 | 1,166 | 179 | 173 | 663 |
Subsequent treatments ($) | 290 | 334 | 335 | 334 | 302 | 334 |
Hospitalization ($) | 8,651 | 11,080 | 10,684 | 11,080 | 10,684 | 11,080 |
Drug administration (infusion) ($) | 39 | 808 | 660 | 233 | 273 | 290 |
Other medical resources ($) | 127 | 112 | 113 | 112 | 113 | 112 |
Productivity ($) | 0 | 0 | 0 | 0 | 0 | 0 |
LY = life-year; QALY = quality-adjusted life-year.
Table 12: PSA Results for the Total Population (Mean* and 95% Confidence Interval), QALYs
Drug | Total QALYs — mean | Total QALYs — lower 95% confidence Interval | Total QALYs — upper 95% confidence interval |
|---|---|---|---|
Dalbavancin IV | 0.4435 | 0.3599 | 0.5385 |
Vancomycin IV | 0.4428 | 0.3595 | 0.5373 |
Linezolid IV | 0.4429 | 0.3595 | 0.5375 |
Cefazolin IV | 0.4428 | 0.3595 | 0.5373 |
Ceftriaxone IV | 0.4429 | 0.3595 | 0.5375 |
Daptomycin IV | 0.4428 | 0.3595 | 0.5373 |
PSA = probabilistic sensitivity analysis; QALY = quality-adjusted life-year.
*Mean results were calculated based on 1,000 Monte Carlo iterations as noncongruence tests showed that it was sufficient for the model to converge.
Table 13: PSA Results for the Total Population (Mean* and 95% Confidence Interval), Costs
Drug | Total costs — mean ($) | Total costs — lower 95% confidence interval ($) | Total costs — upper 95% confidence interval ($) |
|---|---|---|---|
Dalbavancin IV | 15,437 | 13,471 | 17,502 |
Vancomycin IV | 16,139 | 13,560 | 18,888 |
Linezolid IV | 16,539 | 13,890 | 19,384 |
Cefazolin IV | 15,564 | 13,140 | 18,119 |
Ceftriaxone IV | 15,198 | 12,832 | 17,703 |
Daptomycin IV | 16,092 | 13,565 | 18,768 |
PSA = probabilistic sensitivity analysis.
*Mean results were calculated based on 1,000 Monte Carlo iterations as noncongruence tests showed that it was sufficient for the model to converge.
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
CADTH did not conduct any additional pharmacoeconomic analyses in the review of dalbavancin IV.
Appendix 5: Submitted BIA and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 14: Summary of Key Take-Aways
Key take-aways of the BIA |
|---|
|
Summary of Sponsor’s BIA
The sponsor submitted a budget impact analysis (BIA) estimating the incremental budget impact of reimbursing dalbavancin for use by patients aged 18 years and older with ABSSSIs, caused by susceptible isolates of the following gram-positive micro-organisms: Staphylococcus aureus (including methicillin-susceptible and methicillin-resistant strains), Streptococcus pyogenes, Streptococcus agalactiae, Streptococcus dysgalactiae, Streptococcus anginosus group (including Streptococcus anginosus, Streptococcus intermedius, Streptococcus constellatus) and Enterococcus faecalis (vancomycin-susceptible strains).28 The BIA was undertaken from the perspective of the Canadian public drug plans over a 3-year time horizon, and the sponsor’s pan-Canadian estimates reflect the aggregated results from provincial budgets (excluding Quebec). Key inputs to the BIA are documented in Table 15.
The sponsor estimated the number of eligible patients using an epidemiologic approach. The sponsor adopted an average population growth rate of 1.4% and an incidence rate of 0.16% in estimating the total number of ABSSSI cases over the time horizon.29 Comparators included both IV and orally administered drugs. The treatment cost of dalbavancin IV and comparators was calculated by multiplying the cost per milligram with the number of units, frequency, and treatment duration. The sponsor assumed a proportion of drug over the treatment course would be purchased in pharmacy (dalbavancin IV = 0%, other IV treatments = 78% and oral antibiotics = 89%). The estimated proportion of drug purchased in pharmacy was calculated using the average length of stay in hospital for patients with ABSSSIs in the accompanying sponsor-submitted CUA. The total treatment duration was 14 days for all comparators. The sponsor further assumed that 30% of cost of treatment purchased in pharmacy would be borne by public drug plans in Canada. The sponsor included markups and dispensing fees for the proportion of treatment purchased in pharmacy. Drug costs were obtained from public formularies and dosing was obtained from respective product monographs. No drug wastage was assumed.
The market shares for the treatment of ABSSSIs were estimated based on the feedback received from the sponsor’s consulted clinical experts. In estimating the market share of dalbavancin IV, the sponsor assumed 75% of market share of dalbavancin IV would be captured from vancomycin IV. The remaining market share would be captured from other IV and oral treatments for ABSSSIs.
Table 15: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1/year 2/year 3 if appropriate) |
|---|---|
Target population | |
Proportion of patients covered in hospital | 100% |
Proportion of patients covered by public drug plan | 30%a |
Proportion of adults | 81%a |
Growth rate | 1.4%a |
Incidence of ABSSSIs | 0.16% |
Number of patients eligible for drug under review | 40,353 / 40,937 / 41,531 |
Market uptake (3 years) | |
Uptake (reference scenario) Vancomycin IV Linezolid IV Ceftriaxone IV Cefazolin IV Daptomycin IV Clindamycin IV Cloxacillin IV Penicillin IV Ertapenem IV Linezolid PO Clindamycin PO Cephalexin PO Cloxacillin PO Sulfamethoxazole / Trimethoprim PO Clavulin PO | 22.2% / 22.2% / 22.2% 0.0% / 0.0% / 0.0% 11.2% / 11.2% / 11.2% 9.8% / 9.8% / 9.8% 1.6% / 1.6% / 1.6% 1.7% / 1.7% / 1.7% 3.3% / 3.3% / 3.3% 0.8% / 0.8% / 0.8% 0.3% / 0.3% / 0.3% 4.5% / 4.5% / 4.5% 1.6% / 1.6% / 1.6% 22.2% / 22.2% / 22.2% 2.5% / 2.5% / 2.5% 10.3% / 10.3% / 10.3% 8.2% / 8.2% / 8.2% |
Uptake (new drug scenario) Dalbavancin Vancomycin IV Linezolid IV Ceftriaxone IV Cefazolin IV Daptomycin IV Clindamycin IV Cloxacillin IV Penicillin IV Ertapenem IV Linezolid PO Clindamycin PO Cephalexin PO Cloxacillin PO Sulfamethoxazole/Trimethoprim PO Clavulin PO | 0.8% / 2.3% / 5.0% 21.5% / 20.5% / 18.4% 0% / 0% / 0% 11.2% / 11.1% / 11.0% 9.7% / 9.7% / 9.6% 1.6% / 1.6% / 1.6% 1.6% / 1.6% / 1.6% 3.3% / 3.3% / 3.2% 0.8% / 0.8% / 0.8% 0.3% / 0.3% / 0.3% 4.5% / 4.5% / 4.4% 1.6% / 1.6% / 1.6% 22.1% / 22.0% / 21.8% 2.5% / 2.4% / 2.4% 10.2% / 10.2% / 10.1% 8.2% / 8.1% / 8.1% |
Cost of treatment (per patient) | |
Cost of treatment over courseb Dalbavancin Vancomycin IV Linezolid IV Ceftriaxone IV Cefazolin IV Daptomycin IV Clindamycin IV Cloxacillin IV Penicillin IV Ertapenem IV Linezolid PO Clindamycin PO Cephalexin PO Cloxacillin PO Sulfamethoxazole / Trimethoprim PO Clavulin PO | $2,871.50 $295.80 $2,484.72 $140.42 $73.33 $1,370.43 $409.77 $164.49 $31.72 $447.82 $524.17 $15.19 $6.84 $19.19 $4.64 $15.21 |
PO = per os (by mouth).
aAverage weighted by province size is presented.
bTreatment duration is 3 days for dalbavancin and 14 days for all other comparators. The costs for Alberta are presented.
Summary of the Sponsor’s BIA Results
The sponsor estimated the net 3-year budget impact of introducing dalbavancin for treatment of patients aged 18 years and older with severe and nonsevere ABSSSIs, caused by susceptible isolates of gram-positive micro-organisms to be $8,827,239 (Year 1: $877,218; Year 2: $2,451,153; Year 3: $5,498,868).
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The proportion of patients covered by public drug plans is uncertain: The sponsor assumed that dalbavancin IV would be provided in the hospital and funded using the hospital budget. As such, no cost would be borne by the public drug plans. The clinical expert consulted for this review anticipated that dalbavancin IV would be mostly administered in the emergency room or outside of the hospital. The parenteral antibiotic therapy (OPAT) programs allow administration of IV antibiotics at home using a home-care health nurse. Cefazolin IV is currently available through home parenteral programs that facilitate administration of IV treatment at home and the drug cost is covered by the public formulary in Alberta.10 The clinical expert also noted that the proportion of patients accessing dalbavancin IV in various outpatient settings, such as in skilled nursing facility, outpatient infusion centre or at home, would depend on the establishment of OPAT programs.
In estimating treatment costs, the sponsor adjusted drug costs to reflect that a proportion of drug is purchased in pharmacy (dalbavancin IV = 0%, other IV treatments = 78% and oral antibiotics = 89%). For the proportion of drugs purchased in pharmacy, the sponsor assumed a pan-Canadian average public coverage rate of 30%. Given there are programs established to administer IV treatments in outpatient settings, a more appropriate approach would have been to estimate the number of patients eligible for public coverage, rather than adjust costs by public coverage rate. The sponsor’s approach makes it difficult to explore uncertainty in the source of public funding (hospital versus public drug plans) and the proportion of patients covered by public drug plans.
CADTH could not address this limitation.
The market share of dalbavancin IV may be underestimated: The sponsor consulted a small group of clinical experts (n = 3) to estimate the market share of ABSSSI treatments. The methods used to estimate the market share of dalbavancin IV were not described in the sponsor-submitted clinical expert technical document. As such, CADTH was unable to appraise the survey question(s), consistency in the response(s) of clinical expert(s) consulted by the sponsor and method of estimating an average value. The clinical expert consulted by CADTH found the sponsor’s estimate that 5% of patients with ABSSSIs would be prescribed dalbavancin IV by year 3 was an underestimate, as administering a single dose is more convenient and uptake would be much higher in clinical practice. The clinical expert noted all patients with ABSSSIs could receive dalbavancin IV but some factors such as drug cost, patient preference and physician preference may hinder 100% uptake of dalbavancin IV. As such, there is notable uncertainty in the estimated market share of dalbavancin IV, which may have underestimated the budget impact.
In scenario analysis, the impact of doubling the market share of dalbavancin IV on the BIA was explored by CADTH (year 1: 1.6%, year 2: 4.6%, year 3: 10%).
The number of eligible patients is uncertain: The sponsor assumed an incidence rate of 0.16% using the BIA report submitted to Quebec for linezolid. There is limited epidemiological data on the incidence and prevalence of ABSSSIs in Canada. As such, using an epidemiologic approach to estimate the number of ABSSSI cases introduces significant uncertainty in the estimated number of eligible patients. A recent study assessing the burden of skin and soft tissue infections in the US estimated an incidence rate of 48.5 infections per 1,000 patient-years and reported that 59.7% of skin and soft tissue infections are gram-positive.30 Should the incidence of ABSSSIs be higher than estimated, the number of eligible patients and budget impact may have been underestimated.
In scenario analysis, CADTH explored the impact of estimating the number of eligible patients using epidemiology estimates from the Kaye et al., 2019 study (incidence rate = 48.5 per 1,000 patient-years, proportion of gram-positive skin and soft tissue infections = 59.7%).30
The treatment duration of comparators is uncertain: The sponsor assumed a treatment duration of 14 days for comparators. The clinical expert consulted by CADTH noted treatment duration is higher individualized and subjective in clinical practice. It depends on patient response and physician discretion. The Practice Guidelines for the Diagnosis and Management of Skin and Soft Tissue Infections, 2014, strongly recommended a treatment duration of 5 days for antimicrobial therapy based on high-quality evidence and a treatment duration of 7 to 14 days for most bacterial skin and soft tissue infections based on moderate-quality evidence.13
In scenario analysis, CADTH explored the impact of adopting a treatment duration of 10 days based on published guidelines.13
Additional limitations were identified but were not considered to be key limitations. These limitations include misalignment of comparators between the BIA and CUA, as well as estimating health care resource use based on the treatment paradigm modelled in the CUA. The treatment paradigm in the CUA had several limitations that carried over into the BIA, which makes the budget impact estimated from the health care payer perspective uncertain.
CADTH Reanalyses of the BIA
CADTH did not undertake a base-case reanalysis, as CADTH could not identify more appropriate assumptions to the sponsor’s base case to address uncertainty in the market uptake of dalbavancin IV and the estimated number of eligible patients.
CADTH conducted scenario analyses to address uncertainty in the BIA.
Assuming an increase in the market share of dalbavancin IV (year 1: 1.6%, year 2: 4.6%, year 3: 10%).
Using epidemiology estimates from a US study,30 the proportion of population with ABSSSIs was calculated to be 2.9% by multiplying the incidence rate (48.5 infections per 1,000 patient-years) with the proportion of gram-positive infections (59.7%).
Assuming a treatment duration of 10 days for comparators.
Assuming price reduction of 97%, at which dalbavancin IV is at the same price ($32) as the least costly comparator, cefazolin (250 mg, 500 mg, or 1,000 mg every 8 hours).
Results are provided in Table 16, along with a detailed breakdown of the sponsor’s base-case results. The budget impact was sensitive to uncertainty in the estimated number of patients eligible for dalbavancin IV treatment. In scenarios adopting a higher market share and incidence rate, the estimated 3-year budget impact increased to $17,654,478 and $155,846,519, respectively.
Table 16: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | 3-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $5,735,602 | $5,815,916 | $5,897,436 | $5,980,181 | $17,693,534 |
New drug | $5,735,602 | $6,693,134 | $8,348,589 | $11,479,049 | $26,520,773 | |
Budget impact | $0 | $877,218 | $2,451,153 | $5,498,868 | $8,827,239 | |
CADTH scenario analysis: increased market share of dalbavancin IV (year 1: 1.6%, year 2: 4.6%, year 3: 10%) | Reference | $5,735,602 | $5,815,916 | $5,897,436 | $5,980,181 | $17,693,534 |
New drug | $5,735,602 | $7,570,352 | $10,799,743 | $16,977,918 | $35,348,012 | |
Budget impact | $0 | $1,754,435 | $4,902,306 | $10,997,737 | $17,654,478 | |
CADTH scenario analysis: proportion of population with ABSSSIs was 2.9% | Reference | $101,263,104 | $102,681,066 | $104,120,319 | $105,581,192 | $312,382,578 |
New drug | $101,263,104 | $118,168,505 | $147,395,875 | $202,664,717 | $468,229,097 | |
Budget impact | $0 | $15,487,438 | $43,275,555 | $97,083,525 | $155,846,519 | |
CADTH scenario analysis: treatment duration of 10 days | Reference | $4,101,384 | $4,158,813 | $4,217,105 | $4,276,272 | $12,652,190 |
New drug | $4,101,384 | $5,057,209 | $6,727,403 | $9,907,750 | $21,692,362 | |
Budget impact | $0 | $898,396 | $2,510,298 | $5,631,478 | $9,040,172 | |
CADTH scenario analysis: dalbavancin IV price reduction of 97% | Reference | $5,735,602 | $5,815,916 | $5,897,436 | $5,980,181 | $17,693,534 |
New drug | $5,735,602 | $5,770,356 | $5,770,243 | $5,695,090 | $17,235,689 | |
Budget impact | $0 | –$45,560 | –$127,193 | –$285,091 | –$457,844 |
BIA = budget impact analysis.
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.