CADTH Reimbursement Review
Avatrombopag (Doptelet)
Sponsor: Sobi Canada Inc.
Therapeutic area: Chronic immune thrombocytopenia
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Clinical Review
Abbreviations
AE
adverse event
CI
confidence interval
CrI
confidence incredible
EQ-5D-3L
3-Level EQ-5D
FAS
full analysis set
HRQoL
health-related quality of life
ICH
intracranial hemorrhage
IRR
incidence rate ratio
ITC
indirect treatment comparison
ITP
immune thrombocytopenia
IVIG
IV immune globulin
LOCF
last observation carried forward
LOR
loss of response
NMA
network meta-analysis
OC
observed case
PDSA
Platelet Disorder Support Association
RCT
randomized controlled trial
RhD
rhesus D
SAE
serious adverse event
SD
standard deviation
SF-36
36-item Short-Form Health Survey
TEAE
treatment-emergent adverse event
TPO-RA
thrombopoietin receptor agonist
VAS
visual analogue scale
WDAE
withdrawal due to adverse event
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Avatrombopag (Doptelet), 20 mg, tablets, oral |
Indication | Proposed: the treatment of thrombocytopenia in adult patients with chronic ITP who have had an insufficient response to a previous treatment |
Reimbursement request | Per indication |
Health Canada approval status | pre-NOC |
Health Canada review pathway | Standard review |
NOC date | TBD |
Sponsor | Sobi Canada Inc. |
ITP = immune thrombocytopenia; NOC = Notice of Compliance; TBD = to be determined.
Introduction
Immune thrombocytopenia (ITP) is an autoimmune disorder characterized by low platelet counts and an increased bleeding risk.1 Primary ITP is not triggered by a specific condition or event.1 Chronic ITP refers to symptoms persisting more than 12 months after diagnosis.1 In Canada, the prevalence of ITP is estimated to be 9.5 cases per 100,000 population, and the incidence is estimated to be 1.6 to 3.9 per 100,000 persons per year.2,3 Approximately 76% of all cases of ITP in Canada are primary.2
Patients with ITP may be asymptomatic, but sometimes bleeding can be severe or critical, such as intracranial hemorrhage (ICH) or gastrointestinal bleeding.1 Indeed, severe or critical bleeding is a major concern among patients with ITP. The rate of fatal hemorrhage among patients with ITP has been estimated to be between 0.016 and 0.039 cases per patient-year, and this rate increases with age.4 Patients with ITP have a reduced quality of life, resulting from fatigue, bleeding, and ITP treatments.1,2,5-8
The main goals of therapy for patients with ITP are to prevent severe or critical bleeding, reduce or eliminate symptoms, minimize the adverse effects of treatments, and ultimately improve quality of life.9 There are no specific treatment guidelines for ITP in Canada. American and international guidelines recommend corticosteroids or IV immune globulin (IVIG) for first-line therapy in patients with newly diagnosed ITP.9,10 There are multiple second-line and third-line treatments available for patients with ITP who experience a relapse, such as splenectomy, rituximab, thrombopoietin receptor agonists (TPO-RAs) (e.g., romiplostim or eltrombopag), fostamatinib, and immunosuppressants. The choice of treatment should be individualized based on the severity of disease, comorbidities, age, medical and social support networks, patient values and preferences, as well as access (such as cost and availability).6
Avatrombopag (Doptelet) (20 mg/tablet) is an orally bioavailable, small-molecule TPO-RA that stimulates the proliferation and differentiation of megakaryocytes from bone marrow progenitor cells, resulting in an increased production of platelets.11 On November 3, 2023, avatrombopag was approved by Health Canada for the treatment of adult patients with chronic ITP who have had an insufficient response to a previous treatment.11 The reimbursement request by the sponsor for avatrombopag is the same as the proposed Health Canada indication. It is recommended that avatrombopag be initiated at a starting dose of 20 mg once daily. Dose adjustments are based on platelet count response. The maximum daily dose for avatrombopag is 40 mg (2 tablets).
The objective of this review is to evaluate the beneficial and harmful effects of avatrombopag at a starting dose of 20 mg daily for the treatment of thrombocytopenia in adult patients with chronic ITP who have had an insufficient response to a previous treatment.
Stakeholder Perspectives
This section summarizes the input provided by the patient groups that responded to CADTH’s call for patient input and from the clinical expert CADTH consulted for the purpose of this review.
Patient Input
The 1 response to CADTH’s call for patient input for the avatrombopag submission came from the Platelet Disorder Support Association (PDSA). The PDSA is a nonprofit that provides advocacy, education, research, and support for patients with ITP in the US and Canada. Nine comments from patients regarding their experience with avatrombopag were gathered from PDSA’s ITP support group Facebook page. The patients reported experiencing an increase and/or stabilization in platelet counts and few side effects while on avatrombopag.
The PDSA noted that patients with ITP face a complex set of challenges due to the heterogeneity of ITP’s pathophysiology and disease course. Living with ITP can be difficult and unpredictable, despite several available therapies with different mechanisms of action. In addition to the risk of life-threatening bleeding, patients with ITP may experience elevated levels of fatigue, anxiety, depression, physical pain, and sleep disturbances. The PDSA noted that the goal of treatment is to increase platelet counts, which reduces the risk of bleeding and improves patients’ quality of life. The input indicated that many currently available treatments have a high burden of toxicity and that avatrombopag is more convenient to use than weekly injections that require a visit to a clinic or doctor’s office, high-dose steroids that cause mood issues and physical side effects, or splenectomy. The PDSA also suggested that avatrombopag should be available as an alternative treatment option for patients who do not respond or stop responding to another TPO-RA.
Clinician Input
Input From Clinical Expert Consulted by CADTH
The clinical expert indicated that not all patients respond to available therapies, and even if remission is initially achieved, long-term remission is not guaranteed. For currently available treatments, challenges exist in terms of accessibility, reimbursement criteria, costs, ease of administration, and treatment-related adverse effects or complications.
Given the lack of comparative efficacy data, the influence of patient-specific factors on decisions, and the current reimbursement landscape, it is a challenge to identify the optimal place in the therapeutic algorithm for avatrombopag. The clinical expert stated that the safety profile of avatrombopag and the fact that it is administered orally suggest it might be considered a reasonable second-line therapy. Regardless of where it sits in the therapeutic algorithm, however, the addition of avatrombopag as a treatment option would be advantageous for specific patients.
The expert noted that it is difficult to determine which specific patients will respond best to avatrombopag and which are most susceptible to the adverse effects. However, the clinical expert agreed that having avatrombopag as an option for patients would be desirable, regardless of where they are in their disease course.
In practice, clinicians rely on platelet response to monitor disease severity and assess the treatment effect. In general, an increase in platelet count can be seen as early as 2 weeks into treatment with avatrombopag. If a response is observed, clinicians would likely continue to use the treatment for the long-term, with monthly monitoring. A sustained response would generally be considered a platelet count of 30,000/μL to 50,000/μL for the duration of a treatment cycle (e.g., 24 weeks). If a response has not been seen by around 12 weeks, clinicians would generally consider the treatment to be not working and discontinue it. If there are issues related to safety or tolerability, treatment would generally be discontinued earlier, particularly if it is impacting a patient’s quality of life.
Clinician Group Input
One clinician representing the Canadian Hematology Society provided input for this review. The information was gathered from the perspective of hematologists in Canada, as well as from a review of the literature and current clinical practice guidelines.
In general, this input is not contrary to that provided by the clinical expert consulted by CADTH. The input stated that it is vital to improve the quality of life of patients by balancing bleeding prevention and minimizing treatment toxicities. Among patients with ITP, the greatest unmet need is in those who have persistent or chronic ITP. Such patients require additional treatments after first-line therapy because of continued or recurrent severe thrombocytopenia, which is linked to an increased risk of bleeding. Avatrombopag is 1 of the TPO-RAs and is not an immunosuppressant. The input suggested that patients in the earlier stage of the disease course would have a better response to avatrombopag. Therefore, when it is used as a second-therapy, a patient will benefit from more a favourable response and limited exposure to the complications and toxicities of other lines of therapy, such as splenectomy and the associated surgical complications and long-lasting immunosuppression, and rituximab, which can cause immunosuppression and vaccine failures. For patients who experience multiple relapses or who have refractory disease, avatrombopag may fill a gap because other TPO-RAs are not currently available and avatrombopag has more favourable bioavailability and less hepatic toxicities than eltrombopag.
The input indicated that in practice, a clinically meaningful response would be to achieve and maintain a platelet count above 30 × 109/L. This would be correlated to a negligible risk of serious bleeding, an improved quality of life, less fatigue, and the avoidance of hospitalization or reduction in clinic visits for most patients.
Drug Program Input
In response to questions raised in the drug program input regarding considerations for initiation therapy, the clinical expert consulted by CADTH indicated that patients should not be required to have a bleeding complication or to have undergone splenectomy before receiving treatment with avatrombopag.
For questions related to re-treatment with avatrombopag, the clinical expert suggested that if patients have responded to the drug previously, they can be re-treated with the same drug as soon as a relapse occurs.
For questions about how to define loss of response or absence of clinical benefit, the clinical expert indicated that, in general, platelet counts are monitored in practice to determine the treatment effect. Loss of response or absence of clinical benefit can be determined if a treated patient fails to maintain a platelet count above 30 × 109/L at the maximum dose. The observation period should be the 3 months after treatment initiation.
Clinical Evidence
Pivotal Studies and Protocol-Selected Studies
Description of Studies
Study 302 (N = 49) was a multicentre, phase III, double-blind, randomized controlled trial (RCT) that compared the efficacy and safety of avatrombopag with placebo in patients with chronic ITP who had received previous ITP treatment and who had a baseline platelet count below 30 × 109/L. Study 302 consisted of 3 phases: prerandomization, randomization (core phase), and extension. The prerandomization phase had a screening period of up to 4 weeks. The core phase had 6 periods and lasted for 26 weeks. Patients who met all the eligibility requirements and who were willing and able entered the extension phase. Patients who discontinued the core phase early because of a lack of treatment effect remained eligible to continue in the extension phase. All patients who entered the extension phase had a starting dose of 20 mg avatrombopag. During the core phase, 32 patients were randomized to avatrombopag 20 mg (starting dose) and 17 to matching placebo. The primary efficacy end point was the cumulative number of weeks of platelet response (platelet count of 50 × 109/L or higher) without rescue therapy for bleeding.
In Study 302, the median baseline age was similar in the avatrombopag and placebo arms (45 years versus 43 years), but there were more females in the avatrombopag arm than in the placebo arm (72% versus 47%). The vast majority of patients were white in the avatrombopag and placebo arms (97% versus 88%). More patients in the avatrombopag arm than in the placebo arm had undergone splenectomy (34% versus 29%). The baseline platelet count was higher in the avatrombopag arm than in the placebo arm (12.5 × 109/L versus 9.5 × 109/L). At baseline, more patients in the avatrombopag arm than in the placebo arm had received prior ITP medications (47% versus 35%) or were taking concomitant ITP medications (47% versus 41%).
Efficacy Results
In Study 302, the incidence of any bleeding event during the 6 months of treatment in the core phase was 43.8% in the avatrombopag group and 52.9% in the placebo group. This was an exploratory outcome and the between-group difference was not statistically significant. No patients in the placebo group had a bleeding event that was higher than WHO grade 1. Two patients in the avatrombopag group experienced WHO grade 2 bleeding events and 1 patient in the avatrombopag group experienced a WHO grade 3 bleeding event (epistaxis). In the combined core phase and extension phase, 3 patients in the avatrombopag group reported grade 3 or 4 bleeding events.
The results of Study 302 also showed that 6 months of treatment with avatrombopag leads to favourable platelet responses compared to placebo. According to the clinical expert, the between-group differences in platelet response were considered clinically important in the following cases:
The median (range) cumulative number of weeks with a platelet count of 50 × 109/L or higher was 12.4 weeks (0 to 25 weeks) in the avatrombopag group and 0 weeks (0 to 2 weeks) in the placebo group (P < 0.0001).
The number of patients with a platelet count of 50 × 109/L or higher at day 8 was 21 patients (65.63%) in the avatrombopag group and 0 patient in the placebo group, and the difference between the avatrombopag and placebo treatment groups was 65.63% (95% confidence interval [CI], 49.17% to 82.08%; P < 0.0001).
A durable platelet response, defined as the proportion of patients who achieved platelet responses with at least 6 of 8 weekly treatments during the final 8 weeks of treatment over the 6-month treatment period in the absence of rescue therapy, was reported in 11 patients (34.38%) in the avatrombopag group and in 0 patients in the placebo group. The between-group difference between avatrombopag and placebo was 34.38% (95% CI, 17.92% to 50.83%). However, durable platelet response was an exploratory outcome and should be interpreted with consideration of the increased possibility of false-positive conclusions.
The median platelet count in the avatrombopag group appeared to be higher than that in the placebo group during the 6-month core phase starting from day 8; platelet response in the core phase was generally maintained throughout the extension until around week 36.
The treatment effect of avatrombopag that improves patients’ health-related quality of life (HRQoL), reduces the use of concomitant ITP medications or need for rescue therapy, or reduces emergency department visits and/or hospitalization due to thrombocytopenia episodes compared with placebo remain uncertain.
The proportion of patients who needed rescue therapy was 21.9% in the avatrombopag group and 11.8% in the placebo group (P = 0.4668).
A reduction in the use of concomitant ITP medication was achieved by 5 of 15 patients (33.3%) in the avatrombopag group and by 0 of 7 patients in the placebo group (P = 0.1348).
Due to the high discontinuation rate in the study and the low event rates for some of these outcomes (e.g., HRQoL, hospitalization, or emergency department visit), it was not possible to assess whether there were any differences between avatrombopag and placebo in the study population. It was also a challenge to base treatment decisions or draw meaningful conclusions from subgroup analyses.
A posthoc analysis of Study 302 was performed to provide additional information related to avatrombopag treatment. The results suggested that during the open-label extension phase, response (defined as a platelet count ≥ 50 × 109/L) was achieved at 96.1% of the extension phase visits and a complete response (defined as a platelet count ≥ 100 × 109/L) was achieved at 60.1% of extension phase visits. The durable response rate (defined as a platelet count ≥ 30 × 109/L for 6 of the final 8 weeks of the core phase) was reported by 64.0% of patients in the avatrombopag group and by0% in the placebo group. In addition, in the core and extension phases, more than half of the patients who needed corticosteroids at baseline reduced or discontinued corticosteroid therapy.
Harms Results
During the core phase, 31 (96.9%) patients in the avatrombopag group and 10 (58.8%) patients in the placebo group reported any adverse events (AEs). Patients in the avatrombopag group reported higher-grade AEs than those in the placebo group. There were 6 (18.8%) patients in the avatrombopag group who reported an AE of grade 3 or 4 and none in the placebo group. The most commonly reported AEs were headache, contusion, upper respiratory tract infection, arthralgia, epistaxis, fatigue, gingival bleeding, and petechiae.
Table 2: Summary of Key Results From Study 302
Results | Avatrombopag (N = 32) | Placebo (N = 17) |
|---|---|---|
Efficacy | ||
Bleeding events | ||
n | 32 | 17 |
Yes, n (%) | 14 (43.8) | 9 (52.9) |
No, n (%) | 18 (56.3) | 8 (47.1) |
P value (chi-square distribution)a | 0.5394 | |
Cumulative number of weeks with a platelet count ≥ 50 × 109/L | ||
n | 32 | 17 |
Median (range) | 12.4 (0 to 25) | 0.0 (0 to 2) |
P value (Wilcoxon rank sum test) | < 0.0001 | |
% of patients with a platelet count ≥ 50 × 109/L at day 8 | ||
n | 32 | 17 |
Yes, n (%; 95% CI)b | 21 (65.63; 49.17 to 82.08) | 0 |
Between-group difference (95% CI)b | 65.63 (49.17 to 82.08) | |
P value (Fisher’s exact test) | < 0.0001 | |
Durable platelet response ratec | ||
n | 32 | 17 |
Yes, n (%; 95% CI)b | 11 (34.38; 17.92 to 50.83) | 0 |
Between-group difference (95% CI)b | 34.38 (17.92 to 50.83) | |
P value (Fisher’s exact test)a | 0.0090 | |
Change in platelet count, median (range) | ||
n at baseline | 32 | 17 |
Baseline, × 109/L | 12.5 (1.0 to 31.5) | 9.5 (4.0 to 27.0) |
n at week 26 | 22 | 1 |
Platelet count at week 26, × 109/L | 53.95 (3.0 to 187.0) | 31.0 (31.0 to 31.0) |
Change from baseline, × 109/L (95% CI)b | 35.25 (–4.0 to 177.5) | 4.0 (4.0 to 4.0) |
Complete response (by IWG criteria,d a platelet count ≥ 100 × 109/L and absence of bleeding) at week 26 | ||
n | 22 | 1 |
Yes, n (%) | 4 (18.2) | 0 (0.0) |
Complete response (by IWG criteria,d a platelet count ≥ 30 × 109/L and absence of bleeding) at week 26 | ||
n | 22 | 1 |
Yes, n (%) | 12 (54.6) | 0 |
Need for rescue therapy during the 6-month treatment period | ||
n | 32 | 17 |
Yes, n (%; 95% CI)b | 7 (21.88; 7.55 to 36.20) | 2 (11.76; 0.00 to 27.08) |
Between-group difference (95% CI)b | 10.11 (–10.86 to 31.08) | |
P value (Fisher’s exact test)a | 0.4668 | |
Reduction in use of concomitant ITP medications from baseline | ||
n | 15 | 7 |
Yes, n (%) | 5 (33.33) | 0 (0.0) |
Between-group difference (95% CI)b | 33.33 (9.48 to 57.19) | |
P value (Fisher’s exact test) | 0.1348 | |
Safety | ||
N | 32 | 17 |
Patients with ≥ 1 AE, n (%) | 31 (96.9) | 10 (58.8) |
Patients with ≥ 1 SAE, n (%) | 9 (28.1) | 1 (5.9) |
Patients with ≥ 1 WDAE, n (%) | 3 (9.4) | 0 (0.0) |
Deaths, n (%) | 0 (0.0) | 0 (0.0) |
Notable harm, n (%) | ||
Thromboembolic events | 3 (9.4) | 0 (0.0) |
Neoplastic events | 1 (3.1) | 0 (0.0) |
Recurrence of thrombocytopenia | 1 (3.1) | 0 (0.0) |
AE = adverse event; CI = confidence interval; ITP = immune thrombocytopenia; IWG = International Working Group; SAE = serious adverse event; WDAE = withdrawal due to adverse event.
aThe statistical testing for this outcome was not adjusted for multiple comparisons.
bThe 95% CI was calculated based on normal approximation.
cThe durable platelet response was defined as the proportion of patients who had at least 6 of 8 weekly platelet responses during the final 8 weeks of treatment over the 6-month treatment period of the core phase in the absence of rescue therapy.
dA platelet response according to IWG criteria was defined as a platelet count ≥ 100 × 109/L and in the absence of bleeding, or a platelet count of 30 × 109/L and at least a 2-fold increase in baseline platelet count in the absence of bleeding. A platelet count that occurs in the 8 weeks after rescue therapy was considered to be a nonresponse.
Source: Clinical Study Report (CSR) for Study 302.12
Nine patients (28.1%) in the avatrombopag group and 1 patient (5.9%) in the placebo group reported any treatment-emergent serious adverse events (SAEs). Three patients (9.4%) in the avatrombopag group and none in the placebo group reported AEs leading to discontinuation of the study drug (cerebrovascular accident, headache, and polyserositis). No deaths were reported during the study.
For notable harms in the avatrombopag group, 3 patients (9.4%) reported thromboembolic events, 1 patient (3.1%) reported a neoplastic event, and 1 patient (3.1%) reported a recurrence of thrombocytopenia. No patient in the placebo group reported any treatment-emergent adverse event (TEAE) of special interest.
The incidences of AEs, SAEs and AEs leading to discontinuation of the study drug during the extension phase were similar to those reported in the avatrombopag group during the core phase.
Critical Appraisal
Internal Validity
Study 302 was a small, phase III, double-blind, placebo-controlled RCT. Some relatively large baseline imbalances between groups were observed, which could suggest selection bias but is most likely the result of the small sample of patients randomized. The degree to which this may have an impact on data interpretation and bias the results is unclear. The rate of study discontinuation was high in Study 302 and was imbalanced between treatment arms: 22% of patients in the avatrombopag group and 88% of patients in the placebo group withdrew from the study because of inadequate therapeutic effect. The median exposure duration was much shorter with placebo than with avatrombopag. This affected the assessment of the clinically relevant outcomes of bleeding events and rescue medication; no clear conclusions about the effect of avatrombopag on these outcomes could be drawn. The high dropout rate also had a substantial impact on patient-reported outcomes, such as HRQoL. At the end of the core phase, only 1 patient in the placebo group provided data from the 36-item Short-Form Health Survey (SF-36) and EQ-5D. It is not possible to draw meaningful conclusions about the effect of the study drug on HRQoL due to the limited amount of data as a result of study discontinuation. In Study 302, the cumulative number of weeks with a platelet count of 50 × 109/L or higher was the primary outcome measure. In practice, platelet count is considered a surrogate for the risk of bleeding events and survival, although the results of an RCT evaluating the effect of prophylactic platelet transfusion dose on the risk of bleeding in patients with hypoproliferative thrombocytopenia suggest that the relation between bleeding events and platelet count is not well known.13 Gains in the number of weeks with a platelet response may be correlated to a reduction in the risk of bleeding or improved quality of life in the study population. According to the clinical expert consulted by CADTH, a threshold of 30 × 109/L or lower is used by clinicians to determine treatment response and the risk of subsequent bleeding. This is consistent with recommendations from clinical practice guidelines, which indicate that treatment should maintain a target platelet level of at least 20 × 109/L to 30 × 109/L for symptomatic patients (risk for major bleeding increases below this level).9 Although a threshold of 50 × 109/L for platelet response in patients with ITP was used to assess the treatment effect in Study 302, there were limited or no data on patient-important outcomes such as bleeding rates, use of concomitant ITP medications, need for rescue therapy, symptoms, and HRQoL.
Baseline patient characteristics in the population of Study 302 is broadly comparable to patients with ITP in Canada and, thus, the study findings are likely generalizable to Canada. One challenge with Study 302 is that the comparator is placebo. For patients with chronic ITP whose platelet counts are lower than 20 × 109/L, treatment would be warranted. However, Study 302 has provided no information on how the efficacy and safety of avatrombopag may differ from other available treatments. In addition, patients could receive some allowed concomitant ITP therapies; however, the study was not designed to assess the role of any combination therapy (e.g., avatrombopag in combination with corticosteroids), and the effect of any combination therapy is uncertain.
Indirect Comparisons
Description of Studies
The sponsor submitted a systematic review and indirect treatment comparison (ITC) report14 in which avatrombopag was compared to 2 TPO-RAs (eltrombopag and romiplostim), fostamatinib, and rituximab in patients with chronic or persistent ITP.
In this ITC, durable platelet response, need for rescue therapy, use of concomitant ITP medications, bleeding events, WHO grade 2 to 4 bleeding events, and AEs were assessed. Network meta-analyses (NMAs) were conducted within a Bayesian framework.
In total, 9 RCTs were included and contributed evidence. In the trials included in the ITC, the number of enrolled patients ranged from 11 to 135. According to the baseline characteristics of patients presented in the report, differences were observed across trials for the proportion of patients who had undergone splenectomy (0% to 50%), the proportion who used concomitant ITP medication at baseline (13% to 48%), and duration of ITP (median, 0.25 to 8.70 years). There was a noticeable between-trial heterogeneity in the proportion of patients prematurely discontinuing the allocated treatment (range, 0% to 100%).
Efficacy Results
In the sponsor-submitted ITC, results for durable platelet outcome, need for rescue therapy, use of concomitant ITP medication, and higher-grade bleeding events were very imprecise, with credible intervals (CrIs) including the potential for no difference between treatments or for either treatment to be favoured. Avatrombopag was favoured over eltrombopag, romiplostim, and rituximab in the incidence of any bleeding events.
Harms Results
Results of the NMA for AEs were very imprecise, with CrIs including the potential for no difference between treatments or for either treatment to be favoured.
Critical Appraisal
In the sponsor-submitted ITC, trial characteristics and patient baseline characteristics of the studies included in the systematic review and ITC were reported. Based on the data presented, potential sources of heterogeneity with respect to baseline characteristics were identified, such as the proportion of patients who had undergone splenectomy (0% to 50%), the proportion who used concomitant ITP medication at baseline (13% to 48%), and the duration of chronic ITP (median, 0.25 to 8.70 years). Other patient characteristics should also be considered when addressing clinical heterogeneity across the included trials, such as cycles and doses of prior corticosteroid therapy, previous lines of therapy, and the severity of previous bleeding events. Such data were not provided in the ITC, and from the available data, it appears likely that the transitivity assumption was violated. Furthermore, there was a significant between-trial heterogeneity in the proportion of patients prematurely discontinuing the allocated treatment (range, 0% to 100%), which would have an impact on the total exposure time of the study drug in the included trials and could have affected the results for relative efficacy and safety (e.g., by decreasing the chance of bleeding events or AEs in the placebo group). However, the authors of the ITC adjusted for this by summarizing the data using incidence rate ratios (IRRs) that accounted for the duration of exposure. The definitions of durable platelet response and bleeding episodes were measured using different approaches. The inconsistency in outcome definitions could bias the comparisons across the trials. Due to the small evidence base and potential heterogeneity across all trials leading to imprecision, the results of the NMA were largely noninformative.
Other Relevant Evidence
Description of Studies
Two additional studies were included in the sponsor’s submission to CADTH that provided supportive evidence regarding the safety and efficacy of avatrombopag. Study 00315 was a phase II, double-blind, placebo-controlled randomized trial of avatrombopag taken orally once daily for 28 days in adult patients with chronic ITP. A total of 5 patients were randomized to the placebo group and 15 to the avatrombopag 20 mg/day group. Two patients, both in the avatrombopag group, discontinued treatment due to an increase in platelet count of 500 × 109/L or more.
Study 00416 was a phase II, long-term extension study, with avatrombopag administered to patients with chronic ITP who completed Study 003 for an additional 6 months. Of the 53 patients enrolled in Study 004, 13 received the maximum dose of 20 mg/day allowed in Study 003 (10 responders and 3 nonresponders). Four (30.8%) of these patients discontinued Study 004 — 2 from each of the responder and nonresponder groups — with each patient discontinuing for a different reason.
A retrospective observational study assessing the effect of patients switching from other TPO-RAs to avatrombopag was provided by the sponsor to provide evidence for patients with chronic ITP who had been heavily treated.17,18 In this study, the median duration of avatrombopag exposure was 9.2 months (range, 2.8 to 17.2 months).
Efficacy Results
In Study 003, 80% of patients (n = 12) in the avatrombopag group and no patients in the placebo group achieved a treatment response on day 28. Patients were considered responders if they had a baseline platelet count of less than 30 × 109/L and achieved a platelet count of at least 50 × 109/L on day 28, or if they had a baseline platelet count of at least 30 × 109/L but less than 50 × 109/L and were receiving steroids and achieved an increase from baseline of at least 20 × 109/L. The median (range) change in platelet count from baseline to day 28 was 84 × 109/L (–10 × 109/L to 1,012 × 109/L) in the avatrombopag group and –2 × 109/L (–12 × 109/L to 9 × 109/L) in the placebo group. No patients in the placebo group and 12 patients (80.0%) in the avatrombopag group had a platelet count of 50 × 109/L or higher and 8 patients (53.3%) in the avatrombopag group had a platelet count of 100 × 109/L or higher on day 28. Using the last observation carried forward (LOCF) method, 13 patients (86.7%) in the avatrombopag group and 1 patient (20.0%) in the placebo group had their platelet count at least doubled on day 28.
The median (range) change in platelet count from baseline in Study 003 to week 24 in Study 004 was 124 × 109/L (–11 × 109/L to 205 × 109/L) among responders (n = 7) and 199 × 109/L (not applicable) among nonresponders (n = 1). At week 24, a total of 6 (86.7%) responders and 1 (100.0%) nonresponder achieved a response-level platelet count. A total of 6 (60.0%) responders and 1 (33.3%) nonresponder achieved a durable platelet response. Of the 6 responders and 1 nonresponder initially treated with corticosteroids, 2 (33.3%) responders and 1 (100.0%) nonresponder permanently discontinued steroid use during the final 8 weeks of treatment in Study 004.
Results of the retrospective study (n = 44) suggest that a platelet response was achieved by 93% of patients and a complete platelet response was achieved by 86% after switching. Among responders, response was maintained for 84% of their time on treatment. Among the patients who received concomitant ITP medications, 57% discontinued 1 or more concomitant medications after initiating avatrombopag. For patients who were taking concomitant corticosteroids, 63% discontinued the corticosteroids and 32% reduced their dose. Rescue therapy was required by 21% of patients after switching to avatrombopag; in the year before switching, 34% of patients on eltrombopag or romiplostim required rescue.
Harms Results
Safety results were presented for the combined study periods in Study 003 and Study 004. All 20 patients in mean daily dose group of 13.5 mg or higher experienced at least 1 TEAE. The most common TEAEs were fatigue, headache, and epistaxis, each of which occurred in 8 patients (40.0%). A total of 3 patients (15.0%) withdrew due to an AE. Three patients reported at least 1 SAE, which included 2 patients who experienced serious recurrent thrombocytopenia. No deaths occurred during the studies.
Critical Appraisal
Study 003 had patients centrally randomized to treatment groups using simple block randomization (block size of 13) without stratification factors. Patients and study personnel involved in patient care or outcome assessment were blinded to treatment, and the sponsor noted no partial unblinding at the time of the database lock. Therefore, the findings are unlikely to be affected by bias due to deviation from the intended interventions or measurement of the outcome. The study was not powered to detect statistically significant changes in outcomes and analyses were not adjusted for multiplicity, so definitive conclusions cannot be drawn. Study 004 enrolled patients who successfully completed Study 003, which could have resulted in a population of patients who were more tolerant of avatrombopag and could have led to biased estimates of efficacy and safety. The use of concomitant steroid medications among patients throughout the study may have increased the risk of additional side effects not attributable to avatrombopag alone. In terms of external validity, doses of avatrombopag administered throughout the studies to some patients were less than the recommended starting dose of 20 mg/day approved by Health Canada, which limits the generalizability of the results. There was no examination of HRQoL outcomes in either study that were deemed to be important to both patients and clinical experts.
Although findings of the retrospective observational study by Al-Samkari et al. suggest that switching to avatrombopag is associated with an increased platelet response and reduced concomitant ITP medications in patients who had previously been treated with TPO-RAs, the outcomes are limited by concerns about internal validity (specifically in terms of the retrospective observational study design), the lack of a comparator, the small sample size, as well as the external validity in terms of generalizability of the study findings to the patient population in Canada.
Conclusions
The management of chronic ITP is a challenge as patients frequently relapse or are refractory to treatments. Therefore, patients commonly cycle through multiple ITP treatments. Treatment is complicated by a lack of evidence on the comparative efficacy and safety of second-line and subsequent-line treatment options, access issues, and the safety and/or tolerability of available options. In 1 double-blind RCT (Study 302), treatment with avatrombopag, another TPO-RA, led to an improvement in platelet count response compared to placebo among patients with pretreated, primary, chronic ITP. There were limited or no data on patient-important outcomes, such as bleeding rates, use of concomitant ITP medications, need for rescue therapy, symptoms, and HRQoL. These outcomes were exploratory in Study 302, so the impact of avatrombopag on these outcomes remains unclear. Subgroup analyses were not able to provide insight into which patient groups (e.g., based on previous lines of therapy) are most likely to respond to treatment. Further, it remains difficult to draw conclusions about the efficacy of avatrombopag compared to other ITP treatments. One ITC study was included in this review, suggesting that avatrombopag may have favourable efficacy compared to other TPO-RAs and rituximab in terms of the incidence of bleeding events. However, there were important limitations in this study, and it is a challenge to draw firm conclusions around comparative efficacy based on its results. Based on Study 302, avatrombopag appeared to lead to a higher rate of adverse effects such headache, contusion, upper respiratory tract infection, arthralgia, epistaxis, fatigue, gingival bleeding, and petechiae than placebo.
Overall, this review suggests that avatrombopag is another potential treatment option for patients with chronic, pretreated, primary ITP. Avatrombopag leads to a platelet count response in the target population and is generally well tolerated compared to placebo, although its efficacy and safety compared to other ITP treatments and its effect on patient-important clinical outcomes remain unclear. In addition, the study findings may not be generalizable to patients with secondary ITP.
Introduction
Disease Background
ITP is an autoimmune disorder characterized by low platelet counts and increased bleeding risk.1 It is thought to be caused by antibodies directed against platelet antigens, leading to increased platelet destruction.1 ITP can be classified as primary or secondary.1 Primary ITP is not triggered by a specific condition or event, whereas secondary ITP is caused by or associated with another condition, such as chronic lymphocytic leukemia, systemic lupus erythematous, antiphospholipid syndrome.1 ITP is also defined by duration; acute or newly diagnosed ITP refers to the first 3 months after diagnosis, persistent ITP lasts 3 to 12 months after diagnosis, and chronic ITP refers to symptoms persisting more than 12 months after diagnosis.1 In Canada, the prevalence of ITP is estimated to be 9.5 cases per 100,000 population, and the incidence is estimated to be 1.6 to 3.9 per 100,000 persons per year.2,3 The incidence of ITP is reported to increase with age.1 Data from a single centre suggest that approximately 76% of all cases of ITP in Canada are primary.2 Furthermore, it is estimated that 80% of these patients have chronic ITP.19
Patients with ITP may be asymptomatic, although some can experience bleeding and other symptoms.20 Bleeding can be mild; for example, patients may experience petechiae, purpura, or nosebleeds.20 Bleeding can also be more severe or critical, such as ICH or gastrointestinal bleeding.1 Indeed, severe or critical bleeding is a major concern among patients with ITP. Predictors of critical bleeding include platelet count (less than 10,000/μL or 20,000/μL), previous bleeding, and chronic ITP (more than 12 months in duration).1 The rate of fatal hemorrhage among patients with ITP has been estimated to be between 0.016 and 0.039 cases per patient-year, and this rate increases with age.4 The predicted 5-year all-cause mortality rate for patients aged 60 years and older with persistent low platelet counts was 48% in 1 study of 1,817 patients.4 The authors of that study estimated that a 30-year-old woman with ITP and persistent low platelet counts would lose 15 quality-adjusted life-years from her life expectancy.4 Patients with ITP also commonly experience fatigue.9,21 Patients with ITP have a reduced quality of life, resulting from fatigue, bleeding, and ITP treatments.1,2,5-8
ITP is considered a diagnosis of exclusion; thus, the diagnostic evaluation primarily concerns excluding other possible causes of a low platelet count and/or identifying other potential conditions leading to a low platelet count (i.e., secondary ITP).1,9 ITP is an isolated thrombocytopenia, meaning that patients do not have anemia or leukopenia.1 Diagnosis involves a patient history (questions regarding bleeding, symptoms), physical examination, and laboratory testing (e.g., complete blood count, peripheral blood smear).1,9 Bone marrow examination may be needed in patients who relapse after remission, in those not responding to initial treatment options, when splenectomy is considered, or if other abnormalities are detected in the blood count or morphology.9
Standards of Therapy
The need for treatment to increase platelet count among people with ITP is based on assessment of bleeding (site, acuity, severity), platelet count, bleeding risk factors, and previous treatments. Treatment to increase platelet count is generally recommended if the platelet count is lower than 20,000/μL to 30,000/μL and/or if the patient is experiencing bleeding. It is recommended that patients with severe or critical bleeding receive urgent treatment to stop the bleeding and raise the platelet count.9
The main goals of therapy in ITP are to prevent severe or critical bleeding, reduce or eliminate patients’ symptoms, minimize adverse effects from treatments, and ultimately improve patient quality of life.9 Treatments are recommended for patients with platelet levels above 20,000/μL to 30,000/μL, which appear to reduce the risk of major bleeding.9,10 There are no specific treatment guidelines for ITP in Canada; American and international guidelines recommend that for the initial treatment of newly diagnosed ITP, high-dose corticosteroids (e.g., dexamethasone 40 mg/day for 4 days) or IVIG (1 mg/kg to 2 mg/kg over 2 to 5 days) be used as first-line therapy (anti-D immune globulin is another alternative in patients who test positive for the rhesus D (RhD) antigen).9,10 Long-term corticosteroid treatment is generally not a recommended treatment option, as the harms outweigh the benefits.9,10
After corticosteroids or IVIG are stopped, many patients experience a relapse in their condition, manifesting as worsening thrombocytopenia, which may be accompanied by increased bleeding symptoms. Among adults with primary ITP, approximately one-third will relapse in the first year and up to 80% will relapse in the 5 years after initial treatment.9,10 Once patients have relapsed, the optimal choice of subsequent treatment is not well established.9,10 Although there are multiple second-line and third-line treatments available for ITP, there is a lack of comparative efficacy data to provide evidence on a clear sequential treatment pathway. Possible treatment options include splenectomy, rituximab, TPO-RAs (such as romiplostim or eltrombopag), fostamatinib, and various other immunosuppressants (such as vincristine, azathioprine, and cyclophosphamide). The International Consensus Report on the Investigation and Management of Primary ITP highlights these various therapies as treatment options in the subsequent-line treatment of ITP but does not express a preferred pathway among the options.9,10 These guidelines state that the recommended option is generally based on available resources and patient preferences. The guidelines further note that in the subsequent-line treatment of ITP, TPO-RAs, rituximab, and fostamatinib have “robust” evidence supporting their use.9,10 The American Society of Hematology guidelines for ITP state that the choice of treatment should be individualized based on severity of disease (such as duration, frequency of bleeding episodes requiring hospitalization or rescue medication), comorbidities, age, medical and social support networks, patient values and preferences, as well as access (such as cost and availability).9,10 These guidelines also acknowledge the very-low-certainty evidence on comparative efficacy, and state that shared decision-making based on factors previously outlined are important in identifying the appropriate subsequent-line ITP treatment.10
In the Canadian context, the choice of subsequent-line treatment depends on patient-specific factors (e.g., increased susceptibility to adverse effects of a treatment, contraindications, preferences), as well as access (i.e., whether a treatment is listed on a provincial drug formulary and/or whether the patient meets the criteria for reimbursement). Thus, some options recommended by guidelines for a particular patient may not be available to them. Further, many of the second-line and third-line treatment options carry a risk of important harms. For example, some patients may not be surgical candidates due to comorbidities, and even if a splenectomy can be safely performed, the long-term risk of bacterial infection must be considered. Similarly, although rituximab has been shown to be effective in achieving a platelet response, idiopathic and occasionally life-threatening infusion reactions have been reported, as have hepatitis B reactivation and progressive multifocal leukoencephalopathy. In addition, rituximab is an immunosuppressant and has been found to be associated with reduced immune response to vaccinations, including the COVID-19 vaccines.22 TPO-RAs, which are often effective, generally require long-term treatment, and although they are not immunosuppressive (unlike splenectomy and rituximab), they do carry a risk of bone marrow reticulin fibrosis and of both arterial and venous thrombosis.9,10,22 Options also differ in terms of their administration. For example, rituximab is given as an infusion at a clinic or hospital over several weeks, whereas eltrombopag is a daily continuous oral medication that cannot be taken for several hours after the ingestion of calcium. In summary, the potential chance of achieving a platelet response must be considered against the potential harms of the different drugs, administration factors, and access issues.9,10
The clinical expert noted that even when an optimal second-line or third-line therapy has been identified after consideration of these factors, response (including subsequent relapse among initial responders) cannot always be predicted, meaning that patients may cycle through several different treatment regimens over the course of their disease.
Drug
Avatrombopag is an orally bioavailable, small-molecule TPO-RA that stimulates the proliferation and differentiation of megakaryocytes from bone marrow progenitor cells, resulting in an increased production of platelets.11 On November 3, 2023, avatrombopag was approved by Health Canada for the indication of thrombocytopenia in adult patients with chronic ITP who have had an insufficient response to a previous treatment.11 The reimbursement request by the sponsor for avatrombopag is the same as the proposed Health Canada indication.
Avatrombopag is available as an oral tablet of 20 mg. According to the product monograph, avatrombopag should not be administered to patients with chronic ITP to normalize platelet counts.11 During treatment, platelet counts need to be monitored and the dosing guidelines should be followed to achieve target platelet counts.11 The product monograph recommends that avatrombopag be initiated at a starting dose of 20 mg (1 tablet) once daily and be taken orally with food. After initiating therapy with avatrombopag, platelet counts should be assessed weekly until a stable platelet count greater than or equal to 50 × 109/L has been achieved, and platelet counts should be measured monthly thereafter to maintain platelet counts between 50 × 109/L and 200 × 109/L. Dose adjustments are based on platelet count response. The maximum daily dose of avatrombopag is 40 mg (2 tablets). Avatrombopag should be discontinued if the platelet count is less than 50 × 109/L after 4 weeks of dosing at the maximum dose of 40 mg once daily, or if the platelet count is greater than 250 × 109/L after 2 weeks of dosing at 20 mg weekly. After the discontinuation of avatrombopag, platelet counts must be monitored weekly for at least 4 weeks.11
Avatrombopag is a TPO-RA and carries a warning about potential increased thrombotic risk when administered to patients with known risk factors for thromboembolism. Patients receiving avatrombopag should be monitored for signs and symptoms of thromboembolic events. The product monograph also notes the effects of avatrombopag on the risk of bone marrow reticulin fibrosis and progression of existing hematological malignancies.11
Table 3 provides details regarding the mechanism of action, indication, route and dose of administration, and adverse effects of avatrombopag, eltrombopag, romiplostim, and rituximab.
Table 3: Key Characteristics of Avatrombopag, Eltrombopag, Romiplostim, and Rituximab
Criteria | Avatrombopag | Eltrombopag | Romiplostim | Rituximab |
|---|---|---|---|---|
Mechanism of action | Stimulates platelet production by initiating a signalling cascade at the thrombopoietin receptor | Depletion of CD20 antigens | ||
Indicationa | For the treatment of thrombocytopenia in adult patients with chronic ITP who have had an insufficient response to a previous treatment | For the treatment of chronic ITP to increase platelet counts in adult and pediatric patients 1 year and older who have had an insufficient response to corticosteroids or immunoglobulins | To increase platelet levels in adult patients with ITP who have not undergone splenectomy and have had an inadequate response or are intolerant to corticosteroids and/or immunoglobulins; or who have undergone splenectomy and have had an inadequate response to splenectomy | Not indicated for the treatment of ITP |
Route of administration | Oral | Oral | Subcutaneous | IV |
Recommended dose | Initial dose is 20 mg once daily, then adjusted based on platelet counts | Initial dose is 25 mg once daily, then adjusted based on platelet counts | Initial dose is 1 mcg/kg of actual body weight once weekly, then adjusted based on platelet count | 375 mg/m2 once a week for 5 weeks or 100 mg once a week for 4 weeks |
Serious adverse effects or safety issues | Contraindicated in patients who are hypersensitive to the drug or any ingredient in the formulation; has been associated with thrombotic and thromboembolic complications in patients with chronic ITP, therefore patients receiving avatrombopag should be monitored for signs and symptoms of thromboembolic events and treatment should be instituted promptly | Contraindicated in patients with severe hepatic impairment or who are hypersensitive to the product or any of its excipients; should be used with caution in patients with chronic hepatitis C and cirrhosis | Contraindicated in patients who are hypersensitive to the drug or any ingredient in the formulation; should not be used in patients with myelodysplastic syndromes; recurrence of thrombocytopenia to below pretreatment levels and serious life-threatening or fatal bleeding after discontinuation have been reported | Serious adverse effects include infusion reactions, progressive multifocal leukoencephalopathy, tumour lysis syndrome, hepatitis B reactivation, and infections; contraindicated in people with type 1 hypersensitivity reactions or anaphylactic reactions to murine proteins, Chinese hamster ovary cell proteins, or any component of the product, and in patients who have had progressive multifocal leukoencephalopathy |
ITP = immune thrombocytopenia.
aHealth Canada–approved indication.
Sources: Product monographs for avatrombopag,11 eltrombopag,23 romiplostim,24 and rituximab.25
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups.
The PDSA submitted the patient input for this review. The PDSA is a nonprofit that provides advocacy, education, research, and support for patients with ITP in the US and Canada. Nine comments from patients regarding their experience with avatrombopag were gathered from PDSA’s ITP support group Facebook page. These patients reported experiencing an increase in platelet counts and few side effects while on avatrombopag. The PDSA highlighted that in addition to the risk of life-threatening bleeding, patients with ITP may experience elevated levels of fatigue, anxiety, depression, physical pain, and sleep disturbances. The PDSA indicated that many currently available treatments have a high burden of toxicity, and stated that avatrombopag is more convenient than having to attend a clinic or doctor’s office for a weekly injection, taking high-dose steroids that cause mood issues and physical side effects, or undergoing a splenectomy. The PDSA also suggested that avatrombopag should be available as an alternative treatment option for patients who do not respond or stop responding to another TPO-RA. The input stated that patients want a treatment that has little to no side effects, works for more than a 1-week period, and improves their quality of life. A copy of the patient input from the PDSA is presented in the Patient Input section.
Clinician Input
Input From the Clinical Expert Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol; assisting in the critical appraisal of clinical evidence; interpreting the clinical relevance of the results; and providing guidance on the potential place in therapy). The following input was provided by 1 clinical specialist with expertise in the diagnosis and management of ITP.
Current Treatments
Standard first-line therapy for ITP includes corticosteroids, and IVIG (or Rh-immune globulin in patients who test positive for RhD) is often added when an immediate increase in platelets is required, although its effect is often transient. A significant proportion of patients will not respond to steroids, and of those who do, many will relapse once the steroids are tapered. At this point, the historical second-line therapy is splenectomy if patients are suitable candidates; however, subsequently, rituximab has emerged as an alternative second-line therapy. If both splenectomy and rituximab have failed (or are contraindicated), a large number of third-line therapies are available, including immunosuppressant medications such as azathioprine and cyclophosphamide. More recently, TPO-RAs such as eltrombopag or romiplostim have emerged as treatment options and are considered by a growing number of specialists to be equivalent or even superior to splenectomy or rituximab. However, there is very little evidence to guide the selection of second-line or third-line therapy due to the lack of direct comparisons between each approach. Furthermore, decisions are as much dependent on local reimbursement considerations as on patient-specific factors.
Treatment Goals
Broad treatment goals are to reduce bleeding and prolong life. Increasing the platelet count is generally considered to be a reasonable surrogate for those 2 goals. Improving quality of life is also an important goal and must be considered alongside the inconvenience and side effects of the treatments used (e.g., fatigue, cognition, mood, interference with daily life, frequent hospital visits), which many clinicians may overlook in their focus on the patient’s platelet count. However, quality-of-life measures are generally less standardized for patients with ITP than platelet count or bleeding symptoms, and are not consistently incorporated into the design of clinical studies.
Unmet Needs
There are myriad challenges with the current treatment paradigm for ITP. Not all patients respond to available therapies, and even if remission is achieved, long-term remission is not guaranteed. Therefore, durable remission of ITP remains a challenge. Further, although corticosteroids and IVIG are generally accessible for patients, accessibility to appropriate second-line and third-line therapies can be a challenge. This is because not all options are reimbursed in every province or because reimbursement criteria differ among provinces. For example, in Ontario, patients must fail 2 or more therapies after steroids and IVIG before being eligible for TPO-RAs, meaning that these drugs are not available to many patients until later in the treatment pathway. The administration of existing therapies can also be a challenge; for example, the administration of rituximab requires the patient to travel to a hospital or clinic. Oral TPO-RAs can also be difficult to adhere to because they must be administered on an empty stomach. There are also adverse effects with existing treatments; splenectomy carries short-term perioperative risks, as well as longer-term risks of thrombosis and infections with encapsulated bacteria, whereas rituximab increases susceptibility to hepatitis B reactivation and increases vulnerability to other opportunistic infections. The availability of therapies with demonstrated efficacy, convenience of administration, and a low risk of adverse effects would fill an unmet need for the treatment of ITP.
Place in Therapy
Contemporary ITP guidelines suggest that, in general, splenectomy or rituximab can be considered as second-line therapy. There are several third-line options available; however, the comparative efficacy of these drugs is unclear. Therefore, it can be difficult to know what the best treatment option is for a particular patient, and there is often no single clearly defined treatment pathway. Decisions end up largely being driven by access. Given the lack of comparative efficacy data, the influence of patient-specific factors on decisions, and the current reimbursement landscape, it is a challenge to identify the optimal place in the therapeutic algorithm for avatrombopag, a novel TPO-RA for the treatment of ITP. The clinical expert consulted by CADTH noted that rituximab or splenectomy are reasonable second-line choices (TPO-RAs may also be considered a second-line choice for some patients). The safety profile of avatrombopag and the fact that it is administered orally suggest that it might be considered a reasonable second-line therapy. Regardless of where it sits in the therapeutic algorithm, however, the addition of avatrombopag as a treatment option would be advantageous for clinicians to have for specific patients.
Patient Population
The ITP population is very heterogenous, and it is generally not possible, with the available data and current understanding of ITP pathophysiology, to determine which specific patients will respond best and which are most susceptible to adverse effects. However, the clinical expert agreed that having avatrombopag as an option for patients would be desirable, regardless of where they are in their disease course.
Assessing Response to Treatment
Bleeding is a very important outcome in the treatment of ITP, and ultimately any treatment should reduce the occurrence of clinically important bleeding while improving quality of life. In practice, clinicians rely on platelet response, which is assumed to correlate with the risk of clinically relevant bleeding, and, as such, reduces the need for rescue therapy. There are no HRQoL scales used in practice that are specific to ITP. In general, an increase in platelet count can be seen as early as 2 weeks into treatment with avatrombopag, although some patients may not respond until week 12. If a response is observed, clinicians would likely continue to use the treatment in the long-term, with monthly monitoring. A sustained response would generally be considered a platelet count of 30 000/μL to 50 000/μL for the duration of a treatment cycle (e.g., 24 weeks). If a response has not been seen by around 12 weeks, clinicians would generally consider the treatment to have not worked and would discontinue it. If there are issues related to safety or tolerability, treatment would generally be discontinued earlier, particularly if it is impacting a patient’s quality of life.
Prescribing Conditions
The initial management of ITP, such as the use of corticosteroids and IVIG, is frequently undertaken by clinicians practising general internal medicine. However, once patients require second-line treatment, they are often referred to a hematologist. Patients with longstanding ITP who have tried multiple therapies are often seen by multiple hematologists. Although hematologists usually take responsibility for selecting the treatment for patients with ITP, primary care physicians may share responsibility of monitoring patients for AEs.
Clinician Group Input
One clinician representing the Canadian Hematology Society provided input for this review. The information was gathered from the perspective of Canadian hematologists, as well as a review of the literature and current clinical practice guidelines.
In general, this input was not contrary to that provided by the clinical expert consulted by CADTH. The input stated that it is vital to improve the quality of life of patients by balancing bleeding prevention with the need to minimize treatment toxicities. Among patients with ITP, the greatest unmet need is for those who have persistent or chronic ITP. Such patients require additional treatments after first-line therapy because of continued or recurrent severe thrombocytopenia, which is linked to an increased risk of bleeding. Avatrombopag is 1 of the TPO-RAs and is not an immunosuppressant. The input suggested that patients in the earlier stage of their disease course would have a better response to avatrombopag. Therefore, when it is used as a second-line therapy, patients will benefit from a more favourable response and limited exposure to the complications and toxicities of other lines of therapy, such as splenectomy (and the associated surgical complications and long-lasting immunosuppression) and rituximab (which can cause immunosuppression and vaccine failures). For patients who experience multiple relapses or who have refractory disease, avatrombopag may fill a gap when other TPO-RAs are not available, and avatrombopag has more favourable bioavailability and fewer hepatic toxicities than eltrombopag.
The input indicated that, in practice, a clinically meaningful response would be to achieve and maintain a platelet count above 30 × 109/L. This would be correlated with a negligible risk of serious bleeding, improved quality of life, less fatigue, and avoidance of hospitalization or fewer clinic visits for most patients.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
Would you consider the most relevant comparators for avatrombopag to be other TPO-RAs, such as eltrombopag and romiplostim? | The clinical expert confirmed that other TPO-RAs are the most relevant comparators for avatrombopag. |
Considerations for initiation of therapy | |
Other TPO-RAs funded in some jurisdictions require patients to have bleeding complications in addition to a diagnosis of chronic ITP.
| The clinical expert indicated that patients should not be required to have a bleeding complication before they are prescribed avatrombopag. Clinicians will not wait for the occurrence of bleeding events to start a treatment. If the patient lost response or never achieved clinical benefit from previous treatment with a different TPO-RA, the clinician would be unlikely to try avatrombopag. If a patient cannot tolerate adverse events from other TPO-RAs, however, then it would be reasonable to attempt a different drug in the same class of medication. |
Other TPO-RAs funded in some jurisdictions require splenectomy and the failure of a specified number of first-line (e.g., corticosteroids, IVIG) and second-line (e.g., azathioprine, cyclosporine, cyclophosphamide, danazol, dapsone, mycophenolate, rituximab) drugs. The sponsor is requesting reimbursement for patients who have only failed first-line drugs (corticosteroids and/or IVIG).
Note: Approximately 30% of patients in the pivotal trial had previously received 5 or more ITP medications, and 67.3% had not undergone splenectomy. | The clinical expert suggested that a patient should not be required to have a splenectomy before switching to treatment with avatrombopag. However, the issue is that the reimbursement criteria for avatrombopag may need to be aligned with other TPO-RAs, for which a splenectomy is required. A re-evaluation of the role of splenectomy should therefore be considered for all TPO-RAs, not just avatrombopag. The expert stated that the patient does not need to fail rituximab or other immunosuppressants before getting avatrombopag. In terms of what is considered an adequate trial of a therapeutic approach, this varies by treatment. For example, at least 4 infusions of rituximab should be pursued before deeming a patient nonresponsive, whereas repeat attempts at splenectomy (i.e., to assess for residual tissue) are not usually recommended. |
Can the drug be given again to patients who relapsed while off therapy? If so, what would be the appropriate timing of re-treatment? | The expert agreed that if the patient responded to the drug before, re-treatment with the same drug should be initiated as soon as a relapse occurs. |
Considerations for discontinuation of therapy | |
How should loss of response or absence of clinical benefit be defined for the purposes of treatment discontinuation? Are there any specific measures and time frames that need to be considered? | Loss of response or absence of clinical benefit is defined as failure to maintain a platelet count above a level of 30 × 109/L at the maximum dose of a certain drug. In general, platelet counts are monitored in practice to determine the treatment effect. The observation period should be in the 3 months after treatment initiation. |
Generalizability | |
The sponsor is requesting reimbursement for chronic ITP (defined as occurring more than 12 months after diagnosis), which aligns with the population studied in the pivotal trial. However, jurisdictions may receive requests for funding for patients with persistent ITP (defined as thrombocytopenia that remains 3 to 12 or more months after diagnosis), who were included in the supportive trial and for whom treatment with TPO-RAs is recommended in clinical guidelines. Should reimbursement be provided for patients who have persistent ITP? | The expert thought that it would be reasonable to provide avatrombopag to patients with persistent as well as chronic ITP. |
ITP = immune thrombocytopenia; IVIG = IV gamma globulin; TPO-RA = thrombopoietin receptor agonist.
Clinical Evidence
The clinical evidence included in the review of avatrombopag is presented in 3 sections. The first section, the Systematic Review, includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as studies that were selected according to an a priori protocol. The second section includes indirect evidence from the sponsor and indirect evidence selected from the literature that met the selection criteria specified in the review. The third section includes sponsor-submitted long-term extension studies and additional relevant studies that were considered to address important gaps in the evidence included in the Systematic Review.
Systematic Review (Pivotal and Protocol-Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of avatrombopag 20 mg for the treatment of adult patients with chronic ITP who have had an insufficient response to a previous treatment.
Methods
Studies selected for inclusion in the Systematic Review include pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 5. The CADTH review protocol includes outcomes considered important to patients, clinicians, and drug plans.
Of note, the systematic review protocol presented here was established before the granting of a Notice of Compliance from Health Canada.
Table 5: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Population | Adult patients with chronic ITP who have had an insufficient response to a previous treatment Subgroups:
|
Intervention | Avatrombopag at a starting dose of 20 mg (1 tablet) orally once daily with food. Dose adjustments are based on platelet count response; the lowest dose needed to achieve and maintain a platelet count ≥ 50 × 109/L is used. The maximum dose is 40 mg (2 tablets) per day. |
Comparator | A combination of 1 or more of the following:
|
Outcomes | Efficacy outcomes:
Harms outcomes:
Notable harms: thromboembolic events, hepatotoxicity, hypersensitivity reaction, neoplastic events, gastric atrophy events, bone marrow pathology |
Study designs | Published and unpublished phase III and IV RCTs |
AE = adverse event; HRQoL = health-related quality of life; ITP = immune thrombocytopenia; IVIG = IV gamma globulin; RCT = randomized controlled trial; SAE = serious adverse event; WDAE = withdrawal due to adverse event.
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy, according to the PRESS Peer Review of Electronic Search Strategies checklist.26
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946–) via Ovid and Embase (1974–) via Ovid. All Ovid searches were run simultaneously as a multifile search. Duplicates were removed using Ovid deduplication for multifile searches, followed by manual deduplication in Endnote. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concept was avatrombopag. The following clinical trials registries were searched: the US National Institutes of Health’s clinicaltrials.gov, WHO’s International Clinical Trials Registry Platform (ICTRP) search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
No filters were applied to limit retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. Refer to Appendix 1 for the detailed search strategies.
The initial search was completed on March 3, 2022. Regular alerts updated the search until the meeting of the CADTH Canadian Drug Expert Committee (CDEC) on June 22, 2022.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool For Searching Health-Related Grey Literature checklist.27 Included in this search were the websites of regulatory agencies (FDA and European Medicines Agency). Google was used to search for additional internet-based materials. Refer to Appendix 1 for more information on the grey literature search strategy.
These searches were supplemented by reviewing the bibliographies of key papers.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
In addition to the sponsor-provided indirect evidence, additional indirect evidence that includes the patients, interventions, comparators, and outcomes specified in Table 5 was summarized and critically appraised, if considered relevant by CADTH. A focused literature search for NMAs dealing with avatrombopag, TPO-RAs, or immune thrombocytopenia was run in MEDLINE All (1946-) via Ovid on March 3, 2022. No limits were applied.
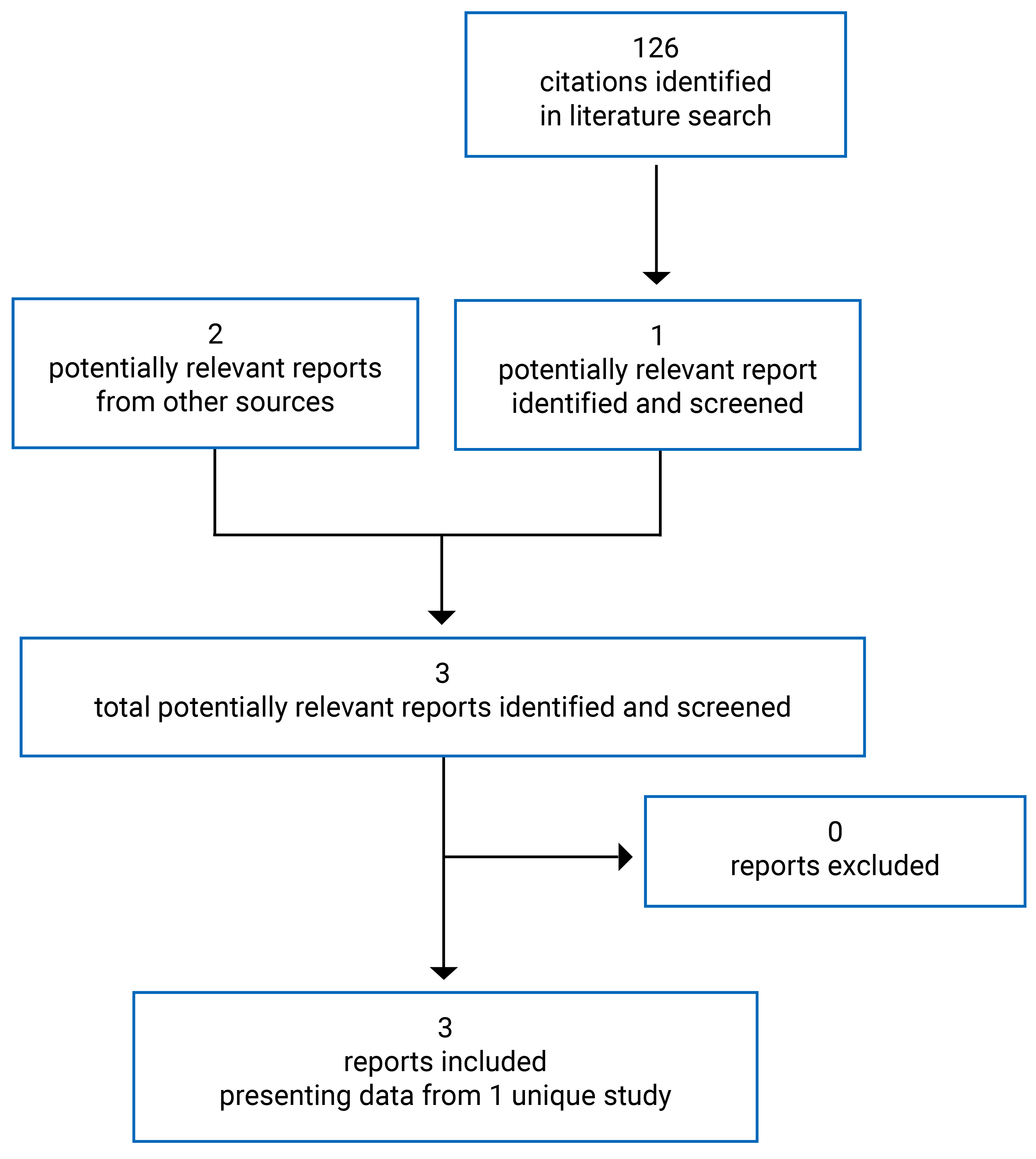
Findings From the Literature
A total of 126 studies were identified from the literature for potential inclusion in the Systematic Review, and 3 reports of a single study were included (Figure 1). The included study is summarized in Table 6.
Table 6: Details of the Included Study
Detail | Study 302 |
|---|---|
Designs and populations | |
Study design | Phase III, DB, RCT |
Locations | 27 sites in Europe, Asia, Africa, Australia, and New Zealand |
Patient enrolment date | February 6, 2012 (first informed consent form received); end date is April 9, 2015 |
Randomized (N) | 49 |
Inclusion criteria | Core phase:
Extension:
|
Exclusion criteria | Core phase:
Extension:
|
Drugs | |
Intervention | Avatrombopag, oral tablet, started at a dose of 20 mg, with dose titration down to 5 mg or up to 40 mg. Maximum daily dose was 40 mg |
Comparator(s) | Placebo |
Duration | |
Phase | |
Screening | Up to 4 weeks |
Double-blind | 26 weeks |
Follow-up | 30 days for patients who did not continue into the extension phase |
Open-label extension | Up to 104 weeks |
Outcomes | |
Primary end point | Cumulative number of weeks of platelet response (platelet count ≥ 50 × 109/L) without rescue therapy for bleeding |
Other end points | Secondary:
Exploratory:
Safety:
Extension phase:
|
Notes | |
Publications | Jurczak et al. (2018)28 Al-Samkari et al. (2022)29 Al-Samkari et al. (2022)18 Al-Samkari et al. (2023)17 Jain et al. (2023)30 |
AE = adverse event; ALT = alanine aminotransferase; ANC = absolute neutrophil count; AST = aspartate aminotransferase; DB = double-blind; EQ-5D-3L = 3-Level EQ-5D; H2 = histamine; HRQoL = health-related quality of life; INR = international normalized ratio; ITP = immune thrombocytopenia; IWG = International Working Group; MDS = myelodysplastic syndrome; PPI = proton pump inhibitor; RCT = randomized controlled trial; SAE = serious adverse event; SF-36 = 36-item Short-Form Health Survey; TPO-RA = thrombopoietin receptor agonist; ULN = upper limit of norm; WBC = white blood count; WDAE = withdrawal due to adverse event.
Note: Two additional reports were included: Submission,31 European Medicines Agency report.32
Source: CSR for Study 302.12
Description of Studies
Study 302 was a multicentre, multinational, randomized, double-blind, placebo-controlled, parallel-group study of avatrombopag administered to adult patients with chronic ITP. The primary objective of the core phase was to demonstrate that the efficacy of avatrombopag (with standard care) was superior to placebo for the treatment of adult patients with chronic ITP, as demonstrated by the cumulative weeks of platelet response. Patients were centrally stratified at the time of randomization by splenectomy status, baseline platelet count (≤ 15 × 109/L or > 15 × 109/L to < 30 × 109/L), and use of concomitant ITP medication (yes or no) at baseline, and were randomized by an Interactive Voice and Web Response System to receive either avatrombopag or placebo in a 2:1 ratio in a double-blind fashion. During the randomization phase, the patients and all personnel involved with the conduct and interpretation of the study, including the investigators, investigational site personnel, and sponsor staff, were blinded to the assigned treatment.
The primary objective of the open-label extension phase was to evaluate the safety and tolerability of long-term therapy with avatrombopag in patients with chronic ITP.
Study 302 consisted of 3 phases (Figure 2):
Prerandomization lasted up to 4 weeks and had 1 screening period during which eligibility and platelet counts were assessed.
Randomization (core phase) had 6 periods and lasted for 26 weeks
Baseline and/or randomization (1 day).
Titration of the study drug (6 weeks) was performed from the initial starting dose in accordance with protocol-specified titration guidelines to find the minimum dose required to maintain platelet counts of at least 50 × 109/L and no more than 150 × 109/L. No downward titration of concomitant ITP medication was permitted during this period unless there was a safety concern.
During the concomitant ITP medication reduction period (12 weeks), downward titration of concomitant ITP medication was permitted in accordance with the ITP concomitant downward titration guidelines. This may have required additional study drug dose adjustments before and after the concomitant ITP medication downward titration.
During maintenance (8 weeks), the primary end point was assessed. Patients continued treatment to maintain platelet counts of at least 50 × 109/L and no more than 150 × 109/L. No downward titration of concomitant ITP medication was permitted during this period unless there was a safety concern. At the end-of-treatment visit (visit 22), patients could choose to enter the extension phase and receive open-label avatrombopag therapy. The patients who were unable or unwilling to continue in the extension phase of the study entered the dose tapering and follow-up periods.
Dose tapering (up to 4 weeks) was required only for patients not continuing into the extension phase. The study drug was down-titrated 1 dose level per week until discontinuation. During this period, subsequent upward titration or addition of concomitant ITP medication was considered, at the investigator’s discretion, to prevent the recurrence of thrombocytopenia.
Follow-up (30 days) was required only for patients not continuing into the extension phase. Platelet counts were monitored for the recurrence of thrombocytopenia. Subsequent upward titration or the addition of concomitant ITP medication was considered, at the investigator’s discretion, to prevent the recurrence of thrombocytopenia.
Extension was for patients who met all the eligibility requirements and who were willing and able to enter this phase of the study. Patients who discontinued the core phase early because of a lack of treatment effect (i.e., did not achieve a platelet count of at least 30 × 109/L during the randomization phase despite upward titration to the maximum dose, or required rescue therapy more than 3 times or continuous rescue therapy for more than 3 weeks during the core phase) remained eligible to continue into the extension phase. Patients who entered the extension phase received a starting dose of 20 mg once daily of open-label avatrombopag and underwent dose titration according to titration guidelines.
Efficacy of the treatment was assessed by measuring platelet counts at each visit. The primary outcome measure was the cumulative number of weeks of platelet response over 6 months of treatment. The WHO bleeding scale was selected to assess the incidence and severity of bleeding events.
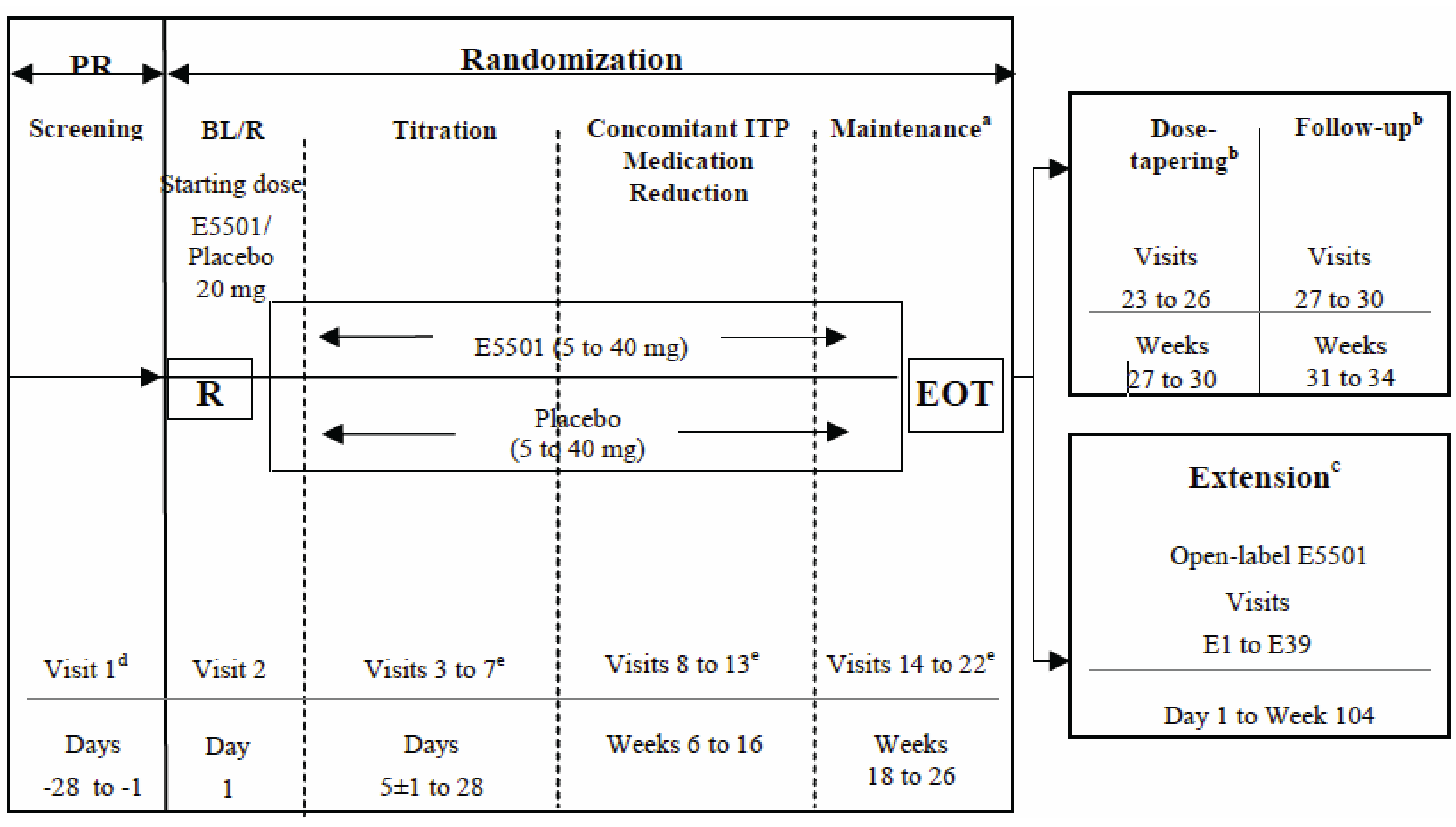
BL = baseline; E5501 = avatrombopag; EOT = end of treatment; ITP = immune thrombocytopenia; PR = prerandomization; R = randomization.
a At the EOT visit (visit 22), patients could choose to enter the extension phase and to receive open-label avatrombopag therapy. Patients who did not continue into the extension phase entered the dose tapering and follow-up periods.
b Only for patients who did not enter the extension phase.
c The extension phase consisted of 4 periods: conversion (6 weeks), maintenance period and/or concomitant ITP medication reduction period (90 weeks), dose tapering (up to 4 weeks), and follow-up (30 days).
d The screening visit and day 1 baseline and/or randomization visit platelet counts were averaged to obtain the baseline platelet count value. The 2 samples were obtained ≥ 48 hours and ≤ 2 weeks apart, and the results were available before randomization. Therefore, an additional screening platelet count may have been required due to issues with scheduling.
e Patients who discontinued early who met the criteria for a lack of treatment effect may have moved directly into the open-label extension.
Source: CSR for Study 302.12
Populations
Inclusion and Exclusion Criteria
In Study 302, patients older than 18 years who had a diagnosis of chronic ITP and who had received at least 1 prior treatment for ITP were enrolled to the core phase. Patients had to have an average of 2 platelet counts lower than 30 × 109/L. Patients in Study 302 had have had no known etiology for their ITP, so patients with secondary ITP were excluded. Further, patients were excluded if they had significant medical conditions that could impact their safety; had a history of myelodysplastic syndrome, pernicious anemia, or arterial or venous thrombosis or more than 2 risk factors for this thrombosis; significant cardiovascular disease or cirrhosis; or had received certain ITP treatments (medications or surgery) in the weeks before randomization.
Patients who had completed 6 months of the study drug in the randomization phase were allowed to enter the extension phase, including those who discontinued the core phase early due to a lack of treatment effect.
Baseline Characteristics
Median baseline age was similar in the avatrombopag and placebo arms (45 years versus 43 years), and there were more female patients in the avatrombopag arm (72% versus 47%). The vast majority of patients were white in both the avatrombopag and placebo arms (97% versus 88%). A higher proportion of patients in the avatrombopag arm than in the placebo arm had undergone splenectomy (34% versus 29%). The baseline platelet count was higher in the avatrombopag arm than the placebo arm (12.5 × 109/L versus 9.5 × 109/L). Patients in the avatrombopag arm were more likely to have received prior ITP medications than those in the placebo arm (47% versus 35%), and to be taking concomitant ITP medications at baseline (47% versus 41%) (Table 7).
Table 7: Summary of Baseline Characteristics — Core Phase, FAS
Characteristic | Study 302 | |
|---|---|---|
Avatrombopag (N = 32) | Placebo (N = 17) | |
Age, median (range) years | 45 (20 to 69) | 43 (18 to 65) |
Sex, n (%) | ||
Male | 9 (28.1) | 9 (52.9) |
Female | 23 (71.9) | 8 (47.1) |
Race, n (%) | ||
White | 31 (96.9) | 15 (88.2) |
Black or African American | 0 (0.0) | 1 (5.9) |
Japanese | 0 (0.0) | 0 (0.0) |
Chinese | 1 (3.1) | 1 (5.9) |
Other Asian | 0 (0.0) | 0 (0.0) |
American Indian or Alaskan Native | 0 (0.0) | 0 (0.0) |
Native Hawaiian or Other Pacific Islander | 0 (0.0) | 0 (0.0) |
Other | 0 (0.0) | 0 (0.0) |
BMI, kg/m2, median (range) | 28.05 (18.7 to 52.1) | 27.35 (19.2 to 46.0) |
Baseline platelet count, × 109/L, median (range) | 12.50 (1.0 to 31.5) | 9.50 (4.0 to 27.0) |
Splenectomy status, n (%) | ||
Yes | 11 (34.4) | 5 (29.4) |
No | 21 (65.6) | 12 (70.6) |
Use of concomitant ITP medication at baseline, n (%) | 15 (46.9) | 7 (41.2) |
Anabolic steroids (danazol) | 0 (0.0) | 1 (5.9) |
Immunosuppressants (azathioprine, ciclosporin) | 0 (0.0) | 2 (11.8) |
Hemostatics (etamsylate) | 3 (9.4) | 3 (17.6) |
Corticosteroids for systemic use | 14 (43.8) | 7 (41.2) |
At least 1 prior ITP medication, n (%) | 15 (46.9) | 6 (35.3) |
Anabolic steroids (danazol) | 1 (3.1) | 0 (0.0) |
Immunoglobulins | 5 (15.6) | 1 (5.9) |
Immunosuppressants (mycophenolic acid) | 1 (3.1) | 0 (0.0) |
Antifibrinolytics (aminocaproic acid, tranexamic acid) | 5 (15.6) | 2 (11.8) |
Hemostatics (eltrombopag, etamsylate, romiplostim) | 4 (12.5) | 1 (5.9) |
Corticosteroids for systemic use | 5 (15.6) | 3 (17.6) |
BMI = body mass index; FAS = full analysis set; ITP = immune thrombocytopenia.
Source: CSR for Study 302.12
Interventions
At randomization, patients received blinded therapy at a starting dose of 20 mg avatrombopag or matching placebo tablet, taken orally once daily with food. Patients were allowed to have their dose titrated up (to a maximum dose of 40 mg for avatrombopag or matching placebo) or down (to a minimum dose 5 mg for avatrombopag or matching placebo) in accordance with their individual response to the study drug; a placebo titration was used to maintain the blind. The overall goal of any dose modification was to maintain a platelet count of at least 50 × 109/L and no more than 150 × 109/L, and to decrease the need for concomitant ITP medications, if possible.
The following concomitant background therapies were allowed in Study 302:
corticosteroids and/or azathioprine taken at a stable dose for 4 weeks before randomization
mycophenolate mofetil or danazol taken at a stable dose for at least 12 weeks before randomization
cyclosporine A (because it is a P-glycoprotein-mediated transport inhibitor, it was to be avoided unless deemed medically necessary) could be taken at a stable dose for at least 12 weeks before randomization.
Patients were allowed to take rescue therapy during the 6-month treatment period when there was an urgent need to increase their platelet count (e.g., platelet count < 10 × 109/L, major bleed, clinical signs or symptoms suggestive of bleeding). Rescue therapy was defined as the addition of any new ITP medication or medication to treat thrombocytopenia, or any increase in the baseline dose of a concomitant ITP medication. These could include corticosteroids, IVIG therapy, anti-D therapy, mycophenolate mofetil, azathioprine, danazol, dapsone, cyclosporine A, and platelet transfusion. Note that TPO-RAs were not allowed as rescue therapy.
Outcomes
A list of efficacy end points identified in the CADTH review protocol that were assessed in the clinical trials and included in this review is provided in Table 8 and subsequently summarized. A detailed discussion of the validity of the WHO bleeding scale33 and HRQoL measures34-36 is provided in Appendix 2.
The primary outcome of Study 302 was the cumulative number of weeks of platelet response, which was defined as the total number of weeks in which the platelet count was greater than 50 × 109/L during 6 months of treatment in the core phase in the absence of rescue therapy (i.e., patients using rescue therapy at any time during the 6-month treatment period were considered not to have any platelet responses at all subsequent scheduled time points after rescue therapy). The proportion of platelet responders at day 8 was a secondary outcome and was defined as the proportion of patients who had a platelet count of at least 50 × 109/L at day 8 in the absence of rescue therapy on or before day 8. Patients with missing platelet counts at day 8 were considered to be nonresponders. Platelet count was assessed at baseline and every 2 weeks thereafter. Platelet counts were performed by local laboratories affiliated with the clinical sites enrolling patients. The other secondary end point was the proportion of patients with a reduction in concomitant ITP medications, which was defined as patients using concomitant medications at baseline who had no use of rescue therapy and had at least 1 concomitant medication dose reduced. The use of concomitant medications was assessed at each study visit.
Table 8: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure | Study 302 |
|---|---|
Bleeding events | Exploratory |
Platelet response: Cumulative number of weeks of platelet response (≥ 50 × 109/L) Proportion of patients with a platelet response at day 8 Platelet count by visit Durable platelet response Complete response Maximum duration of continuous response | Primary Secondary Exploratory Exploratory Exploratory Exploratory |
HRQoL (SF-36, EQ-5D-3L) | Exploratory |
Symptoms (e.g., fatigue, excessive bruising) | NR |
Treatment-free remission | NR |
Need for rescue therapy | Exploratory |
Proportion of patients with a reduction in concomitant ITP medications | Secondary |
Need for monitoring | NR |
Emergency department visits or hospitalization due to a thrombocytopenia episode | Exploratory |
Mortality | Safety |
Safety: AE SAE WDAE Notable harm | Safety |
AE = adverse event; EQ-5D-3L = 3-Level EQ-5D; HRQoL = health-related quality of life: NR = not reported; SAE = serious adverse event; SF-36 = 36-item Short-Form Health Survey.
Exploratory efficacy outcomes in Study 302 included the incidence and severity of bleeding events, change in platelet count from baseline, durable platelet response (i.e., the proportion of patients who had at least 6 of 8 weekly platelet responses during the final 8 weeks of treatment over the 6-month treatment period of the core phase in the absence of rescue therapy), complete response according to International Working Group criteria (platelet count ≥ 100 × 109/L in the absence of bleeding, or ≥ 30 × 109/L and at least a 2-fold increase in the baseline platelet count in the absence of bleeding), maximum duration of continuous platelet response (i.e., consecutive weeks with a platelet count ≥ 50 × 109/L during the core phase in the absence of rescue therapy), HRQoL, need for rescue therapy, and emergency department visits and hospitalization due to a thrombocytopenia episode. Bleeding assessments were performed using the WHO bleeding scale at baseline and every 2 weeks throughout the study. The WHO bleeding scale defines severity of bleeding from 0 (no bleeding) to 4 (debilitating blood loss); bleeding events were considered to include a WHO bleeding grade of 1 or higher. HRQoL was measured using the SF-36 and the EQ-5D, which are commonly used generic self-reported HRQoL questionnaires.
Safety outcomes included AEs, SAEs, withdrawal due to adverse events (WDAEs), and deaths, and were monitored throughout the study and up to 30 days after the last dose of the study drug with clinical laboratory tests (e.g., hematology, coagulation, chemistry, urinalysis, gastric biomarkers), measurement of vital signs, and physical exams.
Statistical Analysis
Power Calculation
Based on data from previous research15 that included a 4-week treatment period, and assuming a 15% dropout rate (all dropout patients were considered to have 0 weeks of platelet response), a total sample size of 45 patients (30 in the avatrombopag group and 15 in the placebo group) would have more than 95% power to detect a treatment difference between avatrombopag and placebo in the cumulative number of weeks of platelet response during the 4-week treatment period using the Wilcoxon rank sum test at a 2-sided alpha significance level of 0.05. Conservatively, assuming that a treatment difference in the cumulative number of weeks of platelet response would be preserved with longer duration of treatment, the sample size of 45 patients would have more than 95% power to detect a treatment difference in the cumulative number of weeks of platelet response during 6 months of treatment between avatrombopag and placebo using the Wilcoxon rank sum test at a 2-sided alpha significance level of 0.05.
Efficacy Analyses
The primary efficacy outcome of Study 302 was the cumulative number of weeks of platelet response, as defined previously. The primary analysis of the cumulative number of weeks of platelet response was performed between avatrombopag and placebo using the Wilcoxon rank sum test at the 2-sided alpha significance level of 0.05. There was no adjustment of covariates for the primary efficacy analysis. Subgroup analysis of the primary efficacy end point by baseline platelet count (≤ 15 × 109/L and > 15 109/L to < 30 × 109/L), splenectomy status (yes and no), and use of concomitant ITP medication at baseline (yes and no) were performed.
The key secondary efficacy outcomes included the number and proportion of platelet responders (platelet count ≥ 50 × 109/L) at day 8 and the proportion of patients with a reduction in concomitant ITP medications. The comparisons of the 2 key secondary efficacy end points between avatrombopag and placebo occurred in a sequential manner to control for type I error. For the 2 key secondary end points, multiplicity was adjusted in a fixed sequential fashion: the comparison of platelet response at day 8 between avatrombopag and placebo was performed first with a 2-sided alpha of 0.05. If this testing was significant, then the comparison of the proportion of patients with a reduction in use of concomitant ITP medications from baseline was performed with a 2-sided alpha of 0.05. The number and proportion of patients with a platelet response at day 8 were summarized descriptively, with 95% CIs calculated using normal approximation. The null hypothesis was tested using the Cochran-Mantel-Haenszel test, adjusted for splenectomy status and baseline platelet count. Fisher’s exact test was used alternatively if any cells had a value of 0. Only patients who used concomitant ITP medications at baseline were included in the analysis of reduction in the use of these medications, and those who withdrew during the treatment period were considered failures. The null hypothesis for a reduction in ITP medications was analyzed in the same way as described for platelet response at day 8.
All other efficacy end points were analyzed without multiplicity adjustment. Exploratory end points were analyzed as follows:
Continuous variables were analyzed with analyses of variance or covariance or the Wilcoxon rank sum test (durable platelet response, duration of response). In addition, the incidence and severity of bleeding events were summarized by treatment group and WHO bleeding scale score. The maximum duration of continuous response was summarized by treatment group using the mean, median, standard deviation, and range.
Categorical variables were tested using the Cochran-Mantel-Haenszel test, chi-square distribution, or Fisher’s exact test (use of rescue therapy). In addition, rescue therapy was summarized by treatment group using frequency distribution, and the results were presented as number and percent.
For analysis of the efficacy end points related to platelet response — including the maximum cumulative number of weeks of platelet response, platelet response at day 8, durable platelet response, and maximum duration of platelet response — a missing platelet assessment at a specific visit was considered a nonresponse at that visit, and a platelet count that occurred in the 8 weeks after rescue therapies were used was also considered a nonresponse. Patients who discontinued the study or who were lost to follow-up before the end of the 6-month treatment period had all subsequent unobserved scheduled platelet assessments at the scheduled visits documented as having missing platelet values.
There were no sensitivity analyses reported in Study 302.
Safety Analyses
The investigators classified AEs according to standardized MedDRA terms (version 16.0 or higher), and reported the number and proportion of patients with at least 1 AE, SAE, or WDAE, as well as deaths during the core and extension phases.
Analysis Populations
The CORE phase included the following:
The full analysis set (FAS) of all patients who were randomized into the study, and all participants were analyzed as randomized. The FAS was used as the primary population for all efficacy analyses.
The safety analysis set included all patients who received at least 1 dose of the study drug and had a postdose safety assessment. The safety analysis set was analyzed as treated. In Study 302, the FAS and the safety analysis set were identical.
The extension phase included the following:
The modified FAS included all patients who received study medication and who provided at least 1 platelet count to derive at least 1 effectiveness assessment during the extension phase.
The safety analysis set included all patients who received at least 1 dose of avatrombopag (either in the core phase or extension phase) and who had at least 1 postdose safety assessment.
Results
Patient Disposition
In Study 302, 100 patients were screened and 49 (49%) patients were randomized, 32 to the avatrombopag group and 17 to the placebo group. A total of 10 patients (31%) in the avatrombopag arm discontinued the study early, as did 16 patients (94%) in the placebo arm. Reasons for discontinuation are provided in Table 9. In Study 302, 22% of patients in the avatrombopag arm discontinued early due to a lack of response, as did 88% in the placebo arm. Three (9%) patients in the avatrombopag arm discontinued early due to AEs, as did 0 in the placebo arm.
Table 9: Patient Disposition — Core and Extension Phases
Disposition | Study 302 | ||
|---|---|---|---|
Avatrombopag | Placebo | ||
Screened, N | 100 | ||
Screen failure | 51 | 42 failed to meet inclusion or exclusion criteria | |
6 failed for the reason of “other” | |||
2 failed due to AE | |||
1 excluded for withdrawal of consent | |||
Randomized and entered core phase, N | 32 | 17 | |
Completed core phase, n (%) | 22 (68.8) | 1 (5.9) | |
Discontinued from core phase, n (%) | 10 (31.3) | 16 (94.1) | |
Reason for discontinuation, n (%) | |||
AEs | 3 (9.4) | 0 (0.0) | |
Lost to follow-up | 0 (0.0) | 0 (0.0) | |
Patient choice | 0 (0.0) | 0 (0.0) | |
Inadequate therapeutic effect | 7 (21.9) | 15 (88.2) | |
Withdrawal of consent | 0 (0.0) | 1 (5.9) | |
Other | 0 (0.0) | 0 (0.0) | |
Entered extension phase, n | 39 | 0 (0.0) | |
Completed extension phase, n (%) | 29 (74.4) | ||
Discontinuation from extension phase, n (%) | 9 (23.1) | ||
AEs | 3 (7.7) | ||
Lost to follow-up | 1 (2.6) | ||
Patient choice | 3 (7.7) | ||
Inadequate therapeutic effect | 2 (5.1) | ||
Withdrawal of consent | 0 (0.0) | ||
Other | 0 (0.0) | ||
FAS, N (core phase) | 32 | 17 | |
Safety, N (core phase) | 32 | 17 | |
mFAS, N (extension phase) | 39 | 0 | |
Safety, N (extension phase) | 47 | 0 | |
AE = adverse event; FAS = full analysis set; mFAS = modified full analysis set.
Source: CSR for Study 302.12
A total of 31 patients (63.3%) had major protocol deviations during the core phase. A conservative definition was used for major protocol deviations, and included noncompliance with protocol procedures. The primary reason for major deviations (25 patients [80.6% of those with deviations]) included informed consent procedural issues, missed visits, and missed laboratory tests. The most common protocol procedural deviations were missed visits and laboratory tests, which were the result of the long duration of the study (median exposure, 43.9 weeks) and high frequency of visits. There were no critical protocol deviations (i.e., protocol deviations that substantially impacted the primary end point or safety assessments). Although a number of deviations from the protocol occurred, these deviations were not considered to have significantly affected the overall evaluation of efficacy or safety.
Exposure to Study Treatments
During the core phase, the median duration of exposure was 26 weeks in the avatrombopag group and 6 weeks in the placebo group; the short duration of exposure in the placebo group was due to the high early discontinuation in this group as a result of a lack of efficacy. Median daily doses were 19.34 mg (range, 5.9 to 37.6 mg) and 33.33 mg (range, 21.3 to 55.6 mg) for the avatrombopag and placebo groups, respectively.
During the extension phase, the median duration of exposure was 44 weeks.
Table 10: Exposure to Treatment — Safety Analysis Set
Exposure | Study 302 | |
|---|---|---|
Avatrombopag (N = 32) | Placebo (N = 17) | |
Core phase | ||
Exposure weeks, median (range)a | 26.0 (3.7 to 31.1) | 6.0 (2.1 to 29.9) |
Daily dose, mg, median (range) | 19.3 (5.9 to 37.6) | 33.3 (21.3 to 55.6) |
Combined core and extension phases | ||
n | 47 | 0 |
Duration of exposure, weeks, median (range)a | 43.9 (7.9 to 75.7) | 0 |
aDuration of exposure = date of last dose of study drug – date of first dose of study drug + 1.
Source: CSR for Study 302.12
Efficacy
Only efficacy outcomes and analyses of subgroups identified in the review protocol are reported here.
Bleeding Events (Incidence, Severity)
The incidence of any bleeding event (i.e., WHO bleeding grade ≥ 1) during the 6 months of treatment in the core phase was 43.8% in the avatrombopag group and 52.9% in the placebo group. Among patients who had any bleeding event, the majority (34.4% in avatrombopag and 52.9% in placebo groups) had WHO grade 1 bleeding events. No patients in the placebo group had a bleeding event that was higher than WHO grade 1. In the avatrombopag 9group, 2 patients had WHO grade 2 bleeding events and 1 patient had a WHO GRADE 3 bleeding event (epistaxis) (Table 11).
In the combined core and extension phases, a total of 3 patients reported grade 3 or 4 bleeding events.
Platelet Count Response
Cumulative Number of Weeks With Platelet Response
The median of the cumulative number of weeks with a platelet count of at least 50 × 109/L during the 6-month treatment period was 12.4 weeks (range, 0 to 25 weeks) for avatrombopag and 0 (range, 0 to 2) weeks for placebo (P < 0.0001).
The results for the subgroup analysis of the primary efficacy end point, except for subgroups with a very small number of patients, were consistent with the primary efficacy analysis, which showed a significant treatment difference favouring avatrombopag (data not shown).
Table 11: Efficacy — Bleeding Events, Core Phase, FAS
Efficacy | Study 302 | |
|---|---|---|
Avatrombopag (N = 32) | Placebo (N = 17) | |
Incidence of bleeding events during 6-month treatment period | ||
Yes | 14 (43.8) | 9 (52.9) |
No | 18 (56.3) | 8 (47.1) |
P value (chi-square distribution)a | 0.5394 | |
Incidence of bleeding events during 6-month treatment period by WHO bleeding scale, n (%) | ||
Grade 1 | 11 (34.4) | 9 (52.9) |
Grade 2 | 2 (6.3) | 0 (0.0) |
Grade 3 | 1 (3.1) | 0 (0.0) |
Grade 4 | 0 (0.0) | 0 (0.0) |
FAS = full analysis set.
aThe statistical testing for this outcome was not adjusted for multiple comparisons.
Source: CSR for Study 302.12
The Proportion of Patients With a Platelet Count of at Least 50 × 109/L At Day 8
The proportion of patients with platelet count of at least 50 × 109/L at day 8 favoured avatrombopag (65.63% [95% CI, 49.17 to 82.08%] for avatrombopag and 0.00% for placebo). The difference between avatrombopag and placebo was 65.63% (95% CI, 49.17% to 82.08%;, P < 0.0001).
Durable Platelet Response
The durable platelet response rate was 34.38% for avatrombopag and 0.00% for placebo. The between-group difference between avatrombopag and placebo was 34.38% (95% CI, 17.92% to 50.83%).
Change in Platelet Count From Baseline
During the core phase, the median platelet count in the avatrombopag group appeared to be higher than that in the placebo group (which was unchanged) over the 6-month treatment period, starting from day 8 (Table 12). The platelet response gained in the core phase was generally maintained throughout the extension phase until around week 36. Beyond week 38, the platelet response was noted to be lower and considerably more variable (Table 13).
Complete Response According to International Working Group Criteria
At week 26, the complete response rate was 18.2% (using a platelet count ≥ 100 × 109/L as the threshold) in the avatrombopag group and 0% in the placebo group. If a placebo count of at least 30 × 109/L was used as the threshold, the complete response rate was 54.6% in the avatrombopag group and0% in the placebo group.
Duration of Platelet Response
The median maximum duration of continuous response was 4.4 weeks (range, 0 to 25 weeks) in the avatrombopag group and 0 weeks (range, 0 to 2 weeks) in the placebo group.
Detailed results related to platelet response are presented in Table 12 and Table 13.
Results of a posthoc analysis of Study 302 reported additional efficacy end points related to platelet response.29 Based on this analysis, a response (platelet count ≥ 50 × 109/L) was achieved at 96.1% of the extension phase visits and a complete response (platelet count ≥ 100 × 109/L) was achieved at 60.1% of extension phase visits. The percent of visits in which a response or complete response was achieved for each patient in that treatment arm was averaged together, then divided by the number of patients in that arm to give an average percentage of time that patients in that arm achieved a response or complete response. A durable clinically relevant response (defined as a platelet count ≥ 30 × 109/L for 6 of the final 8 weeks of the core phase) occurred in 64.0% of patients in the avatrombopag group and0% in the placebo group.
Table 12: Efficacy — Platelet Response, Core Phase, FAS
Efficacy | Study 302 | |
|---|---|---|
Avatrombopag (N = 32) | Placebo (N = 17) | |
Cumulative number of weeks with a platelet count ≥ 50 × 109/L | ||
n | 32 | 17 |
Median (range) | 12.4 (0 to 25) | 0.0 (0 to 2) |
P value (Wilcoxon rank sum test) | < 0.0001 | |
Percent of patients with a platelet count ≥ 50 × 109/L at day 8 | ||
n | 32 | 17 |
Yes, n (%; 95% CI)a | 21 (65.63; 49.17 to 82.08) | 0 |
Between-group difference (95% CI) | 65.63 (49.17 to 82.08) | |
P value (Fisher’s exact test) | < 0.0001 | |
Durable platelet response rateb | ||
n | 32 | 17 |
Yes, n (%; 95% CI)a | 11 (34.38; 17.92 to 50.83) | 0 |
Between-group difference (95% CI) | 34.38 (17.92 to 50.83) | |
P value (Fisher’s exact test)c | 0.0090 | |
Change in platelet count, × 109/L, median (range) | ||
n at baseline | 32 | 17 |
Platelet count at baseline | 12.5 (1.0 to 31.5) | 9.5 (4.0 to 27.0) |
n at week 26 | 22 | 1 |
Platelet count at week 26 | 53.95 (3.0 to 187.0) | 31.0 (31.0 to 31.0) |
Change from baseline | 35.25 (–4.0 to 177.5) | 4.0 (4.0 to 4.0) |
Complete response (by IWG criteria,d a platelet count ≥ 100 × 109/L and absence of bleeding) at week 26 | ||
n | 22 | 1 |
Yes, n (%) | 4 (18.2) | 0 (0.0) |
Complete response (by IWG criteria,d a platelet count ≥ 30 × 109/L and absence of bleeding) at week 26 | ||
n | 22 | 1 |
Yes, n (%) | 12 (54.6) | 0 |
Maximum duration of continuous platelet response | ||
n | 32 | 17 |
Median (range), weeks | 4.4 (0 to 25) | 0.0 (0 to 2) |
P value (Wilcoxon rank sum test)b | < 0.0001 | |
CI = confidence interval; FAS = full analysis set; IWG = International Working Group.
a95% CI calculated based on normal approximation.
bDurable platelet response was defined as the proportion of patients who had at least 6 out of 8 weekly platelet responses during the final 8 weeks of treatment over the 6-month treatment period of the core phase in the absence of rescue therapy.
cStatistical testing for these outcomes was not adjusted for multiple comparisons.
dA platelet response according to IWG criteria was defined as a platelet count ≥ 100 × 109/L in the absence of bleeding, or 30 × 109/L and at least a 2-fold increase in baseline platelet count in the absence of bleeding. A platelet count that occurs in the 8 weeks after rescue therapy was considered a nonresponse.
Source: CSR for Study 302.12
Another posthoc analysis of Study 302 evaluated platelet response to avatrombopag during the core phase in different subgroups and the durability of response in patients who responded to avatrombopag during both the core phase and the combined core and extension phases.30 The subgroups of interest included the number of prior ITP therapies (< 3 versus ≥ 3), concomitant ITP medication use at baseline (yes versus no), and baseline platelet count (< 15 × 109/L versus ≥ 15 × 109/L). The proportion of patients with no loss of response (LOR), defined as a platelet count of less than 30 × 109/L over 2 consecutive scheduled visits, was 72.2% for patients who had received fewer than 3 prior ITP medications and 38.5% for those had received at least 3 prior ITP medications. The rate of patients with no LOR was 72.7% for patients with no concomitant ITP medication use at baseline versus 31.8% for those with concomitant ITP medication use at baseline. The proportion of patients with no LOR was 78.9% for those with a baseline platelet count of at least 15 × 109/L and 32.0% for those with a baseline platelet count of less than 15 × 109/L.
Health-Related Quality of Life
At week 26, there were 21 patients in the avatrombopag group and 1 patient in the placebo group providing HRQoL data. The change from baseline in the median physical component score and the median mental component score of the SF-36 and the visual analogue scale (VAS) score of the 3-Level EQ-5D (EQ-5D-3L) appeared to be minor (with a high level of variability) in the avatrombopag group; there were too few patients remaining at 26 weeks in the placebo group to draw conclusions.
Details are shown in Table 14.
Table 13: Efficacy — Change in Platelet Count, Extension Phase, Modified FAS
Efficacy | Study 302 |
|---|---|
Avatrombopag (N = 39) | |
Change in platelet count, median (range) | |
n at first day of extension (baseline) | 39 |
Platelet count at baseline, × 109/L | 9.5 (1.0 to 31.5) |
n at week 2 of extension | 38 |
Platelet count at week 2 of extension, × 109/L | 46.5 (3.9 to 481.0) |
Change from baseline, × 109/L | 38.8 (–15.0 to 462.0) |
n at week 4 of extension | 37 |
Platelet count at week 4 of extension, × 109/L | 65.0 (2.0 to 391.0) |
Change from baseline, × 109/L | 50.5 (–18.0 to 370.0) |
n at week 8 of extension | 35 |
Platelet count at week 8 of extension, × 109/L | 47.0 (3.0 to 388.0) |
Change from baseline, × 109/L | 31.0 (–8.0 to 367.0) |
n at week 16 of extension | 33 |
Platelet count at week 16 of extension, × 109/L | 48.0 (4.0 to 437.0) |
Change from baseline, × 109/L | 33.0 (–5.2 to 432.0) |
n at week 24 of extension | 25 |
Platelet count at week 24 of extension, × 109/L | 64.0 (3.0 to 231.0) |
Change from baseline, × 109/L | 43.5 (–4.0 to 216.5) |
n at week 32 of extension | 21 |
Platelet count at week 32 of extension, × 109/L | 61.0 (4.0 to 191.0) |
Change from baseline, × 109/L | 43.5 (–4.5 to 181.5) |
n at week 40 of extension | 13 |
Platelet count at week 40 of extension, × 109/L | 21.0 (3.0 to 156.0) |
Change from baseline, × 109/L | 11.0 (–3.5 to 131.9) |
n at week 48 of extension | 6 |
Platelet count at week 48 of extension, × 109/L | 55.7 (30.0 to 83.0) |
Change from baseline, × 109/L | 39.4 (20.5 to 68.5) |
n at week 72 of extension | 1 |
Platelet count at week 72 of extension, × 109/L | 11.0 (11.0 to 11.0) |
Change from baseline, × 109/L | 6.0 (6.0 to 6.0) |
Source: CSR for Study 302.8
Table 14: Efficacy — HRQoL, Core Phase, FAS
Efficacy | Study 302 | |
|---|---|---|
Avatrombopag (N = 32) | Placebo (N = 17) | |
SF-36, physical component score, median (range) | ||
Baseline | 47.8 (24.4 to 62.4) | 48.4 (20.8 to 57.9) |
n at week 26 | 21 | 1 |
Week 26 | 49.8 (29.1 to 64.0) | 41.9 (41.9 to 41.9) |
Change from baseline | 3.3 (–18.7 to 15.0) | 2.6 (2.6 to 2.6) |
SF-36, mental component score, median (range) | ||
Baseline | 48.5 (23.0 to 62.2) | 46.8 (26.6 to 58.1) |
n at week 26 | 21 | 1 |
Week 26 | 49.8 (34.3 to 59.6) | 34.4 (34.4 to 34.4) |
Change from baseline | 8.6 (–22.4 to 19.3) | 7.8 (7.8 to 7.8) |
EQ-5D-3L, VAS score, median (range) | ||
Baseline | 80.0 (45.0 to 100.0) | 70.0 (55.0 to 100.0) |
n at week 26 | 21 | 1 |
Week 26 | 80.0 (50.0 to 100.0) | 40.0 (40.0 to 40.0) |
Change from baseline | 0.0 (–20.0 to 10.0) | –30.0 (–30.0 to –30.0) |
EQ-5D-3L = 3-Level EQ-5D; FAS = full analysis set; HRQoL = health-related quality of life; SF-36 = 36-item Short-Form Health Survey; VAS = visual analogue scale.
Source: CSR for Study 302.12
Symptoms
Not assessed.
Treatment-Free Remission
Not assessed.
Need for Rescue Therapy
The proportion of patients who needed rescue therapy was 21.9% in the avatrombopag group and 11.8% the placebo group. This should be interpreted in light of the 2.6-fold lower exposure and the high discontinuation rate in the placebo group.
Use of Concomitant ITP Medications
For the proportion of patients with a reduction in the use of concomitant ITP medication from baseline, the 22 patients using ITP medication at baseline were included in the analysis. The observed treatment difference between avatrombopag and placebo was 33.33% (33.33% in the avatrombopag and 0.00% in the placebo group; 95% CI, 9.48% to 57.19%; P = 0.1348).
The results of a posthoc analysis of Study 302 showed that 57.1% of patients who needed corticosteroids at baseline reduced or discontinued corticosteroid therapy in the core phase and extension phase (including 7 of 14 patients originally randomized to avatrombopag therapy, and 5 of 7 patients originally randomized to placebo but who switched to avatrombopag therapy during the extension phase).29
Need for Monitoring
Not assessed.
Table 15: Efficacy — Need for Rescue Therapy, Core Phase, FAS
Efficacy | Study 302 | |
|---|---|---|
Avatrombopag (N = 32) | Placebo (N = 17) | |
Need for ≥ 1 rescue therapy during 6-month treatment | ||
Yes, n (%; 95% CI)a | 7 (21.88; 7.55 to 36.20) | 2 (11.76; 0.00 to 27.08) |
Dapsone, n (%) | 1 (3.1) | 0 |
Immunoglobulin human normal, n (%) | 2 (6.3) | 1 (5.9) |
Methylprednisolone, n (%) | 2 (6.3) | 0 |
Prednisolone, n (%) | 1 (3.1) | 0 |
Prednisone, n (%) | 2 (6.3) | 0 |
Platelet transfusion, n (%) | 1 (3.1) | 1 (5.9) |
Between-group difference (95% CI) | 10.11 (–10.86 to 31.08) | |
P value (Fisher’s exact test)b | 0.4668 | |
CI = confidence interval; FAS = full analysis set.
aThe 95% CI was calculated based on normal approximation.
bStatistical testing for this outcome was not adjusted for multiple comparisons.
Source: CSR for Study 302.12
Table 16: Efficacy — Reduction in Use of Concomitant ITP Medications, Core Phase, FAS
Efficacy | Study 302 | |
|---|---|---|
Avatrombopag (N = 32) | Placebo (N = 17) | |
Reduction in use of concomitant ITP medications from baseline | ||
n | 15 | 7 |
Yes, n (%) | 5 (33.33) | 0 (0.0) |
Between-group difference (95% CI)a | 33.33 (9.48 to 57.19) | |
P value (Fisher’s exact test) | 0.1348 | |
CI = confidence interval; FAS = full analysis set; ITP = immune thrombocytopenia.
aThe 95% CI was calculated based on normal approximation.
Source: CSR for Study 302.12
Emergency Department Visits or Hospitalization Due to Thrombocytopenia Episode
Hospitalization was required for patients in the avatrombopag group at day 8 (1 patient), week 3 (2 patients), week 10 (1 patient), week 16 (1 patient), week 21 (1 patient), and week 25 (1 patient).
Hospitalization was required for patients in the placebo group at week 16 (1 patient), week 19 (1 patient), week 20 (1 patient), and week 21 (1 patient).
Emergency visits were required for patients in the avatrombopag group at week 3 (1 patient) and week 16 (1 patient).
Emergency visits were required for patients in the placebo group at week 19 (1 patient) and week 20 (1 patient).
Mortality
No deaths were reported during study.
Harms
Only harms identified in the review protocol are reported here. Refer to Table 17 for detailed harms data.
Adverse Events
During the core phase, there were 31 (96.9%) patients in the avatrombopag group and 10 (58.8%) patients in the placebo group who reported 1 or more TEAEs. There were 6 (18.8%) patients in the avatrombopag group who reported a grade 3 or 4 AEs, compared to none in the placebo group. The most commonly reported (i.e., in 10% of patients or more) AEs were headache, contusion, upper respiratory tract infection, arthralgia, epistaxis, fatigue, gingival bleeding, and petechiae.
Among the 47 patients enrolled in extension phase, 45 (95.7%) reported 1 or more TEAEs. The most commonly reported AEs during the core phase and extension phase were contusion (40.4%), headache (29.8%), upper respiratory tract infection (23.4%), thrombocytopenia (19.1%), epistaxis (17.0%), gingival bleeding (17.0%), fatigue (14.9%), petechiae (14.9%), pharyngitis (12.8%), arthralgia (10.6%), hypertension (10.6%), nasopharyngitis (10.6%).
Serious Adverse Events
During the core phase, there were 9 patients (28.1%) in the avatrombopag group and 1 patient (5.9%) in the placebo group that reported 1 or more treatment-emergent SAEs. The most commonly reported treatment-emergent SAEs (occurring in more than 1 patient) were gastrointestinal disorders in 3 patients (9.4%) in the avatrombopag group (vomiting occurred in 2 patients [6.3%] and nausea in 1 patient [3.1%]) and none in the placebo group, and nervous system disorders in 3 patients (9.4%) in the avatrombopag group (headache occurred in 2 patients [6.3%]) and none in the placebo group.
During the combined core and extension phases, 15 of 47 (31.9%) patients reported any treatment-emergent SAE. The most commonly reported treatment-emergent SAEs were gastrointestinal disorders (in 5 [10.6%] patients) and nervous system disorders (in 4 [8.5%] patients).
Withdrawal Due to Adverse Events
During the core phase, 3 patients (9.4%) in the avatrombopag group and none in the placebo group reported TEAEs leading to discontinuation of the study drug (cerebrovascular accident, headache, and polyserositis).
During the combined core and extension phases, 6 of 47 (12.8%) patients in the avatrombopag group discontinued the study drug with any TEAE. Three additional events occurred in the extension phase: chronic myelomonocytic leukemia, gastritis hemorrhagic, and erosive duodenitis dizziness.
Mortality
No deaths were reported during the study.
Notable Harms
During the core phase, in the avatrombopag group, 3 patients (9.4%) reported thromboembolic events, 1 patient (3.1%) reported neoplastic events, and 1 patient (3.1%) reported the recurrence of thrombocytopenia. No patient in the placebo group reported notable harms.
A total of 4 of 47 (8.5%) patients reported thromboembolic events in the combined core and extension phases (3 in the core phase and 1 in the extension phase). Of note, 7 (14.9%) patients reported a recurrence of thrombocytopenia (1 patient in the core phase and 6 patients in the extension phase), and 2 (4.3%) patients reported neoplastic events (1 in the core phase and 1 in the extension phase).
Table 17: Summary of Harms — Core Phase, Safety Analysis Set
Safety | Study 302 | |
|---|---|---|
Avatrombopag (N = 32) | Placebo (N = 17) | |
Patients with ≥ 1 AE | ||
n (%) | 31 (96.9) | 10 (58.8) |
Most common events,a n (%) | ||
Headache | 12 (37.5) | 2 (11.8) |
Contusion | 10 (31.3) | 4 (23.5) |
Upper respiratory tract infection | 6 (18.8) | 1 (5.9) |
Arthralgia | 4 (12.5) | 0 (0.0) |
Epistaxis | 4 (12.5) | 3 (17.6) |
Fatigue | 4 (12.5) | 1 (5.9) |
Gingival bleeding | 4 (12.5) | 0 (0.0) |
Petechiae | 4 (12.5) | 1 (5.9) |
Patients with ≥ 1 SAE | ||
n (%) | 9 (28.1) | 1 (5.9) |
Events | ||
Idiopathic thrombocytopenia purpura | 0 | 1 |
Thrombocytopenia | 1 | 0 |
Vomiting | 2 | 0 |
Nausea | 1 | 0 |
Headache | 2 | 0 |
Cerebrovascular accident | 1 | 0 |
Food poisoning | 1 | 0 |
Mouth hemorrhage | 1 | 0 |
Polyserositis | 1 | 0 |
UTI | 1 | 0 |
Increased platelet count | 1 | 0 |
Urinary hemorrhage | 1 | 0 |
Epistaxis | 1 | 0 |
Petechiae | 1 | 0 |
Deep vein thrombosis | 1 | 0 |
WDAEs | ||
n (%) | 3 (9.4) | 0 (0.0) |
Events | ||
Cerebrovascular accident | 1 | 0 |
Headache | 1 | 0 |
Polyserositis | 1 | 0 |
Deaths | ||
n (%) | 0 (0.0) | 0 (0.0) |
Notable harms, n (%) | ||
Core phase | ||
Thromboembolic events | 3 (9.4) | 0 (0.0) |
Cerebrovascular accident | 1 (3.1) | 0 (0.0) |
Deep vein thrombosis | 1 (3.1) | 0 (0.0) |
Pulmonary embolism | 1 (3.1) | 0 (0.0) |
Neoplastic events | 1 (3.1) | 0 (0.0) |
Myelofibrosis | 1 (3.1) | 0 (0.0) |
Recurrence of thrombocytopenia | 1 (3.1) | 0 (0.0) |
Thrombocytopenia | 1 (3.1) | 0 (0.0) |
Hypersensitivity reaction | 2 (6.3) | 0 (0.0) |
Clinically significant liver test abnormalities | NR | NR |
Bone marrow pathology | NR | NR |
Gastric atrophy events | NR | NR |
Core and extension phases (n = 47) | ||
Thromboembolic events | 4 (8.5) | |
Cerebrovascular accident | 1 (2.1) | |
Deep vein thrombosis | 1 (2.1) | |
Jugular vein thrombosis | 1 (2.1) | |
Pulmonary embolism | 1 (2.1) | |
Neoplastic events | 2 (4.3) | |
Myelofibrosis | 1 (2.1) | |
Chronic myelomonocytic leukemia | 1 (2.1) | |
Recurrence of thrombocytopenia | 7 (14.9) | |
Thrombocytopenia | 7 (14.9) | |
Hypersensitivity reaction | NR | |
Clinically significant liver test abnormalities | 2 (4.3) | |
Increased alanine aminotransferase | 1 (2.1) | |
Increased aspartate aminotransferase | 1 (2.1) | |
Increased gamma-glutamyl transferase | 1 (2.1) | |
Increased hepatic enzyme | 1 (2.1) | |
Bone marrow pathology | NR | |
Gastric atrophy events | NR | |
AE = adverse event; NR = not reported; SAE = serious adverse event; UTI = urinary tract infection; WDAE = withdrawal due to adverse event.
aFrequency ≥ 10%.
Source: CSR for Study 302.12
Critical Appraisal
Internal Validity
Study 302 was a small (N = 49), phase III, double-blind (core phase), placebo-controlled RCT. Appropriate methods were used to randomize patients to treatments and to conceal treatment allocation (i.e., central randomization with stratification by splenectomy status, baseline platelet count, and use of concomitant ITP medications). There were some relatively large baseline imbalances between the avatrombopag and placebo groups, such as sex (female, 72% versus 47%), baseline platelet count (12.5 × 109/L versus 9.5 × 109/L), concomitant use of ITP medication at baseline (47% versus 41%), and prior ITP medication (47% versus 35%). This could suggest selection bias, but is most likely the result of the small sample of patients randomized. The imbalanced characteristics might bias the results against placebo, especially for the outcomes related to platelet response (such as platelet response at study end points, proportion of responders, duration of response, need for rescue therapy, and bleeding events), although it is unclear to what extent.
During the core phase, all patients and personnel (including investigators, site personnel, and sponsor staff) were blinded to the treatments received (but not dose); therefore, it is unlikely that outcomes data were affected by performance or detection biases. The extension phase was open-label, which may have impacted the reporting of safety data (i.e., a higher number of AEs might have been reported) or patient-reported HRQoL outcomes.
In Study 302, after testing of the primary outcome, multiplicity was adjusted for 2 secondary efficacy outcomes (the proportion of patients who achieved a platelet response at day 8 and the use of concomitant ITP medications) using a gatekeeping approach. Outcomes outside of this testing procedure, such as HRQoL, durable platelet response, and bleeding events (exploratory outcomes in Study 302), were considered to be exploratory and should be interpreted with consideration for the increased possibility of false-positive conclusions.
The rate of study discontinuation was high in Study 302 and was imbalanced between treatment arms; 22% of patients in the avatrombopag group and 88% of patients in the placebo group withdrew from the study because of an inadequate therapeutic effect. These patients were deemed to be nonresponders. It is reasonable to expect that these findings were affected by the substantially shorter length of exposure in the placebo group than in the avatrombopag group (median [range], 6.0 [2.1 to 9.9] versus 26.0 [3.7 to 31.1] weeks). This affected assessment of the clinically relevant outcomes of bleeding events and rescue medication; no clear conclusions about the effects of avatrombopag on these outcomes could be drawn. The high dropout rate also had a substantial impact on patient-reported outcomes, such as HRQoL. At the end of the core phase, only 1 patient in the placebo group provided data for the SF-36 and EQ-5D. It is not possible to draw meaningful conclusions about the effect of the study drug on patients’ HRQoL due to the limited amount of data as a result of study discontinuation.
Prespecified subgroup analyses were conducted for the primary efficacy outcome, and the results were generally consistent with the primary analysis. However, given the small sample size, these analyses are considered to be exploratory and were likely underpowered to detect differences between treatment groups. No conclusions can be drawn from these subgroup analyses.
For platelet count outcomes, missing platelet assessments at a specific visit were considered to be a nonresponse at that visit. Patients who discontinued the study or who were lost to follow-up before the end of the 6-month treatment period had all subsequent unobserved scheduled platelet assessments at the scheduled visits documented as missing platelet values. For the HRQoL outcomes, it appears that a complete case analysis was performed, but it only included patients who had no missing HRQoL data.
In Study 302, the cumulative number of weeks with a platelet count of at least 50 × 109/L was the primary outcome measure. However, the relation between bleeding events and platelet count, based on the results of an RCT evaluating the effect of prophylactic platelet transfusion dose on the risk of bleeding in patients with hypoproliferative thrombocytopenia,13 could not be determined because bleeding can occur over a broad spectrum of platelet counts; therefore, the threshold at which the platelet count affects the risk of bleeding is not clear. In practice, platelet count is considered a surrogate for the risk of bleeding events and survival, according to the patient input and clinician input. Gains from the number of weeks with a platelet response may be correlated to a reduction in the risk of bleeding or improved quality of life. According to the clinical expert consulted by CADTH, a threshold of 30 × 109/L or lower is often used in practice to determine treatment response and the risk of subsequent bleeding, whereas a threshold of 50 × 109/L is more commonly used in patients undergoing invasive procedures. The clinical practice guidelines indicate that treatment should maintain a target platelet level of 20 × 109/L to 30 × 109/L, at least for symptomatic patients (because the risk for major bleeding increases below this level). The guidelines also state that a platelet count of at least 50 × 109/L should be obtained before certain procedures, such as splenectomy, obstetric analgesia and anesthesia, or the administration of anticoagulation.9 Although in Study 302 a threshold of 50 × 109/L for platelet response was used to assess the treatment effect, there were limited or no data on patient-important outcomes, such as bleeding rates, use of concomitant ITP medications, need for rescue therapy, symptoms, and HRQoL.
External Validity
Although the population in Study 302 may be highly selective (about half failed screening), the clinical expert consulted by CADTH indicated that, according to baseline patient characteristics, the population of Study 302 is broadly comparable to patients with ITP in Canada and, thus, the study findings are likely generalizable in Canada. The long duration of ITP, multiple previous treatments, and concomitant ITP medication use among patients in Study 302 mirror what is commonly seen for patients with chronic ITP in clinical practice in Canada. Patients with secondary ITP were excluded from the study, so the generalizability of the study results to this particular patient population may be limited. It is not possible to draw meaningful conclusions about patient-important outcomes, such as HRQoL, due to high dropout rates.
Another challenge with Study 302 is that the comparator is placebo. Treatment would be warranted for patients with chronic ITP who have platelet counts lower than 20 × 109/L. However, Study 302 has provided no information on how the efficacy and safety of avatrombopag may differ from other available treatments.
Indirect Evidence
Objectives and Methods for the Summary of Indirect Evidence
Because there was no direct evidence comparing avatrombopag to other active therapies for the treatment of thrombocytopenia in adult patients who have had an insufficient response to a previous treatment, a review of indirect evidence was undertaken. In addition to reviewing the sponsor’s submission, CADTH conducted a literature search to identify potentially relevant ITCs of patients with chronic ITP. A focused literature search for NMAs dealing with avatrombopag, thrombopoietin receptor agonists, or immune thrombocytopenia was run in MEDLINE All (1946-) via Ovid on March 3, 2022. No limits were applied. Titles, abstracts, and full-text articles were screened for inclusion by 1 reviewer based on the population, intervention, comparator, and outcome criteria outlined in Appendix 1.
One sponsor-submitted ITC was included in this review.14 An ITC by Wojciechowski et al.(2021)37 was identified in the CADTH literature search. This study used the same methods for NMA and reported similar results as the sponsor-submitted ITC; therefore, only the sponsor-submitted ITC is presented in the current review.
Description of Indirect Comparisons
The sponsor-submitted ITC included a systematic review of the literature and an ITC that compared the current pharmaceutical treatments for chronic ITP with each other. In the sponsor-submitted ITC, avatrombopag was compared to 2 TPO-RAs (eltrombopag and romiplostim), fostamatinib, and rituximab.
Methods From the Sponsor-Submitted ITC
Objectives
The objective of the sponsor-submitted report for patients with chronic ITP was to conduct a systematic review and, if possible, an ITC to evaluate the relative efficacy and safety of avatrombopag versus other treatments currently available to this patient population.
Study Selection Methods
A systematic literature search was performed in March 2020 to identify all clinical evidence relevant to the NMA of avatrombopag in patients with chronic ITP. Multiple electronic databases were searched for trials of avatrombopag, eltrombopag, romiplostim, and fostamatinib; clinicaltrials.gov and a multiple technology appraisal conducted by the National Institute for Health and Care Excellence (NICE)38 were also used as sources of studies. Trials of rituximab were identified from another recently published NMA.39 The inclusion and exclusion criteria used for study selection to inform the NMA are presented in Table 18It is unclear whether 2 reviewers independently performed study selection and data extraction. The risk of bias and the quality of the included studies were not assessed.
In this report, chronic ITP was required to have a duration of at least 12 months by definition; however, some included trials may have been designed and conducted before the current definition of chronic ITP was developed, so patients with a shorter disease duration (e.g., at least 6 months) were recruited. Such studies were deemed to be eligible for this analysis if all other inclusion criteria for the NMA were met and the average duration of the disease was at least 12 months.
Inclusion and exclusion criteria for the clinical studies used in the sponsor-submitted ITC are presented in Table 18.
Table 18: Study Selection Criteria and Methods for the ITC
Variable | Sponsor-submitted ITC |
|---|---|
Population | Adult patients (≥ 18 years) with chronic ITP (defined as > 6 months) |
Intervention | Avatrombopag (initial dose, 20 mg once daily) Eltrombopag (initial dose, 50 mg once daily) Romiplostim (initial dose, 1 mcg/kg) Fostamatinib (initial dose, 100 mg twice daily) Rituximab |
Comparator | Another intervention (any), placebo |
Outcome | Platelet count and duration of platelet count response rate and duration of response rate Need for rescue treatments for bleeding Reduction in use of concomitant ITP treatments Bleeding events AEs, SAEs, dropouts due to AEs Mortality |
Study design | RCT |
Exclusion criteria | Studies exclusively of Asian patients Dose regimens not approved by the EMA Non-RCTs Studies with a treatment period < 9 weeks |
Databases searched | Embase, MEDLINE, CENTRAL, Cochrane Database of Systematic Reviews, clinicaltrials.gov, and the multiple technology appraisal of the comparison between avatrombopag, eltrombopag, romiplostim, and fostamatinib developed by NICE Trials of rituximab identified via another recently published NMA.39 |
Selection process | Not specified |
Data extraction process | Not specified |
Quality assessment | Not reported |
AE = adverse event; EMA = European Medicines Agency; ITC = indirect treatment comparison; ITP = immune thrombocytopenia; NICE = National Institute for Health and Care Excellence; NMA = network meta-analysis; RCT = randomized controlled trial; SAE = serious adverse event.
Source: Sponsor-submitted ITC.14
Analysis Methods From the Sponsor-Submitted ITC
The NMA was carried out in a Bayesian framework, using a Markov Chain Monte Carlo (MCMC) method, with vague prior distributions used for the model parameters. The outcomes were reported as odds ratios or IRRs, with corresponding 95% Crls. The IRR was used for the need for rescue therapy, the incidence of bleeding events, and AEs to account for the high number of dropouts and the resulting difference in mean exposure time in some trials. Fixed-effects and random-effects models were fitted, and the model fit was examined based on the deviance information criterion. The fixed-effects model was preferred; it was the simpler model and had a lower number of estimable parameters, although the random-effects model could be selected if the corresponding deviance information criterion value was more than 5 points lower than that for the fixed-effects model. A difference in the mean change from the baseline was considered statistically significant when the associated 95% CrI did not include 0. An odds ratio or IRR was considered statistically significant when the associated 95% CrI did not include 1. The convergence of models has been assessed with 2 diagnostics tools: the trace plot, and the Brooks-Gelman-Rubin diagnostic tool. The consistency of this NMA was assessed by examining the closed loops formed by different studies.
Three chains were run for each analysis, with 25,000 or 50,000 burn-in iterations for the fixed-effects and random-effects models, respectively, followed by 25,000 iterations.
Details of the analysis methods used in this ITC are summarized in Table 19.
Table 19: ITC Analysis Methods
Analysis Methods | Sponsor-submitted ITC |
|---|---|
ITC methods | Bayesian framework using a Markov Chain Monte Carlo method |
Priors | Vague priors were set for model parameters. Noninformative priors were used for nuisance parameters, treatment effect parameters, and heterogeneity parameters. |
Assessment of model fit | Deviance information criterion |
Assessment of consistency | No closed loops |
Assessment of convergence | 2 diagnostic tools were used:
|
Outcomes | Durable platelet response Need for rescue therapy Use of concomitant ITP medication Any bleeding events WHO grade 2 to 4 bleeding events AEs |
Follow-up time points | 26 to 36 weeks |
Construction of nodes | NR |
Sensitivity analyses | None |
Subgroup analysis | None |
Methods for pairwise meta-analysis | NA |
AE = adverse event; ITC = indirect treatment comparison; ITP = immune thrombocytopenia; NA = not available; NR = not reported.
Source: Sponsor-submitted ITC.14
Results of Sponsor-Submitted ITC
Summary of Included Studies
Nine RCTs were included in the NMA. Avatrombopag was assessed in 2 studies: Study 302 comparing avatrombopag versus placebo, and Study 305 comparing avatrombopag with eltrombopag. Study 305 was terminated early due to significant enrolment challenges, but it was included in the NMA for safety outcomes. All other trials compared a single treatment to placebo; there was 1 trial of eltrombopag versus placebo, 2 trials of romiplostim versus placebo, 2 trials of fostamatinib versus placebo, and 2 trials of rituximab versus placebo. Study 302 and Study 305 were the only trials that enrolled patients with ITP lasting longer than 12 months (Table 20).
Figure 3 presents the network of evidence in the NMA in patients with chronic or persistent ITP in the sponsor-submitted ITC. The trials formed a star-shaped network, with 1 closed loop formed by avatrombopag, eltrombopag, and placebo.
The heterogeneity of the included RCTs was assessed to evaluate whether an NMA would have been possible. According to the baseline characteristics presented in the report, age, sex, and race distributions were comparable across trials. Differences were observed for the proportion of patients who had undergone splenectomy (0% to 50%), the proportion of patients who used concomitant ITP medication at baseline (13% to 48%), and the duration of ITP (median, 0.25 to 8.7 years). There was substantial between-trial heterogeneity in the proportion of patients prematurely discontinuing allocated treatment (range, 0% to 100%).
Furthermore, efficacy outcomes for the included trials were defined differently. For example, the outcome of durable response was measured based on data from 4 of 6 visits that occurred from week 14 to week 24 in the FIT1 and FIT2 studies, but this outcome was measured based on data from at least 6 of the final 8 weeks of treatment in the other trials. Bleeding events were assessed using different scales (WHO bleeding scale, National Cancer Institute Common Toxicity Criteria for Adverse Events [CTCAE] scale, ITP bleeding score, unnamed scales, or no specific scale across the trials). To compare bleeding events between all treatments, the authors of this ITC report assumed that WHO grade 2 to grade 4 bleeds were equivalent to grade 2 to grade 5 bleeds described by Kuter (2008),41 to significant bleeds described by Arnold (2012)42 and moderate to severe bleeds in the FIT1 and FIT2studies.44
Efficacy
Durable Platelet Response
There were 6 trials (comprising 458 patients and 5 treatment regimens: avatrombopag, eltrombopag, romiplostim, fostamatinib, and placebo) included in the NMA.
Results of the NMA demonstrated that all active treatments were associated with significantly higher odds of a durable response than placebo. No significant differences regarding the proportion of patients with a durable response were observed between avatrombopag and the active comparators. However, all effects were associated with serious imprecision; the CrIs included the potential for no difference or for either treatment to be favoured in each comparison.
Need for Rescue Therapy
There were 7 trials (comprising 688 patients and 6 treatment regimens: avatrombopag, eltrombopag, romiplostim, fostamatinib, rituximab, and placebo) included in the NMA.
Results of the NMA demonstrated that all active treatments were associated with a significantly lower incidence of the need for rescue therapy than placebo, except the difference between avatrombopag and placebo was not statistically significant. No significant differences regarding the estimated incidence of the need for rescue therapy were observed between avatrombopag and the active comparators. However, all effects were associated with serious imprecision; the CrIs included the potential for no difference or for either treatment to be favoured in each comparison.
Reduction in the Use of Concomitant ITP Medication
There were 4 trials (comprising 155 patients and 4 treatment regimens: avatrombopag, eltrombopag, romiplostim, and placebo) included in the NMA.
Results of the NMA demonstrated that all active treatments were associated with significantly higher odds of a reduction in the use of concomitant ITP medications than placebo. No significant differences regarding the proportion of patients with a reduction in the use of concomitant ITP medications were observed between avatrombopag and the active comparators. However, all effects were associated with serious imprecision; the CrIs included the potential for no difference or for either treatment to be favoured in each comparison.
Any Bleeding Events
There were 7 trials (comprising 712 patients and 6 treatment regimens: avatrombopag, eltrombopag, romiplostim, fostamatinib, rituximab, and placebo) included in the NMA.
Results of the NMA demonstrated that avatrombopag and fostamatinib were associated with a significantly lower incidence of any bleeding than placebo. There were no statistically significant differences in the incidence of any bleeding between eltrombopag and placebo, romiplostim and placebo, or rituximab and placebo. Avatrombopag was associated with a significantly lower incidence rate of any bleeding than eltrombopag, romiplostim, and rituximab (IRR = 0.38 [95% CrI, 0.19 to 0.75], IRR = 0.38 [95% CrI, 0.17 to 0.86], and IRR = 0.44 [95% CrI, 0.21 to 0.94], respectively). The magnitude of the difference between avatrombopag and the active comparators (eltrombopag, romiplostim, rituximab) is unclear due to wide CrIs. No significant difference was observed for avatrombopag compared to fostamatinib (IRR = 0.69 [95% CrI, 0.28 to 1.67]). However, the comparison was associated with serious imprecision; the CrI included the potential for no difference or for either treatment to be favoured.
Figure 3: Network of Evidence in the Sponsor-Submitted ITC
AVA = avatrombopag; ELT = eltrombopag; FOS = fostamatinib; ITC = indirect treatment comparison; PLC = placebo; ROM = romiplostim; RTX = rituximab.
Source: Sponsor-submitted ITC.14
Table 20: Summary of Trials Included in the Sponsor-Submitted NMA
Study | Design | Intervention vs. comparison | Dose regimens, intervention vs. comparator | Number of randomized patients | Length of follow-up | Primary outcome | Duration of ITP |
|---|---|---|---|---|---|---|---|
Study 30228 | Phase III, MC, RAND, DB; 35 centres in multiple countries | AVA vs. PLC | 20 mg once daily | 32 vs. 17 | 26 weeks | Number of weeks with a PC ≥ 50 × 109/L during the 6-month treatment period | ≥ 12 months |
Study 305 | Phase III, MC, RAND, DB; 72 centres in 10 countries (terminated early) | AVA vs. ELT | 20 mg once daily vs. 50 mg once daily | 12 vs. 11 | Terminated early; number of weeks unknown | Change from baseline in PC for the 6-month treatment period, measured by local labs | ≥ 12 months |
RAISE40 | Phase III, MC, RCT, DB; 75 centres in 23 countries | ELT vs. PLC | 50 mg once daily | 135 vs. 62 | 30 weeks (26-week intervention + 4-week follow-up) | Percentage of responders at 6 months | ≥ 6 months |
Kuter 2008 (NCT00102323)41 | phase III, MC, RCT, DB, 35 sites in the US and Europe | ROM vs. PLC | 1 g/kg | 42 vs. 21 | 36 weeks (24 intervention + 12 follow-up) | Durable platelet response during the final 8 weeks of treatment and other platelet response parameters | ≥ 6 months |
Kuter 2008 (NCT00102336)41 | 41 vs. 21 | No restrictions | |||||
Arnold 2012 (NCT00372892)42 | RCT; 7 centres in Canada | RTX vs. PLC | 375 mg/m2 | 32 vs. 26 | 24 weeks | Treatment failure, defined as the composite of:
| Median, 1 year (IQR, 0 to 3.5, with 28 newly diagnosed patients) |
Ghanima 201543 | RCT,14 centres in Norway, Tunisia, and France | RTX vs. PLC | 375 mg/m2 | 55 vs. 54 | 78 weeks | Rate of treatment failure within 78 weeks — a composite of splenectomy and of meeting the criteria for splenectomy after week 12 if splenectomy was not done | Median (IQR): 37 (8 to 288) weeks vs. 50 (14 to 211) weeks for RTX vs. PLC (with 30 newly diagnosed patients) |
FIT 1, FIT 244 | Phase III, MC, RCT, DB | FOS vs. PLC | 100 mg twice daily | 101 vs. 49 | 24 weeks | Stable response (response on at least 4 of the last 6 visits between 14 and 24 weeks) | ≥ 3 months |
AVA = avatrombopag; DB = double-blind; ELT = eltrombopag; FOS = fostamatinib; IQR = interquartile range; ITP = immune thrombocytopenia; MC = multicentre; NMA = network meta-analysis; PC = platelet count; PLC = placebo; RAND = randomized; RCT = randomized controlled trial; ROM = romiplostim; RTX = rituximab.
Source: Sponsor-submitted ITC.14
Bleeding Events of WHO Grade 2 to Grade 4
There were 8 trials (comprising 712 patients and 6 treatment regimens: avatrombopag, eltrombopag, romiplostim, fostamatinib, rituximab, and placebo) included in the NMA.
Results of the NMA demonstrated that romiplostim and fostamatinib were associated with a significantly lower incidence of WHO grade 2 to grade 4 bleeding than placebo. There were no statistically significant differences in the incidence of WHO grade 2 to grade 4 bleeding between avatrombopag and placebo, eltrombopag and placebo, or rituximab and placebo. No significant differences regarding the proportion of patients with WHO grade 2 to grade 4 bleeding were observed between avatrombopag and the active comparators. However, all effects were associated with serious imprecision; the CrIs included the potential for no difference or for either treatment to be favoured in each comparison.
Safety
Adverse Events
There were 6 trials (comprising 542 patients and 6 treatment regimens: avatrombopag, eltrombopag, romiplostim, fostamatinib, rituximab, and placebo) included in the NMA.
Results of the NMA showed that there were no statistically significant differences in the incidence rate of AEs for the comparisons between any active treatments and placebo. No significant differences regarding the estimated incidence of any AEs were observed between avatrombopag and eltrombopag, romiplostim, or fostamatinib. However, all effects were associated with serious imprecision; the CrIs included the potential for no difference or for either treatment to be favoured in each comparison. Avatrombopag was associated with a reduced incidence of AEs compared to rituximab (IRR = 0.49, 95% CrI, 0.25 to 0.95]); the magnitude of the difference is unclear due to substantial imprecision.
Details of the results are presented in Table 21.
Critical Appraisal of the Sponsor-Submitted ITC
The analysis of the efficacy and safety data presented was limited by the size of the evidence base.
Trials were identified by searching multiple databases for prespecified inclusion and exclusion criteria. It is unclear whether 2 independent reviewers performed the study selection based on specific inclusion and exclusion criteria and data extraction; therefore, there is a small possibility that errors were introduced. In the research protocol for the current review, a number of comparators for avatrombopag have been identified (Table 5). Some of them were not included in the ITC. These comparators could have added additional information to the network and improved inferences among treatments. Quality or risk of bias assessment of the included studies was not presented; therefore, it is unclear whether any trials were affected by a high risk of bias, or the direction of any bias introduced.
The trial characteristics and patient baseline characteristics of the studies included in the systematic review and ITC were reported. Based on the data presented, potential sources of heterogeneity with respect to the baseline characteristics were identified, such as the proportion of patients who had undergone splenectomy (0% to 50%), the proportion of patients who used concomitant ITP medication at baseline (13% to 48%), and the duration of chronic ITP (median, 0.25 to 8.7 years). Other patient characteristics should also be considered when addressing clinical heterogeneity across the included trials, such as cycles and doses of prior corticosteroids therapy, previous lines of therapy, and severity of previous bleeding events. Such data were not provided in the ITC. Therefore, adjustment for their potential treatment effect modification was not feasible, and it is likely that the transitivity assumption was not met. Furthermore, there was a significant between-trial heterogeneity in the proportion of patients prematurely discontinuing allocated treatment (range, 0% to 100%), which would have had an impact on the total exposure time of the study drug in the included trials and could have affected the results for relative efficacy and safety, for example, by decreasing the chance of bleeding events or AEs in the placebo group. However, the authors of the ITC adjusted for this by using IRRs, which accounted for the duration of exposure.
Some of the clinical outcome measures were defined differently across the included trials. The outcome of need for rescue therapy was comparable across the included trials. However, durable platelet response was defined using a different observation period.
Bleeding episodes were measured using different approaches (assuming that WHO grade 2 to grade 4 bleeds [reported in all trials] were equivalent to the grade 2 to grade 5 bleeds reported in Kuter (2008) study, significant bleeds reported in the Arnold (2012) study, and the moderate-severe bleeds reported in the FIT1 and FIT2 trials). The inconsistency in outcome definitions could limit comparisons across the trials. Sensitivity analyses based on different definitions of the study outcomes were not performed to examine the robustness of the results from the primary analysis.
In addition, due to the small sample size of the included trials, sparse network, and low event rate for some of the efficacy outcomes, such as durable platelet response and reduction in the use of concomitant ITP medication, all analyses were associated with substantial imprecision; therefore, few comparisons reached statistical significance and conclusions about the comparative effectiveness and safety of avatrombopag versus other treatments are associated with substantial uncertainty. Furthermore, the network was star shaped (1 closed loop was applicable to the AEs only); therefore, consistency in the ITC analyses could not be tested. All comparisons are therefore informed only by indirect evidence, which increases the level of uncertainty.
HRQoL is 1 of the important patient outcomes. This outcome was not included in the NMA; therefore, the relative treatment effect of avatrombopag versus other active treatments on patients’ HRQoL remain unknown.
Table 21: OR IRR (95% CrI) for Avatrombopag Versus Other Treatments for Efficacy and Safety Outcomes in the Sponsor-Submitted ITC
Comparison between AVA and other treatments | Durable platelet response OR (95% CrI) | Reduction in use of concomitant ITP drug OR (95% CrI) | Need for rescue therapy IRR (95% CrI) | Incidence of any bleeding events IRR (95% CrI) | Incidence of WHO grade 2 to grade 4 bleeding events IRR (95% CrI) | Any adverse events IRR (95% CrI) |
|---|---|---|---|---|---|---|
vs. ELT | 7.06 (0.21 to 185,017.47) | 16.08 (0.37 to 579,939.95) | 1.61 (0.30 to 8.51) | 0.38 (0.19 to 0.75) | 0.74 (0.20 to 2.82) | 0.64 (0.36 to 1.13) |
vs. ROM | 2.16 (0.03 to 69,340.75) | 3.71 (0.06 to 149,100.00) | 2.10 (0.39 to 11.46) | 0.38 (0.17 to 0.86) | 1.14 (0.23 to 5.60) | 0.63 (0.32 to 1.25) |
vs. FOS | 9.10 (0.12 to 279,100.00) | NA | 1.98 (0.37 to 10.37) | 0.69 (0.28 to 1.67) | 1.32 (0.24 to 6.98) | 0.91 (0.46 to 1.80) |
vs. RTX | NA | NA | 1.33 (0.25 to 7.12) | 0.44 (0.21 to 0.94) | 0.59 (0.13 to 2.62) | 0.49 (0.25 to 0.95) |
vs. placebo | 102.80 (3.87 to 2,796,448.59) | 48.75 (1.34 to 1,769,074.94) | 0.73 (0.15 to 3.53) | 0.34 (0.18 to 0.66) | 0.50 (0.12 to 2.00) | 0.63 (0.36 to 1.10) |
AVA = avatrombopag; CrI = credible interval; ELT = eltrombopag; FOS = fostamatinib; IRR = incidence rate ratio; ITP = immune thrombocytopenia; NA = not available; OR = odds ratio; ROM = romiplostim; RTX = rituximab.
Notes: fixed-effects models were adopted.
Results are shown in bold when avatrombopag was favoured over the comparator.
Source: Sponsor-submitted ITC.14
Summary
Based on the results of the sponsor-submitted ITC, the comparative effectiveness of avatrombopag compared to other TPO-RAs in achieving a durable platelet response, reducing the need for rescue therapy, reducing the use of concomitant ITP medication, decreasing higher-grade bleeding events, and lowering the incidence of AEs is uncertain due to small sample sizes and the sparsity of the network, which resulted in substantial imprecision in the comparative estimates. Treatment with avatrombopag was favoured over eltrombopag, romiplostim, and rituximab for the incidence of any bleeding events in patients with chronic ITP, but the magnitude of these differences was uncertain, due to substantial imprecision. Additionally, this study has a number of limitations that had an impact on internal and external validity, such as potential intransitivity, reliance solely on indirect data (except for AEs), lack of presentation of risk of bias appraisals across the included studies and their impact on the study results, missing comparators that may have added important information to the network, and a lack of other important outcomes that are important to patients (e.g., HRQoL).
Other Relevant Evidence
This section examines a submitted phase II trial (Study 00315) and a phase II, long-term extension study (Study 00416) included in the sponsor’s submission to CADTH that were considered to provide supportive evidence to the pivotal trial.
In addition, the sponsor submitted a retrospective observational study of adult patients with chronic ITP who switched from other TPO-RAs to avatrombopag to describe the effect of avatrombopag in patients who did not have an adequate response to previous TPO-RAs therapy.18
Study 003
Study 003 provides efficacy and safety data that supplements evidence from the pivotal trial (Study 302).
Methods
Study 003 was a phase II, multicentre, double-blind, placebo-controlled, randomized, dose-ranging trial of avatrombopag taken orally once daily for 28 days by adult patients with chronic ITP. A total of 64 patients were randomized to treatment groups of 2.5 mg/day, 5 mg/day, 10 mg/day, and 20 mg/day of avatrombopag, or placebo in a 3:3:3:3:1 ratio, respectively. The recommended Health Canada starting dose of avatrombopag is 20 mg/day, which is the dose examined in this report. Relevant efficacy outcomes included the platelet count response rate and changes in peripheral blood platelet counts from baseline over time. Relevant safety outcomes included the reporting of TEAEs, SAEs, WDAEs, and deaths.
Populations
Baseline characteristics varied somewhat between the placebo group (n = 5) and the avatrombopag group (n = 15); however, the sample size was too small to meaningfully examine any relevant intergroup differences. Most patients were white (n = 14; 70%), with a median age of 44 years (range, 22 to 77 years) for those in the avatrombopag group and 29 years (range, 23 to 73 years) for patients in the placebo group. A total of 10 patients (66.7%) in the avatrombopag group and 3 patients (60.0%) in the placebo group had been using steroids for ITP at baseline. Forty percent of patients in each group had undergone splenectomy before study entry. Four patients in the avatrombopag group (26.7%) and 2 patients (40.0%) in the placebo group had a baseline platelet count of 15 × 109/L or lower.
Table 22: Summary of Baseline Characteristics for Study 003 (FAS)
Characteristic | Avatrombopag 20 mg (N = 15) | Placebo (N = 5) |
|---|---|---|
Age, years, median (range) | 44.0 (22 to 77) | 29.0 (23 to 73) |
Females, n (%) | 11 (73.3) | 3 (60.0) |
Race, n (%) | ||
Black, African heritage | 2 (13.3) | 0 |
Asian | 1 (6.7) | 0 |
Hispanic | 1 (6.7) | 1 (20.0) |
White | 10 (66.7) | 4 (80.0) |
Other | 1 (6.7) | 0 |
History of splenectomy, n (%) | 6 (40.0) | 2 (40.0) |
Baseline steroid use, n (%) | 10 (66.7) | 3 (60.0) |
Baseline platelet count ≤ 15 × 109/L, n (%) | 4 (26.7) | 2 (40.0) |
ITP medications before study entry,b n (%) | ||
Danazol | 2 (13.3) | 2 (40.0) |
Platelets | 2 (13.3) | 0 |
Prednisonea | 14 (93.3) | 5 (100.0) |
Immunoglobins, normal human | 7 (46.7) | 3 (60.0) |
Rituximab | 7 (46.7) | 2 (40.0) |
Anti-Rh human immunoglobulin | 3 (20.0) | 2 (40.0) |
Concomitant prednisone,a,b n (%) | 10 (66.7) | 2 (40.0) |
ITP = idiopathic thrombocytopenia purpura.
aPrednisone, a systemically acting steroid, was miscoded. The database was not reopened to correct this error.
bUsed by ≥ 10% in either treatment group.
Source: CSR for Study 003.15
Interventions
Patients enrolled in Study 003 were assigned to receive avatrombopag 20 mg or matching placebo, taken orally once daily for 28 days. Patients were allowed certain concomitant medications. Patients receiving maintenance doses of corticosteroids could be enrolled in the study, provided the corticosteroids had been administered at a stable dose for at least 2 weeks before the screening visit and were maintained over the entire duration of the study. Patients receiving corticosteroids had to have a baseline platelet count of less than 50 × 109/L at their first and before their second screening visit. Patients not receiving corticosteroids had to have a baseline platelet count of less than 30 × 109/L at their first and before their second screening visit. Patients receiving stable dosages of cyclosporine A, mycophenolate mofetil, azathioprine, or danazol could be enrolled. The doses of these medications must have been stable for at least 3 months before the study drug administration.
Outcomes
The primary efficacy end point for this study was the responder rate; patients were considered responders if they achieved a platelet count of at least 50 × 109/L on day 28 and had a baseline platelet count of less than 30 × 109/L, or if they were receiving steroids and had an increase from baseline of at least 20 × 109/L and a baseline platelet count of at least 30 × 109/L but less than 50 × 109/L. Secondary efficacy end points included the number of patients who maintained responder status from day 7 to day 28, changes in peripheral blood platelet count from baseline, the proportion of patients who achieved a platelet count of at least 50 × 109/L or at least 100 × 109/L on day 28, and the proportion of patients who doubled their platelet count from baseline to day 28. Relevant safety outcomes included the reporting of AEs, SAEs, WDAEs, and deaths.
Statistical Analysis
Primary efficacy analyses were performed using the FAS, defined as all patients who were randomly assigned to a treatment and who had at least 1 postbaseline platelet count. The safety population included all patients who received at least 1 dose of the study drug and had at least 1 safety assessment. Both the FAS and the safety population were the same as the intention-to-treat population. The LOCF approach was used to impute missing platelet counts on day 28 for all analyses; an observed case (OC) analysis was also performed, in which missing values were not imputed. There was no imputation of other missing data. Because Study 003 was designed as a hypothesis-generating trial, the analyses were exploratory and all P values were nominal without adjustment for multiplicity. Subgroup analyses by baseline platelet count, splenectomy status, and the number of lines of prior treatment were planned, but meaningful findings could not be presented due to the small sample size.
Patient Disposition
A total of 109 patients from the US were screened for Study 003, 64 of whom were randomized into the study. Of these patients, 5 were randomized into the placebo group and 15 into the avatrombopag 20 mg/day group. Only 2 patients discontinued, both in the avatrombopag group, due to an increase in their platelet count to 500 × 109/L or more.
Table 23: Patient Disposition in Study 003
Disposition | Avatrombopag 20 mg | Placebo |
|---|---|---|
Screened, n | 109 | |
Randomized, n | 15 | 5 |
Completed study, n (%) | 13 (86.7) | 5 (100.0) |
Discontinued study, n (%) | 2 (13.3) | 0 |
Platelet count increase to ≥ 500 × 109/L | 2 (13.3) | NA |
Full analysis set | 15 | 5 |
Safety population | 15 | 5 |
NA = not applicable.
Source: CSR for Study 003.15
Exposure to Study Treatments
The median (range) days of exposure to treatment was 29 (28 to 31) in the placebo group and 29 (14 to 30) in the avatrombopag group. A total of 4 patients missed treatment doses in the avatrombopag group.
Efficacy
Platelet Count: Proportion of Responders
A total of 80% of patients (n = 12) in the avatrombopag group and no patients in the placebo group achieved a treatment response on day 28 using the LOCF method. Findings were similar when using the OC method.
Duration of Response
Among those who completed treatment (n = 13 in the avatrombopag and n = 5 in the placebo group), || |||||||| ||||||| in the avatrombopag group had a sustained response between day 7 and day 28. || |||||||| in the placebo group had a response between day 7 and day 28.
Change in Platelet Levels From Baseline
Using the LOCF method, in the avatrombopag group, median (range) platelet count was 22 × 109/L (8 × 109/L to 50 × 109/L) at baseline and 101 × 109/L (18 × 109/L to 1,031 × 109/L) on day 28, and median (range) change from baseline was 84 × 109/L (–10 × 109/L to 1,012 × 109/L). Four patients in this group developed a platelet count higher than ||||||. In the placebo group, median (range) platelet count was 19 × 109/L (9 × 109/L to 46 × 109/L) at baseline and 20 × 109/L (9 × 109/L to 34 × 109/L) at day 28, and median (range) change from baseline was –2 × 109/L (–12 × 109/L to 9 × 109/L). The OC method yielded similar findings.
Patients Who Achieved a Platelet Count of 50 × 109/L or Higher on Day 28
Using the LOCF method, 12 patients (80%) in the avatrombopag group and no patients in the placebo group had a platelet count of 50 × 109/L or higher on day 28. Similar results were found using the OC method.
Patients Who Achieved a Platelet Count of 100 × 109/L or Higher on Day 28
Using the LOCF method, 8 patients (53.3%) in the avatrombopag group and no patients in the placebo group and had a platelet count of 100 × 109/L or higher on day 28. Similar results were found using the OC method.
Patients Who Doubled Their Platelet Count From Baseline on Day 28
Using the LOCF method, 13 patients (86.7%) in the avatrombopag group and 1 patient (20.0%) in the placebo group had their platelet counts at least doubled on day 28. Similar results were found using the OC method.
Harms
A total of || |||||||| ||||||| in the avatrombopag 20 mg/day group and |||||||| ||||||| in the placebo group experienced at least 1 TEAE. The most common AEs in the avatrombopag group were headache and platelet count increase, each of which occurred in |||||||| |||||||. Increases in platelet count were not associated with thrombotic or thromboembolic events. A total of |||||||| ||||||| in the avatrombopag group withdrew from the study due to an AE. There were no reported SAEs in either group, and no deaths occurred in the study.
Table 24: Efficacy Outcomes in Study 003 Through Day 28 (FAS), LOCF Method
Outcome | Avatrombopag 20 mg (N = 15) | Placebo (N = 5) | P valuea |
|---|---|---|---|
Responder rate, n (%) | 12 (80.0) | 0 (0.0) | 0.0036 |
Sustained response between day 7 and day 28b | 10 (76.9) | 0 (0.0) | NA |
Change in platelet countc from baseline, median (range) | |||
Baseline | 22.0 (8 to 50) | 19.0 (9 to 46) | NA |
Day 28 | 101.0 (18 to 1,031) | 20.0 (9 to 34) | NA |
Change from baseline | 84.0 (–10 to 1,012) | –2.0 (–12 to 9) | NR |
Platelet count ≥ 50 × 109/L, n (%) | 12 (80.0) | 0 (0.0) | 0.0036 |
Platelet count ≥ 100 × 109/L, n (%) | 8 (53.3) | 0 (0.0) | 0.0547 |
Platelet count doubled, n (%) | 13 (86.7) | 1 (20.0) | 0.0139 |
FAS = full analysis set; LOCF = last observation carried forward; NA = not applicable; NR = not reported.
aP values were based on Fisher’s exact test. The analyses were not adjusted for multiplicity.
bThe denominator is the number of patients who completed treatment (n = 13 in the avatrombopag group and n = 5 in the placebo group).
cUnit for platelet count is K × 109/L.
Source: CSR for Study 003.15
Table 25: Summary of TEAEs in Study 003 (Safety Population)
Adverse event | Avatrombopag 20 mg (N = 15) | Placebo (N = 5) |
|---|---|---|
Patients with ≥ 1 TEAE, n (%) | || |||||| | |||||| |
Common TEAEs,a n (%) | ||
Headache | |||||| | |||||| |
Migraine | |||||| | |||||| |
Epistaxis | |||||| | |||||| |
Petechiae | |||||| | |||||| |
Increased platelet count | |||||| | |||||| |
Diarrhea | |||||| | |||||| |
Vomiting | |||||| | |||||| |
Fatigue | |||||| | |||||| |
Patients with ≥ 1 SAE, n (%) | |||||| | |||||| |
Patients with WDAEs, n (%) | |||||| | |||||| |
Deaths, n (%) | 0 (0.0) | 0 (0.0) |
SAE = serious adverse event; TEAE = treatment-emergent adverse event; WDAE = withdrawal due to adverse event.
aObserved in ≥ 2 patients.
Source: CSR for Study 003.15
Critical Appraisal
In this phase II study, patients were centrally randomized to treatment groups using simple block randomization (block size of 13) without stratification factors. Baseline characteristics were somewhat imbalanced between the groups of interest, but this is likely a result of the small sample size, and risk of bias arising from the randomization process is not suspected. The study drug and placebo were provided in double-blinded box kits and were indistinguishable. Patients and study personnel involved in patient care or outcome assessment were blinded to treatment, and the sponsor noted no partial unblinding at the time of the database lock. Therefore, the findings are unlikely to be affected by bias due to deviation from the intended interventions or measurement of the outcome. The study was not powered to detect statistically significant changes in outcomes and analyses were not adjusted for multiplicity. As a result, all findings are considered to be hypothesis-generating, and definitive conclusions cannot be drawn. The LOCF method to impute missing data for platelet counts on day 28 appeared to be reasonable for the groups of interest, but may have resulted in somewhat higher platelet counts than reality, given the true trajectory of the platelet counts, which appeared to peak around day 14. However, findings were similar using the OC method, so concerns about bias introduced by this method are minimal. The small sample size limits the credibility of the findings and their generalizability to the patient population in Canada. In addition, there were numerous screen failures (||||| ||| || ||| |||||||| ||||||||||), indicating that the population may be narrower than what would be seen in clinical practice. There was no examination of HRQoL outcomes, which were deemed to be important to both patients and clinical experts.
Study 004
Study 004 was a long-term extension study conducted to evaluate the efficacy, safety, and tolerability of avatrombopag.
Methods
Study 004 was a phase II, rollover study of avatrombopag administered for an additional 6 months to patients with chronic ITP who completed Study 003. Patients who met the efficacy response criteria on day 28 in Study 003 continued their previous daily blinded dose (placebo or 2.5 mg, 5 mg, 10 mg, or 20 mg of avatrombopag) at study entry. Patients who did not meet the efficacy response criteria began treatment in this study with open-label avatrombopag at a dose of 10 mg/day.
Because none of the patients receiving placebo in Study 003 were responders, no patients in Study 004 received placebo.
The study allowed open-label dose escalation of avatrombopag for all patients in 10 mg/day increments to achieve and maintain a platelet response. The recommended Health Canada starting dose of avatrombopag is 20 mg/day, so lower doses are not the focus of this report.
The primary end point for this study was the safety and tolerability of avatrombopag after 6 additional months of treatment upon completion of Study 003. Relevant efficacy outcomes included platelet count data, and relevant safety outcomes included the reporting of TEAEs, SAEs, WDAEs, and deaths.
Populations
A total of 53 patients continued into Study 004, 13 of whom were receiving 20 mg/day of avatrombopag in Study 003 (10 responders and 3 nonresponders, per the primary response criteria). Baseline characteristics have been summarized using data from the baseline visit of Study 003, and are shown in Table 26. Most patients were white (n = 8 [61.5%]), with a median age of 44 years (range, 44 to 58 years) among nonresponders and 41.5 years (range, 22 to 77 years) among responders. One patient (33.3%) in the nonresponder group and 7 patients (70.0%) in the avatrombopag responder group had been using steroids for ITP at baseline. Two patients (66.7%) in the nonresponder group and 4 patients (40.0%) in the responder group had undergone splenectomy before study entry. About 30% of patients in both groups had a baseline platelet count of 15 × 109/L or lower.
Table 26: Summary of Baseline Characteristics for Study 004 (FAS)
Characteristic | Avatrombopag 20 mg responder (N = 10) | Avatrombopag 20 mg nonresponder (N = 3) |
|---|---|---|
Age, years, median (range) | 41.5 (22 to 77) | 44.0 (44 to 58) |
Females, n (%) | 8 (80.0) | 3 (100.0) |
Race, n (%) | ||
Black, African heritage | 2 (20.0) | 0 (0.0) |
Asian | 1 (10.0) | 0 (0.0) |
Hispanic | 1 (10.0) | 0 (0.0) |
White | 5 (50.0) | 3 (100.0) |
Other | 1 (10.0) | 0 (0.0) |
History of splenectomy, n (%) | 4 (40.0) | 2 (66.7) |
Baseline steroid use, n (%) | 7 (70.0) | 1 (33.3) |
Baseline platelet count ≤ 15 × 109/L, n (%) | 3 (30.0) | 1 (33.3) |
FAS = full analysis set.
Source: CSR for Study 004.16
Interventions
Patients who were responders in Study 003 initially continued to receive blinded 20 mg avatrombopag as a tablet orally once per day. Nonresponders initially received open-label 10 mg avatrombopag in a similar manner. The study allowed open-label dose escalation of avatrombopag for all patients in 10 mg/day increments. Patients who had 2 consecutive platelet counts below the protocol-defined response level could participate in a dose modification plan. Responders were allowed up to 20 mg/day of open-label avatrombopag in addition to their blinded dose, for a maximum possible dose of 40 mg/day, and nonresponders were allowed a maximum open-label dose of 40 mg/day. Patients who did not respond to the maximum dose increase after 14 days were withdrawn from the study.
Patients maintained on a stable dose of corticosteroids at study entry could continue to receive corticosteroids during the study. Patients receiving cyclosporine A, mycophenolate mofetil, azathioprine, or danazol at study entry could continue receiving those concomitant medications during the study. The concurrent dose of maintenance corticosteroids or another drug could be decreased during the study if the patient’s platelet count was consistently above 200 × 109/L.
Outcomes
The primary end point for this study was the safety and tolerability of avatrombopag. Relevant efficacy measurements were assessed as secondary end points, and included the change in platelet count from baseline (before day 1 in Study 003). Other efficacy outcomes included the proportion of patients who achieved or maintained a response-level platelet count, as defined in Study 003; the proportion who achieved a durable, transient, or overall response; the proportion who achieved a platelet count of 100 × 109/L or higher; and the proportion who were able to permanently discontinue or decrease their use of concomitant steroid medication.
A durable response was defined as patients who had at least 3 platelet count values measured in the final 14 weeks in Study 004 and whose platelet count was at a response level for at least 75% of those measured values. A transient response was defined as patients whose platelet counts were at a response level at least 2 consecutive analysis windows during the 24-week treatment period of Study 004, without having achieved a durable response. An overall response consisted of all patients who achieved either a durable or transient response.
Relevant safety outcomes included the reporting of AEs, SAEs, WDAEs, and deaths for the combined study periods of Study 003 and Study 004.
Statistical Analysis
Only descriptive statistics were provided for Study 004; no formal statistical testing was performed. Efficacy analyses were performed using the FAS, defined as patients who participated in both Study 003 and Study 004 and provided data to derive at least 1 efficacy assessment in Study 004. All efficacy analyses were performed using OC methodology, and missing values were not imputed. The safety population included all patients who received at least 1 dose of the study drug and had at least 1 safety assessment in either study. Safety results were grouped by mean daily dosage of avatrombopag; with the upper third of the dose group consisting of patients who received a mean daily dose of at least 13.5 mg during the combined active treatment periods of Study 003 and Study 004.
Of the 64 patients who were randomized in Study 003, 53 enrolled in Study 004. Of these patients, 13 received the maximum 20 mg/day dose in Study 003 (10 responders and 3 nonresponders). Four (30.8%) of these patients discontinued Study 004, 2 each from the responder and nonresponder groups, with each patient discontinuing for a different reason.
Disposition | Avatrombopag 20 mg responder | Avatrombopag 20 mg nonresponder |
|---|---|---|
Enrolled in study, n | 53 | |
Enrolled in treatment group, n | 10 | 3 |
Completed study, n (%) | 8 (80.0) | 1 (33.3) |
Discontinued study, n (%) | 2 (20.0) | 2 (66.7) |
Adverse event | 1 (10.0) | 0 (0.0) |
Laboratory abnormality | 1 (10.0) | 0 (0.0) |
Investigator decision | 0 (0.0) | 1 (33.3) |
Other | 0 (0.0) | 1 (33.3) |
Source: CSR for Study 004.16
Exposure to Study Treatments
The median (range) days of exposure to treatment was 195.5 (14 to 227) among the 20 patients in the upper third of the dose group (mean daily dose, at least 13.5 mg/day). ||||| |||||||| ||||| in the upper third of the dose group had open-label dose increases throughout Study 004.
Efficacy
Change in Platelet Count From Baseline
The median (range) change in platelet count from baseline in Study 003 to week 24 in Study 004 was 124 × 109/L (–11 × 109/L to 205 × 109/L) among responders (n = 7) and 199 × 109/L (not applicable) among nonresponders (n = 1).
Achieved a Response-Level Platelet Count at Week 24
Of the 7 responders and 1 nonresponder in the study at week 24, a total of 6 (85.7%) responders and 1 (100.0%) nonresponder in the avatrombopag 20 mg/day group achieved a response-level platelet count at week 24.
Maintained a Response-Level Platelet Count
Patients who maintained their platelet count at a response level were defined as those who had been responders in Study 003, who had no increase in dose of the study drug in Study 004, and who achieved a durable response. A total of |||||||||| ||||| in the avatrombopag 20 mg/day group maintained a response-level platelet count.
Achieved a Durable, Transient, and Overall Response
A total of ||||||| responders and ||||||| nonresponder in the avatrombopag 20 mg/day group achieved a durable platelet response. ||||| ||||||| responders and none of the nonresponders achieved a transient response. This led to an overall response among ||||||||| ||||| and |||||||||||| ||||||| in Study 004.
Permanent Discontinuation of Steroids
Of the 6 responders and 1 nonresponder initially treated with corticosteroids, a total of 2 (33.3%) responders and 1 (100.0%) nonresponder permanently discontinued steroid use during the final 8 weeks of treatment in Study 004.
Reduction in Steroid Use by 50% or More
Of | responders and | nonresponder initially treated with corticosteroids, a total of | responders and | nonresponder reduced their steroid use by 50% or more in the final 8 weeks of treatment.
Table 28: Efficacy Outcomes in Study 004 Through Week 24 (FAS)
Outcomes | N | Avatrombopag 20 mg responder | N | Avatrombopag 20 mg nonresponder |
|---|---|---|---|---|
Platelet counta change from baseline, × 109/L, median (range) | ||||
Baselineb | 10 | 23.0 (8 to 50) | 3 | 22.0 (11 to 28) |
Week 24 | 7 | 154.0 (38 to 227) | 1 | 210.0 (210 to 210) |
Change from baseline | 7 | 124.0 (–11 to 205) | 1 | 199.0 (NA) |
Platelet count ≥ 100 × 109/L at week 24, n (%) | 7 | 6 (85.7) | 1 | 1 (100.0) |
Achieved response-levelc platelet count at week 24, n (%) | 7 | 6 (85.7) | 1 | 1 (100.0) |
Maintainedd a response-levelc platelet count, n (%) | || | |||||| | || | |||||| |
Achieved a durable response at week 24, n (%) | || | |||||| | || | |||||| |
Achieved a transient response at week 24, n (%) | || | |||||| | || | |||||| |
Achieved an overall response at week 24, n (%) | || | |||||| | || | |||||| |
Permanent discontinuation of steroids,e n (%) | 6 | 2 (33.3) | 1 | 1 (100.0) |
Reductionf of steroid use by ≥ 50%, n (%) | || | |||||| | || | |||||| |
FAS = full analysis set; NA = not applicable.
aUnit for platelet count is K × 109/L.
bBaseline platelet count was the last observation before the first dose of the study drug in Study 003.
cResponse level of platelet count is defined as a platelet count ≥ 50 × 109/L for patients with a baseline platelet count < 30 × 109/L or a platelet count ≥ 20 × 109/L above the baseline platelet count for patients receiving steroids with a baseline platelet count ≥ 30 × 109/L but < 50 × 109/L.
dPatients maintained a platelet response if they were responders in Study 003, had no dose titrated up, and achieved durable response.
ePatients who used steroids during the 2-week period before Study 004 were considered to have permanently discontinued steroid use if they had no steroid use during the final 8 weeks of the treatment period of Study 004.
fPatients who used steroids during the 2-week period before Study 004 were considered to have decreased steroid use by ≥ 50% if they had permanently discontinued steroids or if no dose of steroid was > 50% of their baseline steroid dose during the final 8 weeks of the treatment period of Study 004.
Source: CSR for Study 004.16
Harms
Safety results are presented for the combined study periods in Study 003 and Study 004. All 20 patients in the mean daily dose group of 13.5 mg or higher experienced at least 1 TEAE. The most common AEs were fatigue, headache, and epistaxis, each of which occurred in 8 patients (40.0%). A total of 3 patients (15.0%) withdrew from the study due to an AE. Three patients reported at least 1 SAE, 2 of whom experienced serious recurrent thrombocytopenia. No deaths occurred during the studies.
Table 29: Summary of TEAEs in Study 003 and Study 004 (Safety Population)
Adverse event | Avatrombopag (mean daily dose of ≥ 13.5 mg) (N = 20) |
|---|---|
Patients with ≥ 1 TEAE, n (%) | || ||||| |
Common TEAEs,a n (%) | |
||||||||| | |||||| |
|||||||| |||||||| | |||||| |
||||||| | |||||| |
||||||||| | |||||| |
|||||||| | |||||| |
||||||||| | |||||| |
Patients with ≥ 1 SAE, n (%) | |||||| |
Patients with WDAEs, n (%) | |||||| |
Deaths, n (%) | 0 (0.0) |
SAE = serious adverse event; TEAE = treatment-emergent adverse event; WDAE = withdrawal-associated adverse event.
aObserved in ≥ 5 patients.
Sources: CSRs for Study 00315 and Study 004.16
Critical Appraisal
Study 004 allowed for the investigation of the long-term efficacy and harms of avatrombopag for up to 24 weeks. All analyses were descriptive in nature and were limited to a few patients. The absence of an active comparator and the lack of control of confounders means that causal conclusions cannot be drawn. An additional limitation is the open-label titration of avatrombopag, which can bias the reporting of subjective end points (i.e., harms). Furthermore, all missing data were excluded from the analyses, increasing the risk of attrition bias; however, the direction of bias is unclear. Study 004 enrolled patients who had successfully completed Study 003, which could have resulted in a population of patients who were more tolerant of avatrombopag, which can lead to biased estimates of efficacy and safety. The use of concomitant steroid medications by patients throughout the study may have increased the risk of additional side effects not attributable to avatrombopag alone. In terms of external validity, doses of avatrombopag administered throughout the study to some patients in Study 004 were less than the recommended starting dose of 20 mg/day approved by Health Canada, which limits the generalizability of the results. Also, the small sample size limits the credibility of the findings and the generalizability of the results to the patient population in Canada. There was no examination of HRQoL outcomes, which were deemed to be important to both patients and clinical experts.
Study by Al-Samkari et al.
Al-Samkari et al. (2022,18 202317) conducted a multicentre, retrospective, observational study to describe the effectiveness of avatrombopag in adult patients with chronic ITP who had been treated with previous TPO-RAs.17,18
Methods
In this study, patients with ITP who switched from eltrombopag or romiplostim to avatrombopag for any reason from July 2019 through December 2020 were evaluated. Data were retrospectively collected. Platelet response, LOR, and durability of response were evaluated. Other evaluated outcomes include the use of concomitant ITP medications and the need for rescue therapy before and after switching.
Populations
A total of 44 patients 18 years or older with primary or secondary ITP at 4 US tertiary ITP referral centres were included in this study. Patients were ineligible if they were initiated on another TPO-RA while on avatrombopag, had received any experimental therapy for ITP in the 30 days before a switch, or had developed thrombocytopenia unrelated to ITP.
In this study, 25 patients (57%) had primary ITP and 19 (43%) had secondary ITP. Forty-eight percent were male, 68% were white, and the median age was 61 years (range, 21 to 87 years). The patients had received a median of 4 (range, 2 to 10) prior ITP therapies before receiving avatrombopag, including romiplostim (75%), eltrombopag (23%), and a combination of romiplostim and eltrombopag simultaneously in a single patient (2%). Reasons for switching included the greater convenience of avatrombopag (52%), the insufficient effectiveness of prior TPO-RAs (32%), and AEs with prior TPO-RAs (16%). In general, patients switching from eltrombopag transitioned immediately from eltrombopag on 1 day to avatrombopag the following day, and patients switching from romiplostim began avatrombopag approximately 7 days after their final romiplostim dose. In patients switching from romiplostim, the median dose administered before switching was 4 mcg/kg per week, and in patients switching from eltrombopag, the median dose administered before switching was 75 mg/day.
Interventions
Avatrombopag treatment had to be initiated in the month after stopping eltrombopag or romiplostim, and had to be continued for at least 2 months to allow for full-dose titration. The manner in which the patients transitioned from romiplostim or eltrombopag was at the investigators’ discretion.
Outcomes
Platelet response was defined as the achievement of a given platelet count on at least 1 occasion without the need for rescue therapy. The achievement of a platelet response was defined as a platelet count of at least 50 × 109/L and of a complete platelet response was at least 100 × 109/L. LOR was defined as 2 consecutive platelet counts, at least 7 days apart, of less than 50 × 109/L. The durability of response was evaluated as the total number of days a platelet response was achieved compared with the total number of days of exposure to avatrombopag.
The median platelet count before and after switching was calculated separately as the median of the final 3 platelet counts for patients on either romiplostim or eltrombopag and the most recent 3 platelet counts for patients on avatrombopag.
The use of concomitant ITP medications (medications prescribed for long-term use, including chronic corticosteroids) was evaluated both before switching to and during treatment with avatrombopag. Rescue therapy was defined as the acute administration of corticosteroids (either initiation or an increase from a prior stable chronic dose), IVIG, anti-RhD immune globulin, or platelet transfusion.
Statistical Analysis
Descriptive statistics were reported for the study outcomes. Median platelet counts for patients on romiplostim or eltrombopag were compared with median platelet counts for patients on avatrombopag using the Wilcoxon signed rank test. The following subgroups were analyzed in the study by Al-Samkari et al.: patients who switched to avatrombopag because of the insufficient effectiveness of prior TPO-RA therapy, the improved convenience of avatrombopag therapy, or AEs related to prior TPO-RA therapy; patients with primary or secondary ITP; and the TPO-RA therapy before the switch (romiplostim versus eltrombopag).
Patient Disposition
During the observation period, 6 of 44 patients (14%) discontinued avatrombopag, 1 patient each due to attempted remission, formulary limitations, lack of response, AE (headache, portal-vein thrombosis), patient preference, and initiation of rituximab for autoimmune hemolytic anemia. Thirty-eight patients remained on avatrombopag at the end of the observation period.
Exposure to Study Treatments
The median duration of treatment with avatrombopag was 9.2 months (range, 2.8 to 17.2 months). The median weekly dose of avatrombopag was 140 mg (range, 20 to 280 mg).
Efficacy
Platelet Response
A platelet response (platelet count of ≥ 50 × 109/L) was achieved in 41 patients (93%), and a complete platelet response (platelet count of ≥ 100 × 109/L) was achieved in 38 patients (86%) after switching, at least once during the study. Among the responders, the platelet response was maintained for 84% of their time on avatrombopag: 88% among 36 patients who did not require rescue therapy, and 55% among 5 patients who required rescue therapy. A durable response was achieved by 31 patients (84%) who switched from romiplostim, 9 patients (81%) who switched from eltrombopag, and 1 patient (100%) who switched from both romiplostim and eltrombopag.
Platelet Count
The median platelet count for patients on eltrombopag or romiplostim was 45 × 109/L and for patients on avatrombopag was 114 × 109/L (P < 0.0001 by Wilcoxon signed rank test).
Concomitant ITP Medications
Among the 28 patients who were receiving concomitant ITP medications before switching, 16 (57%) discontinued 1 or more concomitant medications after initiating avatrombopag. Among the patients who were receiving concomitant ITP medications after switching, 7% required the addition of a concomitant ITP medication after starting avatrombopag.
Among the 19 patients who were receiving concomitant chronic corticosteroids, 12 (63%) discontinued corticosteroids and 6 (32%) had their corticosteroid dose reduced.
Need for Rescue Therapy
Rescue therapy was required by 9 patients (21% of 44 patients) after switching to avatrombopag and by 15 patients (34%) who were on eltrombopag or romiplostim before switching.
Critical Appraisal
The study by Al-Samkari et al. was limited by several design issues. The retrospective, observational, uncontrolled design is 1 of the key limitations of this evidence. The retrospective study is susceptible to selection bias because it is unclear how the 4 tertiary centres that were the source of patients for the study were chosen. In addition, although the platelet response rates (93% for platelet response and 86% for complete platelet response) were high and durable, without a control arm or control for confounding, it is not possible to assess the relative treatment effect of avatrombopag versus other TPO-RAs or to directly attribute the effect to avatrombopag. Six patients (14%) discontinued treatment with avatrombopag during the observation period. There was no description of how missing data were handled. The direction and magnitude of the potential bias from the discontinuation of avatrombopag on the observed treatment effect was unknown. Subgroup analyses were conducted based on prior treatments, reasons for switching, and types of ITP in this study. It was unclear whether these were prespecified subgroup analyses. Furthermore, result interpretation was a challenge in the underpowered subgroup analyses.
The patients were all identified from ITP referral centres in the US. The generalizability of the study findings to populations in Canada may be limited because there are no specific treatment guidelines for ITP in Canada, and treatment patterns may be different than those in the US. Also, the small sample size limits the credibility of the findings and the generalizability of the results to the patient population in Canada. Clinically meaningful outcomes, such as HRQoL, the occurrence of bleeding events, and the safety of avatrombopag, were not assessed in this study.
Discussion
Summary of Available Evidence
Study 302 (N = 49) was a multicentre, phase III, double-blind, placebo-controlled RCT that evaluated the efficacy and safety of avatrombopag versus placebo in patients with chronic ITP who had received previous ITP treatment and who had a baseline platelet count below 30 × 109/L. In this study, 32 patients were randomized to avatrombopag 20 mg and 17 to matching placebo. The primary efficacy end point was the cumulative number of weeks of platelet response (platelet count of 50 × 109/L or higher) without rescue therapy for bleeding. Key limitations of Study 302 include the small sample size and high discontinuation rates (especially in the placebo group). Patient characteristics were imbalanced at baseline, and some outcomes were likely to be affected by the reduced duration of exposure in the placebo group. The clinical relevance of the primary efficacy outcome and its relationship with other clinically important outcomes, such as bleeding events and HRQoL, is uncertain. In addition, a lack of comparative evidence between avatrombopag and the other active treatments is limitation.
The sponsor submitted a systematic review and ITC in which avatrombopag was compared to 2 other TPO-RAs (eltrombopag and romiplostim), fostamatinib, and rituximab in patients with chronic or persistent ITP. Nine RCTs were included and contributed evidence. The following outcomes were assessed: durable platelet response, the use of concomitant ITP medication, the need for rescue therapy, WHO bleeding events, and AEs. There were important limitations of this ITC due to the limited size of the evidence base and the heterogeneity in patient populations in the included trials; therefore, it provided limited additional insight into the efficacy of avatrombopag compared to other active ITP treatments. Furthermore, some active treatments of interest were not included in this ITC analysis, and the comparative efficacy and safety of avatrombopag versus these drugs cannot be examined.
Two additional studies provided supportive evidence regarding the safety and efficacy of avatrombopag. Study 003 was a phase II double-blind, placebo-controlled, randomized trial of avatrombopag taken orally once daily for 28 days by adult patients with chronic ITP. A total of 5 patients were randomized to placebo and 15 were randomized to avatrombopag 20 mg/day. Study 004 was a phase II, long-term extension study in which avatrombopag was administered for an additional 6 months in patients with chronic ITP who completed Study 003. A total of 53 patients were enrolled in Study 004, of whom 13 received the maximum 20 mg/day dose in Study 003 (10 responders and 3 nonresponders).
Interpretation of Results
Efficacy
The results of Study 302 showed that the incidence of any bleeding event during the 6 months of treatment was 43.8% in the avatrombopag group and 52.9% in the placebo group. The between-group difference was not statistically significant. No patients in the placebo group had a bleeding event that was higher than WHO grade 1. In the avatrombopag group, there were 2 patients with WHO grade 2 bleeding events and 1 with a WHO grade 3 bleeding event (epistaxis). These findings should be interpreted with consideration of the substantially shorter duration of exposure in the placebo group, as most patients discontinued the study early due to a lack of efficacy. In addition, the results suggest that 6 months of treatment with avatrombopag leads to a better platelet response (longer duration with platelet response, more patients achieved a platelet response at day 8) than placebo in a group of pretreated patients with primary chronic ITP. According to the clinical expert, the between-group differences in platelet response can be considered clinically important. The treatment effect of avatrombopag, compared with placebo, remains uncertain in terms of improving patients’ HRQoL, reducing the use of concomitant ITP medications or the need for rescue therapy, and reducing emergency department visits and/or hospitalizations due to thrombocytopenia episodes. The results of a posthoc analysis of Study 302 supported the primary analysis, suggesting that treatment with avatrombopag results in higher platelet response rates than placebo, and platelet response was maintained in the extension study, when all patients received open-label avatrombopag treatment. The use of avatrombopag was also related to more dose reduction or discontinuation of concomitant corticosteroid therapy in the study population. Due to the high and imbalanced discontinuation rates in the study and the low event rates for some of these outcomes, it was not possible to assess whether there were any differences between avatrombopag and placebo in the study population. In addition, most of these outcomes were uncontrolled for multiplicity, so are considered exploratory. It is also a challenge to base treatment decisions or draw meaningful conclusions from subgroup analyses due to the small study size.
In Study 003, 80% of patients (n = 12) in the avatrombopag group and no patients in the placebo group achieved a treatment response on day 28. The median (range) change in platelet count from baseline to day 28 was 84 × 109/L (–10 × 109/L to 1,012 × 109/L) in the avatrombopag group and –2 × 109/L (–12 × 109/L to 9 × 109/L) in the placebo group. Thirteen patients (86.7%) in the avatrombopag group and 1 patient (20.0%) in the placebo group had their platelet count at least doubled on day 28. The median (range) change in platelet count from baseline in Study 003 to week 24 in Study 004 was 124 × 109/L (–11 × 109/L to 205 × 109/L) among responders (n = 7) and 199 × 109/L (not applicable) among nonresponders (n = 1). At week 24, a total of 6 (85.7%) responders and 1 (100.0%) nonresponder achieved a response-level platelet count. A total of 6 (60.0%) responders and 1 (33.3%) nonresponder achieved a durable platelet response. Of the 6 responders and 1 nonresponder initially treated with corticosteroids, 2 (33.3%) responders and 1 (100.0%) nonresponder permanently discontinued steroid use during the final 8 weeks of treatment in Study 004.
Results of a retrospective observational study by Al-Samkari et al. (n = 44) suggested that in adult patients with chronic ITP who had been treated with prior eltrombopag or romiplostim, a durable platelet response and a reduction or discontinuation of concomitant ITP medications (including chronic corticosteroids) were observed for those who switched to avatrombopag. The findings of this study suggested the potential benefit of avatrombopag in patients who had previously received other TPO-RAs. However, the results should be interpreted with caution due to the major limitations of this small, retrospective study.
There is a lack of direct evidence on the comparative efficacy and safety of avatrombopag and currently available active treatments for thrombocytopenia in patients with chronic ITP. The sponsor submitted an NMA that suggested that avatrombopag was superior to eltrombopag, romiplostim, and rituximab in lowering the incidence of bleeding, but the magnitude of the difference was uncertain. The evidence of the comparative efficacy and safety of avatrombopag for other outcomes among patients with persistent or chronic ITP was associated with serious imprecision and is therefore considered to be uncertain. Clinical heterogeneities across eligible studies mean that the transitivity assumption that underlies the NMA is likely to have been violated, and the impact on the study results was not addressed. There were important limitations in this ITC due to heterogeneity in baseline characteristics and the definition of chronic ITP across trials, which is likely to have introduced intransitivity; a lack of consideration of key comparators, which may have added information to the network; differences in some of the outcome definitions across trials; a lack of information on the risk of bias of the included trials; a reliance on solely indirect evidence (except for 1 comparison of AEs); and the small sample size of the included studies and sparseness of the network, which resulted in serious imprecision across all estimates. As a result, the estimates derived from the NMA are very uncertain and the formulation of definitive conclusions is infeasible, and the ITC provided limited additional insight into the efficacy of avatrombopag compared to other active ITP treatments. Furthermore, some active treatments of interest were not included in this ITC analysis, and the comparative efficacy and safety of avatrombopag versus these drugs cannot be examined.
The lack of comparative efficacy and safety data, as well as uncertainty around the optimal treatment pathway in the second-line and subsequent-line treatment of ITP is reflected in guidelines for ITP.9,10 These guidelines highlight the very low certainty of the evidence on ITP treatment options, making it difficult to weigh options against 1 another. The guidelines, therefore, acknowledge that the individualization of therapy and shared decision-making are important in the treatment of ITP, as is the incorporation of duration of ITP, comorbidities, age of the patient, access to medications (cost, availability), and patient preferences.10 This was also echoed by the clinical expert, who noted that it was a challenge to compare avatrombopag to other second-line or subsequent-line ITP treatment options but that the modest efficacy with respect to platelet count response means it represents an additional treatment option among heavily pretreated patients who are in need of options for the management of ITP.
The clinical expert and patients highlighted how reducing bleeding risk and improving symptoms and quality of life are particularly important in chronic ITP, considering how these factors negatively affect patients. The patient group emphasized that symptom and quality-of-life improvements are likely more important to patients than platelet counts. Clinicians and patients also reported that ITP treatments should ideally be convenient and easy to administer for a patient. Unfortunately, the available evidence provides limited insight on outcomes important to patients and clinicians, as eligible and relevant trials (and ITC) focused primarily on platelet counts. Although some outcomes important to patients were assessed, such as bleeding and HRQoL, they were exploratory outcomes with low event rates and/or limited outcomes data and were not included in the statistical hierarchy for multiplicity adjustment; thus, it was not possible to draw conclusions about avatrombopag’s effect on these outcomes. Although eltrombopag is an oral medication, convenience and adherence were not compared in any of the relevant evidence, and, as such, the extent to which it leads to improvements in these measures, compared to existing treatments, is unclear. Platelet response is the main way clinicians assess treatment's effect in clinical practice. It is worth noting that the threshold of 50 × 109/L used in Study 302 for platelet response in patients with ITP may be somewhat arbitrary. The clinical expert noted that platelet response is expected to correlate with reduced bleeding risk, but acknowledged that available evidence for avatrombopag provides limited insight regarding its effect on quality of life, symptoms, and bleeding outcomes.
Harms
In Study 302, avatrombopag was demonstrated to be generally well tolerated. The most common AEs were headache, contusion, upper respiratory tract infection, arthralgia, epistaxis, fatigue, gingival bleeding, and petechiae. The rate of SAEs was higher in the avatrombopag arm than in the placebo arm (28.1% versus 5.9%). The rate of WDAE was higher in the avatrombopag arm than in the placebo arm (9.4% versus 0%). The clinical expert stated that the 3 thromboembolic events associated with avatrombopag is of particular interest, especially given the baseline exclusion of patients at high risk of thrombosis, and emphasized the importance in ensuring that patients do not develop thrombocytosis while on treatment. Otherwise, there are no other major concerns regarding the safety of avatrombopag.
The ITC was not able to provide information on the difference between avatrombopag and other active treatments in the incidence of any AEs because of serious imprecision in the estimates, as well as other important limitations highlighted previously. Therefore, there are limited data on the safety and tolerability of avatrombopag compared to other ITP treatments.
Conclusions
The management of chronic ITP is a challenge, as patients frequently relapse or are refractory to treatments. Therefore, patients commonly cycle through multiple ITP treatments. Treatment is complicated by a lack of evidence on the comparative efficacy and safety of second-line and subsequent-line treatment options, access issues, and the safety and/or tolerability of available options. In 1 double-blind RCT (Study 302), treatment with avatrombopag, a TPO-RA, improved platelet count response compared to placebo among patients with pretreated, primary, chronic ITP. There were limited or no data on patient-important outcomes, such as bleeding rates, the use of concomitant ITP medications, the need for rescue therapy, symptoms, and HRQoL. These outcomes were exploratory in Study 302, so the impact of avatrombopag on these outcomes remains unclear. Subgroup analyses were not able to provide insight into which patient groups (e.g., based on previous lines of therapy) are most likely to respond to treatment. Further, it remains difficult to draw conclusions about the comparative efficacy of avatrombopag versus other ITP treatments. The 1 ITC study included in this review suggested that avatrombopag may have a favourable efficacy compared to other TPO-RAs and to rituximab in terms of the incidence of bleeding events. However, there were important limitations of this study, and it is a challenge to draw firm conclusions about comparative efficacy based on its results. In Study 302, avatrombopag appeared to lead to a higher rate of AEs, such headache, contusion, upper respiratory tract infection, arthralgia, epistaxis, fatigue, gingival bleeding, and petechiae than placebo.
Overall, this review suggests that avatrombopag is another potential treatment option for patients with chronic, pretreated, primary ITP. It leads to a platelet count response in the target population and is generally well tolerated compared to placebo, although its comparative efficacy and safety versus other ITP treatments, and its effect on patient-important clinical outcomes, remains unclear. In addition, the study findings may not be generalizable to the patients with secondary ITP.
References
1.Arnold DM, Leung LKL, Tirnauer JS. Immune thrombocytopenia (ITP) in adults: clinical manifestations and diagnosis. In: Post TW, ed. UpToDate. Waltham (MA): UpToDate; 2021: www.uptodate.com Accessed 2024 May 1.
2.Nazaryan H, Liu Y, Sirotich E, et al. Second-line therapy for immune thrombocytopenia: real-world experience in Canada. Canadian Journal of General Internal Medicine. 2020;15(4):28-35.
3.Platelet Disorder Support Association. ITP in adults. 2022; https://www.pdsa.org/adults.html. Accessed 2022 May 2.
4.Cohen YC, Djulbegovic B, Shamai-Lubovitz O, Mozes B. The bleeding risk and natural history of idiopathic thrombocytopenic purpura in patients with persistent low platelet counts. Arch Intern Med. 2000;160(11):1630-1638. PubMed
5.Trotter P, Hill QA. Immune thrombocytopenia: improving quality of life and patient outcomes. Patient Relat Outcome Meas. 2018;9:369-384. PubMed
6.Hill QA, Newland AC. Fatigue in immune thrombocytopenia. Br J Haematol. 2015;170(2):141-149. PubMed
7.Portielje JE, Westendorp RG, Kluin-Nelemans HC, Brand A. Morbidity and mortality in adults with idiopathic thrombocytopenic purpura. Blood. 2001;97(9):2549-2554. PubMed
8.Rodeghiero F, Stasi R, Gernsheimer T, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood. 2009;113(11):2386-2393. PubMed
9.Provan D, Arnold DM, Bussel JB, et al. Updated international consensus report on the investigation and management of primary immune thrombocytopenia. Blood Adv. 2019;3(22):3780-3817. PubMed
10.Neunert C, Terrell DR, Arnold DM, et al. American Society of Hematology 2019 guidelines for immune thrombocytopenia. Blood Adv. 2019;3(23):3829-3866. PubMed
11.Doptelet (avatrombopag tablets): 20 mg avatrombopag (as maleate), oral [product monograph]. Stockholm (SE): Swedish Orphan Biovitrum AB (publ); 2023 Nov 3.
12.Clinical Study Report: E5501-G000-302. A phase 3, multicenter, randomized, double-blind, placebo-controlled, parallel-group trial with an open-label extension phase to evaluate the efficacy and safety of oral E5501 plus standard care for the treatment of thrombocytopenia in adults with chronic immune thrombocytopenia (idiopathic thrombocytopenic purpura)[internal sponsor's report]. Woodcliff Lake (NJ): Eisai Inc; 2016 Jan.
13.Uhl L, Assmann SF, Hamza TH, Harrison RW, Gernsheimer T, Slichter SJ. Laboratory predictors of bleeding and the effect of platelet and RBC transfusions on bleeding outcomes in the PLADO trial. Blood. 2017;130(10):1247-1258. PubMed
14.Network meta-analysis of efficacy and safety of avatrombopag versus comparators in adult patients with chronic immune thrombocytopenia who have had an insufficient response to a previous treatment [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Doptelet (avatrombopag), 20 mg oral tablets. Oakville (ON): Sobi Canada Inc; 2020.
15.Clinical Study Report:501-CL-003. A phase 2, double-blind, randomized, dose-ranging, placebo-controlled, parallel-group study of AKR-501 tablets taken orally once daily for 28 days in patients with chronic idiopathic thrombocytopenia purpura (ITP [internal sponsor's report]. Woodcliff Lake (NJ): Eisai Inc; 2010 Oct 10.
16.Clinical Study Report: 501-CL-004. A phase 2, parallel-group, rollover study of AKR-501 in patients with chronic idiopathic thrombocytopenia purpura (ITP) who completed 28 days’ of study treatment in protocol AKR-501-CL-003 [internal sponsor's report]. Woodcliff Lake (NJ): Eisai Inc; 2010 Oct 18.
17.Al-Samkari H, Jiang D, Gernsheimer T, et al. Durability of platelet response after switching to avatrombopag from eltrombopag or romiplostim in immune thrombocytopenia. Res Pract Thromb Haemost. 2023;7(3):100134. PubMed
18.Al-Samkari H, Jiang D, Gernsheimer T, et al. Adults with immune thrombocytopenia who switched to avatrombopag following prior treatment with eltrombopag or romiplostim: a multicentre US study. Br J Haematol. 2022;18:18. PubMed
19.Kistangari G, McCrae KR. Immune thrombocytopenia. Hematol Oncol Clin North Am. 2013;27(3):495-520. PubMed
20.Swinkels M, Rijkers M, Voorberg J, Vidarsson G, Leebeek FWG, Jansen AJG. Emerging concepts in immune thrombocytopenia. Front Immunol. 2018;9:880. PubMed
21.Cooper N, Kruse A, Kruse C, et al. Immune thrombocytopenia (ITP) World Impact Survey (iWISh): patient and physician perceptions of diagnosis, signs and symptoms, and treatment. Am J Hematol. 2021;96(2):188-198. PubMed
22.Arnold DM, Cuker A. Second-line and subsequent therapies for immune thrombocytopenia (ITP) in adults. In: Post TW, ed. UpToDate in Waltham (MA): UpToDate; 2022: www.uptodate.com Accessed 2024 May 1.
23.Revolade (eltrombopag tablets): 12.5 mg, 25 mg, 50 mg and 75 mg eltrombopag (as eltrombopag olamine) tablets [product monograph]. Dorval (QC): Novartis Pharmaceuticals Canada Inc; 2019: https://pdf.hres.ca/dpd_pm/00049522.PDF. Accessed 2024 May 1.
24.Nplate (romiplostim for injection): 250 mcg/0.5 mL and 500 mcg/1 mL lyophilized powder for solution [product monograph]. Mississauga (ON): Amgen Canada Inc; 2021: https://www.amgen.ca/-/media/Themes/CorporateAffairs/amgen-ca/amgen-ca/documents/products/en/nplate_pm.pdf. Accessed 2024 May 1.
25.Rituxan (rituximab for injection): 10 mg/mL intravenous infusion [product monograph]. Mississauga (ON): Hoffmann-La Roche Ltd; 2023: https://assets.roche.com/f/173850/x/5af6aeabe9/rituxaniv_pm_e.pdf. Accessed 2024 May 1.
26.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. J Clin Epidemiol. 2016;75:40-46. PubMed
27.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2022 Feb 28.
28.Jurczak W, Chojnowski K, Mayer J, et al. Phase 3 randomised study of avatrombopag, a novel thrombopoietin receptor agonist for the treatment of chronic immune thrombocytopenia. Br J Haematol. 2018;183(3):479-490. PubMed
29.Al-Samkari H, Nagalla S. Efficacy and safety evaluation of avatrombopag in immune thrombocytopenia: analyses of a phase III study and long-term extension. Platelets. 2022;33(2):257-264. PubMed
30.Jain S, Gernsheimer T, Kolodny S, Bernheisel C, Vredenburg M, Panch SR. Additional efficacy analysis of avatrombopag phase III data for the treatment of adults with immune thrombocytopenia. Platelets. 2023;34(1):2195016. PubMed
31.Drug Reimbursement Review sponsor submission: Doptelet (avatrombopag), 20 mg oral tablets [internal sponsor's package]. Oakville (ON): Sobi Canada Inc; 2022 Feb 3.
32.Product information: Doptelet (avatrombopag tablets). (European public assessment report). Amsterdam (NL): European Medicines Agency; 2019: https://www.ema.europa.eu/en/documents/product-information/doptelet-epar-product-information_en.pdf. Accessed 2024 May 1.
33.Fogarty PF, Tarantino MD, Brainsky A, Signorovitch J, Grotzinger KM. Selective validation of the WHO Bleeding Scale in patients with chronic immune thrombocytopenia. Curr Med Res Opin. 2012;28(1):79-87. PubMed
34.Ware J, Kosinski M, Bjorner JB, Turner-Bowker DM, Gandek B, Maruish ME. Determining important differences in scores. User's manual for the SF-36v2 health survey. 2nd ed. Lincoln (RI): Quality Metric Inc; 2007.
35.Brooks R. EuroQol: the current state of play. Health Policy. 1996;37(1):53-72. PubMed
36.EuroQol Group. EuroQol--a new facility for the measurement of health-related quality of life. Health Policy. 1990;16(3):199-208. PubMed
37.Wojciechowski P, Wilson K, Nazir J, et al. Efficacy and safety of avatrombopag in patients with chronic immune thrombocytopenia: a systematic literature review and network meta-analysis. Adv Ther. 2021;38(6):3113-3128. PubMed
38.Avatrombopag and lusutrombopag for treating thrombocytopenia in people with chronic liver disease needing an elective procedure [ID1520]. (Multiple technology appraisal). London (GB): National Institute for Health and Care Excellence; 2019: https://www.nice.org.uk/guidance/ta617/documents/committee-papers. Accessed 2024 May 1.
39.Puavilai T, Thadanipon K, Rattanasiri S, et al. Treatment efficacy for adult persistent immune thrombocytopenia: a systematic review and network meta-analysis. Br J Haematol. 2020;188(3):450-459. PubMed
40.Cheng G, Saleh MN, Marcher C, et al. Eltrombopag for management of chronic immune thrombocytopenia (RAISE): a 6-month, randomised, phase 3 study. Lancet. 2011;377(9763):393-402. PubMed
41.Kuter DJ, Bussel JB, Lyons RM, et al. Efficacy of romiplostim in patients with chronic immune thrombocytopenic purpura: a double-blind randomised controlled trial. Lancet. 2008;371(9610):395-403. PubMed
42.Arnold DM, Heddle NM, Carruthers J, et al. A pilot randomized trial of adjuvant rituximab or placebo for nonsplenectomized patients with immune thrombocytopenia. Blood. 2012;119(6):1356-1362. PubMed
43.Ghanima W, Khelif A, Waage A, et al. Rituximab as second-line treatment for adult immune thrombocytopenia (the RITP trial): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet. 2015;385(9978):1653-1661. PubMed
44.Bussel J, Arnold DM, Grossbard E, et al. Fostamatinib for the treatment of adult persistent and chronic immune thrombocytopenia: Results of two phase 3, randomized, placebo-controlled trials. Am J Hematol. 2018;93(7):921-930. PubMed
45.Miller AB, Hoogstraten B, Staquet M, Winkler A. Reporting results of cancer treatment. Cancer. 1981;47(1):207-214. PubMed
46.Saleh MN, Cheng G, Bussel JB, et al. EXTEND study update: safety and efficacy of eltrombopag in adults with chronic immune thrombocytopenia (ITP) from June 2006 to February 2010. Blood. 2010;116(21):67.
47.Hays RD, Sherbourne CD, Mazel RM. The RAND 36‐item health survey 1.0. Health Econ. 1993;2(3):217-227. PubMed
48.Signorovitch J, Brainsky A, Grotzinger KM. Validation of the FACIT-fatigue subscale, selected items from FACT-thrombocytopenia, and the SF-36v2 in patients with chronic immune thrombocytopenia. Qual Life Res. 2011;20(10):1737-1744. PubMed
49.EuroQol Research Foundation. EQ-5D-3L user guide 2018; https://euroqol.org/publications/user-guides. Accessed 2022 Apr 1.
50.EuroQol Research Foundation. EQ-5D-5L user guide. 2019; https://euroqol.org/publications/user-guides. Accessed 2022 Apr 1.
51.Snyder CF, Mathias SD, Cella D, Isitt JJ, Wu AW, Young J. Health-related quality of life of immune thrombocytopenic purpura patients: results from a web-based survey. Curr Med Res Opin. 2008;24(10):2767-2776. PubMed
52.Sanz MA, Aledort L, Mathias SD, Wang X, Isitt JJ. Analysis of EQ-5D scores from two phase 3 clinical trials of romiplostim in the treatment of immune thrombocytopenia (ITP). Value Health. 2011;14(1):90-96. PubMed
53.Leung YY, Ho K, Zhu T, Tam L-S, Kun E, Li E. Testing scaling assumptions, reliability and validity of medical outcomes study short-form 36 health survey in psoriatic arthritis. Rheumatology (Oxford). 2010;49:1495-1501. PubMed
54.EQ-5D-5L user guide: basic information on how to use the EQ-5D-5L instrument. Rotterdam (NL): EuroQol Research Foundation; 2019: https://euroqol.org/wp-content/uploads/2021/01/EQ-5D-5LUserguide-08-0421.pdf. Accessed 2022 Apr 1.
Appendix 1: Literature Search Strategy
Clinical Literature Search
Overview
Interface: Ovid
Databases:
MEDLINE All (1946-present)
Embase (1974-present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: March 4, 2022
Alerts: Bi-weekly search updates until project completion
Search filters applied: No filters were applied to limit the retrieval by study type.
Limits:
No date or language limits were used
Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
MeSH | Medical Subject Heading |
.fs | Floating subheading |
exp | Explode a subject heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Author keyword heading word (MEDLINE) |
.dq | Candidate term word (Embase) |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multidatabase Strategy
(avatrombopag* or Doptelet* or AKR 501 or AKR501 or AS 1670542 or AS1670542 or E 5501 or E5501 or YM 477 or YM477 or 570406-98-3 or 677007-74-8 or 3H8GSZ4SQL or GDW7M2P1IS).ti,ab,kf,ot,hw,nm,rn.
1 use medall
* avatrombopag/ or (avatrombopag* or Doptelet* or AKR 501 or AKR501 or AS 1670542 or AS1670542 or E 5501 or E5501 or YM 477 or YM477).ti,ab,kf,ot,dq.
3 use oemezd
4 not (conference review or conference abstract).pt.
2 or 5
remove duplicates from 6
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
[Search -- Studies with results | Doptelet OR avatrombopag]
WHO ICTRP
International Clinical Trials Registry Platform, produced by the WHO. Targeted search used to capture registered clinical trials.
[Search terms -- Doptelet OR avatrombopag]
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
[Search terms -- Doptelet OR avatrombopag]
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
[Search terms -- Doptelet OR avatrombopag]
Grey Literature
Search dates: February 22, 2022-February 23, 2022
Keywords: Doptelet, avatrombopag, thrombocytopenia
Limits: None
Updated: Search updated before the completion of stakeholder feedback period
Relevant websites from the following sections of the CADTH grey literature checklist https://www.cda-amc.ca/grey-matters-practical-tool-searching-health-related-grey-literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Health Statistics
Internet Search.
Appendix 2: Description and Appraisal of Outcome Measures
Note this appendix has not been copy-edited.
Aim
To describe the following outcome measures and review their measurement properties (validity, reliability, responsiveness to change, and MID):
WHO (WHO) Bleeding Scale
Short-Form Health Survey Version 2 (SF-36v2)
EQ-5D-3L
Findings
Table 31: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
WHO Bleeding Scale | The WHO Bleeding Scale is an instrument used to classify bleeding on a five-point ordinal scale. The scale grading follows as: 0 (no bleeding), 1 (petechiae), 2 (mild blood loss), 3 (gross blood loss), and 4 (debilitating blood loss).33,45 Classifications can be made based on self-report and/or clinical examination. | Construct validity: Similarity was observed between the WHO Bleeding Scale and IBLS, showing a similar association between platelet counts and severity of bleeding. During known group comparison assessment, significant associations (P < 0.05) were observed between the WHO Bleeding Scale and many clinical outcomes.33 Test-retest reliability: IQRs were from - 0.8% to + 1.3% with ICC of 0.75, and from - 4.7% to + 3.4% with ICC of 0.70 for the WHO Bleeding Scale in the RAISE40 and EXTEND46 studies.33 Responsiveness: Moderate responsiveness; effect size, standardized response and responsiveness statistic were 0.714, 0.745 and 0.560 in RAISE,40 and 0.622, 0.487 and 0.588 in EXTEND46 studies.33 | Ranged from 0.33 to 0.44.33 |
36-item Short-Form Health Survey Version 2 | The SF-36 is a generic self-reported HRQoL measure consisting of 8 subdomains. The SF-36 provides 2 component summaries, PCS and MCS. The 8 subdomains are each measured on a scale of zero to 100, with an increase in score indicating improvement in health status.47 | Construct validity: supported by moderate to strong score correlations with related scales and clinical outcomes.48 Internal consistency Reliability: SF-36 item-to-domain score correlations > 0.20, and Cronbach alpha values for SF-36v2 domains ≥ 0.75, for all items and if each item was deleted from scale.48 Test-retest reliability: ICCs in clinically stable patients were > 0.7 in both RAISE40 and EXTEND46 studies.48 Responsiveness: SF-36v2 had been reported to be less responsive compared to the disease-specific measures of fatigue based on the ability to capture change.48 | Not assessed in ITP patients. |
EQ-5D-3L | Generic self-reported preference based HRQoL scale consisting of a VAS with values between 100 (best imaginable health) and 0 (worst imaginable health). Results in a composite index score of 5 dimensions: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression with 3 response levels ranging from experiencing no problems to extreme problems.49,50 | Construct validity: Illustrated using the known-groups approach, showing a significant difference in adjusted VAS scores between adult patients with ITP (65.5) compared to healthy controls (82.3) (P = 0.002).51 Reliability: Not assessed in ITP patients. Responsiveness: The adjusted mean change (SE) in VAS scores from baseline to the final visit were numerically greater for patients on romiplostim 6.42 (2.08) vs. placebo 0.48 (2.59) potentially indicating a sensitivity to change over time.52 | Not assessed in ITP patients. |
HRQoL = health-related quality of life; IBLS = Immune Thrombocytopenic Purpura Bleeding Scale; ICC = intraclass correlation coefficient; IQRs = interquartile ranges; ITP = immune thrombocytopenia; MCS = mental component score; MID = minimal important difference; PCS = physical component score; SE = standard error; SF-36 = 36-Item Short-Form Survey; VAS = visual analogue scale; WHO = World Health Organization.
WHO Bleeding Scale
The WHO Bleeding Scale is an instrument that may be used to classify bleeding on a 5-point ordinal scale. The scale grading follows as: 0 (no bleeding), 1 (petechiae), 2 (mild blood loss), 3 (gross blood loss), and 4 (debilitating blood loss).33 The ratings can be based on physical examination and/or patients’ verbal responses to questions. Originally developed for bleeding assessment among cancer patients,45 the performance of WHO Bleeding Scale had been evaluated among chronic ITP patients treated with eltrombopag in 2 long-term, phase III clinical trials: the RAISE40 (n = 189) and EXTEND46 (n = 154) studies.33
Intraclass correlation coefficients (ICCs) for test-retest reliability at 2 consecutive patient visits with the least absolute change in platelet count (mean 7.2 days apart; SD = 2 days). The interquartile ranges (IQRs) were from - 0.8% to + 1.3% with an ICC of 0.75, and from - 4.7% to + 3.4% with an ICC of 0.70 for the WHO Bleeding Scale in the RAISE40 and EXTEND46 studies, respectively.33
Construct validity was assessed in 2 ways, first by determining interinstrument correlations (item-to-item and item-to-domain correlations) between the WHO Bleeding Scale and the ITP Bleeding Scale (IBLS) and between the WHO Bleeding Scale and platelet counts, Second, a known-groups comparison was used to describe the relationship between the scale and clinical outcomes.33 Similarity was observed between the WHO Bleeding Scale and IBLS, showing a similar association between platelet counts and severity of bleeding with both scales. Moreover, during known group comparison assessment, significant associations (P < 0.05) were observed between the WHO Bleeding Scale and many clinical outcomes.
The responsiveness was assessed by calculating the differences in grades from baseline to last-on-treatment evaluation among patients with a platelet count response.33 The responsiveness among patients were computed using 3 indices: effect size = D/SD0, standardized response mean = D/ SD*, and responsiveness statistic = D/SD#. Here D denotes the mean score change of interest (i.e., mean change from baseline among patients with platelet count response), SD0 denotes the standard deviation (SD) of scores at baseline, SD* is the SD of D, and SD# is the SD of D among patients with no response to treatment. In the RAISE40 study (n = 129) the effect size, standardized response, and responsiveness statistic were 0.714, 0.745 and 0.560, respectively, indicating moderate responsiveness. In the EXTEND46 (n = 71) the effect size, standardized response, and responsiveness statistic were 0.622, 0.487 and 0.588, respectively, showcasing moderate responsiveness for 2 responsiveness indices, and just below the 0.50 threshold for moderate responsiveness for 1 index.33
The estimated MID for the WHO Bleeding Scale were calculated using distributional methods, calculated as half of the SD of scores at baseline and at the last on-treatment assessment. The MID was also estimated using a clinical anchor-based approach. With this approach, the mean change from baseline in bleeding grade associated with a doubling of platelet count, as well as an increase to above 50x109/L were identified. Confidence intervals were estimated using generalized estimating equations. Using these methods, MIDs ranged from 0.33 to 0.44.33
36-item Short-Form Health Survey (SF-36)
The SF-36 is a generic self-reported health assessment questionnaire that has been used in clinical trials to study the impact of chronic disease on HRQoL. The SF-36 consists of 8 domains: physical functioning, role physical, bodily pain, general health, vitality, social functioning, role emotional, and mental health. The SF-36 also provides 2 component summaries: the physical component summary (SF-36 PCS) and the mental component summary (SF-36 MCS), which are created by aggregating scores on the 8 domains. The SF-36 PCS, SF-36 MCS and 8 domains are each measured on a scale of 0 to 100, with an increase in score indicating improvement in health status. In general use of the SF-36, a change of 2 to 4 points in each domain or 2 to 3 points in each component summary indicates a clinically meaningful improvement as determined by the patient.34 The summary scales are scored using norm-based methods, with regression weights and constants derived from the general US population. Both the PCS and MCS scales are transformed to have a mean of 50 and a standard deviation (SD) of 10 in the general US population. Therefore, all scores above or below 50 are considered above or below average for the general US population.53
The validity, reliability and responsiveness of SF-36v2 has been assessed in 2 clinical trials, RAISE40 and EXTEND,46 prescribing eltrombopag to patients previously treated for chronic ITP.48 RAISE was a 6-month, phase III, randomized, double-blind, placebo-controlled study with 197 ITP patients, whereas EXTEND was an open-label extension study containing 154 patients. In the RAISE40 study, SF-36v2 PCS and MCS mean scores were below but within 1 SD of the US population standardized mean, as well as the mean scores for the 7 out of 8 domains. Internal consistency reliability was assessed using interitem correlations, item-to-total score correlations, and Cronbach alpha. Sufficient and acceptable internal consistency reliability was reported, demonstrating all SF-36 item-to-domain score correlations > 0.20, and Cronbach alpha values for SF-36v2 domains ≥ 0.75, for all items and when each item was deleted from the scale, in both RAISE and EXTEND studies. More specifically, Cronbach alpha values for SF-36v2 were between 0.75 and 0.94 at baseline and between 0.83 and 0.95 at the last assessment in RAISE, and between 0.78 and 0.94 at baseline and between 0.79 and 0.96 at the last assessment in EXTEND.
Test-retest reliability was assessed by calculating ICCs for scores from consecutive pairs of visits corresponding to the minimal absolute percent change in platelet counts. ICCs for test-retest reliability evaluation in clinically stable patients were > 0.7 in both RAISE and EXTEND studies.48 In RAISE, this value was applicable for physical function, general health, and vitality domains (n = 50 to 55), and in EXTEND for all domains of SF-36, except bodily pain and emotional role (n = 126 to 132). During sensitivity analyses, ICCs were calculated using a subgroup of patients with ≤ 15% change in platelet counts between 2 consecutive visits (mean of 49 to 52 days for RAISE and 45 to 50 days in EXTEND). For sensitivity analysis, ICCs in clinically stable patients were ≥ 0.72 for all domains and summary measures of SF-36-v2, except social function and emotional role.
Construct validity of SF-36v2 was assessed by testing hypotheses about relationships (Pearson correlations) with other instruments and with clinical outcomes and was supported by moderate to strong score correlations between scores at baseline, and between the change scores of the PRO measures in both studies.48 While evaluating the longitudinal construct validity of measures by stratifying patients into responders or nonresponders and comparing the change score on each measure between groups based on magnitude of effect, a statistically significant difference between responders and nonresponders was observed. Regarding the responsiveness, SF-36v2 had been reported to be less responsive compared to the disease-specific measures of fatigue based on the ability to capture change.48
No MID has been formally assessed for SF-36v2 among ITP patients.
EQ-5D-3L
The EQ-5D-3L is a generic HRQoL instrument that may be applied to a wide range of health conditions and treatments.35,36 The first of 2 parts of the EQ-5D-3L consists of a descriptive system that classifies respondents based on the following 5 dimensions: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression. There are 3 response levels of severity (no problems, some problems, and extreme problems) in each of the 5 existing dimensions.54 The second part is a 20 cm visual analogue scale (EQ-VAS) that has end points labelled 0 and 100, with respective anchors of “worst imaginable health state” and “best imaginable health state.” Respondents are asked to rate their health by drawing a line from an anchor box to the point on the EQ-VAS which best represents their health on that day.
A cross-sectional study by Snyder et al., (2008)51 compared VAS scores between 1,002 ITP patients and 1,031 age- and gender-matched healthy controls. Patients with ITP scored significantly worse on the VAS with an adjusted score of 65.5 compared to controls with a score of 82.3, indicating construct validity using the known-groups approach (P = 0.002). In terms of EQ-5D-3L dimensions, significantly more ITP patients reported ‘some problems’ for usual activities and for anxiety/depression than controls, but significantly more controls reported ‘many problems’ for pain/discomfort. Results found no significant differences in EQ-5D-3L scores between ITP patients who had undergone splenectomy versus those that had not. Another study examined responsiveness to change on the VAS among 125 ITP patients (83 on romiplostim and 42 on placebo) in 2 24-week clinical trials.52 Results found that the adjusted mean change (SE) in EQ-VAS scores from baseline to the final visit were numerically greater for patients on romiplostim 6.42 (2.08) versus placebo 0.48 (2.59) potentially indicating a sensitivity to change over time, however this difference was not statistically significant (P = 0.066) and an effect size was not calculated.
Pharmacoeconomic Review
Abbreviations
AE
adverse event
BIA
budget impact analysis
ED
emergency department
GI
gastrointestinal
HRQoL
health-related quality of life
ICER
incremental cost-effectiveness ratio
ICH
intracranial hemorrhage
ITP
immune thrombocytopenia
IVIG
IV immunoglobulin
NICE
National Institute for Health and Care Excellence
NIHB
Non-insured Health Benefits
NMA
network meta-analysis
OR
odds ratio
PDSA
Platelet Disorder Support Association
QALY
quality-adjusted life-year
TPO-RA
thrombopoietin receptor agonist
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Avatrombopag (Doptelet), 20 mg, tablets, oral |
Submitted price | Avatrombopag, 20 mg, tablet: = $115.00 |
Indication | Proposed: for the treatment of thrombocytopenia in adult patients with chronic ITP who have had an insufficient response to a previous treatment |
Health Canada approval status | Under review (pre-NOC) |
Health Canada review pathway | Standard |
NOC date | Anticipated: May 2, 2022 |
Reimbursement request | As per indication |
Sponsor | Sobi Canada Inc. |
Submission history | No |
ITP = immune thrombocytopenia; NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov model |
Target population | Adult patients with chronic ITP who have had an insufficient response to a previous treatment |
Treatment | Avatrombopag |
Comparators | Eltrombopag Romiplostim Rituximab Watch and rescue, consisting of no active treatment Scenario analysis: Small-molecule drugs, consisting of azathioprine, cyclosporine, cyclophosphamide, mycophenolate, danazol, dapsone |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (56 years) |
Key data sources | Study 302, a phase III, randomized, double-blind trial (avatrombopag vs. watch and rescue); sponsor’s submitted NMA (response rates for avatrombopag vs. eltrombopag, romiplostim); NICE submission (response rate for avatrombopag vs. rituximab). |
Submitted results | ICER = $84,217 per QALY gained vs. rituximab (incremental costs = $140,383; incremental QALYs = 1.67). All other treatments are dominated. |
Key limitations |
|
CADTH reanalysis results |
|
ICER = incremental cost-effectiveness ratio; ITP = immune thrombocytopenia; LY = life-year; NICE = National Institute for Health and Care Excellence; NMA = network meta-analysis; QALY = quality-adjusted life-year; TPO-RA = thrombopoietin receptor agonist.
Conclusions
The CADTH clinical review found that based on Study 302, avatrombopag led to an improvement in platelet count response compared to placebo among patients with pretreated, primary, chronic immune thrombocytopenia (ITP). There were limited or no data on patient-important outcomes, such as bleeding rates, the use of concomitant ITP medications, the need for rescue therapy, symptoms, and health-related quality of life (HRQoL). These outcomes were exploratory in Study 302, so the impact of avatrombopag on these outcomes remains unclear. The sponsor’s submitted network meta-analysis (NMA) found that there was no significant difference in the proportion of patients with durable platelet response between avatrombopag and other thrombopoietin receptor agonists (TPO-RAs). The NMA suggested that avatrombopag may have favourable efficacy compared to other TPO-RAs and to rituximab in terms of the incidence of bleeding events. The CADTH clinical review noted that, due to important limitations of the sponsor’s NMA, estimates derived from the NMA are very uncertain, and the formulation of definitive conclusions around the comparative efficacy and safety of avatrombopag versus other ITP treatments is infeasible.
CADTH undertook reanalyses to address limitations related to the lack of comparative efficacy data on the response rates for avatrombopag versus rituximab; the uncertain indirect comparative efficacy for avatrombopag and other TPO-RAs; adjusting the response rate for TPO-RAs to reflect the response rate for avatrombopag observed in Study 302; the incorporation of dose adjustments for TPO-RAs; the removal of a utility decrement associated with being a nonresponder from health states with bleeds; and adjusting health care resource use for bleed management to better reflect clinical practice in Canada. In CADTH’s base-case reanalysis, all TPO-RAs, including avatrombopag, yielded an equal number of quality-adjusted life-years (QALYs). All differences in total costs were derived from treatment-acquisition costs; costs for all other health care resource use were equal among TPO-RAs. Avatrombopag was associated with higher treatment-acquisition costs compared with eltrombopag ($7,462), but lower treatment-acquisition costs compared with romiplostim ($148,287). Given that the most relevant comparators for avatrombopag are other TPO-RAs and given the uncertainty in clinical estimates from the sponsor’s NMA, there is limited clinical evidence to support a price premium for avatrombopag over other TPO-RAs. Additionally, as there were no comparative efficacy data for the response rate of avatrombopag versus rituximab, there is no clinical evidence to support a price premium for avatrombopag over rituximab.
Watch and rescue (assumed to be equal to the placebo arm of Study 302) is the only comparator for which there is direct comparative evidence against avatrombopag. For this comparison, the incremental cost-effectiveness ratio (ICER) for avatrombopag compared with watch and rescue was $98,150 per QALY gained. To achieve an ICER of $50,000 per QALY gained compared with watch and rescue, a price reduction of at least 32% is required.
CADTH was unable to address limitations of the sponsor’s model regarding the lack of direct or indirect evidence informing response rate for avatrombopag compared to other relevant comparators (e.g., rituximab and immunosuppressants), the use of surrogate outcomes (platelet counts) to model ITP, the uncertainty of the sponsor’s health-state utility values, the response assumptions, or the fact that treatment sequencing did not reflect clinical practice in Canada.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
CADTH received 1 patient input submission from the Platelet Disorder Support Association (PDSA) for this review. Patient comments were collected from the PDSA Facebook page (the proportion of responses from Canada was not reported). Comments from patients that were included in the submission noted increasing and stabilized platelet counts while using avatrombopag. Some patients reported that they experienced no or minimal manageable side effects while taking avatrombopag, whereas others reported side effects, such as anemia and migraines, which resulted in either treatment discontinuation or dose reductions. Comments from patients noted that previous therapies trialled included fostamatinib, IV immunoglobulin (IVIG), splenectomy, and TPO-RAs; 1 patient noted that these medications either did not work and/or resulted in side effects or had inconvenient administration (i.e., weekly injections). Additional ITP therapies noted by PDSA include prednisone, anti-Rho(D) immune globulin, and rituximab. Feedback from PDSA indicated that ITP impacts the overall HRQoL of patients and their families, with a constant risk of life-threatening bleeding, elevated levels of fatigue, anxiety, depression, physical pain, sleep disturbances, and feelings of isolation and inadequacy due to activity restrictions. PDSA feedback noted that treatment goals were to achieve a platelet count that reduced the risk of bleeding and improved or maintained quality of life. PDSA noted that avatrombopag can be taken daily with food and without dietary restrictions, whereas the metabolism of other TPO-RAs (i.e., eltrombopag) can be influenced by foods such as dairy. Avatrombopag was also noted as being more convenient to use than romiplostim, which requires clinic infusions. PDSA input also noted that patients often have restrictions on their choice in treatment, as they may not respond well to some therapies and may be unable to afford others.
Registered clinician feedback was received from 1 group: the Canadian Hematology Society. Feedback noted that for patients with platelet counts less than 30 × 109/L, first-line therapy is steroids, with IVIG added on for bleeding or severely reduced platelet counts in high-risk patients. Upon relapse after first-line therapy, preferred second-line options include TPO-RAs, rituximab, or splenectomy. Other second-line options include immunosuppressive medications, such as azathioprine, cyclophosphamide, cyclosporine, and mycophenolate; and danazol and dapsone. ITP treatment goals include raising platelet counts to reduce bleeding risk, improving quality of life, minimizing treatment toxicity, and avoiding side effects common to many ITP treatments (i.e., immunosuppression). Canadian Hematology Society feedback noted that current TPO-RAs are perceived to be inconvenient because they involve dietary limitations (eltrombopag) and injections (romiplostim). If avatrombopag becomes available, the feedback noted, it would likely be used as a second-line therapy for patients who failed 1 or more courses of treatment with corticosteroids and IVIG, and as an option for patients who are resistant or refractory to multiple other therapies, including other TPO-RAs. Avatrombopag is expected to cause a shift in the treatment paradigm by enabling access to TPO-RAs in second-line therapy.
Drug plan input noted that in jurisdictions that currently fund TPO-RAs, patient eligibility is contingent on the failure of a specified number of first-line and second-line therapies, and eligibility may require patients to have bleeding complications and/or to have undergone splenectomy, whereas the proposed indication for avatrombopag is for those with an insufficient response to a previous treatment. The drug plans noted that TPO-RAs are funded in more jurisdictions than noted in the sponsor’s budget impact analysis (BIA).
Several of these concerns were addressed in the sponsor’s model:
HRQoL is captured in the model.
The model was based on platelet counts (although thresholds for response in the model were greater than those referenced in clinician feedback).
An administration cost for romiplostim and rituximab was incorporated.
CADTH addressed some of these concerns, as follows:
CADTH added reference scenario market shares for TPO-RAs to the BIA in jurisdictions that had TPO-RA public claims.
CADTH was unable to address the following concerns raised from stakeholder input:
Differential adverse event (AE) rates among TPO-RAs were not incorporated.
The use of TPO-RAs after rituximab and the trialling of TPO-RAs by patients intolerant to an initial TPO-RA was not incorporated.
A decrement to quality of life associated with IV or subcutaneous treatments was not incorporated.
Loss of response or duration of response for TPO-RAs was not directly modelled; instead, this was assumed to be equal to time on treatment.
Economic Review
The current review is for avatrombopag for adult patients with chronic ITP who have had an insufficient response to a previous treatment.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis comparing avatrombopag with eltrombopag, romiplostim, rituximab, and watch and rescue in adult patients with chronic ITP who have had an insufficient response to a previous treatment.
Avatrombopag is available as a 20 mg tablet. The recommended dose of avatrombopag is 20 mg once daily initially, with dose adjustments made based on platelet counts, which could lead to a minimum recommended dose of 20 mg once weekly or a maximum recommended dose of 40 mg daily. At the sponsor’s submitted price of $115.00 per 20 mg tablet, the annual cost of treatment with avatrombopag would be $41,975 if patients remain on a 20 mg once-daily dose for a full year. Eltrombopag had a daily cost of $130.00 in the model, resulting in an annual cost of $47,450 if patients remain on a 50 mg once-daily dose for a full year. Romiplostim costs were based on a dose of 3 mcg/kg weekly, resulting in a daily cost of $145.97 and an annual cost of $53,279 if patients remain on a dose of 3 mcg/kg or less weekly for a full year. Rituximab costs were based on patients receiving a dose of 375 mg/m2 once weekly for 4 weeks, resulting in a daily cost of $308.07, or a cost per 4-week course of $8,626.04.
The clinical outcomes of interest were QALYs and life-years. The economic analysis was undertaken over a lifetime (56-year) time horizon from the perspective of a Canadian public health care payer. Discounting (1.5% per annum) was applied to both costs and outcomes.
Model Structure
A Markov model with 4 health states was submitted by the sponsor, with 4-week cycle lengths (Figure 1).1 The model is based on response, which is defined as a platelet count equal to or greater than 50 × 109/L. All patients who receive an ITP therapy begin in the treatment and no response health state, which consists of 7 tunnel states that are used to model time to response. While in the tunnel states, patients are considered to be nonresponders. If patients in the treatment and no response state respond, they transition to the treatment and response state, where they remain until they discontinue treatment, which leads to a loss of response and a transition back to the treatment and no response health state. Patients can also transition from the treatment and no response state to the no-treatment state if they do not respond after the maximum treatment duration in the treatment and no response state (24 weeks) or if they lose their response in the treatment and response state. Patients remain in the no-treatment state unless they experience a bleed, in which case they can enter the treatment and no response state again and receive another active ITP treatment. Patients who do not receive an active ITP treatment remain in the no-treatment health state.
Model Inputs
The model’s baseline population characteristics were characterized by Study 302, a phase III, randomized, double-blind trial designed to evaluate the efficacy of avatrombopag compared with placebo in adults with chronic ITP and a platelet count less than 30 × 109/L. The sponsor assumed that the Study 302 population (baseline characteristics: mean age = 44.6 years; 36.7% male; mean body surface area = 1.94 m2; mean weight 82.97 kg; 32.7% had undergone splenectomy) reflected the population in Canada.2
Patient movement from the treatment and no response state to the treatment and response and the no-treatment health states was primarily based on time taken to respond, the response rate, and the duration of response. Response in the model was based on platelet count, with patients considered responders if their platelet count was equal to or greater than 50 × 109/L. Time to response informed the time spent in the treatment and no response health state, and was based on the assumption that it would take 24 weeks for avatrombopag, eltrombopag, and romiplostim, and 8 weeks for rituximab. Response rates informed the proportion of patients who transition to the treatment and response health state after the time to response. Response rates for avatrombopag, eltrombopag, and romiplostim were based on the sponsor’s NMA (Table 12).3 The response rate for rituximab was based on the romiplostim submission to the National Institute for Health and Care Excellence (NICE) (Table 12).4 Duration of response informed the probability of remaining in the treatment and response health state and was based on duration of treatment. The sponsor derived the average time on treatment for eltrombopag (434 weeks, or approximately 8.4 years) from a 2013 study,5 and assumed that the time on treatment (i.e., duration of response) was equal for avatrombopag, eltrombopag, and romiplostim. The duration of response for rituximab was also based on the duration of treatment, but was assumed to be 76 weeks.
Bleeding rates for outpatient and inpatient bleeds informed patient transitions from the no-treatment state to the treatment and no response state, where patients could receive an additional active ITP treatment. Patients could receive up to 3 active ITP treatment cycles; after this, patients would receive no active therapy (i.e., watch and rescue), and remain in the treatment and no response health state for the remainder of the model time horizon. Patients who started on 1 of the active comparators in the model (i.e., avatrombopag, eltrombopag, romiplostim, rituximab) could only receive watch and rescue and small-molecule medications (azathioprine, mycophenolate mofetil, cyclosporine, danazol, dapsone, cyclophosphamide, vincristine, vinblastine) in subsequent treatment lines.
Clinical events in the model, included bleeding, the need for rescue medications, the use of concomitant ITP medications, and treatment-related AEs were used to inform health-state costs and utilities. Patients were at risk of bleeding in all alive health states, and bleeding event frequency was dependent on platelet count, with a higher risk of bleeding associated with states in which platelet counts were less than 50 × 109/L (all health states except for treatment and response). Bleeding rates were obtained from the NICE eltrombopag submission and were based on platelet count and type of bleed (Table 9).6 Three types of inpatient bleeds were considered (intracranial hemorrhage, gastrointestinal [GI], and other bleeds), with different distributions of bleeds by platelet count (Table 10). It was assumed that once patients have tried 3 treatment lines of therapy and enter the no-treatment state, their rate of inpatient bleeding doubles.
Rescue therapies are used when there is a need to increase platelet counts urgently due to bleeding or low platelet counts. The proportion of patients requiring rescue therapy was sourced from Study 302 and informed by the proportion of patients requiring rescue therapy for reasons other than bleeding (44%).2 Rescue therapies received by patients included IVIG (80%), IV steroids (31%), and anti-D (46%).4 In the sponsor’s base-case analysis, because the majority of patients use rescue therapy for a bleeding event, rescue therapy was incorporated in the model as a part of bleeding costs.
Concomitant medications were used for all comparators, with the proportion of patients receiving medications dependent on response (45% for patients without a response to active treatment, 36% for those with a response).2 The sponsor assumed that 16% of patients who use concomitant medications after achieving a response will receive a reduced dose. The distribution of concomitant medications used was based on baseline concomitant medication use in Study 302 and was the same for all comparators.2
Specific AEs were not modelled; rather, the sponsor incorporated a frequency of occurrence for serious AEs and other AEs. Rates of AEs (3% serious, 31% other) were assumed to be the same for avatrombopag, eltrombopag, and romiplostim. AE rates for all comparators were based on the NICE romiplostim submission.4
The model considered 2 types of mortality: all-cause mortality (based on Canadian life tables) for all health states, and bleeding-related mortality. This is incorporated by applying a bleed-type–specific mortality rate7 to bleeds requiring hospitalizations.
Costs in the model included drug and administration costs, bleeding costs, and follow-up care costs. Treatment costs for avatrombopag and the comparators were based on recommended initial doses. Doses for the rescue therapies and active treatments received in subsequent treatment lines were sourced from the eltrombopag NICE submission.6 Administration costs were applied to romiplostim and rituximab.8 Bleeding costs were based on the type of bleed and included the costs of hospital admission, surgery, diagnostic imaging and blood tests, and hematology follow-up. As in the sponsor’s base case, rescue medications were included in the bleed costs; the cost of rescue medications were also applied by bleed type. Follow-up care included monthly hematology consultations and blood work.
Health-state utility values were based on Canadian utility norms.9 Depending on the health state, disutilities associated with splenectomy status, outpatient bleed type, and responder status were added to baseline utilities to derive health-state values. These disutilities were calculated using EQ-5D data from Study 302.2 Separate utility values sourced from the literature were applied for intracranial hemorrhage (ICH)10 and GI inpatient bleeds;11 other bleeds treated in inpatients were assumed to have the same utility as GI bleeds. These health-state utility values were applied for the presumed duration of these events (16 weeks for ICH; 4 weeks for GI and other inpatient bleeds). The values for treatment-emergent AEs disutilities were based on assumptions.
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (1,000 iterations). The deterministic and probabilistic results were similar. The probabilistic findings are presented here.
Base-Case Results
Avatrombopag was associated with a QALY gain of 1.67 at an additional cost of $140,383, resulting in an ICER of $84,217 compared with rituximab (Table 3). Watch and rescue and romiplostim were dominated (i.e., less effective and more expensive) by rituximab and avatrombopag, respectively. Eltrombopag was extendedly dominated by avatrombopag. At a willingness-to-pay threshold of $50,000 per QALY gained, there was a 0% probability of avatrombopag being cost-effective compared to rituximab.
Treatment costs as a proportion of total costs were higher for romiplostim (46%) and avatrombopag (41%) than for eltrombopag (31%), rituximab (22%), and watch and rescue (21%). Bleeding costs accounted for the majority of total costs, but vastly differed by comparator (55% of total costs for avatrombopag, and 65%, 50%, 74%, and 75% for eltrombopag, romiplostim, rituximab, and watch and rescue, respectively). Disaggregated QALYs by health state were not provided. Of the 14.96 QALYs associated with avatrombopag, 0.29 (2%) are accrued during the trial period (i.e., 98% of the QALYs for avatrombopag are gained during the extrapolation period).
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Total LYs | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|---|
Rituximab | 846,303 | 20.21 | 13.29 | Reference |
Avatrombopag | 986,686 | 21.79 | 14.96 | 84,217 |
Dominated and/or extended dominated treatments | ||||
Watch and rescue | 855,274 | 19.88 | 12.89 | Dominated by rituximab |
Eltrombopag | 923,961 | 21.18 | 13.64 | Extendedly dominated by avatrombopag |
Romiplostim | 1,129,871 | 21.22 | 14.41 | Dominated by avatrombopag |
ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor assessed several model parameters in probabilistic scenario analyses. In a scenario that assumed equal efficacy in terms of response rate for all TPO-RAs, the deterministic results have equal total QALYs for all TPO-RAs, but avatrombopag had the lowest total costs, resulting in eltrombopag and romiplostim being dominated. If it is assumed that patients do not use rescue therapies for reasons other than bleeds, total incremental costs for avatrombopag increase compared with rituximab, resulting in an ICER of $99,761. Using median dosing from Study 302, as opposed to product monograph dosing, increases avatrombopag total costs, leading to an ICER of $104,234 compared to rituximab. A subgroup analysis that explored all patients who had and all patients who had not undergone splenectomy separately showed that the results were not sensitive to splenectomy status.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
No conclusions can be made regarding the comparative efficacy between avatrombopag and other active ITP treatments in terms of response rate. In the sponsor’s model, the majority of parameters that inform patient transitions between health states and clinical events were largely based on assumptions of equal efficacy or on previous drug submissions to NICE. Specifically, time to response was based on an assumption, and was assumed to be the same for all TPO-RAs. Duration of response was assumed to be equal to time on treatment, and was informed by a romiplostim cost-effectiveness study.5 Duration of response was also assumed to be the same for all TPO-RAs. Finally, bleeding rates specific to platelet counts were obtained from the NICE eltrombopag submission.6 The only differential comparative clinical efficacy parameter included in the model was response rates for avatrombopag, eltrombopag, and romiplostim, which were informed by the sponsor’s NMA. As the response rate for rituximab was not an outcome included in the NMA, its response rate was naively incorporated from the NICE romiplostim submission.4 The sponsor used NMA-derived odds ratios (ORs) (refer to Table 11) and applied them to a placebo response rate to derive the response rates for TPO-RAs used in the model (Table 12).1 The OR-derived response rate for avatrombopag (73%) differed substantially from the rate of durable response for avatrombopag observed in Study 302 (34%).2
The ORs derived from the sponsor’s NMA for durable platelet response for avatrombopag compared with placebo and TPO-RAs (refer to Table 11) had serious imprecision, with extremely wide credible intervals that included the potential for no difference or for either active treatment to be favoured in each comparison. Despite this imprecision, avatrombopag was associated with significantly higher odds of a durable platelet response than placebo. However, the CADTH clinical review concluded that there is no evidence that avatrombopag is superior to other TPO-RAs. The clinical review also noted uncertainty in the sponsor’s NMA, resulting from known and unknown differences in baseline characteristics between trials. Additionally, according to the clinical expert consulted by CADTH for this review, based on the indirect evidence presented by the sponsor, no comparative efficacy conclusions could be made for avatrombopag and other TPO-RAs.
Immunosuppressants (equal proportions of azathioprine, cyclosporine, cyclophosphamide, mycophenolate, danazol, and dapsone) were included as comparators by the sponsor in a scenario analysis. Although these are appropriate comparators, the efficacy assumptions for these comparators were informed by the NICE eltrombopag (time to response) and romiplostim (response rate and duration of response) submissions.4,6 Immunosuppressant medications were not included in the sponsor’s submitted NMA; as such, the comparative efficacy versus avatrombopag is unknown.
Finally, there is also no direct or indirect long-term comparative evidence for avatrombopag versus TPO-RAs. All parameters in the model that are derived from Study 302 were based on the 26-week trial period; none were populated using the extension period. Overall, there is uncertainty in the long term comparative evidence for avatrombopag versus placebo, and the comparative evidence for avatrombopag versus other TPO-RAs is so imprecise that any conclusion is unknown. This adds uncertainty to the overall analysis because the majority of the total QALYs were accrued in the posttrial period.
Because the response rate for rituximab was excluded from the sponsor’s NMA and because the response rate for rituximab was naively derived, there is no direct or indirect evidence informing the comparative rates of durable response for avatrombopag compared to rituximab. As a result, rituximab has been removed as a comparator in the CADTH base case. Additionally, because there is also no direct or indirect evidence informing the comparative efficacy of avatrombopag compared to immunosuppressants, the cost-effectiveness of avatrombopag compared with rituximab and with immunosuppressants is unknown.
Because there is no direct evidence informing the comparative efficacy between TPO-RAs, and because the indirect evidence used by the sponsor to inform response rates was associated with significant imprecision and methodological uncertainty, all TPO-RAs were assumed to be equally efficacious in the CADTH base case.
The response rate used in the sponsor’s equal-efficacy assumption was the average of response rates for TPO-RAs derived using ORs from the sponsor’s NMA. Due to the uncertainty in the sponsor’s NMA, the resulting response rates calculated using the ORs from the NMA are highly uncertain. As such, CADTH used the response rate for avatrombopag observed in Study 302 as the TPO-RA response rate in the base-case analysis.
CADTH was unable to address limitations regarding the uncertainty of the long-term comparative evidence.
Dosing used in the sponsor’s base case does not reflect dose adjustments. Dosing for all TPO-RAs in the sponsor’s base case was based on the initial dose specified in their respective product monographs.1 Therefore, the sponsor assumed that there would be no dose adjustments for TPO-RAs in the model, which was deemed inappropriate. The draft product monograph for avatrombopag indicates that dose adjustments can be made based on platelet levels. These include increasing the dose if the platelet response remains less than 50 × 109/L after 2 weeks of treatment, decreasing the dose if platelet counts are between 150 × 109/L and 250 × 109/L, and stopping treatment if platelet counts are greater than 250 × 109/L or if platelets remain less than 50 × 109/L after 4 weeks of receiving avatrombopag at an increased dose of 40 mg once daily.12 Evidence from Study 302 indicated that patients received a range of avatrombopag doses, with approximately 34% of patients receiving a mean daily dose greater than 30 mg.2 This is not accounted for in the doses used to calculate treatment-acquisition costs in the model, and has likely resulted in an underestimation of avatrombopag costs. Feedback from the clinical expert consulted by CADTH for this review also indicated that a proportion of patients is likely to require dose adjustments, in accordance with product-monograph dosing, and it is not expected that all patients will remain on their initial TPO-RA dose.
In the model, the sponsor specified an alternative approach to dosing based on the mean dose observed in the respective clinical trials of TPO-RAs. For avatrombopag, the mean dose (22.52 mg)2 was assumed to be the same, regardless of the week, whereas dosing for eltrombopag and romiplostim, sourced from the NICE eltrombopag submission, was based on the mean dose observed each week.6 These mean daily doses for all TPO-RAs were higher than their product-monograph doses. Further, these doses were deemed by the clinical expert consulted for this review to be more reflective of doses that would be received in clinical practice. Although using the mean daily dose observed in the trial does not directly demonstrate the distribution of patients receiving a given dose of avatrombopag over time, it provides a more accurate reflection of actual drug exposure that is associated with the observed response rates in the trial.
In the CADTH reanalysis, dosing for TPO-RAs was based on trial dosing.
The use of surrogate outcomes. The health states in the model are defined by blood platelet counts, with responders having platelet counts of at least 50 × 109/L, as defined by Study 302. In the model, nonresponders were those with platelet counts of less than 50 × 109/L. The sponsor assumed that the risk of bleeding would be higher in states with platelet counts of less than 50 × 109/L (i.e., in nonresponder health states).13 In the model, responders and nonresponders have differential risks of bleeds (which in turn results in differential mortality risks costs, and quality of life); use of rescue therapy (which impacts costs); use of concomitant medications (which impact costs); and health-state utility values. Therefore, platelet counts serve as a surrogate outcome for survival, HRQoL, and total costs. The clinical expert consulted by CADTH indicated that platelet count may be an appropriate proxy for bleeding risk, but the threshold at which platelet count corresponds to bleeding risk is uncertain and nonlinear. This introduces additional uncertainty into the sponsors model in the way effective disease control translates into estimates of life-years and QALYs.
In addition, although the sponsor incorporated minor bleed rates based on platelet count from Study 302, CADTH was unable to validate the proportion of patients experiencing minor bleeds by response, as this was not presented in the sponsor’s Clinical Study Report.2 Rates of inpatient and outpatient bleeds by platelet response were sourced from the NICE eltrombopag submission (Table 10); however, CADTH was also unable to validate these bleed rates from the NICE report.6 Taken together, there is uncertainty regarding the relationship between bleeding rates and platelet counts.
CADTH was unable to address this limitation in its reanalysis.
The health-state utility values used are uncertain and lack face validity. The health-state utility values in the model were based on Canadian utility norms, with utility decrements subtracted to measure the impact of response, bleeding, and splenectomy.1,9 These decrements were derived using EQ-5D data from Study 302.2 This included a utility decrement for not having a splenectomy of 0.0676, meaning that patients who had undergone splenectomy had a higher quality of life than patients who had not. According to the clinical expert consulted by CADTH for this review, this is the opposite of what is clinically expected; patients who have undergone splenectomy and are seeking ITP treatment are expected to have a worse quality of life than patients who have not, based on the lifelong risk of being immunocompromised after splenectomy.
Patients in nonresponse health states had a utility decrement of 0.0407, associated with being a nonresponder, subtracted from all nonresponder health states, including those with bleeds. According to the clinical expert consulted for this review, responders may experience an improvement in their quality of life associated with reduced anxiety related to the anticipation of a bleed; however, the magnitude of the quality-of-life benefit for responders compared to nonresponders was likely overestimated and is not expected to persist in states in which responders experience bleeds. That is, when bleeding, there will be a negligible difference in quality of life for patients with platelet counts less than or greater than 50 × 109/L. The clinical expert also indicated that the utility decrement estimated by the sponsor using Study 302 EQ-5D data for minor and outpatient bleeds was likely overestimated.
Due to the low number of inpatient bleeds in Study 302, the sponsor was unable to estimate utilities using EQ-5D study data; instead, inpatient bleed utilities were based on the NICE eltrombopag submission.6 According to the clinical expert consulted for this review, these utility values likely overestimated the quality-of-life impact of these bleeds. For example, the utility for ICH (0.038) closely approximated death, which was deemed to be inappropriate because ICHs can vary in type and severity. For example, an ICH can include bleed types like subdural bleeds, which, although serious, rarely result in a quality of life that is close to death.
In CADTH base case, the utility decrement for nonresponders was removed from all bleeding health states (i.e., utility values for the minor and outpatient bleed health states were equal for responders and nonresponders).
Despite the uncertainty and lack of clinical plausibility for some of the other utility estimates in the model, the efficacy of TPO-RAs in the model were assumed to be equal, so the adjustment of health-state utility values is unlikely to influence model results among TPO-RAs. However, CADTH noted the added uncertainty (e.g., the magnitude of health-state utility values) in estimates with a nonactive comparator, such as watch and rescue.
The assumptions informing response are uncertain. In the sponsor’s model, time to response for all TPO-RAs was assumed to be 24 weeks, and for rituximab was 8 weeks.1 Time to response was not an outcome in Study 302, nor the sponsor’s NMA. According to the clinical expert consulted by CADTH for this review, clinicians would be unlikely to continue treating someone for more than 3 months without seeing a response because at that point, patients should have reached the maximum dose.
The duration of response (the time patients spend in the treatment and response health state) was assumed to be equal to the average time on treatment for eltrombopag,5 and was assumed to be equal for all TPO-RAs. CADTH was unable to validate the sponsor’s estimate of 109 cycles (approximately 8.4 years), which was derived from the study by Lee et al. (2013).5 The duration of response in Study 302 was 4.4 weeks for avatrombopag, and the median time on treatment was reported as the median exposure time and was 26 weeks for avatrombopag during the core phase period.2 For the combined core and extension phases, the median duration of exposure to avatrombopag was 43.9 weeks.2 In the romiplostim extension study, treated adults maintained platelet counts with dose adjustments for up to 277 weeks.2 The eltrombopag extension study demonstrated a median duration of exposure of 121 weeks, with a range of up to 285 weeks.2
According to the clinical expert consulted by CADTH for this review, time on treatment may not be an adequate proxy for response duration, as some patients could discontinue treatment and maintain their response. It would have been more appropriate to use a survival curve to model duration of response, which would account for the proportion of responders who maintain their response over time. Kaplan-Meier curves for duration of response and treatment duration for avatrombopag were not available from Study 302.
There is no direct or indirect evidence informing time to response or duration of response among TPO-RAs; therefore, CADTH maintained the sponsor’s assumption that these would be equal across treatments. Because these are considered equal, adjustment of time to response or duration of response is unlikely to influence model results versus active comparators.
The sequencing of treatments in the model may not reflect clinical practice. In the sponsor’s model, patients can only receive 1 active comparator (consisting of the TPO-RAs or rituximab) across the model time horizon. If they do not respond to initial therapy with TPO-RA and/or rituximab or they lose their response, they can receive another active treatment with immunosuppressant medications. According to the clinical expert consulted for this review, it is appropriate to not provide a second TPO-RA to patients who have lost their response or to patients who did not initially respond. However, if patients discontinued a TPO-RA due to intolerance, the expert noted that it may be appropriate to trial a different TPO-RA for response. Additionally, the expert noted that if someone maintained a response after the tapering of a TPO-RA and then had platelet levels decline while off treatment, it would be appropriate to reinitiate that or a different TPO-RA. Neither of these clinical scenarios was modelled, and the direction and magnitude of the effect of this is unknown. In addition, the expert indicated that assumptions around only receiving 1 active comparator do not apply to clinical practice, as patients who initially received rituximab for their second-line treatment would be treated with a TPO-RA, if available.
CADTH was unable to address potential re-treatment with TPO-RAs due to inflexibilities in the sponsor’s model structure. Because rituximab was excluded as a comparator from the CADTH base case due to a lack of comparative evidence, its use after or before a TPO-RA does not influence the model results.
The assumption that bleeding rates will double after 4 lines of treatment is unsubstantiated. The sponsor’s base case assumed that a doubling of the risk of inpatient bleeding will occur among patients in the no-treatment state after they have received 4 lines of therapy. According to the clinical expert consulted for this review, patients with refractory disease are at higher risk of bleeding, but the magnitude of that risk is uncertain.
CADTH was unable to address this limitation in its reanalysis. This assumption will not impact model results versus active comparators, because bleeding rates are equal among TPO-RAs.
Additional limitations were identified but were not considered to be key limitations. These limitations are outlined subsequently.
Parameter uncertainty does not accurately reflect uncertainty around the ICER. Parameter uncertainty was not incorporated per CADTH guidelines,14 as the sponsor did not source uncertainty estimates for most of the parameters; instead, the majority of model parameters used an arbitrary standard error set at 10% of the mean. This is also the case for model inputs derived from the sponsor’s clinical trial, such as bleed rates and the use of concomitant ITP medications. Parameter uncertainty was inappropriately included for some parameters, such as those based on an assumption (such as time to response) and some unit costs (e.g., costs sourced from the Schedule of Benefits). Given this arbitrary incorporation of parameter uncertainty, it is unclear whether cost-effectiveness outcomes are underestimated or overestimated; however, improper incorporation of uncertainty is likely to bias the cost-effectiveness outcomes.
CADTH corrected the sponsor base case by setting all parameters as fixed in a probabilistic analysis that used a standard error estimate of 10%.
AEs were inappropriately modelled. The sponsor’s model included 2 types of AEs — serious and other — with rates taken from the romiplostim NICE submission.4 AE rates were assumed to be equal for all TPO-RAs. The approach of considering serious and other AEs rather than the specific AEs experienced by patients was deemed to be inappropriate, as labelling an AE as serious does not indicate the impact of that AE has on costs and outcomes. Consequently, the disutility for AEs was an assumed value, taken from the NICE romiplostim submission.4 This assumed value is even more uncertain, because without explanation, a larger disutility was applied for serious AEs in patients receiving immunosuppressant medications than in those receiving TPO-RAs and rituximab. CADTH determined that a more appropriate approach would be to source AE rates from the NMA or clinical trials, and to source a disutility specific to the given AE (e.g., anemia).
CADTH did not address this limitation in its reanalyses. Because the efficacy of TPO-RAs in the model was assumed to be equal, the adjustment of AE event rates and disutilities will not influence model results versus active comparators.
Some costs of bleeding management may have been overestimated. The sponsor assumed that all patients who experience a light bleed would require both an emergency department (ED) visit and outpatient care. According to the clinical expert consulted by CADTH for this review, patients with light bleeds would not require ED admission or outpatient care. Rather, 80% of patients with light bleeds can be managed with an outpatient visit and 20% would be treated in the ED. The sponsor also assumed that 60% of patients with light bleeds and 100% of patients with serious and life-threatening bleeds would require an MRI, which the clinical expert noted to be an overestimate.
CADTH adjusted the distribution of patients with light bleeds who require ED (20%) and outpatient (80%) care. CADTH also removed MRIs for light bleeds and assumed 15% of those with outpatient and life-threatening bleeds would need an MRI.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
Watch and rescue was assumed to be equivalent to the placebo arm of Study 302. | Uncertain. Study 302 compared avatrombopag to placebo, not watch and rescue.2 However, patients in both arms were allowed to receive concomitant ITP treatments.2 According to the CADTH Clinical Review Report, patients entered Study 302 on various levels of concomitant background therapies. |
The use of rescue therapies is primarily for bleeding events. | Appropriate, according to expert consulted by CADTH for this review. |
Concomitant medications may be reduced or discontinued with a treatment response. | Appropriate, according to expert consulted by CADTH for this review. |
The type of concomitant medications for all comparator arms was assumed to be the same as those observed at baseline in Study 302.2 | Likely appropriate; unlikely to influence model results. |
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
CADTH reanalyses addressed several limitations of the economic model, summarized in Table 5. The CADTH base case was derived by making changes in model parameter values and assumptions, in consultation with clinical experts. CADTH was unable to address limitations regarding the lack of direct or indirect comparative evidence for avatrombopag compared with rituximab or immunosuppressant medications; the uncertainty related to using platelet count as a surrogate for bleeding risk; the incorporation of trials of TPO-RAs; re-treatment with the initial TPO-RA if response is lost when the patient is off treatment or the use of a TPO-RA after the patient fails to respond or loses the response to rituximab; the incorporation of AEs based on clinical event type instead of severity; and the incorporation of treatment-specific AE rates.
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Correctionsa to sponsor’s base case | ||
1. PSA includes parameters for which uncertainty is explored using a 10% standard error | Yes | No |
2. Comparator unit costsb | Eltrombopag 25 mg = $65.0000 Romiplostim 250 mcg = $1,021.7900 Cyclosporine 25 mg = $0.9952 Danazol 100 mg = $1.5156 Dapsone 100 mg = $1.4061 Vinblastine price per mg = $1.2500 | Eltrombopag 25 mg = $55.2500 Romiplostim 250 mcg = $1,033.0200c Cyclosporine 25 mg = $0.7870 Danazol 100 mg = $1.5323 Dapsone 100 mg = $0.7031 Vinblastine price per mg = $5.2430d |
Changes to derive the CADTH base case | ||
1. Base-case comparators | Rituximab Watch and rescue Eltrombopag Romiplostim | Watch and rescue Eltrombopag Romiplostim |
2a. Source of efficacy for TPO-RA response rates | Differential response rates; NMA derived | Equal efficacy among TPO-RAs assumed |
2b. Equal efficacy response rate | 52% | 36% |
3. Source for drug dosing | Product monograph | Median dose from respective clinical trials |
4. Utility decrement for nonresponse present in bleeding health states | Yes | No |
5. Resource use for bleed management | Light bleeds visits:
| Light bleeds visits:
|
MRI rates for:
| MRI rates for:
| |
CADTH base case | — | 1 + 2 + 3 + 4 + 5 |
ED = emergency department; NMA = network meta-analysis; PSA = probabilistic sensitivity analysis; TPO-RA = thrombopoietin receptor agonist.
aCorrections are minor errors (e.g., transcription errors between report and model, misapplication of distributions, or standard errors in probabilistic analyses) that are not identified as limitations.
bComparator unit costs were updated to reflect August 2023 pricing from the Ontario Drug Benefit Formulary, unless otherwise indicated.15
cSource: Ontario Drug Benefit Exceptional Access Program price (accessed August 2023).16
dSource: IQVIA Delta PA pricing (accessed August 2023).17
The results of CADTH’s stepped analysis are presented in Table 13. CADTH’s base-case reanalysis demonstrated that all TPO-RAs yielded an equal number of QALYs (Table 13). Because all TPO-RA treatments were equally efficacious, differences in total costs were solely driven by differences in treatment-acquisition costs (Table 6). Avatrombopag is associated with $7,462 in incremental treatment-acquisition costs, compared with eltrombopag, leading it to have higher overall total costs than eltrombopag. Romiplostim is associated with $148,287 in incremental treatment-acquisition costs, compared with avatrombopag, leading avatrombopag to have lower overall total costs than romiplostim.
Table 6: Disaggregated Summary of Discounted Costs in CADTH’s Economic Evaluation
Cost category | Avatrombopag | Eltrombopag | Romiplostim | Incremental avatrombopag vs. eltrombopag | Incremental avatrombopag vs. romiplostim |
|---|---|---|---|---|---|
Treatment acquisition | $308,799 | $301,337 | $457,086 | $–7,462 | $148,287 |
Administration | $15,258 | $15,258 | $15,258 | 0 | 0 |
Monitoring | $27,207 | $27,207 | $27,207 | 0 | 0 |
Bleeding | $477,332 | $477,332 | $477,332 | 0 | 0 |
Total | $828,596 | $821,134 | $976,883 | $–7,462 | $148,287 |
Watch and rescue (assumed to be equal to the placebo arm of Study 302) is the only comparator for which there is direct comparative evidence against avatrombopag. Compared with watch and rescue, avatrombopag yielded 0.90 greater QALYs and cost $88,662 more, leading to an ICER of $98,150 per QALY gained (Table 13).
Scenario Analysis Results
Given the uncertainty in estimates derived from the sponsor’s NMA, equal efficacy was assumed between avatrombopag and other TPO-RAs in the CADTH base case; there is limited evidence to support a price premium for avatrombopag. As such, in jurisdictions that currently reimburse other TPO-RAs for ITP, avatrombopag should be priced such that drug costs are similar to other TPO-RAs. Because TPO-RAs may not be reimbursed in all jurisdictions, CADTH undertook price-reduction analyses and found that a 32% reduction in price would be required for avatrombopag to be cost-effective compared to watch and rescue.
Issues for Consideration
Patients were excluded from Study 302 if they had known secondary ITP, so the cost-effectiveness of avatrombopag for the treatment of secondary chronic ITP is unknown.
The CADTH clinical review concluded that subgroup analyses were not able to provide insight into which patient groups (e.g., based on previous lines of therapy) are most likely to respond to treatment with avatrombopag. Therefore, the cost-effectiveness of avatrombopag compared to other ITP treatments in different lines of therapy (e.g., second-line versus third-line) is unknown. The proposed Health Canada indication for avatrombopag includes its use as a second-line therapy. Drug program input indicated that due to restricted access, other TPO-RAs are currently funded as third-line treatments. If avatrombopag is recommended for use in line with the proposed indication, avatrombopag could be used before other TPO-RAs, making it the dominant TPO-RA. Based on clinical expert feedback, because avatrombopag demonstrated no evidence of a benefit over other TPO-RAs, the reimbursement of avatrombopag should be similar to that of other TPO-RAs.
Overall Conclusions
The CADTH clinical review found that based on Study 302, avatrombopag led to an improvement in platelet count response compared to placebo among patients with pretreated, primary, chronic ITP. There were limited or no data on patient-important outcomes, such as bleeding rates, the use of concomitant ITP medications, the need for rescue therapy, symptoms, and HRQoL. These outcomes were exploratory in Study 302, so the impact of avatrombopag on these outcomes remains unclear. The sponsor’s submitted NMA found that there was no significant difference in the proportion of patients with durable platelet response between avatrombopag and TPO-RAs. The NMA suggested that avatrombopag may have favourable efficacy compared to other TPO-RAs and rituximab in terms of the incidence of bleeding events. The CADTH clinical review noted that, due to important limitations of the sponsor’s NMA, estimates derived from the NMA are very uncertain and the formulation of definitive conclusions around the comparative efficacy and safety of avatrombopag versus other ITP treatments is infeasible.
CADTH undertook reanalyses to address limitations related to the lack of comparative efficacy data for the response rate for avatrombopag versus rituximab; the uncertain indirect comparative efficacy for avatrombopag and other TPO-RAs; adjusting the response rate for TPO-RAs to reflect the response rate for avatrombopag observed in Study 302; the incorporation of dose adjustments for TPO-RAs; the removal of a utility decrement associated with being a nonresponder from health states with bleeds; and the adjustment to health care resource use for bleed management to better reflect clinical practice in Canada. In CADTH’s base-case reanalysis, all TPO-RAs, including avatrombopag, yielded an equal number of QALYs. All differences in total costs were derived from treatment-acquisition costs; costs for all other health care resource use were equal among TPO-RAs. Avatrombopag was associated with higher treatment-acquisition costs compared with eltrombopag ($7,462), but lower treatment-acquisition costs compared with romiplostim ($148,287). Given that the most relevant comparators for avatrombopag are other TPO-RAs, and given the uncertainty about clinical estimates from the sponsor’s NMA, there is limited clinical evidence to support a price premium for avatrombopag compared with other TPO-RAs. Additionally, as there were no comparative efficacy data for the response rate of avatrombopag versus rituximab, there is no clinical evidence to support a price premium for avatrombopag over rituximab.
Watch and rescue (assumed to be equal to the placebo arm of Study 302) is the only comparator for which there is direct comparative evidence against avatrombopag. For this comparison, the ICER for avatrombopag compared with watch and rescue was $98,150 per QALY gained. To achieve an ICER of $50,000 per QALY gained compared with watch and rescue, a price reduction of at least 32% is required.
CADTH was unable to address limitations with the sponsor’s model regarding the lack of direct or indirect evidence informing response rate for avatrombopag compared to other relevant comparators (e.g., rituximab and immunosuppressants), the use of surrogate outcomes (platelet counts) to model ITP, the uncertainty of the sponsor’s health-state utility values, response assumptions, or the treatment sequencing not reflecting clinical practice in Canada.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Doptelet (avatrombopag), 20 mg oral tablets. Oakville (ON): Sobi Canada Inc; 2022 Feb 3.
2.Clinical Study Report: E5501-G000-302. A phase 3, multicenter, randomized, double-blind, placebo-controlled, parallel-group trial with an open-label extension phase to evaluate the efficacy and safety of oral E5501 plus standard care for the treatment of thrombocytopenia in adults with chronic immune thrombocytopenia (idiopathic thrombocytopenic purpura)[internal sponsor's report]. Woodcliff Lake (NJ): Eisai Inc; 2016 Jan.
3.Network meta-analysis of efficacy and safety of avatrombopag versus comparators in adult patients with chronic immune thrombocytopenia who have had an insufficient response to a previous treatment [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Doptelet (avatrombopag), 20 mg oral tablets. Oakville (ON): Sobi Canada Inc; 2022 Feb 3.
4.Single technology appraisaL (STA): romiplostim for the treatment of chronic immune or idiopathic thrombocytopenic purpura (ITP). London (GB): National Institute for Health and Care Excellence; 2008.
5.Lee D, Thornton P, Hirst A, Kutikova L, Deuson R, Brereton N. Cost effectiveness of romiplostim for the treatment of chronic immune thrombocytopenia in Ireland. Appl Health Econ Health Policy. 2013;11(5):457-469. PubMed
6.Single technology appraisal (STA): eltrombopag for adult patients with chronic immune thrombocytopenic purpura (cITP). London (GB): National Institute for Health and Care Excellence; 2012: https://www.nice.org.uk/guidance/ta293/documents/thrombocytopenic-purpura-eltrombopag-rev-ta205-glaxosmithkline4. Accessed 2022 Mar 25.
7.Danese MD, Lindquist K, Gleeson M, Deuson R, Mikhael J. Cost and mortality associated with hospitalizations in patients with immune thrombocytopenic purpura. Am J Hematol. 2009;84(10):631-635. PubMed
8.Schedule of benefits for physician services under the Health Insurance Act: effective October 1, 2021. Toronto (ON): Ontario Ministry of Health; 2021: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf. Accessed 2022 Mar 25.
9.Guertin JR, Feeny D, Tarride J-E. Age-and sex-specific Canadian utility norms, based on the 2013–2014 Canadian Community Health Survey. CMAJ. 2018;190(6):E155-E161. PubMed
10.Szende A, Brazier J, Schaefer C, Deuson R, Isitt JJ, Vyas P. Measurement of utility values in the UK for health states related to immune thrombocytopenic purpura. Curr Med Res Opin. 2010;26(8):1893-1903. PubMed
11.Leontiadis G, Sreedharan A, Dorward S, et al. Systematic reviews of the clinical effectiveness and cost-effectiveness of proton pump inhibitors in acute upper gastrointestinal bleeding. Health Technol Assess. 2007;11(51):iii-iv, 1. PubMed
12.Doptelet (avatrombopag tablets): 20 mg avatrombopag (as maleate), oral [product monograph]. Stockholm SE): Swedish Orphan Biovitrum AB (publ); 2023 Nov 3.
13.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Doptelet (avatrombopag), 20 mg oral tablets. Oakville (ON): Sobi Canada Inc; 2022 Feb 3.
14.Guidelines for the economic evaluation of health technologies: Canada. 4th ed. Ottawa (ON): CADTH; 2017: https://www.cadth.ca/guidelines-economic-evaluation-health-technologies-canada-4th-edition. Accessed 2022 Apr 20.
15.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2021; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2023 Aug 31.
16.Exceptional Access Program (EAP). Toronto (ON): Ontario Ministry of Health; Ontario Ministry of Long-Term Care; 2021: http://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx. Accessed 2023 Aug 31.
17.DeltaPA. Ottawa (ON): IQVIA; 2021: https://www.iqvia.com/. Accessed 2023 Aug 31.
18.Provan D, Arnold DM, Bussel JB, et al. Updated international consensus report on the investigation and management of primary immune thrombocytopenia. Blood Adv. 2019;3(22):3780-3817. PubMed
19.PharmaStat. Ottawa (ON): IQVIA; 2021: https://www.iqvia.com/. Accessed 2022 Apr 21.
20.Platelet Disorder Support Association. ITP in adults. 2020: https://www.pdsa.org/adults.html. Accessed 2022 Apr 21.
21.Nazaryan H, Liu Y, Sirotich E, Duncan J, Nazy I, Arnold D. Second-line therapy for immune thrombocytopenia: real-world experience in Canada. Canadian Journal of General Internal Medicine. 2020;15(4):28-35.
22.Kistangari G, McCrae KR. Immune thrombocytopenia. Hematol Oncol Clin North Am. 2013;27(3):495-520. PubMed
Appendix 1: Cost Comparison Table
Table 7: CADTH Cost Comparison Table for Adult Patients With Chronic ITP Who Have Had an Insufficient Response to a Previous Treatment
Treatment | Strength / concentration | Form | Price | Recommended dosage | Daily cost | Annual cost |
|---|---|---|---|---|---|---|
Avatrombopag (Doptelet) | 20 mg | Tablet | 115.0000a | 20 mg once daily | 115.00 | 41,975 |
TPO-RAs | ||||||
Eltrombopag (Revolade, generics) | 25 mg 50 mg | Tablet | 55.2500b 110.5000b | Initially 50 mg once daily (25 mg in Asian patients). After 2 weeks, increase dose by 25 mg if platelet count < 50 × 109/L, up to a maximum of 75 mg daily. Reduce dose when platelet count is above 200 × 109/L | 110.50 initially (55.25 for 25 mg initial dose), up to 165.75 | 40,333 initially (20,166 for 25 mg initial dose), up to 60,499c |
NPLATE (Romiplostim) | 250 mcg 500 mcg | Vial of lyophilized powder for solution | 1,033.0200b 2,066.0700b | 1 mcg per kgd once weekly, adjusting by increments of 1 mcg/kg until platelet count ≥ 50 × 109/L. Do not exceed 10 mcg/kg. | 147.57 initially, up to 442.73 | 53,865 initially, up to 161,595 |
CD20 Inhibitor (off-label) | ||||||
Rituximab (Truxima, Riximyo, Ruxience) | 10 mg/mL | 10 mL 50 mL Single use vials | 297.0000 1,485.0000 | 375 mg/m2 IV once per week for 4 weekse,f | NA | Cost per four-week course: 8,316 |
100 mg IV once per week for 4 weekse | NA | Cost per four-week course: 1,188 | ||||
Immunosuppressants (off-label) | ||||||
Azathioprine (generic) | 50 mg | Tablet | 0.2405 | 1 to 2 mg/kg daily, maximum 150 mg/dayd,e | 0.48 to 0.72 | 176 to 263 |
Cyclosporin (Neoral, generic) | 10 mg 25 mg 50 mg 100 mg | Capsule | 0.7115 0.7870 1.5350 3.0720 | 5 mg/kg/day for 6 days, then 2.5 to 3 mg/kg/dayd,e | Initial: 12.29 Then: 6.14 to 7.68 | 2,279 to 2,830 |
100 mg/mL | Oral solution | 5.7410 | Initial: 22.96 Then: 11.48 to 13.78 | 4,260 to 5,084 | ||
Cyclophosphamide (Procytox) | 25 mg 50 mg | Tablet | 0.3545 0.4773 | 1 to 2 mg/kg daily for at least 16 weeksd,e | 0.83 to 1.43 | 304 to 523 |
200 mg 500 mg 1,000 mg 2000 mg | Vial for injection 20mg/mL | 74.2300g 101.7100g 184.3600g 339.2000g | 0.3 to 1 g/m2 IV every 2 to 4 weeks for 1 to 3 dosese,f | NA | Per course: 176 to 1,018 | |
Danazol (Cyclomen) | 50 mg 100 mg 200 mg | Capsule | 1.0325 1.5323 2.4486 | 200 mg 2 to 4 times dailye | 4.90 to 9.79 | 1,787 to 3,575 |
Dapsone (generic) | 100 mg | Tablet | 0.7031 | 100 mg per daye | 0.70 | 257 |
Mycophenolate mofetil (generic) | 250 mg 500 mg | Capsule | 0.3712 0.7423 | 1.5 to 2 g/day for at least 12 weekse | 2.23 to 2.97 | 813 to 1,084 |
Vincristine (generic) | 1 mg/mL | Solution for injection | 30.6000 | 6 mg total at 1 to 2 mg per weekly infusione | NA | Per course: 184 |
Vinblastine (generic) | 10 mg/10 mL | Solution for injection | 52.4300g | 30 mg total at 10 mg per weekly infusione | NA | Per course: 157 |
The comparators presented in the above have been deemed to be appropriate based on feedback from clinical expert. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Note: All prices are from the Ontario Drug Benefit Formulary (accessed August 2023),15 unless otherwise indicated, and do not include dispensing fees.
aSponsor’s submitted price1
bOntario Drug Benefit Exceptional Access Program price (accessed August 2023)16
cMaximum annual cost would only be reached if patients never reach a platelet count over 200 × 109/L.
dWeight used is from Study 302 (82.97 kg)2
eOff-label use, dosing from the 2019 Updated international consensus on investigation and management of primary immune thrombocytopenia, including supplemental information.18
fBody surface area used was from Study 302 (1.94m2)2
gIQVIA Delta PA pricing (Accessed August 2023)17
Note this table has not been copy-edited.
Appendix 2: Submission Quality
Note this table was not copy-edited.
Description | Yes/No | Commentsa |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | Yes | No comment. |
Model has been adequately programmed and has sufficient face validity | No | Refer to the limitation “Use of surrogate outcomes.” |
Model structure is adequate for decision problem | No | Refer to the limitation: “Sequencing of treatments in the model may not reflect clinical practice.” |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | Refer to the limitation: “Parameter uncertainty does not accurately reflect uncertainty around the ICER.” |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | Refer to the limitation “Parameter uncertainty does not accurately reflect uncertainty around the ICER.” |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note this figure and tables were not copy-edited.
N t = no treatment; T NR = treatment, no response; T R = treatment, response.
Source: Sponsor’s pharmacoeconomic submission.1
Detailed Results of the Sponsor’s Base Case
Table 9: Bleeding Rates by Platelet Level and Type
Bleeding type | Platelets ≥ 50x109/L | Platelets < 50x109/L | Source |
|---|---|---|---|
Minor bleed | 10.0% | 17.1% | Patient-level data from Study 3022 |
Outpatient bleed | 7.1% | 45.5% | Eltrombopag NICE submission6 |
Inpatient bleed | 0.0% | 4.3% | Eltrombopag NICE submission6 |
Source: Sponsor’s pharmacoeconomic submission.1
Table 10: Types and Frequencies of Inpatient Bleeds
Bleed Type | Platelets ≥ 50x109/L | Platelets < 50x109/L |
|---|---|---|
Intracranial hemorrhage | 0% | 19% |
Gastrointestinal | 29% | 19% |
Other bleeds | 71% | 63% |
Source: Eltrombopag NICE submission.6
Table 11: Odds Ratios for Avatrombopag Versus Other Treatments in the Sponsor-Submitted NMA
Avatrombopag vs. | Mean odds ratio | Lower confidence interval | Upper confidence interval |
|---|---|---|---|
Placebo | 102.80 | 3.87 | 2,796,448.59 |
Eltrombopag | 7.06 | 0.21 | 185,017.47 |
Romiplostim | 2.16 | 0.03 | 69,340.75 |
Source: sponsor-submitted NMA.3
Table 12: Sponsor Response Rates
Drug | Response rate |
|---|---|
Avatrombopaga | 73% |
Eltrombopaga | 27% |
Romiplostima | 55% |
Rituximabb | 58% |
Watch and rescuec | 0% |
aResponse rate derived by applying the odds ratio for placebo vs. active comparator to the placebo response rate from the sponsor’s NMA
bResponse rate naively derived from NICE romiplostim submission.4
cPlacebo response rate in Study 302 was 0%.
Source: Sponsor’s pharmacoeconomic submission.1
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note this appendix has not been copy-edited.
Table 13: Summary of the Stepped Analysis of the CADTH Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
Sponsor’s base case (probabilistic) | Rituximab | 846,303 | 13.29 | Ref. |
Watch and rescue | 855,274 | 12.89 | Dominated | |
Eltrombopag | 923,961 | 13.64 | Ext. Dominated | |
Avatrombopag | 986,686 | 14.96 | 84,217 | |
Romiplostim | 1,129,871 | 14.41 | Dominated | |
Sponsor’s base case (deterministic) | Rituximab | 846,766 | 13.31 | Ref. |
Watch and rescue | 855,860 | 12.90 | Dominated | |
Eltrombopag | 924,996 | 13.67 | Ext. Dominated | |
Avatrombopag | 983,000 | 14.90 | 85,431 | |
Romiplostim | 1,011,653 | 14.42 | Dominated | |
Sponsor’s corrected base case (deterministic) | Rituximab | 846,259 | 13.31 | Ref. |
Watch and rescue | 855,365 | 12.90 | Dominated | |
Eltrombopag | 907,312 | 13.67 | Ext. Dominated | |
Avatrombopag | 982,470 | 14.90 | 85,417 | |
Romiplostim | 1,013,701 | 14.42 | Dominated | |
CADTH reanalysis 1: Removing rituximab as a comparator | Watch and rescue | 855,365 | 12.90 | Ref. |
Eltrombopag | 907,311 | 13.67 | Ext. Dominated by avatrombopag | |
Avatrombopag | 982,468 | 14.90 | 63,457 | |
Romiplostim | 1,013,699 | 14.42 | Dominated by avatrombopag | |
CADTH reanalysis 1 + 2a: Efficacy response rates equal among TPO-RAs | Watch and rescue | 855,365 | 12.90 | Ref. |
Eltrombopag | 942,593 | 14.33 | 61,019 | |
Avatrombopag | 949,422 | 14.33 | Dominated by eltrombopag | |
Romiplostim | 1,005,473 | 14.33 | Dominated by eltrombopag | |
CADTH reanalysis 1 + 2a + 2b: Efficacy response rates equal among TPO-RAs; trial derived response rate | Watch and rescue | 855,365 | 12.90 | Ref. |
Eltrombopag | 917,149 | 13.85 | 64,890 | |
Avatrombopag | 921,911 | 13.85 | Dominated by eltrombopag | |
Romiplostim | 961,088 | 13.85 | Dominated by eltrombopag | |
CADTH reanalysis 1 + 3: Dosing | Watch and rescue | 855,365 | 12.90 | Ref. |
Eltrombopag | 917,827 | 13.67 | Ext. Dominated by avatrombopag | |
Avatrombopag | 1,012,455 | 14.90 | 78,427 | |
Romiplostim | 1,250,215 | 14.42 | Dominated by avatrombopag | |
CADTH reanalysis 1 + 4: Removing utility decrement for nonresponse from bleeding health states | Watch and rescue | 855,365 | 13.39 | Ref. |
Eltrombopag | 907,312 | 14.13 | Ext. Dominated by avatrombopag | |
Avatrombopag | 982,470 | 15.32 | 66,016 by avatrombopag | |
Romiplostim | 1,013,701 | 14.85 | Dominated | |
CADTH reanalysis 1 + 5: Resource use for bleed management | Watch and rescue | 731,385 | 12.90 | Ref. |
Eltrombopag | 789,267 | 13.67 | Ext. Dominated by avatrombopag | |
Avatrombopag | 873,518 | 14.90 | 70,961 | |
Romiplostim | 901,179 | 14.42 | Dominated by avatrombopag | |
CADTH base case 1 + 2 + 3 + 4 + 5 (deterministic) | Watch and rescue | 731,385 | 13.39 | Ref. |
Eltrombopag | 813,099 | 14.31 | 89,310 | |
Avatrombopag | 820,559 | 14.31 | Dominated by eltrombopag | |
Romiplostim | 1,000,566 | 14.31 | Dominated by eltrombopag | |
CADTH base case 1 + 2 + 3 + 4 + 5 (probabilistic) | Watch and rescue | 739,934 | 13.36 | Ref. |
Eltrombopag | 821,134 | 14.26 | 89,889 | |
Avatrombopag | 828,596 | 14.26 | Dominated by eltrombopag | |
Romiplostim | 976,883 | 14.26 | Dominated by eltrombopag |
ICER = incremental cost-effectiveness ratio; QALYs = quality-adjusted life-years; TPO-RAs = thrombopoietin receptor agonists.
Table 14: Disaggregated Summary of CADTH’s Economic Evaluation Results
Treatment | Component | Value | Incremental (vs. reference) | Incremental (sequential) |
|---|---|---|---|---|
Discounted QALYs | ||||
Watch and rescue | Health-state utility | 13.38 | NA | NA |
Disutilities | −0.03 | NA | NA | |
Total | 13.36 | NA | NA | |
Eltrombopag | Health-state utility | 14.39 | 1.01 | NA |
Disutilities | −0.13 | 0.11 | NA | |
Total | 14.26 | 0.90 | NA | |
Avatrombopag | Health-state utility | 14.39 | 1.01 | 0.00 |
Disutilities | −0.13 | 0.11 | 0.00 | |
Total | 14.26 | 0.90 | 0.00 | |
Romiplostim | Health-state utility | 14.39 | 1.01 | 0.00 |
Disutilities | −0.13 | 0.11 | 0.00 | |
Total | 14.26 | 0.90 | 0.00 | |
Discounted costs ($) | ||||
Watch and rescue | Treatment acquisition | 181,689 | NA | NA |
Administration | 16,184 | NA | NA | |
Monitoring | 25,755 | NA | NA | |
Bleeding | 516,306 | NA | NA | |
Total | 739,934 | NA | NA | |
Eltrombopag | Treatment acquisition | 301,337 | 119,648 | NA |
Administration | 15,258 | −926 | NA | |
Monitoring | 27,207 | 1,453 | NA | |
Bleeding | 477,332 | −38,974 | NA | |
Total | 821,134 | 81,200 | NA | |
Avatrombopag | Treatment acquisition | 308,799 | 127,110 | 7,462 |
Administration | 15,258 | −926 | 0.00 | |
Monitoring | 27,207 | 1,453 | 0.00 | |
Bleeding | 477,332 | −38,974 | 0.00 | |
Total | 828,596 | 88,662 | 7,462 | |
Romiplostim | Treatment acquisition | 457,086 | 275,397 | 148,287 |
Administration | 15,258 | −926 | 0.00 | |
Monitoring | 27,207 | 1,453 | 0.00 | |
Bleeding | 477,332 | −38,974 | 0.00 | |
Total | 976,883 | 236,949 | 148,287 | |
LY = life-year; NA = not applicable; QALY = quality-adjusted life-year; vs. = versus.
Table 15: CADTH’s Economic Evaluation Results
Drug | ICER vs. reference ($) | Sequential ICER ($/QALY) |
|---|---|---|
Watch and rescue | Ref. | Ref. |
Eltrombopag | 89,889 | 89,889 |
Avatrombopag | 98,150 | Dominated by eltrombopag |
Romiplostim | 262,305 | Dominated by eltrombopag |
ICER = incremental cost-effectiveness ratio; Ref. = reference; vs. = versus.
Appendix 5: Submitted BIA and CADTH Appraisal
Note this appendix has not been copy-edited.
Table 16: Summary of Key Takeaways
Key Takeaways of the BIA |
|---|
|
Summary of Sponsor’s BIA
The sponsor submitted a BIA estimating the incremental budget impact of reimbursing avatrombopag for the treatment of adult patients with chronic ITP who have had an insufficient response to a previous treatment. The BIA was undertaken from a publicly funded drug plan perspective considering only drug costs in the base-case analysis. The analytic framework, which used an epidemiology-based approach, leveraged data from multiple sources in the literature and assumptions based on clinical expert input to determine the estimated population size (refer to Table 16). New patients were added to the BIA by projecting jurisdictional population sizes based on historical trends from 2014 to 2020.
The sponsor compared a reference scenario in which avatrombopag is not reimbursed for chronic ITP with a new drug scenario, where avatrombopag is funded as per the proposed Health Canada indication. The reference scenario stratified the target population by currently available treatment options which included eltrombopag, romiplostim, rituximab and watch and rescue. The proportion receiving watch and rescue in the reference scenario was assumed to be 10% across all years. Reference scenario market shares were dependent on whether the jurisdiction currently reimburses TPO-RAs (refer to Table 16). In jurisdictions where TPO-RAs are currently funded (Ontario and Saskatchewan), reference scenario markets shares were based on public claims data and extrapolated to year 1, 2 and 3 based on historical trends from 2016 to 2021 using linear extrapolation.19 In the new drug scenario, avatrombopag market uptake was based on an assumption and was assumed to be the same in all jurisdictions regardless of current TPO-RA funding.
Drug costs were informed by median doses reported in the clinical trials or product monographs and did not account for dose adjustments. Key inputs to the BIA are documented in Table 17.
Table 17: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1, year 2, year 3 if appropriate) |
|---|---|
Target population | |
Prevalence of ITP Percentage of ITP cases that are primary Percentage of primary ITP cases that are chronic | 9.5 per 100,00020 76%21 80%22 |
Number of patients eligible for drug under review | 1,462 / 1,482 / 1,502 |
Market Uptake (3 years) | |
Uptake (reference scenario) - Jurisdictions where TPO-RAs are not fundeda Eltrombopag Romiplostim Rituximab Watch and rescue | 0% / 0% / 0% 0% / 0% / 0% 90% / 90% / 90% 10% / 10% / 10% |
Uptake (reference scenario)-Ontario Eltrombopag Romiplostim Rituximab Watch and rescue | 13% / 14% / 15% 9% / 9% / 9% 68% / 67% / 65% 10% / 10% / 10% |
Uptake (reference scenario)-Saskatchewan Eltrombopag Romiplostim Rituximab Watch and rescue | 60% / 66% / 73% 0% / 0% / 0% 30% / 24% / 17% 10% / 10% / 10% |
Uptake (new drug scenario) - Jurisdictions where TPO-RAs are not fundeda Avatrombopag Eltrombopag Romiplostim Rituximab Watch and rescue | 10% / 15% / 20% 0% / 0% / 0% 0% / 0% / 0% 81% / 77% / 72% 9% / 9% / 8% |
Uptake (new drug scenario)-Ontario Avatrombopag Eltrombopag Romiplostim Rituximab Watch and rescue | 10% / 15% / 20% 12% / 12% / 12% 8% / 8% / 7% 61% / 57% / 52% 9% / 9% / 8% |
Uptake (new drug scenario)-Saskatchewan Avatrombopag Eltrombopag Romiplostim Rituximab Watch and rescue | 10% / 15% / 20% 54% / 56% / 58% 0% / 0% / 0% 27% / 20% / 14% 9% / 9% / 8% |
Cost of treatment (per patient) | |
Cost of treatment over 1 year Avatrombopag Eltrombopag Romiplostim Rituximab Watch and rescue | $42,004 $47,483 $53,316 $8,672 $0 |
ITP-immune thrombocytopenia; NIHB = Non-insured Health Benefits; TPO-RA = thrombopoietin receptor agonist.
aJurisdictions where TPO-RAs are not funded include British Columbia, Alberta, Manitoba, New Brunswick, Nova Scotia, Prince Edward Island, Newfoundland and Labrador, and NIHB.
Summary of the Sponsor’s BIA Results
The sponsor estimated the net budget impact of introducing avatrombopag for the treatment of adult patients with chronic ITP who have had an insufficient response to a previous treatment would be $4,219,555 in year 1, $6,339,930 in year 2 and $8,467,370 in year 3 for a total budget impact $19,026,855 over 3 years.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
There is uncertainty in the sponsor’s approach to estimating the reference scenario’s market shares. The sponsor used Pharmastat claims data to calculate the reference scenario’s market shares, by taking total annual claims for a product and dividing that by an annual per patient treatment cost to estimate the number of patients on treatment from 2016 to 2021.13,17 They then predicted patient numbers from 2022 to 2025 using linear trends during the observed period. To obtain the reference scenario market share, the sponsor divided the total number of patients projected to be on a currently available TPO-RA in a given jurisdiction by the market size for that year in that jurisdiction. Resulting market shares are in Table 16. This approach to estimating reference scenario market shares is uncertain for several reasons. First, annual per patient treatment costs are not fixed (refer to Table 7), as product monographs specify dose adjustments to reach a target platelet count. This is also evidenced by the change in the pharmacoeconomic analysis, which demonstrated that mean trial daily doses exceeded the initial product-monograph dose. Additionally, because the sponsor did not incorporate a proportion of patients eligible for public coverage, the denominator (market size) in the sponsor’s analysis may be overestimated, and the reference scenario market shares would therefore be underestimated.
Additionally, in the sponsor’s base case, TPO-RAs were only assumed to be available in Ontario and Saskatchewan. Drug plan input indicated that some additional jurisdictions do fund TPO-RAs. Pharmastat data demonstrated public claims for romiplostim in British Columbia, Ontario and Nova Scotia; also, public claims for eltrombopag were noted in British Columbia, NIHB, Saskatchewan, Ontario, and New Brunswick.
CADTH was able to validate the claims used to derive market shares for Ontario and Saskatchewan in the sponsor’s base case, but noted total claims from 2019 were overestimated in Saskatchewan and Ontario. CADTH used the sponsor’s approach to calculating market shares in the additional jurisdictions noted to have claims (i.e., adding eltrombopag claims to for New Brunswick, NIHB, and British Columbia, and adding romiplostim claims to Nova Scotia and British Columbia). CADTH validated resulting reference scenario market shares with the clinical expert who noted that these largely appeared appropriate and consistent across jurisdictions.
CADTH corrected 2019 claims in Saskatchewan and Ontario for eltrombopag and romiplostim, as needed.
CADTH used the sponsor’s approach to estimating market shares using claims data for jurisdictions that had claims for TPO-RAs that were not included in the sponsor’s reference scenario.
The uptake of avatrombopag is not aligned with clinical expert expectations. In the sponsor’s base case, it was assumed that 10% / 15% / 20% of eligible patients would uptake avatrombopag, should it become available, including in jurisdictions where other TPO-RAs are not available. The clinical expert consulted for this review noted that in these jurisdictions, rituximab is currently the only therapy available for second-line treatment. Due to rituximab’s IV administration and immunosuppressant side effects, if avatrombopag became available for second-line therapy, many patients would prefer avatrombopag over rituximab. This is reflected in the clinician input which indicated that avatrombopag is expected to cause a shift in treatment paradigm by enabling access to TPO-RAs in second-line therapy.
In addition, according to the clinical expert consulted by CADTH for this review, if patients fail to achieve, or lose, their response on an initial TPO-RA, it would not be appropriate to trial a different TPO-RA. Therefore, it is not expected for avatrombopag to capture market shares from other TPO-RAs in jurisdictions that fund TPO-RAs; rather, the majority of avatrombopag’s market capture is expected to come from other active treatments (i.e., rituximab).
In CADTH reanalysis, the proportion of eligible patients who will use avatrombopag in year 1, 2 and 3 was changed to 20% / 30% / 40%, respectively. Additionally, the introduction of avatrombopag is expected to capture market shares from rituximab only, even in jurisdictions currently funding TPO-RAs.
The sponsor’s estimated eligible population does not reflect the proposed Health Indication. The proposed indication for avatrombopag is for the treatment of thrombocytopenia in adult patients with chronic ITP who have had an insufficient response to a previous treatment. The sponsor’s approach to calculating the size of the eligible population only considered those with primary chronic ITP; however, the indication does not specify that only primary ITP patients would be eligible. It is uncertain whether avatrombopag would be used in patients with secondary ITP because the clinical evidence informing its efficacy was solely based in the primary population; however, the experts noted that a proportion of secondary chronic ITP patients may use avatrombopag if approved in some circumstances (e.g., if there are no available treatments for the underlying condition).
In a scenario analysis, CADTH explored the budget impact if avatrombopag is approved for use beyond primary ITP patients.
Doses for TPO-RAs used in the BIA are not aligned with dosing used in the pharmacoeconomic analysis. The pharmacoeconomic analysis adjusted the source for TPO-RA dosing in the base case to reflect mean trial doses, as opposed to the initial dose specified in respective product monographs.
Dosing was changed to being based on mean trial doses to align with the pharmacoeconomic analysis.
CADTH Reanalyses of the BIA
CADTH revised the sponsor’s base case by: correcting 2019 annual claims for Saskatchewan and Ontario; adding annual claims for eltrombopag and romiplostim to derive reference scenario market shares in jurisdictions with public claims for comparators from 2016 to 2021; increasing avatrombopag uptake and having all of its market capture come from rituximab; and, adjusting dosing for TPO-RAs to reflect trial dosing. Table 17 notes the assumptions used by the sponsor in comparison to those used by CADTH in its reanalysis.
Table 18: CADTH Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Correctionsa to sponsor’s base case | ||
1. SK total 2019 eltrombopag 50 mg claims, and resulting reference scenario share by year | $3,043,281; 60% / 66% / 73% | $355,346; 29% / 33%/ 36% |
2. ON total 2019 eltrombopag 50 mg claims, and resulting reference scenario share by year | $1,558,968; 13% / 14% / 15% | $1,325,085; 13% / 14% / 15% |
3. ON total 2019 romiplostim claims, and resulting reference scenario share by year | 250 mcg: $1,992,636.69 500 mcg: $1,482,611.23 Market shares: 9% / 9% / 9% | 250 mcg: $1,875,315.24 500 mcg: $1,432,793 Market shares: 9% / 9% / 9% |
Changes to derive the CADTH base case | ||
1. Eltrombopag market shares in:
| 0% / 0% / 0% 0% / 0% / 0% 0% / 0% / 0% | 7% / 7% / 7% 19% / 20% / 21% 11% / 11% / 11% |
Romiplostim market shares in:
| 0% / 0% / 0% 0% / 0% / 0% | 11% / 12% / 13% 11% / 13% / 16% |
2. Avatrombopag uptake; capturing from rituximab | 10% / 15% / 20% | 20% / 30% / 40% |
3. Source for drug dosing | Product monograph | Mean dose from respective clinical trials |
CADTH base case | 1 + 2 + 3 | |
NIHB = Non-insured Health Benefits; ON = Ontario; SK = Saskatchewan.
aCorrections are minor errors (e.g., transcription errors between report and model, misapplication of distributions or SEs in probabilistic analyses) that are not identified as limitations.
The results of the CADTH step-wise reanalysis are presented in summary format in Table 18 and a more detailed breakdown is presented in Table 19.
In the CADTH reanalysis, the three-year budget impact of reimbursing avatrombopag for the treatment of adult patients with chronic ITP who have had an insufficient response to a previous treatment, was $51,688,953 (year 1: $11,292,967; year 2: $17,171,433; year 3: $23,204,554).
Table 19: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total |
|---|---|
Submitted base case | $19,026,855 |
Corrected base case | $22,651,596 |
CADTH reanalysis 1-Adding eltrombopag to NB, NIHB and BC and romiplostim to NS and BC | $18,080,510 |
CADTH reanalysis 1 + 2-Avatrombopag uptake; capture from rituximab | $44,588,957 |
CADTH reanalysis 3-Trial dosing | $21,078,690 |
CADTH base case | $51,668,953 |
BC = British Columbia; BIA = budget impact analysis; NB = New Brunswick; NIHB = Non-insured Health Benefits; NS = Nova Scotia.
CADTH also conducted additional scenario analyses to address remaining uncertainty, using the CADTH base case. Results are provided in Table 23:
Reduced the price of avatrombopag to the value in which it would be cost-effective at a $50,000 per QALY threshold compared with watch and rescue (32%).
Included both primary and secondary ITP patients to explore the budget impact if avatrombopag was available for all chronic ITP, which would align with the sponsor’s proposed Health Canada indication.
Table 20: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $18,440,162 | $19,210,457 | $19,980,753 | $20,751,048 | $59,942,258 |
New drug | $18,440,162 | $23,430,012 | $26,320,683 | $29,218,418 | $78,969,113 | |
Budget impact | $0 | $4,219,555 | $6,339,930 | $8,467,370 | $19,026,855 | |
CADTH base case | Reference | $25,760,792 | $27,059,542 | $28,358,292 | $29,657,042 | $85,074,877 |
New drug | $25,760,792 | $38,352,509 | $45,529,725 | $52,861,597 | $136,743,830 | |
Budget impact | $0 | $11,292,967 | $17,171,433 | $23,204,554 | $51,668,953 | |
CADTH scenario analysis: 32% price reduction | Reference | $25,760,792 | $27,059,542 | $28,358,292 | $29,657,042 | $85,074,877 |
New drug | $25,760,792 | $33,927,346 | $38,801,078 | $43,768,864 | $116,497,289 | |
Budget impact | $0 | $6,867,804 | $10,442,786 | $14,111,822 | $31,422,412 | |
CADTH scenario analysis: avatrombopag used for primary and secondary ITP | Reference | $29,314,760 | $30,662,857 | $32,010,954 | $33,359,051 | $96,032,863 |
New drug | $29,314,760 | $45,522,024 | $54,604,945 | $63,891,359 | $164,018,328 | |
Budget impact | $0 | $14,859,166 | $22,593,990 | $30,532,308 | $67,985,465 |
BIA = budget impact analysis; ITP = immune thrombocytopenia.
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.