CADTH Reimbursement Review
Pitolisant Hydrochloride (Wakix)
Sponsor: Paladin Labs Inc.
Therapeutic area: Narcolepsy
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
AASM
American Academy of Sleep Medicine
AE
adverse event
ANCOVA
analysis of covariance
BDI-SF-13
13-item Beck Depression Inventory-Short Form
BMI
body mass index
BOCF
baseline observation carried forward
CGI-C
Clinical Global Impression of Change
CGI-S
Clinical Global Impression of Severity
CI
confidence interval
CNS
central nervous system
CSF
cerebrospinal fluid
CUP
compassionate use program
DNS
disturbed nocturnal sleep
DSM-5
Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition
ECG
electrocardiogram
EDS
excessive daytime sleepiness
EIT
extended intention-to-treat
ESS
Epworth Sleepiness Scale
H3
histamine 3
HRQoL
health-related quality of life
IQR
interquartile range
ITT
intention-to-treat
LOCF
last observation carried forward
MID
minimally important difference
MSLT
Multiple Sleep Latency Test
MWT
Maintenance of Wakefulness Test
NIM
noninferiority margin
OR
odds ratio
PGO
patient global opinion
PP
per protocol
RCT
randomized controlled trial
REM
rapid eye movement
RR
relative risk
SAE
serious adverse event
SART
Sustained Attention to Response Task
SD
standard deviation
SNRI
selective norepinephrine reuptake inhibitors
SOC
standard of care
SOREMP
sleep-onset REM period
SSRI
selective serotonin reuptake inhibitors
TCA
tricyclic antidepressant
TEAE
treatment-emergent adverse event
VAS
visual analogue scale
WCR
weekly cataplexy rate
WUN
Wake Up Narcolepsy
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Pitolisant hydrochloride (Wakix), up to 40 mg daily, 5 mg and 20 mg tablets, oral |
Indication | Treatment of EDS or cataplexy in adults with narcolepsy |
Reimbursement request | Per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | May 25, 2021 |
Sponsor | Paladin Labs Inc. |
EDS = excessive daytime sleepiness; NOC = Notice of Compliance.
Introduction
Narcolepsy is a chronic neurologic condition that is caused by an imbalanced sleep-wake cycle or sleep-wake instability.1 It is characterized by chronic, excessive episodes of drowsiness during the day, also known as excessive daytime sleepiness (EDS).2 Type 1 narcolepsy is classified as EDS with cataplexy, whereas type 2 narcolepsy consists of EDS alone.1 Cataplexy is defined as a sudden episode of partial or complete paralysis of voluntary muscles, triggered by strong emotion.3 Approximately 60% to 70% of patients with narcolepsy have cataplexy (type 1 disease).4 Approximately 1 in 2,000 individuals in Canada are affected by narcolepsy.2 This prevalence is considered an underestimate, given the possibility of misdiagnosis and the limited availability of health care providers with experience in narcolepsy.5-7
Narcolepsy can affect all aspects of life in work and social settings, and a patient’s day-to-day functioning, health-related quality of life (HRQoL), and productivity.8 Patients can experience EDS during common situations in the day, such as work or driving, and during sedentary periods.3 Narcolepsy is associated with an increased risk in comorbid conditions, including depression, anxiety, obesity, cardiovascular disease, and overall mortality.8 In Canada, the current treatment standard for EDS in narcolepsy is modafinil, which is thought to improve wakefulness by reducing dopamine reuptake.
Pitolisant hydrochloride is an inverse agonist/antagonist of the histamine 3 (H3) receptor. The human H3 receptor functions as a presynaptic autoreceptor on histamine-containing neurons.9 H3 antagonists promote wakefulness by increasing histamine synthesis and release. By binding competitively to H3 autoreceptors on presynaptic histaminergic neurons, pitolisant hydrochloride blocks the normal negative-feedback mechanisms for histamine release, increasing histaminergic transmission and resulting in enhanced histamine synthesis and release.10-12 Pitolisant hydrochloride is administered orally, up to 40 mg daily, with 5 mg and 20 mg tablets. It is indicated for the treatment of EDS or cataplexy in adults with narcolepsy. It received Notice of Compliance on May 25, 2021, after undergoing standard review. The reimbursement request is per indication.
The objective of this clinical review is to perform a systematic review of the beneficial and harmful effects of pitolisant hydrochloride oral tablets (5 mg and 20 mg), with a daily dose up to 40 mg, for the treatment of EDS or cataplexy in adults with narcolepsy.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient groups that responded to CADTH’s call for patient input and from clinical experts consulted by CADTH for the purpose of this review. The complete patient submission is found at the end of this report.
Patient Input
One patient group, Wake Up Narcolepsy (WUN), submitted patient input for this review. WUN is a patient advocacy nonprofit organization established in 2008 that aims to accelerate research, increase awareness of narcolepsy, and provide supportive services to patients. The input was based on a survey of 19 patients in Canada who have a narcolepsy diagnosis or are undiagnosed but living with narcolepsy symptoms. Most patients were aged 18 to 34 years (66%) and female (72%), and none had experience with the treatment under review.
Respondents reported EDS to be the most troubling symptom of narcolepsy, with 39% of respondents giving it a rating of 6 on a scale of 1 (not at all bothersome) to 7 (completely bothersome). The second-most troublesome symptom reported was disturbed nocturnal sleep (DNS), followed by hallucinations when falling asleep or waking up, cataplexy, and sleep paralysis. The negative impacts of narcolepsy on respondents’ lives include mental health and emotional symptoms (mood swings, anger, depression, and anxiety), missing out on social activities, difficulty managing career and job tasks, depending on others for support for daily activities, and difficulty maintaining physical health and wellness (weight gain). Treatments that respondents reported currently using for their narcolepsy include stimulants (56%), antidepressants (33%), sodium oxybate (13%), and modafinil or armodafinil (13%). Some respondents reported that the physical side effects (28%) and mental side effects (39%) of their current treatment options were moderately or extremely challenging.
Respondents would like a new drug or treatment to be more effective for symptoms of sleepiness, cataplexy, and DNS. Respondents indicated a desire to have a treatment that is easy to swallow and does not cause nausea, weight gain, or affect mood or personality. Respondents want a treatment with an extended release that allows them to stay awake longer during the day without having to take additional doses. A copy of the patient input is presented in Appendix 1.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of narcolepsy.
A number of factors make the diagnosis extremely challenging. Patients often first visit family doctors or pediatricians who may not immediately recognize this condition. Patients are misdiagnosed frequently, and more than 70% of patients with narcolepsy are undiagnosed. Although existing medications treat the underlying symptoms of narcolepsy, including primarily daytime sleepiness and cataplexy, it is believed that none of these treatment options address the fundamental underlying neurochemical abnormality of loss of hypocretin cells and secondary absence or reduction of available central nervous system (CNS) hypocretin associated with narcolepsy.
Several problems persist with existing treatment options. Not all patients respond to treatment with selective serotonin reuptake inhibitors (SSRIs), and selective norepinephrine reuptake inhibitors (SNRIs), or tricyclic antidepressants (TCAs), and some become tolerant to treatment. Tolerance to the rapid eye movement (REM)-suppressing effects of SSRI, SNRI, and TCA medications occurs frequently, leading to persistent cataplexy. Side effects of these drugs, such as stomach upset, night sweats, sexual side effects, and headaches, can be problematic, and can include sedation during the day, which can be a problem despite the anticataplectic effects of drugs. With stimulants, daytime sleepiness may not be fully resolved and/or drugs may or may not wear off at inopportune times, leading to EDS in the evenings and/or insomnia at night. The side effects of stimulants, such as appetite suppression, anxiety, increased blood pressure, cardiac effects, allergic reaction, reduced seizure threshold, fetal defects, inactivation of birth control, and hair loss, can be problematic. There can be potential for misuse or drug diversion; most patients with narcolepsy have a low likelihood of misusing existing treatment options even though they may require high doses, but for some, there could be temptation to obtain the drug for purposes not intended by the prescriber.
The consistent use of anticataplectic treatments with pitolisant hydrochloride may mask and/or minimize the potential benefits pitolisant hydrochloride might have for cataplexy, and if the benefits of pitolisant hydrochloride are minimal for cataplexy, it would be difficult to assess. In short, it is difficult to properly assess the potential benefits of pitolisant hydrochloride for cataplexy with use of ongoing anticataplectic treatments.
Based on the efficacy of pitolisant hydrochloride shown in early studies, its novel mechanism of action as a H3 antagonist/inverse agonist, and its relatively favourable side-effect profile, it is likely to become an early treatment option. It received a strong recommendation from the American Academy of Sleep Medicine (AASM) in their most recent (2021) guidelines for the treatment of hypersomnolence disorders.12 It will be an early drug to consider for the treatment of narcolepsy. It may find a niche as an adjunct treatment to be used in combination with other therapies to boost efficacy, and may also become a drug of choice for patients in whom stimulant and/or other therapies are contraindicated because it has no effect on the efficacy of birth control (unlike modafinil) and no significant known cardiovascular effects (unlike other stimulants). Patients most in need of intervention include those who cannot tolerate stimulant therapies, those concerned about getting pregnant, and those with a history of drug abuse. Jurisdictions should continue to provide coverage for therapies currently considered standard of care (SOC) when used in combination with pitolisant hydrochloride because the mechanism of action of pitolisant hydrochloride is quite different than any currently available drug, which is an exciting prospect for patients living with this debilitating condition.
Primary outcomes in clinical practice will likely be a reduction in EDS, a reduction in this report, treatment goals are primarily to improve quality of life. Although narcolepsy is not lethal, symptoms of EDS and cataplexy can be debilitating if left uncontrolled. In severe circumstances, sleep attacks can occur while a patient is eating or even talking to someone. Uncontrolled, these symptoms limit a patient’s ability to perform basic daily activities, such as driving, working, and interacting with people. Cataplexy (which occurs in 60% to 70% of patients with narcolepsy) is equally, if not more, debilitating when left uncontrolled. Patients cannot drive or walk outside safely because surprises can trigger a cataplectic attack. Basic daily activities, such as showering and bathing, dressing, and eating, can be dangerous and/or challenging for an untreated patient. Without treatment, most patients have very limited, if any, work options, and may not be able to attend school. The symptoms can lead to isolation, anxiety, and depression. Treatment is aimed at reducing EDS and cataplexy so that patients are not dependent on caregivers for support and can interact and be functional members of society. Treatment can significantly improve alertness and daytime abilities, allowing patients to be functional members of society. Diagnosis is often delayed, often occurring 10 years or longer after symptom onset, potentially leading to significant suffering. However, if appropriate treatment is initiated, tolerated, and maintained, up to 80% of functional capacity could be retained. the frequency, intensity, and duration cataplexy episodes, and the ability to predict episodes. Clinically meaningful responses to treatment include a reduction in the frequency, severity, and intensity of cataplexy episodes. Although frequency is easier to assess systematically, the intensity and severity of spells, as well as the perceived predictability of episodes, are more of a clinical assessment. For example, patients may describe certain emotions that no longer trigger episodes in the way they had before. Other parameters for assessment could include a reduction in other REM intrusion phenomena, if present, and the degree to which patients can resume normal functioning and daily activities.
Outcomes typically assessed in most clinical trials include the degree of reduction in EDS and in the frequency of cataplexy spells. The use of Epworth Sleepiness Scale (ESS) scores in clinical practice to determine coverage of pitolisant hydrochloride may not be ideal. The ESS is very subjective and could easily manipulate scores. In addition, there can be significant differences in the way male and female patients score their results, further skewing potential for the determination of coverage. In research trials, it is ideal if patients are blinded to what they are being offered and there is no incentive to report better or worse scores. An ESS score of 10 or lower would indicate that sleepiness is no different than in the general population. As a comparison, an ESS score for patients with narcolepsy would typically be higher than 18 (on the 24-point scale), indicating severe sleepiness; a score of 15 to 17 indicates moderate sleepiness, and a score of 11 to 14 indicates mild sleepiness.
There is very little data available to define what represents an effective reduction of cataplexy. Trials of sodium oxybate demonstrated a more than 90% reduction in cataplexy episodes. Driving is not recommended if a patient has experienced a cataplexy episode in the previous year. A minimum reduction in cataplexy episodes of 50% would be meaningful. Depending on severity and frequency, fewer than 1 episode per week would be a reasonable standard.
At this time, pitolisant hydrochloride may not be suitable for patients who wish to get pregnant or who are breastfeeding. In addition, because of a lack of data on its use in children and in older adults, it should be used with caution in those populations. Patients who are on multiple medications (particularly drugs affecting the QTc interval or those that are significant CYP2D6 inhibitors, which have more potential for drug interactions) and patients who have a history of significant kidney or liver failure also may not be ideal candidates for pitolisant hydrochloride (because of a difficult-to-predict metabolism). Patients who have had adverse reactions to opioids (including hives), or who have a history of urticaria or another skin condition, might be predisposed to allergic reactions to pitolisant hydrochloride. Ongoing treatment will be determined by lack of response and/or excess adverse side effects, like most medications. Whether it will continue to be used as an adjunct if abandoned as monotherapy is unclear. Excess adverse side effects or drug-drug interactions may necessitate withdrawal. Similarly, if a patient wishes to become pregnant, withdrawal may also be necessary.
As with other drugs for narcolepsy, there should be close follow-up of the patient in the first months of therapy. The first follow-up should occur 1 month after the initiation of pitolisant hydrochloride, then every 1 to 2 months for the next several months, and then intermittent follow-up after that, with at least yearly follow-up, at a minimum, in the long-term. Medical supervision in an outpatient setting with a physician trained in sleep medicine would be appropriate for patients with narcolepsy being treated with pitolisant hydrochloride. In the future, psychiatrists will likely become interested in using this medication for conditions and symptoms outside of narcolepsy. At this time, because the indication for pitolisant hydrochloride is only for narcolepsy, with a conditional recommendation for idiopathic hypersomnia, prescribing should be limited to those with specialty training or certification in sleep medicine.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 5.
Clinical Evidence
Pivotal Studies and Protocol-Selected Studies
Description of Studies
Three double-blind, phase III, placebo-controlled, randomized controlled trials (RCTs) met the inclusion criteria for the Systematic Review.13-15 In all 3 trials, patients were included if they had narcolepsy with cataplexy. The HARMONY 1 (NCT01067222) and HARMONY 1bis (NCT01638403) trials also included patients without cataplexy (type 1 narcolepsy). The HARMONY 1 and HARMONY 1bis trials required patients to have an ESS score of at least 14 during the baseline period, whereas the HARMONY CTP trial required an ESS score of at least 12. The HARMONY CTP (NCT01800045) trial included patients with at least 3 cataplexy attacks weekly. In all trials, patients with severe cataplexy were permitted stable doses of anticataplectic medications (except TCAs) that were administrated for at least 1 month before the start of the trial.
The HARMONY 1 and HARMONY 1bis trials were 8-week trials that assessed the superiority of pitolisant hydrochloride to placebo with regard to EDS in patients with narcolepsy. An additional efficacy objective was a noninferiority comparison between pitolisant hydrochloride and modafinil. The HARMONY CTP trial was a 7-week randomized, double-blind placebo-controlled study comparing pitolisant hydrochloride to placebo. It focused on the safety and efficacy of pitolisant hydrochloride in decreasing the frequency of cataplexy attacks in patients who had narcolepsy with cataplexy. The maximum daily dosages of pitolisant hydrochloride were 20 mg in the HARMONY 1bis trial and 40 mg in the HARMONY 1 and HARMONY CTP trials. Titration of the study drug was at the discretion of study investigators, which could have affected efficacy and potentially threatened blinding to treatment arms. Patients on anticataplectic medications represented 35% of all patients in the HARMONY 1 trial, |||||| of all patients in the HARMONY 1bis trial, and 10% of all patients in the HARMONY CTP trial.
Efficacy Results
Excessive Daytime Sleepiness
In the HARMONY 1 trial, the adjusted mean difference in the final ESS score between pitolisant hydrochloride and placebo was –3.10 (95% confidence interval [CI], –5.73 to –0.46; P = 0.022), as shown in Table 2. Sensitivity analyses of the per-protocol (PP) population, without accounting for the centre effect, showed similar results. Because the superiority of pitolisant hydrochloride over placebo for EDS was demonstrated at the a priori level of significance of alpha = 0.025, the noninferiority of pitolisant hydrochloride to modafinil was tested. The adjusted mean difference in the final ESS score between pitolisant hydrochloride and modafinil was 0.09 (95% CI, –2.31 to 2.30); thus, pitolisant hydrochloride was judged to not be noninferior to modafinil at the prespecified noninferiority margin (NIM) of 2. A patient was considered a responder when the final ESS score was less than 10. Based on this threshold, the responder rates were 13.3% in the placebo group, 45.2% in the pitolisant hydrochloride group, and 45.3% in the modafinil group. The adjusted odds ratio (OR) of response for pitolisant hydrochloride compared with placebo was 7.86 (95% CI, 1.59 to 38.86). The adjusted OR of response for pitolisant hydrochloride compared with modafinil was 1.09 (95% CI, 0.31 to 3.81).
In the HARMONY 1bis trial, the mean ESS score reductions from baseline (standard deviation [SD]) were |||||||||| in the placebo group, |||||||||| in the pitolisant hydrochloride group, and |||||||||| in the modafinil group (Table 2). The adjusted mean difference in the final ESS score between pitolisant hydrochloride and placebo was –2.19 (95% CI, –4.17 to –0.22; P = 0.030). Sensitivity analyses without reallocation by centre, and without adjustment for baseline ESS score, or after adjustment for baseline following the mean change, and the mean change over baseline methods showed similar results. Because the superiority of pitolisant hydrochloride over placebo for EDS was demonstrated at the a priori alpha level of 0.05, the noninferiority of pitolisant hydrochloride to modafinil was tested. The adjusted mean difference in the final ESS score between pitolisant hydrochloride and modafinil was 2.75 (95% CI, 1.02 to 4.48); thus, pitolisant hydrochloride was judged to not be noninferior to modafinil at the prespecified NIM of 2. A patient was considered a responder when the final ESS score was 10 or lower or the change from baseline was at least 3 points. The response proportions were |||||||||||||||||||| for the placebo, pitolisant hydrochloride, and modafinil groups, respectively. The adjusted relative risk (RR) for the difference between pitolisant hydrochloride and placebo was ||||||||||||||||||||. The adjusted RR for the difference between pitolisant hydrochloride and placebo was ||||||||||||||||||||||||||||||.
Table 2: Sleepiness and Cataplexy— HARMONY 1 (ITT Population) and HARMONY 1bis (ITT Population) Trials
Variable | HARMONY 1 | HARMONY 1bis | ||||||
|---|---|---|---|---|---|---|---|---|
Placebo N = 30 | Pitolisant hydrochloride N = 31 | Modafinil N = 33 | Placebo N = 32 | Pitolisant hydrochloride N = 66 | Modafinil N = 65 | |||
ESS score | ||||||||
Baseline, mean (SD)a | 18.9 (2.5) | 17.8 (2.5) | 18.5 (2.7) | 18.2 (2.3) | 18.2 (2.4) | 18.1 (2.8) | ||
Final, mean (SD)b | 15.6 (4.7) | 11.8 (6.1) | 11.6 (6.0) | 14.5 (5.9) | 13.7 (5.4) | 10.4 (6.0) | ||
Change from baseline, mean (SD) | –3.3 (4.1) | –6.0 (6.1) | –6.9 (6.1) | –3.6 (5.6) | –4.6 (4.6) | –7.8 (5.9) | ||
Adjusted mean difference in final score, pitolisant hydrochloride vs. placebo (95% CI)c | –3.10 (–5.73 to –0.46) | NA | –2.19 (–4.17 to –0.22) | NA | ||||
P value for test of superiorityc | 0.022 | NA | 0.030 | NA | ||||
Adjusted mean difference in final score, pitolisant hydrochloride vs. modafinil (95% CI)d | NA | 0.09 (–2.11 to 2.30) | NA | –2.75 (1.02 to 4.48) | ||||
P value for test of noninferiorityd | NA | 0.932 | NA | 0.002 | ||||
Complete + partial cataplexy episodes (episodes per day) | ||||||||
Baseline, ne | 30 | 31 | 33 | |||||||||| | |||||||||| | |||||||||| | ||
Baseline, mean (SD) | 0.43 (0.74) | 0.79 (1.53) | 0.76 (1.68) | |||||||||| | |||||||||| | |||||||||| | ||
Final, nf | 28 | 30 | 31 | |||||||||| | |||||||||| | |||||||||| | ||
Final, mean (SD) | 0.68 (1.66) | 0.28 (1.11) | 0.65 (1.62) | |||||||||| | |||||||||| | |||||||||| | ||
Daily rates of complete and partial cataplexy episodes, exposed population | ||||||||
Patients contributing to analysisg | 14 | 20 | 23 | |||||||||| | |||||||||| | |||||||||| | ||
Baseline, geometric mean (95% CI) | 0.4 (0.2 to 1.0) | 0.5 (0.3 to 1.0) | 0.4 (0.2 to 0.8) | |||||||||| | |||||||||| | |||||||||| | ||
Final, geometric mean (95% CI) | 0.4 (0.1 to 1.1) | 0.2 (0.1 to 0.4) | 0.3 (0.1 to 0.5) | |||||||||| | |||||||||| | |||||||||| | ||
RR (95% CI) of cataplexy at end of treatment, pitolisant hydrochloride vs. placeboh | 0.38 (0.15 to 0.93) | NA | |||||||||| | |||||||||| | |||||||||| | |||
P valuei | 0.034 | |||||||||| | |||||||||| | |||||||||| | ||||
RR (95% CI) of cataplexy at end of treatment, pitolisant hydrochloride vs. modafinilh | NA | 0.54 (0.24 to 1.23) | |||||||||| | |||||||||| | |||||||||| | |||
P valuei | NA | 0.138 | |||||||||| | |||||||||| | |||||||||| | |||
Daily rate of cataplexy for patients with cataplexy at baseline or during treatment, final 7 days | ||||||||
Number of patients contributing to the analysis | NR | NR | NR | |||||||||| | |||||||||| | |||||||||| | ||
Baseline, geometric mean (95% CI)j | NR | NR | NR | |||||||||| | |||||||||| | |||||||||| | ||
Final, geometric mean (95% CI)j,k | NR | NR | NR | |||||||||| | |||||||||| | |||||||||| | ||
Least squares mean (CI)l | NR | NR | NR | |||||||||| | |||||||||| | |||
P valuei | NR | NR | NR | |||||||||| | |||||||||| | |||
CI = confidence interval; ESS = Epworth Sleepiness Scale; ITT = intention-to-treat; NA = not applicable; NR = not reported; RR = relative risk; SD = standard deviation.
aMean of the first 2 weeks on treatment. In the HARMONY 1bis trial, when ESS at visit (V)2 was missing, ESS baseline was calculated as the average at V1 and V3.
bMean of the last 2 available post-baseline values.
cLinear mixed model, including final ESS score and groups as fixed effects and centres as random effects to test the superiority of pitolisant hydrochloride vs. placebo. In the HARMONY 1bis trial, linear mixed-effects model, featuring analysis of covariance on final ESS adjusted on baseline with treatment considered as a fixed factor and reallocated centre as a random effect.
dLinear mixed model including final ESS score and groups as fixed effects and centres as random effect to test the noninferiority of pitolisant hydrochloride vs. modafinil. In HARMONY 1bis, noninferiority test by considering the NIM of 2.
eBaseline = (all episodes at V2 and V3) / (number of days at V2 and V3). For patients with no cataplexy at baseline or during the treatment period, imputation value was calculated as 0.5 / number of days.
fFinal = (all episodes at V7 and V9) / (number of days at V7 and V9).
gDaily cataplexy rate was calculated as the ratio of the number of crises during 1 period to the number of days of this period. For these calculations, shortness of the exposures in the baseline, treatment, and final periods were calculated in the following way: for the patients with no observed crisis during these periods, the rate is, at the most, the reciprocal of the duration (1 / number of days) and, at the least, 0; thus, the imputation value was approximated by the mean between the 2 extremes (0.5 / number of days).
hAnalysis conducted on patients who had at least 1 cataplexy episode at baseline or during the study treatment.
iP values have not been adjusted for multiple testing (i.e., the type I error rate has not been controlled).
jGeometric mean is based on base 10 logarithm of titre.
kDaily rate of cataplexy for patients with cataplexy at baseline or during treatment (final 7 days:V6 to V7). Sums of cataplexy equal to 0 have been replaced with 0.1.
lQuasi-Poisson model on the daily cataplexy rate (ratio of final to baseline in geometric mean based on natural logarithm [GMT] of the number of cataplexy episodes on the number of exposed days), final 7 days, adjusted on DCR baseline, with treatment considered as a fixed factor and reallocated centre as a random effect. For all the tests, pitolisant hydrochloride was compared with placebo and modafinil with a superiority test.
Sources: Clinical Study Reports for HARMONY 1 and HARMONY 1bis.13,14
In the HARMONY CTP trial, the observed mean changes in ESS score over baseline were –1.9 (SD = 4.3) and –5.4 (SD = 4.3) in the placebo and pitolisant hydrochloride arms, respectively (Table 3). The adjusted mean difference in the change from baseline between pitolisant hydrochloride and placebo was –3.42 (95% CI, –4.96 to –1.87). Sensitivity analyses using the last observation carried forward (LOCF), the baseline observation carried forward (BOCF), and the PP population were consistent with the main analysis. A patient was considered a responder when the final ESS score was 10 or lower or the change from baseline was at least 3 points. The response proportions were 34.0% and 68.6% for the placebo and pitolisant hydrochloride groups, respectively. The adjusted OR for the difference between pitolisant hydrochloride and placebo was 4.26 (95% CI, 1.72 to 10.52).
Maintenance of Wakefulness Test
In the HARMONY 1 trial, the adjusted mean difference in final Maintenance of Wakefulness Test (MWT) score between placebo and pitolisant hydrochloride was 1.47 (95% CI, 1.01 to 2.14), and the adjusted mean difference in final MWT score between pitolisant hydrochloride and modafinil was 0.77 (95% CI, 0.52 to 1.13). This was consistent with the findings of the HARMONY 1bis trial, in which the adjusted mean difference between placebo and pitolisant hydrochloride was 1.46 (95% CI, 1.06 to 2.01). The adjusted mean difference in final MWT score between pitolisant hydrochloride and modafinil was ||||||||||||||||||||||||||||||. In the HARMONY CTP trial, the geometric mean of ratios (final divided by baseline) was 1.78 (95% CI, 1.22 to 2.60). Sensitivity analyses for all trials using the PP population were consistent with the main analysis.
Table 3: Sleepiness and Cataplexy — HARMONY CTP (ITT Population) Trial
Variable | Placebo (N = 51) | Pitolisant hydrochloride (N = 54) | |
|---|---|---|---|
ESS score | |||
Baseline, na | 51 | 54 | |
Baseline, mean (SD)a | 17.3 (3.2) | 17.4 (3.3) | |
Final, nb | 50 | 51 | |
Final, mean (SD)b | 15.4 (5.0) | 12.0 (5.4) | |
Change from baseline, mean (SD) | –1.9 (4.3) | –5.4 (4.3) | |
Adjusted mean difference in change from baseline, pitolisant hydrochloride vs. placebo (95% CI)c | –3.42 (–4.96 to –1.87) | ||
P valued | < 0.0001 | ||
Weekly rate of cataplexy | |||
Baseline, geometric mean (95% CI)e | 7.31 (6.02 to 8.87) | 9.15 (7.60 to 11.01) | |
Stable-dose period, geometric mean (95% CI)f | 4.51 (2.90 to 7.02) | 2.27 (1.51 to 3.41) | |
Ratio of geometric means, stable period / baseline (95% CI) | 0.62 (0.43 to 0.90) | 0.25 (0.17 to 0.36) | |
Ratio of geometric means during stable-dose period, pitolisant hydrochloride / placebo (95% CI)g | 0.5123 (0.4351 to 0.6033) | ||
P valueg | < 0.0001 | ||
High frequency of cataplexy episodes (> 15) | |||
Baseline, n (%)h | |||
≤ 15 | 42 (82.4) | 39 (72.2) | |
> 15 | 9 (17.6) | 15 (27.8) | |
Stable-dose period, n (%)i | |||
≤ 15 | 39 (76.5) | 51 (94.4) | |
> 15 | 12 (23.5) | 3 (5.6) | |
OR of frequency (95% CI)j | 0.035 (0.0035 to 0.352) | ||
P valued,j | 0.0044 | ||
CI = confidence interval; ESS = Epworth Sleepiness Scale; ITT = intention-to-treat; OR = odds ratio; SD = standard deviation.
aMean of values at visits 1 and 2.
bMean of values at visits 5 and 6. For missing values, the LOCF approach was used.
cLinear mixed model (analysis of covariance) adjusted for baseline ESS and for centre heterogeneity (i.e., including centre as a random factor).
dP values have not been adjusted for multiple testing (i.e., the type I error rate has not been controlled).
eMean of week 1 and week 2 values.
fMean of values during the stable-dose period (i.e., week 6 through week 9). For patients terminating the trial before completion, the final value was calculated as the mean of the 2 last known periods (LOCF).
gANCOVA via mixed nonlinear model featuring a possibly overdispersed Poisson distribution and taking into account centre heterogeneity by using centre as a random factor.
hMeasured at visit 2.
iMeasured at visit 6.
jiNonlinear mixed model taking into account centre heterogeneity.
Source: Clinical Study Report for HARMONY CTP.15
Sustained Attention to Response Task
In the HARMONY 1 trial, the adjusted mean difference in the Sustained Attention to Response Task (SART) between the pitolisant hydrochloride and placebo treatment arms was 0.82 (95% CI, 0.67 to 0.99) for NOGO scores, 0.80 (95% CI, 0.57 to 1.13) for GO scores, and 0.79 (95% CI, 0.64 to 0.99) for TOTAL scores. The adjusted mean difference between the pitolisant hydrochloride and modafinil treatment arms was 1.03 (95% CI, 0.83 to 1.28) for NOGO scores, 1.03 (95% CI, 0.56 to 1.15) for GO scores, and 0.90 (95% CI, 0.70 to 1.14) for TOTAL scores. Sensitivity analyses using the PP population was consistent with the main analysis. In the HARMONY 1bis trial, the ratio of mean change between pitolisant hydrochloride and placebo was significant (0.83; 95% CI, 0.69 to 0.99), whereas the ratio of mean change between pitolisant hydrochloride and modafinil was ||||||||||||||||||||||||||||||||||||||||||||||||||.
Clinical Global Impression of Change on EDS
In the HARMONY 1 and HARMONY 1bis trials, the Clinical Global Impression of Change (CGI-C) score for EDS was improved in a higher proportion of patients in the pitolisant hydrochloride and modafinil groups than in the placebo group. However, the change in CGI-C score was similar in the pitolisant hydrochloride and modafinil arms. In the HARMONY 1 trial, CGI-C scores for EDS improved in the subgroup of patients with a history of cataplexy, but a greater proportion of patients reported an improvement in the modafinil arm. In the HARMONY CTP trial, the mean reduction in CGI-C score for pitolisant hydrochloride compared with placebo was –0.95 (95% CI, –1.36 to –0.54). Mean CGI-C score was 3.5 (SD = 1.1) in the placebo group and 2.6 (SD = 1.1) in the pitolisant hydrochloride group. Similar results were observed for the PP population, with a mean reduction of –0.86 (95% CI, –1.29 to –0.43).
Frequency and Severity of Cataplexy Attacks
In the HARMONY 1 trial, the final mean of complete and partial cataplexy episodes (per day) was 0.68 (SD = 1.66), 0.28 (SD = 1.11), 0.65 (SD = 1.62) in the placebo, pitolisant hydrochloride, and modafinil groups, respectively. In the exposed population, the RR of daily rates of complete and partial cataplexy episodes at the end of treatment for pitolisant hydrochloride compared to placebo was 0.38 (95% CI, 0.15 to 0.93). The RR of daily rates of complete and partial cataplexy episodes at the end of treatment for pitolisant hydrochloride compared to modafinil was 0.54 (95% CI, 0.24 to 1.23). In the HARMONY 1bis trial, the mean least square of daily cataplexy rate for those with cataplexy between the final 7 days of treatment and baseline was |||||||||||||||||||||||||||||| for pitolisant hydrochloride compared to placebo.
The primary end point in the HARMONY CTP trial was the measure of anticataplectic efficacy. During the stable-dose period, the geometric means of the weekly cataplexy rate (WCR) at the end of treatment decreased to 4.51 (95% CI, 2.90 to 7.02) in the placebo group and 2.27 (95% CI, 1.51 to 3.41) in the pitolisant hydrochloride group. The ratio of geometric means during the stable-dose period was 0.51 (95% CI, 0.43 to 0.60; P < 0.0001) for pitolisant hydrochloride compared to placebo. Similar results were observed for the PP population, with a ratio of 0.50 (95% CI, 0.34 to 0.74; P < 0.0001) for pitolisant hydrochloride compared to placebo. The effect of pitolisant hydrochloride on the WCR remained consistent at 20 mg and 40 mg doses. The proportion of patients with a high frequency of weekly cataplexy episodes (> 15) during the stable-dose period was 5.6% in the pitolisant hydrochloride group and 17.6% in the placebo group (OR, 0.035; 95% CI, 0.0035 to 0.352). The effect remained consistent regardless of whether or not patients were taking permitted anticataplectic medications during the trial.
Clinical Global Impression of Change on Cataplexy
In the HARMONY 1 trial, the mean final CGI-C score was 3.4 (SD = 1.4), 2.9 (SD = 1.5), 3.0 (SD = 1.6) in the placebo, pitolisant hydrochloride, and modafinil arms, respectively. The number of patients who improved compared to baseline was 6 (24.0%) in the placebo group, 9 (34.6%) in the pitolisant hydrochloride group, and 8 (28.6%) in the modafinil group. The number of patients who reported no change compared to baseline was 15 (57.7%) in the placebo group, 15 (57.7%) in the pitolisant hydrochloride group, and 16 (57.1%) in the modafinil group. There were 2 (8.0%) patients reporting worsened CGI-C scores in the placebo arm and 1 (3.6%) in the modafinil arm.
In the HARMONY 1bis trial, the number of patients who improved compared to baseline was |||||||||| in the placebo group, |||||||||| in the pitolisant hydrochloride group, and |||||||||| in the modafinil group. The number of patients who reported no change compared to baseline was |||||||||| in the placebo group, |||||||||| in the pitolisant hydrochloride group, and |||||||||| in the modafinil group. There were |||||||||| patients reporting worsened CGI-C scores in the placebo group, |||||||||| in the pitolisant hydrochloride group, and |||||||||| in the modafinil arm.
In the HARMONY CTP trial, the mean reduction in CGI-C score for pitolisant hydrochloride compared with placebo was –0.95 (95% CI, –1.36 to –0.54). The mean CGI-C score was 3.5 (SD = 1.1) with placebo and 2.6 (SD = 1.1) with pitolisant hydrochloride. Similar results were observed for the PP population, with a mean change of –0.86 (95% CI, –1.29 to –0.43).
Harms
In the HARMONY 1 trial, adverse events (AEs) after initiation of treatment were reported by 66.7% of patients in the placebo group, 64.5% in the pitolisant hydrochloride group, and 69.7% in the modafinil arm. In the HARMONY 1bis trial, approximately |||||| of patients in the pitolisant hydrochloride and modafinil groups reported AEs, as did |||||| of patients in the placebo group. In the HARMONY CTP trial, approximately 35% of patients experienced an AE. For HARMONY 1 ||||||||||||||||||||, there was a greater percentage of nervous system disorders in the pitolisant hydrochloride arm, but in in the HARMONY CTP trial, the placebo arm had more nervous system disorders .
In the HARMONY 1 trial, pyelonephritis and hemorrhoids were reported as serious adverse events (SAEs) in the pitolisant hydrochloride arm. |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| HARMONY CTP reported 1 SAE in the pitolisant hydrochloride arm only.
In the HARMONY 1 trial, 1 patient in the pitolisant hydrochloride arm discontinued treatment because of pregnancy. Another patient in the pitolisant hydrochloride arm temporarily discontinued the study, but the study code was not broken and treatment was resumed so the study resumed. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. In the HARMONY CTP trial, 1 patient receiving pitolisant hydrochloride discontinued due to severe nausea, which was characterized as a treatment-emergent adverse event (TEAE). No deaths were reported in any of the trials.
Critical Appraisal
All included trials were double-blinded, placebo-controlled studies with a short duration (7-week or 8-week treatment phase). All trials had a small sample size (between 96 to 164 patients), which can limit the power to detect significant changes in the efficacy outcomes. The allocation sequence was random and balanced for all trials and remained concealed for the duration of the trial. HARMONY 1 |||||||||||||||||||||||||||||| had between-group study differences for previous medication use and for the proportion of patients with cataplexy, which could suggest differences in disease severity. In the HARMONY 1bis trial, |||||||||| of patients had a history of cataplexy in the pitolisant hydrochloride group, as did |||||||||| in the placebo group. In the HARMONY 1 trial, patients taking at least 1 chronic medication in the 3 months before inclusion ranged from 70.0% (modafinil group) to 85.2% (placebo and pitolisant hydrochloride groups). The maximum daily dosage of pitolisant hydrochloride was 20 mg in the HARMONY 1bis trial and 40 mg in the HARMONY 1 and HARMONY CTP trials . Titration of the study drug was at the discretion of the study investigators, which could have had an effect on efficacy and could have potentially threatened blinding to treatment.
All studies authorized patients to remain on stable doses of anticataplectic medications. Patients on anticataplectic medications represented 35% of all patients in the HARMONY 1 trial, |||||||||| of all patients in the HARMONY 1bis trial, and 10% of all patients in the HARMONY CTP trial. In the HARMONY 1 and HARMONY CTP trials, there were between-group differences in the proportion of patients on anticataplectic medications during the trial. In the HARMONY 1 trial, 33.3% of placebo patients remained on authorized medications during the study, compared to 40.7% of patients in the pitolisant hydrochloride group and 56.7% of patients in the modafinil group. In the HARMONY CTP trial, 16% of patients in the placebo group remained on anticataplectic medication, compared with 7% in the pitolisant hydrochloride group. Inconsistency in concomitant anticataplectic medications between trials cannot be clearly explained. The interactions between pitolisant hydrochloride and the concomitant treatments are unknown. Although the trials were double-blinded, some patients who received modafinil previously may have recognized the study drug.
The primary efficacy outcome in the HARMONY 1 and HARMONY 1bis trials, change in EDS, was measured using the validated ESS. ESS is a subjective, self-administered questionnaire widely used in narcolepsy trials. The primary outcome in the HARMONY CTP trial was the weekly rate of cataplexy captured in patient diaries. All primary outcomes were assessed using unvalidated tools. Other secondary end points that assessed EDS were not validated, such as the CGI-C and patient global opinion (PGO) tools. The MWT and SART outcomes were validated, but the statistical analyses did not adjust for multiplicity. Patient diaries were completed daily and reviewed by the investigators for completion, which may have biased future outcome assessments. The primary outcome of the HARMONY CTP trial was the change in WCR, which was recorded in daily patient diaries. A reduction in cataplexy episodes was also reported in the placebo group. This could be related to the use of concomitant treatments or a placebo effect.
Missing values for all trials were imputed for ESS and cataplexy outcomes. Any missing values at the end of treatment were imputed using LOCF or BOCF. It is unclear whether these would be reflective of the true trajectory of the outcomes. Sensitivity analyses using the PP population were provided, which can minimize potential bias. In addition, for all outcomes other than the primary outcome in all trials, there was no adjustment for multiplicity which increases the risk of type I error and limits the ability to draw conclusions. Subgroups were outlined a priori. Conclusions could not be drawn for the subgroups due to the lack of adjustment for multiplicity and were therefore considered exploratory analyses.
The NIM was calculated using historical trials of ESS, which were not specified, that set the minimal important difference (MID) at 3. To remain less than the MID and the proportion of difference between placebo and pitolisant hydrochloride, a NIM of 2 was chosen. In addition, sample-size calculations assumed that the effects of pitolisant hydrochloride and modafinil were similar.
All trials noted protocol amendments. A major amendment in the HARMONY 1 trial included a change from the assessment of superiority of pitolisant hydrochloride over modafinil to a noninferiority analysis. The change in type of analysis would not bias the results because the noninferiority analysis was reported appropriately for both the intention-to-treat (ITT) and PP populations.
According to the clinical experts consulted for this review, the baseline characteristics of study patients are reflective of patients with narcolepsy in Canada seeking further treatment options. The drug titration would be reflective of clinical practice. The primary outcome measures in the trials are used by physicians in clinical practice and measured outcomes important to patients (EDS and cataplexy). Patients were allowed to combine conventional narcolepsy medication with the drug under study. The clinical experts noted that it is common for combination therapy to be used in clinical practice; however, the interactions between concomitant medications and pitolisant hydrochloride are unknown. On that note, TCAs were not allowed as concurrent medications, despite them being common anticataplectic drugs, the clinical experts reported. This may decrease the generalizability of the trial population. Adherence to treatment remained high, at more than 80%, in all trials.
Other Relevant Evidence
The open-label HARMONY III extension study16,17 provides long-term safety and efficacy data that supplements evidence from the RCTs in the Systematic Review.
Description of Studies
The HARMONY III (NCT01399606) long-term, open-label, uncontrolled extension study was conducted to evaluate the efficacy and safety of pitolisant hydrochloride at daily doses of 5 mg, 10 mg, 20 mg, and 40 mg for the treatment of EDS in patients with narcolepsy with or without cataplexy for up to 5 years. Of the 102 patients enrolled in the HARMONY III extension trial, 86 were pitolisant hydrochloride–naive or secondary-naive (the naive group) and were not receiving pitolisant hydrochloride at the time of study enrolment and 16 were patients from a French compassionate use program (CUP) who were being treated with pitolisant hydrochloride in the 2 weeks preceding study enrolment. Of the 86 patients in the naive group, 73 had never been treated with pitolisant hydrochloride and 13 had been treated with pitolisant hydrochloride when they participated in single-blind or double-blind trials, including HARMONY 1,13 HARMONY II,18 and HARMONY 1bis.14
At study inclusion, CUP patients could continue at their established pitolisant hydrochloride dose (20 mg per day or 40 mg per day) without up-titration. The I patients began pitolisant hydrochloride treatment with a 1-month individual up-titration scheme that started at 5 mg per day and increased to up to 40 mg per day. Patients recruited from France who had received at least 1 dose of pitolisant hydrochloride and completed the initial 1-year period in the HARMONY III trial were eligible to continue treatment in a follow-up period for up to 5 more years.
A total of 102 patients with narcolepsy from 8 centres in France (n = 77) and Hungary (n = 25) were enrolled in the extension study, HARMONY III, with the first enrolment occurring in June 2011. After the initial 12-month treatment period, 48 patients from France continued in the 5-year follow-up period. Patients were required to have an ESS score of at least 12 to enrol in the extension study. Overall, the mean age of all participants was 38.0 (SD = 14.9) years, and slightly more than half were female (55.9%). About 75% of naive patients and CUP patients reported a history of cataplexy. Patients in the extension study could take concomitant medications for narcolepsy, including anticataplectics and/or psychostimulants. At inclusion, 35.3% of all patients were taking concomitant medications, with more CUP than naive patients taking concomitant medications (56.3% versus 31.4%). Overall, the baseline characteristics of the patients enrolled in the HARMONY III trial were generally consistent with the baseline characteristics of patients randomized in the pivotal trials. Characteristics of the French patients who continued in the 5-year follow-up period were similar to those of the total study population.
Efficacy Results
Sleepiness, Alertness, Severity of Daytime Sleepiness
In the HARMONY III extension study at year 1, the mean change from baseline in ESS score was –3.99 (SD = 4.56). Fifty-eight (58.2%) patients were considered responders, defined as having an ESS score no higher than 10 or a change from baseline of at least 3Its. Among naive patients, the mean change from baseline in ESS score was –4.30 (SD = 4.47). Forty-nine (59.8%) patients were considered responders. CUP patients, who were already receiving pitolisant hydrochloride treatment at inclusion, had a lower mean ESS score at baseline, and the mean change from baseline in ESS score was –2.38 (SD = 0.79). Eight (50.0%) patients were considered responders.
For patients taking concomitant narcolepsy treatments, the mean change from baseline in ESS score was –3.15 (SD = 4.01), –3.64 (SD = 4.55), and –4.00 (SD = 2.35) for patients taking psychostimulants (n = 26), anticataplectics (n = 14), and both psychostimulants and anticataplectics (n = 13), respectively. For the 45 patients taking pitolisant hydrochloride only (i.e., no concomitant treatments), the mean change from baseline was –4.67 (SD = 5.27). Thirteen (50.0%), 8 (57.1%), and 10 (76.9%) patients taking psychostimulants, anticataplectics, and psychostimulants plus anticataplectics, respectively, were considered responders. Twenty-six (57.8%) patients taking pitolisant hydrochloride only (i.e., no concomitant treatments) were considered responders.
The changes from baseline in ESS scores remained similar during the long-term follow-up in the French cohort. Among French patients who continued in the long term follow-up, the ESS mean change from baseline was –4.41 (SD = 5.38) at year 2 (n = 45), –4.45 (SD = 6.16) at year 3 (n = 38), –4.76 (SD = 5.73) at year 4 (n = 34), and –6.07 (SD = 7.19) at year 5 (n = 14). At 5 years, the mean change from baseline in ESS score was –8.17 (SD = 8.93) and –4.50 (SD = 5.71) for naive (n = 6) and CUP (n = 8) patients, respectively. Of the 14 patients remaining at 5 years, 10 (71.4%) were considered responders, 5 (83.3%) of whom were naive patients and 5 (62.5%) of whom were CUP patients.
For patients taking concomitant narcolepsy treatments, the mean change from baseline in ESS score after 5 years was –5.67 (SD = 6.11), –6.33 (SD = 7.77), and –5.50 (SD = 3.87) for patients taking psychostimulants (n = 3), anticataplectics (n = 3), and both psychostimulants and anticataplectics (n = 4), respectively. For the 4 patients taking pitolisant hydrochloride only (i.e., no concomitant treatments), the mean change from baseline was –6.75 (SD = 11.95). All patients remaining at 5 years, regardless of concomitant treatment, were considered responders.
A total of 71.7% of the 67 patients who completed the initial 1-year treatment period reported a CGI-C score of 1 (very much improved) or 2 (much improved), 22.4% reported a score of 3 (minimally improved), and 6.0% reported a score of 4 (no change). Three-quarters (73.1%) of naive patients and 66.7% of CUP patients were at least much improved, 21.2% and 26.7%, respectively, were minimally improved, and 5.8% and 6.7%, respectively, reported no change. Among French patients who continued in the long term follow-up, the proportion of patients who reported a much improved or very much improved CGI-C score compared to baseline was 77.3% at 2 years (n = 44), 84.2% at 3 years (n = 38), 73.5% at 4 years (n = 34), and 64.3% at 5 years (n = 14). At 5 years, 83.4% of naive patients (n = 5) and 50.0% of CUP patients (n = 4) were at least much improved, 16.7% of naive patients (n = 1) and 37.5% of CUP patients (n = 3) were minimally improved, 12.5% of CUP patients (n = 1) reported no change.
A total of 75.0% of patients (75.0% naive and 75.1% CUP) rated the effect of pitolisant hydrochloride as moderate or marked on the PGO test after 1 year of treatment. Among French patients who continued in the long term follow-up, the proportion of patients who reported a moderate or marked effect of pitolisant hydrochloride on the PGO test was 72.8% at 2 years (n = 44), 84.2% at 3 years (n = 38), 84.4% at 4 years (n = 32), and 64.3% at 5 years (n = 14) of treatment. At 5 years, 83.4% of naive and 50.0% of CUP patients rated the effect of pitolisant hydrochloride as moderate or marked.
Frequency and Severity of Cataplexy Attacks
At the end of the initial 1-year study period, the mean change in total cataplexy from baseline was a –0.25 (SD = 1.37) for all 44 patients who completed the sleep diary; for naive patients, the mean change was –0.25 (SD = 1.38), and for CUP patients it was 0.00 (SD = NA). The mean change in partial cataplexy from baseline was –0.49 (SD = 1.94) for all patients, –0.49 (SD = 1.96) for naive patients, and 0.53 (SD = NA) for CUP patients.
Health-Related Quality of Life
The mean EQ-5D visual analogue scale (VAS) score for all patients was 65.5 (SD = 16.1) at baseline and 72.4 (SD = 16.2) at 1 year, with a mean change of 6.8 (SD = 15.4) from baseline. For naive patients, the mean EQ-5D VAS score was 64.3 (SD = 15.9) at baseline and 73.5 (SD = 17.5) at 1 year; with a mean change of 9.2 (SD = 15.4) from baseline. For CUP patients, the EQ-5D VAS score was 69.6 (SD = 16.7) at baseline and 68.8 (SD = 11.4) at 1 year; with a mean change of –0.8 (SD = 12.7) from baseline.
Among French patients who continued in the long term follow-up, the mean of the EQ-5D VAS was 70.5 (SD = 15.9) at 2 years (n = 44), 69.5 (SD = 13.2) at 3 years (n = 38), 72.2 (SD = 13.3) at 4 years (n = 33), and 75.0 (SD = 12.2) at 5 years (n = 14). At 5 years, the EQ-5D VAS score was 80.5 (SD = 12.5) for naive patients and 70.9 (SD = 10.9) for CUP patients, with a change of 13.8 (SD = 15.5) and 2.4 (SD = 12.5) from baseline, respectively.
Sleep Attacks
At the end of the initial 1-year study period, among patients who completed the sleep diary (n = 44), the mean change in the daily number of sleep attacks from baseline was –0.37 (SD = 1.41) for all patients, –0.39 (SD = 1.42) for naive patients, and 0.47 (SD = NA) for CUP patients. The mean (SD) change in the duration of diurnal involuntary sleep attacks from baseline was –0.37 (SD = 1.41) minutes for all patients, –0.39 (SD = 1.42) minutes for naive patients, and 0.47 (SD = NA) minutes for CUP patients.
Nocturnal Sleep Properties
Among patients who completed the sleep diary (n = 44), the mean change in daily number of nocturnal awakenings from baseline to the 1-year visit was –0.42 (SD = 1.18) for all patients, –0.42 (SD = 1.19) for naive patients, and –0.14 (SD = NA) for CUP patients. The mean change in the duration of nocturnal awakening from baseline to the 1-year visit was –0.09 (SD = 0.73) hours for all patients, –0.10 (SD = 0.73) hours for naive patients, and 0.18 (SD = NA) hours for CUP patients. The mean change in the duration of nocturnal sleep from baseline to the 1-year visit was –0.10 (SD = 1.19) hours for all patients, –0.09 (SD = 1.21) hours for naive patients, and –0.37 (SD = NA) hours for CUP patients.
Number of Hallucinations
At the end of the initial 1-year study period, among patients who completed the sleep diary (n = 44), the mean change in the frequency of hallucinations from baseline was –0.06 (SD = 0.25) for all patients, –0.06 (SD = 0.20) for naive patients, and 0.0 (SD = NA) for CUP patients.
Concomitant Medication Use
The proportion of patients taking a concomitant treatment for narcolepsy or cataplexy changed from 35.3% at baseline to 52.9% over the course of the initial year. A total of 31.4% of naive and 56.3% of CUP patients were taking concomitant treatment at baseline and, over the course of the initial year, 51.2% of naive and 62.5% of CUP patients were taking concomitant medications. The most frequent treatments over the course of the study were methylphenidate (22.5%), modafinil (17.6%), and venlafaxine (13.7%). Eleven patients (10.8%) took sodium oxybate. In the French subset, the proportion of patients taking allowed concomitant treatment for narcolepsy or cataplexy in addition to pitolisant hydrochloride changed from 44.2% at baseline to 70.1% over the 5-year period. A total of 70.5% of naive and 68.8% of CUP patients were taking concomitant treatments over the 5-year period, respectively. The most frequent treatments were methylphenidate (31.2%), modafinil (29.9%), venlafaxine (19.5%), and sodium oxybate (16.9%).
Harms Results
All combinations of concomitant medications for narcolepsy or cataplexy were well tolerated, except there was a greater frequency of insomnia in the subgroup of patients taking concomitant modafinil (55%; n = 5) during the follow-up period in the French subset of patients.
During the initial year, 58 patients (56.9%) reported 168 TEAEs, the most common being headache (11.8% of patients), insomnia (8.8%), weight gain (7.8%), anxiety (6.9%), depression (4.9%), and nausea (4.9%). In the French subset of patients, over the 5-year period, 72.7% of patients reported 296 TEAEs, the most common being headache (19.5%), weight increase (18.2%), nausea (11.7%), anxiety (11.7%), insomnia (11.7%), and depression (11.7%).
A total of 16 patients reported SAEs during the 5-year period in the French subset, with the most common being depression (3.9%) and pregnancy (3.9%). All SAEs were considered unrelated to the study drug, except the 1 spontaneous abortion experienced by a patient who discontinued the study drug and permanently withdrew from the trial. One death was reported in the follow-up period. The clinical study report indicated that the death was determined to be not related to the study medication.
Among all patients, the mean 13-item Beck Depression Inventory-Short Form (BDI-SF-13) score was 4.1 (SD = 3.5) at baseline and 3.8 (SD = 4.1) at the 1-year visit. The mean BDI-SF-13 score among the 12 patients in the French subset at the year-5 visit was 2.4 (SD = 2.8). At each time point, no more than 1 patient experienced severe depression.
Critical Appraisal
The HARMONY III extension study allowed for investigation of the long-term efficacy and harms of pitolisant hydrochloride for up to 5 years. Limitations of the study include the absence of an active comparator, which limits causal conclusions. An additional limitation is the open-label study design and the unblinding of the study drug in the follow-up period, which could bias the reporting of end points. There was no sample-size calculation or statistical testing for changes from baseline, making it difficult to detect a clinically relevant treatment effect. All the end points in the HARMONY III trial were subjective, so it is possible that efficacy outcomes and known harms could have been overestimated. Findings are at a high risk of confounding because of the use of concomitant treatments and a lack of control for confounding variables. None of the P values were adjusted for multiplicity and should be considered hypothesis-generating.
Subgroup analyses were descriptive and often limited to few patients, reducing the chance of detecting a true effect. Interpretation of these patient-reported outcomes are also limited by the large amount of missing data due to attrition. More than one-third of patients discontinued the extension study in the first year, mainly because of AEs or a lack of perceived efficacy. This attrition could have resulted in a population of patients that were more tolerant of pitolisant hydrochloride, as those not responding to treatment may have been less likely to continue into the follow-up period. The presence of patients more tolerant of pitolisant hydrochloride can also lead to biased estimates of efficacy and AEs, potentially resulting in greater efficacy and fewer AEs being reported. The use of concomitant psychostimulant and/or anticataplectic drugs by patients during the HARMONY III extension study may have increased the risk of additional side effects not attributable to pitolisant hydrochloride alone. Furthermore, for the primary efficacy outcome of ESS score at the 1-year time point, LOCF was used for patients without final values, which may have biased the efficacy results, as these values may not be reflective of the true trajectory of this outcome.
External Validity
With respect to external validity, although no patients living in Canada were enrolled in the HARMONY III extension study, the characteristics of the patients enrolled in the trials were representative of patients with narcolepsy in Canada, according to the clinical experts consulted. Doses of pitolisant hydrochloride administered were in line with what would be expected in clinical practice.
Conclusions
Pitolisant hydrochloride has shown benefits over placebo in the treatment of EDS and cataplexy in patients with narcolepsy. However, because of limited comparisons and the failure of noninferiority testing, it is not possible to conclude that pitolisant hydrochloride is similar or noninferior to modafinil or other active drugs used for the treatment of EDS or cataplexy. In addition, the interaction between pitolisant hydrochloride and concomitant medications for narcolepsy is unclear. Pitolisant hydrochloride appeared to be well tolerated and no major safety concern was identified. In alignment with clinical expert input, pitolisant hydrochloride may provide an additional option for the treatment of narcolepsy because of its superiority to placebo. However, its place in therapy relative to other medications will be a challenge to elucidate from the trial results.
Introduction
Disease Background
Narcolepsy is a chronic neurologic condition that is caused by an imbalanced sleep-wake cycle or sleep-wake instability.1 It is characterized by chronic, excessive episodes of drowsiness during the day, also known as EDS.2 According to the International Classification of Sleep Disorders, Third Edition, diagnostic criteria, there are 2 types of narcolepsy: type 1 is classified as EDS with cataplexy, and type 2 is classified as EDS alone.1 Cataplexy is defined as a sudden episode of partial or complete paralysis of voluntary muscles, triggered by strong (often positive) emotion.3 Approximately 60% to 70% of patients with narcolepsy have cataplexy (type 1 disease).4 Other symptoms, including automatic behaviours in which actions are performed in a semiconscious way without awareness, such as walking, eating, driving, hallucinations, and sleep paralysis. Symptoms can vary for each patient.19 Approximately 1 in 2,000 individuals in Canada are affected by narcolepsy.2 The prevalence is likely underestimated because of misdiagnosis and the limited availability of health care providers with experience in narcolepsy.5-7
Type 1 narcolepsy is thought to occur because of a loss hypocretinergic cells in the lateral hypothalamus, likely due to an autoimmune etiology, and 90% of these patients have evidence of hypocretin deficiency.1 Type 2 narcolepsy is associated with significant EDS but no cataplexy or evidence of hypocretin deficiency.1 There is likely significant overlap between type 2 narcolepsy and idiopathic hypersomnia.1 The typical onset of symptoms happens in a person’s teens or early adulthood, but diagnosis can be delayed many years.5-7 Hypocretin (or orexin) is thought to stabilize the state between sleep and wakefulness. Consequently, when such cells are lost, the boundary between sleep and wakefulness becomes more porous or fluid, leading to sleep phenomena intruding on daytime functioning.1 Sleep paralysis involves the paralysis of REM sleep intruding on wakefulness at sleep onset or offset).1 Hypnagogic (sleep onset) and/or hypnopompic (sleep offset) hallucinations are dream-like phenomena that occur during the transition between sleep and wake, that intrude on daytime functioning. Insomnia is wakefulness of the day that intrudes on sleep at night.1
Narcolepsy can affect a patient’s HRQoL by reducing productivity and the ability to work and function in day-to-day life and social settings.8 Patients can experience EDS during common situations in the day, such as work or driving, and often during sedentary periods.3 Narcolepsy is also associated with an increased risk for comorbid conditions, including depression, anxiety, obesity, cardiovascular disease, and an increased overall mortality risk8
Updated diagnostic criteria include the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5) and International Classification of Sleep Disorders, Third Edition.1,20-22 DSM-5 criteria require EDS in association with any 1 of the following: cataplexy; cerebrospinal fluid (CSF) hypocretin deficiency; REM sleep latency of no more than 15 minutes on nocturnal polysomnography; or mean sleep latency of no more than 8 minutes on multiple sleep latency testing (MSLT) with at least 2 sleep-onset REM periods (SOREMPs).21 International Classification of Sleep Disorders, Third Edition relies on objective data in addition to EDS, somewhat complicating the diagnostic criteria: cataplexy and either positive MSLT or polysomnography findings or CSF hypocretin deficiency; MSLT criteria similar to DSM-5, except that a SOREMP on polysomnography may count as 1 of the SOREMPs required on MSLT; and distinct division of narcolepsy into type 1, which requires the presence of cataplexy or documented CSF hypocretin deficiency, and type 2, in which cataplexy is absent and CSF hypocretin levels are either normal or undocumented.23
Standards of Therapy
Based on input from the clinicians consulted by CADTH for this review, the treatment of narcolepsy includes a biopsychosocial approach. Education about the illness is important for patients, who need to understand it is a primary sleep disorder. According to the clinical experts, strategic naps and/or the appropriate use of countermeasures for sleepiness, such as caffeine, can offer modest benefit.
According to the most recent (2021) guidelines from the AASM for the treatment of hypersomnolence disorders, initial pharmacologic treatments for EDS associated with narcolepsy include modafinil and the stimulant methylphenidate (Table 4).12,24 If response is suboptimal, more potent stimulants, including lisdexamfetamine, dextroamphetamine, and/or dextroamphetamine salts can be considered. Solriamfetol is also a recommended drug for treatment of narcolepsy.12,24 For symptoms related to REM intrusion, including cataplexy, SSRIs and SNRIs are generally considered first-line therapies (e.g., fluoxetine and venlafaxine).12,24 TCAs, such as protriptyline, imipramine, and clomipramine, can also be considered for such symptoms.12,24 Sodium oxybate exits as an alternative treatment option; however, it received a Do Not List recommendation from CADTH and is not reimbursed by any public drug plans participating in the CADTH process. It is a powerful gamma-aminobutyric acid-B agonist that improves symptoms of EDS and cataplexy but has significant potential for side effects.12,24 All of the above-mentioned medications are thought to treat the underlying symptoms of narcolepsy, including primarily daytime sleepiness and cataplexy, if present. The AASM recommends pitolisant hydrochloride, sodium oxybate, and dextroamphetamine as the only medications indicated for cataplexy.12,24
According to the clinical experts consulted for this review, the current treatment standard in Canada for EDS in patients with narcolepsy is modafinil, which is thought to improve wakefulness by reducing dopamine reuptake. Stimulants such as methylphenidate (as well as dextroamphetamine and similar amphetamines) have been used off-label for narcolepsy; however, abuse and diversion of these medications does occur. SSRIs are also used off-label for the treatment of cataplexy in patients with narcolepsy.
Drug
Pitolisant hydrochloride is an inverse H3 receptor antagonist/inverse agonist. The human H3 receptor functions as a presynaptic autoreceptor on histamine-containing neurons.9,11,25 H3 antagonists promote wakefulness by increasing histamine synthesis and release. By binding competitively to H3 autoreceptors on presynaptic histaminergic neurons, pitolisant hydrochloride blocks the normal negative-feedback mechanisms for histamine release, increasing histaminergic transmission and resulting in enhanced histamine synthesis and release.10 Pitolisant hydrochloride is administered orally in doses up to 40 mg daily and is available as 5 mg and 20 mg tablets.26,27 It is indicated for the treatment of EDS or cataplexy in adults with narcolepsy. It received a Notice of Compliance from Health Canada on May 25, 2021, after undergoing standard review. The reimbursement request is per the indication.27 Pitolisant hydrochloride has not previously been reviewed by CADTH for any indication.
Table 4: Key Characteristics of Pitolisant Hydrochloride and Other Comparators
Characteristic | Pitolisant hydrochloride | Modafinil | Amphetamines | Methylphenidate | SSRIs | SNRIs | Tricyclic antidepressants |
|---|---|---|---|---|---|---|---|
Mechanism of action | H3 receptor antagonist/reverse agonist increases the synthesis and release of histamine and other neurotransmitters that promote wakefulness | Central nervous system stimulant | Wake-promoting actions by increasing dopamine | Wake-promoting actions by increasing dopamine | Suppress REM sleep | Suppress REM sleep | Suppress REM sleep |
Indicationa | Treatment of EDS or cataplexy in adults with narcolepsy | Treatment of EDS in adults with narcolepsy | Treatment of EDS in adults with narcolepsy and cataplexy | Not indicated for narcolepsy | Not indicated for narcolepsy | Not indicated for narcolepsy | Not indicated for narcolepsy |
Route of administration | Oral tablet | Oral tablet | Oral tablet | Oral tablet | Oral tablet or capsule | Oral tablet or capsule | Oral tablet or capsule |
Recommended dose | Initially 10 mg once daily, then titrated as necessary to a maximum of 40 mg once daily | 200 mg to 400 mg daily | Varies by drug | Varies by drug | Varies by drug | Varies by drug | Varies by drug |
Serious adverse effects or safety issues | Warnings for use in older adults and patients with renal impairment Warnings in patients with severe hepatic impairment, end-stage renal disease, or known QT prolongation Warnings for pregnant or breastfeeding patients Precautions for use with CYP2D6 inhibitors and CYP3A4 inducers | Warning for use in older adults Warning for use in patients with renal impairment Contraindicated in patients with severe hepatic impairment Some potential for psychotic episodes Caution for use in patients with cardiovascular comorbidities Contraindicated for pregnant or breastfeeding individuals | Some potential for psychotic episodes Caution for use in patients with cardiovascular comorbidities | Some potential for psychotic episodes Caution for use in patients with cardiovascular comorbidities | Rebound cataplexy with cessation of treatment Various side effects (e.g., sexual, headache, weight gain) | Rebound cataplexy with cessation of treatment Various side effects (e.g., sexual, headache, weight gain) | Rebound cataplexy with cessation of treatment Various side effects (e.g., headache, lightheadedness, sedation, weight gain) |
EDS = excessive daytime sleepiness; REM = raid eye movement; SSRI = selective serotonin reuptake inhibitor (e.g., escitalopram, sertraline); SNRI = selective noradrenergic reuptake inhibitor (e.g., fluoxetine, venlafaxine, atomoxetine).
aHealth Canada–approved indication.
Sources: Pitolisant hydrochloride, methylphenidate, modafinil product monographs. 12,24 to 30
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups. The full patient group submission can be found at the end of this report in the Stakeholder Feedback section.
One patient group, WUN, submitted patient input for this review. WUN is a nonprofit patient advocacy organization established in 2008 that aims to accelerate research, increase awareness of narcolepsy, and provide supportive services to patients. The input was based on a survey of 19 individuals: 18 in Canada who have a narcolepsy diagnosis or are undiagnosed but living with narcolepsy symptoms, and 1 caregiver. Most patients were aged 18 to 44 years (77%) and female (72%), and none had experience with the treatment under review.
Respondents reported EDS to be the most troubling symptom of narcolepsy, with 39% of respondents giving it a rating of 6 on a scale of 1 (not at all bothersome) to 7 (completely bothersome). The second-most troublesome symptom was DNS, followed by hallucinations when falling asleep or waking up, cataplexy, and sleep paralysis. The negative impacts of narcolepsy on respondents’ lives include mental health and emotional symptoms (mood swings, anger, depression, and anxiety), missing out on social activities, difficulty managing career and job tasks, depending on others for support for daily activities, and difficulty maintaining physical health and wellness (weight gain). Treatments that respondents were currently using for their narcolepsy include stimulants (56%), antidepressants (33%), sodium oxybate (13%), and modafinil or armodafinil (13%). Some respondents reported that the physical side effects (28%) and mental side effects (39%) of their current treatment options were moderately or extremely challenging.
Respondents would like a new drug or treatment to be more effective in treating symptoms of sleepiness, cataplexy, and DNS. Respondents indicated a desire to have a treatment that is easy to swallow and does not cause nausea, weight gain, or affect mood or personality. Respondents want a treatment with an extended-release formulation that allows them to stay awake longer during the day without having to take additional doses.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). The following input was provided by 2 clinical specialists with expertise in the diagnosis and management of narcolepsy.
Unmet Needs
Although existing medications treat the underlying symptoms of narcolepsy, including primarily daytime sleepiness and cataplexy, it is believed that none of the available treatment options address the fundamental underlying neurochemical abnormality of loss of hypocretin cells or the secondary absence or reduction of available CNS hypocretin associated with narcolepsy.
According to the clinical experts consulted for this report, treatment goals are primarily to improve quality of life. Although narcolepsy is not lethal, symptoms of EDS and cataplexy can be debilitating if left uncontrolled. In severe circumstances, sleep attacks can occur when a person is eating or even talking to someone.3,7,31 If left uncontrolled, these symptoms limit the ability of patients to perform basic daily activities, such as driving, working, and interacting with people. Cataplexy (which occurs in 60% to 70% of patients with narcolepsy) is equally, if not more, debilitating when left uncontrolled. Patients cannot drive or walk outside safely because surprises could trigger a cataplectic attack. Basic daily activities, such as showering and bathing, dressing, and eating can be dangerous and/or challenging when patients are untreated.3,7,31 Without treatment, most patients have very limited, if any, work options, and may not be able to attend school.3,7,31 These symptoms can lead to isolation, anxiety, and depression.3,7,31 Treatment is aimed at reducing EDS and cataplexy potential, so that patients are not dependent on caregivers for support and can interact and be functional members of society. Treatment can significantly improve alertness and the ability of patients to be functional members of society during the day. Diagnosis is often delayed, often occurring 10 years or longer after symptom onset, potentially leading to significant suffering, but if appropriate treatment is initiated, tolerated, and maintained, up to 80% of functional capacity could be retained.32
According to the clinical experts consulted for this review, several problems persist with existing treatment options. Not all patients respond to treatment with SSRIs, SNRIs, or TCAs, and some can become tolerant to treatment. Tolerance to the REM-suppressing effects of SSRI, SNRI, and TCA medications frequently occurs, leading to persistent cataplexy. Side effects can be problematic and can include stomach upset, night sweats, sexual side effects, and headaches. Side effects can include excessive sedation during the day, which can be a problem despite anticataplectic effects. Stimulants may not fully resolve daytime sleepiness and drugs may wear off at inopportune times, leading to EDS in the evenings and/or insomnia difficulties at night. Problematic side effects can include appetite suppression, anxiety, increased blood pressure, cardiac effects, allergic reaction, reduced seizure threshold, fetal defects, inactivation of birth control, and hair loss. There can be the potential for abuse or sequestration; usually patients with narcolepsy have a low abuse potential even though they may require high doses, but some patients could experience sequestration temptation.
Place in Therapy
Based on the efficacy of pitolisant hydrochloride in early studies, its novel mechanism of action as a H3 antagonist/inverse agonist, and its relatively favourable side-effect profile, it is likely to become an early treatment option. It received a strong recommendation in the most recent AASM (2021) guidelines for the treatment of hypersomnolence disorders.12 Although pitolisant hydrochloride does not address the underlying abnormality associated with most cases of narcolepsy related to the loss of hypocretinergic neurons, no other drugs available for the treatment of narcolepsy work through histamine. Hypocretin neurons are known to project to the tuberomammillary nucleus and contribute to wakefulness, and patients with narcolepsy have been shown to have lower histamine levels.
It will be an early drug to consider for the treatment of narcolepsy. It may also find a niche as an adjunct treatment to boost the efficacy of other drugs. It may also become a drug of choice for patients in whom stimulant and/or other therapies are contraindicated because it has no effect on birth control efficacy (unlike modafinil) and no significant known cardiovascular effects (unlike other stimulants). Because efficacy studies have shown pitolisant hydrochloride to be not visibly superior to modafinil, this drug will not change the trajectory of the treatment algorithm for narcolepsy. The main reason other medications may be used first is increased familiarity and the availability of longer-term data for other medications, such as modafinil and methylphenidate. However, it also seems quite expensive compared to methylphenidate and modafinil, which may limit its utility as an early drug. Solriamfetol might also be considered an early treatment option, but in some ways has issues similar to pitolisant hydrochloride, including cost, lack of long-term data, and minimal doctor familiarity with the medication. Patients most in need of intervention include those who cannot tolerate stimulant therapies, those who are concerned about getting pregnant, and those with a history of drug abuse.
A trial of SOC would not be required to initiate a trial of pitolisant hydrochloride. Pitolisant hydrochloride will be able to be considered as a first-line or near first-line therapy. If SOC were required, then yes, it would be an issue for jurisdictions that would only have such medications available for attention-deficit/hyperactivity disorder. Jurisdictions should continue to provide coverage for prior therapies that are currently considered SOC when used in combination with pitolisant hydrochloride because the mechanism of action of pitolisant hydrochloride is quite different than any currently available drug, which represents an exciting prospect for patients living with this debilitating condition.
The consistent use of ongoing anticataplectic treatments in combination with pitolisant hydrochloride may mask and/or minimize the potential benefits of pitolisant hydrochloride for cataplexy. However, if pitolisant hydrochloride has minimal benefits for cataplexy, it this would be difficult to assess. In short, it will be difficult to properly assess the potential benefits of pitolisant hydrochloride for cataplexy when it is used in combination with ongoing anticataplectic treatments.
It is difficult to predict which symptoms or disease characteristics will best respond to pitolisant hydrochloride. A patient with mild narcolepsy with cataplexy is most likely to respond because the hypocretin neurons are connected directly to the tuberomammillary nucleus to help promote wakefulness. Because the overall effect of pitolisant hydrochloride is comparable to that of modafinil, patients with more severe symptoms of narcolepsy will require more aggressive therapies; however, even those patients may benefit significantly from pitolisant hydrochloride as an adjunct.
Patient Population
A number of factors make diagnosis extremely challenging. Patients often first visit a family doctor or pediatrician, who may not immediately recognize this condition. Patients are frequently misdiagnosed, and narcolepsy is undiagnosed in more than 70% of patients.
Specific expertise and testing are needed to make the diagnosis, which often can occur 10 or more years after symptom onset. The MSLT administered by a specialist with training in sleep medicine is most commonly used to diagnose narcolepsy. One MSLT is thought to have a sensitivity of 85%; running it twice will bring the sensitivity to more than 95%. Patients should be off all psychiatric and stimulant medications for at least 2 weeks (some for > 1 month) before undergoing testing and should have completed sleep logs before the study. Ideally, diagnosis would involve an analysis of CSF to assess hypocretin levels, but this is technically challenging and not routinely available. Antigen testing can also be used for the diagnosis of narcolepsy with cataplexy.
Sleep medicine was recognized in Canada as an Area of Focused Competence in 2021. Most current sleep medicine specialists obtained subspecialty certification training in the US, which contributes to the shortage of physicians trained in sleep medicine. Furthermore, Quebec does not recognize sleep medicine practised by those outside of respirology, except in very few circumstances, further contributing to the probable underrecognition of this condition. Pitolisant hydrochloride will be considered for off-label use for mood disorders associated with hypersomnolence or hypersomnolence related to other conditions.
At this time, pitolisant hydrochloride may not be suitable for patients who wish to become pregnant or breastfeed. In addition, because of the lack of data on its use in children and in older adults, it should be used with caution in those populations. Pitolisant hydrochloride may not be ideal for patients who are on multiple medications (with more potential for drug interactions, particularly with those that affect the QTc interval or that are significant CYP2D6 inhibitors) or for patients who have a history of significant kidney or liver failure (with difficult-to-predict metabolism). Patients who have had adverse reactions to opioids (including hives), or who have a history of urticaria or other skin conditions, might be predisposed to an allergic reaction to pitolisant hydrochloride.
Assessing Response to Treatment
It is unclear how to select patients who might be more or most likely to respond to pitolisant hydrochloride. The primary outcomes in clinical practice will likely be a reduction in EDS, a reduction in the frequency, intensity, and duration of cataplexy episodes, and the ability to predict episodes. Clinically meaningful responses to treatment include a reduction in frequency, severity, and intensity of cataplexy episodes. Although frequency is easier to assess systematically, the intensity and severity of spells, as well as the perceived predictability of episodes, are more of a clinical assessment. For example, patients may describe certain emotions that no longer trigger episodes in the way they had before. Other parameters for assessment could include a reduction in other REM intrusion phenomena, if present, and the degree to which patients can resume normal functioning and daily activities.
Outcomes typically assessed in most clinical trials include the degree of reduction of EDS and possibly the degree of reduction in the frequency of cataplexy spells. The use of ESS scores in clinical practice to determine coverage of pitolisant hydrochloride may not be ideal. The ESS is very subjective and could easily manipulate their scores. In addition, there can be significant differences in the way male and female patients score their results, further skewing potential for the determination of coverage. In research trials, it’s ideal if patients are blinded to what they are being offered and there is no incentive to report better or worse scores. A score of 10 or lower on the ESS would indicate that sleepiness is no different than in the general population. As a comparison, an ESS score for patients with narcolepsy would typically be above 18, indicating severe sleepiness; a score of 15 to 17 indicates moderate sleepiness and a score of 11 to 14 indicates mild sleepiness.
There are few data to define an effective reduction of cataplexy. Trials of sodium oxybate demonstrated a reduction in cataplexy episodes of more than 90%. Driving is not recommended if a patient has experienced a cataplexy episode in the previous year. A minimum reduction in cataplexy episodes of 50% would be meaningful. Depending on severity and frequency, fewer than 1 episode per week would be a reasonable standard.
As with other drugs for narcolepsy, there should be close follow-up of the patient in the first months of therapy. The first follow-up should occur 1 month after the initiation of pitolisant hydrochloride, then every 1 to 2 months for the next several months, and then intermittent follow-up after that, with at least yearly follow-up, at a minimum, in the long-term.
Discontinuing Treatment
Ongoing treatment will be determined by the lack of response and/or by excess adverse effects, as with most medications. Whether it will continue to be used as an adjunct therapy if abandoned as monotherapy is unclear. Excess adverse effects, drug interactions, or a patient’s wish to become pregnant may necessitate withdrawal.
Prescribing Conditions
Medical supervision in an outpatient setting with a physician trained in sleep medicine would be appropriate for patients with narcolepsy being treated pitolisant hydrochloride. In the future, psychiatrists will likely become interested in using this medication for conditions and symptoms outside of narcolepsy. At this time, because the indication for pitolisant hydrochloride is only for narcolepsy, with a conditional recommendation for idiopathic hypersomnia, prescribing should be limited to those with specialty training or certification in sleep medicine.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may affect their ability to implement a recommendation. Implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 5.
Table 5: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Relevant comparators | |
Would a trial of the less-expensive alternative, modafinil, be required before a patient with narcolepsy without cataplexy is treated with pitolisant hydrochloride? How would jurisdictions that currently do not list modafinil handle these cases? | The use of pitolisant hydrochloride may be based on clinical preference or the situation. For example, if patient has narcolepsy without cataplexy and is anxious, pitolisant hydrochloride might be a better first-line choice. If a patient has narcolepsy and has significant depression and/or difficulties with motivation, a clinician may be more inclined to start with modafinil. Pitolisant hydrochloride has the potential to eventually displace modafinil as a first-line therapy for narcolepsy with and without cataplexy. |
Considerations for initiation of therapy | |
Would a trial of SOC be required before the initiation of pitolisant hydrochloride or would pitolisant hydrochloride be considered a first-line therapy? | With the current data available, which indicates comparable efficacy to modafinil and a favourable safety profile, pitolisant hydrochloride will be viewed as a potential first-line therapy for the treatment of narcolepsy. |
What impact do ongoing anticataleptic treatments have on the clinical evidence if trial participants were already on an anticataleptic treatment, such as sodium oxybate and antidepressants (excluding tricyclic antidepressants)? | Allowing patients to remain on previous anticataplectic therapies can lessen the noticeable effects of new therapies. Some of the SSRIs and SNRIs, such as venlafaxine, can be activating and can partially promote alertness. Doses above 150 mg/day would be expected to have more noradrenergic activity, which could lessen the effects of pitolisant hydrochloride or modafinil on daytime sleepiness perceived by the patient. This could reduce potential benefits of the wake-promoting drugs of pitolisant hydrochloride and modafinil. If sodium oxybate is used by patients with narcolepsy, the expected response to modafinil or pitolisant hydrochloride would be limited because it is known to be a treatment for both cataplexy and daytime sleepiness. |
Considerations for continuation or renewal of therapy | |
For the HARMONY 1 and HARMONY 1bis trials, the primary end points were ESS scores. Will scoring be required to show response to therapy? If so, what is considered a clinically significant change in ESS score, and is this metric used in Canadian clinical practice? | ESS is not routinely used in clinical practice to follow the efficacy, or lack thereof, of any intervention for narcolepsy. Follow-up is guided by clinical assessment and a patient’s subjective report. The ESS itself has a modest correlation with the objective assessments of wakefulness and sleepiness, which limits its usefulness in clinical practice. A meaningful change would be a change from severe to moderate or mild EDS, with an approximate difference in ESS score of 4 points. |
For the HARMONY CTP trial, the primary end point was the number of cataplexy attacks, and participants had to be experiencing at least 3 attacks per week. Will a specific number of attacks be required in patients with narcolepsy and cataplexy to show a treatment response? If so, what is considered a clinically significant reduction in attacks? | Experts would consider a clinically significant reduction a decrease in either the frequency or severity of attacks. Typically, a reduction of 50% in the frequency of events is used as a benchmark, but the severity of cataplexy attacks should also be considered. It would also be considered clinically meaningful if the frequency is stable but events are no longer debilitating because they are short-lived or don’t result in complete collapse. It is not possible to specify a number of cataplexy attacks based on clinical practice. |
Considerations for discontinuation of therapy | |
When considering the discontinuation of therapy, what would be required to measure the loss of response or absence of clinical benefit? | Discontinuation would occur in the absence of clinically meaningful changes in EDS. This could be further supported by other clinical assessments, including the inability to get or retain a job or perform particularly instrumental activities of daily living. |
Considerations for prescribing of therapy | |
Does this diagnosis require a specialist? | Yes. A study by Kryger et al. (2002) showed that only 55% of neurologists could make a correct diagnosis of narcolepsy, while this number sank to 24% for internists and family doctors, 11% for psychiatrists, and 0% for pediatricians.33 Patients with narcolepsy will most likely initially present to one of these specialists before eventually getting referred to a subspecialist for a diagnosis. The diagnosis is ideally done with the MSLT, which is only routinely offered by sleep specialists. |
Would coverage of prior therapies that are currently considered SOC be continued when used in combination with pitolisant hydrochloride? | Yes, ideally. Pitolisant hydrochloride works in a completely different way than any other pharmacologic therapy on the market. Specialists will try to combine pitolisant hydrochloride with current therapies if there is evidence of treatment resistance and/or a suboptimal response to standard therapies. |
EDS = excessive daytime sleepiness; ESS = Epworth Sleepiness Scale; MSLT = Multiple Sleep Latency Test; SNRI = selective noradrenergic reuptake inhibitor (e.g., fluoxetine, venlafaxine, atomoxetine); SSRI = selective serotonin reuptake inhibitors (e.g., escitalopram, sertraline).
Clinical Evidence
The clinical evidence included in the review of pitolisant hydrochloride is presented in 3 sections. The first section, the Systematic Review, includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as studies that were selected according to an a priori protocol. The second section includes indirect evidence from the sponsor and indirect evidence selected from the literature that met the selection criteria specified in the review. The third section includes sponsor-submitted long-term extension studies and additional relevant studies that were considered to address important gaps in the evidence included in the Systematic Review.
Systematic Review (Pivotal and Protocol-Selected Studies)
Objectives
The objective was to perform a systematic review of the beneficial and harmful effects of oral pitolisant hydrochloride (available in 5 mg and 20 mg tablets) at daily doses of up to 40 mg for the treatment of EDS or cataplexy in adults with narcolepsy.
Methods
Studies selected for inclusion in the Systematic Review included pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 6. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
Table 6: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Patient population | Adults with narcolepsy experiencing EDS or cataplexy Subgroups:
|
Intervention | Pitolisant hydrochloride, up to 40 mg daily dosage orally (5 mg and 20 mg tablets), alone or combined with comparator drugs |
Comparators | Alerting drugs:
Anticataplectics:
No treatment |
Outcomes | Efficacy outcomes:
Harms outcomes: AEs, SAEs, WDAEs, mortality, AEs of special interest (e.g., headaches, insomnia, weight gain, nausea, night sweats, sexual side effects, daytime sedation, increased blood pressure, cardiac effects, allergic reaction, reduced seizure threshold, fetal defects, inactivation of birth control, sleep disordered breathing). Notable harms include cardiovascular issues (QTc prolongation), dependence or tolerance, psychiatric issues (depression, suicidal ideation, anxiety), mild-to-severe renal impairment, and hepatic impairment |
Study design | Published and unpublished phase III and IV RCTs |
AE = adverse events; CGI-C = Clinical Global Impression of Change; CGI-S = Clinical Global Impression of Severity; EDS = excessive daytime sleepiness; ESS = Epworth Sleepiness Scale; FOSQ = Functional Outcomes of Sleep Questionnaire; HRQoL = health-related quality of life; LSEQ = Leeds Sleep Evaluation Questionnaire; MSLT = Multiple Sleep Latency Test; MWT = Maintenance of Wakefulness Test; RCT = randomized controlled trial; SAE = serious adverse event; SART = Sustained Attention to Response Task; SNRI = selective noradrenergic reuptake inhibitor; SSRI = selective serotonin reuptake inhibitors; TCA = tricyclic antidepressant; WDAE = withdrawal due to adverse event.
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy in accordance with the Peer Review of Electronic Search Strategies (PRESS) checklist.34
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946–) via Ovid and Embase (1974–) via Ovid. All Ovid searches were run simultaneously as a multifile search. Duplicates were removed using Ovid deduplication for multifile searches, followed by manual deduplication in Endnote. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concept was Wakix (pitolisant hydrochloride). The following clinical trials registries were searched: the US National Institutes of Health’s clinicaltrials.gov, WHO’s International Clinical Trials Registry Platform (ICTRP) search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
No filters were applied to limit the retrieval by study type. Where possible, retrieval was limited to the human population. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. Refer to Appendix 1 for the detailed search strategies.
The initial search was completed on February 4, 2022. Regular alerts updated the search until the meeting of the CADTH Canadian Drug Expert Committee (CDEC) on June 22, 2022.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool For Searching Health-Related Grey Literature checklist.35 Included in this search were the websites of regulatory agencies (FDA and European Medicines Agency). Google was used to search for additional internet-based materials. Refer to Appendix 1 for more information on the grey literature search strategy.
These searches were supplemented by reviewing bibliographies of key papers and through contacts with appropriate experts. In addition, the manufacturer of the drug was contacted for information regarding unpublished studies. A focused literature search for network meta-analyses dealing with Wakix (pitolisant hydrochloride) and narcolepsy was run in MEDLINE All (1946–) on February 4, 2022. No limits were applied to the search.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
Indirect Evidence
In addition to the sponsor-provided indirect evidence, additional indirect evidence that includes the patients, interventions, comparators, and outcomes specified in Table 6 may be summarized and critically appraised, if considered relevant by CADTH.
Other Relevant Studies
Additional studies not meeting the selection criteria of the protocol may be considered for inclusion in the report in this section.
Findings From the Literature
A total of 5 reports from 3 studies were identified from the literature for inclusion in the Systematic Review (Figure 1).13-15,36,37 The included studies are summarized in Table 7. A list of excluded studies is presented in Appendix 2.
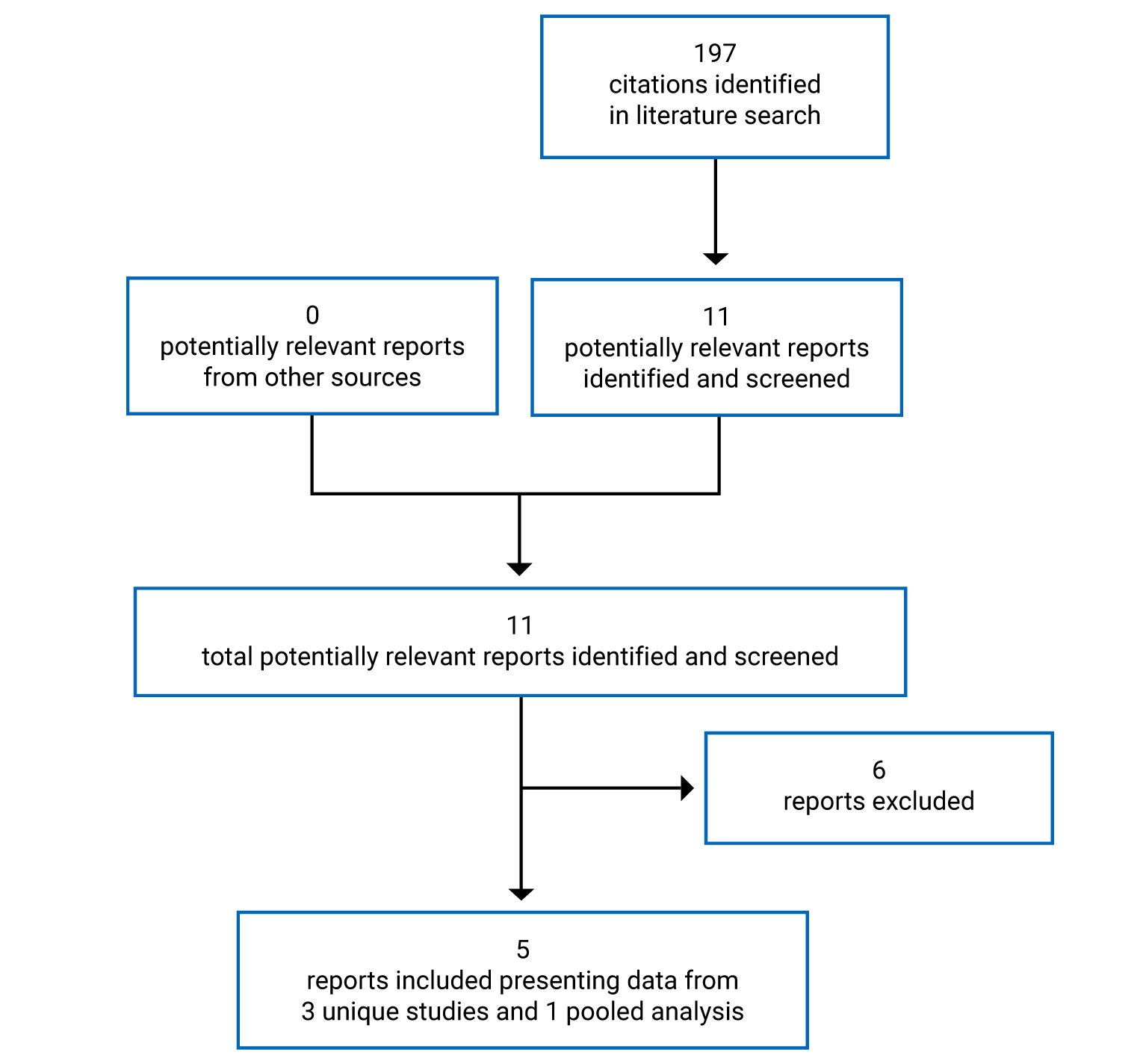
Table 7: Details of Included Studies
Characteristic | HARMONY 1 | HARMONY 1bis | HARMONY CTP |
|---|---|---|---|
Designs and populations | |||
Study design | Multicentre, RCT, DB (participants, investigator, clinical staff) | Multicentre, RCT, DB (participants, investigator, clinical staff) | Multicentre, RCT, DB (participants, investigator, clinical staff) |
Locations (sites) | Europe | Europe, South America | Asia, Europe |
Patient enrolment dates | Date of first patient enrolled: May 26, 2009 Date of last patient completed: June 30, 2010 | Date of first patient enrolled: October 25, 2010 Date of last patient completed: July 24, 2012 | Date of first patient enrolled: April 19, 2013 Date of last patient completed: January 28, 2015 |
Randomized (N) | 95 | 164 | 106 |
Inclusion criteria |
|
| |
Exclusion criteria |
|
|
|
Drugs | |||
Intervention | Pitolisant hydrochloride capsules,10 mg to 40 mg orally, taken twice daily (in the morning and at noon) | Pitolisant hydrochloride capsules, 5 mg to 20 mg orally, taken twice daily (in the morning and at noon) | Pitolisant hydrochloride capsules, 5 mg to 40 mg orally, taken before breakfast |
Comparator(s) | Modafinil capsules, 100 mg to 400 mg orally taken in the morning and at noon Placebo (oral capsule identical in appearance to the intervention) and modafinil capsules taken twice daily (in the morning and at noon) | Modafinil capsules, 100 mg to 400 mg orally taken in the morning and at noon Placebo (oral capsule identical in appearance to the intervention) and modafinil capsules taken twice daily (in the morning and at noon) | Placebo (oral capsule identical in appearance to the intervention), taken once daily before breakfast |
Duration | |||
Phase | |||
Run-in | 2-week washout period; 1-week baseline period | 2-week washout period; 1-week baseline period | 1 week washout period; 2 weeks baseline period |
DB | 8 weeks (a 3-week dose-adjustment period and a 5-week stable-dose period) | 8 weeks (a 3-week dose-adjustment period and a 5-week stable-dose period) | 7 weeks (a 3-week dose-adjustment period and a 4-week stable-dose period) |
Follow-up | 1 week (placebo withdrawal phase) | 1 week (placebo withdrawal phase) | 1 week (placebo withdrawal phase) |
Outcomes | |||
Primary end point | Daytime sleepiness assessed with ESS, compared with placebo | Daytime sleepiness assessed with ESS, compared with placebo | Number of cataplexy attacks per week during the 4-week stable-dose period |
Secondary and exploratory end points | Secondary: Daytime sleepiness assessed with ESS, compared with modafinil; daytime sleepiness assessed with ESS, MWT, patient sleep diaries, and SART; frequency of cataplexy assessed with patient sleep diaries; abuse potential assessed by evaluating DSM-IV amphetamine-like withdrawal symptoms; clinical laboratory tests and adverse reactions; effect of treatment assessed with CGI-S, CGI-C, and PGO; HRQoL assessed with the EQ-5D | Secondary: Number of cataplexy attacks per week during the 2 weeks of the end-of-treatment period; frequency of cataplexy crisis assessed with patient sleep diaries. daytime sleepiness assessed with ESS, MWT, patient sleep diaries, and SART; number of days with hallucinations; clinical laboratory tests and adverse reactions (including duration of nocturnal awakenings; tolerance; drug dependence; BDI-SF-13; and PGO of safety and quality of sleep); effect of treatment assessed with CGI-S, CGI-C, and PGO; HRQoL assessed with the EQ-5D;interaction of study-treatment effect with anticataleptic medication; treatment dose (proportion of patients at a 20 mg and 40 mg once daily stable dose) | |
Notes | |||
Publications | Dauvilliers et al. (2013)36 | — | Szakacs et al. (2017)37 |
BDI-SF-13 = 13-item Beck Depression Inventory-Short Form; CGI-C = Clinical Global Impression of Change; CGI-S = Clinical Global Impression of Severity; CNS = central nervous system; DB = double-blind; DSM-IV = Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition; EDS = excessive daytime sleepiness; ESS = Epworth Sleepiness Scale; HRQoL = health-related quality of life; MWT = Maintenance of Wakefulness Test; PGO = patient global opinion; RCT = randomized controlled trial; SART = Sustained Attention to Response Task; SSRI = selective serotonin reuptake inhibitors; TCA = tricyclic antidepressant.
Sources: Clinical Study Reports for HARMONY 1, HARMONY 1bis, and HARMONY CTP.13-15
Description of Studies
Three double-blind, placebo-controlled, phase III, RCTs met the inclusion criteria for the Systematic Review. In all 3 trials, patients were included if they had narcolepsy with cataplexy. The HARMONY 1 and HARMONY 1bis trials also included patients without cataplexy (type 2 narcolepsy).
The HARMONY 1 trial was a 12-week, double-blind, RCT that assessed the superiority of pitolisant hydrochloride to placebo with regard to EDS (based on the ESS) in patients with narcolepsy. An additional efficacy objective was to evaluate the noninferiority of pitolisant hydrochloride to modafinil for EDS (based on the ESS). Patients were enrolled from 24 sites in Europe, beginning on May 26, 2009. The trial consisted of a 2-week washout period (during which patients discontinued treatments for EDS) and a 1-week baseline period, after which patients who remained eligible were randomized to pitolisant hydrochloride (10 mg to 40 mg), modafinil (100 mg to 400 mg), or placebo. Patients underwent dose titration for 3 weeks and a final 5-week maximum dose period (highest dose reached and tolerated) (Figure 2). The treatment period was followed by a 1-week withdrawal period, during which all patients received placebo. The study randomized 95 patients (32 to pitolisant hydrochloride, 30 to placebo, and 33 to modafinil).
The HARMONY 1bis trial was similar to the HARMONY 1 trial in design, objectives, and end points. The double-blind, RCT assessed the superiority of pitolisant hydrochloride to placebo with respect to EDS (based on the ESS) in patients with narcolepsy. An additional efficacy objective was to evaluate the noninferiority of pitolisant hydrochloride to modafinil for EDS (based on the ESS). Patients were enrolled from 32 sites in Europe beginning on October 25, 2021. There was a 2-week washout period during which prohibited treatments (particularly psychostimulants) were discontinued and a 1-week baseline period, after which patients who met the selection criteria were randomized into 1 of 3 treatment groups: placebo, pitolisant hydrochloride (5 mg to 20 mg), or modafinil (100 mg to 400 mg). Patients underwent dose titration for 3 weeks and a final 5-week maximum dose period (highest dose reached and tolerated) (Figure 3). The treatment period was followed by a 1-week withdrawal period, during which all patients received placebo. A total of 166 patients were randomized (33 to placebo, 67 to pitolisant hydrochloride, and 66 to modafinil).
The HARMONY CTP trial was an 11-week, double-blind, RCT comparing pitolisant hydrochloride to placebo. It focused on the safety of pitolisant hydrochloride and its efficacy in decreasing the frequency of cataplexy attacks in patients who had narcolepsy with cataplexy. Secondary objectives included exploring the efficacy of pitolisant hydrochloride in reducing EDS and analyzing the tolerance profile of pitolisant hydrochloride in patients who had narcolepsy with cataplexy. Patients were enrolled from 16 sites in Europe and Turkey, beginning on April 19, 2013. There was a 1-week washout period, during which prohibited treatments were discontinued (Figure 4). This was followed by a 2-week period during which baseline tests were conducted, after which patients were randomized to pitolisant hydrochloride or placebo. There was 7-week treatment period that consisted of 3 weeks of dose titration up to a maximum of 40 mg and 4 weeks of stable dosing. All patients then received 1 week of placebo as the withdrawal phase. A total of 105 patients were randomized to receive either placebo (n = 51) or pitolisant hydrochloride (n = 54). A total of 13 sites were enrolled outside of Canada. The first patient was enrolled on April 19, 2013, and the last patient completed the study on January 28, 2015.
Figure 2: Study Design of the HARMONY 1 Trial
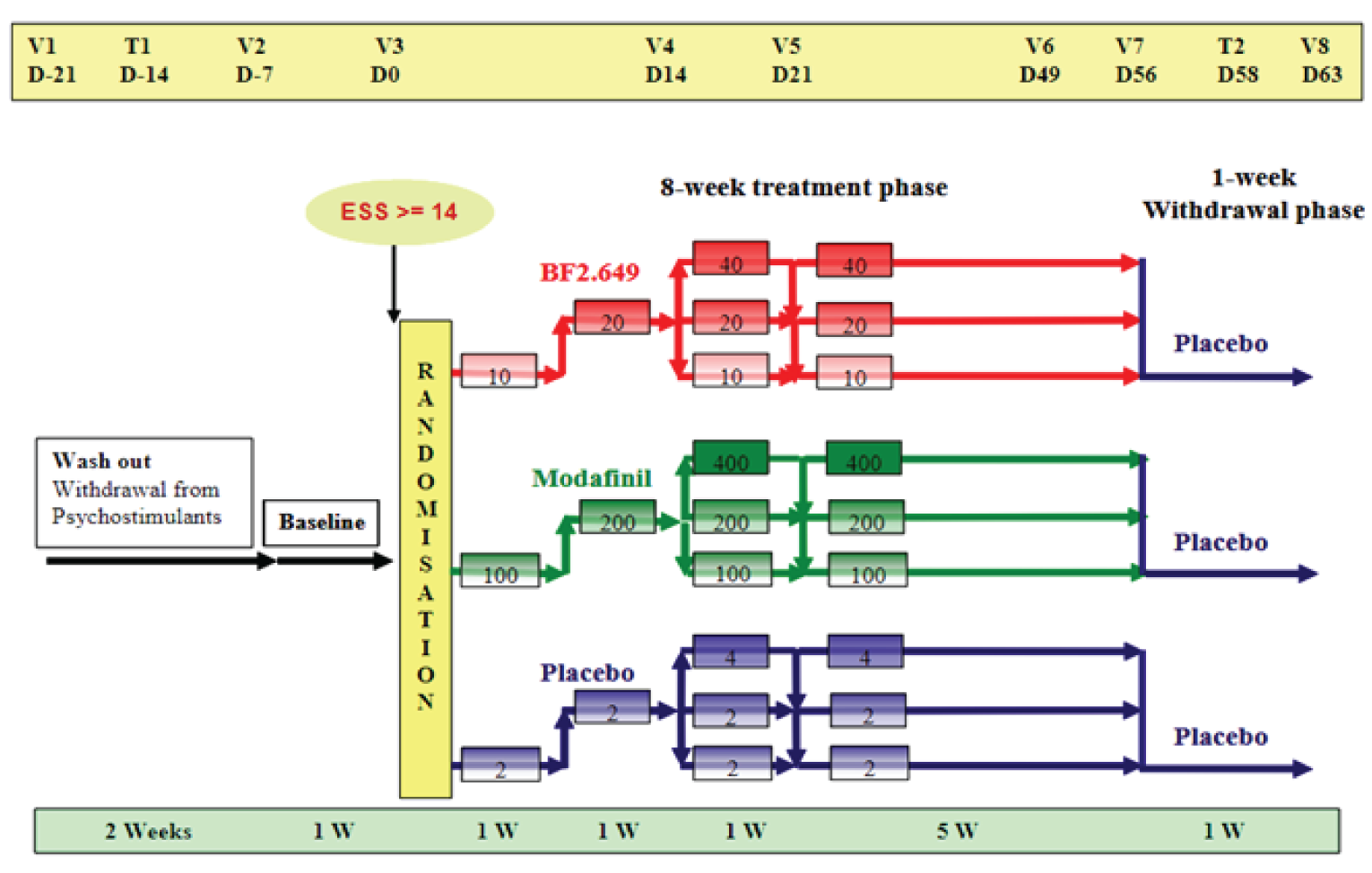
BF2.649 = pitolisant hydrochloride; D = day; ESS = Epworth Sleepiness Scale; t = telephone contact; V = visit; W = week.
Source: Clinical Study Report for HARMONY 1.13
Figure 3: Study Design of the HARMONY 1bis Trial
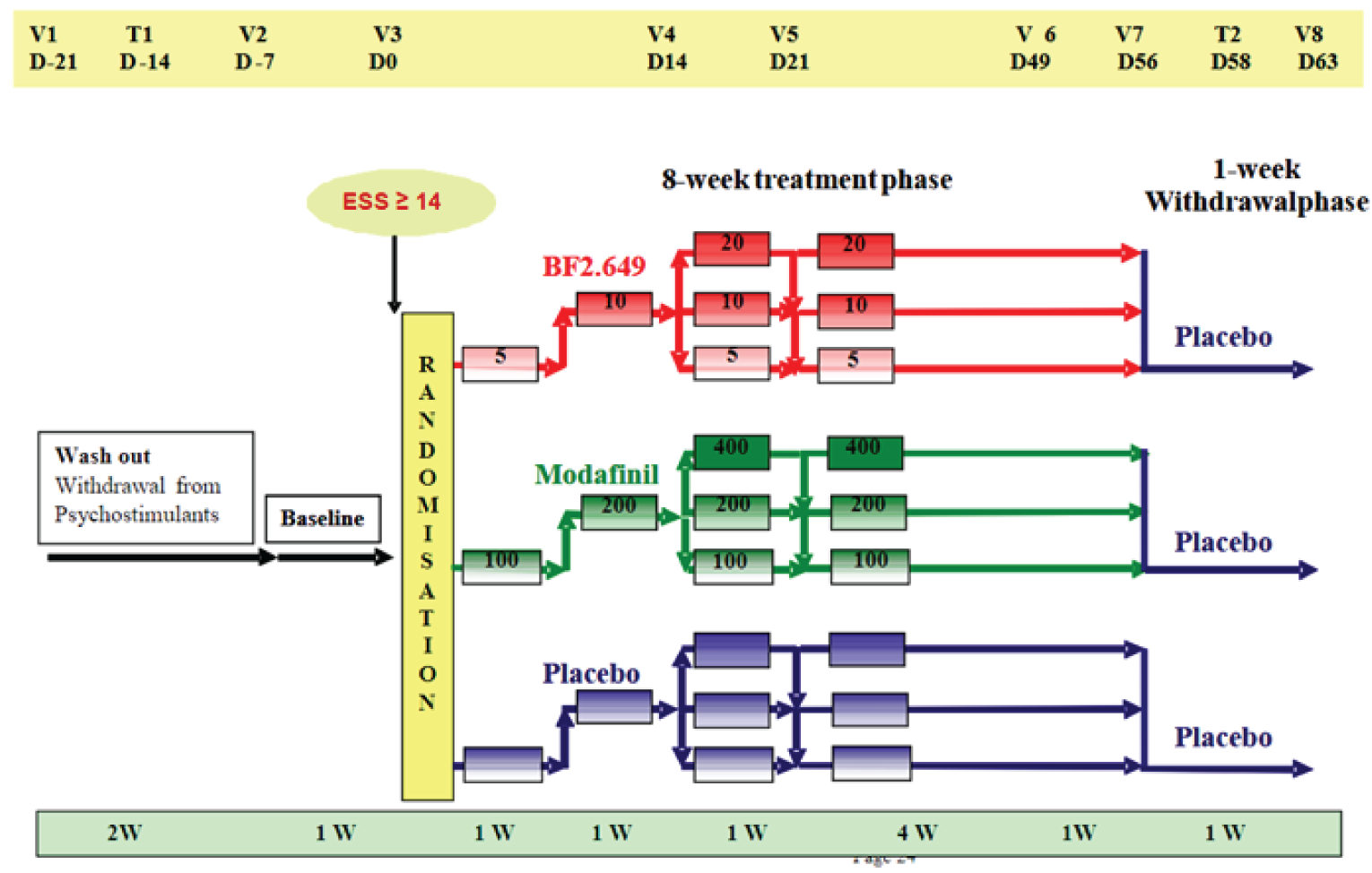
BF2.649 = pitolisant hydrochloride; D = day; ESS = Epworth Sleepiness Scale; t = telephone contact; V = visit; W = week.
Source: Clinical Study Report for HARMONY 1bis.14
Figure 4: Study Design of the HARMONY CTP Trial
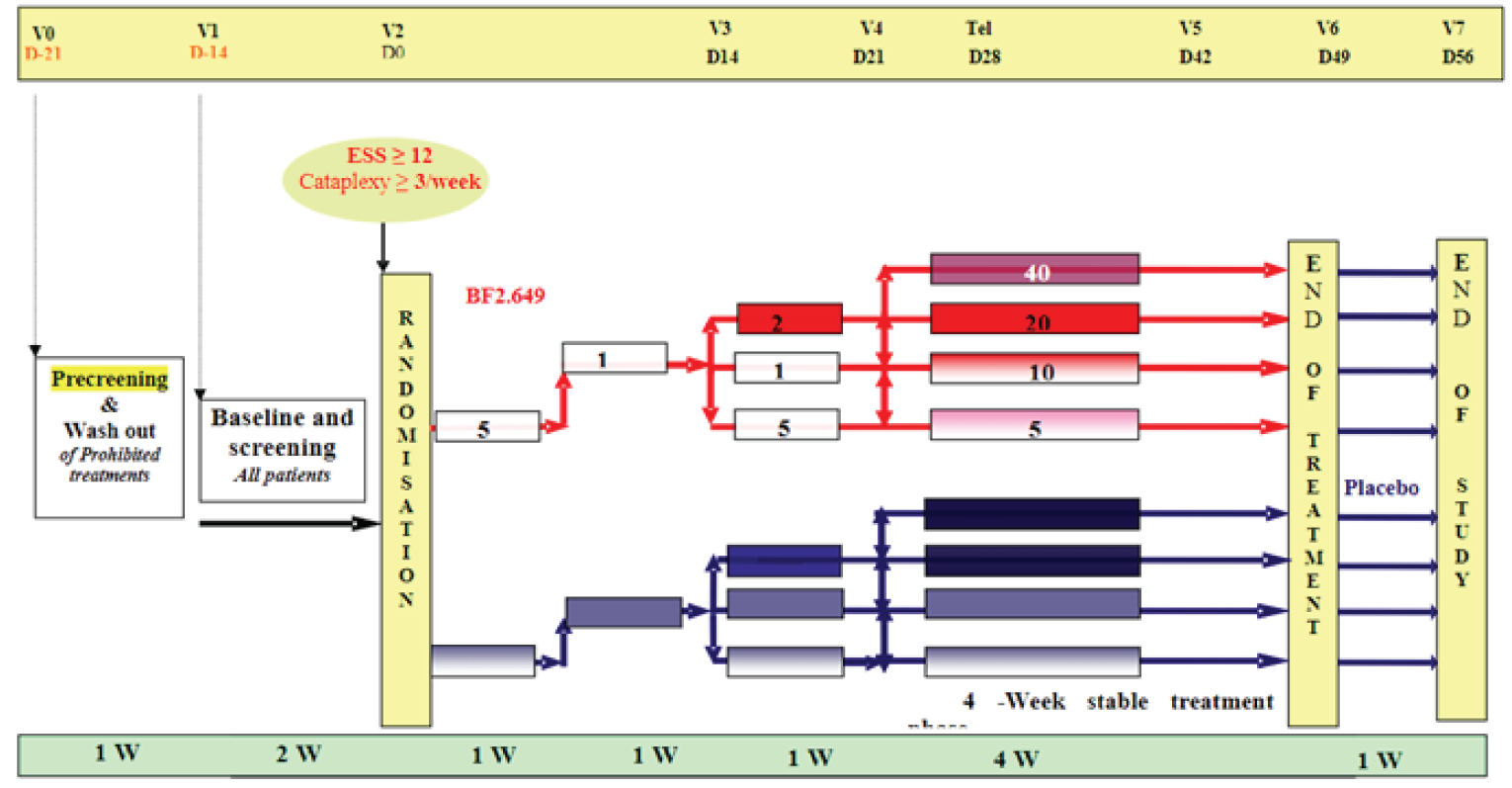
BF2.649 = pitolisant hydrochloride; D = day; ESS = Epworth Sleepiness Scale; Tel = telephone contact; V = visit; W = week.
Populations
Inclusion and Exclusion Criteria
In all 3 trials, patients were included if they had narcolepsy with cataplexy. The HARMONY 1 and HARMONY 1bis trials also included patients without cataplexy (type 2 narcolepsy). The HARMONY 1 and HARMONY 1bis trials required patients to have an ESS score of at least 14 during the baseline period, whereas the HARMONY CTP trial required an ESS score of at least 12. The HARMONY CTP trial included patients with at least 3 cataplexy attacks weekly. In all trials, patients were required to be free of drugs or to have discontinued psychostimulants for at least 7 days (HARMONY 1 and HARMONY 1bis) or 14 days (HARMONY CTP) during baseline. Patients with severe cataplexy were permitted stable doses of anticataplectic medications (except TCAs) that were administrated for at least 1 month before the trial. Doses of anticataplectic medications remained stable throughout the trials.
Patients with narcolepsy were excluded if they had other conditions that could have been the primary cause of EDS. Patients with a history of substance abuse or dependence disorder in the previous 1 year, psychiatric and neurologic disorders, prior severe adverse reactions to CNS stimulants, inability to continue daily activities safely without treatment for EDS, or any clinically significant illness that would interfere with the completion of the study were excluded.
Baseline Characteristics
In the HARMONY 1 trial, there were no clinically relevant differences between groups for any of the characteristics examined (Table 8). The median age of the patients ranged from 33 to 40 years across the 3 treatment groups. Approximately half of the patients were male (43% to 65% across groups). More than 90% of the patients in each group were white. The median time since diagnosis of narcolepsy was 15.2 (interquartile range [IQR], 9.2 to 25.3) years in the placebo group, 11.2 (IQR, 8.2 to 18.0) years in the pitolisant hydrochloride group, and 12.2 (IQR, 5.7 to 20.3) years in the modafinil group. Overall, approximately 80% of the patients in each group reported a history of cataplexy. More than half of all patients reported histories of sleep paralysis (48% to 67% across groups), hallucinations (58% to 64% across groups), or dyssomnia (47% to 61% across groups). Less than half of all patients (30% to 49% across groups) reported automatic behaviour. The MSWT was performed on 54 of the 98 patients at baseline, and the mean latency time appeared to be shorter in the pitolisant hydrochloride group (3.7 [SD = 2.6] minutes) than in the placebo (5.4 [SD = 2.0] minutes) and modafinil (4.9 [SD = 2.4] minutes) groups. The number of patients who took at least 1 chronic medication for the treatment of narcolepsy during the 3-month period before inclusion was 85% in the placebo and pitolisant hydrochloride groups and 70% in the modafinil group. Thirty-nine percent of patients were treated with modafinil before baseline (43% in the placebo group, 42% in the pitolisant hydrochloride group, and 33% in the modafinil group). Across all treatment arms, 33 patients with severe cataplexy were permitted to remain on their anticataplectic medications at stable doses for the duration of the trial (8 taking sodium oxybate and 25 taking antidepressants). The ESS scores at baseline ranged from 17.8 to 18.9 across treatment groups. At baseline across groups, patients with cataplexy reported approximately 1 complete or partial cataplexy episode per day.
Table 8: Summary of Baseline Characteristics — HARMONY 1 (ITT Population) Trial
Characteristic | Placebo (N = 30) | Pitolisant hydrochloride (N = 31) | Modafinil (N = 33) |
|---|---|---|---|
Age in years, median (range) | 39.5 (30.0 to 52.0) | 33.0 (21.0 to 49.0) | 40.0 (25.1 to 48.0) |
Weight in kg, mean (SD) | 81.0 (20.7) | 90.9 (21.0) | 81.0 (16.3) |
Height in cm, mean (SD) | 168.8 (10.4) | 173.9 (9.8) | 171.0 (8.5) |
Body mass index, kg/m2, mean (SD) | 28.2 (6.0) | 30.4 (8.3) | 27.7 (5.3) |
Sex, males, n (%) | 13 (43.3) | 20 (64.5) | 18 (54.5) |
2 years postmenopause or sterilized, n (% of females)a | 6 (35.3) | 3 (27.3) | 4 (26.7) |
Ethnic origin (white), n (%) | 28 (93.3) | 29 (93.5) | 32 (97.0) |
Duration of narcolepsy in years at screening, median (25th % to 75th %) | 15.2 (9.2 to 25.3) | 11.1 (8.2 to 18.0) | 12.2 (5.7 to 20.3) |
Multiple sleep latency test in minutes, mean (SD)a | 5.4 (2.0) | 3.7 (2.6) | 4.9 (2.4) |
History of cataplexy, n (%) | 24 (80.0) | 25 (80.6) | 27 (81.8) |
History of associated symptoms, n (%) | |||
Sleep paralysis | 15 (50.0) | 15 (48.4) | 22 (66.7) |
Hypnagogic hallucinations | 19 (63.3) | 18 (58.1) | 21 (63.6) |
Automatic behaviour | 9 (30.0) | 15 (48.4) | 16 (48.5) |
Bad night-time sleep or dyssomnia | 14 (46.7) | 18 (58.1) | 20 (60.6) |
Patients with at least 1 long-term course of medication ≥ 3 months before inclusion, n (%) | 13 (43.3) | 14 (45.2) | 16 (48.5) |
Patients with at least 1 chronic medication in the 3 months before inclusion, n (%) | 23 (85.2) | 23 (85.2) | 21 (70.0) |
Patients treated with modafinil before baseline, n (%) | 13 (43.0) | 13 (42.0) | 11 (33.3) |
ESS score at baseline, mean (SD) | 18.9 (2.5) | 17.8 (2.5) | 18.5 (2.7) |
SART-NOGO at baseline, GMTb (95% CI)a | 8.0 (6.5 to 10.0) | 9.1 (7.3 to 11.3) | 9.0 (7.3 to 11.0) |
SART-GO at baseline, GMTb (95% CI) | 3.5 (2.4 to 5.1) | 3.6 (2.6 to 4.9) | 3.3 (2.3 to 4.8) |
SART-TOTAL at baseline, GMTb (95% CI)a | 11.4 (8.8 to 14.9) | 12.5 (9.9 to 15.7) | 11.4 (8.9 to 14.5) |
MWT at baseline, GMTb (95% CI) | 8.4 (5.7 to 12.4) | 7.4 (5.4 to 10.1) | 8.8 (6.3 to 12.2) |
Complete + partial cataplexy episodes at baselinec (episodes per day), mean (SD)a | 0.92 (0.87) | 1.2 (1.8) | 1.1 (1.9) |
Sleep paralysis episodes at baselinec (daily rate), mean (SD)a | 0.33 (0.35) | 0.29 (0.42) | 0.27 (0.27) |
Hallucination episodes at baselinec (daily rate), mean (SD)a | 0.73 (1.73) | 0.15 (0.22) | 0.32 (0.50) |
CI = confidence interval; ESS = Epworth Sleepiness Scale; GMT = geometric mean based on natural logarithm; ITT = intention-to-treat; MWT = Maintenance of Wakefulness Test; SART = Sustained Attention to Response Task; SD = standard deviation.
Note: Screening is visit 1. ESS baseline is defined as (visit 2 + visit 3) / 2.
aThere were missing data for some baseline variables. Sample sizes for the placebo, pitolisant hydrochloride, and modafinil groups, respectively, were 17, 11, and 15 for the percent of postmenopause or sterilized females; 18, 20, and 20 for the MSLT; 30, 29, and 33 for SART-NOGO; 30, 30, and 33 for SART-TOTAL; 14, 20, and 23 for the number of cataplexy episodes; 9, 8, and 14 for the number of sleep paralysis episodes, and 13, 11, and 15 for the number of hallucination episodes.
bGMT is the geometric mean based on natural logarithm. The geometric mean was used as the data were of a log-normal distribution. The mean values of log were compared between treatment groups with a Student t-test, and converted as a geometric mean to estimate the geometric mean ratio with the corresponding 95% CI.
cBaseline = (all episodes at visit 2 and visit 3) / (number of days at visit 2 and visit 3).
Source: Clinical Study Report for HARMONY 1.13
In the HARMONY 1bis trial, the 3 treatment groups were comparable for the baseline demographic characteristics of age, weight, height, body mass index (BMI), sex, and ethnicity (Table 9). The median age ranged from 37 to 43 years across the 3 treatment groups. Nearly half of patients were male (47% to 48% across groups). The median time since diagnosis of narcolepsy was 11 (range, 0 to 62) years in the placebo arm, 15 (range, 0 to 47) years in the pitolisant hydrochloride arm, and 10 (range, 0 to 59) years in the modafinil arm. Overall, approximately 80% of the patients reported a history of cataplexy (75% to 81% across groups). Patients reported histories of sleep paralysis (45% to 69% across groups), hallucinations (52% to 63% across groups), or dyssomnia (25% to 40% across groups). MSLT was performed on 129 of the 163 patients at baseline, and the mean latency time appeared comparable. The mean baseline ESS score was approximately 18 in each group at baseline. Eighty-one percent of patients in the placebo group and 75% of patients in each of the pitolisant hydrochloride and modafinil groups had a history of cataplexy. The proportion of patients who took at least 1 chronic medication before inclusion was 52% in the placebo group, 56% in the pitolisant hydrochloride group, and 61% in the modafinil group.
In the HARMONY CTP trial, both treatment groups had comparable baseline demographic characteristics (Table 10). The median age was 39 (range, 18 to 66) years in the placebo group and 34 (range, 18 to 64) years in the pitolisant hydrochloride group. Approximately half of the patients in each group were male. The mean time since diagnosis of narcolepsy was not reported. At baseline, the mean number of cataplexy attacks per week was 9.2 (SD = 8.8) and 11.0 (SD = 8.9) in the placebo and pitolisant hydrochloride groups, respectively. The severity of cataplexy was rated on the CGI-S scale as moderate to severe by most patients (92% in the placebo group and 89% in the pitolisant hydrochloride group). The proportion of patients with associated symptoms (sleep paralysis, hallucinations, automatic behaviour, and dyssomnia) were similar in the treatment groups, except for ongoing sleep paralysis (44% in pitolisant hydrochloride group and 59% in placebo group). In the 3 months preceding inclusion, 66 (63%) patients had received anticataplectic medication (69% in the placebo group and 52% in the pitolisant hydrochloride group).
||||||||||||||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||| | |||||||||||||||||||||||||||||| |
|---|---|---|---|
|||||||||||||||||||||||||||||| | |||||||||| | |||||||||| | |||||||||| |
|||||||||||||||||||||||||||||| | |||||||||| | |||||||||| | |||||||||| |
|||||||||||||||||||||||||||||| | |||||||||| | |||||||||| | |||||||||| |
|||||||||||||||||||||||||||||| | |||||||||| | |||||||||| | |||||||||| |
|||||||||||||||||||||||||||||| | |||||||||| | |||||||||| | |||||||||| |
|||||||||||||||||||||||||||||| | |||||||||| | |||||||||| | |||||||||| |
|||||||||||||||||||||||||||||| | |||||||||| | |||||||||| | |||||||||| |
|||||||||||||||||||||||||||||| | |||||||||| | |||||||||| | |||||||||| |
|||||||||||||||||||||||||||||| | |||||||||| | |||||||||| | |||||||||| |
|||||||||||||||||||||||||||||| | |||
|||||||||||||||||||||||||||||| | |||||||||| | |||||||||| | |||||||||| |
|||||||||||||||||||||||||||||| | |||||||||| | |||||||||| | |||||||||| |
|||||||||||||||||||||||||||||| | |||||||||| | |||||||||| | |||||||||| |
|||||||||||||||||||||||||||||| | |||||||||| | |||||||||| | |||||||||| |
|||||||||||||||||||||||||||||| | |||||||||| | |||||||||| | |||||||||| |
|||||||||||||||||||||||||||||| | |||||||||| | |||||||||| | |||||||||| |
|||||||||||||||||||||||||||||| | |||||||||| | |||||||||| | |||||||||| |
|||||||||||||||||||||||||||||| | |||||||||| | |||||||||| | |||||||||| |
|||||||||||||||||||||||||||||| | |||||||||| | |||||||||| | |||||||||| |
|||||||||||||||||||||||||||||| | |||||||||| | |||||||||| | |||||||||| |
|||||||||||||||||||||||||||||| | |||||||||| | |||||||||| | |||||||||| |
|||||||||||||||||||||||||||||| | |||||||||| | |||||||||| | |||||||||| |
|||||||||||||||||||||||||||||| | |||||||||| | |||||||||| | |||||||||| |
Note that some redacted rows have been deleted.
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Table 10: Summary of Baseline Characteristics— HARMONY CTP (ITT Population) Trial
Characteristic | Placebo (N = 51) | Pitolisant hydrochloride (N = 54) |
|---|---|---|
Age in years, median (range) | 39 (18 to 66) | 34 (18 to 64) |
Weight in kg, mean (SD) | 85 (18.3) | 80.1 (17.8) |
Height in cm, mean (SD) | 172 (10.7) | 171.4 (9.1) |
Body mass index, kg/m2, mean (SD) | 28.8 (6) | 27.2 (5.2) |
Sex, male, n (%) | 27 (53) | 26 (48) |
History of associated symptoms, n (%) | ||
Hallucinations | 32 (63) | 36 (67) |
Ongoing hallucinations | 27 (53) | 32 (59) |
Automatic behaviour | 14 (27) | 16 (30) |
Ongoing automatic behaviour | 13 (26) | 13 (24) |
Dyssomnia | 32 (63) | 37 (69) |
Ongoing dyssomnia | 31 (61) | 33 (61) |
Sleep paralysis | 32 (63) | 32 (59) |
Ongoing sleep paralysis | 30 (59) | 24 (44) |
Patients with ≥ 1 cataplexy medication in previous 3 months, n (%) | 35 (68.6) | 28 (51.9) |
Patients continuing cataplexy medications during the trial, n (%) | 8 (16) | 4 (7) |
Cataplexy episodes per week at prescreening, mean (SD) | 9.2 (8.8) | 11 (8.9) |
ESS score at screening, mean (SD) | 17.1 (3.4) | 17.3 (3.3) |
MWT at baseline, GMT (95% CI) | 4.3 (3.0 to 6.2) | 3.7 (2.7 to 5.2) |
BDI-SF-13 item score at screening, mean (SD) | 5.3 (4.3) | 5.3 (4.1) |
Sleep latency time at prescreening, mean (SD) | 7.8 (7.8) | 6.9 (7.7) |
CGI-S EDS at screening, n (%) | ||
Mildly ill | 1 (2.0) | 1 (1.9) |
Moderately ill | 13 (25.5) | 16 (29.6) |
Markedly ill | 24 (47.1) | 20 (37.0) |
Severely ill | 12 (23.5) | 16 (29.6) |
Among the most extremely ill patients | 1 (2.0) | 1 (1.9) |
CGI-S cataplexy at screening, n (%) | ||
Mildly ill | 4 (7.8) | 6 (11.1) |
Moderately ill | 15 (29.4) | 20 (37.0) |
Markedly ill | 20 (39.2) | 21 (38.9) |
Severely ill | 9 (17.6) | 5 (9.3) |
Among the most extremely ill patients | 3 (5.9) | 2 (3.7) |
BDI-SF-13 = 13-item Beck Depression Inventory-Short Form; CGI-S = Clinical Global Impression of Severity; CI = confidence interval; EDS = excessive daytime sleepiness; ESS = Epworth Sleepiness Scale; GMT = geometric mean based on natural logarithm; ITT = intention-to-treat; MWT = Maintenance of Wakefulness Test; SD = standard deviation.
Source: Clinical Study Report for HARMONY CTP.15
Interventions
All 3 trials were double-blind and investigational treatments were provided in capsules that were identical in appearance for all treatments and provided in blister packs. In the HARMONY 1 and HARMONY 1bis trials, patients were randomized to receive placebo, pitolisant hydrochloride, or modafinil, whereas in the HARMONY CTP trial, patients were randomized to placebo or pitolisant hydrochloride. For the HARMONY 1 trial, patients were instructed to take 4 capsules per day, orally, with a glass of water (2 in the morning and 2 at noon). For the HARMONY 1bis trial, patients were instructed to take 2 capsules per day, orally, with a glass of water (1 in the morning and 1 at noon). For the HARMONY CTP trial, patients were instructed to take 1 capsule per day, orally, with a glass of water before breakfast, at around 8:00 a.m. For all trials, the individual dosage of pitolisant hydrochloride and modafinil was determined by investigators, according to clinical efficacy and tolerance criteria (individual dose titration). The maximum dosage of pitolisant hydrochloride was 40 mg per day in the HARMONY 1 trial (range, 10 mg to 40 mg) and the HARMONY CTP trial (range, 5 mg to 40 mg), whereas in the HARMONY 1bis trial, the maximum dosage was 20 mg daily (range, 5 mg to 20 mg). The maximum dosage of modafinil in the HARMONY 1 and HARMONY 1bis trials was 400 mg (range, 100 mg to 400 mg).
Both the HARMONY 1 and HARMONY 1bis trials had a dose-titration period of 3 weeks and a stable-dose period of 5 weeks, whereas the HARMONY CTP trial had a dose-titration period of 3 weeks and a stable-dose period of 4 weeks. In the HARMONY 1 trial, patients were asked to take a low dose of pitolisant hydrochloride (10 mg) or modafinil (100 mg) for the first 7 days and a medium dose of pitolisant hydrochloride (20 mg) or modafinil (200 mg) for the next 7 days. At the beginning of the maximal-dose period, doses were adjusted for each patient after assessment of efficacy and tolerance by the investigator, according to a predefined dose adjustment of up to 20 mg daily for pitolisant hydrochloride. Patients continued at their assigned stable dose for an additional 4 weeks. From the 1-week withdrawal phase up to the final study visit, all patients received placebo. In the HARMONY 1bis trial, patients took a low dose (5 mg of pitolisant hydrochloride or 100 mg of modafinil or placebo) for 7 days, then a middle dose (10 mg of pitolisant hydrochloride or 200 mg of modafinil or placebo) for the next 7 days. At the beginning of the maximal-dose period, doses were adjusted for each patient after assessment of efficacy and tolerance by the investigator, according to a predefined dose adjustment of up to 20 mg daily for pitolisant hydrochloride. The dose remained stable for a 5-week period. Treatment was stopped at day 56. From the 1-week withdrawal phase up to the final study visit, all patients received placebo. During the first week of the HARMONY CTP trial, all patients took a low dose of pitolisant hydrochloride (5 mg), followed by a second week of a middle dose of pitolisant hydrochloride (10 mg) or placebo. At day 14, doses were adjusted for each patient after assessment of clinical efficacy and safety by investigators, and each patient was assigned an individual optimum dose for the following 4 weeks.
Concomitant Medications Permitted
Across all trials, patients discontinued psychostimulant medications before the start of the baseline period (14 days prior for HARMONY 1 and HARMONY 1bis; 7 days prior for HARMONY CTP). Patients with severe cataplexy who were on stable doses for at least 1 month before the trial were authorized to continue anticataplectic treatments at stable doses throughout the trial. TCAs were not authorized. In the HARMONY 1 trial, no definitive list of authorized medications was provided; however, sodium oxybate and antidepressive drugs like SSRIs and SNRIs were indicated. In the HARMONY 1bis and HARMONY CTP trials, authorized medications included sodium oxybate, sertraline, fluoxetine, venlafaxine, atomoxetine, fluvoxamine, femoxetine, citalopram, paroxetine, viloxazine, reboxetine, and any other SSRI or SNRIs.
Outcomes
A list of efficacy end points identified in the CADTH review protocol that were assessed in the clinical trials included in this review is provided in Table 11 and summarized after Table 14. The timing of efficacy evaluations in the HARMONY 1, HARMONY 1bis, and HARMONY CTP trials are shown in Table 12, Table 13, and Table 14, respectively. A detailed discussion and critical appraisal of the outcome measures is provided in Appendix 4.
Table 11: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure | HARMONY 1 | HARMONY 1bis | HARMONY CTP |
|---|---|---|---|
Sleepiness, alertness, severity of daytime sleepiness | Primary: ESS Secondary: MWT, SART, sleep diary, CGI-S | Primary: ESS Secondary: MWT, SART, sleep diary, CGI-S | Secondary: ESS, MWT, sleep diary, CGI-S |
Frequency and severity of cataplexy attacks | Secondary: sleep diary, CGI-S, CGI-C | Secondary: sleep diary, CGI-S, CGI-C | Primary: sleep diary, CGI-S, CGI-C |
Health-related quality of life | Secondary: 5-Level EQ-5D | Secondary: 5-Level EQ-5D | Secondary: 5-Level EQ-5D, EQ-5D VAS |
Functional activity | NR | NR | NR |
Mental health | Safety: BDI-SF-13 | Safety: BDI-SF-13 | Safety: BDI-SF-13 |
Sexual function | NR | NR | NR |
Sleep attacks (frequency and duration) | NR | NR | NR |
Nocturnal sleep properties | Secondary: sleep diary | Secondary: sleep diary | Safety |
Number of hallucinations | Secondary: sleep diary | Secondary: sleep diary | Secondary: sleep diary |
Concomitant medication use | Monitored, but not an efficacy outcome | Monitored, but not an efficacy outcome | Monitored, but not an efficacy outcome |
Patient satisfaction, ease of use | Secondary: PGO | Secondary: PGO | Secondary: PGO |
Adherence | Monitored, but not an efficacy outcome | Monitored, but not an efficacy outcome | Monitored, but not an efficacy outcome |
Health care resource utilization | NR | NR | NR |
BDI-SF-13 = 13-item Beck Depression Inventory-Short Form; CGI-S = Clinical Global Impression of Severity; ESS = Epworth Sleepiness Scale; MWT = Maintenance of Wakefulness Test; NR = not reported; PGO = patient global opinion; SART = Sustained Attention to Response Task; VAS = visual analogue scale.
Table 12: Overall Time and Events Schedule for the HARMONY 1 Trial
Visit | Screening V1 | Phone contact | Baseline V2 | Inclusion V3 | Titration V4 | Titration V5 | Control V6 | End point V7 | Phone contact | Withdraw V8 | Premature drop out |
|---|---|---|---|---|---|---|---|---|---|---|---|
Study day | 21 | 14 + 1 | 7 + 2 | 0 + 2 | 14 ± 2 | 21 ± 2 | 49 ± 2 | 56 ± 2 | 58 ± 1 | 63 ± 2 | + 3 |
ESS | Yes | — | Yes | Yes if mean ≥ 14 | Yes | Yes | Yes | Yes | — | Yes | Yes |
CGI-S or CGI-C EDS + cataplexy | — | — | Yes | Yes | Yes | Yes | Yes | Yes | — | Yes | Yes |
SART | — | — | — | Yes | — | — | — | Yes | — | — | Yes |
40-minute MWT | — | — | — | Yes | — | — | — | Yes | — | — | Yes |
EQ-5D | — | — | Yes | Yes | — | Yes | — | Yes | — | Yes | Yes |
Withdrawal symptoms | — | — | — | — | — | — | — | — | Yes | Yes | Yes |
PGO | — | — | — | — | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
CGI-C = Clinical Global Impression of Change; CGI-S = Clinical Global Impression of Severity; EDS = excessive daytime sleepiness; ESS = Epworth Sleepiness Scale; MWT = Maintenance of Wakefulness Test; PGO = patient global opinion; SART = Sustained Attention to Response Task; V = study visit.
Source: Clinical Study Report for HARMONY 1.13
Table 13: Overall Time and Events Schedule for the HARMONY 1bis Trial
Visits | Screening V1 | Phone contact | Baseline V2 | Inclusion V3 | Titration V4 | Titration V5 | Control V6 | End point V7 | Phone contact | Withdraw V8 | Premature drop out |
|---|---|---|---|---|---|---|---|---|---|---|---|
Study day | 21 | 14 ± 1 | 7 ± 2 | 0 ± 2 | 14 ± 2 | 21 ± 2 | 49 ± 2 | 56 ± 2 | 58 ± 1 | 63 ± 2 | + 3 |
Polysomnography | — | — | — | Yes | — | — | — | Yes | — | — | — |
ESS | Yes | — | Yes | Yes if mean ≥ 14 | Yes | Yes | Yes | Yes | — | Yes | Yes |
CGI-S or CGI-C EDS + cataplexy | — | — | Yes | Yes | Yes | Yes | Yes | Yes | — | Yes | Yes |
40-minute MWT (4 sessions) | — | — | — | Yes | — | — | — | Yes | — | — | — |
SART | — | — | — | Yes | — | — | — | Yes | — | — | Yes |
EQ-5D | — | — | Yes | Yes | Yes | Yes | — | Yes | Yes | ||
PGO | — | — | — | — | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
CGI-C = Clinical Global Impression of Change; CGI-C = Clinical Global Impression of Severity; EDS = excessive daytime sleepiness; ESS = Epworth Sleepiness Scale; MWT = Maintenance of Wakefulness Test; PGO = patient global opinion; SART = Sustained Attention to Response Task; V = study visit.
Source: Clinical Study Report for HARMONY 1bis.14
Table 14: Overall Time and Events Schedule for the HARMONY CTP Trial
Visit | V0 Prescreening and washout period (treated patients) | V1 Screening and baseline visit | V2 Treatment start | V3 | V4 | Phone contact | V5 | V6 End of treatment | V7 End of study |
|---|---|---|---|---|---|---|---|---|---|
Study day | 21 | 14 | 0 | 14 ± 2 days | 21 ± 2 days | 28 | 42 ± 2 days | 49 ± 2 days | 56 ± 2 days |
Vital signs | — | Yes | Yes | Yes | Yes | — | Yes | Yes | Yes |
ESS | — | Yes | Yes | Yes | Yes | — | Yes | Yes | Yes |
MWT 40 minute (4 sessions) | — | — | Yes | — | — | — | — | Yes | — |
CGI-S on EDS and cataplexy | — | Yes | Yes | — | — | — | — | — | — |
CGI-C on EDS and cataplexy | — | — | — | Yes | Yes | — | Yes | Yes | Yes |
EQ-5D | — | — | Yes | Yes | — | — | Yes | — | |
PGO | — | — | — | — | Yes | — | Yes | Yes | Yes |
Withdrawal symptoms questionnaire | — | — | — | — | — | — | — | — | Yes |
CGI-C = Clinical Global Impression of Change; CGI-S = Clinical Global Impression of Severity; EDS = excessive daytime sleepiness; ESS = Epworth Sleepiness Scale; MWT = Maintenance of Wakefulness Test; PGO = patient global opinion; V = study visit.
Source: Clinical Study Report for HARMONY CTP.15
Sleepiness, Alertness, and Severity of Daytime Sleepiness
Epworth Sleepiness Scale
In the HARMONY 1 and HARMONY 1bis trials, the primary outcome of interest was EDS, assessed with the ESS. EDS assessed with the ESS was a secondary outcome of interest in the HARMONY CTP trial. The ESS is a self-administered questionnaire that evaluates the chance of dozing in 8 different situations often encountered in daily life, ranging from “would never doze” to “high chance of dozing.”38,39 The scores are summed to yield a total score from 0 to 24, with higher scores representing greater sleepiness. A score greater than 10 is considered to indicate abnormal sleepiness. The ESS was completed by the patient at each visit and reviewed by the investigator.
In all 3 trials, the baseline value was calculated as the mean of the first 2 weeks of treatment, whereas the end point value was calculated as the mean of the final 2 weeks on treatment. Responders were defined as those with an ESS score of 10 or lower at the end of treatment.
Maintenance of Wakefulness Test
The MWT is used to assess a patient’s ability to stay awake while resisting pressure to fall asleep. For all trials, patients were required to have sufficient nocturnal sleep (minimum of 6 hours) and not drink alcohol the night before the test. Patients were required to recline in a quiet, dimly lit room and instructed to stay awake as long as possible. Sleep onset was defined as either 3 consecutive 30-second epochs of stage 1 sleep or any single 30-second epoch of stage 2, 3, 4, or REM sleep. In all 3 trials, the MWT was performed 4 times at 2-hour intervals at the inclusion visit and at end point visit.
Sustained Attention to Response Task
The SART is used to evaluate a patient’s vigilance and attention. Patients were required to have sufficient nocturnal sleep (minimum of 6 hours) and not drink alcohol the night before the SART.40 While seated in front of a computer screen, patients were shown numbers, from 1 to 9, 225 times in quasi-random order. Patients were asked to respond to the appearance of each number by pressing a button, except when the number was 3. The SART error score is the total number of errors (i.e., the button was pressed when 3 was presented or the button was not pressed when it should have been). In the HARMONY 1 and HARMONY 1bis trials, SART was performed 4 times at 2-hour intervals at the inclusion visit and at the end point visit.
Patient Diaries
Patient diaries (electronic or paper) were used in the HARMONY 1 and HARMONY 1bis trials to capture EDS using the ESS score and the number and duration of diurnal sleep and sleepiness episodes, and in the HARMONY CTP trial to capture sleepiness more generally. During the initial visit, patients were instructed by the investigators on the use of the diary and provided a detailed review of definitions for the information to be collected. Patients were required to fill in the sleep diary every morning or evening for each 24-hour period in the 7 or 14 days preceding the scheduled visit.
Clinical Global Impression
The severity of EDS was measured by the investigator with the CGI-S and CGI-C. In the HARMONY 1 and HARMONY 1bis trials, during the 2 baseline visits before randomization, CGI-S was rated by the investigator using a 6-grade scale (no sign of illness, borderline ill, slightly ill, moderately ill, markedly ill, among the most extremely ill patients). At each post-baseline visit, a patient’s change in EDS from baseline was rated by the same investigator using the CGI-C, a 7-grade scale (very much improved, much improved, minimally improved, no change, minimally worse, much worse, very much worse).
In the HARMONY CTP trial, during the 2 baseline visits before randomization, CGI-S was rated by the investigator using a 7-point scale (no sign of illness, borderline ill, mildly ill, moderately ill, markedly ill, severely ill, among the most extremely ill patients). At each post-baseline visit, a patient’s change in EDS from baseline was rated by the same investigator with the CGI-C using a 7-grade scale (very much improved, much improved, minimally improved, no change, minimally worse, much worse, very much worse).
Number, Frequency, and Severity of Cataplexy Attacks
The frequency and severity of cataplexy attacks was a primary end point in the HARMONY CTP trial and a secondary end point in the HARMONY 1 and HARMONY 1bis trials. In all 3 trials, the frequency and severity of complete and partial cataplexy attacks were recorded by patients in sleep diaries, as previously described for EDS. The definitions of total and partial cataplexy attacks were provided to the patient. Cataplexy was defined as a sudden muscle weakness triggered by emotional factors; the patient must have remained fully lucid and aware during the attack. Total cataplexy was defined as a cataplexy attack where all striated muscles were affected, with a loss of posture and falling to the ground. Partial cataplexy was defined as a cataplexy attack that was limited to facial muscles or to the upper or lower limbs, leading to head drop, jaw opening, knees unblocking, or dropping of objects.14,15 In all 3 trials, the severity of cataplexy was also rated by the investigator using the CGI-S and CGI-C, as previously described for EDS.
Health-Related Quality of Life
In all 3 trials, the 5-Level EQ-5D was used to measured HRQoL. The 5-Level EQ-5D is a validated, self-reported, generic questionnaire of HRQoL.41 Aspects of HRQoL were assessed with the 5 following dimensions: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression. A VAS was used by patients to rate their own health state (the best state was rated 100 and the worst state was rated 0). In the HARMONY 1 trial, the 5-Level EQ-5D was completed at the baseline visit, the inclusion visit, and the end point visit. In the HARMONY 1bis trial, the 5-Level EQ-5D was completed at the baseline visit, the inclusion visit, the stable-dose visit, the end point visit, and the withdrawal visit. In the HARMONY CTP trial, the 5-Level EQ-5D was completed at treatment start, the stable-dose visit, and the end point visit.
Nocturnal Sleep Properties
Duration of Nocturnal Awakenings, Diurnal Involuntary Sleep Attacks and Severe Sleepiness, Sleep Paralysis
In the HARMONY 1 and HARMONY 1bis trials, the number and duration of nocturnal awakenings, diurnal involuntary sleep attacks and severe sleepiness, total duration of nocturnal sleep, and incidence of sleep paralysis (defined as being unable to move) were assessed with patients’ sleep diaries, as previously described for EDS. In the HARMONY CTP trial, bedtime and wake-up times were the only sleep properties investigated, also with patients’ sleep diaries.
Polysomnography
In the HARMONY 1bis trial, polysomnography (a standard full-night sleep study) was performed at baseline for the first 20 patients enrolled in 3 selected centres and at the end of the study to evaluate the effect of the interventions on various sleep parameters. The polysomnography was scored manually, according to standard criteria. Episodes of apnea were defined as a complete cessation of airflow for more than 10 seconds. Episodes of hypopnea were defined as a decrease in oronasal airflow of more than 50% lasting for at least 10 seconds, a decrease of more than 30% associated with a decrease in oxygen saturation of more than 3%, or a microarousal.14
Number of Hallucinations
In all 3 trials, the frequency of hallucinations was evaluated with the patients’ sleep diaries, as previously described for EDS. Patients were asked to record and estimate the number of hallucinations that occurred in the previous 24 hours, defined as “episodes when the patient imagines seeing or hearing people, animals, objects, or frightening events.”13-15
Patient Satisfaction, Ease of Use
For all trials, at each visit after the onset of the treatment, patients evaluated the global effect of their treatment by comparing the 1-week period before that visit with their condition before study entry. The following 6-level scale was used: marked effect (complete or nearly complete remission of EDS), moderate effect (partial remission of EDS), minimal effect (slight decrease in EDS that does not substantially alter the status of the patient), no change, minimally worse (slight increase in EDS), much worse (substantial increase in EDS). The PGO tool is not validated.
Adherence
In all 3 trials, compliance with treatment was evaluated at each visit during the treatment period. Compliance was not considered an efficacy outcome. Patients were asked to bring back all their used blister packs, including empty ones, at each visit. Details of the quantities of medication dispensed were entered into an accountability form and patients were asked whether they had taken the treatment as prescribed. Any diversions from the prescribed treatment (as reported by patients or evidenced by the blister packs) were recorded.
Harms
The following safety criteria were monitored during the course of all trials: AEs, SAEs, and adverse drug reactions (including frequency, severity, relationship to study drug, incidence, and occurrence). Safety was monitored throughout the trials via changes in vital signs (heart rate, blood pressure, body weight), physical examinations, sleep quality and narcolepsy symptoms based on sleep-diary analysis, electrocardiogram (ECG) parameters, and laboratory abnormalities.
In all 3 trials, the BDI-SF-13 was used to measure depressive symptoms as part of the safety evaluation upon request of the German and Swiss sites to minimize the risk of suicidality.42 The protocol was amended accordingly. In the HARMONY 1 trial, patients completed the BDI-SF-13 at all visits. In the HARMONY 1bis trial, patients completed the BDI-SF-13 at the screening visit, baseline visit, inclusion visit, during treatment, at the end of treatment, and after the withdrawal period. In the HARMONY CTP trial, patients completed the BDI-SF-13 at the screening visit, at randomization, before the stable-dose period, at the end of treatment, and after the withdrawal period. In all 3 trials, withdrawal symptoms were evaluated after the withdrawal period using a trial-specific questionnaire. Withdrawal was defined as dysphoria and at least 2 other symptoms, including fatigue, vivid and unpleasant dreams, insomnia or hypersomnia, increased appetite, and psychomotor retardation or agitation.13-15,23
Statistical Analysis
HARMONY 1 Trial
The primary efficacy criterion was daytime somnolence, assessed using the ESS. To reduce intra-individual variability, ESS score at baseline was calculated as the mean of the pre-baseline visit and end-of-baseline visit values. The final ESS value was calculated as the summary mean of the final 2 visits on treatment. For patients with a premature interruption, the LOCF method was used to impute the missing values. The final ESS value was imputed as the mean of the final 2 available measures, or as the baseline value when no post-baseline values were available. The primary efficacy analysis was the comparison, using a linear mixed-effect model, of the difference in final ESS value between the pitolisant hydrochloride and placebo groups, adjusted for ESS at baseline and using treatment and centre as fixed and random effects, respectively. The significance of pitolisant hydrochloride compared with placebo was assessed by analysis of covariance (ANCOVA) on the final ESS adjusted for ESS at baseline. ANCOVA was conducted with a mixed linear model that took centre heterogeneity into account.
Owing to multiple comparisons of treatments, the multiplicity of type I error for the evaluation of EDS was taken into account using a step-down approach. The test for noninferiority was performed in 2 steps: the null hypothesis (i.e., no difference) for pitolisant hydrochloride versus placebo had to be rejected at an alpha level of 0.025 and the noninferiority of pitolisant hydrochloride to modafinil was only tested if the null hypothesis was rejected; and the noninferiority of pitolisant hydrochloride to modafinil was tested at an alpha level of 0.025. The main ANCOVA model assumed no treatment-baseline interaction term.
For secondary efficacy outcomes, a descriptive analysis for each variable was performed for each treatment group separately. Two-way comparisons between treatment groups were evaluated with ANCOVA, with baseline adjustment on associated baseline values as appropriate. For outcomes involving duration of time, standard survival analysis was undertaken, with adjustment on baseline values as appropriate. For MWT and SART, the significance of treatment difference was tested using the Mann–Whitney U test.
Mean changes from baseline in the daily cataplexy rate were calculated as the ratio of the geometric mean change of the final period from baseline. For patients who had no cataplexy episodes during the baseline or treatment periods, the daily cataplexy rate was imputed as the worst-case value, defined as the reciprocal of the number of days of exposure. The significance of the differences among placebo, pitolisant hydrochloride, and modafinil was tested by comparing the geometric mean of the daily rate of cataplexy among the 3 treatments, and the ratio was tested with a t-test on log-transformed values.
The clinical relevance of the difference between placebo and pitolisant hydrochloride, and between pitolisant hydrochloride and modafinil, for efficacy variables other than EDS was tested by calculating the proportion of patients for whom the increase from baseline to the end of treatment exceeded a predetermined minimum clinical relevance using the absolute risk difference (95% CI). Because BMI, sex, age, and duration of narcolepsy could affect the outcomes, these variables were entered in a stepwise model to identify significant predictors of the studied end point. Any significant predictors identified were added to the main analysis to test the treatment effect.
The determination of sample size was designed under the following hypotheses derived from unspecified historical trials: the minimum clinically relevant difference on ESS was 3, with a SD assumed to be 5 and estimated coefficient of correlation of 0.65, and compound symmetry for the repeated measurements. The NIM was estimated as a small proportion of the difference between the reference and placebo and less than the minimum clinically important difference. The difference, based on meta-analytical results on historical trials of modafinil, was 4.12 (95% CI, 0.14 to 7.09). The NIM of 2 was chosen. Using the power function of ANCOVA, the sample size was determined to achieve a difference of 3 (with the following parameters: 2-sided alpha = 0.05; pre-visits = 2; post-visits = 2; r = 0.65), with a power of at least 95% detected once the sample size exceeded 30 patients per group. Assuming that modafinil and pitolisant hydrochloride have the same efficacy, the probability to reject at a predetermined fixed NIM of 2 (associated with the following parameters: alpha = 0.025; pre-visits = 2; post-visits = 2; r = 0.65) will be at least 80% once the sample size exceeds 30 patients per group. Thus, to satisfy the requirement of the 2 tests, 30 patients per group were planned.
HARMONY 1bis Trial
As in the HARMONY 1 trial, the primary efficacy criterion was daytime somnolence assessed using the ESS, with the same calculations for baseline and final ESS values. The primary efficacy analysis was the comparison, using a linear mixed-effect model, of the difference in final ESS value between the pitolisant hydrochloride and placebo groups, adjusted for ESS at baseline and using treatment and centre as fixed and random effects, respectively. The significance of pitolisant hydrochloride compared with placebo was assessed with ANCOVA on the final ESS adjusted for ESS at baseline. ANCOVA was conducted with a mixed linear model that took centre heterogeneity into account.
Owing to multiple comparisons of treatments, the multiplicity of type I errors for the evaluation of EDS was taken into account using a step-down approach. The test for noninferiority was performed in 2 steps: the null hypothesis (i.e., no difference) for pitolisant hydrochloride versus placebo had to be rejected at an alpha level of 0.05 and the noninferiority of pitolisant hydrochloride to modafinil was only tested if the null hypothesis was rejected; and the noninferiority of pitolisant hydrochloride to modafinil was tested at an alpha level of 0.05. The main ANCOVA model assumed no treatment-baseline interaction term.
For secondary efficacy outcomes, a descriptive analysis of each variable was performed for each treatment group separately. Two-way comparisons between treatment groups were evaluated with ANCOVA, with baseline adjustment on associated baseline values as appropriate. For outcomes involving duration of time, standard survival analysis was undertaken, with adjustment on baseline values as appropriate. For MWT and SART, the significance of treatment difference was calculated as the ratio of the geometric mean change of the final period from baseline. The geometric mean at baseline and the study end was calculated for all treatment groups. The geometric mean was used, as the data were of a log-normal distribution. The mean values of log were compared between treatment groups with a Student t-test and converted as a geometric mean to estimate the geometric mean ratio with the corresponding 95% CI.
Mean changes from baseline in the daily cataplexy rate were calculated as the ratio of the geometric mean change of the final period from baseline. For patients who had no cataplexy episodes during the baseline or treatment periods, the daily cataplexy rate was imputed as the worst-case value, defined as the reciprocal of the number of days of exposure. The significance of the differences among placebo, pitolisant hydrochloride, and modafinil was tested by comparing the geometric mean of the daily rate of cataplexy among the 3 treatments, and the ratio was tested with a t-test on log-transformed values.
The clinical relevance of the difference between placebo and pitolisant hydrochloride, and between pitolisant hydrochloride and modafinil, for efficacy variables other than EDS was tested by calculating the proportion of patients for whom the increase from baseline to the end of treatment exceeded a predetermined minimum clinical relevance using the absolute risk difference (95% CI). Because BMI, sex, age, and duration of narcolepsy could affect the outcomes, these variables were entered in a stepwise model to identify significant predictors of the studied end point. Any significant predictors identified were added to the main analysis to test the treatment effect.
The determination of sample size was informed by the HARMONY 1 trial. The sample-size calculation initially took into account the strategy of ANCOVA under a step-down analysis, using 2 pre-treatment and 2 post-treatment measures. The sample size was determined by separately examining the superiority of pitolisant hydrochloride to placebo with a difference of at least 3, with the following parameters: 2-sided alpha = 0.05; pre-visits = 2; post-visits = 2; r = 0.7; sigma = 5. It would be detected with a power of 95% once the sample size exceeds 20 patients per group. The noninferiority of pitolisant hydrochloride to modafinil was calculated assuming that the 2 drugs had the same efficacy. The probability to refuse noninferiority at a predetermined fixed NIM of 2 (associated with the following parameters: alpha = 0.05; pre-visits = 2; post-visits = 2; r = 0.7; sigma = 5) would be at least 80% once the sample size exceeded 40 patients per group. To satisfy the requirement of the 2 tests, suggested sample sizes of 20 for placebo, 40 for modafinil, and 40 for pitolisant hydrochloride were suggested. The choice of an initial 1:2:2 randomization ratio was reduced because the noninferiority test requires more patients. However, as noninferiority had to be concluded both on ITT and PP bases, it was necessary to increase the sample sizes by 20%. Under these conditions, the sample sizes were initially 25 for placebo, 50 for pitolisant hydrochloride, and 50 for modafinil. An increase in the sample size was necessary after preliminary blind analyses were carried out on the first 50 patients in the HARMONY 1 trial.
HARMONY CTP Trial
The main end point was the measure of anticataplectic efficacy assessed by the change in the average number of cataplexy attacks per week from the 2-week baseline period to the 4-week stable-treatment period. The 2 groups were compared on the WCR. For patients who terminated the study with missing final values, the final value was calculated as the mean of the 2 last known periods. For patients without post-baseline values, the final value was assimilated with baseline (LOCF). For sensitivity purposes, baseline carried forward allocation was also used, meaning that the unknown period for WCR was imputed from the baseline value. The significance of pitolisant hydrochloride compared with placebo was assessed with an ANCOVA on WCR during the 4-week stable-treatment phase adjusted for baseline. ANCOVA was conducted with a mixed nonlinear model featuring a possibly overdispersed Poisson distribution and taking into account centre heterogeneity by using centre as a random factor. In case of overdispersion (a coefficient greater than 2), use of a quasi-Poisson correction was planned. The null hypothesis (i.e., no difference) for pitolisant hydrochloride versus placebo had to be rejected at a 2-sided alpha level of 0.025.
For secondary efficacy outcomes, a descriptive analysis of each variable was performed for each treatment group separately. A 2-way comparison between treatment groups was evaluated with ANCOVA, with baseline adjustment on associated baseline values as appropriate. For outcomes involving duration of time, standard survival analysis was undertaken with adjustment on baseline values as appropriate. For ESS, the treatment effect was tested with ANCOVA after adjustment for baseline severity and centre heterogeneity. The difference between the mean value during baseline (average of first and second visits) and the mean value during the end of treatment period (average of fifth and sixth visits) was calculated. For MWT, the significance of the treatment difference was calculated as the ratio of the geometric mean change of the final period from baseline. The geometric mean at baseline and the study end was calculated for all treatment groups. The geometric mean was used as the data were of a log-normal distribution. The mean values of log were compared between treatment groups with a Student t-test and were converted as a geometric mean to estimate the geometric mean ratio with the corresponding 95% CI.
The sample-size calculation was based on the assumption of a Poisson regression where a rate ratio (ratio of the mean number of cataplexy episodes in pitolisant hydrochloride compared with placebo) was fixed to a rate ratio of 0.75. As the selection of patients is based on a minimum WCR of 3 and because of the expected strong effect of placebo, a placebo effect leading to a reduction of 50% of the baseline value was assumed resulting in a WCR of 1.5 and the corresponding expected mean number of 6 cataplexy crises during the month. Following the sample size calculation, we calculated that an effect of the studied drug was calculated as large as a rate ratio of 0.75 and a mean number of cataplexies of 6 during the studied period (1 month) in the control group should be detected at a 0.05 2-sided level with a power of at least 1 – beta = 0.9 from an equal sample size per group of at least 47.
Subgroup Analysis
Subgroups of patients with a history of cataplexy were reported in the HARMONY 1 trial. Subgroups with a high frequency of cataplexy episodes (> 15) and patients on anticataplectic treatments during the study were reported in the HARMONY CTP trial for the cataplexy frequency outcome.
Analysis Populations
For the HARMONY 1bis and HARMONY CTP trials, the ITT population consisted of all randomized patients who received at least 1 treatment dose and had at least 1 post-dose value to compute the primary end point. However, the definition of ITT used in the trials does not fit the classic definition, as it should include all patients who were randomized, regardless of actual treatment, and was reflected in the extended intention-to-treat (EIT) populations in the trials. For the HARMONY 1 trial, the ITT population consisted of all randomized patients who received at least 1 treatment dose and had missing post-baseline values computed from the baseline value. The ITT population was used for both the primary and secondary analyses in the HARMONY 1 trial. The HARMONY 1 and HARMONY 1bis trials included an EIT population that consisted of all randomized patients, regardless of whether treatment was initiated and irrespective of outcome. For the HARMONY 1 and HARMONY 1bis trials, the PP population consisted of all patients in the ITT population who remained in the study until at least visit 6 (second-to-last visit on treatment) and who had no major protocol deviations related to primary end point. In the HARMONY CTP trial, the PP population was defined as a subset of the ITT sample that included all patients who finished the trial on time according to the protocol, with no major deviations, and in conformity with the prescribed regimen. For all trials, the safety population was composed of all randomized patients who took at least 1 dose of the study drug.
Results
Patient Disposition
Patient disposition in the 3 studies is provided in Table 15 and Table 16. In the HARMONY 1 trial, patients were randomized to placebo, pitolisant hydrochloride, or modafinil for 56 (HARMONY 1 and HARMONY 1bis) and 49 (HARMONY CTP) days. Eighty-six percent of screened patients were randomized. Across all treatment arms, approximately 16% of patients discontinued the study, with AEs and lack of efficacy being the most common reasons. In the HARMONY 1 trial, the 94 patients (31 in the pitolisant hydrochloride group, 30 in the placebo group, and 33 in the modafinil group) in the ITT analysis and the safety population had at least 1 dose of the study drug and provided at least 1 post-baseline value. The EIT population in the HARMONY 1 trial included an additional patient in the pitolisant hydrochloride arm who did not take the study treatment and did not go to the visits after randomization. In the HARMONY 1bis trial, 91% of screened patients were randomized. There were 165 patients in the safety population (33 in the placebo group, 67 in the pitolisant hydrochloride group, and 65 in the modafinil group) and 163 patients in the ITT population (32 in the placebo group, 66 in the pitolisant hydrochloride group, and 65 in the modafinil group). A total of 153 patients completed the study. More patients in the pitolisant hydrochloride group than in the modafinil and placebo groups discontinued the study, with AEs being the most common reason. The EIT population in the HARMONY 1bis trial included an additional patient in the pitolisant hydrochloride arm who had premature withdrawal at day 14 after only 1 treatment. In the HARMONY CTP trial, 91% of screened patients were randomized. A total of 105 patients were treated in the ITT population and the safety population, 54 in the pitolisant hydrochloride group and 51 in the placebo group. Ninety-eight patients completed the study. Discontinuation from the trial was low, at 6% in the placebo group and 8% in the pitolisant hydrochloride group.
Table 15: Patient Disposition in the HARMONY 1 and HARMONY 1bis Trials
Patient disposition | HARMONY 1 | HARMONY 1bis | ||||
|---|---|---|---|---|---|---|
Placebo | Pitolisant hydrochloride | Modafinil | Placebo | Pitolisant hydrochloride | Modafinil | |
Screened, N | 110 | |||||||||| | ||||
Randomized, N (%) | 30 (31.9) | 32 (33.0) | 33 (35.1) | |||||||||| | |||||||||| | |||||||||| |
Discontinued from study, N (%) | 5 (16.7) | 5 (16.1) | 5 (15.2) | |||||||||| | |||||||||| | |||||||||| |
Reason for discontinuation, N (%) | |||||||||| | |||||||||| | |||||||||| | |||
Adverse event | 4 (13.3) | 0 | 4 (12.1) | |||||||||| | |||||||||| | |||||||||| |
Lost to follow-up | 0 | 1 (3.1) | 0 | |||||||||| | |||||||||| | |||||||||| |
Lack of efficacy | 1 (3.3) | 3 (9.4) | 0 | |||||||||| | |||||||||| | |||||||||| |
Administrative | 0 | 1 (3.1) | 0 | |||||||||| | |||||||||| | |||||||||| |
Premature discontinuation | 0 | 1 (3.1) | 1 (3.0) | |||||||||| | |||||||||| | |||||||||| |
EIT population, N | 30 | 32 | 33 | |||||||||| | |||||||||| | |||||||||| |
ITT population, N | 30 | 31 | 33 | |||||||||| | |||||||||| | |||||||||| |
PP population, N | 25 | 26 | 28 | |||||||||| | |||||||||| | |||||||||| |
Safety population, N | NA | NA | NA | |||||||||| | |||||||||| | |||||||||| |
EIT = extended intention-to-treat; ITT = intention-to-treat; NA = not applicable; PP = per protocol.
Sources: Clinical Study Reports for HARMONY 1 and HARMONY 1bis.13,14
Table 16: Patient Disposition in the HARMONY CTP Trial
Patient disposition | Placebo | Pitolisant hydrochloride |
|---|---|---|
Screened, N | 117 | |
Randomized, N (%) | 52 (49.1) | 54 (50.9) |
Discontinued from study, N (%) | 3 (5.8) | 4 (7.4) |
Reason for discontinuation, N (%) | ||
Adverse events | 0 | 1 (1.9) |
Lost to follow-up | 0 | 0 |
Lack of efficacy | 1 (2.0) | 1 (1.9) |
Administrative | 1 (2.0) | 0 |
Withdrawal by patient | 1 (2.0) | 2 (3.7) |
ITT population, N | 51 | 54 |
PP population, N | 42 | 49 |
Safety population, N | 51 | 54 |
ITT = intention-to-treat; PP = per protocol.
Source: Clinical Study Report for HARMONY CTP.15
Exposure to Study Treatments
In the HARMONY 1 trial, among the 31 patients in the pitolisant hydrochloride group, 8 (26%) were treated for the final 6 weeks at the stable medium dose of 20 mg and 19 (61%) at the stable high dose of 40 mg. Among the 33 patients of the modafinil group, 4 (12%) were treated for the final 6 weeks at the stable medium dose and 24 (73%) at the stable high dose. No evaluation of drug dose, drug concentration, or relationship to response was planned in the HARMONY 1bis trial. The average dose taken in each treatment arm was not reported, with a presumed maximum dose of 20 mg daily. In the HARMONY CTP trial, by the end of the study, 32 (59.3%) of 54 patients in the pitolisant hydrochloride group were receiving 40 mg, 11 (20.4%) were receiving 20 mg, and 7 (13.0%) were receiving 10 mg. The stable dose during the stable-dose period was 40 mg for 35 (64.8%) of 54 patients in the pitolisant hydrochloride group, 20 mg for 9 (16.7%) patients, and 10 mg for 7 (13.0%) patients.
In the HARMONY 1 trial, 33 patients with severe cataplexy were permitted to remain on their anticataplectic medications at stable doses for the duration of the trial; 8 were taking sodium oxybate at the previous stable dosage and the others were taking antidepressants used as anticataplectic medication. Prohibited treatments were continued during the trial in 4 cases. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. In the HARMONY CTP trial, 8 (15.7%) patients in the placebo group were treated with an anticataplectic during the study (5 with venlafaxine, 1 with sodium oxybate, 1 with reboxetine, and 1 with escitalopram). There were 4 patients (7.4%) in the pitolisant hydrochloride group treated with an anticataplectic during the study (1 with citalopram, 2 with fluoxetine, 1 with sodium oxybate). Three of 51 patients (5.9%) took at least 1 authorized medication in the placebo group, as did 5 of 54 patients (9.3%) in the pitolisant hydrochloride group.
Table 17: Concomitant Medication Use — HARMONY 1 (ITT Population) and HARMONY 1bis (ITT Population) Trials
Concomitant treatments | HARMONY 1 | HARMONY 1bis | HARMONY CTP | |||||
|---|---|---|---|---|---|---|---|---|
Placebo N = 30 | Pitolisant hydrochloride N = 31 | Modafinil N = 33 | Placebo N = 32 | Pitolisant hydrochloride N = 66 | Modafinil N = 65 | Placebo N = 51 | Pitolisant hydrochloride N = 54 | |
At least 1 chronic medication in the 3 months before inclusion, n (%) | 23 (85.2) | 23 (85.2) | 21 (70.0) | |||||||||| | |||||||||| | |||||||||| | 35 (68.6) | 28 (51.9) |
At least 1 authorized medication during the study, n (%) | 9 (33.3) | 11 (40.7) | 17 (56.7) | |||||||||| | |||||||||| | |||||||||| | 3 (5.9) | 5 (9.3) |
At least 1 unauthorized medication during the study, n (%) | 1 (3.7) | 1 (3.7) | 1 (3.3) | |||||||||| | |||||||||| | |||||||||| | 0 | 0 |
ITT = intention-to-treat.
Sources: Clinical Study Reports for HARMONY 1, HARMONY 1bis, and HARMONY CTP.13-15
Efficacy
Only efficacy outcomes and analyses of subgroups identified in the review protocol are reported here. Refer to Appendix 3 for detailed efficacy data.
Sleepiness, Alertness, and Severity of Daytime Sleepiness
Epworth Sleepiness Scale
HARMONY 1 Trial: The adjusted mean difference in the final ESS score between pitolisant hydrochloride and placebo was –3.10 (95% CI, –5.73 to –0.46; P = 0.022) (Table 18, Figure 5). Sensitivity analyses on the PP population and without accounting for the centre effect showed similar results. Because the superiority of pitolisant hydrochloride to placebo for EDS was demonstrated at the a priori alpha level of 0.025, the noninferiority of pitolisant hydrochloride to modafinil was tested. The adjusted mean difference in the final ESS score between pitolisant hydrochloride and modafinil was 0.09 (95% CI, –2.31 to 2.30); thus, pitolisant hydrochloride was judged to not be noninferior to modafinil at the prespecified NIM of 2.
A patient was considered a responder when the final ESS score was below 10. On this basis, the responder rates were 13.3% in the placebo group, 45.2% in the pitolisant hydrochloride group, and 45.3% in the modafinil group. The adjusted OR of response for pitolisant hydrochloride compared with placebo was 7.86 (95% CI, 1.59 to 38.86). The adjusted OR of response for pitolisant hydrochloride compared with modafinil was 1.09 (95% CI, 0.31 to 3.81).
Maintenance of Wakefulness Test
In the HARMONY 1 trial, the adjusted mean difference in final score between placebo and pitolisant hydrochloride was 1.47 (95% CI, 1.01 to 2.14) and the adjusted mean difference in final score between pitolisant hydrochloride and modafinil was 0.77 (95% CI, 0.52 to 1.13). This was consistent with the findings of the HARMONY 1bis trial, where the adjusted mean difference between placebo and pitolisant hydrochloride was 1.46 (95% CI, 1.069 to 2.010) and the adjusted mean difference in final score between pitolisant hydrochloride and modafinil was ||||||||||||||||||||||||||||||||||||||||||. In the HARMONY CTP trial, the geometric mean of ratios (final divided by baseline) was 1.78 (95% CI, 1.22 to 2.60). Sensitivity analyses for all trials using the PP population were consistent with the main analysis.
Sustained Attention to Response Task
In the HARMONY 1 trial, the adjusted mean difference between the pitolisant hydrochloride and placebo treatment arms was 0.82 (95% CI, 0.67 to 0.99) for NOGO, 0.80 (95% CI, 0.57 to 1.13) for GO, and 0.79 (95% CI, 0.64 to 0.99) for TOTAL SART scores. The adjusted mean difference between the pitolisant hydrochloride and modafinil treatment arms was 1.03 (95% CI, 0.83 to 1.28) for NOGO, 1.03 (95% CI, 0.56 to 1.15) for GO, and 0.90 (95% CI, 0.70 to 1.14) for TOTAL SART scores. Sensitivity analyses using the PP population were consistent with the main analysis.
In the HARMONY 1bis trial, the ratio of mean change between the pitolisant hydrochloride and placebo groups was significant, at 0.83 (95% CI, 0.69 to 0.99; P = 0.043), whereas the difference between the pitolisant hydrochloride and modafinil groups was ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.
CGI-S and CGI-C on EDS
In the HARMONY 1 and HARMONY 1bis trials, CGI-C scores for EDS improved in a higher proportion of patients in the pitolisant hydrochloride and modafinil groups than in the placebo group. However, the change in CGI-C scores was similar in the pitolisant hydrochloride and modafinil arms. In the HARMONY 1 trial, CGI-C scores for EDS improved in the subgroup of patients with a history of cataplexy, but a greater proportion reported an improvement in the modafinil arm. In the HARMONY CTP trial, the mean reduction in CGI-score for pitolisant hydrochloride compared with placebo was –0.95 (95% CI, –1.36 to –0.54; P < 0.0001). Mean CGI-C score was 3.5 (SD = 1.1) with placebo and 2.6 (SD = 1.1) with pitolisant hydrochloride. Similar results were observed in the PP population, with a mean reduction of –0.86 (95% CI, –1.29 to –0.43; P < 0.0001).
Patient Diaries
In the HARMONY 1 trial, the mean drowsiness episodes per day was 0.58 (SD = 1.36), 0.88 (SD = 2.13), and 0.46 (SD = 1.20) at the final visit for placebo, pitolisant hydrochloride, and modafinil, respectively. Drowsiness was not reported in the HARMONY 1bis or HARMONY CTP trials.
Frequency and Severity of Cataplexy Attacks
In the HARMONY 1 trial, the final mean of complete and partial cataplexy episodes (episodes per day) was 0.68 (SD = 1.66), 0.28 (SD = 1.11), 0.65 (SD = 1.62) in the placebo, pitolisant hydrochloride, and modafinil groups, respectively. In the exposed population, the RR of daily rates of complete and partial cataplexy episodes at the end of treatment for pitolisant hydrochloride compared to placebo was 0.38 (95% CI, 0.15 to 0.93). The RR of daily rates of complete and partial cataplexy episodes at the end of treatment for pitolisant hydrochloride compared to modafinil was 0.54 (95% CI, 0.24 to 1.23). In the HARMONY 1bis trial, the mean least squares of daily cataplexy rate for those with cataplexy between the final 7 days of treatment and baseline was |||||||||||||||||||||||||||||||||||||||||||||||||||||||| for pitolisant hydrochloride compared to placebo.
Figure 5: ESS Change in the HARMONY 1 Trial
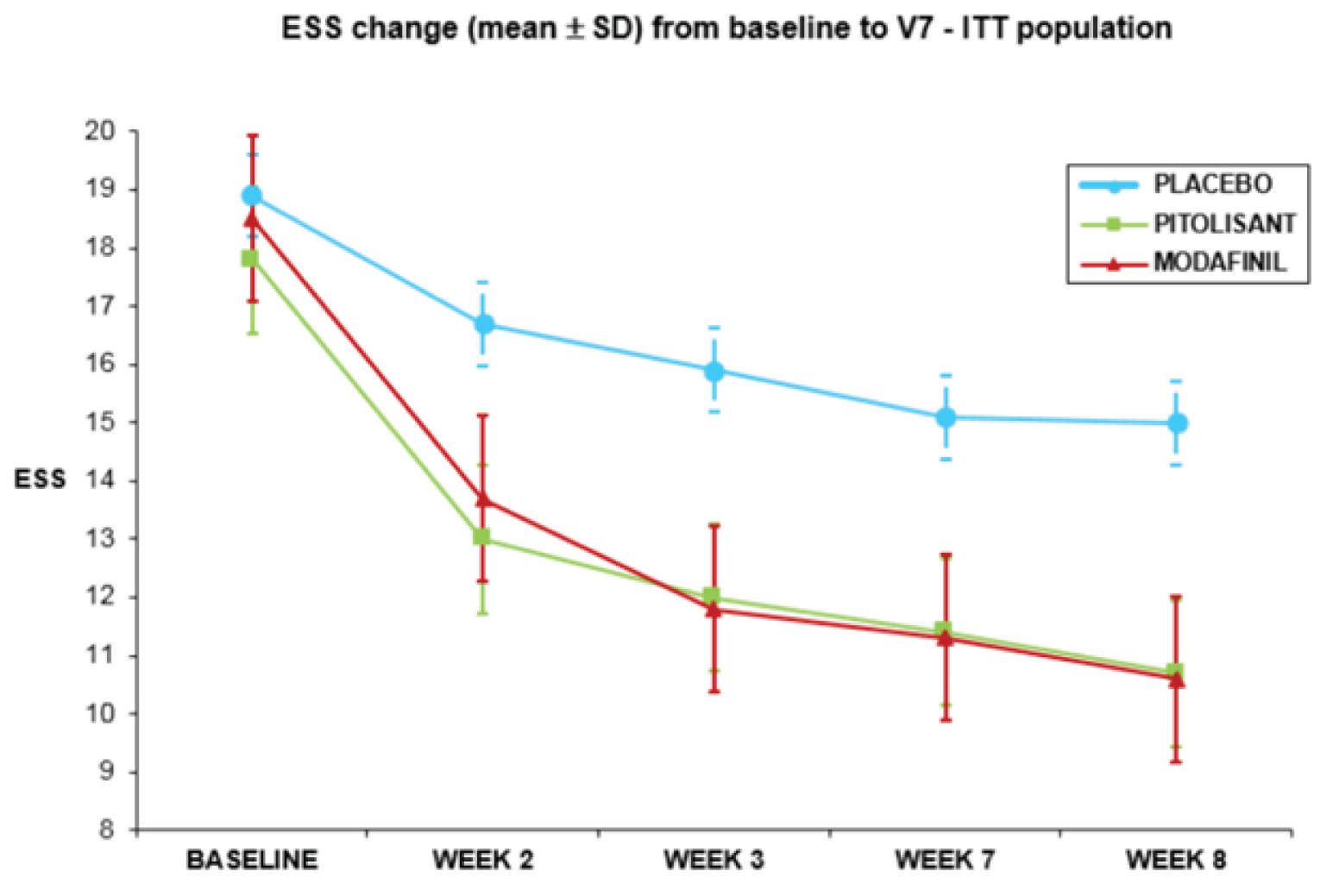
ESS = Epworth Sleepiness Scale; ITT = intention-to-treat; SD = standard deviation; V = visit.
Source: Clinical Study Report for HARMONY 1.13
HARMONY 1bis Trial: By the study end, mean ESS score reductions from baseline (SD) were |||||||||| in the placebo group, |||||||||| in the pitolisant hydrochloride group, and |||||||||| in the modafinil group (Table 18). The adjusted mean difference in the final ESS score for pitolisant hydrochloride compared with placebo was –2.19 (95% CI, –4.17 to –0.22; P = 0.030). Sensitivity analyses without reallocation by centre, without adjustment for baseline ESS, or after adjustment for baseline following the mean change, and the mean change from baseline methods showed similar results. Because the superiority of pitolisant hydrochloride to placebo for EDS was demonstrated at the a priori alpha level of 0.05, the noninferiority of pitolisant hydrochloride to modafinil was tested. The adjusted mean difference in the final ESS score for pitolisant hydrochloride compared with modafinil was 2.75 (95% CI, 1.02 to 4.48); thus, pitolisant hydrochloride was judged to not be noninferior to modafinil at the prespecified NIM of 2.
A patient was considered a responder when the final ESS was 10 or lower or the change from baseline was at least 3 points. The response proportions were |||||||||||||||||||||||||||| for the placebo, pitolisant hydrochloride, and modafinil groups, respectively. The adjusted RR for the difference between pitolisant hydrochloride and placebo ||||||||||||||||||||||||||||||||||||||||||. The adjusted RR for the difference between pitolisant hydrochloride and placebo was ||||||||||||||||||||||||||||||||||||||||||.
HARMONY CTP Trial: The observed mean changes in ESS from baseline were –1.9 (SD = 4.3) and –5.4 (SD = 4.3) in the placebo and pitolisant hydrochloride arms, respectively (Table 19). The adjusted mean difference in the change from baseline for pitolisant hydrochloride compared with placebo was –3.42 (95% CI, –4.96 to –1.87). Sensitivity analyses using LOCF and BOCF and the PP population were consistent with the main analysis. A patient was considered a responder when the final ESS score was 10 or lower or the change from baseline was at least 3 points. The response proportions were 34.0% and 68.6% for placebo and pitolisant hydrochloride, respectively. The adjusted OR for the difference between pitolisant hydrochloride and placebo was 4.26 (95% CI, 1.72 to 10.52).
Table 18: Sleepiness — HARMONY 1 (ITT Population) and HARMONY 1bis (ITT Population) Trials
Characteristic | HARMONY 1 | HARMONY 1bis | ||||
|---|---|---|---|---|---|---|
Placebo N = 30 | Pitolisant hydrochloride N = 31 | Modafinil N = 33 | Placebo N = 32 | Pitolisant hydrochloride N = 66 | Modafinil N = 65 | |
ESS scores | ||||||
Baseline, mean (SD)a | 18.9 (2.5) | 17.8 (2.5) | 18.5 (2.7) | 18.2 (2.3) | 18.2 (2.4) | 18.1 (2.8) |
Final, mean (SD)b | 15.6 (4.7) | 11.8 (6.1) | 11.6 (6.0) | 14.5 (5.9) | 13.7 (5.4) | 10.4 (6.0) |
Change from baseline, mean (SD) | –3.3 (4.1) | –6.0 (6.1) | –6.9 (6.1) | –3.6 (5.6) | –4.6 (4.6) | –7.8 (5.9) |
Adjusted mean difference in final score, pitolisant hydrochloride vs. placebo (95% CI)c | –3.10 (–5.73 to –0.46) | NA | –2.19 (–4.17 to –0.22) | NA | ||
P value for test of superiorityc | 0.022 | NA | 0.030 | NA | ||
Adjusted mean difference in final score, pitolisant hydrochloride vs. modafinil (95% CI)d | NA | 0.09 (–2.11 to 2.30) | NA | 2.75 (1.02 to 4.48) | ||
Proportion of responderse | ||||||
n (%) | 4 (13.3) | 14 (45.2) | 15 (45.5) | |||||||||||||| | |||||||||||||| | |||||||||||||| |
Adjusted OR (95% CI), pitolisant hydrochloride vs. placebof | 7.86 (1.59 to 38.86) | NA | |||||||||||||| | |||||||||||||| | ||
Adjusted RR (95% CI), pitolisant hydrochloride vs. placebof | NA | NA | |||||||||||||| | |||||||||||||| | ||
P valuef | 0.013 | NA | |||||||||||||| | |||||||||||||| | ||
Adjusted OR (95% CI), pitolisant hydrochloride vs. modafinilf | NA | 1.09 (0.31 to 3.81) | |||||||||||||| | |||||||||||||| | ||
Adjusted RR (95% CI), pitolisant hydrochloride vs. modafinilf | NA | NA | |||||||||||||| | |||||||||||||| | ||
P valuef | NA | 0.892 | |||||||||||||| | |||||||||||||| | ||
MWT scores | ||||||
Baseline, geometric mean (95% CI) | 8.44 (5.74 to 12.42) | 7.37 (5.38 to 10.10) | 8.78 (6.31 to 12.22) | |||||||||||||| | |||||||||||||| | |||||||||||||| |
Final, geometric mean (95% CI)g | 7.57 (5.14 to 11.16) | 9.73 (6.76 to 14.00) | 9.73 (6.76 to 14.00) | |||||||||||||| | |||||||||||||| | |||||||||||||| |
Adjusted mean difference in final score, pitolisant hydrochloride vs. placebo (95% CI)h | 1.47 (1.01 to 2.14) | NA | |||||||||||||| | |||||||||||||| | ||
P value | 0.044 | NA | |||||||||||||| | |||||||||||||| | ||
Adjusted mean difference in final score, pitolisant hydrochloride vs. modafinilh | NA | 0.77 (0.52 to 1.13) | |||||||||||||| | |||||||||||||| | ||
P value | NA | 0.173 | |||||||||||||| | |||||||||||||| | ||
SART-NOGO scores | ||||||
Baseline, geometric mean (95% CI) | 8.04 (6.46 to 10.02) | 9.13 (7.34 to 11.34) | 8.98 (7.34 to 10.98) | |||||||||||||| | |||||||||||||| | |||||||||||||| |
Final, geometric mean (95% CI)g | 8.08 (6.22 to 10.50) | 7.49 (5.88 to 9.54) | 7.15 (5.74 to 8.89) | |||||||||||||| | |||||||||||||| | |||||||||||||| |
Adjusted mean difference in final score, pitolisant hydrochloride vs. placebo (95% CI)h | 0.82 (0.67 to 0.99) | NA | |||||||||||||| | |||||||||||||| | ||
P value | 0.042 | NA | |||||||||||||| | |||||||||||||| | ||
Adjusted mean difference in final score, pitolisant hydrochloride vs. modafinilh | NA | 1.03 (0.83 to 1.28) | |||||||||||||| | |||||||||||||| | ||
P value | NA | 0.780 | |||||||||||||| | |||||||||||||| | ||
SART-GO scores | ||||||
Baseline, geometric mean (95% CI) | 3.51 (2.43 to 5.07) | 3.61 (2.64 to 4.92) | 3.31 (2.28 to 4.80) | |||||||||||||| | |||||||||||||| | |||||||||||||| |
Final, geometric mean (95% CI)g | 2.68 (1.94 to 3.70) | 2.20 (1.71 to 2.83) | 2.51 (1.81 to 3.47) | |||||||||||||| | |||||||||||||| | |||||||||||||| |
Adjusted mean difference in final score, pitolisant hydrochloride vs. placebo (95% CI)h | 0.80 (0.57 to 1.13) | NA | |||||||||||||| | |||||||||||||| | ||
P value | 0.202 | NA | |||||||||||||| | |||||||||||||| | ||
Adjusted mean difference in final score, pitolisant hydrochloride vs. modafinilh | NA | 1.03 (0.56 to 1.15) | |||||||||||||| | |||||||||||||| | ||
P value | NA | 0.233 | |||||||||||||| | |||||||||||||| | ||
SART-TOTAL scores | ||||||
Baseline, geometric mean (95% CI) | 11.45 (8.78 to 14.93) | 12.48 (9.90 to 15.73) | 11.40 (8.94 to 14.54) | |||||||||||||| | |||||||||||||| | |||||||||||||| |
Final, geometric mean (95% CI)g | 10.27 (7.79 to 13.53) | 8.89 (6.90 to 11.45) | 9.07 (6.97 to 11.81) | |||||||||||||| | |||||||||||||| | |||||||||||||| |
Adjusted mean difference in final score, pitolisant hydrochloride vs. placebo (95% CI)h | 0.79 (0.64 to 0.99) | NA | |||||||||||||| | |||||||||||||| | ||
P value | 0.041 | NA | |||||||||||||| | |||||||||||||| | ||
Adjusted mean difference in final score, pitolisant hydrochloride vs. modafinilh | NA | 0.90 (0.70 to 1.14) | |||||||||||||| | |||||||||||||| | ||
P value | NA | 0.363 | |||||||||||||| | |||||||||||||| | ||
CGI-S scores on EDS | ||||||
Number of patients contributing to the analysis | 25 | 26 | 28 | |||||||||||||| | |||||||||||||| | |||||||||||||| |
Baseline, mean (SD) | 5.3 (0.8) | 5.2 (0.9) | 5.2 (1.2) | |||||||||||||| | |||||||||||||| | |||||||||||||| |
Final, mean (SD)i | 3.3 (1.5) | 2.5 (1.3) | 2.1 (1.2) | |||||||||||||| | |||||||||||||| | |||||||||||||| |
Change from baseline, n (%) | |||||||||||||| | |||||||||||||| | |||||||||||||| | |||
Improvedj | 14 of 25 (56.0) | 19 of 26 (73.1) | 24 of 28 (85.7) | |||||||||||||| | |||||||||||||| | |||||||||||||| |
No changej | 7 of 25 (28.0) | 5 of 26 (19.2) | 3 of 28 (10.7) | |||||||||||||| | |||||||||||||| | |||||||||||||| |
Worsenedj | 4 of 25 (16.0) | 2 of 26 (7.7) | 1 of 28 (3.6) | |||||||||||||| | |||||||||||||| | |||||||||||||| |
P value for change from baselinek | 0.051 | |||||||||||||| | ||||
CGI-C scores on EDS | ||||||
Number of patients contributing to the analysis | 30 | 31 | 33 | |||||||||||||| | |||||||||||||| | |||||||||||||| |
Baseline, mean (SD) | |||||||||||||| | |||||||||||||| | |||||||||||||| | |||
Final, mean (SD)i | 3.8 (1.4) | 3.1 (1.3) | 3.4 (1.8) | |||||||||||||| | |||||||||||||| | |||||||||||||| |
Change from baseline, n (%) | ||||||
Improvedj | 9 of 25 (36.0) | 17 of 26 (65.4) | 16 of 28 (57.1) | |||||||||||||| | |||||||||||||| | |||||||||||||| |
No changej | 9 of 25 (36.0) | 4 of 26 (15.4) | 4 of 28 (14.3) | |||||||||||||| | |||||||||||||| | |||||||||||||| |
Worsenedj | 7 of 25 (28.0) | 5 of 26 (19.2) | 8 of 28 (28.6) | |||||||||||||| | |||||||||||||| | |||||||||||||| |
P value for change from baselinek | 0.208 | |||||||||||||| | ||||
CGI-C scores on EDS for patients with history of cataplexy | ||||||
Number of patients contributing to the analysis | 24 | 25 | 27 | |||||||||||||| | |||||||||||||| | |||||||||||||| |
Baseline, mean (SD) | NR | NR | NR | |||||||||||||| | |||||||||||||| | |||||||||||||| |
Final, mean (SD)i | 3.4 (1.5) | 2.6 (1.2) | 2.2 (1.3) | |||||||||||||| | |||||||||||||| | |||||||||||||| |
Change from baseline, n (%) | |||||||||||||| | |||||||||||||| | |||||||||||||| | |||
Improvedj | 11 of 21 (52.4) | 15 of 20 (75.0) | 19 of 23 (82.6) | |||||||||||||| | |||||||||||||| | |||||||||||||| |
No changej | 6 of 21 (28.6) | 4 of 20 (20.0) | 3 of 23 (13.0) | |||||||||||||| | |||||||||||||| | |||||||||||||| |
Worsenedj | 4 of 21 (19.0) | 1 of 20 (5.0) | 1 of 23 (4.3) | |||||||||||||| | |||||||||||||| | |||||||||||||| |
P value for change from baselinek | 0.064 | |||||||||||||| | ||||
CI = confidence interval; CGI-C = Clinical Global Impression of Change; CGI-S = Clinical Global Impression of Severity; EDS = excessive daytime sleepiness; ESS = Epworth Sleepiness Scale; ITT = intention-to-treat; MWT = Maintenance of Wakefulness Test; NA = not applicable; OR = odds ratio; RR = relative risk; SART = Sustained Attention to Response Task; SD = standard deviation.
Notes: The HARMONY 1bis ITT population consisted of all randomized patients who took at least 1 dose of drug and provided at least 1 value after baseline. One participant in the pitolisant hydrochloride had a premature withdrawal after 1 treatment. Aside from the ESS score outcome, the P values were not controlled for type I errors (i.e., multiple testing).
aMean of the first 2 weeks on treatment. In the HARMONY 1bis trial, when ESS at visit 2 was missing, then ESS baseline was calculated as the average at visit 1 and visit 3.
bMean of last 2 available post-baseline values.
cLinear mixed model, including final ESS score and groups as fixed effects and centres as random effect to test the superiority of pitolisant hydrochloride to placebo. In the HARMONY 1bis trial, linear mixed-effects model, featuring ANCOVA on the final ESS score adjusted on baseline with treatment considered as a fixed factor and reallocated centre as a random effect.
dLinear mixed model, including final ESS score and groups as fixed effects and centres as random effect to test the noninferiority of pitolisant hydrochloride to modafinil. In the HARMONY 1bis trial, noninferiority test considered the NIM of 2.
eResponders were defined as patients with a final ESS score ≤ 10 in both trials and if there was a difference in final and baseline ESS score of ≥ 3 points in the HAROMONY 1bis trial.
fLogistic regression model adjusted for baseline ESS score. In the HARMONY 1bis trial, analysis was conducted using a Poisson regression model on final ESS score adjusted on baseline ESS score, with treatment considered as a fixed factor and centre as a random effect.
gFinal value at visit 7 or at baseline (if the value at visit 7 was missing).
hAdjusted for baseline value and 95% CI.
IAs measured at the end of the treatment period (visit 7).
jImproved defined as “very much improved, much improved, or minimally improved.” Worsened defined as “very much worse, much worse, or minimally worse.”
kCochran-Mantel-Haenszel test (ANOVA statistic) with modified ridit scores.
Sources: Clinical Study Reports for HARMONY 1 and HARMONY 1bis.13,14
Table 19: Sleepiness — HARMONY CTP (ITT Population) Trial
Variable | Placebo (N = 51) | Pitolisant hydrochloride (N = 54) |
|---|---|---|
ESS scores | ||
Baseline, na | 51 | 54 |
Baseline, mean (SD)a | 17.3 (3.2) | 17.4 (3.3) |
Final, nb | 50 | 51 |
Final, mean (SD)b | 15.4 (5.0) | 12.0 (5.4) |
Change from baseline, mean (SD) | –1.9 (4.3) | –5.4 (4.3) |
Adjusted mean difference in change from baseline, pitolisant hydrochloride vs. placebo (95% CI)c | –3.42 (–4.96 to –1.87) | |
P value | < 0.0001 | |
Proportion of responders | ||
Responders, n (%)d | 17 of 50 (34.0) | 35 of 51 (68.6) |
Adjusted OR of response, pitolisant hydrochloride vs. placebo (95% CI)e | 4.26 (1.72 to 10.52) | |
P value | 0.0002 | |
MWT scoresf | ||
Number of patients contributing to the analysis | 51 | 54 |
Baseline, geometric mean (95% CI) | 4.3 (3.0 to 6.2) | 3.7 (2.7 to 5.2) |
Final, geometric mean (95% CI) | 4.6 (3.1 to 6.8) | 7.1 (4.9 to 10.3) |
Ratio of geometric means, final / baseline (95% CI) | 1.1 (0.8 to 1.4) | 1.9 (1.4 to 2.5) |
Ratio of geometric means (final / baseline), pitolisant hydrochloride / placebo (95% CI)g | 1.78 (1.22 to 2.60) | |
P value | 0.0032 | |
CGI-C scores on EDS | ||
Baseline, nh | 51 | 54 |
Baseline CGI-S score, n (%)h | ||
Normal (not ill at all) | 0 (0.0) | 0 (0.0) |
Borderline mentally ill | 0 (0.0) | 0 (0.0) |
Mildly ill | 0 (0.0) | 3 (5.6) |
Moderately ill | 13 (25.5) | 12 (22.2) |
Markedly ill | 27 (52.9) | 23 (42.6) |
Severely ill | 9 (17.6) | 14 (25.9) |
Among the most extremely ill patients | 2 (3.9) | 2 (3.7) |
Final, ni | 50 | 50 |
Final CGI-S score, n (%)i | ||
Very much improved | 7 (14.0) | 10 (20.0) |
Much improved | 2 (4.0) | 12 (24.0) |
Minimally improved | 3 (6.0) | 15 (30.0) |
No change | 32 (64.0) | 12 (24.0) |
Minimally worse | 3 (6.0) | 1 (2.0) |
Much worse | 1 (2.0) | 0 (0.0) |
Very much worse | 2 (4.0) | 0 (0.0) |
Final CGI-S, mean score (SD) | 3.7 (1.4) | 2.6 (1.1) |
Difference in final mean score (95% CI)j | –0.99 (–1.46 to –0.52) | |
P valuej | < 0.0001 | |
Therapy responder, n (%)k | 12 (23.5) | 37 (68.5) |
OR of response, pitolisant hydrochloride vs. placebo (95% CI)l | 7.07 (2.55 to 19.6) | |
P valuel | < 0.0001 | |
CGI-C = Clinical Global Impression of Change; CGI-S = Clinical Global Impression of Severity; CI = confidence interval; EDS = excessive daytime sleepiness; ESS = Epworth Sleepiness Scale; ITT = intention-to-treat; MWT = Maintenance of Wakefulness Test; OR = odds ratio; SD = standard deviation.
Note: P values were not controlled for type I errors (i.e., multiple testing).
aMean of values at visits 1 and 2.
bMean of values at visits 5 and 6. For missing values, the LOCF approach was used.
cLinear mixed model (ANCOVA) adjusted for baseline ESS and for centre heterogeneity (i.e., including centre as a random factor).
dResponse was defined as a final ESS score ≤ 10 or a change from baseline of ≥ 3 points.
eLogistic regression model adjusted for ESS at baseline and featured by a mixed nonlinear model, taking into account centre heterogeneity.
fMissing values were estimated using the linear relationship between MWT and ESS.
gCalculated as the ratio or geometric means (final/baseline) for pitolisant hydrochloride / ratio of geometric means (final/baseline) for placebo.
hMeasured at visit 2.
iMeasured at visit 6.
jLinear mixed-effects model.
kResponders were patients with CGI-C ≤ 3 at visit 6.
lNonlinear mixed model taking into account centre heterogeneity.
Source: Clinical Study Report for HARMONY CTP.15
Table 20: Drowsiness From Sleep Diary — HARMONY 1 (ITT Population) Trial
Measure | Placebo (N = 30) | Pitolisant hydrochloride (N = 31) | Modafinil (N = 33) |
|---|---|---|---|
Drowsiness episodes per day | |||
Baseline, n | 30 | 31 | 33 |
Baseline, mean (SD)a | 0.92 (1.77) | 0.96 (1.88) | 0.88 (1.75) |
Final, n | 28 | 30 | 31 |
Final, mean (SD)b | 0.58 (1.36) | 0.88 (2.13) | 0.46 (1.20) |
ITT = intention-to-treat; SD = standard deviation.
aAll episodes at visit 2 and visit 3 divided by number of days at visit 2 and visit 3.
bAll episodes at visit 4 to visit 7 and at visit 9 divided by number of days at visit 4 to visit 7 and at visit 9.
Source: Clinical Study Report for HARMONY 1.13
Table 21: Cataplexy — HARMONY 1 (ITT Population) and HARMONY 1bis (ITT Population) Trials
Variable | HARMONY 1 | HARMONY 1bis | ||||
|---|---|---|---|---|---|---|
Placebo N = 30 | Pitolisant hydrochloride N = 31 | Modafinil N = 33 | Placebo N = 32 | Pitolisant hydrochloride N = 66 | Modafinil N = 65 | |
Complete and partial cataplexy episodes per day | ||||||
Baseline, na | 30 | 31 | 33 | NR | NR | NR |
Baseline, mean (SD) | 0.43 (0.74) | 0.79 (1.53) | 0.76 (1.68) | NR | NR | NR |
Final, nb | 28 | 30 | 31 | NR | NR | NR |
Final, mean (SD) | 0.68 (1.66) | 0.28 (1.11) | 0.65 (1.62) | NR | NR | NR |
Daily rates of complete and partial cataplexy episodes, exposed population | ||||||
Patients contributing to analysisc | 14 | 20 | 23 | NR | NR | NR |
Baseline, geometric mean (95% CI) | 0.4 (0.2 to 1.0) | 0.5 (0.3 to 1.0) | 0.4 (0.2 to 0.8) | NR | NR | NR |
Final, geometric mean (95% CI) | 0.4 (0.1 to 1.1) | 0.2 (0.1 to 0.4) | 0.3 (0.1 to 0.5) | NR | NR | NR |
RR (95% CI) of cataplexy at end of treatment, pitolisant hydrochloride vs. placebod | 0.38 (0.15 to 0.93) | NA | NR | NR | NR | |
P value | 0.034 | — | NR | NR | NR | |
RR (95% CI) of cataplexy at end of treatment, pitolisant hydrochloride vs. modafinild | NA | 0.54 (0.24 to 1.23) | NR | NR | NR | |
P value | NA | 0.138 | NR | NR | NR | |
Daily rate of cataplexy for patients with cataplexy at baseline or during treatment, final 7 days | ||||||
Number of patients contributing to the analysis | NR | NR | NR | 18 | 37 | 39 |
Baseline, geometric mean (95% CI)e | NR | NR | NR | |||||||||||||| | |||||||||||||| | |||||||||||||| |
Final, geometric mean (95% CI)e,f | NR | NR | NR | |||||||||||||| | |||||||||||||| | |||||||||||||| |
Least square mean (CI)g | NR | NR | NR | |||||||||||||| | NA | |
P value | NR | NR | NR | |||||||||||||| | NA | |
CGI-C scores on cataplexy | ||||||
Number of patients contributing to the analysis | 25 | 26 | 28 | |||||||||||||| | |||||||||||||| | |||||||||||||| |
Baseline, mean (SD) | 3.6 (1.5) | 3.6 (1.1) | 3.7 (1.2) | NR | NR | NR |
Final, mean (SD)h | 3.4 (1.4) | 2.9 (1.5) | 3.0 (1.6) | NR | NR | NR |
Change from baseline, n (%) | ||||||
Improvedi | 6 (24.0) | 9 (34.6) | 8 (28.6) | |||||||||||||| | |||||||||||||| | |||||||||||||| |
No changei | 15 (60.0) | 15 (57.7) | 16 (57.1) | |||||||||||||| | |||||||||||||| | |||||||||||||| |
Worsenedi | 2 (8.0) | 0 (0.0) | 1 (3.6) | |||||||||||||| | |||||||||||||| | |||||||||||||| |
P value for change from baselinej | 0.607 | 0.075 | — | — | — | — |
CGI-C = Clinical Global Impression of Change; CI = confidence interval; ITT = intention-to-treat; NR = not reported; OR = odds ratio; RR = relative risk; SD = standard deviation.
Note: P values were not controlled for type I error (i.e., multiple testing).
aBaseline = (all episodes at visit 2 and visit 3) / (number of days at visit 2 and visit 3). For patients with no cataplexy at baseline or during treatment period, imputation value was determined by 0.5/number of days.
bFinal = (all episodes at visit 7 and visit 9) / (number of days at visit 7 and visit 9).
cDaily cataplexy rate was calculated as the ratio of the number of crises during 1 period on the number of days of this period. For these calculations, we accounted for the shortness of the exposures in the baseline, treatment, and final periods in the following way: for patients characterized as having no observed crisis during these periods, the rate is, at the most, the reciprocal of the duration (1 / number of days) and 0, at the least; thus, the imputation value was approximated from the mean between the 2 extremes (0.5 / number of days).
dAnalysis conducted on patients who had at least 1 cataplexy episode at baseline or during study treatment.
eGeometric mean based on base 10 logarithm of titre.
fDaily rate of cataplexy for patients with cataplexy at baseline or during treatment, final 7 days: visit 6 to visit 7. Sums of cataplexy equal to 0 have been replaced with 0.1.
gQuasi-Poisson model on daily cataplexy rate (the ratio final/baseline in geometric mean based on natural logarithm [GMT] of the number of cataplexy episodes on the number of exposed days), final 7 days, adjusted on DCR baseline with treatment considered as a fixed factor and reallocated centre as a random effect. For all the tests, pitolisant hydrochloride was compared with placebo and modafinil with a superiority test.
hAs measured at the end of the treatment period (visit 7).
jImproved defined as “very much improved, much improved, or minimally improved.” Worsened defined as “very much worse, much worse, or minimally worse.”
jCochran-Mantel-Haenszel test (ANOVA statistic) with modified ridit scores.
Sources: Clinical Study Reports for HARMONY 1 and HARMONY 1bis.13,14
Table 22: Cataplexy — HARMONY CTP (ITT Population) Trial
Variable | Placebo (N = 51) | Pitolisant hydrochloride (N = 54) | ||
|---|---|---|---|---|
Weekly cataplexy rate | ||||
Baseline, geometric mean (95% CI)a | 7.31 (6.02 to 8.87) | 9.15 (7.60 to 11.01) | ||
Stable-dose period, geometric mean (95% CI)b | 4.51 (2.90 to 7.02) | 2.27 (1.51 to 3.41) | ||
Ratio of geometric means, stable period / baseline (95% CI) | 0.62 (0.43 to 0.90) | 0.25 (0.17 to 0.36) | ||
Ratio of geometric means during stable-dose period, pitolisant hydrochloride / placebo (95% CI)c | 0.5123 (0.4351 to 0.6033) | |||
P valuec | < 0.0001 | |||
High frequency of cataplexy episodes (> 15) | ||||
Baseline, n (%)d | ||||
≤ 15 | 42 (82.4) | 39 (72.2) | ||
> 15 | 9 (17.6) | 15 (27.8) | ||
Stable-dose period, n (%)e | ||||
≤ 15 | 39 (76.5) | 51 (94.4) | ||
> 15 | 12 (23.5) | 3 (5.6) | ||
OR of frequency (95% CI)f | 0.035 (0.0035 to 0.352) | |||
P valuef | 0.0044 | |||
CGI-C scores on cataplexy | ||||
Number of patients contributing to the analysis | 51 | 54 | ||
Baseline, n (%)g | ||||
Normal (not ill at all) | 0 | 0 | ||
Borderline mentally ill | 0 | 0 | ||
Mildly ill | 5 (9.8) | 6 (11.1) | ||
Moderately ill | 16 (31.4) | 19 (35.2) | ||
Markedly ill | 20 (39.2) | 21 (38.9) | ||
Severely ill | 8 (15.7) | 6 (11.1) | ||
Among the most extremely ill patients | 2 (3.9) | 2 (3.7) | ||
Final, nh | 50 | 50 | ||
Final CGI-S, mean score (SD)h | 3.5 (1.1) | 2.6 (1.1) | ||
Final CGI-S, n (%)h | ||||
Very much improved | 4 (8.0) | 10 (20.0) | ||
Much improved | 3 (6.0) | 16 (32.0) | ||
Minimally improved | 10 (20.0) | 10 (20.0) | ||
No change | 31 (62.0) | 14 (28.0) | ||
Minimally worse | 0 | 0 | ||
Much worse | 2 (4.0) | 0 | ||
Very much worse | 0 | 0 | ||
Difference in final mean score (95% CI)i | –0.95 (–1.36 to –0.54) | |||
P valuei | < 0.0001 | |||
Therapy responder, n (%)j | 17 (33.3) | 36 (66.7) | ||
OR of response, pitolisant hydrochloride vs. placebo (95% CI)k | 4.00 (1.54 to 10.38) | |||
P valuek | 0.0044 | |||
Cataplexy frequency stratified on anticataplectic medications | ||||
+ Anticataplectics | Alone | + Anticataplectics | Alone | |
Any ongoing anticataplectic medications, n (%) | 8 (15.7) | 43 (84.3) | 4 (7.4) | 50 (92.6) |
Baseline, geometric meana | 11.57 | 6.71 | 6.49 | 9.40 |
Stable-dose period, geometric meanb | 9.54 | 6.32 | 6.40 | 3.13 |
Ratio of geometric means, stable period / baseline | 0.91 | 0.77 | 0.60 | 0.37 |
Sodium oxybate, n (%) | 1 (2.0) | 50 (98.0) | 1 (1.9) | 53 (98.1) |
Baseline, geometric meana | 18.50 | 6.71 | 5.00 | 9.40 |
Stable-dose period, geometric meanb | 18.00 | 6.32 | 4.50 | 3.13 |
Ratio of geometric means, stable period / baselinel | 0.97 | 0.77 | 0.92 | 0.37 |
SSRIs, n (%) | 1 (2.0) | 50 (98.0) | 3 (5.6) | 51 (94.4) |
Baseline, geometric meana | 18.50 | 5.98 | 7.08 | 9.40 |
Stable-dose period, geometric meanb | 18.50 | 6.32 | 7.62 | 3.13 |
Ratio of geometric means, stable period / baselinel | 2.05 | 0.77 | 0.52 | 0.37 |
Other anticataplectics, n (%) | 6 (11.8) | 45 (88.2) | 0 | 54 (100.0) |
Baseline, geometric meana | 11.27 | 6.71 | NA | 9.40 |
Stable-dose period, geometric meanb | 7.68 | 6.32 | NA | 3.13 |
Ratio of geometric means, stable period / baselinel | 0.78 | 0.77 | NA | 0.37 |
CGI-C = Clinical Global Impression of Change; CGI-S = Clinical Global Impression of Severity; CI = confidence interval ITT = intention-to-treat; OR = odds ratio; SD = standard deviation; SSRI = selective serotonin reuptake inhibitor.
Note: Aside from the WCR outcome, P values were not controlled for type I errors (i.e., multiple testing).
aMean of week 1 and week 2 values.
bMean of values during the stable-dose period (i.e., week 6 through week 9). For patients terminating the trial before completion, the final value was calculated as the mean of the 2 last known periods (LOCF).
cANCOVA via mixed nonlinear model featuring a possibly overdispersed Poisson distribution and taking into account centre heterogeneity by using centre as a random factor.
dMeasured at visit 2.
eMeasured at visit 6.
fNonlinear mixed model taking into account centre heterogeneity.
gMeasured at visit 2.
hMeasured at visit 6.
iLinear mixed-effects model.
jResponders were patients with CGI-C ≤ 3 at visit 6.
kNonlinear mixed model taking into account centre heterogeneity.
lMean ratio of cataplexy rate final / baseline was compared between study treatment and concomitant anticataplectic permitted medication.
Source: Clinical Study Report for HARMONY CTP.15
The primary end point of the HARMONY CTP trial was the measure of anticataplectic efficacy. The geometric means of the WCR at the end of treatment decreased to 4.51 (95% CI, 2.90 to 7.02) in the placebo group and 2.27 (95% CI, 1.51 to 3.41) in the pitolisant hydrochloride group during the stable-dose period (Figure 6). The ratio of geometric means during the stable-dose period was 0.51 (95% CI, 0.43 to 0.60; P < 0.0001) for pitolisant hydrochloride compared to placebo. Similar results were observed in the PP population, with a ratio of 0.50 (95% CI, 0.34 to 0.74; P < 0.0001) for pitolisant hydrochloride compared to placebo. The effect of pitolisant hydrochloride on the WCR remained consistent at 20 mg and 40 mg doses. The proportion of patients with a high frequency of weekly cataplexy episodes (> 15) during the stable-dose period was 5.6% in the pitolisant hydrochloride group and 17.6% in the placebo group (OR, 0.035; 95% CI, 0.0035 to 0.352). The effect remained consistent regardless of whether patients were taking permitted anticataplectic medications during the trial.
Figure 6: Weekly Cataplexy Rates — HARMONY CTP Trial
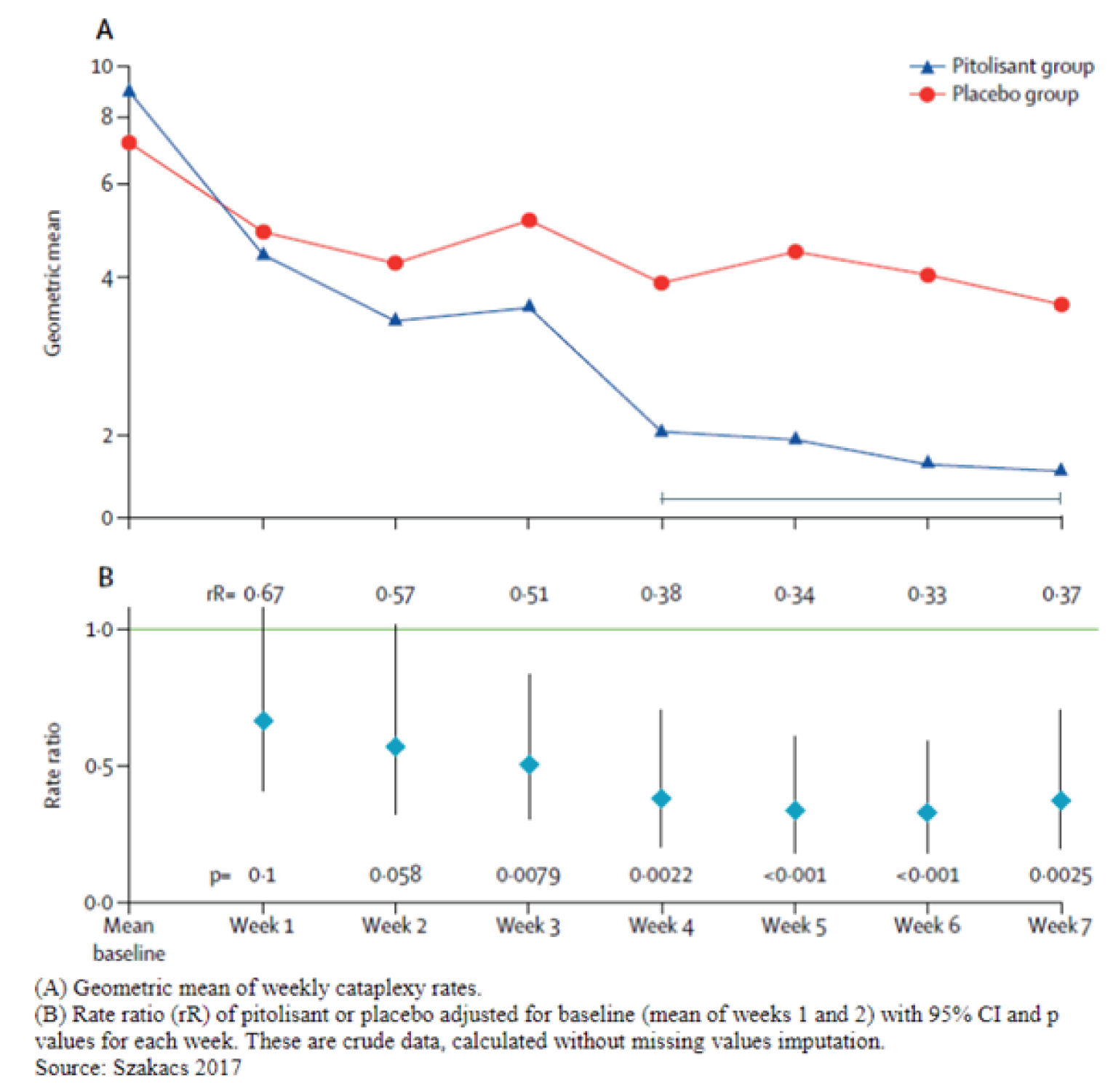
CI = confidence interval.
Source: Clinical Study Report for HARMONY CTP.15
CGI-S and CGI-C on Cataplexy
In the HARMONY 1 trial, the mean final CGI-C score was 3.4 (SD = 1.4), 2.9 (SD = 1.5), 3.0 (SD = 1.6) in the placebo, pitolisant hydrochloride, and modafinil arms, respectively. The number of patients who improved compared to baseline was 6 (24.0%) in the placebo group, 9 (34.6%) in the pitolisant hydrochloride group, and 8 (28.6%) in the modafinil group. The number of patients who reported no change from baseline was 15 (57.7%) in the placebo group, 15 (57.7%) in the pitolisant hydrochloride group, and 16 (57.1%) in the modafinil group. There were 2 (8.0%) patients reporting worsened CGI-C scores in the placebo arm and 1 (3.6%) patient in the modafinil arm.
In the HARMONY 1bis trial, the number of patients who improved from baseline was |||||||||||||| for placebo, |||||||||||||| for pitolisant hydrochloride, and |||||||||||||| for modafinil. The number of patients who reported no change from baseline was |||||||||||||| for placebo, |||||||||||||| for pitolisant hydrochloride, and |||||||||||||| for modafinil. There were |||||||||||||| patients reporting worsened CGI-C scores in the placebo arm, |||||||||||||| in the pitolisant hydrochloride arm, and |||||||||||||| in the modafinil arm.
In the HARMONY CTP group, the mean reduction in CGI-C score for pitolisant hydrochloride compared with placebo was –0.95 (95% CI, –1.36 to –0.54). Mean CGI-C score was 3.5 (SD = 1.1) with placebo versus 2.6 (SD = 1.1) with pitolisant hydrochloride. Similar results were observed for the PP population, with a mean reduction of –0.86 (95% CI, –1.29 to –0.43).
Health-Related Quality of Life
In the HARMONY 1 trial, mean 5-Level EQ-5D scores at baseline were 64.0 (SD = 19.2), 65.3 (SD = 21.3), and 58.7 (SD = 19.4) in the placebo, pitolisant hydrochloride, and modafinil arms, respectively (Table 23). By the end of the treatment period, mean 5-Level EQ-5D scores were 70.2 (SD = 17.7), 43.8 (SD = 17.8), and 72.6 (SD = 16.5) in the placebo, pitolisant hydrochloride, and modafinil groups, respectively. However, there were no differences among the 3 treatment arms and modafinil demonstrated a greater improvement from baseline. In the HARMONY 1bis trial, mean 5-Level EQ-5D scores were |||||||||||||||||||||||||||||||||||||||||||||||||||||||| in the placebo, pitolisant hydrochloride, and modafinil groups, respectively. In the HARMONY CTP trial, no difference between groups was demonstrated for the overall score of this questionnaire (sum of the 5 dimensions) or the corresponding VAS (Table 24).
Table 23: HRQoL — HARMONY 1 (ITT Population) and HARMONY 1bis (ITT Population) Trials
EQ-5D VAS scores | HARMONY 1 | HARMONY 1bis | ||||
|---|---|---|---|---|---|---|
Placebo | Pitolisant hydrochloride | Modafinil | Placebo | Pitolisant hydrochloride | Modafinil | |
Baseline, n | 29 | 31 | 32 | |||||||||||||| | |||||||||||||| | |||||||||||||| |
Baseline, mean (SD)a | 64.0 (19.2) | 65.3 (21.3) | 58.7 (19.4) | |||||||||||||| | |||||||||||||| | |||||||||||||| |
Final, n | 25 | 26 | 27 | |||||||||||||| | |||||||||||||| | |||||||||||||| |
Final, mean (SD)b | 70.2 (17.7) | 73.8 (17.8) | 72.6 (16.5) | |||||||||||||| | |||||||||||||| | |||||||||||||| |
HRQoL = health-related quality of life; ITT = intention-to-treat; SD = standard deviation; VAS = visual analogue scale.
aMeasured at visit 3.
bMeasured at visit 7.
Sources: Clinical Study Reports for HARMONY 1 and HARMONY 1bis.13,14
Table 24: HRQoL — HARMONY CTP (ITT Population) Trial
HRQoL scores | Placebo (N = 51) | Pitolisant hydrochloride (N = 54) |
|---|---|---|
EQ-5D scores | ||
Baseline, n | 51 | 54 |
Baseline score, mean (SD)a | 6.5 (1.2) | 6.4 (1.2) |
Final, n | 50 | 50 |
Final score, mean (SD)b | 6.4 (1.4) | 6.0 (1.1) |
Change from baseline score, mean (SD) | –0.1 (1.0) | –0.4 (1.2) |
Mean difference in change from baseline, pitolisant hydrochloride vs. placebo (95% CI)c | –0.33 (–0.697 to 0.034) | |
P valuec | 0.075 | |
EQ-5D VAS scores | ||
Baseline, n | 51 | 54 |
Baseline score, mean (SD)a | 64.3 (22.6) | 60.6 (25.0) |
Final, n | 50 | 50 |
Final score, mean (SD)b | 71.3 (15.5) | 68.7 (19.5) |
Change from baseline score, mean (SD) | 7.3 (18.9) | 8.6 (18.4) |
Difference in change from baseline, pitolisant hydrochloride vs. placebo (95% CI)c | –0.94 (–6.19 to 4.3) | |
P valuec | 0.723 | |
EQ-5D VAS scores for a high-frequency (> 15) WCR | ||
Baseline WCR ≤ 15, n | 39 | 51 |
Baseline score, mean (SD)a | 67.1 (20.7) | 61.2 (24.6) |
Final WCR ≤ 15, n | 38 | 47 |
Final score, mean (SD)b | 75.8 (10.8) | 69.5 (19.0) |
Baseline WCR > 15, n | 12 | 3 |
Baseline score, mean (SD)a | 55.2 (27.1) | 51.0 (37.3) |
Final WCR > 15, n | 12 | 3 |
Final score, mean (SD)b | 57.1 (19.5) | 56.0 (26.5) |
Change from baseline score, WCR > 15, mean (SD) | 1.9 (16.0) | 5.0 (26.5) |
Change from baseline score, WCR ≤ 15, mean (SD) | 9.0 (19.6) | 8.9 (18.1) |
Difference in change from baseline, pitolisant hydrochloride vs. placebo (95% CI)c | –1.1713 (–27.3615 to 25.0188) | |
P valuec | 0.9072 | |
CI = confidence interval; HRQoL = health-related quality of life; ITT = intention-to-treat; SD = standard deviation; VAS = visual analogue scale; WCR = weekly cataplexy rate.
Note: P values were not controlled for type I errors (i.e., multiple testing).
aMeasured at visit 2.
bMeasured at visit 6.
cLinear mixed-effects model.
Source: Clinical Study Report for HARMONY CTP.15
Functional Activity
This outcome was not reported outside of the PGO score (refer to the Patient Satisfaction, Ease of Use section).
Mental Health
Refer to the Harms section for the mental health outcomes reported.
Sexual Function
This outcome was not reported.
Sleep Attacks
This outcome was reported as part of the Patient Diaries section, immediately preceding Table 20.
Nocturnal Sleep Properties
The duration of nocturnal awakening was reported in the HARMONY 1 trial (Table 25). All treatment arms reported a final duration of 0.36 hours, or 31.6 minutes (SD = 0.03, 0.05, and 0.08 in placebo, pitolisant hydrochloride, and modafinil groups, respectively). Nocturnal sleep properties were not reported in the HARMONY 1bis trial. In the HARMONY CTP trial, the mean stable-dose period duration was 47.0 (SD = 44.6) minutes for placebo and 41.5 (SD = 58.6) minutes for pitolisant hydrochloride (Table 26). The mean change from baseline to the stable-dose period was –20.3 (SD = 94.6) minutes in the placebo group and –8.2 (SD = 31.5) minutes in the pitolisant hydrochloride group. The adjusted difference in change from baseline was –0.631 (95% CI, –21.939 to 20.677).
Sleep paralysis was decreased across all treatment arms in the HARMONY 1 trial. The final mean number of patients reporting sleep paralysis was 0.03 (SD = 0.11), 0.01 (SD = 0.05), and 0.08 (SD = 0.34) in the placebo, pitolisant hydrochloride, and modafinil groups, respectively.
Polysomnography results are provided in Appendix 3.
Number of Hallucinations
The number of days with hallucinations in the HARMONY 1 trial significantly decreased with pitolisant hydrochloride in comparison to placebo (Table 25). The adjusted ratio rate was 0.46 (95% CI, 0.27 to 0.79). The number of days of hallucinations for patients with any reported hallucinations in the HARMONY CTP trial was significantly lower in the pitolisant hydrochloride arm than in the placebo arm, with an adjusted difference of –0.7766 (–1.3143 to –0.2389).
Table 25: Nocturnal Sleep Properties and Hallucinations — HARMONY 1 (ITT Population) Trial
Measure | Events | ||
|---|---|---|---|
Placebo | Pitolisant hydrochloride | Modafinil | |
Hallucinations | |||
Number of patients contributing to the analysis | 30 | 31 | 33 |
Baseline, mean (SD)a | 0.32 (1.17) | 0.05 (0.14) | 0.15 (0.37) |
Final, n | 28 | 30 | 31 |
Final, mean (SD)b | 0.36 (1.86) | 0.01 (0.06) | 0.07 (0.22) |
Diurnal involuntary sleep attacks and severe sleepiness (hours per day) | |||
Number of patients contributing to the analysis | 30 | 31 | 33 |
Baseline, mean (SD)a | 1.52 (1.04) | 1.83 (1.29) | 1.71 (1.67) |
Final, n | 28 | 30 | 31 |
Final, mean (SD)b | 1.46 (1.37) | 1.32 (1.34) | 1.35 (1.54) |
Duration of nocturnal awakenings (hours per day) | |||
Number of patients contributing to the analysis | 30 | 31 | 33 |
Baseline, mean (SD)a | 0.36 (0.05) | 0.38 (0.06) | 0.35 (0.05) |
Final, n | 28 | 30 | 31 |
Final, mean (SD)b | 0.36 (0.03) | 0.36 (0.05) | 0.36 (0.08) |
Sleep paralysis | |||
Number of patients contributing to the analysis | 30 | 31 | 33 |
Baseline, mean (SD)a | 0.10 (0.24) | 0.07 (0.24) | 0.12 (0.22) |
Final, n | 28 | 30 | 31 |
Final, mean (SD)b | 0.03 (0.11) | 0.01 (0.05) | 0.08 (0.34) |
CI = confidence interval; ITT = intention-to-treat; SD = standard deviation.
aAll episodes at visit 2 and visit 3 / number of days at visit 2 and visit 3.
bAll episodes at visit 4 to visit 7 and at visit 9 / number of days at visit 4 to visit 7 and at visit 9.
Sources: Clinical Study Reports for HARMONY 1 and HARMONY 1bis.13,14
Table 26: Sleepiness and Nocturnal Sleep Properties From Sleep Diary — HARMONY CTP (ITT Population) Trial
Event | Placebo (N = 51) | Pitolisant hydrochloride (N = 54) |
|---|---|---|
Duration of nocturnal awakenings (minutes) | ||
Baseline, na | 48 | 52 |
Baseline duration in minutes, mean (SD) | 67.5 (99.9) | 56.0 (63.7) |
Stable-dose period, nb | 43 | 49 |
Stable-dose period duration in minutes, mean (SD) | 47.0 (44.6) | 41.5 (58.6) |
Change from baseline to stable-dose period in minutes, mean (SD) | –20.3 (94.6) | –8.2 (31.5) |
Adjusted difference in change from baseline (95% CI)a | –0.631 (–21.939 to 20.677) | |
P valuec | 0.949 | |
CI = confidence interval; ITT = intention-to-treat; SD = standard deviation.
Note: P values were not controlled for type I errors (i.e., multiple testing).
aMeasured at visit 2.
bMeasured at visit 7.
cLinear mixed-effect model adjusted for baseline value.
Source: Clinical Study Report for HARMONY CTP.15
Concomitant Medication Use
Concomitant medication use was reported for all trials and described in the Exposure to Study Treatments section, which follows Table 35.
Patient Satisfaction, Ease of Use
In the HARMONY 1 trial, patients were classified as improving (PGO score ≥ 3) or not improving (PGO score < 3) on efficacy after 8 weeks of treatment, and improvement in symptoms was reported by 56.0% of patients in the placebo group, 80.8% in the pitolisant hydrochloride group, and 85.7% in the modafinil group (Table 27). The mean final PGO score was 3.1 (SD = 1.4), 2.3 (SD = 1.2), 2.0 (SD = 1.3) in the placebo, pitolisant hydrochloride, and modafinil arms, respectively. The number of patients who reported improvement from baseline was 14 (56.0%) in the placebo group, 21 (80.8%) in the pitolisant hydrochloride group, and 24 (85.7%) in the modafinil group. The number of patients who reported no change from baseline was 7 (28.0%) in the placebo group, 4 (15.4%) in the pitolisant hydrochloride group, and 2 (7.1%) in the modafinil group. Worsened PGO scores were reported by 4 (16.0%) patients in the placebo arm, 1 (3.8%) patient in the pitolisant hydrochloride arm, and 2 (7.1%) patients in the modafinil arm.
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.
In the HARMONY CTP trial, more patients noted an effect for efficacy (marked, moderate, or minimal) in the pitolisant hydrochloride group (39 of 50 patients, or 78%) than in the placebo group (13 of 48 patients, or 27%) (Table 28).
Adherence
Overall, more than 91% of the patients in the ITT population had adherence to treatment of at least 80% (Table 29). There was no significant difference between treatment groups with respect to adherence. In the HARMONY 1bis trial, almost all the patients were 80% to 120% adherent, with the exception of 5 (7.6%) patients in the pitolisant hydrochloride group (1 patient who took more than 120% of prescribed treatment and 4 patients who took less than 80%). In the HARMONY CTP trial, only 1 patient in the pitolisant hydrochloride group took less than 80% or more than 120% of the prescribed treatment (Table 30).
Health Care Utilization
This outcome was not reported.
Table 27: PGO — HARMONY 1 (ITT Population) and HARMONY 1bis (ITT Population) Trials
Measure | HARMONY 1 | HARMONY 1bis | ||||
|---|---|---|---|---|---|---|
Placebo N = 30 | Pitolisant hydrochloride N = 31 | Modafinil N = 33 | Placebo N = 32 | Pitolisant hydrochloride N = 66 | Modafinil N = 65 | |
PGO | ||||||
Visit 4 (2 weeks on treatment), na | 25 | 26 | 28 | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Visit 4 (2 weeks on treatment), mean (SD) | 3.6 (1.2) | 2.7 (1.2) | 2.4 (1.1) | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Final, nb | 25 | 26 | 28 | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Final, mean (SD) | 3.1 (1.4) | 2.3 (1.2) | 2.0 (1.3) | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
PGO at end of treatment, n (%) | ||||||
Improved | 14 (56.0) | 21 (80.8) | 24 (85.7) | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
No change | 7 (28.0) | 4 (15.4) | 2 (7.1) | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Worsened | 4 (16.0) | 1 (3.8) | 2 (7.1) | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
ITT = intention-to-treat; PGO = patient global opinion; SD = standard deviation.
aMeasured at Visit 4 (2 weeks on treatment).
bMeasured at visit 7.
Sources: Clinical Study Reports for HARMONY 1 and HARMONY 1bis.13,14
Table 28: PGO — HARMONY CTP (ITT Population) Trial
Measure | Placebo (N = 51) | Pitolisant hydrochloride (N = 54) |
|---|---|---|
PGO | ||
End of treatment, na | 48 | 50 |
Efficacy, n (%) | ||
Marked effect | 5 (10.4) | 15 (30.0) |
Moderate effect | 6 (12.5) | 12 (24.0) |
Minimal effect | 7 (14.6) | 12 (24.0) |
No change | 26 (54.2) | 9 (18.0) |
Minimally worse | 2 (4.2) | 0 (0.0) |
Much worse | 2 (4.2) | 2 (4.0) |
Treatment responder, n (%)b | 13 (25.5) | 29 (53.7) |
P value for difference in response ratec | 0.0012 | |
Safety, n (%) | ||
Good | 40 (83.3) | 45 (90.0) |
Moderate | 8 (16.7) | 5 (10.0) |
Bad | 0 (0.0) | 0 (0.0) |
Treatment responder, n (%)d | 44 (86.3) | 47 (87.0) |
P value for difference in response ratec | 0.487 | |
Quality of sleep, n (%) | ||
Good | 22 (45.8) | 31 (62.0) |
Moderate | 18 (37.5) | 16 (32.0) |
Bad | 8 (16.7) | 3 (6.0) |
Treatment responder, n (%)d | 25 (49.0) | 30 (55.6) |
P value for difference in response ratec | 0.315 | |
ITT = intention-to-treat; PGO = patient global opinion; PGS = polysomnography.
Note: P values were not controlled for type I errors (i.e., multiple testing).
aMeasured at visit 7.
bDefined as an efficacy score < 3.
cChi-square distribution.
dDefined as a safety or sleep score < 2.
Source: Clinical Study Report for HARMONY CTP.15
Table 29: Treatment Adherence — HARMONY 1 (ITT Population) and HARMONY 1bis (ITT Population) Trials
Measure | HARMONY 1 | HARMONY 1bis | ||||
|---|---|---|---|---|---|---|
Placebo N = 30 | Pitolisant hydrochloride N = 31 | Modafinil N = 33 | Placebo N = 32 | Pitolisant hydrochloride N = 66 | Modafinil N = 65 | |
Treatment adherence | ||||||
< 80% of prescribed treatment, n (%) | 2 (6.7) | 2 (6.5) | 2 (6.1) | 1 (3.1) | 3 (4.6) | 0 |
80% to 120% of prescribed treatment, n (%) | 27 (90.0) | 28 (90.3) | 31 (93.9) | 31 (96.9) | 61 (92.4) | 65 (100.0) |
≥ 120% of prescribed treatment, n (%) | 1 (3.3) | 1 (3.2) | 0 | 0 | 1 (1.5) | 0 |
P value for Fisher’s exact test | 0.939 | 0.222 | ||||
ITT = intention-to-treat.
Note: P values were not controlled for type I errors (i.e., multiple testing).
Sources: Clinical Study Reports for HARMONY 1 and HARMONY 1bis.13,14
Table 30: Treatment Adherence — HARMONY CTP (ITT Population) Trial
Measure | Placebo (N = 51) | Pitolisant hydrochloride (N = 54) |
|---|---|---|
Treatment adherence | ||
< 80% of prescribed treatment, n (%) | 0 | 1 (1.9) |
80% to 120% of prescribed treatment, n (%) | 51 (100.0) | 53 (98.1) |
≥ 120% of prescribed treatment, n (%) | 0 | 0 |
P value for chi-square distribution | 0.329 | |
ITT = intention-to-treat.
Note: P values were not controlled for type I errors (i.e., multiple testing).
Source: Clinical Study Report for HARMONY CTP.15
Harms
Only harms identified in the review protocol are reported here. Refer to Table 31 for detailed harms data. AEs by type were selected based on the total number of patients being at least 3 across treatment arms.
Adverse Events
In the HARMONY 1 trial, AEs after initiation of treatment were reported by 66.7% of patients in the placebo group, 64.5% in the pitolisant hydrochloride group, and 69.7% in the modafinil group (Table 31). In the HARMONY 1bis trial, approximately |||||||||||| of the patients in the pitolisant hydrochloride and modafinil groups reported AEs, whereas ||||||||||| of placebo patients reported AEs. In the HARMONY CTP trial, approximately 31.4% of patients in the placebo group and 35.0% of patients in the pitolisant hydrochloride group experienced an AE. In the HARMONY 1 trial |||||||||||||||, there was a greater percentage of nervous system disorders in the pitolisant hydrochloride arm, but in the HARMONY CTP trial, there was a greater percentage in the placebo group .
Serious Adverse Events
In the HARMONY 1 trial, 12.9% of patients in the pitolisant hydrochloride group reported SAEs, compared with 24.2% in the modafinil group and 10.0% in the placebo group. In the pitolisant hydrochloride arm, pyelonephritis and hemorrhoids were reported as SAEs. In the HARMONY 1bis trial, 15.2% of patients in the pitolisant hydrochloride arm experienced SAEs, compared with 6.5% in the modafinil group. There were no SAEs in the placebo group. In the HARMONY CTP trial, there was 1 (1.9%) SAE in the pitolisant hydrochloride arm.
Withdrawals due to Adverse Events
In the HARMONY 1 trial, 1 patient in the pitolisant hydrochloride arm discontinued because of pregnancy. An additional patient in the pitolisant hydrochloride arm temporarily discontinued the study, but the study code was not broken and treatment was resumed so the study resumed. ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||. In the HARMONY CTP trial, 1 patient receiving pitolisant hydrochloride discontinued because of severe nausea as a TEAE.
Mortality
No deaths were reported in any of the trials.
Table 31: Summary of Harms for the HARMONY 1 (ITT Population), HARMONY 1bis (Safety Population), and HARMONY CTP (Safety Population) Trials
Harms | HARMONY 1 Placebo N = 30 | HARMONY 1 Pitolisant hydrochloride N = 31 | HARMONY 1 Modafinil N = 33 | HARMONY 1bis Placebo N = 33 | HARMONY 1bis Pitolisant hydrochloride N = 67 | HARMONY 1bis Modafinil N = 65 | HARMONY CTP Placebo N = 51 | HARMONY CTP Pitolisant hydrochloride N = 54 |
|---|---|---|---|---|---|---|---|---|
Patients with ≥ 1 TEAE | ||||||||
Overall, n (%) | 20 (66.7) | 20 (64.5) | 23 (69.7) | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| | 16 (31.4) | 19 (35.2) |
Nervous system disorders | 8 (26.7) | 16 (51.6) | 1 (3.0) | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| | 11 (21.6) | 6 (11.1) |
Gastrointestinal disorders | 2 (6.67) | 8 (25.8) | 11 (33.3) | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| | 0 | 3 (5.6) |
Psychiatric disorders | 3 (10.0) | 7 (22.6) | 5 (15.2) | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| | 4 (7.8) | 8 (14.8) |
Infections and infestations | 6 (20.0) | 6 (19.4) | 5 (15.2) | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| | 0 | 2 (3.7) |
Renal and urinary disorders | 1 (3.3) | 4 (12.9) | 1 (3.0) | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| | 0 | 0 |
Metabolism and nutrition disorders | 1 (3.3) | 2 (6.5) | 3 (9.0) | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| | 0 | 0 |
Patients with ≥ 1 SAE | ||||||||
Overall, n (%) | 3 (10) | 4 (12.9) | 8 (24.2) | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| | 0 | 1 (1.9) |
Before study treatment administered, n (%) | 1 (3.3) | 1 (3.2) | 0 | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| | NR | NR |
After study treatment administered, n (%) | 2 (6.7) | 3 (9.7) | 8 (24.2) | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| | NR | NR |
Patients with AE leading to withdrawal | ||||||||
n (%) | 4 (13.3) | 0 | 4 (12.1) | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| | 0 | 1 (1.9) |
Deaths | ||||||||
n (%) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
AE = adverse event; ITT = intention-to-treat; NR = not reported; SAE = serious adverse event; TEAE = treatment-emergent adverse event.
Sources: Clinical Study Reports for HARMMONY I, HARMONY 1bis, and HARMONY CTP.13-15
Notable Harms
In the HARMONY 1 trial, no cardiovascular (QTc prolongation) AEs were noted. There were patients with psychiatric disorders: 3 (10.0%) in the placebo arm, 7 (22.6%) in the pitolisant hydrochloride arm, and 5 (15.2%) in the modafinil arm. Infections and infestations were reported by 6 (20.0%) patients in the placebo arm, 6 (19.4%) patients in the pitolisant hydrochloride arm, and 5 (15.2%) patients in the modafinil arm.
In the HARMONY 1bis trial, there was 1 abnormality in the pitolisant hydrochloride treatment arm (supraventricular extrasystoles) in 1 (1.5%) patient at the end of treatment, and there were 2 (6.0%) abnormal ECGs in the placebo arm and 4 (6.2%) abnormal ECGs in the modafinil arm. There was a greater percentage of TEAEs in the pitolisant hydrochloride arm than in the placebo arm for cardiac disorders, gastrointestinal disorders, hepatobiliary disorders, and psychiatric disorders.
In the HARMONY CTP trial, 1 QTcF variation over 650 ms was reported in 1 patient who received pitolisant hydrochloride from randomization to the end of treatment. No AEs related to dependence or tolerance were noted.
Mental Health
The BDI-SF-13 final score in the HARMONY 1 trial was 2.6 (SD = 2.5) for placebo, 3.3 (SD = 3.1) for pitolisant hydrochloride, and 5.6 (SD = 6.5) for modafinil (Table 32). In the HARMONY 1bis trial, the mean difference in final score from baseline was –1.1 (SD: 3.0) for placebo, –1.7 (SD = 2.6) for pitolisant hydrochloride, and –1.3 (SD = 2.8) for modafinil. In the HARMONY CTP trial, the mean difference in final score from baseline was –0.8 (SD = 2.4) for placebo and –1.8 (SD = 2.8) for pitolisant hydrochloride (Table 33). The adjusted mean difference in final score from baseline for placebo and pitolisant hydrochloride was 0.172 (95% CI, –0.268 to 0.612).
Table 32: BDI-SF-13 — HARMONY 1 (ITT Population) and HARMONY 1bis (ITT Population) Trials
Measure | HARMONY 1 | HARMONY 1bis | ||||
|---|---|---|---|---|---|---|
Placebo | Pitolisant hydrochloride | Modafinil | Placebo | Pitolisant hydrochloride | Modafinil | |
Beck Depression Score | ||||||
Baseline, n | 10 | 12 | 8 | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Baseline, mean (SD)a | 4.4 (3.1) | 4.6 (4.0) | 5.9 (4.8) | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Final, n | 8 | 9 | 5 | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Final, mean (SD)b | 2.6 (2.5) | 3.3 (3.1) | 5.6 (6.5) | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Change from baseline, mean (SD) | NR | NR | NR | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
BDI-SF-13 = 13-item Beck Depression Inventory-Short Form; ITT = intention-to-treat; SD = standard deviation.
aMeasured at visit 3.
bMeasured at visit 8 (following withdrawal period). In the HARMONY 1bis trial, measured at visit 7.
Sources: Clinical Study Reports for HARMONY 1 and HARMONY 1bis.13,14
Table 33: BDI-SF-13 — HARMONY CTP (ITT Population) Trial
Measure | Placebo | Pitolisant hydrochloride |
|---|---|---|
Beck Depression Score | ||
Baseline, na | 51 | 54 |
Baseline score, mean (SD)a | 5.3 (4.4) | 4.8 (4.0) |
Final, nb | 50 | 50 |
Final score, mean (SD)b | 4.3 (4.3) | 2.8 (3.2) |
Change from baseline score, mean (SD) | –0.8 (2.4) | –1.8 (2.8) |
Adjusted difference in change from baseline (95% CI)c | 0.172 (–0.268 to 0.612) | |
BDI-SF-13 = 13-item Beck Depression Inventory-Short Form; CI = confidence interval; ITT = intention-to-treat; SD = standard deviation.
aMeasure at visit 2.
bMeasured at visit 6.
cRepeated measures linear mixed-effects model adjusted for baseline score.
Source: Clinical Study Report for HARMONY CTP.15
Withdrawal Symptoms Questionnaire
In the HARMONY 1 trial, the following withdrawal symptoms were reported for the placebo, pitolisant hydrochloride, and modafinil groups, respectively: dysphoria (0, 1 [3.8%], 4 [14.3%]), increased appetite (0, 0, 2 [7.1%]), fatigue (3 [12.0%], 10 [38.5%], 12 [42.9%]), insomnia or hypersomnia (3 [12.0%], 10 [38.5%], 9 [32.1%]), psychomotor retardation or agitation (0, 1 [3.8%], 3 [10.7%]), and vivid and unpleasant dreams (1 [4.0%], 3 [11.5%], 4 [14.3%]).
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
In the HARMONY CTP trial, the following withdrawal symptoms were reported for the placebo and pitolisant hydrochloride groups, respectively: dysphoria (3 [6.3%], 0), increased appetite (1 [2.1%], 0), fatigue (2 [4.2%], 2 [4.0%]), psychomotor retardation or agitation (1 [2.1%], 1 [ 2.0%]), vivid and unpleasant dreams (1 [2.1%], 0), and insomnia or hypersomnia (1 [2.1%], 2 [4.0%]).
Critical Appraisal
Internal Validity
All included trials were double-blinded, placebo-controlled, and had a short duration (7-week or 8-week treatment phase). The allocation sequence was random and remained concealed to patients, investigators, and clinical staff for the duration of the trial. The HARMONY 1 and HARMONY 1bis trials had between-group baseline imbalances for previous medication use and proportion with cataplexy, which could suggest differences in disease severity. In the HARMONY 1bis trial, ||||||| of patients had a history of cataplexy in the pitolisant hydrochloride group compared to ||||||| in the placebo group. In the HARMONY 1 trial, patients who were taking at least 1 chronic medication in the 3 months before inclusion ranged from 70.0% (modafinil) to 85.2% (placebo and pitolisant hydrochloride). The maximum dosages for pitolisant hydrochloride were 20 mg daily in the HARMONY 1bis trial, whereas in the HARMONY 1 and HARMONY CTP trials, the maximum daily dosage was 40 mg. Titration of the study drug was at the discretion of the study investigators, which could affect efficacy and potentially threaten blinding to treatment.
All studies authorized patients to remain on stable doses of anticataplectic medications. Patients on anticataplectic medications represented 35% of all patients in the HARMONY 1 trial, ||||||| of all patients in the HARMONY 1bis trial, and 10% of all patients in the HARMONY CTP trial. There were between-group differences in the HARMONY 1 and HARMONY CTP trials for the proportion of patients on anticataplectic medications during the trial. In the HARMONY 1 trial, 33.3% of placebo patients compared to 40.7% pitolisant hydrochloride patients and 56.7% modafinil patients remained on authorized medications during the study. In the HARMONY CTP trial, 16% of those in the placebo group remained on anticataplectic medication, compared with 7% in the pitolisant group. Inconsistency in concomitant anticataplectic medications between trials cannot be clearly explained. The interactions between pitolisant hydrochloride and the concomitant treatments are unknown. Although the trials were double-blinded, some patients who received modafinil previously may have recognized the study drug.
The primary efficacy outcome for the HARMONY 1 and HARMONY 1bis trials, change in EDS, was measured using the validated ESS, which is a subjective, self-administered questionnaire that it is widely used in narcolepsy trials. The primary outcome for the HARMONY CTP trial was the WCR captured by patient diaries. All primary outcomes were assessed using unvalidated tools. Other secondary end points that assessed EDS were not validated, such as the CGI-C and PGO tools. The MWT and SART outcomes were validated, but the statistical analyses did not adjust for multiplicity so there was an increased risk of type I errors and conclusions could not be drawn. Patient diaries were completed daily and reviewed by the investigators for completion, which may have biased future outcome assessments.
Missing values for all trials were imputed for ESS and cataplexy outcomes. Any missing values at the end of treatment were imputed using LOCF or BOCF. It is unclear whether these would be reflective of the true trajectory of the outcomes. In addition, for all outcomes other than the primary outcome in all trials, there was no adjustment for multiplicity, which increases the risk of type I errors and limits the ability to draw conclusions. Subgroups were outlined a priori. Conclusions could not be drawn for the subgroups because of the lack of adjustment for multiplicity in those analyses, so they were therefore considered exploratory.
The appropriateness of the NIM is uncertain. The NIM was calculated on the basis of an unpublished meta-analysis of historical trials (in narcolepsy, obstructive sleep apnea, and Parkinson disease) of ESS, which set the MID as 3. To remain below the MID and to maintain the proportion of difference between placebo and pitolisant hydrochloride, a NIM of 2 was chosen. In addition, sample-size calculations assumed that the effects of pitolisant hydrochloride and modafinil were similar.
All trials noted protocol amendments. A major amendment in the HARMONY 1 trial was the change from assessment of the superiority of pitolisant compared with modafinil to a noninferiority analysis. Still, noninferiority was not demonstrated in either the HARMONY 1 trial or HARMONY 1bis trial.
External Validity
According to the clinical experts consulted for this review, the baseline characteristics of study patients are reflective of patients in Canada with narcolepsy seeking further treatment options. The drug titration would be reflective of clinical practice. The primary outcome measures used in the trials are used by physicians in clinical practice and measure outcomes important to patients (EDS and cataplexy). Patients were allowed to combine conventional narcolepsy medication with the drug under study. The clinical experts noted that it is common for combination therapy to be used in clinical practice; however, the interactions between concomitant medications and pitolisant hydrochloride are unknown. On that note, TCAs were not allowed as concurrent medications, despite them being common anticataplectic drugs, according to the clinical experts. This may decrease the generalizability of results from the trial population. Adherence to treatment remained high, at more than 80%, in all trials.
Other Relevant Evidence
This section examines the long-term extension study included in the sponsor’s submission to CADTH that was considered to address important gaps in the evidence covered in the Systematic Review.
Long-Term Extension Study
The open-label extension study, HARMONY III,16,17 provides long-term safety and efficacy data that supplements evidence from the RCTs in the Systematic Review.
Methods
HARMONY III is a long-term, open-label, uncontrolled extension study conducted to evaluate the efficacy and safety of pitolisant hydrochloride at a daily dose of 5 mg, 10 mg, 20 mg, or 40 mg for the treatment of EDS in patients with narcolepsy with or without cataplexy for up to 5 years. Of the 102 patients enrolled in the HARMONY III trial, 86 were pitolisant hydrochloride–naive or secondary-naive and were not receiving pitolisant hydrochloride at the time of study enrolment and 16 were patients from the French CUP who were treated with pitolisant hydrochloride in the 2 weeks preceding the study. Of the 86 naive patients, 73 had never been treated with pitolisant hydrochloride and 13 had been previously treated with pitolisant hydrochloride during single-blind or double-blind trials, including HARMONY 1,13 HARMONY II,18 and HARMONY 1bis.14 At study inclusion, CUP patients could continue at their established pitolisant hydrochloride dose (20 mg per day or 40 mg per day) without up-titration. Naive patients began pitolisant hydrochloride treatment with a 1-month individual up-titration scheme starting at 5 mg per day and increasing to up to 40 mg per day. Relevant efficacy outcomes assessed included ESS score, CGI-C, PGO of efficacy for EDS, HRQoL measures from the 5-Level EQ-5D questionnaire, and patient sleep-diary outcomes. Safety assessments consisted of reporting all AEs, including TEAEs, SAEs, and AEs of special interest, and measuring depressive symptoms with the BDI-SF-13. Patients recruited from France who had received at least 1 dose of pitolisant hydrochloride and completed the initial year of the HARMONY III trial were eligible to continue treatment in a follow-up period until the market authorization of pitolisant hydrochloride was granted in France in 2016 (up to 5 years for some patients).
Populations
A total of 102 patients with narcolepsy from 8 centres in France (n = 77) and Hungary (n = 25) were enrolled in the extension study, HARMONY III, with the first patient enrolled in June 2011. After the initial 12-month treatment period, 48 patients from France continued in the 5-year follow-up period.
Patients were required to have had an ESS score of at least 12 to enrol into the extension study. Overall, the mean age for all participants was 38.0 (SD = 14.9) years, and slightly more than half were female (55.9%). About 75% of each of the naive and CUP patients reported a history of cataplexy. Patients in the extension study could take concomitant medications for narcolepsy, including anticataplectics and/or psychostimulants. At inclusion, 35.3% of all patients were taking concomitant medications, and more CUP patients were taking concomitant medications than naive patients (56.3% versus 31.4%). At inclusion, a greater proportion of CUP patients than naive patients were taking sodium oxybate (25.0% versus 3.4%) or mazindol (12.5% versus 1.2%). More CUP patients reported a history of dyssomnia than naive patients (87.5% versus 46.5%). At inclusion, median ESS score was lower in CUP patients than naive patients (13.0 [range, 0 to 22] versus 17.0 [range, 0 to 24]). On the CGI-S scale, more naive patients than CUP patients were defined as at least markedly ill (82.4% versus 66.7%). Overall, the baseline characteristics of patients enrolled in the HARMONY III trial were generally consistent with the baseline characteristics of the patients randomized in the pivotal trials. Characteristics of the French patients who continued in the 5-year follow-up period were similar to those in the total study population. Refer to Table 34 for a summary of baseline characteristics of patients enrolled in the HARMONY III trial.
Table 34: Summary of Baseline Characteristics in the HARMONY III Extension Study (ITT Population)
Characteristic | Total patients | |
|---|---|---|
N | N = 102 | |
Age (years), median (range) | 102 | 35.9 (18.2 to 69.4) |
Males, n (%) | 102 | 57 (55.9) |
Female mode of contraception, n (%) | ||
Estrogen and progesterone | 43 | 26 (60.5) |
IUD | 43 | 8 (18.6) |
Other method | 43 | 7 (16.3) |
None | 43 | 2 (4.7) |
History of cataplexy, n (%) | 102 | 75 (73.5) |
Ongoing treatments for narcolepsy or cataplexy, n (%) | ||
Methylphenidate | 102 | 13 (12.7) |
Modafinil | 102 | 10 (9.8) |
Venlafaxine | 102 | 9 (8.8) |
Sodium oxybate | 102 | 7 (6.9) |
Mazindol | 102 | 3 (2.9) |
Citalopram | 102 | 1 (1.0) |
Fluoxetine | 102 | 1 (1.0) |
Complaint of EDS occurring almost daily for ≥ 3 months, n (%) | 102 | 102 (100.0) |
MSLT at inclusion (minutes), median (range) | 91 | 5.1 (0.1 to 13.0) |
ESS score at inclusion, median (range) | 102 | 17.0 (0 to 24) |
CGI-S of illness at inclusion, n (%) | ||
Mildly ill | 97 | 4 (4.1) |
Moderately ill | 97 | 15 (15.5) |
Markedly ill | 97 | 29 (29.9) |
Severely ill | 97 | 46 (47.4) |
Among the most extremely ill patients | 97 | 3 (3.1) |
History of cataplexy, n (%) | 102 | 75 (73.5) |
History of associated symptoms, n (%) | ||
Sleep paralysis | 102 | 50 (49.0) |
Hallucinations | 102 | 60 (58.8) |
Automatic behaviour | 102 | 46 (45.1) |
Dyssomnia | 102 | 54 (52.9) |
Sleep-diary measures at screening, median (range) | ||
Daily number of diurnal involuntary sleep attacks | 71 | 0.9 (0 to 6) |
Duration of diurnal involuntary sleep attacks, minutes | 62 | 37.3 (0 to 375) |
Daily number of episodes of severe sleepiness | 71 | 1.6 (0.0 to 7.3) |
Duration of episodes of severe sleepiness, minutes | 67 | 60.0 (3.3 to 368.6) |
Daily number of total cataplexy episodes | 71 | 0 (0.0 to 10.4) |
Daily number of partial cataplexy episodes | 71 | 0 (0.0 to 11.9) |
Duration of nocturnal awakening, hours | 66 | 0.6 (0.0 to 3.9) |
Daily number of nocturnal awakening episodes | 71 | 2.0 (0.0 to 6.4) |
Duration of nocturnal sleep, hours | 71 | 8.9 (5.3 to 11.8) |
Frequency of sleep paralysis | 71 | 0 (0.0 to 1.9) |
Frequency of hallucinations | 71 | 0 (0.0 to 1.8) |
3-Level EQ-5D VAS score at inclusion, median (range) | 96 | 70.0 (20 to 95) |
BDI-SF13 score at inclusion, median (range) | 96 | 3.0 (0 to 24) |
BDI-SF13 = 13-item Beck Depression Inventory-Short Form; CGI-S = Clinical Global Impression of Severity; EDS = excessive daytime sleepiness; ESS = Epworth Sleepiness Scale; ITT = intent-to-treat; IUD = intrauterine device; MSLT = Multiple Sleep Latency Test; VAS = visual analogue scale.
Source: Clinical Study Report for HARMONY III.16
Interventions
At study inclusion, CUP patients could continue at their established pitolisant hydrochloride dose (20 mg per day or 40 mg per day) without up-titration. At 1 month, CUP patients could have their dose adjusted to 20 mg per day or 40 mg per day. This dose remained stable for a 2-month period. Naive patients began treatment with pitolisant hydrochloride with a 1-month individual up-titration scheme, starting at 5 mg per day for 7 days, followed by 10 mg per day for 7 days (Figure 7). After 14 days, the dose could be increased to 20 mg per day if safety and tolerability were good. At day 21, the daily dose could be adjusted to 5 mg, 10 mg, or 20 mg, depending on individual risk and benefit. At 1 month, the daily dose could be adjusted again to 5 mg, 10 mg, 20 mg, or 40 mg. This dose remained stable for a 2-month period. For all patients, daily dose adjustments could occur again at the 3-month, 6-month, 9-month, and 12-month visits to 5 mg, 10 mg, 20 mg, or 40 mg. French patients who continued the treatment beyond the initial 1-year period could have their daily dose adjusted to 5 mg, 10 mg, 20 mg, or 40 mg at follow-up visits every 6 months until the end of the 5 years of follow-up, in September 2016 (Figure 8).
Figure 7: Study Design for Naive Patients in the HARMONY III Extension Study
D = day; EOS = end-of-study visit; Fup = follow-up; mth = month; t = telephone contact; V = visit.
Figure 8: Study Design for French CUP Patients in the HARMONY III Extension Study
ATU = Authorisation Temporaire d’Utilization; CUP = compassionate use program; D = day; EOS = end-of-study visit; Fup = follow-up; mth = month; t = telephone contact; V = visit.
Source: Clinical Study Report for HARMONY III.16
Outcomes
The efficacy outcomes assessed in the long term extension study were consistent with those assessed in the pivotal trials. The primary efficacy outcome was the mean change from baseline to 1 year in ESS score, used to measure daytime subjective somnolence. Secondary efficacy end points included narcolepsy symptoms recorded in patient diaries (number of cataplexy attacks, number of hallucinations, number of sleep paralysis episodes, number and duration of diurnal sleepiness and sleep episodes, number and duration of nocturnal awakenings, and duration of nocturnal sleep), the CGI-S to measure the severity of a patient's illness at baseline and the CGI-C to document a patient’s perceived change in illness from baseline, the 6-item PGO scale, the 3-Level EQ-5D questionnaire to measure a patient’s HRQoL, and measures of compliance. Safety outcomes included TEAEs, SAEs, withdrawals due to AEs, and depressive symptoms measured with the BDI-SF-13.
Statistical Analysis
All analyses were descriptive for the HARMONY III extension study. Efficacy analyses were conducted on the ITT population and safety analyses were conducted on the safety population, both of which consisted of enrolled patients who received at least 1 dose of the study medication (102 in the initial 1-year study period and 77 in the French subset through year 5). Analyses included mean change from baseline for ESS score and patient sleep-diary outcomes; within-group changes from baseline were assessed using the Wilcoxon signed rank test and Student t-test. Missing values for ESS scores were replaced using the LOCF method for the 1-year follow-up.
Patient Disposition
Patient disposition in the extension study is summarized in Table 35. Of the 102 patients enrolled, 73 were de novo patients, 16 were French CUP patients, and 13 were patients treated with pitolisant hydrochloride during previous single-blind or double-blind studies (including HARMONY 1,13 HARMONY II,18 or HARMONY 1bis14). Overall, 34 naive patients (33.3%) withdrew from the extension study during year 1, with 22.5% withdrawing voluntarily and 10.8% discontinuing due to an AE. Of the 77 patients recruited from France, the 50 who had received at least 1 dose of pitolisant hydrochloride and completed the initial year of the HARMONY III trial were eligible to continue treatment for up to 5 years. Of the 49 French patients enrolled in the follow-up period, 16 (33.3%) discontinued before year 5. Reasons for discontinuation included AEs (6.3%), lack of efficacy (6.3%), a death considered unrelated to the study drug (2.1%), or other reasons (18.8%). During the follow-up period, the number of patients remaining enrolled after 2 years was 45, after 3 years was 38, after 4 years was 34, and after 5 years was 14.
Table 35: Patient Disposition in the HARMONY III Extension Study (ITT Population)
Patient disposition | Population |
|---|---|
1-year follow-up | |
Screened, n | 106 |
Enrolled, n | 104 |
Treated, n | 102 |
Completed year 1, n (%) | 68 (66.7) |
Discontinued during year 1, n (%) | 34 (33.3) |
Reason for discontinuation, n (%) | |
Adverse event | 11 (10.8) |
Lack of efficacy | 20 (19.6) |
Other | 3 (2.9) |
5-year follow-up perioda | |
Enrolled in follow-up period, n | 49 |
Completed follow-up period, n (%) | 32 (65.3) |
Discontinued before year 5, n (%) | 16 (32.7) |
Reason for discontinuation, n (%) | |
Adverse event | 3 (6.1) |
Lack of efficacy | 3 (6.1) |
Death | 1 (2.0) |
Other | 9 (18.4) |
ITT = intent-to-treat.
aPatients recruited from France who had received at least 1 dose of pitolisant hydrochloride and completed the initial 52-week period of the HARMONY III trial were eligible to continue treatment for up to 5 years.
Sources: Clinical Study Reports for HARMONY III.16,17
Exposure to Study Treatments
In the first year of the HARMONY III trial, the median duration of treatment exposure was 358 (range, 1 to 162) days. The median duration of treatment exposure among naive patients was 351 (range, 1 to 436) days and among CUP patients was 757 (range, 408 to 1,162). Among the French subset of patients who continued into the 5-year follow-up period, the median length of treatment exposure was 1,276 (range, 1 to 2,488) days. The median length of treatment exposure for naive patients was 708 (range, 1 to 1,841) days and for CUP patients was 2,013 (range, 514 to 2,488) days. Median duration of treatment exposure for CUP patients included the period in which they received pitolisant hydrochloride through the program before enrolment in the HARMONY III extension study.
At the end of the initial 1-month titration period, 83.7% of patients were receiving 20 mg per day of pitolisant hydrochloride, including 95.1% of naive patients and 25.0% of CUP patients. By the 1-year visit, 83.9% of patients were receiving 40 mg per day, including 87.2% of naive patients and 73.3% of CUP patients. At the end of the 5-year follow-up period, most of the French subset of patients, 78.6% of all enrolled French patients, were receiving the highest dose of 40 mg per day, including 83.3% of naive patients and 75.0% of CUP patients.
Efficacy
Sleepiness, Alertness, and Severity of Daytime Sleepiness
In the HARMONY III extension study, EDS was measured by ESS score (Table 36). At year 1, the mean change from baseline for the ESS score was –3.99 (SD = 4.56). Fifty-seven (58.2%) patients were considered responders, defined as having an ESS score no higher than 10 or a change from baseline of at least 3 points. Among naive patients, the mean change from baseline was –4.30 (SD = 4.47). Forty-nine (59.8%) patients were considered responders. For CUP patients, who were already receiving pitolisant hydrochloride treatment at inclusion and had lower mean ESS scores at baseline, the mean change from baseline for the ESS score was –2.38 (SD = 4.79). Eight (50.0%) patients were considered responders.
Regarding patients taking concomitant narcolepsy treatments, the mean change from baseline was –3.15 (SD = 4.01), –3.64 (SD = 4.55), and –4.00 (SD = 2.35) for patients taking psychostimulants (n = 26), anticataplectics (n = 14), and both psychostimulants and anticataplectics (n = 13), respectively. For the 45 patients taking pitolisant hydrochloride only (i.e., no concomitant treatments), the mean change from baseline was –4.67 (SD = 5.27). Thirteen (50.0%), 8 (57.1%), and 10 (76.9%) patients taking psychostimulants, anticataplectics, and psychostimulants and anticataplectics, respectively, were considered responders. Twenty-six (57.8%) patients taking pitolisant hydrochloride only (i.e., no concomitant treatments) were considered responders.
Changes from baseline in ESS scores remained similar during the long-term follow-up period in the French cohort. Among French patients who continued the in the long term follow-up, the mean change from baseline in ESS score was –4.41 (SD = 5.38) at year 2 (n = 45), –4.45 (SD = 6.16) at year 3 (n = 38), –4.76 (SD = 5.73) at year 4 (n = 34), and –6.07 (SD = 7.19) at year 5 (n = 14). At 5 years, the mean change from baseline in ESS score was –8.17 (SD = 8.93) and –4.50 (SD = 5.71) for naive (n = 6) and CUP (n = 8) patients, respectively. Of the 14 patients remaining at 5 years, 10 (71.4%) were considered responders: 5 (83.3%) naive patients and 5 (62.5%) CUP patients.
For patients taking concomitant narcolepsy treatments, the mean change from baseline in ESS score after 5 years was –5.67 (SD = 6.11), –6.33 (SD = 7.77), and –5.50 (SD = 3.87) for patients taking psychostimulants (n = 3), anticataplectics (n = 3), and both psychostimulants and anticataplectics (n = 4), respectively. For the 4 patients taking pitolisant hydrochloride only (i.e., no concomitant treatments, the mean change from baseline was –6.75 (SD = 11.95). All patients remaining at 5 years, regardless of concomitant treatment, were considered responders.
CGI-C was used to document a patient’s perceived overall change in illness from baseline. It should be noted that this overall change in illness is multifaceted and likely includes considerations other than daytime sleepiness. Of the 67 patients who completed the initial 1-year treatment period, 71.7% reported a CGI-C score of 1 (very much improved) or 2 (much improved), 22.4% reported a score of 3 (minimally improved), and 6% reported a score of 4 (no change). About 3-quarters (73.1%) of naive patients and 66.7% of CUP patients were at least much improved, whereas 21.2% and 26.7%, respectively, were minimally improved, and 5.8% and 6.7%, respectively, reported no change. Among French patients who continued in the long term follow-up, the proportion of patients who reported a much improved or very much improved CGI-C score from baseline was 77.3% at 2 years (n = 44), 84.2% at 3 years (n = 38), 73.5% at 4 years (n = 34), and 64.3% at 5 years (n = 14). At 5 years, 83.4% of naive patients (n = 5) and 50.0% of CUP patients (n = 4) were at least much improved; 16.7% of naive patients (n = 1) and 37.5% of CUP patients (n = 3) were minimally improved, and 12.5% of CUP patients (n = 1) reported no change.
A total of 75.0% of patients (75.0% of naive and 75.1% of CUP patients) rated the effect of pitolisant hydrochloride as moderate or marked on the PGO test after 1 year of treatment. Among French patients who continued in the long term follow-up, the proportion of patients who reported a moderate or marked effect of pitolisant hydrochloride on the PGO test was 72.8% at 2 years (n = 44), 84.2% at 3 years (n = 38), 84.4% at 4 years (n = 32), and 64.3% at 5 years (n = 14). At 5 years, 83.4% of naive and 50.0% of CUP patients rated the effect of pitolisant hydrochloride as moderate or marked.
Frequency and Severity of Cataplexy Attacks
At the end of the initial 1-year study period, the mean change in total cataplexy from baseline for the 44 patients who completed the sleep diary was –0.25 (SD = 1.37); for naive patients, mean change was –0.25 (SD = 1.38), and for CUP patients it was 0.00 (SD = NA). The mean change in partial cataplexy from baseline was –0.49 (SD = 1.94) for all patients, –0.49 (SD = 1.96) for naive patients, and 0.53 (SD = NA) for CUP patients.
Health-Related Quality of Life
The mean EQ-5D VAS score for all patients was 65.5 (SD = 16.1) at baseline and 72.4 (SD = 16.2) at 1 year, with a mean change of 6.8 (SD = 15.4) from baseline. For naive patients, the mean EQ-5D VAS score was 64.3 (SD = 15.9) at baseline and 73.5 (SD = 17.5) at 1 year, with a mean change of 9.2 (SD = 15.4) from baseline. For CUP patients, the EQ-5D VAS score was 69.6 (SD = 16.7) at baseline and 68.8 (SD = 11.4) at 1 year, with a mean change of –0.8 (SD = 12.7) from baseline.
Among French patients who continued in the long term follow-up, the mean EQ-5D VAS score was 70.5 (SD = 15.9) at 2 years (n = 44), 69.5 (SD = 13.2) at 3 years (n = 38), 72.2 (SD = 13.3) at 4 years (n = 33), and 75.0 (SD = 12.2) at 5 years (n = 14). At 5 years, the EQ-5D VAS score was 80.5 (SD = 12.5) for naive patients and 70.9 (SD = 10.9) for CUP patients, with a change of 13.8 (SD = 15.5) and 2.4 (SD = 12.5) from baseline, respectively.
Functional Activity
Functional activity was not measured or reported in the HARMONY III trial.
Sexual Function
Sexual function was not measured or reported in the HARMONY III trial.
Sleep Attacks
At the end of the initial 1-year study period, among the 44 patients who completed the sleep diary, the mean change in the daily number of sleep attacks from baseline was –0.37 (SD = 1.41) for all patients, –0.39 (SD = 1.42) for naive patients, and 0.47 (SD = NA) for CUP patients. The mean change in the duration of diurnal involuntary sleep attacks from baseline was –0.37 (SD = 1.41) minutes for all patients, –0.39 (SD = 1.42) minutes for naive patients, and 0.47 (SD = NA) minutes for CUP patients.
Nocturnal Sleep Properties
Among the 44 patients who completed the sleep diary, the mean change in daily number of nocturnal awakenings from baseline to the 1-year visit was –0.42 (SD = 1.18) for all patients, –0.42 (SD = 1.19) for naive patients, and –0.14 (SD = NA) for CUP patients. The mean change in the duration of nocturnal awakening from baseline to the 1-year visit was –0.09 (SD = 0.73) hours for all patients, –0.10 (SD = 0.73) hours for naive patients, and 0.18 (SD = NA) hours for CUP patients. The mean change in the duration of nocturnal sleep from baseline to the 1-year visit was –0.10 (SD = 1.19) hours for all patients, –0.09 (SD = 1.21) hours for naive patients, and –0.37 (SD = NA) hours for CUP patients.
Number of Hallucinations
At the end of the initial 1-year study period, among the 44 patients who completed the sleep diary, the mean change in the frequency of hallucinations from baseline was –0.06 (SD = 0.25) for all patients, –0.06 (SD = 0.20) for naive patients, and 0.0 (SD = NA) for CUP patients.
Concomitant Medication Use
The proportion of patients taking a concomitant treatment for narcolepsy or cataplexy changed from 35.3% at baseline to 52.9% over the course of the initial year. A total of 31.4% of naive and 56.3% of CUP patients were taking concomitant medications at baseline and 51.2% of naive and 62.5% of CUP patients were taking concomitant medications during the initial year. The most frequent treatments over the course of the study were methylphenidate (22.5%), modafinil (17.6%), and venlafaxine (13.7%). Eleven patients (10.8%) took sodium oxybate. In the French subset, the proportion of patients taking allowed concomitant treatment for narcolepsy or cataplexy in addition to pitolisant hydrochloride changed from 44.2% at baseline to 70.1% over the 5-year period. A total of 70.5% of naive and 68.8% of CUP patients were taking concomitant treatments over the 5-year period. The most frequent treatments were methylphenidate (31.2%), modafinil (29.9%), venlafaxine (19.5%), and sodium oxybate (16.9%).
Patient Satisfaction and Ease of Use
Patient satisfaction and ease of use were not measured or reported in the HARMONY III trial.
Adherence
Adherence was not measured or reported in the HARMONY III trial.
Health Care Resource Use
Health care resource use was not measured or reported in the HARMONY III trial.
Table 36: Efficacy Outcomes in the HARMONY III Extension Study in Year 1 and Year 5 (ITT Population)
Efficacy outcomes | N | Baseline, mean (SD) | N | End of treatment, mean (SD) | Change from baseline | P valuea |
|---|---|---|---|---|---|---|
ESSb | ||||||
Year 1 | 98 | 16.8 (3.3) | 98 | 12.8 (5.0) | –4.0 (4.6) | NR |
Year 5 | 14 | 16.7 (4.4) | 14 | 10.6 (4.6) | –6.1 (7.2) | NR |
Daily number of total cataplexy episodes | ||||||
Year 1 | 43 | 0.3 (1.6) | 43 | 0.1 (0.3) | –0.3 (1.4) | 0.051c |
Year 5 | 2 | 0.0 (0.0) | 2 | 0.0 (0.0) | 0.0 (0.0) | NA |
Daily number of partial cataplexy episodes | ||||||
Year 1 | 43 | 0.8 (2.0) | 43 | 0.3 (0.5) | –0.5 (1.9) | 0.083c |
Year 5 | 2 | 0.2 (0.3) | 2 | 0.0 (0.0) | –0.2 (0.3) | NA |
Daily number of diurnal involuntary sleep attacks | ||||||
Year 1 | 44 | 1.4 (1.4) | 44 | 1.0 (0.9) | –0.4 (1.4) | 0.100c |
Year 5 | NR | NR | NR | NR | NR | NR |
Duration of diurnal involuntary sleep attacks, minutes | ||||||
Year 1 | 38 | 59.9 (67.8) | 38 | 39.5 (56.1) | –20.3 (75.5) | 0.056c |
Year 5 | NR | NR | NR | NR | NR | NR |
Daily number of episodes of severe sleepiness | ||||||
Year 1 | 44 | 2.0 (1.6) | 44 | 1.6 (1.5) | –0.5 (1.7) | 0.027c |
Year 5 | NR | NR | NR | NR | NR | NR |
Duration of episodes of severe sleepiness, minutes | ||||||
Year 1 | 39 | 87.1 (76.5) | 39 | 52.8 (49.2) | –34.3 (72.9) | < 0.001c |
Year 5 | NR | NR | NR | NR | NR | NR |
Daily number of nocturnal awakenings | ||||||
Year 1 | 44 | 2.0 (1.6) | 44 | 1.6 (1.6) | –0.4 (1.2) | < 0.001c |
Year 5 | 1 | 0.3 (NA) | 1 | 1.4 (NA) | 1.1 (NA) | NA |
Duration of nocturnal awakenings, hours | ||||||
Year 1 | 40 | 0.8 (0.9) | 40 | 0.7 (0.8) | –0.1 (0.7) | 0.416d |
Year 5 | 2 | 1.09 (0.42) | 2 | 0.73 (0.11) | –0.4 (0.3) | NR |
Duration of nocturnal sleep, hours | ||||||
Year 1 | 44 | 8.9 (1.5) | 44 | 8.8 (1.7) | –0.1 (1.2) | 0.597d |
Year 5 | 2 | 8.0 (0.1) | 2 | 7.4 (2.2) | –0.6 (2.1) | NR |
Frequency of sleep paralysis | ||||||
Year 1 | 44 | 0.2 (0.4) | 44 | 0.1 (0.2) | –0.1 (0.3) | 0.023c |
Year 5 | 2 | 0.0 (0.0) | 2 | 0.0 (0.0) | 0.0 (0.0) | NR |
Frequency of hallucinations | ||||||
Year 1 | 44 | 0.1 (0.4) | 44 | 0.1 (0.2) | −0.1 (0.3) | 0.182c |
Year 5 | 2 | 0.0 (0.0) | 2 | 0.0 (0.0) | 0.0 (0.0) | NR |
3-Level EQ-5D VAS score | ||||||
Year 1 | 68 | 65.5 (16.1) | 68 | 72.4 (16.3) | 6.8 (15.4) | 0.4760e |
Year 5 | 14 | 67.7 (16.8) | 14 | 75.0 (12.2) | 7.3 (14.5) | NR |
ESS = Epworth Sleepiness Scale; ITT = intention-to-treat; NA = not applicable; NR = not reported; SD = standard deviation; VAS = visual analogue scale.
aP values have not been adjusted for multiple testing (i.e., the type I error rate has not been controlled).
bBased on the LOCF method.
cP value from Wilcoxon signed rank test.
dP value from Student t-test.
eP value from mixed model.
Sources: Clinical Study Reports for HARMONY III.16,17
Table 37: Responders, CGI-C, and PGO Outcomes in the HARMONY III Extension Study in Year 1 and Through Year 5 (ITT Population)
Characteristic | n | Patients in year 1 N = 102 | N | French subset of patientsa through year 5 N = 77 |
|---|---|---|---|---|
Responders, ESS ≥ 3 or ESS ≤ 10, n (%) | 73 | 38 (52.1) | 14 | 10 (71.4) |
CGI-C of illness, n (%) | ||||
Very much improved | 49 | 11 (22.4) | 14 | 2 (14.3) |
Much improved | 49 | 25 (51.0) | 14 | 7 (50.0) |
Minimally improved | 49 | 10 (20.4) | 14 | 4 (28.6) |
No change | 49 | 3 (6.1) | 14 | 1 (7.1) |
PGO, n (%) | ||||
No change | 68 | 5 (7.4) | 14 | 1 (7.1) |
Minimal effect (slight decrease in EDS) | 68 | 12 (17.6) | 14 | 4 (28.6) |
Moderate effect (partial remission of EDS) | 68 | 34 (50.0) | 14 | 6 (42.9) |
Marked effect (complete or nearly complete remission of EDS) | 68 | 17 (25.0) | 14 | 3 (21.4) |
CGI-C = Clinical Global Impression of Change; EDS = excessive daytime sleepiness; ESS = Epworth Sleepiness Scale; ITT = intention-to-treat; PGO = patient global opinion.
Sources: Clinical Study Reports for HARMONY III.16,17
Harms
All combinations of concomitant medications for narcolepsy or cataplexy were well tolerated, except there was a higher frequency of insomnia in the subgroup of patients taking concomitant modafinil (55%, n = 5) in the follow-up period among the French subset of patients.
The summary of TEAEs at the interim analysis are presented in Table 38. During the initial 1-year treatment period, 58 patients (56.9%) reported 168 TEAEs, the most common being headache (11.8%), insomnia (8.8%), weight gain (7.8%), anxiety (6.9%), depression (4.9%), and nausea (4.9%). In the French subset of patients over the 5-year period, 72.7% of patients reported 296 TEAEs, the most common being headache (19.5%), weight increase (18.2%), nausea (11.7%), anxiety (11.7%), insomnia (11.7%), and depression (11.7%).
A total of 16 patients reported SAEs in the 5-year period among the French subset of patients, with the most common being depression (3.9%) and pregnancy (3.9%). All SAEs were considered unrelated to the study drug, except for 1 spontaneous abortion in a patient who discontinued the study drug and permanently withdrew from the trial. One death was reported in follow-up period . The clinical study report indicated that the death was determined to be not related to the study medication.
Among all patients, the mean BDI-SF-13 score was 4.1 (SD = 3.5) at baseline and 3.8 (SD = 4.1) at the 1-year visit. The mean BDI-SF-13 score among the French subset of 12 patients at the year-5 visit was 2.4 (SD = 2.8) . At each time point, no more than 1 patient had a severe depression.
Table 38: Summary of TEAEs in the HARMONY III Extension Study (Safety Population)
Adverse event | Patients in year 1 N = 102 | French subset of patientsa through year 5 N = 77 |
|---|---|---|
Any TEAE, n (%) | 58 (56.9) | 56 (72.7) |
Common TEAEs,b n (%) | ||
Insomnia | 9 (8.8) | 9 (11.7) |
Anxiety | 7 (6.9) | 9 (11.7) |
Depression | 5 (4.9) | 9 (11.7) |
Irritability | 4 (3.9) | 4 (5.2) |
Headache | 12 (11.8) | 15 (19.5) |
Nausea | 5 (4.9) | 9 (11.7) |
Abdominal pain | 2 (2.0) | 5 (6.5) |
Vomiting | 4 (3.9) | 5 (6.5) |
Weight increased | 8 (7.8) | 14 (18.2) |
Weight decreased | 3 (2.9) | 4 (5.2) |
Increased level of coagulation factor V | 0 | 4 (5.2) |
Patients with ≥ 1 severe TEAE, n (%) | 15 (14.7) | 23 (29.9) |
Patients with drug-related TEAE, n (%) | 43 (42.2) | 48 (62.3) |
Patients with ≥ 1 SAE, n (%) | 7 (6.9) | 16 (20.8) |
Common SAEs,c n (%) | ||
Depression | 2 (2.0) | 3 (3.9) |
Pregnancy | 2 (2.0) | 3 (3.9) |
Patients with WDAEs, n (%) | 19 (18.6) | 26 (33.8) |
Deaths | 0 | 1 (1.3) |
SAE = serious adverse event; TEAE = treatment-emergent adverse event; WDAE = withdrawal due to adverse event.
aFrench patients who completed the first year of the study are counted in both columns.
bObserved in ≥ 5% of patients.
cObserved in ≥ 2 patients.
Sources: Clinical Study Reports for HARMONY III.16,17
Critical Appraisal
Internal Validity
The long-term extension study allowed for the investigation of long-term efficacy and harms of pitolisant hydrochloride for up to 5 years. Limitations of the extension study include the absence of an active comparator, which limits causal conclusions. An additional limitation is the open-label study design, and the unblinding of the study drug during the follow-up period can bias the reporting of end points. There was no sample-size calculation or statistical testing for changes from baseline, making it difficult to detect a clinically relevant treatment effect. All the end points in the HARMONY III trial were subjective, so it is possible that efficacy outcomes and known harms have been overestimated. Findings are at a high risk of confounding because of the use of concomitant treatments and a lack of control for confounding variables. None of the P values were adjusted for multiplicity and should be considered hypothesis-generating.
Subgroup analyses were descriptive and often limited to few patients, reducing the chance of detecting a true effect. Interpretation of these patient-reported outcomes is also limited by the large amount of missing data due to attrition. More than one-third of patients discontinued the extension study in the first year, mainly because of AEs or a lack of perceived efficacy. This attrition could have resulted in a population of patients that was more tolerant of pitolisant hydrochloride, as those not responding to treatment may have been less likely to continue participation in the extension study. Having patients more tolerant of pitolisant hydrochloride can also lead to biased estimates of efficacy and AEs, potentially resulting in greater efficacy and fewer AEs being reported. The use of concomitant psychostimulant and/or anticataplectic drugs by patients during the extension study may have increased the risk of additional side effects not attributable to pitolisant hydrochloride alone. Furthermore, for the 1-year time point, for the primary efficacy outcome of ESS, LOCF was used for those without final values, which may have biased the efficacy results, as these values may not be reflective of the true trajectory of this outcomes.
External Validity
With respect to external validity, although no patients from Canada were enrolled in the extension study, the characteristics of the patients enrolled in the trials were representative of patients with narcolepsy in Canada, according to the clinical experts consulted. Doses of pitolisant hydrochloride administered were in line with what would be expected in clinical practice.
Discussion
Summary of Available Evidence
Three double-blind, phase III, placebo-controlled, RCTs met the inclusion criteria for the Systematic Review. In all 3 trials, patients were included if they had narcolepsy with cataplexy. The HARMONY 1 and HARMONY 1bis trials also included patients without cataplexy (type 2 narcolepsy). The HARMONY 1 and HARMONY 1bis trials required patients to have an ESS score of at least 14 during the baseline period, whereas the HARMONY CTP trial required an ESS score of at least 12. The HARMONY CTP trial included patients with at least 3 cataplexy attacks weekly. In all trials, patients with severe cataplexy were permitted stable doses of anticataplectic medications (except TCAs) that were administrated for at least 1 month before the trial.
The HARMONY 1 and HARMONY 1bis trials were 8-week trials with dose-adjusting and withdrawal phases that assessed the superiority of pitolisant hydrochloride to placebo with regard to EDS in patients with narcolepsy. An additional efficacy objective in both trials was to test the noninferiority of pitolisant hydrochloride to modafinil. The HARMONY CTP trial was a 7-week randomized, double-blind, placebo-controlled study comparing pitolisant hydrochloride to placebo. It focused on the safety of pitolisant hydrochloride and its efficacy in decreasing the frequency of cataplexy attacks in patients who had narcolepsy with cataplexy. The maximum dosages for pitolisant hydrochloride were 20 mg daily in the HARMONY 1bis trial and 40 mg in the HARMONY 1 and HARMONY CTP trials. Titration of the study drug was at the discretion of the study investigators, which could have affected efficacy and potentially threaten blinding to treatment.
An additional trial was summarized in this report as it provided longer-term evidence. HARMONY III is a long-term, open-label, uncontrolled, extension study conducted to evaluate the efficacy and safety of pitolisant hydrochloride at daily doses of 5 mg, 10 mg, 20 mg, or 40 mg for the treatment of EDS in patients with narcolepsy with or without cataplexy for up to 5 years. Of the 102 patients enrolled the HARMONY III trial, 86 were naive patients who were not receiving pitolisant hydrochloride at the time of study enrolment and 16 were treated with pitolisant hydrochloride in the 2 weeks preceding the study. Of the naive patients, 73 had never been treated with pitolisant hydrochloride and 13 patients were previously treated with pitolisant hydrochloride during single-blind or double-blind trials, including HARMONY 1,13 HARMONY II,18 and HARMONY 1bis.14 At study inclusion, patients could continue at their established pitolisant hydrochloride dose (20 mg per day or 40 mg per day) without up-titration for up to 5 years. Naive patients began pitolisant hydrochloride treatment with a 1-month individual up-titration scheme, starting at 5 mg per day and increasing up to 40 mg per day.
Interpretation of Results
Efficacy
In the HARMONY 1 and HARMONY 1bis trials, pitolisant hydrochloride resulted in a statistically significant and clinically meaningful reduction in ESS score compared with placebo in the ITT population. There was a statistically significant increase in the proportion of EDS score responders with pitolisant hydrochloride compared with placebo. Consistent with these findings, the secondary end point in the HARMONY CTP trial demonstrated an improvement in EDS with pitolisant hydrochloride compared with placebo; however, the P value for this secondary outcome was not adjusted for multiplicity, so there is an increased risk of a false-positive conclusions.
In the HARMONY 1 trial, the mean change in ESS from baseline was compared between the pitolisant hydrochloride and the modafinil treatment arms, but failed to demonstrate noninferiority. In the HARMONY 1bis trial, the test to determine noninferiority between pitolisant hydrochloride and modafinil also failed because of the high upper bound of the 95% CI. Overall, neither pivotal trial was able to demonstrate the noninferiority of pitolisant hydrochloride to modafinil in terms of EDS. Because no other comparisons with an active drug were performed, the comparative efficacy and safety of pitolisant hydrochloride remains unknown. It is worth noting that the clinical experts considered modafinil to be a drug of modest effectiveness for patients with narcolepsy, with other psychostimulants, such as methylphenidate, performing better. Based on this observation and the trial results, it is unknown whether pitolisant hydrochloride would show efficacy equal to or better than other drugs for the treatment of EDS in narcolepsy.
The primary end point in the HARMONY CTP trial was the reduction in WCRs for pitolisant hydrochloride compared to placebo. There was a statistically significant decrease in the WCR episodes at the end of treatment. The effect of pitolisant hydrochloride on the WCR was consistent for each subgroup of patients receiving 20 mg or 40 mg as a stable dose. The WCR was analyzed by studying the interaction effect of other permitted anticataplectic medication. A total of 12 patients (8 in the placebo group and 4 in the pitolisant hydrochloride group) received concomitant anticataplectic treatment during the study. The incremental anticataplectic effect of pitolisant hydrochloride compared with placebo was higher when used as monotherapy, but remained positive when used with other permitted anticataplectic drugs, although definitive conclusions cannot be made with such small patient numbers and without statistical testing.
Consistent with these results, in the HARMONY 1 trial, the daily rates of complete and partial cataplexy episodes, a secondary analysis, were lower with pitolisant hydrochloride than with placebo. In contrast, results from the HARMONY 1bis trial showed that the mean difference in the daily cataplexy rate for patients with cataplexy between the final 7 days of treatment and baseline was not different between the pitolisant hydrochloride and placebo groups. There was no difference in either trial between the pitolisant hydrochloride and modafinil groups. According to the experts consulted for this review, modafinil does not show efficacy against cataplexy. Overall, the inconsistent results, reduced number of patients with cataplexy in the trials, lack of formal statistical testing, and lack of adjustment for multiplicity preclude the drawing of definitive conclusions about cataplexy in the HARMONY 1 and HARMONY 1bis trials.
The unvalidated CGI-C tool appeared to demonstrate an improvement in EDS in the pitolisant hydrochloride and modafinil groups in the HARMONY 1 and HARMONY 1bis trials, and in the pitolisant hydrochloride group in the HARMONY CTP trial. In the HARMONY 1 trial, the subgroup of patients with a history of cataplexy appeared to have improved CGI-C scores for EDS, as well as an apparent improvement, in the modafinil arm. For all CGI-C outcomes and subgroup analyses, the lack of statistical testing between groups does not allow conclusions to be made.
Pitolisant hydrochloride led to better MWT values than placebo across all trials, and there was no noticeable difference compared with modafinil. In the HARMONY 1 trial, there was no difference between pitolisant hydrochloride and placebo or modafinil for SART, whereas in the HARMONY 1bis trial, the ratio of mean change for SART between pitolisant hydrochloride and placebo favoured the former. There was no noticeable difference in SART between pitolisant hydrochloride and modafinil in the HARMONY 1bis trial. The 5-Level EQ-5D values appeared similar in all 3 groups across trials. For CGI-C, MWT, HRQoL, PGO, and SART secondary outcomes, formal statistical testing was not performed because of the absence of adjustment for multiplicity, so results were considered descriptive only. Treatment adherence was high; the majority of patients took between 80% and 120% of their recommended daily dose.
Harms
In the HARMONY 1 trial, AEs after the initiation of treatment were reported by 66.7% of patients in the placebo group, 64.5% in the pitolisant hydrochloride group, and 69.7% in the modafinil arm. In the HARMONY 1bis trial, approximately ||||||| of the patients in the pitolisant hydrochloride and modafinil groups reported AEs, as did ||||||| of patients in the placebo group. In the HARMONY CTP trial, approximately 31% of placebo patients and 35% pitolisant hydrochloride patients experienced an AE. There were few SAEs across the trials. For HARMONY 1 ||||||||||||||, there was a greater percentage of nervous system disorders in the pitolisant hydrochloride arm, but for the HARMONY CTP trial, the placebo arm had more patients with nervous system disorders . No deaths were reported across the trials. The clinical experts indicated that the proportion of patients who withdrew from any of the trials and the number of AEs seemed low for the patient population of interest. There were no concerning results for BDI-SF-13 scores or withdrawals across the trials.
Conclusions
Pitolisant hydrochloride has shown benefits over placebo in the treatment of EDS and cataplexy in patients with narcolepsy. However, because of limited comparisons and the failure of noninferiority testing, it is not possible to conclude that pitolisant hydrochloride is similar or noninferior to modafinil or other active drugs used to treat EDS or cataplexy. In addition, the interaction between pitolisant hydrochloride and concomitant medications for narcolepsy is unclear. Pitolisant hydrochloride appeared well tolerated, and no major safety concern was identified. In alignment with clinical expert input, pitolisant hydrochloride may provide an additional option for the treatment of narcolepsy because of its superiority to placebo. However, its place in therapy relative to other medications will be a challenge to elucidate based on the trial results.
References
1.Ito E, Inoue Y. [The International Classification of Sleep Disorders, third edition. American Academy of Sleep Medicine. Includes bibliographies and index]. Nihon Rinsho. 2015;73(6):916-923. PubMed
2.Hublin C, Partinen M, Kaprio J, Koskenvuo M, Guilleminault C. Epidemiology of narcolepsy. Sleep. 1994;17(8 Suppl):S7-12. PubMed
3.Golden EC, Lipford MC. Narcolepsy: diagnosis and management. Cleve Clin J Med. 2018;85(12):959-969. PubMed
4.Slowik JM, Collen JF, Yow AG. Narcolepsy. StatPearls. Island (FL): StatPearls Publishing; 2022.
5.Scheer D, Schwartz SW, Parr M, Zgibor J, Sanchez-Anguiano A, Rajaram L. Prevalence and incidence of narcolepsy in a US health care claims database, 2008-2010. Sleep. 2019;42(7). PubMed
6.Spruyt K. Narcolepsy presentation in diverse populations: an update. Curr Sleep Med Rep. 2020:1-12. PubMed
7.Quaedackers L, Pillen S, Overeem S. Recognizing the symptom spectrum of narcolepsy to improve timely diagnosis: a narrative review. Nat Sci Sleep. 2021;13:1083-1096. PubMed
8.Jennum P, Ibsen R, Petersen ER, Knudsen S, Kjellberg J. Health, social, and economic consequences of narcolepsy: a controlled national study evaluating the societal effect on patients and their partners. Sleep Med. 2012;13(8):1086-1093. PubMed
9.Scammell TE, Jackson AC, Franks NP, Wisden W, Dauvilliers Y. Histamine: neural circuits and new medications. Sleep. 2019;42(1). PubMed
10.Haas HL, Sergeeva OA, Selbach O. Histamine in the nervous system. Physiol Rev. 2008;88(3):1183-1241. PubMed
11.Williams RH, Chee MJ, Kroeger D, et al. Optogenetic-mediated release of histamine reveals distal and autoregulatory mechanisms for controlling arousal. J Neurosci. 2014;34(17):6023-6029. PubMed
12.Maski K, Trotti LM, Kotagal S, et al. Treatment of central disorders of hypersomnolence: an American Academy of Sleep Medicine clinical practice guideline. J Clin Sleep Med. 2021;17(9):1881-1893. PubMed
13.Clinical Study Report: P07-03 [HARMONY 1]. Prospective, randomized double-blind study, placebo-controlled, parallel-group, multi-center trial assessing the effects of BF2.649 in treatment of excessive daytime sleepiness in narcolepsy (HARMONY I) [internal sponsor's report]. Paris (FR): Bioprojet; 2018 Oct.
14.Clinical Study Report: P09-15 [HARMONY 1 BIS]. Randomized, double-blind, placebo and comparator-controlled, parallel-group, multicenter trial assessing the effects of BF2.649 in the treatment of excessive daytime sleepiness in narcolepsy (“HARMONY 1 BIS”) [internal sponsor's report]. Paris (FR): Bioprojet; 2018 Oct.
15.Clinical Study Report: P11-05 [HARMONY CTP]. A randomized, double blind study comparing BF2.649 (pitolisant) to placeboin two parallel groups on the weekly frequency of cataplexy attacks and excessive daytime sleepiness in narcoleptic patients with cataplexy (“HARMONY CTP”) [internal sponsor's report]. Paris (FR): Bioprojet; 2018 Oct.
16.Clinical Study Report: P09-10 [HARMONY III]. An open-label naturalistic pragmatic study to assess the long-term safety of pitolisant (BF2.649) in the treatment of excessive daytime sleepiness (EDS) in narcolepsy (“HARMONY III”). Part I: 12-month analysis [internal sponsor's report]. Paris (FR): Bioprojet; 2018 May 30.
17.Clinical Study Report: P09-10 [HARMONY III ADDENDUM]. An open-label naturalistic pragmatic study to assess the long-term safety of BF2.649 (pitolisant) in the treatment of excessive daytime sleepiness (EDS) in narcolepsy (“HARMONY III”). Part II: extension of the study up to 5 years [internal sponsor's report]. Paris (FR): Bioprojet; May 2018.
18.Clinical Study Report: P07-07 [HARMONY II]. Prospective, randomized, double-blind study, parallel-group, multi-center trial assessing the effects of escalating doses of BF2.649 and BF2.649 add on modafinil on cataplexy in patients with narcolepsy (Harmony II) [internal sponsor's report]. Paris (FR): Bioprojet; 2011 Oct 4.
19.Thorpy MJ, Shapiro C, Mayer G, et al. A randomized study of solriamfetol for excessive sleepiness in narcolepsy. Ann Neurol. 2019;85(3):359-370. PubMed
20.Sateia MJ. International classification of sleep disorders-third edition: highlights and modifications. Chest. 2014;146(5):1387-1394. PubMed
21.Diagnostic and statistical manual of mental disorders: DSM-5. 5th ed. Washington (DC): American Psychiatric Publishing; 2013: https://franklin.library.upenn.edu/catalog/FRANKLIN_9959555493503681. Accessed 2022 May 25.
22.Lee EK, Kazaglis L. Hypersomnolence disorders in DSM-5: a review for clinicians. Psychiatr Ann. 2015;45(1):25-29.
23.Ruoff C, Rye D. The ICSD-3 and DSM-5 guidelines for diagnosing narcolepsy: clinical relevance and practicality. Curr Med Res Opin. 2016;32(10):1611-1622. PubMed
24.Vignatelli L, D'Alessandro R, Candelise L. Antidepressant drugs for narcolepsy. Cochrane Database Syst Rev. 2008;2008(1):CD003724. PubMed
25.Scammell TE. Narcolepsy. N Engl J Med. 2015;373(27):2654-2662. PubMed
26.Wakix (pitolisant hydrochloride): 5 mg and 20 mg tablets [product monograph]. St-Laurent (QC): Paladin Labs Inc; 2021 Aug 9.
27.Drug Reimbursement Review sponsor submission: Wakix (pitolisant hydrochloride), 5 and 20 mg oral tablets [internal sponsor's package]. St-Laurent (QC): Paladin Labs Inc; 2022 Jan 17.
28.Mar-modafinil (modafinil): 100 mg tablets, USP [product monograph]. Ottawa (ON): Marcan Pharmaceuticals Inc; 2019 Oct 3: https://pdf.hres.ca/dpd_pm/00053403.PDF. Accessed 2022 May 25.
29.Turner M. The treatment of narcolepsy with amphetamine-based stimulant medications: a call for better understanding. J Clin Sleep Med. 2019;15(5):803-805. PubMed
30.Mignot EJ. A practical guide to the therapy of narcolepsy and hypersomnia syndromes. Neurotherapeutics. 2012;9(4):739-752. PubMed
31.Raggi A, Plazzi G, Ferri R. Health-related quality of life in patients with narcolepsy: a review of the literature. J Nerv Ment Dis. 2019;207(2):84-99. PubMed
32.Baumann CR, Mignot E, Lammers GJ, et al. Challenges in diagnosing narcolepsy without cataplexy: a consensus statement. Sleep. 2014;37(6):1035-1042. PubMed
33.Kryger MH, Walld R, Manfreda J. Diagnoses received by narcolepsy patients in the year prior to diagnosis by a sleep specialist. Sleep. 2002;25(1):36-41. PubMed
34.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. J Clin Epidemiol. 2016;75:40-46. PubMed
35.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2022 Jan 27.
36.Dauvilliers Y, Bassetti C, Lammers GJ, et al. Pitolisant versus placebo or modafinil in patients with narcolepsy: a double-blind, randomised trial. Lancet Neurol. 2013;12(11):1068-1075. PubMed
37.Szakacs Z, Dauvilliers Y, Mikhaylov V, et al. Safety and efficacy of pitolisant on cataplexy in patients with narcolepsy: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017;16(3):200-207. PubMed
38.Patel S, Kon SSC, Nolan CM, et al. The Epworth Sleepiness Scale: minimum clinically important difference in obstructive sleep apnea. Am J Respir Crit Care Med. 2018;197(7):961-963. PubMed
39.Lee JL, Chung Y, Waters E, Vedam H. The Epworth sleepiness scale: reliably unreliable in a sleep clinic population. J Sleep Res. 2020;29(5):e13019. PubMed
40.van der Heide A, van Schie MK, Lammers GJ, et al. Comparing treatment effect measurements in narcolepsy: the sustained attention to response task, epworth sleepiness scale and maintenance of wakefulness test. Sleep. 2015;38(7):1051-1058. PubMed
41.EuroQol Research Foundation. EQ-5D-5L user guide: basic information on how to use the EQ-5D-5L instrument. 2019: https://euroqol.org/wp-content/uploads/2021/01/EQ-5D-5LUserguide-08-0421.pdf. Accessed 2022 Mar 10.
42.Beck AT, Beck RW. Screening depressed patients in family practice: a rapid technic. Postgrad Med. 1972;52(6):81-85. PubMed
43.Johns MW. A new method for measuring daytime sleepiness: the Epworth Sleepiness Scale. Sleep. 1991;14(6):540-545. PubMed
44.Kendzerska TB, Smith PM, Brignardello-Petersen R, Leung RS, Tomlinson GA. Evaluation of the measurement properties of the Epworth Sleepiness Scale: a systematic review. Sleep Med Rev. 2014;18(4):321-331. PubMed
45.Johns MW. Sleepiness in different situations measured by the Epworth Sleepiness Scale. Sleep. 1994;17(8):703-710. PubMed
46.Johns MW. Reliability and factor analysis of the Epworth Sleepiness Scale. Sleep. 1992;15(4):376-381. PubMed
47.Crook S, Sievi NA, Bloch KE, et al. Minimum important difference of the Epworth Sleepiness Scale in obstructive sleep apnoea: estimation from three randomised controlled trials. Thorax. 2019;74(4):390-396. PubMed
48.Peebles D, Bothell D. Modelling performance in the sustained attention to response task. In: Lovett M, Schunn C, Lebiere C, Munro P, eds. Proceedings of the Sixth International Conference on Cognitive Modeling. Pittsburgh (PA): Lawrence Elbaum Associates Publishers; 2004: http://act-r.psy.cmu.edu/wordpress/wp-content/uploads/2012/12/542Pebbles.pdf. Accessed 2022 May 25.
49.Robertson IH, Manly T, Andrade J, Baddeley BT, Yiend J. Oops!': performance correlates of everyday attentional failures in traumatic brain injured and normal subjects. Neuropsychologia. 1997;35(6):747-758. PubMed
50.Doghramji K, Mitler MM, Sangal RB, et al. A normative study of the maintenance of wakefulness test (MWT). Electroencephalogr Clin Neurophysiol. 1997;103(5):554-562. PubMed
51.Busner J, Targum SD. The clinical global impressions scale: applying a research tool in clinical practice. Psychiatry (Edgmont). 2007;4(7):28. PubMed
52.EuroQol Research Foundation. EQ-5D-3L user guide. Basic information on how to use the EQ-3L instrument. 2018: https://euroqol.org/wp-content/uploads/2021/01/EQ-5D-3LUserguide-14-0421.pdf. Accessed 2022 Mar 10.
53.The Epworth Sleepiness Scale. About the ESS. 2022: https://epworthsleepinessscale.com/about-the-ess/. Accessed 2022 Mar 15.
54.Rosenberg R, Babson K, Menno D, et al. Test-retest reliability of the Epworth Sleepiness Scale in clinical trial settings. J Sleep Res. 2021:e13476. PubMed
55.Schweitzer PK, Rosenberg R, Zammit GK, et al. Solriamfetol for excessive sleepiness in obstructive sleep apnea (TONES 3). A randomized controlled trial. Am J Respir Crit Care Med. 2019;199(11):1421-1431. PubMed
56.Rozgonyi R, Dombi I, Janszky J, Kovacs N, Faludi B. Low test-retest reliability of the Epworth Sleepiness Scale within a substantial short time frame. J Sleep Res. 2021;30(4):e13277. PubMed
57.Grewe FA, Roeder M, Bradicich M, et al. Low repeatability of Epworth Sleepiness Scale after short intervals in a sleep clinic population. J Clin Sleep Med. 2020;16(5):757-764. PubMed
58.Mitler MM, Gujavarty KS, Browman CP. Maintenance of wakefulness test: a polysomnographic technique for evaluation treatment efficacy in patients with excessive somnolence. Electroencephalogr Clin Neurophysiol. 1982;53(6):658-661. PubMed
59.Brooks R. EuroQol: the current state of play. Health Policy. 1996;37(1):53-72. PubMed
60.EuroQol Group. EuroQol--a new facility for the measurement of health-related quality of life. Health Policy. 1990;16(3):199-208. PubMed
Appendix 1: Literature Search Strategy
Note that this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases:
MEDLINE All (1946 to present)
Embase (1974 to present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: February 4, 2022
Alerts: Weekly search updates until project completion
Search filters applied: No filters were applied to limit the retrieval by study type
Limits:
Publication date limit: none
Humans
Language limit: none
Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
MeSH | Medical Subject Heading |
exp | Explode a subject heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Keyword heading word |
.dq | Candidate term word (Embase) |
.pt | Publication type |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multi-Database Strategy
(Wakix* or pitolisant* or tiprolisant* or ciproxidine* or ozawade* or hbs-101 or hbs101 or "bf 2649" or bf2649 or 4BC83L4PIY or YV33CH63HI).ti,ab,kf,ot,hw,nm,rn.
1 use medall
*pitolisant/ or (Wakix* or pitolisant* or tiprolisant* or ciproxidine*OR ozawade* or hbs-101 or hbs101 or "bf 2649" or bf2649).ti,ab,kf,dq.
3 use oemezd
(conference review or conference abstract).pt.
4 not 5
2 or 6
exp animals/
exp animal experimentation/ or exp animal experiment/
exp models animal/
nonhuman/
exp vertebrate/ or exp vertebrates/
or/8-12
exp humans/
exp human experimentation/ or exp human experiment/
or/14-15
13 not 16
7 not 17
remove duplicates from 18
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
Search: Wakix (pitolisant), narcolepsy
WHO ICTRP
International Clinical Trials Registry Platform, produced by the World Health Organization. Targeted search used to capture registered clinical trials.
Search: Wakix (pitolisant), narcolepsy
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
Search: Wakix (pitolisant), narcolepsy
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
Search: Wakix (pitolisant), narcolepsy
Grey Literature
Search dates: January 27, 2022, to February 7, 2022
Keywords: Wakix (pitolisant), narcolepsy
Limits: Publication years: none
Updated: Search updated prior to the completion of stakeholder feedback period
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Internet Search
Open Access Journals
Appendix 2: Excluded Studies
Note that this appendix has not been copy-edited.
Reference | Reason for exclusion |
|---|---|
BASSETTI, CLA, et al. European guideline and expert statements on the management of narcolepsy in adults and children. J Sleep Res. 2021;30(6):e13387. | Guideline article |
BOLIN K, et al. The cost utility of pitolisant as narcolepsy treatment. Acta Neurol Scand. 2020;141(4):301-310. | Economic article |
DAUVILLIERS Y, et al. Long-term use of pitolisant to treat patients with narcolepsy: Harmony III study. Sleep. 2019;42(11):zs174. | Long-term extension study |
LEHERT P, et al. Multiple treatment comparison in narcolepsy: a network meta-analysis. Sleep. 2018;41(12):zsy185. | Not randomized trial |
LEHERT P, et al. Comparison of modafinil and pitolisant in narcolepsy: a non-inferiority meta analytical approach. Drugs Context. 2020;9:2020-6-2. | Review article |
MASKI K, et al. Treatment of central disorders of hypersomnolence: an American Academy of Sleep Medicine systematic review, meta-analysis, and GRADE assessment. J Clin Sleep Med. 2021;17(9):1895-1945. | Review article |
Appendix 3: Detailed Outcome Data
Note that this appendix has been redacted.
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Appendix 4: Description and Appraisal of Outcome Measures
Note that this appendix has not been copy-edited.
Aim
To describe the following outcome measures and review their measurement properties (validity, reliability, responsiveness to change, and minimal important difference [MID]):
Epworth Sleepiness Scale (ESS)
Sustained Attention to Response Task (SART)
Maintenance of Wakefulness Test (MWT)
Clinical Global Impression of Severity (CGI-S) and Clinical Global Impression of Change (CGI-C)
3-Level EQ-5D
Beck Depression Inventory (BDI-SF-13)
Findings
The findings about the validity, reliability, responsiveness, and MID of each outcome measure are summarized in Table 41.
Table 41: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
HRQoL and symptoms | |||
Epworth Sleepiness Scale (ESS) | A measure of EDS in which patients rate their likelihood of falling asleep in 8 common daily situations on a scale from 0 (would never doze) to 3 (high chance of dozing).43 | In studies including patients with sleep disorders and healthy individuals: Construct validity: Weak pooled Spearman correlation between ESS and MSLT across 4 studies of people with and without sleep disorders: r = –0.27 (95% CI, –0.36 to –0.18).44 Moderate pooled Spearman correlation between the ESS and MWT across 3 studies of patients with sleep apnea: r = –0.43 (95% CI, –0.52 to –0.34).44 Known-group validity: Mean/median differences in ESS scores between healthy people and patients with narcolepsy across 3 studies ranged from 5.5 to 11.6.44 Reliability: Good test-retest reliability with ICC values ranging from 0.6539 in adult patients with suspected OSA to 0.87 in adult narcolepsy patients across studies, with other studies reporting values in between. Good internal consistency with Cronbach alpha values ranging from 0.74 to 0.88 across 4 studies.44 Moderate to strong test-retest reliability among healthy students with Pearson correlation values ranging from r = 0.31 for the item “sitting and talking to someone” to r = 0.67 for the item “sitting quietly after lunch without alcohol.” Repeated total ESS scores had a strong significant Pearson correlation of r = 0.81. All P values < 0.001.45,46 | In studies with patients with OSA: MID estimates in a review of 3 randomized controlled trials using the SF-36 and FOSQ as anchors led to estimates from –1.74 to –4.21. Triangulation of all estimates led to an MID of 2 points.47 MID estimates from one prospective study using both anchor and distribution-based methods ranged from –2 to –3.38 |
Sustained Attention to Response Task (SART) | A computer-based task designed to measure a person's ability to withhold key press responses to infrequent and unpredictable targets (i.e., the digit 3) during a period of rapid and rhythmic responding to frequently presented non-targets (i.e., all other digits from 1 to 9).48,49 | Reliability: Moderate to good test-retest reliability, with ICC values ranging from 0.56 (total error count) to 0.71 (commission errors) among patients with narcolepsy in HARMONY 1.40 | Not assessed in patients with sleep disorders. |
Maintenance of Wakefulness Test (MWT) | Measures the time a patient can stay awake, while withstanding pressure to fall asleep during soporific circumstances.50 | Reliability: Moderate to good test-retest reliability, with ICC values ranging from 0.76 (first session) to 0.93 (initial 4 sessions combined) among patients with narcolepsy in HARMONY 1.40 | Not assessed in patients with sleep disorders. |
Clinical Global Impression of Severity (CGI-S) and Clinical Global Impression of Change (CGI-C) | Two companion 7-grade scales used to rate severity (CGI-S) and change in severity (CGI-C) of disease from the initiation of treatment. Scales range from “no sign of illness of disease,” to “among the most extremely ill patients” for the CGI-S and “very much improved” to “very much worse” for the CGI-C.51 | Not assessed in patients with sleep disorders. | Not assessed in patients with sleep disorders. |
3-Level EQ-5D | Generic preference based HRQoL scale consisting of a VAS with values between 100 (best imaginable health) and 0 (worst imaginable health) as judged by the patient. A composite index score of 5 dimensions: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression with 3 response levels ranging from experiencing no problems to extreme problems.41,52 | Not assessed in patients with sleep disorders. | Not assessed in patients with sleep disorders. |
Beck Depression Inventory (BDI-SF-13) | A shortened version of the original 21-item BDI questionnaire with 13 items to indicate probable severity of depression; scores ≥ 16 indicate severe depression.42 | Not assessed in patients with sleep disorders. | Not assessed in patients with sleep disorders. |
BDI-SF-13 = Beck Depression Inventory-Short Form, 13 items; CGI-C = Clinical Global Impression of Change; CGI-S = Clinical Global Impression of Severity; CI = confidence interval; EDS = excessive daytime sleepiness; ESS = Epworth Sleepiness Scale; FOSQ = Functional Outcomes of Sleep Questionnaire; HRQoL = health-related quality of life; ICC = intraclass correlation coefficient; MID = minimally important difference; MSLT = multiple sleep latency test; MWT = Maintenance of Wakefulness Test; OSA = obstructive sleep apnea; SF-36: 36-Item Short Form Health Survey; VAS = visual analogue scale.
A search of the literature was conducted to examine the psychometric properties of the instruments among patients with sleep disorders.
Epworth Sleepiness Scale (ESS)
Description and Scoring
The Epworth Sleepiness Scale (ESS) measures self-reported (subjective) daytime sleepiness.43 Patients are asked to score their sleepiness in 8 everyday situations requiring different levels of attention (sitting and reading; watching the television; sitting inactive in a public place (cinema, theatre, meetings); as passenger in a car or in public transportation for at least 1 hour without stop; lying down to rest in the afternoon in conditions allowing to have rest; sitting and talking to someone; sitting quietly after lunch; in a car when stopped for a few minutes in a traffic jam). Item-level ESS scores range from 0 (would never doze) to 3 (high chance of dozing); the final ESS score is the sum of all item-level scores. A final score of 0 to 10 is in the normal range, a score of 10 to 12 is borderline, and a score of 12 to 24 indicates EDS. Patients responding to the questionnaire rate their chances of having fallen asleep in the aforementioned scenarios “in recent times” although recall periods (i.e., over the last month) may be specified under certain circumstances.53 The sponsor-provided trials for this submission included patients with an ESS ≥ 12 (HARMONY CTP15 and HARMONY III16) or ≥ 14 (HARMONY 113 and HARMONY 1bis14) and did not specify a recall period in their ESS questionnaires. The change in ESS score from baseline to the stable-treatment period (average score for the final 2 weeks of treatment before the withdrawal phase) was a primary outcome in HARMONY 113 and HARMONY 1bis,9 and a secondary outcome in HARMONY CTP.15 The recall period for each ESS measurement was the immediate past week. In the long-term extension study, HARMONY III,16 ESS was measured every 3 months until 1 year and every 6 months thereafter.
Psychometric Properties
Validity: A 2014 systematic review by Kendzerska et al., evaluated the psychometric properties of the ESS in adults across 35 relevant studies.44 In examining convergent validity, the reviewers estimated moderate Spearman correlations between the ESS to each of the Maintenance of Wakefulness Test (MWT) and the MSLT. Between the ESS and MWT, the pooled Spearman correlation coefficient (95% CI) across 3 studies was –0.43 (–0.52 to –0.34) indicating a moderate correlation. Between the ESS and MSLT a weak association was found across 4 studies with a pooled Spearman correlation coefficient (95% CI) of –0.27 (–0.36 to –0.18), likely because the 2 instruments measure different aspects of sleepiness. A total of 3 studies assessed the differences in mean or median ESS scores between healthy subjects and patients with narcolepsy and found differences in scores ranging from 5.5 to 11.6, indicating strong evidence for known-group validity.
Reliability: Test-retest reliability of each item of the ESS was assessed among a group of Australian medical students (n = 87) over a period of 5 months.45,46 Results found a moderate to strong significant correlations between the matched items ranging from r = 0.31 (P < 0.001) for the item “sitting and talking to someone” to r = 0.31 (p < 0.001) for the item “sitting quietly after lunch without alcohol.” The repeated total ESS scores had a strong significant correlation of r = 0.81 (p < 0.001).
Various studies assessed the test-rest reliability of the ESS questionnaire taken in shorter timeframes. In the sponsor-submitted pivotal trials the ESS questionnaires were completed at every visit, approximately weekly or bi-weekly.13-15 Good test-retest reliability was noted in the study by Rosenberg et al. (2021),54 which assessed the reliability of the scale among patients in North America with narcolepsy (ESS ≥ 10) in 2 different 12-week randomized clinical trials (n = 231).19,55 Post-baseline time point pairs were used to estimate intraclass correlation coefficient (ICC) values. The ICC (95% CI) values were: 0.83 (0.79 to 0.87) for ESS questionnaires completed in weeks 4 and 8 (n = 199), 0.87 (0.83 to 0.90) for questionnaires completed in weeks 8 and 12 (n = 196), and 0.81 (0.76 to 0.85) for questionnaires completed in weeks 4 and 12 (n = 196), indicating moderate reliability. Rozgonyi et al. (2021)56 examined the test-retest reliability of a validated Hungarian translation of the ESS among 100 patients referred to a sleep laboratory with a 1-hour interval between the tests and found a good correlation with Pearson correlation coefficient of 0.76; however, Lin's concordance coefficient was 0.748, indicating poor reproducibility potentially due to the highly subjective interpretation of the items being examined.
Another study examined the short-term repeatability of the ESS in adult patients with suspected OSA where patients completed 2 ESS questionnaires either on the same-day (n = 20) or within the same week (n = 20).57 The mean difference (95% CI) between first and second ESS scores in the entire population was 1.93 (−3.81 to 7.66), indicating variability. The ICC (95% CI) values indicated moderate reliability ranging from 0.65 (0.31 to 0.84) for the same-day group to 0.81 (0.58 to 0.92) for the same-week group. Similar ICC (95% CI) values of 0.73 (0.61 to 0.82) indicating moderate reliability were found in a study among 108 adult patients in a sleep clinic in Australia with a median retest interval of 64 days.39
Van der Heide et al. (2015)40 assessed outcomes among 95 patients in HARMONY 1 . The study calculated the ICC by dividing the within-patient variability (squared) by the total variability (i.e., the within-patient variability [squared] plus the variability of the studied outcome measure [squared]). The study reported an ICC of 0.85 for the ESS, indicating strong reliability.
Kendzerska et al., reported good internal consistency reliability with Cronbach alpha ranging from 0.74 to 0.88 across 4 studies among adults with sleep disorders.44
Responsiveness: No studies evaluating the responsiveness of the instrument in patients with sleep disorders were identified.
Minimally Important Difference: A study by Crook et al. (2019)47 estimated the MID of the ESS among patients with obstructive sleep apnea (OSA) using data from 639 patients across 3 RCTs using both anchor- and distribution-based approaches. The changes in domains of the Functional Outcomes of Sleep Questionnaire (FOSQ) and 36-Item Short Form Health Survey (SF-36) were used as anchors. MID estimates (95% CI) using the SF-36 as an anchor among a pooled sample of 572 patients was –1.74 (–2.18 to –1.30) for the energy/vitality domain and –2.66 (–3.19 to –2.13) for the physical component domain. MID estimates (95% CI) using the FOSQ as an anchor among 264 patients ranged from –3.03 (–3.71 to –2.35) for the FOSQ total score to –4.21 (–5.05 to –3.37) for the FOSQ general productivity domain. Distribution-based estimates ranged from –1.46 to –2.36. The triangulation of all estimates led to a final MID estimate of 2 points.
Patel et al. (2018) estimated the MID for the ESS among 125 patients with OSA in the UK. Distribution-based methods found the MID ranged from –2.21 (SE of measurement method) to –2.65 (0.5 multiplied by the SD method).38 An anchor-based method used the mean change in ESS for those reporting feeling a “little less sleepy” with continuous positive airway pressure (CPAP). This method led to an MID estimate of –2.5. Receiver operating characteristic curves were plotted to determine the ESS change cut-off that best determines those who reported at least a little improvement in sleepiness and found that the MID was between an ESS change of –2 to –3 points.
Sustained Attention to Response (SART)
Description and Scoring
The SART is a computer-based task designed to measure a person's ability to withhold key press responses to infrequent and unpredictable targets (i.e., the digit 3) during a period of rapid and rhythmic responding to frequently presented non-targets (i.e., all other digits from 1 to 9).48,49 In the pivotal trials,13-15 the digits from 1 to 9 were shown 225 times in white on a black computer screen in a quasi-random way in a period over 4 minutes. The SART error score was the total number of errors, consisting of the sum of all omission errors and commission errors. Omission errors consist of key presses when a key should not have been pressed (i.e., pressing the digit 3), called “a no-go trial.” Commission errors consist of an absence of key presses when a key should have been pressed (i.e., after any digit except 3), called a “go trial.” Errors on no-go trials had a maximum count of 25 and errors on go trials had a theoretical maximum count of 200.40 SART was reported as a secondary efficacy outcome in HARMONY 1 and HARMONY 1bis, measured at inclusion and at study end point.13,14
Psychometric Properties
Reliability: With respect to test-retest reliability, Van der Heide et al. (2015) reported a moderate ICC of 0.56 for the log-transformed SART omission errors, 0.65 for the log-transformed SART total error count, and 0.71 for the log-transformed SART commission errors among patients with narcolepsy in HARMONY 1.40
No studies evaluating the validity, responsiveness, or MID of the SART among patients with sleep disorders were identified.
Maintenance of Wakefulness Test (MWT)
Description and Scoring
The MWT is a daytime polysomnographic daytime procedure that measures the ability to stay awake under conditions conducive for sleep for a given length of time.50 The MWT is derived from the MSLT but differs in that it measures sleep latency among patients instructed to remain awake instead of asked to fall asleep.58 Patients are monitored in 20 or 40 minute sessions (or until sleep onset) throughout the day with up to 4 or 5 sessions. Sleep latency is objectively measured and is defined as either of the following: (a) 3 consecutive 30 second epochs of stage 1; or (b) any single, 30 second epoch of stage 2, 3, 4, or REM sleep.58 In the pivotal trials,13-15 four 40-minute MWT sessions were conducted at both the inclusion and end-of-study visits and was reported as a secondary efficacy outcome.
Psychometric Properties
Reliability: With respect to test-retest reliability, Van der Heide et al. (2015) reported a moderate ICC of 0.76 for the log-transformed MWT for the first session. The ICC was good for the first 2, 3 and all 4 combined sessions with values of 0.87, 0.91, and 0.93, respectively.40 The study noted a celling effect for the MWT in HARMONY 1 , caused by the maximum score of 40 minutes, suggesting the tool be used with caution in statistical analyses.
No studies evaluating the validity, responsiveness, or MID of the MWT in patients with sleep disorders were identified.
Clinical Global Impression of Severity (CGI-S) And Clinical Global Impression of Change (CGI-C)
Description and Scoring
The CGI provides a stand-alone subjective evaluation of the clinician's view of a patient's global functioning which takes into account information including patient's history, psychosocial circumstances, symptoms, behaviour, and the impact of the symptoms on the patient's ability to function.51 The CGI consists of 2 companion 1-item measures evaluating the severity of illness (CGI-S) and change in severity of illness from the initiation of treatment (CGI-C) on similar 7-point scales.51
In 2 of the pivotal trials,13,14 a 6-point scale was used for the CGI-S scale: 1 = no sign of illness, 2 = borderline ill, 3 = slightly ill, 4 = moderately ill, 5 = markedly ill, and 6 = among the most extremely ill patients. In HARMONY CTP15 a 7-point scale was used for the CGI-S: 1 = no sign of illness, 2 = borderline ill, 3 = mildly ill, 4 = moderately ill, 5 = markedly ill, 6 = severely ill, and 7 = among the most extremely ill patients.
A 7-point scale was used for the CGI-C in all 3 pivotal trials13-15: 1 = very much improved, 2 = much improved, 3 = minimally improved, 4 = no change, 5 = minimally worse, 6 = much worse, 7 = very much worse. In the pivotal trials, the CGI-C was a secondary outcome and measured at every visit after baseline through to the end point visit.
Psychometric Properties
No studies examining the psychometric properties of the CGI-S or CGI-C among patients with sleep disorders were identified.
3-Level EQ-5D
Description and Scoring: The 3-Level EQ-5D is a generic HRQoL instrument that may be applied to a wide range of health conditions and treatments.59,60 The first of 2 parts of the 3-Level EQ-5D consist of a descriptive system that classifies respondents based on the following 5 dimensions: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression. There are 3 response levels of severity (no problems, some problems, and extreme problems) in each of the 5 existing dimensions.41 The second part is a 20 cm visual analogue scale (EQ-VAS) that has end points labelled 0 and 100, with respective anchors of “worst imaginable health state” and “best imaginable health state.” Respondents are asked to rate their health by drawing a line from an anchor box to the point on the EQ-VAS which best represents their health on that day. In the pivotal trials,13-15 the 3-Level EQ-5D was a secondary outcome and measured at each alternate visit after baseline through to the end point visit.
Psychometric Properties
No studies examining the psychometric properties of the 3-Level EQ-5D among patients with sleep disorders were identified.
Beck Depression Inventory, Short Form-13 items
Description and Scoring: The BDI-SF-13 is a shortened version of the original 21-item BDI questionnaire with 13 items to indicate probable severity of depression.42 Each item has a scores from 0 (absence of symptom) to 3 (severe symptom). A total score of 0 to 4 is considered minimal range, 5 to 7 is mild, 8 to15 is moderate, and 16 to 39 represents severe depression. In the pivotal trials,13-15 the BDI-SF-13 was reported as a safety outcome and measured at screening, baseline, inclusion, and each alternate visit thereafter.
Psychometric Properties
No studies examining the psychometric properties of the BDI-SF13 among patients with sleep disorders were identified.
Pharmacoeconomic Review
Abbreviations
AE
adverse event
BIA
budget impact analysis
CGI-C
Clinical Global Impressions of Change
EDS
excessive daytime sleepiness
ESS
Epworth Sleepiness Scale
HRQoL
health-related quality of life
ICER
incremental cost-effectiveness ratio
QALY
quality-adjusted life-year
SOC
standard of care
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Pitolisant hydrochloride (Wakix), 5 mg and 20 mg oral tablets |
Submitted price | Pitolisant hydrochloride: 5 mg = $16.63 per tablet 20 mg = $16.63 per tablet |
Indication | For the treatment of EDS or cataplexy in adult patients with narcolepsy |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | May 25, 2021 |
Reimbursement request | Per indication |
Sponsor | Paladin Labs Inc. |
Submission history | Previously reviewed: No |
EDS = excessive daytime sleepiness; NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Decision tree during the trial period, followed by a Markov model |
Target population | Adults with narcolepsy, assessed as 2 subgroups:
|
Treatment | Pitolisant hydrochloride |
Comparators | EDS without cataplexy:
EDS with cataplexy:
|
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs and LYs |
Time horizon | Lifetime (70 years) |
Key data source | Clinical efficacy was modelled using evidence from HARMONY I, HARMONY CTP, and HARMONY III trials |
Submitted results | EDS without cataplexy: ICER vs. SOC = $516,553 per QALY (incremental QALYs = 0.09; incremental costs = $45,098) EDS with cataplexy: ICER vs. SOC = $115,254 per QALY (incremental QALYs = 0.58; incremental costs = $66,720) |
Key limitations |
|
CADTH reanalysis results |
|
CGI-C = Clinical Global Impression of Change; EDS = excessive daytime sleepiness; ICER = incremental cost-effectiveness ratio; LY = life-year; QALY = quality-adjusted life-year; SOC = standard of care.
Conclusions
The CADTH clinical review concluded that pitolisant hydrochloride is superior to placebo for the treatment of excessive daytime sleepiness (EDS) and cataplexy in patients with narcolepsy. However, conclusions about the efficacy of pitolisant hydrochloride in comparison with modafinil could not be drawn because of the considerable uncertainty about the available clinical evidence of improvement in EDS or cataplexy. The pivotal clinical trials failed to show the noninferiority of pitolisant hydrochloride compared to modafinil across all primary outcomes assessing EDS, and no outcomes could be assessed when cataplexy was considered. In addition, a considerable proportion of patients in the pivotal trials remained on anticataplectic treatments and the interaction between pitolisant hydrochloride and concomitant medications is unknown. Furthermore, there was no direct or indirect comparative evidence to suggest that pitolisant hydrochloride is as efficacious as or superior to other standard of care (SOC) treatments for the treatment of EDS with and without cataplexy. These limitations with the available clinical evidence affect the interpretability of the sponsor’s pharmacoeconomic submission.
Because of limitations with the available comparative evidence, CADTH reanalyses assumed no difference in treatment effects (i.e., equal quality-adjusted life-years [QALYs]) between pitolisant hydrochloride and all other SOC drugs for the treatment of EDS with and without cataplexy, and conducts a cost comparison to assess annual drug costs. The annual cost of pitolisant hydrochloride is $12,147 per patient per year for the most common dose, which is more costly than all other stimulants and all stimulant plus anticataplectic drug combinations for the treatment of EDS without cataplexy and EDS with cataplexy, respectively, when considering publicly available list prices. The submitted price of pitolisant hydrochloride would need to be reduced by at least 97% to 99% to be equivalent to the lowest-priced generic stimulant (methylphenidate hydrochloride) at the upper and lower recommended doses, respectively, for the treatment of EDS without cataplexy. For the treatment of EDS with cataplexy, the submitted price of pitolisant hydrochloride would need to be reduced by at least 96% to 99% to be equivalent to the lowest-priced generic stimulant plus anticataplectic drug combination (methylphenidate HCl plus venlafaxine) at the upper and lower recommended doses, respectively.
Based on the CADTH clinical and economic reviews, there is no evidence to support a price premium for pitolisant hydrochloride over other available stimulants for the treatment of EDS without cataplexy, or over stimulants in combination with an anticataplectic drug for the treatment of EDS with cataplexy. CADTH notes that the cost-comparison analysis does not take into account potential treatment sequencing or combination use of pitolisant hydrochloride with other drugs, and that uncertainty remains about the comparative efficacy of pitolisant hydrochloride with relevant comparators. Therefore, any incremental benefit with pitolisant hydrochloride may be negative, given the lack of justification to support an assumption of equal efficacy between pitolisant hydrochloride and SOC drugs, and pitolisant hydrochloride could be dominated (more costly, less effective) by SOC. Consequently, a price reduction of even 100% would not make pitolisant hydrochloride cost-effective. CADTH could not fully explore this uncertainty because of the lack of available evidence and, therefore, the possibility that pitolisant hydrochloride generates fewer QALYs at a higher cost than SOC drugs at any price reduction should be considered.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups and drug plans that participated in the CADTH review process. No registered clinician input was received for this review.
CADTH received patient input from Wake Up Narcolepsy, a nonprofit patient advocacy organization. Wake Up Narcolepsy collected patient input through a survey of both patients with narcolepsy residing in Canada and their caregivers. Patients reported that the most disruptive symptoms of narcolepsy include EDS, disturbed nocturnal sleep, and hallucinations. Cataplexy was rated as a somewhat bothersome symptom. It was noted that narcolepsy affects every aspect of life, and that quality of life is highly affected by an inability to drive and concentrate, maintain productivity and physical wellness, and provide childcare. All patients were on different medication regimens: 33% reported taking tricyclic antidepressants, serotonin and norepinephrine reuptake inhibitors, or selective serotonin reuptake inhibitors; 12.5% reported taking modafinil or armodafinil; 55.5% reported taking stimulants; and 12.5% reported taking sodium oxybate. Half of all patients (50%) used multiple medications and the remaining 50% used individual drugs that fell under the permitted categories. Patients reported challenges with access to treatment and costs, including having to take time off work for treatment, as well as physical and mental side effects related to treatments. Patients noted that they would like a more effective drug for both EDS and cataplexy that reduced nocturnal disturbances and required less-frequent dosing (e.g., extended-release formulations).
Drug plans expressed concerns about whether the comparators used in the pivotal clinical trials were relevant to cataplexy, as well as about benefit status and criteria across jurisdictions in Canada. It was noted that modafinil is considerably less expensive than pitolisant hydrochloride. Additionally, the impact of ongoing anticataplectic treatments in the pivotal clinical trials was of concern because of restrictions of certain SOC treatments to other indications, such as attention-deficit/hyperactivity disorder. Drug plans were also concerned about the lack of clarity surrounding eligibility for patients who have received prior treatment and the monitoring of therapeutic response for continuation of treatment. Similarly, drug plans also highlighted lack of clarity surrounding the way loss of response would inform discontinuation of therapy, and noted potential issues with the limited access to specialists for narcolepsy in certain jurisdictions. Finally, drug plans expressed uncertainty surrounding the use of 1 or more medications considered to be SOC in combination with pitolisant hydrochloride.
Several of these concerns were addressed in the sponsor’s model:
The sponsor’s model compared pitolisant hydrochloride to stimulants and anticataplectic drugs for the treatment of EDS with and without cataplexy in adults with narcolepsy.
In addition, CADTH addressed some of these concerns, as follows:
CADTH conducted a cost comparison between pitolisant hydrochloride and its comparators for the treatment of EDS with and without cataplexy to assess the necessary price reduction required to reach that of the lowest-priced stimulant for EDS without cataplexy and the lowest-priced stimulant plus anticataleptic drug combination for EDS with cataplexy, assuming no differences in treatment benefits (i.e., no difference in total QALYs), given limitations with the available clinical evidence.
CADTH was unable to address the following concerns that arose from stakeholder input:
The impact of adverse events (AEs) on the cost-effectiveness of pitolisant hydrochloride compared to SOC treatments could not be explored in the submitted model.
The impact of concomitant anticataplectic use in the pivotal clinical trials on the cost-effectiveness of pitolisant hydrochloride could not be assessed in the sponsor’s submission.
Other aspects of narcolepsy related to disturbed nocturnal sleep and hallucinations could not be assessed, as they were not modelled.
Economic Review
The current review evaluates pitolisant hydrochloride for the treatment of EDS or cataplexy in adults with narcolepsy.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis comparing pitolisant hydrochloride to SOC and to no treatment, assessed separately for EDS in adults with narcolepsy with and without cataplexy. The target population was aligned with the Health Canada indication and reimbursement request for this submission.1
Pitolisant hydrochloride is available as 5 mg and 20 mg tablets. The recommended dose of pitolisant hydrochloride is 10 mg to 40 mg once daily.2 At the submitted price of $16.63 per tablet ($498.86 per 30-tablet bottle), the annual cost of pitolisant hydrochloride ranges from $6,074 to $12,147.2 SOC for EDS without cataplexy consisted of a weighted-basket comparator of stimulants used to treat EDS, either on- or off-label, including dextroamphetamines, lisdexamfetamine dimesylate, methylphenidate, and modafinil. SOC for cataplexy consisted of a weighted-basket comparator of EDS SOC drugs in combination with anticataplectic drugs used off-label, including imipramine, desipramine, clomipramine, fluoxetine, and venlafaxine.1 The annual cost of stimulants used for the treatment of EDS in all patients with narcolepsy ranges from $81 to $2,227, depending on individual patient dosage and drug used (Appendix 1). The additional annual costs of anticataplectic drugs ranged from $70 to $372, depending on the individual dosage and drug (Appendix 1). In its submission, the sponsor calculated SOC comparator costs as a weighted average, based on cost per mg, and estimated dose as the midpoint of expected dose ranges and the proportion of use, according to clinical expert opinion. The weighted average daily cost for the treatment of EDS was estimated to be $2.67, and the weighted average daily cost for EDS and cataplexy was estimated to be $3.25.
The submitted model reported both QALYs and life-years over a lifetime time horizon of 70 years in the modelled population. The base-case analyses were conducted from the perspective of the Canadian public health care payer, with discounting (1.5% per annum) applied to both costs and outcomes.1
Model Structure
The sponsor submitted a hybrid model that consisted of a decision tree followed by a Markov state-transition model, with health states defined by response rate (responder or nonresponder). Two independent models run in parallel were submitted, with 1 assessing a patient population with EDS without cataplexy and the other assessing a patient population with EDS with cataplexy. Patients are assigned to pitolisant hydrochloride, SOC, or no treatment at model entry and continue to receive their assigned treatment for the entire 8-week decision-tree phase of the model. During this initial model phase, all patients are assigned a nonresponder utility. At the end of the 8-week observation period, patients are defined as responders or nonresponders, according to response rates derived from the HARMONY I and HARMONY CTP trials.3
In the second phase of the model, patients enter the Markov model in the responder or nonresponder health state, determined in the decision-tree phase. Patients who enter the Markov model as responders can remain responders until death or transition to the nonresponder health state after treatment discontinuation for reasons such as AEs and lack of efficacy, where they remain until death. Patients who enter the Markov model as nonresponders are assumed to have withdrawn from treatment and incur no treatment-related costs aside from being assigned the utility of a nonresponder. Patients in any health state in the Markov model can die from causes unrelated to narcolepsy. The sponsor’s submitted model structure can be found in Appendix 3.
Model Inputs
The modelled patient characteristics for the sponsor’s submission were based on the HARMONY I and HARMONY CTP trials (mean age = 38 years; 52% male).3
Treatment response for those receiving pitolisant hydrochloride or no treatment in the subgroup of patients with EDS without cataplexy was based on a subsequent analysis of the HARMONY I and HARMONY CTP trials comparing pitolisant hydrochloride to placebo in which the proportion of patients with a final Epworth Sleepiness Scale (ESS) score of 10 or lower was pooled.3 All treatments included as part of the weighted SOC comparator were assumed to have the same efficacy and safety profile as modafinil, which was assumed to have the same response rate as pitolisant hydrochloride, based on an assumption of noninferiority derived from results of the Dauvilliers et al. (2013) study.4
Treatment response rates for patients receiving pitolisant hydrochloride or no treatment in the subgroup of EDS with cataplexy were based on the proportion of patients with a Clinical Global Impression Change (CGI-C) score considered to be very much improved or much improved in the subgroup of patients with a high weekly rate of cataplexy, pooled from the HARMONY I and HARMONY CTP trials.3 The Davis et al. (2021) study did not assess the relative cataplexy response rate of modafinil, and the sponsor estimated a proxy response rate comparing pitolisant hydrochloride, modafinil, and placebo assessed using the CGI-C from the Dauvilliers et al. (2013) study, using the rule of 3.1,3,4
Rates of discontinuation because of AEs, lack of efficacy, or other reasons during the first year of treatment with pitolisant hydrochloride for EDS and cataplexy were based on the Dauvilliers et al. (2019)5 study, which used results from the HARMONY III clinical trial. For subsequent years, discontinuation rates were based on 5-year unpublished results from the HARMONY III extension study.1 The sponsor assumed that discontinuation rates for patients receiving SOC were equal to those for patients receiving pitolisant hydrochloride for EDS with and without cataplexy. Baseline mortality in the model was based on mortality estimates in the general population in Canada.6 Narcolepsy is considered a nonfatal disease in the model. No treatment-related AEs were assumed to occur in the model.
Responders were assumed to have the same utility values as patients in the general population.7 The health-state utility value for nonresponders was determined by mapping Short Form (36) Health Survey estimates of health-related quality of life (HRQoL) for patients with narcolepsy in Scotland7 to mean EQ-5D preference-based scores, using an algorithm described by Ara and Brazier (2008).8 Disutility associated with narcolepsy was calculated as the difference between mapped EQ-5D utility scores for patients with and without narcolepsy. It was assumed that the disutility associated with narcolepsy was applicable for both EDS and cataplexy.
Costs included in the model were drug-acquisition costs and resource utilization costs. Drug-acquisition costs and dosing were consistent with costs and dosing reported in the overview section, with drug costs obtained from the sponsor’s submission and the Ontario Drug Benefit formulary.1,9 The sponsor calculated comparator costs using cost per mg and calculated doses according to clinical expert opinion, presented as weighted averages based on proportions of use for EDS with and without cataplexy. Costs for 2 additional specialist visits were incurred after a lack of response to treatment.10 Relevant costs were inflated to 2021 Canadian dollars.
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (5,000 iterations for the base-case and scenario analyses). The deterministic results did not fully align with the probabilistic results. There was a discrepancy in the estimated incremental QALYs between deterministic and probabilistic results. The deterministic results showed that pitolisant hydrochloride was dominated by SOC for the treatment of EDS without cataplexy because total QALYs were equal, whereas the probabilistic results estimated a small gain in QALYs in favour of pitolisant hydrochloride. Otherwise, the results were generally aligned. The probabilistic findings are presented here.
Base-Case Results
In the EDS without cataplexy subgroup, pitolisant hydrochloride was associated with incremental costs of $45,098 and 0.09 QALYs compared to SOC, resulting in an incremental cost-effectiveness ratio (ICER) of $516,553 per QALY gained (Table 3).
In the subgroup of patients with EDS and cataplexy, pitolisant hydrochloride was associated with incremental costs of $66,720 and 0.58 QALYs compared to SOC, resulting in a sequential ICER of $115,254 per QALY gained (Table 4).
Table 3: Summary of the Sponsor’s Economic Evaluation Results for EDS Without Cataplexy
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
No treatment | 177 | 21.32 | Reference |
SOC | 4,017 | 21.81 | 7,810 vs. no treatment |
Pitolisant | 49,115 | 21.90 | 516,553 vs. SOC |
EDS = excessive daytime sleepiness; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SOC = standard of care.
Note: The submitted analysis is based on publicly available prices of the comparator treatments.
Source: Sponsor’s pharmacoeconomic submission.1
Table 4: Summary of the Sponsor’s Economic Evaluation Results for EDS With Cataplexy
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
No treatment | 177 | 21.29 | Reference |
SOC | 3,998 | 21.67 | 9,886 vs. no treatment |
Pitolisant | 70,718 | 22.25 | 115,254 vs. SOC |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SOC = standard of care.
Note: The submitted analysis is based on publicly available prices of the comparator treatments.
Source: Sponsor’s pharmacoeconomic submission.1
Additional results from the sponsor’s submitted economic evaluation base case are presented in Appendix 3.
Sensitivity and Scenario Analysis Results
Various scenario and sensitivity analyses were conducted. Results of note included analyses that assessed alternate response rates for patients experiencing cataplexy and those that assessed a lower disutility value associated with narcolepsy. The incremental cost-effectiveness of pitolisant hydrochloride, compared to SOC, for cataplexy increased to $159,865 per QALY gained when alternate cataplexy response rates for patients experiencing 3 or more cataplexy attacks were assessed instead of patients experiencing 15 or more attacks.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
The clinical efficacy of pitolisant hydrochloride in comparison to SOC for the treatment patients experiencing EDS with and without cataplexy is highly uncertain: The treatment response rate of pitolisant hydrochloride for EDS was based on achievement of an ESS score of 10 or lower in the study by Davis et al. (2021),3 which included a comparison of pitolisant hydrochloride to placebo. In the absence of direct evidence from this particular analysis, the sponsor assumed that modafinil (one of the wakefulness drugs included in SOC in the sponsor’s submitted analysis) was as effective as pitolisant hydrochloride, based on an analysis conducted by Dauvilliers et al. (2013),4 which included a comparison of pitolisant hydrochloride in addition to placebo. However, the cited publication showed that pitolisant hydrochloride was not noninferior to modafinil, so it cannot be concluded that pitolisant hydrochloride is not worse than modafinil for the treatment of EDS based on ESS score. The sponsor then assumed that all other SOC comparators included as part of the weighted basket for the treatment of EDS (dextroamphetamine sulphate, methylphenidate hydrochloride, and lisdexamfetamine dimesylate) were as effective as modafinil, rather than including each comparator on its own. However, there is no available direct or indirect evidence to support an assumption that pitolisant hydrochloride is as effective as these other comparators, or that these comparators are as effective as modafinil. The clinical experts consulted by CADTH for this review noted that modafinil is typically inferior to these other drugs. As a result, the sponsor’s assessment of the cost-effectiveness of pitolisant hydrochloride compared with modafinil for EDS without cataplexy is associated with considerable uncertainty, and the cost-effectiveness compared with other relevant comparators is unknown.
Similar limitations were observed for comparative-effectiveness estimates in the cataplexy subgroup. In the absence of direct evidence from the sponsor’s analysis of choice (Davis et al. [2021]), the response rate of modafinil, according to CGI-C score, was indirectly estimated using the assumption that the effect size observed for modafinil compared with placebo and pitolisant hydrochloride from an external source (Dauvilliers et al. [2013]) would be applicable to the data for pitolisant hydrochloride and placebo observed in the Davis et al. (2021) study.3,4 The CADTH clinical review team identified limitations of this nonstandard methodology related to the assumption of equal effect sizes and heterogeneity between patient populations. Notably, pooled results from the Davis et al. (2021) study assessed cataplexy in a specific subgroup of patients with a high weekly rate of cataplexy (> 15 attacks) and compared pitolisant hydrochloride to placebo, whereas the Dauvilliers et al. (2013) study assessed patients who experienced 3 or more attacks per week and compared pitolisant hydrochloride to placebo and modafinil.3,4 The CADTH clinical review also noted that there was no evidence to support the superiority of pitolisant hydrochloride to modafinil based on CGI-C score or various other measures, and that conclusions specific to the cataplexy subgroup could only be drawn from a comparison with placebo. As a result, the comparative effectiveness of pitolisant hydrochloride compared with modafinil is highly uncertain for cataplexy. Additionally, the response rates of all anticataplectic drugs used as off-label treatment for cataplexy were assumed to be as effective as modafinil. The clinical experts consulted by CADTH considered this assumption to be inappropriate, given that modafinil is not considered effective for the treatment of cataplexy and it is not used in clinical practice for this purpose. This assumption may have led to underestimation of the efficacy of SOC anticataplectic drugs compared with pitolisant hydrochloride, as these other drugs are more effective in practice than modafinil for the treatment of cataplexy. As a result, the cost-effectiveness of pitolisant hydrochloride compared with modafinil for the treatment of EDS with cataplexy is highly uncertain, and the cost-effectiveness compared with other relevant comparators is unknown.
Concomitant medication use in the HARMONY clinical trials was noted to be a confounding factor by the CADTH clinical review, given the uncertainty of interactions between various anticataplectic medications and pitolisant hydrochloride and modafinil. Although anticataplectic drugs are often used in combination with a stimulant for the treatment of cataplexy, the sponsor assumed that combination use occurred in 98.7% of all patients receiving SOC, which is not reflected in the trial data and limits the generalizability of the sponsor’s estimates. The sponsor also assumed that all treatment combinations are equally effective, which is inappropriate, given the variability in treatment efficacy among SOC comparators. The effectiveness of individual SOC combinations for the treatment of narcolepsy is unknown, particularly for the management of EDS with cataplexy. Furthermore, the sponsor assumed in the model that patients only receive pitolisant hydrochloride as monotherapy, whereas the submitted pivotal trials have patients who were treated with concomitant medication in addition to pitolisant hydrochloride. Therefore, conclusions regarding the efficacy and cost-effectiveness of pitolisant hydrochloride relative to SOC for EDS with and without cataplexy cannot be made. The sponsor’s claims that pitolisant hydrochloride can be used as monotherapy for patients with and without cataplexy is also highly uncertain.
In reanalyses, CADTH assumed that there would be no differences in total QALYs accrued when comparing pitolisant hydrochloride to SOC drugs because of the lack of comparative clinical efficacy data. CADTH notes that this assumption may be conservative and that there is no clinical evidence to suggest that pitolisant hydrochloride is not worse than modafinil or other SOC drugs for EDS with and without cataplexy.
Comparators were not adequately modelled: The assumption that all SOC comparators are equally effective and safe is inappropriate for both subgroups and, as previously noted, there is no comparative efficacy data to inform this assumption. According to the clinical experts consulted by CADTH for this review, there is a clearly defined hierarchy in terms of treatment efficacy and the likely sequence of drugs used. The sponsor’s inclusion of all active comparators as part of a weighted-basket comparator is thus inappropriate. Per CADTH submission requirements, individual comparators should be modelled separately, with relevant AE and discontinuation rates included in the model. Application of treatment sequences was also not explored in the model. The clinical experts further noted that pitolisant hydrochloride appears to have a more favourable side-effect profile, but a potentially lower magnitude of benefit, than stimulants, leading to unknown effects on the ICER. In consideration of the limitations regarding the modelling of SOC, the cost-effectiveness of pitolisant hydrochloride compared with each of the relevant comparators is unknown.
CADTH could not address this limitation in its reanalysis.
The submitted model does not capture key aspects of the treatment paradigm: The sponsor’s submitted model structure was based primarily on response and nonresponse captured by ESS and CGI-C scores for EDS with and without cataplexy, respectively. However, the clinical experts consulted by CADTH expressed concern about using score thresholds to determine a clinically meaningful response and the impact of a drug on patient functioning in the context of EDS with or without cataplexy. For example, the experts noted concerns about patients who achieved a change in ESS score that did not reach a final score of 10 or lower but still led to changes in functional status deemed clinically meaningful by clinicians and led to better HRQoL. Such patients would still continue on their medication and obtain some benefit, even if they did not reach an ESS score of 10 or lower. Therefore, health states that included partial response along with complete response and no response, as opposed to just response and no response, should have been included as separate health states to most accurately represent patient treatment pathways and to appropriately capture treatment costs and patient HRQoL. Furthermore, the clinical experts noted that a general treatment sequence is usually applied in clinical practice that begins with modafinil, methylphenidate, and amphetamines, and is followed by sodium oxybate, when accessible. The exclusion of treatment sequencing from the model leads to concerns regarding the generalizability of the sponsor’s cost-effectiveness estimates. As referenced in previous appraisal points, there are notable differences in efficacy among SOC drugs for the treatment of EDS with and without cataplexy, according to the clinical experts consulted by CADTH for this review. As a result, the cost-effectiveness of pitolisant hydrochloride in the context of its likely use in clinical practice is associated with uncertainty.
CADTH could not address this limitation in its reanalysis.
No treatment is unlikely to be a relevant comparator: Given the availability of various treatments for EDS with and without cataplexy in Canada, no treatment is unlikely to be a relevant comparator for pitolisant hydrochloride. The clinical experts noted there are various treatment options available for patients diagnosed with EDS with and without cataplexy, which allows patients to switch treatments in the case of treatment failure or discontinuation. The placebo comparator used in the pivotal trials is not a clinically relevant intervention and would not be used in practice for patients seeking treatment. The sponsor’s inclusion of no treatment as a comparator for pitolisant hydrochloride may affect the interpretability of the sponsor’s cost-effectiveness analysis.
CADTH’s reanalysis consisted of a cost comparison between pitolisant hydrochloride and SOC. No treatment was excluded from this reanalysis.
Additional limitations were identified but were not considered to be key limitations.
Uncertainty in the sponsor’s derivation of utilities: The sponsor mapped Short Form (36) Health Survey scores from the Teixeira et al. (2004)7 study to obtain EQ-5D estimates for nonresponders who still had symptoms of narcolepsy using the mapping algorithm described by Ara and Brazier (2008).8 However, mapping is not recommended for the derivation of utilities in CADTH Guidelines for the Economic Evaluation of Health Technologies in Canada.11 Mapping is unlikely to successfully capture the utility relationship between 2 measures because of the high variability in predictive values, depending on the instruments being mapped, the algorithm used, and the severity of the health states included.11 Given that the disutility associated with narcolepsy is a key driver in the model, uncertainty in the mapped utility estimates leads to high uncertainty in the sponsor’s estimates of cost-effectiveness.
Inappropriate implementation of uncertainty for response rates in EDS: In its assessment of model behaviour, CADTH found that the sponsor presented a probabilistic sensitivity analysis with nonequivalent draws across interventions that were assumed to be equally effective in the EDS subgroup model. This led to the sponsor’s probabilistic base case producing a gain in QALYs for pitolisant hydrochloride, despite an assumption of equal efficacy. For modafinil to be considered as effective as pitolisant hydrochloride, the draws for response rate must be equal within each probabilistic draw.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (refer to Table 5).
Table 5: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
Patients who respond to treatment have the same utility as the general population. | Uncertain. Clinical experts consulted by CADTH noted that achievement of the same functionality as in the general population is the goal of treatment, but that it is unlikely to be attained in clinical practice. |
Treatment-related AEs were not considered in the model. | Inappropriate. Safety profiles of SOC drugs and pitolisant hydrochloride differed and treatment-related AEs should have been included to reflect treatment experience more accurately. Generalizability of the model is limited by this exclusion. |
Discontinuation rates for SOC comparators and pitolisant hydrochloride were assumed to be equal for the treatment of EDS with and without cataplexy. | Inappropriate. Clinical experts consulted by CADTH noted that discontinuation rates for pitolisant hydrochloride and SOC should vary because of different side-effect profiles and different magnitudes of efficacy. |
Stimulants were assumed to only be effective for the treatment of EDS. | Clinical experts consulted by CADTH noted that stimulants can be used in clinical practice to treat cataplexy in addition to EDS. However, the comparative effectiveness of stimulants and anticataplectic drugs for the treatment of cataplexy is unknown. |
Dose ranges for each SOC drug were estimated by clinical experts. The dose used to calculate drug costs was estimated using the midpoint of the estimated ranges. | Uncertain. The dosing for SOC drugs was noted to vary in individual patients by clinical experts consulted by CADTH. Dose ranges for off-label SOC drugs, provided by clinical experts consulted by CADTH, are presented in Appendix 1. |
AE = adverse event; EDS = excessive daytime sleepiness; SOC = standard of care.
CADTH Reanalyses of the Economic Evaluation
Results
Key limitations related to available clinical efficacy data informing the treatment benefit of pitolisant hydrochloride were identified, and there was a lack of available efficacy and safety data comparing pitolisant hydrochloride to relevant SOC drugs for the treatment of EDS in patients with and without cataplexy in both the short-term and long-term. The CADTH critical appraisal of the clinical evidence concluded that pitolisant hydrochloride is not noninferior to modafinil for improvement in EDS and, because of methodological limitations, the evidence for the cataplexy subgroup in comparison with modafinil is uncertain. Furthermore, there was no direct or indirect comparative evidence to suggest that pitolisant hydrochloride is as effective as or superior to other SOC treatments for EDS with and without cataplexy. Additionally, CADTH identified key limitations related to the model structure, which did not capture key aspects of the treatment of patients with EDS with or without cataplexy, nor did the model consider relevant AEs. In the absence of available evidence in support of a treatment benefit with pitolisant hydrochloride in comparison with relevant SOC comparators, CADTH assumed no difference in treatment effects (i.e., no difference in total QALYs) with pitolisant hydrochloride and compared the annual drug costs of pitolisant hydrochloride with SOC comparators in its reanalysis. CADTH notes that this assumption is highly uncertain and may be conservative because there is no evidence that pitolisant hydrochloride is as effective as other SOC drugs, and it may even be inferior.
In the CADTH reanalysis of the subgroup of patients with EDS without cataplexy, costs of stimulants ranged from $81 to $2,677 per patient per year, whereas the annual cost of pitolisant hydrochloride ranged from $6,074 to $12,147 per patient per year. All stimulants were less expensive than pitolisant hydrochloride, with pitolisant hydrochloride ranging from $9,470 to $12,066 when the upper bound of the cost per patient is considered, as this is associated with the most frequent dosing of pitolisant hydrochloride used in the HARMONY trials and is the most likely dosing, according to the clinical experts consulted by CADTH for this review. Annual costs of each stimulant and the difference in annual drug costs in comparison to pitolisant hydrochloride can be found in Table 6.
Table 6: CADTH Cost Comparison for Pitolisant Hydrochloride and Stimulants
Price-reduction scenarios for each SOC stimulant | Current list price of pitolisant hydrochloride ($)a | Reduction needed (%) | Reduced annual price of pitolisant hydrochloride ($) | Annual reduction from sponsor’s price ($) |
|---|---|---|---|---|
Dextroamphetamine sulphate (generics) SR capsule, 10 mg to 60 mg daily | 12,147 | 88 to 98 | 296 to 1,446 | 11,852 to 10,701 |
Dextroamphetamine sulphate (generics) tablet, 10 mg to 60 mg daily | 12,147 | 82 to 97 | 371 to 2,227 | 11,776 to 9,920 |
Lisdexamfetamine dimesylate (Vyvanse) capsule or tablet, 10 mg to 70 mg daily | 12,147 | 78 to 93 | 832 to 2,677 | 11,316 to 9,470 |
Methylphenidate hydrochloride (generics) tablet, 10 mg to 60 mg daily | 12,147 | 97 to 99 | 81 to 370 | 12,066 to 11,777 |
Modafinil (generics) tablet, 200 mg to 400 mg daily | 12,147 | 89 to 94 | 679 to 1,358 | 11,468 to 10,790 |
SOC = standard of care; SR = sustained release.
Note: Range of prices was calculated using the recommended dose of each stimulant, in accordance with the product monograph or clinical expert opinion. CADTH’s reanalysis is based on publicly available prices of the comparator treatments.
aIt was assumed that patients would receive 2 pills of pitolisant hydrochloride, as 10 mg and 40 mg are the most frequent doses used.
A separate cost comparison of pitolisant hydrochloride with each available stimulant in combination with an anticataplectic drug was considered for the treatment of EDS with cataplexy. The costs of a stimulant plus an anticataplectic drug ranged from $114 to $3,421 per patient per year. Compared with the cost of pitolisant hydrochloride, at $6,074 to $12,147 per patient per year, all stimulant plus anticataplectic drug combinations were less expensive than pitolisant hydrochloride. The costs of pitolisant hydrochloride ranged from $8,726 to $12,033 when the upper bound of the cost per patient, the most frequent dosing, was considered. Annual costs of each combination and costs in comparison pitolisant hydrochloride can be found in Table 7. This analysis only considered pitolisant hydrochloride as monotherapy, which may have underestimated total drug costs, as some patients treated with pitolisant hydrochloride for EDS with cataplexy in the HARMONY trials continued to receive concomitant anticataplectic drugs.
Table 7: CADTH Cost Comparison for Pitolisant Hydrochloride and Stimulant Plus Anticataplectic Combinations
SOC stimulant annual cost and incremental annual cost relative to pitolisant hydrochloride ($12,147)a | Anticataplectic SOC drug | ||||
|---|---|---|---|---|---|
Clomipramine (generic),b 10 mg to 75 mg daily ($) | Desipramine,b 25 to 150 mg daily ($) | Fluoxetine (generic),b 20 to 80 mg daily ($) | Imipramine,b 25 to 150 mg daily ($) | Venlafaxine (generic),b 37.5 to 300 mg daily ($) | |
Dextroamphetamine sulphate (generic) SR capsule, 10 g to 60 mg daily | 403 to 1,801 11,744 to 10,347 | 454 to 2,190 11,693 to 9,957 | 417 to 1,930 11,731 to 10,217 | 390 to 1,996 11,758 to 10,151 | 329 to 1,587 11,818 to 10,560 |
Dextroamphetamine sulphate (generic) tablet, 10 mg to 60 mg daily | 479 to 2,582 11,668 to 9,566 | 530 to 2,971 11,617 to 9,176 | 492 to 2,711 11,655 to 9,437 | 465 to 2,777 11,682 to 9,370 | 405 to 2,368 11,743 to 9,779 |
Lisdexamfetamine dimesylate (Vyvanse) capsule or tablet, 10 mg to 70 mg daily | 939 to 3,032 11,208 to 9,115 | 990 to 3,421 11,157 to 8,726 | 953 to 3,161 11,195 to 8,986 | 926 to 3,228 11,222 to 8,920 | 865 to 2,818 11,282 to 9,329 |
Methylphenidate hydrochloride (generic) tablet, 10 mg to 60 mg daily | 189 to 725 11,959 to 11,423 | 240 to 1,114 11,908 to 11,033 | 202 to 854 11,945 to 11,294 | 175 to 920 11,972 to 11,227 | 114 to 511 12,033 to 11,637 |
Modafinil (generic) tablet, 200 mg to 400 mg daily | 787 to 1,712 11,361 to 10,435 | 838 to 2,102 11,310 to 10,046 | 800 to 1,841 11,347 to 10,306 | 773 to 1,908 11,374 to 10,239 | 712 to 1,498 11,435 to 10,649 |
SOC = standard of care; SR = sustained release.
Note: Range of prices was calculated using the lowest recommended doses for both stimulant and anticataplectic drugs for the lower dose, and the highest recommended dose for both stimulant and anticataplectic drugs for the higher dose. CADTH’s reanalysis is based on publicly available prices of the comparator treatments.
aIt was assumed that patients would receive 2 pills, as 10 mg and 40 mg are the most frequent doses used in patients.
bThe first row in each cell is the cost range of the treatment plus anticataplectic. The second row in each cell is the range in incremental cost difference compared with pitolisant hydrochloride. For example, the annual cost of dextroamphetamine sulphate SR plus clomipramine ranges from $403 to $1,801. The associated incremental cost difference compared with pitolisant hydrochloride ranges from –$11,744 to –$10,347.
Price-Reduction Analyses
In the absence of clinical information to justify a price premium for pitolisant hydrochloride and given that the annual cost of pitolisant hydrochloride is higher than all treatments for the management of EDS with or without cataplexy, price-reduction analyses were conducted to determine the percentage reductions required for the price of pitolisant hydrochloride to be similar to that of relevant comparators.
The price reduction required for the most common dosing of pitolisant hydrochloride (i.e., 2 tablets per day) to be comparative with the lowest-priced generic stimulant (methylphenidate hydrochloride) for the treatment of EDS without cataplexy was considered. The submitted price of pitolisant hydrochloride would need to be reduced by at least 97% to 99% to be equivalent to the lowest-priced generic stimulant at the upper and lower recommended doses of methylphenidate hydrochloride, respectively.
The price reduction required for the most common dosing of pitolisant hydrochloride to be comparative with the lowest-priced combination of a generic stimulant plus anticataplectic drug (methylphenidate hydrochloride plus venlafaxine) for the treatment of EDS with cataplexy was considered. The submitted price of pitolisant hydrochloride would need to be reduced by at least 96% to 99% to be equivalent to the lowest-priced generic stimulant plus anticataplectic drug combination at the upper and lower recommended doses, respectively.
Table 8: CADTH Price-Reduction Analyses
Scenario | Current list price of pitolisant hydrochloride ($)a | Reduction needed (%) | Reduced annual price of pitolisant hydrochloride ($) | Annual reduction from sponsor’s price ($) |
|---|---|---|---|---|
EDS without cataplexy | ||||
Price reduction required for pitolisant hydrochloride to equal the lowest recommended dose of methylphenidate hydrochloride | 12,147 | 99% | 81 | –12,066 |
Price reduction required for pitolisant hydrochloride to equal the highest recommended dose of methylphenidate hydrochloride | 12,147 | 97% | 370 | –11,777 |
EDS with cataplexy | ||||
Price reduction required for pitolisant hydrochloride to equal the lowest recommended dose of methylphenidate hydrochloride plus venlafaxine | 12,147 | 99% | 114 | –12,033 |
Price reduction required for pitolisant hydrochloride to equal the highest recommended dose of methylphenidate hydrochloride plus venlafaxine | 12,147 | 96% | 511 | –11,637 |
EDS = excessive daytime sleepiness.
Note: CADTH’s reanalysis is based on publicly available prices of the comparator treatments.
aIt was assumed that patients would receive 2 pills of pitolisant hydrochloride, as 10 mg and 40 mg are the most frequent doses used in patients.
Issues for Consideration
Sodium oxybate was included as a SOC comparator in a scenario analysis in the sponsor’s model, where it was assumed to have the same efficacy as pitolisant hydrochloride, leading to an additional annual cost of $25,789. At doses of 12 mL to 18 mL nightly, the annual cost of sodium oxybate ranges from $13,646 to $20,468 per patient.12 However, sodium oxybate is not readily accessible in Canada, according to clinical experts, and received a Do Not List recommendation in 2009.13 A survey of drug plans showed that no responding jurisdictions reimbursed the drug. As such, it was deemed to not be a relevant comparator in this submission.
The clinical experts also noted the strong potential for pitolisant hydrochloride to be used in combination with existing wakefulness or anticataplectic drugs off-label. The cost-effectiveness of pitolisant hydrochloride used in combination with existing drugs for EDS or cataplexy is unknown.
Overall Conclusions
The CADTH clinical review concluded that pitolisant hydrochloride is superior to placebo for the treatment of EDS and cataplexy in patients with narcolepsy. However, conclusions about the efficacy of pitolisant hydrochloride in comparison with modafinil could not be drawn because of considerable uncertainty about the available clinical evidence for improvement in EDS or cataplexy. The pivotal clinical trials failed to show the noninferiority of pitolisant hydrochloride to modafinil across all primary outcomes used to assess EDS, and no outcomes could be assessed when cataplexy was considered. A considerable proportion of patients in the pivotal trials also remained on anticataplectic treatments, and the interaction between pitolisant hydrochloride and concomitant medications is unknown. Furthermore, there was no direct or indirect comparative evidence to suggest that pitolisant hydrochloride is as effective as or superior to other SOC treatments for EDS with or without cataplexy. These limitations affect the interpretability of the sponsor’s pharmacoeconomic submission.
Given the number of therapeutic options available to treat EDS (drugs that comprise SOC, as defined by the sponsor), the fact that available clinical information is limited to a comparison with modafinil and no treatment (placebo), and the fact that findings from the HARMONY trials suggest the treatment effects of pitolisant hydrochloride in comparison with modafinil are highly uncertain, with noninferiority to modafinil not established, the CADTH reanalyses assumed no difference in treatment effects (i.e., equal QALYs) between pitolisant hydrochloride and all SOC drugs for EDS with or without cataplexy, and a cost comparison assessing annual drug costs was conducted.
The annual cost of the most common dose of pitolisant hydrochloride is $12,147 per patient per year, which is more than all stimulants and all stimulant plus anticataplectic drug combinations (according to publicly available list prices) for the treatment of EDS without cataplexy and EDS with cataplexy, respectively. The submitted price of pitolisant hydrochloride would need to be reduced by at least 97% to 99% to be equivalent to the lowest-priced generic stimulant (methylphenidate hydrochloride) at the upper and lower recommended doses, respectively, for the treatment of EDS without cataplexy. For the treatment of EDS with cataplexy, the submitted price of pitolisant hydrochloride would need to be reduced by at least 96% to 99% to be equivalent to the lowest-priced generic stimulant plus anticataplectic drug combination (methylphenidate hydrochloride plus venlafaxine) at the upper and lower recommended doses, respectively. CADTH notes that an assumption of equal efficacy between pitolisant hydrochloride and SOC drugs is likely conservative, and there is no available evidence to show that pitolisant hydrochloride is not worse than SOC for the treatment of EDS with or without cataplexy. In addition, CADTH’s price reductions are potentially an underestimation, given the concomitant use of anticataplectic drugs with pitolisant hydrochloride observed in clinical trials.
Based on the CADTH clinical and economic reviews, there is no evidence to support a price premium for pitolisant hydrochloride over other available stimulants for the treatment of EDS without cataplexy, or stimulants in combination with an anticataplectic drug for the treatment of EDS with cataplexy. CADTH notes that the cost-comparison analysis does not take into account potential treatment sequencing or the combination use of pitolisant hydrochloride with other drugs, and that uncertainty remains about the comparative efficacy of pitolisant hydrochloride and relevant comparators. Therefore, the incremental benefit with pitolisant hydrochloride may be negative, given the lack of justification to support an assumption of equal efficacy between pitolisant hydrochloride and SOC drugs, and pitolisant hydrochloride could be dominated (more costly, less effective) by SOC. Consequently, a price reduction of even 100% would not make pitolisant hydrochloride cost-effective. CADTH could not fully explore this uncertainty because of the lack of available evidence and, therefore, the possibility that pitolisant hydrochloride generates fewer QALYs at a higher cost than SOC drugs at any price reduction should be considered.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Wakix (pitolisant hydrochloride), 5 and 20 mg oral tablets [internal sponsor's package]. St-Laurent (QC): Paladin Labs Inc; 2022 Jan 17.
2.Wakix (pitolisant hydrochloride): 5 mg and 20 mg tablets [product monograph]. St-Laurent (QC): Paladin Labs Inc; 2021 Aug 9.
3.Davis CW, Kallweit U, Schwartz JC, Krahn LE, Vaughn B, Thorpy MJ. Efficacy of pitolisant in patients with high burden of narcolepsy symptoms: pooled analysis of short-term, placebo-controlled studies. Sleep Med. 2021;81:210-217. PubMed
4.Dauvilliers Y, Bassetti C, Lammers GJ, et al. Pitolisant versus placebo or modafinil in patients with narcolepsy: a double-blind, randomised trial. Lancet Neurol. 2013;12(11):1068-1075. PubMed
5.Dauvilliers Y, Arnulf I, Szakacs Z, et al. Long-term use of pitolisant to treat patients with narcolepsy: Harmony III study. Sleep. 2019;42(11):21. PubMed
6.Statistics Canada. Life expectancy and other elements of the life table, three-year estimates, Canada, all provinces except Prince Edward Island: table: 13-10-0114-01. 2022; https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011401. Accessed 2022 Feb 23.
7.Teixeira VG, Faccenda JF, Douglas NJ. Functional status in patients with narcolepsy. Sleep Med. 2004;5(5):477-483. PubMed
8.Ara R, Brazier J. Deriving an algorithm to convert the eight mean SF-36 dimension scores into a mean EQ-5D preference-based score from published studies (where patient level data are not available). Value Health. 2008;11(7):1131-1143. PubMed
9.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2021; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2022 Feb 20.
10.Canadian Institute for Health Information. Physicians in Canada report. Summary report. 2019; https://www.cihi.ca/sites/default/files/document/physicians-in-canada-report-en.pdf. Accessed 2022 Feb 23.
11.Guidelines for the economic evaluation of health technologies: Canada. 4th ed. Ottawa (ON): CADTH; 2017: https://www.cadth.ca/guidelines-economic-evaluation-health-technologies-canada-4th-edition. Accessed 2022 Feb 23.
12.DeltaPA. Ottawa (ON): IQVIA; 2021: https://www.iqvia.com/. Accessed 2022 Feb 23.
13.CADTH Canadian Expert Drug Advisory Committee (CEDAC) final recommendation on reconsideration and reasons for recommendation: sodium oxybate resubmission (Xyrem - Valean Canada Ltd.). Ottawa (ON): CADTH; 2009 Jan 21: https://www.cadth.ca/sites/default/files/cdr/complete/cdr_complete_Xyrem%20Resubmission_January-28-2009.pdf. Accessed 2022 Apr 11.
14.Saskatchewan Drug Plan: search formulary. 2022; https://formulary.drugplan.ehealthsask.ca/SearchFormulary. Accessed 2022 Feb 23.
15.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Wakix (pitolisant hydrochloride), 5 and 20 mg oral tablets. St-Laurent (QC): Paladin Labs Inc; 2022 Jan 17.
16.Scheer D, Schwartz SW, Parr M, Zgibor J, Sanchez-Anguiano A, Rajaram L. Prevalence and incidence of narcolepsy in a US health care claims database, 2008-2010. Sleep. 2019;42(7). PubMed
17.Statistics Canada. Population estimates on July 1st by age and sex: table: 17-10-0005-01 (formerly CANSIM 051-0001) 2021; https://www150.statcan.gc.ca/t1/tbl1/en/cv.action?pid=1710000501. Accessed 2022 Feb 23.
18.Narcolepsy fast facts. Lynnwood (WA): Narcolepsy Network, Inc; 2015: https://narcolepsynetwork-org.s3.us-east-2.amazonaws.com/wp-content/uploads/2019/05/FastFacts-website.pdf. Accessed 2022 Feb 23.
Appendix 1: Cost-Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 9: CADTH Cost-Comparison Table for EDS
Treatment | Strength/concentration | Form | Price ($) | Recommended dosagea | Daily cost ($) | Annual drug cost ($) |
|---|---|---|---|---|---|---|
Pitolisant hydrochloride (Wakix) | 5 mg 20 mg | Tablet | 16.6287 | 10 mg to 40 mg, once daily | 16.63 to 33.26 | 6,074 to 12,147 |
Stimulants | ||||||
Dextroamphetamine sulphate (generics) | 10 mg 15 mg | SR Capsule | 0.8096 0.9898 | 10 mg to 60 mg, once daily | 0.81 to 3.96 | 296 to 1,446 |
5 mg | Tablet | 0.5081 | 10 mg to 60 mg, once daily | 1.02 to 6.10 | 371 to 2,227 | |
Methylphenidate HCl (generics) | 10 mg 20 mg | Tablet | 0.2216 0.3376 | 10 mg to 60 mg daily | 0.22 to 1.01 | 81 to 370 |
20 mg | ER Tablet | 0.2820 | In place of tablets when 8-hour dosage of ER tablet corresponds to titrated 8-hour dosage of tablets | 0.28 to 0.85 | 103 to 309 | |
Modafinil (generics) | 100 mg | Tablet | 0.9293 | 200 mg to 400 mg daily | 1.86 to 3.72 | 679 to 1,358 |
Stimulants (not specifically indicated) | ||||||
Lisdexamfetamine dimesylate (Vyvanse) | 10 mg 20 mg 30 mg 40 mg 50 mg 60 mg | Capsule or chewable tablet | 2.2769 2.8322 3.3875 3.9429 4.4982 5.0535 | 10 mg to 70 mg, once daily | 2.28 to 7.33 | 832 to 2,677 |
EDS = excessive daytime sleepiness; ER = extended release; SR = sustained release.
Note: All prices are from the Ontario Drug Benefit Formulary (accessed February 2022) unless otherwise indicated, and do not include dispensing fees.9 All annual costs were determined by multiplying daily costs by 365.25.
aDosing information for off-label use of lisdexamfetamine dimesylate for management of EDS was based on feedback from clinical experts consulted by CADTH.
Table 10: CADTH Cost-Comparison Table for Cataplexy
Treatment | Strength / concentration | Form | Price ($) | Recommended dosagea | Daily cost ($) | Annual drug cost ($) |
|---|---|---|---|---|---|---|
Pitolisant hydrochloride (Wakix) | 5 mg 20 mg | Tablet | 16.6287 | 10 mg to 40 mg once daily | 16.63 to 33.26 | 6,074 to 12,147 |
Stimulants | ||||||
Dextroamphetamine sulphate (generics) | 10 mg 15 mg | SR Capsule | 0.8096 0.9898 | 10 mg to 60 mg once daily | 0.81 to 3.96 | 296 to 1,446 |
5 mg | Tablet | 0.5081 | 10 mg to 60 mg once daily | 1.02 to 6.10 | 371 to 2,227 | |
Methylphenidate HCl (generics) | 10 mg 20 mg | Tablet | 0.2216 0.3376 | 10 mg to 60 mg daily | 0.22 to 1.01 | 81 to 370 |
20 mg | ER Tablet | 0.2820 | In place of tablets when 8-hour dosage of ER tablet corresponds to titrated 8-hour dosage of tablets | 0.28 to 0.85 | 103 to 309 | |
Modafinil (generics) | 100 mg | Tablet | 0.9293 | 200 mg to 400 mg daily | 1.86 to 3.72 | 679 to 1,358 |
Stimulants (not specifically indicated) | ||||||
Lisdexamfetamine dimesylate (Vyvanse) | 10 mg 20 mg 30 mg 40 mg 50 mg 60 mg | Capsule or chewable tablet | 2.2769 2.8322 3.3875 3.9429 4.4982 5.0535 | 10 mg to 70 mg once daily | 2.28 to 7.33 | 832 to 2,677 |
Anticataplectic agents (not specifically indicated) | ||||||
Clomipramine (generics) | 10 mg 25 mg 50 mg | Tablet | 0.2949 0.3417 0.6291 | 10 mg to 75 mg once daily | 0.29 to 0.97 | 108 to 355 |
Desipramine | 25 mg 50 mg 75 mg | Tablet | 0.4345 0.7659 1.0185 | 25 mg to 150 mg once daily | 0.43 to 2.04 | 159 to 744 |
Fluoxetine (generics) | 10 mg 20 mg | Capsule | 0.3404 0.3311 | 20 to 80 mg once daily | 0.33 to 1.32 | 121 to 484 |
Imipramine | 10 mg 25 mg 50 mg | Tablet | 0.1397 0.2573 0.5021 | 25 mg to 150 mg once daily | 0.26 to 1.51 | 94 to 550 |
Venlafaxine (generics) | 37.5 mg 75 mg 150 mg | ER Capsule | 0.0913 0.1825 0.1927 | 37.5 to 300 mg once daily | 0.091 to 0.39 | 33 to 141 |
ER = extended release; SR = sustained release.
Note: All prices are from the Ontario Drug Benefit Formulary (accessed February 2022) unless otherwise indicated, and do not include dispensing fees.9 The price of fluoxetine was sourced from the Saskatchewan Formulary Database.14 All annual costs were determined by multiplying daily costs by 365.25
aDosing information for off-label use of anticataplectic agents for management of cataplexy and the off-label use of lisdexamfetamine dimesylate for management of EDS was based on feedback from clinical experts consulted by CADTH.
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | CADTH identified limitations with the weighted basket of comparators representing standard of care. Individual standard of care treatments should have been modelled separately. Refer to CADTH appraisal section. |
Model has been adequately programmed and has sufficient face validity | No | Nonequivalent draws were presented in the probabilistic sensitivity analysis for interventions that were assumed to be equally effective in the excessive daytime sleepiness subgroup. Refer to CADTH appraisal section. |
Model structure is adequate for decision problem | No | The submitted model structure was found to be inadequate in capturing clinically meaningful response and patient function in cases where partial response would have been appropriate. Treatment sequence was also unable to be assessed due to constraints with the modelling of comparators. Refer to CADTH appraisal section. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | CADTH identified limitations with the derivation of treatment response for modafinil, particularly for the treatment of cataplexy. Refer to CADTH appraisal section. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | No | CADTH identified several limitations with the submission related to transparency. The reporting of derivation of treatment response was not adequate to explain the methodology. There are typographical errors, such as the mislabeling of columns in Table 19 (daily dose, rather than combined costs of stimulant and anticataplectic drug). The submission states that pitolisant hydrochloride was found to be noninferior to modafinil but the cited publication states that it was not found to be noninferior. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
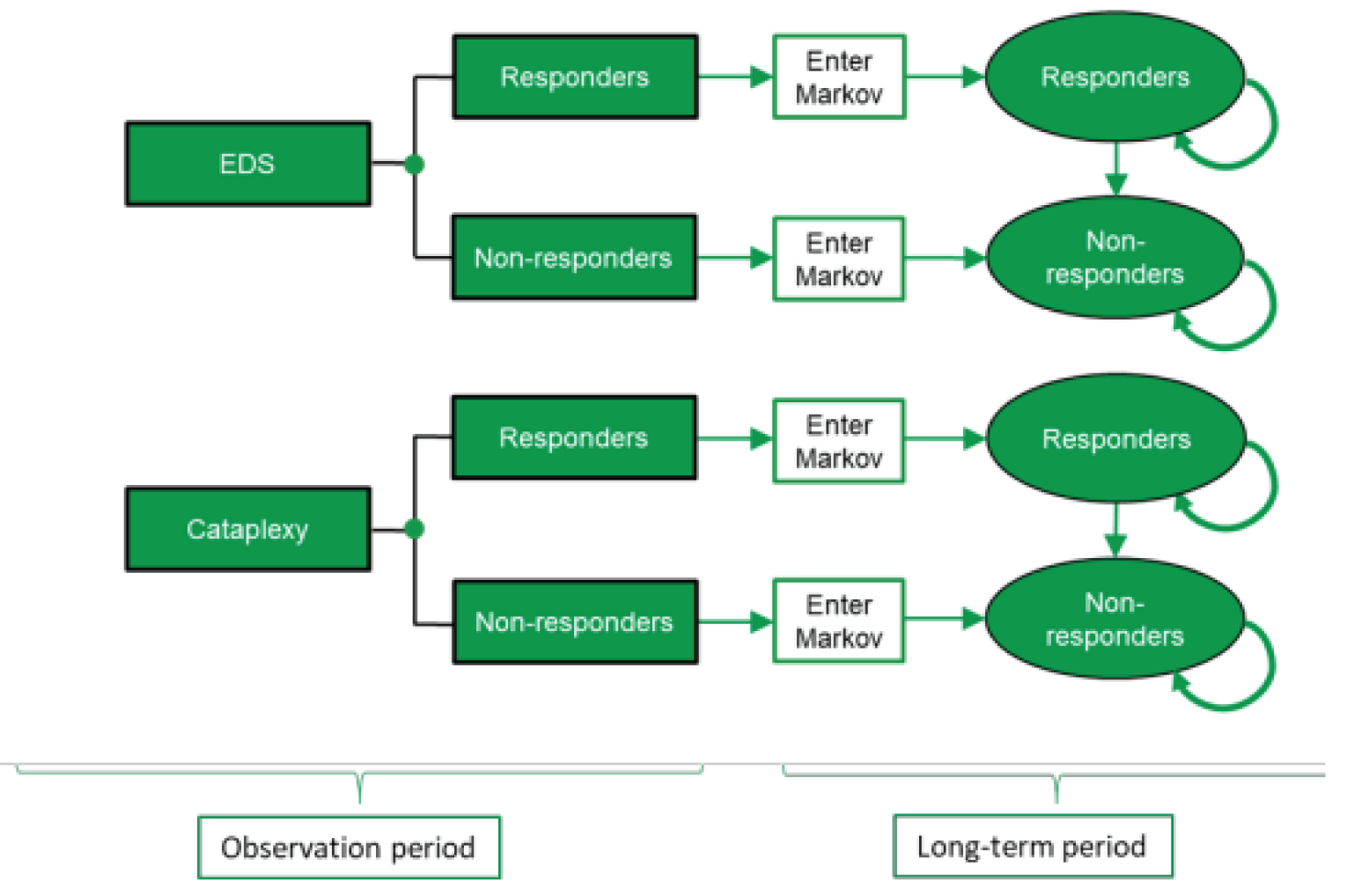
Detailed Results of the Sponsor’s Base Case
Table 12: Disaggregated Summary of the Sponsor’s Economic Evaluation Results for EDS Without Cataplexy
Treatment | Component | Value | Incremental (vs. reference) | Incremental (sequential) |
|---|---|---|---|---|
Discounted LYs | ||||
No treatment | Responders | 1.10 | NA | NA |
Nonresponders | 31.30 | NA | NA | |
Total | 32.40 | NA | NA | |
SOC | Responders | 3.80 | 2.69 | NA |
Nonresponders | 28.60 | –2.69 | NA | |
Total | 32.40 | 0 | NA | |
Wakix | Responders | 4.28 | 3.17 | 0.48 |
Nonresponders | 28.13 | –3.17 | –0.48 | |
Total | 32.40 | 0 | 0 | |
Discounted QALYs | ||||
No treatment | Responders | 0.95 | NA | NA |
Nonresponders | 20.37 | NA | NA | |
Total | 21.32 | NA | NA | |
SOC | Responders | 3.28 | 2.33 | NA |
Nonresponders | 18.53 | –1.84 | NA | |
Total | 21.81 | 0.49 | NA | |
Wakix | Responders | 3.67 | 2.72 | 0.39 |
Nonresponders | 18.23 | –2.14 | –0.30 | |
Total | 21.90 | 0.58 | 0.09 | |
Discounted costs ($) | ||||
No treatment | Drug | 0 | NA | NA |
Other resource costs | 177 | NA | NA | |
Total | 177 | NA | NA | |
SOC | Drug | 3,849 | 3,849 | NA |
Other resource costs | 167 | –10 | NA | |
Total | 4,017 | 3,840 | NA | |
Wakix | Drug | 48,952 | 48,952 | 45,102 |
Other resource costs | 163 | –14 | –4 | |
Total | 49,115 | 48,938 | 45,098 | |
Treatment | ICER vs. reference ($) | Sequential ICER ($) | ||
No treatment | Reference | Reference | ||
SOC | 7,810 | 7,810 | ||
Wakix | 84,532 | 516,553 | ||
EDS = excessive daytime sleepiness; ICER = incremental cost-effectiveness ratio; LY = life-year; NA = not applicable; QALY = quality-adjusted life-year; SOC = standard of care.
Table 13: Disaggregated Summary of the Sponsor’s Economic Evaluation Results for EDS With Cataplexy
Treatment | Component | Value | Incremental (vs. reference) | Incremental (sequential) |
|---|---|---|---|---|
Discounted LYs | ||||
No treatment | Responders | 0.96 | NA | NA |
Nonresponders | 31.45 | NA | NA | |
Total | 32.42 | NA | NA | |
SOC | Responders | 3.07 | 2.11 | NA |
Nonresponders | 29.34 | –2.11 | NA | |
Total | 32.42 | 0 | NA | |
Wakix | Responders | 6.23 | 5.27 | 3.16 |
Nonresponders | 26.19 | –5.26 | –3.15 | |
Total | 32.42 | 0 | 0 | |
Discounted QALYs | ||||
No treatment | Responders | 0.83 | NA | NA |
Nonresponders | 20.45 | NA | NA | |
Total | 21.29 | NA | NA | |
SOC | Responders | 2.66 | 1.83 | NA |
Nonresponders | 19.01 | –1.44 | NA | |
Total | 21.67 | 0.38 | NA | |
Wakix | Responders | 5.35 | 4.52 | 2.69 |
Nonresponders | 16.90 | –3.55 | –2.11 | |
Total | 22.25 | 0.96 | 0.58 | |
Discounted costs ($) | ||||
No treatment | Drug | 0 | NA | NA |
Other resource costs | 177 | NA | NA | |
Total | 177 | NA | NA | |
SOC | Drug | 3,828 | 3,828 | NA |
Other resource costs | 170 | –7 | NA | |
Total | 3,998 | 3,821 | NA | |
Wakix | Drug | 70,562 | 70,562 | 66,734 |
Other resource costs | 156 | –21 | –14 | |
Total | 70,718 | 70,541 | 66,720 | |
Treatment | ICER vs. reference ($) | Sequential ICER ($) | ||
No treatment | Reference | Reference | ||
SOC | 9,886 | 9,886 | ||
Wakix | 73,073 | 115,254 | ||
EDS = excessive daytime sleepiness; ICER = incremental cost-effectiveness ratio; LY = life-year; NA = not applicable; QALY = quality-adjusted life-year; SOC = standard of care.
Price-Reduction Analysis Based on Sponsor’s Submitted Base Case
Table 14: CADTH Price-Reduction Analyses for EDS Without Cataplexy
Analysis | ICERs for Wakix vs. SOC | |
|---|---|---|
Price reduction | Sponsor base case | CADTH reanalysis |
No price reduction | Dominated | NA |
10% | Dominated | NA |
50% | Dominated | NA |
90% | Dominated | NA |
92% | Dominant | NA |
EDS = excessive daytime sleepiness; ICER = incremental cost-effectiveness ratio; NA = not applicable.
Table 15: CADTH Price-Reduction Analyses for EDS With Cataplexy
Analysis | ICERs for Wakix vs. SOC | |
|---|---|---|
Price reduction | Sponsor base case | CADTH reanalysis |
No price reduction | $115,423 | NA |
10% | $103,052 | NA |
20% | $90,681 | NA |
30% | $78,310 | NA |
40% | $65,939 | NA |
50% | $53,568 | NA |
53% | $49,857 | NA |
EDS = excessive daytime sleepiness; ICER = incremental cost-effectiveness ratio; NA = not applicable.
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
CADTH did not conduct any additional pharmacoeconomic analyses in the review of pitolisant hydrochloride.
Appendix 5: Submitted Budget Impact Analysis and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 16: Summary of Key Takeaways
Key Takeaways of the Budget Impact Analysis |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
The submitted budget impact analysis (BIA) estimated the costs of reimbursing pitolisant hydrochloride for the treatment of EDS in narcolepsy with and without cataplexy in adult patients.15 The analysis took the perspective of Canadian public drug plans using an epidemiological approach and incorporated drug-acquisition costs. A time of horizon of 3 years between June 2022 to May 2025 was taken, with June 2021 through May 2022 being the base year of the model. The target population size was estimated using the prevalence16 of narcolepsy in adults covered by public health plans in Canada. The model assumed a stable incidence of narcolepsy, meaning that market size increased in each subsequent year in accordance with population growth rates.17 Further specifications of population size included the removal of patients not being treated for narcolepsy, which was estimated to be 75% of all eligible adult patients.18 The sponsor then assumed that 17.6% of treated patients would be treated for EDS with cataplexy.16 The reference case scenario included dextroamphetamine sulphate, methylphenidate HCl, modafinil, and lisdexamfetamine dimesylate for treatment of EDS without cataplexy. The reference case scenario for treatment of EDS with cataplexy included additional anticataplectic treatment given in combination with each stimulant for EDS. The new drug scenario for EDS without cataplexy included pitolisant hydrochloride and the stimulant comparators. Similarly, the new drug scenario for EDS with cataplexy included pitolisant hydrochloride and each stimulant given in combination with anticataplectic treatment. The anticataplectic treatment cost was calculated as a weighted average of imipramine, desipramine, clomipramine, fluoxetine, venlafaxine, and no anticataplectic based on cost per mg, estimated dosing, and proportion of use estimated by clinical experts. Key inputs to the BIA are documented in Table 18.
Table 17: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1/year 2/year 3 if appropriate) |
|---|---|
Target population | |
CADTH-participating pan-Canadian Population17 | 30,190,187 |
Proportion of those under public coverage by drug plans Newfoundland and Labrador Prince Edward Island Nova Scotia New Brunswick Ontario Manitoba Saskatchewan Alberta British Columbia Non-Insured Health Benefits | 28.9% 40.7% 42.1% 19.6% 28.1% 16.3% 21.9% 21.1% 29.1% 100% |
Prevalence of narcolepsy16 | 0.06% |
Proportion treated for narcolepsy18 | 25% |
Proportion of patients with narcolepsy treated for cataplexy16 | 17.6% |
Number of patients eligible for treatment in the reference scenario | 1,252 / 1,271 / 1,291 |
Number of patients eligible for drug under review in the new drug scenarioa | 1,258 / 1,284 / 1,317 |
Market uptake (3 years) | |
Uptake for EDS (reference scenario): Pitolisant hydrochloride Dextroamphetamine sulphate Methylphenidate HCl Modafinil Lisdexamfetamine dimesylate | 0% / 0% / 0% 29.4% / 29.4% / 29.4% 24.3% / 24.3% / 24.3% 43.7% / 43.7% / 43.7% 29.4% / 29.4% / 29.4% |
Uptake for EDS with cataplexy (reference scenario): Pitolisant hydrochloride Dextroamphetamine sulphate + anticataplectic Methylphenidate HCl + anticataplectic Modafinil + anticataplectic Lisdexamfetamine dimesylate + anticataplectic | 0% / 0% / 0% 28.0% / 28.0% / 28.0% 24.0% / 24.0% / 24.0% 45.3% / 45.3% / 45.3% 2.7% / 2.7% / 2.7% / 2.7% |
Uptake for EDS (new drug scenario): Pitolisant hydrochloride Dextroamphetamine sulphate Methylphenidate HCl Modafinil Lisdexamfetamine dimesylate | 1.2% / 3.7% / 7.0% 29.0% / 28.3% / 27.3% 24.0% / 23.4% / 22.6% 43.2% / 42.1% / 40.7% 2.5% / 2.4% / 2.4% |
Uptake for EDS with cataplexy (new drug scenario): Pitolisant hydrochloride Dextroamphetamine sulphate + anticataplectic Methylphenidate HCl + anticataplectic Modafinil + anticataplectic Lisdexamfetamine dimesylate + anticataplectic | 1.8% / 5.8% / 11.0% 26.4% / 24.9% / 27.5% 22.6% / 21.4% / 23.6% 42.7% / 40.3% / 44.5% 2.5% / 2.4% / 2.6% |
Cost of treatment (per patient) | |
Cost of treatment over 1 year for EDS: Pitolisant Dextroamphetamine sulphate Methylphenidate HCl Modafinil Lisdexamfetamine dimesylate | $11,054 $1,160 $374 $417 $2,038 |
Additional cost of anticataplectic treatment over 1 year for patients experiencing EDS with cataplexyb | $251 |
EDS = excessive daytime sleepiness; HCl = hydrochloride.
aIn the new drug scenario, the sponsor applies an estimated growth rate specific to each province in the absence of data on the evolution of narcolepsy in Canada.15
bFor the treatment of narcolepsy with cataplexy, the additional cost of anticataplectic treatment is added to dextroamphetamine sulphate, methylphenidate HCl, modafinil, and lisdexamfetamine dimesylate. The anticataplectic treatment cost is a weighted average of imipramine, desipramine, clomipramine, fluoxetine, venlafaxine, and no anticataplectic treatment based on cost per mg and proportion of use as per clinical expert opinion.1
Summary of the Sponsor’s BIA Results
The sponsor’s estimated budget impact of funding pitolisant hydrochloride for the treatment of EDS in narcolepsy with and without cataplexy in adult patients was $170,782 in year 1, $536,849 in year 2, and $1,043,297 in year 3, for a 3-year total of $1,750,929.
Sensitivity analyses conducted by the sponsor demonstrated that the budget impact of pitolisant hydrochloride is most impacted by changes to market share, prevalence of narcolepsy, and proportion of patients treated for EDS with and without cataplexy.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Anticipated uptake of pitolisant hydrochloride is likely underestimated: The sponsor anticipated a gradual uptake of pitolisant hydrochloride from 1.2% to 7.0% for EDS and from 1.8% to 11.0% for EDS with cataplexy in years 1 to 3 in the new drug scenario. Given the lack of available treatments that are indicated for cataplexy and the potentially favourable safety profile and mechanism of action of pitolisant hydrochloride in comparison to other comparators such as amphetamines, clinical experts noted that the market share estimates for pitolisant hydrochloride were likely underestimated and that pitolisant hydrochloride would likely become a first-line treatment option for narcolepsy. Clinician input indicated that pitolisant hydrochloride uptake would be rapid if it was to be made available, likely taking up to 40% of the market for EDS without cataplexy by year 3. Clinical experts also indicated that pitolisant hydrochloride would likely take up 60% of the market for EDS with cataplexy by year 3.
CADTH increased the market shares of pitolisant hydrochloride for treating EDS to reach 40% by year 3 and 60% for the treatment of EDS with cataplexy by year 3, as anticipated by clinical experts.
Proportion of patients receiving treatment for narcolepsy is likely underestimated: The sponsor estimates only 25% of patients with narcolepsy receive treatment, which likely underestimates the target population eligible for pitolisant hydrochloride. Clinical experts consulted by CADTH expected this number to be closer to 30% due to increased awareness in recent years regarding narcolepsy.
CADTH increased the proportion of patients receiving treatment for narcolepsy to 30% as per clinical expert input.
Lack of clarity regarding treatment discontinuation: Drug plan and clinician input indicated uncertainty in stopping rules due to lack of efficacy or other reasons following treatment with pitolisant hydrochloride. Although there are no clear discontinuation criteria for pitolisant hydrochloride, increases in discontinuation rates would affect the budget impact estimates of pitolisant hydrochloride.
CADTH could not address this limitation in reanalysis.
CADTH Reanalyses of the BIA
The results of the CADTH step-wise reanalysis are presented in summary format in Table 19 and a more detailed breakdown is presented in Table 20. Based on the CADTH base case, the budget impact of the reimbursement of pitolisant hydrochloride for the treatment of EDS in narcolepsy with and without cataplexy in adult patients is expected to be $1,790,647 in year 1, $4,297,152 in year 2, and $6,946,649 in year 3. The 3-year total budget impact for pitolisant hydrochloride is $13,034,448.
Table 18: CADTH Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption | |
|---|---|---|---|
None | — | — | |
Changes to derive the CADTH base case | |||
1. Market shares underestimated for the uptake scenario | EDS without cataplexy: 1.2% / 3.7% / 7.0% EDS with cataplexy: 1.8% / 5.8% / 11.0% | EDS without cataplexy: 10% / 25% / 40% EDS with cataplexy: 20% / 40% / 60% | |
2. Proportion of treated patients underestimated | 25% | 30% | |
CADTH base case | Reanalysis 1 + 2 | ||
EDS = excessive daytime sleepiness.
Table 19: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | 3-year total |
|---|---|
Submitted base case | $1,750,929 |
CADTH reanalysis 1 | $10,862,040 |
CADTH reanalysis 2 | $2,101,115 |
CADTH base case | $13,034,448 |
BIA = budget impact analysis.
Table 20: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | 3-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $1,086,093 | $1,103,282 | $1,120,755 | $1,138,516 | $3,362,554 |
New drug | $1,086,093 | $1,274,064 | $1,657,605 | $2,181,814 | $5,113,483 | |
Budget impact | $0 | $170,782 | $536,849 | $1,043,297 | $1,750,929 | |
CADTH base case | Reference | $1,086,093 | $1,323,939 | $1,344,906 | $1,366,220 | $4,035,065 |
New drug | $1,086,093 | $3,114,586 | $5,642,058 | $8,312,869 | $17,069,513 | |
Budget impact | $0 | $1,790,647 | $4,297,152 | $6,946,649 | $13,034,448 |
BIA = budget impact analysis.
Stakeholder Input
Patient Input
Wake Up Narcolepsy, Inc.
About Wake Up Narcolepsy, Inc.
Wake Up Narcolepsy, Inc. (WUN), established in 2008, is a patient advocacy non-profit organization that aims to accelerate narcolepsy research, increase awareness of narcolepsy and provide supportive services to people with narcolepsy and their loved ones.
Website: https:/www.wakeupnarcolepsy.org
Information Gathering
WUN gathered the perspectives through a survey of nineteen individuals residing in Canada. The respondents either have a diagnosis of narcolepsy, are undiagnosed with the medial disorder living with symptoms and one caregiver. Eleven percent of the respondents are between the ages of 18-24; 33% are between 25-34 years old; 33% are 35-44 years old; 5.5% are between 45-54 years old and 16.5% are 65+. Seventy two percent are female and 28% are male.
Disease Experience
The data showed that the most troubling symptom of narcolepsy is the excessive daytime sleepiness (EDS). On a scale of 1-7, with 7 being “completely bothersome” and 1 being “not at all bothersome”, the highest score was 39% of the respondents rated excessive daytime sleepiness as 6 out of 7 as the most disruptive symptom of narcolepsy.
Twenty-two percent rated it a “4” and “5” on this scale 1-7. The next most disruptive symptom of narcolepsy with this group was “disturbed nocturnal sleep (DNS)” and on the same rating scale 1-7, 16.7% of the respondents rated it a “7”, completely bothersome, while 22% rated it a 6, and 16.7% rated it “4” and “5”. Hallucinations while falling asleep or waking up was the third highest bothersome symptom of narcolepsy. This symptom was rated a “4” by 28% of the respondents. Cataplexy, another common symptom of narcolepsy, was rated a somewhat bothersome symptom.
Lastly, sleep paralysis was also rated somewhat bothersome.
According to the results, the most area of one’s life impacted by narcolepsy is managing the related mental health and emotional symptoms such as mood swings, anger, depression and anxiety. Missing out on social activities, vacations or hobbies with friends and family was reported as second highest impact on the patient’s life. Difficulty keep up with career and job tasks, dependency on others for support for daily activities such as driving, childcare and, finally, keeping up with one’s physical health and wellness (weight gain) were reported as other areas where narcolepsy impact’s one life! Below are details explaining the impact of these areas in the patients’ lives.
Since narcolepsy affects every aspect of one’s life, it has a major impact on the quality of life. Some individuals with narcolepsy may not drive can only drive for very limited periods of time. They claim not to have adequate concentration or focus while doing quiet, sitting activities. Narcolepsy affects social life and respondents claim that they don’t have the stamina to attend social activities in the evenings and often fall asleep during work meetings in the afternoon.
Making and keeping friends, takes time and effort, something that individuals with narcolepsy don’t have in surplus. Some respondents state they are exhausted all the time which affects their mood and they don’t have energy to spend time with their kids or friends. Another respondent states, that they need naps throughout the day and some days at work are highly unproductive. This person feels as though they are constantly being monitored and criticized for their lack of productivity at work. Napping at the workspace is a necessity, but causes embarrassment. This medical disorder severely impacts one’s academic success. Individuals reported that they experienced sleep attacks while writing exams at university and has cause failure in classes. One respondent states that the untreated symptoms of narcolepsy have caused low self-esteem and the failure to compete any post-secondary education. Fear of career advancement is an issue. Individuals state they know they are capable of achieving more in life, but narcolepsy presents too many challenges. Several respondents state that narcolepsy has completely destroyed their lives and they cannot hold a job or attend school. If they do attend school, they have to self-teach due to the frequent sleep attacks during lectures.
Experiences With Currently Available Treatments
Everyone with narcolepsy is on a different regime of medication to treat symptoms. Thirty three percent currently tricyclic antidepressants, SNRIs or SSRIs; 12.5% currently use Provigil/Nuvigil; 55.5% currently use stimulants; 12.5% use sodium oxybates; 50% stated individual drugs that fall into one of the listed categories.
Eleven percent of respondents reported that the physical side effects they experience from current treatments are extremely challenging and 17% report these side effects as moderately challenging; 11% reported mental side effects as extremely challenging and 28% as moderately challenging. Ease of access to the treatment and cost are reported as other challenges experienced by patients. Some respondents must take time off from work as a result of the treatment currently used.
All patients would like to see a more effective drug for both the sleepiness and cataplexy. Frequent dosing is reported as an area for improvement. The patient would like to have less frequent doses with extended release. A drug that helps with the nocturnal disturbances would help patients. Respondents would like a treatment that is easy to swallow, won’t cause nausea and doesn’t affect mood or personality. In the same theme, respondents want a medication that helps them stay awake longer in the day without having to take more or treatments that cause weight gain.
Improved Outcomes
All patients would like to see a more effective drug for both the sleepiness and cataplexy. Frequent dosing is reported as an area for improvement. The patient would like to have less frequent doses with extended release. A drug that helps with the nocturnal disturbances would help patients. Respondents would like a treatment that is easy to swallow, won’t cause nausea and doesn’t affect mood or personality. In the same theme, respondents want a medication that helps them stay awake longer in the day without having to take more or treatments that cause weight gain.
Experience With Drug Under Review
Not applicable.
Companion Diagnostic Test
Not applicable.
Anything Else?
Not applicable.
Conflict of Interest Declaration — Wake Up Narcolepsy, Inc.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
No.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
Table 1: Conflict of Interest Declaration for Wake Up Narcolepsy, Inc.
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Paladin/Endo Labs – for patient education & support (webinar, support groups) | — | X | — | — |
We receive funding from six other corporations/pharmaceuticals companies for educational purposes | — | X | X | X |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.