CADTH Reimbursement Review
Nusinersen (Spinraza)
Sponsor: Biogen Canada Inc.
Therapeutic area: Spinal Muscular Atrophy
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
6MWT
6-minute walk test
ACSA
Anamnestic Comparative Self-Assessment
ADI-12
ALS Depression Inventory 12 Items
AE
adverse event
ALSFRS-R
Amyotrophic Lateral Sclerosis Functional Rating Scale Revised
CDEC
CADTH Canadian Drug Expert Committee
CHOP-ATEND
Children’s Hospital of Philadelphia–Adult Test of Neuromuscular Disorders
CI
confidence interval
CSMAC
Cure SMA Canada
CT
computed tomography
FEV1
forced expiratory volume in 1 second
FSS
Fatigue Severity Scale
FVC
forced vital capacity
HFMSE
Hammersmith Functional Motor Scale Expanded
HRQoL
health-related quality of life
ISMAR
International SMA Registry
IV
intravenous
LOCF
last observation carried forward
MDC
Muscular Dystrophy Canada
MRC
Medical Research Council
QoL
quality of life
RCT
randomized controlled trial
RULM
Revised Upper Limb Module
SAE
serious adverse event
SD
standard deviation
SF-36
Short Form 36 Health Survey
SMA
spinal muscular atrophy
SMAFRS
Spinal Muscular Atrophy Functional Rating Scale
SMN
survival motor neuron
SMN1
survival motor neuron 1 gene
SMN2
survival motor neuron 2 gene
SSQ
Sydney Swallow Questionnaire
WDAE
withdrawal due to adverse event
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Nusinersen (Spinraza), 2.4 mg/mL solution for intrathecal injection |
Indication | Nusinersen is indicated for the treatment of 5q SMA |
Reimbursement request | The sponsor requests that the previous CADTH-recommended criteria (project SR0576 to 000) for nusinersen be expanded to include SMA type II and type III patients older than 18 years, regardless of ambulatory status |
Health Canada approval status | NOC |
Health Canada review pathway | Priority review |
NOC date | June 29, 2017 |
Sponsor | Biogen Canada Inc. |
NOC = Notice of Compliance; SMA = spinal muscular atrophy.
Introduction
Spinal muscular atrophy (SMA) is a severe neuromuscular disease and is the leading genetic cause of infant death.1,2 It is characterized by the degeneration of alpha motor neurons in the anterior horn of the spinal cord, leading to progressive muscle weakness.1 The most common form of SMA, 5q SMA, makes up more than 95% of all cases and is an autosomal recessive disorder caused by homozygous deletion or deletion and mutation of the alleles of the survival motor neuron (SMN) 1 gene (SMN1).3,4 A second survival motor neuron gene (SMN2) acts in a capacity similar to SMN1, but is usually not sufficient on its own to maintain motor neurons. The number of SMN2 genes usually determines the severity of SMA.1-3,5 Genetic testing provides a definitive diagnosis of 5q SMA, and the first step is to test for an SMN1 gene deletion.1
SMA is a rare disease, and estimates of its incidence and prevalence vary in different studies. Currently, the incidence of SMA in Canada is unknown, although it is estimated that SMA affects 1 in every 6,000 to 10,000 live births.4,6-8 However, a recent review reported estimates of 700 to 2,140 active cases of SMA in Canada, with approximately 35 new cases per year.9
SMA presents in various ways, depending on age at onset. Adult-onset SMA presents as mild proximal muscle weakness, and it is more severe in the lower limbs than in the upper limbs.1,2 SMA is divided into 4 clinical subtypes that vary in age at onset, highest motor milestone achieved, and prognosis. Of interest to this review are type II and type III SMA. In SMA type II, age at onset is 6 to 18 months, and patients have delayed motor milestones, respiratory issues, and the possibly of a shortened life expectancy. Patients with SMA type II achieve the milestone of sitting unsupported, but never walk independently.10 Patients with type II SMA make up about 20% to 30% of all SMA cases, and most patients with SMA type II have 3 copies of SMN2.11 Onset of SMA type III occurs in patients from 18 months to 18 years of age. Type III SMA makes up about 10% to 20% of all SMA cases.4 These patients are able to walk independently at some point in their lives and typically have a normal life expectancy.10
In Canada, treatment options for 5q SMA consist of disease-modifying therapies (nusinersen [Spinraza], risdiplam [Evrysdi]), which stimulate the production of the SMN protein, and gene-replacement therapy (onasemnogene abeparvovec), which is a 1-time IV (IV) infusion that replaces missing or faulty SMN1 genes.
Nusinersen is an antisense oligonucleotide that increases the proportion of exon 7 inclusion in SMN2 messenger ribonucleic acid (RNA) transcripts by binding to a specific site in the SMN2 pre-messenger RNA, leading to the translation of the messenger RNA into functional full-length SMN protein.12
The recommended dose of nusinersen is 12 mg, administered by intrathecal injection via lumbar puncture at day 0, day 14, and day 8, and day 63, followed by maintenance doses every 4 months.12
Nusinersen was granted a Health Canada Notice of Compliance for the indication of 5q SMA on June 29, 2017. When nusinersen was initially reviewed by CADTH in 2017, it was recommended for reimbursement for patients with 5q SMA with 2 copies of the SMN2 gene and for those with a disease duration of less than 26 weeks and an onset of clinical signs and symptoms consistent with SMA from 1 week to 7 months of age.13 In 2019, nusinersen was reviewed as a resubmission, and a conditional positive recommendation was granted for patients with 5q SMA with 2 or 3 copies of the SMN2 gene, for patients with a disease duration of less than 6 months and symptom onset from 1 week to 7 months of age, and for patients 12 years and younger with symptom onset after 6 months of age who never achieved the ability to walk independently.14 Across Canada, most provinces and drug plans only reimburse nusinersen if initiated in patients 18 years or younger, with the exception of British Columbia, where nusinersen is reimbursed if initiated in patients 12 years or younger. It should be noted that patients are not expected to stop treatment with nusinersen when they reach 12 or 18 years of age.
The sponsor has submitted nusinersen for reassessment to expand reimbursement conditions to include adults with type II and type III SMA who are older than 18 years, regardless of ambulatory status. The objective of the current reassessment is to perform a systematic review of the beneficial and harmful effects of nusinersen in adults (≥ 18 years of age) with type II or type III 5q SMA.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient and clinician groups that responded to CADTH’s call for input and from clinical experts consulted by CADTH for the purpose of this review.
Patient Input
Patient input for the CADTH reassessment of nusinersen was received from 3 groups — Cure SMA Canada (CSMAC), Muscular Dystrophy Canada (MDC), and the Love for Lewiston Foundation — all of which are registered charities.
Patients who responded to surveys and participated in interviews conducted by CSMAC and MDC noted that as they approached adulthood, they experienced a decline in physical abilities, highlighting that they have lost the ability to, or are just barely able to walk as adults. Along with the loss of gross motor skills, patients noted a significant impact on activities of daily living due to a progressive loss of life skills and overall independence, including loss of the ability to dress themselves, feed themselves, swallow, turn over in bed, and transfer for the purpose of toileting. Additionally, patients reported a lack of energy and loss of strength in their voice, making communication difficult and affecting the ability to maintain employment, and an increase in hospitalizations and need for supportive equipment. The devastation of disease progression and loss of function in patients with full mental capacity has a severe negative impact on mental health and well-being. Patient groups noted that, coupled with the continued inability to access effective treatments, they experienced a significant increase in anxiety, depression, and self-harm, requiring additional mental health support. Last, with the loss of physical function, patients require alterations to their homes for accessibility, which has a considerable financial impact. Patients hope the treatment will stop disease progression and even reverse muscle atrophy, which they consider to be an improvement in terms of quality and quantity of life.
Patient and caregiver responders identified an unmet need for access to treatments in the adult population that offer stability and improved quality of life (QoL) through greater independence, improved strength (primarily in the arms and respiratory function), and halting of progression. With improvements in these facets, patients believe they can achieve greater independence and a better QoL. Patients also noted that some of the largest barriers to treatment and challenges with currently available treatment are unreasonable costs, the mode of delivery with intrathecal therapy, and potential harms.
Given the few options for adults with SMA, treatment is limited to nusinersen, risdiplam, alternative management of the disease, or no treatment. In the CSMAC survey, 41 (47%) patients provided information about their experience with SMA treatments. Of those, 32 (78%) were receiving nusinersen and 9 (22%) were receiving risdiplam. Many of the patients who participated in the CSMAC and MDC surveys and interviews were treatment-naive because of limited access to SMA treatments in Canada. Of the patients receiving nusinersen, 79% reported that they experienced improvements in respiratory function, endurance, upper limb and core strength, and voice, and 15% reported stabilization of their disease. The remaining 6% reported no stabilization or improvement. Patients were receiving treatment with nusinersen for 1 to 3.5 years. Negative experiences reported by patients receiving nusinersen included a wearing-off of treatment and a drop in function shortly before the next maintenance dose; both were rectified after subsequent treatment. Additional negative experience included temporary headaches, discomfort from intrathecal injections, and the travel and time off work required for treatment. Regardless, patients felt that the benefit of nusinersen, including gains in function, improved strength and energy, and disease stabilization, far outweighs the negative aspects of the treatment. Several patients from the MDC interviews revealed that they switched to risdiplam after the initiation of nusinersen because of limited access, financial constraints, and difficulties with intrathecal administration. Patients also reported seeking alternative ways to manage their SMA, such as physiotherapy, exercise, and traditional Chinese medicine.
Clinician Input
Input from Clinical Experts Consulted by CADTH
In adults with type II and III SMA, the clinical-expert panel identified an unmet need for treatments that can change the natural course of the disease, including the ability to reverse the weakness associated with motor neuron degeneration, as there are currently no disease-modifying treatments available for adults. Experts agree that the goals of treatment are dependent on the type of SMA, given the high degree of heterogeneity in the disease and disability, and that treatment should be individualized to specific manifestations of the disease.
Currently, the mainstay of disease-modifying treatment for treatment-naive adults with type II or III SMA is nonpharmacologic, and includes occupational therapy, physiotherapy, and speech-language pathology, which are aimed at supporting function, mobility, and independence, as well as supportive measures, such as ventilation, nutritional assistance, and assistive devices. If nusinersen is recommended for treatment-naive adults with type II or III SMA, the clinical-expert panel noted that it could become a first-line treatment. They also noted that risdiplam has recently been given a positive CADTH Canadian Drug Expert Committee (CDEC) recommendation in younger adults, so the treatment paradigm may shift in the future. As noted by 1 expert, nusinersen has received funding in Quebec for the treatment of adults with type II or type III SMA and is currently being used in this population. Regardless, the clinical experts suggested that for the treatment-naive adult population, there is no guidance on whether other medications should be tried before nusinersen.
The experts highlighted the lack of higher-level evidence (i.e., from randomized controlled trials [RCTs]) in this population to determine which adults with SMA are most likely to respond to treatment with nusinersen. The experts hypothesized that patients with higher functioning and who are ambulatory may demonstrate better responses because they have more nerves, leading to better function. As well, the clinical experts believe that patients without complex spines are more likely to have a better risk-benefit profile. However, most of the experts stressed that the earlier treatment is administered (i.e., in pre-symptomatic children), the greater the benefit observed, although there was some disagreement among panel members about whether to consider age a factor when assessing response. Conversely, the clinical-expert panel noted that patients least suitable for treatment with nusinersen are those with complex spines as a result of spinal fusion surgery, those who cannot tolerate lumbar puncture, and those who have previously been treated as infants or children (there is no evidence of benefit in these patients).
Clinically, numerous outcomes and measures are used to assess response to treatment. The clinical experts agreed that — given the variation in response to treatment and individualized treatment goals — several outcome measures can be used to assess the benefits of treatment, including motor-function and respiratory outcomes and outcomes such as bulbar function, strengthening of speech, and functional independence. The experts noted that in patients with type II or III SMA, disease progression occurs slowly, over the course of years; thus, the impact of treatment on these outcome measures is not likely to be seen over a short period of time.
The clinical-expert panel agreed that the main reasons for discontinuation would be progression or worsening of disease and any major complications or adverse events (AEs) related to therapy. One clinical expert noted that — based on experience with nusinersen in the adult population — the most common reason for discontinuation is a patient’s desire to stop because of lack of improvement or inability to tolerate the treatment. The panel agreed that all patients with SMA should receive care at a tertiary centre that has a variety of neuromuscular specialists, a multidisciplinary team, access to interventional radiology or neurosurgery, and the ability to admit patients who experience potential procedural or treatment-related complications.
Clinician-Group Input
CADTH received clinician-group input from the Neuromuscular Disease Network for Canada, a pan-Canadian network launched in 2020 to bring together clinical, scientific, technical, and patient expertise in neuromuscular disease, with the aim of improving the care, research, and treatment of neuromuscular diseases for all Canadians. Eight clinicians with experience treating SMA patients provided input.
The clinician group highlighted the 3 main disease-modifying treatments for SMA: nusinersen, risdiplam, and onasemnogene abeparvovec. The clinician group agreed that treatment goals for later-onset SMA would be to maintain current levels of motor function and strength, achieve disease stabilization (including the avoidance of ventilation), promote independence, and improve overall health-related quality of life (HRQoL). The clinician group highlighted the fact that risdiplam may be the only other treatment option for these patients. In this case, they noted that either nusinersen or risdiplam could be tried first. The clinician group explained that younger patients are most likely to derive benefit from nusinersen, and noted that it may be difficult to accurately identify which adults are most likely to derive benefit from nusinersen. The clinician group stated that a clinically meaningful response to treatment in adults is likely to consist of stabilization of motor and respiratory function, maintenance of independence, and a reduction in hospitalizations. Moreover, they noted that maintaining the ability to speak and avoiding the need for ventilatory support have profound impacts on patient QoL, autonomy, and the ability to maintain vocational and social roles. The clinician group emphasized that current provincial monitoring requirements are too frequent and there is significant variation between provinces. They agreed that patients should be assessed at treatment initiation, at 6 months, and annually thereafter to reduce patient burden and strain on health care resources, given the slow progression in functional decline. Last, the clinician group noted that nusinersen must be administered by or under the direction of health care providers experienced in performing lumbar puncture at designated treatment centres.
Drug-Program Input
The drug programs identified the following jurisdictional implementation issues: relevant comparators, considerations for initiation of therapy, considerations for continuation or renewal of therapy, considerations for discontinuation of therapy, generalizability, and system and economic issues. The clinical experts consulted by CADTH weighed evidence from the key studies submitted by the sponsor and clinical expertise to provide responses to drug-program implementation questions. Refer to Table 3 for more details.
Clinical Evidence
Pivotal Studies and Protocol Selected Studies
Description of Studies
As part of the reassessment for nusinersen, the sponsor provided CADTH with 5 observational studies, 1 open-label extension study, and 1 critical review and meta-analysis. Four studies were included in the report15-19 and the critical review and meta-analysis was summarized in the Other Relevant Evidence section.20 The other 2 studies described in the report were considered outside the scope of this review and not discussed any further.21,22
The study by Hagenacker et al. (2020)17 was a prospective, German, multi-centre, noncomparative observational study that evaluated the safety and effectiveness of nusinersen in 124 adults with 5q SMA. The study by Maggi et al. (2020)18 was an Italian, retrospective, noncomparative cohort study that evaluated the safety and effectiveness of nusinersen in 116 patients with type II or type III SMA. The EU registry study15,16 was an observational, registry-based cohort study of combined data that evaluated the safety and effectiveness of nusinersen in 252 adults with 5q SMA from 3 prospective and retrospective European registries (SMArtCARE, CuidAME, and the International SMA Registry [ISMAR]) in 2 subcohorts: a before-and-after treatment comparison with nusinersen in 74 patients with type III SMA and 1 patient with type IV SMA; and a comparative dataset from 252 adults with type III SMA (235 who had been treated with nusinersen and 17 who had not). The study by Pera et al. (2021)19 was an observational, noncomparative, registry-based study from ISMAR in Italy of 144 ambulant and nonambulant type III SMA patients treated with nusinersen.
All studies included treatment-naive adults with SMA. Across studies, most patients had type III SMA (62% to 100%), with mainly 3 or 4 copies of SMN2. Type II SMA was infrequently reported, with only 45 and 13 type II patients in Hagenacker et al. (2020)17 and Maggi et al. (2020),18 respectively. No type II patients were included in the EU registry study15,16 or in the study by Pera et al. (2021).19 Across studies, 37% to 56% of patients were considered ambulatory. Baseline motor-function scores were high, with mean Hammersmith Functional Motor Scale Expanded (HFMSE) scores at baseline ranging from 20.74 to 30.75, mean Revised Upper Limb Module (RULM) scores at baseline ranging from 20.87 to 27.57, and mean 6-minute walk test (6MWT) distance at baseline ranging from 300.87 m to 323.03 m.15-19
Effectiveness Results
HFMSE
In Hagenacker et al. (2020),17 the mean change from baseline in HFMSE scores was 1.73 points (95% confidence interval [CI], 1.05 to 2.41) at 6 months (n = 124), 2.58 points (95% CI, 1.76 to 3.39) at 10 months (n = 92), and 3.12 points (95% CI, 2.06 to 4.19) at 14 months (n = 57). The proportion of patients with an increase of 3 points in HFMSE score were 28%, 35%, and 40% at 6 months, 10 months, and 14 months, respectively. In Maggi et al. (2020),18 the mean change from baseline in HFMSE scores was 1.48 points (standard deviation [SD] = 2.28), 2.44 points (SD = 2.8), and 2.85 points (SD = 2.93) at 6 months (n = 103), 10 months (n = 75), and 14 months (n = 46), respectively, for all type III SMA patients. In all SMA patients at 6, 10, and 14 months, increases of 3 or more points in HFMSE score occurred in 28%, 38%, and 49% of patients, respectively. In the EU registry study,15,16 the slope for the change in HFMSE score per week was –0.00006 points per week (95% CI, –0.00955 to 0.009428) before treatment with nusinersen, and was 0.2575 points per week (95% CI, 0.01038 to 0.04112) after treatment with nusinersen (n = 75). In the analysis comparing nusinersen-treated patients with untreated patients, the slope for the change in HFMSE score was 0.02907 points per week (95% CI, 0.01930 to –0.03884) in nusinersen-treated patients (n = 235), compared with −0.01129 points per week (95% CI, −0.03289 to 0.01031) in untreated patients (n = 17).15,16 In the study by Pera et al. (2021),19 at 12 months, the mean change from baseline in HFMSE was 0.79 points (95% CI, –0.29 to 1.87) (n = 45), with the HFMSE results showing a decline in 11.1% of patients, stability in 53.3% of patients, and improvement in 35.6% of patients.
RULM
In Hagenacker et al. (2020),17 the mean change from baseline in RULM scores was 0.66 points (95% CI, 0.26 to 1.05) at 6 months (n = 120), 0.59 points (95% CI, 0.15 to 1.03) at 10 months (n = 90), and 1.09 points (95% CI, 0.62 to 1.55) at 14 months (n = 58). An increase of at least 2 points in RULM score was observed in 28 (23%) patients at 6 months, whereas 74 (64%) showed no meaningful change and 28 (23%) showed a decline. In Maggi et al. (2020),18 the mean change from baseline in RULM scores was 0.31 points (SD = 1.97), 0.61 points (SD = 2.08), and 0.86 points (SD = 2.18) at 6 months (n = 102), 10 months (n = 71), and 14 months (n = 44), respectively, for all type III SMA patients. Patients with type II SMA had a numerically greater change in mean RULM scores than those with type III with scores, at 0.8 points (SD = 1.95) at 6 months (n = 12), 1.67 points (SD = 1.8) at 10 months (n = 9), and 1.6 points (SD = 1.52) at 14 months (n = 5). A 2-point change in RULM score in all SMA patients at 6, 10, and 14 months was shown in 21%, 28%, and 35% of patients, respectively. In the EU registry study,15,16 the slope for the change in RULM score was –0.00745 points per week (95% CI, –0.01401 to 0.0009) before treatment with nusinersen, and was 0.002569 points per week (95% CI, −0.00533 to 0.01047) after treatment with nusinersen (n = 75). In the analysis comparing nusinersen-treated with untreated patients, the slope for the change in RULM score was 0.01168 points per week (95% CI, 0.004957 to 0.01841) in nusinersen-treated patients (n = 235), compared with 0.003936 points per week (95% CI, −0.01030 to 0.01817) in untreated patients (n = 17). In the study by Pera et al. (2021), the mean change from baseline in RULM at 12 months was 0.07 points (95% CI, –0.48 to 0.63) (n = 55), with the RULM results showing a decline in 13.0% of patients, stability in 75.9% of patients, and improvement in 15.6% of patients.19
6MWT
In Hagenacker et al. (2020),17 the mean change from baseline in 6MWT was 22.12 m (95% CI, 8.7 to 35.6) at 6 months (n = 47), 31.14 m (95% CI, 15.2 to 47.1) at 10 months (n = 37), and 45.96 m (95% CI, 25.4 to 66.6) at 14 months (n = 25). In Maggi et al. (2020),18 change from baseline in 6MWT was only available for type III SMA walkers, who demonstrated a mean change in 6MWT of 14.66 m (SD = 27.57) at 6 months (n = 48), 26.45 m (SD = 34.6) at 10 months (n = 35), and 23.11 m (SD = 51.2) at 14 months (n = 24). The proportion of patients that achieved a minimum 30-metre improvement in 6MWT was 29% at 6 months, 46% at 10 months, and 42% at 14 months. In the EU registry study,15,16 the slope for the change in 6MWT was –0.03399 m per week (95% CI, –0.4373 to 0.3694) after treatment with nusinersen (n = 75). In the analysis comparing nusinersen-treated with untreated patients, the slope for the change in 6MWT score was 0.2633 m per week (95% CI, 0.09922 to 0.42740) in nusinersen-treated patients (n = 235), compared with –0.7148 m per week (95% CI, –1.2789 to –0.1506) in untreated patients (n = 17). Mean change from baseline in 6MWT for adults in Pera et al. (2021)19 at 12 months was 0.52 m (95% CI, –19.85 to 20.89) (n = 17).
Other Effectiveness Outcomes
Respiratory outcomes of forced vital capacity (FVC)/forced expiratory volume in 1 second (FEV1) were only evaluated in Maggi et al. (2020),18 with a mean change in percent-predicted FVC from baseline for all SMA type III patients at 14 months of 6.47% (SD = 9.22). Mean change in percent-predicted FVC at 14 months was not available for patients with type II SMA because of sample-size constraints. The mean change from baseline at 14 months in percent-predicted FEV1 was 5.86% (SD = 9.22) for all type III SMA patients. The mean change in percent-predicted FEV1 at 14 months was not available for type II SMA patients.
Other outcomes of interest to this review, including bulbar function, survival, hospitalization, HRQoL, anatomic-related outcomes, and caregiver burden, were not assessed in the included studies.
Harms Results
Harms were infrequently reported in the included studies, with all but 1 study (Pera et al. [2021]19) reporting the frequency of harms. When reported, the frequency of AEs in the included studies ranged from 30.0% to 41.4% and were considered mild to moderate by the investigators. The frequency of serious adverse events (SAEs) was low in all studies, when reported.
In Hagenacker et al. (2020),17 2 patients withdrew from treatment at 10 months because of adverse drug reactions. In Maggi et al. (2020),18 nusinersen treatment was stopped in 2 (1.7%) type III SMA patients after 6 months because of a lack of subjective benefit and poor tolerability of repeated lumbar puncture. Withdrawals due to AEs (WDAEs) were not reported in the EU registry study15,16 or in the study by Pera et al. (2021).19
The most frequently reported notable harms of interest to this review were lumbar puncture-related AEs; however, these were not reported in the EU registry study15,16 or in the study by Pera et al. (2021).19 Post-procedural complications of headache (35% and 37.1%) and back pain (22% and 8.6%) were the most frequently reported AEs in the Hagenacker et al. (2020)17 and Maggi et al. (2020)18 studies, respectively. The frequency of headache and back pain was not reported in the study by Pera et al. (2021),19 but they were noted as the most frequently occurring AEs. Frequency of other notable harms, including serious infections, renal toxicities, and coagulation abnormalities, was infrequently reported in the included studies.
No deaths were noted in any of the studies.
Critical Appraisal
No RCTs focusing on treatment-naive, adult, type II or III SMA patients were identified as part of the CADTH literature search, and all available and included studies were of observational design, focusing on real-world data, which have more limitations than RCTs.
The studies included in this reassessment are associated with lower internal validity because of the limitations in design, enrolled patient populations, and statistical analyses. The included studies shared a common limitation pertaining to the study design: they were noncomparative, so the results observed cannot be attributed to treatment with nusinersen. However, the EU registry study15,16 included an untreated comparison group, albeit with a sample size of 17 patients. The studies included in this reassessment also suffer from a high level of selection bias, reporting bias, and information bias. In all studies, patients with SMA had to be able to complete at least 6 months of treatment with nusinersen, which selected for patients who were able to complete the induction dosing and who were able to tolerate and/or receive doses. Moreover, included patients were mostly SMA type III, with a seemingly higher functional status at baseline, according to ambulatory status and baseline motor scale scores. No or limited techniques were used to adjust for potential selection biases across studies. In all studies, important potential confounders and treatment-effect modifiers that were not identified or considered, which may have influenced the results, include training for the outcomes of interest, routine exercise and close observation, other routine care (such as physiotherapy and occupational therapy), as well as the placebo effect, and the extent to which uncontrolled confounders and treatment-effect modifiers influenced the results is unclear. In all studies, no protocol was identified, and it was not possible to determine whether sample sizes (ranging from 67 to 252 patients) were appropriate for the research question and objectives of each study. The EU registry study15,16 conducted analyses on 2 groups: the first consisted of 235 nusinersen-treated patients and 17 untreated patients; and the second consisted of 75 patients assessed before and after treatment. Because of the limited population in the untreated group, the results observed could not be attributed to treatment with nusinersen. The proportion of patients lost to follow-up was infrequently reported in the included studies, although there was a notable proportion of patients with a lack of longer-term follow-up at 14 months, compared with earlier times of assessment. With the exception of the EU registry study,15,16 no imputation of missing data was conducted, so missing data affected the validity of the results.
As previously mentioned, selection bias in the included populations was noted as a key limitation. Patients enrolled in the included studies consisted of mainly type III SMA (62.0% to 100.0%), with few type II patients (11.2% to 36.0%), which was noted by the clinical experts to be higher than what they see in clinical practice. The SMA patients included in the 4 studies were considered to be higher functioning, based on the high prevalence of type III disease, with most patients having 3 or 4 copies of SMN2, and the proportion of ambulatory patients (37.0% to 56.03%). Moreover, baseline motor-function scores were considered high, suggesting a population with less severe disease. As such, the included study populations were unrepresentative of the reimbursement request (lack of type II SMA patients), and the results may not be generalizable to adults with type II or III SMA in Canada. Given that up to half of all patients across studies were nonambulatory, the HFMSE and 6MWT may not be appropriate outcome measures in all patients, the clinical experts report, which further limits the evaluable population and the generalizability of the results. HRQoL and other patient-reported outcomes, which were outcomes important to patients, were not assessed in the included studies; therefore, the effect of nusinersen with respect to these outcomes remains unknown. The maximum follow-up time across studies was 14 months, which was considered insufficient to assess any clinically meaningful change in outcomes in adults with type II or III SMA because of the slowly progressing nature of the disease, as well as its natural history.
Indirect Comparisons
No indirect evidence was included in the sponsor’s submission to CADTH or identified in the literature search that matched the inclusion and exclusion criteria of this review.
Other Relevant Evidence
Other Sponsor Submitted Evidence
As part of the reassessment for nusinersen, the sponsor submitted a publicly available critical review and meta-analysis of patients with type II and III SMA. The objective of the meta-analysis by Coratti et al. (2021)20 was to critically review literature that reported real-world data on motor function in type II and III SMA patients treated with nusinersen to establish possible patterns of efficacy by subdividing the results by SMA type, age (children versus adults), and type of assessment. Only results related to the adult population with type II or III SMA were of interest in this reassessment.
The meta-analysis was informed by a systematic review of existing literature. A total of 14,627 articles were identified. After study selection, 19 papers reporting data on efficacy in nusinersen-treated and untreated patients using structured assessments in type II and III SMA were selected. Pooled analyses were conducted at multiple levels. First, a rough evaluation on the overall benefit of treatment versus no treatment was run that included the largest available evidence, even if heterogeneous. Next, the effect size was estimated using random-effect models, and heterogeneity among studies was quantified by the I2 coefficient. Then, meta-regression analysis was undertaken to identify possible sources of heterogeneity among studies. Motor-function outcomes included HFMSE, RULM, 6MWT, Medical Research Council (MRC) scale for muscle strength, and Children’s Hospital of Philadelphia–Adult Test of Neuromuscular Disorders (CHOP-ATEND).20 Meta-regression was not conducted for the MRC and CHOP-ATEND outcomes and are not summarized. Meta-regression analyses were employed with a random-effects model using aggregate-level data. Only studies with complete data available (sample size, mean, SD, or 95% CI) were included in the meta-analysis.20
In the meta-regression analysis, pooled mean changes from baseline in HFMSE score, RULM score, and 6MWT in the adult population were 1.87 points (95% CI, 1.05 to 2.68), 0.64 points (95% CI, 0.27 to 1.01), and 20.28 m (95% CI 1.17 to 39.40), respectively.20
The meta-analysis was based on an adequately conducted and reproducible systematic literature search. It was unclear if the inclusion and exclusion criteria for population, outcomes, and study design were pre-specified. A quality assessment of the included studies was conducted using the Risk of Bias Assessment Tool for Nonrandomized Studies). No interpretation on the quality of studies was conducted; however, as all studies were observational, most studies were considered to suffer from a high level of bias in the selection of participants. All publicly available studies summarized in the review conducted by CADTH (Hagenacker et al. [2020],17 Maggi et al. [2020],18 and Pera et al. [2021]19) were included in the submitted meta-analysis. Outcomes included in the meta-analysis were appropriate and relevant to the Canadian context, with HFMSE, RULM, and 6MWT most commonly included in studies, although there were differences in reporting and time of assessment. Most of the included studies had a follow-up time ranging from 10 to 14 months, which was considered by the clinical experts consulted by CADTH to be insufficient to observe clinically meaningful changes in the motor function of adults. There was considerable heterogeneity in the studies, given the inclusion of both ambulant and nonambulant type II and type III SMA patients, as well as the inclusion of both adults and children. Additionally, the included studies had small sample sizes, limiting the conclusions that can be drawn from individual studies. Overall, there was a moderate to considerable level of heterogeneity in the included studies across outcomes, with I2 values ranging from 43% to 71%. Pooled estimates of mean change for motor-function outcomes favoured nusinersen treatment in the adult population; however, the pooled estimates generally displayed wide 95% CIs, particularly for the 6MWT, and in many cases crossed the zero meridian, indicating a high level of variability in these cohorts and substantial imprecision in estimates of treatment effect. Additionally, the change from baseline in motor-function outcomes was minor, and in discussion with the clinical experts, there is uncertainty about what constitutes a clinically meaningful change in the adult population for these outcome measures. Moreover, given the nature of the included studies and the limitations defined for the studies included in both the systematic review conducted by CADTH and the meta-analysis, the observed effects cannot be attributed to nusinersen.
Evidence Identified From the Literature
Eight non-comparative, observational studies were identified in the literature search that met all inclusion criteria of the systematic review, with the exception of study design, as they consisted of descriptive observational studies. As with the studies provided by the sponsor, the effectiveness of nusinersen in these studies is highly uncertain due to the noncomparative study design, selection bias, and relatively small sample sizes of adults with type II and III SMA.
Conclusions
Four noncomparative, observational studies were included in the reassessment for nusinersen in adults with type II or III SMA. The observational nature and lack of a well-defined concurrent comparator in the included studies significantly limits the ability to establish causal relationships in treatment effect with nusinersen.
In all of the studies, selection bias in the included populations and relatively small sample sizes were noted as key limitations. All studies included mostly type III SMA adults with higher physical functioning at baseline because of their SMA type, number of SMN2 copies, ambulatory status, and higher baseline scores for motor-function outcomes. Input from clinical experts noted the populations were not reflective of the reimbursement request, particularly due to the lack of type II SMA patients, or their clinical practice.
Although there was a generally consistent positive effect of nusinersen on motor-function outcomes, the magnitude of the treatment effect with nusinersen was variable and often not clinically meaningful and, given the limitations of these studies in study design, statistical analysis, duration, and the heterogeneous natural history of adults with SMA, results in all studies were considered highly uncertain and may not be generalizable to the Canadian population. Harms associated with nusinersen were generally mild to moderate in severity, and AEs related to the lumbar puncture procedure were the most frequently reported. However, the reporting of AEs was inconsistent and infrequent, and based on the study design and associated biases, may be under-reported.
Although the amount of real-world data for nusinersen is relatively high, the overall quality of studies remained a concern. Most of the identified evidence could not provide conclusive evidence demonstrating the effectiveness of nusinersen in adults with type II or III SMA. Overall, it remains unclear if nusinersen resulted in clinically meaningful improvements or disease stabilization, which were considered important outcomes to patients. Additionally, since HRQoL was not assessed in any of the studies, the effect of nusinersen on this important outcome in adults is unknown.
Introduction
Disease Background
SMA is a severe neuromuscular disease and is the leading genetic cause of infant death.1,2 It is characterized by the degeneration of alpha motor neurons in the anterior horn of the spinal cord, leading to progressive muscle weakness.1 The most common form of SMA, 5q SMA, makes up more than 95% of all cases and is an autosomal recessive disorder caused by homozygous deletion or deletion and mutation of the alleles of the survival motor neuron (SMN) 1 gene (SMN1).3,4 Whereas deletion or mutation of the SMN1 gene results in SMN protein deficiency, a second nearby gene, the SMN2 gene, produces a small amount of functional SMN protein. The number of available SMN2 gene copies and the extent of the expression of these genes modulates the severity of the disease.1-3,5
SMA is a rare disease, and estimates of its incidence and prevalence vary between studies. Currently, the incidence of SMA in Canada is unknown, although it is estimated that SMA occurs in 1 of every 6,000 to 10,000 live births.4,6-8 One study that examined the Cure SMA database (a voluntary registry that is 1 of the largest patient-reported repositories in the world) reported the birth prevalence in the US at about 1 in 20,000 live births.23 A recent review reported estimates of 700 to 2,140 active cases of SMA in Canada, with approximately 35 new cases per year.9
SMA presents in various ways, depending on age of onset. Infants present with severe hypotonia and feeding difficulties, whereas later onset in young children may present as difficulty with stairs and frequent falls.10 Adult-onset SMA presents as mild proximal muscle weakness.2 Genetic testing gives a definitive diagnosis for 5q SMA. The first step is to test for SMN1 gene deletion.1 In 2020, the Government of Canada and Muscular Dystrophy Canada, in collaboration with Novartis Canada, developed a National Newborn Screening in SMA program to help improve early diagnosis and timely access to treatment for infants born with SMA.24
Deficiency in SMN results in defects in multiple components of the motor system, including the motor neurons.2 Electrophysiological studies and clinical findings in patients with SMA show that patients typically experience a sharp decline in motor function, with motor-unit loss soon after symptom onset, followed by a long plateau period of relative stability in motor function.2,25 Clinical-expert input indicated to CADTH that motor-function decline is irreversible, aside from possible gains in strength and gross motor abilities in infants still undergoing normal muscle hypertrophy in the first 2 years of life. Muscle weakness tends to be symmetric, proximal rather than distal, and more severe in the lower limbs than in the upper limbs.1
SMA is divided into 4 clinical subtypes that vary in age of onset, highest motor milestone achieved, and prognosis. Although the subtypes provide a convenient means of classifying patients, it should be noted that patients exist along a continuum of disease severity, with overlap of symptoms between subtypes. This spectrum is represented in Figure 1.
Figure 1: Continuous Spectrum of SMA Phenotype
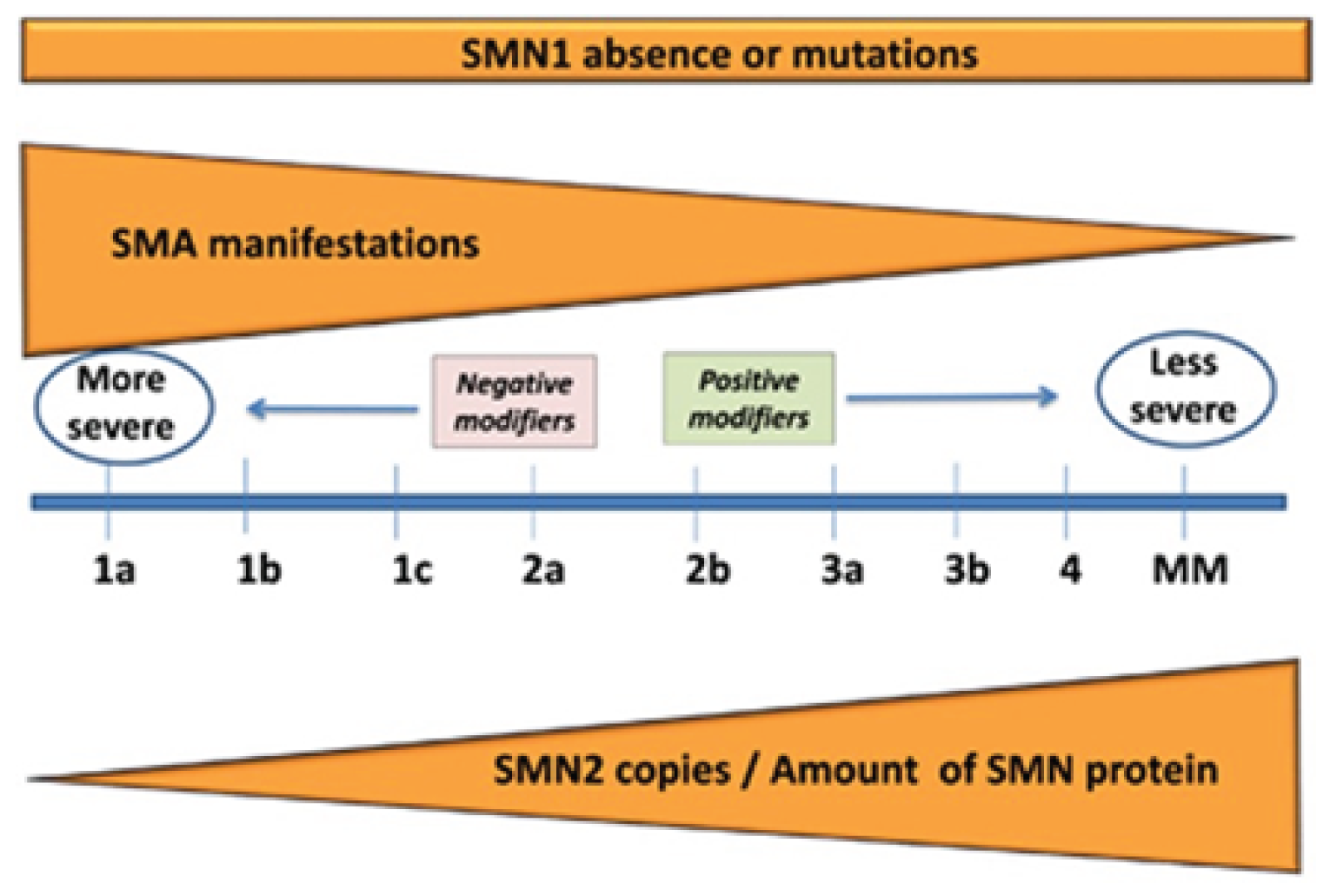
MM = minimal manifestations; SMA = spinal muscular atrophy; SMN = survival motor neuron.
Source: Talbot K, Tizzano EF. The clinical landscape for SMA in a new therapeutic era. Gene Ther. 2017; 24:529 to 533. Licensed under: https://creativecommons.org/licenses/by-nc-nd/4.0/.
Type I SMA presents by the age of 6 months and is the most common genetic cause of infant mortality. These patients never achieve the motor milestone of sitting unsupported, and generally do not survive past 2 years of age, owing to respiratory failure.1-3 Almost all patients with SMA type I have 2 or 3 copies of SMN2, giving rise to a broad range of phenotypes.11
In SMA type II, age of onset is 6 to 18 months and patients have delayed motor milestones, respiratory issues, and the possibly of a shortened life expectancy. Patients with type II SMA achieve the milestone of sitting unsupported, but never walk independently. Symptoms generally appear 6 to 18 months after birth, and most patients survive past the age of 25,5,10 with life expectancy improved by aggressive supportive care.10 Patients with type II SMA represent about 20% to 30% of SMA cases, and most patients with SMA type II have 3 copies of SMN2.11 In addition to the inability to walk independently, common symptoms are fine tremors of the upper extremities, tongue fasciculation, difficulty swallowing, joint contractures, and scoliosis.1 Scoliosis and weak intercostal muscles can cause restrictive lung disease.3 There is a range in severity, and weaker patients require noninvasive ventilation.1 Difficulty swallowing is less common than in patients with type I SMA, and difficulty with feeding comes from masticatory muscle weakness. In a study that examined 1,966 patients in the Cure SMA database (with data available from 2010 to 2016), median survival for those with type II SMA was 59.9 years.23
Patients with SMA type III have an age of onset from 18 months to 18 years and experience muscle weakness. Type III SMA makes up about 10% to 20% of SMA cases.4 These patients are able to walk independently at some point in their lives and typically have a normal life expectancy.10 Most patients with type III SMA have 3 or 4 copies of SMN2.11 Patients with SMA type III have little or no respiratory weakness.3 Ambulatory patients may exhibit abnormal gait characteristics due to proximal weakness,10 and patients who lose the ability to walk often develop scoliosis.1
SMA type IV is a very rare form of adult-onset SMA with various degrees of muscle weakness. These patients retain the ability to walk, have a normal life expectancy, and do not suffer from respiratory or nutritional issues.1 Common to all types of SMA is a progressive decline in muscle function.
Standards of Therapy
There is no cure for SMA, and treatment options are dependent on SMA type and extent of symptoms. Treatment options for 5q SMA available in Canada consist of disease-modifying therapies (nusinersen, risdiplam), which stimulate the production of the SMN protein, and gene-replacement therapy (onasemnogene abeparvovec), which is a 1-time IV (IV) infusion that replaces missing or faulty SMN1 genes.
In addition to treatment with disease-modifying therapies, current standards of practice involve clinical monitoring and surveillance, anticipatory management of symptoms, and attempts to improve overall QoL. SMA patients are monitored for growth, gastrointestinal function, nutrition, respiratory complications, and orthopedic complications (i.e., scoliosis and/or contractures). These standards of practice include respiratory management, secretion mobilization in patients with weak cough, management of swallowing difficulties, and various multidisciplinary strategies to manage gross motor functions and spinal deformities (e.g., physiotherapeutic, orthopedic, surgical).
Drug
Nusinersen is an antisense oligonucleotide that increases the proportion of exon 7 inclusion in SMN2 messenger RNA transcripts made, through binding to a specific site in the SMN2 pre-messenger RNA. This leads to the translation of the messenger RNA into functional full-length SMN protein.12
Nusinersen 2.4 mg/mL solution is administered via intrathecal injection by lumbar puncture. The recommended dose of nusinersen is 12 mg in a 5 mL solution, administered as nusinersen sodium.
It is administered in a regimen of 4 loading doses, with the first 3 administered at 14-day intervals (day 0, day 14, and day 28), with the fourth loading dose approximately 30 days after the third loading dose (day 63). After the fourth loading dose, a maintenance dose should be administered once every 4 months.12
Nusinersen was granted a Health Canada Notice of Compliance for the indication of 5q SMA on June 29, 2017. Nusinersen was initially reviewed by CADTH in 2017, and was recommended for reimbursement for patients with 5q SMA with 2 copies of the SMN2 gene and for those with a disease duration of less than 26 weeks and an onset of clinical signs and symptoms consistent with SMA from 1 week to 7 months of age.13 In 2019, nusinersen was reviewed as a resubmission, and a conditional positive recommendation was granted for patients with 5q SMA with 2 or 3 copies of the SMN2 gene, for patients with a disease duration of less than 6 months and symptom onset from week 1 to 7 months of age, and for patients 12 years or younger with symptom onset after 6 months of age who never achieved the ability to walk independently.14 Across Canada, most provinces and drug plans only reimburse nusinersen if initiated in patients 18 years or younger, with the exception of British Columbia, where reimbursement for nusinersen is only provided when initiated in patients 12 years or younger. It should be noted that patients are not expected to stop treatment with nusinersen upon reaching 12 or 18 years of age.
As part of the reassessment for nusinersen, the sponsor is requesting that the reimbursement criteria for nusinersen be expanded to include adults (> 18 years) with type II or type III SMA, regardless of ambulatory status.
Recently, the European Medicines Agency recognized real-world clinical findings that support the use of nusinersen for stabilization or improvement in motor function for some adults with type II and III SMA.26
The characteristics of treatments for 5q SMA available in Canada are summarized in Table 2.
Table 2: Key Characteristics of Nusinersen and Risdiplam
Characteristics | Nusinersen (Spinraza) | Risdiplam (Evrysdi) |
|---|---|---|
Mechanism of action | Nusinersen is an ASO that binds to a specific site in the SMN2 pre-mRNA to increase the proportion of exon 7 inclusion in SMN2 mRNA transcripts, increasing functional SMN protein levels | Pre-mRNA splicing modifier of SMN2, shifting the balance from exon 7 exclusion to exon 7 inclusion in the mRNA transcript, leading to increased production in functional and stable SMN protein |
Indicationa | Nusinersen is indicated for the treatment of 5q SMA | Risdiplam is indicated for the treatment of SMA in patients 2 months and older |
Route of administration | Intrathecal injection via lumbar puncture | Powder for oral solution |
Recommended dose | 12 mg (5 mL) in 4 loading doses (day 0, day 14, day 28, day 63), followed by a maintenance dose every 4 months | The recommended once-daily dose of risdiplam is dependent on age and body weight:
|
SAEs or safety issues |
|
|
ASO = antisense oligonucleotide; mRNA = messenger ribonucleic acid; SAE = serious adverse effects; SMA = spinal muscular atrophy; SMN = survival motor neuron.
aHealth Canada–approved indication.
Sources: Nusinersen product monograph,12 Risdiplam product monograph.27
Stakeholder Perspectives
Patient-Group Input
This section was prepared by CADTH staff based on the input provided by patient groups.
Patient input for the CADTH reassessment of nusinersen was received from 3 groups — CSMAC, MDC, and the Love for Lewiston Foundation — all of which are registered charities. The information provided by CSMAC was collected with an online survey conducted in November 2020 that consisted of open-ended questions, rating scales, and forced-choice options, and semi-structured interviews conducted in December 2021 with 88 adults from Ontario (35%), Quebec (24%), British Columbia (19%), Alberta (12%), Saskatchewan (4%), Manitoba (4%), New Brunswick (1%), and Nova Scotia (1%); in addition, 1 respondent resided outside of Canada. The information provided by MDC was gathered through a health care experience survey conducted by MDC neuromuscular service support staff who completed semi-structured virtual interviews (e.g., phone or Zoom sessions) from December 2021 to January 4, 2022 with 60 adults with SMA; 20 participants resided in Quebec and the rest resided in Alberta, British Columbia, Manitoba, New Brunswick, Nova Scotia, Ontario, Prince Edward Island, and Saskatchewan. The information provided by the Love for Lewiston Foundation, which works to identify SMA individuals and families that require funding due lack of support and adequate resources, highlights personal experiences and opinions of families affected by SMA.
Respondents to the CSMAC and MDC surveys and interviews noted that as they approached adulthood, they experienced a decline in physical abilities, highlighted by a complete or partial loss in the ability to walk. Along with the loss of gross motor skills, patients noted a significant impact on activities of daily living due to a progressive loss of life skills and overall independence, including a loss of the ability to dress themselves, feed themselves, swallow, turn over in bed, and transfer for the purpose of toileting. Additionally, patients reported a lack of energy and a loss the strength in their voice, making communication difficult and affecting their ability to maintain employment, and an increase in hospitalizations and the need for supportive equipment. The devastation of disease progression and loss of function in a person with full mental capacity has a severe negative impact on mental health and well-being. Patient groups noted that, coupled with the continued inability to access effective treatments, these patients experience a significant increase in anxiety, depression, and self-harm, requiring additional mental health support. Last, with the loss of physical function, patients must alter their homes for accessibility, which has a considerable financial impact. Patients hope the treatment will stop disease progression and even reverse muscle atrophy, which they consider is an improvement in terms of quality and quantity of life.
Patient and caregiver responders identified an unmet need for treatments in the adult population that offer stability and improved QoL through greater independence, improved strength (primarily in the arms and respiratory function), and a halting of progression. Patients believe, with improvements in these facets, they can achieve greater independence and a better QoL. Patients also noted that some of the largest barriers to treatment and challenges with currently available treatment are the unreasonable costs, the mode of delivery with intrathecal therapy, and the potential harms of treatment.
Given the few treatment options available for adults with SMA, experience with treatment was limited to nusinersen, risdiplam, alternative management of the disease, or no treatment. In the CSMAC survey, 41 (47%) patients provided information about their experience with SMA treatments; of those, 32 (78%) were receiving nusinersen and 9 (22%) were receiving risdiplam. Many of the patients from the CSMAC and MDC surveys and interviews were treatment-naive as a result of limited access to SMA treatments in Canada. Of the patients receiving nusinersen, 79% reported that they experienced improved respiratory function, endurance, upper limb and core strengths, and voice, with 15% reporting stabilization of their disease. The remaining 6% reported no stabilization or improvement. Patients were receiving treatment with nusinersen for 1 to 3.5 years. Negative experiences reported by patients receiving nusinersen included a wearing-off of treatment and a drop in function shortly before the next maintenance dose, which was subsequently rectified after treatment. Additional negative experience included temporary headaches, discomfort from intrathecal injections, as well as the travel and time off work required to receive treatment. Regardless, patients felt that the benefit of nusinersen, including gains in function, improved strength and energy, and disease stabilization, far outweighs the negative aspects of this treatment. Several patients from the MDC interviews revealed that they switched to risdiplam after the initiation of nusinersen because of limited access, financial constraints, and difficulties with the intrathecal administration. Patients also reported seeking alternative ways to manage their SMA, such as physiotherapy, exercise, and traditional Chinese medicine.
Clinician Input
Input From Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, and providing guidance on the potential place in therapy). In addition, as part of the nusinersen review, a panel of 4 clinical experts from across Canada was convened to characterize unmet therapeutic needs, assist in identifying and communicating situations where there are gaps in the evidence that could be addressed with the collection of additional data, promote the early identification of potential implementation challenges, gain further insight into the clinical management of patients living with a condition, and explore the potential place in therapy of the drug (e.g., potential reimbursement conditions). A summary of this panel discussion follows.
Unmet Needs
All the clinical experts agree that the main limitation and unmet need in adult type II and type III SMA patients is that there are no disease-modifying treatments available that can change the natural course of the disease, such as the ability to reverse the weakness associated with motor neuron degeneration and, as such, cannot address key outcomes for these patients. Currently, only nonpharmacologic treatments are mostly available in Canada, and are aimed at supporting function, mobility, and independence. Supportive measures, including ventilation and nutritional assistance, can help prolong survival, and assistive devices can help manage weakness; however, the effect is limited and there is a need for treatments that go beyond the benefits provided by the current standard of care.
The clinical experts noted the high degree of heterogeneity in the disease and disability. They agreed that the goals of treatment are dependent on the type of SMA and should, therefore, be individualized to the specific manifestations of the disease. Patients with type II SMA are by definition nonambulatory, with more significant and rapid progression than patients with type III SMA, and a shortened lifespan. Hence, prolonging life would be an important outcome metric for patients with type II SMA. Type III patients generally have a normal lifespan, although motor disability remains. The loss of motor function in the lower limbs translates to the loss of ambulation, resulting in a transition from walking to requiring assistive devices (such as a cane, walker, and subsequently a wheelchair) and, in more affected patients, loss of the use of the upper limbs may occur; both have a major impact on QoL. The clinical experts noted that there is no 1 outcome measure that fits all patients, and ambulation should not be considered the gold standard of an unmet need for type II and III patients. For some severely disabled, nonambulatory patients, being able to use the upper limbs can mean being able to use noninvasive ventilation (i.e., BiPAP), being able to use a computer or cell phone and communicate with the outside world, or even being employed. Additional parameters of importance include the ability to speak and swallow independently.
Place in Therapy
Currently, the treatment of adults with type II or III SMA is similar to that of patients with other neuromuscular disorders and consists of a multidisciplinary approach, including the provision of assistive devices, as needed (such as canes, walkers, and wheelchairs), physiotherapy to prevent contractures, and speech-language pathology for patients with dysphagia.
Given that there are currently no other disease-modifying drugs reimbursed by public drug plans for adults in Canada with type II or III SMA, the approach to the treatment of adults varied among the panel members. If nusinersen is recommended in this population, it could be considered a first-line treatment. Clinical experts noted that risdiplam has recently been given a positive CDEC recommendation for younger adults, so the treatment paradigm may shift in the future. One clinical expert noted that nusinersen is reimbursed for the treatment of adults with type II or type III SMA and is currently being used in this population in Quebec based on observational studies from Germany and Italy.
The clinical experts noted that nusinersen would not be used in patients who cannot tolerate the lumbar puncture due to pain or discomfort. As well, logistical issues, including care, travel, and numerous testing requirements may prevent some patients from being able to receive nusinersen. Moreover, it was noted that some patients may have already undergone spinal fusion surgery, impeding access to the intrathecal sac and making treatment with nusinersen increasingly complicated, although interventional radiology has assisted in making treatment more accessible to patients with complex spines.
The clinical experts suggested that for the adult, treatment-naive population, there is no guidance on whether other medications should be tried before nusinersen.
Patient Population
The diagnosis of 5q SMA requires molecular genetic profiling to identify biallelic variants in SMN1 or increases in SMN2 copy number. The experts highlighted the fact that early diagnostic tests often do not provide accurate results on SMN2 copy number, so repeat genetic results to confirm the SMN2 copy number should be considered.
The experts highlighted the lack of higher-level evidence (i.e., from RCTs) in this population to determine which adults with SMA are most likely to respond to treatment with nusinersen. The experts hypothesized that patients with higher functioning and who are ambulatory may demonstrate better responses because they have more nerves, leading to better function. As well, the clinical experts believed that patients without complex spines are more likely to have a better risk-benefit profile. However, most of the experts stressed that the earlier treatment is administered (i.e., in pre-symptomatic children), the greater the benefit observed, as shown in the CHERISH and SUNFISH studies for nusinersen and risdiplam, although there was some disagreement among the panel members as to whether age should be considered when determining response.
Conversely, the patients least suitable for treatment with nusinersen are those with complex spines due to spinal fusion surgery, those who cannot tolerate lumbar puncture, and those who have previously been treated as infants or children, as there is no evidence of benefit in these patients.
Assessing Response to Treatment
Clinically, there are numerous outcomes and measures to determine response to treatment. The experts noted that there is heterogeneity in the way patients are treated and assessed in Canada. Smaller centres may only be equipped to conduct standard neurologic examinations and may not have the personnel or equipment necessary to conduct the battery of validated functional tests conducted in tertiary neuromuscular centres.
The clinical experts agreed that — given the variation in response to treatment and individualized treatment goals — several outcome measures should be used to determine the benefits of treatment. The most common outcomes used in adults with SMA include the HFMSE, RULM, 6MWT, MRC strength score, and respiratory function (FEV1, FVC, maximal inspiratory pressure, maximal expiratory pressure); all have been validated and have concordance in clinical practice and trials. They noted that other outcomes, such as bulbar function, strengthening of speech, or functional independence, may be important but are not routinely assessed.
The experts noted that in patients with type II and III SMA, disease progression occurs slowly, over the course of years, so the impact of treatment on these outcome measures is not likely to be seen over a short period of time. As such, they agreed that patients should be seen annually, and that more frequent visits would place an undue burden on the patient and not provide a meaningful assessment of change that could be used to inform clinical decisions. It was noted, however, that at the initiation of treatment, patients may be seen every 6 months.
Discontinuing Treatment
The clinical-expert panel agreed that the main reasons for discontinuation would be progression or worsening of disease, as well as any major complications or AEs related to therapy. The experts explained that trying to determine whether patients have achieved stabilization is difficult, given that SMA progresses so slowly that it can be unclear whether progression is due to a lack of therapeutic efficacy or the natural history of the disease. One clinical expert noted that, based on experience, the most common reason for discontinuation of nusinersen was a patient’s desire to stop, owing to a lack of improvement or inability to tolerate the treatment due to the time investment and overall burden associated with painful injections, risk of bleeding or infection, or nerve damage or meningitis.
Prescribing Conditions
The panel agreed that all patients with SMA should be referred to a tertiary centre with a variety of neuromuscular specialists, a multidisciplinary team, and access to interventional radiology or neurosurgery, as most patients will have complex spines. The clinical experts also noted that these centres would have to be able to admit patients who experience potential procedural or treatment-related complications. Additionally, respiratory therapists, physiotherapists, occupational therapists, and nutritionists are essential members of the care team and can help with nonpharmacological therapies and assess appropriate outcome measures.
One panel member contended that once patients have passed the diagnosis and initial-treatment phases, it may be reasonable to provide some services (i.e., physiotherapy and occupational therapy) at less specialized centres during the more chronic phase of the disease; however, patients would still be required to attend a specialized treatment centre to receive injections.
Clinician-Group Input
This section was prepared by CADTH staff based on the input provided by clinician groups.
CADTH received clinician-group input from the Neuromuscular Disease Network for Canada, a pan-Canadian network launched in 2020 to bring together clinical, scientific, technical, and patient expertise in neuromuscular disease with the aim of improving the care, research, and treatment of neuromuscular diseases for all Canadians. Eight clinicians with experience treating SMA patients provided input to this submission.
The clinician group highlighted the 3 main disease-modifying treatments for SMA: nusinersen, risdiplam, and onasemnogene abeparvovec. The clinician group agreed that treatment goals for later-onset SMA would be to maintain current levels of motor function and strength, achieve disease stabilization (including the avoidance of ventilation), promote independence, and improve overall HRQoL. The clinician group highlighted the fact that risdiplam may be the only other treatment option for these patients, and that either nusinersen or risdiplam could be tried first. The clinician group explained that younger patients are most likely to derive benefit from nusinersen, and noted that it may be difficult to accurately identify which adults are most likely to derive benefit from nusinersen. The clinician group stated that a clinically meaningful response to treatment in adults is likely to consist of stabilization of motor and respiratory function, maintenance of independence, and a reduction in hospitalizations. Moreover, they noted that maintaining the ability to speak and avoiding ventilatory support have profound impacts on patient QoL, autonomy, and the ability to maintain vocational and social roles. The clinician group emphasized that current provincial monitoring requirements are too frequent and that there is significant variation among provinces. They agreed that patients should be assessed at treatment initiation, at 6 months, and then annually thereafter to reduce patient burden and strain on health care resources, given the slowly progressive functional decline that takes place over years. Last, the clinician group noted that nusinersen must be administered by or under the direction of health care providers experienced in performing lumbar puncture at designated treatment centres.
The clinician group provided anecdotal reports of 12 patients from Quebec with type II or III SMA. The CADTH clinical team has reviewed these reports, but they are not included in this report as they do not meet the review protocol criteria.
Drug-Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 3.
Table 3: Summary of Drug-Plan Input and Clinical-Expert Response
Implementation issues | Clinical-expert response |
|---|---|
Relevant comparators | |
Risdiplam is likely to be a comparator for the 18- to 25-year age group in the future. However, patients started on risdiplam at 18 to 25 years will likely continue on therapy for life, so risdiplam would be a reasonable comparator and should be considered as such. | No response required. For CDEC consideration. |
The sponsor identified an unmet need in adults with SMA for a treatment to help them achieve stabilization or improve functional status. The sponsor provided an estimate of disease prevalence per 100,000 across the various types: 0.17 for type I, 0.62 for type II, and 0.85 for type III SMA. Further, 4% of type I patients, 37% of type II patients, and 64% of type III patients were 18 years and older. In different jurisdictions, how will implementation of the initiation and renewal criteria be guided? Is it appropriate to treat patients older than 18 years with nusinersen after risdiplam use or in combination with risdiplam? | Patients with type II SMA represent a higher proportion of patients than type III in Canada. Patients with type II and III disease generally progress very slowly, and measures and scoring methods to assess disease activity and treatment response are limited in this population. Moreover, there is significant heterogeneity in the population of type II and type III SMA patients, based on disease presentation and individual ability. It was the opinion of the clinical experts that it would not be appropriate to treat patients with nusinersen after treatment with risdiplam, as nusinersen has not demonstrated efficacy in the adult population in phase III RCTs. There is currently no evidence related to the sequencing of nusinersen and risdiplam, nor is there evidence demonstrating an additive effect of nusinersen when combined with risdiplam. |
Considerations for initiation of therapy | |
Would any prior therapies preclude eligibility for nusinersen, such as prior onasemnogene abeparvovec-xioi (Zolgensma) use? | Yes, the use of onasemnogene abeparvovec at the appropriate time would preclude eligibility for nusinersen, as patients would have received the necessary SMN1 gene when it was essential. As such, providing the additional SMN2 with nusinersen would not be required. |
If a patient was treated with nusinersen as a child but stopped, is there any reason it couldn’t be restarted in adulthood? | There is insufficient evidence to indicate that nusinersen would be useful in the adult population, regardless of whether it was received in the pediatric or adolescent setting. |
Considerations for continuation or renewal of therapy | |
What is the best assessment tool to measure an SMA patient’s condition? What is the best assessment tool to measure a response to nusinersen for the indicated population? | There are currently no tools specifically indicated for use in the population of adults with type II or III SMA. Treatment response in patients with type II or III SMA would generally be measured with HFMSE, RULM, or 6MWT scores; however, there are concerns with these measures in all SMA patients. Tools or measures that are resistant to training or learning would be most appropriate. |
Considerations for discontinuation of therapy | |
Are there clear discontinuation criteria for nusinersen for this indication? | Patients would be discontinued if there were no significant improvements in RULM, 6MWT, or FVC, or if there were any drug- or procedure-related AEs that could not be tolerated. |
Generalizability | |
Owing to the extremely low prevalence of SMA, the assessment of new treatments in adult SMA patients is a challenge. The evidence base for this medication in adults with SMA consists of real-world evidence to inform treatment evaluation. Is there adequate real-world evidence from the 6 studies to inform this decision? Is there an upper limit for this drug? | Based on experience in other rare diseases areas, as well as the availability of a large number of patients in this population (as evidenced by the number of observational studies), a global, multi-centre RCT in this patient population would have been feasible. |
System and economic issues | |
Funding for adults with type II or III SMA is estimated to result in incremental costs of $69 million in the first 3 years of reimbursement. The number of patients is estimated to be 42, 66, and 78 in the first 3 years of funding. Is the sponsor’s budget impact analysis realistic? Clinicians is British Columbia estimate that there are 45 adults with SMA in that province alone. | No response required. For CDEC consideration. |
6MWT = 6-minute walk test; AEs = adverse events; CDEC = CADTH Canadian Drug Expert Committee; FVC = forced vital capacity; HFMSE = Hammersmith Functional Motor Scale Expanded; RCT = randomized controlled trial; RULM = Revised Upper Limb Module; SMA = spinal muscular atrophy.
Clinical Evidence
The clinical evidence included in the review of nusinersen is presented in 3 sections. The first section, the systematic review, includes key studies provided in the sponsor’s submission to CADTH, as well as studies that were selected according to an a priori protocol. The second section includes indirect evidence selected from the literature that met the selection criteria specified in the review. No indirect evidence was submitted by the sponsor. The third section includes any additional relevant studies that were considered to address important gaps in the evidence included in the systematic review.
Systematic Review (Pivotal and Protocol Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of nusinersen 2.4 mg/mL solution for intrathecal injection for the treatment of adults (> 18 years) with type II and type III 5q SMA.
Methods
Studies selected for inclusion in the systematic review included the key studies provided in the sponsor’s submission to CADTH and those meeting the selection criteria presented in Table 4. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
Table 4: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Population | Adults (> 18 years) with type II or type III SMA Subgroups:
|
Intervention | Nusinersen 2.4 mg/mL intrathecal injection |
Comparator |
|
Outcomes | Efficacy or effectiveness outcomes:
Harms outcomes:
|
Study designs | Published and unpublished phase II, III and IV RCTs |
AEs = adverse events; FEV1 = forced expiratory volume in 1 second; FVC = forced vital capacity; HRQoL = health-related quality of life; RCTs = randomized controlled trials; SAEs = serious adverse events; SMA = spinal muscular atrophy; WDAEs = withdrawal due to adverse events.
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy according to the PRESS Peer Review of Electronic Search Strategies checklist.28
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946‒) via Ovid, and Embase (1974‒) via Ovid. All Ovid searches were run simultaneously as a multi-file search. Duplicates were removed using Ovid deduplication for multi-file searches, followed by manual deduplication in Endnote. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concept was nusinersen. The following clinical trials registries were searched: the US National Institutes of Health’s clinicaltrials.gov, the WHO’s International Clinical Trials Registry Platform (ICTRP) search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
No filters were applied to limit the retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. Refer to Appendix 1 for the detailed search strategies.
The initial search was completed on January 6, 2022. Regular alerts updated the search until the meeting of the CDEC on April 27, 2022.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool For Searching Health-Related Grey Literature checklist.29 Included in this search were websites of regulatory agencies (FDA and European Medicines Agency). Google was used to search for additional internet-based materials. See Appendix 1 for more information on the grey literature search strategy.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the pre-determined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
A focused literature search for network meta-analyses dealing with SMA was run in MEDLINE All (1946–) on January 6, 2022. No limits were applied to the search.
Findings From the Literature
A total of 6 studies were identified for inclusion in the systematic review (Figure 2). The included studies are summarized in Table 5. A list of excluded studies is presented in Appendix 2.
Table 5: Details of Included Studies
Details | Hagenacker et al. (2020) | Maggi et al. (2020) | EU registry study (2020) | Pera et al. (2021) | Konersman et al. (2021) | SHINE (Study CS11) | Coratti et al. (2021) |
|---|---|---|---|---|---|---|---|
Designs and populations | |||||||
Study design | Prospective, multi-centre, observational study | Retrospective cohort study | Registry-based study of prospective and retrospective data from 3 registries | Registry-based study from the ISMAR | Retrospective chart review | Open-label extension study | Critical review and meta-analysis |
Locations | Germany | Italy | SMArtCARE: Germany ISMAR: Italy, UK, US CuidAME: Spain | Italy, UK, US | US | Global | N/A |
Study duration | July 13, 2017, to May 1, 2019 | NR | NR | NR | April 2017-June 2019 | Ongoing. Planned duration of 5 years. | N/A |
Enrolled (n) | 139 | 116 | 252 | 144 | 35 | 28 | N/A |
Inclusion criteria |
|
| NR |
| NR | Patients with SMA who completed Index Study CS3A, CS3B, CS4, CS12, or SM202 |
|
Exclusion criteria | NR | NR | NR | NR | NR |
| NR |
Drugs | |||||||
Treatment | 12 mg nusinersen administered intrathecally on days 1, 14, 28, and 63, with repeated maintenance injections every 4 months | 12 mg nusinersen at baseline, day 14, day 28, and day 63, followed by maintenance doses every 4 months | Nusinersen | Nusinersen | Nusinersen (dose not specified) first 3 doses every 2 weeks, fourth dose 30 days after third dose, followed by maintenance doses every 4 months thereafter | 12 mg (5 mL) of nusinersen administered as intrathecal injections via lumbar puncture | Nusinersen dose and regimen not specified |
Comparator(s) | None | None | None | None | NA | NA | N/A |
Outcomes | |||||||
Primary end point | Change from baseline in total HFMSE score at months 6, 10, and 14 | Change from baseline in HFMSE, RULM, and 6MWT at months 6, 10, and 14 | Change from baseline in HFMSE, RULM, 6MWT at 5 and 11 months | Change in HFMSE, RULM, and 6MWT from baseline to 12 months | Motor outcome measures (HFMSE, HINE, RULM, CHOP-ATEND) | Long-term safety and tolerability |
|
Secondary and exploratory end points | Change from baseline to months 6, 10, and 14 in RULM score, and 6MWT |
| Safety and tolerability | NR | NR |
| N/A |
Notes | |||||||
Publications | Hagenacker et al. (2020)17 | Maggi et al. (2020)18 | Pera et al. (2021)19 | Konersman et al. (2021)22 | Coratti et al. (2021)20 | ||
6MWT = 6-minute walk test; CHOP-ATEND = Children’s Hospital of Philadelphia–Adult Test of Neuromuscular Disorders; ECG = electrocardiogram; FEV1 = forced expiratory volume in 1 second; FVC = forced vital capacity; HFMSE Hammersmith Functional Motor Scale Expanded; HINE = Hammersmith Infant Neurologic Examination; ISMAR = International SMA Registry; NA = not applicable; NR = not reported; RULM = Revised Upper Limb Module; SMA = spinal muscular atrophy.
Sources: Hagenacker et al. (2020),17 Maggi et al. (2020),18 sponsor submission,15,16 Pera et al. (2021),19 Konersman et al. (2021),22 Coratti et al. (2021).20
Description of Studies
No RCTs focusing on treatment-naive adults with type II or III SMA were identified as part of the CADTH literature search, and all available and included studies were of observational design and focused on real-world data; such studies have more limitations than RCTs.
As part of the reassessment for nusinersen, the sponsor provided CADTH with 7 studies: 1 prospective noncomparative cohort study (Hagenacker et al. [2020]17), 1 retrospective noncomparative cohort study (Maggi et al. [2020]18), 1 real-world cohort comprised of prospectively and retrospectively collected data from of 3 European registries, 1 retrospective chart review (Konersman et al. [2021]22), 1 retrospective registry-based review (Pera et al. [2021]19), 1 open-label extension of trials investigating nusinersen (SHINE trial), and 1 critical review and meta-analysis of patients with type II or III SMA (Coratti et al. [2020]20). No additional studies meeting the inclusion criteria for the systematic review were identified by the CADTH review team.
Hagenacker et al. (2020)
The study by Hagenacker et al. (2020)17 was a prospective, multi-centre, noncomparative observational study of 124 patients with 5q SMA treated with nusinersen from 10 German neurologic centres that aimed to investigate the safety and effectiveness of nusinersen in adults with 5q SMA over a 6-, 10-, and 14-month period. There was no funding source reported for this study.
Maggi et al. (2020)
Maggi et al. (2020)18 was a retrospective, noncomparative cohort study of 18 Italian secondary or tertiary care centres for SMA aimed at investigating the safety and effectiveness of nusinersen on motor function in a cohort of 116 adults with type II or type III SMA. No funding source from public, commercial, or not-for-profit sectors was reported for this study.
EU Registry Study
This observational, registry-based cohort study was conducted using combined data from 252 adults from 3 European registries (SMArtCARE from German-speaking countries; CuidAME from Spain; and ISMAR from Italy, UK, and US) with the aim of assessing the safety and effectiveness of nusinersen in adults with 5q SMA and at least 6 months of follow-up, and informing data analyses of the impact of nusinersen on motor function.15,16 Two subcohorts were used for the statistical analyses: a before-and-after nusinersen treatment group of 75 patients with type III or IV SMA; and a comparative dataset from 252 adults with type III SMA (235 who had been treated with nusinersen and 17 who had not). Further details on the analysis populations are provided in the summary of the Statistical Analysis section.
Pera et al. (2021)
The study by Pera et al. (2021)19 was a noncomparative, longitudinal registry-based study of ISMAR in Italy, the UK, and the US, and aimed at reporting treatment outcomes in a cohort of 144 ambulant and nonambulant type III SMA patients treated with nusinersen.
Multiple foundations and organizations were acknowledged in the funding information, with support from the sponsor to the International SMA Consortium registry.
Although the enrolled population for this study included children and adolescents, details for the adult population were provided, so results for this group were presented and summarized in this report.
Konersman et al. (2021)
The study by Konersman et al. (2021)22 was a noncomparative retrospective chart review of 35 older SMA patients (aged 5 to 58 years) in the US conducted from April 2017 to June 2019. The objective of this study was to determine the effectiveness of nusinersen at improving motor function, to report on methods of administration, and to detail the AEs seen in adolescents and adults with SMA.
No stratified or subgroup analyses were conducted for the population of interest defined in Table 4. Therefore, this study will not be further summarized because the relevant population for the reassessment could not be evaluated.
SHINE
The SHINE trial (Study CS11)21 was identified as a key study by the sponsor. It is an ongoing open-label extension study of SMA patients who previously participated in investigational studies to evaluate the long-term safety, tolerability, and efficacy of nusinersen. All participants received 12 mg (5 mL) of nusinersen. The study consisted of a screening phase, a treatment period that included a loading dose and maintenance treatment, a post-treatment follow-up period, and an end-of-study evaluation for all patients who completed an index study of nusinersen. A modified maintenance dosing regimen was used in Study CS11; the index studies had different maintenance schedules. After the loading-dose period, nusinersen is administered intrathecally once every 4 months to all participants. The planned duration of participation in the study is 5 years after administration of the modified maintenance dosing regimen. As of the data cut-off, 190 patients were included in the ongoing SHINE trial as part of the later-onset analysis group; data from 28 of these adults informed the efficacy analysis.
Although a subgroup of later-onset adults with type II or III SMA was used in a post hoc analysis, these patients had previously been enrolled in index studies for nusinersen (studies CS2 and CS12) and, therefore, received treatment with nusinersen when they were younger than 18 years. Results provided in this analysis are confounded by the age at which treatment was initiated and cannot be used to assess treatment benefit in the treatment-naive adult population. Although identified as a key study by the sponsor, the SHINE later-onset analysis will not be further summarized or appraised because it is not relevant to the reassessment of nusinersen.
Coratti et al. (2021)
Coratti et al. (2021)20 was a critical review and meta-analysis of patients with type II and III SMA treated with nusinersen that was included in the list of key studies submitted by the sponsor. This study is further summarized in the Other Relevant Evidence section. This study was also identified in the literature search; however, the study design did not meet the eligibility criteria of the systematic review conducted by CADTH because the population combined children (< 18 years) and adults (≥ 18 years), even though a subgroup analysis for adults was presented.
It is worth noting that all primary studies of the population of interest included in the meta-analysis that were publicly available were also identified in the CADTH systematic review (Hagenacker et al. [2020],17 Maggi et al. [2020],18 Konersman et al. [2021],22 and Pera et al. [2021]19).
Populations
Inclusion and Exclusion Criteria
Limited information on inclusion and exclusion criteria were provided for each study. In all studies submitted by the sponsor, a common inclusion criterion was a confirmed diagnosis of SMA, with genetic documentation of 5q SMA homozygous deletions of exons 7, 8, or both, or with compound heterozygous mutations.15-19
In the studies by Hagenacker et al. (2020)17 and Maggi et al. (2020),18 patients were required to be adults (≥ 18 years), with nusinersen treatment initiation after they reached the age of 18 years. However, in the EU registry study15,16 and Pera et al. (2021)19 studies, patients younger than 18 years were also included.
Additionally, when reported, patients were required to have type II or type III SMA (Maggi et al. [2020]18 and Pera et al. [2021]19). In Pera et al. (2021),19 type III SMA was further subdivided into type IIIA or IIIB, depending on age at symptom onset (before or after 3 years). In the Hagenacker et al. (2020)17 and EU registry study,15,16 cases of type I and type IV SMA were included. In Hagenacker et al. (2020),17 1 patient with type I SMA and 1 patient with type IV SMA were enrolled, but they were only included in the 10-month analysis. In the EU registry study15,16 submitted by the sponsor, patients of all SMA types were included; however, the analysis only included patients with type III or IV SMA, and only the adult population with type III SMA was of interest for this review, so only results for this population will be summarized. In most cases, to be eligible for inclusion in the analyses, patients had to have undergone a minimum of 6 months of treatment.
Baseline Characteristics
Hagenacker et al. (2020)
Baseline characteristics for patients in the Hagenacker et al. (2020)17 study were provided separately for patients with 6, 10, and 14 months follow-up; these are summarized in Table 6. A total of 139 patients completed the 6-month assessment. The primary end point analysis included 124 (89%) patients with a treatment period of at least 6 months, 92 (66%) patients with a treatment period of 10 months, and 57 (41%) patients with a treatment period of 14 months. Patient with 6 months of follow-up ranged in age from 16 to 65 years, with a mean age of 36 years (SD = 12). Most patients were SMA type III (77 [62%]) or type II (45 [36%]). In the 6-month analysis, 2 patients (2%) were SMA type I, whereas in the 10-month analysis, 1 patient (1%) each was included in the type I and type IV SMA groups. Most patients had 3 (48 [39%]) or 4 (41 [33%]) SMN2 copies, but SMN2 status was unknown in 24 (19%) patients. Only 46 (37%), 35 (38%), and 23 (40%) patients were ambulant at baseline in the 6-, 10-, and 14-month analyses, respectively, and 20% to 25% had prior spondylodesis. According to the authors, most patients had low HFMSE scores at baseline, defined as a score of less than 35 points (69% at 6 months, 64% at 10 months, and 61% at 14 months).
Table 6: Summary of Baseline Characteristics (Hagenacker et al. [2020])
Characteristic | Included in 6-month analysis, n = 124 | Included in 10-month analysis, n = 92 | Included in 14-month analysis, n = 57 |
|---|---|---|---|
Sex | |||
Female | 57 (46%) | 39 (42%) | 20 (35%) |
Male | 67 (54%) | 53 (58%) | 37 (65%) |
Age at treatment, years | 36 (12;16 to 65) | 37 (12; 16 to 65) | 33 (11; 16 to 59) |
SMN2 copy number | |||
2 | 7 (6%) | 7 (8%) | 4 (7%) |
3 | 48 (39%) | 33 (36%) | 21 (37%) |
4 | 41 (33%) | 31 (34%) | 21 (37%) |
5 | 2 (2%) | 1 (1%) | 0 |
6 | 2 (2%) | 0 | 0 |
Unknown | 24 (19%) | 20 (22%) | 11 (19%) |
SMA type | |||
I | 2 (2%) | 1 (1%) | 0 |
II | 45 (36%) | 30 (33%) | 20 (35%) |
III | 77 (62%) | 60 (65%) | 37 (65%) |
IV | 0 | 1 (1%) | 0 |
Ambulant | 46 (37%) | 35 (38%) | 23 (40%) |
Previous spondylodesis | 28 (23%) | 18 (20%) | 14 (25%) |
Baseline HFMSE score, (out of 66) | 20.74 (21.39) | 22.95 (21.66) | 24.65 (21.83) |
High, ≥ 35 points | 39 (31%) | 33 (36%) | 22 (39%) |
Low, < 35 points | 85 (69%) | 59 (64%) | 35 (61%) |
Baseline RULM score (out of 37) | 20.87 (13.27) | 23.00 (12.80) | 23.85 (12.16) |
Baseline 6MWT distance, m | 321.76 (217.66) | 353.03 (218.46) | 371.43 (210.34) |
6MWT = 6-minute walk test; HFMSE = Hammersmith Functional Motor Scale Expanded; RULM = Revised Upper Limb Module; SD = standard deviation; SMA = spinal muscular atrophy.
Source: Hagenacker et al. (2020).17
Maggi et al. (2020)
A summary of baseline characteristics of patients in the study by Maggi et al. (2020)18 are summarized in Table 7. Of the 116 patients enrolled, 103 (88.8%) had type III SMA — equally distributed between type III sitters (n = 51) and type III walkers (n = 52) — and 13 (11.2%) had type II SMA. Enrolled patients had a median age at onset of 3 years (range = 0 to 17) and a median age at the beginning of treatment (T0) of 34 years (range = 18 to 72). Median age at treatment initiation was 40 years for sitters and 33 years for walkers. The majority of patients were male (58.62%), and most patients had 3 (n = 36 [31%]) or 4 (n = 54 [46.6%]) copies of SMN2, but some had 2 copies (n = 5 [4.3%]) and some copy numbers were unknown (n = 21 [18.1%]).
Ventilatory support at treatment initiation was required by 21 (18.1%) patients; of these, 10 (76.9%) had type II SMA, 8 (15.7%) were type III sitters, and 3 (5.8%) were type III walkers. Prior surgery for scoliosis was reported in 16 (13.8%) patients overall, and in 61.5% of type II patients, 13.7% of type III sitters, and 1.9% of type III walkers.18
Table 7: Summary of Baseline Characteristics (Maggi et al. [2020])
Variable | na | All SMA | na | SMA type II | na | SMA type III sitters | na | SMA type III walkers |
|---|---|---|---|---|---|---|---|---|
Age at onset, years, median (min to max) | 116 | 3 (0 to 17) | 13 | 0.8 (0.5 to 12) | 51 | 3 (0.3 to 15) | 52 | 8 (0 to 17) |
Age at T0, years, median (min to max) | 116 | 34 (18 to 72) | 13 | 24 (19 to 41) | 51 | 40 (18 to 72) | 52 | 33 (18 to 68) |
Disease duration at T0, years, median (min to max) | 116 | 29 (3 to 63) | 13 | 22.5 (7 to 40.5) | 51 | 37 (14 to 63) | 52 | 26 (3 to 51) |
Sex, female/male | 116 | 48/68 | 13 | 3/10 | 51 | 15/36 | 52 | 30/22 |
SMN2 copies,b | 116 | 13 | 51 | 52 | ||||
2 copies, n (%) | 5 (4.3) | 3 (23.1) | 2 (3.9) | 0 (0) | ||||
3 copies, n (%) | 36 (31.0) | 6 (46.2) | 16 (31.4) | 14 (26.9) | ||||
4 copies, n (%) | 54 (46.6) | 2 (15.4) | 21 (41.2) | 31 (59.6) | ||||
Unknown, n (%) | 21 (18.1) | 2 (15.4) | 12 (23.5) | 7 (13.5) | ||||
Salbutamol, n (%) | 116 | 27 (23.3) | 13 | 5 (38.5) | 51 | 9 (17.8) | 52 | 13 (25.0) |
Ventilatory support at T0, n (%) | 116 | 21 (18.1) | 13 | 10 (76.9)c | 51 | 8 (15.7)d | 52 | 3 (5.8) |
Surgery for scoliosis, n (%) | 116 | 16 (13.8) | 13 | 8 (61.5) | 51 | 7 (13.7) | 52 | 1 (1.9)e |
Clinical assessments, median (min to max) | ||||||||
HFMSE score | 116 | 22.5 (0 to 64) | 13 | 0 (0 to 9) | 51 | 9 (0 to 40) | 52 | 50.5 (17 to 64) |
RULM score | 114 | 29 (0 to 37) | 12 | 2.5 (0 to 22) | 51 | 20 (0 to 34) | 51 | 37 (25 to 37) |
6MWT, m | N/A | N/A | 0 | N/A | 0 | N/A | 48 | 322 (14 to 588) |
Rise from floor, s–1 | N/A | N/A | 0 | N/A | 0 | N/A | 28 | 0.1 (0.01 to 0.33) |
Rise from chair, s–1 | N/A | N/A | 0 | N/A | 0 | N/A | 31 | 0.25 (0.06 to 1) |
Climb 4 steps, steps/s | N/A | N/A | 0 | N/A | 0 | N/A | 35 | 0.8 (0.17 to 2) |
Run/walk 10 m, m/s | N/A | N/A | 0 | N/A | 0 | N/A | 40 | 1.12 (0.09 to 2.08) |
FVC, % of predicted | 86 | 88.5 (11 to 139) | 7 | 20 (11 to 74) | 40 | 83 (30 to 128) | 39 | 102 (40 to 139) |
FEV1, % of predicted | 76 | 92.5 (16 to 134) | 5 | 20 (16 to 55) | 35 | 84.3 (35 to 120) | 36 | 103 (47 to 134) |
6MWT = 6-minute walk test; FEV1 = forced expired volume in 1 second; FVC = forced vital capacity; HFMSE = Hammersmith Functional Motor Scale Expanded; max = maximum; min = minimum; N/A = not available; RULM = Revised Upper Limb Module; SMA = spinal muscular atrophy; T0 = treatment initiation.
Note: All patients carried homozygous SMN1 exon 7 deletions, except 3: 1 with a nonsense and 2 with a missense mutation on the other allele.
aHFMSE was available for all patients; remaining assessments were not available for all patients.
bSMN2 copy number was not available in 21 patients.
cA further patient stopped ventilatory support before T0 due to poor tolerance.
dTwo patients used ventilatory support due to obstructive sleep apnea and a further patient refused ventilatory support although indicated.
ePatient able to walk for few steps with cane.
Source: Maggi et al. (2020).18
EU Registry Study
Baseline characteristics for the cohort of patients from the EU registry study15,16 are summarized by registry and the pooled population in Table 8. Baseline characteristics for all patients in each registry for all ages and for SMA types III and IV were provided; however, as previously mentioned, only characteristics for adult type III populations were summarized.
All adult type III SMA patients in the SMArtCARE registry were treated with nusinersen (n = 151). Adult type III patients who were treated with nusinersen and who were untreated were included in the ISMAR (n = 53, n = 5, respectively) and CuidAME (n = 24, n = 9, respectively) registries.15,16
The mean age of type III adults treated with nusinersen across registries was 35 to 39 years (range = 18 to 71 years). In untreated type III SMA patients, the mean age was 36 years in the CuidAME registry and 46 years in the ISMAR registry. The age at diagnosis of SMA was not reported. In general, most patients were male across registries, particularly those treated with nusinersen (60% to 63%); in the untreated population, the majority were female (60% in ISMAR, 56% in CuidAME). In type III adults, most had 4 copies of SMN2 (38% to 67% in treated and untreated patients across registries). Across registries, most patients were not fully ambulatory (54% of nusinersen-treated patients and 29% of untreated patients), most did not have scoliosis (75% to 100% of treated and untreated patients), and few required noninvasive ventilation (6% to 7%). No information on feeding support was provided in the EU registry study.15,16
Table 8: Summary of Baseline Characteristics (EU Registry Study)
Characteristic | SMArtCARE registry | ISMAR registry | CuidAME registry | Pooled populationa | Piecewise linear mixed model (before-after design) | |||
|---|---|---|---|---|---|---|---|---|
Adult type III population (N = 151) | Adult type III treated (N = 53) | Adult type III untreated (N = 5) | Adult type III treated (N = 24) | Adult type III untreated (N = 9) | Adult type III treated (N = 228) | Adult type III untreated (N = 14) | Adult population (N = 75)b | |
Demographic characteristics | ||||||||
Age, months | ||||||||
Age at symptom onset, mean (SD) | 79.62 (65.49) | 77.42 (66.33) | 52.58 (17.92) | 88.87 (67.30) | 117 (73.15) | 80.10 (65.65) | 93.99 (66.46) | 85.46 (73.17) |
Age at treatment initiation, mean (SD) | 419.68 (150.26) | 467.42 (155.15) | N/A | 472.08 (142.53) | N/A | 436.29 (151.78) | N/A | 450.55 (153.60) |
Mean age (SD) | 419.68 (150.26) | 467.42 (155.15) | 548.42 (245.48) | 472.08 (142.53) | 427.73 (197.90) | 436.29 (151.78) | 470.83 (215.04) | 450.55 (153.60) |
Median age (range) | 396 (216 to 852) | 490.88 (236.15 to 817.25) | 485.72 (273.60 to 827.80) | 477.5 8 (250.0 to 762.02) | 396 (216.00 to 805.15) | 414 (216 to 852) | 401.05 (216.00 to 827.80) | 441.17 (216.0 to 888.56) |
Disease duration, mean months (SD) | 340.07 (153.34) | 384.29 (150.27) | 495.83 (230.73) | 383.20 (174.46) | 310.73 (205.72) | 354.63 (155.67) | 376.84 (225.60) | 363.39 (153.91) |
Sex, n (%) | ||||||||
Female | 56 (37.09) | 21 (39.62) | 3 (60.00) | 9 (37.50) | 5 (55.56) | 86 (37.72) | 8 (57.14) | 29 (38.67) |
Male | 95 (62.91) | 32 (60.38) | 2 (40.00) | 15 (62.50) | 4 (44.44) | 142 (62.28) | 6 (42.86) | 46 (61.33) |
Disease characteristics | ||||||||
SMA type, n (%) | ||||||||
Type I | N/A | N/A | N/A | N/A | N/A | N/A | N/A | NR |
Type II | N/A | N/A | N/A | N/A | N/A | N/A | N/A | NR |
Type III | 151 (100) | 53 (100) | 5 (100) | 24 (100) | 9 (100) | 228 (100) | 14 (100) | NR |
Type IV | N/A | N/A | N/A | N/A | N/A | N/A | N/A | NR |
Number of SMN2 copies, n (%) | ||||||||
2 | 11 (7.28) | 5 (9.43) | 0 | 2 (8.33) | 0 | 18 (7.89) | 0 | 5 (6.67) |
3 | 37 (24.50) | 12 (22.64) | 1 (20.00) | 11 (45.83) | 3 (33.33) | 60 (26.32) | 4 (28.57) | 23 (30.67) |
4 | 71 (47.02) | 20 (37.74) | 2 (40.00) | 11 (45.83) | 6 (66.67) | 102 (44.74) | 8 (57.14) | 33 (44.00) |
> 4 | 5 (3.31) | 0 | 0 | 0 | 0 | 5 (2.19) | 0 | 1 (1.33) |
Unknown | 26 (17.22) | 16 (30.19) | 2 (40.00) | 0 | 0 | 42 (18.42) | 2 (14.29) | 13 (17.33) |
Ambulatory status | ||||||||
Ambulatory | 77 (50.99) | 21 (39.62) | 1 (20.00) | 8 (33.33) | 9 (100.00) | 106 (46.49) | 10 (71.43) | 34 (45.33) |
Nonambulatory | 74 (49.01) | 32 (60.38) | 4 (80.00) | 16 (66.67) | 0 | 122 (53.51) | 4 (28.57) | 41 (54.67) |
Wheelchair users | 95 (60.51) | 37/52 (71.15) | 4 (80.00) | 8/15 (53.33) | 1/6 (20.00) | 140/218 (64.22) | 5/10 (50.00) | 43/69 (62.32) |
Scoliosis | ||||||||
Yes | 16 (10.60) | 30/52 (57.69) | 0 | 10 (41.67) | 0 | 56/227 (24.67) | 0 | 29 (38.67) |
No | 135 (89.40) | 22/52 (42.31) | 5 (100.00) | 14 (58.33) | 9 (100.00) | 171/227 (75.33) | 14 (100.00) | 46 (61.33) |
Any prior surgery | NR | 6 (11.32) | 0 | NR | NR | 6/53 (11.32) | 0 | 3/64 (4.69) |
Respiratory status | ||||||||
Respiratory events | 0 | 1 (1.89) | 0 | NR | NR | 1/204 (0.49) | 0 | 0 |
Ventilatory support | NR | NR | NR | NR | NR | NR | NR | NR |
Invasive ventilation | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Noninvasive ventilation | 3 (1.25) | 8 (15.09) | 1 (20.00) | 3 (12.50) | 0 | 14 (6.14) | 1 (7.14) | 7 (9.33) |
Baseline disease assessments | ||||||||
Motor-function assessments | ||||||||
HFMSE | ||||||||
Mean (SD) | 29.29 (20.23) | 29.25 (18.76) | 20.60 (17.40) | 27.67 (20.18) | 52.44 (13.18) | 29.09 (19.78) | 41.07 (21.23) | 29.15 (51.51) |
Median (range) | 29 (0 to 64) | 29 (0 to 63) | 16 (0 to 41) | 21.5 (0 to 59) | 56 (25 to 66) | 28 (0 to 64) | 44.5 (0 to 66) | 25 (0 to 64) |
RULM | ||||||||
Mean (SD) | 27.59 (9.54) | 27.92 (9.20) | 32.50 (6.10) | 26.71 (10.03) | 35.44 (4.67) | 27.57 (9.47) | 32.79 (6.10) | 26.15 (10.25) |
Median (range) | 31 (0 to 37) | 30 (1 to 37) | 37 (23 to 37) | 29 (1 to 37) | 37 (23 to 37) | 30 (0 to 37) | 37 (23 to 37) | 27 (1 to 37) |
6MWT, m | ||||||||
Mean (SD) | 283 (196.47) | 307.48 (179.48) | 280 | 317.57 (152.55) | 479.83 (106.34) | 300.87 (186.87) | 451.29 (123.00) | 383.40 (154.36) |
Median (range) | 334.5 (0 to 600) | 340 (75 to 597) | 280 | 374 (62 to 484) | 479 (325 to 605) | 361 (0 to 600) | 443 (280 to 605) | 428 (0 to 591) |
6MWT = 6-minute walk test; HFMSE = Hammersmith Functional Motor Scale Expanded; N/A = not applicable; NR = not reported; RULM = Revised Upper Limb Module; SD = standard deviation; SMA = spinal muscular atrophy.
aAdult type III treated: 151 (66.23%) patients from the German registry, 53 (23.25%) patients from the Italian registry, and 24 (10.53%) from the Spanish registry. Adult type III untreated: 0 patients in the German registry, 5 (35.71%) patients in the Italian registry, and 9 (64.23%) patients in the Spanish registry.
b26 (34.67%) patients from the German registry, 38 (50.67%) patients from the Italian registry, and 11 (11.67%) patients from the Spanish registry.
Pera et al. (2021)
Baseline characteristics for the study by Pera et al. (2021)19 are summarized in Table 9. Only 67 patients (47%) in the Pera et al. (2021)19 cohort were adults (≥ 18 years). Baseline characteristics for the adult population were not reported in the article.
Treatment Exposure
As the studies were observational in nature, none had an intervention. Instead, the exposure of interest in these studies was nusinersen. Given the design of all the studies submitted by the sponsor, the only treatment in all studies was nusinersen 12 mg administered by intrathecal injection, although the specific dose was not mentioned in the study by Pera et al. (2021).19 The dosing regimen for nusinersen consisted of induction injections on days 1, 14, 28, and 63, with maintenance injections every 4 months thereafter, in accordance with the product label, and was only reported in the studies by Hagenacker et al. (2020)17 and Maggi et al. (2020).18 Dosing regimen was not described in the EU registry study15,16 or in Pera et al. (2021).19
In Hagenacker et al. (2020),17 intrathecal injections were administered by a trained neurologist or neuroradiologist via conventional, fluoroscopy-guided or CT (CT)-guided lumbar puncture.17 In Maggi et al. (2020),18 intrathecal injections were primarily performed with standard lumbar access in 85 (82.5%) patients, CT-guided procedures in 4 (3.9%) patients, and X-ray-guided procedures in 14 (13.6%) patients with type III SMA. A total of 7 (8.2%) patients with type III SMA shifted from manual to imaging-guided techniques during treatment. Only 1(7.7%) patient with type II SMA (N = 13) was managed without imaging guidance, whereas 7 (53.8%) received intrathecal injection via CT-guided procedures and 5 (38.5%) received intrathecal injection with X-ray-guided approaches.18
In the EU registry study,15,16 a comparison between nusinersen-treated and untreated adults was made in 1 subcohort; however, no information on the intrathecal administration or procedures were provided. No information on nusinersen dosing or administration was provided in the study by Pera et al. (2021).19
Limited information on concomitant medications and cointerventions was available.
Outcomes
A list of effectiveness end points identified in the CADTH review protocol that were assessed in the studies included in this review is provided in Table 10. These end points are further summarized in the following sections, when information was available. A detailed discussion and critical appraisal of the outcome measures is provided in Appendix 4.
In both Hagenacker et al. (2020)17 and Maggi et al. (2020),18 available outcomes were assessed at months 6, 10, and 14. Outcomes in Pera et al. (2021)19 were evaluated at 12 months. All patients included in the EU registry study15,16 had to have a treatment duration of at least 6 months; however, the specific time of assessment of effectiveness outcomes were not reported.
Table 9: Summary of Baseline Characteristics (Pera et al. [2021])
Characteristic | All SMA | SMA type IIIA | SMA type IIIB |
|---|---|---|---|
N | 144 | 74 | 70 |
Sex, n (%) | |||
Male | 84 (58.33) | 40 (54.05) | 44 (62.86) |
Female | 60 (41.67) | 34 (45.95) | 26 (37.14) |
Age at baseline in years, median (first to third quartile) | 16.42 (9.14 to 35.69) | 12.60 (5.5 to 26.27) | 23.22 (13.07 to 43.94) |
Age < 18 years, n (%) | 77 (53.47) | 51 (68.92) | 26 (37.14) |
Median age in pediatric population in years (first to third quartile) | 9.50 (5.50 to 13.43) | 8.01 (4.40 to 13.11) | 11.74 (9.24 to 15.08) |
Age ≥ 18 years, n (%) | 67 (46.53) | 23 (31.08) | 44 (62.86) |
Median age in adult population in years (first to third quartile) | 36.60 (26.27 to 47.08) | 35.40 (27.10 to 39.00) | 38.84 (25.51 to 49.35) |
Disease duration in years, median (first to third quartile) | 12.10 (4.4 to 28.89) | 10.41 (3.51 to 25.11) | 13.33 (4.66 to 31.43) |
SMN2 copy number, n (%) | |||
1 | 0 (0.00) | 0 (0.00) | 0 (0.00) |
2 | 11 (7.64) | 4 (5.41) | 7 (10.00) |
3 | 56 (38.88) | 40 (54.05) | 16 (22.85) |
4 | 14 (9.72) | 8 (10.81) | 6 (8.57) |
4+ | 29 (20.14) | 6 (8.11) | 23 (32.86) |
Unknown | 34 (23.61) | 16 (21.62) | 18 (25.71) |
SMA function, n (%) | |||
Nonsitter | 3 (2.08) | 3 (4.05) | 0 (0.00) |
Sitter | 62 (43.06) | 40 (54.05) | 22 (31.43) |
Walker | 79 (54.86) | 31 (41.89) | 48 (68.57) |
Baseline HFMSE score, median (first to third quartile) | 41 (23 to 54) | 32.50 (15 to 50) | 48.5 (28.0 to 58.5) |
n = 130 | n = 66 | n = 64 | |
Baseline RULM score, median (first to third quartile) | 31 (24 to 37) | 27 (22 to 32) | 35.5 (29.5 to 37.0) |
n = 116 | n = 56 | n = 60 | |
Baseline 6MWT, m, median (first to third quartile) | 321.5 (166 to 425) | 283 (107 to 397) | 356 (236 to 434) |
n = 62 | n = 23 | n = 39 | |
Follow-up, years, mean (SD) | 1.83 (0.61) | 1.91 (0.63) | 1.75 (0.58) |
Number of visits, median (range) | 5 (2 to 11) | 6 (2 to 11) | 5 (3 to 11) |
6MWT = 6-minute walk test; HFMSE = Hammersmith Functional Motor Scale Expanded; RULM = Revised Upper Limb Module; SD = standard deviation; SMA = spinal muscular atrophy.
Source: Pera et al. (2021).19 This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 International Licence. Full text available here: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8351459/
Table 10: Outcomes Evaluated in the Included Studies
Outcome | Study | |||
|---|---|---|---|---|
Hagenacker et al. (2020) | Maggi et al. (2020) | EU registry study | Pera et al. (2021) | |
Motor-function outcome | ||||
HFMSE | — | — | — | — |
RULM | — | — | — | — |
6MWT | — | — | — | — |
Respiratory-related outcome | ||||
FEV1/FVC | X | — | X | X |
Bulbar function | X | X | X | X |
Survival | X | X | X | X |
Hospitalization | X | X | X | X |
HRQoL | X | X | X | X |
Anatomic-related outcome | X | X | X | X |
Caregiver burden | X | X | X | X |
Safety outcome | ||||
AEs, SAEs | — | — | — | X |
6MWT = 6-minute walk test; AEs = adverse events; FEV1 = forced expiratory volume in 1 second; FVC = forced vital capacity; HFMSE = Hammersmith Functional Motor Scale Expanded; HRQoL = health-related quality of life; RULM = Revised Upper Limb Module; SAEs = serious adverse events.
Sources: Hagenacker et al. (2020),17 Maggi et al. (2020),18 Sponsor submission,15,16 Pera et al. (2021).19
Motor-Function Outcomes
Motor function was the most commonly reported outcome in the included studies, with the HFMSE, RULM, and 6MWT the most frequently used measures to evaluate motor function in type II and III SMA adults.
The HFMSE consists of 33 itemized motor functions that assess activities of daily living. Each item is scored on a scale from 0 to 2, with higher scores indicating better motor function, up to a maximum of 66 points. According to Hagenacker et al. (2020)17 and Maggi et al. (2020),18 a score change of at least 3 points is considered a clinically meaningful improvement and indicated a response to treatment. Additional information on the measurement properties and minimally important difference in HFMSE results are described in Table 26 of Appendix 4.
The RULM consists of 20 items with a maximum of 37 points, with higher scores indicating better arm function. Score changes of at least 2 points were considered to be clinically meaningful and indicated a response to treatment, according to the Hagenacker et al. (2020)17 and Maggi et al. (2020).18 Additional information on the measurement properties and minimally important difference in RULM results are described in Table 26 of Appendix 4.
The 6MWT measures the maximum distance a patient can walk in 6 minutes. In Maggi et al. (2020),18 responders were defined as patients who improved from baseline by at least 30 m. Additional information on the measurement properties and minimally important difference in 6MWT results are described in Table 26 of Appendix 4.
Additional motor-function outcomes assessed included timed-function tests (timed run or walk 10 m, timed rise from floor, timed rise from chair, and timed climb 4 standard steps) in Maggi et al. (2020),18 with all results expressed as velocities; however, no information on these methods of assessment were provided.
Respiratory Outcomes
Respiratory outcomes were not frequently assessed in the included studies, with only Maggi et al. (2020)18 evaluating pulmonary function with the percent-predicted FEV1 and FVC. No description of these outcomes was provided.
Other Outcomes of Interest
The included studies did not provide information on any additional outcomes of interest defined in Table 4, including bulbar function, feeding requirements, survival, hospitalization, HRQoL, anatomic-related outcomes, or caregiver burden.
Safety Outcomes
Safety was not assessed using defined measures in any of the studies. In Hagenacker et al. (2020),17 AEs were reported as adverse drug reactions according to MedDRA (version 21.1). In Maggi et al. (2020),18 safety evaluations included vital signs, clinical and laboratory findings, and patient-reported AEs, all categorized by severity and relationship to nusinersen.18 The EU registry strudy15,16 reported the frequency of AEs, specifically in the adult population; however, AEs were not recorded in a standardized manner and the MedDRA classification was not used. AEs were not recorded in the study by Pera et al. (2021).19
Statistical Analysis
Hagenacker et al. (2020)
Statistical analyses were based on mean comparisons from baseline to months 6, 10, and 14 for primary and secondary end points, with the corresponding 95% CIs, and with the Wilcoxon signed-rank test. A difference of 0.31 or greater in a treatment-effect size can be detected with a power of 80% and with a 2-sided alpha of 0.05. This estimation was considered suitable by the authors for the primary end point and for the descriptive analysis of the secondary end points. A mixed model was used to estimate the treatment effect on the HFMSE score. The model used sex, time, age, spondylodesis, and HFMSE baseline score as fixed effects and the patient as a random effect. Outliers were not removed because there were no indications of incorrect measurements. No imputation of missing data was done for the 6-month, 10-month, or 14-month analyses. No alpha adjustment was done for potentially inflated type I error in the secondary end point analyses.17
A protocol for this study could not be located; however, in the article by Hagenacker et al. (2020),17 it was stated that preplanned subgroup analyses included analyses by HFMSE baseline score (≥ 35 versus < 35) or previous spondylodesis (yes versus no). Post hoc subgroup analyses were based on ambulant versus nonambulant status, and on SMA type (II versus III). Subgroup analyses were done by Mann–Whitney U test.
Sensitivity analyses were conducted by replacing missing baseline values for the primary end point using the existing values at later time points, such that these values were included in the analysis with a change from baseline of 0, and thus replaced conservatively. An additional sensitivity analysis also considered replacing the missing baseline values with values 5 points higher than at a later time points.17 The results and methods of these sensitivity analyses were not reported.17
Maggi et al. (2020)
No formal power calculation was presented for this study. The magnitude of change in outcomes from baseline at months 6, 10, and 14 was assessed in SMA type II and type III groups, including 2 type III subgroups of sitters and walkers. Walkers were defined as patients able to take at least a few steps independently or with aids (e.g., cane) but without the assistance of others. Responders to treatment with nusinersen were defined as patients who improved from baseline by at least 3 points on the HFMSE, 2 points on the RULM total score, or 30 m on the 6MWT. Responders in at least 1 of the 3 outcomes were defined as overall responders to treatment with nusinersen.18
No protocol for this study could be located. Change from baseline in study outcomes were summarized as mean (SD) or median (range), as appropriate. For comparisons of quantitative and ordinal variables between times of assessment, the Wilcoxon-Mann–Whitney test or Student’s t-test was used. For correlations between quantitative and/or ordinal variables, Spearman’s method was used. Distributions of categorical variables were compared with the Chi-square test. The effect of age, sex, and SMN2 copy number on treatment response was explored with logistic regression. Statistical significance was set at P < 0.05. No adjustments for multiplicity were done.18
No pre-specified sensitivity analyses were reported.
EU Registry Study
No protocol was provided for this study. No formal power calculation was reported for this study, and sample size was based on enrolment in the registries.
Two sets of data analyses were performed for the registry-based cohorts: a piecewise linear mixed model of pre- and post-treatment initiation comparisons; and an analysis of treated versus untreated patients based on a mixed-effect model. The first set of analyses explored the effects of treatment in the group of 75 adults with type III or IV SMA with data available before and after treatment initiation, and used piecewise linear mixed modelling of pre- and post-treatment outcomes to assess changes in HFMSE and RULM. For this analysis, the slope before the start of treatment was compared with the slope after treatment initiation in the treated cohort of patients (time spline). A piecewise linear mixed model was developed to consider the impact of treatment on functional ability scores, which estimated slopes of change over time separately in each treatment group, permitting assessment of whether the trajectory of the outcome over time differed between treated and untreated patients. Results were expressed as estimated change in points/week (95% CI). From the registries, 75 of 235 (31.91%) treated type III or type IV adults with at least 1 visit before the start of treatment and with at least 6 months of follow-up after treatment initiation were retained in the piecewise mixed models. These included:
38 patients from the Italian registry
26 patients from the German registry
11 patients from the Spanish registry.
The second set of analyses explored the effects of treatment by comparing outcomes among 235 patients treated with nusinersen and 17 untreated patients, using linear mixed-effects modelling to assess changes in disease progression, measured by changes in HFMSE and RULM scores over time. Results were presented for the overall type III population and the adult population (type III and IV SMA). Subgroup analyses were presented for type III fully ambulatory and type III not fully ambulatory patients. No pre-specified sensitivity analyses were reported.
The imputation of missing values for HFMSE and RULM were based on the scale total scores. Any missing baseline values were imputed using median within stratu, considering nonmissing baseline records. For post-baseline visits, flanked by nonmissing visits, missing values were imputed using linear interpolation. Only actual visits with nonmissing data were imputed for each patient. For the HFMSE, if more than 6 item scores were missing for an individual, then the total score imputed was as if all the 33 items were missing. For the RULM, if 3 or more items were missing for an individual, then the total score imputed was as if all 19 items were missing.15 At the final assessment, if the date was available and at least 1 item was nonmissing, the last observation carried forward (LOCF) method was used to impute missing HFMSE, RULM, and 6MWT scores. The multiple imputation method was also used, in which baseline missing data were handled with a chained equation with 10 imputations and stratified by registry.
The dependency in the data due to repeated measures was accounted for by a random intercept per individual, and an autoregressive covariance R matrix was used as correlation structure. The default estimation method (restricted maximum likelihood) was used for the covariance parameters. The Kenward-Roger method was used to compute degrees of freedom for the tests of fixed effects. The structure of the models was kept uniform with regard to the fixed- and random-effects structure. In consultation with 2 clinical experts, the EU registry study15,16 investigators included the following potential confounders in the regression model: age at onset of symptoms (onset at ≥ 3 years versus onset at < 3 years), sex (male versus female), number of SMA gene copies, motor function (not fully ambulatory or fully ambulatory), disease duration, registry (Germany, Italy, or Spain), age at baseline, and baseline score value.
Pera et al. (2021)
No protocol for this study could be located. Two analyses were conducted. First, all patients with a follow-up at 1 year of treatment were analyzed to evaluate 12-month changes in functional measures. Comparisons of measurements from baseline to 12 months were performed using estimates and 95% CI of before-and-after differences and the Wilcoxon signed-rank test. Twelve-month changes were also analyzed, subdividing the cohort by functional status (sitters versus walkers), SMA type III subtype (IIIA or IIIB), and age (children younger than 18 years versus adults). Details on 12-month trajectories grouped changes as stable (± 2 points), improved (more than 2 points), or declined (more than –2 points).19 For this review, only results for the overall adult population and for subgroup data for patients older than 20 years are presented.
A second analysis was added as no imputation was performed on missing data, and a number of patients failed the 12-month assessment because of restricted access to hospitals during the COVID-19 pandemic or other reasons. The second analysis consisted of a mixed model to estimate changes in the whole type III population and exclude possible selection bias. The model was set up with measurements at baseline (age, sex, time, disease duration, SMN2 copy number, disease onset, and SMA function) as fixed effects and the patient as a random effect. To make inferences about mean slopes by age at onset, the model was expanded to include appropriate main-effect and interaction terms in the model.19
Sensitivity analyses were performed for HFMSE and RULM after exclusion of the 3 nonsitter patients. An additional sensitivity analysis was conducted after the exclusion of 27 patients with baseline RULM scores equal to 37.19 No further information on the sensitivity analyses were provided.
Results
Patient Disposition
Hagenacker et al. (2020)
Of the 173 patients assessed for eligibility, 139 (80%) completed the 6-month assessment, 105 (61%) completed the 10-month assessment, and 61 (35%) completed the 14-month assessment at the time of analysis. Fifteen patients were excluded from the 6-month time of assessment because of missing baseline or 6-month time-of-assessment values, resulting in 124 patients available for the 6-month analysis. Thirty-four patients were subsequently excluded before 10 months — 30 who had not yet reached the 10-month assessment and 4 who withdrew (2 because of AEs and 2 who chose to) — and 13 were missing baseline values, resulting in a total of 92 patients in the 10-month assessment. A further 48 patients were excluded from the 14-month assessment — 44 and 4 did not have 14-month or baseline values, respectively, resulting in a total of 57 patients in the 14-month analysis.17
Maggi et al. (2020)
Of the 149 patients screened, 33 were excluded, resulting in a total of 116 included patients. Patients were excluded due to age (n = 5), disease onset after 18 years of age (n = 4), inability to complete the treatment loading phase because of side effects (n = 2), clinical trial enrolment (n = 1), no 6-month follow-up at the time of data collection (n = 21).18
EU Registry Study
A quality assessment of the individual registries was conducted; however, no information was provided on the quality of the databases in these registries. Data from the SMArtCARE, ISMAR, and CuidAME registries were combined to create a cohort of SMA patients from multiple countries. The SMArtCARE registry is an indication-specific registry that was initiated before the approval of nusinersen in Europe, but it did not start enrolment until the launch of nusinersen. The SMArtCARE registry aimed to evaluate all people with 5q SMA, regardless of their current treatment. Data collection took place as part of a patient’s regular, clinically recommended routine visits, which depended on the treatment they were receiving. Standardized results were collected during routine visits at regular intervals of 4 (nusinersen treatment) or 6 months (maximum time frame recommended by guidelines). The ISMAR registry is an ongoing collaboration between 3 large national networks in 16 locations in Italy, the UK, and the US to gain understanding of the disease and response to treatments. The data for the registry were collected as part of regular, clinically recommended routine visits, depending on the treatment the patient was receiving. The CuidAME registry collected data from 6 clinics relevant to the care of people diagnosed with 5q SMA, and included all patients with SMA, regardless of the treatment they were receiving.
The SMArtCARE registry included 240 type III SMA patients, the ISMAR registry included 114 type III SMA patients, and the CuidAME registry included 55 type III SMA patients. Overall, in the cross-registry analysis, 252 adults with type III SMA (235 treated with nusinersen and 17 untreated) were included in the analysis. Full details of the patient flow in the registries can be found in Figure 3.
Figure 3: Patient Flow in the EU Registry Study
FU = follow-up; RCT = randomized controlled trial; SMA = spinal muscular atrophy.
Note: a) Patient flow from the SMArtCARE registry in Germany; b) Patient flow from the ISMAR registry in Italy, the UK, and the US; c) Patient flow from the CuidAME registry in Spain.
Pera et al. (2021)
A summary of patient disposition was not provided in the study by Pera et al. (2021).19 A total of 144 SMA type III patients were enrolled in the study based on data collected from ISMAR.
Exposure to Study Treatments
Reporting of drug exposure varied in the included studies, given the study designs. However, patients all received nusinersen 12 mg according to standard protocols for at least 6 months. The follow-up duration in the Hagenacker et al. (2020)17 and Maggi et al. (2020)18 studies was up to 14 months, with assessments conducted at 6, 10, and 14 months.
In the EU registry study15,16 submitted by the sponsor, patients were required to have at least 6 months of treatment with nusinersen. In the SMArtCARE registry, the median length of follow-up in adults with type III SMA (n = 151) was 329 days (10.82 months; range = 168 to 930 days), with a mean of 5.01 (SD = 1.53) visits. In the ISMAR registry, the median length of follow-up in adults with type III SMA was 489 days (16.08 months; range = 161 to 757 days) in treated patients (n = 53), and 567 days (18.64 months; range = 520 to 700 days) in untreated patients (n = 5). Treated patients had a mean of 9.40 (SD = 5.88) visits, whereas untreated patients had a mean of 8.20 (SD = 8.29) visits during the entire follow-up period. In the CuidAME registry, the median follow-up in treated (n = 24) and untreated (n = 9) adults with type II SMA was 412 days (13.55 months; range = 167 to 706 days) and 413 days (13.58 months; range = 189 to 1,462 days), respectively. The mean number of visits was 4.96 (SD = 1.60) in treated patients and 4.22 (SD = 1.86) in untreated patients.
In Pera et al. (2021),19 patients treated with nusinersen were required to have a minimum of 12 months of follow-up, and the mean duration of follow-up was 1.83 years (SD = 0.61).
Effectiveness
Only effectiveness outcomes and analyses of subgroups identified in the review protocol are reported here. Refer to Appendix 3 for detailed effectiveness data.
Motor-Function Outcomes
HFMSE
Hagenacker et al. (2020): Results for HFMSE in the Hagenacker et al. (2020)17 study are summarized in Table 11. The mean HFMSE scores at all time points were greater than at baseline, with mean differences of 1.73 points (95% CI, 1.05 to 2.41) at 6 months (n = 124), 2.58 points (95% CI, 1.76 to 3.39) at 10 months (n = 92), and 3.12 points (95% CI, 2.06 to 4.19) at 14 months (n = 57). Compared with baseline, 35 (28%) of 124 patients at 6 months, 33 (35%) of 92 patients at 10 months, and 23 (40%) of 57 patients at 14 months had a greater than 3-point increase in HFMSE score, which the authors determined to be clinically meaningful.
Subgroup analyses of mean change in HFMSE score from baseline were consistent with the primary analysis at all time points in patients in all subgroups. However, changes were numerically larger in patients with type III SMA, ambulatory patients, and those without prior spondylodesis (Refer to Table 12).
In the subgroup analyses, more type III patients than type II patients had a greater than 3-point increase in HFMSE scores at all time points (23 [30%] versus 1 [2%] at 6 months, 19 [32%] versus 2 [7%] at 10 months, and 15 [41%] versus 1 [5%] at 14 months).
Results for sensitivity analyses, in which missing baseline values were replaced with existing values at later time points and missing baseline values were replaced with values 5 points higher than the values at later time points, remained consistent with the primary analysis (not shown).
Maggi et al. (2020): Results for HFMSE from the study by Maggi et al. (2020)18 are summarized in Table 13. In SMA type III patients, the mean change from baseline in HFMSE score at 6 months (n = 103), 10 months (n = 75), and 14 months (n = 46) was 1.48 points (SD = 2.28), 2.44 points (SD = 2.8), and 2.85 points (SD = 2.93), respectively. In patients with type II SMA (n = 13), the mean change from baseline in HFMSE score reached a high of 1.2 points (SD = 2.7) at 14 months; however, the median HFMSE change was zero at all assessment time points.
Table 11: Changes in HFMSE, RULM, and 6MWT Scores vs. Baseline (Hagenacker et al. [2020])
6-month analysis | 10-month analysis | 14-month analysis | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
n | Mean score (SD) | Mean difference vs. baseline (95% CI) | P value | n | Mean score (SD) | Mean difference vs. baseline (95% CI) | P value | n | Mean score (SD) | Mean difference vs. baseline (95% CI) | P value | |
HFMSE score | 124 | 22.47 (22.41) | 1.73 (1.05 to 2.41) | < 0.0001 | 92 | 25.52 (22.97) | 2.58 (1.76 to 3.39) | < 0.0001 | 57 | 27.77 (23.47) | 3.12 (2.06 to 4.19) | < 0.0001 |
RULM score | 120 | 21.53 (13.28) | 0.66 (0.26 to 1.05) | 0.0007 | 90 | 23.27 (12.46) | 0.59 (0.15 to 1.03) | 0.0014 | 58 | 23.95 (12.42) | 1.09 (0.62 to 1.55) | < 0.0001 |
6MWT distance, m | 47 | 366.8 (200.8) | 22.1 (8.7 to 35.6) | 0.0022 | 37 | 363.2 (224.2) | 31.1 (15.2 to 47.1) | < 0.0001 | 25 | 403.0 (225.7) | 46·0 (25.4 to 66.6) | < 0.0001 |
6-minute walk test; CI = confidence interval; HFMSE = Hammersmith Functional Motor Scale Expanded; RULM = Revised Upper Limb Module; SD = standard deviation; vs. = versus.
Notes: P values not adjusted for type I error.
Source: Hagenacker et al. (2020).17
Table 12: Subgroup Analysis of Changes in HFMSE and RULM Scores vs. Baseline (Hagenacker et al. [2020])
Subgroup | 6-month analysis | 10-month analysis | 14-month analysis | ||||||
|---|---|---|---|---|---|---|---|---|---|
n | Mean difference vs. baseline* | P value† | n | Mean difference vs. baseline* | P value† | n | Mean difference vs. baseline* | P value† | |
HFMSE score | |||||||||
SMA type | |||||||||
II | 45 | 0.6 (1.4; 0.2 to 1.1) | 0.0010 | 30 | 0.8 (1.5; 0.2 to 1.4) | 0.0054 | 20 | 1.1 (1.4; 0.4 to 1.7) | 0.0059 |
III | 77 | 2.4 (4.6; 1.4 to 3.5) | < 0.0001 | 60 | 3.4 (4.4; 2.2 to 4.5) | < 0.0001 | 37 | 4.2 (4.5; 2.7 to 5.7) | < 0.0001 |
Ambulant | |||||||||
Yes | 46 | 3.0 (4.7) | < 0.0001 | 35 | 4.3 (3.7) | < 0.0001 | 23 | 4.6 (4.4) | < 0.0001 |
No | 78 | 1.0 (3.0) | 0.0006 | 57 | 1.5 (3.0) | < 0.0001 | 34 | 2.1 (3.4) | < 0.0001 |
Baseline HFMSE score | |||||||||
≥ 35 | 39 | 2.4 (4.5) | 0.0002 | 33 | 3.6 (4.1) | < 0.0001 | 22 | 4.6 (4.2) | < 0.0001 |
< 35 | 85 | 1.4 (3.5) | < 0.0001 | 59 | 2.0 (3.7) | < 0.0001 | 35 | 2.2 (3.7) | < 0.0001 |
Spondylodesis | |||||||||
Yes | 28 | 0.8 (1.1) | 0.0024 | 18 | 1.2 (1.6) | 0.0059 | 14 | 1.4 (1.3) | 0.0078 |
No | 96 | 2.0 (4.3) | < 0.0001 | 74 | 2.9 (4.3) | < 0.0001 | 43 | 3.7 (4.4) | < 0.0001 |
RULM score | |||||||||
SMA type | |||||||||
II | 43 | 1.1 (2.4; 0.3 to 1.8) | 0.0005 | 30 | 1.1 (1.7; 0.5 to 1.7) | 0.0010 | 20 | 1.6 (2.0; 0.7 to 2.5) | 0.0049 |
III | 74 | 0.4 (2.1; –0.1 to 0.9) | 0.1371 | 58 | 0.4 (2.0; –0.1 to 0.9) | 0.0702 | 38 | 0.7 (1.7; 0.2 to 1.3) | 0.0100 |
CI = confidence interval; HFMSE = Hammersmith Functional Motor Scale Expanded; RULM = Revised Upper Limb Module; SD = standard deviation.; vs. = versus.
Note: P values were not adjusted for type I error.
*Data are mean difference (SD; 95% CI) or mean difference (SD).
†For 6-month, 10-month, or 14-month values vs. baseline.
Source: Hagenacker et al. (2020).17
Table 13: Summary of Clinical Assessment Outcomes (Maggi et al. [2020])
Variable | SMA type II | SMA type III sitters | SMA type III walkers | SMA type III total | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
n | Mean (SD) | Median (range) | Paired Wilcoxon | n | Mean (SD) | Median (range) | Paired Wilcoxon | n | Mean (SD) | Median (range) | Paired Wilcoxon P value | n | Mean (SD) | Median (range) | Paired Wilcoxon P value | |||
T0 to T6 change | ||||||||||||||||||
HFMSE | 13 | 0.15 (2.08) | 0 (–5 to 5) | NS | 51 | 1.37 (2.02) | 1 (–4 to 6) | < 0.0001 | 52 | 1.58 (2.52) | 1 (–5 to 8) | < 0.0001 | 103 | 1.48 (2.28) | 1 (–5 to 8) | < 0.0001 | ||
RULM | 12 | 0.8 (1.95) | 0 (–1 to 6) | NS | 51 | 0.63 (2.48) | 0 (–8 to 6) | 0.056 | 51 | 0 (1.23) | 0 (–4 to 3) | NS | 102 | 0.31 (1.97) | 0 (–8 to 6) | 0.093 | ||
6MWT | 0 | NA | NA | NA | 0 | NA | NA | NA | 48 | 14.66 (27.57) | 11 (–42.2 to 96) | 0.0005 | NA | NA | NA | NA | ||
FVC% | 4 | −0.25 (2.06) | 0 (–3 to 2) | NS | 19 | 0 (9.04) | 1 (–19 to 28) | NS | 16 | 1.16 (6.16) | 0.5 (–9 to 16) | NS | 35 | 0.53 (7.77) | 1 (–19 to 28) | NS | ||
HFMSE | 9 | 1 (2) | 0 (0 to 6) | NS | 35 | 2.51 (2.94) | 1 (–3 to 9) | < 0.0001 | 40 | 2.38 (2.71) | 2 (–3 to 8) | < 0.0001 | 75 | 2.44 (2.8) | 2 (–3 to 9) | < 0.0001 | ||
RULM | 9 | 1.67 (1.8) | 2 (0 to 5) | 0.057 | 33 | 1 (2.45) | 1 (–6 to 5) | 0.21 | 38 | 0.26 (1.66) | 0 (–4 to 6) | NS | 71 | 0.61 (2.08) | 0 (–6 to 6) | 0.011 | ||
6MWT | 0 | NA | NA | NA | 0 | NA | NA | NA | 35 | 26.45 (34.6) | 25 (–53 to 90) | 0.00019 | NA | NA | NA | NA | ||
FVC% | 4 | 0.75 (2.5) | 0.5 (–2 to 4) | NS | 7 | 3.3 (7.83) | 4.1 (–10 to 16) | NS | 10 | 5.8 (14.26) | 4.5 (–10 to 39) | NS | 17 | 4.77 (11.79) | 4.1 (–10 to 39) | NS | ||
HFMSE | 5 | 1.2 (2.68) | 0 (0 to 6) | NS | 19 | 3.53 (3.67) | 3 (–3 to 11) | 0.0014 | 27 | 2.37 (2.22) | 2 (–2 to 6) | 0.00016 | 46 | 2.85 (2.93) | 3 (–3 to 11) | < 0.0001 | ||
RULM | 5 | 1.6 (1.52) | 2 (0 to 3) | NS | 19 | 1.47 (2.5) | 2 (–6 to 5) | 0.018 | 25 | 0.4 (1.83) | 0 (–3 to 6) | NS | 44 | 0.86 (2.18) | 0.5 (–6 to 6) | 0.012 | ||
6MWT | 0 | NA | NA | NA | 0 | NA | NA | NA | 24 | 23.11 (51.2) | 20 (–101 to 111) | 0.016 | NA | NA | NA | NA | ||
FVC% | 0 | NA | NA | NA | 8 | 4.25 (8.55) | 1 (–4 to 19) | NS | 7 | 9 (9.95) | 7 (–1 to 29) | 0.031 | 15 | 6.47 (9.22) | 4 (–4 to 29) | 0.020 | ||
6WMT = 6-minute walk test distance (m); FVC% = percent-predicted forced vital capacity; HFMSE = Hammersmith Functional Motor Scale Expanded; NA = not available; NS = not significant; RULM = Revised Upper Limb Module (score); SD = standard deviation; SMA = spinal muscular atrophy; T0 = treatment initiation; T6 = 6 months of treatment; T10 = 10 months of treatment; T14 = 14 months of treatment.
Note: Significant P values are highlighted in bold. P values were not adjusted for type I error. The original publication by Maggi et al. (2020)18 reports both nonsignificant P values (> 0.05) and “NS” for other nonsignificant P values.
Source: Maggi et al. (2020).18
Clinically meaningful improvements in HFMSE (i.e., a 3-point change), as concluded by the authors, at all time points are summarized in Figure 4. The proportion of patients who had a 3-point or greater change from baseline in HFMSE score at 6 to 14 months ranged from 28% to 49% for all SMA patients, 8% to 20% for patients with type II SMA, 31% to 52% for patients with type III SMA, 27% to 58% for type III SMA sitters, and 35% to 48% for type III SMA walkers.
Figure 4: Responder Analyses for Change From Baseline in HFMSE, RULM, and 6MWT Scores in Adults With type II or type III SMA (Maggi et al. [2020])
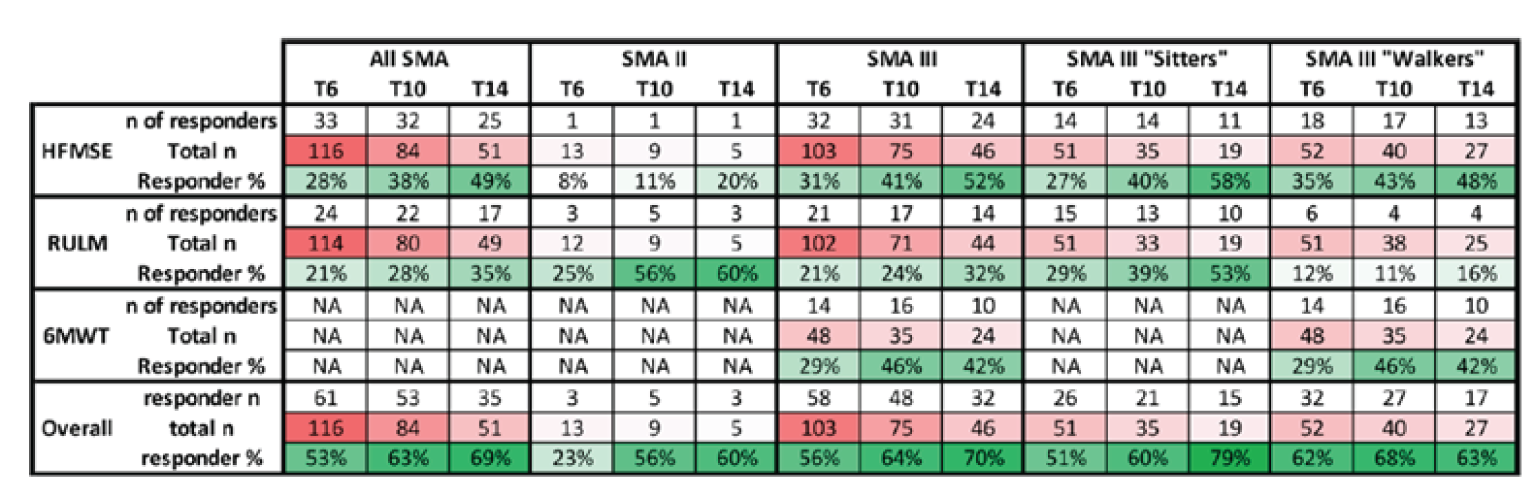
6MWT = 6-minute walk test; HFMSE = Hammersmith Functional Motor Scale Expanded; NA = not applicable; RULM = Revised Upper Limb Module; SMA = spinal muscular atrophy; T6 = 6 months of treatment; T10 = 10 months of treatment; T14 = 14 months of treatment.
Note: Responders were defined as having a change in HFMSE score from baseline of at least 3 points, a change in RULM score from baseline of at least 2 points, and a change in 6MWT distance from baseline of at least 30 m. “Overall” response was defined as a clinically meaningful response in at least 1 measure.
Source: Maggi et al. (2020).18
EU Registry Study: Results for the change in the HFMSE total score for the pre- and post-treatment nusinersen analysis are summarized in Table 14. The model results showed that before the start of nusinersen treatment, the HFMSE score decreased by an average of 0.00006 points per week. After treatment with nusinersen, the slope for the change in score per week was 0.2575 (95% CI, 0.01038 to 0.04112) (n = 75).15,16
Results for the change in the HFMSE total score between nusinersen-treated (n = 235) and untreated (n = 17) patients are summarized in Table 15. The slope for the change in nusinersen-treated patients was 0.02907 per week (95% CI, 0.01930 to 0.03884), compared with −0.01129 per week (95% CI, −0.03289 to 0.01031) in untreated patients.15,16 The slopes were similar regardless of the imputation method for missing data (LOCF or multiple imputation).
Pera et al. (2021): Results for the change in HFMSE in adults in Pera et al. (2021)19 are summarized in Table 16. In the primary analysis, the results for the mean change in HFMSE score from baseline for the adult population at 12-months (n = 45) was 0.79 points (95% CI, –0.29 to 1.87). Supplemental results based on descriptive statistics for all adults older than 20 years with type IIIA and type IIIB SMA varied, with mean changes from baseline at 12 months of 0.26 points (SD = 2.66) for ambulant patients (n = 19), and 1.58 points (SD = 3.91) for nonambulant patients (n = 26).
Table 14: Pre- and Post-Treatment With Nusinersen (n = 75; EU Registry Study)
Motor-function score | Pre-treatmenta | Post-treatmenta |
|---|---|---|
HFMSE | ||
Change in score in points/week (95% CI) | –0.00006 (–0.00955 to 0.009428) | 0.02575 (0.01038 to 0.04112) |
RULM | ||
Change in score in points/week (95% CI) | –0.00745 (–0.01401 to 0.0009) | 0.002569 (−0.00533 to 0.01047) |
6MWT | ||
Change in score in metres/week (95% CI) | –0.2393 (–0.349 to –0.1297) | –0.03399 (–0.4373 to 0.3694) |
6MWT = 6-minute walk test; CI = confidence interval; HFMSE = Hammersmith Functional Motor Scale Expanded; RULM = Revised Upper Limb Module.
aPiecewise linear mixed models adjusted for age at onset of symptoms (onset ≥ 3 years vs. onset < 3 years), sex (male vs. female), number of SMA gene copies, motor SMA function (not fully ambulatory or fully ambulatory), disease duration, registry (Germany, Italy, or Spain), age at baseline, and baseline score value.
Source: Sponsor submission.15,16
Table 15: Nusinersen-Treated vs. Untreated (Best Supportive Care Alone) Patients (EU Registry Study)
Motor-function score | Nusinersen-treated (n = 235)a | Untreated (n = 17)a |
|---|---|---|
HFMSE | ||
Change in score in points/week (95% CI) | 0.02907 (0.01930 to 0.03884) | −0.01129 (−0.03289 to 0.01031) |
RULM | ||
Change in score in points/week (95% CI) | 0.01168 (0.004957 to 0.01841) | 0.003936 (−0.01030 to 0.01817) |
6MWT | ||
Change in score in metres/week (95% CI) | 0.2633 (0.09922 to 0.42740) | –0.7148 (–1.2789 to –0.1506) |
6MWT = 6-minute walk test; CI = confidence interval; HFMSE = Hammersmith Functional Motor Scale Expanded; RULM = Revised Upper Limb Module.
aStandard Linear Mixed Model adjusted for age at onset of symptoms (Onset ≥ 3 years vs. Onset < 3 years), sex (Male vs. Female), number of SMA gene copies, motor SMA function (non-fully ambulatory; fully ambulatory), disease duration, registry (Italy; Germany; Spain), age at baseline, and baseline score value
Source: Sponsor submission.15,16
Table 16: Changes in HFMSE (12 Months vs. Baseline) in the Overall Adult and the Greater Than 20 Year SMA type III Populations (Pera et al. [2021])
Population | Baseline HFMSE score | 12-month HFMSE score | Mean difference | P value |
|---|---|---|---|---|
Primary analysis, adults, mean (95% CI) | ||||
Adults, n = 45 | 30.75 (29.97 to 31.53) | 31.54 (30.79 to 32.30) | 0.79 (–0.29 to 1.87) | 0.148 |
Supplementary descriptive statistics, > 20 years, mean (SD) | ||||
All SMA, n = 104 | ||||
All SMA > 20, n = 45 | 30.13 (18.3) | 31.16 (18) | 1.02 (3.47) | N/A |
Type IIIA > 20, n = 11 | 21.91 (18.12) | 22.45 (17.23) | 0.55 (4.59) | N/A |
Type IIIB > 20, n = 34 | 32.79 (17.81) | 33.97 (17.56) | 1.18 (3.09) | N/A |
Nonambulant (n = 44) | ||||
All SMA > 20, n = 26 | 17.85 (11.94) | 19.42 (12.65) | 1.58 (3.91) | N/A |
Type IIIA > 20, n = 9 | 16.33 (14.56) | 16.33 (14.56) | 1.33 (4.72) | N/A |
Type IIIB > 20, n = 17 | 18.65 (10.71) | 20.35 (11.72) | 1.71 (3.57) | N/A |
Ambulant (n = 60) | ||||
All SMA > 20, n = 19 | 46.95 (10.21) | 47.21 (9.92) | 0.26 (2.66) | N/A |
Type IIIA > 20, n = 2 | 47 (7.07) | 44 (8.49) | –3 (1.41) | N/A |
Type IIIB > 20, n = 17 | 47.59 (10.24) | 47.59 (10.24) | 0.65 (2.52) | N/A |
CI = confidence interval; HFMSE = Hammersmith Functional Motor Scale Expanded; N/A = not applicable; SD = standard deviation; SMA = spinal muscular atrophy.
Source: Pera et al. (2021).19 This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 International Licence. Full text available here: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8351459/
Stability of HFMSE scores at 12 months in adults older than 20 years are summarized in Table 19. In all adults (> 20 years), the HFMSE results declined in 11.1% of patients, remained stable in 53.3%, and improved in 35.6%, based on the definitions used to categorize stability. These results were similar in walkers and sitters.
In an additional comparison with an external untreated cohort, patients older than 20 years treated with nusinersen (n = 45) had a mean 12-month change in HFMSE of 1.02 points (SD = 3.47), compared with –1.65 points (SD = 3.42) in the untreated external cohort (n = 49).
RULM
Hagenacker et al. (2020): Results for RULM from the Hagenacker et al. (2020)17 study are summarized in Table 11. The mean RULM scores at all time points were similar to those at baseline, with mean differences of 0.66 points (95% CI, 0.26 to 1.05) at 6 months (n = 120), 0.59 points (95% CI, 0.15 to 1.03) at 10 months (n = 90), and 1.09 points (95% CI, 0.62 to 1.55) at 14 months (n = 58). At the 6-month follow-up, a greater than 2-point increase in RULM score, which was considered clinically meaningful by the authors, was observed in 28 (23%) patients, whereas 74 (64%) patients showed no meaningful change and 18 (15%) and 10 (8%) showed a decline of 1 point or more, or a decline of 2 points or more, respectively.
Subgroup analysis for RULM scores in type II and type III patients is presented in Table 12.
Maggi et al. (2020): Results for RULM scores in Maggi et al. (2020)18 are summarized in Table 13. In SMA type III patients, the mean change from baseline in RULM score at 6 months (n = 102), 10 months (n = 71), and 14 months (n = 44) was 0.31 points (SD = 1.97), 0.61 points (SD = 2.08), and 0.86 points (SD = 2.18), respectively. Patients with type II SMA had changes in mean RULM scores of 0.8 points (SD = 1.95) at 6 months (n = 12), 1.67 points (SD = 1.8) at 10 months (n = 9), and 1.6 points (SD = 1.52) at 14 months (n = 5).
Clinically meaningful improvements in HFMSE, according to the authors, at all time points are summarized in Figure 4. The proportion of patients with a 2-point change in RULM score at 6 to 14 months ranged from 21% to 35% for all SMA patients, 25% to 60% for patients with type II SMA, 21% to 32% for patients with type III SMA, 29% to 53% for type III SMA sitters, and 11% to 16% for type III SMA walkers.
EU Registry Study: Results for the change in RULM score from the pre- and post-treatment analysis in the EU registry study15,16 are summarized in Table 14. The model results showed that before the start of nusinersen treatment, the RULM score decreased by an average of 0.00745 points per week (95% CI, –0.01401 to 0.0009). After treatment with nusinersen, the slope for the change in score per week was 0.002569 points per week (95% CI, −0.00533 to 0.01047). The same trend was seen when patients with a ceiling effect were excluded from the analysis (not shown).
Results comparing the change in RULM between nusinersen-treated (n = 235) and untreated (n = 17) patients are summarized in Table 15. The slope in nusinersen-treated patients was 0.01168 points per week (95% CI, 0.004957 to 0.01841), compared with 0.003936 points per week (95% CI, −0.01030 to 0.01817) in untreated patients.
Pera et al. (2021): Results for the change in RULM scores in adults in Pera et al. (2021)19 are summarized in Table 17. In the primary analysis, the mean change from baseline in RULM score in adults (n = 55) at 12-months was 0.07 points (95% CI, –0.48 to 0.63). In the subgroup of nonambulant patients older than 20 years (n = 33), the mean change from baseline in RULM score at 12 months was 0.48 points (SD = 2.12). In the subgroup of ambulant type III SMA patients 20 years and older (n = 21), the mean change from baseline at 12 months in RULM score was –0.62 points (SD = 2.11).
Stability of RULM scores at 12 months are summarized in Table 19. In all adults (> 20 years), RULM scores declined in 13.0% of patients, remained stable in 75.9%, and improved in 15.6%. These results were generally similar in ambulant and nonambulant patients.
Table 17: Changes in RULM (12 Months vs. Baseline) in the Overall Adult and the Greater Than 20 Year SMA type III Populations (Pera et al. [2021])
Population | Baseline RULM score | 12-month RULM score | Mean difference | P value |
|---|---|---|---|---|
Primary analysis, adults, mean (95% CI) | ||||
Adults, n = 55 | 27.31 (26.91 to 27.71) | 27.38 (26.99 to 27.78) | 0.07 (–0.48 to 0.63) | 0.792 |
Adults with baseline RULM score < 37, n = 42 | 24.38 (23.87 to 24.88) | 24.62 (24.12 to 25.11) | 0.24 (–0.47 to 0.94) | 0.505 |
Supplementary descriptive statistics, > 20 years, mean (SD) | ||||
All SMA, n = 100 | ||||
All SMA > 20, n = 54 | 27.07 (9.64) | 27.13 (9.29) | 0.06 (2.17) | N/A |
Type IIIA > 20, n = 18 | 22 (9.56) | 22.83 (9.94) | 0.83 (1.79) | N/A |
Type IIIB > 20, n = 36 | 29.61 (8.75) | 29.28 (8.27) | –0.33 (2.26) | N/A |
Nonambulant (n = 47) | ||||
All SMA > 20, n = 33 | 22.67 (9.53) | 23.15 (9.27) | 0.48 (2.12) | N/A |
Type IIIA > 20, n = 16 | 20.38 (8.82) | 21.25 (9.36) | 0.88 (1.89) | N/A |
Type IIIB > 20, n = 17 | 24.82 (9.93) | 24.94 (9.1) | 0.12 (2.32) | N/A |
Ambulant (n = 53) | ||||
All SMA > 20, n = 21 | 34 (4.34) | 33.38 (4.89) | –0.62 (2.11) | N/A |
Type IIIA > 20, n = 2 | 35 (2.83) | 35.5 (2.12) | 0.5 (0.71) | N/A |
Type IIIB > 20, n = 19 | 33.89 (4.51) | 33.16 (5.08) | –0.74 (2.18) | N/A |
CI = confidence interval; N/A = not applicable; RULM = Revised Upper Limb Module; SD = standard deviation; SMA = spinal muscular atrophy.
Source: Pera et al. (2021).19 This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 International Licence. Full text available here: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8351459/
Table 18: Changes in 6MWT (12 Months vs. Baseline) in the Overall Adult and the Greater Than 20 Year SMA type III Populations (Pera et al. [2021])
Population | Baseline 6MWT score | 12-month 6MWT score | Mean difference | P value |
|---|---|---|---|---|
Primary analysis, adults, mean (95% CI) | ||||
Adults, n = 17 | 323.03 (308.23 to 337.83) | 323.55 (309.55 to 337.55) | 0.52 (–19.85 to 20.89) | 0.959 |
Supplementary descriptive statistics, > 20 years, mean (SD) | ||||
All SMA, n = 51 | ||||
All SMA > 20, n = 16 | 313 (173.21) | 310.31 (176.69) | –2.69 (42.35) | N/A |
Type IIIA > 20, n = 2 | 196.5 (126.57) | 171 (135.76) | –25.5 (9.19) | N/A |
Type IIIB > 20, n = 14 | 329.64 (176.06) | 330.21 (176.61) | 0.57 (44.4) | N/A |
6MWT = 6-minute walk test; CI = confidence interval; N/A = not applicable; SD = standard deviation; SMA = spinal muscular atrophy.
Source: Pera et al. (2021).19 This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 International Licence. Full text available here: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8351459/
Table 19: 12-Month Trajectories Grouped as Stable ( + 2 Points), Improved (> + 2), or Declined (< −2) HFMSE and RULM Scores in Patients Greater Than 20 Years (Pera et al. [2021])
Population | Declined (< –2), n (%) | Stable (−2.2), n (%) | Improved (> 2), n (%) |
|---|---|---|---|
HFMSE, n (%) | |||
All SMA, n = 104 | |||
All SMA > 20, n = 45 | 5 (11.1) | 24 (53.3) | 16 (35.6) |
Type IIIA > 20, n = 11 | 2 (18.2) | 6 (54.5) | 3 (27.3) |
Type IIIB > 20, n = 34 | 3 (8.8) | 18 (52.9) | 13 (38.2) |
Nonsitter, n = 1 | |||
Type IIIA > 20, n = 1 | 0 | 1 (100) | 0 |
Sitter, n = 43 | |||
All SMA > 20, n = 25 | 2 (8.0) | 12 (48.0) | 11 (44.0) |
Type IIIA > 20, n = 8 | 1 (12.5) | 4 (50.0) | 3 (37.5) |
Type IIIB > 20, n = 17 | 1 (5.9) | 8 (47.1) | 8 (47.1) |
Walker, n = 60 | |||
All SMA > 20, n = 19 | 3 (15.8) | 11 (57.9) | 5 (26.3) |
Type IIIA > 20, n = 2 | 1 (50.0) | 1 (50.0) | 0 |
Type IIIB > 20, n = 17 | 2 (11.8) | 10 (58.8) | 5 (29.4) |
RULM, n (%) | |||
All SMA, n = 100 | |||
All SMA > 20, n = 47 | 7 (13.0) | 41 (75.9) | 6 (11.1) |
Type IIIA > 20, n = 18 | 0 | 15 (83.3) | 3 (16.7) |
Type IIIB > 20, n = 36 | 7 (19.4) | 26 (72.2) | 3 (8.3) |
Nonsitter, n = 2 | |||
Type IIIA > 20, n = 1 | 0 | 1 (100) | 0 |
Sitter, n = 46 | |||
All SMA > 20, n = 32 | 2 (6.3) | 25 (78.1) | 5 (15.6) |
Type IIIA > 20, n = 15 | 0 | 12 (80.0) | 3 (20.0) |
Type IIIB > 20, n = 17 | 2 (11.8) | 13 (76.5) | 2 (11.8) |
Walker, n = 53 | |||
All SMA > 20, n = 21 | 5 (23.8) | 15 (71.4) | 1 (4.8) |
Type IIIA > 20, n = 2 | 0 | 2 (100.0) | 0 |
Type IIIB > 20, n = 19 | 5 (26.3) | 13 (68.4) | 1 (5.3) |
CI = confidence interval; HFMSE = Hammersmith Functional Motor Scale Expanded; RULM = Revised Upper Limb Module; SD = standard deviation; SMA = spinal muscular atrophy.
Source: Pera et al. (2021).19 This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 International Licence. Full text available here: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8351459/
6MWT
Hagenacker et al. (2020): Results for the change in 6MWT in Hagenacker et al. (2020)17 are summarized in Table 11. The mean changes from baseline in 6MWT were 22.12 m (95% CI, 8.7 to 35.6) at 6 months (n = 47), 31.14 m (95% CI, 15.2 to 47.1) at 10 months (n = 37), and 45.96 m (95% CI, 25.4 to 66.6) at 14 months (n = 25).
Maggi et al. (2020): Results for the change in 6MWT in Maggi et al. (2020)18 are summarized in Table 13. Results for SMA type II patients, type III sitters, and all-type III SMA patients were not available, Walkers with type III SMA had a mean change in 6MWT of 14.66 m (SD = 27.57) at 6 months (n = 48), 26.45 m (SD = 34.6) at 10 months (n = 35), and 23.11 m (SD = 51.2) at 14 months (n = 24). The proportion of patients achieving a minimum improvement in 6MWT of 30 m was 29% at 6 months, 46% at 10 months, and 42% at 14 months (Figure 4).
Results for the rise from chair timed function test showed an increase in velocity at 6 months (0.02 second–1), 10 months (0.04 second–1), and 14 months (0.06 second–1). Ten-metre run or walk speed increased only at 6 months ( + 0.07 m/s).
EU Registry Study: Results for the change in 6MWT from the pre- and post-treatment analysis in the EU registry study15,16 are summarized in Table 14. The model results showed that before the start of nusinersen treatment, the 6MWT distance decreased by 0.2393 m per week (95% CI, –0.349 to –0.1297). After treatment with nusinersen, the slope for the change in 6MWT was –0.03399 m per week (95% CI, −0.4373 to 0.3694).
Results comparing the change in 6MWT between nusinersen-treated and untreated patients are summarized in Table 15. The slope in nusinersen-treated patients (n = 235) was 0.2633 m per week (95% CI, 0.09922 to 0.4272), compared with –0.7148 m per week (95% CI, −1.2789 to –0.1506) in untreated patients (n = 17).
Pera et al. (2021): Changes in 6MWT for adults in Pera et al. (2021)19 are summarized in Table 18. The mean 6MWT distance from baseline to 12 months for adults (n = 17) was 0.52 m (95% CI, –19.85 to 20.89).
Respiratory Outcomes
FVC/FEV1
Hagenacker et al. (2020): Changes in lung function were not evaluated in the Hagenacker et al. (2020)17 study.
Maggi et al. (2020): Results for respiratory outcomes in the study by Maggi et al. (2020)18 are summarized in Table 13. The mean change in percent-predicted FVC in all type III SMA patients was 0.53% (SD = 7.77) at 6 months (n = 35), 4.77% (SD = 11.79) at 10 months (n = 17), and 6.47% (SD = 9.22) at 14 months (n = 15). In SMA type II patients, the mean change in FVC was –0.25% (SD = 2.06) at 6 months, 0.75% (SD = 2.5) at 10 months, and was not available at 14 months due to small sample sizes. Mean change in percent-predicted FVC showed an increase over time for type III sitters and walkers.
In all SMA patients, the percent-predicted FEV1 change from baseline was 0.01% (SD:8.45) at 6 months (n = 32), 6.93% (SD = 18.37) at 10 months (n = 18), and 5.86% (SD = 9.22) at 14 months (n = 14). Change from baseline in the percent-predicted FEV1 at 6 months in subgroups of type II SMA patients, type III SMA patients, type III sitters, and type III walkers showed varied results at the 3 evaluation time points.
EU Registry Study: Changes in lung function were not evaluated in the EU registry study.15,16
Pera et al. (2021): Changes in lung function were not evaluated in the Pera et al. (2021)19 study.
Bulbar Function
Outcomes related to bulbar function or feeding support were not included in the studies.
Survival
Outcomes related to survival were not included in the studies.
Hospitalization
Outcomes related to hospitalization were not included in the studies.
HRQoL
Outcomes related to HRQoL were not included in the studies.
Anatomic-Related Outcomes
Anatomic-related outcomes were not included in the studies.
Caregiver Burden
Outcomes related to caregiver burden were not included in the studies.
Harms
Only harms identified in the review protocol are reported in the following sections.
AEs and SAEs
Hagenacker et al. (2020)
A total of 82 (47%) patients who received at least 1 injection had AEs due to drug reactions or procedure-related complications (Table 20). Throughout the 14-month period, the most frequently reported AEs were headache (35%), back pain (22%), and nausea (11%).17
No SAEs were reported.
Table 20: Adverse Drug Reactions and Procedure-Related Complications Recorded in Participants (n = 173), Classified According to MedDRA Version 21.1 (Hagenacker et al. [2020])
Day 1, injection 1; n = 173 | Day 14, injection 2; n = 170 | Day 28, injection 3; n = 165 | Day 63, injection 4; n = 158 | Month 6, injection 5; n = 139 | Month 10, injection 6; n = 105 | Month 14, injection 7; n = 61 | |
|---|---|---|---|---|---|---|---|
Total adverse reactions, n | 68 | 58 | 31 | 23 | 20 | 14 | 8 |
Total patients with adverse reactions, n (%) | 52 (30) | 40 (24) | 26 (16) | 20 (13) | 17 (12) | 13 (12) | 6 (10) |
Headache, n (%) | 35 (20) | 27 (16) | 19 (12) | 12 (8) | 7 (5) | 4 (4) | 4 (7) |
Back pain, n (%) | 16 (9) | 16 (9) | 7 (4) | 5 (3) | 7 (5) | 3 (3) | 2 (3) |
Nausea, n (%) | 12 (7) | 6 (4) | 3 (2) | 2 (1) | 3 (2) | 1 (1) | 1 (2) |
Vertigo, n (%) | 3 (2) | 5 (3) | 2 (1) | 0 | 1 (1) | 2 (2) | 0 |
Upper airway infection, n (%) | 1 (1) | 0 | 0 | 2 (1) | 0 | 2 (2) | 0 |
Constipation, n (%) | 0 | 2 (1) | 0 | 1 (1) | 2 (1) | 0 | 1 (2) |
Diffuse pain, n (%) | 0 | 0 | 0 | 0 | 0 | 1 (1) | 0 |
Bladder disorder not otherwise specified, n (%) | 0 | 0 | 0 | 1 (1) | 0 | 0 | 0 |
Tinnitus aggravated, n (%) | 0 | 0 | 0 | 0 | 0 | 1 (1) | 0 |
Infection, n (%) | 0 | 1 (1) | 0 | 0 | 0 | 0 | 0 |
Meningitis aseptic, n (%) | 0 | 1 (1) | 0 | 0 | 0 | 0 | 0 |
Source: Hagenacker et al. (2020).17
Maggi et al. (2020)
Overall, AEs were reported in 48 (41.4%) patients, with the most frequently reported AE being post-procedural headache (n = 43 [37.1%]). Headache was orthostatic, mild to moderate in intensity, and spontaneously resolved in a few days, except in 5 patients who required hospitalization. Two patients with type III SMA reported transient worsening of existing hand tremor. The investigators generally considered AEs mild or moderate and to be related to nusinersen administration procedures.18
EU Registry Study
AEs and SAEs at 6 and 11 months in the EU registry study15,16 are summarized in Table 21. In patients who received nusinersen, 78 (34.67%) experienced at least 1 AE at 6 months, compared with 86 (38.22%) at 11 months. At least 1 SAE occurred in 8 (3.79%) and 10 (4.74%) patients at 6 and 11 months, respectively.
Table 21: Safety in the Adult Population (EU Registry Study)
Population | With treatment | Without treatment | Without vs. with treatment | ||||
|---|---|---|---|---|---|---|---|
N | Patients with event, n (%) | N | Patients with event, n (%) | IRR | 95% CI | P value | |
6 monthsa | |||||||
Patients with ≥ 1 AE | 225 | 78 (34.67) | 12 | 0 | 316.21 | –605.94 to 633.59 | 0.965 |
Patients with ≥ 1 SAE | 211 | 8 (3.79) | 6 | 0 | b | b | b |
11 monthsa | |||||||
Patients with ≥ 1 AE | 225 | 86 (38.22) | 12 | 0 | 316.20 | –605.73 to 633.73 | 0.965 |
Patients with ≥ 1 SAE | 211 | 10 (4.74) | 6 | 0 | 104.80 | –196.88 to 213.93 | 0.935 |
AE = adverse event; CI = confidence interval; IRR = incidence rate ratio; n = number of patients with event; N = number of patients assessed; SAE = serious adverse event.
aDifference in time ≤ 10%.
bModel did not converge.
Source: Sponsor submission.15
Pera et al. (2021)
The most frequent AEs were all related to the procedure, and included headache, nausea, and back pain,19 although frequency was not reported. No SAEs were reported at the time of data collection.
WDAEs
WDAEs were minimal in the included studies. In Hagenacker et al. (2020),17 2 patients withdrew from treatment at 10 months because of adverse drug reactions. In Maggi et al. (2020),18 nusinersen treatment was stopped in 2 (1.7%) type III SMA patients after 6 months due to lack of subjective benefit and poor tolerability of repeated lumbar puncture. WDAEs were not reported in the EU registry study15,16 or the study by Pera et al. (2021).19
Mortality
Survival and mortality were not reported in any of the included studies.
Notable Harms
Serious Infections
Serious infections were not reported in any of the included studies; however, as noted in Table 20, 1 (1%) patient experienced an infection at injection 2, and upper airway infections were experienced by 1 (1%) patient at injection 1 and by 2 (1%) patients each at injections 4 and 6 in the Hagenacker et al. (2020)17 study, although the severity was unknown.
Lumbar-Puncture-Related AEs
Hagenacker et al. (2020): After lumbar puncture, headache (61 [35%]) and back pain (38 [22%]) were the most frequently reported AEs in the Hagenacker et al. (2020)17 study at 14 months, occurring in up to 1-fifth of patients.
Maggi et al. (2020): As mentioned previously, 2 (1.7%) patients with type III SMA discontinued treatment due to poor tolerability of repeated lumbar puncture.18 Additionally, post-procedural headache was reported in 43 (37.1%) patients. Headaches were generally characterized as mild to moderate in intensity and resolved spontaneously; however, hospitalization was required in 5 patients. Headache was associated with manual procedures in 34 cases and with imaging-guided techniques in 9 cases, with no difference in headache probability between the 2 approaches. Lumbar pain was reported by 10 (8.6%) patients, 7 of whom underwent imaging-guided lumbar puncture.
The frequency of lumbar-puncture-related AEs was not reported in the EU registry study15,16 or in the study by Pera et al. (2021).19
Coagulation Abnormalities
No coagulation abnormalities were reported in the included studies.
Renal Toxicity
In the study by Maggi et al. (2020),18 1 case of renal colic requiring hospitalization was reported in a patient with type II SMA after the 10-month assessment. No other included studies reported events of renal toxicity.
Critical Appraisal
No RCTs focusing on treatment-naive adults with type II or III SMA were identified as part of the CADTH literature search, and all available and included studies were of observational design, focusing on real-world data, which have more limitations than RCTs.
The studies included in this reassessment consisted of 1 prospective (Hagenacker et al. [2020]17) and 3 retrospective, observational, noncomparative studies, comprising cohorts of patients who had been treated with nusinersen at individual centres or who were enrolled in a registry. Overall, the included studies suffer from high levels of selection bias, reporting bias, and information bias. There is a high potential for selection bias because of the observational design of the studies and the way patients were chosen, and a risk of reporting and information bias because of the methods used for data collection. All studies were also noncomparative in nature, negating the ability to draw a statistical association between the reported results and nusinersen because of small sample sizes (although this is reasonable for a rare disease), the slow and minimal progression of SMA in adults, and the restriction to mostly high-functioning type III SMA patients. Additional details on the internal and external validity of the included studies follow.
Internal Validity
Hagenacker et al. (2020)
The study by Hagenacker et al. (2020)17 was a prospective, multi-centre, uncontrolled, noncomparative, observational cohort study. Given the noncomparative study design, there was no control group for comparison, so the results observed cannot be attributed to treatment with nusinersen. Moreover, there was a lack of blinding of both patients and outcome assessors. There is a risk of bias because of the subjective nature of the outcome measurements, as patients and providers were aware of the treatment.
This was a prospective cohort study, and selection criteria limited patients to those with confirmed 5q SMA and prior nusinersen treatment for at least 6 months. This selected for patients who were able to complete the induction dosing and who were able to tolerate and/or receive doses. No other selection criteria were defined, and the authors noted that as nusinersen is approved for the treatment of a severe, chronic, progressive disease without limitations on age or disease classification, further controls were not warranted. Evidence of selection biases was noted in baseline characteristics — the study included higher-functioning, ambulatory, type III SMA patients — and is discussed in detail in the External Validity section. As such, the risk of selection bias is likely, and no adjustment techniques were applied to correct for potential selection biases.
Missing data also impacted the validity of the results. A total of 173 patients were screened for enrolment. The proportion of patients lost to follow-up was not reported, although of the 139 patients enrolled, only 57 (41.0%) were available at 14-month follow-up. Thus, there was a high level of variation in the number patients available at different follow-up times, resulting in a lack of data at later time points. No imputation of missing data was done for the 6-month, 10-month, or 14-month analyses, nor for the amount of missing data during the observation period for each outcome reported. Assessment of attrition was not conducted, and it is unclear whether attrition was random (i.e., whether nusinersen was discontinued due to lack of efficacy or AEs); therefore, there is an increased risk that the treatment effect of nusinersen is overestimated.
Methods for statistical analysis of pre- and post-treatment comparisons for the primary outcome of HFMSE using nonparametric tests at selected time points, and the conducted subgroups analyses, were reportedly pre-specified, although no protocol was identified. A list of potential confounders was not described; however, a mixed model that included sex, time, age, spondylodesis, and baseline HFMSE score as a fixed effect and the patient as random effect was only used to estimate effect on the primary outcome. No rationale for these variables was provided. Other potential confounders and treatment-effect modifiers that were not identified or considered, even though they may influence outcomes, include training for the outcomes of interest, routine exercise and observation, other routine care (such as physiotherapy and occupational therapy), and the placebo effect. The extent to which uncontrolled confounders and treatment-effect modifiers influenced the results is unclear.
Maggi et al. (2020)
The study by Maggi et al. (2021)18 was a retrospective, multi-centre, noncomparative, observational cohort study. The aim and outcome measures of this study were clearly described, although no protocol was identified. There was no comparator group in this study, so the results observed cannot be attributed to treatment with nusinersen. Because patients and outcome assessors were not blinded to treatment, there is a risk of bias in outcome assessment.
As with the Hagenacker et al. (2020)17 study, a key limitation with the Maggi et al. (2020)18 study was the potential for selection bias. A similar population of eligible patients was included (i.e., adults with confirmed 5q SMA), and reasons for exclusion were noted; however, given that it was a retrospective study, patients were likely selected after treatment with nusinersen and, therefore, the study is at high risk of selection bias. It was not clear whether or how the potential for selection bias was assessed, and it was not reported whether adjustment techniques were applied to correct for potential selection biases. There was potential for information bias in this study because of the retrospective design and the lack of reporting on data quality and accuracy. No consideration of potential information bias was noted in this study and, therefore, it is unclear what effect this may have had on the results. No information was provided on the methods used to account for missing data, nor were amounts of missing data reported. The authors noted that missing data were mostly limited to timed and pulmonary function tests.
A total of 116 patients were enrolled in the study; however, just 17% to 65% of patients (depending on the analysis group and outcome measure) completed outcome assessments at 14-month follow-up. There were also notably small sample sizes for certain subgroups, particularly patients with type II SMA (n = 13), making it very difficult to interpret results for these patients.
Both parametric and nonparametric tests were conducted on the population. No consideration was given to potential confounders in this study. Logistic regression was used to identify the effects of predictor variables (age, sex, SMN2 copy number) on treatment response; however, no further information on this process (e.g., selection of variables in the model) or the way results of the regression were used was provided. Potential confounders and treatment-effect modifiers that were not identified or considered, even though they may influence outcomes, include training for the outcomes of interest, routine exercise and observation, other routine care (such as physiotherapy and occupational therapy), and the placebo effect. The extent to which uncontrolled confounders and treatment-effect modifiers influenced the results is unclear.
EU Registry Study
Data from the EU registry study15,16 submitted by the sponsor was based on data from 3 registries — SMArtCARE, ISMAR, and CuidAME — which collected data from multiple centres in Germany, Italy, Spain, the UK, and the US. Data in these registries were collected both prospectively and retrospectively. Although registries can provide large collections of data on real-world patients and settings, there are concerns with the quality of data from registries due to limitations in the availability of key elements (patient characteristics, exposures, outcomes), as well as the reliability of data accuracy, completeness, provenance, or traceability, and no information on accuracy was reported.
A key issue is the way patients are enrolled in the registry, which can increase the potential for selection bias. It was unclear if these registries represented patients with different self-care practices, socioeconomic backgrounds, or levels of supportive care than patients in the entire population of interest for the reassessment (type II and III SMA). According to the sponsor, the inclusion and exclusion criteria for patients in each registry were identical, including a genetically confirmed diagnosis of 5q SMA. Patient were enrolled in each registry through SMA clinics; however, information on the enrolment process was not provided. It was unclear if patients received nusinersen before or after enrolment in the registries. Additional selection biases were noted in the baseline characteristics — the study included ambulatory, high-functioning, type III SMA patients — and are discussed in detail in the External Validity section.
The sponsor conducted a quality assessment of each individual registry using the IQWiG tool, an abbreviated version of the Reporting of Studies Conducted Using Observational Routinely-Collected Data (RECORD) tool, and the National Institutes of Health Quality Assessment Tool for Observational Cohort and Cross-Sectional Studies, although no accuracy or agreement between the registry sample and reabstracted records was provided. The results of this quality assessment were not appraised. No overarching protocol was submitted for this study; however, the sponsors provided individual publications for the development of each of the registries. The ISMAR registry did not share a single protocol at all centres, so there may be differences in collection methods or procedures across institutions that are unaccounted for.
A statistical analysis plan for the EU registry study15,16 was submitted in response to a request from CADTH. The sponsor pooled patients from the 3 registries into 2 analysis groups (one that compared 235 treated patients with 17 untreated patients, and 1 pre- and post-treatment analysis that consisted of 75 patients); however, no information on the pooling methods or justification for the pooling of patients from registries was provided. Regardless of pooling, differences in treated (N = 235) and untreated (N = 17) populations — the German SMArtCARE registry had no untreated patients, the Italian ISMAR registry had 5 untreated type III adults, and the Spanish CuidAME registry had 9 untreated type III adults — highlighting the uncertainty around how patients were enrolled in the registries and potential bias in the selection of patients. Due to the limited number of patients in the untreated groups, the results observed cannot be attributed to nusinersen.
Two separate analyses were presented for the EU registry study.15,16 In an unpublished report, the sponsor provided baseline data and motor-function outcome results for all type III SMA patients and for adults with type III SMA, with subgroups of fully ambulatory and not fully ambulatory patients. First, a standard mixed-effect model used LOCF and multiple-imputation methods to account for missing data, with interaction terms between treatment and time in the model. However, no information on the modelling approach was provided. No information was presented on the amount of missing data. The LOCF method of imputing missing data may have increased the chance of bias because it cannot be confirmed, based on the available information, whether missing data occurred at random. Also, the lack of details reported about the amount of missing data and the time contribution of each patient make it difficult to determine the effects of LOCF imputation. The sponsor also conducted a multiple imputation model on the missing, pooled baseline characteristics, as well as on the outcomes of interest; however, no information on the amount of missing data was provided and, therefore, it is unclear which missing data were considered. Overall, results using the LOCF and multiple-imputation methods were similar, suggesting that there were minimal differences related to missing data; however, the CADTH review team noted that despite the lack of details on how the multiple imputation was conducted, this method is preferred. The sponsor noted that these analyses were adjusted for any potential predefined confounders, although it is unclear to what extent uncontrolled confounders and treatment effect-modifiers influenced the results.
In a separate, draft manuscript, the second analysis consisted of a piecewise linear mixed-effect model, which was used to evaluate pre-and post-treatment comparisons. The model included relevant confounders, including age, sex, number of SMN2 copies, ambulatory status, disease duration, baseline scores, and registry. Other potential confounders and treatment-effect modifiers that were not identified or considered, even though they may influence outcomes, include training for the outcomes of interest, routine exercise and observation, other routine care (such as physiotherapy and occupational therapy), and the placebo effect. The population used for the model included 75 SMA adults — 74 with type III SMA and 1 with type IV — with at least 6 months of follow-up, although the actual assessment times for these patients was unknown. Missing values at baseline (HFMSE, RULM, and 6MWT) were imputed using linear interpolation based on pre-treatment measures.
In the unpublished draft manuscript, results for the standard mixed-effect model and piecewise linear mixed model were presented as the difference between slopes in points per week for each measure for the populations before and after treatment (n = 75), as well as in 235 treated (228 with type III and 7 with type IV SMA) and 17 untreated patients (14 with type III and 3 with type IV SMA), using a time-spline methodology. Using this method, the difference between the slope before treatment start and the slope after treatment start (time spline) was assessed. In both the standard linear-effects model and the piecewise linear mixed model, although weeks were used as the time variable, it was unclear what the duration of follow-up was. The CADTH review team, as well as the clinical experts consulted by CADTH, considered weeks as the unit of measure to be inappropriate for type II and III SMA. The sponsor attempted to extrapolate the results over 1 year, but CADTH considered the results of this extrapolation to be highly uncertain and inappropriate, given the already short duration of follow-up.
Pera et al. (2021)
Pera et al. (2021)19 was a noncomparative, registry-based analysis of SMA patients collected from ISMAR data. No published protocol was specified or identified, and no sample size calculation was provided. Minimal information was provided on eligibility criteria for these patients, and data were based on what was available in the registry. No information on the quality or accuracy of the registry was provided. This study enrolled patients outside of the reassessment reimbursement request; patients younger than 18 years were included. Only 67 (47%) patients were in the population of interest for this review. The authors attempted to reduce any selection bias by conducting a mixed-model assessment to estimate changes in the entire type III population, using baseline measurements (age, sex, time, disease duration, SMN2 copy number, disease onset, and SMA function) as fixed effects and the patient as a random effect. Other potential confounders and treatment-effect modifiers that were not identified or considered include training for the outcomes of interest, routine exercise and observation, other routine care (such as physiotherapy and occupational therapy), and the placebo effect. The extent to which uncontrolled confounders and treatment-effect modifiers influenced the results is unclear.
Relevant subgroups based on SMA type (type IIIA and IIIB), age (children versus adult), and functional status (sitters versus walkers) appeared to be pre-specified; however, no information on the analysis of these subgroups was provided. No imputation of missing data was performed, and the amount of missing data was not reported, so the impact of missing data on the results could not be determined. Given the noncomparative nature of the study, the results observed cannot be attributed to nusinersen.
No baseline characteristics were reported solely for the adult population; thus, specific selection biases in this population could not be determined.
External Validity
Hagenacker et al. (2020)
Hagenacker et al. (2020)17 enrolled 139 adults with genetically confirmed 5q SMA; mostly type II or type III SMA. The proportion of patients with type III SMA was 62% to 65%, most had 3 SMN2 gene copies (36% to 39%), and 37% to 40% were ambulant. These values were higher than what the clinical experts see in clinical practice. As well, there was a lower-than-expected proportion of patients with prior spinal fusion surgery (20% to 25%). As such, the baseline characteristics of the included population suggested a selective population of patients that limits the generalizability of the results. The clinical experts consulted by CADTH reported that these proportions did not reflect the type II and III SMA adult population they see in practice; instead, they would expect more type II patients who are nonambulatory (approximately 75%) and more patients (approximately 65% to 70%) who have undergone spinal fusion surgery. Overall, the CADTH review team and clinical experts considered the patients included in the study by Hagenacker et al. (2020)17 to be of lower disease severity, reducing the generalizability of this study.
There was potential survivorship bias in this study, as evidenced by the percent of ambulant patients with type III SMA and the higher baseline HFMSE, RULM, and 6MWT scores at baseline in the 14-month group than in the 6-month group. The follow-up duration of this study was only 14 months. In discussion with the clinical experts, it was determined that the follow-up duration of 14 months was inadequate to capture the true effects of treatment in type II and III SMA patients, although the direction of bias is unclear.
In this study, all nusinersen injections were conducted by trained neurologists or neuroradiologists with conventional, fluoroscopy-guided or CT-guided lumbar puncture. It was unclear how many patients required guided intrathecal injections, which may have implications on clinical practice in Canada, given the spinal complexity of these patients.
The outcomes chosen for this study were based on validated measures of motor function and are routinely used in studies of SMA and in Canadian clinical practice. All patients were evaluated according to the recommendations of the SMArtCARE real-world data collection initiative. As such, all evaluators were properly trained, minimizing potential variation in the way motor-function outcomes were measured. However, the HFMSE scale was the primary outcome of this study and, by definition, is to be used in patients who can both sit and walk. Considering that only 37% to 40% of the population was ambulatory, neither HFMSE nor 6MWT may be an appropriate measure of improvement in the patients studied. HRQoL and other patient-reported outcomes, which were outcomes important to patients, were not assessed in the study by Hagenacker et al. (2020).17
As noted by the authors, the natural disease course of SMA might be influenced by the type and level of supportive care provided to the patient, and all included patients had access to supportive care before and during nusinersen treatment, although it was unclear what supportive care consisted of. The clinical experts emphasized the importance of patients with SMA receiving care from a multi-dimensional care centre that employs physiotherapists and other health providers. However, it is not clear what proportion of patients in Canada have access to and receive this level of care, so the results may not be generalizable to these patients.
Maggi et al. (2020)
As with the Hagenacker et al. (2020)17 study, although the enrolled population consisted of adults with type II and III SMA, they were a selective cohort of patients. The study by Maggi et al. (2020)18 enrolled 116 adults with type II and III SMA, but only 13 (11.2%) had type II SMA. As noted by the experts, type II patients represent the majority of adults they see in practice; thus, the population in this study is less generalizable to type II patients in Canada because of the limited inclusion in the study. Approximately half of all type III patients included were sitters; the other half were walkers. Most patients had 4 copies of SMN2 (46.6%), which was noted by the clinical experts to be unreflective of the eligible population. Compared with type III walkers, more patients who were considered sitters required ventilatory support (15.7% versus 5.8%) and had undergone surgery for scoliosis (13.7% versus 1.9%). The clinical experts consulted by CADTH indicated that most patients would likely be not fully ambulatory, and most would have undergone prior spinal surgery (including spinal fusion); thus, this population may not be reflective of Canadian patients. Additionally, baseline motor-function scale scores were considered to be higher than expected, indicating better baseline function in these patients, particularly SMA type III walkers, suggesting a bias in the patients selected for inclusion in this study. Most ventilatory support at baseline occurred in type II (76.9%) and type III sitter (15.7%) patients. However, it was unclear whether this referred to the proportion of patients that required night-time BiPap, or if some other method of ventilation was required. As in the Hagenacker et al. (2020)17 study, the CADTH review team and clinical experts considered the included population to have less severe disease than most adults with type II and III SMA.
Nusinersen was administered by trained professionals, mostly with standard lumbar access, which, according to the clinical experts consulted by CADTH, reflects the type III nature of patients; however, they noted that this may not be reflective of the general SMA population, because most of these patients would have complex spines, requiring interventional radiology to administer doses.
The outcomes selected for this study were appropriate and were in line with standard practice; however, there was significant attrition in the proportion of patients completing the outcome measures between baseline and 14 months, resulting in uncertainty and limited generalizability of the results. As previously noted, HFMSE and 6MWT may not be appropriate outcome measures in all patients, considering that half of all patients were nonambulatory. HRQoL and patient-reported outcomes, which were outcomes important to patients, were not assessed in the study by Maggi et al. (2020)18 and, therefore, the effect of nusinersen on these outcomes remains unknown.
This study was a retrospective cohort study that was limited to 14 months of follow-up. As in the Hagenacker et al. (2020)17 study, this duration of follow-up was considered to be too short to observe the true effects of treatment in type II and III SMA patients due to the slowly progressing nature of the disease.
EU Registry Study
As previously mentioned, the EU registry study15,16 enrolled patients from 3 registries from multiple centres in Germany, Italy, Spain, the UK, and the US. Overall, there were differences in the proportions of patients from each registry, as 157 (67%), 54 (23%), and 24 (10%) treated patients, and 0, 6 (35%), and 11 (65%) untreated patients came from the German SMArtCARE, Italian ISMAR, and Spanish CuidAME registries, respectively. As such, there may be differences in disease management for treated and untreated patients across regions, which may affect the generalizability of results, given the larger contributions of patients from specific countries and registries. Although all patients were likely similar with respect to indicated disease, based on registry enrolment, there was considerable heterogeneity across registries in baseline characteristics with regard to age, age at symptom onset, ambulatory status, and proportion of patients with scoliosis. Nearly all patients included in the study were adults, had type III SMA, and had 4 copies of SMN2 (45%), which was noted by the clinical experts consulted by CADTH to be unreflective of practice. Despite the fact that included populations were generally evenly split between ambulatory (48%) and nonambulatory (52%) type III patients, many of the patients included in the registry analysis did not have scoliosis (76%) or prior spinal fusion surgery. Additionally, only 15 (6.2%) patients required noninvasive ventilatory support, although it was unclear what this consisted of. Overall, there was considerable selection bias for higher-functioning patients, based on baseline scale scores that were considered high, indicating greater motor function and less severe disease. The CADTH review team and clinical experts consulted by CADTH considered there to be a high degree of selection bias in the patients included in the EU registry study,15,16 which may affect generalizability to Canadian adults with type II or III SMA.
Most patients in the analysis of treated and untreated adults had type III SMA (n = 228); however, there were also 10 type IV patients, which was not reflective of the reimbursement request. Comparative analysis of treated and untreated patients only included 17 patients in the untreated group, 3 of whom had type IV SMA. The characteristics and small sample size of the untreated group make it very difficult to generalize the results or draw conclusions from the analysis.
A total of 74 adults with type III SMA and 1 patient with type IV SMA were included in the pre- and post-treatment analysis. Although the few type IV SMA patients included are unlikely to affect the results of either analysis, the small sample size limits the conclusions that can be drawn. Furthermore, there were no patients with type II SMA included in the study, so any results cannot be generalized to the type II population.
There was no information provided on the method of administration of nusinersen, the dosing regimen in the registries, or background care, so the CADTH review team was unable to determine whether how and where patients were treated had any potential impact on outcomes.
It was unclear what the overall time of assessment was for the EU registry study.15,16 Regardless, the CADTH review team, as well as the clinical experts consulted by CADTH, considered weeks as the unit of measure to be inappropriate for type II and III SMA. The sponsor attempted to extrapolate the results over 1 year; however, CADTH considered the results of this extrapolation to be highly uncertain and inappropriate, given the short duration of follow-up relative to the natural history of SMA and the unverified assumptions required to make this extrapolation.
Outcomes included in the EU registry study15,16 were appropriate and matched other published studies; however, as previously mentioned, the conclusions that can be drawn from the HFMSE and 6MWT measures are limited, as nearly half of the patients were nonambulatory. HRQoL and patient-reported outcomes, which were outcomes important to patients, were not assessed in the EU registry study, so the effect of nusinersen with respect to these outcomes remains unknown.
Pera et al. (2021)
The study by Pera et al. (2021)19 was based on the data collected from the ISMAR registry, so there may be some overlap in patients included in this study, the EU registry study,15,16 and the Maggi et al. (2020)18 study. It is unclear what effect this may have on the interpretation of the results. The Pera et al. (2021)19 study included 144 patients with type III SMA, but also included children and adults. Overall, only 67 patients included in this study met the reimbursement request criteria of adults with type II or III SMA. No baseline characteristics of the target population were included, so CADTH is unable to assess the potential for selection bias or external validity in these patients.
No information on the dose, regimen, or setting of nusinersen administration were provided, so it was unclear whether the recommended dosing administration procedure was followed. Outcome measures were consistent with the other studies, including HFMSE, RULM, and 6MWT, and were administered by trained clinical evaluators. In the overall population (including adults and children), nearly half of all patients were sitters; thus, as previously mentioned, the HFMSE and 6MWT may not be appropriate. As in the other included studies, baseline scores for HFMSE, RULM, and 6MWT were considered high for the adult type III population, which may not be reflective of the Canadian population, and did not include type II SMA patients. HRQoL and patient-reported outcomes, which were outcomes important to patients, were not assessed in the study by Pera et al. (2021),19 so the effect of nusinersen with respect to these outcomes remains unknown.
The time of assessment of this study was 12 months, which was considered too short to assess changes in adults with type II or III SMA and generally too short to notice any meaningful change.
Indirect Evidence
No indirect evidence was included in the sponsor’s submission to CADTH or identified in the literature search conducted by CADTH that matched the review protocol criteria.
Other Relevant Evidence
Other Sponsor-Submitted Evidence
As part of the reassessment for nusinersen, the sponsor submitted a publicly available critical review and meta-analysis of patients with type II and III SMA. The objective of this section is to summarize and critically appraise methods and findings of the submitted meta-analysis, which evaluated efficacy patterns of nusinersen on motor function in the existing literature in real-world datasets of patients with type II and III SMA.20
Description of Study
The sponsor submitted a publicly available critical review and meta-analysis, informed by a systematic review that evaluated motor-function outcomes in patients with type II or III SMA. A total of 14,627 articles were preliminarily selected based on title. A total of 30 full-text articles were included in the analysis.20
Methods
Objectives
The objective of the meta-analysis by Coratti et al. (2021)20 was to critically review the literature reporting real-world data on motor function in type II and III patients treated with nusinersen to establish possible patterns of efficacy by subdividing results by SMA type, age (children versus adults), and type of assessment. Only results related to the adult population with type II or III SMA were of interest to this reassessment.
Study Selection Methods
The meta-analysis was informed by a systematic review of existing literature conducted up to January 2021. PRISMA guidelines were applied to the critical review, including research on online databases for peer-reviewed journal (PUBMED, MEDLINE, Web of Science, CINAHL, PsycINFO, and Embase) and manual research on reference lists of included articles. Eligible publications reporting data on nusinersen-treated patients were grouped into categories by SMA type (type II, type III, or a combination of the 2 types) or age (children versus adult). A similar approach was used to classify data from untreated patients. When needed, recalculation of the mean and SD for age or motor outcome was performed from papers reporting full data.20
Screening and data collection were conducted by 5 authors. Full-text review was conducted by that group and by the senior author to determine the full eligibility of articles. Data extracted included outcome measures, target population (size, type, age category, age range at treatment, mean age at treatment), magnitude of changes at 10, 12, or 14 months from baseline (mean, SD or 95% CI). Studies reporting data on type I SMA only were excluded. All electronic searches were limited to the English language.20 No additional information on the PICOS framework or on inclusion criteria for the review and meta-analysis were provided.
Analysis Methods
Data looking at reported changes in individual motor-function outcome measures in the different treated groups, subdivided by age category (adults, children), motor function (ambulant, nonambulant), and SMA type were analyzed. Pooled analyses were conducted at multiple levels: first, a rough comparison of the overall benefit of treatment versus no treatment was run, which included the largest amount of available evidence, even if heterogeneous. The effect size was estimated using random-effect models, and heterogeneity among studies was quantified with the I2 coefficient. Subgroup analyses were conducted to verify and estimate the influence of different categories (age, SMA type, and motor function) on the pooled results of the treated population. Meta-regression analysis was undertaken to identify possible sources of heterogeneity among studies. Meta-regression analyses were employed, with a random-effects model using aggregate-level data. Only studies with complete data available (sample size, mean, SD or 95% CI) were included in the meta-analysis.20
Results
Summary of Included Studies
The PRISMA flow diagram for study selection in the Coratti et al. (2021)20 meta-analysis is summarized in Figure 5. A total of 14,627 articles were selected based on title, 788 of which were related to nusinersen treatment. After the review of the abstracts, 9,221 were excluded. After the review of 55 full-text papers, 30 publications were selected and analyzed. Fifteen studies reported on adults treated with nusinersen and 10 reported on natural history in adults.
After the exclusion of reviews, commentaries, and individual case reports, 19 papers reporting data on efficacy that used structured assessments in type II and III SMA were selected. In 4 of the 19 papers, type I SMA patients were also included, with 1 of the 4 papers describing data on type I SMA separately. According to the authors, none of the data for outcomes of interest (HFMSE, RULM, and 6MWT) included type I patients. Data from clinical trials were not included in the review. The 19 papers were reviewed using the Risk of Bias Assessment Tool for Nonrandomized Studies. Twelve papers reported data in untreated patients.20
A summary of specific demographic or baseline patient characteristics from the included trials was not provided, so assessment of heterogeneity related to patients, outcomes, or study design was not possible.
Figure 5: Search and Selection Process (PRISMA Framework)
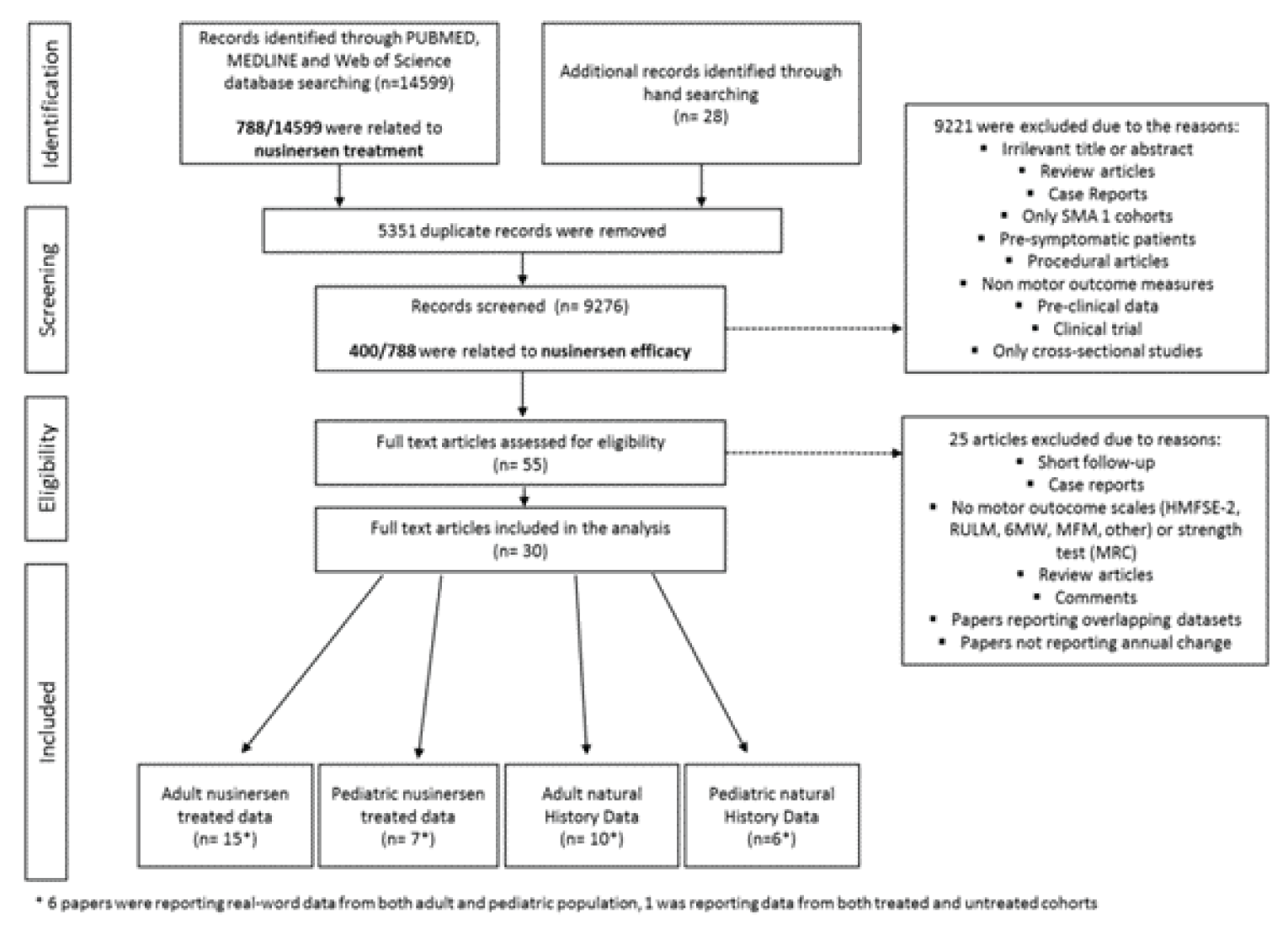
6MW = 6-minute walk; HFMSE = Hammersmith Functional Motor Scale Expanded-2; MFM = motor-function measurement; MRC = Medical Research Council; RULM = Revised Upper Limb Module; SMA = spinal muscular atrophy.
Source: Coratti et al. (2021).20 This work is licensed under the Attribution 4.0 International Licence (https://creativecommons.org/licenses/by/4.0/). Full text available here: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8515709/
Results of Meta-Analysis
Motor-function outcomes for the adult subgroup in the meta-analysis include HFMSE, RULM, 6MWT, MRC Scale for Muscle Strength, and CHOP-ATEND.20 Meta-regression was not conducted for the MRC and CHOP-ATEND outcomes and are not summarized.
HFMSE: Of the 13 publications that reported HFMSE results for patients treated with nusinersen, 9 focused on adults, 2 focused on adult and pediatric populations, and 2 focused only on the pediatric population. Five publications reported HFMSE results in untreated patients. Results for change in HFMSE score in the individual publications are summarized in Figure 6. Individual data on HFMSE were available in 3 papers; therefore, to subdivide the populations according to SMA type or age group, and to eliminate the problem of missing data, the authors recalculated mean change over time.20
Figure 6: HFMSE Results by Reporting Author in the Adult Population
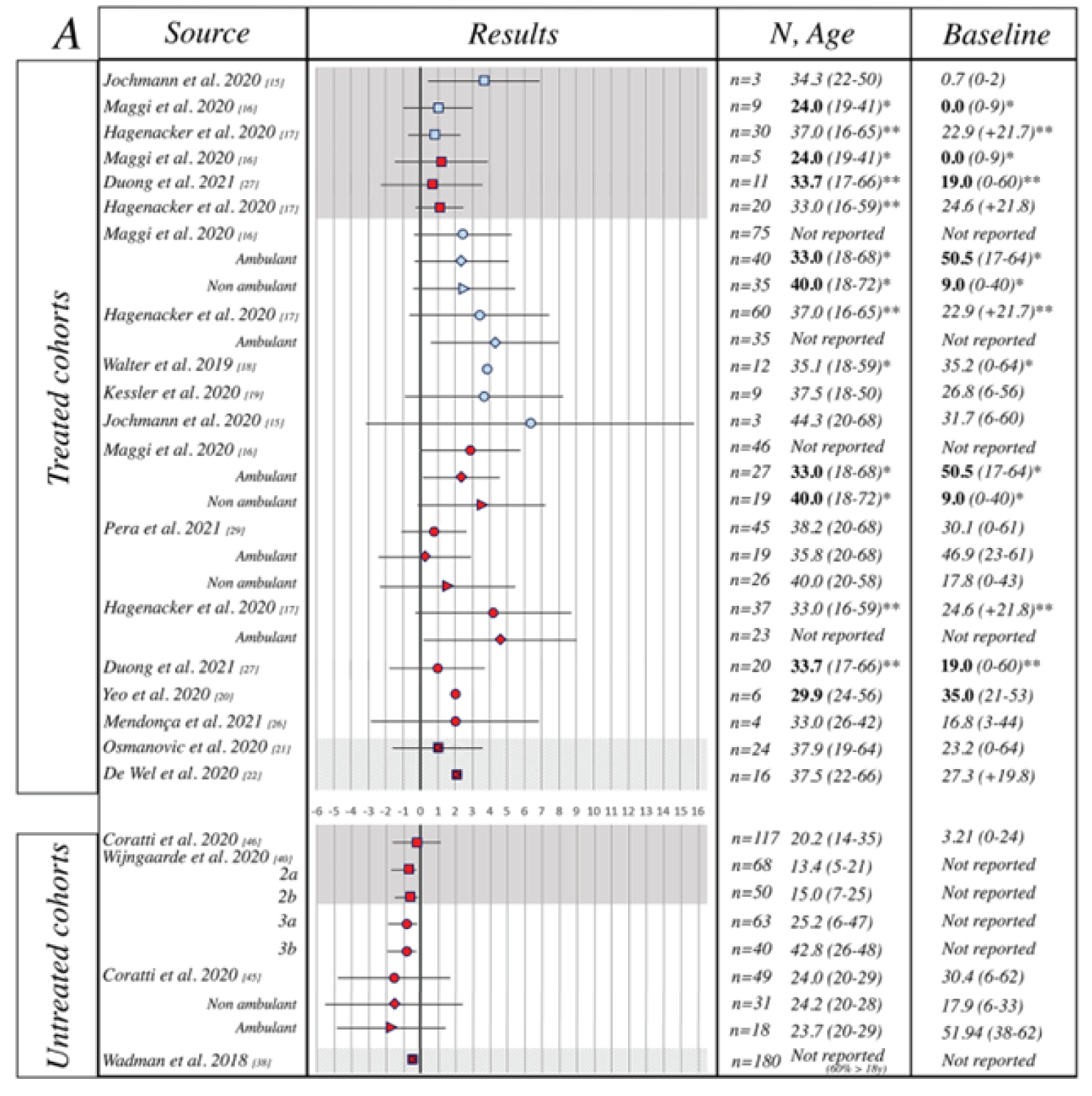
Dashed line = 95% confidence interval; Plain line = standard deviation; Square = SMA type II; Circle = SMA type III, Diamond = ambulant SMA type III; Triangle = nonambulant SMA type III; Square + circle + triangle = mixed phenotypes; Bold font = median value; italic = mean value; Light blue = approximately 10 months from initiation of drug; Red = approximately 12 months from initiation of drug; Grey shade = SMA type II; White shade = SMA type III; Striped shade = mixed phenotypes.
* Mean/median values of the baseline population, non excluding drop-outs at 10, 14, or 24 months of follow-up.
** Mean/median values of the baseline population of both SMA type II and III combined.
Source: Coratti et al. (2021).20 This work is licensed under the Attribution 4.0 International Licence (https://creativecommons.org/licenses/by/4.0/). Full text available here: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8515709/
Results of the meta-regression analysis for the mean change in HFMSE in the adult population are summarized in Figure 7. Heterogeneity in the included studies for adults was moderate (I2 = 68%). The pooled mean change in HFMSE score in adults was 1.87 points (95% CI, 1.05 to 2.68).20
RULM: Thirteen papers reported RULM results for patients treated with nusinersen: 9 in adults only, 2 in children only, and 2 in adults and children. Five publications reported RULM results in untreated patients. Individual data on RULM were available in just 1 paper; therefore, to eliminate the problem of missing data, the authors have recalculated mean change over time.20 Results for change in RULM score in the individual publications are summarized in Figure 8.
Results of the meta-regression analysis for the mean change in RULM in the adult population are summarized in Figure 9. Heterogeneity in the included studies was moderate (I2 = 43%). The pooled mean change in RULM score in adults was 0.64 points (95% CI, 0.27 to 1.01).20
6MWT: Eight publications were identified that reported data on patients treated with nusinersen, and 1 publication reported data from untreated patients. Of the 8 publications on nusinersen, 6 focused on adults, 1 focused on children only, and 1 focused on both adults and children.20 Results for change in 6MWT score in the individual publications are summarized in Figure 10.
Results of the meta-regression analysis for the mean change in 6MWT in the adult, ambulatory population are summarized in Figure 11. Heterogeneity in the included studies was considerable (I2 = 71%). The pooled mean change in 6MWT score in adults was 20.28 m (95% CI, 1.17 to 39.40).20
Figure 7: Meta-Regression Analysis of Change in HFMSE Score From Baseline
CI = confidence interval; HFMSE = Hammersmith Functional Motor Scale Expanded.
Source: Coratti et al. (2021).20 This work is licensed under the Attribution 4.0 International Licence (https://creativecommons.org/licenses/by/4.0/). Full text available here: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8515709/
Figure 8: RULM Results by Reporting Author in the Adult Population
Dashed line = 95% confidence interval; Plain line = standard deviation; Square = SMA type II; Circle = SMA type III, Diamond = ambulant SMA type III; Triangle = nonambulant SMA type III; Square + circle + triangle = mix phenotypes; Bold font = median value; Italic = mean value; Light blue = approximately 10 months from initiation of drug; Red = approximately 12 months from initiation of drug; Grey shade = SMA type II; White shade = SMA type III; Striped shade = mixed phenotypes.
* Mean/median values of the baseline population, non excluding drop-outs at 10, 14, or 24 months of follow-up.
** Mean/median values of the baseline population of both SMA type II and III combined.
Source: Coratti et al. (2021).20 This work is licensed under the Attribution 4.0 International Licence (https://creativecommons.org/licenses/by/4.0/). Full text available here: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8515709/
Figure 9: Meta-Regression Analysis of Change in RULM Score from Baseline
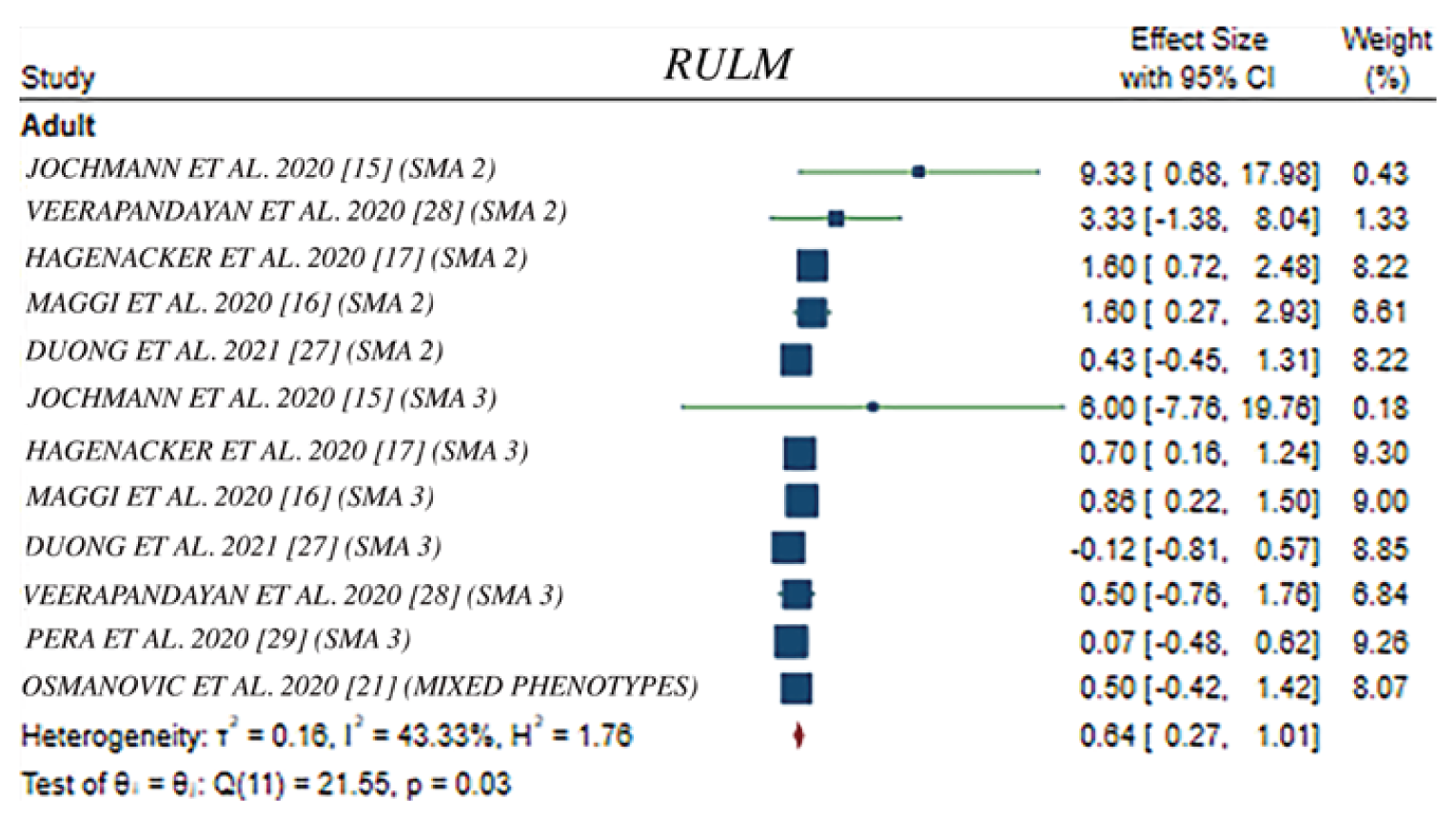
CI = confidence interval; RULM = Revised Upper Limb Module.
Source: Coratti et al. (2021).20 This work is licensed under the Attribution 4.0 International Licence (https://creativecommons.org/licenses/by/4.0/). Full text available here: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8515709/
Figure 10: 6MWT Results by Reporting Author in the Adult Population
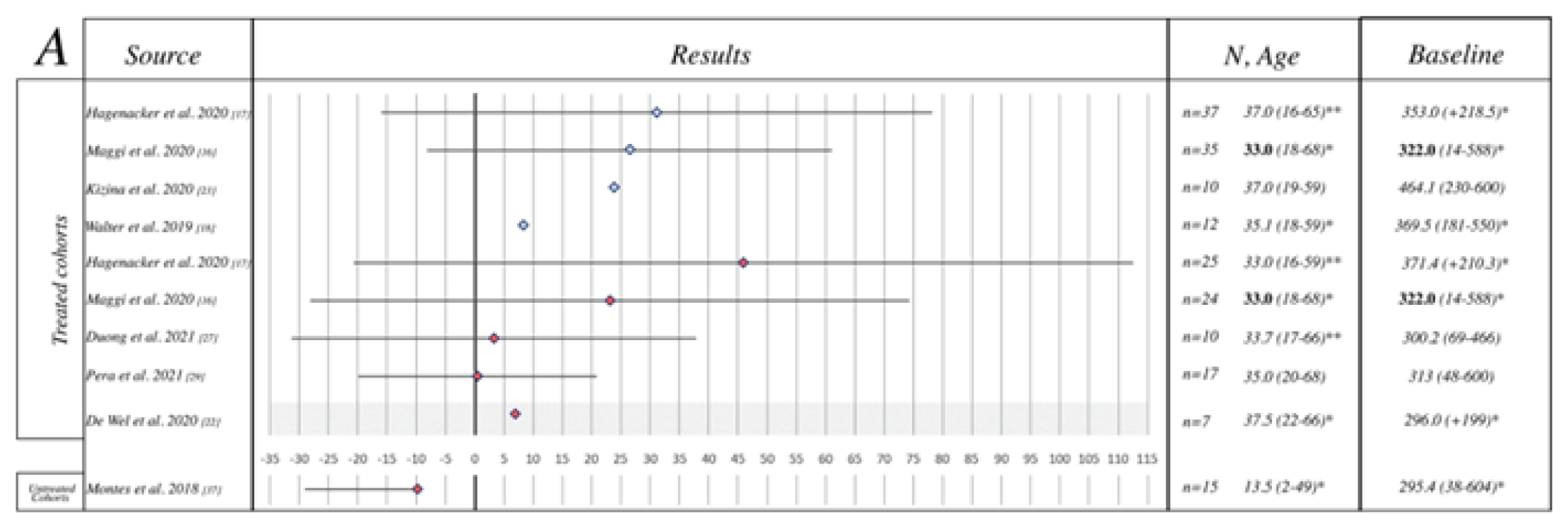
Dashed line = 95% confidence interval; Plain line = standard deviation; Bold font = median value; Italic = mean value; Light blue = approximately 10 months from initiation of drug; Red = approximately 12 months from initiation of drug.
* Mean/median values of the baseline population, non excluding drop-outs at 10, 14, or 24 months of follow-up.
** Mean/median values of the baseline population of both SMA type II and III combined.
Source: Coratti et al. (2021).20 This work is licensed under the Attribution 4.0 International Licence (https://creativecommons.org/licenses/by/4.0/). Full text available here: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8515709/
Figure 11: Meta-Regression Analysis of Change in 6MWT Score From Baseline
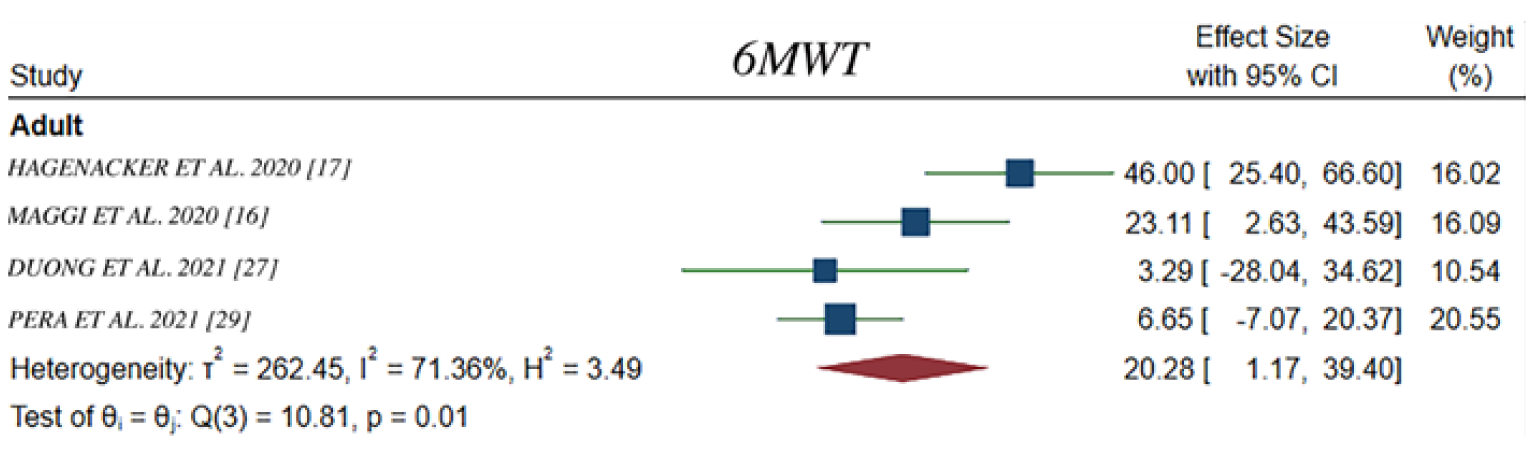
6MWT = 6-minute walk test; CI = confidence interval.
Source: Coratti et al. (2021).20 This work is licensed under the Attribution 4.0 International Licence (https://creativecommons.org/licenses/by/4.0/). Full text available here: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8515709/
Critical Appraisal
The meta-analysis was based on an adequately conducted and reproducible systematic literature search that included planned searches of multiple databases and hand searches of references. Information on screening and data collection was provided, but it was unclear if studies were selected independently or in duplicate. Moreover, it was unclear if the inclusion and exclusion criteria related to population, outcomes, and study design were pre-specified. A quality assessment of the included studies was conducted using the Risk of Bias Assessment Tool for Nonrandomized Studies. No interpretation on the quality of studies was conducted; however, as all studies were observational, most were noted to suffer from a high level of bias in the selection of participants. The publicly available studies summarized previously (Hagenacker et al. [2020],17 Maggi et al. [2020],18 and Pera et al. [2021]19) were included in the submitted meta-analysis.
Outcomes included in the meta-analysis were appropriate and relevant to the Canadian context, with HFMSE, RULM, and 6MWT (in ambulant patients) most commonly included in studies, although there were differences in reporting and time of assessment. The authors noted that details on respiratory function or safety concerns were not systematically addressed in all the studies, so the impact of nusinersen on these outcomes cannot be determined. Most of the included studies had follow-up times ranging from 10 to 14 months. As previously mentioned, in discussion with the clinical experts consulted by CADTH, this short follow-up duration is insufficient to observe clinically meaningful changes in motor-function outcomes, so there is uncertainty in the magnitude of benefit.
Key features of the study design, inclusion or exclusion criteria, and baseline characteristics in the included studies were not provided, so a complete assessment of potential sources of clinical and methodological heterogeneity was not possible. The authors noted that there was considerable heterogeneity in the studies, given the inclusion of both ambulant and nonambulant type II and type III SMA patients, as well as the inclusion of both adults and children. Additionally, the included studies had small sample sizes, limiting the conclusions that can be drawn from individual studies. To explore the sources of heterogeneity, meta-regression was used, generally adjusted by age, SMA type, and treatment. Overall, there was a considerable level of heterogeneity in the overall cohorts, including both adult and pediatric populations, with I2 ranging from 68% to 90%. In the adult subgroup, there was a moderate to considerable level of heterogeneity in the included studies across outcomes, with I2 values ranging from 43% to 71%. No further information on sources of clinical, methodological, or statistical heterogeneity were explored.
Results and effect sizes for each outcome were presented for each study, with effect size estimated with random-effect models, which was considered appropriate because of the high level of between-study heterogeneity. Pooled estimates of mean change for motor-function outcomes — 1.87 points (95% CI, 1.05 to 2.68) for HFMSE, 0.64 points (95% CI, 0.27 to 1.01) for RULM, and 20.28 m (95% CI, 1.17 to 39.40) for 6MWT — favoured nusinersen treatment. Although there is a general positive association between nusinersen treatment and motor-function outcomes, it is unclear to what extent these changes are clinically meaningful. There was variation in what the included studies considered a clinically meaningful change, with some defining clinically meaningful improvements as a 3-point change in HFMSE score, a 2- to 3-point change in RULM score, and a 30-metre change in 6MWT, so the results of the meta-analysis may not demonstrate clinically meaningful improvements in motor-function outcomes. As previously mentioned, in discussion with the clinical experts, there is uncertainty about what constitutes a clinically meaningful change in the adult population for these outcome measures. Moreover, as previously discussed, given the nature of the included studies and the limitations defined for the studies included in both the systematic review conducted by CADTH and the meta-analysis, the observed effects cannot be attributed to nusinersen; therefore, the results of the meta-analysis are uncertain. Last, the estimates generally displayed wide 95% CIs, particularly for the 6MWT, and in many cases crossed the zero meridian, indicating a high level of variation in these cohorts and substantial imprecision in estimates of treatment effect. In addition, the cause of the wide CIs was unclear, but it is believed to be due to unaccounted-for heterogeneity and low sample sizes, particularly for the subgroups of interest that were evaluated (i.e., adults only). No sensitivity analyses were conducted. The authors noted the high number of missing study details on baseline functional status, and scores and other variables did not allow for detailed statistical analysis or meta-analysis, which the authors noted would have helped them to better understand the possible treatment effect of a number of variables, such as age, SMN2 copies, and functional ability at baseline. Although suggesting that the benefit of nusinersen is favourable in motor function, the results of the meta-analysis are unclear and provide imprecise estimation of the true effect of nusinersen on these outcomes in adults with type II or III SMA.
Evidence Identified From the Literature
Eight studies were identified in the literature search that met all inclusion criteria of the systematic review except study design, as they were all noncomparative observational studies. These 8 studies are briefly summarized in the following sections.
Brakemeier et al. (2021)
The study by Brakemeier et al. (2021)30 was a prospective, noncomparative, German, single-centre, observational study that included 22 patients with molecularly confirmed 5q SMA who had been on therapy for at least 6 months. Included patients had type II or III SMA, documented bulbar dysfunction before treatment initiation, and no percutaneous enteroscopic gastrostoma. Patients were treated with nusinersen 12 mg on days 1, 14, 28 and 63, and then consecutively at 4-month intervals, in accordance with the product monograph. The study aimed to assess bulbar function in adults with type II and III SMA at 6 and 14 months. Patients ranged in age from 20 to 72 years, with a mean age of 38.5 years (SD = 14.2). Most patients were male (13 [59%]), had type II SMA (12 [54%]), had 3 copies of the SMN2 gene (14 [64%]), and had no presence of spondylodesis (12 [54%]). Symptom duration ranged from 14 to 52 years, with a mean duration of 34.3 years (SD = 11.9).
The primary outcomes were pre- and post-treatment comparisons of the bulbar subscore of the Amyotrophic Lateral Sclerosis Functional Rating Scale Revised (ALSFRS-R) and the Sydney Swallow Questionnaire (SSQ) scores at baseline, after 6 months of therapy, and after 14 months of therapy. The article stated that pre- and post-treatment comparisons in outcomes were made using the Wilcoxon signed-rank test with an alpha of 0.05 or less. There were 16, 20, and 18 patients measured using ALSFRS-R at each time point, respectively. The mean bulbar subscores from the ALSFRS-R were 10.50 (SD = 1.21) at baseline, 10.50 (SD = 1.32) at 6 months, and 10.94 (SD = 0.97) at 14 months. No significant change in bulbar function measured with the bulbar subscore of the ALSFRS-R was found between baseline and 6 months (z = –0.302; P = 0.763), or between baseline and 14 months (z = –1.406; P = 0.160). A total of 18, 15, and 13 patients completed the SSQ at baseline, 6 months, and 14 months, respectively. The mean SSQ scores were 23.59 (SD = 18.85) at baseline, 24.12 (SD = 19.32) at 6 months, and 19.95 (SD = 17.86) at 14 months. No significant change was found in the SSQ score between baseline and 6 months (z = –0.210; P = 0.834), or between baseline and 14 months (z = –0.392; P = 0.695). No significant changes in bulbar function were observed in subgroup analyses in which pre- and post-treatment comparisons of the 2 bulbar scores for type II and III SMA patients were calculated separately. Secondary outcomes included pre- and post-treatment comparisons of HFMSE and RULM scores. Improvement in HFMSE was observed from baseline to 6 months (z = –2.236; P = 0.025), but the change from baseline to 14 months was not significant (z = –1.248; P = 0.212). Similarly, no significant pre- and post-treatment differences were observed for the change in RULM score between baseline and 6 months for 22 patients (z = –1.932; P = 0.053), or between baseline and 14 months for 18 patients (z = –0.256; P = 0.798).30
The study used an observational, noncomparative design. A clear hypothesis to be tested was not stated, so a direct cause-and-effect relationship could not be established for nusinersen. It was unclear if the study had a pre-specified protocol, and there was no mention of how patients were identified or selected to participate in the study, which prevented assessment of the population enrolled for selection bias and external validity. The study did not provide clear information regarding patients who discontinued treatment or withdrew from the study. The number of patients completing outcome measures at various time points was provided, but reasons for drop out or noncompletion were not reported. The approach for handling missing data was not mentioned, so the impact of attrition bias and missing data should be considered when interpreting the study results. The study included patients with type II and III SMA who had been on nusinersen for at least 6 months, and 19 patients had been on therapy for 14 months at the time of data analysis; however, there was no reporting of the time patients had contributed before and after the data analysis, or whether the patients’ time had been considered into the analysis. Moreover, the study did not provide details of the therapy patients received; for example, there was no mention of whether there were other treatments or whether such treatments were administered before nusinersen, at the time nusinersen was started, or during nusinersen treatment. Thus, it is unclear whether the patients were truly naive to disease-modifying therapy, what supportive treatments they received, or whether the time patients’ contributed was considered in data analysis. Last, the funding source for this study was Biogen.
Duong et al. (2021)
Duong et al. (2021)31 was a prospective, noncomparative, multi-centre, observational study that included 42 patients with genetic confirmation and clinical symptoms of SMA in the US. Included patients were 17 years or older at the start of nusinersen treatment. Patients were treated intrathecally with doses of 12 mg of nusinersen, based on the recommended schedule (baseline, day 14, day 28, day 58, and every 4 months thereafter). The study aimed to evaluate changes in clinical outcome measures and to obtain safety information related to nusinersen treatment. Patient ages ranged from 17.7 to 66.1 years, with a mean age of 33.7 years (SD = 12.6). Most patients were male (24 [57.1%]), had type III SMA (24 [57.2%]), had 3 copies of SMN2 (26 [72.2%]), were nonambulant (17 [40.5%] nonsitter and 14 [33.3%] sitter), and had no presence of spinal fusion (26 [61.9%]). Patients had a symptom duration that ranged from 12.1 to 60.6 years, with a mean duration of 30.9 years (SD = 12.3).
Motor function was measured with HFMSE, RULM, CHOP-ATEND, Spinal Muscular Atrophy Functional Rating Scale (SMAFRS), 6MWT, and timed up and go scores. Baseline assessments occurred in the 5 months after the first dose, and post-treatment assessments were done up to 24 months after nusinersen initiation. The study used linear mixed-effects models to estimate the mean annual rate of change (slope) for each outcome.
The slopes of all motor-function measures showed change in a positive direction. However, greater changes were observed in CHOP-ATEND (slope = 3.59 points per year [95% CI, 0.67 to 6.51]; n = 24) and SMAFRS (slope = 1.44 points per year [95% CI, 0.04 to 2.83]; n = 31] than in HFMSE (slope = 0.86 points per year [95% CI, −0.52 to 2.24]; n = 31), RULM (slope = 0.11 points per year [95% CI, −0.45 to 0.67]; n = 39), 6MWT (slope = 3.29 m per year [95% CI, −28.04 to 34.62]; n = 10), and timed up and go (log slope = −0.10 [95% CI, −0.21 to 0.01]; n = 8). Ventilatory function was measured using percent-predicted FVC, maximal expiratory pressure, and maximal inspiratory pressure. The slopes for the changes from baseline were 0.75% per year (95% CI, −1.87 to 3.38) for FVC, 6.38 cm H2O per year (95% CI, 2.52 to 10.25) for maximal expiratory pressure, and −5.50 cm H2O per year (95% CI, −11.47 to 0.47) for maximal inspiratory pressure. In the subgroup analysis, greater changes were observed in patients who were SMA type II, nonsitters, and had 3 copies of SMN2 than in other subgroups in general. For example, greater scores changes were observed in CHOP-ATEND in patients who were SMA type II (slope = 3.75 points per year [95% CI, −0.16 to 7.66]), nonsitters (slope = 6.44 points per year [95% CI, 2.25 to 10.62]), and had 3 copies of SMN2 (slope = 3.62 points per year [95% CI, −0.10 to 7.35]) than in patients who were SMA type III (slope = 3.26 points per year [95% CI, −1.34 to 7.86]), had 4 copies of SMN2 (slope = 3.05 points per year [95% CI, −5.96 to 12.05]), and sitters (slope = −0.29 points per year [95% CI, −5.07 to 4.50]).
The most frequently reported AE was post-lumbar-puncture headache (n = 6 [14.3%]). Other less frequent AEs included injection-site pain, nausea and/or vomiting, lightheadedness, and anxiety. No cases of hydrocephalus, bleeding and/or bruising, or renal compromise were reported.31
The primary limitation of the study was the noncomparative observational design. The study recruited participants through the Pediatric Neuromuscular Clinical Research network; however, there was no mention of inclusion or exclusion criteria for study participants. In addition, the study included participants with good prognostic factors; for example, the majority of patients were nonsitters (40.5%) and ambulant (26.2%), with 3 copies of SMN2 (72.2%) and no spinal fusion (61.9%). Therefore, the study was at high risk for selection bias. The study lacked a comparator group, making it difficult to determine whether the observed effects in the study population can be attributed to nusinersen. The study did not specify any hypotheses and did not report any sample size or power calculations; thus, it is not clear whether the sample size of 42 was sufficient to detect changes in the targeted population, or in subgroup analyses of patients with different SMA types, SMN2 copy numbers, ambulatory status, or spinal fusion status. The baseline clinical and disease characteristics (SMN2 copy number, proportion of ambulatory patients, and presence of spinal fusion) were suggestive of an over-representation of patients with higher-functioning disease and may compromise the representativeness of the study sample to the general population of adults with SMA. Thus, the primary results and results of subgroup analyses must be considered with caution and may not be generalizable. The lack of blinding in this study may lead to expectation bias in favour of nusinersen, as both patients and investigators were aware of the assigned treatment. The SMA Foundation and Cure SMA were the funding sources for this study.
Elsheikh et al. (2021)
Elsheikh et al. (2021)32 was a prospective, open-label, noncomparative, observational study conducted in the US that assessed the safety and treatment effect of nusinersen in 19 nonambulatory adults with confirmed 5q SMA. All patients were reportedly given intrathecal nusinersen in accordance with product-monograph dosing. At baseline, mean age of the patients was 39.7 years (SD = 13.9; range = 21.3 to 64.8). Only 7 patients (36.8%) were male. Most patients (16 [84.2%]) had 3 copies of SMN2, and 1 (5.3%) and 2 patients (10.4%) had 2 and 4 copies, respectively. Nearly half (9 [47%]) of the included patients had type II SMA, and 10 (53%) had SMA type III. The mean age at SMA onset was 27 years (SD = 34). The mean age at loss of ambulation in SMA type III patients was 25.8 years (SD = 18.3). A total of 10 (52.6%) patients had spinal fusion, and 12 (63.2%) were on ventilatory support at baseline.
The primary outcome of the study was the mean change from baseline in FVC at 2 months, 6 months, 10 months, and 14 months, which were –0.02 L (95% CI, –0.11 to 0.08), –0.02 L (95% CI, –0.11 to 0.07), –0.02 L (95% CI, –0.11 to 0.07), and 0.02 L (95% CI, –0.09 to 0.12), respectively. Key secondary outcomes were measured at the same time points and included mean change from baseline in HFMSE (0.77 points [95% CI, –0.29 to 1.83]; 0.74 points [95% CI, –0.3 to 1.78]; 0.32 points [95% CI, –0.73 to 1.36]; and 0.11 points [95% CI, –1.11 to 1.32], respectively), mean change from baseline in RULM (1.31 points [95% CI, 0.24 to 2.39]; 0.89 points [95% CI, –0.16 to 1.95]; 0.95 points [95% CI, –0.1 to 2], and 0.27 points [95% CI, –0.96 to 1.5], respectively).
AEs reported include transient headache, nausea, dizziness, back and neck pain, urinary tract infection, and upper respiratory infection. There was no report of clinically significant vital-sign abnormalities. High baseline protein/creatinine ratio was seen in 4 patients, but no significant change related to treatment was noted.32
A key limitation of the study was the noncomparative observational design. In the absence of a comparator group, it is difficult to determine whether the observed effects in the study population can be attributed to nusinersen. Furthermore, given that no blinding was in place, patients and outcome assessors were aware of the interventions and outcomes of the study. Expectations of treatment may interfere with performance or assessment of outcomes. There were also concerns with selection bias in this study. It was unclear if the sampling method was determined a priori, or how patients were chosen. There was about an equal proportion of SMA type II and type III patients enrolled in the study, which appeared to be inconsistent with the observation that SMA type II is the predominant subtype in adults in Canada. Type III patients generally have better baseline function and are more responsive to interventions than type II patients. Thus, the higher proportion of type III SMA patients may result in bias toward nusinersen. The study also had a significant loss to follow-up. Of the 19 patients enrolled, only 12 patients were studied at all time points for most outcomes. Reasons for loss to follow-up were not mentioned. In addition, when assessing the HFMSE score, the study assigned a HFMSE score of 0 to 13 patients who were not scorable due to phenotypic severity. This imputation method may lead to bias in favour of the intervention, as these patients had no change in score throughout the study and were assumed to be stable.
Mix et al. (2021)
The study by Mix et al. (2021)33 was a prospective, noncomparative, observational study of 26 patients with genetically confirmed 5q SMA in Germany. Included patients were a mix of adults (n = 14) and children (n = 12) treated with nusinersen. In addition, the investigators included a comparator group of 22 untreated individuals who were neurologically healthy. The patients and the comparator group were mean-matched for sex, age, and education. The study aimed to assess physical function and psychological well-being, including QoL and depressiveness in patients with SMA at baseline, day 60, and day 180 (6 months) of nusinersen treatment. For the 14 adults, ages ranged from 21.9 to 60.8 years, with a median age of 44.2 years. Most patients were male (10 [71%]), had type III SMA (9 [64%]), and were nonambulant (6 [43%] nonsitters and 4 [29%] sitters). Age at disease onset ranged from 0 to 17 years, with a median age of 1 year.
Well-being and physical-function measurements were compared between patients and the comparator group at baseline and 6 months. Psychological well-being was measured with the Anamnestic Comparative Self-Assessment (ACSA) for global QoL and the 36-item Short Form Health Survey (SF-36) for HRQoL. Depressive symptoms were assessed with the 12-item ALS Depression Inventory (ADI-12). For adults, no significant differences in ACSA were observed in the comparator group, and there were no data reported on the baseline comparisons of ADI-12 and SF-36 between adults and comparator group. Four adults (28.6%) showed at least mild depressive symptoms on a scale from 25.0% (least depressive) to 100% (most depressive) at baseline. At 6 months, no difference was observed in ACSA and ADI-12 between adults and the comparator group. Concerning SF-36 scores in adults, changes were observed in 3 of 9 dimensions, including role-limitations due to physical health (P = 0.034), general health (P = 0.021), and health transition (P = 0.002) over 6 months. Physical function was evaluated with the ALSFRS-R for all patients and with the HFMSE for patients with type II or III SMA. For motor-function assessments, no data were reported on the baseline and 6-month comparison of ALSFRS-R and HFMSE between adults and matched patients in the comparator group.33
No inclusion and exclusion criteria were specified in the study by Mix et al. (2021),33 and 50% of the participants had type III SMA, 50% were nonsitters, and 23% were ambulant (walkers), which increases the potential for selection bias. Moreover, there was no mention of the demographic characteristics of the comparator group, or how they were identified and selected to participate in the study. The study did not report any sample size or power calculations that tied to a hypothesis; thus, it is not clear whether the sample size was sufficient to detect changes in the targeted population and in subgroup analyses of patients with different ages. As such, primary and subgroup analyses must be considered with caution. In addition, patients reported their psychological well-being status in a retrospective manner (from the previous 2 to 4 weeks); this approach increases the risk of recall bias. The study had a relative short follow-up period (6 months), which is insufficient to observe the longer-term benefits and harms of nusinersen treatment in patients with a chronic and heterogeneous disease. As such, there is limited confidence in the ability to extrapolate these results in the longer term. Furthermore, there was limited reporting of within-group and between-group changes in outcome measures. For example, measurements of ACSA, SF-36, ADI-12, HFMSE, and ALSFRS-R scores were taken at baseline, day 60, and day 180, but the study only reported Chi-square and P values, with brief summaries of the trend in change from baseline to day 180; outcome data from day-60 assessments were not reported. The study was funded by various founding agencies, such as a clinician scientist fellowship sponsored by the Charcot Foundation for ALS research, the Bundesministerium für Bildung und Forschung, and the Deutsche Forschungsgemeinschaft.
Kizina et al. (2020)
Kizina et al. (2020)34 was a prospective, observational, cohort study that assessed fatigue in 28 adults with genetically confirmed 5q type II and III SMA in Germany. All patients had 3 to 5 copies of the SMN2 gene and a report of disease progression over the 12 months before treatment with nusinersen was started. Patients with psychosocial stress, a flu-like infection during the previous week, or depression were excluded, as were those using sedative medication. Intrathecal nusinersen was administered to all patients in accordance with the product monograph. Fatigue severity scale (FSS) and motor-function scores (6WMT and HFMSE) were measured at baseline, and at 6 and 10 months. The correlation between the change in FSS and the change in motor-function scores were also assessed. At baseline, the mean age of patients was 37 years (SD = 12). A total of 18 (64%) patients were male, 10 (36%) had type II SMA, and 18 (64%) had type III SMA. Most patients (18 [64%]) had 3 or 4 copies (9 [32%]) of the SMN2 gene. Most patients were nonambulatory (18 [64%]), but 10 (36%) were ambulatory.
The mean difference in FSS from baseline was –0.69 points (SD = 1.10) at 6 months and –0.70 points (SD = 1.56) at 12 months. In the subgroup analyses, the mean difference in FSS from baseline at 6 months and at 12 months was –0.93 points (SD = 1.15) and –0.89 points (SD = 1.86), respectively, in ambulatory patients, and –0.56 points (SD = 1.08) and –0.59 points (SD = 1.40), respectively, in nonambulatory patients. The correlation coefficient between the difference in FSS from baseline and the difference in HFMSE from baseline was –0.19567 (P = 0.3183) at 6 months and –0.18663 (P = 0.3513) at 10 months. The correlation coefficient between the differences in FSS and the differences in 6WMT from baseline was –0.68294 (P = 0.0295) at 6 months and –0.52727 (P = 0.1173) at 10 months.34
A key limitation of the study was the lack of a comparator. The study did not control for known prognostic factors or effect modifiers of SMA in the analyses, so it is impossible to establish a causal link between the treatment and outcomes. It is also noteworthy that the distribution of SMA subtypes (type II vs III) in the study was not in line with the distribution in Canada, according to feedback from the clinical experts consulted by CADTH. The majority of patients had SMA type III. It appeared that the study population did not reflect a random sampling of the SMA population, suggesting a risk of selection bias. Another limitation was that FSS has not been validated in the SMA population. The scale is subject to a high risk of reporting bias, given that FSS is a patient-reported outcome,35 and fatigue is highly subjective. Because no blinding was in place, a patient’s experience of fatigue can be influenced by a perception of the treatment. In addition, given that the scale used a recall period of 1 week and was administered only 3 times during the study — at baseline, 6 months, and 10 months — it is unclear if the data are reflective of patients’ treatment experiences throughout the study.
Moshe-Lilie et al. (2020)
Moshe-Lilie et al. (2020)36 was a retrospective chart review that examined the change in motor function in 22 adults with genetically confirmed type II and III SMA between 2017 and 2019 at a medical centre in the US. Ten patients received treatment with nusinersen and 12 were untreated. The sum of MRC scores of upper and lower limb strength was recorded at baseline and at 4, 6, 12, and 24 months. The patients had a median age of 26 years (range = 20 to 71). Most patients were female (15 [68%]), nonambulatory (20 [91%]), had 3 copies of the SMN2 gene (13 [59%]), and symptom onset in childhood (14 [64%]). Most patients had type III SMA (13 [59%]) and the remainder had type II (9 [41%]). Scoliosis was present in 17 (77%) patients, and ventilator support was required by 10 (45%) patients. It was unclear if the patients in the nusinersen group had received any prior nusinersen treatment.
The change from baseline in mean % MRC was the primary outcome of interest. In the treated group, the mean % MRC change from baseline was 2.5% at 12 months and 3.9% at 24 months. In the untreated group, the mean % MRC change was not reported but, according to the authors, % MRC was stable in most untreated patients. A decline of 2.5% to 3.8% in % MRC was noted in 3 nusinersen-treated patients. Of the 3 treated patients for whom HFMSE scores were available, 2 remained stable and 1 had a 12-point improvement at 12 months. In terms of safety, lumbar-puncture headache was observed in 5 patients, 2 of whom required a blood patch. One patient was hospitalized with bacterial meningitis and required long-term antibiotics. One patient died of respiratory failure in the setting of pneumonia shortly after treatment initiation. Three patients discontinued treatment for reasons that included recurrent pneumonia, lack of improvement, and proteinuria.36
The primary limitation of the study was the risk of selection bias. All charts reviewed were sourced from 1 neuromuscular group. Having a single source may introduce selection bias, as the patients under the care of this team may share common characteristics, (such as treatment history, disease severity, and level of supportive care) that can bias estimation of the treatment effect. It is unlikely that the selected patients were representative of the SMA patient population, considering that the majority (59%) had SMA type III, which does not align with the comment from the clinical experts that SMA type II is the predominant SMA subtype seen in clinical practice. Furthermore, there was no mention whether any matching or adjustments for plausible prognostic factors were made between treated and untreated patients. It is impossible to assess whether there were imbalances in the baseline characteristics between the 2 groups, as only pooled data were presented. Any imbalances in prognostic variables will likely result in a biased conclusion. Another limitation was that there was very limited reporting of outcome data. Other than a trajectory plot to illustrate the % MRC of each patient over time, most outcome data were incomplete and were summarized descriptively. For example, the difference in mean % MRC from baseline, which was the key outcome, was reported in the treated arm but not in the untreated arm. Minimal information on measurements of HFMSE and RULM scores was provided. The qualitative descriptions provided limited value for objective interpretation of the outcomes. The risk of reporting bias was high, given the limited availability of data and the presence of selective reporting.
Yeo et al. (2020)
The study by Yeo et al. (2020)37 was a prospective, noncomparative, observational study that included 6 adults with molecularly confirmed 5q type III SMA from a single centre in the US. Patients with limb contractures, a history of spinal fusion, spine surgery or significant scoliosis that affected function, or respiratory insufficiency that required more than nocturnal BiPAP or continuous positive airway pressure were excluded from the study. Patients were treated with nusinersen, with loading doses of 12 mg at baseline, 2, 4, and 8 weeks, followed by maintenance doses of 12 mg at 4-month intervals (120 days). The study aimed to determine the impact of nusinersen on motor outcomes in a targeted cohort of adults with type III SMA for at least 12 months. Primary outcomes were HFMSE and RULM scores. Secondary outcomes were the PedsQL Multidimensional Fatigue Scale, the SMAFRS, and the 6-minute and 10-metre walk tests. Outcome measures and evaluations were conducted every 3 to 4 months, before each maintenance dose. The mean follow-up duration was 17 months, with a range of 14 to 21 months. Patient ages ranged from 24.9 to 56.5 years, with a median age of 29.9 years. Most patients were male (5 [83%]), had 3 copies of SMN2 (3 [50%]), and were ambulant (4 [67%]). Age at symptom onset ranged from 1 to 14 years, with a median of 8 years. Other patient characteristics included a median HFMSE score of 35 (range = 21 to 53), and a median RULM score of 31.5 (range = 22 to 37).
Mean changes in HFMSE and RULM scores over 14 months were 2 points (range = 1 to 5) and 1.8 points (range = 0 to 3), respectively. Over the course of the study (at various points until 21 months), individual changes greater than 2 points were observed in the HFMSE score of 3 (50%) patients and in the RULM score of 2 (33%) patients. Of the 4 ambulatory patients able to complete the 6MWT and 10-metre walk test, no statistically or clinically meaningful changes were observed in either test from baseline to the final study visit. The study reported a decline in the SMAFRS total score and no changes in the PedsQL. A total of 12 AEs were reported: 4 patients developed post-lumbar-puncture headache, 2 developed vertigo, 4 experienced fall-related injuries (including 1 SAE that required hospitalization), and 2 developed recurrent pressure sores (including 1 SAE that required hospitalization).37
The main limitations of the study are the noncomparative, observational design and the lack of reporting on any sample size or power calculations tied to a hypothesis; thus, it is not clear whether the sample size was sufficient to detect changes in the targeted population, which may limit the conclusions and generalizations that can be drawn from this study. All patients included in the study had SMA type III, and patients with worse prognostic factors (such as limb contractures, spinal fusion or scoliosis, and respiratory insufficiency) were excluded, which resulted in a higher-functioning population and compromised the representativeness of the study sample to the general adult SMA population. The study did not deploy any method of blinding, so both the patients and investigators were aware of the nusinersen treatment. The expectation of treatment-induced improvements may have influenced the behaviour of patients or outcome assessors, leading to expectation bias in favour of nusinersen. The study reported limited data on outcome measures using line graphs and brief description, and the investigators didn’t provide statistical analysis of outcomes. For example, HFMSE, RULM, PedsQL, SMAFRS, 6MWT, and 10-metre walk test were assessed at various time points until 21 months, but the study only reported the mean change and range for HFMSE and RULM from baseline to 14 months and brief summaries of the trend of change for PedsQL, SMAFRS, 6MWT, and 10-metre walk test from baseline to final visits; there was no mention of data on outcome measures at other time points. Although the study described the statistical analysis in the analysis plan section, it provided no statistical test results. Moreover, limited data were presented for outcomes of interest at various time points. The selective reporting of data may lead to distortion, and prevent readers from interpreting the study results objectively. Therefore, the study reported incomplete data and was subjected to a high risk of reporting bias.
Walter et al. (2019)
Walter et al. (2019)38 was a prospective, single-centre, observational study that evaluated the safety and treatment effects of nusinersen in 19 adults with 5q SMA type III in Germany. Patients were treated with intrathecal nusinersen regimen, per the product monograph, for up to 300 days. Patients were monitored within the SMArtCARE registry. Motor-function scores, pulmonary function, and laboratory biomarkers were assessed at baseline and days 63, 180, and 300. The mean age at baseline was 35.1 years (SD = 11.72; range = 18 to 59). The mean disease duration was 24.3 years (SD = 12.1). The mean age at symptom onset was 10.8 years (SD = 9.2). Most patients had 4 copies of SMN2 (79%), and most patients were ambulatory (12 [63%]); only 7 (37%) patients were wheelchair-dependent and only 3 (15.7%) patients had scoliosis. The study did not indicate whether the patients had received any prior nusinersen treatment.
The mean RULM score improved from 32.32 (SD = 7.39) at baseline to 33.06 (SD = 7.33) on day 300. The mean HFMSE score was 35.16 (SD = 21.14) at baseline and 39.50 (SD = 20.58) on day 300. The mean 6WMT increased from 369.50 m (SD = 126.62) at baseline to 377.75 m (SD = 156.60) on day 300. The mean percent-predicted FVC at baseline (94.54 [SD = 15.45]) was similar to the level on day 300 (99.54% [SD = 12.42]). Among the 11 patients who reported AEs, there were 4 cases of post-lumbar-puncture headache, 1 case of fatigue, and 7 cases of lower back pain. Laboratory tests were normal and there was no evidence of nephrotoxicity.38
As in the other studies, the noncomparative design was a key limitation. All enrolled patients were treated with nusinersen; there was no comparison arm. No definitive conclusion can be drawn regarding the treatment effect of nusinersen, as the outcomes were confounded by factors such as physical exercise and learning effect, which are known effect modifiers of SMA. It was also unclear if random sampling of patients was involved; the sampling method was not described. It is, therefore, unclear whether the patients selected were reflective of the type III SMA population. Although the study captured a group of patients with large variations in disease duration (range = 6 to 52 years), the distribution was not even across the spectrum. Of the 19 patients, 14 (73.7%) had less than 30 years of SMA history. Having a high proportion of patients with shorter disease history can result in biased results, as these patients generally have more functional reserves, which allow for the detection of any potential treatment responses. Moreover, the baseline motor-function scores in this study were high, suggesting that patients enrolled had less severe disease. Another limitation was that the duration of follow-up was only up to 300 days, which was inadequate to capture meaningful changes in functional outcomes because SMA patients generally have very slow disease progression.
Discussion
Summary of Available Evidence
As part of the reassessment for nusinersen, 7 studies were submitted to CADTH, of which 4 were summarized and appraised; Hagenacker et al. (2020)17 was a prospective, multi-centre, noncomparative observational study; Maggi et al. (2020)18 was a retrospective, noncomparative cohort study; Pera et al. (2021)19 was a noncomparative, registry-based study of the ISMAR; and the EU registry study15,16 submitted by the sponsor was a noncomparative, registry-based cohort study from 3 European registries.
There was a high degree of selection bias in the included patients. Overall, the studies enrolled mostly adults with type III SMA (62% to 100%), with very few type II patients. Moreover, across studies, the majority of patients had 4 or more copies of SMN2, with a high degree of variation in the proportion of ambulatory and nonambulatory patients. The primary outcome in all studies was change in HFMSE over time; other measures included RULM and 6MWT. Baseline functional measures for the HFMSE, RULM, and 6MWT in all studies were considered high, indicating a high level of motor functionality in included patients. The median follow-up for each study was not frequently reported; however, most patients were followed for a minimum of 6 months and up to 14 months, which was considered immature for SMA. In addition, the noncomparative nature of these studies prevents the determination of any association between nusinersen and the reported results.
In addition to the key studies reviewed, CADTH identified 8 studies that included the relevant population; however, they did not meet the study design criteria for inclusion in the systematic review.
Interpretation of Results
Effectiveness
Nusinersen has previously been reviewed by CADTH in 2017 and 2019, and the CDEC recommended reimbursement of the drug with conditions for patients with 5q SMA with 2 or 3 copies of the SMN2 gene and have had a disease duration of less than 6 months and symptom onset from 1 week to 7 months of age, and for patients who are 12 years or younger with symptom onset after 6 months of age who never achieved the ability to walk independently on the basis of multiple comparative and noncomparative RCTs (ENDEAR, CHERISH, and NURTURE).13,14 The clinical experts and patients identified an important unmet need for treatments in adults with SMA naive to disease-modifying therapy that will reverse damage, halt or stabilize muscle weakness related to disease progression, and maintain independence and QoL by improving strength.
Overall, the numerous limitations in the reviewed studies, identified in the critical appraisal, prevent generalization of the results to adults who have type II or III SMA and who have never been treated with nusinersen. The key limitations were the relatively small sample sizes (ranging from 67 to 252 patients), selection bias leading to study populations unrepresentative of the target population or the reimbursement request (e.g., a lack of type II SMA patients), and the generally noncomparative study designs that did not allow for the establishment of cause and effect or associations between nusinersen and the observed changes in outcomes measures. As well, the notable rates of drop-outs for key outcomes between baseline and 14 months and the relatively short duration of follow-up for a chronic, slowly progressing disease were important limitations that hindered interpretation of the results.
Most of the experts agreed that there is limited evidence in this population and highlighted the lack of RCTs, noting that all available evidence comes from observational studies, precluding the use of nusinersen in adults. Despite the small sample sizes, the clinical experts consulted by CADTH considered it reasonable to conduct a comparative, placebo-controlled RCT in adults with type II and III SMA. However, several of the clinical-expert panel members discussed the lack of a clear biologic mechanism through which SMN2-modifying therapy such as nusinersen could benefit adults with SMA in whom the production of the SMN protein had already decreased significantly, making the reversal of muscle function impossible.
Most of the included studies did not include a comparator, other than evaluating pre- and post-treatment outcomes. An analysis of nusinersen-treated and untreated patients was conducted in the EU registry study15,16; however, only 17 SMA patients (14 type III, and 3 type IV) were included in the untreated group, which was considered insufficient to determine whether nusinersen demonstrated an effect in these patients.
All of the main studies provided by the sponsor and summarized in this reassessment shared a common outcome of change from baseline in motor function, measured with the HFMSE, RULM, or 6MWT. These measures are well known and validated in the SMA population and are regularly used in Canadian clinical practice. Although the HFMSE and 6MWT are appropriate measures to determine motor function in SMA, they are generally limited to ambulatory patients who can stand and sit. Given that in all the included studies there was heterogeneity in the proportion of ambulant and nonambulant patients, these measures are subject to even smaller sample sizes, affecting the interpretation of the results across studies. In addition to outcomes that evaluated motor function, clinically important outcomes identified by patients and clinicians, including the maintenance of independence and QoL, were not evaluated in any of the included studies; therefore, the effect of nusinersen on these outcomes remains uncertain. Other important outcomes, including vocalization, swallowing issues, ventilation, or nutritional requirements, were not available in the included studies, which many patients and clinicians cited as having a significant impact on HRQoL and daily life. Only Brakemeier et al. (2021)30 (summarized in the Other Relevant Evidence section) evaluated the effect of nusinersen on bulbar function.
Most patients enrolled in the studies had type III SMA with 3 or 4 copies of the SMN2 gene, and appeared to have higher functioning on baseline scale scores and ambulatory status. As hypothesized in the clinical-expert input, this population may be more likely to experience a benefit due to an elevated number of motor units and nerves; however, it remains unclear if clinical significance was reached, and the magnitude of effect is still uncertain. Combined with the identified limitations in the data, there is a high degree of uncertainty about the benefit of nusinersen in the requested population.
Across studies, the baseline scores for motor-function outcomes in nusinersen-treated patients — considered to be high by the clinical experts consulted by CADTH, based on the patients seen in their practice — ranged from 20.74 in Hagenacker et al. (2020)17 to 30.75 in Pera et al. (2021)19 for the HFMSE, from 20.87 in Hagenacker et al. (2020)17 to 29.0 in Pera et al. (2021)19 for the RULM, and from 300.87 m in the EU registry study15,16 to 323.03 m in Pera et al. (2021),19 suggesting that the enrolled patients had less severe disease and greater physical function. Changes from baseline in these motor-function outcomes varied across studies, with a mean change from baseline in HFMSE score ranging from 0.79 points (95% CI, –0.29 to 1.87) at 12 months in Pera et al. (2021)19 to 3.12 points (95% CI, 2·06 to 4·19) at 14 months in Hagenacker et al. (2020).17 In the EU registry study,15,16 the change in HFMSE was 0.02907 points per week (95% CI, 0.01930 to 0.03884) in nusinersen-treated patients, compared with –0.01129 (95% CI, −0.03289 to 0.01031) in untreated patients, and 0.2575 points per week (95% CI, 0.01038 to 0.04112) after the initiation of nusinersen; however, before initiation of nusinersen, the HFMSE was declining at –0.00006 points per week (95% CI, –0.0096 to 0.0094). As such, there is uncertainty as to whether patients were truly declining before nusinersen, and whether the slope improved in a meaningful way after nusinersen treatment. For the RULM, mean change from baseline ranged from 0.07 in Pera et al. (2021)19 at 12 months to 1.09 points in Hagenacker et al. (2020)17 at 14 months. In the EU registry study,15,16 the change in RULM was 0.01168 points per week (95% CI, 0.004957 to 0.01841) in nusinersen-treated patients, with a post-treatment change of 0.002569 points per week (95% CI, −0.00533 to 0.01047). The post-treatment results were not statistically significant, and it is unclear whether this change represents a clinically meaningful improvement in the change in RULM score after treatment. Moreover, the comparison group consisted of only 17 untreated patients, making it difficult to interpret these comparisons. For the 6MWT, the mean change from baseline ranged from 0.52 m in Pera et al. (2021)19 at 12 months to 46.0 m in Hagenacker et al. (2020)17 at 14 months. In the EU registry study,15,16 the change in 6MWT was 0.2633 points per week (95% CI, 0.09922 to 0.42740) in nusinersen-treated patients, with a post-treatment change of –0.03399 points per week (95% CI, –0.4373 to 0.3694). Despite extrapolation of the EU registry study15,16 results to a 1-year timespan, it could not be concluded that these results were due to treatment with nusinersen, and they were not considered clinically meaningful by the clinical experts consulted by CADTH. In all studies, the mean changes in motor-function scores over the short study durations were considered small, and according to the clinical-expert panel, are likely to be noise due to the natural history of the disease. Coupled with the wide CIs, the motor-function results are considered imprecise, and do not represent meaningful change that can be attributed to nusinersen.
Despite the limitations related to study design, sample sizes, and selection bias, the totality of evidence identified and submitted consistently suggests a positive association in motor-function outcomes in adults treated with nusinersen throughout the studies. However, it is uncertain whether any of the observed changes can be attributed to nusinersen or whether they can be considered clinically meaningful. As to the natural history of the disease, the clinical experts consulted by CADTH noted that individual variations in disease progression are observed among adults, with phases of clinical worsening but also periods of stabilization, during which patients may retain vital motor functions; for some patients, this translates into preservation of functional independence. The reported interpretations in each study that nusinersen led to improvement or stabilization cannot be made without a clear hypothesis statement or a design or analysis to test this outcome specifically; thus, any changes seen in patients across studies must consider the individual variance and heterogeneity, and must be interpreted with the utmost caution. Furthermore, the maximum time of assessment across studies was 14 months, which was considered immature to determine the effectiveness of nusinersen or capture clinical changes in adults with type II or III SMA because of individual disease heterogeneity, natural history, and periods of disease stabilization.
The clinical experts consulted by CADTH noted that in SMA or any neuromuscular disease, there are many additional factors to consider when interpreting scale scores. Potential confounders and treatment-effect modifiers that are likely to influence outcomes and that are routinely seen in clinical practice, according to the clinical experts consulted by CADTH, were not considered, including training for the outcomes of interest, routine exercise and observation, and other routine care (such as physiotherapy and occupational therapy). It has been well documented in other chronic diseases that there are notable learning and encouragement effects associated with the motor-function measures, particularly the 6MWT,39-42 although no training effect has been described for 6MWT in natural-history studies,38 and the clinical experts stressed that routine exercise is known to have a profound effect on the maintenance of neuronal connectivity and muscle. These factors are expected to heavily influence the results of the included motor-function outcome measures, limiting the generalizability of results to Canadian patients. It should also be noted that in Canada, many SMA patients are potentially under a system of renewal for nusinersen, whereby reimbursement is contingent on the demonstration of improvements in motor-function scales; thus, patients are motivated to show improvements, which may further influence the outcome results. Another main concern of the clinical experts consulted by CADTH was evidence for the use of nusinersen in treatment-naive adults with type II or III SMA; the studies did not consider pathobiology or the natural history of the disease. The experts noted that nusinersen works best in young children with SMA, and emphasized the lack of data in older patients with more advanced disease. The experts highlighted the well-established pathobiology of SMA, particularly the demonstrated post-natal progression of motor denervation, reflected by compound muscle action potential and motor unit number estimation values in all SMA types in early ages by Swoboda et al. (2005).25 The experts also noted that these results were compounded by the subgroup analysis for the CHERISH study, which demonstrated no statistically significant functional benefit with nusinersen in HFMSE scores in patients older than 4 years.43 As such, the clinical experts consulted by CADTH indicated that pathobiology, natural history, trial data, and the often difficult administration of nusinersen make it unclear whether it (or any other currently available medications) would have a clinically meaningful effect in adults with type II or III SMA.
Clinically meaningful improvements in the motor-function outcomes have been established; however, there is some uncertainty about what constitutes a clinically meaningful improvement in adults with type II or III SMA. The experts also noted that there is a discrepancy in the reflection of the actual limitations of these measures on daily life; improvements in the scales may be shown, but might not translate into changes in functional ability. As noted in Appendix 4, an increase in HFMSE score greater than 2 points is unlikely in untreated patients with type II or III SMA.44 However, patients and caregivers consider a 1-point increase meaningful,45 and Pera et al. (2017)19 considered a 3-point change to be clinically meaningful.46 With RULM, 2 points was considered to be a clinically meaningful improvement, and with 6MWT, an improvement of 23 to 45 m is considered to be clinically meaningful in other chronic conditions. The clinical experts consulted by CADTH noted that there is some heterogeneity in what can be considered a meaningful change in motor function, and explained that these minor changes in HFMSE or RULM would only result in minimal improvements, which may not cross important disease-severity thresholds that affect patient function or QoL.
A single meta-analysis of published evidence submitted by the sponsor evaluated the effect of nusinersen treatment on key motor-function outcomes. Although there was a general positive association between nusinersen and HFMSE, RULM, and 6MWT scores, the results were imprecise because of wide 95% CIs, and there was notable heterogeneity in the studies included in the meta-analysis with regard to study design (all observational), the included populations (both adults and children with type II and III SMA of various ambulatory ability), and small sample sizes (which limited the ability to draw any conclusions about the effectiveness of nusinersen). Moreover, it was uncertain if the pooled point estimates for the adult subgroup were considered clinically meaningful.
Eight studies were identified in the literature search that met all inclusion criteria of the systematic review except study design; all were noncomparative observational studies. As with the studies provided by the sponsor, the effectiveness of nusinersen in these studies is highly uncertain because of the noncomparative study design, selection bias, and relatively small sample sizes of adults with type II and III SMA.
Harms
The safety of nusinersen has been examined in the children and adolescents with SMA, as noted in the previous submissions to CADTH.13,14 However, it was important for this review to consider the harms of nusinersen associated with the intrathecal administration method, given that patients with type II and III SMA generally have complex spines (some had already undergone spinal-fusion surgery).47,48 It was not expected that harms would vary considerably by SMA type, but patients with type II SMA are more likely to have complex spines due to the inability to walk.48 Because of the smaller sample sizes in the studies, separate analyses of AEs by SMA type were not conducted in any study.
Harms results varied and were infrequently reported in the available evidence. Across studies, the overall frequency of AEs was low, occurring in up to 47% of patients across studies, and the reported AEs were mostly related to procedural complications (most frequently headache and back pain), which are relatively common with this procedure. It should be noted that AE reporting is not standardized in registries, so these results should be interpreted with caution and may be biased. SAEs were rarely reported. As noted by the clinical experts, any AEs that occur with nusinersen will likely occur immediately after administration.
The frequency of attrition in the included studies likely affected the reporting of AEs and complications. As previously noted, survivorship bias may be present in the study populations, in which patients who are more likely to tolerate treatment remain on treatment for the duration of the study. Overall, only 4 patients across all included studies withdrew due to AEs; however, given the study designs, it is unclear whether more patients withdrew due to AEs but were not reported. In terms of safety, the duration of follow-up of 14 months for most studies is likely sufficient to observe nusinersen-related AEs; however, the effect of long-term exposure in adults with type II or III SMA is unknown.
Notable harms of interest for this reassessment were guided, in part, by the product monograph for nusinersen, and included serious infections, lumbar-puncture-related AEs, coagulation abnormalities, and renal toxicity, which are generally considered to be important in this patient population receiving disease-modifying therapy. Minimal results were provided for the notable harms of interest to this review, or for harms in general, which may be limited due to the design and aims of the included studies, as well as availability of information.
Overall, the clinical experts consulted by CADTH indicated that the potential for harms in adults related to the method of administration and the frequent need for interventional-radiology-guided administration in practice is a key part of their decision to use nusinersen in adults. The experts noted that there are limited intraspinous spaces available for lumbar puncture, which reduces the area for the various nusinersen injections. Additionally, the complexity of the spine can be a barrier for the intrathecal administration of nusinersen. Procedural complications in patients with complex spines were not reported in any of the included studies, and the proportion of patients requiring guided administration was only reported in 1 study (Maggi et al. [2020]18). The experts emphasized that the administration of nusinersen in patients with complex spines increases the difficulty and risk of the procedure. Administration of nusinersen using interventional-radiology-guided techniques allows for patients with complex spines to receive treatment, but the experts expressed concern about the frequency of exposure to ionizing radiation in patients who requiring X-ray-, CT-, or fluoroscopy-guided injections. Although the cumulative radiation is known to be low in the intrathecal, interventional-radiology-guided administration of nusinersen, clinical experts expressed concern that the effect of long-term exposure due to repeated administration over a significant period of time is unknown, and they acknowledged a need for more safety data in this area.
Conclusions
Four noncomparative, observational studies were included in the reassessment of nusinersen for adults with type II or III SMA. The observational nature and lack of a well-defined concurrent comparator in the included studies significantly limits the ability to establish causal relationships between treatment effects and nusinersen.
In all of the studies, selection bias in the included populations and relatively small sample sizes were noted as key limitations. All studies included mostly type III SMA adults with higher physical functioning at baseline based on SMA type, a higher number of SMN2 copies, better ambulatory status, and higher baseline scores for motor-function outcomes. Input from clinical experts noted that the populations were not reflective of the reimbursement request, particularly due to the lack of type II SMA patients, or to their clinical practice.
Although there was generally a consistent positive effect of nusinersen on motor-function outcomes, the magnitude of the treatment effect with nusinersen varied and was often not clinically meaningful. Given the limitations in study design, statistical analysis, duration, and the heterogeneous natural history of adults with SMA, results in all studies were considered highly uncertain and may not be generalizable to the Canadian population. Harms associated with nusinersen were generally mild to moderate in severity and were related to the administration procedure, with lumbar-puncture-related AEs the most frequently reported. However, the reporting of AEs was inconsistent and infrequent and, because of study designs and associated biases, may be under-reported.
Although the amount of real-world data for nusinersen is relatively high, the overall quality of studies remained a concern. Most of the identified evidence could not provide conclusive evidence demonstrating the effectiveness of nusinersen in adults with type II or III SMA. Overall, it remains unclear if nusinersen resulted in clinically meaningful improvements or disease stabilization, which were considered important outcomes by patients. Additionally, since HRQoL was not assessed in any studies, the effect of nusinersen on this important outcome in adults is unknown.
References
1.D’Amico A, Mercuri E, Tiziano FD, Bertini E. Spinal muscular atrophy. Orphanet J Rare Dis. 2011;6:71. PubMed
2.Tisdale S, Pellizzoni L. Disease mechanisms and therapeutic approaches in spinal muscular atrophy. J Neurosci. 2015;35(23):8691-8700. PubMed
3.Kolb SJ, Kissel JT. Spinal muscular atrophy. Neurol Clin. 2015;33(4):831-846. PubMed
4.Verhaart IEC, Robertson A, Wilson IJ, et al. Prevalence, incidence and carrier frequency of 5q-linked spinal muscular atrophy - a literature review. Orphanet J Rare Dis. 2017;12(1):124. PubMed
5.Farrar MA, Park SB, Vucic S, et al. Emerging therapies and challenges in spinal muscular atrophy. Ann Neurol. 2017;81(3):355-368. PubMed
6.Prior TW, Snyder PJ, Rink BD, et al. Newborn and carrier screening for spinal muscular atrophy. Am J Med Genet A. 2010;152a(7):1608-1616. PubMed
7.Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008;371(9630):2120-2133. PubMed
8.Ogino S, Wilson RB. Genetic testing and risk assessment for spinal muscular atrophy (SMA). Hum Genet. 2002;111(6):477-500. PubMed
9.McMillan HJ, Gerber B, Cowling T, et al. Burden of spinal muscular atrophy (SMA) on patients and caregivers in Canada. J Neuromuscul Dis. 2021;8(4):553-568. PubMed
10.Arnold WD, Kassar D, Kissel JT. Spinal muscular atrophy: diagnosis and management in a new therapeutic era. Muscle Nerve. 2015;51(2):157-167. PubMed
11.Wadman RI, Stam M, Gijzen M, et al. Association of motor milestones, SMN2 copy and outcome in spinal muscular atrophy types 0-4. J Neurol Neurosurg Psychiatry. 2017;88(4):365-367. PubMed
12.Spinraza (nusinersen): solution for intrathecal injection 2.4 mg/mL nusinersen as nusinersen sodium [product monograph]. Mississauga (ON): Biogen Canada Inc.; 2020 Apr 20.
13.CADTH Drug Reimbursement Expert Review Committee final recommendation: nusinersen (Spinraza — Biogen Canada Inc.). Ottawa (ON): CADTH; 2017: https://www.cadth.ca/sites/default/files/cdr/complete/SR0525_Spinraza_complete_Dec_22_17.pdf. Accessed 2022 Apr 4.
14.CADTH Drug Reimbursement Expert Review Committee final recommendation: nusinersen (Spinraza — Biogen Canada Inc.). Ottawa (ON): CADTH; 2019: https://www.cadth.ca/sites/default/files/cdr/complete/SR0576-Spinraza-Resubmission-Mar-1-19.pdf Accessed 2022 Apr 4.
15.Drug Reimbursement Review sponsor submission: Spinraza (nusinersen injection), solution for intrathecal injection 2.4 mg/mL nusinersen as nusinersen sodium [internal sponsor’s package]. Toronto (ON): Biogen Canada Inc.; 2021 Dec 1.
16.Bovis F, Sormani MP. Registry analyses spinal muscular atrophy. In: Drug Reimbursement Review sponsor submission: Spinraza (nusinersen injection), solution for intrathecal injection 2.4 mg/mL nusinersen as nusinersen sodium [internal sponsor’s package]. Toronto (ON): Biogen Canada Inc.; 2021 Dec 1.
17.Hagenacker T, Wurster CD, Gunther R, et al. Nusinersen in adults with 5q spinal muscular atrophy: a non-interventional, multicentre, observational cohort study. Lancet Neurol. 2020;19(4):317-325. PubMed
18.Maggi L, Bello L, Bonanno S, et al. Nusinersen safety and effects on motor function in adult spinal muscular atrophy type 2 and 3. J Neurol Neurosurg Psychiatry. 2020;91(11):1166-1174. PubMed
19.Pera MC, Coratti G, Bovis F, et al. Nusinersen in pediatric and adult patients with type III spinal muscular atrophy. Ann Clin Translat Neurol. 2021;8(8):1622-1634. PubMed
20.Coratti G, Cutrona C, Pera MC, et al. Motor function in type 2 and 3 SMA patients treated with Nusinersen: a critical review and meta-analysis. Orphanet J Rare Dis. 2021;16(1):430. PubMed
21.Clinical Study Report: ISIS 396443-CS11. An open-label extension study for patients with spinal muscular atrophy who previously participated in investigational studies of ISIS 396443 [internal sponsor’s report]. Cambridge (MA): Biogen MA Inc.; 2021 Jan 4.
22.Konersman CG, Ewing E, Yaszay B, Naheedy J, Murphy S, Skalsky A. Nusinersen treatment of older children and adults with spinal muscular atrophy. Neuromuscul Disord. 2021;31(3):183-193. PubMed
23.Belter L, Jarecki J, Reyna SP, et al. The Cure SMA membership surveys: highlights of key demographic and clinical characteristics of individuals with spinal muscular atrophy. J Neuromuscul Dis. 2021;8(1):109-123. PubMed
24.Muscular Dystrophy Canada. MDC Statement. 2020; https://muscle.ca/2020/12/04/mdc-statement/. Accessed 2022 Apr 4.
25.Swoboda KJ, Prior TW, Scott CB, et al. Natural history of denervation in SMA: relation to age, SMN2 copy number, and function. Ann Neurol. 2005;57(5):704-712. PubMed
26.Prodcut information: Spinraza (nusinersen): solution for intrathecal injection 2.4 mg/mL nusinersen as nusinersen sodium. (European public assessment report). London (GB): European Medicines Agency; 2022: https://www.ema.europa.eu/en/documents/product-information/spinraza-epar-product-information_en.pdf. Accessed 2022 Apr 4.
27.EVRYSDI: risdiplam powder for oral solution 60 mg/bottle (0.75 mg/mL after reconstitution), oral or enteral [product monograph]. Mississauga (ON): Hoffmann-La Roche Limited; 2021 Apr 14.
28.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. J Clin Epidemiol. 2016;75:40-46. PubMed
29.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2022 Apr 8.
30.Brakemeier S, Stolte B, Thimm A, et al. Assessment of bulbar function in adult patients with 5q-SMA type 2 and 3 under treatment with nusinersen. Brain sci. 2021;11(9):20. PubMed
31.Duong T, Wolford C, McDermott MP, et al. Nusinersen treatment in adults with spinal muscular atrophy. Neurol. 2021;11(3):e317-e327. PubMed
32.Elsheikh B, Severyn S, Zhao S, et al. Safety, tolerability, and effect of nusinersen in non-ambulatory adults with spinal muscular atrophy. Front Neurol. 2021;12:650532. PubMed
33.Mix L, Winter B, Wurster CD, et al. Quality of life in SMA patients under treatment with nusinersen. Front Neurol. 2021;12:626787. PubMed
34.Kizina K, Stolte B, Totzeck A, et al. Fatigue in adults with spinal muscular atrophy under treatment with nusinersen. Sci Rep. 2020;10(1):11069. PubMed
35.Mattsson M, Moller B, Lundberg I, Gard G, Bostrom C. Reliability and validity of the Fatigue Severity Scale in Swedish for patients with systemic lupus erythematosus. Scand J Rheumatol. 2008;37(4):269-277. PubMed
36.Moshe-Lilie O, Visser A, Chahin N, Ragole T, Dimitrova D, Karam C. Nusinersen in adult patients with spinal muscular atrophy: Observations from a single center. Neurology. 2020;95(4):e413-e416. PubMed
37.Yeo CJJ, Simeone SD, Townsend EL, Zhang RZ, Swoboda KJ. Prospective cohort study of nusinersen treatment in adults with spinal muscular atrophy. J Neuromuscul Dis. 2020;7(3):257-268. PubMed
38.Walter MC, Wenninger S, Thiele S, et al. Safety and treatment effects of nusinersen in longstanding adult 5q-SMA Type 3 - a prospective observational study. J Neuromuscul Dis. 2019;6(4):453-465. PubMed
39.Chan WLS, Pin TW. Practice effect and cueing of 2-minute walk test, 6-minute walk test and 10-metre walk test in frail older adults with and without dementia - Recommendations to walk tests protocols. Exp Gerontol. 2019;124:110648. PubMed
40.Morales Mestre N, Audag N, Caty G, Reychler G. Learning and encouragement effects on six-minute walking test in children. J Pediatr. 2018;198:98-103. PubMed
41.Spencer L, Zafiropoulos B, Denniss W, Fowler D, Alison J, Celermajer D. Is there a learning effect when the 6-minute walk test is repeated in people with suspected pulmonary hypertension? Chron Respir Dis. 2018;15(4):339-346. PubMed
42.Wu G, Sanderson B, Bittner V. The 6-minute walk test: how important is the learning effect? Am Heart J. 2003;146(1):129-133. PubMed
43.Mercuri E, Darras BT, Chiriboga CA, et al. Nusinersen versus sham control in later-onset spinal muscular atrophy. N Engl J Med. 2018;378(7):625-635. PubMed
44.Mercuri E, Finkel R, Montes J, et al. Patterns of disease progression in type 2 and 3 SMA: Implications for clinical trials. Neuromuscul Disord. 2016;26(2):126-131. PubMed
45.McGraw S, Qian Y, Henne J, Jarecki J, Hobby K, Yeh WS. A qualitative study of perceptions of meaningful change in spinal muscular atrophy. BMC Neurol. 2017;17(1):68. PubMed
46.Pera MC, Coratti G, Forcina N, et al. Content validity and clinical meaningfulness of the HFMSE in spinal muscular atrophy. BMC Neurol. 2017;17(1):39. PubMed
47.Fujak A, Raab W, Schuh A, Richter S, Forst R, Forst J. Natural course of scoliosis in proximal spinal muscular atrophy type II and IIIa: descriptive clinical study with retrospective data collection of 126 patients. BMC Musculoskelet Disord. 2013;14:283-283. PubMed
48.Wijngaarde CA, Brink RC, de Kort FAS, et al. Natural course of scoliosis and lifetime risk of scoliosis surgery in spinal muscular atrophy. Neurology. 2019;93(2):e149-e158. PubMed
49.O’Hagen JM, Glanzman AM, McDermott MP, et al. An expanded version of the Hammersmith Functional Motor Scale for SMA II and III patients. Neuromuscul Disord. 2007;17(9-10):693-697. PubMed
50.Pera MC, Coratti G, Forcina N, et al. Content validity and clinical meaningfulness of the HFMSE in spinal muscular atrophy. BMC Neurol. 2017;17(1):39, 2017.
51.Glanzman AM, O’Hagen JM, McDermott MP, et al. Validation of the expanded Hammersmith Functional Motor Scale in spinal muscular atrophy type II and III. J Child Neurol. 2011;26(12):1499-1507. PubMed
52.Glanzman AM, Mazzone ES, Young SD, et al. Evaluator training and reliability for SMA global nusinersen trials1. J Neuromuscul Dis. 2018;5(2):159-166. PubMed
53.Stolte B, Bois JM, Bolz S, et al. Minimal clinically important differences in functional motor scores in adults with spinal muscular atrophy. Eur J Neurol. 2020;27(12):2586-2594. PubMed
54.King MT. A point of minimal important difference (MID): a critique of terminology and methods. Expert Rev Pharmacoecon Outcomes Res. 2011;11(2):171-184. PubMed
55.Revicki D, Hays RD, Cella D, Sloan J. Recommended methods for determining responsiveness and minimally important differences for patient-reported outcomes. J Clin Epidemiol. 2008;61(2):102-109. PubMed
56.Mazzone ES, Mayhew A, Montes J, et al. Revised upper limb module for spinal muscular atrophy: development of a new module. Muscle Nerve. 2017;55(6):869-874. PubMed
57.Hobart J, Cano S. Improving the evaluation of therapeutic interventions in multiple sclerosis: the role of new psychometric methods. Health Technol Assess. 2009;13(12):iii, ix-x, 1-177.
58.Glanzman AM, Mazzone ES, Young SD, et al. Evaluator training and reliability for SMA global nusinersen trials. J Neuromuscul Dis. 2018;5(2):159-166. PubMed
59.Pera MC, Coratti G, Mazzone ES, et al. Revised upper limb module for spinal muscular atrophy: 12 month changes. Muscle Nerve. 2019;59(4):426-430. PubMed
60.ATS statement: guidelines for the six-minute walk test. Am J Respir Crit Care Med. 2002;166(1):111-117. PubMed
61.Slayter J, Hodgkinson V, Lounsberry J, et al. A Canadian adult spinal muscular atrophy outcome measures toolkit: results of a National Consensus using a Modified Delphi Method. J Neuromuscul Dis. 2021;8(4):579-588. PubMed
62.Elsheikh B, King W, Peng J, et al. Outcome measures in a cohort of ambulatory adults with spinal muscular atrophy. Muscle Nerve. 2020;61(2):187-191. PubMed
63.Dunaway Young S, Montes J, Kramer SS, et al. Six-minute walk test is reliable and valid in spinal muscular atrophy. Muscle Nerve. 2016;54(5):836-842. PubMed
64.Montes J, McDermott MP, Martens WB, et al. Six-Minute Walk test demonstrates motor fatigue in spinal muscular atrophy. Neurology. 2010;74(10):833-838. PubMed
Appendix 1: Literature Search Strategy
Note that this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases:
MEDLINE All (1946-present)
Embase (1974-present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: January 6, 2022
Alerts: Bi-weekly search updates until project completion
Search filters applied: No filters were applied to limit the retrieval by study type.
Limits: Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
MeSH | Medical Subject Heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Author keyword heading word (MEDLINE) |
.dq | Candidate term word (Embase) |
.pt | Publication type |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multi-Database Strategy
(Spinraza* or Nusinersen or ASO-10-27 or ASO1027 or ISIS 396443 or ISIS396443 or ISIS SMN?Rx? or ISISSMN?Rx? or IONIS SMN?Rx? or IONISSMN?Rx? or 5Z9SP3X666 or "biib 058" or biib058 or 4CHB7QQU1Q).ti,ab,kf,ot,hw,rn,nm.
1 use medall
*nusinersen/
(Spinraza* or Nusinersen or ASO-10-27 or ASO1027 or ISIS 396443 or ISIS396443 or ISIS SMN?Rx? or ISISSMN?Rx? or IONIS SMN?Rx? or IONISSMN?Rx? or "biib 058" or biib058).ti,ab,kf,dq.
3 or 4
(conference review or conference abstract).pt.
5 not 6
7 use oemezd
2 or 8
remove duplicates from 9
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
[Search -- Studies with results | nusinersen or spinraza]
WHO ICTRP
International Clinical Trials Registry Platform, produced by the World Health Organization. Targeted search used to capture registered clinical trials.
[Search -- Studies with results | nusinersen or spinraza]
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
[Search -- Studies with results | nusinersen or spinraza]
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
[Search -- Studies with results | nusinersen or spinraza]
Grey Literature
Search dates: December 7-9, 2021
Keywords: nusinersen, spinraza, SMA, spinal muscular atrophy
Limits: none
Updated: Search updated prior to the meeting of the CADTH Canadian Drug Expert Committee (CDEC)
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Health Statistics
Internet Search
Open Access Journals.
Appendix 2: Excluded Studies
Reference | Reason for Exclusion |
|---|---|
Bonanno S, Zanin R, Bello L, et al. Quality of life assessment in adult spinal muscular atrophy patients treated with nusinersen. J Neurol. 2022;03:03. | Study Design: Not phase II-IV RCT. Study Population: Includes 2 type IV patients evaluated within the group of type III patients without stratified results. |
Carson VJ, Young M, Brigatti KW, et al. Nusinersen by subcutaneous intrathecal catheter for symptomatic spinal muscular atrophy patients with complex spine anatomy. Muscle Nerve. 2022;65(1):51-59. | Study Design: Not phase II-IV RCT. Study Population: Some patients received nusinersen prior to 18 years of age. No stratified results by age were presented. |
Arnold WD, Severyn S, Zhao S, et al. Persistent neuromuscular junction transmission defects in adults with spinal muscular atrophy treated with nusinersen. BMJ Neurol. 2021;3(2):e000164. | Study Design: Not phase II-IV RCT. Study Outcomes: No outcomes of interest from the pre-specified protocol. |
Becker LL, Weis C, Tietze A, Martiny V, Kaindl AM. Lumbar Puncture Opening Pressure in Patients with Spinal Muscular Atrophy. Neuropediatrics. 2021;52(3):219-223. | Study Design: Not phase II-IV RCT. Study Outcomes: No outcomes of interest from the pre-specified protocol. |
Binz C, Schreiber-Katz O, Kumpe M, et al. An observational cohort study on impact, dimensions and outcome of perceived fatigue in adult 5q-spinal muscular atrophy patients receiving nusinersen treatment. J Neurol. 2021;268(3):950-962. | Study Design: Not phase II-IV RCT. Study Population: Includes 1 type IV patient, however no results stratified. |
Brakemeier S, Stolte B, Thimm A, et al. Assessment of Bulbar Function in Adult Patients with 5q-SMA Type 2 and 3 under Treatment with Nusinersen. Brain Sci. 202120;11(9):20. | Study Design: Not phase II-IV RCT. |
De Wel B, Goosens V, Sobota A, et al. Nusinersen treatment significantly improves hand grip strength, hand motor function and MRC sum scores in adult patients with spinal muscular atrophy types 3 and 4. J Neurol. 2021;268(3):923-935. | Study Design: Not phase II-IV RCT. Study Population: Includes 2 type IV patients without stratified results. |
Duong T, Wolford C, McDermott MP, et al. Nusinersen Treatment in Adults With Spinal Muscular Atrophy. Neurol. 2021;11(3):e317-e327. | Study Design: Not phase II-IV RCT. |
Elsheikh B, Severyn S, Zhao S, et al. Safety, Tolerability, and Effect of Nusinersen in Non-ambulatory Adults With Spinal Muscular Atrophy. Front Neurol. 2021;12:650532. | Study Design: Not phase II-IV RCT. |
Freigang M, Wurster CD, Hagenacker T, et al. Serum creatine kinase and creatinine in adult spinal muscular atrophy under nusinersen treatment. Ann Clin Transl Neurol. 2021;8(5):1049-1063. | Study Design: Not phase II-IV RCT. Study Outcomes: Measures correlation between biomarkers and SMA progression. |
Goedeker NL, Gibbons JL, Varadhachary AS, Connolly AM, Zaidman CM. Laboratory monitoring of nusinersen safety. Muscle Nerve. 2021;63(6):902-905. | Study Design: Not phase II-IV RCT. Study Population: Includes patients of all ages and types of SMA without stratified results. |
Hiebeler M, Abicht A, Reilich P, Walter MC. Effect of Discontinuation of Nusinersen Treatment in Long-Standing SMA3. J Neuromuscul Dis. 2021;8(4):537-542. | Study Design: Not phase II-IV RCT. Case report of a single patient with type III SMA. |
Mendonca RH, Fernandes HDS, Pinto RBS, et al. Managing intrathecal administration of nusinersen in adolescents and adults with 5q-spinal muscular atrophy and previous spinal surgery. Arq Neuropsiquiatr. 2021;79(2):127-132. | Study Design: Not phase II-IV RCT. Study Population: Includes 2 patients < 18 years of age. No stratified results. |
Meyer T, Maier A, Uzelac Z, et al. Treatment expectations and perception of therapy in adult patients with spinal muscular atrophy receiving nusinersen. Eur J Neurol. 2021;28(8):2582-2595. | Study Design: Not phase II-IV RCT. Study Population: Includes patients < 18 years of age, with no stratified results. Study Outcomes: Treatment expectations and perceptions were not outcomes of interest to this review. |
Milella G, Introna A, D’Errico E, et al. Cerebrospinal Fluid and Clinical Profiles in Adult Type 2-3 Spinal Muscular Atrophy Patients Treated with Nusinersen: An 18-Month Single-Centre Experience. Clin Drug Investig. 2021;41(9):775-784. | Study Design: Not phase II-IV RCT. Study Outcomes: No aggregate data presented. Only individual patient data. |
Mix L, Winter B, Wurster CD, et al. Quality of Life in SMA Patients Under Treatment With Nusinersen. Front Neurol. 2021;12:626787. | Study Design: Not phase II-IV RCT. |
Osmanovic A, Ranxha G, Kumpe M, et al. Treatment expectations and patient-reported outcomes of nusinersen therapy in adult spinal muscular atrophy. J Neurol. 2020;267(8):2398-2407. | Study Design: Not phase II-IV RCT. Study Population: Includes SMA types II-IV. Results presented for type II, however, type III and IV are grouped. |
Osmanovic A, Schreiber-Katz O, Petri S. Nusinersen Wearing-Off in Adult 5q-Spinal Muscular Atrophy Patients. Brain Sci. 2021;11(3):13. | Study Design: Not phase II-IV RCT. Study Population: Includes patients with type IV SMA. Study Outcomes: Measurement of perception of treatment wearing off is not of interest. |
Osmanovic A, Ranxha G, Kumpe M, et al. Treatment satisfaction in 5q-spinal muscular atrophy under nusinersen therapy. Ther Adv Neurol Disord. 2021;14:1756286421998902. | Study Design: Not phase II-IV RCT. Study Population: Includes patients < 18 years of age and patients with types I and IV SMA with no stratified results for the population of interest. |
Osredkar D, Jilkova M, Butenko T, et al. Children and young adults with spinal muscular atrophy treated with nusinersen. Eur J Paediatr Neurol. 2021;30:1-8. | Study Design: Not phase II-IV RCT. Study Population: Population is made up of young adults (< 18 years of age) |
Sansone VA, Coratti G, Pera MC, et al. Sometimes they come back: New and old spinal muscular atrophy adults in the era of nusinersen. Eur J Neurol. 2021;28(2):602-608. | Study Design: Not phase II-IV RCT. Study Population: Includes all SMA types (1 type I, 1 type IV), not stratified results. |
Tanaka R, Fukushima F, Motoyama K, Kobayashi C, Izumi I. Nusinersen improved respiratory function in spinal muscular atrophy type 2. Pediatr Int. 2021;63(8):973-974. | Study Design: Not phase II-IV RCT. Study Population: Not adults (< 18 years) |
Wataya T, Takasaki S, Hoshino M, Makioka H, Nakamura G, Matsuda N. Real-world safety of nusinersen in Japan: results from an interim analysis of a post-marketing surveillance and safety database. Int J Neurosci. 2021:1-13. | Study Design: Not phase II-IV RCT. Study Population: Includes types II-IV and patients of all ages. No stratification by groups of interest. |
Weaver JJ, Hallam DK, Chick JFB, et al. Transforaminal intrathecal delivery of nusinersen for older children and adults with spinal muscular atrophy and complex spinal anatomy: an analysis of 200 consecutive injections. J Neurointerv Surg. 2021;13(1):75-78. | Study Design: Not phase II-IV RCT. Study Population: Includes both adults, and children, however, results not stratified by groups of interest. |
Barp A, Carraro E, Albamonte E, et al. Muscle MRI in two SMA patients on nusinersen treatment: A two years follow-up. J Neurol Sci. 2020;417:117067. | Study Design: Not phase II-IV RCT. Case report of 2 patients. |
Faravelli I, Meneri M, Saccomanno D, et al. Nusinersen treatment and cerebrospinal fluid neurofilaments: An explorative study on Spinal Muscular Atrophy type 3 patients. J Cell Mol Med. 2020;24(5):3034-3039. | Study Design: Not phase II-IV RCT. Study Population: Includes pediatric and adult population, however, results not stratified. |
Farrar MA, Kiernan MC. Treating adults with spinal muscular atrophy with nusinersen. J Neurol Neurosurg Psychiatry. 2020;91(11):1139. | Study Design: Not phase II-IV RCT (Review). |
Jochmann E, Steinbach R, Jochmann T, et al. Experiences from treating seven adult 5q spinal muscular atrophy patients with Nusinersen. Ther Adv Neurol Disord. 2020;13:1756286420907803. | Study Design: Not phase II-IV RCT. Study Outcomes: No aggregate data presented. Only individual patient data. |
Kim AR, Lee JM, Min YS, et al. Clinical Experience of Nusinersen in a Broad Spectrum of Spinal Muscular Atrophy: A Retrospective Study. Ann Indian Acad Neurol. 2020;23(6):796-801. | Study Design: Not phase II-IV RCT. Study Population: Includes both pediatric and adult patients with types I and II SMA, however, results are not stratified for groups of interest. |
Kizina K, Stolte B, Totzeck A, et al. Fatigue in adults with spinal muscular atrophy under treatment with nusinersen. Sci Rep. 2020;10(1):11069. | Study Design: Not phase II-IV RCT. |
Lam K, Wu A. Clinical Outcome of Adult Spinal Muscular Atrophy Patients Treated with Nusinersen: A Case Series Review. Perm J. 2020;25:1. | Study Design: Not phase II-IV RCT. Case series of 4 patients. Study Population: SMA type unknown. |
McMillan HJ. Nusinersen: Evidence of sustained clinical improvement and lessened fatigue in older ambulatory patients with spinal muscular atrophy. Muscle Nerve. 2020;61(1):1-2. | Study Design: Not phase II-IV RCT (Review). |
Mercuri E, Sansone V. Nusinersen in adults with spinal muscular atrophy: new challenges. Lancet Neurol. 2020;19(4):283-284. | Study Design: Not phase II-IV RCT (Review). |
Moshe-Lilie O, Riccelli LP, Karam C. Possible recurrent aseptic meningitis associated with nusinersen therapy. Muscle Nerve. 2020;62(5):E79-E80. | Study Design: Not phase II-IV RCT. Case report of 1 adult patient with type II SMA. |
Moshe-Lilie O, Visser A, Chahin N, Ragole T, Dimitrova D, Karam C. Nusinersen in adult patients with spinal muscular atrophy: Observations from a single center. Neurology. 2020;95(4):e413-e416. | Study Design: Not phase II-IV RCT. |
Shah JS, Rubin DI, Dimberg EL, et al. Two Years of Improved Neurological Function With Nusinersen in a 48-Year-Old Patient With Spinal Muscular Atrophy Type 3. Neurologist. 2020;25(5):141-143. | Study Design: Not phase II-IV RCT. Case report of 1 adult with type III SMA. |
Veerapandiyan A, Eichinger K, Guntrum D, et al. Nusinersen for older patients with spinal muscular atrophy: A real-world clinical setting experience. Muscle Nerve. 2020;61(2):222-226. | Study Design: Not phase II-IV RCT. Study Population: Includes pediatric and adult patients with type III SMA, however, results were not stratified for groups of interest. |
Yeo CJJ, Simeone SD, Townsend EL, Zhang RZ, Swoboda KJ. Prospective Cohort Study of Nusinersen Treatment in Adults with Spinal Muscular Atrophy. J Neuromuscul Dis. 2020;7(3):257-268. | Study Design: Not phase II-IV RCT. |
Walter MC, Wenninger S, Thiele S, et al. Safety and Treatment Effects of Nusinersen in Longstanding Adult 5q-SMA Type 3 - A Prospective Observational Study. J Neuromuscul Dis. 2019;6(4):453-465. | Study Design: Not phase II-IV RCT. |
RCT = randomized controlled trial; SMA = spinal muscular atrophy.
Appendix 3: Detailed Outcome Data
Note that this appendix has not been copy-edited.
Table 24: Motor-Function Scores (EU Registry-Based Analysis)
Motor-function Scores | LOCF: Standard Mixed Effect Model | Multiple Imputation: Standard Mixed Effect Model | ||
|---|---|---|---|---|
Estimate (SE) | P Value | Estimate (SE) | P Value | |
HFMSE Score | ||||
Registry | ||||
Italy | 0 | — | 0 | — |
Germany | 0.9879 (0.473) | 0.0379 | -0.1144 (0.8571) | 0.8939 |
Spain | 1.3427 (0.6683) | 0.0456 | 1.2638 (1.228) | 0.3045 |
Overall | ||||
Slope Treated | 0.02442 (0.004299) | <0.0001 | 0.02304 (0.006695) | 0.0006 |
Slope Untreated | -0.01092 (0.008623) | 0.2059 | -0.00946 (0.01402) | 0.5002 |
RULM Score | ||||
Registry | ||||
Italy | 0 | — | 0 | — |
Germany | 0.2198 (0.2972) | 0.4604 | -0.4052 (0.5154) | 0.4326 |
Spain | 0.4824 (0.4187) | 0.2503 | 0.08569 (0.7361) | 0.9074 |
Overall | ||||
Slope Treated | 0.01042 (0.002896) | 0.0003 | 0.004122 (0.004055) | 0.3096 |
Slope Untreated | -0.00533 (0.005577) | 0.3392 | -0.00622 (0.008288) | 0.4533 |
6MWT Distance | ||||
Registry | ||||
Italy | 0 | — | 0 | — |
Germany | 7.8305 (7.5799) | 0.3039 | -48.305 (20.5734) | 0205 |
Spain | 26.1753 (12.4403) | 0.0375 | -35.068 (37.1349) | 0.3463 |
Overall | ||||
Slope Treated | 0.2168 (0.06696) | 0.0013 | 0.9071 (0.173) | <0.0001 |
Slope Untreated | 0.00445 (0.1191) | 0.9702 | -0.1872 (0.3735) | 0.6166 |
6MWT = 6-minute walk test; HFMSE = Hammersmith Functional Motor Scale Expanded; LOCF = last observation carried forward; RULM = Revised Upper Limb Module; SE = standard error.
Source: Sponsor submission.15,16
Table 25: Summary of Findings of Key Included Clinical Studies
Main Study Findings | Authors’ Conclusions |
|---|---|
Hagenacker et al. 2020 | |
| “Despite the limitations of the observational study design and a slow functional decline throughout the natural disease course, our data provide evidence for the safety and efficacy of nusinersen in the treatment of adults with 5q SMA, with clinically meaningful improvements in motor function in a real-world cohort.” |
Maggi et al. 2020 | |
SMA type II
SMA type III
Subgroup results By ambulatory status – SMA type III sitters
By ambulatory status – SMA type III walkers
Safety
| “Our data provide further evidence of nusinersen safety and efficacy in adult SMA2 and SMA3, with the latter appearing to be cumulative over time. In patients with extremely advanced disease, effects on residual motor function are less clear.” |
EU Registry Study | |
Pre-vs. post-treatment with nusinersen
Nusinersen-treated vs. untreated patients
Safety
| “Despite the limitations of the observational study design and a slow functional decline throughout the natural disease course, the data provides evidence for the safety and efficacy of nusinersen in the treatment of adults with 5q SMA, with clinically meaningful improvements in motor function in a real-world cohort.” |
Pera et al. 2021 | |
Change in motor function scores in adults (over the age of 20)
By ambulatory status – non-ambulatory adults over the age of 20
By ambulatory status – ambulatory adults over the age of 20
The change in HFMSE from baseline at 12 months in treated (study) population vs external untreated control who were over the age of 20
| “Our results expand the available data on the effect of Nusinersen on type III patients, so far mostly limited to data from adult type III patients.” |
6MWT = 6-minute walk test; AE = adverse event; CI = confidence interval; HFMSE = Hammersmith Functional Motor Scale Expanded; RULM = Revised Upper Limb Measure; SMA = spinal muscular atrophy.
Appendix 4: Description and Appraisal of Outcome Measures
Note that this appendix has not been copy-edited.
Aim
To describe the following outcome measures and review their measurement properties (validity, reliability, responsiveness to change, and MID):
HFMSE
RULM
6MWT
Findings
Table 26: Summary of Outcome Measures and their Measurement Properties
Outcome Measure | Type | Conclusions about Measurement Properties | MID |
|---|---|---|---|
HFMSE | A set of 33 tasks to measure motor function in patients with type II and III SMA with limited mobility, a 3-point ordinal scale for each item. | Validity: Content and construct validity were adequate in patients with SMA. Reliability: Test-retest and intra-rater reliability were adequate in patients with SMA. Responsiveness: No literature was identified that assessed responsiveness in patients with SMA. | An increase of > 2 points in total score is unlikely in untreated patients with type II and III SMA. Patient and caregivers consider a 1-point increase meaningful. Standard error of measurement MID estimated to be a 4.3-point change for all patients with SMA. |
RULM | A set of 19 tasks to measure motor function in non-ambulatory SMA patients, with a 3-point ordinal scale for each item. | Validity: No literature was identified that assessed validity in patients with SMA. Reliability: Internal consistency, inter- and intra-rater reliability were adequate. Responsiveness: No literature was identified that assessed responsiveness in patients with SMA. | A change of 2 points in total is considered clinically meaningful in patients with type II and III SMA who were aged 15 years or older. Standard error of measurement MID estimated to be 2.9-point change for all patients with SMA. |
6MWT | A clinical exercise test measures the distance an ambulatory SMA patient can walk on a flat, hard surface within 6 minutes. | Validity: Construct validity were adequate in patients with SMA. Reliability: Test-retest reliability were adequate in patients with SMA. Responsiveness: No literature was identified that assessed responsiveness in patients with SMA. | A change of 24 metres is unlikely be due to measurement error in patients with SMA type III. Standard error of measurement MID estimated to be 55.5-metre change for all patients with SMA. |
6MWT = 6-minute walk test; HFMSE = Hammersmith Functional Motor Scale Expanded; MID = minimal important difference; RULM = Revised Upper Limb Module.
Hammersmith Functional Motor Scale Expanded (HFMSE)
The HFMSE was designed to measure motor function in patients with type II and III SMA with limited mobility.49 The HFMSE builds upon the HFMS by adding 13 items from the Gross Motor Function Measure (GMFM), an instrument designed for patients with cerebral palsy and previously validated in children with SMA. The HFMSE is intended for use in patients with type II and III SMA and captures higher functioning skills. It consists of 33 activities that can be scored one of 3 ways: 0 for unable to perform, 1 for performs with modification/adaptation, and 2 for performs without modification. The item scores are summed to give a total score with a maximum of 66. The higher the total score, the greater the patient’s motor functioning.
Clinical evaluators deemed the items added from the GMFM to be clinically meaningful and focus groups and interviews established content validity of all of the HFMSE items.45,50 Focus groups with caregivers (n = 30) and patients (n = 25) of type II and III SMA were able to relate each item to at least one relevant activity of daily living.50 A similar sample of patients and caregivers indicated in focus groups and interviews that the items on HFMSE were relevant to their life and that improvements in any of the items would translate to greater independence.45
Construct validity was assessed using both convergent validity and known-group comparisons in 2 studies in patients with type II and III SMA and ages ranging from 2 to 45 years.49,51 Hypotheses regarding the strength of correlations with other measures were not stated. HFMSE score had strong (Spearman rank correlation coefficient ρ > 0.80) positive associations with the GMFM (both with and without the items that were added to the HFMSE), as well as a simple, 10-point functional rating score ranging from “unable to sit” to “age-appropriate in motor skills” (ρ ranging from 0.88 to 0.98).49,51 Further convergent validity was established through positive correlations with forced vital capacity as a percentage of predicted normal value (ρ = 0.98), knee flexion and extension strength (Pearson correlation coefficient r = 0.74 for both), and elbow flexion strength (r = 0.77).51 Known-group comparisons showed statistically significant differences in median HFMSE score between those receiving BiPAP for less than and greater than 8 hours/day (23 versus 3, P < 0.0001), those who are able and unable to walk (52 versus 8, P < 0.0001), and those who have type II and III SMA (49 versus 8, P < 0.0001). There were also statistically significant differences in median scores between patients with different SMN2 copy numbers (Kruskal-Wallis test: P = 0.0007).
In one trial where examiners were extensively trained on the administration and interpretation of the HFMSE (in 2 global phase III clinical trials that examined nusinersen in patients with type 1 SMA), the intra-rater reliability was acceptable according to the 0.7 threshold (ICC[1, 1] = 0.0.959 and by video review, with ICC[1,1] ranging between 0.987 and 0.994).52
Reliability and change over time have also been studied. The HFMSE demonstrated adequate test-retest reliability when administered two months apart in patients with type II and III SMA (ICC = 0.98).51 A natural history study measured HFMSE score over time in patients with type II and III SMA (n = 268, age range of 2.5 to 55.5 years).44 Over 75% of the patients had a change in score from baseline to 12 months of -2 to +2 points. Only 7.84% experienced an increase of more than 2 points, and this was most likely to occur in children below 5 years of age. Focus groups and interviews with patients, parents, and clinicians representing SMA types 1 to 3 revealed that increases in the HFMSE scale as little as 1 point would represent meaningful change and that the scale increments may not be sensitive enough to capture small functional changes that are noticeable to patients.45
In a study of 51 adult patients with type II and III SMA (n = 15 and 36, respectively), Stolte et al. calculated MIDs based on the standard error of measurement (SEM), 1/2 standard deviation (1/2 SD), and 1/3 standard deviation (1/3 SD) using previously published test-retest reliability values.53 The SEM provided the smallest MID for all patients at 4.3 compared to 7.0 and 10.6 for 1/2 SD and 1/3 SD MIDs. A smaller MID range was calculated for patients with SMA type II (0.5 to 1.2) compared to those with SMA type 3 (4.3 to 10.7). The MID ranges were similar between ambulatory (n = 16) and non-ambulatory patients (n = 35) at 1.8 to 4.3 vs. 1.5 to 3.8, respectively. A floor effect can be observed with HFMSE for patients with type II SMA resulting in a low MID score and may potentially limit its use in assessing patients who are weaker. The distribution-based approach to MID estimation that was used by Stolte et al. is generally less favoured than an anchor-based method.54,55 The MID may differ based on context and population or method of estimation.
Revised Upper Limb Module (RULM)
The original Upper Limb Module was designed to capture upper limb function in non-ambulatory SMA patients, especially in young children, and was previously validated in this population.56 Due to ceiling effects, it was revised and renamed the RULM. Some items in the RULM were incorporated from other upper limb scales, particularly the Performance of Upper Limb scale for Duchenne muscular dystrophy. During the revision process, the RULM was well-tolerated with no refusals to participate noted, even in young children, with a test duration of 5 to 20 minutes. It consists of 19 items reflecting different functional domains that are graded on a 3-point scale. With the exception of one activity with a binary score, the possible scores are: 0 (unable), 1 (able, with modification), and 2 (able, no difficulty), giving a maximum total score of 37. The patient chooses one arm with which to perform the tasks.
Adequate inter-rater reliability was established using 3 video assessments of the RULM that were evaluated by 17 physiotherapists (ICC = 0.928).56 Rasch analysis was conducted on RULM assessments of 134 ambulatory and non-ambulatory SMA patients aged 2 to 52 years (median age of 9 years). Item and person locations revealed no floor or ceiling effects and only small gaps in measurement accuracy. The threshold map indicated that response categories for each item functioned as intended. The Person Separation Index (PSI), an indicator analogous to Cronbach’s alpha that assesses the ability of a set of items to separate the sample, demonstrated adequate internal consistency reliability (0.954).56,57 Indicators of fit demonstrated that the observed data overall did not differ from the expected responses as predicted by the Rasch model and that total RULM score is a suitable measurement of a single concept.56,57 Two pairs of items had correlated residuals, but their presence did not inflate the PSI. Scale performance did not differ between males and females, though it was not tested for groups expected to score differently.56 In another trial where examiners were extensively trained on the administration and interpretation of the RULM (in 2 global phase III clinical trials that examined nusinersen in patients with SMA type 1), the intra- and inter-rater reliability for the overall score were acceptable according to the 0.7 threshold (ICC [1, 1] = 0.948 and by video review, with ICC [1,1] ranging between 0.966 and 0.990, respectively).58 Associations with other measures of motor function and test-retest reliability were not found for the RULM.
A study of the 12-month RULM changes in 27 patients with type II and III SMA who were aged 15 years or older demonstrated a mean change of –0.6 (SD 2.3) in all patients and 21(78%) patients had a changed with 2 points. Subgroup analyses revealed that there was a mean change of –1.7 (SD 2.4) for non-ambulatory individuals with type III SMA aged 15 years or older (n=6) and a mean change of –1.4(SD 2.7) those who were ambulatory in the same age group (n = 7), the study suggested a clinically meaningful cut-off point of 2 points.59 In the same study evaluating the HFMSE, Stolte et al. calculated MIDs based on the standard error of measurement (SEM), 1/2 standard deviation (1/2 SD), and 1/3 standard deviation (1/3 SD) for a group of 51 adult patients with types II or III SMA (n = 15 or 36, respectively) using previously published test-retest reliability values.53 The SEM provided the smallest MID for all patients at 2.9 compared to 4.3 and 6.4 for 1/2 SD and 1/3 SD MIDs. A smaller MID range was calculated for patients with SMA type II (1.2 to 2.7) compared to those with SMA type III (2.7 to 5.9). Likewise, the calculated MID range was lower for ambulatory patients (n = 16) than for non-ambulatory patients (n = 35) (0.4 to 0.8 versus 2.0 to 4.4, respectively). It is worth noting that a ceiling effect can be observed with RULM for ambulant patients resulting in low MID scores which may limit its use in these populations. The distribution-based approach to MID estimation that was used by Stolte et al. is generally less favoured than an anchor-based method.54,55 The MID may differ based on context and population or method of estimation.
6-Minute Walk Test (6MWT)
The 6MWT was developed by the American Thoracic Society to measure the distance a participant can walk on a flat, hard surface within 6 minutes. The 6MWT should be performed indoors using a 30-metre hallway preferably with turnaround points marked with a cone. A starting line highlighted using bright colours is needed to mark the beginning and end of each 60-metre lap. There are no exercise equipment or advanced training for technicians. The participants can choose their own intensity of exercise and are allowed to stop and rest during the test.60 Longer walking distance indicates higher ambulatory capacity.
The 6MWT has been deemed suitable for ambulatory SMA patients. A group of clinicians (n = 10) with experience of treating adults with SMA in Canada included the 6MWT in a proposed toolkit of outcome measures that is appropriate for assessment of adults with SMA. The censuses were reached using 2 rounds of modified Delphi method.61 Elsheikh et al. conducted the 6MWT in 30 ambulatory adults with SMA and found that 97% of patients were able to complete the test, suggesting feasibility for assessment in the targeted population.62
Construct validity was assessed using convergent validity in 3 studies in 65 ambulatory patients with type II and III SMA and ages ranging from 4 to 55.3 years.62-64 Only one study mentioned the null hypotheses of no correlations with other measures.63 The 6MWT had strong to moderate positive associations with HFMSE (Pearson correlation coefficient r ranging from 0.755 to 0.83), maximal voluntary isometric contraction testing (r = 0.83), stride length (r = 0.789), manual muscle testing of lower extremities (r = 0.676), the SMA Functional Rating Scale (r = 0.65), knee flexion hand-held dynamometry (r = 0.62), ulnar compound muscle action potential (r = 0.47), and a strong negative association with 10-metre walk/run time (r ranging from –0.937 to –0.87) and moderate negative associations with fatigue (r = –0.505),Timed Up & Go test (r = –0.535).62-64 The associations with forced vital capacity (r ranging from 0.246 to 0.35) and knee extension HHD (r ranging from 0.377 to 0.36) were relatively weaker.63,64 Further discriminative validity of the 6MWT was established through differences observed between patients with types IIIa and IIIb SMA (F = 5.707; P = 0.024).63 Criterion validity was assessed through a moderate association between the 6MWT and peak oxygen uptake (r = 0.558) of the 14 participants who performed exercise tolerance testing.63
The 6MWT demonstrated good test-retest reliability when administered one months apart in 17 patients with type II and III SMA with the ICC of 0.992 (95% CI, 0.979–0.997). The mean difference between the 6MWT distance at baseline and 1 month was 2.294 metres (SD = 30.796).63 Elsheikh conducted the 6MWT in 30 adult SMA patients within a 6-week period and reported a high test-retest reliability (ICC = 0.85).62
Dunaway Young conducted the 6MWT in 30 ambulatory patients diagnosed with SMA type III and suggested 24 metres is the minimum detectable change, which is unlikely to be due to measurement error. The study did not provide a minimum important difference but suggested that in other chronic conditions a minimum important difference of 23 to 45 metres has been defined.63 In the same study evaluating the HFMSE and RULM, Stolte et al. calculated MIDs based on the standard error of measurement (SEM), 1/2 standard deviation (1/2 SD), and 1/3 standard deviation (1/3 SD) for a group of 51 adult patients with types 2 or 3 SMA (n = 15 or 36, respectively) using previously published test-retest reliability values.53 The 1/3 SD provided the smallest MID for all patients at 47.8 metres compared to 55.5 metres and 71.1 metres for SEM and 1/2 SD MIDs. It is worth noting that, unlike HFMSE and RULM, the SEM was positioned between 1/2 SD and 1/3 SD for 6MWT, this is because data on the MCID values of the 6MWT were not sufficient to produce adequate test-retest reliability.53 Moreover, the distribution-based approach to MID estimation that was used by Stolte et al. is generally less favoured than an anchor-based method.54,55 The MID may differ based on context and population or method of estimation.
Pharmacoeconomic Review
Abbreviations
BIA
budget impact analysis
HFMSE
Hammersmith Functional Motor Scale Expanded
HRQoL
health-related quality of life
ICER
incremental cost-effectiveness ratio
LY
life-year
QALY
quality-adjusted life-year
RWC
real-world care
SMA
spinal muscular atrophy
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Nusinersen (Spinraza), 2.4 mg/mL solution for intrathecal injection |
Submitted price | Nusinersen, 2.4 mg/mL: $118,000 per 5 mL vial |
Indication | For the treatment of 5q SMA |
Health Canada approval status | NOC |
Health Canada review pathway | Priority review |
NOC date | June 29, 2017 |
Reimbursement request | The sponsor requests that the previous CADTH-recommended criteria (project SR0576 to 000) for nusinersen be expanded to include adult type II and type III patients older than 18 years, regardless of ambulatory status |
Sponsor | Biogen Canada Inc. |
Submission history | Previously reviewed: Yes Indication: SMA (resubmission) Recommendation date: February 27, 2019 Recommendation: Reimburse with clinical criteria and/or conditions (note: changes to initiation and administration criteria in comparison with prior submission in 2017) Indication: SMA Recommendation date: December 22, 2017 Recommendation: Reimburse with clinical criteria and/or conditions |
NOC = Notice of Compliance; SMA = spinal muscular atrophy
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Markov model |
Target population | Adults with SMA |
Treatment | Nusinersen in combination with RWC (respiratory, nutritional, and orthopedic care for type II and III SMA) |
Comparators | RWC alone Risdiplam in combination with RWC was considered in a scenario analysis |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (60 years) |
Key data source | |
Submitted results |
|
Key limitations | The available clinical studies were primarily noncomparative in nature and could not provide any conclusive evidence in support of a clinical benefit with nusinersen over RWC alone or risdiplam in the short term with regard to motor-function milestones, HRQoL, or any other outcomes important to patients, nor were there any long-term data available in the target population of adults with SMA type II or III who had not received prior treatment. The submitted model based on motor-function milestones does not capture all key aspects of SMA in adults (e.g., loss of functional status, bulbar status, and requirement of nutritional support) expected to affect their health-related quality of life. The submitted model has technical limitations and produces results that lack face validity (i.e., cannot produce equal QALYs when equal efficacy is assumed for nusinersen and RWC alone), which introduces uncertainty into the sponsor’s estimates of cost-effectiveness. The impact of treatment-related adverse events and the mode of treatment administration on patient quality of life were not captured in the sponsor’s model. Clinician and patient input indicates that complications and additional harms related to intrathecal injections are of concern. Minor limitations identified include the exclusion of risdiplam from the base-case analysis and the inclusion of caregiver utilities that overestimate the incremental benefit associated with nusinersen. |
CADTH reanalysis results | Given the key limitations with the available clinical evidence, no conclusions can be drawn regarding the comparative clinical effects of nusinersen, RWC alone, or risdiplam in adults with SMA type II or III. In addition, given the issues related to the model structure and programming that could not be addressed, CADTH could not derive a base case. Assuming equal efficacy for nusinersen, RWC alone, and risdiplam, nusinersen is associated with higher drug-acquisition costs. However, this assumption does not account for treatment-related adverse events, including those related to the intrathecal mode of administration, which could result in reduced QALYs for those on nusinersen. Based on the available clinical information, there is no evidence available to suggest the cost of nusinersen should be higher than the cost of risdiplam, with a greater price reduction likely necessary to offset costs associated with intrathecal administration and its complications. Compared with RWC alone, a price reduction of at least 100% would be necessary for nusinersen to be considered cost-effective. |
HR-QoL = health-related quality of life; ICER = incremental cost-effectiveness ratio; inc. = incremental; LY = life-year; QALY = quality-adjusted life-year; RWC = real-world care; SMA = spinal muscular atrophy.
Conclusions
The CADTH clinical review found limited clinical-effectiveness data for motor function or disease stabilization associated with nusinersen (Spinraza), compared with relevant comparators, in adults with SMA type II or III, with most evidence being noncomparative in nature. Furthermore, the identified studies could not provide conclusive evidence demonstrating the effectiveness of nusinersen in adults with SMA type II or III, or any evidence related to an improvement in patient’s health-related quality of life (HRQoL). Therefore, there is no evidence of benefit with nusinersen in comparison with relevant comparators.
The purpose of this submission was to reassess the use of nusinersen in adults with SMA type II or III, regardless of ambulatory status, with a view toward expanding the population covered in the currently available listing criteria. There is a significant lack of evidence to support an assessment of the comparative clinical and cost-effectiveness of nusinersen in this context. Given this issue, as well as key limitations with the model structure and validity, CADTH was unable to derive a base case, and the cost-effectiveness of nusinersen in adults compared with real-world care (RWC) alone or risdiplam in this population is unknown.
In the absence of comparative clinical evidence, when exploring the assumption of equal efficacy for nusinersen, RWC alone, and risdiplam — based on the conclusions of the CADTH clinical review and feedback from the clinical experts consulted by CADTH, and considering only drug-acquisition costs — nusinersen is associated with higher costs than RWC alone or risdiplam. When compared with RWC alone, a price reduction of 100% would be required before the cost of nusinersen could be considered equal to that of RWC alone. This is aligned with findings from the price-reduction analyses conducted on the sponsor’s base case, which indicated that a price reduction greater than 99% was necessary for nusinersen to be cost-effective at a $50,000 per QALY willingness-to-pay threshold (this assumes there is a benefit to patients and caregivers with nusinersen, despite no evidence in support of such an assumption). When considering only drug-acquisition costs (based on public list prices), nusinersen is more costly than risdiplam because of the higher costs associated with the initial loading doses.
The exploratory cost comparisons discussed do not take into account costs associated with intrathecal-injection administration and the impact of administration complications on patient quality of life. Where these aspects are included, assuming equivalent efficacy, administration would increase the health care costs associated with nusinersen and reduce the associated benefits. This means that nusinersen would be even less likely to be a cost-effective option and may necessitate further price reductions to account for these impacts.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process.
CADTH received patient input from Cure SMA Canada, Muscular Dystrophy Canada, and the Love for Lewiston Foundation. The approaches used to collect patient input differed across organizations, but the results were similar, although CADTH notes that the input received included information from caregivers and from patients outside the reimbursement request (i.e., type I patients). Patient-expressed treatment goals were a delay in disease progression and improvements in quality of life, reflected through improvements in muscle strength and independence. For patients receiving nusinersen, the majority noted improvements in endurance and arm strength; the remainder experienced either disease stability or worsening. Side effects or negative aspects of nusinersen were related to the method of drug administration, which was described as invasive and requiring multiple visits and/or travel. Patients with spinal fusion rods or complex spines expressed concern about the difficulty of intrathecal injections, noting that oral administration is preferred because it is a less invasive, easier method of delivery that does not require patients from rural areas to travel. Patients also noted that costs were a considerable barrier to treatment access. However, nusinersen was considered by many patients, particularly those older than 25 years, to help address the high unmet need for treatment.
CADTH received registered clinician input from the Neuromuscular Disease Network for Canada. The current pathway of care for adults up to 25 years with SMA is risdiplam. Treatment goals for those with early-onset SMA include the preservation of motor neurons, improved survival, improved motor function, delayed disease progression, and a reduced burden on caregivers. Treatment goals for those with late-onset SMA include maintenance of motor function and strength at the current level, disease stabilization, and improved quality of life. Clinicians stated that patients least suitable for treatment with nusinersen include those who have contraindications to the drug or procedure or difficulty of lumbar punctures; those who have deteriorated or not benefited from treatment over a reasonable period of time; bed-ridden, fully ventilated patients; and asymptomatic patients.
Drug plans expressed concerns about the initiation criteria and renewal criteria for certain jurisdictions. They also noted that risdiplam is likely to be a reasonable comparator for adults in 18- to 25-year age group. The plans expressed interest in the discontinuation criteria, and noted a lack of clarity around the definition of clinical benefit, which they anticipate will be a challenge for some jurisdictions to implement. Furthermore, they expressed concerns about whether re-treatment would be feasible for adults who received nusinersen as a child. Last, the drug plans noted that the evidence base for nusinersen in the adult population consists of real-world evidence due to the low prevalence of SMA, which could make the assessment of new treatment options difficult.
Several of these concerns were addressed in the sponsor’s model:
The sponsor’s model compared nusinersen with RWC alone for the treatment of type II and type III SMA in the adult population. Risdiplam was considered in a scenario analysis.
In addition, CADTH addressed some of these concerns as follows:
CADTH adjusted the market shares of nusinersen in the budget impact analysis (BIA) to reflect clinical-expert opinion on the anticipated use of nusinersen in adults with SMA type II or type III.
CADTH was unable to address the following concerns raised in stakeholder’s input:
The impact of intrathecal injections was excluded from the submitted model
Re-treatment and treatment discontinuation criteria could not be explored in the submitted model.
Economic Review
The current review examines nusinersen in adults with SMA type II and III, regardless of ambulatory status. CADTH notes that this submission is a reassessment of the adult type II and III subgroup, which is part of the Health Canada indication for nusinersen.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis comparing nusinersen with RWC alone in adults with SMA type II and III, regardless of ambulatory status. The target population was aligned with a subgroup that met the Health Canada indication and with the reimbursement request for this reassessment. CADTH has previously reviewed nusinersen for its broader indication. The sponsor’s submission also included a scenario analysis comparing nusinersen with risdiplam, both in combination with RWC, in the target population.
Nusinersen is a solution for intrathecal use in lumbar puncture that is available in 12 mg/5 mL single-dose vials.1 The recommended dose of nusinersen is 12 mg over 1 to 3 minutes, and consists of 4 loading doses (3 doses of 12 mg administered at 14-day intervals, with the final loading dose administered 30 days after the third loading dose) followed by maintenance dosing (12 mg) every 4 months.1 At the sponsor’s submitted price of $118,000 per 12 mg vial, the cost per loading or maintenance dose is $118,000.2 Patients receive 6 injections in the first year of treatment, at an annual cost of $708,000 (Table 5). In subsequent years, patients will require 3 maintenance dose injections per year, at an annual cost of $354,000, with additional administration costs required for lumbar puncture. The RWC alone comparator consisted of respiratory, nutritional, and orthopedic care for SMA type II and III; these costs were applied to patients receiving nusinersen and to those receiving RWC alone. The cost of risdiplam is $11,638 per 60 mg of powder, which is reconstituted into an oral solution by a health care provider before being dispensed.3 The recommended dose depends on the age and body weight of the patient and is administered by oral syringe. For patients 2 years and older weighing at least 20 kg, the recommended daily dose is 5 mg.4 At a cost of $194/mg, the total annual cost is $354,000.
The submitted model reported both quality-adjusted life-years (QALYs) and life-years (LYs) over a lifetime time horizon of 60 years in the SMA type II and III population. QALYs were reported for patients and caregivers, with caregiver utilities applied as a decrement. Base-case analyses were conducted from the perspective of the Canadian public health care payer, with discounting (1.5% per annum) applied to both costs and outcomes.
Model Structure
The sponsor submitted a Markov model with 7 health states, defined by motor-function milestones, aligned with the Hammersmith Functional Motor Scale Expanded (HFMSE) (sits but does not roll, sits and rolls independently, sits and crawls, stands or walks with assistance, stands unaided, and walks unaided) and death.5 Patients entered the model based on their health state at baseline, according to Hagenacker et al. (2020).6 Patients receiving nusinersen could remain in the same health state or transition to a better or worse health state at 6, 10, and 14 months, corresponding to clinical assessments observed in Hagenacker et al. (2020).5,6 In subsequent cycles, which were 4 months in length to correspond with the timing of nusinersen maintenance doses, patients could experience an increase in HFMSE score and improve to a better motor-function milestone state, remain in the same state, or worsen. For RWC alone, patients transitioned to worse health states over the lifetime of the model. Patients could transition to the death state from any of the other 6 health states. The sponsor’s submitted model structure can be found in Appendix 3.
Model Inputs
The modelled patient characteristics for the sponsor’s submission were based on Hagenacker et al. (2020)6 (mean age = 33 to 36 years; 54% to 65% male). Patient counts of health state occupation by HFMSE score was derived from Hagenacker et al. (2020)6 to determine the proportion of patients in the 6 motor-function health states at baseline, and was applied to all patients, regardless of treatment. At baseline, 51% of patients met the “sits without support but does not roll” milestone, 16% met the “sits and rolls independently” milestone, 1% met the “sits and crawls with hands and knees” milestone, 2% met the “stands or walks with assistance” milestone, 2% met the “stands unaided” milestone, and 28% met the “walks unaided” milestone.”5
Transition probabilities between motor-function milestone health states were informed by different data sources for nusinersen and RWC alone, and were noncomparative in nature. The transition probabilities for motor-function milestone health states in the initial 14-month period of the model for nusinersen were based on individual patient-level data from Hagenacker et al. (2020).6 All patients in the Hagenacker et al. (2020)6 cohort were assigned total HFMSE scores, which were then mapped to the various model health states. For model cycles after the initial 14-month trial follow-up period, 95% of patients in the nusinersen arm were assumed to improve and move to better a health state; the mean increase (0.22 points per month) in HFMSE scores observed from Hagenacker et al. (2020)6 was used to calculate the transition probability to the next health state, which represented a gain in motor-function milestones. The remaining 5% of patients, assumed to worsen on treatment, transitioned to the next worse health state, based on the rate of mean monthly decline from Kaufmann et al. (2012).7 Treatment discontinuation only occurred in patients who remained in the “sits without support but does not roll” health state for 1 year. No treatment discontinuation was assumed to occur in any other cases.
In the absence of comparative data, the probabilities of transitioning between health states for patients receiving RWC alone were calculated based on annual change in HFMSE score from an observational study by Kaufmann et al. (2012).7 All patients receiving RWC alone were assumed to gradually decline over time and transition to worse health states. In the scenario analysis comparing nusinersen with risdiplam, the sponsor assumed that risdiplam was no different than nusinersen in terms of clinical efficacy.
Baseline mortality in the model was based on mortality estimates in the general Canadian population. The risk of death in health states associated with SMA type II (i.e., sitting health states) was determined by applying a hazard ratio from Zerres et al. (1997)8 in patients with SMA type II to that of the general population.8 Patients in health states consistent with type III SMA (i.e., standing or walking health states) were assumed to have a mortality risk identical to that in the general population.9 No treatment-related adverse events were assumed to occur in the model.
Health-state utility values for patients and caregivers were obtained from multiple sources. Patient utility values by health state were sourced from an SMA review conducted by the National Institute for Health and Care Excellence evidence review group, with utility values for each motor milestone health state derived using the EuroQoL 5 Dimension 3 Level (EQ-5D-3L) parent proxy in children and adolescents.5 Caregiver utility values, derived by Biogen, measured the HRQoL of caregivers using the EuroQoL 5 Dimension 5 Level (EQ-5D-5L) questionnaire, and were applied as a disutility in the sponsor’s base case.10
Costs included in the model were drug-acquisition costs, health-state costs, administration costs, and end-of-life costs. Drug-acquisition costs and dosing were consistent with those reported in the overview section, with drug costs for nusinersen obtained from the sponsor’s submission. Administration costs varied by inpatient, outpatient, or day-case lumbar puncture.11 Health-state costs were obtained from a 2017 real-world data survey that reflected total direct costs of respiratory, nutritional, and orthopedic care for SMA type I, II, and III.11 Costs per year and per type of care were assumed to be equivalent for nusinersen and RWC alone. End-of-life costs were obtained from Zwicker et al. (2019)12 for patients with amyotrophic lateral sclerosis in their final year of life, assumed to be a proxy for SMA patients. Relevant costs were adjusted to 2021 Canadian dollars.
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (1,000 iterations for the base-case and scenario analyses). The deterministic results did not fully align with the probabilistic results. There was a discrepancy between total QALYs across results; however, the overall conclusions of the deterministic and probabilistic results aligned. The probabilistic findings are presented below.
Base-Case Results
Nusinersen was associated with incremental costs of $5,915,675 and 1.66 QALYs, compared with RWC alone, resulting in an incremental cost-effectiveness ratio (ICER) of $3,568,727 per QALY gained (Table 3).
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs | Incremental costs | Total QALYs | Incremental QALYs | ICER vs. RWC alone |
|---|---|---|---|---|---|
RWC alone | 337,386 | Ref. | 5.43 | Ref. | Ref. |
Nusinersen | 6,253,061 | 5,915,675 | 7.09 | 1.66 | 3,568,727 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; Ref. = reference; vs. = versus; RWC = real-world care.
Note: The submitted analysis is based on publicly available prices of the comparator treatments.
Source: Sponsor’s pharmacoeconomic submission.5
Additional results from the sponsor’s submitted economic evaluation base case are presented in Table 7, Appendix 3.
Sensitivity and Scenario Analysis Results
In a key analysis comparing nusinersen with risdiplam as a comparator, rather than RWC alone, nusinersen was dominated by risdiplam. Both treatments were considered equally effective (i.e., identical total QALYs), but risdiplam cost $338,642 less than nusinersen.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
Clinical efficacy of nusinersen and clinical efficacy and safety in comparison with RWC alone and with risdiplam in adults with SMA type II or III is highly uncertain: The sponsor’s submission consisted of a reassessment of nusinersen with a view to expand its listing to adults with SMA type II and III, regardless of ambulatory status. The treatment effect of nusinersen in this population was based on the change in HFMSE scores from Hagenacker et al. (2020),6 which was a 14-month noncomparative observational study. The CADTH clinical review identified several limitations to the submitted clinical data that informed the treatment effect of nusinersen in the target population: selection bias for type III patients, with a low proportion having undergone spinal fusion surgery; a disease severity lower than expected in the Canadian population; survivorship bias that reduces generalizability; inadequate duration of follow-up; limited applicability of HFMSE and 6-minute walk test scores to nonambulatory patients; lack of assessment of HRQoL; and uncertainty about what constitutes a clinically meaningful improvement in adults with SMA type II or type III. The clinical experts consulted by CADTH noted that the observed improvement in HFMSE scores for those receiving nusinersen could be due to a training effect in the absence of a control group, that 14 months was unlikely to be sufficient to assess clinically meaningful changes related to treatment in the target population, and that, because of these limitations, improvements in HFMSE scores observed in the available studies could not be attributed to treatment with nusinersen.
The sponsor also extrapolated 14 months of data from Hagenacker et al. (2020)6 to assume continuous improvement in HFMSE scores with nusinersen over 60 years in the submitted model. In addition to there being no evidence of treatment benefit of nusinersen in the short-term, there are no clinical data available to suggest that treatment with nusinersen leads to long-term benefits over a patient’s lifetime. In addition, an assumption of continued improvement in motor function over the patient’s lifetime was deemed to be implausible, and there is no evidence in support of disease stabilization over the patient’s lifetime. The clinical experts also discussed the lack of a clear biologic mechanism by which SMN2-modifying therapy such as nusinersen could benefit adults with SMA, and explained that production of the SMN protein would already be decreased significantly, making a reversal of muscle function impossible. As noted by the clinical experts consulted by CADTH for this review, this was supported by clinical data from the CHERISH study, which showed no statistically significant functional benefit with nusinersen, measured by HFMSE scores, in patients older than 4 years.2 It is therefore unlikely that adults would benefit from nusinersen in the short-term or continuously during their lifetime.
In the absence of direct comparative evidence assessing nusinersen with RWC alone in the population of adults with SMA type II or III, the sponsor used external data from a real-world observational study of children and adolescents with SMA type II and III (mean age = 11.3 years) to estimate decline during the course of the disease, which likely do not reflect the adult patient population. This was noncomparative in nature, making the comparative clinical efficacy of nusinersen and RWC alone highly uncertain; no data were available for the comparison of nusinersen with risdiplam. Overall, limitations of the available clinical evidence in the target population mean that the efficacy of nusinersen and its relative treatment effect, compared with RWC alone, do not support the sponsor’s estimate of a gain in incremental QALYs. The overall conclusions drawn from the analysis comparing nusinersen with risdiplam are less at risk of bias, given an assumption of equal efficacy for risdiplam and nusinersen, although it remains unclear whether nusinersen and risdiplam are truly equally effective in the absence of available evidence.
CADTH was unable to address this limitation in reanalysis.
The sponsor’s submitted model likely does not capture key aspects of the disease most relevant to adults with SMA type II or III: The sponsor’s submitted model structure was based primarily on patient achievement of motor-function milestones, assessed with the HFMSE. However, such a model structure may not adequately capture all disease-related aspects that are most relevant to patients, including the key outcomes most likely to affect their HRQoL, according to feedback from clinical experts consulted by CADTH. The clinical experts indicated that quality of life in adults with SMA type II and III is not determined solely by motor-function ability, and that other aspects — such as maintenance of independence, requirement of nutritional support, bulbar function, ventilation, necessity of assisted living, and loss of functional status — would also have a significant impact on health care costs and HRQoL. These events, which are meaningful to patients, were not explicitly captured in the sponsor’s model and are not always fully correlated with motor function.
Furthermore, the clinical experts consulted by CADTH noted that the applicability of HFMSE is limited in adults with SMA type II or III; the scale is better suited to children. Outcomes more meaningful to adults are better captured by the Revised Upper Limb Module for SMA. The HFMSE is also incapable of measuring small changes in the natural course of disease, as noted by the clinical experts consulted by CADTH for this review. The meaningfulness of the observed HFMSE score increase in Hagenacker et al. (2020)6 and the sponsor’s transformation of HFMSE scores to motor milestones are, therefore, highly uncertain. The incremental cost-effectiveness of nusinersen as it relates to improvements in meaningful outcomes in adults with SMA type II and III is therefore unknown.
CADTH was unable to address this limitation in reanalysis.
The submitted model has technical limitations and produces results that lack face validity: In its assessment of model behaviour, CADTH found that the model could not produce identical total LYs or QALYs when all available efficacy and mortality parameters were considered to be equal in a comparison of nusinersen and RWC alone. These results do not meet face validity and introduce uncertainty to the results produced by the sponsor’s model.
CADTH was unable to address this limitation.
Inappropriate exclusion of treatment-related adverse events and impact of mode of administration on patient quality of life: The sponsor assumed there would be no treatment-related adverse events with nusinersen, and that there would be no impact from, or complications related to, nusinersen’s intrathecal mode of administration that would affect patient quality of life. As a result, no disutilities for treatment-related adverse events or for complications from intrathecal administration were included in the model. The clinical experts consulted by CADTH for this review noted that nusinersen can be a challenge to administer, particularly in patients with complex spines, which are common in this population, and that complications from repeated intrathecal administration can arise. Further, stakeholder input from clinicians and patients indicated that complications related, in particular, to the intrusive nature of intrathecal administration can result in additional harm, and that an oral mode of administration is generally preferred. The exclusion of key complications related to treatment with nusinersen, including its administration, leads to an overestimation of incremental QALYs in favour of nusinersen in the sponsor’s cost-effectiveness estimates. The sponsor’s model is not generalizable to patients with complex spines.
CADTH was unable to address this limitation in reanalyses.
The following additional limitations were identified but not considered to be key limitations:
Exclusion of risdiplam as a comparator in the base case: The sponsor used RWC alone as the sole comparator for the treatment of SMA type II and III in their base case, which does not capture all relevant comparators for the decision problem. To meet the CADTH submission requirements, the base case must include all relevant comparators (i.e., treatments currently reimbursed by at least 1 participating drug plan for the indication under review, reimbursed treatments that are currently used off-label in Canadian practice, and treatments that have previously received a recommendation for reimbursement from CADTH for the indication under review). Risdiplam has received a positive listing recommendation.13 The clinical experts consulted by CADTH for this review noted that although there is limited evidence, risdiplam would likely be prescribed to adults with SMA type II or III, if available, and thus would be an appropriate comparator. Therefore, the sponsor’s scenario analysis with risdiplam as a comparator is as important in considering the cost-effectiveness of nusinersen as the comparison with RWC alone. This is supported by input received from drug plans.
CADTH was unable to conduct a base-case analysis due to other limitations of the model. Risdiplam is considered an important comparator, alongside RWC alone, and the cost-effectiveness of nusinersen should be compared with both treatment options.
Inclusion of caregiver utilities leads to an overestimate of the incremental benefit associated with nusinersen: Caregiver utilities were included for SMA patients with type II or type III in the sponsor’s base case. CADTH acknowledges that caregiver burden is significant with SMA and that motor-function improvement in patients is likely to lead to a gain in caregiver quality of life. However, CADTH requirements for CADTH Common Drug Review submissions note that the base case should be aligned with the Health Canada–indicated population, which is specific to patients with SMA. The inclusion of caregiver quality of life would be appropriate to include in a scenario analysis, but should be excluded from the base-case analysis. The inclusion of caregiver disutilities increases the incremental benefit observed with nusinersen, compared with RWC alone, contributing to 41% of total QALYs gained and 21% of incremental benefits observed.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (refer to Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
The sponsor has assumed that 95% of patients on nusinersen will continue to experience a treatment benefit, based on the mean HFMSE score increase from the 14-month Hagenacker et al. (2020)6 trial. The remaining 5% of patients are assumed to have worsened. | Inappropriate. There is no reported treatment response rate in the Hagenacker et al. (2020)6 trial, and the sponsor’s assumed rate of improvement was determined to be overly optimistic and inappropriate by clinical experts consulted by CADTH. The sponsor’s use of results from the Hagenacker et al. (2020)6 trial and assumptions surrounding response to therapy over a patient’s lifetime is associated with considerable uncertainty. |
The sponsor has assumed that patients will continue treatment indefinitely unless their motor function remains limited (sits without support but does not roll) for more than 12 months. | Inappropriate. Clinical experts consulted by CADTH noted that patients who worsen continuously in other health states or do not show improvement may discontinue treatment with nusinersen as well. Patients may also discontinue for reasons such as treatment-related adverse events and complications related to intrathecal injections. |
The utility values used in the sponsor’s model are obtained from an unpublished analysis provided for Biogen Inc. The EQ-5D-3L score was used by a parent proxy to estimate HRQoL for children and adolescents. | Inappropriate. The applicability of these utilities is uncertain when applied to the adult patient population. |
The sponsor has assumed that patients receiving RWC alone will experience a constant rate of decline in HFMSE scores every 4 months, corresponding to a constant rate of decline in motor function. | Inappropriate. According to the clinical experts consulted by CADTH for this review, the assumption of a constant rate of decline is unlikely to be appropriate, given the heterogeneity in patient decline observed in clinical practice. Furthermore, the sponsor’s mean HFMSE score decrease is taken from a population of patients predominantly younger than 12 years, which was not thought to be generalizable to the target population under review. The observed change in motor function is also typically an all-or-none change, as opposed to a minute decrease in HFMSE score at a constant rate over the lifetime, as assumed by the sponsor. Consequently, the sponsor’s application of patient decline does not meet face validity and introduces uncertainty into the sponsor’s cost-effectiveness analyses. |
The risk of mortality for SMA type II and type III differs greatly; mortality for type II patients was determined by applying a hazard ratio of 26.4 to the mortality risk in the general population, and mortality for type III patients was considered to be equal to that of the general population. | Inappropriate. These mortality assumptions alone are appropriate; however, because of the sponsor’s modelling of consistent improvement in motor milestone achievements, type II patients can improve and achieve motor milestones consistent with type III patients over their lifetime, and therefore experience a significant reduction in mortality risk. There is no clinical evidence to suggest a benefit related to reduced mortality with nusinersen in adults with SMA type II or type III. Therefore, the sponsor’s modelling approach results in an underestimation of mortality associated with SMA type II. |
EQ-5D-3L = EuroQoL 5 Dimension 3 Level; HFMSE = Hammersmith Functional Motor Scale Expanded; HRQoL = health-related quality of life; SMA = spinal muscular atrophy.
CADTH Reanalyses of the Economic Evaluation
Several key limitations were identified, with available clinical efficacy data informing the treatment benefit of nusinersen, and there was a lack of available efficacy and safety data comparing nusinersen with RWC alone or with risdiplam, in both the short and long-term, in adults with SMA type II or type III. The CADTH critical appraisal of the clinical evidence concluded that results observed in studies assessing nusinersen could not be attributed to the drug because of study-design and selection-bias issues, and that there was therefore no evidence of benefit with nusinersen, compared with relevant comparators.
Additionally, CADTH identified key limitations with the model structure, which did not align with the key factors affecting adults with SMA type II or III (including HRQoL), and with the model validity. As such, CADTH could not derive a base case and did not conduct any reanalyses.
Where similar clinical efficacy between nusinersen and RWC alone or risdiplam is assumed, nusinersen would be more costly than RWC alone and risdiplam, primarily as a result of greater drug-acquisition costs.
Compared with RWC alone, nusinersen would require a 100% price reduction to be considered comparative in cost and effectiveness. This is aligned with findings from price-reduction analyses conducted on the sponsor’s base case, which indicated that a price reduction greater than 99% would be necessary for nusinersen to be cost-effective at a $50,000 per QALY willingness-to-pay threshold, assuming there is a benefit with nusinersen to patients and their caregivers (Appendix 4).
At the publicly available list price, the cost of nusinersen is $708,000 during the first year of treatment because of the additional loading doses (6 injections per year) and $354,000 in subsequent years (3 injections per year) (Table 5). The treatment costs for risdiplam are identical to those for nusinersen in subsequent years, at $354,000 annually; however, costs in the first year of treatment are greater for nusinersen (this does not include administration costs, which would be greater for nusinersen).
These simplistic comparisons do not take into account the potential impact of complications associated with the intrathecal administration of nusinersen, which may lead to fewer benefits (and QALYs) and greater costs than with RWC alone or risdiplam.
Issues for Consideration
Risdiplam has recently received a positive CADTH reimbursement recommendation under specific clinical and pricing conditions.13 Risdiplam is an orally administered drug that does not require intrathecal injection or diagnostic radiotherapy. The cost-effectiveness of risdiplam, in comparison with nusinersen, is uncertain, but drug-acquisition costs are higher for nusinersen than for risdiplam at publicly listed prices in the first year of treatment and the same in subsequent years.
Patients and clinicians expressed interest in the treatment sequence of risdiplam and nusinersen, particularly upon failure or discontinuation. Combined or sequential use of risdiplam and nusinersen is not supported by clinical evidence, and the cost-effectiveness of nusinersen in this context is unknown.
Nusinersen has previously been reviewed by CADTH for the treatment of 5q SMA of any type, including pre-symptomatic patients and patients of any age at the submitted price of $118,000 per 12 mg vial.11 The CADTH reimbursement recommendation was positive, conditional upon a price reduction and clinical conditions related to initiation, administration, and renewal criteria.14 Only the publicly available list price was considered in this submission.
Overall Conclusions
The CADTH clinical review found limited comparative clinical-effectiveness data for motor function or disease stabilization associated with nusinersen or relevant comparators in adults with SMA type II or III, with most evidence being noncomparative in nature. Furthermore, the identified studies could not provide conclusive evidence demonstrating the effectiveness of nusinersen in adults with SMA type II or III, or any evidence related to an improvement in patient HRQoL. Therefore, there is no evidence of benefit with nusinersen over with relevant comparators.
In addition to a lack of short- and long-term comparative clinical evidence, CADTH identified several other key limitations in the sponsor’s pharmacoeconomic submission: the model does not capture key aspects of the condition most relevant to adults with SMA type II or III; technical limitations of the model lead to issues with the validity of the results; and the impact of treatment-related adverse events, including those related to its administration with intrathecal injection, have not been considered. None of these assumptions could be addressed in reanalysis. As a result, CADTH was unable to derive a base case for the assessment of nusinersen versus RWC alone or risdiplam for the treatment of SMA type II and III in adults, and the cost-effectiveness of nusinersen in adults is unknown.
The purpose of this submission was to reassess the use of nusinersen specifically in adults with SMA type II or III, regardless of ambulatory status, with a view to expanding the population in the currently available listing criteria. There is a significant lack of evidence to support an assessment of the comparative clinical and cost-effectiveness of nusinersen in this context.
In the absence of comparative clinical evidence, when exploring the assumption of equal efficacy for nusinersen, RWC alone, and risdiplam, nusinersen is associated with higher costs than RWC alone or risdiplam, based on conclusions of the CADTH clinical review, feedback from the clinical experts consulted by CADTH, and consideration of only drug-acquisition costs. Compared with RWC alone, a price reduction of 100% would be required for nusinersen to be considered in the same cost range as RWC alone. This is aligned with findings from price-reduction analyses conducted in the sponsor’s base case, which indicated that a price reduction greater than 99% was necessary for nusinersen to be cost-effective at a $50,000 per QALY willingness-to-pay threshold (assuming there is a benefit to patients and their caregivers with nusinersen, despite no evidence to support such an assumption). When considering the public list prices of nusinersen and risdiplam and only drug-acquisition costs, nusinersen is more costly than risdiplam because of the higher costs associated with the initial loading doses.
The exploratory cost comparisons discussed do not take into account the costs associated with the administration of intrathecal injections and the impact of associated complications on patient quality of life. Where these aspects are included, and assuming equivalent efficacy, they would increase the health care costs associated with nusinersen and reduce associated benefits. Nusinersen would be even less likely to be a cost-effective option, and could require further price reductions to account for these impacts.
References
1.Spinraza (nusinersen): solution for intrathecal injection 2.4 mg/mL nusinersen as nusinersen sodium [product monograph]. Mississauga (ON): Biogen Canada Inc.; 2020 Apr 20.
2.DeltaPA. [Ottawa (ON)]: IQVIA; 2021: https://www.iqvia.com/. Accessed 2022 Apr 5.
3.CADTH Drug Reimbursement Reviews: risdiplam. Ottawa (ON): CADTH; 2021: www.cadth.ca/risdiplam. Accessed 2022 Apr 8.
4.EVRYSDI: risdiplam powder for oral solution 60 mg/bottle (0.75 mg/mL after reconstitution), oral or enteral [product monograph]. Mississauga (ON): Hoffmann-La Roche Limited; 2021 Apr 14.
5.Pharmacoeconomic evaluation [internal sponsor’s report]. In: Drug Reimbursement Review sponsor submission: Spinraza (nusinersen injection), solution for intrathecal injection 2.4 mg/mL nusinersen as nusinersen sodium. Toronto (ON): Biogen Canada Inc.; 2021 Dec 1.
6.Hagenacker T, Wurster CD, Gunther R, et al. Nusinersen in adults with 5q spinal muscular atrophy: a non-interventional, multicentre, observational cohort study. Lancet Neurol. 2020;19(4):317-325. PubMed
7.Kaufmann P, McDermott MP, Darras BT, et al. Prospective cohort study of spinal muscular atrophy types 2 and 3. Neurology. 2012;79(18):1889-1897. PubMed
8.Zerres K, Rudnik-Schöneborn S, Forrest E, Lusakowska A, Borkowska J, Hausmanowa-Petrusewicz I. A collaborative study on the natural history of childhood and juvenile onset proximal spinal muscular atrophy (type II and III SMA): 569 patients. J Neurol Sci. 1997;146(1):67-72. PubMed
9.Table: 13-10-0114-01. Life expectancy and other elements of the life table, Canada, all provinces except Prince Edward Island. Ottawa (ON): Statistics Canada; 2021: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011401. Accessed 2022 Apr 5.
10.Lopez-Bastida J, Pena-Longobardo LM, Aranda-Reneo I, Tizzano E, Sefton M, Oliva-Moreno J. Social/economic costs and health-related quality of life in patients with spinal muscular atrophy (SMA) in Spain. Orphanet J Rare Dis. 2017;12(1):141. PubMed
11.Drug Reimbursement Review pharmacoeconomic report: nusinersen (Spinraza - Biogen Canada Inc.) for the treatment of patients with 5q SMA. Ottawa (ON): CADTH; 2018 Jan: https://www.cadth.ca/sites/default/files/cdr/pharmacoeconomic/SR0525_Spinraza_PE_Report.pdf. Accessed 2022 Apr 5.
12.Zwicker J, Qureshi D, Talarico R, et al. Dying of amyotrophic lateral sclerosis: Health care use and cost in the last year of life. Neurology. 2019;93(23):e2083-e2093. PubMed
13.CADTH Canadian Drug Expert Committee (CDEC) final recommendation: risdiplam (Evrysdi). Ottawa (ON): CADTH; 2021: https://www.cadth.ca/sites/default/files/DRR/2021/SR0661%20Evrysdi%20Recommendation%20Final.pdf. Accessed 2022 Apr 5.
14.CADTH Drug Reimbursement Expert Review Committee final recommendation: nusinersen (Spinraza — Biogen Canada Inc.). Ottawa (ON): CADTH; 2019 Feb 27: https://www.cadth.ca/sites/default/files/cdr/complete/SR0576-Spinraza-Resubmission-Mar-1-19.pdf Accessed 2022 Apr 5.
15.Norwood FL, Harling C, Chinnery PF, Eagle M, Bushby K, Straub V. Prevalence of genetic muscle disease in Northern England: in-depth analysis of a muscle clinic population. Brain. 2009;132(Pt 11):3175-3186. PubMed
16.Thieme A, Mitulla B, Schulze F, Spiegler AW. Epidemiological data on Werdnig-Hoffmann disease in Germany (West-Thuringen). Hum Genet. 1993;91(3):295-297. PubMed
17.Tassie B, Isaacs D, Kilham H, Kerridge I. Management of children with spinal muscular atrophy type 1 in Australia. J Paediatr Child Health. 2013;49(10):815-819. PubMed
18.Tangsrud SE, Halvorsen S. Child neuromuscular disease in southern Norway. Prevalence, age and distribution of diagnosis with special reference to “non-Duchenne muscular dystrophy”. Clin Genet. 1988;34(3):145-152. PubMed
19.Chung B, Wong V, Ip P. Prevalence of neuromuscular diseases in Chinese children: a study in southern China. J Child Neurol. 2003;18(3):217-219. PubMed
20.Budget Impact Analysis [internal sponsor’s report]. In: Drug Reimbursement Review sponsor submission: Spinraza (nusinersen injection), solution for intrathecal injection 2.4 mg/mL nusinersen as nusinersen sodium. Toronto (ON): Biogen Canada Inc.; 2021 Dec 1.
21.Table 17-10-0057-01. Projected population, by projection scenario, age and sex, as of July 1 (x1,000). Ottawa (ON): Statistics Canada; 2019: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710005701. Accessed 2022 Apr 5.
22.Lally C, Jones C, Farwell W, Reyna SP, Cook SF, Flanders WD. Indirect estimation of the prevalence of spinal muscular atrophy Type I, II, and III in the United States. Orphanet J Rare Dis. 2017;12(1):175. PubMed
Appendix 1: Cost Comparison Table
Note that this appendix has not been copy-edited
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 5: CADTH Cost Comparison Table for Spinal Muscular Atrophy
Treatment | Strength | Form | Price | Age and weight | Recommended dosagea | Daily cost | Annual cost |
|---|---|---|---|---|---|---|---|
Nusinersen (Spinraza) first year subsequent years | 12 mg / 5 mL | Injection | $118,000.0000b | NA | Six injections per year | $1,939.73 | $708,000 |
Three injections per year | $969.86 | $354,000 | |||||
Drug Comparator | |||||||
Risdiplam (EVRYSDI) | 60 mg | Powder for oral solution | $11,638.3500c | ≥ 2 years and ≥ 20 kg | 5 mg daily | $969.86 | $354,000 |
kg = kilograms; mg = milligrams; mL = millilitres; NA = not available.
Note: Prices do not include dispensing fees. Annual prices are based on 365 days per year.
aRecommended dosages are from the respective product monographs, unless otherwise indicated.1,4
bSponsor’s submitted price.5
cCADTH Reimbursement Review of Risdiplam.3
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | The model does not include important outcomes to patients, as noted in the key limitations section. |
Model has been adequately programmed and has sufficient face validity | No | In assessing model behaviour, CADTH found technical limitations with the sponsor’s model, as noted in the key limitations section. |
Model structure is adequate for decision problem | No | The applicability of the model structure to the decision problem is of concern, as noted in the key limitations section. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | No | The model was not user-friendly, with issues identified regarding face validity as noted in the key limitations section. Several sheets were poorly organized with additional information remaining from previous use of the model for another submission (i.e., infantile SMA cells and large sections of sheets that are unused but retained). The probabilistic sensitivity analysis inputs were not reported in the written submission and information was not easy to locate. There was a lack of clear and transparent reporting as well as technical documentation in the report. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited

Detailed Results of the Sponsor’s Base Case
Table 7: Disaggregated Summary of the Sponsor’s Economic Evaluation Results
Parameter | Nusinersen | RWC alone | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total | 15.88 | 14.78 | 1.09 |
Sits without support but does not roll | 6.02 | 7.80 | −1.78 |
Sits and rolls independently | 1.73 | 1.32 | 0.41 |
Sits and crawls with hands and knees | 0.63 | 0.77 | −0.14 |
Stands/Walks with assistance | 0.38 | 0.42 | −0.04 |
Stands unaided | 1.91 | 1.74 | 0.17 |
Walks unaided | 5.21 | 2.73 | 2.48 |
Discounted QALYs | |||
Total (patients and caregivers) | 7.09 | 5.43 | 1.66 |
Sits without support but does not roll | 3.62 | 4.69 | −1.07 |
Sits and rolls independently | 1.04 | 0.80 | 0.25 |
Sits and crawls with hands and knees | 0.38 | 0.46 | −0.09 |
Stands/Walks with assistance | 0.28 | 0.32 | −0.03 |
Stands unaided | 1.63 | 1.48 | 0.15 |
Walks unaided | 4.44 | 2.33 | 2.11 |
Caregiver | −4.30 | −4.63 | 0.34 |
Discounted costs ($) | |||
Total | 6,253,061 | 337,386 | 5,915,675 |
Acquisition | 5,869,846 | NA | 5,869,846 |
Administration | 44,247 | NA | 44,247 |
General disease management, sits without support but does not roll | 149,068 | 193,042 | −43,974 |
General disease management, sits and rolls independently | 42,773 | 32,699 | 10,074 |
General disease management, sits and crawls with hands and knees | 15,489 | 19,045 | −3,557 |
General disease management, Stands/Walks with assistance | 9,359 | 10,401 | −1,042 |
General disease management, Stands unaided | 29,104 | 26,451 | 2,653 |
General disease management, Walks unaided | 79,381 | 41,662 | 37,719 |
End-of-life costs | 13,794 | 14,086 | −291 |
ICER ($/QALY) | 3,568,727 | — | — |
ICER = incremental cost-effectiveness ratio; LY = life-year; NA = not applicable; QALY = quality-adjusted life-year; RWC = real-world care.
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Price Reduction Analysis
Table 8: CADTH Price Reduction Analyses
Analysis | ICERs for nusinersen vs. RWC alone | |
|---|---|---|
Price reduction | Sponsor base case | CADTH reanalysis |
No price reduction | $3,044,962 | NA |
10% | $2,742,859 | NA |
20% | $2,440,757 | NA |
30% | $2,138,655 | NA |
40% | $1,836,552 | NA |
50% | $1,534,450 | NA |
60% | $1,232,348 | NA |
70% | $930,245 | NA |
80% | $628,143 | NA |
90% | $326,041 | NA |
99% | $54,149 | NA |
ICER = incremental cost-effectiveness ratio; NA = not applicable; RWC = real-world care; vs. = versus.
Appendix 5: Submitted BIA and CADTH Appraisal
Table 9: Summary of Key Take-Aways
Key Take-Aways of the BIA |
|---|
|
Summary of Sponsor’s BIA
The submitted budget impact analysis (BIA) estimated the costs of reimbursing nusinersen for the treatment of adult patients with SMA type II and III. The analysis took the perspective of Canadian public drug plans using a top-down epidemiological approach and incorporated drug-acquisition costs. A time horizon of 3 years between 2023 to 2025 was taken, with 2022 being the base year of the model. The target population size was estimated using the prevalence of SMA by type and the proportion of adult patients stratified by type.15-20 Further specifications of population size were derived from SMA 360 data to determine the proportion of adult patients covered under public plans if nusinersen were reimbursed.20 The sponsor assumed that for drug plans with no data on adult patients, the pan-Canadian estimate of 86% public coverage was applicable. The reference case scenario included real world care (i.e., non-active therapy) and use of nusinersen through case-by-case reimbursement estimated using SMA 360 data.20 The new drug scenario included nusinersen and real-world care. A scenario analysis including risdiplam as a comparator was included for use in patients aged 18 to 25 years. Key inputs to the BIA and the sponsor’s methodology in calculating target population are documented in Table 10.
Table 10: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1 / year 2 / year 3 if appropriate) |
|---|---|
Target population | |
CADTH-participating pan-Canadian Population21 | 30,883,226 |
Prevalence of SMA (per 100,000)15-19 Type II Type III | 0.62 0.85 |
Proportion of adult patients20 Type II Type III | 37% 64% |
Proportion of those under public coverage by drug plansa Newfoundland and Labrador Prince Edward Island Nova Scotia New Brunswick Ontario Manitoba Saskatchewan Alberta British Columbia Non-Insured Health Benefits | 86% 86% 100% 63% 88% 86% 100% 89% 86% 86% |
Number of total patients eligible for nusinersen Type II Type III | || / || / || ||| / ||| / ||| |
Market Uptake (3 years) | |
Uptake (reference scenario) Type II Nusinersen Real world care Type III Nusinersen Real world care | || / || / || ||| / ||| / ||| || / || / || ||| / ||| / ||| |
Uptake (new drug scenario) Type II Nusinersen Real world care Type III Nusinersen Real world care | ||| / ||| / ||| ||| / ||| / ||| ||| / ||| / ||| ||| / ||| / ||| |
Cost of treatment (per patient) | |
Cost of annual treatment (year 1: 6 injections) Nusinersen Real world care Cost of annual treatment (year 2 or beyond: 3 injections) Nusinersen Real world care | $708,000 $0 $354,000 $0 |
SMA = spinal muscular atrophy.
aPortion of adult public patients covered were derived using data from SMA 360. When no adults were treated in specific jurisdictions, 86% public coverage was assumed as per the pan-Canadian estimate.20
Summary of the Sponsor’s BIA Results
The sponsor’s estimated budget impact of funding nusinersen for the treatment of adult patients with SMA type II and III was $17,464,831 in year 1, $24,375,677 in year 2, and $27,115,418 in year 3, for a 3-year total of $68,955,926.
The sponsor’s estimated budget impact of funding nusinersen when including costs of administration (i.e., outpatient lumbar administration) was $17,596,480 in year 1, $24,559,420 in year 2, and $27,319,813 in year 3, for a 3-year total of $69,475,714. The sponsor’s estimated budget impact of funding nusinersen when considering risdiplam use for patients aged 18 to 25 years was $12,717,562 in Year 1, $20,033,761 in Year 2, and $22,180,845 in Year 3, for a 3-year total of $54,932,168.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Anticipated uptake of nusinersen is likely underestimated: The sponsor anticipated a gradual uptake of nusinersen from 19% to 36% in years 1 to 3 in the new drug scenario. Given the lack of available treatments for adult patients with SMA type II and III, clinical experts noted that the market share estimates for nusinersen were likely underestimated. Clinician and patient input indicated that nusinersen uptake would be rapid if it was to be made available given there are at present no other treatment options available for adults with SMA type II or III, likely taking up 75% of the market by year 3. The clinical experts consulted by CADTH noted that in the scenario where risdiplam is available, the anticipated uptake of these agents would reach a combined 75%, distributed as 80% captured by risdiplam, due to its preferred oral route of administration, and the remaining 20% by nusinersen.
CADTH increased the market shares of nusinersen in the base case to reach 75% by year 3, as anticipated by clinical experts consulted by CADTH. A scenario analysis was included in which 80% of the total 75% uptake were captured by risdiplam with the remaining 20% being captured by nusinersen.
Uncertainty in deriving target population: The prevalence of SMA type II and III in Canada is not known, leading to uncertainty in the target population derived by the sponsor. While current estimates in literature vary, estimates of prevalence assessed by CADTH appear to be higher than the estimates used by the sponsor. An increase or decrease in target population will lead to large fluctuations in the anticipated budget impact for nusinersen and an underestimated prevalence rate led to an underestimation of target population. The sponsor estimated that the prevalence rates (per 100,000) were 0.62 for type II and 0.85 for type III. CADTH considered alternate prevalence rates in a scenario analysis where total cases of SMA were estimated to be 5 per 100,000, of which 29% will develop type II and 13% will develop type III SMA in the United States.22 The prevalence rates applied in the scenario analysis were therefore 1.45 for type II and 0.65 for type III per 100,000.
CADTH assessed the impact of increasing the prevalence of SMA type II and III in a scenario analysis.
Lack of clarity regarding treatment discontinuation: Drug plan and clinician input indicated uncertainty in stopping rules due to lack of efficacy or other reasons following treatment with nusinersen. Although there are no clear discontinuation criteria for nusinersen, increases in discontinuation rates would affect the budget impact of nusinersen.
CADTH could not address this concern in reanalysis.
CADTH Reanalyses of the BIA
Table 11: CADTH Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
None | — | — |
Changes to derive the CADTH base case | ||
Market shares underestimated for the uptake scenario | Nusinersen = ||| / ||| / ||| RWC alone = ||| / ||| / ||| | Nusinersen = 25% / 50% / 75% RWC alone = 75% / 50% / 25% |
CADTH base case | Reanalysis 1 | |
RWC = real-world care.
The results of the CADTH stepwise reanalysis are presented in summary format in Table 12 and a more detailed breakdown is presented in Table 13. Based on the CADTH base case, the budget impact of the reimbursement of nusinersen for the treatment of adult patients with SMA type II and III is expected to be $23,240,632 in year 1, $44,044,233 in year 2, and $65,387,990 in year 3. The 3-year total budget impact for nusinersen is $132,672,855.
CADTH also conducted 3 additional scenario analyses. A scenario analysis assessing the budget impact if the price of the drug under review reflected the price in which the ICER would be potentially cost-effective resulted in a 3-year budget impact of $1,326,729. An additional scenario analysis assessing the budget impact of nusinersen if risdiplam was available to all patients as a comparator led to a 3-year budget impact of $17,082,166. Lastly, a scenario analysis assessing the impact of increasing the prevalence of SMA type II and type III led to a 3-year budget impact of $166,468,890. The submitted analysis is based on the publicly available prices of the comparator treatments.
Table 12: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total |
|---|---|
Submitted base case | $68,955,926 |
CADTH reanalysis 1 | $132,672,855 |
CADTH base case | $132,672,855 |
BIA = budget impact analysis.
Table 13: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $3,186,000 | $3,244,169 | $3,422,349 | $3,555,989 | $10,222,507 |
New drug | $3,186,000 | $20,709,000 | $27,798,026 | $30,671,407 | $79,178,433 | |
Budget impact | $0 | $17,464,831 | $24,375,677 | $27,115,418 | $68,955,926 | |
CADTH base case | Reference | $3,186,000 | $3,244,169 | $3,422,349 | $3,555,989 | $10,222,507 |
New drug | $3,186,000 | $26,484,801 | $47,466,582 | $68,943,978 | $142,895,361 | |
Budget impact | $0 | $23,240,632 | $44,044,233 | $65,387,990 | $132,672,855 | |
CADTH scenario analysis: 99% price reduction | Reference | $31,860 | $32,442 | $34,223 | $35,560 | $102,225 |
New drug | $31,860 | $264,848 | $474,666 | $689,440 | $1,428,954 | |
Budget impact | $0 | $232,406 | $440,442 | $653,880 | $1,326,729 | |
CADTH sensitivity analysis: risdiplam included (80% of 75% total uptake) | Reference | $3,186,000 | $18,218,988 | $20,271,111 | $22,295,635 | $60,785,735 |
New drug | $3,186,000 | $18,997,380 | $26,342,079 | $32,528,442 | $77,867,900 | |
Budget impact | $0 | $778,391 | $6,070,967 | $10,232,807 | $17,082,166 | |
CADTH sensitivity analysis: increased prevalence | Reference | $3,186,000 | $3,244,169 | $3,422,349 | $3,555,989 | $10,222,507 |
New drug | $3,186,000 | $33,052,239 | $58,569,084 | $85,070,074 | $176,691,397 | |
Budget impact | $0 | $29,808,070 | $55,146,735 | $81,514,085 | $166,468,890 |
BIA = budget impact analysis.
Stakeholder Input
Patient Input
Cure SMA Canada
About Cure SMA Canada
Cure SMA Canada is the national registered charity supporting families and individuals affected by Spinal Muscular Atrophy from the point of diagnoses, through the life course and even after loss of life. Cure SMA Canada also funds critical Canadian research projects with the aim of affecting accessible treatments for SMA. Cure SMA Canada provides support, advocacy, information and resources to families, communities and health professionals through its various initiatives. These initiatives include but are not limited to national conferences, SMA camps, and direct patient support.
Information Gathering
This submission summarizes the perspectives of individuals and caregivers affected by spinal muscular atrophy (SMA) who are over the age of 18, collected through semi-structured interviews and survey created by Cure SMA Canada (CSMAC). The interview data provided rich in-depth understanding of the impact of spinal muscular atrophy on the patient and the family. The patient experiences and perspectives were also derived from the survey. The online survey consisted of open-ended questions, rating scales, and forced-choice options. The individual participants were recruited through Cure SMA Canada. The links to the survey was distributed through the CSMAC database in November 2020, emailed to its membership with request for secondary distribution to other patients and caregivers. The survey was available in English and French. The interviews were conducted in December 2021 in English and French and all French responses were translated to English for the purpose of this submission.
Among the 88 respondents, 13% identified SMA Type I, 58% SMA Type II, 27% SMA Type III, and 2% responding unsure or other. In terms of relation to SMA, 67% self-identified as a patient, and 33%, were parents or caregivers... In terms of age, the largest cohort (58%) were in the range of 18 to 35 years; the second and third cohorts were in the age range of 36 to 50 years (29%) and 51 to 65 years (13%). In terms of experience with treatment, 47% of the respondents were receiving treatment and alternatively 53% of the respondents were treatment naïve. 78% of the respondents with treatment access are receiving Spinraza while 22% are receiving Risdiplam.
In terms of residence, 100% of respondents are Canadian citizens, with only 1 respondent living outside of Canada. There was good representation across the provinces, with 35% from Ontario, 19% from BC, 24% from Quebec and 12% from Alberta, 4% from Saskatchewan, another 4% from Manitoba, and 1% each from New Brunswick, 1% Nova Scotia.
Disease Experience
Respondents were asked to describe “how the disease impacts” their daily lives or those of their caregivers. Regardless of the type of SMA or the age of diagnosis, a diagnosis of SMA was experienced as “overwhelming” and “devastating “; not only to the patient, but to the patient’s family as well. Patients who are over the age of 18 have experienced a decline in their physical abilities over the years. Respondents who were previously ambulatory, have lost the ability or are “just barely able to walk” as adults. Along with the loss of gross motor skills, patients continue to experience a progressive loss of life skills such as dressing themselves, feeding themselves and transferring for the purpose of toileting. Non-ambulatory patients experience losses such as feeding themselves, the ability to swallow or turn over in bed. They lose the strength in their voice making communication difficult. Patients lose their stamina which impacts their ability to meet the requirements to maintain employment and experience an increase in hospitalizations and need for supportive equipment. As patients lose the ability to maintain physical function, they require alterations to their homes for accessibility which has a considerable financial impact. Patients slowly lose their independence as they lose function, and with that physical loss, the need for mental health support increases, not only for the patient but for the patient’s extended family as well. As the patient experiences the loss of function, the greater the burden of care, financial, emotional and psychological burden is placed on the family unit.
“I went from having an invisible disability to a visible disability. 5 years ago, I could climb stairs, care for my child, walk completely unassisted, just a bit slower. Today I cannot stand from a seated position on my own, I can’t independently dress myself. I have been to urgent care often due to falls.”
“I honestly am worried that one day I won’t want to live, not because I have SMA, but because I’m progressing and I cannot access treatment to stop it.”
“In the last 2 years, I have stopped being able to write and draw which was so important to me, breathing has become harder.”
“The financial cost has been overwhelming, each year we have spent an average of $20,000 for things such like equipment and therapies, it’s been very hard on our family.”
“I have almost completed achieving my teaching degree, during my years at university, I have lost significant use of my arms and hands, I’m very worried that I won’t be able to work in the career I have worked so hard for. I was so sure I would be receiving treatment by now. I’m terrified what my life will be like without treatment.”
The progressive loss of function, in some respects, can be even more challenging psychologically than not having these capabilities in early life.
“It is so hard to watch my son fall. It hurts him as much emotionally as it does physically. The worst is knowing there is a treatment that can help him but he hasn’t been able to receive it. Every month without treatment is one where he loses strength.”
“My daughter’s loss of function over the years has been incredibly difficult to bear. With a progressive disease, a loss of function is like going through the shock of diagnoses all over again, learning to live with the new norm. As a family, the emotional pain behind closed doors is immense. What will she lose next? Now, even more devastating is that there is treatment that can halt the progression, that can save her life, but she has not been able to receive it.”
“He suffers bouts of depression, he has talked to MAID (Medical Assistance in Dying) which impacts me as a caregiver greatly.”
“Weakening has quickened, swallowing is getting worse. I want to stabilize before I lose too much and have higher health risk and bigger burden to my aging parents.”
Experiences With Currently Available Treatments
47% of the respondents are receiving treatment, of those, 78% are receiving Spinraza, the remaining 22% (9 patients) are receiving Risdiplam.
Of the patients receiving Risdiplam, 8 of the 9 (89%) are experiencing improved energy, stronger voice and cough as well as gains in strength. One patient has only received treatment for 3 months and has not experienced any improvement at this time. 5 of the 9 patients receiving Risdiplam have been on treatment for 3 – 8 months. The remaining Risdiplam patients have been receiving treatment for 1 - 4 years.
Patients report that negative experiences with this treatment ranged from diarrhea for one week after initiation of treatment, the taste of the product and minor tremors that rectified itself with time, temperature control of product.
Please see Question 6 for responses related to Spinraza.
Improved Outcomes
Caregivers and patients who are experiencing an unmet need feel treatment would offer them stability and an improved quality of life with access to treatment. They expect greater independence and improved strength, which equates to a higher quality of life. They are hoping minimally for a stop in progression of the disease, because that alone is an improvement in terms of quality and quantity of life.
The SMA community has been struggling to offer continued justification for access to effective innovative available treatment. Real world evidence and patient reported experience is readily available for this purpose. This treatment offers a future for the group of patients who are still waiting. It offers the ability to change the trajectory of the disease and potentially to increase their lifespan.
Experience With Drug Under Review
The majority of respondents accessed Spinraza through provincial reimbursement, most of those within the province of Quebec, however several respondents participated in clinical trial or through approval by private insurance.
78% (32 patients) of the reporting patients receiving treatment, are receiving Spinraza. Of these patients, 73% were in the age range of 18 – 35 years. 15% are in the age range of 36 – 50years and 12% were in the age range of 51-65 years.
Of those patients, 79% reported that they experienced marked improvement since initiating treatment. The improvements included fewer respiratory illnesses, improved endurance, increased strength in arms, head control, stronger cough, improved lung function, increased voice strength, increased core strength.
15% of the patients receiving Spinraza reported that they experience stabilization of their disease.
Only 6% of the respondents who received Spinraza are experiencing no stabilization or improvement.
Of the 18 – 35 age cohort, 79% experienced marked improvement, 17% experienced stabilization, one patient experienced no improvement or stabilization. Of the 36 – 50 aged cohort, 3 patients or 60% experienced marked improvement, 1 patient experienced stabilization and 1 experienced no benefit. Of the 51 – 65 cohort, all 4 respondents experienced marked improvement of their disease.
Of the patients receiving Spinraza, all have been receiving treatment between 1 – 3.5 years.
Respondents were asked to provide “open” comments about treatment effectiveness:
“I have more strength, more energy, I’m able to do physical tasks that were impossible before.”
“I improved 7 points on my Motor Function Test.” (52 year old)
“I have small improvements in a variety of ways such as improved neck and core strength.”
“He has not been sick in 18 months since starting Spinraza.”
“Others can understand him now because he speaks louder. More social opportunities because he can be heard in a social setting. He now sees a future for himself.”
“Muscle pain has stopped completely and has a big boost in energy levels.”
“Increased energy levels, less exhaustion. Tasks are easier to do. Gained back my ability to drive!”
“For the first time in 25 years, I’m able to roll over on my own!” (41 year old)
“I used to have regular sleep apnea episodes and haven’t had those issues for years since starting Spinraza.”
“I’m feeling more confident, I really feel like I have my life back.”
“I’m not as exhausted so I can go and take part in more family functions where before I would have to say no because of the exhaustion.”
In response to the query of negative experiences with Spinraza, respondents reported several, including a drop in function shortly before receiving their next maintenance dose which was subsequently rectified after receiving the dose. Some patients experience temporary headaches after receiving their dosing. Travel and time off work to receive their injection are also reported as a negative aspect along with discomfort of receiving an intrathecal injection.
While the majority of patients do not experience negative side effects, the majority of patients do feel that the gains in function and disease stabilization that Spinraza provides far outweighs the negative aspects of receiving this treatment. The benefits of receiving treatment has had profound positive impact on patient’s general health, strength and mental health. The benefits are experienced by the patient as well as their extended families.
Companion Diagnostic Test
Not applicable.
Anything Else?
53% of the respondents were treatment naïve. These patients are experiencing continued loss of function as well as a decline in their mental health. It is important to understand that the knowledge that patients in other jurisdictions are accessing treatment and are experiencing an improvement in quality of life, is increasingly difficult. The devastation of progression with a disease with full mental capacity is one that results in dangerous lows in mental health. For these patients, the future is bleak. From the perspective of a patient group, we are seeing a spike in patients who are considering self harm due to the continued inability to access effective treatment. The answer is not simply to address the consequences, it is to address the cause which is unmet need and the ability to effect change.
As our existing treated patients age, treatment will continue to be available for them in conjunction with stopping rules. It can not be deemed as just to forgo the needs of treatment naïve patients who would benefit as well, to initiate treatment at their present age with the same stopping rules should they not prove disease stabilization.
Conflict of Interest Declaration — Cure SMA Canada
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
No outside help was obtained for this submission. All research, interviews, compilation of data and submission preparation was performed by Cure SMA Canada.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
No outside help was obtained for this submission. All research, interviews, compilation of data and submission preparation was performed by Cure SMA Canada.
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
Table 1: Financial Disclosures — Cure SMA Canada
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Biogen | — | — | — | X |
Novartis | — | — | — | X |
Roche | — | — | — | X |
I hereby certify that I have the authority to disclose all relevant information with respect to any matter involving this patient group with a company, organization, or entity that may place this patient group in a real, potential, or perceived conflict of interest situation.
Position: Executive Director
Patient Group: Cure SMA Canada
Date: January 4, 2022
Muscular Dystrophy Canada
About Muscular Dystrophy Canada
Muscular Dystrophy Canada is registered with CADTH. www.muscle.ca
Muscular Dystrophy Canada (MDC) supports people affected by hereditary and immune-mediated conditions where the primary effect is on the muscles, the neuromuscular junction and/or peripheral nerves. These include the following disorder groups:
Primary disorders of the muscle (muscular dystrophies, hereditary or immune-mediated myopathies),
Neuromuscular junctions disorders (hereditary or immune mediated myasthenic conditions)
Primary disorders of the peripheral nervous system (hereditary motor and sensory neuropathies; immune- mediated neuropathies; lower motor neuron disorders)
The specific types of muscle affected, the severity, and age at which symptoms begin to show depend on factors such as the individuals’ exact diagnosis. Commonly persons living with neuromuscular disorders experience some level of muscle weakness. This may affect their arms and legs, and in some disorders the muscles needed for eating, speaking, breathing, heart and eye function maybe affected as well. Some neuromuscular disorders have multisystem effects and might affect other parts of the body such as the endocrine system, cognitive function, and gastrointestinal system. For a very small subset of neuromuscular disorders, like Spinal Muscular Atrophy, life-changing treatments are now available but access is limited; the majority of neuromuscular disorders have no definitive cures.
Since 1954, Muscular Dystrophy Canada has been the leading health charity and voice of the neuromuscular community in Canada. MDC’s mission is to enhance the lives of those impacted by neuromuscular disorders by continually working to provide ongoing support and resources while relentlessly searching for cures through well-funded research. MDC represents over 50,000 registered individuals including those impacted by neuromuscular disorders themselves, family members/caregivers, healthcare professionals, and researchers. MDC supports individuals impacted by neuromuscular disorders by investing in research, delivering critical programs and services, and challenging public policy. Our services and programs play a crucial role in informing and supporting members of the neuromuscular community by funding equipment and assistive technologies to improve daily life, hosting family and caregiver retreats, providing emotional and educational support, and providing access to vital resources and support systems.
Funded by Canadians from coast to coast, our investment in the research community is advancing the development of important new treatments. Our programs and services play a critical role in improving access to vital resources and support systems and ultimately, affecting quality of life. Our advocacy efforts focus on enhancing public policy at all levels of government to bring about positive change. We are currently working to bring new treatments and trials to Canada. Advances in medicine have resulted in individuals with neuromuscular disorders living longer but not necessarily living better. As their disorder progresses and changes, so do their needs and financial strains. MDC recently completed its own health economics research study to best determine the costs patients incur during their neuromuscular journey. These “hidden costs” include all out-of-pocket expenses (e.g. genetic testing, home modifications, mobility devices), any loss of income due to loss of employment or forced early retirement, or any of the time provided by family care-givers. The results of our study reinforce the value proposition for access to life-changing treatments and supports.
Our desire is to provide support to an individual and family through all stages of disease progression by providing the tools, resources and support to live a full and rich life and at the same time, invest in research and real-world evidence generation to support public policies and influence positive change.
About SMA
Spinal Muscular Atrophy (5q SMA) is a neuromuscular disorder caused by biallelic mutations in the SMN1 gene. There are different clinical subtypes of SMA based on age of onset, function, and outcome. The most common subtype is SMA Type 1, which presents with symptoms in the first 6 months of life, and untreated results in death typically before the age of 2 (refer to: https://www.youtube.com/watch?v=EG8zMxZeOOs). SMA subtype corresponds with the number of genetic copies of a modifier gene called SMN2. Both SMN1 and SMN2 encode for SMN protein. Patients with SMA Type 1 most commonly have two copies of SMN2, while patients with Type 2 typically have 3 copies, though can have 2 or 4. Therapeutic strategies for SMA have centred on increasing the amount of SMN protein, either by acting on SMN2 (Spinraza, Risdiplam) or SMN1 (Zolgensma). Nusinersen (Spinraza), has been approved by Health Canada since July 2017 for the treatment of 5q spinal muscular atrophy. This treatment has been recommended for reimbursement by Canadian Drug Expert Committee CDEC and MDC has previously submitted patient input submissions in support of providing access and reimbursement for nusinersen. To date, most children with SMA registered with MDC are receiving nusinersen as a part of their treatment regimen; adults affected by SMA in Quebec are also receiving nusinersen as part of their treatment regimen. Since our patient input submissions in 2017/2018, emerging evidence and real-world data have expanded our knowledge on safety and efficacy of the drug in a much larger population of SMA patients than those reported in the initial studies.
We hope this submission will demonstrate the unmet need for treatment – particularly for those above the age of 25 affected by SMA – and will reinforce the importance of access to this life-changing treatment. Please note, in addition to this written submission, MDC has conducted short video-interviews with adults in Quebec who have received consistent access to Spinraza; these are available for your review and consideration.
Information Gathering
Muscular Dystrophy Canada has Neuromuscular Service Support Staff in all provinces across Canada. As part of the System Navigation Program, the Neuromuscular Service Support Staff provide front-line support to thousands of Canadians affected by neuromuscular disorders. The program operates on collaboration and patient engagement principles. Neuromuscular Service Support Staff work directly with patients and family members to identify non-medical needs (e.g., housing, transportation, access to equipment) and provide them access to the right resources in a personalized customized manner. Neuromuscular Service Support Staff work in partnership with patients and their families to address barriers, network and make connections with others in the community, share education materials and resources, enhance life skills and self-coping strategies, embrace inclusion and ultimately provide supports to help positively improve the overall well-being and quality of life of the patient and their family members.
The Neuromuscular Service Support Staff identified and contacted adults living with spinal muscular atrophy to participate in a healthcare experience survey (available in English and French) and semi-structured virtual (phone, Zoom) interviews.
The following submission reflects data from 60 individuals (age 18+) impacted by spinal muscular atrophy. Each respondent had a confirmed diagnosis of 5q SMA, as signed off/confirmed by a neuromuscular specialist on their registration form. The respondents included 31 males and 29 females. Respondents were between ages of 19 – 80, with responses from adults affected by SMA in Quebec (n=20), Ontario, British Columbia, Alberta, Manitoba, Saskatchewan, New Brunswick, Nova Scotia and Prince Edward Island. The responses were collected from December 2021 to January 4, 2022.
We sought the opinion on the value of having Spinraza approved for adults affected by SMA in Canada. A qualitative descriptive approach, employing the technique of constant comparison, was used to produce a thematic analysis. We have included patients’ quotes to ensure their voices are captured in this report and to provide context for quantitative elements. A report capturing all patient comments is also available for review.
Disease Experience
In response to the question posed by MDC: "Can you describe how SMA impacts your day-to-day life and quality of life?”- the following 5 key themes were identified (in order of frequently reported): 1- significant impact on independence; 2- significant impact on activities of daily living; 3- negative impact on mental health and well-being; 4- negative impact on energy levels (fatigue); 5- negative impact on work participation. The below quotes from individuals affected by SMA highlight that the impact of SMA on adults is not purely physical, but that the condition impacts mental health, quality of life and the well-being of families.
Impact on Independence
“Unable to live day to day life independently. Extreme muscle weakness, fatigue, pain and endless tests and doctors’ appointments while trying to live like every other member of society. It is impossible to have a true quality of life without treatment, knowing any day could be your last.”
“SMA atrophies muscles and day to day quality of life. It atrophies independence and challenges life in every aspect. The fact it is degenerative you never know what is going to be lost the next day.”
“SMA has taken a lot of physical abilities away from me. I can no longer feed myself, dress myself, brush my own teeth, or grab a simple glass of water. I do have a good quality of life thanks to my amazing family that does not let my disability stop me from anything, but it is an exhausting disease nonetheless. It’s tiring relying on other people 24/7 and going through the emotional roller-coaster of losing function (prior to treatment).”
“Spinal muscular atrophy (type 2) greatly limits my independence. It is difficult, or perhaps impossible, to point to an aspect of my life that is untouched by this disease. Feeding, transfers, personal care, and transportation are all severely different than those without this disease. My daily life is also greatly changed as I require help with most simple tasks (drinking, completing school-work, showering, toileting, dressing, etc.). The activities that I can engage in, and places I can visit, are strictly limited by the accessibility of buildings/areas (for example: the height of my wheelchair limits my ability to sit at tables of regular height, I can only sit at tables that are bar height).
“SMA contributes to body muscle weakness and that involves being confined to a wheelchair at all times. It also means that over my lifetime my muscles will atrophy and grow weaker because I do not produce the protein for muscle development. I require assistance for most aspects of my day-to-day living, and that level of assistance will only continue to increase as I get older because I will get weaker. If I did not have a stable family unit and comfortable household income my quality of life would most definitely be a struggle, as the amount of health care aide I require exceeds what the government can supply for me. I'm fortunate, but there are many in my position who have to live in full-time care facilities to have their basic needs met. As you can imagine, someone's quality of life in that type of situation would not be very high.”
“I require 24hr support of a caregiver for all aspects of my day-to-day life. From dressing to meal prep and assistance using the washroom. SMA makes it so that every step in my day is a little more challenging to achieve a healthy quality of life.”
“It's hard to get comfortable while sleeping because I can't move around easy. I need a hospital bed to raise me up enough to stand. I need to use two canes to walk, which is increasingly becoming harder and I worry I'm about to lose my ability to do so. I moved back in with my parents 4 years ago after living alone for 14 years because I was getting to weak to live on my own, and have since become a lot weaker. I now require help doing daily activities that I didn't need help with even a couple months ago, like getting out of the shower or off the toilet. I don't leave the house much more than once or twice a month because I get tired too easily now. Over-use of my "good" limbs that have gotten weaker causes a lot of pain, including bicep tendinitis and rotator cuff issues in my right arm, which is the only arm I can lift.”
“I require assistance with all aspects of my life by specially trained caregivers. I am ventilator dependent and unable to swallow. Presently I am able to move my thumbs.”
“SMA has affected my ability to eat, walk, dress and toilet independently. I rely on someone else for all aspects of personal care and cannot live independently. I have chronic pain and my mental health has been impacted due to SMA.”
“I have very little movement in my body so I am 100% dependent. I can't feed myself or even move my arms. Swallowing is tricky so eating takes a great deal of time. I have pain if my body is not in the right position. The biggest issue is my breathing which is extremely shallow. I have stayed in my house outside of the summer season for most of my life because of risk of infection from viruses. My social life is all virtual because of this.”
“Every single moment of every single day I am affected by SMA in one way or another. I cannot get into bed on my own and rely on home care to come every morning to get me dressed and showered.”
“I need to have a caregiver around all the time, or at least someone who is willing to do my care. In high school I couldn’t just go to a friends house for a sleepover, because I need to be repositioned at night and use my bipap. In college, I couldn’t just go on a blind date on my own, because I need help with feeding / drinking. As an adult, I struggle to have autonomy in my life and career because I am relying on someone else’s schedule 100% of the time.”
“I cannot drive, get to Dr appointments, get groceries, do laundry, cook myself a meal, work a normal job, clear secretions to keep myself from choking or truly have any type of independence outside my home.”
“SMA has really affected my independence because I was able to walk and now I’m stuck in an electric wheelchair and requiring help all the time. I’m too weak to drive. I don’t feel comfortable anymore going outside or doing an activity by myself anymore because going to the washroom has become almost impossible. Being an electric chair causes so much problems because you can’t fold up a power chair it’s really bulky heavy. In addition, because there’s no funding for manual chairs, you have to pick one. I’ve always wanted to slow dance with a beautiful woman standing and that will never happen.”
“It has affected my ability to do virtually anything independently, other than drive my specialized wheelchair and type on my iPhone.”
“I have no independence, I am completely dependent on other people which restricts my ability to move out, travel, and forces me to arrange my life around other people (caretakers getting sick, needing vacation, etc).”
“I can't do my personal care. It's becoming more difficult to go out on my own especially in winter (because of extra clothes).”
“I can't live alone anymore. I need help getting into and out of the shower. I need help getting off the toilet. I need help getting items out of the freezer or cupboards to make my own meals. I need to be driven everywhere by one of my parents, and even if I could drive on my own, it'd be too expensive to ever buy one of those vehicles. I need help going to the bathroom in public places. I need help transferring to the eye doctor or dentist chair. I need help with shopping because I can't carry too much or reach some items. I need help plugging in my wheelchair to charge it because my hands have become too weak. I need someone to do my laundry for me and make my bed and put my clothes away. Basically, shortly after turning 30, I started to get increasingly weaker and lost a lot of my abilities and nearly all of my independence. I've lost most of my abilities just in the last 4 years since Spinraza was approved by Health Canada but not accessible to type 3 adults. For example, in 2017 I was able to babysit my infant niece of my own for short periods of time, lift her on my own, get her out of her crib on my own, take her outside to play on my own, cook for her and feed her, change her diaper etc. Now, I can barely hold my new 5 month old niece on my own while sitting down without my arms giving out, and sure as heck can't lift her, change her diaper or anything of the sort.”
“I cannot live independently and rely on others for assistance. I need someone to accompany me to appointments and community activities.”
“I am in a motorized wheelchair. I require a caregiver to perform personal tasks. I also have specialized equipment to help me at work.”
Impact on Activities of Daily Living
“I require the use of an electric wheelchair, I have to have attendant care for even the basic things like going to the washroom bathing changing transferring. As I get weaker, I require more care and less I can do.”
“It is hard for him to me to work on the computer. Everything, toileting dressing, transfers and even eating is getting more difficult because it is harder to hold utensils.”
“SMA affects everything about it. Everything is a chore for me. I am at the point right now where I have a hard time getting out of chairs. It affects me constantly. I am okay and can get around, but if I sit in a chair with no sides I have a hard time getting up.”
“Having SMA my entire life and in a wheelchair since I was 15, it is hard in just about every way. The biggest impact is the progressive nature that I cannot count on being the same person in any type of future. My body, mobility, strength, and function in the future is unknown.”
“I need more and more help in doing all my daily activities. My medical needs and appointments are consuming a lot of my time and resources. I decided to go through surgeries to manager my bladder and bowels without transferring to the toilet. The structural changes improved my life, but they certainly come with some issues.”
“I depend every day on a caregiver, my wife need help to use bathroom, shower, getting dress, meals. I cannot walk, I have a powered wheelchair and lifts.”
“It limits me in most capacities through my day including waking up, getting dressed, preparing meals, caring for my son, cleaning my home, attending my appointment, basically every aspect of my life is carefully planned out and coordinated so that I'm able to continue to continue to be a healthy human. My disease affects my personal finances greatly, personal relationships and most importantly do to my last bout of progression my mental health has been severely compromised due to the lack of an accessible treatment for my disease which is absolutely disheartening considerate than a 4 year battle to get access to Spinraza and I'm still unable to access it.”
“SMA impacts all areas of my day to day life. I need assistance with dressing, showering, transferring, grooming. I use non invasive ventilation during sleep, and a cough assist machine when sick. I am unable to maintain nutrition orally because of a weakened swallow, so most of my nutrition is by g-tube.”
“SMA impacts my quality of life greatly every single day from the moment I wake up; to the moment I go to bed. I need assistance with transfers, lifting objects, toileting, showering, dressing, and generally doing most things.”
Impact on Mental Health & Well-Being
“SMA imparts all aspects of my life, from the mobility in my body limiting my ability to dress, eat, bathe, or perform other essential tasks of daily living. However, further to this, it affects my mental health as well. Knowing that my body is constantly failing me rapidly gives me anxiety around my place in society and self-worth. The knowledge that there is a drug available to help that is still out of reach because of government mandates makes me feel less than my younger counterparts.”
“Mental health is a huge issue and I have dealt with depression for most of my life. I deal with discrimination a lot of the time and ignorance from people in the public.”
“I am a very sociable person. I find people see the chair and judge me because I am in a chair as opposed to not knowing who I am as a human being.”
“Emotional is having my family watch me degrade. It is hard for them to watch this, they feel bad, they are unsure of the future. Having to tell people and share medical information because it is something that is not hidden anymore. It has to be explained anywhere I go why I can't do things.”
“The mental toll this disease takes is just as cruel as the physical toll it takes. Living with the knowledge that your body is wasting away to the point of death. Grieving the loss of each new ability. Grieving the loss of a future that will never be. All of this grief and anger and anxiety is exasperated by the fact that a treatment exists that could save your life but the government does not think you’re worth it.”
“Loss of independence despair; financially reliant for all housing care for all personal support.”
“Because of me getting weaker I’ve become depressed, I’ve been suicidal even taking my life once and being rescued by paramedics. People treat you differently when you become weaker. Or people think you’re the same strength when you’re really not. It is so disappointing when all your friends want to go to a place and you really can’t go because either you’re tired or it’s not accessible or your chair can’t fit in in the car or going to your family members house and it’s not accessible because most houses are not and you missed out on a lot.”
“Very embarrassing, people laugh about the fact that I cannot get up.”
“It keeps you from going to things. It is starting to confine me more. I am becoming more wanting to stay home because it takes a lot from me. I really don't like to be embarrassed. When I went for my booster, I told them I couldn't sit down and they thought I was joking. It is very limiting.”
“My constant need for help is emotionally trying and I have a hard time doing things that others take for granted.”
“The need to rely on my elderly parents is emotionally taxing because they have their own issues. I am less comfortable going out on my own to social gathering, and I do not want to have my caregivers with me everywhere I go. I cannot enjoy my baby niece as I would love too because my arms are getting weaker and weaker. She is my biggest joy in life but I cannot lift her to smell her and kiss her.”
“The one that immediately comes to mind is finding and maintaining a romantic relationship. It's hard for many to see past a wheelchair and obvious body deformity but that's not to say it's impossible either. I have maintained strong social and emotional connections with friends and partners, but I think I might be the exception to the rule on that one. I've had conversations with others who have SMA and they have told me they felt ostracized for much of their life. Living with SMA means you have to be able to overcome many social hurdles before being able to form a connection with people. I was able to do this, but many are not.”
“Knowing that I'll continue to get weaker and the anxiety that comes with knowing that to the point that I've had to go on an SSRI medication for anxiety and depression. Constantly thinking of all the things I used to be able to do and can't any more is emotionally draining. I don't really have a social life or friends, I rarely leave the house because it's too tiring and I have to rely on my parents to drive me because I can no longer live on my own in the city. Knowing that I'm dependent on my parents and feeling like a burden at times is frustrating and I wish I didn't have to put that responsibility on them. I've always felt ashamed/embarrassed about not having a job after college, which made me avoid old friends and acquaintances. I've never been on a date or had a romantic partner and I'll never have a family of my own. I worry about the future a lot and what I'll do when my parents die or can no longer take care of me.”
“Feeling excluded from events or social situations because of a lack of accessibility.” “Severe depression and fatigue.”
“I am not always able to do activities I enjoy because of my energy and health, and the lack of financial resources (for hiring caregivers). As a result, I have bad mental health.”
“Life is a roller coaster. You never know what you are going to lose next. You constantly are fighting battles of some sort or another. It's extremely wearing on not only myself but those around me.”
“Growing up, seeing your friends gain more independence while you remain the same. As an adult, this is slightly less but the same thing does happen. Being left out of things because you cannot participate. The mental/emotional impacts of realizing that you lost the ability to do something as the condition progresses.”
“Fatigue, inability to feed oneself, reliability on others for personal care can greatly hinder the ability to participate in social activities.”
“For me, the isolation from in person contact is depressing. I have to avoid crowds and I do not even leave my home for 9 months per year. I have a good virtual life but it's not the same.”
“I feel isolated around everyone because my life and body is so different from theirs.”
“I have often felt very socially isolated. What lockdown was like during the beginning of the pandemic that is what most winters are like for me.”
“The social and emotional consequences of SMA are almost more powerful to me than the physical ones. I often struggle with feelings of being a burden, being unworthy of love, being unable to participate in society in meaningful ways, limited freedom of choice in terms of where I can live, where I can go, and who I can spend time with. As a 30-year-old, as very much affected dating, where people often see me wheelchair-first and gives me profound feelings as emptiness and loneliness.”
Impact on Energy Level (Fatigue)
“I am very active person so managing my energy is almost like a full-time job. I have to choose what I do so I am not too tired or get weaker.”
“I deal daily with shortness of breath, weakness, fatigue and cramps.”
“I'm having less and less energy. I'm less able to participate actively in the community and enjoy my social life.”
“Quite literally every aspect even down to the task of breathing is affected. The tasks that the majority of people don’t even think about their body doing, such as breathing, takes a lot of energy and focus for my body.”
“SMA makes it difficult to do tasks and I get tired very easily.”
“My energy levels are low so I often cannot do much before needing to rest. Because of having to be full-time in a power wheelchair I have extremely sore hips and back and struggle often with pressure sores.”
“The fatigue limits my ability to socialize, and restricts my functional time to work and study. Additionally, having to rely on other people 24/7 is a crushing weight on my mental health and self-worth, as well places a strain on these relationships.”
“I hold myself back from activities and experiences because it will be too much energy usage to do it.”
Impact on Work Participation
“I have very limited use of my four limbs. My dexterity is extremely difficult to even bend my fingers. I used to work for the Toronto star for 25 years. When I turned 51 I became very fatigued so I had to go on long-term disability. I am in a motorized chair and I do have PSW's come in to help in the morning and night.”
“It has been progressive. I am a father of three and especially over the last two years, I cannot do any long distance walks (a block), I can no longer climb stairs. I had to advise work of my condition. I had to share with everyone that in the new future I would need some assistance. The impact is the slow degradation and loss of the ability to do things. Also, the inability to do things with my family.”
SMA affects every aspect of my life. Living confined to a wheelchair with extremely weak muscles limits my ability to do anything normally. I rely on others 24 hours a day to help me with most aspects of my life. I am able to continue to hold down a job and function fairly well at it with help. As my muscles weaken, I don’t know how long that will continue.”
“Maintaining a career or position outside of my home is incredibly difficult, as I would have to pay someone to be with me most of the time. So I have chosen not to pursue a typical nine to five job and take advantage of opportunities from my home. I was able to complete a bachelor’s degree in Arts and have the option of pursuing a masters or PhD, but again this was only possible due to my generational wealth and healthy familial unit.”
“I haven't worked in 9 years. It can be very depressing.”
“In the past, I have had to fight my employer in order to have basic needs not in order to keep employment.” “Due to exhaustion and decline in my ability to type, I am on long-term disability with my employer until I retire.”
Experiences With Currently Available Treatments
In response to the question posed by MDC: “How are you managing SMA with currently available treatments or therapies. For each therapy what are the benefits seen, and side effects experienced? Do you have any difficulties accessing these treatments?” - the following 3 key themes emerged: no treatment experience; positive benefits of Risdiplam observed; opted for alternative ways to manage SMA.
The below quotes from individuals affected by SMA highlight the significant treatment gap for adults.
No Treatment Experience
“No types of medication.”
“No therapies at the moment for me.”
“I have not had any treatment experiences. I live in Ontario, my brother lives in Quebec and he started treatment with Spinraza a year ago because of my age. This is very frustrating. I am hearing that they are seeing improvements in my brother and the Ontario government is saying it doesn't help.”
“I am an adult and currently no treatment is available for me. It’s criminal. Especially when most of the world covers SMA drugs for adults.”
“No experience with treatment.”
“I feel as though I have not had any treatment my whole life as any procedures that I have had done are due to the progression of my disease, rather than a treatment for improvements.”
“No treatment. No medication for adults like me affected by SMA.” “I've never been granted an opportunity for treatment.”
“I have had no treatments.”
“I haven't had any drug experiences.”
“I wasn't able to access any SMA treatments.”
“To date no treatment options have been available.”
Positive Treatment Experience With Spinraza Initially; Switched to Risdiplam
“I have done 4 Spinraza doses and Risdiplam for 3.5 years. They both worked well. Only reason I switched was because Spinraza was not funded and I got accepted into a clinical trial for Risdisplam. I was on a trial for Cytokinetics (a muscle activator) I saw muscle improvements with this medication. I was on this study for 4 months, which was the length of the study.”
“I have been fortunate enough to receive both Spinraza (7 doses, I believe) and now Risdiplam. On Spinraza I noticed a MASSIVE energy boost after each shot. It was truly unbelievable. I went from needing 12 hours of sleep at night to 7. I was working a full time (in office) internship for school at the time and I had absolutely no issues keeping up or maintain energy. In fact, I actually had a second summer job on the weekends! During this time, my trunk and strength also significantly improved, as well as hand movements. The only downside was that the injections were painful and due to my scoliosis, it was very challenging for them to access my spinal fluid. On my last dose, the radiologist tried for about 2 hours but was unable to get to the right spot. This is when we made the decision to switch over to Risdiplam, since it’s oral. On Risdiplam, I’ve been seeing positive improvements too. Most noticeably is my increased lung capacity; I can now nap without my bipap (which I lost the ability to do in elementary school), I cough and sneeze so much stronger, and my voice is so loud and clear. I also gained the ability to whistle! Physically, I can now wiggle my toes and I have much better neck control overall. I believe my grip in my hands is stronger too.”
“I am currently on Risdiplam/Evrysdi and have seen slightly improvements in my energy and endurance. My swallowing is slightly better as well.”
“I have been taking Risdiplam for a few months. I've seen moderate improvements and had no side effects.”
Opted for Alternative Ways to Manage SMA
“Traditional Chinese medicine improved circulation. Heel cord lengthening and mobility aids have made it easier to get around.”
“I took creatine for about 5 years in early 2000’s with marginal benefits.” “Besides physio, I've never been treated.”
"Currently I am not receiving any drug treatments for SMA. The only “treatment” I am receiving is the use of a BiPap at night and Cough Assist during the day to try to maintain my lung functioning for as long as possible. These practices have so far been able to slow the loss of lung functioning, but act as more of a deterrent and less of a solution. These machines are in my own home, I am required to see a doctor/respiratory therapist once a year to maintain them.”
“As far as treatment goes, there hasn't been much. I received an operation when I was 10 to implant metal rods in my spine for support. Without this operation, my chest cavity would have declined to the point of crushing my organs I'm bending my spine. This was a major operation and a lot for a 10-year-old, I don't even think I realized the scope of what was happening to me at the time. But it worked and prolonged my life. The only other thing I could consider treatment is physiotherapy, and while it helps, it's simply a means of prolonging the inevitable.”
“To be honest, I have never really treated SMA. I never took medicine for SMA. I used to do physical exercise but it didn't show improvement and eventually became more hassle and pain then it was worth. Swimming was good but again, eventually became too difficult and/or painful.”
“Because of COVID I have not seen my specialist in some time. I find staying active to be the best thing for me. Unfortunately I wasn’t offered any treatment, for example physiotherapy until I broke my femur and fibula bones in my right leg. This began a journey back to strengthening my core and hope ultimately to be able to stand and walk again. I had resigned to my diagnosis and hadn’t exercised. I wish this had been offered or even suggested to me.”
Improved Outcomes
Improvements that patients and caregivers would like to see in a new treatment can be categorized as those that (1) promote muscle strength (primarily in the arms and with respiratory function) and (2) slow down progression of disease (or reverse damage).
Improve Muscle Strength (Primarily Arm and Respiratory)
“Arm strength and dexterity is the most important thing that I'd like to see. It would give me more freedom. It wouldn't give me all the freedom I'd like, but a much better life.”
“I think that both Spinraza and risdiplam meet the goal of treatment. The only thing I would like to see as a secondary treatment would be a muscle activator to the gene therapy.”
“If I was able to get a little bit of strength back in my arms I can hold my baby niece and I keep playing the sport I love.”
“A faster reversal of lost strength / motor neurons would of course be nice, but honestly I’m incredibly happy with what it’s already given me. I never would have expected one treatment (never mind several) to come out in my lifetime.”
“To be able to walk up stairs and to get up off the floor or to garden.”
“The fine motor skills would be a big thing to improve his hands and neck. They are little improvements, but it would make a big difference.”
“I would like to get some of my strength. I would re gain my social life. I would hope to be stronger and continue with my life without being in a facility. That would be the best. My life is slipping away from me.”
“More strength in arms.”
“More impact with respiratory strength.” “More strength, less pain and less stiff joints.”
“I wish there was something I was able to access that would improve my strength or maintain what I have.” “Energy and upper body strength are the most important to me.”
“I would like to see anything that can safely increase or stabilize the muscles regarding the lungs. Even a 10% increase in breathing would be a godsend. Secondary, improvement in arm movement would be amazing as well.”
Slow Down Progression of Disease
“Firstly to stop the progression of the disease. Secondly to stabilize. Thirdly to repair the atrophied muscles.”
“Just stabilize what I have would be a huge benefit. In addition, it would give me hope for even newer drugs that could potentially fully cure me. If I can get more energy, I could do so much more.”
“I would like to see a stop in progression, and any regain of function would be a big bonus. Neither of these are attainable with the current treatments that I am on. Daily life would become easier, I would be able to spend less time on current treatments as well as possibly regain some independence. The greatest difference would be made years down the line, as a new treatment may be the only thing preventing me from being bedridden and permanently trached.”
“Improvement in breathing and further slowing down of muscular weakness.” “A complete stop and reversal of symptoms.”
“A drug that is able to replicate or allow my body to create the protein for muscle development would be huge. That is really the only way I can see my quality of life remaining stable and/or improving. I don't see robotics and brain bandwidth computing reaching the same level of quality of life improvement in my lifetime. Medical equipment and wheelchairs can always be improved, but that's no substitute for a declining body.”
“I would love to be able to regenerate muscles or at least stop the progression.”
“I would absolutely love to see they're being a drug or treatment that will slowly reverse the damage done. I know that's asking so much and most likely not going to happen. But one can dream.”
“Any improvement inability would be amazing, but to maintain what I currently have would also be a win.”
In response to “how might daily life and quality of life for patients, caregivers, and families be different if the new treatment provided those desired improvements?” the respondents shared:
“Especially in the winter I rarely go out. Only to do groceries with someone with me. I would like to go out more often, but I have to put on a coat and gloves etc and I need assistance to do that. With this treatment if I can get dexterity and arm strength I can do those things on my own and not rely on someone to do it for me. Also cooking, without my arm strength I shy away from it. I live by myself so I have to get everything done before the PSW leaves at 11am, but there's not a lot I can do.”
“It would give me a huge sense of security. Right now, I am trying to decide how to handle the next few years. Am I going to leave my job, which I love because I can't walk? For me, it would keep me as an active participant in the workforce and able to do the things I can currently do. If the progression doesn't stop soon it is unlikely I can come back from it.”
“Maintain and or gain independence emotional well being.”
“If treatments could reverse damage to the point where we could be independent in some or all of our daily life, that would have massive impact. Less caregiver burnout, better mental health, more independence and autonomy for the patient.”
“I would gain more independence with increased strength and reduced atrophy, which would improve all aspects of my life, including my mental health and relationships with caretakers and family. The other people in my life would have more time for themselves.”
“It would make everything simpler. It would make everything better. Everything gets better. My whole life-social, mental to physical it would help me.”
“I would hope to become more independent. My family wouldn’t have to do as much for me.”
“If it works, just being able to stay stable with a strength level or an ability level that I can count on would change a lot. I could make plans for the future and know that I can reasonably pull of the plan whether it is travel etc. Right now I can plan a trip in a year, but I have no idea what kind of shape I will be in.”
“I will rely less on them. They will worry less about me. They will live their life more freely. The relationship between the family members will be more familial than caregiver-care recipient relationship. This will also grant us more dignity and independence.”
“The level of improvement would be hard to quantify, but it would be huge. We're talking about needing less round-the- clock care and more independence for myself. Sure I won't be walking or achieving complete independence, but the reduction in cost to health care services would be astronomical on its own. Possibly being able to drive myself, continue to feed myself and even prepare my own meals, being able to be left alone without worry of an emergency happening in my home. The reduction in stress to my family members alongside the growth of independence for myself is immeasurable if you ask me, and that's not even taking into account the reduced financial strain to the government over the course of my lifetime. I should also mention that treatment such as Spinraza is also potentially lifesaving, as I'm less likely to succumb to sickness such as pneumonia in my elder years. A longer life filled with fewer complications sounds like a pretty good boost to quality of life to me.”
“My husband wouldn't worry so much. I have a two-year-old grand daughter and there's not much can do with her. I can't lift her up because the added weight may make me fall.”
“Patients would be less dependent on families and caregivers, health care, equipment, renovations. Patients and even their caregivers would have way more independence, freedom, happiness, a sense of purpose and way less stress, anxiety, depression.”
“Quality of life would be drastically improved. I'd be more optimistic for the future and consider starting a family.” “I would be able to work more and have more social interactions with my friends and family.”
“Any increase in independence would have tremendous emotional improvements on the patient. It would also free up time for families/caregivers, and provide an emotional boost for them as well.”
“This would be a huge impact to clients and caregivers, families and other supports. This would also be a big impact on the healthcare system in a positive way as it would be Less costly (medical equipment, medical procedures, strain on the medical system for hospital stays etc.)”
“If my breathing increased enough so a common cold wouldn't be life threatening, then that would completely change my life. I wouldn't be so nervous around people and I would be able to go more places more often. I would have more energy and not need my caregivers to be so careful with my position as some positions make my breathing too difficult. If my arm movement increased enough then perhaps I could once again feed myself. That would be amazing.”
In response to “trade-offs which patients and families consider when choosing therapy” the respondents shared:
Costs
“The largest barrier to treatment is the price tag, it is unreasonable to believe that any family can afford such preposterously priced drugs. Outside of this, there are no trade-offs I can think of that individuals and their families wouldn’t hurry to embrace.”
“As far as pain, that is fine. As far as money it is my life. The money issue isn't up to me, but as far as anything else I am not concerned.”
“Costs mostly.”
“Hospital admissions, financial resources, time.” “The only thing now would be cost. That's it.”
“Cost is a big one. I could not access Risdiplam in Canada so I had to access a trial in the U.S which was a lot of travelling. I was the only Canadian accessing it. For receiving treatment, finding a doctor who is willing to administer the drug was big. Administering Spinraza with scoliosis and a rod sometimes required multiple appointments.”
“There are no difficulties except financial. Many patients are unable to sustain permanent full time employment.”
“Currently the Ontario government does not cover any treatment. I’m on ODSP and make little money. So travel costs money for the treatment itself would be all difficult.”
“Cost, provincial access, travel to clinic, missed work hours, uncomfortable treatment options.”
Potential Harm
“Does this treatment help my life more than it harms it? Personally, preventing muscle loss via treatment is the most important thing I could do for my health. So, in my opinion, a treatment would have to be really harming me to opt for no treatment.”
“I understand that there ma be some pain involved in the surgery and in the recovery. I would be afraid if there was a chance of paralysis from the treatment. I wouldn't want to lose that.”
“Clearly I don't want something worse than the benefits. If the medication is going to destroy organs or something it would have to be a substantial problem for me to reconsider treatment.”
“If the side effects will be worse than any gains made.”
“Nothing that negatively affects my breathing is even considered. It must not do that. If the therapy gets rid of my arm movement entirely, then also no. Those are the 2 most important things to me. It also goes without saying that negatively affecting my mental capabilities is out of the question. Also, the pain involved must be minimal.”
Mode of Therapy Delivery
“I would say that the method of delivery. I prefer the oral drugs instead of the intrathecal because I can do it from home and it is less medical procedures for me. It is also cheaper.”
“Ease of use. I live in a rural area which would be to get Spinraza I would probably have to travel 4 +plus hours to get it. I also have rods in my back.”
“With Spinraza, it was just accessing my spinal fluid for to my spinal fusion rods and scoliosis. I have no difficulties with Risdiplam at all.”
High Desire to Opt for Therapy Regardless of Trade-Offs
“As the patients age they are willing to trade almost anything to have new hope.” “Honestly I would do anything for therapy.”
“It doesn't matter as long as I can do the things I want to do.”
“Not many. If it will stop my body from atrophying I'll likely do it no matter the cost.”
“Willing to drive a long distance. I would do whatever I could if there was something that would help me maintain what I have or stop the progression.”
“Wouldn't choosing a therapy be a wonderful privilege?”
“I have never been eligible for any other treatment for SMA. So when Spinraza became available to me, I was willing to take the risks of the method of administration, and side effects of the drug if it meant I might attain some kind of improvement.”
“At this point there is nothing that I would not try as long as it was reasonably safe.” “I am willing to do almost whatever it takes to get any sort of treatment.”
“The inconvenience of the procedure and the after effects would be part of it. I would need help with the financial part as well.”
Experience With Drug Under Review
23 adults indicated they received the drug under review. The majority received the drug under review in Quebec; others received it through either federal health insurance plans or exceptional access. The majority reported positive benefits as it relates to strength, energy levels and slowing progression. The side effects or negative aspects were related to drug administration (invasive, requires multiple visits/travel).
“We fundraised and my parents and grandparents all took out loans to pay for it out of pocket to qualify for the clinical trial. Benefits was a quick increase in energy, stamina and strength, but the downside was the downward climb before the next dose. I found I progressed back to where I was by the time I was due for another dose. I received some nerve damage from the intrathecal process. The cost of Spinraza was also a large disadvantage.”
“I received it through OHIP. I was never on any previous treatments so I cannot compare to anything else taken previously. Although I will admit it was a little painful, I tolerated it no problem and I would 100% recommend it to anyone with SMA wanting to go on treatment.”
“The only therapy I have ever used has been Spinraza. My private insurance approved Spinraza for me in October 2019. I have noticed improvements in stamina, I’m able to do daily tasks with more ease and for longer amounts of time. The only side effect I have experienced has been from the administration of the drug and not Spinraza itself. I have had Lumbar Puncture Headaches on a few occasions which are managed by laying flat.”
“I have access to Spinraza. I applied to the government of Saskatchewan, which is providing coverage on a case-by-case basis. I was approved, luckily 2 and 1/2 years ago. I am extremely proud to say that I am a resident of Saskatchewan.”
“I currently have access as my province allows patients over 18 on a case-by-case basis. I have demonstrated measurable improvements and maintained abilities. No other treatment has done anything like this. The disadvantage is that it is invasive and painful, but that is worth it. I also need to travel to receive treatment.”
“The only treatment I have experience with has been Spinraza. I started the drug in October 2019. I have noticed small improvements in my stamina. I’m able to do things for longer and with more ease. At my last physiotherapy occupational therapy assessment I had gained 11 points!”
“I have been on Spinraza for 2 and 1/2 years and it has completely changed my life for the good! Not only has my disease stopped but I've also regained a little bit of what I lost many years ago. I will say that side effects are definitely spinal headaches and being off of my feet for a few days. Usually 3 days of rest in bad is how I manage the headaches. And of course with that comes the sore back due to the lumbar puncture. But I would do it every single day if I had to and if it meant that I would maintain the strength and abilities that I currently have.”
“I have been fortunate enough to be receiving Spinraza since June 2019. The results are undeniable. I received a significant increase in my strengths following my loading doses, and my abilities have not declined in the following years.”
“Spinraza has helped me maintain my strength.”
“I have a lot of benefits to receive Spinraza. Significant motor gains have been observed through post-treatment testing. My dexterity has doubled. My energy level has returned. My neck is stronger therefore less dangerous in transport to injure me. I regained the hope of living a normal life without degeneration, despite my diagnosis. My morale has increased.”
“After the first dose of Spinraza I had a lot of difficulty with heartache and headache and also a lot of vomiting. For the benefits I gained a lot of strength and autonomy.”
“The first injections a lot of pain in my neck and my spine for 10 days. I felt sick I could not eat and after those days I felt fine. My body was getting used to the injections and on the day of the injection I had side effects but after that I felt great. It’s like a battery that we charge after the treatment (we are weak before the treatment and after the treatment we regain strength) I stopped in June because of my fat and I lost strength I am more weak in my arms and struggling to raise my glass.”
“The Spinraza treatment greatly stopped the degeneration.”
“Spinraza resulted in better breathing, less burnout on his caregivers and less stress for them - Generally less stressful for the family due to the stabilization of capacities and no degeneration.”
“Spinraza was causing me tremendous muscle cramps in my buttocks and quadriceps about 1-2 weeks after treatment. I had temporary energy surges that lasted for about 1-2 months and dropped drastically before the next treatment. I didn't have any permanent gains. I felt the dose was not strong enough for an adult. Whether it was me or a toddler, we had the same dose.”
“The disadvantages are minimal. The treatment is complex. We have to go to the hospital. It's not super enjoyable, but the payoffs are well worth it. The trips to go there are complex, I have to go to Montreal. The last two weeks there is fatigue. I had big side effects at first, but not anymore. I only have a little constipation. For family and caregivers, there is no disadvantage, there are only advantages. There was just too much expectation from those around me and there were a lot of improvements at the start, but afterwards there was a plateau effect. This may be due to the pool shutdown. I do not know. When I stopped swimming, I had been taking Spinraza for a year. At the beginning, I was more independent, so that freed my relatives a little. They trusted me more, they were less afraid that I would fall, that I would be alone.”
“The advantages: I regained some strength not in my arms and legs. With the help of physiotherapy, I regained strength in the buttocks, the abdominals which allows me to be more comfortable, more stable and to tolerate more long journeys in the car. In the neck too, I have less pain and it helps me to eat a little. I no longer have to take Voltaren and Tylenol for the pain. I have no side effects. The afternoon of my treatment I'm a little bumpy, but I don't have headaches and nothing else. The next day, I leave like a bomb. 2-3 weeks before the next treatment, I feel a drop. If it was closer I probably wouldn't feel that way. For my partner, who is also my caregiver, our life has changed because I have more energy. Life is better. It's less stressful for him too because my swallowing has improved so I'm less likely to choke. In my transfers I am stronger, there is a difference.”
“I reacted positively to the first treatment. I saw a marked difference. Eating and brushing my teeth had become more difficult before Spinraza. Since Spinraza, this is extremely rare. I really hit a peak and I had more core strength to support myself in my chair, my posture has improved, I no longer have shoulder pain. This is also my most significant advantage. I have had the treatments for 2 years now. There is a small gradual decline, but I have kept several benefits. I have had a little less strength and energy since the last year. It still remained at a very good level. I also have a small drop at the end of the 4 month. I still consider that I feel a benefit. I haven't had any side effects from the treatment itself, but I have a little question about asthma. I have developed asthma. Whether it is related to Spinraza or not, doctors cannot tell. It could be due to allergies. Only once the puncture was more difficult and I had a migraine the next day. For those close to me, the Spinraza treatments have no real impact on my family except for trips to the hospital for the treatments. My family is really happy that the Spinraza works, that it relieves me, that I have improvements. The only thing was my grandma kept telling me that I was easier to understand and spoke louder and looked really fit and energized. Another beneficial effect is that I eat faster than before which gives me time to do other things.”
Companion Diagnostic Test
100% reported that they did have diagnostic testing completed with a muscle biopsy and/or blood test. The majority (67%) found it to be a relatively easy process, but a timely process riddled with misdiagnoses. Below are quotes that further highlight the experiences of patients and caregivers with the testing:
“It took 8 months from me starting to show symptoms to the time of diagnosis and 5 different doctors who thought I was a lazy baby. I was diagnosed through muscle biopsy, which was the only option at the time. The first biopsy was inconclusive and so they did a second one. The tests were covered because I was symptomatic and the tests were ordered by a neurologist.”
“Could not get an answer in Calgary. Needed to travel to the US for genetic testing. Was told at the time would be eligible for Spinraza shot and have been waiting for over two years without a shot or any treatment. Paid $7,000 out of pocket. The testing very painful, testing was done 3 times in Calgary - went to Mayo clinic and paid $7,000 had to travel by train - 3 days.”
“My genetic testing has since come back inconclusive, and they are wondering if I have a very rare and undiagnosed type. The tests that I have had done however were paid for by health care and a recent test was covered by a company that was offering testing for a certain type of MD.”
“I was misdiagnosed for the first 12 years of my life. They thought it was nerve damage during pregnancy or birth. It wasn't until my dad did a bunch of research and figured out it was SMA. I don't remember much but I did get tested in Vancouver after my dad found SMA. All I remember is that they gave me a sedative because it would be painful. I just have flashes of them poking me with something. I believe that my parents were also tested for each having the recessive gene. We were nearby so no travel costs. I don't know who paid for it. I don't believe treatment changed after knowing the disability.”
“I was diagnosed through muscle biopsies in the 80s and 90s, which was very painful.” “I was diagnosed with a muscle biopsy as a child.”
“I was diagnosed at age 2, via a muscle biopsy.”
“I was diagnosed at the age of four and I am 33 now, at first the doctor thought it was just bad parenting because I was walking on my tippy toes, it took the luck of a neurologist to be on duty that day and saw me walking and thought that I had muscular dystrophy. Some tests were done everything was covered and it was confirmed that I had spinal muscular atrophy.”
“I was 20 when I was diagnosed. My mother had been taking me to the doctor since I was 2. They kept saying I would grow out of it. I had a muscle biopsy, nerve tests, a few neurologists to get to one that diagnosed me. No out of pocket tests.”
“I was diagnosed in the early 70s and as such I’ve never really had genetic testing so far. I am looking to have this done I was initially diagnosed in Quebec. There was not a lot of experience around it. I was just told this is what it is and there's nothing you can do about it. I didn't have a document that said I had SMA, just a verbal diagnosis at first. When I moved to Ontario they did genetic testing and gave me an official diagnosis. Genetic testing was no problem.”
“I do know that SMA was not very common and there was not a lot of information provided to my parents at the time of diagnosis. From my understanding, they had to travel from Saskatoon to Calgary for testing. I do not know if there were any adverse effects associated with the testing. They did have to pay out of pocket though to get the testing done, meaning travel to another province. I was diagnosed through a muscle biopsy when I was approximately 3 years old.”
“I was born in Canada, but my family moved to New York when I was young. When I was 2-3 the family doctor did some testing and discovered I had Muscular Dystrophy and nothing after that until 14. Then they did the Muscle Biopsy and they told me I would live until the age of 30. When I was 20 I moved to Ottawa and the Civic Hospital did the EKG and the test where they did the EMG test. That is when they indicated I had SMA type II. Since then it is just my family doctor. I have been to Sunnybrook in Toronto and the doctor there I communicate with once a year and they monitor at a distance.”
“My parents noticed that I wasn't moving and crawling normally as a baby. So when they took me to the doctor at the age of two they got a muscle biopsy. From what I understand it was fairly straightforward and easy with no adverse side effects. I think most of the test was covered by the government.”
“I was 10. It was paid by OHIP. It was a blood test. I was tested because my brother is type 2 and the doc was curious because I walked on my toes. We drove 2 hours and spent the entire day at the hospital for the diagnosis.”
“I was diagnosed 33 years ago at the age of 18 months. From what I understand, I was admitted to a children’s hospital for testing where they did blood tests and a muscle biopsy. I don’t believe any of this was out of pocket. There was no treatment available at that time.”
“Muscle biopsy at 10 months old. To my knowledge, this test was covered.”
“I had to have my diagnosis confirmed by blood test before I could begin treatment. This was done at my local hospital. I did not have any out-of-pocket expenses.”
Anything Else?
“I am a person who wants to remain mobile as long as possible. I don’t want to burden the system anymore than I have to. It seems from personal correspondence that Spinraza for adults has either slowed /stopped progression of SMA and in some cases shown some improvement. I don’t know what else I can do as an individual to get access to Spinraza , but the roll out seems to be inconsistent and even discriminatory in some provinces.”
“It’s imperative that you make this recommendation because without it, provincial governments will use it as an excuse not to cover this wonderful drug that will stabilize and in some cases even improve persons with SMA. Importance of stabilization is massive for a person with a SMA. Like myself, I find myself getting weaker year-by-year. Key important things like dressing, bathing, going to the washroom Independently, playing hockey with my friends, being able to work full time, standing, walking and being able to hold a baby in my arms etc. Now these abilities are gone or fading away. I find my life becoming harder and harder to function. By stabilizing my abilities now will buy me time for future cures and better planning for my health going forward. The fear of me getting weaker will be gone. Knowing that I will be able to have the same Energy levels throughout the day is life changing! The fear of becoming so weak and becoming a bigger burden to my family, to my wife and my caregivers haunts me daily.”
“If you deny coverage for the drug you’ll be causing so much harm to all adult SMA patients. It is a fundamental right in Canada by law to be able to be treated from an illness without discrimination. The province of Quebec understood this and made coverage for this drug available for everyone. Please follow suit.”
“Please don’t sentence me to a death sentence. I need this drug to live an healthier and productive life.”
“I missed out on childhood because of this rare genetic disorder and know that adulthood will be better with this life- changing treatment. We are hoping that CADTH will expand the coverage of Spinraza to include Type 2 and 3 adult patients. I do not want to lose his basic motor skills of eating, breathing, and walking. My health and future, like many other adults with SMA, depend on this treatment. We are witnessing patients in other provinces who have access to this treatment which is improving their quality of life while theirs continue to decline. As Canadians, all patients deserve to have equal access to treatment that will improve their lives.”
“People with SMA are fighting for their life everyday and shouldn't have to fight this hard for an accessible treatment that is proven to work.”
“I want treatment immediately, I am desperate and the coverage is very expensive to and to have funding coverage.”
“If this drug would work to help maintain or improve muscle strength everyone should have access! Progression is going on a faster pace. Delay means more loses: physically, emotionally, socially and financially. Eventually I'll need more of the government services and healthcare system which is going to be costly too. I feel dehumanized by having the treatment out there, but not allowing me to access it although I need it badly. I've been waiting since I was 2 years old when I was first diagnosed.”
“This drug could potentially be the difference between me being able to hold my newborn baby and not being able to. This could potentially be the difference between me being able to feed myself in the future or not being able to. Do the right thing. Do it for all adults living with SMA.’
“Both Spinraza and Risdiplam have changed my life in unimaginable ways. I went from living my life in a steady (and fast) decline to a steady incline. I’m happy, I’m stronger, and most of all I’m so much healthier. I hope that every one in Canada gets access to this treatment, regardless of age, type of SMA, or geographic location.”
“My entire life I have had this. I have seen countless neurologists and doctors who say there's nothing you can do. Suddenly they say, there's something you can do, but you can't afford it. That is the single most frustrating thing I have ever experienced. It is inaccessible and the government doesn't want to pitch in and its cost prohibitive. This is time sensitive. I feel myself deteriorating, any type of messing around on their part, for me that is the difference between keeping what I have and ending up with a lot less than I have.”
“This isn't about being able to walk. This is a revolution in my quality of life that might as well be life-saving. I completely understand why those just recently being born with SMA have been favored to receive drugs like Spinraza. But people need to realize that it's just as life-changing for us adults too. Put me in front of anyone and I'd be happy to explain to them what this means for people like me.”
“Access to this drug, regardless of age or type is essential to the SMA community. I am 34 and have had improvements and stability since starting treatment when I would surely have been experiencing a steep decline.”
“My life is in your hands regarding treatment.”
Conflict of Interest Declaration — Muscular Dystrophy Canada
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
None.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
None.
List any companies or organizations that have provided your group with financial payment over the past 2 years AND who may have direct or indirect interest in the drug under review.
Table 2: Financial Disclosures — Muscular Dystrophy Canada
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Biogen Canada | — | — | — | Xa |
aAll funds were for educational initiatives (e.g., webinars, patient conferences) or for community engagement events; no funds received were directly or indirectly related to drug under review)
I hereby certify that I have the authority to disclose all relevant information with respect to any matter involving this patient group with a company, organization, or entity that may place this patient group in a real, potential, or perceived conflict of interest situation.
Position: Vice-President, Research & Public Policy
Patient Group: Muscular Dystrophy Canada
Date: January 4, 2022
The Love for Lewiston Foundation
About The Love for Lewiston Foundation
As you read this submission our ask is for open hearts, open minds and a willingness to learn first hand the impact of the drug Spinraza. Let us say that we do not envy your role and the difficult job that you have tasked in front of you. As you sift through stacks of paper, documents, medical information, and patience testimonials do not let this letter fall to the bottom of the pile. Do not let this opportunity to change lives and provide hope be missed.
My name is ||||||||||||||||||||||||||||||. My journey with SMA started in 2007 when I said to a friend that I would never want to work with handicap kids cause they disgust me (Feel free to slap me across the face if we ever meet in person). A week later after saying that out loud a little boy named Ishan was put in the middle of my path. He was 6 and living his life in a wheelchair. What is interesting is that looking back on this moment, not for one second did he disgust me. He was the wake up call I needed to start using my voice and to start using it well.
Often we wait our whole life to find our purpose, to find out what work we should be focused on. I am fortunate enough that I found mine in my early 20’s. I cared for not only that little boy but also for his sister. They changed my life and gave me a perspective so great I will never be able to tell them just how thankful I am for the lessons they taught me. After one long day of caring for both this brother and sister team who were living with Spinal Muscular Atrophy (SMA) I got into my car, closed the door, pounded the steering wheel with my fist and started bawling. Hot tears streaming down my face. I bawled uncontrollably because up until that moment I had taken the majority of my life for granted. The ability to walk to the bathroom when I needed, the ability to eat without fear of choking, the simple task of picking my nose when a booger was bothering me. The ability to put myself to bed and simply roll over when I needed to get comfy. It is the smallest of tasks that often are the SMA’s communities biggest challenges.
Years later as these siblings grew up, they would roll down my wedding aisle as ring bearer and a flower girl, they would celebrate the birth of their first child, our second and third. You could say that they were there for the mountain top moments but also in our valleys.
In 2016 they would be present for one of our darkest and hardest battles. They would attend our son Lewiston’s funeral after his passing of the very same rare genetic disease that they lived with. Our second child Lewiston James Olstad would ironically get diagnosed with that very same disease they lived with but instead of SMA 2, our little boy had Spinal Muscular Atrophy Type 1. It took our whole family by surprise. No history of any muscular disease, only the experience of helping a brother and sister for the past 9 years navigate the challenges SMA brings. Lewiston’s life was short, but fighting those 6 months felt like the longest 6 months to our family. Our whole world was rocked. Again I would be reminded of my purpose and that my voice is needed.
Lewiston was just 2 months old when he was diagnosed that sunny summer day August 5, 2016. When our pediatric neurologist delivered the news that she was sorry that there were no treatments available yet and no cure. Their role would be to make Lewiston as comfortable as possible until his death, which they believed would happen prior to him turning one.
Lewiston passed on November 22, 2016 after a courageous fight with SMA.
Four months after his passing, we decided to not have a pity party but to celebrate our son’s first birthday even though he was not with us. We decided to raise money and help those struggling with SMA. Since that choice we have started The Love for Lewiston Foundation and have raised over 1.3 Million dollars for Alberta Children’s Hospital, Critical research and programs and helped provide practical needs like physiotherapy, wheelchairs, and accessibility aids.
Fifteen years ago when I started caring for a brother and sister with SMA there was no hope, no cure, no sign of a life changing drug. The landscape has shifted and we exist to create awareness for the SMA community and provide financial support in areas that are often overlooked as they are deemed “not crucial”. We exist to fight for the SMA Community so we don’t lose another Lewiston.
Approving Gene Therapy drugs like Spinraza or Zolgensma equips those individuals who presented SMA symptoms before gene therapy existed, a fighting chance at becoming contributing members to society and improve their quality of life. Spinraza can turn back the hands of time and give older SMA individuals the energy they once had; the ability to move independently; to feed themselves without assistance; to take a bath; or simply to open their front door.
We watched Lewiston go from a healthy, happy infant to taking his last breath in a matter of months. We have seen two young robust, energetic children go from walking and playing to now fighting for every moment. Scared that a common cold and now Covid could be the one thing that puts them 6 feet under.
I watched my baby boy go from living to fighting for his life in a matter of days. He went from achieving milestones to rapid muscle decline, and completely losing any ability to even hold up his head. We fought hard to try and understand what had happened to our once healthy baby boy. Even told by a Pediatric Neurologist he was 99% certain it was not SMA. His 1% uncertainty led us to press for testing.
Lewiston would require 24 hour care, since he did not have the ability to cough or clear his throat on his own, which could kill him. We spent the majority of his life in the hospital and then Hospice at Alberta Children’s Hospital to ensure proper care. What we know to be true is that Lewiston could still be here. Lewiston could still be here had we been able to access treatment immediately. At the time of Lewiston’s diagnosis in 2016, Spinraza was not available in Alberta. Ontario and BC were the only provinces that had Clinical Trial sites in Canada.
The team at AB Children’s Hospital fought hard to get Lewiston on a clinical trial - he was not stable enough to travel. After climbing through hoops and all of the appropriate signs off were completed, Lewiston became the first person in AB to access Spinraza. Unfortunately it was too late. The deterioration was too significant and he was weakening rapidly after catching a simple cold. Lewiston took his last breath on November 22, 2016 in my arms with my husband by my side.
What hurts and stings to this day is that another infant just weeks apart from Lewiston, with the same diagnosis, with the same deterioration, was able to access this treatment about a month before Lewiston. That little boy is 5 now - this year he will be celebrating his 6th birthday. His life looks totally different. While we still celebrate our son’s birthday we do it without him physically here. We do it as a fundraiser to help those diagnosed with SMA. Imagine having to celebrate your child’s birthday without them there.
I saw Spinraza work. I saw it before my very eyes and in a matter of days, improvement was drastic. I can only imagine what would have happened had he accessed the treatment earlier. Imagine treatment just after birth.
I choose not to sit in the why? Why did this happen. I choose not to stay stuck over this chapter but choose to write a new one. The question our family asks ourselves is not WHY - BUT SO NOW WHAT! We can not rewrite the past but we can write a better future. I want the next chapter we write to be one that says “You Matter”. I want a future that doesn't require hoop jumping and more red tape then Santa would need at Christmas. I dream of a future where treatment options are provided for all.
Spinraza has the ability to change the course of not just infants' lives, but also those inflicted by the disease and suffering from gradual decline. How do I know this. I have seen it time and time again.
Young adults without SMA find the transition from teen to young adulthood incredibly challenging. Now imagine a teen that has been outside of the norm their whole life. Struggling to achieve their dreams with a rare genetic diagnosis and a complete loss of independence. They struggle to fit in, they struggle to find proper accessibility and now they struggle with a transition into adulthood. Our system says the moment you turn 18 you should have it all figured out - when we all know in fact that when we turned 18 we were far from having it all together.
No one wants to feel like a burden to those they love, nevermind feeling like they are a burden in society simply because the world was built without them in mind.
I have witnessed these individuals living with SMA attempt to gain independence while losing theirs. I have watched them try to navigate a grocery store with a wheelchair that doesn’t lift them to the correct shelf height so they can get what they need. I have watched them struggle time and time again. It is easy to sit around a table or a zoom call to deny a drug because of paper evidence. It is easy to deny or make treatment not accessible because you don’t have to look them in the eyes, or understand their challenges and struggles. It is easy to deny access to Spinraza when you do not know the incredible character and depth of this community.
It seems unnecessary that there is a drug that could help those with SMA achieve more independence, regain motor abilities and more importantly have hope for a future. Yet here we are debating if they can access a drug that is life changing. These hurdles I fear will delay the limited resource of time they have left. Will they suffer the same fate as Lewiston or can things change before it's too late for them?
CureSMA and MDC is an ally and has composed testimonies from the SMA community in which we serve. CureSMA and MDC compliment our foundation as they advocate for treatments and policy change on issues to improve the lives of those living with SMA. While Love for Lewiston Foundation works together to identify SMA individuals and families that require funding due lack of support and adequate resources. Please refer to the testimonials submitted by CureSMA and MDC to hear first-hand accounts of our communities experiences with available treatments and ways that Spinraza has changed their lives. As well as with respect to what their experience has been from taking Spinraza.
With my 15 years of experience with this disease I have witnessed 2 adults who have personally accessed this drug because of their insane efforts to privately purchase the drug. Their families have sold investments, added years of work rather than retiring as once planned, taken out 2nd and 3 mortgages to provide an opportunity at a fuller, healthier, more vibrant energetic life. It should not have to be this way. We live in the best country possible. Canada should be leading the way for treatment options.
I have been in the treatment room during the procedure, I have cared post treatment and I have witnessed first hand the incredible gains Spinraza provides them. From increased energy, longer stamina, better head control, a stronger voice, ability to move muscles that once lay limp and a confidence that allowed them to continue to show up when many of us would have wanted to just throw in the towel.
The last few years have been challenging for the whole world. We have been stretched, pushed and then stretched some more when we didn't think it was possible because of a Global Pandemic. Everyone’s mental health has been challenged and tested. Everyone is fighting a battle. We know that to be true. As we go through life battles will be won and lost.
As I fight this battle of changing the landscape of SMA for those living with the disease. I truly believe that this is a battle that we can win and change lives. You have the opportunity to tell the SMA community that they matter. You have the ability to say we will stand in this fight for your life with you and give you every opportunity.
This is my goal, our families goal and our foundation's goal. It is to give those living with this rare disease the opportunity Lewiston did not have. Please partner with us in this fight against SMA and approve Spinraza for those 18 years and older. Let us bring joy, hope and a solid future for this community.
The Love for Lewiston Foundation raises funds and allocates funding to improve the quality of life for individuals living with SMA. We believe that every child should be regularly screened for SMA during routine newborn screening. Should those infants be diagnosed with SMA, then we believe they should have access to life changing drugs like Spinraza or Zolgenzma. We advocate for the implementation of new provincial policies around SMA newborn screening. We advocate for early SMA detection, we advocate for provincial and federal governments to take proactive measures in preventing the onset of this debilitating and life threatening disease.
I personally know and have cared for infants, youth, teens and young adults diagnosed with Spinal Muscular Atrophy. It is brutal. Just imagine having to sit waiting for someone to take you to the bathroom, waiting to get you a tissue so you can simply blow your nose. These are the simplest of daily tasks we all take for granted. It is a disease that robs an individual from everything they once were able to do independently and it cruelly deteriorates the body unless life saving treatment is given.
Our experience with SMA began in 2007 when I volunteered for a family with two children ages 2 and 6 with Spinal Muscular Atrophy. I was their caregiver and helped provide support. I have seen these two intelligent, outgoing, energetic, capable beings go from walking and feeding themselves independently to now managing their energy levels while trying to attend University in a wheelchair in a world that is still very much inaccessible to this community. I have watched these two slowly lose motor functions at an incredibly slow cruel pace. I have been in situations where I didn’t know if they would survive. That cold that is so easily fought off by a healthy immune system, would take them time and time again to the ICU. That cold would interfere with their academic studies and stress their bodies, increasing the deterioration and taxing our health care system.
I’ve witnessed a child’s spine go from straight to having a 70% curvature and having a 25% lung capacity.I have watched them slowly be stripped of their motor skills, no longer able to use their hands and at times and waiting for aids to be able to simple put a glass of water up to their lips. I’ve seen them wet themselves in public as grown young adults simply because they didn't time their water intake for the day properly because they had limited care due to the lack of support. I’ve seen them struggle to get out their doors to get outside to go for a walk or an important appointment. I have seen the toll it took on this once united family, to lead to separation and divorce. I now witness the struggles of these two in adulthood, with the pressures of a pandemic, isolation and working towards their future that at times seems insignificant and unattainable. Why bother, why live. For what? They have contemplated. They have struggled so significantly with mental health, it has felt so consuming they said why even bother being here.
This is what the system is saying to them. YOU DON’T EVEN MATTER.
I built my life on hope. I hope and believe that their fight for independence and a healthy fulfilling life will not be in vain.
Conflict of Interest Declaration — The Love for Lewiston Foundation
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
The Love for Lewiston Foundation was notified about the CADTH submission from Biogen. They ask |||||| to contribute and share her experience with SMA as she is the Executive Director of the Love for Lewiston Foundation; was a pillar in getting Newborn Screening funded & approved in Alberta; had a son who passed from complications due to SMA type 1 (received Spinraza); and was a personal care volunteer for two SMA type 2 children.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
Table 3: Conflict of Interest Declaration for The Love for Lewiston Foundation
Company | $0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 |
|---|---|---|---|---|
Biogen | — | X | — | — |
Novartis | — | — | X | — |
Blakes | 0 | — | — | — |
BeSpoke | 0 | — | — | — |
Radical Gentleman Creative | 0 | — | — | — |
Southbase Creative | 0 | — | — | — |
Wrinkle & Crease | — | X | — | — |
Leading Outdoor | 0 | — | — | — |
Tourmaline Oil Corp | X | — | — | — |
Burnet, Duckworth & Palmer LLP | X | — | — | — |
RiverWalk Dental | X | — | — | — |
Marvel Cabinetry | X | — | — | — |
Hesco | X | — | — | — |
Lux Windows | X | — | — | |
WINDY CREEK FARSM INC. | X | — | — | — |
Pinnacle Foods | X | — | — | — |
Blue Rock Construction | X | — | — | — |
Mercedes Benz Country Hills | X | — | — | — |
Dansons | — | X | — | |
CES Energy | X | — | — | — |
WINDY CREEK FARSM INC. | X | — | — | — |
10 Foot Henry | X | — | — | — |
Cardel Homes | X | — | — | — |
Dream Homes | X | — | — | — |
I hereby certify that I have the authority to disclose all relevant information with respect to any matter involving this patient group with a company, organization, or entity that may place this patient group in a real, potential, or perceived conflict of interest situation.
Position: Executive Director
Patient Group: Love for Lewiston Foundation
Date: Jan 11, 2022
Clinician Input
The Neuromuscular Disease Network for Canada
About The Neuromuscular Disease Network for Canada
The Neuromuscular Disease Network for Canada (NMD4C) is the new pan-Canadian network that brings together the country’s leading clinical, scientific, technical, and patient expertise to improve care, research, and collaboration in neuromuscular disease.
Launched in January 2020 with funding from the Canadian Institutes of Health Research (CIHR) and Muscular Dystrophy Canada (MDC), NMD4C builds on existing national initiatives such as the Canadian Neuromuscular Disease Registry (CNDR), the Canadian Pediatric Neuromuscular Group (CPNG), and the former neuromuscular network CAN-NMD. The mission of NMD4C is to improve the care, research and treatment of NMDs for all Canadians. Its vision is to be a comprehensive, inclusive, open and enduring network through which Canadian stakeholders can share expertise and data and collaborate on joint activities and research for the benefit of Canadian patients.
The network’s goals are to:
Formalize and sustain a network of NMD stakeholders united around a cohesive three-year work plan
Train and educate the next generation of NMD stakeholders (clinicians, scientists, and patient advocates)
Raise the standard of care for NMD and access to therapies across Canada
Strengthen biomedical and clinical infrastructure to build research capacity in Canada
Information Gathering
Clinicians with experience treating SMA, including clinicians with experience with nusinersen were asked to contribute to this submission. These expert clinicians contribute to the knowledge of SMA and its treatments and are involved in clinical and observational research, clinical guidelines development and health technology assessment. The clinicians contributing herein are familiar with the data from clinical trials on treatments for SMA, and, specifically, for nusinersen. In section 7, Canadian real world data has been provided by a Canadian physician member of NMD4C.
Current Treatments
5q SMA (hereafter referred to as SMA) is a genetic disorder caused by biallelic mutations in the SMN1 gene. There are different clinical subtypes of SMA based on age of onset, function, and outcome. The most common subtype is SMA Type 1, which presents with symptoms in the first 6 months of life, and untreated results in death typically before the age of 2. SMA subtype is correlated with the number of genetic copies of a modifier gene called SMN2. Both SMN1 and SMN2 encode for SMN protein. Patients with SMA Type 1 most commonly have 2 copies of SMN2, while patients with Type 2 typically have 3 copies, though can have 2 or 4. Therapeutic strategies for SMA have centred on increasing the amount of SMN protein, either by acting on SMN2 (Spinraza, Risdiplam) or SMN1 (Zolgensma).
Nusinersen (Spinraza), has been approved by Health Canada since July 2017 for the treatment of 5q spinal muscular atrophy (SMA). It is a synthetic anti-sense oligonucleotide (a type of genetic material) that enables the SMN2 gene to produce more full-length SMN protein thereby correcting the molecular abnormality of the disease which is necessary to help relieve the symptoms of the disease. If commenced early enough, it is possible that it may prevent the severe loss of motor neuron function and profound progressive weakness. This treatment has been recommended for reimbursement by Canadian Drug Expert Committee CDEC and as such most children with SMA are receiving nusinersen as a part of their treatment regimen. Nusinersen is injected into the spinal fluid every four months after an initial four loading doses that occur closer together during the first two months of treatment. The procedure is typically done under sedation at an experienced pediatric centre.
One main study of nusinersen, involving 121 babies (of an average age of 7 months at treatment onset) with SMA, showed that it is effective in improving motor function, reducing the need for assisted ventilation, and greatly extending survival, when compared to placebo (sham injection).
SMA-treating clinicians consider the approval of nusinersen to be a significant advancement in the treatment of patients with SMA.
In Quebec nusinersen was re-evaluated by INESSS in 2018 and subsequently recommended for expanded access beyond paediatric Type 1 patients to include Type 2 and Type 3 patients of any age and regardless of ambulatory status.
Onasemnogene abeparvovec (Zolgensma) is an adeno-associated virus (AVV) vector-based gene therapy for the treatment of children less than 2 years of age with spinal muscular atrophy (SMA) with bi-allelic mutations in the survival motor neuron 1 (SMN1) gene and with two copies of SMN2 gene. This treatment is currently under review by the Common Drug Review and has been granted Priority Review status by Health Canada. There is a managed access plan globally and a very limited number of Canadian children have been able to access this drug via that pathway.
Onasemnogene abeparvovec is a one-time intravenous infused treatment that has been shown to reduce the need for assisted ventilation and extend survival in infants with SMA type 1. It has also been shown to help improve motor developmental, including sitting without support for at least 30 seconds, a motor milestone not obtained in untreated babies with SMA type 1.
Risdiplam (Evrysdi), in August 2021, received a positive recommendation for reimbursement by CADTH for the treatment of SMA in patients 2 months and older provided that eligible patients are under the care of a specialist in the diagnosis and management of SMA, and that risdiplam not be used in combination with Onasemnogene abeparvovec or nusinersen. Further, CADTH recommended that risdiplam should only be reimbursed to treat patients aged 2 months to 7 months with genetic documentation of 2 or 3 copies of the survival motor neuron 2 (SMN2) gene or non-ambulatory patients aged 8 months to 25 years with genetic documentation of 2 or 3 copies of the SMN2 gene. Patients are ineligible if they currently require permanent invasive ventilation.
Treatment Goals
For patients with early onset SMA, preservation of motor neurons, improving survival, improving motor function, delaying or alleviating the need for assisted ventilation, delaying or alleviating loss of ability to speak, and delaying other secondary complications (such as failure to thrive, scoliosis, recurrent pulmonary infections, etc.), and reducing burden on caregivers are goals that new treatments would ideally address.
For individuals with late onset SMA, treatment goals would be to maintain current level of motor function and strength (prevent further loss of motor function), achieve disease stabilization (prevent disease progression, including avoidance of need for ventilation), promote independence, and improve overall health-related quality of life.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
Adult SMA patients have a high unmet need for treatment.
Also, while treatments that address the root cause of SMA by producing the missing protein are changing care, other therapies that help improve muscle functions are greatly needed.
Which patients have the greatest unmet need for an intervention such as the drug under review?
Adult SMA patients - particularly those above the age of 25 - have a high unmet need for treatment. While we recognize that there is limited evidence available for assessment of this population, real-world data have expanded our knowledge on safety and efficacy of the drug in a much larger population of SMA patients than those reported in the pivotal studies.
NM4DC points CADTH to a paper whereby a critical review was conducted on the literature reporting real world data on motor function in type 2 and 3 patients (n=659) treated with Nusinersen: Motor function in type 2 and 3 SMA patients treated with Nusinersen: a critical review and meta-analysis (Coratti 2021). That review highlights that improved motor function can be observed in all the type 2 and 3 cohorts of nusinersen treated patients including adults, in contrast to the negative changes found in studies reporting untreated cohorts. This held true, with very few exceptions, both when considering the overall results of the studies in heterogeneous cohorts or smaller groups subdivided according to age, type or functional status.
Further, in section 7 (Additional Information) of this clinician input submission, a report detailing the clinical real world experience of Dr. Xavier Rodrigue is provided. Dr. Rodrigue provides an analysis of 12 patients who initiated nusinersen after the age of 17 and have had at least 12 months of follow-up post-nusinersen initiation. Functional testing, respiratory testing, and patient-reported outcomes were performed at each clinical visit where possible. In this analysis of Canadian adult patients there is demonstrated positive benefits of nusinersen in either stabilization or functional gains regardless of age of therapy initiation, ambulatory status, spinal fusion status, or SMA type/SMN2 copy number.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Providing access to nusinersen to adult type 2 & type 3 patients older than 18 years of age (regardless of ambulatory status) will position many provinces to catch up with Quebec and over 43 other countries where this treatment is available for a broad population of SMA patients.
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective.
There is only one other treatment approved for (some) adults, and that is risdiplam for patients 18-25 years of age. Currently available data does not suggest that risdiplam works better in these adults than nusinersen. Convenience or patient preference may be reasons for some adult patients to try risdiplam as the first treatment for SMA.
How would this drug affect the sequencing of therapies for the target condition?
While both compounds (risdiplam and nusinersen) work through more efficient splicing of SMN2 into SMN protein, they have different modes of action on how they increase the SMN protein levels in the cell. They also have different routes of administration, different distribution in the body, and different tolerability.
If one of these drugs does not have the desired clinical effect, it is possible that the other drug may have the desired clinical effect.
Both sequences - i) risdiplam first, then nusinersen ii) nusinersen first, then risdiplam - would be reasonable treatment sequencing approaches.
Which patients would be best suited for treatment with the drug under review?
For many adults living with SMA, their life course with the disease is associated with progressive loss of motor function, leading to deterioration in health and reduced independence. Stabilization of the disease can mean retention of a vital motor function(s), avoidance of ventilator dependency, continued ability to speak and swallow, and survival. For some patients, this translates into preservation of functional independence. As such, Nusinersen would be appropriate for adult type II & type III patients older than 18 years of age (regardless of ambulatory status).
How would patients best suited for treatment with the drug under review be identified?
The diagnosis is secured through genetic testing and confirmed by the absence of normal copies of the SMN1 gene. A major disease modifier is the number of SMN2 gene copies, with fewer copies associated with earlier-onset and more severe SMA.
Which patients would be least suitable for treatment with the drug under review?
Patients least suitable for treatment with the drug under review:
patients who have contraindications to the drug or the procedure, or difficulty of lumbar punctures
patients who have clearly not benefited or markedly deteriorated under the treatment for a reasonable time
bed-ridden, fully ventilated patients
asymptomatic patients
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review?
While it may be difficult to accurately identify (adult) patients who are most likely to derive benefit from nusinersen, in general, younger patients should benefit more. It is important to define a therapeutic goal together with the patient at the start of treatment, with treatment response assessed at regular intervals (see 6.10).
What outcomes are used to determine whether a patient is responding to treatment in clinical practice?
Outcomes tests vary according to age and functional state and include lung function (Forced Vital Capacity), Revised Upper Limb Module (RULM) and 6-Minute-Walk-Test (6MWT). In small children also there are tests that have different sensitivity with respect to the achievement of motor milestones. Revised Hammersmith Scale (RHS) is a specifically designed outcome measure for people affected by SMA. However, because that scale might lack sensitivity and has “floor effect” the 32-item Motor Function Measure (MFM32) may be a preferred measure in younger individuals particularly aged 2–5 years, and in non-ambulant individuals with Types 2 or 3 SMA, aged 2–25 years. However, in Canadian clinical practice the RHS is widely used and accepted as the tool to measure motor function. The tests will vary according to age and functional state and include lung function (Forced Vital Capacity), Revised Upper Limb Module (RULM) and 6-Minute-Walk-Test (6MWT). In small children tests will also vary on whether they achieve motor milestones. These measures require trained practitioners and are outside of the bounds of a traditional clinic visit.
Clinician experts generally agree that Patient Reported Outcomes (PRO) instruments would be useful, however, while there are no internationally validated and agreed upon PRO instruments yet, data is being developed to inform the selection of such a measure.
In 2020, Canadian experts in adults with SMA undertook a modified Delphi process exercise to determine a consensus-based recommendation for outcomes measures to be used in adults with SMA at different functional stages. Through the CNDR and the NMD4C, it is anticipated that all clinics can prospectively collect such measures, allowing a rich pool of real-world outcomes data. Below is the abstract from the manuscript published in the Journal of Neuromuscular Diseases (J Neuromuscul Dis 2021;8:579-588)
What would be considered a clinically meaningful response to treatment?
In adults: stabilisation of motor and respiratory function, less disability with maintenance of independence and fewer hospitalisations. Maintaining ability to speak and avoiding need for ventilation support have profound impacts on patient quality of life, autonomy, ability to maintain vocational and social roles.
How often should treatment response be assessed?
With new therapies for SMA being introduced into clinical practice, it is important to monitor their effectiveness and to collect evidence to help determine which therapy should be chosen for any given patient. However, the current provincial government monitoring requirements are too frequent and there is significant variability between the provinces. In addition, quantitative outcome measures require specially trained practitioners, are time consuming, and are not currently covered with provincial funding as part of the expense of therapy.
A group of Canadian neuromuscular disease specialists, most of whom are involved in NMD4C, have written a letter to the provincial governments outlining their concerns and recommending an alternative timeline for outcome measurements in patients receiving SMA therapies. This content has also been published in the Canadian Journal of Neurological Sciences (Can J Neurol Sci. 2021 Mar;48(2):201-203. PMID: 32713403)
What factors should be considered when deciding to discontinue treatment?
Factors to consider when deciding to discontinue treatment:
Accelerated deterioration in clinical status while on nusinersen for at least 12-18 months
Allergic reaction and critical SAEs.
What settings are appropriate for treatment with the drug under review?
Intrathecal therapy with nusinersen should be administered at designated treatment centers by or under the direction of a qualified healthcare provider (HCP) experienced in performing lumbar punctures.
For non-oncology drugs, is a specialist required to diagnose, treat, and monitor patients who might receive the drug under review?
Neurologists/Physiatrists are specialists who lead the diagnosis, treatment and monitoring of persons with SMA, typically in an interdisciplinary specialized clinic.
Additional Information
Dr. Xavier Rodrigue is a physiatrist at the Institut de réadaptation en déficience physique de Québec, Centre intégré universitaire de santé et de services sociaux de la Capitale-Nationale (CIUSSSCN). Dr. Rodrigue has been treating SMA patients for 8 years, and currently sees 27 adult patients with spinal muscular atrophy at his clinic: 16 being treated with nusinersen, 7 being treated with risdiplam, and 4 patients not receiving drug therapy.
The clinician group provided anecdotal reports of 12 Canadian patients from Quebec with type II and III SMA. The CADTH clinical team has reviewed these reports but are not included in this report as they do not meet the review protocol.
Conflict of Interest Declarations — The Neuromuscular Disease Network for Canada
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
No. This submission was completed exclusively by NMD4C.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No. Information and data was analyzed with researchers and clinicians associated with NMD4C and the Canadian Neuromuscular Disease Registry (CNDR).
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician who contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
Declaration for Clinician 1
Name: Hanns Lochmüller
Position: Senior Scientist, Professor of Neurology
Date: 04-01-2022
Table 4: Conflict of Interest Declaration for NMD4C — Clinician 1
Company | Check appropriate dollar range | |||
|---|---|---|---|---|
$0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In excess of $50,000 | |
Biogen | — | X | — | — |
Roche | — | X | — | — |
Declaration for Clinician 2
Name: Victoria Hodgkinson, PhD
Position: Scientific Director, Canadian Neuromuscular Disease Registry
Date: 22-12-2021
Table 5: Conflict of Interest Declaration for NMD4C — Clinician 2
Company | Check appropriate dollar range | |||
|---|---|---|---|---|
$0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In excess of $50,000 | |
Novartis | X | — | — | — |
Biogen | X | — | — | — |
Roche | X | — | — | — |
Declaration for Clinician 3
Name: Xavier Rodrigue
Position: Physiatre, Institut de réadaptation en déficience physique de Québec, CIUSSSCN
Date: 22-12-2021
Table 6: Conflict of Interest Declaration for NMD4C — Clinician 3
Company | Check appropriate dollar range | |||
|---|---|---|---|---|
$0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In excess of $50,000 | |
Novartis | X | — | — | — |
Biogen | X | — | — | — |
Roche | X | — | — | — |
Declaration for Clinician 4
Name: Aaron Izenberg
Position: Neurologist, Sunnybrook Health Sciences Centre
Date: Jan 5, 2022
Table 7: Conflict of Interest Declaration for NMD4C — Clinician 4
Company | Check appropriate dollar range | |||
|---|---|---|---|---|
$0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In excess of $50,000 | |
Biogen | — | X | — | — |
Roche | — | X | — | — |
Mitsubishi Tanabe | X | — | — | — |
Alnylam | X | — | — | — |
Takeda | X | — | — | — |
Alexxion | X | — | — | — |
Amylyx | X | — | — | — |
Declaration for Clinician 5
Name: Jean K. Mah
Position: Pediatric Neurologist, Alberta Children’s Hospital, Calgary, Alberta
Date: 30-12-2021
Table 8: Conflict of Interest Declaration for NMD4C — Clinician 5
Company | Check appropriate dollar range | |||
|---|---|---|---|---|
$0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In excess of $50,000 | |
Biogen – Research grant for clinical trial | — | — | X | — |
Biogen – Consulting / Speakers fee | X | — | — | — |
Roche – Research grant for clinical trial | — | — | X | — |
Roche – Consulting fee | X | — | — | — |
Declaration for Clinician 6
Name: Colleen O’Connell
Position: Medical Director, Stan Cassidy Centre for Rehabilitation, Fredericton New Brunswick
Date: 29-12-21
Table 9: Conflict of Interest Declaration for NMD4C — Clinician 6
Company | Check appropriate dollar range | |||
|---|---|---|---|---|
$0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In excess of $50,000 | |
Biogen | X | — | — | — |
Roche | — | — | X | — |
Declaration for Clinician 7
Name: Dr. Jiri Vajsar
Position: Neurologist, The Hospital for Sick Children, Toronto
Date: January 3, 2022
Table 10: Conflict of Interest Declaration for NMD4C — Clinician 7
Company | Check appropriate dollar range | |||
|---|---|---|---|---|
$0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In excess of $50,000 | |
Novartis | X | — | — | — |
Declaration for Clinician 8
Name: Jodi Warman Chardon
Position: Neurologist, The Ottawa Hospital/Children’s Hospital of Eastern Ontario
Date: Jan 2, 2022
Table 11: Conflict of Interest Declaration for NMD4C — Clinician 8
Company | Check appropriate dollar range | |||
|---|---|---|---|---|
$0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 9
Name: Kerri Schellenberg
Position: Associate Professor, Neurology. University of Saskatchewan
Date: 05-01-2022
Table 12: Conflict of Interest Declaration for NMD4C — Clinician 9
Company | Check appropriate dollar range | |||
|---|---|---|---|---|
$0 to $5,000 | $5,001 to $10,000 | $10,001 to $50,000 | In excess of $50,000 | |
Biogen | X | — | — | — |
Roche | X | — | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.