CADTH Reimbursement Review
Ospemifene (Osphena)
Sponsor: Duchesnay Inc.
Therapeutic area: Dyspareunia, vaginal dryness
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
ANCOVA
analysis of covariance
AE
adverse event
BMI
body mass index
CI
confidence interval
CrI
credible interval
DHEA
dehydroepiandrosterone
DVT
deep vein thrombosis
FSFI
Female Sexual Function Index
HRQoL
health-related quality of life
ITC
indirect treatment comparison
ITT
intention to treat
LOCF
last observation carried forward
LS
least squares
LTSE
long-term safety extension
MBS
most bothersome symptom
MD
mean difference
MI
Maturation Index
MID
minimal important difference
mITT
modified intention to treat
MMRM
mixed-effects model for repeated measures
NMA
network meta-analysis
PP
per protocol
RCT
randomized controlled trial
RR
relative risk
SAE
serious adverse event
SD
standard deviation
SE
standard error
SERM
selective estrogen receptor modulator
SOGC
Society of Obstetricians and Gynaecologists of Canada
TEAE
treatment-emergent adverse event
UDI-6
Urinary Distress Inventory – Short Form
UTI
urinary tract infection
VVA
vulvar and vaginal (or vulvovaginal) atrophy
WHC
Women’s Health Coalition Alberta
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Ospemifene (Osphena), 60 mg tablets for oral administration |
Indication | Indicated in post-menopausal women for the treatment of moderate to severe dyspareunia and/or vaginal dryness, symptoms of vulvar and vaginal atrophy, a component of genitourinary syndrome of menopause |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | July 16, 2021 |
Sponsor | Duchesnay Inc. |
NOC = Notice of Compliance.
Introduction
Hormonal changes, particularly a decrease in estrogen, are often associated with signs and symptoms in post-menopausal women that have an impact on health-related quality of life (HRQoL) as well as physical, mental, and sexual health.1 Many post-menopausal women experience vulvar and vaginal atrophy (also called vulvovaginal atrophy) (VVA) due to physiologic changes in the female genital anatomy that result from aging and a lack of ovarian estrogen production in menopause. Women with VVA will typically present with vaginal dryness, pruritis, burning, pain, or dyspareunia as self-reported symptoms. Genitourinary syndrome of menopause is a newer, broader term that encompasses VVA as well as other genitourinary symptoms and may not be limited to patients who are sexually active.2 Symptoms of genitourinary syndrome of menopause can be grouped as genital symptoms, sexual symptoms, and urinary symptoms.1 While no report of incidence of VVA among Canadians is available, a study that included 1,016 Canadians reported a prevalence of 34%3; however, the prevalence of VVA is likely significantly underreported because many patients assume that the symptoms experienced during menopause are normal changes associated with aging. Previous literature suggests that 60% to 90% of post-menopausal patients may suffer from VVA and experience significant deficits in their quality of life because of it.4
The Society of Obstetricians and Gynaecologists of Canada (SOGC) recommends vaginal lubricants or moisturizers as first-line management options for genitourinary syndrome of menopause, particularly if the patient’s concerns are limited to vaginal dryness or dyspareunia.2 Treatment for VVA typically includes estrogen hormonal therapies administered vaginally as creams, tablets, capsules, or a ring. The clinical expert consulted for this review indicated that the majority of women do not get adequate relief from vulvovaginal symptoms from systemic estrogen alone; local vaginal estrogen is still required.
The drug under review by CADTH is ospemifene 60 mg tablets for oral administration.5 Ospemifene is a selective estrogen receptor modulator (SERM) that binds to estrogen receptors, eliciting both antagonistic and agonistic effects and increasing the cellular maturation and mucification of the vaginal epithelium.5 In Canada, ospemifene is indicated in post-menopausal women for the treatment of moderate to severe dyspareunia and/or vaginal dryness, which are symptoms of VVA, a component of genitourinary syndrome of menopause. The sponsor has requested that ospemifene be reimbursed according to the approved Health Canada indication. The objective of this CADTH Reimbursement Review is to perform a systematic review of the beneficial and harmful effects of ospemifene (60 mg) for the treatment of moderate to severe dyspareunia and/or vaginal dryness, which are symptoms of VVA in post-menopausal women.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient groups that responded to CADTH’s call for patient input and from a clinical expert consulted by CADTH for the purpose of this review.
Patient Input
Input was received from 1 patient group, Women’s Health Coalition Alberta (WHC). WHC advocates, raises awareness, and provides education about the urogynecological and reproductive health of patients of all ages. WHC noted the overall lack of awareness and understanding of urogynecological health, the limited therapeutic options for peri- and post-menopausal conditions (e.g., post-menopausal VVA), and the potential inequity in accessing preferred treatments when they are not reimbursed by public drug plans. WHC emphasized that the clinical and psychological impacts caused by untreated menopausal conditions are often overlooked and dismissed. Further, it expressed the expectation that a suitable treatment option for patients would improve their health outcomes and potentially raise clinician awareness of the importance of treating menopausal conditions.
To provide additional background on lived experience, values, and preferences of patients with VVA, patient group websites were searched for information about original experiences of patients with VVA. Healthtalk.org is a non-profit organization that has collected hundreds of stories from patients with any health condition.6 Information from video interviews with 13 British patients about VVA was available through Healthtalk.org, and obtained, assessed, and synthesized by the CADTH review team. The interviewed patients reported vaginal dryness, decline in libido (contributing to a decline in sexual activity), and urinary problems as some of the common complications they experienced after entering menopause. Interviewed patients also described the importance of sex in a relationship and how decreased sexual activity attributed to VVA symptoms may add significant complications to a relationship over time. In the interviews, some patients indicated they were made aware of the lack of knowledge regarding the effects of hormone replacement therapies, and of the fact that treatment with such therapies may not prevent thinning of the vaginal wall. The thinning of vaginal tissue was reported as causing severe discomfort for many patients, resulting in vaginal tears and bleeding. Patients also described how the decline in estrogen they had experienced affected the pelvic floor, bladder, uterus, vagina, or bowel, sometimes leading to urinary and bowel problems. Patients also reported difficulties with incontinence. Negative impacts on quality of life were experienced by many patients.
Clinician Input
Input From Clinical Experts Consulted by CADTH
The following input was provided by a clinical specialist with expertise in the diagnosis and management of post-menopausal patients with VVA.
The clinical expert indicated that women with VVA will typically present with vaginal dryness, pruritis, burning, pain, or dyspareunia; the goal of treatment is to provide relief of these symptoms. In the experience of the clinical expert, in most cases, currently available treatments are effective in providing relief of VVA symptoms. Because most available treatments for the symptoms of VVA are administered intravaginally, ospemifene offers an alternative route of administration, as an orally administered tablet; however, the clinical expert also noted that some patients may prefer a local therapy over systemic therapy due to hesitancy around the use of hormonal treatments.
The clinical expert consulted for this review noted that despite the utility of vaginal moisturizers and lubricants, women with VVA will generally experience more effective symptomatic relief from local vaginal estrogen. The clinical expert noted that additional therapeutic options have recently become available for the treatment of genitourinary syndrome of menopause, such as intravaginal prasterone and orally administered ospemifene (a SERM). These treatments represent a departure from traditional estrogen-based management strategies and substantially widen the scope of options available to women with VVA. The clinical expert relayed that the main adverse effect of ospemifene is hot flashes, which may be a significant barrier to widespread use in women with post-menopausal symptoms. The clinical expert suggested that taking this and other factors into account, it is unlikely that ospemifene will become first-line therapy.
The clinical expert consulted for this review felt that the majority of patients with genitourinary syndrome of menopause are anticipated to benefit from a therapeutic drug with estrogen receptor agonist properties, such as ospemifene. Feedback from the expert indicated that the patients most in need of intervention are those with more severe symptoms, and ospemifene provides an additional option versus traditional vaginal estrogen therapy. Additionally, the clinical expert felt that the oral route of administration for ospemifene may be especially suited for women who are unable to self-administer vaginal medication, such as due to severe pain or mobility limitations.
The clinical expert stated that women who do not report symptoms would generally not be diagnosed with VVA in clinical practice, and that patients will generally self-identify based on their description of symptoms (clinical history). Alternatively, patients seen for urogynecological issues, such as vaginal prolapse or urinary incontinence, may have VVA identified by history and visual inspection. The expert noted that clinical history and visual inspection of the vulva during a physical exam would be the usual methods for identifying patients with VVA; however, given the rise in telemedicine, the expert anticipated that more diagnoses will be made based on clinical history alone, and that this may also be considered a reasonable approach.
The clinical expert did not identify a specific subgroup of patients who would be less suited for treatment with ospemifene beyond those with any contraindication to ospemifene.
The clinical expert consulted by CADTH indicated that in clinical practice, subjective patient-reported improvement in symptoms is the primary outcome used to determine whether a patient is responding to treatment. The expert noted that improvement of symptoms is most often correlated with visual inspection at examination, although subjective symptoms are clinically more meaningful than appearance. Moreover, any improvement in vulvovaginal symptoms would be considered a clinically meaningful response, according to the clinical expert, who noted that this may include decrease in the sensation of vaginal dryness, decreased vaginal burning/pain, decreased frequency of urinary tract infections or bladder urgency or irritation, and decreased dryness and pain during intercourse. Histologic examination is generally not performed or required, based on the experience of the clinical expert.
Based on feedback from the clinical expert, there is no strict schedule for when treatment response needs to be assessed. The expert suggested that it would be reasonable to assess response approximately 3 months to 6 months after initiating treatment, then at 6 months to 12 months, and yearly thereafter.
Regarding discontinuation of treatment, the clinical expert stated that a patient may discontinue treatment if they wish, though symptoms may return after some time. They noted that the assessment of the risks and benefits is subjective, given that the condition is ultimately a quality of life issue.
The clinical expert indicated that ospemifene would most likely be prescribed in an outpatient ambulatory clinic setting by a family physician or gynecologist, with patients self-administering the medication at home. Because genitourinary syndrome of menopause is very common, the clinical expert believed that a vast majority of clinicians with experience in the treatment of women’s health issues would be suitable prescribers for pharmacologic treatment.
Clinician Group Input
Input from clinician groups was not received for the review of ospemifene.
Drug Program Input
Input was obtained from the drug programs that participate in the CADTH reimbursement review process. The following were identified as key factors that could potentially affect the implementation of a CADTH recommendation for ospemifene:
considerations for initiation of therapy
considerations for continuation or renewal of therapy
considerations for discontinuation of therapy
considerations for prescribing of therapy
generalizability of trial populations to the broader populations in the jurisdictions
system and economic issues.
Clinical Evidence
Pivotal Studies and Protocol Selected Studies
Description of Studies
A total of 5 phase III, double-blind, placebo-controlled, randomized controlled trials (RCTs) that assessed ospemifene 60 mg were included in the systematic review: Study 310 (N = 544, excluding the ospemifene 30 mg treatment group), Study 821 (N = 919), Study 231 (N = 631), Study 718 (N = 426), and Study 310X (N = 118 patients who continued from Study 310). Studies 310, 821, and 231 were designed to assess the efficacy and safety of ospemifene 60 mg over 12 weeks; Study 718 was designed to assess the efficacy and long-term safety of ospemifene 60 mg over 52 weeks; and Study 310X was a 52-week, long-term safety extension (LTSE) of Study 310 that assessed only safety outcomes. Most of the trials were conducted from 2006 to 2009 (studies 310, 821, and 718); Study 231 was conducted between 2016 and 2017. The trials primarily recruited patients in the US. No patients were studied in Canada. All of the studies enrolled post-menopausal women between 40 years and 80 years of age who had 5% or fewer superficial cells in the Maturation Index (MI) of the vaginal smear and a vaginal pH level greater than 5.0. In addition, studies 310, 821, and 231 included patients who identified at least 1 moderate to severe symptom of VVA that was considered the most bothersome.
Studies 310, 821, 231, and 718 included the following as co-primary end points assessed at week 12: percentage of vaginal superficial and vaginal parabasal cells on a vaginal smear and vaginal pH. Studies 310, 821, and 231 also included severity of the most bothersome symptom (MBS) of VVA as a co-primary end point. Secondary end points assessed in the 12-week studies included urinary symptoms using the Urinary Distress Inventory – Short Form (UDI-6) and sexual function (studies 821 and 231) using the Female Sexual Function Index (FSFI). HRQoL, mental health-related outcomes, bone mineral density, and adherence were identified as outcomes of interest to this review, but were not assessed in any of the included studies. The majority of patients included in studies 310, 821, 231, and 718 were 55 years of age and older and White. The proportion of patients who had previous experience with hormonal treatment varied significantly between the studies, ranging from 3% to 61% of patients. Of the 544 patients in Study 310, 222 (41%) reported vaginal dryness as the MBS and 242 (44%) reported vaginal pain with sexual activity (dyspareunia) as the MBS. In Study 821, 314 (34%) of patients reported vaginal dryness as their MBS and 605 (66%) reported dyspareunia as their MBS. Study 231 required patients to have vaginal dryness as their MBS. Study 718 did not report assessments of MBS at baseline. Baseline characteristics for Study 310X were limited to demographic information.
Efficacy Results
The efficacy of ospemifene was presented by 4 of the 5 included studies (all except Study 310X). A summary of key efficacy results is provided in Table 2.
Table 2: Summary of Key Efficacy Results From Pivotal and Protocol Selected Studies
Outcome | Study 310 | Study 821 (dryness stratum) | Study 821 (dyspareunia stratum) | Study 231 | Study 718 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
OSP 60 mg N = 276 | PBO N = 268 | OSP 60 mg N = 463 | PBO N = 456 | OSP 60 mg N = 463 | PBO N = 456 | OSP 60 mg N = 313 | PBO N = 314 | OSP 60 mg N = 363 | PBO N = 63 | |
Change from baseline in vaginal dryness as the MBS of VVAa | ||||||||||
N | 118 | 104 | 160 | 154 | NA | NA | 277 | 281 | NR | NR |
Baseline, mean (SD) | 2.42 (0.56) | 2.38 (0.51) | 2.5 (0.50) | 2.5 (0.50) | NA | NA | 2.53 (0.50) | 2.54 (0.50) | NR | NR |
Week 12, mean (SD) | 1.15 (0.98) | 1.55 (1.03) | 1.2 (1.03) | 1.4 (1.03) | NA | NA | NR | NR | NR | NR |
Change, mean (SD) | –1.26 (1.03) | –0.84 (1.00) | –1.3 (1.08) | –1.1 (1.02) | NA | NA | –1.29 (1.01) | –0.91 (0.96) | NR | NR |
OR (95% CI); P value, OSP vs. PBO | NA | — | NA | — | NA | NA | 2.23 (95% CI, 1.62 to 3.06); P < 0.0001 | — | NR | NR |
CMH P value, OSP vs. PBO | 0.021 | — | 0.080 | — | NA | NA | NA | — | NR | NR |
Change from baseline in dyspareunia as the MBS of VVAa | ||||||||||
N | 120 | 122 | NA | NA | 303 | 302 | NR | NR | NR | NR |
Baseline, mean (SD) | 2.6 (0.7) | 2.7 (0.6) | NA | NA | 2.7 (0.47) | 2.7 (0.49) | NR | NR | NR | NR |
Week 12, mean (SD) | 1.4 (1.2) | 1.8 (1.2) | NA | NA | 1.1 (1.08) | 1.5 (1.16) | NR | NR | NR | NR |
Mean change (SD) | –1.2 (1.3) | –0.9 (1.1) | NA | NA | –1.5 (1.08) | –1.2 (1.12) | NR | NR | NR | NR |
CMH P value, OSP vs. PBO | 0.023 | — | NA | NA | 0.0001 | — | NR | NR | NR | NR |
Change from baseline in percentage of parabasal cellsb,c | ||||||||||
N | 272 | 261 | 160 | 151 | 303 | 302 | 216 | 233 | 363 | 63 |
Baseline, mean (SD) | 39.3 (38.98) | 38.5 (37.60) | 45.9 (40.70) | 45.6 (40.54) | 51.1 (38.21) | 50.6 (39.87) | 25.8 (33.3) | 28.3 (33.1) | NA | NA |
Baseline, median (range) | NA | NA | NA | NA | NA | NA | NA | NA | 50 (0 to 100) | 48 (0 to 100) |
Week 12, mean (SD) | 8.78 (19.31) | 42.7 (37.22) | 14.2 (27.27) | 42.2 (36.47) | 11.0 (21.86) | 50.6 (38.81) | NR | NR | NA | NA |
Week 12, median (range) | NA | NA | NA | NA | NA | NA | NA | NA | 0 (0 to 100) | 70 (0 to 100) |
Change, mean (SD) or LS mean (SE)j | –30.1 (37.93) | 3.98 (35.21) | –31.7 (2.11)j | –3.9 (2.18)j | –40.3 (1.56)j | –0.4 (1.57)j | –23.7 (1.4)j | –1.9 (1.4)j | NA | NA |
Change, median (range; 95% distribution-free CI) | NA | NA | NA | NA | NA | NA | NA | NA | –40 (–100 to 75; –55.0 to –30.0) | 0 (–90 to 98; 0.0 to 10.0) |
Mean difference (95% CI), OSP vs. PBO | NA | — | –27.8 (–33.75 to –21.90) | — | –39.9 (–44.15 to –35.63) | — | –21.8 (–25.7 to –18.0) | — | NA | NA |
P value, OSP vs. PBO | < 0.001,d < 0.001e | — | < 0.0001 | — | < 0.0001 | — | P < 0.0001 | — | < 0.0001 | — |
Change from baseline in percentage of superficial cellsc,f,g | ||||||||||
N | 272 | 261 | 160 | 151 | 303 | 302 | 306 | 308 | 363 | 63 |
Baseline, mean (SD) | 1.04 (3.37) | 0.91 (2.64) | NA | NA | NA | NA | 3.0 (7.6) | 2.8 (6.9) | NA | NA |
Baseline, median (range) | NA | NA | 0.0 (0 to 35) | 0.0 (0 to 11) | 0.0 (0 to 9) | 0.0 (0 to 21) | NA | NA | 0 (0 to 5) | 0 (0 to 5) |
Week 12, mean (SD) | 12.1 (15.85) | 3.09 (8.62) | NA | NA | NA | NA | NR | NR | NA | NA |
Week 12, median (range) | NA | NA | 8.5 (0 to 67) | 1.0 (0 to 57) | 7.0 (0 to 79) | 0.0 (0 to 85) | NA | NA | 5 (0 to 60) | 0 (0 to 30) |
Change, mean (SD) or LS mean (SE)j | 10.8 (15.66) | 2.18 (8.39) | 12.4 (15.36) | 3.3 (9.02) | 12.3 (14.77) | 1.7 (6.88) | 7.8 (0.7)j | 0.6 (0.7)j | NA | NA |
Change, median (range) or (range; 95% distribution-free CI)j | NA | NA | 7.0 (–4 to 65) | 0.0 (–11 to 57) | 7.0 (–6 to 79) | 0.0 (–5 to 85) | NA | NA | 5 (–5 to 60; 5.0 to 7.0)j | 0 (–5 to 28; 0.0 to 0.0)j |
Mean difference (95% CI), OSP vs. PBO | NA | — | NA | — | NA | — | 7.2 (5.2 to 9.1); P < 0.0001 | — | NA | NA |
P value, OSP vs. PBO | < 0.001,d < 0.001e | — | < 0.0001 | — | < 0.0001 | — | < 0.0001 | — | < 0.0001 | — |
Change from baseline in vaginal pHh | ||||||||||
N | 276 | 268 | 160 | 154 | 303 | 302 | 277 | 280 | 363 | 63 |
Baseline, mean (SD) | 6.4 (0.8) | 6.3 (0.7) | 6.24 (0.80) | 6.26 (0.75) | 6.31 (0.77) | 6.31 (0.76) | 6.11 (0.70) | 6.14 (0.73) | 6.23 (0.73) | 6.20 (0.75) |
Week 12, mean (SD) | 5.4 (0.9) | 6.2 (0.9) | 5.32 (0.91) | 6.02 (0.93) | 5.37 (0.89) | 6.25 (0.96) | NR | NR | 5.03 (0.72) | 6.04 (0.89) |
Change, mean (SD) or LS mean (SE)j | –1.0 (1.1) | –0.1 (0.8) | –0.95 (0.07)j | –0.25 (0.07)j | –0.94 (0.05)j | –0.07 (0.05)j | –1.01 (0.04)j | –0.29 (0.04)j | –1.21 (0.91) | –0.16 (0.95) |
Mean difference (95% CI), OSP vs. PBO; P value | NA | — | NR | — | NR | — | –0.72 (–0.84 to –0.59); P < 0.0001 | — | –0.97 (–1.17 to –0.77); P < 0.0001 | — |
P value, OSP vs. PBO | < 0.001,d < 0.001e | — | < 0.0001i | — | < 0.0001i | — | NA | — | NA | — |
CI = confidence interval; CMH = Cochran-Mantel-Haenszel; LS = least squares; MMRM = mixed-effects model for repeated measures; N = number of patients contributing to the analysis; NA = not applicable; NR = not reported; OR = odds ratio; OSP = ospemifene; PBO = placebo; SD = standard deviation; SE = standard error; vs. = versus.
Note: Studies 821, 231, and 718 did not control for multiplicity.
aStudy 310: P values for treatment comparisons (each active vs. placebo) from the CMH row mean score test controlling for uterine status (intact uterus or hysterectomized) and pooled centre. Study 821: P value was computed using CMH row mean score test controlling for centre. Study 231: Odds ratio: exponential of the mean of cumulative log odds ratio. To calculate the odds ratio, 95% CI, and P value, the generalized estimating equations method was used.
bIn Study 310 and Study 231, the LS means, SE, and P values for the percentages of parabasal cells, percentages of superficial cells, and vaginal pH were computed using MMRM. The P value for vaginal dryness was computed using the generalized estimating equations method. In Study 821, the P value was computed using ANCOVA, where change from baseline is the response variable, baseline assessment is the covariate, and treatment and centre are fixed effects.
cIn Study 718, the P value is from the CMH model.
dThe P values for treatment comparisons (each active vs. placebo) from rank-based analysis of variance are stratified by uterus status (intact or hysterectomized).
eThe P values for treatment comparisons (each active vs. placebo) from rank-based analysis of variance are stratified by pooled centre.
fIn Study 231, the LS means, SE, and P values for the percentages of parabasal cells, the percentages of superficial cells, and vaginal pH were computed using MMRM. The P value for vaginal dryness was computed using the generalized estimating equations method.
gIn Study 821, the P value was computed using rank-based analysis of variance, stratifying by study centre.
hIn Study 231, to calculate LS means, SE, and P value, the MMRM was used. In Study 718, the estimated difference, CI of the difference, and P value comparing the treatments are model-based from ANCOVA.
iThe P value was computed using ANCOVA, where change from baseline was the response variable, baseline assessment was the covariate, and treatment and centre were fixed effects.
jData corresponds to the reported LS means (SE) rather than the mean (SD).
Change in the severity of symptoms of VVA following 12 weeks of treatment was measured using the VVA questionnaire and evaluated in studies 310, 821, and 231 as a co-primary end point. Further, a formal minimal important difference (MID) was not identified in the published literature; however, the clinical expert consulted by CADTH indicated that any reduction in symptom severity was considered clinically meaningful because that is a primary goal of treatment. Each of these studies evaluated the change in vaginal dryness in patients who identified it as the MBS of VVA. In Study 310, the mean changes in severity of vaginal dryness at week 12 were –1.26 (standard deviation [SD] = 1.03) and –0.84 (SD = 1.00) for the ospemifene and placebo treatment groups, respectively, indicating that patients randomized to ospemifene reported a greater reduction in symptom severity compared to patients randomized to placebo (P = 0.021). |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| All patients included in Study 231 reported moderate or severe vaginal dryness as the MBS of VVA at baseline. The mean changes in symptom severity from baseline to week 12 were –1.29 (SD = 1.01) for ospemifene and –0.91 (SD = 0.96) for placebo (P < 0.0001). In Study 821, change from baseline in vaginal dryness was assessed in the dryness stratum (patients indicating vaginal dryness as the MBS of VVA at baseline). In contrast to the results of Study 310 and 231, Study 821 did not demonstrate a statistically significant difference in the reduction of severity of vaginal dryness compared to placebo, based on mean differences of –1.3 (SD = 1.08) for ospemifene and –1.1 (SD = 1.02) for placebo (P = 0.080) at week 12. Given the failure to demonstrate an improvement based on vaginal dryness as the MBS of VVA, the efficacy of ospemifene could not be concluded in the dryness stratum.
Patients who identified dyspareunia as the MBS of VVA were also enrolled in Study 310 and Study 821. In Study 310, this included 142 patients of 544 patients (26%) from the overall intention-to-treat (ITT) population. This population informed the analysis of the change from baseline to week 12 in severity of dyspareunia as the MBS of VVA as part of the co-primary end point for severity of the MBS of VVA. In Study 310, the mean changes from baseline to week 12 in severity of dyspareunia were –1.2 (SD = 1.3) in the ospemifene treatment group and –0.9 (SD = 1.1) in the placebo treatment group, which corresponded to a greater reduction in the severity of dyspareunia with ospemifene compared to placebo (P = 0.023). In Study 821, the co-primary end points in the dyspareunia stratum were analyzed independently from those in the dryness stratum. In Study 821, the mean changes from baseline in severity of dyspareunia were –1.5 (SD = 1.08) in the ospemifene treatment group and –1.2 (SD = 1.12) in the placebo treatment group. Therefore, a greater reduction in the severity of dyspareunia with ospemifene compared to placebo was demonstrated (P = 0.0001).
Cytology measurements included the percentage of parabasal cells and the percentage of superficial cells from a vaginal smear. The changes from baseline to week 12 in the percentages of parabasal cells and superficial cells were co-primary end points in studies 310, 821, 231, and 718. These outcomes provide an objective assessment of the signs of VVA and are considered standard in clinical trials; however, the clinical expert consulted by CADTH indicated that they are not particularly relevant to clinicians, given that they are rarely assessed in clinical practice. A reduction in the percentage of parabasal cells and increase in the percentage of superficial cells correspond with an improvement in VVA. Studies 310, 821 (dryness stratum and dyspareunia stratum), 231, and 718 demonstrated differences in the change in the percentages of parabasal cells and superficial cells in favour of ospemifene compared to placebo. Because these outcomes are not typically used in clinical practice, the clinical expert was unable to quantify a clinically meaningful improvement. Further, a formal MID was not identified in published literature.
The changes from baseline to week 12 in the percentages of parabasal cells and superficial cells were reported as follows:
In Study 310, the mean changes from baseline in the percentages of parabasal cells were –30.1 (SD = 37.93) and 3.98 (SD = 35.21) for the ospemifene and placebo treatment groups, respectively, in favour of ospemifene (P < 0.001). The mean changes in the percentages of superficial cells were 10.8 (SD = 15.66) for the ospemifene group and 2.18 (SD = 8.39) for the placebo group, in favour of ospemifene (P < 0.001).
In Study 231, the least squares (LS) mean changes in the percentages of parabasal cells were –23.7 (standard error [SE] = 1.4) and –1.9 (SE = 1.4) for the ospemifene and placebo treatment groups, respectively, in favour of ospemifene; the treatment-group difference was –21.8 (95% confidence interval [CI], –25.7 to –18.0; P < 0.0001). The LS mean changes in the percentages of superficial cells were 7.8 (SE = 0.7) and 0.6 (SE = 0.7) for the ospemifene and placebo treatment groups, respectively, in favour of ospemifene; the treatment-group difference was 7.2 (95% CI, 5.2 to 9.1; P < 0.0001).
In the dryness stratum of Study 821, the LS mean changes in the percentages of parabasal cells were –31.7 (SE = 2.11) for ospemifene and –3.9 (SE = 2.18) for placebo, respectively; the treatment-group difference was –27.8 (95% CI, –33.75 to –21.90; P < 0.0001). The change in the percentage of superficial cells was reported as a median (range) because the analysis of covariance (ANCOVA) assumptions were not met. The median changes at week 12 were 7.0 (range = –4 to 65) for ospemifene and 0.0 (range = –11 to 57) for placebo in favour of ospemifene (P < 0.0001).
In the dyspareunia stratum of Study 821, the LS mean changes in the percentages of parabasal cells for ospemifene and placebo were –40.3 (SE = 1.56) and –0.4 (SE = 1.57), respectively; the treatment-group difference was –39.9 (95% CI, –44.15 to –35.63; P < 0.0001). The change in the percentage of superficial cells was reported as a median (range) because the ANCOVA assumptions were not met. The median changes at week 12 were 7.0 (range = –6 to 79) for ospemifene and 0.0 (range = –5 to 85) for placebo, in favour of ospemifene (P < 0.0001).
A non-parametric method of analysis was used in Study 718 because the assumptions of ANCOVA were not met. The medians of the changes in the percentages of parabasal cells were –40 (95% distribution-free CI, –55.0 to –30.0) for ospemifene and 0 (95% distribution-free CI, 0.0 to 10.0) for placebo, in favour of ospemifene (P < 0.0001). The medians of the changes in the percentages of superficial cells were 5 (95% CI, 5.0 to 7.0) for ospemifene and 0 (0.0 to 0.0) for placebo, in favour of ospemifene (P < 0.0001).
Study 718 also assessed the percentages of parabasal and superficial cells at weeks 26 and 52 as secondary outcomes. The results of both outcome assessments were similar to the results at week 12; however, the study was not powered to detect a difference in secondary outcomes, and the assessments were not controlled for multiplicity.
Vaginal pH was assessed in studies 310, 821, 231, and 718 as the change from baseline to week 12. This was a co-primary end point in each of the 4 studies. As with cytology assessments, vaginal pH is often measured in clinical trials, but is not particularly relevant to clinicians because it is rarely assessed in clinical practice. The clinical expert consulted by CADTH was unable to quantify a clinically meaningful improvement vaginal pH, and a formal MID was not identified in published literature. However, vaginal pH greater than 5.0 is an indicator of vaginal atrophy; therefore, a reduction in pH is suggestive of an improvement in VVA.
In Study 310, the mean changes from baseline to week 12 in vaginal pH were –1.0 (SD = 1.1) for ospemifene and –0.1 (SD = 08) for placebo, in favour of ospemifene (P < 0.001).
In Study 231, the LS mean changes from baseline to week 12 were –1.01 (SE = 0.04) for ospemifene and –0.29 (SE = 0.04) for placebo, corresponding to a treatment-group difference of –0.72 (95% CI, –0.84 to –0.59; P < 0.0001) in favour of ospemifene.
In Study 821, the LS mean changes from baseline to week 12 in vaginal pH were –0.95 (SE = 0.07) and –0.94 (SE = 0.05) for ospemifene in the dryness and dyspareunia strata, respectively. The LS mean changes from baseline in the placebo treatment groups were –0.25 (SE = 0.07) and –0.07 (SE = 0.05) in the dryness and dyspareunia strata, respectively. The difference in the change in vaginal pH was in favour of ospemifene for both strata (P < 0.0001).
In Study 718, the mean changes from baseline to week 12 in vaginal pH were –1.21 (SD = 0.912) for ospemifene and –0.16 (SD = 0.945) for placebo, corresponding to a treatment-group difference of –0.97 (95% CI, –1.17 to –0.77; P < 0.0001) in favour of ospemifene. The analyses at week 26 and week 52 were based on observed cases, which yielded similar results to those reported at week 12.
Urinary symptoms were assessed as a secondary outcome using the UDI-6 in studies 310 and 821 by domain score and total score, and by the total score in Study 231. No change in urinary symptoms, as measured by the UDI-6, were observed in any of the analyses. Sexual function was assessed as a secondary outcome in studies 821 and 231 using the FSFI. The FSFI is commonly used in clinical trials and is a validated tool for the measurement of women’s overall sexual function. The clinical expert consulted by CADTH indicated that the domains of the FSFI are clinically relevant, but sexual function is typically evaluated informally in clinical practice. Overall, the results of the FSFI were inconsistent between studies or did not demonstrate an improvement in sexual function compared to placebo, with the exception of the pain domain. The treatment-group differences in the change from baseline to week 12 for the pain domain were 0.58 (95% CI, 0.327 to 0.838) in Study 821 (all patients) and 0.45 (95% CI, 0.11 to 0.80) in Study 231, suggesting an improvement in favour of ospemifene. This result is aligned with a reduction in severity of dyspareunia demonstrated in the trials.
Harms Results
A summary of key safety results is provided in Table 3. No deaths were reported in any of the included studies, and specific serious adverse events (SAEs) were infrequently reported. No SAEs were reported by patients who received ospemifene in Study 310; 1.5% of patients who received placebo reported at least 1 SAE. The proportions of patients reporting at least 1 SAE in studies 821 and 231 were similar between treatment groups (1.3% versus 1.5% in Study 821 and 1.6% versus 1.0% in Study 231 for ospemifene versus placebo). In Study 718, 4.9% of patients in the ospemifene group and 6.5% of patients in the placebo treatment group reported at least 1 SAE. During the 12-week treatment period of studies 310, 821, and 231, patients who received ospemifene reported adverse events (AEs) at a similar or slightly higher frequency than patients who received placebo (60%, 63%, and 35% of patients who received ospemifene versus 52%, 51%, and 33% of patients who received placebo in studies 310, 821, and 231, respectively). Similar results were observed during the 52-week treatment period of Study 718, although the frequency of AEs was higher overall than in the 12-week studies. In addition, AEs were reported more frequently by those who received ospemifene compared to placebo (64% versus 45%) during the 52-week treatment period of Study 310X (including 12 weeks in Study 310), although this is likely biased in favour of placebo due to the high rate of discontinuation from study in the placebo treatment group. Specific AEs were not reported in more than 9% of patients in the 12-week studies or 13% of patients in Study 718. The most commonly reported AE in each of the 4 studies was hot flashes. Hot flashes were consistently reported more frequently by patients who received ospemifene (i.e., by 6% to 8% of patients who received ospemifene and by 3% to 3% of patients who received placebo). Vaginal infections, vaginal discharge, and muscle spasms were also reported as AEs more frequently by those in the ospemifene group versus the placebo group. Overall, patients who withdrew from treatment due to an AE were similar between treatment groups in the 12-week trials (2% to 5% for ospemifene and 3% to 5% for placebo). In studies 718 and 310X, withdrawals due to AEs were more frequent in the ospemifene treatment groups (14% and 6%, respectively) than in the placebo groups (10% and 2%, respectively). The rates of specific AEs leading to discontinuation were infrequent; however, hot flashes were the only AEs that led to treatment discontinuation for at least 1 patient who received ospemifene in every study.
The following notable ha rms were included in the CADTH systematic review protocol: vaginal hemorrhage, abnormal genital bleeding, cervical dysplasia, breast mass, endometrial hyperplasia, uterine polyps, cardiovascular disorders (e.g., thromboembolic and hemorrhagic stroke, coronary heart disease), breast cancer, uterine cancer, deep vein thrombosis (DVT), and pulmonary embolism. In studies 310, 821, 231, and 718, a total of |||||||| patients in the ospemifene treatment groups and |||||||| patients in the placebo treatment groups reported vaginal hemorrhage. Uterine polyps were reported by 6 patients and 1 patient for ospemifene and placebo, respectively. Cervical dysplasia was reported in |||||||| patients for ospemifene and |||||||| patients for placebo, and breast mass was reported in 7 patients in both the ospemifene and placebo groups. Endometrial hyperplasia was reported in 1 patient who received ospemifene, and breast cancer was reported in 1 patient who received placebo. A total of 2 patients reported DVT, both of whom were in ospemifene treatment groups. No patients reported experiencing abnormal genital bleeding, uterine cancer, pulmonary embolism, or other cardiovascular disorders (e.g., thromboembolic and hemorrhagic stroke, coronary heart disease). In Study 310X, vaginal hemorrhage, |||||||||||||||||||||||| and breast mass were reported by 1 patient (each) in the ospemifene treatment group. No other notable harms were reported.
Table 3: Summary of Key Safety Results From Pivotal and Protocol Selected Studies
Outcome | Study 310 | Study 821 (all patients) | Study 231 | Study 718 | Study 310X | |||||
|---|---|---|---|---|---|---|---|---|---|---|
OSP N = 276 | PBO N = 268 | OSP N = 463 | PBO N = 456 | OSP N = 317 | PBO N = 310 | OSP N = 364 | PBO N = 62 | OSP N = 69 | PBO N = 49 | |
Harms, n (%) (safety population) | ||||||||||
AEs | 164 (59.4) | 140 (52.2) | 290 (62.6) | 232 (50.9) | 112 (35.3) | 103 (33.2) | 308 (84.6) | 47 (75.8) | 44 (63.8) | 22 (44.9) |
SAEs | 0 | 4 (1.5) | 6 (1.3) | 7 (1.5) | 5 (1.6) | 3 (1.0) | 18 (4.9) | 4 (6.5) | 5 (7.2) | 1 (2.0) |
WDAEs (from study treatment) | 13 (4.7) | 13 (4.9) | 26 (5.6) | 15 (3.3) | 6 (1.9) | 10 (3.2) | 49 (13.5) | 6 (9.7) | 4 (5.8) | 1 (2.0) |
Deaths | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Notable harms,a n (%) | ||||||||||
Vaginal hemorrhage (genital hemorrhage) | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| | 1 (1.4) | 0 |
Cervical dysplasia | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| |
Breast mass | 4 (1.4) | 1 (0.4) | 3 (0.6) | 5 (1.1) | 0 | 1 (0.3) | 0 | 4 (1.4) | 1 (1.4) | 0 |
Endometrial hyperplasia | 0 | 0 | 0 | 0 | 0 | 0 | 1 (0.3) | 0 | 0 | 0 |
Uterine polyps | 0 | 0 | 1 (0.2) | 1 (0.2) | 0 | 0 | 5 (1.4) | 0 | 0 | 0 |
Breast cancer | 0 | 1 (0.4) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Deep vein thrombosis | 0 | 0 | 1 (0.2) | 0 | 0 | 0 | 1 (0.3) | 0 | 0 | 0 |
AE = adverse event; OSP = ospemifene; PBO = placebo; SAE = serious adverse event; WDAE = withdrawal due to adverse event.
aNo patients reported abnormal genital bleeding, cardiovascular disorders (e.g., thromboembolic and hemorrhagic stroke, coronary heart disease), uterine cancer, or pulmonary embolism.
Source: Clinical study reports.7-11
Critical Appraisal
Each study used a mix of objective clinical outcomes and subjective patient-reported outcomes. Objective outcomes included cytology assessments (percentage of parabasal and superficial cells) and vaginal pH based on clinical results obtained from a vaginal smear. While commonly used in clinical trials, the objective outcomes are not typically used in clinical practice, according to feedback from the clinical expert. Subjective outcomes were patient reported and included the VVA questionnaire to assess the symptoms of VVA, the UDI-6 to assess urinary symptoms, and the FSFI to assess sexual function. Although the clinical expert consulted by CADTH indicated that the self-reported outcomes are considered clinically relevant in practice to measure treatment response, published MIDs were not identified for these outcome measures in post-menopausal women. Therefore, it is unclear whether the reported between-group differences are clinically meaningful. Further, evidence of validity, reliability, and responsiveness of the VVA questionnaire was not identified for this review; nor was the validity of treating the ordinal data as continuous. These factors make it difficult to interpret the results. Additionally, secondary outcomes (UDI-6, FSFI, and any outcomes reported after week 12) were not controlled for multiplicity; therefore, they are subject to type I error.
In all studies, the primary efficacy analyses were performed using the ITT population, and supportive analyses were performed in the per-protocol (PP) and modified intention-to-treat (mITT) populations (Study 231 only). All of the supportive analyses performed were consistent with the primary analyses, with the exception of vaginal dryness as the MBS of VVA in Study 310, for which statistical significance was not demonstrated in the PP population. The sponsor attributed the lack of statistical significance for the supportive analysis to the small sample size, which is likely a contributing factor; however, the results of the analysis of vaginal dryness as the MBS of VVA in Study 310 remain uncertain.
In Study 718, patients with VVA were identified based on MI and vaginal pH without a requirement for self-reported symptoms of VVA. This introduces uncertainty about the generalizability of the patient population to post-menopausal patients with moderate to severe vaginal dryness or dyspareunia. Otherwise, the eligibility criteria used in the included studies were generally considered appropriate and reflective of post-menopausal patients with VVA, although restrictive (70% of patients in Study 231 failed screening; data were not reported in the other included studies). Most notably, patients with comorbidities, such as a history of cancer or cardiovascular disorders, were excluded from the trials, leading to uncertainty regarding the generalizability of the safety results. Lastly, evidence informing the efficacy of ospemifene is primarily based on patients receiving treatment for up to 12 weeks. Supportive efficacy data based on clinical outcomes were available for up to 52 weeks; however, the evidence is weak and not based on clinically relevant outcomes (symptom severity), causing uncertainty in the long term efficacy. Additionally, safety evidence in patients who received treatment for up to 52 weeks was available, but subject to high and imbalanced discontinuation rates. Moreover, patients are expected to continue treatment for more than 1 year, but there is no evidence of safety beyond this time point.
Indirect Comparisons
Description of Studies
The sponsor-submitted indirect treatment comparison (ITC)12 was included in this review along with an additional ITC (Li et al.) identified in the literature search.13 Both ITCs conducted a systematic review and network meta-analysis (NMA) to evaluate the comparative efficacy and safety of ospemifene versus other alternative therapies in the treatment of VVA. Both ITCs used a Bayesian framework for NMA analysis.
In the sponsor-submitted ITC, 27 RCTs were eligible, 5 of which involved ospemifene. Other treatments investigated included a conjugated estrogens vaginal cream (Premarin), an estradiol vaginal insert (Vagifem), an estradiol soft gel vaginal insert (Imex), an estradiol vaginal ring (Estring), and a prasterone vaginal ovule (Intrarosa). The sample size of the included trials ranged from 21 patients to 826 patients, and the mean age ranged from 56 years to 63 years. The eligible RCTs recruited primarily post-menopausal women with moderate to severe genitourinary symptoms, and the majority of the trials were 12 weeks in duration (range = 12 weeks to 14 weeks). For the NMA, the sponsor only included RCTs with the following treatments: ospemifene 60 mg oral daily (Osphena), estradiol vaginal cream 0.02 mg (Estrace), estradiol transdermal patch 14 mcg (Estradiol patch), estradiol vaginal cream 2 mg and 7.5 mg (Estring), estriol vaginal pessary 0.5 mg (Estriol pessary), estradiol vaginal capsule 4 mcg and 10 mcg (Imvexxy), dehydroepiandrosterone (DHEA) vaginal suppository 6.5 mg (Intrarosa), lubricants, conjugated estrogens vaginal cream 0.3 mg or 0.63 mg (Premarin), promestriene vaginal cream 10 mg, or estradiol vaginal insert 10 mcg (Vagifem). The sponsor noted that the majority of trials were at low risk of bias; however, 4 RCTs were at high risk of bias from blinding.
In the Li et al. ITC, 29 RCTs were eligible, with 8,311 participants (sample sizes ranged from 180 patients to 909 patients). Five treatments were investigated: laser therapy, vaginal estrogen (vaginal estrogen therapies pooled together), ospemifene, vaginal DHEA, and moisturization and/or lubrication. The mean age of participants ranged from 51 years to 65 years, and the duration of the trials ranged from 6 weeks to 52 weeks. Neither the severity nor the duration of symptoms was described by the authors.
Efficacy Results
Sponsor-Submitted Indirect Treatment Comparison
For the outcome of mean difference (MD) in change from baseline to follow-up in MBS score for vaginal dryness, ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
For the outcome of MD in change from baseline to follow-up in MBS score for dyspareunia, ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
For the outcome of MD in change from baseline to follow-up for combined MBS score for vaginal dryness and dyspareunia, ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
For the outcome of MD in change in percentage of parabasal cells, ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
For the outcome of MD in change in percentage of superficial cells, ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
For the outcome of MD in reduction of vaginal pH, ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Li et al. Indirect Treatment Comparison
In the Li et al. ITC, there was no difference between ospemifene and vaginal estrogens for the outcomes of mean difference in change in vaginal dryness (MD = –2.9; 95% credible interval [CrI], –13 to 8.1), dyspareunia (MD = 8.0; 95% CrI, 0.2 to 17), or sexual function (MD = 1.5; 95% CrI, –2.7 to 5.6). The reduction in vaginal pH was smaller for ospemifene versus vaginal estrogens (MD = 0.31; 95% CrI, 0.05 to 0.58). There was no difference in the reduction in percentage of parabasal cells for ospemifene compared with vaginal estrogens (MD = 2.2; 95% CrI, –9.5 to 15).
Harms Results
In the sponsor-submitted ITC, there was no difference in the risk of treatment-emergent adverse events (TEAEs) for ospemifene versus conjugated estrogens vaginal cream (relative risk [RR] = 1.07; 95% CrI, 0.93 to 1.24) or versus estradiol vaginal tablet (RR = 1.11; 95% CrI, 0.95 to 1.28). There was no difference in the risk of serious TEAEs for ospemifene versus conjugated estrogens vaginal cream (RR = 0.75; 95% CrI, 0.02 to 31) or versus estradiol vaginal tablet (RR = 0.87; 95% CrI, 0.15 to 4.17). There was no difference in the risk of urinary tract infection (UTI) between ospemifene and estradiol vaginal tablet (RR = 2.55; 95% CrI, 0.23 to 35). The risk of headaches was lower for ospemifene compared with estradiol vaginal ring (RR = 0.00; 95% CrI, 0.00 to 0.04), while there was no difference compared with conjugated estrogens vaginal cream (RR = 0.74; 95% CrI, 0.38 to 1.42) or estradiol vaginal tablet (RR = 1.43; 95% CrI, 0.24 to 8.50). There was no difference in the risk of discontinuation due to AEs for ospemifene versus estradiol vaginal ring (RR = 1.26; 95% CrI, 0.28 to 1.52), conjugated estrogens vaginal cream (RR = 0.97; 95% CrI, 0.31 to 2.69), or estradiol vaginal tablet (RR = 0.94; 95% CrI, 0.31 to 2.45).
Critical Appraisal
The sponsor-submitted ITC provided a clear rationale and objective, and was generally well conducted aside from the following limitations of note. Heterogeneity in effect sizes (based on I2 > 50%) was observed for some comparisons; however, it was not explored further through meta-regression with suspected effect modifiers. The extent to which eligible studies satisfied the similarity assumption was unclear. While patient and study characteristics were broadly similar, the appropriateness of combined different doses in nodes, differences in placebo use across trials, and the unclear extent of prior VVA treatment make it challenging to assess the similarity of the eligible studies. Subgroup or sensitivity analyses did not result in different results than the base case and were generally not able to explain heterogeneity, although decisions regarding the methodology were not adequately described. In the analysis of safety outcomes, there were wide CrIs and low event rates (resulting in extremely low RRs) for some comparisons; these outcomes make it challenging to assess comparative safety (e.g., for headache or UTI). Further, for some efficacy outcomes, there were a limited number of trials for some nodes, resulting in wide and overlapping CrIs. This makes it difficult to draw conclusions around comparative efficacy for ospemifene and relevant comparators.
The Li et al. ITC described the study objective and study selection process. Concerns were identified with respect to study selection. Specific eligibility criteria were not provided (e.g., based on severity of symptoms), and the authors did not provide explicit criteria around specific relevant interventions or comparators. Information about disease severity and duration of symptoms was not extracted, making it challenging to assess whether the similarity assumption was satisfied. Further, since severity of symptoms was not provided, it is unclear how relevant the population was for the present review. The authors converted the continuous outcomes into a 0 to 100 scale because different outcome scales were used across studies; however, they did not provide details about how this was carried out or whether it was appropriate. Not all comparators in the Li et al. ITC were relevant to this review. The comparison of ospemifene to vaginal estrogens was relevant. However, the Li et al. ITC combined all vaginal estrogens into 1 node (including different drugs and dosage forms; for example, conjugated estrogens, estradiol 4 mcg or 10 mcg vaginal capsule). Some of the vaginal estrogens included in the vaginal estrogen node were not comparators of interest for this review (e.g., estriol cream). Given that there may be differences between different vaginal estrogen products in terms of efficacy and safety, the appropriateness of combining these treatments into 1 node is uncertain. It further makes it challenging to draw conclusions around the comparative efficacy and safety of ospemifene versus individual relevant treatments. A description of model fit was not provided; therefore, it is unclear if model fit was adequate. Network diagrams were not provided in the Li et al. ITC, and it was unclear how many studies contributed to specific comparisons. Heterogeneity (I2 > 50%) was observed for some outcomes involving ospemifene (dyspareunia, vaginal pH, parabasal cells), which could be explained by age (vaginal pH) or dose (change in the percentage of parabasal cells), but could not be explained for other outcomes.
Other Relevant Evidence
Description of Studies
Study 312, a multi-centre, open-label, phase III LTSE of Study 310, has been summarized to provide additional evidence regarding the long-term safety of oral daily doses of ospemifene 60 mg for the treatment of VVA in post-menopausal women without a uterus. During this extension study, all patients received ospemifene 60 mg per day irrespective of their treatment assignment in the initial 12-week Study 310. The duration of treatment was 52 weeks followed by a 4-week post-treatment follow-up period, for a total of 68 weeks (including the initial 12 weeks of Study 310).14,15 The baseline characteristics of those who continued into the LTSE were similar to those in the core study in terms of age, race, ethnicity, and body mass index (BMI).
Of the 826 post-menopausal women randomized to Study 310, 301 women (36.4%) enrolled in the open-label extension study, Study 312. Overall, 117 patients (38.7%) discontinued from the study. The most common reasons for discontinuation were patient decision or withdrawal of consent (13.2%), AEs (12.3%), and loss to follow-up (5.6%).
Efficacy Results
Efficacy was not assessed in Study 312.
Harms Results
During the 52-week treatment period, 73.1% of patients reported at least 1 TEAE, and 4% of patients reported at least 1 SAE. The most common AEs were sinusitis (8%), UTI (8.6%), and hot flashes (10.3%). None of the specific SAEs were reported in more than 2 patients. AEs leading to treatment discontinuation were reported in 34 patients (11.3%); hot flashes, reported by 2% of patients, were the most frequent AE leading to discontinuation. ||||||||||||||||||||||||||||||||||||||||||||||||||||||| breast mass (n = 1), and hemorrhagic stroke (n = 1) were the only notable harms reported in Study 312, all of which were infrequent.
Critical Appraisal
Study 312 had several limitations resulting from the overall design, including the lack of a comparison group to provide context and control for potential confounders. Additionally, the open-label design may influence the perception of improvement by patients and clinicians, which could affect the reporting of harms. Among the enrolled patients, 117 patients (38.7%) discontinued prematurely from the study, which may have resulted in safety outcomes being reported. Because the patients who took part in Study 312 were originally from the parent studies, and the eligibility criteria remained the same, it is reasonable to expect that the same limitations to generalizability are relevant to the open-label extension study. For instance, given that the participants were predominantly White (92.4%), the results from these trials may not be generalizable to other racial groups that may commonly be seen at some centres in Canada. Since this open-label extension safety study focuses on a very specific patient population (post-menopausal women with no uterus), it would be best to compare the safety results with similar studies to get a more accurate idea of the safety profile among the general population. The treatment duration was 52 weeks, which might not be a sufficient time frame over which to observe and note all potential safety issues.
Conclusions
Five studies were summarized as part of the CADTH systematic review: 4 phase III, double-blind RCTs that assessed the efficacy and safety of ospemifene 60 mg compared to placebo over 12 weeks (studies 310, 821, 231) and over 52 weeks (Study 718) in post-menopausal women with VVA, as well as a double-blind, placebo-controlled LTSE of Study 310 (Study 310X) that provided evidence of safety over up to 52 weeks of treatment. The studies included in this review demonstrated a beneficial effect of ospemifene compared to placebo over a 12-week treatment period in post-menopausal women who had self-reported moderate to severe vaginal dryness or dyspareunia VVA symptoms. Although the efficacy of ospemifene versus placebo in relieving vaginal dryness was demonstrated in Study 231, there was inconsistency in this finding across the studies due to lack of statistical significance for this outcome in the primary analysis of Study 821, as well as in a supportive analysis performed in the PP population of Study 310. Across the included studies, the clinical benefit was estimated using a patient-reported outcome, the VVA questionnaire. The exact clinical interpretation of the difference in score of the VVA questionnaire is unclear, particularly due to the lack of a recognized MID, a lack of sufficient validation of the questionnaire, and the nature of an ordinal score; however, the magnitude of observed change in symptom scores was similar between the different trial populations of similar eligibility criteria. The self-assessment of individual patients may suffer from recall bias in both the ospemifene and placebo groups. However, the observed clinical benefit was supported by objective measures of VVA, namely a reduction in the percentage of parabasal cells, an increase in the percentage of superficial cells, and a reduction in pH.
Evidence from 2 ITCs summarized for this review suggests there is no difference between ospemifene and other treatments for symptoms of VVA in terms of comparative effectiveness. This finding should be interpreted with caution due to uncertainty associated with the ITCs; however, it highlights the limitation of a lack of direct comparative evidence for active treatment options. Other gaps in the evidence include the absence of assessments of both HRQoL, and symptom relief beyond 12 weeks of treatment. Evidence assessing the safety of ospemifene was available for up to 52 weeks on treatment. No deaths were reported, and SAEs were reported infrequently. Overall, the safety profile of ospemifene was acceptable based on the included trials, with the exception of the frequency of hot flashes and uncertainty around the risk of thromboembolic events. Further study is warranted to obtain long-term safety data, including evidence of safety beyond 1 year.
Introduction
Disease Background
Hormonal changes, particularly a decrease in estrogen, are often associated with signs and symptoms in post-menopausal women that have an impact on HRQoL as well as physical, mental, and sexual health.1 Many post-menopausal women experience VVA due to physiologic changes in the female genital anatomy that result from aging and a lack of ovarian estrogen production in menopause. The vulva and vagina are particularly susceptible to changes related to menopause because there are estrogen receptors in the vulva, vaginal, bladder, urethra, and muscles of the pelvic floor.2 The clinical expert consulted by CADTH indicated that with menopause, the vulva loses much of its collagen and adipose tissue, and glandular secretions are diminished; the vaginal surface thins, loses its elasticity, and is more easily injured, with decreased fluid secretion; and changes in urethral and vaginal flora and pH can render menopausal women more susceptible to UTIs and vaginal infections. Women with VVA will typically present with vaginal dryness, pruritis, burning, pain, and dyspareunia as self-reported symptoms.
Genitourinary syndrome of menopause is a newer, broader term that encompasses VVA as well as other genitourinary symptoms and may not be limited to patients who are sexually active.2 Symptoms of genitourinary syndrome of menopause can be grouped as: genital symptoms, including dryness, burning, itching, irritation, and bleeding; sexual symptoms, including dyspareunia and other sexual dysfunctions; and urinary symptoms, including urgency, dysuria, and recurrent UTIs.1 Women may present with some or all of the signs and symptoms of genitourinary syndrome of menopause.16 Signs of genitourinary syndrome of menopause can be observed through physical examination conducted by an experienced health care provider, given that there may be changes in the colour, size, and integrity of the anatomy of the vagina. There may also be signs of decreased lubrication and an increase in vaginal pH; typically, a pH of greater than 5.0 would be considered abnormal.2 The clinical expert consulted by CADTH indicated that assessments of vaginal pH, parabasal cells, and superficial cells are not typical in Canadian clinical practice, although these are often evaluated in clinical trials. In women with vaginal atrophy, an increase in parabasal cells and decrease in superficial cells can be observed. Moreover, as women age, the proportion of parabasal cells will continue to increase, and the MI may eventually consist entirely of parabasal cells.16 According to guidelines published by SOGC, in the absence of treatment, genitourinary syndrome of menopause will evolve chronically in most women and progress to functional and structural urogenital tissue changes that can be difficult to reverse even with treatment.2
Of note, genitourinary syndrome of menopause has been described as being defined by the presence of symptoms; however, not all women with signs of atrophy identified through pelvic examination are symptomatic.16 While no report of the incidence of VVA among Canadians is available, a study that included 1,016 Canadians reported a prevalence of 34%.3 However, estimates regarding the prevalence of patients who suffer from VVA or genitourinary syndrome of menopause may be underreported. Many patients will not report changes they experience during menopause because they will associate changes to normal aging. Previous literature suggests that 60% to 90% of post-menopausal patients may suffer from VVA and experience significant deficits in their quality of life because of it.4 Due to underreporting, it may be important for health care providers to take the initiative and ask post-menopausal patients about symptoms related to genitourinary syndrome of menopause to identify the condition as early as possible and provide optimal care.2
Standards of Therapy
An ideal treatment for genitourinary symptoms of menopause would provide complete symptomatic relief from the urogenital and/or vulvovaginal changes experienced by women in menopause, thereby improving patients’ quality of life, with minimal adverse effects and long-term health risks. SOGC recommends that patients try vaginal lubricants and/or vaginal moisturizers as first-line management options for genitourinary syndrome of menopause, particularly if patient concerns are limited to vaginal dryness or dyspareunia.2 The clinical expert indicated that these treatments may include hyaluronic acid gel or polycarbophil gel. Second-line treatment for VVA typically includes the administration of local estrogen to reverse the effects of estrogen withdrawal, or non-hormonal treatments to counter the effects of VVA.2 Several formulations exist for local vaginal estrogen, including creams, a hormone-releasing ring, and tablets. The clinical expert indicated that the majority of women who receive systemic estrogen for other menopausal symptoms do not get adequate relief from vulvovaginal symptoms from systemic estrogen alone; local vaginal estrogen is still required. The clinical expert noted that some alternative supplements, such as phytoestrogens, black cohosh, and dong quai, have been studied, but have not been found to be effective compared with placebo. They also noted that the benefit of vitamin D supplementation is unclear.
Estrogen treatment favourably alters patients’ physiology to treat the underlying disease and targets disease symptoms. The clinical expert noted that estrogen improves blood supply to vulvovaginal tissues, restoring normal vaginal flora and pH, improving symptoms, and reducing the risk of urogenital infections. However, Health Canada has issued a black box warning for vaginal estrogen therapies for several disease risks (myocardial infarction, stroke, invasive breast cancer, pulmonary emboli, and DVT, for most products) based on evidence for oral estrogen-plus-progestin therapy and oral estrogen-alone therapy. These therapies are also contraindicated in patients with known or suspected estrogen-dependent malignant neoplasia and patients with a known, suspected, or past history of breast cancer, also based on evidence for systemic therapies. According to the clinical expert, it is possible for some patients with these contraindications to be treated with vaginal estrogen; however, the product monograph warnings can lead to hesitancy. Estrogen hormonal therapies may be administered to patients vaginally as creams, tablets, capsules, or a ring. Some patients may prefer products other than vaginal creams, because they can be messy.
Drug
The drug under review by CADTH is ospemifene (60 mg) tablets for oral administration. It is recommended that ospemifene be administered consistently, once daily with food.5 In Canada, ospemifene is indicated in post-menopausal women for the treatment of moderate to severe dyspareunia and/or vaginal dryness, which are symptoms of VVA, a component of genitourinary syndrome of menopause. The sponsor has requested that ospemifene be reimbursed according to the approved Health Canada indication.
Ospemifene is a SERM that acts by binding to estrogen receptors. In some tissues, ospemifene acts as an agonist that activates estrogenic pathways; in other tissues, it acts as an antagonist by causing a blockade of estrogenic pathways.5 Ospemifene has an effect on estrogen receptors in the vagina, increasing the cellular maturation and mucification of the vaginal epithelium.
Table 4: Key Characteristics of Ospemifene and Vaginal Estrogen Therapies
Characteristic | Ospemifene | Vagifem, tablet | Estring, ring | Premarin, cream |
|---|---|---|---|---|
Mechanism of action | SERM, which acts by binding to estrogen receptors as an agonist of estrogenic pathways in some tissues, and an antagonist in others In the vagina, ospemifene has an effect on estrogen receptors that increases the cellular maturation and mucification of the vaginal epithelium. | Estrogen therapy for estrogen deficiency | Estrogen therapy for estrogen deficiency | Estrogen therapy for estrogen deficiency |
Indicationa | Indicated in post-menopausal women for the treatment of moderate to severe dyspareunia and/or vaginal dryness, which are symptoms of vulvar and vaginal atrophy, a component of genitourinary syndrome of menopause | Treatment of the symptoms of vaginal atrophy due to estrogen deficiency | For post-menopausal urogenital complaints due to estrogen deficiency, such as feeling of dryness in the vagina, with or without pruritus vulvae, dyspareunia, dysuria, or urinary urgency | Treatment of atrophic vaginitis, dyspareunia, and kraurosis vulvae |
Route of administration | Oral | Vaginal | Vaginal | Vaginal |
Recommended dose | 60 mg once daily with food | Initial dose: 10 mcg, 1 insert daily for 2 weeks Maintenance dose: 1 insert twice weekly with a 3-day to 4-day interval between doses The tablet is inserted into the vagina as far as it can comfortably go without force, using an applicator. | The ring (2 mg) should be left in place continuously for 90 days and then, if continuation of therapy is deemed appropriate, replaced by a new ring. The ring should be inserted into the upper third of the vaginal vault. | The cream should be administered cyclically for short-term use only. Low dose: 0.5 g administered intravaginally or topically twice weekly Maximum dose: women should be started at 0.5 g daily. Dosage adjustment (0.5 g to 2 g) may be made based on individual response. |
Serious adverse effects or safety issues | Serious warnings:
Contraindications:
| Serious warnings:
Contraindications:
| Serious warnings:
Contraindications are the same as those for Vagifem, in addition to:
| Serious warnings:
Contraindications are the same as those for Vagifem, in addition to:
|
CEE = conjugated equine estrogen; CHD = coronary heart disease; DVT = deep vein thrombosis; MPA = medroxyprogesterone acetate; SERM = selective estrogen receptor modulator.
aHealth Canada–approved indication
Source: Product monographs for ospemifene,5 Vagifem,17 Estring,18 and Premarin.19
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups.
Input was received from 1 patient group, WHC. WHC advocates, raises awareness, and provides education about the urogynecological and reproductive health of patients of all ages. WHC noted the overall lack of awareness and understanding of urogynecological health, the limited therapeutic options for peri- and post-menopausal conditions (e.g., post-menopausal VVA), and the potential inequity in accessing preferred treatments when they are not reimbursed by public drug plans. WHC emphasized that the clinical and psychological impacts caused by untreated menopausal conditions are often overlooked and dismissed. Further, it expressed the expectation that a suitable treatment option for patients would improve their health outcomes and potentially raise clinician awareness of the importance of treating menopausal conditions.
To provide additional background on the lived experiences, values, and preferences of patients with VVA, patient group websites were sought. Healthtalk.org is a non-profit organization that has collected hundreds of stories from patients with any health condition.6 Information from video interviews with 13 British patients about VVA was available through Healthtalk.org and was obtained, assessed, and synthesized by the CADTH review team. The interviewed patients reported vaginal dryness, decline in libido (contributing to a decline in sexual activity), and urinary problems as some of the common complications they experienced after entering menopause. Interviewed patients also described the importance of sex in a relationship and how decreased sexual activity (attributed to VVA symptoms) can add significant complications to a relationship over time. In the interviews, some patients indicated they were made aware of the lack of knowledge regarding the effects of hormone replacement therapies, and that treatment with hormone replacement therapies may not prevent thinning of the vaginal wall. Thinning of vaginal tissue was reported as causing severe discomfort for many patients, resulting in vaginal tears and bleeding. Patients also described how the decline of estrogen they experienced had affected their pelvic floor, bladder, uterus, vagina, and bowel, sometimes leading to urinary and bowel problems. Patients also reported difficulties with incontinence. Negative impacts on quality of life were experienced by many patients.
Clinician Input
Input From the Clinical Expert Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol; assisting in the critical appraisal of clinical evidence; interpreting the clinical relevance of the results; and providing guidance on the potential place in therapy). The following input was provided by 1 clinical specialist with expertise in the diagnosis and management of post-menopausal patients with VVA.
Unmet Needs
According to the clinical expert consulted by CADTH, treatment for VVA typically includes the administration of local estrogen to reverse the effects of estrogen withdrawal or of non-hormonal treatments to counter the effects of VVA. The clinical expert indicated that women with VVA will typically present with vaginal dryness, pruritis, burning, pain, and/or dyspareunia; the goal of treatment is to provide relief of these symptoms. In the experience of the clinical expert, in most cases, currently available treatments are effective in providing relief of VVA symptoms. Because most available treatments for symptoms of VVA are administered intravaginally, ospemifene offers an alternative route of administration (as an orally administered tablet); however, the clinical expert also noted that some patients may prefer a local therapy over a systemic therapy due to hesitancy around the use of hormonal treatments.
Place in Therapy
The clinical expert consulted for this review noted that despite the utility of vaginal moisturizers and lubricants, women with VVA will generally experience more effective symptomatic relief from vaginal local estrogen. While estrogen can be delivered through oral, transdermal, or vaginal routes of administration, the vaginal route has traditionally been the most effective for vulvovaginal symptoms. The clinical expert noted that some studies have indicated that the majority of women receiving systemic estrogen (alone) for other menopausal symptoms do not get adequate relief of VVA symptoms; local vaginal estrogen is still required. They also indicated that several formulations exist for local vaginal estrogen, including creams, a hormone-releasing ring, and tablets. The clinical expert noted that more recently, additional therapeutic options have become available for the treatment of genitourinary syndrome of menopause, such as intravaginal prasterone and orally administered ospemifene (a SERM). These represent a departure from traditional estrogen-based management strategies and substantially widen the scope of options available to women with VVA.
The clinical expert consulted by CADTH relayed that ospemifene is a SERM that has specific estrogen receptor agonist activity on vaginal tissues as well as on the bones, with partial agonist activity on the uterus. The clinical expert indicated that by targeting the estrogen receptors, ospemifene provides symptomatic relief from VVA caused by the decline in estrogen levels in menopause. It was also noted that hot flashes represent the main adverse effect of ospemifene; the clinical expert believed that this could be a significant barrier to widespread use in women with menopausal symptoms. The clinical expert suggested that, considering this factor and others, it is unlikely that ospemifene will become first-line therapy.
Patient Population
The clinical expert consulted for this review believed that the majority of patients with genitourinary syndrome of menopause are anticipated to benefit from a therapeutic drug with estrogen receptor agonist properties, such as ospemifene. Feedback from the expert indicated that the patients most in need of intervention are those with more severe symptoms, and that ospemifene provides an additional option to traditional vaginal estrogen therapy. Additionally, the clinical expert believed that the oral route of administration for ospemifene may be especially suited for women who are unable to self-administer vaginal medication (e.g., due to severe pain or mobility limitations).
The clinical expert stated that women who do not report symptoms would generally not be diagnosed with VVA in clinical practice. The expert indicated that patients will generally self-identify based on their description of symptoms during clinical history. Alternatively, patients seen for urogynecological issues, such as vaginal prolapse or urinary incontinence, may have VVA identified at the time of assessment by history and visual inspection. The expert noted that clinical history and visual inspection of the vulva on physical exam would be the usual methods for identifying patients with VVA; however, given the rise in telemedicine, the expert anticipated that diagnoses will increasingly be made based on clinical history alone, which may also be considered a reasonable approach.
The clinical expert stated that patients with any contraindication to ospemifene would not be suitable for treatment with this medication. The clinical expert noted that these patients include those with a hypersensitivity or allergy to the medication or its components, undiagnosed vaginal bleeding, active venous or arterial thromboembolic disease or history of such disease, and those with a known or suspected estrogen-dependent tumour.
Assessing Response to Treatment
The clinical expert consulted by CADTH indicated that in clinical practice, subjective patient-reported improvement in symptoms is the primary outcome used to determine whether a patient is responding to treatment. The expert noted that improvement of symptoms is most often correlated with visual inspection at examination; however, subjective symptoms are more clinically meaningful than appearance. Moreover, any improvement in vulvovaginal symptoms would be considered a clinically meaningful response, according to the clinical expert, who noted that this may include decrease in sensation of vaginal dryness, decreased vaginal burning and/or pain, decreased frequency of UTIs or bladder urgency and/or irritation, and decreased dryness and pain during intercourse. Histologic examination is generally not performed or required, based on the experience of the clinical expert.
Based on feedback from the clinical expert, there is no strict schedule for when treatment response needs to be assessed. The expert suggested that it would be reasonable to assess response to treatment approximately 3 months to 6 months after initiation, then again at 6 months to 12 months, and yearly thereafter.
Discontinuing Treatment
The clinical expert consulted by CADTH stated that a patient may discontinue treatment if she wishes; however, symptoms may return after some time. The expert noted that the assessment of the risks and benefits is subjective, given that the treatment is meant to improve quality of life. In less common cases, VVA may be very severe, with erosion and ulceration of the vaginal mucosa. In such cases, pharmacologic treatment will be strongly recommended, according to the clinical expert, and may be particularly important in women with pessaries.
Prescribing Conditions
According to the clinical expert consulted by CADTH, ospemifene would most likely be prescribed in an outpatient ambulatory clinic setting by a family physician or gynecologist. The clinical expert indicated patients would self-administer the medication at home. Given that genitourinary syndrome of menopause is very common, the clinical expert believed that the vast majority of clinicians with experience in the treatment of women’s health issues would be suitable prescribers.
Additional Considerations
The clinical expert noted that vaginal prasterone is also a recent addition to traditional vaginal estrogen treatments, and along with ospemifene, may cause a shift in the management approach, as described previously.
Clinician Group Input
Input for the review of ospemifene was not received from any clinician groups.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 5.
Table 5: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Considerations for continuation or renewal of therapy | |
Compared with placebo, in long-term studies there were no significant estrogen-related or clinically important AEs related to endometrial or breast tissue for patients treated with ospemifene over 52 weeks; however, the product monograph indicates that Osphena is a medicine that works like estrogen in the lining of the uterus, and may increase the chance of endometrial cancer. Should consideration be given to monitoring parameters (e.g., endometrial sampling in the event that breakthrough bleeding or spotting occurs)? | The clinical expert indicated that endometrial sampling, most commonly by endometrial biopsy, is generally required in patients who experience post-menopausal bleeding, and that this would be important, especially in patients taking medication with agonist effects on the uterus, such as estrogen, tamoxifen, and (similarly) ospemifene. |
Considerations for prescribing of therapy | |
Has consideration been given to the concomitant use of other medications for the treatment of hot flashes resulting from the use of Osphena? As per the product monograph, Osphena should not be use concomitantly with estrogens and estrogen receptor agonists and/or antagonists. The safety of concomitant use of Osphena with estrogens and estrogen receptor agonists and/or antagonists has not been studied. | The clinical expert indicated that vasomotor symptoms are common in menopausal women, and that it is anticipated that many women may require treatment for both vasomotor symptoms and vulvovaginal symptoms. That ospemifene should not be used with other estrogens or estrogen agonists and/or antagonists represents a limitation to its use in women with concomitant vasomotor symptoms; the increase in vasomotor symptoms from the use of ospemifene is a further complicating consideration. Ospemifene may still be used in women who are using non–estrogen-based therapies for vasomotor symptoms, including antidepressants, gabapentinoids, clonidine, oxybutynin, and lifestyle management strategies. |
Generalizability | |
Genitourinary syndrome of menopause describes various menopausal symptoms and signs associated with physical changes of the vulva, vagina, and lower urinary tract. Genitourinary syndrome of menopause includes not only genital symptoms (such as dryness, burning, and irritation) and sexual symptoms (such as lack of lubrication, discomfort or pain, and impaired function), but also urinary symptoms (such as urgency, dysuria, and recurrent urinary tract infections). Has consideration been given to using Osphena for non-vaginal symptoms, given that it is a systemic drug vs. a locally administered product like Vagifem or Premarin? | The clinical expert indicated that ospemifene is used for genitourinary symptoms of menopause (as described previously), not for systemic symptoms of menopause, such as vasomotor symptoms. Further, the use of ospemifene has been shown to be associated with an increase in vasomotor symptoms of menopause. |
AE = adverse event.
Clinical Evidence
The clinical evidence included in the review of ospemifene is presented in 3 sections. The first section, the Systematic Review, includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those studies that were selected according to an a priori protocol. The second section includes indirect evidence from the sponsor and indirect evidence selected from the literature that met the selection criteria specified in the review. The third section includes sponsor-submitted LTSEs and additional relevant studies that were considered to address important gaps in the evidence included in the systematic review.
Systematic Review (Pivotal and Protocol Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of ospemifene (60 mg) for the treatment of moderate to severe dyspareunia and/or vaginal dryness — which are symptoms of VVA, a component of genitourinary syndrome of menopause — in post-menopausal women.
Methods
Studies selected for inclusion in the systematic review include pivotal studies provided in the sponsor’s submission to CADTH and Health Canada as well as those meeting the selection criteria presented in Table 6. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
Table 6: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Population | Patients who are post-menopausal with moderate to severe dyspareunia and/or vaginal dryness, which are symptoms of vulvar and vaginal atrophy, a component of genitourinary syndrome of menopause Subgroups:
|
Intervention | Ospemifene (60 mg tablet for oral administration) administered with food once daily |
Comparator | Vaginal estrogen therapy (cream, tablet, or sustained-release ring)
|
Outcomes | Efficacy outcomes: Symptoms (e.g., vulvar and vaginal pain, vaginal dryness, dyspareunia, vaginal and/or vulvar irritation and/or itching, incontinence, genitourinary prolapse) HRQoL Sexual function Mental health-related outcomes (e.g., anxiety, depression, mood, cognition) Bone mineral density Cytology (e.g., % of superficial cells, % of parabasal cells) Vaginal pH Adherence Harms outcomes: AEs, SAEs, WDAEs, mortality Notable harms: vaginal hemorrhage, abnormal genital bleeding, cervical dysplasia, breast mass, endometrial hyperplasia, uterine polyps, cardiovascular disorders (e.g., thromboembolic and hemorrhagic stroke, coronary heart disease), breast cancer, uterine cancer, DVT, pulmonary embolism |
Study designs | Published and unpublished phase III and IV RCTs |
AE = adverse event; DVT = deep vein thrombosis; HRQoL = health-related quality of life; RCT = randomized controlled trial; SAE = serious adverse event; vs. = versus; WDAE = withdrawal due to adverse event.
aThese outcomes were identified as being of particular importance to patients in the input received by CADTH from patient groups.
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy according to the PRESS Peer Review of Electronic Search Strategies checklist.20
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946—) through Ovid and Embase (1974—) through Ovid. All Ovid searches were run simultaneously as a multi-file search. Duplicates were removed using Ovid deduplication for multi-file searches, followed by manual deduplication in Endnote. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concept was ospemifene (Osphena/Senshio). The following clinical trial registries were searched: the US National Institutes of Health’s clinicaltrials.gov, WHO’s International Clinical Trials Registry Platform search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
No filters were applied to limit the retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. See Appendix 1 for the detailed search strategies.
The initial search was completed on November 22, 2021. Regular alerts updated the search until the meeting of the CADTH Canadian Drug Expert Committee on March 23, 2022.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the CADTH tool Grey Matters: A Practical Tool For Searching Health-Related Grey Literature.21 Included in this search were the websites of regulatory agencies (US FDA and European Medicines Agency). Google was used to search for additional internet-based materials. See Appendix 1 for more information on the grey literature search strategy.
The drug sponsor was contacted for information regarding unpublished studies.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
Findings From the Literature
A total of 5 studies were identified from the literature for inclusion in the systematic review (Figure 1). The included studies are summarized in Table 7. A list of excluded studies is presented in Appendix 3.
Table 7: Details of Included Studies
Detail | Study 310 | Study 821 | Study 231 | Study 718 |
|---|---|---|---|---|
Designs and populations | ||||
Study design | Phase III, placebo-controlled, DB RCT | Phase III, placebo-controlled, DB RCT | Phase III, placebo-controlled, DB RCT | Phase III, placebo-controlled, DB RCT |
Locations | US (76 sites) | US (112 centres randomized at least 1 patient) | US (68 sites) | Belgium, Denmark, Finland, and Sweden (23 centres) |
Patient enrolment dates | January 16, 2006, to November 19, 2007 | August 4, 2008, to July 30, 2009 | January 26, 2016, to July 5, 2017 | November 26, 2007, to June 26, 2009 |
Randomized (N) | 826 | 919 | 631 | 426 |
Common inclusion criteria |
Post-menopausal, defined as:
| |||
Other inclusion criteria | At least 1 moderate or severe symptom of VVA | Moderate to severe vaginal dryness or dyspareunia as the self-reported MBS of VVA | Moderate to severe vaginal dryness as the self-reported MBS of VVA | An intact uterus |
Common exclusion criteria | The studies excluded patients who had:
The studies also excluded patients who used the following within the specified number of days before the initial screening visit:
| |||
Other exclusion criteria | Patients who had:
| Patients who had:
| Patients who had:
| Patients who had:
|
Drugs | ||||
Intervention | Ospemifene 60 mg tablets and non-hormonal vaginal lubricant as needed Ospemifene was administered orally, once daily, in the morning, with food. | Ospemifene 60 mg tablets and non-hormonal vaginal lubricant as needed Ospemifene was administered orally, once daily, in the morning, with food. | Ospemifene 60 mg tablets and non-hormonal vaginal lubricant for vaginal dryness as needed Ospemifene was administered orally, once daily, in the morning, with food. | Ospemifene 60 mg tablets Ospemifene was administered orally, once daily, in the morning, with food. |
Comparator(s) | Placebo tablets and non-hormonal vaginal lubricant Placebo was administered orally, once daily, in the morning, with food. | Placebo tablets and non-hormonal vaginal lubricant Placebo was administered orally, once daily, in the morning, with food. | Placebo tablets and non-hormonal vaginal lubricant Placebo was administered orally, once daily, in the morning, with food. | Placebo was administered orally, once daily, in the morning, with food. |
Duration | ||||
Phase | ||||
Screening | 6 weeks | 6 weeks | 4 weeks | 6 weeks |
Double blind | 12 weeks | 12 weeks | 12 weeks | 52 weeks |
Follow-up | 4 weeks | 4 weeks | 2 weeks | 4 weeks |
Outcomes | ||||
Primary end points | 4 co-primary efficacy end points Change from baseline to week 12 in:
| 4 co-primary efficacy end points Change from baseline to week 12 in:
| 4 co-primary efficacy end points Change from baseline to week 12 in:
| 3 co-primary efficacy end points Change from baseline to week 12 in:
|
Secondary and exploratory end points | Secondary Efficacy End Points Change from baseline (screening) to week 4 in:
Change from baseline to week 4 and week 12 in:
Change from baseline in:
Change from baseline (screening) to week 12 in:
| Change from baseline to week 4 in:
Change from baseline to week 4 and week 12 in:
Change from baseline to week 12 in:
| Change from baseline to week 4 and week 8 in:
Change from baseline over 12 weeks in:
Change from baseline in markers of bone metabolism | Change from baseline to weeks 12, 26, and 52 in:
|
Notes | ||||
Publications | Portman (2013),24 Portman (2014),25 Nappi (2015),23 Constantine (2015)26 | Goldstein (2014)29 | ||
BMI = body mass index; DB = double blind; DBP = diastolic blood pressure; ECG = electrocardiogram; FSFI = Female Sexual Function Index; FSH = follicle stimulating hormone; MBS = most bothersome symptom; MI = Maturation Index; RCT = randomized controlled trial; SBP = systolic blood pressure; SERM = selective estrogen receptor modulator; SIL = squamous intraepithelial lesion; UDI-6 = Urinary Distress Inventory – Short Form; VVA = vulvovaginal atrophy.
Note: Eight additional reports were included: Bachmann (2010),22 Nappi (2015),23 Portman (2013),24 Portman (2014),25 Constantine (2015),26 Archer (2019),27 Goldstein (2019),28 and Goldstein (2014).29
aStudies 310, 821, and 231 specified that the mammogram was obtained at screening or within 9 months before randomization.
bStudies 821 and 231 specified that endometrial thickness was determined by a centrally read ultrasound. (Note for Study 231: mammograms obtained within 9 months of screening were to be available.)
cPatients in Study 718 were required to have an intact uterus.
dIn studies 231 and 718, if there was any uncertainty about the time of the last spontaneous bleeding, the post-menopausal status was confirmed with FSH levels > 40 IU/L (both studies) and estradiol levels < 0.20 nmol/L (Study 718 only).
eExcluded Bethesda system (2001) classifications included: atypical squamous cells of undetermined significance; HPV high-risk positive; atypical squamous cells — could not exclude high-grade SIL; atypical glandular cells (endocervical, endometrial, not otherwise specified); low-grade SIL; high-grade SIL; carcinoma; unsatisfactory specimen.
fStudy 231: Patients were excluded if they had a vaginal infection and refused treatment or the infection did not respond to treatment.
gEstimated glomerular filtration rate of less than 60 mL/min/1.73 m3 based on the Modification of Diet in Renal Disease Study.
hProhibited medications included: dietary supplements and herbal therapies with assumed clinically significant estrogenic vaginal effects; any form of local vaginal hormonal products; any form of oral or transdermal estrogen or progestin; any form of progestin implants (subdermal or intrauterine) or estrogen implants (pellets); any form of estrogen-alone or progestin-alone injectable drug therapy; sex hormones or medications that were expected to affect clinically significant sex hormone levels; any SERMs; any vaginal lubricant or moisturizer other than that provided by the sponsor for use in the study; systemic fluconazole, rifampicin, rifabutin, carbamazepine, phenytoin, or St John’s wort.
iMaturation value (MV) is defined as MV = (S × 1) + (I × 0.5) + (P × 0), where S = % of superficial cells; I = % of intermediate cells; and P = % of parabasal cells.
Source: Clinical Study Reports.7-11
Table 8: Details of Included Studies — Extension Study 310X
Detail | Study 310X |
|---|---|
Designs and populations | |
Study design | Phase III, randomized, placebo-controlled, DB, LTSE |
Locations | US (51 sites) |
Patient enrolment dates | May 16, 2006, to September 18, 2008 |
Randomized (N) | 180 |
Inclusion criteria | Patients who met the following criteria at week 12 of Study 310:
|
Exclusion criteria | Patients who:
|
Drugs | |
Intervention | Ospemifene 60 mg tablets once daily, orally, each morning with food. |
Comparator(s) | Ospemifene 30 mg tablets once daily or placebo tablets once daily. Treatment was administered orally, each morning with food. |
Durations | |
Phase | |
Preceding study (Study 310) | 12 weeks |
Double-blind safety extension | 40 weeks |
Follow-up | 4 weeks |
Outcomes | |
Primary end point and secondary and exploratory end points | NA (efficacy was not evaluated). |
Safety end points | Adverse events from the signing of informed consent of protocol 15 to 50310X through the follow-up period Treatment compliance, assessed by number of doses taken Frequency and reason for early discontinuation At weeks 26 and 52:
At week 52:
|
Notes | |
Publications | Simon (2013)30 |
BMI = body mass index; DB = double blind; LTSE = long-term safety extension; NA = not available.
Note: One additional report was included: Simon (2013).30
Source: Clinical Study Report.11
Description of Studies
The primary objective of studies 310, 821, and 231 was to assess the efficacy, safety, and tolerability of ospemifene 60 mg once daily compared to placebo in the treatment of symptoms of VVA in post-menopausal women. More specifically, Study 310 assessed the treatment of symptoms of VVA broadly; Study 821 assessed the treatment of moderate to severe vaginal dryness and moderate to severe vaginal pain associated with sexual activity; and Study 231 assessed the treatment of vaginal dryness as a symptom of VVA due to menopause. The primary objective of Study 718 was to assess the long-term safety of ospemifene 60 mg once daily in the treatment of VVA in post-menopausal women with an intact uterus.
All of the included studies were phase III, double-blind, placebo-controlled RCTs that enrolled post-menopausal women with VVA. Studies 310, 821, and 718 were conducted from 2006 to 2009, whereas Study 231 was conducted from 2016 to 2017. The 4 pivotal trials were conducted primarily in the US, with the exception of Study 718, which was conducted in Europe (Belgium, Denmark, Finland, and Sweden). No study sites were located in Canada. A total of 2,798 patients were enrolled in the 4 pivotal trials, Study 310 (N = 826), Study 821 (N = 919), Study 231 (N = 627), and Study 718 (N = 426). Study 310 included 3 treatment arms (ospemifene 30 mg, ospemifene 60 mg, and placebo); however, the ospemifene 30 mg treatment arm was not reported in this review because the dosage does not align with the Health Canada–approved dose. Patients in studies 310, 821, and 231 were randomized to ospemifene 60 mg once daily or placebo at a 1:1 ratio, respectively, using a randomization code (Study 310 and 821) or web- or voice-based interactive response technology (Study 231). Randomization was stratified by uterine status in Study 310, by MBS (vaginal dryness or vaginal pain associated with sexual activity) reported on the vaginal atrophy symptom questionnaire taken at randomization (Study 821), and by severity of vaginal dryness (moderate or severe) and uterine status (Study 231). In Study 718, patients were randomized to ospemifene 60 mg once daily or placebo at a 6:1 ratio, respectively, using permuted block randomization stratified by centre.
The duration of the double-blind treatment period was 12 weeks in studies 310, 821, and 231, and it was 52 weeks in Study 718. Each study was preceded by a 4-week or 6-week screening period during which pre-defined study entry criteria were reviewed, and each included a 2-week or 4-week follow-up period. Study 231 originally included a 92-week safety assessment period following week 12; however, this phase of the study was discontinued following a protocol amendment on November 2, 2016. Study 821 also originally planned to include a long-term open-label extension study, but this was not initiated.
Study 310X was an LTSE study following Study 310. Patients with an intact uterus who completed the protocol for Study 310 were eligible. Patients without an intact uterus had the option of continuing into Study 312. Study 312 is summarized in the Other Relevant Evidence section. Excluding the ospemifene 30 mg treatment arm, a total of 118 patients were included. Patients remained on the treatment they were assigned to in Study 310, and blinding was maintained. The treatment period was 52 weeks in total (which included 12 weeks of the core study), followed by a 4-week follow-up period.
Populations
Inclusion and Exclusion Criteria
Patients included in the 4 pivotal trials were required to be between the ages of 40 years and 80 years and post-menopausal, to have 5% or fewer superficial cells in the MI of the vaginal smear, and to have a vaginal pH greater than 5.0. In Study 310, patients were included if they had at least 1 moderate or severe symptom of VVA (vaginal dryness, dyspareunia, vaginal and/or vulvar irritation or itching, difficult and/or painful urination, or vaginal bleeding with sexual activity). In Study 821, patients were included if they self-reported moderate to severe vaginal dryness or dyspareunia as the MBS of VVA. In Study 231, patients were included if they self-reported moderate to severe vaginal dryness as the MBS of VVA. Patients were not required to report VVA as the MBS to be included in Study 718. Patients in studies 310, 821, and 231 were required to have been hysterectomized or have an intact uterus with a double-layer endometrial thickness of less than 4 mm at screening. Patients in Study 718 were required to have an intact uterus.
Patients were excluded from the 4 pivotal studies if they had evidence of hyperplasia, cancer, or other pathology from an endometrial biopsy at screening, clinically significant gynecological findings other than vaginal atrophy (e.g., vaginal prolapse of grade 2 or higher), or history or evidence of malignancy, or if they consumed more than 14 drinks containing alcohol per week. Additionally, patients were excluded if they used certain treatments within a specified number of days before the initial screening visit (Table 7).
In Study 310, 812 patients were excluded if they had a BMI of greater than or equal to 37 kg/m2; in Study 231, they were excluded if they had a BMI greater than or equal to 38.5 kg/m2; and in Study 718, they were excluded if they had a BMI greater than or equal to 30 kg/m2. In studies 310, 812, and 718, patients were excluded if they were currently using heparin, itraconazole, ketoconazole, or digitalis alkaloids. Additionally, patients in Study 812 and Study 718 were excluded if they were currently using HIV antivirals, clarithromycin, telithromycin, or nefazodone.
To be eligible for Study 310X, patients were required to have an intact uterus, to have met the inclusion and exclusion criteria for Study 310, and to have completed the protocol for Study 310. Patients were excluded if they had clinically significant abnormal findings at the week 12 end-of-study visit.
Baseline Characteristics
A summary of baseline characteristics is provided in Table 9. In studies 310, 821, 231, and 718, the mean age of patients was between 58.5 years (SD = 6.4) and 62.9 years (SD = 6.5), and the majority of patients were over the age of 55 years (54.0% to 58.3% were 55 years to 64 years; 16.9% to 34.9% were at least 65 years). Most patients in the included studies were White (84.7% to 100.0%), with Black patients representing 0% to 12.1% of patients; Asian patients represented fewer than 3% of patients in the 4 studies. Further, in Study 718, 49% of patients were from Finland, 28% were from Belgium, 15% were from Denmark, and 8.0% were from Sweden. The mean BMI ranged from 26.0 kg/m2 (SD = 4.4) to 27.3 kg/m2 (SD = 4.5) in studies 310, 821, and 231, and was not reported in Study 718. From 40.9% to 56.8% of patients in studies 310, 821, and 231 had an intact uterus; all patients were required to have an intact uterus in Study 718. Information about hot flashes was not reported in Study 718, but in studies 310 and 821, 59.0% to 63.0% of patients reported experiencing hot flashes at baseline. In contrast, 8.3% to 8.9% of patients in Study 231 reported experiencing hot flashes at baseline. The majority of patients (86.6% to 90.7%) in studies 310 and 821 did not report a UTI in the past 6 months. Information about UTIs at baseline was not reported in Study 231 or Study 718.
The proportion of patients who had previous experience with systemic or vaginal hormonal treatment varied between the studies. In the ospemifene and placebo treatment groups, respectively, 19.6% and 18.7% of patients in Study 310, 61.3% and 55.0% of patients in Study 821, 2.9% and 2.5% of patients in Study 231, and 57.0% and 52.4% of patients in Study 718 had previously received hormonal treatment. The proportions of patients in Study 821 who had previously used vaginal hormone products in the ospemifene and placebo treatment groups were 31.3% and 24.1%, respectively, as well as 34.2% and 39.7% in the ospemifene and placebo treatment groups in Study 718, respectively.
Baseline measurements for the MBS of VVA, cytology, and vaginal pH are provided in Table 10. Of the 544 patients in Study 310, 222 patients (41%) reported vaginal dryness as the MBS; 242 patients (44%) reported vaginal pain with sexual activity (dyspareunia) as the MBS; 67 patients (12%) reported vaginal and/or vulvar irritation or itching as the MBS; 4 patients (1%) reported difficult and/or painful urination as the MBS; and 5 patients (1%) reported vaginal bleeding with sexual activity as the MBS. Vaginal dryness was reported as moderate by 52% and 60% of patients in the ospemifene and placebo treatment groups, respectively, and as severe by 45% and 40% of patients in the ospemifene and placebo treatment groups, respectively. Most of the patients (70% and 71% in the ospemifene and placebo treatment groups, respectively) reported severe dyspareunia. Additionally, irritation or itching was reported as moderate by 57% and 65% of patients in the ospemifene and placebo treatment groups, respectively, and as severe by 30% and 24% of patients in the ospemifene and placebo treatment groups, respectively.
In Study 821, 314 patients (34%) reported vaginal dryness as their MBS, and 605 patients (66%) reported dyspareunia as their MBS. In Study 231, patients were required to have vaginal dryness as their MBS. Study 718 did not report assessments of MBS at baseline. More than 50% of patients in both studies reported severe vaginal dryness, and treatment groups were balanced by severity of MBS. In Study 821, treatment groups were balanced by severity of dyspareunia, with 66% (ospemifene) and 67% (placebo) of patients reporting severe dyspareunia.
The mean percentage of parabasal cells, mean percentage of superficial cells, and vaginal pH were also reported in the 4 studies and were balanced between treatment groups.
The baseline characteristics available for Study 310X were limited (Table 11); however, the characteristics of those who continued into the LTSE were similar to those in the core study in terms of age, race, and BMI.
Table 9: Summary of Baseline Characteristics (ITT Population)
Characteristic | Study 310 | Study 821 | Study 231 | Study 718 | ||||
|---|---|---|---|---|---|---|---|---|
OSP 60 mg N = 276 | PBO N = 268 | OSP 60 mg N = 463 | PBO N = 456 | OSP 60 mg N = 313 | PBO N = 314 | OSP 60 mg N = 363 | PBO N = 63 | |
Age (years), mean (SD) | 58.6 (6.3) | 58.9 (6.1) | 58.7 (6.6) | 58.5 (6.4) | 59.7 (6.6) | 59.8 (7.2) | 61.7 (6.2) | 62.9 (6.5) |
Age distribution, n (%) | ||||||||
< 45 years | NR | NR | 8 (1.7) | 7 (1.5) | 3 (1.0) | 7 (2.2) | 0 | 0 |
45 years to 54 years | NR | NR | 113 (24.4) | 106 (23.2) | 66 (21.1) | 58 (18.5) | 45 (12.4) | 5 (7.9) |
55 years to 64 years | NR | NR | 260 (56.2) | 266 (58.3) | 171 (54.6) | 174 (55.4) | 196 (54.0) | 36 (57.1) |
≥ 65 years | NR | NR | 82 (17.7) | 77 (16.9) | 73 (23.3) | 75 (23.9) | 122 (33.6) | 22 (34.9) |
Race, n (%) | ||||||||
White | 249 (90.2) | 242 (90.3) | 409 (88.3) | 396 (86.8) | 273 (87.2) | 266 (84.7) | 361 (99.4) | 63 (100.0) |
Black or African American | 18 (6.5) | 14 (5.2) | 28 (6.0) | 35 (7.7) | 38 (12.1) | 32 (10.2) | 1 (0.3) | 0 |
Asian | 4 (1.4) | 6 (2.2) | 8 (1.7) | 3 (0.7) | 1 (0.3) | 3 (1.0) | 1 (0.3) | 0 |
Other | 5 (1.8) | 5 (1.9) | 18 (3.9) | 22 (4.8) | 1 (0.3) | 13 (4.1) | 0 | 0 |
Missing | 0 | 1 (0.4) | 0 | 0 | 0 | 0 | 0 | 0 |
BMI (kg/m2), mean (SD) | 26.0 (4.4) | 26.1 (4.4) | 26.2 (4.3) | 26.2 (4.3) | 27.3 (4.5) | 27.1 (4.8) | NR | NR |
Intact uterus, n (%) | 128 (46.4) | 122 (45.5) | 242 (52.3) | 245 (53.8) | 128 (40.9) | 132 (42.0) | NA (100%) | NA (100%) |
Intact cervix, n (%) | 133 (48.2) | 129 (48.1) | 259 (55.9) | 259 (56.8) | NR | NR | NR | NR |
Both ovaries removed, n (%) | NR | NR | 151 (32.6) | 126 (27.6) | NR | NR | 4 (1.1) | 0 |
Number of pregnancies, mean (SD) | 2.4 (1.6) | 2.4 (1.5) | 2.5 (1.7) | 2.4 (1.7) | NR | NR | NR | NR |
Number of vaginal births, mean (SD) | 1.7 (1.5) | 1.6 (1.4) | 1.7 (1.4) | 1.7 (1.5) | NR | NR | NR | NR |
None | NR | NR | NR | NR | NR | NR | 51 (14.0) | 8 (12.7) |
1 | NR | NR | NR | NR | NR | NR | 53 (14.6) | 11 (17.5) |
2 | NR | NR | NR | NR | NR | NR | 147 (40.5) | 30 (47.6) |
3 or more | NR | NR | NR | NR | NR | NR | 112 (30.9) | 14 (22.2) |
Number of UTIs in the past 6 months, n (%) | ||||||||
0 | 239 (86.6) | 242 (90.3) | 420 (90.7) | 405 (88.8) | NR | NR | NR | NR |
1 | 24 (8.7) | 17 (6.3) | 31 (6.7) | 37 (8.1) | NR | NR | NR | NR |
2 | 9 (3.3) | 8 (3.0) | 7 (1.5) | 7 (1.5) | NR | NR | NR | NR |
3 | 2 (0.7) | 1 (0.4) | 4 (0.9) | 4 (0.9) | NR | NR | NR | NR |
4 or more | 1 (0.4) | 0 | 1 (0.2) | 3 (0.7) | NR | NR | NR | NR |
Missing | 1 (0.4) | 0 | 0 | 0 | NR | NR | NR | NR |
Currently experiencing hot flashes, n (%) | 174 (63.0) | 155 (57.8) | 273 (59.0) | 281 (61.6) | 26 (8.3) | 28 (8.9) | NR | NR |
Number of days with hot flashes per month | ||||||||
Mean (SD) | 19.6 (11.5) | 19.6 (11.1) | 18.6 (11.1) | 16.9 (11.6) | NR | NR | NR | NR |
Median (range) | 25.5 (1 to 31) | 20.0 (1 to 31) | 20.0 (1 to 31) | 15.0 (0 to 31) | NR | NR | NR | NR |
Previous hormone treatment, n (%) | 54 (19.6) | 50 (18.7) | 284 (61.3) | 251 (55.0) | 9 (2.9) | 8 (2.5) | 210 (57.9) | 33 (52.4) |
HRT (excluding vaginal) | NR | NR | 232 (50.1) | 210 (46.1) | 5 (1.6) | 5 (1.6) | NR | NR |
Vaginal hormone products | NR | NR | 145 (31.3) | 110 (24.1) | 4 (1.3) | 3 (1.0) | 124 (34.2) | 25 (39.7) |
Uterine prolapse | n = 355 | n = 351 | ||||||
Gr. 0 – normally positioned cervix or vaginal apex | NR | NR | 327 (92.1) | 327 (93.2) | NR | NR | 319 (88.6) | 57 (93.4) |
Gr. 1 – less than halfway to the hymenal ring | NR | NR | 28 (7.9) | 24 (6.8) | NR | NR | 41 (11.4) | 4 (6.6) |
Vaginal prolapse | — | — | n = 462 | n = 456 | — | — | — | — |
Gr. 0 – normal | NR | NR | 381 (82.5) | 384 (84.2) | NR | NR | 285 (79.2) | 53 (86.9) |
Gr. 1 – some bulging during Valsalva, no symptoms | NR | NR | 80 (17.3) | 72 (15.8) | NR | NR | 75 (20.8) | 8 (13.1) |
Gr. 2 – size approximately hen’s egg | NR | NR | 1 (0.2) | 0 | NR | NR | 0 | 0 |
Duration of VVA (years) | ||||||||
Mean (SD) | NR | NR | NR | NR | 8.4 (6.9) | 9.0 (7.8) | NR | NR |
Median (range) | NR | NR | NR | NR | 6.0 (0.2 to 38.0) | 6.0 (0.1 to 42.0) | NR | NR |
BMI = body mass index; Gr. = grade; HRT = hormone replacement therapy; ITT = intention to treat; NA = not available; NR = not reported; OSP = ospemifene; PBO = placebo; SD = standard deviation; UTI = urinary tract infection; VVA = vulvovaginal atrophy.
Source: Clinical Study Reports.7-10
Table 10: Summary of Baseline Measurements (ITT Population)
Characteristic | Study 310 | Study 821 | Study 231 | Study 718 | ||||
|---|---|---|---|---|---|---|---|---|
OSP N = 276 | PBO N = 268 | OSP N = 463 | PBO N = 456 | OSP N = 313 | PBO N = 314 | OSP N = 363 | PBO N = 63 | |
Most bothersome symptom at baseline | ||||||||
Vaginal dryness, N | 118 | 104 | 160 | 154 | 313 | 314 | NR | NR |
Mild | 4 (3.4) | 1 (1.0) | 0 | 0 | 0 | 0 | NR | NR |
Moderatea | 61 (51.7) | 62 (59.6) | 78 (48.8) | 75 (48.7) | 148 (47.3) | 143 (45.5) | NR | NR |
Severea | 53 (44.9) | 41 (39.4) | 82 (51.3) | 79 (51.3) | 165 (52.7) | 171 (54.5) | NR | NR |
Vaginal pain with sexual activity, N | 120 | 122 | 303 | 302 | — | — | — | — |
None | 4 (3.3) | 2 (1.6) | 0 | 0 | NA | NA | NR | NR |
Mild | 3 (2.5) | 1 (0.8) | 0 | 0 | NA | NA | NR | NR |
Moderatea | 29 (24.2) | 33 (27.0) | 102 (33.7) | 98 (32.5) | NA | NA | NR | NR |
Severea | 84 (70.0) | 86 (70.5) | 201 (66.3) | 203 (67.2) | NA | NA | NR | NR |
Vaginal and/or vulvar irritation or itching, N | 30 | 37 | NA | NA | NA | NA | NR | NR |
None | 0 | 1 (2.7) | NA | NA | NA | NA | NR | NR |
Mild | 4 (13.3) | 3 (8.1) | NA | NA | NA | NA | NR | NR |
Moderate | 17 (56.7) | 24 (64.9) | NA | NA | NA | NA | NR | NR |
Severe | 9 (30.0) | 9 (24.3) | NA | NA | NA | NA | NR | NR |
Difficult and/or painful urination, N | 2 | 2 | NA | NA | NA | NA | NR | NR |
Moderate | 1 (50.0) | 2 (100) | NA | NA | NA | NA | NR | NR |
Severe | 1 (50.0) | 0 | NA | NA | NA | NA | NR | NR |
Vaginal bleeding with sexual activity, N | 5 | 0 | NA | NA | NA | NA | NR | NR |
None | 1 (20.0) | 0 | NA | NA | NA | NA | NR | NR |
Moderate | 1 (20.0) | 0 | NA | NA | NA | NA | NR | NR |
Severe | 3 (60.0) | 0 | NA | NA | NA | NA | NR | NR |
Cytology | ||||||||
N | 272 | 261 | Dry = 160 Dys = 303 | Dry = 151 Dys = 302 | 306 | 308 | 363 | 63 |
% parabasal cells, mean (SD) | 39.3 (39.0) | 38.5 (37.6) | Dry = 45.9 (40.7) Dys = 51.1 (38.2) | Dry = 45.6 (40.5) Dys = 50.6 (39.9) | 25.8 (33.3) | 28.3 (33.1) | 52.9 (41.1) | 47.8 (40.4) |
% superficial cells, mean (SD) | 1.0 (3.4) | 0.9 (2.6) | Dry = 1.2 (3.2) Dys = 0.7 (1.4) | Dry = 0.9 (1.7) Dys = 0.8 (1.8) | 3.0 (7.6) | 2.8 (6.9) | 0.7 (1.3) | 0.7 (1.3) |
Vaginal pH | ||||||||
N | 276 | 268 | Dry = 160 Dys = 303 | Dry = 154 Dys = 302 | 313 | 314 | 363 | 63 |
Mean (SD) | 6.4 (0.8) | 6.3 (0.7) | Dry = 6.2 (0.8) Dys = 6.3 (0.8) | Dry = 6.3 (0.8) Dys = 6.3 (0.8) | 6.11 (0.7) | 6.14 (0.7) | 6.23 (0.7) | 6.20 (0.7) |
Dry = dryness stratum; Dys = dyspareunia stratum; ITT = intention to treat; NA = not applicable; NR = not reported; OSP = ospemifene; PBO = placebo; SD = standard deviation.
aIn Study 821, vaginal dryness as the most bothersome symptom at baseline was assessed in the dryness stratum; vaginal pain with sexual activity as the most bothersome symptom at baseline was assessed in the dyspareunia stratum.
Source: Clinical Study Reports.7-10
Table 11: Summary of Baseline Characteristics — Extension Study 310X (ITT Population)
Characteristic | OSP 60 mg N = 69 | PBO N = 49 |
|---|---|---|
Age (years), mean (SD) | 57.7 (5.9) | 58.2 (4.0) |
Race, n (%) | ||
White | 61 (88.4) | 43 (87.8) |
Black or African American | 4 (5.8) | 2 (4.1) |
Asian | 2 (2.9) | 1 (2.0) |
Other | 2 (2.8) | 2 (4.1) |
Missing | 0 (0) | 1 (2.0) |
BMI (kg/m2), mean (SD) | 24.8 (4.1) | 25.0 (3.8) |
BMI = body mass index; ITT = intention to treat; OSP = ospemifene; PBO = placebo; SD = standard deviation.
Source: Clinical Study Report.11
Interventions
In each of the included studies, the intervention employed was ospemifene 60 mg tablets, administered orally once daily in the morning with food. In studies 310, 821, and 231, the duration of treatment was 12 weeks and patients were instructed to use non-hormonal vaginal lubricant as needed. Patients were provided with a vaginal lubricant (K-Y jelly) and asked to record their use of it in a diary. Of note, patients in Study 821 were advised not to use vaginal lubricant and to refrain from sexual intercourse within 24 hours before any clinic visit. In Study 718, the duration of treatment was 52 weeks, and patients were not permitted to use non-hormonal vaginal lubricant during the first 12 weeks of treatment, but they were able to use it freely after week 12.
The studies were double blind and placebo controlled, using matching placebo tablets for oral administration. The ospemifene and placebo tablets were identical in size, weight, and colour, and could not be visually differentiated.
A summary of concomitant medication use reported by patients in the included studies is presented in Table 12. The proportions of patients reporting any concomitant medication use were not available for Study 310 or Study 231. In the dryness stratum of Study 821, 94% and 88% patients randomized to ospemifene and placebo, respectively, reported any concomitant medication use. In the dyspareunia stratum of Study 821, 93% and 94% of patients randomized to ospemifene and placebo, respectively, reported concomitant medication use. The most frequently reported concomitant medications in studies 310 and 821 were multivitamins, calcium, fish oil, acetylsalicylic acid, ibuprofen, and levothyroxine sodium. In Study 231, the most frequently reported concomitant medications were levothyroxine, omeprazole, acetylsalicylic acid, and ibuprofen. In Study 718, which reported prior and concomitant medication use, 91% and 87% of patients randomized to ospemifene and placebo, respectively, reported any concomitant medication use. The most frequently reported medications used by patients in Study 718 were ibuprofen, acetaminophen, simvastatin, and estradiol.
Outcomes
A list of efficacy end points identified in the CADTH review protocol that were assessed in the clinical trials included in this review is provided in Table 13. These end points are further summarized in the discussion that follows. A detailed discussion and critical appraisal of the outcome measures are provided in Appendix 4.
Table 12: Concomitant Medications (ITT Population)
Concomitant medication | Study 310 | Study 821 – dryness stratum | Study 821 – dyspareunia stratum | Study 231 | Study 718b | |||||
|---|---|---|---|---|---|---|---|---|---|---|
OSP N = 276 | PBO N = 268 | OSP N = 160 | PBO N = 154 | OSP N = 303 | PBO N = 302 | OSP N = 313 | PBO N = 314 | OSP N = 363 | PBO N = 63 | |
Any medication, n (%) | NR | NR | 150 (93.8) | 135 (87.7) | 283 (93.4) | 283 (93.7) | NR | NR | 331 (91.2) | 55 (87.3) |
Most commonly reported concomitant medications,a n (%) | ||||||||||
Ascorbic acid | 26 (9.4) | 27 (10.1) | 21 (13.1) | 17 (11.0) | < 10% | < 10% | < 10% | < 10% | NR | NR |
Alendronate sodium | 28 (10.1) | 19 (7.1) | < 10% | < 10% | < 10% | < 10% | < 10% | < 10% | < 10% | 0 |
Calcium | 56 (20.3) | 55 (20.5) | 28 (17.5) | 27 (17.5) | 69 (22.8) | 54 (17.9) | < 10% | < 10% | < 10% | < 10% |
Calcium D | < 10% | < 10% | 18 (11.3) | 10 (6.5) | < 10% | < 10% | < 10% | < 10% | < 10% | < 10% |
Multivitamins | 88 (31.9) | 79 (29.5) | 56 (35.0) | 45 (29.2) | 112 (37.0) | 112 (37.1) | < 10% | < 10% | < 10% | 0 |
Fish oil | 29 (10.5) | 38 (14.2) | 26 (16.3) | 17 (11.0) | 54 (17.8) | 45 (14.9) | < 10% | < 10% | < 10% | 0 |
Tocopherol (vitamin E) | 17 (6.2) | 29 (10.8) | < 10% | < 10% | < 10% | < 10% | < 10% | < 10% | < 10% | 0 |
Acetylsalicylic acid | 34 (12.3) | 37 (13.8) | 26 (16.3) | 31 (20.1) | 49 (16.2) | 42 (13.9) | 37 (11.8) | 37 (11.8) | < 10% | < 10% |
Ibuprofen | 42 (15.2) | 38 (14.2) | 24 (15.0) | 26 (16.9) | 43 (14.2) | 50 (16.6) | 37 (11.8) | 36 (11.5) | 70 (19.3) | 12 (19.0) |
Levothyroxine | < 10% | < 10% | < 10% | < 10% | < 10% | < 10% | 51 (16.3) | 50 (15.9) | < 10% | < 10% |
Levothyroxine sodium | 39 (14.1) | 29 (10.8) | < 10% | < 10% | 39 (12.9) | 33 (10.9) | NR | NR | < 10% | < 10% |
Vitamin D NOS | < 10% | < 10% | 17 (10.6) | 15 (9.7) | 44 (14.5) | 33 (10.9) | NR | NR | 0 | < 10% |
Paracetamol (acetaminophen) | < 10% | < 10% | 18 (11.3) | 12 (7.8) | < 10% | < 10% | < 10% | < 10% | 61 (16.8) | 10 (15.9) |
Omeprazole | < 10% | < 10% | < 10% | < 10% | < 10% | < 10% | 28 (8.9) | 40 (12.7) | < 10% | < 10% |
Simvastatin | < 10% | < 10% | < 10% | < 10% | < 10% | < 10% | < 10% | < 10% | 53 (14.6) | 10 (15.9) |
Estradiol | < 10% | 0 | 0 |||||||| | 0 |||||| | 0 | 0 | < 10% | < 10% | 61 (16.8) | 7 (11.1) |
OSP = ospemifene; ITT = intention to treat; NOS = not otherwise specified; NR = not reported; PBO = placebo.
aReported by greater than or equal to 10.0% of patients in either treatment group.
bReported as prior and concomitant medications.
Source: Clinical Study Reports.7-10
Table 13: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Time of assessment | Outcome measure | Study 310 | Study 821 | Study 231 | Study 718 |
|---|---|---|---|---|---|
Week 12 | Cytology: change from baseline in the percentage of parabasal cells in the Maturation Index of the vaginal smear | Co-primary | Co-primary | Co-primary | Co-primary |
Week 12 | Cytology: change from baseline in the percentage of superficial cells in the Maturation Index of the vaginal smear | Co-primary | Co-primary | Co-primary | Co-primary |
Week 12 | Vaginal pH: change from baseline in vaginal pH | Co-primary | Co-primary | Co-primary | Co-primary |
Week 12 | Symptoms: change from baseline in the severity of the most bothersome symptoma of VVA | Co-primary | Co-primary | Co-primary | NA |
Weeks 4 and 12 | Symptoms: severity of VVA symptoms (by symptom) in patients reporting the symptom as moderate or severe at baseline | Secondary | Secondary | NA | NA |
Weeks 4 and 12 | Symptoms: severity of the MBS by symptom (except for vaginal dryness and vaginal pain associated with sexual activity at week 12) | Secondary | NA | NA | NA |
Weeks 4 and 12 | Symptoms: severity of VVA symptoms (all) | Secondary | Secondary | NA | NA |
Week 12 | Symptoms: severity of VVA symptoms other than vaginal dryness (i.e., dyspareunia, vulvar and/or vaginal itching and/or irritation, dysuria, and/or vaginal bleeding associated with intercourse) | NA | NA | Secondary | NA |
Weeks 4 and 8b | Symptoms: severity of the MBSc | NA | Secondary | Secondary | NA |
Week 4d Week 8b Week 12 Week 26e Week 52e | Cytology: percentage of parabasal cells in the Maturation Index | Secondary | Secondary | Secondary | Secondary |
Week 4d Week 8b Week 12 Week 26e Week 52e | Cytology: percentage of superficial cells in the Maturation Index | Secondary | Secondary | Secondary | Secondary |
Week 4d Week 8b Week 12 Week 26e Week 52e | Vaginal pH | NA | Secondary | Secondary | Secondary |
Weeks 4 and 12 | Sexual function: total score and the domains of the FSFI | NA | Secondary | Secondary | NA |
Weeks 4 and 12 | Symptoms: urinary symptoms assessed by the UDI-6 | Secondary | Secondary | Secondaryf | NA |
FSFI = Female Sexual Function Index; MBS = most bothersome symptom; NA = not applicable; UDI-6 = Urinary Distress Inventory – Short Form; VVA = vulvovaginal atrophy.
aFor the co-primary outcome, the MBS was vaginal dryness or dyspareunia in Study 310 and Study 821. In Study 231, the MBS was vaginal dryness. Of note, Study 310 included patients reporting any of the symptoms of VVA (vaginal dryness, vaginal and/or vulvar irritation or itching, dysuria, dyspareunia, and vaginal bleeding associated with sexual activity) as moderate or severe. The Study 821 inclusion criteria indicated that patients must report either vaginal dryness or dyspareunia as the MBS, and in Study 231, the MBS had to be vaginal dryness.
bWeek 8 assessments were conducted in Study 231 only.
cAs noted, the MBS reported by patients was either vaginal dryness (Study 821 and 231) or dyspareunia (Study 821).
dWeek 4 assessments were conducted in studies 310, 821, and 231 only.
eWeek 24 and week 52 assessments were conducted in Study 718 only.
fReported as a total score.
Source: Clinical Study Reports.7-10
Symptoms
VVA and urogenital symptoms were identified as outcomes of importance to patients and the clinical expert consulted by CADTH. Specifically, the following symptoms were of interest to this review: vulvar and vaginal pain, vaginal dryness, dyspareunia, vaginal and/or vulvar irritation and/or itching, incontinence, and genitourinary prolapse.
VVA Symptoms
Vulvar and vaginal symptoms (i.e., vaginal dryness, vaginal and/or vulvar irritation or itching, dysuria, dyspareunia, and vaginal bleeding associated with sexual activity) were assessed using the VVA questionnaire. The VVA questionnaire is completed by the patient and based on a 1-month recall or recall since the last study visit. The first part of the questionnaire asks patients if they have had any of the symptoms of VVA in the past month (or since the last visit); if they have, it asks them to indicate the severity of the most severe episode using 1 of 4 options: none, mild, moderate, or severe, which correspond to scores of 0, 1, 2, or 3, respectively. In the second part of the questionnaire, patients are asked to identify which symptom of the symptoms they rated as moderate or severe in the first part of the questionnaire is the most bothersome. In Study 310 and 821, the investigator or qualified study personnel reviewed the questionnaire with patients at study visits. Study 231 did not specify whether the questionnaire was reviewed with patients.
In the studies that used the VVA questionnaire (studies 310, 821, and 231), symptoms were assessed based on:
the severity of the MBS of VVA identified by the patient (note: to be eligible for Study 821, the patient must have reported moderate or severe vaginal dryness or dyspareunia as their MBS at screening and randomization; to be eligible for Study 231, the patient must have reported moderate to severe vaginal dryness at screening and have indicated vaginal dryness as their MBS at screening and randomization)
the severity of each symptom of VVA.
The change from baseline in the most bothersome VVA symptom was assessed in Study 310 (any VVA symptom at week 4 and week 12), Study 821 (vaginal dryness or dyspareunia at week 4 and week 12), and Study 231 (vaginal dryness at weeks 4, 8, and 12). The changes from baseline in all symptoms reported were also assessed in these studies (at weeks 4 and 12 in Study 310 and 821, and at weeks 4, 8, and 12 in Study 231). The VVA questionnaire was not employed in Study 718.
All assessments of the change in VVA symptoms were made using the scores associated with the symptom severity rating. A change from baseline of –3, –2, –1, 0, or 1 was interpreted to indicate the following changes in symptom severity:
–3 indicated severe (3) to none (0)
–2 indicated severe (3) to mild (1), or moderate (2) to none (0)
–1 indicated severe (3) to moderate (2), moderate (2) to mild (1), or mild (1) to none (0)
0 indicated no change
+ 1 indicated none (0) to mild (1), mild (1) to moderate (2), or moderate (2) to severe (3).
The use of the MBS approach involving patients’ self-reported and -rated VVA symptoms is recommended by the FDA for standardizing patient-reported outcome measures for clinical studies of VVA treatments for post-menopausal women.31 No evidence of the validity, reliability, and responsiveness of the MBS approach using the VVA questionnaire in post-menopausal women was identified. Additionally, a formal MID for the VVA questionnaire was not identified.
Urinary Symptoms
The presence or absence of urinary symptoms (urgency, frequency, incontinence, retention, and pain or discomfort) were assessed using the UDI-6. Patients completed the questionnaire at study visits, and study personnel entered the date of completion and responses in the electronic case report form. The questionnaire asked patients if they experienced the following symptoms (yes/no): frequent urination, urine leakage related to the feeling of urgency, urine leakage related to physical activity, coughing, sneezing, small amounts of urine leakage (drops), difficulty emptying their bladder, and pain or discomfort in the lower abdominal or genital area. If the patient selected yes, they were asked to rate how much the symptom bothered them based on a 4-point scale: not at all (1), slightly (2), moderately (3), or greatly (4). If the symptom was absent, it was assigned a score of 0. The scores of the 6 items were summed to obtain a total score. No evidence of validity, reliability, responsiveness, or MID was estimated for the UDI-6 in post-menopausal patients with VVA-associated symptoms.
The changes from baseline in urinary symptoms were assessed at week 4 and week 12 in studies 310 and 821. In Study 231, the change from baseline in urinary symptoms was reported as a total score from the questionnaire, which ranged from 0 to 26, where lower values indicated less urinary distress.
Health-Related Quality of Life
HRQoL was not assessed in any of the included studies.
Sexual Function
Sexual function was evaluated in Study 821 and Study 231 using the FSFI. The FSFI (Rosen et al., 2000) was developed as a brief, multi-dimensional self-report instrument for assessing key dimensions of sexual function in women. The scale consists of 19 items that assess sexual function over the past 4 weeks and yield domain scores in 6 areas: sexual desire, arousal, lubrication, orgasm, satisfaction, and pain. Of the 19 items, 2 questions correspond to the desire domain, 4 correspond to arousal, 4 correspond to lubrication, and 3 correspond to each of orgasm, satisfaction, and pain.
A domain score is obtained by summing the score of each of the individual items of a domain, which is then multiplied by the domain factor. The domain factor for desire is 0.6; arousal and lubrication have a domain factor of 0.3 each; and the remaining domains (orgasm, satisfaction, and pain) have a domain factor of 0.4 each. The total score of the FSFI is derived from the sum of the 6 domain scores. The total score for the FSFI can range from 2.0 to 36.0 points. For each FSFI domain and total score, a higher score indicates a better rating of sexual function.
In these studies, patients completed the questionnaire at study visits. The questionnaires were then reviewed by study personnel, who entered the responses into the electronic case report form.
The FSFI has been translated into more than 20 languages and adapted in more than 30 countries.32,33 It has also been studied for use with multiple populations, including women in different age groups with diverse medical conditions and various sexual dysfunctions.32,33 Evidence of its validity and reliability has been demonstrated; however, none of the studies was specifically designed to test the psychometric properties of the FSFI in post-menopausal women with symptoms of VVA. The generalizability of the FSFI to this population is uncertain. Additionally, no evidence of responsiveness and no formal MID were identified.
Mental Health-Related Outcomes
Outcomes related to mental health, such as depression, anxiety, mood, and cognition, were not evaluated in any of the included studies.
Bone Mineral Density
Studies 310, 821, 231, and 718 did not assess outcomes related to bone mineral density.
Cytology
All of the included studies evaluated the change from baseline to week 12 in the percentage of parabasal cells in the MI of the vaginal smear. The changes from baseline to week 12 in the percentage of superficial cells were evaluated as well. Vaginal smear samples were taken from middle third of the lateral vaginal wall and obtained by the investigator throughout the study. Vaginal smear samples were evaluated at a central pathology laboratory by a qualified pathologist, who performed the cell count that was used to determine the percentage of parabasal cells and superficial cells per sample.
Vaginal pH
The change in vaginal pH from baseline to week 12 was evaluated in all of the included studies. It was also evaluated as a change from baseline to week 4 (Study 821 and Study 231), week 8 (Study 231), and weeks 26 and 52 (Study 718), as indicated in Table 13. Vaginal pH was measured by the investigator using a pH indicator strip by pressing the indicator strip against the middle third of the vaginal wall. In Study 821 and Study 231, 2 types of pH strips with different pH ranges were used. The first had a pH range from 4 to 7; if the measurement was outside of that range, the second pH strip (with a pH range from 2 to 9) was to be used. Patients were advised not to have sexual intercourse in the 24 hours before the measurement of vaginal pH. Additionally, patients in Study 821 were advised not to use vaginal lubricant 24 hours before measurement.
Adherence
None of the included studies assessed outcomes related to adherence as an efficacy outcome.
Harms
Safety outcomes were evaluated in all included studies. Study 310X was an LTSE of Study 310 that evaluated only safety outcomes.
Statistical Analysis
General Considerations
In Study 310 and Study 821, all efficacy and safety analyses were conducted in the ITT population. In addition, the primary efficacy analyses were performed in the PP population and were considered supportive. In Study 231, all efficacy analyses were conducted in the ITT population. The primary efficacy analyses were also conducted in mITT and PP populations and considered supportive. In Study 231, safety analyses were conducted in the safety population. In Study 718, efficacy analyses were conducted in the ITT population and safety analyses were conducted in the safety population.
Primary Outcomes
Studies 310, 821, and 231 employed 4 co-primary end points evaluated as the change from baseline to week 12 in the:
percentage of parabasal cells in the MI of the vaginal smear
percentage of superficial cells in the MI of the vaginal smear
vaginal pH
severity of the MBS of VVA.
In Study 310, the severity of the MBS of VVA end point was evaluated by symptom. The change in the severity of vaginal dryness was evaluated in patients who reported vaginal dryness as their MBS. Similarly, the change in the severity of dyspareunia was evaluated in patients who reported dyspareunia as their MBS. The evaluation of the co-primary variable of MBS of VVA was performed using the evaluation of these 2 symptoms (vaginal dryness and dyspareunia).
In Study 821, patients were stratified by their self-reported MBS (vaginal dryness or dyspareunia) at randomization and each stratum was analyzed as a separate experiment. To demonstrate a statistically significant improvement of ospemifene compared to placebo for the treatment of vaginal dryness, each co-primary end point was required to be in favour of ospemifene when analyzed in patients included in the dryness stratum. Similarly, to demonstrate a statistically significant improvement of ospemifene compared to placebo for the treatment of dyspareunia, each co-primary end point was required to be in favour of ospemifene when analyzed in patients who were included in the dyspareunia stratum.
Study 231 included only patients who reported vaginal dryness as their MBS; therefore, the co-primary end point of severity of the MBS of VVA was specific to vaginal dryness as a VVA symptom.
Study 718 included the same co-primary end points, with the exception of severity of MBS of VVA, for a total of 3 co-primary end points.
Power Calculation
A summary of the sample size and power calculations for studies 310, 821, 231, and 718 is provided in Table 14. Studies 310, 821, and 231 based the power calculations on the assumption of a 2-sided alpha level of 5%. The alpha level was not reported for Study 718. Additionally, Study 310 assumed a dropout rate of 15%; dropout rate assumptions were not reported in the other studies.
In Study 310, a sample size of 795 was required for greater than 99% power to detect a difference of 20% (SD = 40.3) in the change in parabasal cells, for 98% power to detect a difference of 5% (SD = 12.4) in the change in superficial cells, for 99% power to detect a difference of 0.5 (SD = 1.1) in vaginal pH, and for 81% power to detect a difference of 0.4 (SD = 0.94) in the change in severity of the MBS of VVA.
Sample size and power calculations were conducted separately for the dryness and dyspareunia strata in Study 821. A sample size of 250 patients in the vaginal dryness stratum and 500 patients in the dyspareunia stratum was required for 99% power to detect a difference of 34.0% (SD = 35.2) in the change of parabasal cells, a difference of 8.6% (SD = 12.1) in the change of superficial cells, and a difference of 0.90 (SD = 0.98) in vaginal pH. The sample size of 250 patients provided 90% power to detect a difference of 0.42 (SD = 0.99) in the MBS of vaginal dryness in vaginal dryness stratum. In the dyspareunia stratum, 500 patients provided 80% power to detect a difference of 0.30 (SD = 1.18) in the MBS of dyspareunia.
In Study 231, a sample size of 600 patients was required for greater than or equal to 90% power to detect a difference in each of the co-primary end points. The magnitude of the difference that could be detected was not reported; however, the effect size was available (Table 14). In Study 718, 350 patients (50 to receive placebo and 300 to receive ospemifene) were considered sufficient to provide supportive evidence of efficacy for the primary efficacy end points. Additional information was not provided.
End point | Sample size | Power | Difference in means (OSP vs. PBO) | Standard deviation |
|---|---|---|---|---|
Study 310 | ||||
Parabasal cells | 795 patients (265 per treatment group), assuming a 2-sided alpha level of 5% and a dropout rate of 15% | > 99% | 20% | 40.3 |
Superficial cells | 98% | 5% | 12.4 | |
Vaginal pH | 99% | 0.5 | 1.1 | |
Severity of MBS of VVAa | 81% | 0.4 | 0.94 | |
Study 821 | ||||
Parabasal cells | 250 patients (125 per treatment group) in the vaginal dryness stratum and 500 patients (250 per treatment group) in the dyspareunia stratum, both assuming a 2-sided alpha level of 5% | 99%b | 34.0% | 35.2 |
Superficial cells | 99%b | 8.6% | 12.1 | |
Vaginal pH | 99%b | 0.90 | 0.98 | |
MBS of vaginal dryness | 90% | 0.42 | 0.99 | |
MBS of dyspareunia | 80% | 0.30 | 1.18 | |
Study 231c | ||||
Parabasal cells | 600 patients (300 per treatment group) assuming a 2-sided alpha level of 5% | > 99% | Effect size: 0.83 | NR |
Superficial cells | > 99% | Effect size: 0.68 | NR | |
Vaginal pH | > 99% | Effect size: 0.70 | NR | |
MBS of vaginal dryness | 91% | Effect size: 0.27 | NR | |
Study 718 | ||||
Parabasal cells | 350 patients (50 to receive placebo and 300 to receive ospemifene) | NRc | NR | NR |
Superficial cells | NRc | NR | NR | |
Vaginal pH | NRc | NR | NR | |
MBS = most bothersome symptom; NR = not reported; OSP = ospemifene; PBO = placebo; vs. = versus; VVA = vulvovaginal atrophy.
aIt was estimated that 40% of patients would declare vaginal dryness as their MBS.
bPower calculations were performed for each stratum. Both the vaginal dryness stratum and the dyspareunia stratum had 99% power to detect the indicated differences based on the same size for each stratum.
cThe sample size calculation was based on an estimated effect size rather than difference in means.
dThe power was not reported, but the Clinical Study Report stated that “the sample size is considered to be sufficient to provide supportive evidence of efficacy for the primary efficacy end points.”
Source: Clinical Study Reports.7-10
Statistical Test or Model
A summary of the statistical analysis of efficacy end points is described in Table 15. In Study 310 and Study 821, the co-primary outcomes regarding the change in parabasal cells, change in superficial cells, and change in vaginal pH were analyzed using ANCOVA unless the assumptions were violated, in which case a non-parametric approach was used. Treatment and study centre were included as fixed effects for the ANCOVA model in Study 310 and Study 821, as well as uterus status in Study 310 only. The change in severity of the MBS of VVA was analyzed using the Cochran-Mantel-Haenszel approach, controlling for study centre and uterus status in Study 310, with only study centre in Study 821. In Study 231, the change in parabasal cells, change in superficial cells, and change in vaginal pH were analyzed using a mixed-effects model for repeated measures (MMRM), with treatment, week, treatment by week interaction, and study centre as fixed effects and baseline value as a covariate. The change in severity of the MBS of VVA in Study 231 was analyzed using a generalized estimating equations approach, with the same fixed effects and covariate as the MMRM analysis. In Study 718, the changes in parabasal and superficial cells were analyzed using the Cochran-Mantel-Haenszel approach with centre as a stratification factor. Further, the change in vaginal pH was analyzed using the ANCOVA model with treatment and study centre as fixed effects and baseline value as a covariate.
In Study 310, multiplicity was addressed using a step-down approach. Initially, ospemifene 60 mg versus placebo with vaginal dryness as the MBS was evaluated. If statistical significance was demonstrated for each of the 4 co-primary outcomes, dyspareunia as the MBS was evaluated at 60 mg. To demonstrate effectiveness of ospemifene in the treatment of VVA, the 4 co-primary outcomes must demonstrate statistically significant improvements compared to placebo. The evaluation was stopped if statistical significance was not achieved for any of the co-primary outcomes. Study 231 and Study 718 also did not report any methods to control for multiplicity. In Study 821, to demonstrate effectiveness, all 4 co-primary outcomes were required to demonstrate statistical significance of ospemifene 60 mg versus placebo. Each stratum was analyzed independently and in a similar manner.
Secondary outcomes were not controlled for multiplicity in any of the 4 pivotal studies.
Data Imputation Methods
In Study 310 and Study 821, a last observation carried forward (LOCF) approach was used for missing data in the ITT population when conducting efficacy analyses, with the exception of urinary symptoms (only observed cases were reported). Baseline assessments were carried forward for patients lost to follow-up after the baseline visit. For patients without a baseline measurement, a change score of 0 was used. Study 821 also noted that patients without a baseline or post-baseline measurement were excluded from the corresponding analysis.
In Study 310, missing week 4 data for patients who discontinued the study early with missing week 4 records were handled as follows:
If the early termination visit occurred at or before 35 days from randomization visit, the missing week 4 records were replaced by early termination records.
If the early termination visit occurred after 35 days from the randomization visit, the missing week 4 records were not replaced.
An LOCF approach was also used in Study 718 for the analysis of the co-primary end points when data were missing due to patient discontinuation or other causes. Patients with no post-baseline observations were excluded from the analysis (LOCF was not used).
All analyses performed in Study 231 were based on observed data, given that missing data were not imputed.
Subgroup Analyses
None of the included studies analyzed the data by the subgroups of interest to this review (i.e., by severity of atrophy or by prior treatment experience).
Sensitivity Analyses
In Study 231, a sensitivity analysis of the co-primary end point of MBS of vaginal dryness was conducted using an MMRM method in which categorical data were treated as continuous data. The sensitivity analysis was performed to evaluate the robustness of the generalized estimating equations approach used for the primary analysis.
Sensitivity analyses were not performed in studies 310, 821, 718, or 310X.
Secondary Outcomes of the Studies
A summary of the statistical analysis of secondary efficacy end points is included in Table 15. Secondary end points were analyzed in a similar manner to the co-primary end points in studies 310, 821, 231, and 718, with the following exceptions.
In Study 821, secondary end points were analyzed by stratum and as a combined dataset. Continuous secondary end points were analyzed using ANCOVA. The analyses were performed in a similar manner to the primary end point when analyzed by stratum. When analyzed as a combined dataset, the model included stratum rather than study centre. Additionally, an analysis including treatment by stratum interaction was also conducted for generalizability across the strata. For categorical secondary end points, or the change in severity of MBS by symptom, analyses were conducted using a Cochran-Mantel-Haenszel row mean score controlling for study centre (for analyses by stratum) or for stratum (for the combined dataset).
In Study 310 and Study 821, urinary symptoms were reported descriptively as a change from baseline and based on observed values for each time point. In Study 231, ANCOVA with baseline score as a covariate was used to analyze the UDI-6 as a change from baseline in total score and the FSFI as a change from baseline in total score and domain scores.
Table 15: Statistical Analysis of Efficacy End Points
End points | Statistical model | Adjustment factors | Sensitivity analyses | Handling of missing data |
|---|---|---|---|---|
Change in parabasal cells, change in superficial cells, and change in vaginal pH | ANCOVA If the assumptionsa of ANCOVA were severely violated, a non-parametric approach (rank-based analysis of variance method) was used. | Fixed effects: treatment, uterus status (intact uterus or not) and study centre Covariate: baseline value The non-parametric approach was stratified by study centre and uterus status, separately | None | LOCF |
Study 310 | ||||
Change in severity of VVA symptoms (MBS, any symptom, symptoms reported as moderate or severe at baseline) | Cochran-Mantel-Haenszel | Controlling for study centre and uterus status | None | LOCF |
Study 821 | ||||
Change in parabasal cells, change in superficial cells, and change in vaginal pH | ANCOVA If the assumptions of ANCOVA were severely violated, a rank-based analysis of variance method was used. | Fixed effects: treatment and study centre (when analyzed by stratum) or stratum (when analyzed as a combined dataset) Covariate: baseline value | None | LOCF (week 12) |
Change in FSFI total score and domain scores | Same as the previous outcomes (change in parabasal cells, change in superficial cells, and change in vaginal pH) | Same as the previous outcomes | Same as the previous outcomes | Same as the previous outcomes |
Change in severity of VVA symptoms (MBS, any symptom, symptoms reported as moderate or severe at baseline) | Cochran-Mantel-Haenszel | Controlling for study centre (when analyzed by stratum) or stratum (when analyzed as a combined dataset) | None | LOCF (week 12) |
Study 231 | ||||
Change in parabasal cells, change in superficial cells, and change in vaginal pH | MMRM | Fixed effects: treatment, week, treatment by week, interaction, and study centre Covariate: baseline value | None | None |
Change in MBS, change in VVA symptoms, other than vaginal dryness | Generalized estimating equations | Same as above | MMRM in which ordered categorical data are treated as continuous | None |
Change from baseline in domain scores and total FSFI score | ANCOVA | Covariate: baseline value | None | None |
Change from baseline in the UDI-6 | Same as the previous outcomes (change from baseline in domain scores and total FSFI score) | Same as the previous outcomes | None | None |
Study 718 | ||||
Change in parabasal cells and change in superficial cells | Cochran-Mantel-Haenszel | Centre as a stratification factor | None | LOCF |
Change in vaginal pH | ANCOVA | Fixed effects: treatment and study centre Covariate: baseline value | None | LOCF |
ANCOVA = analysis of covariance; FSFI = Female Sexual Function Index; LOCF = last observation carried forward; MBS = most bothersome symptom; MMRM = mixed-effects model for repeated measures; UDI-6 = Urinary Distress Inventory – Short Form; VVA = vulvovaginal atrophy.
aAssumptions: normality of errors, homogeneity of variances, and equality of slopes among treatment groups.
Source: Clinical Study Reports.7-10
Analysis Populations
In studies 310, 821, 231, and 718, all efficacy analyses were conducted on the ITT population. The ITT population included all randomized patients who received at least 1 dose of study medication (ospemifene or placebo).
An mITT population was used in Study 231 to conduct a supportive analysis of the primary end points. The mITT population included ITT patients who also met the following inclusion criteria: 5% or fewer superficial cells in the MI at screening, vaginal pH greater than 5.0 at screening and at randomization, and moderate to severe vaginal dryness as the self-reported MBS of VVA at screening and at randomization.
In studies 310, 821, and 231, the primary efficacy analysis was also performed on the PP population as supportive analyses. The PP population was determined before breaking the study blind. The PP population included patients from the ITT population who had completed at least 10 weeks of treatment and the end-of-study assessments, had taken at least 85% of the study medication (Study 231 specified within 12 weeks), did not have a vaginal infection or any other medical condition that would confound the primary efficacy assessment, and did not have any other major protocol violations (Study 231 specified within 12 weeks). Major protocol violations were reviewed on a case-by-case basis to determine eligibility for the PP dataset. An example of a violation that would exclude a patient is the use of concomitant hormonal medication.
In Study 310 and Study 821, all safety analyses were conducted on the ITT population using an as-treated approach; i.e., patients were assessed based on the treatment they received rather than on the treatment group to which they were randomized. For this review, these populations will be referred to as the safety population. Similarly, in Study 231 and Study 718, safety analyses were performed in the safety population, which was defined as all randomized patients who received at least 1 dose of a study drug; the analyses were performed according to the treatment received. In addition, it was noted that patients who received both ospemifene and placebo were included in the ospemifene treatment group.
Study 310X
All analyses were done on the ITT population. The ITT population was defined as all patients who entered the study and received at least 1 dose of study medication in Study 310X. All 180 enrolled patients were included in the ITT population.
Results
Patient Disposition
The disposition of patients in studies 310, 821, 231, and 718 is summarized in Table 16. Approximately 15% of patients in Study 310, 11% of patients in Study 821, and 11% of patients in Study 231 discontinued from the studies. Discontinuation rates were similar between the ospemifene and placebo treatment groups. In each of these studies, AEs were the most common reasons for discontinuation, followed by patient decision or withdrawal of consent. The proportion of patients who withdrew from the study was balanced by reason between the treatment groups, except for those withdrawing due to AEs (5.4% versus 3.1% for ospemifene versus placebo, respectively) and, in Study 821, patient decision (1.7% versus 4.2% for ospemifene versus placebo, respectively). In Study 718, 18% of patients discontinued, and the proportion of patients who withdrew was lower in the placebo group (12.7%) compared with the ospemifene group (19.0%). The most common reasons for discontinuing were AEs (9.5% versus 13.2%, placebo versus ospemifene, respectively) and patient request (1.6% versus 3.9%). Of the 14 patients who withdrew by patient request, 2 patients felt better with hormone replacement therapy, 4 patients had unresolved hot flashes, 2 patients complained of lack of effect, and 1 patient had surgery to remove an ovarian cyst and did not want further gynecological examinations. The other 5 patients withdrew for personal reasons or did not wish to provide further comment.
The number of patients in each study population analyzed is summarized in Table 16. In Study 310, reasons for exclusion from the PP population were limited to protocol deviations. This included no moderate or severe MBS at baseline (15 patients and 9 patients randomized to ospemifene and placebo, respectively), more than 5% superficial cells at baseline (7 patients and 10 patients randomized to ospemifene and placebo, respectively), and inadequate washout of medications before study (6 patients and 1 patient randomized to ospemifene and placebo, respectively). Other reasons included pH of 5 or less at baseline, and taking prohibited, hormonally active medications during study, baseline endometrial thickness of 4 mm or greater, homogenous or heterozygous for Factor V Leiden, a BMI of 37 or greater at baseline, and drug dispensing errors.
In Study 821, 11.4% of patients overall were excluded from the PP population. The most common reasons were less than 85% compliance with the treatment protocol (11.7% and 11.2% of patients in the ospemifene and placebo treatment groups, respectively) and treatment durations of less than 70 days (8.9% and 7.7% of patients in the ospemifene and placebo treatment groups, respectively). Other reasons for exclusion from the PP were infrequent and balanced between the treatment groups, with the exception of foreign organisms detected in the vaginal smear at baseline or visit 4, which occurred in 3.2% of patients in the ospemifene treatment group and 0.2% of patients in the placebo treatment group.
In Study 231, 14% and 16% of patients randomized to ospemifene and placebo, respectively, were excluded from the mITT set because they had more than 5% superficial cells in the MI at baseline or no measurement at baseline. Other reasons for exclusion were infrequent, occurring in fewer than 1% of patients. This warranted exclusion from the PP as well. Other reasons for exclusion from the PP included: treatment duration less than 10 weeks (9% of patients in both treatment groups), vaginal infection (4% and 2% of patients randomized to ospemifene and placebo, respectively), and incorrect dispensing of the study drug at any visit (1% and 2% of patients randomized to ospemifene and placebo, respectively).
Category | Study 310 | Study 821 | Study 231 | Study 718 | ||||
|---|---|---|---|---|---|---|---|---|
OSP | PBO | OSP | PBO | OSP | PBO | OSP | PBO | |
Screened, N | NR | NR | NR | NR | 2,058 | NR | NR | |
Randomized, N (%) | 276 | 268 | 463 | 456 | 316 | 315 | 363 | 63 |
Discontinued from study, N (%) | 42 (15.2) | 38 (14.2) | 47 (10.2) | 53 (11.6) | 33 (10.4) | 36 (11.4) | 69 (19.0) | 8 (12.7) |
Reason for discontinuation, N (%) | ||||||||
Adverse events | 13 (4.7) | 11 (4.1)a | 25 (5.4) | 14 (3.1) | 6 (1.9) | 10 (3.2) | 48 (13.2) | 6 (9.5) |
Lost to follow-up | 6 (2.2) | 4 (1.5) | 9 (1.9) | 9 (2.0) | 7 (2.2) | 7 (2.2) | 1 (0.3) | 0 |
Patient decision or withdrawal of consent | 14 (5.1) | 12 (4.5) | 8 (1.7) | 19 (4.2) | 13 (4.1) | 16 (5.1) | 14 (3.9) | 1 (1.6) |
Major protocol violation | 6 (2.2) | 7 (2.6) | 1 (0.2) | 2 (0.4) | 3 (0.9) | 0 (0) | 4 (1.1) | 1 (1.6) |
Other | 3 (1.1) | 4 (1.5) | 4 (0.9) | 9 (2.0) | 4 (1.3) | 3 (1.0) | 2 (0.6) | 0 |
Study populations | ||||||||
ITT, N | 276 | 268 | 463 | 456 | 313 | 314 | 363 | 63 |
mITT, N | NA | NA | NA | NA | 269 | 263 | NA | NA |
PP, N | 177 | 194 | 382 | 388 | 231 | 227 | NA | NA |
Safety, N | See ITT | See ITT | See ITT | See ITT | 317b | 310c | 363 | 63 |
ITT = intention to treat; mITT = modified intention to treat; OSP = ospemifene; NR = not reported; PBO = placebo; PP = per protocol.
aThis count does not include 2 patients in the placebo treatment group who discontinued due to treatment-emergent adverse events and who are accounted for in the summary of harms.
bThree hundred and 16 patients were randomized to ospemifene. Three patients did not receive the study drug and 4 patients who were randomized to placebo received ospemifene in error.
cThree hundred and 15 patients were randomized to placebo, and 4 received ospemifene in error. Additionally, 1 patient was excluded from the safety population because they were enrolled at 2 different sites at the same time.
Source: Clinical Study Reports.7-10
The safety population was identical to the ITT population in Study 718.
A summary of the patient disposition in Study 310X is available in Table 17. Of the 464 patients who completed Study 310, 118 patients (25%) enrolled in extension Study 310X. Overall, 23% of patients discontinued from study. The proportion of patients who withdrew was lower in the ospemifene group (17%) compared with the placebo group (31%). The most common reasons for discontinuation were patient decision or withdrawal of consent (11% overall) and AEs (4% overall). The proportion of patients who decided to discontinue was lower in the ospemifene group (7%) compared than in the placebo group (16%). The proportion of patients who discontinued due to AEs was greater in the ospemifene group (6%) compared with the placebo group (2%). Other reasons for discontinuing were reported in fewer than 3% of patients overall, including loss to follow-up, major protocol violation, significant non-compliance with treatment or study procedures, and other.
Exposure to Study Treatments
A summary of duration of exposure and compliance with study treatments is provided for studies 310, 821, 231, and 718 in Table 18, and for Study 310X in Table 19. In each of the 12-week studies (studies 310, 821, and 231), patients were exposed to treatment for approximately 80 days to 82 days, and the exposures were similar between treatment groups. In the 52-week study (Study 718), the median number of days on which patients were exposed to ospemifene was 362 (range = 8 to 392); for patients in the placebo group, the median number was 363 (range = 61 to 378).
In studies 310, 821, 231, and 718, the compliance rate, defined as the number of doses taken divided by the number of doses that should have been taken for the duration of the treatment period, was greater than 92% in all treatment groups. The compliance rates in studies 310 and 718 were lower in the ospemifene treatment groups relative to the placebo treatment groups (93% versus 96% in Study 310, and 95% versus 99% in Study 718).
In Study 310X, the mean durations of treatment were 254 days (SD = 70) in the ospemifene treatment group and 232 days (SD = 93) in the placebo treatment group. Further, the mean compliance rates were 85% (SD = 22) in the ospemifene treatment group and 93% (SD = 17) in the placebo treatment group. When analyzed as medians (ranges), the reported treatment durations and compliance rates were similar between treatment groups.
Table 17: Patient Disposition — Extension Study 310X
Patient disposition | OSP 60 mg | PBO |
|---|---|---|
Screened, N | NR | NR |
Randomized, N (%) | 69 | 49 |
Discontinued from study, N (%) | 12 (17.4) | 15 (30.6) |
Reason for discontinuation, N (%) | ||
Adverse events | 4 (5.8) | 1 (2.0) |
Loss to follow-up | 0 | 3 (6.1) |
Patient decision or withdrawal of consent | 5 (7.2) | 8 (16.3) |
Major protocol violation | 1 (1.4) | 2 (4.1) |
Significant non-compliance with treatment or study procedures | 2 (2.9) | 0 |
Other | 0 | 1 (2.0) |
ITT, N | 69 | 49 |
ITT = intention to treat; NR = not reported; OSP = ospemifene; PBO = placebo.
Source: Clinical Study Report.11
Table 18: Exposure to Study Treatments (ITT Population)
Category | Study 310 | Study 821 | Study 231 | Study 718 | ||||
|---|---|---|---|---|---|---|---|---|
OSP N = 276 | PBO N = 268 | OSP N = 463 | PBO N = 456 | OSP N = 313 | PBO N = 314 | OSP N = 363 | PBO N = 63 | |
Duration of treatment (days)a | ||||||||
N | 271 | 263 | 463 | 456 | 317 | 310 | 361 | 62 |
Mean (SD) | 80.4 (21.3) | 79.9 (18.4) | 80.4 (17.3) | 80.8 (17.3) | 81.6 (16.8) | 81.0 (17.2) | 321.5 (97.1) | 339.3 (74.9) |
Median (range) | 84.0 (–79 to 121) | 84.0 (4 to 117) | 84.0 (1 to 118) | 84.0 (1 to 103) | 85.0 (1 to 121) | 85.0 (4 to 134) | 362 (8 to 392) | 363 (61 to 378) |
Compliance rate (%) | ||||||||
N | 261 | 253 | 451 | 437 | 313 | 314 | 359 | 62 |
Mean (SD) | 92.8 (40.0) | 95.6 (20.4) | 95.0 (13.7) | 96.0 (10.3) | 98.8 (4.4) | 99.0 (2.2) | 94.9 (14.7) | 99.1 (13.6) |
Median (range) | 100 | 100 | 99.0 (3.6 to 120.5) | 99.0 (18.2 to 122.0) | 100.0 (32.9 to 100.0) | 100.00 (75.0 to 100.0) | 99.2 (8.0 to 196.4) | 99.4 (64.0 to 191.6) |
ITT = intention to treat; OSP = ospemifene; PBO = placebo; SD = standard deviation.
aIn Study 231, the duration of treatment was reported in the safety population. All other studies reported duration of treatment in the ITT population.
Source: Clinical Study Reports.7-10
Table 19: Exposure to Study Treatments (ITT Population) — Study 310x
Category | OSP 60 mg N = 69 | PBO N = 49 |
|---|---|---|
Duration of treatment (days)a | ||
N | 68 | 44 |
Mean (SD) | 253.6 (69.8) | 232.4 (93.0) |
Median (range) | 280.0 (23 to 312) | 278.5 (1 to 305) |
Compliance rate (%) | ||
N | 67 | 44 |
Mean (SD) | 84.6 (21.6) | 93.4 (17.2) |
Median (range) | 94.8 (8.3 to 105.3) | 98.8 (32.5 to 128.8) |
ITT = intention to treat; OSP = ospemifene; PBO = placebo; SD = standard deviation.
Source: Clinical Study Report.11
Efficacy
Only the efficacy outcomes and analyses of subgroups identified in the review protocol are reported in this section. See Appendix 3 for detailed efficacy data.
Symptoms
In studies 310, 821, 231, and 718, the VVA questionnaire was used to assess the symptoms of VVA; however, the symptoms were analyzed using different approaches. The change from baseline in symptoms of VVA reported as moderate or severe at baseline, the change from baseline in symptoms of VVA reported as the MBS at baseline, and in general, the change from baseline in symptoms of VVA (broadly) were considered clinically relevant and have been summarized. Further, although the VVA questionnaire evaluated the 5 symptoms of VVA and they were reported in the included studies, the results summarized in this section focus on 3 of the symptoms (vaginal dryness, dyspareunia, and vaginal irritation or itching), as per the CADTH systematic review protocol.
Most Bothersome Symptoms of VVA (Vaginal Dryness, Vaginal Pain Associated With Sexual Activity, Vaginal and/or Vulvar Irritation and/or Itching)
One of the co-primary end points in studies 310, 821, and 231 was the change from baseline in severity of vaginal dryness, reported as the self-reported MBS of VVA, assessed at week 12. The MBS of VVA was not assessed in Study 718. A summary of the results is provided in Table 20.
In Study 310, the analysis of the co-primary end point for MBS of vaginal dryness included patients in the ITT population who identified vaginal dryness as the MBS of VVA at baseline. Of this subset of patients, 51.7% and 59.6% of patients in the ospemifene and placebo treatment groups, respectively, reported moderate vaginal dryness as the MBS of VVA at baseline. Similarly, 44.9% of patients on ospemifene and 39.4% on placebo reported severe vaginal dryness as the MBS at baseline. At week 12, the mean changes in the severity of the symptom were –1.26 (SD = 1.03) and –0.84 (SD = 1.00) for the ospemifene and placebo treatment groups, respectively, indicating a greater reduction in symptom severity with ospemifene compared to placebo (P = 0.021). |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| Of note, 29.7% of patients in the ospemifene group and 18.3% in the placebo group reported no symptoms of vaginal dryness at week 12.
Patients in Study 821 were stratified at baseline by MBS. The analysis of the co-primary end point for MBS of vaginal dryness included only patients from the dryness stratum. At baseline in both the ospemifene and placebo treatment groups of the dryness stratum, 49% of patients reported moderate vaginal dryness as the MBS and 51% reported severe vaginal dryness as the MBS. At week 12, there was no difference between treatment groups in the change in severity of vaginal dryness as the MBS of VVA, based on a mean change of –1.3 (SD = 1.08) for ospemifene and –1.1 (SD = 1.02) for placebo (P = 0.080).
All patients included in Study 231 reported moderate or severe vaginal dryness as the MBS of VVA at baseline. The proportions of patients reporting moderate dryness were 47.3% and 45.5% for ospemifene and placebo, respectively. Severe dryness at baseline was reported by 52.7% of patients in the ospemifene group and 54.5% in the placebo group. The mean change in symptom severity from baseline to week 12 was –1.29 (SD = 1.01) for ospemifene and –0.91 (SD = 0.96) for placebo. This corresponded to an odds ratio of 2.23 (95% CI, 1.62 to 3.06; P < 0.0001).
The supportive analyses performed in Study 821 (PP population) and Study 231 (PP population and mITT population) were consistent with the primary analysis (P = 0.0143 and P < 0.0001, respectively).
Table 20: Change From Baseline in Vaginal Dryness, Reported as the MBS of VVA at Baseline (ITT Population)
Category | Study 310 | Study 821 (dryness stratum) | Study 231 | |||
|---|---|---|---|---|---|---|
OSP 60 mg N = 276 | PBO N = 268 | OSP 60 mg N = 463 | PBO N = 456 | OSP 60 mg N = 313 | PBO N = 314 | |
Baseline | ||||||
N | 118 | 104 | 160 | 154 | 313 | 314 |
Mild (1) | 4 (3.4) | 1 (1.0) | 0 | 0 | 0 | 0 |
Moderate (2) | 61 (51.7) | 62 (59.6) | 78 (48.8) | 75 (48.7) | 148 (47.3) | 143 (45.5) |
Severe (3) | 53 (44.9) | 41 (39.4) | 82 (51.3) | 79 (51.3) | 165 (52.7) | 171 (54.5) |
Mean (SD) | 2.42 (0.56) | 2.38 (0.51) | 2.5 (0.50) | 2.5 (0.50) | 2.53 (0.50) | 2.54 (0.50) |
Week 12 | ||||||
N | 118 | 104 | 160 | 154 | NR | NR |
None (0) | 35 (29.7) | 19 (18.3) | 48 (30.0) | 34 (22.1) | NR | NR |
Mild (1) | 43 (36.4) | 32 (30.8) | 51 (31.9) | 48 (31.2) | NR | NR |
Moderate (2) | 27 (22.9) | 30 (28.8) | 39 (24.4) | 44 (28.6) | NR | NR |
Severe (3) | 13 (11.0) | 23 (22.1) | 22 (13.8) | 28 (18.2) | NR | NR |
Mean (SD) | 1.15 (0.98) | 1.55 (1.03) | 1.2 (1.03) | 1.4 (1.03) | NR | NR |
Change from baseline | ||||||
N | 118 | 104 | 160 | 154 | 277 | 281 |
–3 | 14 (11.9) | 5 (4.8) | 23 (14.4) | 14 (9.1) | 39 (14.1) | 15 (5.3) |
–2 | 36 (30.5) | 23 (22.1) | 51 (31.9) | 39 (25.3) | 73 (26.4) | 63 (22.4) |
–1 | 38 (32.2) | 32 (30.8) | 39 (24.4) | 52 (33.8) | 94 (33.9) | 94 (33.5) |
0 | 27 (22.9) | 38 (36.5) | 44 (27.5) | 44 (28.6) | 70 (25.3) | 99 (35.2) |
+ 1 | 3 (2.5) | 6 (5.8) | 3 (1.9) | 5 (3.2) | 1 (0.4) | 10 (3.6) |
Mean (SD) | –1.26 (1.03) | –0.84 (1.00) | –1.3 (1.08) | –1.1 (1.02) | –1.29 (1.01) | –0.91 (0.96) |
OR (95% CI); P value, OSP vs. PBO | NA | NA | 2.23 (95% CI, 1.62 to 3.06); P < 0.0001 | |||
CMH P value, OSP vs. PBO | 0.021 | 0.080 | NA | |||
CI = confidence interval; CMH = Cochran-Mantel-Haenszel; ITT = intention to treat; MBS = most bothersome symptom; OR = odds ratio; OSP = ospemifene; PBO = placebo; SD = standard deviation; vs. = versus; VVS = vulvovaginal atrophy.
Notes: In Study 310, P values for treatment comparisons (each active vs. placebo) are from the CMH row mean score test controlling for uterine status (intact or hysterectomized) and pooled centre.
In Study 821, the P value was computed using CMH row mean score test controlling for centre.
In Study 231, odds ratio was exponential of the mean of cumulative log odds ratio. To calculate the odds ratio, 95% CI, and P value, the generalized estimating equations method was used.
Source: Clinical Study Reports.7-9
In studies 310 and 821, the change from baseline to week 12 in the MBS of dyspareunia was also a co-primary end point. This was evaluated in patients who reported dyspareunia as their MBS. In Study 310, the statistical significance for the assessment of vaginal dryness (described previously) needed to be demonstrated before performing the analysis for dyspareunia as the MBS of VVA. A summary of the results is provided in Table 21. Of note, Study 231 included only patients whose MBS was vaginal dryness; therefore, this outcome does not apply. As previously noted, Study 718 did not assess the MBS of VVA.
In Study 310 and Study 821, the majority of patients included in the analyses rated dyspareunia as severe at baseline (Study 310: 70% and 71% for ospemifene and placebo, respectively; Study 821: 66.3% and 67.2% for ospemifene and placebo, respectively). In Study 310, the mean change from baseline to week 12 in the severity of dyspareunia was –1.2 (SD = 1.3) in the ospemifene treatment group and –0.9 (SD = 1.1) in the placebo treatment group. In Study 821, the mean change from baseline in the severity of dyspareunia was –1.5 (SD = 1.08) in the ospemifene treatment group and –1.2 (SD = 1.12) in the placebo treatment group. The difference in the change in severity of dyspareunia was greater for patients in the ospemifene group than for those in the placebo group in both studies (Study 310, P = 0.023; Study 821, P = 0.0001). Additionally, 28.3% and 18.9% of patients in the ospemifene and placebo treatment groups of Study 310, respectively, reported no symptoms of dyspareunia at week 12. In Study 821, 38.0% of patients in the ospemifene group and 28.1% of patients in the placebo group reported no symptoms of dyspareunia at week 12.
The supportive analyses performed in the PP populations of Study 310 and Study 821 were consistent with the primary analyses (P = 0.004 and P = 0.0004, respectively).
Table 21: Change From Baseline in Dyspareunia, Reported as the MBS of VVA at Baseline (ITT Population)
Category | Study 310 | Study 821 (dyspareunia stratum) | ||
|---|---|---|---|---|
OSP 60 mg N = 276 | PBO N = 268 | OSP 60 mg N = 463 | PBO N = 456 | |
Baseline | ||||
N | 120 | 122 | 303 | 302 |
None (0) | 4 (3.3) | 2 (1.6) | 0 (0.0) | 1 (0.3) |
Mild (1) | 3 (2.5) | 1 (0.8) | 0 (0.0) | 0 (0.0) |
Moderate (2) | 29 (24.2) | 33 (27.0) | 102 (33.7) | 98 (32.5) |
Severe (3) | 84 (70.0) | 86 (70.5) | 201 (66.3) | 203 (67.2) |
Mean (SD) | 2.6 (0.7) | 2.7 (0.6) | 2.7 (0.47) | 2.7 (0.49) |
Week 12 | ||||
N | 120 | 122 | 303 | 302 |
None (0) | 34 (28.3) | 23 (18.9) | 115 (38.0) | 85 (28.1) |
Mild (1) | 35 (29.2) | 28 (23.0) | 76 (25.1) | 58 (19.2) |
Moderate (2) | 18 (15.0) | 24 (19.7) | 68 (22.4) | 80 (26.5) |
Severe (3) | 33 (27.5) | 47 (38.5) | 44 (14.5) | 79 (26.2) |
Mean (SD) | 1.4 (1.2) | 1.8 (1.2) | 1.1 (1.08) | 1.5 (1.16) |
Change from baseline | ||||
N | 120 | 122 | 303 | 302 |
–3 | 23 (19.2) | 13 (10.7) | 67 (22.1) | 47 (15.6) |
–2 | 26 (21.7) | 23 (18.9) | 93 (30.7) | 70 (23.2) |
–1 | 33 (27.5) | 30 (24.6) | 82 (27.1) | 76 (25.2) |
0 | 32 (26.7) | 49 (40.2) | 55 (18.2) | 102 (33.8) |
+ 1 | 3 (2.5) | 7 (5.7) | 6 (2.0) | 7 (2.3) |
+ 2 | 1 (0.8) | 0 | 0 | 0 |
+ 3 | 2 (1.7) | 0 | 0 | 0 |
Mean (SD) | –1.2 (1.3) | –0.9 (1.1) | –1.5 (1.08) | –1.2 (1.12) |
CMH P value, OSP vs. PBO | 0.023 | 0.0001 | ||
CMH = Cochran-Mantel-Haenszel; ITT = intention to treat; OR = odds ratio; OSP = ospemifene; PBO = placebo; SD = standard deviation; vs. = versus.
Study 310: The P values for treatment comparisons (each active vs. placebo) are from the CMH row mean score test controlling for uterine status (intact uterus or hysterectomized) and pooled centre.
Study 821: The P value was computed using CMH row mean score test controlling for centre.
Source: Clinical Study Reports.7,8
Symptoms of VVA (Vaginal Dryness, Dyspareunia) Reported as Moderate or Severe at Baseline
The change from baseline in VVA symptoms reported as moderate or severe at baseline was assessed in Study 310 and Study 821. These were secondary outcomes in both studies and not adjusted for multiplicity. Summaries of the results for vaginal dryness and dyspareunia are provided in Table 22 and Table 23, respectively. The change from baseline in VVA symptoms reported as moderate or severe at baseline was also assessed in Study 231, but was reported as the MBS of VVA, which was a requirement for inclusion in the study. The results for Study 231 are summarized in Table 20.
In Study 310 and Study 821, |||||||||||||||||||||||| of the patients included in the analysis of change from baseline in vaginal dryness (Table 22) reported vaginal dryness as moderate at baseline, and |||||||||||||||| reported it as severe at baseline. The proportion of patients reporting moderate dryness was |||||||| in Study 310 |||||||||||||||| for ospemifene and placebo, respectively) than in Study 821 ||||||||||||||| for ospemifene and placebo, respectively) and Study 231 (|||||||||||||||| for ospemifene and placebo, respectively).
In Study 310, the change from baseline |||||||||||||||| in the severity of vaginal dryness was |||||||| in patients in the ospemifene treatment group than in the placebo treatment group ||||||||, based on a mean change of |||||||||||||||| for ospemifene and |||||||||||||||| for placebo. |||||||||||||||||||||||||||||||| of patients in the ospemifene treatment group and |||||||| of patients in the placebo treatment group reported ||||||||||||||||||||||||||||||||||||||||.
In Study 821, the change from baseline |||||||||||||||| in the severity of vaginal dryness was |||||||||||||||| for ospemifene and |||||||||||||||| for placebo (ospemifene versus placebo, |||||||||||||||||||||||||||||||| of patients in the ospemifene treatment group and |||||||| of patients in the placebo treatment group reported ||||||||||||||||||||||||||||||||||||||||.
Table 22: Change From Baseline in Vaginal Dryness, Reported as Moderate or Severe at Baseline (ITT Population)
Category | Study 310 | Study 821 | ||
|---|---|---|---|---|
OSP 60 mg N = 276 | PBO N = 268 | OSP 60 mg N = 463 | PBO N = 456 | |
Baseline | ||||
N | |||||||| | |||||||| | |||||||| | |||||||| |
Moderate (2) | |||||||| | |||||||| | |||||||| | |||||||| |
Severe (3) | |||||||| | |||||||| | |||||||| | |||||||| |
Mean (SD) | |||||||| | |||||||| | |||||||| | |||||||| |
Week 12 | ||||
N | |||||||| | |||||||| | |||||||| | |||||||| |
None (0) | |||||||| | |||||||| | |||||||| | |||||||| |
Mild (1) | |||||||| | |||||||| | |||||||| | |||||||| |
Moderate (2) | |||||||| | |||||||| | |||||||| | |||||||| |
Severe (3) | |||||||| | |||||||| | |||||||| | |||||||| |
Mean (SD) | |||||||| | |||||||| | |||||||| | |||||||| |
Change from baseline | ||||
N | |||||||| | |||||||| | |||||||| | |||||||| |
–3 | |||||||| | |||||||| | |||||||| | |||||||| |
–2 | |||||||| | |||||||| | |||||||| | |||||||| |
–1 | |||||||| | |||||||| | |||||||| | |||||||| |
0 | |||||||| | |||||||| | |||||||| | |||||||| |
+ 1 | |||||||| | |||||||| | |||||||| | |||||||| |
Mean (SD) | |||||||| | |||||||| | |||||||| | |||||||| |
CMH P value,a OSP vs. PBO | |||||||| | |||||||| | ||
CMH = Cochran-Mantel-Haenszel; ITT = intention to treat; LOCF = last observation carried forward; OSP = ospemifene; PBO = placebo; SD = standard deviation; vs. = versus.
Note: Missing values were replaced through the LOCF for ITT patients.
aP values for treatment comparisons (OSP vs. PBO) are from the CMH row mean score test controlling for stratum (Study 310 = uterus status [intact or hysterectomized uterus] and pooled centre; Study 821 = dryness stratum and dyspareunia stratum). P values were not adjusted for multiplicity (i.e., type I error rate was not controlled for).
Source: Clinical Study Reports.7,8
The results for the change from baseline in dyspareunia reported as moderate or severe at baseline, as reported in studies 310, 821, and 231, are provided in Table 23. Of the patients included in the analysis in Study 310 and Study 821, |||||||| of patients reported |||||||| dyspareunia at baseline. In Study 231, |||||||||||||||| of patients reported |||||||| dyspareunia at baseline. Of note, Study 310 included patients if they had at least 1 moderate or severe symptom of VVA (vaginal dryness, dyspareunia, vaginal and/or vulvar irritation or itching, difficult and/or painful urination, or vaginal bleeding with sexual activity), and Study 821 included patients with moderate to severe vaginal dryness or dyspareunia as the MBS of VVA.
In Study 310, the change in severity of dyspareunia from baseline ||||||||||||||||||||||||||| in the ospemifene treatment group and |||||||||||||||||||||||| in the placebo treatment group ||||||||||||||||. The proportions of patients included in the analysis reporting no symptoms of dyspareunia at week 12 were |||||||||||||||| in the ospemifene and placebo treatment groups, respectively. In Study 821, the change in severity of dyspareunia from baseline |||||||||||||||||||||||||||||||||||| in the ospemifene treatment group and |||||||||||||||||||||||| in the placebo treatment group. The proportions of patients included in the analysis reporting no symptoms of dyspareunia at week 12 were |||||||||||||||||||||||| in the ospemifene and placebo treatment groups, respectively.
In Study 231, the change from baseline in severity of dyspareunia was |||||||||||||||||||||||||||||||||||||||| in the ospemifene treatment group and |||||||||||||||||||||||||||||||| in the placebo treatment group. This corresponded to ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||.
Table 23: Change From Baseline in Dyspareunia, Reported as Moderate or Severe at Baseline (ITT Population)
Category | Study 310 | Study 821 | Study 231 | |||
|---|---|---|---|---|---|---|
OSP 60 mg N = 276 | PBO N = 268 | OSP 60 mg N = 463 | PBO N = 456 | OSP 60 mg N = 313 | PBO N = 314 | |
Baseline | ||||||
N | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| |
Moderate (2) | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| |
Severe (3) | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| |
Mean (SD) | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| |
Week 12 | ||||||
N | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| |
None (0) | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| |
Mild (1) | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| |
Moderate (2) | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| |
Severe (3) | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| |
Mean (SD) | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| |
Change from baseline | ||||||
N | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| |
–3 | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| |
–2 | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| |
–1 | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| |
0 | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| |
+ 1 | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| |
Mean (SD) | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| |
OR (95% CI),a P value, OSP vs. PBO | |||||||| | |||||||| | |||||||| | |||
CMH P value,b OSP vs. PBO | |||||||| | |||||||| | |||||||| | |||
CI = confidence interval; CMH = Cochran-Mantel-Haenszel; ITT = intention to treat; LOCF = last observation carried forward; OR = odds ratio; OSP = ospemifene; PBO = placebo; SD = standard deviation; vs. = versus.
Note: Missing values are replaced through the LOCF for ITT patients in Study 310 and Study 821. The analysis in Study 231 was performed using observed cases.
aOdds ratio: exponential of the mean of cumulative log odds ratio. To calculate the odds ratio, 95% CI, and P value, the generalized estimating equations method was used. The reported P value was not adjusted for multiplicity (i.e., type I error rate was not controlled for).
bP values for treatment comparisons (OSP vs. PBO) are from the CMH row mean score test controlling for stratum (Study 310 = uterus status [intact or hysterectomized uterus] and pooled centre; Study 821 = dryness stratum and dyspareunia stratum). P values were not adjusted for multiplicity (i.e., type I error rate was not controlled for).
Source: Clinical Study Reports.7-9
Other Symptoms of VVA (Vaginal Irritation and/or Itching)
Study 821 and 231 assessed the severity of vaginal irritation or itching in patients with moderate or severe vaginal dryness (both studies) or dyspareunia (Study 821). Study 821 assessed vaginal irritation or itching in all included patients, while Study 231 was limited to patients who reported moderate or severe symptoms at baseline. The results are summarized in Table 24. Vaginal irritation or itching as a symptom of VVA was also assessed in Study 310; however, the analyses were not limited to the patient population of interest (patients reporting moderate or severe vaginal dryness or dyspareunia at baseline). Therefore, the change from baseline in vaginal irritation or itching among patients who reported this symptom as the MBS of VVA in Study 310 was not reported for this review.
In Study 821, |||||||| in the ospemifene group and |||||||| in the placebo group reported experiencing vaginal irritation or itching to varying degrees of severity. Symptoms were reported |||||||||||||||||||||||||||||||| of patients, |||||||||||||||| of patients, and |||||||||||||||||||||||||||||||| of patients in the ospemifene and placebo treatment groups, respectively. At week 12, the mean change from baseline in symptom severity was |||||||||||||||||||||||||||||||| for ospemifene and |||||||||||||||| for placebo. Of note, |||||||||||||||||||||||| of patients in the ospemifene and placebo treatment groups reported |||||||||||||||| vaginal irritation or itching at week 12.
Of the patients included in the analysis for Study 231, |||||||| of patients in both treatment groups reported |||||||||||||||| vaginal irritation or itching at baseline and |||||||| of patients in both treatment groups reported |||||||| symptoms. The mean change in severity of symptoms from baseline to week 12 was |||||||||||||||| for ospemifene |||||||||||||||||||||||| for placebo, corresponding to an ||||||||||||||||||||||||||||||||||||||||.
Table 24: Change From Baseline in Vaginal Irritation or Itching (ITT Population)
Category | Study 821 | Study 231 | ||
|---|---|---|---|---|
OSP 60 mg (N = 463) | PBO (N = 456) | OSP 60 mg (N = 313) | PBO (N = 314) | |
Baseline | ||||
N | |||||||| | |||||||| | |||||||| | |||||||| |
None (0) | |||||||| | |||||||| | |||||||| | |||||||| |
Mild (1) | |||||||| | |||||||| | |||||||| | |||||||| |
Moderate (2) | |||||||| | |||||||| | |||||||| | |||||||| |
Severe (3) | |||||||| | |||||||| | |||||||| | |||||||| |
Mean (SD) | |||||||| | |||||||| | |||||||| | |||||||| |
Week 12 | ||||
N | |||||||| | |||||||| | |||||||| | |||||||| |
None (0) | |||||||| | |||||||| | |||||||| | |||||||| |
Mild (1) | |||||||| | |||||||| | |||||||| | |||||||| |
Moderate (2) | |||||||| | |||||||| | |||||||| | |||||||| |
Severe (3) | |||||||| | |||||||| | |||||||| | |||||||| |
Mean (SD) | |||||||| | |||||||| | |||||||| | |||||||| |
Change from baseline | ||||
N | |||||||| | |||||||| | |||||||| | |||||||| |
–3 | |||||||| | |||||||| | |||||||| | |||||||| |
–2 | |||||||| | |||||||| | |||||||| | |||||||| |
–1 | |||||||| | |||||||| | |||||||| | |||||||| |
0 | |||||||| | |||||||| | |||||||| | |||||||| |
+ 1 | |||||||| | |||||||| | |||||||| | |||||||| |
+ 2 | |||||||| | |||||||| | |||||||| | |||||||| |
+ 3 | |||||||| | |||||||| | |||||||| | |||||||| |
Mean (SD) | |||||||| | |||||||| | |||||||| | |||||||| |
CMH P value,c OSP vs. PBO | |||||||| | |||||||| | ||
OR (95% CI); P value,d OSP vs. PBO | |||||||| | |||||||||||||||||||||||||||||||||||||||||||||||| | ||
CI = confidence interval; CMH = Cochran-Mantel-Haenszel; ITT = intention to treat; OR = odds ratio; OSP = ospemifene; PBO = placebo; SD = standard deviation; vs. = versus.
aThe analysis included only patients who reported vaginal itching or irritation of moderate or severe severity at baseline.
bCalculated using the number of patients accounted for in the change from baseline values reported; N was not reported in the Clinical Study Report.
cP values for treatment comparisons (OSP vs. PBO) are from the CMH row mean score test controlling for stratum. The reported P value was not adjusted for multiplicity (i.e., type I error rate was not controlled for).
dOdds ratio: exponential of the mean of cumulative log odds ratio. To calculate the odds ratio, 95% CI, and P value, the generalized estimating equations (GEEs) method was used. The GEE model has the terms for treatment group, time, treatment by time, and study centre as fixed effects and baseline value as covariate. The reported P value was not adjusted for multiplicity (i.e., type I error rate was not controlled for).
Source: Clinical Study Reports.8,9
Urinary Symptoms
Urinary symptoms were assessed using the UDI-6 in studies 310, 821, and 231. Studies 310 and 821 reported the UDI-6 by each of the 6 domains (Table 25); Study 231 reported urinary symptoms as the total score for the UDI-6 (Table 26). Urinary symptoms were not reported in Study 718.
Results of the UDI-6 were reported descriptively. In Study 310 and Study 821, the majority of patients reported no change in each of the domains of the UDI-6 after 12 weeks of treatment. Further, the proportion of patients who reported improvement in each of the domains of the UDI-6 was similar between treatment groups.
In Study 231, the mean total scores at baseline were 5.3 (SD = 5.1) for ospemifene and 5.0 (SD = 4.9) for placebo. The LS means for the changes from baseline to week 12 were –1.3 (SD = 0.2) for ospemifene and –1.6 (SD = 0.2) for placebo, corresponding to an LS MD of 0.3 (95% CI, –0.2 to 0.9).
Table 25: Change From Baseline to Week 12 in Urinary Symptoms Assessed by the UDI-6 (ITT Population)
Category | Study 310 | Study 821 | ||
|---|---|---|---|---|
OSP 60 mg (N = 276) | PBO (N = 268) | OSP 60 mg (N = 463) | PBO (N = 456) | |
Frequent urination | ||||
N | 222 | 216 | 413 | 408 |
Improved | 63 (28.4) | 60 (27.8) | 97 (23.5) | 80 (19.6) |
Worsened | 25 (11.3) | 22 (10.2) | 53 (12.8) | 61 (15.0) |
No change | 134 (60.4) | 134 (62.0) | 263 (63.7) | 267 (65.4) |
Urine leakage related to feeling of urgency | ||||
N | 223 | 216 | 414 | 409 |
Improved | 53 (23.8) | 57 (26.4) | 90 (21.7) | 80 (19.6) |
Worsened | 26 (11.7) | 32 (14.8) | 50 (12.1) | 71 (17.4) |
No change | 144 (64.6) | 127 (58.8) | 274 (66.2) | 258 (63.1) |
Urine leakage related to physical activity | ||||
N | 221 | 216 | 414 | 409 |
Improved | 56 (25.3) | 47 (21.8) | 83 (20.0) | 92 (22.5) |
Worsened | 23 (10.4) | 27 (12.5) | 50 (12.1) | 48 (11.7) |
No change | 142 (64.3) | 142 (65.7) | 281 (67.9) | 269 (65.8) |
Small amount of urine leakage | ||||
N | 221 | 216 | 414 | 410 |
Improved | 55 (24.9) | 58 (26.9) | 91 (22.0) | 95 (23.2) |
Worsened | 32 (14.5) | 27 (12.5) | 61 (14.7) | 61 (14.9) |
No change | 134 (60.6) | 131 (60.6) | 262 (63.3) | 254 (62.0) |
Difficulty emptying bladder | ||||
N | 223 | 216 | 414 | 410 |
Improved | 26 (11.7) | 24 (11.1) | 48 (11.6) | 27 (6.6) |
Worsened | 11 (4.9) | 15 (6.9) | 28 (6.8) | 21 (5.1) |
No change | 186 (83.4) | 177 (81.9) | 338 (81.6) | 362 (88.3) |
Pain or discomfort in the lower abdominal or genital area | ||||
N | 223 | 216 | 414 | 410 |
Improved | 41 (18.4) | 37 (17.1) | 68 (16.4) | 57 (13.9) |
Worsened | 13 (5.8) | 16 (7.4) | 31 (7.5) | 39 (9.5) |
No change | 169 (75.8) | 163 (75.5) | 315 (76.1) | 314 (76.6) |
ITT = intention to treat; OSP = ospemifene; PBO = placebo; UDI-6 = Urinary Distress Inventory – Short Form.
Source: Clinical Study Reports.7,8
Table 26: Change From Baseline in Urinary Symptoms Assessed by the UDI-6 (ITT Population) in Study 231
Total score ouctomea | OSP 60 mg (N = 313) | PBO (N = 314) |
|---|---|---|
Baseline | ||
n | 311 | 314 |
Mean (SD) | 5.3 (5.1) | 5.0 (4.9) |
Median (range) | 4.0 (0 to 22) | 4.0 (0 to 24) |
Change from baseline to week 12 | ||
n | 275 | 280 |
LS mean (SE) | –1.3 (0.2) | –1.6 (0.2) |
Difference of LS mean (95% CI) | 0.3 (–0.2 to 0.9) | |
P valueb | 0.2448 | |
ANCOVA = analysis of covariance; CI = confidence interval; ITT = intention to treat; LS = least squares; OSP = ospemifene; PBO = placebo; SD = standard deviation; SE = standard error; UDI-6 = Urinary Distress Inventory – Short Form.
aTo calculate LS means, SE, 95% CI, and P value, the ANCOVA model was used. The ANCOVA model has the terms for treatment group as a fixed effect and baseline value as a covariate.
bThe reported P value was not adjusted for multiplicity (i.e., type I error rate was not controlled for).
Source: Clinical Study Report.9
Health-Related Quality of Life
HRQoL was not evaluated in any of the included studies.
Sexual Function
Sexual function was assessed using the FSFI in Study 821 and Study 231 and reported as the change from baseline to week 12 in each of the 6 domains as well as total score. The results are summarized in Table 27. Sexual function was not assessed in Study 310 or 718. To interpret the results of the FSFI, note that a higher score indicates a better rating of sexual function for each domain score and the total score. Additionally, secondary outcomes, including the FSFI, were not adjusted for multiplicity.
In Study 821, the changes from baseline to week 12 in the desire, arousal, orgasm, and satisfaction domains were negative, indicating a declining rating of sexual function. The magnitude of the changes was small, with an LS mean change of less than 1.0 in both the ospemifene and placebo treatment groups. The treatment-group differences between ospemifene and placebo were –0.25 (95% CI, –0.377 to –0.117) in the desire domain, –0.10 (95% CI, –0.308 to 0.115) in the arousal domain, 0.11 (95% CI, –0.076 to 0.292) in the orgasm domain, and –0.17 (95% CI, –0.324 to –0.007) in the satisfaction domain. The changes from baseline to week 12 in the lubrication and pain domains were positive, indicating an improvement in the rating of sexual function. The treatment-group differences between ospemifene and placebo were 0.14 (95% CI, –0.036 to 0.323) for the lubrication domain and 0.58 (95% CI, 0.327 to 0.838) for the pain domain.
In Study 231, the change from baseline to week 12 was positive for all domains of the FSFI, indicating an improvement in the rating of sexual function, although the magnitude of the changes was small. The treatment-group differences between ospemifene and placebo were 0.16 (95% CI, –0.02 to 0.34) in the desire domain, 0.20 (95% CI, –0.10 to 0.49) in the arousal domain, 0.40 (95% CI, 0.07 to 0.73) in the lubrication domain, 0.16 (95% CI, –0.16 to 0.48) in the orgasm domain, 0.16 (95% CI, –0.10 to 0.41) in the satisfaction domain, and 0.45 (95% CI, 0.11 to 0.80) in the pain domain.
The total score for the FSFI was associated with a treatment-group difference for ospemifene compared to placebo of 0.21 (95% CI, –0.470 to 0.883) in Study 821 and 1.59 (95% CI, 0.08 to 3.09) in Study 231.
Table 27: Change From Baseline in Sexual Function Assessed by the FSFIa (ITT Population)
Category | Study 821 | Study 231 | ||
|---|---|---|---|---|
OSP 60 mg (N = 463) | PBO (N = 456) | OSP 60 mg (N = 313) | PBO (N = 314) | |
Desire | ||||
Number of patients contributing to the analysis | 462 | 456 | 276 | 281 |
Baseline, mean (SD) | 4.81 (1.062) | 4.65 (1.160) | 2.50 (1.11) | 2.60 (1.18) |
Week 12, mean (SD) | 4.25 (1.230) | 4.39 (1.197) | NR | NR |
Change from baseline, LS mean (SE) | –0.52 (0.048) | –0.27 (0.048) | 0.56 (0.07) | 0.39 (0.06) |
Treatment-group difference (95% CI) OSP vs. PBO | –0.25 (–0.377 to –0.117) | — | 0.16 (–0.02 to 0.34) | — |
P valueb | 0.0002 | — | 0.0752 | — |
Arousal | ||||
Number of patients contributing to the analysis | 462 | 454 | 276 | 280 |
Baseline, mean (SD) | 3.55 (1.929) | 3.45 (1.887) | 2.17 (1.64) | 2.30 (1.71) |
Week 12, mean (SD) | 3.04 (1.692) | 3.10 (1.809) | NR | NR |
Change from baseline, LS mean (SE) | –0.52 (0.078) | –0.42 (0.079) | 0.64 (0.11) | 0.44 (0.11) |
Treatment-group difference (95% CI) OSP vs. PBO | –0.10 (–0.308 to 0.115) | — | 0.20 (–0.10 to 0.49) | — |
P valueb | 0.3696 | — | 0.1867 | — |
Lubrication | ||||
Number of patients contributing to the analysis | 463 | 456 | 275 | 281 |
Baseline, mean (SD) | 2.98 (1.627) | 3.02 (1.584) | 1.65 (1.43) | 1.65 (1.41) |
Week 12, mean (SD) | 3.23 (1.472) | 3.11 (1.569) | NR | NR |
Change from baseline, LS mean (SE) | 0.20 (0.066) | 0.06 (0.067) | 1.29 (0.12) | 0.89 (0.12) |
Treatment-group difference (95% CI) OSP vs. PBO | 0.14 (–0.036 to 0.323) | — | 0.40 (0.07 to 0.73) | — |
P valueb | 0.1180 | — | 0.0161 | — |
Orgasm | ||||
Number of patients contributing to the analysis | 463 | 454 | 276 | 281 |
Baseline, mean (SD) | 2.97 (1.785) | 2.98 (1.728) | 2.08 (1.89) | 2.03 (1.81) |
Week 12, mean (SD) | 3.01 (1.534) | 2.91 (1.656) | NR | NR |
Change from baseline, LS mean (SE) | –0.01 (0.068) | –0.12 (0.069) | 0.78 (0.12) | 0.63 (0.11) |
Treatment-group difference (95% CI) OSP vs. PBO | 0.11 (–0.076 to 0.292) | — | 0.16 (–0.16 to 0.48) | — |
P valueb | 0.2486 | — | 0.3400 | — |
Satisfaction | ||||
Number of patients contributing to the analysis | 454 | 446 | 262 | 271 |
Baseline, mean (SD) | 3.69 (1.287) | 3.69 (1.356) | 2.81 (1.42) | 2.84 (1.45) |
Week 12, mean (SD) | 2.96 (1.360) | 3.11 (1.374) | NR | NR |
Change from baseline, LS mean (SE) | –0.74 (0.059) | –0.58 (0.059) | 0.78 (0.09) | 0.62 (0.09) |
Treatment-group difference (95% CI) OSP vs. PBO | –0.17 (–0.324 to –0.007) | — | 0.16 (–0.10 to 0.41) | — |
P valueb | 0.0410 | — | 0.2195 | — |
Pain | ||||
Number of patients contributing to the analysis | 462 | 454 | 275 | 279 |
Baseline, mean (SD) | 1.57 (1.484) | 1.61 (1.456) | 1.65 (1.70) | 1.57 (1.56) |
Week 12, mean (SD) | 3.29 (2.253) | 2.74 (2.181) | NR | NR |
Change from baseline, LS mean (SE) | 1.68 (0.094) | 1.10 (0.095) | 1.47 (0.12) | 1.01 (0.12) |
Treatment-group difference (95% CI) OSP vs. PBO | 0.58 (0.327 to 0.838) | — | 0.45 (0.11 to 0.80) | — |
P valueb | 0.0000 | — | 0.0103 | — |
Total Score | ||||
Number of patients contributing to the analysis | 453 | 445 | 261 | 271 |
Baseline, mean (SD) | 19.84 (5.965) | 19.55 (6.070) | 13.05 (7.40) | 13.13 (7.29) |
Week 12, mean (SD) | 19.93 (5.675) | 19.57 (5.749) | NR | NR |
Change from baseline, LS mean (SE) | 0.11 (0.252) | –0.10 (0.253) | 5.71 (0.55) | 4.13 (0.54) |
Treatment-group difference (95% CI) OSP vs. PBO | 0.21 (–0.470 to 0.883) | — | 1.59 (0.08 to 3.09) | — |
P valueb | 0.5493 | — | 0.0392 | — |
ANCOVA = analysis of covariance; CI = confidence interval; FSFI = Female Sexual Function Index; ITT = intention to treat; LS = least squares; NR = not reported; OSP = ospemifene; PBO = placebo; SD = standard deviation; SE = standard error; vs. = versus.
aIn Study 821, P values were computed using ANCOVA, where change from baseline was the response variable, baseline assessment was the covariate, and treatment and stratum were fixed effects. In Study 231, the ANCOVA model was used to calculate LS means, SE, 95% CI, and P value. The ANCOVA model has the terms for treatment group as a fixed effect and baseline value as a covariate.
bThe reported P values were not adjusted for multiplicity (i.e., type I error rate was not controlled for).
Source: Clinical Study Reports.8,9
Mental Health-Related Outcomes
Outcomes related to mental health, such as depression, anxiety, mood, and cognition, were not evaluated in any of the included studies.
Bone Mineral Density
Markers of bone metabolism were reported in Study 231. Studies 310, 821, and 718 did not assess outcomes related to bone mineral density.
Cytology
Measurements of cytology include the percentage of parabasal cells and the percentage of superficial cells from a vaginal smear. The changes from baseline to week 12 in the percentages of parabasal and superficial cells were co-primary end points in studies 310, 821, 231, and 718.
The results for Study 310 and Study 231 are summarized in Table 28. In Study 310, the means of the changes in the percentages of parabasal cells from baseline to week 12 were –30.1 (SD = 37.93) and 3.98 (SD = 35.21) for the ospemifene and placebo treatment groups, respectively, in favour of ospemifene (P < 0.001). In Study 231, the LS means of the changes in the percentages of parabasal cells from baseline to week 12 were –23.7 (SE = 1.4) and –1.9 (SE = 1.4) for the ospemifene and placebo treatment groups, respectively, corresponding to a treatment-group difference of –21.8 (95% CI, –25.7 to –18.0) in favour of ospemifene (P < 0.0001). In Study 310, the changes from baseline to week 12 in the percentages of superficial cells were 10.8 (SD = 15.66) for the ospemifene group and 2.18 (SD = 8.39) for the placebo group, respectively, in favour of ospemifene (P < 0.001). In Study 231, the LS means of the changes in the percentages of superficial cells from baseline to week 12 were 7.8 (SE = 0.7) and 0.6 (SE = 0.7) for the ospemifene and placebo treatment groups, respectively, corresponding to a treatment-group difference of 7.2 (95% CI, 5.2 to 9.1) in favour of ospemifene (P < 0.0001).
The supportive analyses performed in Study 310 (PP population) and Study 231 (PP population and mITT population) were consistent with the primary analysis (P < 0.0001).
Table 28: Change From Baseline in the Percentage of Parabasal and Superficial Cells (Study 310 and Study 231, ITT Population)
Category | Study 310 | Study 231 | ||
|---|---|---|---|---|
OSP 60 mg (N = 276) | PBO (N = 268) | OSP 60 mg (N = 313) | PBO (N = 314) | |
% parabasal cells | ||||
Baseline, N | 272 | 261 | 306 | 308 |
Mean (SD) | 39.3 (38.98) | 38.5 (37.60) | 25.8 (33.3) | 28.3 (33.1) |
Week 12, N | 276 | 268 | 216 | 233 |
Mean (SD) | 8.78 (19.31) | 42.7 (37.22) | NR | NR |
Change from baseline | ||||
Mean (SD) | –30.1 (37.93) | 3.98 (35.21) | NA | NA |
LS mean (SE) | NA | NA | –23.7 (1.4) | –1.9 (1.4) |
Treatment-group difference (95% CI), OSP vs. PBOc | NA | NA | –21.8 (–25.7 to –18.0) | |
P value | < 0.001,a < 0.001b | < 0.0001 | ||
% superficial cells | ||||
Baseline, N | 272 | 261 | 306 | 308 |
Mean (SD) | 1.04 (3.37) | 0.91 (2.64) | 3.0 (7.6) | 2.8 (6.9) |
Week 12, N | 276 | 268 | 216 | 233 |
Mean (SD) | 12.1 (15.85) | 3.09 (8.62) | NR | NR |
Change from baseline | ||||
Mean (SD) | 10.8 (15.66) | 2.18 (8.39) | NA | NA |
LS mean (SE) | NA | NA | 7.8 (0.7) | 0.6 (0.7) |
Treatment-group difference (95% CI), OSP vs. PBOc | NA | NA | 7.2 (5.2 to 9.1) | |
P value | < 0.001,a < 0.001b | < 0.0001c | ||
CI = confidence interval; ITT = intention to treat; LS = least squares; NA = not applicable; NR = not reported; OSP = ospemifene; PBO = placebo; SD = standard deviation; SE = standard error; vs. = versus.
aThe P values for treatment comparisons (each active vs. placebo) from the rank-based analysis of variance were stratified by uterus status (intact or hysterectomized).
bThe P values for treatment comparisons (each active vs. placebo) from the rank-based analysis of variance were stratified by pooled centre.
cLS means, SE, and P values for the percentage of parabasal cells, the percentage of superficial cells, and vaginal pH were computed using a mixed-effects model for repeated measures. The P value for vaginal dryness was computed using the generalized estimating equations method.
Source: Clinical Study Reports.7,9
Study 821 analyzed cytology assessments by dryness stratum and dyspareunia stratum (Table 29). In the dryness stratum, the LS mean changes in the percentages of parabasal cells from baseline to week 12 for ospemifene and placebo were –31.7 (SE = 2.11) and –3.9 (SE = 2.18), respectively, corresponding to a treatment-group difference of –27.8 (95% CI, –33.75 to –21.90; P < 0.0001). In the dyspareunia stratum, the LS mean changes in the percentages of parabasal cells from baseline to week 12 for ospemifene and placebo were –40.3 (SE = 1.56) and –0.4 (SE = 1.57), respectively, with a treatment-group difference of –39.9 (95% CI, –44.15 to –35.63; P < 0.0001). Both analyses were in favour of ospemifene when compared to placebo.
In both the dryness stratum and dyspareunia stratum, the change from baseline to week 12 in the percentage of superficial cells was reported as a median (range) because the ANCOVA assumptions were not met. In the dryness stratum, the median of the change from baseline was 7.0 (range = –4 to 65) for ospemifene and 0.0 (range = –11 to 57) for placebo. In the dyspareunia stratum, the change from baseline to week 12 in the percentage of superficial cells was 7.0 (range = –6 to 79) for ospemifene and 0.0 (range = –5 to 85) for placebo. Both analyses were in favour of ospemifene compared to placebo (P < 0.0001 for both strata).
The supportive analyses performed in the PP population were consistent with the primary analyses (P < 0.0001).
Table 29: Change From Baseline in the Percentage of Parabasal and Superficial Cells (Study 821 by Stratum, ITT Population)
Category | Study 821 – dryness stratum | Study 821 – dyspareunia stratum | ||
|---|---|---|---|---|
OSP 60 mg (N = 160) | PBO (N = 154) | OSP 60 mg (N = 303) | PBO (N = 302) | |
% parabasal cells | ||||
Baseline, N | 160 | 151 | 303 | 302 |
Mean (SD) | 45.9 (40.70) | 45.6 (40.54) | 51.1 (38.21) | 50.6 (39.87) |
Week 12, N | 160 | 153 | 303 | 302 |
Mean (SD) | 14.2 (27.27) | 42.2 (36.47) | 11.0 (21.86) | 50.6 (38.81) |
Change from baseline | ||||
LS mean (SE) | –31.7 (2.11) | –3.9 (2.18) | –40.3 (1.56) | –0.4 (1.57) |
Treatment-group difference (95% CI), OSP vs. PBO | –27.8 (–33.75 to –21.90) | –39.9 (–44.15 to –35.63) | ||
P valuea | < 0.0001 | < 0.0001 | ||
% superficial cells | ||||
Baseline, N | 160 | 151 | 303 | 302 |
Median (range) | 0.0 (0 to 35) | 0.0 (0 to 11) | 0.0 (0 to 9) | 0.0 (0 to 21) |
Week 12, N | 160 | 153 | 303 | 302 |
Median (range) | 8.5 (0 to 67) | 1.0 (0 to 57) | 7.0 (0 to 79) | 0.0 (0 to 85) |
Change from baseline | ||||
Median (range) | 7.0 (–4 to 65) | 0.0 (–11 to 57) | 7.0 (–6 to 79) | 0.0 (–5 to 85) |
P valueb | < 0.0001 | < 0.0001 | ||
CI = confidence interval; ITT = intention to treat; LS = least squares; OSP = ospemifene; PBO = placebo; SD = standard deviation; SE = standard error; vs. = versus.
aThe P value was computed using ANCOVA, where change from baseline is the response variable, baseline assessment is the covariate, and treatment and centre are fixed effects.
bANCOVA assumptions were not met; therefore, the P value was computed using rank-based analysis of variance, stratifying by study centre.
Source: Clinical Study Report.8
Study 718 assessed the change from baseline to weeks 12, 26, and 52 in the percentage of parabasal and superficial cells (Table 30), with the assessment at week 12 as 2 of the 3 co-primary end points for the study. The assumptions of ANCOVA were not met; therefore, a non-parametric approach was used for the analysis of change in the percentage of parabasal and superficial cells. The medians of the changes in the percentages of parabasal cells from baseline to week 12 were –40 (95% CI, –55.0 to –30.0) for ospemifene and 0 (95% CI, 0.0 to 10.0) for placebo in favour of ospemifene (P < 0.0001). The results for the median of the change from baseline to week 26 and week 52 were similar to the results at week 12 (P < 0.0001). The median of the change from baseline to week 12 in the percentage of superficial cells was 5 (95% CI, 5.0 to 7.0) for ospemifene and 0 (95% CI, 0.0 to 0.0) for placebo in favour of ospemifene (P < 0.0001). The results for the median of the change from baseline to week 26 and week 52 in the percentage of superficial cells were similar to the results for week 12 (P < 0.0001).
Table 30: Change From Baseline in the Percentage of Parabasal and Superficial Cells (Study 718, ITT Population)
Category | OSP 60 mg (N = 363) | PBO (N = 63) | P valuea |
|---|---|---|---|
% parabasal cells | |||
Baseline, N | 363 | 63 | — |
Median (range) | 50 (0 to 100) | 48 (0 to 100) | — |
Week 12, N | 331 | 60 | |
Median (range) | 0 (0 to 100) | 70 (0 to 100) | — |
Change from baseline | |||
Week 12, median (range) | –40 (–100 to 75) | 0 (–90 to 98) | < 0.0001 |
95% distribution-free CI | –55.0 to –30.0 | 0.0 to 10.0 | — |
Week 26, median (range) | –45 (–100 to 90) | 0 (–80 to 100) | < 0.0001 |
95% distribution-free CI | –55.0 to –30.0 | 0.0 to 5.0 | — |
Week 52, median (range) | –45 (–100 to 82) | 4 (–60 to 97) | < 0.0001 |
95% distribution-free CI | –55.0 to –30.0 | 0.0 to 11.0 | — |
% superficial cells | |||
Baseline, N | 363 | 63 | — |
Median (range) | 0 (0 to 5) | 0 (0 to 5) | — |
Week 12, N | 331 | 60 | — |
Median (range) | 5 (0 to 60) | 0 (0 to 30) | — |
Change from baseline | |||
Week 12, median (range) | 5 (–5 to 60) | 0 (–5 to 28) | < 0.0001 |
95% distribution-free CI | 5.0 to 7.0 | 0.0 to 0.0 | — |
Week 26, median (range) | 4 (–5 to 55) | 0 (–5 to 20) | < 0.0001 |
95% distribution-free CI | 3.0 to 5.0 | 0.0 to 0.0 | — |
Week 52, median (range) | 2 (–5 to 50) | 0 (–4 to 8) | < 0.0001 |
95% distribution-free CI | 1.0 to 3.0 | 0.0 to 0.0 | — |
CI = confidence interval; CMH = Cochran-Mantel-Haenszel; ITT = intention to treat; OSP = ospemifene; PBO = placebo; SE = standard error.
aThe P value is from the CMH model.
Source: Clinical Study Report.10
Vaginal pH
The change from baseline to week 12 in vaginal pH was a co-primary end point in studies 310, 821, 231, and 718. The results for Study 310 and 231 are presented in Table 31; the results for Study 821 are presented by stratum in Table 32; and the results for Study 718 are presented in Table 33.
In Study 310, the mean changes from baseline to week 12 in vaginal pH were –1.0 (SD = 1.1) for ospemifene and –0.1 (SD = 08) for placebo in favour of ospemifene (P < 0.001). In Study 231, the LS mean changes from baseline to week 12 were –1.01 (SE = 0.04) for ospemifene and –0.29 (SE = 0.04) for placebo, corresponding to a treatment-group difference of –0.72 (95% CI, –0.84 to –0.59; P < 0.0001) in favour of ospemifene.
The supportive analyses performed in Study 310 (PP population) and Study 231 (PP population and mITT population) were consistent with the primary analysis (P < 0.0001).
In Study 821, the LS mean changes from baseline to week 12 in vaginal pH were –0.95 (SE = 0.07) and –0.94 (SE = 0.05) for ospemifene in the dryness and dyspareunia strata, respectively. The LS mean changes from baseline in the placebo treatment groups were –0.25 (SE = 0.07) and –0.07 (SE = 0.05) in the dryness and dyspareunia strata, respectively. The difference in the change in vaginal pH was in favour of ospemifene for both strata (P < 0.0001).
The supportive analyses performed in the PP population were consistent with the primary analyses (P < 0.0001).
Table 31: Change From Baseline in Vaginal pH (Study 310 and Study 231, ITT Population)
Category | Study 310 | Study 231 | ||
|---|---|---|---|---|
OSP 60 mg (N = 276) | PBO (N = 268) | OSP 60 mg (N = 313) | PBO (N = 314) | |
Baseline, N | 276 | 268 | 313 | 314 |
Mean (SD) | 6.4 (0.8) | 6.3 (0.7) | 6.11 (0.70) | 6.14 (0.73) |
Week 12, N | 276 | 268 | 277 | 280 |
Mean (SD) | 5.4 (0.9) | 6.2 (0.9) | NR | NR |
Change from baseline to week 12 | ||||
Mean (SD) | –1.0 (1.1) | –0.1 (0.8) | NA | NA |
LS mean (SE) | NA | NA | –1.01 (0.04) | –0.29 (0.04) |
Treatment-group difference (95% CI),a OSP vs. PBO | NA | NA | –0.72 (–0.84 to –0.59) | |
P value | < 0.001b < 0.001c | < 0.0001a | ||
ITT = intention to treat; NA = not applicable; NR = not reported; OSP = ospemifene; PBO = placebo; SD = standard deviation; SE = standard error; vs. = versus.
aA mixed-effects model for repeated measures was used to calculate LS means, SE, and P value.
bThe P values for treatment comparisons (each active vs. placebo) are from the rank-based analysis of variance stratified by uterine status (intact or hysterectomized).
cThe P values for treatment comparisons (each active vs. placebo) are from the rank-based analysis of variance stratified by pooled centre.
Source: Clinical Study Reports.7,9
Table 32: Change From Baseline to Week 12 in Vaginal pH (Study 821 by Stratum, ITT Population)
Category | Study 821 – dryness stratum | Study 821 – dyspareunia stratum | ||
|---|---|---|---|---|
OSP 60 mg | PBO | OSP 60 mg | PBO | |
Baseline, N | 160 | 154 | 303 | 302 |
Mean (SD) | 6.24 (0.80) | 6.26 (0.75) | 6.31 (0.77) | 6.31 (0.76) |
Week 12, N | 160 | 154 | 303 | 302 |
Mean (SD) | 5.32 (0.91) | 6.02 (0.93) | 5.37 (0.89) | 6.25 (0.96) |
Change from baseline | 160 | 154 | 303 | 302 |
LS mean (SE) | –0.95 (0.07) | –0.25 (0.07) | –0.94 (0.05) | –0.07 (0.05) |
P valuea | < 0.0001 | < 0.0001 | ||
ANCOVA = analysis of covariance; ITT = intention to treat; LS = least squares; OSP = ospemifene; PBO = placebo; SD = standard deviation; SE = standard error.
aThe P value was computed using ANCOVA where change from baseline was the response variable, baseline assessment was the covariate, and treatment and centre were fixed effects.
Source: Clinical Study Report.8
In Study 718, the mean changes from baseline to week 12 in vaginal pH were –1.21 (SD = 0.912) for ospemifene and –0.16 (SD = 0.945) for placebo, corresponding to a treatment-group difference of –0.97 (95% CI, –1.17 to –0.77; P < 0.0001) in favour of ospemifene. The analyses at week 26 and week 52 were based on observed cases, and yielded similar results to those reported at week 12.
Table 33: Change From Baseline to Week 12, 26, and 52 in Vaginal pH (Study 718, ITT Population)
Category | OSP 60 mg | PBO | Treatment-group difference, | P valuea |
|---|---|---|---|---|
Baseline, N | 363 | 63 | — | — |
Mean (SD) | 6.23 (0.728) | 6.20 (0.749) | NA | NA |
Week 12 (LOCF), N | 330 | 60 | — | — |
Mean (SD) | 5.03 (0.715) | 6.04 (0.887) | NA | NA |
Week 26 (OC), N | 313 | 58 | — | — |
Mean (SD) | 4.88 (0.752) | 6.18 (0.859) | NA | NA |
Week 52 (OC), N | 294 | 56 | — | — |
Mean (SD) | 4.92 (0.763) | 6.14 (0.997) | NA | NA |
Change from baseline | ||||
Week 12 (LOCF), mean (SD) | –1.21 (0.912) | –0.16 (0.945) | –0.97 (–1.17 to –0.77) | < 0.0001 |
Week 26 (OC), mean (SD) | –1.36 (0.981) | –0.02 (0.917) | –1.32 (–1.53 to –1.11) | < 0.0001 |
Week 52 (OC), mean (SD) | –1.30 (0.972) | –0.07 (1.210) | –1.21 (–1.44 to –0.98) | < 0.0001 |
ANCOVA = analysis of covariance; CI = confidence interval; ITT = intention to treat; LOCF = last observation carried forward; NA = not applicable; OC = observed case; OSP = ospemifene; PBO = placebo; SD = standard deviation; vs. = versus.
aThe estimated difference, CI of the difference, and P value comparing the treatments are model-based from ANCOVA.
Source: Clinical Study Report.10
Adherence
Adherence was not evaluated in any of the included studies.
Harms
Only those harms identified in the review protocol are reported in this section. See Table 34 and Table 35 for detailed harms data. Of note, all AEs reported herein were TEAEs.
Adverse Events
During the12-week period of studies 310, 821, and 231, 60%, 63%, and 35% of patients who received ospemifene reported at least 1 AE, respectively. In the placebo treatment groups, 52%, 51%, and 33% of patients in studies 310, 821, and 231 reported at least 1 AE, respectively. The most commonly reported AEs in all 3 studies were hot flashes (reported by 6% to 8% of patients who received ospemifene and 3% to 3% of patients who received placebo) and UTIs (reported by 2% to 8% of patients who received ospemifene and 3% to 5% of patients who received placebo); both AEs were reported in a greater proportion of patients in the ospemifene groups than in the placebo groups. In Study 310, fungal infections (likely vaginal infections), vulvovaginal mycotic infection, muscle spasms, and vaginal discharge were reported more frequently in the ospemifene treatment groups than in the placebo groups. In Study 821, vulvovaginal mycotic infection, vaginal candidiasis, and vaginal discharge were reported more frequently in the ospemifene treatment groups than in the placebo groups. In Study 310 and 821, 3% and 4% of patients who received ospemifene and 5% of patients in both studies who received placebo reported headaches.
In Study 718, the 52-week safety study, 85% of patients who received ospemifene and 76% of patients who received placebo reported at least 1 AE. The most commonly reported AEs, which were reported in a greater proportion of patients who received ospemifene, were nasopharyngitis, vaginal candidiasis, cystitis, muscle spasms, back pain, vaginal discharge, and hot flashes. The most commonly reported AEs that were reported in a greater proportion of patients who received placebo were UTIs, influenza-like illnesses, hyperhidrosis, and hypercholesterolemia. Headaches were also a commonly reported AE, but were reported in similar proportions of patients in both treatment groups.
The AEs reported in Study 310X are summarized in Table 35. AEs were reported by 64% and 45% of patients in the ospemifene and placebo treatment groups, respectively. The most common AEs were sinusitis and nasopharyngitis, which were reported more commonly in the placebo treatment group, as well as hot flashes, hypercholesterolemia, and pharyngolaryngeal pain, which were reported more frequently in the ospemifene group. UTIs were also common AEs, reported at a similar frequency in both treatment groups.
Serious Adverse Events
A summary of SAEs reported in studies 310, 821, 231, and 718 is provided in Table 34; the SAEs reported in Study 310X are summarized in Table 35. No SAEs were reported by patients who received ospemifene in Study 310; 1.5% of patients who received placebo reported at least 1 SAE. The proportions of patients reporting at least 1 SAE in studies 821 and 231 were similar between treatment groups (1.3% versus 1.5% in Study 821 and 1.6% versus 1.0% in Study 231 for ospemifene versus placebo). In Study 718, 4.9% of patients in the ospemifene group and 6.5% of patients in the placebo treatment group reported at least 1 SAE. Appendicitis, reported in 2 patients from the ospemifene group in Study 821, was the only SAE reported in more than 1 patient in any of the studies included in the systematic review.
In Study 310X, SAEs were reported by 5 patients (7.2%) and 1 patient (2.0%) in the ospemifene and placebo treatment groups, respectively (Table 35). Patients who received ospemifene reported the following SAEs (n = 1 for all): gastritis, non-cardiac chest pain, herpes encephalitis, meningitis candida, dehydration, chronic obstructive pulmonary disease, and breast prothesis implantation. The single SAE reported by a patient in the placebo treatment group was due to breast cancer in situ.
Withdrawals Due to Adverse Events
Withdrawals due to AEs in studies 310, 821, 231, and 718 are summarized in Table 34. The proportions of patients who discontinued treatment due to AEs were 5% for both ospemifene and placebo in Study 310, 6% for ospemifene and 3% for placebo in Study 821, and 2% for ospemifene and 3% for placebo in Study 231. In Study 718, 14% of patients in the ospemifene group and 10% of patients in the placebo group discontinued treatment due to AEs. Hot flashes were among the most common AEs that resulted in treatment discontinuation in studies 310, 821, 231, and 718, and were more frequently reported in the ospemifene treatment group in all studies except Study 310, in which they were reported by 1 patient who received ospemifene and 2 patients who received placebo. In Study 821, 3 patients in the ospemifene group and 1 patient in the placebo group discontinued treatment due to headaches. In Study 718, 5 patients in the ospemifene group and 1 patient in the placebo group discontinued treatment due to muscle spasms.
As summarized in Table 35, 6% and 2% of patients who received ospemifene and placebo, respectively, stopped treatment due to AEs. None of the specific AEs leading to study drug discontinuation were reported in more than 2 patients in Study 310X.
In Study 310X, patients discontinued treatment due to the following AEs: upper abdominal pain, meningitis candida, post-procedural complication, and hyperlipidemia in the ospemifene treatment group, and hypersensitivity in the placebo treatment group.
Mortality
No deaths were reported in any of the included studies.
Notable Harms
The following notable harms were included in the CADTH systematic review protocol: vaginal hemorrhage, abnormal genital bleeding, cervical dysplasia, breast mass, endometrial hyperplasia, uterine polyps, cardiovascular disorders (e.g., thromboembolic and hemorrhagic stroke, coronary heart disease), breast cancer, uterine cancer, DVT, and pulmonary embolism. In studies 310, 821, 231, and 718 (Table 34), a total of |||||||| patients in the ospemifene treatment groups and |||||||| patients in the placebo treatment groups reported vaginal hemorrhage. Uterine polyps were reported by 6 patients on ospemifene and 1 patient on placebo, respectively. Cervical dysplasia was reported in |||||||| patients in the ospemifene group and |||||||| patients in the placebo group, and breast mass was reported in 7 patients in each of the ospemifene and placebo treatment groups. Endometrial hyperplasia was reported in 1 patient who received ospemifene, and breast cancer was reported in 1 patient who received placebo. A total of 2 patients reported DVT, both in the ospemifene treatment groups. No patients experiencing abnormal genital bleeding, cardiovascular disorders, uterine cancer, or pulmonary embolism were reported.
In Study 310X (Table 35), vaginal hemorrhage, |||||||||||||||||||||||||||||||| and breast mass were each reported by 1 patient in the ospemifene treatment group. No other notable harms were reported.
Table 34: Summary of Harms (Safety)
Category | Study 310 | Study 821 | Study 231 | Study 718 | ||||
|---|---|---|---|---|---|---|---|---|
OSP N = 276 | PBO N = 268 | OSP N = 463 | PBO N = 456 | OSP N = 317 | PBO N = 310 | OSP N = 364 | PBO N = 62 | |
Patients with ≥ 1 adverse event | ||||||||
n (%) | 164 (59.4) | 140 (52.2) | 290 (62.6) | 232 (50.9) | 112 (35.3) | 103 (33.2) | 308 (84.6) | 47 (75.8) |
Most common events,a n (%) | ||||||||
Infection and infestations | ||||||||
Upper respiratory tract infection | — | — | — | — | 7 (2.2) | 11 (3.5) | — | — |
Urinary tract infection | 20 (7.2) | 8 (3.0) | 37 (8.0) | 23 (5.0) | 7 (2.2) | 10 (3.2) | 20 (5.5) | 7 (11.3) |
Urinary tract infection (enterococcal, staphylococcal or pseudomonal) | — | — | — | — | — | — | 7 (1.9) | 4 (6.5) |
Fungal infectionb | 9 (3.3) | 1 (0.4) | — | — | — | — | — | — |
Sinusitis | 7 (2.5) | 10 (3.7) | — | — | — | — | — | — |
Vulvovaginal mycotic infection | 11 (4.0) | 2 (0.7) | 17 (3.7) | 2 (0.4) | — | — | — | — |
Nasopharyngitis | — | — | 20 (4.3) | 16 (3.5) | — | — | 36 (9.9) | 4 (6.5) |
Vaginal candidiasis | — | — | 18 (3.9) | 2 (0.4) | — | — | 28 (7.7) | 1 (1.6) |
Cystitis | — | — | — | — | — | — | 19 (5.2) | 0 (0.0) |
Nervous system disorders | ||||||||
Headache | 7 (2.5) | 14 (5.2) | 16 (3.5) | 21 (4.6) | — | — | 33 (9.1) | 6 (9.7) |
Insomnia | — | — | — | — | — | — | 19 (5.2) | 0 (0.0) |
Musculoskeletal and connective tissue disorders | ||||||||
Muscle spasms | 11 (4.0) | 4 (1.5) | — | — | — | — | 31 (8.5) | 4 (6.5) |
Back pain | — | — | — | — | — | — | 24 (6.6) | 2 (3.2) |
Reproductive system and breast disorders | ||||||||
Vaginal Discharge | 11 (4.0) | 1 (0.4) | 21 (4.5) | 3 (0.7) | — | — | 20 (5.5) | 0 (0.0) |
Vascular disorders | ||||||||
Hot flashes | 23 (8.3) | 9 (3.4) | 32 (6.9) | 15 (3.3) | 20 (6.3) | 8 (2.6) | 46 (12.6) | 4 (6.5) |
General disorders and administration site conditions | ||||||||
Influenza-like illness | — | — | — | — | — | — | 14 (3.8) | 4 (6.5) |
Hyperhidrosis | — | — | — | — | — | — | 22 (6.0) | 5 (8.1) |
Metabolism and nutrition disorders | ||||||||
Hypercholesterolemia | — | — | — | — | — | — | 6 (1.6) | 4 (6.5) |
Patients with ≥ 1 SAE | ||||||||
n (%) | 0 | 4 (1.5) | 6 (1.3) | 7 (1.5) | 5 (1.6) | 3 (1.0) | 18 (4.9) | 4 (6.5) |
Most common eventsc n (%) | ||||||||
Appendicitis | — | — | 2 (0.4) | 0 | — | — | — | — |
Patients who stopped treatment due to adverse events | ||||||||
n (%) | 13 (4.7) | 13 (4.9) | 26 (5.6) | 15 (3.3) | 6 (1.9) | 10 (3.2) | 49 (13.5) | 6 (9.7) |
Most common events,c n (%) | ||||||||
Hot flashes | 1 (0.4) | 2 (0.7) | 4 (0.9) | 1 (0.2) | 3 (0.9) | 1 (0.3) | 8 (2.2) | 0 |
Muscle spasms | — | — | — | — | — | — | 5 (1.4) | 1 (1.6) |
Headaches | — | — | 3 (0.6) | 1 (0.2) | — | — | — | — |
Vaginal discharge | — | — | 2 (0.4) | 0 | — | — | — | — |
Drug hypersensitivity | — | — | 2 (0.4) | 0 | — | — | — | — |
Rash | — | — | 2 (0.4) | 0 | — | — | — | — |
Abdominal pain | 0 | 2 (0.7) | — | — | — | — | — | — |
Cough | 0 | 2 (0.7) | — | — | — | — | — | — |
Deaths | ||||||||
n (%) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Notable harms, n (%) | ||||||||
Vaginal hemorrhage (genital hemorrhage) | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| |
Abnormal genital bleeding | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Cervical dysplasia | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| | |||||||| |
Breast mass | 4 (1.4) | 1 (0.4) | 3 (0.6) | 5 (1.1) | 0 | 1 (0.3) | 0 | 0 |
Endometrial hyperplasia | 0 | 0 | 0 | 0 | 0 | 0 | 1 (0.3) | 0 |
Uterine polyps | 0 | 0 | 1 (0.2) | 1 (0.2) | 0 | 0 | 5 (1.4) | 0 |
Cardiovascular disordersd | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Breast cancer | 0 | 1 (0.4) | 0 | 0 | 0 | 0 | 0 | 0 |
Uterine cancer | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Deep vein thrombosis | 0 | 0 | 1 (0.2) | 0 | 0 | 0 | 1 (0.3) | 0 |
Pulmonary embolism | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
OSP = ospemifene; PBO = placebo; SAE = serious adverse event.
aFrequency greater than or equal to 3% for studies 310, 821, 231. Frequency greater than or equal to 5% for Study 718.
bThe Clinical Study Report indicated that all “fungal infections” were likely vaginal infections.
cFrequency greater than or equal to 2 patients.
dCardiovascular disorders (e.g., thromboembolic and hemorrhagic stroke, coronary heart disease).
Source: Clinical Study Reports.7-10
Table 35: Summary of Harms (Study 310x, Indirect Treatment Comparison)
Category | OSP 60 mg (N = 69) | PBO (N = 49) |
|---|---|---|
Patients with ≥ 1 adverse event | ||
n (%) | 44 (63.8) | 22 (44.9) |
Most common events,a n (%) | ||
Infection and infestations | ||
Urinary tract infection | 6 (8.7) | 4 (8.2) |
Sinusitis | 2 (2.9) | 2 (4.1) |
Nasopharyngitis | 1 (1.4) | 3 (6.1) |
Vascular disorders | ||
Hot flash | 5 (7.2) | 2 (4.1) |
Metabolism and nutrition disorders | ||
Hypercholesterolemia | 4 (5.8) | 1 (2.0) |
Respiratory, thoracic, and mediastinal disorders | ||
Pharyngolaryngeal pain | 4 (5.8) | 0 |
Patients with ≥ 1 SAEb | ||
n (%) | 5 (7.2) | 1 (2.0) |
Patients who stopped treatment due to adverse eventsb,c | ||
n (%) | 4 (5.8) | 1 (2.0) |
Deaths | ||
n (%) | 0 | 0 |
Notable harms, n (%) | ||
n (%) | ||
Vaginal hemorrhage | 1 (1.4) | 0 |
Abnormal genital bleeding | 0 | 0 |
Cervical dysplasia | |||||||| | |||||||| |
Breast mass | 1 (1.4) | 0 |
Endometrial hyperplasia | 0 | 0 |
Uterine polyps | 0 | 0 |
Cardiovascular disordersd | 0 | 0 |
Breast cancer | 0 | 0 |
Uterine cancer | 0 | 0 |
DVT | 0 | 0 |
Pulmonary embolism | 0 | 0 |
AE = adverse event; DVT = deep vein thrombosis; OSP = ospemifene; PBO = placebo; SAE = serious adverse event; WDAE = withdrawal due to adverse event.
aFrequency greater than or equal to 5%.
bFrequency greater than or equal to 2 patients; however, specific SAEs and WDAEs were not reported in more than 1 patient.
cThese counts included AEs that were ongoing from the parent study. At each level of summarization, patients reporting more than 1 AE are counted only once (under the strongest severity or causality reported).
dCardiovascular disorders (e.g., thromboembolic and hemorrhagic stroke, coronary heart disease).
Source: Clinical Study Report.11
Critical Appraisal
Internal Validity
Studies 310, 821, 231, and 718 used appropriate methods to randomize patients, and the double-blind study design was adequate for allocation concealment. Overall, study baseline characteristics were roughly balanced between treatment groups in all 4 studies. In Study 821, there was a difference between the ospemifene and placebo groups in terms of the proportion of patients who had had both ovaries removed (33% versus 28% for ospemifene versus placebo); in Study 821 and Study 718, more patients had received previous hormone treatment in the ospemifene groups compared to the placebo groups (61% versus 55% for ospemifene versus placebo in Study 821 and 58% versus 52% for ospemifene versus placebo in Study 718). There were also more patients with a grade 1 uterine prolapse and a grade 1 vaginal prolapse in the ospemifene treatment group versus the placebo group of Study 718 (uterine prolapse: 11% versus 7%; vaginal prolapse: 21% versus 13%). The clinical expert consulted by CADTH believed that differences in the number of vaginal births, grade 1 uterine or vaginal prolapse, and prior hormone treatment would not bias the efficacy results of the trials. In general, VVA symptoms indicated as the MBS at baseline were balanced between the treatment groups. In Study 310, 45% of patients in the ospemifene treatment group reported severe vaginal dryness at baseline compared to 40% of patients in the placebo treatment group. This difference may have biased the efficacy results against ospemifene.
In studies 310, 821, and 231, the duration of treatment was 12 weeks and patients were instructed to use non-hormonal vaginal lubricant as needed. In Study 718, the duration of treatment was 52 weeks and patients were not permitted to use non-hormonal vaginal lubricant during the first 12 weeks of treatment, but were able to use it freely following week 12. Of note, patients in Study 821 were advised not to use vaginal lubricant and to refrain from sexual intercourse within 24 hours before any clinic visit. Lubricant use was slightly more frequent in the placebo arm than in the ospemifene group in Study 310. Lubricant use was similar between treatment groups in Study 821 and Study 231, and was not reported in Study 718. Overall, concomitant medication use and non-hormonal lubricant use were not seen as having a differential effect on outcomes between treatment groups.
The rates of discontinuation from the studies ranged from 10% to 20% across trials and were similar between treatment groups, with the exception of Study 718, in which 19% of patients discontinued from ospemifene and 13% discontinued from placebo. The most common reasons for discontinuation in all studies were AEs or patient decision. The rate of discontinuation due to AEs was slightly higher in the ospemifene group than in the placebo group in Study 718. Although affecting fewer than 3% of patients in any treatment group, hot flashes were the most frequently reported AE leading to discontinuation from treatment. Further, hot flashes as an AE were consistently reported at a greater frequency in the ospemifene treatment groups than in the placebo treatment groups.
The LOCF approach was used to handle missing data in studies 310, 821, and 718. This was likely a conservative approach, given that with treatment, improvement was expected over time. Study 231 did not employ any methods to account for missing data; all analyses that were performed were based on observed data using MMRM. In Study 231, 10% to 20% of the data were missing for assessments of the co-primary end points at week 12. Although discontinuation rates were similar between treatment groups, it is unclear whether the missing-at-random assumption was upheld.
In all studies, efficacy analyses were performed using the ITT population. In studies 310 and 821, the primary efficacy analyses were also performed in the PP population and were considered supportive. In Study 231, the primary efficacy analyses were performed in the mITT and PP populations, and were also considered supportive. There was a notable difference in the number of patients included in the PP or mITT populations compared to the ITT populations; however, given that the reasons for patients’ exclusion from these datasets were similar between treatment groups, the exclusion was unlikely to cause bias. All of the supportive analyses performed were consistent with the primary analyses, with the exception of vaginal dryness as the MBS of VVA in Study 310; in that analysis, statistical significance was not demonstrated in the PP population. The sponsor attributed this to the small sample size, which is likely a contributing factor; however, the results of the analysis of vaginal dryness as the MBS of VVA in Study 310 remain uncertain.
Each of the studies enrolled enough patients to meet the requirement for the calculated sample size to adequately power the studies to detect a difference in each of the co-primary end points. The co-primary end points in Study 310 were controlled for multiplicity using a step-down approach. In Study 821, to claim effectiveness, demonstration of statistical significance was required for all 4 co-primary end points. Methods to control for multiplicity were not used in Study 231 or 718, although both studies reported statistical significance for all co-primary end points. None of the studies was powered to detect a difference in secondary outcomes, nor were secondary outcomes controlled for multiplicity.
Subgroup analyses were not described in any of the included studies, but a subset of the ITT population was used to perform analyses of outcomes for VVA symptoms. In Study 310, the co-primary end point of vaginal dryness or dyspareunia as the MBS of VVA was assessed in patients who reported vaginal dryness or dyspareunia as the MBS at baseline; however, this was not treated as a subgroup analysis and did not include the full ITT population. A protocol amendment in April 2007 changed the co-primary end point from change from baseline to week 12 in “most bothersome VVA symptom” to “severity of the most bothersome VVA symptom,” noting that symptoms of vaginal dryness and dyspareunia were evaluated within the context of the primary variable. This is logical from a practical perspective, but the study did not stratify patients by symptom or account for this in the sample size calculation, which is a limitation of the study design. Study 821 was stratified by MBS at baseline: dyspareunia or vaginal dryness.
Each of the studies used a mix of objective clinical outcomes and subjective patient-reported outcomes that were of interest to this review. The objective outcomes included cytology assessments (percentages of parabasal and superficial cells) and vaginal pH, based on clinical results. Subjective outcomes included the VVA questionnaire used to assess the symptoms of VVA, the UDI-6 to assess urinary symptoms, and the FSFI to assess sexual function. A MID was not identified for any of the outcomes. As a result, it is difficult to interpret the clinical significance of the results of the patient-reported outcomes. No evidence of validity, reliability, or responsiveness for the VVA questionnaire was identified. Categorical results of the VVA questionnaire used to assess the severity of symptoms were equated to a numerical value (e.g., patients who had none, mild, moderate, and severe symptoms were equated to scores of 0, 1, 2, and 3, respectively). The severity scores were used to calculate a mean, which was used to evaluate the change form baseline in symptom severity. In the absence of evidence to validate this approach, it is difficult to interpret ordinal data of the VVA using a mean. Moreover, recall bias is also a significant concern (e.g., patients may not remember their past experiences accurately). Consequently, there is still significant uncertainty with regard to the clinical relevance of the observed improvements in VVA symptoms.
Study 310X was a 52-week LTSE study of Study 310. The LTSE assessed safety outcomes only and reported them descriptively. Patients remained on the treatment they were assigned to in Study 310, with blinding maintained. Excluding the ospemifene 30 mg treatment arm (because 30 mg is not a Health Canada–approved dose) leaves 118 patients in the LTSE, with 69 patients in the ospemifene 60 mg treatment group and 49 patients in the placebo treatment group. The treatment groups were similar based on the characteristics that were reported, which were limited to age, race, ethnicity, and BMI. During the 52-week study, 17% and 31% of patients discontinued from the study in the ospemifene and placebo treatment groups, respectively, which is a limitation of this study. The most common reasons for discontinuation were patient decision or withdrawal of consent (7% and 16% for ospemifene and placebo, respectively) followed by AEs (6% and 2% for ospemifene and placebo, respectively) and loss to follow-up (0% for ospemifene and 6% for placebo). The imbalance in the rate of discontinuation may suggest knowledge of treatment received or that blinding was compromised, which introduces uncertainty with regard to the safety outcomes reported.
External Validity
In the studies included in the CADTH systematic review, a diagnosis of VVA was determined by the MI, vaginal pH, and self-reported symptoms of VVA, except in Study 718, which did not require patients to report a symptom of VVA. The clinical expert consulted by CADTH indicated that in clinical practice, post-menopausal patients with VVA are identified primarily through self-reported symptoms, although identification of patients using less subjective assessments, such as the MI and vaginal pH, were likely sufficient for use in clinical trials. Despite this, symptoms of VVA were not reported at baseline in Study 718, which introduces uncertainty about the generalizability of the patient population based on diagnosis. In Study 310, the eligibility criteria for symptoms of VVA were not limited to vaginal dryness or dyspareunia because patients who self-reported any of the 5 symptoms of VVA as the MBS were included; however, 85% of patients reported vaginal dryness or dyspareunia as their MBS at baseline. The other eligibility criteria of the included studies were generally considered appropriate and reflective of post-menopausal patients with VVA; however, these were restrictive — as is typical of clinical trial eligibility criteria — and ultimately not representative of all patients in clinical practice. This is evidenced by the large number of patients who were considered to have failed screening (70% of patients in Study 231; percentages not reported in the other included studies). Patients with comorbidities were excluded from the included studies. Patients with a BMI of 30 or greater in Study 718, or a BMI of 37 or greater in the 12-week studies, were also excluded. The clinical expert consulted by CADTH indicated that patients with a high BMI may be better suited to an oral treatment, such as ospemifene, due to the difficulty of administering therapies that are applied intravaginally. The impact of treatment on patients with comorbidities is not clear. Based on comments from the clinical expert consulted by CADTH for this review, the findings are likely to be generalizable to patients seen in Canadian clinical practice.
The trial protocols specified that patients were permitted to use non-hormonal lubricants as needed, with the exception of Study 718, which permitted lubricant use after 12 weeks. It was noted by the clinical expert consulted by CADTH for this review that in practice, patients may use ospemifene in combination with non-hormonal lubricants; therefore, its use in the trials was appropriate.
The duration of treatment in studies 310, 821, and 231 was 12 weeks. The clinical expert consulted by CADTH indicated that assessment at 3 months (12 weeks) following initiation of treatment was appropriate, and was a reasonable amount of time after which to observe a treatment effect for ospemifene. However, the clinical expert also indicated that in clinical practice, treatment with ospemifene would extend beyond 12 weeks. Given that the assessments of efficacy in the trials were short, the long-term benefits are uncertain, and patients who are prescribed ospemifene in clinical practice are likely to take it for longer than 12 weeks. Study 718 did provide data for up to 52 weeks, but did not report on symptom severity. Study 310 included an LTSE, Study 310X, which was 52 weeks in duration; however, it was subject to the limitations described earlier. In addition, Study 310X included only patients with an intact uterus who had completed the protocol for Study 310.
The 4 co-primary end points of studies 310, 821, and 231 were change in the percentage of parabasal cells, change in percentage of superficial cells, and change in vaginal pH, as well as change in the severity of the MBS of VVA. The 4 co-primary end points also align with FDA recommendations that specify that these outcomes should be used for studies of this indication.31 Study 718 included the same co-primary end points, with the exception of MBS of VVA, for a total of 3 co-primary end points. Other secondary end points included analyses of other symptoms of VVA, urinary symptoms assessed using the UDI-6, and sexual function assessed using the FSFI. The clinical expert consulted by CADTH for this review agreed that these outcomes were important to patients and clinicians for consideration in the treatment of post-menopausal VVA. While commonly used in clinical trials, the objective outcomes, such as the change in parabasal and superficial cells and vaginal pH, are not typically assessed in clinical practice, as per feedback from the clinical expert. Therefore, the end points that assess symptoms and their severity are more relevant to patients, given that self-reported symptoms are the predominant method of assessment in clinical practice, as per feedback from the clinical expert consulted by CADTH.
Indirect Evidence
There is a lack of direct evidence on the efficacy and safety of ospemifene compared to other treatments (e.g., topical estrogen therapies) for genitourinary syndrome of menopause. Thus, a review of ITCs was conducted. In addition to evaluating the sponsor-submitted ITC,12 CADTH conducted a literature search for NMAs related to the management of dyspareunia and/or vaginal dryness or symptoms of VVA among post-menopausal women. The search was run in MEDLINE (1946–) on November 19, 2021. No limits were applied. Titles, abstracts, and full-text articles were screened for inclusion by 2 reviewers based on the inclusion criteria for the main systematic review (Table 6). Two relevant NMAs were identified in the literature search: a study by Li et al.13 and a study by Bruyniks et al.34 The Bruyniks ITC was excluded from this review because the methodology was poorly reported, hindering the interpretability of the ITC findings. One ITC (Lee et al. [2018]35) was submitted by the sponsor for consideration, but excluded because it did not contain any relevant comparators.
Description of Indirect Treatment Comparisons
The sponsor-submitted ITC was an NMA that compared ospemifene to various treatments for genitourinary syndrome of menopause. The study by Li et al. was an NMA that compared the efficacy and safety of various treatments for genitourinary symptoms of menopause.
Methods of Eligible Indirect Treatment Comparisons
Objectives
The objective of the sponsor-submitted ITC was to evaluate the relative efficacy and safety of ospemifene to treat VVA. The objective of Li et al. ITC was to evaluate the efficacy and safety of different treatment options for genitourinary symptoms of menopause.
Study Selection Methods
Study selection criteria and methods for both ITCs are in Table 36.
Sponsor-Submitted Indirect Treatment Comparison
The sponsor-submitted ITC aimed to determine the relative efficacy and safety of ospemifene compared to current therapies for VVA. In the sponsor-submitted ITC, a systematic review was conducted in August 2021 to identify eligible studies. MEDLINE and Embase were searched, and 2 reviewers independently screened titles, abstracts, and extracted data. The Cochrane Risk of Bias tool was used for quality assessment. The sponsor-submitted ITC included studies with any of the following outcomes: change in severity of MBS of vaginal dryness, change in severity of MBS of dyspareunia, change in the percentage of parabasal cells, change in the percentage of superficial cells, TEAEs, serious TEAEs, headaches, UTIs, hot flashes, discontinuation due to AEs, and endometrial thickness.
Li et al. Indirect Treatment Comparison
The Li et al. ITC examined the safety and efficacy of treatments for genitourinary syndrome of menopause. In the Li et al. ITC, a systematic review was conducted in March 2020 to identify eligible studies. The following databases were searched: Pubmed, Embase, Scopus, Cochrane Library, Web of Science, and ScienceDirect. Two independent reviewers screened titles, abstracts, and extracted data. The Cochrane Risk of Bias tool was used for quality assessment. Studies exploring the following outcomes were included: symptoms (dryness, burning sensation, itching, urinary incontinence), vaginal pH, sexual function index, percentage of parabasal cells, and AEs. For efficacy outcomes, the authors converted the outcomes to a 0 to 100 scale because the studies used different scales.
Table 36: Study Selection Criteria and Methods for Indirect Treatment Comparisons
Criteria | Sponsor ITC12 | Li et al. (2021)13 |
|---|---|---|
Population | Post-menopausal women with moderate to severe symptoms of dyspareunia and/or vaginal dryness, symptoms of VVA | Women with genitourinary syndrome of menopause (dryness, burning sensation, irritation, sexual symptoms, urinary symptoms); women in studies must have been diagnosed with atrophic vaginitis and vaginal or vulvovaginal or urogenital atrophy by a clinician |
Intervention | Ospemifene | Trials comparing 2 different therapies for GSM (not further specified) |
Comparators | Conjugated estrogens vaginal cream Estrone vaginal cream Estradiol vaginal insert/vaginal ring Other local estrogen formulations Prasterone vaginal ovule Vaginal lubricants Moisturizers | Trials comparing 2 different therapies for GSM (not further specified) |
Outcomes |
|
|
Study design | Phase III controlled clinical trials | RCTs |
Exclusion criteria | Clinical trials with crossover design Phase I and II clinical trials Non-original publications Language other than English or French | Studies involving patients with underlying genitourinary comorbidities, such as pelvic organ prolapse, bacterial vaginosis |
Databases searched | MEDLINE, Embase (to August 2021) | Pubmed, Embase, Scopus, Cochrane Library, Web of Science, ScienceDirect (to March 2020) |
Selection process | Two reviewers independently screened titles and abstracts | Two independent reviewers screened titles and abstracts |
Data extraction process | Two reviewers extracted information using a piloted form; a third reviewer validated data | Not described |
Quality assessment | Cochrane Risk of Bias tool for RCTs | Cochrane Risk of Bias tool for RCTs |
Reference | Sponsor ITC12 | Li et al. (2021)13 |
AE = adverse event; GSM = genitourinary syndrome of menopause; MBS = most bothersome symptom; ITC = indirect treatment comparison; RCT = randomized controlled trial; TEAE = treatment-emergent adverse event; UTI = urinary tract infection; VVA = vulvovaginal atrophy.
Source: Sponsor-submitted ITC12; Li et al. (2021).13
Indirect Treatment Comparison Analysis Methods
The ITC analysis methods are described in Table 37.
Sponsor-Submitted Indirect Treatment Comparison
The sponsor-submitted ITC used a Bayesian framework NMA for analysis, with an initial burn-in of 50,000 samples. A fixed-effects or random-effects model was used, depending on which 1 produced a lower deviance information criterion. Inconsistency was to be assessed using a node-splitting approach, but this was not performed due to limited evidence in the network. The Brooks-Gelman-Rubin method was used to assess model convergence. Informative priors for the heterogeneity parameter (τ2) were used if there were 10 or fewer studies in the model (log-normal [mean = –3.02; SD = 1.852] for semi-objective outcomes and log-normal [mean = –2.13; SD = 1.582] for subjective outcomes; the definition of semi-objective and subjective outcomes was not provided). These priors were based on previous studies. Assessment of heterogeneity was based on the I2 statistic for direct comparison for each pairwise treatment comparison in the network. Any heterogeneity (I2 > 50%) was investigated by conducting subgroup analyses based on study characteristics or baseline characteristics, with no further details provided. For each drug (e.g., vaginal estrogen ring, conjugated estrogens vaginal cream), all doses were pooled into 1 node. Sensitivity analyses were conducted based on the quality assessment.
Li et al. Indirect Treatment Comparison
The Li et al. ITC also used a Bayesian framework NMA for analysis. A random-effects model was chosen for all analyses because it was deemed more conservative than a fixed-effects model and could account for between-trial heterogeneity. Markov Chain Monte Carlo simulations with 50,000 iterations were used for analysis, with the first 20,000 iterations as burn-ins. Vague priors were used for the overall mean effect and between-study SD. The authors used the Brooks-Gelman-Rubin method for assessing model convergence, but did not describe how model fit was assessed. The authors used a node-splitting approach to assess consistency. The assessment of heterogeneity was based on the I2 statistic for direct comparison for each pairwise treatment comparison in the network. As a sensitivity analysis, the authors used meta-regression to investigate possible differences based on demographics and dosages. A subgroup analysis was conducted to investigate any sources of heterogeneity. A frequentist framework with pooled estimates was used to conduct pairwise meta-analysis. All vaginal estrogens, regardless of dose, were pooled into 1 node, and both doses of ospemifene were pooled into 1 node.
Table 37: Indirect Treatment Comparison Analysis Methods
Methods | Sponsor-submitted ITC12 | Li et al. ITC13 |
|---|---|---|
ITC methods | Bayesian network meta-analysis with Markov Chain Monte Carlo simulations using either an FE or RE model (model choice depended on lowest deviance information criterion score); an identity link function was performed for continuous outcomes and a log link function for binary outcomes; initial burn-in of 50,000 samples. | Bayesian network meta-analysis using RE model; Markov Chain Monte Carlo simulations with 50,000 iterations and first 20,000 iterations as a burn-in |
Priors | Informative priors were used in RE models of outcomes with 10 or fewer studies | Vague priors for the overall mean effect and between-study variation |
Assessment of model fit | Lowest deviance information criterion score | Not described |
Assessment of consistency | Not performed | Node-splitting model comparing deviance between study variance and calculating a Bayesian P value to estimate conflict between direct and indirect evidence |
Assessment of convergence | Brooks-Gelman-Rubin method | Brooks-Gelman-Rubin method |
Follow-up time points | 12 weeks to 14 weeks | 6 weeks to 52 weeks |
Construction of nodes | Both doses of estradiol vaginal ring, conjugated estrogens vaginal cream pooled into 1 node | All vaginal estrogens pooled together into 1 node; both doses of ospemifene pooled into 1 node |
Sensitivity analyses | Based on Cochrane Risk of Bias assessment and model selection (i.e., FE vs. RE) | Meta-regression was used to investigate differences in demographics and dosages. |
Subgroup analyses | Based on differences in study characteristics and baseline characteristics, if relevant and possible | To identify sources of variation and heterogeneity |
Methods for pairwise meta-analysis | Reported results using both FE and RE models | Frequentist framework with pooled estimates quantified based on odds ratios or mean differences; Bayesian pairwise analysis performed with RE model and inverse variance weights |
Reference | Sponsor-submitted ITC12 | Li et al. ITC13 |
FE = fixed effects; ITC = indirect treatment comparison; RE = random effects; vs. = versus.
Source: Sponsor-submitted ITC12; Li et al. (2021).13
Results of Indirect Treatment Comparison 1
Summary of Included Studies
Sponsor-Submitted Indirect Treatment Comparison
In the sponsor-submitted ITC, 27 RCTs (with dates ranging from 1994 to 2019) were eligible, 5 of which involved ospemifene. Other investigated treatments included a conjugated estrogens vaginal cream (Premarin), an estradiol vaginal tablet (Vagifem), an estradiol soft gel vaginal insert (Imvexxy), an estradiol vaginal ring (Estring), and a prasterone vaginal ovule (Intrarosa). The sample size ranged from 21 patients to 826 patients, and the mean age ranged from 56 years to 63 years. The eligible RCTs primarily recruited post-menopausal women with moderate to severe genitourinary symptoms, and the majority of the trials were 12 weeks in duration (range = 12 weeks to 14 weeks). For the NMA, the sponsor included only RCTs with the following treatments (because these were noted to be marketed treatments in Canada): ospemifene 60 mg oral daily (Osphena), estradiol vaginal cream 0.02 mg (Estrace), estradiol transdermal patch 14 mcg (Estradiol patch), estradiol vaginal cream 2 mg and 7.5 mg (Estring), estriol vaginal pessary 0.5 mg (Estriol pessary), estradiol vaginal capsule 4 mcg and 10 mcg (Imvexxy), DHEA vaginal suppository 6.5 mg (Intrarosa), lubricants, conjugated estrogens vaginal cream 0.3 mg or 0.63 mg (Premarin), promestriene vaginal cream 10 mg, or estradiol vaginal insert 10 mcg (Vagifem). The sponsor noted that the majority of trials were at low risk of bias; however, 4 RCTs were at high risk of bias from blinding. Characteristics of the included RCTs are in Table 38.
Table 38: Characteristics of Studies Included in the Sponsor-Submitted Indirect Treatment Comparison
First author and publication date | Sample size, N | Age (years), mean (SD) | MBS vaginal dryness at baseline, mean (SD) | MBS dyspareunia at baseline, mean (SD) | Treatment 1 | Treatment 2 |
|---|---|---|---|---|---|---|
Archer (2015) | 253 | 58 (NR) | 2.3 (NR) | 2.6 (NR) | Prasterone 3.25 mg/6.5 mg vaginal ovule daily | Placebo |
Archer (2018) | 573 | 60 (6) | 2.5 (0.5) | 2.1 (1) | Estradiol vaginal cream 0.015 mg twice weekly | Placebo |
Archer (2019) | 627 | 60 (7) | 2.5 (0.5) | 2.5 (0.5) | Ospemifene 60 mg daily | Placebo |
Ayton (1996) | 194 | 59 (7) | NR | NR | Estradiol vaginal ring in situ for 12 weeks | Conjugated estrogens vaginal cream 0.625mg/g, 21 days on and 7 days off |
Bachmann (2008) | 230 | NR | NR | Estradiol 10 mcg/25 mcg vaginal insert twice weekly | Placebo | |
Bachmann (2009) | 423 | 58 (6) | 1.6 (NR) | 2.2 (NR) | Conjugated estrogens vaginal cream 0.3 mg/g, 21 days on and 7 days off | Placebo |
Bachmann (2010) | 826 | 58 (6) | 2.4 (0.5) | 2.7 (0.6) | Ospemifene 30 mg/60 mg daily | Placebo |
Bouchard (2015) | 441 | 58 (NR) | 2.4 (0.04) | 2.6 (0.05) | Prasterone 3.25 mg/6.5mg vaginal ovule twice weekly | Placebo |
Bydeman (1996) | 39 | NR | NR | NR | Dienestrol vaginal cream daily for 2 weeks, then 3 times weekly | Replens vaginal gel 3 times weekly |
Casper (1999) | 67 | NR | NR | NR | Estradiol vaginal ring in situ for 12 weeks | Placebo |
Constantine (2017) | 747 | 60 (6) | NR | 2.7 (0.5) | Estradiol soft gel vaginal insert 4 mcg/10 mcg/25 mcg twice weekly | Placebo |
Freedman (2009) | 305 | 60 (7) | NR | NR | Conjugated estrogens 0.625 mg/g vaginal cream twice weekly | Placebo |
Goldstein (2014) | 426 | 62 (6) | NR | NR | Ospemifene 60 mg daily | Placebo |
Gupta (2008) | 165 | 56 (4) | NR | NR | Estradiol vaginal ring in situ for 12 weeks | Estradiol transdermal patch for 12 weeks |
Henriksson (1994) | 548 | 59 (7) | NR | NR | Estradiol vaginal ring in situ for 12 weeks | Estradiol vaginal pessary 0.5 mg twice weekly |
Kroll (2018) | 482 | 59 (6) | 2.3 (0.7) | 2.7 (0.5) | Estradiol vaginal cream 0.015 mg 3 times weekly | Placebo |
Labrie (2009) | 216 | 58 (NR) | NR | NR | Prasterone vaginal ovule 3.25 mg/6.5 mg/13 mg daily | Placebo |
Labrie (2011) | 216 | NR | NR | 2.8 (0.08) | Prasterone vaginal ovule 3.25 mg/6.5 mg/13 mg daily | Placebo |
Labrie (2018) | 482 | 60 (NR) | 2.3 (0.03) | 2.55 (0.04) | Prasterone vaginal ovule 6.5 mg daily | Placebo |
Mitchell (2018) | 302 | 61 (4) | 2.3 (NR) | 2.5 (NR) | Estradiol vaginal insert 10 mcg plus placebo gel twice weekly or every 3 days | Placebo vaginal insert plus Replens vaginal gel twice weekly or every 3 days |
Natchigall (1994) | 30 | NR | NR | NR | Replens vaginal gel 3 times per week | Estrogens cream daily |
Politano (2019) | 72 | 57 (5) | NR | NR | Promestriene vaginal cream 10 mg 3 times weekly | Lubricant as needed |
Portman (2014) | 314 | 60 (7) | NR | NR | Ospemifene 60 mg daily | Placebo |
Portman (2013) | 605 | 58 (6) | NR | 2.7 (0.5) | Ospemifene 60 mg daily | Placebo |
Simon (2008) | 309 | 58 (5) | NR | NR | Estradiol vaginal insert 10 mcg twice weekly | Placebo |
MBS = most bothersome symptom; NR = not reported; SD = standard deviation.
Source: Sponsor-submitted ITC.12
Li et al. Indirect Treatment Comparison
In the Li et al. ITC, 29 RCTs were eligible, with 8,311 participants (sample sizes ranged from 180 patients to 909 patients, and dates ranged from 1992 to 2020). Five treatments were investigated: laser therapy, vaginal estrogen, ospemifene, vaginal prasterone, and moisturization and/or lubrication. The mean age of patients ranged from 51 years to 65 years, and the duration of the trials ranged from 6 weeks to 52 weeks. The severity or duration of symptoms was not described by the authors. The authors reported that 11% of eligible studies included patients with a history of breast or gynecological cancer. The authors noted that the majority of trials were at low or unclear risk of bias based on the Cochrane Risk of Bias tool for RCTs; however, 4 studies had high risk of bias for random sequence generation, 3 studies had high risk of bias for allocation concealment, 6 studies had high risk of bias for blinding, and 3 studies were at high risk of bias for incomplete outcome data.
Results
Sponsor-Submitted Indirect Treatment Comparison
For the sponsor-submitted ITC, the evidence networks for relevant outcomes are displayed in Figure 2. The NMA results are displayed in Table 39 and Table 40. A random-effects model was used for the analyses of all outcomes except the MBS score for vaginal dryness, which used a fixed-effects model.
Figure 2: Network Diagram for MBS Score Reduction of Vaginal Dryness at 12 Weeks in Sponsor-Submitted ITC
Estrace = estradiol vaginal cream; ITC = indirect treatment comparison; Intrarosa = prasterone; MBS = most bothersome symptom; Osphena = ospemifene; Premarin = conjugated estrogens vaginal cream; Vagifem = estradiol vaginal tablet.
Source: Sponsor-submitted ITC.12
Figure 3: Network Diagram for MBS Score Reduction of Dyspareunia at 12 Weeks in Sponsor-Submitted ITC
Estrace = estradiol vaginal cream; Imvexxy = estradiol vaginal capsule; ITC = indirect treatment comparison; Intrarosa = prasterone; MBS = most bothersome symptom; Osphena = ospemifene; Premarin = conjugated estrogens vaginal cream; Vagifem = estradiol vaginal tablet.
Source: Sponsor-submitted ITC.12
Figure 4: Network Diagram for MBS Score of Vaginal Dryness and Dyspareunia Combined at 12 Weeks in Sponsor-Submitted ITC
Estrace = estradiol vaginal cream; Imvexxy = estradiol vaginal capsule; ITC = indirect treatment comparison; Intrarosa = prasterone; MBS = most bothersome symptom; Osphena = ospemifene; Premarin = conjugated estrogens vaginal cream; Vagifem = estradiol vaginal tablet.
Source: Sponsor-submitted ITC.12
Figure 5: Network Diagram for Reduction in Percentage of Parabasal Cells at 12 Weeks in Sponsor-Submitted ITC
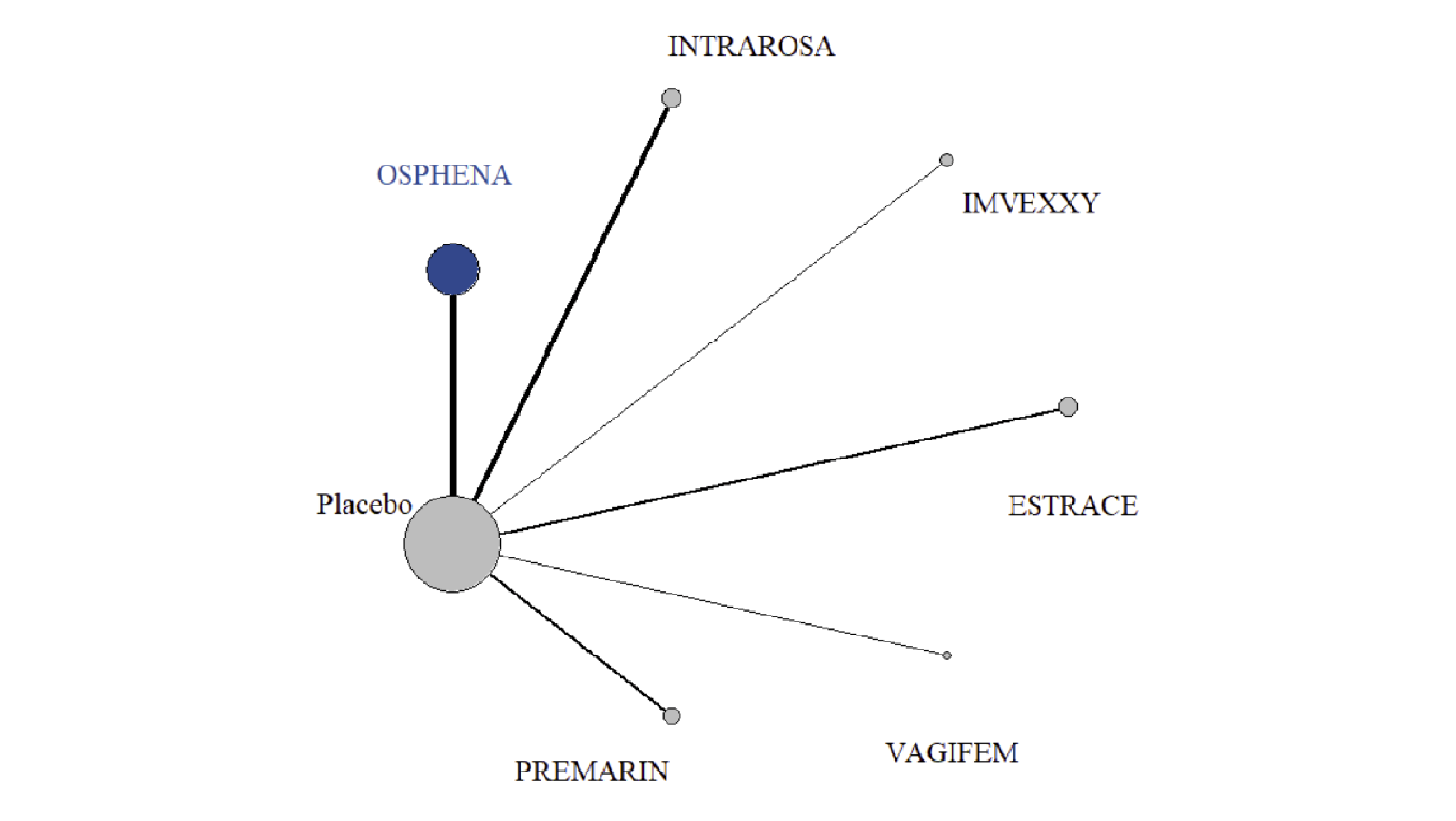
Estrace = estradiol vaginal cream; Imvexxy = estradiol vaginal capsule; Intrarosa = prasterone; ITC = indirect treatment comparison; Osphena = ospemifene; Premarin = conjugated estrogens vaginal cream; Vagifem = estradiol vaginal insert.
Source: Sponsor-submitted ITC.12
Figure 6: Network Diagram for Increase in Percentage of Superficial Cells at 12 Weeks in Sponsor-Submitted ITC
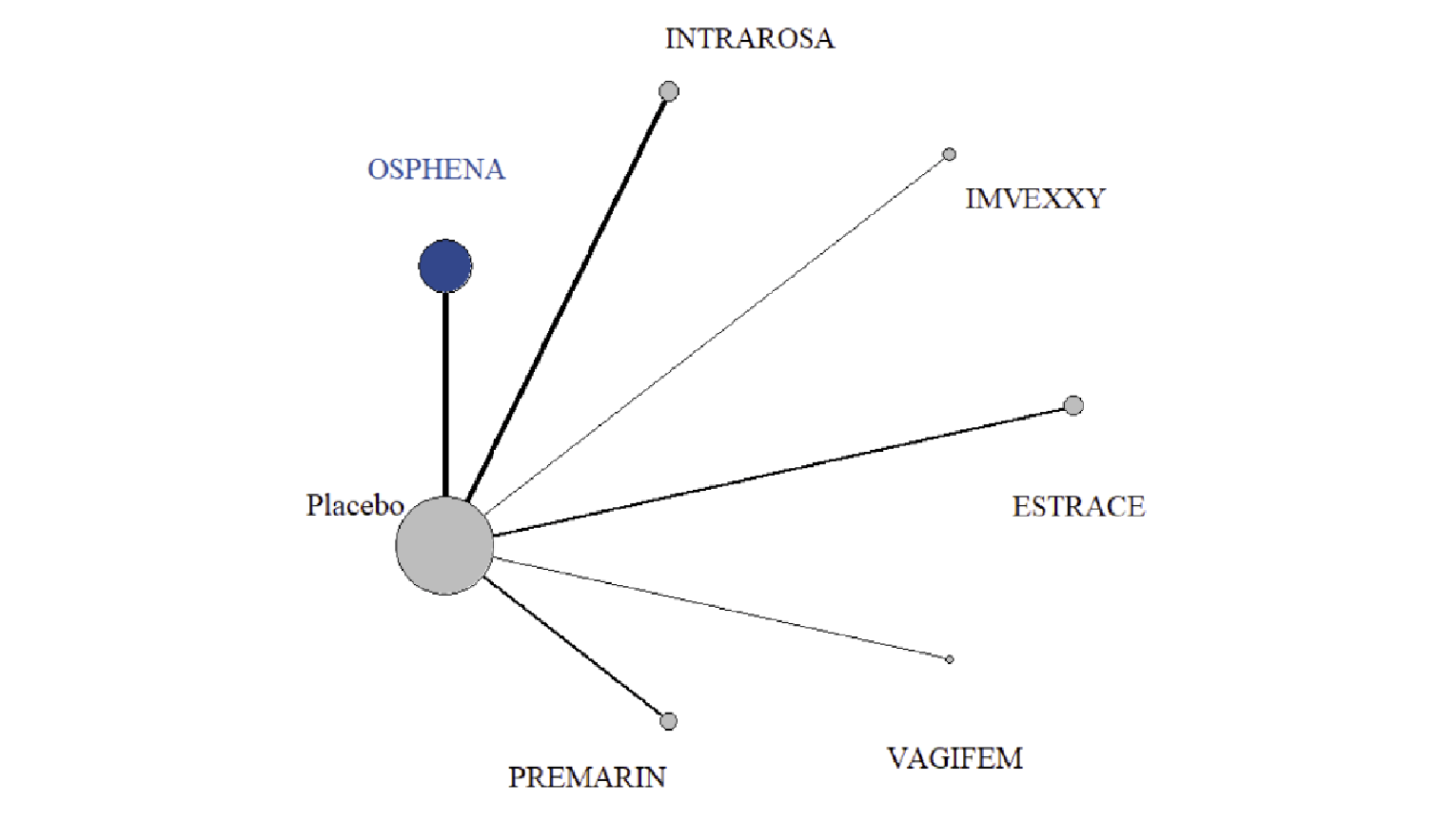
Estrace = estradiol vaginal cream; Imvexxy = estradiol vaginal capsule; Intrarosa = prasterone; ITC = indirect treatment comparison; Osphena = ospemifene; Premarin = conjugated estrogens vaginal cream; Vagifem = estradiol vaginal tablet.
Source: Sponsor-submitted ITC.12
Figure 7: Network Diagram for Reduction in Vaginal pH at 12 Weeks in Sponsor-Submitted ITC
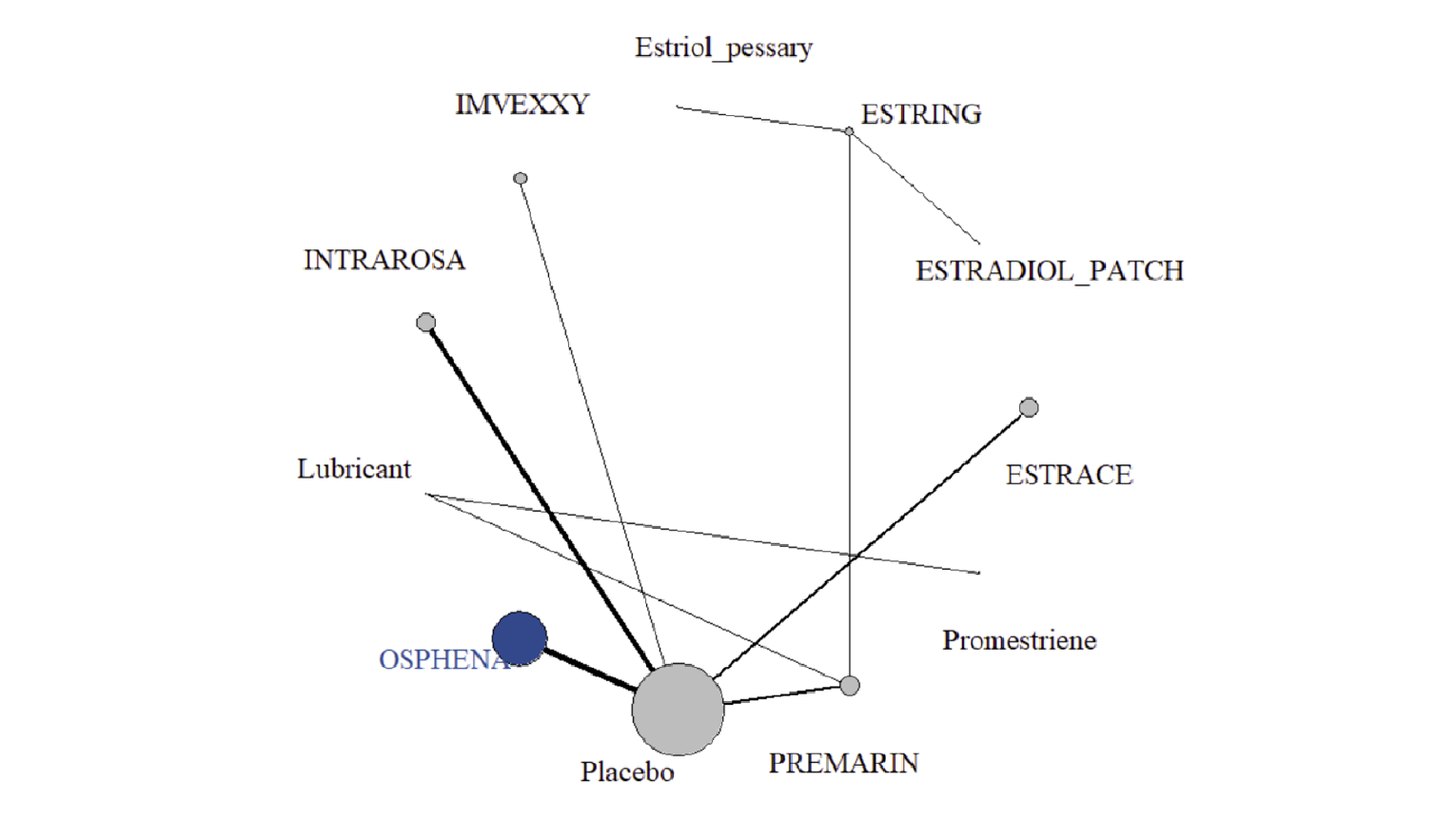
Estrace = estradiol vaginal cream; Estring = estradiol vaginal ring; Imvexxy = estradiol vaginal capsule; Intrarosa = prasterone; ITC = indirect treatment comparison; Osphena = ospemifene; Premarin = conjugated estrogens vaginal cream; Vagifem = estradiol vaginal tablet.
Source: Sponsor-submitted ITC.12
For the outcome of MD in change from baseline to follow-up in MBS score for vaginal dryness, ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
For the outcome of MD in change from baseline to follow-up in MBS score for dyspareunia, ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
For the outcome of MD in change from baseline to follow-up for the combined MBS score for vaginal dryness and dyspareunia, ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
For the outcome of MD in change in percentage of parabasal cells, ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
For the outcome of MD in change in percentage of superficial cells, ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
For the outcome of MD in reduction of vaginal pH, ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
There was no difference in the risk of TEAEs for ospemifene versus conjugated estrogens vaginal cream (RR = 1.07; 95% CrI, 0.93 to 1.24) or versus estradiol vaginal tablet (RR = 1.11; 95% CrI, 0.95 to 1.28). There was no difference in the risk of serious TEAEs for ospemifene versus conjugated estrogens vaginal cream (RR = 0.75; 95% CrI, 0.02 to 31) or versus estradiol vaginal tablet (RR = 0.87; 95% CrI, 0.15 to 4.17). There was no difference in the risk of UTI between ospemifene and estradiol vaginal tablet (RR = 2.55; 95% CrI, 0.23 to 35). The risk of headaches was lower for ospemifene compared with estradiol vaginal ring (RR = 0.00; 95% CrI, 0.00 to 0.04), while there was no difference between ospemifene and conjugated estrogens vaginal cream (RR = 0.74; 95% CrI, 0.38 to 1.42) or estradiol vaginal tablet (RR = 1.43; 95% CrI, 0.24 to 8.50). There was no difference in the risk of discontinuation due to AEs for ospemifene versus estradiol vaginal ring (RR = 1.26; 95% CrI, 0.28 to 1.52), conjugated estrogens vaginal cream (RR = 0.97; 95% CrI, 0.31 to 2.69), or estradiol vaginal tablet (RR = 0.94; 95% CrI, 0.31 to 2.45).
Table 39: Summary of ITC Results for Relevant Comparisons in the Sponsor-Submitted ITC
Treatments | MD (95% CrI) in MBS reduction: vaginal dryness | MD (95% CrI) in MBS reduction: dyspareunia | MD (95% CrI) in combined MBS reduction: dyspareunia and vaginal dryness | MD (95% CrI) in % reduction in parabasal cells | MD (95% CrI) in % increase in superficial cells | MD (95% CrI) in reduction in vaginal pH |
|---|---|---|---|---|---|---|
Number of studies | 10 studies (N = 3,686) | 13 studies (N = 4,685) | 14 studies (N = 8,371) | 15 studies (N = 5,653) | 15 studies (N = 5,653) | 19 studies (n = 5,891) |
|||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| |
||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| |
||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| | ||||||||||||||||||||||||| |
CrI = credible interval; ITC = indirect treatment comparison; MBS = most bothersome symptom; MD = mean difference.
Source: Sponsor-submitted ITC.12
Table 40: Summary of Relative Effects From NMA for Safety Outcomes in Sponsor-Submitted ITC
Treatments | Relative risk (95% CrI) TEAEs | Relative risk (95% CrI) serious TEAEs | Relative risk (95% CrI) headaches | Relative risk (95% CrI) UTIs | Relative risk (95% CrI) hot flashes | Relative risk (95% CrI) DAEs |
|---|---|---|---|---|---|---|
Number of studies (N) | 14 studies (N = 6,271) | 11 studies (N = 4,557) | 9 studies (N = 3,572) | 8 studies (N = 3,767) | 6 studies (N = 2,923) | 16 studies (N = 5,962) |
Ospemifene vs. estradiol vaginal ring | NA | NA | 0.00 (0.00 to 0.04) | NA | NA | 1.26 (0.28 to 5.12) |
Ospemifene vs. conjugated estrogens vaginal cream | 1.07 (0.93 to 1.24) | 0.75 (0.02 to 31) | 0.74 (0.38 to 1.42) | NA | NA | 0.97 (0.31 to 2.69) |
Ospemifene vs. estradiol vaginal insert | 1.11 (0.95 to 1.28) | 0.87 (0.15 to 4.17) | 1.43 (0.24 to 8.50) | 2.55 (0.23 to 35) | NA | 0.94 (0.31 to 2.45) |
Comments | FE model | FE model | FE model | RE model | FE model | FE model |
CrI = credible interval; DAE = discontinuation due to adverse event; FE = fixed effects; ITC = indirect treatment comparison; RE = random effects; NA = not applicable; NMA = network meta-analysis; TEAE = treatment-emergent adverse event; UTI = urinary tract infection; vs. = versus.
Source: Sponsor-submitted ITC.12
Li et al. Indirect Treatment Comparison
In the Li et al. ITC, no evidence network diagrams were provided, and the number of studies contributing to each node or analysis was unclear. There was no difference between ospemifene and vaginal estrogens for the outcomes of mean difference in change in vaginal dryness (MD = –2.9; 95% CrI, –13 to 8.1), dyspareunia (MD = 8.0; 95% CrI, 0.2 to 17), or sexual function (MD = 1.5; 95% CrI, –2.7 to 5.6). The reduction in vaginal pH was smaller for ospemifene versus vaginal estrogens (MD = 0.31; 95% CrI, 0.05 to 0.58). There was no difference between ospemifene and vaginal estrogens for reduction in percentage of parabasal cells (MD = 2.2; 95% CrI, –9.5 to 15).
Critical Appraisal of Indirect Treatment Comparisons
Sponsor-Submitted Indirect Treatment Comparison
The sponsor-submitted ITC provided a clear rationale and objective. The systematic review methods were described, including the eligibility criteria, information sources, search strategy, study selection, and data extraction. Study selection methods were pre-specified and generally appropriate. Only 2 databases were searched, and there was no grey literature search, so it is possible that relevant literature was missed. Outcome measures were clearly outlined. Outcomes included both efficacy and safety outcomes, and these were relevant to this review. Comparators investigated in the sponsor-submitted ITC included those relevant to standard of care in Canada (e.g., Vagifem, Estring, Premarin). Quality assessment was performed and eligible RCTs were mostly of high quality (except for 4 RCTs for the outcome of vaginal pH, which were at high risk of bias due to blinding). The analysis methods were described adequately, including model selection and effect measures. The sponsor-submitted ITC included a justification of the model selection (fixed effects versus random effects), which was based on an assessment of model fit. Inconsistency was planned but not assessed, while sensitivity and subgroup analyses were performed to investigate sources of heterogeneity (e.g., due to baseline characteristics, study characteristics, drug dose). Heterogeneity within pairwise comparisons was assessed using the I2 statistic.
Eligible studies were described in terms of study population and length of follow-up. All eligible studies had similar mean ages (56 years to 62 years) and lengths of follow-up (12 weeks to 14 weeks). The sponsor-submitted ITC included only studies among women with moderate to severe symptoms. Durations of symptoms were reported, but the baseline MBS scores were generally similar across the studies that reported them. The extent to which eligible studies satisfied the similarity assumption was unclear. While patient and study characteristics were broadly similar, the combining of different doses in nodes and differences in placebo across trials may have introduced heterogeneity across pairwise comparisons. Also, the unclear extent of prior VVA treatment and use of concomitant interventions make it challenging to assess the similarity of the eligible studies.
Evidence network figures were provided, and the results of both pairwise comparisons and results of the NMA were presented. Heterogeneity in effect sizes (based on I2 > 50%) was observed for some pairwise comparisons. Subgroup or sensitivity analyses did not produce different results than those of the base case, and were generally not able to explain heterogeneity; however, heterogeneity assessment methodology decisions were not well described in the methods section. In the analysis of safety outcomes, there were wide CrIs and low event rates (resulting in extremely low RRs) for some comparisons, which makes it challenging to assess the comparative safety (e.g., for headache or UTI). Further, for some efficacy outcomes, there were a limited number of trials for some nodes, resulting in wide and overlapping CrIs for comparisons between ospemifene and other treatments. This similarly makes it difficult to draw conclusions about the comparative efficacy of ospemifene versus relevant comparators.
Li et al. Indirect Treatment Comparison
The Li et al. ITC described the study objective and study selection process. Systematic review methods were outlined; however, there were some concerns with respect to study selection. The population of interest was broadly described, but explicit eligibility criteria were not provided (e.g., based on severity of symptoms). Similarly, the authors did not provide explicit criteria around specific relevant interventions or comparators. Outcomes of interest were listed by authors and were relevant for this review. Ospemifene data were available for certain outcomes only, not for all outcomes of interest in this ITC. The authors converted to continuous outcomes to a 0 to 100 scale because different outcome scales had been used across the studies; however, the authors did not provide details about how this was carried out or whether it was appropriate. Not all comparators in the Li et al. ITC were relevant to this review. The comparison of ospemifene to vaginal estrogens was relevant. However, the Li et al. ITC combined all vaginal estrogens into 1 node (including different drugs and dosage forms, such as conjugated estrogens, estradiol 4 mcg, and a 10 mcg vaginal capsule). Some of the vaginal estrogens included in the vaginal estrogen node were not comparators of interest for this review (e.g., estriol cream). Given that there may be differences between vaginal estrogen products in terms of efficacy and safety, the appropriateness of combining these treatments into 1 node is uncertain. It further makes it challenging to draw conclusions about the comparative efficacy and safety of ospemifene versus individual relevant treatments. Analysis methods were described in the Li et al. ITC, including model selection, assessment of consistency, and assessment of convergence; however, data on convergence were not provided. A description of model fit was not provided; therefore, it is unclear if model fit was adequate. The authors used a node-splitting approach to check for inconsistency and evaluated heterogeneity using the I2 statistic. Heterogeneity was further investigated using meta-regression and subgroup analysis.
Li et al. provided a table with the characteristics of included studies, which included age, treatments, history of gynecologic cancer, and treatment duration. These characteristics were considered effect modifiers in meta-regression and subgroup analyses; however, it was unclear whether effect modifiers were identified a priori. Network diagrams were not provided in the Li et al. ITC, and it was unclear how many studies contributed to specific comparisons. The follow-up durations ranged from 4 weeks to 52 weeks, and participants’ ages were broadly similar across the studies. Information about disease severity and duration of symptoms was not extracted, making it challenging to assess whether the similarity assumption was satisfied. Further, placebos differed across trials, the extent of prior VVA treatments was not described, and the appropriateness of pooling different products and/or doses was not described. Since the severity of symptoms was not provided, it is unclear how relevant the population was for the present review. Heterogeneity (I2 > 50%) was observed for some outcomes involving ospemifene (dyspareunia, vaginal pH, parabasal cells), which could be explained by age (vaginal pH) or dose (change in the percentage of parabasal cells), but could not be explained for other outcomes.
Other Relevant Evidence
This section includes an open-label LTSE study included in the sponsor’s submission to CADTH that was considered to address important gaps in the evidence included in the systematic review.
Long-Term Safety Extension Study
One multi-centre, open-label, phase III LTSE study, Study 312, which was a follow-up of Study 310, has been summarized to provide additional evidence regarding the long-term safety of oral daily doses of ospemifene 60 mg for the treatment of VVA in post-menopausal women without a uterus. Data for this summary were presented in the Clinical Study Report dated May 08, 2006, with the completion of the last participant on December 22, 2008.14
Methods
A summary of the details of Study 312 is provided in Table 41.
Study 312 was a multi-centre, open-label, phase III LTSE follow-up study of Study 310 conducted at 59 sites in the US. Its objective was to assess the long-term safety of oral daily doses of ospemifene 60 mg in the treatment of VVA in post-menopausal women without a uterus. For the phase III, placebo-controlled, randomized, double-blind Study 310, a total of 826 post-menopausal women with and without a uterus were randomized to receive ospemifene 30 mg/day, ospemifene 60 mg/day, or placebo (1:1:1) for 12 weeks. Post-menopausal women without a uterus who completed the protocol for Study 310 were eligible to continue to Study 312.15 Consent for Study 312 was obtained from patients at the week 12 study visit of Study 310. During this extension study, all patients were treated with ospemifene 60 mg once daily irrespective of their treatment assignment in Study 310. The treatment period of Study 312 was 52 weeks, followed by a 4-week post-treatment follow-up period, for a total of 68 weeks (including the initial 12 weeks of Study 310).14,15
Statistical Analysis
In Study 312, all safety outcomes were reported descriptively in the ITT population, defined as all patients who entered the study and received at least 1 dose of study medication. A total of 301 patients were enrolled and included in the ITT population. The most frequently occurring AEs, SAEs, and AEs that led to study drug discontinuation, as well as notable harms, are summarized for this study.
Table 41: Details of Study 312
Detail | Description |
|---|---|
Designs and populations | |
Study design | Multi-centre, long-term, open-label, long-term safety extension |
Locations | US (59 sites) |
Patient enrolment dates | May 08, 2006, to December 22, 2008 |
Patients included (ITT) | 301 |
Inclusion criteria | Patients who met the following criteria at week 12 of Study 310:
|
Exclusion criteria | Clinically significant abnormal findings at the week 12 end-of-study visit for Study 310 Any physical or mental condition that, in the opinion of the investigator, may have interfered with the patient’s ability to comply with the study procedures |
Drugs | |
Intervention | Ospemifene 60 mg tablets once daily, administered orally each morning with food |
Comparator | None |
Duration | |
Phase | |
Preceding study (Study 310) | 12 weeks |
LTSE | 52 weeks |
Follow-up | 4 weeks |
Outcomes | |
Primary end point and secondary and exploratory end points | NA (efficacy was not evaluated) |
Safety end points | Adverse events (from signing of informed consent for Study 312 through follow-up period) Adherence to study medication Concomitant medications |
Notes | |
Publications | Simon (2014)15 |
ITT = intention to treat; LTSE = long-term safety extension; NA = not applicable.
Source: Clinical Study Report14 and Simon (2014).15
Populations
Inclusion and Exclusion Criteria
Patients were eligible to participate in Study 312 if they:
met the inclusion and exclusion criteria for Study 310
had no uterus
had completed the protocol for Study 310
provided written informed consent, including an agreement to follow the dosing regimens and attend all study visits.
Patients were not eligible to participate in Study 312 if they:
had clinically significant abnormal findings at the week 12 end-of-study visit for Study 310
had any physical or mental condition that, in the opinion of the investigator, may have interfered with their ability to comply with the study procedures.
Baseline Characteristics
There were limited baseline characteristics available for Study 312 (Table 42); however, the characteristics of the patients who continued into the LTSE were similar to those of patients in the core study in terms of age, race, ethnicity, and BMI. Most patients were White (92.4%) with a mean age of 59.4 years and a mean BMI of 26.9 kg/m2.
Table 42: Summary of Baseline Characteristics — Open-Label Extension Study 312 (ITT Population)
Characteristic | OSP 60 mg (N = 301) |
|---|---|
Age (years), mean (SD) | 59.4 (6.7) |
Race, n (%) | |
White | 278 (92.4) |
Black or African American | 11 (3.7) |
Asian | 6 (2.0) |
American Indian or Alaska Native | 3 (1.0) |
Other | 3 (1.0) |
Ethnicity, n (%) | |
Not Hispanic or Latino | 280 (93.0) |
Hispanic or Latino | 21 (7.0) |
BMI (kg/m2), mean (SD) | 26.9 (4.4) |
BMI = body mass index; ITT = intention to treat; OSP = ospemifene; SD = standard deviation.
Source: Clinical Study Report.14
Results
Patient Disposition
A summary of the patient disposition in Study 312 by the treatment group patients were assigned to in Study 310 is available in Table 43. Of note, ospemifene 30 mg once daily is not a Health Canada–approved dose; as such, it is not discussed in detail in the summary of Study 312. Of the 826 post-menopausal women with and without a uterus who were randomized in Study 310, 302 patients (36.6%) signed the consent form, and 301 patients (36.4%) enrolled in the open-label extension study, Study 312. Overall, 117 (38.7%) patients discontinued from the study. The proportion of patients who discontinued from the study was greater among those who received ospemifene 30 mg (42.9%) or placebo (41.1%) in Study 310 compared to those who previously received ospemifene 60 mg (32.0%).
Table 43: Patient Disposition — Open-Label Extension Study 312
Category | OSP 30 mg in Study 310 | OSP 60 mg in Study 310 | PBO in Study 310 | Total (OSP 60 mg in Study 312) |
|---|---|---|---|---|
Number in ITT population, N | 97 | 97 | 107 | 301 |
Discontinued from study, N (%) | 42 (42.9) | 31 (32.0) | 44 (41.1) | 117 (38.7) |
Reason for discontinuation, N (%) | ||||
Adverse events | 10 (10.2) | 13 (13.4) | 14 (13.1) | 37 (12.3) |
Lost to follow-up | 6 (6.1) | 5 (5.2) | 6 (5.6) | 17 (5.6) |
Patient decision or withdrawal of consent | 18 (18.4) | 8 (8.2) | 14 (13.1) | 40 (13.2) |
Major protocol violation | 1 (1.0) | 0 | 2 (1.9) | 3 (1.0) |
Significant non-compliance with treatment or study procedures | 2 (2.0) | 4 (4.1) | 6 (5.6) | 12 (4.0) |
Use of concomitant medication compromising safety | 1 (1.0) | 0 | 0 | 1 (0.3) |
Other | 4 (4.1) | 1 (1.0) | 2 (1.9) | 7 (2.3) |
ITT = intention to treat; OSP = ospemifene; PBO = placebo.
Source: Clinical Study Report.14
Table 44: Exposure to Study Treatments — Open-Label Extension Study 312 (ITT Population)
Category | OSP 60 mg (N = 301) |
|---|---|
Duration of treatment (days) | |
N | 266 |
Mean (SD) | 309.2 (113) |
Median (range) | 364.0 (2 to 629) |
Compliance rate (%) | |
N | 266 |
Mean (SD) | 86.7 (30) |
Median (range) | 95.0 (0 to 375) |
ITT = intention to treat; OSP = ospemifene; SD = standard deviation.
Source: Clinical Study Report.14
The most common reasons for discontinuation were patient decision or withdrawal of consent (13.2%), AEs (12.3%), and loss to follow-up (5.6%). The proportion of patients who discontinued due to an AE was similar between treatment groups based on treatment received in Study 310 (ospemifene 30 mg = 10.2%; ospemifene = 60 mg, 13.4%; placebo = 13.1%). However, the proportion of patients who discontinued due to patient decision or withdrawal of consent differed between the ospemifene 60 mg group (8.2%) and the placebo group (13,1%), reflecting the fact that those who were in the placebo group in Study 310 had more withdrawal of consent than those who were taking ospemifene. Other reasons for discontinuation were major protocol violation, use of a concomitant medication, significant non-compliance, and other.
Exposure to Study Treatments
In Study 312, the mean duration of treatment for the ITT population was 309.2 days (SD = 113), with a mean compliance rate of 86.7% (SD = 30), and a median compliance rate of 95.0%.
Efficacy
Efficacy was not assessed in Study 312.
Safety
The AEs reported in Study 312 are summarized in Table 45. Among the 301 ITT patients, 220 (73.1%) experienced at least 1 AE during the study. The most common AEs were sinusitis (8%), UTIs (8.6%), and hot flashes (10.3%). AEs leading to study discontinuation happened for 34 patients (11.3%). Hot flashes (2%) were the most common among the AEs leading to study discontinuation. Vaginal hemorrhage (0.7%), cervical dysplasia (0.3%), breast mass (0.3%), and hemorrhagic stroke (0.3%) were the notable harms reported. No deaths were reported.
A summary of harms that developed in the ITT population during the 52-week extension study is available in Table 45.
Table 45: Summary of Harms in Study 312 (Intention-to-Treat Population)
Event, n (%) | OSP 60 mg (N = 301) |
|---|---|
Patients with ≥ 1 adverse event | |
n (%) | 220 (73.1) |
Most frequent AEsa | |
Urinary tract infection | 26 (8.6) |
Sinusitis | 24 (8.0) |
Hot flashes | 31 (10.3) |
Patients with ≥ 1 SAE | |
n (%) | 13 (4.3) |
Non-cardiac chest pain | 2 (0.7) |
Patients who stopped treatment due to adverse events | |
n (%) | 34 (11.3) |
Nausea | 3 (1.0) |
Muscle spasms | 2 (0.7) |
Headache | 3 (1.0) |
Hyperhidrosis | 2 (0.7) |
Rash | 2 (0.7) |
Hot flashes | 6 (2.0) |
Deaths | |
n (%) | 0 |
Notable harms, n (%) | |
Vaginal hemorrhage (genital hemorrhage) | |||||||||||||| |
Cervical dysplasia | |||||||||||||| |
Breast mass | 1 (0.3) |
Cardiovascular disordersb | 1 (0.3) |
AE = adverse event; OSP = ospemifene; SAE = serious adverse event.
aOccurred in greater than or equal to 5% of patients in any prior treatment group.
bCardiovascular disorders included thromboembolic and hemorrhagic stroke and coronary heart disease. One patient was reported as having experienced hemorrhagic stroke.
Source: Clinical Study Report14 and Simon (2014).15
Critical Appraisal
Internal Validity
The open-label safety extension Study 312 had 2 main limitations imposed by the overall design: the lack of a comparison group to provide context and control for potential confounders and the open-label design. An open-label design may influence the perception of improvement by patients and clinicians and affect the reporting of harms. As part of the eligibility criteria for the LTSE study, patients had to complete the prior Study 310, potentially allowing for selection bias. Out of the total population of patients who entered Study 310 (N = 826), almost half (N = 447) had no uterus.15 However, 301 patients with no uterus (67.3%) ultimately enrolled in the LTSE study, meaning that almost 1-third of potential participants were lost from the parent study. This may have added to the selection bias as well; however, some of the losses may be due to discontinuations in Study 310. The latter is more likely to bias the reporting of outcomes (i.e., patients who tolerated treatment well or found it efficacious would have been more likely to continue versus those who did not). Additionally, among the entered participants, 117 (38.7%) discontinued prematurely from the study. The proportion of patients who withdrew prematurely was greater among those who received ospemifene 30 mg (42.9%) or placebo (41.1%) in the parent study compared to those who received 60 mg (32.0%). This could imply that those who were already on the same dose of ospemifene in the prior study experienced better tolerability than the other group. This would generate the potential for survival bias, because the other 2 groups might have lost more participants due to tolerability issues. It is also important to note that discontinuation rates were high in general; therefore, AEs were probably underreported. Any lack of follow-up after patients discontinued Study 312 could also mean that important long-term safety data are missing. Among the ITT population, the mean adherence rate to the study drug was 86.7%.
External Validity
Because the patients who took part in Study 312 were originally from the parent study (Study 310) and the eligibility criteria remained the same, it is reasonable to expect that the same limitations to generalizability are relevant to the LTSE study. For instance, since the participants were predominantly White (92.4%), the results from these trials may not be generalizable to other racial groups that may commonly be seen at some centres in Canada. The population in this LTSE study was limited to patients without a uterus; however, the clinical expert consulted by CADTH indicated that uterine status was unlikely have a differential effect on safety. The study duration was a total of 68 weeks, including the baseline of 12 weeks in Study 310 and 4 weeks of follow-up. This time frame might not allow sufficient time to observe certain safety issues of interest to this review, such as cancer, dysplasia, or hyperplasia.
Discussion
Summary of Available Evidence
A total of 5 phase III, double-blind, placebo-controlled RCTs that assessed ospemifene 60 mg were included in the CADTH systematic review: Study 310 (N = 544, excluding the ospemifene 30 mg treatment group), Study 821 (N = 919), Study 231 (N = 631), Study 718 (N = 426), and Study 310X (N = 118, who continued from Study 310). Studies 310, 821, and 231 were designed to assess the efficacy and safety of ospemifene 60 mg over 12 weeks; Study 718 assessed the efficacy and long-term safety of ospemifene 60 mg over 52 weeks; and Study 310X was a 52-week LTSE of Study 310 that assessed only safety outcomes. All of the studies enrolled post-menopausal women between 40 years and 80 years of age who had 5% or fewer superficial cells in the MI of the vaginal smear and a vaginal pH greater than 5.0. In addition, studies 310, 821, and 231 included patients who had identified at least 1 moderate to severe symptom of VVA that was considered the most bothersome. Studies 310, 821, 231, and 718 included the following as co-primary end points assessed at week 12: vaginal pH and the percentage of vaginal superficial and vaginal parabasal cells on a vaginal smear. Studies 310, 821, and 231 also included severity of the MBS of VVA as a fourth co-primary end point. Secondary end points assessed in the 12-week studies included urinary symptoms using the UDI-6 and sexual function (Study 821 and Study 231 only) using the FSFI. HRQoL, mental health-related outcomes, bone mineral density, and adherence were identified as outcomes of interest to this review, but were not assessed in any of the included studies.
None of the 4 studies included sites in Canada. The majority of patients included in studies 310, 821, 231, and 718 were 55 years of age or older and White. The proportion of patients who had previous experience with hormonal treatments varied between the studies (ranging from 3% to 61% of patients). Among the 544 patients in Study 310, 222 patients (41%) reported vaginal dryness as their MBS, while 242 patients (44%) reported vaginal pain with sexual activity (dyspareunia) as their MBS. In Study 821, 314 patients (34%) reported vaginal dryness as their MBS, and 605 (66%) reported dyspareunia as their MBS. In Study 231, patients were required to have vaginal dryness as their MBS. Study 718 did not report assessments of MBS at baseline. Baseline characteristics for Study 310X were limited to demographic information. The major limitations of the included studies were the lack of an active comparator, the use of an unvalidated questionnaire to assess VVA symptoms, and the potential for recall bias with respect to clinical symptoms. The absence of outcomes related to HRQoL is also a major gap in the evidence.
Other available evidence summarized as part of this CADTH Reimbursement Review include an open-label, single-arm, 52-week LTSE of Study 310 (Study 312,14 N = 301), a sponsor-submitted NMA,12 and an NMA published by Li et al.13
Interpretation of Results
Efficacy
After 12 weeks of treatment, studies 310 and 231 met their objectives by demonstrating improvement in favour of ospemifene 60 mg compared to placebo on the 4 co-primary end points: change from baseline to week 12 in the percentage of parabasal cells, superficial cells, vaginal pH, and severity of the MBS of VVA. Study 718 met its objectives by demonstrating improvement in favour of ospemifene 60 mg compared to placebo on the 3 co-primary end points: change from baseline to week 12 in the percentage of parabasal cells, superficial cells, and vaginal pH. Study 821 was evaluated based on the dryness stratum and dyspareunia stratum, which were treated as 2 independent assessments. In Study 821, an improvement in favour of ospemifene 60 mg compared to placebo was demonstrated on all 4 co-primary end points analyzed in patients reporting dyspareunia as the MBS at baseline; however, the analysis of the severity of the MBS, vaginal dryness (in the vaginal dryness stratum) failed to demonstrate an improvement with ospemifene treatment compared to placebo. Therefore, Study 821 failed to demonstrate efficacy in patients who identified vaginal dryness as the MBS of VVA. Additionally, a supportive analysis of the change in severity of vaginal dryness as the MBS of VVA in Study 310 that was performed in the PP population did not demonstrate benefit with ospemifene compared to placebo, a finding that was inconsistent with the primary analysis. As a result, the evidence for the efficacy of ospemifene in terms of relief of vaginal dryness as a symptom of VVA is associated with notable uncertainty. The Health Canada Reviewer’s report stated that Study 310 and Study 821 supported FDA approval of the indication for dyspareunia, and Study 231 supported the approval of the indication for vaginal dryness.36
The clinical expert consulted by CADTH indicated that because vaginal pH and cytology assessments — such as of the percentages of parabasal and superficial cells — are not routinely performed in post-menopausal women in clinical practice, these are not clinically relevant. However, assessment of these outcomes is recommended as co-primary end points by the FDA, along with the change in severity of moderate to severe symptoms identified by the patient as most bothersome.31 For context, a vaginal pH of 5 or greater in the absence of other causes can be considered an indicator of vaginal atrophy due to estrogen deficiency.16 In premenopausal women with adequate estrogen levels, the MI is typically 40% to 70% intermediate cells, 30% to 60% superficial cells, and 0% parabasal cells.16 In women with vaginal atrophy, an increase in parabasal cells and a decrease in superficial cells are observed. Moreover, as women age, the proportion of parabasal cells will continue to increase, and the MI may eventually consist entirely of parabasal cells.16 Therefore, the reduction in vaginal pH, reduction in the percentage of parabasal cells, and increase in the percentage of superficial cells likely support an improvement in VVA and may be useful in clinical trials, but do not necessarily represent a clinically relevant improvement, according to the clinical expert consulted by CADTH.
The VVA questionnaire was used to assess the change in severity of VVA symptoms in studies 310, 821, and 231. Evidence of validity, reliability, and responsiveness of the VVA questionnaire was not identified, nor was a MID. The clinical expert consulted by CADTH indicated that a formal assessment of symptoms (using a questionnaire) is not typically performed in clinical practice, but that any reduction in symptoms is clinically meaningful. Despite the limitations of the analysis of the change in symptom severity, a statistically significant reduction in the severity of dyspareunia was reported for patients who identified dyspareunia as their MBS at baseline in studies 310 and 821 (dyspareunia stratum). This is supported by the proportion of patients who experience a change in severity of less than 0 (i.e., a change from baseline of –1, –2 or –3), which would indicate a reduction in severity in the majority of patients; however, a notable response was also observed in the placebo treatment groups, albeit not as great a response as that observed among patients in the ospemifene treatment group. (For ospemifene versus placebo, 68% versus 54% of patients in Study 310 and 80% versus 64% of patients in Study 821 reported a change of less than 0.) The assessment of the change in severity of vaginal dryness, assessed in patients who reported dryness as the MBS at baseline, yielded similar results in Study 310 and Study 231. A study by Ettinger et al.37 compared the change in severity of VVA symptoms in 3 populations: all treated women, those who rated their symptoms as moderate or severe at baseline, and those who reported a moderate or severe symptom as their MBS at baseline. The effect size was greatest in the analysis of MBS, followed by symptoms rated as moderate or severe at baseline, and lastly, in all treated patients. The study also highlighted the uncertainty of the clinical relevance of the MBS; it suggests that an improvement in symptoms is more likely to be observed when patients with more severe symptoms are analyzed. Considering the subjective nature of the outcome, this is likely to overestimate the treatment effect in trials compared to real-world settings, in which more patients likely experience moderate or less severe VVA symptoms. The changes in severity of VVA symptoms rated as moderate or severe (not necessarily as most bothersome) at baseline were assessed as secondary end points in Study 310 (vaginal dryness and dyspareunia), Study 821 (vaginal dryness, dyspareunia, and vaginal itching or irritation), and Study 231 (dyspareunia and vaginal itching or irritation) and are considered supportive evidence. The results for vaginal dryness and dyspareunia were aligned with the primary analyses, but an improvement in vaginal itching or irritation was not observed.
Urinary symptoms, such as incontinence and genitourinary prolapse, were identified as outcomes of interest to this review. Urinary symptoms were assessed as a secondary outcome using the UDI-6 in studies 310, 821, and 231. Studies 310 and 821 reported the UDI-6 descriptively by each of the 6 domains of the outcome, and Study 231 reported the total score. No notable differences in urinary symptoms as measured by the UDI-6 were observed between ospemifene and placebo. Sexual function was assessed as a secondary outcome in studies 821 and 231 using the FSFI. The FSFI is commonly used in clinical trials and is a validated tool for the measurement of women’s overall sexual function. The clinical expert consulted by CADTH indicated that the domains of the FSFI assess clinically relevant components of sexual function; however, a formal assessment of sexual function is typically not performed in clinical practice. In general, the results of the FSFI were inconsistent between studies or did not demonstrate an improvement in sexual function for ospemifene compared to placebo, with the exception of the pain domain. The treatment-group difference in the change from baseline to week 12 for the pain domain suggested an improvement in favour of ospemifene, which is aligned with the demonstrated reduction in severity of dyspareunia. Despite this, an MID was not identified in post-menopausal women with symptoms of VVA, and the analysis of the FSFI was not controlled for multiplicity; therefore, the treatment effect of ospemifene on urinary symptoms largely remains uncertain.
HRQoL, mental health-related outcomes, bone mineral density, and adherence were identified as outcomes of interest to this review, but were not assessed in any of the included studies.
All of the included trials used placebo as a comparator, despite the availability of a number of therapies for the treatment of symptoms of VVA, including estrogen therapies. In particular, Vagifem was identified as being available to Canadian patients and covered under most public insurance plans. The lack of head-to-head comparative evidence is a key limitation of the evidence available for ospemifene.
Indirect evidence for ospemifene was available in a sponsor-submitted NMA and a published ITC. For the most part, |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| In the published ITC by Li et al., there were no differences found between ospemifene and vaginal estrogens with respect to vaginal dryness, sexual function, or change in the percentage of parabasal cells, while vaginal estrogens were favoured with respect to reduction in vaginal pH and dyspareunia. In both ITCs, the findings were uncertain due to wide 95% CrIs. The limited number of studies in the network for many of the analyses and heterogeneity within pairwise comparisons likely contributed to the uncertainty in the estimates. There were concerns about the assumption of transitivity; and consistency between direct and indirect evidence could not be assessed due to the lack of closed loops, leading to further uncertainty about the efficacy and safety of ospemifene compared with other relevant treatments for patients with post-menopausal VVA.
The long-term efficacy of ospemifene is an important consideration. Based on feedback from the clinical expert consulted by CADTH, patients are likely to continue with treatment as long as they need it or until they wish to discontinue, assuming there are no concerns about safety (however, symptoms may return following discontinuation). The expert noted that the impact of treatment on HRQoL is a significant consideration for discontinuation. As a result, the absence of HRQoL data are a major limitation of the available evidence for this review. One of the included studies, Study 718, provided evidence of efficacy in terms of the percentage of parabasal cells, superficial cells, and vaginal pH for up to 52 weeks. Assessments beyond 12 weeks were included as secondary outcomes, not controlled for multiplicity, and subject to missing data, but the results at week 26 and week 52 were consistent with the results at week 12, suggesting maintenance of the treatment effect for up to 1 year. Evidence of long-term efficacy relief from symptoms of VVA was not assessed in any of the included studies, and was another limitation of the studies, given the clinical relevance of self-reported symptom assessments.
Harms
No deaths were reported in any of the included studies, and specific SAEs were infrequently reported. During the 12-week treatment period of studies 310, 821, and 231, patients who received ospemifene reported AEs at a similar or slightly higher frequency than patients who received placebo. Similar results were observed during the 52-week treatment period of Study 718, although the frequency of AEs was higher overall in this study than in the 12-week studies. This was not seen as a concern by the clinical expert consulted by CADTH because it is likely due to the longer duration of exposure to treatment. During the 52-week treatment period of Study 310X (including 12 weeks in Study 310), AEs were reported more frequently by those who received ospemifene compared to placebo (64% versus 45%); however, this is likely biased in favour of placebo due to the high rate of discontinuation from the study. The frequencies of specific AEs were not reported for more than 9% of patients in the 12-week studies or for 13% of patients in Study 718; however, the most commonly reported AEs in each of the 4 studies were hot flashes and UTIs. Hot flashes were consistently reported more frequently by patients who received ospemifene. These were also identified as a known adverse reaction to ospemifene in the Health Canada Reviewers Report and the Health Canada–approved product monograph.5,36 Vaginal infections, vaginal discharge, and muscle spasms were also commonly reported AEs and were more reported more frequently with ospemifene than placebo.
Overall, the proportion of patients who withdrew from treatment due to an AE were similar between treatment groups and specific AEs that lead to discontinuation were infrequent; however, hot flashes led to treatment discontinuation for at least 1 patient on ospemifene in every study. The clinical expert consulted by CADTH suggested that this was a notable concern for ospemifene, which may lead to ospemifene being reserved as a second-line treatment option. Further, ospemifene is not recommended for use with other estrogens or estrogen agonists or antagonists due to drug interactions. The clinical expert consulted by CADTH relayed that this represents a limitation in the use of ospemifene in women with concomitant vasomotor symptoms; the increase in vasomotor symptoms (such as hot flashes) from the use of ospemifene further complicates this issue. The clinical expert noted that ospemifene may still be used in women who are using non-estrogen-based therapies for vasomotor symptoms, including antidepressants, gabapentinoids, clonidine, oxybutynin, and lifestyle management strategies.
Notable harms were identified in the CADTH systematic review protocol in consultation with the clinical expert for this review. These included: vaginal hemorrhage, abnormal genital bleeding, cervical dysplasia, breast mass, endometrial hyperplasia, uterine polyps, cardiovascular disorders (e.g., thromboembolic and hemorrhagic stroke, coronary heart disease), breast cancer, uterine cancer, DVT, and pulmonary embolism. In general, notable harms were reported infrequently (i.e., by fewer than 2% of patients in any treatment group), but uterine polyps (reported in 6 patients on ospemifene and 1 patient on placebo across the 5 included studies) and DVT (reported in 2 patients on ospemifene across the 5 included studies) were reported more frequently by patients who received ospemifene. Although evidence of treatment with ospemifene for up to 52 weeks was available, this is likely not a long enough time frame over which to assess certain safety outcomes that tend to be observed beyond 1 year. In addition, patients included in the ospemifene studies were generally healthy due to restrictive exclusion criteria; as a result, the safety of ospemifene may be underestimated in the general population. A 5-year, post-authorization safety study was conducted at the request of international regulatory agencies to address ospemifene safety concerns; the sponsor concluded that the results did not change the risk-benefit profile of ospemifene.36 However, the results of the post-authorization safety study were not reviewed or critically appraised by CADTH for this review.
It should be noted that endometrial cancer and cardiovascular disorders were listed as serious warnings on the product monograph for ospemifene.5 This was due in part to the risk of endometrial cancer in women with a uterus who use unopposed estrogens. In addition, DVT was identified in 2 patients in the trials; increased thrombotic risk is a known effect of SERMs as a drug class.5,36 The product monograph states that ospemifene should not be used in women with known or suspected breast cancer because its use has not been adequately studied in this population.5
As noted in the discussion of efficacy results, direct evidence of ospemifene was limited to placebo-controlled comparisons. The 2 ITCs identified for this review suggested that the safety profile of ospemifene was similar to that of Vagifem and Premarin. However, this comparison was subject to notable limitations, causing uncertainty in these results.
Other Considerations
The indication for ospemifene approved by Health Canada and the sponsor’s reimbursement request for ospemifene align with the FDA-approved indication.38 The indication approved by the European Medicines Agency initially restricted ospemifene to second-line therapy due to potential safety concerns associated with the drug class,39,40 but it has since been revised to the following: for the treatment of moderate to severe symptomatic VVA in post-menopausal women.41 The Health Canada Reviewers Report noted that a post-authorization safety study to further evaluate the cardiovascular risks associated with ospemifene treatment was submitted as part of a risk management plan for ospemifene.36
Conclusions
Five studies were summarized as part of the CADTH systematic review: 4 phase III, double-blind RCTs that assessed the efficacy and safety of ospemifene 60 mg compared to placebo over 12 weeks (studies 310, 821, and 231) and over 52 weeks (Study 718) in post-menopausal women with VVA, as well as a double-blind, placebo-controlled LTSE of Study 310 (Study 310X) that provided evidence of safety with up to 52 weeks of treatment. The studies included in this review demonstrated a beneficial effect of ospemifene compared to placebo over a 12-week treatment period in post-menopausal women who self-reported moderate to severe VVA symptoms of vaginal dryness or dyspareunia. Although the efficacy of ospemifene versus placebo in relieving vaginal dryness was demonstrated in Study 231, there was inconsistency in this finding across the studies due to the lack of statistical significance for this outcome in the primary analysis of Study 821, as well as in a supportive analysis performed in the PP population of Study 310. Across the included studies, the clinical benefit was estimated using a patient-reported outcome, the VVA questionnaire. The exact clinical interpretation of the difference in the VVA questionnaire score is unclear, particularly due to the lack of a recognized MID, lack of sufficient validation of the questionnaire, and the nature of an ordinal score; however, the magnitude of the observed change in symptom scores was similar between different trial populations with similar eligibility criteria. Self-assessments from individual patients may suffer from recall bias in both the ospemifene and placebo treatment groups. Nonetheless, the observed clinical benefit was supported by objective VVA measures, namely a reduction in the percentage of parabasal cells, an increase in the percentage of superficial cells, and a reduction in pH.
The evidence from 2 ITCs summarized for this review suggests there is no difference between ospemifene and other treatments for symptoms of VVA in terms of comparative effectiveness. This suggestion should be interpreted with caution due to the uncertainty associated with the ITCs; however, it highlights the limitations associated with a lack of direct comparative evidence to active treatment options. Other gaps in the evidence include the absence of assessments of both HRQoL and symptom relief beyond 12 weeks of treatment. Evidence assessing the safety of ospemifene was available for up to 52 weeks of treatment. No deaths were reported, and SAEs were reported infrequently. Overall, the safety profile of ospemifene was acceptable based on the included trials, with the exception of the frequency of hot flashes and uncertainty around the risk of thromboembolic events. Further study is warranted to obtain long-term safety data, including evidence of safety beyond 1 year.
References
1.Nappi RE, Martini E, Cucinella L, et al. Addressing vulvovaginal atrophy (VVA)/genitourinary syndrome of menopause (GSM) for healthy aging in women. Front Endocrinol (Lausanne). 2019;10:561. PubMed
2.Johnston S, Bouchard C, Fortier M, Wolfman W. Guideline no. 422b: menopause and genitourinary health. J Obstet Gynaecol Can. 2021;43(11):1301-1307.e1301. PubMed
3.Nappi RE, Kokot-Kierepa M. Women's voices in the menopause: results from an international survey on vaginal atrophy. Maturitas. 2010;67(3):233-238. PubMed
4.Nappi RE, Palacios S, Bruyniks N, Particco M, Panay N, on behalf of the ESi. The burden of vulvovaginal atrophy on women's daily living: implications on quality of life from a face-to-face real-life survey. Menopause. 2019;26(5):485-491. PubMed
5.Osphena® (ospemifene tablets): tablets, 60mg, oral [product monograph]. Blainville (QC): Duchesnay Inc.; 2021 Jul 15.
6.Dipex Charity. Healthtalk.org. 2019; https://healthtalk.org/. Accessed 2022 Feb 28.
7.Clinical Study Report: 15-50310. Efficacy and safety of ospemifene in the treatment of vulvar and vaginal atrophy (VVA) in postmenopausal women: a 12-week, randomized, double-blind, placebo-controlled, parallel-group study comparing oral ospemifene 30 mg and 60 mg daily doses with placebo [internal sponsor's report]. Ann Arbor (MI): QuatRx Pharmaceuticals Company; 2009 Apr 20.
8.Clinical Study Report: 15-50821. Efficacy and safety of ospemifene in the treatment of moderate to severe vaginal dryness and vaginal pain associated with sexual activity, symptoms of vulvar and vaginal atrophy (VVA), associated with menopause: a 12-week, randomized, double-blind, placebo-controlled, parallel-group study comparing oral ospemifene 60 mg daily dose with placebo in postmenopausal women [internal sponsor's report]. Ann Arbor (MI): QuatRx Pharmaceuticals Company; 2010 Apr 13.
9.Clinical Study Report: 1517I0231. A phase 3, randomized, double-blind, placebo-controlled multicenter study to evaluate the efficacy and safety of ospemifene in patients with moderate to severe vaginal dryness, a symptom of vulvovaginal atrophy (VVA) due to menopause [internal sponsor's report]. Florham Park (NJ): Shionogi Inc.; 2018 Jan 18.
10.Clinical Study Report: 15-50718. Efficacy and long-term safety of ospemifene in the treatment of vulvar and vaginal atrophy (VVA) in postmenopausal women: a 52-week, randomized, double-blind, placebo-controlled, parallel-group study comparing 60 mg oral daily dose of ospemifene with placebo [internal sponsor's report]. Ann Arbor (MI): Hormos Medical Ltd.; QuatRx Pharmaceuticals Company; 2011 Aug 15.
11.Clinical Study Report: 15-50310X. Long-term safety of 30 mg and 60 mg oral daily doses of ospemifene in the treatment of vulvar and vaginal atrophy (VVA) in postmenopausal women with an intact uterus: a 40 week randomized, double blind, placebo controlled, follow-up to protocol 15-50310 [internal sponsor's report]. Ann Arbor (MI): QuatRx Pharmaceuticals Company; 2009 Jun 22.
12.Osphena® for the treatment of vulvovaginal atrophy: systematic literature review and network meta-analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Osphena (ospemifene), oral tablets, 60 mg. Blainville (QC): Duchesney Inc.; 2021 Oct 20.
13.Li B, Duan H, Chang Y, Wang S. Efficacy and safety of current therapies for genitourinary syndrome of menopause: A Bayesian network analysis of 29 randomized trials and 8311 patients. Pharmacol Res. 2021;164:105360. PubMed
14.Clinical Study Report: 15-50312. Long-term safety of ospemifene 60 mg oral daily dose for the treatment of vulvar and vaginal atrophy (VVA) in postmenopausal women without a uterus: a 52-week open-label follow-up to protocol 15-50310 [internal sponsor's report]. Ann Arbor (MI): QuatRx Pharmaceuticals Company; 2009 Sep 28.
15.Simon J, Portman D, Mabey RG, Jr., Ospemifene Study G. Long-term safety of ospemifene (52-week extension) in the treatment of vulvar and vaginal atrophy in hysterectomized postmenopausal women. Maturitas. 2014;77(3):274-281. PubMed
16.Bachmann G, Santen RJ. Genitourinary syndrome of menopause (vulvovaginal atrophy): clinical manifestations and diagnosis. In: Barbieri RL, Chakrabarti A, eds. Waltham (MA): UpToDate; 2020: http://www.uptodate.com (subscription required). Accessed 2021 Aug 30.
17.Vagifem (estradiol vaginal inserts USP): 10 ug estradiol [product monograph]. Mississauga (ON): Novo Nordisk Canada Inc; 2017 Mar 20: https://www.novonordisk.ca/content/dam/nncorp/ca/en/products/Vagifem_10_PM_English.pdf. Accessed 2021 Aug 27.
18.Estring (17 β-estradiol): 2 mg vaginal ring [product monograph]. Kirkland (QC): Pfizer Canada Inc; 2017 Nov 16: https://pdf.hres.ca/dpd_pm/00042172.PDF. Accessed 2021 Aug 27.
19.Premarin (conjugated estrogens): 0.3 mg, 0.625 mg, and 1.25 mg tablets [product monograph]. Kirkland (QC): Pfizer Canada Inc., Licensee; 2012 Jan 10: https://pdf.hres.ca/dpd_pm/00015274.PDF. Accessed 2021 Aug 27.
20.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. Journal of clinical epidemiology. 2016;75:40-46. PubMed
21.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2021 Nov 11.
22.Bachmann GA, Komi JO, Ospemifene Study G. Ospemifene effectively treats vulvovaginal atrophy in postmenopausal women: results from a pivotal phase 3 study. Menopause. 2010;17(3):480-486. PubMed
23.Nappi RE, Panay N, Bruyniks N, Castelo-Branco C, De Villiers TJ, Simon JA. The clinical relevance of the effect of ospemifene on symptoms of vulvar and vaginal atrophy. Climacteric. 2015;18(2):233-240. PubMed
24.Portman DJ, Bachmann GA, Simon JA, Ospemifene Study G. Ospemifene, a novel selective estrogen receptor modulator for treating dyspareunia associated with postmenopausal vulvar and vaginal atrophy. Menopause. 2013;20(6):623-630. PubMed
25.Portman D, Palacios S, Nappi RE, Mueck AO. Ospemifene, a non-oestrogen selective oestrogen receptor modulator for the treatment of vaginal dryness associated with postmenopausal vulvar and vaginal atrophy: a randomised, placebo-controlled, phase III trial. Maturitas. 2014;78(2):91-98. PubMed
26.Constantine G, Graham S, Portman DJ, Rosen RC, Kingsberg SA. Female sexual function improved with ospemifene in postmenopausal women with vulvar and vaginal atrophy: results of a randomized, placebo-controlled trial. Climacteric. 2015;18(2):226-232. PubMed
27.Archer DF, Goldstein SR, Simon JA, et al. Efficacy and safety of ospemifene in postmenopausal women with moderate-to-severe vaginal dryness: a phase 3, randomized, double-blind, placebo-controlled, multicenter trial. Menopause. 2019;26(6):611-621. PubMed
28.Goldstein I, Simon JA, Kaunitz AM, et al. Effects of ospemifene on genitourinary health assessed by prospective vulvar-vestibular photography and vaginal/vulvar health indices. Menopause. 2019;26(9):994-1001. PubMed
29.Goldstein SR, Bachmann GA, Koninckx PR, et al. Ospemifene 12-month safety and efficacy in postmenopausal women with vulvar and vaginal atrophy. Climacteric. 2014;17(2):173-182. PubMed
30.Simon JA, Lin VH, Radovich C, Bachmann GA, Ospemifene Study G. One-year long-term safety extension study of ospemifene for the treatment of vulvar and vaginal atrophy in postmenopausal women with a uterus. Menopause. 2013;20(4):418-427. PubMed
31.Guidance for industry: estrogen and estrogen/progestin drug products to treat vasomotor symptoms and vulvar and vaginal atrophy symptoms — recommendations for clinical evaluation [draft guidance]. Silver Spring (MD): U.S. Food and Drug Administration; 2003: https://www.fda.gov/media/71359/download. Accessed 2022 Feb 28.
32.Stephenson KR, Toorabally N, Lyons L, C MM. Further validation of the Female Sexual Function Index: specificity and associations with clinical interview data. J Sex Marital Ther. 2016;42(5):448-461. PubMed
33.Neijenhuijs KI, Hooghiemstra N, Holtmaat K, et al. The Female Sexual Function Index (FSFI)-A Systematic Review of Measurement Properties. J Sex Med. 2019;16(5):640-660. PubMed
34.Bruyniks N, Biglia N, Palacios S, Mueck AO. Systematic indirect comparison of ospemifene versus local estrogens for vulvar and vaginal atrophy. Climacteric. 2017;20(3):195-204. PubMed
35.Lee A, Kim TH, Lee HH, et al. Therapeutic approaches to atrophic vaginitis in postmenopausal women: A systematic review with a network meta-analysis of randomized controlled trials. J Menopausal Med. 2018;24(1):1-10. PubMed
36.Health Canada reviewer's report: Osphena (ospemifene) [internal sponsor's report] In: Drug Reimbursement Review sponsor submission: Osphena (ospemifene), oral tablets, 60 mg. Blainville (QC): Duchesney Inc.; 2021.
37.Ettinger B, Hait H, Reape KZ, Shu H. Measuring symptom relief in studies of vaginal and vulvar atrophy: the most bothersome symptom approach. Menopause. 2008;15(5):885-889. PubMed
38.Highlights of prescribing information: OSPHENA® (ospemifene) tablets, for oral use. Silver Spring (MD): U.S. Food and Drug Administration; 2019 Jan: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/203505s015lbl.pdf. Accessed 2021 Feb 28.
39.Shionogi. Summary of product characteristics. Product information: Senshio 60 mg film-coated tablets. (European public assessment report). Amsterdam (Netherlands): European Medicines Agency; 2021: https://www.ema.europa.eu/en/documents/product-information/senshio-epar-product-information_en.pdf. Accessed 2022 Feb 28.
40.Committee for Medicinal Products for Human Use. Assessment report: Senshio (ospemifene). (European public assessment report). Amsterdam (Netherlands): European Medicines Agency: https://www.ema.europa.eu/en/documents/assessment-report/senshio-epar-public-assessment-report_en.pdf. Accessed 2022 Feb 28.
41.Committee For Medicinal Products For Human Use. CHMP post-authorisation summary of positive opinion for Senshio (II-0041). EMA/CHMP/22550/2022. Amsterdam (Netherlands): European Medicines Agency; 2022 Jan 28: https://www.ema.europa.eu/en/medicines/human/summaries-opinion/senshio. Accessed 2022 Mar 9.
42.Rosen R, Brown C, Heiman J, et al. The Female Sexual Function Index (FSFI): a multidimensional self-report instrument for the assessment of female sexual function. J Sex Marital Ther. 2000;26(2):191-208. PubMed
43.Meston CM. Validation of the Female Sexual Function Index (FSFI) in women with female orgasmic disorder and in women with hypoactive sexual desire disorder. J Sex Marital Ther. 2003;29(1):39-46. PubMed
44.Wiegel M, Meston C, Rosen R. The Female Sexual Function Index (FSFI): cross-validation and development of clinical cutoff scores. J Sex Marital Ther. 2005;31(1):1-20. PubMed
45.Simon JA, Reape KZ, Wininger S, Hait H. Randomized, multicenter, double-blind, placebo-controlled trial to evaluate the efficacy and safety of synthetic conjugated estrogens B for the treatment of vulvovaginal atrophy in healthy postmenopausal women. Fertil Steril. 2008;90(4):1132-1138. PubMed
46.Cohen J. A power primer. Psychol Bull. 1992;112(1):155-159. PubMed
47.Reed SD, Mitchell CM, Joffe H, et al. Sexual function in women on estradiol or venlafaxine for hot flushes: a randomized controlled trial. Obstet Gynecol. 2014;124(2 Pt 1):233-241. PubMed
48.Reeve BB, Wyrwich KW, Wu AW, et al. ISOQOL recommends minimum standards for patient-reported outcome measures used in patient-centered outcomes and comparative effectiveness research. Qual Life Res. 2013;22(8):1889-1905. PubMed
Appendix 1: Literature Search Strategy
Note that this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid.
Databases
MEDLINE All (1946-present)
Embase (1974-present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: November 22, 2021
Alerts: Biweekly search updates until project completion.
Search filters applied: No filters were applied to limit the retrieval by study type.
Limits: Conference abstracts: excluded.
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
MeSH | Medical Subject Heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
.ti | Title |
.ab | Abstract |
.dq | Candidate term word (Embase) |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Author keyword heading word (MEDLINE, Embase) |
.nm | Name of substance word (MEDLINE) |
.ot | Original title |
.pt | Publication type |
.rn | Registry number |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multi-Database Strategy
(ospemifene* or Osphena* or Senshio* or FC-1271 or FC-1271a or FC1271 or FC1271a or CCRIS-9205 or CCRIS9205 or HSDB 8281 or HSDB8281 or G03XC05 or deaminohydroxytoremifene or deamino-hydroxytoremifene or B0P231ILBK).ti,ab,kf,ot,hw,rn,nm.
1 use medall
*ospemifene/
(ospemifene* or Osphena* or Senshio* or FC-1271 or FC-1271a or FC1271 or FC1271a or CCRIS-9205 or CCRIS9205 or HSDB 8281 or HSDB8281 or G03XC05 or deaminohydroxytoremifene or deamino-hydroxytoremifene).ti,ab,kf,dq.
3 or 4
5 use oemezd
6 not (conference abstract or conference review).pt.
2 or 7
remove duplicates from 8
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
[Search terms – ospemifene OR Osphena OR Senshio OR FC-1271 OR FC-1271a OR FC1271 OR FC1271a OR CCRIS-9205 OR CCRIS9205 OR HSDB 8281 OR HSDB8281 OR G03XC05 OR deaminohydroxytoremifene OR deamino-hydroxytoremifene OR B0P231ILBK]
WHO International Clinical Trials Registry Platform ICTRP
International Clinical Trials Registry Platform, produced by the WHO. Targeted search used to capture registered clinical trials.
[Search terms – (ospemifene* OR Osphena* OR Senshio*) NOT NCT*]
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
[Search terms – ospemifene, Osphena, Senshio]
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
[Search terms – ospemifene or Osphena or Senshio]
Grey Literature
Search dates: November 11 to 17, 2021
Keywords: ospemifene (Osphena / Senshio), genitourinary syndrome of menopause, vulvovaginal atrophy, dyspareunia, or vaginal dryness in menopause.
Limits: None.
Updated: Search updated before the completion of stakeholder feedback period.
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Internet Search
Appendix 2: Excluded Studies
Note that this appendix has not been copy-edited.
Reference | Reason for Exclusion |
|---|---|
Constantine G, Graham S, Koltun WD, Kingsberg SA. Assessment of ospemifene or lubricants on clinical signs of VVA. J Sex Med. 2014 Apr;11(4):1033 to 1041. | Study design: pooled, post hoc analysis |
Simon JA, Altomare C, Cort S, Jiang W, Pinkerton JV. Overall Safety of Ospemifene in Postmenopausal Women from Placebo-Controlled phase 2 and 3 Trials. J Womens Health (Larchmt). 2018 01;27(1):14 to 23. | Study design: pooled analysis of phase II and phase III studies |
Cui Y, Zong H, Yan H, Li N, Zhang Y. The efficacy and safety of ospemifene in treating dyspareunia associated with postmenopausal vulvar and vaginal atrophy: a systematic review and meta-analysis. J Sex Med. 2014 Feb;11(2):487 to 497. | Study design: systematic review and meta-analysis |
Bruyniks N, Nappi RE, Castelo-Branco C, de Villiers TJ, Simon J. Effect of ospemifene on moderate or severe symptoms of vulvar and vaginal atrophy. Climacteric. 2016;19(1):60 to 65. | Study objective and outcomes |
Appendix 3: Description and Appraisal of Outcome Measures
Note that this appendix has not been copy-edited.
Aim
To describe the following outcome measures and review their measurement properties (validity, reliability, responsiveness to change, and MID):
The Patient Self-assessment of Vulvar and Vaginal Atrophy Questionnaire was included in the trial as both primary and secondary end points.
The MI was included in the trials as both primary and secondary end points.
The FSFI was included in the trials as secondary end points.
The UDI-6 was included in the trials as secondary end points.
Findings
Table 48: Summary of outcome measures and their measurement properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
VVA Questionnaire | A patient-reported, 5-item questionnaire, assessing 5 VVA symptoms - vaginal dryness, vaginal and/or vulvar irritation/itching, difficult or painful urination/dysuria, vaginal pain associated with sexual activity/dyspareunia, and vaginal bleeding associated with sexual activities. The questionnaire has 2 parts; in the second part participants are asked about MBS, evaluated by assigning a numerical value to none (0), Mild (1), Moderate (2), or severe (3), and then been reported the change from baseline as a numerical change in this trial as follows: −3 (severe to none) −2 (severe to mild or moderate to none) −1 (severe to moderate, moderate to mild, or mild to none) 0 (no change) 1 (none to mild, mild to moderate, or moderate to severe) The improvement of the MBS was defined by the decrease in severity. | Evidence of validity, reliability, or responsiveness were not identified for this outcome. | Unknown |
Percentage of parabasal cells and percentage of superficial cells based on the Maturation Index of Vaginal Epithelium | The Maturation Index of the vaginal epithelium was determined from the vaginal smear samples taken from the middle third of the lateral vaginal wall. | Evidence of validity, reliability, or responsiveness were not identified for this outcome. | Unknown |
FSFI | A self-completed, 19-item Likert-type scale that consists of 6 domains measuring female sexual function: desire, arousal, lubrication, orgasm, satisfaction, and pain. Score ranges for each domain and full scale:
Higher score indicates better sexual function | Validity: Content validity has been ensured by panel selection of initial items, pre-testing with healthy volunteers, and consultation with experts.42 Discriminant validity has been demonstrated between control groups and patients with various sexual dysfunctions, e.g., FSAD, FSOD, HSDD, and other types (all P ≤ 0.001).42-44 Divergent validity has been shown between FSFI and Locke-Wallace Marital Adjustment Test in control groups and patients with FSAD, FSOD, HSDD, in which Pearson correlation coefficients (r) ranged from 0.03 to 0.72, where satisfaction domain showed relatively higher correlation compared to other domains.42,43 Reliability: Pearson correlation coefficients (r) for test-retest reliability were high for all domains (r = 0.79 to 0.86) and full scale (r = 0.88) in a sample population composed of controls and patients with FSAD (n = 259).42 Internal consistency was acceptable (Cronbach alpha ≥ 0.7) for all 6 domains and full scale in control and various patient groups, e.g., FSAD, FSOD, HSDD, and other types, except for the desire domain in patients with HSSD (alpha = 0.58).42-44 Responsiveness to change: No data were located. Note: evidence of validity, reliability, and responsiveness of the FSFI used for post-menopausal women was not identified. | Unknown FSFI total score of 26.55 is a cut-off to differentiate patients with or without sexual dysfunction |
UDI-6 | A questionnaire used to assess the presence or absence of 6 urinary symptoms: (1) frequent urination; (2) urine leakage related to the feeling of urgency; (3) urine leakage related to physical activity, coughing, or sneezing; (4) small amounts of urine leakage; (5) difficulty emptying bladder; and (6) pain or discomfort in the lower abdominal or genital area. When a symptom was present, the degree to which a patient was bothered was rated on a 4-point scale (not at all, slightly, moderately, and greatly). | Evidence of validity, reliability, or responsiveness were not identified for this outcome. | Unknown |
FSAD = female sexual arousal disorder; FSFI = female sexual function index; FSOD = female sexual orgasm disorder; HSSD = hypoactive sexual desire disorder; MBS = most bothersome symptom; MID = minimal important difference; UDI-6 = Urinary Distress Inventory – Short Form; VASQ = vaginal atrophy symptom questionnaire; VVA = vulvovaginal atrophy.
Patient Self-assessment of Vulvar and Vaginal Atrophy Questionnaire
The VVA questionnaire is used to assess 5 VVA symptoms - vaginal dryness, vaginal and/or vulvar irritation/itching, difficult or painful urination/dysuria, vaginal pain associated with sexual activity/dyspareunia, and vaginal bleeding associated with sexual activities. Patients themselves completed the questionnaire, and the investigator or qualified study personnel reviewed the responses.
The VVA questionnaire was divided into 2 parts. In the first part, patients were asked, “Have you had the following symptoms in the past month? (or since the last visit?)” The severity of each symptom was assessed as “none/no,” “mild,” “moderate,” or “severe” by the patients. For the second part, the patients were asked – “Which symptom is the most bothersome to you?” Based on the symptoms that were indicated in the first part as “moderate” or “severe.” If a patient reported moderate to severe vaginal dryness and/or vaginal pain associated with sexual activity as her MBS at screening and at randomization, she was considered eligible for the study. The MBS has been evaluated by assigning a numerical value to none (0), Mild (1), Moderate (2), or severe (3), and then been reported the change from baseline as a numerical change in this trial as followed:
−3 (severe to none)
−2 (severe to mild or moderate to none)
−1 (severe to moderate, moderate to mild, or mild to none)
0 (no change)
1 (none to mild, mild to moderate, or moderate to severe)
The improvement of the MBS was defined by the decrease in severity.
The use of the MBS approach involving patients’ self-reported and rated VVA symptoms is recommended by the FDA with for standardizing patient-reported outcome measures for clinical studies of VVA treatments for post-menopausal women.31 However, evidence of validity, reliability, and responsiveness MBS approach using the VVA questionnaire in post-menopausal women was identified.
In addition, Ettinger et al.37 assessed the usefulness and importance of this MBS approach in evaluating the treatment for VVA among 310 women in a double-blind, placebo-controlled multi-centre study in the US. This study used data from the Simon et al. (2008) study.45 For the statistical analyses, the severity change from baseline was calculated as the post-baseline value (0 = no symptom present, 1 = mild, 2 = moderate, 3 = severe) minus the baseline value for each symptom. Analysis of dyspareunia was restricted to women reporting sexual intercourse at both baseline and at end of treatment. ANCOVA was used to compare mean changes in symptom severity between placebo and active treatment groups and to determine the variance of those changes. Standardized treatment effect was calculated as the MD in change between the 2 groups divided by the S (SD) of that difference. A standardized effect size of 0.2 was considered small, 0.5 was medium, and 0.8 was large for the statistical analyses.37,46
In the Simon et al. (2008) study,45 the primary objective was to evaluate the safety and efficacy of SCE-B for the treatment of signs and symptoms of vaginal atrophy, whereas in the Ettinger et al.37 study, further analyses were done to determine whether the magnitude of treatment effect was influenced by the MBS definition and whether other expressions of the symptom data lead to different conclusions about treatment efficacy. Vaginal dryness and dyspareunia were the most frequently reported moderate to severe symptoms and were also most frequently classified as the MBS (44.4% and 30.2% of participants, respectively). Because these 2 symptoms were most commonly reported as moderate to severe and also most commonly chosen as MBS, the analysis was focused specifically on these 2 symptoms. In all cases for both symptoms, a strongly statistically significant (P ≤ 0.001, adjusted for baseline severity) effect favouring SCE-B over placebo was observed, despite the substantial decrease in available sample size when the moderate to severe and MBS symptom definitions were used. Changes in the standardized effect size was similar to the findings for the treatment effect, as the greatest standardized effect size was observed for the MBS. Compared with the standardized effect sizes for all women, those calculated from the MBS were 49% and 62% greater for dyspareunia and dryness, respectively. Standardized effect sizes were large (≥ 1.0) for both dryness and dyspareunia when reported as the MBS. Moreover, compared with the variance calculated from changes of all women, the variances for the MBS were 24% higher for dyspareunia and 22% lower for dryness.
It was reported in the Ettinger el. al.37 study that using the MBS increased the effect size and allowed statistically significant treatment effects to be shown in relatively small subgroups, observed from a further analysis of the clinical study of treatment for VVA. However, the clinical relevance of the MBS was not proven very clearly. Moreover, a lack of reliable distributional characteristics was identified in the MBS metric, along with an overstatement of the expectations from the treatment and a selected patient population. Lastly, none of these trial results demonstrated any clinically meaningful differences using the MBS approach or the VVA questionnaire.
Maturation Index: Percentage of Parabasal Cell and Superficial Cells
The MI of the vaginal epithelium was determined from the vaginal smear samples taken from the middle third of the lateral vaginal wall. Maturation value in this study was defined as MV = (S x 1) + (I x 0.5) + (P x 0), where “S” was the percentage of superficial cells, “I” was the percentage of intermediate cells, and “P” was the percentage of parabasal cells. The MI is the proportion of parabasal, intermediate, and superficial cells in each 100 cells counted on a smear of the upper 2-thirds of the vagina. It is used to quantify the proportions of cell types of the vaginal epithelium.16 It is usually done in a laboratory experienced in running this test. In premenopausal women with adequate estrogen levels, intermediate and superficial cells predominate. The MI for these women is typically 40 to 70 intermediate cells, 30 to 60superficial cells, and 0 parabasal cells. In women with vaginal atrophy, an increase in parabasal cells and a decrease in superficial cells are observed. Women in early menopause typically have a MI of 65 parabasal cells, 30 intermediate cells, and 5 superficial cells. As women age, parabasal cells will continue to increase, and the MI may eventually consist entirely of parabasal cells.16
Evidence of a clinically meaningful change in the percentage of parabasal cells or percentage of superficial cells was not identified in the literature.
Female Sexual Function Index
The FSFI is a self-reported, multi-dimensional, 19-item measure of female sexual function consisting of 6 domains: desire, arousal, lubrication, orgasm, satisfaction, and pain. Following initial usability testing among 30 female volunteers, the FSFI was tested for validity and reliability with a sample of 259 control females from the general population (age 21 to 68 years) and an age-matched clinical sample of patients who met DSM-IV-TR criteria for Female Sexual Arousal Disorder (FSAD).42 Later, the instrument was tested among females with other sexual dysfunction diagnoses, e.g., Female Sexual Orgasm Disorder (FSOD), Hypoactive Sexual Desire Disorder (HSDD), dyspareunia / vaginismus (pain), and multiple sexual dysfunctions.43,44 The questionnaire was designed to be used to assess female sexual function and quality of life in clinical trials or epidemiological studies.42
FSFI provides an overall sexual function score on a Likert-type scale. Specifically, each item is scored from 0 to 5 except for questions 1 and 2 (for desire domain), 15 to 16 (in satisfaction domain), which are scored from 1 to 5. The individual domain scores and full scale (overall) score of the FSFI can be derived from the computational formula (Table 49). For individual domain scores, scores of the individual items that comprise the domain are added and multiplied by the corresponding domain factor. Full scale score is obtained by adding 6 domain scores together. Score ranges for the arousal, lubrication, orgasm, and pain domains are from 0 to 6.0; for desire and satisfaction domains ranges are from 1.2 to 6.0 and from 0.8 to 6.0, respectively. The total FSFI score ranges from 2.0 to 36.0. Higher scores indicate a greater level of sexual function. The recall period is the past 4 weeks. A domain score of zero indicates that the patient reported having no sexual activity during the past month.47 An FSFI total score of 26.55 is taken to be the cut score for differentiating women with and without sexual dysfunction.44
Table 49: FSFI Domain Scores and Full Scale Score
Domain (Total 6) | Questions (Total 19) | Score Range | Factor | Minimum Score | Maximum Score |
|---|---|---|---|---|---|
Desire | 1, 2 (2) | 1 to 5 | 0.6 | 1.2 | 6.0 |
Arousal | 3 to 6 (4) | 0 to 5 | 0.3 | 0 | 6.0 |
Lubrication | 7 to 10 (4) | 0 to 5 | 0.3 | 0 | 6.0 |
Orgasm | 11 to 13 (3) | 0 to 5 | 0.4 | 0 | 6.0 |
Satisfaction | 14 to 16 (3) | 0 (or 1) – 5* | 0.4 | 0.8 | 6.0 |
Pain | 17 to 19 (3) | 0 to 5 | 0.4 | 0 | 6.0 |
Full Scale Score Range | 2.0 | 36.0 | |||
*Range for item 14 = 0 to 5; range for items 15 and 16 = 1 to 5
Source: Reed, et al.,47 Rosen et al.42
The FSFI has been translated into more than 20 languages and has been adapted in more than 30 countries.32,33 Also, it has been studied for use with multiple populations, including women from different age groups, with diverse medical conditions, and with various sexual dysfunctions.32,33
Reliability
Rosen et al.42 assessed test-retest reliability at 2 separate visits 2 to 4 weeks apart, with 131 general population controls and 128 age-matched patients diagnosed with FSAD at 5 research centres in the US. It is unclear if the post-menopausal patients included in the study had symptoms attributable to VVA. Overall test-retest reliability was acceptable (r ≥ 0.70)48 for all of the domains (Pearson’s product-moment correlation [r] = 0.79 to 0.86) and for the total scale (r = 0.88) among the full sample. In general, higher test-retest reliability of domain scores was obtained for the control group than for the FSAD group. For the FSAD group, the domain of desire showed the highest test-retest reliability (r = 0.80), with the other domains showing moderate to high correlations (r = 0.62 to 0.71).
In addition, Rosen et al.42 demonstrated acceptable internal consistency (Cronbach alpha ≥ 0.70)48 for all 6 domains (Cronbach alpha ≥ 0.82 for all 6 domains). Meston43 conducted further internal consistency testing among women with FSOD (n = 71) or HSDD (n = 44), as well as control patients (n = 71) who were age-matched to FSOD patients. Meston found that inter-item correlations remained high for all the domain scores among women with FSOD (Cronbach alpha ≥ 0.84), control women (alpha ≥ 0.83), and women with HSDD (alpha ≥ 0.74), except for the desire domain in patients with HSSD (alpha = 0.58). The moderate Cronbach alpha value of 0.58 suggests that the 2-item FSFI desire composite may not be a reliable indicator of sexual desire among this population.
Lastly, Wiegel et al.44 combined data from Rosen et al. (n = 255) and Meston (n = 138) with an additional sample population (n = 134) to assess internal consistency in women with or without sexual dysfunction. Internal consistency for subscales and total score was acceptable (alpha ≥ 0.7)48 for all sample populations and all domains regardless of sexual dysfunction status indicating that questionnaire items remained highly related within each domain or all scale in women with or without sexual dysfunction.
Validity
Validity reflects the degree to which the instrument measures what it aims to measure.
Content validity has been ensured by Rosen et al.42 in the population as described in the reliability section. Briefly, the FSFI was developed in a series of stages, including panel selection of the initial items, pre-testing with 30 healthy, female volunteers at 3 investigational sites, followed by linguistic and conceptual validation with a panel of expert consultants.
Discriminant validity was assessed by Rosen et al.42 by comparing the mean responses of patients with FSAD (n = 126 to 128) with those of the controls without FSAD (n = 129 to 131). Significant differences (for all P ≤ 0.001) between the groups were observed for all 6 domains (not included in this report) and the full scale score (19.2 ± SD 6.63 in 126 patients with FSAD versus 30.5 ± SD 5.29 in 129 control patients). Moreover, Meston43 assessed the discriminant validity in additional clinical samples by comparing the mean responses of women with FSOD (n = 71) and HSDD (n = 44) to those of the age-matched control women (n = 71 and 44, respectively). The results from between groups analyses of variance revealed significant differences between women with sexual dysfunction (FSOD or HSDD) and their controls on each of the FSFI domain and total scores (for all P ≤ 0.001). As expected, the largest differences between women with FSOD and controls were noted for orgasm and arousal domains (effect size estimated using Cohen D = 1.69, 1.58, respectively), and the largest differences between women with HSDD and controls were seen for the arousal and desire domains (ES estimated using Cohen D = 1.85, 1.69, respectively). Lastly, discriminant validity has been confirmed by Wiegel et al.44 with the sample populations described in the reliability section. Evidence for discriminant validity was observed for the total score (MANOVA and univariate tests, P < 0.001) and individual domain scores (univariate tests, P < 0.001) between patients with and without sexual dysfunction diagnoses.
Divergent validity was tested by Rosen et al.42 by specifically measuring the construct under study (i.e., sexual function) compared to an instrument that assesses a different, albeit partially related, construct (e.g., marital satisfaction). The Pearson product-moment correlation between the Locke-Wallace Marital Adjustment Test and the total FSFI score was moderate for the control group (r = 0.53) and low for the FSAD group (r = 0.22). This result indicates that for patients with sexual dysfunction, FSFI scores appeared to have a greater degree of independence with the marital adjustment effect compared to control group. To extend divergent validity testing in other clinical samples, Meston43 calculated Pearson correlations between the FSFI scores and the Locke-Wallace Marital Adjustment Test score in patients with FSOD or HSDD. Correlations between the Locke-Wallace Marital Adjustment Test and total FSFI scores were low for women with FSOD (r = 0.22) or HSDD (r = 0.16), and moderate for controls (r = 0.52). Of note, the satisfaction domain showed moderate to high level of correlation (r = 0.40 to 0.72) between FSFI and Locke-Wallace across all the samples.
Responsiveness to Change
There was no evidence located to support the FSFI’s responsiveness to clinical or health status changes among post-menopausal patients over time.
MID
MID for FSFI has not been estimated in post-menopausal patients with VVA-associated symptoms.
Other Considerations and Limitations
The 3 studies, i.e., Rosen et al., Meston, Wiegel et al., assessed validity in control and sexually dysfunctional populations that contain various numbers of post-menopausal women. Based on the age of populations included in these studies, the number of post-menopausal women seems small, For example, Rosen et al.42 study population had mean age ± SD ages of the FSAD group (n = 128) and control group (n = 131) 40.5 ± 12.98 years and 39.7 ± 13.15 years, respectively. Meston study43 included women between 18 and 53 years of age with mean ± SD for patients with FSOD (n = 71) 29.4 ± 8.76 years, HSDD (n = 44) 33.0 ± 10.42 years, and controls (n = 71) 29.2 ± 7.9 years, respectively. Lastly, in Wiegel et al., the mean age of the combined sample was 36.2 ± 13.2 years and ranged from 18 to 72 years. Only 3.6% (n = 20) patients in Wiegel et al. study were peri- or post-menopausal. Taken together, no study is specifically designed to test psychometric properties of FSFI in post-menopausal women.
Also, these studies were conducted with patients diagnosed with sexual dysfunctions rather than those with symptoms of post-menopausal VVA. Some manifestations of sexual dysfunctions and post-menopausal VVA may overlap, for example, dyspareunia. However, the extent of overlap is unclear. It makes it difficult to apply the validity results to population other than those with sexual dysfunction diagnoses. Given the limitations, we can say that the results for validity and reliability can be generalized to post-menopausal patients with VVA.
Urinary Distress Inventory-Short Form
The UDI-6 questionnaire was used to assess the presence or absence of 6 urinary symptoms. These 6 symptoms included: (1) frequent urination; (2) urine leakage related to the feeling of urgency; (3) urine leakage related to physical activity, coughing, or sneezing; (4) small amounts of urine leakage; (5) difficulty emptying bladder; and (6) pain or discomfort in the lower abdominal or genital area. When a symptom was present, the degree to which a patient was bothered was rated on a 4-point scale (not at all, slightly, moderately, and greatly).
There was no evidence of validity, reliability, responsiveness, and MIDs estimated in post-menopausal patients with VVA-associated symptoms.
Pharmacoeconomic Review
Abbreviations
ET
estrogen therapy
GSM
genitourinary syndrome of menopause
ICER
incremental cost-effectiveness ratio
LY
life-year
MBS
most bothersome symptom
MRS
Menopause Rating Scale
NMA
network meta-analysis
QALY
quality-adjusted life-year
SOC
standard of care
VVA
vulvar and vaginal (or vulvovaginal) atrophy
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Ospemifene (Osphena) tablets |
Submitted price | Ospemifene 60 mg tablets: $1.5540 per tablet |
Indication | In post-menopausal patients for the treatment of moderate to severe dyspareunia and/or vaginal dryness, symptoms of vulvar and vaginal atrophy, a component of genitourinary syndrome of menopause |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | July 16, 2021 |
Reimbursement request | As per indication |
Sponsor | Duchesnay Inc. |
Submission history | Previously reviewed: No |
NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis |
Target population | Post-menopausal patients for the treatment of moderate to severe dyspareunia and/or vaginal dryness, symptoms of VVA, a component of GSM |
Treatment | Ospemifene plus SOC, defined as over-the-counter lubricants and moisturizers |
Comparator | A mixed basket of local ETs (Premarin and Estragyn vaginal creams, Vagifem vaginal insert, and Estring slow-release ring) plus SOC |
Perspective | Canadian publicly funded health care payer |
Outcome | QALYs |
Time horizon | 10 years |
Key data source | Efficacy data were obtained from an NMA of direct and indirect evidence for ospemifene and local ETs |
Submitted results | ICER = $27,733 per QALY gained compared with SOC (incremental costs: $213; incremental QALYs: 0.008) |
Key limitations |
|
CADTH reanalysis results |
|
EQ-5D-3L = EQ-5D 3-Levels; ET = estrogen therapy; GSM = genitourinary syndrome of menopause; ICER = incremental cost-effectiveness ratio; LY = life-year; MBS = most bothersome symptom; MRS = Menopause Rating Scale; NMA = network meta-analysis; QALY = quality-adjusted life-year; SOC = standard of care; VVA = vulvovaginal atrophy.
Conclusions
According to the CADTH Clinical Review, ospemifene is an effective therapy for reducing symptom severity in vulvar and vaginal (also called vulvovaginal) atrophy (VVA) in post-menopausal patients whose most bothersome symptoms (MBS) is dyspareunia when compared to placebo. Although the efficacy of ospemifene versus placebo in relieving vaginal dryness was demonstrated, there was inconsistency in this finding across studies. The severity of symptoms was evaluated using the VVA questionnaire, which is not a validated outcome measure and is not associated with a formal minimally important difference based on the available evidence; thus, there is uncertainty about the magnitude of benefit observed in patients treated with ospemifene in the included trials. A network meta-analysis (NMA) comparing ospemifene with local estrogen therapies (ETs) was submitted by the sponsor; however, key limitations in the comparative evidence indicate that there is uncertainty in the comparative efficacy and safety, and that there is no difference in the comparative efficacy and safety of ospemifene versus other currently available local ETs. Health-related quality of life was an important outcome that was not assessed in any of the trials.
CADTH undertook reanalyses to address limitations in the sponsor’s pharmacoeconomic submission, including assuming no difference in clinical efficacy between ospemifene and local ETs (i.e., mean combined MBS score reduction); assuming treatment discontinuation rates are the same for all treatments; and correcting the prices of 2 comparators. CADTH’s reanalyses found that ospemifene was dominated; thus, it was less effective (–0.001 quality-adjusted life-years [QALYs]) and more costly ($175) than the mixed basket of local ETs. The results are driven primarily by marginal differences in efficacy due to differential treatment discontinuation and by differences in drug acquisition costs, with ospemifene being more costly than other available local ETs. A reduction of 93% in the price for ospemifene would be required for ospemifene’s costs to be considered cost-neutral versus the lowest-cost local ET.
Based on the clinical evidence, and as seen in the CADTH reanalysis results, there is no evidence to support a price premium for ospemifene over other available local ETs used to treat symptoms of VVA, a component of genitourinary syndrome of menopause (GSM).
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process (specifically, information that pertains to the economic submission). Clinician input was not received.
One patient group, the Women’s Health Coalition Alberta, provided input. Patient input noted that there are very limited therapeutic options to treat the conditions associated with menopause, and that reimbursement restrictions limit their accessibility to patients with private health coverage and/or personal wealth. Patient input indicated that the clinical and psychological effects of untreated menopausal conditions are overlooked and commonly dismissed.
Feedback from the drug plans indicated that currently available treatment options for the indicated population include local estrogen products, such as Premarin cream, the Estring vaginal ring, and Vagifem vaginal inserts, although accessibility and funding for some relevant treatments (i.e., Vagifem and Estring) vary across jurisdictions. The drug plans identified additional costs that patients may incur when they use concomitant medication for the vasomotor symptoms (e.g., hot flashes and urinary tract infections) that are side effects of ospemifene. Drug plans raised concerns about the confidential nature of prices for Vagifem, Premarin, and Estring; due to a lack of evidence demonstrating the superiority of ospemifene versus standard of care (SOC) treatments, drug plans anticipate that consideration should be given to a pricing condition that drug plan costs for ospemifene not exceed the drug plan costs of the least costly vaginal estrogen product. Drug plans were also unclear on whether patients with non-vaginal symptoms would be eligible for ospemifene. Lastly, drug plans indicated that consideration should be given to discontinuation criteria in the event of a thromboembolic or hemorrhagic stroke.
Several of these concerns were addressed in the sponsor’s model:
The sponsor’s base-case analysis compared ospemifene to a mixed basket of ETs that reflect current clinical practice, based on drug plan input.
The sponsor captured disutilities associated with treatment-related adverse events, including urinary tract infections, headaches, and hot flashes.
CADTH was unable to address the following concerns raised in the stakeholder input:
While the sponsor captured medication adherence through treatment-specific discontinuation rates in its base-case analysis, the sponsor did not consider discontinuation criteria in the event of thromboembolic or hemorrhagic stroke, as noted by drug plans.
Economic Review
The current review is for ospemifene (Osphena) for post-menopausal patients for the treatment of moderate to severe dyspareunia and/or vaginal dryness, symptoms of VVA, a component of GSM.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis comparing ospemifene plus SOC to a mixed basket of local ETs plus SOC (i.e., over-the-counter vaginal lubricants and moisturizers) for the treatment of moderate to severe dyspareunia and/or vaginal dryness, which are symptoms of VVA, a component of GSM. The modelled population was aligned with the Health Canada–indicated population.1
The recommended dose for ospemifene is an oral administration of 60 mg once daily. At the submitted price of $1.5540 per 60 mg tablet ($139.86 per 90-count bottle or $46.62 per 30-count pack), the annual cost of ospemifene is $567. The sponsor modelled 4 comparator estrogen treatments as part of the mixed basket of local ETs, including 17-beta-estradiol (as a non-ring vaginal insert); vaginal creams, such as estrone and estrogen; and 17-beta-estradiol (as a ring insert).1 The mixed basket of comparators was modelled according to the products’ recommended dosages based on their respective product monographs, and their drug costs per treatment cycle were weighted according to the distribution of their relative use among patients, which were assumed to be 37.6%, 2.1%, 57.1%, and 3.2%, respectively. Relative use among patients was based on claims data from the US.1 Altogether, the weighted cost of the mixed basket of local therapies based on this distribution was $73.69 per cycle. The individual cost of treatment for each of the comparators considered within the mixed basket was calculated by CADTH and can be found in Appendix 1. Drug administration costs were not included because all treatments are self-administered. Full drug adherence was assumed in the sponsor’s base case for all treatments.1
The clinical outcomes of interest were QALYs and life-years (LYs). The economic analysis was conducted from the perspective of the Canadian public health care payer over a 10-year time horizon.1 Discounting (1.5% per annum) was applied to both costs and outcomes.1
Model Structure
A Markov model structure was developed to capture the long-term costs and outcomes associated with the treatment of moderate to severe VVA symptoms of dyspareunia and/or vaginal dryness. Each model cycle was 12 weeks in duration. The model consisted of 4 health states. These were classified according to the MBS scale, a self-reported measure of vaginal dryness and dyspareunia symptom severity (no symptoms [MBS = 0], mild symptoms [MBS = 1], moderate symptoms [MBS = 2], and severe symptoms [MBS = 3]), and a death state. The sponsor applied a cohort simulation technique to randomly assign patients to either ospemifene plus SOC or ETs plus SOC in either no, mild, moderate, or severe VVA symptom health states, corresponding to their mean baseline MBS scores at model entry and at 12 weeks after receiving their first active treatment based on their respective mean reduction in MBS score.
Patients could transition between health states based on the mean reduction of the MBS score of each treatment and treatment-specific discontinuation rates over time. After the first 12 weeks on treatment (first cycle), patients whose VVA symptoms improved from baseline (i.e., those who entered or remained in the no- or mild-symptoms health state after receiving their first active treatment) were considered responders and could either continue to respond to their first active treatment combined with SOC (and remain in the same health state) or discontinue their first active treatment (in the first year or subsequent year of treatment) and continue to receive only SOC for the remainder of the time horizon. Patients whose VVA symptoms did not improve significantly from baseline were considered non-responders (i.e., those who transitioned to or remained in the moderate or severe VVA symptoms health states after receiving their first active treatment); these patients transitioned to a second active treatment consisting of any available local ET (e.g., Premarin) plus continued SOC. Of those patients who did not respond to their second active treatment, 75% stopped treatment and transitioned to receiving SOC alone for the remainder of the model time horizon. However, the other 25% continued a second active treatment each cycle and remained in the moderate or severe VVA symptom health states following that second active treatment, as informed by published literature.2 Barring no treatment discontinuation, patients who continued to receive their first or second active treatment each cycle were assumed to be in a state of symptom maintenance (i.e., existing symptoms could neither improve nor worsen). A constant 12-week treatment discontinuation rate was applied each cycle for patients on any active treatment in either the no-symptoms or mild-symptoms health state. Finally, patients in any state could transition to death at any point in time.
Model Inputs
The patient cohort comprised post-menopausal patients with moderate to severe dyspareunia and/or vaginal dryness, aligned with the indicated population. On average, the cohort was 60 years old, which was reflective of patients included in the ospemifene clinical trials.1
In the economic model, treatment response was based on MBS scores for ospemifene and local ETs. The comparative efficacy of ospemifene versus local ETs was based on the relative mean reduction in MBS score and was derived from the sponsor’s submitted NMA, which comprised studies that reported a reduction of MBS scores at 12 weeks.
The sponsor derived 12-week treatment discontinuation rates from a retrospective database study using integrated medical and pharmacy claims data from the US IQVIA Real World Data claims database.1 Annual treatment discontinuation rates for local ETs were initially obtained from Faught et al. (2019)3 and converted into 12-week discontinuation rates reflecting the model cycle. A treatment discontinuation rate of 28.8% was applied to patients on ospemifene each cycle, and a treatment-weighted discontinuation rate of 39.7% was applied to patients on local ETs (49.8% for Premarin; 47.1% for Estragyn; 34.0% for Vagifem; and 17.4% for Estring).3
The health state utility values used in the model were derived from a cross-sectional study of post-menopausal patients aged 40 years to 75 years in the US and Europe that examined the association between VVA symptom severity (none, mild, moderate, severe) assessed using the Menopause Rating Scale (MRS) and quality of life assessed through the EQ-5D 3-Levels questionnaire. Utility scores were predicted through a generalized linear regression model. The sponsor also included disutility values associated with adverse events due to treatment with ospemifene based on published literature.4-6
Costs captured in the economic model included drug acquisition costs, health care resource utilization and monitoring costs associated with follow-up, and costs related to adverse events.1,7 Drug acquisition costs for ospemifene and comparator treatments were obtained from publicly listed prices on several provincial drug formularies, and drug costs were calculated based on the recommended dose per product monograph. Real-world utilization data for each local ET available and reimbursed in Canada were used to calculate a weighted average of the unit costs for local ETs. Full treatment adherence was assumed in drug cost calculations, and no administration costs were considered because all treatments are self-administered.1
Summary of Sponsor’s Economic Evaluation Results
The sponsor’s cost-effectiveness analysis was run probabilistically (i.e., 5,000 iterations for the base-case and scenario analyses). The sponsor’s deterministic results differed from the probabilistic findings with respect to differences in QALYs and costs. The deterministic analysis predicted greater incremental LYs and QALYs compared to the probabilistic analysis (9.11 LYs and 8.69 LYs, respectively) even though the deterministic incremental cost-effectiveness ratio (ICER) ($26,888 per QALY) was similar to the probabilistic ICER ($27,733 per QALY).1
Base-Case Results
In the sponsor’s base case, ospemifene was associated with an incremental cost of $213 and gains of 0.008 QALYs over a 10-year time horizon (Table 3).
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total QALYs | Incremental QALYs | ICER vs. SOC ($/QALY) |
|---|---|---|---|---|---|
SOC | 1,844 | Reference | 6.75 | Reference | Reference |
Ospemifene | 2,057 | 213 | 6.76 | 0.008 | 27,733 |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; SOC = standard of care; vs. = versus.
Source: Sponsor’s pharmacoeconomic submission.1
Sensitivity and Scenario Analysis Results
The sponsor conducted several sensitivity and scenario analyses. These included testing treatment adherence based on real-world evidence data from Faught et al. (2019)3; assuming that non-responders do not receive a second active treatment; assuming no treatment discontinuation (0%) in subsequent years; undertaking a societal perspective; varying the time horizon to 1 year and 50 years; and varying the discount rate to 0% and 3%. The sponsor’s results were most affected by the scenarios testing the shortened time horizon of 1 year (ICER = $61,392 per QALY), 0% discontinuation in subsequent years (ICER = $16,498 per QALY), and treatment adherence based on real-world evidence data from Faught et al. (2019)3 (ICER = $12,000 per QALY).
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis:
The comparative efficacy and safety of ospemifene versus local ETs is uncertain, and there is no benefit of ospemifene over local ETs in reducing patients’ MBS scores: The sponsor submitted an NMA to compare the relative efficacy and safety of ospemifene to current therapies for the treatment of VVA, including local ETs. In the economic model, the comparative efficacy of ospemifene versus local ETs was based on the relative mean reduction in MBS score, which was informed by the sponsor’s submitted NMA. In the pharmacoeconomic submission, the sponsor reported that ospemifene was associated with a ||||||||| relative reduction in MBS score at 12 weeks compared to local ETs (credible interval 95%, |||||||||||, indicating no significant difference.
Several limitations were identified with the sponsor’s submitted NMA. First, the small number of studies reporting on efficacy outcomes led to results that were highly uncertain, as evidenced by wide and overlapping credible intervals when compared to ospemifene. This made it difficult to draw conclusions around comparative efficacy for ospemifene and relevant comparators. Additionally, there was heterogeneity across pairwise comparisons that could not be explained, and a limited number of trials contributing to the evidence network for safety outcomes (e.g., headaches and urinary tract infections). Due to all these limitations, the CADTH Clinical Review affirmed that the findings of the sponsor’s NMA suggested that there was no difference between ospemifene and local ETs for the treatment of moderate to severe symptoms of VVA in post-menopausal patients.
CADTH addressed this limitation by assuming no difference in the reduction of MBS scores between ospemifene and local ETs at 12 weeks. This assumption aligns with the NMA results, which indicate no significant difference.
The claims data used to model treatment discontinuation rates are highly uncertain: In the economic model, the sponsor modelled treatment discontinuation by applying a 12-week treatment discontinuation rate in each cycle derived from a retrospective US claims database. There are several limitations with the sponsor’s approach to modelling treatment discontinuation for Canadian patients using US claims data. First, treatment discontinuation rates were derived from a claims database in the absence of direct clinical evidence. As noted in Faught et al. (2019),3 claims data are primarily derived from the coordination of health benefit payments and are not designed to answer specific research questions. For instance, the use of claims data in cost-effectiveness analyses may produce results that are not generalizable to the population of interest, due to differences in characteristics between patients in the claims database and others from published journal articles using clinical trials or cohorts.8 Second, in this model, adherence was calculated using the proportion of days covered over a fixed 12-month period, but proportion of days covered is a proxy measure of adherence and is of lower quality than direct trial results. This is evidenced by most claims databases being unable to distinguish between nonadherence and death or nonadherence and a switch to another insurance plan. Finally, the study population in Faught et al. (2019)3 consisted of nationally insured US patients who access health care within a system that is not directly comparable to the Canadian setting because of many underlying differences (e.g., public health care system, patient co-payments, drug costs, billing and administrative costs, eligibility criteria for drug coverage, and so on). This may have resulted in a higher adherence to ospemifene (or lower treatment discontinuation relative to other treatments) than might be observed in Canadian clinical practice. As such, the use of claims data from the US for an economic evaluation in the Canadian context results in significant uncertainty and may have limited external validity.
CADTH addressed this limitation by assuming treatment discontinuation rates are the same for all treatments.
The derivation of health state utility values used in the economic model is highly uncertain: In the economic model, health state utility values were derived from a cross-sectional survey of post-menopausal patients aged 40 years to 75 years in the US and Europe.9 The study assessed the relationship between VVA symptom severity (none, mild, moderate, severe) using the MRS and predicted quality of life using a general linear model with the UK value set for the EQ-5D 3-Levels questionnaire. The sponsor implicitly assumed that the MRS categories for symptom severity were directly linked to the symptom severity categories found on the MBS scale (i.e., vaginal dryness or vaginal pain associated with sexual activity), which is how the sponsor structured the economic model to correspond to each health state in the model (i.e., health state by VVA symptom severity). According to the clinical expert consulted by CADTH, neither the MRS nor the MBS is consistently used to measure VVA symptoms in Canadian clinical practice, given that these scales are less pragmatic in clinical practice; nor are they used by prescribers. The association between the 2 scales is unknown, and no justification regarding their interchangeable nature was provided by the sponsor. Further, clinical experts indicated that clinical decisions about how to address VVA symptoms are assessed on an individual basis — according to patients’ feelings or descriptions of symptoms — rather than based on results from research scales. Therefore, the impact of ospemifene on a patient’s quality of life remains uncertain.
CADTH was unable to address this limitation in its reanalysis.
The sponsor inappropriately calculated local ET costs from highly uncertain claims data: In the economic model, the sponsor compared the costs of ospemifene to local ETs by calculating an aggregated (or weighted average) drug cost for local ETs based on individual treatments. There are 2 key issues with the sponsor’s approach to calculating these costs. First, a direct head-to-head comparison of costs should have been facilitated between ospemifene and each individual local estrogen product rather than comparing ospemifene with a mixed basket of treatments. This would ensure generalizability and transparency, given that the relative use of each product can differ between jurisdictions. Second, there is uncertainty around the market share distribution assumed for each estrogen product. As such, aggregating costs makes it difficult to assess the cost-effectiveness of ospemifene versus relevant comparators and allow for an appropriate interpretation of the ICER.10
In the scenario analysis section of this report, given the equal efficacy of all treatments, CADTH compared the annual cost of ospemifene to the cost of each individual comparator.
Additional limitations were identified, but were not considered to be key.
The list prices of conjugated estrogen creams and the estradiol ring vary across jurisdictions: The sponsor’s submitted cost-utility analysis based the unit price of conjugated estrogen cream ($0.84 per 0.625 mg) and an estradiol ring ($89.21 per 2 mg ring) on the Ontario Drug Benefit Formulary.11 However, the costs of these comparators are lower in other jurisdictions, where the lowest listed prices are $0.76 per 0.625 mg for conjugated estrogen cream11 and $74.67 per 2 mg for an estradiol ring.11
CADTH addressed this limitation by revising the costs for conjugated estrogen cream and the estradiol ring based on the lowest publicly available list prices.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
A 10-year time horizon was applied based on the rationale that although menopause symptoms last for 1 to 2 years after onset in most patients, they may continue for up to 10 years. | Inappropriate. The time horizon does not reflect that VVA is a chronic condition. The clinical experts consulted by CADTH noted that approximately 10% to 20% of patients on treatment will likely continue treatment beyond 10 years. |
The baseline distribution of the economic model’s patients into each health state (according to MBS score), which reflects the severity of VVA symptoms, was generated by a microsimulation of a hypothetical patient cohort that randomly assigned patients to each health state. | Appropriate. According to the clinical experts consulted by CADTH, most patients would have moderate to severe symptoms, and the target population would be approximately equally split between moderate and severe symptoms. |
For patients who initially received local ETs plus SOC, the sponsor assumed that the mean MBS score reduction attributed to the second active treatment is equivalent to that of the first active treatment, despite failing the first active treatment. As such, patients’ symptoms could continue to improve on local ETs plus SOC as second-line therapy (i.e., a proportion of patients either no longer experienced symptoms or had mild symptoms). | Uncertain. The clinical experts consulted by CADTH noted that patients whose symptoms are difficult to treat with their first active treatment will likely continue to be difficult to treat for their second active treatment. Because symptoms are treated on an individual basis, it is difficult to tell whether patients would have no symptoms (i.e., an improvement in symptoms) after 2 lines of hormonal therapy. |
VVA symptoms are not likely to affect the mortality of patients. Patients could die at any point in time, based on the mortality rate of the general Canadian population. | Appropriate, according to the clinical experts consulted by CADTH. |
Rates of AEs (i.e., urinary tract infections, hot flashes, and headaches) for ospemifene, local ETs, and SOC alone were derived from the sponsor’s submitted ITC. | Uncertain. While the AEs considered in the economic model were appropriate, according to the CADTH Clinical Review, the methods used to calculate the rates of AEs in each group based on the NMA were sparsely documented. CADTH was unable to validate any of the sponsor's assumptions related to AEs; however, the impact of AEs on model results is minimal. |
AE = adverse event; ET = estrogen therapy; ITC = indirect treatment comparison; NMA = network meta-analysis; SOC = standard of care; VVA = vulvovaginal atrophy.
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
CADTH undertook the reanalyses outlined in Table 5 to address, where possible, the limitations within the sponsor’s submitted economic model. The CADTH base case was derived by making changes in model parameter values and assumptions in consultation with clinical experts.
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | None | None |
Changes to derive the CADTH base case | ||
1. Treatment discontinuation rates | Differed by each comparator treatment | Assumed that the rate of treatment discontinuation for ospemifene was the same for all comparators (i.e., a constant rate) |
2. Comparative efficacy of ospemifene to local estrogen therapies | ||||||||||||| | 0.00 |
3. Price of conjugated estrogen cream and the estradiol ring | Conjugated estrogen cream: $0.8423 17-beta-estradiol: $89.2100 | Conjugated estrogen cream: $0.7510 17-beta-estradiol: $74.6655 |
CADTH base case | Reanalyses 1 + 2 + 3 | |
The results of these deterministic stepwise analyses can be found in Table 6. The probabilistic analysis of the CADTH base case found that ospemifene was dominated (i.e., was more costly and less effective) by local ETs. The results were driven primarily by CADTH’s assumption of equal treatment discontinuation rates for all treatments, indicating that the limitations associated with the US claims database used to derive adherence remain a key model driver.
Table 6: Summary of the Stepped Analysis of the CADTH Reanalysis Results
Stepped analysisa | Drug | Total costs ($) | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
Sponsor’s base case: probabilistic | Local estrogen therapies | 1,844 | 6.747 | Reference |
Ospemifene | 2,057 | 6.755 | 27,733 | |
Sponsor’s base case: deterministic | Local estrogen therapies | 1,896 | 7.075 | Reference |
Ospemifene | 2,105 | 7.083 | 26,888 | |
CADTH reanalysis 1 | Local estrogen therapies | 1,946 | 7.084 | Reference |
Ospemifene | 2,116 | 7.082 | Dominated | |
CADTH reanalysis 2 | Local estrogen therapies | 1,897 | 7.075 | Reference |
Ospemifene | 2,105 | 7.083 | 25,811 | |
CADTH reanalysis 3 | Local estrogen therapies | 1,892 | 7.075 | Reference |
Ospemifene | 2,105 | 7.083 | 27,306 | |
CADTH base case: deterministic (reanalyses 1 + 2 + 3) | Local estrogen therapies | 1,941 | 7.084 | Reference |
Ospemifene | 2,115 | 7.082 | Dominated | |
CADTH base case: probabilistic (reanalyses 1 + 2 + 3) | Local estrogen therapies | 1,861 | 6.746 | Reference |
Ospemifene | 2,036 | 6.745 | Dominated |
ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year.
aDeterministic results are presented for all stepwise reanalyses.
Scenario Analysis Results
CADTH undertook the following additional scenario analyses of the CADTH base case to explore the uncertainty associated with the treatment discontinuation rates in the model (scenarios 1 and 2, Table 11):
setting the treatment discontinuation rate for all non-ring comparators as equal to Vagifem (34%), with a 10% improvement for ospemifene (30.6%)
setting the treatment discontinuation rate for all non-ring comparators as equal to Vagifem (34%), with a 20% improvement for ospemifene.
In both scenarios, ospemifene was assumed to have an advantage over other available alternatives (i.e., non-ring comparators); however, any such advantage is highly uncertain due to the fact that there is neither Canadian nor trial evidence to inform these estimates. Thus, they are exploratory. The results of these exploratory scenario analyses indicated that ospemifene was associated with incremental costs and QALYs compared with local ETs, resulting in an ICER of $167,930 per QALY in scenario 1 and an ICER of $40,427 per QALY in scenario 2.
Price Reduction Analyses
Given that the CADTH reanalysis found no difference in comparative efficacy between ospemifene and local ETs, CADTH compared the annual cost of all treatments using their list prices. All local ETs ranged in annual cost based on their recommended dosage regimen. Ranked in ascending cost of local ET, based on their lowest dosage, the costs were $39 to $411 for estrogen (Premarin cream); $103 to $827 for estrone (Estragyn); $105 to $299 for 17-beta-estradiol (Estring); and $448 to $491 for 17-beta-estradiol (Vagifem). Ospemifene’s annual cost of $567 was higher than all comparators except the highest dosage of estrone (Estragyn).
Based on the CADTH reanalysis, a price reduction analysis was conducted to determine the price point at which ospemifene would be equal to the price of each local ET. A price reduction of 93% would be required for ospemifene’s drug costs to be cost-neutral when compared to the least costly ET (i.e., Premarin at its lowest dose). However, the price reduction varies from 13% to 93% based on the local ET and its dosage (Table 7).
Table 7: CADTH Price Reduction Analyses
Scenario | Sponsor’s submitted price ($) | Reduction needed (%) | Reduced price ($) | Annual savingsa ($) |
|---|---|---|---|---|
Price reduction required to equal least costly estrogen therapy (Premarin cream at lowest dose) | 1.55 | 93.1 | 0.11 | 528 |
Price reduction required to equal estrogen (Premarin cream) | 1.55 | 27.5 to 93.1 | 0.11 to 1.13 | 156 to 528 |
Price reduction required to equal estrone (Estragyn) | 1.55 | 0 to 81.7 | 0.84 to 1.55 | 0 to 464 |
Price reduction required to equal 17-beta-estradiol (Estring) | 1.55 | 47.3 to 73.6 | 0.41 to 0.82 | 269 to 418 |
Price reduction required to equal estrogen 17-beta-estradiol (Vagifem) | 1.55 | 13.2 to 20.8 | 1.23 to 1.35 | 75 to 118 |
Note: Reanalyses are based on CADTH calculated prices of the comparator treatments.
aSavings from the sponsor list price per patient per year.
Issue for Consideration
Additional comparators will soon be available: Two additional comparators, the vaginal insert Imvexxy (17-beta-estradiol) and the vaginal ovule Intrarosa (prasterone), have been recently approved by Health Canada for dyspareunia and VVA symptoms, respectively. Both products were recently reviewed by CADTH. The final recommendation of the CADTH Canadian Drug Expert Committee for Imvexxy stated that Imvexxy should be reimbursed for the treatment of post-menopausal moderate to severe dyspareunia in a similar manner to currently funded vaginal estrogen products12 — specifically, that the cost could be negotiated to provide savings relative to the least costly local ET reimbursed for the indication. Neither of these products is currently funded by any Canadian public drug plan. Nor are they currently used in Canadian clinical practice for the treatment of VVA symptoms.
Overall Conclusions
According to the CADTH Clinical Review, compared to placebo, ospemifene is an effective therapy for reducing symptom severity of VVA in post-menopausal patients whose MBS is dyspareunia. Although the efficacy of ospemifene versus placebo in relieving vaginal dryness was demonstrated, there was inconsistency in this finding across studies. The severity of symptoms was evaluated using the VVA questionnaire, which is not a validated outcome measure and is not associated with a formal minimally important difference based on the available evidence; thus, there is uncertainty about the magnitude of benefit observed in patients treated with ospemifene in the included trials. An NMA comparing ospemifene with local ETs was submitted by the sponsor; however, key limitations in the comparative evidence indicate that there is uncertainty with the comparative efficacy and safety, and that there is no difference in the comparative efficacy and safety of ospemifene versus other currently available local ETs. Health-related quality of life was an important outcome that was not assessed in any of the trials.
CADTH undertook reanalyses to address limitations in the sponsor’s pharmacoeconomic submission, including assuming no difference in clinical efficacy between ospemifene and local ETs (i.e., mean combined MBS score reduction); assuming treatment discontinuation rates are the same for all treatments; and correcting the prices of 2 comparators. CADTH’s reanalyses found that ospemifene was dominated; thus, it was less effective (–0.001 QALYs) and more costly ($175) than the mixed basket of local ETs. The results are driven primarily by marginal differences in efficacy due to differential treatment discontinuation and by differences in drug acquisition costs, with ospemifene being more costly than other available local ETs. A reduction of 93% in the price of ospemifene would be required for its cost to be considered cost-neutral versus the lowest-cost local ET.
Based on the clinical evidence, and as seen in the CADTH reanalysis results, there is no evidence to support a price premium for ospemifene over other available local ETs used to treat symptoms of VVA.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Osphena (ospemifene), oral tablets, 60 mg [internal sponsor's package]. Blainville (QC): Duchesnay Inc.; 2021 Oct 20.
2.Dymond A, Holmes H, McMaster J, et al. Economic evaluation of Senshio(®) (Ospemifene) for the treatment of vulvovaginal atrophy in Scotland. Appl Health Econ Health Policy. 2021;19(1):123-132. PubMed
3.Faught BM, Soulban G, Yeaw J, et al. Ospemifene versus local estrogen: adherence and costs in postmenopausal dyspareunia. J. 2019;8(13):1111-1123.
4.Shingler S, Fordham B, Evans M, et al. Utilities for treatment-related adverse events in type 2 diabetes. J Med Econ. 2015;18(1):45-55. PubMed
5.Hux M, Ng C, Ortega GL, Ferrazzi S, Goeree R. Utility values for pre-menopausal women suffering from symptomatic uterine fibroids. Expert Rev Pharmacoecon Outcomes Res. 2015;15(1):181-189. PubMed
6.Sullivan PW, Slejko JF, Sculpher MJ, Ghushchyan V. Catalogue of EQ-5D scores for the United Kingdom. Med Decis Making. 2011;31(6):800-804. PubMed
7.Ministry of Health and Long Term Care. Health Data Branch Web Portal - OCC - Cost Analysis Tool. 2021: https://hsim.health.gov.on.ca/hdbportal/. Accessed 2021 Aug 26.
8.Shih Y-CT, Liu L. Use of claims data for cost and cost-effectiveness research. Semin Radiat Oncol. 2019;29(4):348-353. PubMed
9.DiBonaventura M, Luo X, Moffatt M, Bushmakin AG, Kumar M, Bobula J. The association between vulvovaginal atrophy symptoms and quality of life among postmenopausal women in the United States and Western Europe. J Womens Health. 2015;24(9):713-722. PubMed
10.Drummond M, Sculpher M. Common methodological flaws in economic evaluations. Med Care. 2005;43(7 Suppl):5-14. PubMed
11.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2021; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2021 Dec 1.
12.CADTH Canadian Drug Expert Committee (CDEC) final recommendation: estradiol (Imvexxy). Ottawa (ON): CADTH; 2022: https://www.cadth.ca/sites/default/files/DRR/2022/SR0694%20Imvexxy%20-%20CADTH%20Final%20Rec%20KH.pdf. Accessed 2022 Feb 28.
13.CADTH Canadian Drug Expert Committee (CDEC) reimbursement review: estradiol (Imvexxy). Ottawa (ON): CADTH; 2021: https://www.cadth.ca/estradiol. Accessed 2022 Feb 28.
14.Nova Scotia Formulary. Formulary. Nova Scotia Department of Health; 2022: https://novascotia.ca/dhw/pharmacare/documents/formulary.pdf. Accessed 2021 Dec 1.
15.Government of Saskatchewan. Saskatchewan Drug Formulary. 2021: https://formulary.drugplan.ehealthsask.ca/SearchFormulary/BG/455680. Accessed 2021 Dec 1.
16.Government of Alberta. Interactive Drug Benefit List (iDBL). 2021: https://www.ab.bluecross.ca/dbl/idbl_main1.php. Accessed 2021 Dec 1.
17.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Osphena (ospemifene), oral tablets, 60 mg [internal sponsor's package]. Blainville (QC): Duchesney Inc.; 2021 Oct 20.
18.Naumova I, Castelo-Branco C. Current treatment options for postmenopausal vaginal atrophy. International journal of women's health. 2018;10:387-395. PubMed
19.Palacios S, Nappi RE, Bruyniks N, Particco M, Panay N. The European Vulvovaginal Epidemiological Survey (EVES): prevalence, symptoms and impact of vulvovaginal atrophy of menopause. Climacteric. 2018;21(3):286-291. PubMed
20.Cardozo L, Bachmann G, McClish D, Fonda D, Birgerson L. Meta-analysis of estrogen therapy in the management of urogenital atrophy in postmenopausal women: second report of the Hormones and Urogenital Therapy Committee. Obstet Gynecol. 1998;92(4 Pt 2):722-727. PubMed
Appendix 1: Cost Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical experts and public drug plans. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 8: CADTH Cost Comparison Table for the Treatment of Dyspareunia and/or Vaginal Dryness (Symptoms of Vulvar and Vaginal Atrophy)
Treatment | Strength / concentration | Dosage form | Price ($) | Recommended dosage regimen | Average daily cost ($) | Annual cost |
|---|---|---|---|---|---|---|
Ospemifene | 60 mg | Oral tablet | 1.5540a | 60 mg tab once daily | 1.55 | 567 |
Insert comparator | ||||||
17-beta-estradiol (Vagifem) | 10 mcg | Vaginal tablet insert | 4.3089 | Initial dose: 1 vaginal insert daily for 2 weeks Maintenance dose: 1 vaginal insert twice a week with a 3- or 4-day interval between doses | First year: 1.35 Subsequent years: 1.22 | First year: 491 Subsequent years: 448 |
Cream comparators | ||||||
Estrone (Estragyn) | 0.1% w/w | gram | 0.7576c | 0.5 to 4 grams per day taken intravaginally, adjusted to the lowest amount that controls symptoms. Administration should be cyclic (e.g., 3 weeks on one week off). | 0.28 to 2.27 | 103 to 827 |
Estrogen (Premarin cream) | 0.625 mg/g | gram | 0.7510d | Ranging from 0.5 grams twice per week, to 2 grams daily for 21 days and then off for 7 days.e | 0.11 to 1.13 | 39 to 411 |
Ring comparator | ||||||
17-beta-estradiol (Estring) | 2 mg | Vaginal ring | 74.6655f | One Estring is to remain in place continuously for 3 months, after which it is to be removed and, if continuation of therapy is deemed appropriate, replaced by a new ring. The need to continue treatment should be assessed at 3- or 6-month intervals. | 0.41 to 0.82 | 150 to 299 |
Note: All prices are from the Ontario Drug Benefit Formulary (accessed November 18, 2021),11 unless otherwise indicated, and do not include dispensing fees. Reanalyses are based on publicly available prices of the comparator treatments. Annual period assumes 365 days or 52 weeks for all comparators.
aSponsor’s submission for Osphena.1
bSponsor’s submission for Imvexxy.13
cNova Scotia Drug Formulary,14 accessed December 1, 2021.
dSaskatchewan Drug Formulary,15 accessed December 1, 2021.
eAs per product monograph, CADTH’s calculation for Estrogen (Premarin cream) calculation is based on the low dose while the sponsor’s submitted model is based on a maximum dose.
fAlberta Drug Formulary,16 accessed December 1, 2021.
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | Yes | No comment. |
Model has been adequately programmed and has sufficient face validity | Yes | No comment. |
Model structure is adequate for decision problem | Yes | No comment. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | No | Incorrect pricing for Premarin and Estring. The MBS score reduction from the NMA was incorrect and did not align with the sponsor’s ITC technical report. See Key Limitations for the CADTH critical appraisal of the sponsor’s economic evaluation. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | No comment. |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
![Diagram outlining the Markov model structure that captures the long-term costs and outcomes associated with the treatment of moderate to severe VVA symptoms of dyspareunia and/or vaginal dryness. The model consisted of 4 health states. These were classified according to the MBS scale, a self-reported measure of vaginal dryness and dyspareunia symptom severity (no symptoms [MBS = 0], mild symptoms [MBS = 1], moderate symptoms [MBS = 2], and severe symptoms [MBS = 3]), and a death state. Patients could transition between health states based on the mean reduction of the MBS score of each treatment and treatment-specific discontinuation rates over time.](https://canjhealthtechnol.ca/index.php/cjht/article/download/SR0709r/version/417/846/2811/SR0709PE-fig01.png)
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Table 10: Disaggregated Summary of CADTH’s Economic Evaluation Results
Parameter | Ospemifene | Local estrogen therapies | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total | 8.68 | 8.68 | 0 |
Discounted QALYs | |||
Total | 6.745 | 6.746 | |
QALYs generated within trial period (SD) | 0.19 | 0.19 | 0 |
QALYs generated after trial period (SD) | 6.60 | 6.60 | 0 |
QALYs in No Symptom - 1st Active Treatment (SD) | 0.20 | 0.20 | 0 |
QALYs in Mild Symptoms - 1st Active Treatment (SD) | 0.31 | 0.31 | 0 |
QALYs in Moderate Symptoms - 1st Active Treatment (SD) | 0.03 | 0.03 | 0 |
QALYs in Severe Symptoms - 1st Active Treatment (SD) | 0.01 | 0.01 | 0 |
QALYs in No Symptom - 2nd Active Treatment (SD) | 0.04 | 0.04 | 0 |
QALYs in Mild Symptoms - 2nd Active Treatment (SD) | 0.07 | 0.07 | 0 |
QALYs in Moderate Symptoms - 2nd Active Treatment (SD) | 0.01 | 0.01 | 0 |
QALYs in Severe Symptoms - 2nd Active Treatment (SD) | 0.00 | 0.00 | 0 |
QALYs in No Symptom - SoC (SD) | 0.00 | 0.00 | 0 |
QALYs in Mild Symptoms - SoC (SD) | 0.16 | 0.16 | 0 |
QALYs in Moderate Symptoms - SoC (SD) | 3.31 | 3.31 | 0 |
QALYs in Severe Symptoms - SoC (SD) | 2.63 | 2.64 | 0 |
Disutility Associated with Treatment-Related AEs (SD) | 0.040 | 0.039 | 0.001 |
Discounted costs ($) (SD) | |||
Total | 2,036 | 1,861 | 175 |
Drug Cost | 431 | 260 | 171 |
1st Active Treatment | 379 | 207 | 171 |
2nd Active Treatment | 52 | 52 | 0 |
SoC Treatment | 0 | 0 | 0 |
Health Care Cost | 0 | 0 | 0 |
Follow-Up | 1,577 | 1,577 | 0 |
Adverse Events | 29 | 24 | 5 |
ICER ($/QALY) | Dominated | ||
ICER = incremental cost-effectiveness ratio; LY= life-year; QALY = quality-adjusted life-year.
Scenario Analyses
Table 11: Scenario Analyses for Osphena Versus Local ETs
Scenario analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|---|
Scenario 1: Treatment discontinuation rate for Vagifem applied to all comparators except Estring, and a 10% benefit for Ospemifene. | Local ETs | 1,820 | 6.746 | Reference |
Ospemifene | 1,996 | 6.747 | 167,930 | |
Scenario 2: Treatment discontinuation rate for Vagifem applied to all comparators except Estring, and a 20% benefit for Ospemifene. | Local ETs | 1,863 | 6.777 | Reference |
Ospemifene | 2,073 | 6.782 | 40,427 |
ET = estrogen therapy.
Appendix 5: Submitted Budget Impact Analysis and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 12: Summary of Key Take-Aways
Key Take-aways of the BIA |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
The sponsor assessed the budget impact of the introduction of ospemifene for post-menopausal patients experiencing moderate to severe dyspareunia and/or vaginal dryness, from the perspective of the public drug plans in Canada (excluding Quebec), over a 3-year time horizon. The sponsor included drug acquisition costs, mark-ups, dispensing fees, and co-payments in their base case.
In the reference scenario, the sponsor assumed that patients received various ETs including conjugated estrogen vaginal cream, an estradiol vaginal insert, a 17B-estradiol vaginal ring, and an estrone vaginal cream. In the new drug scenario, ospemifene was assumed to capture market share from comparator treatments proportionally. Drug costs of ospemifene and individual treatments were calculated assuming full (100%) adherence, and according to the recommended dosages from their respective product monographs.
The sponsor used an epidemiological approach to identify the total population eligible for treatment with ospemifene. The sponsor estimated the proportion of post-menopausal patients between ages 45 and 60 within the pan-Canadian population, based on Statistics Canada data.17 The number of post-menopausal patients experiencing VVA was estimated according to a study of post-menopausal patients with VVA symptoms in the US and Western Europe by DiBonaventura (2015).9 Among these patients, the proportion of those with moderate to severe dyspareunia and/or vaginal dryness was estimated based on an international study of post-menopausal patients with VVA symptoms, between ages 40 and 75. The sponsor further estimated the proportion of those who sought medical help based on a European study by Naumova (2018) that described current treatment options for post-menopausal vaginal atrophy.18
The sponsor’s BIA also included the following key assumptions:
An annual treatment discontinuation rate was considered for each individual treatment, based on a retrospective study assessing medical and pharmacy claims data in the US.3 Treatment discontinuation rates were applied for only the first year of treatment.
Key inputs to the BIA are documented in Table 13.
Table 13: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as Year 1 / Year 2 / Year 3 if appropriate) |
|---|---|
Target population | |
Proportion of post-menopausal patients with VVA symptoms | 44.3% |
Proportion of post-menopausal patients with VVA symptoms, who have moderate to severe symptoms | 55.0% |
Proportion of patients who seek medical help | 25.0% |
Proportion of patients included <65 years old | 81.5% |
Percentage of patients covered by a publicly funded drug plan | 59.6% |
Proportion of patients with VVA ≥65 years old | 18.5% |
Percentage of patients covered by a publicly funded drug plan | 93.6% |
Number of patients eligible for drug under review | 195,472 / 196,395 / 197,247 |
Market uptake (3 years) | |
Uptake (reference scenario) | |
Conjugated estrogen vaginal cream | ||||||||||| |
Estradiol vaginal insert | ||||||||||| |
17-beta-estradiol vaginal ring | ||||||||||| |
Estrone vaginal cream | ||||||||||| |
Uptake (new drug scenario) | |
Ospemifene | ||||||||||| |
Conjugated estrogen vaginal cream | ||||||||||| |
Estradiol vaginal insert | ||||||||||| |
17-beta-estradiol vaginal ring | ||||||||||| |
Estrone vaginal cream | ||||||||||| |
Cost of treatment (per patient) | |
Cost of treatment per month | |
Ospemifene | $47.27 |
Conjugated estrogen vaginal cream | $9.61 |
Estradiol vaginal insert | $37.45 |
17-beta-estradiol vaginal ring | $30.15 |
Estrone vaginal cream | $34.57 |
Summary of the Sponsor’s Budget Impact Analysis Results
Results of the sponsor’s submitted base case suggested that the introduction of ospemifene for the treatment of post-menopausal patients, between the ages of 45 and 69, experiencing moderate to severe dyspareunia and/or vaginal dryness, would result in an incremental budget of $649,020 in Year 1, $1,269,622 in Year 2, and $1,950,318 in Year 3, for a total incremental budget impact of $3,868,960 over the 3-year time horizon.
CADTH Appraisal of the Sponsor’s Budget Impact Analysis
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Annual treatment discontinuation rates were based claims data and likely underestimates drug costs associated with ospemifene. In the submitted BIA, the sponsor calculated annual treatment discontinuation rates for ospemifene and all comparator treatments based on a retrospective claims analysis by Faught et al. 2019.3 It found ospemifene had the lowest rate of discontinuation among non-ring treatments (77.1% for ospemifene versus 95.0%, 83.6%, 93.7%, and 56.4%, for conjugated estrogen cream, estradiol vaginal insert, estradiol cream, and estradiol ring, respectively). Several methodological limitations were identified regarding the use of claims data from the US to model treatment discontinuation, as noted in CADTH’s Appraisal of the Sponsor’s Economic Evaluation. Lower treatment discontinuation rates for ospemifene likely underestimates total drug costs of ospemifene compared to all other treatments, and thus, total incremental costs may be lower than estimated.
CADTH addressed this limitation by applying the annual treatment discontinuation rate for ospemifene (77.1%) to all comparator treatments, thus aligning with the CADTH pharmacoeconomic model’s base case.
The estimated number of patients eligible for treatment with ospemifene is uncertain: The sponsor undertook an epidemiological approach to estimate the size of the population eligible for ospemifene. This required deriving inputs from the published literature and applying several assumptions to derive estimates for the target population in a multi-step approach. The clinical experts consulted by CADTH indicated that the estimated population size appeared to be reasonable, however there may be some uncertainty in the final estimated eligible population due to some of the published literature informing population size estimates were outdated. Specifically, the sponsor cited a study by Naumova et al. 2018 which reported that 75% of patients with clinical manifestations of VVA do not seek help from specialists, and thus, assumed that only 25% of post-menopausal patients with moderate to severe VVA symptoms would seek medical help. The CADTH pharmacoeconomic review team reviewed this evidence and noted that the original source for this estimate traced back to a 1998 meta-analysis.18-20 Given this, there is uncertainty around this estimate as women’s health has gained more awareness in recent decades. Additionally, the sponsor assumed that 93.6% of Canadian patients 65 years and older, and 59.5% of patients less than 65 years of age would receive drug coverage by a publicly funded drug plan and this estimate was used as part of deriving the final population size; however, a more appropriate approach is for the population size to be strictly based on the number of patients eligible, rather than be influenced by any potential eligibility criteria for drug coverage.
CADTH addressed this limitation by assuming 100% of patients would receive drug coverage across jurisdictions. In a scenario analysis, CADTH also explored the impact of a higher proportion of patients seeking medical help, by assuming a value of 50%.
The anticipated market share of ospemifene is uncertain: The sponsor assumed that with the introduction of ospemifene in the new drug scenario, ospemifene will have a market share of approximately 4%, 5% and 6% in years 1 to 3. The clinical experts consulted by CADTH indicated that these estimates are uncertain and the anticipated market share of ospemifene in the new drug scenario is difficult to determine, as there are several factors influencing a prescriber’s practice.
CADTH was unable to address this limitation.
Additional limitations were identified but were not considered to be key limitations. These limitations include:
List prices of conjugated estrogen cream and an estradiol ring varies across jurisdictions: The sponsor’s submitted BIA is based on the unit price of conjugated estrogen cream ($0.84 per 0.625 mg) and the estradiol ring ($89.21 per 2 mg ring) on the Ontario Drug Benefit Formulary.11 The cost of these comparators varies across jurisdictions, where the lowest list price for conjugated estrogen cream is $0.76 per 0.625 mg11 and for an estradiol ring is $74.67 per 2 mg ring.11 According to CADTH guidelines, jurisdiction-specific inputs (i.e., costs) should be applied for a pan-Canadian analysis. However, the sponsor did not apply jurisdiction-specific costs and instead, equated the cost of these comparators to their publicly listed price in Ontario. CADTH revised these costs to reflect the lowest dispensable pricing applied in the CADTH pharmacoeconomic analysis.
CADTH addressed this limitation by revising the costs for conjugated estrogen cream and the estradiol ring based on the lowest publicly available list prices.
CADTH Reanalyses of the Budget Impact Analysis
Table 14: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Correctionsa to sponsor’s base case | ||
None | None | None |
Changes to derive the CADTH base case | ||
1. Annual treatment discontinuation rates | Ospemifene: 77.1% Conjugated estrogen cream: 95.0%, Estradiol vaginal insert: 83.6%, Estradiol cream: 93.7% Estradiol ring: 56.4% | Ospemifene: 77.1% Conjugated estrogen cream: 77.1% Estradiol vaginal insert: 77.1% Estradiol cream: 77.1% Estradiol ring: 77.1% |
2. Proportion of patients assumed to receive drug coverage | Less than 65 years: 59.6% 65 years and over: 93.6% | Less than 65 years: 100% 65 years and over: 100% |
3. Lowest publicly available list prices. | Conjugated estrogen cream: $0.8423 Estradiol cream: $89.2100 | Conjugated estrogen cream: $0.7510 Estradiol cream: $74.6655 |
CADTH base case | Reanalysis 1 + 2 + 3 | |
aCorrections are minor errors (e.g., transcription errors between report) that are not identified as limitations.
Applying the changes in Table 14 resulted in a decrease in the estimated budget impact under the drug plan perspective to $2,822,184 over 3 years. The results of the CADTH stepwise reanalyses are presented in summary format in Table 15 and a more detailed breakdown is presented in Table 16.
Table 15: Summary of the CADTH Reanalyses of the Budget Impact Analysis
Stepped analysis | Three-year total |
|---|---|
Submitted base case | $3,868,960 |
CADTH reanalysis 1 | $2,772,242 |
CADTH reanalysis 2 | $5,961,972 |
CADTH reanalysis 3 | $3,891,515 |
CADTH base case | $4,294,925 |
BIA = budget impact analysis.
Table 16: Detailed Breakdown of the CADTH Reanalyses of the Budget Impact Analysis
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $10,069,320 | $10,140,225 | $18,485,700 | $25,437,449 | $54,063,375 |
New drug | $10,069,320 | $10,789,245 | $19,755,323 | $27,387,768 | $57,932,335 | |
Budget impact | $0 | $649,020 | $1,269,622 | $1,950,318 | $3,868,960 | |
CADTH base case | Reference | $22,834,980 | $22,967,151 | $40,801,774 | $54,668,436 | $118,437,362 |
New drug | $22,834,980 | $23,687,703 | $42,230,299 | $56,889,546 | $122,807,547 | |
Budget impact | $0 | $720,551 | $1,428,525 | $2,221,110 | $4,370,185 |
BIA = budget impact analysis.
CADTH conducted one additional scenario analysis from the drug plan perspective (Table 17). Specifically, CADTH applied an alternate assumption that approximately 50% of patients will seek medical help, instead of the sponsor’s estimated 25%.
Table 17: CADTH Scenario Analyses
Stepped analysis | Budget impact | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
CADTH scenario analysis 1: 50% of patients seeking medical help | Reference | $29,616,971 | $29,819,863 | $53,010,460 | $71,078,310 | $153,908,633 |
New drug | $29,616,971 | $30,745,865 | $54,847,438 | $73,936,629 | $159,529,932 | |
Budget impact | $0 | $926,002 | $1,836,978 | $2,858,319 | $5,621,299 |
Stakeholder Input
Patient Group Input
Women’s Health Coalition of Alberta Society
About the Women’s Health Coalition of Alberta Society
The Women’s Health Coalition of Alberta Society (WHC) is committed to creating a movement that empowers people to speak openly, learn and engage with purpose to address barriers, gaps, policies and unconscious- bias, that impact women’s menstrual, reproductive, and sexual health. They are enabling advocacy, awareness and education in gynecological, uro-gynecological, menstrual, uterine, and reproductive health, through all the ages and stages of a woman’s life.
The WHC is highly committed to ensure that women have access to the right treatment and support at the right time, for improved health outcomes. Uro-gynecological health is not well understood, is underserved in the health system and offers very limited therapeutic options for conditions associated with menopause (peri and post). In addition, when reimbursement is not available, it can result in preferred treatments only being accessible to women with private health coverage and/or personal wealth.
The clinical and psychological effects of untreated menopausal conditions are overlooked and commonly dismissed. Any improvement in therapeutic choice/access, in the treatment of menopausal conditions, will benefit women physically and wholistically. Recommendation of new treatments will not only improve treatment options, choice, and access for women facing menopausal conditions, it may raise clinician awareness of the importance of treating menopausal conditions.
We welcome the opportunity to address this matter with you in greater detail. Should you wish to speak with us, please e-mail us at ||||||||||||||||||||||||||||||.
The WHC is committed to creating a movement that empowers people to speak openly, learn and engage with purpose to address barriers, gaps, and policies that impact women’s menstrual, reproductive, and sexual health. The WHC is raising awareness, conducting research and advocating to address gender bias in health delivery. They are a network of women who have faced health challenges, people who care about, and for women, health care professionals, and business leaders motivated to improving women’s health.
We are enabling advocacy, awareness and education in gynecological, uro-gynecological, menstrual, uterine, and reproductive health, through all the ages and stages of a woman’s life.
We are a network of women who have faced health challenges, people who care about, and for women, health care professionals, and business leaders motivated to improving women’s health.
Information Gathering
The WHC collects patient experiences through formally submitted stories, participation in webinars, and by conducting patient collaboration forums. Challenges faced by women in dealing with menopause (peri and post) is the most common testimony shared with the WHC and the most commonly requested topic for forum discussions.
Clinical information for menopause, hormone replacement therapy, and incontinence is provided to the WHC and members by qualified clinicians including gynecologists, nurse practitioners, physical therapists specializing in pelvic floor health.
Disease Experience
In member/ patient reported experiences, clinical support is most often provided by a family physician whereby, menopause and uro-gynecological health is not well understood, and referrals are discouraged.
Women are often embarrassed and are not comfortable discussing their symptoms if not effectively prompted by the practitioner to do so.
Symptoms are treated as a normal process of aging and monitoring is the most common response.
Experiences With Currently Available Treatments
The WHC is highly committed to ensuring that women have access to the right treatment and support at the right time, for improved health outcomes.
Members shared experiences of the benefits of early treatment of vaginal dryness and urinary incontinence, including comfortable intercourse, and improved urinary control.
Peer support dialogue has provided some reference to tolerance of treatments and encouragement to persevere to find the best option ‘for you’. Choice is paramount to addressing individual needs.
Improved Outcomes
Early treatment of urinary incontinence may postpone or eliminate the need for more invasive/disruptive treatments.
Comfortable sexual relations is important to physical, psychological and family health and wellbeing.
Vaginal itching, dryness and/or discharge, is very uncomfortable and distracting to deal with for a prolonged period.
Urinary incontinence and persistent vaginal discharge requires feminine hygiene and leak protection products that carry significant financial burden.
Anything Else?
Recommendation of ospemifene/Osphena will improve treatment options, choice, and access for women facing menopausal conditions. Increasing options in the treatment of menopause related conditions may also raise clinician awareness of the importance of treating menopausal conditions.
The clinical and psychological effects of untreated menopausal conditions are overlooked and commonly dismissed.
Menopause, vaginal and urinary health in older women is underserved and predisposed to perfunctory treatment options.
When reimbursement is not available, it can result in preferred treatments only being accessible to women with private health coverage and/or personal wealth.
Patient Group Conflict of Interest Declaration
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
No.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
Table 1: Conflict of Interest Declaration for the Women’s Health Coalition of Alberta Society
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Allergan | — | X | — | — |
Hologic | — | X | — | — |
I hereby certify that I have the authority to disclose all relevant information with respect to any matter involving this patient group with a company, organization, or entity that may place this patient group in a real, potential, or perceived conflict of interest situation.
Patient Group: Women’s Health Coalition of Alberta
Date: November 8, 2021
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.