CADTH Reimbursement Review
Avalglucosidase Alfa (Nexviazyme)
Sponsor: Sanofi Genzyme, a division of Sanofi-Aventis Canada Inc.
Therapeutic area: Pompe disease
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
6MWT
6-minute walk test
AE
adverse event
ANCOVA
analysis of covariance
BMI
body mass index
CI
confidence interval
e-CRF
electronic case report form
EQ-5D-5L
EQ-5D 5-Levels
FEV1
forced expiratory volume in 1 second
FVC
forced vital capacity
GAA
acid alpha-glucosidase
GMFCS
Gross Motor Function Classification System
GMFM-88
Gross Motor Function Measure-88
GSGC
gait, stair, Gower’s manoeuvre, and chair
HHD
hand-held dynamometry
HRQoL
health-related quality of life
ICC
intraclass correlation coefficient
IOPD
infantile-onset Pompe disease
ITT
intention-to-treat population
LOPD
late-onset Pompe disease
LS
least squares
M6P
mannose-6-phosphate
MedDRA
medical dictionary for regulatory activity
MEP
maximum expiratory pressure
MeSH
Medical Subject Headings
mg
milligram
MID
minimal important difference
MIP
maximum inspiratory pressure
mITT
modified intention-to-treat
MMRM
mixed model repeated measures
PP
per protocol
QMFT
Quick Motor Function Test
RCT
randomized controlled trial
rhGAA
recombinant human acid alpha-glucosidase
SAE
serious adverse event
SD
standard deviation
SE
standard error
SF-12
12-item Short Form Health Survey
SF-36
36-item Short Form Health Survey
SMQ
Standardized MedDRA Queries
TEAE
treatment-emergent adverse event
WDAE
withdrawal due to adverse event
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Avalglucosidase alfa (Nexviazyme), 100 mg/vial, 20 mg/kg of body weight, administered every other week by IV infusion |
Indication | Nexviazyme (avalglucosidase alfa) is an enzyme-replacement therapy indicated for the long-term treatment of patients with LOPD (acid alpha-glucosidase deficiency) |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | November 12, 2021 |
Sponsor | Sanofi Genzyme, a division of Sanofi-Aventis Canada Inc. |
LOPD = late-onset Pompe disease; NOC = Notice of Compliance.
Introduction
Pompe disease is a rare, autosomal recessive disorder caused by pathogenic variants in the acid alpha-glucosidase (GAA) gene, resulting in dysfunctional GAA enzymes.1 With Pompe disease, the defect in the enzyme allows glycogen to accumulate in cells, leading to impaired cellular function and tissue damage.1,2 Patients with late-onset Pompe disease (LOPD) have variable and reduced enzyme function (between 2% and 40% of normal),3 whereas patients with infantile-onset Pompe disease (IOPD) have minimal or no enzyme activity.1,3 Pompe disease is usually diagnosed with molecular testing or enzymatic analysis of white blood cells or dried blood spots; however, in some cases, a biopsy of skin or muscle tissue can be performed and may show glycogen accumulation, but this method is more invasive. Gene sequencing is the preferred method to confirm a diagnosis and is both noninvasive and routinely available. The presence of 2 pathogenic variants of the GAA gene confirms a diagnosis of Pompe disease.
The rate of disease progression varies among patients, and disease severity is inversely correlated with residual GAA activity.3 Additionally, disease severity is associated with disease duration, and patients who have symptom onset at a younger age have more severe disease.1 It has been estimated that the 5-year post-diagnosis survival for untreated patients with LOPD is 95%, and 30-year post-diagnosis survival is 40%.1 It has been reported that patients treated with enzyme-replacement therapy have a mean age at death of less than 60 years,1 although this varies with rate of progression, extent of muscle involvement, and comorbidities.3 For instance, early involvement of the diaphragm is followed by respiratory failure and death during the second or third decade of life.1 In general, earlier diagnosis and treatment can improve outcomes. Clinical features vary from a slowly progressive myopathy, which might be preceded by an asymptomatic interval, to a much more rapid and progressive myopathy that results in wheelchair and ventilatory dependence and early death.1 It is also a common and unique feature of Pompe disease to have early involvement of the diaphragm and respiratory accessory muscles that is not observed with most other myopathies. This can lead to respiratory failure, which is a major cause of morbidity and mortality in patients with LOPD.1,3,4 The progression of symptoms often leads to new or increased use of respiratory support and mobility aids, and patients with LOPD can also experience respiratory failure while still ambulatory.3
The clinical expert CADTH consulted for this review estimated that a prevalence of 1 in 40,000 for all Pompe disease would be reasonable. For LOPD specifically, a study from the Netherlands estimated a prevalence of 1 in 57,000.5 The sponsor indicated that there were |||| patients with LOPD in Canada receiving treatment with alglucosidase alfa (|||| adults and |||| children) as of December 2020.6 The incidence of all Pompe disease (both LOPD and IOPD) has been estimated to be between 1 in 14,000 and 1 in 300,000, depending geographic location and ethnicity,3 whereas the incidence of LOPD has been estimated to be 1.75 in 100,000 births.7 A study using data from births between 1969 and 1996 in British Columbia estimated the incidence of Pompe disease to be 1 in 115,091.8 It is expected that this is an underestimate of the true number of patients with LOPD in Canada because many would be have been undiagnosed at the time of the study. No updated prevalence or incidence data specific to Canada have been identified.
Clinicians consulted by CADTH for the purpose of this review indicated that enzyme-replacement therapy with alglucosidase alfa, a recombinant human GAA (rhGAA), at a dose of 20 mg/kg by IV infusion every 2 weeks, is the standard treatment and the only specific treatment for LOPD, although it is not a cure for Pompe disease. Aside from enzyme-replacement therapy, supportive care includes continued monitoring of pulmonary function and motor performance to assess new or increased need for ventilatory support and mobility aids. Canadian guidelines for the diagnosis and management of Pompe disease state that there is no evidence for the use of enzyme-replacement therapy in patients who have confirmed Pompe disease but are otherwise asymptomatic.9
Supportive therapies also include exercise and dietary changes, and new disease-specific therapies include other forms of enzyme-replacement therapy, and gene therapies are in development. Other interventions, such as physical therapy, occupational therapy, speech therapy, and assistive technological devices, can be used to support respiratory and motor function and attempt to improve health-related quality of life (HRQoL).3,4 Input from the clinical expert indicated that beta-2 agonists have been used off-label to try to increase the efficacy of enzyme-replacement therapy, although this is outside of the indication approved by Health Canada. The clinical expert emphasized that the main goals of currently available forms of treatment are to stabilize and/or improve motor and respiratory function, as well as to prevent further disease progression. New treatments should improve immune tolerance, have a low risk of treatment-related reactions, and be less of a burden on patients; current infusions are frequent and require hours to complete.
Avalglucosidase alfa is a rhGAA that provides an exogenous source of GAA enzyme-replacement therapy with an approximate 15-fold increase in mannose-6-phosphate, compared with alglucosidase alfa.10 The increased mannose-6-phosphate moieties provide a mechanism to drive uptake of avalglucosidase alfa into the diaphragm and other skeletal muscle through the cation-independent receptor.10 In Canada, avalglucosidase alfa is indicated for the long-term treatment of patients with LOPD. Avalglucosidase alfa underwent a standard review at Health Canada and reimbursement for the approved Health Canada indication has been requested. Avalglucosidase alfa has not been previously reviewed by CADTH.
The objective of this report is to perform a systematic review of the beneficial and harmful effects of avalglucosidase alfa administered once every other week as an IV infusion of 20 mg/kg of body weight for the treatment of LOPD.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient groups who responded to CADTH’s call for patient input and from clinical experts consulted by CADTH for the purpose of this review.
Patient Input
Muscular Dystrophy Canada, in partnership with the Canadian Association for Pompe, conducted a survey and semi-structured phone or Zoom teleconference interviews with adults or the parents and caregivers of children living with Pompe disease. In total, 41 individuals affected by Pompe disease provided information for the submission.
Respondents frequently reported that Pompe disease negatively affected motor ability (including mobility, strength, balance, and energy) and breathing. Quality of life was also important to the patient group, and the detrimental effects of the disease on patients’ social health, mental health, and ability to participate in daily activities, and on their families, were identified as key issues related to Pompe disease.
Some respondents reported having no experience with medications for Pompe disease and were focusing on physical therapy, whereas others described being on enzyme-replacement therapy for years. Some patients treated with alglucosidase alfa described minor improvements followed by a plateauing of effect; others reported major improvements. Patients and caregivers said they would like new treatments to improve strength, breathing function, and prevent disease progression. They would also like a better mode of delivery, fewer side effects, a treatment that has a continuous effect in the body, and greater accessibility without the need to travel.
Two adults who reported clinical trial experience with avalglucosidase alfa had been receiving the enzyme-replacement therapy for 2 to 3 years. During this time, they noticed improvements in mobility, balance, and endurance, with the most significant benefits being improvements in daily living and mental health.
All patients from the group submission reported undergoing diagnostic testing with blood tests, and some also underwent confirmatory biopsies. In general, patients did not have to pay for testing, although some did incur costs associated with travelling for appointments. Some respondents experienced no delays in testing, but others faced multiple tests or significant wait times, and many patients recalled the stress of being misdiagnosed.
Clinician Input
Input From Clinical Experts Consulted by CADTH
The clinical expert consulted by CADTH explained that the most important goals of currently available treatments are to stabilize and/or improve motor and respiratory function and to prevent further disease progression. Although reversal of muscle involvement at the time of diagnosis would be ideal, novel tools to target muscle cell growth and regeneration will need to be developed before this goal can be achieved. Therapies should also have a minimal burden on patients and a low risk of infusion-related reactions.
The clinical expert expects that avalglucosidase alfa will replace alglucosidase alfa as first-line treatment for Pompe disease and that all patients who meet the criteria for treatment will receive the new drug. This would include patients who have never received enzyme-replacement therapy; those already being treated with alglucosidase alfa would be switched over to avalglucosidase alfa.
Patients with Pompe disease are identified with enzymatic testing and genetic testing. The clinical expert noted that a free multigene panel provided by the drug manufacturer includes testing for Pompe disease. This has allowed clinicians to screen and identify potential patients before they qualify for genetic testing, which is funded in some jurisdictions.
According to the clinical expert, any patients with symptomatic disease should be treated with enzyme-replacement therapy. The heterogeneity in clinical presentation of LOPD precludes treatment in the primary-prevention setting, and patients without symptoms should be closely monitored to detect early signs of disease progression. Patients with very advanced disease, such as those who are wheelchair-bound and on permanent invasive ventilation, might be least suited for avalglucosidase alfa, although the clinical expert added that clinical context should be considered on a case-by-case basis.
Canadian evidence-based guidelines9 for the treatment of LOPD emphasize the importance of having and meeting clearly defined, objective outcomes and tracking progression, the clinical expert reported. Assessments for skeletal muscle function (e.g., 6-minute walk test [6MWT], quantitative muscle strength scoring) and respiratory muscle function (e.g., forced vital capacity [FVC], maximum inspiratory pressure [MIP], maximum expiratory pressure [MEP], change in FVC between upright and supine positions) were noted as relevant outcomes in clinical trials. Testing at individual clinics may vary. It is recommended that patients are followed at least annually by a regional centre of excellence. Patients who begin a new therapy should initially be assessed every 6 months, whereas patients on long-term treatment who remain stable should be assessed at least annually. For patients who live in remote areas, it may be acceptable to have a detailed annual assessment at an expert centre along with visits every 6 months with a local physician.
The clinical expert stated that most patients are treated with enzyme-replacement therapy until they develop end-stage disease, which could include the need for a wheelchair and full-time invasive ventilation. Anaphylactic reactions to the medication that cannot be managed with premedications and comorbidities that significantly reduce lifespan (e.g., cancer) might be reasons to discontinue treatment.
According to the clinical expert, new patients often start their treatment in a hospital clinical setting and, once stable, transition to home infusions. Post-infusion follow-up would always be performed by a centre with expertise in the management of patients with Pompe disease.
Clinician Group Input
Clinician input was provided by the Neuromuscular Disease Network for Canada and 8 clinicians with experience treating Pompe disease.
Input from the clinician group was similar to that given by the clinical expert consulted by CADTH.
Drug Program Input
The drug programs had questions about age eligibility and switching from alglucosidase alfa to avalglucosidase alfa. The drug programs also noted a preference for uniform initiation, renewal, and discontinuation criteria for avalglucosidase alfa among jurisdictions. Responses to the questions are in Table 4.
Clinical Evidence
Pivotal Studies and Protocol Selected Studies
Description of Studies
One multi-centre, double-blind, active-control, phase III, randomized controlled trial (RCT) was included in the CADTH review for avalglucosidase alfa. The COMET trial was designed to evaluate the efficacy and safety of avalglucosidase alfa 20 mg/kg body weight administered every other week for the treatment of LOPD. The study consisted of a screening period of up to 14 days, a double-blind treatment period of 49 weeks, an open-label extension phase of up to 240 weeks, and follow-up for up to 4 weeks. At the end of the double-blind phase, patients in the alglucosidase alfa arm switched to the avalglucosidase alfa arm for the duration of the open-label treatment phase. The primary outcome of FVC (% predicted) in the upright position was used to test the noninferiority of avalglucosidase alfa to alglucosidase alfa, using a noninferiority margin of –1.1%. Sequential testing continued with superiority testing for FVC (% predicted) followed by the key secondary outcome of distance walked and % predicted on the 6MWT. Patients older than 3 years were eligible to participate in the COMET trial if they had confirmed GAA enzyme deficiency from any tissue source and/or 2 confirmed GAA gene mutations. Patients with known Pompe-specific cardiac hypertrophy or who had severe disease (e.g., wheelchair-dependent, requiring invasive ventilation) were excluded from the study. Previous treatment with alglucosidase alfa or other investigational treatments for Pompe disease were also reasons for exclusion.
In total, 100 patients were randomized in a 1:1 ratio to either avalglucosidase alfa or alglucosidase alfa. The mean age of the patients in the COMET trial was 48 years (standard deviation [SD] = 14), and patients were older in the alglucosidase alfa group. Patients were predominantly White (94%), and there was a similar number of male and female patients within and between treatment groups. The baseline mean distance walked on the 6MWT was numerically higher for the avalglucosidase alfa group (399.3 m; SD = 110.9) than for the alglucosidase alfa group (378.1 m; SD = 116.2). Also, more patients reported using no mobility aids in the avalglucosidase alfa arm. Use of a rolling walker or a single crutch was higher in the comparator arm (6.1% and 4.1%, respectively) than in the avalglucosidase alfa arm (0.0% and 0.0%, respectively). The mean age at diagnosis for Pompe disease was lower for patients in the avalglucosidase alfa group (44.73 years; SD = 14.74) than in the alglucosidase alfa group (48.16 years; SD = 14.64). The time between diagnosis and first infusion of the study drug was shorter in the avalglucosidase alfa group (15.60 months; SD = 32.06) than in the alglucosidase alfa group (26.52 months; SD = 59.86).
Efficacy Results
Patients in the modified intention-to-treat (mITT) population, which was equivalent to the intention-to-treat (ITT) population, demonstrated a least squares (LS) mean change in FVC (% predicted) in the upright position from baseline to week 49 of 2.89% (95% confidence interval [CI] = 1.13 to 4.65) in the avalglucosidase alfa arm and 0.46% (95% CI = –1.39 to 2.31) in the alglucosidase alfa arm. The mean difference of change between treatment groups was 2.43% (95% CI = –0.13 to 4.99), for which the lower bound of the 95% CI did not exceed the noninferiority margin of –1.1%, indicating that the criteria for noninferiority of avalglucosidase alfa to alglucosidase was demonstrated (P = 0.0074). The P value for superiority testing was not statistically significant (P = 0.0626), so statistical testing was stopped for all subsequent efficacy outcomes. Analysis of the per protocol (PP) population had similar results, with a FVC (% predicted) LS mean change from baseline to week 49 of 2.87% (95% CI = 1.02 to 4.73) and 0.19% (95% CI = –1.83 to 2.21) for the avalglucosidase alfa and alglucosidase alfa groups, respectively. The mean difference of change between treatment groups was 2.69% (95% CI = –0.06 to 5.44; P for noninferiority = 0.0076 and P for superiority = 0.0555).
The mean change from baseline to week 49 for the 6MWT distance was 32.21 m (95% CI = 12.47 to 51.94) for the avalglucosidase alfa group and 2.19 m (95% CI = –18.48 to 22.86) for the alglucosidase alfa group. The mean difference of change between treatment groups was 30.01 m (95% CI = 1.33 to 58.69), which was numerically greater for avalglucosidase alfa, with a CI that excluded the null. The mean change from baseline for the 6MWT (% predicted) was 5.02% (95% CI = 1.95 to 8.09) for the avalglucosidase alfa group and 0.31% (95% CI = –2.90 to 3.52) for the alglucosidase alfa group. The mean difference of change was 4.71% (95% CI = 0.25 to 9.17) between treatments.
Harms Results
During the double-blind phase, 44 patients (86.3%) who received avalglucosidase alfa and 45 patients (91.8%) who received alglucosidase alfa experienced an adverse event (AE). The most frequently reported events were nasopharyngitis (12 patients, 23.5%), back pain (12 patients, 23.5%), and headache (11 patients, 21.6%) for the avalglucosidase alfa group, and headache (16 patients, 32.7%), nasopharyngitis (12 patients, 24.5%), and falls (10 patients, 20.4%) for the alglucosidase alfa group. Overall, SAEs were infrequent among either treatment group. Serious AEs (SAE) were reported in 8 (15.7%) patients who received avalglucosidase alfa, compared to 12 (24.5%) patients who received alglucosidase alfa. Four patients (8.2%) in the alglucosidase alfa group withdrew from the study because of the following AEs: acute myocardial infarction, arthritis, dyspnea, and urticaria. No patients withdrew from the avalglucosidase alfa group because of AEs. One death (2%), from acute myocardial infarction, was reported in the alglucosidase alfa group.
Treatment-emergent anaphylactic reactions (pruritus and rash) were reported in 2 patients (approximately 4.0%) in each of the treatment arms during the double-blind phase. Treatment-emergent hypersensitivity reactions occurred in 12 patients (23.5%) and 15 patients (30.6%) in the avalglucosidase alfa and alglucosidase alfa groups, respectively, with pruritus and rash being the most frequently reported. Treatment-emergent infusion-associated reactions occurred in 13 patients (25.5%) in the avalglucosidase alfa and in 16 patients (32.7%) in the alglucosidase alfa group, with pruritus and nausea being the most frequently reported infusion-associated reactions. Treatment-emergent immune-mediated reactions occurred in 12 patients (23.5%) who received avalglucosidase alfa and 15 patients (30.6%) who received alglucosidase alfa, with arthralgia and myalgia being the most common. Nearly all patients were positive for treatment-emergent anti-drug antibodies. Overall, 10 patients (19.6%) in the avalglucosidase alfa group and 16 patients (33.3%) in the alglucosidase alfa group had peak titre levels greater than 12,800. Acute cardiorespiratory failure was not reported in the COMET study.
Table 2: Summary of Key Results From the COMET Study
Outcome | Avalglucosidase alfa, N = 51 | Alglucosidase alfa, N = 49 |
|---|---|---|
Primary efficacy outcome | ||
FVC, % predicted, in upright position: mITT population | ||
Patients contributing to analysis, n | 49 | 43 |
Baseline, mean (SD) | 62.55 (14.39) | 61.56 (12.40) |
End of double-blind phase, week 49; mean (SD) | 65.49 (17.42) | 61.16 (13.49) |
LS mean change from baselinea (SE) [95% CI] | 2.89 (0.88) [1.13 to 4.65] | 0.46 (0.93) [–1.39 to 2.31] |
Group difference, treatment – control (SE) [95% CI] | 2.43 (1.29) [–0.13 to 4.99] | Reference |
P value for noninferiorityb | 0.0074 | Reference |
P value for superiority | 0.0626 | Reference |
FVC, % predicted, in upright position: PP population | ||
Patients contributing to analysis, n | 46 | 39 |
Baseline, mean (SD) | 63.13 (14.65) | 61.46 (13.02) |
End of double-blind phase, week 49; mean (SD) | 66.15 (17.27) | 61.38 (13.91) |
LS mean change from baselinea (SE) [95% CI] | 2.87 (0.93) [1.02 to 4.73] | 0.19 (1.02) (–1.83 to 2.21) |
Group difference, treatment – control (SE) [95% CI] | 2.69 (1.38) [–0.06 to 5.44] | Reference |
P value for noninferiorityb | 0.0076 | Reference |
P value for superiority | 0.0555 | Reference |
Key secondary efficacy outcome | ||
6MWT, distance in m): mITT population | ||
Patients contributing to analysis, n | 48 | 43 |
Baseline, mean (SD) | 399.30 (110.93) | 378.09 (116.22) |
End of double-blind phase, week 49; mean (SD) | 441.31 (109.77) | 383.56 (141.09) |
Change from baseline,c mean (SE) [95% CI] | 32.21 (9.93) [12.47 to 51.94] | 2.19 (10.40) [–18.48, 22.86] |
Group difference, treatment – control (SE) (95% CI) | 30.01 (14.43) [1.33 to 58.69] | Reference |
P valued | 0.0405 | Reference |
6MWT, % predicted: mITT population | ||
Patients contributing to analysis, n | 48 | 43 |
Baseline, mean (SD) | 57.32 (14.97) | 55.29 (16.64) |
End of double-blind phase, week 49; mean (SD) | 63.73 (15.13) | 55.39 (19.15) |
Change from baseline,c mean (SE) [95% CI] | 5.02 (1.54) [1.95 to 8.09] | 0.31 (1.62) [–2.90 to 3.52] |
Group difference, treatment – control (SE) [95% CI] | 4.71 (2.24) [0.25 to 9.17] | Reference |
P valued | 0.0386 | Reference |
Harms, n (%): safety population | ||
TEAEs | 44 (86.3) | 45 (91.8) |
SAEs | 8 (15.7) | 12 (24.5) |
WDAE, from study treatment | 0 | 4 (8.2) |
Deaths | 0 | 1 (2.0) |
Notable harms, n (%) | ||
Treatment-emergent anaphylactic reactions by SMQ, broad and narrow combined | 2 (3.9) | 2 (4.1) |
Treatment-emergent hypersensitivity reactions by SMQ, broad and narrow combined | 12 (23.5) | 15 (30.6) |
Treatment-emergent infusion-associated reactions | 13 (25.5) | 16 (32.7) |
Treatment-emergent immune-mediated reactions | 12 (23.5) | 15 (30.6) |
Acute cardiorespiratory failure | NR | NR |
6MWT = 6-minute walk test; CI = confidence interval; FVC = forced vital capacity; LS = least squares; mITT = modified intention-to-treat; NA = not applicable; NR = not reported; PP = per protocol; SAE = serious adverse events; SD = standard deviation; SE = standard error; SMQ = Standardized MedDRA Queries; TEAE = treatment-emergent adverse events; WDAE = withdrawal due to adverse events.
aMMRM model with fixed, continuous effects of baseline FVC (% predicted) and age (in years at baseline), and fixed, categorical effects of sex, treatment group, visit, and interaction term between treatment group and visit.
bNoninferiority margin is –1.1%. P value has been adjusted for multiple testing (i.e., the type I error rate has been controlled).
cMMRM model for 6MWT distance with fixed effects of baseline FVC (% predicted), baseline 6MWT (distance walked in m), age (in years at baseline), sex, treatment group, visit, and treatment-by-visit interaction.
dP value has not been adjusted for multiple testing (i.e., the type I error rate has not been controlled).
Source: COMET Clinical Study Report.11
Critical Appraisal
A key limitation to the COMET trial was the difference in baseline characteristics between the treatment arms. The avalglucosidase alfa group had a younger age at baseline, younger age at diagnosis, shorter time between diagnosis and treatment, greater 6MWT mean distance, and fewer patients who used a mobility aid during the 6MWT. Of note, the time between diagnosis and first infusion of the study drug was different between the treatment groups and was not adjusted for in the statistical analysis, which might have confounded the results. The clinical expert consulted for this review stated that the differences may be a result of the small patient numbers, but noted that the differences in baseline characteristics in most cases tend to cause biases in the results in favour of the avalglucosidase alfa group. Patients who present at an early age are likely progressing at an accelerated rate, and earlier treatment is expected to result in better outcomes, but the direction and magnitude of the biases caused by the differences in these factors in the baseline characteristics are unclear. All 5 patients (10.2%) who discontinued treatment during the double-blind phase were from the alglucosidase alfa group, and it is unknown what impact these losses had on the results, considering the small patient numbers. Nearly all outcomes reported in this review had missing data, and methods for handling missing data were lacking, which must be considered when interpreting the results. Missing data were not imputed for the primary outcome, and it was assumed that data were missing at random. This assumption may bias 1 treatment over another, although sensitivity analyses were performed to assess the impact of missing data, which supported the missing-at-random assumption for the primary outcome. To control for multiplicity, a sequential testing strategy was used, and statistical testing was stopped at the first nonsignificant outcome (superiority testing of the primary outcome). As a result, all secondary and tertiary outcomes were not controlled for type I error and should be interpreted as supportive of the primary outcome. Subgroup analyses for the primary outcome were specified a priori; although there was no control for multiplicity, each subgroup had a small number of patients, and the wide 95% CIs indicated imprecision with the estimates.
The noninferiority margin of –1.1% was based on data from the double-blind, placebo-controlled LOTS trial for alglucosidase alfa 20 mg/kg every other week. The noninferiority margin of –1.1% retained approximately half of the lower bound of the 80% CI of the estimated treatment effect of alglucosidase alfa over placebo for FVC (% predicted) in the LOTS trial at 12 months (i.e., 2.14). The clinical expert believes that this is a reasonable approach to estimate the margin, and that it is also a reasonable choice of margin from a clinical perspective. Retention of half the comparator’s treatment effect is consistent with FDA guidance for noninferiority trials.12 Although FDA guidance12 indicates that a 95% CI is commonly used, the COMET publication13 stated that the CI was lowered to 80% in response to suggestions by regulatory bodies. The rationale for this was not described. The constancy assumption is such that the effect of the active comparator (i.e., alglucosidase alfa) in the current noninferiority trial is the same as the effect observed in past trials and requires the trials be sufficiently similar.14
The similarities in study design, eligibility criteria, treatment doses, and key outcomes between the COMET and LOTS trials support the constancy assumption. Furthermore, the pre-specified constancy-assumption analysis estimated an effect of 2.87 for alglucosidase alfa, compared with placebo, in the COMET trial, and an effect of 3.02 in the LOTS trial based on the predictive model.13 The investigators considered the difference in effect to be small (–0.15) compared with the noninferiority margin (1.1%).13 Considering that Pompe disease is rare, the clinical expert noted that the patients who received alglucosidase alfa were mostly similar in the 2 studies. However, key differences in baseline characteristics (e.g., older baseline age, older age at symptom onset, higher FVC, and better 6MWT scores) were noted for patients who received alglucosidase alfa in the COMET trial compared to the LOTS trial. Although these differences should bias the results in favour of alglucosidase alfa in the COMET study, the clinical expert did not feel the differences explained why the patients in the COMET trial did not respond as well as those in the LOTS trial. Consequently, the lower-than-expected improvements in patients in the alglucosidase alfa arm in COMET study could bias the interpretation of results in favour of treatment with avalglucosidase alfa. Although these concerns may have impacted interpretation of the trend toward superiority in the COMET trial, it is the opinion of the clinical expert that the concerns do not affect the statistically significant conclusion of the trial: that avalglucosidase alfa is noninferior to alglucosidase alfa.
In general, the patients in the COMET study resembled those seen in clinical practice in Canada. Despite limited evidence for the treatment of children with LOPD, the clinical expert consulted for this review stated that the results were generalizable to pediatric patients, but not to patients with IOPD, and highlighted the urgent need for additional data in the IOPD population. The clinical expert noted that clinical practice, background care, and reporting of AEs can vary among countries, which may confound results. Canadian guidance9 for the management of patients with Pompe disease suggests that assessments be performed at least every 6 months, which is less frequent than in the COMET trial. The greater access to health care resources and attention from clinicians should be considered when the results are generalized to real-world practice.
Given that Pompe disease is a lifelong condition, the 1 year of data for avalglucosidase alfa from the double-blind phase of the COMET trial is somewhat limiting, although the open-label extension phase is ongoing. The literature search conducted to inform the description and appraisal of outcome measures showed there was a lack of evidence supporting the validity, reliability, or responsiveness to change for some of the trial outcomes. Therefore, there is uncertainty around the use of measures such as MEP, SF-12, and GMFM-88 to assess treatment for patients with LOPD. Furthermore, a literature search did not find any minimally important differences (MIDs) for populations with Pompe disease.
Indirect Comparisons
Indirect treatment evidence for avalglucosidase alfa was not identified in this review.
Other Relevant Evidence
Description of Studies
The NEO1 study was a phase I, multi-centre, open-label, ascending-dose study conducted to determine safety and tolerability, pharmacokinetic parameters, and the pharmacodynamic effects of avalglucosidase alfa in patients with LOPD who were treatment-naïve (group 1) and in patients with LOPD who were previously treated with alglucosidase alfa for at least 9 months (group 2). Patients received IV infusions of avalglucosidase alfa every other week, for a total of 13 infusions at 1 of the following doses: 5 mg/kg, 10 mg/kg, or 20 mg/kg. Study participants had to be at least 18 years of age with a confirmed GAA enzyme deficiency and/or a confirmed GAA gene mutation, no known cardiac hypertrophy, FVC in the upright position of 50% predicted or more, and be able to ambulate 50 m without stopping and without an assistive device.
The NEO-EXT study is the long-term extension of NEO1. All patients who completed treatment with the 5 mg/kg or 10 mg/kg dose were invited to enrol in the extension and receive the 20 mg/kg dose for up to 6 years. The results summarized in this review focused on the Health Canada–approved 20 mg/kg dose.
Efficacy Results
All efficacy results in the NEO1 study were considered exploratory in nature.
In the NEO1 study, baseline mean FVC (% predicted) for patients who received avalglucosidase alfa 20 mg/kg were 63.4% (SD = 17.84) and 70.4% (SD = 16.40) in group 1 and group 2, respectively. At week 25, mean FVC (% predicted) changed to 69.5% (SD = 20.63) and 69.9% (SD = 16.92) in group 1 and group 2, respectively, with a mean change from baseline of 6.2% (SD = 3.15) and 1.4% (SD = 5.71) for the respective groups. In the NEO-EXT study, baseline mean FVC (% predicted) were 69.2% (SD = 19.27) and 77.3% (SD = 16.45) in combined (5 mg/kg, 10 mg/kg, or 20 mg/kg doses) group 1 and combined group 2, respectively. At week 286, mean FVC (% predicted) changed to 65.7% (SD = 30.07) and 74.5% (SD = 21.24) in the combined group 1 and combined group 2, respectively. Results beyond week 286 were available, but reduced patient numbers resulted in uninformative data.
In the NEO1 study, baseline mean 6MWT % predicted for patients who received avalglucosidase alfa 20 mg/kg were 75.2% (SD = 9.80) and 72.8% (SD = 20.59) in group 1 and group 2, respectively. At week 25, mean 6MWT % predicted changed to 79.1% (SD = 12.55) and 65.6% (SD = 12.03) in group 1 and group 2, respectively, with mean changes from baseline of 3.9% (SD = 3.45) and –1.3% (SD = 8.94) for the respective groups. In the NEO-EXT study, 6MWT results were 64.9% (SD = 28.05) and 69.1% (SD = 21.37) in group 1 and group 2 at week 286, respectively. Results beyond week 286 were available, but reduced patient numbers resulted in uninformative data.
Harms Results
In the initial NEO1 study period, 1 of the 3 patients in group 1 who received the 20 mg/kg dose experienced an AE: namely, nasopharyngitis and erythema. All 6 patients in group 2 who received the 20 mg/kg dose experienced an AE, but arthralgia and musculoskeletal pain were the only 2 AEs to be reported in multiple patients (33.3%). In the NEO-EXT trial, all 24 patients, including those who switched to 20 mg/kg, experienced an AE. The most commonly reported AEs were nasopharyngitis (15 patients, 62.5%), fall (12 patients, 50.0%), diarrhea (11 patients, 45.8%), headache (10 patients, 41.7%), and muscle spasms (10 patients, 41.7%). None of the patients who received the 20 mg/kg dose reported a SAE in the NEO1 study. In the NEO-EXT study, 9 patients (37.5%) reported a SAE, but no individual SAE was reported in more than 1 patient. In the NEO1 study period, no patients who received the 20 mg/kg dose reported an AE that led to treatment discontinuation. In the NEO-EXT study, 1 patient (4.2%) discontinued treatment due to an AE. No deaths related to AEs were reported in either the NEO1 or NEO-EXT study.
Notable harms — including anaphylactic reactions, hypersensitivity, infusion-associated reactions, and immune-mediated reactions — were less common in the NEO1 study period, but occurrence increased in NEO-EXT. In NEO-EXT, 17 patients (70.8%) experienced a treatment-emergent hypersensitivity reaction, 12 patients (50.0%) experienced a treatment-emergent infusion-associated reaction, 2 patients (8.3%) experienced a treatment-emergent anaphylactic reaction, and no patients experienced a treatment-emergent immune-mediated reaction.
Critical Appraisal
In the NEO1 and NEO-EXT studies, efficacy outcomes were considered strictly exploratory. Inherent in phase I trials are the issues of a low number of patients, the lack of a comparator arm, and the lack of randomization. As a result, it is not possible to determine a causal relationship between the study drug and outcomes observed. The baseline demographics varied between patients receiving different doses, likely because of the low number of patients.
Inclusion of the long-term extension of the phase I NEO1 study in the sponsor’s submission allows for greater generalizability of the safety and tolerability data, beyond the time points presented in the pivotal trials; however, the study design greatly limits the generalizability of any findings.
Conclusions
In the COMET trial, the study’s main objective was achieved, and avalglucosidase alfa met the criteria for noninferiority, compared with alglucosidase alfa. This comparison was made at the noninferiority margin of –1.1%, based on the primary outcome of FVC (% predicted) in the upright position for the first 49 weeks of treatment in patients with LOPD. Based on the evidence from the COMET trial, treatment with avalglucosidase alfa appeared to prevent further respiratory deterioration during the first year, which is 1 of the main goals of currently available forms of treatment, according to the clinical expert. The results from the study were not statistically significant when testing for the superiority of avalglucosidase alfa over alglucosidase alfa for FVC (% predicted). Statistical testing was stopped for secondary outcomes and results should be interpreted as supportive of the primary outcome. Most patients experienced at least 1 AE during the first year of treatment, but fewer experienced a SAE. Notable harms related to anaphylactic reactions occurred with the same frequency among the treatment groups; however, hypersensitivity reactions, infusion-associated reactions, and immune-mediated reactions were numerically lower in patients who received avalglucosidase alfa.
Key limitations include the small number of patients enrolled in the studies and the lack of data available for pediatric patients. The small number of patients limits the ability to accurately quantify the differences in outcomes, beyond the primary outcome, in long-term benefits and in potential harms between the treatments. Moreover, it will be beneficial to have continued long-term efficacy and safety data (including anti-drug antibody assessments) for avalglucosidase alfa, which the extension phase of the COMET study will provide. Last, these results apply only to patients with LOPD; trials evaluating avalglucosidase alfa in patients with IOPD are ongoing.
Introduction
Disease Background
Pompe disease (also known as GAA deficiency, or glycogen storage disease type II) was the first lysosomal storage disease to be identified.1 It is a rare, autosomal recessive disorder caused by pathogenic variants in the GAA gene, resulting in dysfunctional GAA enzymes. In the low pH of the lysosome, GAA breaks down the alpha-1,4- and the alpha-1,6-glycosidic links of glycogen molecules. With Pompe disease, the defect in the enzyme allows glycogen to accumulate, leading to impaired cellular function and tissue damage.1,2 Patients with LOPD have variable and reduced enzyme function (between 2% and 40% of normal),3 whereas patients with IOPD have minimal or no enzyme activity.1
The diagnosis of Pompe disease can be a challenge because symptoms resemble those of other neuromuscular disorders.4 Pompe disease might be suspected in children and adults who show progressive proximal limb weakness and significantly reduced FVC.1 It is diagnosed with molecular testing or enzymatic analysis of white blood cells or dried blood spots, which is typically available in clinical biochemical and genetic diagnostic labs. In some cases, a biopsy of skin or muscle tissue can be performed, and may show glycogen accumulation. Gene sequencing is the preferred method for confirming a diagnosis and is noninvasive and routinely available. Genetic sequencing can also be used to rule out false-positive results that arise from a homozygous pseudodeficiency allele (leading to low GAA activity). The presence of 2 pathogenic variants of the GAA gene confirms a diagnosis of Pompe disease. Newborn screening for GAA deficiency has been implemented in some countries, using a tiered system that consists of an enzymatic assay followed by molecular genetic testing. Differential diagnosis might be necessary to distinguish Pompe disease from other myopathies using age at symptom onset, high creatine kinase levels, and absence of metabolic abnormalities (e.g., hypoglycemia, lactic acidosis, metabolic acidosis). In general, earlier diagnosis and treatment can improve outcomes.
In terms of the natural history of LOPD, the rate of disease progression varies among patients and disease severity is inversely correlated with residual GAA activity.3 Additionally, disease severity is associated with disease duration, and patients who have symptom onset at a younger age have more severe disease.1 It has been estimated that 5-year post-diagnosis survival for untreated patients with LOPD is 95%, and 30-year post-diagnosis survival is 40%.1,3 It has been reported that patients treated with enzyme-replacement therapy have a mean age at death of less than 60 years,1 although this varies with rate of progression, extent of muscle involvement, and comorbidities.3 For instance, early involvement of the diaphragm is followed by respiratory failure and death during the second or third decade of life.1
Patients with LOPD do not develop hypertrophic cardiomyopathy (a characteristic of IOPD), and clinical presentation can be at any age, even among individuals with the same genetic variant, indicating that there are other factors that influence clinical outcomes.1 Moreover, clinical features vary from a slowly progressive myopathy, which may be preceded by an asymptomatic interval, to a much more rapid and progressive myopathy that results in wheelchair and ventilatory dependence and early death. Typically, there is greater weakness in the proximal muscles than the distal muscles, and the pelvic girdle is affected more than the shoulder girdle.4 Early involvement of the diaphragm and respiratory accessory muscles is a common and unique feature of Pompe disease that is not observed with most other myopathies. This can lead to respiratory failure, which is a major cause of morbidity and mortality in patients with LOPD.1,3,4 Patients may also have reduced lung volume, impaired ability to cough, and poor breathing during sleep,3 and respiratory dysfunction can be identified through testing that shows reduced FVC in upright and supine positions, as well as lower MIP and MEP measurements.4 It has been estimated that 60% of patients have a slight reduction in vital capacity (< 80% predicted), and 30% to 40% have moderate reduction in vital capacity (< 60% predicted).3 The progression of symptoms often leads to new or increased use of respiratory support and mobility aids, and patients with LOPD can experience respiratory failure while still ambulatory. Patients may also have vascular and/or gastrointestinal complications, and muscle weakness can cause difficulties chewing and swallowing foods, leading to poor nutrition.3,4
The clinical expert CADTH consulted for this review estimated that a prevalence of 1 in 40,000 for all Pompe disease would be reasonable. For LOPD specifically, a study from the Netherlands estimated a prevalence of 1 in 57,000.5 The sponsor indicated that there were |||| patients with LOPD in Canada receiving treatment with alglucosidase alfa (|||| adults and |||| children) as of December 2020.6 The incidence of all Pompe disease (both LOPD and IOPD) has been estimated to be between 1 in 14,000 and 1 in 300,000, depending on geographic location and ethnicity,3 whereas the incidence of LOPD has been estimated to be 1.75 in 100,000 births.7 A study using data from births between 1969 and 1996 in British Columbia estimated the incidence of Pompe disease to be 1 in 115,091.8 It is expected that this is an underestimate of the true number of patients with LOPD in Canada because many would be have been undiagnosed at the time of the study. No updated prevalence or incidence data specific to Canada have been identified.
Standards of Therapy
Clinicians consulted by CADTH for the purpose of this review indicated that enzyme-replacement therapy with alglucosidase alfa, a rhGAA, at a dose of 20 mg/kg by IV infusion every 2 weeks, is the standard therapy and the only specific treatment for Pompe disease and LOPD, although it is not a cure. The exogenous source of GAA helps to break down glycogen, alleviate symptoms, and slow disease progression. The clinician input describes potential concerns about immune-related intolerance and reduced efficacy related to enzyme-replacement therapy. Aside from enzyme-replacement therapy, supportive care includes continued monitoring of pulmonary function and motor performance to assess new or increased need for ventilatory support and mobility aids. Canadian guidelines for the diagnosis and management of Pompe disease state that there is no evidence for the use of enzyme-replacement therapy in patients who have confirmed Pompe disease but are otherwise asymptomatic.9
Supportive therapies also include exercise and dietary changes, and new disease-specific therapies include other forms of enzyme-replacement therapy, and gene therapies are in development. Evidence has indicated that submaximal exercise may be beneficial3,4 for preserving muscle function, under careful guidance from a patient’s health care team (in particular, a physical therapist and pulmonologist), but strenuous exercise should be avoided.4 Although there are no established guidelines, it has been suggested that a high-protein diet and nutritional optimization may also be beneficial. Other interventions, such as physical therapy, occupational therapy, speech therapy, and assistive technological devices, can be used to support respiratory and motor function and attempt to improve HRQoL.3,4 Input from the clinical expert indicated that beta-2 agonists have been used off-label to try to increase the efficacy of enzyme-replacement therapy, although this is outside of the indication approved by Health Canada.
The clinical expert emphasized that the main goals of currently available forms of treatment are to stabilize and/or improve motor and respiratory function, as well as to prevent further disease progression. New treatments should improve immune tolerance, have a low risk of treatment-related reactions, and be less of a burden on patients; current infusions are frequent and require hours to complete.
As highlighted in the clinician input, a multidisciplinary team of specialists — including neurologists, metabolic geneticists, internists, orthopedists, cardiologists, respirologists, dieticians, and physical therapists — is required for the management of Pompe disease.
Drug
The key characteristics of avalglucosidase alfa and alglucosidase alfa are summarized in Table 3. Avalglucosidase alfa is a rhGAA used to break down lysosomal glycogen.10 The drug provides an exogenous source of GAA enzyme-replacement therapy with an approximate 15-fold increase in mannose-6-phosphate, compared with alglucosidase alfa. The increased mannose-6-phosphate moieties provide a mechanism to drive uptake of avalglucosidase alfa into the diaphragm and other skeletal muscle through the cation-independent receptor.
Avalglucosidase alfa received a Health Canada Notice of Compliance on November 12, 2021. The drug is intended for long-term, chronic use under the guidance and supervision of a health care professional who is knowledgeable in the treatment of Pompe disease. Avalglucosidase alfa is available as a lyophilized powder in a 100 mg vial that is reconstituted, diluted, and administered via IV infusion by a health care professional in a hospital or an appropriate outpatient-care setting. The recommended dose for patients with LOPD is 20 mg/kg body weight every other week. The product monograph recommends that infusions be administered over approximately 4 to 7 hours at an initial rate of 1 mg/kg per hour, which is increased by 2 mg/kg per hour every 30 minutes to a maximum rate of 7 mg/kg per hour if there are no signs of infusion-associated reactions and the patient responds well and is comfortable. An infusion should be immediately stopped if anaphylaxis, severe hypersensitivity reaction, or severe infusion-associated reaction occurs. The infusion rate may be slowed or temporarily stopped if mild-to-moderate hypersensitivity reactions or infusion-associated reactions occur. Patients may be pre-treated with antihistamines, antipyretics, and/or corticosteroids to prevent or reduce allergic reaction.
Avalglucosidase alfa underwent a standard review at Health Canada, and the manufacturer has requested reimbursement for the approved Health Canada indication. Avalglucosidase alfa has not been previously reviewed by CADTH.
Table 3: Key Characteristics of Avalglucosidase Alfa and Alglucosidase Alfa
Characteristic | Avalglucosidase alfa | Alglucosidase alfa |
|---|---|---|
Mechanism of action | Avalglucosidase alfa is a rhGAA that provides an exogenous source of GAA that cleaves alpha-1,4 and alpha-1,6 linkages of glycogen in the lysosome. Avalglucosidase alfa is a modification of alglucosidase alfa in which approximately 7 hexamannose structures, each containing 2 terminal M6P moieties, are conjugated to oxidized sialic acid residues on alglucosidase alfa, resulting in a 15-fold increase in M6P moieties, compared with alglucosidase alfa. This facilitates greater uptake into the diaphragm and other skeletal muscle via the cation-independent M6P receptor. | Alglucosidase alfa is a rhGAA that provides an exogenous source of GAA that cleaves alpha-1,4 and alpha-1,6 linkages of glycogen in the lysosome |
Indicationa | For the long-term treatment of patients with late-onset Pompe disease | For use in patients with Pompe disease (GAA deficiency) |
Route of administration | IV infusion | IV infusion |
Recommended dose | 20 mg/kg of body weight administered every other week | 20 mg/kg of body weight administered every other week |
SAEs or safety issues |
|
|
GAA = acid alpha-glucosidase; M6P = mannose-6-phosphate; rhGAA = recombinant human acid alpha-glucosidase; SAE = serious adverse event.
aHealth Canada–approved indication.
Source: Avalglucosidase alfa product monograph10; alglucosidase alfa product monograph.15
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups. Refer to Appendix 1 for the full Patient Group Input.
Muscular Dystrophy Canada, in partnership with the Canadian Association for Pompe, conducted a survey (available in both English and French) and semi-structured phone or Zoom video interviews with adults or the parents or caregivers of children living with Pompe disease. In total, 41 individuals affected by Pompe disease provided information for the submission. Of the respondents, 26 were patients (12 males and 14 females) and 11 identified as parents or caregivers. Patients were 4 to 81 years of age.
The negative impact Pompe disease has on motor ability (including mobility, strength, balance, and energy) and breathing were the issues most frequently reported by respondents. Quality of life was also important to the patient group, and the detrimental effects on patients’ social health, mental health, and ability to participate in daily activities, and on their families, were identified as key issues related to Pompe disease.
Some respondents reported having no experience with medications for Pompe disease (because of allergic reaction to treatment) and focusing on physical therapy, whereas others described being on enzyme-replacement therapy for years. Some patients treated with alglucosidase alfa described minor improvements followed by a plateauing of effect, whereas others reported major improvements. A few respondents also mentioned making high-protein and low-carbohydrate dietary changes.
Patients and caregivers would like new treatments to improve strength and breathing function and prevent disease progression, without the plateauing effect many have described. They would also like a better mode of delivery (e.g., oral medication, shorter infusion time), fewer side effects, a treatment that has a continuous effect in the body, and a reduction in costs associated with treatment and travel.
Two adults who reported clinical trial experience with avalglucosidase alfa had been receiving enzyme-replacement therapy for 2 to 3 years. During this time, they noticed improvements in mobility, balance, and endurance. Neither reported significant breathing issues before treatment. One patient reported no side effects, other than a single allergic reaction after the first few treatments, which was treated with medication and did not recur. The other patient reported no side effects related to treatment, but took medications for nausea and pain and an antihistamine before infusion. The greatest benefits of enzyme-replacement therapy on the patients’ lives were improvements in daily living and mental health. One patient reported having to travel out of province for treatment, which had an impact on their education for a couple of years.
All patients from the group submission reported undergoing diagnostic testing with blood testing, and some also underwent confirmatory biopsies. In general, patients did not have to pay for testing, although some did incur costs associated with having to travel to appointments in different cities. Some respondents described no delays in testing, but others faced multiple tests or significant wait times before receiving a diagnosis, and many patients recalled the stress of being misdiagnosed.
Clinician Input
Input From the Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol; assisting in the critical appraisal of clinical evidence; interpreting the clinical relevance of the results; and providing guidance on the potential place in therapy). The following input was provided by 1 clinical specialist with expertise in the diagnosis and management of LOPD.
Unmet Needs
The clinical expert consulted by CADTH stated that, given the biology of the disease, the most important goals of currently available forms of treatment are to stabilize and/or improve motor and respiratory function, as well as to prevent disease progression. Although it would be ideal if muscle functional deficits present at the time of diagnosis could be completely reversed, novel forms of therapy that affect cell growth and regenerative capacity will be required to meet this goal. Therapies should also have minimal burden on patients (e.g., shorter infusion time) and have a low risk of infusion-related reactions, which are unaddressed by alglucosidase alfa, the currently available enzyme-replacement therapy for Pompe disease.
Place in Therapy
The clinical expert expects that avalglucosidase alfa will replace alglucosidase alfa as the first-line treatment for LOPD, and that all patients who meet the criteria for treatment will receive the new drug. This would include patients who have never received enzyme-replacement therapy, and those already being treated with alglucosidase alfa would be switched over to avalglucosidase alfa. The clinical expert agrees that evidence from the COMET trial supports the noninferiority conclusion of avalglucosidase alfa, compared with alglucosidase alfa, and that short-term data show that avalglucosidase alfa has lower levels of immunogenicity.
Patient Population
Patients with Pompe disease are identified with genetic testing and enzymatic testing. The clinical expert noted that enzymatic testing that uses dried blood spots is more accessible than tests using white blood cells, although the former can produce false-positive results, which emphasizes the importance of additional genetic testing for confirmation. The clinical expert also indicated that a free multigene panel provided by the drug manufacturer includes testing for Pompe disease. This has allowed clinicians to screen and identify potential patients before they develop symptoms that are specific to Pompe disease, which may facilitate diagnosis at an earlier stage in the disease. It is hoped that easy access to such multigene panels for the diagnosis of myopathies will allow clinicians to speed up the diagnostic process.
According to the clinical expert, any patients with symptomatic disease should be treated with enzyme-replacement therapy. Although the heterogeneity of LOPD clinical presentation precludes treatment in the primary-prevention setting, patients without symptoms should be closely monitored for early signs of disease progression. This is important because response to treatment is expected to be better in patients who have less-severe disease. Patients with very advanced disease, such as those who are wheelchair-bound and on permanent invasive ventilation, may be least suited for avalglucosidase alfa, although the clinical expert added that clinical context should be considered on a case-by-case basis.
Assessing Response to Treatment
Canadian evidence-based guidelines9 for the treatment of LOPD emphasize the importance of having and meeting clearly defined, objective outcomes and tracking progression, the clinical expert reported. Assessments for skeletal muscle function (e.g., 6MWT, quantitative muscle strength scoring) and respiratory muscle function (e.g., FVC, MIP, MEP, change in FVC between upright and supine positions) were noted as relevant outcomes in clinical trials. However, the clinical expert acknowledged that not all tests can be performed in all clinics, particularly if they are smaller clinics or in remote locations. Consequently, it is recommended that patients are followed at least annually at a regional centre of excellence.
The clinical expert emphasized that stabilization of current mobility and pulmonary function is the main goal of currently available forms of therapy. Improved survival, use of mobility aids, ability to perform activities of daily living, frequency of and complications from falls, time lost from work or school, hospitalizations for pulmonary complications, and the need for ventilatory support are considered clinically meaningful measures, if available. Alternatively, surrogate outcomes such as the 6MWT, which can be easily measured, and changes in pulmonary function, which are strongly correlated with pulmonary outcomes, should be tracked.
Patients who begin a new therapy should initially be evaluated every 6 months, whereas patients on long-term treatment who remain stable should be assessed at least annually. For patients who live in remote areas, it may be acceptable to have a detailed annual assessment at an expert centre along with visits every 6 months with a local physician. Additionally, those who switch treatment from alglucosidase alfa to avalglucosidase alfa should be evaluated every 6 months to ensure they are stable on the new medication. Patients who have a high antibody titre or who are dosed based on body mass index (BMI) may require more frequent follow-ups to assess efficacy.
Discontinuing Treatment
Most patients are treated with enzyme-replacement therapy until they develop end-stage disease, which could include wheelchair requirements and full-time invasive ventilation. Anaphylactic reaction to the medication that cannot be managed with premedications, as well as comorbidities that significantly reduce lifespan (e.g., cancer), might be reasons to discontinue treatment.
Prescribing Conditions
According to the clinical expert, new patients often start treatment in a hospital clinical setting and, once stable, transition to home infusions. Patients may also be treated at infusion centres or started immediately with home infusions, depending on patient characteristics, available resources, and the physician’s comfort with the medication. The clinical expert expects that the same approach would be used for avalglucosidase alfa, although some patients who switch from avalglucosidase alfa may be closely monitored and receive infusions in the centres. Post-infusion follow-up would always be performed at a centre with expertise in the management of patients with Pompe disease.
In addition to a patient’s primary care physician, the multidisciplinary team may include neurologists, respirologists, lysosomal disease experts, physiotherapists, relevant specialists for the treatment of related complications, and medical geneticists who can assist with genetic counselling and the screening of family members.
Additional Considerations
The clinical expert highlighted the importance and use of consistent criteria across Canada for the treatment of LOPD, particularly for symptomatic patients, as there is currently a lack of uniformity in this regard.
Clinician Group Input
This section was prepared by CADTH staff based on the input provided by clinician groups. Refer to Appendix 2 for the full Clinician Group Input.
CADTH received a submission from the Neuromuscular Disease Network for Canada and from clinicians who treat Pompe disease. The 8 authors consisted of doctors who specialize in neurology, respirology, pediatrics, and clinical biochemical or metabolics genetics who practice in Alberta, British Columbia, Ontario, Québec, and Saskatchewan. The Neuromuscular Disease Network for Canada strives to be a comprehensive, inclusive, and enduring network for Canadian stakeholders working to improve the care, research, and treatment of neuromuscular diseases for all Canadians.
Unmet Needs
Ideally, new treatments would effectively stabilize and/or improve mobility and breathing and stop disease progression. The input highlighted that patients with LOPD have varied responses to enzyme-replacement therapy, and that most show a small improvement, plateau effect of variable duration, and eventual disease progression. The clinician group suggested that although the clinical evidence does not immediately rule out any particular group of patients, those who do not tolerate or progress while on alglucosidase alfa should have priority access to avalglucosidase alfa.
Place in Therapy
The group suggested that avalglucosidase alfa would likely become the first-line standard of care, replacing alglucosidase alfa, for the treatment of all patients with LOPD.
Patient Population
Patients who are best suited for treatment with avalglucosidase alfa can be identified through DNA analysis and GAA enzyme activity. Patients who are least suited to avalglucosidase alfa may be those who experience a new AE or SAE after switching from alglucosidase alfa; a switch back to alglucosidase alfa might be considered for these patients. Furthermore, the clinician input noted that evidence from the NEO1 and NEO-EXT studies showed that patients younger than 50 years with LOPD could have similar or greater improvements with avalglucosidase alfa than with alglucosidase alfa for key outcomes, such as motor and respiratory function, safety, and HRQoL. Although any patient with Pompe disease could benefit from the new therapy, the clinician group drew attention to the need for effective treatment for patients who have progressed on alglucosidase alfa.
Assessing Response to Treatment
The 6MWT, manual muscle testing, and hand-held dynamometry (HHD), and FVC (% predicted) were identified as common outcome measures used in clinical practice to follow patients with Pompe disease. Improvement or stability in 6MWT and FVC measures would be considered clinically meaningful responses to treatment. According to the clinician group, treatment response should be assessed every 6 to 12 months, depending on clinical severity.
Discontinuing Treatment
The clinician group suggested that in the case of clinical decline to the point of severe motor or respiratory disability (e.g., nonambulatory or noninvasive ventilation while awake), when the patient no longer derives benefit from treatment, or severe AE(s), discontinuation of treatment can be considered.
Prescribing Conditions
The clinician group stated that outpatient infusion clinics or home infusion would be appropriate settings for treatment administration. The group also noted that treatment and monitoring should be conducted by a clinical biochemical or metabolics geneticist or a neuromuscular specialist, and acknowledged that patients in remote areas could be monitored by local physicians with regular evaluations by a neuromuscular specialist.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that might impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Initiation | |
The product monograph refers to patients 6 months and older. Would patients who are younger than 6 months be treated with avalglucosidase alfa? | The clinical expert consulted by CADTH indicated that in practice, avalglucosidase alfa might be prescribed for infants younger than 6 months, owing to the drug’s possible lower immunogenicity and possible improved efficacy, compared with alglucosidase alfa; however, the clinical expert acknowledged that this would represent off-label use, and highlighted the urgent need for data on the product in the IOPD population. |
Should patients unresponsive to alglucosidase alfa be considered for avalglucosidase alfa therapy? | Patients who progress while receiving alglucosidase alfa may be switched to avalglucosidase alfa and may still respond on the new treatment. |
Would treatment with avalglucosidase alfa be lifelong? | Treatment with avalglucosidase alfa is expected to continue until the patient has declined to the point that there is no longer benefit (e.g., nonambulatory with permanent invasive ventilation). |
In the recommendation for alglucosidase alfa, CDEC recommended that drug plans develop specific criteria for the monitoring and stopping alglucosidase alfa, in consultation with experts in the management of lysosomal storage disease. Should the recommendation for avalglucosidase alfa be consistent with that issued by CEDAC for alglucosidase alfa? | The clinical expert stated that the criteria used for avalglucosidase alfa should be consistent with alglucosidase alfa, as outlined in the remaining sections of this table. |
Continuation or renewal | |
Currently listed agents do not require assessment of response for continued therapy. There are no renewal criteria provided by CADTH for alglucosidase alfa. Should there be renewal criteria for avalglucosidase alfa? | Consistent with clinical practice guidelines9 for the diagnosis and management of Pompe disease, the clinical expert noted that a trial of enzyme-replacement therapy should be offered to patients with LOPD who demonstrate clinical signs and symptoms of the disease, are ambulatory, and are either nonventilated or on noninvasive ventilation when asleep. Furthermore, a trial of enzyme-replacement therapy may be considered for patients with LOPD who are nonambulatory and/or receive noninvasive ventilation while awake, or invasive ventilation, if there are predefined skeletal muscle outcomes that can be evaluated, which, if achieved, would improve the functional status of the patient. If the trial does not result in the pre-specified outcomes, then the trial should be discontinued. The guidelines also suggest that patients be monitored for the development of respiratory complications and undergo regular pulmonary function tests every 6 to 12 months. Additionally, the goals of care should be reviewed on a regular basis and with interval changes in health. When disease control is no longer an objective, discontinuation of enzyme-replacement therapy, supportive care, and palliative measures should be available to patients. |
Discontinuation | |
Alglucosidase alfa does not have discontinuation criteria. Should there be discontinuation criteria for avalglucosidase alfa? | The clinical expert noted that jurisdictions currently have different discontinuation criteria for alglucosidase alfa, and emphasized that it would be beneficial to have uniform renewal and discontinuation criteria for avalglucosidase alfa across Canada. Consistent with clinical practice guidelines9 for the diagnosis and management of Pompe disease, the clinical expert agreed that discontinuation may be considered if a patient has severe infusion-related reactions that are not amenable to therapy and that compromise patient safety; has an estimated life expectancy (e.g., owing to comorbidities or advanced disease stage); is noncompliant with infusions and recommended assessments; and has a rate of decline in skeletal and/or pulmonary function after enzyme-replacement therapy initiation that is similar to that seen before the use of enzyme-replacement therapy. The clinical expert also noted that there may be situations in which patients can be reviewed on a case-by-case basis. |
Prescribing | |
Who should be able to prescribe avalglucosidase alfa? Should the prescribing criteria for avalglucosidase alfa be aligned with the prescribing criteria for alglucosidase alfa criteria? | Prescription of avalglucosidase alfa should be restricted to those with experience treating lysosomal storage diseases or other types of neuromuscular diseases. The clinical expert stated that prescribing criteria for avalglucosidase alfa should be aligned with that for alglucosidase alfa, and for LOPD rather than IOPD. |
Generalizability | |
Are data available regarding switching from alglucosidase alfa to avalglucosidase? | Data are available from the phase III COMET trial (and the open-label extension with data up to 97 weeks) for patients who received alglucosidase alfa for 1 year and switched to avalglucosidase alfa. Data for switching treatments are also available from the phase I NEO1 and phase II NEO-EXT studies, in which a subset of patients who had experience with alglucosidase alfa before enrolment were given avalglucosidase alfa, with data up to approximately 240 weeks. |
CDEC = Canadian Drug Expert Committee; CEDAC = Canadian Expert Drug Advisory Committee; IOPD = infantile-onset Pompe disease; LOPD = late-onset Pompe disease.
Clinical Evidence
The clinical evidence included in the review of avalglucosidase alfa is presented in 2 sections. The first section, the systematic review, includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those studies that were selected in accordance with an a priori protocol. The second section includes sponsor-submitted long-term extension studies and additional relevant studies that address important gaps in the evidence included in the systematic review. No indirect evidence was identified from the literature that met the selection criteria specified in the review.
Systematic Review (Pivotal and Protocol Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of avalglucosidase alfa administered once every other week as an IV infusion of 20 mg/kg of body weight for the treatment of LOPD.
Methods
Studies selected for inclusion in the systematic review included pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 5. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
Table 5: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Population | Patients with LOPD Subgroups:
|
Intervention | Avalglucosidase alfa every other week as an IV infusion, given at 20 mg/kg of body weight |
Comparator |
|
Outcomes | Efficacy outcomes:
Harms outcomes:
|
Study designs | Published and unpublished phase III and IV RCTs |
6MWT = 6-minute walk test; AE = adverse events; FVC = forced vital capacity; HRQoL = health-related quality of life; RCT = randomized controlled trial; SAE = serious adverse events; SF-12 = 12-item Short Form Health Survey; WDAE = withdrawal due to adverse events.
The literature search for clinical studies was performed by an information specialist who used a peer-reviewed search strategy in accordance with the PRESS Peer Review of Electronic Search Strategies tool.16
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946—) via Ovid, and Embase (1974—) via Ovid. All Ovid searches were run simultaneously as a multi-file search. Duplicates were removed using Ovid deduplication for multi-file searches, followed by manual deduplication in Endnote. The search strategy comprised controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concepts were Nexviazyme (avalglucosidase alfa). The following clinical trials registries were searched: the US National Institutes of Health’s clinicaltrials.gov, WHO’s International Clinical Trials Registry Platform search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
No filters were applied to limit the retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. See Appendix 3 for the detailed search strategies.
The initial search was completed on October 29, 2021. Regular alerts updated the search until the meeting of the CADTH Canadian Drug Expert Committee on February 23, 2022.
Grey literature (literature that is not commercially published) was identified through searches of relevant websites from the Grey Matters: A Practical Tool For Searching Health-Related Grey Literature tool.17 Included in this search were the websites of regulatory agencies (FDA and European Medicines Agency). Google was used to search for additional internet-based materials. See Appendix 3 for more information on the grey literature search strategy.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, in accordance with the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
Findings From the Literature
A total of 6 studies were identified from the literature for inclusion in the systematic review (Figure 1). The included studies are summarized in Table 6. A list of excluded studies is presented in Appendix 2.
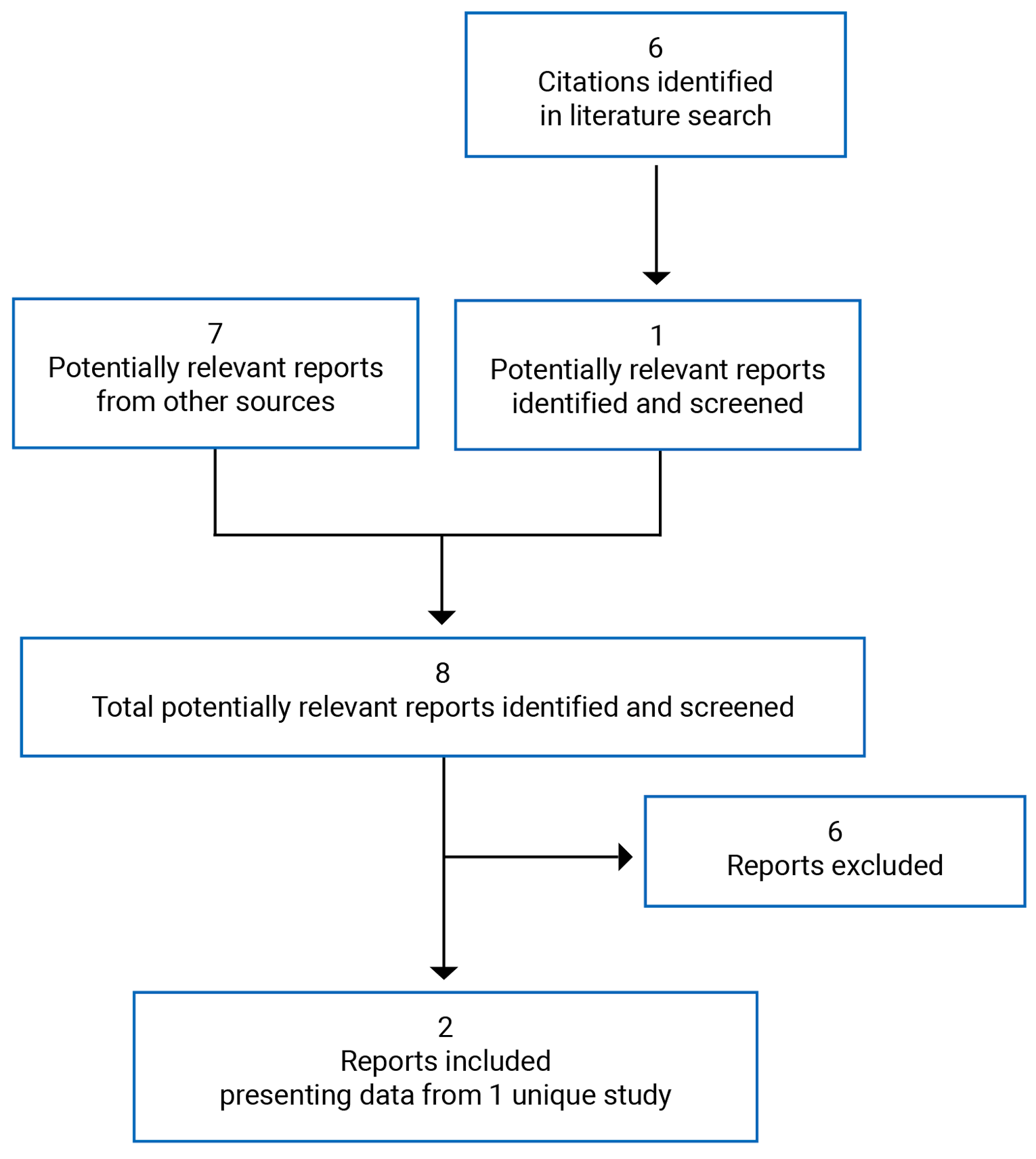
Table 6: Details of Included Studies
Detail | COMET |
|---|---|
Designs and populations | |
Study design | Phase III, double-blind RCT with active-control and parallel groups |
Locations | 55 centres in 26 countries in North America (including Canada), Asia, Australia, Europe, and South America |
Patient enrolment dates | November 2, 2016, to March 19, 2020 |
Randomized (N) | N = 100
|
Inclusion criteria |
|
Exclusion criteria |
|
Drugs | |
Intervention | Avalglucosidase alfa 20 mg/kg of body weight every other week as an IV infusion |
Comparator(s) | Alglucosidase alfa 20 mg/kg of body weight every other week as an IV infusion |
Duration | |
Screening phase | Up to 14 days |
Double-blind phase | 49 weeks |
Open-label extension phase (alglucosidase alfa crossover to avalglucosidase alfa) | Up to 240 weeks |
Post-treatment follow-up phase | Up to 4 weeks |
Outcomes | |
Primary end point | Change from baseline to week 49 in FVC (% predicted) in the upright position |
Secondary and exploratory end points | Secondary:
Tertiary:
Exploratory:
|
Notes | |
Publications | Diaz-Manera et al. (2021)13 |
6MWT = 6-minute walk test; EQ-5D-5L = 5-Level-EuroQoL; FVC = forced vital capacity; GAA = acid alpha-glucosidase; GMFCS = Gross Motor Function Classification System; GMFM-88 = Gross Motor Function Measure-88; GSGC = gait, stair, Gower’s maneuver, and chair; HHD = hand-held dynamometry; HRQoL = health-related quality of life; MEP = maximum expiratory pressure; MIP = maximum inspiratory pressure; QMFT = Quick Motor Function Test; RCT = randomized controlled trial; SF-12 = 12-item Short Form Health Survey.
aKey secondary efficacy outcome.
Note: One additional report was included (COMET Clinical Study Report).
Source: COMET Clinical Study Report,11 Diaz-Manera et al. (2021).13
Description of Studies
One pivotal study was included in the CADTH review for avalglucosidase alfa. The COMET trial was a multi-centre, double-blind, active-control, phase III RCT designed to evaluate the efficacy and safety of avalglucosidase alfa 20 mg/kg body weight given every other week for the treatment of LOPD. The COMET trial was conducted internationally, with 2 study centres in Canada. The study design is outlined in Figure 2: Diagram of COMET Trial and consisted of a screening period of up to 14 days, a double-blind treatment period of 49 weeks, an open-label extension phase of up to 240 weeks, and follow-up for up to 4 weeks.
In the COMET trial, 100 patients were randomized, using a centralized treatment allocation system in a 1:1 ratio, to either avalglucosidase alfa or alglucosidase alfa. Randomization was stratified based on baseline FVC (% predicted: < 55% versus ≥ 55%), sex, age (< 18 years versus ≥ 18 years), and country (Japan versus not Japan). To control the number of patients with high baseline FVC (% predicted), the number of patients with baseline FVC (% predicted) between 80% and 85% was capped at 15% of the total population. Patients, study investigators, and site personnel were blinded to the treatment assignment until the primary analysis was completed. Independent pharmacists or designees who prepared the study drugs were unblinded. In the event of an AE, the randomization code was broken when knowledge of the study drug was deemed necessary to treat the patient. When the treatment assignment was revealed, the date, time, and reason were documented. Furthermore, the patient was required to withdraw from study drug administration.
The interim clinical study report data cut-off date was March 19, 2020, which included all data from the double-blind phase, followed by a database lock on April 23, 2020. At the end of the double-blind phase, patients in the alglucosidase alfa arm were switched to avalglucosidase alfa for the duration of the open-label treatment phase. There was no washout period for patients who switched treatments.
||||| |||| | |||||||||| || ||| |||||||| |||| ||||||| ||||||| || ||| ||||||||||| |||||||| ||| |||||||| ||| ||||||| ||| ||| || |||||||||| |||| ||| || |||| || | ||||||| ||| |||||| |||| ||||||||| |||| || |||||||| || || ||||||||| | ||||| ||||||||| |||||||| ||| |||| ||||| ||||| || || ||| ||||| || ||||| |||||||| || |||||||| ||||||||| ||||||||| || ||||| ||||||||||||||| |||| ||| |||||||| ||| ||| || |||| |||||||| ||||| |||||||||| |||||||| |||||||| |||||| |||||||||| || ||||||||| |||||||| |||| ||| |||| ||||| ||||| |||| || ||| |||||||||| |||||| ||||||| ||||||||||||| |||||||| |||| ||||||||| |||||| ||| |||| ||||| ||||||||| |||||| ||| ||||||| ||||||| |||||||| ||||||||||||| ||| ||||||||||| ||||||| ||| ||||||| || |||||| ||| |||||||||||||| |||| || | ||||||||||| |||| ||| ||| |||| ||||| || |||||||| |||| |||||||||| ||||||||| || |||| || |||||| ||||||||||| ||||||| ||| ||| |||| || ||||||| |||| |||| ||||||||||| ||||| ||||||||||| |||| || |||||| |||||||| ||| |||||||| || |||||||| ||||| ||||| ||| || ||||||||||| |||||||||| |||| ||| |||||||||||| ||| ||||||| || ||||||| |||||
Figure 2: Diagram of COMET Trial
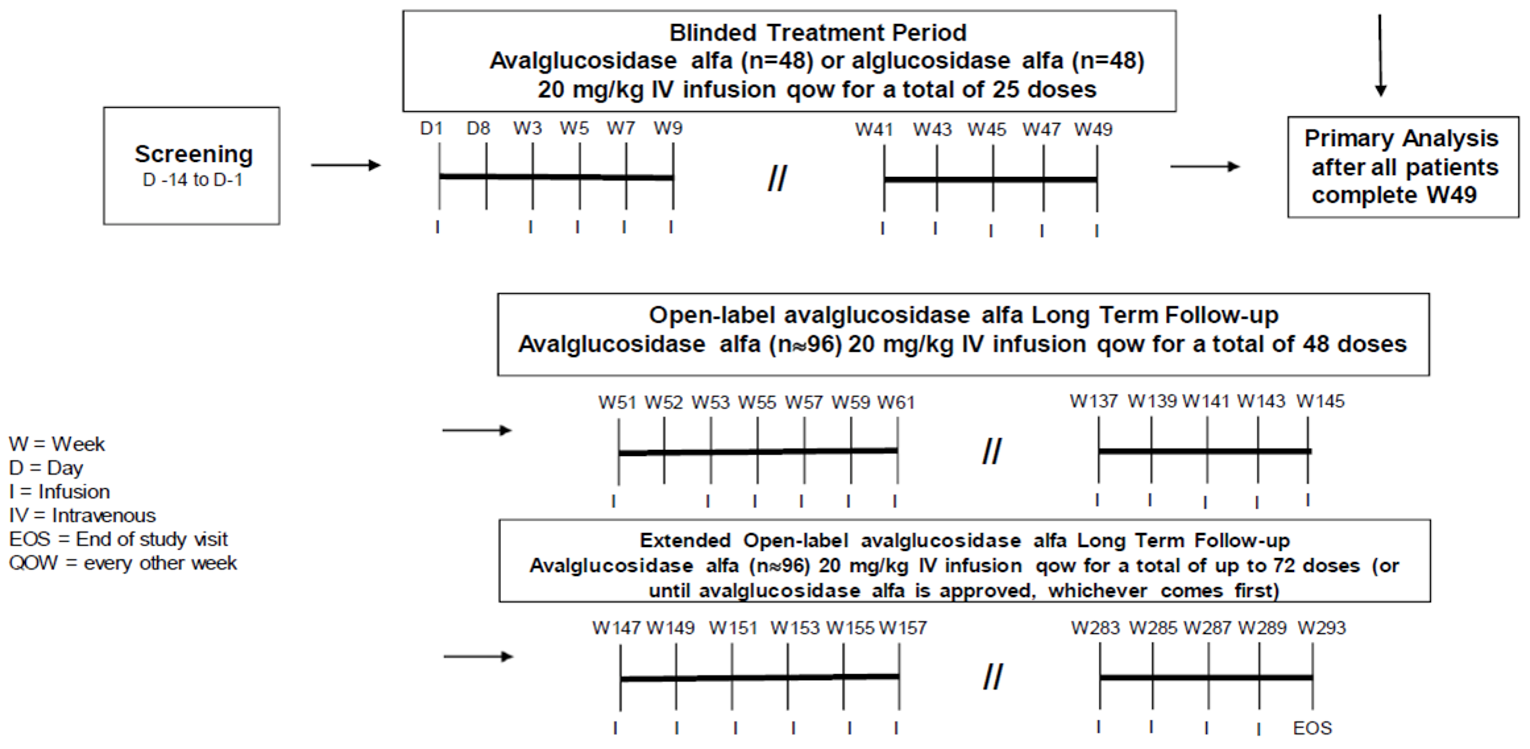
D = day; EOS = end-of-study; I = infusion; qow = every other week; W = week.
Source: COMET Clinical Study Report.11
Populations
Inclusion and Exclusion Criteria
Patients older than 3 years were eligible to participate in the COMET trial if they had a confirmed GAA enzyme deficiency from any tissue source and/or 2 confirmed GAA gene mutations. Patients with known Pompe-specific cardiac hypertrophy or with severe disease (wheelchair dependence, unable to ambulate 40 m without stopping and without an assistive device, requiring invasive ventilation, or unable to perform repeated FVC [% predicted] measurements between 30% and 85%) were excluded from the study. Previous treatment with alglucosidase alfa or other investigational treatments for Pompe disease were also reasons for exclusion. Patients were ineligible if they had significant organic disease or had previously used or were currently using immune tolerance induction therapy.
Baseline Characteristics
Patient baseline characteristics are summarized in Table 7. Overall, the mean age of the patients in the study was 48 years (SD = 14), and patients were older in the alglucosidase alfa group than in the avalglucosidase alfa group. Patients were predominantly White (94 patients [94%]), and there was a similar number of male and female patients within and between treatment arms.
Baseline disease characteristics appeared to be similar among the treatment groups, with a few exceptions. The baseline mean distance walked on the 6MWT was numerically higher for the avalglucosidase alfa group (399.3 m; SD = 110.9) than for the alglucosidase alfa group (378.1 m; SD = 116.2). Also, more patients reported using no mobility aids in the avalglucosidase alfa arm. Use of a rolling walker or a single crutch was higher in the comparator arm (6.1% and 4.1%, respectively) than in the avalglucosidase alfa group (0.0% and 0.0%, respectively). In contrast, other measurements were lower in the avalglucosidase alfa group than in the alglucosidase alfa group for mean MEP % predicted (65.77%; SD = 38.97 versus 74.83%; SD = 35.22) and HHD lower extremity composite scores (1,330.45; SD = 625.44 versus 1,466.16; SD = 604.91).
Patient medical history was similar between the study arms, with a few notable differences. The mean age at diagnosis for Pompe disease was lower for patients in the avalglucosidase alfa group (44.73 years; SD = 14.74) than for those in the alglucosidase alfa group (48.16 years; SD = 14.64). The time between diagnosis and first infusion of the study drug was shorter for the avalglucosidase alfa group (15.60 months; SD = 32.06) than for the alglucosidase alfa group (26.52 months; SD = 59.86).
Table 7: Summary of Baseline Characteristics — COMET mITT Population
Characteristic | Avalglucosidase alfa, N = 51 | Alglucosidase alfa, N = 49 |
|---|---|---|
Demographics | ||
Age (years) | NA | NA |
Mean (SD) | 46.0 (14.5) | 50.3 (13.7) |
Median (range) | 47.7 (16 to 78) | 48.9 (20 to 78) |
Sex, n (%) | NA | NA |
Male | 27 (52.9) | 25 (51.0) |
Female | 24 (47.1) | 24 (49.0) |
Race, n (%) | NA | NA |
White | 47 (92.2) | 47 (95.9) |
Asian | 3 (5.9) | 0 |
Black | 1 (2.0) | 2 (4.1) |
Weight, kg, mean (SD) | 77.8 (22.1) | 79.3 (18.2) |
BMI, kg/m2, mean (SD) | 26.39 (6.79) | 26.69 (5.42) |
Randomization strata, n (%) | NA | NA |
Age < 18 | | ||||| | ||||| |
Age ≥ 18, Japan | | ||||| | ||||| |
Age ≥ 18, male and FVC < 55%, ex-Japan | || |||||| | || |||||| |
Age ≥ 18, female and FVC < 55%, ex-Japan | | ||||| | | |||||| |
Age ≥ 18, male and FVC ≥ 55%, ex-Japan | || |||||| | || |||||| |
Age ≥ 18, female and FVC ≥ 55%, ex-Japan | || |||||| | || |||||| |
Baseline disease characteristics | ||
FVC % predicted, upright, mean (SD) | 62.5 (14.4) | 61.6 (12.4) |
6MWT | NA | NA |
Distance walked, m, mean (SD) | 399.3 (110.9) | 378.1 (116.2) |
% predicted, mean (SD) | 57.3 (15.0) | 55.3 (16.6) |
Use of mobility aid, n (%) | NA | NA |
None | 44 (86.3) | 39 (79.6) |
Straight cane | 4 (7.8) | 3 (6.1) |
Wide-based cane | 1 (2.0) | 1 (2.0) |
Rolling walker | 0 | 3 (6.1) |
One crutch | 0 | 2 (4.1) |
Other | 2 (3.9) | 1 (2.0) |
MIP % predicted, upright, mean (SD) | 59.9 (47.1) | 60.6 (41.0) |
MEP % predicted, upright, mean (SD) | 65.77 (38.97) | 74.83 (35.22) |
HHD (lower extremity), mean (SD) | NA | NA |
Composite score | 1,330.45 (625.44) | 1,466.16 (604.91) |
% predicted | 40.05 (21.76) | 45.02 (23.27) |
QMFT total score, mean (SD) | 41.29 (10.15) | 42.30 (10.58) |
SF-12, mean (SD) | NA | NA |
Physical component summary | 35.95 (7.82) | 36.76 (9.40) |
Mental component summary | 48.31 (10.11) | 50.58 (8.69) |
Medical history | ||
Age at diagnosis of Pompe disease, years, mean (SD) | 44.73 (14.74) | 48.16 (14.64) |
Time from diagnosis to first infusion of the study drug, months, mean (SD) | 15.60 (32.06) | 26.52 (59.86) |
Ear, nose, throat history, n (%) | NA | NA |
Enlarged tongue | | ||||| | | ||||| |
Respiratory history, n (%) | NA | NA |
Tracheostomy | | ||||| | ||||||| |
Pneumonia | || |||||| | | |||||| |
Sleep apnea | || |||||| | || |||||| |
Use of noninvasive respiratory support | || |||||| | || |||||| |
Musculoskeletal history, n (%) | NA | NA |
Muscle weakness in upper extremities | || |||||| | || |||||| |
Muscle weakness in lower extremities | || |||||| | || |||||| |
Ambulatory | || |||||| | || |||||| |
6MWT = 6-minute walk test; BMI = body mass index; FVC = forced vital capacity; HHD = hand-held dynamometry; MEP = maximum expiratory pressure; MIP = maximum inspiratory pressure; mITT = modified intention-to-treat; NA = not applicable; QMFT = Quick Motor Function Test; SD = standard deviation; SF-12 = 12-item Short Form Health Survey.
Source: COMET Clinical Study Report.11
Interventions
In the COMET trial, patients were randomized to receive either avalglucosidase alfa or alglucosidase alfa. Avalglucosidase alfa was supplied as a sterile, lyophilized product in single-use vial that contained 100 mg of avalglucosidase alfa. Alglucosidase alfa was supplied as a sterile, lyophilized cake or powder in a single-use vial that contained 50 mg of alglucosidase alfa. The number of vials needed depended on the patient’s most recent body weight, if it was measured in the previous 1 month for pediatric patients or in the previous 3 months for adult patients. Both drugs were reconstituted and diluted to a dose of 20 mg/kg body weight and administered by IV infusion every other week during the double-blind phase (up to week 49). The rate of infusion was started at a slow initial rate and gradually increased to a maximum rate if no signs of infusion-associated reactions were observed. The length of infusion depended on the dose of medication administered, and patients remained at the hospital or infusion centre for at least 2 hours after the infusion to monitor AEs. During the open-label extension phase, patients who received alglucosidase alfa were switched to avalglucosidase alfa for the remainder of their treatment (up to 120 doses) or until avalglucosidase alfa was approved in their country.
To maintain blinding of patients and investigators, an independent unblinded pharmacist who was not involved in AE assessments and who did not have access to efficacy data prepared the study drugs on site.
Infusions were scheduled in 14-day increments ± 7 days for infusions and safety assessments and ± 14 days for other assessments, but there had to be at least 7 days between any 2 infusions. Modifications to the dose or frequency of administration were only permitted when resulting from an AE, and doses above 20 mg/kg body weight every other week were not allowed.
Home infusions were permitted during the open-label extension phase, in accordance with local and national regulations, and patients must have met specified eligibility criteria. The investigator had to agree that home infusion was appropriate, the patient had to be willing and able to adhere to home infusion procedures and be trained in the process, and the patient had to be clinically stable on avalglucosidase alfa, with no moderate or severe infusion-associated reactions for at least 12 months on a stable dose. Patients could not have ongoing SAEs, and if a patient experienced a moderate or severe infusion-associated reaction at home, they returned to the study site for infusions until no infusion-associated reactions occurred for at least 3 months. Patients were to be monitored by trained staff for at least 2 hours after infusion.
Concomitant medications, preinfusion medications, and assistive mobility devices were recorded in the electronic case report form (e-CRF). The use of mechanical ventilation was reported on the e-CRF ventilator use form. Although the use of routine pre-treatment with antihistamines, antipyretics, and/or corticosteroids was not recommended, particularly for patients who experienced prior immunoglobulin E-mediated hypersensitivity reactions, pre-treatment was at the discretion of the investigator. It was noted that antihistamines could mask symptoms of hypersensitivity reactions and prevent intervention by infusion staff. Immunomodulating treatments (e.g., methotrexate, rituximab, immunoglobulins, and other immunosuppressants) that would interfere with the evaluation of the immunogenic potential of avalglucosidase alfa were prohibited.
Outcomes
A list of efficacy end points identified in the CADTH review protocol that were assessed in the clinical trials included in this review is provided in Table 8. These end points are further summarized in the text that follows the table. A detailed discussion and critical appraisal of the outcome measures is provided in Appendix 4.
Table 8: Summary of Outcomes of Interest Identified in the CADTH Review Protocol — COMET Trial
Outcome measure | Outcome |
|---|---|
Survival-related outcomes | Deaths noted in harms outcomes |
Respiratory-related outcomes | NA |
FVC (% predicted) | Primary |
Need for ventilation | Not an outcome |
MEP (% predicted) | Secondary |
MIP (% predicted) | Secondary |
Motor-related outcomes | NA |
6MWT (distance and % predicted) | Secondary |
Use of mobility aids | Not an outcome |
HHD (lower extremity muscle groups) | Secondary |
QMFT | Secondary |
GSGC | Tertiary |
GMFM-88 | Tertiary |
GMFCS | Tertiary |
HHD (upper extremity muscle groups) | Tertiary |
HRQoL | NA |
SF-12 | Secondary |
Pediatric Quality of Life Inventory | Tertiary |
EQ-5D-5L | Tertiary |
Patient-reported outcomes | NA |
Pompe Disease Impact Scale | Exploratory |
Pompe Disease Symptom Scale | Exploratory |
Rasch-built Pompe-specific Activity scale | Exploratory |
Patient Global Impression of Change | Exploratory |
Harms | Secondary |
6MWT = 6-minute walk test; EQ-5D-5L = 5-Level-EuroQoL; FVC = forced vital capacity; GMFCS = Gross Motor Function Classification System; GMFM-88 = Gross Motor Function Measure-88; GSGC = gait, stair, Gower’s maneuver, and chair; HHD = hand-held dynamometry; HRQoL = health-related quality of life; MEP = maximum expiratory pressure; MIP = maximum inspiratory pressure; NA = not applicable; QMFT = Quick Motor Function Test; SF-12 = 12-item Short Form Health Survey.
Efficacy Outcomes
FVC is the total volume of air that can be forcibly exhaled after maximal inspiration.11 Pulmonary function testing (including FVC) was performed locally at the study centre and measured by a central laboratory using a standardized protocol that met American Thoracic Society/European Respiratory Society 2005 quality standards. Patients were required to have at least 3 acceptable FVC manoeuvres with acceptable repeatability. Assessments were made before infusions and performed up to at least week 49. FVC in the upright position was reported in litres, and percent of predicted normal values based on age, sex, race, and height used Global Lung Initiative 2012 reference equations. Normal FVC values range from 80% to 120%;a lower FVC value may indicate abnormal lung function.18 Acceptable validity has been demonstrated in patients with Pompe disease.19 Good evidence of reliability and weak evidence of responsiveness were found for patients with pulmonary disorders, although no information was identified for patients with Pompe disease.20 No MID was identified from the literature for patients with Pompe disease.
The 6MWT is the distance a patient can walk on a hard, flat surface in 6 minutes, and was used to assess functional capacity.11 The distance covered was measured in m, and the amount of time walked was recorded (as a measure of endurance) for patients who did not complete the full 6-minute walk. Equipment and administration methods were standardized across study sites and testing was performed before infusions. The percent predicted value was calculated using reference equations applicable to the study population. Patients were permitted to use a walking device, which was recorded in the e-CRF, and any change in the use of mobility devices was also recorded. Validity and reliability of the 6MWT have been established in various patient populations.21-31 The 6MWT distance showed validity and responsiveness to change, compared with Rotterdam Handicap Scale and Rasch-built Pompe-specific Activity scale instruments, for patients with Pompe disease.19 No information on reliability and no MIDs were identified from the literature for patients with Pompe disease.
MIP and MEP are measures of the maximum negative or positive pressures from an inspiratory or expiratory effort, respectively. MIP and MEP (% predicted) measurements in the upright position were included with pulmonary function testing (described earlier) for the COMET trial and were performed and assessed in a similar manner. Evidence of validity and reliability for both MIP and MEP was found for patients with pulmonary disorders32 and neuromuscular disorders.33,34 For patients with Pompe disease, MIP showed acceptable validity and reliability,35,36 although no information was found that assessed responsiveness to change. No evidence of validity, reliability, or responsiveness was found for MEP for this patient population. No MID was identified from the literature for either MIP or MEP for patients with Pompe disease.
HHD for lower extremity muscle strength was conducted using the make technique.11 Equipment was standardized across sites and testing was performed before infusions. To prevent the use of compensatory muscles, stabilization procedures for all muscle groups were followed. During testing, the examiner holds the dynamometer stationary as the patient exerts a force that gradually increases to a maximal force, which is held for 4 to 5 seconds. Testing was conducted by the same physical therapist or trained assessor each time. The lower extremity muscle groups included hip flexion, hip extension, hip abduction, hip adduction, knee flexion, knee extension, ankle dorsiflexion, and ankle plantar flexion. Limb tests were completed bilaterally to account for differences between dominant and nondominant limbs. Muscle groups were tested twice and the higher value was reported in the e-CRF. Noninvasive ventilation was permitted during testing. Acceptable validity and reliability have been established in other patient populations.37-41 Scores for the HHD, Rotterdam Handicap Scale, and Rasch-built Pompe-specific Activity scale were significantly correlated among patients with Pompe disease, demonstrating evidence of validity among the outcome measures.19 Information that assessed reliability and responsiveness was not found for HHD, and no MID was identified from the literature for patients with Pompe disease.
The QMFT was used to evaluate changes in motor function concurrent with the GMFM-88, and testing was performed before infusions.11 The QMFT is an observer-administered assessment that consists of 16 items individually scored on a 5-point ordinal scale (ranging from 0 to 4). The total score for all items ranges from 0 to 64 points, and a higher score indicates greater motor function. Evidence of validity and reliability has been established among patients with Pompe disease,42 but no information that evaluated responsiveness was found. No MID was identified from the literature for patients with Pompe disease.
The SF-12, version 2.0, is a questionnaire that consists of 4 2-item health domain scales (physical functioning, role-physical, role-emotional, and mental health) and 4 1-item health domain scales (bodily pain, general health, vitality, and social functioning).11 Similar to the original 36-item Short Form Health Survey (SF-36), the items on the SF-12 are used to reproduce physical component summary and mental component summary scores, which are computed as weighted sums and range from 0 to 100, with higher scores indicating better HRQoL.43 In the COMET trial, the SF-12 was used to assess HRQoL in patients 18 years and older and was measured before infusions, when possible. Validity, reliability, and responsiveness have been demonstrated in various patient populations,44-46 although no evidence was found for patients with Pompe disease. No MID was identified from the literature for patients with Pompe disease.
The GSGC consists of 4 functional tests — walk 10 m (gait), climb 4 stairs (stair), stand from sitting on the floor (Gower’s maneuver), and stand from sitting position (chair) — used to evaluate functional performance.11 The first 3 tests are scored from 1 to 7, whereas the last test (chair) is scored from 1 to 6. A total score, computed from the sum of the 4 tests, ranges from 4 (normal performance) to 27 (poor functional score). The 4 functional tests were also evaluated for the time required to complete each task. Assessments were performed before infusions. Evidence of validity and correlation with the 6MWT and Walton and Gardner-Medwin scale (a measure of functional status) has been established in patients with LOPD.47 No information on reliability or responsiveness and no MID was identified from the literature for patients with Pompe disease.
The GMFM-88 was used to evaluate changes in gross motor function, using 2 of the 5 dimensions: dimension D (standing; 13 items) and dimension E (walking, running, and jumping; 24 items).11 Each item was scored on a 4-point Likert scale (0 = cannot complete, 1 = initiates [< 10% of the task], 2 = partially completes [10% to < 100% of the task], and 3 = task completed). The score for each dimension was expressed as a percentage of the maximum score for the dimension, and a total score was computed from the sum of the percentage scores for each dimension and dividing by the number of dimensions (i.e., each dimension contributes equally to the total score). A higher score indicates greater motor function. Testing was performed before infusions. Validity, reliability, and responsiveness have been established in children with cerebral palsy,48 but no evidence was found for patients with Pompe disease. No MID was identified from the literature for patients with Pompe disease.
Because tertiary and exploratory outcomes are descriptive and hypothesis-generating, the Gross Motor Function Classification System (GMFCS), HHD (upper extremity muscle strength), and 5-Level-EuroQoL (EQ-5D-5L) were not reviewed in the CADTH report. A summary of the data for these outcomes has been included in Table 24. Upon review of the limited available data and discussion with the clinical expert CADTH consulted, the description, data, and critical appraisal of the following tertiary and exploratory outcomes have not been included in this report: Pediatric Quality of Life Inventory, Pompe Disease Impact Scale, Pompe Disease Symptom Scale, Rasch-built Pompe-specific Activity scale, and Patient Global Impression of Change.
Harms Outcomes
Incidence and seriousness of AEs, withdrawals due to AEs, and deaths were reported for the double-blind phase for all patients. AEs, SAEs, and AEs related to hypersensitivity and anaphylactic reactions were described based on the preferred term and associated system organ class, using the most recent version of the standardized medical dictionary for regulatory activity (MedDRA) Query (SMQ). Infusion-associated reactions were defined as AEs of special interest that occurred during the infusion or post-infusion observation period that were possibly related to the study drug.
For immunogenicity testing of anti-drug antibodies, samples were taken from patients every month until week 73 and every 3 months thereafter for the duration of the study. Samples were also collected from all patients at week 2 and week 52 when patients switched from alglucosidase alfa to avalglucosidase alfa. Patients were tested for anti-drug antibodies to the study drug they received during the double-blind phase; patients who switched treatment were tested for anti-drug antibodies to both alglucosidase alfa and avalglucosidase alfa during the open-label phase until week 145, after which testing was performed for anti-drug antibodies to avalglucosidase alfa only.
Statistical Analysis
The statistical analysis of efficacy end points conducted in the COMET trial is summarized in Table 9.
Table 9: Statistical Analysis of Efficacy End Points — COMET Trial
End point | Statistical model | Adjustment factors | Sensitivity analyses |
|---|---|---|---|
Change from baseline FVC % predicted | MMRM |
|
|
Change from baseline in total distance walked in 6MWT | MMRM |
|
|
Change from baseline in MIP % predicted | MMRM |
|
|
Change from baseline in MEP % predicted | MMRM |
|
|
Change from baseline in HHD lower extremity muscle strength composite score | MMRM |
|
|
Change from baseline in QMFT total score | MMRM |
|
|
Change from baseline SF-12 physical component summary | MMRM |
|
|
Change from baseline SF-12 mental component summary | MMRM |
|
|
6MWT = 6-minute walk test; ANCOVA = analysis of covariance; FVC = forced vital capacity; HHD = hand-held dynamometry; MEP = maximum expiratory pressure; MIP = maximum inspiratory pressure; MMRM = mixed model for repeated measures; QMFT = Quick Motor Function Test; SF-12 = 12-item Short Form Health Survey.
Source: COMET Clinical Study Report.11
Primary Outcome Analysis and Noninferiority Margin
Summary statistics for continuous and categorical variables were presented. Efficacy analyses were performed on the mITT population, and corresponding analysis of the primary outcome was performed for the PP population. A sensitivity analysis was only performed if the ITT population (all randomized patients) was different from the mITT population (randomized patients who received at least 1 infusion).
In the COMET trial, a mixed model repeated measures (MMRM) model assuming data were missing at random was used for the primary outcome. The model included fixed effects of baseline FVC (% predicted) age, sex, treatment group, visit, and treatment-by-visit interaction.
To estimate a noninferiority margin, studies investigating alglucosidase alfa were identified and included. Three studies that enrolled treatment-naïve patients with LOPD were assessed (LOTS, EMBASSY, and TDR12857). The LOTS trial was a placebo-controlled, double-blind RCT that investigated 20 mg/kg alglucosidase alfa every other week (N = 90). Key inclusion criteria for the LOTS study were being 8 years of age or older, being able to walk 40 m or more on the 6MWT, having a FVC (% predicted) upright between 30% and 80%, and having confirmed GAA enzyme deficiency and/or 2 GAA gene mutations. Because of the similar study design and eligibility criteria between the LOTS and COMET studies, the LOTS study was the only 1 used to inform the choice of the noninferiority margin of the COMET trial. Estimated treatment effects of alglucosidase alfa from the single-arm EMBASSY and the open-label TDR12857 multi-dose studies were used for power calculations in the COMET trial.
The estimated treatment effect of alglucosidase alfa, compared with placebo, for FVC (% predicted) in the LOTS trial at 12 months was 3.64% (SD = 4.98; 80% CI = 2.14 to 5.15). A noninferiority margin of 1.1% retained approximately 50% of the treatment effect, based on the lower bound of the 80% CI (i.e., 2.14). The CI was lowered from 95% to 80% in the COMET trial as suggested and agreed upon by regulatory bodies.13 Furthermore, the sponsor noted that a recent literature review revealed that 2-thirds of studies using alglucosidase alfa to treat patients with LOPD showed that changes from baseline in FVC (% predicted) were above or within the MID of 2.0% to 6.0% estimated for populations with idiopathic pulmonary fibrosis, and added that 1.1% was less than this range.
Power Calculation and Determination of Sample Size
The sample size calculations were based on noninferiority testing of the primary efficacy end point of change from baseline to week 49 in FVC (% predicted) upright position with the following assumptions:
The primary end point is normally distributed with a common SD of 5.1% predicted, which was estimated from the phase III, placebo-controlled LOTS trial data.
A mean difference (avalglucosidase alfa – alglucosidase alfa) of 2.0% predicted was assumed based on a conservative estimate from the LOTS and TDR12857 studies.
A 2-sided 5% significance level was used.
Missing data up to 10% was used, based on the LOTS and EMBASSY studies.
A noninferiority margin of 1.1% predicted was used, which was based on the estimated alglucosidase alfa effect from the LOTS study.
Based on retention rates from the LOTS study (95% of patients remaining at 12 months) and the amount of evaluable data from EMBASSY study (94% of patients with evaluable data at 6 months), the sponsor estimated a 10% dropout rate for the COMET trial and sample size of 96 patients. This sample size provides approximately 80% power to demonstrate noninferiority of avalglucosidase alfa versus avalglucosidase alfa if the true treatment difference was 2.0% predicted. If the noninferiority conclusion was met, a test for superiority was performed. If the true difference was 3.5% predicted, the study would have more than 85% power to demonstrate the superiority of avalglucosidase alfa over alglucosidase alfa.
Multiple Testing Procedure and Control for Multiplicity
To control for the type I error rate, a sequential testing strategy was used with all tests performed at a 2-sided 5% significance level. All analyses compared the avalglucosidase alfa treatment group with the alglucosidase alfa treatment group at 49 weeks of treatment. Testing was stopped at the first nonsignificant outcome. Elements of the sequential testing strategy consisted of the following:
FVC (% predicted) tested for noninferiority at a noninferiority margin of –1.1%
FVC (% predicted) (superiority test)
6MWT total distance walked (superiority test)
MIP (% predicted) (superiority test)
MEP (% predicted) (superiority test)
HHD lower extremity strength summary score (superiority test).
Missing Data and Data Imputation
Missing data for the primary outcome of FVC (% predicted) were not imputed and were assumed to be missing at random.
||| || ||||||||||| |||||||||| |||| ||| ||| || ||||||||| ||||||||||| || ||||||| ||||||||||| |||| ||| |||||||| ||| |||||||| || |||||||| ||||| |||||| ||| ||||||| ||| ||||| || ||| || ||| || ||||| ||| |||||||| ||| ||||| ||||| ||| |||||||||| |||||||| || | ||||||| ||| ||| |||||||| ||| || ||||| || ||| |||||| | ||||||| |||| |||||||| |||||| ||| |||| || ||||| |||||||| ||||||| |||| |||||| ||| |||| || ||||||| ||| |||||||| ||||||||| ||||||| ||| |||||| ||||||||| ||||||| ||||||| ||| ||| |||||| |||||| |||||| |||||| ||||||||| |||||||||||| |||||||||||||| ||||||||||||||| |||||| |||||||| | ||||||| |||| ||| ||||||||| ||||| || ||| |||||||| || || |||||||| ||||| || |||| ||||| |||| |||||||| ||| ||||| ||| |||||||||| |||||||| ||| ||| | |||||| ||||||| || ||| |||||| |||| ||| ||| |||||||||| ||| ||||| ||| |||||||||| |||||||| || |||| |||| | ||||| ||| |||||||| ||||||| ||| |||||||| ||||||||| ||||||| ||| |||||| ||||||||| ||||||| ||| |||||||||| ||||||||||||| ||| |||||||| ||||||||||| ||||| |||| |||| |||| ||||||||| || ||||||||| ||| |||||||| ||||||||| ||||||| ||||| ||||| ||| |||||| |||||| ||||| |||| |||| |||| ||||||||| || ||||||||| ||| |||||| ||||||||| ||||||| |||||| ||| ||| ||||| || | ||||||||||| |||| ||| |||||||| ||| ||||| ||||| ||| |||||||||| |||||||| | ||||||| |||| ||||| ||| |||| |||| ||| || ||| ||||||| ||| ||||| |||| || |||||||| ||| |||| |||||| ||||||| ||||| || | || ||| ||| ||| |||||||| || || |||| ||| |||||||| ||| ||||||||| ||||| ||| |||||||||| ||||||||
Subgroup Analyses
Subgroups in the CADTH systematic review protocol include previous treatment with alglucosidase alfa versus no treatment, age, baseline respiratory function, and baseline motor function.
The COMET trial included pre-specified subgroup analyses for FVC (% predicted) for age (< 18 years, ≥ 18 years and < 45 years, or ≥ 45 years), sex, baseline FVC (< 55% or ≥ 55%), race, and ethnicity. Three MMRM models were used to assess the sex, age, and baseline FVC interactions with treatment. The first MMRM model included baseline FVC (% predicted, as continuous), age (as continuous), sex, treatment group, visit, treatment-by-sex interaction, and treatment-by-visit interaction as fixed effects. The second MMRM model included baseline FVC (% predicted, as continuous), age (< 18 years and ≥ 18 years), sex, treatment group, visit, treatment-by-age interaction, and treatment-by-visit interaction as fixed effects. The last MMRM model included baseline FVC (% predicted, as continuous), age (as continuous), sex, treatment group, visit, treatment-by-FVC (categorical) interaction, and treatment-by-visit interaction as fixed effects. The LS mean difference between treatment groups, 95% CI, and P value for interaction were reported. Multiplicity was not controlled for in the subgroup analyses.
Exploratory subgroup analyses were performed for region (US versus non-US), baseline use of walking device during the 6MWT, baseline 6MWT distance, duration of disease, use of walking device, use of respiratory support, and angiotensin-converting-enzyme genotype; only findings related to the 6MWT were summarized in this report.
Sensitivity Analyses
A tipping-point method was used to test how large a difference from the missing-at-random assumption would need to be to change the conclusions of the primary analysis. If it was implausible that the LS mean difference between avalglucosidase alfa and alglucosidase alfa would change the results from statistically significant to not statistically significant, the results were considered robust.
A second set of sensitivity analyses were performed using alternative models and different distribution assumptions. An ANCOVA with baseline FVC, age, sex, and treatment group as covariates was performed for the primary end point. A Wilcoxon-Mann–Whitney test was used on the primary end point to test the robustness of results when there was a deviation in the normality assumption for the MMRM model. Additional MMRM models were tested with covariates of baseline FVC, age, sex, treatment group, visit, and treatment-by-visit interaction as fixed effects. The primary outcome was also evaluated using a linear mixed effects model with fixed effects for age, sex, treatment, time, and the treatment-by-time interaction.
The constancy assumption that the effect of alglucosidase alfa, compared with placebo, in the COMET trial was consistent with the effect observed in the LOTS trial was tested by comparing the change from baseline FVC (% predicted) at week 49 between the 2 trials. An ANCOVA model was fitted to the LOTS data with covariates of treatment, age, sex, race, disease duration, baseline FVC, baseline 6MWT, respiratory support device use at baseline, and treatment interaction with sex, baseline FVC, and respiratory support device use at baseline to estimate the change in FVC (% predicted) from baseline to week 49. The constancy assumption may have been violated if the difference of the estimate of effect of alglucosidase alfa versus placebo between COMET and LOTS exceeded the noninferiority margin of 1.1%.
Supportive Analyses
Supportive analyses for the primary outcome included a responder analysis (responders were defined as having a relative change from baseline of ≥ 5%, ≥ 10%, or ≥ 15% at week 49), change in use of respiratory device from baseline, and correlation between FVC (% predicted) change; in addition, other efficacy parameters, patient-reported outcomes, and safety end points were explored.
Secondary, Tertiary, and Exploratory Outcome Analyses
Secondary outcomes were analyzed using a MMRM model in a manner similar to the primary outcome for the mITT population.
If superiority of avalglucosidase alfa was established for FVC (% predicted), tests for superiority for the 6MWT distance walked, MIP (% predicted), MEP (% predicted), and HHD lower extremity strength summary score at week 49 were performed at a 2-sided 5% significance level, as previously described.
Supportive analyses were presented for the 6MWT, including % predicted change from baseline, categorical response for relative change from baseline, patients who completed the test (excluding those who did not complete the 6 minutes), responder analysis (based on 3 responder thresholds), and change in use of assistive walking device.
Raw measurements for change from baseline and % predicted MIP, MEP, and HHD were summarized descriptively based on the mITT population. Item scores for QMFT and change from baseline in the SF-12 physical component summary and mental component summary scores for adults were summarized descriptively based on the mITT population. Tertiary and exploratory outcomes were analyzed in a method similar to the primary outcome and were summarized descriptively based on the mITT population.
Open-Label Extension phase
All efficacy data for patients who continued receiving treatment in the open-label phase were summarized descriptively.
The effect of switching from alglucosidase alfa to avalglucosidase alfa was assessed in 2 analyses. First, piecewise linear mixed effect models for FVC (% predicted) and 6MWT were fitted for all patients who switched from alglucosidase alfa and for patients who continued with avalglucosidase alfa after the double-blind phase. Model assumption on the linearity of the treatment effect before and after 49 weeks was assessed. Second, FVC (% predicted) was compared between week 49 and week 97, using a within-patient test for those who switched to avalglucosidase alfa at the end of the double-blind phase. A MMRM model with a fixed term for visit and random term for subject was used to calculate the change in FVC (% predicted) and 95% CI between the time points.
Analysis Populations
The mITT population included randomized patients who received at least 1 infusion and was the main population used for efficacy analyses. Analyses with the mITT population were performed according to the treatment arm assigned at randomization, regardless of the treatment received.
The PP population included mITT patients who met the eligibility criteria, received at least 80% of the planned number of doses, had a valid FVC (% predicted) assessment at week 49, and had no major protocol deviations. Major protocol deviations included receiving a dose 2 times greater than that specified in the protocol, receiving a treatment different from the randomized assignment during the double-blind phase of the study, and having the treatment assignment unblinded for reasons other than safety before completion of the double-blind phase. The PP population was used for sensitivity analyses of the primary efficacy end point during the double-blind phase.
The safety population was analyzed according to treatment received during either the double-blind or open-label phase of the study and included randomized patients who received at least 1 infusion during those phases. The overall safety of avalglucosidase alfa was assessed in patients who received at least 1 infusion during either double-blind or open-label phase.
The anti-drug antibody evaluable population included patients from the safety population who had at least 1 valid anti-drug antibody sample taken after baseline.
Results
Patient Disposition
Patient disposition is summarized in Table 10. Of the 146 patients screened, 100 were randomized to receive either avalglucosidase alfa or alglucosidase alfa. The most frequently reported reasons for screening failures included being unable to perform repeated FVC measurements in an upright position between 30% and 85% predicted (37 patients) or between 40% and 85% predicted (5 patients). Overall, 95% of patients completed the double-blind phase of the COMET trial, and 5 patients discontinued alglucosidase alfa because of an AE (8.2%) or other reason (2.0%). Patients who completed the double-blind phase entered the open-label phase, during which 3 patients discontinued avalglucosidase alfa because of an AE (3.9%) or other reason (2.0%) and 1 patient discontinued alglucosidase alfa due to an AE (2.0%).
Major protocol deviations were reported for 11 patients — 5 from the avalglucosidase alfa arm and 6 from the alglucosidase alfa arm — all of whom were excluded from the PP analysis. Reasons for the protocol deviations included use of protocol prohibited medication (n = 6), pulmonary function test not being performed during the protocol specified time (n = 2), not following the sequence of study procedures (n = 1), patient not meeting inclusion criterion for Pompe disease diagnosis (n = 1), and visit data not being performed during the protocol specified time (n = 1).
Table 10: Patient Disposition — COMET Trial
Patient disposition | Avalglucosidase alfa | Alglucosidase alfa |
|---|---|---|
Screened, N | 146 | |
Randomized,a N (%) | 51 (34.9) | 49 (33.6) |
Completed double-blind phase, n (%) | 51 (100) | 44 (89.8) |
Discontinued double-blind phase, n (%) | 0 | 5 (10.2) |
Reason for discontinuation, n (%) | NA | NA |
AE | 0 | 4 (8.2) |
Other | 0 | 1 (2.0) |
Entered open-label phase, n (%) | 51 (100) | 44 (89.8) |
Treatment in open-label phase ongoing,b n (%) | 48 (94.1) | 43 (87.8) |
Discontinued open-label phase, n (%) | 3 (5.9) | 1 (2.0) |
Reason for discontinuation, n (%) | NA | NA |
AE | 2 (3.9) | 1 (2.0) |
Other | 1 (2.0) | 0 |
mITT,c n (%) | 51 (100) | 49 (100) |
PP,c n (%) | 46 (90.2) | 39 (79.6) |
Safety analysis set,c n (%) | 51 (100) | 49 (100) |
Anti-drug antibody evaluable population,c n (%) | 51 (100) | 48 (98.0) |
AE = adverse event; mITT = modified intention-to-treat; NA = not applicable; PP = per protocol.
aFor any subject randomized more than once, only the data associated with the first randomization will be used in any analysis population.
bOngoing as of the primary completion date of the COMET clinical study report (March 19, 2020).
cFor the double-blind phase of the COMET study.
Source: COMET Clinical Study Report.11
Exposure to Study Treatments
In the double-blind phase of the COMET trial, the mean duration of exposure to avalglucosidase alfa was 11.56 months (SD = 0.14; median = 10.58 months [range = 0.5 to 27.6]). Patients in this arm had a mean of 24.8 infusions (SD = 0.5; median = 25.0 infusions [range = 23 to 25]). The mean duration of exposure to alglucosidase alfa was 10.69 months (SD = 2.69). Patients had a mean of 22.8 infusions (SD = 5.8; median = 25.0 infusions (range = 1 to 25).
Four patients (8.2%) in the alglucosidase alfa group missed at least 2 consecutive infusions during the double-blind phase of the study. No patients in the avalglucosidase alfa missed more than 2 consecutive doses.
In the open-label phase of the COMET trial (after week 49), the mean duration of exposure to avalglucosidase alfa was 12.11 months (SD = 8.74; median = 10.58 months [range = 0.5 to 27.6]) for patients who received avalglucosidase alfa in both the double-blind and open-label phases. Patients in this arm had a mean of 26.2 infusions (SD = 19.0; median = 23.0 infusions [range = 1 to 60]). The mean duration of exposure to avalglucosidase alfa was 12.17 months (SD = 9.3; median = 10.46 months [range = 0.5 to 29.0]) for patients who received alglucosidase alfa in double-blind phase and avalglucosidase alfa in the open-label phase. Patients in this arm had a mean of 26.0 infusions (SD = 19.8; median = 22.0 infusions [range = 1 to 63]).
Of the patients who received avalglucosidase alfa for both the double-blind and open-label phases of the COMET trial, 3 (5.9%) missed at least 2 consecutive infusions during the open-label phase. No patients who received alglucosidase alfa during the double-blind phase missed more than 2 consecutive doses.
Efficacy
Only efficacy outcomes and subgroup analyses identified in the CADTH review protocol are reported in the subsequent sections.
Survival-Related Outcomes
The 1 death (2.0%) that occurred during the double-blind phase in the alglucosidase alfa treatment group was caused by an acute myocardial infarction.
Respiratory-Related Outcomes
Forced Vital Capacity
Data for the primary efficacy outcome during the double-blind phase are summarized in Table 11. Overall, patients in the mITT population demonstrated a LS mean change in FVC (% predicted) in the upright position from baseline to week 49 of 2.89% (95% CI = 1.13 to 4.65) for the avalglucosidase alfa arm and 0.46% (95% CI = –1.39 to 2.31) for the alglucosidase alfa arm. The mean difference of change between treatment groups was 2.43% (95% CI = –0.13 to 4.99), for which the lower bound of the 95% CI did not exceed the noninferiority margin of –1.1%, indicating that the criteria for the noninferiority of avalglucosidase alfa to alglucosidase alfa was demonstrated (P = 0.0074). The P value for superiority testing was not statistically significant (P = 0.0626), so statistical testing was stopped for all subsequent efficacy outcomes.
Analysis of the PP population showed similar results, with a FVC (% predicted) LS mean change from baseline to week 49 of 2.87% (95% CI = 1.02 to 4.73) and 0.19% (95% CI = –1.83 to 2.21) for the avalglucosidase alfa and alglucosidase alfa groups, respectively. The mean difference of change between treatment groups was 2.69% (95% CI = –0.06 to 5.44; P for noninferiority = 0.0076 and P for superiority = 0.0555).
Based on the tipping-point method used to assess the data-missing-at-random assumption, a 16% reduction in FVC (% predicted) for the avalglucosidase alfa group and a 2% increase in FVC (% predicted) for the alglucosidase alfa group indicated that the results would remain robust for the primary outcome.
Subgroup analyses for the primary outcome are summarized in Table 23. Since the COMET trial permitted only patients who were treatment-naïve, there were no subgroups of patients previously treated with alglucosidase alfa that could be compared with a no-treatment group. Among adult patients 18 to 45 years of age, there was a mean difference between groups of 2.99% (95% CI = –1.52 to 7.49). The results were similar for adults 45 years and older, with a mean between-group difference of 2.46% (95% CI = –0.84 to 5.77). Analysis by baseline FVC (% predicted) showed that patients with a FVC of less than 55% had a mean between-group difference of –0.76% (95% CI = –5.23 to 3.71), whereas patients with a FVC of at least 55% had a mean between-group difference of 4.10% (95% CI = 0.95 to 7.26) for avalglucosidase alfa. Patients who used a walking device on the 6MWT at baseline had a mean difference of change between treatment groups of 4.44% (95% CI = –1.00 to 9.88), whereas those who did not use a walking device had a mean between-group difference of 2.19% (95% CI = –0.76 to 5.13). For analysis of baseline 6MWT distance, patients who walked less than 403.5 m at baseline had a mean difference of change between treatment groups of 1.27% (95% CI = –3.03 to 5.57), and patients who travelled at least 403.5 m had a mean between-group difference of 3.58% (95% CI = 0.36 to 6.81).
Long-term data (week 97) for the primary and key secondary efficacy outcomes from the open-label phase of the COMET trial are summarized in Table 25, and no statistical testing was conducted for these end points. For FVC (% predicted) at week 97, mean change from study baseline was 1.60% (SD = 7.72) for patients who received only avalglucosidase alfa and 1.64% (SD = 8.97) for those who switched from alglucosidase alfa to avalglucosidase alfa. Note that these results are from a preliminary analysis of patients with data at week 97 and do not represent to entire enrolled population.
Good evidence of reliability and weak evidence of responsiveness for FVC measurements were found for patients with pulmonary disorders, although no information was identified for patients with Pompe disease.20 No MID was identified from the literature for patients with Pompe disease.
Need for Ventilation
Changes in the need for ventilatory support were not included as an outcome in the COMET trial, although new use of ventilatory support was reported. During the double-blind phase of the COMET trial, 3 patients (6%) in the avalglucosidase alfa group and 4 patients (8%) in the alglucosidase alfa group were reported to have started using a new respiratory device.
Maximum Expiratory Pressure
Results for the double-blind phase are summarized in Table 12. For MEP (% predicted), the LS mean changes from baseline were 10.89% (95% CI = 5.24 to 16.54) and 8.38% (95% CI = 2.49 to 14.26) for the avalglucosidase alfa and alglucosidase alfa arms, respectively. The mean difference of change between treatment groups was 2.51% (95% CI = –5.70 to 10.73).
No evidence of validity, reliability, or responsiveness was found for MEP for this patient population. No MID was identified from the literature for MEP for patients with Pompe disease.
Maximum Inspiratory Pressure
Results for the double-blind phase are summarized in Table 12. For MIP (% predicted), the LS mean changes from baseline were 8.70% (95% CI = 4.54 to 12.85) and 4.29% (95% CI = –0.07 to 8.65) for the avalglucosidase alfa and alglucosidase alfa arms, respectively. The mean difference of change between treatment groups was 4.40% (95% CI = –1.63 to 10.44).
For patients with Pompe disease, MIP showed acceptable validity and reliability,35,36 although no information was found on the assessment of responsiveness to change. No MID was identified from the literature for MIP for patients with Pompe disease.
Motor-Related Outcomes
6-Minute Walk Test
Results for the double-blind phase are summarized in Table 12. The mean change from baseline to week 49 for the 6MWT distance was 32.21 m (95% CI = 12.47 to 51.94) for the avalglucosidase alfa group and 2.19 m (95% CI = –18.48 to 22.86) for the alglucosidase alfa group. The mean difference of change between treatment groups was 30.01 m (95% CI = 1.33 to 58.69). The mean change from baseline for the 6MWT (% predicted) was 5.02% (95% CI = 1.95 to 8.09) for the avalglucosidase alfa group and 0.31% (95% CI = –2.90 to 3.52) for the alglucosidase alfa group. The mean difference of change between treatment groups was 4.71% (95% CI = 0.25 to 9.17).
Long-term data (week 97) for the 6MWT from the open-label phase of the COMET trial are summarized in Table 25. No statistical testing was conducted for these end points. The mean change from baseline for distance travelled during the 6MWT was 37.34 m (SD = 68.41) for the avalglucosidase alfa and 25.71 m (SD = 71.31) for the switch group (i.e., patients previously treated with alglucosidase alfa). At week 97, patients had a mean change from baseline for 6MWT (% predicted) of 6.01% (SD = 10.18) for the avalglucosidase alfa group and 4.23% (SD = 11.17) for the switch group.
Validity and reliability of the 6MWT have been established in various patient populations.21-31 The 6MWT distance showed validity and responsiveness to change, compared with the Rotterdam Handicap Scale and Rasch-built Pompe-specific instruments for patients with Pompe disease.19 No information on reliability and no MID were identified from the literature for patients with Pompe disease.
Use of Mobility Aids
Change in the use of mobility aids was not included as a main outcome in the COMET trial, although change in use of mobility aids was reported. During the double-blind phase of the COMET trial, 3 patients (6%) in the avalglucosidase alfa group and 1 patient (2%) in the alglucosidase alfa group reported new or increased use of assistive walking devices. However, 8 patients (16%) in the avalglucosidase alfa group and no patients in the alglucosidase alfa group reported decreased use of assistive walking devices.
HHD (Lower Extremity Composite Score)
Results for the double-blind phase are summarized in Table 12. For HHD (composite score), the LS mean change from baseline was 260.69 (95% CI = 169.11 to 352.27) for the avalglucosidase alfa group and 153.72 (95% CI = 57.22 to 250.22) for the alglucosidase alfa group. The mean difference of change between treatment groups was 106.97 (95% CI = –26.56 to 240.50).
Scores for the HHD, Rotterdam Handicap Scale, and Rasch-built Pompe-specific Activity scale instruments were found to be significantly correlated among patients with Pompe disease, demonstrating evidence of validity among the outcome measures.19 Information assessing reliability and responsiveness was not found for HHD, and no MID was identified from the literature for patients with Pompe disease.
Quick Motor Function Test
Results for the double-blind phase are summarized in Table 12. The LS mean change for QMFT (total score) from baseline was 3.98 (95% CI = 2.72 to 5.23) for the avalglucosidase alfa group and 1.89 (95% CI = 0.52 to 3.26) for the alglucosidase alfa group. The mean difference of change between treatment groups was 2.08 (95% CI = 0.22 to 3.95).
Evidence of validity and reliability has been established for the QMFT among patients with Pompe disease,42 but no information was found that evaluated responsiveness. No MID was identified from the literature for patients with Pompe disease.
Gait, Stair, Gower’s Maneuver, and Chair
Results for the double-blind phase are summarized in Table 12. The mean changes from baseline for the GSGC were –1.57 (SD = 2.72) and –0.38 (SD = 1.81) for the avalglucosidase alfa and alglucosidase alfa groups, respectively. The mean difference of change between treatment groups was –1.31 (95% CI = –0.37 to –2.25).
Evidence of validity and correlation of the GSGC with the 6MWT and Walton and Gardner-Medwin scale (a measure of functional status) has been established in patients with LOPD.47 No information on reliability or responsiveness and no MID was identified from the literature for patients with Pompe disease.
Gross Motor Function Measure-88
Results for the double-blind phase are summarized in Table 12. The mean changes from baseline for the Gross Motor Function Measure (GMFM)-88 dimension D (standing) were 4.29 (SD = 7.37) and 1.77 (SD = 5.79) for the avalglucosidase alfa and alglucosidase alfa groups, respectively. For dimension E (walking, running, and jumping), the mean changes from baseline were 5.33 (SD = 8.02) and 2.81 (SD = 5.38) for the 2 groups, respectively. The mean difference of change between treatment groups 2.58 (95% CI = –0.02 to 5.18) for dimension D and 2.54 (95% CI = –0.09 to 5.18) for dimension E.
Validity, reliability, and responsiveness have been established for the GMFM-88 in children with cerebral palsy,48 but no evidence was found for patients with Pompe disease. No MID was identified from the literature for patients with Pompe disease.
GMFCS and HHD (Upper Extremity Muscle Strength)
Results for the GMFCS and HHD (upper extremity muscle strength) from the COMET trial were not described in the CADTH review of avalglucosidase alfa. Results for the double-blind phase are summarized in Table 24.
Disease-Related Symptoms and Impact Outcomes
Pompe Disease Symptom Scale, Pompe Disease Impact Scale, Rasch-Built Pompe-Specific Activity Scale
Results for the Pompe Disease Symptom Scale, Pompe Disease Impact Scale, and Rasch-built Pompe-specific Activity scale from the COMET trial were not described in the CADTH review of avalglucosidase alfa.
Health-Related Quality of Life
12-item Short Form Health Survey
Results for the double-blind phase are summarized in Table 12. For the SF-12 physical component summary, the LS mean changes from baseline were 2.37 (95% CI = 0.40 to 4.34) and 1.60 (95% CI = –0.52 to 3.72) for the avalglucosidase alfa and alglucosidase alfa arms, respectively. The mean difference of change between treatment groups was 0.77 (95% CI = –2.13 to 3.67). For the SF-12 mental component summary, the mean changes from baseline were 2.88 (95% CI = 0.47 to 5.30) and 0.76 (95% CI = –1.86 to 3.39) for the avalglucosidase alfa and alglucosidase alfa arms, respectively. The mean difference of change between treatment groups was 2.12 (95% CI = –1.46 to 5.69).
Validity, reliability, and responsiveness for the SF-12 have been demonstrated in various patient populations,44-46 although no evidence was found for patients with Pompe disease. No MID was identified from the literature for patients with Pompe disease.
EQ-5D-5L and Pediatric Quality of Life Inventory
Results for the EQ-5D-5L and Pediatric Quality of Life Inventory from the COMET trial were not described in the CADTH review of avalglucosidase alfa. Results for the double-blind phase are summarized in Table 24.
Other Patient-Reported Outcomes
Patient Global Impression of Change
Results for the Patient Global Impression of Change scale from the COMET trial were not described in the CADTH review of avalglucosidase alfa.
Table 11: Primary Efficacy Outcome During the Double-Blind phase — COMET Trial
Outcome | Avalglucosidase alfa, N = 51 | Alglucosidase alfa, N = 49 |
|---|---|---|
FVC (% predicted) in upright position: mITT population | ||
Patients contributing to analysis, n | 49 | 43 |
Baseline, mean (SD) | 62.55 (14.39) | 61.56 (12.40) |
End of double-blind phase, week 49, mean (SD) | 65.49 (17.42) | 61.16 (13.49) |
LS mean change from baselinea (SE) [95% CI] | 2.89 (0.88) [1.13 to 4.65] | 0.46 (0.93) [–1.39 to 2.31] |
Group difference (treatment – control) (SE) [95% CI] | 2.43 (1.29) [–0.13 to 4.99] | Reference |
P value for noninferiorityb | 0.0074 | Reference |
P value for superiority | 0.0626 | Reference |
FVC (% predicted) in upright position: PP population | ||
Patients contributing to analysis, n | 46 | 39 |
Baseline, mean (SD) | 63.13 (14.65) | 61.46 (13.02) |
End of double-blind phase, week 49, mean (SD) | 66.15 (17.27) | 61.38 (13.91) |
LS mean change from baselinea (SE) [95% CI] | 2.87 (0.93) [1.02 to 4.73] | 0.19 (1.02) [–1.83 to 2.21] |
Group difference (treatment – control) (SE) [95% CI] | 2.69 (1.38) [–0.06 to 5.44] | Reference |
P value for noninferiorityb | 0.0076 | Reference |
P value for superiority | 0.0555 | Reference |
CI = confidence interval; FVC = forced vital capacity; LS = least squares; mITT = modified intention-to-treat; NA = not applicable; PP = per protocol; SD = standard deviation; SE = standard error.
aMMRM model with fixed, continuous effects of baseline FVC (% predicted) and age (in years at baseline), and fixed, categorical effects of sex, treatment group, visit, and interaction term between treatment group and visit.
bNoninferiority margin is –1.1%. P value has been adjusted for multiple testing (i.e., the type I error rate has been controlled).
Source: COMET Clinical Study Report.11
Table 12: Secondary and Selected Tertiary Efficacy Outcomes During the Double-Blind phase — COMET Trial mITT Population
Outcome | Avalglucosidase alfa, N = 51 | Alglucosidase alfa, N = 49 |
|---|---|---|
Secondary outcomes | ||
6MWT (distance in m) | ||
Patients contributing to analysis, n | 48 | 43 |
Baseline, mean (SD) | 399.30 (110.93) | 378.09 (116.22) |
End of double-blind phase, week 49, mean (SD) | 441.31 (109.77) | 383.56 (141.09) |
Change from baseline,a mean (SE) (95% CI) | 32.21 (9.93) [12.47 to 51.94] | 2.19 (10.40) [–18.48 to 22.86] |
Group difference (treatment – control) (SE) [95% CI] | 30.01 (14.43) [1.33 to 58.69] | Reference |
P valueb | 0.0405 | Reference |
6MWT (% predicted) | ||
Patients contributing to analysis, n | 48 | 43 |
Baseline, mean (SD) | 57.32 (14.97) | 55.29 (16.64) |
End of double-blind phase, week 49, mean (SD) | 63.73 (15.13) | 55.39 (19.15) |
Change from baselinea, mean (SE) [95% CI] | 5.02 (1.54) [1.95 to 8.09] | 0.31 (1.62) [–2.90 to 3.52] |
Group difference (treatment – control) (SE) [95% CI] | 4.71 (2.24) [0.25 to 9.17] | Reference |
P valueb | 0.0386 | Reference |
MIP (% predicted)c | ||
Patients contributing to analysis, n | 49 | 42 |
Baseline, mean (SD) | 59.88 (47.10) | 60.65 (41.05) |
End of double-blind phase, week 49, mean (SD) | 61.20 (30.37) | 57.65 (22.34) |
Change from baseline,d mean [95% CI] | 8.70 [4.54 to 12.85] | 4.29 (–0.07 to 8.65) |
Group difference (treatment – control) [95% CI] | 4.40 [–1.63 to 10.44] | Reference |
P valueb | 0.6451 | Reference |
MEP (% predicted)c | ||
Patients contributing to analysis, n | 49 | 42 |
Baseline, mean (SD) | 65.77 (38.97) | 74.83 (35.22) |
End of double-blind phase, week 49, mean (SD) | 71.24 (29.69) | 78.60 (28.13) |
Change from baseline,e mean [95% CI] | 10.89 [5.24 to 16.54] | 8.38 (2.49 to 14.26) |
Group difference (treatment – control) [95% CI] | 2.51 [–5.70 to 10.73] | Reference |
P valueb | 0.6557 | Reference |
HHD (composite score) | ||
Patients contributing to analysis, n | 46 | 42 |
Baseline, mean (SD) | 1,330.45 (625.44) | 1,466.16 (604.91) |
End of double-blind phase, week 49, mean (SD) | 1,557.71 (628.19) | 1,639.24 (669.27) |
Change from baseline,f mean (SE) [95% CI] | 260.69 (46.07) [169.11 to 352.27] | 153.72 (48.54) [57.22 to 250.22] |
Group difference (treatment – control) (SE) [95% CI] | 106.97 (67.17) [–26.56 to 240.50] | Reference |
P valueb | 0.1150 | Reference |
QMFT (total score) | ||
Patients contributing to analysis, n | 49 | 42 |
Baseline, mean (SD) | 41.29 (10.15) | 42.30 (10.58) |
End of double-blind phase, week 49, mean (SD) | 45.49 (9.94) | 43.67 (11.99) |
Change from baseline,g mean (SE) [95% CI] | 3.98 (0.63) [2.72 to 5.23] | 1.89 (0.69) [0.52 to 3.26] |
Group difference (treatment – control) (SE) [95% CI] | 2.08 (0.94) [0.22 to 3.95] | Reference |
P valueb | 0.0288 | Reference |
SF-12 physical component summary | ||
Patients contributing to analysis, n | 50 | 43 |
Baseline, mean (SD) | 35.95 (7.82) | 36.76 (9.40) |
End of double-blind phase, week 49, mean (SD) | 38.55 (8.17) | 38.31 (10.89) |
Change from baseline,h mean (SE) [95% CI] | 2.37 (0.99) [0.40 to 4.34] | 1.60 (1.07) [–0.52 to 3.72] |
Group difference (treatment – control) (SE) [95% CI] | 0.77 (1.46) [–2.13 to 3.67] | Reference |
P valueb | 0.5996 | Reference |
SF-12 mental component summary | ||
Patients contributing to analysis, n | 50 | 43 |
Baseline, mean (SD) | 48.31 (10.11) | 50.58 (8.69) |
End of double-blind phase, week 49, mean (SD) | 51.67 (8.19) | 51.28 (11.67) |
Change from baseline,h mean (SE) [95% CI] | 2.88 (1.22) [0.47 to 5.30] | 0.76 (1.32) [–1.86 to 3.39] |
Group difference (treatment – control) (SE) [95% CI] | 2.12 (1.80) [–1.46 to 5.69] | Reference |
P valueb | 0.2427 | Reference |
Selected tertiary outcomes | ||
GSGC | ||
Patients contributing to analysis, n | 51 | 42 |
Baseline, mean (SD) | 12.92 (4.60) | 13.55 (5.25) |
End of double-blind phase, week 49, mean (SD) | 11.35 (4.66) | 13.17 (5.43) |
Change from baseline, mean (SD) | –1.57 (2.72) | –0.38 (1.81) |
P valueb, h | 0.0205 | Reference |
GMFM-88 dimension D (standing) | ||
Patients contributing to analysis, n | 49 | 42 |
Baseline, mean (SD) | 80.49 (14.21) | 79.28 (15.03) |
End of double-blind phase, week 49, mean (SD) | 85.19 (11.06) | 80.95 (15.84) |
Change from baseline, mean (SD) | 4.29 (7.37) | 1.77 (5.79) |
P valueb, h | 0.0613 | Reference |
GMFM-88 dimension E (walking, running, and jumping) | ||
Patients contributing to analysis, n | 49 | 42 |
Baseline, mean (SD) | 81.02 (15.72) | 78.85 (18.49) |
End of double-blind phase, week 49, mean (SD) | 86.48 (13.47) | 82.18 (18.84) |
Change from baseline, mean (SD) | 5.33 (8.02) | 2.81 (5.38) |
P valueb, h | 0.0475 | Reference |
6MWT = 6-minute walk test; CI = confidence interval; FVC = forced vital capacity; GMFM-88 = Gross Motor Function Measure-88; GSGC = gait, stair, Gower’s maneuver, and chair; HHD = hand-held dynamometry; MEP = maximum expiratory pressure; MIP = maximum inspiratory pressure; mITT = modified intention-to-treat; NA = not applicable; QMFT = Quick Motor Function Test; SD = standard deviation; SE = standard error; SF-12 = 12-item Short Form Health Survey.
aMMRM model for 6MWT distance with fixed effects of baseline FVC (% predicted), baseline 6MWT (distance walked in m), age (in years at baseline), sex, treatment group, visit, and treatment-by-visit interaction.
bP value is nominal. P value has not been adjusted for multiple testing (i.e., the type I error rate has not been controlled).
cValues for change from baseline and group difference were provided by the sponsor based on the Diaz-Manera et al. (2021)13 publication (without SE). Baseline and week 49 values reflect the data available from the COMET Clinical Study Report.11
MMRM model for MIP % predicted with fixed effects of MIP (% predicted) at baseline, age (in years, at baseline), sex, treatment group, visit, and treatment-by-visit interaction.
dMMRM model for MEP % predicted with fixed effects of MEP (% predicted) at baseline, age (in years, at baseline), sex, treatment group, visit, and treatment-by-visit interaction.
eMMRM model for HHD lower extremity muscle strength composite score with fixed effects for summary HHD lower extremity score at baseline, baseline FVC (% predicted), age (in years, at baseline), sex, treatment group, visit, and treatment-by-visit interaction.
fMMRM model for QMFT total score with fixed effects for QMFT total score at baseline, baseline FVC (% predicted), age (in years, at baseline), sex, treatment group, visit, and treatment-by-visit interaction.
gMMRM model for SF-12 (physical component summary and mental component summary) with fixed effects for baseline score (physical component summary or mental component summary), baseline FVC (% predicted), age (in years, at baseline), sex, treatment group, visit, and treatment-by-visit interaction.
hP values are from the Wilcoxon-Mann–Whitney stratified by stratification factors.
Source: COMET Clinical Study Report,11 Diaz-Manera et al. (2021).13
Harms
Only harms identified in the CADTH review protocol are reported below. Harms data are summarized in Table 13 for the double-blind phase and Table 26 for the open-label phase.
Adverse Events
During the double-blind phase of the COMET trial, 44 patients (86.3%) who received avalglucosidase alfa and 45 patients (91.8%) who received alglucosidase alfa experienced an AE. The most frequently reported events were nasopharyngitis (12 patients, 23.5%), back pain (12 patients, 23.5%), and headache (11 patients, 21.6%) for the avalglucosidase alfa group, and headache (16 patients, 32.7%), nasopharyngitis (12 patients, 24.5%), and falls (10 patients, 20.4%) for the alglucosidase alfa group. In general, the events were similar between the treatment groups, although influenza and back pain were numerically higher in the avalglucosidase alfa arm and headache was numerically higher in the alglucosidase alfa arm.
AEs in the open-label phase of the COMET study were similar for patients who received avalglucosidase alfa during the double-blind phase and those who received alglucosidase alfa during the double-blind phase but switched to avalglucosidase alfa, and most patients experienced at least 1 AE. Nasopharyngitis (8 patients, 15.7%) and nausea (7 patients, 13.7%) were the most common AEs among patients who continued receiving avalglucosidase alfa, whereas headache (11 patients, 25.0%), nasopharyngitis (10 patients, 22.7%), and diarrhea (10 patients, 22.7%) were the most common AEs among those who switched from alglucosidase alfa.
Serious Adverse Events
Overall, SAEs were infrequent in the 2 treatment groups, with 8 patients (15.7%) reporting a SAE in the avalglucosidase alfa arm and 12 (24.5%) reporting a SAE in the alglucosidase alfa arm. No SAE occurred in more than 1 patient who received avalglucosidase alfa, and only dyspnea occurred in 2 patients who received alglucosidase alfa during the double-blind phase.
During the open-label phase of the COMET trial, 8 patients (15.7%) who continued receiving avalglucosidase alfa reported a SAE, as did 5 patients (11.4%) who switched from alglucosidase alfa. Similarly, no SAE occurred in more than 1 patient for either treatment group.
Withdrawals Due to Adverse Events
Four patients (8.2%) from the alglucosidase alfa group withdrew from the double-blind phase of the COMET study because of the following AEs: acute myocardial infarction, arthritis, dyspnea, and urticaria. No patients from the avalglucosidase alfa group withdrew during the double-blind phase due to AEs.
During the open-label phase, 2 patients (3.9%) who continued receiving avalglucosidase alfa withdrew due to AEs (ocular hyperemia, acute myocardial infarction, and erythema).
Mortality
One death (2.0%) due to acute myocardial infarction was reported in the alglucosidase alfa group during the double-blind phase.
One death (2.3%) due to adenocarcinoma pancreas was reported during the open-label phase for a patient who had received alglucosidase alfa during the double-blind phase.
Notable Harms
Hypersensitivity Reactions (Including Anaphylaxis)
Treatment-emergent anaphylactic reactions (pruritus and rash) were reported for 2 patients (4%) in each of the treatment arms during the double-blind phase.
Treatment-emergent hypersensitivity reactions were generally balanced between treatments (12 patients [23.5%] in the avalglucosidase alfa and 15 patients [30.6%] in the alglucosidase alfa group). In the avalglucosidase alfa group, 4 patients (7.8%) reported experiencing pruritus, 3 patients (5.9%) reported erythema, and 3 patients (5.9%) reported urticaria. In the alglucosidase alfa group, 4 patients (8.2%) each reported experiencing pruritus or rash, and 3 patients (6.1%) each reported erythema or flushing.
No data on treatment-emergent anaphylactic reactions and hypersensitivity reactions were available for the open-label phase.
Infusion-Associated Reactions
Treatment-emergent infusion-associated reactions were similar in the 2 treatment arms: 13 patients (25.5%) in the avalglucosidase alfa and 16 patients (32.7%) in the alglucosidase alfa group. Among those who received avalglucosidase alfa, pruritus (4 patients, 7.8%) and urticaria (3 patients, 5.9%) were the most frequently reported infusion-associated reactions. Among those who received alglucosidase alfa, pruritus (4 patients, 8.2%), nausea (4 patients, 8.2%), and flushing (3 patients, 6.1%) were the most frequently reported infusion-associated reactions.
For the open-label phase, infusion-associated reactions continued to be less frequently reported in the avalglucosidase alfa group (6 patients; 11.8%) than in the group that switched from alglucosidase alfa (15 patients, 34.1%).
Immunogenicity and Anti-Drug Antibodies
Treatment-emergent immune-mediated reactions occurred in || |||||||| ||||||| who received avalglucosidase alfa and || |||||||| ||||||| who received alglucosidase alfa. Arthralgia (5 patients, 9.8%), myalgia (5 patients, 9.8%), and lymphadenopathy (2 patients, 3.9%) were reported among patients who received avalglucosidase alfa, whereas only arthralgia (8 patients, 16.3%) and myalgia (7 patients, 14.3%) were reported among those who received alglucosidase alfa.
No data for treatment-emergent immune-mediated reactions were available for the open-label phase.
Nearly all patients were positive for treatment-emergent anti-drug antibodies (96.1% and 95.8% in the avalglucosidase alfa and alglucosidase alfa groups, respectively) (Table 14). The peak titre is the highest anti-drug antibody titre after baseline for patients who seroconverted or developed anti-drug antibodies de novo. In the avalglucosidase alfa group, 21.3% of patients had a peak titre greater than 12,800, whereas in the alglucosidase alfa group, 36.4% of patients did.
Acute Cardiorespiratory Failure
Acute cardiorespiratory failure was not reported during the double-blind or open-label phases of the COMET study.
Table 13: Summary of Harms During the Double-Blind phase — COMET Trial, Safety Population
Event | Avalglucosidase alfa, N = 51 | Alglucosidase alfa, N = 49 |
|---|---|---|
Patients with ≥ 1 TEAE by primary system organ class and preferred term,a n (%) | 44 (86.3) | 45 (91.8) |
Infections and infestations | 30 (58.8) | 25 (51.0) |
Nasopharyngitis | 12 (23.5) | 12 (24.5) |
Influenza | 9 (17.6) | 2 (4.1) |
Upper respiratory tract infection | 4 (7.8) | 2 (4.1) |
Cystitis | 3 (5.9) | 0 |
Nervous system disorders | 20 (39.2) | 21 (42.9) |
Headache | 11 (21.6) | 16 (32.7) |
Dizziness | 5 (9.8) | 4 (8.2) |
Paresthesia | 3 (5.9) | 2 (4.1) |
Respiratory, thoracic, and mediastinal disorders | 12 (23.5) | 16 (32.7) |
Dyspnea | 3 (5.9) | 4 (8.2) |
Gastrointestinal disorders | 16 (31.4) | 22 (44.9) |
Diarrhea | 6 (11.8) | 8 (16.3) |
Nausea | 6 (11.8) | 7 (14.3) |
Vomiting | 4 (7.8) | 3 (6.1) |
Dyspepsia | 3 (5.9) | 3 (6.1) |
Skin and subcutaneous tissue disorders | 9 (17.6) | 15 (30.6) |
Pruritus | 4 (7.8) | 4 (8.2) |
Erythema | 3 (5.9) | 3 (6.1) |
Urticaria | 3 (5.9) | 1 (2.0) |
Musculoskeletal and connective tissue disorders | 29 (56.9) | 26 (53.1) |
Back pain | 12 (23.5) | 5 (10.2) |
Pain in extremity | 8 (15.7) | 7 (14.3) |
Arthralgia | 5 (9.8) | 8 (16.3) |
Myalgia | 5 (9.8) | 7 (14.3) |
Muscle spasms | 3 (5.9) | 5 (10.2) |
General disorders and administration-site conditions | 21 (41.2) | 24 (49.0) |
Fatigue | 9 (17.6) | 7 (14.3) |
Influenza like illness | 3 (5.9) | 1 (2.0) |
Noncardiac chest pain | 3 (5.9) | 0 |
Edema peripheral | 3 (5.9) | 3 (6.1) |
Injury, poisoning, and procedural complications | 13 (25.5) | 15 (30.6) |
Fall | 7 (13.7) | 10 (20.4) |
Contusion | 5 (9.8) | 4 (8.2) |
Patients with ≥ 1 SAE by primary system organ class and preferred term, n (%) | 8 (15.7) | 12 (24.5) |
Dyspnea | 1 (2.0) | 2 (4.1) |
Pneumonia | 1 (2.0) | 1 (2.0) |
Syncope | 1 (2.0) | 0 |
Hypoventilation | 1 (2.0) | 0 |
Respiratory failure | 1 (2.0) | 0 |
Calculus urinary | 1 (2.0) | 0 |
Hydronephrosis | 1 (2.0) | 0 |
Renal colic | 1 (2.0) | 0 |
Breast cyst | 1 (2.0) | 0 |
Sepsis | 0 | 1 (2.0) |
Inappropriate antidiuretic hormone secretion | 0 | 1 (2.0) |
Brain stem stroke | 0 | 1 (2.0) |
Cerebellar ischemia | 0 | 1 (2.0) |
Dizziness | 0 | 1 (2.0) |
Visual impairment | 0 | 1 (2.0) |
Acute myocardial infarction | 0 | 1 (2.0) |
Angina pectoris | 0 | 1 (2.0) |
Supraventricular tachycardia | 0 | 1 (2.0) |
Hypotension | 0 | 1 (2.0) |
Diaphragmatic paralysis | 0 | 1 (2.0) |
Pulmonary embolism | 0 | 1 (2.0) |
Abdominal upper pain | 0 | 1 (2.0) |
Gastrointestinal hemorrhage | 0 | 1 (2.0) |
Cold sweat | 0 | 1 (2.0) |
Nephrolithiasis | 0 | 1 (2.0) |
Chills | 0 | 1 (2.0) |
Hemoglobin decreased | 0 | 1 (2.0) |
WDAE, n (%) | 0 | 4 (8.2) |
Acute myocardial infarction | 0 | 1 (2.0) |
Arthritis | 0 | 1 (2.0) |
Dyspnea | 0 | 1 (2.0) |
Urticaria | 0 | 1 (2.0) |
Deaths, n (%) | 0 | 1 (2.0) |
Acute myocardial infarction | 0 | 1 (2.0) |
Notable harms,b n (%) | NA | NA |
Treatment-emergent anaphylactic reactions by SMQ, broad and narrow combined, n (%) | 2 (3.9) | 2 (4.1) |
Pruritus | 2 (3.9) | 0 |
Rash | 1 (2.0) | 2 (4.1) |
Treatment-emergent hypersensitivity reactions by SMQ, broad and narrow combined, n (%) | 12 (23.5) | 15 (30.6) |
Pruritus | 4 (7.8) | 4 (8.2) |
Erythema | 3 (5.9) | 3 (6.1) |
Urticaria | 3 (5.9) | 1 (2.0) |
Rash | 2 (3.9) | 4 (8.2) |
Rhinitis allergic | 2 (3.9) | 0 |
Rash erythematous | 1 (2.0) | 1 (2.0) |
Flushing | 0 | 3 (6.1) |
Treatment-emergent infusion-associated reactions, n (%) | 13 (25.5) | 16 (32.7) |
Pruritus | 4 (7.8) | 4 (8.2) |
Urticaria | 3 (5.9) | 1 (2.0) |
Rash | 2 (3.9) | 2 (4.1) |
Headache | 2 (3.9) | 1 (2.0) |
Diarrhea | 2 (3.9) | 0 |
Nausea | 1 (2.0) | 4 (8.2) |
Dyspnea | 1 (2.0) | 2 (4.1) |
Erythema | 1 (2.0) | 2 (4.1) |
Chills | 1 (2.0) | 2 (4.1) |
Flushing | 0 | 3 (6.1) |
Dizziness | 0 | 2 (4.1) |
Feeling hot | 0 | 2 (4.1) |
Treatment-emergent immune-mediated reactions, n (%) | || |||||| | || |||||| |
Arthralgia | 5 (9.8) | 8 (16.3) |
Myalgia | 5 (9.8) | 7 (14.3) |
Lymphadenopathy | 2 (3.9) | 0 |
Acute cardiorespiratory failure | NR | NR |
NA = not applicable; NR = not reported; SAE = serious adverse events; SMQ = Standardized MedDRA Queries; TEAE = treatment-emergent adverse events; WDAE = withdrawal due to adverse events.
aFrequency of AE ≥ 5% per treatment group.
bFrequency of notable harm ≥ 2 patients total for both treatment groups.
Source: COMET Clinical Study Report.11
Table 14: Summary of Immunogenicity Outcomes During the Double-Blind phase — COMET Trial, Anti-Drug Antibody Evaluable Population
Event | Avalglucosidase alfa,a N = 51 | Alglucosidase alfa,b N = 48 |
|---|---|---|
Treatment-emergent anti-drug antibodies,a n (%) | 49 (96.1) | 46 (95.8) |
Anti-drug antibody status, n (%) | NA | NA |
Always negative | 2 (3.9) | 2 (4.2) |
Ever positive with negative baseline | 47 (92.2) | 44 (91.7) |
Positive at baseline | 2 (3.9) | 2 (4.2) |
Peak titreb for treatment-induced anti-drug antibodies, n (%) | NA | NA |
Negative | 0 | 0 |
100 to 800 | 17 (33.3) | 8 (16.7) |
1,600 to 6,400 | 20 (39.2) | 20 (41.7) |
≥ 12,800 | 10 (19.6) | 16 (33.3) |
NA = not applicable.
aTreatment-emergent anti-drug antibody incidence is defined as the sum of treatment boosted and treatment-induced anti-drug antibody positive patients divided by the number of evaluable patients multiplied by 100.
bPeak titre is the highest anti-drug antibody titre after baseline for patients who seroconverted or developed anti-drug antibodies de novo.
Source: COMET Clinical Study Report.11
Critical Appraisal
Internal Validity
Overall, the COMET study was well designed, with a 95% completion rate for the double-blind phase. Randomization was successful at balancing most prognostic factors at baseline, although it is worth noting a few key differences between the 2 arms. Patients who received avalglucosidase alfa had a younger mean age (46 years versus 50 years), a greater 6MWT mean distance (399 m versus 378 m), a younger mean age at diagnosis (45 years versus 48 years), a shorter mean time from diagnosis to first infusion of the study drug (16 months versus 27 months), and fewer used a mobility aid during the 6MWT (44 patients versus 39 patients). Conversely, those randomized to receive alglucosidase alfa had better baseline MEP (% predicted) (75% versus 66%) and HHD (lower extremity) scores (1,466 versus 1,330). Of note, the time between diagnosis and first infusion of the study drug was different between the treatment groups and was not adjusted for in the statistical analysis, which may confound the results. The clinical expert noted that the differences in baseline characteristics in most cases tended to cause biases in the results in favour of the avalglucosidase alfa group. Patients who have symptom onset at a younger age have more severe disease,1 and earlier treatment is expected to result in better outcomes, but it is unclear the direction and magnitude of the biases caused by the differences in baseline characteristics. No baseline information was reported on the duration of noninvasive respiratory support (e.g., BiPap at night versus day, and how many hours), which the clinical expert suggested has a large impact on a patient’s quality of life, and can also be used to measure treatment efficacy.
Blinding of patients, investigators, and study personnel to treatment assignment and allocation concealment — through the use of independent pharmacists who prepared the study drugs and a centralized treatment allocation system — appeared to be adequate. Based on the harms profiles of avalglucosidase alfa and alglucosidase alfa, there appeared to be little risk of unblinding on the basis of AEs. All 5 (10.2%) patients who discontinued treatment during the double-blind phase were from the alglucosidase alfa arm, and it is unknown what impact these losses had on the results, considering the small patient numbers at the start of the study.
The ITT population was defined as all randomized patients, and the mITT population was defined as all randomized patients who received at least 1 infusion of the study drug. The 2 populations were the same, and no sensitivity analysis was performed. Although the ITT analysis is considered conservative for superiority trials, this is not the case for noninferiority trials.12 Thus, the presentation of both mITT and PP analyses for the primary outcome helped to confirm that the analyses produced similar estimates of the treatment effect, which allowed the results to be interpreted with greater confidence.
Sample size and study power calculations were based on 3 previous studies that investigated the use of alglucosidase alfa for treatment-naïve patients with LOPD — a patient population that is generally consistent with the COMET trial, although specific eligibility criteria and study designs differed. A 10% dropout rate was estimated based on 2 of the 3 studies (LOTS and EMBASSY), resulting in a necessary sample size of at least 96 patients for the COMET trial to have approximately 80% power. In total, 100 patients were enrolled in the COMET study, of whom 95 completed the double-blind phase. The MMRM statistical models with fixed effects for baseline FVC, age, sex, treatment group, visit, and treatment-by-visit interaction used were considered acceptable for the primary analysis. Missing data were not imputed for the primary outcome, and it was assumed that data were missing at random. This assumption may bias 1 treatment over another, although sensitivity analyses were performed to assess the impact of missing data, which supported the missing-at-random assumption for the primary outcome. Procedures for handling missing data were described for some secondary (e.g., 6MWT, MEP, MIP, and HHD) and tertiary outcomes. Data were missing for nearly all outcomes; thus, the mITT population was not fully represented, and it is unclear what impact the missing data had on the results and conclusions. As these were secondary outcomes, and statistical testing was stopped, there is a high degree of uncertainty when interpreting these results. Sensitivity analyses (both a priori and post hoc analyses) indicated that patients who were excluded for various reasons (e.g., protocol deviation, implausible MEP or MIP values) did not change the noninferiority conclusion. To control for multiplicity, a sequential testing strategy was used. Avalglucosidase alfa showed noninferiority to alglucosidase alfa at the –1.1% margin for the primary outcome, but failed to show superiority, which stopped all subsequent statistical testing. Because testing stopped at the primary outcome, results for all secondary and tertiary outcomes for efficacy and HRQoL were not controlled for type I errors and should be interpreted as supportive of the primary outcome. Subgroup analyses for the primary outcome were specified a priori; although there was no control for multiplicity, each subgroup had a small number of patients and the wide 95% CIs indicated imprecision with the estimates.
The noninferiority margin was based on the double-blind, placebo-controlled LOTS trial for alglucosidase alfa 20 mg/kg every other week, with eligibility criteria similar to that of the COMET trial. The noninferiority margin of –1.1% retained approximately half of the lower bound of the 80% CI of the estimated treatment effect of alglucosidase alfa over placebo for FVC (% predicted) in the LOTS trial at 12 months (i.e., 2.14). The clinical expert believed that this was a reasonable approach to estimate the margin and that it was also a reasonable choice of margin from a clinical perspective. Retention of half of the comparator’s treatment effect is consistent with FDA guidance for noninferiority trials.12 Although FDA guidance12 indicates that a 95% CI is commonly used, the COMET publication13 stated that the CI was lowered to 80% in response to a suggestion from regulatory bodies. The rationale for this was not further described.
The constancy assumption is such that the effect of the active comparator (i.e., alglucosidase alfa) in the current noninferiority trial is the same as the effect observed in past trials and requires the trials to be sufficiently similar.14 The study designs, eligibility criteria, treatment doses, and key outcomes were similar to or the same as those in the COMET and LOTS trials, which supports the constancy assumption. Furthermore, the pre-specified constancy-assumption analysis estimated an effect of 2.87 for alglucosidase alfa, compared with placebo, in the COMET trial, and an effect of 3.02 in the LOTS trial, based on the predictive model.13 The investigators considered the difference in effect to be small (–0.15) compared with the noninferiority margin (1.1%).13 Pompe disease is rare, but the clinical expert noted that the patients who received alglucosidase alfa were similar overall in the COMET and LOTS trials. However, there were key differences in baseline characteristics — such as older baseline age, older age at symptom onset, higher FVC, and better 6MWT — for patients who received alglucosidase alfa in the COMET trial and LOTS trial. Although these differences should bias the results in favour of alglucosidase alfa treatment in the COMET study, the clinical expert did not feel they explained why the patients in the COMET trial did not respond as well as those in the LOTS trial. Consequently, the lower-than-expected improvements in the alglucosidase alfa arm in the COMET study could bias interpretation of the results in favour of treatment with avalglucosidase alfa. Although these concerns may affect interpretation of the trend toward superiority noted in the COMET trial, it is the opinion of the clinical expert that the concerns do not affect the statistically significant conclusions of the trial that avalglucosidase alfa is noninferior to alglucosidase alfa.
External Validity
In general, patients in the COMET study resembled those seen in clinical practice in Canada. The eligibility criteria included patients with LOPD as young as 3 years. However, only 1 pediatric patient was enrolled in the COMET study; all the other participants were adult patients. Despite the limited evidence for treatment of pediatric patients with LOPD, the clinical expert consulted on this review was of the opinion that the results are generalizable to pediatric patients, but not to patients with IOPD, and highlighted the urgent need for additional data in the IOPD population.
The multi-centre trial took place in 26 countries and included 2 Canadian sites. The clinical expert noted that clinical practice, background care, and reporting of AEs can vary in different countries, which may confound the results. Canadian guidance for the management of patients with Pompe disease suggests regular assessments at least every 6 months, which is less frequent than the study visits in the COMET trial. The greater access to health care resources and attention from clinicians during clinical trials should be considered when generalizing the results to real-world practice.
The avalglucosidase alfa dose of 20 mg/kg body weight every other week is consistent with both the product monograph and the alglucosidase alfa dose used to treat patients with LOPD. Alglucosidase alfa is the only available enzyme-replacement therapy for Pompe disease and, thus, is an appropriate comparator. Home infusions were allowed during the open-label phase when national regulations permitted, which is consistent with Canadian clinical practice.
Given that LOPD is a lifelong condition, the 1 year of data available from the double-blind phase provides only a short-term look at the efficacy and safety results of treatment with avalglucosidase alfa. Long-term efficacy and safety are still unknown and the extension phase of the COMET trial is ongoing (median time was 11 months as of March 19, 2020, the COMET trial primary completion date). When the open-label extension phase began after week 49, patients who started on avalglucosidase alfa continued to receive the medication and patients who had been receiving alglucosidase alfa switched to avalglucosidase alfa. Although it was not part of the primary analysis, the treatment switch meant a loss of the control arm, and the open-label design meant that patients were aware they were receiving avalglucosidase alfa, which could have affected subjective outcomes (e.g., SF-12). The benefit is that the treatment switch allows for the collection of data from patients who started on alglucosidase alfa and changed to avalglucosidase alfa after 1 year.
The literature search conducted to inform the description and appraisal of outcome measures showed there was a lack of evidence supporting the validity, reliability, or responsiveness to change for some of the trial outcomes. Therefore, there is uncertainty around the use of some measures, such as MEP, SF-12, and GMFM-88, to assess treatment for patients with LOPD. Furthermore, a literature search did not find any MIDs for populations with Pompe disease. The sponsor’s submission compared the noninferiority margin with a MID for FVC (% predicted) between 2% and 6%, based on patients with idiopathic pulmonary fibrosis, although the clinical expert believes that idiopathic pulmonary fibrosis and Pompe disease may not be directly comparable because of different physiologies and disease processes. Upon consultation with the clinical expert, in general, the outcome measures were deemed to be clinically relevant to the assessment of treatment in patients with Pompe disease. Moreover, the trial end points aligned with the most important outcomes identified by the patient group submission (i.e., motor- and respiratory-related and HRQoL outcomes). Patient input highlighted concern with how Pompe disease affected social and mental health, activities of daily living, and families or caregivers, but these factors were not the focus of the COMET study or this review. The clinical expert noted that both the primary outcome of FVC and the key secondary outcome of 6MWT have available data for patients with Pompe disease, although some clinics may be restricted in their ability to conduct the tests. For instance, a clinic must have assessors trained to evaluate the 6MWT, as well as the physical space to perform the test.
Although there may be some limitations to generalizability, as discussed here, the results of the COMET trial appear to be generalizable to Canadian clinical practice for the treatment of patients with LOPD.
Indirect Evidence
Objectives and Methods for the Summary of Indirect Evidence
A focused literature search for network meta-analyses dealing with Pompe disease was run in MEDLINE All (1946–) on October 28, 2021. No limits were applied to the search. Indirect treatment evidence for avalglucosidase alfa was not identified in this review.
Other Relevant Evidence
This section includes 1 open-label, dose-escalation study and 1 long-term extension of the same open-label, dose-escalation study included in the sponsor’s submission to CADTH that were considered to address important gaps in the evidence included in the systematic review. Safety and exploratory efficacy were evaluated within the sponsor’s submission. A summary and critical appraisal of the additional evidence are presented in this section.
Dose-Escalation Study and Long-Term Extension
TDR12857 (NEO1) was a phase I, multi-centre, open-label, ascending-dose study to determine safety and tolerability, pharmacokinetic parameters, and pharmacodynamic effects of avalglucosidase alfa in treatment-naïve patients with LOPD and in patients with LOPD previously treated with alglucosidase alfa. Three dosing regimens were analyzed in the study; however, only the 20 mg/kg regimen is approved by Health Canada.
LTS13769 (NEO-EXT) is the long-term extension of NEO1. All patients who completed treatment with the 5 mg/kg dose or 10 mg/kg dose were invited to enrol in the extension, where they would receive 20 mg/kg. Both studies will be presented alongside each other in this section. The results will focus on the Health Canada–approved 20 mg/kg dose.
Methods
Two groups of patients were included in the study. Group 1 included treatment-naïve patients with LOPD and group 2 included patients with LOPD who were previously treated with alglucosidase alfa for a minimum of 9 months. Both groups received IV infusions of avalglucosidase alfa every other week for a total of 13 infusions. Three dosing regimens — 5 mg/kg, 10 mg/kg, and 20 mg/kg — were included. After a 90-day screening period, patients receiving the 5 mg/kg dose began the 24-week treatment period, which was followed by post-treatment evaluation at week 27 and an end-of-study visit at week 29. Successive escalating-dosage groups were initiated 5 weeks after initiation of the previous dose.
After completion of NEO1, patients were invited to enrol in the extension study, where either continued to receive the 20 mg/kg dose or were switched to the 20 mg/kg dose. In the extension study, the planned duration for each patient was 6 years. Patients continued in the study until the patient withdrew, the investigator withdrew the patient, or the sponsor terminated the study.
Populations
Patients enrolled in NEO1 were required to be at least 18 years of age with a confirmed GAA enzyme deficiency and/or confirmed GAA gene mutation, without known cardiac hypertrophy. To be enrolled in group 2, patients must have been treated with alglucosidase alfa for at least 9 months. FVC in the upright position of at least 50% of predicted and the ability to ambulate 50 m without stopping and without an assistive device were also requirements for enrolment. Exclusion criteria for NEO1 included dependence on a wheelchair, a need for invasive ventilation, and the unlikelihood that the patient, in the opinion of the investigator, would adhere to the requirements of the study.
A summary of baseline characteristics is shown in Table 15. Mean age was 49.1 (SD = 25.61) years in group 1 patients who received the 20 mg/kg dose and 43.8 (SD = 17.05) years in group 2 patients who received the 20 mg/kg dose. All patients in group 1 and group 2 who received 20 mg/kg were White, and their mean weight was 67.6 kg (SD = 12.55) and 78.1 kg (SD = 23.91), respectively. One-third of group 1 patients who received the 20 mg/kg dose required an assistive walking device for community ambulation, but no group 2 patients who received the 20 mg/kg dose required such assistance.
The NEO-EXT trial included only patients from the NEO1 trial; therefore, baseline characteristics were identical.
Table 15: Summary of Baseline Characteristics — NEO1, Full Analysis Set
Characteristic | Group 1: 5 mg/kg, N = 4 | Group 1: 10 mg/kg, N = 3 | Group 1: 20 mg/kg, N = 3 | Group 2: 5 mg/kg, N = 4 | Group 2: 10 mg/kg, N = 4 | Group 2: 20 mg/kg, N = 6 |
|---|---|---|---|---|---|---|
Demographics | ||||||
Age, years | NA | NA | NA | NA | NA | NA |
Mean (SD) | 55.8 (14.67) | 26.0 (8.23) | 49.1 (25.61) | 50.6 (13.89) | 47.2 (12.13) | 43.8 (17.05) |
Median (range) | 56.0 (38.2 to 73.0) | 22.8 (19.8 to 35.3) | 38.5 (30.5 to 78.3) | 50.7 (33.6 to 67.5) | 44.6 (36.4 to 63.3) | 41.4 (20.5 to 67.1) |
Sex, n (%) | NA | NA | NA | NA | NA | NA |
Male | 1 (25.0) | 0 | 2 (66.7) | 2 (50.0) | 3 (75.0) | 4 (66.7) |
Female | 3 (75.0) | 3 (100.0) | 1 (33.3) | 2 (50.0) | 1 (25.0) | 2 (33.3) |
Race, n (%) | NA | NA | NA | NA | NA | NA |
White | 4 (100.0) | 1 (33.3) | 3 (100.0) | 4 (100.0) | 3 (75.0) | 6 (100.0) |
Black | 0 | 0 | 0 | 0 | 1 (25.0) | 0 |
Multiple | 0 | 1 (33.3) | 0 | 0 | 0 | 0 |
Other | 0 | 1 (33.3) | 0 | 0 | 0 | 0 |
Weight in kg, mean (SD) | 63.8 (3.24) | 63.9 (15.76) | 67.6 (12.55) | 72.8 (8.22) | 78.6 (6.15) | 78.1 (23.91) |
BMI in kg/m2, mean (SD) | 23.7 (0.91) | 21.9 (4.59) | 20.9 (3.87) | 23.7 (2.25) | 24.3 (1.07) | 25.4 (5.49) |
Age at diagnosis of Pompe disease, years, mean (SD) | 65.7 (9.33) | 23.0 (9.97) | 48.6 (25.99) | 48.1 (NE) | 36.3 (6.50) | 34.0 (21.79) |
Baseline disease characteristics | ||||||
Assistive walking devices and orthoses, n (%) | NA | NA | NA | NA | NA | NA |
None | 3 (75.0) | 3 (100.0) | 2 (66.7) | 2 (50.0) | 3 (75.0) | 6 (100.0) |
Rolling walker | 0 | 0 | 1 (33.3) | 0 | 1 (25.0) | 0 |
Straight cane | 0 | 0 | 0 | 2 (50.0) | 0 | 0 |
Other: 2 walking poles | 1 (25.0) | 0 | 0 | 0 | 0 | 0 |
BMI = body mass index; NA = not applicable; NE = not evaluable; SD = standard deviation.
Note: Group 1 includes patients with LOPD who were treatment-naïve. Group 2 includes patients with LOPD who were previously treated with alglucosidase alfa.
Source: NEO1 Clinical Study Report.49
Interventions
In NEO1, avalglucosidase alfa was administered to all patients at a dose of 5 mg/kg, 10 mg/kg, or 20 mg/kg every other week for a total of 13 infusions. Infusion length was dependent on the dose received by the patient. Infusion administration began at a slow initial rate and increased if there were no signs of infusion-associated reactions. In NEO-EXT, all patients received the 20 mg/kg dose.
Outcomes
The primary outcomes of NEO1 were related to pharmacodynamics and were not relevant to this report. Assessment of safety and tolerability was another main objective of the study. An AE was defined as any untoward medical occurrence in a patient that did not necessarily have a causal relationship with the study drug. A SAE was defined as any untoward medical occurrence that resulted in death, was life-threatening, required inpatient hospitalization, resulted in persistent or significant disability, was a congenital anomaly, or was considered medically important. AEs of special interest included infusion-associated reactions, pregnancy, and alanine aminotransferase, aspartate transaminase, and serum creatinine abnormalities. All efficacy outcomes were considered exploratory. These measures included pulmonary function tests, 6MWT, GSGC, GMFM-88, QMFT, HHD, and the Pediatric Quality of Life Inventory; only the pulmonary function tests and 6MWT are presented in detail in this report. Results will only be presented for patients who received the Health Canada–approved 20 mg/kg dose.
Statistical Analysis
There was no comparator arm in this phase I trial and, as such, all outcomes are descriptive in nature; no formal statistical testing was conducted. Treatment-naïve patients are referred to as group 1 and patients previously treated with alglucosidase alfa are referred to as group 2.
Patient Disposition
A summary of patient disposition in NEO1 is shown in Table 16. Nearly all patients who received the 20 mg/kg dose completed the trial, with 1 patient in group 1 discontinuing due to SAE and 2 patients in group 2 choosing to withdraw from the study.
Of the 21 patients who completed NEO1, 19 enrolled in NEO-EXT. At the time of the February 27, 2020, data cut, 2 patients had discontinued from the extension study, so 17 patients remained on the study drug. Reasons cited for discontinuation of the extension study were patient’s wishes to discontinue and other.
Table 16: Patient Disposition — NEO1, Full Analysis Set
Patient disposition | Group 1: 5 mg/kg, N = 4 | Group 1: 10 mg/kg, N = 3 | Group 1: 20 mg/kg, N = 3 | Group 2: 5 mg/kg, N = 4 | Group 2: 10 mg/kg, N = 4 | Group 2: 20 mg/kg, N = 6 |
|---|---|---|---|---|---|---|
Full analysis set, n (%) | 4 (100) | 3 (100) | 3 (100) | 4 (100) | 4 (100) | 6 (100) |
Safety analysis set, n (%) | 4 (100) | 3 (100) | 3 (100) | 4 (100) | 4 (100) | 6 (100) |
Treated, n (%) | 4 (100) | 3 (100) | 3 (100) | 4 (100) | 4 (100) | 6 (100) |
Completed, n (%) | 3 (75.0) | 3 (100) | 3 (100) | 3 (75.0) | 4 (100) | 5 (83.3) |
Discontinued treatment, n (%) | 1 (25.0) | 0 | 0 | 1 (25.0) | 0 | 1 (16.7) |
Reason for discontinuation, n (%) | NA | NA | NA | NA | NA | NA |
AE | 1 (100) | 0 | 0 | 0 | 0 | 0 |
Noncompliant | 0 | 0 | 0 | 0 | 0 | 0 |
Chose to withdraw | 0 | 0 | 0 | 1 (100) | 0 | 1 (100) |
Lost to follow-up | 0 | 0 | 0 | 0 | 0 | 0 |
Other | 0 | 0 | 0 | 0 | 0 | 0 |
AE = adverse event; NA = not applicable.
Note: Group 1 includes patients with LOPD who were treatment-naïve. Group 2 includes patients with LOPD who were previously treated with alglucosidase alfa.
Source: NEO1 Clinical Study Report.49
Exposure to Study Treatments
A summary of treatment exposure in NEO1 is presented in Table 17. Mean duration on avalglucosidase alfa was 175.3 days (SD = 1.15) in group 1 patients who received the 20 mg/kg dose and 161.5 days (SD = 29.95) in group 2 patients who received the 20 mg/kg dose. All patients in group 1 who received the 20 mg/kg dose received the full 13 infusions, whereas group 2 patients who received the 20 mg/kg dose received a mean of 12.2 (SD = 2.04) infusions. In the extension study, mean duration on the study drug was 221.4 weeks (SD = 136.9) in group 1 and 242.3 weeks (SD = 124.5) in group 2.
Table 17: Exposure to Treatment — NEO1 and NEO-EXT, Full Analysis Set
Exposure to treatment | NEO1 | NEO-EXT | ||||||
|---|---|---|---|---|---|---|---|---|
Group 1: 5 mg/kg, N = 4 | Group 1: 10 mg/kg, N = 3 | Group 1: 20 mg/kg, N = 3 | Group 2: 5 mg/kg, N = 4 | Group 2: 10 mg/kg, N = 4 | Group 2: 20 mg/kg, N = 6, | Group 1 combined N = 10 | Group 2 combined, N = 14 | |
Duration on study druga | NA | NA | NA | NA | NA | NA | NA | NA |
Mean (SD) | 156.5 (32.25) | 168.7 (0.58) | 175.3 (1.15) | 170.3 (1.26) | 168.3 (2.06) | 161.5 (29.94) | 221.4 (136.9) | 242.3 (124.5) |
Median (range) | 168.0 (109.0 to 181.0) | 169.0 (168.0 to 169.0) | 176.0 (174.0 to 176.0) | 170.0 (169.0 to 172.0) | 168.0 (166.0 to 171.0) | 172.0 (102.0 to 183.0) | 293.3 (17.0 to 329.0) | 304.4 (16.0 to 340.0) |
Number of study drug infusions | NA | NA | NA | NA | NA | NA | NA | NA |
Mean (SD) | 12.0 (2.00) | 13.0 (0.00) | 13.0 (0.00) | 12.8 (0.50) | 13.0 (0.00) | 12.2 (2.04) | 104.3 (64.4) | 117.3 (60.7) |
Median (range) | 13.0 (9.0 to 13.0) | 13.0 (13.0 to 13.0) | 13.0 (13.0 to 13.0) | 13.0 (12.0 to 13.0) | 13.0 (13.0 to 13.0) | 13.0 (8.0 to 13.0) | 137.5 (9.0 to 162.0) | 151.0 (8.0 to 164.0) |
Cumulative study drug (mg/kg) | NA | NA | NA | NA | NA | NA | NA | NA |
Mean (SD) | 60.0 (10.00) | 130.0 (0.00) | 260.0 (0.00) | 63.8 (2.50) | 130.0 (0.00) | 243.3 (40.82) | NR | NR |
Median (range) | 65.0 (45.0 to 65.0) | 130.0 (130.0 to 130.0) | 260.0 (260.0 to 260.0) | 65.0 (60.0 to 65.0) | 130.0 (130.0 to 130.0) | 260.0 (160.0 to 260.0) | NR | NR |
NA = not applicable; NR = not reported; SD = standard deviation.
aDuration in days for NEO1 study. Duration in weeks for NEO-EXT study.
Note: Group 1 includes patients with LOPD who were treatment-naïve. Group 2 includes patients with LOPD who were previously treated with alglucosidase alfa.
Source: NEO1 Clinical Study Report,49 NEO-EXT Clinical Study Report.50
Efficacy
A summary of respiratory efficacy outcomes in NEO1 and NEO-EXT are presented in Table 18. All efficacy results in NEO1 were considered exploratory in nature. Only results in patients who received the Health Canada–approved dose of 20 mg/kg have been presented.
Survival-Related Outcomes
Survival-related outcomes were not reported in the NEO1 or NEO-EXT trials.
Respiratory-Related Outcomes
Respiratory-related efficacy measures in the NEO1 trial included FVC, MEP, and MIP. Baseline mean FVC was 63.4% (SD = 17.84) and 70.4% (SD = 16.40) in group 1 and group 2, respectively. At week 25, mean FVC changed to 69.5% (SD = 20.63) and 69.9% (SD = 16.92), in group and group 2, respectively, with a mean change from baseline of 6.2% (SD = 3.15) and 1.4% (SD = 5.71) for the respective groups. Baseline mean MEP was 66.1% (SD = 18.51) and 84.9% (SD = 25.21) in group 1 and group 2, respectively. At week 25, mean MEP changed to 78.1% (SD = 22.26) and 85.6% (SD = 17.71), in group 1 and group 2, respectively. Baseline mean MIP was 50.2% (SD = 19.89) and 74.2% (SD = 17.01) in group 1 and group 2, respectively. At week 25, mean MIP changed to 58.1% (SD = 17.97) and 72.8% (SD = 19.00), in group 1 and group 2, respectively.
In the extension study, baseline mean FVC was 69.2% (SD = 19.27) and 77.3% (SD = 16.45) in combined group 1 and combined group 2, respectively. At week 286, mean FVC changed to 65.7% (SD = 30.07) and 74.5% (SD = 21.24), in the combined group 1 and combined group 2, respectively. Baseline mean MEP was 75.7% (SD = 18.06) and 80.1% (SD = 29.47) in the combined group 1 and combined group 2, respectively. At week 286, mean MEP changed to 80.9% (SD = 33.57) and 82.8% (SD = 35.31), in the combined group 1 and combined group 2, respectively. Baseline mean MIP was 67.9% (SD = 30.52) and 67.2% (SD = 23.29) in the combined group 1 and combined group 2, respectively. At week 286, mean MEP changed to 64.8% (SD = 40.20) and 72.3% (SD = 20.22), in the combined group 1 and combined group 2, respectively. Results beyond week 286 were available; however, reduced patient numbers resulted in uninformative data.
Table 18: Respiratory Efficacy Outcomes — NEO1 and NEO-EXT
Outcome | NEO1 | NEO-EXT | ||
|---|---|---|---|---|
Group 1: 20 mg/kg, N = 3 | Group 2: 20 mg/kg, N = 6 | Group 1 combined, N = 10 | Group 2 combined, N = 14 | |
FVC (% predicted) in upright position | ||||
Baseline, mean (SD) | 63.4 (17.84) | 70.4 (16.40) | 69.2 (19.27) | 77.3 (16.45) |
Week 25, mean (SD) | 69.5 (20.63) | 69.9 (16.92) | NA | NA |
Change from baseline, mean (SD) | 6.2 (3.15) | 1.4 (5.71) | NA | NA |
Week 286, mean (SD) [N] | NA | NA | 65.7 (30.07) [7] | 74.5 (21.24) [9] |
Change from baseline, mean (SD) | NA | NA | –1.0 (13.16) | –6.0 (7.67) |
MEP (% predicted) in upright position | ||||
Baseline, mean (SD) | 66.1 (18.51) | 84.9 (25.21) | 75.7 (18.06) | 80.1 (29.47) |
Week 25, mean (SD) | 78.1 (22.26) | 85.6 (17.71) | NA | NA |
Change from baseline, mean (SD) | 12.0 (4.05) | 6.0 (21.80) | NA | NA |
Week 286, mean (SD) [N] | NA | NA | 80.9 (33.57) [7] | 82.8 (35.31) [7] |
Change from baseline, mean (SD) | NA | NA | 12.6 (24.76) | 9.8 (5.54) |
MIP (% predicted) in upright position | ||||
Baseline, mean (SD) | 50.2 (19.89) | 74.2 (17.01) | 67.9 (30.52) | 67.2 (23.93) |
Week 25, mean (SD) | 58.1 (17.97) | 72.8 (19.00) | NA | NA |
Change from baseline, mean (SD) | 7.9 (15.73) | –0.2 (6.85) | NA | NA |
Week 286, mean (SD) [N] | NA | NA | 64.8 (40.20) [7] | 72.3 (20.22) [7] |
Change from baseline, mean (SD) | NA | NA | 0.5 (22.48) | 1.5 (8.77) |
FVC = forced vital capacity; MEP = maximum expiratory pressure; MIP = maximum inspiratory pressure; NA = not applicable; SD = standard deviation.
Note: Group 1 includes patients with LOPD who were treatment-naïve. Group 2 includes patients with LOPD who were previously treated with alglucosidase alfa.
Source: NEO1 Clinical Study Report,49 NEO-EXT Clinical Study Report.50
Motor-Related Outcomes
Selected motor-related efficacy outcomes are presented in Table 19. Baseline mean 6MWT % predicted was 75.2% (SD = 9.80) and 72.8% (SD = 20.59) in group 1 and group 2, respectively. At week 25, mean 6MWT % predicted changed to 79.1% (SD = 12.55) and 65.6% (SD = 12.03), in group 1 and group 2, respectively, with a mean change from baseline of 3.9% (SD = 3.45) and –1.3% (SD = 8.94) for the respective groups. In the extension study, 6MWT was 64.9% (SD = 28.05) and 69.1% (SD = 21.37) at week 286, in group 1 and group 2, respectively. Results beyond week 286 were available; however, reduced patient numbers resulted in uninformative data.
Table 19: Motor-Related Efficacy Outcomes — NEO1 and NEO-EXT
6MWT (% predicted) outcome | NEO1 | NEO-EXT | ||
|---|---|---|---|---|
Group 1: 20 mg/kg, N = 3 | Group 2: 20 mg/kg, N = 6 | Group 1 combined, N = 10 | Group 2 combined, N = 14 | |
Baseline, mean (SD) | 75.2 (9.80) | 72.8 (20.59) | 65.5 (15.54) | 62.2 (17.63) |
Week 25, mean (SD) | 79.1 (12.55) | 65.6 (12.03) | NA | NA |
Change from baseline, mean (SD) | 3.9 (3.45) | –1.3 (8.94) | NA | NA |
Week 286, mean (SD) [N] | NA | NA | 64.9 (28.05) [6] | 69.1 (21.37) [8] |
Change from baseline, mean (SD) | NA | NA | 1.5 (10.22) | 1.3 (8.12) |
6MWT = 6-minute walk test; NA = not applicable; SD = standard deviation.
Note: Group 1 includes patients with LOPD who were treatment-naïve. Group 2 includes patients with LOPD who were previously treated with alglucosidase alfa.
Source: NEO1 Clinical Study Report,49 NEO-EXT Clinical Study Report.50
GSGC, GMFM, QMFT, and HHD were reported in the NEO1 and NEO-EXT studies but are not described in this report.
Health-Related Quality of Life
The Pediatric Quality of Life Inventory was reported in the NEO1 and NEO-EXT studies but not described in this report.
Harms
A summary of harms is presented in Table 20. In the initial study period of the NEO1 trial, 1 of the 3 patients in group 1 who received the 20 mg/kg dose experienced an AE, namely nasopharyngitis and erythema. All 6 patients in group 2 who received the 20 mg/kg dose experienced an AE; arthralgia and musculoskeletal pain were the only 2 AEs to be reported in multiple patients (33.3%). In NEO-EXT, all 24 patients, including those who switched to 20 mg/kg, experienced an AE. The most commonly reported AEs were nasopharyngitis (15 patients, 62.5%), fall (12 patients, 50.0%), diarrhea (11 patients, 45.8%), headache (10 patients, 41.7%), and muscle spasms (10 patients, 41.7%).
No patients who received the 20 mg/kg dose reported a SAE in the initial NEO1 study. In the NEO-EXT study, 9 patients (37.5%) reported a SAE, and there was no individual SAE that was reported in more than 1 individual patient. In the initial NEO1 study period, no patients who received the 20 mg/kg dose reported an AE that led to treatment discontinuation. In the NEO-EXT study, there was 1 patient (4.2%) who discontinued treatment due to an AE. No deaths due to AE were reported in either the NEO1 or NEO-EXT studies.
Notable harms, including anaphylactic reactions, hypersensitivity, infusion-associated reactions, and immune-mediated reactions, were less common during the initial NEO1 study period, and occurrence increased in the NEO-EXT study. In the NEO-EXT study, 17 patients (70.8%) experienced a treatment-emergent hypersensitivity AE, 12 patients (50.0%) experienced a treatment-emergent infusion-associated reaction, 2 patients (8.3%) experienced a treatment-emergent anaphylactic reaction, and no patients experienced a treatment-emergent immune-mediated reaction.
Table 20: Summary of Harms — NEO1 and NEO-EXT
Event | NEO1 | NEO-EXT | |
|---|---|---|---|
Group 1: 20 mg/kg, N = 3 | Group 2: 20 mg/kg, N = 6 | Combined,a N = 24 | |
Patients with ≥ 1 AE by primary system organ class and preferred term,a n (%) | 1 (33.3) | 6 (100.0) | 24 (100.0) |
Eye disorders | 0 | 1 (16.7) | NA |
Vision blurred | 0 | 1 (16.7) | NA |
Gastrointestinal disorders | 0 | 1 (16.7) | 19 (79.2) |
Abdominal pain | 0 | 1 (16.7) | 6 (25.0) |
Nausea | 0 | 0 | 9 (37.5) |
Diarrhea | 0 | 0 | 11 (45.8) |
General disorders and administration-site conditions | 0 | 4 (66.7) | 18 (75.0) |
Asthenia | 0 | 1 (16.7) | NA |
Facial pain | 0 | 1 (16.7) | NA |
Fatigue | 0 | 1 (16.7) | 7 (29.2) |
Injection-site reaction | 0 | 1 (16.7) | NA |
Pyrexia | 0 | 1 (16.7) | 6 (25.0) |
Immune system disorders | 0 | 1 (16.7) | NA |
Hypersensitivity | 0 | 1 (16.7) | NA |
Infections and infestations | 1 (33.3) | 4 (66.7) | 22 (91.7) |
Nasopharyngitis | 1 (33.3) | 0 | 15 (62.5) |
Acute tonsilitis | 0 | 1 (16.7) | NA |
Influenza | 0 | 0 | 6 (25.0) |
Gastroenteritis viral | 0 | 1 (16.7) | 6 (25.0) |
Rhinitis | 0 | 1 (16.7) | NA |
Upper respiratory tract infection | 0 | 1 (16.7) | 8 (33.3) |
Tooth abscess | 0 | 1 (16.7) | NA |
Injury, poisoning, and procedural complications | 0 | 1 (16.7) | 15 (62.5) |
Chest injury | 0 | 1 (16.7) | NA |
Fall | 0 | 1 (16.7) | 12 (50.0) |
Musculoskeletal and connective tissue disorders | 0 | 4 (66.7) | 19 (79.2) |
Arthralgia | 0 | 2 (33.3) | 7 (29.2) |
Back pain | 0 | 1 (16.7) | 9 (37.5) |
Myalgia | 0 | 0 | 9 (36.7) |
Muscle spasms | 0 | 0 | 10 (41.7) |
Muscle tightness | 0 | 1 (16.7) | NA |
Musculoskeletal pain | 0 | 2 (33.3) | 8 (33.3) |
Neck pain | 0 | 1 (16.7) | NA |
Pain in extremity | 0 | 1 (16.7) | 7 (29.2) |
Nervous system disorders | 0 | 1 (16.7) | 16 (66.7) |
Headache | 0 | 1 (16.7) | 10 (41.7) |
Dizziness | 0 | 0 | 7 (29.2) |
Somnolence | 0 | 1 (16.7) | NA |
Syncope | 0 | 1 (16.7) | NA |
Reproductive, system, and breast disorders | 0 | 1 (16.7) | NA |
Balanoposthitis | 0 | 1 (16.7) | NA |
Respiratory, thoracic, and mediastinal disorders | 0 | 3 (50.0) | 16 (66.7) |
Oropharyngeal pain | 0 | 0 | 6 (25.0) |
Epistasis | 0 | 1 (16.7) | NA |
Painful respiration | 0 | 1 (16.7) | NA |
Upper respiratory tract congestion | 0 | 1 (16.7) | NA |
Skin and subcutaneous tissue disorders | 1 (33.3) | 2 (33.3) | 14 (58.3) |
Erythema | 1 (33.3) | 1 (16.7) | NA |
Rash | 0 | 1 (16.7) | 8 (33.3) |
Pruritis | 0 | 1 (16.7) | NA |
Vascular disorders | 0 | 1 (16.7) | 12 (50.0) |
Hematoma | 0 | 1 (16.7) | NA |
Patients with ≥ 1 SAE by primary system organ class and preferred term, n (%) | 0 | 0 | 9 (37.5) |
WDAE, n (%) | 0 | 0 | 1 (4.2) |
Deaths, n (%) | 0 | 0 | 0 |
Notable harms, n (%) | NA | NA | NA |
Treatment-emergent anaphylactic reactions by SMQ, broad and narrow combined, n (%) | 0 | 0 | 2 (8.3) |
Treatment-emergent hypersensitivity reactions by SMQ, broad and narrow combined, n (%) | 1 (33.3) | 2 (33.3) | 17 (70.8) |
Treatment-emergent infusion-associated reactions, n (%) | 1 (33.3) | 1 (16.7) | 12 (50.0) |
Treatment-emergent immune-mediated reactions, n (%) | ||||||| | | |||||| | ||||||| |
AE = adverse events; NA = not applicable; SAE = serious adverse events; SMQ = Standardized MedDRA Queries; WDAE = withdrawal due to adverse events.
aAEs in NEO-EXT were reported only for those exceeding 25%.
Note: Group 1 includes patients with LOPD who were treatment-naïve. Group 2 includes patients with LOPD who were previously treated with alglucosidase alfa.
Source: NEO1 Clinical Study Report,49 NEO-EXT Clinical Study Report.50
Critical Appraisal
Internal Validity
The phase I NEO1 dose-escalation study was conducted to assess the safety and tolerability of avalglucosidase alfa in patients with LOPD and to characterize the pharmacodynamic and pharmacokinetic profiles. The long-term extension, NEO-EXT, followed these patients for more than 4 years to assess safety and tolerability over a longer period. Efficacy outcomes were considered strictly exploratory. The number of patients who discontinued the trial was acceptable, and therefore there is little concern that results are biased in favour of avalglucosidase alfa for this reason. Inherent to phase I trials are the issues of a low number of patients enrolled, lack of a comparator arm, and lack of randomization. As a result, it is not possible to determine a causal relationship between the study drug and outcomes observed. The baseline demographics varied between patients receiving different doses, likely due to the low number of patients. Specifically, the mean age of group 1 patients receiving 10 mg/kg was 26 years, which is far younger than the rest of the study population. As the clinical expert explained, patients diagnosed earlier in life tend to progress more rapidly than those who present at a later age, and it would be expected that the age difference between groups 1 and 2 would bias the findings against the younger group 1 patients.
External Validity
The inclusion of the long-term extension of the phase I NEO1 study in the sponsor’s submission allows for greater assessment of the safety and tolerability data beyond the time points presented in the pivotal trials. However, the study design greatly limits the generalizability of any findings.
Discussion
Summary of Available Evidence
One multi-centre, active comparator, double-blind, phase III study was included in the CADTH systematic review. The COMET study was designed to assess the efficacy and safety of avalglucosidase alfa 20 mg/kg administered by IV infusion every other week for the treatment of patients with LOPD. Patients received either avalglucosidase alfa or alglucosidase alfa at a dose of 20 mg/kg body weight. The double-blind phase consisted of 49 weeks of treatment; after that, patients who were receiving alglucosidase alfa were switched to avalglucosidase alfa for the open-label phase, which lasted up to 240 weeks. To be included, patients had to be treatment-naïve, 3 years or older, have confirmed GAA enzyme deficiency from any tissue source and/or 2 confirmed GAA gene mutations, and no known Pompe-specific cardiac hypertrophy. Patients must also have been able to ambulate at least 40 m without stopping and without assistive device, be able to successfully perform repeated FVC measurements in the upright position between 30% and 85% predicted, and not require invasive ventilation. In the COMET study, avalglucosidase alfa was compared against alglucosidase alfa for the primary outcome of change from baseline to week 49 in FVC (% predicted) upright. Secondary outcomes included the 6MWT (total distance and % predicted), MEP, MIP, HHD (lower extremity composite score), QMFT, SF-12, and safety. The study included several tertiary and exploratory outcomes, of which the GSGC and GMFM-88 (dimensions D and E) were included in this report.
In total, 100 patients with LOPD were randomized to either avalglucosidase alfa (n = 51) or alglucosidase alfa (n = 49). Patients were between 16 and 78 years of age (mean = 48 years), 52% were male, 94% were White, and 34% were from North America. The mean age of patients who received avalglucosidase alfa was slightly younger than that of those who received alglucosidase alfa, their age at diagnosis was younger, and they had a shorter time between diagnosis and treatment. The mean baseline FVC for all patients was 62.1% predicted, which was similar among the treatment groups. The mean distance on the 6MWT was 388.9 m and 56.3% predicted, both of which were slightly greater in the avalglucosidase alfa group.
The major limitations of the COMET study were the small number of patients and lack of data for pediatric patients. The small number of patients limits the ability to accurately quantify the differences in outcomes, beyond the primary outcome, in long-term benefits and in potential harms between the treatments. Furthermore, the differences in baseline age and time from diagnosis to treatment may result in bias, although the direction and magnitude are unclear. The 5 patients who discontinued treatment during the double-blind phase were from the alglucosidase alfa group, and it is unknown what impact these losses had on the results. Data for week 49 were missing for primary and secondary outcomes, with a lack of information on how missing values were handled for many of the outcomes, which must be considered when interpreting the results. There was also a lack of evidence to support the validity, reliability, or responsiveness for outcome measures, and no MIDs for populations with Pompe disease were identified. Currently, there are limited long-term data available for the COMET study, as the open-label phase is still ongoing.
Two other relevant studies were summarized for this review that provided long-term data for the use of avalglucosidase alfa for the treatment of patients with LOPD. The studies included the open-label, phase I NEO1 and NEO-EXT studies that evaluated patients (N = 19) who were either treatment-naïve or had previously been treated with alglucosidase alfa. NEO1 consisted of treatment for up to 25 weeks, whereas in NEO-EXT, treatment was for up to 6 years. The major limitations included the small number of patients, lack of comparator arm, and lack of randomization, which hinder interpretation of the results and generalizability to clinical practice. No indirect treatment comparisons were identified for this review.
Interpretation of Results
Efficacy
In general, the results of the COMET study indicated that patients responded to both avalglucosidase alfa and alglucosidase alfa, compared with the natural history of Pompe disease (i.e., without treatment). The clinical expert emphasized that, given the progressive nature of the disease, improvement in a patient’s condition is desirable, but the goal of treatment is to prevent further decline. Furthermore, the clinical expert noted that patients who are diagnosed with Pompe disease at an earlier stage of muscle involvement and treated earlier can be maintained at a higher level of muscle function, which impacts patient morbidity.
The mean difference of change between treatment groups for FVC (% predicted) of both mITT and PP analyses at week 49 favoured avalglucosidase alfa (2.43% and 2.69%, respectively). Moreover, neither of the lower bounds of the 95% CIs (–0.13 and –0.06, respectively) exceeded the noninferiority margin of –1.1%, supporting the noninferiority conclusion (P = 0.0074 and P = 0.0076, respectively). Superiority testing followed, but neither P value from the mITT and PP analyses were statistically significant (P = 0.0626 and P = 0.0555, respectively); thus, superiority of avalglucosidase alfa could not be established and statistical testing stopped for all subsequent outcomes. Because no MID was identified from the literature for populations with Pompe disease, it was difficult to assess how meaningful the changes observed in the study were. Nevertheless, the clinical expert noted that any improvement in FVC was beneficial, compared with the natural and constant decline seen in untreated patients.
Because of inherent limitations in subgroup analyses, the overall design of the study, the small sample size, and the demonstrated wide confidence intervals in the results of the subgroups analyses, there is so much uncertainty in these results that they make the use of this information unhelpful for decision-making.
The results for the secondary and tertiary outcomes were generally as expected, in that treated patients did not show a major decline, compared with what would be expected and observed in untreated patients. Statistical testing was stopped after avalglucosidase alfa failed to show superiority for the primary outcome. The direction of estimates for all secondary and tertiary outcomes included in the results section of the CADTH report are in support of avalglucosidase alfa over alglucosidase alfa, except for MEP (% predicted) and GSGC. The clinical expert noted that changes from baseline in HRQoL, as measured by the SF-12, were small. Despite enzyme-replacement therapy possibly being effective and patients feeling better and responding well to treatment, the burden of treatment administration should not be overlooked because infusions are frequent and require hours to complete, which can have a large impact on patients’ HRQoL. Administration of avalglucosidase alfa is the same as for alglucosidase alfa and may not offer notable benefits to patients in this regard. Because tertiary and exploratory outcomes tend to be descriptive and hypothesis-generating, the outcomes assessed that were deemed not to be pivotal to the review were not included. Moreover, the high proportion of missing data for these outcomes reduced their use for the purposes of the CADTH review.
The long-term effects on FVC (% predicted) and 6MWT (distance and % predicted) appear to be sustained throughout the open-label phase of the COMET trial. The clinical expert expected that the best result of current treatment would be for patients to remain stable over time, rather than show continual improvement in measured outcomes, but reinforced the fact that stability is contrary to the natural history of the disease in untreated patients, so resulting benefits would be in considerable over time. Long-term data are limited to 97 weeks for the COMET trial, and it is difficult to draw any conclusions on treatment efficacy with certainty. Moreover, no statistical testing was conducted for end points during the open-label phase and are strictly supportive in nature.
Although there were few patients in the NEO1 and NEO-EXT studies (9 and 19 patients received avalglucosidase alfa 20 mg/kg, respectively), the baseline characteristics in these studies were similar to those of patients in the COMET study. In general, data for FVC (% predicted) showed slight improvements from baseline to the end of the NEO1 study (week 25). The results at week 286 of the extension study showed small decreases from baseline. Published literature studying the natural history of disease progression in patients with Pompe disease report annual declines in FVC (% predicted) in the sitting position between 1.04%51 and 4.60%52 among adults from the Netherlands and Belgium51 and patients 8 years and older from the US and Europe.52 Results for the 6MWT were similar from baseline to the end of the NEO1 study and week 286 of the NEO-EXT study. Other respiratory outcomes for MEP and MIP showed improvements from baseline to week 25 of the NEO1 study. In the NEO-EXT study, improvements were observed from baseline to week 286 for MEP, whereas baseline MIP values were sustained at week 286.
The differences in baseline characteristics (e.g., younger mean age at baseline and at diagnosis, better 6MWT at baseline, and shorter time between diagnosis and treatment start for patients who received avalglucosidase alfa) may bias and limit interpretations of the study results. Only 1 pediatric patient was included in the COMET trial, which limits the generalizability to patients with LOPD younger than 18 years, although the clinical expert was of the opinion that the results were generalizable to pediatric patients, but not to patients with IOPD. No MIDs were identified in populations with Pompe disease, and evidence of validity, reliability, and responsiveness was limited for this patient population; thus, there is greater uncertainty around the interpretation of the results.
Although the outcomes measured in the COMET trial align with the most important outcomes identified by the patient input submission (i.e., motor, respiratory, and HRQoL), the data do not support statistical superiority of avalglucosidase alfa over alglucosidase alfa for the outcomes assessed in the COMET study.
Harms
Overall, 44 patients (86.3%) in the avalglucosidase alfa group and 45 patients (91.8%) in the alglucosidase alfa group reported at least 1 AE during the COMET trial. SAEs were infrequent (8 patients [15.7%] and 12 patients [24.5%] in the avalglucosidase alfa and alglucosidase alfa arms, respectively. The 4 withdrawals due to AEs (WDAEs) and the 1 death were all in the alglucosidase alfa group. The clinical expert was of the opinion that there were no significant safety concerns associated with avalglucosidase alfa treatment. When there were differences in the incidence of some AEs between the groups, the clinical expert suggested they could be due to the small patient numbers or a result of the study being conducted at various international sites with different reporting procedures for AEs.
Notable harms identified in the CADTH systematic review protocol included anaphylactic reactions, hypersensitivity reactions, infusion-associated reactions, immune-mediated reactions, and acute cardiorespiratory failure. Frequencies of notable harms were the same for anaphylactic reactions between the treatment groups or numerically lower for hypersensitivity reactions, infusion-associated reactions, and immune-mediated reactions among patients who received avalglucosidase alfa. Acute cardiorespiratory failure was not reported during the COMET trial.
According to the clinical expert, the association between anti-drug antibody titres and loss of efficacy in LOPD are not well established. The clinical expert explained that high titres of anti-drug antibodies do not necessarily indicate that a patient will have an infusion-associated reaction, but the treating clinician may use more caution in withdrawing premedication before infusions. In the COMET study, numerically fewer number of patients had a peak titre of at least 12,800 in the avalglucosidase alfa group than the alglucosidase alfa group, which the clinical expert suggested could indicate that the former is less immunogenic than the latter. It will be necessary to collect data from more patients and for a longer time on treatment to confirm whether avalglucosidase alfa is less immunogenic than alglucosidase alfa.
The types of AEs in the NEO-EXT study (median duration on study treatment was around 300 days) were different than those in the COMET study. More than 1-third of patients reported an SAE in the NEO-EXT study, 1 patient withdrew due to an AE, and no deaths were reported.
When only data from the double-blind phase of the COMET study were considered, avalglucosidase alfa and alglucosidase alfa appeared to be similar in terms of type of AE, although, because of the small numbers of patients, it is unclear if the new treatment offers significant benefits in terms of reduced harms or lower immunogenicity.
Conclusions
In the COMET trial, the study’s main objective was achieved and avalglucosidase alfa met the criteria for noninferiority, compared with alglucosidase alfa. This comparison was made at the noninferiority margin of –1.1%, based on the primary outcome of FVC (% predicted) in the upright position for the first 49 weeks of treatment in patients with LOPD. Based on the evidence from the COMET trial, treatment with avalglucosidase alfa appeared to prevent further respiratory deterioration during the first year, which is 1 of the main goals of currently available forms of treatment, according to the clinical expert. The results from the study were not statistically significant for the superiority of avalglucosidase alfa over alglucosidase alfa for FVC (% predicted). Statistical testing was stopped for secondary outcomes and results should be interpreted as supportive of the primary outcome. Most patients experienced at least 1 AE during the first year of treatment, and fewer experienced a SAE. Notable harms for anaphylactic reactions occurred with the same frequency among the treatment groups, whereas hypersensitivity reactions, infusion-associated reactions, and immune-mediated reactions were numerically lower for patients who received avalglucosidase alfa.
Key limitations include the small number of patients enrolled in the studies and the lack of data available for pediatric patients. The small number of patients limits the ability to accurately quantify the differences in outcomes, beyond the primary outcome, in long-term benefits, and in potential harms between the treatments. Moreover, it will be beneficial to have continued long-term efficacy and safety data (including anti-drug antibody assessments) for treatment with avalglucosidase alfa, which is expected from the extension phase of the COMET study. Last, these results apply only to patients with LOPD and do not extend to patients with IOPD, for which there are ongoing trials evaluating avalglucosidase alfa.
References
1.Hahn S. Lysosomal acid alpha-glucosidase deficiency (Pompe disease, glycogen storage disease II, acid maltase deficiency). In: Firth H, TePas E, eds. UpToDate. Waltham (MA): UpToDate; 2020: http://www.uptodate.com. Accessed 2021 Oct 19.
2.NIH Genetic and Rare Diseases Information Center. Glycogen storage disease type 2. 2018; https://rarediseases.info.nih.gov/diseases/5714/glycogen-storage-disease-type-2#:~:text=Glycogen%20storage%20disease%20type%202%2C%20also%20known%20as%20Pompe%20disease,is%20an%20inherited%20metabolic%20disorder%20. Accessed 2021 Dec 10, 2021.
3.Kishnani PS, Steiner RD, Bali D, et al. Pompe disease diagnosis and management guideline. Genet Med. 2006;8(5):267-288. PubMed
4.Cupler EJ, Berger KI, Leshner RT, et al. Consensus treatment recommendations for late-onset Pompe disease. Muscle Nerve. 2012;45(3):319-333. PubMed
5.Ausems MG, Verbiest J, Hermans MP, et al. Frequency of glycogen storage disease type II in The Netherlands: implications for diagnosis and genetic counselling. Eur J Hum Genet. 1999;7(6):713-716. PubMed
6.Drug Reimbursement Review sponsor submission: Nexviazyme (avalglucosidase alfa),100 mg/vial lyophilized powder for injection [internal sponsor's package]. Mississauga (ON): Sanofi Genzyme; 2021 Sep 30.
7.Orphanet. Prevalence and incidence of rare diseases: bibliographic data. Orphanet Report Series, Rare Diseases collection, January 2019, number 1: Diseases listed in alphabetical order. 2019; https://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_diseases.pdf. Accessed 2021 Dec 10.
8.Applegarth DA, Toone JR, Lowry RB. Incidence of inborn errors of metabolism in British Columbia, 1969-1996. Pediatrics. 2000;105(1):e10. PubMed
9.Tarnopolsky M, Katzberg H, Petrof BJ, et al. Pompe disease: Diagnosis and management. Evidence-based guidelines from a Canadian Expert Panel. Can J Neurol Sci. 2016;43(4):472-485. PubMed
10.Nexviazyme (avalglucosidase alfa): lyophilized powder 100 mg/vial for intravenous infusion [product monograph]. Mississauga (ON): Sanofi Genzyme; 2021 Nov 12.
11.Clinical Study Report: EFC14028. A phase 3 randomized, multicenter, multinational, double-blinded study comparing the efficacy and safety of repeated biweekly infusions of avalglucosidase alfa (neoGAA, GZ402666) and alglucosidase alfa in treatment-naïve patients with late onset Pompe disease (COMET) [internal sponsor's report]. Mississauga (ON): Sanofi Genzyme; 2020 Sep 1.
12.U.S. Department of Health and Human Services Food and Drug Administration. Non-inferiority clinical trials to establish effectiveness: Guidance for industry. Silver Spring (MD): Center for Drug Evaluation and Research, U.S. Food and Drug Administration; 2016: https://www.fda.gov/media/78504/download. Accessed 2021 Dec 14.
13.Diaz-Manera J, Kishnani PS, Kushlaf H, et al. Safety and efficacy of avalglucosidase alfa versus alglucosidase alfa in patients with late-onset Pompe disease (COMET): a phase 3, randomised, multicentre trial. Lancet Neurol. 2021;20(12):1012-1026. PubMed
14.Koopmeiners JS, Hobbs BP. Detecting and accounting for violations of the constancy assumption in non-inferiority clinical trials. Stat Methods Med Res. 2018;27(5):1547-1558. PubMed
15.Myozyme (alglucosidase alfa), lyophilized powder, 50 mg vial [product monograph]. Mississauga (ON): Sanofi Genzyme; 2016 Dec 16: https://pdf.hres.ca/dpd_pm/00037590.PDF. Accessed 2022 Feb 3.
16.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. J Clin Epidemiol. 2016;75:40-46. PubMed
17.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2021 Oct 18.
18.Barreiro TJ, Perillo I. An approach to interpreting spirometry. Am Fam Physician. 2004;69(5):1107-1114. PubMed
19.Yuan M, Andrinopoulou ER, Kruijshaar ME, et al. Positive association between physical outcomes and patient-reported outcomes in late-onset Pompe disease: a cross sectional study. Orphanet J Rare Dis. 2020;15(1):232. PubMed
20.Berger KI, Kanters S, Jansen JP, et al. Forced vital capacity and cross-domain late-onset Pompe disease outcomes: an individual patient-level data meta-analysis. J Neurol. 2019;266(9):2312-2321. PubMed
21.Tappen RM, Roach KE, Buchner D, Barry C, Edelstein J. Reliability of physical performance measures in nursing home residents with Alzheimer's disease. J Gerontol A Biol Sci Med Sci. 1997;52(1):M52-55. PubMed
22.Rastelli E, Montagnese F, Massa R, Schoser B. Towards clinical outcome measures in myotonic dystrophy type 2: a systematic review. Curr Opin Neurol. 2018;31(5):599-609. PubMed
23.Harada ND, Chiu V, Stewart AL. Mobility-related function in older adults: assessment with a 6-minute walk test. Arch Phys Med Rehabil. 1999;80(7):837-841. PubMed
24.Steffen TM, Hacker TA, Mollinger L. Age- and gender-related test performance in community-dwelling elderly people: Six-Minute Walk Test, Berg Balance Scale, Timed Up & Go Test, and gait speeds. Phys Ther. 2002;82(2):128-137. PubMed
25.Mossberg KA. Reliability of a timed walk test in persons with acquired brain injury. Am J Phys Med Rehabil. 2003;82(5):385-390; quiz 391-382. PubMed
26.van Loo MA, Moseley AM, Bosman JM, de Bie RA, Hassett L. Test-re-test reliability of walking speed, step length and step width measurement after traumatic brain injury: a pilot study. Brain Inj. 2004;18(10):1041-1048. PubMed
27.Fulk GD, Echternach JL. Test-retest reliability and minimal detectable change of gait speed in individuals undergoing rehabilitation after stroke. J Neurol Phys Ther. 2008;32(1):8-13. PubMed
28.Flansbjer UB, Holmbäck AM, Downham D, Patten C, Lexell J. Reliability of gait performance tests in men and women with hemiparesis after stroke. J Rehabil Med. 2005;37(2):75-82. PubMed
29.Steffen T, Seney M. Test-retest reliability and minimal detectable change on balance and ambulation tests, the 36-item short-form health survey, and the unified Parkinson disease rating scale in people with parkinsonism. Phys Ther. 2008;88(6):733-746. PubMed
30.Ries JD, Echternach JL, Nof L, Gagnon Blodgett M. Test-retest reliability and minimal detectable change scores for the timed “up & go” test, the six-minute walk test, and gait speed in people with Alzheimer disease. Phys Ther. 2009;89(6):569-579. PubMed
31.Wevers LE, Kwakkel G, van de Port IG. Is outdoor use of the six-minute walk test with a global positioning system in stroke patients' own neighbourhoods reproducible and valid? J Rehabil Med. 2011;43(11):1027-1031. PubMed
32.Terzano C, Ceccarelli D, Conti V, Graziani E, Ricci A, Petroianni A. Maximal respiratory static pressures in patients with different stages of COPD severity. Respir Res. 2008;9(1):8. PubMed
33.Bourke SC, Tomlinson M, Williams TL, Bullock RE, Shaw PJ, Gibson GJ. Effects of non-invasive ventilation on survival and quality of life in patients with amyotrophic lateral sclerosis: a randomised controlled trial. Lancet Neurol. 2006;5(2):140-147. PubMed
34.Trojan DA, Arnold DL, Shapiro S, et al. Fatigue in post-poliomyelitis syndrome: association with disease-related, behavioral, and psychosocial factors. PM&R. 2009;1(5):442-449. PubMed
35.Jalan NS, Daftari SS, Retharekar SS, Rairikar SA, Shyam AM, Sancheti PK. Intra- and inter-rater reliability of maximum inspiratory pressure measured using a portable capsule-sensing pressure gauge device in healthy adults. Can J Respir Ther. 2015;51(2):39-42. PubMed
36.van der Ploeg AT, Barohn R, Carlson L, et al. Open-label extension study following the Late-Onset Treatment Study (LOTS) of alglucosidase alfa. Mol Genet Metab. 2012;107(3):456-461. PubMed
37.Bohannon RW. Test-retest reliability of hand-held dynamometry during a single session of strength assessment. Phys Ther. 1986;66(2):206-209. PubMed
38.Bohannon RW, Andrews AW. Interrater reliability of hand-held dynamometry. Phys Ther. 1987;67(6):931-933. PubMed
39.Crompton J, Galea MP, Phillips B. Hand-held dynamometry for muscle strength measurement in children with cerebral palsy. Dev Med Child Neurol. 2007;49(2):106-111. PubMed
40.May LA, Burnham RS, Steadward RD. Assessment of isokinetic and hand-held dynamometer measures of shoulder rotator strength among individuals with spinal cord injury. Arch Phys Med Rehabil. 1997;78(3):251-255. PubMed
41.Taylor NF, Dodd KJ, Graham HK. Test-retest reliability of hand-held dynamometric strength testing in young people with cerebral palsy. Arch Phys Med Rehabil. 2004;85(1):77-80. PubMed
42.van Capelle CI, van der Beek NA, de Vries JM, et al. The quick motor function test: a new tool to rate clinical severity and motor function in Pompe patients. J Inherit Metab Dis. 2012;35(2):317-323. PubMed
43.Ware J, Kosinski M, Keller S. How to score the SF-12 physical and mental summary scales. Boston (MA): The Health Institute, New England Medical Centre; 1995.
44.Gandek B, Ware JE, Aaronson NK, et al. Cross-validation of item selection and scoring for the SF-12 Health Survey in nine countries: results from the IQOLA Project. International Quality of Life Assessment. J Clin Epidemiol. 1998;51(11):1171-1178. PubMed
45.Jenkinson C, Layte R, Jenkinson D, et al. A shorter form health survey: can the SF-12 replicate results from the SF-36 in longitudinal studies? J Public Health Med. 1997;19(2):179-186. PubMed
46.Ware J, Jr., Kosinski M, Keller SD. A 12-Item Short-Form Health Survey: construction of scales and preliminary tests of reliability and validity. Med Care. 1996;34(3):220-233. PubMed
47.Angelini C, Semplicini C, Ravaglia S, et al. New motor outcome function measures in evaluation of late-onset Pompe disease before and after enzyme replacement therapy. Muscle Nerve. 2012;45(6):831-834. PubMed
48.Ko J, Kim M. Reliability and responsiveness of the gross motor function measure-88 in children with cerebral palsy. Phys Ther. 2013;93(3):393-400. PubMed
49.Clinical Study Report: TDR12857. An open-label, multicenter, multinational, ascending dose study of the safety, tolerability, pharmacokinetics, pharmacodynamics, and exploratory efficacy of repeated biweekly infusions of neoGAA in naïve and alglucosidase alfa treated late-onset Pompe disease patients (NEO1) [internal sponsor's report]. Mississauga (ON): Sanofi Genzyme; 2015 Jul 28.
50.Clinical Study Report: LTS13769. An open-label, multicenter, multinational extension study of the long-term safety and pharmacokinetics of repeated biweekly infusions of avalglucosidase alfa (neoGAA, GZ402666) in patients with Pompe disease (NEO-EXT) [internal sponsor's report]. Mississauga (ON): Sanofi Genzyme; 2020 Aug 12.
51.van der Beek NA, de Vries JM, Hagemans ML, et al. Clinical features and predictors for disease natural progression in adults with Pompe disease: a nationwide prospective observational study. Orphanet J Rare Dis. 2012;7:88. PubMed
52.Wokke JH, Escolar DM, Pestronk A, et al. Clinical features of late-onset Pompe disease: a prospective cohort study. Muscle Nerve. 2008;38(4):1236-1245. PubMed
53.Phillips D, Tomazos IC, Moseley S, L'Italien G, Gomes da Silva H, Lerma Lara S. Reliability and Validity of the 6-Minute Walk Test in Hypophosphatasia. JBMR Plus. 2019;3(6):e10131. PubMed
54.du Bois RM, Weycker D, Albera C, et al. Forced vital capacity in patients with idiopathic pulmonary fibrosis: test properties and minimal clinically important difference. Am J Respir Crit Care Med. 2011;184(12):1382-1389. PubMed
55.Patel AS, Siegert RJ, Keir GJ, et al. The minimal important difference of the King's Brief Interstitial Lung Disease Questionnaire (K-BILD) and forced vital capacity in interstitial lung disease. Respir Med. 2013;107(9):1438-1443. PubMed
56.Storm FA, Petrarca M, Beretta E, et al. Minimum clinically important difference of gross motor function and gait endurance in children with motor impairment: A comparison of distribution-based approaches. Biomed Res Int. 2020;2020:2794036. PubMed
57.Abizanda P, Navarro JL, García-Tomás MI, López-Jiménez E, Martínez-Sánchez E, Paterna G. Validity and usefulness of hand-held dynamometry for measuring muscle strength in community-dwelling older persons. Arch Gerontol Geriatr. 2012;54(1):21-27. PubMed
58.Hébert LJ, Maltais DB, Lepage C, Saulnier J, Crête M, Perron M. Isometric muscle strength in youth assessed by hand-held dynamometry: a feasibility, reliability, and validity study. Pediatr Phys Ther. 2011;23(3):289-299. PubMed
59.Katz-Leurer M, Rottem H, Meyer S. Hand-held dynamometry in children with traumatic brain injury: within-session reliability. Pediatr Phys Ther. 2008;20(3):259-263. PubMed
60.ATS statement: guidelines for the six-minute walk test. Am J Respir Crit Care Med. 2002;166(1):111-117. PubMed
61.van Hedel HJ, Wirz M, Dietz V. Assessing walking ability in subjects with spinal cord injury: validity and reliability of 3 walking tests. Arch Phys Med Rehabil. 2005;86(2):190-196. PubMed
62.Scivoletto G, Tamburella F, Laurenza L, Foti C, Ditunno JF, Molinari M. Validity and reliability of the 10-m walk test and the 6-min walk test in spinal cord injury patients. Spinal Cord. 2011;49(6):736-740. PubMed
63.Redelmeier DA, Bayoumi AM, Goldstein RS, Guyatt GH. Interpreting small differences in functional status: the Six Minute Walk test in chronic lung disease patients. Am J Respir Crit Care Med. 1997;155(4):1278-1282. PubMed
64.O'Keeffe ST, Lye M, Donnellan C, Carmichael DN. Reproducibility and responsiveness of quality of life assessment and six minute walk test in elderly heart failure patients. Heart. 1998;80(4):377-382. PubMed
65.Lachmann R, Schoser B. The clinical relevance of outcomes used in late-onset Pompe disease: can we do better? Orphanet J Rare Dis. 2013;8:160. PubMed
66.Díaz-Arribas MJ, Fernández-Serrano M, Royuela A, et al. Minimal clinically important difference in quality of life for patients with low back pain. Spine (Phila Pa 1976). 2017;42(24):1908-1916. PubMed
67.Linder-Lucht M, Othmer V, Walther M, et al. Validation of the Gross Motor Function Measure for use in children and adolescents with traumatic brain injuries. Pediatrics. 2007;120(4):e880-886. PubMed
68.Versteegh T, Beaudet D, Greenbaum M, Hellyer L, Tritton A, Walton D. Evaluating the reliability of a novel neck-strength assessment protocol for healthy adults using self-generated resistance with a hand-held dynamometer. Physiother Can. 2015;67(1):58-64. PubMed
69.Kodesh E, Laufer Y. The reliability of hand-held dynamometry for strength assessment during electrically induced muscle contractions. Physiother Theory Pract. 2015;31(1):61-66. PubMed
70.Spink MJ, Fotoohabadi MR, Menz HB. Foot and ankle strength assessment using hand-held dynamometry: reliability and age-related differences. Gerontology. 2010;56(6):525-532. PubMed
71.Wadsworth C, Nielsen DH, Corcoran DS, Phillips CE, Sannes TL. Interrater reliability of hand-held dynamometry: effects of rater gender, body weight, and grip strength. J Orthop Sports Phys Ther. 1992;16(2):74-81. PubMed
72.Nitschke JE, McMeeken JM, Burry HC, Matyas TA. When is a change a genuine change? A clinically meaningful interpretation of grip strength measurements in healthy and disabled women. J Hand Ther. 1999;12(1):25-30. PubMed
73.Farrero E, Antón A, Egea CJ, et al. Guidelines for the management of respiratory complications in patients with neuromuscular disease. Sociedad Española de Neumología y Cirugía Torácica (SEPAR). Arch Bronconeumol. 2013;49(7):306-313. PubMed
74.Evans JA, Whitelaw WA. The assessment of maximal respiratory mouth pressures in adults. Respir Care. 2009;54(10):1348-1359. PubMed
75.ATS/ERS Statement on respiratory muscle testing. Am J Respir Crit Care Med. 2002;166(4):518-624. PubMed
76.Mendoza M, Gelinas DF, Moore DH, Miller RG. A comparison of maximal inspiratory pressure and forced vital capacity as potential criteria for initiating non-invasive ventilation in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2007;8(2):106-111. PubMed
77.Schoser B, Fong E, Geberhiwot T, et al. Maximum inspiratory pressure as a clinically meaningful trial endpoint for neuromuscular diseases: a comprehensive review of the literature. Orphanet J Rare Dis. 2017;12(1):52. PubMed
78.Cheah BC, Boland RA, Brodaty NE, et al. INSPIRATIonAL--INSPIRAtory muscle training in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2009;10(5-6):384-392. PubMed
79.Pena LDM, Barohn RJ, Byrne BJ, et al. Safety, tolerability, pharmacokinetics, pharmacodynamics, and exploratory efficacy of the novel enzyme replacement therapy avalglucosidase alfa (neoGAA) in treatment-naive and alglucosidase alfa-treated patients with late-onset Pompe disease: A phase 1, open-label, multicenter, multinational, ascending dose study. Neuromuscul Disord. 2019;29(3):167-186. PubMed
80.Committee for Medicinal Products for Human Use. Assessment report: Myozyme (alglucosidase alfa). (European public assessment report). Amsterdam (Netherlands): European Medicines Agency; 2013: https://www.ema.europa.eu/en/documents/variation-report/myozyme-h-c-636-ii-0041-epar-assessment-report-variation_en.pdf. Accessed 2022 Feb 3.
Appendix 1: Literature Search Strategy
Note that this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases:
MEDLINE All (1946-present)
Embase (1974-present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: October 29, 2021
Alerts: Bi-weekly search updates until project completion
Search filters applied: No filters were applied to limit the retrieval by study type.
Limits:
No date or language limits were used
Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
MeSH | Medical Subject Heading |
exp | Explode a subject heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Author keyword heading word (MEDLINE) |
.kw | Author keyword (Embase) |
.dq | Candidate term word (Embase) |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multi-Database Strategy
(Nexviazyme* or avalglucosidase* or Nexviadyme* or gz 402666 or gz402666 or neoGAA or neo GAA or ATB-200 or ATB200 or EO144CP0X9).ti,ab,kf,ot,hw,rn,nm.
1 use medall
avalglucosidase alfa/
((Nexviazyme* or avalglucosidase* or Nexviadyme* or gz 402666 or gz402666 or neoGAA or neo GAA or ATB-200 or ATB200).ti,ab,kf,dq.
3 or 4
5 use oemezd
6 not (conference review or conference abstract).pt.
2 or 7
remove duplicates from 8
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
[Search -- Studies with results | Nexviazyme or avalglucosidase alfa]
WHO ICTRP
International Clinical Trials Registry Platform, produced by the WHO. Targeted search used to capture registered clinical trials.
[Search terms -- Nexviazyme or avalglucosidase alfa]
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
[Search terms -- Nexviazyme or avalglucosidase alfa]
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
[Search terms -- Nexviazyme or avalglucosidase alfa]
Grey Literature
Search dates: October 18, 2021 – October 22, 2021
Keywords: Nexviazyme, avalglucosidase alfa, Pompe disease
Limits: None
Updated: Search updated before the completion of stakeholder feedback period.
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Health Statistics
Internet Search
Appendix 2: Excluded Studies
Note that this appendix has not been copy-edited.
Reference | Reason for exclusion |
|---|---|
Pena, L. D. M., et al. (2019). “Safety, tolerability, pharmacokinetics, pharmacodynamics, and exploratory efficacy of the novel enzyme-replacement therapy avalglucosidase alfa (neoGAA) in treatment naive and alglucosidase alfa-treated patients with late-onset Pompe disease: A phase I, open-label, multicenter, multinational, ascending-dose study.” Neuromuscular Disorders 29(3): 167 to 186. | Study design |
Appendix 3: Detailed Outcome Data
Note that this appendix has not been copy-edited.
Table 23: Subgroup Analyses for the Primary Efficacy Outcome – COMET Trial, mITT Population
Outcome | COMET | |
|---|---|---|
avalglucosidase alfa (N = 51) | alglucosidase alfa (N = 49) | |
Subgroup analysis by baseline agea | ||
Patients aged ≥ 18 and < 45 years old | ||
Patients, n | 23 | 19 |
Change from baseline to Week 49b, mean (SE) (95% CI) | 3.78 ||||||||||||| ||||| | 0.80 |||||||||||||| ||||| |
Group difference (treatment – control) (SE) (95% CI) | 2.99 |||||| (−1.52, 7.49) | Reference |
Patients aged ≥ 45 years old | ||
Patients, n | 27 | 30 |
Change from baseline to Week 49b, mean (SE) (95% CI) | 2.32 |||||||||||||| ||||| | −0.14 |||||||||||||| ||||| |
Group difference (treatment – control) (SE) (95% CI) | 2.46 |||||| (−0.84, 5.77) | Reference |
Subgroup analysis by baseline FVC (% predicted) | ||
Patients with FVC (% predicted) < 55% | ||
Patients, n | 16 | 19 |
Change from baseline to Week 49b, mean (SE) (95% CI) | 0.85 |||||||||||||| ||||| | 1.61 |||||||||||||| ||||| |
Group difference (treatment – control) (SE) (95% CI) | −0.76 |||||| (−5.23, 3.71) | Reference |
Patients with FVC (% predicted) ≥ 55% | ||
Patients, n | 35 | 30 |
Change from baseline to Week 49b, mean (SE) (95% CI) | 3.99 ||||||||||||| ||||| | −0.11 |||||||||||||| ||||| |
Group difference (treatment – control) (SE) (95% CI) | 4.10 |||||| (0.95, 7.26) | Reference |
Subgroup analysis by baseline walking device use or not used on 6MWT | ||
Patients who used a walking device on 6MWT | ||
Patients, n | 7 | 10 |
Change from baseline to Week 49b, mean (SE) (95% CI) | 4.77 ||||||||||||| ||||| | 0.32 |||||||||||||| ||||| |
Group difference (treatment – control) (SE) (95% CI) | 4.44 |||||| (−1.00, 9.88) | Reference |
Patients who did not use a walking device on 6MWT | ||
Patients, n | 44 | 39 |
Change from baseline to Week 49b, mean (SE) (95% CI) | 2.69 ||||||||||||| ||||| | 0.51 |||||||||||||| ||||| |
Group difference (treatment – control) (SE) (95% CI) | 2.19 |||||| (−0.76, 5.13) | Reference |
Subgroup analysis by baseline 6MWT median distance | ||
Patients with 6MWT median distance < 403.5 m | ||
Patients, n | 22 | 28 |
Change from baseline to Week 49b, mean (SE) (95% CI) | 1.99 |||||||||||||| ||||| | 0.72 |||||||||||||| ||||| |
Group difference (treatment – control) (SE) (95% CI) | 1.27 |||||| (−3.03, 5.57) | Reference |
Patients with 6MWT median distance ≥ 403.5 m | ||
Patients, n | 29 | 21 |
Change from baseline to Week 49b, mean (SE) (95% CI) | 3.77 ||||||||||||| ||||| | 0.19 |||||||||||||| ||||| |
Group difference (treatment – control) (SE) (95% CI) | 3.58 |||||| (0.36, 6.81) | Reference |
6MWT = 6 minute walk test; CI = confidence interval; FVC = forced vital capacity; mITT = modified intention-to-treat; NA = not applicable; PP = per protocol; SE = standard error.
aAdditional subgroup for patients aged < 18 years included 1 patient. Data were not included in this summary.
bBased on MMRM model that includes baseline FVC (% predicted, as continuous), sex, age (in years at baseline), treatment group, visit, interaction term between treatment group and visit as fixed effects. For subgroup analyses, the fixed effect factor is excluded from the model if it is the same as subgroup factor.
Source: COMET Clinical Study Report.11
Table 24: Additional Tertiary Efficacy and Health-Related Quality of Life Outcomes During the Double-Blind phase – COMET Trial mITT Population
Outcome | COMET | |
|---|---|---|
avalglucosidase alfa (N = 51) | alglucosidase alfa (N = 49) | |
||||| | ||
Patients contributing to analysis, n | |||||| | |||||| |
Baseline, patients at each level, n | |||||| | |||||| |
Level I | |||||| | |||||| |
Level II | |||||| | |||||| |
Level III | ||||| | ||||| |
End of double-blind phase (Week 49), patients at each level, n | |||||| | |||||| |
Level I | |||||| | ||||| |
Level II | |||||| | |||||| |
Level III | ||||| | ||||| |
||| |||||| ||||||||||| ||||||||| |||||| | ||
Patients contributing to analysis, n | |||||| | |||||| |
Baseline, mean (SD) | ||||||| |||||||| | ||||||| |||||||| |
End of double-blind phase (Week 49), mean (SD) | ||||||| |||||||| | ||||||| |||||||| |
Change from baseline, mean (SD) | |||||| |||||||| | ||||| |||||||| |
P valuea, b | |||||| | ||||||||| |
|||||||| | ||
Patients contributing to analysis, n | |||||| | |||||| |
Baseline, mean (SD) | |||| |||||| | |||| |||||| |
End of double-blind phase (Week 49), mean (SD) | |||| |||||| | |||| |||||| |
Change from baseline, mean (SD) | ||||| |||||| | ||||| |||||| |
P valuea, b | |||||| | ||||||||| |
CI = confidence interval; EQ-5D-5L = 5-Level-EuroQoL; GMFCS = gross motor function classification system; HHD = hand-held dynamometry; mITT = modified intention-to-treat; NA = not applicable; SD = standard deviation.
aP value is nominal. P value has not been adjusted for multiple testing (i.e., the type I error rate has not been controlled).
bP values are from the Wilcoxon-Mann–Whitney stratified by stratification factors.
Source: COMET Clinical Study Report.11
Table 25: Long-term Primary and Key Secondary Efficacy Outcomes to Week 97 — COMET Trial, mITT Population
Outcome | COMET | |
|---|---|---|
avalglucosidase alfa (N = 51) | alglucosidase alfa to avalglucosidase alfa (N = 49) | |
FVC (% predicted) in upright positiona | ||
Baseline | NA | NA |
Patients, n | 51 | 49 |
Mean (SD) | 62.55 (14.39) | 61.56 (12.40) |
Week 49 (end of double-blind phase) | NA | NA |
Patients, n | 49 | 43 |
Change from baselineb, mean (SD) | 3.0 (6.8) | −0.0 (5.8) |
Week 97 (during open-label phase) | NA | NA |
Patients, n | 24 | 21 |
Change from baselineb, mean (SD) | |||| |||||| | |||| |||||| |
6MWT (distance in m)c | ||
Baseline | NA | NA |
Patients, n | 51 | 49 |
Mean (SD) | 399.30 (110.93) | 378.09 (116.22) |
Week 49 (end of DB phase) | NA | NA |
Patients, n | 48 | 43 |
Change from baselineb, mean (SD) | 37.9 (52.8) | −1.7 (85.2) |
Week 97 (during open-label phase) | NA | NA |
Patients, n | 24 | 22 |
Change from baselineb, mean (SD) | ||||| ||||||| | ||||| ||||||| |
6MWT (% predicted)c | ||
Baseline | NA | NA |
Patients, n | 51 | 49 |
Mean (SD) | 57.32 (14.97) | 55.29 (16.64) |
Week 49 (end of double-blind phase) | NA | NA |
Patients, n | 48 | 43 |
Change from baselineb, mean (SD) | |||| |||||| | ||||| ||||||| |
Week 97 (during open-label phase) | NA | NA |
Patients, n | 24 | 22 |
Change from baselineb, mean (SD) | |||| ||||||| | |||| ||||||| |
6MWT = 6 minute walk test; CI = confidence interval; FVC = forced vital capacity; mITT = modified intention-to-treat; NA = not applicable; SD = standard deviation; SE = standard error.
aPrimary efficacy outcome.
bChange from baseline values have not been adjusted for using statistical models as in Table 11 and Table 12.
cKey secondary efficacy outcome.
Source: COMET Clinical Study Report.11
Table 26: Summary of Harms During Open-Label phase – COMET Trial, Safety Population
Event | COMET | |
|---|---|---|
avalglucosidase alfa (N = 51) | alglucosidase alfa to avalglucosidase alfa (N = 44) | |
|||||||| |||| | | |||| || ||||||| |||||| ||||| ||||| ||| ||||||||| |||||| | ||| | || |||||| | || |||||| |
|||||||||| ||| |||||||||||| | || |||||| | || |||||| |
||||||||||||||| | | |||||| | || |||||| |
||||||||| | | ||||| | | ||||| |
||||| ||||||||||| ||||| ||||||||| | | ||||| | | |||||| |
|||||||||| | ||||| | | ||||| |
NA = not applicable; NR = not reported; SAE = serious adverse events; SMQ = Standardized MedDRA Queries; TEAE = treatment-emergent adverse events; WDAE = withdrawal due to adverse events.
aFrequency of AE ≥ 5% per treatment group.
bFrequency of SAE ≥ 2 patients total for both treatment groups.
cFrequency of notable harm ≥ 2 patients total for both treatment groups.
Note: Redacted rows have been deleted.
Source: COMET Clinical Study Report.11
Appendix 4: Description and Appraisal of Outcome Measures
Note that this appendix has not been copy-edited.
Aim
To describe the following outcome measures used in the COMET trial (Table 27) and review their measurement properties (validity, reliability, responsiveness to change, and MID; Table 28).
Table 27: List of Outcome Measures Used in the COMET Trial
Measure | Type |
|---|---|
6-minute walk test (6MWT) | Secondary |
12-Item Short Form Health Survey (SF-12) | Secondary |
Forced vital capacity (FVC) | Primary |
Gait, Stair, Gower’s Maneuver, and Chair (GSGC) | Tertiary |
Gross motor function measure-88 (GMFM-88) | Tertiary |
Hand-held dynamometry (HHD) | Secondary |
Maximum expiratory pressure (MEP) | Secondary |
Maximum inspiratory pressure (MIP) | Secondary |
Quick Motor function (QMFT) | Secondary |
Findings
Table 28: Summary of Outcome Measures and Their Measurement Properties
Instrument | Types | Conclusions about Measurement Properties | MID |
|---|---|---|---|
6MWT | A supervised test that measures the distance a patient can walk on a hard-flat surface over a 6 minute period. The 6MWT is a commonly used test to evaluate global function of organ systems involved in exercise, namely the heart, lungs, peripheral circulation, blood, nervous system, muscles, bones, and joints during walking, a self-paced activity. | Validity: Good construct validity was demonstrated in various patient populations. In patients with a confirmed diagnosis of Pompe disease, 6MWT scores were significantly associated with better Rotterdam Handicap Scale and the Rasch-built Pompe-specific Activity scale.19 Reliability: Excellent test-retest reliability and interrater reliability has been demonstrated across various adult patient populations21-31 and among patients with pediatric hypophosphatasia.53 No evidence found for patients with Pompe disease. Responsiveness: Measure was found to be associated with Rotterdam Handicap Scale and Rasch-built Pompe-specific Activity scale in patients with Pompe disease.19 | No reported MID was found for patients with Pompe disease. |
FVC | A measure of the volume of air that can be forcibly exhaled from the lungs after taking the deepest breath possible. Typically reported as the percentage of the volume predicted for a person of the same size, age, and sex. | Validity: Construct validity was demonstrated in the supine and upright positions in patients with Pompe disease.19 Reliability: Demonstrated good test-retest reliability in patients with idiopathic pulmonary fibrosis. No evidence found for patients with Pompe disease. Responsiveness: Weak responsiveness in patients with idiopathic pulmonary fibrosis.54 No evidence found for patients with Pompe disease. | 2% to 6% among patients with idiopathic pulmonary fibrosis and interstitial lung disease.54,55 No reported MID was found for patients with Pompe disease. |
GSGC | A composite test that includes quantitative measures of 4 main motor performances: 1. Walking for 10 m; 2. Climbing 4 steps on stairs; 3. Gower’s maneuver; and 4. Rising from a chair as well as a qualitative global assessment of the manner to accomplish them. | Validity: Found to be significantly correlated with the 6MWT and the Walton and Gardner-Medwin (a measure that evaluates change in functional state) in patients with LOPD.47 Reliability: No evidence found for patients with Pompe disease. Responsiveness: No evidence found for patients with Pompe disease. | No reported MID was found for patients with Pompe disease. |
GSFM-88 | An assessment tool that was designed to evaluate changes in gross motor function over time or with intervention in children with cerebral palsy. The 88 items of the GMFM-88 measure gross motor activities in 5 dimensions: 1. Lying and rolling; 2. Sitting; 3. Crawling and kneeling; 4. Standing; and 5. Walking, running, and jumping. | Validity: Demonstrated good content and construct validity in children with cerebral palsy.48 No evidence found for patients with Pompe disease. Reliability: Excellent relative reliability in terms of test-retest reliability, interrater, intrarater, and internal reliability in children with cerebral palsy.48 No evidence found for patients with Pompe disease. Responsiveness: Responsive to changes in performances in functional tasks in children with cerebral palsy.48 No evidence found for patients with Pompe disease. | 1.1% to 5.3% among patients a cohort of children with acquired brain injury, and values of 0.1% to 3% for children with cerebral palsy.56 No reported MID was found for patients with Pompe disease. |
HHD | A series of assessments developed to assess muscle strength of various body points and provides a quantified measurement of force. | Validity: Good convergent validity demonstrated across a variety of patient populations, including healthy adults over the age of 65,57 patients with various neurologic dysfunction,37,38,40 healthy children and adolescents,58 children with cerebral palsy,39,41 and traumatic brain injury.59 Among patients with Pompe disease, better HHD scores were found to be significantly associated with better Rotterdam Handicap Scale and Rasch-built Pompe-specific Activity scale, accounting for the effects of sex, disease duration, use of wheelchair and ventilator support.19 Reliability: Excellent reliability demonstrated across a variety of patient populations (e.g., healthy adults over the age of 65,57 patients with various neurologic dysfunction,37,38,40 healthy children and adolescents,58 and children with cerebral palsy39,41 and traumatic brain injury59) for both upper and lower extremities. No evidence found for patients with Pompe disease. Responsiveness: No evidence found for patients with Pompe disease. | No reported MID was found for patients with Pompe disease. |
MEP | A measure of the maximum positive pressure that can be generated from an expiratory effort starting from total lung capacity or function residual capacity. | Validity: Accurately assesses respiratory muscle weakness and positively correlated with maximal static inspiratory pressure, FEV1, FVC, peak expiratory flow, and total lung capacity in patients with chronic obstructive pulmonary disease.32 No evidence found for patients with Pompe disease. Reliability: No evidence found for patients with Pompe disease. Responsiveness: No evidence found for patients with Pompe disease. | No reported MID was found for patients with Pompe disease. |
MIP | A measure of the maximum negative pressure that can be generated from an inspiratory effort starting from functional residual capacity or residual volume. | Validity: Accurately assesses respiratory muscle weakness in healthy adults.35 Positively correlated with maximal static inspiratory pressure, FEV1, FVC, peak expiratory flow, and total lung capacity in patients with chronic obstructive pulmonary disease,32 and correlated with improvements in SF-36 mental component summary, the sleep apnea quality-of-life index symptom domain, and the Multidimensional Fatigue Inventory in patients with neuromuscular disorders.33,34 Significantly correlated with the 6MWT in patients with Pompe disease.36 Reliability: Excellent intrarater and interrater reliability in healthy aduts.35 No evidence found for patients with Pompe disease. Responsiveness: No evidence found for patients with Pompe disease. | No reported MID was found for patients with Pompe disease. |
QMFT | A functional motor scale designed specifically for Pompe disease. | Validity: Strongly correlated with the HHD and manual testing and able to discriminate between different Pompe disease severities.42 Reliability: Good interrater and intrarater reliability in patients with Pompe disease.42 Responsiveness: No evidence found for patients with Pompe disease. | No reported MID was found for patients with Pompe disease. |
SF-12 | A self-reported outcome measure assessing the impact of health on an individual’s everyday life on 8 domains: 1. Limitations in physical activities because of health problems; 2. Limitations in social activities because of physical or emotional problems; 3. Limitations in usual role activities because of physical problems; 4. Bodily pain; 5. General mental health (psychological distress and well-being); 6. Limitations in usual role activities because of emotional problems; 7. Vitality (energy and fatigue); and 8. General health perceptions. | Validity: Good construct and criterion validity demonstrated to the SF-36.44,46 No evidence found for patients with Pompe disease. Reliability: No evidence found for patients with Pompe disease. Responsiveness: Responsive to change in health over time among patients with congestive heart failure, sleep apnea, and inguinal hernia.45 No evidence found for patients with Pompe disease. | No reported MID was found for patients with Pompe disease. |
6MWT = 6 minute walk test; FEV1 = forced expiratory volume in 1 second; FVC = forced vital capacity; GMFM-88 = gross motor function measure-88; GSGC = gait, stair, Gowers’ maneuver, and chair; HHD = hand-held dynamometry; LOPD = late-onset Pompe disease; MEP = maximum expiratory pressure; MID = minimal important difference; MIP = maximum inspiratory pressure; MRC = Medical Research Council-skeletal muscle strength; QMFT = quick motor function test; SF-12 = 12-item Short Form Health Survey; SF-36 = 36-item Short-Form Health Survey.
Six Minute Walk Test
Description
The 6 minute walk test (6MWT) is a supervised test that measures the distance a patient can walk on a hard, flat surface over a 6 minute period.60 The American Thoracic Society provides guidelines for standardization of this test to maximize reliability.60 Walk tests aim to evaluate global function of organ systems involved in exercise, namely the heart, lungs, peripheral circulation, blood, nervous system, muscles, bones, and joints while walking. Individuals self-pace their walk, resting as needed as they traverse back and forth along a marked walkway. The 6MWT was developed in frail elderly patients 60 to 90 years of age referred to a geriatric hospital, and it targets community-dwelling elder adults. Since its development, the test has been used in adult and pediatric populations with a variety of chronic conditions, as well as in healthy adults.
Scoring
The primary outcome is the distance covered in metres or converted measure over 6 minutes. A lower score, reflecting a shorter distance covered in 6 minutes, indicates worse function.
Psychometric Properties
Across various adult patient populations, the 6MWT has demonstrated excellent test-retest reliability (intraclass correlation coefficient [ICC] = 0.86 to 0.99),21-31 and excellent interrater (ICC = 0.97 to 0.99) and intrarater reliability (ICC = 0.76 to 0.99).21,61,62 In patients with pediatric hypophosphatasia, the 6MWT was found to have excellent test-retest reliability (r = 0.81 for adolescents and r = 0.95 for children) and showed concurrent validity with relevant measures of skeletal disease and parent-reported function.53
In a cohort of Dutch patients with a confirmed diagnosis of Pompe disease (n = 132), 6MWT scores were significantly positively associated with the Rotterdam Handicap Scale (a measure that assesses activities of daily living) and the Rasch-built Pompe-specific Activity scale (a Pompe disease-specific measure of activities of daily living) with a standardize estimate of 0.485 (95% CI = 0.232, 0.738) and 0.495 (95% CI = 0.217, 0.773), respectively.19 A 1%-point higher 6MWT corresponded to a 0.348 unit (95% CI = 0.15, 0.55; P = 0.01) higher Rasch-built Pompe-specific Activity scale score; accounting for sex, disease, and the use of wheelchair and ventilator.
MID
In the adult population, the MIDs for the 6MWT distances were reported for chronic obstructive pulmonary disease (54 m)60,63 and heart failure (43 m).60,64 Among pediatric patients, the most conservative MIDs were 31 m in children and 43 m in adolescents.53 Among children with acquired brain injury, the MID range was 20 to 38 m, and 6 to 23 m among children with cerebral palsy.56
A literature search was conducted to identify MIDs of the 6MWT in patients with Pompe disease; none were identified.
A systematic review of the clinical relevance of outcomes used in LOPD attempted to determine the clinical relevance of the 6MWT among patients with LOPD and compared the parameters to those used in studies of other neuromuscular disorders.65 In particular, the authors assessed if patients noticed change was below, within, or above the MID set for the 6MWT among patients with respiratory diseases. The review identified 10 studies that used the 6MWT and found that in 9 of the studies, patients noticed changes were above or within the MID established in respiratory disease.65 The authors cautioned that applying MIDs from studies of chronic respiratory diseases to LOPD has several limitations, and the relevance of MIDs established for chronic respiratory diseases to LOPD is unclear.65
12-Item Short-Form Health Survey
Description
The 12-item Short-Form Health Survey (SF-12) is a self-reported outcome measure assessing the impact of health on an individual’s everyday life. It is often used as a measure of HRQoL. The SF-12 is a shortened version of the 36-item Short-Form Health Survey (SF-36) and includes the same 8 domains as the SF-36:
Limitations in physical activities because of health problems (physical functioning);
Limitations in social activities because of physical or emotional problems (social functioning);
Limitations in usual role activities because of physical problems (role-physical);
Bodily pain;
General mental health (psychological distress and well-being);
Limitations in usual role activities because of emotional problems (role-emotional);
Vitality (energy and fatigue); and
General health perceptions.
Scoring
Scores of the SF-12 range from 0 to 100, which creates 2 summary scores: the physical component summary and mental component summary.43 Higher scores indicate better HRQoL. A score of 50 or less on the physical component summary-12 has been recommended as a cut-off to determine a physical condition while a score of 42 or less on the mental component summary-12 is considered indicative of ‘clinical depression.’43 Scores are created according a manual by Ware et al. (1995).43
Psychometric Properties
The validity of the SF-12 has been assessed in multiple patient populations in various age, physical and mental health. When compared to the SF-36, the SF-12 scores were similar46 and correlated,44 but almost always had larger standard errors.46 The SF-12 has been shown to be responsive to change in health over time among patients with congestive heart failure, sleep apnea, and inguinal hernia.45
A literature search was conducted to identify validation information with Pompe disease; none were identified.
MID
Among adults with back pain, the MIDs for the physical and mental components of the SF-12 were 3.29 and 3.77, respectively.66
A literature search was conducted to identify the MIDs of the SF-12 in patients with Pompe disease; none were identified.
Forced Vital Capacity
Description
FVC is the total volume of air that can be forcibly exhaled from the lungs after taking the deepest breath possible during a forced expiratory volume (FEV) test. FVC helps distinguish obstructive lung disease from restrictive lung disease, as well as helps assess the progression of lung disease and evaluates the effectiveness of treatment.
Scoring
FVC is usually reported as the percentage of the volume predicted for a person of the same size, age, and sex, and is reported in 2 ways:
As an absolute value, reported as number in litres (L); and
On a linear graph to chart the dynamics of exhalation.
The normal FVC range for adults is between 3.0 and 5.0 L.
Psychometric Properties
A meta-analysis of 15 studies found that among patients with LOPD, FVC was positively associated with LOPD measures and outcomes across multiple domains.20 Specifically, patients with 10% higher FVC were associated with 4.75% higher Medical Research Council-skeletal muscle strength score and with slopes for the 6MWT and the SF-36 physical component summary at 33.2 m and 1.2%, respectively. Longitudinal analyses found that a 10% incremental increase in predicted FVC was associated with an average increase of 4.12% (95% CI = 1.29, 6.95) in MRC score, 35.6 m (95% CI = 19.9, 51.6) in the 6MWT and 1.34% (95% CI = 0.08, 2.6) in SF-36.
In a cohort of Dutch patients with a confirmed diagnosis of Pompe disease (N = 132), higher FVC in supine and upright positions was found to be significantly associated with better Rotterdam Handicap Scale and the Rasch-built Pompe-specific Activity scale, accounting for the effects of sex, disease duration, and use of wheelchair and ventilator support.19
MID
A decline of 2% to 6% predicted FVC was estimated as the MID in adult patients with idiopathic pulmonary fibrosis and interstitial lung disease.54,55
A literature search was conducted to identify the MIDs of the FVC in patients with Pompe disease; none were identified.
A systematic review of the clinical relevance of outcomes used in LOPD attempted to determine the clinical relevance of the FVC among patients with LOPD and compared the parameters to those used in studies of other neuromuscular disorders.65 In particular, the authors assessed if patients noticed changed were below, within, or above the MID set for the FVC among patients with respiratory diseases. The review identified 9 studies that used the FVC, and found that in 6 of the studies, patients noticed changes were above or within the MID established in respiratory disease.65 The difference was perceived as either an improvement or stabilization by patients.65 The authors cautioned that applying MIDs from studies of chronic respiratory diseases to LOPD has several limitations, and the relevance of MIDs established for chronic respiratory disease to LOPD is unclear.65
Gait, Stair, Gowers’ Maneuver, and Chair
Description
The gait, stair, Gower’s maneuver, and chair (GSGC) is a composite test that includes quantitative measures of time in seconds to complete 4 main motor tasks, and a qualitative global assessment of the manner to accomplish each:
Gait:
Quantitative component: Time to walk 10 m
Qualitative component: 7-point Likert type scale to assess task with anchors 1: normal and 7: confined to wheelchair
Stairs
Quantitative component: Time to climb 4 steps on stairs
Qualitative component: 7-point Likert type scale to assess task with anchors 1: climbing without assistance and 7: unable to climb steps
Gower’s maneuver
Quantitative component: Time to standing from laying
Qualitative component: 7-point Likert type scale to assess task with anchors 1: normal and 7: unable to arise
Chair: Rising from a chair
Quantitative component: Time to standing from sitting in a chair
Qualitative component: 6-point Likert type scale to assess task with anchors 1: normal and 6: not possible
Scoring
The final score is obtained by adding the scores attributed to each function test according to how the individual performed it. Scores may vary from a minimum of 4 (normal performance) to a maximum of 27 (worst score).
Psychometric Properties
In a cohort of 40 patients with LOPD, the time to walk 10 m component of the GSGC was moderately correlated with the 6MWT (r = 0.67, P < 0.0001).47 Moreover, the qualitative components of the GSGC score were moderately correlated with both the 6MWT (r = 0.71, P < 0.001) and the Walton and Gardner-Medwin (r = 0.62, P < 0.001) which is an assessment that measures changes in functional states.47
MID
A literature search was conducted to identify MIDs of the SF-12 in patients with Pompe disease; none were identified.
Gross Motor Function Measure-88
Description
The gross motor function measure-88 (GMFM-88) is an assessment tool that was designed to evaluate changes in gross motor function over time or with intervention in children with cerebral palsy. The items of the GMFM-88 measure gross motor activities in 5 dimensions:
A. Lying and rolling (17 items);
B. Sitting (20 items);
C. Crawling and kneeling (14 items);
D. Standing (13 items); and
E. Walking, running, and jumping (24 items).
The motor skills captured in these domains are ones that are typical of normal developmental milestones. It may be assumed that the GMFM-88 may be useful for other diagnostic populations.
Scoring
Each item is scored using a 4-point Likert type scale rating an individual’s ability to complete the task where a higher score indicates better motor function. Scores correspond to the following:
0 = Does not initiate;
1 = Initiates;
2 = Partially completes; and
3 = Completes.
The scoring key also includes option 9, which corresponds to ‘not tested.’
The scores for each dimension are added to create a total dimension score. Total dimension scores are then converted into a dimension percentage score which are then combined to create a total summary score (sum of percent score for each dimension divided by the number of goal areas assessed). The GMFM-88 provides an option to calculate a summary score based on the use of aids or orthoses.
Only dimensions D (standing) and E (walking, running, jumping) were assessed in the pivotal study.
Psychometric Properties
The GMFM-88 has been validated in children from 5 months to 16 years of age. The GMFM-88 demonstrated excellent relative reliability in terms of test-retest reliability, interrater, intrarater, and internal reliability (ICC ranged from 0.95 to 1.0).48 The content and construct validity of the test was found to be good and showed concurrent validity with the Pediatric Evaluation of Disability Inventory,48 a scale that measures functional skills, level of independence, and the extent of modifications required to perform functional activities in young children. Additionally, the GMFM-88 is responsive to changes in performances in functional tasks.48 Although the instrument was developed for children with cerebral palsy, it has been validated in children with Down Syndrome and acquired brain damage.67 Currently, there are no published references of use of the GMFM-88 in adult populations.
A literature search was conducted to identify validation information in patients with Pompe disease; none were identified.
MID
For total GMFM-88 score, MIDs from 1.1% to 5.3% were estimated for a cohort of children with acquired brain injury and values of 0.1% to 3% were estimated for children with cerebral palsy.56
A literature search was conducted to identify MIDs of the GMFM-88 in patients with Pompe disease; none were identified.
Hand-Held Dynamometry
Description
HHD assessments were developed to measure muscle strength of the hand and forearm muscles and provide a quantified measurement of force. The patient holds the dynamometer in the hand being tested with the arm at right angles and the elbow by the side of the body. The handle of the instrument is adjusted as necessary to ensure the base rests on the first metacarpal, while the handle rests on the middle of the 4 fingers. The patient is then instructed to squeeze the dynamometer with maximum isometric effort for at least 5 seconds. Depending on the position of the arm and hand, different results can be achieved. The various positions include having the arm hanging by the side and the extended arm swung above the head and then out to the side during the squeezing motion. A patient will perform several trials of the test, resting for 15 minutes between each test.
The dynamometer may also be used to assess muscle groups of the lower extremities including hip, knee, and ankle. For measurement of hip flexion, patients are seated with feet hanging above floor level. With the test thigh held 10 cm above the table surface, the dynamometer is positioned 5 cm proximal to the patella and the contralateral limb remains neutral. The assessor applies a downward force on the test thigh while the patient resists until resistance cannot longer be maintained. For measurement of hip abduction, patients are placed in a side-lying position with the test leg outstretched and raised 20 cm above the surface bench and the dynamometer is positioned 10 cm proximal to the lateral malleolus. Measurements are repeated bilaterally, in triplicate for hip flexors and hip abductor, with no recovery period between trials.
Scoring
The HHD is scored using force production in kilograms (range = 0 to 90) or pounds (range = 0 to 200). The best result of each trial is recorded and the average of the best scores is calculated. Scores are rated on a range from very poor (low values) to excellent (high values) based on sex-specific norms. Normative data for the upper extremities are found in Table 29.
Table 29: Normative Values for the Upper Extremities for Hand-Held Dynamometry Assessment
Rating | Males | Females | ||
|---|---|---|---|---|
lbs | kg | lbs | kg | |
Excellent | > 141 | > 65 | > 84 | > 38 |
Very good | 123 to 141 | 56 to 64 | 75 to 84 | 34 to 38 |
Above average | 114 to 122 | 52 to 55 | 66 to 74 | 30 to 33 |
Average | 105 to 113 | 48 to 51 | 57 to 65 | 26 to 29 |
Below average | 96 to 104 | 44 to 47 | 49 to 56 | 23 to 25 |
Poor | 88 to 95 | 40 to 43 | 44 to 48 | 20 to 22 |
Very poor | < 88 | < 40 | < 44 | < 20 |
Psychometric Properties
Reliability and validation of the HHD has been reported in community-dwelling elderly adults,57 adolescents,58 and healthy young adults.68-70 In a cross-section of 281 adults over the age of 65, the HHD demonstrated good test-retest reliability and good concurrent validity with a variety of functional tests.57 Excellent test-retest reliability (ICC = 0.84 to 0.99) has also been demonstrated in patients with various neurologic dysfunctions including stroke, traumatic brain injury, incomplete spinal cord injury, and peripheral neuropathy.37,38 In terms of construct validity, the HHD was found to have excellent convergent validity of myometry and isokinetic testing in patients with paraplegia (r = 0.86 to 0.88) and adequate convergent validity in patients with tetraplegia (r = 052 to 0.56).40 Among a group of healthy children and adolescents, the HHD demonstrated good to excellent intra- and interrater reliability (ICC range 0.75 to 0.98) with respect to measurement of maximal isometric torque of a variety of upper and lower muscle groups, except for ankle dorsiflexor.58 Within specific pediatric disorders, the HHD demonstrated excellent test-retest reliability for children with cerebral palsy (r = 0.8 to 0.98)39,41 and traumatic brain injury (r = 0.91 to 0.99).59
In a cohort of Dutch patients with a confirmed diagnosis of Pompe disease (N = 132), better HHD scores were found to be significantly associated with better Rotterdam Handicap Scale and Rasch-built Pompe-specific Activity scale, accounting for the effects of sex, disease duration, and use of wheelchair and ventilator support.19
Several considerations should be noted with the use of HHD: 1. Proper stabilization of the devise must occur to improve reliability39; 2. Sex, body weight, and grip strength can affect a rater’s ability to stabilize an instrument and may influence reliability when “smaller” testers are testing stronger muscle groups71; and 3. Examiners must be strong enough to hold against isometric contraction or overcome for eccentric contraction for the patients being tested.
MID
In a group of healthy female participants (N = 42), a change of more than 6 kg (13.2 pounds) is necessary to detect a genuine change in grip strength 95% of the time with the HHD.72
A literature search was conducted to identify validation information and MIDs of the HHD in patients with Pompe disease; none were identified.
Maximum Expiratory Pressure and Maximum Inspiratory Pressure
Description
MEP and MIP are direct measures of respiratory muscle strength. Maximum expiratory pressure measures the maximum positive pressure that can be generated from 1 expiratory effort starting from total lung capacity or function residual capacity, while MIP measures the maximum negative pressure that can be generated from 1 inspiratory effort starting from functional residual capacity or residual volume. Both are noninvasive tests in which patients are asked to perform a forceful inspiration after an expiration to residual volume level (MIP) or expiration after a full inspiration to total lung capacity (MEP) with an open glottis against an occluded mouthpiece.73-75
Psychometric Properties
Both MIP and MEP measurements can accurately assess respiratory muscle weakness. In a group of patients with chronic obstructive pulmonary disorder of varying severity, MEP and MIP were found to be positively correlated with maximal static inspiratory pressure and forced expiratory volume in 1 second (FEV1), FVC, peak expiratory flow, and total lung capcity.32
MIP may predict diaphragm weakness before a significant change in spirometry end points (e.g., FVC).76 A systematic review evaluating MIP as a clinically meaningful trial end point for neuromuscular disorders found MIP to be a clinically relevant outcome measure in chronic diseases when respiratory failure is secondary to respiratory muscle weakness.77 In patients with neuromuscular disorders, MIP was found to be correlated with improvements in SF-36 mental component summary, the sleep apnea quality-of-life index symptom domain, and the Multidimensional Fatigue Inventory.33,34
Among patients with Pompe disease, MIP was significantly correlated with the 6MWT.36 In terms of its reliability, the MIP was found to have excellent intrarater (ICC = 0.96) and interrater (ICC = 0.92) reliability.35
It should be noted, however, that diminished MEP and MIP do not always reliably confirm inspiratory muscle weakness due to measurement errors such as submaximal effort, poor transmission of intrathoracic pressure to the extrathoracic airways,74 as well as neuromuscular disorder patient-device interface issues, or additional chest wall alternations.78
MID
A literature search was conducted to identify validation information and MIDs of MEP and MIP in patients with Pompe disease; none were identified.
Quick Motor Function Test
Description
The quick motor function test (QMFT) is a functional motor scale designed specifically for Pompe disease.42 The QMFT was constructed on the basis of the clinical expertise of physicians involved in the care of patients with Pompe disease, the GMFM, and the International Pompe Association/Erasmus MC Pompe survey (an ongoing international survey study on the effects of Pompe disease on patients’ lives and how these may change with treatment).42 The test consists of 16 items, including:
Raising the torso;
Neck flexion;
Hand across the midline;
Hip and knee flexion;
Extending the legs;
Sit up;
Extending the arms;
Standing up from a chair;
Standing up from half-knee;
Squatting;
Standing up form a squatting position;
Picking up an object;
Standing on 1 leg;
Walking 10 m;
Jumping; and
Walking up steps.
Scoring
Items on the QMFT are individually scored on a 5-point ordinal scale (ranging from 0 = unable to complete to 4 = completes without difficulty).79,80 The total score for all items ranges from 0 to 64 points where a higher score indicates greater motor function.
Psychometric Properties
The validity and test reliability of the QMFT were assessed in a cohort of 91 patients with Pompe disease, ranging from 5 to 76 years of age.42 The QMFT was found to have excellent internal consistency (Cronbach’s alpha = 0.94), good interrater reliability (ICC = 0.91 for total scale; individual items range from 0.76 to 0.98), good intrarater reliability (ICC = 0.95 for total scale; individual items range from 0.78 to 0.98), and excellent test-retest reliability (ICC = 0.9 for total scale; ICC for individual items range from 0.84 to 1.0). In addition, the QMFT was strongly correlated with the HHD (r = 0.81) and manual testing (r = 0.89) and was able to discriminate between different disease severities.42
MID
A literature search was conducted to identify MIDs of the QMFT in patients with Pompe disease; none were identified.
Pharmacoeconomic Review
Abbreviations
BIA
budget impact analysis
FVC
forced vital capacity
INESSS
Institut national d'excellence en santé et en services sociaux
IOPD
infant-onset Pompe disease
LOPD
late-onset Pompe disease
NOC
Notice of Compliance
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Avalglucosidase alfa (Nexviazyme), lyophilized powder for IV injection |
Submitted price | Avalglucosidase alfa, 100 mg vial: $1,596.59 |
Indication | Nexviazyme (avalglucosidase alfa) is an enzyme-replacement therapy indicated for the long-term treatment of patients with LOPD (acid alpha-glucosidase deficiency) |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | November 12, 2021 |
Reimbursement request | As per indication |
Sponsor | Sanofi Genzyme, a division of Sanofi-Aventis Canada Inc. |
Submission history | Previously reviewed: No |
LOPD = late-onset Pompe disease; NOC = Notice of Compliance.
Table 2: Summary of Economic Information
Component | Description |
|---|---|
Type of economic evaluation | Cost-minimization analysis |
Target population | Patients with LOPD |
Treatment | Avalglucosidase alfa |
Comparator | Alglucosidase alfa |
Perspective | Canadian publicly funded health care payer |
Time horizon | 1 year |
Key data source | |
Costs considered | Drug acquisition costs |
Submitted results | At an estimated cost of $524,563 per patient per year, avalglucosidase alfa was $27,397 less costly than alglucosidase alfa ($551,960 per patient per year). |
Key limitations |
|
CADTH reanalysis results | CADTH did not conduct a base-case reanalysis |
LOPD = late-onset Pompe disease.
Conclusions
The sponsor submitted a cost-minimization analysis based on an assumption of equal efficacy and safety between avalglucosidase alfa and the comparator product, alglucosidase alfa. A head-to-head study determined that avalglucosidase alfa was noninferior to alglucosidase alfa in terms of forced vital capacity (FVC [percent predicted]), although superiority was not demonstrated and additional outcomes were not statistically tested. At the submitted price of $1,597 per 100 mg vial, avalglucosidase is 5% less expensive per mg than the publicly available price of alglucosidase ($840 per 50 mg vial). According to the sponsor’s analysis, and as the recommended dosing of both products is identical, the average annual cost of avalglucosidase alfa ($518,539 per patient) resulted in a savings of $27,292 per patient per year compared to that of alglucosidase alfa ($545,831 per patient). This analysis does not consider any confidential price discounts that may exist for alglucosidase alfa. Of note, the patent for alglucosidase alfa expired in 2021. Should a biosimilar alglucosidase alfa product become available, the relative cost of avalglucosidase alfa is likely to become considerably less attractive. Overall, to ensure cost-effectiveness, avalglucosidase alfa should be no more costly to the health system than alglucosidase alfa, as incremental clinical benefit was not demonstrated.
Economic Review
The current review is for avalglucosidase alfa (Nexviazyme) for the long-term treatment of patients with late-onset Pompe disease (LOPD).
Economic Information
Summary of Sponsor’s Economic Information
The sponsor submitted a cost-minimization analysis of avalglucosidase alfa compared with alglucosidase alfa (Myozyme) for the treatment of patients with LOPD over a 1-year time horizon and from the perspective of a Canadian public health system.3
The safety and efficacy of avalglucosidase alfa was assumed to be equal to alglucosidase alfa on the basis of the COMET trial,2 a double-blind, phase III randomized controlled trial of patients with LOPD who were naïve to enzyme therapy and which reported avalglucosidase alfa to be noninferior to alglucosidase alfa for the primary outcome of change from baseline to week 49 in FVC percentage predicted, although superiority could not be concluded and statistical testing stopped for all subsequent outcomes (see CADTH Clinical Review Report). Results for secondary and tertiary outcomes were generally as expected, in that patients did not show major clinical decline when treated with either drug, compared with no treatment.
Only drug acquisition costs were included in the model, as all other costs were assumed to be equal between treatments. ||||||| percent of patients were assumed to be children with a mean body weight of 30 kg,4 whereas the other ||% patients were assumed to be adults with a mean body weight of 76 kg.5 Drug wastage was considered, with doses rounded to the nearest whole vial. Patients with LOPD were assumed to be ||% adherent. Patient weight, the proportion of patients who were adults, and the adherence rate were all varied probabilistically, and a standard error of 10% of the mean was assumed.
At a submitted price of $1,596.59 per 100 mg vial and the product monograph recommended dosing of 20 mg/kg every 2 weeks, the weighted average annual cost per patient of avalglucosidase alfa for the population previously described was $524,563, a savings of $27,397 per patient per year compared with the average annual cost per patient for alglucosidase alfa of $551,960 (see Table 3). Deterministic results were similar.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total 1-year drug costs ($) | Incremental drug costs ($) |
|---|---|---|
Alglucosidase alfa | 551,960 | Reference |
Avalglucosidase alfa | 524,563 | –27,397 |
Source: Sponsor’s economic submission.3
The sponsor conducted a number of sensitivity analyses that considered infant-onset Pompe disease (IOPD), excluded drug wastage, assumed all patients were adults or children, and varied the average patient weight. Avalglucosidase alfa remained cost saving compared to alglucosidase alfa in all scenarios.
CADTH Appraisal of the Sponsor’s Economic Information
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
Clinical equivalence is uncertain: In the COMET trial, avalglucosidase alfa was noninferior to alglucosidase alfa for the primary outcome of FVC (percent predicted) for the first 49 weeks of treatment in patients with LOPD.1,2 Avalglucosidase alfa appeared to be at least as effective as alglucosidase alfa in preventing further deterioration, though superiority was not demonstrated and statistical testing was stopped for secondary outcomes. No new safety signals were identified. These results begin to address, at least partially, some of the most important outcomes identified in the patient group submission, which included motor, respiratory, and health-related quality of life measures. Key limitations of this trial include the small number of patients and the lack of data available for the pediatric population. Long-term differences between these 2 enzyme-replacement therapies may be better demonstrated once the extension phase of COMET is complete.
CADTH was unable to account for limitations associated with the trial and lack of long-term efficacy data in reanalyses.
Uncertainty in mean pediatric patient weight: The sponsor’s model used a mean pediatric patient weight of 30 kg, in accordance with the 2016 Institut national d'excellence en santé et en services sociaux (INESSS) review of imiglucerase for Gaucher disease,4 as a proxy for the mean pediatric patient weight for LOPD. However, the cited report provides a range of 10 kg to 40 kg for their patient population, rather than a mean.
CADTH explored this uncertainty in scenario analyses by varying pediatric patient weight from 10 kg to 40 kg.
Uncertainty in the impact of adherence: The sponsor included an adherence input that assumed that ||% of doses of both enzyme-replacement therapies would be received by patients with LOPD. Although the clinical expert considered this figure to be plausible, it is based on the sponsor’s internal data, which CADTH was unable to validate.
CADTH conducted a scenario analysis assuming 100% adherence to both alglucosidase alfa and avalglucosidase alfa.
CADTH Reanalyses of the Economic Information
CADTH did not undertake a base-case reanalysis. Given the simplicity of the model, the direct evidence supporting the noninferiority of avalglucosidase alfa to alglucosidase alfa, the reduced cost per unit of avalglucosidase alfa, and the expected similarities between avalglucosidase alfa and alglucosidase alfa for all nondrug costs, CADTH reviewers considered the sponsor’s cost-minimization analysis to be adequate to address the decision problem.
CADTH conducted several scenario analyses to test the impact of varying the mean weight of pediatric patients with LOPD from 10 kg to 40 kg, and by assuming 100% adherence to therapy for both enzyme-replacement products. The results showed a similar degree of cost savings, ranging from $25,984 to $29,665 per patient per year. See Table 5 in Appendix 1 for details.
Issues for Consideration
Patent for alglucosidase alfa recently expired: The patent for Myozyme (alglucosidase alfa) expired in July 2021.6 As such, it is possible that a biosimilar alglucosidase alfa product may become available in the next few years at a price lower than the submitted price of avalglucosidase alfa. This would make the cost of avalglucosidase alfa at the submitted price less attractive to public drug plans.
Comparator pricing based on publicly available prices: The modelled price of alglucosidase alfa is based on publicly accessible list prices and does not reflect any confidential pricing that may have been negotiated by public plans. The comparative price of avalglucosidase alfa is likely less attractive than estimated if there are existing confidential discounts on alglucosidase alfa.
Potential for increased drug wastage owing to different package sizes: Alglucosidase alfa is available in 50 mg vials,7 whereas avalglucosidase alfa will be available in 100 mg vials.8 At costs of approximately $16 to $17 per mg, it is important to minimize product wastage while ensuring patients receive an appropriate dose. The clinical expert consulted by CADTH indicated that dosing of alglucosidase is rounded to the nearest whole vial, when possible, to ensure minimal wastage of excess product, with more experienced clinics adjusting individual doses over time to adhere to an appropriate average dose. However, the availability of avalglucosidase alfa in a larger vial size than that of alglucosidase alfa may make such rounding more difficult and, thus, may potentially increase drug wastage and decrease estimated savings.
Potential for off-label use for patients with IOPD: Avalglucosidase alfa is only indicated for the long-term treatment of patients with LOPD,8 whereas alglucosidase alfa is indicated for Pompe disease in general, including IOPD.7 Potentially, some patients with IOPD could receive off-label treatment with avalglucosidase alfa. However, because alglucosidase alfa is often used at a higher dose (40 mg/kg) in clinical practice than indicated in the product monograph, according to the clinical expert consulted by CADTH, the use of avalglucosidase alfa for these patients at a similarly higher dose would result in increased cost savings, compared with LOPD patients of an equivalent weight.
Potential for 1-time increase in administration costs in switching patients: The clinical expert consulted by CADTH did not express concern about switching patients previously treated with alglucosidase alfa to avalglucosidase alfa, but did indicate that for patients who typically receive their infusions at home, the first infusion with avalglucosidase alfa might instead be performed in an infusion clinic or hospital as a precaution against unexpected reactions, which may be associated with a small 1-time increase in costs. However, the clinical expert also indicated that the sponsor often pays infusion costs for alglucosidase alfa and may continue to do so for avalglucosidase alfa; therefore, it is unlikely this potential increase in costs will be tangible. The clinical expert consulted by CADTH otherwise agreed that all other costs associated with administration, as well as other nondrug costs, would be similar between the 2 enzyme-replacement therapies.
Conclusions
The sponsor submitted a cost-minimization analysis based on an assumption of equal efficacy and safety between avalglucosidase alfa and the comparator product, alglucosidase alfa. A head-to-head study determined that avalglucosidase alfa was noninferior to alglucosidase alfa in terms of FVC (percent predicted), although superiority was not demonstrated and additional outcomes were not statistically tested. At the submitted price of $1,597 per 100 mg vial, avalglucosidase is 5% less expensive per mg than the publicly available price of alglucosidase ($840 per 50 mg vial). According to the sponsor’s analysis, and as the recommended dosing of both products is identical, the average annual cost of avalglucosidase alfa ($518,539 per patient) resulted in a savings of $27,292 per patient per year compared to that of alglucosidase alfa ($545,831 per patient). This analysis does not consider any confidential price discounts that may exist for alglucosidase alfa. Of note, the patent for alglucosidase alfa expired in 2021. Should a biosimilar alglucosidase alfa product become available, the relative cost of avalglucosidase alfa is likely to become considerably less attractive. Overall, to ensure cost-effectiveness, avalglucosidase alfa should be no more costly to the health system than alglucosidase alfa, as incremental clinical benefit was not demonstrated.
References
1.Clinical Study Report: EFC14028. A phase 3 randomized, multicenter, multinational, double blinded study comparing the efficacy and safety of repeated biweekly infusions of avalglucosidase alfa (neoGAA, GZ402666) and alglucosidase alfa in treatment-naïve patients with late onset Pompe disease (COMET) [internal sponsor's report]. Mississauga (ON): Sanofi Genzyme; 2020 Sep 1.
2.Diaz-Manera J, Kishnani PS, Kushlaf H, et al. Safety and efficacy of avalglucosidase alfa versus alglucosidase alfa in patients with late-onset Pompe disease (COMET): a phase 3, randomised, multicentre trial. Lancet Neurol. 2021;20(12):1012-1026. PubMed
3.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Nexviazyme (avalglucosidase alfa),100 mg/vial lyophilized powder for injection. Mississauga (ON): Sanofi Genzyme; 2021 Sep 30.
4.Cerezyme – Maladie de Gaucher: avis de refus d’inscription à la liste établissements. Québec City (QC): INESSS; 2016: https://www.inesss.qc.ca/fileadmin/doc/INESSS/Inscription_medicaments/Avis_au_ministre/Octobre_2016/Cerezyme_2016_10.pdf. Accessed 2021 Nov 18.
5.Avis aux fabricants de médicaments: conventions relatives au poids moyen et à la surface corporelle moyenne des patients. Québec City (QC): INESSS; 2019: https://www.inesss.qc.ca/fileadmin/doc/INESSS/Inscription_medicaments/Avis_fabricants/20190806_Avis_fabricants_Analyses_economiques.pdf. Accessed 2021 Nov 18.
6.Health Canada. DIN 02284863, alglucosidase alfa, Myozyme, Patent no. 2416492. Search for expired patents [online database]. Ottawa (ON): Government of Canada; 2021: https://pr-rdb.hc-sc.gc.ca/pr-rdb/search_expiration-recherche_expires.do. Accessed 2021 Nov 18.
7.Myozyme (alglucosidase alfa): lyophilized powder 50 mg/vial [product monograph]. Mississauga (ON): Sanofi Genzyme; 2016: https://pdf.hres.ca/dpd_pm/00037590.PDF. Accessed 2021 Nov 18.
8.Nexviazyme (avalglucosidase alfa): lyophilized powder 100 mg/vial for intravenous infusion [product monograph]. Mississauga (ON): Sanofi Genzyme; 2021 Nov 12.
9.Exceptional Access Program (EAP). Toronto (ON): Ontario Ministry of Health; Ontario Ministry of Long-Term Care; 2021: https://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx. Accessed 2021 Nov 18.
10.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Nexviazyme (avalglucosidase alfa),100 mg/vial lyophilized powder for injection. Mississauga (ON): Sanofi Genzyme; 2021 Sep 30.
11.Wencel M, Shaibani A, Goyal NA, et al. Investigating late-onset Pompe prevalence in neuromuscular medicine academic practices: the IPaNeMA study. Neurol Genet. 2021;7(6):e623.
12.Table: 17-10-0005-01. Population estimates on July 1st, by age and sex, Canada. Ottawa (ON): Statistics Canada; 2020: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710000501. Accessed 2021 Jul 20.
Appendix 1: Additional Economic Information
Note that this appendix has not been copy-edited.
Cost Comparison Table
The comparators presented in the following table have been deemed appropriate based on feedback from a clinical expert. Comparators may be recommended based on appropriate practice or actual practice. Confidential discounts based on potential Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 4: CADTH Cost Comparison Table for LOPD
Treatment | Strength / concentration | Form | Price ($) | Recommended dosage | Daily drug cost | Annual cost |
|---|---|---|---|---|---|---|
Avalglucosidase alfa (Nexviazyme) | 10 mg/mL | 100 mg vial | 1,596.5890a | 20 mg/kg IV infusion every other week. | 30 kg patient: $684.25 75 kg patient: $1,710.63 | 30 kg patient: $249,752 75 kg patient: $624,381 |
Alglucosidase alfa (Myozyme) | 5 mg/mL | 50 mg vial | 840.3100 | 20 mg/kg IV infusion every other week. | 30 kg patient: $720.27 75 kg patient: $1,800.66 | 30 kg patient: $262,897 75 kg patient: $657,242 |
LOPD = Late-Onset Pompe Disease.
Note: All prices are public list prices from the Ontario Drug Benefit Formulary Exceptional Access Program (accessed November 2021),9 unless otherwise indicated, and include wastage of excess medication in vials but do not include markups or dispensing fees. Annual cost assumes a 365-day year.
aSponsor’s submitted price.3
Scenario Analyses
CADTH did not conduct a base-case reanalysis in the review of avalglucosidase alfa. Several scenario reanalyses were conducted with minimal impact on incremental results. See Table 5.
Table 5: Summary of the CADTH Scenario Reanalysis Results (Deterministic)
Scenario | Total 1-year drug costs: alglucosidase alfa ($) | Total 1-year drug costs: avalglucosidase alfa ($) | Incremental costs ($) |
|---|---|---|---|
Sponsor’s deterministic base case | 545,831 | 518,539 | −27,292 |
CADTH Scenario: pediatric patient weight 10 kg | 519,683 | 493,699 | −25,984 |
CADTH Scenario: Pediatric patient weight 40 kg | 558,905 | 530,960 | −27,945 |
CADTH Scenario: 100% adherence | 593,295 | 563,630 | −29,665 |
aRange of pediatric patient weights was as reported in a 2016 INESSS review regarding children with Gaucher’s disease.4
Appendix 2: Submitted Budget Impact Analysis and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 6: Summary of Key Take-Aways
Key Take-aways of the BIA |
|---|
|
BIA = budget impact analysis.
Summary of Sponsor’s Budget Impact Analysis
In the submitted budget impact analysis (BIA),10 the sponsor assessed the reimbursement of avalglucosidase alfa for patients with LOPD. The BIA was conducted from a Canadian public drug payer perspective over a 3-year time horizon using a claims-based approach and including only drug acquisition costs.
Data for the model was obtained mostly from internal sponsor data, including the number of expected patients, the growth in patient numbers over time, the adherence rate, and the predicted market uptake of avalglucosidase alfa.10 Patient weight and adherence were as estimated in the cost comparison above. Key inputs to the BIA are documented in Table 7.
The sponsor’s submission included the following key assumptions:
Avalglucosidase alfa will be used in the same patients who would otherwise receive alglucosidase alfa.
No wastage nor rounding of doses to the nearest vial occurs.
Table 7: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as Year 1 / Year 2 / Year 3 if appropriate) |
|---|---|
Target population | |
Number of reimbursed LOPD patients aged < 18 years (30 kg)a | | |b |
Number of reimbursed LOPD patients aged 18+ years (76 kg)a | | |b |
Market Uptake – Reference Scenario (all patients, 3 years) | |
Alglucosidase alfa | 100% / 100% / 100% |
Market Uptake – New Drug Scenario (switching patients, 3 years) | |
Avalglucosidase alfa | || ||| |
Alglucosidase alfa | || | |
Market Uptake – New Drug Scenario (new patients, 3 years) | |
Avalglucosidase alfa | 100% / 100% / 100% |
Alglucosidase alfa | 0% / 0% / 0% |
Adherence rate | |
Adherence for patients with LOPD | | |c |
Cost of treatment (per patient older than 1 year) | |
Alglucosidase alfa (aged < 18 / 18+) | $241,865 / $612,725 |
Avalglucosidase alfa (aged < 18 / 18+) | $229,772 / $582,089 |
LOPD = late-onset Pompe disease.
aPediatric patient weight was assumed to be 30 kg as that is within the 10 kg to 40 kg range accepted by INESSS in a report on children with Gaucher’s disease.4 Adult patient weight is the overall average weight recommended by INESSS for the adult population (all genders).5
bProjected from the sponsor’s data on file of patients reimbursed for alglucosidase alfa by CDR-participating plans.10
cBased on sponsor’s internal data.10
Summary of the Sponsor’s Budget Impact Analysis Results
Results of the sponsor’s base-case BIA suggest that the yearly incremental savings associated with the reimbursement of avalglucosidase alfa, excluding dispensing fees and markup, for patients with LOPD were expected to be $746,548 in Year 1, $1,046,458 in Year 2, and $1,285,095 in Year 3, for a 3-year cumulative total of $3,078,101. When dispensing fees and markups are included, the sponsor’s model reports an incremental budget savings of $747,308 in Year 1, $1,047,591 in Year 2, and $1,286,483 in Year 3, for a 3-year cumulative total of $3,081,382. The sponsor conducted scenario analyses varying the patient population by 25%, increasing adherence to 100%, varying the average weight of pediatric patients, and rounding the number of vials used per dose down. All scenarios had 3-year total cumulative savings between $2.3 and $3.8 million.
CADTH Appraisal of the Sponsor’s Budget Impact Analysis
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
Uncertainty in the number of patients with LOPD in Canada: The sponsor’s analysis is based on a total of | | patients with LOPD being publicly reimbursed for alglucosidase alfa in the base year, rising to | | patients by Year 3 of the analysis as new patients are identified. This figure is based on the sponsor’s internal data on the known number of patients with LOPD receiving commercial alglucosidase alfa who were reimbursed by CDR-participating drug plans. Prevalence data in other countries vary by location and ethnicity, with France reporting a prevalence of 1 in approximately 70,000 people while data from the Netherlands indicates a prevalence of 1 in 57,000.11 Canadian-specific data were not found, yet with a population of approximately 29.4 million people in 2020 outside of Quebec and assuming a lower prevalence of 1 in 100,000 people, the expected number of LOPD patients would be almost 300.12 While not all patients in Canada (outside of Quebec) would be reimbursed by public plans, given current general public drug coverage proportions and the high annual cost of alglucosidase alfa, it is likely that a larger proportion of patients would be publicly reimbursed than the sponsor’s estimate. However, information from 2 public plans indicates similar numbers of patients using alglucosidase alfa as reported by the sponsor. The source of this apparent discrepancy is unknown, however given that avalglucosidase alfa is less expensive than alglucosidase alfa, increases in assumed population size would increase the resulting budgetary savings.
CADTH considered a scenario analysis in which the number of publicly reimbursed patients with LOPD was doubled.
Wastage was not considered: Unlike in the sponsor’s cost-minimization analysis, the submitted BIA base case did not consider wastage of excess medication in vials nor rounding to the nearest full vial. Alglucosidase alfa is available in 50 mg vials,7 while avalglucosidase alfa will be available in 100 mg vials.8 The clinical expert consulted by CADTH indicated that dosing of alglucosidase is rounded when possible to ensure minimal wastage of excess product, with more experienced clinics adjusting individual doses over time to adhere to an appropriate average dose while further minimizing wastage. However, the availability of avalglucosidase alfa in a larger vial size than that of alglucosidase alfa may make such rounding more difficult and thus may potentially increase drug wastage and thus decrease estimated savings.
CADTH considered wastage in its base-case reanalysis, rounding doses to the nearest full vial.
Uncertainty in impact of adherence: The sponsor included an adherence input assuming that | |% of doses of both enzyme-replacement therapies would be received by patients with LOPD. While the clinical expert considered this figure to be plausible, it is based on the sponsor’s internal data, which CADTH was unable to validate.
CADTH assumed 100% adherence to both alglucosidase alfa and avalglucosidase alfa in a scenario reanalysis.
Uncertainty in mean pediatric patient weight: The sponsor’s model reportedly used the mean pediatric patient weight of 30 kg, cited as being from the 2016 INESSS review of imiglucerase for Gaucher disease,4 as a proxy for the mean pediatric patient weight for LOPD. However, the cited report appears to give a range of 10 kg to 40 kg for their patient population, rather than reporting a mean.
CADTH explored this uncertainty in scenario analyses by varying pediatric patient weight from 10 kg to 40 kg.
CADTH Reanalyses of the Budget Impact Analysis
CADTH revised the sponsor’s base case by rounding doses to the nearest vial to consider potential wastage. Table 8 outlines the parameters used by the sponsor in comparison to those used by CADTH.
Table 8: CADTH Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
1. None | — | — |
Changes to derive the CADTH base case | ||
1. Wastage | Wastage not considered. | Wastage considered. Doses rounded to the nearest full vial rather than rounded down as programmed by the sponsor. |
CADTH base case | 1 | |
BIA = budget impact analysis.
Applying this change reduced the total 3-year budgetary savings associated with reimbursing avalglucosidase alfa for patients with LOPD to $3,041,419 when markups and dispensing fees are excluded, or $3,044,660 when they are included. The results of the CADTH reanalysis are presented in summary format in Table 9 and a more detailed breakdown is presented in Table 10.
Table 9: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total |
|---|---|
Submitted base case | -$3,078,101 |
CADTH reanalysis 1 and base case: Wastage | -$3,041,419 |
BIA = budget impact analysis.
CADTH also conducted additional scenario analyses to address remaining uncertainty:
A. The number of patients treated for LOPD was doubled to explore the impact of uncertainty in the eligible population size.
B. Adherence was assumed to be 100%.
C. Pediatric patients were assumed to weigh 10 kg.
D. Pediatric patients were assumed to weigh 40 kg.
Table 10: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $20,445,674 | $22,525,715 | $23,993,031 | $25,702,211 | $72,220,957 |
New drug | $20,445,674 | $21,779,166 | $22,946,573 | $24,417,116 | $69,142,855 | |
Budget impact | $0 | -$746,548 | -$1,046,458 | -$1,285,095 | -$3,078,101 | |
CADTH Base Case | Reference | $20,195,746 | $22,251,601 | $23,702,792 | $25,395,849 | $71,350,242 |
New drug | $20,195,746 | $21,513,921 | $22,668,831 | $24,126,071 | $68,308,823 | |
Budget impact | $0 | -$737,680 | -$1,033,962 | -$1,269,777 | -$3,041,419 | |
CADTH Scenario Analysis A: Population size doubled | Reference | $40,391,493 | $44,503,202 | $47,405,584 | $50,791,698 | $142,700,484 |
New drug | $40,391,493 | $43,027,841 | $45,337,661 | $48,252,143 | $136,617,646 | |
Budget impact | $0 | -$1,475,360 | -$2,067,923 | -$2,539,555 | -$6,082,838 | |
CADTH Scenario Analysis B: 100% Adherence | Reference | $21,951,898 | $24,186,523 | $25,763,905 | $27,604,184 | $77,554,611 |
New drug | $21,951,898 | $23,384,696 | $24,640,033 | $26,223,991 | $74,248,721 | |
Budget impact | $0 | -$801,826 | -$1,123,871 | -$1,380,193 | -$3,305,890 | |
CADTH Scenario Analysis C: Pediatric patients weigh 10 kg | Reference | $19,228,286 | $21,122,896 | $22,412,844 | $23,783,414 | $67,319,155 |
New drug | $19,228,286 | $20,433,589 | $21,443,379 | $22,594,257 | $64,471,226 | |
Budget impact | $0 | -$689,308 | -$969,465 | -$1,189,157 | -$2,847,929 | |
CADTH Scenario Analysis D: Pediatric patients weigh 40 kg | Reference | $20,679,477 | $22,815,953 | $24,347,766 | $26,202,066 | $73,365,785 |
New drug | $20,679,477 | $22,054,087 | $23,281,556 | $24,891,979 | $70,227,622 | |
Budget impact | $0 | -$761,866 | -$1,066,210 | -$1,310,088 | -$3,138,164 |
BIA = budget impact analysis.
Stakeholder Input
Patient Group Input
Muscular Dystrophy Canada
About Muscular Dystrophy Canada
Muscular Dystrophy Canada is registered with CADTH. www.muscle.ca
Muscular Dystrophy Canada (MDC) supports people affected by muscular dystrophies and related muscle diseases. Together, these rare conditions are referred to as “neuromuscular disorders.” Neuromuscular disorders are a group of diseases that weaken the body’s muscles. The causes, symptoms, age of onset, severity and progression vary depending on the exact diagnosis and the individual.
Since 1954, Muscular Dystrophy Canada has been the leading health charity and voice of the neuromuscular community in Canada. MDC represents 30,896 Canadians impacted by neuromuscular disorders including 12,047 persons with neuromuscular disorders, and 19,155 family members/caregivers.
MDC’s mission is to enhance the lives of those impacted by neuromuscular disorders by continually working to provide ongoing support and resources while relentlessly searching for a cure through well-funded research.
Muscular Dystrophy Canada offers a range of critical programs and services that include: systems navigation, education and knowledge translation, access to financial supports for critical life- changing equipment and services to improve quality of life, peer-to-peer networking, emotional support, evidence- based information for new treatments, medical advances, and clinical trials and advocacy.
Funded by Canadians from coast to coast, our investment in the research community is advancing the development of important new treatments. Our programs and services play a critical role in informing and supporting members of the neuromuscular community by funding equipment to improve daily life; hosting family and caregiver retreats; providing emotional and educational support; and with providing access to vital resources and support systems. Our advocacy efforts focus on enhancing public policy at all levels of government to bring about positive change. We are currently working to bring new treatments and trials to Canada. Advances in medicine have resulted in individuals with neuromuscular disorders living longer but not necessarily living better. As their disorder progresses and changes, so do their needs and financial strains.
Our desire is to provide support through all stages of disease progression by providing the tools, resources and support individuals need to live a full and rich life.
Pompe disease is one of the neuromuscular disorders that falls under MDC’s umbrella.
Pompe disease is caused by the lack or deficiency of a single enzyme, lysosomal acid alpha- glucosidase, leading to severe respiratory and skeletal muscle myopathy due to progressive accumulation of glycogen, which builds up to abnormal levels in tissues, particularly in muscles, ultimately causing the disease’s symptoms. It is a rare condition that is identified in about 1 in 40,000 births. Pompe disease occurs from a defect in the GAA gene leading to the accumulation of lysosomal glycogen and, depending on the form and severity, can result in cardiomyopathy, progressive muscle weakness, respiratory failure, and heart failure.
Information Gathering
Muscular Dystrophy Canada has Neuromuscular Service Support Staff in all provinces across Canada. As part of the System Navigation Program, the Neuromuscular Service Support Staff provide front-line support to thousands of Canadians affected by neuromuscular disorders. The program operates on collaboration and patient engagement principles. Neuromuscular Service Support Staff work directly with patients and family members to identify non-medical needs (e.g., housing, transportation, access to equipment) and provide them access to the right resources in a personalized customized manner. Neuromuscular Service Support Staff work in partnership with patients and their families to address barriers, network and make connections with others in the community, share education materials and resources, enhance life skills and self-coping strategies, embrace inclusion and ultimately provide supports to help positively improve the overall well-being and quality of life of the patient and their family members.
The Neuromuscular Service Support Staff identified and contacted parents whose child (children) have Pompe disease or adults living with Pompe disease to participate in a healthcare experience survey (available in English and French) and semi-structured virtual (phone, Zoom) interviews. The Canadian Association for Pompe (CAP) was instrumental in supporting the dissemination of the survey and call for feedback. CAP has 50 parent/patient society members affected by Pompe; they shared the survey with their constituents by e-blasts and personalized invites.
The following submission reflects data from a total of 41 individuals impacted by Pompe disease; this is remarkable as there is expected to be 60 Canadians affected by Pompe disease at present. Our submission reflects 68% of the population under consideration, and 2 individuals had been treated with Nexviazyme. The respondents included 12 males between ages 26 to 81; 14 females between ages 23 to 75; and 11 parents/caregivers of individuals between the ages of 4 to 63 (7 males; 4 females).
We sought the opinion on the value of having Nexviazyme approved for use in Canada. A qualitative descriptive approach, employing the technique of constant comparison, was used to produce a thematic analysis. We have included patients’ quotes to ensure their voices are captured in this reportand to provide context for quantitative elements. A report capturing all patient comments is also available for review.
Disease Experience
In response to the question posed in the MDC survey: "Can you describe how Pompe disease impacts your (or your child’s) day-to-day life and quality of life? Are there any aspects of Pompe disease that are more important to control than others?" - the following 5 key themes were identified (in order of frequently reported): 1- significant impact on mobility, strength, balance and energy levels; 2- significant impact on breathing; 3- negative impact on mental health; 4- reduced ability to participate in daily activities; 5- negative impact on the family. The below quotes from individuals affected by Pompe disease highlight that the impact of Pompe is not purely physical, but that the condition impacts mental health, quality of life and the well-being of families.
Significant Impact on Mobility, Strength, Balance and Energy Levels
“Having Pompe has caused me to struggle with my movements and balance. I am currently dependant on a wheelchair for mobility. I currently experience a lot of pain mostly in my hips, back and shoulders. I have experienced lots of falls and can not help myself off the floor. This has affected my respiratory system whereas I have been very fatigued. I use to require a trach but no longer require this. But I am dependant on oxygen and require a c-pap machine. I also get frequent headaches. I am required to exercise regularly to keep my strength and range of motion.”
“Pompe disease has caused some mobility issues for me. The most obvious symptom is that my knees are weak, I get very tired from climbing stairs, and I cant stand up without using my hands if I’m sitting on the ground. I also can't do sit ups. My arms are also weaker, so I can't carry heavy things.”
“I have some mobility issues which makes some things more challenging. I still try to continue to do as much as I can but it can be frustrating.”
“It affects the strength of my proximal muscles. In consequence, my balance is affected; my breathing while laying down; my overall strength especially when using stairs and walking uphill.”
“I need a walker as I have poor balance. I can't do things in the kitchen as I have to hold onto the counter with one hand.”
“As a Pompe patient with mild to moderate symptoms, I am no longer able to take part in most physical activities as I no longer have the body strength necessary to do so.”
“The largest impact Pompe has on my day to day life is related to my mobility and confidence.”
“I am not able to move my body in ways that most others find easy. Walking a flight of stairs can be taxing, having to stand up quickly isn’t an option, my balance is not great, and I suffer from sore muscles daily.”
“With the limited movement range and relatively low energy, I am unable to do simple tasks such as long period of standing and walking, physical tasks like heavy lifting, any chores that requires moderate core and lower back strength, even simple chore such as bring laundry up and down a few flights of stairs proved to be difficult.”
“I am bound to a wheelchair and on a ventilator due to late onset Pompe ( diagnosed at 28 months old ) Pompe hinders my day-to-day life by limiting my ability to move and do the simplest of things. I need assistance with all my personal care but still have the ability to drive my chair, eat and drink on my own.”
“Daily activities tax stamina and they must pick and choose so as not to over-expend their energy, which affects their social lives.”
“My son and daughter both have Pompe... they require a lot of sleep. Each day requires planning of activities so as not to over-do things and pay the price after. A too high energy expenditure results in several days of extreme fatigue...the inability to do much at all. Appetite is an issue. Both struggle to eat enough to keep weight on...both extremely thin and always trying to put on weight.”
“I'm fortunate not to have significant pain, but I do have frequent fatigue. It causes me to take longer to do anything more than basic tasks. I avoid doing things that I know I won't be able to handle.”
“Muscle weakness affects breathing and walking, two basics of being able to have energy to accomplish basic tasks.”
“General fatigue means an inability to plan ahead. never knowing how they will feel each day.
Often having to cancel social plans. Limited ability to engage in physical activities and hesitance to divulge to their peers their health issues.”
Significant Impact on Breathing
“Both my children affected by Pompe battle anxiety and depression, muscle pain, spasms and weakness.”
“I am limited in what I can do with my children (unsteady on my feet, can't run, can't lie on my back without breathing assistance, can't swim in deep water) and at work (I'm a cook, I have a hard time lifting things and get pretty worn out being on my feet all day).”
“I have breathing issues and my diaphragm muscle don't work. My posture is very bent as well.” “Breathing is labored most of the time and always using a BPAP at night.”
“I use a Bi-Pap machine for sleeping at night.”
“Pompe has affected my breathing, I have to wear a respirator at night to help me breathe when laying down. I cannot sleep without it. Because of poor muscle strength, I wake each time I move in my sleep. My quality of sleep is definitely worse than it was before the onset of symptoms. If I sleep too long I get quite sore, so it's a balance. I take care when getting out of bed or I might strain a leg, hip or abdominal muscle. I take that kind of care when I do many things.”
Negative Impact on Mental Health
“There are also days where my mental health is affected as I do feel down from time to time that I have to deal with this illness.”
“It negatively impacts my self-esteem to know that I am not going to be able to be the one that helps my children with a lot of things in their life.”
“The biggest negative effect that disease has on me is unavoidable stress linked to “There are a few things I'm unable to do such as sweeping and washing floors and walking with a walker all the time. Need help all of the time.”
“Pompe impacts my confidence to perform daily tasks, attempt athletic activities or try things like hiking with those I don’t know well. I sometimes feel like a burden when friends want to do physical activities or colleagues participate in a sports based activity.”
“The physical limitations and challenges that comes with Pompe disease has also created a negative impact on my mental health. Having to second guess and be careful of what I can and cannot do limits my past time activities, ability to go out and hanging out with friends, and having to carefully plan ahead every time I want to leave the house can really make me shy away from being outside so much, or partake in any activities and accept invitations from friends.”
“Pompe has brought an extra layer of stress into my life. As a female hoping to start a family I question my ability to carry a child. I wonder for how long will I be able to keep up with them, will I be able to play with them outside, etc.”
“He is doing virtual school right now but he is very conscious of the way he looks. He is sitting in a wheelchair during school and worries about what others think about him.”
“Because of Pompe, I suffer from stomach issues which I have heard many others with this disease complain about. The unpredictability and urgency of bowel movements can be extremely nerve wracking. This impacts my ability to do leisure activities such as hike or boat as I need to ensure I can get to a washroom quickly should I experience a flair up.”
“On any given day I don't feel good. Family has accepted but emotionally it's hard.”
“Because of the disease, I always see flaws in things I do. I always wonder how it would be if I was born without the disease and it makes me sad to think about it.”
“It has changed everything in my daily routines and can't do the things I use to do. I can no longer work and help people like I use to. I have struggled with depression and suicidal thoughts. It has impacted my personal relationships and I feel that nobody will want to be in an intimate relationship with me as a result of my disease.”
“Both battle anxiety and depression to varying degrees, off/on. They both live with a sense of not knowing what kind of future they have, how long they will live. For their age, they do quite well with this, but sometimes it weighs heavier than others.”
“The frustration that comes with being limited in what I can do with my children.”
“For me it is just that sometimes I feel down and when I do I don’t much feel like being social.”
“He has been significantly affected from a social perspective. He would get teased because of the way he walks. He would be called "weird" a lot He hates being in a wheelchair and this causes him much distress He doesn’t want to leave the house because he is in a wheelchair. When he was not as verbal, his mental health was exhibited through irritable behaviors. He tends to get agitated more because he can’t do what others kids can do.”
Reduced Ability to Participate in Daily Activities
“It takes time to do everything. We went to a farm this past weekend, but I had to stop and research the different farms to find which ones are most accessible - one that can maneuver his wheelchair. As his mother, I have to assist him in the shower because he is using a bath chair. Independence is very limited and this impacts a lot of what he can on a daily basis.”
“It definitely affects every aspect of everyday life. Just being able to use the toilet can be a challenge, being able to shower, brush your teeth and just begin able to get out bed.”
“It impacts my life and quality of life negatively in almost every way possible.”
“Pompe disease has impacted my day-to-day life tremendously. It limited my movement ability and the energy I have throughout the day. There are days I feel very tired even with adequate amount of rest, this has been made worse by the COVID pandemic, preventing me from visiting the gym, which is essential for keeping myself healthy and slowing down the progression of Pompe.”
“I no longer work, I can not do stairs, basic every day chores are getting harder to do.” “All activities must be planned. There is no spontaneity in life.”
Impact on the Family
“My mother is a Pompe patient. As a result, I assist her in her day-to-day activities, attend doctor appointments, and assist in lifting her up after falls and being an advocate when needed in respect to the healthcare system.”
“I am able to get around by using walkers, wheel-chairs, and scooters. I have a partner who is able and willing to assist me whenever needed. I have a personally modified bathroom, stairs, and chairs in our home.”
“Have to be with someone 24/7 in case something happens to my ventilator.”
“My son is 4 years old. He has low muscle tone, he cannot run or jump or keep up with his peers. He has low oral muscle tone which makes eating more challenging, sometimes he gags and/or vomits. He is hyper-nasal which makes it difficult for people to understand him so I need to translate for him. We have also experienced delays in potty training (he isn't fully trained yet). We are more isolated from socializing for fear of him getting sick. I am his full-time care giver. We are busy with appointments either at the hospital or virtual, homework from physiotherapy, speech therapy and occupational therapy as well as weekly 7 hour long infusions (not including prep and wait times). We need to adapt for his lack of stamina which changes the way we go for walks, bike rides, has limited our ability to hike and removed the possibility for other sports.”
Experiences With Currently Available Treatments
In response to the question posed by MDC: “How are you managing Pompe with currently available treatments or therapies. For each therapy what are the benefits seen, and side effects experienced? Do you have any difficulties accessing these treatments?” - the following 3 key themes emerged: no treatment experience, but focus on rehabilitation; minimal (or plateau) effect of Myozyme observed; positive benefits of enzyme-replacement therapy with minimal side effects observed.
The below quotes from individuals affected by Pompe highlight that while enzyme replacement therapy (Myozyme) has contributed to positive health outcomes, there remains significant concerns over long-term/sustained benefits.
No treatment experience, but focused on rehabilitation
“I am allergic to the standard IV treatment for Pompe and could not take it due to a rash and swelling. I complete physical therapy regularly which helps.”
“I didn't have significant symptoms when diagnosed but declined in a few years. I have received enzyme replacement therapy for over 11 years. At the time I started that treatment, my breathing and mobility were declining rapidly. The decline stopped and I my breathing and mobility both improved. 11 years later they are still better than before treatment. I have experienced gradual decline in the last 5 years, but not as rapid as before. The only side effect that I've seen is difficulty sleeping the day of and some fatigue the next day. I've tried inspiratory muscle training but had to discontinue. My diaphragm is partially paralyzed so I experienced too much discomfort. I've tried CoEnzyme Q10 with no benefit. I've tried salbutamol, which was studied at Duke University and found to help with strength for those treated with Myozyme. Unfortunately, severe muscle cramps caused me to discontinue.”
Minimal (or plateau) effect of Myozyme observed
“I was put on the Myozyme for 18 months all together drugs and stopped them partially because of my decision because they didn't have a big effect. It was expensive and I did not think it was worthwhile carrying on. I am not currently on any medication for Pompe. Not since 2015.”
“I am Myozyme patient with zero side effect. It worked really well in the beginning but I plateaued and I've been regressing since.”
“I follow Myozyme treatment. To my understanding it is supposed to slow down progression of the disease. I have not experienced improvement in my condition and the effect on progression reduction are impossible to evaluate as there are no baselines. I have not experienced any side effects that I am aware of other that the stress related to the infusion procedure as I am have a severe fear of needles.”
“He has received ERT. We find that in the first few years, he was thriving but now he has plateaued. We don’t see any improvements but also not much decline.”
“Myozyme is the only treatment that's been available in our area, it definitely slowed the down progression of his disease but the longer he was on it the more it affected his mental health. The only way to manage it was to stop the Myozyme.”
“I tried enzyme replacement therapy. It seemed to make me more tired and I didn't get any stronger or even stop the weakness from progressing much, if at all. I'm on a high protein, low carb diet. I am hoping a new treatment will work for me before I die.”
“Before the development of Myozyme doctors had only suggested dietary changes to my mother (patient) which ultimately changed my diet as well. Once Myozyme was developed and available, we saw a slight improvement in her mobility (less falls and could walk greater distances before fatigue). However after a few years the results started to plateau and falls were happening more frequently. Vibration therapy was another therapy suggested by her doctor. This therapy requires her to stand on a teeter-totter style vibration plate for a few minutes every day. This therapy improved muscle tone present already, and increased the bone density back to normal levels. However, this therapy requires consistent use and for safety measures requires someone to be present while she completes it in case of falls which is not always possible with mine and my father's work schedules.”
“I am currently doing Myozyme treatment, just finished #4, go to the hospital every two weeks. It’s a 13hour day, I get very exhausted and drained. Haven't gotten any side effects and no results yet. Can't wait till I can do the treatments at home because getting to hospital and the environment makes me really tired.”
“Since I had a robust physical exercise program which started prior to my diagnosis in 2011 and lasted until 2020, there was no dramatic impact that ERT provided when I was approved for it in 2016. I believe that ERT provided initial support for my general condition, though it did not improve my mobility. I have not experienced any side effects. I am very pleased with the availability of Home infusions and would like to see infusions also available internationally which would allow more flexibility in travel.”
Positive benefits to enzyme-replacement therapy with minimal side effects
“Prior to starting ERT I would have several downfalls a month, now I have one or two a year.” “Drastically reduced inflammation in my back and legs.”
“I have experience with Myozyme for 10+ years No side effects Benefits - I find my face starts to droop before my infusion and once I have it, I am tired, but it gives me a boost. It was so good at the beginning, but now not going to the gym or rehab, has not made the effects as good. I have lost a lot of muscle. Hard to tell if Myozyme effect is wearing off or if it’s the time/progression/ageing.”
“My treatments seem to be going okay. I have port that is accessed. The treatments are helping me as I do as much as I can each day and it helps me to keep moving, Side effects are on the following day my face gets red and hot around 10:00 in a.m. and is cleared up by about 4:00p.m. I take a Benadryl, an allergy pill and 2 Tylenol for it.”
“The treatment I have been receiving has helped tremendously. I do notice a steady, but slow negative progression of the symptoms and impacts, which in my knowledge is as good as it can get. There were not many side effects I have experienced with the current treatment, with only one mild reaction to the medication which seems to have been caused by a dramatic increase in infusion speed.”
“ERT had been amazing. I do wish that it took less time as it can feel like you lose a day every second week and to a young teacher that can seem like a lot. I luckily haven’t experienced any negative side effects from my treatment but have found my exhaustion level decreased significantly.”
“Besides exercise the only real treatment I have been on is Myozyme, and well that saved my life, so I think that has been a huge benefit.”
“Very fortunate that ERT has worked well, few side effects; I received them in the hospital which is 10 minutes away. I now have access with Handi-Trans.”
“My son has only been on Myozyme. First 20mg every 2 weeks, then weekly, now 40mg weekly. With the increased doses he has shown an increase in energy and oral capability. He was initially failure to thrive leading to tube feeding which increased his energy. His left lung was collapsed by his enlarged heart so he was on BiPap for a while until his heart returned to a relatively normal size. That also increased his energy however it disrupted his sleep and caused him to vomit if he was sick.”
“I have been fortunate to have experienced excellent treatment in the two ways I have received treatment. The first was with hospital care in which I would go to the hospital to receive my infusion on a designated day every two weeks. The care was exemplary and professional. I could arrive at the hospital at a regular time and be done at a regular time. This was during my first 10 years. When the pandemic required home infusions, I received excellent care there as well. Everything went well and was professional. I had no side effects which made management unnecessary.”
“My treatment experience has always been great I had have no complications through my whole life I have been receiving the treatments.”
“Treatment is keeping me somewhat stable, at the beginning, I would get itchy but that has gone away. I get a bit tired after treatment but the next day I am good.”
“I have been on ERT since 2004 and was stable for a while but find myself declining. Treatment has made great and huge improvement to me, my ability to do sports has increased and I have more strengths in both legs and arms. I haven't have much side effects yet.”
“Getting treatment has been great. When I was more mobile I was at a clinic and they were great. Now I'm getting home infusions and that's been great so far. I've been able to access physicians and help when needed.”
Improved Outcomes
Improvements that patients and caregivers would like to see in a new treatment can be categorized as those that (1) promote strength and breathing function; (2) slow down progression without a plateau effect; and is (3) delivered in a different mode that saves time.
Regaining strength and breathing function
“I would like to be able to stand again.”
“Continue to improve with muscle strength and keep my lung function as is without any decrease.”
“Compared to Myozyme, I need this to give me better strength I know this drug is not a cure, but I need it to help with my fatigue and give me muscle strength.”
“Regaining strength.”
“I would like to see more strength and easier breathing.” “Muscle strengthening breathing improvement.” “Breathing capacity , additional leg muscle mobility.”
“Independence. Breathing. Being able to walk. Being able to work. Being able to mother.”
“Recovery of muscle strength and respiratory capacity would be wonderful but, realistically, I'd like to see a more effective halt in decline. I would consider that a win.”
“I would like to get better with my breathing and walking.”
“I would like to see positive improvement in muscle strength.”
“I would love to see something that got rid of the glycogen from my muscles and repaired the damage to them so I could get stronger again. So the muscles around my arteries could repair themselves.”
“Stronger effectiveness for oral muscles.”
Slow down progression without plateau effect
“Realistically, I would like to see even further slowing of the negative impacts and progression of the disease. Optimistically, I would like to see it preventing any further impacts or damage cause by the disease. Very optimistically, I hope for it to reverse the damages caused by the disease.”
“I would love to see a treatment that stops all deterioration and my dream would to see one that rejuvenates your muscle to become normal.”
“If possible get some of the muscle loss back and not decline after a few years.”
“It would be particularly beneficial to me if the new treatment would specifically slow the progression of the Pompe disease, and especially my legs and core muscles.”
Different mode of drug delivery
“Something in pill form.”
“Faster infusion time would be great!”
“Any other mode of treatment other than infusion.”
“Something that could be taken orally or doesn't take hours to infuse.” “A more rapid method of delivery.”
“Maybe less process time the medicine needs to get in the body not really sure.” “Less time involvement, effective at addressing general fatigue.”
“General greater effectiveness that would lead to fewer infusions and shorter infusion times.”
“Better absorption into the muscle tissue, longer active periods in the body (current medications only last 72 hours before excretion), no plateau in chronic usage, assistance in regenerating muscular tissue to replace that which is degenerated already.”
When considered therapy, patients, families and caregiver consider mode of delivery, side effects, time, frequency of treatments, convenience and impact on finances (cost). It was consistently noted that low invasiveness, limited hospital visits, safety/low side effects and low costs were highly valued when considering a treatment. Not requiring the hospital to administer the drug. Having the ability to take medication at home would simplify the process by allowing persons affected to have more control. A treatment that has continuous presence in the system may provide with a more constant response. Less time in hospitals was indicated as highly valued and welcomed especially in the era of COVID-19. If families were faced with the decision to choose a different therapy, they would consider potential side effects reported by the “new” versus “current” therapy. They would consider the ease of accessibility of treatment and whether private/provincial insurance would cover costs.
Experience With Drug Under Review
Two adults indicated they received the drug under review as part of the clinical trial. Detailed video interviews were conducted with both individuals where they shared their treatment experiences (please see links attached to this submission). In short, the individuals shared:
“I am on the clinical trial. I have not done any other therapies. I have not had any side affects I have seen huge improvements.”
“I was in the clinical trial for it. It was my first treatment so I don't have any comparison. It gave me a huge improvement with my muscle strength however the site is out of my province so it is very time consuming for me to go there (and my caregiver). There were no side effects for me.”
Companion Diagnostic Test
100% reported that they did have diagnostic testing completed with at least a blood test; but many also had biopsies to confirm diagnosis. The vast majority found it to be a cost-effective but lengthy process. Below are quotes that further highlight the experiences of patients and caregivers with the testing:
Simple blood test and/or muscle biopsy
“I had to see a specialist in my hometown and went for blood work. I then went to Hamilton to see a specialist after being diagnosed here. I was able to access treatments at my local hospital. I received travel grants for any travel.”
“I believe it was paid for my Ontario Medicare. Testing was set up for me and it was all requested by the doctor at the hospital. I did not have any concerns with the testing. It was no worse than having blood taken. I was diagnosed with muscle biopsy after having elevated liver enzymes. It took few months.”
“The doctor that diagnosed a family member organized a genetic blood test for me that confirmed my diagnosis; I was not exposed to any costs.”
“It only took a blood test. The family later had genetic testing done. There was no out of pocket costs except for recommended yearly visits to Halifax (5 hours’ drive) to a rare disease specialist.”
“Government covered all costs, genetic testing was blood tests done at the doctors request. We also completed muscle biopsy at this point. No delay in treatment from testing.”
“I had back pain was lucky to have a great walk in clinic who got the process going fast once I had been sent to children’s had a biopsy right away to collect piece of my thigh, after that I was diagnosed with Pompe and go on the list for treatment hoping to get approved and I did!”
“I just did some blood tests; we went to many doctor’s and finally got a referral to a specialist. After hearing my symptoms, the specialist was pretty sure I had Pompe disease.”
Misdiagnoses
“Testing was part of the diagnostic process from the get-go. Was initially diagnosed with muscular dystrophy, but got correct diagnosis within a few week waiting period for test to be done at lab in Quebec.”
“I had repeated misdiagnoses. I kept getting worse and worse. Tests upon tests.”
“I was diagnosed with Muscular Dystrophy at first, however, my (at the time) pediatric specialist was not satisfied with the result and did further testing. I was then scheduled to have a muscle biopsy, which properly diagnosed me with Pompe.”
“Biggest delay in diagnosis was being ignored. Misdiagnosed as B12 deficient due to being vegan (B12 was fine), blood came back hypo-thyroid, abdominal ultrasound showed enlarged heart; finally sent to Metabolics after 2 weeks. Blood was sent from BC to Quebec and South Carolina but 14 days from meeting Metabolics we had the first treatment. Testing was covered by BC.”
“I was diagnosed with Limb Girdle Muscular Dystrophy at age 12. Because I was getting different symptoms I asked my doctor to have me retested. They did muscle testing, bloodwork and it came back as Pompe Disease.”
“Spent years thinking I had Limb Girdle Muscular Dystrophy. New symptoms lead to new testing. Breathing test led to muscle bi posy. Hospital for breathing local (20km), main testing Ottawa (60km). Appointment for genetics was easy to get. No cost for test. Just travel, gas and parking.”
“Following a rigorous physical examination in my doctor's office in 2010, I began a series of tests which while inconclusive by Dec 2010, suggested that I had Limb Girdle Muscular Dystrophy. When my younger brother was properly diagnosed with Pompe Disease in February 2011, I immediately was given a blood spot test and had a muscle biopsy done to confirm my Pompe disease in late February 2011. At the time I was living in the Lower mainland of BC so I had no difficulty in travelling to the VGH for testing. I had a 5 year delay in receiving ERT due to the report from the provincial health rare disease committee that I was too healthy. All the costs associated with testing for Pompe Disease were covered by my BC health plan.”
Lengthy diagnostic process: multiple tests
“It took a long time to get diagnosed I kept going to the doctor. It wasn’t until I went to Physiotherapy that they told me more was going on. After much more testing with a neurologist they finally did a biopsy to diagnose Pompe. There was no cost to me.”
“I had an awful genetic testing experience. It took them almost 30 years. Delay in diagnosis. Took them about a year and half until finally I got the right test. I got bloodwork. I was tested with leukemia and had to get bone marrow. The tests were paid for but lots of travel to different docs.”
“I travelled from Thunder Bay to Hamilton for testing, Because they diagnosed my brother with Pompe, they called me and asked me to go down with him as he was already having his first treatment
“They covered all cost for me to go. It was about a month after being diagnosed that I started my treatments. I was identified as a potential patient because a family member was diagnosed after a great length of time.”
“It took about 3.5 weeks to diagnose him. There was a lot of testing and process of elimination. There was bloodwork. There wasn’t much information given along the way. We didn’t pay for any of the testing, it was conducted through the testing. The results were explained...but NOT well. I was at a loss for words. Unexpected diagnosis. I had never heard of Pompe.”
“We had to travel about 90 minutes for testing and doctors’ appointments; he had a few blood tests done and a MRI done for the diagnosis. Our provincial health insurance covered all the appointments and tests, social assistance helped with mileage and meals.”
“The doctors did tons of tests for a myriad of different diseases before they found out what it was. I was the first diagnosed juvenile in Canada. I'm in Canada so we didn't have to pay for anything out of pocket. I was 14 when diagnosed, so I was still with my parents.”
“It was my mother that fought for the diagnosis as I was 28 months old. She went to many doctors for many tests; most just pushed her away saying things like she was a lazy mother. It was the muscle biopsy that helped with making the right diagnosis.”
“It took years to finally be referred to the proper doctor and department. But once we were, testing and diagnosis went fairly smoothly and was expedited as fast as possible. Still took many months to receive clear answers. Cost covered by Alberta Health Care.”
“Testing took a very long time.”
“I was misdiagnosed for a while but after seeing a few physicians they got to the bottom of it. Being persistent with my family doctor helped. I had a muscle biopsy and then seen another doctor who said I didn’t have Pompe so ordered a DNS test. It took about 2 years for the final result. Treatment was not available to me at first and the provincial government refused to pay for it. Took 10 years from diagnosis until getting treatment.”
“I was referred to a metabolic specialist to look at unusual blood test results. I was also seen by a neuromuscular specialist in the clinic as well. I was otherwise healthy. I was tested for many things, eventually being diagnosed with Pompe disease after about four years. I had so many tests, some taking some time to get results for. I've had a liver biopsy and a muscle biopsy. Both were painful for a few days. Genetic testing wasn't done. It was never made clear to me which test method they used. When I was diagnosed I was still mostly asymptomatic, so the delay in diagnosis didn't delay treatment for me. For the last two years of testing, I was living about 400kms from the clinic, so there were travel costs. I can imagine that for some people that would have been an issue.”
“I saw a doctor in Toronto who wasted 10 months testing and not reporting the results quickly. After the dry blood test she said it might be Pompe and she walked away. She came back and said we might go to see a doctor in Hamilton but it would take a long time to get an appointment and it would be very expensive. We already had an appointment for the next day. He diagnosed me on the spot and my first infusion was 2 weeks later. We didn't pay for anything except one test which was $75 and that was with the first doctor.”
Emotional experience
“It was an extremely stressful and emotional time. A lot of uncertainty.”
“It was probably the most stressful decision in my life as to this day it remains one that is mostly based on hope and faith.”
“A shocking and overwhelming process.”
“I wasn't fully aware of the impact of what happened at the time, and I cannot recall many of the negative feelings I experienced.”
“I was overwhelmed and don't remember much but we had a great team who helped us through everything.”
“I had a muscle biopsy with the first doctor and an intern did it. He hit a nerve which was horrible. It was some time before we got the results, and that was cause for anxiety. The whole time was very tense as we didn't know what to expect.”
“Lots of anxiety waiting for results and then knowing the results with no cure led to depression. Then when there was a treatment here and couldn’t get it, it made me furious.”
Anything Else?
“Anything that can help us is so important to our quality of life.”
“I hope this new medication will be available to everyone and that it will improve everyone's well-being.”
“This disease SUCKS for lack of better terms. It is devastating. While we can frame it in positives such as appreciating life, but the challenges it comes with - it’s difficult as a single parent. It’s a lot. The financial support is limited but the needs are a lot. Any drug or therapy that can reduce the impact is worthwhile. It not only takes a toll on the person diagnosed, but on family members: I had to quit my job, my daughter had to pick and choose where she works because she is mindful of her brother's condition and all the choices we make.”
“I think this drug under review really changed my life. I cannot imagine after being diagnosed and knowing that my muscle will weaken as I age, but having no solution to it. Even though the drug is not a cure for the disease, it slows down the process and even had a huge improvement for me. I am really thankful and I really hope this drug can be approved in Canada so all patients can have access to it.”
“From what I've been able to gather, Nexviazyme looks like an improvement over the current enzyme replacement therapy. Even modestly reducing decline can be very significant over a patient's lifetime. I am looking forward to access to this drug myself.
“This new drug offers a window of opportunity to positively affect my health. If the drug effectively slows the progression of the disease, I will be pleased.”
Conflict of Interest Declaration for the Muscular Dystrophy Canada
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
None.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
We worked in partnership with the Canadian Association for Pompe to ensure we captured the voices and experiences of Canadians affected by Pompe. The Canadian Association for Pompe was instrumental in sharing the survey with their members and helping to refer individuals to MDC for semi-structured phone interviews.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
Table 1: Conflict of Interest Declaration for Muscular Dystrophy Canada
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Sanofi-Genzyme | — | — | — | $101, 500 – all for educational initiatives:
|
I hereby certify that I have the authority to disclose all relevant information with respect to any matter involving this patient group with a company, organization, or entity that may place this patient group in a real, potential, or perceived conflict of interest situation.
Patient Group: Muscular Dystrophy Canada
Date: October 25, 2021
Clinician Group Input
The Neuromuscular Disease Network for Canada and Other Pompe Disease-Treating Clinicians
About The Neuromuscular Disease Network for Canada and Other Pompe Disease-Treating Clinicians
The Neuromuscular Disease Network for Canada (NMD4C) is the new pan-Canadian network that brings together the country’s leading clinical, scientific, technical, and patient expertise to improve care, research, and collaboration in neuromuscular disease.
https://neuromuscularnetwork.ca
Launched in January 2020 with funding from the Canadian Institutes of Health Research (CIHR) and Muscular Dystrophy Canada (MDC), NMD4C builds on existing national initiatives such as the Canadian Neuromuscular Disease Registry (CNDR), the Canadian Pediatric Neuromuscular Group (CPNG), and the former neuromuscular network CAN-NMD. The mission of NMD4C is to improve the care, research and treatment of NMDs for all Canadians. Its vision is to be a comprehensive, inclusive, open and enduring network through which Canadian stakeholders can share expertise and data, and collaborate on joint activities and research for the benefit of Canadian patients.
The network’s goals are to:
Formalize and sustain a network of NMD stakeholders united around a cohesive three-year work plan
Train and educate the next generation of NMD stakeholders (clinicians, scientists, and patient advocates)
Raise the standard of care for NMD and enable access to therapies across Canada
Strengthen biomedical and clinical infrastructure to build research capacity in Canada
Information Gathering
Clinicians with experience treating Pompe disease, including clinicians with experience with avalglucosidase alfa were asked to contribute to this submission. These expert clinicians contribute to the knowledge of Pompe disease and its treatments and are involved in clinical and observational research, clinical guidelines development and health technology assessment. The clinicians contributing herein are familiar with the data from clinical trials on treatments for Pompe disease.
Current Treatments
Describe the current treatment paradigm for the disease.
Pompe disease is caused by the lack or deficiency of a single enzyme, lysosomal acid alpha- glucosidase, leading to severe respiratory and skeletal muscle myopathy due to progressive accumulation of glycogen, which builds up to abnormal levels in tissues, particularly in muscles, ultimately causing the disease’s symptoms.
Enzyme replacement therapy (ERT) is an approved treatment for patients with Pompe disease. It involves the intravenous administration of recombinant human acid alpha-glucosidase (rhGAA), a version of the dysfunctional enzyme, allowing glycogen to be broken down; it alleviates symptoms and slows down the progression of the disease. But it may lead to immune intolerance with reduced potency of the treatment.
The management of patients with Pompe disease typically involves the coordinated efforts of a team of specialists typically led by neurologists that may involve pediatricians, metabolic geneticists, internists, orthopedists, cardiologists, respirologists, dieticians, physical therapists and other healthcare professionals.
While ERT is not curative, it is the only pharmacologic treatment to date that has been shown to modify the disease course in patients with Pompe disease.
Additional management of Pompe disease is symptomatic and supportive. Respiratory support may be required, as most patients have some deterioration of respiratory function, with serious respiratory insufficiency being highly prevalent in patients with Late Onset Pompe Disease (LOPD). Some patients may need mechanical ventilation. Physical therapy and a high protein diet may be used to strengthen skeletal and respiratory muscles.
Treatment Goals
What are the most important goals that an ideal treatment would address?
An ideal treatment would be curative, defined as fully and permanently arresting or reversing disease, and there is research underway that is examining a gene therapy approach for Pompe disease that uses a virus to deliver a healthy copy of the gene that is mutated in Pompe, the GAA gene, into the patients' liver cells that would then become a continuous source of circulating enzyme available for muscle uptake.
In the near term, therapy improvements for Pompe disease should seek to stabilize and/or improve motor and respiratory function, halt disease progression and prevent the loss of skeletal muscle strength. Additionally, a new treatment should provide improved immune tolerance.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
Current therapy can improve or stabilize motor and respiratory function. However, LOPD is characterized by progressive skeletal and respiratory muscle weakness which can result in progressive respiratory and motor disability. LOPD patients demonstrate a great deal of variability in how they respond to ERT, with some patients continuing to respond well up to 8–10 years after initiation of therapy, to those who do not respond at all to treatment (Pompe Disease: New Developments in an Old Lysosomal Storage Disorder doi:10.3390/biom10091339 https://www.mdpi.com/2218-273X/10/9/1339). On the whole, most patients on ERT experience a small improvement, followed by plateau phase of highly variable length, and then resumed progression.
Which patients have the greatest unmet need for an intervention such as the drug under review?
It is not immediately obvious based on the data available that any Pompe disease patient would not be suitable for the drug under review (avalglucosidase alfa), however, as a priority, patients who do not tolerate alglucosidase alfa, or who get worse under treatment with alglucosidase alfa, should have priority for accessing this new treatment.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Avalglucosidase alfa, the treatment under review was compared (in the phase 3 COMET trial) to the currently available enzyme-replacement predecessor alglucosidase alfa in 100 patients.
At the 49-week mark, avalglucosidase alfa patients scored 2.4 points higher on a standard lung function test (forced vital capacity) used to measure respiratory muscle weakness than those on alglucosidase alfa. Additionally, patients on avalglucosidase alfa were able to walk 30 meters farther than those in the alglucosidase alfa cohort at 49 weeks, based on the results of the six-minute walk test used to measure functional endurance.
Importantly, in the NEO1 and NEO-EXT studies, avalglucosidase alfa also demonstrated a more favorable safety and tolerability profile compared to alglucosidase alfa.
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective.
Avalglucosidase alfa is a next-generation enzyme replacement therapy for late-onset Pompe disease designed to improve the delivery of acid alpha-glucosidase (GAA) enzyme to muscle cells. In the near term this treatment could be an important option for LOPD patients who do not, or no longer, respond to alglucosidase alfa, or who develop immune intolerance to alglucosidase alfa.
This treatment will likely replace alglucosidase alfa as the standard of care first line therapy for all patients with Pompe disease.
How would this drug affect the sequencing of therapies for the target condition?
This treatment may become the new standard treatment in Pompe disease.
Which patients would be best suited for treatment with the drug under review?
The data offer clinical evidence that all patients with LOPD, and especially those under the age of 50 years (NEO1/NEO-EXT), could see improvement with avalglucosidase alfa over alglucosidase alfa related to prevention of deterioration of respiratory and motor function, and functional endurance, as well as improved safety and health-related quality of life.
For Infant Onset Pompe Disease (IOPD), in recognition that Pompe disease is very rare, and that it is unrealistic to expect extensive data, we have to rely on the data we have in addition to the data in adults. In Pompe disease the biology in IOPD and LOPD is the same, but the extent of the pathology is just much worse in IOPD.
Clinical scientists in Canada who have done pre-clinical work, participated in clinical trials and who treat adults and infants with Pompe disease observe from the available data that the safety of avalglucosidase alfa appears the same or better as with alglucosidase alfa.
The (limited) data for children with IOPD shows avalglucosidase alfa is at least as efficacious (as alglucosidase alfa) and likely more so.
For LOPD all the data in adults shows avalglucosidase alfa is at least as efficacious and likely a better treatment than alglucosidase alfa.
How would patients best suited for treatment with the drug under review be identified?
The diagnosis of Pompe disease is challenging given the heterogeneous presentation of symptoms. This is particularly true in patients with LOPD. Diagnosis is usually established using DNA analysis of the GAA gene to find deficiency in acid alpha-glucosidase enzyme activity.
Patients best suited for treatment with avalglucosidase alfa would be identified in the same way as patients previously identified for treatment with alglucosidase alfa.
Which patients would be least suitable for treatment with the drug under review?
It is not immediately obvious based on the data available that any Pompe disease patient would not be suitable for the drug under review (avalglucosidase alfa), as the side effect profile was favourable to the current standard therapy, and the indications are the same. In theory, a patent who is switched from alglucosidase alfa to avalglucosidase alfa and experiences a clinical deterioration or new adverse event may not be suitable and consideration would be made to switch the patient back to alglucosidase alfa if still available. Patients who have had a severe adverse reaction to a component of the drug under review would perhaps be the least suitable for ongoing treatment with the drug under review.
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review?
Based on the data from the clinical trial, in theory any patient with Pompe disease could be expected to respond and derive benefit from the drug under review. However, one patient population that might be of particular importance in trying avalglucosidase would be those Pompe patients who have exhibited decline or possibly lack of improvement on alglucosidase alfa.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice?
In clinical practice, the 6MWT, manual muscle testing (MRC grading), hand grip dynamometry, and FVC (% predicted) are all commonly used to follow patients with Pompe disease, and align nicely with the outcomes used in the clinical trials.
What would be considered a clinically meaningful response to treatment?
Improvement in clinical parameters described above (6MWT and FVC) or clinical stability (lack of progression in those parameters).
How often should treatment response be assessed?
Treatment response should be assessed every 6 – 12 months depending on clinical severity.
What factors should be considered when deciding to discontinue treatment?
If a patient declines to the point of severe motor or respiratory disability (e.g. non-ambulatory or non- invasive ventilation while awake) and the patient is no longer deriving any benefit, then consideration of discontinuation of therapy is appropriate. Severe adverse event(s) may also preclude ongoing therapy.
What settings are appropriate for treatment with the drug under review?
Outpatient infusion clinic or home infusion.
For non-oncology drugs, is a specialist required to diagnose, treat, and monitor patients who might receive the drug under review?
Ideally, treatment and monitoring should be via a clinical biochemical (metabolics) geneticist or neuromuscular specialist. Some of the monitoring for patients in remote areas can be performed by their local physicians, with episodic evaluation by a neuromuscular specialist depending on the clinical circumstances (e.g. in the midst of a pandemic).
Additional Information
Is there any additional information you feel is pertinent to this review?
N/A
Conflict of Interest Declarations
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
This submission content was completed exclusively by the named authors of this submission, with the Neuromuscular Disease Network for Canada (NMD4C) facilitating collaboration and editing.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No outside help was used to collect or analyze information used in this submission.
See Information Gathering section for details on how information in this submission was developed.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician that contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
Declaration for Clinician 1
Name: Charles Kassardjian
Position: Staff Neurologist, Assistant Professor, St Michael’s Hospital, University of Toronto
Date: September 21, 2021
Table 2: Declaration for the NMD4C and Other Pompe Disease-Treating Clinicians — Clinician 1
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Sanofi Genzyme | X | — | — | — |
Alexion | X | — | — | — |
Declaration for Clinician 2
Name: Anna Lehman
Position: Medical Director, Adult Metabolic Diseases Clinic, Vancouver General Hospital
Date: October 24, 2021
Table 3: Declaration for the NMD4C and Other Pompe Disease-Treating Clinicians — Clinician 2
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Sanofi Genzyme | — | — | X | — |
Amicus | — | — | X | — |
Takeda Shire | — | — | X | — |
Horizon | — | X | — | — |
Biomarin | — | X | — | — |
Declaration for Clinician 3
Name: Angela Genge
Position: Neurologist, Montreal Neurological Hospital
Date: October 22, 2021
Table 4: Declaration for the NMD4C and Other Pompe Disease-Treating Clinicians — Clinician 3
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Sanofi Genzyme (Advisory Board) | — | X | — | — |
Declaration for Clinician 4
Name: Basil Petrof
Position: Professor of Medicine, McGill University
Date: October 20, 2021
Table 5: Declaration for the NMD4C and Other Pompe Disease-Treating Clinicians — Clinician 4
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Sanofi Genzyme (Advisory board and educational talks) | — | X | — | — |
Declaration for Clinician 5
Name: Lochmuller, Hanns
Position: Professor of Neurology, Senior Scientist
Date: October 20, 2021
Table 6: Declaration for the NMD4C and Other Pompe Disease-Treating Clinicians — Clinician 5
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Sanofi-Genzyme advisory board | X | — | — | — |
Declaration for Clinician 6
Name: Kerri Schellenberg
Position: Assistant Professor of Neurology, University of Saskatchewan
Date: September 24, 2021
Table 7: Declaration for the NMD4C and Other Pompe Disease-Treating Clinicians — Clinician 6
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Speaker’s Honoraria: Genzyme, EMD Serono, Akcea, Takeda, Roche, Biogen | X | — | — | — |
Advisory Board: Mitsubishi-Tanabe, Alexion, Roche, Biogen, Akcea, Amylyx, Sanofi-Genzyme | X | — | — | — |
Declaration for Clinician 7
Name: Dr. Shailly Jain
Position: Clinical genetics and metabolics specialist, University of Alberta Hospital, Edmonton
Date: October 15, 2021
Table 8: Declaration for the NMD4C and Other Pompe Disease-Treating Clinicians — Clinician 7
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Genzyme-Sanofi advisory board for Avalglucosidase | X | — | — | — |
Declaration for Clinician 8
Name: Ramona Salvarinova
Position: Clinical Associate Professor
Date: October 15, 2021
Table 9: Declaration for the NMD4C and Other Pompe Disease-Treating Clinicians — Clinician 8
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Horizon Pharma | X | — | — | — |
Alexion advisory board 2019 | X | — | — | — |
Sanofi Genzyme ( travel and accommodation for Gaucher meeting 2019 | X | — | — | — |
Alexion advisory board 2020 | X | — | — | — |
Ultragenyx virtual advisory board | X | — | — | — |
Cycle advisory board meeting 202 | X | — | — | — |
Ultragenyx presentation honoraria Sept 2020 | X | — | — | — |
Sanofi Genzyme advisory board 2020 | X | — | — | — |
Utragenyx advisory board March/April 2021 | X | — | — | — |
Cycle ad board meeting 2021 | X | — | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.