CADTH Reimbursement Review
Ruxolitinib (Jakavi)
Sponsor: Novartis Pharmaceutical Canada Inc.
Therapeutic area: Acute graft versus host disease
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
AE
adverse event
aGvHD
acute graft-versus-host disease
alloSCT
allogeneic stem cell transplant
ANC
absolute neutrophil count
ATG
antithymocyte globulin
BAT
best available therapy
BOR
best overall response
cGvHD
chronic graft-versus-host disease
CI
confidence interval
CIBMTR
Center for International Blood and Marrow Transplant Research
CLL
chronic lymphocytic leukemia
CML
chronic myelogenous leukemia
CNI
calcineurin inhibitors
CR
complete response
CTCAE
Common Terminology Criteria for Adverse Events
CTTC
Cell Therapy Transplant Canada
DOR
duration of response
EBMT
European Society for Blood and Marrow Transplantation
ECP
extracorporeal photopheresis
EFS
event-free survival
EOT
end of treatment
EQ-5D-3L
3-Level EQ-5D
EQ-5D-5L
5-Level EQ-5D
FACT-BMT
Functional Assessment of Cancer Therapy-Bone Marrow Transplant
FFS
failure-free survival
GI
gastrointestinal
GvHD
graft-versus-host disease
GVT
graft-versus-tumour
HLA
human leukocyte antigen
HR
hazard ratio
HRQoL
health-related quality of life
HSCT
hematopoietic stem cell transplants
IBMTR
International Bone Marrow Transplant Registry
IPD
individual participant data
JAK
Janus-associated kinase
KM
Kaplan-Meier
MID
minimally important difference
MMF
mycophenolate mofetil
MSC
mesenchymal stem cell
mTOR
mammalian target of rapamycin
NE
not evaluable
NIH
National Institutes of Health
NRM
nonrelapse mortality
ORR
overall response rate
OS
overall survival
PR
partial response
PV
polycythemia vera
RCT
randomized controlled trial
RDI
relative dose intensity
SAE
serious adverse event
SCT
stem cell transplant
SD
standard deviation
SR
steroid-refractory
TEAE
treatment-emergent adverse event
VAS
visual analogue scale
VGPR
very good partial response
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Ruxolitinib (Jakavi), tablet 5 mg, 10 mg, 15 mg, 20 mg, oral |
Indication | The treatment of SR or dependent aGvHD in adults and children 12 years and older |
Reimbursement request | The treatment of aGvHD in patients 12 years and older who have an inadequate response to corticosteroids or other systemic therapiesa |
Health Canada approval status | NOC |
Health Canada review pathway | Project Orbis |
NOC date | May 23, 2022 (target date) |
Sponsor | Novartis Pharmaceutical Canada Inc. |
aGvHD = acute graft-versus-host disease; NOC = Notice of Compliance; SR = steroid refractory.
aThe reimbursement request differs from the Health Canada–approved indication, in that it states “steroid refractory or dependent” rather than “inadequate response to corticosteroids or other systemic therapies.”
Source: Product monograph.1
Introduction
Hematopoietic stem cell transplants (HSCTs) provide stem cells to patients whose bone marrow has been destroyed by disease, chemotherapy, or radiation.2 The 2 main types of stem cell transplants (SCTs) are autologous and allogeneic (alloSCTs). Although alloSCT has curative potential, there is a risk that the donor’s stem cells could die or be destroyed by the patients’ body before settling in the patient’s bone marrow, or the donor’s immune cells may attack healthy cells in the patient’s body; the latter is called graft-versus-host disease (GvHD).2 GvHD is a multi-system disorders in which donor-derived immune cells initiate an adverse immune reaction to tissues, cells, and organs in the transplant recipient, leading to tissue damage, organ failure, or death.3 GvHD has been found to be the leading cause of morbidity and nonrelapse mortality (NRM) in patients after alloSCT, and affects up to 70% of patients who receive SCTs.3,4 From 2008 to 2019, an estimated 13,033 transplants were performed in Canada, of which 5,672 were alloSCTs.5
Acute GvHD (aGvHD) typically occurs within 100 days after alloSCT, and typically affects the skin, liver, and intestines.6 aGvHD occurs in 30% to 50% of patients who undergo alloSCT, with 14% to 36% developing severe aGvHD (i.e., grade III to IV).7 Prognosis among patients with aGvHD is less favourable, with an estimated 3-year survival rate of 54%.8 Common symptoms of aGvHD are skin rash, burning and redness of the skin on the palms of the hands or soles of the feet, blisters and peeling skin, diarrhea, persistent nausea and vomiting, cramping or abdominal pain, enlarged liver, liver tenderness, abnormal liver enzymes or liver failure, and increased levels of serum bilirubin.9
The choice of initial treatment for patients with aGvHD depends on multiple factors, including the severity of symptoms, the type of prophylactic regimen used, and the importance of a graft-versus-tumour (GVT) effect.7 Grade I aGvHD (i.e., skin involvement of 50% or less of the body surface area without liver or gastrointestinal [GI] tract involvement) is often managed with topical treatments (e.g., tropical steroids) and adjustment of prophylactic treatments (e.g., calcineurin inhibitors [CNIs], such as cyclosporine in combination with methotrexate). Patients with grade II or higher aGvHD receive high-dose systemic glucocorticoids (e.g., methylprednisolone or prednisone) in addition to the care received by patients with grade I aGvHD. In patients who respond, steroid treatment is continued for several weeks until gradually being tapered to prevent a flare of aGvHD. Approximately 25% to 40% of patients achieve complete responses with glucocorticoids7; however, treatment is associated with significant side effects, which affect patients’ quality of life and increase susceptibility to infection.10 Approximately 25% to 50% of patients become refractory to steroids and are considered to have steroid-refractory (SR)-aGvHD. There is currently no standardized second-line therapy for patients with aGvHD who have an inadequate response to steroids in Canada.
In the absence of sufficient evidence to guide second-line treatment selection, factors that influence the choice include the experience of the treating physician, the types of aGvHD prophylaxis used, and the risk of potential toxicities and worsening pre-existing comorbidities.7 According to the clinical experts consulted by CADTH, available second-line options in Canada include extracorporeal photopheresis (ECP), mycophenolate mofetil (MMF), etanercept, infliximab, mammalian target of rapamycin (mTOR) inhibitors (i.e., everolimus or sirolimus), antithymocyte globulin (ATG), and interleukin-2 receptors. It was noted by the clinical experts that ATG is also often used as prophylaxis rather than aGvHD treatment. The clinical experts expressed challenges with currently available therapies in this heavily pre-treated target population, including no responses or only PRs (response rate was estimated by the clinical experts to be approximately 50%, but the durable response rate, in which patients still show a response at about 2 months, to be only 10% to 30%). According to the clinical experts, responses in this patient population are important because they enable the tapering of steroids, which mitigates long-term side effects (e.g., osteoporosis, hypertension, hyperglycemia, diabetes, and bone and joint health) and risk of infection. It was emphasized by the clinical experts that opportunistic infection and organ damage from aGvHD are major causes of NRM in SR-aGvHD.
There was consensus among the clinical experts that there is an unmet need for effective therapies with acceptable toxicity profile that improve health related quality of life (HRQoL), reduce disease symptoms of aGvHD, enhance patient’s performance status, and improve overall survival. They noted that a convenient oral route of administration would help improve adherence and reduce hospital-based resource use.
Ruxolitinib is a Janus-associated kinase (JAK) inhibitor which has received market authorization by Health Canada for the treatment of SR or dependent aGvHD in adult and pediatric patients 12 years and older. Ruxolitinib underwent review by Health Canada through the expedited pathway (Orbis). The sponsor’s requested reimbursement criteria for ruxolitinib for aGvHD differ from the Health Canada indication in that the sponsor’s criteria state, “inadequate response to corticosteroids or other systemic therapies” rather than “steroid refractory or dependent” aGvHD.
This current CADTH review focuses on aGvHD. Ruxolitinib is concurrently being reviewed by CADTH in the chronic GvHD (cGvHD) setting for the treatment of cGvHD in patients 12 years and older who have an inadequate response to corticosteroids or other systemic therapies. Ruxolitinib received a positive conditional CADTH recommendation in March 2016 for the treatment of patients with polycythemia vera (PV) who are resistant to or intolerant of hydroxyurea, according to the modified European LeukemiaNet Criteria used in the RESPONSE trial, and who have a good performance status. Ruxolitinib is available as 5 mg, 10 mg, 15 mg, and 20 mg tablets. The recommended starting dose for ruxolitinib for aGvHD is 5 mg administered orally twice daily. An increase in dose to 10 mg twice daily is recommended after at least 3 days of treatment if the absolute neutrophil count (ANC) and platelet count are not decreased by 50% or more relative to the first day of dosing with ruxolitinib. The product monograph states that tapering of ruxolitinib in the setting of aGvHD may be considered in patients with a response and after they have discontinued corticosteroids. A tapering of ruxolitinib — reducing the dose to 50% every 2 months — is recommended; in the event that signs or symptoms of GvHD reoccur during or after the taper, re-treatment with ruxolitinib should be considered.1
The objective of this CADTH report is to perform a systematic review of the beneficial and harmful effects of ruxolitinib (10 mg twice daily oral tablets) for the treatment of aGvHD in patients 12 years and older who have an inadequate response to corticosteroids or other systemic therapies.
According to the information in the product monograph and confidential information in the Health Canada Reviewer Report, Health Canada considered reviewing REACH 1 as the pivotal study and using the safety data from REACH 2 as supportive evidence for the aGvHD indication.1,11 This decision is the result of uncertainties around the REACH 2 trial that were identified by the FDA upon the review of raw data from REACH 2 as part of a sponsor-proposed label update of the FDA-approved ruxolitinib indication for aGvHD, which was based on REACH 1 trial data. The sponsor subsequently withdrew the supplemental New Drug Application for the REACH 2 update; in its submission, the sponsor noted that the proposed label update could not be completed based on REACH 2 trial data. Specifically, the 3 issues identified by the FDA concerned the following: missing and incomplete information in the case report forms related to organ staging of aGvHD, which precluded the FDA from conducting their own analyses and confirming the investigators’ efficacy assessments; the frequency of protocol deviations related to investigator-determined organ staging; and the information regarding prior therapies (i.e., prophylactic versus therapeutic aGvHD treatments). Health Canada concluded that as a result of the “integrity issues in the REACH 2 study” identified by the FDA, the efficacy results of REACH 2 could not be “verified.”11 Therefore, Health Canada considered REACH 1 to be the pivotal study, and safety and pharmacokinetic data from REACH 2 to be supportive evidence in their assessment of the aGvHD indication. The sponsor clarified with CADTH that the initial decision to submit the REACH 2 trial as the pivotal trial for the proposed CADTH reimbursement request remains unchanged.3 This CADTH review report does not include a review or critical appraisal of the issues raised by the FDA, as CADTH has no access to the FDA’s assessment, and reviewing raw data is not in the mandate of CADTH.
Health Canada based its efficacy assessment of the REACH 1 trial on the FDA-evaluable population (49 patients who failed steroids alone), which is a subset of the REACH 1 trial’s full efficacy-evaluable patients (N = 71). According to the Health Canada Clarifax, the following were excluded from the FDA-evaluable population: 9 patients who failed steroids plus additional therapies before randomization, and 13 patients who did not meet the SR criteria because of suboptimal dosing or duration of corticosteroid treatment before randomization.12 The HC Clarifax noted that the available data are considered insufficient to support the proposed ruxolitinib indication for patients with aGvHD who failed 1 systemic treatment in addition to corticosteroids (± CNIs).12 Of note, this CADTH clinical review does not report on the subset of 49 patients in the FDA-evaluable population; it presents results for all 71 efficacy-evaluable patients from the REACH 1 trial. Results for the 49 patients in the FDA-evaluable population are reported in the product monograph.1
The Health Canada Reviewer Report stated that the assessment of the safety of ruxolitinib for aGvHD was informed by the full safety-evaluable population (N = 71) of the REACH 1 trial and by safety results observed in the REACH 2 trial.1 Safety results reported in this CADTH clinical review are based on the 71 patients in the REACH 1 trial and on safety results from the REACH 2 trial. The Health Canada Reviewer Report noted that patients with SR or steroid-dependent aGvHD who meet the eligibility criteria in REACH 1 are considered acceptable.11 The REACH 1 eligibility criteria for SR and steroid-dependent aGvHD align with the current National Comprehensive Cancer Network guidelines for hematopoietic cell transplant, version 2.2020,13 the Alberta Bone Marrow and Blood and Cell Transplant Program Standard Practice Manual (2021),14 and the European Society for Blood and Marrow Transplantation (EBMT)-National Institutes of Health (NIH)-Center for International Blood and Marrow Transplant Research (CIBMTR) Task Force position statement,15 with minor variations. Steroid dependence has been defined as the inability to taper prednisone under 2 mg/kg per day after initially successful treatment for at least 7 days or as the recurrence of aGvHD activity during steroid taper.11
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient groups that responded to CADTH’s call for patient input and by clinical experts consulted by CADTH for the purpose of this review.
Patient Input
Eight patient groups — Lymphoma Canada, the Lymphoma & Leukemia Society of Canada, Chronic Lymphocytic Leukemia Canada, Myeloma Canada, the Aplastic Anemia & Myelodysplasia Association of Canada, Canadian Myeloproliferative Neoplasms Research Foundation, the Canadian Chronic Myelogenous Leukemia (CML) Network, and Cell Therapy Transplant Canada (CTTC) — created 1 joint patient input document for this review. The input was based on an online survey, and responses from 68 patients were reported in the patient input. Sixty patients reported having received an SCT, 6 patients reported not having received an SCT, and 2 patients did not answer the question. Of the 60 patients who received an SCT, 49 reported having received an alloSCT. Fifty-three patients had experienced GvHD after their SCT. Data on the type of GvHD were available for 45 of the 53 patients with GvHD: 13% experienced aGvHD, 24% experienced cGvHD, and 62% experienced both acute and chronic GvHD. Twenty patients reported receiving ruxolitinib treatment.
Overall, survey respondents indicated that they had long-lasting GvHD symptoms (3 to 5 years for 26% of respondents, and more than 5 years for 28% of respondents). To manage their GvHD, the respondents reported requiring numerous medical consultations, hospital stays, and nights away from home. Respondents reported a range of GvHD symptoms that had a significant impact on daily activities and were detrimental to quality of life. Respondents highlighted problems with interruption of life goals and accomplishments (career, school), difficulty sleeping, and impacts on their mental health (stress, anxiety, worry, and concentration problems), and finances. Other common symptoms experienced by respondents included burning and redness of the skin on the palms of the hands or soles of the feet, rashes that could spread over the entire body, blisters and peeling skin, skin problems (such as dryness, rash, itching, peeling, darkening, hard texture, and feeling tight), enlarged liver, liver tenderness, abnormal liver enzymes or liver failure, jaundice, dry eyes that may have a burning or gritty feeling, dry mouth with or without mouth ulcers, diarrhea, loss of appetite, stomach cramps, vomiting, weight loss, pain in muscles and joints, mobility issues, infections, and difficulty breathing.
According to the patient input received, respondents expected new drugs or treatments to address the following key outcomes: OS, GvHD symptoms, quality of life, and severity of side effects. Additionally, the administration of treatment in the outpatient setting (rather than requiring an overnight hospital stay), access to treatment locally (rather than requiring extensive travel), coverage of treatment by insurance or drug plans, and treatments recommended by health care professionals were perceived to be very important by respondents. Respondents who had direct experience with ruxolitinib indicated that, overall, ruxolitinib was an effective treatment, improved their quality of life, and had tolerable side effects, and they would take again if recommended by their physician and would recommend it to other patients.
Clinician Input
Input From Clinical Experts Consulted by CADTH
The clinical experts consulted by CADTH noted that there are currently no Health Canada–authorized standard-care regimens specifically for patients with SR-aGvHD in Canada. According to the clinical experts consulted by CADTH, available second-line options in Canada include ECP, MMF, etanercept, infliximab, mTOR inhibitor (e.g., sirolimus or sirolimus), and ATG. It was noted by the clinical experts that ATG was often used as prophylaxis rather than a treatment for aGvHD. There was consensus among the clinical experts that there is an unmet need for effective therapies with acceptable toxicity profiles that improve HRQoL, reduce symptoms of aGvHD, enhance performance status, and improve OS. They added that a convenient oral route of administration would help improve adherence and reduce hospital-based or ambulatory centre resource use. They stated that ruxolitinib is used as add-on to immunosuppressive regimens of corticosteroids with or without CNIs in patients 12 years and older with grades II to IV SR-aGvHD as per the REACH 2 trial. They agreed that ruxolitinib, as a therapy for SR-aGvHD, would likely shift the current treatment paradigm. The clinical experts consulted by CADTH agreed that patients who meet the inclusion and exclusion criteria from the REACH 2 trial should be eligible for ruxolitinib therapy. The clinical experts identified patients with grade IV aGvHD, who have the highest risk of death from aGvHD, as being most in need of ruxolitinib therapy. Patient subgroups that would potentially benefit least from ruxolitinib may include patients with refractory vomiting or ileus who are not able to take an oral drug such as ruxolitinib, and patients with thrombocytopenia, especially those with clinical bleeding, who may be a challenge to treat with ruxolitinib and may receive an alternative second-line drug instead. Patients with active uncontrolled infections or non-aGvHD cytopenia are a challenge to treat with ruxolitinib or with other available second-line therapy options; ruxolitinib should be used with caution and may require dose adjustment in these patients. The clinical experts consulted by CADTH felt that it would be reasonable to generalize the REACH 2 trial results to patients who received 2 or more systemic treatments for aGvHD and to leave it to the discretion of the treating physician to apply some flexibility in terms of using ruxolitinib in patients with overlap syndrome and patients with grade I aGvHD.
In the opinion of the clinical experts consulted by CADTH, an accurate assessment of response in patients with aGvHD is based on the NIH consensus criteria, which was used in the REACH 2 trial. Response to treatment is usually assessed daily for inpatients and weekly for outpatients. The clinical experts indicated that the most clinically meaningful responses to treatment include improvements in OS (survival beyond 1 year after alloSCT), overall response (complete or partial response), improvements in HRQoL and performance status, and the ability to taper corticosteroids.
Clinician Group Input
Two clinician groups provided input: CTTC, which contained input from 8 clinicians; and Ontario Health Cancer Care Ontario Complex Malignant Hematology, which contained input from 2 clinicians. The views of the clinician groups were, overall, consistent with the clinical experts consulted by CADTH, indicating that, based on the evidence from the REACH 2 trial, ruxolitinib would likely become the dominant first-line therapy for SR-aGvHD. The outcomes assessed in the REACH 2 trial were judged to be applicable to Canadian clinical practice and reflective of clinically meaningful responses. It was noted by both input groups that ruxolitinib is not as immunosuppressive as other available therapies. Clinicians from Ontario Health Cancer Care Ontario noted the following drawbacks of currently available therapies: IV administration, which requires patients to be at the hospital; side effects and broad immune suppression; and the high price and delivery costs of treatment. Input from CTTC noted that a Health Canada–approved and provincially funded therapy for SR-aGvHD would be an important step forward in the target setting, because existing therapies offer low response rates and high rates of toxicity. According to input from CTTC, experience with ruxolitinib (accessible through compassionate access programs) and real-world effectiveness appear similar to what was observed in the REACH 2 trial, which had low rates of toxicity.
Drug Program Input
The Formulary Working Group identified the following jurisdictional implementation issues: relevant comparators, consideration for initiation of therapy, consideration for discontinuation of therapy, considerations for prescribing of therapy, and system issues and economic considerations. The clinical experts consulted by CADTH weighed evidence from the REACH 2 trial and considered the REACH 1 trial and other clinical considerations to provide responses to the Provincial Advisory Group’s drug program implementation questions. Refer to Table 4 for more details.
Clinical Evidence
Pivotal Studies and Protocol Selected Studies
Description of Studies
The REACH 2 trial is a completed, international, multi-centre, open-label, randomized, phase III trial that compared ruxolitinib (10 mg administered orally twice daily) with investigator’s choice of best available therapy (BAT) — i.e., ATG, ECP, mesenchymal stem cells (MSCs), methotrexate, MMF, mTOR inhibitors (everolimus or sirolimus), etanercept, or infliximab — in patients 12 years and older with grade II to IV SR-aGvHD. Patients continued to receive their systemic immunosuppressive regimen of corticosteroids with or without CNIs. Staging of aGvHD was based on the NIH criteria of Harris et al. (2016).16 A total of 309 patients were randomized in a 1:1 ratio to receive ruxolitinib or BAT. The primary outcome was overall response rate (ORR) at day 28, and the key secondary outcome was the rate of durable ORR at day 56. Additional secondary outcomes were OS, failure-free survival (FFS), ORR at day 14, duration of response (DOR), best overall response (BOR), HRQoL assessed with the Functional Assessment of Cancer Therapy-Bone Marrow Transplant (FACT-BMT) and the 5-level EQ-5D (EQ-5D-5L) instruments, event-free survival (EFS), NRM, incidence of malignancy relapse or progression, cumulative steroid dose up to day 56, incidence of cGvHD, and safety.
The REACH 1 trial is a completed, open-label, single-arm, multi-centre phase II trial that evaluated the efficacy and safety of ruxolitinib in combination with corticosteroids in patients with grade II to IV SR-aGvHD. The severity grading of aGvHD was based on the NIH criteria by Harris et al. (2016).16 A total of 71 patients were enrolled to received ruxolitinib 5 mg orally twice daily; then, if hematologic parameters were stable and no treatment-related toxicity was observed after the first 3 days of treatment, the dose could be increased to 10 mg orally twice daily. The primary outcome was ORR at day 28 and the key secondary outcome was DOR at month 6. Additional secondary outcomes were OS, FFS, ORR at day 14, DOR at month 3, NRM, incidence of malignancy relapse or progression, relapse rate, relapse-related mortality rate, and safety.
The REACH 2 trial enrolled male and female patients 12 years and older who had undergone alloSCT, had evidence of myeloid and platelet engraftment (ANC > 1,000/mm3 and platelet count > 20,000/mm3), and were diagnosed with grade II to IV aGvHD, defined by the NIH consensus criteria, that was determined to be corticosteroid-refractory, according to the protocol-defined criteria. Overall, the REACH 1 trial had similar inclusion criteria. However, there were slight variations in the definition of corticosteroid refractoriness (criterion C, as outlined in Table 4) and engraftment. Both studies excluded patients who had received more than 1 systemic treatment for SR-aGvHD, a clinical presentation resembling de novo overlap syndrome (defined by Jagasia et al. [2015])17 or active uncontrolled infection. REACH 2 explicitly excluded patients with multi-focal leuko-encephalopathy, whereas REACH 1 did not. In the REACH 2 trial, the mean ages for the ruxolitinib and BAT groups were, respectively, 48.1 (standard deviation [SD] = 16.30) and 50.9 (SD = 14.9) years. The majority of patients (ruxolitinib versus BAT groups) were 18 to 65 years of age (83.1% versus 81.3%), but a few were 12 years to younger than 18 years (3.2% versus 2.6%). Most patients were male (59.7% versus 58.7%). The aGvHD grade at baseline was mostly grade III (44.2% versus 43.9%), followed by grade II (32.5% versus 34.8%) and grade IV (19.5% versus 20.6%). The most common criteria for SR-aGvHD were failure to achieve a response after 7 days (46.8% versus 40.6%), failure on steroid taper (30.5% versus 31.6%), and progression after at least 3 days (22.7% versus 27.7%).18 The ruxolitinib group had a higher proportion of patients with aGvHD organ involvement at baseline of the skin (60.4% versus 47.7%) and liver (24.0% versus 16.1%), and a lower proportion of patients with aGvHD organ involvement at baseline of the upper GI tract (18.2% versus 23.9%) and lower GI tract (62.3% versus 74.2%). Demographic characteristics and disease and alloSCT history at baseline in the REACH 1 trial were similar, overall, to those in the REACH 2 trial. As in the REACH 2 trial, the majority of patients in the REACH 1 trial were 18 to 65 years (81.7%), and the distribution of aGvHD grades was similar in the 2 trials, with the majority of patients having grade III aGvHD (46.5%), followed by grade II (31.0%) and grade IV (22.5%). As in REACH 2 trial, the most common criteria for SR-aGvHD were no aGvHD improvement after 7 days of primary treatment (40.8%), failing corticosteroid taper (36.2%), and progression after 3 days or primary treatment (23.9%). Most patients in the 2 trials received grafts from identical human leukocyte antigen (HLA)-matched donors (60.2% in REACH 2 and 63.4% in REACH 1).19
This CADTH review is based on REACH 2 data from the primary analysis (July 25, 2019), the updated secondary analysis (January 6, 2020), and the final analysis (April 23, 2021), which was conducted once all patients had completed the study.20 The REACH 1 data in this CADTH review are based on the final data cut-off date of June 5, 2019, which is also the study completion date.21
Efficacy Results
The key efficacy results from the REACH 2 and REACH 1 trials are summarized in Table 2.
As of the primary analysis, the median duration of follow-up for OS in the REACH 2 trial was 5.04 months in the ruxolitinib group and 3.58 months in the BAT group. Median OS was 11.14 months, or 339 (95% confidence interval [CI], 186 to not evaluable [NE]) days, in the ruxolitinib group, compared with 6.47 months, or 197 (95% CI, 114 to 458) days, in the BAT group, with a stratified hazard ratio (HR) of 0.83 (95% CI, 0.60 to 1.15).21 The OS results at the secondary analysis were, overall, consistent with those in the primary analysis of the REACH 2 trial. In the REACH 1 trial, median OS was 232.0 (95% CI, 93.0 to 675.0) days at the final analysis.19
As of the primary analysis in the REACH 2 trial, the number of patients who experienced a FFS event (i.e., hematologic disease relapse or progression, NRM, or the addition of new systemic aGvHD treatment) was 84 (54.5%) and 119 (76.8%) in the ruxolitinib and BAT groups, respectively. Median FFS was 4.99 months and 1.02 months in the ruxolitinib and BAT groups, respectively, with a HR of 0.46 (95% CI, 0.35 to 0.60). The FFS results of the secondary analyses were, overall, consistent with those of the primary analysis in the REACH 2 trial.18 In the REACH 1 trial, 60 (84.5%) patients experienced an event (i.e., underlying malignancy relapse or progression [n = 3], death [n = 22], addition of new systemic aGvHD treatment [n = 28], or signs or symptoms of cGvHD [n = 7]). The median FFS was 85.0 (95% CI, 42.0 to 158.0) days.19
In the REACH 2 trial, ORR at day 28 was only analyzed at the primary analysis and was not reassessed at the secondary or final analyses. As of the primary analysis, the REACH 2 trial met its primary objective. The proportion of patients who achieved an overall response at day 28 was 62.3% (n = 96) (95% CI, 54.2% to 70.0%) in the ruxolitinib group and 39.4% (n = 61) (95% CI, 31.6% to 47.5%) in the BAT group, with a stratified odds ratio of 2.64 (95% CI, 1.65 to 4.22). The proportion of patients who achieved a complete response (CR) or particle response (PR) was 34.4% (n = 53) and 27.9% (n = 43), respectively, in the ruxolitinib group, and 19.4% (n = 30) and 20.0% (n = 31), respectively, in the BAT group.18 The REACH 1 trial met the predetermined threshold for a positive study outcome (lower limit of the 95% CI for ORR at day 28 ≥ 40%). The proportion of patients who achieved an overall response at day 28 was 56.3% (n = 40) (95% CI, 44.0% to 68.1%). The proportion of patients who achieved a CR, a very good partial response (VGPR), or a PR was 19 (26.8%), 6 (8.5%), or 15 (21.1%), respectively.19
In the REACH 2 trial, durable ORR at day 56 was only analyzed at the primary analysis and was not reassessed at the secondary, or final analyses. As of the primary analysis, the proportion of patients who achieved a durable ORR at day 56 was 39.6% (n = 61) in the ruxolitinib group and 21.9% (n = 34) in the BAT group, with a stratified odds ratio of 2.38 (95% CI, 1.43 to 3.94) in favour of ruxolitinib.18 Durable ORR at day 56 was not assessed in the REACH 1 trial.19
As of the primary analysis in the REACH 2 trial, among the patients who achieved a CR or PR at or before day 28, median DOR was 168 days (range = 22 to 423) in the ruxolitinib group and 101 days (range = 10 to 289) in the BAT group. Results for DOR at the secondary and final analyses were consistent with DOR results at the primary analysis.18 In the REACH 1 trial, the median DOR for responding patients at any time point was 345.0 (95% CI, 154.0 to not NE) days, with a median follow-up time of 128.5 days (range = 3 to 805 days).19 The 6-month event-free probability for DOR in responding patients (i.e., PR, VGPR, or CR) at any time point was 62.1% (95% CI, 45.8% to 74.8%).19
In the REACH 2 trial, BOR was only analyzed at the primary analysis and was not reassessed at the secondary or final analyses. At the primary analysis, the proportion of patients who had achieved BOR by day 28 in the ruxolitinib group was 81.8% (95% CI, 74.8% to 87.6%) and 60.6% (95% CI, 52.5% to 68.4%) in the BAT group, with an odds ratio of 3.07 (95% CI, 1.80 to 5.25).18 In the REACH 1 trial, the proportion of patients who achieved BOR at any time point was 76.1% (95% CI, 64.5% to 85.4%).19
In the REACH 2 trial, as of the primary analysis, a higher proportion of patients had tapered off corticosteroids in the ruxolitinib group (21.4%; 95% CI, 15.2% to 28.8%) than in the BAT group (14.8%; 95% CI, 9.6% to 21.4%).18 The proportions of patients with a relative dose intensity (RDI) of 50% or less (ruxolitinib and BAT group) were 29.2% and 24.5%, and with a RDI of more than 50% were 68.8% and 74.8%.18 The results for cumulative steroid dosing until day 56 at the secondary and final analyses were, overall, consistent with those at the primary analysis in the REACH 2 trial.18 In the REACH 1 trial, the proportion of patients who were still receiving ruxolitinib and who had tapered off (discontinued) corticosteroids was 6.9% at day 56, 34.8% at day 100, and 61.1% at day 180. The proportion of patients with a decrease of 50% or more in corticosteroid dose relative to day 1 (or day 2) increased from 23.2% on day 14 to 55.8% on day 28 and 100.0% on day 100.19
Harms Results
The key harms outcomes reported in the REACH 2 (secondary analysis) and REACH 1 (final analysis) trials are summarized in Table 2.
In the REACH 2 trial, as of the secondary analysis, the percentage of patients reporting at least 1 treatment-emergent adverse event (TEAE) was 99.3% in the ruxolitinib group and 98.7% in the BAT group. The most commonly reported TEAEs (ruxolitinib versus BAT groups) were anemia (40.1% versus 32%), thrombocytopenia (36.8% versus 20.7%), cytomegalovirus infection (30.9% versus 26.7%), neutropenia (24.3% versus 14.7%), and edema peripheral (24.3% versus 21.3%). In the REACH 1 trial, as of the final analysis, all patients in the REACH 1 trial experienced at least 1 TEAE (100.0%). The most commonly reported TEAEs in REACH 1 were similar to those reported in REACH 2, and included anemia (64.8%), thrombocytopenia (62.0%), hypokalemia (49.3%), neutropenia (47.9%), and peripheral edema (46.5%).19
In the REACH 2 trial, the percentage of patients who experienced at least 1 TEAE of grade 3 or higher in the ruxolitinib and BAT groups was 91.4% and 87.3%, respectively. The most commonly reported TEAEs of grade 3 or higher (ruxolitinib versus BAT groups) were anemia (35.5% versus 24.0%), thrombocytopenia (33.6% versus 16.7%), neutropenia (21.7% versus 12.0%), platelet count decrease (17.8% versus 15.3%), and white blood cell count decrease (13.2% versus 8.7%).18 In the REACH 1 trial, TEAEs of grade 3 or higher occurred in 97.2% of patients. The most commonly reported TEAEs of grade 3 or higher in REACH 1 were similar to those reported in REACH 2, and included thrombocytopenia (53.5%), anemia (50.7%), neutropenia (42.3%), and hyperglycemia (19.7%).19
In the REACH 2 trial, the percentage of patients experiencing at least 1 serious TEAE was 66.4% in the ruxolitinib group and 53.3% in the BAT group. The most common serious TEAEs were (ruxolitinib versus BAT) sepsis (7.9% versus 7.3%), pyrexia (6.6% versus 4.0%), septic shock (6.6% versus 5.3%), and diarrhea (5.3% versus 2.0%).18 In the REACH 1 trial, the percentage of patients experiencing serious TEAEs was 83.1%. The most commonly reported serious TEAEs in REACH 1 were similar to those reported in REACH 2, and included sepsis (12.7%), pyrexia (11.3%), respiratory failure (11.3%), and lung infection (7.0%).19
In the REACH 2 trial, the percentage of patients who discontinued treatment due to TEAEs in the ruxolitinib group was 27.0% and in the BAT groups was 9.3%. The most commonly cited TEAEs contributing to treatment discontinuation were neutropenia (n = 4; 2.6%), sepsis (n = 4; 2.6%), anemia (n = 3; 2.0%), and thrombocytopenia (n = 3; 2.0%) in the ruxolitinib group, and sepsis (n = 1; 0.7%), anemia (n = 1; 0.7%), thrombocytopenia (n = 1; 0.7%), and platelet count decrease (n = 1; 0.7%) in the BAT group.18 In the REACH 1 trial, TEAEs led to discontinuation of ruxolitinib treatment in 32.4% of patients. The most commonly reported TEAEs leading to discontinuation of ruxolitinib were sepsis (5.6%), acute kidney injury (2.8%), and respiratory failure (2.8%).19
In the REACH 2 trial, on-treatment deaths occurred in 28.3% and 24.0% of patients in the ruxolitinib and BAT groups, respectively. The most common cause of death was the study indication of aGvHD (including aGvHD and related complications) in 21 (13.8%) and 21 (14.0%) patients in the ruxolitinib and BAT groups, respectively.18 In the REACH 1 trial, there were 35.2% (n = 25) of patients who had died during treatment with ruxolitinib or within 30 days of their last dose. The most common cause of death was “other” (25.4%, n = 18), which included underlying GvHD, multi-organ failure, pulseless electrical activity arrest, and respiratory failure, many of which were counted as fatal TEAEs.19
In the REACH 2 trial, serious infections were reported in 38.2% and 30.0% of patients in the ruxolitinib and BAT groups, respectively, and serious infections of grade 3 or higher in 38.2% and 28.7% of patients, respectively. The percentage of patients experiencing at least 1 infection TEAE of any grade was 80.9% and 69.3% in the ruxolitinib and BAT groups, respectively.18 In the REACH 1 trial, there were 58 patients (81.7%) with at least 1 TEAE infections and infestation, 36 of whom experienced serious TEAE infections and infestations.19
In the REACH 2 trial, 1 patient in each of the ruxolitinib and BAT groups reported experiencing bradycardia of any grade. No patients reported bradycardia of grade 3 or higher.18 In the REACH 1 trial, 2 patients reported experiencing bradycardia of any grade, and 1 patient-reported bradycardia of grade 3 or higher.19
In the REACH 2 trial, cytopenia TEAEs of any grade (ruxolitinib versus BAT group) included anemia (40.8% versus 34.0%), thrombocytopenia (56.6% versus 36.7%), leukopenia (46.7% versus 32.0%), and other cytopenias (8.6% versus 6.0%). Cytopenia TEAEs grade 3 or higher were of special interest (ruxolitinib versus BAT group), and included anemia (36.2% versus 25.3%), thrombocytopenia (50.7% versus 32.0%), leukopenia (42.8% versus 27.3%), and other cytopenias (5.9% versus 4.7%).18 In the REACH 1 trial, cytopenia TEAEs of any grade included anemia (64.8%), neutropenia (47.9%), and thrombocytopenia (62.0%). Cytopenia TEAEs of grade 3 or higher included anemia (50.7%), neutropenia (42.2%), and thrombocytopenia (53.5%).19
In the REACH 2 trial, lipid abnormality events of any grade were reported in 9.9% and 7.3% of patients in the ruxolitinib and BAT groups. Lipid abnormality events of grade 3 or higher were reported in the ruxolitinib and BAT groups by 3.9% and 2.7% of patients, respectively.18 Lipid abnormalities were not reported in the REACH 1 trial.
In the REACH 2 trial, the safety profile for the 9 adolescents was, overall, similar to that of the study safety set.18 REACH 1 did not include any adolescents.19
Table 2: Summary of Key Results From Pivotal and Protocol Selected Studies
Data cut-off date | REACH 2 | REACH 1 | |
|---|---|---|---|
Ruxolitinib N = 154 | BAT N = 155 | Ruxolitinib N = 71 | |
July 25, 2019 | June 5, 2019 | ||
OS | |||
Median OS follow-up time, months | 5.04 | 3.58 | NRa |
Median OS | |||
Months | 11.14 | 6.47 | NA |
Days (95% CI) | 339 | 197 | 232.0 (93.0 to 675.0) |
Events, death, n (%) | 72 (46.8) | 79 (51.0) | 44 (62.0) |
Censored, n (%) | 82 (53.2) | 76 (49.0) | 27 (38.0)b |
HR (95% CI)c | 0.83 (0.60 to 1.15) | NA | |
P value | 0.2648 | NA | |
FFS | |||
Median FFS | |||
Months | 4.99 | 1.02 | NA |
Days (95% CI) | NA | NA | 85.0 (42.0 to 158.0) |
Patients with events, n (%) | 84 (54.5) | 119 (76.8) | 60 (84.5) |
Patients with competing risk, n (%) | 30 (19.5) | 14 (9.0) | NA |
Patients censored, n (%) | 40 (26.0) | 22 (14.2) | 11 (15.5) |
HRc (95% CI) | 0.46 (0.35 to 0.60) | NA | |
P valued | 0.0001 | NA | |
ORR at day 28 | |||
Patients with overall response, n (%) | 96 (62.3) | 61 (39.4) | 40 (56.3) |
95% CIe | (54.2 to 70.0) | (31.6 to 47.5) | (44.0 to 68.1) |
CR | 53 (34.4) | 30 (19.4) | 19 (26.8) |
VGPR | NA | NA | 6 (8.5) |
PR | 43 (27.9) | 31 (20.0) | 15 (21.1) |
Odds ratio for ruxolitinib vs. BAT (95% CI)f | 2.64 (1.65 to 4.22) | NA | |
P value | < 0.0001 | NA | |
Rate of durable ORR at day 56 | |||
Patients with overall response, n (%) | 61 (39.6) | 34 (21.9) | NA |
95% CIe | 31.8 to 47.8 | 15.7 to 29.3 | NA |
CR | 41 (26.6) | 25 (16.1) | NA |
PR | 20 (13.0) | 9 (5.8) | NA |
Odds ratio for ruxolitinib vs. BAT (95% CI)f | 2.38 (1.43 to 3.94) | NA | |
P value | 0.0005 | NA | |
DOR in patients with CR or PR at or before day 28 | |||
Response at or before day 28, n | 96 | 61 | NA |
Patients with eventsg , n (%) | 9 (9.4) | 21 (34.4) | NA |
Patients with competing risks, n (%) | 53 (55.2) | 23 (37.7) | NA |
Patients censored, n (%) | 34 (35.4) | 17 (27.9) | NA |
DOR, days | |||
Medianh | 168.0 | 101.0 | NA |
6-month DOR in patients with PR, VGPR, or CR assessed once all patients had completed the day 180 visit | |||
Event-free probability estimate, % (95% CI) | — | — | — |
Month 6 | NA | NA | 62.1 (45.8 to 74.8) |
BOR by day 28 | |||
Patients with overall response, n (%) | 126 (81.8) | 94 (60.6) | NA |
95% CIe | 74.8 to 87.6 | 52.5 to 68.4 | NA |
Odds ratio (95% CI)f | 3.07 (1.80 to 5.25) | NA | |
P value | 0.0001 | NA | |
Cumulative steroid dosing until day 56 | |||
Completely tapered off by day 56, n (%) [95% CI] | 33 (21.4) [15.2 to 28.8] | 23 (14.8) [9.6 to 21.4] | NA |
≤ 50% RDI,i n (%) [95% CI] | 45 (29.2) [22.2 to 37.1] | 38 (24.5) [18.0, 32.1] | NA |
> 50% RDI,i n (%) [95% CI] | 106 (68.8) [60.9 to 76.0] | 116 (74.8) [67.2 to 81.5] | NA |
Patients with ongoing ruxolitinib who had discontinued corticosteroids by day 56, n = 29, n (%) | NA | NA | 2 (6.9) |
Harms, safety set, n (%) | |||
Data cut-off date | January 6, 2020 | June 5, 2019 | |
Safety set, N | 152 | 150 | 152 |
Total TEAEs | 151 (99.3) | 148 (98.7) | 71 (100.0) |
Total SAE | 101 (66.4) | 80 (53.3) | 59 (83.1) |
WDAE | 41 (27.0) | 14 (9.3) | 25 (35.2) |
BOR = best overall response; CI = confidence interval; CR = complete response; DOR = duration of response; FFS = failure-free survival; HR = hazard ratio; NA = not applicable; NE = not evaluable; NR = not reported; ORR = overall response rate; OS = overall survival; PR = partial response; RDI = relative dose intensity; SAE = serious adverse event; TEAE = treatment-emergent adverse event; VGPR = very good partial response; vs. = versus; WDAE = withdrawal due to adverse event.
aUpon request to the sponsor, the median OS follow-up time was not provided.
bParticipants with no observed death or loss to follow-up were censored at the last date they were known to be alive.
cHR and 95% CI are obtained from the stratified Cox proportional hazards model using the Wald test.
dP value nominal.
eThe 95% CI for the response rate was calculated using the Clopper-Pearson exact method.
fOdds ratio and 95% CI are calculated using the stratified Cochran-Mantel-Haenszel test.
gThe event was defined as the progression of aGvHD or the addition of systemic therapies for aGvHD after day 28. The competing risks included death without prior observation of aGvHD progression and onset of cGvHD.
hMedian and quartiles are provided using Kaplan–Meier method.
iRDI includes days of zero dose in the calculation.
Sources: Clinical Study Report (REACH 2),18 Clinical Study Report (REACH 1).19
Critical Appraisal
There are insufficient data to describe how the requested reimbursement criteria match the patient population in the REACH 2 trial. The sponsor was asked for clarification on the number of patients in the REACH 2 trial who had an inadequate response to corticosteroids, an inadequate response to other systemic therapies, or an inadequate response to corticosteroids and other systemic therapies, but reported that such data are not available.22 Because an inadequate response to corticosteroids was an eligibility criterion of the REACH 2 trial, it follows that all patients in the trial had an inadequate response to corticosteroids; it also follows that data for patients who only had an inadequate response to other systemic therapies and not to steroids were not available from the REACH 2 trial. However, the number of patients who had an inadequate response to other systemic therapies in addition to an inadequate response to corticosteroids remains unclear. It is not known if patients who are refractory to 1 therapy, as opposed to multiple therapies, would respond differently to ruxolitinib. The clinical experts consulted by CADTH agreed that the difference between patients who either have an inadequate response to corticosteroids alone or to multiple therapies would be unlikely to influence the treatment effect of ruxolitinib.
The REACH 2 trial had an open-label design, so the investigator and the study participants were aware of their treatment status, which increases the risk of detection and performance bias. This had the potential to bias results and outcomes in favour of ruxolitinib if the assessor (investigator or patient) believed the study drug was likely to provide a benefit. Subjective outcomes (i.e., adverse outcomes and patient-reported outcomes) may be at particular risk of bias with an open-label design. Furthermore, the underlying complexity of aGvHD has been acknowledged as a key challenge for the design and analysis of clinical trials in the current target setting, and may contribute to subjective inter-physician variability in response assessments. To mitigate the impact of this bias, the investigators used standardized criteria (i.e., aGvHD disease evaluation and response-assessment criteria were done in accordance with the standard NIH criteria of Harris et al. [2016])16 to evaluate responses. However, no independent review committee was used to evaluated responses. Overall, the magnitude and direction of this bias remain unclear. Although imbalances were noted for a few baseline characteristics (e.g., prior therapy of steroids plus CNIs plus an aGvHD prophylaxis; organ involvement of the skin, liver, and upper and lower GI tracts; time from diagnosis of underlying disease to transplant and time from diagnosis of underlying disease to screening), they were unlikely to influence clinical outcomes, according to the clinical experts consulted by CADTH. Patients in the BAT group who experienced disease progression, mixed response, or no response were allowed to add or initiate a new systemic therapy up to day 28 without proceeding to discontinuation; however, this was considered a failure of initial BAT. The clinical experts consulted by CADTH noted that changing or initiating new systemic aGvHD therapies is reflective of clinical practice. It was felt by the clinical experts that changes to the BAT treatment up the day 28 were unlikely to affect OS results, given the similar efficacy and similar responses achieved with various BAT therapies. Addition to or change of systemic therapy was treated as treatment failure and, therefore, did not affect ORR at day 28 or the FFS outcomes. Crossover of patients in the BAT group to the ruxolitinib group after day 28 may have biased the OS and EFS outcomes. Patients in the BAT group could cross over to the ruxolitinib group if they failed to meet the primary end point (CR or PR at day 28), lost the response thereafter, and met the criteria for progression, mixed response, or no response, which necessitated new additional systemic immunosuppressive treatment. Overall, 49 patients in the BAT group crossed over to the ruxolitinib group. Crossover of patients in the BAT group may have prolonged survival beyond what would have occurred had the patients only received their randomized study treatment. During the randomized treatment phase (i.e., the period from day 1 to week 24 or end of treatment [EOT]), the median duration of treatment was close to twice as long with ruxolitinib as with BAT, at 82.5 days (range = 8 to 396) and 45.5 days (range = 2 to 218), respectively. A safety comparison between the study groups over that period may have been biased against ruxolitinib. Additionally, the investigator’s choice of BAT may have influenced the safety profile in the BAT group, as the toxicity profiles of BAT treatments differ. The interpretation of results for the EQ-5D-5L and the FACT-BMT scales (i.e., the ability to assess trends over time and to make comparisons across treatment groups) is limited by the significant decline in patients available to provide assessments over time. It was noted that few patients in the trial were younger than 18 years. The clinical experts supported the generalization of the study results to patients younger than 18 years, as the management of these patients is similar to the management of adults in clinical practice, the safety profile of ruxolitinib in these patients was similar to the overall safety set, and there is no biologic rational to assume that outcomes with ruxolitinib would be different between adult and adolescents with SR-aGvHD.
REACH 1
The sponsor was asked for clarification on the number of patients in the REACH 1 trial who had an inadequate response to corticosteroids, an inadequate response to other systemic therapies, or an inadequate response to corticosteroids and other systemic therapies, and reported that 42 patients were refractory to steroids alone and 29 patients were refractory to steroids and 1 additional systemic therapy (i.e., 1 systemic treatment in addition to corticosteroids (± CNIs) for aGvHD was allowed in the REACH 1 trial).22 The sponsor was asked about the specific types of additional systemic therapies received by the 29 patients in REACH 1 who were refractory to 1 additional systemic therapy, but no additional data were provided beyond the information shown in Table 16.22 It is not known if patients who are refractory to 1 therapy, as opposed to multiple therapies, would respond differently to ruxolitinib. The clinical experts consulted by CADTH agreed that the difference between patients who have an inadequate response to corticosteroids alone or to multiple therapies would be unlikely to have an impact on the treatment effect of ruxolitinib.
Phase II (randomized or nonrandomized) trials document safety outcomes and investigate whether the estimate of effect for a new drug is large enough to use it in confirmatory phase III trials. Phase II trials may not accurately predict harm and/or the effectiveness of treatments. There are numerous examples of phase III trials with results that did not support the phase II trial results.23 Interpretation of time‐to‐event end points, such as OS, is limited in single‐arm studies. The nonrandomized design makes it a challenge to interpret OS events attributable to ruxolitinib, because all patients received the same treatment. The noncomparative design of the REACH 1 trial precludes the ability to compare the relative therapeutic benefit or safety of ruxolitinib with currently available therapies in Canadian clinical practice. All patients in the REACH 1 trial received at least 1 concomitant medication. For instance, CNIs and glucocorticoids were received by 88.7% and 45.1% of patients, respectively. Given the uncontrolled design of the REACH 1 trial, the effect of concomitant treatments on the overall study outcome cannot be determined. Outcomes such as observed responses, durability of responses, and survival may have been influenced by the concomitant use of steroids or other therapies. The REACH 1 trial had an open-label design in which the investigator and the study participants were aware of their treatment status, which increased the risk of detection and performance bias. This had the potential to bias results in favour of ruxolitinib if the assessor (investigator or patient) believed the study drug was likely to provide a benefit. Furthermore, the underlying complexity of aGvHD and its nonspecific presentation have been acknowledged as a key challenge in the design and analysis of clinical trials in the current target setting, and may contribute to subjective inter-physician variability in response assessments. To mitigate the impact of this bias, the investigators used standardized criteria (i.e., aGvHD disease evaluation and response-assessment criteria were done in accordance with the standard NIH criteria of Harris et al. [2016]16) to evaluate responses. No formal statical significance and hypotheses testing was performed, so no P values were reported. Point estimates with 95% CIs were reported to estimate the magnitude of the treatment effect. The REACH 1 trial did not collect data on patient-reported outcomes. The input provided by the patient advocacy and registered clinician groups, as well as by the clinical experts consulted by CADTH, agreed that improvements in HRQoL and aGvHD symptom severity are important treatment goals for the target population. aGvHD has been found to be the leading cause of morbidity in patients who have undergone alloSCT and who have a multitude of symptoms and various degrees of severity.3
Indirect Comparisons
No indirect treatment comparisons were included in the sponsor’s submission to CADTH or identified in the literature search.
Other Relevant Evidence
The section on other relevant evidence included:
1 additional relevant study (Moiseev et al. [2020]24) included in the sponsor’s submission to CADTH that reported results for ruxolitinib in adults and children with SR-aGvHD
a brief summary of methods and results of post hoc analyses of the REACH 2 trial that were applied in the submitted pharmacoeconomic model
a list of ongoing trials, presented in Table 43.
Moiseev et al. (2020) Study
Description of the Study
Moiseev et al. (2020)24 was a prospective, single-centre, open-label study conducted in Russia that included 75 patients with acute (n = 32) or chronic (n = 43) SR-GvHD. In the study sample of adults and children, about half the participants were children (53% had acute and 39% had chronic GvHD). The median ages in the acute and chronic GvHD groups were 17 years (range = 1 to 67) and 21 years (range = 2 to 62), respectively. Adult and children weighing more than 40 kg received ruxolitinib at a starting dose of 10 mg twice a day, children weighing less than 40 kg received 0.15 mg/kg twice a day. Previous treatments were continued if the attending physician considered it necessary. Ruxolitinib was stopped if there were signs of GvHD progression. The primary end point was ORR. ORR for acute and chronic GvHD was assessed in accordance with the joint statement criteria of Martin et al. (2009)25 and the NIH criteria of Lee et al. (2015),26 respectively. The secondary end points included OS, toxicity, relapse, and infection complications.
Efficacy Results
The ORR was 75% (95% CI, 57% to 89%) in the aGvHD group and 81% (95% CI, 67% to 92%) in the cGvHD group. OS was 59% (95% CI, 49% to 74%) in the aGvHD group and 85% (95% CI, 70% to 93%) in the cGvHD group. In patients with aGvHD and cGvHD, there were no significant differences between adults and children in any of the outcomes, including ORR (aGvHD P = 0.31; cGvHD P = 0.35) and survival (aGvHD P = 0.44; cGvHD P = 0.12).
Harms Results
The most common adverse event (AE) was hematologic toxicity, with 79% and 44% of grade III to IV neutropenia occurring in the acute and chronic GvHD groups, respectively. There were no significant differences in toxicity between adults and children.
Critical Appraisal
Given the single-arm observational design, interpretation of the study results is limited. Because of the lack of a comparator group and blinding, it is difficult to determine the effectiveness of the treatment on the study outcomes. Given the relatively small sample size of patients with aGvHD (n = 32), the generalizability of these results may be limited. Moreover, as this trial was conducted in Russia, there may be limitations to the generalizability of these findings to the Canadian context.
Relevance for CADTH Review
In the REACH 3 trial, patients 12 to 18 years represented a small proportion of the study sample (3.6%). In the study by Moiseev et al. (2020),24 approximately 50% of the study sample was younger than 18 years. Hence, this additional study supplements the evidence for ruxolitinib in patients younger than 18 years.
Post Hoc Analyses of the REACH 2 Trial
Several post hoc analyses of the REACH 23 trial were conducted, and the results were applied to the submitted pharmacoeconomic model. High-level summaries of the methods and results of the post hoc analyses were provided by the sponsor and were summarized by CADTH, and key critical appraisal points were added by the CADTH review team. The post hoc analyses included OS by response, DOR by response at day 28, duration of treatment by response at day 28, duration of treatment by individual initial BAT, duration of treatment from randomization, and resource use by study group for initial hospitalization and response at day 28 for readmissions. The CADTH review team was unable to conduct a rigorous evaluation of the conduct and reporting of the post hoc analyses, as only a high-level summary of methods was provided by the sponsor. Overall, the CADTH methods team concluded that results from post hoc analyses are considered exploratory and hypotheses-generating only. Because of the lack of formal inferential statistical testing, the ability to interpret results of such analyses is significantly limited.
Conclusions
One phase III, open-label, multi-centre randomized controlled trial (RCT) (REACH 2) and 1 single-arm phase II trial (REACH 1) were included in this CADTH review. The REACH 2 trial demonstrated statistically significant improvements in ORR at day 28 and in the rate of durable ORR at day 56 in patients treated with ruxolitinib, compared those treated with BAT. The improvements in the response outcomes of the magnitude observed in the REACH 2 trial were considered clinically meaningful by the clinical experts consulted by CADTH. Other secondary outcomes — DOR, BOR, FFS, and steroid use — were supportive of the observed ORR day 28 benefit with ruxolitinib. The open-label design of the trial and reliance on the assessment of trial outcomes by local investigators may have introduced bias that is difficult to quantify. The results of HRQoL, EQ-5D-5L, and FACT-BMT measures remain uncertain because of several important limitations. The actual degree of OS benefit with ruxolitinib is uncertain, given the risk of potential bias that arises from the crossover of patients in the BAT group to the ruxolitinib group and the limited follow-up time. The clinical experts consulted by CADTH noted that no new safety concerns were observed with ruxolitinib. Although the REACH 1 trial achieved the predetermined threshold for a positive outcome (lower limit of the 95% CI for ORR at day 28 ≥ 40%) in patients who received ruxolitinib, there was uncertainty regarding the magnitude of clinical benefit directly attributable to ruxolitinib because of limitations associated with the study design, including the single-arm, open-label design with no formal statical significance testing and the relatively small sample size (N = 71).
Introduction
Disease Background
HSCTs provide stem cells to patients whose bone marrow has been destroyed by disease, chemotherapy, or radiation.2 The 2 main types of SCT are autologous and allogeneic transplants. alloSCTs use stem cells from a matched related or unrelated donor, whereas autologous SCTs use the patient’s own stem cells.2 Between 2006 and 2014, nearly 1 million SCTs were performed worldwide, approximately 40% of which were alloSCTs.3 alloSCT can be used for the treatment of malignant and nonmalignant hematologic diseases.3 According to a Canadian population-based cohort study,27 547 alloSCTs were performed in Ontario between 2012 and 2015. Between 2008 and 2019, an estimated 13,033 transplants were performed in Canada, 5,672 of which were alloSCTs5; in the year from 2018 to 2019, 2,843 transplants were recorded in Canada.5 Most transplants were conducted in patients with plasma cell disorders (30.28%), non-Hodgkin’s lymphoma (20.07%), and acute myeloid leukemia (15.93%). Although alloSCT has curative potential, there are risks that the donor’s stem cells will die or be destroyed by the patient’s body before settling in the patient’s bone marrow or that the donor’s immune cells will attack healthy cells in the patient’s body; the latter is called GvHD.2,3
GvHD is a multi-system disorder in which the donor-derived immune cells initiate an adverse immune reaction in the transplant recipient’s tissues, cells, and organs, leading to tissue damage, organ failure, or death. GvHD has been found to be the leading cause of morbidity and NRM in patients after alloSCT,4 affecting up to 70% of patients who receive HSCTs.3 GvHD is estimated to be responsible for 21% to 31% and 31% to 40% of deaths in patients who received a transplant from a HLA-matched sibling and from an unrelated donor, respectively.3
GvHD has a multitude of syndromes that are defined by clinical manifestations, according to NIH consensus criteria, rather than the time of onset (i.e., before or after day 100 of transplant, as was used previously).28,29 GvHD is typically classified as acute or chronic, depending on the set of distinct clinical manifestations. Overlap syndrome may also occur, in which diagnostic or distinctive features of aGvHD and cGvHD appear together.6 cGvHD typically occurs 100 days or more after alloSCT and can last a few months or a lifetime, affecting almost any part of the body.6 aGvHD typically occurs within 100 days after alloSCT and often affects the skin, liver, and intestines.6
Acute GvHD occurs in 30% to 50% of patients who undergo alloSCT, with 14% to 36% developing severe aGvHD (i.e., grade III).7 Prognosis among patients with aGvHD is poor, with an estimated 3-year survival rate of 54%8; only 25% to 30% of patients with grade III aGvHD and 1% to 2% of patients with grade IV aGvHD experience long-term (> 2 years) survival.7 Grade I aGvHD has not been found to significantly affect long-term survival.3 It has been suggested that patients with SR-aGvHD are at an elevated mortality risk, with an estimated 2-year survival rate of 17%. Patients with SR-GvHD have a high mortality from infections.7 In a retrospective study of 127 patients with SR-aGvHD, 4-year infection-related mortality and OS were 46% and 15%, respectively; the 1-year incidence of bacterial, viral, and fungal infections was 74%, 65%, and 14%, respectively.7
Among the several factors thought to affect the incidence and severity of aGvHD, 1 of the most important is alloSCT from HLA-nonidentical or unrelated donors.30 Additional risk factors include the advanced age of patient and/or donor, a female donor for male recipient, peripheral blood as the stem cell source, the type of GvHD prophylaxis, and recipient seropositivity for cytomegalovirus.7
Clinical manifestations of aGvHD typically affect the skin, GI tract, and liver. Common symptoms of aGvHD are skin rash, burning and redness of the skin on the palms of the hands or soles of the feet, blisters and peeling skin, diarrhea, persistent nausea and vomiting, cramping or abdominal pain, enlarged liver, liver tenderness, abnormal liver enzymes or liver failure, and increased levels of serum bilirubin.9 Commonly, 1 of the first clinical symptoms of aGvHD is a maculopapular skin rash with gradually worsening manifestations.31 The degree of skin involvement is graded according to the degree and severity of lesions as stage 1 to 4. GI symptoms typically involve the upper and lower tracts. Diagnosis is typically confirmed upon review of tissues with upper endoscopy, rectal biopsy, or colonoscopy. The degree of GI involvement is graded according to the severity of diarrhea as stage 1 to 4. Liver involvement is commonly observed in patients who also exhibit symptoms of skin and GI aGvHD. Liver biopsy is used to confirm GvHD of the liver. Early signs of hepatic involvement include abnormal liver function tests, such as a serum liver rise in conjugated bilirubin and alkaline phosphatase. The degree of liver involvement is graded according to the serum total bilirubin levels as stage 1 to 4. The stages of skin, GI, and liver involvement are combined to determine the overall severity grade of aGvHD.31
Over the past 40 years, several grading systems for aGvHD have been developed; the most commonly used include the initial Glucksberg grade (grades I to IV)32 and the International Bone Marrow Transplant Registry (IBMTR) grading system (grades A to D).33,31 These grading systems have developed over time; for example, the Glucksberg system has been updated by an NIH working group to include persistent nausea with histologic evidence of GvHD in stage 1 upper GI aGvHD (Przepiorka et al. [1995]34). A more recent widely adopted grading system is the NIH consensus grading,16 which allows measurement of the frequency of stools or the stool volume when staging lower GI involvement (stages 0 to 4). The NIH criteria have been refined and tested for clarity and ease of use by an international GvHD research consortium (the Mount Sinai Acute GvHD Consortium) to standardize the compilation of clinical data from multiple organ systems for clinical research (Harris et al. [2016]16) (see Table 46 in Appendix 3 for the updated NIH criteria).3 Standardized approaches to grading of aGvHD that have been developed through international expert consensus, such as the NIH criteria, increase the uniformity of aGvHD symptom capture and may help to reduce variability in the diagnosis and grading of aGvHD between transplant centres.7,31
Diagnosis of aGvHD may be straightforward in patients who present with classical symptoms, such as rash, diarrhea with abdominal cramps, and rising serum bilirubin concentration in the first few months after alloSCT.31 However, in many cases, aGvHD presents with less obvious signs and symptoms, and competing causes of abnormalities must be excluded. For example, skin rash can be caused by a variety of drugs that these patients are often treated with, diarrhea may be caused by infection, and hyperbilirubinemia is a common side effect of multiple drugs.31 Alternative diagnoses may be excluded on biopsy of the involved tissue; however, biopsies may lack sensitivity and specificity,7 and liver biopsy may pose a significant risk of major bleeding, given that most patients with aGvHD are thrombocytopenic.31 Currently, there are no established biomarkers for the diagnosis or prognosis of aGvHD.31 The diagnosis of aGvHD comes down to a careful integration of all available clinical information.3
About 5% of all patients with aGvHD are 12 to 18 years.3 The management and treatment of aGvHD is similar in adolescents and adults.3 aGvHD is a major cause of morbidity and mortality after alloSCT in patients of all ages.35 Adults and children with grade III to IV aGvHD have a 2-year survival rate of just 27% to 35%.3 The clinical experts consulted by CADTH agreed that there is a significant unmet need in adolescents with aGvHD.
Standards of Therapy
Treatment options generally aim for the immunosuppression of donor T cells, which can cause aGvHD. However, the same cells are likely responsible for elimination of residual malignant cells’ GVT effect). The management of patients with aGvHD must therefore achieve a balance between the benefits of reducing GvHD and the potential harms from a reduced GVT effect.7 The choice of initial treatment for patients with aGvHD depends on multiple factors, including the severity of symptoms, the type of prophylactic regimen used, and the importance of a GVT effect.7 Grade I aGvHD (i.e., skin involvement of 50% or less of the body surface area without liver or GI tract involvement) is managed with topical treatments (e.g., tropical steroids) and adjustment of prophylactic treatments (e.g., MMF or CNIs such as cyclosporine). Patients with grade II or higher aGvHD (i.e., skin involvement of greater than 50% of the body surface area with liver or GI tract involvement) receive high-dose systemic glucocorticoids (e.g., methylprednisolone or prednisone) in addition to the care that patients with grade I aGvHD receive. In patients who respond, steroid treatment is continued over several weeks until it is gradually tapered to prevent a flare of aGvHD. Approximately 25% to 40% of patients achieve CRs with glucocorticoids7; however, treatment is associated with significant side effects that can affect a patient’s quality of life and increase susceptibility to infection.10 Approximately 25% to 50% of patients become refractory to steroids and are considered to have SR-aGvHD. In general, patients whose disease progresses 3 to 5 days after the initial start of systemic steroids or who show no response after 5 to 7 days are considered to have SR-aGvHD; the exact definition of SR-aGvHD may vary by centre.7
There is currently no standardized second-line therapy for patients with aGvHD who have an inadequate response to steroids in Canada. Evidence from patients with SR-aGvHD has mostly been obtained in retrospective, single-arm, phase II studies. Comparisons of data across studies is a challenge because of the small number of enrolled patients, the heterogenous patient populations, and the lack of standardized end points.7 The weighted average 6-month survival estimate across 25 studies that evaluated second-line therapies for aGvHD was reported to be 0.49 by the American Society of Blood and Morrow Transplantation; however, given the significant heterogeneity across study populations and designs, interpretation of this estimate is significantly limited.36 In the absence of sufficient evidence to guide the selection of second-line treatment, factors that influence the treatment choice include the experience of the treating physician, the type of aGvHD prophylaxis used, and the risk of potential toxicities and worsening pre-existing comorbidities.7 According to the clinical experts consulted by CADTH, available second-line options in Canada include ECP, MMF, etanercept, infliximab, mTOR inhibitors (i.e., everolimus or sirolimus), ATG, and interleukin-2 receptors. The clinical experts noted that available therapies are currently used off-label. In the absence of proven treatment options, there is inter-province variability in standard practices and access to therapies. The clinical experts explained the challenges with currently available therapies in this heavily pre-treated target population, including no responses or only PRs (response rate was estimated by the clinical experts to be approximately 50%, but the durable response rate, in which patients still show a response at about 2 months, to be only 10% to 30%). According to the clinical experts, responses in this patient population are important to enable the tapering of steroids to mitigate long-term side effects (e.g., osteoporosis, hypertension, hyperglycemia, diabetes, and bone or joint health) and the risk of infection. It was emphasized by the clinical experts that opportunistic infection and organ damage related to aGvHD are major causes of NRM in patients with SR-aGvHD.
There was consensus among the clinical experts that there is an unmet need for effective therapies with acceptable toxicity profiles that improve HRQoL, reduce the symptoms of aGvHD, enhance performance status, and improve OS. They added that a convenient oral route of administration would help improve adherence and reduce hospital-based resource use.
Drug
Ruxolitinib is a JAK inhibitor that mediates the signalling of a number of cytokines and growth factors important for hematopoiesis and immune function.1 Ruxolitinib binds to and inhibits protein tyrosine kinases JAK 1 and JAK2, which may lead to a reduction in inflammation and an inhibition of cellular proliferation.37 Ruxolitinib has received Health Canada market authorization for the treatment of SR or dependent aGvHD in patients 12 years and older. The sponsor’s requested reimbursement criteria for ruxolitinib for aGvHD differ from the Health Canada indication, in that the sponsor’s criteria specify “inadequate response to corticosteroids or other systemic therapies,” rather than “steroid refractory or dependent” aGvHD. Concurrent with this CADTH review for aGvHD, ruxolitinib is being reviewed by CADTH in the cGvHD setting for the treatment of cGvHD in patients 12 years and older who have an inadequate response to corticosteroids or other systemic therapies. Ruxolitinib has 2 Health Canada–approved indications: the treatment of splenomegaly and/or its associated symptoms in adults with primary myelofibrosis (also known as chronic idiopathic myelofibrosis), post-PV myelofibrosis, or post-essential thrombocythemia myelofibrosis; and the control of hematocrit in adults with PV resistant to or intolerant of a cytoreductive drug. Ruxolitinib received a positive conditional CADTH recommendation in March 2016 for the treatment of patients with PV who are resistant to or intolerant of hydroxyurea, according to the modified European LeukemiaNet Criteria used in the RESPONSE trial, and who have a good performance status. Ibrutinib is the only Health Canada–approved therapy for GvHD3; it is indicated in the chronic setting for the treatment of patients with steroid-dependent or SR-cGvHD.38 However, ibrutinib has not been reviewed by CADTH and is currently not publicly reimbursed in Canada for the current target indication; it is available through private drug insurance.
After being granted priority review with orphan product designation, the FDA-approved ruxolitinib in September 2021 for cGvHD after the failure of 1 or 2 lines of systemic therapy in patients 12 years and older, based on evidence from the phase III REACH 3 trial.39 In May 2019, after granting priority review, the FDA-approved ruxolitinib for SR-aGvHD in patients 12 years and older based on evidence from the phase II REACH 1 trial.40 In addition to the GvHD setting, the FDA has approved ruxolitinib for intermediate or high-risk myelofibrosis, including primary myelofibrosis, post-PV myelofibrosis, and post-essential thrombocythemia myelofibrosis in adults, and for PV in adults who have had an inadequate response to or are intolerant of hydroxyurea. After being granted priority review with orphan product designation, the FDA-approved belumosudil in July 2021 for patients 12 years and older with cGVHD after failure of at least 2 lines of systemic therapy based on evidence from the phase II KD025 to 213 trial.41 The European Medicines Agency has approved ruxolitinib for the treatment of disease-related splenomegaly or symptoms in adults with primary myelofibrosis (also known as chronic idiopathic myelofibrosis), post-PV myelofibrosis, or post-essential thrombocythemia myelofibrosis, and for the treatment of adults with PV who are resistant to or intolerant of hydroxyurea.
Ruxolitinib is available as 5 mg, 10 mg, 15 mg, and 20 mg tablets. The recommended starting dose for ruxolitinib for aGvHD is 5 mg administered orally twice daily. A dose increase to 10 mg twice daily is recommended after at least 3 days of treatment if ANCs and platelet counts are not decreased by at least 50% relative to the first day of dosing with ruxolitinib. The product monograph also states that tapering of ruxolitinib may be considered in patients with a response after they have discontinued corticosteroids. Tapering of ruxolitinib, by reducing the dose to 50% every 2 months, is recommended; in the event that signs or symptoms of GvHD recur during or after the taper, re-treatment with ruxolitinib should be considered.1 Table 3 summarizes key characteristics of ruxolitinib.
According to information in the product monograph and confidential information in the Health Canada Reviewer Report, Health Canada considered reviewing REACH 1 as the pivotal study, and using safety data from the REACH 2 trial as supportive evidence for the aGvHD indication.1,11 This decision is the result of uncertainties that were identified by the FDA when it reviewed raw data from the REACH 2 trial as part of a sponsor-proposed label update for the FDA-approved indication of aGvHD, which had already been approved based on REACH 1 data. The sponsor’s submission noted that the proposed label update based on REACH 2 trial data could not be completed, and the sponsor subsequently withdrew the supplemental New Drug Application for REACH 2. Specifically, the 3 issues identified by the FDA concerned the following: missing and incomplete information in the case report forms related to organ staging of aGvHD, which precluded the FDA from conducting their own analyses and confirming the investigators’ efficacy assessments; the frequency of protocol deviations related to investigator-determined organ staging; and information regarding prior therapies (i.e., prophylactic versus therapeutic aGvHD treatment). Health Canada concluded that as a result of “integrity issues in the REACH 2 study” identified by the FDA, the efficacy results of REACH 2 could not be “verified.”11 Therefore, Health Canada upheld its initial decision to consider REACH 1 as the pivotal study, and to use safety and pharmacokinetic data from the REACH 2 trial as supportive evidence in their assessment of the aGvHD indication. The sponsor clarified with CADTH that the initial decision to submit the REACH 2 trial as the pivotal trial for the proposed CADTH reimbursement request remains unchanged.3 This CADTH report does not include a review or critical appraisal of the issues raised by the FDA, as CADTH has no access to the FDA’s assessment and reviewing raw data are not in the mandate of CADTH.
Health Canada based their efficacy assessment of the REACH 1 trial on the FDA-evaluable population (49 patients who failed steroids alone), which is a subset of the REACH 1 trial’s full efficacy-evaluable patients (N = 71). According to the Health Canada Clarifax, the following subsets of patients were excluded from the FDA-evaluable population: 9 patients who failed steroids plus additional therapies before randomization, and 13 patients who did not meet the SR criteria because of suboptimal dosing or duration of corticosteroid treatment before randomization.12 The Health Canada Clarifax noted that the available data are considered insufficient to support the proposed ruxolitinib indication for patients with aGvHD who failed 1 other systemic treatment in addition to corticosteroids (± CNIs).12 This CADTH clinical review report does not report on the subset of 49 patients in the FDA-evaluable population; it presents results for all 71 efficacy-evaluable patients from the REACH 1 trial. Results for the 49 patients in the FDA-evaluable population are reported in the product monograph.1
The Health Canada Reviewer Report stated that assessment of the safety of ruxolitinib for aGvHD was informed by the full safety-evaluable population (N = 71) of the REACH 1 trial and by safety results observed in the REACH 2 trial.1 Safety results reported in this CADTH clinical review are based on 71 patients in the REACH 1 trial and safety results from the REACH 2 trial. The Health Canada Reviewer Report noted that patients with SR or steroid-dependent aGvHD as per the eligibility criteria in REACH 1 are considered acceptable.11 The REACH 1 eligibility criteria for SR and steroid-dependent aGvHD align with the current National Comprehensive Cancer Network guidelines for hematopoietic cell transplantation, version 2.2020,13 the Alberta Bone Marrow and Blood and Cell Transplant Standard Practice Manual (2021),14 and the EBMT-NIH-CIBMTR Task Force position statement,15 with minor variations. Steroid dependence has been defined as the inability to taper prednisone under 2 mg/kg per day after initially successful treatment for at least 7 days or as the recurrence of aGvHD activity during steroid taper.11
Table 3: Key Characteristics of Ruxolitinib
Characteristic | Ruxolitinib |
|---|---|
Mechanism of action | Ruxolitinib is a JAK inhibitor that mediates the signalling of a number of cytokines and growth factors important for hematopoiesis and immune function.1 Ruxolitinib binds to and inhibits protein tyrosine kinases JAK1 and JAK2, which may lead to a reduction in inflammation and an inhibition of cellular proliferation.37 |
Indicationa | For the treatment of SR or dependent aGvHD in patients 12 years and older. |
Route of administration | Oral. |
Recommended dose | 5 mg twice daily; increase the dose to 10 mg twice daily after at least 3 days of treatment if ANCs and platelet counts are not decreased by 50% or more relative to the first day of dosing with ruxolitinib. |
SAEs or safety issues | Serious infections have been reported in patients treated with ruxolitinib, some of which were life-threatening or led to death. |
aGvHD = acute graft-versus-host disease; ANC = absolute neutrophil count; JAK = Janus-associated kinase; SAE = serious adverse event; SR = steroid refractory.
aHealth Canada–approved indication.
Sources: Product monograph,1 National Library of Medicine.37
Stakeholder Perspectives
Patient Group Input
Eight patient groups — Lymphoma Canada, the Lymphoma & Leukemia Society of Canada, Chronic Lymphocytic Leukemia Canada, Myeloma Canada, the Aplastic Anemia & Myelodysplasia Association of Canada, the Canadian Myeloproliferative Neoplasms Research Foundation and the CML Network, MPN Canadian Research Foundation, and CTTC — created 1 joint patient input for this review. The input was based on an online survey with 68 respondents. Sixty patients reported having received an SCT, 6 patients reported not having received an SCT, and 2 patients did not answer the question. Of the 60 patients who received an SCT, 49 reported having received an alloSCT. Fifty-three patients had experienced GvHD after their SCT. Data on type of GvHD were available for 45 of the 53 patients with GvHD: 13% experienced aGvHD, 24% experienced cGvHD, and 62% experienced both acute and chronic GvHD. Twenty patients reported receiving ruxolitinib.
Respondents indicated that they had long-lasting GvHD symptoms (3 to 5 years for 26% of respondents and more than 5 years for 28% of respondents). To manage their GvHD, the respondents reported requiring numerous medical consultations, hospital stays, and nights away from home. Respondents reported a range of GvHD symptoms that had a significant impact on daily activities and were detrimental to quality of life. Respondents highlighted the interruption of life goals and accomplishments (career, school), difficulty sleeping, and impacts on their mental health (stress, anxiety, worry, and concentration problems), and finances. Other commonly experienced symptoms reported by respondents included burning and redness of the skin on the palms of the hands or soles of the feet, rashes that could spread over the entire body, blisters and peeling skin, skin problems (such as dryness, rash, itching, peeling, darkening, hard texture, and feeling tight), enlarged liver, liver tenderness, abnormal liver enzymes or liver failure, jaundice, dry eyes that may have a burning or gritty feeling, dry mouth with or without mouth ulcers, diarrhea, loss of appetite, stomach cramps, vomiting, weight loss, pain in muscles and joints, mobility issues, infections, and difficulty breathing.
According to the patient input received, respondents expected new drugs or treatments to improve the following key outcomes: OS, GvHD symptoms, quality of life, and severity of side effects. Additionally, the ability to receive treatment in the outpatient setting (rather than requiring an overnight hospital stay), having access to treatment locally (rather than requiring extensive travel), coverage of treatment by insurance or drug plans, and treatments recommended by health care professionals were perceived to be very important by respondents. Respondents who had direct experience with ruxolitinib indicated that, overall, ruxolitinib was an effective treatment, improved their quality of life, had tolerable side effects, and they would take again if recommended by their physician and would recommend it to other patients.
Clinician Input
Input From the Clinical Experts Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise in the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol; assisting in the critical appraisal of clinical evidence; interpreting the clinical relevance of the results; and providing guidance on the potential place in therapy). The following input was provided by 3 clinical specialists with expertise in the diagnosis and management of aGvHD.
Unmet Needs
The clinical experts consulted by CADTH noted that there are currently no standard-care regimens specific to patients with SR-aGvHD in Canada. The clinical experts noted that, in the absence of proven treatment options, there is inter-province variability in standard practices and access to therapies. Available therapies are currently used off-label and, because of the lack of large-scale, positive, randomized, prospective studies, no consensus could be reached on the optimal second-line therapy for SR-aGvHD.3 There was consensus among the clinical experts that there is an unmet need for effective therapies with acceptable toxicity profiles that improve HRQoL, reduce the symptoms of aGvHD, enhance performance status, and improve OS. The added that a convenient oral route of administration would help improve adherence and reduce hospital-based resource use. The clinical experts expressed challenges with currently available therapies in this heavily pre-treated target population, including no responses or only PRs (response rate was estimated by the clinical experts to be approximately 50%, but the durable response rate, in which patients still show a response at about 2 months, to be only 10% to 30%) and intolerance to currently available treatments. According to the clinical experts, responses in this patient population are important to enable the tapering of steroids to mitigate long-term side effects (e.g., osteoporosis, hypertension, hyperglycemia, diabetes, and bone or joint health) and the risk of opportunistic infection. It was emphasized by the clinical experts that opportunistic infection and organ damage from aGvHD are major causes of NRM in SR-aGvHD.
Place in Therapy
Ruxolitinib can be used as an add-on to an immunosuppressive regimen of corticosteroids with or without CNIs in patients 12 years and older with grade II to IV SR-aGvHD, according to NIH criteria used in the REACH 2 trial. The clinical experts agreed that ruxolitinib would likely shift the current treatment paradigm. They noted that ruxolitinib works as a JAK inhibitor and blocks the JAK-STAT pathway, and that its mechanism of action is novel in the context of other immunosuppressives used in the management of aGvHD, potentially offering synergy with other therapies. The clinical experts highlighted the differential effects of ruxolitinib on different T cell subsets, which help prevent post-transplant relapse. The trial excluded patients who received more than 1 systemic treatment for SR-aGvHD, patients with overlap syndrome, and patients with grade I SR-aGvHD. The clinical experts consulted by CADTH felt that it would be reasonable to generalize the REACH 2 trial results to patients who received 2 or more systemic treatments for aGvHD. They noted that ruxolitinib has a novel mechanism of action in the context of other second-line immunosuppressives, with the potential to offer synergy with other therapies. As well, given the acceptable safety profile of ruxolitinib, they felt that it would be reasonable to leave it to the discretion of the treating physician to apply some flexibility in terms of using ruxolitinib in patients with overlap syndrome and patients with grade I aGvHD.
Patient Population
Overall, the clinical experts consulted by CADTH agreed that patients who meet the inclusion and exclusion criteria of the REACH 2 trial should be eligible for ruxolitinib. Although they agreed that there is currently insufficient evidence to identify which patient subgroups would most likely show a response to ruxolitinib, the clinical experts identified patients with grade IV aGvHD, who have the highest risk of death from aGvHD, as the subgroup most in need of ruxolitinib therapy. Subgroups that would potentially benefit least from ruxolitinib may include patients with refractory vomiting or ileus who are not able to take an oral drug such as ruxolitinib, and patients with thrombocytopenia, especially those with clinical bleeding, who may be a challenge to treat with ruxolitinib and may receive an alternative second-line drug instead. Patients with active uncontrolled infections or non-aGvHD cytopenia are a challenge to treat with ruxolitinib or other available second-line therapy options; ruxolitinib should be used with caution and may require dose adjustment in these patients.
The clinical experts agreed that patients considered to have SR-aGvHD would be possible candidates for ruxolitinib. They noted that aGvHD is a complex clinical diagnosis made on a daily basis by experienced bone marrow transplant physicians. In general, the experts agreed that the diagnosis of aGvHD is made on clinical grounds (patients with grade II to IV aGvHD are generally not asymptomatic), although tissue diagnosis (biopsy) confirmation may be pursued. They noted that SCT programs are highly specialized and well supported by histopathologists. The clinical experts agreed that a potential for misdiagnosis exists, given the underlying complexity of aGvHD and the nonspecific presentation of aGvHD, so a differential diagnosis must be considered. However, misdiagnosis is minimized in Canada because patients who have undergone alloSCT are followed in specialized clinics.
Assessing Response to Treatment
In the opinion of the clinical experts consulted by CADTH, an accurate assessment of response in aGvHD is based on the NIH consensus criteria used in the REACH 2 trial. Given the underlying complexity of aGvHD evaluation, the experts noted that treatment response can be formally assessed, but it is a challenge to undertake outside the structure of a clinical trial. Clinical assessments in Canadian clinical practice to evaluate response to treatment examine the following areas: skin rash; nausea or vomiting; stool consistency, diarrhea volumes, or abdominal pain; liver function tests; and performance status. In addition, the ability to reduce the corticosteroid dose without exacerbating symptoms and signs of aGvHD is indictor of response to treatment.
The experts noted that response to treatment is usually assessed daily for inpatients and weekly for outpatients.
The clinical experts indicated that the most clinically meaningful responses to treatment include improvements in OS (survival more than 1 year after alloSCT) overall response (CR or PR), improvements in HRQoL and performance status, and the ability to taper corticosteroids.
Discontinuing Treatment
In the opinion of the clinical experts consulted by CADTH, ruxolitinib should be discontinued if a patient experiences aGvHD disease progression, lacks response after 4 to 6 weeks on treatment and either an alternative treatment is being introduced or further treatment is deemed futile and a palliative care approach is pursued, or experiences intolerable toxicity (e.g., severe and refractory thrombocytopenia, cytopenia, or severe neurologic toxicity). The clinical experts agreed that, based on their clinical experience, ruxolitinib generally works quickly (within a few weeks).
Prescribing Conditions
In the opinion of the clinical experts consulted by CADTH, ruxolitinib is an oral drug that can be self-administered in a patient’s home. Patients are assessed and managed in the SCT follow-up clinic. All assessments and prescriptions should be managed by providers familiar with GvHD. Generally, patients with aGvHD are medically unwell to the point of requiring hospitalization in a bone marrow transplant unit in the hospital. Occasionally, patients may be managed as an outpatient, such as with higher doses of steroids and a second-line drug (like ruxolitinib). Patients who respond to treatment are generally able to transition to outpatient care.
Clinician Group Input
This section was prepared by CADTH staff based on the input provided by clinician groups.
The information in this section is a summary of 2 inputs provided by the registered clinician groups that responded to CADTH’s call for clinician input for the purpose of this review. The full clinician inputs received can be found in Appendix 6.
Two clinician group inputs were provided: 1 from CTTC (which contained input from 8 clinicians) and 1 from Ontario Health Cancer Care Ontario Complex Malignant Hematology (which contained input from 2 clinicians). The views of the clinician groups were, overall, consistent with the clinical experts consulted by CADTH, indicating that, based on the evidence from the REACH 2 trial, it is likely that ruxolitinib will become the dominant first-line therapy for SR-aGvHD. The outcomes assessed in the REACH 2 trial were judged to be applicable to Canadian clinical practice and reflective of clinically meaningful responses. It was noted by both input groups that ruxolitinib is not as immunosuppressive as other available therapies. The clinicians from Ontario Health Cancer Care Ontario noted the drawbacks of currently available therapies, such as the IV administration (which requires patients to be at the hospital), side effects and broad immune suppression, and the high price and delivery costs of treatments. The CTTC noted that a Health Canada–approved and provincially funded therapy for SR-aGvHD would be an important step forward in the present target setting, because existing therapies offer low response rates and high rates of toxicity. According to input from the CTTC, experience with ruxolitinib (accessible through compassionate access programs) and real-world effectiveness appear similar to what was observed in the REACH 2 trial, with low rates of toxicity.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Drug program implementation questions | Clinical expert response |
|---|---|
Is there a patient population that would require a combination of 1 of the off-label comparator treatments and ruxolitinib for SR-aGvHD? | As responses to second-line drugs are not as rapid and complete, 2 drugs might be used simultaneously (e.g., ruxolitinib + ECP, ruxolitinib + etanercept, or ruxolitinib + MMF) if the manifestations are particularly concerning. |
What is the definition of inadequate response to corticosteroids or steroid refractoriness in patients with aGvHD? | Criteria for inadequate response to corticosteroids or steroid refractoriness in aGvHD are defined in the EBMT-NIH-CIBMTR Task Force position statement15 that was used in the REACH 2 trial. In the REACH 2 trial, patients on high-dose systemic corticosteroids (methylprednisolone 2 mg/kg per day [or an equivalent prednisone dose of 2.5 mg/kg per day]), given alone or in combination with a CNI were defined as SR in each of the following scenarios. A. Progression based on organ assessment after at least 3 days, compared with organ stage at the time of initiation of high-dose systemic corticosteroids ± CNIs for the treatment of grade II to IV aGvHD OR B. Failure to achieve a PR, at a minimum, based on organ assessment after 7 days, compared with organ stage at the time of initiation of high-dose systemic corticosteroids ± CNIs for the treatment of grade II to IV aGvHD OR C. Corticosteroid taper failure, defined as the fulfillment of 1 of the following criteria:
OR
In the REACH 1 trial, criteria A and B were the same as in the REACH 2 trial. There were small variations in criterion C between the REACH 1 and REACH 2 trials; however, the clinical experts felt that these would be unlikely to influence treatment effects. |
Ruxolitinib is indicated for the treatment of aGvHD in patients 12 years and older who have an inadequate response to corticosteroids or other systemic therapies. What other systemic therapies are specified in the reimbursement request for aGvHD? | Treatments that might be used BEFORE second-line ruxolitinib include:
Upon inadequate response to these treatments, ruxolitinib may be attempted. The sponsor was asked for clarification on the number of patients in the REACH 2 trial who had an inadequate response to corticosteroids, an inadequate response to other systemic therapies, or an inadequate response to corticosteroids and other systemic therapies, but noted that such data are not available.22 Because an inadequate response to corticosteroids was an eligibility criterion of the REACH 2 trial, it follows that all patients had an inadequate response to corticosteroids; it also follows that data for patients who only had an inadequate response to other systemic therapies and not to steroids were not available from the REACH 2 trial. However, the proportion of patients with an inadequate response to other systemic therapies in addition to an inadequate response to corticosteroids remains unclear. The sponsor was asked for clarification on the number of patients in the REACH 1 trial who had an inadequate response to corticosteroids, an inadequate response to other systemic therapies, or an inadequate response to corticosteroids and other systemic therapies, and noted that 42 patients were refractory to steroids alone and 29 patients were refractory to steroids and 1 additional systemic therapy (i.e., receipt of 1 systemic treatment in addition to corticosteroids [± CNIs] for aGvHD was allowed in the REACH 1 trial).22 The sponsor was asked about the specific types of additional systemic therapies received by the 29 patients in REACH 1 who were refractory to 1 additional systemic therapy, but provided no additional data beyond the information found in Table 16.22 It is not known if patients who are refractory to 1 therapy, as opposed to multiple therapies, would respond differently to ruxolitinib. The clinical experts consulted by CADTH agreed that the difference between patients who either have an inadequate response to corticosteroids alone or to multiple therapies would be unlikely to impact the treatment effect of ruxolitinib. |
Part of the safety outcomes in the REACH 2 trial were AEs leading to treatment discontinuation. What specific AEs would lead to treatment discontinuation in patients with aGvHD? | It is a challenge to be definitive about the specific AEs that would lead to treatment discontinuation in patients with aGvHD. Examples include thrombocytopenia (especially if associated with clinical bleeding or in patients refractory to platelet transfusion), severe anemia, rarely acute kidney injury (likely multi-factorial), and severe neurologic sequelae. Furthermore, the clinical experts speculated that rare but serious congestive heart failure may be observed in patients treated with ruxolitinib as more real-world data are collected. |
What specialist or prescriber would be required to initiate and monitor ruxolitinib for this indication? | Ruxolitinib is an oral drug that is self-administered in a patient’s home. Patients are assessed and managed in the SCT follow-up clinic. All assessments and prescriptions should be managed by providers familiar with GvHD. Generally, patients with aGvHD are medically unwell to the point of requiring hospitalization in a BMT unit in the hospital. Occasionally, patients can be managed as an outpatient, such as with higher doses of steroids and a second-line drug (like ruxolitinib). With response to treatment, patients are generally able to transition to outpatient care. |
AE = adverse event; aGvHD = acute graft-versus-host disease; ATG = antithymocyte globulin; BMT = bone marrow transplant; CIBMTR = Center for International Blood and Marrow Transplant Research; CNI = calcineurin inhibitor; EBMT = European Society for Blood and Marrow Transplantation; ECP = extracorporeal photopheresis; GvHD = graft-versus-host disease; IL 2r = interleukin-2 receptor; MMF = mycophenolate mofetil; NIH = National Institutes of Health; PR = partial response; SCT = stem cell transplant; SR = steroid refractory.
Clinical Evidence
The clinical evidence included in the review of ruxolitinib is presented in 3 sections. The first section, the systematic review, includes clinical pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as studies that were selected according to an a priori protocol. For the second section, no indirect evidence that met the selection criteria specified in the review was identified. The third section includes additional relevant studies that were considered to address important gaps in the evidence in the systematic review.
Systematic Review (Pivotal and Protocol Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of ruxolitinib (10 mg twice daily oral tablets) for the treatment of aGvHD in patients 12 years and older who have an inadequate response to corticosteroids or other systemic therapies.
Methods
Studies selected for inclusion in the systematic review included pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 5. Outcomes included in the CADTH review protocol reflect outcomes considered to be important by patients, clinicians, and drug plans.
Of note, the systematic review protocol presented here was established before the granting of a Notice of Compliance from Health Canada.
Table 5: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Population | Patients 12 years and older with aGvHD who have an inadequate response to corticosteroids or other systemic therapies Subgroups:
|
Intervention | Ruxolitinib 10 mg given orally twice daily |
Comparator |
|
Outcomes |
Harms outcomes: AEs, SAEs, WDAEs, deaths Notable harms or harms of special interest:
|
Study designs | Published and unpublished phase III and IV RCTs |
AE = adverse event; aGvHD = acute graft-versus-host disease; ATG = antithymocyte globulin; BOR = best overall response; cGvHD = chronic graft-vs.-host disease; ECP = extracorporeal photopheresis; EFS = event-free survival; FFS = failure-free survival; HRQoL = health-related quality of life; IL-2r = interleukin-2 receptor; MMF = mycophenolate mofetil; mTOR = mammalian target of rapamycin; NRM = nonrelapse mortality; ORR = overall response rate; OS = overall survival; PR = partial response; RCT = randomized controlled trial; SAE = serious adverse event; SR = steroidrefractory; vs. = versus; WDAE = withdrawal due to adverse event.
aThese outcomes were identified as being of particular importance to patients in the input received by CADTH from a patient group.
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy, according to the PRESS Peer Review of Electronic Search Strategies checklist.42
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946—) via Ovid and Embase (1974—) via Ovid. All Ovid searches were run simultaneously as a multi-file search. Duplicates were removed using Ovid deduplication for multi-file searches, followed by manual deduplication in Endnote. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concepts were ruxolitinib and GvHD. Clinical trials registries were searched: the NIH’s clinicaltrials.gov, WHO’s International Clinical Trials Registry Platform (ICTRP) search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
No filters were applied to limit retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. See Appendix 1 for the detailed search strategies.
The initial search was completed on September 2, 2021. Regular alerts updated the search until the meeting of the CADTH Canadian Drug Expert Committee on April 27, 2022.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool For Searching Health-Related Grey Literature resource.43 Included in this search were the websites of regulatory agencies (FDA and European Medicines Agency). Google was used to search for additional internet-based materials. See Appendix 1 for more information on the grey literature search strategy.
These searches were supplemented by a review of bibliographies of key papers and through contacts with appropriate experts. In addition, the manufacturer of the drug was contacted for information regarding unpublished studies.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
A focused literature search for network meta-analyses was run in MEDLINE All (1946–) for GvHD. No limits were applied.
Findings From the Literature
A total of 245 studies were identified from the literature for inclusion in the systematic review (Figure 1). The included studies are summarized in Table 6. A list of excluded studies is presented in Appendix 2.
Table 6: Details of Included Studies
Detail | REACH 2 | REACH 1 |
|---|---|---|
Design and population | ||
Study design | Phase III, completed, open-label, randomized, multi-centre RCT | Phase II, completed, open-label, single-arm, multi-centre trial |
Locations | Patients randomized at 103 sites in 22 countries:
| Patients enrolled at 26 centres in 17 American states |
Patient enrolment dates | First patient enrolled: April 12, 2017 Enrolment end date: May 30, 2019 | First patient enrolled: December 27, 2016 Enrolment end date: December 28, 2017 |
Data cut-off dates | Primary analysis: July 25, 2019 Secondary analysis: January 6, 2020 Final analysis: April 23, 2021 | Interim analysis (futility analysis): October 2017 Updated analysis: July 2, 2018 Final analysis: June 5, 2019 Actual study completion date (last patient’s final visit): June 5, 2019 |
Patients randomized and enrolled | Randomized: 309 patients
| Enrolled: 71 patients
|
Inclusion criteria |
A. Progression based on organ assessment after at least 3 days, compared with organ stage at the initiation of high-dose systemic corticosteroids ± CNIs for the treatment of grade II to IV aGvHD OR B. Failure to achieve at a minimum PR based on organ assessment after 7 days, compared with organ stage at the initiation of high-dose systemic corticosteroids ± CNIs for the treatment of grade II to IV aGvHD, OR C. Failed corticosteroid taper, defined as the fulfillment of 1 of the following criteria: 1. requirement for an increase in the corticosteroid dose to methylprednisolone ≥ 2 mg/kg per day (or equivalent prednisone dose of ≥ 2.5 mg/kg per day) OR 2. failure to taper the methylprednisolone dose to < 0.5 mg/kg per day (or equivalent prednisone dose of < 0.6 mg/kg per day) for a minimum 7 days. |
A. Progression based on organ assessment after 3 days, compared with organ stage at the initiation of high-dose systemic corticosteroids ± CNIs treatment OR B. Failure to achieve aGvHD improvement based on organ assessment after 7 days (decrease in stage in at least 1 involved organ system) after 7 days, compared with organ stage at the initiation of high-dose systemic corticosteroids ± CNIs OR C. Failure of corticosteroid taper, defined as either: 1. patients who cannot tolerate a corticosteroid taper (i.e., patients who begin corticosteroids at 2.0 mg/kg per day, demonstrate a response, but progress before a 50% decrease from the initial starting dose of corticosteroids is achieved) 2. patients who previously began corticosteroid therapy at a lower dose (at least 1 mg/kg methylprednisolone) for the treatment of skin GvHD or skin GvHD accompanied by upper GI GvHD who develop new GvHD in another organ system. |
Exclusion criteria |
|
|
Drugs | ||
Intervention | Ruxolitinib
Treatment with ruxolitinib started on day of randomization (day 1) and continued until week 24, unless patient required additional systemic therapy for aGvHD progression and/or lack of response at day 28 or crossover day 28, development of cGvHD, recurrence or relapse of underlying disease, unacceptable toxicity, death, or study withdrawal. In the first 28 days, patients who met the criteria for aGvHD disease progression, had mixed response, or no response could move to a new systemic chosen by the investigator. Requirement of a new systemic therapy was considered a treatment failure. Taper: Starting no earlier than day 56, tapering of ruxolitinib could be attempted as needed at the time of documented CR or PR, conditional on patients being off corticosteroids. Tapering consisted of a 50% dose reduction (from 10 mg to 5 mg twice daily) every 2 months (56 days). For patients who experienced no worsening of aGvHD signs and symptoms, ruxolitinib was tapered with a second 50% dose reduction, to 5 mg once daily for an additional 2 months (56 days), before ruxolitinib discontinuation. Taper should be completed no later than week 24 unless prolonged tapering was indicated because of an aGvHD flare or other safety concern, in which case taper of ruxolitinib had to be started no later than week 24 and be completed by no later than the end of the study for the patient (up to approximately 2 years from randomization). | Ruxolitinib
Taper: Starting no earlier than day 180, tapering of ruxolitinib could be attempted conditional on patients having achieved CR or VGPR and corticosteroids having been discontinued for at least 8 weeks. |
Comparator(s) | Investigator’s choice of BATh BAT consisted of ATG, ECP, MSCs, MTX, MMF, mTOR inhibitors (everolimus or sirolimus), etanercept, or infliximab. No other types or combinations of BAT were permitted in this study.
During the treatment period, patients randomized to BAT may be eligible to crossover and receive ruxolitinib between day 28 and week 24 if they: 1. failed to meet the primary end point response definition (CR or PR) at day 28 OR 2. lost the response thereafter AND met criteria for progression, mixed response, or no response, necessitating new additional systemic immunosuppressive treatment for aGvHD. AND 3. did not have signs or symptoms of cGvHD (overlap syndrome, progressive, or de novo cGvHD). The crossover EOT visit was at crossover week 24, or earlier if the patient met any of the criteria for discontinuation of study treatment.i | NA |
Standard of care: corticosteroids and CNIs | Use of systemic corticosteroids, CNIs (cyclosporine or tacrolimus), and topical corticosteroid therapy was permitted, per institutional guidelines. Patients were allowed to receive other systemic medication for aGvHD after randomization only if used for aGvHD prophylaxes (i.e., started before the diagnosis of aGvHD). Taper:
| Use of anti-infective medications, GvHD prophylaxis medications (including CNIs), transfusion support, and topical steroid therapy was permitted. Administration of either oral prednisone or IV methylprednisolone for corticosteroid treatment could begin at the investigator's discretion. CNIs or other systemic medications for aGVHD could be continued only if used as aGvHD prophylaxis (i.e., started before the diagnosis of aGvHD). Taper:
|
Outcomes | ||
Primary end point | ORR at day 28 | ORR at day 28 |
Secondary and exploratory end points | Secondary:
Safety: Safety and tolerability were assessed by monitoring the frequency, duration, and severity of AEs (including the occurrence of any second primary malignancy or infection) and by performing physical exams and evaluating changes in vital signs from baseline, routine serum chemistry, hematology results, and coagulation profile Exploratory:
| Secondary:
Safety: Safety was assessed by monitoring the frequency and severity of AEs; measuring vital signs, monitoring ECOG PS; and performing physical examinations, 12-lead ECGs, and clinical laboratory assessments Exploratory:
|
Notes | ||
Publicationsi | Zeiser et al. (2020)10; primary analysis (data cut-off date of July 25, 2019) | Jagasia et al. (2020)44; final analysis (data cut-off date of July 2, 2018) |
AE = adverse event; aGvHD = acute graft-versus-host disease; alloSCT = allogeneic stem cell transplant; ANC = absolute neutrophil count; ATG = antithymocyte globulin, BAT = best available therapy; cGVHD = chronic graft-vs.-host disease; CMV = cytomegalovirus; CNI = calcineurin inhibitor; CR = complete response; DLI = donor lymphocyte infusion; DOR = duration of response; ECG = electrocardiogram; ECOG PS = Eastern Cooperative Oncology Group performance status; ECP = extracorporeal photopheresis; EFS = event-free survival; EOT = end of treatment; EQ-5D-5L = 5-level EQ-5D; FACT-BMT = Functional Assessment of Cancer Therapy-Bone Marrow Transplant; GI = gastrointestinal; GvHD = graft-versus-host disease; ICU = intensive care unit; JAK = Janus-associated kinase; MAGIC = Mount Sinai Acute GvHD Consortium; MMF = mycophenolate mofetil; MSC = mesenchymal stromal cell; mTOR = mechanistic (formerly mammalian) target of rapamycin; MTX = methotrexate; NA = not applicable; NIH = National Institutes of Health; NRM = nonrelapse mortality; ORR = overall response rate; PR = partial response; RCT = randomized controlled trial; SR = steroid refractory; vs. = versus.
aRecipients of nonmyeloablative, myeloablative, and reduced-intensity conditioning are eligible.
bRecipients of nonmyeloablative and myeloablative transplants are eligible.
cUse of growth factor supplementation and transfusion support was allowed during the trial. Evident myeloid and platelet engraftment was to be confirmed in the 48 hours before study treatment start.
dUse of growth factor supplementation and transfusion support was allowed during the trial.
eActive uncontrolled infection included significant bacterial, fungal, viral, or parasitic infection requiring treatment. Infections are considered controlled if appropriate therapy has been instituted and, at the time of screening, no signs of progression are present. Progression of infection is defined as hemodynamic instability attributable to sepsis, new symptoms, worsening physical signs, or radiographic findings attributable to infection. Persisting fever without other signs or symptoms will not be interpreted as progressing infection.
fAn active uncontrolled infection is defined as hemodynamic instability attributable to sepsis or new symptoms, worsening physical signs, or radiographic findings attributable to infection. Persistent fever without signs or symptoms will not be interpreted as an active uncontrolled infection.
gInfections are considered controlled if appropriate therapy has been instituted and, at the time of screening, no signs of progression are present. Progression of infection is defined as hemodynamic instability attributable to sepsis, new symptoms, worsening physical signs, or radiographic findings attributable to infection. Persist fever without other signs or symptoms will not be interpreted as progressing infection.
hConcomitant use of CNIs and steroids is allowed. Medications used for aGvHD prophylaxis (i.e., started before the diagnosis of aGvHD) that failed to prevent aGvHD in a patient before randomization could not be selected as BAT in the same patient.18
iPatients who met crossover criteria and received ruxolitinib could continue corticosteroids and CNIs for aGvHD treatment, per standard of care, with discontinuation required of any other systemic immunosuppressive treatment before crossover, unless it was used for aGvHD prophylaxis (i.e., started before the diagnosis of aGvHD).
jTwo additional reports were included: the clinical study reports for the REACH 218 and REACH 119 trials from the submission to CADTH.
Sources: Zeiser et al. (2020),10 Clinical Study Report (Reach 2),18 Clinical Study Report (REACH 1),19 Jagasia et al. (2020),44 ClinicalTrials.gov (REACH 1),45 ClinicalTrials.gov (REACH 2),20 Protocol (REACH 2),3 Protocol (REACH 1),3 Statistical analysis plan (REACH 1),3 additional information request.21
Description of Studies
REACH 2
The REACH 2 trial is a completed, international, multi-centre, open-label, randomized, phase III trial comapring ruxolitinib (10 mg administered orally twice daily) with investigator choice of BAT — i.e., ATG, ECP, MSCs, methotrexate, MMF, mTOR inhibitors everolimus or sirolimus), etanercept, or infliximab — in patients 12 years and older with grade II to IV SR-aGvHD. Patients continued to receive their systemic immunosuppressive regimen of corticosteroids with or without CNIs. Staging of aGvHD was based on the NIH criteria of Harris et al. (2016)16 (see Table 46 in Appendix 3 for NIH consensus criteria for aGvHD grading). This CADTH review is based on data from the primary analysis (July 25, 2019), the updated secondary analysis (January 6, 2020), and the final analysis (April 23, 2021). The final analysis was conducted once all patients had completed the study. The REACH 2 trial was sponsored by Novartis.3
The primary end point of the trial was the ORR at day 28. Patients in this international trial were randomized at 105 sites in 22 countries, which are listed in Table 6. The majority of sites were in Europe. REACH 2 enrolled 14 patients who were randomized at 8 Canadian sites. A total of 309 patients were randomized (using an interactive voice response system) in a 1:1 ratio to receive ruxolitinib or BAT. Enrolment occurred between April 12, 2017 and May 30, 2019.10 Randomization was stratified by aGvHD grade (grade II versus grade III versus grade IV).18
The study design is depicted in Figure 2. The study consisted of 4 main periods: the screening period (lasting for 28 days), the treatment period (day 1 to week 24 or EOT), the long-term follow-up period (from EOT to month 24), and the safety follow-up.18 The end of the study was to occur when all patients had completed month 24 (i.e., the end of the long-term follow-up observation period), unless the patient withdrew consent. During the treatment period, the EOT visit was to occur at week 24, or earlier if the patient met any study treatment discontinuation criteria.18
The primary end point, ORR, was assessed at day 28. Between day 28 and week 24, patients in the BAT group could cross over to the ruxolitinib group if they failed to meet the primary end point or lost their response after having achieved ORR and met the criteria for progression, mixed response, or no response, requiring new additional systemic immunosuppressive treatment for aGvHD, and did not show any signs or symptoms of cGvHD. During the randomized treatment period, aGvHD disease and safety assessments were planned every week from day 1 to day 56, and every 28 days thereafter until week 24.3
Figure 2: Study Design of REACH 2 Trial
aGvHD = acute graft-versus-host disease; BAT = best available therapy; EOT = end of treatment; ICF = informed consent form; SR = steroid refractory.
Source: Protocol (REACH 2).3
REACH 1
The REACH 1 trial is a completed, open-label, single-arm, multi-centre phase II trial that evaluated the efficacy and safety of ruxolitinib in combination with corticosteroids in patients with grade II to IV SR-aGvHD. The severity grading of aGvHD was based on the NIH criteria of Harris et al. (2016)16 (see Table 46 in Appendix 3). This CADTH review is based on the final data cut-off date of June 5, 2019, and the study was completed on June 5, 2019.21 The REACH trial was sponsored by Incyte-Corporation.19
The primary end point was ORR at day 28. Patients in this multi-centre trial were enrolled at 26 centres in 17 American states. A total of 71 patients were enrolled to received ruxolitinib (5 mg orally twice daily; if hematologic parameters were stable and no treatment-related toxicity was observed after the first 3 days of treatment, the dose could be increased to 10 mg orally twice daily). Enrolment occurred from December 2016 to December 2017.19
The study consisted of 3 phases: the screening phase (lasting up to 28 days), the treatment phase, and the follow-up phase. During the follow-up, patients were followed for safety (final follow-up was 30 to 35 days after EOT) and for survival until death, withdrawal of consent, or the end of the study, whichever occurred first. The end of the study was to occur when 75% of patients achieved 2-year NRM, the patient died, or the patient was lost to follow-up.19
Assessments for aGvHD staging were performed weekly for the first 8 weeks after enrolment, every 28 days thereafter, on days 100, 180, and 365, and at the EOT visit.19 Safety was evaluated from screening though to 30 to 35 days after EOT by monitoring the incidence and severity of AEs and serious adverse events (SAEs).3
Populations
Inclusion and Exclusion Criteria
The key inclusion and exclusion criteria used in the REACH 2 and REACH 1 trials are described in Table 6.
Briefly, the REACH 2 trial enrolled male or female patients 12 years and older who had undergone alloSCT, had evidence of myeloid and platelet engraftment (ANC > 1,000/mm3 and platelet count > 20,000/mm3), and were diagnosed with grade II to IV aGvHD, as defined by the NIH consensus criteria, and which was determined to be corticosteroid-refractory per protocol-defined criteria.
The REACH 1 trial had, overall, similar inclusion criteria. However, corticosteroid refractoriness (criterion C in Table 4) included a slight variation, in that failure to taper corticosteroids in REACH 1 included patients who began corticosteroids at a lower dose (at least 1 mg/kg methylprednisolone) but developed new GvHD, as opposed to patients who failed to taper the methylprednisolone dose to less than 0.5 mg/kg per day (or equivalent prednisone dose < 0.6 mg/kg per day) for a minimum 7 days in REACH 2. The clinical experts consulted by CADTH noted that criteria A and B are most commonly used in clinical practice and were consistent in REACH 2 and REACH 1. According to the clinical experts, the differences in criterion C would be unlikely influence overall study results. There was a slight variation in the definition of engraftment; patients in REACH 1 did not require adequate platelet engraftment, but patients in REACH 2 did. Patients in both trials required evidence of myeloid engraftment.
Both studies excluded patients who had received more than 1 systemic treatment for SR-aGvHD, presented with a clinical presentation resembling de novo overlap syndrome (defined by Jagasia et al. [2015]17) or active uncontrolled infection. REACH 2 explicitly excluded patients with multi-focal leuko-encephalopathy, whereas REACH 1 did not.
Baseline Characteristics
The demographic characteristics and disease and alloSCT history at baseline of patients in the REACH 2 trial are summarized in Table 7, Table 8, Table 9, and Table 10.
REACH 2
The mean ages for the ruxolitinib and BAT groups were, respectively, 48.1 (SD = 16.30) and 50.9 (SD = 14.9) years. The majority of patients (ruxolitinib versus BAT) were 18 to 65 years of age (83.1% versus 81.3%), but there were a few patients 12 to 18 years or younger (3.2% versus 2.6%). Most patients were male (59.7% versus 58.7%) and identified as White (72.1% versus 65.8%). The most common primary diagnosis was malignant leukemia or myelodysplastic syndrome (83.8% versus 78.1%), and most patients had received a peripheral blood alloSCT (87.0% versus 76.1%). The aGvHD grade at baseline was mostly grade III (44.2% versus 43.9%), followed by grade II (32.5% versus 34.8%) and grade IV (19.5% versus 20.6%). The most common criterion for SR-aGvHD was failure to achieve a response after 7 days (46.8% versus 40.6%), followed by failure on steroid taper (30.5% versus 31.6%), and progression after at least 3 days (22.7% versus 27.7%).18
In terms of prior aGvHD therapy, most patients had received steroids plus CNIs (49.4% versus 49.0%), followed by steroids plus CNIs plus another systemic aGvHD treatment (37.0% versus 31.6%). The ruxolitinib group had a higher proportion of patients who received steroids plus CNIs plus an aGvHD prophylaxis as prior therapy (26.6% versus 19.4%) and patients with aGvHD organ involvement at baseline in the skin (60.4% versus 47.7%) and liver (24.0% versus 16.1%), and a lower proportion of patients with aGvHD organ involvement at baseline in upper GI (18.2% versus 23.9%) and lower GI (62.3% versus 74.2%). Patients in the ruxolitinib group had a longer mean time from diagnosis of underlying disease to screening (2.16 years versus 1.72 years) and from diagnosis of underlying disease to transplant (713.07 days versus 553.29 days).18
Table 7: Summary of Demographic Baseline Characteristics in REACH 2, Full Analysis Set
Characteristic | Ruxolitinib N = 154 | BAT N = 155 | All patients N = 309 |
|---|---|---|---|
Age, years | |||
n | 154 | 155 | 309 |
Mean (SD) | 48.1 (16.30) | 50.9 (14.97) | 49.5 (15.69) |
Median | 52.5 | 54.0 | 54.0 |
Age category, n (%) | |||
12 to < 18 years | 5 (3.2) | 4 (2.6) | 9 (2.9) |
18 to 65 years | 128 (83.1) | 126 (81.3) | 254 (82.2) |
> 65 years | 21 (13.6) | 25 (16.1) | 46 (14.9) |
Sex, n (%) | |||
Female | 62 (40.3) | 64 (41.3) | 126 (40.8) |
Male | 92 (59.7) | 91 (58.7) | 183 (59.2) |
Race, n (%) | |||
White | 111 (72.1) | 102 (65.8) | 213 (68.9) |
Black or African American | 0 | 1 (0.6) | 1 (0.3) |
Asian | 19 (12.3) | 29 (18.7) | 48 (15.5) |
Other | 8 (5.2) | 4 (2.6) | 12 (3.9) |
Unknown | 16 (10.4) | 19 (12.3) | 35 (11.3) |
Weight, kg | |||
n | 150 | 152 | 302 |
Mean (SD) | 67.5 (14.04) | 66.2 (14.78) | 66.9 (14.41) |
Height, cm | |||
n | 148 | 144 | 292 |
Mean (SD) | 169.7 (9.86) | 170.0 (10.16) | 169.9 (9.99) |
Body mass index, kg/m2 | |||
n | 146 | 142 | 288 |
Mean (SD) | 23.4 (4.24) | 22.7 (4.15) | 23.1 (4.20) |
BAT = best available therapy; SD = standard deviation.
Source: Clinical Study Report (REACH 2).18
Table 8: Summary of Disease History by Treatment in REACH 2, Full Analysis Set
Disease history | Ruxolitinib N = 154 | BAT N = 155 | All patients N = 309 |
|---|---|---|---|
Primary diagnosis classification, n (%) | |||
Malignant leukemia or MDS | 129 (83.8) | 121 (78.1) | 250 (80.9) |
Malignant lymphoproliferative | 18 (11.7) | 26 (16.8) | 44 (14.2) |
Nonmalignant | 1 (0.6) | 5 (3.2) | 6 (1.9) |
Other | 6 (3.9) | 3 (1.9) | 9 (2.9) |
Diagnosis of underlying malignant disease, n (%) | |||
Acute lymphoblastic leukemia | 25 (16.2) | 16 (10.3) | 41 (13.3) |
Acute myelogenous leukemia | 58 (37.7) | 63 (40.6) | 121 (39.2) |
Chronic myelogenous leukemia | 6 (3.9) | 2 (1.3) | 8 (2.6) |
Excess blasts 2, developed from Fanconi syndrome | 1 (0.6) | 0 | 1 (0.3) |
Hodgkin’s lymphoma | 6 (3.9) | 2 (1.3) | 8 (2.6) |
Multiple myeloma | 2 (1.3) | 5 (3.2) | 7 (2.3) |
MDS | 26 (16.9) | 29 (18.7) | 55 (17.8) |
Non-Hodgkin’s lymphoma | 9 (5.8) | 19 (12.3) | 28 (9.1) |
Other acute leukemia | 4 (2.6) | 3 (1.9) | 7 (2.3) |
Other leukemia | 6 (3.9) | 8 (5.2) | 14 (4.5) |
Other | 4 (2.6) | 0 | 4 (1.3) |
Diagnosis of underlying nonmalignant disease, n (%) | |||
Histiocytic disorders | 0 | 1 (0.6) | 1 (0.3) |
Sickle cell disease | 1 (0.6) | 1 (0.6) | 2 (0.6) |
Other | 0 | 3 (1.9) | 3 (1.0) |
Diagnosis of underlying disease, other, n (%) | |||
Blastic neoplasm of plasmacytoid dendritic cells | 0 | 1 (0.6) | 1 (0.3) |
Multiple myeloma and secondary acute myeloid leukemia | 0 | 1 (0.6) | 1 (0.3) |
Myelofibrosis | 2 (1.3) | 0 | 2 (0.6) |
Myeloma | 0 | 1 (0.6) | 1 (0.3) |
Myeloproliferative neoplasm | 1 (0.6) | 0 | 1 (0.3) |
Post-PV myelofibrosis | 1 (0.6) | 0 | 1 (0.3) |
Primary myelofibrosis | 1 (0.6) | 0 | 1 (0.3) |
Septic granulomatosis | 1 (0.6) | 0 | 1 (0.3) |
Time from diagnosis of underlying disease to screening, years | |||
n | 154 | 154 | 308 |
Mean (SD) | 2.16 (3.2) | 1.72 (2.2) | 1.94 (2.7) |
Median | 1.04 | 0.86 | 0.94 |
Range | 0.2 to 25.7 | 0.2 to 15.1 | 1 0.2 to 25.7 |
CIBMTR risk assessment, n (%) | |||
Low | 46 (29.9) | 46 (29.7) | 92 (29.8) |
Intermediate | 43 (27.9) | 48 (31.0) | 91 (29.4) |
High | 61 (39.6) | 55 (35.5) | 116 (37.5) |
Unknown | 4 (2.6) | 6 (3.9) | 10 (3.2) |
BAT = best available therapy; CIBMTR = Center for International Blood and Marrow Transplant Research; MDS = myelodysplastic syndrome; PV = polycythemia vera; SD = standard deviation.
Source: Clinical Study Report (REACH 2).18
Table 9: Summary of Transplant-Related History by Treatment in REACH 2, Full Analysis Set
Transplant-related history | Ruxolitinib N = 154 | BAT N = 155 | All patients N = 309 | |
|---|---|---|---|---|
Conditioning regimen type, n (%) | ||||
Myeloablative | 85 (55.2) | 65 (41.9) | 150 (48.5) | |
Nonmyeloablative | 31 (20.1) | 41 (26.5) | 72 (23.3) | |
Reduced intensity | 38 (24.7) | 49 (31.6) | 87 (28.2) | |
Total HCT-specific comorbidity index score, n (%) | ||||
0 | 70 (45.5) | 63 (40.6) | 133 (43.0) | |
1 | 30 (19.5) | 27 (17.4) | 57 (18.4) | |
2 | 24 (15.6) | 19 (12.3) | 43 (13.9) | |
3 | 9 (5.8) | 26 (16.8) | 35 (11.3) | |
4 | 12 (7.8) | 6 (3.9) | 18 (5.8) | |
≥ 5 | 6 (3.9) | 6 (3.9) | 12 (3.9) | |
Missing | 3 (1.9) | 8 (5.2) | 11 (3.6) | |
Time from diagnosis of underlying disease to transplant, days | ||||
n | 154 | 154 | 308 | |
Mean (SD) | 713.07 (1,156.4) | 553.29 (786.1) | 633.18 (990.4) | |
Median | 276.00 | 213.50 | 234.00 | |
Range | 45.0 to 9,003.0 | 28.0 to 5,426.0 | 28.0 to 9,003.0 | |
Time from transplant to randomization, days | ||||
n | 154 | 155 | 309 | |
Mean (SD) | 84.34 (71.9) | 81.52 (66.8) | 82.93 (69.3) | |
Median | 58.50 | 52.00 | 56.00 | |
Range | 14.0 to 386.0 | 14.0 to 439.0 | 14.0 to 439.0 | |
Stem cell type, n (%) | ||||
Bone marrow | 19 (12.3) | 30 (19.4) | 49 (15.9) | |
Peripheral blood | 134 (87.0) | 118 (76.1) | 252 (81.6) | |
Single cord blood | 1 (0.6) | 7 (4.5) | 8 (2.6) | |
CMV status at time of transplant, n (%) | ||||
Negative | 73 (47.4) | 68 (43.9) | 141 (45.6) | |
Positive | 81 (52.6) | 87 (56.1) | 168 (54.4) | |
BAT = best available therapy; CMV = cytomegalovirus; HCT = hematopoietic cell transplantation; SD = standard deviation.
Source: Clinical Study Report (REACH 2).18
Table 10: Summary of aGvHD Disease History by Treatment in REACH 2, Full Analysis Set
aGvHD disease history | Ruxolitinib N = 154 | BAT N = 155 | All patients N = 309 |
|---|---|---|---|
Time from transplant to diagnosis of aGvHD grade ≥ II, days | |||
n | 154 | 155 | 309 |
Mean (SD) | 54.77 (62.8) | 58.25 (58.7) | 56.52 (60.7) |
Median | 33.50 | 34.00 | 34.00 |
Minimum to maximum | 3.0 to 380.0 | 6.0 to 412.0 | 3.0 to 412.0 |
Time from diagnosis of aGvHD grade ≥ II to randomization, days | |||
n | 154 | 155 | 309 |
Mean (SD) | 29.57 (43.6) | 23.26 (31.4) | 26.41 (38.0) |
Median | 14.00 | 14.00 | 14.00 |
Minimum to maximum | 2.0 to 332.0 | 1.0 to 293.0 | 1.0 to 332.0 |
aGvHD grade at diagnosis of grade ≥ II, n (%) | |||
Grade II | 68 (44.2) | 74 (47.7) | 142 (46.0) |
Grade III | 68 (44.2) | 62 (40.0) | 130 (42.1) |
Grade IV | 18 (11.7) | 19 (12.3) | 37 (12.0) |
SR-aGvHD criteria met, n (%) | |||
Progression after at least 3 days | 35 (22.7) | 43 (27.7) | 78 (25.2) |
Failure to achieve a response after 7 days | 72 (46.8) | 63 (40.6) | 135 (43.7) |
Failure on steroid taper | 47 (30.5) | 49 (31.6) | 96 (31.1) |
Time from SR-aGvHD to randomization, day | |||
n | 154 | 155 | 309 |
Mean (SD) | 3.38 (5.9) | 3.14 (5.1) | 3.26 (5.5) |
Median | 1.00 | 1.00 | 1.00 |
Range | 0.0 to 47.0 | 0.0 to 24.0 | 0.0 to 47.0 |
Time from diagnosis of aGvHD grade ≥ II to SR-aGvHD, day | |||
n | 154 | 155 | 309 |
Mean (SD) | 26.19 (43.1) | 20.13 (30.8) | 23.15 (37.5) |
Median | 11.00 | 10.00 | 10.00 |
Range | 1.0 to 331.0 | 1.0 to 293.0 | 1.0 to 331.0 |
Overall aGvHD grade at randomization, n (%) | |||
Grade 0 | 4 (2.6) | 1 (0.6) | 5 (1.6) |
Grade I | 2 (1.3) | 0 | 2 (0.6) |
Grade II | 50 (32.5) | 54 (34.8) | 104 (33.7) |
Grade III | 68 (44.2) | 68 (43.9) | 136 (44.0) |
Grade IV | 30 (19.5) | 32 (20.6) | 62 (20.1) |
aGvHD organ involvement at randomization, n (%) | |||
Skin | 93 (60.4) | 74 (47.7) | 167 (54.0) |
Liver | 36 (23.4) | 26 (16.8) | 62 (20.1) |
Upper GI | 28 (18.2) | 37 (23.9) | 65 (21.0) |
Lower GI | 96 (62.3) | 115 (74.2) | 211 (68.3) |
Missing | 4 (2.6) | 1 (0.6) | 5 (1.6) |
Steroid dose at randomization, mg/day | |||
n | 149 | 151 | 300 |
Mean (SD) | 132.29 (90.9) | 126.50 (73.1) | 129.37 (82.3) |
Median | 120.00 | 120.00 | 120.00 |
Range | 16.0 to 1,000.0 | 16.0 to 680.0 | 16.0 to 1,000.0 |
Prior aGvHD therapy, n (%) | |||
Steroids only | 12 (7.8) | 18 (11.6) | 30 (9.7) |
Steroids + CNIs | 76 (49.4) | 76 (49.0) | 152 (49.2) |
Steroids + CNIs + other systemic aGvHD treatment | 57 (37.0) | 49 (31.6) | 106 (34.3) |
Steroids + CNIs + only aGvHD prophylaxis | 42 (27.3) | 30 (19.4) | 72 (23.3) |
Steroids + CNIs + only aGvHD treatment | 8 (5.2) | 12 (7.7) | 20 (6.5) |
Steroids + CNIs + both aGvHD prophylaxis and treatment | 7 (4.5) | 7 (4.5) | 14 (4.5) |
Steroids + other systemic aGvHD treatment | 9 (5.8) | 12 (7.7) | 21 (6.8) |
Steroids + only aGvHD prophylaxis | 8 (5.2) | 8 (5.2) | 16 (5.2) |
Steroids + only aGvHD treatment | 1 (0.6) | 4 (2.6) | 5 (1.6) |
aGvHD = acute graft -vs.-host disease; BAT = best available therapy; CNI = calcineurin inhibitors; GI = gastrointestinal; SD = standard deviation; SR = steroid refractory.
Source: Clinical Study Report (REACH 2).18
REACH 1
Patients’ demographic characteristics and disease and alloSCTs history at baseline in the REACH 1 trial were, overall, similar to those in the REACH 2 trial and are summarized in Table 11, Table 12, Table 13, and Table 14. As in the REACH 2 trial, the majority of patients in the REACH 1 trial were 18 to 65 years of age (81.7%), identified as White (93.0%), had received a peripheral blood alloSCT (80.3%), and the most common primary diagnosis was malignant leukemia or myelodysplastic syndrome (71.9%). The distribution of aGvHD grades was similar in the 2 trials, with grade III aGvHD in the majority of patients (46.5%), followed by grade II (31.0%) and grade IV (22.5%) in REACH 1. As in REACH 2 trial, the most common criterion for SR-aGvHD in REACH 1 was no aGvHD improvement after 7 days of primary treatment (40.8%), followed by failing corticosteroid taper (36.2%) and progression after 3 days or primary treatment (23.9%). The mean time from diagnosis of underlying disease to screening was similar in the 2 trials; it was 2.15 years in the REACH 1 trial, and 2.16 years versus 1.72 years in the ruxolitinib and BAT groups in the REACH 2 trial, respectively. The mean time from transplant to randomization was 82.93 days in the REACH 2 trial and 110.8 days in the REACH 1 trial. Most patients in the 2 trials received grafts from identical HLA-matched donors (60.2% in the REACH 2 trial and 63.4% in the REACH 1 trial).19 Differences in patient’s baseline characteristics between the REACH 2 and REACH 1 trials were noted for the following categories. The mean age of patients was slightly higher in the REACH 1 trial, at 52.9 years (SD = 14.18), than in the overall REACH 2 trial population, at 49.5 years (SD = 15.69). No patient in the REACH 1 trial was younger than 18 years, but in the REACH 2 trial overall, 2.9% of patients were 12 years to younger than 18 years. Most patients in the REACH 2 trial were male (59.2%), compared with 49.3% in the REACH 1 trial.19
In terms of prior aGvHD therapy, as in the REACH 2 trial, all patients had received corticosteroids as prior anti-GvHD treatment. However, the percentage of patients who had additionally received prior CNIs as aGvHD treatment was lower in the REACH 1 trial (23.9%) than in the REACH 2 trial (50.0% in the ruxolitinib group and 49.0% in the BAT group). In the REACH 1 trial, all patients had received treatment for GvHD prophylaxis before enrolment, compared with 72.1% of patients in the REACH 2 trial. The REACH 1 trial had a higher proportion of patients who had received prior prophylactic treatment with CNIs (97.2%) than the REACH 2 trial (65.6% in the ruxolitinib group and 60.0% in the BAT group).19
Table 11: Summary of Demographic Baseline Characteristics in REACH 1, Efficacy-Evaluable Population
Characteristic | Ruxolitinib N = 71 |
|---|---|
Age, years | |
n | 71 |
Mean (SD) | 52.9 (14.18) |
Age category, n (%) | |
< 65 years | 58 (81.7) |
≥ 65 years | 13 (18.3) |
Sex, n (%) | |
Female | 36 (50.7) |
Male | 35 (49.3) |
Height, cm | |
n | 66 |
Mean (SD) | 170.2 (10.64) |
Weight, kg | |
n | 71 |
Mean (SD) | 78.64 (21.65) |
BMI, kg/m2 | |
n | 66 |
Mean (SD) | 26.83 (6.19) |
BSA, m2 | |
n | 66 |
Mean (SD) | 1.91 (0.30) |
ECOG PS at baseline, n (%) | |
0 | 3 (4.2) |
1 | 24 (33.8) |
2 | 25 (35.2) |
3 | 17 (23.9) |
4 | 1 (1.4) |
5 | 0 |
Missing | 1 (1.4) |
BMI = body mass index; BSA = body surface area; ECOG PS = Eastern Cooperative Oncology Group performance status; SD = standard deviation.
Source: Clinical Study Report (REACH 1).19
Table 12: Summary of Baseline aGvHD Disease Characteristics in REACH 1, Efficacy-Evaluable Population
Characteristic | Ruxolitinib N = 71 |
|---|---|
Skin rash, %BSA | |
Mean (SD) | 30.23 (35.31) |
Median | 2.0 |
Minimum to maximum | 0 to 100 |
Skin rash, stage, n (%) | |
Stage 0 | 35 (49.3) |
Stage 1 | 4 (5.6) |
Stage 2 | 7 (9.9) |
Stage 3 | 22 (31.0) |
Stage 4 | 3 (4.2) |
Lower GI, stage, n (%) | |
Stage 0 | 20 (28.2) |
Stage 1 | 11 (15.5) |
Stage 2 | 8 (11.3) |
Stage 3 | 19 (26.8) |
Stage 4 | 13 (18.3) |
Upper GI, stage, n (%) | |
Stage 0 | 49 (69.0) |
Stage 1 | 22 (31.0) |
Liver, stage, n (%) | |
Stage 0 | 55 (77.5) |
Stage 1 | 3 (4.2) |
Stage 2 | 5 (7.0) |
Stage 3 | 7 (9.9) |
Stage 4 | 1 (1.4) |
MAGIC criteria grade, n (%) | |
Grade 0 | 0 |
Grade I | 0 |
Grade II | 22 (31.0) |
Grade III | 33 (46.5) |
Grade IV | 16 (22.5) |
SR criteria, n (%) | |
Progressive aGvHD after 3 days of primary treatment | 17 (23.9) |
aGvHD that had not improved after 7 days of primary treatment | 29 (40.8) |
Previously began steroid therapy at a lower dose but developed new aGvHD in another organ system | 10 (14.1) |
Patients that cannot tolerate steroid taper | 15 (21.1) |
aGvHD = acute graft-versus-host disease; BSA = body surface area; GI = gastrointestinal; MAGIC = Mount Sinai Acute GvHD Consortium; SD = standard deviation; SR = steroid refractory.
Source: Clinical Study Report (REACH 1).19
Table 13: Summary of Cancer History in REACH 1, Efficacy-Evaluable Population
Characteristic | Ruxolitinib N = 71 |
|---|---|
Underlying malignancy, n (%) | |
AML | 20 (28.2) |
ALL | 8 (11.3) |
CLL | 3 (4.2) |
Lymphoma | 9 (12.7) |
MDS | 20 (28.2) |
Othera | 11 (15.5) |
Time since diagnosis of underlying malignancy, years | |
n | 71 |
Mean (SD) | 2.15 (3.29) |
Median | 1.08 |
Minimum to maximum | 0.3 to 26.3 |
Disease status at the time of transplant, n (%) | |
CR | 50 (70.4) |
PR | 8 (11.3) |
SD | 5 (7.0) |
Relapsed or refractory | 6 (8.5) |
Unknown | 2 (2.8) |
ALL = acute lymphoblastic leukemia; AML = acute myeloid leukemia; CLL = chronic lymphocytic leukemia; CR = complete response; MDS = myelodysplastic syndrome; PR = partial response; SD = standard deviation.
a“Other” includes multiple myeloma, mixed B-cell myeloid acute leukemia, unclassifiable acute leukemia, diffuse large B-cell lymphoma (with subsequent T-cell prolymphocytic leukemia), chronic myelogenous leukemia, chronic myelomonocytic leukemia, plasma cell leukemia, essential thrombocythemia (with secondary myelofibrosis), and myeloproliferative neoplasm.
Source: Clinical Study Report (REACH 1).19
Table 14: Summary of alloSCT History in REACH 1, Efficacy-Evaluable Population
Characteristic | Ruxolitinib N = 71 |
|---|---|
Type of allogeneic transplant, n (%) | |
Bone marrow | 13 (18.3) |
PBSC | 57 (80.3) |
Cord blood | 1 (1.4) |
Other | 0 (0.0) |
Time since stem cell transplant, daysa | |
n | 71 |
Mean (SD) | 110.8 (83.1) |
Median | 74 |
Range | 25 to 357 |
Best response at time of transplant, n (%) | |
Complete remission | 48 (67.6) |
Active disease | 14 (19.7) |
Other | 9 (12.7) |
Time since diagnosis of GvHD, daysb | |
n | 71 |
Mean (SD) | 40.1 (56.27) |
Median | 17.0 |
Minimum to maximum | 4 to 286 |
Donor source, n (%) | |
Identical | 0 |
Sibling | 26 (36.6) |
Related (nonsibling) | 3 (4.2) |
Unrelated | 41 (57.7) |
Other | 1 (1.4) |
Donor CMV status, n (%) | |
Positive | 31 (43.7) |
Negative | 39 (54.9) |
Missing | 1 (1.4) |
Patient CMV status, n (%) | |
Positive | 41 (57.7) |
Negative | 30 (42.3) |
HLA matching, n (%) | |
Identical | 45 (63.4) |
Haploidentical | 13 (18.3) |
Mismatched | 13 (18.3) |
alloSCT = allogeneic stem cell transplant; CMV = cytomegalovirus; GvHD = graft-versus-host disease; HLA = human leukocyte antigen; PBSC = peripheral blood stem cell; SD = standard deviation.
aTime since stem cell transplant = first date of ruxolitinib – transplant date + 1.
bTime since diagnosis of GVHD = first date of ruxolitinib – GVHD diagnosis date + 1.
Source: Clinical Study Report (REACH 1).19
Prior aGvHD Treatment
REACH 2
Almost all patients in the full analysis set received treatment for aGvHD before enrolment in the trial, and the proportions and types of therapies were balanced across groups, as summarized in Table 15.
The most common prior systemic treatments (ruxolitinib versus BAT) were glucocorticoids (mainly methylprednisolone, prednisolone, prednisone, and methylprednisolone sodium succinate) (96.8% versus 96.1%), followed by corticosteroids (weak; mainly methylprednisolone, prednisolone, and methylprednisolone sodium succinate) (87.7% versus 89.0%;), corticosteroids (plain; mainly methylprednisolone, prednisolone, and prednisone) (87.0% versus 80.6%), and corticosteroid combinations for the treatment of acne (mainly methylprednisolone and methylprednisolone sodium succinate) (68.2% versus 76.1%). The most commonly used CNIs (50.6% versus 49.0%) included ciclosporin (37.0% versus 27.7%) and tacrolimus (14.9% versus 21.3%).18
The sponsor was asked for clarification on the number of patients in the REACH 2 trial who had an inadequate response to corticosteroids, an inadequate response to other systemic therapies, or an inadequate response to corticosteroids and other systemic therapies, and noted that such data are not available.22 Because an inadequate response to corticosteroids was an eligibility criterion in the REACH 2 trial, it follows that all patients had an inadequate response to corticosteroids. However, the proportion of patients who had an inadequate response to other systemic therapies in addition to having an inadequate response to corticosteroids remains unclear (for more details, refer to the REACH 2 internal validity section in Critical Appraisal).
REACH 1
As in the REACH 2 trial, all patients in the REACH 1 trial received prior systemic therapy for aGvHD (Table 16), and all patients received prior systemic therapy with glucocorticoids (81.7% of patients received methylprednisolone and 57.7% received prednisone). The number of patients who received CNIs (predominantly tacrolimus [19.7%], with or without methotrexate) as prior systemic therapy was lower in the REACH 1 trial (23.9%) than in the REACH 2 trial.19
The sponsor was asked for clarification on the number of patients in the REACH 1 trial who had an inadequate response to corticosteroids, an inadequate response to other systemic therapies, or an inadequate response to corticosteroids and other systemic therapies, and reported that 42 patients were refractory to steroids alone and 29 patients were refractory to steroids and 1 additional systemic therapy (receipt of 1 systemic treatment in addition to corticosteroids [± CNIs] for aGvHD was allowed in the REACH 1 trial).22 The sponsor was asked about the specific types of additional systemic therapies received by the 29 patients in the REACH 1 trial who were refractory to 1 additional systemic therapy, but no information beyond that in Table 16 was provided.22
Table 15: Prior aGvHD Treatment in REACH 2, Full Analysis Set
ATC class, preferred term | Ruxolitinib N = 154 | BAT N = 155 |
|---|---|---|
Any ATC class, n (%) | ||
Total | 152 (98.7) | 149 (96.1) |
ATC not coded, n (%) | ||
Total | 0 | 3 (1.9) |
ECP | 0 | 2 (1.3) |
MSCs | 0 | 1 (0.6) |
Drugs for dermatitis, excluding corticosteroids, n (%) | ||
Total | 24 (15.6) | 33 (21.3) |
Tacrolimus | 23 (14.9) | 33 (21.3) |
Tacrolimus monohydrate | 1 (0.6) | 0 |
Bile acid preparations, n (%) | ||
Total | 1 (0.6) | 0 |
Ursodeoxycholic acid | 1 (0.6) | 0 |
CNIs, n (%) | ||
Total | 78 (50.6) | 76 (49.0) |
Ciclosporin | 57 (37.0) | 43 (27.7) |
Tacrolimus | 23 (14.9) | 33 (21.3) |
Tacrolimus monohydrate | 1 (0.6) | 0 |
Corticosteroids, n (%) | ||
Total | 46 (29.9) | 36 (23.2) |
Hydrocortisone sodium succinate | 1 (0.6) | 0 |
Prednisolone | 42 (27.3) | 31 (20.0) |
Prednisolone sodium succinate | 4 (2.6) | 6 (3.9) |
Corticosteroids acting locally, n (%) | ||
Total | 70 (45.5) | 56 (36.1) |
Hydrocortisone sodium succinate | 1 (0.6) | 0 |
Prednisolone | 42 (27.3) | 31 (20.0) |
Prednisone | 32 (20.8) | 29 (18.7) |
Corticosteroids for local oral treatment, n (%) | ||
Total | 43 (27.9) | 31 (20.0) |
Hydrocortisone sodium succinate | 1 (0.6) | 0 |
Prednisolone | 42 (27.3) | 31 (20.0) |
Corticosteroids, combinations for treatment of acne, n (%) | ||
Total | 105 (68.2) | 118 (76.1) |
Methylprednisolone | 83 (53.9) | 85 (54.8) |
Methylprednisolone sodium succinate | 23 (14.9) | 39 (25.2) |
Corticosteroids, plain, n (%) | ||
Total | 134 (87.0) | 125 (80.6) |
Hydrocortisone sodium succinate | 1 (0.6) | 0 |
Methylprednisolone | 83 (53.9) | 85 (54.8) |
Prednisolone | 42 (27.3) | 31 (20.0) |
Prednisolone sodium succinate | 4 (2.6) | 6 (3.9) |
Prednisone | 32 (20.8) | 29 (18.7) |
Corticosteroids, potent (group III), n (%) | ||
Total | 83 (53.9) | 85 (54.8) |
Methylprednisolone | 83 (53.9) | 85 (54.8) |
Corticosteroids, weak (group I), n (%) | ||
Total | 135 (87.7) | 138 (89.0) |
Hydrocortisone sodium succinate | 1 (0.6) | 0 |
Methylprednisolone | 83 (53.9) | 85 (54.8) |
Methylprednisolone sodium succinate | 23 (14.9) | 39 (25.2) |
Prednisolone | 42 (27.3) | 31 (20.0) |
Prednisolone sodium succinate | 4 (2.6) | 6 (3.9) |
Glucocorticoids, n (%) | ||
Total | 149 (96.8) | 149 (96.1) |
Hydrocortisone sodium succinate | 1 (0.6) | 0 |
Methylprednisolone | 83 (53.9) | 85 (54.8) |
Methylprednisolone sodium succinate | 23 (14.9) | 39 (25.2) |
Prednisolone | 42 (27.3) | 31 (20.0) |
Prednisolone sodium succinate | 4 (2.6) | 6 (3.9) |
Prednisone | 32 (20.8) | 29 (18.7) |
Interleukin inhibitors, n (%) | ||
Total | 0 | 3 (1.9) |
Basiliximab | 0 | 3 (1.9) |
Other immunosuppressants, n (%) | ||
Total | 0 | 3 (1.9) |
Remestemcel-L | 0 | 3 (1.9) |
Other ophthalmologicals, n (%) | ||
Total | 57 (37.0) | 43 (27.7) |
Ciclosporin | 57 (37.0) | 43 (27.7) |
Protein kinase inhibitors | ||
Total | 1 (0.6) | 1 (0.6) |
Everolimus | 1 (0.6) | 1 (0.6) |
Selective immunosuppressants, n (%) | ||
Total | 10 (6.5) | 10 (6.5) |
ATG | 0 | 1 (0.6) |
ATG, rabbit | 1 (0.6) | 1 (0.6) |
Everolimus | 1 (0.6) | 1 (0.6) |
Mycophenolate mofetil | 7 (4.5) | 6 (3.9) |
Mycophenolate sodium | 1 (0.6) | 0 |
Mycophenolic acid | 0 | 1 (0.6) |
aGvHD = acute graft-versus-host disease; ATC = Anatomical Therapeutic Chemical; ATG = antithymocyte immunoglobulin; BAT = best available therapy; CNI = calcineurin inhibitor; ECP = extracorporeal photopheresis; MSC = mesenchymal stromal cell.
Note: A medication or therapy can appear in more than 1 ATC class.
Source: Clinical Study Report (REACH 2).18
Table 16: Prior aGvHD Treatment in REACH 1, Efficacy-Evaluable Population
Variable | Ruxolitinib N = 71 |
|---|---|
Participants who received prior anti-GvHD therapy, n (%) | 71 (100.0) |
Duration of prior corticosteroid exposure, daysa | |
N | 71 |
Mean (SD) | 31.9 (46.45) |
Median | 16.0 |
Range | 3.0 to 285.0 |
CNIs, n (%) | 17 (23.9) |
Ciclosporin | 3 (4.2) |
Tacrolimus | 14 (19.7) |
Corticosteroids acting locally, n (%) | 7 (9.9) |
Budesonide | 7 (9.9) |
Corticosteroids, moderately potent (group II), n (%) | 3 (4.2) |
Triamcinolone | 3 (4.2) |
Corticosteroids, very potent (group IV), n (%) | 1 (1.4) |
Clobetasol | 1 (1.4) |
Corticosteroids, weak (group I), n (%) | 1 (1.4) |
Hydrocortisone | 1 (1.4) |
Folic acid analogues, n (%) | 8 (11.3) |
Methotrexate | 7 (9.9) |
Methotrexate sodium | 1 (1.4) |
Glucocorticoids, n (%) | 71 (100.0) |
Beclometasone dipropionate | 7 (9.9) |
Methylprednisolone | 58 (81.7) |
Methylprednisolone sodium succinate | 7 (9.9) |
Prednisone | 41 (57.7) |
Gonadotropins, n (%) | 1 (1.4) |
Chorionic gonadotrophin | 1 (1.4) |
Interleukin inhibitors, n (%) | 1 (1.4) |
Basiliximab | 1 (1.4) |
Other antihistamines for systemic use, n (%) | 1 (1.4) |
Hydroxyzine hydrochloride | 1 (1.4) |
Other therapeutic products, n (%) | 1 (1.4) |
Other therapeutic products | 1 (1.4) |
Psoralens for systemic use, n (%) | 1 (1.4) |
Methoxsalen | 1 (1.4) |
Selective immunosuppressants, n (%) | 10 (14.1) |
Abatacept | 2 (2.8) |
ATG, rabbit | 1 (1.4) |
Mycophenolate mofetil | 3 (4.2) |
Sirolimus | 2 (2.8) |
Vedolizumab | 2 (2.8) |
Tumour necrosis factor alpha inhibitors, n (%) | 1 (1.4) |
Etanercept | 1 (1.4) |
aGvHD = acute graft-versus-host disease; ATG = antithymocyte immunoglobulin; CNI = calcineurin inhibitor; SD = standard deviation.
aDate of last exposure to corticosteroids as prior anti-GVHD therapy – date of first exposure to corticosteroids as prior anti-GVHD therapy + 1; includes methylprednisolone, methylprednisolone sodium succinate, and prednisone.
Source: Clinical Study Report (REACH 1).19
Prior aGvHD Prophylaxis Treatment
REACH 2
Approximately 2-thirds of patients (72.1%) in the full analysis set of the REACH 2 trial received aGvHD prophylaxis before enrolment into the trial, and the proportions and types of therapies were balanced across groups, as summarized in Table 17. The most common prior prophylactic treatments (ruxolitinib versus BAT) were CNIs (mainly ciclosporin) (65.6% versus 60.0%), followed by other ophthalmologicals (mainly ciclosporin) (56.5% versus 42.6%), glucocorticoids (mainly methylprednisolone) (19.5% versus 20.0%), and corticosteroids (plain; mainly methylprednisolone) (18.8% versus 13.5%).18
REACH 1
In the REACH 1 trial, all patients received GvHD prophylaxis before enrolment, compared with 72.1% of patients in the REACH 2 trial (see Table 18). The REACH 1 trial had a higher proportion of patients who received prior prophylactic treatment with CNIs (97.2%) than the REACH 2 trial (ruxolitinib versus BAT = 65.6% versus 60.0%). After CNIs, the most common prior prophylactic therapies in the REACH 1 trial were selective immunosuppressants (mainly MMF) (71.8%) and folic acid analogues (mainly methotrexate) (22.5%).19
Table 17: Prior aGvHD Prophylaxis Treatment in REACH 2, Full Analysis Set
ATC class, preferred term | Ruxolitinib N = 154 | BAT N = 155 |
|---|---|---|
Any ATC class, n (%) | ||
Total | 111 (72.1) | 100 (64.5) |
Drugs for dermatitis, excluding corticosteroids, n (%) | ||
Total | 19 (12.3) | 31 (20.0) |
Tacrolimus | 19 (12.3) | 31 (20.0) |
CNIs, n (%) | ||
Total | 101 (65.6) | 93 (60.0) |
Ciclosporin | 85 (55.2) | 64 (41.3) |
Tacrolimus | 19 (12.3) | 31 (20.0) |
Corticosteroids, n (%) | ||
Total | 7 (4.5) | 7 (4.5) |
Dexamethasone | 1 (0.6) | 1 (0.6) |
Prednisolone | 4 (2.6) | 5 (3.2) |
Prednisolone sodium succinate | 3 (1.9) | 1 (0.6) |
Corticosteroids acting locally, n (%) | ||
Total | 12 (7.8) | 9 (5.8) |
Prednisolone | 4 (2.6) | 5 (3.2) |
Prednisone | 9 (5.8) | 4 (2.6) |
Corticosteroids for local oral treatment, n (%) | ||
Total | 5 (3.2) | 6 (3.9) |
Dexamethasone | 1 (0.6) | 1 (0.6) |
Prednisolone | 4 (2.6) | 5 (3.2) |
Corticosteroids, combinations for treatment of acne, n (%) | ||
Total | 19 (12.3) | 24 (15.5) |
Dexamethasone | 1 (0.6) | 1 (0.6) |
Methylprednisolone | 17 (11.0) | 12 (7.7) |
Methylprednisolone sodium succinate | 1 (0.6) | 11 (7.1) |
Corticosteroids, moderately potent (group II), n (%) | ||
Total | 1 (0.6) | 1 (0.6) |
Dexamethasone | 1 (0.6) | 1 (0.6) |
Corticosteroids, plain, n (%) | ||
Total | 29 (18.8) | 21 (13.5) |
Dexamethasone | 1 (0.6) | 1 (0.6) |
Methylprednisolone | 17 (11.0) | 12 (7.7) |
Prednisolone | 4 (2.6) | 5 (3.2) |
Prednisolone sodium succinate | 3 (1.9) | 1 (0.6) |
Prednisone | 9 (5.8) | 4 (2.6) |
Corticosteroids, potent (group III), n (%) | ||
Total | 17 (11.0) | 12 (7.7) |
Methylprednisolone | 17 (11.0) | 12 (7.7) |
Corticosteroids, weak (group I), n (%) | ||
Total | 22 (14.3) | 26 (16.8) |
Methylprednisolone | 17 (11.0) | 12 (7.7) |
Methylprednisolone sodium succinate | 1 (0.6) | 11 (7.1) |
Prednisolone | 4 (2.6) | 5 (3.2) |
Corticosteroids, weak (group I), n (%) | ||
Prednisolone sodium succinate | 3 (1.9) | 1 (0.6) |
Folic acid analogues, n (%) | ||
Total | 7 (4.5) | 7 (4.5) |
Methotrexate | 6 (3.9) | 7 (4.5) |
Methotrexate sodium | 1 (0.6) | 0 |
Glucocorticoids, n (%) | ||
Total | 30 (19.5) | 31 (20.0) |
Dexamethasone | 1 (0.6) | 1 (0.6) |
Methylprednisolone | 17 (11.0) | 12 (7.7) |
Methylprednisolone sodium succinate | 1 (0.6) | 11 (7.1) |
Prednisolone | 4 (2.6) | 5 (3.2) |
Prednisolone sodium succinate | 3 (1.9) | 1 (0.6) |
Prednisone | 9 (5.8) | 4 (2.6) |
Nitrogen mustard analogues, n (%) | ||
Total | 2 (1.3) | 3 (1.9) |
Cyclophosphamide | 2 (1.3) | 3 (1.9) |
Other gynecologicals, n (%) | ||
Total | 6 (3.9) | 7 (4.5) |
Methotrexate | 6 (3.9) | 7 (4.5) |
Other immunosuppressants, n (%) | ||
Total | 7 (4.5) | 7 (4.5) |
Methotrexate | 6 (3.9) | 7 (4.5) |
Other immunosuppressants, n (%) | ||
Methotrexate sodium | 1 (0.6) | 0 |
Other ophthalmologicals, n (%) | ||
Total | 87 (56.5) | 66 (42.6) |
Ciclosporin | 85 (55.2) | 64 (41.3) |
Sirolimus | 2 (1.3) | 2 (1.3) |
Other therapeutic products, n (%) | ||
Total | 2 (1.3) | 2 (1.3) |
Sirolimus | 2 (1.3) | 2 (1.3) |
Protein kinase inhibitors, n (%) | ||
Total | 2 (1.3) | 0 |
Everolimus | 2 (1.3) | 0 |
Selective immunosuppressants, n (%) | ||
Total | 32 (20.8) | 22 (14.2) |
ATG | 3 (1.9) | 1 (0.6) |
ATG, rabbit | 0 | 2 (1.3) |
Everolimus | 2 (1.3) | 0 |
MMF | 25 (16.2) | 18 (11.6) |
Mycophenolate sodium | 7 (4.5) | 1 (0.6) |
Mycophenolic acid | 4 (2.6) | 0 |
Sirolimus | 2 (1.3) | 2 (1.3) |
aGvHD = acute graft-versus-host disease; ATC = Anatomical Therapeutic Chemical; ATG = antithymocyte immunoglobulin; BAT = best available therapy; CNI = calcineurin inhibitor; MMF = mycophenolate mofetil.
Source: Clinical Study Report (REACH 2).18
Table 18: Prior aGvHD Prophylaxis Treatment in REACH 1, Efficacy-Evaluable Population
Medication class or standardized medication name | Ruxolitinib N = 71 |
|---|---|
Patients with GvHD prophylaxis, n (%) | 71 (100.0) |
CNIs, n (%) | 69 (97.2) |
Ciclosporin | 6 (8.5) |
Tacrolimus | 65 (91.5) |
Folic acid analogues, n (%) | 16 (22.5) |
Methotrexate | 15 (21.1) |
Methotrexate sodium | 1 (1.4) |
Selective immunosuppressants, n (%) | 51 (71.8) |
Abatacept | 3 (4.2) |
ATG | 11 (15.5) |
ATG, rabbit | 4 (5.6) |
Mycophenolate mofetil | 34 (47.9) |
Sirolimus | 15 (21.1) |
Vedolizumab | 2 (2.8) |
aGvHD = acute graft-versus-host disease; ATG = antithymocyte immunoglobulin; CNI = calcineurin inhibitor; GvHD = graft-versus-host disease.
Source: Clinical Study Report (REACH 1).19
Interventions
REACH 2
Patients randomized to the REACH 2 trial were allocated to either the ruxolitinib group or the BAT group. Treatments and doses are described in Table 19. For treatment management based on patient response at day 28, refer to Table 20.
During the randomized treatment period in the REACH 2 trial, aGvHD disease and safety assessments were planned every week from day 1 up to day 56, and every 28 days thereafter until week 24. In case the taper of ruxolitinib was not completed by week 24 because of aGvHD flare or safety concerns, patients continued to be assessed (every 8 weeks from week 24 to week 48, and every 12 weeks thereafter) until taper of ruxolitinib was complete. All patients who discontinued treatment (regardless of when it occurred) entered the long-term survival period and were followed for long-term data on survival, any relapse or progression of the underlying hematologic disease, NRM, any occurrence of graft failure, EFS, any occurrence of cGvHD, and occurrence of any second primary malignancies. During the long-term survival follow-up period, follow-up occurred at 6, 9, 12, 18, and 24 months from randomization (day 1), as applicable, once the treatment period was completed. During the crossover period, patients followed the same treatment and taper schedule as patients originally randomized to ruxolitinib treatment; assessment occurred every week up to day 56 after ruxolitinib initiation, and every 28 days thereafter until week 24. A 30-day safety follow-up assessment was completed after the final dose of ruxolitinib or BAT for all patients.3
REACH 1
All patients enrolled in the REACH 1 trial received ruxolitinib. Treatments and doses are described in Table 19.
Assessments for aGvHD staging were performed weekly for the first 8 weeks after enrolment, every 28 days thereafter, on days 100, 180, and 365, and at the EOT visit. Unscheduled visits were allowed at the investigator's discretion.3 Safety was evaluated from screening to 30 to 35 days after EOT by monitoring the incidence and severity of AEs and SAEs.3 The treatment period started on the day the patient received the first dose of the study drug and ended when the patient permanently discontinued the study treatment, per the investigator’s decision. Patients who experienced disease progression or started a new GvHD therapy, entered the survival follow-up period and were assessed at least every 8 weeks (± 7 days) for new GvHD therapy, survival, withdrawal of consent, or the end of the study, whichever occurred first.3 Patients who terminated study treatment for reasons other than progression of GvHD entered the disease status follow-up period and were assessed every 28 days to monitor disease status until GvHD progression, relapse of malignancy, death, or the end of the study.19
Table 19: Treatment Regimens in the REACH 2 and REACH 1 Trials
Treatment | REACH 2 | REACH 1 | |
|---|---|---|---|
Ruxolitinib | BAT | Ruxolitinib | |
Dose |
In the first 28 days, patients who experienced disease progression, mixed response, or no response were allowed to move to new systemic treatment (investigator’s choice), but had to terminate ruxolitinib and were considered treatment failures.
|
BAT included (no other types or combinations of BATs were permitted in this study):
Medications used for aGvHD prophylaxis (i.e., started before the diagnosis of aGvHD, before randomization) that failed to prevent aGvHD in a patient could not be chosen as BAT for that same patient. In the first 28 days, patients who experienced disease progression, mixed response, or no response were allowed to move to treatment with another BAT but were considered treatment failures on initial BAT.
|
Patients were treated for as long as benefit was observed and/or treatment withdrawal criteria were not met.
Administration of either oral prednisone or IV methylprednisolone for corticosteroid treatment could begin at the investigator's discretion. CNIs or other systemic medications for aGVHD could be continued only if used as aGvHD prophylaxis (i.e., started before the diagnosis of aGvHD). |
Treatment discontinuation | Treatment was discontinued when the following criteria were met.b
| Same as ruxolitinib group. | Treatment was discontinued when the following criteria were met.c
|
AE = adverse events; aGvHD = acute graft-versus-host disease; ATG = antithymocyte globulin; BAT = best available therapy; cGvHD = chronic graft-vs.-host disease; CNI = calcineurin inhibitor, CR = complete response, DLI = donor lymphocyte infusion; ECP = extracorporeal photopheresis; EOT = end of treatment; IEC = independent ethics committee; IRB = institutional review board; MSC = mesenchymal stromal cell; mTOR = target of rapamycin; PR = partial response.
aStable hematologic parameters are defined as the absence of a ≥ 50% decrease in platelet counts and/or ANC relative to day 1.
bPatients on ruxolitinib were allowed to continue ruxolitinib outside the study if they met the study discontinuation criteria, responded to ruxolitinib at day 28 (or crossover day 28), and were assessed, per investigator, to be deriving clinical benefit from ruxolitinib; these patients do not enter the long-term follow-up period.
cA patient may be withdrawn from study treatment as follows:
• If, during the course of the study, a subject is found not to have met eligibility criteria, then the medical monitor, in collaboration with the investigator, will determine whether the subject should be withdrawn from the study.
• If a patient is noncompliant with study procedures or study drug administration in the investigator's opinion, the sponsor should be consulted for instruction on handling the patient.
Source: Protocol (REACH 2).3
Table 20: Treatment Management Based on Patient Response at Day 28 in REACH 2
End point | Patient management |
|---|---|
Primary end point met at day 28 | Patients responding to ruxolitinib will continue ruxolitinib until day 56. These patients may be tapered off ruxolitinib as needed, starting no earlier than day 56. The dose-tapering strategy should be based on the condition of the patient, current dosing regimen, and the clinical judgment of the investigator.
|
Patients responding to BAT will be managed per institutional practices. These patients may cross over to the ruxolitinib group between day 28 and week 24 if they meet crossover criteria after an initial response.
| |
Primary end point not met at day 28 | Patients who are randomized to ruxolitinib will discontinue study treatment and be treated per investigator’s judgment. These patients will then enter the long-term follow-up period. |
Patients who are randomized to BAT and who do not meet crossover criteria at day 28 will have their EOT visit, safety follow-up visit, and enter the long-term follow-up period. | |
Patients who are randomized to BAT and who meet crossover criteria at or after day 28 may cross over to the ruxolitinib treatment arm. Patients who cross over at day 28 or later will follow the same treatment duration and taper schedule as patients originally randomized to ruxolitinib. Corticosteroids and CNIs for aGvHD treatment can be continued, with cessation of any other systemic immunosuppressive treatment required before crossover, unless used for aGvHD prophylaxis (i.e., started before the diagnosis of aGvHD).
Should a tapering strategy not be in the best interest of the patient, or should the taper be completed before crossover week 24, the patient must still follow the assigned visit evaluation schedule, including all safety and efficacy assessments, until crossover week 24. |
aGvHD = acute graft-versus-host disease; BAT = best available therapy; CNI = calcineurin inhibitor, CR = complete response, EOT = end of treatment; PR = partial response.
Source: Protocol (REACH 2).3
Tapering of Therapies
REACH 2
Tapering of corticosteroids, CNIs, and ruxolitinib was done in 2 steps: first systemic corticosteroids were tapered upon documented CR or PR, then CNIs and ruxolitinib were tapered.
Taper of corticosteroids could be initiated at day 7 in patients who achieved a CR or PR. Taper consisted of a 10% dose reduction every 5 days up to approximately day 56 to allow a 7 to 8 week taper. It was anticipated that the taper of immunosuppressive medications would be complete by week 24. If the taper occurred before day 56, the dose of corticosteroids could be re-escalated at the investigator’s discretion and was not considered a treatment failure as long as the criteria for treatment failure were not met (criteria on the treatment of aGvHD flare are listed in the following). Taper of corticosteroids was to be repeated once patients achieved PR or CR, in which case the taper of corticosteroids could be prolonged beyond day 56, delaying the taper of CNIs and of ruxolitinib.3
Tapering guidelines per the study protocol for CNIs indicate that taper could be initiated once patients with documented CR or PR were off systemic corticosteroids. Taper consisted of a 25% dose reduction per month, or could follow a taper schedule per institutional practice.3
Taper guidelines for ruxolitinib indicated that, starting at day 56, taper could be initiated as required once patients with documented CR or PR were off systemic corticosteroids. Taper consisted of a 50% dose reduction every 2 months (56 days) from 10 mg to 5 mg administered orally twice daily. In patients with systemic aGvHD response (i.e., no worsening of aGvHD signs or symptoms), further taper included a second 50% dose reduction, to 5 mg orally once daily, for an additional 2 months (56 days), before discontinuation of ruxolitinib. The taper of ruxolitinib was to be complete by week 24 (or crossover week 24), except if prolonged tapering was indicated because of an aGvHD flare or other safety concerns, in which case the taper would be initiated no later than week 24 (or crossover week 24) and completed by the end of the patient’s study (no later than week 96; up to approximately 2 years from randomization). If GvHD flare occurred during ruxolitinib taper after day 56, patients could have had the ruxolitinib dose increased back to initial dose, and ruxolitinib taper repeated as needed until the patient’s end of study (up to approximately 2 years form randomization). Should a tapering strategy not be in the best interest of the patient, or should the taper be completed before crossover week 24, the patient must still follow the assigned visit evaluation schedule, including all safety and efficacy assessments, until crossover week 24.3
REACH 1
Tapering of corticosteroids and ruxolitinib was done in 2 steps: first, systemic corticosteroids were tapered per instructional guidelines at a rate appropriate for the resolution of GvHD manifestation; and second, taper of ruxolitinib could be attempted.3
Corticosteroid taper was done per instructional guidelines at a rate that was appropriate for the resolution of GvHD manifestation. Although there were no protocol guidelines for corticosteroid taper, recommendations were provided (see Table 21).3
Per-protocol guidelines for ruxolitinib indicated that after day 180, a taper of 1 dose level (e.g., 10 mg twice daily to 5 mg twice daily to 5 mg once daily) could be initiated once patients who achieved either a CR or VGPR were off systemic corticosteroids for at least 8 weeks. Ruxolitinib taper could be initiated during ongoing, concomitant use of ani-infective medication, GvHD prophylaxis medications (including CNIs), transfusion support, and topical steroid therapy. A subsequent dose level reduction of ruxolitinib was allowed after an additional 56 days during which patients did not experience any grade 2 or higher hematologic toxicity related to ruxolitinib or symptoms of an active infection.3
Investigators who wished to initiate taper or ruxolitinib earlier than proposed in the guidelines could do so conditional on consultation with and approval from the sponsor’s medical monitor.3
Table 21: Recommended Corticosteroid Administration in REACH 1, Days 1 to Day 28
Study days | Dose |
|---|---|
1 to 5 | Prednisone 2.5 mg/kg once daily orally (or methylprednisolone 2.0 mg/kg per day IV) |
6 to 10 | Prednisone 2.0 mg/kg once daily orally (or methylprednisolone 1.6 mg/kg per day IV) |
11 to 15 | Prednisone 1.5 mg/kg once daily orally (or methylprednisolone 1.2 mg/kg per day IV) |
16 to 20 | Prednisone 1.0 mg/kg once daily orally (or methylprednisolone 0.8 mg/kg per day IV) |
21 to 25 | Prednisone 0.5 mg/kg once daily orally (or methylprednisolone 0.4 mg/kg per day IV) |
26 to 28 | Prednisone 0.25 mg/kg once daily orally (or methylprednisolone 0.2 mg/kg per day IV) |
Source: Protocol (REACH 1).3
Treatment of aGvHD Flare
REACH 2
aGvHD flare was defined as any increase in symptoms of aGvHD sustained for more than 24 hours after an initial response (CR or PR) that required re-escalation of immunosuppressive therapy for aGvHD (e.g., corticosteroids, CNIs, BAT, and/or ruxolitinib dosing). An aGvHD flare was not considered a treatment failure as long as no change or addition of another systemic treatment was required. If addition or initiation of a new systemic therapy was required because it was not possible to taper corticosteroids to a dose below methylprednisolone 0.5 mg/kg per day (or equivalent < 0.6 mg/kg per day of prednisone) for a minimum 7 days or to re-escalate corticosteroids to methylprednisolone more than 2 mg/kg per day (or equivalent > 2.5 mg/kg per day of prednisone), patients were considered to have experienced treatment failure. See the Tapering of Therapies section for instruction on the treatment of aGvHD flare.3 The sponsor was asked about the number of aGvHD flares experienced by patients in the trial, but explained that those data were not available.21
REACH 1
Upon aGvHD flare, the dose of corticosteroids could be re-escalated at the investigator’s discretion. An aGvHD flare was not considered a treatment failure as long as the dose did not exceed the initial starting dose, the flare was not unresponsive to the re-escalation, and there were no multiple flares.3
Upon aGvHD flare, the dose of ruxolitinib could be re-escalated by incremental dose levels (e.g., 5 mg once daily to 5 mg twice daily to 10 mg twice daily), provided hematologic thresholds were met. An aGvHD flare was not considered a treatment failure as long as no additional systemic therapy was required (including the restarting of corticosteroids). Upon treatment failure, ruxolitinib would be discontinued.3
If aGvHD signs or symptoms recurred at a later time, after patients had completely tapered off ruxolitinib, ruxolitinib could be restarted at the discretion of the investigator and the patient would resume the assessment schedule using the original day 1 as a reference point.3
Dose Modifications
REACH 2
Dose interruptions or reductions were permitted for patients who did not tolerate the protocol-specified dosing schedule. For ruxolitinib, patients could not receive less than 5 mg once daily and not more than 10 mg twice daily. Once a dose or schedule modification occurred, titration back up to the original dose or schedule was permitted. If a ruxolitinib or BAT dose interruption exceeded 14 days, the study treatment had to be discontinued or the sponsor’s medical monitor contacted.3
REACH 1
Dose reductions or modifications of ruxolitinib were allowed based on AEs, clinical evaluation, and laboratory assessments. Patients could not receive less than 5 mg once daily and not more than 10 mg twice daily. If the ruxolitinib dose interruption exceeded 14 days, the study treatment had to be discontinued or the sponsor’s medical monitor contacted.3
Concomitant Medications
REACH 2
Routine administration of transfusion support and systemic immunosuppressive regimens of corticosteroids, CNIs (cyclosporine or tacrolimus), and topical corticosteroid therapy were allowed and were administered per institutional guidelines. Routine administration of antibiotics, anti-infectives, and immunizations as prophylactic therapy was allowed per institutional guidelines. Other systemic medications for aGvHD could be continued only if used as aGvHD prophylaxis (i.e., started before the diagnosis of aGvHD).3
REACH 1
Routine administration of systemic immunosuppressive regimens of corticosteroids, CNIs (cyclosporine or tacrolimus), and topical corticosteroid therapy was allowed per institutional guidelines. Routine administration of antibiotics, anti-infectives, and immunizations as prophylactic therapy was allowed per institutional guidelines. Other systemic medications for aGvHD could be continued only if used as aGvHD prophylaxis (i.e., started before the diagnosis of aGvHD).3
Outcomes
A list of end points identified in the CADTH review protocol that were assessed in the clinical trials included in this review is provided in Table 22. These end points are summarized in the following text. A detailed discussion and critical appraisal of the outcome measures is provided in Appendix 4.
Table 22: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure | REACH 2 | REACH 1 |
|---|---|---|
OS | Other secondary | Secondary |
FFS | Other secondary | Secondary |
ORR at day 28 | Primary | Primary |
ORR at day 14 | Other Secondary | Secondary |
Rate of durable ORR at day 56 | Key secondary | NR |
DOR | Other secondary | Key secondary: DOR at month 6 Secondary: DOR at month 3 |
BOR | Other secondary | Additional analysis of the primary end point |
HRQoL | ||
FACT-BMT | Other secondary | NR |
EQ-5D-5L | Other secondary | NR |
Symptom severity | NR | NR |
EFS | Other secondary | NR |
NRM | Other secondary | Secondary |
Incidence of malignancy relapse or progression | Other secondary | Secondary |
Relapse rate | NR | Secondary |
Relapse-related mortality rate | NR | Secondary |
Cumulative steroid dose up to day 56 | Other secondary | Exploratory |
Incidence of cGvHD | Other secondary | Exploratory |
Resource use: time to discharge, number of readmissions to hospital, admission to inpatient unit for any reason by inpatient setting (e.g., ICU, general ward), duration of readmissions by inpatient setting | Exploratory | NR |
Safety: frequency, duration and severity of AEs | Other secondary | Secondary |
AE = adverse event; BOR = best overall response; cGvHD = chronic graft-vs.-host disease; DOR = duration of response; EFS = event-free survival; EQ-5D-5L = 5-level EQ-5D; FACT-BMT = Functional Assessment of Cancer Therapy-Bone Marrow Transplant; FFS = failure-free survival; HRQoL = health-related quality of life; ICU = intensive care unit; NR = not reported; NRM = nonrelapse mortality; ORR = overall response rate; OS = overall survival.
Sources: Statistical Analysis Plan (REACH 2),3 Statistical Analysis Plan (REACH 1).3
Overall Survival
REACH 2
OS was a secondary outcome in the REACH 2 trial and was defined as the time from date of randomization to time of death from any cause.18
REACH 1
OS was a secondary outcome in the REACH 1 trial and was defined as the time from date of enrolment (first date of ruxolitinib treatment) to death from any cause.3
Failure-Free Survival
REACH 2
FFS was a secondary outcome in the REACH 2 trial and was defined as the time from date of randomization to date of hematologic disease relapse or progression, NRM, or addition of new systemic aGvHD treatment. For this analysis, the local investigator’s review of hematologic disease relapse or progression, according to protocol-defined criteria, was used.3
REACH 1
FFS was a secondary outcome in the REACH 1 trial and was defined as the interval from date of the first dose of ruxolitinib to date of underlying malignancy relapse or progression, death, addition of new systemic aGvHD treatment, or signs or symptoms of cGvHD.19
ORR at Day 28
REACH 2
ORR at day 28 was the primary outcome of the REACH 2 trial. ORR at day 28 was defined as the proportion of patients with CR or PR, according to the standard NIH criteria of Harris et al. (2016),16 at day 28.3 Response was assessed relative to the disease evaluation of aGvHD at baseline. aGvHD disease grading by the investigator was used for randomization and all analyses.3
CR was defined as a score of 0 for aGvHD grading (see Table 46 in Appendix 3 for aGvHD staging criteria of Harris et al. [2016]16) in all evaluable organs, which indicates complete resolution of all signs and symptoms of aGvHD in all evaluable organs without administration of additional systemic therapies for any earlier progression, mixed response, or nonresponse of aGvHD.3
PR was defined as a improvement of 1 stage, per staging criteria of Harris et al. (2016),16 in 1 or more organs involved with aGvHD signs or symptoms without progression in other organs or sites and without administration of additional systemic therapies for an earlier progression, mixed response, or nonresponse of aGvHD.3
Mixed response was defined as improvement of at least 1 stage, per aGvHD staging criteria of Harris et al. (2016),16 in the severity of aGvHD in at least 1 organ, accompanied by progression in another organ or development of signs or symptoms of aGvHD in a new organ.3
Progression was defined as worsening in 1 or more organs by 1 or more stages, per aGvHD staging criteria of Harris et al. (2016),16 without improvement in any involved organ.3
A patient was not considered a responder at day 28 in the event of any of the following:18
missing aGvHD assessment at baseline or day 28
no CR or PR at day 28
additional systemic therapy for aGvHD before day 28.
REACH 1
ORR at day 28 was the primary end point in the REACH 1 trial. ORR at day 28 was defined as the proportion of patients with CR, VGPR, or PR, per Center for International Blood and Marrow Transplant Research (CIBMTR) modifications to the International Bone Marrow Transplant Registry response index (CIBMTR [2009],46 Martin et al. [2009],25 Harris et al. [2016]16) at the day 28 response assessment (± 2 days) and before the start of new anti-aGvHD therapy, if applicable. Response was assessed relative to the disease evaluation of aGvHD at study day 1. aGvHD disease grading by the investigator was used for randomization and all analyses.3,19
CR is defined as a score of 0 for the aGvHD grading in all evaluable organs without administration of additional systemic therapies for any earlier progression, mixed response, or nonresponse of aGvHD.3,19
VGPR is defined as follows:
for skin, no rash or residual erythematous rash involving less than 25% of the body surface, without bullae (residual faint erythema and hyperpigmentation do not count)
for liver, total serum bilirubin concentration below 2 mg/dL or below 25% of baseline at enrolment
for gut —
tolerance of food or enteral feeding
predominantly formed stools
no overt GI bleeding or abdominal cramping
no more than occasional nausea or vomiting.
PR is defined as improvement of 1 stage in 1 or more organs involved with aGvHD symptoms without progression in other organs or sites without administration of additional systemic therapies for any earlier progression, mixed response, or nonresponse of aGvHD.19
Mixed response is defined as improvement in 1 or more organs with deterioration in another organ that manifests symptoms of aGvHD or development of symptoms of aGvHD in a new organ.19
Progression of disease is defined as deterioration in at least 1 organ without any improvement in others.19
A patient was not considered a responder at day 28 in the event of any of the following:19
missing aGvHD assessment at baseline or day 28
no CR, VGPR, or PR at day 28
additional systemic therapy for aGvHD before day 28 (including the need to re-escalate steroid dose above the day 1 dose).
Overall, the definition for ORR at day 28 was similar in the REACH 2 and REACH 1 trials. However, the REACH 1 trial assessed VGPR based on Martin et el. (2009),25 which defined a response state that approximated CR, with some qualifications. Martin et al. (2009)25 suggests that a benefit of the VGPR outcome is the inclusion of minor intermittent clinical abnormalities that may not be due to aGvHD, as opposed to CR that is strictly defined as a resolution of all signs and symptoms of aGvHD. The Martin et al. (2009)25 notes that VGPR should not be a substitute for CR in clinical trials and that it’s use will have to be validated through experience.25
ORR at Day 14
REACH 2
ORR at day 14 was a secondary outcome in the REACH 2 trial. ORR at day 14 was defined as the proportion of patients with CR or PR at day 14. Definitions of CR, PR, mixed response, and progression are the same as for ORR at day 28. ORR at day 14 and at day 56 was derived as it was for ORR at day 28. The local investigator’s review of aGvHD assessment data was used for this analysis.18
REACH 1
ORR rate at days 14, 56, and 100 was a secondary outcome in the REACH 1 trial. ORR at days 14, 56, and 100 was defined as the proportion of patients achieving a CR, VGPR, or PR at days 14, 56, and 100, respectively. Definitions of CR, VGPR, and PR are the same as for ORR at day 28. The investigator’s review of aGvHD assessment data was used for this analysis.19
Rate of Durable ORR at Day 56
REACH 2
Rate of durable ORR at day 56 was the key secondary outcome of the REACH 2 trial. Rate of durable ORR at day 56 was defined as the proportion of all patients in each group who achieved a CR or PR at day 28 and maintained a CR or PR at day 56. Response was assessed relative to the last disease evaluation of aGvHD before or at the start of crossover treatment (ruxolitinib). The local investigator’s review of aGvHD assessment data was used for this analysis.18
A patient was not considered a durable responder at day 56 in the event of the following:18
not a responder at day 28
missing aGvHD assessment at day 56
no CR or PR at day 56
additional systemic therapy for aGvHD before day 56.
REACH 1
Rate of durable ORR at day 56 was not reported in the REACH 1 trial.19
Duration of Response
REACH 2
DOR, a secondary outcome in the REACH 2 trial, was assessed in responders (CR or PR at day 28) only, and was defined as time from first response until aGvHD progression or until the addition of systemic therapies for aGvHD on or after day 28.18
The first documented response of CR or PR (i.e., the start date of response) could have occurred before or on day 28. If the start date of response was before day 28, there should have been no progression or addition of systemic therapies for aGvHD between the response start date and day 28. The end date of the DOR interval was defined as the date of aGvHD progression or the date of addition of systemic therapies for aGvHD after day 28.3
REACH 1
DOR at month 6 was a key secondary outcome in the REACH 1 trial and was performed once all patients had completed the day 180 visit. This end point was assessed in patients who had at least 1 response measurement (i.e., PR, VGPR, or CR on or before the start of new anti-GvHD therapy, if appliable). The DOR interval was defined as the difference between the end of response (progression or death) and the start of response (PR or better). Definitions of CR, VGPR, PR, and progressive disease are the same as for ORR at day 28. The investigator’s review of aGvHD assessment data was used for this analysis.19
DOR at month 3 was a secondary outcome in the REACH 1 trial and was performed once all patients had completed the day 84 visit. DOR at month 3 was derived in the same way as DOR at month 6.19
Additional analyses were performed as reported in the Clinical Study Report for the DOR using the day 28 response (i.e., patients who had CR, VGPR, or PR at the day 28 assessment or other response assessments within 2 days prior or after day 28, before the start of new anti-GvHD therapy, if applicable) and time to first response; however, these supportive analyses were not pre-specified a priori in the statistical analysis plan of the REACH 1 trial.19
Best Overall Response
REACH 2
BOR was designated a secondary outcome in the REACH 2 trial and defined as the proportion of patients who achieved overall response (CR or PR) at any time point up to and including day 28 and who had no additional systemic therapy for aGvHD before the time point.18
REACH 1
The BOR rate was not pre-specified a priori in the statistical analysis plan of the REACH 1 trial, but was assessed as an additional supportive analysis of the primary end point. It was defined as the proportion of patients who achieved a CR, VGPR, or PR at any time point before starting a new anti-aGvHD therapy.19
Health-Related Quality of Life
REACH 2
The HRQoL outcomes measured in the REACH 2 trial included the FACT-BMT and EQ-5D-5L instruments as secondary outcomes. Neither an analysis plan or objective nor a minimally important difference (MID) for the FACT-BMT and EQ-5D-5L instruments were specified a priori in the statistical analysis plan; it was noted, however, that the scores for each scale were calculated according to the respective user’s guides47,48 of the instruments. Scores for each scale (mean, SD, median, minimum, and maximum) and changes from baseline to each visit were measured and summarized descriptively.3 The FACT-BMT and EQ-5D-5L questionnaires were administered at baseline and every week in the first 2 months, and every 4 weeks thereafter until EOT. During the crossover period (after completion of all assessments at cycle 7 day 1), disease assessments were planned at the same frequency as during the randomized treatment period. The FACT-BMT was not administered to patients younger than 18 years.3 A detailed discussion and critical appraisal of the HRQoL measures is provided in Appendix 4.
The FACT-BMT instrument is a self-administered questionnaire that combines assessments of 2 tools: the Functional Assessment of Cancer Therapy-General (FACT-G) questionnaire, which assesses the effects of cancer therapy on physical, social/family, emotional, and functional well-being on 23 items; and the bone marrow transplant subscale, which assesses specific bone marrow transplant-related concerns on 23 items.3,49 The higher the score, the better the quality of life.50
No study was found that assessed psychometric properties related to the validity or reliability of the 3-level EQ-5D (EQ-5D-3L) in patients with aGvHD. A MID for the FACT-BMT was not identified in the literature for patients with aGvHD.
The EQ-5D is a generic, utility-based measure of HRQoL. The EQ-5D is a self-administered 2-part questionnaire, consisting of the EQ-5D descriptive system and the EQ-5D visual analogue scale (VAS).47 For the descriptive system of the EQ-5D, respondents are asked to indicate their health status that day (i.e., a 1-day recall) on 5 dimensions (mobility, self-care, usual activities, pain/discomfort, and anxiety/depression). The rating on each dimension is combined to create a descriptive health profile. The EQ-5D-5L was created by the EuroQol Group in 2009 to enhance the instrument’s sensitivity and to reduce the ceiling effects of the EQ-5D-3L.47 The EQ-5D VAS is a distinct component of the EQ-5D questionnaire. The VAS score is determined by asking respondents to rate their health that day on a vertical line, with anchors (end points) labelled “Worst imaginable health state” at 0 and “Best imaginable health state” at 100. Although the EQ-5D index score reflects societal preferences for the health state, the VAS captures the individual’s own value or judgment of his or her current health state. EQ-5D VAS scores are not used to create utility scores but provide complementary information to the EQ-5D-5L index score.47
No study was found that assessed psychometric properties related to the validity or reliability of the EQ-5D-3L in patients with aGvHD. A MID for the EQ-5D-5L was not identified in the literature for patients with aGvHD.
REACH 1
HRQoL outcomes were not included in the REACH 1 trial.
Event-Free Survival
REACH 2
EFS was a secondary outcome in the REACH 2 trial and was defined as the date of randomization to the date of hematologic disease relapse or progression, graft failure, or death from any cause. The local investigator’s review of hematologic disease relapse or progression, according to protocol-defined criteria, was used for this analysis.3
REACH 1
EFS was not assessed in the REACH 1 trial.
Nonrelapse Mortality
REACH 2
NRM was a secondary outcome in the REACH 2 trial and was defined as the time from date of randomization to date of death not preceded by hematologic disease relapse or progression.18
REACH 1
NRM was a secondary outcome in the REACH 1 trial and was defined as the proportion of patients whose death was the result of causes other than malignancy relapse at months 6, 9, 12, and 24.19
Incidence of Malignancy Relapse or Progression
REACH 2
The incidence of malignancy relapse or progression was a secondary outcome in the REACH 2 trial and was defined as the time from date of randomization to hematologic malignancy relapse or progression. Malignancy relapse or progression was assessed according to local institutional practices.3
REACH 1
The incidence of malignancy relapse or progression was not assessed in the REACH 1 trial. However, relapse rate and relapse-related mortality rate were 2 secondary outcomes in the REACH 1 trial and were defined as the proportion of patients who experienced underlying malignancy relapse and the proportion of patients who experienced malignancy relapse with a fatal outcome, respectively.3
Cumulative Steroid Dosing Until Day 56
REACH 2
Cumulative steroid dosing until day 56 was a secondary outcome in the REACH 2 trial and assessed the cumulative steroid dose up to day 56 or discontinuation of randomized treatment for each patient.18
REACH 1
Average and cumulative steroid dosing on days 28, 56, 100, and 180 was an exploratory outcome in the REACH 1 trial and assessed the cumulative steroid dose up to days 28, 56, 100, and 180 or corticosteroid discontinuation.
In addition, the use of immunosuppressive medications during ruxolitinib treatment was an exploratory outcome in the REACH 1 trial and assessed the proportion of patients taking immunosuppressive medications at certain time points while on ruxolitinib.3
Incidence of cGvHD
REACH 2
The incidence of cGvHD was a secondary outcome in the REACH 2 trial and was defined as the time from randomization to onset of cGvHD.18
REACH 1
The incidence of cGvHD was an exploratory outcome in the REACH 1 trial and was defined as the proportion of patients who experienced cGvHD.3
Resource Use
REACH 2
The assessment of resource use was a secondary outcome in the REACH 2 trial and was captured for use in a post-study health economics analysis. Resource-use data collected included time to discharge (only for patients starting treatment while hospitalized), measured as the start of treatment to discharge), the number of readmissions to a hospital inpatient unit for any reason by inpatient setting (e.g., intensive care unit, general ward) measured as the number of accesses to hospital for any reason that required at least 1 overnight stay and the duration of readmissions by inpatient setting measured as the number of overnight stays for each access to hospital that required an admission.3,18
REACH 1
Resource use was not assessed in the REACH 1 trial.
Safety
REACH 2
AEs that occurred or worsened after patients’ informed consent were to be recorded in the AE case report forms. Abnormal laboratory values or test results observed in patients only constituted AEs if they were associated with clinical signs or symptoms, were considered clinically meaningful, required therapy (e.g., hematologic abnormality requiring transfusion or hematological stem cell support), or required changes to the study drug. Components of study end points (i.e., worsening of study indication [aGvHD], including occurrence of aGvHD flare, occurrence of cGvHD, or relapse or recurrence of underlying disease [including fatal outcomes]) were not reported as SAEs and were reported on electronic case report forms other than AE electronic case report forms.3
AEs were assessed and graded according to the Common Terminology Criteria for Adverse Events (CTCAE), version 4.03. If a toxicity was not included in the CTCAE v4.03 criteria, it was graded on a scale of 1 to 5, as follows: 1 = mild, 2 = moderate, 3 = severe, 4 = life-threatening, and 5 = death related to the AE.3
All AEs were documented by the investigator from the date a patient signed informed consent to at least 30 days after the final dose of the study treatment.
The REACH 2 trial included the following parameters for the analysis of AEs: AEs by system organ class and preferred term, severity (based on CTCAE grade), type of AE, relationship to study treatment, seriousness (SAEs and non-SAEs), AEs of special interest, death, AEs leading to treatment discontinuation, AEs leading to dose interruption or adjustment, and AEs leading to fatal outcome.3
The presentation of AEs was performed for the following 4 mutually exclusive categories: pre-treatment period, on-randomized treatment period, on-crossover treatment period, and post-treatment period.3
REACH 1
Safety was designated a secondary outcome in the REACH 1 trial. A TEAE was any AE that was described for the first time or involved the worsening of a pre-existing event after the first dose of the study drug until 30 days after the final dose of the study drug.3
AEs were assessed and graded according to the CTCAE v4.03. If a toxicity was not included in the CTCAE v4.03 criteria, it was graded on a scale of 1 to 4 as follows: 1 = mild, 2 = moderate, 3 = severe, 4 = life-threatening.3
The REACH 2 trial included the following outcomes for the analysis of AEs: AEs by MedDRA preferred term and system organ class, incidence of AEs, relationship to study treatment, seriousness (SAEs and non-SAEs), AEs of special interest, deaths, AEs leading to treatment discontinuation, AEs leading to dose interruption or adjustment, and AEs leading to fatal outcome.3 Safety was assessed with vital signs, Eastern Cooperative Oncology Group (ECOG) performance status, physical examination, 12-lead electrocardiogram, and clinical laboratory assessments.19
Statistical Analysis
Details related to statistical analyses of efficacy end points are summarized in Table 23 for the REACH 2 trial and Table 24 for the REACH 1 trial.
Sample-Size Determination
REACH 2
The trial sample size of 308 patients was determined based on the primary end point of ORR at day 28.3 Assuming an odds ratio of 1.63 or higher for the primary outcome, the study would have 90% power to demonstrate a statistically significant difference at a 1-sided alpha of 0.025. Several assumptions drove the sample-size determination. First, ORR at day 28 for the BAT group was expected to be 58%, as suggested in the study by Martin et al. (2012),36 based on aggregated results of 29 studies that evaluated available secondary therapies for aGvHD. Second, for the BAT group, it was expected that stratum-specific rates were 69%, 59%, and 50% for grades II, III and IV, respectively, based on the assumed aGvHD grade II:III:IV ratio of 0.2:0.4:0.4. No rationale was provided in the sponsor’s submission for the assumption of the aGvHD grade ratio. Third, it was expected that treatment with ruxolitinib would result in an 18% improvement in ORR, corresponding to an increase in ORR to 75%. No rational was provided in the sponsor’s submission for the assumed ORR benefit with ruxolitinib. If the response rates in grades II, III, and IV in the BAT group were, respectively, 69%, 59%, and 50% (57% overall), corresponding response rates of 78%, 70%, and 62% (68% overall) or more in the ruxolitinib group would achieve statistical significance.3
For the analysis of the key secondary outcome — durable ORR at day 56 — a total of 308 patients was considered sufficient. Assuming an odds ratio of 1.59 or more in the ruxolitinib group, the study would have 90% power to demonstrate a statistically significant difference at a 1-sided alpha of 0.025. Several assumptions drove the sample-size determination. First, durable ORR at day 58 for the BAT group was expected to be approximately 35%, based on a study by van Groningen et al. (2016)51 that evaluated responses with second-line therapy in 21 patients with severe aGvHD. Second, for the BAT group, it was expected that stratum-specific rates would be 45%, 36%, and 30% for grades II, III, and IV, respectively, based on the assumed aGvHD grade II:III:IV ratio of 0.2:0.4:0.4. No rationale was provided in the sponsor’s submission for the assumption of the aGvHD grade ratio. Third, it was expected that treatment with ruxolitinib would result in a 20% improvement in ORR, corresponding to an increase in durable ORR to 55%. No rationale was provided in the sponsor’s submission for the assumed ORR benefit with ruxolitinib. If the response rates in grades II, III, and IV in the BAT group were, respectively, 45%, 36%, and 30% (35% overall), corresponding response rates of at least 57%, 47%, and 41% (47% overall) in the ruxolitinib group would achieve statistical significance.3
REACH 1
The trial sample size of 70 patients was determined based on the primary end point of ORR at day 28. Assuming an ORR of 60%, the study would have a greater than 90% probability of having a 95% CI with a lower limit of 40% or more. If 37 or more of 70 patients responded (i.e., if the lower limit of the 95% CI for ORR at day 28 exceeded 40%), it was predetermined that the trial results would be considered positive. The minimum clinically meaningful proportion of patients with an objective response was considered to be a 20% improvement over historic data. No rationale was provided in the sponsor’s submission for the assumed minimum clinically meaningful improvement, and no benchmark for historic ORR was provided.3,19
Interim Analysis
REACH 2
No formal interim analysis was planned a priori in the statistical analysis plan.3 A hierarchical testing procedure was applied for the primary (ORR at day 28) and key secondary end points (ORR at day 56) in the primary analysis (July 25, 2019 data cut-off date), which was planned for when all patients had completed the day 56 visit or discontinued earlier.18 Formal statistical significance testing with full alpha was only planned a priori for the primary analysis. An updated secondary analysis for secondary end points (January 6, 2020, data cut-off date) was planned for when all patients had completed approximately 6 months of treatment or discontinued earlier. The end of the study occurred when all patients had completed the study (up to 24 months from randomization), unless the patient withdrew consent; at the time of the final analysis (April 23, 2021),52 secondary end points were analyzed.
REACH 1
An interim analysis for futility was planned after 35 patients completed the day 28 visit. Enrolment in the REACH 1 trial could have been terminated if the lower boundary of futility was crossed. To calculate the lower boundary of futility, a group sequential-design method for 1 sample binary outcome data and the spending function of Hwang-Shih-DeCani (−4) were used. The study was to be terminated if no more than 15 patients responded at the time of the interim analyses (for the null hypothesis, P = 0.4; for the alternative hypothesis, P = 0.6, and a 1-sided binomial test with alpha of 0.025). This was to provide a 70.03% probability for the response rate of 40% at the interim analysis.3 At the interim analysis, 21 of 35 patients (60.0%) achieved an overall response at day 28, and the study proceeded as planned.19 No formal data monitoring committee was to review the results of the futility analysis.3
If 37 or more patients responded at the final analysis, it was predetermined that the trial results would be positive, based on 89.88% power for the response rate of 60% at the final analysis, with a type I error of 0.0189.3
The final analysis, when the predetermined threshold (i.e., lower limit of the 95% CI for ORR at day 28 was 40% or greater) was to be assessed, was planned for when 75% of patients had achieved 2-year NRM, died, or were lost to follow-up, whichever occurred first. The final analysis was conducted at the data cut-off date of June 5, 2019. The results of the final analysis have been provided to CADTH by the sponsor and are the focus of this CADTH review.
An earlier data analysis occurred at the July 2, 2018, data cut-off date, which was not pre-specified a priori in the statical analysis plan of the REACH 1 trial. According to the sponsor, the July 2, 2018 data cut-off date was requested by the FDA to provide data for 6-month DOR and 6-month NRM once all patients had completed the 6-month (day 180) study visit, started a new treatment for aGVHD, or discontinued the study treatment.21 The results of this earlier data cut-off date were published by Jagasia et al. (2020).44 This CADTH review will focus on the results of the final analysis.
Primary Outcome
REACH 2
The primary outcome in the REACH 2 trial was ORR at day 28. A brief overview of the statistical methods used for the primary outcome is provided in Table 23.
To compare ORRs between the 2 study groups, a Cochrane-Mantel-Haenszel chi-square test, stratified by the randomization factor (i.e., aGvHD grade II versus III versus IV) was used at a 1-sided 2.5% level of significance. The primary analysis was based on the full analysis set, per the intention-to-treat principle. ORR was also summarized using descriptive statistics (N, %) and 2-sided exact binomial 95% CIs (Clopper and Pearson [1934]53). P value, odds ratio, and 95% Wald confidence limits were calculated using the stratified Cochran-Mantel-Haenszel test. Patients with missing assessments were considered nonresponders.3
Supportive analyses for ORR at day 28 were planned as follows:
ORR at day 28 assessed with the same analysis conventions as the primary efficacy analysis, using all patients in the per-protocol set
a detailed description of organ-specific response for all organs at day 28, using shift tables of aGvHD stage by organ and treatment group to compare baseline and day 28 value
logistic regression model to estimate treatment effect adjusted for key baseline and prognostic factors (covariates may include age, sex, race, aGvHD grade, source of graft, criteria for SR-aGvHD, and prior aGvHD therapy in addition to treatment).
A sensitivity analysis was performed using the full analysis set to assess the impact of stratification by comparing the study groups with the Fisher’s exact test. ORR was summarized using descriptive statistics (N, %) and 2-sided exact binomial 95% CIs (Clopper and Pearson [1934]53).3
In a supportive analysis, ORR at crossover day 28 was summarized descriptively and defined as the proportion of crossover patients with CR or PR at crossover day 28, according to the standard NIH criteria of Harris et al. (2016).16 Using the local investigator’s review of aGvHD assessment, summary statistics (N, %) with 2-sided exact binomial 95% CIs were presented.3
REACH 1
The primary outcome in the REACH 1 trial was ORR at day 28. A brief overview of statistical methods used for the primary outcome is provided in Table 24.
No formal statistical tests were performed. The 95% CI for ORR was estimated using the exact method for binomial distribution. The primary analysis was performed when the final patient completed the day 28 visit or withdrew from the study. Patients with insufficient response data at day 28 (e.g., death, discontinuation, missing visit) were considered nonresponders.3
An additional analysis was performed, as reported in the Clinical Study Report, for the BOR; however, this supportive analysis was not pre-specified in the statistical analysis plan of the REACH 1 trial. The 95% CI for BOR was estimated using the exact method for binomial distribution.3
Key Secondary Outcome
REACH 2
Durable ORR at day 56 was the key secondary outcome in the REACH 2 trial. A hierarchical testing procedure was applied in which the durable ORR at day 56 was only tested if the ORR at day 28 was statistically significant. The key secondary outcome was tested at the same time as the primary outcome (i.e., at the primary analyses, when all patients had completed their day 56 visit or discontinued the study earlier). To compare durable ORRs at day 56 in the 2 study groups, a Cochrane-Mantel-Haenszel chi-square test, stratified by the randomization factor (i.e., aGvHD grade II versus III versus IV) was used at a 1-sided 2.5% level of significance. The analysis was based on the full analysis set, per the intention-to-treat principle. Durable ORR at day 56 was also summarized using descriptive statistics (N, %) and 2-sided exact binomial 95% CIs (Clopper and Pearson [1934]53). P value, odds ratio, and 95% Wald confidence limits were calculated using the stratified Cochran-Mantel-Haenszel test. Patients with missing assessments were considered nonresponders. Missing data were handled the same way they were in the primary outcome.
As an additional analysis, durable ORR at crossover day 56 was analyzed and defined as the proportion of all crossover patients who achieved a CR or PR at crossover day 28 and maintained a CR or PR at crossover day 56. Response was assessed relative to the last assessment of aGvHD before or at the start date of crossover treatment (ruxolitinib). Durable ORR at crossover day 56 was summarized using descriptive statistics (N, %) and 2-sided exact binomial 95% CIs (Clopper and Pearson [1934]53), based on the crossover set.3
REACH 1
DOR at month 6 was the key secondary outcome in the REACH 2 trial. Six-month DOR was assessed when all patients had completed the day 180 visit. A Kaplan–Meier (KM) plot of DOR was presented with its 95% CI; 95% CI was estimated using the method of Brookmeyer and Crowley (1982).54 Reasons for censoring included the patient still responding at the data cut-off date and discontinuation at the time of last valid response assessment.3
Additional analyses were performed, as reported in the Clinical Study Report, for the DOR using the day 28 response and time to first response; however, these supportive analyses were not pre-specified a priori in the statistical analysis plan of the REACH 1 trial.3
Secondary Outcomes
REACH 2
All analyses for other secondary end points were noncomparative in nature and were not included in formal hypothesis testing. All analyses were based on the full analysis set.
The ORR at day 14 was a secondary outcome in the REACH 2 trial. ORR at day 14 was also summarized using descriptive statistics (N, %) by treatment group and 2-sided exact binomial 95% CIs (Clopper and Pearson [1934]53). Odds ratio and 95% Wald confidence limits were calculated using the stratified Cochran-Mantel-Haenszel test.18
DOR was a secondary outcome in the REACH 2 trial and was calculated for all patients who had CR or PR at day 28. The cumulative incidence rates and associated 95% CIs at 1, 2, 6, 18, and 24 months were assessed for each study group and the cumulative incidence curve was provided. Reasons for censoring included not having experienced events or competing risks before or at the data cut-off date. Censoring occurred at the last response assessment before or at the analysis cut-off date.18
Cumulative steroid dosing up to day 56 was a secondary outcome in the REACH 2 trial. Overall and weekly cumulative steroid doses for each patient up to day 56 or at discontinuation of randomized treatment were summarized. RDI by week was analyzed relative to the starting dose of corticosteroids, and classified as a complete reduction in which patients are tapered off corticosteroids by day 56, a RDI of no more than 50%, and a RDI of more than 50%. The proportion of patients in each aforementioned group with associated 95% CIs were presented by study group. For weeks ending on days 14, 28, 56, 84, and 168, the average weekly corticosteroid dose was also assessed and plotted.18
OS was a secondary outcome in the REACH 2 trial. The analysis for OS was conducted according to the randomized treatment group, stratified by aGvHD grade (i.e., aGvHD grade II versus III versus IV). A KM plot of OS was presented by treatment group. Medians and KM estimated hazard risk and corresponding 95% CIs (Brookmeyer and Crowley [1982]54) at 1, 2, 6, 12, 18, and 24 months were presented. The HR and 95% CIs were estimated from a stratified Cox proportional hazards regression model. Patients were censored at the latest date a patient was known to be alive (on or before the data cut-off date). Patients who crossed over to ruxolitinib from the BAT group were included in the OS analysis for the BAT group.3
The following exploratory sensitivity analyses were planned a priori if the primary analysis is significant and sufficient OS events have occurred:3
Rank-preserving structural failure time model (Robins and Tsiatis [1991]55 and Korhonen et al. [1999]56) to estimate the treatment effect, taking into account the switch from the BAT group to the ruxolitinib group.
Stratified Cox regression model adjusted for prognostic factors, using a stepwise selection process, if appropriate. Goodness-of-fit of the model was examined.
The following exploratory analyses were planned a priori to be performed as necessary if the primary analysis is significant.3
Relationship between ORR at day 28 and OS will be performed in the ruxolitinib group in the form of a landmark analysis, using day 28 as the landmark time (patients who died before day 28 were excluded). The null hypothesis was that survival after day 28 would not depend on response status at day 28. KM curve and log-rank test were to be used to compare the survival of responders and nonresponders.
EFS was a secondary outcome in the REACH 2 trial. It was analyzed using the same statistical methods as OS. Reasons for censoring included no known experience of any event. Patients were censored at the latest date a patient was known to be alive (on or before the data cut-off date). Patients who crossed over to ruxolitinib from the BAT group were included in the OS analysis for the BAT group. A sensitivity analysis was planned in which aGvHD progression was included as an event.3
FFS was a secondary outcome in the REACH 2 trial. The cumulative incidence of FFS at 1, 2, 6, 12, 18, and 24 months with associated 95% CIs was assessed with onset of chronic GvHD as the competing risk. The cumulative incidence of each of the 3 components (i.e., date of hematologic disease relapse or progression, NRM, and addition of new systemic aGvHD treatment), taking the other 2 components as a competing risk, was also calculated. The cumulative incidence curves were presented for each study group. A sensitivity analyses in which aGvHD progression was considered an event was planned.3
NRM was a secondary outcome in the REACH 2 trial and was analyzed according to randomized treatment group and strata assigned at randomization. The cumulative incidence of NRM at 1, 2, 6, 12, 18, and 24 months was assessed with underlying disease relapse or progression as the competing events. Reasons for censoring included no confirmed death and the experience of the competing event. Censoring occurred at the latest date the patient was known to be alive (on or before the cut-off date). Sensitivity analyses were planned in which the cumulative incidence curve of NRM and estimates at 1, 2, 6, 12, 18, and 24 months with 95% CIs were presented for patients with underlying hematologic malignant disease in each study group.18
The incidence of malignancy relapse or progression was a secondary outcome in the REACH 2 trial. The cumulative incidence of malignancy relapse or progression at 1, 2, 6, 12, 18, and 24 months with associated 95% CIs was assessed, with underlying hematologic malignant disease, accounting for NRM, as the competing risk. The proportion of patients who had hematologic malignancy relapse or progression and the associated 95% CI at 1, 2, 6, 12, 18, and 24 months were presented by study group for patients with underlying hematologic malignant disease. Odds ratio and 95% Wald confidence limits were also calculated using the stratified Cochran-Mantel-Haenszel test. Reasons for censoring included no known relapse or progression. Censoring occurred at the latest date the patient was known to be alive (on or before the cut-off date).18
The incidence of cGvHD was a secondary outcome in the REACH 2 trial. The cumulative incidence of cGvHD at 1, 2, 6, 12, 18, and 24 months with associated 95% CIs was assessed with deaths without prior onset of cGvHD and hematologic disease relapse or progression as the competing risks. Reasons for censoring included no known event or competing risks. Censoring occurred at the latest date of the patient was known to be alive (on or before the cut-off date).18
BOR response was a secondary outcome in the REACH 2 trial. BOR was summarized using descriptive statistics (N, %) by treatment group and 2-sided exact binomial 95% CIs (Clopper and Pearson [1934]53). Odds ratio and 95% Wald confidence limits were calculated using the stratified Cochran-Mantel-Haenszel test.18
Resource use was a secondary outcome in the REACH 2 trial. Summary statistics will be provided for each resource component (i.e., time to discharge, readmissions to hospital, duration of readmission to hospital, and proportion of patients with readmission to hospital). The 95% CIs will be presented for the proportion of patients with readmission to hospital, by study group. No further analyses were specified a priori in the statistical analysis plan. Data related to resource use were to be collected for the purpose of the economic evaluation and were analyzed and reported separately.3
REACH 1
OS was a secondary outcome in the REACH 1 trial. The KM plot of OS was presented with its 95% CI; 95% CI was estimated using the method of Brookmeyer and Crowley (1982).54 Survival rate at month 6 was estimated. Reasons for censoring included still being alive or being lost to follow-up at the data cut-off date, and occurred at the last date a patient was known to be alive. Additional analyses were performed, as reported in the Clinical Study Report, for OS by response status, deaths by response status, and deaths while still receiving ruxolitinib or within 30 days of the last ruxolitinib dose; however, these analyses were not pre-specified in the statistical analysis plan of the REACH 1 trial.3
FFS was a secondary outcome in the REACH 1 trial. The KM plot of FFS was presented with its 95% CI; 95% CI was estimated using the method of Brookmeyer and Crowley (1982).54 Survival rate at month 6 was estimated. Reasons for censoring included still being alive, being lost to follow-up, having no relapse or progression of the underlying malignancy, requiring no additional therapy for aGvHD, and having no demonstrated signs or symptoms of cGvHD.3
ORR at days 14, 56, and 100 was a secondary outcome in the REACH 1 trial. Statistical methods were the same as those used for the primary outcome of ORR at day 28.3
DOR at month 3 was a secondary outcome in the REACH 1 trial and assessed when all patients completed the day 84 visit. Statistical methods were the same as those used for the key secondary outcome of DOR at month 6.3
NRM was a secondary end point in the REACH 1 trial. The cumulative incidence of NRM at 6, 9, 12, and 24 months was assessed with relapse-related mortality as the competing event. Reasons for censoring included no confirmed death. Censoring occurred at the latest date the patient was known to be alive. Per the statistical analysis plan, the 95% CI for NRM was estimated using the exact method for binomial distribution. However, according the Clinical Study Report, changes to the planned analyses included using Marubini and Valsecchi's method (1995)57 and the delta method (Hosmer et al. [2008])58 with log-log transformation, respectively, for the calculation of cumulative incidence rates and 95% CIs.3,19
Relapse rate was a secondary end point in the REACH 1 trial. The 95% CI for the relapse rate was estimated using the exact method for binomial distribution.3
Relapse-related mortality rate was a secondary end point in the REACH 1 trial. The 95% CI was estimated using the exact method for binomial distribution.3
Exploratory End Points
REACH 1
Average and cumulative steroid dosing on days 28, 56, 100, and 180 were exploratory end points in the REACH 1 trial. However, according the Clinical Study Report, changes to the planned analyses included replacing cumulative steroid dosing with a summary of the average and relative (to initial dose) corticosteroid dose at selected time points during ruxolitinib treatment.19
The incidence of cGvHD was an exploratory outcome and the proportion of patients with cGvHD was calculated.
Subgroup Analyses
REACH 2
For each subgroup, the point estimate and 2-sided exact binominal 95% CI (Clopper and Pearson [1934]53) were calculated. The odds ratio was calculated with 95% CI, using a logistic regression model with covariates (i.e., treatment and stratification factors). A forest plot was presented. No formal statistical tests of hypotheses were conducted. Subgroup analyses were planned a priori in the statistical analyses plan. Planned subgroup analyses were to be conducted for the primary end point if statistically significant results were observed:18
age (12 years to younger than 18 years, 18 to 65 years, older than 65 years)
sex
race
Europe plus Australia and Canada, Asia excluding Japan, Japan
aGvHD grade (grade II, III, IV)
source of grafts (related, unrelated)
criteria for SR-aGvHD (progression after at least 3 days, failure to achieve a response after 7 days, flare failure during taper)
prior aGvHD therapy (steroids ± CNIs, steroids ± other systemic aGvHD treatment, steroids ± CNIs ± other systemic aGvHD treatment).
The following subgroups, planned a priori in the statistical analysis plan, aligned with the subgroups pre-specified in the protocol for this CADTH review: overall aGvHD grade, organ involvement for aGvHD, disease underlying aGvHD, age, criteria for SR-aGvHD, prior aGvHD therapy, and stem cell source. Only subgroups identified in the CADTH review protocol are reported in the efficacy section.18
REACH 1
Additional supportive analyses for the primary end point were performed, as reported in the Clinical Study Report; however, none of the following supportive analyses were pre-specified in the statistical analysis plan of the REACH 1 trial19:
day 28 ORR by baseline aGvHD grade
day 28 ORR by baseline SR subcategory
day 28 ORR by use of immunosuppressive medications
day 28 ORR by use of CNIs
day 28 ORR by average reported daily ruxolitinib dose from day 1 to day 28
day 28 ORR by age, sex, race, and baseline GvHD organ involvement (liver, upper GI, lower GI, and skin).
Multiplicity
REACH 2
Apart from the pre-specified hierarchical testing of ORR at day 28 and durable ORR at day 56, no adjustments for multiplicity were performed to control the type I error rate.
REACH 1
Not applicable.
Amendments
REACH 2
The protocol of the REACH 2 trial was amended twice (Amendment 1 on May 31, 2017, and Amendment 2 on June 21, 2018). Amendment 1 included changes to exclusion criterion 5, other eligibility criteria, and administrative changes. Amendment 2 included changes to allow for more flexibility in the tapering of corticosteroids, CNIs, and ruxolitinib, and to allow for the complete taper to occur beyond week 24, if required. Amendment 2 also implemented a post-trial-access commitment by Novartis, in which patients who meet certain protocol treatment discontinuation criteria or who were still receiving ruxolitinib at their end of study (approximately 2 years from randomization) and were judged by the investigator to be deriving clinical benefit from ruxolitinib were given the option to continue ruxolitinib outside the study. Further, to align with the clinical management of adolescents and to increase their enrolment in the study, other systemic medications for aGvHD prophylaxis could be continued after randomization for all patients. It was noted in the protocol that the impact of this change on overall patient homogeneity was judged to be limited. The secondary end point, BOR, was added to align with aGvHD publications. A data monitoring committee was added to uphold blinding of members of the study steering committee during their review of pooled safety data. In the decision to add a data monitoring committee, no efficacy or safety data from the study were considered.3
REACH 1
The protocol of the REACH 1 trial was amended twice (Amendment 1 on September 12, 2016, and Amendment 2 on October 4, 2016). Both amendments occurred before the first patient was enrolled in December 2016. Amendment 1 included changes to eligibility criteria, secondary end points, GvHD staging and grading criteria, the starting dose of ruxolitinib, and the ruxolitinib dose in participants with liver GvHD, and added an interim analysis and the ability of investigators to taper ruxolitinib. Amendment 1 addressed regulatory feedback. Key changes included:19
adding the assessment of DOR as a key secondary end point
changing the starting dose to 5 mg twice daily and providing guidance to investigators for escalating the dose to 10 mg twice daily after 3 days if hematologic parameters were stable and no treatment-related toxicity was observed
changing GvHD staging and grading criteria to Mount Sinai Acute GvHD Consortium guidelines
expanding eligibility to participants 12 years and older, clarifying requirements for patients defined as SR, expanding eligibility to participants with ANC ≥ 0.5 × 109/L, removing the requirement for platelet engraftment, removing the Eastern Cooperative Oncology Group performance status requirement, clarifying the definition of severe organ dysfunction, and expanding eligibility with respect to prior use of JAK inhibitors
adding language to reflect a planned interim analysis for futility once 35 participants completed the day 28 visit
adding toxicity management guidelines to provide guidance on the appropriate management of bilirubin elevations in participants with and without liver GvHD, given the potential for pre-existing cytopenia and liver function test abnormalities
removing a statement about reducing the total daily dose of ruxolitinib by approximately 50% when administering ruxolitinib with strong CYP3A4 inhibitors or dual moderate inhibitors of CYP2C9 and CYP3A4 (e.g., fluconazole). As azole antifungal support is routinely administered to GvHD patients as a part of their prophylaxis regimen, and as available retrospective ruxolitinib data indicated that starting doses of 5 mg to 10 mg twice daily with concomitant azole use did not result in significant changes in safety, reducing the starting dose of ruxolitinib in patients receiving concurrent azole therapy as is done in other indications was not required in this study.
Amendment 2 included changes to secondary end points, dose modifications of ruxolitinib in participants with liver GvHD, the ability of investigators to taper ruxolitinib, and guidance on corticosteroid tapering. Amendment 2 addressed regulatory feedback received after Amendment 1. Key changes included:19
changing the key secondary end point from 3-month DOR to 6-month DOR, and 3-month DOR was added as a secondary end point
modifying toxicity management guidelines to provide guidance on the appropriate management of bilirubin elevations in participants with and without liver GvHD based on the upper limit of the normal range instead of total bilirubin concentration, given the potential for different ranges for adolescents
revising instructions for tapering ruxolitinib to permit tapering after day 180 for participants achieving CR or VGPR; allowing investigators to continue treating participants with CNIs and other GvHD prophylaxis medications as appropriate; allowing investigators to initiate an earlier taper of ruxolitinib, with sponsor approval; and allowing participants who have completely tapered off ruxolitinib to restart treatment when GvHD symptoms reappear
deleting specific guidance on corticosteroid tapering; corticosteroids were to be tapered per institutional guidelines at a rate commensurate with the resolution of GvHD manifestations.
Table 23: Statistical Analysis of Efficacy End Points in REACH 2 (Outcomes Are Presented in Order of Priority as Identified in the CADTH Review Protocol)
End point | Statistical model | Adjustment factors | Sensitivity analyses |
|---|---|---|---|
OS (secondary end point) | KM survival method to estimate median, 1-, 2-, 6-, 12-, 18, and 24-month survival probabilities, and 2-sided 95% CIs (Brookmeyers and Crowley [1982]54) HRs and 95% CIs for the difference between treatment groups were derived using a stratified Cox proportional hazard regression model | Stratification factors:
| The following sensitivity analyses were planned a priori if the primary analysis was significant and sufficient OS events occurred:
|
FFS (key secondary outcome) | Cumulative incidence curve for FFS; estimates at 1, 2, 6, 12, 18, and 24 months and 95% CIs Cumulative incidence of each of the 3 components, with the other 2 components as a competing risk, were estimated (onset of chronic GvHD was considered a competing risk for all 3 types of failure) Cumulative incidence curves were plotted for each treatment group | None | aGvHD progression included as an event |
ORR at day 28 (primary end point) | Cochran-Mantel-Haenszel chi-square test was used to compare ORR between the 2 study groups at the 1-sided 2.5% level of significance One-sided P value, odds ratio and 95% Wald confidence limits were calculated from stratified Cochran-Mantel-Haenszel test ORR was summarized using descriptive statistics (N, %) and 2-sided exact binomial 95% CIs (Clopper and Pearson [1934]53) | Stratification factors:
| Supportive analyses:
Sensitivity analyses:
|
ORR at day 14 (secondary end point) | ORR was summarized using descriptive statistics (N, %) and 2-sided exact binomial 95% CIs (Clopper and Pearson [1934]53) Odds ratio and 95% Wald confidence limits were calculated from stratified Cochran-Mantel-Haenszel test | None | None |
Durable ORR at day 56 (key secondary end point) | Tested hierarchically; if the ORR at day 28 is statistically significant, the durable ORR at day 56 will be tested, and if the ORR at day 28 is not statistically significant, the durable ORR at day 56 will not be tested If the primary end point was significant, the Cochran-Mantel-Haenszel chi-square test was used to compare ORRs in the 2 study groups at the 1-sided 2.5% level of significance Summarized using descriptive statistics (N, %) and 2-sided exact binomial 95% CIs (Clopper and Pearson [1934]53) P value, odds ratio, and 95% Wald confidence limits were calculated from stratified Cochran-Mantel-Haenszel test | Stratification factors:
| Durable ORR at crossover day 56b was summarized using descriptive statistics (N, %) and 2-sided exact binomial 95% CIs (Clopper and Pearson [1934]53), based on crossover analysis set |
DOR (secondary end point) | Cumulative incidence rates and 95% CIs at 1, 2, 6, 12, 18 and 24 months for each study group and cumulative incidence curve were assessed DOR was summarized by study group | None | None |
BOR (secondary end point) | BOR was summarized using descriptive statistics (N, %) and 2-sided exact binomial 95% CIs (Clopper and Pearson [1934]53) P value, odds ratio, and 95% Wald confidence limits were calculated from stratified Cochran-Mantel-Haenszel test | None | None |
FACT-BMT (secondary outcome) Definition: A 50-item self-reported questionnaire with questions relevant to BMT patients (domains include physical, functional, social/family, emotional well-being, and additional concerns) | Responses to FACT-BMT were generated per its scoring manual50 Descriptive statistics (mean, SD, median, minimum, and maximum) were used to summarize scores at each assessment point; change from baseline in scores at the time of each assessment were summarized | None | None |
EQ-5D-5L (secondary outcome) Definition: Self-reported generic measure of health, including a descriptive system with 5 health dimensions (mobility, self-care, usual activities, anxiety/depression, and pain/discomfort) and a VAS | Responses to the EQ-5D-5L were generated per its scoring manual50 Descriptive statistics (mean, SD, median, minimum, and maximum) were used to summarize scores at each assessment point; change from baseline in scores at the time of each assessment was summarized | None | None |
EFS (secondary end point) | Same as OS | Stratification factors:
| Sensitivity analysis: aGvHD progression included as an event |
NRM (secondary end point) | Cumulative incidence of NRM and derived probabilities at months 1, 2, 6, 12, 18, and 24 with 95% CIs were evaluated; underlying disease relapse or recurrence was considered as a competing event | Stratification factors:
| Sensitivity analysis: NRM in patients with underlying hematologic malignant disease in each treatment group, using same statistical analysis as for initial NRM |
Malignancy relapse or recurrence (secondary end point) | For patients with underlying hematological malignant disease, cumulative incidence curve and estimates at 1,2, 6, 12, 18 and 24 months with 95% CIs were assessed, with NRM as the competing risk Proportion of patients who had hematologic malignancy relapse or recurrence and 95% CIs at 1,2, 6, 12, 18 and 24 months were presented by study group for patients with underlying hematologic malignant disease Odds ratio and 95% Wald confidence limits were calculated from stratified Cochran-Mantel-Haenszel test | None | None |
Cumulative steroid dosing until day 56 (secondary end point) | Cumulative steroid dose (overall and weekly) for each patient up to day 56 or discontinuation of randomized treatment was tabulated; RDI by week relative to the starting dose of corticosteroids was categorized as a complete reduction (patients were tapered off corticosteroids by day 56), a RDI ≤ 50%, or a RDI > 50%; the proportion of patients in each category with 95% CIs were presented by study group; and average corticosteroid dose for weeks ending on days 14, 28, 56, 84, and 168 was presented. | None | None |
Incidence of cGvHD (secondary end point) | Cumulative incidence of cGvHD and estimates at 1, 2, 6, 12, 18 and 24 months with 95% CIs were assessed, accounting for competing risks; cumulative incidence curves were presented | None | None |
Resource use | Summary statistics were provided for each study group. For patients starting treatment while hospitalized:
For all patients on study:
| None | None |
aGvHD = acute graft-versus-host disease; BMT = bone marrow transplant; BOR = best overall response; cGvHD = chronic graft-vs.-host disease; CI = confidence interval; DOR = duration of response; EFS = event-free survival; EQ-5D-5L = 5-level EQ-5D; FACT-BMT = Functional Assessment of Cancer Therapy-Bone Marrow Transplant; FFS = failure-free survival; HR = hazard ratio; KM = Kaplan–Meier; NRM = nonrelapse mortality; ORR = overall response rate; OS = overall survival; RDI = relative dose intensity; SD = standard deviation; SR = steroid refractory; VAS = visual analogue scale; vs. = versus.
aORR at crossover is defined as the proportion of crossover patients with a CR or PR at crossover day 28, according to the standard criteria of Harris et al. (2016).16 Response is relative to the last assessment of aGvHD before or at the start of crossover treatment (ruxolitinib).
bDurable ORR at crossover day 56 was defined as the proportion of all crossover patients who achieve a CR or PR at crossover day 28 and maintained a CR or PR at crossover day 56. Response was assessed relative to the last assessment of aGvHD before or at the start of crossover treatment (ruxolitinib).3
Sources: Statistical analysis plan (REACH 2),3 Study Protocol (REACH 2).3
Table 24: Statistical Analysis of Efficacy End Points in REACH 1 (Outcomes Are Presented in Order of Priority as Identified in the CADTH Review Protocol)
End point | Statistical model | Adjustment factors | Sensitivity analyses |
|---|---|---|---|
OS (secondary end point) | KM survival method to estimate median, 6-month survival probability, and 95% CIs (Brookmeyers and Crowley [1982]54) | None | Additional analyses:
|
FFS (secondary outcome) | Same as OS | None | None |
ORR at day 28 (primary end point) | Summarized using descriptive statistics (N, %) and exact binomial 95% CIs; the primary analysis was performed when the last patient completed the day 28 visit or withdrew from the study | None | Additional supportive analyses for the primary end point:
Analysis of BOR rate defined as the proportion of patients achieving a CR, VGPR, or PR any time before starting a new anti-aGvHD therapy |
ORR at days 14, 56, and 100 (secondary end point) | Same as ORR at day 28 | None | None |
DOR at month 6 (key secondary end point) | KM survival method to estimate median and 95% CIs (Brookermeyers and Crowley [1982]54); 6-month DOR was assessed when all patients completed the day 180 visit | None | Additional analyses:
|
DOR at month 3 (secondary end point) | Same as DOR at 6 months; however, 3-month DOR was assessed when all patients completed the day 84 visit | None | None |
NRM (secondary end point) | Cumulative incidence of NRM and derived probabilities at months 6, 9, 12, and 24 with exact binomial 95% CIs were evaluated, considering relapse-related mortality as competing events | None | None |
Relapse rate (secondary end point) | Primary disease relapse rate at month 6 with exact binomial 95% CIs and cumulative incidence rate were evaluated | None | None |
Relapse-related mortality rate (secondary end point) | Same as relapse rate | None | None |
Average and cumulative steroid dosing on days 28, 56, 100, and 180 (exploratory outcome) | Number and percentage of patients receiving corticosteroids while still receiving ruxolitinib at certain time points, and average and relative (to initial dose) corticosteroid dose were assessed | None | None |
Incidence of cGvHD (exploratory end point) | Proportion of patients with cGvHD was calculated | None | None |
aGvHD = acute graft-versus-host disease; BOR = best overall response; cGvHD = chronic graft-vs.-host disease; CI = confidence interval; CNI = calcineurin inhibitor; CR = complete response; DOR = duration of response; FFS = failure-free survival; GI = gastrointestinal; GvHD = graft-vs.-host disease KM = Kaplan–Meier; NRM = nonrelapse mortality; ORR = overall response rate; OS = overall survival; SR = steroid refractory; PR = partial response; VGPR = very good partial response; vs. = versus.
aParticipants who had progressive GvHD after 3 days of primary treatment, GvHD that had not improved after 7 days of primary treatment, previously started steroid therapy at a lower dose but developed new GvHD in another organ system, and who could not tolerate a steroid taper.
Sources: Statistical analysis plan (REACH 1),3 Clinical Study Report (REACH 1).19
Analysis Populations
REACH 2
At the time of the primary analysis, the primary end point and all secondary end points were analyzed using the full analysis set, as defined in Table 25. In supportive analyses at the primary analysis, ORR at day 28 was analyzed using the per-protocol set, as defined in Table 25. The crossover analysis set included patients randomized to the BAT group who crossed over to ruxolitinib treatment between day 28 and week 24. This set was used for all analyses for the crossover patients. Analyses of safety were performed using the safety population.
REACH 1
At the final analysis, all efficacy analyses were analyzed using the efficacy-evaluable population, as defined in Table 25. Analysis of safety was performed using the safety-evaluable population.
Table 25: Analysis Populations in the REACH 2 and REACH 1 Trials
Analysis population | Description |
|---|---|
REACH 2 | |
Full analysis set | All patients who were randomized, regardless of whether they received the study treatment or not per the ITT principle |
Per-protocol set | Patients who met the requirements of the trial protocol and experienced none of the following protocol deviations:
|
Crossover analysis set | Patients randomized to BAT who then crossed over and received at least 1 dose of ruxolitinib |
Safety population | All randomized patients who received at least 1 dose of the study drug |
REACH 1 | |
Efficacy-evaluable population | All enrolled patients |
Safety-evaluable population | All enrolled patients who received at least 1 dose of the study drug |
aGvHD = acute graft-versus-host disease; BAT = best available therapy; CNI = calcineurin inhibitor; ITT = intention-to-treat; SR = steroid refractory.
Sources: Clinical Study Report (REACH 2),18 Clinical Study Report (REACH 1).19
Results
Patient Disposition
REACH 2
Details of the patient disposition in the REACH 2 trial are summarized in Table 26. A total of 620 patients were screened and, of those, 49.8% (N = 309) of patients were randomized to receive ruxolitinib (n = 154) or BAT (n = 155). Reasons for not being randomized included not meeting the inclusion criteria (n = 296), death (n = 7), patient chose not to participate (n = 4), and other reasons (n = 3). The 302 (97.7%) randomized patients who were treated included 152 of 154 (98.7%) patients in the ruxolitinib group and 150 of 154 (96.8%) patients in the BAT group. As of the primary analysis (July 25, 2019, data cut-off date), 111 (72.1%) patients in the ruxolitinib group and 132 (85.2%) patients in the BAT group had discontinued treatment. The main reasons for discontinuation of the assigned treatment were (ruxolitinib versus BAT) lack of efficacy (20.8% versus 43.9%), AEs (16.9% versus 3.2%), and death (16.2% versus 14.2%). At the primary analysis, 12 (7.8%) patients in the ruxolitinib group and 6 (3.9%) patients in the BAT group were still on randomized treatment.18
As of the secondary analysis (January 6, 2020, data cut-off date), 116 (75.3%) patients in the ruxolitinib group and 134 (86.5%) patients in the BAT group had discontinued treatment. The main reasons for discontinuation of the assigned treatment were (ruxolitinib versus BAT) lack of efficacy (20.8% versus 44.5%), AEs (17.5% versus 3.9%), and death (16.2% versus 13.5%). At the secondary analysis, 3 (1.9%) patients in the ruxolitinib group and 0 patients in the BAT group were still on randomized treatment.18
At the final analysis, no patient was on ongoing treatment, and 22.7% and 12.9% of patients in the ruxolitinib and BAT groups, respectively, had completed the treatment period (see Table 26).
Table 26: Patient Disposition in REACH 2, Full Analysis Set (Data Cut-Off Dates of July 25, 2019, January 6, 2020, and April 23, 2021)
Variable | Ruxolitinib | BAT | |||||||
|---|---|---|---|---|---|---|---|---|---|
Data cut-off date | July 25, 2019 | January 6, 2020 | April 23, 2021 | ||||||
Treatment | Ruxolitinib | BAT | Ruxolitinib | BAT | Ruxolitinib | BAT | |||
Screened, n | 620 | ||||||||
Enrolled, n | 309 | ||||||||
Randomized, n | 154 | 155 | |||||||
Treated, n (%) | 152 (98.7) | 150 (96.8) | 152 (98.7) | 150 (96.8) | 152 (98.7) | 150 (96.8) | |||
Not treated, n (%) | 2 (1.3) | 5 (3.2) | 2 (1.3) | 5 (3.2) | 2 (1.3) | 5 (3.2) | |||
Treatment ongoinga | 12 (7.8) | 6 (3.9) | 3 (1.9) | 0 | 0 | 0 | |||
Completed treatment period | 31 (20.1) | 17 (11.0) | 35 (22.7) | 21 (13.5) | 35 (22.7) | 20 (12.9) | |||
Discontinued from treatment period, n (%) | 111 (72.1) | 132 (85.2) | 116 (75.3) | 134 (86.5) | 119 (77.3) | 135 (87.1) | |||
Reason for discontinuation from treatment phase, n (%) | |||||||||
Lack of efficacy | 32 (20.8) | 68 (43.9) | 32 (20.8) | 69 (44.5) | 32 (20.8) | 69 (44.5) | |||
AEs | 26 (16.9) | 5 (3.2) | 27 (17.5) | 6 (3.9) | 27 (17.5) | 5 (3.2) | |||
Death | 25 (16.2) | 22 (14.2) | 25 (16.2) | 21 (13.5) | 25 (16.2) | 22 (14.2) | |||
Failure to meet protocol continuation criteria | 10 (6.5) | 9 (5.8) | 12 (7.8) | 9 (5.8) | 13 (8.4) | 10 (6.5) | |||
Disease relapse | 7 (4.5) | 12 (7.7) | 8 (5.2) | 13 (8.4) | 8 (5.2) | 13 (8.4) | |||
Physician decision | 6 (3.9) | 8 (5.2) | 6 (3.9) | 9 (5.8) | 8 (5.2) | 9 (5.8) | |||
Patient or guardian decision | 4 (2.6) | 7 (4.5) | 4 (2.6) | 6 (3.9) | 4 (2.6) | 6 (3.9) | |||
Graft loss | 1 (0.6) | 0 | 2 (1.3) | 0 | 2 (1.3) | 0 | |||
Lost to follow-up | 0 | 0 | 0 | 0 | 0 | 0 | |||
Pregnancy | 0 | 0 | 0 | 0 | 0 | 0 | |||
Protocol deviation | 0 | 0 | 0 | 0 | 0 | 0 | |||
Study terminated by sponsor | 0 | 0 | 0 | 0 | 0 | 0 | |||
Technical problems | 0 | 0 | 0 | 1 (0.6) | 0 | 1 (0.6) | |||
Continued to next phase at the end of randomized treatment, n (%) | |||||||||
Crossover treatment | 0 | 49 (31.6) | 0 | 49 (31.6) | 0 | 49 (31.6) | |||
Entered long-term follow-up | 87 (56.5) | 45 (29.0) | 45 (29.0) | 51 (32.9) | 102 (66.2) | 51 (32.9) | |||
Analysis sets (all randomized patients) | |||||||||
Full analysis set, n (%) | 154 (100.0) | 155 (100.0) | |||||||
Per-protocol set, n (%) | 97 (63.0) | 87 (56.1) | |||||||
Crossover analysis set, n (%) | 0 | 49 (31.6) | |||||||
Safety, n (%) | 152 (98.7) | 150 (96.8) | |||||||
AE = adverse event; BAT = best available therapy.
aOngoing at the time of the data cut-off date.
Source: Clinical Study Report (REACH 2).18
About 1-third of patients (n = 49; 31.6%) randomized to the BAT group crossed over to the ruxolitinib group between day 28 and week 24. At the time of the primary and secondary analyses, respectively, 10 (20.4%) and 11 (22.4%) patients had completed the crossover treatment period with ruxolitinib. At the primary and secondary analyses, 6 (12.2%) and 2 (4.1%) patients, respectively, were still receiving ruxolitinib (see Table 27). Of the patients who crossed over to ruxolitinib, 33 (67.3%) and 36 (73.5%) patients discontinued the crossover treatment period at the primary and secondary analyses, respectively. Reasons for discontinuing ruxolitinib treatment (primary analysis and secondary analysis) included AEs (20.4% and 24.5%), death (16.3% and 16.3%), and lack of efficacy (12.2% and 12.2%). At the primary and secondary analyses, respectively, 24 (49.0%) and 27 (55.1%) patients entered long-term survival follow-up.18
Table 27: Patient Disposition in REACH 2, Crossover Analysis Set (Data Cut-Off Dates of July 25, 2019, and January 6, 2020)
Variable | Ruxolitinib N = 49 | |
|---|---|---|
Data cut-off date | July 25, 2019 | January 6, 2020 |
Treated, n (%) | 49 (100.0) | 49 (100.0) |
Treatment ongoinga | 6 (12.2) | 2 (4.1) |
Completed crossover treatment period | 10 (20.4) | 11 (22.4) |
Discontinued from crossover treatment, n (%) | 33 (67.3) | 36 (73.5) |
Reason for discontinuation from crossover treatment, n (%) | ||
AEs | 10 (20.4) | 12 (24.5) |
Death | 8 (16.3) | 8 (16.3) |
Lack of efficacy | 6 (12.2) | 6 (12.2) |
Physician decision | 3 (6.1) | 3 (6.1) |
Disease relapse | 2 (4.1) | 2 (4.1) |
Failure to meet protocol continuation criteria | 2 (4.1) | 3 (6.1) |
Patient or guardian decision | 2 (4.1) | 2 (4.1) |
Graft loss | 0 | 0 |
Lost to follow-up | 0 | 0 |
Pregnancy | 0 | 0 |
Protocol deviation | 0 | 0 |
Study terminated by sponsor | 0 | 0 |
Technical problems | 0 | 0 |
Continued to next phase at the end of crossover treatment | ||
Entered long-term follow-up | 24 (49.0) | 27 (55.1) |
AE = adverse event.
aOngoing at the data cut-off date.
Source: Clinical Study Report (REACH 2).18
REACH 1
Details of patient disposition in the REACH 1 trial are summarized in Table 28. A total of 85 patients were screened and, of those, 71 patients were enrolled. The 14 patients not enrolled failed to meet the trial eligibility criteria, and 1 of those failed to meet written informed consent. All patients enrolled in the trial received ruxolitinib. As of the final analysis (June 5, 2019), 24 (33.8%) and 68 (95.8%) patients had discontinued treatment on or before day 28 and at the end of the study, respectively. The most common reasons for treatment discontinuation were physician decision and AEs.19
At the June 5, 2019, data cut-off date, 47 (66.2%) patients had discontinued the study treatment. The main reason for study discontinuation was death (44 patients) and withdrawal from the study (3 patients). Of the 24 patients (33.8%) who remained in the study, 3 patients were continuing the study treatment (they were transferred to a commercial product outside the study) and the other 21 patients had discontinued any study follow-up assessments.19
Table 28: Patient Disposition in REACH 1, All Enrolled Patients (Data Cut-Off Date of June 5, 2019)
Variable | Ruxolitinib |
|---|---|
Enrolled patients, n (%) | 71 (100.0) |
Treated patients, n (%) | 71 (100.0) |
Patients who discontinued ruxolitinib treatment on or before day 28, n (%) | 24 (33.8) |
Primary reason for ruxolitinib treatment discontinuation on or before day 28, n (%) | |
Physician decision | 10 (14.1) |
AEs | 8 (11.3) |
Progression of GvHD | 4 (5.6) |
Death | 1 (1.4) |
Withdrawal by participant | 1 (1.4) |
Patients who discontinued ruxolitinib treatment, n (%) | 68 (95.8) |
Primary reason for ruxolitinib treatment discontinuation, n (%) | |
Physician decisiona | 23 (32.4) |
AEs | 20 (28.2) |
Death | 7 (9.9) |
Progression of GvHD | 7 (9.9) |
Otherb | 5 (7.0) |
Relapse of underlying malignancy | 3 (4.2) |
Withdrawal by participant | 3 (4.2) |
Patients continuing treatment, n (%)c | 3 (4.2) |
Patients who discontinued the study by day 28, n (%) | 10 (14.1) |
Primary reason for discontinuation on or before day 28, n (%) | |
Death | 10 (14.1) |
Patients who remained in the study, n (%) | 24 (33.8) |
Patients who discontinued the study, n (%) | 47 (66.2) |
Primary reason for discontinuation from study, n (%) | |
Death | 44 (62.0) |
Withdrawal by patient | 3 (4.2) |
Analysis sets | |
Efficacy-evaluable patients, n (%) | 71 (100.0) |
Safety-evaluable patients, n (%) | 71 (100.0) |
AE = adverse event; GvHD = graft-vs.-host disease.
aIncludes 6 participants who discontinued ruxolitinib treatment because of clinical improvement: 4 achieved a CR, 1 achieved a VGPR, and 1 had experienced malignancy relapse at the EOT visit.
bIncludes 2 participants who discontinued ruxolitinib treatment because of clinical improvement (both had a CR at the EOT visit).
cParticipants were transferred to a commercial product.
Source: Clinical Study Report (REACH 1).19
Protocol Deviations
REACH 2
Protocol deviations were reported for the primary and secondary analyses (see Table 29). The type and frequency of deviations were similar at the analyses cut-off points. As of the January 6, 2020, data cut-off date, protocol deviations occurred in 131 (85.1%) and 135 (87.1%) patients in the ruxolitinib and BAT groups, respectively. Patients may have had more than 1 violation. The overall type and frequency of protocol deviations appeared balanced between the treatment groups, except the deviation of investigational study treatment dispensing error, which occurred more frequently in the BAT group (n = 61; 39.4%) than in the ruxolitinib group (n = 43; 27.9%).18 When asked, the sponsor explained that dispensing error referred to a variety of protocol deviations in which the study treatment was received differently than set out in the clinical trial plan.21
The most commonly reported protocol deviation was other deviations (63.0% for ruxolitinib versus 63.9% for BAT), which included differences in aGvHD overall grades used for randomization between electronic case report forms and Interactive Response Technology (that includes Interactive Voice Response System and Interactive Web Response System); missing 2 consecutive monthly scheduled viral load tests; missing aGvHD assessment at day 28 or day 56; organ staging assessment done per investigator criteria or judgment rather than the criteria of Harris et al. (2016);16 response assessment done per investigator criteria or judgment rather than the protocol definition; and the implementation of protocol Amendment 2 before a patient's re-consent was obtained. When asked, the sponsor reported that a more detailed breakdown of other deviations was not available.21 Inclusion criteria deviations were observed in 36.4% of patients in the ruxolitinib group and 38.7% of patients in the BAT group; exclusion criteria deviations were observed in 2.6% of patients in the ruxolitinib group and 2.6% of patients in the BAT groups. Deviations associated with prohibited drugs taken occurred in 20.8% and 18.1% of patients in the ruxolitinib and BAT groups, respectively.3,18
REACH 1
Protocol deviations were reported for the final analysis (June 5, 2019) (see Table 30) and occurred in 64 (90.1%) patients. None of the protocol deviations were related to inclusion or exclusion criteria. It was noted in the Clinical Study Report that no protocol deviations had a significant impact on the completeness, accuracy, and/or reliability of the study data or the study conclusions. Key deviations included:19
informed consent (1 participant signed an outdated version of the informed consent form)
concomitant medication (1 patient started chemotherapy during ruxolitinib treatment and discontinued ruxolitinib less than a week later)
study procedure (1 patient missed 5 study visits and study assessment were not performed; 1 patient missed daily ruxolitinib doses 9 times, with no negative impact on aGvHD response; 1 patient took ruxolitinib for 3 successive days with a platelet count below 50% baseline and an ANC < 1.0 × 109/L).
Table 29: Protocol Deviations in REACH 2, Full Analysis Set (Data Cut-Off Dates of July 25, 2019, and January 6, 2020)
Protocol deviations, n (%) | Ruxolitinib N = 154 | BAT N = 155 | Ruxolitinib N = 154 | BAT N = 155 |
|---|---|---|---|---|
Data cut-off date | July 25, 2019 | January 6, 2020 | ||
Any protocol deviation | 131 (85.1) | 135 (87.1) | 132 (85.7) | 134 (86.5) |
Any exclusion criteria deviation | 4 (2.6) | 3 (1.9) | 4 (2.6) | 4 (2.6) |
Exclusion criteria of concomitant medications not met | 2 (1.3) | 0 | 2 (1.3) | 0 |
Exclusion criterion of absence of cGvHD — de novo or overlap syndrome — met | 1 (0.6) | 1 (0.6) | 1 (0.6) | 1 (0.6) |
Exclusion criteria of absence of significant or uncontrolled cardiac disease not met | 1 (0.6) | 0 | 1 (0.6) | 0 |
Exclusion criteria of prior systemic aGvHD therapy met | NR | NR | 0 | 1 (0.6) |
Exclusion criteria of absence of severely impaired renal function not met | 0 | 1 (0.6) | 0 | 1 (0.6) |
Exclusion criteria of use of JAK therapy not met | NR | NR | 0 | 1 (0.6) |
Exclusion criteria of absence of relapsed primary malignancy not met | 0 | 1 (0.6) | 0 | 1 (0.6) |
Any inclusion criteria deviation | 56 (36.4) | 61 (39.4) | 56 (36.4) | 60 (38.7) |
Viral load assessment at screening or day 1 is beyond protocol-acceptable window | 26 (16.9) | 25 (16.1) | 26 (16.9) | 24 (15.5) |
Chimerism at screening or day 1 is beyond protocol-acceptable window | 20 (13.0) | 18 (11.6) | 20 (13.0) | 18 (11.6) |
Study informed consent not obtained | 8 (5.2) | 7 (4.5) | 8 (5.2) | 7 (4.5) |
SR-aGvHD criteria not met | 7 (4.5) | 15 (9.7) | 7 (4.5) | 15 (9.7) |
aGvHD grade criteria not met | 6 (3.9) | 3 (1.9) | 6 (3.9) | 3 (1.9) |
Myeloid platelet engraftment not confirmed | 3 (1.9) | 6 (3.9) | 3 (1.9) | 6 (3.9) |
Screening informed consent not obtained | 2 (1.3) | 0 | 2 (1.3) | 0 |
Other deviation | 95 (61.7) | 101 (65.2) | 97 (63.0) | 99 (63.9) |
Prohibited concomitant medication | 31 (20.1) | 28 (18.1) | 32 (20.8) | 28 (18.1) |
Patient not withdrawn, per protocol | 10 (6.5) | 19 (12.3) | 13 (8.4) | 21 (13.5) |
Study treatment continued because a withdrawal criterion was met | 6 (3.9) | 0 | 7 (4.5) | 1 (0.6) |
Study treatment withdrawal criteria not met | 5 (3.2) | 19 (12.3) | 7 (4.5) | 20 (12.9) |
Treatment deviation | 44 (28.6) | 60 (38.7) | 44 (28.6) | 61 (39.4) |
Investigational study treatment dispensing error | 43 (27.9) | 60 (38.7) | 43 (27.9) | 61 (39.4) |
Criteria for dose reduction or interruption not followed | 3 (1.9) | 3 (1.9) | 3 (1.9) | 3 (1.9) |
aGvHD = acute graft-versus-host disease; BAT = best available therapy; JAK = Janus-associated kinase; NR = not reported; SR = steroid refractory.
Note: A patient can have more than 1 deviation. Protocol deviation of “Study Informed Consent not obtained” should be “Screening Informed Consent obtained after screening procedures were performed,” per corrected study specification document.
Source: Clinical Study Report (REACH 2).18
Table 30: Protocol Deviations in REACH 1, Efficacy-Evaluable Population (Data Cut-Off Date of June 5, 2019)
Protocol deviation category, n (%) | Ruxolitinib N = 71 |
|---|---|
Patients with any deviation | 64 (90.1) |
AEs | 0 |
Informed consent | 2 (2.8) |
Entry criteria | 1 (1.4) |
Concomitant medications | 0 |
Noncompliance with study | 8 (11.3) |
Noncompliance with study procedure, out of window assessment | 46 (64.8) |
Safety, vital signs | 5 (7.0) |
Safety, physical exam | 5 (7.0) |
Safety, clinical lab tests | 10 (14.1) |
Safety, ECG | 3 (4.2) |
Efficacy, evaluation | 2 (2.8) |
Pharmacokinetic or correlative sampling | 30 (42.3) |
Other | 11 (15.5) |
Noncompliance with study procedure, missed assessment | 52 (73.2) |
Safety, vital signs | 14 (19.7) |
Safety, physical exam | 4 (5.6) |
Safety, clinical lab tests | 34 (47.9) |
Safety, ECG | 6 (8.5) |
Efficacy, evaluation | 6 (8.5) |
Pharmacokinetic or correlative sampling | 10 (14.1) |
Other | 25 (35.2) |
Other, n (%) | 16 (22.5) |
AE = adverse event; ECG = electrocardiogram.
Note: Patients can have more than 1 deviation.
Source: Clinical Study Report (REACH 1).19
Exposure to Study Treatments
REACH 2
Exposure to ruxolitinib and BAT as of the primary and secondary analyses is summarized in Table 31. Treatment duration and exposure at the secondary analysis remained consistent with the primary analysis. As of the July 25, 2019, data cut-off date, the median duration of treatment with ruxolitinib of 82.5 (range = 8 to 396) days was close to twice that of the median treatment duration with BAT of 45.5 (range = 2 to 218) days. More patients had discontinued treatment at or before day 28 in the BAT group (n = 43; 28.7%) than in the ruxolitinib group (n = 16; 10.5%). The median duration of exposure in the ruxolitinib group of 63 (range = 6 to 396) days was approximately twice that of the median duration of exposure in the BAT group of 29 (range = 1 to 188) days.
Exposure to ruxolitinib in the crossover analysis set at the primary and secondary analyses is summarized in Table 32. As of the July 25, 2019 data cut-off date, the median duration of treatment with ruxolitinib was 63.0 (range = 2.0 to 266.0) days.18
For the adolescents enrolled in the REACH 2 trial, the median duration of exposure was longer in the ruxolitinib group (163.0 days; range = 11.0 to 242.0 days) than in the BAT group (58.0 days; range = 2.0 to 162.0 days).18
The median dose intensity and median RDI of ruxolitinib up to day 28 was high, indicating good treatment adherence. The median dose intensity for ruxolitinib was 20.0 mg/day (mean = 18.2 mg/day; SD = 2.96; range = 8.4 mg/day to 21.0 mg/day) up to the day 28 visit; the median RDI for ruxolitinib was 100.0% (mean = 91.2%; SD = 14.81; range = 42.0% to 104.9%) up to the day 28 visit. The median dose intensity for ruxolitinib up to day 56 was 19.2 mg/day (mean = 17.3 mg/day; SD = 3.25; range = 8.4 mg/day to 20.7 mg/day); the median RDI for ruxolitinib was 95.8% (mean = 86.7%; SD = 16.24; range = 42.0% to 103.4%). The median dose intensity for ruxolitinib up to the end of the randomized treatment period was 16.8 mg/day (mean = 15.8 mg/day; SD = 3.82; range = 8.3 mg/day to 20.0 mg/day).18
During the crossover period (49 patients), the median dose intensity for ruxolitinib was 17.0 mg/day (mean = 15.8 mg/day; SD = 4.15; range = 4.7 mg/day to 20.0 mg/day).18
Table 31: Duration of Randomized Treatment Period in REACH 2, Safety Set (Data Cut-Off Dates of July 25, 2019, and January 6, 2021)
Categories | Ruxolitinib N = 152 | BAT N = 150 | Ruxolitinib N = 152 | BAT N = 150 |
|---|---|---|---|---|
Data cut-off date | July 25, 2019 | January 6, 2020 | ||
Duration of treatment period, days | ||||
Mean (SD) | 110.2 (79.81) | 71.7 (56.14) | 118.8 (93.56) | 73.6 (59.12) |
Median | 82.5 | 45.5 | 85.5 | 45.5 |
Duration of treatment period categories, n (%) | ||||
≤ 28 days | 16 (10.5) | 43 (28.7) | 16 (10.5) | 43 (28.7) |
> 28 to 56 days | 33 (21.7) | 41 (27.3) | 33 (21.7) | 42 (28.0) |
> 56 to 112 days | 45 (29.6) | 30 (20.0) | 43 (28.3) | 26 (17.3) |
> 112 to 168 days | 14 (9.2) | 19 (12.7) | 12 (7.9) | 17 (11.3) |
> 168 to 336 days | 41 (27.0) | 17 (11.3) | 41 (27.0) | 22 (14.7) |
> 336 to 672 days | 3 (2.0) | 0 | 7 (4.6) | 0 |
> 672 days | 0 | 0 | 0 | 0 |
Duration of exposure, days | ||||
Mean (SD) | 88.2 (76.94) | 45.8 (42.40) | 94.9 (89.73) | 46.3 (44.64) |
Median | 63.0 | 29.0 | 63.0 | 29.0 |
Duration of exposure categories, n (%) | ||||
≤ 28 days | 43 (28.3) | 71 (47.3) | 43 (28.3) | 73 (48.7) |
> 28 to 56 days | 28 (18.4) | 38 (25.3) | 28 (18.4) | 37 (24.7) |
> 56 to 112 days | 30 (19.7) | 27 (18.0) | 29 (19.1) | 24 (16.0) |
> 112 to 168 days | 27 (17.8) | 10 (6.7) | 24 (15.8) | 11 (7.3) |
> 168 to 336 days | 23 (15.1) | 4 (2.7) | 25 (16.4) | 5 (3.3) |
> 336 to 672 days | 1 (0.7) | 0 | 3 (2.0) | 0 |
> 672 days | 0 | 0 | 0 | 0 |
BAT = best available therapy; SD = standard deviation.
Source: Clinical Study Report (REACH 2).18
Table 32: Duration of Crossover Treatment Period in REACH 2, Crossover Analysis Set (Data Cut-Off Dates of July 25, 2019, and January 6, 2020)
Categories | Ruxolitinib N = 49 | |
|---|---|---|
Data cut-off date | July 25, 2019 | January 6, 2020 |
Duration of treatment period, days | ||
Mean (SD) | 97.5 (70.52) | 112.6 (91.51) |
Median | 63.0 | 79.0 |
Duration of treatment period categories, n (%) | ||
≤ 28 days | 7 (14.3) | 7 (14.3) |
> 28 to 56 days | 11 (22.4) | 10 (20.4) |
> 56 to 112 days | 14 (28.6) | 13 (26.5) |
> 112 to 168 days | 5 (10.2) | 4 (8.2) |
> 168 to 336 days | 12 (24.5) | 13 (26.5) |
> 336 to 672 days | 0 | 2 (4.1) |
> 672 days | 0 | 0 |
Duration of exposure, days | ||
Mean (SD) | 81.1 (67.79) | 91.4 (84.20) |
Median | 61.0 | 61.0 |
Duration of exposure categories, n (%) | ||
≤ 28 days | 14 (28.6) | 14 (28.6) |
> 28 to 56 days | 10 (20.4) | 10 (20.4) |
> 56 to 112 days | 10 (20.4) | 8 (16.3) |
> 112 to 168 days | 7 (14.3) | 6 (12.2) |
> 168 to 336 days | 8 (16.3) | 10 (20.4) |
> 336 to 672 days | 0 | 1 (2.0) |
> 672 days | 0 | 0 |
SD = standard deviation.
Source: Clinical Study Report (REACH 2).18
Exposure to BAT Treatment
Initial BAT treatment and the number of lines of BAT treatments started in the first 28 days are summarized in Table 33. The majority of patients (79.3%) in the BAT group received 1 BAT treatment, but 18.0% of patients received 2 BAT treatments and 2.7% of patients received more than 2 lines of BAT treatments. Most patients (27.3%) received ECP as initial BAT treatment, followed by MMF (16.7%) and etanercept (14.7%). The most frequently administered second-line BAT treatments were etanercept and MMF (administered to more than 5% of patients who received at least 2 lines of BAT before day 28).18
The median duration of exposure to ECP was 47.5 days (range = 2 to 173) and the mean was 58.8 (SD = 46.69) days. The median treatment exposure to MMF was 28.0 days (range = 5 to 188) and the mean was 43.1 (SD = 41.63) days. The median treatment exposure with etanercept was 28.0 days (range = 1 to 179) and the mean was 40 (SD = 44.22) days.18
Table 33: Initial BAT and Number of BAT Treatments in REACH 2, Safety Set (Data Cut-Off Dates of July 25, 2019, and January 6, 2020)
BAT therapies, n (%) | BAT N = 150 | |
|---|---|---|
Data cut-off date | July 25, 2019 | January 6, 2020 |
Initial BAT | ||
ECP | 41 (27.3) | 41 (27.3) |
MMF | 25 (16.7) | 25 (16.7) |
Etanercept | 22 (14.7) | 22 (14.7) |
ATG | 20 (13.3) | 20 (13.3) |
Infliximab | 17 (11.3) | 17 (11.3) |
MSCs | 15 (10.0) | 15 (10.0) |
Low-dose methotrexate | 5 (3.3) | 5 (3.3) |
Sirolimus | 3 (2.0) | 3 (2.0) |
Everolimus | 2 (1.3) | 2 (1.3) |
Number of BATs, (%) | ||
1 | 119 (79.3) | 119 (79.3) |
2 | 27 (18.0) | 27 (18.0) |
> 2 | 4 (2.7) | 4 (2.7) |
ATG = antithymocyte globulin; BAT = best available therapy; ECP = extracorporeal photopheresis; MMF = mycophenolate mofetil; MSC = mesenchymal stromal cell.
Source: Clinical Study Report (REACH 2).18
Exposure to Immunosuppressive Therapy
During the treatment period, the overall duration of concurrent treatment with systemic steroids was similar in the 2 study groups. The median duration of concurrent systemic steroid treatment was 56.0 (mean = 72.9; SD = 54.85; range = 8 to 246) days in the ruxolitinib group and 41.5 (mean = 64.7; SD = 52.28; range = 2 to 199) days in the BAT group, which was consistent with the discontinuation of patients from the randomized treatment period being earlier in the BAT group than in the ruxolitinib group. The median duration of concurrent systemic steroid treatment was 54.0 (mean = 62.6; SD = 49.33; range = 1 to 208) days during the crossover treatment period.18
During the treatment period, the duration of treatment with CNIs was longer in the ruxolitinib group (median = 73.5 days; mean = 94.1 days; SD = 74.69; range = 1 to 396 days) than in the BAT group (median = 32.5 days; mean = 61.2 days; SD = 53.17; range = 1 to 199 days), which was consistent with the discontinuation of patients from the randomized treatment period being earlier in the BAT group than in the ruxolitinib group. The median duration of CNI treatment was 61.0 (mean = 79.1; SD = 62.70; range = 6 to 218) days during the crossover treatment period.18
Dose of Steroids
As of the primary data cut-off date, the average daily dose of steroids was similar in the ruxolitinib and BAT study groups up to day 28 (mean ± SD = 102.23 mg/day ± 44.71 mg/day versus 109.84 mg/day ± 66.60 mg/day). Patients in the ruxolitinib group had a lower maximum daily dose (270.0 mg/day) than those in the BAT group (493.2 mg/day) up to day 28. The mean RDI up to day 28 was similar in the ruxolitinib and BAT groups (72.86% versus 75.81%). Average daily doses up to day 56 for the ruxolitinib and BAT groups were (mean ± SD) 86.9 mg/day ± 44.17 mg/day and 98.2 ± 65.72 mg/day, respectively, and up to the primary data cut-off date were 78.9 mg/day ± 46.41 mg/day and 91.6 mg/day ± 67.99 mg/day, respectively.18
The results at the secondary data cut-off date were, overall, consistent with the primary data cut-off date. The average daily dose of steroids up to day 28 in the ruxolitinib and BAT groups was (mean ± SD) 101.95 mg/day ± 44.72 mg/day and 109.71 mg/day ± 66.56 mg/day, respectively. Patients in the ruxolitinib group had a lower maximum daily dose (270.0 mg/day) than those in the BAT group (493.2 mg/day) up to day 28. The mean RDI up to day 28 was similar in the ruxolitinib and BAT study groups (72.62% versus 75.99%). Average daily doses up to day 56 in the ruxolitinib and BAT groups were (mean ± SD) 86.61 mg/day ± 44.07 mg/day and 98.13 mg/day ± 65.68 mg/day, respectively, and up to the secondary data cut-off date were 78.44 mg/day ± 46.41 mg/day and 91.55 ± 68.06 mg/day, respectively.18
REACH 1
Exposure to ruxolitinib at the final analysis is summarized in Table 34. As of the June 5, 2019 data cut-off date, the median duration of treatment with ruxolitinib was 46.0 (range = 4 to 811) days, and the median average reported daily dose was 10.21 mg/day (range = 5.1 mg/day to 19.7 mg/day).19
On day 1, all except 2 patients enrolled in the REACH 1 trial received ruxolitinib 5 mg twice daily, per protocol (see Table 34). By day 7, all patients except 4 were still receiving ruxolitinib; of those, approximately half received ruxolitinib 10 mg twice daily, per protocol. The rest of the patients received less than the per-protocol dose, with the majority receiving 5 mg twice daily. By day 28, 43 patients were still on treatment with ruxolitinib, 20 (46.5%) of whom received ruxolitinib 10 mg twice daily and the remainder of whom received less (most commonly ruxolitinib 5 mg twice daily).19
Table 34: Summary of Ruxolitinib Exposure and Compliance in REACH 1, Safety-Evaluable Population (Data Cut-Off Date of June 5, 2019)
Variable | Ruxolitinib N = 71 |
|---|---|
Duration of treatment, daysa | |
n | 71 |
Mean (SD) | 132.0 (183.63) |
Median | 46.0 |
Exposure period categories, days, n (%) | |
≤ 28 | 29 (40.8)b |
> 28 to 56 | 14 (19.7) |
> 56 to 100 | 5 (7.0) |
> 100 to 180 | 5 (7.0) |
> 180 | 18 (25.4) |
Average reported daily dose, mg/dayc | |
n | 71 |
Mean (SD) | 11.89 (4.408) |
Median | 10.21 |
Compliance (%)d | |
n | 71 |
Mean (SD) | 99.38 (1.654) |
Median | 100.00 |
Prescribed dose of ruxolitinib on day 1, n (%) | |
Total, n | 71 |
5 mg once daily | 2 (2.8) |
5 mg twice daily | 69 (97.2) |
Prescribed dose of ruxolitinib on day 7, n (%) | |
Total, n | 67 |
5 mg once daily | 2 (3.0) |
10 mg once daily | 1 (1.5) |
15 mg once daily | 1 (1.5) |
5 mg twice daily | 28 (41.8) |
10 mg twice daily | 35 (52.2) |
Prescribed dose of ruxolitinib on day 28, n (%) | |
Total, n | 43 |
0 mg | 2 (4.7) |
5 mg once daily | 7 (16.3) |
10 mg once daily | 0 |
15 mg once daily | 1 (2.3) |
5 mg twice daily | 13 (30.2) |
10 mg twice daily | 20 (46.5) |
SD = standard deviation.
aFor patients with missing data for date of last dose, the treatment duration was computed as minimum (EOT date, end of study date, death date, cut-off date) – date of first dose + 1.
bIncludes 1 participant whose last dose was on day 28.
cAverage reported daily dose (mg/day) = [total reported dose (mg)] / [duration of treatment (days)].
dCompliance (%) = 100 × [total reported dose taken (mg)] / [total prescribed dose (mg)].
Source: Clinical Study Report (REACH 1).19
Exposure to Corticosteroids
Exposure to corticosteroids at the final analysis is summarized in Table 35. As of the June 5, 2019 data cut-off date, the median duration of treatment with corticosteroids was 45.0 days (range = 4 to 341 days); the median initial dose was 156.25 mg/day (range = 50.0 to 300.0 mg/day). By day 28, 43 patients were still receiving corticosteroids and, for those, the median average dose (median of each patient’s average weekly dose) was 62.50 mg/day.
Table 35: Summary of Corticosteroid Exposure in REACH 1, Safety-Evaluable Population (Data Cut-Off Date of June 5, 2019)
Variable | Ruxolitinib N = 71 |
|---|---|
Duration of treatment, daysa | |
n | 71 |
Mean (SD) | 71.7 (77.56) |
Median | 45.0 |
Exposure period categories, days, n (%) | |
≤ 28 | 26 (36.6) |
> 28 to 56 | 17 (23.9) |
> 56 to 100 | 12 (16.9) |
> 100 to 180 | 8 (11.3) |
> 180 | 8 (11.3) |
Total dose of corticosteroids, mgb | |
n | 71 |
Mean (SD) | 3,369.49 (2,115.351) |
Median | 2,800.00 |
Initial corticosteroid dose, mg/dayc | |
n | 71 |
Mean (SD) | 157.25 (62.232) |
Median | 156.25 |
Average corticosteroid dose, in mg/day, during ruxolitinib treatment for the week ending on day 28d | |
n | 43e |
Mean (SD) | 62.25 (32.112) |
Median | 62.50 |
SD = standard deviation.
aFor subjects with missing value of date of last dose, the treatment duration was computed as minimum (EOT date, end of study date, death date, cut-off date) – date of first dose + 1.
bCorticosteroid dose (mg) = methylprednisolone dose (mg) × 1.25 + prednisone dose (mg).
cDay 1 dose when available or day 2 dose if the day 1 dose was missing.
dFor participants who were still receiving ruxolitinib treatment, average corticosteroid dose (mg/day) = total corticosteroid dose (mg) for the week / 7.
eAll 43 participants who were still receiving ruxolitinib on day 28 were also receiving corticosteroids.
Source: Clinical Study Report (REACH 1).19
Dose Modifications (Interruption, Reduction)
REACH 2
Patients who received ruxolitinib and experienced an AE that was considered to be related to ruxolitinib required dose modifications or interruptions, as specified in the protocol. Patients in the BAT group did not have protocol-specified criteria for dose adjustment or interruptions, but were allowed to follow the investigator’s discretion, institutional guidelines, or per-product label.18
As of the primary analysis (July 25, 2019, data cut-off date), 126 (82.9%) patients in the ruxolitinib group in the safety population required at least 1 dose change or interruption and 118 (77.6%) patients required a dose change. The main reason for dose change or interruption was AEs (57.2%), followed by per protocol (39.5%), dose tapering (28.9%), and physician decision (19.1%).18
During the crossover treatment period, all patients who crossed over to receive ruxolitinib required at least 1 dose change (n = 49) and 31 (63.3%) patients required at least 1 dose interruption. The main reason for dose change or interruption was physician decision (91.8%), dose tapering (69.4%), AEs (61.2%), per protocol (46.9%), and re-escalation (38.8%).18
Results at the secondary data cut-off date were, overall, consistent with the results at the primary data cut-off date.18
REACH 1
As of the final analysis (June 5, 2019), 29 (40.8%) and 25 (35.2%) patients had at least 1 TEAE leading to a ruxolitinib dose interruption and reduction, respectively. The most common TEAE leading to dose interruption was thrombocytopenia (9%), followed by neutropenia (8.5%) and sepsis (5.6%). The most commonly reported TEAE leading to a ruxolitinib dose reduction was thrombocytopenia (18.3%), followed by neutropenia (15.5%) and erythropenia (5.6%).19
No TEAE led to the interruption of the corticosteroid dose. There were 19 (26.8%) patients with at least 1 TEAE that led to a corticosteroid dose reduction. The most common TEAE leading to a dose reduction was muscular weakness (4.2%).19
Concomitant Medication
REACH 2
Concomitant medications, as of the July 25, 2019, data cut-off date, were generally similarly administered in the ruxolitinib and BAT treatment groups and were reported for almost all patients (98.7% versus 100.0%). In addition to corticosteroids and CNIs, medications for the treatment of infections, gastric motility enhancers, and electrolytes were among those most commonly used. The most frequently reported Anatomic Therapeutic Chemical classes during the randomized treatment period are summarized in Table 36 and included (ruxolitinib versus BAT): nucleosides and nucleotides, excluding reverse transcriptase inhibitors (92.8% versus 90.7%), proton pump inhibitors (84.9% versus 86.7%), antivirals (80.3% versus 78.0%), triazole derivatives (77.0% versus 70.7%), and glucocorticoids (76.3% versus 76.0%). The most frequently reported concomitant medications by preferred term during the randomized treatment period are summarized in Table 36 and included (ruxolitinib versus BAT): trimethoprim plus sulfamethoxazole (Bactrim) (65.1% versus 56.7%), acyclovir (60.5% versus 56.7%), ursodeoxycholic acid (56.6% versus 56.7%), posaconazole (52.0% versus 46.0%), and paracetamol (44.7% versus 49.3%). The ruxolitinib group had a lower proportion of patients who received furosemide (44.7% versus 57.3%), budesonide (33.6% versus 42.7%), and albumin human (31.6% versus 42.0%). The frequency and type of concomitant medication from randomization to day 28 were similar to the frequency and type at the primary data cut-off date (see Table 36).18
At the primary data cut-off date, 85.5% of patients in the ruxolitinib group and 82.0% in the BAT group received CNIs. The most frequently administered CNIs in the ruxolitinib and BAT groups was cyclosporin (61.2% and 54.7%, respectively).18
During the crossover period, the frequency and type of concomitant medications were similar to those at the primary data cut-off date.18
The profile of concomitant medications at the secondary analyses was similar in the 2 study groups to that at the primary analysis.18
REACH 1
All patients in the REACH 1 trial received at least 1 concomitant medication (see Table 57 in Appendix 3). At the final analysis (June 5, 2019), the most frequently reported Anatomic Therapeutic Chemical classes were nucleosides and nucleotides excluding reverse transcriptase inhibitors (97.2%), CNIs (88.7%), proton pump inhibitors (84.5%), and electrolyte solutions (84.5%). The most frequently reported concomitant medications by preferred term included tacrolimus (84.5%), acyclovir (77.5%), paracetamol (73.2%), pantoprazole (66.2%), potassium chloride (32.4%), ursodeoxycholic acid (64.8%), diphenhydramine (62.0%), and ondansetron (62.0%).19
With regard to concomitant immunosuppressive medications, the most frequently reported CNI was tacrolimus (84.5%) and the most commonly reported selective immunosuppressants were MMF (26.8%) and sirolimus (16.9%). Glucocorticoids were received by 45.1% of patients, with the most common being hydrocortisone (15.5%).19
Table 36: Concomitant Therapies in REACH 2 (Data Cut-Off Date of July 25, 2019)
Concomitant medication | Randomized treatment phase | Randomization to day 28 | ||
|---|---|---|---|---|
Ruxolitinib N = 152 | BAT N = 150 | Ruxolitinib N = 152 | BAT N = 150 | |
ATC class,a % | ||||
Nucleosides and nucleotides excluding reverse transcriptase inhibitors | 92.8 | 90.7 | 91.4 | 88.0 |
Proton pump inhibitors | 84.9 | 86.7 | 83.6 | 84.7 |
Antivirals | 80.3 | 78.0 | 77.0 | 74.0 |
Triazole derivatives | 77.0 | 70.7 | 70.4 | 65.3 |
Glucocorticoids | 76.3 | 76.0 | 56.6 | 68.0 |
Other ophthalmologicals | 75.7 | 74.7 | 66.4 | 72.0 |
Antibiotics | 75.0 | 68.0 | NR | NR |
Combinations of sulfonamides plus trimethoprim, including derivatives | 71.1 | 66.7 | 66.4 | 64.7 |
Corticosteroids acting locally | 67.1 | 68.7 | 49.3 | 62.0 |
Electrolyte solutions | 66.4 | 67.3 | 61.8 | 64.7 |
Corticosteroids, potent (group III) | 65.8 | 66.7 | 51.3 | 62.7 |
Corticosteroids | 65.8 | 66.7 | 52.0 | 62.7 |
Solutions affecting electrolyte balance | 64.5 | 65.3 | 58.6 | 62.0 |
Preferred term,b % | ||||
Sulfamethoxazole plus trimethoprim | 65.1 | 56.7 | 61.2 | 55.3 |
Acyclovir | 60.5 | 56.7 | 57.9 | 54.0 |
Ursodeoxycholic acid | 56.6 | 56.7 | 52.0 | 55.3 |
Posaconazole | 52.0 | 46.0 | NR | NR |
Paracetamol | 44.7 | 49.3 | NR | NR |
Furosemide | 44.7 | 57.3 | NR | NR |
Potassium chloride | 43.4 | 46.0 | 36.2 | 44.0 |
Budesonide | 33.6 | 42.7 | 31.6 | 40.0 |
Albumin human | 31.6 | 42.0 | NR | NR |
ATC = Anatomical Therapeutic Chemical; NR = not reported.
aAt least 60% in each treatment group.
bAt least 40% in each treatment group.
Source: Clinical Study Report (REACH 2).18
Efficacy
Only efficacy outcomes and analyses of subgroups identified in the review protocol are reported in the following. See Appendix 3 for detailed efficacy data.
REACH 2
Although the primary outcome (ORR at day 56), the key secondary outcome (durable ORR at day 56), and other secondary outcomes (ORR at day 14 and BOR at any time up to day 28) were only analyzed at the primary data cut-off date (July 25, 2019), subgroup analyses of ORR at day 28 were reanalyzed at the updated data cut-off date (January 6, 2020) and the final data cut-off date (April 23, 2021), as were the following secondary outcomes: DOR, cumulative steroid dosing until day 56, OS, EFS, FFS, NRM, incidence of malignancy relapse or progression, incidence of cGvHD, HRQoL, and safety.18
REACH 1
All efficacy outcomes were reported for the final data cut-off date of June 5, 2019, with a median follow-up time of 160 (95% CI, 9 to 826) days.
Overall Survival
REACH 2
The OS results for the REACH 2 trial for the ruxolitinib and BAT groups are summarized in Table 37 for the primary (July 25, 2019) and secondary (January 6, 2020) data cut-off dates. A detailed version of Table 37 is available in Table 56 (Appendix 3), and shows OS probabilities. As of the July 25, 2019, data cut-off date, 151 death events had occurred: 72 (46.8%) in the ruxolitinib group and 79 (51.0%) in the BAT group. The median duration of follow-up for OS was 5.04 months in the ruxolitinib group and 3.58 months in the BAT group. Median OS was 11.14 months, or 339 (95% CI, 186 to NE) days, in the ruxolitinib group, and 6.47 months, or 197 (95% CI, 114 to 458) days, in the BAT group, with a stratified HR of 0.83 (95% CI, 0.60 to 1.15).21 The KM curves are depicted in Figure 26 (Appendix 3). The probability of patients surviving at 1 month and 6 months was 90.00 (95% CI, 84.02 to 93.87) and 59.54 (95% CI, 50.92 to 67.14), respectively, in the ruxolitinib group, and 85.48 (95% CI, 78.79 to 90.19) and 50.36 (95% CI, 41.61 to 58.47), respectively, in the BAT group.18
The results at the secondary analysis (January 6, 2020) were, overall, consistent with the results at the primary analysis. As of the secondary analysis, 82 (53.2%) and 88 (56.8%) death events had occurred in the ruxolitinib and BAT groups, respectively. The median duration of follow-up for OS was 7.34 months in the ruxolitinib group and 3.81 months in the BAT group. Median OS was 10.71 months, or 326 (95% CI, 182 to 547) days, in the ruxolitinib group, compared with 5.82 months, or 177 (95% CI, 115 to 392) days, in the BAT group, with a stratified HR of 0.83 (95% CI, 0.62 to 1.13).21 The KM curves are depicted in Figure 4. The probability of patients surviving at 1 month and 6 months was 90.00 (95% CI, 84.02 to 93.87) and 58.27 (95% CI, 49.90 to 65.73), respectively, in the ruxolitinib group, and 85.48 (95% CI, 78.79 to 90.19) and 49.42 (95% CI, 40.89 to 57.37), respectively, in the BAT group.18
The results at the final analysis (April 23, 2021) were, overall, consistent with the results at the primary analysis. The median duration of follow-up for OS was 8.23 months in the ruxolitinib group and 3.81 months in the BAT group. Median OS was 10.71 months in the ruxolitinib group and 5.82 months in the BAT group, with a stratified HR of 0.83 (95% CI, 0.63 to 1.14). Detailed OS results at the final analysis can be found in Table 58 (Appendix 3).18
REACH 1
The OS results for the REACH 1 trial are summarized in Table 37. As of the final analysis (June 5, 2019), 44 (62.0%) patients had died. The median follow-up time was 160 days.52 Median OS was 232.0 (95% CI, 93.0 to 675.0) days. The KM curve is depicted in Figure 27 (Appendix 3). The probability of patients surviving at 3 months and 6 months was 63.3 (95% CI, 51.0 to 73.3) and 51.3 (95% CI, 38.9 to 62.3), respectively. The OS times at the 25th, 50th, and 75th percentiles were 42.0 (95% CI, 30.0 to 62.0) days, 232.0 (95% CI, 93.0 to 675.0) days, and NE (95% CI, 675.0 to NE), respectively.19
Table 37: Summary of Efficacy End Points in the REACH 2 Full Analysis Set and the REACH 1 Efficacy-Evaluable Population (Outcomes Are Presented in Order of Priority as Identified in the CADTH Review Protocol)
Outcome Data cut-off date | REACH 2 | REACH 1 | |||
|---|---|---|---|---|---|
Ruxolitinib N = 154 | BAT N = 155 | Ruxolitinib N = 154 | BAT N = 155 | Ruxolitinib N = 71 | |
July 25, 2019 | January 6, 2020 | June 5, 2019 | |||
OS | |||||
Median OS follow-up time | |||||
Months | 5.04 | 3.58 | 7.34 | 3.81 | NA |
Days | NA | NA | NA | NA | 160 |
Median OS | |||||
Months | 11.14 | 6.47 | 10.71 | 5.82 | NA |
Days, (95% CI) | 339 (186 to NE) | 197 (114 to 458) | 326 (182 to 547) | 177 (115 to 392) | 232.0 (93.0 to 675.0) |
Events, death, n (%) | 72 (46.8) | 79 (51.0) | 82 (53.2) | 88 (56.8) | 44 (62.0) |
Censored, n (%) | 82 (53.2) | 76 (49.0) | 72 (46.8) | 67 (43.2) | 27 (38.0)a |
HR (95% CI)b | 0.83 (0.60 to 1.15) | 0.83 (0.6 to 1.13) | NA | ||
P value | 0.2648 | 0.2331 | NA | ||
FFS | |||||
Median FFS | |||||
Months | 4.99 | 1.02 | 4.86 | 1.02 | NA |
Days, (95% CI) | NA | NA | NA | NA | 85.0 (42.0 to 158.0) |
Number of patients with events, n (%) | 84 (54.5) | 119 (76.8) | 91 (59.1) | 121 (78.1) | 60 (84.5) |
Number of patients with competing risk, n (%) | 30 (19.5) | 14 (9.0) | 36 (23.4) | 15 (9.7) | NA |
Number of patients censored, n (%) | 40 (26.0) | 22 (14.2) | 27 (17.5) | 19 (12.3) | 11 (15.5) |
HR (95% CI)b | 0.46 (0.35 to 0.60) | 0.49 (0.37 to 0.63) | NA | ||
P valuec | 0.0001 | 0.0001 | NA | ||
ORR at day 28 | |||||
Patients with overall response, n (%) | 96 (62.3) | 61 (39.4) | NA | NA | 40 (56.3) |
95% CId | 54.2 to 70.0 | 31.6 to 47.5 | NA | NA | 44.0 to 68.1 |
CR, n (%) | 53 (34.4) | 30 (19.4) | NA | NA | 19 (26.8) |
VGPR, n (%) | NA | NA | NA | NA | 6 (8.5) |
PR n (%) | 43 (27.9) | 31 (20.0) | NA | NA | 15 (21.1) |
Odds ratio (ruxolitinib/BAT) (95% CI)d | 2.64 (1.65 to 4.22) | NA | NA | NA | |
P value | < 0.0001 | NA | NA | NA | |
Nonresponders, n (%) | |||||
No response | 7 (4.5) | 10 (6.5) | NA | NA | 2 (2.8) |
Mixed response | 10 (6.5) | 17 (11.0) | NA | NA | 3 (4.2) |
Progression | 4 (2.6) | 13 (8.4) | NA | NA | 2 (2.8) |
Other | 1 (0.6) | 7 (4.5) | NA | NA | 1 (1.4) |
Unknown | 36 (23.4) | 47 (30.3) | NA | NA | NA |
Death | 15 (9.7) | 22 (14.2) | NA | NA | 10 (14.1) |
Early discontinuation | 17 (11.0) | 16 (10.3) | NA | NA | 12 (16.9) |
Missing visits | 4 (2.6) | 9 (5.8) | NA | NA | 1 (1.4) |
ORR at day 14 | |||||
Patients with overall response, n (%) | 97 (63.0) | 73 (47.1) | NA | NA | 44 (62.0) |
95% CId | 54.8 to 70.6 | 39.0 to 55.3 | NA | NA | 49.7 to 73.2 |
CR, n (%) | 32 (20.8) | 18 (11.6) | NA | NA | 14 (19.7) |
VGPR, n (%) | NA | NA | NA | NA | 6 (8.5) |
PR, n (%) | 65 (42.2) | 55 (35.5) | NA | NA | 24 (33.8) |
Odds ratio (ruxolitinib/BAT) (95% CI)d | 1.98 (1.24 to 3.17) | NA | NA | NA | |
P valuec | 0.0029 | NA | NA | NA | |
Nonresponders, n (%) | NA | NA | |||
No response | 18 (11.7) | 26 (16.8) | NA | NA | 6 (8.5) |
Mixed response | 9 (5.8) | 18 (11.6) | NA | NA | 2 (2.8) |
Progression | 10 (6.5) | 16 (10.3) | NA | NA | 7 (9.9) |
Other | 1 (0.6) | 7 (4.5) | NA | NA | 1 (1.4) |
Unknown | 19 (12.3) | 15 (9.7) | NA | NA | NA |
Death | 6 (3.9) | 8 (5.2) | NA | NA | 5 (7.0) |
Early discontinuation | 6 (3.9) | 5 (3.2) | NA | NA | 5 (7.0) |
Missing visits | 7 (4.5) | 2 (1.3) | NA | NA | 1 (1.4) |
Rate of durable ORR at day 56 | |||||
Overall response, n (%) | 61 (39.6) | 34 (21.9) | NA | NA | NA |
95% CId | 31.8 to 47.8 | 15.7 to 29.3 | NA | NA | NA |
CR, n (%) | 41 (26.6) | 25 (16.1) | NA | NA | NA |
PR, n (%) | 20 (13.0) | 9 (5.8) | NA | NA | NA |
Odds ratio (ruxolitinib/BAT) (95% CI)d | 2.38 (1.43 to 3.94) | NA | NA | NA | |
P value | 0.0005 | NA | NA | NA | |
DOR in patients with CR or PR at or before day 28 | |||||
Response at or before day 28, n | 96 | 61 | 97 | 62 | NA |
Patients with events,e n (%) | 9 (9.4) | 21 (34.4) | 9 (9.3) | 22 (35.5) | NA |
Number of patients with competing risks, n (%) | 53 (55.2) | 23 (37.7 | 66 (68.0) | 26 (41.9) | NA |
Death, n (%) | 28 (29.2) | 12 (19.7) | 34 (35.1) | 14 (22.6) | NA |
Incidence of cGvHD, n (%) | 25 (26.0) | 11 (18.0) | 32 (33.0) | 12 (19.4) | NA |
Number of patients censored, n (%) | 34 (35.4) | 17 (27.9) | 22 (22.7) | 14 (22.6) | NA |
DOR | |||||
Medianf | 168.0 | 101.0 | 163.0 | 101.0 | NA |
Q1 to Q3f | 78.0 to 225.0 | 46.0 to 170.0 | 78.0 to 246.0 | 46.0 to 181.0 | NA |
Range | 22.0 to 423.0 | 10.0 to 289.0 | 22.0 to 623.0 | 10.0 to 456.0 | NA |
6-month DOR in patients with PR, VGPR, or CR assessed once all patients had completed the day 180 visit | |||||
Event-free probability estimate, % (95% CI) | — | — | — | — | — |
Month 6 | NA | NA | NA | NA | 62.1 (45.8 to 74.8) |
3-month DOR in patients with PR, VGPR, or CR assessed once all patients had completed the day 84 visit | |||||
Event-free probability estimate, % (95% CI) | — | — | — | — | — |
Month 3 | NA | NA | NA | NA | 75.6 (61.0 to 85.4) |
BOR by day 28 | |||||
Patients with overall response, n (%) | 126 (81.8) | 94 (60.6) | NA | NA | NA |
95% CId | 74.8 to 87.6 | 52.5 to 68.4 | NA | NA | NA |
Odds ratio (95% CI)d | 3.07 (1.80 to 5.25) | NA | NA | NA | |
P value | 0.0001 | NA | NA | NA | |
EFS | |||||
Median EFS, months | 8.28 | 4.17 | 8.18 | 4.17 | NA |
Events,g n (%) | 77 (50.0) | 86 (55.5) | 87 (56.5) | 95 (61.3) | NA |
Censored, n (%) | 77 (50.0) | 69 (44.5) | 67 (43.5) | 60 (38.7) | NA |
HR (95% CI) | 0.80 (0.58 to 1.08) | 0.80 (0.60 to 1.08) | NA | ||
P valuec | 0.1466 | 0.1431 | NA | ||
NRM | |||||
Number of patients with events, n (%) | 60 (39.0) | 66 (42.6) | 69 (44.8) | 70 (45.2) | 40 (56.3) |
Number of patients with competing risks, n (%) | 15 (9.7)h | 20 (12.9)h | 17 (11.0)h | 25 (16.1)h | NR |
Number of patients censored, n (%) | 79 (51.3) | 69 (44.5) | 68 (44.2) | 60 (38.7) | NR |
Malignancy relapse or progression | |||||
Number of patients in full analysis set | 147 | 147 | 147 | 147 | NA |
Number of patients with events, n (%) | 14 (9.5) | 20 (13.6) | 16 (10.9) | 25 (17.0) | NA |
Number of patients with competing risks, n (%) | 56 (38.1) | 62 (42.2) | 65 (44.2) | 66 (44.9) | NA |
Number of patients censored, n (%) | 77 (52.4) | 65 (44.2) | 66 (44.9) | 56 (38.1) | NA |
Relapse rate | |||||
Number of patients with relapse of underlying malignancy, n (%) [95% CI] | NA | NA | NA | NA | 5 (7.0) [2.3 to 15.7] |
Relapse mortality rate | |||||
Number of patients with relapse of underlying malignancy and a fatal outcome, n (%) [95% CI] | NA | NA | NA | NA | 4 (5.6) [1.6 to 13.8] |
Cumulative steroid dosing until day 56 | |||||
Completely tapered off by day 56, n (%) [95% CI] | 33 (21.4) [15.2 to 28.8] | 23 (14.8) [9.6 to 21.4] | 34 (22.1) [15.8 to 29.5] | 23 (14.8) [9.6 to 21.4] | NA |
≤ 50% RDI,i n (%) [95% CI] | 45 (29.2) [22.2 to 37.1] | 38 (24.5) [18.0 to 32.1] | 45 (29.2) [22.2 to 37.1] | 37 (23.9) [17.4 to 31.4] | NA |
> 50% RDI,i n (%) [95% CI] | 106 (68.8) (60.9 to 76.0) | 116 (74.8) [67.2 to 81.5] | 106 (68.8) [60.9 to 76.0] | 117 (75.5) [67.9 to 82.0] | NA |
Number of patients with ongoing ruxolitinib who had discontinued corticosteroids by day 56 [n = 29], n (%) | NA | NA | NA | NA | 2 (6.9) |
Incidence of cGvHD | |||||
Number of patients with events, n (%) | 38 (24.7) | 26 (16.8) | 45 (29.2) | 29 (18.7) | 11 (15.5) |
Number of patients with competing risk, n (%) | 69 (44.8) | 78 (50.3) | 79 (51.3) | 85 (54.8) | NA |
Death | NR | NR | 62 (40.3) | 63 (40.6) | NA |
Hematologic disease relapse or progression, n (%) | NR | NR | 17 (11.0) | 22 (14.2) | NA |
Number of patients censored, n (%) | 47 (30.5) | 51 (32.9) | 30 (19.5) | 41 (26.5) | NA |
BAT = best available therapy; BOR = best overall response; cGVHD = chronic graft-vs.-host disease; CI = confidence interval; CR = complete response; DOR = duration of response; EFS = event-free survival; FFS = failure-free survival; HR = hazard ratio; NA = not applicable; NE = not evaluable; NRM = nonrelapse mortality; OS = overall survival; PR = partial response; RDI = relative dose intensity; VGPR = very good partial response; vs. = versus.
aParticipants with no observed death or loss to follow-up were censored at the last date they were known to be alive.
bHR and 95% CI are obtained from the stratified Cox proportional hazards model using the Wald test.
cThe 95% CI for the response rate was calculated using the Clopper-Pearson exact method.
cP value nominal.
dOdds ratio and 95% CI are calculated using a stratified Cochran-Mantel-Haenszel test.
eThe event was defined as the progression of aGvHD or addition of systemic therapies for aGvHD after day 28. The competing risks included death without prior observation of aGvHD progression and onset of cGvHD.
fMedian and quartiles are calculated using KM method.
gThe event includes hematologic disease relapse or progression, graft failure, or death from any cause.
hThe competing risk included hematologic disease relapse or progression.
iRDI includes days of 0 dose in the calculation.
Sources: Clinical Study Report (REACH 2),18 Clinical Study Report (REACH 1),19 Sponsor’s response to additional information.52
Figure 3: KM Curves of OS in REACH 2, Full Analysis Set (January 6, 2020, Data Cut-Off Date)
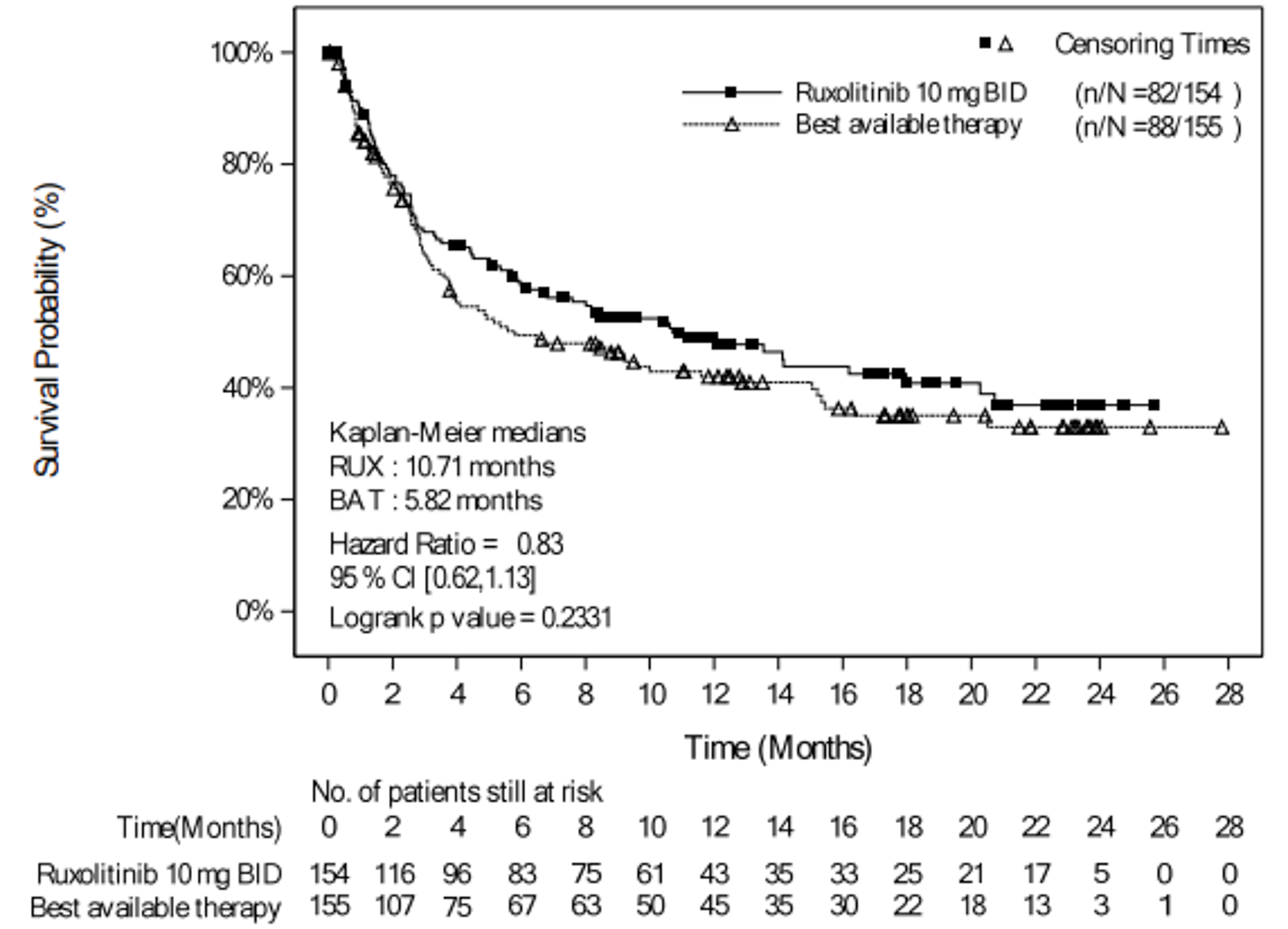
BID = twice daily; BAT = best available therapy; CI = confidence interval; KM = Kaplan–Meier; OS = overall survival; RUX = ruxolitinib.
Source: Clinical Study Report (REACH 2).18
Failure-Free Survival
REACH 2
The FFS results of the REACH 2 trial for the ruxolitinib and BAT groups are summarized in Table 37 for the primary and secondary analyses. A detailed version of Table 37 is available in Table 56 (Appendix 3), showing estimated cumulative incidence. As of the primary analysis (July 25, 2019 data cut-off date), the number of patients who experienced an event (i.e., hematologic disease relapse or progression, NRM, or addition of new systemic aGvHD treatment) was 84 (54.5%) and 119 (76.8%) in the ruxolitinib and BAT groups, respectively. The number of patients who were censored was 40 (26.0%) and 22 (14.2%), respectively. The median FFS was 4.99 months and 1.02 months in the ruxolitinib and BAT groups, respectively, with a HR of 0.46 (95% CI, 0.35 to 0.60). The KM curves are depicted in Figure 28 (Appendix 3). The 1-month and 6-month cumulative incidence of events were 18.47% (95% CI, 12.74% to 25.04%) and 52.85% (95% CI, 44.24% to 60.74%), respectively, in the ruxolitinib group, and 49.13% (95% CI, 40.94% to 56.80%) and 80.86% (95% CI, 72.95% to 86.67%), respectively, in the BAT group. The competing risk (cGvHD) was low in both study groups (30 of 154 patients and 14 of 155 patients in the ruxolitinib and BAT groups, respectively). The cumulative incidence of FFS is depicted in Figure 21 (Appendix 3).18
Results at the secondary analysis (January 6, 2020) were, overall, consistent with the results at the primary analysis. As of the secondary analysis (January 6, 2020, data cut-off date), the number of patients who experienced an event was 91 (59.1%) and 121 (78.1%) in the ruxolitinib and BAT groups, respectively. The number of patients who were censored was 27 (17.5%) and 19 (12.3%), respectively. The median FFS was 4.86 months and 1.02 months in the ruxolitinib and BAT groups, respectively, with a HR of 0.49 (95% CI, 0.37 to 0.63). The KM curves are depicted in Figure 4. The 1- and 6-month cumulative incidence of events were 18.47% (95% CI, 12.74% to 25.04%) and 54.07% (95% CI, 45.69% to 61.71%), respectively, in the ruxolitinib group, and 49.13% (95% CI, 40.94% to 56.80%) and 80.17% (95% CI, 72.5% to 85.90%), respectively, in the BAT group. The competing risk (cGvHD) was low in both study groups (36 of 154 patients and 15 of 155 patients in the ruxolitinib and BAT groups, respectively). The cumulative incidence of FFS is depicted in Figure 22 (Appendix 3).
Results at the final analysis were, overall, consistent with the results at the primary analysis. The median FFS was 4.86 months and 1.02 months in the ruxolitinib and BAT groups, respectively, with a HR of 0.51 (95% CI, 0.39 to 0.66). Detailed results for FFS at the final analysis are described in Table 58 (Appendix 3).18
REACH 1
The FFS results of the REACH 1 trial are summarized in Table 37. As of the June 5, 2019, data cut-off date, the number of patients who experienced an event (i.e., underlying malignancy relapse or progression [n = 3], death [n = 22], addition of new systemic aGvHD treatment [n = 28], or signs or symptoms of cGvHD [n = 7]) was 60 (84.5%). The number of patients who were censored was 11 (15.5%). The median FFS was 85.0 (95% CI, 42.0 to 158.0) days. The KM curve is depicted in Figure 29 (Appendix 3). The 3- and 6-month FFS probabilities were 49.1% (95% CI, 37.1% to 60.1%) and 33.8% (95% CI, 22.9% to 45.0%), respectively. The FFS times at the 25th, 50th, and 75th percentiles were 23.0 (95% CI, 15.0 to 37.0) days, 85.0 (95% CI, 42.0 to 158.0) days, and 331.0 (95% CI, 165.0 to 602.0) days, respectively.19
Figure 4: KM Curves of FFS in REACH 2, Full Analysis Set (January 6, 2020, Data Cut-Off Date)
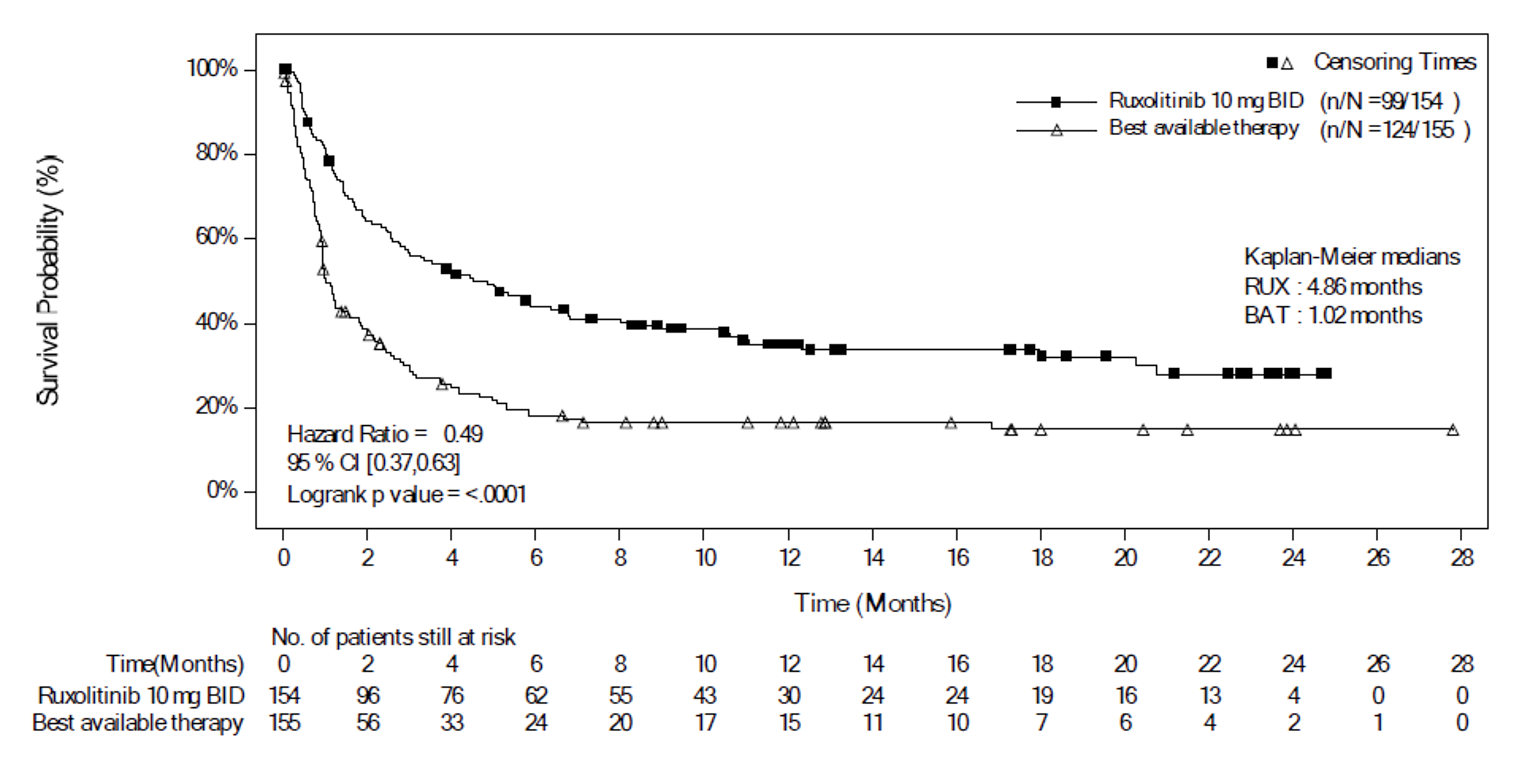
BAT = best available therapy; BID = twice daily; CI = confidence interval; FFS = failure-free survival; KM = Kaplan–Meier; RUX = ruxolitinib.
Note: P value is obtained from the log-rank test. The event included hematologic disease relapse or progression, NRM, or addition of new systemic aGvHD treatment.
Source: Clinical Study Report (REACH 2).18
ORR at Day 28
REACH 2
The ORR at day 28 (assessed by local investigator review according to NIH criteria) was the primary end point in the REACH 2 trial, and results are summarized in Table 37 for the primary analysis (July 25, 2019 data cut-off date). ORR at day 28 was only analyzed at the July 25, 2019 data cut-off date and was not reassessed at the secondary analysis. As of the July 25, 2019 data cut-off date, the REACH 2 trial met its primary objective. The proportion of patients who achieved an overall response at day 28 was 62.3% (n = 96) (95% CI, 54.2% to 70.0%) in the ruxolitinib group and 39.4% (n = 61) (95% CI, 31.6% to 47.5%) in the BAT group, with a stratified odds ratio of 2.64 (95% CI, 1.65 to 4.22). The proportions of patients with CR and PR were 34.4% (n = 53) and 27.9% (n = 43), respectively, in the ruxolitinib group, and 19.4% (n = 30) and 20.0% (n = 31), respectively, in the BAT group. At the day 28 supportive analysis, the ORR in the per-protocol analysis set showed results consistent with the ORR in the full analysis set. ORR at day 28 was achieved by 63.9% (n = 62) (95% CI, 53.5% to 73.4%) of patients in the ruxolitinib group and 39.1% (n = 34) (95% CI, 28.8% to 50.1%) of patients in the BAT group, with a stratified odds ratio of 2.86 (95% CI, 1.55 to 5.27). Additional supportive analyses were conducted to present the shift tables of aGvHD stage by organ and treatment group to compare baseline with day 28 values (see Figure 23 in Appendix 3).18
A sensitivity analysis was conducted to assess the impact of stratification on the primary end point. The 2 treatment groups were compared using Fisher’s exact test, which confirmed the results of the primary analysis, with an odds ratio of 2.55 (95% CI, 1.61 to 4.03). In addition, results for ORR at crossover day 28, defined as the proportion of crossover patients with CR or PR at crossover day 28, were consistent with results of ORR at day 28 in the ruxolitinib group (see Table 48 in Appendix 3).18
The ORR at day 28 by subgroups of interest, as specified in the protocol for this CADTH review, are summarized in Table 38. The treatment effect on ORR at day 28 was consistent with the primary analysis across patient subgroups, except for the subgroup of prior steroids plus CNIs plus other systemic aGvHD treatment as both aGvHD prophylaxis and treatment, and for the subgroup of patients older than 65 years. Of note, the sample sizes of these subgroups were small (less than 30 patients in each study group). Several other subgroups (i.e., 12 years ≤ 18 years, older than 65 years, grade IV aGvHD, the SR-aGvHD criteria of progression after at least 3 days and of failure on steroid taper, and all subcategories of prior aGvHD therapy except steroids plus CNIs) had relatively small sample sizes (< 50 patients in either group). The wide CIs in subgroups reflected uncertainty in the effect estimates. Subgroup results at the secondary analysis (January 6, 202) were consistent with those at the primary analysis.18 Subgroup results at the final analysis (April 23, 2021) were, overall, consistent with the results at the primary analysis; however, there was an improved odds ratio (favouring ruxolitinib) for patients who received steroids plus CNIs plus other systemic aGvHD treatment as prophylaxis and/or treatment, from 0.57 (95% CI, 0.03 to 11.58) to 2.60 (95% CI, 0.37 to 18.42).18
ORR at day 28 was not assessed at the secondary analysis (January 6, 2020, data cut-off date) or at the final analysis (April 23, 2021).
Table 38: Subgroup Results for ORR at Day 28 in REACH 2, Full Analysis Set (Data Cut-Off Date of July 25, 2019)
Subgroups | Ruxolitinib, N = 154, n/N (%) | BAT, N = 155, n/N (%) | Ruxolitinib vs. BAT, Odds ratio (95% CI) |
|---|---|---|---|
ORR at cycle 7 day 1 | |||
Age | |||
12 to ≤ 18 years | 4/5 (80.0) | 3/4 (75.0) | NE (NE to NE) |
18 to 65 years | 83/128 (64.8) | 48/126 (38.1) | 3.12 (1.84 to 5.30) |
> 65 years | 9/21 (42.9) | 10/25 (40.0) | 0.99 (0.28 to 3.45) |
aGvHD grade at randomization | |||
Grade II | 40/53 (75.5) | 27/53 (50.9) | 2.96 (1.3 to 6.76) |
Grade III | 40/71 (56.3) | 27/72 (37.5) | 2.15 (1.10 to 4.20) |
Grade IV | 16/30 (53.3) | 7/30 (23.3) | 3.76 (1.24 to 11.38) |
Criteria for SR-aGvHD | |||
Progression after at least 3 days | 24/35 (68.6) | 14/43 (32.6) | 5.04 (1.85 to 13.75) |
Failure to achieve a response after 7 days | 41/72 (56.9) | 24/63 (38.1) | 2.04 (1.00 to 4.16) |
Failure on steroid taper | 31/47 (66.0) | 23/49 (46.9) | 2.19 (0.95 to 5.05) |
Prior aGvHD therapy | |||
Steroids + CNIs + other systemic aGvHD treatment, only for aGvHD prophylaxis | 28/41 (68.3) | 13/30 (43.3) | 3.00 (1.11 to 8.13) |
Steroids + other systemic aGvHD treatment, only for aGvHD prophylaxis | 5/8 (62.5) | 4/8 (50.0) | 2.63 (0.25 to 27.09) |
Steroids only | 5/12 (41.7) | 6/18 (33.3) | 1.55 (0.31 to 7.71) |
Steroids + CNIs | 48/77 (62.3) | 31/76 (40.8) | 2.41 (1.24 to 4.68) |
Steroids + CNIs + other systemic aGvHD treatment, only for aGvHD treatment | 5/8 (62.5) | 3/12 (25.0) | 7.60 (0.67 to 86.67) |
Steroids + CNIs + other systemic aGvHD treatment, for both aGvHD prophylaxis and aGvHD treatment | 4/7 (57.1) | 3/7 (42.9) | 0.57 (0.03 to 11.58) |
Steroids + other systemic aGvHD treatment, only for aGvHD treatment | 1/1 (100.0) | 1/4 (25.0) | NE (NE to NE) |
aGvHD = acute graft-versus-host disease; BAT = best available therapy; CI = confidence interval; CNI = calcineurin inhibitor; NE = not evaluable; ORR = overall response rate; SR = steroid refractory; vs. = versus.
Note: The 95% CI for the response rate is calculated using the Clopper-Pearson exact method. Odds ratio and 95% CI are calculated using a stratified Cochran-Mantel-Haenszel test.
Source: Clinical Study Report.18
REACH 1
The ORR at day 28 (assessed by investigator review, per the CIBMTR modifications to the International Bone Marrow Transplant Registry response index) was the primary end point in the REACH 1 trial, and results are summarized in Table 37 for the final analysis (June 5, 2019). The study met the predetermined threshold for a positive study outcome (lower limit of the 95% CI for ORR at day 28 ≥ 40%). The proportion of patients who achieved an overall response at day 28 was 56.3% (n = 40) (95% CI, 44.0% to 68.1%). The number of patients with CR, VGPR, and PR was 19 (26.8%), 6 (8.5%), and 15 (21.1%), respectively.19
Additional supportive subgroup analyses for the primary end point were performed, but only those consistent with the pre-specified subgroups in the protocol for this CADTH review are reported. Results for ORR at day 28 by baseline aGvHD grade were, overall, consistent with the result for all grades combined; ORR at day 28 was 81.8% for grade II, 45.5% for grade III, and 43.8% for grade IV (see Table 47 in Appendix 3). Results for ORR at day 28 by baseline SR subcategory were, overall, consistent with the results for all subcategories combined; ORR at day 28 was 64.7% for progressive GvHD after 3 days of primary treatment, 48.3% for GvHD not improved after 7 days of primary treatment, and 50.0% for previously began steroid therapy at a lower dose but developed new GvHD in another organ system (see Table 50 in Appendix 3). For results of ORR at day 28 by baseline organ involvement, the following differences were noted: 63.6% and 31.3% of patients achieved ORR at day 28 with baseline liver involvement of stage 0 and of all other stages, respectively; and 75.0% and 49.0% of patients achieved ORR at day 28 with baseline lower GI involvement of stage 0 and of all other stages, respectively (see Table 49 in Appendix 3).19 Of note, the sample sizes of the subgroups were small (less than 35 patients).
ORR at Day 14
REACH 2
The ORR at day 14 results in the REACH 2 trial at the primary analysis (July 25, 2019, data cut-off date) are summarized in Table 37. As of the primary analysis (July 25, 2019), the proportion of patients who achieved an overall response at day 14 was 63.0% (n = 97) (95% CI, 54.8% to 70.6%) in the ruxolitinib group and 47.1% (n = 73) (95% CI, 39.0% to 55.3%) in the BAT group, with a stratified odds ratio of 1.98 (95% CI, 1.24 to 3.17).
ORR at day 14 was not assessed at the secondary analysis (January 6, 2020, data cut-off date).
REACH 1
The ORR at day 14 results in the REACH 1 trial at the final analysis (June 9, 2019, data cut-off date) are summarized in Table 37. ORR at days 56, 100, and 180 are summarized in Table 51 (Appendix 3). Analyses at day 180 were not pre-specified a priori in the statistical analysis plan. The proportion of patients achieving an overall response at days 14, 56, 100, and 180 were 62.0% (95% CI, 49.7% to 73.2%), 36.6% (95% CI, 25.5% to 48.9%), 32.4% (95% CI, 21.8% to 44.5%), and 21.1% (95% CI, 12.3% to 32.4%), respectively.19
Durable ORR at Day 56
REACH 2
Durable ORR at day 56 was a key secondary end point in the REACH 2 trial, and results for the primary analysis (July 25, 2019 data cut-off date) are summarized in Table 37. A detailed version of Table 37 is available in Table 56 (Appendix 3), showing the number of nonresponders by their response status. As of the primary analysis, the proportion of patients who achieved a durable ORR at day 56 was 39.6% (n = 61) in the ruxolitinib group and 21.9% (n = 34) in the BAT group, with a stratified odds ratio of 2.38 (95% CI, 1.43 to 3.94) in favour of ruxolitinib. At the day 56 supportive analysis, the durable ORR in the per-protocol analysis set showed a trend similar to that in the full analysis set. Durable ORR at day 56 was achieved by 36.1% (n = 35) (95% CI, 26.6% to 46.5%) of patients in the ruxolitinib group and by 26.4% (n = 23) (95% CI, 17.6% to 37.0%) of patients in the BAT group, with a stratified odds ratio of 1.58 (95% CI, 0.83 to 3.01).
Results for durable ORR at crossover day 56 were consistent with results of durable ORR at day 56, with 40.8% (n = 20) (95% CI, 27.0% to 55.8%) of patients achieving a durable ORR at crossover day 56.18
Durable ORR at day 56 was not assessed at the secondary analysis (January 6, 2020, data cut-off date).
REACH 1
Durable ORR at day 56 was not assessed in the REACH 1 trial.
Duration of Response
REACH 2
The DOR results for the REACH 2 trial for the primary and secondary analyses are summarized in Table 37. A more detailed version of Table 37 is available in Table 56 (Appendix 3), showing the cumulative incidence. At the primary analysis, among the patients who achieved a CR or PR at or before day 28, median DOR was 168 (range = 22 to 423) days in the ruxolitinib group and 101 (range = 10 to 289) days in the BAT group. The number of patients who experienced an event (i.e., aGvHD progression or addition of systemic therapy for aGvHD) was 9 of 96 (9.4%) patients and 21 of 61 (34.4%) patients in the ruxolitinib and BAT groups, respectively. The number of patients censored was 34 of 96 (35.4%) and 17 or 61 (27.9%), respectively. The 1- and 6-month cumulative incidence of events was 2.08% (95% CI, 0.40% to 6.65%) and 9.65% (95% CI, 4.39% to 17.40%), respectively, in the ruxolitinib group, and 11.54% (95% CI, 5.03% to 21.03%) and 38.98% (95% CI, 25.54% to 52.19%), respectively, in the BAT group. The cumulative incidence of DOR is depicted in Figure 25 (Appendix 3).18
DOR results at the secondary analysis were consistent with those at the primary analysis. As of the secondary analysis, median DOR was 163 (range = 22 to 623) days in the ruxolitinib group and 101 (range = 10 to 456) days in the BAT group. The cumulative incidence of events at 1 month and 6 months was 2.06% (95% CI, 0.39% to 6.58%) and 8.73% (4.03% to 15.68%), respectively, in the ruxolitinib group, and 12.97% (95% CI, 6.01% to 22.66%) and 37.34% (95% CI, 24.95% to 49.71%), respectively, in the BAT group. The cumulative incidence of DOR is depicted in Figure 5.18
Results for DOR at the final analysis were consistent with those at the primary analysis. As of the final analysis, median DOR was 167 (range = 22 to 677) days in the ruxolitinib group and 106 (range = 10 to 526) days in the BAT group. Detailed DOR results at the final analysis can be found in Table 58 (Appendix 3).18
REACH 1
Results for 3-month and 6-month DORs are summarized in Table 37. The 3- and 6-month event-free probabilities for DOR in responding patients (i.e., those who achieved PR, VGPR, or CR) at any time point were 75.6% (95% CI, 61.0% to 85.4%) and 62.1% (95% CI, 45.8% to 74.8%), respectively. The median DOR for responding patients at any time point was 345.0 (95% CI, 154.0 to NE) days, with a median follow-up time of 128.5 (range = 3 to 805) days (see Figure 30 in Appendix 3).19
Additional analyses for DOR for patients who had a response (PR, VGPR, or CR) on day 28 (n = 40; 56.3%) were performed, and the median DOR was 669.0 (95% CI, 159.0 to NE) days, with a median follow-up time of 195.0 (range = 7 to 805) days (see Figure 31 in Appendix 3); 3- and 6-month event-free probabilities for DOR were 84.5% (95% CI, 68.7% to 92.7%) and 68.2% (95% CI, 49.6% to 81.2%), respectively. Results for DOR by the day 28 response for patients who had CR, VGPR, and PR at day 28, respectively, are described in Table 52 (Appendix 3).19
Figure 5: Cumulative Incidence Curve of DOR in REACH 2, Full Analysis Set (Data Cut-Off Date of January 6, 2020)
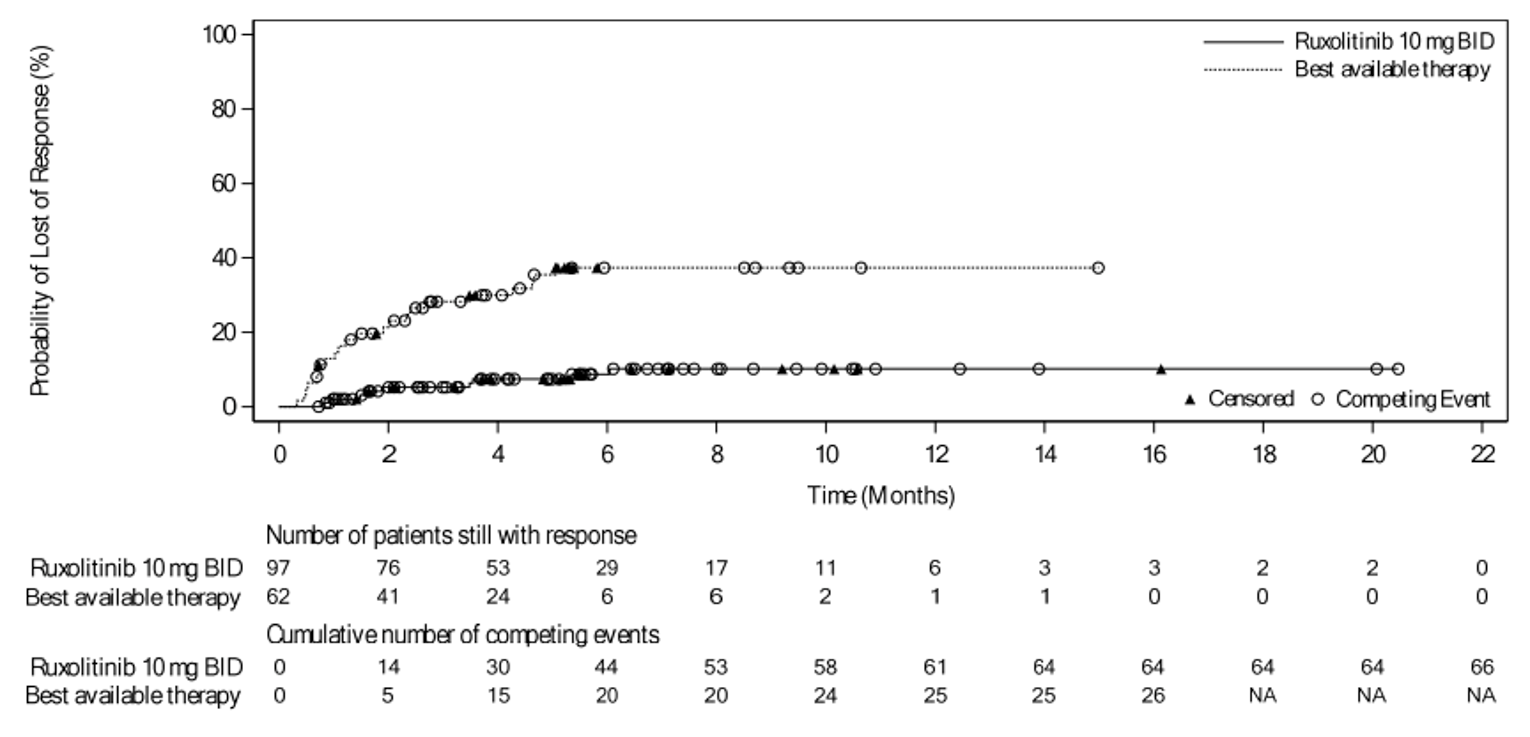
BID = twice daily; DOR = duration of response; NA = not applicable.
Note: The competing risks include death without prior observation of aGvHD progression and onset of cGvHD.
Source: Clinical Study Report (REACH 2).18
Best Overall Response
REACH 2
The BOR results in the REACH 2 trial for the primary analysis (July 25, 2019, data cut-off date) are summarized in Table 37. A detailed version of Table 37 is available in Table 56 (Appendix 3), showing the number of nonresponders by their response status. At the primary analysis, the proportion of patients who had achieved BOR by day 28 was 81.8% (95% CI, 74.8% to 87.6%) in the ruxolitinib group and 60.6% (95% CI, 52.5% to 68.4%) in the BAT group, with an odds ratio of 3.07 (95% CI, 1.80 to 5.25). By day 28, a lower proportion of patients experienced no response in the ruxolitinib group than in the BAT group (8.4% versus 13.5%).18
BOR was not assessed at the secondary analysis (January 6, 2020, data cut-off date).18
REACH 1
BOR was assessed as an additional supportive analysis of the primary end point. At the final analysis, the proportion of patients who had achieved BOR at any time point was 76.1% (95% CI, 64.5% to 85.4%). The proportion of patients who achieved a CR was 57.7% and who achieved a VGPR was 5.6% (see Table 53 in Appendix 3).19
Health-Related Quality of Live
REACH 2
FACT-BMT
The completion rates declined over time in both study groups. At baseline, week 4, and week 8, respectively, 71.1%, 61.1%, and 37.6% of patients in the ruxolitinib group were available for completion, whereas that number declined more rapidly in the BAT group, with 62.9%, 47.7%, and 22.5% of patients available at baseline, week 4, and week 8, respectively.21 Overall observed scores from baseline to day 28 (week 4) and to the end of randomized treatment (week 24) fluctuated, but overall improvement over time was suggested in all aspects of the FACT-BMT questionnaire in both treatment groups. For the FACT-BMT trial outcome index, the mean change from baseline up to day 28 (week 4) was 1.59 (SD = 12.352) in the ruxolitinib group (n = 76) and – 0.11 (SD = 11.750) in the BAT group (n = 61), and the mean change from baseline to week 24 was 10.31 (SD = 15.871) in the ruxolitinib group (n = 26) and 7.00 (SD = 10.365) in the BAT group (n = 8). However, because of the significant decline in the number of patients available for assessment over time, it was not possible the identify trends over time, so the results remain inconclusive. During the crossover period, observed scores for patients in the BAT group who crossed over to ruxolitinib were similar to those during the randomized treatment period. For the FACT-BMT trial outcome index, the mean change from baseline up to day 28 (week 4) was 1.17 (SD = 9.843) (n = 23) and from baseline to week 24 was 8.60 (SD = 14.347) (n = 10). However, because of the significant decline in the number of crossover patients available for assessment over time, it was not possible the identify trends over time, so the results remain inconclusive. Results at the secondary analysis (January 6, 2020, data cut-off date) were, overall, consistent with results at the primary analysis.18 Results at the final analysis were, overall, consistent with results at the primary analysis.18
EQ-5D-5L
The completion rates declined over time in both study groups. At baseline, week 4, and week 8, respectively, 79.2%, 64.9%, and 41.6% of patients in the ruxolitinib group were available for completion, whereas the number of patients declined more rapidly in the BAT group, with 76.1%, 52.3%, and 25.2% of patients available at baseline, week 4, and week 8, respectively.21 Overall observed scores from baseline to day 28 (week 4) and to end of randomized treatment (week 24) varied across all dimensions of the EQ-5D-5L questionnaire in both treatment groups. The mean change in EQ-5D-5L score from baseline up to day 28 (week 4) was 0.03 (SD = 0.282) in the ruxolitinib group (n = 85) and 0.01 (SD = 0.308) in the BAT group (n = 69), whereas the mean change from baseline to week 24 was 0.15 (SD = 0.297) in the ruxolitinib group (n = 29) and 0.09 (SD = 0.124) in the BAT group (n = 8). Because of the significant decline in number of patients available for assessment over time, it was not possible the identify trends over time, so results remain inconclusive. During the crossover period, observed scores for patients in the BAT group who crossed over to ruxolitinib were similar to those during the randomized treatment period; the mean change in EQ-5D-5L score from baseline up to day 28 (week 4) was –0.01 (SD = 0.280) (n = 31), and from baseline to week 24 was 0.01 (0.226) (n = 9). However, because of the significant decline in number of patients available to for assessment over time, it was not possible the identify trends over time, so results remain inconclusive. Results at the secondary analysis (January 6, 2020, data cut-off date) were, overall, consistent with results at the primary analysis.18 Results at the final analysis were, overall, consistent with results at the primary analysis.18
REACH 1
HRQoL was not assessed in the REACH 1 trial.
Event-Free Survival
REACH 2
The EFS results in the REACH 2 trial for the ruxolitinib and the BAT groups are summarized in Table 37 (primary and secondary analyses). A detailed version of Table 37 is available in Table 56 (Appendix 3), showing the survival probabilities. As of the primary analysis (July 25, 2019 data cut-off date), the median EFS was 8.28 months in the ruxolitinib group and 4.17 months in the BAT group, with a stratified HR of 0.80 (95% CI, 0.58 to 1.08). The KM curves are depicted in Figure 6. The probability of patients surviving event-free at 1 month and 6 months was 89.38 (95% CI, 83.24 to 93.35) and 54.77 (95% CI, 46.16 to 62.58), respectively, in the ruxolitinib group, and 82.83 (95% CI, 75.81 to 87.97) and 44.04 (95% CI, 35.49 to 52.26), respectively, in the BAT group. A sensitivity analysis in which aGvHD progression was included as an event showed consistent results with the primary analysis.18
Results for EFS at the secondary analysis were consistent with EFS results at the primary analysis. As of the secondary analysis (January 6, 2020, data cut-off date), the median EFS was 8.18 months in the ruxolitinib group and 4.17 months in the BAT group, with a stratified HR of 0.80 (95% CI, 0.60 to 1.08). The KM curves are depicted in Figure 7. The probability of patients surviving event-free at 1 month and 6 months was 89.38% (95% CI, 83.24% to 93.35%) and 44.21% (95% CI, 35.88% to 52.20%), respectively, in the ruxolitinib group, and 82.83% (95% CI, 75.81% to 87.97%) and 44.14% (95% CI, 35.82% to 52.13%), respectively, in the BAT group. A sensitivity analysis in which aGvHD progression was included as an event showed consistent results with the primary analysis.
Results for EFS at the final analysis were consistent with EFS results at the primary analysis. The median EFS was 8.18 months in the ruxolitinib group and 4.17 months in the BAT group, with a stratified HR of 0.85 (95% CI, 0.64 to 1.13). Detailed EFS results at the final analysis are described in Table 58 (Appendix 3).18
REACH 1
EFS was not assessed in the REACH 1 trial.
Figure 6: KM Curves of EFS in REACH 2, Full Analysis Set (July 25, 2019, Data Cut-Off Date)
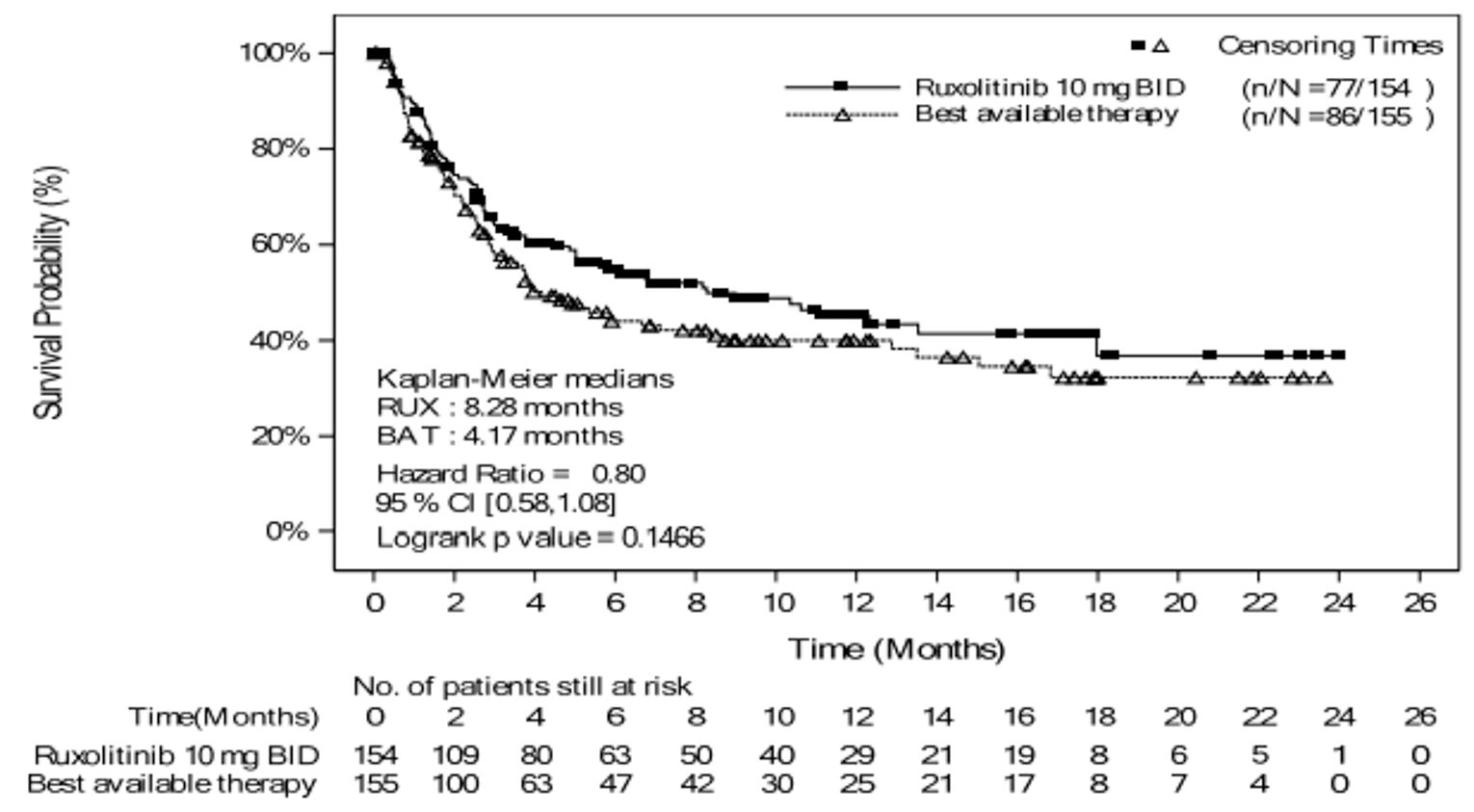
BAT = best available therapy; BID = twice daily; CI = confidence interval; EFS = event-free survival; KM = Kaplan–Meier; RUX = ruxolitinib.
Source: Clinical Study Report (REACH 2).18
Figure 7: KM Curves of EFS in REACH 2, Full Analysis Set (January 6, 2020, Data Cut-Off Date)
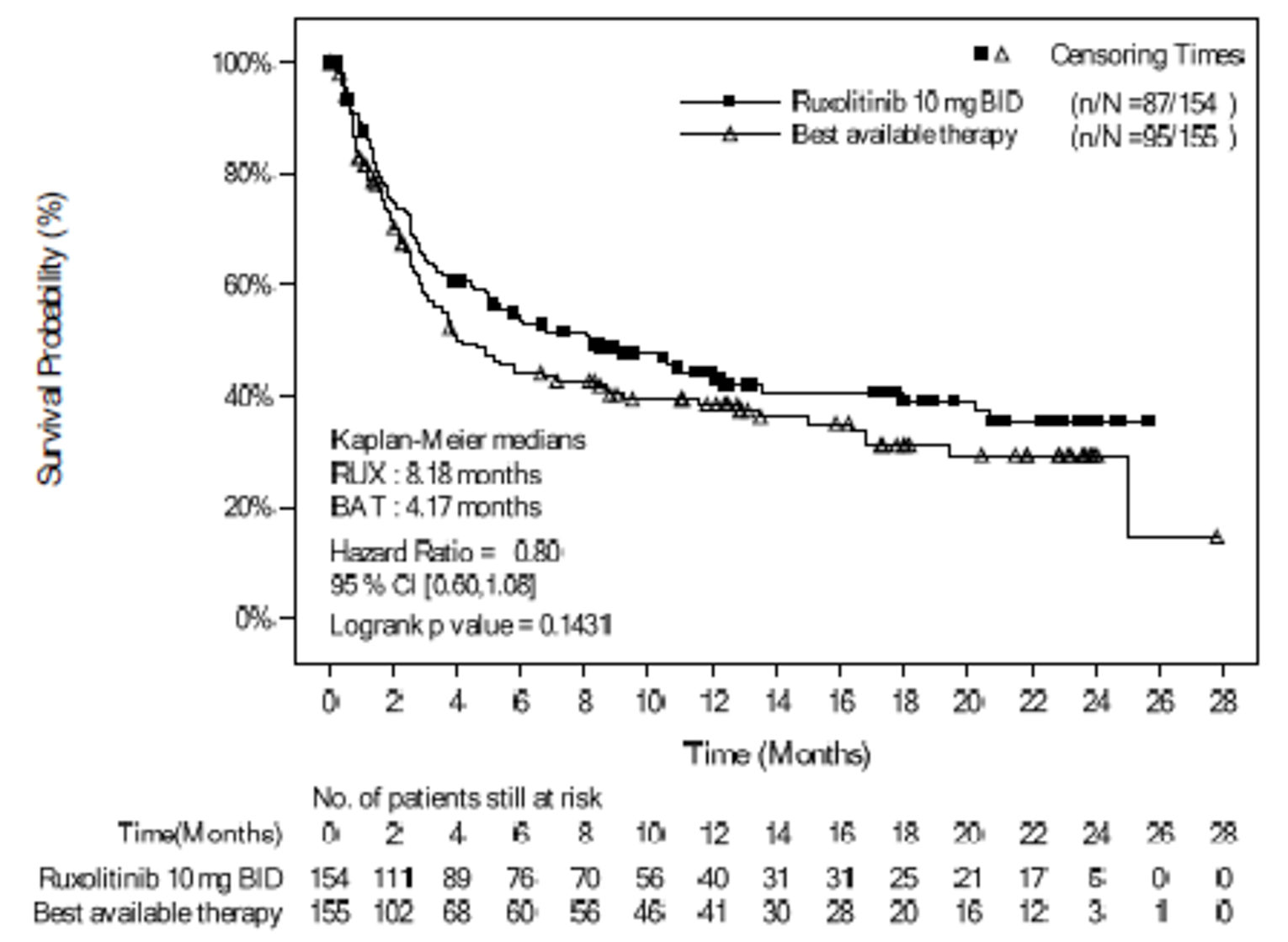
BAT = best available therapy; BID = twice daily; CI = confidence interval; EFS = event-free survival; KM = Kaplan–Meier; RUX = ruxolitinib.
Note: P value is obtained from the log-rank test. The event includes hematologic disease relapse or progression, graft failure, or death from any cause.
Source: Clinical Study Report (REACH 2).18
Nonrelapse Mortality
REACH 2
The results for NRM in the REACH 2 trial are summarized in Table 37. A detailed version of Table 37 is available in Table 56 (Appendix 3), showing the cumulative incidence. As of the primary analysis (July 25, 2019), the number of patients who experienced NRM was 60 (39.0%) in the ruxolitinib group and 66 (42.6%) in the BAT group. The number of patients who were censored was high, with 79 (51.3%) and 69 (44.5%) patients in the ruxolitinib and BAT groups, respectively. The probability of NRM at 1 month and 6 months was 9.96% (95% CI, 5.83% to 15.39%) and 36.18% (95% CI, 28.28% to 44.12%), respectively, in the ruxolitinib group, and 14.52% (95% CI, 9.45% to 20.64%) and 43.34 (95% CI, 34.89% to 51.48%), respectively, in the BAT group. The competing risk (hematologic disease relapse or progression) was low in both study groups, at 15 of 154 patients and 20 of 155 patients in the ruxolitinib and BAT groups, respectively. The cumulative incidence of NRM is depicted in Figure 32 (Appendix 3). The curves for both study groups were overlapping, which indicates similar event rates over time. A sensitivity analysis in which patients with underlying hematologic malignant disease were included showed results similar to those in the primary analysis.18
As of the secondary analysis (January 6, 2020), results were consistent with the primary data cut-off date and are depicted in Table 37 and Figure 33 (Appendix 3). A sensitivity analysis in which patients with underlying hematologic malignant disease were included showed results similar to those at the primary analysis.18
The results for NRM at the final analysis were, overall, consistent with the primary data cut-off date. As of the final analysis, the number of patients who experienced NRM was 72 (46.8%) in the ruxolitinib group and 71 (45.8%) in the BAT group. Detailed results for NRM at the final analysis are described in Table 58 (Appendix 3).18
REACH 1
The results for NRM in the REACH 1 trial are summarized in Table 37. As of the final analysis (June 5, 2019), the number of patients who experienced NRM was 56.3% (n = 40). The cumulative incidence of NRM is depicted in Figure 34 (Appendix 3).19
Malignancy Relapse or Recurrence
REACH 2
The results for malignancy relapse or recurrence in the REACH 2 trial are summarized in Table 37. A detailed version of Table 37 is available in Table 56 (Appendix 3), showing the cumulative incidence. At baseline, 147 patients in each of the ruxolitinib and BAT groups had underlying malignant disease. As of the July 25, 2019, data cut-off date, the number of patients who had events of malignancy relapse or recurrence was 14 (9.5%) in the ruxolitinib group and 20 (13.6%) in the BAT group. The number of patients who were censored was high, at 77 and 65 patients in the ruxolitinib and BAT groups, respectively. The probability of malignancy relapse or recurrence at 1 month and 2 months was 0.69% (95% CI, 0.06% to 3.51%) and 4.23% (95% CI, 1.73% to 8.49%), respectively, in the ruxolitinib group, and 2.80% (95% CI, 0.92% to 6.54%) and 4.30% (95% CI, 1.76% to 8.63%), respectively, in the BAT group. The competing risk (hematologic disease relapse or progression) was relatively low in both study groups, at 15 of 147 patients and 20 of 155 patients in the ruxolitinib and BAT groups, respectively.18
As of the secondary analysis (January 6, 2020, data cut-off date), there were few patients with events of malignancy relapse or recurrence, at 16 (10.9%) and 25 (17.0%) patients, respectively, in the ruxolitinib and BAT groups. In the ruxolitinib and BAT groups, deaths (44.2% versus 44.9%) and censoring (44.9% versus 38.1%) were high. The probability of malignancy relapse or recurrence at 1 month and 2 months was low, at 0.69% (95% CI, 0.06% to 3.51%) and 4.21% (95% CI, 1.73% to 8.46%), respectively, in the ruxolitinib group, and 2.80% (95% CI, 0.92% to 6.54%) and 4.29% (95% CI, 1.75% to 8.60%), respectively, in the BAT group. The number of patients with competing risk (death with NRM for patients with underlying hematologic malignant disease) was 65 (44.2%) and 66 (44.9%) in the ruxolitinib and BAT groups, respectively.18
Results for malignancy relapse or recurrence at the final analysis were, overall, consistent with results at the primary analysis. As of the final data cut-off date (April 23, 2021), the number of patients who had events of malignancy relapse or recurrence was 20 (13.6%) in the ruxolitinib group and 25 (17.0%) in the BAT group. Detailed results for malignancy relapse or recurrence at the final analysis are described in Table 58 (Appendix 3).18
REACH 1
The incidence of malignancy relapse or progression was not assessed in the REACH 1 trial.
However, the relapse rate and relapse-related mortality rate were assessed and are summarized in Table 37. As of June 5, 2019, 5 (7.0%) patients had a relapse of the underlying malignancy and, of those, 4 (5.6%) patients had fatal outcomes.19
Cumulative Steroid Dosing Until Day 56
REACH 2
Results for the cumulative steroid dosing until day 56 outcome are summarized in Table 37. As of the July 25, 2019 data cut-off date, a higher proportion of patients had tapered off corticosteroids in the ruxolitinib group (21.4%; 95% CI, 15.2% to 28.8%) than in the BAT group (14.8%; 95% CI, 9.6% to 21.4%).18
The mean corticosteroid dose intensity at week 1 was 135.9 mg/day in the ruxolitinib group and 148.5 mg/day in the BAT group. In the week ending on the day 28 visit, the mean average corticosteroid dose (i.e., the mean of each participant's weekly average dose) had decreased to 74.8 mg/day and 75.8 mg/day in ruxolitinib and BAT groups, respectively. At each subsequent time point, mean average corticosteroid doses continued to decrease in both study groups (see Figure 8).18
The proportion of patients with a RDI of 50% or less was 29.2% and 24.5% in the ruxolitinib and BAT groups, respectively, and the proportion with a RDI of at least 50% was 68.8% and 74.8%, respectively (see Table 37).18
Results at the January 6, 2020, data cut-off date were consistent with those at the primary data cut-off date, and are summarized in Table 37. A higher proportion of patients had tapered off corticosteroids in the ruxolitinib group (22.1%; 95% CI, 15.8% to 29.5%) than in the BAT group (14.8%; 95% CI, 9.6% to 21.4%), with an odds ratio of 1.63 (95% CI, 0.91 to 2.92).18
The mean corticosteroid dose intensity at week 1 was 135.9 mg/day in the ruxolitinib group and 151.4 mg/day in the BAT group. In the week ending on the day 28 visit, the mean average corticosteroid dose (i.e., the mean of each participant's weekly average dose) had decreased to 73.9 mg/day and 75.5 mg/day in the ruxolitinib and BAT groups, respectively. At each subsequent time point, the mean average corticosteroid dose continued to decrease in both study groups.18
The proportions of patients with a RDI of 50% or less was 29.2% and 23.9% in the ruxolitinib and BAT groups, respectively, and the proportion with a RDI of at least 50% was 68.8% and 75.5%, respectively (see Table 37).18
Results at the final analysis were consistent with those at the primary data cut-off date. A higher proportion of patients had tapered off corticosteroids by day 56 in the ruxolitinib group (22.1%; 95% CI, 15.8% to 29.5%) than in the BAT group (14.8%; 95% CI, 9.6% to 21.4%), with an odds ratio of 1.63 (95% CI, 0.91 to 2.92). The proportion of patients with a RDI of 50% or less was 29.2% and 23.9% in the ruxolitinib and BAT groups, respectively, and the proportion with a RDI of at least 50% was 68.8% and 75.5%, respectively (see Table 58 in Appendix 3).18
At the final analysis, cumulative steroid dosing was assessed until EOT, and the trend was similar to that seen at day 56. A higher proportion of patients had tapered off corticosteroids in the ruxolitinib group (43.5%; 95% CI, 35.5% to 51.7%) than in the BAT group (31.6%; 95% CI, 24.5% to 39.6%), with an odds ratio of 1.67 (95% CI, 1.05 to 2.65). The proportion of patients with any dose reduction of corticosteroids by EOT was 92.2% and 87.1% in the ruxolitinib and BAT groups, respectively, and the proportion with a reduction of at least 50% was 77.3% and 74.2%, respectively. The dose reduction of corticosteroids achieved at EOT was greater in the ruxolitinib group (–61.6%) than the BAT group (–50.7%).18
REACH 1
The proportion of patients still receiving ruxolitinib who had tapered off (discontinued) corticosteroids was 6.9% at day 56 (see Table 37), 34.8% at day 100, and 61.1% day 180 (see Table 54 in Appendix 3).19
The mean average corticosteroid dose continued to decrease from day 1 (or day 2 if the day 1 dose was missing) (157.25 mg/day) to day 28 (62.25 mg/day), and then to day 56 (27.43 mg/day), day 100 (16.06 mg/day), and day 180 (8.57 mg/day). The proportion of patients with at least a 50% decrease in corticosteroid dose relative to the day 1 (or day 2 dose) continued to increase, from 23.2% on day 14 to 55.8% on day 28 and 100.0% on day 100. Figure 9 depicts the daily corticosteroid dose over time.19
Figure 8: Average Weekly Steroid Dosing in REACH 2, Full Analysis Set (Data Cut-Off Date of July 25, 2019)
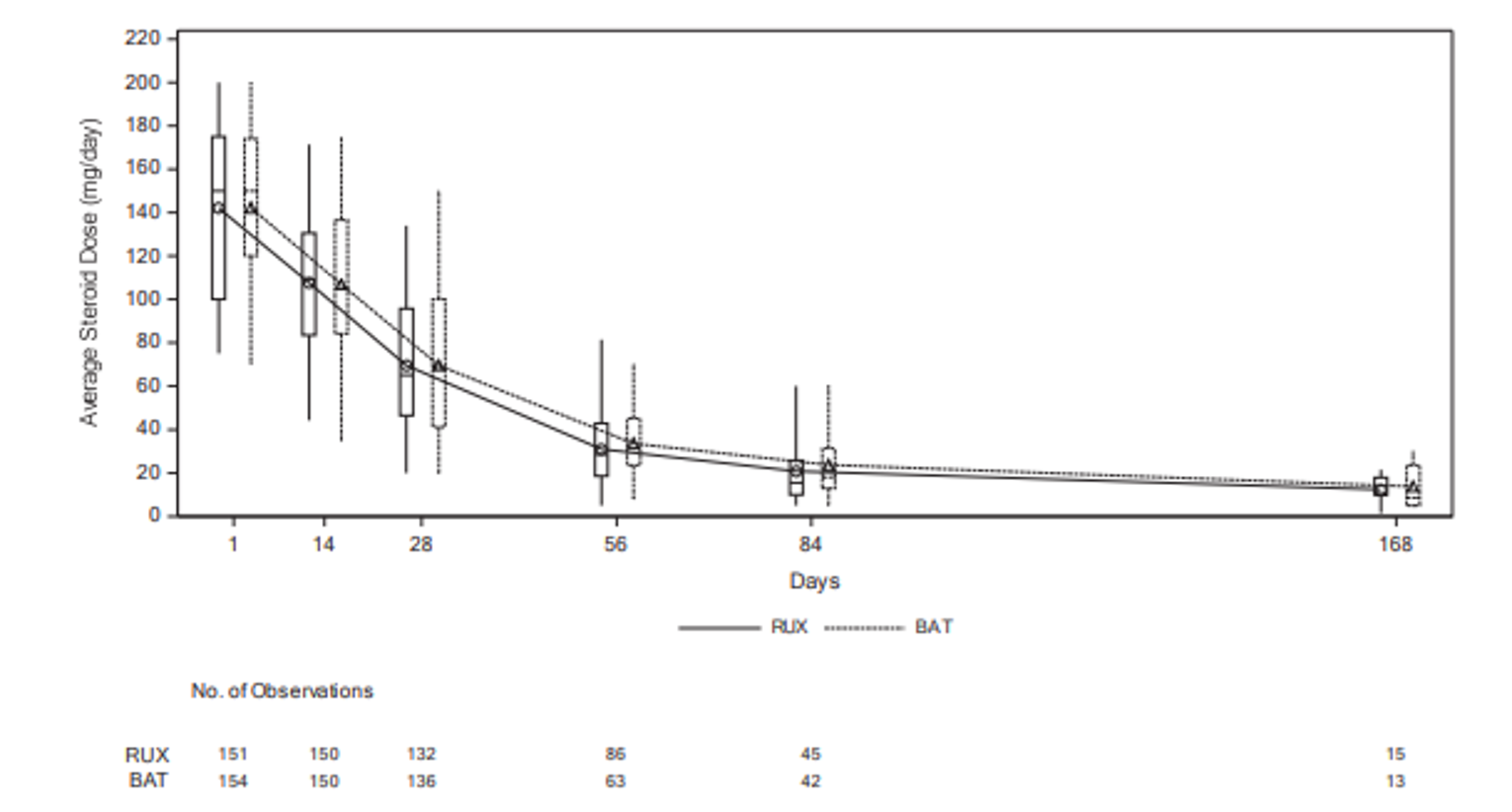
BAT = best available therapy; RUX = ruxolitinib.
Source: Clinical Study Report (REACH 2).18
Figure 9: Box Plot of Daily Corticosteroid Dose Over Time in REACH 1, Efficacy-Evaluable Patients (June 5, 2019, Data Cut-Off Date)
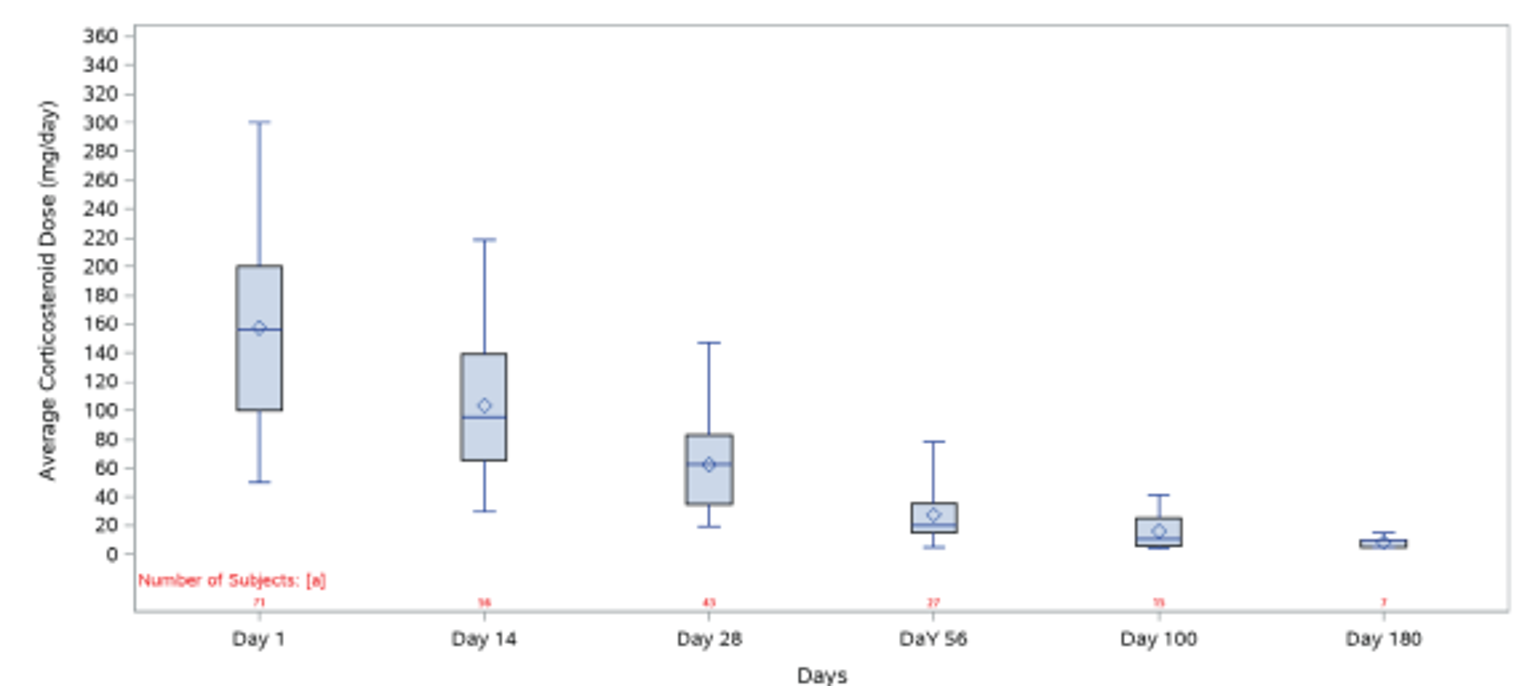
Source: Clinical Study Report (REACH 1).19
Incidence of cGvHD
REACH 2
Results for the incidence of cGvHD are summarized in Table 37. A detailed version of Table 37 is available in Table 56 (Appendix 3), showing the cumulative incidence. As of the July 25, 2019 data cut-off date, the proportion of patients who had been diagnosed with cGvHD was higher in the ruxolitinib group (24.7%; n = 38) than in the BAT group (16.8%; n = 26).18 The number of patients who were censored was 47 (30.5%) and 51 (32.9%) in the ruxolitinib and BAT groups, respectively. At 6 and 12 months, the probability of cGvHD incidence increased over time in both groups, from 14.85% (95% CI, 9.30% to 21.63%) to 32.28% (95% CI, 23.74% to 41.10%), respectively, in the ruxolitinib group, and from 13.28% (95% CI, 8.04% to 19.84%) to 22.19% (95% CI, 14.83% to 30.50%), respectively, in the BAT group, although the rates were higher in the ruxolitinib group. The number of patients with competing risk (hematologic malignancy relapse or progression and death without prior onset of cGvHD) was 69 (44.8%) and 78 (50.3%) for the ruxolitinib and BAT groups, respectively.18
Results at the January 6, 2020, data cut-off date were consistent with those at the primary data cut-off date and are summarized in Table 37. The proportion of patients who had been diagnosed with cGvHD was higher in the ruxolitinib group (29.2%; n = 45) than in the BAT group (18.7%; n = 29). At 12 and 18 months, the probability of cGvHD was higher in the ruxolitinib group, at 29.95% (95% CI, 22.53% to 37.71%) and 32.36% (95% CI, 24.47% to 40.48%), respectively, than in the BAT group, at 20.27% (95% CI, 13.87% to 27.54%) and 22.80% (95% CI, 15.75% to 30.65%), respectively. The median onset of cGvHD was longer in the ruxolitinib group (181.0 days) than in the BAT group (142.0 days). Most of the cGvHD events were mild at the time of onset in both study groups, and fewer patients had severe cGvHD in the ruxolitinib group (4 patients) than in the BAT group (7 patients). The cumulative incidence of cGvHD at the January 6, 2020, data cut-off date is depicted in Figure 35 (Appendix 3).
Results at the final analysis were, overall, consistent with those at the primary data cut-off date. The proportion of patients who had been diagnosed with cGvHD was higher in the ruxolitinib group (33.8%; n = 52) than in the BAT group (21.9%; n = 34). Detailed results for the incidence of cGvHD are described in Table 58 (Appendix 3).18
REACH 1
Results for the incidence of cGvHD are summarized in Table 37 and Table 55 (Appendix 3). As of the June 5, 2019, data cut-off date, the proportion of patients who had been diagnosed with cGvHD in the ruxolitinib group was 15.5% (n = 11; 95% CI, 8.0% to 26.0%). The interval between the initiation of ruxolitinib and diagnosis of cGvHD was less than 100 days for 1 patient, between 100 and 180 days for 3 patients, and more than 180 days for 7 patients. The numbers of patients who discontinued ruxolitinib before developing cGvHD, received ongoing ruxolitinib treatment, or had discontinued ruxolitinib treatment were 6, 2, and 2, respectively.19
Resource Use
REACH 2
At the final analysis, the percentage of patients who started study treatment at the same time as being hospitalized was similar in the ruxolitinib and BAT groups (16.9% and 12.9%, respectively). Overall, patients in the BAT group had longer hospital stays (median = 42 days; interquartile range = 24 to 67 days) than patients in the ruxolitinib group (median = 32.5 days; interquartile range = 8 to 53 days). The most reported types of facility for hospitalization were (ruxolitinib versus BAT) the transplant unit (16.9% versus 12.9%) and the general ward (14.9% versus 20.6%). The proportion of patients readmitted to hospital, the median duration of stay at readmission, and the number of readmissions were similar in the study groups.18
REACH 1
Resource use data were not collected in REACH 1.
Harms
Only harms identified in the review protocol are reported in the following.
REACH 2
See Table 39 for detailed harms data in the REACH 2 trial. There were minimal differences in the harms data presented at the primary, secondary, and final analyses (at the final analysis, 1 additional patient in the ruxolitinib group was reported to have AEs; at the secondary analysis, 1 additional patient in ruxolitinib group and 2 additional patients in BAT group were reported to have AEs).18 This CADTH clinical report presents harms data for the secondary data cut-off date (January 6, 2020).18
REACH 1
See Table 40 for detailed harms data in the REACH 1 trial. This CADTH clinical report presents harms data for the final analysis cut-off date (June 5, 2019).
Adverse Events
REACH 2
Up to the data cut-off date of January 6, 2020, almost all patients in the ruxolitinib and BAT study groups experienced at least 1 TEAE (99.3% and 98.7%, respectively). The most commonly reported TEAEs in the ruxolitinib and BAT groups, respectively, were anemia (40.1% versus 32%), thrombocytopenia (36.8% versus 20.7%), cytomegalovirus infection (30.9% versus 26.7%), neutropenia (24.3% versus 14.7%), and peripheral edema (24.3% versus 21.3%). The percentage of patients who experienced at least 1 TEAE of grade 3 or higher was 91.4% and 87.3% in the ruxolitinib and BAT groups, respectively. The most commonly reported TEAEs of grade 3 or higher in the ruxolitinib and BAT groups, respectively, were anemia (35.5% versus 24.0%), thrombocytopenia (33.6% versus 16.7%), neutropenia (21.7% versus 12.0%), decreased platelet count (17.8% versus 15.3%), and decreased white blood cell count (13.2% versus 8.7%).18
The percentage of patients reporting at least 1 TEAE up to day 28 was similar to that in the period up to the data cut-off date, with slightly more patients reporting at least 1 TEAE in the ruxolitinib group than in the BAT group (96.1% versus 94.7%, respectively). The most commonly reported TEAEs up to day 28 were similar to TEAEs up to the data cut-off date in the ruxolitinib and BAT groups, and included anemia (30.3% versus 28.0%), thrombocytopenia (32.9% versus 18.7%), cytomegalovirus infection (25.7% versus 20.7%), peripheral edema (18.4% versus 17.3%), and neutropenia (15.8% versus 12.7%). The percentage of patients who experienced at least 1 TEAE of grade 3 or higher was 78.3% and 79.3% in the ruxolitinib and BAT groups, respectively. The most commonly reported TEAEs of grade 3 or higher in the ruxolitinib and BAT groups, respectively, were anemia (22.4% versus 18.7%), thrombocytopenia (27.0% versus 16.0%), platelet count decreased (14.5% versus 13.3%), and neutropenia (13.2% versus 9.3%).18
REACH 1
All patients in the REACH 1 trial experienced at least 1 TEAE (100.0%) (see Table 40). The most commonly reported TEAEs were similar in the REACH 1 and REACH 2 trials, and included anemia (64.8%), thrombocytopenia (62.0%), hypokalemia (49.3%), neutropenia (47.9%), and peripheral edema (46.5%).19
TEAEs of grade 3 or higher occurred in 97.2% of patients. The most commonly reported TEAEs of grade 3 or higher were similar in the REACH 1 and REACH 2 studies, and included thrombocytopenia (53.5%), anemia (50.7%), neutropenia (42.3%), and hyperglycemia (19.7%).19
Serious Adverse Events
REACH 2
Up to the data cut-off date of January 6, 2020, the percentage of patients experiencing at least 1 serious TEAE was 66.4% in the ruxolitinib group and 53.3% in the BAT group. The most common serious TEAEs (ruxolitinib versus BAT) were sepsis (7.9% versus 7.3%), pyrexia (6.6% versus 4.0%), septic shock (6.6% versus 5.3%), and diarrhea (5.3% versus 2.0%).18
Up to day 28, the percentage of patients reporting serious TEAEs was lower than in the period up to the data cut-off date for the ruxolitinib and BAT groups (37.5% and 34.0%, respectively). The most commonly reported serious TEAEs (ruxolitinib versus BAT) were sepsis (5.3% versus 2.0%), pyrexia (2.0% versus 1.3%), diarrhea (3.3% versus 0.7%), and septic shock (2.6% versus 2.7%).18
REACH 1
The percentage of patients experiencing serious TEAEs was 83.1% in the REACH 1 trial. The most commonly reported serious TEAEs were similar in the REACH 1 and REACH 2 trials, and included sepsis (12.7%), pyrexia (11.3%), respiratory failure (11.3%), and lung infection (7.0%).19
Withdrawals due to Adverse Events
REACH 2
Up to the data cut-off date of January 6, 2020, the percentage of patients who discontinued treatment due to TEAEs was 27.0% in the ruxolitinib group and 9.3% in the BAT group. The most commonly cited TEAEs contributing to treatment discontinuation in the ruxolitinib group were neutropenia (n = 4; 2.6%), sepsis (n = 4; 2.6%), anemia (n = 3; 2.0%), and thrombocytopenia (n = 3; 2.0%), and in the BAT group were sepsis (n = 1; 0.7%), anemia (n = 1; 0.7%), thrombocytopenia (n = 1; 0.7%), and decreased platelet count (n = 1; 0.7%).18
Up to day 28, the percentage of patients discontinuing study treatment due to TEAEs was lower than in the period up to the data cut-off date (11.2% and 4% in the ruxolitinib and BAT groups, respectively). As in the period up to the data cut-off date, the following TEAEs were reported as reasons for treatment discontinuation in the ruxolitinib group: anemia (n = 3; 2.0%), thrombocytopenia (n = 3; 2.0%), pancytopenia (n = 2; 1.3%), sepsis (n = 1; 0.7%), and leukopenia (n = 1; 0.7%). In the BAT group, 1 patient discontinued treatment due to anemia (0.7%) and 1 discontinued due to septic shock (0.7%).18
REACH 1
AEs led to discontinuation of ruxolitinib in 32.4% of patients in the REACH 1 trial. The most commonly reported TEAEs leading to discontinuation of ruxolitinib were sepsis (5.6%), acute kidney injury (2.8%), and respiratory failure (2.8%).19
Mortality
REACH 2
On-treatment deaths up to the data cut-off date of January 6, 2020, occurred in 28.3% and 24.0% of patients in the ruxolitinib and BAT groups, respectively. On-treatment deaths were defined as deaths that occurred from the first administration of the randomized treatment up to 30 days after the final administration of the randomized treatment. The most common cause of death was the study indication of aGvHD (including aGvHD and related complications) in 21 (13.8%) and 21 (14.0%) patients in the ruxolitinib and BAT groups, respectively. The number of on-treatment deaths suspected to be related to the study treatment was 10 and 4 in the ruxolitinib and BAT groups, respectively.18
Up to the data cut-off date of January 6, 2020, TEAEs leading to death were similar in the ruxolitinib and BAT groups, at 21.7% and 21.3%, respectively. The most commonly occurring TEAEs leading to death in the ruxolitinib and BAT groups, respectively, were sepsis (5.3% versus 2.7%), septic shock (4.6% versus 2.5%), pneumonia (1.3% versus 2.7%), multiple organ dysfunction syndrome (1.3% versus 2.0%), and respiratory failure (0.7% versus 2.7%).18
Up to day 28, on-treatment deaths occurred in 9.9% of patients in the ruxolitinib and 14.0% of patients in the BAT group. The primary cause of death was the study indication of aGvHD (including aGvHD and related complications) in the ruxolitinib group (n = 9; 5.9%) and the BAT group (n = 17; 11.3%).18
Up to day 28, 7.9% of patients in the ruxolitinib group experienced a TEAE that led to death, compared with 11.3% in the BAT group. The most commonly occurring TEAEs leading to death in the ruxolitinib and BAT groups, respectively, were sepsis (2.0% versus 1.3%), septic shock (2.0% versus 2.0%), pneumonia (0.7% versus 2.0%), and respiratory failure (0.7% and 2.0%).18
REACH 1
As of the June 5, 2019, data cut-off date, 35.2% (n = 25) of patients had died during treatment with ruxolitinib or in the 30 days after their final dose. The most common cause of death was other (25.4%, n = 18), which included underlying GvHD, multi-organ failure, pulseless electrical activity arrest, and respiratory failure; many of t were counted as fatal TEAEs. The remainder of patients (9.9%, n = 7) died of GvHD progression. No patients died from relapse of the underlying malignancy.19
The percentage of patients who had at least 1 TEAE that led to death was 39.4% (n = 28). The most commonly occurring TEAE leading to death was respiratory failure (8.5%, n = 6). Other fatal TEAEs occurring in more than 1 patient included sepsis, disease progression, multiple organ dysfunction syndrome, and hepatic failure (2.8% for each; n = 2 for each). Respiratory failure was reported to have several potential causes, including medical history, prior or concomitant medications, pulmonary infections, or renal failure. None of the fatal TEAEs was attributed to ruxolitinib treatment alone.19
Notable Harms
Notable harms specified in the CADTH review protocol included serious infections, heart rate decrease and electrocardiographic PR-interval prolongation, cytopenia, and lipid abnormalities.
Infections
REACH 2
Up to the data cut-off date of January 6, 2020, serious infections were reported in 38.2% and 30.0% of patients in the ruxolitinib and BAT groups, respectively, and serious infections of grade 3 or higher were reported in 38.2% and 28.7% of patients, respectively. The percentage of patients who experienced at least 1 infection TEAE of any grade was 80.9% and 69.3% in the ruxolitinib and BAT groups, respectively. In the ruxolitinib group, viral infections were most common (58.6%), followed by bacterial infections (47.4%) and fungal infections (17.1%). In the BAT group, bacterial infections were most common (46.0%), followed by viral infections (44.7%) and fungal infections (9.3%).18
Up to day 28, serious infections occurred in 21.7% and 17.3% of patients in the ruxolitinib and BAT groups, respectively, and serious infections of grade 3 or higher occurred in 21.1% and 17.3% of patients, respectively. The percentage of patients who experienced at least 1 infection TEAE of any grade was 61.2% and 55.3% in the ruxolitinib and BAT groups, respectively. In the ruxolitinib group, viral infections were most common (42.8%), followed by bacterial infections (29.6%) and fungal infections (8.6%). In the BAT group, viral infections were most common (33.3%), followed by bacterial infections (32.7%) and fungal infections (4.7%).18
REACH 1
As of the final analysis (June 5, 2019, data cut-off date), there were 58 patients (81.7%) with at least 1 TEAE infection or infestation, 36 of whom experienced serious TEAE infections and infestations. The most commonly reported infections and infestations were sepsis (14.1%), cytomegalovirus infection (12.7%), upper respiratory tract infection (9.9%), and bacteremia (9.9%); the most commonly reported serious TEAE of infections and infestations was bacteremia (5.6%).19
Heart Rate Decrease and PR-Interval Prolongation
REACH 2
One patient in the ruxolitinib group and 1 patient in the BAT reported bradycardia of any grade up to the data cut-off date of January 6, 2020, and up to day 28. No patients reported bradycardia of grade 3 or higher.18
REACH 1
Two patients reported experiencing bradycardia of any grade up to the final data cut-off date of June 5, 2019. One patient-reported bradycardia of grade 3 or higher.19
Cytopenias
REACH 2
Up to the data cut-off date of January 6, 2020, cytopenia TEAEs of special interest of any grade in the ruxolitinib and BAT groups, respectively, included anemia (40.8% versus 34.0%), thrombocytopenia (56.6% versus 36.7%), leukopenia (46.7% versus 32.0%), and other cytopenias (8.6% versus 6.0%). Grade 3 or higher cytopenia TEAEs of special interest in the ruxolitinib and BAT groups, respectively, included anemia (36.2% versus 25.3%), thrombocytopenia (50.7% versus 32.0%), leukopenia (42.8% versus 27.3%), and other cytopenias (5.9% versus 4.7%).18
Up to day 28, cytopenia TEAEs of special interest of any grade in the ruxolitinib and BAT groups included anemia (30.3% versus 29.3%), thrombocytopenia (50.0% versus 32.7%), leukopenia (32.9% versus 26.7%), and other cytopenias (5.9% versus 4.7%). Grade 3 or higher cytopenia TEAEs of special interest in the ruxolitinib and BAT groups, respectively, included anemia (22.4% versus 20.0%), thrombocytopenia (41.4% versus 29.3%), leukopenia (28.9% versus 22.0%), and other cytopenias (4.6% versus 3.3%).
REACH 1
Up to the final data cut-off date, cytopenia TEAEs of any grade included anemia (64.8%), neutropenia (47.9%), and thrombocytopenia (62.0%). Grade 3 or higher cytopenia TEAEs included anemia (50.7%), neutropenia (42.2%), and thrombocytopenia (53.5%).19
Lipid Abnormalities
REACH 2
Up to the cut-off date of January 6, 2020, lipid abnormality events of any grade were reported in 9.9% and 7.3% of patients in the ruxolitinib and BAT groups, respectively, and lipid abnormality events of grade 3 or higher were reported in 3.9% and 2.7% of patients.18
Up to day 28, the percentage of patients reporting lipid abnormalities was similar in the 2 groups. Lipid abnormality events of any grade were reported in 3.9% and 4.0% of patients in the ruxolitinib and BAT groups, respectively. Two patients in each group reported lipid abnormality events of grade 3 or higher.18
REACH 1
Lipid abnormalities were not reported in the REACH 1 trial.
Harms in Adolescents
REACH 2
The safety profile of the 9 adolescent participants was, overall, similar to that of the study safety set.18
REACH 1
REACH 1 did not include any adolescents.19
Harms in the Crossover Set
REACH 2
Overall, the safety profile of the 49 patients in the crossover set was similar to that observed in the ruxolitinib group at the secondary data cut-off date. The most commonly reported TEAEs of any grade were anemia (30.6%), thrombocytopenia (30.6%), hypokalemia (22.4%) neutropenia (20.4%), cytomegalovirus infection (18.4%), sepsis (18.4%), pyrexia (18.4%), thrombocytopenia (18.4%), decreased platelet count (12.2%), leukopenia (6.1%), and pancytopenia (6.1%). The most common SAEs were sepsis (14.3%) and respiratory failure (12.2%). The percentage of patients experiencing AEs leading to study drug discontinuation was 36.7% (n = 18). A total of 15 (30.6%) patients were reported to have experienced fatal SAEs, 4 of which were considered to be related to the study drug.18
Table 39: Summary of Harms in REACH 2, Safety Population (January 6, 2020, Data Cut-Off Date)
Harms | Up to data cut-off date of January 6, 2020 | Up to day 28 | ||
|---|---|---|---|---|
Ruxolitinib N = 152 | BAT N = 150 | Ruxolitinib N = 152 | BAT N = 150 | |
Patients with at least 1 TEAE | ||||
n (%) | 151 (99.3) | 148 (98.7) | 146 (96.1) | 142 (94.7) |
Most common events, n (%)a | ||||
Anemia | 61 (40.1) | 48 (32.0) | 46 (30.3) | 42 (28.0) |
Thrombocytopenia | 56 (36.8) | 31 (20.7) | 50 (32.9) | 28 (18.7) |
CMV infection | 47 (30.9) | 40 (26.7) | 39 (25.7) | 31 (20.7) |
Neutropenia | 37 (24.3) | 22 (14.7) | 24 (15.8) | 19 (12.7) |
Peripheral edema | 37 (24.3) | 32 (21.3) | 28 (18.4) | 26 (17.3) |
Hypokalemia | 34 (22.4) | 28 (18.7) | 21 (13.8) | 25 (16.7) |
Pyrexia | 34 (22.4) | 26 (17.3) | 16 (10.5) | 18 (12.0) |
Decreased platelet count | 31 (20.4) | 24 (16.0) | 26 (17.1) | 21 (14.0) |
Nausea | 30 (19.7) | 17 (11.3) | NR | NR |
Vomiting | 25 (16.4) | 17 (11.3) | NR | NR |
Diarrhea | 24 (15.8) | 22 (14.7) | 14 (9.2) | 15 (10.0) |
Hypomagnesemia | 23 (15.1) | 24 (16.0) | 16 (10.5) | 19 (12.7) |
Hypertension | 21 (13.8) | 19 (12.7) | 16 (10.5) | 14 (9.3) |
Decreased white blood cell count | 21 (13.8) | 16 (10.7) | NR | NR |
Abdominal pain | 20 (13.2) | 13 (8.7) | NR | NR |
Acute kidney injury | 19 (12.5) | 11 (7.3) | NR | NR |
Decreased neutrophil count | 19 (12.5) | 16 (10.7) | 10 (6.6) | 15 (10.0) |
Hypoalbuminemia | 17 (11.2) | 19 (12.7) | 16 (10.5) | 16 (10.7) |
Increased alanine aminotransferase | 16 (10.5) | 11 (7.3) | NR | NR |
Cough | 16 (10.5) | 12 (8.0) | NR | NR |
Pneumonia | 16 (10.5) | 14 (9.3) | NR | NR |
Sepsis | 16 (10.5) | 19 (12.7) | NR | NR |
Urinary tract infection | 16 (10.5) | 9 (6.0) | NR | NR |
Hypophosphatemia | 15 (9.9) | 15 (10.0) | NR | NR |
Hypocalcemia | 14 (9.2) | 16 (10.7) | NR | NR |
Hyperglycaemia | 10 (6.6) | 15 (10.0) | NR | NR |
Increased blood bilirubin | 9 (5.9) | 15 (10.0) | NR | NR |
Patients with at least 1 TEAE of grade 3 or higher | ||||
n (%) | 139 (91.4) | 131 (87.3) | 119 (78.3) | 119 (79.3) |
Most common events, n (%)a | ||||
Anemia | 54 (35.5) | 36 (24.0) | 34 (22.4) | 28 (18.7) |
Thrombocytopenia | 51 (33.6) | 25 (16.7) | 41 (27.0) | 24 (16.0) |
Neutropenia | 33 (21.7) | 18 (12.0) | 20 (13.2) | 14 (9.3) |
Decreased platelet count | 27 (17.8) | 23 (15.3) | 22 (14.5) | 20 (13.3) |
Decreased white blood cell count | 20 (13.2) | 13 (8.7) | NR | NR |
Decreased neutrophil count | 17 (11.2) | 14 (9.3) | 10 (6.6) | 12 (8.0) |
Hypokalemia | 15 (9.9) | 18 (12.0) | 9 (5.9) | 9 (6.0) |
CMV infection | 14 (9.2) | 18 (12.0) | 11 (7.2) | 12 (8.0) |
Sepsis | 13 (8.6) | 18 (12.0) | NR | NR |
Diarrhea | 11 (7.2) | 8 (5.3) | 7 (4.6) | 5 (3.3) |
Pneumonia | 11 (7.2) | 13 (8.7) | NR | NR |
Hypertension | 10 (6.6) | 8 (5.3) | 9 (5.9) | 6 (4.0) |
Hypoalbuminemia | 8 (5.3) | 11 (7.3) | 6 (3.9) | 10 (6.7) |
Increased alanine aminotransferase | 7 (4.6) | 5 (3.3) | NR | NR |
Hypophosphatemia | 7 (4.6) | 7 (4.7) | NR | NR |
Acute kidney injury | 6 (3.9) | 7 (4.7) | NR | NR |
Urinary tract infection | 6 (3.9) | 5 (3.3) | NR | NR |
Hypocalcemia | 5 (3.3) | 6 (4.0) | NR | NR |
Hyperglycaemia | 5 (3.3) | 9 (6.0) | NR | NR |
Increased blood bilirubin | 5 (3.3) | 9 (6.0) | NR | NR |
Pyrexia | 4 (2.6) | 4 (2.7) | 2 (1.3) | 2 (1.3) |
Vomiting | 4 (2.6) | 2 (1.3) | NR | NR |
Abdominal pain | 4 (2.6) | 5 (3.3) | NR | NR |
Peripheral edema | 3 (2.0) | 3 (2.0) | 2 (1.3) | 1 (0.7) |
Hypomagnesemia | 2 (1.3) | 2 (1.3) | 0 | 1 (0.7) |
Nausea | 1 (0.7) | 4 (2.7) | NR | NR |
Cough | 1 (0.7) | 0 | NR | NR |
Patients with at least 1 serious TEAE | ||||
n (%) | 101 (66.4) | 80 (53.3) | 57 (37.5) | 51 (34.0) |
Most common events, n (%)b | ||||
Sepsis | 12 (7.9) | 11 (7.3) | 8 (5.3) | 3 (2.0) |
Pyrexia | 10 (6.6) | 6 (4.0) | 3 (2.0) | 2 (1.3) |
Septic shock | 10 (6.6) | 8 (5.3) | 4 (2.6) | 4 (2.7) |
Diarrhea | 8 (5.3) | 3 (2.0) | 5 (3.3) | 1 (0.7) |
Pneumonia | 7 (4.6) | 8 (5.3) | 3 (2.0) | 3 (2.0) |
CMV infection | 6 (3.9) | 8 (5.3) | 4 (2.6) | 5 (3.3) |
Respiratory failure | 6 (3.9) | 6 (4.0) | 4 (2.6) | 4 (2.7) |
Acute kidney injury | 3 (2.0) | 5 (3.3) | NR | NR |
Increased blood bilirubin | 3 (2.0) | 0 | NR | NR |
CMV colitis | 3 (2.0) | 0 | NR | NR |
Febrile neutropenia | 3 (2.0) | 2 (1.3) | NR | NR |
Neutropenia | 3 (2.0) | 3 (2.0) | NR | NR |
Pancytopenia | 3 (2.0) | 0 | 3 (2.0) | 3 (2.0) |
Pseudomonal sepsis | 3 (2.0) | 0 | NR | NR |
Respiratory distress | 3 (2.0) | 0 | NR | NR |
Multiple organ dysfunction syndrome | 2 (1.3) | 3 (2.0) | NR | NR |
Renal failure | 2 (1.3) | 3 (2.0) | NR | NR |
Abdominal pain | 1 (0.7) | 3 (2.0) | NR | NR |
Confusional state | 1 (0.7) | 3 (2.0) | NR | NR |
Bacteremia | 1 (0.7) | 4 (2.7) | NR | NR |
Acute respiratory failure | 0 | 3 (2.0) | NR | NR |
GvHD | 0 | 3 (2.0) | NR | NR |
Decreased platelet count | 0 | 3 (2.0) | NR | NR |
Patients who stopped treatment due to TEAEs | ||||
n (%) | 41 (27.0) | 14 (9.3) | 17 (11.2) | 6 (4.0) |
Most common events, n (%)c | ||||
Neutropenia | 4 (2.6) | 0 | NR | NR |
Sepsis | 4 (2.6) | 1 (0.7) | 1 (0.7) | 0 |
Anemia | 3 (2.0) | 1 (0.7) | 3 (2.0) | 1 (0.7) |
Thrombocytopenia | 3 (2.0) | 1 (0.7) | 3 (2.0) | 0 |
Bacterial sepsis | 2 (1.3) | 0 | NR | NR |
CMV colitis | 2 (1.3) | 0 | NR | NR |
Graft loss | 2 (1.3) | 0 | NR | NR |
Leukopenia | 2 (1.3) | 0 | 1 (0.7) | 0 |
Pancytopenia | 2 (1.3) | 0 | 2 (1.3) | 0 |
Decreased platelet count | 2 (1.3) | 1 (0.7) | NR | NR |
Septic shock | 0 | 2 (1.3) | 0 | 1 (0.7) |
Deaths, n (%) | ||||
On-treatment deathsd | 43 (28.3) | 36 (24.0) | 15 (9.9) | 21 (14.0) |
SAEs with fatal outcome | 33 (21.7) | 32 (21.3) | 12 (7.9) | 17 (11.3) |
Most common SAEs with fatal outcome, n (%)b | ||||
Sepsis | 8 (5.3) | 4 (2.7) | 3 (2.0) | 2 (1.3) |
Septic shock | 7 (4.6) | 4 (2.7) | 3 (2.0) | 3 (2.0) |
Pneumonia | 2 (1.3) | 4 (2.7) | 1 (0.7) | 3 (2.0) |
Multiple organ dysfunction syndrome | 2 (1.3) | 3 (2.0) | 0 | 1 (0.7) |
Cardiac arrest | 2 (1.3) | 0 | 1 (0.7) | 0 |
Abnormal general physical condition | 2 (1.3) | 0 | 1 (0.7) | 0 |
Pseudomonal sepsis | 2 (1.3) | 0 | 1 (0.7) | 0 |
Respiratory failure | 1 (0.7) | 4 (2.7) | 1 (0.7) | 3 (2.0) |
aGvHD | 0 | 2 (1.3) | 0 | 1 (0.7) |
GvHD | 0 | 3 (2.0) | 0 | 3 (2.0) |
Notable harms, n (%) | ||||
Infections | ||||
Serious TEAE infection | 58 (38.2) | 45 (30.0) | 33 (21.7) | 26 (17.3) |
Serious infection, grade ≥ 3 | 58 (38.2) | 43 (28.7) | 32 (21.1) | 24 (16.0) |
Heart rate decrease and PR-interval prolongation | ||||
Bradycardia, any grade TEAE | 1 (0.7) | 1 (0.7) | 1 (0.7) | 1 (0.7) |
Bradycardia, grade ≥ 3 | 0 | 0 | 0 | 0 |
Cytopenia | ||||
Anemia, any grade TEAE | 62 (40.8) | 51 (34.0) | 46 (30.3) | 44 (29.3) |
Anemia, grade ≥ 3 TEAE | 55 (36.2) | 38 (25.3) | 34 (22.4) | 30 (20.0) |
Thrombocytopenia, any grade TEAE | 86 (56.6) | 55 (36.7) | 76 (50.0) | 49 (32.7) |
Thrombocytopenia, grade ≥ 3 | 77 (50.7) | 48 (32.0) | 63 (41.4) | 44 (29.3) |
Leukopenia, any grade TEAE | 71 (46.7) | 48 (32.0) | 50 (32.9) | 40 (26.7) |
Leukopenia, grade ≥ 3 | 65 (42.8) | 41 (27.3) | 44 (28.9) | 33 (22.0) |
Other cytopenia, any grade TEAE | 13 (8.6) | 9 (6.0) | 9 (5.9) | 7 (4.7) |
Other cytopenia, grade ≥ 3 | 9 (5.9) | 7 (4.7) | 7 (4.6) | 5 (3.3) |
Lipid abnormalities | ||||
Lipid abnormality, any grade TEAE | 15 (9.9) | 11 (7.3) | 6 (3.9) | 6 (4.0) |
Lipid abnormality, grade ≥ 3 | 6 (3.9) | 4 (2.7) | 2 (1.3) | 2 (1.3) |
aGvHD = acute graft-versus-host disease; BAT = best available therapy; CMV = cytomegalovirus; GvHD = graft-vs.-host disease; NR = not reported; SAE = serious adverse event; TEAE = treatment-emergent adverse event.
Note: A patient with multiple severity grades for an AE is only counted under the maximum grade. AEs occurring outside the on-randomized-treatment period are not summarized. MedDRA version 22.1, CTCAE version 4.03.
aFrequency: > 10% of patients in either treatment group (all grades).
bFrequency: ≥ 2% of patients in either treatment group.
cFrequency: > 1% of patients in either treatment group (all grades).
dDeaths from date of first administration of randomized treatment to 30 days after the final administration of randomized treatment. Deaths occurring outside the on-randomized-treatment period or after day 31 are not summarized.
Source: Clinical Study Report (REACH 2).18
Table 40: Summary of Harms in REACH 1, Safety-Evaluable Population (June 5, 2019, Data Cut-Off Date)
Harms | Ruxolitinib N = 152 |
|---|---|
Patients with at least 1 TEAEa | |
n (%) | 71 (100.0) |
Most common events, n (%)b | |
Erythropenia (anemia) | 46 (64.8) |
Thrombocytopeniac | 44 (62.0) |
Hypokalemia | 35 (49.3) |
Neutropeniad | 34 (47.9) |
Peripheral edema | 33 (46.5) |
Muscular weakness | 25 (35.2) |
Dyspnea | 24 (33.8) |
Hypomagnesemia | 24 (33.8) |
Nausea | 23 (32.4) |
Fatigue | 22 (31.0) |
Hypocalcemia | 22 (31.0) |
Diarrhea | 21 (29.6) |
Decreased white blood cell count | 21 (29.6) |
Increased alanine aminotransferase | 19 (26.8) |
Hypophosphatemia | 19 (26.8) |
Increased aspartate aminotransferase | 18 (25.4) |
Hyperglycemia | 18 (25.4) |
Vomiting | 18 (25.4) |
Acute kidney injury | 17 (23.9) |
Back pain | 17 (23.9) |
Pyrexia | 17 (23.9) |
Decreased appetite | 16 (22.5) |
Hypertension | 16 (22.5) |
Hypotension | 16 (22.5) |
Abdominal pain | 15 (21.1) |
Cough | 15 (21.1) |
Headache | 15 (21.1) |
Hyponatremia | 15 (21.1) |
Fall | 14 (19.7) |
Sinus tachycardia | 14 (19.7) |
Arthralgia | 13 (18.3) |
Increased blood creatinine | 13 (18.3) |
Constipation | 13 (18.3) |
Hypoalbuminemia | 13 (18.3) |
Hematuria | 12 (16.9) |
Pain in extremity | 12 (16.9) |
Abdominal distension | 11 (15.5) |
Increased blood bilirubin | 11 (15.5) |
Depression | 11 (15.5) |
Dry eye | 11 (15.5) |
Dry mouth | 11 (15.5) |
Flatulence | 11 (15.5) |
Lower GI hemorrhage | 11 (15.5) |
Anxiety | 10 (14.1) |
Confusional state | 10 (14.1) |
Dizziness | 10 (14.1) |
Hypoglycemia | 10 (14.1) |
Insomnia | 10 (14.1) |
Pollakiuria | 10 (14.1) |
Respiratory failure | 10 (14.1) |
Sepsis | 10 (14.1) |
Increased blood alkaline phosphatase | 9 (12.7) |
CMV infection | 9 (12.7) |
Hypertriglyceridemia | 9 (12.7) |
Pleural effusion | 9 (12.7) |
Maculopapular rash | 9 (12.7) |
Chills | 8 (11.3) |
Epistaxis | 8 (11.3) |
Hyperkalemia | 8 (11.3) |
Decreased lymphocyte count | 8 (11.3) |
Nasal congestion | 8 (11.3) |
Vision blurred | 8 (11.3) |
Patients with at least 1 grade ≥ 3 TEAE | |
n (%) | 69 (97.2) |
Most common events, n (%)e | |
Thrombocytopeniae | 38 (53.5) |
Erythropenia (anemia) | 36 (50.7) |
Neutropeniac | 30 (42.3) |
Hyperglycaemia | 14 (19.7) |
Hypokalemia | 13 (18.3) |
Hypophosphatemia | 12 (16.9) |
Decreased white blood cell count | 12 (16.9) |
Hyponatremia | 11 (15.5) |
Fatigue | 10 (14.1) |
Hypertension | 10 (14.1) |
Hypoalbuminemia | 10 (14.1) |
Hypotension | 10 (14.1) |
Respiratory failure | 10 (14.1) |
Sepsis | 9 (12.7) |
Hypocalcemia | 8 (11.3) |
Decreased lymphocyte count | 8 (11.3) |
Muscular weakness | 8 (11.3) |
Peripheral edema | 8 (11.3) |
Bacteremia | 7 (9.9) |
Increased blood bilirubin | 7 (9.9) |
Acute kidney injury | 6 (8.5) |
Decreased appetite | 6 (8.5) |
Dyspnea | 6 (8.5) |
Lower GI hemorrhage | 6 (8.5) |
Lung infection | 6 (8.5) |
Abdominal pain | 5 (7.0) |
Increased alanine aminotransferase | 5 (7.0) |
Diarrhea | 5 (7.0) |
Hypoxia | 5 (7.0) |
Pneumonia | 5 (7.0) |
Increased blood alkaline phosphatase | 4 (5.6) |
CMV infection | 4 (5.6) |
Enterococcal infection | 4 (5.6) |
Hyperkalemia | 4 (5.6) |
Nausea | 4 (5.6) |
Pneumatosis intestinalis | 4 (5.6) |
Septic shock | 4 (5.6) |
Staphylococcal infection | 4 (5.6) |
Patients with at least 1 serious TEAE | |
n (%) | 59 (83.1) |
Most common events, n (%)e | |
Sepsis | 9 (12.7) |
Pyrexia | 8 (11.3) |
Respiratory failure | 7 (9.9) |
Lung infection | 5 (7.0) |
Pneumonia | 5 (7.0) |
Acute kidney injury | 4 (5.6) |
Bacteremia | 4 (5.6) |
Pneumatosis intestinalis | 4 (5.6) |
Diarrhea | 3 (4.2) |
Mental status change | 3 (4.2) |
Septic shock | 3 (4.2) |
Thrombocytopenia | 3 (4.2) |
Device-related infection | 2 (2.8) |
Disease progression | 2 (2.8) |
Erythropenia (anemia) | 2 (2.8) |
Failure to thrive | 2 (2.8) |
Headache | 2 (2.8) |
Hepatic failure | 2 (2.8) |
Hyponatremia | 2 (2.8) |
Hypotension | 2 (2.8) |
Influenza | 2 (2.8) |
Lower GI hemorrhage | 2 (2.8) |
Multiple organ dysfunction syndrome | 2 (2.8) |
Muscular weakness | 2 (2.8) |
Myocardial infarction | 2 (2.8) |
Nausea | 2 (2.8) |
Staphylococcal bacteremia | 2 (2.8) |
Vomiting | 2 (2.8) |
Patients who stopped treatment due to TEAEs | |
n (%) | 23 (32.4) |
Most common events, n (%)e | |
Sepsis | 4 (5.6) |
Acute kidney injury | 2 (2.8) |
Respiratory failure | 2 (2.8) |
Deaths | |
On-treatment deaths,f n (%) | 25 (35.2) |
SAEs with fatal outcome, n (%) | 28 (39.4) |
Most common SAEs with fatal outcome,h n (%) | |
Disease progression | 2 (2.8) |
Multiple organ dysfunction syndrome | 2 (2.8) |
Hepatic failure | 2 (2.8) |
Sepsis | 2 (2.8) |
Respiratory failure | 6 (8.5) |
Notable harms | |
Infections, n (%) | |
Serious TEAE infection | 36 (50.7) |
Infection, grade ≥ 3 | 46 (64.8) |
Heart rate decrease and PR-interval prolongation, n (%) | |
Bradycardia, any grade TEAE | 2 (2.8) |
Bradycardia, grade ≥ 3 | 1 (1.4) |
Cytopenia, n (%) | |
Anemia, any grade TEAE | 46 (64.8) |
Anemia, grade ≥ 3 TEAE | 36 (50.7) |
Neutropenia, any grade TEAE | 34 (47.9) |
Neutropenia, grade ≥ 3 TEAE | 30 (42.2) |
Thrombocytopenia, any grade TEAE | 44 (62.0) |
Thrombocytopenia, grade ≥ 3 | 38 (53.5) |
Lipid abnormalities, n (%) | NR |
CMV = cytomegalovirus; GI = gastrointestinal; NR = not reported; SAE = serious adverse event; TEAE = treatment-emergent adverse event.
Note: A patient with multiple severity grades for an AE is only counted under the maximum grade. AEs occurring outside the on-randomized-treatment period are not summarized. MedDRA version 22.1, CTCAE version 4.03.
aTEAE reported for the first time or the worsening of a pre-existing event after the first dose of the study drug up to 30 days after the final dose of the study drug.
bFrequency: > 10% of patients (all grades).
cIncludes preferred terms of thrombocytopenia (14 participants) and decreased platelet count (32 participants); participants who had both terms reported are counted only once.
dIncludes preferred terms of neutropenia (6 participants), febrile neutropenia (3 participants), and decreased neutrophil count (28 participants); participants who had more than 1 of these terms reported are counted only once.
eFrequency: > 5% of patients (grade 3 or higher).
fIncludes preferred terms of thrombocytopenia (10 participants) and decreased platelet count (29 participants); participants who had both terms reported are counted only once.
gFrequency: ≥ 2% of patients in either treatment group.
hDeaths from start of ruxolitinib dosing to date of final dose of ruxolitinib plus 30 days.
Source: Clinical Study Report (REACH 1).19
Critical Appraisal
Given the heterogeneity in the 2 trials across study designs and populations, the CADTH review team noted that the results of the REACH 1 trial and the ruxolitinib group in the REACH 2 trial are not directly comparable. The key differences between the REACH 2 and REACH 1 trials — study design (phase III versus phase II), prior therapies, concomitant treatments, outcome definitions, ruxolitinib dosing, and tapering of treatments — are detailed here.
There were some notable differences in the percentage of patients who had received prior treatments. The REACH 1 trial had a higher proportion of patients who had received prior prophylactic treatment with CNIs (97.2%) than the REACH 2 trial ruxolitinib (65.6%) and BAT (60.0%) groups, and a lower percentage of patients who had received prior CNIs as aGvHD treatment (23.9%) than the REACH 2 trial ruxolitinib (50.0%) and BAT (49.0%) groups. The clinical experts consulted by CADTH noted that in Canadian clinical practice, most patients would receive prophylactic treatment with CNIs that is almost always continued after patients develop aGvHD. Although concomitant treatments received by patients in the 2 trials appeared similar, overall, fewer patients in the REACH 1 trial received concomitant glucocorticoids (45.1%) than in the REACH 2 trial ruxolitinib (76.3%) and BAT (76.0%) groups. The dosing of ruxolitinib differed in the 2 studies, in that the starting dose was 10 mg twice daily in the REACH 2 trial, and 5 mg twice daily with the option to increase to 10 mg twice daily after the first 3 days in the REACH 1 trial. In the REACH 1 trial, of the 43 patients who were still on treatment with ruxolitinib on day 28, only 20 received a dose of 10 mg twice daily. There were differences with regard to tapering guidelines in the 2 trials. Although the REACH 2 trial had mandatory per-protocol guidelines for the tapering of corticosteroids and ruxolitinib, the REACH 1 trial had specific guidance for the tapering of ruxolitinib, but corticosteroid dose-tapering recommendations were not mandatory. As there are inter-patient variations in response and tolerability to steroids, the lack of tight control on steroid management had the potential to lead to various taper schedules in the REACH 1 trial. As well, in the REACH 2 trial, tapering of ruxolitinib could start at day 56 once patients with documented CR or PR were off systemic corticosteroids, whereas in the REACH 1 trial, tapering of ruxolitinib could start after day 180 once patients who achieved either CR or VGPR were off systemic corticosteroids. The following differences were noted in outcome definitions in the REACH 2 and REACH 1 trials:
FFS — In the REACH 1 trial, an event for the FFS outcome included, among others, signs or symptoms of cGvHD, whereas in the REACH 2 trial, this event was not included.
ORR at day 28 — In the REACH 1 trial, ORR at day 28 included patients with CR, VGPR, or PR, whereas in the REACH 2 trial, VGPR was not defined.
DOR in responders at day 28 — In the REACH 2 trial, responses that occurred before or at the day 28 assessment were included; whereas in the REACH 1 trial, responses that occurred at the day 28 assessment or within 2 days of day 28 were incorporated.
BOR — In the REACH 2 trial, patients with ORR at any time up to and including day 28 were included if they had not started a new anti-aGvHD therapy, whereas in the REACH 1 trial, patients with ORR at any time were included if they had not started new anti-aGvHD therapy.
REACH 2
Internal Validity
Reimbursement request: The reimbursement criteria for this CADTH review are for the treatment of GvHD in patients 12 years and older who have an inadequate response to corticosteroids or other systemic therapies.
There are insufficient data to determine how the requested reimbursement criteria match the patient population in the REACH 2 trial. The sponsor was asked for clarification on the number of patients in the REACH 2 trial who had an inadequate response to corticosteroids, an inadequate response to other systemic therapies, or an inadequate response to corticosteroids and other systemic therapies, but noted that such data are not available.22 Because an inadequate response to corticosteroids was an eligibility criterion of the REACH 2 trial, it follows that all patients in the trial had an inadequate response to corticosteroids; it also follows that data for patients who only had an inadequate response to other systemic therapies and not to steroids were not available in the REACH 2 trial. The clinical experts noted that it would be very rare for patients in Canadian clinical practice not to receive steroids and to receive only other systemic therapies, given that steroid treatment is the standard of care. Per the trial criteria, patients were allowed to have received 1 prior systemic treatment for SR-aGvHD. However, the proportion of patients who had an inadequate response to other systemic therapies in addition to having an inadequate response to corticosteroids remains unclear. It is not known if patients who are refractory to 1 therapy, as opposed to multiple therapies, would respond differently to ruxolitinib. The clinical experts consulted by CADTH agreed that the difference between patients who have an inadequate response to corticosteroids alone and patients who have an inadequate response to multiple therapies would be unlikely to have an impact on the treatment effect of ruxolitinib. The clinical experts noted that once patients are SR, management of the disease would be a challenge and responses to any currently available second-line therapy are dismal. The clinical experts noted that the lack of data in the REACH 2 trial regarding differences between patients with inadequate responses to corticosteroids alone and patients with inadequate responses to multiple therapies may have been due to responses to systemic drugs not captured in the trial’s case report forms. The clinical experts consulted by CADTH were of the opinion that the type of response to second-line treatment (whether patients responded briefly, partially, or were refractory) would be of little clinical importance.
Baseline characteristics: A stratified randomization procedure was based on a known prognostic factor, severity of aGvHD, to minimize potential imbalances between study groups that might lead to biased results. Imbalances were noted for a few baseline characteristics (e.g., prior therapy of steroids plus CNIs plus an aGvHD prophylaxis; organ involvement of the skin, liver, and upper and lower GI; time from diagnosis of underlying disease to transplant and time from diagnosis of underlying disease to screening), and their impact on treatment outcomes is unknown. The clinical experts consulted by CADTH were of the opinion that the imbalances observed were unlikely to influence clinical outcomes.
Open-label design: The REACH 2 trial had an open-label design in which the investigator and the study participants were aware of their treatment status, which increased the risk of detection and performance bias. This had the potential to bias results and outcomes in favour of ruxolitinib if the assessor (investigator or patient) believed the study drug was likely to provide a benefit. Furthermore, the underlying complexity of aGvHD and its nonspecific presentation have been acknowledged as a key challenge for the design and analysis of clinical trials in the current target setting, and may contribute to subjective inter-physician variability in response assessments. The sponsor’s submission noted that, because of the various treatment modalities of comparator treatments (e.g., tablets, cellular therapy, and photopheresis) and to accommodate modifications and dose adjustments, depending on patients’ responses, a double-blind design would have been operationally impossible.3 To mitigate the impact of this bias, the investigators used aGvHD disease evaluation and response-assessment criteria in accordance with the standard NIH criteria of Harris et al. (2016)16 to evaluate responses. However, no independent review committee was used to evaluated responses. The sponsor was asked about a reliable real-time review of the complex GvHD response assessment by an independent review committee, but explained that such a review was not considered feasible. The sponsor’s response mentioned that adjudication committees would be feasible in disease settings in which objective measures are available (e.g., nuclear magnetic resonance, labs, urine analysis, endoscopy recordings, CT). However, according to the sponsor, GvHD required subjective assessments performed by investigators in the REACH 2 trial, which would have been difficult to record and share with a central committee. The sponsor noted that strategies (e.g., detailed description in protocol, investigator training, and regular review of entered data by blinded data reviewers) were implemented to ensure that the aGvHD response assessments were consistent with the NIH guidelines.22
Furthermore, subjective outcomes (i.e., adverse outcomes and patient-reported outcomes) may be biased, owing to the open-label design. For example, if study personnel and patients knew that the treatment was ruxolitinib (which is known to cause thrombocytopenia, anemia, and other AEs), their reporting of harms could have been influenced. Overall, the magnitude and direction of this bias remain unclear.
OS and EFS of ruxolitinib: The crossover of patients from BAT to ruxolitinib after day 28 may have biased the OS and EFS outcomes. Patients in the BAT group could cross over to the ruxolitinib group if they failed to meet the primary end point (CR or PR at day 28), lost the response after day 28, and met the criteria for progression, mixed response, or no response, necessitating new additional systemic immunosuppressive treatment. Overall, 49 patients in the BAT group crossed over to the ruxolitinib group. The crossover of patients in the BAT group may have prolonged survival beyond what would have occurred had the patients received only their randomized study treatment.
Exposure to the study drug: During the randomized treatment phase (i.e., the period from day 1 to week 24 or EOT), the median duration of treatment was close to twice as long with ruxolitinib as with BAT, at 82.5 (range = 8 to 396) days and 45.5 (range = 2 to 218) days, respectively. An imbalance in exposure between study groups was not unexpected, given the crossover from BAT to ruxolitinib after day 28. However, a safety comparison between the study groups during that period may have been biased against ruxolitinib. Additionally, the investigator’s choice of BAT treatment may have influenced the safety profile in the BAT group, as the toxicity profile of BAT treatments differs. For example, it was noted in the sponsor’ submission that ECP may have a different safety profile than other types of BATs, such as immunosuppressants. The investigator’s choice of BAT may have been influenced by factors such as risk of infection, prior clinical experience, and patient access.
Change of BAT treatment up to day 28: Patients in the BAT group who experienced disease progression, mixed response, or no response were allowed to add or initiate a new systemic therapy up to day 28 without proceeding to discontinuation; however, this was considered a failure of the initial BAT treatment. The clinical experts consulted by CADTH noted that changing or initiating new systemic aGvHD therapies is reflective of clinical practice. As responses to second-line drugs are not as rapid and complete, 2 drugs might be used simultaneously if the manifestations are particularly concerning. It was felt by the clinical experts that changes to the BAT treatment up the day 28 were unlikely to have an impact on OS results, given the similar efficacy and responses achieved with various BAT therapies. The addition or change of systemic therapy was treated as a treatment failure and, therefore, did not have an impact on ORR at day 28 or FFS outcomes.
HRQoL assessments: Interpretation of results from the EQ-5D-5L and FACT-BMT instruments (i.e., the ability to assess trends over time and to compare treatment groups) is limited by the significant decline in patients available for assessment over time. In addition, selection bias over time should be considered when interpreting results, as patients who remain on treatment longer and those available to provide patient-reported outcomes assessments tend to be the healthier. As noted previously, subjective outcomes may be biased by the open-label design. For example, a patient’s belief that ruxolitinib is likely to provide a benefit may influence the reporting of patient-reported outcomes in favour of ruxolitinib.
Assessments of the psychometric properties for the EQ-5D-5L and the FACT-BMT instruments in patients with aGvHD were not found in the literature. Estimates for MIDs in the literature were not found for EQ-5D-5L or FACT-BMT in patients with aGvHD. Therefore, it is unclear if the changes in the EQ-5D-5L or FACT-BMT instruments in the REACH 2 trial are reflective of a clinically meaningful change in patients with SR-aGvHD. Overall, the methodologic issues noted limit the ability to interpret results from the EQ-5D-5L and FACT-BMT instruments.
Protocol deviation: The majority of patients experienced protocol deviations in the REACH 2 trial in the ruxolitinib (n = 131; 85.1%) and BAT (n = 135; 87.1%) groups. The overall type and frequency of protocol deviations appeared balanced between the treatment groups, and supportive analyses of the primary end point, ORR at day 28, in the per-protocol set (including 63.0% and 56.1% of patients in the ruxolitinib and BAT groups, respectively) showed consistent results with ORR results for the full analysis set. However, the per-protocol set did not exclude all patients with protocol deviations; for example, it appears that patients with aGvHD staging or response assessments done using investigator criteria or judgment rather than the NIH criteria of Harris et al. (2016),16 per protocol, were not excluded from the per-protocol set. The post hoc sensitivity analysis conducted at the final data cut-off date (April 23, 2021) that excluded patients with protocol deviations of organ staging and/or aGvHD assessed by investigator judgment at day 28 suggested results for the primary outcome (odds ratio = 2.87; 95% CI, 1.77 to 4.65) that were similar to those observed at the primary analysis.18 A large number of protocol deviations could raise concerns about the quality of the clinical trial. However, in the setting of aGvHD, the potential for protocol deviations has been recognized as a challenge in the literature. For example, mandated taper schedules of steroids to ensure balanced effects of steroid treatment between study groups (mandated taper schedules were used in the REACH 2 trial) may cause protocol deviations because of inter-patient variability in response and tolerability to steroids.25 Overall, the magnitude and impact of a large number of protocol deviations on the study results remain unclear.
Follow-up time: Given that there was insufficient time to follow patients for EFS, NRM, and incidence of malignancy relapse or progression outcomes, the ability to interpret these analyses remains limited.
External Validity
The REACH 2 trial was an international, multi-centre trial. Although the majority of patients in the trial were enrolled at trial sites in Europe, according to the clinical experts consulted by CADTH, the population enrolled in the trial was consistent with the population likely to be treated for SR-aGvHD in Canadian clinical practice. The clinical experts agreed that no different treatment effect would be expected based on different disease management practices across countries. It was noted that few patients in the trial were younger than 18 years. The clinical experts supported generalizing the study results to patients younger than 18 years, as they are managed in a similar way as adults in clinical practice, the safety profile of ruxolitinib in these patients appeared to be similar to the overall safety set, and there is no biologic rationale to assume that outcomes with ruxolitinib would be different between adults and adolescents with SR-aGvHD. Prior aGvHD therapies and prior aGvHD prophylactic therapies received by patients were generally balanced across study groups. It was agreed by the clinical experts that the NIH consensus criteria used in the trial for aGvHD disease and response assessment, per protocol, as well as the tapering schedule for treatments applied in the trial, were, overall, reflective of Canadian clinical practice. The proportion of patients with aGvHD disease staging of grade II, III, and IV and the proportion of patients meeting the SR-aGvHD criteria (A versus B versus C, as defined in Table 4) were reflective of patients seen in clinical practice.
Relevance of trial efficacy outcomes: The primary outcome in the REACH 2 trial was ORR at day 28, and the key secondary outcome was rate of durable ORR at day 56. According to the clinical experts consulted by CADTH, ORR at day 28 and durable ORR at day 56 are clinically meaningful end points for patients with SR-aGvHD. According to the clinical experts, responses in this patient population are important to improve patients’ well-being and to enable the tapering of steroids to mitigate long-term side effects (e.g., osteoporosis, hypertension, hyperglycemia, diabetes, and bone or joint health) and risk of opportunistic infection. It was emphasized by the clinical experts that infectious complications are a leading cause of NRM in SR-aGvHD. The interval from the start of therapy up to day 28 was considered by the clinical experts to be a clinically relevant and reasonable time point for the assessment of ORR. In Canadian clinical practice, patients who have not shown a response about 4 weeks after the initiation of treatment will receive alternative or additional treatment. Furthermore, the clinical experts noted that aGvHD is associated with a reduced HRQoL and high symptom burden, which are compounded by a lack of response and increased disease severity.
Excluded patients: The REACH 2 trial excluded patients who received 2 or more systemic treatments for aGvHD in addition to corticosteroids with or without CNIs for aGvHD, patients with overlap syndrome, and patients with grade I aGvHD. The clinical experts consulted by CADTH considered that it would be reasonable to generalize the REACH 2 trial results to patients who received 2 or more systemic treatments for aGvHD in addition to corticosteroids with or without CNIs. The clinical experts noted that ruxolitinib has a novel mechanism of action in the context of other seconds-line immunosuppressives, with the potential to offer synergy with other therapies. As well, given the manageable safety profile of ruxolitinib, it was felt by clinical experts that it would be reasonable to leave it to the discretion of the treating physician to apply some flexibility in terms of using ruxolitinib in patients with grade I aGvHD and with overlap syndrome.
REACH 1
Internal Validity
Reimbursement request: The reimbursement criteria for this CADTH review are for the treatment of GvHD in patients 12 years and older who have an inadequate response to corticosteroids or other systemic therapies.
The sponsor was asked for clarification on the number of patients in the REACH 1 trial who had an inadequate response to corticosteroids, an inadequate response to other systemic therapies, or an inadequate response to corticosteroids and other systemic therapies, and noted that 42 patients were refractory to steroids alone and 29 patients were refractory to steroids and 1 additional systemic therapy (the receipt of 1 systemic treatment in addition to corticosteroids with or without CNIs for aGvHD was allowed in the REACH 1 trial).22 The sponsor was asked about the specific types of additional systemic therapies received by patients in the REACH 1 trial who were refractory to 1 additional systemic therapy (n = 29), but no additional data were provided beyond the information found in Table 16.22 It is not known if patients who are refractory to 1 therapy, as opposed to multiple therapies, would respond differently to ruxolitinib. The clinical experts consulted by CADTH agreed that the difference between patients who have an inadequate response to corticosteroids alone and those who have an inadequate response to multiple therapies would be unlikely to have an impact on the treatment effect of ruxolitinib.
Phase II design: phase II (randomized or nonrandomized) trials document safety outcomes and investigate whether the estimate of effect for a new drug is large enough to use it in confirmatory phase III trials. Phase II trials may not accurately predict harm or the effectiveness of treatment. There are numerous examples of trials in which phase III trial results did not support phase II trial results.23
Limited interpretation of time-to-event end points: Interpretation of time‐to‐event end points, such as OS, is limited in single‐arm studies. The nonrandomized design makes it a challenge to interpret OS events attributable to ruxolitinib, because all patients received the same treatment. The extent to which observed survival is due to the natural history of the disease or to the intervention remains unclear.59
Concomitant therapies: All patients in the REACH 1 trial received at least 1 concomitant medication. For instance, CNIs and glucocorticoids were received by 88.7% and 45.1% of patients, respectively. Given the uncontrolled design of the REACH 1 trial, the effect of concomitant treatments on overall study outcome cannot be determined. Outcomes such as observed response, DOR, and survival may have been influenced by concomitant steroids or by other concomitant therapies. The extent to which the observed REACH 1 trial outcome was due to concomitant medications or to ruxolitinib remains unclear.
Open-label design: The REACH 1 trial had an open-label design in which the investigator and the study participants were aware of their treatment status, which increased the risk of detection and performance bias. This had the potential to bias results in favour of ruxolitinib if the assessor (investigator or patient) believed the study drug was likely to provide a benefit. Furthermore, the underlying complexity of aGvHD and its nonspecific presentation have been acknowledged as key challenges in the design and analysis of clinical trials in the current target setting, and may contribute to subjective inter-physician variability in response assessments. To mitigate the impact of this bias, the investigators used aGvHD disease evaluation and response-assessment criteria in accordance with the standard NIH criteria of Harris et al. (2016)16 to evaluate responses. Furthermore, subjective outcomes (e.g., adverse outcomes) may be biased by the open-label design. For example, if study personnel and patients knew that the treatment was ruxolitinib (which is known to cause thrombocytopenia, anemia, and other AEs), the reporting of harms could have been affected. Overall, the magnitude and direction of this bias remain unclear.
Statistical analyses: No formal statical significance or hypotheses testing were performed, so no P values were reported. Point estimates with 95% CIs were reported to estimate the magnitude of treatment effect. A greater than 90% probability of a 95% CI for ORR at day 28 with a lower limit greater than 40% was the basis for sample-size determination and was regarded as the threshold for a positive study outcome. Results for ORR at day 28 appeared consistent with the sample-size assumptions, and the study recruited the intended number of patients.
Small sample size: There were a limited number of patients in the efficacy-evaluable dataset (n = 71). The magnitude of the treatment-effect estimates observed in a small study sample may not be replicable in a larger study sample or generalizable to the target population in real-world clinical practice.
Lack of HRQoL and symptom severity assessments: The REACH 1 trial did not collect data on patient-reported outcomes. The input provided by the patient advocacy groups and registered clinician groups, as well as the clinical experts consulted by CADTH, agreed that improvements in HRQoL and aGvHD symptom severity are important treatment goals for the current target population. aGvHD has been found to be the leading cause of morbidity in patients after alloSCT, with a multitude of symptoms with various degrees of severity.3
External Validity
The REACH 1 trial exclusively enrolled patients from centres in the US. The clinical experts agreed that no difference in treatment effect would be expected based on different disease management practices in the US and Canada. It was noted that none of the patients in the trial were younger than 18 years; therefore, there are insufficient data to guide recommendations on the generalizability of the treatment effect observed with ruxolitinib in the REACH 1 trial to adolescents. Almost all patients in the REACH 1 trial received prior aGvHD treatment, and about 2-thirds of patients received prior aGvHD prophylaxis treatment. The clinical experts consulted by CADTH agreed that the type and frequency of prior treatments were reflective of prior treatments received by patients in Canadian clinical practice. The experts also noted that the add-on study design, in which second-line aGvHD treatment is administered in addition to ongoing treatment with steroids with or without CNIs, is reflective of Canadian clinical practice. It was agreed by the clinical experts that the NIH consensus criteria used in the trial for aGvHD disease and response assessment, as well as for the tapering of treatments, were overall reflective of Canadian clinical practice. The proportion of patients with aGvHD of grade II, III, and IV, as well as the proportion of patients meeting the various SR-aGvHD criteria, were reflective of patients seen in clinical practice.
Noncomparative design: The noncomparative design of the REACH 1 trial precludes the assessment of the relative therapeutic benefit or safety of ruxolitinib compared with currently available therapies in Canadian clinical practice. The REACH 2 trial is the only available phase III RCT that compares ruxolitinib with currently used therapies in Canada in the current target population.
Relevance of trial efficacy outcomes: The primary outcome in the REACH 1 trial was ORR at day 28, and the key secondary outcome was DOR once all patients had completed the day 180 visit. According to the clinical experts consulted by CADTH, ORR at day 28 and DOR are clinically meaningful end points for patients with SR-aGvHD. According to the clinical experts, responses in this patient population are important to improve patients’ well-being and to enable the tapering of steroids to mitigate long-term side effects (e.g., osteoporosis, hypertension, hyperglycemia, diabetes, and bone or joint health) and risk of opportunistic infection. It was emphasized by the clinical experts that infectious complications are a leading cause of NRM in patients with SR-aGvHD. The interval from the initiation of therapy up to day 28 was considered by the clinical experts to be a clinically relevant and reasonable time point for the assessment of ORR. In Canadian clinical practice, patients who have not shown a response about 4 weeks after the initiation of treatment will receive alternative or additional treatment. Furthermore, the clinical experts noted that aGvHD is associated with a reduced HRQoL and high symptom burden, which are compounded by a lack of response and increased disease severity.
Excluded patients: Same as for REACH 2.
Indirect Evidence
No indirect treatment comparisons were included in the sponsor’s submission to CADTH or identified in the literature search.
Other Relevant Evidence
This section includes:
1 additional relevant study (Moiseev et al. [2020]60) included in the sponsor’s submission to CADTH reported results for ruxolitinib in adults and children with SR-aGvHD
a list of ongoing trials (Table 43)
a brief summary of the methods and results of post hoc analyses of the REACH 2 trial3 that were applied in the submitted pharmacoeconomic model.
Moiseev et al. (2020) Study
Moiseev et al. (2020)60 was a prospective, single-centre, open-label study conducted in Russia that included 75 patients with either acute (n = 32) or chronic (n = 43) SR-GvHD. Patients were recruited from 2016 to 2018 from the First Pavlov Medical University. Half of the study sample comprised children (53% in the acute and 39% in the chronic group). Study participants received ruxolitinib at a starting dose of 10 mg twice a day for adults, 10 mg twice a day for children weighing more than 40 kg, and 0.15 mg/kg twice a day for children weighing less than 40 kg. Previous treatments were continued if the attending physician considered it necessary. Ruxolitinib was stopped if there were signs of GvHD progression. The primary end point was ORR, and secondary end points included OS, toxicity, relapse, and infection complications.
The ORR was 75% (95% CI, 57% to 89%) in patients with aGvHD and 81% (95% CI, 67% to 92%) in patients with cGvHD. OS was 59% (95% CI, 49% to 74%) for aGvHD and 85% (95% CI, 70% to 93%) for cGvHD. The most common complication was hematological toxicity, with 79% and 44% of grade III to IV neutropenia occurring in the acute and chronic groups, respectively. There was no significant difference between adults and children on any of the outcomes, including ORR (P = 0.31 and P = 0.35, respectively), survival (P = 0.44 and P = 0.12, respectively), and toxicity (P > 0.93). The study demonstrated that ruxolitinib can be used both in adults and children and has comparable response and survival rates.60
Alternative Population
In the REACH 3 trial, the number of patients 12 years to 18 years made up a small proportion of the study sample (3.6%). In the study by Moiseev et al. (2002),60 more than 50% of the study sample was made up of patients younger than 18 years. Hence, this additional relevant study provides greater insight into the efficacy of ruxolitinib in adolescence and children.
Key Critical Appraisal Points
Given the single-arm observational design, interpretation of the study results is limited. Because of the lack of a comparator group and blinding, it is difficult to determine the effectiveness of treatment on the study outcomes. Given the relatively small sample of patients with aGvHD (n = 32), the generalizability of these results may be limited. Moreover, as this trial was conducted in Russia, there may be limitations to the generalizability of these findings to the population in Canada.
Post Hoc Analyses of the REACH 2 trial
OS by Response
A post hoc analysis of the REACH 2 trial on OS by response was conducted and the results were applied to the submitted pharmacoeconomic model. The following section presents the sponsor-provided high-level summary of the methods and results of the post hoc analysis.
Methods
The sponsor’s submission noted that, based on individual participant data (IPD) from the January 6, 2020, data cut-off date, a post hoc analysis was conducted to assess OS by response (ORR, CR, PR, no response) at day 28 from the time of randomization for all 309 patients randomized in the trial (154 in the ruxolitinib group and 155 in the BAT group). A time-to-event analysis with KM survival methods was used in this analysis (see Figure 10 and Figure 11). The sponsor’s submission reported that a landmark was made at day 28 (i.e., patients who died or discontinued before the day 28 response-assessment time point were excluded from the analysis). No further details on assessment methods were provided. KM curves were fitted with parametric survival functions to extrapolate OS by response beyond the available trial data in the pharmacoeconomic model.
Figure 10: REACH 2 OS by Response From Randomization; ORR vs. No Response (January 6, 2020, Data Cut-Off Date)

Note: This figure has been redacted at the request of the sponsor.
Source: Submission materials.3
Figure 11: REACH 2 OS by Response From Randomization; Complete Responders vs. Partial Responders vs. Nonresponders (January 6, 2020, Data Cut-Off Date)

Note: This figure has been redacted at the request of the sponsor.
Source: Submission materials.3
DOR by Response at Day 28
A post hoc analysis of the REACH 2 trial on DOR by response at day 28 was conducted and results were applied to the submitted pharmacoeconomic model. The following section presents the sponsor-provided high-level summary of the methods and results of the post hoc analysis.
Methods
The sponsor’s submission noted that, based on IPD data from the January 6, 2020, data cut-off date, a post hoc analysis was conducted to assess DOR in patients based on their response status at day 28 (ORR, CR, PR) for each study group in the safety analysis set population (n = 302) (see Figure 12, Figure 13, Figure 14). Analyses to obtain cumulative incidence of loss-of-response estimates were performed that accounted for the competing risks of death without prior observation of aGvHD progression and the onset of cGvHD. The sponsor’s submission noted that these analyses were performed in a manner similar to the analysis in the Clinical Study Report for each response status at day 28 (ORR, CR, PR). Subsequently, the probability of maintaining response was calculated as 1 minus the cumulative incidence at each time point. According to the sponsor, the acquired probability of maintaining responses that accounted for competing risk could be considered to be avoidance of loss of response or the competing risks defined.
Figure 12: REACH 2 DOR by Response at Day 28 (ORR) (January 6, 2020, Data Cut-Off Date)

Note: This figure has been redacted at the request of the sponsor.
Source: Submission materials.3
Figure 13: REACH 2 DOR by Response at Day 28 (CR) (January 6, 2020, Data Cut-Off Date)

Note: This figure has been redacted at the request of the sponsor.
Source: Submission materials.3
Figure 14: REACH 2 DOR by Response at Day 28 (PR) (January 6, 2020, Data Cut-Off Date)

Note: This figure has been redacted at the request of the sponsor.
Source: Submission materials.3
Duration of Treatment by Response at Day 28
A post hoc analysis of the REACH 2 trial on duration of treatment by response at day 28 was conducted and results were applied to the submitted pharmacoeconomic model. The following section presents the sponsor-provided high-level summary of the methods and results of the post hoc analysis.
Methods
The sponsor’s submission noted that, based on IPD data from the January 6, 2020, data cut-off date, a post hoc analysis was conducted to assess duration of treatment by response at day 28 in patients based on their response status at day 28 (ORR, CR, PR, NR) for each study group in the safety analysis set population (n = 302). A time-to-event analysis with KM methods was used in the post hoc analysis to obtain results for the duration of initial treatment (as randomized) from day 28 by response at day 28 for each study group (see Figure 15, Figure 16, Figure 17, and Figure 18). The duration of treatment for patients randomized to BAT was defined as the duration of exposure to initial BAT. The sponsor’ submission reported that a landmark was made at day 28 by subtracting 28 days from each patient’s duration-of-treatment time (i.e., patients with negative or 0 time after landmark adjustments [patients who discontinued treatment before day 28] were removed from analyses). Two analyses were conducted: in 1, deaths were counted as events; and in another, deaths were censored. KM curves were fitted with parametric survival functions to extrapolate data beyond the available trial data in the pharmacoeconomic model.
Figure 15: REACH 2 KM Curves for DOR by ORR at Day 28 (ORR) (Death = Censor) (January 6, 2020, Data Cut-Off Date)

Note: This figure has been redacted at the request of the sponsor.
Source: Submission materials.3
Figure 16: REACH 2 KM Curves for DOR by CR at Day 28 (Death = Censored) (January 6, 2020, Data Cut-Off Date)

Note: This figure has been redacted at the request of the sponsor.
Source: Submission materials.3
Figure 17: REACH 2 KM Curves for DOR by PR at Day 28 (Death = Censored) (January 6, 2020, Data Cut-Off Date)

Note: This figure has been redacted at the request of the sponsor.
Source: Submission materials.3
Figure 18: REACH 2 KM Curves for DOR by No Response at Day 28 (Death = Censored) (January 6, 2020, Data Cut-Off Date)

Note: This figure has been redacted at the request of the sponsor.
Source: Submission materials.3
Duration of Treatment From Randomization by Individual Initial BAT
A post hoc analysis of the REACH 2 trial on duration of treatment by initial BAT was conducted and results were applied to the submitted pharmacoeconomic model. The following section presents the sponsor-provided high-level summary of the methods.
Methods
The sponsor’s submission noted that, based on IPD data from the January 6, 2020, data cut-off date, a post hoc analysis was conducted to assess the duration of initial BAT treatment in the BAT group of the safety analysis set population (n = 302) (see Figure 19). A time-to-event analysis with KM methods was used in the post hoc analysis to obtain results for the duration of treatment of the initial BAT from randomization by individual BAT (i.e., ATG, etanercept, everolimus, ECP, infliximab, methotrexate, MSCs, MMF, and sirolimus). The events of interest in the duration of treatment analysis included treatment discontinuation (based on treatment discontinuation data provided in Table 10 to 1 of the Clinical Study Report) and death. In an alternative scenario, death was censored to capture the proportion of patients who are still alive and on treatment with each BAT at each model cycle. KM curves were fitted with parametric survival functions to extrapolate data beyond the available trial data in the pharmacoeconomic model.
Figure 19: REACH 2 KM Curves for Duration of Treatment for Each Individual Initial BAT (Death = Censored) (January 6, 2020, Data Cut-Off Date)

Note: This figure has been redacted at the request of the sponsor.
Source: Submission materials.3
Duration of Treatment From Randomization
A post hoc analysis of the REACH 2 trial on duration of treatment from randomization was conducted and results were applied to the submitted pharmacoeconomic model. The following section presents the sponsor-provided high-level summary of the methods and results of the post hoc analysis.
Methods
The sponsor’s submission noted that, based on IPD data from the January 6, 2020, data cut-off date, a post hoc analysis was conducted to assess duration of initial treatment from randomization based on a time-to-event analysis, with KM methods used to obtain results for each study group in the safety analysis set population (n = 302) (see Figure 20). Definitions for duration of treatment in the ruxolitinib group were based on assessments provided in the Clinical Study Report for the duration of exposure. Duration of treatment for patients randomized to the BAT group was defined as duration of exposure to initial BAT. The events of interest in the duration-of-treatment analysis included treatment discontinuation (based on treatment discontinuation data provided in Clinical Study Report) and death. In an alternative scenario, death was censored. KM curves were fitted with parametric survival functions to extrapolate data beyond the available trial data in the pharmacoeconomic model.
Figure 20: REACH 2 KM Curves for Duration of Treatment From Randomization by Treatment Group (Death = Censor) (January 6, 2020, Data Cut-Off Date)

Note: This figure has been redacted at the request of the sponsor.
Source: Submission materials.3
Resource Use by Study Group for Initial Hospitalizations and Response at Day 28 for Readmissions
A post hoc analysis of the REACH 2 trial on resource use by study group for initial hospitalization and response at day 28 for readmissions was conducted and results were applied to the submitted pharmacoeconomic model. The following section presents the sponsor-provided high-level summary of the methods and results of the post hoc analysis.
Methods
The sponsor’s submission noted that, based on IPD data from the May 8, 2020, data cut-off date, post hoc analyses were conducted for the full analysis set population (n = 309) to:
obtain resource use data up to 6 months
compare resource use by response status to better align with the model health states
correctly adjust for the time period of data capture (at the patient level) to prevent biases in analyses (to ensure that there was no bias related to nonresponders being more likely to die or discontinue treatment or having shorter follow-up times for CR, PR, and ORR than responders).
For the post hoc analyses, the duration and frequency of initial hospitalizations and readmission by health care facility type (e.g., bone marrow transplant, emergency department, intensive care unit, general ward) were tabulated and statistics were produced by response at day 28 (ORR, CR, PR, no response) and by study group. Admissions (initial and readmissions) occurring between the start of study treatment and the end of study participation were included (admissions that started before the initiation of study treatment were included if the discharge date was after the start of study treatment; however, length of stay was adjusted to reflect time since the start of study treatment.
Analyses conducted for initial hospitalizations by treatment group included the following (for results, see Table 41):
the number of patients with at least 1 admission involving each facility type
per-admission lengths of stay for each facility type (the duration, in days, for each facility type was recorded and summarized for all admissions and all patients.
Analyses conducted for readmissions by response status included the following (for results, see Table 42):
the per-patient annualized rate of admissions for each facility type (for each patient, the number of admissions involving a given facility type were divided by the length of follow-up, in years, defined as the difference in time between the start of study treatment and end of study participation [i.e., the observation window defined and applied for these analyses]; if patients did not have any admissions associated with a particular facility type, they were assigned an annualized rate of 0, which ensured the inclusion of all patients in the summary statistics for this analysis
per-admission lengths of stay for each facility type (the duration, in days, for each facility type was recorded and summarized for all admissions and all patients.
Table 41: REACH 2 Results — Initial Hospitalizations
||||||||||||||||||| | ||||||||||||| | ||||||||||||| |
|---|---|---|
|||||||||||||||||||||||||| | ||||||||||||| | ||||||||||||| |
|||||||||||||||||||||||||| | ||||||||||||| | ||||||||||||| |
|||||||||||||||||||||||||| | ||||||||||||| | ||||||||||||| |
|||||||||||||||||||||||||| | ||||||||||||| | ||||||||||||| |
Note: This table has been redacted at the request of the sponsor.
Source: Submission materials.3
Table 42: REACH 2 Results — Readmissions
||||||||||||||||||| | ||||||||||||| | ||||||||||||| |
|---|---|---|
|||||||||||||||||||||||||| | ||||||||||||| | ||||||||||||| |
|||||||||||||||||||||||||| | ||||||||||||| | ||||||||||||| |
|||||||||||||||||||||||||| | ||||||||||||| | ||||||||||||| |
|||||||||||||||||||||||||| | ||||||||||||| | ||||||||||||| |
Note: This table has been redacted at the request of the sponsor.
Source: Submission materials.3
Key Critical Appraisal Points of the Post Hoc Analyses by the CADTH Review Team
The CADTH review team was unable to conduct a rigorous evaluation of the conduct and reporting of the post hoc analyses, as only a high-level summary of methods was provided by the sponsor. Overall, the CADTH review team concluded that results from post hoc analyses are considered exploratory and hypotheses-generating only. Because of the lack of formal inferential statistical testing, the ability to interpret results of such analyses is significantly limited.
Ongoing Studies
A number of ongoing studies were provided by the sponsor (see Table 43). However, these could not be evaluated due to the lack of available results.
Table 43: Ongoing Studies of Ruxolitinib for the Treatment of Patients With GvHD
Study Sponsor | Study ID | Title of Study |
|---|---|---|
Novartis Pharmaceuticals | NCT03774082 CINC424G12201 2018 to 003296 to 35 | A phase II Open-label, Single-Arm, Multicenter Study of Ruxolitinib Added to Corticosteroids in Pediatric Subjects with Moderate and Severe Chronic Graft vs. Host Disease After Allogeneic Stem Cell Transplantation |
National Heart, Lung, and Blood Institute Blood and Marrow Transplant Clinical Trials Network NIH National Marrow Donor Program | NCT04934670 BMT CTN 2022 2021 to 000343 to 53 5U24HL138660- | Phase III, Randomized, Open-Label, Multicenter Study to Compare T-Guard to Ruxolitinib for the Treatment of Patients with grade III or IV Steroid Refractory Acute Graft-vs.-Host Disease (SR-aGvHD) |
University of Nebraska | NCT03616184 333 to 18 | A Singe Arm, Open Label, phase II Study of Ruxolitinib in Sclerotic Chronic Graft-vs.-Host Disease After Failure of Systemic Glucocorticoids |
Zhejiang University | NCT04838704 RCMvsCM | Ruxolitinib with Calcineurin and Methotrexate vs. Calcineurin Plus Methotrexate and Mycophenolate Mofetil as Graft vs. Host Disease Prophylaxis for HLA-haploidentical Hematopoietic Stem Cell Transplantation |
Memorial Sloan Kettering Cancer Center Hackensack Meridian Health Incyte Corporation | NCT03954236 18 to 412 | A Pilot, Prospective, Randomized, Double-Blinded, Vehicle- and Comparator-Controlled Trial on Safety and Efficacy of a Topical Inhibitor of Janus Kinase 1/2 (Ruxolitinib INCB018424 Phosphate 1.5% Cream) for Non-Sclerotic Chronic Cutaneous Graft-vs.-Host Disease |
National Institute of Arthritis and Musculoskeletal and Skin Diseases NIH Clinical Center | NCT03395340 180035 18-AR-0035 | Phase II Study of Topical Ruxolitinib for Cutaneous Chronic Graft vs. Host Disease (cGvHD) |
aGvHD = acute graft-versus-host disease; cGvHD = chronic graft-vs.-host disease; NIH = National Institutes of Health; SR = steroid refractory; vs. = versus.
Discussion
Summary of Available Evidence
The CADTH systematic review included 1 phase III RCT (REACH 2) that met the selection criteria of the CADTH review protocol and 1 single-arm phase II trial (REACH 1) that was provided in the sponsor’s submission to CADTH and Health Canada.
REACH 2 compared the efficacy and safety of ruxolitinib with the investigator’s choice of BAT in patients 12 years and older with grade II to IV SR-aGvHD. Patients continued to receive their systemic immunosuppressive regimen of corticosteroids with or without CNIs. Randomization was centrally performed in a 1:1 ratio and stratified by aGvHD grade (grade II versus III versus IV), based on the NIH criteria of Harris et al. (2016).16 Patients in the BAT group were allowed to cross over to treatment with ruxolitinib between day 28 and week 24. The primary outcome was ORR at day 28 and the key secondary outcome was rate of durable ORR at day 56. Other secondary end points included OS, FFS, ORR at day 14, DOR, BOR, HRQoL (i.e., FACT-BMT and EQ-5D-5L instruments), EFS, NRM, malignancy relapse or progression, cumulative steroid dose up to day 56, incidence of cGvHD, resource use, and safety. The REACH 2 trial enrolled male and female patients 12 years and older who had undergone alloSCT, had evidence of myeloid and platelet engraftment (ANC > 1000/mm3 and platelet count > 20,000/mm3), and were diagnosed with grade II to IV aGvHD that was determined to be refractory to corticosteroids. The majority of patients had grade III SR-aGvHD, met the corticosteroid-refractory criterion (failure to achieve a response after 7 days), and had received steroids plus CNIs as prior systemic aGvHD therapy.
REACH 1 evaluated the efficacy and safety of ruxolitinib in combination with corticosteroids in patients with grade II to IV SR-aGvHD. The severity grading of aGvHD was based on the NIH criteria of Harris et al. (2016).16 The primary outcome was ORR at day 28 and the key secondary outcome was DOR at month 6. Other secondary end points included OS, FFS, ORR at day 14, DOR, BOR, NRM, malignancy relapse or progression, cumulative steroid dose until day 56, incidence of cGvHD, and safety. The REACH 1 trial enrolled male and female patients 12 years and older who had undergone alloSCT, had evidence of myeloid engraftment (e.g., ANC ≥ 0.5 × 109/L for 3 consecutive days if ablative therapy was previously used), and were diagnosed with grade II to IV aGvHD that was determined to be refractory to corticosteroids. The majority of patients had grade III SR-aGvHD and met the corticosteroid-refractory criterion of no aGvHD improvement after 7 days of primary treatment, and all patients received corticosteroids alone or in combination with 1 or more additional drugs as first-line therapy.
No indirect treatment comparisons or other evidence were included in the sponsor’s submission to CADTH or identified in the literature search.
Other relevant evidence included the summary of the open-label, noncomparative, observational study by Moiseev et al. (2020)60 of ruxolitinib in adults and children with acute or chronic SR-GvHD and several post hoc analyses of the REACH 2 trial that were applied to the submitted pharmacoeconomic model.
Interpretation of Results
Efficacy
The REACH 2 trial met its primary end point, demonstrating a statistically significant improvement in ORR at day 28 in favour of ruxolitinib, compared with the BAT group. Results for the subgroups of interest, as specified in the protocol for this CADTH systematic literature review, suggested that the ORR at day 28 benefit consistently favoured ruxolitinib in pre-specified subgroups of patients (except for the subgroups of prior steroid + CNI + other systemic aGvHD treatment for both aGvHD prophylaxis and treatment, and older than 65 years old). However, given that the trial was not designed to detect differences in treatment effects across subgroups, no conclusions can be made from the subgroup results. Results for the key secondary outcome, durable ORR at day 56, were supportive of the ORR at day 28 results and demonstrated statistically significant improvements in favour of ruxolitinib. Other secondary outcomes, such as DOR, BOR, and FFS, were also supportive of the observed ORR day 28 benefit and durability of observed responses with ruxolitinib. The REACH 1 trial achieved the predetermined threshold for a positive ORR at day 28 (lower limit of the 95% CI for ORR ≥ 40%). The key secondary outcome, DOR at month 6, was supportive of the observed ORR at day 28 benefit. The noncomparative design of the REACH 1 trial and the high percentage of patients who received concomitant treatments, make it a challenge to determine the extent to which the observed responses were due to the study intervention or to concomitant therapies, such as steroids. REACH 2 and REACH 1 had open-label designs, which increased the risk of detection and performance. This had the potential to bias results and outcomes in favour of ruxolitinib if the assessor (investigator or patient) believed the study drug was likely to provide a benefit. Furthermore, the underlying complexity of aGvHD and its nonspecific presentation have been acknowledged as a key challenge for the design and analysis of clinical trials in the current target setting and may contribute to inter-physician or inter-assessment-centre variability in aGvHD assessments. The clinical experts noted that responses in this patient population are clinically meaningful for patients’ well-being and to enable the tapering of steroids to mitigate long-term side effects (e.g., osteoporosis and osteonecrosis) and risk of infection. It was emphasized by the clinical experts that infectious complications are a leading cause of NRM in SR-aGvHD. In addition, the clinical experts noted that aGvHD is associated with a reduced HRQoL and high symptom burden, which are compounded by a lack of response and increased disease severity. The experts emphasized the clinical relevance and importance of maintaining even a PR for the prevention of deterioration to a patient’s performance status and a worsening of disease symptoms in this setting. The clinical experts further noted that improvements in ORR and durability of response of the magnitude observed in the REACH 2 and REACH 1 trials are of clinical importance in a patient population for which there is currently no standard treatment.
This view was echoed by the input provided by patient advocacy groups and registered clinician groups, which highlighted improvements in HRQoL and the potential to reduce steroid use as important goals of treatment for patients. Steroid use in the REACH 2 trial was investigated as a secondary outcome, and suggested that a reduction in steroid dose in the ruxolitinib group was slightly (but consistently) higher than that in the BAT group. Similarly, the exploration of the steroid dose in the REACH 1 trial suggested a continued reduction from day 1 of study treatment through to day 180. An overall improvement in FACT-BMT scores and steady scores on the EQ-5D-5L instrument suggested an improvement in HRQoL in both treatment groups. However, given several important limitations — including the noninferential analyses, the significant decline in patients available for assessment over time, and the open-label design of the trial — interpretation of the EQ-5D-5L and FACT-BMT scores is limited.
Input received by the patient advocacy groups, registered clinicians, and the clinical experts consulted by CADTH highlighted OS as an important outcome and treatment goal for patients. Results for OS in the REACH 2 trial may have been biased by the crossover of patients in the BAT group to the ruxolitinib group after day 28. Given the limited follow-up time, the ability to interpret OS results is limited. The nonrandomized design of the REACH 1 trial makes it a challenge to interpret OS events attributable to ruxolitinib, because all patients received the same treatment. Although the clinical experts agreed that, based on the available evidence, it was not possible to determine whether responses would translate into clinical benefits in terms of OS, they felt that durable responses could potentially reduce NRM and result in prolonged survival in this patient population. The clinical experts agreed that, given that the average 6-month survival of patients with SR-aGvHD who receive available second-line therapies has been estimated to be 49%,18 a potential 4- to 5-month increase in OS in favour of ruxolitinib is an encouraging trend and meaningful for patients in this setting.
Although patients recruited in the REACH 2 and REACH 1 trials were, overall, considered to be representative of patients in Canadian clinical practice, the clinical experts noted that it would be reasonable to generalize the trial results to patients younger than 18 years, given that the management of adults and adolescents is similar in clinical practice and the safety profile of ruxolitinib in adolescents appeared to be similar to that in the overall safety set of the REACH 2 trial. As well, the clinical experts consulted by CADTH felt that it would be reasonable to generalize the trial results to patients who received 2 or more systemic treatments for aGvHD in addition to corticosteroids with or without CNIs. The clinical experts noted that ruxolitinib has a novel mechanism of action in the context of other second-line immunosuppressives, with the potential to offer synergy with other therapies. As well, given the manageable safety profile of ruxolitinib, it was felt by the clinical experts that it would be reasonable to leave it to the discretion of the treating physician to apply some flexibility in terms of using ruxolitinib in patients with grade I aGvHD and with overlap syndrome.
The proportion of patients in the ruxolitinib group diagnosed with cGvHD (24.7%; n = 38) was higher than that in the BAT group (16.8%; n = 26).
Because of heterogeneity in the REACH 1 and REACH 2 trials in study designs and populations, the CADTH review team noted that the results of the REACH 1 trial and the ruxolitinib group in the REACH 2 trial are not directly comparable. Key differences in the REACH 2 and REACH 1 trials were observed for study design (i.e., phase III versus phase II), definitions of SR disease, prior therapies, concomitant treatments, outcome definitions, ruxolitinib dosing, and tapering of treatments.
Harms
In the REACH 2 trial, the median duration of treatment was about twice as long with ruxolitinib as with BAT in the main treatment period (82.5 days versus 45.5 days in the ruxolitinib and BAT groups, respectively), which should be considered when reviewing the incidence of TEAEs. Additionally, the single-arm design of the REACH 1 trial made it a challenge to interpret the safety events attributable to ruxolitinib, because all patients received the same treatment.
The great majority of patients in the REACH 2 trial and all patients in the REACH 1 trial experienced at least 1 TEAE, which, according to the clinical experts, was to be expected in this heavily pre-treated and immunocompromised target population. In the REACH 2 trial, the percentage of patients experiencing cytopenia was higher in the ruxolitinib group, which is in line with the expected safety profile of ruxolitinib, based on previously published data.18 Differences in the proportion of patients experiencing TEAEs of grade 3 of higher were mainly driven by thrombocytopenia, anemia, and neutropenia, and differences in the proportion experiencing serious TEAEs were driven by diarrhea and pyrexia. The most commonly reported TEAEs of any grade in the ruxolitinib group and BAT groups, respectively, were anemia, thrombocytopenia, cytomegalovirus infection, neutropenia, peripheral edema, hypokalemia, and pyrexia, and in the BAT group were anemia, cytomegalovirus infection, peripheral edema, thrombocytopenia, hypokalemia, and pyrexia. In the ruxolitinib group, the most commonly reported TEAEs of grade 3 or higher were anemia, thrombocytopenia, and decreased platelet count, and the most common serious TEAEs were sepsis, pyrexia, and septic shock; in the BAT group, the most commonly reported TEAEs of grade 3 or higher were anemia, thrombocytopenia, and decreased platelet count, and the most common serious TEAEs were and sepsis, septic shock, pneumonia, and cytomegalovirus infection. From the review of notable harms, it appeared that toxicities related to ruxolitinib were mostly reported as cytopenia, followed by infections and lipid abnormalities. Deaths due to TEAEs were similar in the ruxolitinib and BAT groups, with 10 and 4 deaths, respectively, suspected to be related to the study treatment.
Although the most common TEAEs of any grade, TEAEs of grade 3 or higher, and serious TEAEs in the REACH 1 trial were similar to those in the REACH 2 trial, the percentage of patients experiencing any degree of toxicity appeared, overall, to be higher in the REACH 1 trial than in the REACH 2 trial. However, because of heterogeneity in the REACH 2 and REACH 1 trials with regard to concomitant treatments and ruxolitinib dosing, a safety comparison of the 2 trials is a challenge. The clinical experts consulted by CADTH noted that most TEAEs associated with ruxolitinib could be managed with dose modifications and best supportive care. The clinical experts noted that the investigator’s choice of BAT treatment in the REACH 2 trial may have influenced the safety profile in the BAT group, as the toxicity profiles of BAT treatments differ. In general, it can be a challenge to report AEs in clinical trials, the clinical experts noted, given the underlying complexity of aGvHD and the similarity between aGvHD symptoms and AEs that result from study treatments in the target setting. Overall, the clinical experts consulted by CADTH agreed that no unexpected safety concerns were observed with ruxolitinib, and patients could be adequately managed in clinical practice. Input received by CADTH from the patient advocacy group stated that side effects were tolerable in patients with experience with ruxolitinib, and indicated that these patients would take ruxolitinib again if recommended by their doctor.
Conclusions
One phase III, open-label, multi-centre RCT (REACH 2) and 1 single-arm phase II trial (REACH 1) were included in this CADTH review. The REACH 2 trial demonstrated statistically significant improvements in ORR at day 28 and in the rate of durable ORR at day 56 in patients who were treated with ruxolitinib, compared to BAT. The improvements in the response outcomes of the magnitude observed in the REACH 2 trial were considered clinically meaningful by the clinical experts consulted by CADTH. Other secondary outcomes — DOR, BOR, FFS, and steroid use — were also supportive of the observed ORR day 28 benefit with ruxolitinib. The open-label design of the trial and reliance on a local investigator’s assessment of trial outcomes may have introduced a bias that is difficult to quantify. The results of HRQoL measures, assessed with the EQ-5D-5L and FACT-BMT instruments, remain uncertain because of several important limitations. The actual degree of OS benefit with ruxolitinib is uncertain, given the risk of potential bias arising from the crossover of patients in the BAT group to the ruxolitinib group and the limited follow-up time. The clinical experts consulted by CADTH noted that no new safety concerns were observed with ruxolitinib. Although the REACH 1 trial achieved the predetermined threshold for a positive outcome (lower limit of the 95% CI for ORR at day 28 ≥ 40%) in patients who received ruxolitinib, there was uncertainty regarding the magnitude of clinical benefit directly attributable to ruxolitinib, owing to the limitations associated with the study, including the single-arm, open-label trial design, the lack of formal statical significance testing, and the relatively small sample size of 71 patients.
References
1.Jakavi (ruxolitinib): tablets, 5 mg, 10 mg, 15 mg and 20 mg, oral [product monograph]. Dorval (QC): Novartis Pharmaceuticals Canada Inc.; 2022 Jun 8.
2.American Cancer Society. Types of stem cell and bone marrow transplants. 2022; https://www.cancer.org/treatment/treatments-and-side-effects/treatment-types/stem-cell-transplant/types-of-transplants.html. Accessed 2022 Jan 11.
3.Drug Reimbursement Review sponsor submission: Jakavi (ruxolitinib): tablets, 5 mg, 10 mg, 15 mg and 20 mg, oral [internal sponsor's package]. Dorval (QC): Novartis Pharmaceuticals Canada Inc.; 2021 Aug 10.
4.DeFilipp Z, Alousi AM, Pidala JA, et al. Nonrelapse mortality among patients diagnosed with chronic GVHD: an updated analysis from the Chronic GVHD Consortium. Blood Adv. 2021;5(20):4278-4284. PubMed
5.Annual report 2020/2021. Vancouver (BC): Cell Therapy Transplant Canada; 2021: https://cdn.ymaws.com/www.cttcanada.org/resource/resmgr/2021/CTTC_2021_Annual_Report.pdf. Accessed 2021 Nov 10.
6.Nelson J Zeiser R. Treatment of chronic graft-versus-host disease. In: Post TW, ed. UpToDate. Waltham (MA): UptoDate; 2021: www.uptodate.com. Accessed 2022 Jan 26.
7.Malard F, Huang X-J, Sim JPY. Treatment and unmet needs in steroid-refractory acute graft-versus-host disease. Leukemia. 2020;34(5):1229-1240. PubMed
8.Khoury HJ, Wang T, Hemmer MT, et al. Improved survival after acute graft-versus-host disease diagnosis in the modern era. Haematologica. 2017;102(5):958-966. PubMed
9.Canadian Cancer Society. Graft-versus-host disease (GVHD). 2022: https://cancer.ca/en/treatments/side-effects/graft-versus-host-disease-gvhd. Accessed 2022 Jan 26.
10.Zeiser R, von Bubnoff N, Butler J, et al. Ruxolitinib for Glucocorticoid-Refractory Acute Graft-versus-Host Disease. N Engl J Med. 2020;382(19):1800-1810. PubMed
11.Health Canada Reviewer Report - Pharmaceutical Safety and Efficacy Assessment: (Supplemental) New Drug Submission. Therapeutic Products Directorate; 2022 May 18.
12.Clarification Request for: Jakavi (Ruxolitinib) SNDS #250022. Therapeutic Products Directorate; 2022 Apr 1.
13.Saad A, de Lima M, Anand S, et al. Hematopoietic Cell Transplantation, Version 2.2020, NCCN Clinical Practice Guidelines in Oncology. Journal of the National Comprehensive Cancer Network J Natl Compr Canc Netw. 2020;18(5):599-634. PubMed
14.Alberta Bone Marrow and Blood Cell Transplant Program: Standard Practice Manual. Guidelines Resource Unit guru@ahs.ca; 2021 Jun 15: https://www.albertahealthservices.ca/assets/info/hp/cancer/if-hp-cancer-guide-bmt-manual.pdf.
15.Schoemans HM, Lee SJ, Ferrara JL, et al. EBMT-NIH-CIBMTR Task Force position statement on standardized terminology & guidance for graft-versus-host disease assessment. Bone Marrow Transplant. 2018;53(11):1401-1415. PubMed
16.Harris AC, Young R, Devine S, et al. International, Multicenter Standardization of Acute Graft-versus-Host Disease Clinical Data Collection: A Report from the Mount Sinai Acute GVHD International Consortium. Biol Blood Marrow Transplant. 2016;22(1):4-10. PubMed
17.Jagasia MH, Greinix HT, Arora M, et al. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: I. The 2014 Diagnosis and Staging Working Group report. Biol Blood Marrow Transplant. 2015;21(3):389-401.e381. PubMed
18.Clinical Study Report: INC424C2301. A phase III randomized open-label multi-center study of ruxolitinib versus best available therapy in patients with corticosteroid-refractory acute graft vs. host disease after allogeneic stem cell transplantation. [internal sponsor's report]. Freiburg (DE): Novartis; 2020 Mar 9.
19.Clinical Study Report: INCB 18424-271. A single-cohort, phase 2 study of ruxolitinib in combination with corticosteroids for the treatment of steroid-refractory acute graft-versus-host disease (REACH-1). [interanl sponsor's resport]. Novartis; 2019 Dec 20.
20.Novartis Pharmaceuticals. NCT02913261: Safety and Efficacy of Ruxolitinib Versus Best Available Therapy in Patients With Corticosteroid-refractory Acute Graft vs. Host Disease After Allogeneic Stem Cell Transplantation (REACH2). ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2021: https://www.clinicaltrials.gov/ct2/show/NCT02913261. Accessed 2022 Jan 26.
21.Novartis Pharmaceuticals Canada Inc. response to February 2, 2022 DRR request for additional information regarding ruxolitinib DRR review [internal additional sponsor's information]. Dorval (QC): Novartis Pharmaceutical Canada Inc; 2022.
22.Novartis Pharmaceuticals Canada Inc. response to February 2, 2022 DRR request for additional information regarding ruxolitinib [internal additional sponsor's information]. Dorval (QC): Novartis Pharmaceutical Canada Inc; 2021.
23.FDA. U.S. Food & Drug Administration. 22 case studies where phase 2 and phase 3 trials had divergent results. 2017: https://www.fda.gov/media/102332/download.
24.Moiseev IS, Morozova EV, Bykova TA, et al. Long-term outcomes of ruxolitinib therapy in steroid-refractory graft-versus-host disease in children and adults. Bone Marrow Transplant. 2020;55(7):1379-1387. PubMed
25.Martin PJ, Bachier CR, Klingemann H-G, et al. Endpoints for Clinical Trials Testing Treatment of Acute Graft-versus-Host Disease: A Joint Statement. Biol Blood Marrow Transplant. 2009;15(7):777-784. PubMed
26.Lee SJ, Wolff D, Kitko C, et al. Measuring therapeutic response in chronic graft-versus-host disease. National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: IV. The 2014 Response Criteria Working Group report. Biol Blood Marrow Transplant. 2015;21(6):984-999. PubMed
27.Hershenfeld SA, Matelski J, Ling V, Paterson M, Cheung M, Cram P. Utilisation and outcomes of allogeneic hematopoietic cell transplantation in Ontario, Canada, and New York State, USA: a population-based retrospective cohort study. BMJ Open. 2020;10(10):e039293. PubMed
28.Chao NJ, Zeiser R. Pathogenesis of graft-versus-host disease (GVHD). In: Post TW, ed. UpToDate. Waltham (MA): UpToDate: https://www.uptodate.com/contents/pathogenesis-of-graft-versus-host-disease-gvhd. Accessed 2022 Jan 11.
29.Vigorito AC, Campregher PV, Storer BE, et al. Evaluation of NIH consensus criteria for classification of late acute and chronic GVHD. Blood. 2009;114(3):702-708. PubMed
30.Cleveland Clinic. Graft vs host disease: an overview in bone marrow transplant. 2022; https://my.clevelandclinic.org/health/diseases/10255-graft-vs-host-disease-an-overview-in-bone-marrow-transplant. Accessed 2022 Jan 11.
31.Chao NJ. Clinical manifestations, diagnosis, and grading of acute graft-versus-host disease. In: Post TW, ed. UpToDate. Waltham (MA): UpToDate; 2021: www.uptodate.com. Accessed 2021 Nov 11.
32.Glucksberg H, Storb R, Fefer A, et al. Clinical manifestations of graft-versus-host disease in human recipients of marrow from HL-A-matched sibling donors. Transplantation. 1974;18(4):295-304. PubMed
33.Rowlings PA, Przepiorka D, Klein JP, et al. IBMTR Severity Index for grading acute graft-versus-host disease: retrospective comparison with Glucksberg grade. Br J Haematol. 1997;97(4):855-864. PubMed
34.Pavletic SZ. Response as an end point in treatment trials for acute GVHD. Bone Marrow Transplant. 2012;47(2):161-163. PubMed
35.Gatza E, Reddy P, Choi SW. Prevention and Treatment of Acute Graft-versus-Host Disease in Children, Adolescents, and Young Adults. Biol Blood Marrow Transplant. 2020;26(5):e101-e112. PubMed
36.Martin PJ, Rizzo JD, Wingard JR, et al. First- and second-line systemic treatment of acute graft-versus-host disease: recommendations of the American Society of Blood and Marrow Transplantation. Biol Blood Marrow Transplant. 2012;18(8):1150-1163. PubMed
37.National Library of Medicine. PubChem: ruxolinib (compound). 2022; https://pubchem.ncbi.nlm.nih.gov/compound/Ruxolitinib. Accessed 2022 Jan 11.
38.Imbruvica (ibrutinib): capsules, 140mg. Toronto (ON): Janssen Inc.; 2018 Jul 24: https://pdf.hres.ca/dpd_pm/00046525.PDF. Accessed 2022 Jan 11.
39.U.S. Food & Drug Administration. FDA approves ruxolitinib for chronic graft-versus-host disease. 2021; https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-ruxolitinib-chronic-graft-versus-host-disease. Accessed 2022 Jan 26.
40.U.S. Food & Drug Administration. FDA approves ruxolitinib for acute graft-versus-host disease. 2019; https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-ruxolitinib-acute-graft-versus-host-disease. Accessed 2022 Jan 11.
41.U.S. Food & Drug Administration. FDA approves belumosudil for chronic graft-versus-host disease. 2022; https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-belumosudil-chronic-graft-versus-host-disease. Accessed 2022 Jan 11.
42.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. J Clin Epidemiol. 2016;75:40-46. PubMed
43.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2021 Aug 30.
44.Jagasia M, Perales MA, Schroeder MA, et al. Ruxolitinib for the treatment of steroid-refractory acute GVHD (REACH1): a multicenter, open-label phase 2 trial. Blood. 2020;135(20):1739-1749. PubMed
45.Incyte Corporation. NCT02953678: A Study of Ruxolitinib in Combination With Corticosteroids for the Treatment of Steroid-Refractory Acute Graft-Versus-Host Disease (REACH-1). ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine; 2021: https://clinicaltrials.gov/ct2/show/NCT02953678. Accessed 2022 Jan 26.
46.Center for International Blood and Marrow Transplant Research (CIBMTR). Clinical trial endpoints for patients with acute GVHD. 2009: https://www.cibmtr.org/Meetings/Materials/GVHDworkshop/GvHD%20Workshop%20Library/08_Alousi_FDAaGVHDwrkshp_May09.pdf. Accessed December 4, 2022.
47.EQ-5D-5L user guide. Rotterdam (NL): EuroQol Research Foundation; 2021: https://euroqol.org/publications/user-guides/. Accessed 2021 Jul 8.
48.FACIT Group. Functional assessment of cancer therapy - bone marrow transplantation. 2021: https://www.facit.org/measures/FACT-BMT. Accessed 2021 Nov 5.
49.Soudy H, Maghfoor I, Elhassan TAM, et al. Translation and validation of the Functional Assessment of Cancer Therapy-Bone Marrow Transplant (FACT-BMT) version 4 quality of life instrument into Arabic language. Health Qual Life Outcomes. 2018;16(1):47. PubMed
50.FACIT.org. FACT-BMT scoring downloads. [no date][no date]: . Accessed 2022 Jan 11.
51.van Groningen LF, Liefferink AM, de Haan AF, et al. Combination Therapy with Inolimomab and Etanercept for Severe Steroid-Refractory Acute Graft-versus-Host Disease. Biol Blood Marrow Transplant. 2016;22(1):179-182. PubMed
52.Novartis Pharmaceuticals Canada Inc. response to March 15, 2022 DRR request for additional information regarding ruxolitinib [internal additional sponsor's information]. Dorval (QC): Novartis Pharmaceuticals Canada Inc.; 2022.
53.Clopper C, Pearson E. The Use of Confidence or Fiducial Limits Illustrated in the Case of the Binomial. Biometrika. 1934;26(4):404-413.
54.Brookmeyer R, Crowley J. A Confidence Interval for the Median Survival Time. Biometrics. 1982;38(1):29-41.
55.Robins JM, Tsiatis AA. Correcting for non-compliance in randomized trials using rank preserving structural failure time models. Commun Stat - Theory Methods. 1991;20(8):2609-2631.
56.Korhonen PA, Laird NM, Palmgren J. Correcting for non-compliance in randomized trials: an application to the ATBC Study. Stat Med. 1999;18(21):2879-2897. PubMed
57.Marubini E, Valsecchi MG. Analysing Survival Data from Clinical Trials and Observational Studies. Wiley; 2004: https://www.wiley.com/en-ca/Analysing+Survival+Data+from+Clinical+Trials+and+Observational+Studies-p-9780470093412. Accessed 2022 Jan 26.
58.Hosmer Jr. DW, Lemeshow S, May S. Applied Survival Analysis: Regression Modeling of Time-to-Event Data, 2nd Edition. Wiley; 2008: https://www.wiley.com/en-us/Applied+Survival+Analysis%3A+Regression+Modeling+of+Time+to+Event+Data%2C+2nd+Edition-p-9780471754992. Accessed 2022 Jan 26.
59.Food and Drug Administration (FDA). Guidance for Industry: Clinical Trial Endpoints for the Approval of Cancer Drugs and Biologics. 2018. Accessed September 10, 2019. 2018: https://www.fda.gov/downloads/Drugs/Guidances/ucm071590.pdf.
60.Moiseev IS, Morozova EV, Bykova TA, et al. Long-term outcomes of ruxolitinib therapy in steroid-refractory graft-versus-host disease in children and adults. Bone Marrow Transplant. 2020;55(7):1379-1387. PubMed
61.Grasso AG, Del Bufalo F, Boccieri E, et al. Use of ruxolitinib to control graft-versus-host-like disease in Omenn syndrome and successfully bridging to HSCT. J Allergy Clin Immunol Pract. 2021;9(6):2531-2533.e2531. PubMed
62.Lauterio A, De Carlis R, Pugliano MT, et al. Complete resolution of a cutaneous grade 2 graft-versus-host disease after liver transplantation using ruxolitinib. Clin Transplant. 2021:e14366. PubMed
63.Singh S. Ruxolitinib for glucocorticoid-refractory acute graft-versus-host disease: A giant leap -To start with baby steps. Indian J Med Paediatr Oncol. 2020;41(5):733-734.
64.Borg MA, Shalabi RA, Childs R, Wells BC. Alopecia Universalis and Chronic Graft-vs-Host Disease Treated With Ruxolitinib. JAMA Dermatol. 2018;154(11):1357-1358. PubMed
65.Sylvine P, Thomas S, Pirayeh E. Infections associated with ruxolitinib: study in the French Pharmacovigilance database. Ann Hematol. 2018;97(5):913-914. PubMed
66.Barabanshikova MV, Moiseev IS, Morozova EV, et al. Posttransplant ruxolitinib combined with cyclophosphamide for graft versus host disease prophylaxis and relapse prevention in patients with myelofibrosis. Cell Ther Transplant. 2016;5(3):15-17.
67.Philippe L. Ruxolitinib: Treatment option in steroid-refractory graft-versus-host disease following hematopoietic stem cell transplantation. Hematologie. 2015;21(6):322-323.
68.EuroQol Research Foundation. EQ-5D-5L: about. 2017; https://euroqol.org/eq-5d-instruments/eq-5d-5l-about/. Accessed 2021 Jul 8.
69.Kilgour JM, Wali G, Gibbons E, et al. Systematic Review of Patient-Reported Outcome Measures in Graft-versus-Host Disease. Biol Blood Marrow Transplant. 2020;26(5):e113-e127. PubMed
70.McQuellon RP, Russell GB, Cella DF, et al. Quality of life measurement in bone marrow transplantation: development of the Functional Assessment of Cancer Therapy-Bone Marrow Transplant (FACT-BMT) scale. Bone Marrow Transplant. 1997;19(4):357-368. PubMed
Appendix 1: Literature Search Strategy
Note that this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases:
MEDLINE All (1946-present)
Embase (1974-present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: September 2, 2021
Alerts: Bi-weekly search updates until project completion
Search filters applied: No filters were applied to limit the retrieval by study type.
Limits:
Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
MeSH | Medical Subject Heading |
exp | Explode a subject heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
adj# | Requires terms to be adjacent to each other within # number of words (in any order) |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Author keyword heading word (MEDLINE) |
.kw | Author keyword (Embase) |
.dq | Candidate term word (Embase) |
.pt | Publication type |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multi-Database Strategy
(ruxolitinib* or Jakafi* or Jakavi* or INC424* or INC 424* or INCA24* or INCB424* or INCB 424* or INCB018424* or INCB 018424* or INCB18424* or INCB 18424* or 82S8X8XX8H or HSDB8259* or HSBD 8259* or 436LRU32H5*).ti,ab,kf,ot,hw,nm,rn.
Graft vs Host Disease/ or Graft vs Host Reaction/ or exp Host vs Graft Reaction/
((graft vs host or graft vs host or graftvshost or graftvs host or graft vshost or graftversushost or graft versus host or graftversus host or graft versushost or graft host* or graft v host* or homologous wast* or runt* or transplant* or allogenic* or allogeneic* or GVH) adj3 (disease* or react* or respons* or reject*)).ti,ab,kf.
(GvHD or aGvHD or taGvHD or overlap syndrome*).ti,ab,kf.
(graft* adj3 (host* or fail* or reject*)).ti,ab,kf.
or/2-5
1 and 6
7 use medall
*ruxolitinib/ or (ruxolitinib* or Jakafi* or Jakavi* or INC424* or INC 424* or INCA24* or INCB424* or INCB 424* or INCB018424* or INCB 018424* or INCB18424* or INCB 18424* or HSDB8259* or HSBD 8259*).ti,ab,kw,dq.
exp graft versus host reaction/ or exp graft rejection/
((graft vs host or graft vs host or graftvshost or graftvs host or graft vshost or graftversushost or graft versus host or graftversus host or graft versushost or graft host* or graft v host* or homologous wast* or runt* or transplant* or allogenic* or allogeneic* or GVH) adj3 (disease* or react* or respons* or reject*)).ti,ab,kw,dq.
(GvHD or aGvHD or taGvHD or overlap syndrome*).ti,ab,kw,dq.
(graft* adj3 (host* or fail* or reject*)).ti,ab,kw,dq.
or/10-13
9 and 14
15 use oemezd
(conference review or conference abstract).pt.
16 not 17
8 or 18
remove duplicates from 19
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
[Search terms – ruxolitinib, Jakavi, Jakafi, graft]
WHO ICTRP
International Clinical Trials Registry Platform, produced by WHO. Targeted search used to capture registered clinical trials.
[Search terms – ruxolitinib, Jakavi, Jakafi, graft]
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
[Search terms – ruxolitinib, Jakavi, Jakafi, graft]
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
[Search terms – ruxolitinib, Jakavi, Jakafi, graft]
Grey Literature
Search dates: August 23 to 30, 2021
Keywords: ruxolitinib, Jakavi, Jakafi, graft versus host disease, GvHD
Limits: None
Updated: Search updated before the completion of stakeholder feedback period
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Health Statistics
Internet Search
Open Access Journals
Appendix 2: Excluded Studies
Note that this appendix has not been copy-edited.
Reference | Reason for Exclusion |
|---|---|
Grasso AG, Del Bufalo F, Boccieri E, et al. Use of ruxolitinib to control graft-versus-host-like disease in Omenn syndrome and successfully bridging to HSCT. J Allergy Clin Immunol Pract. 2021;9(6):2531-2533.e2531.61 | Study design |
Lauterio A, De Carlis R, Pugliano MT, et al. Complete resolution of a cutaneous grade 2 graft-versus-host disease after liver transplantation using ruxolitinib. Clin Transplant. 2021:e14366.62 | Study design |
Singh S. Ruxolitinib for glucocorticoid-refractory acute graft-versus-host disease: A giant leap -To start with baby steps. Indian J Med Paediatr Oncol. 2020;41(5):733-734.63 | Commentary |
Borg MA, Shalabi RA, Childs R, Wells BC. Alopecia Universalis and Chronic Graft-vs-Host Disease Treated With Ruxolitinib. JAMA Dermatol. 2018;154(11):1357-1358.64 | Study design |
Sylvine P, Thomas S, Pirayeh E. Infections associated with ruxolitinib: study in the French Pharmacovigilance database. Ann Hematol. 2018;97(5):913-914.65 | Study design |
Barabanshikova MV, Moiseev IS, Morozova EV, et al. Posttransplant ruxolitinib combined with cyclophosphamide for graft versus host disease prophylaxis and relapse prevention in patients with myelofibrosis. Cell Ther Transplant. 2016;5(3):15-17.66 | Study design |
Philippe L. Ruxolitinib: Treatment option in steroid-refractory graft-versus-host disease following hematopoietic stem cell transplantation. Hematologie. 2015;21(6):322-323.67 | French language |
Appendix 3: Detailed Outcome Data
Note that this appendix has not been copy-edited.
Table 46: Acute GvHD Staging as per NIH Criteria (Harris et al. [2016])16 in REACH 2 and REACH 1
Stage | Skin (Active Erythema Only) | Liver (Bilirubin) | Upper GI | Lower GI (stool output/day) | |
|---|---|---|---|---|---|
Stage 0 | No active (erythematous) | < 2mg/dL | No or intermittent nausea, vomiting, or anorexia | Adult: < 500mL/day or < 3 episodes/day | |
Stage 1 | Maculopapular rash < 25% BSA | 2 to 3 mg/dL | Persistent nausea, vomiting, or anorexia | Adult: 500 to 999 mL/day or 3 to 4 episodes/day | |
Stage 2 | Maculopapular rash < 25% to 50% BSA | 3.1 to 6 mg/dL | NA | Adult: 1000 to 1500mL/day or 5 to 7 episodes/day | |
Stage 3 | Maculopapular rash > 50% BSA | 6.1 to 15 mg/dL | NA | Adult: > 1500mL/day or > 7 episodes/day | |
Stage 4 | Generalized erythroderma (> 50% BSA) plus bullous formation and desquamation > 5% BSA | > 15 mg/dL | NA | Severe abdominal pain with or without ileus or grossly bloody stool (regardless of stool volume) | |
Overall clinical grade (based on most severe target organ involvement): Grade 0: No stage 1 to 4 of any organ. Grade I: Stage 1 to 2 skin without liver, upper GI or lower GI involvement. Grade II: Stage 3 rash and/or stage 1 liver and/or stage 1 upper GI and/or stage 1 lower GI. Grade III: Stage 2 to 3 liver and/or stage 2 to 3 lower GI with stage 0 to 3 skin and/or stage 0 to 1 upper GI. Grade IV: Stage 4 skin, liver or lower GI involvement, with stage 0 to 1 upper GI. | |||||
GvHD = graft vs. host disease; NA = not applicable; NIH = National Institutes of Health
Source: Protocol (REACH 2)18
Figure 21: Failure-Free Survival by Treatment in REACH 2, Full Analysis Set (July 25, 2019, Data Cut-Off Date)
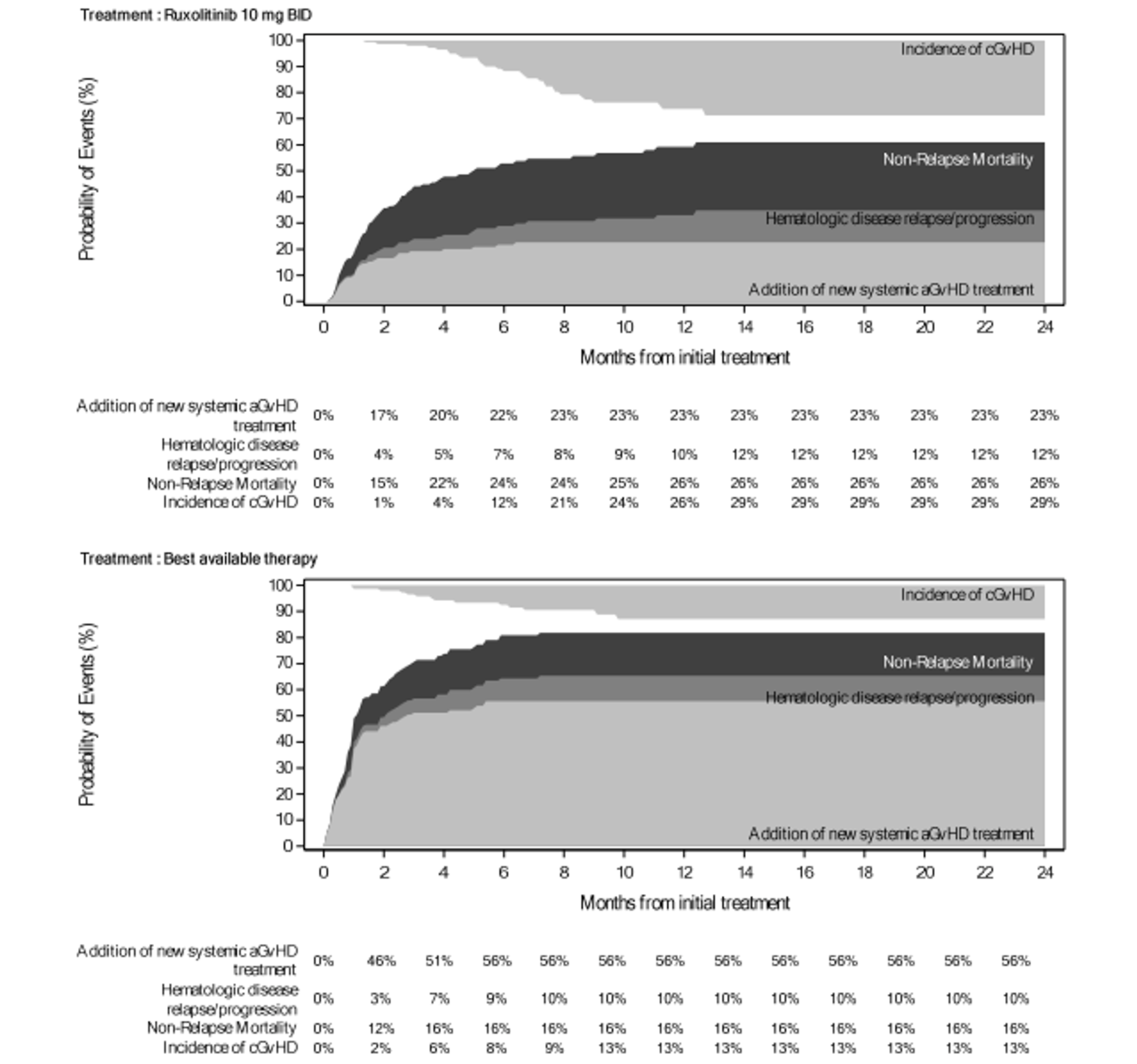
Note: The event included hematologic disease relapse/progression, non-relapse mortality or addition of systemic aGvHD treatment. Onset of cGvHD was a competing risk.
Source: Clinical Study Report (REACH 2)18
Figure 22: Failure-Free Survival by Treatment in REACH 2, Full Analysis Set (January 6, 2020, Data Cut-Off Date)
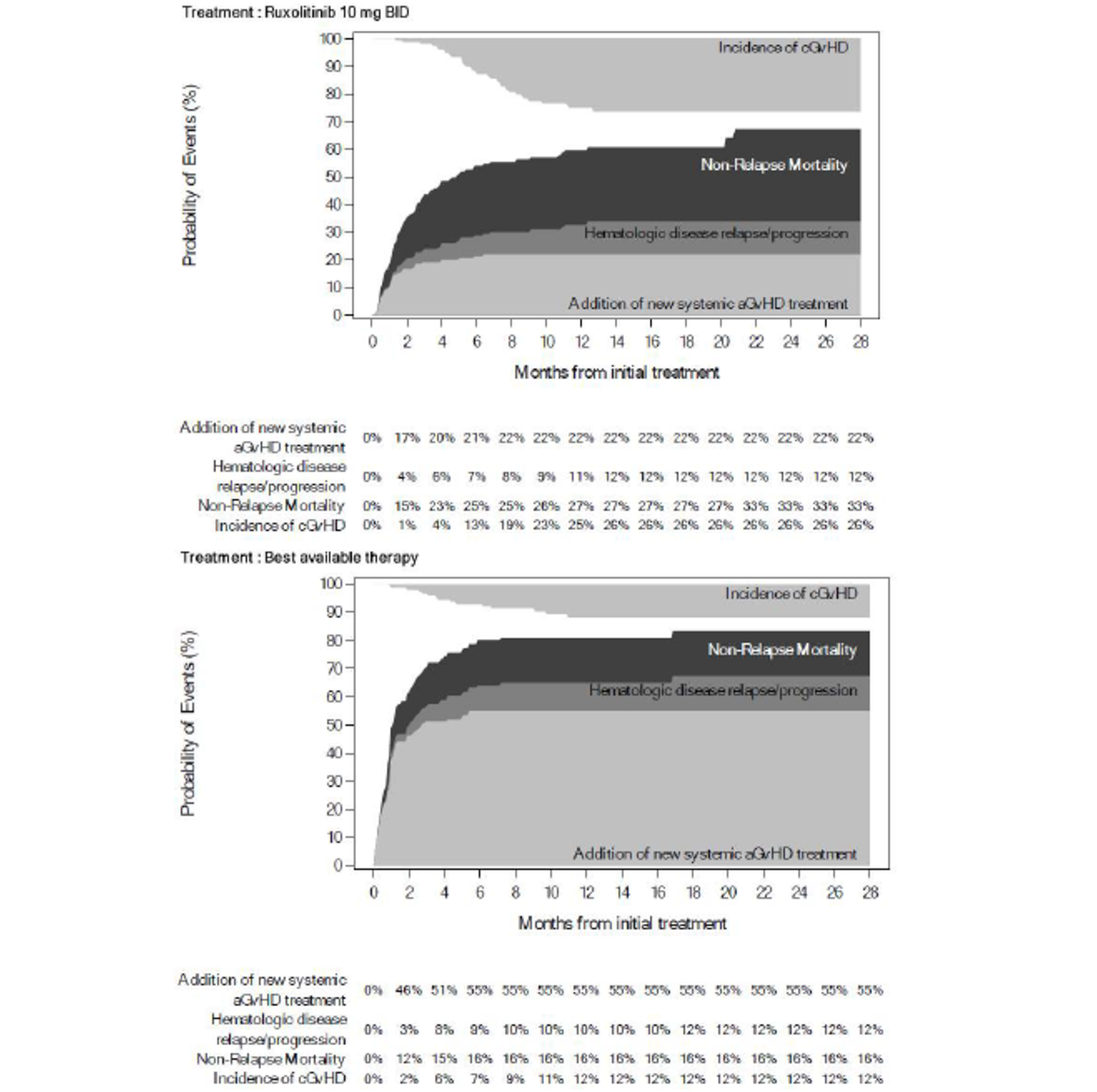
The event includes hematologic disease relapse/progression, non-relapse mortality or addition of systemic aGvHD treatment.
Source: Clinical Study Report (REACH 2)18
Figure 23: Shift in aGvHD Organ Staging From Baseline to Day 28 for Ruxolitinib and BAT in REACH 2 for Skin (Panel A), Liver (Panel B), Upper GI (Panel C), and Lower GI Involvement (Panel D) (July 25, 2019, Data Cut-Off Date)
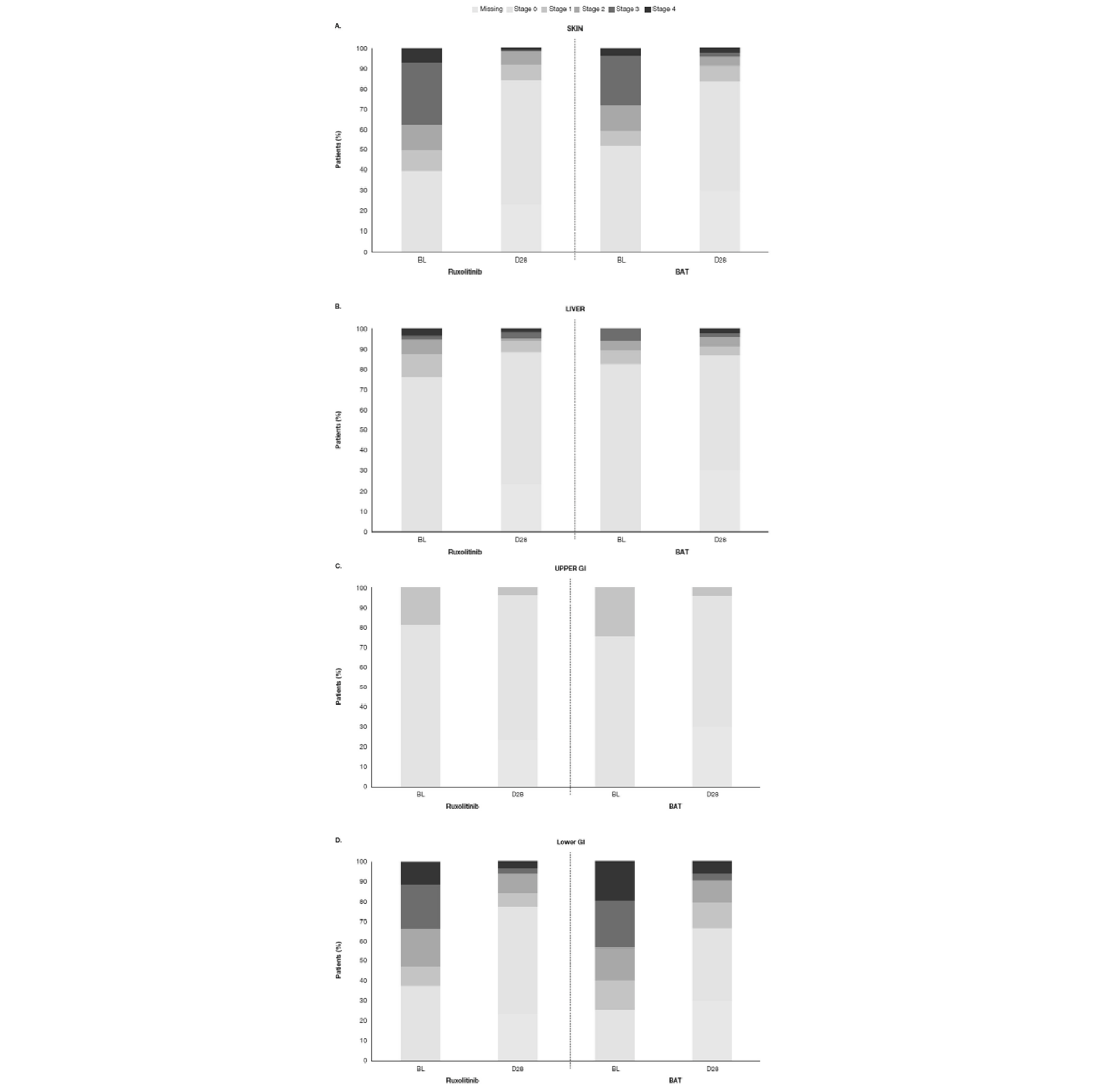
Clinical Study Report (REACH 2)18
Figure 24: Cumulative Incidence of cGvHD in REACH 2, Full Analysis Set (July 25, 2019, Data Cut-Off Date)
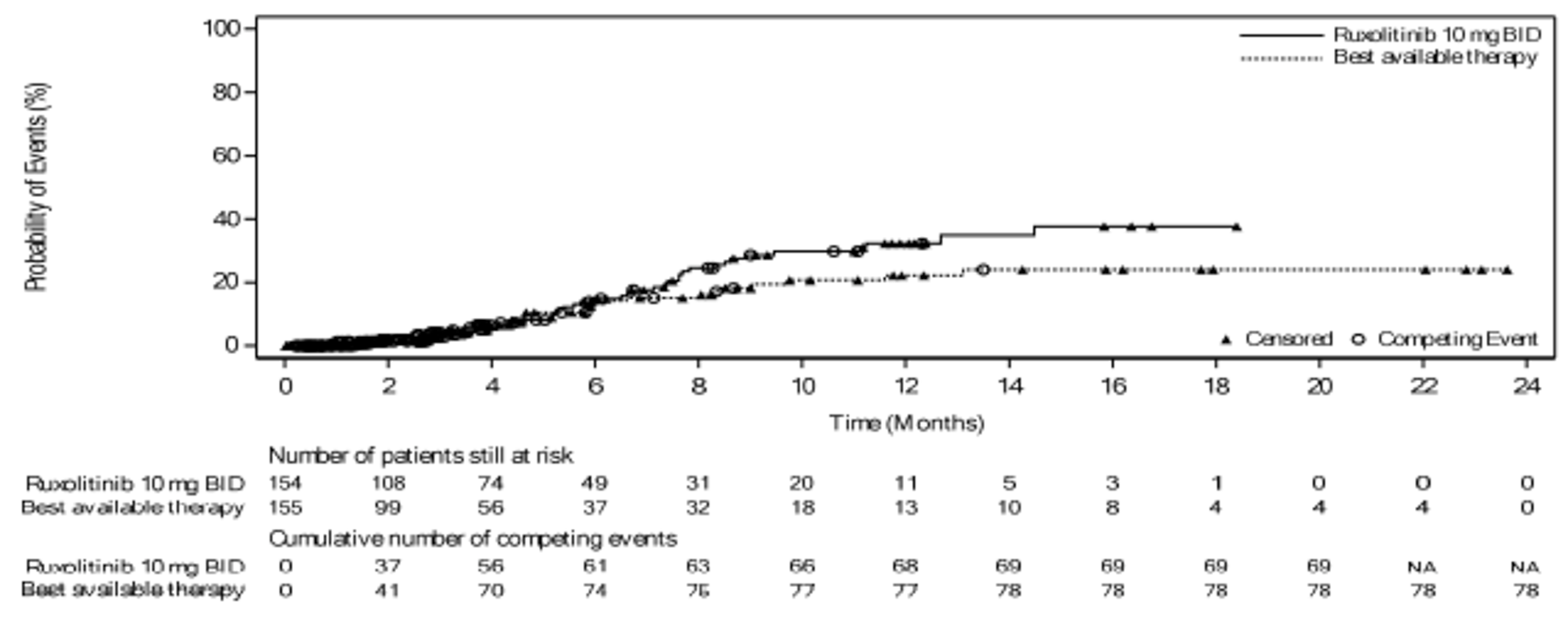
The competing risk includes deaths without prior onset of cGvHD and hematologic disease relapse/progression. NA - Not Applicable
Source: Clinical Study Report (REACH 2)18
Figure 25: Cumulative Incidence Curve of Duration of Response in REACH 2, Full Analysis Set (July 25, 2019, Data Cut-Off Date)
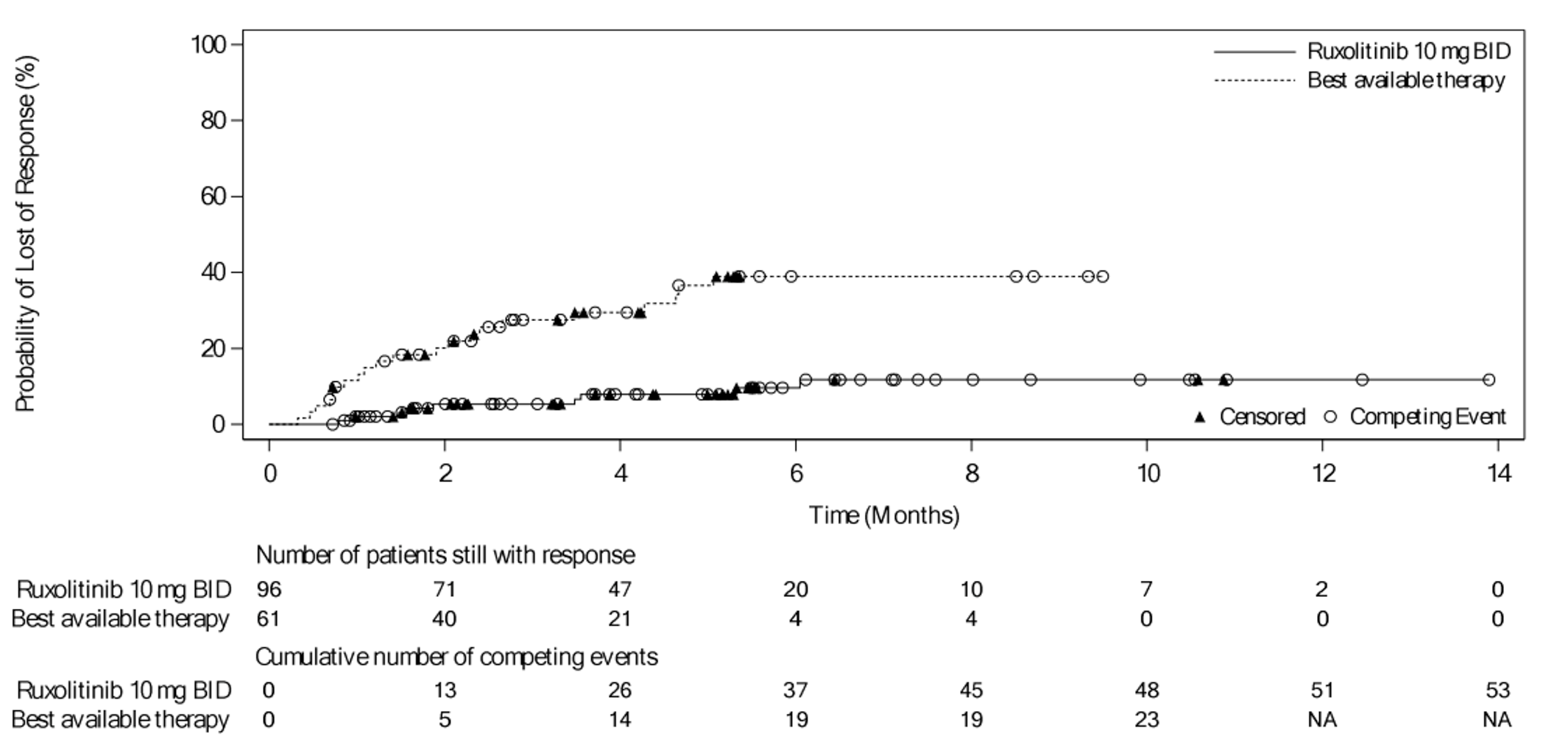
The competing risks include death without prior observation of aGvHD progression and onset of cGvHD. NA - Not Applicable
Source: Clinical Study Report (REACH 2)
Table 47: Summary of Day 28 Overall Response Rate by Baseline Acute GvHD Grade in REACH 1, Efficacy-Evaluable Participants (Data Cut-Off Date: June 5, 2019)
Variable | Grade II (N = 22) | Grade III (N = 33) | Grade IV (N = 16) |
|---|---|---|---|
Number (%) of participants who had an overall responsea | 18 (81.8) | 15 (45.5) | 7 (43.8) |
95% CI for ORR | (59.7, 94.8) | (28.1, 63.6) | (19.8, 70.1) |
Responders | |||
CR | 11 (50.0) | 6 (18.2) | 2 (12.5) |
VGPR | 3 (13.6) | 2 (6.1) | 1 (6.3) |
PR | 4 (18.2) | 7 (21.2) | 4 (25.0) |
Nonresponders | |||
MR | 0 | 3 (9.1) | 0 |
NR | 0 | 2 (6.1) | 0 |
PD | 0 | 1 (3.0) | 1 (6.3) |
Other | 1 (4.5) | 0 | 0 |
Missingb | 3 (13.6) | 12 (26.4) | 8 (50.0) |
Death | 1 (4.5) | 7 (21.2) | 2 (12.5) |
Discontinuation | 1 (4.5) | 5 (15.2) | 6 (37.5) |
Missing visits | 1 (4.5) | 0 | 0 |
CI = confidence interval; CR = complete response; MR = mixed response; NR = no response; PD = progressive disease; PR = partial response; VGPR = very good partial response.
aParticipants that had CR, VGPR, or PR at the Day 28 response assessment or other response assessments within ± 2 days of Day 28, on or before the start of new anti-GvHD therapy (if applicable).
bParticipants with missing assessment were considered nonresponders.
Source: Clinical Study Report (REACH 1)19
Table 48: Overall Response Rate at Crossover Day 28 in REACH 2, Crossover Analysis Set (January 6, 2020, Data Cut-Off Date)
Variable | Ruxolitinib (N = 49) | |
|---|---|---|
n (%) | 95% CI | |
Overall response | ||
Responders | ||
Complete Response | 23 (46.9) | NA |
Partial Response | 10 (20.4) | NA |
Nonresponders | ||
No Response | 4 (8.2) | NA |
Mixed response | 1 (2.0) | NA |
Progression | 1 (2.0) | NA |
Othera | 2 (4.1) | NA |
Unknown | 8 (16.3) | NA |
Death | 5 (10.2) | NA |
Early discontinuation | 3 (6.1) | NA |
Missing visits | 0 | NA |
Overall Response Rate (ORR: CR+PR) | 33 (67.3) | (52.5,80.1) |
CI = confidence interval; CR = complete response; ORR = overall response rate; PR = partial response
N: The total number of patients in the treatment group. It is the denominator for percentage (%) calculation.
n: Number of patients who are at the corresponding category. The 95% CI for the response rate is calculated using the Clopper-Pearson exact method.
aOther: Patients with additional systemic therapies along with CR/PR as per investigator assessment.
Source: Clinical Study Report (REACH 2)18
Table 49: Subgroup Results for ORR at Day 28 by Baseline Organ Involvement in REACH 1, Efficacy-Evaluable Patients (Data Cut-Off Date: June 5, 2019)
Subgroups | Ruxolitinib Number of overall response patients n/N (%) | (95% CI) |
|---|---|---|
Baseline Liver Involvement | ||
Stage 0 | 35/ 55 (63.6) | (49.6, 76.2) |
Other Stages | 5/ 16 (31.3) | (11.0, 58.7) |
Baseline Upper GI Involvement | ||
Stage 0 | 29/ 49 (59.2) | (44.2, 73.0) |
Other Stages | 11/ 22 (50.0) | (28.2, 71.8) |
Baseline Lower GI Involvement | ||
Stage 0 | 15/ 20 (75.0) | (50.9, 91.3) |
Other Stages | 25/ 51 (49.0) | (34.8, 63.4) |
Baseline aGvHD Skin Rash Stage | ||
Stage 0 | 17/ 35 (48.6) | (31.4, 66.0) |
Other Stages | 23/ 36 (63.9) | (46.2, 79.2) |
Source: Clinical Study Report (REACH 1)19
Table 50: Summary of Day 28 Overall Response by Steroid-Refractory Subcategory in REACH 1, Efficacy-Evaluable Patients (June 5, 2019, Data Cut-Off Date)
Variable | Progressive GvHD After 3 Days of Primary Treatment (N = 17) | GvHD Not Improved After 7 Days of Primary Treatment (N = 29) | Previously Began Steroid Therapy at a Lower Dose but Developed New GvHD in Another Organ System (N = 10) | Could not Tolerate a Steroid Taper (N = 15) |
|---|---|---|---|---|
Number (%) of participants who had an overall responsea | 11 (64.7) | 14 (48.3) | 5 (50.0) | 10 (66.7) |
95% CI for ORR | (38.3, 85.8) | (29.4, 67.5) | (18.7, 81.3) | (38.4, 88.2) |
Responders | ||||
CR | 6 (35.3) | 5 (17.2) | 2 (20.0) | 6 (40.0) |
VGPR | 4 (23.5) | 1 (3.4) | 1 (10.0) | 0 |
PR | 1 (5.9) | 8 (27.6) | 2 (20.0) | 4 (26.7) |
Nonresponders | ||||
MR | 1 (5.9) | 0 | 1 (10.0) | 1 (6.7) |
NR | 0 | 2 (6.9) | 0 | 0 |
PD | 0 | 1 (3.4) | 1 (10.0) | 0 |
Other | 0 | 0 | 0 | 1 (6.7) |
Missingb | 5 (29.4) | 12 (41.4) | 3 (30.0) | 3 (20.0) |
Death | 2 (11.8) | 5 (17.2) | 1 (10.0) | 2 (13.3) |
Discontinuation | 3 (17.6) | 6 (20.7) | 2 (20.0) | 1 (6.7) |
Missing visits | 0 | 1 (3.4) | 0 | 0 |
CI = confidence interval; CR = complete response; MR = mixed response; NR = no response; PD = progressive disease; PR = partial response; VGPR = very good partial response.
Note: Steroid-refractory subcategories were based on investigator assessment at baseline.
aParticipants who had a CR, VGPR, or PR at Day 28 response assessment or other response assessments within ± 2 days of Day 28, on or before the start of new anti-GvHD therapy (if applicable).
bParticipants with missing assessment were considered nonresponders.
Source: Clinical Study Report (REACH 1)19
Table 51: Overall Response Rate at Days 14, 56, 100, and 180 in REACH 1, Efficacy-Evaluable Patients (June 5, 2019, Data Cut-Off Date)
Variable | Day 14 (N = 71) | Day 56 (N = 71) | Day 100 (N = 71) | Day 180 (N = 71) |
|---|---|---|---|---|
Number (%) of participants who had an overall responsea | 44 (62.0) | 26 (36.6) | 23 (32.4) | 15 (21.1) |
95% CI for ORR | (49.7, 73.2) | (25.5, 48.9) | (21.8, 44.5) | (12.3, 32.4) |
Responder | ||||
CR | 14 (19.7) | 21 (29.6) | 21 (29.6) | 13 (18.3) |
VGPR | 6 (8.5) | 1 (1.4) | 0 | 2 (2.8) |
PR | 24 (33.8) | 4 (5.6) | 2 (2.8) | 0 |
Nonresponders | ||||
MR | 2 (2.8) | 2 (2.8) | 1 (1.4) | 0 |
NR | 6 (8.5) | 1 (1.4) | 0 | 0 |
PD | 7 (9.9) | 0 | 0 | 0 |
Other | 1 (1.4) | 0 | 0 | 0 |
Missingb | 11 (15.5) | 42 (59.2) | 47 (66.2) | 56 (78.9) |
Death | 5 (7.0) | 24 (33.8) | 27 (38.0) | 34 (47.9) |
Discontinuation | 5 (7.0) | 17 (23.9) | 20 (28.2) | 18 (25.4) |
Missing visits | 1 (1.4) | 1 (1.4) | 0 | 4 (5.6) |
CI = confidence interval; CR = complete response; MR = mixed response; NR = no response; PD = progressive disease; PR = partial response; VGPR = very good partial response.
aParticipants who had a CR, VGPR, or PR at respective response assessment visit or other response assessments with study day within ± 2 days of the expected assessment day, on or before the start of new anti-GvHD therapy (if applicable).
bParticipants with missing corresponding assessment were considered nonresponders.
Source: Clinical Study Report (REACH 1)19
Table 52: Summary of Duration of Response by the Day 28 Overall Response in REACH 1, Efficacy-Evaluable Patients (June 5, 2019, Data Cut-Off Date)
Variable | Ruxolitinib (N = 71) | ||
|---|---|---|---|
CR | VGPR | PR | |
Number (%) of participants who had response at Day 28a | 19 (26.8) | 6 (8.5) | 15 (21.1) |
Number (%) of participants with eventsb | 8 (42.1) | 3 (50.0) | 5 (33.3) |
Progression of disease | 5 (26.3) | 0 | 0 |
Death | 3 (15.8) | 3 (50.0) | 5 (33.3) |
Duration of response (days) (95% CI) | |||
25th percentile | 106.0 (7.0, 669.0) | 144.0 (43.0, NE) | 159.0 (29.0, 326.0) |
50th percentile (median) | 669.0 (106.0, NE) | 154.0 (43.0, NE) | 262.0 (96.0, NE) |
75th percentile | NE (669.0, NE) | NE (144.0, NE) | 326.0 (159.0, NE) |
% Event-free probability estimates (95% CI) | |||
Month 3 | 78.9 (53.2, 91.5) | 83.3 (27.3, 97.5) | 92.9 (59.1, 99.0) |
Month 6 | 73.7 (47.9, 88.1) | 41.7 (5.6, 76.7) | 65.0 (22.8, 88.2) |
Median follow-up time (days) | |||
Median | 428.0 | 149.0 | 96.0 |
Min, Max | 7, 805 | 43, 683 | 24, 349 |
CR = complete response; NE = not evaluable; PR = partial response; VGPR = very good partial response.
aParticipants who had a CR, VGPR, or PR at Day 28 response assessment or other response assessments within ± 2 days of Day 28, on or before the start of new anti-GvHD therapy (if applicable).
bDenominator is the total number of responders.
Source: Clinical Study Report (REACH 1)19
Table 53: Summary of Best Overall Response Rate in REACH 1, Efficacy-Evaluable Patients (June 5, 2019, Data Cut-Off Date)
Variable | Ruxolitinib (N = 71) |
|---|---|
Number (%) of participants who had a response at any timea | 54 (76.1) |
95% CI for best overall response | (64.5, 85.4) |
Responders | |
CR | 41 (57.7) |
VGPR | 4 (5.6) |
PR | 9 (12.7) |
Nonresponders | |
MR | 1 (1.4) |
NR | 10 (14.1) |
PD | 6 (8.5) |
CI = confidence interval; CR = complete response; MR = mixed response; NR = no response; PD = progressive disease; PR = partial response; VGPR = very good partial response.
aParticipants who had CR, VGPR, or PR before the start of new anti-GvHD therapy (if applicable).
Source: Clinical Study Report (REACH 1)19
Table 54: Summary of Corticosteroid Use During Ruxolitinib Treatment in REACH 1, Efficacy-Evaluable Patients (June 5, 2019, Data Cut-Off Date)
Variable | Ruxolitinib (N = 71) | ||||
|---|---|---|---|---|---|
Day 14 | Day 28 | Day 56 | Day 100 | Day 180 | |
Number (%) of participants with ongoing ruxolitinib treatment on the specified study daya | 58 (81.7) | 43 (60.6) | 29 (40.8) | 23 (32.4) | 18 (25.4) |
Number (%) of participants with ongoing ruxolitinib treatment who were taking corticosteroids on the specified study dayb | 56 (96.6) | 43 (100.0) | 27 (93.1) | 15 (65.2) | 7 (38.9) |
Number (%) of participants with ongoing ruxolitinib who had discontinued corticosteroids by the specified dayb | 2 (3.4) | 0 | 2 (6.9) | 8 (34.8) | 11 (61.1) |
Average corticosteroid dose (mg/day) during the week ending on the specified study dayc | |||||
N | 56 | 43 | 27 | 15 | 7 |
Mean daily dose (SD) | 103.37 (48.531) | 62.25 (32.112) | 27.43 (19.088) | 16.05 (13.142) | 8.57 (3.780) |
Median | 95.45 | 62.50 | 20.00 | 10.71 | 10.00 |
Min, max | 30.0, 218.8 | 19.3, 147.1 | 5.0, 78.6 | 4.3, 41.4 | 5.0, 15.0 |
Relative (to initial dose) corticosteroid dose (%) during the week ending on the specified study dayd | |||||
N | 56 | 43 | 27 | 15 | 7 |
Mean daily relative dose (SD) | 68.07 (19.340) | 44.80 (22.251) | 20.17 (15.454) | 10.95 (8.668) | 6.54 (4.690) |
Median | 69.37 | 47.62 | 15.00 | 8.57 | 5.00 |
Min, max | 22.3, 100.0 | 10.2, 100.0 | 3.6, 75.0 | 2.1, 34.9 | 2.2, 16.0 |
Number (%) of participants and proportions of initial corticosteroid dose on the specified study daye | |||||
≤ 25% | 1 (1.8) | 11 (25.6) | 20 (74.1) | 14 (93.3) | 7 (100.0) |
> 25% - 50% | 12 (21.4) | 13 (30.2) | 6 (22.2) | 1 (6.7) | 0 |
> 50% - 75% | 22 (39.3) | 17 (39.5) | 1 (3.7) | 0 | 0 |
> 75% | 21 (37.5) | 2 (4.7) | 0 | 0 | 0 |
SD = standard deviation
Note: Corticosteroid dose (mg) = methylprednisolone dose (mg) × 1.25 + prednisone dose (mg).
aParticipants whose last ruxolitinib treatment was on or after the specified study day.
bDenominator is the number of participants with ongoing ruxolitinib treatment on the specified study day.
cAverage corticosteroid dose (mg/day) = total corticosteroid dose (mg) for the week / 7.
dRelative corticosteroid dose (%) = (total corticosteroid dose [mg] for the week / 7) / Initial corticosteroid dose.
eDenominator is the number of participants with ongoing ruxolitinib treatment who were also receiving corticosteroids on the specified study day.
Source: Clinical Study Report (REACH 1)19
Table 55: Summary of Incidence of Chronic GvHD in REACH 1, Efficacy-Evaluable Patients (June 5, 2019, Data Cut-Off Date)
Variable | Treatment Group Ruxolitinib (N = 71) |
|---|---|
Number (%) of Subjects with Chronic GvHD | 11 (15.5) |
95% CIa | (8.0, 26.0) |
Earliest Chronic GvHD Diagnosis Day - n (%) | |
< = 100 days | 1 (1.4) |
> 100 to 180 | 3 (4.2) |
> 180 days | 7 (9.9) |
aThe 95% CI was calculated based on the exact method for binomial distributions.
Source: Clinical Summary Report (REACH 1)19
Table 56: Summary of Efficacy End points, REACH 2 — Full Analysis Set; and REACH 1 — Efficacy-Evaluable Population (Outcomes Are Presented in Order of Priority as Identified in the CADTH Review Protocol)
Data cut-off date | REACH 2 | REACH 1 | |||
|---|---|---|---|---|---|
Ruxolitinib N = 154 | BAT N = 155 | Ruxolitinib N = 154 | BAT N = 155 | Ruxolitinib N = 71 | |
July 25, 2019 | January 6, 2020 | June 5, 2019 | |||
Overall Survival | |||||
Median OS follow-up | |||||
months | 5.04 | 3.58 | 7.34 | 3.81 | NA |
days | NA | NA | NA | NA | 160 |
Median OS | |||||
months, (95% CI) | 11.14 (NR) | 6.47 (NR) | 10.71 (NR) | 5.82 (NR) | NA |
days, (95% CI) | 339 (186, NE) | 197 (114, 458) | 326 (182, 547) | 177 (115, 392) | 232.0 (93.0, 675.0) |
Events (death), n (%) | 72 (46.8) | 79 (51.0) | 82 (53.2) | 88 (56.8) | 44 (62.0) |
Censored, n (%) | 82 (53.2) | 76 (49.0) | 72 (46.8) | 67 (43.2) | 27 (38.0)Beta |
HR (95% CI)a | 0.83 (0.60, 1.15) | 0.83 (0.6, 1.13) | NA | ||
P value | 0.2648 | 0.2331 | NA | ||
Survival probability at: | |||||
0 to < 1 months, (95% CI) | 90.04 (84.02, 93.87) | 85.48 (78.79, 90.19) | 90.04 (84.02, 93.87) | 85.48 (78.79, 90.19) | NA |
1 to < 2 months, (95% CI) | 77.91 (70.36, 83.75) | 75.62 (67.83, 81.78) | 77.95 (70.42, 83.79) | 75.69 (67.92, 81.83) | NA |
Month 3 | NA | NA | NA | NA | 63.3 (51.0, 73.3) |
2 to < 6 months, (95% CI) | 59.54 (50.92, 67.14) | 50.36 (41.61, 58.47) | 58.27 (49.90, 65.73) | 49.42 (40.89, 57.37) | NA |
Month 6 | NA | NA | NA | NA | 51.3 (38.9, 62.3) |
Month 9 | NA | NA | NA | NA | 48.2 (36.1, 59.4) |
6 to < 12 months, (95% CI) | 48.69 (39.35, 57.38) | 43.64 (34.60, 52.32) | 48.92 (40.43, 56.87) | 42.03 (33.62, 50.19) | NA |
Month 12 | NA | NA | NA | NA | 42.2 (30.4, 53.5) |
12 to < 18 months, (95% CI) | 37.69 (25.24, 50.07) | 36.18 (26.37, 46.05) | 40.84 (31.69, 49.77) | 35.04 (26.54, 43.65) | NA |
18 to < 24 months, (95% CI) | NE (NE, NE) | NE (NE, NE) | 36.95 (27.35, 46.56) | 32.98 (24.18, 42.03) | NA |
24 to < 48 months, (95% CI) | NE (NE, NE) | NE (NE, NE) | NE (NE, NE) | NE (NE, NE) | NA |
Failure-Free Survival | |||||
Median FFS | |||||
months, (95% CI) | 4.99 | 1.02 | 4.86 | 1.02 | NA |
days, (95% CI) | NA | NA | NA | NA | 85.0 (42.0, 158.0) |
Number of patients with events, n (%) | 84 (54.5) | 119 (76.8) | 91 (59.1) | 121 (78.1) | 60 (84.5) |
Number of patients with competing risk | 30 (19.5) | 14 (9.0) | 36 (23.4) | 15 (9.7) | NA |
Number of patients censored, n (%) | 40 (26.0) | 22 (14.2) | 27 (17.5) | 19 (12.3) | 11 (15.5) |
Hazard ratioa (95% CI) | 0.46 (0.35, 0.60) | 0.49 (0.37, 0.63) | NA | ||
P valuel | 0.0001 | 0.0001 | NA | ||
Estimated cumulative incidence (95% CI) | |||||
1 months | 18.47 (12.74, 25.04) | 49.13 (40.94, 56.80) | 18.47 (12.74, 25.04) | 49.13 (40.94, 56.80) | NA |
2 months | 35.83 (28.22, 43.50) | 61.32 (53.00, 68.61) | 35.82 (28.21, 43.48) | 61.32 (53.00, 68.61) | NA |
6 months | 52.85 (44.24, 60.74) | 80.86 (72.95, 86.67) | 54.07 (45.69, 61.71 | 80.17 (72.52, 85.90) | NA |
12 months | 59.20 (50.01, 67.26) | 81.83 (73.93, 87.53) | 59.59 (51.02, 67.14) | 80.97 (73.37, 86.60) | NA |
18 months | 61.02 (51.36, 69.34) | 81.83 (73.93, 87.53) | 60.76 (52.06, 68.38) | 83.41 (74.17, 89.57) | NA |
24 months | NA | NA | NE (NE, NE) | 83.41 (74.17, 89.57) | NA |
Failure-free survival probability (%) (95% CI) | |||||
Month 3 | NA | NA | NA | NA | 49.1 (37.1, 60.1) |
Month 6 | NA | NA | NA | NA | 33.8 (22.9, 45.0) |
Month 9 | NA | NA | NA | NA | 30.7 (20.2, 41.8) |
Month 12 | NA | NA | NA | NA | 21.5 (12.6, 32.0) |
Rate of durable ORR at Day 56 | |||||
Patients with overall response | 61 (39.6) | 34 (21.9) | NA | NA | NA |
95% CIc | (31.8, 47.8) | (15.7, 29.3) | NA | NA | NA |
Complete Response | 41 (26.6) | 25 (16.1) | NA | NA | NA |
Partial Response | 20 (13.0) | 9 (5.8) | NA | NA | NA |
Odds ratio (ruxolitinib/BAT) (95% CI)d | 2.38 (1.43, 3.94) | NA | NA | NA | |
P value | 0.0005 | NA | NA | NA | |
Nonresponders, n (%) | NA | NA | |||
No Response | 1 (0.6) | 1 (0.6) | NA | NA | NA |
Mixed response | 5 (3.2) | 4 (2.6) | NA | NA | NA |
Progression | 0 | 0 | NA | NA | NA |
Otherb | 0 | 1 (0.6) | NA | NA | NA |
Unknown | 29 (18.8) | 21 (13.5) | NA | NA | NA |
Death | 7 (4.5) | 2 (1.3) | NA | NA | NA |
Early discontinuation | 13 (8.4) | 15 (9.7) | NA | NA | NA |
Missing visits | 9 (5.8) | 4 (2.6) | NA | NA | NA |
Duration of Response in patients with CR or PR at or before Day 28 | |||||
Number of patients with eventse, n (%) | 9 (9.4) | 21 (34.4) | 9 (9.3) | 22 (35.5) | NA |
Number of patients with competing risks | 53 (55.2) | 23 (37.7 | 66 (68.0) | 26 (41.9) | NA |
Death | 28 (29.2) | 12 (19.7) | 34 (35.1) | 14 (22.6) | NA |
Incidence of cGvHD | 25 (26.0) | 11 (18.0) | 32 (33.0) | 12 (19.4) | NA |
Number of patients censored, n (%) | 34 (35.4) | 17 (27.9) | 22 (22.7) | 14 (22.6) | NA |
Duration of response, days | |||||
Median | 168.0 | 101.0 | 163.0 | 101.0 | NA |
Q1 – Q3 | 78.0 to 225.0 | 46.0 to 170.0 | 78.0 to 246.0 | 46.0 to 181.0 | NA |
Range | 22.0 to 423.0 | 10.0 to 289.0 | 22.0 to 623.0 | 10.0 to 456.0 | NA |
Estimates cumulative incidence at: | |||||
1 months (95% CI) | 2.08 (0.40, 6.65) | 11.54 (5.03, 21.03) | 2.06 (0.39, 6.58) | 12.97 (6.01, 22.66) | NA |
2 months (95% CI) | 5.37 (1.98, 11.30) | 20.13 (11.02, 31.19) | 5.20 (1.92, 10.96) | 21.38 (12.05, 32.47) | NA |
6 months (95% CI) | 9.65 (4.39, 17.40) | 38.98 (25.54, 52.19) | 8.73 (4.03, 15.68) | 37.34 (24.95, 49.71) | NA |
12 months (95% CI) | 11.76 (5.51, 20.57) | NE (NE, NE) | 10.16 (4.91, 17.64) | 37.34 (24.95, 49.71) | NA |
18 months (95% CI) | NA | NA | 10.16 (4.91, 17.64) | NE (NE, NE) | NA |
Best Overall Response by Day 28 | |||||
Patients with overall response | 126 (81.8) | 94 (60.6) | NA | NA | NA |
95% CIc | (74.8, 87.6) | (52.5, 68.4) | NA | NA | NA |
Complete response | 20 (12.1) | 11 (6.7) | NA | NA | NA |
Partial response | 106 (64.2) | 88 (53.7) | NA | NA | NA |
Odds ratio (95% CI)d | 3.07 (1.80, 5.25) | NA | NA | NA | |
P value | 0.0001 | NA | NA | NA | |
Nonresponders | |||||
No Response | 13 (8.4) | 21 (13.5) | NA | NA | NA |
Mixed response | 7 (4.5) | 14 (9.0) | NA | NA | NA |
Progression | 4 (2.6) | 10 (6.5) | NA | NA | NA |
Unknown | 4 (2.6) | 16 (10.3) | NA | NA | NA |
Death | 2 (1.3) | 6 (3.9) | NA | NA | NA |
Early discontinuation | 2 (1.3) | 4 (2.6) | NA | NA | NA |
Missing visits | 0 | 6 (3.9) | NA | NA | NA |
Event-free survival | |||||
Median EFS, months | 8.28 | 4.17 | 8.18 | 4.17 | NA |
Eventsg, n (%) | 77 (50.0) | 86 (55.5) | 87 (56.5%) | 95 (61.3%) | NA |
Censored, n (%) | 77 (50.0) | 69 (44.5) | 67 (43.5) | 60 (38.7) | NA |
Hazard ratio (95% CI) | 0.80 (0.58, 1.08) | 0.80 (0.60, 1.08) | NA | ||
P value | 0.1466 | 0.1431 | NA | ||
Survival probability at: | |||||
0 to < 1 months, (95% CI) | 89.38 (83.24, 93.35) | 82.83 (75.81, 87.97) | 89.38 (83.24, 93.35) | 82.83 (75.81, 87.97) | NA |
1 to < 1 months, (95% CI) | 74.54 (66.74, 80.78) | 71.65 (63.62, 78.21) | 74.60 (66.82, 80.82) | 71.72 (63.71, 78.26) | NA |
2 to < 6 months, (95% CI) | 54.77 (46.16, 62.58) | 44.04 (35.49, 52.26) | 53.56 (45.21, 61.21) | 44.14 (35.82, 52.13) | NA |
6 to < 12 months, (95% CI) | 45.14 (36.06, 53.78) | 39.99 (31.43, 48.40) | 44.21 (35.88, 52.20) | 38.49 (30.35, 46.56) | NA |
12 to < 18 months, (95% CI) | 36.83 (25.02, 48.66) | 32.29 (22.84, 42.09) | 39.06 (30.38, 47.64) | 31.15 (22.92, 39.71) | NA |
18 to < 24 months, (95% CI) | NE (NE, NE) | NE (NE, NE) | 35.34 (26.22, 44.57) | 29.32 (20.95, 38.17) | NA |
24 to < 48 months, (95% CI) | NE (NE, NE) | NE (NE, NE) | NE (NE, NE) | 14.66 (1.80, 39.94) | NA |
Non relapse mortality | |||||
Number of patients with events, n (%) | 60 (39.0) | 66 (42.6) | 69 (44.8) | 70 (45.2) | 40 (56.3) |
Number of patients with competing risks, n (%) | 15 (9.7)h | 20 (12.9)h | 17 (11.0)h | 25 (16.1)h | NR |
Number of patients censored, n (%) | 79 (51.3) | 69 (44.5) | 68 (44.2) | 60 (38.7) | NR |
Estimate cumulative incidence (%) at: | |||||
1 month, (95% CI) | 9.96 (5.83, 15.39) | 14.52 (9.45, 20.64) | 9.96 (5.83, 15.39) | 14.52 (9.45, 20.64) | NA |
2 months, (95% CI) | 20.75 (14.64, 27.60) | 23.60 (17.09, 30.73) | 20.71 (14.61, 27.54) | 23.54 (17.04, 30.65) | NA |
6 months, (95% CI) | 36.18 (28.28, 44.12) | 43.34 (34.89, 51.48) | 37.68 (29.88, 45.45) | 42.42 (34.18, 50.41) | 44.3 (32.5, 55.5) |
9 months, (95% CI) | NA | NA | NA | NA | 47.3 (35.2, 58.5) |
12 months, (95% CI) | 42.67 (33.84, 51.19) | 45.33 (36.67, 53.57) | 43.80 (35.54, 51.75) | 46.51 (37.98, 54.58) | 53.4 (40.9, 64.3) |
18 months, (95% CI) | 49.38 (36.37, 61.12) | 50.77 (40.73, 59.96) | 47.83 (38.80, 56.30) | 51.23 (42.09, 59.64) | NA |
24 months, (95% CI) | 49.38 (36.37, 61.12) | NE (NE, NE) | 51.60 (41.60, 60.71) | 51.23 (42.09, 59.64) | NA |
Malignancy relapse/ progression | |||||
Number of patients with events, n (%) | 14 (9.5) | 20 (13.6) | 16 (10.9) | 25 (17.0) | NA |
Number of patients with competing risks, n (%) | 56 (38.1) | 62 (42.2) | 65 (44.2) | 66 (44.9) | NA |
Number of patients censored, n (%) | 77 (52.4) | 65 (44.2) | 66 (44.9) | 56 (38.1) | NA |
Estimated cumulative incidence at: | |||||
1 month, (95% CI) | 0.69 (0.06, 3.51) | 2.80 (0.92, 6.54) | 0.69 (0.06, 3.51) | 2.80 (0.92, 6.54) | NA |
2 months, (95% CI) | 4.23 (1.73, 8.49) | 4.30 (1.76, 8.63) | 4.21 (1.73, 8.46) | 4.29 (1.75, 8.60) | NA |
6 months, (95% CI) | 8.28 (4.36, 13.80) | 12.45 (7.40, 18.88) | 8.49 (4.62, 13.83) | 13.49 (8.32, 19.91) | NA |
12 months, (95% CI) | 10.65 (5.84, 17.11) | 14.62 (8.96, 21.60) | 11.12 (6.47, 7.17) | 15.13 (9.61, 21.82) | NA |
18 months, (95% CI) | 12.56 (6.84, 20.08) | 19.04 (11.36, 28.23) | 12.40 (7.29, 8.93) | 19.02 (12.36, 26.79) | NA |
24 months, (95% CI) | 12.56 (6.84, 20.08) | NE (NE, NE) | 12.40 (7.29,18.93) | 20.93 (13.56, 29.40) | NA |
Relapse rate | |||||
Number of patients with relapse of underlying malignancy, n (%) (95% CI) | NA | NA | NA | NA | 5 (7.0) (2.3, 15.7) |
Relapse Mortality Rate | |||||
Number of patients with relapse of underlying malignancy and a fatal outcome, n (%) (95% CI) | NA | NA | NA | NA | 4 (5.6) (1.6, 13.8) |
Incidence of cGvHD | |||||
Number of patients with events, n (%) | 38 (24.7) | 26 (16.8) | 45 (29.2) | 29 (18.7) | 11 (15.5) |
Number of patients with competing risk, n (%) | 69 (44.8) | 78 (50.3) | 79 (51.3) | 85 (54.8) | NA |
Death | NR | NR | 62 (40.3) | 63 (40.6) | NA |
Hematologic disease relapse/ progression | NR | NR | 17 (11.0) | 22 (14.2) | NA |
Number of patients censored, n (%) | 47 (30.5) | 51 (32.9) | 30 (19.5) | 41 (26.5) | NA |
Estimated cumulative incidence at: | |||||
1 month, (95% CI) | 0 | 1.33 (0.26, 4.34) | 0 (NE, NE) | 1.33 (0.26, 4.34) | NA |
2 months, (95% CI) | 1.35 (0.26, 4.40) | 2.03 (0.55, 5.41) | 1.34 (0.26, 4.39) | 2.03 (0.55, 5.41) | NA |
6 months, (95% CI) | 14.85 (9.30, 21.63) | 13.28 (8.04, 19.84) | 15.71 (10.34, 22.11) | 12.19 (7.40, 18.25) | NA |
12 months, (95% CI) | 32.28 (23.74, 41.10) | 22.19 (14.83, 30.50) | 29.95 (22.53, 37.71) | 20.27 (13.87, 27.54) | NA |
18 months, (95% CI) | 37.65 (27.18, 48.07) | 24.05 (16.11, 32.87) | 32.36 (24.47, 40.48) | 22.80 (15.75, 30.65) | NA |
24 months, (95% CI) | Not reported | Not reported | NE (NE, NE) | 22.80 (15.75, 30.65) | NA |
NA = not applicable; NE = not evaluable; NR = not reported
aHazard ratio and 95% CI are obtained from the stratified Cox proportional hazards model using Wald test.
bOther: patient with additional systemic therapies along with CR/PR per investigator assessment.
cThe 95% CI for the response rate was calculated using the Clopper-Pearson exact method.
dOdds ratio and 95% CI are calculated using stratified Cochran-Mantel-Haenszel test.
eThe event was defined as the progression of aGvHD or addition of systemic therapies for aGvHD after Day 28. The competing risks included death without prior observation of aGvHD progression and onset of cGvHD.
fMedian and Quartiles are provided using Kaplan–Meier method.
gThe event includes hematologic disease relapse/progression, graft failure or death due to any cause
hThe competing risk included hematologic disease relapse/progression.
iThe competing risk included hematologic malignancy relapse/progression and deaths without prior onset of cGvHD.
jParticipants with no observed death or loss to follow-up were censored at their last date known to be alive.
kRelapsed-related mortality was considered competing risk
lP value nominal
Source: Clinical Study Report (REACH 2),18 Clinical Study Report (REACH 1)19
Table 57: Summary of Concomitant Medications in REACH 1, Efficacy-Evaluable Population (Medication Class; Received by 40% of Patients in the REACH 1 Trial)
Medication Class | Ruxolitinib (N = 71) |
|---|---|
Number (%) of Subjects with Concomitant Medications | 71 (100) |
Aminoalkyl Ethers | 49 (69.0) |
Anilids | 52 (73.2) |
Antipropulsives | 48 (67.6) |
Benzodiazepine derivatives | 42 (59.2) |
Bile acid preparations | 46 (64.8) |
Calcineurin inhibitors | 63 (88.7) |
Colony stimulating factors | 30 (42.3) |
Corticosteroids acting locally | 30 (42.3) |
Electrolyte solutions | 60 (84.5) |
Fluoroquinolones | 31 (43.7) |
Fourth-generation cephalosporins | 29 (40.8) |
Glucocorticoids | 32 (45.1) |
Glycopeptide antibacterials | 30 (42.3) |
Insulins and analogues for injection, fast-acting | 49 (69.0) |
Natural opium alkaloids | 48 (67.6) |
Nucleosides and nucleotides excel reverse transcriptase inhibitors | 69 (97.2) |
Other agents for local treatment | 29 (40.8) |
Other antiemetics | 32 (45.1) |
Other antimycotics for systemic use | 42 (59.2) |
Proton pump inhibitors | 60 (84.5) |
Serotonin (5HT3) antagonists | 48 (67.6) |
Solution for parenteral nutrition | 38 (53.5) |
Sulfonamides, plain | 39 (54.9) |
Triazole derivatives | 59 (83.1) |
Vitamin D and analogues | 30 (42.3) |
Clinical Study Report (REACH 1)19
Figure 26: Kaplan–Meier Curves of Overall Survival in REACH 2, Full Analysis Set (July 25, 2019, Data Cut-Off Date)
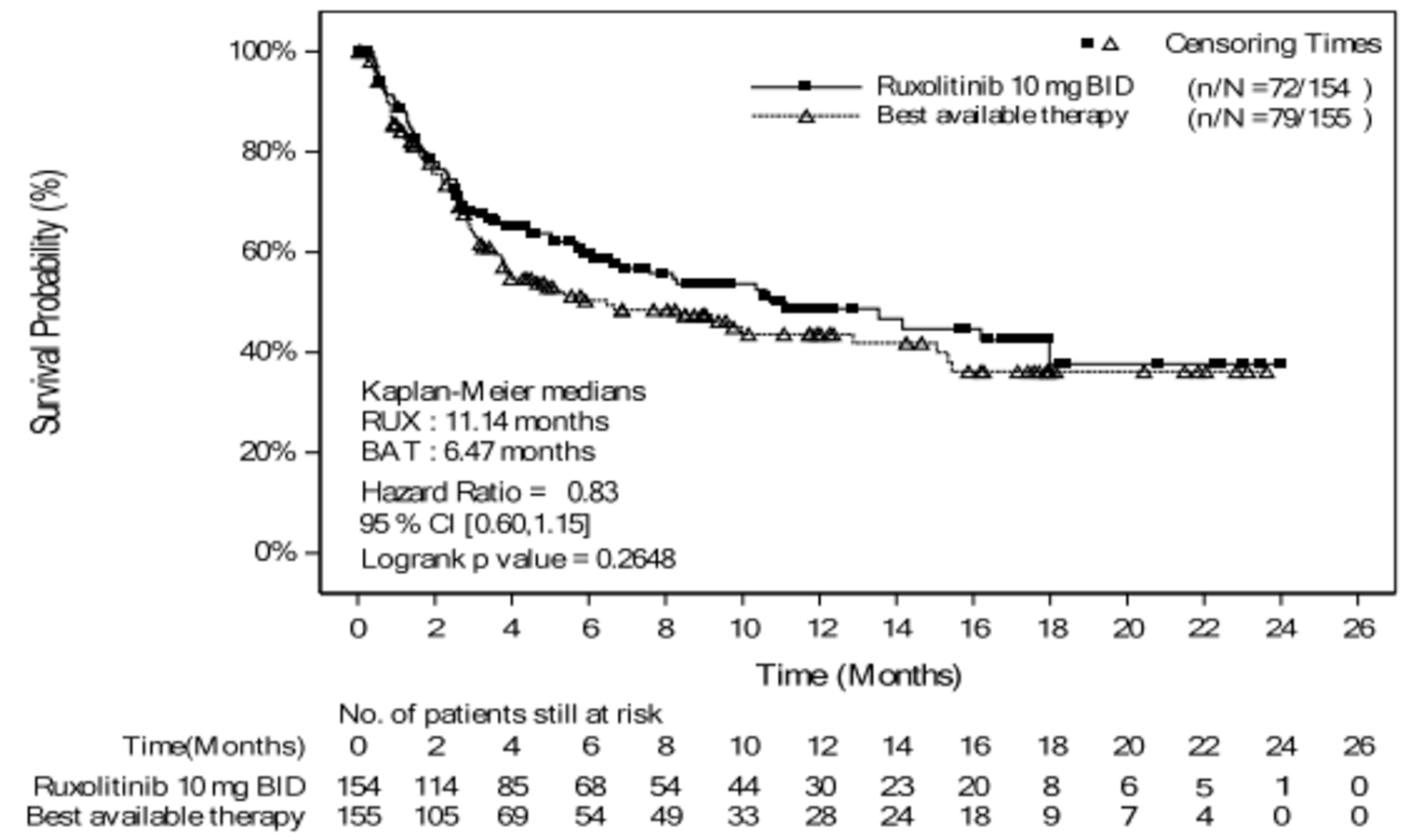
P value is obtained from the log-rank test
Source: Clinical Study Report (REACH 2)18
Figure 27: Kaplan–Meier Curves of Overall Survival in REACH 1, Efficacy-Evaluable Patients (June 5, 2019, Data Cut-Off Date)
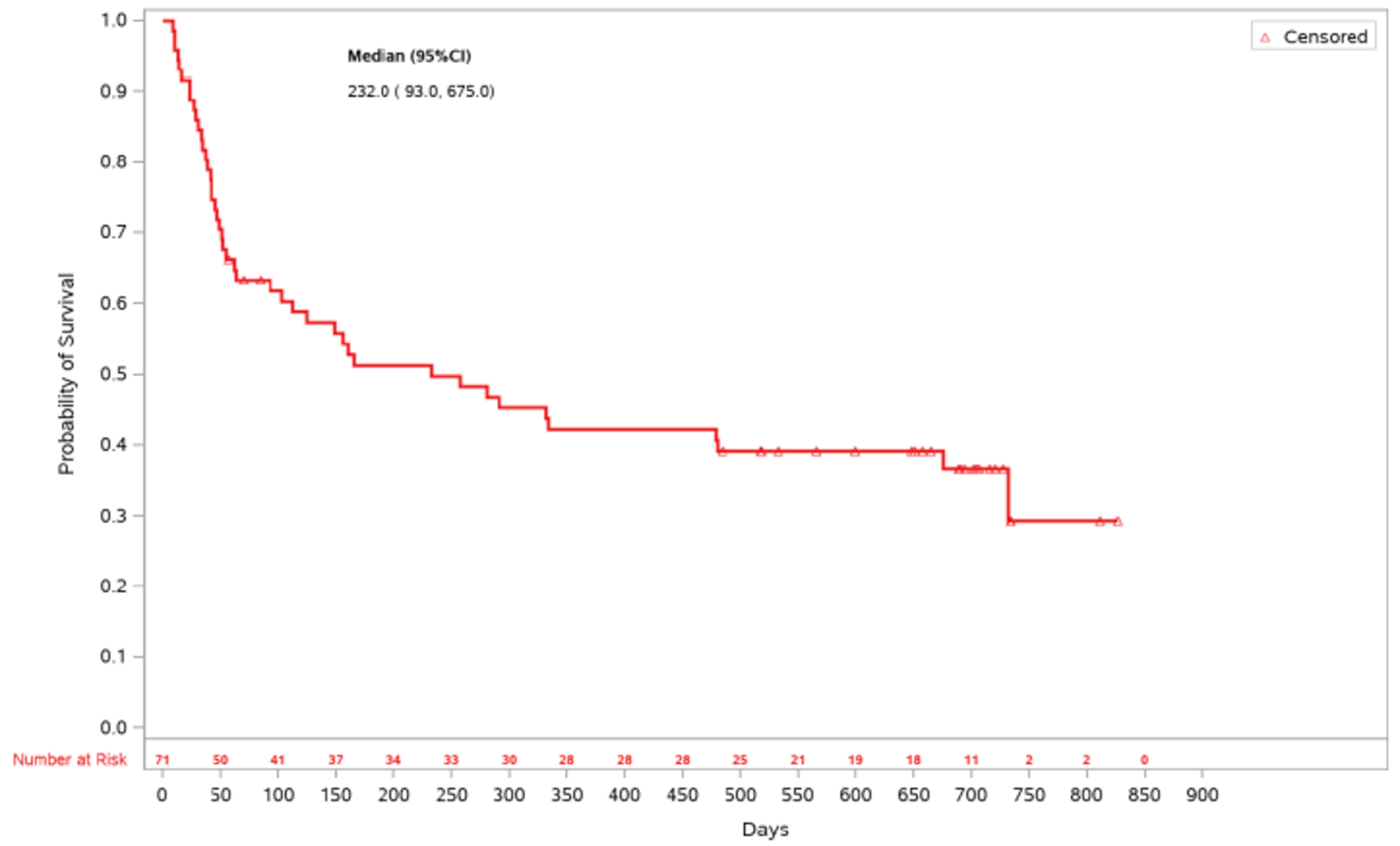
Source: Clinical Study Report (REACH 1)19
Figure 28: Kaplan–Meier Curves of FFS in REACH 2, Efficacy-Evaluable Patients (July 25, 2019, Data Cut-Off Date)
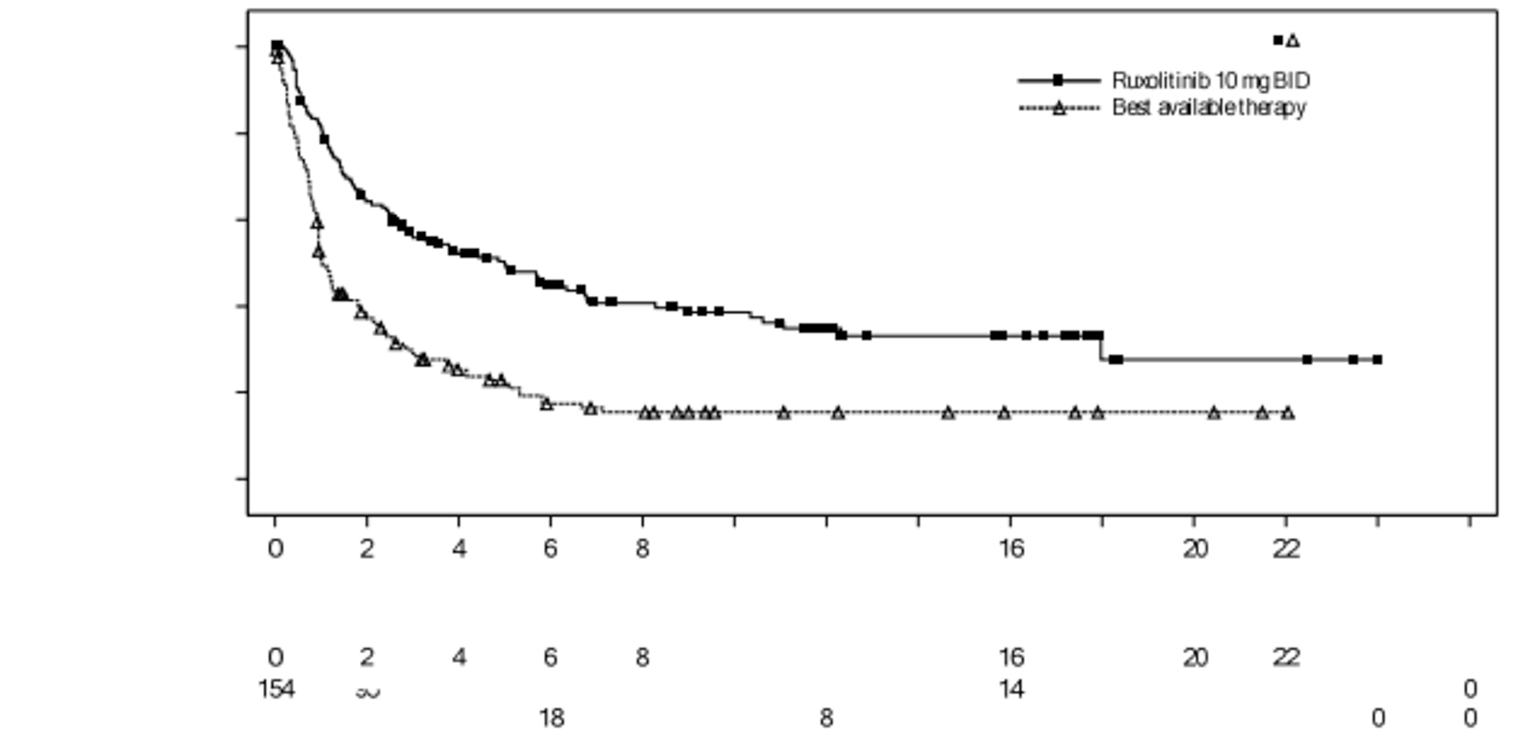
Source: Clinical Study Report (REACH 2)18
Figure 29: Kaplan–Meier Curves of FFS in REACH 1, Efficacy-Evaluable Patients (June 5, 2019, Data Cut-Off Date)
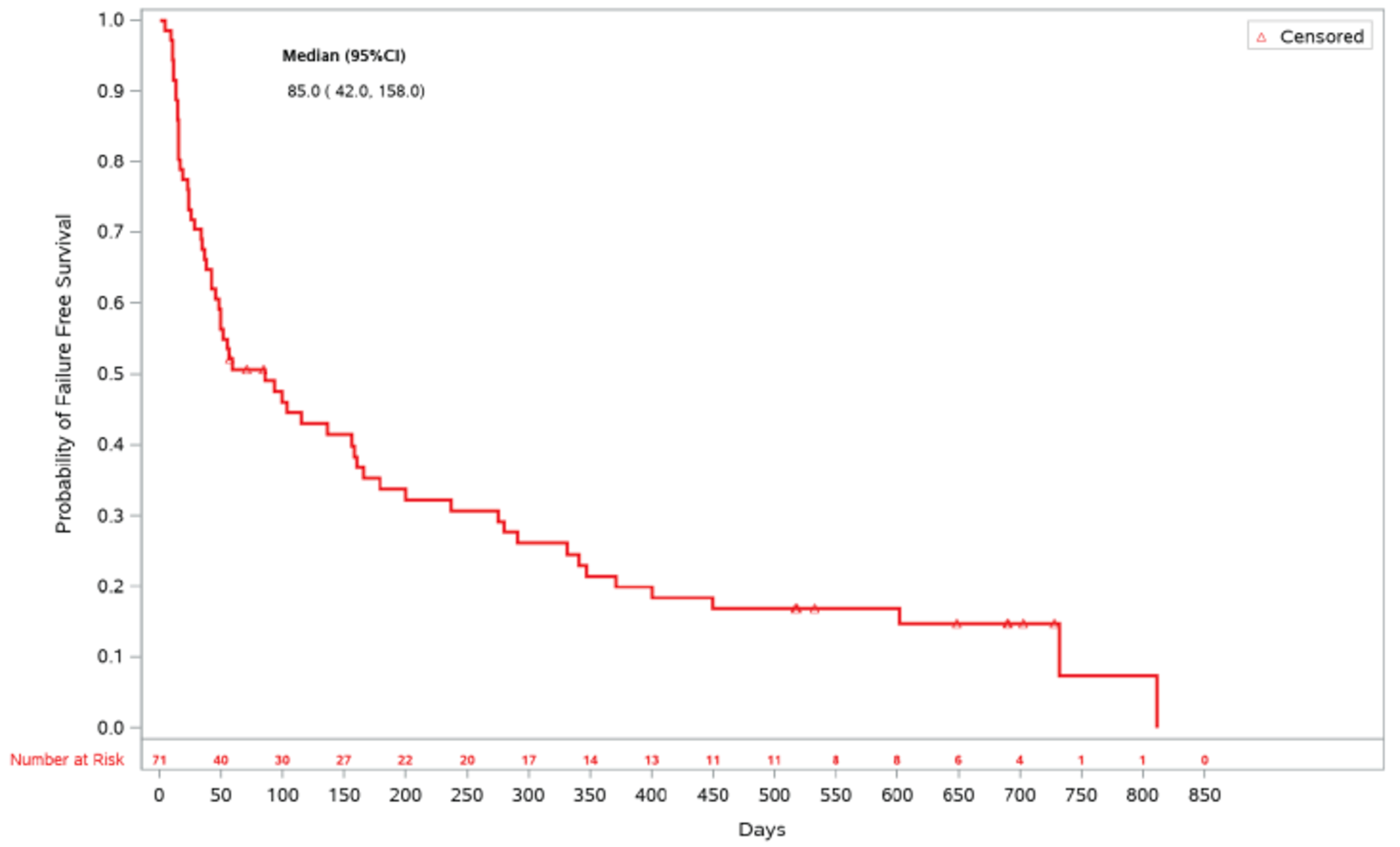
Source: Clinical Study Report (REACH 1)19
Figure 30: Kaplan–Meier Estimate of Duration of Response for Participants Who Had a Response at Any Time point in REACH 1, Efficacy-Evaluable Patients (June 5, 2019, Data Cut-Off Date)
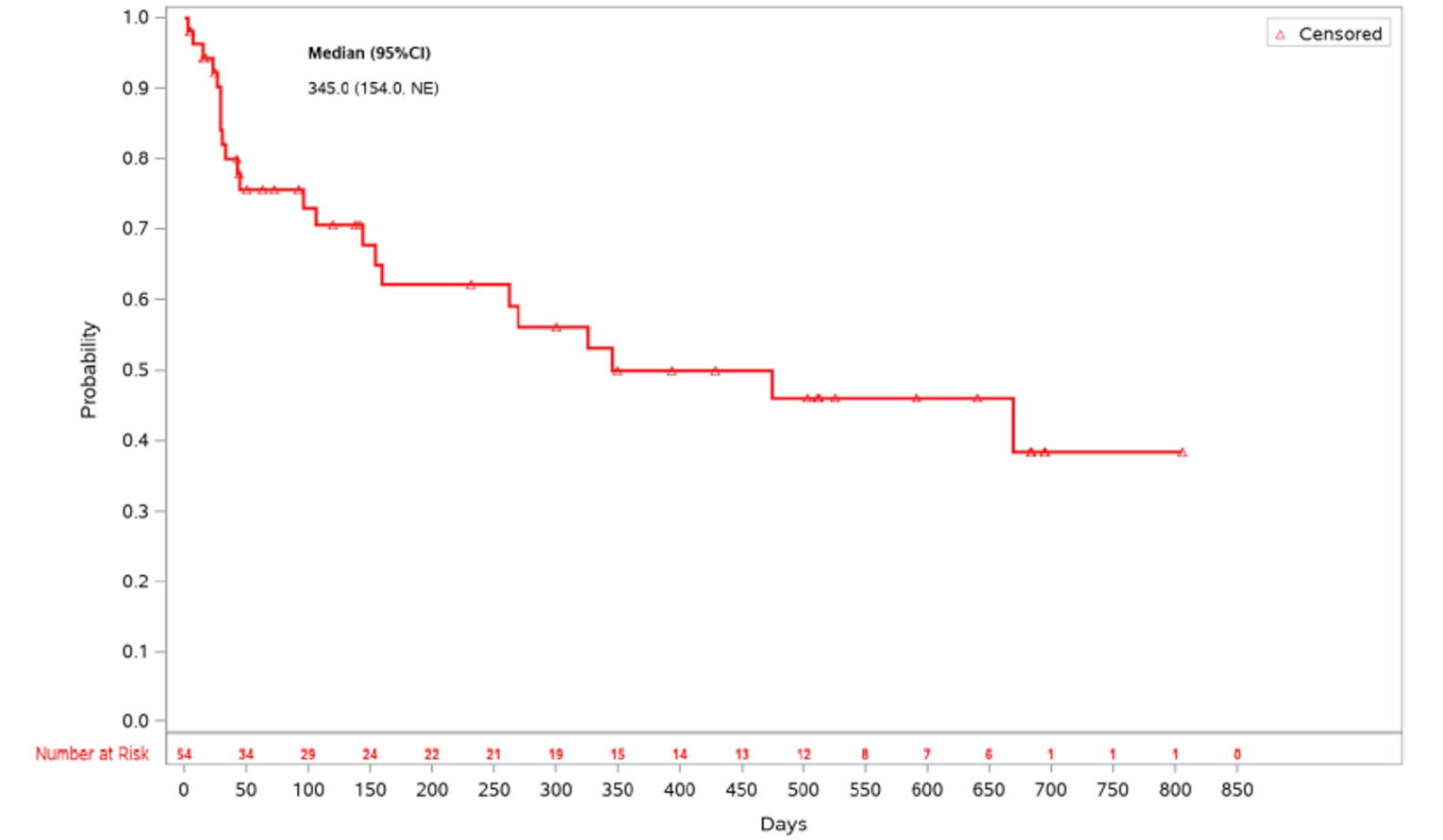
Source: Clinical Study Report (REACH 1)19
Figure 31: Kaplan–Meier Estimate of Duration of Response for Patients Who Had a Response at Day 28 in REACH 1, Efficacy-Evaluable Patients (June 5, 2019, Data Cut-Off Date)
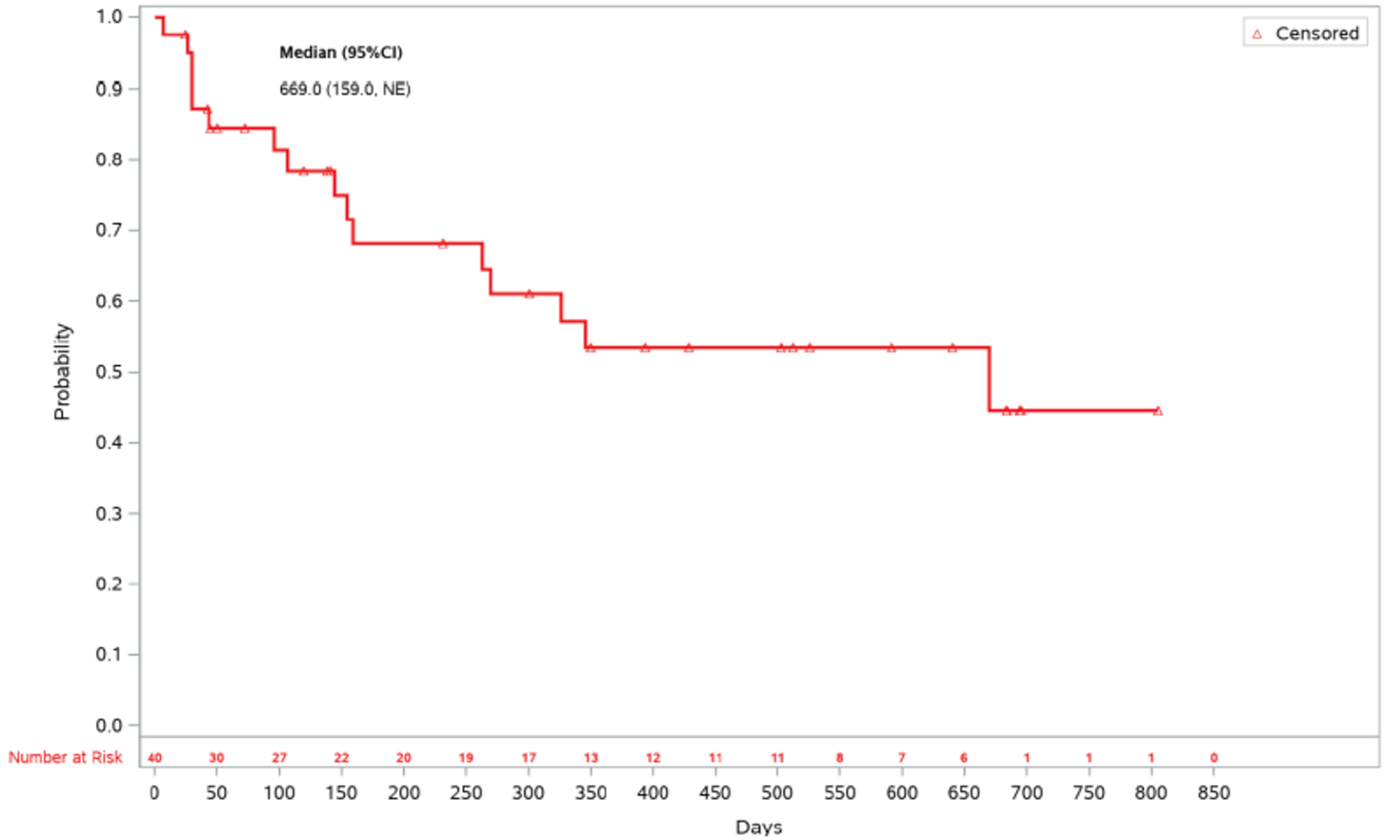
Source: Clinical Study Report (REACH 1)19
Figure 32: Cumulative Incidence Curve of Non-Relapse Mortality in REACH 2, Full Analysis Set (Data Cut-Off Date: July 25, 2019)
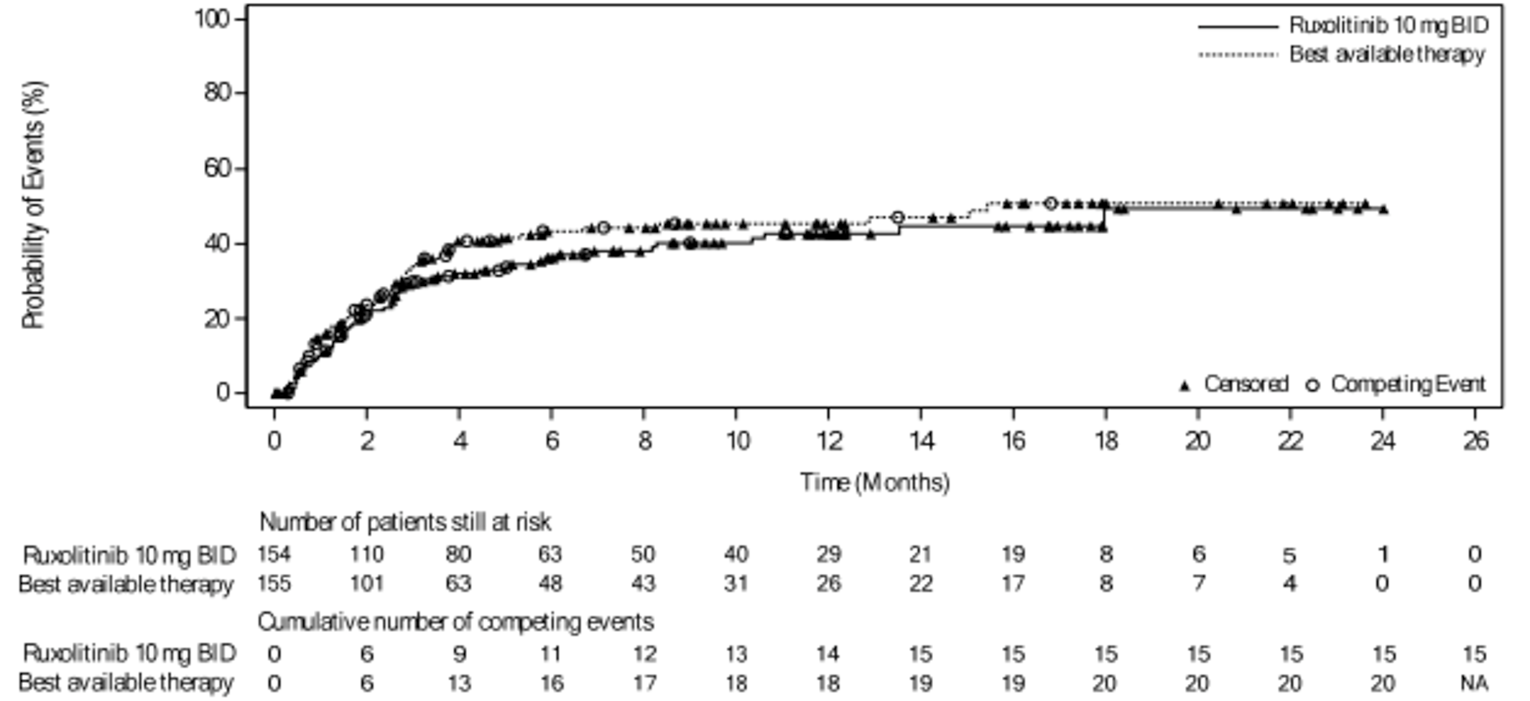
NA = not applicable
Source: Clinical Study Report (REACH 2)18
Figure 33: Cumulative Incidence Curve of Non-Relapse Mortality in REACH 2, Full Analysis Set (Data Cut-Off Date: January 6, 2020)
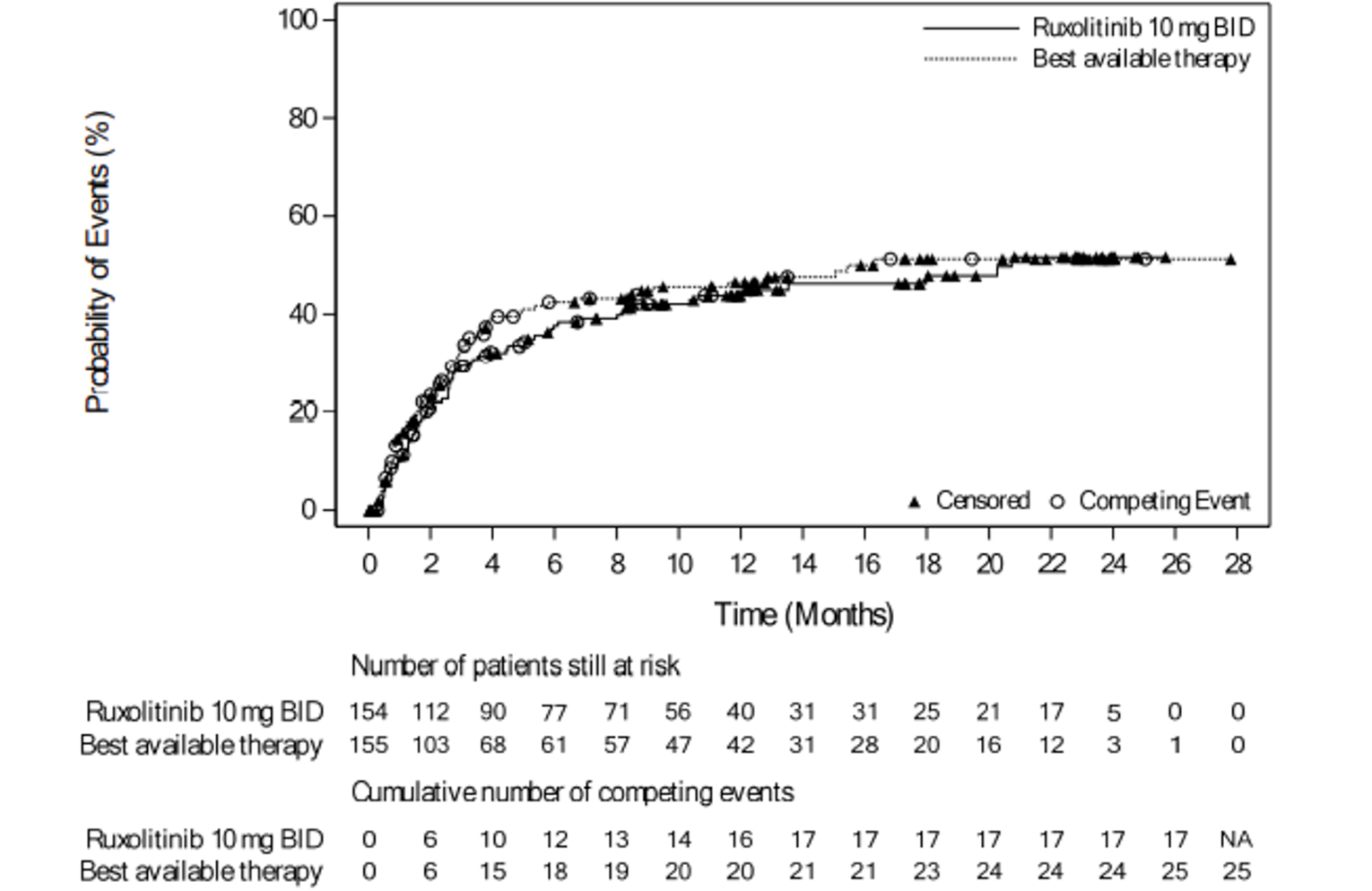
NA = not applicable
Source: Clinical Study Report (REACH 2)18
Figure 34: Cumulative Incidence Function of Non-Relapse Mortality in REACH 1, Efficacy-Evaluable Patients (June 5, 2019, Data Cut-Off Date)
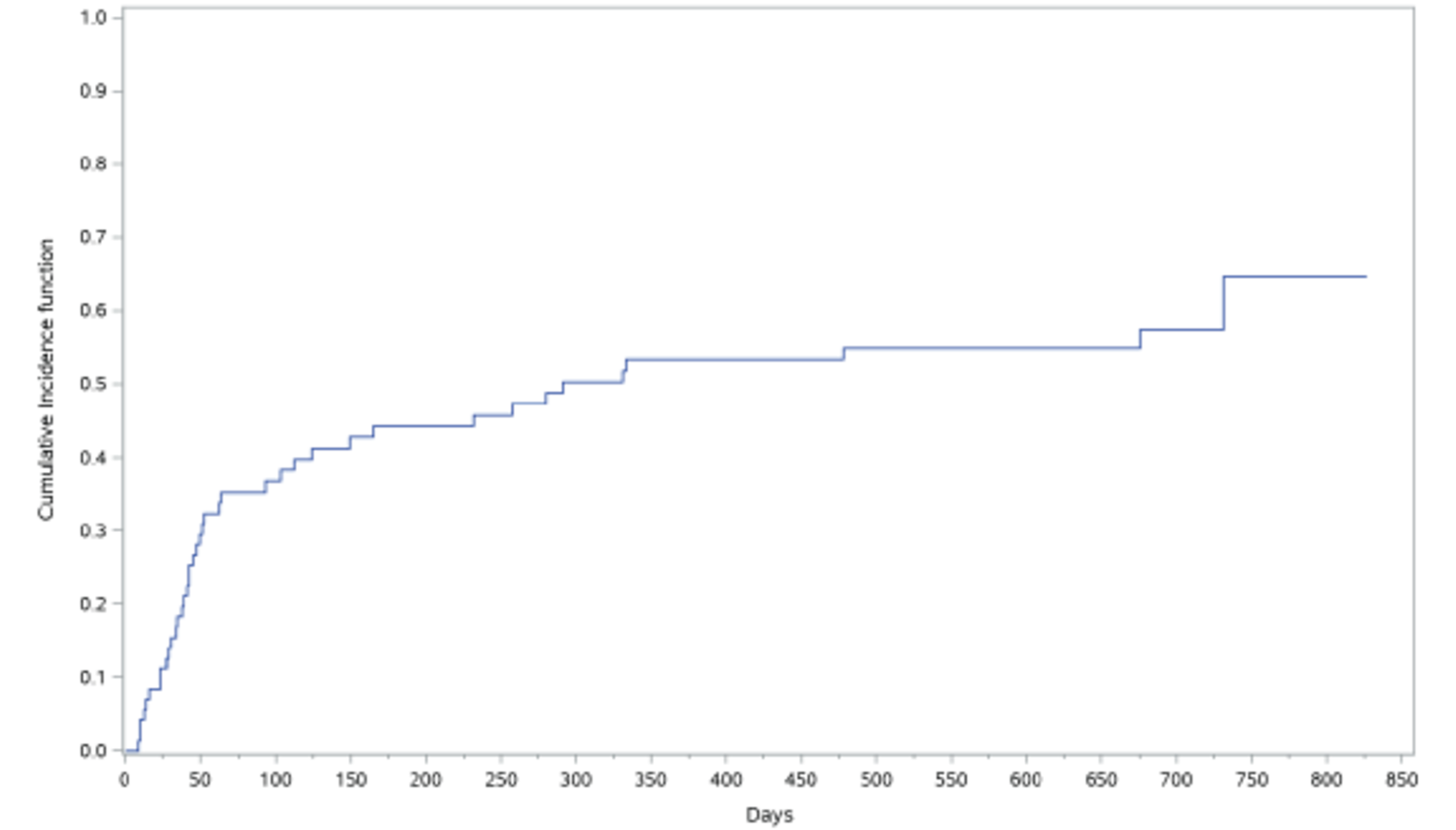
Source: Clinical Study Report (REACH 1)19
Figure 35: Cumulative incidence of cGvHD in REACH 2, Full Analysis Set (Data Cut-Off Date: January 6, 2020)
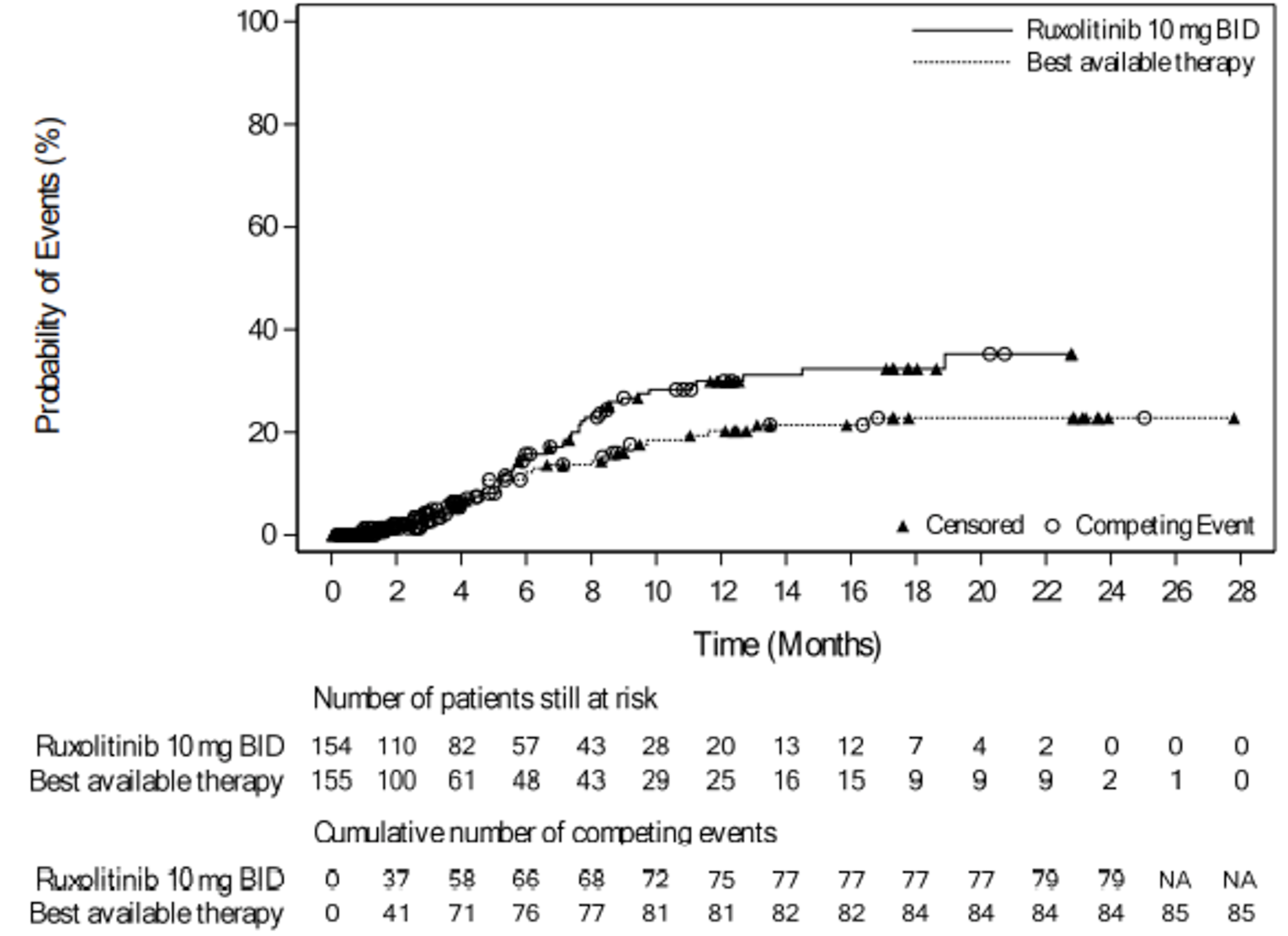
The competing risk includes deaths without prior onset of cGvHD and hematologic disease relapse/progression. NA - Not Applicable
Source: Clinical Study Report (REACH 2)18
Table 58: Summary of Efficacy End points, REACH 2 — Full Analysis Set (July 25, 2019, and April 23, 2021, Data Cut-Off Dates)
Data cut-off date | REACH 2 | |||
|---|---|---|---|---|
Ruxolitinib N = 154 | BAT N = 155 | Ruxolitinib N = 154 | BAT N = 155 | |
July 25, 2019 | April 23, 2021 | |||
Overall Survival | ||||
Median OS follow-up time, months | 5.04 | 3.58 | 8.23 | 3.81 |
Median OS | ||||
months, (95% CI) | 11.14 (NR) | 6.47 (NR) | 10.71 (NR) | 5.82 (NR) |
days, (95% CI) | 339 (186, NE) | 197 (114, 458) | 326 (186, 621) | 177 (115, 392) |
Events (death), n (%) | 72 (46.8) | 79 (51.0) | 89 (57.8) | 91 (58.7) |
Censored, n (%) | 82 (53.2) | 76 (49.0) | 65 (42.2) | 64 (41.3) |
Hazard ratio (95% CI)a | 0.83 (0.60, 1.15) | 0.85 (0.63, 1.14) | ||
P value | 0.2648 | NR | ||
Failure-Free Survival | ||||
Median FFS | ||||
months, (95% CI) | 4.99 (NR) | 1.02 (NR) | 4.86 (NR) | 1.02 (NR) |
days, (95% CI) | NA | NA | NA | NA |
Number of patients with events, n (%) | 84 (54.5) | 119 (76.8) | 91 (59.1) | 121 (78.1) |
Number of patients with competing risk | 30 (19.5) | 14 (9.0) | 42 (27.3) | 16 (10.3) |
Number of patients censored, n (%) | 40 (26.0) | 22 (14.2) | 21 (13.6) | 18 (11.6) |
Hazard ratioa (95% CI) | 0.46 (0.35, 0.60) | 0.51 (0.39, 0.66) | ||
P valuee | 0.0001 | 0.0001 | ||
Duration of Response in patients with CR or PR at or before Day 28 | ||||
Number of patients with response at or before Day 28 | 96 | 61 | 98 | 62 |
Number of patients with eventsb, n (%) | 9 (9.4) | 21 (34.4) | 10 (10.2) | 22 (35.5) |
Number of patients with competing risks | 53 (55.2) | 23 (37.7 | 74 (75.5) | 27 (43.5) |
Death | 28 (29.2) | 12 (19.7) | 35 (35.7) | 14 (22.6) |
Incidence of cGvHD | 25 (26.0) | 11 (18.0) | 39 (39.8) | 13 (21.0) |
Number of patients censored, n (%) | 34 (35.4) | 17 (27.9) | 14 (14.3) | 13 (21.0) |
Duration of response, days | ||||
Median | 168.0 | 101.0 | 167.0 | 106.0 |
Q1 – Q3 | 78.0 to 225.0 | 46.0 to 170.0 | 80.0 to 288.0 | 46.0 to 259.0 |
Range | 22.0 to 423.0 | 10.0 to 289.0 | 22.0 to 677.0 | 10.0 to 526.0 |
Event-free survival | ||||
Median EFS, months | 8.28 | 4.17 | 8.28 | 4.17 |
Eventsc, n (%) | 77 (50.0) | 86 (55.5) | 94 (61.0) | 96 (61.9) |
Censored, n (%) | 77 (50.0) | 69 (44.5) | 60 (39.0) | 59 (38.1) |
Hazard ratio (95% CI) | 0.80 (0.58, 1.08) | 0.85 (0.64, 1.13) | ||
P valuee | 0.1466 | NR | ||
Non relapse mortality | ||||
Number of patients with events, n (%) | 60 (39.0) | 66 (42.6) | 72 (46.8) | 71 (45.8) |
Number of patients with competing risks, n (%) | 15 (9.7) | 20 (12.9) | 21 (13.6) | 25 (16.1) |
Number of patients censored, n (%) | 79 (51.3) | 69 (44.5) | 61 (39.6) | 59 (38.1) |
Malignancy relapse/ progression | ||||
Number of patients with events, n (%) | 14 (9.5) | 20 (13.6) | 20 (13.6) | 25 (17.0) |
Number of patients with competing risks, n (%) | 56 (38.1) | 62 (42.2) | 68 (46.3) | 67 (45.6) |
Number of patients censored, n (%) | 77 (52.4) | 65 (44.2) | 59 (40.1) | 55 (37.4) |
Cumulative steroid dosing until Day 56 | ||||
Completely tapered off by Day 56, n (%) (95% CI) | 33 (21.4) (15.2, 28.8) | 23 (14.8) (9.6, 21.4) | 34 (22.1) (15.8, 29.5) | 23 (14.8) (9.6, 21.4) |
≤ 50% RDId, n (%) (95% CI) | 45 (29.2) (22.2, 37.1) | 38 (24.5) (18.0, 32.1) | 45 (29.2) (22.2, 37.1) | 37 (23.9) (17.4, 31.4) |
> 50% RDId, n (%) (95% CI) | 106 (68.8) (60.9, 76.0) | 116 (74.8) (67.2, 81.5) | 106 (68.8) (60.9, 76.0) | 117 (75.5) (67.9, 82.0) |
Incidence of cGvHD | ||||
Number of patients with events, n (%) | 38 (24.7) | 26 (16.8) | 52 (33.8) | 34 (21.9) |
Number of patients with competing risk, n (%) | 69 (44.8) | 78 (50.3) | 80 (51.9) | 85 (54.8) |
Death | NR | NR | 62 (40.3) | 64 (41.3) |
Hematologic disease relapse/ progression | NR | NR | 18 (11.7) | 21 (13.5) |
Number of patients censored, n (%) | 47 (30.5) | 51 (32.9) | 22 (14.3) | 36 (23.2) |
NA = not applicable; NE = not evaluable; NR = not reported.
Note: Outcomes are presented in order of priority as identified in the CADTH review protocol.
aHazard ratio and 95% CI are obtained from the stratified Cox proportional hazards model using Wald test.
bThe event was defined as the progression of aGvHD or addition of systemic therapies for aGvHD after Day 28. The competing risks included death without prior observation of aGvHD progression and onset of cGvHD.
cThe event includes hematologic disease relapse/progression, graft failure or death due to any cause
dRelative dose intensity includes days of zero dose in the calculation.
eP value nominal
Source: Clinical Study Report (REACH 2).18
Appendix 4: Description and Appraisal of Outcome Measures
Note that this appendix has not been copy-edited.
Aim
To describe the following outcome measures and review their measurement properties (validity, reliability, responsiveness to change, and MID):
The Functional Assessment of Cancer Therapy – Bone Marrow Transplant (FACT-BMT)
5-level EQ-5D US English Version 4.4 (EQ-5D-5L)
Findings
Table 59: Summary of Outcome Measures and Their Measurement Properties
Outcome Measure | Type | Conclusions about Measurement Properties | MID |
|---|---|---|---|
FACT-BMT | 50-item self-report questionnaire that measures the effect of a therapy on domains including physical, functional, social/family, and emotional well-being, together with additional concerns relevant for bone marrow transplantation patients. Comprised of the FACT-G questionnaire and a 12-item BMT-specific subscale. | Validity No relevant literature was identified in patients with aGvHD. Reliability No relevant literature was identified in patients with aGvHD. Responsiveness No relevant literature was identified in patients with aGvHD. | No relevant literature identified in patients with aGvHD. No MID was provided in the sponsor’s submission. |
EQ-5D-5L | Generic, utility-based measure of HRQoL, consisting of an index score and a VAS. Index score: The tool consists of 5 dimensions: mobility, self-care, usual activity, pain/ discomfort and anxiety/ depression; each dimension has 5 levels: no problems, slight problems, moderate problems, severe problems and extreme problems. VAS: The tool assessed patient’s self-rated health on a vertical visual analogue scale.68 | Validity No relevant studies found in patients with aGvHD. Reliability No relevant studies found in patients with aGvHD. Responsiveness No relevant studies found in patients with aGvHD. | No relevant literature identified in patients with aGvHD. No MID was provided in the sponsor’s submission. |
EQ-5D-5L = European Quality of Life 5-Five Dimensions 5-Levels; FACT-BMT = The Functional Assessment of Cancer Therapy – Bone Marrow Transplant; HRQoL = health-related quality of life; MID = minimal important difference; VAS = visual analogue scale.
FACT-BMT
The Functional Assessment of Cancer Therapy – Bone Marrow Transplant (FACT-BMT) version 4.0 is 50-item self-report questionnaire that measures the effect of a therapy on domains including physical, functional, social/family, and emotional well-being, together with additional concerns relevant for bone marrow transplantation patients.48 The FACT-BMT consists of the general 27-item FACT (FACT-G) questionnaire and a 23-item BMT subscale, which focuses on concerns of patients who have undergone bone marrow and other transplant procedures.69 The questions are based on 5-point Likert scale, where 0 corresponds to ‘not at all’ and 4 correspond to ‘very much’.69 The recall period is 7 days for this version of the scale.48 The higher the final score, the better the quality of life.69,70 The FACT-BMT is the second most frequently used PROM in clinical studies.69
Psychometric properties
No relevant literature was identified that assessed validity, reliability, or responsiveness in patients with aGvHD. No MID information was identified in populations with aGvHD.
EQ-5D-5L
Description
The EQ-5D is a generic, utility-based measure of HRQoL. The EQ-5D is a 2-part questionnaire, consisting of the EQ-5D descriptive system and the EQ-5D VAS.47
For the descriptive system of the EQ-5D, respondents are asked to indicate their health status that day (i.e., a 1-day recall) on 5 dimensions (mobility, self-care, usual activities, pain/ discomfort, and anxiety/depression). The EQ-5D-5L was created by the EuroQol Group in 2009 to enhance the instrument’s sensitivity and to reduce ceiling effects, as compared to the EQ-5D-3L.47
The 5-level version of the EQ-5D has response options for each of the 5 dimensions that reflect 3 possible levels of functioning.
Level 1: No problems
Level 2: Slight problems
Level 3: Moderate problems
Level 4: Severe problems
Level 5: Extreme problems
The rating on each dimension is combined to create a descriptive health profile (referred to as the health state description) that is a vector of the levels. For example, an individual with no health problems on any dimension would have a health profile of 11111, while a person with extreme problems on all dimensions would have a health profile of 55555. The numerical values assigned to the levels 1, 2, 3, 4, or 5 for each dimension reflect rank order categories of function. There are 3,125 unique health states that exist for the EQ-5D-5L.47 The EQ-5D-5L is available in 150 different languages.47
Scoring
Index Scores
The health profile (health state description or vector) defined by the EQ-5D-5L questionnaire is used to create an overall index score. To create the EQ-5D-5L index score, a scoring algorithm (a mathematical equation termed a utility function) is applied to the vector. Various scoring algorithms for the EQ-5D-5L have been derived by determining the societal preferences for its 3,125 health states (i.e., by assessing how much value society places on each health state) using techniques such as the standard gamble or TTO. In all scoring algorithms of the EQ-5D-5L, a score of 0 represents the health state “dead” and 1.0 reflects “full health”. Negative scores are also possible for those health states that society (not the individual patient) considers to be “worse than dead”.47 In the REACH 2 trial, the EQ-5D-3L health utility index scores were derived using UK population sample weights.22
Visual Analogue Scale Scores
The EQ-5D VAS is a distinct component of the EQ-5D questionnaire. The VAS score is determined by asking respondents to rate their health that day on a vertical line, with anchors (end points) labelled “Worst imaginable health state” at 0 and “Best imaginable health state” at 100. While the EQ-5D index score reflects societal preferences for the health state, the VAS captures the individual’s own value or judgment of his or her present health state. The EQ-5D VAS scores are not used to create utility scores but provide complementary information to the EQ-5D-5L index score.47
Psychometric Properties
No relevant literature was identified that assessed validity, reliability, or responsiveness in patients with aGvHD. No MID information was identified in populations with aGvHD.
Pharmacoeconomic Review
Abbreviations
AE
adverse event
aGvHD
acute graft-versus-host disease
ATG
antithymocyte globulin
BAT
best available therapy
cGvHD
chronic graft-versus-host disease
ECP
extracorporeal photopheresis
ICER
incremental cost-effectiveness ratio
KM
Kaplan-Meier
MMF
mycophenolate mofetil
ORR
overall response rate
OS
overall survival
QALY
quality-adjusted life-year
SR
steroid refractory
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Ruxolitinib (Jakavi), 5 mg, 10 mg, 15 mg and 200 mg tablets, oral |
Submitted price | 5 mg, tablet = $86.63 10 mg, tablet = $87.38 15 mg, tablet = $87.58 20 mg, tablet = $87.64 |
Indication | The treatment of SR or dependent aGvHD in adults and children 12 years and older |
Health Canada approval status | NOC |
Health Canada review pathway | Project Orbis |
NOC date | May 19, 2022 |
Reimbursement request | Per indication |
Sponsor | Novartis Pharmaceutical Canada Inc. |
Submission history | Previously reviewed: yes Indication: myelofibrosis Recommendation date: January 14, 2013 Recommendation: reimburse with clinical criteria and/or conditions Indication: polycythemia vera Recommendation date: March 3, 2016 Recommendation: reimburse with clinical criteria and/or conditions |
aGvHD = acute graft-versus-host disease; NOC = Notice of Compliance; SR = steroid refractory.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Semi-Markov model |
Target population | Patients 12 years or older with SR-aGvHD |
Treatment | Ruxolitinib |
Comparator | BAT, consisting of ATG, ECP, MTX, MMF, SIR, ETA, INF |
Perspective | Canadian publicly funded health care payer |
Outcomes | QALYs, LYs |
Time horizon | Lifetime (15 years) |
Key data source | REACH 2, a multi-centre, randomized, phase III, open-label trial comparing the efficacy and safety of oral ruxolitinib with the investigator’s choice of BAT in patients 12 years and older who developed SR-aGvHD after alloSCT |
Submitted results | Ruxolitinib dominates (i.e., was more effective [incremental QALYs = 0.15] and less costly [$39,934 cost-savings]), compared with BAT |
Key limitations |
|
CADTH reanalysis results |
|
aGvHD = acute graft-versus-host disease; alloSCT = allogeneic stem cell transplantation; ATG = antithymocyte globulin; BAT = best available therapy; cGvHD = chronic graft-versus-host disease; ECP = extracorporeal photopheresis; ETA = etanercept; ICER = incremental cost-effectiveness ratio; INF = infliximab; KM = Kaplan-Meier; LY = life-years; MTX = methotrexate; MMF = mycophenolate mofetil; OS = overall survival; QALYs = quality-adjusted life-years; SIR = sirolimus; SR = steroid refractory.
Conclusions
The CADTH clinical review found that, compared with best available therapy (BAT), ruxolitinib (Jakavi) demonstrates a statistically significant improvement in overall response rates (ORRs) at day 28. The improvements in ORR of the magnitude observed in the REACH 2 trial were considered clinically meaningful by the clinical experts consulted by CADTH. Other secondary outcomes, including duration of response, best overall response, and failure-free survival, were supportive of the observed ORR at day 28. The open-label design of the trial and reliance on local investigators’ assessments of trial outcomes may have introduced a bias that is difficult to quantify. The actual degree of overall survival (OS) benefit for ruxolitinib is uncertain, given potential bias related to the crossover of patients in the BAT group to the ruxolitinib group and the limited follow-up time (7.34 months and 3.81 months of OS follow-up time for ruxolitinib and BAT, respectively).
Given the high degree of uncertainty concerning the post hoc analysis used to populate model parameters and the inappropriateness of the sponsor’s model structure, CADTH was unable to derive a base-case analysis. The exploratory reanalysis performed by CADTH uses more appropriate assumptions, but these estimates remain highly uncertain because the majority of the parameters were based on the post hoc analysis (including duration of response, duration of treatment, resource use, and drug dosing), the model structure did not fully capture the health condition, and the distribution of BAT varied among clinicians. Therefore, the magnitude of benefit for ruxolitinib in the CADTH exploratory reanalysis may be overestimated.
CADTH undertook exploratory reanalyses to address limitations related to the model not capturing the long-term outcomes of steroid-refractory (SR)-aGvHD, leading to uncertain long-term efficacy; use of an approach to OS that aligned with the REACH 2 trial rather than the post hoc analysis; aligning the dosing for ruxolitinib and BAT treatments with the literature; modelling duration of treatment using individual BAT Kaplan-Meier (KM) curves; and aligning the distribution of BAT treatments with clinical expert expectations. Based on the CADTH exploratory reanalysis, conducted over a 1-year time horizon, the incremental cost-effectiveness ratio (ICER) for ruxolitinib, compared with BAT, was estimated to be $21,057 per quality-adjusted life-year (QALY) gained, and ruxolitinib was cost-effective in 52% of iterations at a willingness-to-pay threshold of $50,000 per QALY. When the price of ruxolitinib was reduced by 10% and 25%, the probability of ruxolitinib being cost-effective at a $50,000 per QALY threshold increased to 57% and 62%, respectively. Given the uncertainty of the results and the presence of other limitations that could not be addressed (e.g., the sponsor’s uncertain model structure and inputs derived from the post hoc analysis), price reductions are likely required.
Of note, results from CADTH’s exploratory reanalysis reflect the cost-effectiveness of ruxolitinib over a 1-year time horizon; because of the limitations identified in the sponsor’s modelling approach, the cost-effectiveness of ruxolitinib in aGvHD beyond 1 year is unknown. In the acute indication, the median duration of treatment with ruxolitinib was approximately 11 weeks, and 2% of patients remained on treatment for 52 weeks. The median duration of treatment varied for the individual BATs, but no patients remained on treatment for more than 27 weeks. Another source of uncertainty is the cost-effectiveness of ruxolitinib, compared with individual BATs. As the distribution of BATs reimbursed in jurisdictions may differ, and because the cost-effectiveness of ruxolitinib is dependent on the cost of the comparator treatments, cost-effectiveness conclusions will differ if a different distribution of BATs is used. Finally, the modelled population did not fully align with the proposed Health Canada indication, the sponsor’s reimbursement request did not align with available evidence, and the modelled population, based on REACH 2 data, did not reflect dosing in the product monograph, which was based on REACH 1. The model is specific to aGvHD, therefore based on the submitted evidence in a SR population, the cost-effectiveness of chronic graft-versus-host disease (cGvHD), subgroups of aGvHD, and patients with an inadequate response to systemic therapies is unknown. Also, because the model used efficacy and dosing from REACH 2 (10 mg twice daily), the cost-effectiveness of ruxolitinib at the product-monograph dose (5 mg twice daily) is unknown.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process. Note that patient, registered-clinician, and drug plan input was submitted for both aGvHD and cGvHD indications; therefore, the information here pertains to both indications and may not be specific to aGvHD.
Eight groups collaborated on a single patient-input submission: Lymphoma Canada, Lymphoma and Leukemia Society of Canada, CLL Canada, Myeloma Canada, Aplastic Anemia & Myelodysplasia Association of Canada, Canadian MPN Research Foundation, CML Network, and Cell Therapy Transplant Canada. Information was collected using an anonymous online survey for patients who developed graft-versus-host disease (GvHD) after allogenic stem cell transplantation. Of the 68 respondents, 46 were from Canada, 16 of whom had experience with ruxolitinib. Patients with GvHD reported a wide profile of symptoms associated with the disease, with the most common symptoms being skin problems, dry mouth with or without mouth ulcers, dry eyes, and mobility and joint issues. Skin problems were found to have the most significant impact on patient’s quality of life, followed by dry eyes and mouth issues. Respondents had experience with a range of treatments for GvHD, including steroids, cyclosporine, tacrolimus, tyrosine kinase inhibitors, extracorporeal photopheresis (ECP), mycophenolate mofetil (MMF), methotrexate, monoclonal antibodies, and azathioprine. Patients receiving treatment at the time of the survey noted side effects that affected their quality of life, with the most common severe side effects of current treatments being tiredness, difficulty sleeping, and eye problems. In terms of hopes for a new treatment, patients identified improved survival time as the most important outcome, followed by improved relief of GvHD symptoms. Patients also noted that an improvement in quality of life was important. Among patients with experience using ruxolitinib, 24% reported that the treatment improved all of their GvHD symptoms, whereas 71% noted that it helped with some of their symptoms. In terms of treatment response, 50% of patients experienced a response (11% a complete response and 39% a partial response), and 6% did not experience a response. The side effects of ruxolitinib were reported to be tolerable or very tolerable by 67% of patients. The most common serious side effect was infection, followed by low platelet and/or red blood cell counts.
Registered-clinician input was received from 2 groups: Ontario Health Complex Malignant Hematology and Cell Therapy Transplant Canada. The clinicians noted that there are no Health Canada–indicated treatments for SR-cGvHD, although several treatments (ECP, MMF, sirolimus, everolimus, imatinib, and rituximab) can be used off-label (they did not specify the treatments used specifically for cGvHD). They also noted the need for therapies that are widely available and that improve survival, quality of life, and reduce corticosteroid use and health care costs. If ruxolitinib became available, it would likely become the treatment of choice for SR-cGvHD, and be used as the first-line treatment for patient’s who become steroid-resistant. The clinicians did not specify which groups would be best suited for treatment with ruxolitinib, but did note that patients with significant existing thrombocytopenia may be a challenge to treat.
Input from drug plans noted that ibrutinib, a comparator to ruxolitinib, does have a Health Canada indication for the treatment of adults with SR-cGvHD, but it is not publicly reimbursed. The input also noted that ruxolitinib is self-administered, so it has important patient and health care benefits over treatments that require administration in hospitals or infusion clinics.
Several of these concerns were addressed in the sponsor’s model:
adverse events (AEs), cost, and impact on quality of life were accounted for
OS by responder status was considered
differences in quality of life by responder status were captured
administration costs for relevant BAT therapies were included.
CADTH was unable to address the following concerns raised from stakeholder input:
reductions in corticosteroid use were not used as a model outcome.
Economic Review
The current review is for ruxolitinib for patients with SR-aGvHD.
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
The sponsor submitted a cost-utility analysis of ruxolitinib, compared with BAT, for patients with SR-aGvHD, which aligned with part of the proposed Health Canada indication for ruxolitinib.
Ruxolitinib is available as a 5 mg, 10 mg, 15 mg, and 30 mg tablets. The recommended dose of ruxolitinib is 10 mg twice daily.1 At the sponsor’s submitted price of $87.38 per 10 mg tablet, the cost of ruxolitinib therapy would be $63,786 for a full year of therapy. BAT consisted of multiple comparators, the distribution of which was informed by the sponsor’s survey of clinicians in Canada (Table 11.) The sponsor estimated the mean dose for each BAT treatment by week, using data from REACH 2. Then a single cost for BAT was derived by weighting weekly treatment costs by the distribution of BAT treatments. This resulted in an annual BAT cost of $50,338 for the first year of treatment, and $52,797 for subsequent years. This difference across years is because the dose for subsequent years was based on the doses used at the end of the first year of treatment, not because some treatments were assumed to be time-limited.
The clinical outcomes of interest were QALYs and life-years. The economic analysis was undertaken over a lifetime (15-year) time horizon from the perspective of a Canadian public health care payer. Discounting (1.5% per annum) was applied to both costs and outcomes.
Model Structure
A semi-Markov model with 7 health states was submitted by the sponsor, with weekly cycle lengths (Figure 1). All patients begin in the disease baseline health state, which consists of 4 tunnel states that capture mortality and treatment discontinuation that occurs before the response-assessment time point. Transitions between these tunnel states is unidirectional until the response-assessment time point at day 28. After 28 days of treatment in model cycle 4, efficacy is assessed and patients are stratified into responder or nonresponder health states. In the following model cycles, patients in the responder health state can maintain their initial response and remain in that state, lose their initial response and transition to the nonresponder health state, or transition to the death health state. Patients in the nonresponder state can either remain in that state or transition to death; transitions from the nonresponder state to the responder state are not possible.
Model Inputs
The baseline population characteristics of the model and the clinical efficacy parameters were characterized according to REACH 2, a phase III, randomized, open-label trial comparing the efficacy and safety of oral ruxolitinib with the investigator’s choice of BAT in patients 12 years and older who developed SR-aGvHD after receiving an allogenic stem cell transplant.2 The sponsor assumed that the baseline characteristics of the REACH 2 population (mean age = 49.5 years; proportion of males = 59.2%; mean weight = 66.9 kg; body surface area = 1.8 m2) reflected the Canadian population.2
To populate model outcomes based on treatment response, the sponsor conducted a post hoc analysis on REACH 2 data, which informed the cumulative mortality, OS, duration of response, duration on treatment, health state utility values, weekly dosing, and resource use.3 During the first 28 days of the model, patients who remain alive automatically transition to the subsequent tunnel state. Transitions to death during the first 28 days were informed by cumulative mortality from the sponsor’s post hoc analysis (see Table 12). At the response-assessment time point at day 28, patient assignment to the responder or nonresponder health states were informed by the ORR by treatment at day 28 in the REACH 2 trial.2 The sponsor’s post hoc analysis was used to inform the proportion of patients that remain in the overall responder health state each cycle by treatment (Table 13).3 Duration of response for overall responders was defined as the time from first response to progression or the addition of a new systemic therapy to treat aGvHD.4 Parametric survival functions were fit to the duration-of-response KM curves (Figure 2) to extrapolate duration of response by treatment over the model time horizon.3 The exponential and log-normal curves were selected for ruxolitinib and BAT on the basis of statistical fit, visual fit with REACH 2 KM data, and clinical expectations for duration of response (see Figure 3 for duration-of-response extrapolations).4
Transitions to death from the overall responder and nonresponder health states were informed by the sponsor’s post hoc analysis. The sponsor conducted a time-to-event analysis of OS to estimate OS by responder status from the response-assessment time point at day 28 (see Figure 4 for KM OS curves). Data for ruxolitinib and BAT were combined to generate OS response curves; therefore, in the model, OS for responders is the same regardless of initial treatment. A landmark approach was taken by subtracting 28 days from each patient’s OS time, and patients who had died by day 28 were removed from the analysis.3 Parametric survival functions were then fit to KM OS curves to extrapolate OS for responders and nonresponders for the model time horizon, using an individual-fit approach in which OS was extrapolated separately for responders and nonresponders. The sponsor selected the Weibull function to extrapolate OS for responders and nonresponders on the basis of statistical fit and because it predicted clinically relevant survival (Figure 6).4 General population mortality was used in the model to ensure that OS for all patients could not be better than that for the general population in a given model cycle.
AEs were incorporated in the model as a 1-time average cost and disutility, and the percentage of patients experiencing an AE was based on data from the REACH 2 trial.2 Two main complications of aGvHD — malignancy relapse or progression of underlying disease, and transformation to chronic GvHD — were included as additional events; the proportion of patients receiving each treatment who experienced these complications was derived from REACH 2.2 Cytomegalovirus infection was included as an AE in the model, along with AEs of grade 3 or higher that occurred in at least 10% of patients in either the ruxolitinib or BAT treatment arm in REACH 2.2,4,5 AEs were incorporated in the model as a 1-time cost and disutility, and were based on the time of event occurrence from the initiation of treatment, based on cumulative incidence data from REACH 2.2,5 When the time of event occurrence was unknown, it was assumed to occur halfway to the response assessment (14 days).4 The disutility and cost of each event were assumed to occur only once in the model time horizon and to last for the full cycle in which the event occurs.
Health state utility values were derived from a post hoc analysis of data from the REACH 2 EQ-5D questionnaire. Three sets of health utilities were applied at different time points: disease baseline (applies from disease baseline up to day 28); day 28 (applies from cycle 4 to cycle 11 for each response health state); and week 12 and onward (for each response health state). Disutilities for AEs were sourced from the literature.4 The duration of the disutility was sourced from the Ontario Case Costing Initiative.6
Costs in the model included treatment-acquisition costs, disease management, and complications costs. Drug costs for ruxolitinib and BAT therapies were sourced from public formularies.7,8 The cost for ECP was derived from a report by the Ontario Ministry of Health and Long-Term Care.9 The cost of antithymocyte globulin (ATG) was informed by clinician estimate.4 The sponsor conducted a post hoc analysis of REACH 2 data to determine the mean daily dose on a weekly basis for ruxolitinib and each BAT treatment to capture changes, such as dose tapering. A weekly recurring dose was used for all treatments beyond the first year of the model time horizon. Duration-of-treatment data from a post hoc analysis of REACH 2 were used to adjust drug-acquisition costs over the model time horizon.3 In the sponsor’s base case, KM data (Figure 7) for the probability of remaining on treatment by comparator were derived and parametric survival curves were fit to estimate the duration of treatment, regardless of response status. Administration costs were applied for ATG and infliximab only; these were the only treatments in the model administered intravenously and they included the cost of a physician consultation,10 the cost per hour of administration,11 and the duration of administration.12,13 Subsequent treatment costs were applied to patients in the nonresponder health state and were assumed to be the same regardless of initial treatment. The weekly subsequent therapy was assumed to be the weekly cost of BAT in the first year, weighted by the proportion of patients receiving each BAT and the duration of individual BATs administered in REACH 2.
Disease-management costs were applied by treatment arm for the first 28 days of the model, and then by responder status for the remainder of the time horizon.4 For the first 28 days, the proportion of patients hospitalized and their mean length of stay was derived from REACH 2.2,3 Beyond the assessment time point, disease-management costs were primarily based on hospital readmissions by responder status, which were derived from the sponsor’s post hoc analysis of the REACH 2 data.3 Disease-management costs also included specialist visits, with frequency informed by clinical expert feedback (once weekly during the baseline period for nonresponders; once every 1.5 months for overall responders).4 The cost per day for hospital admissions was informed by a survey of clinicians in Canada conducted by the sponsor; outpatient visit costs were sourced from the Ontario Schedule of Benefits.10 Costs associated with AEs or disease-complication events were sourced from the Ontario Case Costing Initiative, and events were assumed to be treated in the inpatient setting.6
Summary of Sponsor’s Economic Evaluation Results
All analyses were run probabilistically (3,000 iterations for the base-case and scenario analyses). The deterministic and probabilistic results were similar. The probabilistic findings are presented here.
Base-Case Results
Ruxolitinib was associated with a QALY gain of 0.15 at a cost that was $39,934 lower than BAT, resulting in ruxolitinib dominating (i.e., being more effective and less costly) BAT (Table 1). Ruxolitinib was dominant in 93% of iterations. At a willingness-to-pay threshold of $50,00 per QALY gained, there was a 99.87% probability of ruxolitinib being cost-effective compared to BAT.
The majority of cost-savings for ruxolitinib ($56,242) were associated with lower resource-use costs among patients receiving ruxolitinib in the nonresponder health state. The second-largest source of cost-savings ($5,788) came from lower subsequent therapy costs for nonresponders receiving ruxolitinib versus those receiving BAT. Initial treatment-acquisition costs were $3,809 more for ruxolitinib than for BAT. Additional event costs were $4,132 higher for ruxolitinib than for BAT, because patients receiving ruxolitinib were more likely to experience the additional event of progressing from aGvHD to cGvHD. The majority of the QALY gain for ruxolitinib was accrued in the overall responder health state (0.42).
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total costs ($) | Incremental costs ($) | Total LYs | Incremental LYs | Total QALYs | Incremental QALYs | ICER vs. reference ($/QALY) |
|---|---|---|---|---|---|---|---|
BAT | 212,141 | Reference | 1.76 | Reference | 0.92 | Reference | Reference |
Ruxolitinib | 172,207 | –39,934 | 1.93 | 0.17 | 1.07 | 0.15 | Dominant |
BAT = best available therapy; ICER = incremental cost-effectiveness ratio; LYs = life-years; QALYs = quality-adjusted life-years; vs. = versus.
Note: The submitted analysis is based on publicly available prices of the comparator treatments.
Source: Sponsor’s pharmacoeconomic submission.4
Sensitivity and Scenario Analyses Results
The sponsor conducted several scenario analyses to examine uncertainty. Ruxolitinib remained more effective and cost-saving (i.e., dominant) in all scenario analyses. The sponsor’s alternative model structure, which compared complete responders with partial responders and nonresponders (as opposed to their base case, which compared overall responders with nonresponders), led to a slightly greater incremental QALY gain (0.18) and more cost-savings ($43,249) than their base-case analysis. Additionally, using the sponsor’s extrapolation choices for ruxolitinib and BAT OS, the exploration of OS by treatment arm rather than responder status led to a doubling of the QALY gain for ruxolitinib (0.35) and less cost-savings ($14,735) than their base case.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the economic analysis:
The pharmacoeconomic analysis does not reflect ruxolitinib’s product monograph recommended dose. According to information in the product monograph, confidential information in the Health Canada Review Report, and Clarifax supplemented with information provided by the sponsor to CADTH, Health Canada considers REACH 1 to be the pivotal study and safety data from the REACH 2 trial to be supportive evidence for the proposed aGvHD indication.14,15 Additional details explaining the rationale of Health Canada are provided in the CADTH clinical review report. REACH 1 was an open-label phase II, noncomparative clinical study.16 The efficacy parameters informing the pharmacoeconomic analysis were based on the phase III, randomized, open-label REACH 2 trial.2 Because REACH 1 was used by Health Canada for the aGvHD indication, the ruxolitinib dose in the product monograph is aligned with REACH 1’s dosing (5 mg twice daily, with the possibility of increasing the dose to 10 mg twice daily after 3 days).16 This differs from the ruxolitinib dosing used in REACH 2 (10 mg twice daily), and therefore differs from dosing used in the pharmacoeconomic model. As CADTH was not able to incorporate efficacy data from REACH 1 into the pharmacoeconomic analysis, CADTH was unable to use REACH 1 dosing.
CADTH was unable to address this limitation. As such, the cost-effectiveness of a 5 mg twice daily dose for patients with aGvHD is unknown. Cost-effectiveness estimates provided for ruxolitinib are based on a 10 mg twice daily dose, which is different than the dose in the product monograph.
Given the limitations of the REACH 2 data, interpretation of results from the economic model comes with additional uncertainty.
The model’s efficacy parameters are primarily estimated from an uncertain post hoc analysis of REACH 2 trial data. The sponsor conducted post hoc analyses on REACH 2 data to fit the trial data to their model structure, which was based on response, not the treatment arm. The sponsor’s post hoc analysis largely stratified patients based on an outcome (response), and then compared what happened to OS, health-related quality of life, and resource use in responders and nonresponders. Overall, the CADTH review team concluded that results from post hoc analyses are considered exploratory and hypotheses-generating only. Because of the lack of formal inferential statistical testing, the ability to interpret results is significantly limited. CADTH requested the sponsor’s full statistical analysis plan so it could conduct a critical appraisal of the post hoc analyses, but it was not provided. The CADTH review team was unable to conduct a rigorous evaluation of the conduct and reporting of the post hoc analyses, as only a high-level summary of methods was provided by the sponsor.
CADTH was unable to fully address this limitation. Because the data used in the pharmacoeconomic model were different across most parameters than that critically appraised in the CADTH clinical review report, CADTH was unable to assess the validity of the sponsor’s post hoc analysis. As such, the economic analysis is associated with significant uncertainty and is likely biased; therefore, CADTH was unable to conduct a base-case analysis. Instead, a CADTH exploratory reanalysis using more appropriate assumptions was conducted.
Where possible, CADTH opted to parameterize the exploratory reanalysis model using REACH 2 trial data by treatment arm rather than data from the post hoc analysis. This was possible for OS (not used in the sponsor’s base case; the limitation related to the modelling of OS, which did not align with the REACH 2 trial, is discussed elsewhere) and duration of treatment (used in the sponsor’s base case).
The model structure does not fully capture the health condition. To estimate the cost-effectiveness of ruxolitinib compared to BAT, the sponsor used tunnel health states to model what happens to patients in terms of mortality before day 28, followed by responder and nonresponder health states at day 28 onward, which was representative of the point at which response was assessed in the REACH 2 trial. Although responders can lose their response and transition to the nonresponse health state, nonresponders who receive subsequent therapies cannot transition to the responder health state; flow is unidirectional from the responder to the nonresponder health state. This was deemed to be inappropriate by the clinical experts consulted by CADTH for this review. According to clinical experts consulted by CADTH, treatment of aGvHD requires daily monitoring, and patients are expected to respond to treatment in 5 to 10 days. If they do not respond, other treatments will be trialed to see if they elicit a response. Therefore, although patients may not achieve a response on ruxolitinib or a given BAT comparator, they could achieve a response to a subsequent BAT treatment. In this case, they would become responders and accrue the same benefits as initial responders, such as increased quality of life and decreased disease-management costs. Although the sponsor assumes that subsequent therapy costs are accrued by patients in the nonresponder health state, the outcomes of these subsequent therapies are not modelled.
Second, the sponsor’s model was based on response status, which does not accurately reflect the underlying health condition of patients with SR-aGvHD. According to clinical experts consulted by CADTH for this review, aGvHD can affect any organ system, but the most common effects are related to the skin and subcutaneous tissues, liver abnormalities, and gastrointestinal issues, like nausea, vomiting, and diarrhea. The influence of ruxolitinib and comparators on these outcomes were not explicitly modelled. The assumption that all outcomes for patients with aGvHD can be captured by response status is an oversimplification that does not accurately reflect the complexity of the disease, as it assumes that all responders will experience the same costs and consequences and that all nonresponders will have the same costs and consequences. This is because patients who survive with aGvHD do not remain in the same aGvHD responder health state for the remainder of their lives. According to clinical experts, aGvHD lasts up to 100 days after graft infusion. After day 100, patients would no longer be considered to have aGvHD; instead, they would have late acute GvHD (which is defined as having acute-like GvHD after day 100 without having cGvHD) or overlap syndrome, which is the appearance of classic acute and chronic GvHD symptoms together.17 Patients could then also have cGvHD, which is typically characterized by scarring (aGvHD is characterized by inflammation). Parsing these different types of GvHD in patients who remain alive is important to accurately capture the long-term outcomes of aGvHD. For example, the National Institutes of Health consensus criteria suggests that patients with late acute disease have higher mortality rates than those with cGvHD.17 In addition, patients with aGvHD have competing mortality risks. To elaborate, patients could die from aGvHD- or cGvHD-related illnesses (due to the disease itself or complications related to the immunosuppression required to manage the disease, such as infection) or from a relapse of the underlying illness for which they required stem cell transplantation. These mortality rates would differ, depending on where in the disease trajectory a patient is (i.e., acute, late acute, overlap, chronic, or responding with mostly resolved symptoms). Although the model included the risks of transition to cGvHD, malignancy relapse, and progression, such events were captured as 1-time events that incurred a cost and a disutility, and did not adequately capture the appropriate health outcomes, including differential mortality rates, associated with these health states. Given the range of events that could occur if a patient survives aGvHD, the assumption that all relevant outcomes could be captured by the responder and nonresponder health states is an oversimplification.
The clinical experts consulted by CADTH noted that 1 of the most important outcomes for patients with SR-aGvHD is the number of immunosuppressants required. Specifically, experts indicated that outcomes for patients on fewer treatments and for those able to discontinue corticosteroid use would be different than outcomes for patients on higher doses of corticosteroids, despite both groups being considered responders. This aligned with registered-clinician input, which emphasized that a reduction in steroid use was an important outcome in aGvHD. However, neither the number of concurrent treatments nor the discontinuation of steroids was reflected as a cost or health outcome in the model.
CADTH was unable to fully address this limitation. As there are uncaptured costs and consequences associated with both the responder and nonresponder health states, the direction and magnitude of this limitation on cost-effectiveness results is uncertain. Both the sponsor and clinical experts explained that there is no long-term evidence regarding duration of response or duration of treatment for ruxolitinib, and noted the high degree of variability in duration of response for BAT. Because of the inappropriate model structure (designed to capture only aGvHD and only response to treatment achieved in the first 28 days) and uncertainty about the way long-term outcomes for responders and nonresponders were modelled, CADTH used a 1-year time horizon in its exploratory reanalyses.
To address uncertainty in the sponsor’s duration-of-response approach and to more closely match the trial data available (given that the KM data for duration of response for ruxolitinib and BAT is greater than 1 year), the CADTH reanalysis modelled duration of response based on KM data, rather than fitted survival curves.
The proposed Health Canada indication is not fully modelled, and the sponsor’s reimbursement request does not align with available evidence. As the model was based on response to ruxolitinib in an aGvHD population and only incorporated cGvHD as a 1-time cost and disutility, this submission does not reflect the full proposed Health Canada indication for GvHD; rather, the sponsor’s submitted model reflects a subgroup of the full Health Canada indication (i.e., the aGvHD population). Further, in the aGvHD subgroup, there are additional stages of the condition that may not be properly captured by the sponsor’s model structure (e.g., de novo late acute, recurrent late acute, and chronic overlap, as illustrated in Figure 8).
In addition, the sponsor’s reimbursement request for the CADTH review was for GvHD in patients 12 years and older with inadequate responses to corticosteroids and other systemic therapies. According to the CADTH clinical review report, there are insufficient data to describe how the requested reimbursement criteria match the patient population in the REACH 2 trial, as the sponsor did not present data on the number of patients in the REACH 2 trial who had an inadequate response to corticosteroids, an inadequate response to other systemic therapies, or an inadequate response to both corticosteroids and other systemic therapies. As the efficacy of ruxolitinib in patients who have inadequate responses to other systemic therapies is unknown, so too is the cost-effectiveness of ruxolitinib in this patient population. As a REACH 2 eligibility criteria specified that patients must have an inadequate response to corticosteroids, the results of the economic analyses conducted by the sponsor and by CADTH reflect the cost-effectiveness of ruxolitinib in a SR population.
CADTH was unable to address this limitation. Neither the sponsor’s submitted model nor the CADTH reanalysis reflect the cost-effectiveness of ruxolitinib in the full Health Canada indication or the sponsor’s reimbursement request; instead, results only reflect the aGvHD population with an inadequate response to corticosteroids.
The approach to modelling OS did not align with the REACH 2 trial. In the model, mortality before the response-assessment time point (day 28) was based on weekly cumulative mortality rates in patients receiving ruxolitinib and BAT.4 After day 28, survival for patients in all treatment groups was based on responder status. OS by response was derived from the sponsor’s post hoc analysis, which is highly uncertain, as outlined previously. Although the post hoc KM OS curves initially demonstrate higher OS for responders than nonresponders, at around |||||||| months, the OS benefit for responders diminishes, and then at approximately |||||||| months, a small benefit for responders remains but plateaus (see Figure 4). Experts reported that this makes clinical sense because when patients survive the first part of their aGvHD, mortality is more likely to be related to comorbidities associated with GvHD (such as infections due to immunosuppression) or to relapse of the underlying condition. As relapse is largely expected to occur independent of response, experts reported that they would expect to see similar mortality rates in responders and nonresponders over time.
When looking at OS from the full analysis set from the REACH 2 clinical study report, despite ruxolitinib displaying a an improved ORR than BAT, OS from the KM curves for ruxolitinib and BAT were similar for the first 3 months or so, after which ruxolitinib demonstrated better OS than BAT, but the curves nearly merged and then plateaued at approximately 18 months (see Figure 5). Clinical experts reported that this finding is likely due to the competing risk that GvHD patients face, which is relapse of their underlying disease. According to clinical experts, the main driver of death in the medium to long-term is expected to be relapse of the underlying disease. Therefore, OS differences between responders and nonresponders and between patients in the ruxolitinib and BAT groups are expected in the short-term only. As OS by treatment arm reflects trial results and aligns with clinical expert opinion, OS was extrapolated by treatment arm. This approach circumvents the issue of using data from the sponsor’s uncertain post hoc analyses.
Implementation of dosing for ruxolitinib and BAT was uncertain. To calculate ruxolitinib costs in the model, the sponsor conducted a post hoc analysis using individual patient data from the REACH 2 trial to estimate the mean and median daily doses of treatment received on a weekly basis (for example, the mean daily dose of ruxolitinib in week 1, then the mean daily dose of ruxolitinib in week 2, and so on).3 This was also done for each initial BAT treatment. In the base case, the sponsor used the mean daily dose for a given week to calculate ruxolitinib and BAT costs. According to the sponsor’s post hoc analysis, this was done to capture changes in dosing over time, including dose reductions, tapering, and up-dosing.
CADTH was unable to validate the sponsor’s approach to estimating median and mean daily doses on a weekly basis for both comparators, as this was derived in their post hoc analysis and not presented in the REACH 2 clinical study report. CADTH observed that the mean daily dose for ruxolitinib was consistently lower than the dose provided in the product monograph of 10 mg twice daily (see Appendix 1). This was deemed to be inappropriate by CADTH, as the treatment-acquisition costs for ruxolitinib could be vastly underestimated using the sponsor’s mean dosing approach derived from their post hoc analysis.
CADTH also noted discrepancies in dosing for BAT treatments between the sponsor’s analysis and the literature. For example, median etanercept dosing ranged from |||||||| mg to |||||||| mg weekly in the sponsor’s analysis, rather than the 25 mg twice-weekly dose found in the literature.18
Duration of treatment in the sponsor’s model was implemented by treatment arm, by fitting parametric survival extrapolations to ruxolitinib and BAT duration-of-treatment KMs from the sponsor’s post hoc analysis.4,19 The sponsor also included an alternative approach to extrapolate treatment duration by individual BAT. When looking at the sponsor’s duration-of-treatment KMs for individual treatments, CADTH observed a large range in the total duration of treatments among BAT comparators. For example, all patients had discontinued ATG treatment by approximately |||||||| weeks, whereas some patients remained on MMF for up to approximately |||||||| weeks. This variation among BAT comparators is clinically expected, but the sponsor’s approach of extrapolating treatment duration by the overall BAT KM curve resulted in all patients receiving BAT accruing some BAT costs until 36 weeks, thereby overestimating overall BAT costs.
In the CADTH exploratory reanalysis, dosing for ruxolitinib reflected the labelled dose suggested in the draft product monograph for the first 8 weeks of treatment, and then the dose was tapered according to the sponsor’s assumptions using median ruxolitinib dosing from the REACH 2 post hoc analysis.
CADTH adjusted the dose and frequency of administration for the following BAT treatments in its exploratory reanalysis:
Etanercept was initiated at 25 mg twice weekly for 4 weeks, then decreased to 25 mg once weekly for the subsequent 4 weeks.18 The people in the model who remained on etanercept for more than 8 weeks continued to receive 25 mg once weekly while on etanercept.
Infliximab was initiated at 10 mg/kg per week for 8 weeks (it was assumed that those remaining on infliximab after week 4 would repeat another 4-week treatment course), then tapered according to the sponsor’s assumptions.20
The approach to estimating duration of treatment in the CADTH exploratory reanalysis was based on KM curves for individual BAT treatments.
The distribution of BAT therapies is uncertain. To estimate BAT costs, the sponsor derived the distribution of patients across possible treatments in Canadian clinical practice from a survey of 10 clinicians in Canada (see Table 11).21 Detailed results by respondent regarding the distribution of BAT treatments used demonstrate the variability of treatments used in Canada. For example, most respondents reported no or very little use of ATG, but 1 respondent reported use in 25% of their patient population. This highlights the variation in practice and consequent uncertainty in the estimation of BAT costs. When validating the sponsor’s distribution of BAT, the clinical experts consulted by CADTH for this review noted that some therapies in the list were rarely used in their practice, whereas others were used often. CADTH surveyed the clinical experts consulted by CADTH on BAT distributions and similarly noted variation across respondents, highlighting the uncertainty regarding current practice and, consequently, the cost-effectiveness of ruxolitinib compared to what is currently being used for treatment. This adds significant uncertainty to the analysis, as a weighted BAT distribution will not be reflective of a given jurisdiction’s coverage, meaning that the cost-effectiveness of ruxolitinib compared with BAT will vary, depending on the treatments a jurisdiction uses.
In CADTH exploratory reanalyses, the distribution of BAT was revised based on the average responses received from the clinical experts consulted by CADTH for this review.
As a scenario analysis, CADTH explored the cost-effectiveness of ruxolitinib compared with each individual BAT.
The implementation of subsequent treatments is inappropriate. In the sponsor’s model, nonresponders accrue the costs of subsequent treatments for the entire model time horizon. This is because subsequent therapies are only incorporated as an additional cost for nonresponders; the outcomes of subsequent therapies (i.e., response) are not considered. The model structure does not permit nonresponders to become responders, even though a proportion of nonresponders who receive subsequent therapy will respond and discontinue immunosuppressant therapy.
In addition, although experts consulted by CADTH noted that, ideally, responders will not require subsequent therapy, in clinical practice, responders may still require subsequent immunotherapy. However, this has not been modelled, as responders are not assigned subsequent therapy costs in the sponsor’s model.
Given the limitations of the sponsor’s model structure, CADTH was unable to properly incorporate the previously noted consequences of subsequent therapies. As nonresponders accrue the costs of subsequent therapy but derive no benefit from additional treatment, in a scenario analysis, CADTH explored setting subsequent therapy costs for nonresponders to $0.
Additional limitations were identified but not considered key limitations. These limitations are outlined subsequently.
The approach to estimating resource use in the baseline disease period is uncertain. The sponsor estimated resource use by treatment arm from the beginning of the model time horizon until week 24 using individual patient data from REACH 2.2,3 These initial estimates may be biased, as the sponsor included health care encounters that began before the start of study treatment if discharge occurred during the study, with length of stay adjusted to start at day 0 of the study.3,4 CADTH was unable to validate resource-use estimates in the sponsor’s base case, including the frequency of admission for patients receiving ruxolitinib and BAT. A differential frequency of admission by treatment arm when baseline admissions are included is inappropriate, as it does not account for where ruxolitinib and BAT patients may have started receiving their treatment. For example, at the start of study treatment, more patients receiving ruxolitinib than those receiving BAT were admitted to the transplant unit, but more patients receiving BAT were admitted to a general ward. The initial hospitalization rates translated into the disease baseline frequency estimates. This is inappropriate, as these differential frequencies do not reflect a treatment effect, but rather where in the hospital patients were admitted when they began treatment.
In addition, although length of stay for events occurring during the disease baseline period were adjusted to begin at treatment initiation for those who were hospitalized before treatment start date, the length of stay during the baseline period was longer than the baseline period itself for the transplant unit and general ward. Also, although the mean length of stay for BAT was higher than for ruxolitinib (79.27 days versus 55.17 days), the median length of stay was shorter (58 days versus 61 days), indicating that the mean length of stay is being inflated by outliers with long length of stays. In fact, the maximum length of stay for BAT in the general ward was 241 days, which extends beyond the response-assessment time point.
Because of uncertainty in the way the sponsor estimated resource use in the baseline disease period, and because CADTH was unable to validate their approach, the frequency and duration of resource use were assumed to be equal across comparators in the exploratory reanalysis until day 28.
Treatment administration costs are overestimated. The sponsor’s treatment administration costs accounted for an hourly rate of administration and assumed a physician visit for every administration. Physician monitoring visits were captured as part of resource use. As it is unlikely that physician visits are required for each IV administration, and because the hourly infusion rate used by the sponsor incorporated nurse time, CADTH deemed the additional physician cost to be inappropriate.
Physician visits as part of the administration cost have been removed in the CADTH exploratory reanalyses.
Rates of underlying malignancy relapse are not expected to be related to immunosuppressant therapies. As an additional event, the sponsor considered rates of malignancy relapse and progression for the underlying conditions for which patients underwent stem cell transplantation and which led to their SR-aGvHD. As ruxolitinib and BAT are not expected to augment the disease course for patient’s underlying cancer, and because clinical experts consulted for this review reported that relapse is independent of response, CADTH deemed the implementation of differential rates of disease progression to be inappropriate.
Relapse recurrence rates for BAT were set to be equal to those for ruxolitinib in the CADTH exploratory reanalysis.
The cost of ATG is uncertain. The sponsor based the unit price of ATG on expert opinion collected from a sponsor-commissioned survey of 10 clinicians. CADTH obtained the wholesale price of ATG dated 2014 using the IQVIA Delta PA database,22 which is much lower than the price calculated by the sponsor using expert opinion. A higher ATG cost favours ruxolitinib, as it raises treatment costs for the BAT comparator.
CADTH used wholesale pricing from IQVIA for ATG in the exploratory reanalysis.
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (See Table 4).
CADTH Reanalyses of the Economic Evaluation
Base-Case Results
CADTH reanalyses addressed several limitations in the economic model, which are summarized in Table 5. CADTH was unable to fully address limitations related to uncertainty in the sponsor’s post hoc analysis, which was used to populate the majority of model parameters, and concerns related to the model structure not adequately capturing the complexity of SR-aGvHD. As such, the changes below reflect a CADTH exploratory reanalysis, rather than a base-case estimate of the cost-effectiveness for ruxolitinib compared with BAT. The CADTH exploratory reanalysis was derived by making changes to model parameter values and assumptions, in consultation with clinical experts.
The results of CADTH’s stepped analysis are presented in Table 6. CADTH’s exploratory reanalysis demonstrates that, compared with BAT, ruxolitinib yields 0.06 additional QALYs at an incremental cost of $1,279, leading to an ICER of $21,057 per QALY gained (Table 6) over a 1-year time horizon. At a willingness-to-pay threshold of $50,000 per QALY, there was a 52% probability of ruxolitinib being cost-effective. CADTH observed a large difference in incremental costs between the probabilistic and deterministic analyses, which led to differences in their respective ICERs. Upon investigation, this discrepancy is noted to largely be driven by the uncertainty around overall response and nonresponse rates for ruxolitinib and BAT. As a scenario, when these response rates were removed from the probabilistic analysis, incremental costs and the ICER more closely approximated the deterministic analysis (incremental costs = $623; ICER = $10,218 per QALY, compared to BAT) (Table 15).
Table 4: Key Assumptions of the Submitted Economic Evaluation (Not Noted as Limitations to the Submission)
Sponsor’s key assumption | CADTH comment |
|---|---|
The efficacy of BAT based on the distribution in REACH 2 is equivalent to the efficacy of BAT based on the distribution of treatments used in Canada | Appropriate, according to clinical experts consulted by CADTH |
The costs of subsequent treatments for nonresponders were assumed to be equal for patients who received ruxolitinib and BAT | Appropriate, according to clinical experts consulted by CADTH |
BAT = best available therapy.
Table 5: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Correctionsa to sponsor’s base case | ||
None | — | — |
Changes to derive the CADTH base case | ||
1. Time horizon | 15 years | 1 year |
2. Duration of response | Survival curves (exponential and log-normal for ruxolitinib and BAT, respectively) fitted to post hoc KM data | Post hoc KM data |
3. OS approach | By response | By treatment arm |
4. Dosing | Mean dose for ruxolitinib and BAT | |
5. Duration of treatment | Extrapolated by treatment arm | KM curve for individual treatments |
6. Proportion of people receiving each BAT treatment | ATG = ||||||||% ECP = ||||||||% Methotrexate = ||||||||% MMF = ||||||||% Sirolimus = ||||||||% Etanercept = ||||||||% Infliximab = ||||||||% | ATG = 10.00% ECP = 16.67% Methotrexate = 0% MMF = 11.67% Sirolimus = 30.83% Etanercept = 18.33% Infliximab = 12.50% |
7. Disease baseline period resource use | Duration and frequency of visits differ between BAT and ruxolitinib | Duration and frequency of visits do not differ between BAT and ruxolitinib |
8. Relapse and recurrence rates | Treatment specific | Equal for both ruxolitinib and BAT |
9. Treatment administration | Includes costs of physician visit with each administration | Excludes costs of physician visit with each administration |
10. Price of ATG | $12.60/mg | $1.401422 |
CADTH base case | — | 1 + 2 + 3 + 4 + 5 + 6 + 7 + 8 + 9 + 10 |
ATG = antithymocyte globulin; BAT = best available therapy; ECP = extracorporeal photopheresis; KM = Kaplan-Meier; MMF = mycophenolate mofetil; OS = overall survival.
aCorrections are minor errors (e.g., transcription errors between report and model, misapplication of distributions or SEs in probabilistic analyses, etc.) that are not identified as limitations.
Table 6: Summary of the Stepped Analysis of the CADTH Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
Sponsor’s base case (probabilistic) | BAT | 212,141 | 0.92 | Reference |
Ruxolitinib | 172,207 | 1.07 | Dominant | |
Sponsor’s base case (deterministic) | BAT | 209,358 | 0.92 | Reference |
Ruxolitinib | 171,602 | 1.08 | Dominant | |
CADTH reanalysis 1: time horizon | BAT | 201,477 | 0.32 | Reference |
Ruxolitinib | 167,256 | 0.37 | Dominant | |
CADTH reanalysis 2: duration of response by KM + step 1 (1-year time horizon) | BAT | 195,924 | 0.32 | Reference |
Ruxolitinib | 167,526 | 0.37 | Dominant | |
CADTH reanalysis 3: OS | BAT | 205,365 | 0.68 | Reference |
Ruxolitinib | 191,359 | 1.03 | Dominant | |
CADTH reanalysis 4: dosing | BAT | 202,893 | 0.92 | Reference |
Ruxolitinib | 170,578 | 1.08 | Dominant | |
CADTH reanalysis 5: duration of treatment (KM by individual treatment) | BAT | 207,446 | 0.92 | Reference |
Ruxolitinib | 171,583 | 1.08 | Dominant | |
CADTH reanalysis 6: distribution of BAT | BAT | 222,568 | 0.92 | Reference |
Ruxolitinib | 174,643 | 1.08 | Dominant | |
CADTH reanalysis 7: disease baseline period resource use | BAT | 203,666 | 0.92 | Reference |
Ruxolitinib | 171,602 | 1.08 | Dominant | |
CADTH reanalysis 8: relapse and recurrence rates | BAT | 206,915 | 0.92 | Reference |
Ruxolitinib | 171,602 | 1.08 | Dominant | |
CADTH reanalysis 9: treatment administration costs | BAT | 209,033 | 0.92 | Reference |
Ruxolitinib | 171,567 | 1.08 | Dominant | |
CADTH reanalysis 10: PRICE of ATG | BAT | 205,760 | 0.92 | Reference |
Ruxolitinib | 170,764 | 1.08 | Dominant | |
CADTH exploratory reanalysis (deterministic) | BAT | 185,782 | 0.31 | Reference |
Ruxolitinib | 186,250 | 0.37 | 7,760 | |
CADTH exploratory reanalysis (probabilistic) | BAT | 181,920 | 0.31 | Reference |
Ruxolitinib | 183,198 | 0.37 | 21,057 |
ATG = antithymocyte globulin; BAT = best available therapy; ICER = incremental cost-effectiveness ratio; KM = Kaplan-Meier; OS = overall survival; QALY = quality-adjusted life-year.
Shortening the time horizon to 1 year reduced total QALYs for ruxolitinib and BAT, as well as incremental QALYs. Changing the OS approach reduced total QALYs for BAT, whereas QALYs for ruxolitinib remained similar. These were the only steps that changed total QALYs; the remaining steps only influenced total costs. The step resulting in the largest change to total BAT costs was the combined step of changing duration of response to being based on the KM curve plus the 1-year time horizon. Changing the distribution of BAT comparators also resulted in a large increase in total BAT costs. The largest increase in total costs for ruxolitinib occurred by changing the OS approach.
The majority of the incremental QALYs gained for ruxolitinib were accrued in the overall response health state. This is because ruxolitinib had a better response rate and a longer duration of response than BAT (Table 14). The majority of the incremental costs for ruxolitinib were initial treatment-acquisition costs ($8,574 more for ruxolitinib than BAT) (Table 14). For ruxolitinib, the median duration of treatment was approximately 11 weeks, and 2% of patients remained on treatment for 52 weeks; for the individual BATs, the median duration of treatment varied, but no patients remained on treatment for more than 27 weeks. The majority of the cost-savings associated with ruxolitinib was in resource use, which was $12,925 lower for ruxolitinib than for BAT. This is because patients receiving BAT spent more time in the nonresponder health state, which was associated with higher health care resource use (due to a higher frequency of hospitalizations and physician visits, along with longer length of stays for nonresponders) than the overall responder health state.
Scenario Analysis Results
To address remaining uncertainty regarding parameterization of the model, CADTH conducted several scenario analyses. Full results are presented in Table 15. When choosing to model OS by treatment arm and using an intention-to-treat approach (rather than the crossover-corrected approach used in the CADTH exploratory reanalysis), QALYs increased for BAT, along with total costs, leading ruxolitinib to dominate BAT (Table 15). The main source of higher BAT costs in this scenario came from increased resource-use costs, specifically in the nonresponder health state. Patients receiving BAT accrued 0.03 more life-years in the nonresponder state with the intention-to-treat approach than with the crossover-corrected approach; because they are alive longer, they accrue greater resource-use costs.
When no subsequent therapies were included in the analysis, the ICER for ruxolitinib increased to $31,665/QALY. Finally, the cost-effectiveness of ruxolitinib depends largely on the BAT comparator, as illustrated in the scenarios comparing ruxolitinib with all individual BATs. When compared with ATG, ECP, and infliximab, ruxolitinib dominated (i.e., was more effective and less expensive), as these treatments are associated with higher acquisition costs. However, the ICER for ruxolitinib rose to $118,423, $139,249, and $136,469 when ruxolitinib was compared to etanercept, MMF, and sirolimus, respectively. This demonstrates that the overall cost-effectiveness of ruxolitinib is highly dependent on the BAT comparator.
Issues for Consideration
According to clinical experts consulted for this review, ruxolitinib may be given to patients with aGvHD in clinical practice who were excluded from the trial because of steroid dosing and overlap syndrome. As these patients were not included in REACH 2, the cost-effectiveness of ruxolitinib in this expanded patient population is unknown.
Ruxolitinib was concurrently reviewed by CADTH for the same proposed Health Canada indication (the treatment of GvHD in patients 12 years and older who have inadequate responses to corticosteroids or other systemic therapies), but focused on ruxolitinib versus BAT in the cGvHD population. In that review, CADTH similarly conducted an exploratory reanalysis to account for limitations in the sponsor’s model structure and the uncertain nature of the data derived from the sponsor’s post hoc analysis. In the exploratory reanalysis, ruxolitinib was associated with an ICER of $1,062,977 per QALY, compared to BAT.
Ruxolitinib has been previously reviewed by CADTH for myelofibrosis and polycythemia vera and received a recommendation to reimburse with clinical criteria and/or conditions for both indications.23,24 In both reviews, the recommendation concluded that ruxolitinib was not cost-effective at the submitted price.23,24 The submitted price for ruxolitinib ($82.19) in both reviews was lower than the submitted price in the current review (see Appendix 1).23,24
The company did not provide an evidence submission to the National Institute for Health and Care Excellence for the appraisal of ruxolitinib for the treatment of SR-aGvHD; therefore, the topic was suspended and not reviewed by the institute.25
Overall Conclusions
The CADTH clinical review found that, compared to BAT, ruxolitinib demonstrates a statistically significant improvement in ORR at day 28. The improvements in ORR of the magnitude observed in the REACH 2 trial were considered clinically meaningful by the clinical experts consulted by CADTH. Other secondary outcomes, including duration of response, best overall response, and failure-free survival, were supportive of the observed ORR at day 28. The open-label design of the trial and reliance on local investigator’s assessment of trial outcomes may have introduced a bias that is difficult to quantify. The actual degree of OS benefit for ruxolitinib is uncertain, given potential bias from the crossover of patients in the BAT group to the ruxolitinib group and the limited follow-up time (7.34 months and 3.81 months of OS follow-up time for ruxolitinib and BAT, respectively).
Given the high degree of uncertainty concerning the post hoc analysis used to populate model parameters and the inappropriateness of the sponsor’s model structure, CADTH was unable to derive a base-case analysis. The exploratory reanalysis performed by CADTH uses more appropriate assumptions, but these estimates remain highly uncertain because the majority of the parameters were based on the post hoc analysis (including duration of response, duration of treatment, resource use, and drug dosing), the model structure did not fully capture the health condition, and the distribution of BAT treatments that would be used by clinicians varied. Therefore, the magnitude of benefit for ruxolitinib in the CADTH exploratory reanalysis may be overestimated.
CADTH undertook exploratory reanalyses to address limitations related to the following: the model not capturing long-term outcomes of SR-aGvHD and uncertain long-term efficacy; use of an approach to OS that aligned with the REACH 2 trial rather than the post hoc analysis; alignment of dosing for ruxolitinib and BAT treatments with the literature; the modelling of duration of treatment by individual BAT KM curves; and alignment of the distribution of BAT treatments with clinical expert expectations. Based on the CADTH exploratory reanalysis, conducted over a 1-year time horizon, the ICER for ruxolitinib compared with BAT was estimated to be $21,057 per QALY gained, and ruxolitinib was cost-effective in 52% of iterations at a willingness-to-pay threshold of $50,000 per QALY. When the price of ruxolitinib was reduced by 10% and 25%, the probability of ruxolitinib being cost-effective at a $50,000 per QALY threshold increased to 57% and 62%, respectively. Given the uncertainty in the results and the presence of limitations that could not be addressed (e.g., the sponsor’s uncertain model structure and inputs derived from the post hoc analysis), price reductions are likely required.
The majority of the cost-savings associated with ruxolitinib was in resource use, which was $12,925 lower for ruxolitinib than for BAT. This is because patients receiving BAT spent more time in the nonresponder health state, which was associated with higher health care resource use (due to a higher frequency of hospitalizations and physician visits, along with longer hospital stays for nonresponders) than the overall responder health state.
Of note, results from CADTH’s exploratory reanalysis reflect the cost-effectiveness of ruxolitinib over a 1-year time horizon; because of the limitations identified in the sponsor’s modelling approach, the cost-effectiveness of ruxolitinib in aGvHD beyond 1 year is unknown. In the acute indication, the median duration of treatment for ruxolitinib was approximately 11 weeks, and 2% of patients remained on treatment for 52 weeks. The median duration of treatment varied for the individual BATs, but no patients remained on treatment for more than 27 weeks. Another source of uncertainty is the cost-effectiveness of ruxolitinib compared to individual BATs. As the distribution of BATs reimbursed in jurisdictions may differ, and because the cost-effectiveness of ruxolitinib is dependent on the cost of the comparator treatments, cost-effectiveness conclusions will differ if a different distribution of BATs is used. Finally, the modelled population did not fully align with the proposed Health Canada indication and the sponsor’s reimbursement request did not align with available evidence. The model is specific to aGvHD; therefore, based on the submitted evidence in a SR population, the cost-effectiveness of ruxolitinib for cGvHD, subgroups of aGvHD, and patients with an inadequate response to systemic therapies is unknown. Also, since the model used REACH 2 efficacy and dosing (10 mg twice daily), the cost-effectiveness of ruxolitinib using the product-monograph dose (5 mg twice daily) is unknown.
References
1.PrJakavi® (ruxolitinib phosphate): tablets, 5 mg, 10 mg, 15 mg and 20 mg [draft product monograph]. Dorval (QC): Novartis Pharmaceuticals Canada Inc.; 2021 Feb 25.
2.Clinical Study Report: INC424C2301. A phase III randomized open-label multi-center study of ruxolitinib versus best available therapy in patients with corticosteroid-refractory acute graft vs. host disease after allogeneic stem cell transplantation. [internal sponsor's report]. Freiburg (DE): Novartis; 2020 Mar 9.
3.Ruxolitinib for steroid-refractory acute graft-versus-host disease (SR-aGvHD): additional REACH 2 trial IPD analyses to inform the cost-effectiveness model of ruxolitinib for SR-aGvHD [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Jakavi (ruxolitinib): tablets, 5 mg, 10 mg, 15 mg and 20 mg, oral. Dorval (QC): Novartis Pharmaceuticals Inc.; 2021 Aug 10.
4.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Jakavi (ruxolitinib): tablets, 5 mg, 10 mg, 15 mg and 20 mg, oral. Dorval (QC): Novartis Pharmaceuticals Canada Inc.; 2021 Aug 10.
5.Zeiser R, von Bubnoff N, Butler J, et al. Ruxolitinib for glucocorticoid-refractory acute graft-versus-host disease. N Engl J Med. 2020;382(19):1800-1810. PubMed
6.Ontario Case Costing Initiative (OCCI). Toronto (ON): Ontario Health and Long-Term Care; 2017: https://data.ontario.ca/dataset/ontario-case-costing-initiative-occi. Accessed 2021 Oct 26.
7.Exceptional Access Program (EAP). Toronto (ON): Ontario Ministry of Health; Ontario Ministry of Long-Term Care; 2021: https://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx. Accessed 2021 Oct 13.
8.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2021; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2021 Oct 7.
9.Medical Advisory Secretariat. Extracorporeal photophoresis: an evidence-based analysis. Ont Health Technol Assess Ser. 2006;6(6):1-82. PubMed
10.Schedule of benefits for physician services under the Health Insurance Act: (January 24, 2022 (effective November 1, 2021)). Toronto (ON): Ontario Ministry of Health; 2022: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master.pdf. Accessed 2022 Mar 1.
11.Tam VC, Ko YJ, Mittmann N, et al. Cost-effectiveness of systemic therapies for metastatic pancreatic cancer. Curr Oncol. 2013;20(2):90-106. PubMed
12.PrAtgam® (lymphocyte immune globulin, anti-thymocyte globulin [equine]): concentrate for solution for infusion / sterile solution −50 mg/mL [product monograph]. Kirkland (QC): Pfizer Canada Inc.; 2014 Jun 12: https://pdf.hres.ca/dpd_pm/00025436.PDF. Accessed 2021 Feb 11.
13.PrAvsolaTM (infliximab for injection): powder for solution, sterile, Llophilized, 100mg/vial [product monograph]. Mississauga (ON): Amgen Canada Inc.; 2020 Mar 12: https://pdf.hres.ca/dpd_pm/00055391.PDF. Accessed 2021 Feb 11.
14.Jakavi (ruxolitinib): tablets, 5 mg, 10 mg, 15 mg and 20 mg ruxolitinib (as ruxolitinib phosphate), oral [product monograph]. Dorval (QC): Novartis Pharmaceuticals Canada Inc.; 2022 May 19.
15.Health Canada reviewer's report: Jakavi (ruxolitinib) [CONFIDENTIAL internal report]. Ottawa (ON): Therapeutics Products Directorate, Health Canada; 2022.
16.Jagasia M, Perales MA, Schroeder MA, et al. Ruxolitinib for the treatment of steroid-refractory acute GVHD (REACH1): a multicenter, open-label phase 2 trial. Blood. 2020;135(20):1739-1749. PubMed
17.Lee SJ. Classification systems for chronic graft-versus-host disease. Blood. 2017;129(1):30-37. PubMed
18.Hoda D, Pidala J, Salgado-Vila N, et al. SIROLIMUS for treatment of steroid-refractory acute graft-versus-host disease. Bone Marrow Transplant. 2010;45(8):1347-1351. PubMed
19.Government B.C. BC PharmaCare formulary search. 2021; https://pharmacareformularysearch.gov.bc.ca. Accessed 2022 Mar 1.
20.Yalniz FF, Hefazi M, McCullough K, et al. Safety and efficacy of infliximab therapy in the setting of steroid-refractory acute graft-versus-host disease. Biol Blood Marrow Transplant. 2017;23(9):1478-1484. PubMed
21.GvHD Canadian clinician survey responses [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Jakavi (ruxolitinib): tablets, 5 mg, 10 mg, 15 mg and 20 mg, oral. Dorval (QC): Novartis Pharmaceuticals. 2021 Jul 13.
22.DeltaPA. Ottawa (ON): IQVIA; 2021: https://www.iqvia.com/. Accessed 2021 Oct 26.
23.CADTH pCODR Expert Review Committee (pERC) final recommendation: ruxolitinib (Jakavi). Ottawa (ON): CADTH; 2013 Jan 14: https://www.cadth.ca/sites/default/files/pcodr/pcodr-jakavimyelofibro-fn-rec.pdf. Accessed 2021 Nov 22.
24.CADTH pCODR Expert Review Committee (pERC) final recommendation: ruxolitinib (Jakavi). Ottawa (ON): CADTH; 2016 Mar 3: https://www.cadth.ca/sites/default/files/pcodr/pcodr_ruxolitinib_jakavi_pv_fn_rec.pdf. Accessed 2021 Nov 22.
25.Ruxolitinib for treating acute graft versus host disease refractory to corticosteroids in people aged 12 and over. NICE guidance [ID2715]. London (UK): National Institute for Health and Care Excellence; 2020: https://www.nice.org.uk/guidance/indevelopment/gid-ta10760. Accessed 2022 Mar 1.
26.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Jakavi (ruxolitinib): tablets, 5 mg, 10 mg, 15 mg and 20 mg, oral. Dorval (QC): Novartis Pharmaceuticals Canada Inc.; 2021 Aug 10.
27.Nova Scotia Pharmacare: formulary. Halifax (NS): Government of Nova Scotia; 2021: https://novascotia.ca/dhw/pharmacare/formulary.asp. Accessed 2021 Oct 13.
28.Tan Y XH, Wu D, Luo Y, Lan J et al. Combining therapeutic antibodies using basiliximab and etanercept for severe steroid-refractory acute graft-versus-host disease: a multi-center prospective study. Oncoimmunology. 2017. PubMed
29.Xhaard A, Rocha V, Bueno B, et al. STEROID-refractory acute GVHD: lack of long-term improved survival using new generation anticytokine treatment. Biol Blood Marrow Transplant. 2012;18(3):406-413. PubMed
30.Xhaard A, Launay M, Sicre de Fontbrune F, et al. A monocentric study of steroid-refractory acute graft-versus-host disease treatment with tacrolimus and mTOR inhibitor. Bone Marrow Transplant. 2020;55(1):86-92. PubMed
31.Nassar A, Elgohary G, Elhassan T, Nurgat Z, Mohamed SY, Aljurf M. Methotrexate for the treatment of graft-versus-host disease after allogeneic hematopoietic stem cell transplantation. J Transplant. 2014:980301. PubMed
32.Pidala J, Kim J, Perkins J, et al. Mycophenolate mofetil for the management of steroid-refractory acute graft vs host disease. Bone Marrow Transplant. 2010;45(5):919-924. PubMed
33.Das-Gupta E, Greinix H, Jacobs R, et al. Extracorporeal photopheresis as second-line treatment for acute graft-versus-host disease: impact on six-month freedom from treatment failure. Haematologica. 2014;99(11):1746-1752. PubMed
34.Table 17-10-0057-01: projected population, by projection scenario, age and sex, as of July 1 (x 1,000). Ottawa (ON): Statistics Canada; 2021: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710005701&pickMembers%5B0%5D=1.1&pickMembers%5B1%5D=3.1&pickMembers%5B2%5D=4.1&cubeTimeFrame.startYear=2020&cubeTimeFrame.endYear=2027&referencePeriods=20200101%2C20270101. Accessed 2021 Nov 15.
Appendix 1: Cost Comparison Table
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s). Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 7: CADTH Cost Comparison Table for aGvHD
Treatment | Strength | Form | Price ($) | Recommended dosage | Daily cost ($) | Annual cost ($) |
|---|---|---|---|---|---|---|
Jakavi (Ruxolitinib) | 5 mg 10 mg 15 mg 20 mg | Tablet | 86.6275ac 87.3775ab 87.5775ab 87.6375ab | 10 mg twice daily | 174.76 | 63,786 |
aGvHD = acute graft-versus-host disease.
aSponsor’s submitted price.26
bOntario Exceptional Access Formulary,7 accessed October 13, 2021.
cNova Scotia formulary,27 accessed October 13, 2021.
dCourse cost is over a treatment duration of 6 months.
Table 8: CADTH Cost Comparison Table for aGvHD (Not Indicated for GvHD)
Treatment | Strength / concentration | Form | Price ($) | Recommended dosage | Daily cost ($) | Course cost ($) |
|---|---|---|---|---|---|---|
Biological response modifier | ||||||
Etanercept (generics) | 50 mg/mL | 25 mg 50 mg Pre-filled syringe Pre-filled autoinjector | 120.5000 241.0000 241.0000 | 25 mg twice per week for 4 weeks, and then 25 mg once per week for another 4 weeks28,29 | 25.82 | 1,446b |
Infliximab (Inflectra, Renflexis, Avsola) | 100 mg | 100 mg Vial powder for IV infusion | 493.0000 | 10 mg/kg (710 mg) per week for 4 weeks20 | 563.43 | 15,776c |
Antithymocyte globulin | 50 mg/mL | 250 mg Vial solution IV infusion | 1.4014 | 3 mg/kg (213 mg) to 7.5 mg/kg (532.5 mg) daily for 3-5 daysa | 7.01 to 15.42 | 21 to 77d |
mTOR inhibitors | ||||||
Sirolimus (Rapamune) | 1 mg 2 mg 5 mg | Tablets | 8.5220 Not available Not available | Loading dose of 6 mg and then, maintenance dose of 1-2 mg once per day for 12 days18,30 | 12.78 to 21.31 | 153 to 256e |
Systemic Immunosuppressants | ||||||
Methotrexate (generic) | 2.5 mg | Tablet | 0.6325 | 7.5 mg/m2 (13.5 mg) per week for a median of 3 doses31 | 3.80 | 11f |
Mycophenolate mofetil | 250 mg 500 mg | Capsule | 0.3712 0.7423 | 1,000 mg to 1,500 mg twice per day for 28 days32 | 2.97 to 8.91 | 83 to 249g |
aGvHD = acute graft-versus-host disease; GvHD = graft-versus-host disease.
Note: All prices are from the Ontario Drug Benefit Formulary (accessed October 7, 2021), unless otherwise indicated, and do not include dispensing fees. For dosing that depends on weight or body surface area, CADTH assumed mean body weight of 71 kg and mean body surface area was 1.8 m2.
aDose informed by feedback from clinical experts consulted for this review by CADTH.
bCourse cost is over treatment duration of 56 days.
cCourse cost is over treatment duration of 28 days.
dCourse cost is over treatment duration of 3-5 days. Range represents treatment cost with minimum to maximum dose and treatment duration.
eCourse cost is over treatment duration of 12 days.
fCourse cost is over treatment duration of 21 days.
gCourse cost is over treatment duration of 28 days.
Table 9: CADTH Cost Comparison Table of Other Non-Drug Interventions For Chronic GvHD (Not Indicated for GvHD)
Treatment | Strength / concentration | Form | Price ($) | Recommended dosage | Daily cost ($) | Course cost ($) |
|---|---|---|---|---|---|---|
Extracorporeal photopheresis | Not applicable | Not applicable | 1,851.3119a | 2 to 3 treatments per week for first 4 to 6 weeks and then, 1 treatment every 2 weeks33 | 192.74 to 284.04 | 35,175 to 51,837 |
GvHD = graft-versus-host disease.
Note: All prices are from the Ontario Drug Benefit Formulary (accessed October 7, 2021), unless otherwise indicated, and do not include dispensing fees. Course cost assumes treatment duration of 6 months.
aOntario Health Technology Assessment Series,9 accessed November 4, 2021. Cost inflated from 2006 to 2021 CAD.26
bDose obtained from Das-Gupta et al.33
cCourse cost range is based on minimum of 18 treatments and maximum of 22 treatments over a duration of 6 months.
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | Relevant clinical outcomes identified as being important by patients and clinicians were not included in the model. See limitation in critical appraisal, ‘The model structure does not fully capture the health condition.’ |
Model has been adequately programmed and has sufficient face validity | No | Patients not being able to transition from being nonresponders to responders was determined to not have face validity by clinical experts consulted for this review. See limitation in critical appraisal ‘The model structure does not fully capture the health condition.’ |
Model structure is adequate for decision problem | No | The model’s inability to fully capture the health condition and the uncertainty associated with the post hoc analysis means that results obtained from the analysis are highly uncertain. See limitation in critical appraisal, ‘The model structure does not fully capture the health condition’ and ‘The model’s efficacy parameters are primarily estimated from an uncertain post hoc analysis of REACH 2 trial data.’ |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | No comment. |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | No comment. |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | No | The post hoc analysis used to populate model parameters was not clearly and transparently reported. See limitation, ‘ The model’s efficacy parameters are primarily estimated from an uncertain post hoc analysis of REACH 2 trial data.’ |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
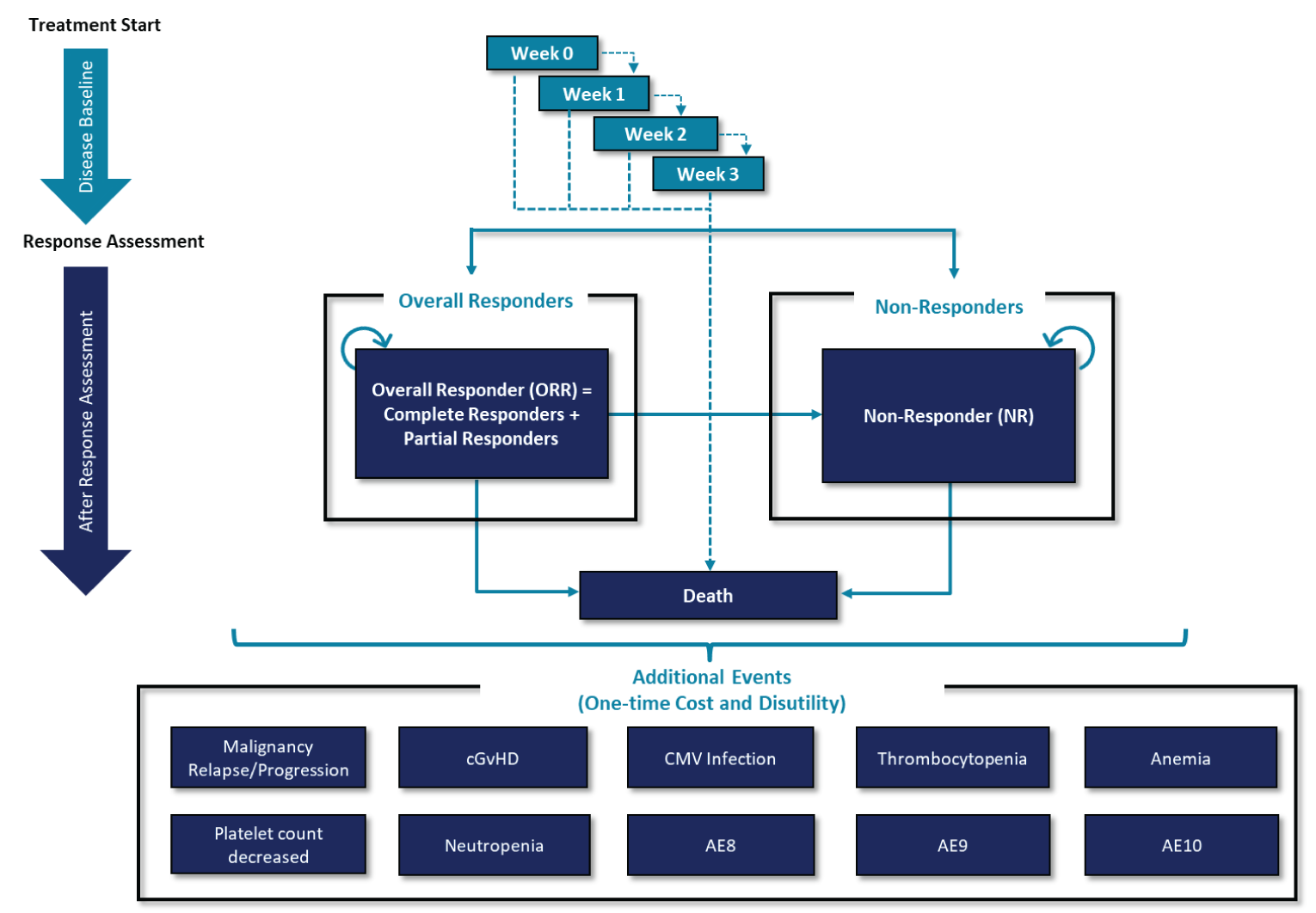
AE = adverse event; cGvHD = chronic graft-versus-host disease; CMV = cytomegalovirus.
Source: Sponsor’s pharmacoeconomic submissions4
Table 11: Treatments Included in Best Available Therapy
Treatment Name | Proportion of Patients Receiving Treatment |
|---|---|
Antithymocyte globulin | ||||||||% |
Extracorporeal photopheresis | ||||||||% |
Mesenchymal stromal cells | ||||||||% |
Methotrexate | ||||||||% |
Mycophenolate mofetil | ||||||||% |
Everolimus | ||||||||% |
Sirolimus | ||||||||% |
Etanercept | ||||||||% |
Infliximab | ||||||||% |
Total | 100.00% |
Source: Sponsor’s pharmacoeconomic submission21
Table 12: REACH 2 Cumulative Mortality Up to Response-Assessment Time Point (Day 28)
Week | Ruxolitinib | BAT |
|---|---|---|
Week 1 | ||||||||% | ||||||||% |
Week 2 | ||||||||% | ||||||||% |
Week 3 | ||||||||% | ||||||||% |
Week 4 | ||||||||% | ||||||||% |
Source: Sponsor’s post hoc analysis.19
Table 13: Response Rate at Day 28
Response Type | Ruxolitinib | BAT |
|---|---|---|
Overall respondera | 62.34% | 39.35% |
Non responderb | ||||||||% | ||||||||% |
Dead (cumulative mortality up to day 168) | ||||||||% | ||||||||% |
BAT=best available therapy
aOverall responders include individuals with a complete and partial response
bNonresponders include all alive patients without a complete or partial response.
Source: Sponsor’s pharmacoeconomic submissions4
Figure 2: Kaplan-Meier Curves for Duration of Response for Overall Responders, by Treatment

Note: This figure has been redacted at the request of the sponsor.
Source: Sponsor’s post hoc analysis.19
Figure 3: Sponsor’s Extrapolations of Duration of Response for Overall Response, by Treatment
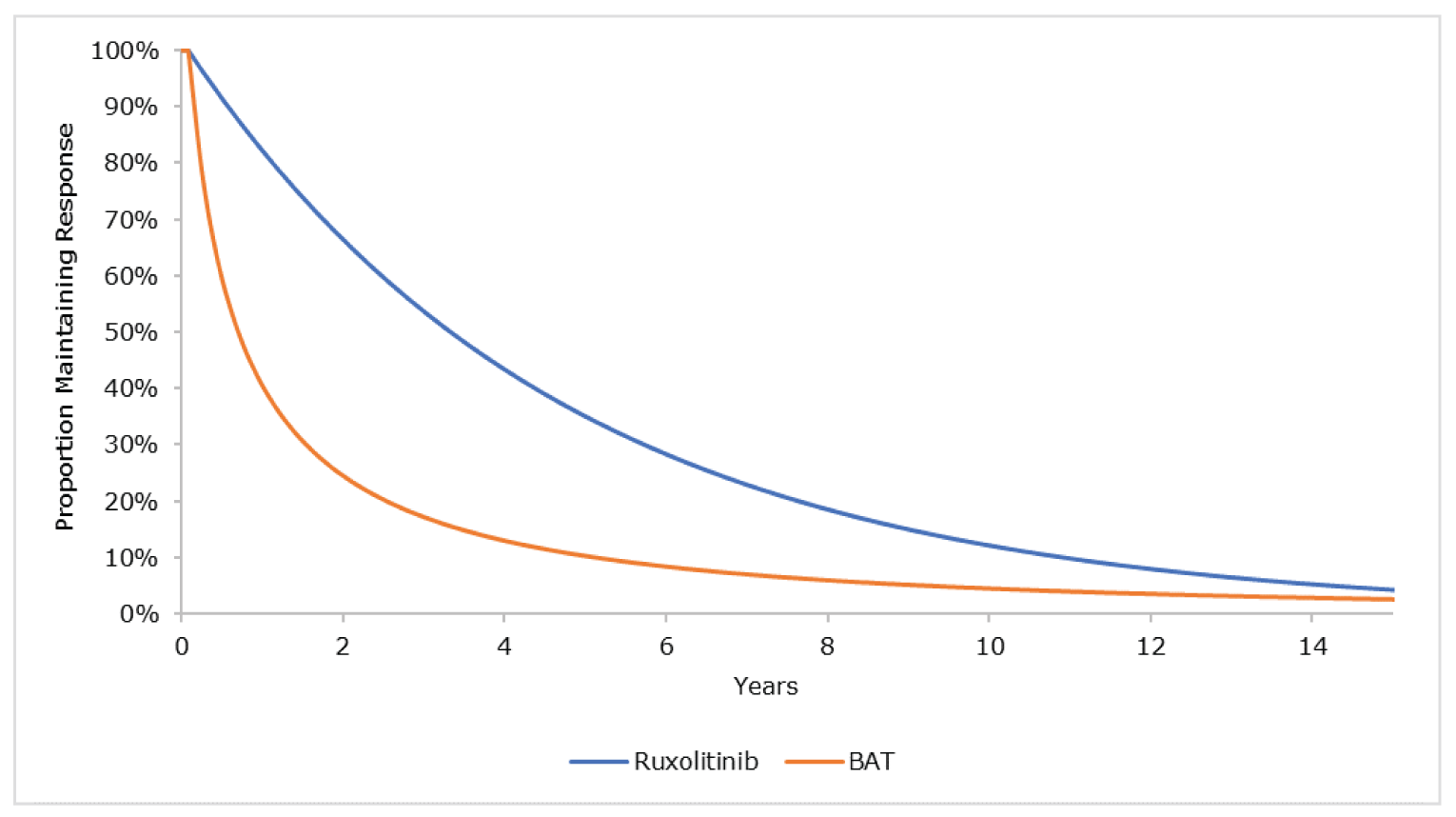
Source: Sponsor’s pharmacoeconomic submission4
Figure 4: Kaplan-Meier Curves for Overall Survival From Day 28, by Responder Status

Note: This figure has been redacted at the request of the sponsor.
Source: Sponsor’s post hoc analysis.19
Figure 5: Kaplan-Meier Curves for Overall Survival by Treatment, Full Analysis Set (July 25, 2019, Data Cut-Off Date)
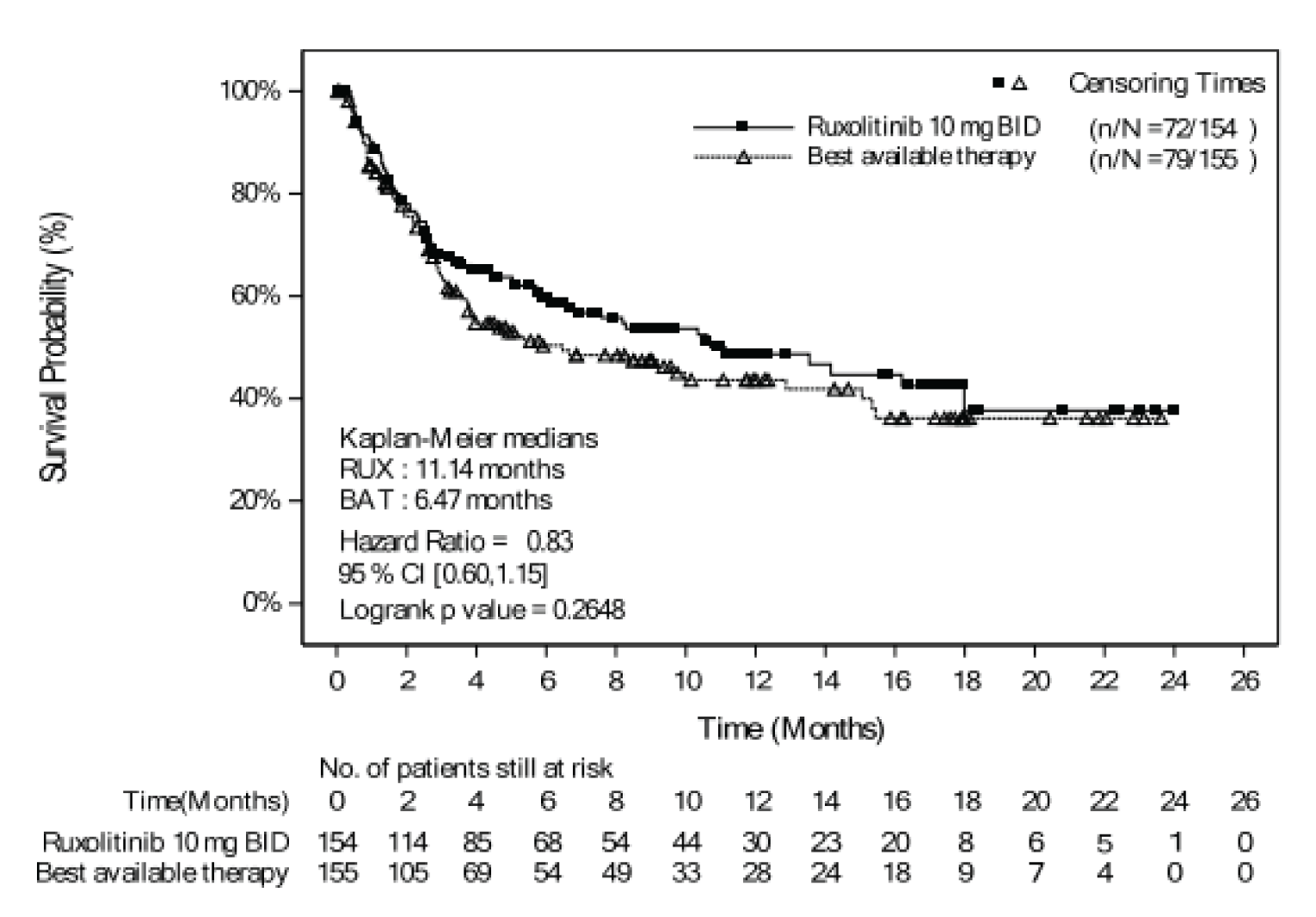
Source: REACH 2 clinical study report.2
Figure 6: Extrapolated OS for Overall Responders Versus Nonresponders
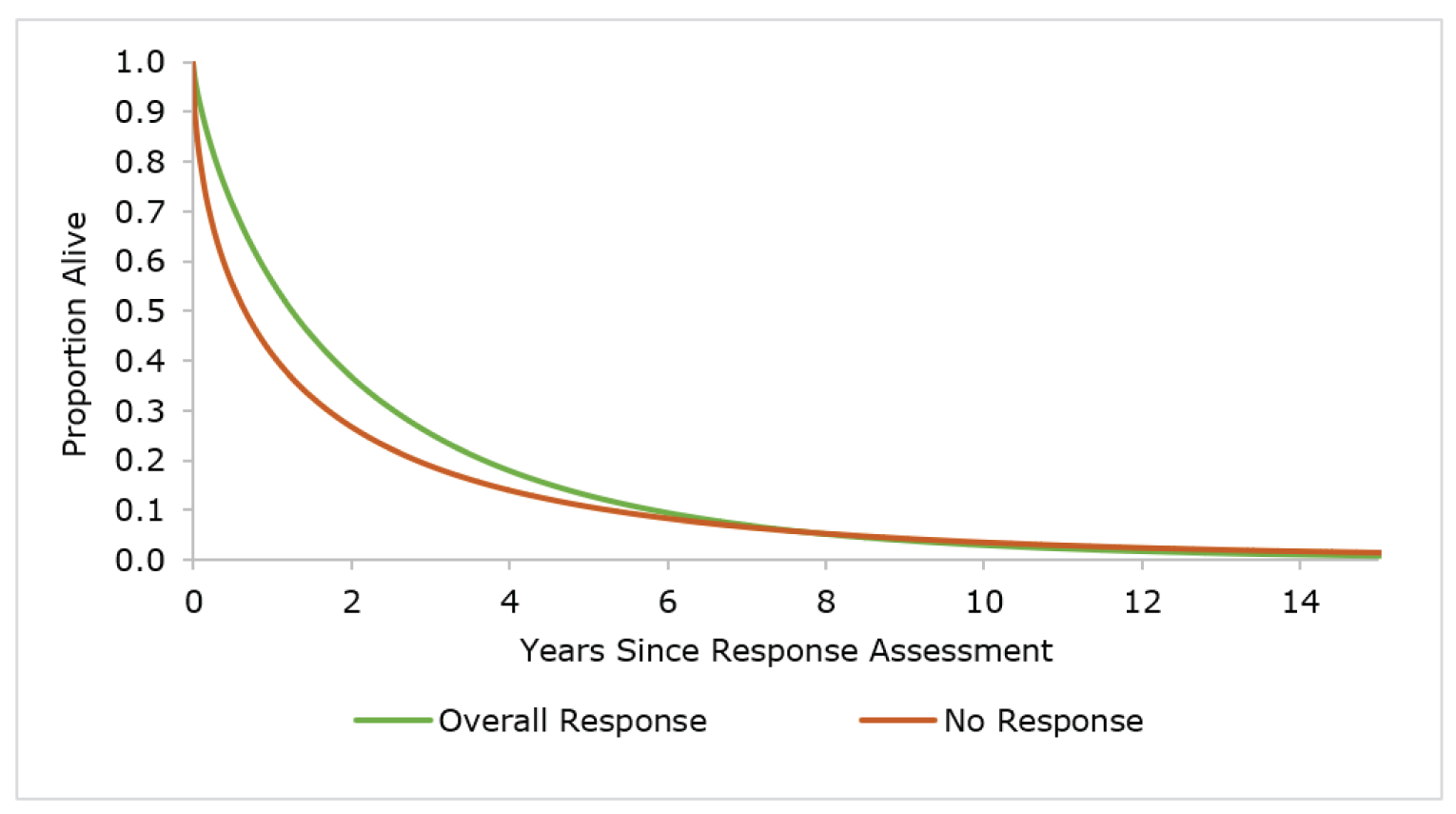
Source: Sponsor’s pharmacoeconomic submission4
Figure 7: Duration of Treatment, by Treatment Arm KM

Note: This figure has been redacted at the request of the sponsor.
Source: Sponsor’s post hoc analysis.19
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
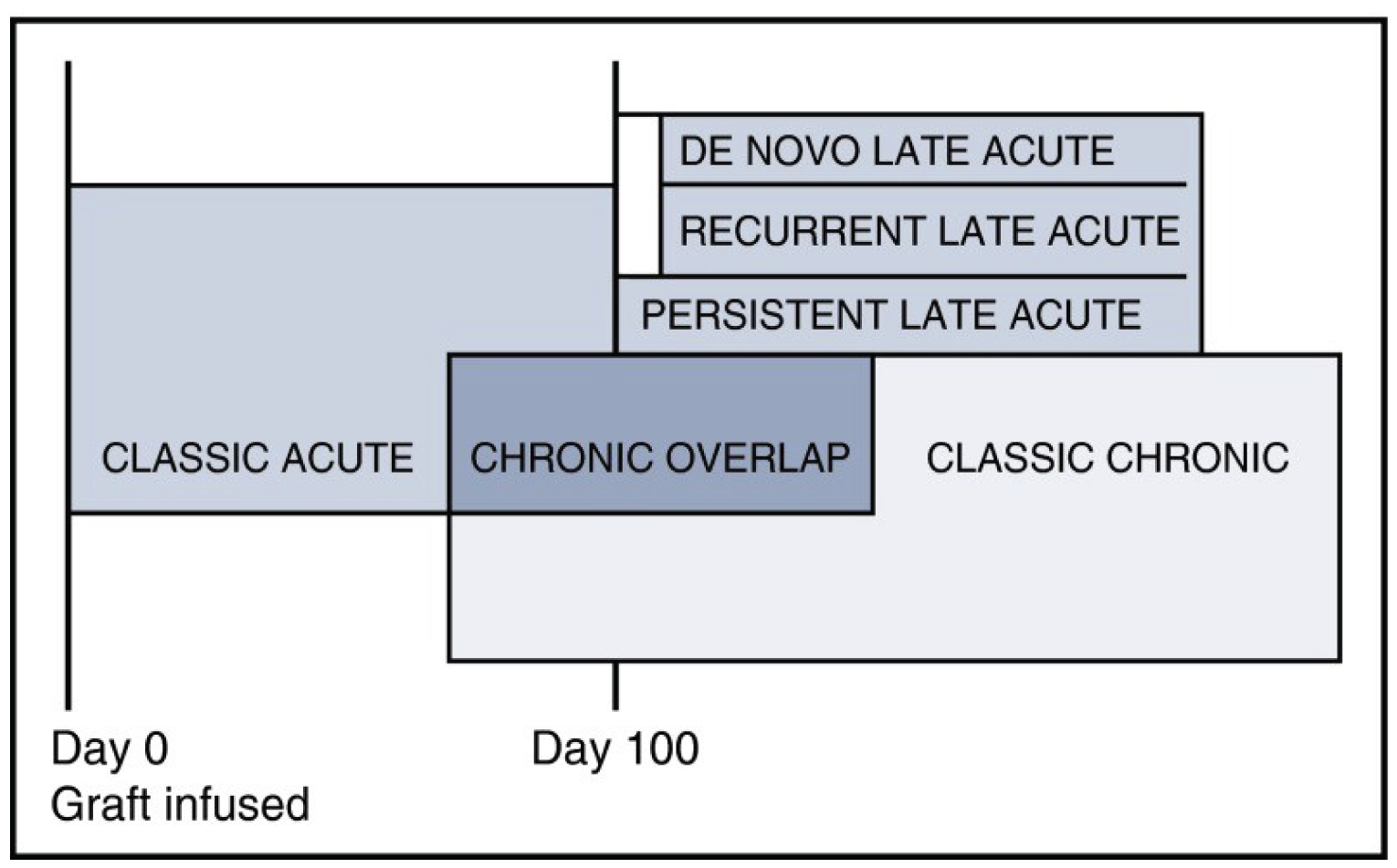
Table 14: Disaggregated Summary of CADTH’s Economic Evaluation Results
Parameter | BAT | Ruxolitinib | Incremental |
|---|---|---|---|
Discounted LYs | |||
Total | 0.61 | 0.70 | 0.09 |
Disease baseline | 0.07 | 0.07 | 0.00 |
Overall responder | 0.21 | 0.39 | 0.18 |
Nonresponder | 0.32 | 0.24 | -0.09 |
Discounted QALYs | |||
Total | 0.31 | 0.37 | 0.06 |
Disease baseline | 0.03 | 0.04 | 0.00 |
Overall responder | 0.12 | 0.22 | 0.10 |
Nonresponder | 0.16 | 0.11 | -0.04 |
All additional and adverse events | 0.00 | 0.00 | 0.00 |
Discounted costs ($) | |||
Total | 181,920 | 183,198 | 1,279 |
Acquisition-initial treatment | 5,525 | 14,099 | 8,574 |
Acquisition-subsequent treatment | 1,992 | 1,454 | -538 |
Administration | 1,524 | 286 | -1,238 |
Resource use | 154,399 | 142,302 | -12,096 |
Additional events and adverse events | 18,480 | 25,057 | 6,577 |
ICER ($/QALY) | 21,057 | ||
BAT=best available therapy; ICER = incremental cost-effectiveness ratio; LY= life-year; QALY = quality-adjusted life-year; [add as required].
Detailed Results of CADTH Base Case
Figure 9: Cost-Effectiveness Plane Comparing Ruxolitinib to BAT From the Probabilistic CADTH Exploratory Reanalysis Results
QALYs = quality-adjusted life-years.
Source: Sponsor’s pharmacoeconomic model, CADTH exploratory reanalysis
Scenario Analyses
Table 15: CADTH Scenario Analyses
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
CADTH exploratory reanalysis | BAT | 181,920 | 0.31 | Ref. |
Ruxolitinib | 183,198 | 0.37 | 21,057 | |
Removing ORR and NR rates from the probabilistic analysis | BAT | 181,868 | 0.31 | Ref. |
Ruxolitinib | 182,491 | 0.37 | 10,218 | |
OS by treatment arm-ITT approach | BAT | 191,992 | 0.33 | Ref. |
Ruxolitinib | 183,204 | 0.37 | Dominant | |
Subsequent therapy costs=$0 | BAT | 179,542 | 0.31 | Ref. |
Ruxolitinib | 181,465 | 0.37 | 31,665 | |
Ruxolitinib vs. ATG | BAT | 184,898 | 0.31 | Ref. |
Ruxolitinib | 183,659 | 0.37 | Dominant | |
Ruxolitinib vs. ECP | BAT | 195,468 | 0.31 | Ref. |
Ruxolitinib | 185,796 | 0.37 | Dominant | |
Ruxolitinib vs. etanercept | BAT | 174,617 | 0.31 | Ref. |
Ruxolitinib | 181,809 | 0.37 | 118,423 | |
Ruxolitinib vs. infliximab | BAT | 201,876 | 0.31 | Ref. |
Ruxolitinib | 187,150 | 0.37 | Dominant | |
Ruxolitinib vs. MMF | BAT | 173,040 | 0.31 | Ref. |
Ruxolitinib | 181,497 | 0.37 | 139,249 | |
Ruxolitinib vs. sirolimus | BAT | 173,243 | 0.31 | Ref. |
Ruxolitinib | 181,531 | 0.37 | 136,469 |
ATG= Antithymocyte globulin; BAT=best available therapy; ECP= extracorporeal photopheresis; ICER = incremental cost-effectiveness ratio; ITT=intent to treat; MMF=mycophenolate mofetil; NR=non response rate; ORR=overall response rate; OS=overall survival QALY = quality-adjusted life-year.
Appendix 5: Submitted Budget Impact Analysis and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 16: Summary of Key Take-Aways
Key Take-aways of the BIA |
|---|
|
BIA = budget impact analysis.
Summary of the Sponsor’s Budget Impact Analysis
The submitted budget impact analysis (BIA)26 assessed the expected budgetary impact of reimbursing ruxolitinib for patients aged 12 years and older with steroid-refractory acute graft-versus-host disease. The BIA was conducted from the perspective of the Canadian public drug plans, over a 3-year time horizon (2022-2025) and included drug-acquisition costs, markup, and dispensing fees. The sponsor’s pan-Canadian estimates reflect the aggregated results from provincial budgets (excluding Quebec), as well as the Non Insured Health Benefits Program. Key inputs to the BIA are documented in Table 17.
The analytic framework, which used an epidemiological-based approach, leveraged data from a survey the sponsor conducted with 10 clinicians, to estimate the size of the treatment-eligible population.26 The sponsor included new patients (incident) with mild to moderate (Grade I-IV) aGvHD. The sponsor estimated the annual number of allogeneic stem cell transplant (alloSCT) cases across jurisdictions based on clinical input. For jurisdictions where clinical input was not received, the sponsor assumed a weighted average number of alloSCT cases based on the annual number of expected alloSCT cases in Canada. To estimate the incident population, the sponsor assumed that |||||||| of alloSCT patients experience a complication and develop GvHD. Of these patients, |||||||| are classified as acute, and |||||||| of acute patients are corticosteroid refractory. The sponsor adopted the same definition of corticosteroid-refraction in patients with aGvHD as the REACH 2 trial.5 The sponsor included all grades of SR-aGvHD; assuming |||||||| are Grade I, |||||||| are Grade II, |||||||| are Grade III and |||||||| are Grade IV. Further details on estimating the size of the eligible population based on a patient flow diagram is shown in Figure 10.
The comparators included ATG, ECP, sirolimus, MMF, MTX, etanercept and infliximab. Costs for each treatment were based on the mean daily doses, dose frequency, and days on treatment using a post hoc analysis of data in the BAT arm of the REACH 2 trial5 and published literature.33 The dose for ruxolitinib was 10 mg twice daily, ATG was ||||||||||||||||||||||||, ECP was ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||, sirolimus was ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||, MMF was ||||||||||||||||, MTX was ||||||||||||||||||||||||, etanercept was |||||||||||||||||||||||||||||||||||||||||||||||||||||||| and infliximab was ||||||||||||||||||||||||.26 The median treatment duration of ruxolitinib was ||||||||, ATG was ||||||||, ECP was on starting dose and |||||||| on maintenance dose, sirolimus was |||||||| on starting dose and |||||||| on maintenance dose, MMF was ||||||||, MTX was ||||||||, etanercept was |||||||| and infliximab was ||||||||.26 Unit costs for each drug was obtained from the IQVIA Delta PA database22 and the cost of ECP therapy was obtained from published literature.9 The total costs for patients with SR-aGvHD were accrued by incident population without tracking patient cohorts into subsequent years due to the short life expectancy of typically less than one year.
Table 17: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as Year 1 / Year 2 / Year 3 if appropriate) |
|---|---|
Target Populationa | |
Total Population34 (Year 1 / Year 2 / Year 3) | 30,876,379 / 31,244,773 / 31,618,067 |
Number of alloSCT (Year 1 / Year 2 / Year 3) | |||||||||||||||||||||||| |
Incidence of GvHD in alloSCT patients (%) | |||||||||||||||||||||||| |
Proportion of aGvHD (%) | |||||||||||||||||||||||| |
Incidence of SR-aGvHD (%) | |||||||||||||||||||||||| |
Proportion covered by Public Payer (%) | |||||||||||||||||||||||| |
Number of patients eligible for drug under review | 65 / 67 / 70 |
Market Uptake (3 years)a | |
Uptake (reference scenario) | |
ATG ECP Etanercept Infliximab MMF MTX Sirolimus | |||||||||||||||||||||||| |||||||||||||||||||||||| |||||||||||||||||||||||| |||||||||||||||||||||||| |||||||||||||||||||||||| |||||||||||||||||||||||| |||||||||||||||||||||||| |
Uptake (new drug scenario) | |
Ruxolitinib ATG ECP Etanercept Infliximab MMF MTX Sirolimus | |||||||||||||||||||||||| |||||||||||||||||||||||| |||||||||||||||||||||||| |||||||||||||||||||||||| |||||||||||||||||||||||| |||||||||||||||||||||||| |||||||||||||||||||||||| |||||||||||||||||||||||| |
Cost of treatment (per patient)b | |
Cost of treatment over year | |
Ruxolitinib ATG ECP Etanercept Infliximab MMF (BC, SK, MB, ON, NB, PEI, NIHB / NF / AB,NS) MTX Sirolimus (ON, NIHB / AB / BC, SK, MB, NB, NS, PEI, NF) | $11,009.57 $9,955.26 $17,389.11 $3,055.00 $42,930.44 $61.69 / $85.68 / $171.35 $31.52 $210.78 / $211.44 / $212.69 |
Note: Sponsor uses tern “incidence” for estimates that are rather proportions, not true incidence.
AB = Alberta; aGvHD = acute Graft-versus-Host Disease; alloSCT = allogeneic stem cell transplant; ATG = antithymocyte globulin; BC = British Columbia; ECP = extracorporeal photopheresis; MB = Manitoba; MMF = mycophenolate mofetil; MTX = methotrexate; NB = New Brunswick; NF = Newfoundland and Labrador; NIHB = Non insured Health Benefit; NS = Nova Scotia; ON = Ontario; PEI = Prince Edward Island; SK = Saskatchewan; SR = steroid refractory
aThe inputs were informed by sponsor’s survey of 11 clinicians, unless otherwise indicated.26
bUnit cost was obtained from the IQVIA Delta PA database, unless otherwise indicated.22
cCost is inflated from 2006 to 2021 CAD and estimates include cost of procedure kit, instrument operator, methoxsalen, saline and supplies.9,26
Summary of the Sponsor’s Budget Impact Analysis Results
The sponsor estimated the net budget impact of introducing ruxolitinib for patients with SR-aGvHD was $151,010 in year 1, $159,076 in year 2, and $171,959 in year 3, for an overall 3-year budget impact of $482,045 to the public drug plans.
CADTH Appraisal of the Sponsor’s Budget Impact Analysis
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
There is uncertainty in the estimated number of eligible patients: The sponsor used an epidemiology-based approach to estimate the population size eligible for treatment with ruxolitinib, primarily based on a sponsor collected survey of a small group of clinical experts (N=10) across Canada. However, there was inconsistency across expert opinions, and the methods used to control for these inconsistencies may have been inappropriate and resulted in an estimate that was not reflective of jurisdictional differences. For example, the sponsor reported excluding some clinician responses that were considered highly uncertain and inaccurate in the presented context when estimating the proportion of patients with acute GvHD who are steroid refractory (i.e., 32.74%). Of note, this estimate was based on clinical expert opinion which ranged from |||||||||| to |||||||||| across jurisdictions. The inconsistency among clinical expert opinion, the small sample size of the survey, and an evidence source more susceptible to bias collectively add uncertainty to the resultant estimates used for deriving the population size and the estimated budget impact.
Further, the sponsor assumed |||||||||| of eligible patients would be covered by payers. However, according to the clinical experts, 100% of eligible patients would be covered under public drug plans if ruxolitinib is publicly reimbursed. Overall, there is notable uncertainty in the estimated proportion of aGvHD individuals who are steroid refractory and the proportion of patients with public coverage, which may have underestimated the population size and budget impact.
CADTH explored the impact of uncertainty in the proportion of eligible patients covered under public drug plans on the budget impact in a scenario analysis assuming a value of 100%, as estimated by expert opinion.
In a scenario analysis, CADTH explored the uncertainty in the proportion of patients with aGvHD who are steroid refractory, by assuming a value of 60%.
There is uncertainty regarding the market share of ruxolitinib and its comparators: The sponsor derived the market share of patients using ruxolitinib in Canadian clinical practice based on a survey of 10 clinicians across Canada. The sponsor assumed ruxolitinib has a market share of |||||||||| in year 1, |||||||||| in year 2, and |||||||||| in year 3. However, according to the clinical experts consulted for this review by CADTH, ruxolitinib is available through compassionate care or exceptional access programs on a case-by-case basis. If reimbursed, the clinical experts anticipate a higher market share and rapid uptake of ruxolitinib for aGvHD, with an initial market share between 70% to 90%.
Further, the sponsor aligned the market share of comparators with the proportions assumed under the REACH 2 trial’s BAT arm, which is also used in the cost-utility analysis. However, according to the clinical experts consulted by CADTH, there is heterogeneity in clinical practice and consequently, uncertainty in estimating the distribution and market share of comparators. For example, the sponsor assumed MTX has a market share of 1.10% at baseline. However, the clinical experts consulted for this review noted MTX is not used in clinical practice for treatment of SR-aGvHD. Further, 2 clinical experts reported using ECP in clinical practice, and one reported not using it at all. The heterogeneity in clinical practice adds uncertainty to market share estimates and the resultant budget impact. As such, CADTH’s distribution of BAT was based on the average responses received by clinical experts consulted for this review.
In the CADTH reanalysis, ruxolitinib had a market share of 80% in year 1, and 90% in year 2 and 3.
In the CADTH reanalysis, the market share of comparators at baseline was revised based on the average responses received by clinical experts consulted by CADTH for this review.
There is uncertainty in the dosing and treatment cost of comparators: To calculate the annual treatment costs of each comparator, except for ECP, the sponsor conducted a post hoc analysis on a 6-month data cut including individual patient information from the REACH 2 trial to estimate the mean and median daily doses, and treatment duration. CADTH was unable to validate and appraise the sponsor’s approach to estimating doses and treatment duration of comparators, as this was conducted in their post hoc analysis and was not presented in the REACH 2 clinical study report.
CADTH noted discrepancies in dosing of comparators from the sponsor’s post hoc estimation, when compared to the dosing found in published literature and indicated by expert opinion. For example, the sponsor adopted a much higher dose of infliximab (||||||||||) than recommended in the published literature (710 mg) for SR-aGvHD. Further, the sponsor adopted a median etanercept dosing ranged from ||||||||||||||||||||||||||||||, rather than 25 mg twice per week, as found in the literature.18 Similarly, the sponsor assumed a dose of ||||||||||||||||||||, but clinical experts consulted for this review by CADTH noted ATG can be dosed up to 7.5 mg/kg for SR-aGvHD. To increase the accuracy of the analysis, CADTH aligned the dosing of comparators with the published literature and expert opinion.
The cost of ATG is also uncertain. The sponsor based the unit price of ATG based on expert opinion collected from a sponsor-commissioned survey of 10 clinicians. CADTH obtained the wholesale price of ATG dated 2014 using IQVIA Delta PA database,22 which is much lower than the price calculated by the sponsor based on expert opinion.
The sponsor obtained the treatment cost of ECP using published literature and inflated the cost of ECP from 2006 to 2021 Canadian dollars. However, treatment reimbursement coverage of ECP is not through provincial oncology or non oncology drug plans. According to the clinical experts consulted by CADTH, ECP is primarily administered in an outpatient setting, which are funded by cancer care programs. In some scenarios, ECP treatment is delivered through the hospital electrophoresis unit, and funding comes from the global hospital budget. CADTH deemed the treatment cost of ECP is unlikely to be applicable to the public drug payer perspective.
In the CADTH reanalysis, the dosing of etanercept, infliximab, MMF and sirolimus were aligned with the published literature, and latest unit price available for ATG was adopted.
In the CADTH reanalysis, the treatment cost of ECP is excluded.
In scenario analyses, CADTH explored the impact of:
including ECP costs through a health care payer perspective.
Inflating ECP treatment cost.
In scenario analysis, CADTH explored the impact of assuming maximum ATG dosing (7.5 mg/kg or 532.5 mg).
CADTH Reanalyses of the Budget Impact Analysis
CADTH revised the sponsor’s base case by adopting a public drug plan perspective (excluding ECP treatment cost), revising market shares of ruxolitinib and comparators based on expert opinion, and aligning dosing of etanercept, infliximab, MMF and sirolimus with the published literature.18,20,28-30,32
Table 18: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Correctionsa to sponsor’s base case | ||
1. Perspective | Health care payer perspective (includes ECP treatment cost) | Public drug payer perspective (exclude ECP treatment cost) |
Changes to derive the CADTH base case | ||
1. Market share of ruxolitinib | |||||||||||||||||||||||||||||||||||||||| | 80.00% / 90.00% / 90.00% |
2. Market share of comparators (year 1 / year 2 / year 3) | ATG: |||||||||| ECP: |||||||||| Etanercept: |||||||||| Infliximab: |||||||||| MMF: |||||||||| MTX: |||||||||| Sirolimus: |||||||||| | ATG: 10.00% ECP: 17.00% Etanercept: 18.00% Infliximab: 12.00% MMF: 12.00% MTX: 0.00% Sirolimus: 31.00% |
3. Dosing | Etanercept: |||||||||||||||||||||||||||||||||||||||||||| Infliximab: |||||||||||||||||||||||||||||||||||||||| MMF: |||||||||||||||||||||||||||||||||||||||||||||||||| Sirolimus: |||||||||||||||||||||||||||||||||||||||||||||||||| | Etanercept: initial dose of 25 mg twice per week and maintenance dose of 25 mg once per week Infliximab: 710 mg once per week MMF: 1,000 mg twice daily Sirolimus: Loading dose of 6 mg and then, maintenance dose of 1 mg once daily |
4. Unit price of ATG | $12.6000 / mg | $1.4014 / mg |
CADTH base case | Reanalysis 1 + 2 + 3 + 4 | |
ATG = Antithymocyte globulin; ECP = extracorporeal photopheresis; MMF = mycophenolate mofetil; MTX = methotrexate
The results of the CADTH step-wise reanalysis are presented in summary format in Table 19 and a more detailed breakdown is presented in Table 20.
In the CADTH reanalysis, the 3-year budget impact of reimbursing ruxolitinib for patients aged 12 years and older with steroid refractory acute graft-versus-host disease was $1,412,268 (Year 1: $419,840; Year 2: $483,866; Year 3: $508,562).
Table 19: Summary of the CADTH Reanalyses of the Budget Impact Analysis
Stepped analysis | Three-year total |
|---|---|
Sponsor Submitted base case | $482,045 |
Corrected sponsor base case | $1,317,115 |
CADTH reanalysis 1 | $1,522,537 |
CADTH reanalysis 2 | $642,092 |
CADTH reanalysis 3 | $1,478,567 |
CADTH reanalysis 4 | $1,317,115 |
CADTH base case | $ 1,412,268 |
BIA = budget impact analysis
CADTH also conducted additional scenario analyses to address remaining uncertainty in nivolumab dosing, using the CADTH base case. Results are provided in Table 20:
Adopting a health care payer perspective, which includes the cost of ECP.
Assuming the average proportion of eligible patients covered under public drug plans is 100%.
Assuming 60% of patients with aGvHD are steroid refractory.
Assuming the maximum dose of ATG.
Adopting non inflated ECP costs from 2006 ($1,528 per treatment) using the health care payer perspective.
Table 20: Detailed Breakdown of the CADTH Reanalyses of the Budget Impact Analysis
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | Three-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $ 497,614 | $ 509,504 | $ 521,964 | $ 548,640 | $ 2,077,722 |
New drug | $ 497,614 | $ 660,513 | $ 681,040 | $ 720,599 | $ 2,559,767 | |
Budget impact | $ 0 | $ 151,010 | $ 159,076 | $ 171,959 | $ 482,045 | |
Corrected sponsor base case | Reference | $ 147,279 | $ 150,797 | $ 154,488 | $ 162,399 | $ 614,963 |
New drug | $ 147,279 | $ 563,395 | $ 589,131 | $ 632,273 | $ 1,932,078 | |
Budget impact | $ 0 | $ 412,598 | $ 434,643 | $ 469,874 | $ 1,317,115 | |
CADTH base case | Reference | $ 187,302 | $ 191,778 | $ 196,468 | $ 206,513 | $ 782,061 |
New drug | $ 187,302 | $ 611,618 | $ 680,334 | $ 715,075 | $ 2,194,329 | |
Budget impact | $ 0 | $ 419,840 | $ 483,866 | $ 508,562 | $ 1,412,268 | |
CADTH scenario analysis 1: Health care payer perspective | Reference | $ 375,218 | $ 384,184 | $ 393,579 | $ 413,688 | $ 1,566,669 |
New drug | $ 375,218 | $ 650,099 | $ 700,045 | $ 735,792 | $ 2,461,154 | |
Budget impact | $ 375,218 | $ 650,099 | $ 700,045 | $ 735,792 | $ 2,461,154 | |
CADTH scenario analysis 2: 100% public drug plan coverage | Reference | $ 234,128 | $ 239,722 | $ 245,585 | $ 258,141 | $ 977,576 |
New drug | $ 234,128 | $ 764,522 | $ 850,418 | $ 893,843 | $ 2,742,911 | |
Budget impact | $ 0 | $ 524,800 | $ 604,832 | $ 635,703 | $ 1,765,335 | |
CADTH scenario analysis 3: 60% of patients with aGvHD are steroid refractory | Reference | $ 343,274 | $ 351,476 | $ 360,073 | $ 378,481 | $ 1,433,304 |
New drug | $ 343,274 | $ 1,120,928 | $ 1,246,867 | $ 1,310,537 | $ 4,021,606 | |
Budget impact | $ 0 | $ 769,452 | $ 886,794 | $ 932,055 | $ 2,588,302 | |
CADTH scenario analysis 4: Maximum dose of ATG | Reference | $ 337,273 | $ 345,332 | $ 353,777 | $ 371,854 | $ 1,408,236 |
New drug | $ 337,273 | $ 642,328 | $ 696,065 | $ 731,609 | $ 2,407,275 | |
Budget impact | $ 0 | $ 296,996 | $ 342,288 | $ 359,755 | $ 999,040 | |
CADTH scenario analysis 5: No inflation of ECP cost | Reference | $ 342,401 | $ 350,582 | $ 359,155 | $ 377,507 | $ 1,429,645 |
New drug | $ 342,401 | $ 643,378 | $ 696,603 | $ 732,174 | $ 2,414,556 | |
Budget impact | $ 342,401 | $ 643,378 | $ 696,603 | $ 732,174 | $ 2,414,556 |
BIA = budget impact analysis
Stakeholder Input
Patient Input
Lymphoma Canada, Lymphoma and Leukemia Society of Canada, CLL Canada, Myeloma Canada, Aplastic Anemia & Myelodysplasia Association of Canada, Canadian MPN Research Foundation, Canadian MPN Network, Chronic Myelogenous Leukemia Network, Cell Therapy Transplant Canada
About Lymphoma Canada, Lymphoma and Leukemia Society of Canada, CLL Canada, Myeloma Canada, Aplastic Anemia & Myelodysplasia Association of Canada, Canadian MPN Research Foundation, Canadian MPN Network, Chronic Myelogenous Leukemia Network, and Cell Therapy Transplant Canada
The organizations involved in this submission are Canadian registered charities that provide support, education, and advocacy for their patient constituents. To learn more about the organizations involved in this submission, you can visit their respective websites:
Lymphoma Canada — https://www.lymphoma.ca/
Lymphoma and Leukemia Society of Canada (LLSC) — https://www.llscanada.org/
CLL Canada — https://cllcanada.org/
Myeloma Canada — https://www.myelomacanada.ca/en
Aplastic Anemia & Myelodysplasia Association of Canada (AAMAC) — https://aamac.ca/
Canadian MPN Research Foundation (CMPNRF) — https://www.cmpnrf.ca/ and the Canadian MPN Network — https://canadianmpnnetwork.ca/
Chronic Myelogenous Leukemia (CML) Network — https://cmlnetwork.ca/
Cell Therapy Transplant Canada (CTTC) — https://www.cttcanada.org/
Information Gathering
The patient organizations in collaboration conducted an anonymous online survey for patients with Graft versus Host Disease (GVHD) following allogeneic stem-cell transplantation between April 8, 2021 – June 26, 2021. Links to access the survey in French and English were sent via e-mail to patients registered through the organizations constituent databases. The survey was also made available via social media outlets as well as patient forums and was further sent to physicians to share with their patients. The survey had a combination of multiple choice, rating and open‐ended questions. Skipping logic was built into the survey so that respondents were asked questions only relevant to them. Open-ended responses that reflected the sentiment of a majority are included verbatim to provide a deeper understanding of the patient experience.
Of the 68 patients that responded to the survey, 53 experienced GVHD and 20 received treatment with Ruxolitinib. The patients without treatment experience provided their experience with GVHD. Of the patients that responded to this survey, (see Tables 1 and 2), 68% live in Canada, 54% are female, and 59% are ≥ 55 years-old.
Table 1: Country of Survey Respondents (68 respondents)
Respondents Ruxolitinib Experience | CAN* | USA | Europe | Asia | Total |
|---|---|---|---|---|---|
WITHOUT | 30 | 9 | 8 | 1 | 48 |
WITH | 16 | 2 | 2 | 0 | 20 |
*patients within Canada provided details on their province location: AB (n=1), BC (n=8), MB (n=1), NFL (n=2), NS (n=2), ON (n=22), SK (n=4), QB (n=6).
Table 2: Gender and Age of Survey Respondents (68 respondents)
Respondents Ruxolitinib Experience | Age Range | Gender | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
<18 | 18-24 | 25-34 | 35-44 | 45-54 | 55-64 | 65-74 | 75+ | Skipped | Female | Male | Prefer not to answer | Skipped | Total | |
WITHOUT | 2 | 0 | 4 | 5 | 5 | 11 | 18 | 2 | 1 | 29 | 18 | 0 | 1 | 48 |
WITH | 1 | 0 | 0 | 5 | 4 | 3 | 6 | 0 | 1 | 8 | 11 | 0 | 1 | 20 |
As GVHD can affect any patient receiving an allo-SCT, patients provided details related to their underlying condition. Of the 66 respondents (out of a total of 68 survey participants) who provided information on their subtype, 24% had Acute Myeloid Leukemia, 18% had Chronic Lymphocytic Leukemia, 21% had Myelodysplasia, 12% had Non-Hodgkin’s lymphoma, and 9% had Aplastic Anemia. The remainder of the patients had other blood-related disorders.
Disease Experience
With certain blood disorders, stem-cell transplantation can be a frontline treatment or treatment in the relapsed/refractory setting. Patients provided details about their treatment experience with SCT and GVHD. Of the 66 respondents, there were 6 patients that did not receive a SCT. For those that did receive an SCT, patients received this treatment options as their frontline treatment (17%), after one line of treatment (28%), after two lines of treatment (35%), after three lines of treatment (7%), after four lines of treatment (7%), or after 5 or more lines of treatment (6%). The majority of patients that had an SCT received one allo-SCT (82%; 49 respondents). Of the patients that underwent a SCT, 53 patients experienced GVHD as a side effect of this treatment. 45 of these 53 patients provided detail regarding the type of GVHD experienced: 13% experienced acute GVHD (within 100 days of transplant), 24% experienced chronic GVHD (later than 100 days after transplant), and 62% experienced both acute and chronic GVHD.
GVHD can have a lasting impact on a patient’s life, as symptoms can continue for months to years. GVHD appeared for patients at different times following their SCT: 1-30 days post-SCT (31%), 31-100 days post-SCT (33%), 101-356 days post-SCT (33%), and over one year following SCT (3%). Figures 1 and 2 provide additional information on the severity of GVHD experienced by patients.
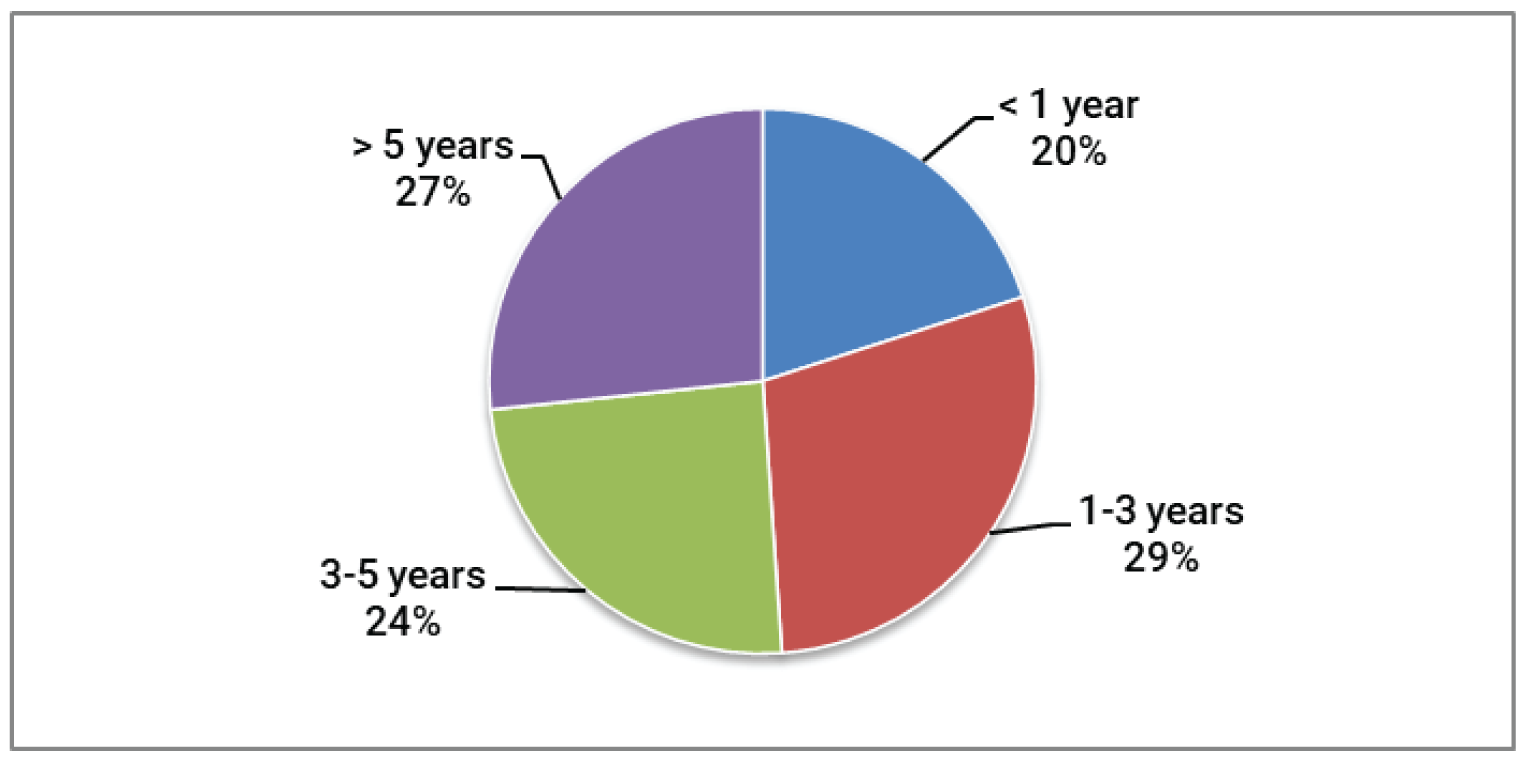
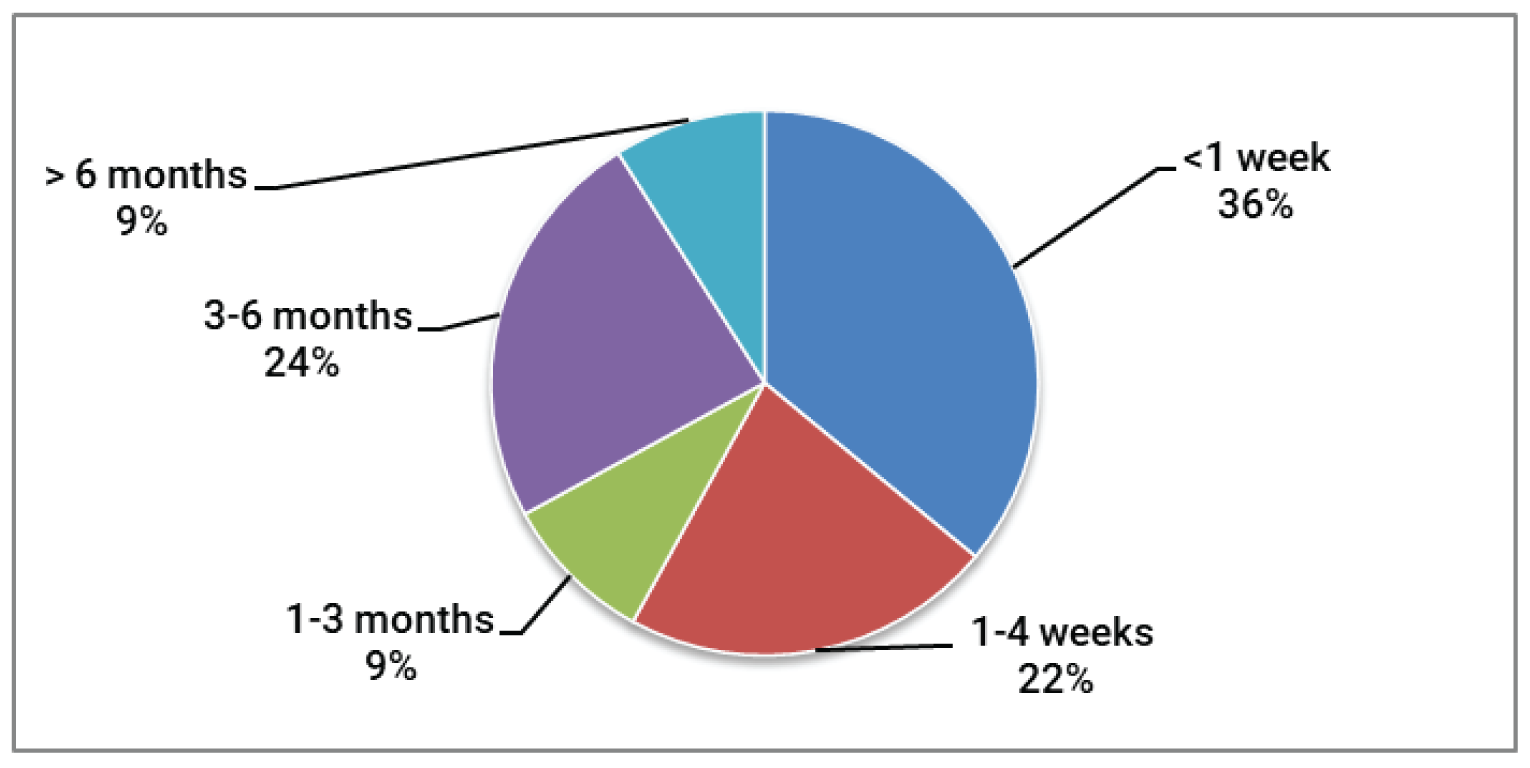
As a result of patients experiencing GVHD following their stem-cell transplant, many patients have had to visit the transplant centre many more times to treat their GVHD (76%), they had to consult with specialists for treatment of their GVHD (67%), had to be re-admitted to the hospital for care for their GVHD (31%), and visited the Emergency Department multiple times due to their GVHD (24%) (45 respondents). There is a wide symptom profile that impacts patients QoL when diagnosed with GVHD. Patients diagnosed with GVHD listed their symptoms and the impact to their QoL (Table 3).
Table 3: Symptom Profile and Severity Related to GVHD and Impacts on QoL
Symptom of GVHD | Did not Experience | Mild-Moderate Impact | Significant Impact | n |
|---|---|---|---|---|
Burning and redness of the skin on the palms of the hands or soles of the feet | 49% | 32% | 19% | 41 |
Rashes that can spread over the entire body | 33% | 38% | 29% | 42 |
Blisters and peeling skin | 60% | 27% | 13% | 40 |
Skin problems such as dryness, rash, itching, peeling, darkening, hard texture and feeling tight | 12% | 38% | 50% | 42 |
Enlarged liver, liver tenderness, abnormal liver enzymes or liver failure | 52% | 29% | 19% | 42 |
Jaundice | 87% | 10% | 3% | 39 |
Dry eyes that may have a burning or gritty feeling | 23% | 34% | 43% | 44 |
Dry mouth with or without mouth ulcers | 14% | 43% | 43% | 44 |
Diarrhea, loss of appetite, stomach cramps, vomiting | 36% | 31% | 33% | 42 |
Weight loss | 40% | 45% | 15% | 40 |
Pain in muscles and joints | 31% | 45% | 24% | 42 |
Mobility issues and difficulties | 29% | 40% | 31% | 42 |
Infections | 35% | 37% | 28% | 43 |
Difficulty breathing | 42% | 34% | 24% | 41 |
Other | 58% | 10% | 32% | 29 |
“Nobody can tell me why most people with gvhd suffer from severe muscle cramping. Doctors seem to be baffled what the cause is and how to remedy it.”
“It’s complicated, difficult to control and manage, unsure if symptoms are GVHD or side effects of medication.”
43 respondents out of 45 indicated that their GVHD had an impact on their quality of life, rating at least one of the following impacts as significant (4) or extremely significant (5), the ability to: work, travel, exercise, spend time with family and friends, continue daily activities, concentrate, maintain intimate relationships, and maintain mental health.
As a result of the symptoms experienced from their GVHD diagnosis, patients described the psychological and social impacts to their life. The top 5 impacts are presented in Table 4, (on a scale of 1 – 5, where 1= No impact and 5 = Significant negative impact). Every one of the 45 respondents experienced at least one social and psychological impact. Only one respondent rated the impacts as mild; 34 out of 45 respondents rated at least one impact as significant (4) or extremely significant (5). The full list of impacts can be found in Table 4. As described by one patient:
“Not being able to play with my child is torture.”
Table 4: Quality of Life Impacts Related to GVHD (43-45 respondents)
Psychological/Social Impact | Mild to Moderate Impact (2-3) | Significant Impact (4-5) |
|---|---|---|
Interruption of life goals/accomplishments (career, schooling) | 33% | 44% |
Difficulty sleeping | 55% | 30% |
Stress/anxiety/worry | 55% | 31% |
Problems concentrating | 47% | 28% |
Financial impacts (cost of travel, inability to work, etc.) | 42% | 38% |
“8 years and it is on-going! It is very frustrating! Most times , gvhd in one place/organ is one of a cluster of symptoms somewhere else... eg lungs /sepsis ; chronic kidney disease / meds”
“Also the uncertainty of life: having a new grandchild in another province.... will I or will I not be well enough to visit in 3 months? Never being able to plan ahead. Always having to make last minute plans depending on health.”
Summary
The symptoms of GVHD are long lasting: 3-5 years for 26% of respondents and more than 5 years for 28% of them.
Respondents required numerous medical consultations, hospital stays and nights away from home.
The symptoms of GVHD are many and varied. They have a significantly reduce the capacity of a majority of respondents to live day to day and to experience the simple pleasures of life.
Experiences With Currently Available Treatments
A treatment like Jakavi, which does not require a visit to a treatment centre, has a clear advantage for patients who live outside major urban areas, even more so to the degree that it controls the GVHD. While the majority of patients (59%) lived less than a hundred kilometers from their treatment centre, 11% lived between 200-400 km away and 11% lived over 400 km from their treatment centre (44 respondents). On a scale 1 (no impact) to 5 (extremely significant impact), patients rated significant impacts (4-5) of not being able to access care locally: extensive cost of travel and accommodations (30%), impact to daily activities/routine (23%), emotional hardship (20%), and not receiving proper care for my GVHD (23%) (30 respondents). A full list of treatments and side effects and their impacts on quality of life can be found in Tables 5 and 6. As described by one GVHD patient:
“Biggest problem is distance. I live 1200 kms (return) from most specialists. I had my transplant 10 years ago and the cost in $$'s, time and disruption both physically and mentally is immense. One 15 minute appt takes 3 days: Day 1... drive for 7 hours, Day 2 ...have a 15 minute appointment Day 3.....drive 7 hours home. Other times I would drive for a 15 minute appt and then an array of tests/other specialists etc would be scheduled and I would not get home for 1 to 2 weeks. Then I would have to arrange for care of my home and find a place to stay, never really knowing how long I would need accommodations.”
Table 5: GVHD Treatment Experience (43 respondents)
Treatment | Percentage | Treatment | Percentage |
|---|---|---|---|
Steroids | 91% | Mycophenolate mofetil (MMF) | 20% |
Cyclosporine | 44% | Methotrexate | 13% |
Tacrolimus | 38% | Monoclonal antibodies | 9% |
Tyrosine kinase inhibitors (TKIs) incl. Ruxolitinib (Jakavi) | 31% | Imuran (Azathioprine) | 9% |
Light treatment (ECP) | 24% | Can’t Remember | 9% |
Table 6: GVHD Treatment-Related Side Effects (45 respondents)
Side Effect | No Impact (1) | Mild-Moderate Impact (2-3) | Significant Impact (4-5) |
|---|---|---|---|
General feeling of being unwell | 18% | 51% | 31% |
Tiredness | 16% | 44% | 40% |
Raised blood pressure | 47% | 31% | 22% |
Shaking hands (tremor) | 44% | 29% | 27% |
Kidney problems | 51% | 27% | 22% |
High blood sugar | 53% | 18% | 29% |
Feeling sick and loss of appetite | 42% | 31% | 27% |
Diarrhea | 42% | 36% | 22% |
Difficulty sleeping | 18% | 49% | 33% |
Itchy skin | 20% | 58% | 22% |
Eye problems | 25% | 33% | 42% |
“Sometimes, the doctors at my transplant center hospital do not understand about GVHD and so are not sure why I see them for some of the issues, such as polymyositis. I have been terminated from meeting with several rheumatologists because they do not know why I am seeing them, as it is GVHD, and then my transplant doctor struggles with this because he is a hematologist oncologist and so has concerns about his competency in treatment of PM GVHD, which is frustrating for him and me.”
“Multiple complications from medications eg prednisone causing hypertension, diabetes, hyper cholesterolemia, osteoporosis, weight gain, cataracts, hips a vascular sclerosis, etc.”
Summary
Patients undergo many treatments to treat the symptoms of their GVHD, but those treatments have their own side effects which necessitate further medical consultations and treatments.
The side effects from treatments have a negative effect on the quality of life of a majority of respondents.
Managing GVHD requires a significant amount of travel and time away from home, particularly for patients residing outside of major urban centres
Improved Outcomes
When making a decision about taking a new GvHD treatment, patients rated the most important factors to them on a scale from 1-5 (1= not important at all, and 5 =extremely important) (Table 7).
Table 7: Important Considerations Related to New GVHD Treatments (43-44 respondents)
Consideration | Rating (4-5) | Consideration | Rating (4-5) |
|---|---|---|---|
That the treatment Improves Quality of Life | 89% | Religious considerations | 2% |
Outpatient treatment (no overnight hospital stay required) | 65% | Severity of side effects | 75% |
Recommended by healthcare team | 84% | The impact to caregiver/partner/family | 50% |
Least amount of travel required for treatment | 55% | Improved length of survival | 95% |
Degree of certainty that it will relieve my GvHD | 91% | Covered by insurance/drug plan | 82% |
Summary
While it is no surprise that improved survival was rated as the most important consideration in a new treatment, quality of life followed close behind.
The degree of certainty that the treatment will relieve GVHD was also highly rated, a sign of the frustration patients feel of having to undergo multiple treatments that are not always effective in treating the symptoms of GVHD and sometimes created their own problems.
Experience With Drug Under Review
“GvHD has been frustrating, as few treatments seem to solve it and it is a condition that doesn't get a lot of attention. During my treatment with Ruxolitinib, my symptoms appeared to improve.”
“C'est lui qui a régler ma GVH (It is this treatment that fixed my GVHD).”
Twenty patients treated with Ruxolitinib shared their experience. Their disease indications for which SCT was needed which then led to GVHD included: Myelofibrosis (4), CLL (2), Myelodysplasia (2), AML (7), ALL (1), Multiple Myeloma (1), NHL (1), Aplastic Anemia (1), and not reported (1).
Patients treated with Ruxolitinib have received this treatment for 1-3 years (47%), 1-6 months (26%), 6-12 months (16%), <1 month (5%), or over 3 years (5%) (19 respondents). 74% of patients were still receiving this treatment while completing the survey; three patients had completed their course of Ruxolitinib, one patient had to stop treatment due to side effects, and one patient stopped as it was not helping their GVHD (19 respondents).
Patients were able to access this treatment through a compassionate use program (32%), it was paid for by a cancer board/agency or government (32%), a clinical trial (16%), private insurance (16%), or paid out of pocket (5%). Patients questioned about the difficulty in obtaining the drug on a scale of 1 to 5 (1=not difficult, 5=extremely difficult) rated getting the drug paid for was the most difficult. Getting a prescription from the physician for this drug, having the drug delivered, and access to a treatment centre were rated as not difficult to mildly difficult. Access is an important consideration for patients living far from treatment centres.
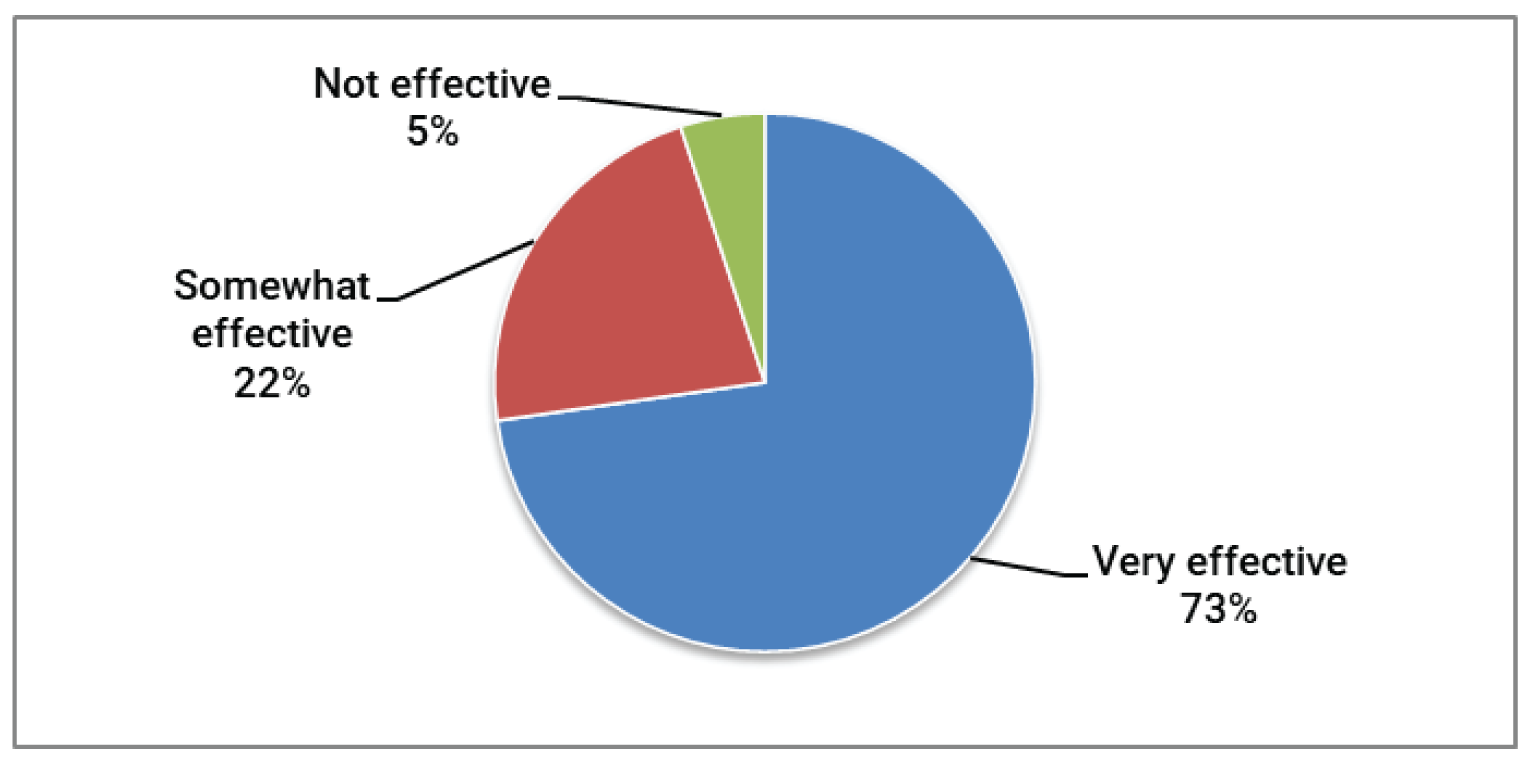
Success of Treatment: Patients were asked if Ruxolitinib helped control all of their GVHD symptoms. Patients rated this on a scale of 1 to 5 (1=did not help any symptoms, 3=helped with some of symptoms, 5=helped with all my symptoms), and found that 24% of patients rated all their GVHD symptoms were managed by this treatment. 71% of patients rated 3 or 4, indicating that some to most of their symptoms were managed by Ruxolitinib (18 respondents). Patients were asked if their GVHD overall responded to the Ruxolitinib treatment. 50% of patients had their GVHD respond completely or partially (39%), while only 6% did not respond (18 respondents). Figures 3 and 4 share the effectiveness and impacts on QoL of treatments.
Side Effects of Ruxolitinib Treatment and Impacts of Quality of Life: Rated on a scale from (1=did not experience side effect, 2-3 = minor to manageable side effect, 5=very serious side effect), the weighted average was below 2, indicating that, overall, the side effects experienced were minor or manageable. The most common serious side effects (4-5) experienced by patients included infection (12%), low platelet/red blood cell count (11%), bruising (6%), diarrhea (11%), and fluid retention (6) (17-18respondents). Based on patients experience with side effects, the majority of patients (67%) rated the side effects experienced from tolerable to very tolerable when rated on a scale from 1-5 (1=not tolerable, 3=tolerable, 5=very tolerable). The full impact of treatments on QoL are described in Table 8.
Table 8: Quality of Life Impacts with Ruxolitinib Treatment (18 respondents)
Impact | Negative Impact (1-2) | No Impact (3) | Positive Impact (4-5) |
|---|---|---|---|
Relationships with family/friends | 0% | 56% | 44% |
Intimate relationships | 6% | 67% | 27% |
Personal image | 6% | 56% | 38% |
Ability to work/go to school/volunteer | 6% | 56% | 38% |
Mental health | 6% | 50% | 44% |
Travel | 0% | 57% | 33% |
Perform daily activities | 0% | 50% | 50% |
Overall Experience: Based on patients experience with Ruxolitinib, 94% of patients would take this treatment again if their doctor recommended it. Similarly, 94% of patients would recommend this treatment to other patients diagnosed with GVHD. As described by patients:
“I find that we are too cautious and wait too long before using the best method... why didn't we go immediately with Jakavi when we saw that other drugs (tacro and syro) didn't work for me... it adds time, fatigue and very painful side effects... and that compromises our quality of life for a long time...”
“Need faster authorization of new drugs that act more quickly and effectively to improve our quality of life during these terrible ordeals of chemotherapy, radiotherapy, autograft, allograft, etc.”
“Jakavi has been helpful in controlling my GVHD, and I appreciate the lack of side effects compared to other treatments (e.g., Prednisone).”
Summary
A large majority of respondents stated that Ruxolitinib:
Was an effective treatment
Improved their quality of life
Had tolerable side effects
Is a treatment they would take again if recommended by their doctor
Is a treatment they would recommend to other patients.
Companion Diagnostic Test
There is no companion diagnostic testing required for this treatment.
Anything Else?
The following are quotes shared by patients through their participation in the survey about their experience with their GVHD and treatments:
GVHD takes over a patient’s life.
I sometimes wonder if I made the right decision to go with a stem cell transplant. If I knew what I know now about how shitty gvhd is, I would have not gone through with the stem cell transplant. Even though it did prolong my life, it hasn't been much of an enjoyable life with gvhd. I don't find that it was worth it. Gvhd slowly wastes us away into shriveled up remnants of a human being. It is not what I want to be reminded as, but I have no choice at the moment.
GVHD is not well known, so it is difficult to find doctors that are sufficiently knowledgeable about it
The doctors in my small Northern Michigan town are outstanding... but no one here knows much about gvhd and most have never even heard if it. As a result, I have been misdiagnosed several times and given a prescription that actually worsened the situation. GVHD can mimic a lot of other maladies and is so unique.
There is a HUGE deficit of doctors who have an interest in, or knowledge of, GVHD. This MUST be rectified !!! My BMT team sent me to an eye doctor who supposedly “specialized” in GVHD - but she misdiagnosed me and mid treated me –
All said: it is better to have chronic GVH then the alternative. (To misquote Mark Twain)
It was a hell of a lifetime… in a bad way!! But I survived so I guess it was worth it! And still is..
Not so much access to treatment, more of how long it would take to receive treatment. A gvhd flare up would be out of control before I would receive treatment.
There just isn’t a lot of answers to our problems. We find that we are our own doctors and if we don’t push for answers they never come
I would hope for one medication and not for 10 different ones and always having to try if it is the right one, or if this one might help now…
Je trouve qu’on y va souvent avec trop de précautions et d’attente avant d’utiliser la meilleure méthode... pourquoi ne pas avoir été immédiatement avec Jakavi lorsque on voyait que tacro et syro ne fonctionnaient pas pour moi... ça rajoute du temps de la fatigue et des effets secondaires très pénibles... et qui hypothèque notre qualité de vie.... pour très longtemps.
Vivement autoriser plus rapidement tout nouveaux médicaments qui agit plus rapidement et plus efficacement afin d’améliorer notre qualité de vie durant ces terribles épreuves de chimiothérapie radiothérapie autogreffe allogreffe etc.
Patient Group Conflict of Interest Declaration
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
There were eight patient groups that participated in the development of the survey and the analysis to develop the final submission.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
Table 9: Conflict of Interest Declaration for Lymphoma Canada
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Novartis | — | — | X | — |
Table 10: Conflict of Interest Declaration for CLL Canada
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Novartis | X | — | — | — |
Table 11: Conflict of Interest Declaration for the Lymphoma and Leukemia Society of Canada
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Novartis | — | — | — | X |
Table 12: Conflict of Interest Declaration for Cell Therapy Transplant Canada
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Novartis | X | — | — | — |
Table 13: Conflict of Interest Declaration for Chronic Myelogenous Leukemia Network
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Novartis | — | — | X | — |
Table 14: Conflict of Interest Declaration for Myeloma Canada
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Novartis | X | — | — | — |
Table 15: Conflict of Interest Declaration for the Aplastic Anemia & Myelodysplasia Association of Canada
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Novartis | — | X | — | — |
Table 16: Conflict of Interest Declaration for the Canadian MPN Research Foundation
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Novartis — No COI | — | — | — | — |
Clinician Input
Cell Therapy Transplant Canada
About Cell Therapy Transplant Canada
Cell Therapy Transplant Canada (CTTC) is a member-led, national, multidisciplinary organization providing leadership and promoting excellence in patient care, research, and education in the field of hematopoietic stem cell transplant and cell therapy. The CTTC advocates, nationally and internationally, for improving the outcomes and accessibility of cellular therapies and transplantation for Canadians. Representation in the CTTC includes physicians, nursing, laboratory and allied health professionals, along with an active family and caregiver group.
Information Gathering
Information was gathered by discussion and approval by two CTTC committees – the CTTC Board of Directors, and the CTTC standing committee of program directors, with representation from all 23 allogeneic stem cell transplant programs across Canada. This report was approved by both committees.
Current Treatments
Describe the current treatment paradigm for the disease.
Response: There are no Health Canada approved therapies for either steroid refractory acute Graft-versus-host disease (aGvHD) or chronic GvHD (cGvHD). The prognosis of both steroid refractory aGvHD and cGvHD is poor resulting in a significant increase in both mortality and morbidity of stem cell transplantation. There is no standard of care as a second line therapy. There are several aGvHD and cGvHD therapies that are currently used off label. Examples include extracorporeal photopheresis, mycophenolate mofetil, sirolimus, everolimus, imatinib, and rituximab. There is some province-to-province variation on standard practice, based on local funding of available options. Comparison of ruxolitinib to these currently used therapies in the REACH-2 and REACH-3 trials found all established therapies were inferior to ruxolitinib.
Treatment Goals
What are the most important goals that an ideal treatment would address?
Response: Based on the REACH trials, ruxolitinib currently represents the best therapeutic option to reduce the mortality and symptom burden associated with both steroid refractory aGvHD and cGvHD. In particular, it has the potential to significantly reduce corticosteroid use and improve quality of life with a low rate of adverse events. Non-response of steroid refractory aGvHD to second line therapies has a high mortality rate [1]. Steroid refractory cGvHD is the primary non-relapse cause of post-transplant mortality. In addition, steroid refractory cGvHD has major morbidity and can include decreased mobility, liver failure, renal failure, gastrointestinal failure, cardiac failure, renal failure, keratoconjunctivitis, and stomatitis [2]. One of the most severe symptoms is development of irreversible bronchiolitis obliterans which has a high mortality rate [3].
Mohty M, Holler E, Jagasia M, Jenq R, Malard F, Martin P, Socié G, Zeiser R. Refractory acute graft-versus-host disease: a new working definition beyond corticosteroid refractoriness. Blood. 2020 Oct 22;136(17):1903-1906.
DeFilipp Z, Couriel DR, Lazaryan A, Bhatt VR, Buxbaum NP, Alousi AM, Olivieri A, Pulanic D, Halter JP, Henderson LA, Zeiser R, Gooley TA, MacDonald KPA, Wolff D, Schultz KR, Paczesny S, Inamoto Y, Cutler CS, Kitko CL, Pidala JA, Lee SJ, Socie G, Sarantopoulos S, Pavletic SZ, Martin PJ, Blazar BR, Greinix HT. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: III. The 2020 Treatment of Chronic GVHD Report Transplant Cell Ther. 2021 Jun 11:S2666-6367(21)00895-2.
Wolff D, Radojcic V, Lafyatis R, Cinar R, Rosenstein RK, Cowen EW, Cheng GS, Sheshadri A, Bergeron A, Williams KM, Todd JL, Teshima T, Cuvelier GDE, Holler E, McCurdy SR, Jenq RR, Hanash AM, Jacobsohn D, Santomasso BD, Jain S, Ogawa Y, Steven P, Luo ZK, Dietrich-Ntoukas T, Saban D, Bilic E, Penack O, Griffith LM, Cowden M, Martin PJ, Greinix HT, Sarantopoulos S, Socie G, Blazar BR, Pidala J, Kitko CL, Couriel DR, Cutler C, Schultz KR, Pavletic SZ, Lee SJ, Paczesny S. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: IV. The 2020 Highly morbid forms report. Transplant Cell Ther. 2021 Jun 10:S2666-6367(21)00949-0.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
Response: Current available treatment options are suboptimal and new therapies are urgently needed. In addition, current aGvHD and cGvHD therapies can increase the risk of relapse these high-risk patients post-allogeneic stem cell transplant. Current therapies still require relatively high doses and the prolonged use of corticosteroids to control disease [2] and drugs that offer the potential to decrease the long-term morbidity of steroids are needed. Ruxolitinib represent one of the best options of currently available drugs. Minimizing the use of prolonged steroids will result in a reduced risk of steroid-induced opportunistic infections, osteoporosis, and avascular necrosis.
Which patients have the greatest unmet need for an intervention such as the drug under review?
Response: All patients with steroid refractory aGvHD or cGvHD would be expected to benefit – there are no specific subpopulations that would be appropriate for this treatment.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Response: Given that this therapy was shown to be superior to current standard of care options, it would become the dominant first line therapy for steroid refractory aGvHD or cGvHD. Other therapies that are currently used off-label would be used for patients that do not respond to ruxolitinib, or patients that are not candidates for ruxolitinib, for example due to significant thrombocytopenia.
Please indicate whether it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective.
Response: Patients would be required to start therapy with corticosteroids, as this remains the initial therapy for both aGvHD and cGvHD, but a second agent in addition to steroids is almost always required. It would not be appropriate to require that patients try other therapies for steroid refractory aGvHD or cGvHD prior to ruxolitinib, given that all these therapies were shown to be inferior to ruxolitinib.
How would this drug affect the sequencing of therapies for the target condition?
Response: This therapy would become the preferred initial therapy for patients with steroid refractory aGvHD or cGvHD.
Which patients would be best suited for treatment with the drug under review?
Response: All patients with steroid refractory aGvHD or cGvHD would be well suited for this therapy, except for patients with significant baseline thrombocytopenia.
How would patients best suited for treatment with the drug under review be identified?
Response: Patients with aGvHD and cGvHD are managed in highly specialized stem cell transplant clinics, at a limited number of tertiary care centres across Canada. These centres have physicians and clinical teams that are experienced at managing GvHD, and we do not expect misdiagnosis to be a significant issue. Patients that are eligible for this therapy will be identified by these teams.
Which patients would be least suitable for treatment with the drug under review?
Response: Patients with baseline thrombocytopenia would be least suitable for this therapy.
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review?
Response: All patients with steroid refractory aGvHD or cGvHD would be good candidates for this therapy. There are no subsets of these patients that would be more or less likely to respond.
What outcomes are used to determine whether a patient is responding to treatment in clinical practice?
Response: The outcomes that were used in the clinical trial are used in clinical practice (overall response rate, corticosteroid dose).
What would be considered a clinically meaningful response to treatment?
Response: Patients will be determined to be responding if their overall symptom burden due to GvHD is decreasing, if their overall quality of life is improving, and if the corticosteroid dose is able to be successfully tapered. The outcomes used in the clinical trial correspond to clinically meaningful responses.
How often should treatment response be assessed?
Response: These patients are followed quite closely by transplant physicians (often weekly or biweekly visits).
What factors should be considered when deciding to discontinue treatment?
Response: In general, the criteria used in the clinical trial to determine lack of response are those used in clinical practice (absence of improvement in GvHD symptoms, worsening of GvHD, or lack of ability to reduce the dose of corticosteroid).
What settings are appropriate for treatment with the drug under review?
Response: This therapy should only be prescribed for this indication by specialists working in a clinic associated with an allogeneic stem cell transplant program. In general, these are in cancer centres associated with tertiary care hospitals in Canada.
For non-oncology drugs, is a specialist required to diagnose, treat, and monitor patients who might receive the drug under review?
Response: See previous statement.
Additional Information
Response: The availability of a Health Canada approved and provincially funded therapy for steroid refractory GvHD would be an important step forward for our community. There is a significant unmet need for this indication, with existing therapies offering low response rates and high rates of toxicity. The completion of a randomized control trial for this indication is a large step forward for our community and our patients. Many of us have experience using ruxolitinib through an available compassionate access program, and real world effectiveness appears similar to that in the clinical trial, with very low rates of toxicity. We feel strongly that this therapy should be readily available for our patients, many of whom suffer with low quality of life due to this debilitating disease.
Conflict of Interest Declarations — Cell Therapy Transplant Canada
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
No help from outside the clinician group was obtained.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No help from outside the clinician group was obtained.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician that contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
Declaration for Clinician 1
Name: Kristjan Paulson
Position: President, CTTC, Hematologist, CancerCare Manitoba, Assistant Professor, University of Manitoba
Date: 19-08-2021
Table 17: Conflict of Interest Declaration for Cell Therapy Transplant Canada — Clinician 1
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 2
Name: Mohamed Elemary
Position: Secretary, CTTC, Hematologist, Saskatoon Cancer Center, Professor, University of Saskatchewan
Date: 19-08-2021
Table 18: Conflict of Interest Declaration for Cell Therapy Transplant Canada — Clinician 2
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Jazz Pharmaceuticals | X | — | — | — |
AbbVie pharmaceuticals | X | — | — | — |
Bristol Myers Squibb | X | — | — | — |
Paladin Labs Inc. | X | — | — | — |
AstraZeneca | X | — | — | — |
Pfizer | X | — | — | — |
Novartis | X | — | — | — |
Declaration for Clinician 3
Name: Wilson Lam
Position: Education Director, CTTC, Hematologist, Princess Margaret Cancer Centre
Date: 22-08-2021
Table 19: Conflict of Interest Declaration for Cell Therapy Transplant Canada — Clinician 3
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 4
Name: Kirk R. Schultz
Position: President-elect, CTTC, Professor of Pediatrics, UBC, Pediatric HSCT physician, BCCH
Date: 24-08-2021
Table 20: Conflict of Interest Declaration for Cell Therapy Transplant Canada — Clinician 4
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 5
Name: Jonas Mattsson
Position: Director allo-BMT, Princess Margaret Cancer Centre, Professor University of Toronto
Date: 01-09-2021
Table 21: Conflict of Interest Declaration for Cell Therapy Transplant Canada — Clinician 5
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 6
Name: Imran Ahmad
Position: Hematologist, Cellular Therapy & Transplantation Program Director, HMR, Université de Montréal
Date: 01-09-2021
Table 22: Conflict of Interest Declaration for Cell Therapy Transplant Canada — Clinician 6
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Novartis (consultancy for Jakavi submission at INESSS) | — | X | — | — |
Declaration for Clinician 7
Name: Gizelle Popradi
Position: Hematologist, Director of the McGill University Hospital Center Stem Cell Transplant Program
Date: 01-09-2021
Table 23: Conflict of Interest Declaration for Cell Therapy Transplant Canada — Clinician 7
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Novartis, speaker and consultancy fees | — | X | — | — |
Declaration for Clinician 8
Name: Mona Shafey
Position: Medical Director, Alberta Blood & Marrow Transplant Program
Date: 02-09-2021
Table 24: Conflict of Interest Declaration for Cell Therapy Transplant Canada — Clinician 8
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Novartis | X | — | — | — |
Ontario Health (Cancer Care Ontario) Complex Malignant Hematology
About Ontario Health (Cancer Care Ontario) Complex Malignant Hematology
An agency of the Ministry of Health, Ontario Health (Cancer Care Ontario) is the Ontario government’s principal advisor on cancer and chronic kidney disease care, as well as access to care for key health services. It is guided by a mission that together we will improve the performance of our health systems in Ontario by driving quality, accountability, innovation and value. Ontario Health (Cancer Care Ontario) manages infrastructure, assets and models to improve the province’s health systems for cancer and chronic kidney disease (through its division the Ontario Renal Network). It also directs and oversees healthcare funds for hospitals and other cancer and chronic kidney disease care providers, enabling them to deliver high-quality, timely services and improved access to care. As an operational service agency of the Government of Ontario, Ontario Health (Cancer Care Ontario) is accountable for conducting a fair and transparent process, providing equal treatment to all qualified parties, in selecting a candidate for the above mentioned role.
Information Gathering
Discussed via emails.
Current Treatments
Describe the current treatment paradigm for the disease.
Response: Half of patients treated with steroids for aGVHD will fail steroids and require second line therapy. Over half of patients with steroid refractory aGVHD eventually die of GVHD or treatment-related toxicity (opportunistic infections and side effects of treating them).
Treatment Goals
What are the most important goals that an ideal treatment would address?
Response: Effective, widely available aGVHD therapy will improve survival, QoL, and decrease health care costs.
Treatment Gaps (Unmet Needs)
Considering the treatment goals, please describe goals (needs) that are not being met by currently available treatments.
Response: This is an area of unmet need. JAKAVI is the only Health Canada approved oral therapy for this indication.
Which patients have the greatest unmet need for an intervention such as the drug under review?
Response: It greatly facilitates care of patients who do not live near transplant centres. Oral therapy facilitates its use and ruxolitinib is not as immunosuppressive as other options.
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
Response: Ruxolitinib will be the treatment of choice for the majority of patients with steroid-refractory aGVHD.
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective.
Response: Once steroid refractory, ruxolitinib would be the next line of therapy for the majority of patients. Our alternatives, used off label, all have drawbacks (IV, require patient to be at hospital, side effects and broad immune suppression, expensive products and related delivery costs). There may be some patients that we would still favour off label use but that will likely change as we gain more experience. One example is primarily moderate-severe lower GI aGVHD that is steroid refractory. We would like continue to start with a TNF inhibitor but would add in JAKAVI and decrease TNF inhibitor doses as they are very immune suppressive.
How would this drug affect the sequencing of therapies for the target condition?
Response: Ruxolitinib would be the preferred option when steroid refractory. When ruxolitinib fails, patients will try other treatment alternatives.
Which patients would be best suited for treatment with the drug under review?
Response: There were no subgroups that benefited less to ruxolitinib. Patients with significant thrombocytopenia may be challenging to treat with ruxolitinib.
How would patients best suited for treatment with the drug under review be identified?
Response: Patients will be followed by transplant centre.
Which patients would be least suitable for treatment with the drug under review?
Response: Ruxolitinib would be challenging for patients with thrombocytopenia who are on full anticoagulation.
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review?
Response: No data to inform
What outcomes are used to determine whether a patient is responding to treatment in clinical practice?
Response: There is established standard GVHD response measurement/scale in practice.
What would be considered a clinically meaningful response to treatment?
Response: Improvement as assessed by validated GVHD scale
How often should treatment response be assessed?
Response: Ruxolitinib generally works quickly (weeks) so we know if we are making progress within a few weeks to a month generally. Patients weaned off JAKAVI can restart the medication if aGVHD flares.
What factors should be considered when deciding to discontinue treatment?
Response: Disease progression, adverse events, lack of response, successful weaning of corticosteroid
What settings are appropriate for treatment with the drug under review?
Response: Ruxolitinib is oral (out-patient therapy). Some patients will require to start ruxolitinib in the hospital.
For non-oncology drugs, is a specialist required to diagnose, treat, and monitor patients who might receive the drug under review?
Response: Yes. These patients are followed in transplant centres.
Additional Information
Is there any additional information you feel is pertinent to this review?
Response: NA
Conflict of Interest Declarations — Ontario Health (Cancer Care Ontario) Complex Malignant Hematology
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (section 6.3) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
OH-CCO provided secretariat support in completing this input.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician that contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
Declaration for Clinician 1
Name: Dr. Tom Kouroukis
Position: Provincial Head, Complex Malignant Hematology, Ontario Health (Cancer Care Ontario)
Date: 9-Sep-2021
Table 25: Conflict of Interest Declaration for Ontario Health (Cancer Care Ontario) Complex Malignant Hematology — Clinician 1
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Declaration for Clinician 2
Name: Dr. Christopher Bredeson
Position: Clinical Lead, Quality Care and Access, Complex Malignant Hematology, Ontario Health (Cancer Care Ontario)
Date: 1-Sep-2021
Table 26: Conflict of Interest Declaration for Ontario Health (Cancer Care Ontario) Complex Malignant Hematology — Clinician 2
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Novartis — No COI | — | — | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines.
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.