CADTH Reimbursement Review
Upadacitinib (Rinvoq)
Sponsor: AbbVie Corporation
Therapeutic area: Atopic dermatitis
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
AAD
American Academy of Dermatology
AD
atopic dermatitis
ADerm-IS
Atopic Dermatitis Impact Scale
ADerm-SS
Atopic Dermatitis Symptom Scale
ADL
activity of daily living
AE
adverse event
AESI
adverse event of special interest
ANOVA
analysis of variance
BE
blinded extension
BMI
body mass index
BSA
body surface area
CDLQI
Children’s Dermatology Life Quality Index
CI
confidence interval
CMH
Cochran-Mantel-Haenszel
CPK
creatine phosphokinase
CrI
credible interval
CSPA
Canadian Skin Patient Alliance
DIC
deviance information criterion
DLQI
Dermatology Life Quality Index
EASI
Eczema Area and Severity Index
EASI 50
at least 50% improvement in Eczema Area and Severity Index total score from baseline
EASI 75
at least 75% improvement in Eczema Area and Severity Index total score from baseline
EASI 90
at least 90% improvement in Eczema Area and Severity Index total score from baseline
EQ-5D-5L
EQ-5D Five-Level
ESC
Eczema Society of Canada
FE
fixed effects
HADS
Hospital Anxiety and Depression Scale
HADS-A
Hospital Anxiety and Depression Scale – Anxiety
HADS-D
Hospital Anxiety and Depression Scale – Depression
HRQoL
health-related quality of life
hsCRP
high-sensitivity C-reactive protein
IGA
Investigator Global Assessment
ICC
intra-class correlation coefficient
ICER
Institute for Clinical and Economic Review
IL
interleukin
ITC
indirect treatment comparison
ITT
intention to treat
JAK
Janus kinase
LS
least squares
MACE
major adverse cardiac event
MAR
missing at random
MCID
minimal clinically important difference
MMRM
mixed-effects model for repeated measures
NMA
network meta-analysis
NICE
National Institute for Health and Care Excellence
NRI-C
non-responder imputation to handle data missing due to COVID-19
NRI-NC
non-responder imputation with no special data handling for data missing due to COVID-19
NRS
Numerical Rating Scale
OC
observed case
OR
odds ratio
PDE 4
phosphodiesterase 4
PGA
Physician Global Assessment
POEM
Patient-Oriented Eczema Measure
PUVA
psoralen and UVA radiation
RCT
randomized controlled trial
RE
random effects
SAE
serious adverse event
SC
subcutaneous
SCORAD
Scoring Atopic Dermatitis
SD
standard deviation
SLR
systematic literature review
TCI
topical calcineurin inhibitor
TCS
topical corticosteroid
TEAE
treatment-emergent adverse event
TSS-7
7-Item Total Symptom Score
vIGA
validated investigator Global Assessment
vIGA-AD
validated Investigator Global Assessment for Atopic Dermatitis
VTE
venous thromboembolic events
WPAI:AD
Work Productivity and Activity Impairment Index: Atopic Dermatitis
WP-NRS
Worst Pruritus Numerical Rating Scale
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Upadacitinib (Rinvoq) oral extended-release tablets, 15 mg and 30 mg |
Indication | For the treatment of adults and adolescents 12 years of age and older with refractory, moderate to severe AD who are not adequately controlled with a systemic treatment (e.g., a steroid or biologic), or when the use of such therapies is inadvisable. Upadacitinib (Rinvoq) can be used with or without topical corticosteroids |
Reimbursement request | For the treatment of patients aged 12 years and older with moderate to severe AD whose disease is not adequately controlled with topical prescription therapies and/or who are refractory to or ineligible for systemic immunosuppressant therapies (i.e., due to contraindications, intolerance, or need for long-term treatment) |
Health Canada approval status | NOC |
Health Canada review pathway | Standard |
NOC date | October 6, 2021 |
Sponsor | AbbVie Corporation |
AD = atopic dermatitis; NOC = Notice of Compliance.
Introduction
Atopic dermatitis (AD), also known as atopic eczema, is an inflammatory, chronic skin disease commonly occurring in families with other allergic conditions. AD is considered among the most common non-communicable skin diseases, affecting 20% of children and 2% to 8% of adults worldwide.1 In Canada, the lifetime prevalence of AD is up to 17% of the population. There is evidence to suggest that the prevalence has increased over the past 30 years.2,3
AD is characterized by severe pruritus and rash as well as scratching that may result in lichenification.4 Secondary skin infections are common due to a compromised skin barrier function and scratching.1,4 AD usually develops before the age of 5 and may persist into adulthood.4 The majority of children outgrow the condition, but it is common for children with AD to develop asthma and/or hay fever — a process commonly referred to as the “atopic march.”5 Symptoms can worsen through the night, resulting in sleep loss and affecting school or work activities. Health-related quality of life (HRQoL) is also altered. Stigma can affect patients physically and mentally. Health care utilization and costs are also affected and usually associated with the severity of the disease.6
The goals of AD management are to prevent flares — and to manage flares effectively when they do occur — by preventing the disease’s progression. While there is no cure for AD, there are several therapeutic options available to patients to manage the condition. The majority of patients treat AD by using general skin care methods and topical anti-inflammatory therapy and by avoiding skin irritants. If these common methods fail to improve AD, patients may use off-label systemic therapy (i.e., immunosuppressant therapy) or other therapies, such as phototherapy.1,4,7
The most common pharmaceutical topical therapies include topical corticosteroid (TCS) and topical calcineurin inhibitors (TCIs).1 In Canada, hydrocortisone 1% (low potency) is the most commonly prescribed type of TCS, followed by triamcinolone or betamethasone valerate (moderate potency).1,4 TCIs are steroid-free, anti-inflammatory, immunosuppressant drugs that can be used long-term. In Canada, the 2 drugs available are pimecrolimus and tacrolimus.4,7 The most common adverse event (AE) associated with TCIs is application site–specific burning and irritation. Crisaborole, a topical phosphodiesterase type 4 inhibitor, is also available. The advantage of the TCIs and crisaborole is that both can be safely applied to the face and in creases, whereas TCS therapies that are more potent than 1% hydrocortisone are inappropriate. Systemic therapy involves the use of antimicrobials, antihistamines, or immunomodulators.2,7 Immunomodulatory drugs include methotrexate, cyclosporine A, mycophenolate mofetil, and azathioprine. These can be used in patients who are not responsive to other treatments.2,4,7 However, these commonly used off-label treatments are administered in the lowest dose and for the shortest duration possible due to the possibility of side effects. Dupilumab (Dupixent) is an interleukin (IL)-4 and IL-13 inhibitor indicated for use in adults and the pediatric population with moderate to severe AD whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable. CADTH has recommended that dupilumab be reimbursed with conditions, and it is currently reimbursed by participating drug programs for patients whose AD is inadequately controlled with topical prescription therapies and who have demonstrated failure on or intolerance to an adequate trial of phototherapy (where available), methotrexate, and cyclosporine A.8 Phototherapy is another second-line therapy that is commonly used after failure of TCS therapies and TCIs. Phototherapy requires 2 or 3 treatments per week for a duration of 8 weeks and is guided by a number of factors, including patient skin type and skin cancer history.9
Upadacitinib is a small-molecule, reversible, Janus kinase (JAK) inhibitor indicated for the treatment of adults and adolescents 12 years of age and older with refractory, moderate to severe AD who are not adequately controlled with a systemic treatment (e.g., a steroid or biologic) or when use of those therapies is inadvisable. Upadacitinib can be used with or without TCS. Upadacitinib is available as 15 mg or 30 mg oral extended-release tablets. The recommended starting dose of upadacitinib for adult patients is 15 mg once daily. If an adequate response (e.g., at least 75% improvement in the Eczema Area and Severity Index score from baseline [EASI 75]) is not achieved, physicians can consider increasing the dosage to 30 mg once daily. For some patients, such as those with severe disease, a starting dose of 30 mg once daily may be appropriate. Upadacitinib should be discontinued if an adequate response is not achieved with the 30 mg dose after 16 weeks of treatment. Patients should use the lowest effective dose needed to maintain response. For patients older than 65 years of age, the 30 mg dose once daily is not recommended. The recommended dosage of upadacitinib is 15 mg once daily for adolescents (from 12 years to 17 years of age) weighing at least 40 kg. Upadacitinib has not been studied in adolescents weighing less than 40 kg.10
The objective of this review is to perform a systematic review of the beneficial and harmful effects of upadacitinib for the treatment of adults and adolescents 12 years and older with moderate to severe AD who are candidates for systemic therapy.
Stakeholder Perspectives
The information in this section is a summary of the input provided by the patient groups who responded to CADTH’s call for patient input and from 1 clinical expert consulted by CADTH for the purpose of this review.
Patient Input
Three patient groups responded to CADTH’s call for patient input: the Eczema Society of Canada (ESC), the Canadian Skin Patient Alliance (CSPA), and Eczéma Québec. The latter 2 organizations provided a joint submission. ESC is a registered Canadian charity dedicated to improving the lives of Canadians living with eczema, and has a mission of support, education, awareness, and research. CSPA is a national, non-profit organization that educates, supports, and advocates for Canadians affected by skin, hair, and nail disorders. Eczéma Québec is a patient advisory committee and registered non-profit organization.
ESC gathered survey data from more than 3,000 adults living with AD and caregivers of children living with AD. Meanwhile, Eczéma Québec and CSPA developed a web-based survey that was distributed through both organizations’ newsletters and social media. There were 56 survey respondents.
The patient groups reported that AD negatively affects patients and their families and can lead to psychological distress. Patients frequently report that itch is their most burdensome symptom; more than half of adult respondents with severe AD reported rarely being able to control their urge to scratch. Itch also significantly affects sleep; patients report being woken frequently and having trouble falling and staying asleep due to their itch. The severity of AD correlates with impacts on HRQoL as well as lost productivity at school and burden on health systems. AD also has significant impacts in terms of the psychosocial burden of symptoms. All respondents experienced itching because of their condition. According to the CSPA survey, other symptoms included redness of the skin (87.88%), repeated rashes (84.85%), frequent scratching (84.85%), cracked skin (84.85%), dry and rough skin (78.79%), disrupted sleep (75.76%), bleeding (69.70%), flaking of the skin (69.70%), pain (69.70%), thickening of the skin (60.61%), oozing (48.48%), swelling (42.42%), lichenification (39.39%), and blistering (36.36%). From the ESC survey, 32% of adult respondents with moderate or severe AD have missed work events due to their condition, and 30% have had to change careers or give up certain activities. Caregivers noted that AD places a significant emotional toll on the entire family, and feelings of guilt, frustration, anger, and sadness are common. Forty-one percent of caregivers reported feeling like failures when they cannot control their children’s flares. Patients and caregivers reported that the mental health impact of AD is a significant aspect of the condition and is often not understood by others or prioritized by health care providers. Uncontrolled chronic AD can lead to feelings of depression and anxiety as well as poor self-esteem, low energy, and — in some extreme cases — suicidal thoughts.
Most patients expressed their dissatisfaction with the treatment options currently available to them. Another source of frustration for these participants was that they didn’t view these treatments as long-term options, but rather as temporary. Respondents also expressed concern about the financial impacts of treatments.
Overall, patients desire a treatment that will help them to manage itch, reduce flares and rashes, and enjoy an improved quality of life and sleep. Patients also want to improve the appearance of their hands and eyes (i.e., with less apparent eczema) and to have better pain relief.
Clinician Input
Input From the Clinical Expert Consulted by CADTH
The clinical expert consulted by CADTH visualizes the ideal treatment for AD as 1 that is available to all Canadians, is cost-effective in the context of a publicly funded health care system, has a proven long-term safety record, and completely reverses the barrier dysfunction and immunologic abnormalities that constitute AD.
Upadacitinib was considered by the clinical expert as a potentially useful addition to the currently available therapeutic options for AD, especially in patients who have contraindications to, experience adverse effects from, or are unresponsive to off-label immunosuppressive drugs. Upadacitinib could also be of value in patients treated with dupilumab who have a suboptimal response, develop severe conjunctivitis or other ocular side effects, or are intolerant to injections (e.g., due to severe injection-site reactions) and prefer an oral drug. Furthermore, the clinical expert noted that all patients with AD treated with upadacitinib would be expected to continue with emollients, TCSs, and/or TCIs.
According to the clinical expert, upadacitinib can be another effective treatment option in the Canadian clinical landscape. Off-label immunosuppressives or dupilumab are not expected to be used in combination with upadacitinib; however, the clinical expert believed that many practitioners would still consider a trial of methotrexate and cyclosporine A before initiating treatment with upadacitinib. The clinical expert suggested that patients less suitable for treatment with upadacitinib would be those with AD who are well controlled with topical therapy, phototherapy, and/or intermittent off-label immunosuppressive therapy, as well as patients well controlled with dupilumab. Upadacitinib should be avoided in patients with potential contraindications to JAK inhibitors, such as those with severe active infections, malignancies, ongoing chemotherapy treatment (including checkpoint inhibitors), severe hepatic disease, severe renal disease, a history of thromboembolic events, or pre-existing hematologic disease. Patients who are pregnant or lactating, or who weigh less than 40 kg, should also avoid upadacitinib.
In general, the outcomes used in clinical practice are aligned with the outcomes typically used in clinical trials of AD treatments. Of these outcome measurements, a rational benchmark was a 75% reduction from baseline in the EASI score at 16 weeks. In the opinion of the clinical expert, patients placed on upadacitinib would be re-evaluated at 16 weeks after initiating treatment. Those who are judged to be responders at this visit would be seen subsequently at 6-month intervals. Those who have not reached response targets at 16 weeks would be re-evaluated at 20 weeks.
According to the clinical expert, patients deemed to have severe symptoms would start on 30 mg for 16 weeks and be assessed for response (e.g., EASI 75); if a response is reached, they would switch to 15 mg. The product monograph approved by Health Canada states that patients who are receiving 15 mg and do not achieve a response after 16 weeks of treatment would be switched to 30 mg. The product monograph also states that if patients do not achieve adequate response (e.g., EASI 75) after 16 weeks of treatment on the 30 mg dose, upadacitinib should be discontinued.
The factors anticipated by the clinical expert to be used as criteria for discontinuation included failure to achieve a clinically meaningful response at 16 weeks to 20 weeks, failure to maintain an adequate response on long-term maintenance, development of a hypersensitivity response judged to be due to upadacitinib, treatment-emergent adverse effects (TEAEs) (e.g., lymphopenia, neutropenia, arterial thrombosis, venous thromboembolism [VTE]), and treatment-emergent severe infections or malignancies.
There are no special challenges for the administration of the drug. However, a specialist would still be required to diagnose, treat, and monitor patients taking upadacitinib. Appropriate specialists include pediatric dermatologists, general dermatologists, or pediatricians with experience in treating patients with AD. Dermatologists are well versed in the appropriate dosing and duration of therapy and appropriate monitoring for potential toxicities.
Clinician Group Input
One clinician group provided input on the reimbursement review of upadacitinib for the treatment of adult and adolescents with moderate to severe AD: the Atlantic Specialist Group Managing Atopic Dermatitis is a group of physicians, including general practitioners, dermatology, and allergy and immunology specialists, who manage patients with AD. The members of the group are located in various clinical settings across Atlantic Canada.
The clinician group indicated that the greatest unmet need is in the subset of patients with moderate to severe AD, who lack access to an effective, convenient, and safe treatment that enables long-term disease control and remission, given that many patients experience flares as soon as they stop their current medication. This cycle of recurrence leads to disease progression, ending in chronic severe AD and severe impacts on HRQoL.
According to the clinician group, the place in therapy for upadacitinib would be after initial treatments for mild AD (e.g., lifestyle measures and topical steroids). In such a case, upadacitinib would replace systemic therapies that are currently used off-label to treat AD, as well as phototherapy. In the clinician group’s opinion, dupilumab addresses some of the concerns and needs of some patients, but upadacitinib may shift the paradigm due to its efficacy and ease of administration. This place-in-therapy judgment differs from the opinion of the clinical expert consulted by CADTH, who considered that upadacitinib should be used after a trial of systemic therapies that are currently used (even if off-label) to treat patients who fail to respond to TCSs, such as methotrexate or cyclosporine A.
The clinician group noted that upadacitinib would be best suited to treat patients with moderate to severe AD who have not responded, are not expected to respond, or have had adverse reactions to long-term use of TCSs. These patients have the greatest need for intervention because they lack long-term treatment options and are at high risk of disease progression.
The outcomes measured in clinical trials, such as the Investigator Global Assessment (IGA), are also used in clinical practice, perhaps with the exception of EASI scores, which are relatively unknown in day-to-day practice. The group mentioned that a clinically meaningful response to upadacitinib would include improvement in patient-reported itch (a 4-point reduction on the numerical rating scale [NRS] or an NRS score of less than 3), a Dermatology Life Quality Index (DLQI) score reduction of greater than or equal to 4 (or an acceptable improvement), patient-reported improvement in sleep quality, fewer AD-related disruptions at school and work, and a Physician Global Assessment (PGA) score 0 or 1. Importantly, a patient should not experience any severe side effects, including over sustained time periods, in order for the response to upadacitinib to be clinically meaningful.
The group suggests that the response to systemic therapy should be reassessed in 12 weeks to 16 weeks after the initiation of treatment. According to input from the clinician group, the decision to discontinue treatment should be assessed if there is a lack of response, significant disease progression (i.e., lichenification, increased affected body surface area [BSA], or increased itching), or deterioration in quality of life, or if the patient experiences adverse reactions or intolerance to the medication that are deemed to be unacceptable by the patient-physician team. Treatment with upadacitinib should be interrupted if a patient develops a serious infection or presents serious abnormal laboratory results (e.g., an absolute lymphocyte count of less than 500 cells/mm3, an absolute neutrophil count of less than 1,000 cells/mm3, or hemoglobin less than 8 g/dL), or if drug-induced liver injury is suspected (based on hepatic transaminases); treatment with upadacitinib may be resumed once levels return to normal. Patients with AD receiving upadacitinib would ideally be managed in any non-emergent setting to which they have access and that has a dermatologist or allergist who is well versed in managing moderate to severe AD. Referring family physicians, nurse practitioners, or other health care providers should be counselled on the appropriate referral process.
Drug Program Input
Input was obtained from the drug programs that participate in the CADTH reimbursement review processes. The following were identified as key factors that could affect the implementation of a CADTH recommendation for upadacitinib:
Access to phototherapy may be limited in some areas of Canada. The clinical expert consulted by CADTH noted that phototherapy is typically accessible in urban areas, but that access may be limited in rural areas. The expert noted that this barrier to phototherapy access should be considered in the reimbursement review decision-making process.
As to whether upadacitinib should be initiated in patients who have failed previous treatment with a biologic drug, the clinical expert’s perspective was that patients who have failed dupilumab plus 1 of the immunomodulators would be candidates to receive upadacitinib, but that this also would apply for those who have failed dupilumab alone. The clinical expert noted that there is limited evidence for the sequential use of upadacitinib after an adequate trial of dupilumab in patients with moderate to severe AD.
On the question of whether patients would require a previous trial of (or be ineligible for) cyclosporine A, methotrexate, and/or phototherapy before initiating upadacitinib, the expert’s opinion was that a trial of at least 2 of the 4 immunomodulators (i.e., methotrexate, cyclosporine A, mycophenolate mofetil, or azathioprine) should be considered before initiating upadacitinib.
On the question of whether the reimbursement criteria that were recommended for dupilumab would be applicable to upadacitinib (e.g., the initiation and renewal criteria), the clinical expert consulted by CADTH noted that the criteria for dupilumab could be applicable for upadacitinib and could be implemented in clinical practice. Dupilumab as prior therapy should not be an initiation criterion. Both drugs would have the same place in therapy in the population for this indication.
Clinical Evidence
Pivotal Studies and Protocol Selected Studies
Description of Studies
Four clinical studies were included in this report evaluating the use of upadacitinib in patients with moderate to severe AD.
Measure Up 1 and Measure Up 2 were 2 similar studies (n = 847 and n = 836, respectively) with a double-blind, placebo-controlled parallel design. Eligible patients were adults and adolescents (≥ 40 kg) with chronic AD and a documented history of systemic treatment or inadequate response to topical AD treatments. Both studies randomized patients to upadacitinib 15 mg, upadacitinib 30 mg, or placebo. The studies evaluated co-primary outcomes, the proportion of responders based on the EASI score (where 75 indicates an improvement of 75% from baseline), and a validated Investigator Global Assessment for Atopic Dermatitis (vIGA-AD) score of 0 or 1 at week 16.
The AD Up study had a similar design to the Measure Up 1 and Measure Up 2 studies, with the same inclusion criteria and population (n = 901). However, it used TCSs in combination with upadacitinib 15 mg, upadacitinib 30 mg, or placebo. It used the same co-primary end points at 16 weeks.
The Heads Up study was a double-blind, double-dummy, active-controlled, randomized study (n = 692) comparing upadacitinib 30 mg to dupilumab 300 mg subcutaneous (SC) in adults (18 years to 75 years old) with chronic AD and a documented history of inadequate response to topical treatments or documented treatment with systemic therapies. This study’s primary end point included the proportion of patients achieving EASI 75 at week 16.
The eligibility criteria for the included randomized controlled trials (RCTs) were similar except for the differences in the age range for the study comparing upadacitinib to dupilumab (i.e., the Heads Up study was limited to adults, while the others included adolescents). All of the trials enrolled patients with moderate to severe AD and an inadequate response to topical AD therapies. This is reflective of the indication that was initially submitted to Health Canada and CADTH; however, the approved Health Canada indication reflects a more restrictive population (i.e., those with refractory, moderate to severe AD and an inadequate response to other systemic drugs). The proportion of patients with prior exposure to at least 1 systemic therapy for AD in the included trials were: Measure Up 1 = 46.4%; Measure Up 2 = 54.5%; AD Up = 66.6%; and Heads Up = |||||.
Efficacy Results
Outcomes of disease severity, such as EASI 75, the vIGA-AD, and the Scoring Atopic Dermatitis (SCORAD) scale, were considered by the clinical expert consulted by CADTH critical for decision-making and clinical practice and mentioned in the patient input received from patient groups. Similarly, among the outcomes of symptoms — as measured with the Worst Pruritus Numerical Rating Scale (WP-NRS), Patient-Oriented Eczema Measure (POEM), and the Atopic Dermatitis Impact Scale (ADerm-IS) and Atopic Dermatitis Symptom Scale (ADerm-SS) — HRQoL, mood, and productivity were considered among the most important across different domains of decision-making.
At 16 weeks of follow-up, upadacitinib showed statistically significant improvements in the co-primary end points of disease severity in the Measure Up, AD Up, and Heads Up studies. In Measure Up 1, more patients reached EASI 75 in the upadacitinib 15 mg group (196 patients of 281 patients [69.6%]) and in the upadacitinib 30 mg group (227 patients of 285 patients [79.7%]) than in the placebo group (46 patients of 281 patients [16.3%]); the adjusted differences versus placebo were 53.3% (95% confidence interval [CI], 46.4 to 60.2; P < 0.001) for the upadacitinib 15 mg group and 63.4% (95% CI, 57.1 to 69.8; P < 0.001) for the upadacitinib 30 mg group.
When assessing for an EASI score of 90, more patients in the upadacitinib 30 mg group (187 patients [65.8%]) and the upadacitinib 15 mg group (149 patients [53.1%]) were responders compared to placebo (23 [8.1%]); the adjusted differences were 57.8% (95% CI, 51.5 to 64.1) and 45.1% (95% CI, 38.6 to 51.7), respectively. Similarly, in Measure Up 2, 166 patients out of 276 patients (60.1%) in the upadacitinib 15 mg group and 206 patients out of 282 patients (2.9%) in the upadacitinib 30 mg group versus 37 patients out of 278 patients (13.3%) in the placebo group reached EASI 75, with adjusted differences in the EASI 75 response rate versus placebo of 46.9% (95% CI, 39.9 to 53.9; P < 0.001) for the upadacitinib 15 mg group and 59.6% (95% CI, 53.1 to 66.2; P < 0.001) for the upadacitinib 30 mg group. When assessing for an EASI score of 90, more patients in the upadacitinib 30 mg group (165 patients [58.5%]) and the upadacitinib 15 mg group (117 patients [42.4%]) were responders compared to placebo (15 patients [5.4%]); the adjusted differences were 53.1% (95% CI, 46.7 to 59.4) and 36.9% (95% CI, 30.6 to 43.3), respectively.
Likewise, in Measure Up 1, a larger proportion of patients achieved a vIGA-AD response at week 16 in the upadacitinib 15 mg group (135 patients [48.1%]) and the upadacitinib 30 mg group (177 patients [62.0%]) than in the placebo group (24 patients [8.4%]); the adjusted differences versus placebo were 39.8% (95% CI, 33.2 to 46.4; P < 0.001) for the upadacitinib 15 mg group and 53.6% (95% CI, 47·2 to 60·0; P < 0.001) for the upadacitinib 30 mg group. In Measure Up 2, 107 patients (38.8%) in the upadacitinib 15 mg group and 147 patients (52.0%) in the upadacitinib 30 mg group achieved a vIGA-AD response at week 16 versus 13 patients (4.7%) in the placebo group; the adjusted differences in vIGA-AD response rate versus placebo were 34.0% (95% CI, 278 to 40.2; P < 0.001) for the upadacitinib 15 mg group and 47.4% (95% CI, 41.0 to 53.7; P < 0.001) for the upadacitinib 30 mg group.
In the AD Up study, at week 16, the proportion of patients who had achieved EASI 75 was statistically significantly higher in the group receiving upadacitinib 15 mg plus TCS (194 out of 300 patients [64.6%]) and in the group receiving upadacitinib 30 mg plus TCS (229 of 297 patients [77.1%]) than in the placebo group (80 of 304 patients [26.4%]); the adjusted differences in the EASI 75 response rate versus placebo were 38.1% (95% CI, 30.8 to 45.4; P < 0.001) for the upadacitinib 15 mg group and 50.6% (95% CI, 43.8 to 57.4; P < 0.001) for the upadacitinib 30 mg group. When assessing for an EASI score of 90, more patients in the upadacitinib 30 mg group (187 patients [63.1%]) and upadacitinib 15 mg group (128 patients [42.8%]) were responders compared to placebo (40 patients [13.2%]); the adjusted differences were 57.8% (95% CI, 51.5 to 64.1) and 45.1% (95% CI, 38.6 to 51.7), respectively. The proportions of patients who achieved a vIGA-AD response at week 16 were statistically significantly higher in the group receiving upadacitinib 15 mg plus TCS (119 patients [39.6%]) and in the group receiving upadacitinib 30 mg plus TCS (174 patients [58.6%]) than in the placebo group (33 patients [10.9%]); the adjusted differences were 28.5% (95% CI, 22.1 to 34.9) for the upadacitinib 15 mg group and 47.6% (95% CI, 41.1 to 54.0) for the upadacitinib 30 mg group (P < 0.0001 for both doses).
In the Heads Up trial, patients in the upadacitinib 30 mg group showed statistically significantly higher rates of achieving EASI 75 (247 patients [71.0%]) than those on dupilumab 300 mg (210 patients [61.1%]) at week 16; the adjusted difference between groups was 10.0% (95% CI, 2.9 to 17.0; P = 0.006). This difference was no longer statistically significant at week 24, with 205 patients in the dupilumab group (59.5%) and 223 patients in the upadacitinib 30 mg group (64.2%) achieving EASI 75 (adjusted difference = 4.6%; 95% CI, –2.6 to 11.9; P = 0.211). When assessing for an EASI score of 90 at week 16, more patients in the upadacitinib 30 mg group (211 patients [60.6%]) were responders compared to those in the dupilumab group (133 patients [38.8%]; the adjusted difference was 21.8% (95% CI, 14.5 to 29.1), and this difference between groups was smaller at week 24, at 4.6% (95% CI, 0.5 to 15.4; P = 0.036).
An assessment of patients with previous systemic therapies showed similar results to the base case in all studies (i.e., the Measure Up 1, Measure Up 2, AD Up, and Heads Up trials) for the primary end points of response based on EASI 75 at week 16 and on the vIGA (except in the Heads Up study, which did not assess vIGA). In the subgroup of patients with prior use of a systemic treatment for AD (e.g., a steroid or biologic), the adjusted differences (95% CI) for upadacitinib 15 mg once daily and upadacitinib 30 mg once daily (respectively) compared with placebo for EASI 75 at week 16 were: for Measure Up 1, |||||||||||||||||| and |||||||||||||||||||||||||||; for Measure Up 2, ||||||||||||||||||||||||||||||||||||||||||||||||||||||; for AD Up, |||||||||||||||||||||||||||||||||||||||||||||. For Heads Up, the proportions of patients reaching EASI 75 at week 16 were ||||||||| and ||||||||| for dupilumab and upadacitinib, respectively; the adjusted difference for upadacitinib 30 mg versus dupilumab 300 mg was |||||||||||||||||||||||||||. For IGA response, the results were: Measure Up 1, ||||||||||||||||||||||||||| and ||||||||||||||||||; Measure Up 2, |||||||||||||||||| and ||||||||||||||||||; AD Up, |||||||||||||||||| and ||||||||||||||||||. Heads Up did not include a vIGA assessment.
Symptoms of AD were also improved. In the Measure Up studies, the proportion of patients achieving a WP-NRS score of greater than or equal to 4 from baseline at week 16 was statistically significantly higher compared to placebo in the upadacitinib 15 mg group (i.e., the absolute risk differences from placebo were 40.5% [95% CI, 33.5 to 47.5] in Measure Up 1 and 32.6% [95% CI, 25.8 to 39.4] in Measure Up 2) and in the upadacitinib 30 mg group (48.2% [95% CI, 41.3 to 55.0] in Measure Up 1 and 50.4% [95% CI, 43.8 to 57.1] in Measure Up 2; P < 0.001) for all comparisons. The proportion of patients achieving a POEM total score improvement (reduction) of at least 4 points from baseline at week 16 was also statistically significantly higher in the upadacitinib 15 mg group, with an absolute risk difference of 52.3% (95% CI, 45.2 to 59.4) in Measure Up 1 and 42.1% (95% CI, 34.5 to 49.8) in Measure Up 2 and in the 30 mg group, with absolute risk differences of 58.6% (95% CI, 51.9 to 65.3) in Measure Up 1 and 54.7% (95% CI, 47.7 to 61.7) in Measure Up 2 versus placebo (P < 0.001 for all comparisons). Improvements were also observed in the ADerm skin pain, sleep, emotional, and daily activities domains (P < 0.001 for all comparisons). No subgroup analyses were performed on symptoms outcomes.
In AD Up, the results were similar to those of Measure Up, in which the proportion of patients achieving a WP-NRS score of greater than or equal to 4 from baseline at week 16 was statistically significantly higher when compared to placebo in the upadacitinib 15 mg group (absolute risk difference from placebo = 36.8% [95% CI, 29.7 to 43.8]) and in the upadacitinib 30 mg group (absolute risk difference from placebo = 48.8% [95% CI, 41.9 to 55.7]); P < 0.001 for all comparisons. The POEM total score improvement of at least 4 points from baseline was also statistically significantly higher in the upadacitinib 15 mg group (absolute risk difference of ||||||||||||||||||||||||||| and 30 mg [|||||||||||||||||||||||||||] against placebo; P < 0.001 for all comparisons). Improvements were also observed in the ADerm skin pain, sleep, emotional, and daily activities domains (P < 0.001 for all comparisons).
When compared to dupilumab (Heads Up study), the proportion of patients achieving a WP-NRS of greater than or equal to 4 at week 16 was statistically significantly higher in the upadacitinib 30 mg group (55.2%) compared to the dupilumab 300 mg group (35.9%), with an absolute risk difference of 19.3% (95% CI, 11.9 to 26.7; P < 0.001). The risk difference decreased at week 24 to 8.3% (95% CI, 0.8 to 15.8), although it was still statistically significant (P = 0.030).
HRQoL, assessed by the DLQI score, was also improved more frequently in the upadacitinib 15 mg and upadacitinib 30 mg groups than in the placebo group in the Measure Up and AD Up studies, but not when assessed using the generic EQ-5D Five-Level (EQ-5D-5L) questionnaire. Mood and work productivity were similarly improved in the upadacitinib groups versus placebo. The absenteeism domains were not significantly statistically different between groups. No subgroup analyses were performed on these outcomes.
Harms Results
Upadacitinib 15 mg and upadacitinib 30 mg doses in all studies were well tolerated compared to placebo at week 16, without significant increases in AEs or serious adverse events (SAEs) up to the latest follow-up of 52 weeks in the blinded extension (BE) studies. The incidence of SAEs and AEs leading to study drug discontinuation were similar among groups except in the Heads Up study. The most frequently reported AEs were acne, upper respiratory tract infection, nasopharyngitis, headache, elevation in creatine phosphokinase (CPK) levels, and AD. No deaths were reported. No subgroup analyses based on prior exposure to systemic treatment (e.g., steroid or biologic) for AD were performed for AE.
In the AD Up study, the most frequently reported AEs (≥ 5% in any treatment group) were acne, nasopharyngitis, upper respiratory tract infection, oral herpes, elevation of blood CPK levels, headache, and AD. Acne was more frequent in the upadacitinib groups (10% to 14% in the 15 mg and 30 mg groups, respectively) than in the placebo group (2%) at week 16. No deaths were reported.
In the Heads Up study, the safety profile of upadacitinib was similar to those of the Measure Up and AD Up studies. The rates of serious AEs and AEs leading to study drug discontinuation were 2.9% and 1.2% for upadacitinib and 1.2% and 1.2% for dupilumab, respectively. One death was reported in an upadacitinib-treated patient due to influenza-associated bronchopneumonia. The most frequently reported AE among patients receiving upadacitinib was acne (15.8%); this AE was reported by only 2.6% of patients receiving dupilumab. The most frequently reported AE with dupilumab was conjunctivitis (8.4%); this AE was reported by only 1.4% of patients on upadacitinib. Other AEs that were more common in the upadacitinib groups were serious infections (1.1% versus 0.6%), eczema herpeticum (0.3% versus 0%), hepatic disorders (2.9% versus 1.2%), and herpes zoster (2.0% versus 0.9%). Also, rates of anemia (2.0% versus 0.3%), neutropenia (1.7% versus 0.6%), and CPK elevations (6.6% versus 2.9%) were higher for upadacitinib than dupilumab.
In the Japan study, through week 16, the rates of AEs observed in the upadacitinib 15 mg and 30 mg groups were higher than those observed in the placebo group overall.
Table 2: Summary of Key Results — Pivotal and Protocol Selected Monotherapy Studies
Result | Placebo | UPA 15 mg q.d. | UPA 30 mg q.d. |
|---|---|---|---|
Measure Up 1 | N = 281 | N = 281 | N = 285 |
EASI 75 at week 16a | |||
N (%) | 281 (100) | 281 (100) | 285 (100) |
Responders, n (%) | 46 (16.3) | 196 (69.6) | 227 (79.7) |
95% CI | 12.0 to 20.7 | 64.2 to 75.0 | 75.0 to 84.4 |
Adjusted difference vs. placebo, % (95% CI)b | Reference | 63.4 (57.1 to 69.8) | 63.4 (57.1 to 69.8) |
P value | Reference | < 0.001 | < 0.001 |
Subgroup: with previous systemic therapies | |||
N (%) | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
Responders, n (%) | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
95% CI | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
Adjusted difference vs. placebo, % (95% CI) | Reference | |||||||||||||||||| | |||||||||||||||||| |
P value against placebo | Reference | |||||||||||||||||| | |||||||||||||||||| |
vIGA-AD response 0 or 1 at week 16a | |||
N (%) | 281 (100) | 281 (100) | 285 (100) |
Responders, n (%) | 24 (8.4) | 135 (48.1) | 177 (62.0) |
95% CI | 5.2 to 11.7 | 42.3 to 54.0 | 56.4 to 67.7 |
Adjusted difference vs. placebo, % (95% CI)b | Reference | 39.8 (33.2 to 46.4) | 53.6 (47.2 to 60.0) |
P value | Reference | < 0.001 | < 0.001 |
Subgroup: with previous systemic therapies | |||
N (%) | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
Responders, n (%) | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
95% CI | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
Adjusted difference vs. placebo, % (95% CI) | Reference | |||||||||||||||||| | |||||||||||||||||| |
P value against placebo | Reference | |||||||||||||||||| | |||||||||||||||||| |
WP-NRS response ≥ 4 from baseline at week 16a | |||
N (%)e | 272 (96.80) | 274 (97.51) | 280 (98.25) |
Responders, n (%) | 32 (11.8) | 143 (52.2) | 168 (60.0) |
% responders – 95% CI | 7.9 to 15.6 | 46.3 to 58.1 | 54.3 to 65.7 |
Adjusted difference vs. placebo, % (95% CI)d | Reference | 40.5 (33.5 to 47.5) | 48.2 (41.3 to 55.0) |
P valued | Reference | < 0.001 | < 0.001 |
Patients with ≥ 1 adverse event | |||
n (%) | 166 (59.1) | 176 (62.6) | 209 (73.3) |
Patients with ≥ 1 SAE | |||
n (%) | 8 (2.8) | 6 (2.1) | 8 (2.8) |
Most common events, n (%)b | |||
Atopic dermatitis | 26 (9) | 9 (3) | 4 (1) |
Notable harms | |||
Opportunistic infection, excluding tuberculosis and herpes zoster | 4 (1.4) | 0 | 3 (1.1) |
Herpes zoster | 0 | 5 (1.8) | 6 (2.1) |
Hepatic disorder | 2 (0.7) | 5 (1.8) | 8 (2.8) |
Neutropenia | 2 (0.7) | 4 (1.4) | 15 (5.3) |
CPK elevation | 7 (2.5) | 16 (5.7) | 16 (5.6) |
Measure Up 2 | N = 278 | N = 276 | N = 282 |
EASI 75 at week 16a | |||
N (%) | 278 (100.00) | 276 (100.00) | 282 (100.00) |
Responders, n (%) | 37 (13.3) | 166 (60.1) | 206 (72.9) |
95% CI | (9.3 to 17.3) | (4.4 to 65.9) | (67.7 to 78.2) |
Adjusted difference vs. placebo, % (95% CI)b | Reference | 46.9 (39.9 to 53.9) | 59.6 (53.1 to 66.2) |
P value | Reference | < 0.001 | < 0.001 |
Subgroup: with previous systemic therapies | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
N (%) | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
Responders, n (%) | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
95% CI | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
Adjusted difference vs. placebo, % (95% CI) | Reference | |||||||||||||||||| | |||||||||||||||||| |
P value against placebo | Reference | |||||||||||||||||| | |||||||||||||||||| |
vIGA-AD response 0 or 1 at week 16a | |||
N (%) | 278 (100.00) | 276 (100.00) | 282 (100.00) |
Responders, n (%) | 13 (4.7) | 107 (38.8) | 147 (52.0) |
95% CI | 2.2 to 7.2 | 33.0 to 44.5 | 46.1 to 57.9 |
Adjusted difference vs. placebo, % (95% CI)b | Reference | 34.0 (27.8 to 40.2) | 47.4 (41.0 to 53.7) |
P value | Reference | < 0.001 | < 0.001 |
Subgroup: with previous systemic therapies | |||
N (%) | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
Responders, n (%) | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
95% CI | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
Adjusted difference vs. placebo, % (95% CI) | Reference | |||||||||||||||||| | |||||||||||||||||| |
P value against placebo | Reference | |||||||||||||||||| | |||||||||||||||||| |
WP-NRS response ≥ 4 from baseline at week 16 | |||
N (%) | 274 (98.56) | 270 (97.83) | 280 (99.29) |
Responders, n (%) | 25 (9.1) | 113 (41.9) | 167 (59.6) |
% responders – 95% CI | 5.7 to 12.5 | 36.0 to 47.7 | 53.9 to 65.4 |
Adjusted difference vs. placebo, % (95% CI)d | Reference | 32.6 (25.8 to 39.4) | 50.4 (43.8 to 57.1) |
P valued | Reference | < 0.001 | < 0.001 |
Patients with ≥ 1 adverse eventc | |||
n (%) | 146 (52.5) | 166 (60.1) | 173 (61.3) |
Patients with ≥ 1 SAE | |||
n (%) | 8 (2.9) | 5 (1.8) | 7 (2.5) |
Most common events, n (%) | |||
Atopic dermatitis | 26 (9) | 9 (3) | 4 (1) |
Notable harms | |||
Herpes zoster | 2 (0.7) | 6 (2.2) | 3 (1.1) |
Hepatic disorder | 4 (1.4) | 2 (0.7) | 4 (1.4) |
Neutropenia | 1 (0.4) | 2 (0.7) | 6 (2.1) |
CPK elevation | 5 (1.8) | 9 (3.3) | 12 (4.3) |
CI = confidence interval; CPK = creatine phosphokinase; EASI = Eczema Area and Severity Index; ITT = intention to treat; q.d. = once daily; SAE = serious adverse event; UPA = upadacitinib; vIGA-AD = validated Investigator Global Assessment for Atopic Dermatitis; vs. = versus; WP-NRS = Worst Pruritus Numerical Rating Scale.
aAssessed in the ITT population with nonresponder imputation incorporating multiple imputation to handle data missing due to COVID-19.
b95% CI for adjusted difference and P values are calculated according to the Cochran-Mantel-Haenszel test adjusted for strata (baseline vIGA-AD score categories and age [adolescent vs. adult]) for the comparison of 2 treatment groups.
cSafety population.
Source: Clinical Study Reports for Measure Up 1 and Measure Up 2.11,12
Table 3: Summary of Key Results — Pivotal and Protocol Selected Combination Therapy Studies
AD Up | Placebo + TCS N = 304 | UPA 15 mg q.d.+ TCS N = 300 | UPA 30 mg q.d. TCS N = 297 |
|---|---|---|---|
EASI 75 at week 16a | |||
N (%) | 304 (100.00) | 300 (100.00) | 297 (100.00) |
Responders, n (%) | 80 (26.4) | 194 (64.6) | 229 (77.1) |
95% CI | (21.5 to 31.4) | (59.1 to 70.0) | (72.3 to 81.9) |
Adjusted difference vs. placebo, % (95% CI)b | Reference | 38.1 (30.8 to 45.4) | 50.6 (43.8 to 57.4) |
P value | Reference | < 0.001 | < 0.001 |
Subgroup: with previous systemic therapies | |||
N (%) | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
Responders, n (%) | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
95% CI | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
Adjusted difference vs. placebo, % (95% CI) | Reference | |||||||||||||||||| | |||||||||||||||||| |
P value against placebo | Reference | |||||||||||||||||| | |||||||||||||||||| |
vIGA-AD response 0 or 1 at week 16a | |||
N (%) | 304 (100.00) | 300 (100.00) | 297 (100.00) |
Responders, n (%) | 33 (10.9) | 119 (39.6) | 174 (58.6) |
95% CI | (7.4 to 14.4) | (34.1 to 45.2) | (53.0 to 64.2) |
Adjusted difference vs. placebo, % (95% CI)b | Reference | 28.5 (22.1 to 34.9) | 47.6 (41.1 to 54.0) |
P value | Reference | < 0.001 | < 0.001 |
Subgroup: with previous systemic therapies | |||
N (%) | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
Responders, n (%) | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
95% CI | |||||||||||||||||| | |||||||||||||||||| | |||||||||||||||||| |
Adjusted difference vs. placebo, % (95% CI) | Reference | |||||||||||||||||| | |||||||||||||||||| |
P value against placebo | Reference | |||||| | |||||| |
WP-NRS response ≥ 4 from baseline at week 16a | |||
N (%) | 294 (96.71) | 288 (96.00) | 291 (97.98) |
Responders, n (%) | 44 (15.0) | 149 (51.7) | 186 (63.9) |
% responders – 95% CI | (10.9 to 19.0) | (46.0 to 57.5) | (58.4 to 69.4) |
Adjusted difference vs. placebo, % (95% CI)b | Reference | 36.8 (29.7 to 43.8) | 48.8 (41.9 to 55.7) |
P valueb | Reference | < 0.001 | < 0.001 |
Patients with ≥ 1 adverse eventc | |||
n (%) | 190 (62.7) | 200 (66.7) | 215 (72.4) |
Patients with ≥ 1 SAE | |||
n (%) | 9 (3.0) | 7 (2.3) | 4 (1.3) |
Most common events, n (%)b | |||
Atopic dermatitis | 20 (7) | 11 (4) | 2 (1) |
Notable harms | |||
Creatine phosphokinase elevation | 7 (2.3) | 13 (4.3) | 18 (6.1) |
Hepatic disorder | 5 (1.7) | 6 (2.0) | 3 (1.0) |
Opportunistic infection, excluding tuberculosis and herpes zoster | 0 | 3 (1.0) | 4 (1.3) |
CI = confidence interval; EASI = Eczema Area and Severity Index; ITT = intention to treat; q.d. = once daily; SAE = serious adverse event; TCS = topical corticosteroid; UPA = upadacitinib; vIGA-AD = validated Investigator Global Assessment for Atopic Dermatitis; vs. = versus; WP-NRS = Worst Pruritus Numerical Rating Scale.
aAssessed in the ITT population with nonresponder imputation incorporating multiple imputation to handle data missing due to COVID-19.
b95% CIs for adjusted difference and P values are calculated according to the Cochran-Mantel-Haenszel test adjusted for strata (baseline vIGA-AD score categories and age [adolescent vs. adult]) for the comparison of 2 treatment groups.
cSafety population.
Source: Clinical Study Report: AD Up.13
Table 4: Summary of Key Results — Pivotal and Protocol Selected Head-to-Head Studies
Heads Up | DUP 300 mg 2.q.w. N = 344 | UPA 30 mg q.d. N = 348 |
|---|---|---|
EASI 75 at week 16a | ||
N (%) | 344 (100.00) | 348 (100.00) |
Responders, n (%) | 210 (61.1) | 247 (71.0) |
95% CI | (55.9 to 66.2) | (66.2 to 75.8) |
Adjusted difference, % (95% CI)b | 10.0 (2.9 to 17.0) | |
P valueb | 0.006 | |
Subgroup: with previous systemic therapies | ||
N (%) | |||||||||||||||||| | |||||||||||||||||| |
Responders, n (%) | |||||||||||||||||| | |||||||||||||||||| |
95% CI | |||||||||||||||||| | |||||||||||||||||| |
Adjusted difference, % (95% CI) | |||||||||||||||||| | |
P value | |||||||||||||||||| | |
EASI 75 at week 24a | ||
N (%) | 344 (100.00) | 348 (100.00) |
Responders, n (%) | 205 (59.5) | 223 (64.2) |
95% CI | (54.4 to 64.7) | (59.1 to 69.2) |
Adjusted difference, % (95% CI)c | |||||||||||||||||| | |
P valueb | |||||||||||||||||| | |
WP-NRS response ≥ 4 from baseline at week 16 | ||
N (%) | 336 (97.67) | 340 (97.70) |
Responders, n (%) | 120 (35.9) | 188 (55.2) |
% responders – 95% CI | (30.7 to 41.0) | (49.9 to 60.5) |
Adjusted difference, % (95% CI)b | |||||||||||||||||| | |
P valueb | < 0.001 | |
Patients with ≥ 1 adverse eventc | ||
n (%) | 216 (62.8) | 249 (71.6) |
Patients with ≥ 1 SAE | ||
n (%) | 4 (1.2) | 10 (2.9) |
Notable harms | ||
Hepatic disorder | 4 (1.2) | 10 (2.9) |
Anemia | 1 (0.3) | 7 (2.0) |
Herpes zoster | 3 (0.2) | 7 (2.0) |
CPK elevation | 10 (2.9) | 23 (6.6) |
2.q.w. = every 2 weeks; CI = confidence interval; CPK = creatine phosphokinase; DUP = dupilumab; EASI = Eczema Area and Severity Index; ITT = intention to treat; q.d. = once daily; SAE = serious adverse event; UPA = upadacitinib; vIGA-AD = validated Investigator Global Assessment for Atopic Dermatitis; vs. = versus; WP-NRS = Worst Pruritus Numerical Rating Scale.
aAssessed in the ITT population with nonresponder imputation incorporating multiple imputation to handle data missing due to COVID-19.
b95% CI for adjusted difference and P values are calculated according to the Cochran-Mantel-Haenszel test adjusted for strata (baseline vIGA-AD score categories and age [adolescent vs. adult]) for the comparison of 2 treatment groups.
cSafety population.
Source: Clinical Study Report: Heads Up.14
Critical Appraisal
Randomization and allocation concealment were properly completed, resulting in a similar distribution of baseline demographics and disease characteristic variables between the treatment groups in each trial, without important imbalances.
Blinding of patients and study personnel was appropriately maintained. However, given that a placebo was used in several studies, it is possible that patients may have potentially become unblinded or aware of their assignments through improvement or lack of improvement (placebo) in their AD symptoms over the study period. There is the possibility that, in the Heads Up trial, certain adverse events (such as injection-site reactions, hypersensitivity reactions, or conjunctivitis) known to be potential risks associated with dupilumab could have resulted in unblinding, which could have biased the results of patient-reported outcomes, such as HRQoL. However, the co-primary end points are relatively objective, and risk of bias would be small.
The co-primary outcomes were based on the vIGA and EASI scores, both of which are reliable and valid for the assessment of the severity and extent of AD. The co-primary end points were analyzed appropriately using the intention-to-treat (ITT) population. Secondary end points were analyzed based on complete case analyses. This is expected to introduce some risk of bias in favour of upadacitinib (given that more complete data were available for upadacitinib due to lower discontinuations and drop-outs) because the groups may no longer be balanced in characteristics, and the data observed from the incomplete cases are discarded (i.e., patients who are responding to treatment and have limited AEs may be more likely to stay in the study and contribute data to the end points). Controlling for multiplicity was appropriate for the primary and secondary end points of all trials; a graphical multiple testing procedure was used. The greatest number of patients who discontinued the intervention were within the placebo groups in the Measure Up and AD Up trials. This introduces the potential for bias against the null (i.e., toward an inflated efficacy of upadacitinib) due to the analytical approaches used because more placebo patients would have been imputed as nonresponders. However, sensitivity analyses were based on multiple imputation, the tipping point approach, and the per-protocol population, with similar conclusions to those reached in the primary analyses. Several subgroup analyses were properly specified a priori and conducted across the trials (e.g., based on baseline vIGA-AD, baseline EASI, previous systemic therapy, age, and sex, among other factors), showing similar results.
The population in the included pivotal studies seems to be generalizable to adults and adolescents in the Canadian population having AD. However, when considering the applicability of the results for the population of patients previously treated with systemic therapies (i.e., the approved indication for upadacitinib), only a proportion of the patients included in the pivotal studies was similar to the approved Health Canada indication. Furthermore, the information from the pivotal studies for the 30 mg dose also represents a proportion of the population, and data to inform the approved indication are lacking — that is, data estimating the effects of upadacitinib in patients with AD who are switched from upadacitinib 15 mg to upadacitinib 30 mg if an adequate response (e.g., EASI 75) is not achieved. In addition, there were no clinical studies that studied a dose de-escalation to upadacitinib 15 mg once daily in patients who achieved a response to upadacitinib 30 mg once daily. This lack of evidence adds uncertainty to the generalizability of the results in the population for whom the indication would be applicable.
In all of the pivotal trials included in this CADTH review, an assessment of the subgroup of patients who had taken previous systemic therapies showed similar results to the base case for the primary end points of response based on EASI 75 at week 16 and vIGA score (except for the Heads Up study, in which no vIGA score was assessed). Although this implies that the beneficial effect of upadacitinib in the previously treated population is reflective of the overall base-case population, such a conclusion should be drawn with caution because it was not an a priori specification for this subgroup analysis and can be underpowered for drawing conclusions. The clinical expert consulted by CADTH suggested that the response to upadacitinib would likely be similar for those with and without prior exposure to a systemic therapy for AD.
The population in Measure Up 1 appeared to have slightly less severe AD than the populations in the rest of the studies. The rest of the baseline and demographic characteristics were similar overall between studies. The results for the adolescent populations analyzed in these included studies (except in the Heads Up trial) mirrored those for the adult populations; however, the adolescent populations were relatively small in the included trials. More evidence will be needed in under-represented populations, such as people of Black/African, First Nations, and Asian descent, compared to the White population. Also, the trial durations might not have been long enough to assess long-term outcomes (harms); and given that patients in the Measure Up and AD Up studies were dupilumab-naive, more evidence is needed to assess the response to upadacitinib in patients who were previously treated with dupilumab.
Indirect Comparisons
Description of Studies
Three indirect treatment comparisons (ITCs) — 2 sponsor-submitted (ITC 1 and ITC 2) and 1 obtained from the CADTH literature search (ITC 3, conducted by the Institute for Clinical and Economic Review [ICER]) — were included to provide an increased perspective on the body of evidence by including indirect comparisons of upadacitinib against dupilumab and other systemic therapies. All ITCs analyzed upadacitinib and its efficacy against other common comparators using Bayesian network meta-analyses (NMAs).
The first is an NMA comparing upadacitinib 15 mg or upadacitinib 30 mg (with or without TCS) versus dupilumab in adults or adolescents with moderate to severe AD with an inadequate response to cyclosporine A or other systemic therapy (i.e., ITC 1, a post-cyclosporine A NMA). The second is an NMA of a comprehensive published RCT evidence base conducted to determine the comparative effectiveness of upadacitinib 15 mg and upadacitinib 30 mg versus other immunomodulators in patients with moderate to severe AD as monotherapy (i.e., in patients not concomitantly receiving TCS) and as combination therapy (i.e., concomitantly receiving TCS). The third is an NMA report from ICER evaluating systemic therapies (abrocitinib, baricitinib, upadacitinib, tralokinumab, and dupilumab) with or without topical therapies in adults and children (≥ 12 years old) with moderate to severe AD.
Efficacy Results
Overall, the results from the 3 ITCs suggest that upadacitinib 30 mg and upadacitinib 15 mg are among the most effective systemic therapies for reducing the severity and symptoms of moderate to severe AD in adults and adolescents, whether as monotherapy or in combination with TCS.
As monotherapy, based on the results from ITC 1, upadacitinib 30 mg and upadacitinib 15 mg demonstrated |||||||||||||||||| against dupilumab 300 mg in improving EASI 75 scores; the NMA detected |||||||||||||||||| between upadacitinib 30 mg and upadacitinib 15 mg. In ITC 2, upadacitinib 30 mg was superior to all comparators (abrocitinib 100 mg, dupilumab 300 mg, baricitinib 2 mg, baricitinib 4 mg, tralokinumab 300 mg) on all outcomes except against abrocitinib 200 mg, versus which no difference was detected in EASI 75 scores and WP-NRS. Upadacitinib 15 mg was superior to abrocitinib 100 mg, dupilumab 300 mg, baricitinib 2 mg, baricitinib 4 mg, and tralokinumab 300 mg, with no difference detected against abrocitinib 200 mg; it was inferior only to upadacitinib 30 mg in both the EASI 75 and IGA scores. The ITC 3 ICER report showed that upadacitinib 30 mg was superior to upadacitinib 15 mg, abrocitinib 100 mg, dupilumab 300 mg, baricitinib 1 mg, baricitinib 2 mg, and tralokinumab 300 mg, with no difference detected against abrocitinib 200 mg when assessing EASI 75 and IGA scores. Upadacitinib 15 mg was superior to abrocitinib 100 mg, baricitinib 1 mg, baricitinib 2 mg, and tralokinumab 300 mg; there was no difference detected against abrocitinib 200 mg and dupilumab 300 mg; and it was inferior only to upadacitinib 30 mg in EASI 75 and IGA scores.
When combination therapy with TCS was assessed, ITC 1 showed no difference between upadacitinib 30 mg and upadacitinib 15 mg or against dupilumab 300 mg in EASI 75 scores. ITC 2 demonstrated that upadacitinib 30 mg was superior to all comparators (upadacitinib 15 mg, baricitinib 2 mg, baricitinib 4 mg, and tralokinumab 300 mg) in EASI 75, IGA, and WP-NRS except dupilumab 300 mg, versus which no difference was detected when evaluating the EASI 75 score. Upadacitinib 15 mg demonstrated superiority only to baricitinib (2 mg and 4 mg) and tralokinumab 300 mg; it was no different from dupilumab 300 mg, and inferior only to upadacitinib 15 mg in terms of EASI 75, IGA, and WP-NRS. ITC 3 demonstrated superiority of upadacitinib 30 mg against upadacitinib 15 mg, abrocitinib 200 mg, abrocitinib 100 mg, dupilumab 300 mg, baricitinib 2 mg, and tralokinumab 300 mg in EASI 75, IGA, and WP-NRS. Only when evaluating the IGA score was no difference detected between abrocitinib 200 mg and upadacitinib 30 mg. Upadacitinib 15 mg was inferior only to upadacitinib 30 mg. It was superior to abrocitinib 100 mg, baricitinib 2 mg, and tralokinumab 300 mg. No difference was detected when compared to dupilumab 200 mg and abrocitinib 200 mg for all outcomes.
Effect estimates from ITC 3 had generally lower OR values when compared to ITC 1 and ITC 2. However, overall, results were similar between the 3 ITCs, demonstrating superiority of upadacitinib 30 mg over upadacitinib 15 mg, dupilumab, and the other comparators, with no difference detected against abrocitinib 200 mg.
Harms Results
No harms data were analyzed in any of the ITCs.
Critical Appraisal
The limitations from the 3 ITCs stem from uncertainty in the effect estimates due to imprecision (wide and overlapping credible intervals [CrIs] among comparisons) and baseline heterogeneity. It is uncertain how upadacitinib relates to other relevant comparators in the population previously treated with systemic therapies (i.e., the approved indication for upadacitinib). Only 1 ITC (ITC 1) evaluated patients previously exposed to systemic therapies (cyclosporine A). Although the comparison in this ITC is exclusively of upadacitinib versus dupilumab, which limits the ITC’s generalizability to other comparisons, the dupilumab comparison is still relevant because that drug is commonly prescribed and reimbursed for AD treatment in Canada. Conclusions regarding the long-term efficacy of upadacitinib compared to the active comparators relevant to this review cannot be drawn because the NMA used study results that were collected over a relatively short time frame, considering the chronic nature of AD. There is also uncertainty due to the inherent heterogeneity across trials in the networks. The robustness of the comparative efficacy was further compromised by the lack of precision in the findings; therefore, results from the ITCs must be interpreted with caution. Moreover, no information was obtained regarding the comparative safety of upadacitinib versus other active comparators. In addition, no conclusion could be drawn regarding the HRQoL outcomes.
Other Relevant Evidence
Description of Studies
Three extension studies of the included studies were reported in the submission. Measure Up 1 to 52, Measure Up 2 to 52, and AD Up 52 are phase III, randomized, double-blind, placebo-controlled, multi-centre studies in adolescents (12 years to 17 years) and adults (18 years to 75 years) with moderate to severe atopic dermatitis. The Measure Up studies included a 35-day screening period, a 16-week double-blind period, a BE period up to week 136, and a 30-day follow-up visit. AD Up (for which the week 52 data cut-off was December 18, 2020) included a 35-day screening period, a 16-week double-blind period, a BE period up to week 136, and a 30-day follow-up visit. At week 16, patients in the placebo group were re-randomized in a 1 to 1 ratio to receive daily oral doses of upadacitinib 30 mg or upadacitinib 15 mg in a blinded fashion up to week 136 in the BE period.
Efficacy Results
Patients in the Measure Up 1 to 52, Measure Up 2 to 52, and AD Up 52 studies maintained response in the co-primary end points. For instance, in Measure Up 1 to 52, 59.2% and 62.5% of the patients who started upadacitinib 15 mg and upadacitinib 30 mg every day, respectively, maintained a vIGA-AD response of 0 or 1 at week 52; and 82% and 84.9% of the patients who started upadacitinib 15 mg and upadacitinib 30 mg once daily, respectively, maintained an EASI 75 response at week 52. In Measure Up 2 to 52, 52.6% and 65.1% of the patients who started upadacitinib 15 mg and upadacitinib 30 mg once daily, respectively, maintained a vIGA-AD response of 0 or 1 at week 52; and 79.1% and 84.3% of the patients who started upadacitinib 15 mg and upadacitinib 30 mg once daily, respectively, maintained a EASI 75 response at week 52. In AD Up 52, 46.3% and 55.7% of the patients who started upadacitinib 15 mg and 30 mg once daily plus TCS, respectively, maintained a vIGA-AD response of 0 or 1 at week 52; and 70.8% and 83.5% of the patients who started upadacitinib 15 mg and 30 mg once daily plus TCS, respectively, maintained an EASI 75 response at week 52.
Harms Results
In Measure Up 1 to 52, a total of ||||| patients had at least 1 AE during the study. The most common AEs (per 100 patient-years) were related to acne (|||||), upper respiratory tract infections (|||||), and nasopharyngitis (|||||). In Measure Up 2 to 52, a total of 606 patients (||||| per 100 patient-years) had at least 1 AE during the study. The most common AE was acne (|||||). In AD Up, a total of ||||| patients had at least 1 TEAE during the study, most commonly related to nasopharyngitis (|||||) or acne (|||||). No deaths were reported. SAEs included CPK elevations. The most common notable harms were hepatic disorder (|||||), herpes zoster (|||||), CPK elevation (|||||), and serious infection (|||||).
Conclusions
Evidence from 3 double-blind, placebo-controlled studies (Measure Up 1, Measure Up 2, and AD Up) shows that compared to placebo, both upadacitinib 15 mg and upadacitinib 30 mg improve disease severity end points in adults with moderate to severe AD, based on EASI 75 and vIGA-AD scores, whether as monotherapy (as in the Measure Up 1 and 2 studies) or in addition to TCS (as in the AD Up study). The evidence from these studies also indicates that upadacitinib 15 mg and upadacitinib 30 mg would likely reduce AD symptoms (WP-NRS, POEM, ADerm-IS), improve HRQoL (DLQI), and improve mood and productivity domains (Hospital Anxiety and Depression Scale – Anxiety [HADS-A] and Work Productivity and Activity Impairment Index: Atopic Dermatitis [WPAI:AD]). The evidence suggests that these effect estimates are similar in the adolescent subpopulation. These results were considered clinically relevant by both clinical experts and patients. Results from 1 head-to-head study (Heads Up) demonstrated superiority of upadacitinib 30 mg in reducing disease severity and symptoms (based on the EASI 75 and WP-NRS) compared to dupilumab at week 16; however, after 24 weeks, this difference was no longer observed, and beyond this time point, the evidence is still uncertain.
Three ITCs support the notion that upadacitinib 15 mg and upadacitinib 30 mg are effective when compared to dupilumab and other systemic immunomodulators, and that upadacitinib may be among the most effective systemic therapies for reducing severity and symptoms in patients with moderate to severe AD, either as monotherapy or in combination with TCS. However, conclusions regarding the long-term efficacy of upadacitinib compared to the active comparators relevant to this review cannot be drawn because the ITCs used study results collected over a relatively short duration, whereas AD is chronic in nature. There is also uncertainty due to the inherent heterogeneity across trials in the networks. The robustness of the comparative efficacy was further compromised by the lack of precision in the findings; therefore, the results from the ITCs must be interpreted with caution. Moreover, no information was obtained regarding comparative safety versus other active comparators. In addition, no conclusion could be drawn on HRQoL outcomes.
All of the trials enrolled patients with moderate to severe AD and an inadequate response to topical and systemic AD therapies. This is reflective of the indication that was initially submitted to Health Canada and CADTH; however, the approved indication reflects a more restrictive population (i.e., patients who are not adequately controlled with a systemic treatment, such as a steroid or biologic, or when use of those therapies is inadvisable). Although there is some similarity in the results between the overall populations and the proportion of patients with prior exposure to a systemic therapy (indicating that prior exposure had little to no effect on benefits and harms compared to the overall population), generalizability of the results from the included studies to the approved indication is uncertain because only a proportion of the populations from the pivotal studies is relevant to the current indication of patients with previous systemic therapies; in addition, there was no evidence for dose escalation to upadacitinib 30 mg once daily in patients with an inadequate response to upadacitinib 15 mg once daily, and there was no clinical evidence for dose de-escalation to upadacitinib 15 mg once daily in patients who achieved a response to upadacitinib 30 mg once daily. The clinical expert consulted by CADTH indicated that the subgroup analyses suggested that the response to upadacitinib would likely be similar for those with and without prior exposure to a systemic therapy for AD.
Overall, upadacitinib was safe and tolerated in all studies. AEs that were more common with upadacitinib included acne and respiratory tract infections. The safety profile of upadacitinib once daily over 52 weeks was consistent with that observed during the 16-week double-blind period, with no unexpected safety signals reported. However, longer-term data will help better characterize the efficacy and safety of upadacitinib in the treatment of this chronic condition.
Introduction
Disease Background
AD, also known as atopic eczema, is an inflammatory, pruritic, chronic or chronically relapsing skin disease commonly occurring in families with other allergic conditions, such as asthma and rhino-conjunctivitis. AD is considered among the most common non-communicable skin diseases, affecting up to 20% of children and 2% to 8% of adults worldwide.1 In Canada, the lifetime prevalence of AD is up to 17% of the population, and there is evidence to suggest that the prevalence has increased over the past 30 years.2,3
AD is characterized by severe pruritus that results in red and swollen skin (a rash). The resulting lesions may appear as fluid-filled vesicles that ooze, crack, and crust. Frequent scratching may result in lichenification (thickening of the skin).4 AD typically affects body creases or flexural areas, such as the popliteal and antecubital fossa, but may also appear on the face, neck, and hands. In patients with AD, secondary skin infections are common due to a compromised skin barrier function plus frequent scratching. Furthermore, the reduced water-holding capacity of the skin produces dryness that requires treatment with specific bathing, cleansing, and moisturizing practices.1,4
AD usually develops in childhood (most cases begin before the age of 5 years) and may persist into adulthood; less frequently, it starts in mid-life or late life.4 The majority of children will outgrow the condition by adolescence. However, it is common for children with AD to develop asthma and/or hay fever — a process commonly referred to as the “atopic march.” AD is often the first step in this sequence of the development of atopic conditions.5
Patients often experience worse itching throughout the night, which may result in sleep loss and ensuing detrimental effects pertaining to school or work. Individuals with AD may also suffer from the social stigma of having a highly visible skin condition. Overall, these patients describe a physically and mentally exhausting condition that can result in anxiety, depression, and decreased quality of life. Health care utilization and costs are also affected and usually associated with the severity of the disease.6
The goals of AD management are to prevent flares and to manage flares effectively when they do occur by preventing AD from progressing. While there is no cure for AD, there are several therapeutic options available to patients to manage the condition. The majority of patients treat AD using general skin care methods, such as by avoiding skin irritants, and by using topical anti-inflammatory therapy. If these common methods fail to improve AD, patients may use off-label systemic therapy (i.e., immunosuppressant therapy) or other therapies, such as phototherapy.4
Standards of Therapy
General Skin Care
General skin care practices for patients with AD include avoiding irritants and managing dry skin. The symptoms of AD may be reduced or prevented by avoiding known skin irritants or triggers.1,4,7 Some common irritants include temperature, humidity, dust, pets (animal dander), smoke, and grass. Using mild detergents to wash clothing (with no bleach or fabric softener) and double-rinsing clothing during laundering have been recommended for those with AD. Dry skin associated with AD can be countered through specific bathing, cleansing, and moisturizing practices. Baths using lukewarm water and emulsifying oil followed by the use of moisturizers is recommended. Limiting the use of soap and fragranced products may also help to reduce symptoms.1,7
Topical Therapy
While a number of non-pharmacological topical therapies exist for treating the symptoms of AD, the most common therapy is the use of moisturizers to combat dry skin through hydration and the prevention of trans-epidermal water loss. Moisturizers are routinely used to provide some barrier protection for the skin from irritants or allergens and can act to soften, reduce itching, and minimize cracking, fissuring, and lichenification. Moisturizers are routinely used frequently throughout the day, preferably after bathing, and can contain a combination of emollients, humectants, and occlusive drugs.2,4,7 Emollients (e.g., glycol and glyceryl stearate, soy sterols) lubricate and soften the skin by smoothing out its surface and filling spaces with droplets. Humectants (e.g., glycerol, lactic acid, urea) attract water and increase the skin’s water-holding capacity. However, humectants sting open skin and are not useful in children with AD. Occlusive drugs (e.g., petrolatum, dimethicone, mineral oil) provide a layer of oil on the surface of the skin to slow trans-epidermal water loss and prevent water loss though evapouration, thereby increasing the moisture content of the skin. The choice of moisturizer depends on the area of the body and the degree of dryness of the skin.2,4,7
The most common pharmaceutical topical therapies include TCSs and TCIs. TCSs act as anti-inflammatory therapies and are considered to be the first-line treatment for AD.1 There are more than 30 different types of TCSs that can take the form of lotions, creams, oily creams, ointments, or gels. These may be combined with other drugs, such as antibiotics. TCSs vary in potency. In Canada, hydrocortisone 1% (low potency) is the most commonly prescribed type for use on the face. For the rest of the body, triamcinolone or betamethasone valerate (moderate potency) are most commonly prescribed. TCSs are applied directly to the area of affected skin before the use of emollients. A response is typically seen within 10 days to 14 days. Side effects associated with the long-term use of TCSs include striae (stretch marks), petechiae (small red or purple spots), telangiectasia (small, dilated blood vessels on the surface of the skin), skin thinning, atrophy, and acne.1,2,4,7 TCSs can also be recommended for use in children, according to the American Academy of Dermatology (AAD), with cautions regarding dosing, given that children have a larger surface area to body mass ratio and that there are mixed results from various studies suggesting that systemic absorption may have an impact on growth.1,4
TCIs are steroid-free, anti-inflammatory, immunosuppressant drugs that can be used over the long-term. In Canada, the 2 second-line drugs available are pimecrolimus and tacrolimus. Pimecrolimus 1% cream can be used for short-term and intermittent long-term therapy for mild to moderate AD and is effective in controlling pruritus.4,7 Topical tacrolimus is an ointment that can be used for short-term and intermittent long-term therapy in moderate to severe AD and demonstrates rapid and sustained AD symptom control. The most common AE associated with TCIs is site-specific burning and irritation. There remains a black box warning for the TCIs regarding lymphoma; however, long-term (10-year) surveillance studies have not found an increased risk of lymphoma versus that of the general pediatric population. Other topical therapies for AD include treatments with diluted bleach baths, which can help reduce the occurrence of secondary skin infections.1,4
Crisaborole, a topical phosphodiesterase type 4 inhibitor, is also available. The advantage of the calcineurin inhibitors and crisaborole is that both can be safely applied to the face and to creases. TCSs more potent than hydrocortisone 1% are inappropriate.
Systemic Therapy
Systemic therapy for the treatment of AD typically involves the use of antimicrobials, antihistamines, or immunomodulators.2,7 Systemic antibiotic treatments can be used to counter widespread secondary bacterial infections. Many patients encounter infection with Staphylococcus aureus, which may cause new inflammation and exacerbate AD symptoms. The choice of systemic antibiotic drug depends upon the skin culture and sensitivity profile. Sedating antihistamines have been used in cases where patients are not achieving adequate sleep due to itching.4
Immunomodulatory drugs include methotrexate, cyclosporine A, mycophenolate mofetil, and azathioprine. These can be used in patients who are not responsive to other treatments.2,4,7 However, these commonly used, off-label treatments are administered in the lowest doses and for the shortest durations possible due to the possibility of side effects. There is limited evidence for the use of methotrexate and azathioprine in the pediatric population; however, a recent 12-week study showed that while methotrexate had a slower onset than low-dose cyclosporine A, it had an increased time before relapse after discontinuation.15 Regarding azathioprine, there is evidence of efficacy in children; however, its use is recommended to be reserved for recalcitrant cases of AD, or in cases where AD is having a significant psychosocial impact.16 The AAD states that mycophenolate mofetil is a relatively safe systemic therapy in all ages; however, its long-term (> 24 months) efficacy and safety in the pediatric population have not been studied.7 With respect to corticosteroids, there is a longstanding understanding that chronic use can affect growth in children.
Dupilumab (Dupixent) is an IL-4 and IL-13 inhibitor indicated for use in adults and pediatrics with moderate to severe AD whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable. CADTH recommended that dupilumab be reimbursed with conditions; it is currently reimbursed by the participating drug programs for patients whose AD is inadequately controlled with topical prescription therapies and who have demonstrated failure on or intolerance to an adequate trial of phototherapy (where available), methotrexate, and cyclosporine A.8
Other Therapy
Phototherapy is another second-line therapy that is commonly used after the failure of TCSs, TCIs, and crisaborole. This therapy is offered over several sessions and its use is guided by a number of factors, including patient skin type and skin cancer history. According to the AAD guidelines, phototherapy is considered to be a safe and effective treatment for AD in children. There are no studies that address its long-term use in children or adults.9
Drug
Upadacitinib is a small-molecule, reversible JAK inhibitor. JAKs are intracellular enzymes that transmit signals arising from cytokine or growth factor receptor interactions on the cellular membrane to influence the cellular processes of hematopoiesis and immune cell function. Upadacitinib is indicated for the treatment of adults and adolescents 12 years of age and older with refractory, moderate to severe AD who are not adequately controlled with a systemic treatment (e.g., a steroid or biologic) or when the use of such therapies is inadvisable. Upadacitinib can be used with or without TCSs. The sponsor’s reimbursement request is for the treatment of patients aged 12 years and older with moderate to severe AD whose disease is not adequately controlled with topical prescription therapies and/or who are refractory to or ineligible for systemic immunosuppressant therapies (i.e., due to contraindications, intolerance, or need for long-term treatment).10
Upadacitinib is available as 15 mg or 30 mg oral extended-release tablets. The recommended starting dose for adult patients is 15 mg once daily. If an adequate response (e.g., EASI 75) is not achieved, the dosage can be increased to 30 mg once daily. For some patients, such as those with severe disease, a starting dose of 30 mg once daily may be appropriate. Upadacitinib should be discontinued if an adequate response is not achieved with the 30 mg dose after 16 weeks of treatment. The lowest effective dose needed to maintain response should be used. For patients older than 65 years, the 30 mg dose once daily is not recommended. The recommended dosage of upadacitinib is 15 mg once daily for adolescents (from 12 years to 17 years of age) weighing at least 40 kg. Upadacitinib has not been studied in adolescents weighing less than 40 kg. The product monograph indicated that treatment should be interrupted if a patient develops a serious infection, and should also be interrupted if laboratory abnormalities (such as in absolute neutrophil count, absolute lymphocyte count, hemoglobin, and hepatic transaminases) are present or develop during treatment.10
Upadacitinib was previously reviewed by CADTH on February 4, 2020, for the indication of adults with moderate to severely active rheumatoid arthritis, after which a final recommendation was issued that it be reimbursed with conditions. Upadacitinib has also been reviewed recently (June 16, 2021) for the indication of adult patients with active psoriatic arthritis who have had an inadequate response or intolerance to methotrexate or other disease-modifying antirheumatic drugs.
The characteristics of upadacitinib and its most common comparators for the purpose of this review are presented in Table 5.
Table 5: Key Characteristics of Upadacitinib and Comparators
Characteristic | Upadacitinib | Dupilumab | Azathioprine | Mycophenolate mofetil | Cyclosporine | Methotrexate |
|---|---|---|---|---|---|---|
Mechanism of action | Janus kinase inhibitor | IL 4 and IL 13 inhibitor | Immunosuppressive drug Antimetabolite (reduces proliferation of lymphocytes) | Immunosuppressive drug Inhibits purine synthesis, reduces lymphocyte proliferation Reduces antibody formation by B lymphocytes | Immune suppressive; inhibits IL 2 and T-cell activation | Immune suppressant |
Indicationa | For the treatment of adults and adolescents 12 years of age and older with refractory moderate to severe AD who are not adequately controlled with a systemic treatment (e.g., a steroid or biologic) or when the use of those therapies is inadvisable (can be used with or without topical corticosteroids) | Treatment of patients aged 6 years and older with moderate to severe atopic dermatitis whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable | Rheumatoid arthritis Prevention of transplant rejection (renal) | Prevention of transplant rejection (renal) | Prevention of transplant rejection Psoriasis Rheumatoid arthritis Nephrotic syndrome | Various neoplasia Psoriasis Rheumatoid arthritis |
Route of administration | Oral | Subcutaneous | Oral | Oral or IV | Oral | Oral Subcutaneous |
Recommended dose | Adults: 15 mg or 30 mg once daily Adolescents (from 12 years to 17 years of age): 15 mg once daily for adolescents weighing at least 40 kg | ≥ 18 years: 600 mg followed by 300 mg 2.q.w. 6 years to 17 years:
| Renal transplant: initial dose 3 mg/kg to 5 mg/kg daily, then dose reduction maintenance level of 1 mg/kg to 3 mg/kg daily Rheumatoid arthritis: initial dose of 1 mg/kg (50 mg to 100 mg) as single dose or twice daily; dose increments of 0.5 mg/kg daily up to a maximum of 2.5 mg/kg/day | 1g orally twice a day 1g IV twice a day | Initial dose for psoriasis: 2.5 mg/kg/day in 2 divided doses, not to exceed 5 mg/kg/day | Varies with indication |
Serious adverse effects or safety issues | Tuberculosis, invasive fungal infections, bacterial, viral, including herpes zoster, and other opportunistic infections, malignancies, thrombosis, lymphopenia, neutropenia | Conjunctivitis, keratitis, hypersensitivity, helminthic infections | Carcinogenic leukopenia, thrombocytopenia, infection, hepatoxicity | Infection, lymphoma, progressive multifocal leukoencepha-lopathy | Infection, malignancy, nephrotoxicity, hypertension, hepatotoxicity, neurotoxicity | Malignancy, serious rash, bone marrow suppression, vomiting, diarrhea, hepatotoxicity, pulmonary toxicity |
Other | Impact of fetal harm unknown | Impact of fetal harm unknown | Fetal harm (mutagenic) | Fetal harm and/or pregnancy loss | Impact of fetal harm unknown | Fetal harm (mutagenic) |
2.q.w. = every 2 weeks; 4.q.w. = every 4 weeks; IL = interleukin.
aHealth Canada–approved indication.
Source: Product monographs for upadacitinib,10 dupilumab,17 azathioprine,18 mycophenolate mofetil,19 cyclosporine A,20 and methotrexate.21
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups.
About the Patient Groups and Information Gathered
Three patient groups responded to CADTH’s call for patient input: ESC, CSPA, and Eczéma Québec. The latter 2 organizations provided a joint submission.
ESC is a registered Canadian charity dedicated to improving the lives of Canadians living with eczema with a mission of support, education, awareness, and research.
CSPA is a national non-profit organization advocating, educating, and supporting Canadians affected by skin, hair, and nail disorders. Its mission is to promote skin health and improve the quality of life of Canadians living with skin disorders through advocacy, education, and awareness, and by supporting research and working with affiliate member organizations that serve specific communities of patient such as those with eczema, melanoma, and psoriasis.
Eczéma Québec was created as a branch of the McGill University Hospital Network Center of Excellence for Atopic Dermatitis. It is a patient advisory committee and registered non-profit organization. It established a network of adult patients with AD and health care practitioners in the field of AD (encompassing specialist clinician dermatologists, general practitioners, nurse practitioners, and more), with a goal of building resources based on international best-practice guidelines. Eczéma Québec works with McGill’s Center of Excellence on knowledge translation tools to improve patient education and care as well as awareness.
ESC gathered survey data from more than 3,000 adults living with AD and from the caregivers of children living with AD. Topics included quality of life impact, experience with systemic treatments, the patient journey, and experience with itch related to AD. Information was also gathered through questionnaires and 1-on-one interviews.
Eczéma Québec and CSPA developed and circulated a web-based survey in English and French using the Survey Monkey platform. The survey was distributed through both organizations’ newsletters, social media, and websites. Eczéma Québec held 30- to 60-minute individual interviews through the Microsoft Teams platform with 3 adult respondents living with moderate to severe AD. There were 56 respondents to the survey. Of the respondents, 91% resided in Quebec, 3.6% in Ontario, 3.6% in New Brunswick, and 1.9% in Manitoba; 76.8% were patients and 12.5% were parents of patients. Most (80.4%) reported their gender assigned at birth as female, and 19.6% reported their assigned gender as male. Of these respondents, 11 identified as male, 43 as female, and 2 as non-binary. The age groups of the respondents were divided as follows: 18 years to 24 years (3.6%), 25 years to 34 years (23.2%), 35 years to 44 years (26.8%), 45 years to 54 years (21.4%), 55 years to 64 years (5.4%), and 65 years and older (16.1%).
Disease Experience
According to the patient input received for this review, AD negatively affects individuals and their families and can lead to psychological distress. Patients frequently report that itch is the most burdensome symptom of AD; more than half of adult respondents with severe AD reported rarely being able to control their urge to scratch. Itch also significantly affects sleep; patients report being woken frequently, and have trouble falling and staying asleep due to their itch.
The severity of AD correlates with impacts on HRQoL as well as lost productivity at school and burden on health systems. Among adult respondents, 54% reported rarely being able to control their urge to scratch. One noted that, “All my life, I have struggled with itch. The constant, debilitating itch that would never leave me alone.” AD also has significant impacts in terms of the psychosocial burden of symptoms, as 1 respondent reflected: “If flaring, [it is] hard to do some things physically and [I’m] self-conscious so tend to stay home.”
One respondent reported “Work stoppage, repeated depression, lack of sleep. It’s hard to participate in social or seasonal activities.” AD can negatively affect mood, work, school, and social interactions. Thirty-two percent of adult survey respondents with moderate or severe AD have missed work events due to their condition, and 30% have had to change careers or give up certain activities. Among the respondents, 18.2% noted that they would miss 1 or 2 days of work per month; 6.1% would miss more than 7 days each month because of their condition.
The respondents noted that the most prevalent areas where they experienced AD were the backs of their hands (63.64%), thighs and/or legs (54.55%), neck (51.52%), the insides of their arms and/or the elbow folds (51.52%), the outsides of their arms and/or the exterior parts of their elbows (51.52%), scalp (48.48%), face (45.45%), ears (45.45%), abdomen (45.45%), the area around the eyes (39.39%), breasts, under breasts, and/or nipples (39.39%), back (39.39%), backs of the knees (36.36%), tops of the feet (30.30%), palms (30.30%), groin area and/or genitalia (24.24%), buttocks (21.21%), front of the knees (21.21%), soles of the feet (21.21%), and armpits (18.18%).
All respondents experienced itching because of their condition. Other symptoms included redness of the skin (87.88%), repeated rashes (84.85%), frequent scratching (84.85%), cracked skin (84.85%), dry and rough skin (78.79%), disrupted sleep (75.76%), bleeding (69.70%), flaking of the skin (69.70%), pain (69.70%), thickening of the skin (60.61%), oozing (48.48%), swelling (42.42%), lichenification (39.39%), and blistering (36.36%). In the ESC survey, 32% of adult respondents with moderate or severe AD said they had missed work events due to their condition, and 30% had to change careers or give up certain activities.
One respondent recounted that, “When I was younger, my mom would wrap my hands so I couldn’t scratch myself in my sleep. My AD was so bad my clothes would stick to my skin during the day, and I had to take a bath in oil just to get clothing, like my tights, off my body.”
Caregivers noted that AD places a significant emotional toll on the entire family, and feelings of guilt, frustration, anger, and sadness are common. Forty-one percent of caregivers reported feeling like a failure when they cannot control their child’s flares. “As a parent, you question everything when your child is suffering, and you are trying to find a solution. You wonder if you are doing enough, or if you are doing something wrong. There needs to be a better way — lives are being destroyed by this condition.” Patients and caregivers reported that the mental health impact of AD is a significant aspect of the condition and is often neither understood by others nor prioritized by health care providers. Uncontrolled chronic AD can lead to feelings of depression and anxiety as well as poor self-esteem, low energy, and — in some extreme cases — suicidal thoughts: “AD is not only exhausting, it is hard on mental health and self-confidence. It is all-consuming for those that suffer from it and for their families.”
Experience With Currently Available Treatments
Patients generally use frequent moisturizing, trigger avoidance, and topical treatments to control their AD flares. Respondents with uncontrolled AD have used systemic treatments that include off-label immunosuppressant medications (such as methotrexate and cyclosporine A), oral corticosteroids (e.g., prednisone), and phototherapy. Oral corticosteroids were the most frequently used systemic treatments. Respondents also noted that they had limited experience with targeted treatments for AD (14.3% had experience with dupilumab and TCIs and 28.6% had experience with cyclosporine A). Respondents mentioned the hurdles associated with the trial-and-error method that many experience before they find an appropriate treatment. One respondent noted that, “I haven’t found the right treatment for me yet. I use diprosone and it doesn’t do much for me unless I am in a major outbreak.” Another mentioned “A lot of trial and error. Now, it's better after looking for 30 years.”
Most patients expressed dissatisfaction with the treatment options available to them and how these addressed the most important symptom of their disease. One respondent stated: “Nothing works,” while another respondent said, “Nothing has stopped the itch.” Another source of frustration for these participants was that they did not view these treatments as long-term options, but rather “temporary.”
Some of the respondents also mentioned the side effects associated with certain treatments. For example, 1 said it was “Alright for a while but had side [e]ffects so discontinued.” Another said, “I started Dupixent a week ago. It's working well so far. It’s unbelievable. I, however, have an intense conjunctivitis in my eyes (side effect of treatment).”
Respondents expressed concern over the financial impact of treatments, with 1 noting, “I had to suspend the use of Protopic until I was on my spouse's insurance. I also needed the Freedom Dupixent program AND my spouse's private insurance. Again, I have a deadline for my [when coverage for my] prescription [ends]. Who can pay $2,500 a month for ONE drug? Not to mention that I must pay for other medications (antidepressants for example).”
Improved Outcomes
Overall, patients desire improvement in managing itch, reducing flares and rashes, and improving their quality of life and sleep. Respondents expressed that they would like a new medication to result in manageable AD in the summer without the use of hydrocortisone, “to improve the appearance of [their] hands and eyes,” to have “less apparent eczema” and more “freedom,” and to provide pain relief. Caregivers want a treatment that will permit them or their child to have a good life, free of itchy skin and painful rashes. Of the respondents, 67.9% preferred daily pills taken by mouth, 50% preferred daily topical medications, and 42.9% preferred injections every other week they could do themselves or with help. The trial-and-error process of cycling through currently available treatments is a common experience among patients, and for those who have experienced repeated failed treatments, the experience is demoralizing, tiring, and causes significant distress, including mental health deterioration. This significant challenge highlights the need for improved treatments for this small population of patients who do not respond to topical treatments.
One respondent noted that, “If you knew how many layers of creams, I had to slather on my body with help.... It was just inhumane.”
On the subject of tolerating side effects, respondents were generally unwilling to accept serious side effects. However, across the spectrum of AD severity, patients and caregivers consistently reported carefully weighing the risks and benefits of any medication, from topical to systemic medications. The willingness of a patient to accept potential serious adverse effects correlates to the severity of their disease and the impact it has on their quality of life. The trial participants who were interviewed reported having had painful and debilitating symptoms of uncontrolled AD. They also shared that they were willing to accept some level of side effects associated with a new treatment and a clinical trial if it meant it would bring them relief from their symptoms.
“[T]he inability to sleep due to the symptoms of eczema is a serious problem in the medium and long term, it must serve as a comparison to the side effects of the drug.”
Experience With Drug Under Review
Respondents who participated in the clinical trial of upadacitinib reported positive experiences, including relief from itch as well as significant and rapid improvement in skin rashes and lesions. One patient reported experiencing significant relief from the itch within days of starting the treatment, and improvement in their skin condition within weeks. A caregiver shared that their child’s rash, which once covered the child’s entire body, was finally able to heal for the first time in their lives. The child no longer had to struggle with constant infections, open sores, and raw, inflamed skin.
One respondent said, “Upadacitinib was extremely helpful in managing my AD. When I think back to where I started, I don’t know where I would be if I hadn’t tried it.” Patients and caregivers also shared that the once-a-day oral pill was an improvement compared to the time-consuming and uncomfortable nature of their previous skin care and topical treatment routines.
Patients and caregivers who had experience with upadacitinib noted that the medication rapidly improved their symptoms and significantly improved their quality of life. It also allowed patients a variety of opportunities, such as regained self-confidence and ability to exercise, better work productivity, improved personal relationships, and the ability to better care for themselves or their loved ones.
Additional Information
Uncontrolled moderate to severe AD can be debilitating and life-altering, and there are significant gaps in treatment for this patient population. The need for more treatment options for uncontrolled AD is critical. The patient groups mention that those living with skin disorders deserve to be treated with respect and dignity by the health care system, which includes embracing new and tailored treatment options.
All respondents noted that the treatment needs to be accessible, with 1 stating that, “Treatments need to be accessible to everyone who needs them and who qualify (i.e., if a doctor deems it helpful). This is not just a skin rash. It is an all-consuming issue that can really affect an individuals’ quality of life — physical and mental. There needs to be knowledge, empathy, and medical support for the physical, mental, and emotional aspects of AD.”
Clinician Input
Input From the Clinical Expert Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol; assisting in the critical appraisal of clinical evidence; interpreting the clinical relevance of the results; and providing guidance on the potential place in therapy). The following input was provided by 1 clinical specialist with expertise in the diagnosis and management of AD.
Treatment Goals
The clinical expert consulted by CADTH visualizes the ideal treatment for AD as 1 that is available to all Canadians, is cost-effective in the context of a publicly funded health care system, has a proven long-term safety record, and completely reverses the barrier dysfunction and immunologic abnormalities that constitute AD. The ideal treatment goal would aim at maintaining complete clearance of AD without additional therapy and would result in a complete elimination of pruritus and resolution of all visible signs of skin inflammation.
Unmet Needs
According to the clinical expert, off-label immunosuppressive drugs are commonly offered to patients who are achieving suboptimal disease control with appropriate disease-specific skin care measures (i.e., irritant avoidance, emollients, and bleach baths), TCSs, calcineurin inhibitors or crisaborole, and phototherapy. In Canada, the most commonly used immunosuppressive drug is methotrexate, followed by cyclosporine A, azathioprine, and mycophenolate mofetil. Because of their potential toxicities, these drugs are generally prescribed as intermittent courses for patients with AD. New drugs could fill this potential treatment gap when toxicities are a concern. There are also patients who do not respond to these drugs. Dupilumab is offered as a second-line systemic therapy alternative to immunosuppressive drugs, but reimbursement for it remains problematic in Canada.
Place in Therapy
Upadacitinib was considered by the clinical expert as a potentially useful addition to the currently available therapeutic options for AD, with special consideration in patients who have contraindications to, or who experience adverse effects from off-label immunosuppressive drugs or who are unresponsive to these. It could also be useful in a subset of patients who respond to off-label immunosuppressive drugs, but still require continuous long-term therapy to control AD.
Upadacitinib could also be of value in patients treated with dupilumab who have a suboptimal response, who develop severe conjunctivitis or other ocular side effects, or who are intolerant to injections (e.g., due to severe injection-site reactions) and prefer an oral drug.
The clinical expert noted that all patients with AD treated with upadacitinib would be expected to continue on emollients, TCSs, and/or TCIs. It is expected that upadacitinib would not be combined with other systemic treatments.
According to the clinical expert consulted by CADTH, upadacitinib is unlikely to cause a significant shift in the current treatment paradigm for AD beyond its inclusion as another effective treatment option in the Canadian clinical landscape. Off-label immunosuppressives or dupilumab (or the new biologics, such as tralokinumab, that are emerging in AD) are not expected to be used in combination with upadacitinib; however, the clinical expert believed that many practitioners would still consider a trial of methotrexate and cyclosporine A before initiating upadacitinib. Dermatologists are well versed in the appropriate dosing and duration of therapy and appropriate monitoring for potential toxicities.
Patient Population
In the opinion of the clinical expert, any patient with moderate to severe AD could respond to treatment with upadacitinib. However, it is still unclear whether this drug can effectively treat patients who have failed methotrexate and/or cyclosporine A. The efficacy of upadacitinib in patients who have failed dupilumab must also be assessed.
AD is, in general, not a diagnostic challenge for a dermatologist. However, there are some differential diagnoses to consider (e.g., psoriasis, ichthyoses, allergic contact dermatitis, irritant contact dermatitis, and cutaneous T-cell lymphoma). Biopsy would usually be reserved for patients recalcitrant to all therapy — for instance, to rule out cutaneous T-cell lymphoma or to differentiate AD from psoriasis.
The clinical expert suggested that patients less suitable for treatment with upadacitinib would be those with AD who are well controlled with topical therapy, phototherapy, and/or intermittent, off-label immunosuppressive therapy, as well as those who are well controlled with dupilumab. Patients with potential contraindications to JAK inhibitors, such as severe active infections (acute or chronic, including latent tuberculosis, deep fungal infections, and opportunistic infections), potential malignancy (including ongoing treatment with chemotherapy, including checkpoint inhibitors), severe hepatic disease, severe renal disease, pregnancy or lactation, a history of thromboembolic events, pre-existing hematologic disease (i.e., lymphopenia and neutropenia), or a weight of less than 40 kg.
Assessing Response to Treatment
In general, the outcomes used in clinical practice are aligned with the outcomes typically used in clinical trials of AD treatments. The clinical expert anticipates that the EASI score could be used as the benchmark for decision-makers. As such, this will be calculated and recorded at each patient visit. Reduction in pruritus will also be noted by clinicians, but perhaps not formally scored. The patient’s impression of their overall improvement will also be important and recorded.
Of these outcome measurements, a rational benchmark response will be EASI 75 at 16 weeks. However, EASI score reductions of 50% to 75% could also be considered clinically meaningful to patients, particularly those who have had severe disease recalcitrant to all previous therapies.
In the opinion of the clinical expert, patients placed on upadacitinib would be re-evaluated 16 weeks after initiating treatment. Those judged to be responders at this visit would be seen subsequently at 6-month intervals. Those who have not reached response targets at 16 weeks would be re-evaluated at 20 weeks following initiation of the drug. The decision to stop upadacitinib or continue on it would be made at the 20-week visit. Bloodwork, including complete blood count and differential, liver function tests, creatinine, lipids, and CPK, would be done monthly before the first follow-up visit and every 3 months thereafter if there are no concerns.
Discontinuing Treatment
The factors anticipated by the clinical expert to be used as criteria for discontinuation included failure to achieve a clinically meaningful response at 16 weeks to 20 weeks, failure to maintain an adequate response on long-term maintenance, development of a hypersensitivity response judged to be due to upadacitinib, TEAEs (e.g., lymphopenia, neutropenia, arterial thrombosis, VTE), and treatment-emergent severe infections or malignancies.
Prescribing Conditions
Because this is an orally administered drug, the clinical expert consulted by CADTH indicated that there are no special administration challenges. However, a specialist would still be required to diagnose, treat, and monitor patients taking upadacitinib. Appropriate specialists will include pediatric dermatologists, general dermatologists, or pediatricians with experience and interest in AD.
According to the clinical expert, patients deemed to have severe symptoms would start on 30 mg for 16 weeks and then be assessed for their response (e.g., EASI 75); if a response is reached, they would switch to a 15 mg dose. The product monograph approved by Health Canada states that patients who are receiving upadacitinib 15 mg and do not achieve a response after 16 weeks of treatment should be switched to upadacitinib 30 mg. The product monograph also states that if patients do not achieve an adequate response (e.g., EASI 75) after 16 weeks of treatment on the 30 mg dose, upadacitinib should be discontinued.
Additional Considerations
The clinical expert considered that some of the comparators included in this review, such as apremilast, retinoids (acitretin, alitretinoin), and ustekinumab, are unlikely to be prescribed in Canada for patients with AD.
Clinician Group Input
This section was prepared by CADTH staff based on the input provided by patient groups.
One clinician group provided input on the reimbursement review of upadacitinib for the treatment of adults and adolescents with moderate to severe AD. The Atlantic Specialist Group Managing Atopic Dermatitis is a group of physicians, including general practitioners, dermatologists, and allergy and immunology specialists, who manage patients with AD. The members of the group are located in various clinical settings across Atlantic Canada.
Unmet Needs
The clinician group considers that improving AD symptoms — such as chronic itch, dry and inflamed skin, and sleep disturbances — and improving quality of life and patient satisfaction (i.e., better sleep and less work or school disruption) are top priorities for treatment goals. Other priorities include flare reduction and disease control, as reflected by the DLQI and PGA scores of clear or almost clear.
The group considers that the greatest unmet need is in the subset of patients with moderate to severe AD, among whom the main unmet need is for an effective, convenient, safe treatment that enables long-term disease control and remission, given that many experience flares as soon as they stop their current medications. This cycle of recurrence leads to disease progression, ending in chronic, severe AD and severe impact on quality of life. Available off-label treatments have poor efficacy and safety profiles that are unacceptable for long-term use. Phototherapy, which is often used in conjunction with systemic therapies, is associated with issues such as poor accessibility, long wait times, low efficacy, and exposure to UV radiation. Suboptimal care of patients with AD commonly leads them to access drugs that are neither effective nor approved for AD and that can be harmful to their health.
According to the clinician group, upadacitinib would allow rapid disease control in patients with moderate to severe AD, addressing the unmet needs that lead to many of the problems experienced by these patients.
Place in Therapy
Upadacitinib would be the first JAK inhibitor approved for AD. The first biologic drug approved for AD, dupilumab, targets IL 14 and IL 13 signalling; therefore, it targets the underlying disease mechanisms of AD through a different mode of action. In the clinician’s opinion, dupilumab addresses some concerns and needs of some patients, but upadacitinib may shift the paradigm due to its efficacy and ease of administration.
The clinician group states that upadacitinib would be used after initial treatments for AD have been implemented, such as lifestyle measures and topical steroids, and after the patient has been diagnosed with moderate to severe AD. In the group’s opinion, upadacitinib would replace both phototherapy and systemic therapies that are currently used off-label to treat moderate to severe AD. If patients fail on or have a contraindication or intolerance to upadacitinib, their treating physician may consider dupilumab as the next therapeutic option. This place-in-therapy judgment differs from that of the clinical expert consulted by CADTH, who considers that upadacitinib should be used after a trial of currently used (even if off-label) systemic therapies, such as methotrexate or cyclosporine A.
Patient Population
According to the clinician group, upadacitinib would be best suited to treat patients with moderate to severe AD who have not responded, are not expected to respond, or have had adverse reactions to long-term use of TCSs. These patients have the greatest need for intervention because they lack long-term treatment options and are at high risk of disease progression. The patients suited for treatment would be treated by a specialist initially, but there are no diagnostic challenges once the patient reaches specialized care; nor are there drug administration challenges. On occasion, a patient with AD will insist on allergy testing due to misperceptions about the link between their condition and allergic reactions. This often leads to an inconclusive diagnosis (with rash or eruption as the descriptor) and patient frustration. Patient and physician education about the pathophysiology of AD is crucial to treatment success. Upadacitinib would be least suitable for patients with mild AD or whose symptoms are well controlled with initial treatments (e.g., TCSs).
Assessing Response to Treatment
The clinician group considers that the outcomes measured in clinical trials, such as the IGA and the proportion of participants achieving a 75% or 90% to 100% improvement on the EASI, are also used in clinical practice (in which the equivalent PGA is used instead of IGA) and are similarly aligned with additional measures, including BSA affected and WP-NRS score (which ranges from 0 [“no itch”] to 10 [“worst imaginable itch”]). EASI scoring is not routinely used in clinical practice. The group mentions that a clinically meaningful response to upadacitinib would include improvement in patient-reported itch (i.e., a 4-point reduction on the WP-NRS or a WP-NRS score of less than 3), a DLQI score reduction of greater than or equal to 4 (or an acceptable improvement), patient-reported improved sleep quality, fewer AD-related disruptions at school and work, and a PGA score of 0 or 1. Importantly, for the response to upadacitinib to be clinically meaningful, a patient should not experience any severe side effects, including over sustained time periods.
The group also suggests that a response to systemic therapy should be reassessed 12 weeks to 16 weeks after the initiation of treatment.
Discontinuing Treatment
According to the clinician group, the decision to discontinue treatment should be assessed based on lack of response, significant disease progression (i.e., lichenification, increased affected BSA and itching) and deterioration in quality of life. Treatment should also be discontinued if the patient experiences adverse reactions or intolerances to the medication that are deemed unacceptable by the patient-physician team.
Treatment with upadacitinib should be interrupted if a patient develops a serious infection, until the infection is controlled. Treatment should also be interrupted to address abnormal laboratory results (i.e., absolute lymphocyte count less than 500 cells/mm3, absolute neutrophil count less than 1,000 cells/mm3, hemoglobin less than 8 g/dL) or if drug-induced liver injury is suspected (based on hepatic transaminases). It may be resumed once levels return to normal.
Prescribing Conditions
Patients with AD who are receiving upadacitinib would ideally be managed in any non-emergent setting to which they have access to that has a dermatologist or allergist well versed in managing moderate to severe AD. Referring family physicians, nurse practitioners, or other health care providers should be counselled on the appropriate referral process.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may affect their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical experts consulted by CADTH are summarized in Table 6.
Table 6: Summary of Drug Plan Input and Clinical Expert Responses
Drug program implementation questions | Clinical expert responses |
|---|---|
Jurisdictional context: relevant comparators and implementation issues | |
Access to phototherapy seems to be limited across Canada. Is this factual or perceived among clinicians and dermatologists? | Phototherapy is mostly accessible in urban areas, but not in rural areas. It is important to consider this barrier in the decision-making process. |
Policy considerations for reimbursement: initiation, continuation, renewal, discontinuation, and prescribing of therapy | |
Would upadacitinib be initiated in patients who have failed previous treatment with a biologic drug? | From the expert’s clinical perspective, patients who have failed dupilumab plus 1 of the immunomodulators would be candidates to receive upadacitinib. This would also apply for those who have failed dupilumab alone; however, there is a high degree of uncertainty in this clinical recommendation due to lack of evidence. |
Should patients be required to have had an adequate trial of (or be ineligible for) cyclosporine A, methotrexate, and phototherapy before initiating upadacitinib? | In the expert’s opinion, a trial of 2 of the 4 immunomodulators (methotrexate, cyclosporine A, mycophenolate mofetil, and azathioprine) should be considered before initiating upadacitinib. |
Would consideration be given to aligning the initiation criteria of upadacitinib with those of dupilumab? The CDEC initiation criteria for dupilumab are: 1. Patients should be 12 years of age or older with moderate to severe AD with disease that is not adequately controlled with topical prescription therapies or when those therapies are not advisable. 2. Patients must have had an adequate trial or be ineligible for each of the following therapies: phototherapy (where available), methotrexate, and cyclosporine A. 3. Patients who have had an adequate trial of phototherapy, methotrexate, and/or cyclosporine A must have documented refractory disease or intolerance. 4. The physician must provide the EASI score and Physician Global Assessment score at the time of initial request for reimbursement. 5. The maximum duration of initial authorization is 6 months. | Yes, these criteria are feasible for upadacitinib. It would be practical also to consider fewer than 6 months for the duration of the initial authorization (i.e., 16 weeks to 20 weeks instead of 24 weeks) and to proceed to assess the continuation or renewal of the indication. |
Will dupilumab (or other biologics approved for AD) be among the prior therapies required in the eligibility criteria for initiation of therapy with upadacitinib? | No. Dupilumab as prior therapy should not be an initiation criterion. Both drugs would have the same place in therapy in the population for this indication. |
Should the renewal criteria for upadacitinib be aligned with those of dupilumab? CDEC renewal criteria for dupilumab are as follows: 1. The physician must provide proof of beneficial clinical effect when requesting continuation of reimbursement, defined as a 75% or greater improvement from baseline in the EASI score (EASI 75) 6 months after treatment initiation. 2. The physician must provide proof of maintenance of EASI 75 response from baseline every 6 months for subsequent authorizations. | The clinical expert consulted by CADTH noted that the renewal criteria are feasible to apply to upadacitinib, although the timing of 6 months (24 weeks) could be long for upadacitinib, and consideration for shorter duration (e.g., 16 weeks to 20 weeks) might be required. |
The included trials had durations of 12 weeks to 16 weeks, with the longest follow-up assessing up to 48 weeks. Based on the available evidence, would you consider that the long-term safety data have been established with certainty? | The clinical expert consulted by CADTH noted that the currently available evidence is not sufficient to establish the long-term safety profile of upadacitinib in the treatment of AD. |
The CDEC recommendation for dupilumab included the following 3 implementation considerations: 1. Based on the trials, moderate to severe AD is defined as an EASI score of 16 points or higher, or an Investigator (or Physician) Global Assessment score of 3 or 4. 2. Adequate control and refractory disease are optimally defined using similar criteria to those used in the dupilumab RCTs, such as achieving EASI 75. 3. Phototherapy may not be available in all jurisdictions. Geographic inability to access phototherapy should not preclude patients from accessing dupilumab if otherwise indicated. Should these 3 implementation considerations also be considered for upadacitinib? | The clinical expert consulted by CADTH noted that these implementation considerations are relevant for the reimbursement of upadacitinib and should be noted in the recommendation. |
Can upadacitinib be used in combination with other JAK inhibitors, biologic DMARDs, phototherapy, or immunosuppressants? | The clinical expert consulted by CADTH noted that upadacitinib should not be used in combination with other systemic treatments for AD. (There is no evidence investigating the safety and efficacy of such combinations.) |
Should upadacitinib be prescribed in consultation with a dermatologist and/or specialist? | A specialist would be required to diagnose, treat, and monitor patients taking upadacitinib. Appropriate specialists would include a pediatric dermatologist, a general dermatologist, or a pediatrician with an interest in atopic dermatitis. |
How would an “adequate trial” be defined in clinical practice for patients with AD who undergo treatment with phototherapy (where available), methotrexate, and cyclosporine A? | The clinical expert consulted by CADTH noted the following:
|
How would “ineligible” be defined in clinical practice for patients with AD who are ineligible to receive therapy with methotrexate or cyclosporine A? | Risk factors or potential adverse reactions from the interventions would make patients ineligible, as mentioned in the clinical input. |
AD = atopic dermatitis; CDEC = CADTH Canadian Drug Expert Committee; DMARD = disease-modifying antirheumatic drug; EASI = Eczema Area and Severity Index; EASI 75 = at least 75% improvement in EASI total score from baseline; JAK = Janus kinase; RCT = randomized controlled trial.
Clinical Evidence
The clinical evidence included in the review of upadacitinib is presented in 3 sections. The first section, the Systematic Review, includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those studies that were selected according to an a priori protocol. The second section includes indirect evidence from the sponsor. The third section includes sponsor-submitted long-term extension studies and additional relevant studies that were considered to address important gaps in the evidence included in the systematic review.
Systematic Review (Pivotal and Protocol Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of upadacitinib for the treatment of adults and adolescents 12 years and older with moderate to severe AD who are candidates for systemic therapy.
Methods
Studies selected for inclusion in the systematic review will include pivotal studies provided in the sponsor’s submission to CADTH and Health Canada as well as those meeting the selection criteria presented in Table 7. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
Of note, the systematic review protocol presented here was established before the granting of a Notice of Compliance from Health Canada.
Table 7: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Population | Patients aged 12 years and older diagnosed with moderate to severe AD whose disease is not adequately controlled with topical prescription therapies, or for whom those therapies are not advisable, or who are refractory to or ineligible for systemic immunosuppressant therapies. Subgroups:
|
Intervention | Upadacitinib 15 mg or 30 mg (extended-release tablets) administered orally once daily in adult patients, with dose selection based on individual patient presentation Upadacitinib 15 mg (extended-release tablets) administered orally once daily in adolescents (12 years to 17 years of age) weighing at least 40 kg Upadacitinib can be used with or without topical corticosteroids. |
Comparator | When used alone or in combination with topical therapy:
|
Outcomes | Efficacy outcomes:
Harms outcomes:
|
Study designs | Published and unpublished phase III and IV RCTs |
AE = adverse event; CPK = creatine phosphokinase; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; IGA = Investigator Global Assessment; MACE = major adverse cardiovascular event; RCT = randomized controlled trial; SAE = serious adverse event; SCORAD = Scoring Atopic Dermatitis; vs. = versus; WDAE = withdrawal due to adverse event.
The literature search was performed by an information specialist using a peer-reviewed search strategy according to the PRESS Peer Review of Electronic Search Strategies checklist.22
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946–) through Ovid and Embase (1974–) through Ovid. The search strategy comprised both controlled vocabulary, such as the National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concept was Rinvoq (upadacitinib). Clinical trial registries were searched: the US National Institutes of Health’s clinicaltrials.gov, WHO’s International Clinical Trials Registry Platform search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
No filters were applied to limit the retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. Refer to Appendix 1 for the detailed search strategies. The initial search was completed on May 11, 2021. Regular alerts updated the search until the meeting of the CADTH Canadian Drug Expert Committee on February 23, 2022.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the CADTH checklist, Grey Matters: A Practical Tool For Searching Health-Related Grey Literature checklist.23 Included in this search were the websites of regulatory agencies (US FDA and European Medicines Agency). Google was used to search for additional internet-based materials. Refer to Appendix 1 for more information on the grey literature search strategy.
These searches were supplemented by reviewing bibliographies of key papers and through contacts with appropriate experts. In addition, the manufacturer of the drug was contacted for information regarding unpublished studies.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to include in the review, and differences were resolved through discussion.
Findings From the Literature
A total of 5 unique studies were identified from the literature for inclusion in the systematic review (Figure 1). The included studies are summarized in Table 8 and Table 9. A list of excluded studies is presented in Appendix 2.
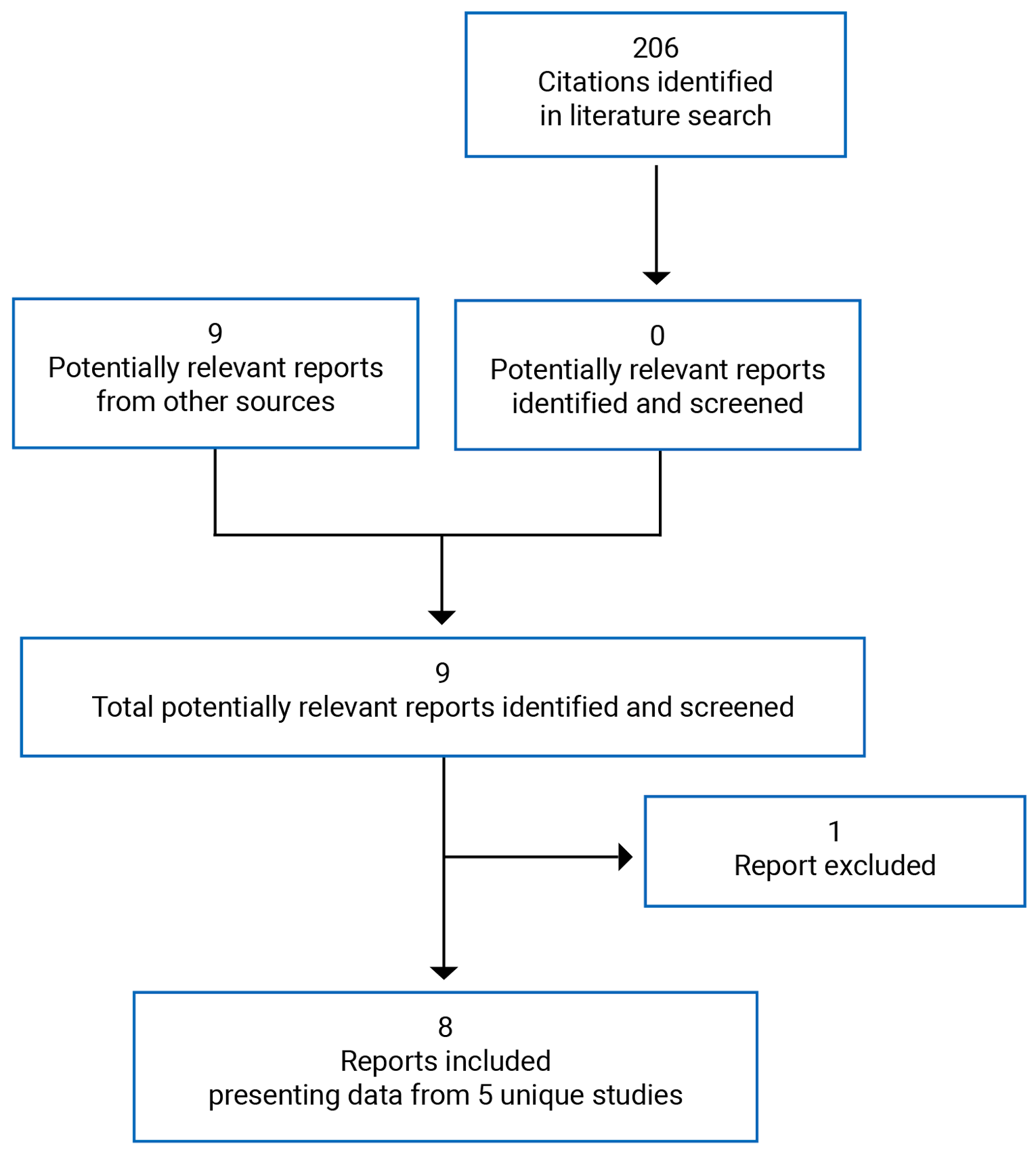
Table 8: Details of Included Studies — Part 1
Study detail | Measure Up 1 M16 to 045 | Measure Up 2 M18 to 891 |
|---|---|---|
Design and populations | ||
Study design | Phase III, DB, placebo-controlled RCT | Phase III, DB, placebo-controlled RCT |
Locations | 151 sites in 24 countries, including the US, Canada, China, and countries in Latin America and Europe | 154 study sites in 23 countries in North America (including Canada), Europe, Asia, and Australia |
Patient enrolment dates | First visit: August 13, 2018 Last visit: December 21, 2020 (week 52) | First visit: July 27, 2018 Last visit (week 52): January 15, 2021 |
Randomized (N) | 847 | 836 |
Inclusion criteria |
| |
Exclusion criteria |
| |
Drugs | ||
Intervention | Arm 1: Upadacitinib 15 mg administered orally once daily Arm 2: Upadacitinib 30 mg administered orally once daily Both administered for 16 weeks (DB period), then for up to 136 weeks (BE period) | Arm 1: Upadacitinib 15 mg administered orally once daily Arm 2: Upadacitinib 30 mg administered orally once daily Both administered for 16 weeks (DB period), then for up to 136 weeks (BE period) |
Comparator | Placebo administered orally once daily for 16 weeks (DB period) | Placebo administered orally once daily for 16 weeks (DB period) |
Duration | ||
Run-in phase | 5-week screening period | 5-week screening period |
Double-blind phase | 16 weeks | 16 weeks |
Follow-up phase | BE period up to 136 weeks | BE period up to 136 weeks |
Outcomes | ||
Primary end point | Co-primary Proportion of patients achieving: 1. At least a 75% reduction in EASI from baseline at week 16 2. vIGA-AD of 0 or 1 (clear or almost clear) with at least 2 grades of reduction from baseline at week 16 | Co-primary 1. Proportion of patients achieving at least a 75% reduction in EASI from baseline at week 16 2. Proportion of patients achieving vIGA-AD of 0 or 1 (clear or almost clear) with at least 2 grades of reduction from baseline at week 16 |
Secondary and exploratory end points | Key secondary:
| Key secondary:
|
Notes | ||
Publications | Guttman-Yassky (2021)24 | Guttman-Yassky (2021)24 |
AD = atopic dermatitis; ADerm-IS = Atopic Dermatitis Impact Scale; ADerm-SS = Atopic Dermatitis Symptom Scale; BE = blinded extension; DB = double blind; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; HADS-A = Hospital Anxiety and Depression Scale – Anxiety; HADS-D = Hospital Anxiety and Depression Scale – Depression; MCID = minimal clinically important difference; POEM = Patient-Oriented Eczema Measure; RCT = randomized controlled trial; SCORAD = Scoring Atopic Dermatitis; TCI = topical calcineurin inhibitor; TCS = topical corticosteroid; TSS-7 = 7-Item Total Symptom Score; vIGA-AD = validated Investigator Global Assessment for Atopic Dermatitis; vs. = versus; WP-NRS = Worst Pruritis Numerical Rating Scale.
Source: Clinical Study Reports for Measure Up 1 and Measure Up 2.11,12
Table 9: Details of Included Studies — Part 2
Study detail | AD Up M16 to 047 | Heads Up M16 to 046 | Japan M17 to 377 |
|---|---|---|---|
Design and populations | |||
Study design | Phase III, DB, placebo-controlled RCT | Phase IIIb, randomized, DB, double-dummy, active-controlled study | Phase III, DB, placebo-controlled RCT |
Locations | 170 sites in 22 countries (including the US, Canada, Australia, and countries in Europe and Asia) | 129 sites in 22 countries (including the US, Canada, and countries in Europe and Asia) | 42 sites in Japan |
Patient enrolment dates | First patient first visit: August 9, 2018 Last patient last visit: December 18, 2020 (week 52) | First patient first visit: February 21, 2019 Last patient last visit: December 9, 2020 | First patient first visit: October 27, 2018 Last patient last visit: December 2, 2019 (week 24) |
Randomized (N) | 901 | 692 | 272 |
Inclusion criteria |
|
|
|
Exclusion criteria |
|
|
|
Drugs | |||
Intervention | Arm 1: upadacitinib 15 mg orally plus TCS administered once daily Arm 2: upadacitinib 30 mg orally plus TCS administered once daily Both administered for 16 weeks (DB period), then for up to 136 weeks (BE period) | Upadacitinib 30 mg orally administered daily (until week 24 visit) plus placebo pre-filled syringe administered SC (2 injections) at baseline followed by placebo pre-filled syringe (1 injection) every other week until the week 22 visit | Arm 1: upadacitinib 15 mg orally plus TCS administered once daily Arm 2: upadacitinib 30 mg orally plus TCS administered once daily Both administered for 16 weeks (DB period), then for up to 136 weeks (BE period) |
Comparator | Placebo administered orally once daily plus TCS for 16 weeks (DB period) | Dupilumab 600 mg (2 × 300 mg SC injection) administered at the baseline visit, followed by dupilumab 300 mg SC injection every other week until the week 22 visit; daily oral doses of placebo tablets from the baseline visit to the week 24 visit | Placebo administered orally once daily plus TCS for 16 weeks (DB period) |
Duration | |||
Run-in phase | 5-week screening period | 5-week screening period | 5-week screening period |
Double blind phase | 16 weeks | 24 weeks | 16 weeks |
Follow-up phase | BE period up to 136 weeks and a 30-day follow-up visit | End-of-treatment follow-up visit at 12 weeks after the last injection for patients who did not enrol in the open-label study | BE period up to 52 weeks and open-label period up to week 136 |
Outcomes | |||
Primary end point | Co-primary Proportion of patients achieving: 1. At least a 75% reduction in EASI from baseline at week 16 2. vIGA-AD of 0 or 1 (clear or almost clear) with at least 2 grades of reduction from baseline at week 16 | Proportion of patients achieving a 75% reduction in EASI from baseline at week 16 | There were no primary or secondary end points. Only as exploratory. Safety end points included:
|
Secondary and exploratory end points | Key secondary end points
| Key secondary end points
| Exploratory end points
|
Notes | |||
Publications | Reich (2021), Silverberg (2021) (unpublished, submitted) | Blauvelt (2021) (unpublished) | None |
AD = atopic dermatitis; BE = blinded extension; BSA = body surface area; DB = double blind; EASI = Eczema Area and Severity Index; EASI 75 = at least 75% improvement in EASI total score from baseline; EASI 90 = at least 90% improvement in EASI total score from baseline; EASI 100 = at least 100% improvement in EASI total score from baseline; PDE 4 = phosphodiesterase 4; RCT = randomized controlled trial; SC = subcutaneous; TCI = topical calcineurin inhibitor; TCS = topical corticosteroid; vIGA-AD = validated Investigator Global Assessment for Atopic Dermatitis; WP-NRS = Worst Pruritis Numerical Rating Scale.
Source: Clinical Study Reports for AD Up, Heads Up, and Japan studies.13,14,25
Description of Studies
Table 10 provides an overview of the studies that were summarized and appraised by CADTH in the current review of upadacitinib.
There were 5 double-blind, phase III RCTs included in the CADTH systematic review: 2 similar multi-centre, placebo-controlled trials conducted with upadacitinib used as monotherapy for AD (Measure Up 1 and Measure Up 2); 1 double-blind, placebo-controlled trial conducted with upadacitinib used as combination therapy with TCSs for AD (AD Up study); 1 double-blind, double-dummy, active-controlled trial comparing upadacitinib versus dupilumab (Heads Up study); and 1 phase III, double-blind, placebo-controlled trial conducted with upadacitinib used as combination therapy with TCSs in a Japanese setting (Japan study) assessing the safety of upadacitinib as the main end point.
Table 10: Details of Included Studies in Systematic Review
Regimen | Study ID | Design | Duration | Status |
|---|---|---|---|---|
Monotherapy | Measure Up 1 | Phase III, DB, placebo-controlled, randomized trial | 16 weeks | Complete |
Measure Up 2 | Phase III, DB, placebo-controlled, randomized trial | 16 weeks | Complete | |
Combination therapy with TCSs | AD Up | Phase III, DB, placebo-controlled, randomized trial | 16 weeks | Complete |
Head to head | Heads Up | Phase IIIb, randomized, DB, double-dummy, active-controlled study | 24 weeks | Complete |
Combination therapy and safety | Japan study | Phase III, DB, placebo-controlled, randomized trial | 16 weeks up to 136 weeks | Complete |
DB = double blind; TCS = topical corticosteroid.
Source: Clinical Study Reports for Measure Up 1 and Measure Up 211,12 and for the AD Up, Heads Up, and Japan studies.13,14,25
Monotherapy Studies
Measure Up 1 and Measure Up 2 were both phase III, double-blind RCTs aimed at evaluating the efficacy and safety of upadacitinib as monotherapy in adolescents (12 years to 17 years of age at the time of the screening visit and body weight ≥ 40 kg) and adults (18 years to 75 years of age) with moderate to severe AD. Both studies included patients with AD whose onset of symptoms was at least 3 years before the baseline screening date. Both studies consisted of a 5-week screening period, a 16-week double-blind treatment phase, and a BE study of a duration of up to 136 weeks and a 30-day follow-up visit.11,12 Eligible patients were randomized in a 1 to 1 to 1 ratio to receive a daily oral dose of upadacitinib 30 mg, upadacitinib 15 mg, or matching placebo every day. Randomization was stratified by baseline disease severity (moderate [vIGA-AD score of 3] versus severe [vIGA-AD score of 4]), by geographic region (US, Puerto Rico, and Canada; Japan; China; and other), and by age (adolescent [ages 12 years to 17 years] versus adult [ages 18 years to 75 years]). At week 16, patients in the placebo group were re-randomized in a 1 to 1 ratio (stratified by week 16 reduction of at least 50% in EASI from baseline [EASI 50], responder status [yes or no], geographic region [US, Puerto Rico, Canada, China, Japan, and other], and age group [adolescent or adult]) to receive daily oral doses of upadacitinib 30 mg or upadacitinib 15 mg in the BE period. Patients originally randomized to upadacitinib were to continue on upadacitinib in the extension period at the same dose. Rescue treatment for AD was permitted at the week 4 visit at the discretion of the investigator if medically necessary and if specified parameters were met. The overall designs of the Measure Up 1 and Measure Up 2 studies are presented in Figure 2.
Measure Up 1 was conducted from August 2018 to December 2020 at 151 sites in 24 countries (US, Canada, China, and countries in Latin America and Europe), while Measure Up 2 was conducted from July 2018 to January 2021 at 154 study sites across 23 countries, including Australia, Canada, the US, and countries in Europe and Asia.
Figure 2: Study Design Schematic of the Measure Up 1 and Measure Up 2 Studies
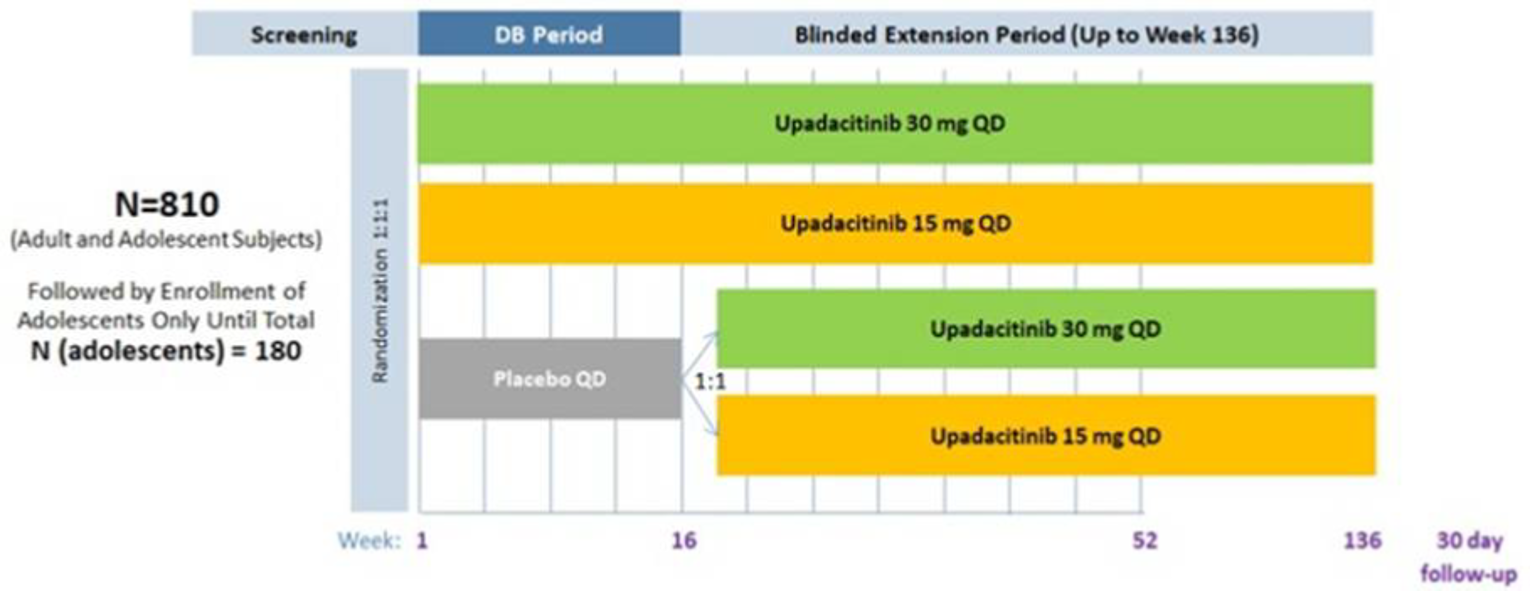
DB = double blind; QD = once daily.
Source: Clinical Study Reports for Measure Up 1 and Measure Up 2.11,12
Combination Therapy
The AD Up study was a phase III, double-blind RCT aimed at evaluating the efficacy and safety of upadacitinib as combination therapy with TCSs in adolescents (12 years to 17 years of age at the time of the screening visit, with body weight ≥ 40 kg) and adults (18 years to 75 years of age) with moderate to severe chronic AD (i.e., the onset of symptoms was at least 3 years before baseline screening). The study consisted of a 5-week screening period, a 16-week double-blind treatment phase, and a BE study lasting up to 136 weeks plus a 30-day follow-up visit. Eligible patients were randomized in a 1 to 1 to 1 ratio to receive a daily oral dose of upadacitinib 30 mg plus TCS, upadacitinib 15 mg plus TCS, or a matching placebo plus TCS. Randomization was stratified by baseline disease severity (moderate [vIGA-AD score of 3] versus severe [vIGA-AD score of 4]), by geographic region (US, Puerto Rico, Canada; Japan; mainland China; other), and by age (adolescent [ages 12 years to 17 years] versus adult [ages 18 years to 75 years]). At week 16, patients in the placebo group were re-randomized in a 1 to 1 ratio (stratified by week16 EASI 50, responder status [yes or no], geographic region [US, Puerto Rico, Canada; mainland China; Japan; other], and age group [adolescent or adult]) to receive daily oral doses of upadacitinib 30 mg plus TCS or upadacitinib 15 mg plus TCS in the BE period. Patients originally randomized to upadacitinib were to continue upadacitinib in the extension period at the same dose. Rescue treatment for AD was permitted from the week 4 visit through week 24, if medically necessary and with an < EASI 50 response at 2 consecutive visits. After week 24, rescue therapy was permitted if medically necessary and with an < EASI 50 response at any visit. The overall design of the AD Up study is presented in Figure 3.
The AD Up study was conducted from August 2018 to December 2020 at 170 sites in 22 countries (including the US, Canada, China, and countries in Europe).
Figure 3: Study Design Schematic of the AD Up Study
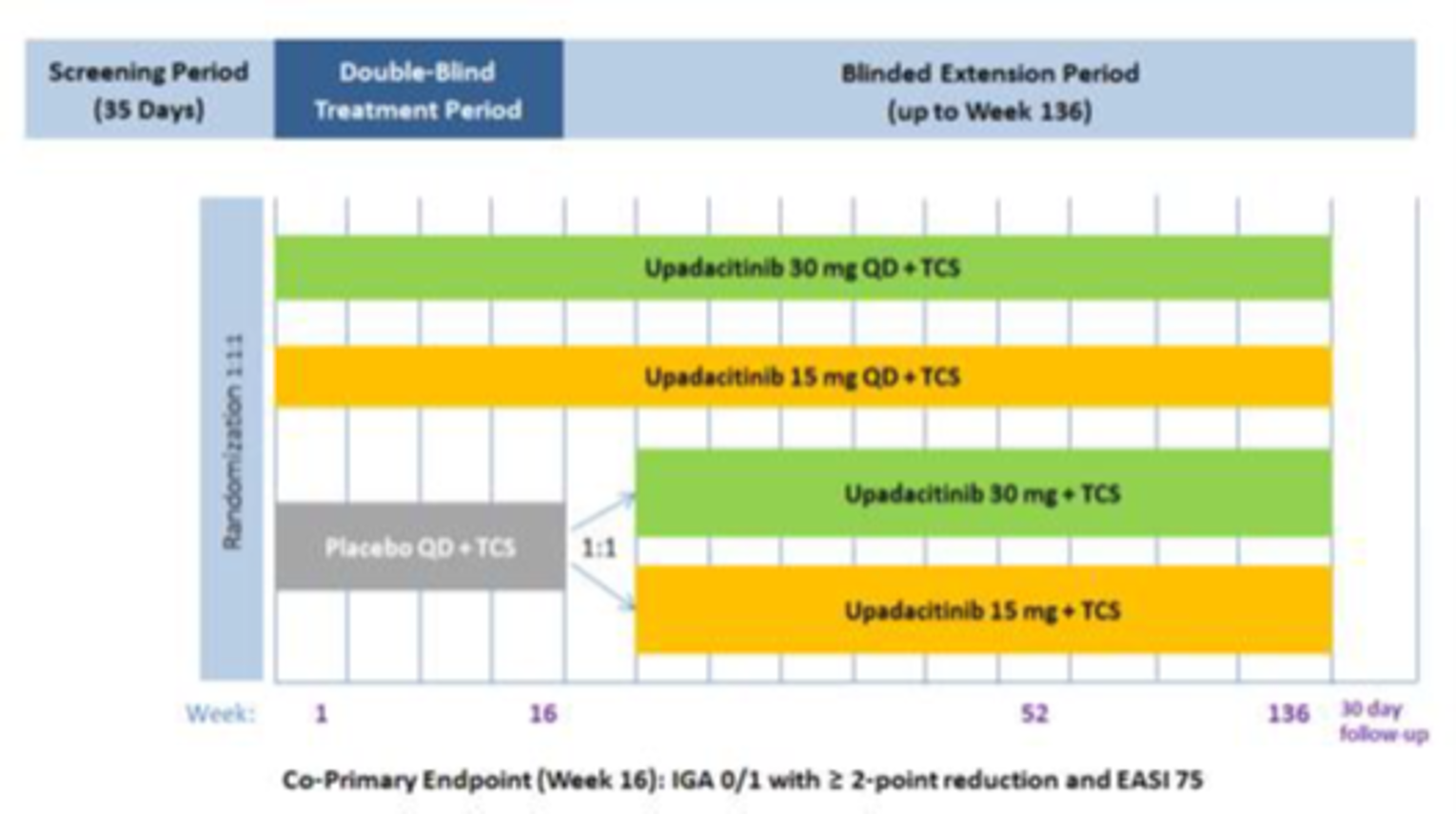
EASI 75 = at least 75% improvement in EASI total score from baseline; IGA = Investigator Global Assessment; QD = every day; TCS = topical corticosteroid.
Source: Clinical Study Report: AD Up.13
Head-to-Head Studies
The Heads Up study was a phase III, double-blind, double-dummy, active-controlled, randomized trial aimed at evaluating the efficacy and safety of upadacitinib versus dupilumab for the treatment of adult patients (18 years to 75 years of age) with moderate to severe chronic AD (i.e., with onset of symptoms at least 3 years before baseline screening) who are candidates for systemic therapy.
The Heads Up study compared the safety and efficacy of upadacitinib 30 mg given once daily as an extended-release tablet until week 24 versus dupilumab 600 mg as an SC injection administered at baseline followed by dupilumab 300 mg as an SC injection every other week from week 2 to week 22. At the end of the double-blind period, patients either entered an open-label upadacitinib extension in which patients were to be treated for an additional 52 weeks or had an end-of-treatment follow-up visit (12 weeks from the last injection). This study consisted of a 5-week screening period, a 24-week double-blind treatment phase, and an end-of-treatment follow-up visit at 12 weeks after the last injection for patients who did not enrol in the open-label study. Eligible patients were randomized in a 1 to 1 ratio and stratified by baseline disease severity (moderate [vIGA-AD = 3] versus severe [vIGA-AD = 4]) and age (< 40 years, ≥ 40 years to < 65 years, ≥ 65 years). Prior use of dupilumab or JAK inhibitors was not allowed. Patients required a documented history of inadequate response to TCSs or TCIs, a documented history of systemic treatment for AD within 6 months, or had to have been medically advised not to use topical AD treatment. Rescue treatment for AD was permitted from the week 4 visit through week 16 if medically necessary.
The overall design of the Heads Up study is presented in Figure 4. The Heads Up study was conducted from February 2019 to December 2020 at 129 sites in 22 countries (including the US, Canada, and countries in Asia and Europe).
Figure 4: Study Design Schematic of the Heads Up Study
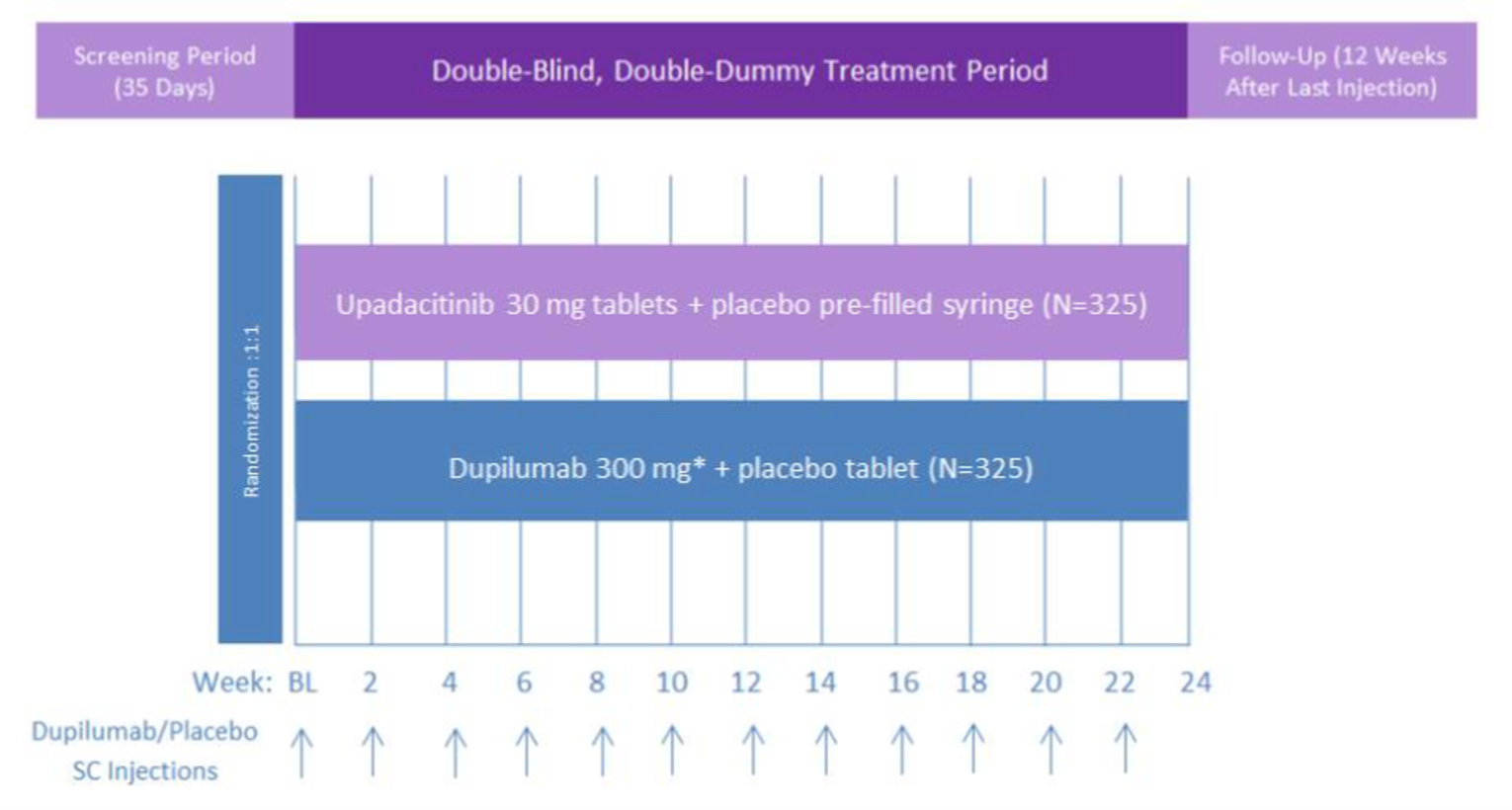
BL = baseline; SC = subcutaneous.
Source: Clinical Study Report: Heads Up.14
Populations
Inclusion and Exclusion Criteria
Monotherapy Studies
Measure Up 1 and Measure Up 2 had similar inclusion criteria among them except with regards to the time since AD diagnosis: the Measure Up 2 study included patients with chronic AD (defined as AD with onset of symptoms at least 3 years before baseline, with patients meeting the Hanifin and Rajka criteria),26 while the Measure Up 1 study included patients with more recent onset of symptoms. Both studies included patients with a documented history of inadequate response to treatment with topical AD treatments or use of systemic treatment for AD who met the following disease activity criteria: EASI score greater than or equal to 16; vIGA-AD score greater than or equal to 3; greater than or equal to 10% BSA of AD involvement at the screening and baseline visits; and a baseline weekly average of daily WP-NRS greater than or equal to 4. Both studies required adolescent patients (≥ 12 years and < 18 years of age) to have a body weight greater than or equal to 40 kg at the baseline visit.
Both studies excluded patients with prior exposure to JAK inhibitors; patients using systemic treatments for AD (e.g., corticosteroids, methotrexate, cyclosporine A, azathioprine, mycophenolate mofetil), phototherapy, laser therapy, tanning, or Chinese medicine within 4 weeks from screening; and those who had used targeted biologic treatments within 12 weeks.
Combination Studies
The AD Up study main difference was the addition of a TCS to each arm of the study, whether in the double-blind or BE period. The AD Up study included patients with characteristics that were similar to those of patients in the Measure Up 2 study: i.e., those with chronic AD (defined as AD with onset of symptoms at least 3 years before baseline) who met the Hanifin and Rajka criteria,26 had a documented history of systemic treatment or inadequate response to topical AD treatments and met the following disease activity criteria: EASI score greater than or equal to 16; vIGA-AD score greater than or equal to 3; greater than or equal to 10% BSA of AD involvement at the screening and baseline visits; and a baseline weekly average of daily WP-NRS greater than or equal to 4. The study required adolescent patients (≥ 12 years and < 18 years of age) to have a body weight greater than or equal to 40 kg at the baseline visit. This study excluded patients with prior exposure to a JAK inhibitor or dupilumab and those with greater than or equal to 30% of AD surface involvement at baseline that could not be treated with a medium- or high-potency TCS. Investigators also excluded patients using any of the following AD treatments within the specified time frame before the baseline visit: systemic therapy for AD, including but not limited to corticosteroids, methotrexate, cyclosporine A, azathioprine, and phosphodiesterase 4 (PDE 4) inhibitors; mycophenolate mofetil within 4 weeks; and oral or parenteral Chinese medicine within 4 weeks.
Head-to-Head Studies
As with the Measure Up 2 and AD Up studies, the Heads Up study (comparing upadacitinib versus dupilumab) inclusion criteria included patients greater than or equal to 18 years old and less than or equal to 75 years old at the screening visit who were in general good health (other than AD) as determined by the principal investigator based on results of medical history, laboratory profile, and physical examination. Patients with chronic AD had an onset of symptoms at least 3 years before baseline and met Hanifin and Rajka criteria. At screening and baseline, they met the following disease activity criteria: EASI score greater than or equal to 16; vIGA-AD score greater than or equal to 3; greater than or equal to 10% BSA of AD involvement; and a baseline weekly average of daily WP-NRS ≥ 4.
Prior use of dupilumab or JAK inhibitors was not allowed. Patients were required to have a documented history of inadequate response to TCSs or TCIs, a documented history of systemic treatment for AD within 6 months, or past medical advice against the use of topical AD treatment.
Baseline Characteristics
The main demographic information, disease characteristics, medical history, and prior medications for the Measure Up 1, Measure Up 2, AD Up, and Heads Up studies included in this review are presented in Table 11, Table 12, Table 13, and Table 14, respectively.
No major differences were observed in any of the demographic or baseline characteristics between the intervention and placebo arms in or among any of the included studies. All 4 studies involved young adult patients (with median ages from 32 years to 37 years) with similar median disease durations since diagnosis, varying from 17 years in the Measure Up studies to 23 years in the Heads Up study. The baseline median EASI scores (ranging from 24 to 27) and the proportions of patients with vIGA-AD scores indicating severe AD (ranging from 44% to 55% of included patients) were also similar within and among the included pivotal studies.
Approximately half of the included patients in the 4 studies had tried a previous systemic therapy (range = 42.7% to 58%) at baseline. Nearly all patients (99% to 100%) had any type of AD therapy before the study, with TCS as the most frequent previous therapy; the use of TCIs ranged from 32% to 47% of patients. While the number of patients who had previously tried immunomodulating drugs ranged from 50% to 57%, previous use of biologics was rare (from 0.6% to 4%).
Table 11: Summary of Baseline Characteristics — Measure Up 1
Characteristic | Placebo (N = 281) | UPA 15 mg (N = 281) | UPA 30 mg (N = 285) |
|---|---|---|---|
Demographics | |||
Sex – n (%) | |||
Male | 144 (51.2) | 157 (55.9) | 155 (54.4) |
Female | 137 (48.8) | 124 (44.1) | 130 (45.6) |
Ethnicity – n (%) | |||
Hispanic or Latino | 33 (11.7) | 35 (12.5) | 41 (14.4) |
Not Hispanic or Latino | 248 (88.3) | 246 (87.5) | 244 (85.6) |
Race – n (%) | |||
White | 182 (64.8) | 182 (64.8) | 191 (67.0) |
Black or African American | 21 (7.5) | 26 (9.3) | 8 (2.8) |
Asian | 69 (24.6) | 63 (22.4) | 71 (24.9) |
American Indian or Alaska Native | 3 (1.1) | 0 | 0 |
Native Hawaiian or other Pacific Islander | 1 (0.4) | 1 (0.4) | 1 (0.4) |
Other | 0 | 0 | 0 |
Multiple | 5 (1.8) | 9 (3.2) | 14 (4.9) |
Age group (years) – n (%) | |||
< 18 | 40 (14.2) | 42 (14.9) | 42 (14.7) |
18 to < 40 | 145 (51.6) | 143 (50.9) | 154 (54.0) |
40 to < 65 | 85 (30.2) | 83 (29.5) | 74 (26.0) |
≥ 65 | 11 (3.9) | 13 (4.6) | 15 (5.3) |
Age (years) | |||
Mean (SD) | 34.4 (15.50) | 34.1 (15.72) | 33.6 (15.84) |
Median | ||||| | ||||| | ||||| |
Range | 12 to 75 | 12 to 74 | 12 to 75 |
Weight (kg) | |||
Mean (SD) | 75.53 (19.935) | 74.22 (19.361) | 73.09 (18.325) |
Median | ||||| | ||||| | ||||| |
Range | 38.3 to 170.1 | 40.0 to 160.6 | 36.3 to 151.0 |
BMI (kg/m2) | |||
N (%) | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Mean (SD) | 26.72 (6.291) | 25.78 (6.147) | 25.61 (5.867) |
Median | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Range | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
BMI (kg/m2) – n (%) | |||
< 25 | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
25 to < 30 | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
≥ 30 | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Tobacco – n (%) | |||
Current | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Former | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Never | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Unknown | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Disease characteristics | |||
Baseline vIGA-AD – n (%) | |||
3 (moderate) | 156 (55.5) | 154 (54.8) | 154 (54.0) |
4 (severe) | 125 (44.5) | 127 (45.2) | 131 (46.0) |
Baseline EASI – n (%) | |||
< Median (25.8) | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
≥ Median (25.8) | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Previous systemic therapy – n (%) | |||
With | 144 (51.2) | 120 (42.7) | 129 (45.3) |
Without | 137 (48.8) | 161 (57.3) | 156 (54.7) |
EASI | |||
Mean (SD) | 28.84 (12.616) | 30.57 (12.759) | 28.98 (11.110) |
Median | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Range | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
BSA in percentage | |||
Mean (SD) | 45.67 (21.600) | 48.52 (22.227) | 47.00 (21.973) |
Median | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Range | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Overall SCORAD score | |||
N (%) | 277 (98.57) | 279 (99.28) | 278 (97.54) |
Mean (SD) | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Median | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Range | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
SCORAD itch | |||
Mean (SD) | 7.8 (1.66) | 7.6 (1.62) | 7.8 (1.52) |
Median | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Range | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
SCORAD sleep | |||
N (%) | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Mean (SD) | 6.6 (2.73) | 6.3 (2.63) | 6.4 (2.72) |
Median | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Range | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
DLQI | |||
N (%) | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Mean (SD) | 17.0 (6.85) | 16.2 (7.00) | 16.4 (6.97) |
Median | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Range | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
CDLQI | |||
N (%) | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Mean (SD) | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Median | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Range | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
HADS total score | |||
N (%) | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Mean (SD) | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Median | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Range | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
HADS-A total score | |||
N (%) | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Mean (SD) | 7.2 (4.35) | 7.5 (4.03) | 7.4 (4.36) |
Median | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Range | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
HADS-D total score | |||
N (%) | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Mean (SD) | 5.0 (4.00) | 5.2 (3.87) | 5.2 (4.22) |
Median | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Range | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
WP-NRS (daily) | |||
N (%) | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Mean (SD) | 7.5 (1.84) | 7.4 (1.84) | 7.5 (1.71) |
Median | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Range | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
ADerm-SS skin pain | |||
N (%) | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Mean (SD) | 6.480 (2.3770) | 6.236 (2.3165) | 6.492 (2.1163) |
Median | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Range | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
ADerm-SS TSS-7 | |||
N (%) | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Mean (SD) | 46.1 (14.48) | 45.7 (13.98) | 46.3 (13.37) |
Median | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Range | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
POEM | |||
N (%) | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Mean (SD) | 21.5 (5.35) | 21.2 (4.76) | 21.4 (5.14) |
Median | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Range | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Disease duration since diagnosis (years) | |||
Mean (SD) | 21.268 (15.2761) | 20.539 (15.8692) | 20.412 (14.2859) |
Median | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Range | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
AD medication history, N (%) | |||
||||||||||||||||||||||||||||||||||||||||||||| | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
||||||||||||||||||||||||||||||||||||||||||||| | ||||||||||||||| | ||||||||||||||| | ||||||||||||||| |
Medical history | |||
Acne | 22 (7.8) | 24 (8.5) | 35 (12.3) |
Asthma | 115 (40.9) | 108 (38.4) | 115 (40.4) |
Chronic sinusitis | 0 | 1 (0.4) | 0 |
Allergic conjunctivitis | 14 (5.0) | 18 (6.4) | 18 (6.3) |
Eosinophilic esophagitis | 3 (1.1) | 1 (0.4) | 0 |
Food allergy | 90 (32.0) | 98 (34.9) | 87 (30.5) |
Nasal polyps | 8 (2.8) | 3 (1.1) | 5 (1.8) |
Allergic rhinitis | 94 (33.5) | 92 (32.7) | 102 (35.8) |
AD = atopic dermatitis; ADerm-SS = Atopic Dermatitis Symptom Scale; BMI = body mass index; BSA = body surface area; CDLQI = Children’s Dermatology Life Quality Index; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; HADS = Hospital Anxiety and Depression Scale; HADS-A = Hospital Anxiety and Depression Scale – Anxiety; HADS-D = Hospital Anxiety and Depression Scale – Depression; max = maximum; min = minimum; POEM = Patient-Oriented Eczema Measure; SCORAD = Scoring Atopic Dermatitis; SD = standard deviation; TSS-7 = 7-Item Total Symptom Score; UPA = upadacitinib; vIGA-AD = validated Investigator Global Assessment for Atopic Dermatitis; WP-NRS = Worst Pruritus Numerical Rating Scale.
Note: Redacted rows have been deleted.
Source: Clinical Study Report: Measure Up 1.12
Table 12: Summary of Baseline Characteristics — Measure Up 2
Characteristic | Placebo (N = 278) | UPA 15 mg (N = 276) | UPA 30 mg (N = 282) |
|---|---|---|---|
Demographics | |||
Sex – n (%) | |||
Male | 154 (55.4) | 155 (56.2) | 162 (57.4) |
Female | 124 (44.6) | 121 (43.8) | 120 (42.6) |
Ethnicity – n (%) | |||
Hispanic or Latino | 31 (11.2) | 24 (8.7) | 23 (8.2) |
Not Hispanic or Latino | 247 (88.8) | 252 (91.3) | 259 (91.8) |
Race – n (%) | |||
White | 195 (70.1) | 184 (66.7) | 198 (70.2) |
Black or African American | 16 (5.8) | 17 (6.2) | 18 (6.4) |
Asian | 56 (20.1) | 65 (23.6) | 62 (22.0) |
American Indian or Alaska Native | 5 (1.8) | 5 (1.8) | 2 (0.7) |
Native Hawaiian or other Pacific Islander | 1 (0.4) | 2 (0.7) | 0 2 (0.4) |
Other | 0 | 0 | 0 |
Multiple | 5 (1.8) | 3 (1.1) | 2 (0.7) |
Age group (years) – n (%) | |||
< 18 | 36 (12.9) | 33 (12.0) | 35 (12.4) |
≥ 18 | 242 (87.1) | 243 (88.0) | 247 (87.6) |
Age (years) | |||
N (%) | 278 (100) | 276 (100) | 282 (100) |
Mean (SD) | |||||||||| | |||||||||| | |||||||||| |
Median | 29.0 | 28.0 | 30.0 |
Range | 13 to 71 | 12 to 74 | 12 to 75 |
Weight (kg) | |||
N (%) | 277 (99.64) | 276 (100) | 282 (100) |
Mean (SD) | 76.66 (19.560) | 73.98 (18.521) | 75.31 (18.355) |
Median | |||||||||| | |||||||||| | |||||||||| |
Range | 41.0 to 175.0 | 37.0 to 136.1 | 37.4 to 142.0 |
BMI (kg/m2) | |||
N (%) | |||||||||| | |||||||||| | |||||||||| |
Mean (SD) | 26.27 (5.661) | 25.78 (5.595) | 25.93 (5.801) |
Median | |||||||||| | |||||||||| | |||||||||| |
Range | |||||||||| | |||||||||| | |||||||||| |
BMI (kg/m2) – n (%) | |||
< 25 | |||||||||| | |||||||||| | |||||||||| |
25 to < 30 | |||||||||| | |||||||||| | |||||||||| |
≥ 30 | |||||||||| | |||||||||| | |||||||||| |
Tobacco – n (%) | |||
Current | |||||||||| | |||||||||| | |||||||||| |
Former | |||||||||| | |||||||||| | |||||||||| |
Never | |||||||||| | |||||||||| | |||||||||| |
Unknown | |||||||||| | |||||||||| | |||||||||| |
Disease characteristics | |||
Baseline vIGA-AD – n (%) | |||
3 (moderate) | 125 (45.0) | 126 (45.7) | 126 (44.7) |
4 (severe) | 153 (55.0) | 150 (54.3) | 156 (55.3) |
Baseline EASI – n (%) | |||
< Median (25.8) | |||||||||| | |||||||||| | |||||||||| |
≥ Median (25.8) | |||||||||| | |||||||||| | |||||||||| |
Previous systemic therapy – n (%) | |||
With | 156 (56.1) | 155 (56.2) | 145 (51.4) |
Without | 122 (43.9) | 121 (43.8) | 137 (48.6) |
EASI | |||
N (%) | |||||||||| | |||||||||| | |||||||||| |
Mean (SD) | 29.08 (12.131) | 28.60 (11.692) | 29.65 (12.194) |
Median | |||||||||| | |||||||||| | |||||||||| |
Range | |||||||||| | |||||||||| | |||||||||| |
BSA in percentage | |||
N (%) | |||||||||| | |||||||||| | |||||||||| |
Mean (SD) | 47.61 (22.694) | 45.12 (22.352) | 47.02 (23.182) |
Median | |||||||||| | |||||||||| | |||||||||| |
Range | |||||||||| | |||||||||| | |||||||||| |
Overall SCORAD score | |||
N (%) | |||||||||| | |||||||||| | |||||||||| |
Mean (SD) | 67.910 (12.0956) | 66.558 (12.4943) | 66.744 (12.9667) |
Median | |||||||||| | |||||||||| | |||||||||| |
Range | |||||||||| | |||||||||| | |||||||||| |
SCORAD itch | |||
N (%) | |||||||||| | |||||||||| | |||||||||| |
Mean (SD) | 7.9 (1.65) | 7.5 (1.69) | 7.6 (1.74) |
Median | |||||||||| | |||||||||| | |||||||||| |
Range | |||||||||| | |||||||||| | |||||||||| |
SCORAD sleep | |||
N (%) | |||||||||| | |||||||||| | |||||||||| |
Mean (SD) | 6.8 (2.65) | 6.2 (2.74) | 6.4 (2.79) |
Median | |||||||||| | |||||||||| | |||||||||| |
Range | |||||||||| | |||||||||| | |||||||||| |
DLQI | |||
N (%) | |||||||||| | |||||||||| | |||||||||| |
Mean (SD) | 17.1 (7.17) | 16.9 (7.04) | 16.7 (6.93) |
Median | |||||||||| | |||||||||| | |||||||||| |
Range | |||||||||| | |||||||||| | |||||||||| |
CDLQI | |||
N (%) | |||||||||| | |||||||||| | |||||||||| |
Mean (SD) | |||||||||| | |||||||||| | |||||||||| |
Median | |||||||||| | |||||||||| | |||||||||| |
Range | |||||||||| | |||||||||| | |||||||||| |
HADS total score | |||
N (%) | |||||||||| | |||||||||| | |||||||||| |
Mean (SD) | |||||||||| | |||||||||| | |||||||||| |
Median | |||||||||| | |||||||||| | |||||||||| |
Range | |||||||||| | |||||||||| | |||||||||| |
HADS-A total score | |||
N (%) | |||||||||| | |||||||||| | |||||||||| |
Mean (SD) | 7.5 (4.29) | 7.2 (4.17) | 7.6 (4.28) |
Median | |||||||||| | |||||||||| | |||||||||| |
Range | |||||||||| | |||||||||| | |||||||||| |
HADS-D total score | |||
N (%) | |||||||||| | |||||||||| | |||||||||| |
Mean (SD) | 5.8 (4.15) | 5.3 (4.18) | 5.9 (4.09) |
Median | |||||||||| | |||||||||| | |||||||||| |
Range | |||||||||| | |||||||||| | |||||||||| |
WP-NRS (daily) | |||
N (%) | |||||||||| | |||||||||| | |||||||||| |
Mean (SD) | 7.5 (1.85) | 7.2 (1.79) | 7.4 (1.69) |
Median | |||||||||| | |||||||||| | |||||||||| |
Range | |||||||||| | |||||||||| | |||||||||| |
ADerm-SS skin pain | |||
N (%) | |||||||||| | |||||||||| | |||||||||| |
Mean (SD) | 6.540 (2.1633) | 6.360 (2.1065) | 6.375 (2.2616) |
Median | |||||||||| | |||||||||| | |||||||||| |
Range | |||||||||| | |||||||||| | |||||||||| |
ADerm-SS TSS-7 | |||
N (%) | |||||||||| | |||||||||| | |||||||||| |
Mean (SD) | 47.2 (13.58) | 46.8 (13.18) | 46.3 (13.80) |
Median | |||||||||| | |||||||||| | |||||||||| |
Range | |||||||||| | |||||||||| | |||||||||| |
POEM | |||
N (%) | |||||||||| | |||||||||| | |||||||||| |
Mean (SD) | 21.9 (5.24) | 21.2 (5.13) | 21.8 (4.76) |
Median | |||||||||| | |||||||||| | |||||||||| |
Range | |||||||||| | |||||||||| | |||||||||| |
Disease duration since diagnosis (years) | |||
N (%) | |||||||||| | |||||||||| | |||||||||| |
Mean (SD) | 21.051 (13.5791) | 18.829 (13.3017) | 20.779 (14.2842) |
Median | |||||||||| | |||||||||| | |||||||||| |
Range | |||||||||| | |||||||||| | |||||||||| |
AD medication history, N (%) | |||
|||||||||||||||||||||||||||||||||||||||||||||||||| | |||||||||| | |||||||||| | |||||||||| |
|||||||||||||||||||||||||||||||||||||||||||||||||| | |||||||||| | |||||||||| | |||||||||| |
Medical history N (%) | |||
Acne | 25 (9.0) | 32 (11.6) | 32 (11.3) |
Asthma | 115 (41.4) | 112 (40.6) | 106 (37.6) |
Chronic sinusitis | 0 | 1 (0.4) | 2 (0.7) |
Allergic conjunctivitis | 7 (2.5) | 10 (3.6) | 13 (4.6) |
Eosinophilic esophagitis | 2 (0.7) | 0 | 1 (0.4) |
Food allergy | 70 (25.2) | 67 (24.3) | 96 (34.0) |
Nasal polyps | 8 (2.9) | 3 (1.1) | 6 (2.1) |
Allergic rhinitis | 101 (36.3) | 88 (31.9) | 95 (33.7) |
AD = atopic dermatitis; ADerm-SS = Atopic Dermatitis Symptom Scale; BMI = body mass index; BSA = body surface area; CDLQI = Children’s Dermatology Life Quality Index; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; HADS = Hospital Anxiety and Depression Scale; HADS-A = Hospital Anxiety and Depression Scale – Anxiety; HADS-D = Hospital Anxiety and Depression Scale – Depression; max = maximum; min = minimum; POEM = Patient-Oriented Eczema Measure; SCORAD = Scoring Atopic Dermatitis; SD = standard deviation; TSS-7 = 7-Item Total Symptom Score; UPA = upadacitinib; vIGA-AD = validated Investigator Global Assessment for Atopic Dermatitis; WP-NRS = Worst Pruritus Numerical Rating Scale.
Note: Redacted rows have been deleted.
Source: Clinical Study Report: Measure Up 2.11
Table 13: Summary of Baseline Characteristics — AD Up
Characteristic | Placebo + TCS (N = 304) | UPA 15 mg + TCS (N = 300) | UPA 30 mg + TCS (N = 297) |
|---|---|---|---|
Demographics | |||
Sex – n (%) | |||
Male | 126 (41.4) | 121 (40.3) | 107 (36.0) |
Female | 178 (58.6) | 179 (59.7) | 190 (64.0) |
Ethnicity – n (%) | |||
Hispanic or Latino | 26 (8.6) | 32 (10.7) | 20 (6.7) |
Not Hispanic or Latino | 278 (91.4) | 268 (89.3) | 277 (93.3) |
Race – n (%) | |||
White | 225 (74.0) | 204 (68.0) | 218 (73.4) |
Black or African American | 18 (5.9) | 19 (6.3) | 13 (4.4) |
Asian | 60 (19.7) | 64 (21.3) | 61 (20.5) |
American Indian or Alaska Native | 1 (0.3) | 2 (0.7) | 3 (1.0) |
Native Hawaiian or other Pacific Islander | 0 | 3 (1.0) | 1 (0.3) |
Other | 0 | 0 | 0 |
Multiple | 0 | 8 (2.7) | 1 (0.3) |
Age group (years) – n (%) | |||
< 18 | 40 (13.2) | 39 (13.0) | 37 (12.5) |
≥ 18 | 264 (86.8) | 261 (87.0) | 260 (87.5) |
Age (years) | |||
N (%) | |||||||||| | |||||||||| | |||||||||| |
Mean (SD) | 34.3 (15.12) | 32.5 (14.02) | 35.5 (15.79) |
Median | |||||||||| | |||||||||| | |||||||||| |
Range | |||||||||| | |||||||||| | |||||||||| |
Weight (kg) | |||
N (%) | |||||||||| | |||||||||| | |||||||||| |
Mean (SD) | |||||||||| | |||||||||| | |||||||||| |
Median | |||||||||| | |||||||||| | |||||||||| |
Range | |||||||||| | |||||||||| | |||||||||| |
BMI (kg/m2) | |||
N (%) | |||||||||| | |||||||||| | |||||||||| |
Mean (SD) | 25.92 (5.667) | 25.81 (6.159) | 25.74 (5.420) |
Median | |||||||||| | |||||||||| | |||||||||| |
Range | |||||||||| | |||||||||| | |||||||||| |
BMI (kg/m2) – n (%) | |||
< 25 | |||||||||| | |||||||||| | |||||||||| |
25 - < 30 | |||||||||| | |||||||||| | |||||||||| |
≥ 30 | |||||||||| | |||||||||| | |||||||||| |
Tobacco – n (%) | |||
Current | |||||||||| | |||||||||| | |||||||||| |
Former | |||||||||| | |||||||||| | |||||||||| |
Never | |||||||||| | |||||||||| | |||||||||| |
Unknown | |||||||||| | |||||||||| | |||||||||| |
Disease characteristics | |||
Baseline vIGA-AD – n (%) | |||
3 (moderate) | 141 (46.4) | 143 (47.7) | 140 (47.1) |
4 (severe) | 163 (53.6) | 157 (52.3) | 157 (52.9) |
Baseline EASI – n (%) | |||
< Median (25.8) | |||||||||| | |||||||||| | |||||||||| |
≥ Median (25.8) | |||||||||| | |||||||||| | |||||||||| |
Previous systemic therapy – n (%) | |||
With | 157 (51.6) | 171 (57.0) | 172 (57.9) |
Without | 147 (48.4) | 129 (43.0) | 125 (42.1) |
EASI | |||
N (%) | |||||||||| | |||||||||| | |||||||||| |
Mean (SD) | 30.26 (12.974) | 29.16 (11.829) | 29.72 (11.781) |
Median | |||||||||| | |||||||||| | |||||||||| |
Range | |||||||||| | |||||||||| | |||||||||| |
BSA in percentage | |||
N (%) | |||||||||| | |||||||||| | |||||||||| |
Mean (SD) | 48.57 (23.106) | 46.68 (21.647) | 48.53 (23.090) |
Median | |||||||||| | |||||||||| | |||||||||| |
Range | |||||||||| | |||||||||| | |||||||||| |
Overall SCORAD score | |||
N (%) | |||||||||| | |||||||||| | |||||||||| |
Mean (SD) | |||||||||| | |||||||||| | |||||||||| |
Median | |||||||||| | |||||||||| | |||||||||| |
Range | |||||||||| | |||||||||| | |||||||||| |
SCORAD itch | |||
N (%) | |||||||||| | |||||||||| | |||||||||| |
Mean (SD) | |||||||||| | |||||||||| | |||||||||| |
Median | |||||||||| | |||||||||| | |||||||||| |
Range | |||||||||| | |||||||||| | |||||||||| |
SCORAD sleep | |||
N (%) | |||||||||| | |||||||||| | |||||||||| |
Mean (SD) | |||||||||| | |||||||||| | |||||||||| |
Median | |||||||||| | |||||||||| | |||||||||| |
Range | |||||||||| | |||||||||| | |||||||||| |
DLQI | |||
N (%) | |||||||||| | |||||||||| | |||||||||| |
Mean (SD) | 16.3 (6.99) | 16.4 (7.20) | 17.1 (7.00) |
Median | |||||||||| | |||||||||| | |||||||||| |
Range | |||||||||| | |||||||||| | |||||||||| |
CDLQI | |||
N (%) | |||||||||| | |||||||||| | |||||||||| |
Mean (SD) | |||||||||| | |||||||||| | |||||||||| |
Median | |||||||||| | |||||||||| | |||||||||| |
Range | |||||||||| | |||||||||| | |||||||||| |
HADS total score | |||
N (%) | |||||||||| | |||||||||| | |||||||||| |
Mean (SD) | |||||||||| | |||||||||| | |||||||||| |
Median | |||||||||| | |||||||||| | |||||||||| |
Range | |||||||||| | |||||||||| | |||||||||| |
HADS-A total score | |||
N (%) | |||||||||| | |||||||||| | |||||||||| |
Mean (SD) | |||||||||| | |||||||||| | |||||||||| |
Median | |||||||||| | |||||||||| | |||||||||| |
Range | |||||||||| | |||||||||| | |||||||||| |
HADS-D total score | |||
N (%) | |||||||||| | |||||||||| | |||||||||| |
Mean (SD) | |||||||||| | |||||||||| | |||||||||| |
Median | |||||||||| | |||||||||| | |||||||||| |
Range | |||||||||| | |||||||||| | |||||||||| |
WP-NRS (weekly average) | |||
N (%) | |||||||||| | |||||||||| | |||||||||| |
Mean (SD) | 7.135 (1.6253) | 7.062 (1.7580) | 7.360 (1.6495) |
Median | |||||||||| | |||||||||| | |||||||||| |
Range | |||||||||| | |||||||||| | |||||||||| |
ADerm-SS skin pain | |||
N (%) | |||||||||| | |||||||||| | |||||||||| |
Mean (SD) | |||||||||| | |||||||||| | |||||||||| |
Median | |||||||||| | |||||||||| | |||||||||| |
Range | |||||||||| | |||||||||| | |||||||||| |
ADerm-SS TSS-7 | |||
N (%) | |||||||||| | |||||||||| | |||||||||| |
Mean (SD) | |||||||||| | |||||||||| | |||||||||| |
Median | |||||||||| | |||||||||| | |||||||||| |
Range | |||||||||| | |||||||||| | |||||||||| |
POEM | |||
N (%) | |||||||||| | |||||||||| | |||||||||| |
Mean (SD) | 21.1 (5.14) | 21.0 (4.98) | 21.5 (5.27) |
Median | |||||||||| | |||||||||| | |||||||||| |
Range | |||||||||| | |||||||||| | |||||||||| |
Disease duration since diagnosis (years) | |||
N (%) | |||||||||| | |||||||||| | |||||||||| |
Mean (SD) | 24.300 (15.2327) | 22.900 (13.8552) | 23.098 (16.1170) |
Median | |||||||||| | |||||||||| | |||||||||| |
Range | |||||||||| | |||||||||| | |||||||||| |
AD medication history, N (%) | |||
|||||||||||||||||||||||||||||||||||||||| | |||||||||| | |||||||||| | |||||||||| |
|||||||||||||||||||||||||||||||||||||||| | |||||||||| | |||||||||| | |||||||||| |
Medical history N (%) | |||
Acne | 21 (6.9) | 26 (8.7) | 21 (7.1) |
Asthma | 138 (45.4) | 130 (43.3) | 140 (47.1) |
Chronic sinusitis | 0 | 1 (0.3) | 2 (0.7) |
Allergic conjunctivitis | 21 (6.9) | 22 (7.3) | 17 (5.7) |
Eosinophilic esophagitis | 0 2 (0.7) | 3 (1.0) | 5 (0.8) |
Food allergy | 89 (29.3) | 112 (37.3) | 101 (34.0) |
Nasal polyps | 3 (1.0) | 5 (1.7) | 7 (2.4) |
Allergic rhinitis | 108 (35.5) | 96 (32.0) | 104 (35.0) |
AD = atopic dermatitis; ADerm-SS = Atopic Dermatitis Symptom Scale; BMI = body mass index; BSA = body surface area; CDLQI = Children’s Dermatology Life Quality Index; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; HADS = Hospital Anxiety and Depression Scale; HADS-A = Hospital Anxiety and Depression Scale – Anxiety; HADS-D = Hospital Anxiety and Depression Scale – Depression; max = maximum; min = minimum; POEM = Patient-Oriented Eczema Measure; SCORAD = Scoring Atopic Dermatitis; SD = standard deviation; TCS = topical corticosteroid; TSS-7 = 7-Item Total Symptom Score; UPA = upadacitinib; vIGA-AD = validated Investigator Global Assessment for Atopic Dermatitis; WP-NRS = Worst Pruritus Numerical Rating Scale.
Note: Redacted rows have been deleted.
Source: Clinical Study Report: AD Up.13
Table 14: Summary of Baseline Characteristics — Heads Up
Characteristic | DUP 300 mg (N = 344) | UPA 30 mg (N = 348) |
|---|---|---|
Demographics | ||
Sex – n (%) | ||
Male | 194 (56.4) | 183 (52.6) |
Female | 150 (43.6) | 165 (47.4) |
Ethnicity – n (%) | ||
Hispanic or Latino | ||||||||||||||||| | ||||||||||||||||| |
Not Hispanic or Latino | ||||||||||||||||| | ||||||||||||||||| |
Race – n (%) | ||
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
Age (years) | ||
N (%) | ||||||||||||||||| | ||||||||||||||||| |
Mean (SD) | 36.9 (14.09) | 36.6 (14.61) |
Median | ||||||||||||||||| | ||||||||||||||||| |
Range | ||||||||||||||||| | ||||||||||||||||| |
Weight (kg) | ||
N (%) | ||||||||||||||||| | ||||||||||||||||| |
Mean (SD) | 75.55 (18.390) | 78.77 (22.283) |
Median | ||||||||||||||||| | ||||||||||||||||| |
Range | ||||||||||||||||| | ||||||||||||||||| |
BMI (kg/m2) | ||
N (%) | ||||||||||||||||| | ||||||||||||||||| |
Mean (SD) | 25.99 (5.718) | 26.99 (6.533) |
Median | ||||||||||||||||| | ||||||||||||||||| |
Range | 15.7 to 54.1 | 15.2 to 60.7 |
||||||||||||||||| | ||
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
Disease characteristics | ||
Baseline vIGA-AD – n (%) | ||
< 4 (clear, almost clear, mild or moderate) | 171 (49.7) | 174 (50.0) |
4 (severe) | 173 (50.3) | 174 (50.0) |
Baseline EASI – n (%) | ||
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
Previous systemic therapy – n (%) | ||
With | ||||||||||||||||| | ||||||||||||||||| |
Without | ||||||||||||||||| | ||||||||||||||||| |
EASI | ||
N (%) | ||||||||||||||||| | ||||||||||||||||| |
Mean (SD) | 28.81 (11.512) | 30.75 (12.538) |
Median | ||||||||||||||||| | ||||||||||||||||| |
Range | ||||||||||||||||| | ||||||||||||||||| |
BSA in percentage | ||
N (%) | ||||||||||||||||| | ||||||||||||||||| |
Mean (SD) | 44.41 (22.833) | 48.20 (23.964) |
Median | ||||||||||||||||| | ||||||||||||||||| |
Range | ||||||||||||||||| | ||||||||||||||||| |
WP-NRS (daily) | ||||||||||||||||| | ||||||||||||||||| |
N (%) | ||||||||||||||||| | ||||||||||||||||| |
Mean (SD) | 7.5 (1.82) | 7.2 (1.85) |
Median | ||||||||||||||||| | ||||||||||||||||| |
Range | ||||||||||||||||| | ||||||||||||||||| |
||||||||||||||||| | ||
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
Disease duration since diagnosis (years) | ||
N (%) | ||||||||||||||||| | ||||||||||||||||| |
Mean (SD) | 25.045 (14.7932) | 23.458 (14.7172) |
Median | ||||||||||||||||| | ||||||||||||||||| |
Range | ||||||||||||||||| | ||||||||||||||||| |
AD medication history, N(%) | ||
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
Medical history N (%) | ||
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
||||||||||||||||| | ||||||||||||||||| | ||||||||||||||||| |
AD = atopic dermatitis; BMI = body mass index; BSA = body surface area; DUP = dupilumab; EASI = Eczema Area and Severity Index; max = maximum; min = minimum; SD = standard deviation; UPA = upadacitinib; vIGA-AD = validated Investigator Global Assessment for Atopic Dermatitis; WP-NRS = Worst Pruritus Numerical Rating Scale.
Note: Redacted rows have been deleted.
Source: Clinical Study Report: Heads Up.14
Interventions
A summary of the randomized study treatments in Measure Up 1, Measure Up 2, AD Up, and Heads Up is presented in Table 15.
Table 15: Summary of Interventions in the Included Studies
Studies | Interventions |
|---|---|
Monotherapy studies | |
Measure Up 1 and Measure Up 2 |
|
Combination therapy studies | |
AD Up |
|
Japan study (safety) | |
Head-to-head study | |
Heads Up |
|
SC = subcutaneous; TCS = topical corticosteroid.
Source: Clinical Study Reports for Measure Up 1 and Measure Up 211,12 and the AD Up, Heads Up, and Japan studies.13,14,25
Monotherapy Studies
The Measure Up 1 and Measure Up 2 studies used upadacitinib 15 mg or 30 mg extended-release tablets or a matching placebo as comparator. The placebo consisted of a film-coated tablet with similar characteristics and packaging. Treatments were started on day 1 (baseline) and taken daily at approximately the same time each day. The study drug can be taken with or without food. All patients who completed the double-blind period on the study drug received either upadacitinib 15 mg or upadacitinib 30 mg in the BE period.
In the Measure Up 1 and Measure Up 2 studies, beginning at the screening visit, twice-daily use of an additive-free, bland emollient was required for at least 7 days before baseline and during the study until week 16. Other JAK inhibitors were not allowed; nor were targeted biologic therapies or other non-biologic systemic therapies (i.e., methotrexate, cyclosporine A, azathioprine, PDE 4 inhibitors, and mycophenolate mofetil). Inhaled ophthalmic drops and nasal corticosteroid formulations were allowed throughout the study. Patients could be treated with systemic corticosteroids for non-AD reasons if medically necessary after week 16. IV, intramuscular, intralesional and oral corticosteroids were prohibited throughout the study for the treatment of AD. UVB or UVA phototherapy — including psoralen and UVA radiation (PUVA) or laser therapy for at least 4 weeks before the baseline visit and during the study — were not allowed. No topical treatments for AD (including TCIs) were to be started through week 16 except for rescue treatment. As described previously, only topical emollients were allowed. Traditional oral or parenteral Chinese medicines were not permitted during the study because these may interfere with upadacitinib metabolism. Any patient who received an oral corticosteroid for more than 2 consecutive weeks (or any other systemic therapy, such as parenteral corticosteroid, methotrexate, mycophenolate mofetil, azathioprine, or dupilumab) would permanently discontinue study drug, regardless of the dosage.
In the Measure Up 1 and Measure Up 2 studies, rescue therapy was permitted from week 4 through week 24 if an EASI response of less than 50 was reported at 2 consecutive visits, or after week 24 with an EASI response of less than 50 at any visit. Investigators were instructed to attempt to limit the first step of rescue therapy to topical medications and to escalate to systemic medications only for those patients who did not respond adequately after at least 7 days of topical treatment. After the week 16 visit, any concomitant topical medication for AD could be administered per investigator discretion and would no longer be considered as rescue therapy. Only systemic treatments for AD were considered as rescue therapy for the purposes of statistical analyses of efficacy. If oral corticosteroids had to be used, rescue treatment was to be limited to prednisone or prednisolone for up to 1 mg/kg for no more than 2 consecutive weeks.
Studies of Combination Therapies
The AD Up study used upadacitinib 15 mg or 30 mg extended-release tablets or a matching placebo as comparator. The placebo consisted of a film-coated tablet with similar characteristics and packaging. Treatments were started on day 1 (baseline) and taken at approximately the same time each day. The study drug could be taken with or without food. All patients who completed the double-blind period on the study drug received upadacitinib 15 mg or upadacitinib 30 mg in the BE period.
Beginning at the screening visit, twice-daily use of an additive-free, bland emollient was required for at least 7 days before baseline and during the study until week 52. Other JAK inhibitors were not allowed; nor were targeted biologic therapies or other non-biologic systemic therapies (e.g., methotrexate, cyclosporine A, azathioprine, PDE 4 inhibitors, and mycophenolate mofetil). Inhaled ophthalmic drops and nasal corticosteroid formulations were allowed throughout the study. Patients could be treated with systemic corticosteroids for non-AD reasons if medically necessary after week 16. IV, intramuscular, intralesional, and oral corticosteroids were prohibited throughout the study for the treatment of AD. UVB and UVA phototherapy, including PUVA or laser therapy, for at least 4 weeks before the baseline visit and during the study were not allowed. No topical treatments (including TCIs and TCSs) were to be started through week 52 except for rescue treatment. As described earlier, only topical emollients were allowed. In the AD Up study, all patients, starting at the baseline and continuing through week 52, were to initiate treatment with a TCS and/or TCI with a step-down regimen from a medium-potency TCS (i.e., triamcinolone cream 0.1% or fluocinolone acetonide ointment 0.025%) to a low-potency TCS (i.e., hydrocortisone 1% cream).
In the AD Up study, rescue therapy was permitted from week 4 through week 24 if an EASI response of less than 50 was reported at 2 consecutive visits, or after week 24 with an EASI response of less than 50 at any visit. The first step of rescue therapy consisted of a high-potency TCS (e.g., mometasone 0.1% ointment) or a super–high-potency TCS (e.g., augmented betamethasone dipropionate 0.05% ointment or clobetasol propionate 0.05% cream) unless the higher-potency TCS was considered unsafe. Alternative topical AD medications could have been used and escalated to systemic medications for only those patients who did not respond adequately after at least 7 days of topical treatment.
Head-to-Head Studies
The Heads Up study used upadacitinib 30 mg extended-release tablets plus a placebo pre-filled syringe for SC injection in the upadacitinib group, or a pre-filled syringe of dupilumab 300 mg for SC injection plus a matching film-coated tablet as placebo. Use of an emollient was required for at least 7 days before baseline and for the duration of the study. Other JAK inhibitors were not allowed; nor were targeted biologic therapies or other non-biologic systemic therapies (e.g., methotrexate, cyclosporine A, azathioprine, PDE 4 inhibitors, and mycophenolate mofetil). Inhaled ophthalmic drops and nasal corticosteroid formulations were allowed throughout the study. Concomitant treatment with systemic corticosteroids (oral, IV, intramuscular) and intralesional corticosteroids was not allowed during treatment with the study drug. However, patients could be treated with systemic corticosteroids for non-AD reasons if medically necessary. UVB or UVA phototherapy, including PUVA or laser therapy, for at least 4 weeks before the baseline visit and during the study were not allowed. No topical treatments for AD (including TCIs and TCSs) were to be started during the study except for rescue treatment.
Rescue treatment for AD was permitted at the week 4 visit and throughout the study if medically necessary. In this study, investigators were advised to attempt to limit the first step of rescue therapy to topical medications and to escalate to systemic medications for only those patients who did not respond adequately after at least 7 days of topical treatment. Patients who received topical rescue treatment during the study treatment period could continue the study drug. If a patient required rescue treatment with a systemic drug (including, but not limited to, corticosteroids, cyclosporine A, methotrexate, mycophenolate mofetil, or azathioprine) or phototherapy, the study drug was permanently discontinued before the initiation of the rescue systemic drug or phototherapy.
Outcomes
A list of efficacy end points identified in the CADTH review protocol that were assessed in the clinical trials included in this review is provided in Table 16. These end points are further summarized in the following section. A detailed discussion and appraisal of the outcome measures is provided in Appendix 4.
Table 16: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure | Measure Up 1 and Measure Up 2 | AD Up | Heads Up | Japan studya |
|---|---|---|---|---|
EASI 75 | Co-primary | Co-primary | Primary | Exploratory |
vIGA-AD | Co-primary | Co-primary | NA | Exploratory |
WP-NRS | Key secondary | Key secondary | Key secondary | Exploratory |
EASI 90 | Key secondary | Key secondary | Key secondary | Exploratory |
EASI % change from BL | Key secondary | Key secondary | Key secondary | Exploratory |
SCORAD | Key secondary | Key secondary | NA | NA |
POEM ≥ 4 from BL | Key secondary | Key secondary | NA | NA |
ADerm-IS | Key secondary | Key secondary | NA | NA |
ADerm-SS | Key secondary | Key secondary | NA | NA |
ADerm-SS TSS-7 | Key secondary | Key secondary | NA | NA |
DLQI | Key secondary | Key secondary | NA | NA |
CDLQI 0 or 1 | Key secondary | Key secondary | NA | NA |
EQ-5D-5L | Key secondary | Key secondary | NA | NA |
HADS-A | Key secondary | Key secondary | NA | NA |
WPAI:AD | Key secondary | Key secondary | NA | NA |
ADerm-IS = Atopic Dermatitis Impact Scale; ADerm-SS = Atopic Dermatitis Symptom Scale; BL = baseline; CDLQI = Children’s Dermatology Life Quality Index; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; EASI 75 = at least 75% improvement in EASI total score from baseline; EASI 90 = at least 90% improvement in EASI total score; EQ-5D-5L = EQ-5D Five-Level; HADS-A = Hospital Anxiety and Depression Scale – Anxiety; NA = not applicable; POEM = Patient-Oriented Eczema Measure; = SCORAD = Scoring Atopic Dermatitis; TSS-7 = 7-Item Total Symptom Score; vIGA-AD = validated Investigator Global Assessment for Atopic Dermatitis; WPAI:AD = Work Productivity and Activity Impairment Questionnaire: Atopic Dermatitis; WP-NRS = Worst Pruritis Numerical Rating Scale.
aThe Japan study aimed to evaluate harms and safety outcomes as primary end points.
Source: Clinical Study Reports for Measure Up 1 and Measure Up 211,12 and the AD Up, Heads Up, and Japan studies.13,14,25
Eczema Area and Severity Index
The EASI is a scale used in clinical trials to assess the severity and extent of AD.27 In EASI, 4 disease characteristics of AD (erythema, infiltration and/or papulation, excoriations, and lichenification) are assessed for severity by the investigator on a scale of 0 (absent) to 3 (severe). The scores are added up for each of the 4 body regions (head, arms, trunk, and legs). The assigned percentages of BSA for each section of the body are 10% for head, 20% for arms, 30% for trunk, and 40% for legs, respectively. Each subtotal score is multiplied by the BSA represented by that region. In addition, the affected area of AD assessed as a percentage by each body region is converted to a score of 0 to 6, where the area is expressed as 0 (none), 1 (1% to 9%), 2 (10% to 29%), 3 (30% to 49%), 4 (50% to 69%), 5 (70% to 89%), or 6 (90% to 100%). Each of the body area scores is multiplied by the area affected.
Therefore, the total EASI score ranges from 0 to 72 points, with the highest score indicating worst severity of AD.28 It is suggested that the severity of AD based on EASI is categorized as follows: 0 = clear; 0.1 to 1.0 = almost clear; 1.1 to 7.0 = mild; 7.1 to 21.0 = moderate; 21.1 to 50.0 = severe; 50.1 to 72.0 = very severe.29 The end points of reduction of EASI 50, EASI 75, EASI 90, and EASI 100 indicate improvements of greater than or equal to 50%, greater than or equal to 75%, greater than or equal to 90%, and 100% improvement from baseline, respectively. The validity and reliability of the EASI were examined in several studies, demonstrating good performance in all these domains.28,30-32 Correlation coefficients for assessing content and construct validity were estimated between EASI and SCORAD,27 with reports of moderate to high correlation (r = 0.84 to 0.93) between these 2 tools. The internal consistency of EASI is adequate, with Spearman and Cronbach alpha values of 0.86 and 0.94, respectively.27 Intra- and Inter-rater reliability have also been examined, with adequate values of test-retest reliability and kappa values of 0.76.27 Responsiveness (sensitivity to change) was also judged as adequate by the systematic review authors. The overall minimal important difference is 6.6, based on results from 1 study.32
Validated Investigator Global Assessment for Atopic Dermatitis
The vIGA-AD is a 5-point scale that provides a global clinical assessment of AD severity ranging from 0 to 4, where 0 indicates clear and 4 indicates severe AD.33 A decrease in score relates to an improvement in signs and symptoms.
The vIGA-AD has been widely used in many AD clinical trials and required by regulatory agencies for drug approval trials.33 However, the instrument has had many issues with variable content validity, definitions, and implementations — the tool has had more than 20 different names and various numbers of scale categories (from 4-point to 7-point scales), as well as content of the scales.33,34 A 2016 systematic review of the literature found no information on the validity and reliability of the IGA instrument in patients with AD as well as no information on what would constitute a MID in patients with AD.33
The studies included in this CADTH submission use the newly validated version of the IGA — based on a recent study to validate the IGA was published by Simpson et al. (2020)35 — known as the vIGA-AD scale with the objective to harmonize outcome assessments in clinical trials. In this study, a 5-point IGA scale (0 to 4) was agreed to be used and content validity was achieved, with strong inter-rater reliability (intra-class correlation coefficient [ICC] = 0.817) and excellent agreement (kappa = 0.857).
No MID is presented. However, the pivotal studies included in this CADTH submission used a value of 0 or 1 in the vIGA-AD to classify a response. The assessment of the overall severity of AD and assignment of a vIGA-AD score and category is described in Table 17.
Table 17: Validated Investigator Global Assessment for Atopic Dermatitis Score
Score | Category | Description |
|---|---|---|
0 | Clear | No inflammatory signs of atopic dermatitis (no erythema, no induration or papulation, no lichenification, no oozing or crusting). Post-inflammatory hyperpigmentation and/or hypopigmentation may be present. |
1 | Almost clear | Barely perceptible erythema, barely perceptible induration or papulation, and/or minimal lichenification. No oozing or crusting. |
2 | Mild | Slight but definite erythema (pink), slight but definite induration or papulation, and/or slight but definite lichenification. No oozing or crusting. |
3 | Moderate | Clearly perceptible erythema (dull red), clearly perceptible induration or papulation, and/or clearly perceptible lichenification. Oozing and crusting may be present. |
4 | Severe | Marked erythema (deep or bright red), marked induration or papulation, and/or marked lichenification. Disease is widespread in extent. Oozing or crusting may be present. |
Notes | 1. In indeterminate cases, use extent to differentiate between scores. 2. Excoriations should not be considered when assessing disease severity. | |
Source: Clinical Study Report.
Worst Pruritus Numerical Rating Scale
The WP-NRS is used to report the intensity of a patient’s itch during a daily recall period. Patients rate their overall (average) and maximum intensity of itch experienced during the past 24 hours based on a scale of 0 to 10 (where 0 = no itch and 10 = worst itch imaginable). The reliability of the WP-NRS is adequate,36 with pooled ICCs in the range of 0.95 to 0.97.36 WP-NRS scores are stable over a period of time. The validity of the WP-NRS has been shown using known-groups approaches, with all above the Cohen threshold of 0.80 for large effect sizes.36 Based on the data from the phase IIb study, using EASI and IGA as anchors, the WP-NRS responder reportedly ranged between 2.2 and 4.2, with the highest estimates based on the most stringent clinical criteria (i.e., EASI 90 to 100 and IGA 0 or 1). Using a Pruritus Categorical Scale as an anchor, the responder was estimated at 2.6 points. These analyses suggested that the most appropriate definition of a responder on the WP-NRS is in the range of 3 to 4 points.36 The investigators from the included studies evaluated in this CADTH submission evaluated the WP-NRS as the proportion of patients achieving an improvement (reduction) in WP-NRS greater than or equal to 4 from baseline for those patients with WP-NRS greater than or equal to4 at baseline at week 16.
Scoring Atopic Dermatitis
The SCORAD was developed to standardize the evaluation of the extent and severity of AD.37 SCORAD is considered a valid and reliable tool for the objective assessment of eczema clinical signs.38 The instrument assesses 3 components of AD: the extent of affected BSA (0 to 100), severity (0 to 18), and symptoms (0 to 20). The extent of AD is assessed as a percentage of each defined body area and reported as the sum of all areas. The score ranges from 0 to 100. The severity of 6 specific signs of AD (redness, swelling, oozing or crusting, excoriation, skin thickening or lichenification, and dryness) is assessed using a 4-point scale (i.e., none = 0, mild = 1, moderate = 2, and severe = 3), with a minimum score of 0 and a maximum score of 18. The subjective symptoms (itch and sleeplessness) are recorded by the patient or relative on a visual analogue scale, with scores ranging from 0 (no symptoms) to 10 (worst imaginable symptoms), with a maximum possible score of 20. The total SCORAD is calculated based on the 3 components, with a maximum possible total score of 103. Higher scores indicate a poorer or a more severe condition.
SCORAD has been found to be valid and reliable, with excellent agreement with global assessments of disease severity.27,39 Content validity has been deemed adequate, with good construct validity (i.e., Spearman correlation coefficient ranging from 0.53 to 0.92) and internal consistency. Sensitivity to change and inter-observer reliability are also adequate; the latter has several measurements of ICC from 0.84 to 0.99. However, intra-observer reliability (test-retest) was unclear.27 The minimal important difference has been estimated using mean changes in the SCORAD scores of patients that showed a relevant improvement based on IGA, defined as an improvement or decline of greater than or equal to 1 point in PGA and IGA; thus, a difference of 8.7 points in SCORAD was estimated as the minimal important difference for patients with AD.32
This tool was used in the Measure Up and AD Up studies as a secondary end point as the percentage change in SCORAD from baseline at week 16.
Patient-Oriented Eczema Measure
This is a 7-item questionnaire used in clinical trials to assess disease symptoms in children and adults. Based on the frequency of occurrence during the preceding week, the 7 items (dryness, itching, flaking, cracking, sleep loss, bleeding, and weeping) are assessed using a 5-point scale. The possible scores for each question range from 0 to 4, where 0 indicates no days, 1 indicates 2 days, 2 indicates 3 to 4 days, 3 indicates 5 to 6 days, and 4 indicates every day. The maximum total score is 28; a high score is indicative of poor quality of life (a total score of 0 to 2 indicates clear or almost clear; 3 to 7 indicates mild eczema; 8 to 16 indicates moderate eczema; 17 to 24 indicates severe eczema; and 25 to 28 indicates very severe eczema).40
The tool has been tested for its validity, reliability, and responsiveness. When compared to the patient-oriented (PO)-SCORAD and DLQI,41 a moderate concurrent validity (Spearman correlation coefficient = 0.56) was detected in adults. There is good convergent validity when compared to the DLQI, but moderate to weak convergent validity when compared to the EASI and NRS. There is poor discriminant validity in predicting self-reported global severity. In other studies including children, content validity was poor to moderate as a measurement of clinical signs of AD.27,40 The same studies have revealed moderate responsiveness and good reliability (ICC = 0.90).41 The minimal important difference has been established in AD as 3.4 points in adults and from 3.0 to 3.9 points in children. Other studies have established 5 points as the minimal important difference for adults using global severity of AD as anchor.41
This tool was used in the Measure Up and AD Up studies as a secondary end point as the proportion of patients achieving an improvement (reduction) of greater than or equal to 4 from baseline at week 16.
Atopic Dermatitis Impact Scale
The ADerm-IS is an AD-specific, patient-reported questionnaire to assess the signs, symptoms, and impacts of moderate to severe AD in adults. The ADerm-IS was developed as an electronic diary that includes 3 items to be completed daily to assess impact over the previous 24 hours, and 8 items completed weekly to assess impacts over the past 7 days. Response categories are assessed over an 11-point Likert scale from 0 (no impact) to 10 (extreme impact).24,42 Thirteen sign and symptom concepts are included: bleeding, blisters, burning, dry skin, fissures, inflammation, itching, pain, rash, redness, scaling, skin thickening, and swelling. Additionally, 43 impact concepts have been identified and organized into 8 domains: activities of daily living (ADLs), cognitive, emotional, financial, physical, sleep, social, and work and/or school. The most frequently reported impacts were sleep disturbances, followed by interruptions to work and/or school activities, social withdrawal, anxiety, feelings of depression, embarrassment, and the inability to participate in ADLs.43 The pivotal studies report the impact score from 3 domains: sleep, emotional state, and daily activities.44 One study supports the content validity of the ADerm-IS,43 but no minimal important difference or additional validity information regarding the ADerm-IS was identified from the literature search for AD in this CADTH review.
This tool was used in the Measure Up and AD Up studies for this CADTH review as a secondary end point as the proportion of patients achieving an improvement (reduction) in ADerm-IS sleep domain score greater than or equal to 12 (minimal clinically important difference [MCID]) from baseline at week 16 for patients who had an ADerm-IS sleep domain score greater than or equal to 12 at baseline.
Atopic Dermatitis Symptom Scale
The ADerm-SS is also an AD-specific, patient-reported questionnaire to assess the signs, symptoms, and impacts of moderate to severe AD in adults. The scale was developed as an electronic diary that includes 3 items to be completed daily, assessing impact over the previous 24 hours, and 8 items completed weekly to assess impacts over the past 7 days. Response categories are assessed over an 11-point Likert scale from 0 (no impact) to 10 (extreme impact).24,42 Thirteen sign and symptom concepts are included: bleeding, blisters, burning, dry skin, fissures, inflammation, itching, pain, rash, redness, scaling, skin thickening, and swelling. Additionally, 43 impact concepts were identified and organized into 8 domains: ADLs, cognitive, emotional, financial, physical, sleep, social, and work and/or school. The most frequently reported impacts were sleep disturbances, followed by interruptions to work and/or school activities, social withdrawal, anxiety, feelings of depression, embarrassment, and the inability to participate in ADLs.43 The pivotal studies report the symptom score from 2 domains: skin pain and 7-Item Total Symptom Score (TSS-7).44 One study supports the content validity of the ADerm-SS.43 No MID or additional validity information regarding the ADerm-SS was identified from the literature search for AD.
This tool was used in the Measure Up and AD Up studies for this CADTH review as a secondary end point as the proportion of patients achieving an improvement (reduction) in ADerm-SS skin pain score greater than or equal to 4 (MCID) from baseline at week 16 for patients who had an ADerm-SS skin pain score greater than or equal to 4 at baseline.
A variant of this measurement is the ADerm-SS TSS-7, defined as the algebraic sum of the responses to items 1 through 7 of the ADerm-SS. It was also used in the Measure Up and AD Up studies as the proportion of patients achieving an improvement (reduction) in ADerm-SS TSS-7 greater than or equal to 28 (MCID) from baseline at week 16 for patients who had an ADerm-SS TSS-7 score greater than or equal to 28 at baseline.
Dermatology Life Quality Index
The DLQI is a widely used, dermatology-specific HRQoL instrument. It is a 10-item questionnaire that assesses 6 different aspects that may affect quality of life.45,46 These aspects are symptoms and feelings, daily activities, leisure, work and school performance, personal relationships, and treatment. The maximum score per aspect is either 3 (with single questions) or 6 (with 2 questions) and the scores for each can be expressed as a percentage of either 3 or 6. Each of the 10 questions is scored from 0 (not at all) to 3 (very much), and the overall DLQI is calculated by summing the score of each question, resulting in a numeric score between 0 and 30 (or a percentage of 30).45,46 The higher the score, the more quality of life is impaired. The meaning of the DLQI scores in a patient’s life is as follows47:
0 to 1 = no effect
2 to 5 = small effect
6 to 10 = moderate effect
11 to 20 = very large effect
21 to 30 = extremely large effect.
The validity of the DLQI has been assessed in patients with eczema39,48-50 with good test-retest reliability (i.e., the correlation between overall DLQI scores was 0.99 [P < 0.0001] and of individual question scores was 0.95 to 0.98 [P < 0.001]),46 internal consistency (reliability) (with Cronbach alpha coefficients ranging from 0.75 to 0.92),47 construct validity,47 and responsiveness.47-49
Estimates of the minimal important difference have ranged from 2.2 to 6.9.45,47 It should be noted that some of the anchors that were used to obtain the DLQI minimal important difference were not patient-based (i.e., Basra et al.47 derived estimates from the Psoriasis Area and Severity Index and PGA anchors as well as using a distribution-based approach); in addition, some limitations of the DLQI include concerns regarding uni-dimensionality and the behaviour of items of the DLQI in different psoriatic patient populations.47 No validity or information about minimal important difference was found for patients with AD.51
This tool was used in the Measure Up and AD Up studies for this CADTH review as a secondary end point as the proportion of patients greater than or equal to 16 years old who achieve a DLQI score of 0 or 1 at week 16.
Children’s Dermatology Life Quality Index
The Children’s Dermatology Life Quality Index (CDLQI) is based on the adult version (DLQI). This is a child-completed questionnaire for children 3 years to 16 years of age. It is designed to measure the impact of any skin disease on quality of life over a recall period of 7 days. It is 1 of the most commonly used instruments for measuring HRQoL in children.42,52,53 The instrument has 10 questions about the impact of a skin disease on the life of the affected child, including symptoms, embarrassment, friendships, clothes, playing, sports, bullying, sleeping, and impact of treatment. Each question is answered on a 4-point Likert scale scored from 0 to 3. These are summed to give a minimum of 0 and maximum of 30. A higher score indicates a greater degree of impairment in HRQoL.
A 2013 systematic review did not identify studies demonstrating content validity.53 In the same review, 3 studies demonstrated concurrent validity, 2 between the CDLQI and the Cardiff Acne Disability Index and 1 between the CDLQI and the Childhood Atopic Dermatitis Impact Scale. The CDLQI was correlated in 10 studies with SCORAD, the primarily sign-based severity scoring system for AD. Forty-five studies demonstrated convergent construct validity, and 6 studies demonstrated divergent construct validity. The same review showed good internal consistency of the CDLQI (examined in 6 studies), with Cronbach alpha values ranging from 0.82 to 0.92. Similarly, test–retest reliability was adequate, with the Spearman correlation coefficient calculated in 4 studies (range = 0.74 to 0.97). One study examined the ICC, finding 0.80. Good responsiveness to change was found in studies using the Wilcoxon signed rank test and repeated analysis of variance measures (ANOVA).
One study conducted in the US and Canada with 202 participants using a distribution-based approach determined the MCID of the CDLQI in psoriasis to be 2.5. However, no specific MID for AD has been established.
This tool was used in the Measure Up and AD Up studies for this CADTH review as a secondary end point as the proportion of patients less than 16 years of age achieving a CDLQI score of 0 or 1 at week 16.
EQ-5D
The EQ-5D is a generic quality of life instrument that has been applied to a wide range of health conditions and treatments, including AD.54,55 The first of 2 parts of the EQ-5D consist of a descriptive system that classifies respondents (aged ≥ 12 years) into 1 of 243 distinct health states. The descriptive system consists of the following 5 dimensions: mobility, self-care, usual activities, pain and/or discomfort, and anxiety and/or depression. Each dimension has 3 possible levels (1, 2, or 3), representing no problems, some problems, and extreme problems, respectively. Respondents are asked to choose 1 level that reflects their own health state for each of the 5 dimensions. A scoring function can be used to assign a value (the EQ-5D index score) to self-reported health states from a set of population-based preference weights.54,55 The second part is a 20 cm EQ-VAS that has end points labelled 0 and 100, with respective anchors of “worst imaginable health state” and “best imaginable health state,” respectively. Respondents are asked to rate their own health by drawing a line from an anchor box to the point on the EQ-VAS that best represents their own health on that day. The third part is the EQ-5D index score, which is generated by applying a multi-attribute utility function to the descriptive system. Different utility functions are available that reflect the preferences of specific populations (e.g., US or UK). Hence, the EQ-5D produces 3 types of data for each respondent.
The lowest possible overall score (corresponding to severe problems on all 5 attributes) varies depending on the utility function that is applied to the descriptive system (e.g., −0.59 for the UK algorithm and −0.109 for the US algorithm). Scores less than 0 represent health states that are valued by society as being worse than dead, while the scores 0 and 1.00 are assigned to the health states “dead” and “perfect health,” respectively. The minimal important difference for the EQ-5D ranges from 0.033 to 0.074. No additional validity or minimal important difference were found in the literature search for EQ-5D in patients with AD.
This tool was used in the Measure Up and AD Up studies for this CADTH review as a secondary end point reported as the change and percentage change from baseline in EQ-5D-5L values.
Hospital Anxiety and Depression Scale – Anxiety
The HADS-A is a widely used, patient-reported questionnaire designed to identify anxiety disorders and depression in patients at non-psychiatric medical institutions. Repeated administration also provides information about changes in a patient’s emotional state.56-58 The questionnaire contains 14 items that assess symptoms experienced in the previous week. Among these, 7 items are related to anxiety and 7 are related to depression. Patients provided responses to each item based on a 4-point Likert scale. Each item is scored from 0 (the best) to 3 (the worst); thus, a person can score between 0 and 21 for each subscale (anxiety and depression). A high score is indicative of a poor state. Scores of 11 or more on either subscale are considered to indicate a “definite case” of psychological morbidity, while scores of 8 to 10 represent a “probable case” and 0 to 7 is likely “not a case.”58 One study59 indicated that HADS-A has good construct validity, with no overall floor or ceiling effects. HADS-A may be useful for the assessment of patients with AD in clinical trials and practice. The author concluded that additional research is needed to confirm the construct validity and to assess content validity and feasibility in research and clinical practice.59 No additional validity and minimal important difference information regarding HADS-A was found from the literature search for patients with AD. This tool was used in the Measure Up and AD Up studies for this CADTH review as a secondary end point reported as the change and percentage change from baseline in HADS-A.
Work Productivity and Activity Index: Atopic Dermatitis
The WPAI:AD is an instrument used to measure loss of productivity at work and impairment in daily activities over the preceding 7 days.60 The questionnaire includes 4 items — absenteeism, presenteeism, overall work impairment, and activity impairment — that range from 0% to 100%, with higher values indicating greater impairment. While absenteeism represents the percentage of work time missed due to AD, presenteeism represents the percentage of impairment while at work due to AD. Overall work impairment represents the total percentage of work time missed due to either absenteeism or presenteeism (given that these are mutually exclusive). Activity impairment represents the percentage of impairment during daily activities other than work. All 4 items are evaluated using an 11-point Likert-type scale from 0 (no effect) to 10 (completely prevented), and the scores are multiplied by 10 to arrive at a percentage. The WPAI:AD has been validated to quantify work impairments for numerous diseases, such as asthma, psoriasis, irritable bowel syndrome, ankylosing spondylitis, and Crohn disease, with established construct validity.61 It has overall good reproducibility, with correlation coefficients ranging from 0.71 to 0.87. However, no minimal important difference for AD has been established.
The WPAI:AD tool was used in the Measure Up and AD Up studies for this CADTH review as a secondary end point reported as the change and percentage change from baseline in WPAI:AD domain scores (absenteeism, presenteeism, activity impairment, and overall work productivity).
Statistical Analysis
Monotherapy Regimen Studies (Measure Up 1 and Measure Up 2)
The co-primary end points for the Measure Up studies were the proportion of patients achieving at least EASI 75 at week 16 and the proportion of patients achieving a vIGA-AD score of 0 or 1 (clear or almost clear) with at least 2 grades of reduction from baseline at week 16. Secondary outcomes were also assessed by controlling for type I error, as described later.
Power and Sample Size
The sample size was determined assuming an EASI 75 response rate of ||||||||%, and a vIGA-AD score of 0 or 1 with at least a 2-point reduction response rate of ||||||||% in the placebo arm. A sample size of 810 patients randomized in a 1:1:1 ratio provides more than 90% power to detect the treatment differences of ||||||||% and ||||||||%, respectively, for these 2 end points simultaneously using 2-sided test at a 0.05 significant level. The assumptions of placebo response rates for EASI 75 and a vIGA-AD of 0 or 1 were based on the maximum placebo rate in an upadacitinib AD phase IIb study and 2 phase III studies of dupilumab as monotherapy (SOLO 1 and SOLO 2).
Statistical Tests
In the Measure Up studies, the co-primary end points were compared between the upadacitinib and placebo groups using the Cochran-Mantel-Haenszel (CMH) test, stratified by vIGA-AD score categories and age (adolescent versus adult) in the ITT population. Continuous variables were analyzed using mixed-effects model for repeated measures (MMRM).
The overall type I error rate of the primary and secondary end points for upadacitinib 15 mg and 30 mg was controlled using a graphical multiple testing procedure following a pre-specified alpha transfer path that includes downstream transfer along the end points sequence within each dose as well as cross-dose transfer. Each of the 2 doses of upadacitinib was tested separately against placebo (i.e., not pooled) at a significance initial alpha (2-sided) for the graphic approach of 0.05. The graphical approach is presented in Figure 37, Appendix 3. In this graph, the arrows specify an alpha transfer path. Once an end point is rejected (i.e., deemed significant) at its assigned significance level, its significance level is transferred to subsequent end point(s) following the arrow(s). If more than 1 arrow originates from an end point, the significance level for this end point (once rejected) will be split between multiple subsequent end points following the arrows. The numbers on the arrows denote the weights for transferring and (possibly) splitting significance levels. Specifically, the weight 1 denotes a 100% transfer of significance level, and the weight 0.5 denotes a 50% splitting of significance level. First, EASI 75 and vIGA-AD at week 16 were tested, and if the tests for both were significant, the significance was transferred to test the 15 mg dose on the same end points, and so on. In addition, within each dose, selected patient-reported outcomes were grouped into blocks and were to be tested using the Hochberg method.
Missing Data and Imputation
Due to the COVID-19, missing values and visits after the rescue were handled as missing at random (MAR), and a nonresponder imputation to handle data missing due to COVID-19 (NRI-C) was used for categorical variables. The NRI-C categorized any patient who did not have an evaluation during a pre-specified visit window (either due to missing assessment or due to early withdrawal from the study) as a nonresponder for the visit.
Also, for categorical variables, multiple imputation methods were used, and the effects of these imputations were assessed in a sensitivity analysis. Several variables were included in an imputation model: i.e., treatment group, major stratum (vIGA-AD score categories, age [adolescent versus adult] if applicable, and regions), gender, baseline, and measurements at each visit up to the end of the analysis period.
Another imputation method for categorical variables (which was also used in a sensitivity analysis) was the “tipping point” analysis conducted on the co-primary end points (EASI 75 and vIGA-AD 0 or 1 at week 16) in the ITT population.
For continuous end points, missing data were handled using MMRM, including observed measurements at all visits. The mixed model included the categorical fixed effects of treatment, visit, and treatment-by-visit interaction, main stratification factors at randomization (i.e., vIGA-AD score categories and age [adolescent versus adult] if applicable), and the continuous fixed covariates of baseline measurement.
Subgroup Analyses
The subgroups identified in the protocol for this CADTH review were:
severity of disease (assessed in Measure Up studies using the EASI and vIGA-AD)
failure to respond, contraindication, or intolerance to 1 or more systemic therapies (not assessed in the Measure Up studies; assessed only as previous use or no previous use of systemic therapies)
age (adolescents versus adults, assessed in the Measure Up studies using a threshold of 18 years of age)
smoking status (not assessed in the Measure Up studies)
obesity (defined in the Measure Up studies as: normal = body mass index [BMI] < 25; overweight = BMI of 25 to < 30; obese = BMI ≥ 30).
In the Measure Up studies, the subgroups performed for the co-primary outcomes were:
age group 1 (< 18 years, ≥ 18 years)
age group 2 (< 18 years, ≥ 18 to < 40 years, ≥ 40 to < 65 years, and ≥ 65 years)
sex (male, female)
BMI (defined as normal = less than 25; overweight = 25 to < 30; obese = 30 or greater)
race (White, Asian, Black, and other)
weight (< median, ≥ median)
geographic regions (US, Puerto Rico, Canada; Japan; mainland China; other)
baseline vIGA-AD (< 4, 4)
baseline EASI (< median, ≥ median)
high-sensitivity C-reactive protein (hsCRP) (< median, ≥ median)
previous systemic therapy (with and without)
patients who reported an intolerance to at least 1 prior TCS or TCI therapy
patients who reported an inadequate response to at least 1 beforepical treatment.
Sensitivity Analyses
Sensitivity analyses were performed to assess the impact of missing data and the robustness of the conclusion. In this case, for the categorical end points in the co-primary outcome and for all categorical secondary end points, investigators used “non-responder imputation with no special data handling for data missing due to COVID-19” (NRI-NC) as a variable for the sensitivity analysis. In addition, sensitivity analyses were conducted based on the multiple imputation methods, the tipping point approach, and the per-protocol population.
Analysis Populations
Three populations were analyzed:
the ITT population, consisting of all patients who were randomized in the overall study
the per-protocol population for the main study, which was defined to exclude patients with major protocol violations such that only those who completed the study intervention were included in the analysis (the primary efficacy end points were also analyzed in the per-protocol population as sensitivity analysis)
the safety population, consisting of all randomized patients who received at least 1 dose of the study drug in the overall study during the double-blind period.
Combination Regimen Studies (AD Up)
In the AD Up study, the co-primary outcomes were the proportion of patients achieving an EASI 75 (i.e., at least a 75% reduction) from baseline at week 16 and the proportion of patients achieving a vIGA-AD score of 0 or 1 with at least 2 grades of reduction from baseline at week 16.
In the Japan study, investigators assessed safety outcomes as the primary end points. These are discussed in the harm outcomes section.
Power and Sample Size
The sample size was determined assuming an EASI 75 response rate of ||||||||% and a vIGA-AD score of 0 or 1 with at least a 2-point reduction response rate of ||||||||% in the placebo arm. A sample size of 810 patients randomized in a 1:1:1 ratio (270 per treatment group) provides more than 90% power to detect the treatment differences of ||||||||% and ||||||||%, respectively, for these 2 end points simultaneously using a 2-sided test at a 0.05 significant level. The assumptions of placebo response rates for EASI 75 and vIGA-AD 0 or 1 were based on the maximum placebo rate in the upadacitinib AD phase IIb study and the dupilumab phase III monotherapy studies (SOLO 1 and SOLO 2), adding the estimation of topical treatment effect, which is also based on the difference between the monotherapy and combination therapy (CHRONOS) studies of dupilumab.
Statistical Tests
In the AD Up study, the co-primary end points were compared between the upadacitinib and placebo groups using the CMH test, stratified by vIGA-AD score categories and age (adolescent versus adult) in the ITT population. Continuous variables were analyzed using MMRM. For continuous end points with only 1 post-baseline assessment in the double-blind period (e.g., WPAI:AD), an analysis of covariance model was applied.
The overall type I error rate of the primary and secondary end points for upadacitinib 15 mg plus TCS and upadacitinib 30 mg plus TCS was controlled using a graphical multiple testing procedure following a pre-specified alpha transfer path that included downstream transfer along the end points sequence within each dose as well as cross-dose transfer. Each of the 2 doses of upadacitinib was tested separately against placebo (i.e., not pooled) at a significance initial alpha of 0.05. The graphical approach is presented in in Figure 37, Appendix 3. In these graphs, the arrows specify the alpha transfer path. Once an end point is rejected (i.e., deemed significant) at its assigned significance level, its significance level is transferred to subsequent end point(s) following the arrow(s). If more than 1 arrow originates from an end point, the significance level for this end point (once rejected) will be split between multiple subsequent end points following the arrows. The numbers on the arrows denote the weights for transferring and (possibly) splitting significance levels. Specifically, the weight 1 denotes 100% transfer of significance level, and the weight 0.5 denotes 50% splitting of significance level. First, the authors tested EASI 75 and vIGA-AD score at week 16, and if the tests for both were significant, the significance was transferred to test the 15 mg dose using the same end points, and so on. In addition, within each dose, selected patient-reported outcomes were grouped into blocks be tested using the Hochberg method.
Missing Data and Imputation
Due to the COVID-19 pandemic, missing values and visits after the rescue were handled as MAR and a nonresponder imputation for categorical variables was used to handle data missing due to COVID-19. The NRI-C categorized any patient who did not have an evaluation during a pre-specified visit window (either due to missing assessment or due to early withdrawal from the study) as a nonresponder for the visit.
Also, for categorical variables, multiple imputation methods were used and the effects these imputations were assessed in a sensitivity analysis. Several variables were included in an imputation model: i.e., treatment group, major stratum (vIGA-AD score categories, age [adolescent versus adult], if applicable, and regions), gender, baseline, and measurements at each visit up to the end of the analysis period.
One last imputation method for categorical variables (which was also used in a sensitivity analysis) was the tipping point analysis conducted on the co-primary end points (EASI 75 and vIGA-AD score of 0 or 1 at week 16) in the ITT population.
For continuous end points, missing data were handled using MMRM, including observed measurements at all visits. The mixed model includes the categorical fixed effects of treatment, visit and treatment-by-visit interaction, main stratification factors at randomization (vIGA-AD score categories and age [adolescent versus adult], if applicable), and the continuous fixed covariates of baseline measurement.
Subgroup Analyses
The subgroups identified in the protocol for this CADTH review were:
severity of disease (assessed in AD Up with EASI and vIGA-AD)
failure to respond, contraindication, or intolerance to 1 or more systemic therapy (not assessed in AD Up; assessed only as previous use or no previous use of systemic therapies)
age (adolescents versus adults; assessed in the AD Up study as a threshold of 18 years)
smoking status (not assessed in AD Up)
obesity (defined in AD Up as normal = BMI < 25; overweight = BMI of ≥ 25 to < 30; obese = BMI ≥ 30)
The primary subgroups analyzed in the AD Up study included:
age group 1 (< 18 years, ≥ 18 years)
age group 2 (< 18 years, ≥ 18 to < 40 years, ≥ 40 to < 65 years, ≥ 65 years)
sex (male, female)
BMI (defined as normal = less than 25; overweight = 25 to < 30; obese = 30 or greater)
race (White, Asian, Black, and other)
weight (< median, ≥ median)
geographic regions (US, Puerto Rico, Canada; Japan; mainland China; other)
baseline vIGA-AD (< 4, 4)
baseline EASI (< median, ≥ median)
hsCRP (< median, ≥ median)
previous systemic therapy (with and without)
patients reporting an intolerance to at least 1 prior TCS or TCI therapy
patients reporting an inadequate response to at least 1 beforepical treatment.
Sensitivity Analyses
Sensitivity analyses were performed to assess the impact of missing data and the robustness of the conclusion. In this case, for categorical end points in the co-primary and all categorical secondary end points, investigators used NRI-NC as a variable for the sensitivity analysis. Also, sensitivity analyses were conducted based on the multiple imputation methods, the tipping point approach, and the per-protocol population.
Analysis Populations
Three populations were analyzed:
the ITT population, consisting of all patients who were randomized in the overall study
the per-protocol population for the main study, defined to exclude patients with major protocol violations such that only those who completed the study intervention were included in the analysis (the primary efficacy end points were also analyzed in the per-protocol population as sensitivity analysis)
the safety population, consisting of all randomized patients who received at least 1 dose of the study drug in the overall study during the double-blind period.
Head-to-Head Regimen Studies
In the Heads Up study, the primary outcome was the proportion of patients achieving at least a 75% reduction in EASI from baseline at week 16.
Power and Sample Size
Investigators calculated that 650 patients (18 years to 75 years old) had to be randomized to upadacitinib 30 mg or dupilumab in a ratio of 1 to 1 (325 patients per treatment group). Assuming an EASI 75 response rate of at most ||||||||% in the dupilumab arm, this sample size would provide more than 80% power to detect at least a ||||||||% treatment difference using a 2-sided test at a 0.05 significant level. The assumptions of dupilumab response rate for EASI 75 at week 16 and ||||||||% treatment difference were based on the pooled response rates of the dupilumab phase III monotherapy studies (SOLO 1 and SOLO 2) and the response rate of patients on upadacitinib 30 mg in the upadacitinib AD phase IIb study.
Statistical Tests
Pairwise comparisons of upadacitinib versus dupilumab were completed using the CMH test with baseline vIGA-AD score categories (i.e., vIGA-AD score of 3 or 4) as the stratification factor. Construction of CIs for the common risk difference was based on the CMH estimate adjusting for stratification factors. Breslow-Day tests were performed to test the homogeneity between strata.
For continuous variables, in the ITT population, the percentage change from baseline in the treatment groups was compared using an MMRM model that included the categorical fixed effects of treatment, visit and treatment-by-visit interaction, and the continuous fixed covariates of baseline measurement, adjusted for stratification factor (i.e., vIGA-AD score categories at randomization). Point estimates, standard error, and 95% CIs of least squares (LS) mean change from baseline within treatment groups, and between upadacitinib group and dupilumab, were provided.
In addition, the primary end point and all key secondary categorical end points were analyzed using multiple imputations as sensitivity analysis. All categorical end points were also analyzed using NRI-NC, as defined in the previous studies and later in this review (and used as a sensitivity analysis approach).
A multiple testing procedure was used to control the type I error rate at alpha = 0.05 (2-sided) across analyses comparing upadacitinib versus dupilumab with respect to the primary end point and ranked secondary end points. Specifically, testing used a sequence of hypothesis testing for the primary end point (EASI 75 at week 16) followed by the ranked secondary end points (WP-NRS at week 16, EASI 100 at week 16, EASI 90 at week 16, WP-NRS at week 4, and so on), beginning with testing the primary end points using an alpha of 0.05 (2-sided) for upadacitinib versus dupilumab. If the primary end point achieved statistical significance, it continued testing following a hierarchical order of the secondary end point. Only the significance of a higher-ranked secondary end point implied the continuation of the next secondary end point.
Missing Data and Imputation
Due to the COVID-19 pandemic, missing values and visits after the rescue were handled as MAR, and a nonresponder imputation for categorical variables was used to handle data missing due to COVID-19. The NRI-NC categorized any patient who did not have an evaluation during a pre-specified visit window (due to either missing an assessment or early withdrawal from the study) as a nonresponder for the visit. NRI-C was the primary approach for handling missing data in the analyses of the primary efficacy end point. The NRI-C and multiple imputation approaches were used as sensitivity analyses.
Also, for categorical variables, multiple imputation methods were used, and the effects of these imputations were assessed in a sensitivity analysis. Several variables were included in an imputation model: i.e., treatment group, major stratum (vIGA-AD score categories, age [adolescent versus adult], if applicable, and regions), gender, baseline, and measurements at each visit up to the end of the analysis period.
For continuous end points, missing data were handled using MMRM, including observed measurements at all visits. The mixed model includes the fixed effects of categorical variable of treatment, visit and treatment-by-visit interaction, main stratification factor at randomization (baseline vIGA-AD score categories), and the continuous variable of baseline measurement.
Subgroup Analyses
The primary efficacy end points were analyzed in the following subgroups that were considered potential effect modifiers:
age group (< 40 years, ≥ 40 to < 65 years, and ≥ 65 years)
sex (male, female)
BMI (defined as normal = less than 25; overweight = 25 to < 30; obese = 30 or greater)
race (White, Asian, Black, and other)
weight (< median, ≥ median)
geographic regions (US, Puerto Rico, Canada; other)
baseline vIGA-AD score (moderate or milder [vIGA-AD ≤ 3], severe [vIGA-AD 4])
baseline EASI (< median, ≥ median)
hsCRP (< median, ≥ median)
previous systemic therapy (with and without).
Sensitivity Analyses
Sensitivity analyses were performed to assess the impact of missing data and the robustness of the conclusion. In this case, for co-primary and all key secondary end points, investigators used NRI-NC as a variable for the sensitivity analysis. In addition, sensitivity analyses were conducted based on the multiple imputation methods and per-protocol population.
Analysis Populations
The following populations were analyzed:
the ITT population, used for all efficacy analyses (patients were included in the analysis according to the treatment groups to which they were randomized)
the per-protocol population for the main study, which excluded patients with major protocol violations, such that the analysis included only those who had completed the study intervention (the primary efficacy end points were also analyzed in the per-protocol population as sensitivity analysis)
the safety population, consisting of all patients who received at least 1 dose of the study drug, including matching placebo.
For the safety population, patients were assigned to a treatment group based on the “as treated” treatment group, regardless of the treatment randomized. The “as treated” is determined by the treatment the patient received during the majority of the patient’s drug exposure time in the analysis period if any mis-dosing occurred.
Results
Patient Disposition
Monotherapy Regimen Studies
In the Measure Up 1 study, a total of 1,093 patients were screened for eligibility to enter the study. Of these, 847 patients were randomized at 151 study sites located in 24 countries, and 246 (22.5%) were considered screening failures. Reasons for screening failure included not meeting the eligibility criteria (76%), withdrawal of consent (19.1%), loss to follow-up (3.3%), and other (1.6%). All 847 patients who proceeded to the study (100%) received the study drug. A total of 778 patients (91.9%) completed the study through the double-blind period (week 16), and 782 patients (92.3%) participated in the study through week 16. There were no differential dropout rates between the 2 upadacitinib arms, but there were more discontinuations in the placebo arm (Table 18).
In the Measure Up 2 study, a total of 1,143 patients were screened for eligibility to enter the study. Of these, 836 patients were randomized at 154 study sites located in 23 countries, and 307 (26.8%) were screening failures. Reasons for screening failure included not meeting the eligibility criteria (84.4%), withdrawal of consent (12.1%), loss to follow-up (1.6%), and other (2%). All 836 patients who proceeded to the study (100%) received the study drug. A total of 764 (91.4%) completed the study (with or without rescue therapy) through the double-blind period (week 16), while 768 patients (91.9%) completed participated in the study through week 16. Sixty-seven patients discontinued the study drug in the double-blind period. The most frequent reasons for discontinuation of the study drug were withdrawal of consent in the upadacitinib 30 mg group, AEs in the upadacitinib 15 mg group, and lack of efficacy in the placebo group (Table 18).
Table 18: Patient Disposition — Measure Up 1 and 2 Studies
Patient disposition | Measure Up 1 | Measure Up 2 | ||||
|---|---|---|---|---|---|---|
Placebo | UPA 15 mg q.d. | UPA 30 mg q.d. | Placebo | UPA 15 mg q.d. | UPA 30 mg q.d. | |
Screened, N | 1,093 | 1,143 | ||||
Randomized, N | 281 | 281 | 285 | 278 | 276 | 282 |
Discontinued study drug, N (%) | 38 (13.5) | 8 (2.8) | 15 (5.3) | 37 (13.3) | 16 (5.8) | 14 (5.0) |
Reason for discontinuation, N (%) | ||||||
Adverse event | 12 (4.3) | 1 (0.4) | 8 (2.8) | 12 (4.3) | 11 (4.0) | 6 (2.1) |
Withdrawal of consent | 15 (5.3) | 2 (0.7) | 4 (1.4) | 11 (4.0) | 3 (1.1) | 5 (1.8) |
Loss to follow-up | 3 (1.1) | 4 (1.4) | 2 (0.7) | 2 (0.7) | 0 | 1 (0.4) |
Lack of efficacy | 16 (5.7) | 2 (0.7) | 0 2 (0.4) | 12 (4.3) | 3 (1.1) | 0 3 (0.5) |
EASI score: worsening of 25% criteria was met | 2 (0.7) | 0 | 0 | 1 (0.4) | 0 | 1 (0.4) |
Protocol-mandated discontinuation due to systemic rescue | 2 (0.7) | 0 | 1 (0.4) | 4 (1.4) | 2 (0.7) | 1 (0.4) |
Other | 3 (1.1) | 0 | 1 (0.4) | 7 (2.5) | 4 (1.4) | 2 (0.7) |
ITT, N (%) | 281 (100) | 281 (100) | 285 (100) | 278 (100) | 276 (100) | 282 (100) |
PP, N (%) | 271 (96.44) | 266 (94.66) | 271 (95.08) | 263 (94.60) | 269 (97.46) | 273 (96.80) |
Safety, N (%) | 281 (100) | 281 (100) | 285 (100) | 278 (100) | 276 (100) | 282 (100) |
EASI = Eczema Area and Severity Index; ITT = intention to treat; PP = per protocol; q.d. = once daily; UPA = upadacitinib.
Source: Clinical Study Reports for Measure Up 1 and Measure Up 2.11,12
Combination Therapy Regimen Studies
A total of 1,160 patients were screened for eligibility to enter the AD Up study. Of these, 901 patients were randomized and 259 (22.3%) were screening failures. Reasons for screening failure included not meeting the eligibility criteria (79.9%), withdrawal of consent (11.6%), loss to follow-up (6.9%), and other (1.5%). Among the 901 randomized patients, 900 patients (including 115 adolescents) were treated with the study drug at 171 sites located in 22 countries. One adolescent was randomized, but not treated.
Almost all patients completed study treatment in the double-blind period (94.8%). The primary reasons for discontinuation for all patients, including adolescents, were AEs; however, few patients were in this category (≤ 5 patients in any treatment group). Six patients who discontinued the study drug in the double-blind period continued to be followed in the study (as permitted by the protocol) while off study drug treatment. A total of 854 patients (97.4%), including 111 adolescents, continued into the BE period.
The disposition of patients in the AD Up study is presented in Table 19.
Table 19: Patient Disposition — AD Up Study
Patient disposition | Placebo + TCS | UPA 15 mg q.d.+ TCS | UPA 30 mg q.d.+ TCS |
|---|---|---|---|
Screened, N | 1,160 | ||
Randomized, N | 304 | 300 | 297 |
Never received study drug, N (%) | 1 (0.03) | 0 | 0 |
Discontinued study drug, N (%) | 21 (6.9) | 11 (3.7) | 10 (3.4) |
Reason for discontinuation, N (%) | |||
Adverse event | 6 (2.0) | 4 (1.3) | 3 (1.0) |
Withdrawal of consent | 5 (1.6) | 5 (1.7) | 2 (0.7) |
Loss to follow-up | 5 (1.6) | 3 (1.0) | 1 (0.3) |
Lack of efficacy | 4 (1.3) | 3 (1.0) | 0 3 (0.5) |
EASI score: worsening of 25% criteria was met | 0 | 0 | 0 |
Protocol-mandated discontinuationa | 1 (0.3) | 1 (0.3) | 0 1 (0.2) |
Other | 4 (1.3) | 1 (0.3) | 6 (2.0) |
ITT, N (%) | 304 (100) | 300 (100) | 297 (100) |
PP, N (%) | 275 (90.46) | 285 (95.00) | 278 (93.60) |
Safety, N (%) | 304 (100) | 300 (100) | 297 (100) |
EASI = Eczema Area and Severity Index; ITT = intention to treat; PP = per protocol; q.d. = once daily; TCS = topical corticosteroid; UPA = upadacitinib.
aDue to systemic rescue in the AD Up study; due to sponsor termination in the Heads Up study.
Head-to-Head Therapy Regimen Studies
In the Heads Up study, a total of 924 patients were screened for eligibility to enter. Of these, a total of 692 patients were randomized at 129 sites located in 22 countries, while 232 (25.1%) were screening failures. Reasons for screening failure included not meeting the eligibility criteria (65.5%), withdrawal of consent (18.1%), loss to follow-up (0.9%), logistical reasons related to COVID-19 (6%), and other (9.5%).
At week 16 (the evaluation of the primary end point), 93.5% of patients had completed 16 weeks of the study drug. At the final visit, (week 24 plus a 12-week follow-up after the last injection, for those who did not enrol in the open-label extension study), 91.8% of patients had completed the study drug. More patients in the upadacitinib group discontinued the study drug due to AEs versus the dupilumab group.
The disposition of patients in the Heads Up study is presented in Table 20.
Table 20: Patient Disposition — Heads Up Study
Patient disposition | Dupilumab 300 mg 2.q.w. | Upadacitinib 30 mg q.d. |
|---|---|---|
Screened, N | 924 | |
Randomized, N | 344 | 348 |
Never received study drug, N (%) | 0 | 0 |
Discontinued study drug, N (%) | 25 (7.3) | 32 (9.2) |
Reason for discontinuation, N (%) | ||
Adverse event | 4 (1.2) | 11 (3.2) |
Withdrawal of consent | 7 (2.0) | 9 (2.6) |
Loss to follow-up | 6 (1.7) | 4 (1.1) |
Lack of efficacy | 3 (0.9) | 6 (1.7) |
EASI score: worsening of 25% criteria was met | 0 | 0 |
Protocol-mandated discontinuationa | 0 | 0 |
Other | 6 (1.7) | 3 (0.9) |
ITT, N (%) | 344 (100) | 348 (100) |
PP, N (%) | 311 (90.40) | 310 (89.08) |
Safety, N (%) | 344 (100) | 348 (100) |
2.q.w. = every 2 weeks; EASI = Eczema Area and Severity Index; ITT = intention to treat; PP = per protocol; q.d. = once daily.
aDue to systemic rescue in the AD Up study; due to sponsor termination in the Heads Up study.
Exposure to Study Treatments
Monotherapy Regimen Studies
In the Measure Up 1 study, during the double-blind period, the mean durations of exposure to study drug were 102.4 days in the placebo group, 110.6 days in the upadacitinib 15 mg group, and 109.2 days in the upadacitinib 30 mg group. The extent of exposure in adolescents was similar to that of the overall population in the double-blind period. Through the data cut-off, for any patient who received at least 1 dose of upadacitinib, the mean duration of exposure was similar between the upadacitinib 30 mg and 15 mg groups. Overall, the extent of exposure to upadacitinib in adolescents was similar to that of the overall population in the main study. An exposure of greater than or equal to 24 weeks occurred in 64.3% and 65.0% of patients in the upadacitinib 15 mg and upadacitinib 30 mg groups, respectively. In total, 139 patients (17.3%), including 20 adolescent patients (16.8%), were treated with upadacitinib for a minimum of 52 weeks. Concomitant medication usage was similar across all treatment groups, with emollients and protectives being the most frequently reported concomitant medications (27.5% and 27.4% of patients in the double-blind and BE periods). Adherence was calculated as the number of tablets actually taken by the patient divided by the number of tablets planned to be taken by the patient during the double-blind and BE periods of the study, respectively. Adherence rates were high, with mean compliance greater than 96% and median compliance greater than 99% in all 3 groups. Rescue medications in the double-blind period were used in 133 (47.3%), 32 (11.4%), and 19 (6.7%) patients in the placebo, upadacitinib 15 mg, and upadacitinib 30 mg groups, respectively.
In the Measure Up 2 study, during the double-blind period, the mean duration of exposure to study drug was 101.4 days in the placebo group, 109.6 days in the upadacitinib 15 mg group, and 109.7 days in the upadacitinib 30 mg group. The extent of exposure in adolescents was similar to the overall population. Through the data cut-off, for any patient who received at least 1 dose of upadacitinib, the mean duration of exposure was similar between the upadacitinib 30 mg and 15 mg groups. An exposure of greater than or equal to 24 weeks occurred in 62.3% and 64.4% of patients in the upadacitinib15 mg and 30 mg groups, respectively. At greater than or equal to 52 weeks duration, 9.1% and 12.0% of patients in the 15 mg and 30 mg groups, respectively, were treated with upadacitinib. Overall, the extent of exposure to upadacitinib in adolescents was numerically higher than for the overall population in the main study. As with the Measure Up 1 study, emollients and protectives were the most frequently reported prior medications (used by 39.2% of patients) and concomitant medications (used by 38.9% and 37.9% of patients in the double-blind and BE period, respectively). Adherence was calculated as the number of tablets actually taken divided by the number of tablets planned to be taken by the patient during the double-blind and BE periods of the study, respectively. Adherence rates were high in the double-blind period, with mean compliance greater than 95% and median compliance greater than 98% in all 3 groups. Rescue medications in the double-blind period were used in 120 (43.2%), 25 (9.1%), and 16 (5.7%) patients in the placebo, upadacitinib 15 mg, and upadacitinib 30 mg groups, respectively.
Combination Therapy Regimen Studies
For the AD Up study, in patients (including adolescents) who received at least 1 dose of upadacitinib, the mean duration of exposure was similar between the groups receiving upadacitinib 30 mg plus TCS and those receiving 15 mg plus TCS. At week 24, the exposures of patients to upadacitinib were 93.5% and 95.2% in the group receiving 15 mg plus TCS and the group receiving 30 mg plus TCS group, respectively. The majority (73.7%) of patients were treated with upadacitinib for 52 weeks, while 274 patients (31.2%), including 45 adolescents, were treated with upadacitinib for a minimum of 72 weeks.
In the AD Up study, adherence was calculated as the number of tablets taken divided by the number of tablets planned to be taken by each patient during the double-blind and BE periods of the study, respectively. Adherence rates in the double-blind period were 94.05%, 95.75%, and 95.85% in the groups receiving placebo plus TCS, upadacitinib 15 mg plus TCS, and upadacitinib 30 mg plus TCS, respectively, while in the BE period, the mean adherence was greater than 92% and the median adherence was greater than 97% in all groups.
More than half of the patients (54.6.%) had received prior non-biologic immunomodulating systemic therapies and only 30 patients (3.3%) had received prior biologic systemic therapies (Table 19). Rescue medication was used in 78 patients (25.7%), 16 patients (5.3%), and 16 patients (5.4%) in the groups receiving placebo plus TCS, upadacitinib 15 mg plus TCS, and upadacitinib 30 mg plus TCS, respectively. The most common medium-potency TCS therapies used during the study were triamcinolone and mometasone. The most common low-potency TCS used during the study was hydrocortisone.
Head-to-Head Therapy Regimen Studies
In the Heads Up study, before week 16, the mean extents of exposure were 110 days (standard deviation [SD] = 12.29) in the upadacitinib group and 108 days (SD = 17.32) in the dupilumab group. The median duration of treatment was 112 days for both groups. Through the end of the study, the mean durations of exposure were 162.5 days (SD = 24.66) in the upadacitinib group and 160.4 days (SD = 29.90) in the dupilumab group.
Adherence was summarized by treatment in the safety population. Dupilumab adherence was defined as the number of dupilumab injections administered during the patient's participation up to week 22 divided by the number of injections planned to be administered by the patient during the study. Upadacitinib compliance was defined as the number of upadacitinib tablets actually taken by the patient divided by the number of tablets planned to be taken by the patient during the study. Adherence rates were similar in both treatment groups. At week 16, mean and median compliance rates of approximately 96% and 98%, respectively, were reached in the upadacitinib group, while mean and median compliance rates of 99% and 100%, respectively, were reached in the dupilumab group. Through the end of study, the mean and median compliance rates were approximately 95% and 98% in the upadacitinib group and 99% and 100% in the dupilumab group, respectively.
Most of the patients (95.1%) entered the study with an inadequate response or loss of response to prior AD therapy. A total of 87.7% of patients had received at least 1 prior systemic therapy (Table 20). Concomitant medications were similarly distributed by class and in the 2 groups of study. Any rescue medication was used by 85 patients (24.7%) and 87 patients (25.0%) in the dupilumab and upadacitinib arms, respectively. Of the patients who received rescue medication, most received a TCS therapy (17.3% before week 16, and 22.3% through the end of the study).
Efficacy
Only those efficacy outcomes and analyses of subgroups identified in the review protocol are reported here. Refer to Appendix 3 for detailed efficacy data in adolescents.
Severity of Disease
Measurements of the severity of AD were considered a critical end point by the clinical expert consulted by CADTH; the measurements proposed in the protocol (i.e., the EASI score, the vIGA-AD score, and SCORAD score) were deemed appropriate to evaluate disease severity, to detect any significant change due to the interventions, and to serve as benchmarks for decision-making. The studies included in this CADTH review assessed the EASI scores together with the vIGA-AD scores as co-primary outcomes, except for the Heads Up and Japan trials, which evaluated these outcomes separately. These co-primary end points were evaluated using the ITT population.
Eczema Area and Severity Index
Monotherapy Regimen Studies
The results of the EASI score measurement at week 16 (as a co-primary outcome in the Measure Up 1 study) are presented in Table 21. A statistically significantly larger proportion of patients in the upadacitinib groups achieved EASI 75 at week 16 compared with the placebo group based on the primary approach of NRI-C. Forty-six patients (16.3%), 196 patients (69.6%), and 227 patients (79.7%) responded in the placebo, upadacitinib 15 mg, and upadacitinib 30 mg groups, respectively, with adjusted differences versus placebo of 63.4% (95% CI, 57.1 to 69.8) and 63.4% (95% CI, 57.1 to 69.8) in the upadacitinib 15 mg and upadacitinib 30 mg groups, respectively (P < 0.001 for all comparisons).
Table 21: Efficacy Outcomes in the Measure Up 1 Study — Co-Primary End Points for All Patients
Outcome | Placebo N = 281 | UPA 15 mg q.d. N = 281 | UPA 30 mg q.d. N = 285 |
|---|---|---|---|
EASI 75 at week 16a | |||
N (%) | 281 (100) | 281 (100) | 285 (100) |
Responders, n (%) | 46 (16.3) | 196 (69.6) | 227 (79.7) |
95% CI | 12.0 to 20.7 | 64.2 to 75.0 | 75.0 to 84.4 |
Adjusted difference vs. placebo, % (95% CI)b | — | 53.3 (46.4 to 60.2) | 63.4 (57.1 to 69.8) |
P value | — | < 0.001 | < 0.001 |
Subgroup: with previous systemic therapies | |||
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
vIGA-AD response 0 or 1 at week 16a | |||
N (%) | 281 (100) | 281 (100) | 285 (100) |
Responders, n (%) | 24 (8.4) | 135 (48.1) | 177 (62.0) |
95% CI | 5.2 to 11.7 | 42.3 to 54.0 | 56.4 to 67.7 |
Adjusted difference vs. placebo, % (95% CI)b | — | 39.8 (33.2 to 46.4) | 53.6 (47.2 to 60.0) |
P value | — | < 0.001 | < 0.001 |
Subgroup: with previous systemic therapies | |||
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
AD = atopic dermatitis; CI = confidence interval; EASI = Eczema Area and Severity Index; EASI 75 = at least 75% improvement in EASI total score; ITT = intention to treat; q.d. = once daily; UPA = upadacitinib; vIGA-AD = validated Investigator Global Assessment; vs. = versus.
aAssessed in the ITT population.
b95% CI for adjusted difference and P values are calculated according to the Cochran-Mantel-Haenszel test adjusted for strata (baseline vIGA-AD score categories and age [adolescent vs. adult]) for the comparison of 2 treatment groups.
Note: Redacted rows have been deleted.
Source: Clinical Study Report: Measure Up 1.12
Similar results were found in the Measure Up 2 study, as shown in Table 22. A statistically significantly larger proportion of patients in the upadacitinib groups achieved EASI 75 at week 16 compared with the placebo group, based on the primary approach of NRI-C. In this study, 37 patients (13.3%), 166 patients (60.1%), and 206 patients (72.9%) in the placebo, upadacitinib 15 mg, and upadacitinib 30 mg groups, respectively, were considered responders based on EASI 75, with adjusted differences versus placebo of 46.9% (95% CI, 39.9 to 53.9) and 59.6% (95% CI, 53.1 to 66.2) in the upadacitinib 15 mg and 30 mg groups, respectively (P < 0.001 for all comparisons).
Table 22: Efficacy Outcomes in the Measure Up 2 Study — Co-Primary End Points for All Patients
Outcome | Placebo N = 278 | UPA 15 mg q.d. N = 276 | UPA 30 mg q.d. N = 282 |
|---|---|---|---|
EASI 75 at week 16a | |||
N (%) | 278 (100.00) | 276 (100.00) | 282 (100.00) |
Responders, n (%) | 37 (13.3) | 166 (60.1) | 206 (72.9) |
95% CI | (9.3 to 17.3) | (54.4 to 65.9) | (67.7 to 78.2) |
Adjusted difference vs. placebo, % (95% CIb | — | 46.9 (39.9 to 53.9) | 59.6 (53.1 to 66.2) |
P value | — | < 0.001 | < 0.001 |
Subgroup: with previous systemic therapies | |||
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
vIGA-AD response 0 or 1 at week 16a | |||
N (%) | 278 (100.00) | 276 (100.00) | 282 (100.00) |
Responders, n (%) | 13 (4.7) | 107 (38.8) | 147 (52.0) |
95% CI | (2.2 to 7.2) | (33.0 to 44.5) | (46.1 to 57.9) |
Adjusted difference vs. placebo, % (95% CIb | — | 34.0 (27.8 to 40.2) | 47.4 (41.0 to 53.7) |
P value | — | < 0.001 | < 0.001 |
Subgroup: with previous systemic therapies | |||
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
CI = confidence interval; EASI = Eczema Area and Severity Index; EASI 75 = at least 75% improvement in EASI total score; ITT = intention to treat; q.d. = once daily; UPA = upadacitinib; vIGA-AD = validated Investigator Global Assessment; vs. = versus.
aAssessed in the ITT population.
b95% CI for adjusted difference and P values are calculated according to the Cochran-Mantel-Haenszel test adjusted for strata (baseline vIGA-AD score categories and age [adolescent vs. adult]) for the comparison of 2 treatment groups.
Note: Redacted rows have been deleted.
Source: Clinical Study Report: Measure Up 2.11
The primary end point results in both studies were supported by all sensitivity analyses, including NRI-NC, multiple imputation, tipping point analysis, and per-protocol analysis. Treatment effects in all pre-specified subgroups (across demographic and baseline characteristics), including in adolescents, consistently favoured both upadacitinib doses versus placebo in EASI 75 (refer to Appendix 3), all with 95% CIs, excluding 0. The overall type I error rate was controlled at the 0.05 (2-sided) level using a pre-specified graphical approach, as described previously.
Combination Therapy Regimen Studies
The results of the EASI 75 measurement at week 16 as a co-primary outcome in the AD Up study are presented in Table 23.
A statistically significantly larger proportion of patients in the upadacitinib groups compared to placebo achieved EASI 75 at week 16 based on the primary approach of NRI-C, with 80 patients (26.4%), 194 patients (64.6%), and 229 patients (77.1%) responding in the groups receiving placebo plus TCS, upadacitinib 15 mg plus TCS, and 30 mg plus TCS, respectively. The adjusted differences versus placebo were 38.1% (95% CI, 30.8 to 45.4) and 50.6% (95% CI, 43.8 to 57.4) in the groups receiving upadacitinib 15 mg plus TCS and 30 mg plus TCS, respectively (P < 0.001 for all comparisons).
This differences in the proportions of patients in the upadacitinib groups achieving better performance on EASI 75 at week 16 compared with the placebo group were based on the primary approach of NRI-NC. The co-primary end point results were supported by all sensitivity analyses, including NRI-NC, multiple imputation, tipping point analysis, and per-protocol analysis. Treatment effects in all pre-specified subgroups (across demographic and baseline characteristics), including in adolescents, consistently favoured both groups receiving upadacitinib plus TCS versus placebo for EASI 75, all with 95% CIs (excluding 0). The results by subgroup are presented in Appendix 3. The overall type I error rate was controlled at the 0.05 (2-sided) level using a pre-specified graphical approach, as described previously.
Head-to-Head Studies
The results of the EASI 75 score measurement at week 16 as a primary outcome in the Heads Up study are presented in Table 24. The analyses were conducted when all ongoing patients completed week 24 (the primary analysis). This was the only and final analysis for efficacy. This analysis used the ITT population. A statistically significantly larger proportion of patients in the upadacitinib group achieved EASI 75 from baseline at week 16 compared with the dupilumab group, with 210 patients (61.1%) and 247 patients (71.0%) classified as responders in the dupilumab and upadacitinib 30 mg groups, respectively. The adjusted difference between groups was 10.0% (95% CI, 2.9 to 17.0; P = 0.006). The primary end point result was supported by sensitivity analyses, including multiple imputation, NRI-NC, and per-protocol population analyses. Treatment effects in the pre-specified subgroups consistently favoured upadacitinib compared to dupilumab in EASI 75 at week 16 (refer to Appendix 3), with the upadacitinib group having a higher numerical response rate.
A multiple testing procedure was used to provide control of the type I error rate at alpha = 0.05 (2-sided) across analyses comparing upadacitinib versus dupilumab for the primary and ranked secondary end points.
Validated Investigator Global Assessment for Atopic Dermatitis
Monotherapy Regimen Studies
The results of the vIGA-AD score measurement at week 16 as a co-primary outcome in the Measure Up 1 study are presented in Table 21. A statistically significantly larger proportion of patients in the upadacitinib groups achieved a vIGA-AD score of 0 or 1 (clear or almost clear) with a clinically meaningful score reduction (defined by the investigator as at least 2 grade reductions from baseline) at week 16 compared with the placebo group, based on the primary approach of NRI-C, with 24 patients (8.4%), 135 patients (48.1%), and 177 patients (62.0%) responding in the groups receiving placebo, upadacitinib 15 mg, and upadacitinib 30 mg, respectively, with an adjusted difference versus placebo of 39.8% (95% CI, 33.2 to 46.4) and 53.6 (95% CI, 47.2 to 60.0) in the upadacitinib 15 mg and 30 mg groups, respectively (P < 0.001 for all comparisons).
Similar results were found in the Measure Up 2 study, as shown in Table 22. Compared with the placebo group, a statistically significantly larger proportion of patients in the upadacitinib groups achieved vIGA-AD scores of 0 or 1 (clear or almost clear), with clinically meaningful score reductions (defined by the investigator as at least 2 grade reductions from baseline), based on the primary approach of NRI-C. In this study, 13 patients (4.7%), 107 patients (38.8%), and 147 patients (52.0%) in the groups receiving placebo, upadacitinib 15 mg, and upadacitinib 30 mg, respectively, were considered responders based on the vIGA-AD score, with an adjusted difference versus placebo of 34.0% (95% CI, 27.8 to 40.2) and 47.4% (95% CI, 41.0 to 53.7) in the upadacitinib 15 mg and upadacitinib 30 mg groups, respectively (P < 0.001 for all comparisons).
The primary end point results in both studies were supported by all sensitivity analyses, including NRI-NC, multiple imputation, tipping point analysis, and per-protocol analysis. Treatment effects in all pre-specified subgroups (across demographic and baseline characteristics), including in adolescents, consistently favoured both upadacitinib doses compared to placebo in vIGA-AD scores (refer to Appendix 3), all with 95% CIs (excluding 0). The overall type I error rate was controlled at the 0.05 (2-sided) level using a pre-specified graphical approach, as described previously.
Combination Therapy Regimen Studies
The results of the vIGA-AD score measurements at week 16 in the AD Up study are presented in Table 23.
Table 23: Efficacy Outcomes in the AD Up Study — Co-Primary End Points
Outcome | Placebo + TCS N = 304 | UPA 15 mg q.d.+ TCS N = 300 | UPA 30 mg q.d. + TCS N = 297 |
|---|---|---|---|
EASI 75 at week 16a | |||
N (%) | 304 (100.00) | 300 (100.00) | 297 (100.00) |
Responders, n (%) | 80 (26.4) | 194 (64.6) | 229 (77.1) |
95% CI | (21.5 to 31.4) | (59.1 to 70.0) | (72.3 to 81.9) |
Adjusted difference vs. placebo, % (95% CI)b | — | 38.1 (30.8 to 45.4) | 50.6 (43.8 to 57.4) |
P value | — | < 0.001 | < 0.001 |
Subgroup: with previous systemic therapies | |||
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
vIGA-AD response 0 or 1 at week 16a | |||
N (%) | 304 (100.00) | 300 (100.00) | 297 (100.00) |
Responders, n (%) | 33 (10.9) | 119 (39.6) | 174 (58.6) |
95% CI | (7.4 to 14.4) | (34.1 to 45.2) | (53.0 to 64.2) |
Adjusted difference vs. placebo, % (95% CI)b | — | 28.5 (22.1 to 34.9) | 47.6 (41.1 to 54.0) |
P value | — | < 0.001 | < 0.001 |
Subgroup: with previous systemic therapies | |||
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
CI = confidence interval; EASI = Eczema Area and Severity Score; EASI 75 = at least 75% improvement in EASI total score; ITT = intention to treat; q.d. = once daily; TCS = topical corticosteroid; UPA = upadacitinib; vIGA-AD = validated Investigator Global Assessment; vs. = versus.
aAssessed in the ITT population.
b95% CI for adjusted difference and P values are calculated according to the Cochran-Mantel-Haenszel test adjusted for strata (baseline vIGA-AD score categories and age [adolescent vs. adult]) for the comparison of 2 treatment groups.
Note: Redacted rows have been deleted.
Source: Clinical Study Report: AD Up.13
Compared with the placebo group, a statistically significantly larger proportion of patients in the upadacitinib groups achieved a vIGA-AD score of 0 or 1 (clear or almost clear) with a clinically meaningful reduction (defined by the investigator as at least 2 grade reductions from baseline) at week 16, based on the primary approach of NRI-C, with 33 patients (10.9%), 119 patients (39.6%), and 174 patients (58.6%) responding in the groups receiving placebo plus TCS, upadacitinib 15 mg plus TCS, and upadacitinib 30 mg plus TCS, respectively. The adjusted differences versus placebo were 28.5% (95% CI, 22.1 to 34.9) and 47.6% (95% CI, 41.1 to 54.0) in the groups receiving upadacitinib 15 mg plus TCS and upadacitinib 30 mg plus TCS, respectively (P < 0.001 for all comparisons). The differences in the proportions of patients in the upadacitinib groups achieving better performances on the vIGA-AD at week 16 compared with the placebo group was based on the primary approach of NRI-C.
The co-primary end point results were supported by all sensitivity analyses, including NRI-NC, multiple imputation, tipping point analysis, and per-protocol analysis.
Treatment effects in all pre-specified subgroups (across demographic and baseline characteristics), including in adolescents, consistently favoured the groups receiving upadacitinib plus TCS versus placebo in terms of vIGA-AD score, with all 95% CIs (excluding 0); the exception was the race subgroup category “other,” which consisted of Black or African American, Indian or Alaska Native, Native Hawaiian or other Pacific Islander, or multiple. In the race subgroup, the 95% CIs of “other” included 0 for both upadacitinib doses (30 mg and 15 mg). In the race subgroup, the 95% CI of “other” included 0 for both upadacitinib doses (30 mg and 15 mg) for EASI 75 and for the upadacitinib 15 mg dose for vIGA-AD score of 0 or 1. It should be noted that the number of patients in this category was low (69 patients). The results by subgroup are presented in Appendix 3. The overall type I error rate was controlled at the 0.05 (2-sided) level using a pre-specified graphical approach, as described previously.
Head-to-Head Studies
The vIGA-AD score was not evaluated as a primary or secondary end point in the Heads Up study.
Table 24: Efficacy Outcomes in the Heads Up Study — Primary End Point
Outcome | DUP 300 mg 2.q.w. N = 344 | UPA 30 mg q.d. N = 348 |
|---|---|---|
EASI 75 at week 16a | ||
N (%) | 344 (100.00) | 348 (100.00) |
Responders, n (%) | 210 (61.1) | 247 (71.0) |
95% CI | (55.9 to 66.2) | (66.2 to 75.8) |
Adjusted difference, % (95% CI)b | 10.0 (2.9 to 17.0) | |
P valueb | 0.006 | |
Subgroup: with previous systemic therapies | ||
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
2.q.w. = every 2 weeks; CI = confidence interval; DUP = dupilumab; EASI = Eczema Area and Severity Index; EASI 75 = at least 75% improvement in EASI total score; ITT = intention to treat; q.d. = once daily; UPA = upadacitinib; vs. = versus.
aAssessed in the ITT population.
b95% CI for adjusted difference and P values are calculated according to the Cochran-Mantel-Haenszel test adjusted for strata (baseline vIGA-AD score categories and age [adolescent vs. adult]) for the comparison of 2 treatment groups.
Note: Redacted rows have been deleted.
Source: Clinical Study Report: Heads Up.14
The following end points were all noted as secondary end points in the trials. Of note, these end points were often assessed only in a specific subset of patients or using a complete case analysis, as noted in the table footnotes.
Scoring Atopic Dermatitis
Monotherapy Regimen Studies
The SCORAD was measured in the Measure Up 1 study as a key secondary end point under the overall type I error control at the 0.05 (2-sided) level using a pre-specified graphical approach, as described previously. The superiority of each upadacitinib dose versus placebo was demonstrated in the measurement of the SCORAD. The results of the SCORAD measurement at week 16 as a key secondary outcome in the Measure Up 1 study is presented in Table 25. The LS mean changes from baseline were –32.68% (95% CI, –37.26 to –28.11), –65.71% (95% CI, –69.20 to –62.23), and –73.07% (95% CI, –76.47 to –69.68) in the groups receiving placebo, upadacitinib 15 mg, and upadacitinib 30 mg, respectively, with mean differences versus the placebo group of –33.03% (95% CI, –38.44 to –27.61) and –40.39% (95% CI, –45.75 to –35.03) in the groups receiving upadacitinib 15 mg and upadacitinib 30 mg, respectively (P < 0.001 for all comparisons). The proportion of patients who achieved SCORAD 50, 75, or 90 continued to increase from week 2 to week 16 in patients on upadacitinib 30 mg and 15 mg compared to placebo. This pattern was consistent with the percentage change in improvements in SCORAD and its individual components (i.e., objective SCORAD, SCORAD itch, and SCORAD sleep). No subgroup analyses or sensitivity analyses were performed for the SCORAD outcome or other key secondary outcomes.
Table 25: Efficacy Outcomes in the Measure Up 1 Study — Key Secondary and Exploratory End Points for All Patients
Outcome | Placebo N = 281 | UPA 15 mg q.d. N = 281 | UPA 30 mg q.d. N = 285 |
|---|---|---|---|
Disease severity | |||
EASI 90 at week 16a | |||
N (%) | 281 (100) | 281 (100) | 285 (100) |
Responders, n (%) | 23 (8.1) | 149 (53.1) | 187 (65.8) |
% responders – 95% CI | 4.9 to 11.3 | 47.2 to 58.9 | 60.2 to 71.3 |
Adjusted difference vs. placebo, % (95% CI) | Reference | 45.1 (38.6 to 51.7) | 57.8 (51.5 to 64.1) |
P value | Reference | < 0.001 | < 0.001 |
Change in EASI score from baseline at week 16b | |||
N (%) | 128 (45.55) | 244 (86.83) | 259 (90.87) |
Baseline mean | 26.47 | 30.39 | 29.29 |
At week 16 mean | 11.33 | 4.87 | 3.38 |
LS mean % change from baseline (95% CI)c,d | –40.71 (–45.18 to –36.23) | –80.24 (–83.99 to –76.49) | –87.74 (–91.42 to –84.06) |
Adjusted LS mean difference from placebo (95% CI)c,d | Reference | –39.53 (–44.91 to –34.15) | –47.03 (–52.37 to –41.70) |
Change in overall SCORAD score from baseline at week 16b | |||
N (%) | 125 (44.48) | 239 (85.05) | 253 (88.77) |
Baseline mean | 64.428 | 68.272 | 67.369 |
At week 16 mean | 40.735 | 21.728 | 17.723 |
LS mean % change from baseline (95% CI)c,d | –32.68 (–37.26 to –28.11) | –65.71 (–69.20 to –62.23) | –73.07 (–76.47 to –69.68) |
Adjusted LS mean difference from placebo (95% CI)c,d | Reference | –33.03 (–38.44 to –27.61) | –40.39 (–45.75 to –35.03) |
Symptoms | |||
WP-NRS response ≥ 4 from baseline at week 16a | |||
N (%)e | 272 (96.80) | 274 (97.51) | 280 (98.25) |
Responders, n (%) | 32 (11.8) | 143 (52.2) | 168 (60.0) |
% responders – 95% CI | (7.9 to 15.6) | (46.3 to 58.1) | (54.3 to 65.7) |
Adjusted difference vs. placebo, % (95% CI)d | Reference | 40.5 (33.5 to 47.5) | 48.2 (41.3 to 55.0) |
P value d | Reference | < 0.001 | < 0.001 |
% Change in WP-NRS from baseline at week 16b | |||
N (%)e | 123 (43.77) | 225 (80.07) | 236 (82.81) |
Baseline mean | 7.256 | 7.232 | 7.290 |
At week 16 mean | 4.886 | 2.518 | 2.010 |
LS mean % change from baseline (95% CI)c,d | –26.06 (–36.66 to –15.46) | –62.79 (–71.60 to | –72.04 (–80.69 to –63.39) |
Adjusted LS mean difference from placebo (95% CI)c,d | Reference | –36.74 (–49.66 to | –45.98 (–58.82 to –33.15) |
POEM total score improvement (≥ 4) from baseline at week 16a | |||
N (%)e | 276 (98.22) | 278 (98.93) | 280 (98.25) |
Responders, n (%) | 63 (22.8) | 209 (75.0) | 228 (81.4) |
% responders – 95% CI | (17.8 to 27.8) | (69.9 to 80.1) | (76.9 to 86.0) |
Adjusted difference vs. placebo, % (95% CI)d | Reference | 52.3 (45.2 to 59.4) | 58.6 (51.9 to 65.3) |
P valued | Reference | < 0.001 | < 0.001 |
ADerm-SS skin pain improvement (≥ 4) from baseline at week 16b | |||
N (%)e | 233 (82.92) | 237 (84.34) | 249 (87.37) |
Responders, n (%) | 35 (15.0) | 127 (53.6) | 158 (63.5) |
% responders – 95% CI | (10.4 to 19.6) | (47.2 to 59.9) | (57.5 to 69.4) |
Adjusted difference vs placebo, % (95% CI)d | Reference | 38.7 (30.9 to 46.5) | 48.6 (41.0 to 56.1) |
P valued | Reference | < 0.001 | < 0.001 |
ADerm-SS TSS-7 improvement (≥ 28) from baseline at week 16b | |||
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
ADerm-IS sleep domain score improvement (≥ 12) from baseline at week 16b | |||
N (%) | 220 (78.29) | 218 (77.58) | 218 (76.49) |
Responders, n (%) e | 29 (13.2) | 120 (55.0) | 144 (66.1) |
% responders – 95% CI | (8.7 to 17.7) | (48.4 to 61.6) | (59.8 to 72.3) |
Adjusted difference vs. placebo, % (95% CI)d | Reference | 41.8 (33.9 to 49.7) | 52.9 (45.2 to 60.6) |
P valued | Reference | < 0.001 | < 0.001 |
ADerm-IS daily activities domain score improvement (≥ 14) from baseline at week 16b | |||
N (%) e | 197 (70.11) | 203 (72.24) | 205 (71.93) |
Responders, n (%) | 40 (20.3) | 132 (65.0) | 150 (73.2) |
% responders – 95% CI | (14.7 to 25.9) | (58.5 to 71.6) | (67.1 to 79.2) |
Adjusted difference vs placebo, % (95% CI)d | Reference | 44.7 (36.2 to 53.2) | 53.1 (44.9 to 61.3) |
P valued | Reference | < 0.001 | < 0.001 |
ADerm-IS emotional state domain score improvement (≥ 11) from baseline at week 16b | |||
N (%)e | 212 (75.44) | 227 (80.78) | 226 (79.30) |
Responders, n (%) | 42 (19.8) | 142 (62.6) | 164 (72.6) |
% responders – 95% CI | (14.4 to 25.2) | (56.3 to 68.9) | (66.7 to 78.4) |
Adjusted difference vs. placebo, % (95%CI)d | Reference | 42.7 (34.4 to 50.9) | 52.5 (44.7 to 60.4) |
P valued | Reference | < 0.001 | < 0.001 |
HRQoL | |||
DLQI improvement (≥ 4) from baseline at week 16b | |||
N (%)e | 250 (88.97) | 254 (90.39) | 256 (89.82) |
Responders, n (%) | 73 (29.0) | 192 (75.4) | 210 (82.0) |
% responders – 95% CI | (23.3 to 34.7) | (70.1 to 80.8) | (77.3 to 86.7) |
Adjusted difference vs. placebo, % (95%CI)d | Reference | 46.7 (39.0 to 54.4) | 53.2 (45.9 to 60.5) |
P valued | Reference | < 0.001 | < 0.001 |
DLQI score of 0 or 1 at week 16b | |||
N (%)e | 252 (89.68) | 258 (91.81) | 261 (91.58) |
Responders, n (%) | 11 (4.4) | 78 (30.3) | 108 (41.5) |
% responders – 95% CI | (1.9 to 7.0) | (24.7 to 35.9) | (35.5 to 47.4) |
Adjusted difference vs. placebo, % (95%CI)d | Reference | 25.9 (19.7 to 32.1) | 37.3 (30.8 to 43.8) |
P valued | Reference | < 0.001 | < 0.001 |
Change in EQ-5D index from baseline at week 16b,c | |||
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
Mood | |||
HADS-A or HADS-D response (< 8) at week 16b | |||
N (%)e | 126 (44.84) | 145 (51.60) | 144 (50.53) |
Responders, n (%) | 18 (14.3) | 66 (45.5) | 71 (49.2) |
% responders – 95% CI | (8.2 to 20.4) | (37.4 to 53.6) | (41.0 to 57.4) |
Adjusted difference vs. placebo, % (95%CI)d | Reference | 31.5 (21.4 to 41.6) | 34.9 (24.8 to 45.1) |
P valued | Reference | < 0.001 | < 0.001 |
Productivity | |||
Change in WPAI:AD (work productivity loss) domain scoresb,f | |||
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
Change in WPAI:AD (absenteeism) domain scoresb,f | |||
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
ADerm-IS = Atopic Dermatitis Impact Scale; ADerm-SS = Atopic Dermatitis Symptom Scale; ANCOVA = analysis of covariance; CI = confidence interval; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; EASI 90 = at least 90% improvement in EASI total score from baseline; HADS-A = Hospital Anxiety and Depression Scale – Anxiety; HADS-D = Hospital Anxiety and Depression Scale – Depression; HRQoL = health-related quality of life; ITT = intention to treat; LS = least squares; POEM = Patient-Oriented Eczema Measure; q.d. = once daily; UPA = upadacitinib; vIGA-AD = validated Investigator Global Assessment for Atopic Dermatitis; SCORAD = Scoring Atopic Dermatitis; TSS-7 = 7-Item Total Symptom Score; vs. = versus; WP-NRS = Worst Pruritus Numerical Rating Scale; WPAI:AD = Work Productivity and Activity Impairment.
aAssessed in the ITT population with nonresponder imputation incorporating multiple imputation to handle data missing due to COVID-19.
bComplete case analysis.
cMixed-effects model for repeated measures with baseline, treatment, visit, treatment-by-visit interaction, and strata (baseline vIGA-AD score categories and age [adolescent vs. adult]) in the model.
d95% CI for adjusted difference and P values are calculated according to the Cochran-Mantel-Haenszel test adjusted for strata (baseline vIGA-AD score categories and age [adolescent vs. adult]) for the comparison of 2 treatment groups.
eThe calculations at each visit are based on nonresponder imputation incorporating multiple imputation to handle data missing due to COVID-19 or nonresponder imputation only if there are no data missing due to COVID-19.
fWithin-group LS mean and 95% CI as well as between-group LS mean, 95% CI, and P value are calculated using ANCOVA with baseline, treatment, and strata (baseline vIGA-AD score categories and age [adolescent vs. adult]) in the model.
Note: Redacted rows have been deleted.
Source: Clinical Study Report: Measure Up 1.12
Similar results were found in the Measure Up 2 study, as shown in Table 26, with a significantly superior percentage change from baseline in the upadacitinib groups versus placebo. The LS mean changes from baseline were –28.43% (95% CI, 33.34 to –23.52), –57.90% (95% CI, –61.84 to –53.97), and –68.44% (95% CI, –72.44 to –64.44) in the groups receiving placebo, upadacitinib 15 mg, and upadacitinib 30 mg, respectively, with mean differences compared to the placebo group of –29.47% (95% CI, –35.24 to –23.69) and –40.01% (95% CI, –45.80 to –34.22) in the groups receiving upadacitinib 15 mg and upadacitinib 30 mg groups, respectively (P < 0.001 for all comparisons). No subgroup analyses or sensitivity analyses were performed for the SCORAD outcome or other key secondary outcomes.
Table 26: Efficacy Outcomes in the Measure Up 2 Study — Key Secondary and Exploratory End Points for All Patients
Outcome | Placebo N = 278 | UPA 15 mg q.d. N = 276 | UPA 30 mg q.d. N = 282 |
|---|---|---|---|
Disease severity | |||
EASI 90 at week 16a | |||
N (%) | 278 (100.00) | 276 (100.00) | 282 (100.00) |
Responders, n (%) | 15 (5.4) | 117 (42.4) | 165 (58.5) |
% responders – 95% CI | (2.7 to 8.1) | (36.6 to 48.2) | (52.7 to 64.2) |
Adjusted difference vs. placebo, % (95% CI) | Reference | 36.9 (30.6 to 43.3) | 53.1 (46.7 to 59.4) |
P value | Reference | < 0.001 | < 0.001 |
Change in EASI score from baseline at week 16b | |||
N (%) | 142 (51.08) | 246 (89.13) | 250 (88.65) |
Baseline mean | 26.98 | 28.70 | 29.63 |
At week 16 mean | 14.79 | 6.52 | 4.37 |
LS mean % change from baseline (95% CI)c,d | –34.51 (–39.60 to –29.42) | –74.13 (–78.45 to –69.82) | –84.65 (–88.93 to –80.37) |
Adjusted LS mean difference from placebo (95% CI)c,d | Reference | –39.62 (–45.79 to –33.46) | –50.14 (–56.28 to –44.00) |
P valuec,d | Reference | < 0.001 | < 0.001 |
Change in overall SCORAD score from baseline at week 16b | |||
N (%) | 136 (48.92) | 245 (88.77) | 241 (85.46) |
Baseline mean | 66.380 | 66.700 | 66.760 |
At week 16 mean | 46.804 | 28.177 | 21.672 |
LS mean % change from baseline (95% CI)c,d | –28.43 (–33.34 to –23.52) | –57.90 (–61.84 to –53.97) | –68.44 (–72.44 to –64.44) |
Adjusted LS mean difference from placebo (95% CI)c,d | Reference | –29.47 (–35.24 to –23.69) | –40.01 (–45.80 to –34.22) |
P value | Reference | < 0.001 | < 0.001 |
Symptoms | |||
WP-NRS response ≥ 4 from baseline at week 16a, e | |||
N (%) | 274 (98.56) | 270 (97.83) | 280 (99.29) |
Responders, n (%) | 25 (9.1) | 113 (41.9) | 167 (59.6) |
% responders – 95% CI | (5.7 to 12.5) | (36.0 to 47.7) | (53.9 to 65.4) |
Adjusted difference vs. placebo, % (95%CI)d | Reference | 32.6 (25.8 to 39.4) | 50.4 (43.8 to 57.1) |
P valued | Reference | < 0.001 | < 0.001 |
% Change in WP-NRS from baseline at week 16b | |||
N (%)e | 119 (42.81) | 224 (81.16) | 235 (83.33) |
Baseline mean | 7.120 | 7.203 | 7.273 |
At week 16 mean | 5.051 | 3.143 | 2.101 |
LS mean % change from baseline (95% CI)c,d | –17.04 (–22.39 to –11.69) | –51.20 (–55.80 to –46.61) | –66.49 (–71.03 to –61.96) |
Adjusted LS mean difference from placebo (95% CI)c,d | Reference | –34.16 (–40.81 to –27.51) | –49.45 (–56.05 to –42.84) |
P valuec,d | Reference | < 0.001 | < 0.001 |
POEM total score improvement from baseline at week 16a,e | |||
N (%) | 268 (96.40) | 268 (97.10) | 269 (95.39) |
Responders, n (%) | 77 (28.7) | 190 (70.9) | 225 (83.5) |
% responders – 95% CI | (23.3 to 34.1) | (65.5 to 76.3) | (79.1 to 88.0) |
Adjusted difference vs. placebo, % (95% CI) | Reference | 42.1 (34.5 to 49.8) | 54.7 (47.7 to 61.7) |
P value | Reference | < 0.001 | < 0.001 |
ADerm-SS skin pain improvement (≥ 4) from baseline at week 16a, e | |||
N (%) | 247 (88.85) | 237 (85.87) | 238 (84.40) |
Responders, n (%) | 33 (13.4) | 117 (49.4) | 155 (65.1) |
% responders – 95% CI | (9.1 to 17.6) | (43.0 to 55.7) | (59.1 to 71.2) |
Adjusted difference vs. placebo, % (95%CI)d | Reference | 35.9 (28.2 to 43.5) | 51.8 (44.4 to 59.1) |
P value d | Reference | < 0.001 | < 0.001 |
ADerm-SS TSS-7 improvement (≥ 28) from baseline at week 16a,e | |||
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
ADerm-IS sleep domain score improvement (≥ 12) from baseline at week 16a,e | |||
N (%) | 233 (83.81) | 219 (79.35) | 228 (80.85) |
Responders, n (%) | 29 (12.4) | 110 (50.2) | 142 (62.3) |
% responders – 95% CI | (8.2 to 16.7) | (43.6 to 56.9) | (56.0 to 68.6) |
Adjusted difference vs. placebo, % (95% CI)d | Reference | 37.9 (30.1 to 45.8) | 49.8 (42.2 to 57.3) |
P valued | Reference | < 0.001 | < 0.001 |
ADerm-IS daily activities domain score improvement (≥ 14) from baseline at week 1a,e | |||
N (%) | 227 (81.65) | 207 (75.00) | 223 (79.08) |
Responders, n (%) | 43 (18.9) | 118 (57.0) | 155 (69.5) |
% responders – 95% CI | (13.8 to 24.0) | (50.3 to 63.7) | (63.5 to 75.5) |
Adjusted difference vs. placebo, % (95% CI) d | Reference | 37.9 (29.5 to 46.3) | 50.6 (42.8 to 58.5) |
P valued | Reference | < 0.001 | < 0.001 |
ADerm-IS emotional state domain score improvement (≥ 11) from baseline at week 16a,e | |||
N (%) | 234 (84.17) | 228 (82.61) | 228 (80.85) |
Responders, n (%) | 39 (16.7) | 130 (57.0) | 163 (71.5) |
% responders – 95% CI | (11.9 to 21.4) | (50.6 to 63.4) | (65.6 to 77.4) |
Adjusted difference vs. placebo, % (95% CI)d | Reference | 40.3 (32.3 to 48.3) | 54.8 (47.2 to 62.3) |
P valued | Reference | < 0.001 | < 0.001 |
HRQoL | |||
DLQI improvement (≥ 4) from baseline at week 16a,e | |||
N (%) | 250 (89.93) | 251 (90.94) | 251 (89.01) |
Responders, n (%) | 71 (28.4) | 180 (71.7) | 195 (77.6) |
% responders – 95% CI | (22.8 to 34.0) | (66.1 to 77.3) | (72.5 to 82.8) |
Adjusted difference vs. placebo, % (95% CI)d | Reference | 42.8 (35.0 to 50.6) | 49.0 (41.4 to 56.5) |
P valued | Reference | < 0.001 | < 0.001 |
DLQI score of 0 or 1 at week 16a,e | |||
N (%) | 257 (92.45) | 252 (91.30) | 256 (90.78) |
Responders, n (%) | 12 (4.7) | 60 (23.8) | 97 (37.9) |
% responders – 95% CI | (2.1 to 7.2) | (18.6 to 29.1) | (32.0 to 43.9) |
Adjusted difference vs. placebo, % (95% CI)d | Reference | 19.1 (13.3 to 24.9) | 33.3 (26.9 to 39.8) |
P valued | Reference | < 0.001 | < 0.001 |
Change in EQ-5D index from baseline at week 16b | |||
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
Mood | |||
HADS-A or HADS-D response (< 8) at week 16b,e | |||
N (%) | 140 (50.36) | 137 (49.64) | 146 (51.77) |
Responders, n (%)e | 16 (11.4) | 63 (46.0) | 82 (56.1) |
% responders – 95% CI | (6.2 to 16.7) | (37.6 to 54.3) | (48.1 to 64.2) |
Adjusted difference vs. placebo, % (95% CI)d | Reference | 34.4 (24.7 to 44.2) | 44.5 (35.0 to 54.1) |
P valued | Reference | < 0.001 | < 0.001 |
ADerm-IS = Atopic Dermatitis Impact Scale; ADerm-SS = Atopic Dermatitis Symptom Scale; CI = confidence interval; COVID-19 = coronavirus disease 2019 pandemic; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; EASI 90 = at least 90% improvement in EASI total score from baseline; HADS-A = Hospital Anxiety and Depression Scale – Anxiety; HADS-D = Hospital Anxiety and Depression Scale – Depression; HRQoL = health-related quality of life; ITT = intention to treat; LS = least squares; POEM = Patient-Oriented Eczema Measure; q.d. = once daily; SCORAD = Scoring Atopic Dermatitis; TSS-7 = 7-item total symptom score; UPA = upadacitinib; vIGA-AD = validated Investigator Global Assessment for Atopic Dermatitis; vs. = versus; WP-NRS = Worst Pruritus Numerical Rating Scale; WPAI:AD = Work Productivity and Activity Impairment.
aAssessed in the ITT population with nonresponder imputation incorporating multiple imputation to handle data missing due to COVID-19.
bComplete case analysis.
cMixed-effects model for repeated measures with baseline, treatment, visit, treatment-by-visit interaction, and strata (baseline vIGA-AD score categories and age [adolescent vs. adult]) in the model.
d95% CI for adjusted difference and P values are calculated according to the Cochran-Mantel-Haenszel test adjusted for strata (baseline vIGA-AD score categories and age [adolescent vs. adult]) for the comparison of 2 treatment groups.
eThe calculations at each visit are based on nonresponder imputation incorporating multiple imputation to handle data missing due to COVID-19 or nonresponder imputation only if there are no data missing due to COVID-19.
fWithin-group LS mean and 95% CI as well as between-group LS mean, 95% CI, and P value are calculated using ANCOVA with baseline, treatment, and strata (baseline vIGA-AD score categories and age [adolescent vs. adult]) in the model.
Note: Redacted rows have been deleted.
Source: Clinical Study Report: Measure Up 2 study.11
Combination Therapy Regimen Studies
In the AD Up study, SCORAD was measured as a key secondary end point under the overall type I error control at the 0.05 (2-sided) level using a pre-specified graphical approach, as described previously and detailed in Table 27. The LS mean changes from baseline were –33.62% (95% CI, –37.34 to –29.89), –61.20% (95% CI, –64.53 to –57.86), and –70.95% (95% CI, –74.32 to –67.59) in the groups receiving placebo plus TCS, upadacitinib 15 mg plus TCS, and upadacitinib 30 mg plus TCS, respectively, with mean differences of –27.58% (95% CI, –32.16 to –23.01) and –37.34% (95% CI, –41.93 to –32.75) versus placebo in the groups receiving upadacitinib 15 mg plus TCS and upadacitinib 30 mg plus TCS, respectively (P < 0.001 for all comparisons). No subgroup analyses or sensitivity analyses were performed for the SCORAD outcome or other key secondary outcomes.
Head-to-Head Studies
The SCORAD was not measured in the Heads Up study as either a primary or secondary end point.
Reduction in Atopic Dermatitis Symptoms
A reduction in AD symptoms was considered an important end point by the clinical expert consulted by CADTH. The measurements proposed in the protocol (i.e., WP-NRS, POEM, and ADerm-IS) for this outcome domain were deemed appropriate to evaluate symptom changes that are important for decision-making, such as in pruritus, pain, and sleep.
Worst Pruritus Numerical Rating Scale
Monotherapy Regimen Studies
The investigators in the Measure Up 1 and Measure Up 2 studies evaluated the WP-NRS as the proportion of patients achieving an improvement (reduction) in WP-NRS of greater than or equal to 4 from baseline for patients who had an WP-NRS greater than or equal to 4 at baseline (week 16). WP-NRS was assessed in the Measure Up studies as a key secondary end point under the overall type I error control at the 0.05 (2-sided) level using a pre-specified graphical approach, as described previously.
In the Measure Up 1 study, upadacitinib 30 mg and 15 mg demonstrated superiority in itch reduction, including improvement (reduction) in WP-NRS of greater than or equal to 4 at day 2, day 3, week 1, week 4, and week 16 (Figure 5), and percentage change in WP-NRS at week 16. This information is presented in Table 25.
Adolescent patients on upadacitinib 30 mg and 15 mg achieved greater itch reduction as measured by WP-NRS compared to placebo (Appendix 3).
Similar results were found in the Measure Up 2 study, as shown in Figure 5 and Table 26, with a significantly superior percentage change from baseline in the upadacitinib groups versus placebo, including improvement (reduction) in WP-NRS greater than or equal to 4 at day 2, day 3, week 1, week 4, and week 16, and percentage change in WP-NRS at week 16. Adolescent patients on upadacitinib 30 mg and 15 mg achieved greater itch reduction as measured by WP-NRS compared to placebo (Appendix 3).
Figure 5: Proportion of Patients Achieving an Improvement (Reduction) in WP-NRS (≥ 4) From Baseline Through Week 16 (NRI-C, ITT Population) in the Measure Up 1 and Measure Up 2 Studies
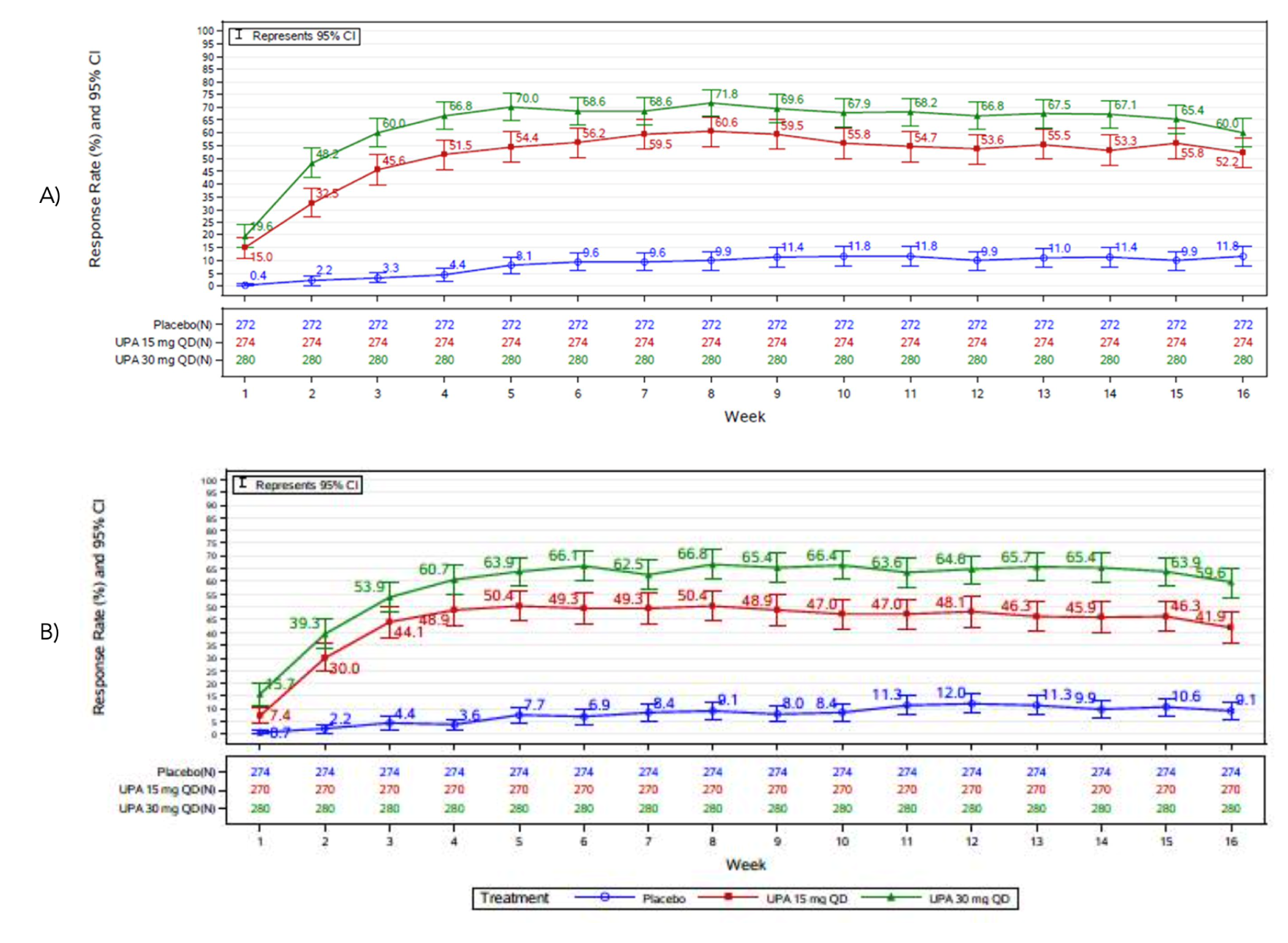
CI = confidence interval; ITT = intention to treat; NRI-C = nonresponder imputation to handle data missing due to COVID-19; QD = once daily; UPA = upadacitinib; WP-NRS = Worst Pruritis Numerical Rating Scale.
Note: (A) refers to the Measure Up 1 study and (B) refers to the Measure Up 2 study. Patients with missing data or who received rescue treatment were considered as non-responders. The weekly rolling average was calculated up to week 16. The 95% CI for response rate was based on the normal approximation to the binomial distribution. The population included only patients with WP-NRS greater than or equal to 4 at baseline.
Source: Clinical Study Reports for Measure Up 1 and Measure Up 2.11,12
Combination Therapy Regimen Studies
In the AD Up study, upadacitinib (30 mg and 15 mg) plus TCS displayed superiority against placebo plus TCS in itch reduction in achieving WP-NRS greater than or equal to 4 at week 1, week 4, and week 16, and in percentage change from baseline in WP-NRS at week 16, as described in Table 27.
Table 27: Efficacy Outcomes in the AD Up Study — Key Secondary and Exploratory End Points for All Patients
Outcome | Placebo + TCS N = 304 | UPA 15 mg q.d.+ TCS N = 300 | UPA 30 mg q.d. TCS N = 297 |
|---|---|---|---|
Disease severity | |||
EASI 90 at week 16a | |||
N (%) | 304 (100.00) | 300 (100.00) | 297 (100.00) |
Responders, n (%) | 40 (13.2) | 128 (42.8) | 187 (63.1) |
% responders – 95% CI | (9.4 to 17.0) | (37.2 to 48.4) | (57.6 to 68.6) |
Adjusted difference vs. placebo, % (95% CI)b | Reference | 29.5 (22.8 to 36.3) | 49.9 (43.3 to 56.4) |
P valueb | Reference | < 0.001 | < 0.001 |
Change in EASI score from baseline at week 16c | |||
N (%) | 206 (67.76) | 275 (91.67) | 276 (92.93) |
Baseline mean | 29.30 | 28.95 | 29.74 |
At week 16 mean | 14.49 | 5.89 | 3.82 |
LS mean % change from baseline (95% CI)d | –45.86 (–50.09 to –41.63) | –77.99 (–81.87 to –74.10) | –87.31 (–91.20 to –83.41) |
Adjusted LS mean difference from placebo (95% CI)d | Reference | –32.13 (–37.35 to –26.91) | –41.45 (–46.68 to –36.22) |
P valued | Reference | < 0.001 | < 0.001 |
Change in overall SCORAD score from baseline at week 16c | |||
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
Symptoms | |||
WP-NRS response ≥ 4 from baseline at week 16a | |||
N (%) | 294 (96.71) | 288 (96.00) | 291 (97.98) |
Responders, n (%) | 44 (15.0) | 149 (51.7) | 186 (63.9) |
% responders – 95% CI | (10.9 to 19.0) | (46.0 to 57.5) | (58.4 to 69.4) |
Adjusted difference vs. placebo, % (95% CI)b | Reference | 36.8 (29.7 to 43.8) | 48.8 (41.9 to 55.7) |
P valueb | Reference | < 0.001 | < 0.001 |
% Change in WP-NRS from baseline at week 16c | |||
N (%) | 184 (60.53) | 260 (86.67) | 247 (83.16) |
Baseline mean | 7.063 | 7.015 | 7.384 |
At week 16 mean | 4.714 | 2.697 | 1.879 |
LS mean % change from baseline (95% CI)d | –25.07 (–31.64 to –18.49) | –58.14 (–64.24 to –52.05) | –66.85 (–72.99 to –60.72) |
Adjusted LS mean difference from placebo (95% CI)d | Reference | –33.08 (–41.72 to –24.44) | –41.79 (–50.46 to –33.11) |
P valued | Reference | < 0.001 | < 0.001 |
POEM total score improvement (≥ 4) from baseline at week 16a | |||
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
ADerm-SS skin pain improvement (≥ 4) from baseline at week 16a | |||
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
ADerm-SS TSS-7 improvement (≥ 28) from baseline at week 16a | |||
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
ADerm-IS sleep domain score improvement (≥ 12) from baseline at week 16a | |||
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
ADerm-IS daily activities domain score improvement (≥ 14) from baseline at week 16a | |||
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
ADerm-IS emotional state domain score improvement (≥ 11) from baseline at week 16a | |||
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
HRQoL | |||
DLQI improvement (≥ 4) from baseline at week 16a | |||
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
DLQI score of 0 or 1 at week 16a | |||
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
Change in EQ-5D-5L index from baseline at week 16c | |||
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
Mood | |||
HADS-A or HADS-D response (< 8) at week 16a | |||
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
Productivity | |||
Change in WPAI:AD (work productivity loss) domain scorec | |||
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
Change in WPAI:AD (absenteeism) domain scoresc | |||
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
AD = atopic dermatitis; ADerm-SS = Atopic Dermatitis Symptom Scale; ADerm-IS = Atopic Dermatitis Impact Scale; ADerm-SS = Atopic Dermatitis Symptom Scale; ANCOVA = analysis of covariance; CI = confidence interval; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; EASI 90 = at least 90% improvement in EASI total score from baseline; EQ-5D-5L = EQ-5D Five-Level; HADS-A = Hospital Anxiety and Depression Scale – Anxiety; HADS-D = Hospital Anxiety and Depression Scale – Depression; HRQoL = health-related quality of life; ITT = intention to treat; LS = least squares; POEM = Patient-Oriented Eczema Measure; q.d. = once daily; SCORAD = Scoring Atopic Dermatitis; TCS = topical corticosteroid; TSS-7 = 7-Item Total Symptom Score; UPA = upadacitinib; vIGA-AD = validated Investigator Global Assessment for Atopic Dermatitis; vs. = versus; WP-NRS = Worst Pruritus Numerical Rating Scale; WPAI:AD = Work Productivity and Activity Impairment.
aAssessed in the ITT population with nonresponder imputation incorporating multiple imputation to handle data missing due to COVID-19.
b95% CI for adjusted difference and P values are calculated according to the Cochran-Mantel-Haenszel test adjusted for strata (baseline vIGA-AD score categories and age [adolescent vs. adult]) for the comparison of 2 treatment groups.
cComplete case analysis.
dMixed-effects model for repeated measures with baseline, treatment, visit, treatment-by-visit interaction, and strata (baseline vIGA-AD score categories and age [adolescent vs. adult]) in the model.
eWithin-group LS means and 95% CI as well as between-group LS means, 95% CI, and P value are calculated using ANCOVA with baseline, treatment, and strata (baseline vIGA-AD score categories and age [adolescent vs. adult]) in the model.
Note: Redacted rows have been deleted.
Source: Clinical Study Report: AD Up.13
Figure 6: Proportion of Patients Achieving an Improvement (Reduction) in WP-NRS (≥ 4) From Baseline Through Week 16 (NRI-C, ITT Population) in the AD Up Study
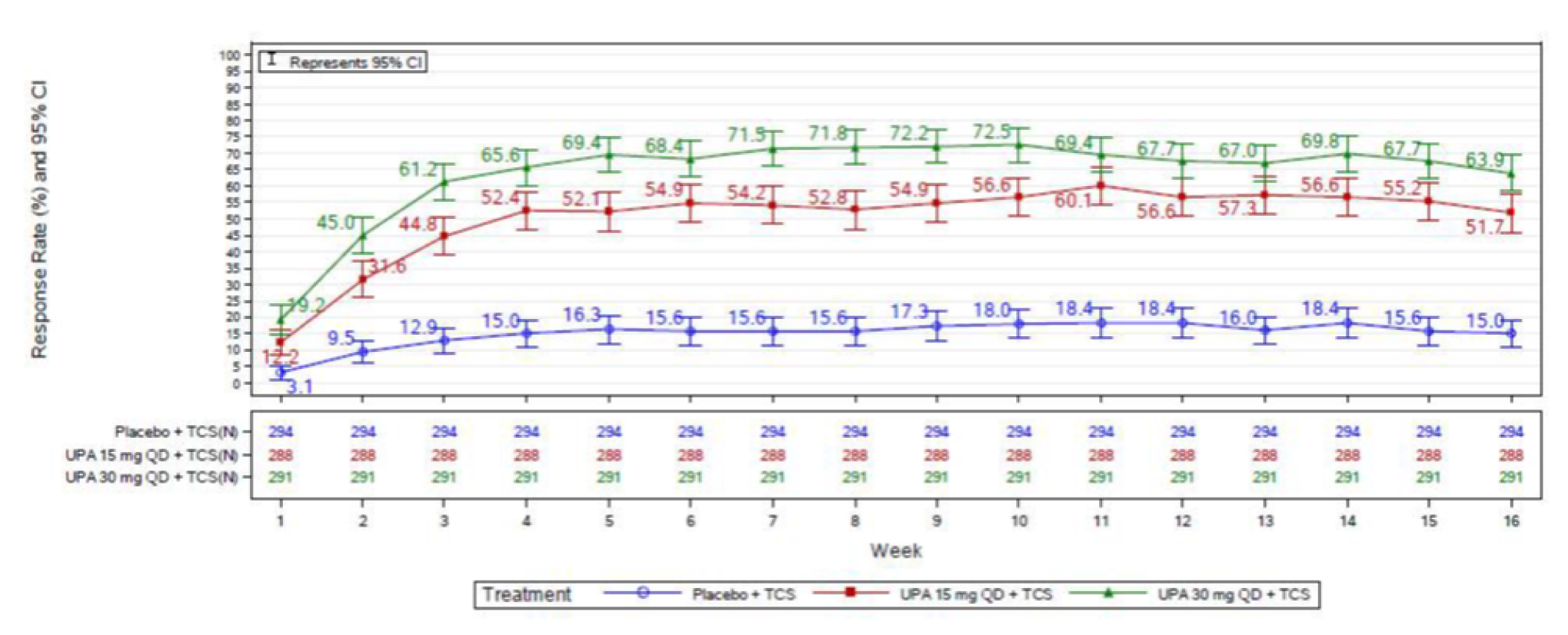
CI = confidence interval; ITT = intention to treat; NRI-C = nonresponder imputation to handle data missing due to COVID-19; QD = every day; TCS = topical corticosteroid; UPA = upadacitinib; WP-NRS = Worst Pruritis Numerical Rating Scale.
Notes: Patients with missing data or who received rescue treatment were considered as non-responders. The weekly rolling average was calculated up to week 16. The 95% CI for response rate was based on the normal approximation to the binomial distribution. The population included only patients with WP-NRS greater than or equal to 4 at baseline.
Source: Clinical Study Report: AD Up study.13
Head-to-Head Studies
WP-NRS was evaluated as a secondary end point, controlling for type I error rate with the multiple testing procedure across analyses comparing upadacitinib versus dupilumab. Patients in the upadacitinib group achieved a greater proportion of improvement in WP-NRS greater than or equal to 4 (Table 28) compared to the dupilumab group at all visits. At week 16, 120 patients (35.9%) in the dupilumab group versus 188 patients (55.2%) in the upadacitinib 30 mg group achieved a WP-NRS greater than or equal to 4 (adjusted difference = 19.3%; 95% CI, 11.9 to 26.7). At week 24, this difference decreased, as shown in Table 28 and Figure 7, with 141 patients (41.9%) in the dupilumab group versus 171 patients (50.2%) in the upadacitinib 30 mg group achieving a WP-NRS greater than or equal to 4, for an adjusted difference of 8.3% (95% CI, 0.8 to 15.8; P = 0.030). The WP-NRS was supported by sensitivity analyses (multiple imputation, NRI-NC).
Table 28: Efficacy Outcomes in the Heads Up Study — Key Secondary and Exploratory End Points
Outcome | DUP 300 mg 2.q.w. N = 344 | UPA 30 mg q.d. N = 348 |
|---|---|---|
Disease severity | ||
EASI 90 at week 16a | ||
N (%) | 344 (100.00) | 348 (100.00) |
Responders, n (%) | 133 (38.8) | 211 (60.6) |
% responders – 95% CI | (33.6 to 43.9) | (55.4 to 65.7) |
Adjusted difference to % (95%CI)b | 21.8 (14.5 to 29.1) | |
P valueb | < 0.001 | |
EASI 75 at week 24a | ||
N (%) | 344 (100.00) | 348 (100.00) |
Responders to n (%) | 205 (59.5) | 223 (64.2) |
% responders – 95% CI | (54.4 to 64.7) | (59.1 to 69.2) |
Adjusted difference to % (95%CI)b | 4.6 (–2.6 to 11.9) | |
P valueb | 0.211 | |
EASI 90 at week 24a | ||
N (%) | 344 (100.00) | 348 (100.00) |
Responders to n (%) | 164 (47.6) | 194 (55.6) |
% responders – 95% CI | (42.3 to 52.9) | (50.4 to 60.8) |
Adjusted difference to % (95%CI)b | 8.0 (0.5 to 15.4) | |
P valueb | 0.036 | |
Symptoms | ||
WP-NRS response ≥ 4 from baseline at week 16a | ||
N (%) | 336 (97.67) | 340 (97.70) |
Responders to n (%) | 120 (35.9) | 188 (55.2) |
% responders – 95% CI | (30.7 to 41.0) | (49.9 to 60.5) |
Adjusted difference to % (95%CI)b | 19.3 (11.9 to 26.7) | |
P valueb | < 0.001 | |
WP-NRS response ≥ 4 from baseline at week 24a | ||
N (%) | 336 (97.67) | 340 (97.70) |
Responders to n (%) | 141 (41.9) | 171 (50.2) |
% responders – 95% CI | (36.6 to 47.1) | (44.8 to 55.5) |
Adjusted difference to % (95%CI)b | 8.3 (0.8 to 15.8) | |
P value b | 0.030 | |
% Change in WP-NRS from baseline at week 16 | ||
N (%)c | 251 (72.97) | 258 (74.14) |
Baseline mean | 7.466 | 7.351 |
At week 16 mean | 3.459 | 2.129 |
LS mean % change from baseline (95% CI)d | –49.04 (–52.87 to –45.22) | –66.88 (–70.59 to –63.17) |
Adjusted LS mean difference (95% CI)d | –17.84 (–23.17 to –12.50) | |
P valued | < 0.001 | |
% Change in WP-NRS from baseline at week 24 | ||
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
2.q.w. = every 2 weeks; CI = confidence interval; DUP = dupilumab; EASI = Eczema Area and Severity Index; EASI 75 = at least 75% improvement in EASI total score from baseline; EASI 90 = at least 90% improvement in EASI total score from baseline; ITT = intention to treat; LS = least squares; q.d. = once daily; UPA = upadacitinib; vIGA-AD = validated Investigator Global Assessment; vs. = versus; WP-NRS = Worst Pruritus Numerical Rating Scale.
aAssessed in the ITT population with nonresponder imputation incorporating multiple imputation to handle missing data due to COVID-19.
b95% CI for adjusted difference and P values are calculated according to the Cochran-Mantel-Haenszel test adjusted for strata (baseline vIGA-AD score categories and age [adolescent vs. adult]) for the comparison of 2 treatment groups.
cComplete case analysis.
dMixed-effects model for repeated measures with baseline, treatment, visit, treatment-by-visit interaction, and strata (baseline vIGA-AD score categories and age [adolescent vs. adult]) in the model.
Note: Redacted rows have been deleted.
Source: Clinical Study Report: Heads Up Study.14
Figure 7: Proportion of Patients Achieving an Improvement (Reduction) in WP-NRS ≥ 4 From Baseline by Visit (ITT Population; NRI-C) in the Heads Up Study
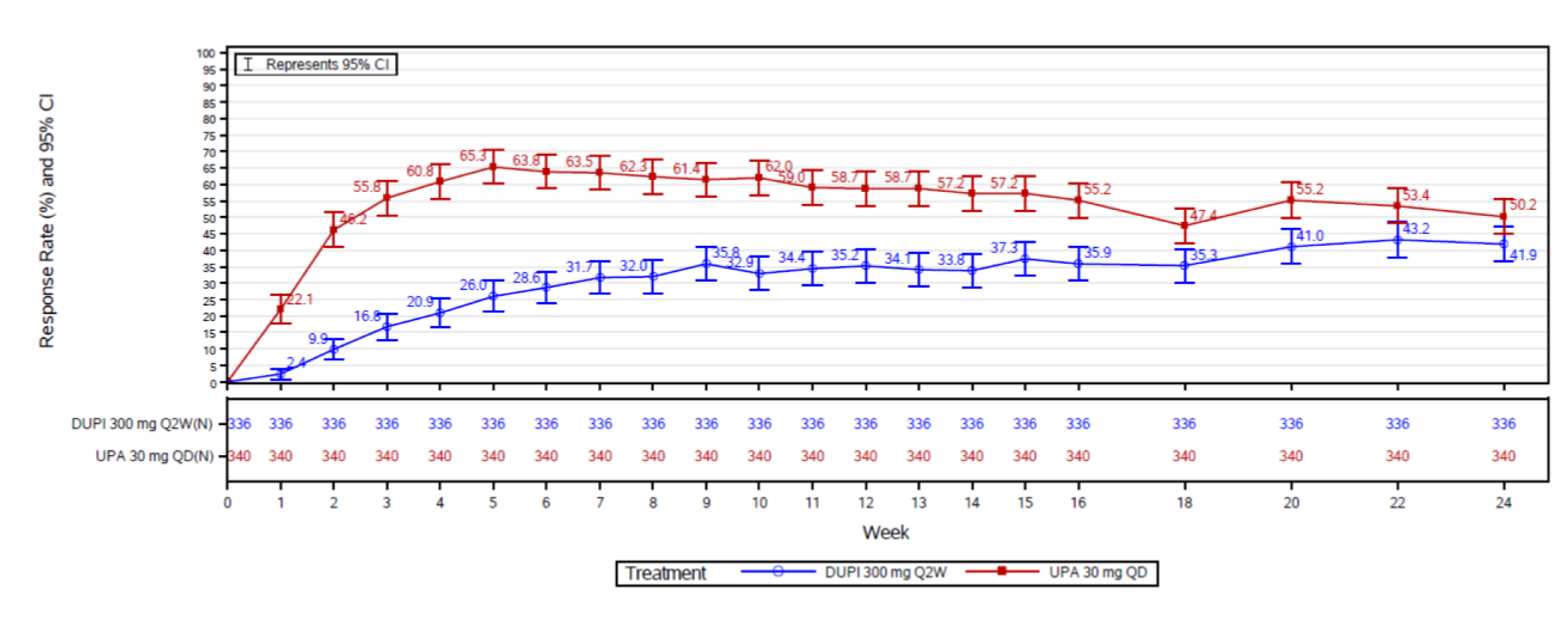
CI = confidence interval; DUPI = dupilumab; ITT = intention to treat; NRI-C = nonresponder imputation to handle data missing due to COVID-19; Q2W = every 2 weeks; QD = once daily; UPA = upadacitinib; WP-NRS = Worst Pruritis Numerical Rating Scale.
Notes: The 95% CI for response rate is the synthetic result based on the student's t-distribution from the PROC MIANALYZE procedure if there are data missing due to COVID-19, or is based on the normal approximation to the binomial distribution if there are no data missing due to COVID-19.
Source: Clinical Study Report: Heads Up.62
Patient-Oriented Eczema Measure
Monotherapy Regimen Studies
The investigators from the Measure Up 1 and Measure Up 2 studies evaluated the POEM, defining a reduction (improvement) as greater than or equal to 4 from baseline. POEM was measured as a key secondary end point under the overall type I error control at the 0.05 (2-sided) level using a pre-specified graphical approach as described before.
In the Measure Up 1 study, a significantly greater proportion of patients on upadacitinib 30 mg and 15 mg achieved a reduction in symptoms of AD, measured as an improvement in POEM of greater than or equal to 4 at week 16 (Table 25), with 63 patients (22.8%), 209 patients (75.0%), and 228 patients (81.4%) in the groups receiving placebo, upadacitinib 15 mg, and upadacitinib 30 mg, respectively, and adjusted differences versus the placebo group of 52.3% (95% CI, 45.2 to 59.4) and 58.6% (95% CI, 51.9 to 65.3) in the groups receiving upadacitinib 15 mg and upadacitinib 30 mg, respectively (P < 0.001 for both differences). Adolescents showed consistent results versus the overall population (refer to Appendix 3).
Similar results were found in the Measure Up 2 study, as shown in Table 26, with 77 patients (28.7%), 190 patients (70.9%), and 225 patients (83.5%) achieving reductions (improvements) of greater than or equal to 4 from baseline in their POEM scores.
Combination Therapy Regimen Studies
In the AD Up study, the POEM score was also evaluated as a key secondary end point adjusted for multiplicity. Upadacitinib (30 mg and 15 mg) plus TCS displayed superiority in improvement based on the POEM score (≥ 4) at week 16, with 117 patients (38.8%), 234 patients (78.9%), and 244 patients (83.7%) achieving an improvement (≥ 4) in the POEM score from baseline at week 16, as presented in Table 27. The adjusted differences versus placebo were 40.2% (95% CI, 33.0 to 47.4) and 44.9% (95% CI, 37.9 to 51.8) in the groups receiving upadacitinib 15 mg plus TCS and upadacitinib 30 mg plus TCS, respectively.
Head-to-Head Studies
In the Heads Up study, POEM scores were not evaluated within the primary or secondary end points.
Atopic Dermatitis Impact Scales
Monotherapy Regimen Studies
The investigators in the Measure Up 1 and Measure Up 2 studies evaluated clinically important outcomes of symptoms and related domains, including skin pain, sleep, and daily activities (which were also pre-specified in this CADTH review protocol) using the ADerm-IS instrument and its subscales. These key secondary end points were evaluated under overall type I error control at the 0.05 (2-sided) level using a pre-specified graphical approach, as described previously.
In the Measure Up 1 study, using the ADerm-SS tool, a skin pain score reduction (improvement) of greater than or equal to 4 from baseline at week 16 was achieved in 35 patients (15.0%), 127 patients (53.6%), and 158 patients (63.5%) in the groups receiving placebo, upadacitinib 15 mg, and upadacitinib 30 mg, respectively, with adjusted differences versus placebo of 38.7% (95% CI, 30.9 to 46.5) and 48.6% (95% CI, 41.0 to 56.1), respectively. The ADerm-SS TSS-7 was also used to measure skin pain and overall symptoms, with a reduction of greater than or equal to 28 (improvement) from baseline as a cut-off value (refer to Table 25), in this end point, 34 (15.0%), 125 (53.6%), and 167 (67.9%) of patients in the groups receiving placebo, upadacitinib 15 mg, and upadacitinib 30 mg, respectively, improved above the cut-off value. Statistically significantly higher proportions of patients also improved in the sleep domain score of the ADerm-IS (defined as values ≥ 12), with 29 patients (13.2%) 120 patients (55.0%), and 144 patients (66.1%) in the groups receiving placebo, upadacitinib 15 mg, and upadacitinib 30 mg, respectively, with adjusted differences versus placebo of 41.8% (95% CI, 33.9 to 49.7) and 52.9% (95% CI, 45.2 to 60.6).
Similar results were found in the Measure Up 2 study, as shown in Table 26. Using the ADerm-SS tool, a skin pain score reduction (improvement) of greater than or equal to 4 from baseline at week 16 was achieved in 33 patients (13.4%), 117 patients (49.4%), and 155 patients (65.1%) in the groups receiving placebo, upadacitinib 15 mg, and upadacitinib 30 mg, respectively, with adjusted differences versus placebo of 35.9% (95% CI, 28.2 to 43.5) and 51.8% (95% CI, 44.4 to 59.1) in the groups receiving upadacitinib 15 mg and upadacitinib 30 mg, respectively. The ADerm-SS TSS-7 was also used to measure overall pain symptoms, with a reduction (improvement) of greater than or equal to 28 from baseline as a cut-off value achieved by 31 patients (12.7%), 122 patients (53.0%), and 155 patients (66.2%) in the groups receiving placebo, upadacitinib 15 mg, and upadacitinib 30 mg, respectively. Statistically significantly higher proportions of patients also improved in the sleep domain score of the ADerm-IS (defined as values ≥ 12). Such improvement was observed in 29 patients (12.4%), 110 patients (50.2%), and 142 patients (62.3%) in the groups receiving placebo, upadacitinib 15 mg, and upadacitinib 30 mg, respectively, with adjusted differences versus placebo of 37.9% (95% CI, 30.1 to 45.8) and 9.8% (95% CI, 42.2 to 57.3).
Combination Therapy Regimen Studies
As shown in Table 27, AD Up study investigators assessed the ADerm-IS and subscales as key secondary end points under the overall type I error control at the 0.05 (2-sided) level using a pre-specified graphical approach, as described previously. A greater proportion of patients on upadacitinib (30 mg or 15 mg) achieved improvement in the additional end points that relate to skin pain (ADerm-SS skin pain and ADerm-SS TSS-7) at week 16 compared with placebo.
A score reduction (improvement) of greater than or equal to 4 in skin pain from baseline at week 16 was achieved in 51 patients (20.0%), 146 patients (57.9%), and 169 patients (68.4%) in the groups receiving placebo plus TCS, upadacitinib 15 mg plus TCS, and upadacitinib 30 mg plus TCS, respectively, with adjusted differences versus placebo of 38.2% (95% CI, 30.5 to 46.0) and 48.3% (95% CI, 40.8 to 55.9), respectively.
Upadacitinib (30 mg and 15 mg) plus TCS also displayed superiority in improvement in the ADerm-SS TSS-7 (defined as a change in score greater than or equal to 28 from baseline at week 16), the ADerm-IS sleep domain (defined as a change in score greater than or equal to 12 from baseline), and the ADerm-IS emotional state, as shown in Table 27.
Head-to-Head Studies
The Heads Up study did not investigate the ADerm-IS and sub-domains within the primary or secondary end points.
Health-Related Quality of Life
Monotherapy Regimen Studies
The investigators in the Measure Up 1 and Measure Up 2 studies included HRQoL outcomes in their assessments as key secondary end points under the overall type I error control described previously. These included the outcomes established in the protocol of this CADTH review: DLQI improvement (≥ 4) from baseline at week 16, CDLQI improvement (evaluated in those under the age of 16 years), and EQ-5D-5L index score change from baseline at week 16.
In the Measure Up 1 study, patients receiving upadacitinib 30 mg and 15 mg showed improvements (reductions) in DLQI greater than or equal to 4 at week 16 compared to placebo, with 73 patients (29.0%), 192 patients (75.4%), and 210 patients (82.0%) in the groups receiving placebo, upadacitinib 15 mg, and upadacitinib 30 mg, respectively, achieving a DLQI greater than or equal to 4, with an adjusted difference versus placebo of 46.7% (95% CI, 39.0 to 54.4) in the 15 mg group and 53.2% (95% CI, 45.9 to 60.5) in the 30 mg group (P < 0.001 for both comparisons) as shown in Table 25. In the EQ-5D-5L index, the 15 mg and 30 mg showed better increases (improvements) in the mean change from baseline although the differences did not reach statistically significance between groups, with mean % change of 2.02 (95% CI, –70.66 to 74.71) for the placebo group, 75.40 (95% CI, 4.75 to 146.05) in the 15 mg, and 70.28 (95% CI, –0.09 to 140.64) in the 30 mg group, and the adjusted mean difference against placebo of 73.38 (95% CI, –15.94 to 162.71) and 68.25 (95% CI, –20.96 to 157.47) in the 15 mg and 30 mg groups, respectively (P = 0.107 and 0.134, respectively), as shown in Table 25.
Similar results were found in the Measure Up 2 study, as shown in Table 26, with 71 patients (28.4%),180 patients (71.7%), 195 patients (77.6%) patients in the groups receiving placebo, upadacitinib 15 mg, and upadacitinib 30 mg, respectively, achieving a DLQI greater than or equal to 4 at week 16, for an adjusted difference versus placebo of 42.8% (95% CI, 35.0 to 50.6) in the 15 mg group and 49.0% (95% CI, 41.4 to 56.5) in the 30 mg group (P < 0.001 for both comparisons). For the EQ-5D-5L index measure, the upadacitinib groups had higher values (improvement); however, these did not reach statistical significance.
In the Measure Up 1 study, a greater proportion of patients (adolescents) achieving CDLQI 0 or 1 compared to placebo was demonstrated as early as week 2 for upadacitinib 30 mg and week 8 for upadacitinib 15 mg. In addition, consistent improvement (reduction) in CDLQI was observed in patients under 16 years old on upadacitinib 30 mg and 15 mg. This difference was not statistically significant in the Measure Up 2 study in adolescents (refer to Appendix 3, Table 62, Table 63).
Combination Therapy Regimen Studies
The HRQoL outcomes in the AD Up study are shown in Table 27. These outcomes were also considered key secondary end points and adjusted for multiplicity. An improvement in the DLQI (≥ 4) from baseline at week 16 was observed in 111 patients (41.3%), 215 patients (80.8%), and 228 patients (84.9%) in the groups receiving placebo plus TCS, upadacitinib 15 mg plus TCS, and upadacitinib 30 mg plus TCS, respectively, with adjusted differences versus placebo of 39.7% (95% CI, 32.2 to 47.2) and 43.7% (95% CI, 36.4 to 51.0) in the groups receiving upadacitinib 15 mg plus TCS and upadacitinib 30 mg plus TCS, respectively (P < 0.001 for both comparisons).
In the EQ-5D index, the groups receiving upadacitinib plus TCS had better scores; however, these did not reach statistical significance, with mean differences from placebo of 31.34% (95% CI, –68.08 to 130.77) in the group receiving upadacitinib 15 mg plus TCS and 36.06% (95% CI, –63.94 to 136.06) in the group receiving upadacitinib 30 mg plus TCS (P = 0.536 and 0.479, respectively).
The CDLQI in the adolescent population reached statistical significance, with improvements in the upadacitinib groups, with adjusted differences versus placebo of 13.6% (95% CI, 1.4 to 25.7) and 19.6% (95% CI, 2.1 to 37.1) in the groups receiving upadacitinib 15 mg plus TCS and upadacitinib 30 mg plus TCS, respectively, with P values of 0.028 for both comparisons (refer to Appendix 3, Table 64).
Head-to-Head Studies
HRQoL outcomes were not evaluated in the Heads Up study within the primary or secondary end points.
Mood and Productivity Outcomes
Hospital Anxiety and Depression Scale
Monotherapy Regimen Studies
The investigators in the Measure Up 1 and Measure Up 2 studies evaluated the clinically important outcomes of mood and productivity (pre-specified in this CADTH review protocol) using the HADS. The HADS was evaluated as a key secondary end point under the overall type I error control at the 0.05 (2-sided) level using a pre-specified graphical approach, as described previously.
In Measure Up 1, a greater improvement (reduction) in HADS was observed in patients on upadacitinib 30 mg and 15 mg compared to those on placebo (Table 25). A response (improvement) in HADS-A or HADS-D scores of less than 8 at week 16 was achieved in 18 patients (14.3%), 66 patients (45.5%), and 71 patients (49.2%) in the groups receiving placebo, upadacitinib 15 mg, and upadacitinib 30 mg, respectively, with adjusted differences versus placebo of 31.5% (95% CI, 21.4 to 41.6) and 34.9% (95% CI, 24.8 to 45.1), respectively (P < 0.001 for both comparisons).
Similar results were found in the Measure Up 2 study, as shown in Table 26. A response (improvement) in the HADS-A or HADS-D scores of less than 8 at week 16 was achieved in 16 patients (11.4%), 63 patients (46.0%), and 82 patients (56.1%) in the groups receiving placebo, upadacitinib 15 mg, and upadacitinib 30 mg, respectively, with adjusted differences versus placebo of 34.4% (95% CI, 24.7 to 44.2) and 44.5% (95% CI, 35.0 to 54.1), respectively (P < 0.001 for both comparisons).
Combination Therapy Regimen Studies
In the AD Up study, investigators assessed the HADS-A and HADS-D as key secondary end points under the overall type I error control at the 0.05 (2-sided) level using a pre-specified graphical approach, as described previously, also with a cut-off point of less than 8 to determine a response (refer to Table 27). In this study, a response (improvement) in the HADS-A or HADS-D score of less than 8 at week 16 was achieved in 39 patients (27.4%), 72 patients (45.7%), and 75 patients (48.4%) in the groups receiving placebo plus TCS, upadacitinib 15 mg plus TCS, and upadacitinib 30 mg plus TCS, respectively, with adjusted differences versus placebo of 18.0% (95% CI, 7.3 to 28.7) and 20.4% (95% CI, 9.7 to 31.2) in the groups receiving upadacitinib 15 mg plus TCS and upadacitinib 30 mg plus TCS, respectively (P < 0.001 for both comparisons).
Head-to-Head Studies
In the Heads Up study, the HADS-A and HADS-D were not evaluated within the primary or secondary end points.
Work Productivity and Activity Impairment
Monotherapy Regimen Studies
The investigators from the Measure Up 1 and Measure Up 2 studies evaluated outcomes related to work productivity and activity impairment, as pre-specified in the CADTH review protocol. The WPAI:AD was evaluated as a key secondary end point under the overall type I error control at the 0.05 (2-sided) level using a pre-specified graphical approach, as described previously. The results were evaluated in observed cases only. The WPAI:AD domain scores were evaluated in fewer than 5 adolescents in the Measure Up studies.
In Measure Up 1, patients on upadacitinib 30 mg and 15 mg achieved a greater improvement (reduction) in WPAI:AD domain scores (work productivity loss, presenteeism, and activity impairment) compared to patients on placebo (refer to Table 25). In the WPAI:AD work loss domain, the mean percentage changes from baseline in the groups receiving placebo, upadacitinib 15 mg, and upadacitinib 30 mg were 3.59 (95% CI, –24.81 to 32.00), –40.14 (95% CI, –65.76 to –14.53), and –48.44 (95% CI, –75.05 to –21.83), respectively, with adjusted mean differences versus placebo of –43.74 (95% CI, –60.36 to –27.11) and –52.03 (95% CI, –68.71 to –35.36) in the groups receiving upadacitinib 15 mg and upadacitinib 30 mg, respectively (P = < 0.001 for both comparisons). Results for the absenteeism scores did not reach statistical significance (P = 0.0529 and P = 0.0611 in the adjusted mean differences versus placebo for upadacitinib 15 mg and upadacitinib 30 mg, respectively).
Similar results were found in the Measure Up 2 study, as shown in Table 26.
Combination Therapy Regimen Studies
In the AD Up study, investigators assessed the WPAI:AD as a key secondary end point under the overall type I error control at the 0.05 (2-sided) level using a pre-specified graphical approach, as described previously. Of note, this analysis did not use the ITT population, but rather the complete case analysis.
Compared to placebo, larger improvements were observed in the groups receiving upadacitinib 15 mg plus TCS and upadacitinib 30 mg plus TCS in the mean percentage change from baseline of the work productivity loss domain score, with –37.87 (95% CI, –58.78 to –16.97), –68.77 (95% CI, –89.43 to –48.11), and –69.40 (95% CI, –89.75 to –49.05) in the groups receiving placebo upadacitinib 15 mg plus TCS, and upadacitinib 30 mg plus TCS, respectively, with adjusted mean differences versus placebo of –30.90 (95% CI, –44.24 to –17.56) and –31.52 (95% CI, –44.91 to –18.14) in the groups receiving upadacitinib 15 mg plus TCS and upadacitinib 30 mg plus TCS, respectively (P < 0.001 for both results), as shown in Table 27.
As with the Measure Up studies, there were no statistically significant differences in the WPAI:AD or absenteeism domain scores.
Head-to-Head Studies
In the Heads Up study, the WPAI:AD score and domains were not evaluated within the primary or secondary end points.
Patients With Severe Disease and Prior Exposure to Oral Systemic Therapy
To better understand the efficacy values of upadacitinib at 30 mg doses in patients with severe AD (i.e., EASI ≥ 21) and prior exposure to oral systemic therapy, a post hoc subgroup analysis of patients with these characteristics is presented based on additional information requested from the sponsor.
The information is depicted for the EASI 75 (Table 29) and vIGA-AD score (Table 30) end points at baseline and at week 16 for the Measure Up 1, Measure Up 2, AD Up, and Heads Up studies.
Overall, response rates in the EASI 75 and vIGA-AD scores among patients with prior exposure to oral systemics and with severe symptoms of AD were aligned with the results in the overall population.
Evidence was also sought from the sponsor in relation to patients with severe AD and partial response by week 16 while on upadacitinib 15 mg who were increasing their dose to 30 mg, as well as for patients with severe disease who were initiating therapy with upadacitinib 30 mg and reducing the dose to 15 mg in those who responded at week 16 to explore and better understand the possible efficacy in these situations.
Table 29: Efficacy Outcomes, EASI Score — Post Hoc Subgroup of Patients With Severe AD (EASI ≥ 21) and Prior Exposure to Oral Systemic Therapy — Redacted
Study | Arms of study | ||
|---|---|---|---|
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
AD = atopic dermatitis; CI = confidence interval; EASI = Eczema Area and Severity Index; SD = standard deviation; vIGA-AD = validated Investigator Global Assessment for Atopic Dermatitis; vs. = versus.
a95% CI for adjusted difference and P value are calculated according to the Cochran-Mantel-Haenszel test adjusted for strata (baseline vIGA-AD score categories and age [adolescent vs. adult]) for the comparison of 2 treatment groups.
Note: Redacted rows have been deleted.
Source: Addendum submitted by the sponsor on the following Clinical Study Reports: Measure Up 1,12 Measure Up 2,11 AD Up, and Heads Up.14
Table 30: Efficacy Outcomes, vIGA-AD Score — Post Hoc Subgroup of Patients With Severe AD (EASI ≥ 21) and Prior Exposure to Oral Systemic Therapy — Redacted
Study | Arms of study | ||
|---|---|---|---|
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
AD = atopic dermatitis; CI = confidence interval; EASI = Eczema Area and Severity Index; SD = standard deviation; vIGA-AD = validated Investigator Global Assessment for Atopic Dermatitis; vs. = versus.
a95% CI for adjusted difference and P value are calculated according to the Cochran-Mantel-Haenszel test adjusted for strata (baseline vIGA-AD score categories and age [adolescent vs. adult]) for the comparison of 2 treatment groups.
Note: Redacted rows have been deleted.
Source: Addendum submitted by the sponsor on the following Clinical Study Reports: Measure Up 1,12 Measure Up 2,11 AD Up,13 and Heads Up.14
The only information obtained comes from a conference presentation63 aimed at assessing the effect of withdrawal and re-treatment with upadacitinib during an 88-week, phase IIb trial in patients with moderate or severe AD who — after a 16-week period of randomly receiving upadacitinib 7.5 mg, upadacitinib 15 mg, upadacitinib 30 mg, or placebo — were re-randomized to placebo or any of the other 3 arms (7.5 mg, 15 mg, or 30 mg). The evidence was reported with descriptive statistics and without any imputation or adjustments for multiplicity. The results depicting the effects of upadacitinib in patients who were re-randomized to upadacitinib 15 mg and then stopped responding and were rescued with the 30 mg dose were available for only 12 patients. Among these 12 patients, 9 were available for assessing the improvement in EASI scores at 8 weeks with upadacitinib 30 mg (mean improvement of 76.2%); 5 of the 9 achieved EASI 75, while 2 reached an IGA of 0 or 1.
No evidence was found for the scenario of patients who would be on 30 mg and responding, then switch to 15 mg of upadacitinib.
Harms
Only those harms identified in the review protocol are reported.
Adverse Events
Monotherapy Regimen Studies
AEs in the Measure Up 1 trial are depicted in Table 31. AEs were evaluated in the safety population and classified as such if they began or worsened in severity after the initiation of the study drug through 30 days following the last dose of the study drug in the respective analysis period. AEs were reported in 59.1%, 62.6%, and 73.3% of patients in the placebo, upadacitinib 15 mg, and upadacitinib 30 mg groups, respectively. The most common AE was acne, found in 2.1%, 6.8%, and 17.2% of patients in the placebo, upadacitinib 15 mg, and upadacitinib 30 mg groups, respectively, in both the overall population and adolescents. Other AEs included upper respiratory tract infections, nasopharyngitis, headache, and elevation of plasma CPK.
Table 31: Summary of Harms in the Measure Up 1 Study
Harms | Placebo N = 281 | UPA 15 mg q.d. N = 281 | UPA 30 mg q.d. N = 285 |
|---|---|---|---|
Patients with ≥ 1 adverse event | |||
n (%) | 166 (59.1) | 176 (62.6) | 209 (73.3) |
Most common events,a n (%) | |||
Acne | 6 (2.1) | 19 (6.8) | 49 (17.2) |
Upper respiratory tract infection | 20 (7.1) | 25 (8.9) | 38 (13.3) |
Nasopharyngitis | 16 (5.7) | 22 (7.8) | 33 (11.6) |
Headache | 12 (4.3) | 14 (5.0) | 19 (6.7) |
Plasma CPK elevation | 7 (2.5) | 16 (5.7) | 16 (5.6) |
Patients with ≥ 1 SAE | |||
n (%) | 8 (2.8) | 6 (2.1) | 8 (2.8) |
Most common events, n (%)b | |||
Atopic dermatitis | 3 (1.1) | 0 | 0 |
Patients who stopped treatment due to adverse events | |||
n (%) | 12 (4.3) | 4 (1.4) | 11 (3.9) |
Most common events,c n (%) | |||
Possible malignancy | 0 | 0 | 2 (0.7) |
Deaths | |||
n (%) | 0 | 0 | 0 |
Notable harms | |||
Serious infection | 0 | 2 (0.7) | 2 (0.7) |
Opportunistic infection excluding tuberculosis and herpes zoster | 4 (1.4) | 0 | 3 (1.1) |
Herpes zoster | 0 | 5 (1.8) | 6 (2.1) |
Active tuberculosis | 0 | 0 | 0 |
Possible malignancy | 0 | 1 (0.4) | 2 (0.7) |
Malignancy | 0 | 1 (0.4) | 2 (0.7) |
NMSC | 0 | 1 (0.4) | 0 |
Malignancy other than NMSC | 0 | 0 | 2 (0.7) |
Lymphoma | 0 | 0 | 0 |
Hepatic disorder | 2 (0.7) | 5 (1.8) | 8 (2.8) |
Anemia | 1 (0.4) | 1 (0.4) | 5 (1.8) |
Neutropenia | 2 (0.7) | 4 (1.4) | 15 (5.3) |
Lymphopenia | 2 (0.7) | 1 (0.4) | 2 (0.7) |
CPK elevation | 7 (2.5) | 16 (5.7) | 16 (5.6) |
MACE | 0 | 0 | 0 |
VTE | 0 | 0 | 0 |
CPK = creatine phosphokinase; MACE = major adverse cardiovascular event; NMSC = non-melanoma skin cancer; q.d. = once daily; SAE = serious adverse event; UPA = upadacitinib; VTE = venous thromboembolism.
aFrequency > 5%.
bFrequency > 1%.
cFrequency > 0.5%.
Source: Clinical Study Report: Measure Up 1.12
AEs in the Measure Up 2 trial, shown in Table 32, were classified and defined as in the Measure Up 1 study. AEs were similarly distributed, reported in 52.5%, 60.1%, and 61.3% of patients in the placebo, upadacitinib 15 mg, and upadacitinib 30 mg groups, respectively. Similarly, the most common AE was acne, experienced by 2.2%, 12.7%, and 14.5% of patients in the placebo, upadacitinib 15 mg, and upadacitinib 30 mg groups, respectively. The distribution was similar in both the overall population and in adolescents. Other AEs included upper respiratory tract infections, nasopharyngitis, and headache.
Table 32: Summary of Harms in the Measure Up 2 Study
Harms | Placebo N = 278 | UPA 15 mg q.d. N = 276 | UPA 30 mg q.d. N = 282 |
|---|---|---|---|
Patients with ≥ 1 adverse event | |||
n (%) | 146 (52.5) | 166 (60.1) | 173 (61.3) |
Most common events,a n (%) | |||
Acne | 6 (2.2) | 35 (12.7) | 41 (14.5) |
Upper respiratory tract infection | 12 (4.3) | 19 (6.9) | 17 (6.0) |
Nasopharyngitis | 13 (4.7) | 16 (5.8) | 18 (6.4) |
Headache | 11 (4.0) | 18 (6.5) | 20 (7.1) |
Patients with ≥ 1 SAE | |||
n (%) | 8 (2.9) | 5 (1.8) | 7 (2.5) |
Most common events,b n (%) | |||
Atopic dermatitis | 3 (1.1) | 1 (0.4) | 0 |
Patients who stopped treatment due to adverse events | |||
n (%) | 12 (4.3) | 11 (4.0) | 7 (2.5) |
Most common events,c n (%) | |||
Atopic dermatitis | 7 (2.5) | 3 (1.1) | 0 |
Eczema | 2 (0.7) | 0 | 1 (0.4) |
Deaths | |||
n (%) | 0 | 0 | 0 |
Notable harms | |||
Serious infection | 2 (0.7) | 1 (0.4) | 2 (0.7) |
Opportunistic infection excluding tuberculosis and herpes zoster | 0 | 3 (1.1) | 0 |
Herpes zoster | 2 (0.7) | 6 (2.2) | 3 (1.1) |
Active tuberculosis | 0 | 0 | 0 |
Possible malignancy | 0 | 2 (0.7) | 2 (0.7) |
Malignancy | 0 | 2 (0.7) | 2 (0.7) |
NMSC | 0 | 2 (0.7) | 1 (0.4) |
Malignancy other than NMSC | 0 | 0 | 1 (0.4) |
Lymphoma | 0 | 0 | 1 (0.4) |
Hepatic disorder | 4 (1.4) | 2 (0.7) | 4 (1.4) |
Adjudicated gastrointestinal perforation | 0 | 0 | 0 |
Anemia | 2 (0.7) | 2 (0.7) | 4 (1.4) |
Neutropenia | 1 (0.4) | 2 (0.7) | 6 (2.1) |
Lymphopenia | 0 | 0 | 1 (0.4) |
CPK elevation | 5 (1.8) | 9 (3.3) | 12 (4.3) |
MACE | 0 | 0 | 0 |
VTE | 1 (0.4) | 0 | 0 |
CPK = creatine phosphokinase; MACE = major adverse cardiovascular event; NMSC = non-melanoma skin cancer; q.d. = once daily; SAE = serious adverse event; UPA = upadacitinib; VTE = venous thromboembolism.
aFrequency > 5%.
bFrequency > 1%.
cFrequency > 0.5%.
Source: Clinical Study Report: Measure Up 2 Study.11
Combination Therapy Regimen Studies
AEs in the AD Up trial are depicted in Table 33. AEs were evaluated in the safety population and classified as such if they began or worsened in severity after the initiation of the study drug through 30 days following the last dose of the study drug in the respective analysis period. AEs were reported in 62.7%, 66.7%, and 72.4% of patients in the groups receiving placebo plus TCS, upadacitinib 15 mg plus TCS, and upadacitinib 30 mg plus TCS, respectively. The most common AE was acne, experienced by 2.0%, 10%, and 13.8% of patients receiving placebo, upadacitinib 15 mg, and upadacitinib 30 mg, respectively. Other AEs included upper respiratory tract infections, nasopharyngitis, and headache.
Adolescents showed a similar pattern of AEs compared to all patients except that they did not report SAEs in any treatment group. Overall, 58 adolescents out of a total of 76 adolescents (76.3%) presented at least 1 AE, including 17 patients (43.6%), 27 patients (69.2%), and 31 patients (83.8%) in the groups receiving placebo plus TCS, upadacitinib 15 mg plus TCS, and upadacitinib 30 mg plus TCS, respectively.
Table 33: Summary of Harms in the AD Up Study
Harms | Placebo + TCS N = 303 | UPA 15 mg q.d.+ TCS N = 300 | UPA 30 mg q.d. TCS N = 297 |
|---|---|---|---|
Patients with ≥ 1 adverse event | |||
n (%) | 190 (62.7) | 200 (66.7) | 215 (72.4) |
Most common events,a n (%) | |||
Acne | 6 (2.0) | 30 (10.0) | 41 (13.8) |
Upper respiratory tract infection | 22 (7.3) | 21 (7.0) | 23 (7.7) |
Nasopharyngitis | 34 (11.2) | 37 (12.3) | 40 (13.5) |
Headache | 15 (5.0) | 15 (5.0) | 14 (4.7) |
Patients with ≥ 1 SAE | |||
n (%) | 9 (3.0) | 7 (2.3) | 4 (1.3) |
Patients who stopped treatment due to adverse events | |||
n (%) | 7 (2.3) | 4 (1.3) | 4 (1.3) |
Most common events,c n (%) | |||
Atopic dermatitis | 2 (0.7) | 2 (0.7) | 0 |
Deaths | |||
n (%) | 0 | 0 | 0 |
Notable harms | |||
Serious infection | 3 (1.0) | 3 (1.0) | 0 |
Opportunistic infection excluding tuberculosis and herpes zoster | 0 | 3 (1.0) | 4 (1.3) |
Herpes zoster | 3 (1.0) | 3 (1.0) | 5 (1.7) |
Active tuberculosis | 0 | 0 | 0 |
Possible malignancy | 0 | 0 | 2 (0.7) |
Malignancy | 0 | 0 | 2 (0.7) |
NMSC | 0 | 0 | 1 (0.3) |
Malignancy other than NMSC | 0 | 0 | 1 (0.3) |
Lymphoma | 0 | 0 | 0 |
Hepatic disorder | 5 (1.7) | 6 (2.0) | 3 (1.0) |
Anemia | 1 (0.3) | 0 | 3 (1.0) |
Neutropenia | 0 | 2 (0.7) | 3 (1.0) |
Lymphopenia | 1 (0.3) | 0 | 0 |
Creatine phosphokinase elevation | 7 (2.3) | 13 (4.3) | 18 (6.1) |
MACE | 0 | 0 | 0 |
VTE | 0 | 0 | 0 |
MACE = major adverse cardiovascular event; NMSC = non-melanoma skin cancer; q.d. = once daily; SAE = serious adverse event; TCS = topical corticosteroid; UPA = upadacitinib; VTE = venous thromboembolism.
aFrequency > 5%.
bFrequency > 1%.
cFrequency > 0.5%.
Source: Clinical Study Report: AD Up.13
Head-to-Head Studies
In the Heads Up trial, AEs (Table 34) were assessed in the safety population and defined as any AE that began or worsened in severity after the initiation of upadacitinib or dupilumab (30 days following the last dose of upadacitinib or 84 days following the last dose of dupilumab). AEs were more common in the upadacitinib 30 mg group (found in 271 patients [77.9%]) than in the dupilumab group (found in 230 patients [66.9%]). Acne was the most frequently reported AE related to upadacitinib (experienced by 3.2% versus 18.4% in the dupilumab versus upadacitinib groups, respectively). Conjunctivitis was the most frequently reported AE related to dupilumab (10.2% versus 1.4% for dupilumab versus upadacitinib, respectively). Other common AEs included upper respiratory tract infections, nasopharyngitis, folliculitis, urinary tract infections, and headache. Of these, folliculitis was more common in the upadacitinib group (6.3%) versus the dupilumab group (1.2%).
Table 34: Summary of Harms in the Heads Up Study
Harms | DUP 300 mg q.2.w. N = 344 | UPA 30 mg q.d. N = 348 |
|---|---|---|
Patients with ≥ 1 adverse event | ||
n (%) | 230 (66.9) | 271 (77.9) |
Most common events,a n (%) | ||
Acne | 11 (3.2) | 64 (18.4) |
Upper respiratory tract infection | 17 (4.9) | 26 (7.5) |
Nasopharyngitis | 27 (7.8) | 23 (6.6) |
Folliculitis | 4 (1.2) | 22 (6.3) |
Urinary tract infection | 15 (4.4) | 19 (5.5) |
Conjunctivitis | 35 (10.2) | 5 (1.4) |
Headache | 24 (7.0) | 17 (4.9) |
Patients with ≥ 1 SAE | ||
n (%)b | 7 (2.0) | 14 (4.0) |
Patients who stopped treatment due to AEs | ||
n (%)b | 4 (1.2) | 11 (3.2) |
Deaths | ||
n (%) | 0 | 1 (0.3) |
Notable harms | ||
Serious infections | 2 (0.6) | 4 (1.1) |
Opportunistic infection excluding tuberculosis and herpes zoster | 0 | 3 (0.9) |
Active tuberculosis | 0 | 0 |
Malignancy | 1 (0.3) | 1 (0.3) |
NMSC | 1 (0.3) | 0 |
Malignancy excluding NMSC | 0 | 1 (0.3) |
Lymphoma | 0 | 0 |
Hepatic disorder | 5 (1.5) | 12 (3.4) |
Anemia | 1 (0.3) | 8 (2.3) |
Neutropenia | 2 (0.6) | 6 (1.7) |
Lymphopenia | 0 | 2 (0.6) |
Herpes zoster | 4 (1.2) | 12 (3.4) |
CPK elevation | 11 (3.2) | 26 (7.5) |
MACE | 0 | 0 |
VTE | 0 | 0 |
2.q.w. = every 2 weeks; AE = adverse event; CPK = creatine phosphokinase; DUP = dupilumab; MACE = major adverse cardiovascular event; NMSC = non-melanoma skin cancer; q.d. = once daily; SAE = serious adverse event; UPA = upadacitinib; VTE = venous thromboembolism.
aFrequency greater than or equal to 5%.
bFrequencies were less than 0.4% for all AEs.
Source: Clinical Study Report: Heads Up Study.14
The Japan study evaluated harms and efficacy outcomes as exploratory end points only. In this study, through week 16, the observed rates of AEs in the upadacitinib 15 mg and 30 mg groups were higher than in the placebo group overall. The percentage of patients with AEs was numerically higher in the upadacitinib 30 mg group than the upadacitinib 15 mg group. The observed rate of AEs with the reasonable possibility of being drug-related was also higher in the upadacitinib 30 mg group than in the upadacitinib 15 mg and placebo groups. Among adolescents through week 16, the rate of AEs was similar across all treatment groups.
Serious Adverse Events
Monotherapy Regimen Studies
SAEs in the Measure Up 1 trial are depicted in Table 31. In the overall population, SAEs were reported in 8 patients (2.8%) in the upadacitinib 30 mg group, 6 patients (2.1%) in the upadacitinib 15 mg group, and 8 patients (2.8%) in the placebo group. AD was reported in 3 patients in the placebo treatment group; all other SAEs were reported in only 1 patient in a treatment group. SAEs were reported in 2 adolescent patients (AD in a patient on placebo and impetigo in a patient on upadacitinib 15 mg). SAEs of chest pain, impetigo, and pharyngeal abscess in patients who received upadacitinib were considered by the investigator to have a reasonable possibility of being related to the study drug.
SAEs in the Measure Up 2 trial are shown in Table 32. In the overall population, SAEs were reported in 7 patients (2.5%) in the upadacitinib 30 mg group, 5 patients (1.8%) in the upadacitinib 15 mg group, and 8 patients (2.9%) in the placebo group. SAEs of AD (worsening of underlying disease) were reported in 3 patients in the placebo group and 1 patient in the upadacitinib 15 mg group; all other SAEs were reported in only 1 patient in a treatment group. Overall, SAEs of eczema herpeticum, orchitis, rhabdomyolysis, bipolar disorder, and suicide attempt in patients receiving upadacitinib (15 mg or 30 mg) were considered by the investigator to have a reasonable possibility of being related to the study drug.
Combination Therapy Regimen Studies
SAEs in the AD Up trial are depicted in Table 33. Serious AEs were reported in 4 patients (1.3%) in the upadacitinib 30 mg group, 7 patients (2.3%) in the upadacitinib 15 mg group, and 9 patients (3.0%) in the placebo group. No treatment group reported more than 1 SAE. No adolescents developed an SAE. Five SAEs were considered by the investigator to have a reasonable possibility of being related to the study drug, including a retinal detachment, appendicitis, pleural effusion, an intentional overdose of upadacitinib 15 mg, and a staphylococcal sepsis infection.
In the Japan study, through week 16, the number of patients with SAEs was low overall, with 1 patient in each of the 3 treatment groups reporting an SAE (cerebellar hemorrhage in the upadacitinib 15 mg group, herpes simplex in the upadacitinib 30 mg group, and cholelithiasis in the placebo group).
Head-to-Head Studies
SAEs in the Heads Up trial are shown in Table 34. In the safety population, 7 patients (2.0%) and 14 patients (4.0%) in the dupilumab and upadacitinib 30 mg groups, respectively, had an SAE. Apart from 2 events of bursitis in the dupilumab group, no SAE was reported more than once.
Withdrawals Due to Adverse Events
Monotherapy Regimen Studies
In the Measure Up 1 study, the numbers of patients with AEs leading to discontinuation of the study drug were 11 (3.9%) in the upadacitinib 30 mg group, 4 (1.4%) in upadacitinib 15 mg group, and 12 (4.3%) in the placebo group. No single event leading to study drug discontinuation was reported in more than 1 patient in the upadacitinib 30 mg or 15 mg group. In the placebo group, 5 patients (1.8%) discontinued due to worsening of AD, and 2 patients (0.7%) discontinued due to drug hypersensitivity.
In the Measure Up 2 study, the numbers of patients with AEs leading to discontinuation of study drug were 7 (2.5%) in the upadacitinib 30 mg group, 11 (4.0%) in the upadacitinib 15 mg group, and 12 (4.3%) in the placebo group. In the upadacitinib 30 mg group, no event leading to discontinuation of the study drug occurred in more than 1 patient. In the upadacitinib 15 mg group, 3 patients discontinued the study drug due to AD (i.e., worsening of underlying disease); no other event leading to discontinuation of the study drug occurred in more than 1 patient. In the placebo group, 7 patients discontinued the study drug due to AD (worsening of underlying disease) and 2 patients discontinued due to eczema. Among adolescents, the numbers of patients with AEs leading to discontinuation of the study drug were 0 in the upadacitinib 30 mg group, 2 (6.1%) in upadacitinib 15 mg group, and 1 (2.8%) in the placebo group.
Combination Therapy Regimen Studies
In the AD Up study, AEs leading to discontinuation of the study drug were reported in 4 patients (1.3%) in each of the upadacitinib treatment groups (30 mg and 15 mg) and in 7 patients (2.3%) in the placebo group. Worsening of AD occurred in more than 1 patient. Two adolescent patients discontinued the study drug (1 patient with AD on placebo and 1 patient with abnormal hepatic function on upadacitinib 15 mg).
In the Japan study, 1 patient in each of the upadacitinib 30 mg and placebo groups and 2 patients in the upadacitinib 15 mg group experienced AEs leading to discontinuation of the study drug. In the adolescent population, no SAEs or AEs leading to discontinuation were reported.
Head-to-Head Studies
In the Heads Up study, the number of AEs leading to study drug discontinuation was higher in the upadacitinib group compared to the dupilumab group. However, apart from influenza and increased alanine aminotransferase in the upadacitinib group, no other AE led to discontinuation of the study drug more than once per treatment group.
Mortality
One death associated with influenza A was reported in the upadacitinib group of the Heads Up study. No other deaths were reported in the rest of the pivotal studies included in this CADTH review.
Notable Harms
Monotherapy Regimen Studies
Notable harms in the Measure Up 1 trial and Measure Up 2 trial are presented in Table 31 and Table 32. The most common AEs specified in this review protocol and recommended by the clinical expert consulted by CADTH for consideration were CPK elevation, herpes zoster infection, hepatic disorder, and neutropenia, which were also numerically more common in the upadacitinib arms than in the placebo arm. Compared with adults, a greater proportion of adolescents had adverse events of special interest (AESIs) for CPK elevation and herpes zoster. Overall, no AESIs of active tuberculosis, lymphoma, adjudicated gastrointestinal perforation, renal dysfunction, adjudicated major adverse cardiac event (MACE), or adjudicated VTE were reported in the double-blind period. In addition, no AESIs of opportunistic infection — excluding tuberculosis, herpes zoster, malignancy, non-melanoma skin cancer, malignancy other than non-melanoma skin cancer, lymphopenia, or renal dysfunction — were reported in the adolescent group.
Combination Therapy Regimen Studies
Notable harms in the AD Up trial are depicted in Table 33. The most frequently reported notable AE was CPK elevation (6.1% on upadacitinib 30 mg, 4.3% on upadacitinib 15 mg, and 2.3% on placebo). The majority of CPK elevations were mild or moderate, were associated with exercise or heavy physical exertion, and were asymptomatic. Only 1 patient withdrew from the study due to CPK elevation. Other AESIs occurred in less than or equal to 2% of patients in any treatment group. No patient in any treatment group had an adjudicated MACE, VTE, gastrointestinal perforation, active tuberculosis, lymphoma, or lymphopenia. One event of arterial stent thrombosis was reported in a patient on placebo. Herpes zoster occurred in 5 patients on upadacitinib 30 mg, in 3 patients on upadacitinib 15 mg, and in 3 patients on placebo. No event of herpes zoster was serious or led to study drug discontinuation; most were mild or moderate and involved a single dermatome. Opportunistic infection occurred in 4 patients on upadacitinib 30 mg, in 3 patients on upadacitinib 15 mg, and in 0 on placebo. All opportunistic infections were cases of eczema herpeticum or the synonymous Kaposi's varicelliform eruption, which is the most prevalent viral complication in the general AD population. All events were non-serious, and none led to study drug discontinuation. The safety profile of adolescents was consistent with that observed for all patients.
Head-to-Head Studies
Notable harms in the Heads Up trial are presented in Table 34. The most frequently reported AESI was creatine CPK elevation, experienced by 11 patients (3.2%) in the dupilumab group and 26 patients (7.5%) in the upadacitinib group. This was followed by hepatic disorder, which was experienced by 5 patients (1.5%) in the dupilumab group versus 12 patients (3.4%) in the upadacitinib group. No patient had any events of active tuberculosis, lymphoma, adjudicated gastrointestinal perforation, MACE, or VTE.
Critical Appraisal
Internal Validity
All studies included in this CADTH review were randomized controlled trials evaluating the efficacy and safety of upadacitinib in patients with AD. The Measure Up 1 and 2, AD Up, and Japan studies were placebo-controlled, while the Heads Up study was a head-to-head randomized trial using a double-dummy, placebo-controlled design comparing upadacitinib versus dupilumab. Each trial was clearly described with specific objectives, end points, and interventions. The randomization was accomplished by an interactive response technology system that assigned a number that encoded each patient's treatment group assignment according to the randomization schedule generated by the statistics department from the sponsor, ensuring that the allocation sequence generation list and allocation concealment were properly attained; thus, there was a low risk of bias arising from the randomization process. This is emphasized by the similar distribution of baseline demographics and disease characteristic variables between the treatment groups in each trial, without important imbalances.
Blinding was adequate in all included studies and measures were taken to ensure that patients, clinicians, and all personnel beyond the statisticians and data monitoring committee were unaware of the assigned intervention during the double-blind periods. The placebos used in the Measure Up and AD Up studies were adequate in terms of similarity to the intervention, providing proper blinding throughout. The studies also used coded drug kits, and the Heads Up study used SC placebo-matched injections. Blinding of end point assessors was also adequate. Given that placebos were used, it is possible that some patients were unblinded or could have become aware of their assignments due to improvement or lack of improvement in their AD symptoms over the study period. The use of rescue medication occurred in a substantially higher number of patients in the placebo arms of the included studies. Although more objective measures would unlikely be affected, some patient-reported subjective measures could have been influenced; however, the degree and direction of the bias is unknown. There is the possibility that, in the Heads Up trial, certain adverse effects (such as injection-site or hypersensitivity reactions or conjunctivitis) that are known to be more likely with dupilumab may have potentially resulted in unblinding, or that patients (or investigators) would become aware or infer that they were assigned to dupilumab when experiencing these harms. These occurrences, which were relatively frequent (10.2% in the dupilumab group versus 1.4% in the upadacitinib group), could have affected blinding in a significant way and also influenced the reporting on more subjective outcomes. However, because the co-primary end points were relatively objective measures, the impact of bias on these end points would be expected to be small.
The co-primary outcomes assessed in all trials (except the Japan study, in which efficacy end points were exploratory and not described in this report) were based on the vIGA-AD and EASI scores. The EASI has been determined to be both reliable and valid for assessing the severity and extent of AD. A lack of minimal important difference for vIGA-AD restricts the ability to determine the clinical relevance of the vIGA-AD outcome for disease severity. The analysis of co-primary end points was conducted on all randomized patients based on the treatment allocated at the time of randomization for each trial. This ITT analysis was appropriate because it preserved statistical power and better reflected clinical practice by including patients who were non-compliant or had violated the protocol. The primary efficacy analysis for each trial was conducted using the CMH test stratified by vIGA-AD score categories and age (adolescent versus adult) in the ITT population. Continuous variables were analyzed using MMRM.
The greatest number of patients who discontinued the intervention were in the placebo groups in the Measure Up and AD Up trials. This introduces the potential for bias against the null (i.e., toward an inflated efficacy of upadacitinib) because more placebo patients would have been imputed as non-responders. The proportions of discontinuations could be considered important (for instance, in the Measure Up studies, these were around 13% and 5% in the placebo and upadacitinib arms, respectively), largely due to lack of efficacy. The investigators have assumed that missing data were MAR, which is not supported by the differential losses to follow-up and reasons for discontinuations, as noted. Moreover, the MMRM approach further assumes that data are MAR and that patients for whom data are missing data continue to behave or change in a similar fashion, as estimated by patients with ongoing data points. This assumption is strong and unverifiable and may — particularly in situations where patients discontinue therapy due to AEs or lack of efficacy, as observed in these trials — increase the bias in the observed results. Although the difference between groups might be considered small, some bias in results would be expected. Although the investigators used multiple imputation (which is appropriate only when the data are truly MAR [i.e., missing completely at random]) and non-responder imputation to account for missing data, neither may fully account for the bias potentially introduced by the missing data. Indeed, given that missing data were more common for placebo patients, the non-responder imputation could have further inflated the efficacy results in favour of upadacitinib in some of the trials. Nonetheless, it is important to note that sensitivity analyses were conducted based on per-protocol populations and a tipping point analysis, supporting the robustness of the conclusions of the primary analyses.
The secondary end points were analyzed based on complete case analyses. For instance, in the change in EASI score from baseline at week 16, only patients with non-missing change-from-baseline values were included in the analysis. This occurred in other end points, such as SCORAD, WP-NRS, HRQoL, HADS, and WPAI. This is expected to introduce some risk of bias in favour of upadacitinib (given that more complete data were available for upadacitinib due to lower discontinuations and drop-outs) because the groups may no longer be balanced in characteristics, and the data observed from the incomplete cases are discarded (i.e., patients who are responding to treatment and have limited AEs may be more likely to stay in the study and contribute data to the end points). Complete case analyses can give valid inferences only when data are missing completely at random, which does not seem to be the case in the studies included in this review.
In the Heads Up study, conducting the primary end point analysis at 16 weeks — at which time the efficacy of dupilumab would not have peaked and would be than that of upadacitinib — could have misinformed the analysis in favour of upadacitinib. At week 24, small to no differences were observed between upadacitinib and dupilumab for the primary (EASI 75) and secondary end points.
The use of a graphical multiple testing procedure to control for multiplicity was appropriate for the primary and secondary end points for upadacitinib 15 mg and upadacitinib 30 mg. The graphical approach provides a transparent and flexible framework to design and implement multiple testing procedures. The procedure was pre-specified in the protocol, supported by previous data, and avoided data-driven decisions.
Sensitivity analyses were based on multiple imputation, the tipping point approach, and the per-protocol population, and yielded conclusions that were similar to those of the primary analyses. Several subgroup analyses were properly specified a priori and conducted across the trials (e.g., based on baseline vIGA-AD, baseline EASI, previous systemic therapy, age, and sex, among others); however, as with most subgroups, interpretation should be done cautiously because most were not included as stratification variables in the randomization scheme, and imbalances between groups may exist within the subgroups.
External Validity
The population in the included pivotal studies was generalizable to adults and adolescents in the Canadian population having AD. However, when considering the applicability of the results, the population of patients previously treated with systemic therapies (i.e., the current indication for upadacitinib) represented only a proportion of the population included in the pivotal studies. Furthermore, the information from the pivotal studies for the 30 mg dose also represents a proportion of the population, and data to inform the approved indication are lacking. (In other words, data estimating the effects of upadacitinib in patients with severe AD who switch from 15 mg to 30 mg if there is no initial response are absent.) This lack of evidence adds uncertainty to the generalizability of the results in the population to whom the indication would apply. Also, no evidence was found for the scenario of patients who would start on 30 mg, respond, and then switch to 15 mg of upadacitinib. The clinical expert consulted by CADTH indicated that these analyses suggested that the response to upadacitinib would likely be similar for those with and without prior exposure to a systemic therapy for AD.
The patient population in Measure Up 1 appeared to have slightly less severe atopic dermatitis than those in the other studies: 45% of the population in Measure Up 1 had vIGA-AD scores indicating severe AD, whereas that figure reached 55% in the Measure Up 2, AD Up, and Heads Up studies. The other baseline and demographic characteristics were overall similar between studies and deemed representative of the Canadian population. One expected issue is the relatively short duration of the studies to evaluate long-term outcomes. Table 35 summarizes the generalizability of the evidence.
The adolescent substudy is ongoing, and results will be reported separately. The adolescent population analyzed in these included studies (except in the Heads Up trial) mirrored the results from the adult population overall (Appendix 3), with some loss in precision in certain outcomes; hence the uncertainty in the estimated effects. However, up-to-date data from these reports are still small in quantity and of short duration for certain long-term outcomes.
Despite the relatively small number of Canadians in these studies, the clinical expert consulted in this review suggested that the study population was generally representative of the Canadian adult and adolescent patients treated in clinical practice. However, more evidence is needed in under-represented populations, such as people of Black or African, First Nations, and Asian descent.
The inclusion and exclusion criteria for each study were clearly described and similar between studies. Only the Heads Up study differed in that it did not include adolescents, due to the use of dupilumab. These inclusion criteria, among others relating to AD therapies used within specific time frames, create a study population that may be consistent with the Canadian population.
The vIGA-AD and EASI scores were part of the co-primary outcomes of the studies. These instruments are standard tools used in clinical trials; however, their use in clinical practice is still limited. According to the clinical expert consulted by CADTH, these are not difficult to implement and do not require specific or complicated training.
Table 35: Assessment of Generalizability of Evidence for Upadacitinib in AD
Domain | Factor | Evidence | CADTH assessment of generalizability |
|---|---|---|---|
Population | Patients with moderate to severe AD not adequately controlled with topical and/or systemic therapies | Inclusion and exclusion criteria for the trials | The eligibility criteria for the included RCTs were similar except for the age ranges in the Heads Up study, which included adults only. All trials enrolled patients with moderate to severe AD and an inadequate response to topical AD therapies. This is reflective of the indication that was initially submitted to Health Canada and CADTH; however, the approved Health Canada indication reflects a more restrictive population (i.e., those with refractory, moderate to severe AD and an inadequate response to other systemic drugs). There are some issues with the small representation of African American or Black and Asian patients vs. White patients. |
Intervention | Upadacitinib 15 mg and 30 mg with or without TCS | Trial protocols | No concerns of generalizability for patients using the Health Canada–approved initial dose of 15 mg once daily. The 30 mg dose seems to be appropriate for patients who do not initially respond to the 15 mg dose, or for some patients whose AD is severe enough to start at 30 mg. However, no evidence was available for assessing generalizability in those cases starting at 30 mg and then reducing the dose to 15 mg once clinical response is achieved. The combination of TCS with upadacitinib was evaluated in the AD Up study; this combination is very likely to be used in clinical practice. |
Comparator | Biologics (e.g., dupilumab) Immunomodulating drugs (e.g., methotrexate, cyclosporine A, azathioprine, mycophenolate mofetil) | Trial protocols | Only the Heads Up study compared upadacitinib against dupilumab, a decision that will — according to the clinical expert — be commonly considered in clinical practice. The rest of the studies evaluated upadacitinib vs. placebo. |
Outcomes |
| Trial protocols | The outcomes are relevant to patients and clinicians and applicable to clinical practice. There are some issues with the short-term duration of the evaluation of outcomes in the placebo trials as well as in the Heads Up trial when comparing dupilumab to upadacitinib. Most of these outcomes were valid and reliable in studies assessing patients with AD (refer to Appendix 4). |
Setting | Outpatient setting | Trial sites | The administration of upadacitinib does not require special inpatient settings. |
AD = atopic dermatitis; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; RCT = randomized controlled trial; SCORAD = Scoring Atopic Dermatitis; TCS = topical corticosteroid; vIGA-AD = validated Investigator Global Assessment for Atopic Dermatitis.
Indirect Evidence
Objectives and Methods for the Summary of Indirect Evidence
The objective of this section is to summarize and appraise the indirect evidence comparing the relative efficacy and safety of upadacitinib in patients with moderate to severe AD against relevant comparators, as established in the protocol of this review.
A supplemental literature search was conducted by CADTH to identify additional ITCs. A focused literature search for ITCs dealing with AD was run. Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers made the final selection of studies included in the review, and differences were resolved through discussion. Based on this literature search conducted by CADTH, 1 additional ITC from ICER was identified and included in this review report.
Description of Indirect Comparisons
Three ITCs were included in this clinical review report. The first is an NMA comparing upadacitinib 15 mg or upadacitinib 30 mg (with or without a TCS) against dupilumab in adults or adolescents with moderate to severe AD with an inadequate response to cyclosporine A or other systemic therapy (i.e., ITC 1, a post-cyclosporine A NMA).64 The second (ITC 2)65 is an NMA of a comprehensive, published RCT evidence base to determine the comparative effectiveness of upadacitinib versus other immunomodulators in patients with moderate to severe AD as monotherapy (i.e., not concomitantly receiving TCS) and as combination therapy (i.e., concomitantly receiving a TCS). The third ITC is an evidence report from ICER66 aimed at evaluating systemic therapies (abrocitinib, baricitinib, upadacitinib, tralokinumab, dupilumab) with or without topical therapies in adults and adolescents (≥ 12 years old) with moderate to severe AD. The characteristics of each ITC and selection criteria are presented in Table 36.
Table 36: Study Selection Criteria and Methods for ITCS
Criteria and methods | ITC 1: post-cyclosporine A NMA | ITC 2: post-topicals NMA | ITC 3: ICER evidence report |
|---|---|---|---|
Population | Adults and adolescents (≥ 12 years) with moderate to severe AD and an inadequate response or intolerance to cyclosporine A or any systemic therapy | Adults and adolescents (≥ 12 years) with moderate to severe AD | Adults and adolescents (≥ 12 years) with moderate to severe AD |
Interventions | Any formulation of the following (with or without combination corticosteroids, concomitant therapies [e.g., emollients], rescue therapy, and/or re-treatment):
| Any formulation of the following (with or without combination corticosteroids, concomitant therapies [e.g., emollients], rescue therapy, and/or re-treatment):
| Abrocitinib Baricitinib Upadacitinib Tralokinumab (All used alone or with topical therapies) |
Comparators | Placebo Active intervention (i.e., head-to-head trials) | Placebo Active intervention (i.e., head-to-head trials) | Dupilumab Placebo Any treatment previously listed (All used alone or with topical therapies) |
Outcomes | EASI (e.g., EASI 50, EASI 75, EASI 90) EASI and DLQI composite (e.g., EASI 50 + DLQI ≥ 4) | EASI vIGA-AD WP-NRS | Efficacy outcomes Patient-reported pruritus or itching EASI 50, EASI 75, EASI 90 vIGA-AD Sleep SCORAD POEM score DLQI score CLDQI score HADS score EQ-5D Measures of productivity Safety/adverse events All-cause mortality |
Study design | RCTs (phase III) | RCTs (phase III and IV) | RCTs (phase III) |
Publication characteristics |
|
| English only |
Exclusion criteria | Excluded studies containing only:
Children (< 12 years) and patients with other active skin diseases, infections | Excluded studies containing only:
Children (< 12 years) and patients with other active skin diseases, infections | Articles indexed as guidelines, letters, editorials, narrative reviews, case reports, or news items |
Databases searched | MEDLINE, Embase, LILACS, PsycINFO, CENTRAL, CDSR, DARE, grey literature | MEDLINE, Embase, LILACS, PsycINFO, CENTRAL, CDSR, DARE, grey literature | MEDLINE, Embase, Cochrane Library (CDSR and CENTRAL) |
Selection process | Articles screened independently by 2 researchers | Articles screened independently by 2 researchers | Full-text articles screened by single reviewer, providing justification for exclusions |
Data extraction process | Two researchers independently extracted data, and disagreements were resolved by discussion or a third reviewer | Two researchers independently extracted data, and disagreements were resolved by discussion or a third reviewer | Not specified |
Quality assessment | Studies were critically appraised for methodological quality using validated tools in accordance with NICE single technology appraisal and highly specialized technologies evaluation: User guide for company evidence submission template | Studies were critically appraised for methodological quality using validated tools in accordance with NICE single technology appraisal and highly specialized technologies evaluation: User guide for company evidence submission template | Criteria published by the US Preventive Services Task Force |
2.q.w. = every 2 weeks; AD = atopic dermatitis; CDLQI = Children’s Dermatology Life Quality Index; CDSR = Cochrane Database of Systematic Reviews; CENTRAL = Cochrane Central Register of Controlled Trials; DARE = Database of Abstracts of Reviews of Effects; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; EASI 50 = at least 50% improvement in EASI total score; EASI 75 = at least 75% improvement in EASI total score; EASI 90 = at least 90% improvement in EASI total score; HADS = Hospital Anxiety and Depression Scale; ICER = Institute for Clinical and Economic Review; ITC = indirect treatment comparison; IL = interleukin; JAK = Janus kinase; LILACS = Latin American and Caribbean Health Sciences Literature; NICE = National Institute for Health and Care Excellence; NMA = network meta-analysis; POEM = Patient-Oriented Eczema Measure; q.d. = once daily; RCT = randomized controlled trial; SCORAD = Scoring Atopic Dermatitis; vIGA-AD = validated Investigator Global Assessment for Atopic Dermatitis; WP-NRS = Worst Pruritis Numerical Rating Scale.
Source: Indirect comparisons submitted by the sponsor and ITC performed by ICER.
Indirect Treatment Comparison 1: Post-Cyclosporine Network Meta-Analysis
Methods
Objectives
This ITC’s main objective was to conduct an NMA of a comprehensive, published, randomized clinical trials evidence base to determine the comparative effectiveness of upadacitinib versus dupilumab in patients with moderate to severe AD who have had an inadequate response or intolerance to cyclosporine A according to concomitant use of TCS among patients (i.e., with TCS versus without TCS). Different approaches on how the use of rescue medication is considered in the trial dataset (primary analysis versus all-observed analysis) are also analyzed.
Study Selection Methods
A clinical, sponsor-run, systematic literature review (SLR) identified phase III RCTs that assessed and reported the clinical efficacy of relevant AD treatments. Data from eligible RCTs were collected using an Excel-based data extraction form. In addition to the outcomes of interest, study design and patient baseline characteristics were extracted to assess the comparability of the studies and identify the presence of heterogeneity. Authors included published and unpublished RCTs evaluating the efficacy and safety of competing interventions used for the treatment of moderate to severe AD in patients with an inadequate response or intolerance to cyclosporine A. Two researchers extracted data independently, and disagreements were resolved by discussion or a third reviewer.
In addition to data gathered as part of the clinical SLR, data were also extracted from the following clinical trials: Measure Up 1, Measure Up 2, AD Up, and Heads Up. For this ITC, beyond the SLR criteria, a narrower set of criteria was established to include a population with an inadequate response or intolerance to cyclosporine A or any systemic therapy.
The available RCTs for AD could be separated into those in which patients received an immunomodulator plus a TCS or placebo plus TCS (i.e., combination therapy) and those in which patients received only an immunomodulator or only placebo (i.e., monotherapy).
Trials that included TCSs defined those therapies to include hydrocortisone 1% cream, triamcinolone acetonide 0.1% cream, and fluocinolone acetonide 0.025% ointment.
Indirect Treatment Comparison Analysis Methods
Two trial populations were explored in the NMA, reflecting different approaches for considering the use of rescue medication in the trial dataset:
all-observed analysis (patients who responded and received rescue medication were considered as responders)
primary analysis (patients who responded and received rescue medication were considered as non-responders).
Furthermore, networks were built based on the use of monotherapy or combination therapy, resulting in the next 4 networks:
all-observed analysis with monotherapy: all included RCTs for monotherapy using all observed data regardless of rescue use to determine response
all-observed analysis with combination therapy: all included RCTs for combination therapy with TCS using all observed data regardless of rescue use to determine response
primary analysis with monotherapy: all included RCTs for monotherapy where patients requiring rescue are considered non-responders
primary analysis with combination therapy: all included RCTs for combination therapy with TCS where patients requiring rescue are considered non-responders.
Outcomes included EASI (i.e., EASI 50, EASI 75, and EASI 90) and the composite of EASI and DLQI (i.e., EASI 50 plus DLQI ≥ 4). These end points were assessed at week 16. An ITT perspective was used, defined whereby the denominator was based on the sample size at randomization. No continuous outcomes were evaluated. All trials (Measure Up, AD Up, Heads Up, SOLO 1, SOLO 2, CHRONOS, and CAFÉ) imputed missing patient values following discontinuation or rescue use as non-responders.
Specific analysis methods are described in Table 37.
Table 37: Indirect Treatment Comparison 1 Analysis Methods
Characteristic | ITC 1: post-cyclosporine A |
|---|---|
ITC methods | An NMA was conducted using 2 approaches:
|
Priors | Vague or flat prior distributions were given to the parameters to be estimated by default. For parameters assumed to be specified on a continuous scale — namely, the relative treatment effects d, trial-specific baselines mu, and baseline adjustment regression term B (for models with baseline risk adjustment) — a normal (0 to 1,002) prior distribution was used. For the between-study standard deviation sigma (for RE models), a uniform (0 to 5) prior distribution was used. |
Assessment of model fit | The models’ global fits were assessed and compared using their overall residual deviance, effective number of parameters, deviance information criteria, and the posterior distribution of the between-study standard deviation (sigma) associated with the RE model. |
Assessment of consistency | FE and RE inconsistency models were used and their fit was compared to corresponding consistency models. Plots of the individual data points’ posterior mean deviance contribution in each of the 2 models along with the line of equality were produced. |
Assessment of convergence | The Brooks-Gelman-Rubin method using the PRSF was used. To reach proper convergence, the PRSF should gradually shrink to 1, with increasing numbers of iterations; a value of < 1.05 was used to indicate a good convergence. |
Outcomes |
|
Follow-up time points | 16 weeks |
Construction of nodes | Four NMAs were conducted for each outcome:
The NMAs were conducted in a generalized linear model framework using Bayesian Markov Chain Monte Carlo simulations using 4 chains with 100,000 runs each. |
Sensitivity analyses | Not reported. |
Subgroup analysis | Baseline characteristics were a priori identified and analyzed as potential effect modifiers. These included: age, gender, duration of disease, and baseline severity (i.e., baseline EASI, baseline vIGA-AD, WP-NRS, baseline DLQI). |
FE = fixed effects; DLQI = Dermatology Life Quality Index;; EASI = Eczema Area and Severity Index; EASI 50 = at least 50% improvement in EASI total score; EASI 75 = at least 75% improvement in EASI total score; EASI 90 = at least 90% improvement in EASI total score; ITC = indirect treatment comparison; NMA = network meta-analysis; PRSF = potential scale reduction factor; RCT = randomized controlled trial; RE = random effects; vIGA-AD = validated Investigator Global Assessment for Atopic Dermatitis; WP-NRS = Worst Pruritis Numerical Rating Scale.
Source: Sponsor-submitted ITC 1.64
Results of Indirect Treatment Comparison 1
Summary of Included Studies
In total, 8 studies were considered potentially relevant for the analysis: 4 that had been identified in the SLR (SOLO1, SOLO2, CAFÉ, CHRONOS), and 4 upadacitinib studies (Measure Up 1, Measure Up 2, AD Up, and Heads Up). The characteristics of the 8 included studies are presented in Table 38.
Table 38: Overview of the Studies Included From the Clinical Systematic Literature Review
Study | Population | Intervention or comparator | Number of patients | Treatment duration (time on treatment and study duration) | Primary outcomes reported |
|---|---|---|---|---|---|
SOLO 1 | Adults ≥ 18 years with moderate to severe AD for which topical TX provided inadequate control or was medically inadvisable | DUP 300 mg q.w. or 2.q.w., SC Placebo q.w., SC | 671 DUP 300 mg 2.q.w.: 224 DUP 300 mg q.w.: 223 PBO: 224 | 16 wks Study: up to 28 wks | IGA 0 or 1 and reduction of ≥ 2 points EASI 75 (co-primary (in EU and Japan only) |
SOLO 2 | Adults ≥ 18 years with moderate to severe AD for which topical TX provided inadequate control or was medically inadvisable | DUP 300 mg q.w. or 2.q.w., SC PBO q.w., SC | 708 DUP 300 mg 2.q.w.: 233 DUP 300 mg q.w.: 239 PBO: 236 | 16 wks Study: up to 28 wks | Same as SOLO 1 |
CAFÉ | Adults ≥ 18 years with moderate to severe AD and IR to TCS, or intolerance and/or unacceptable toxicity to CsA, or CsA-naive PTS for whom CsA TX is contraindicated | DUP SC 300 mg DUP q.w. 300 mg 2.q.w. PBO | 325 DUP 300 mg q.w.: 110 DUP 300 mg 2.q.w.: 107 PBO: 108 | TX: 16 wks Study: 32 wks (Screening: 4 wks; follow-up: 12 wks) | EASI 75 EASI 50 or 90 WP-NRS ≥ 4-point reduction |
CHRONOS | Age ≥ 18 years with moderate to severe AD and a history of inadequate response to medium- to high-potency TCSs (± TCIs) or systemic TX (or both) within the past 6 months | DUP SC: 300 mg q.w. or 300 mg 2.q.w. Loading dose: 600 mg with TCS PBO q.w., SC with TCS | 740 DUP 300 mg q.w. + TCS: 319 DUP 300 mg 2.q.w. + TCS: 106 PBO + TCS: 315 | TX: 52 wks Study: 64 wks | IGA 0 or 1 and ≥ 2-point reduction EASI 75 (co-primary) |
Measure Up 1 | Adolescent and adult patients with moderate to severe atopic dermatitis | UPA 15 mg q.d. UPA 30 mg q.d. PBO | 810 UPA 15 mg q.d.: 270 UPA 30 mg q.d.: 270 PBO: 270 | 16-week double-blind period, 120-week blinded extension period | IGA 0 or 1 EASI 75 |
Measure Up 2 | Adolescent and adult patients with moderate to severe atopic dermatitis | UPA 15 mg q.d. | 810 UPA 15 mg q.d.: 270 UPA 30 mg q.d.: 270 PBO: 270 | 16-week double-blind period, 120-week blinded extension period | IGA 0 or 1 EASI 75 |
AD Up | Adolescent and adult patients with moderate to severe atopic dermatitis | UPA 15 mg q.d. + TCS UPA 30 mg q.d.+ TCS PBO + TCS | 810 UPA 15 mg q.d.: 270 UPA 30 mg q.d.: 270 PBO: 270 | 16-week double-blind period, 120-week blinded extension period | IGA 0 or 1 EASI 75 |
Heads Up | Adult patients with moderate to severe atopic dermatitis | UPA 30 mg q.d. + TCS DUP 300 mg 2.q.w. + TCS | 650 UPA 30 mg q.d.: 325 DUP 300 mg 2.q.w.: 325 | 24-week double-blind period, follow-up visit 12 weeks after last injection | EASI 75 |
2.q.w. = every 2 weeks; AD = atopic dermatitis; CsA = cyclosporine A; DUP = dupilumab; EASI = Eczema Area and Severity Index; EASI 50 = at least 50% improvement in EASI total score; EASI 75 = at least 75% improvement in EASI total score; EASI 90 = at least 90% improvement in EASI total score; IGA = Investigator Global Assessment; IR = inadequate response; ITC = indirect treatment comparison; PBO = placebo; PTS = patient; q.d. = once daily; q.w. = once a week; SC = subcutaneous; TCI = topical calcineurin inhibitor; TCS = topical corticosteroid; TX = treatment; UPA = upadacitinib; wks = weeks; WP-NRS = Worst Pruritis Numerical Rating Scale.
Source: Sponsor-submitted ITC 1.64
Four unique RCTs (Measure Up 1, Measure Up 2, SOLO 1, and SOLO 2) containing 4 arms were included in the monotherapy network, while 4 unique RCTs (AD Up, Heads Up, CAFÉ, and CHRONOS) containing 4 arms were included in the combination therapy network. Studies were critically appraised for methodological quality using validated tools in accordance with National Institute for Health and Care Excellence (NICE) requirements, as specified in the NICE single technology appraisal and highly specialized technologies evaluation: User guide for company evidence submission template. The critical appraisal included 7 specific domains: random sequence generation, allocation concealment, blinding of participants, blinding of investigators, blinding of outcome assessors, incomplete outcome data, selective reporting, and other potential sources of bias that could affect the internal or external validity and generalizability of the study findings to the general population. All 8 studies were assessed as at low risk of bias, according to this tool.
Baseline characteristics identified and measured by the authors to be potential treatment-effect modifiers included age, gender, duration of disease, and baseline severity (i.e., baseline EASI, baseline IGA, baseline WP-NRS, and baseline DLQI).
Overall, patient ages were similar between the monotherapy studies (with mean ages ranging from 31 years to 39 years) and combination therapy studies (with mean ages ranging from 35 years to 38 years). There were more male patients in all monotherapy and combination therapy studies, with proportions ranging from 53% to 78%; the Measure Up 1 study was an exception, with 47% male patients. The proportion of patients with severe AD (IGA = 4) ranged from 54% to 73%, and EASI mean scores were in the range of 30 to 37.
The CAFÉ study was the only RCT conducted for dupilumab that targeted patients with an inadequate response or intolerance to cyclosporine A. Due to the absence of information about the subgroup of patients previously treated with cyclosporine A in the SOLO 1, SOLO 2, and CHRONOS studies, the authors used a NICE single-technology appraisal report of dupilumab for treating moderate to severe AD after topical treatments (TA543). They extracted pooled data from SOLO 1 and SOLO 2 (named SOLO CAFÉ-like data), and pooled data from CAFÉ and CHRONOS (named CAFÉ + CHRONOS CAFÉ-like data). Hence, the extracted data in this NMA included information on patients who showed inadequate efficacy of response to oral cyclosporine A, inadequate efficacy of response or intolerance to oral cyclosporine A, or had not received prior oral cyclosporine A treatment because cyclosporine A was contraindicated or otherwise medically inadvisable.
Results
The network plots of all included RCTs are shown in Figure 8 and Figure 9.
Baseline (placebo) risks for each outcome were assessed across the included RCTs. Heterogeneity was evaluated through meta-analysis for single proportions using generalized linear model regression with a binomial distribution and logit-link function. Results for the I2 statistic and heterogeneity test are summarized in Table 39. Overall, there was moderate heterogeneity across the end points for the all-observed analysis populations and low heterogeneity for the primary analysis populations.
Figure 8: Network Plot of All Included RCTs for Monotherapy (All-Observed and Primary Analyses)
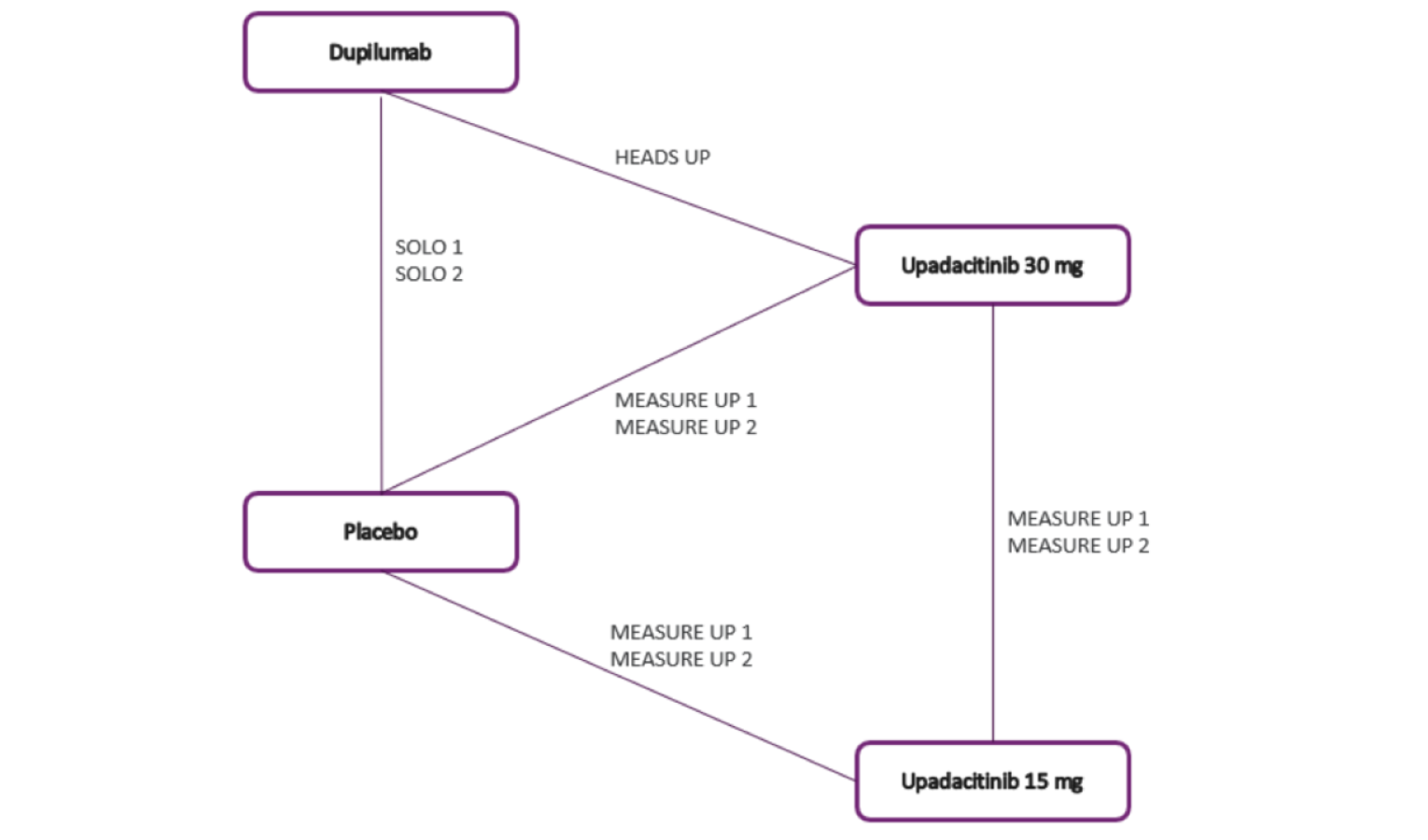
RCT = randomized controlled trial.
Source: Sponsor-submitted ITC 1.64
Figure 9: Network Plot of All Included RCTs for Combination Therapy With TCS (All-Observed and Primary Analyses)
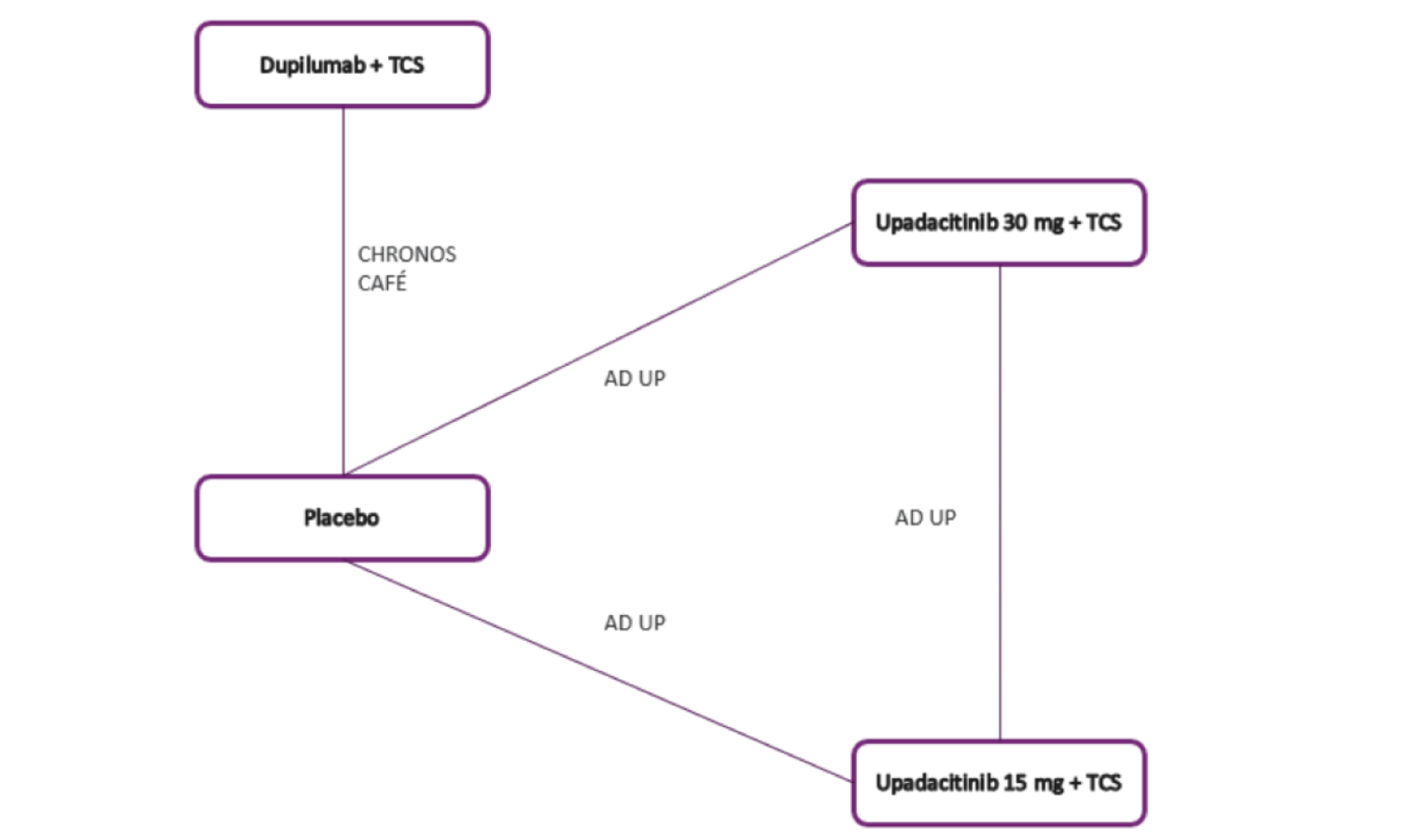
RCT = randomized controlled trial; TCS = topical corticosteroid.
Source: Sponsor-submitted ITC 1.64
Table 39: ITC 1 Assessment of Placebo Response Rate Heterogeneity in the All-Observed and Primary Analysis Populations — Redacted
Network | End point | Response rate (%) | GLMM ln (odds) | GLMM SE ln (odds) | I2 statistic | Heterogeneity P value |
|---|---|---|---|---|---|---|
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; GLMM = generalized linear mixed models; Ln = natural logarithm; ITC = indirect treatment comparison; SE = standard error.
Note: Redacted rows have been deleted.
Source: Sponsor-submitted ITC 1.64
Monotherapy
The fixed-effects (FE) model indicated an appropriate model fit. The FE model presented a smaller deviance information criterion (DIC) and residual deviance compared to the random-effects (RE) model. Individual trial results for the EASI 75 end point in the monotherapy network following the primary and all-observed analysis methodology are summarized in Table 40.
Table 40: Individual Study Data — EASI 75 at Week 16 (EASI 75, Primary and All-Observed Analysis, Monotherapy Network)
Treatment | Study | Responders | |
|---|---|---|---|
N (%) | Sample size | ||
Primary Analysis | |||
Placebo | Measure Up 1 | 5 (12.50) | 40 |
Upadacitinib 15 mg | Measure Up 1 | 23 (58.97) | 39 |
Upadacitinib 30 mg | Measure Up 1 | 23 (71.88) | 32 |
Placebo | Measure Up 2 | 3 (4.69) | 64 |
Upadacitinib 15 mg | Measure Up 2 | 44 (58.67) | 75 |
Upadacitinib 30 mg | Measure Up 2 | 42 (73.68) | 57 |
Dupilumab 300 mg 2.q.w. | SOLO 1 to 2 | 42 (40.38) | 104 |
Placebo | SOLO 1 to 2 | 10 (11.36) | 88 |
All-Observed Analysis | |||
Placebo | Measure Up 1 | 9 (22.50) | 40 |
Upadacitinib 15 mg | Measure Up 1 | 26 (66.67) | 39 |
Upadacitinib 30 mg | Measure Up 1 | 24 (75.00) | 32 |
Placebo | Measure Up 2 | 4 (6.25) | 64 |
Upadacitinib 15 mg | Measure Up 2 | 46 (61.33) | 75 |
Upadacitinib 30 mg | Measure Up 2 | 44 (77.19) | 57 |
Dupilumab 300 mg 2.q.w. | SOLO 1 to 2 | 47 (45.19) | 104 |
Placebo | SOLO 1 to 2 | 15 (17.05) | 88 |
2.q.w. = every 2 weeks; EASI 75 = at least 75% improvement in EASI total score; ITC = indirect treatment comparison.
Source: Sponsor-submitted ITC 1.64
The results presented in the league table for the primary analysis are shown in Table 41, with the all-observed analysis in Table 42. In the primary analysis network, upadacitinib 30 mg was superior as monotherapy against dupilumab to reach EASI 75 (odds ratio [OR] = 6.40; 95% CI, 2.0 to 21.3), and upadacitinib 15 mg was superior as monotherapy against dupilumab to reach EASI 75 (OR = 3.33; 95% CI, 1.08 to 10.63).
When considering the all-observed network, the results were slightly different, yielding lower OR values but remaining overall consistent with the primary analyses, as shown in Table 42.
Table 41: League Table With Odds Ratio and 95% Credible Interval for Pairwise Comparisons (Fixed Effects) (EASI 75, Primary Analysis, Monotherapy Network) — ITC 1 — Redacted
Outcome | Placebo vs. comparators | Upadacitinib 15 mg vs. comparators | Upadacitinib 30 mg vs. comparators | Dupilumab 300 mg 2.q.w. vs. comparators |
|---|---|---|---|---|
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
2.q.w. = every 2 weeks; EASI 75 = at least 75% improvement in EASI total score; ITC = indirect treatment comparison; vs. = versus.
Note: Redacted rows have been deleted.
Source: Sponsor-submitted ITC 1.64
Table 42: League Table With Odds Ratio and 95% Credible Interval for Pairwise Comparisons (Fixed Effects) (EASI 75, All-Observed Analysis, Monotherapy Network) — ITC 1 — Redacted
Outcome | Placebo vs. comparators | Upadacitinib 15 mg vs. comparators | Upadacitinib 30 mg vs. comparators | Dupilumab 300 mg 2.q.w. vs. comparators |
|---|---|---|---|---|
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
2.q.w. = every 2 weeks; EASI 75 = at least 75% improvement in EASI total score; ITC = indirect treatment comparison; vs. = versus.
Note: Redacted rows have been deleted.
Source: Sponsor-submitted ITC 1.64
Combination Therapy
Individual trial results for the EASI 75 end point in the combination therapy network following the primary and all-observed analysis methodology are summarized in Table 43. There were no meaningful differences in the fit (residual deviance) or DIC between the FE and RE NMA models. Thus, the simpler, FE model was chosen.
Table 43: Individual Study Data — EASI 75 at Week 16 (EASI 75, Primary and All-Observed Analysis, Combination Therapy Network)
Treatment | Study | Responders | |
|---|---|---|---|
N (%) | Sample size | ||
Primary Analysis | |||
Placebo | AD Up | 13 (24.07) | 54 |
Upadacitinib 15 mg | AD Up | 39 (67.24) | 58 |
Upadacitinib 30 mg | AD Up | 43 (75.44) | 57 |
Dupilumab 300 mg 2.q.w. | CAFÉ | 67 (62.62) | 107 |
Placebo | CAFÉ | 32 (29.63) | 108 |
Dupilumab 300 mg 2.q.w. | CHRONOS | 16 (69.57) | 23 |
Placebo | CHRONOS | 11 (18.03) | 61 |
All-Observed Analysis | |||
Placebo | AD Up | 16 (29.63) | 54 |
Upadacitinib 15 mg | AD Up | 40 (68.97) | 58 |
Upadacitinib 30 mg | AD Up | 46 (80.70) | 57 |
Dupilumab 300 mg 2.q.w. | CAFÉ | 69 (64.49) | 107 |
Placebo | CAFÉ | 35 (32.41) | 108 |
Dupilumab 300 mg 2.q.w. | CHRONOS | 18 (78.26) | 23 |
Placebo | CHRONOS | 16 (26.23) | 61 |
2.q.w. = every 2 weeks; EASI 75 = at least 75% improvement in EASI total score; ITC = indirect treatment comparison.
Source: Sponsor-submitted ITC 1.64
The results presented in the league table for the primary analysis are shown in Table 44, and in Table 45 for the all-observed analysis. In the primary analysis network, no difference was detected between upadacitinib 30 mg as combination therapy versus dupilumab to reach EASI 75 (OR = 2.01; 95% CI, 0.74 to 5.68) or between upadacitinib 15 mg as combination therapy versus dupilumab to reach EASI 75 (OR = 1.32; 95% CI, 0.50 to 3.63).
Table 44: League Table With Odds Ratio and 95% Credible Interval for Pairwise Comparisons (Fixed Effects) (EASI 75, Primary Analysis, Combination Therapy Network) —ITC 1 — Redacted
Outcome | Placebo | Upadacitinib 15 mg | Upadacitinib 30 mg | Dupilumab 300 mg 2.q.w. |
|---|---|---|---|---|
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
2.q.w. = every 2 weeks; EASI 75 = at least 75% improvement in EASI total score; ITC = indirect treatment comparison.
Note: Redacted rows have been deleted.
Source: Sponsor-submitted ITC 1.64
Table 45: League Table With Odds Ratio and 95% Credible Interval for Pairwise Comparisons (Fixed Effects) (EASI 75, All-Observed Analysis, Combination therapy Network) — ITC 1 — Redacted
Outcome | Placebo | Upadacitinib 15 mg | Upadacitinib 30 mg | Dupilumab 300 mg 2.q.w. |
|---|---|---|---|---|
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
|||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| | |||||||||||||||| |
2.q.w. = every 2 weeks; EASI 75 = at least 75% improvement in EASI total score; ITC = indirect treatment comparison.
Note: Redacted rows have been deleted.
Source: Sponsor-submitted ITC 1.64
Critical Appraisal of ITC 1
ITC 1 is a sponsor-submitted NMA obtained from a primary SLR also performed by the sponsor. Based on the methods detailed in the sponsor SLR, overall the ITC has an adequate search strategy, screening, and appraisal of the risk of bias of the included studies done by 2 reviewers who solved disagreements by discussion.
Overall, the researchers appropriately identified and included all relevant trials for their PICO question except for those not published yet . The networks were adequately constructed with proper modelling and fitting of the models. The individual included studies are of low risk of bias, according to the investigators’ judgments and based on the NICE guidance methods. There were no significant systematic differences in the treatment-effect modifiers detected across the different treatment comparisons in the networks, although not all existent clinically relevant effect modifiers were examined.
Statistical methods were appropriate and aimed at preserving within-study randomization. However, only network estimates are presented, which precludes us from making judgments about the certainty in the effect estimates from direct and indirect comparisons. Furthermore, there is no discussion of possible pairwise heterogeneity, no discussion of transitivity assumption in the overall network, and no presentation of the pairwise comparisons.
The populations addressed by the ITC are relevant, but very specific to the review question; that is, the population of interest aims to include patients with moderate to severe AD with an inadequate response or intolerance to cyclosporine A. In this ITC, the CAFÉ study was the only RCT that targeted patients with an inadequate response or intolerance to cyclosporine A. Data on cyclosporine A intolerance were not available for the other included trials (SOLO 1, SOLO 2, CHRONOS, Measure Up 1, Measure Up 2, AD Up, and Heads Up); therefore, the authors had to use 2 measures. First, they extracted pooled data from a NICE report evaluating patients with moderate to severe AD with an inadequate response to cyclosporine A. Second, because data from the upadacitinib studies (Measure Up 1 and 2, AD Up, and Heads Up) for patients with intolerance to cyclosporine A were not available, the authors extracted data about patients with inadequate response to cyclosporine A; based on consultation with clinical experts, this was considered equivalent to the definition of inadequate response and/or intolerance to cyclosporine A. The focus of this ITC on a very specific population decreases the sample size (i.e., the ITC is underpowered) and begets imprecision; also, the use of different definitions could have introduced indirectness (i.e., issues with applicability) of the populations, issues with heterogeneity of the network, and uncertainty in the final effect estimates.
Only outcomes of efficacy were identified, and EASI 75 was the only outcome relevant to the CADTH protocol and review. No harm outcomes were detected in the SLR or ITC report.
Overall, upadacitinib and dupilumab showed superior effect estimates versus placebo in the measured EASI 75 scores, and upadacitinib was superior to dupilumab when used as monotherapy, but not when used as combination therapy. The certainty in the estimates is low due to imprecision, baseline heterogeneity, and issues of the applicability of the included populations from some of the individual studies to the specific clinical question. The baseline characteristics were overall similar across studies in the variables evaluated. However, some unmeasured heterogeneity in the study and population characteristics might still produce imbalances between groups; for instance, the definition of cyclosporine A use was different across the studies — and furthermore, it was not considered a stratification variable in most, or all, of the available studies in the network. The network of combination therapy (all-observed cases analysis) can potentially bias the effect estimates because it is likely that patients with adequate responses who are free from AEs would contribute more to the results. However, even given these inconsistencies, the primary analysis was generally consistent with the all-observed approach in the effect estimates.
Indirect Treatment Comparison 2: Post-Topicals Network Meta-Analysis
Methods
Objectives
The primary study objective was to conduct an NMA of a comprehensive published RCT evidence base to determine the comparative effectiveness of upadacitinib versus immunomodulators in patients with moderate to severe AD. The analyses were conducted in 2 types of patients with AD:
those on monotherapy (not concomitantly receiving TCS)
those on combination therapy (concomitantly receiving TCS).
Secondary objectives were to assess the number needed to treat results from the NMA using the IGA 0 or 1 and EASI 75 end points, which were the primary end points for the analyses.
Study Selection Methods
Published data from RCTs that were identified and extracted as part of a complete clinical SLR were used in the NMA. The clinical SLR identified RCTs that assessed and reported the clinical efficacy of relevant AD treatments, including upadacitinib, dupilumab, abrocitinib, tralokinumab, and baricitinib.
In addition to data gathered as part of the clinical SLR, data were also extracted from the following upadacitinib clinical trials: Measure Up 1, Measure Up 2, and AD Up.
Data from eligible RCTs were collected using an Excel-based data extraction form. In addition to the outcomes of interest, study design and patient baseline characteristics were extracted to assess the comparability of studies and identify the presence of heterogeneity.
Only studies identified by the clinical SLR that reported outcomes of interest for doses either currently licensed by the European Medicines Agency or FDA, or expected to be licensed for the treatment of moderate to severe AD, were included in the NMA.
Outcomes analyzed included the EASI 50, EASI 75, and EASI 90, the IGA 0 or 1, and WP-NRS ≥ 4. These end points were assessed at the primary time point: week 16 for upadacitinib, dupilumab, tralokinumab, and baricitinib, and week 12 for abrocitinib. An ITT perspective was used so that the sample at randomization was used as the denominator in all analyses.
Outcomes were assessed in all included RCTs at their primary end point for upadacitinib, dupilumab, abrocitinib, tralokinumab, and baricitinib. All trials measured primary end points at week 16, except for abrocitinib, which was measured at week 12.
All NMAs were conducted on the covariate “with TCS” versus “without TCS.” RCTs for AD could be separated into those where patients received an immunomodulator and TCS or placebo and TCS (i.e., combination therapy) and those where patients received only an immunomodulator or only placebo (i.e., monotherapy). As such, 2 NMAs were conducted for each outcome:
NMA of monotherapy RCTs
NMA of combination therapy RCTs.
ITC Analysis Methods
The NMA was developed based on methods considered valid by NICE. Network connectivity of all included RCTs was checked and illustrated using a network plot. The following baseline characteristics were identified a priori from published clinical research to be potential treatment-effect modifiers:
age
gender
duration of disease
baseline severity (i.e., baseline EASI, baseline IGA, baseline WP-NRS).
The mean baseline (placebo) effects across the included RCTs were also assessed. To address any discernable heterogeneity across RCTs, investigators adjusted for baseline risk as a proxy for both measured and unmeasured patient- and study-level characteristics that can collectively influence a patient’s response to treatment.
Model fit and priors, as well as other methods conducted for the NMA, are presented in Table 46.
Table 46: Indirect Treatment Comparison 2 Analysis Methods
Analysis | ITC 2: post-topicals |
|---|---|
ITC methods | For each outcome, 2 NMAs were conducted using Bayesian NMA methods:
NMAs were conducted in a generalized linear model framework with Bayesian Markov Chain Monte Carlo simulations using 4 chains with 100,000 runs each, with a burn-in that was half of the convergence sequence (set size of 10,000). |
Priors | Vague or flat prior distributions were given to the parameters to be estimated by default. For parameters assumed to be specified on a continuous scale, namely the relative treatment effects d, trial-specific baselines mu, and baseline adjustment regression term B (for models with baseline risk adjustment), a normal (0 to 1002) prior distribution was used. For the between-study standard deviation sigma (for RE models), a uniform (0 to 5) prior distribution was used. |
Assessment of model fit | For all networks, both FE and RE models were tested. The models’ global fits were assessed and compared using their overall residual deviance, effective number of parameters, deviance information criteria, leverage plots, and the posterior distribution of the between-study standard deviation (sigma) associated with the RE model. |
Assessment of consistency | To assess inconsistency in the networks, FE and RE inconsistency models (unrelated mean effects) were compared in their fit (leverage plots, overall residual deviance, and deviance information criteria statistics) to corresponding consistency models. |
Assessment of convergence | Convergence was assessed using the Brooks-Gelman-Rubin method with the potential scale reduction factor. The factor should gradually shrink to 1 with increasing numbers of iterations; a value of < 1.05 was used to indicate a good convergence. |
Outcomes |
|
Follow-up time points | 16 weeks |
Construction of nodes | The network connectivity of all included RCTs was checked and illustrated using a network plot. |
Sensitivity analyses | For each selected model, a baseline risk-adjusted sensitivity analysis was conducted that adjusted for differences in mean placebo effect across studies. |
Subgroup analysis | The following baseline characteristics were identified a priori from published clinical research to be potential treatment-effect modifiers:
|
EASI = Eczema Area and Severity Index; EASI 50 = at least 50% improvement in EASI total score from baseline; EASI 75 = at least 75% improvement in EASI total score from baseline; EASI 90 = at least 90% improvement in EASI total score; FE = fixed effects; IGA = Investigator Global Assessment; ITC = indirect treatment comparison; NMA = network meta-analysis; RCT = randomized controlled trial; RE = random effects; TCS = topical corticosteroid; WP-NRS = Worst Pruritis Numerical Rating Scale.
Source: Sponsor-submitted ITC 2.65
For each selected model, a baseline risk-adjusted sensitivity analysis was conducted that adjusted for differences in mean placebo effect across studies. This adjustment aims to detect effect modification, including unmeasured or unknown effects, within a single measure.
Results of Indirect Treatment Comparison 2
Summary of Included Studies
From the SLR, 13 studies — 9 monotherapy RCTs and 4 combination therapy RCTs — were included (Table 47). A total of 22 trial arms and 4,571 patients were included in the monotherapy network. The sample sizes ranged from 77 to 603. A total of 9 trial arms and 1,345 patients were included in the combination therapy network. The sample sizes ranged from 106 to 315. Furthermore, the ITC included data from studies assessing upadacitinib: Measure Up 1, Measure Up 2, and AD Up. There was 1 additional upadacitinib trial available (but not yet published), RISING UP; this trial was excluded on the basis that it did not include any of the outcomes of interest as primary or secondary end points.
Included RCTs employed non-responder imputation to impute missing outcomes. Multiple imputation was used to handle data missing due to COVID-19 when analyzing responses for binary outcomes at week 16 for upadacitinib RCTs.
Table 47: Overview of the Studies Included From the Clinical Systematic Literature Review
Study | Population | Intervention or comparator | Number of patients | Treatment duration | Primary and key secondary outcomes reported |
|---|---|---|---|---|---|
LIBERTY AD SOLO 1 | ≥ 18 years with moderate to severe AD for which topical TX provided inadequate control or was medically inadvisable | DUP 300 mg 2.q.w., SC PBO q.w., SC | 448 DUP 300 mg 2.q.w.: 224 PBO: 224 | 16 wks, study up to 28 wks | IGA 0 or 1, EASI 75, WP-NRS, SCORAD, BSA, DLQI, POEM, HADS |
LIBERTY AD SOLO 2 | ≥ 18 years with moderate to severe AD for which topical TX provided inadequate control or was medically inadvisable | DUP 300 mg 2.q.w., SC PBO q.w., SC | 469 DUP 300 mg 2.q.w.: 233 PBO: 236 | 16 wks, study up to 28 wks | Refer to SOLO1 |
LIBERTY AD CAFÉ | ≥ 18 years with moderate to severe AD and IR to TCS, or intolerance and/or toxicity to CsA, or CsA contraindicated | DUP 300 mg 2.q.w., SC + TCS PBO q.w., SC + TCS | 215 DUP 300 mg 2.q.w. + TCS: 107 PBO: 108 | 16 wks; study: 32 wks | EASI, WP-NRS, SCORAD, GISS, BSA, DLQI, POEM, HADS |
LIBERTY AD CHRONOS | ≥ 18 years; moderate to severe AD, with history of IR to medium- to high-potency TCS (± TCI), or systemic treatment, within the past 6 months, or both | DUP 300 mg 2.q.w., SC + TCS PBO q.w., SC + TCS | 421 DUP 300 mg 2.q.w. + TCS: 106 PBO + TCS: 315 | 52 wks; Study: 64 wks | IGA, EASI 75, WP-NRS, SCORAD, HADS, POEM, DLQI |
JADE Mono-1 | ≥ 12 years with chronic moderate to severe AD and had IR to TX with TCS or TCI, AD TX being considered medically inadvisable, or a history of receiving systemic therapies | ABRO 200 mg q.d., PO ABRO 100 mg q.d., PO PBO q.d., PO | 387 ABRO 200 mg: 154 ABRO 100 mg: 156 PBO: 77 | 12 wks; study up to 16 wks | IGA 0 or 1, EASI 75 (co- primary), EASI 50, EASI 90, WP-NRS, SCORAD |
JADE Mono-2 | ≥ 12 years old with chronic moderate to severe AD and IR to TCS or TCI, topical AD considered medically inadvisable, or a history of receiving systemic therapies for AD; weight ≥ 40 kg | ABRO 200 mg q.d., PO ABRO 100 mg q.d., PO PBO q.d., PO | 391 ABRO 200 mg: 155 ABRO 100 mg: 158 PBO: 78 | 12 wks; study up to 16 wks | IGA, EASI 75 (co- primary), EASI 50, EASI 90, WP-NRS, SCORAD |
BREEZE-AD1 | ≥ 18 years old with moderate to severe AD and history of IR to topical therapies. Failure to respond to systemic therapies within 6 months of screening or clinically significant adverse reactions to TCS | BAR 4 mg q.d., PO BAR 2 mg q.d., PO PBO q.d., PO | 497 BAR 4 mg: 125 BAR 2 mg: 123 PBO: 249 | 16 wks; study: 20 wks | vIGA-AD 0 or 1, EASI 75, EASI 90, WP-NRS ≥ 4-point reduction, SCORAD, ADSS, POEM, DLQI |
BREEZE-AD2 | ≥ 18 years old with moderate to severe AD and IR to topical therapies | BAR 4 mg q.d., PO BAR 2 mg q.d., PO PBO q.d., PO | 490 BAR 4 mg: 123 BAR 2 mg: 123 PBO: 244 | 16 wks; study: 20 wks | vIGA-AD 0 or 1, EASI 75, EASI 90, WP-NRS, SCORAD, ADerm-SS, POEM, DLQI |
BREEZE-AD5 | > 18 years old with moderate to severe AD and documented history of IR or intolerance to topical therapies | BAR 2 mg q.d., PO PBO q.d., PO | 293 BAR 2 mg: 146 PBO: 147 | 16 wks | EASI 75, vIGA-AD 0 or 1, EASI 90, SCORAD, WP-NRS, ADerm-SS, BSA, POEM, HADS, DLQI, WPAI:AD, EQ-5D-5L |
BREEZE-AD7 | ≥ 18 years old with moderate to severe AD and documented history of IR to topical therapies | BAR 4 mg q.d., PO + TCS BAR 2 mg q.d., PO + TCS PBO q.d., PO + TCS | 329 BAR 4 mg with TCS: 111 BAR 2 mg with TCS: 109 PBO: 109 | 16 wks | vIGA-AD 0 or 1, EASI 75, EASI 90, SCORAD, WP-NRS ≥ 4, ADerm-SS, POEM, HADS, DLQI, WPAI:AD |
ECZTRA 1 | ≥ 18 years old with AD diagnosis, AD involvement of ≥ 10% BSA, and history of IR to topical therapies | TRALO 300 mg 2.q.w., SC PBO 2.q.w., SC | 802 TRALO: 603 PBO: 199 | 16 wks | IGA 0 or 1 with EASI 75 (co- primary) SCORAD, WP-NRS ≥ 4, DLQI |
ECZTRA 2 | ≥ 18 years old with moderate to severe AD and documented history of IR to topical therapies | TRALO 300 mg 2.q.w., SC PBO 2.q.w., SC | 794 TRALO: 593 PBO: 201 | 16 wks; study: 30 wks | IGA 0 or 1 with EASI 75 (co-primary), SCORAD, WP-NRS ≥ 4, DLQI, EASI 50, SCORAD |
ECZTRA 3 | ≥ 18 years old with AD diagnosis, AD involvement of ≥ 10% BSA, and history of IR to topical therapies | TRALO 300 mg 2.q.w., SC+TCS PBO 2.q.w., SC +TCS | 380 TRALO: 253 PBO: 127 | 16 wks; study: 32 wks | IGA 0 or 1 with ≥ 2-point reduction EASI 75 (co-primary) SCORAD, WP-NRS ≥ 4-point reduction, DLQI |
Measure Up 1 | Adolescent and adult patients with moderate to severe AD | UPA 15 mg q.d. UPA 30 mg q.d. PBO | 810 UPA 15 mg q.d.: 270 UPA 30 mg q.d.: 270 PBO: 270 | 16-week double-blind, 120-week blinded extension period | IGA 0 or 1 EASI 75 |
Measure Up 2 | Adolescent and adult patients with moderate to severe AD | UPA 15 mg q.d. UPA 30 mg q.d. PBO | 810 UPA 15 mg q.d.: 270 UPA 30 mg q.d.: 270 PBO: 270 | 16-week double-blind, 120-week blinded extension period | IGA 0 or 1 EASI 75 |
AD Up | Adolescent and adult patients with moderate to severe AD | UPA 15 mg q.d. + TCS UPA 30 mg q.d.+ TCS PBO + TCS | 810 UPA+ TCS 15 mg q.d.: 270 UPA+TCS 30 mg q.d.: 270 PBO+TCS: 270 | 16-week double-blind, 120-week blinded extension period | IGA 0 or 1 EASI 75 |
2.q.w. = every 2 weeks; ABRO = abrocitinib; AD = atopic dermatitis; ADerm-SS = Atopic Dermatitis Symptom Scale; BAR = baricitinib; BSA = body surface area; CsA = cyclosporine A; DLQI = Dermatology Life Quality Index; DUP = dupilumab; EASI = Eczema Area and Severity Index; EASI 50 = at least 50% improvement in EASI total score from baseline; EASI 75 = at least 75% improvement in EASI total score from baseline; EASI 90 = at least 90% improvement in EASI total score; EQ-5D-5L = EQ-5D Five-Level; HADS = Hospital Anxiety and Depression Scale; IGA = Investigator Global Assessment; IR = inadequate response; PBO = placebo; PO = orally; POEM = Patient-Oriented Eczema Measure; SC = subcutaneous; SCORAD = Scoring Atopic Dermatitis; TCI = topical calcineurin inhibitor; TCS = topical corticosteroid; q.d. = once daily; TRALO = tralokinumab; TX = treatment; UPA = upadacitinib; vIGA-AD = validated Investigator Global Assessment for Atopic Dermatitis; WP-NRS = Worst Pruritus Numerical Rating Scale; WPAI:AD = Work Productivity and Activity Impairment: Atopic Dermatitis.
Source: Sponsor-submitted ITC 2.65
Results
The generic network plots of all included RCTs for the monotherapy network are shown in Figure 10, while network plots of all included RCTs for the combination therapy network are shown in Figure 11. Overall, the combination therapy networks have less data and fewer trials than the monotherapy networks.
Figure 10: ITC 2 Network Plot of Monotherapy RCTs for All End Points
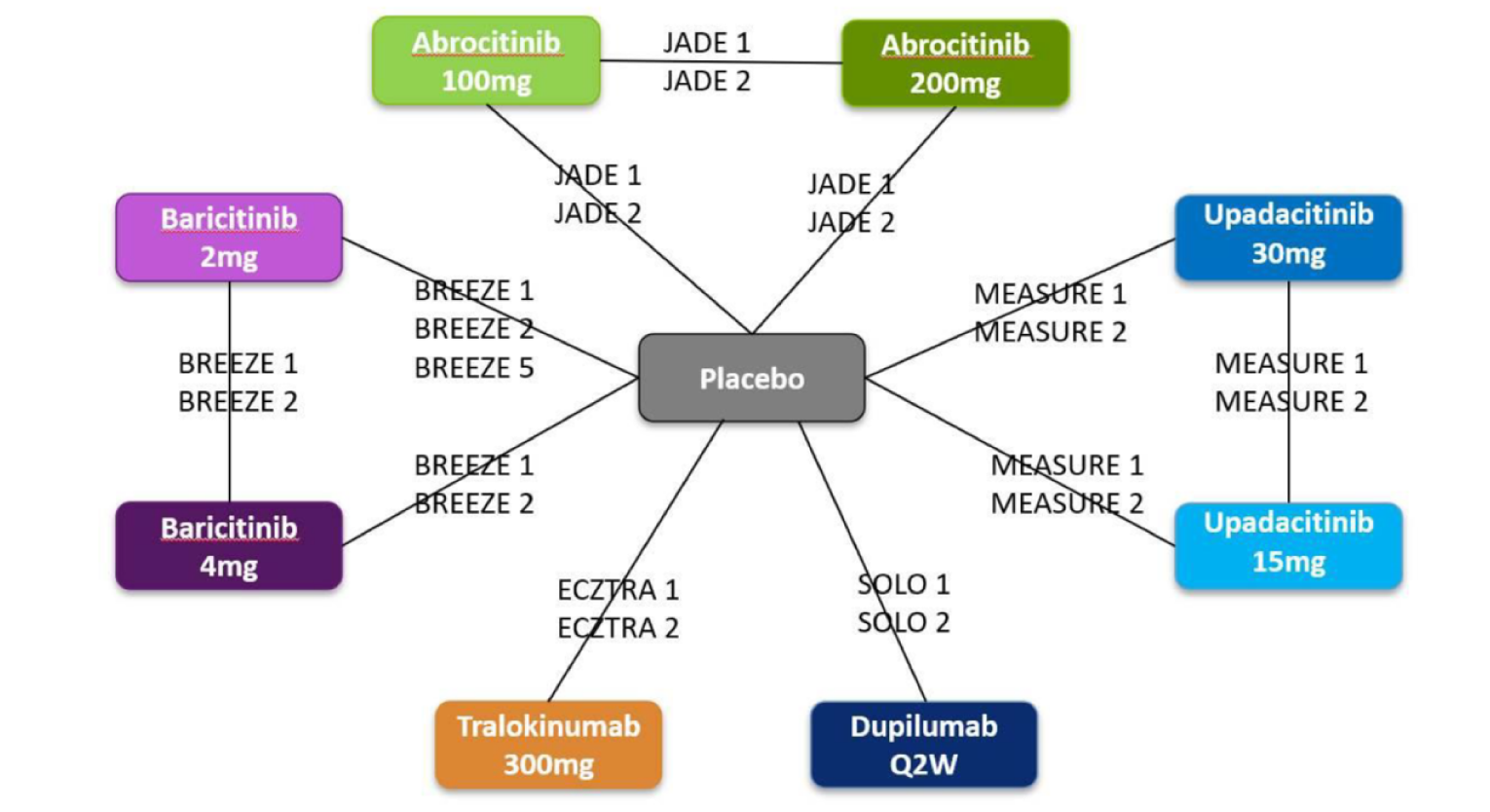
ITC = indirect treatment comparison; Q2W = every 2 weeks; RCT = randomized controlled trial.
Source: Sponsor-submitted ITC 2.65
Figure 11: ITC 2 Network Plot of Combination Therapy RCTs for All End Points
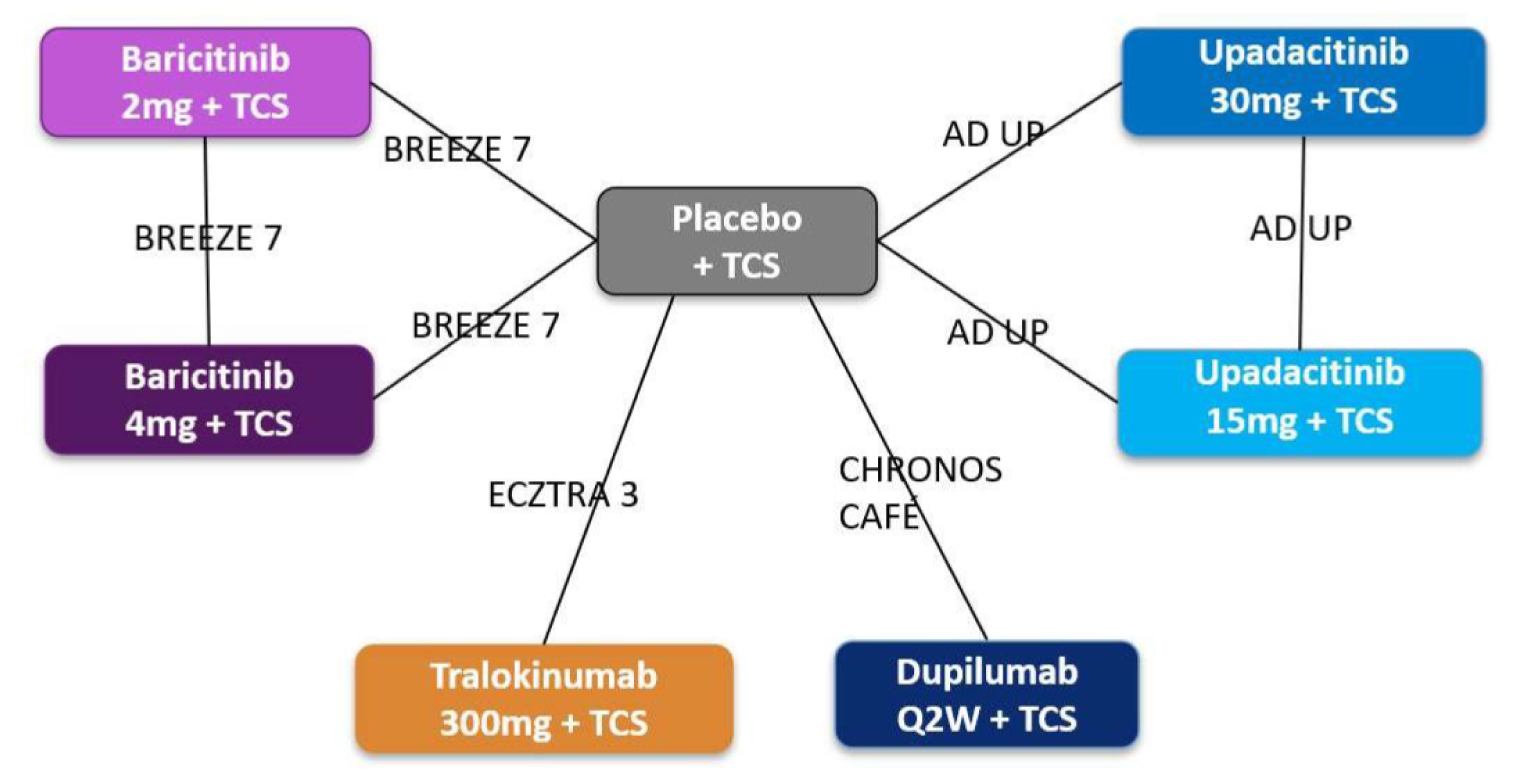
ITC = indirect treatment comparison; Q2W = every 2 weeks; RCT = randomized controlled trial; TCS = topical corticosteroid.
Source: Sponsor-submitted ITC 2.65
Overall, baseline demographic and clinical characteristics did not differ importantly across trials in terms of age, baseline IGA score, or baseline EASI score; however, they differed in terms of gender, disease duration, and WP-NRS at baseline. These differences were the rationale for estimating baseline risk-adjusted models, a form of meta-regression, for all results whenever possible.
In addition to potential treatment-effect modifiers, the baseline risks of each outcome were assessed using placebo response rates across the included RCTs. CIs were calculated based on reported or imputed standard errors. In multiple cases, the CIs for a given end point did not overlap when comparing different trials. There were differences in 1 or both networks in placebo response rates for all outcomes, including IGA 0 or 1, EASI 75, EASI 90, EASI 50 and change in WP-NRS ≥ 4. Given the heterogeneity in baseline (placebo) risk observed across the trials in the monotherapy and combination therapy networks, as well as the differences in baseline patient characteristics, in some cases, baseline risk-adjusted models were included in the analysis.
For the relative treatment effects of binary outcomes, median ORs and 95% CrIs are presented. The FE model was selected in all analyses because it was generally more parsimonious than the random-effects models. The baseline risk-adjusted models could not be estimated in most of the combination therapy networks; in the monotherapy networks, these were ruled out, given that the CrIs overlapped.
The results of the NMAs are shown here, organized by combination therapy and monotherapy networks, with the network disposition and league table of each efficacy end point (IGA, EASI 75, and NRS).
Monotherapy
Overall, upadacitinib was superior to the other systemic interventions for patients with moderate to severe AD. The network of the IGA outcome is presented in Figure 12. The IGA 0 or 1 monotherapy NMA evidence base includes 8 targeted immunomodulators plus best supportive care or standard of care, 11 studies (none with 0 events), 6,254 patients, and 36 possible pairwise comparisons, 11 with direct data.
Figure 12: ITC 2 IGA Monotherapy Network
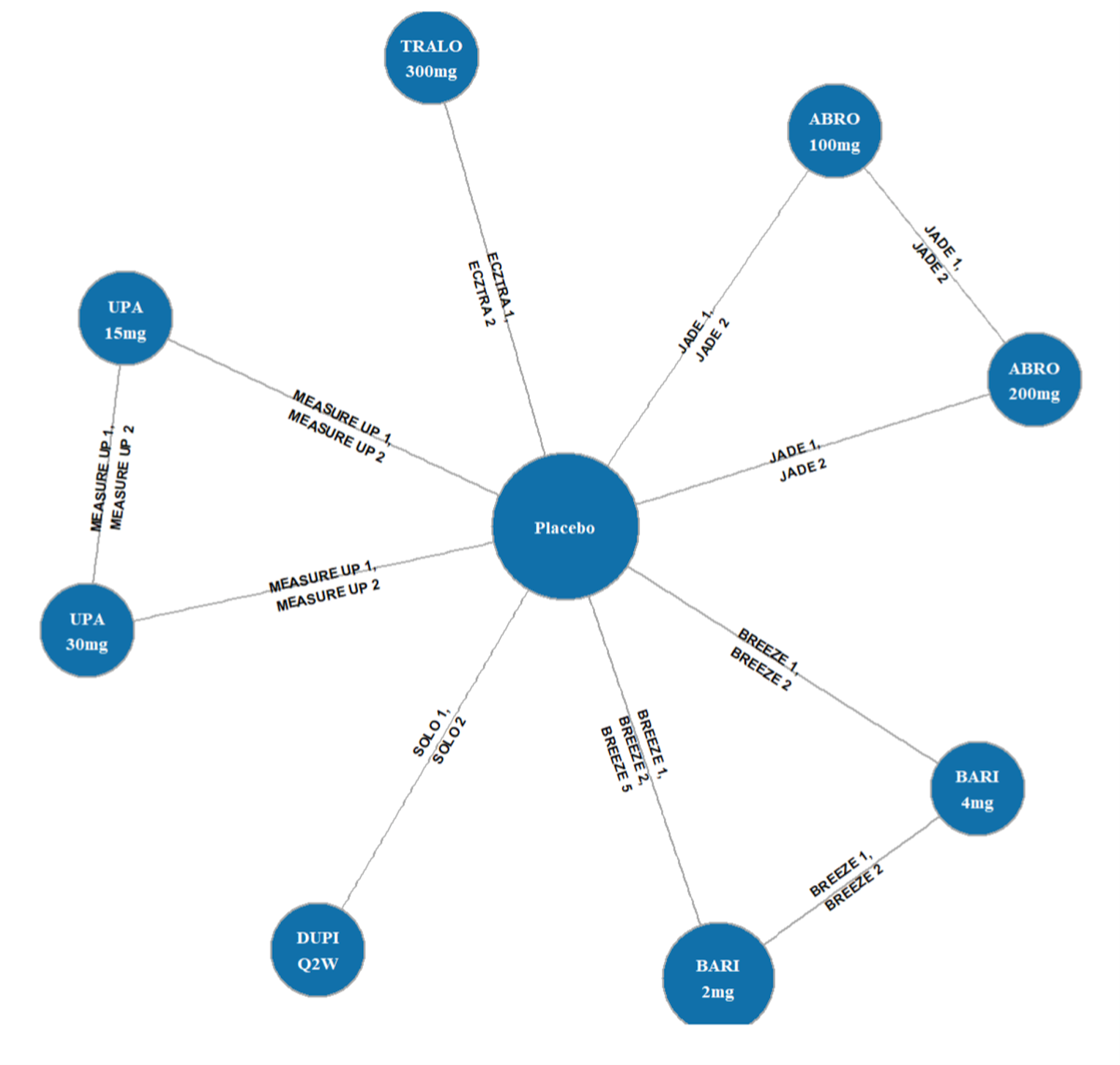
ABRO = abrocitinib; BARI = baricitinib; DUPI = dupilumab; ITC = indirect treatment comparison; Q2W = every 2 weeks; TRALO = tralokinumab; UPA = upadacitinib.
Source: Sponsor-submitted ITC 2.65
The fixed- and random-effects NMAs were overall consistent and found upadacitinib 30 mg to be superior to placebo and all comparators in terms of ORs. Upadacitinib was favoured over placebo and all comparators except for abrocitinib 200 mg, as shown in Figure 13.
Figure 13: ITC 2 Odds Ratio League Table — IGA 0 or 1, Monotherapy Network, Fixed-Effects Model
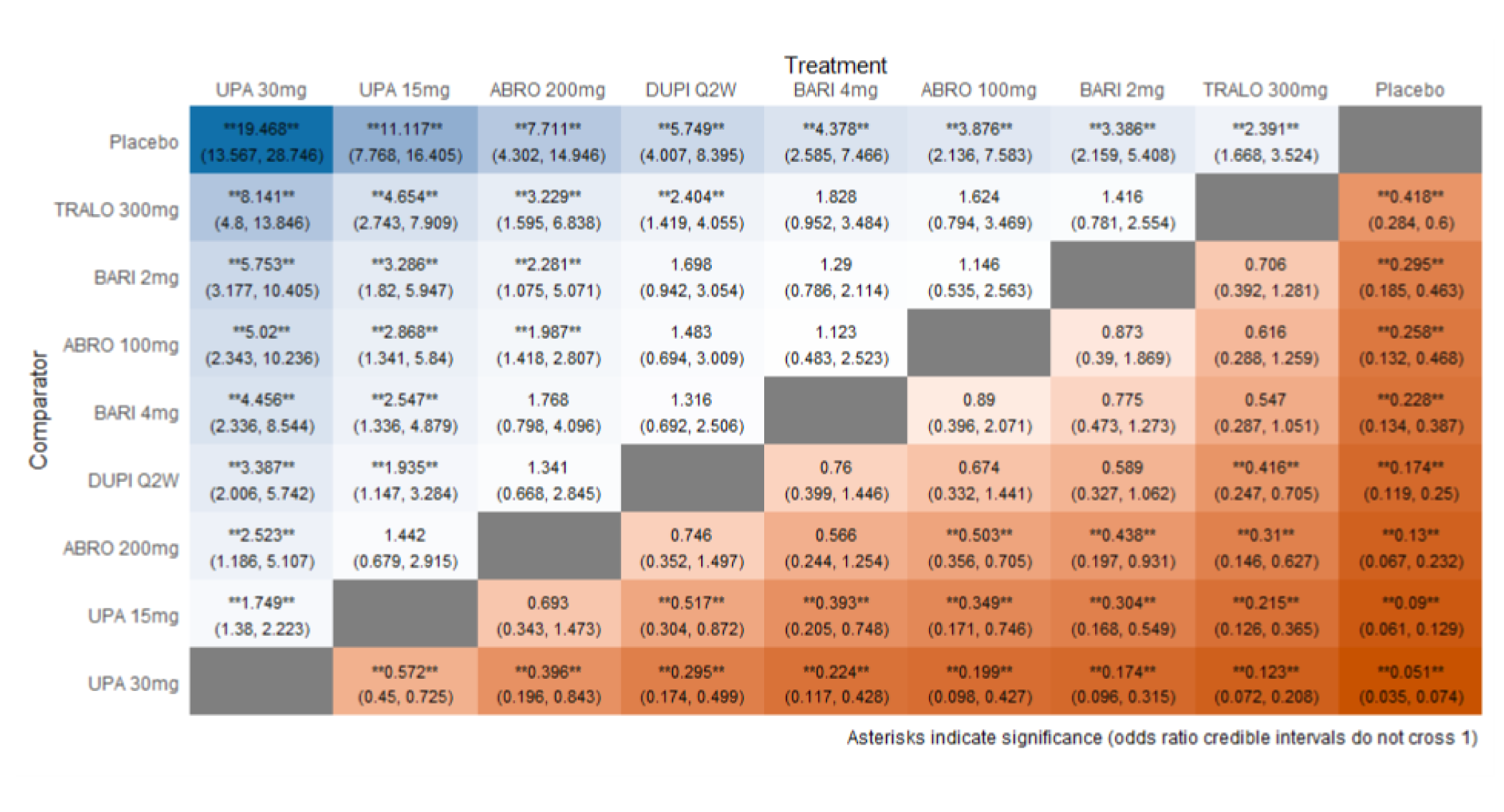
ABRO = abrocitinib; BARI = baricitinib; DUPI = dupilumab; IGA = Investigator Global Assessment; ITC = indirect treatment comparison; Q2W = every 2 weeks; TRALO = tralokinumab; UPA = upadacitinib.
Source: Sponsor-submitted ITC 2.65
The EASI 75 monotherapy network evidence base includes 8 targeted immunomodulators plus standard of care, 11 studies (none with 0 events), 6,254 patients, and 36 possible pairwise comparisons, 11 with direct data, as shown in Figure 14.
Figure 14: ITC 2 EASI 75 Monotherapy Network
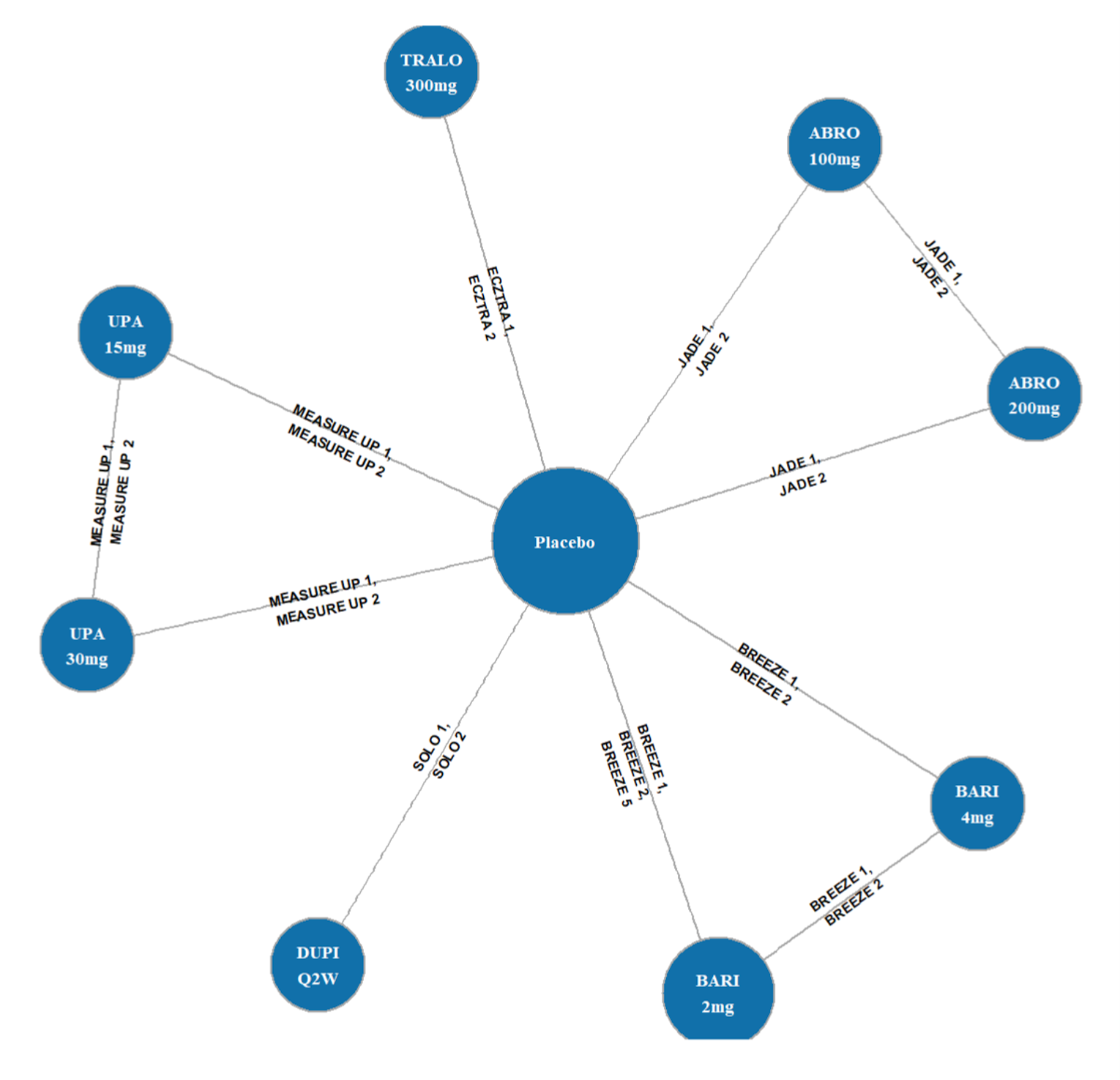
ABRO = abrocitinib; BARI = baricitinib; DUPI = dupilumab; EASI 75 = at least 75% improvement in EASI total score from baseline; ITC = indirect treatment comparison; Q2W = every 2 weeks; TRALO = tralokinumab; UPA = upadacitinib.
Source: Sponsor-submitted ITC 2.65
The fixed- and random-effects NMAs were consistent and found upadacitinib 30 mg to be favoured over placebo and all other comparators in terms of OR. Upadacitinib 15 mg was favoured over placebo and all other comparators except abrocitinib 100 mg and abrocitinib 200 mg in the same analysis, as shown in the league table (Figure 15).
Figure 15: ITC 2 Odds Ratio League Table — EASI 75, Monotherapy Network, Fixed-Effects Model
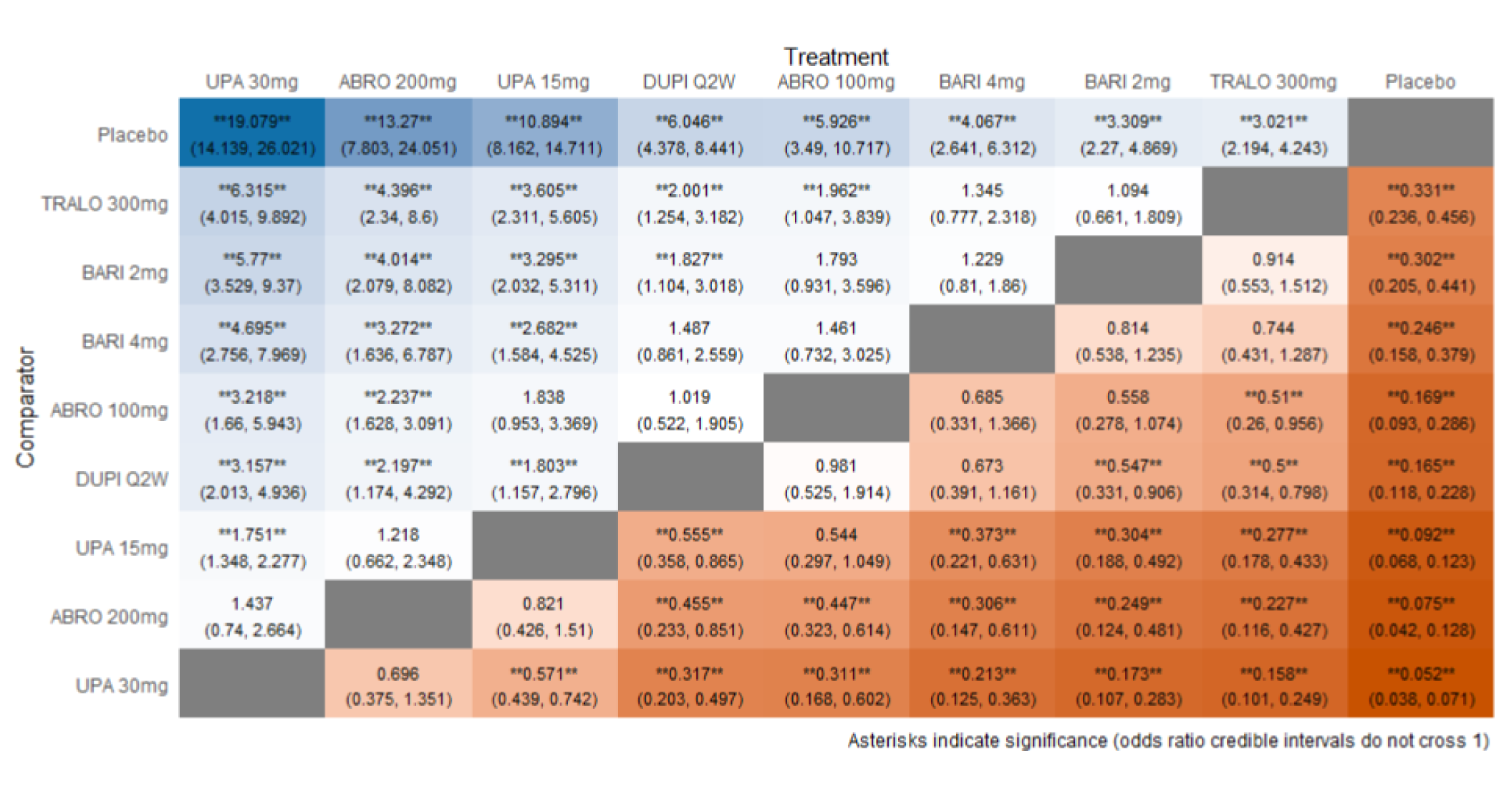
ABRO = abrocitinib; BARI = baricitinib; DUPI = dupilumab; EASI 75 = at least 75% improvement in EASI total score from baseline; ITC = indirect treatment comparison; Q2W = every 2 weeks; TRALO = tralokinumab; UPA = upadacitinib.
Source: Sponsor-submitted ITC 2.65
For the outcome of WP-NRS, the monotherapy NMA evidence base includes 8 immunomodulators plus standard of care, 11 studies (none with 0 events), 6,254 patients, and 36 possible pairwise comparisons, 11 with direct data. The network is presented in Figure 16.
Figure 16: ITC 2 — WP-NRS (≥ 4) Monotherapy Network
ABRO = abrocitinib; BARI = baricitinib; DUPI = dupilumab; Q2W = every 2 weeks; ITC = indirect treatment comparison; TRALO = tralokinumab; UPA = upadacitinib; WP-NRS = Worst Pruritis Numerical Rating Scale.
Source: Sponsor-submitted ITC 2.65
The FE and RE NMAs were consistent overall, and found that upadacitinib 30 mg was superior to placebo and all other comparators in terms of OR except for abrocitinib 200 mg. Upadacitinib 15 mg was superior to placebo and all comparators except that it was less favoured than abrocitinib 200 mg and similar to abrocitinib 100 or dupilumab. The league table with the results is shown in Figure 17.
Figure 17: ITC 2 Odds Ratio League Table — Change in WP-NRS (≥ 4), Monotherapy Network, Fixed-Effects Model
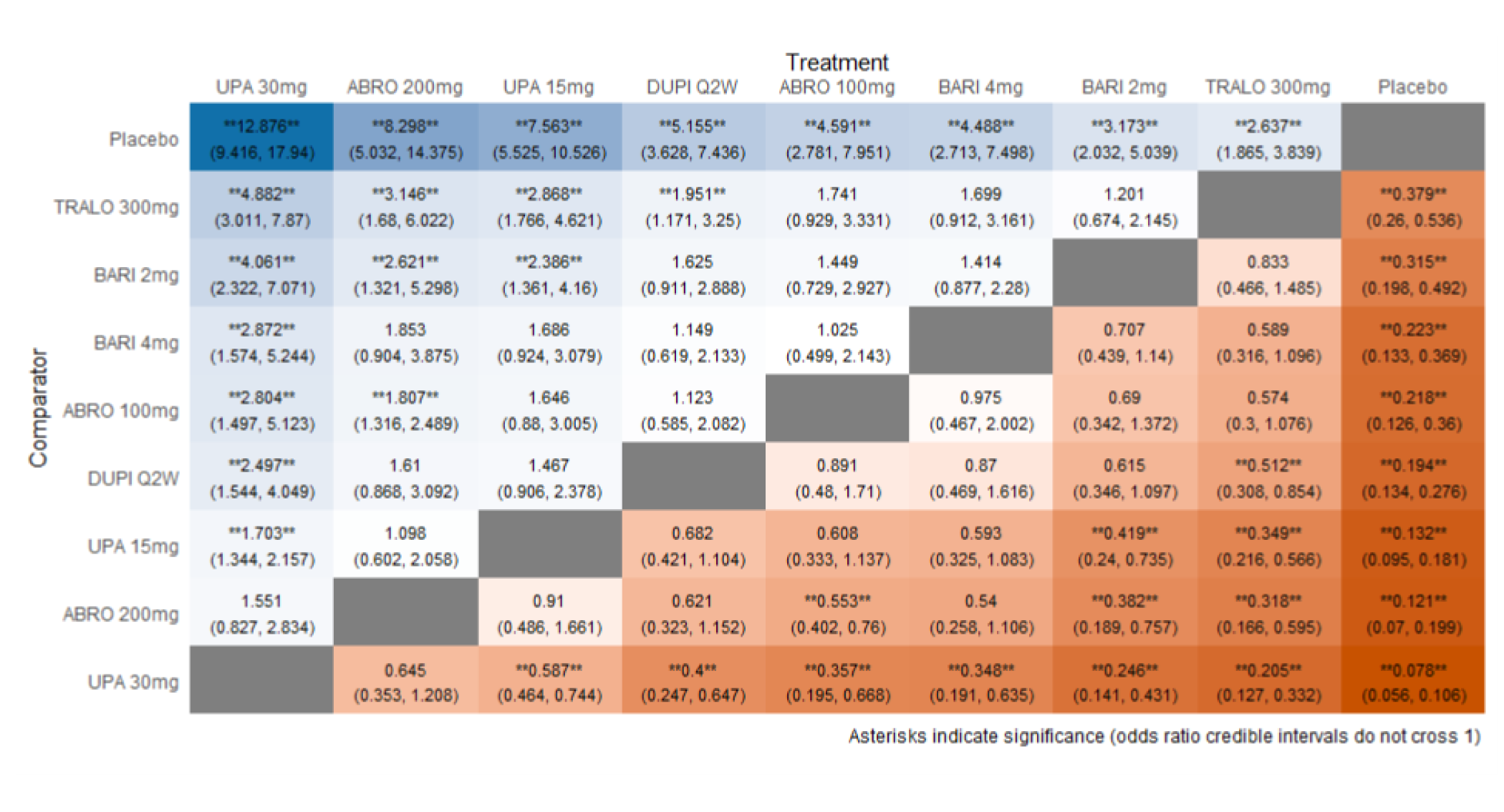
ABRO = abrocitinib; BARI = baricitinib; DUPI = dupilumab; ITC = indirect treatment comparison; Q2W = every 2 weeks; TRALO = tralokinumab; UPA = upadacitinib; WP-NRS = Worst Pruritis Numerical Rating Scale.
Source: Sponsor-submitted ITC 2.65
Combination Therapy
For the combination therapy NMA, the evidence-base for the IGA of 0 or 1 includes 6 targeted immunomodulators plus best supportive care, 5 studies (none with 0 events), 2,246 patients, and 21 possible pairwise comparisons, 8 with direct data, as shown in Figure 18.
Figure 18: ITC 2 IGA Combination Therapy Network
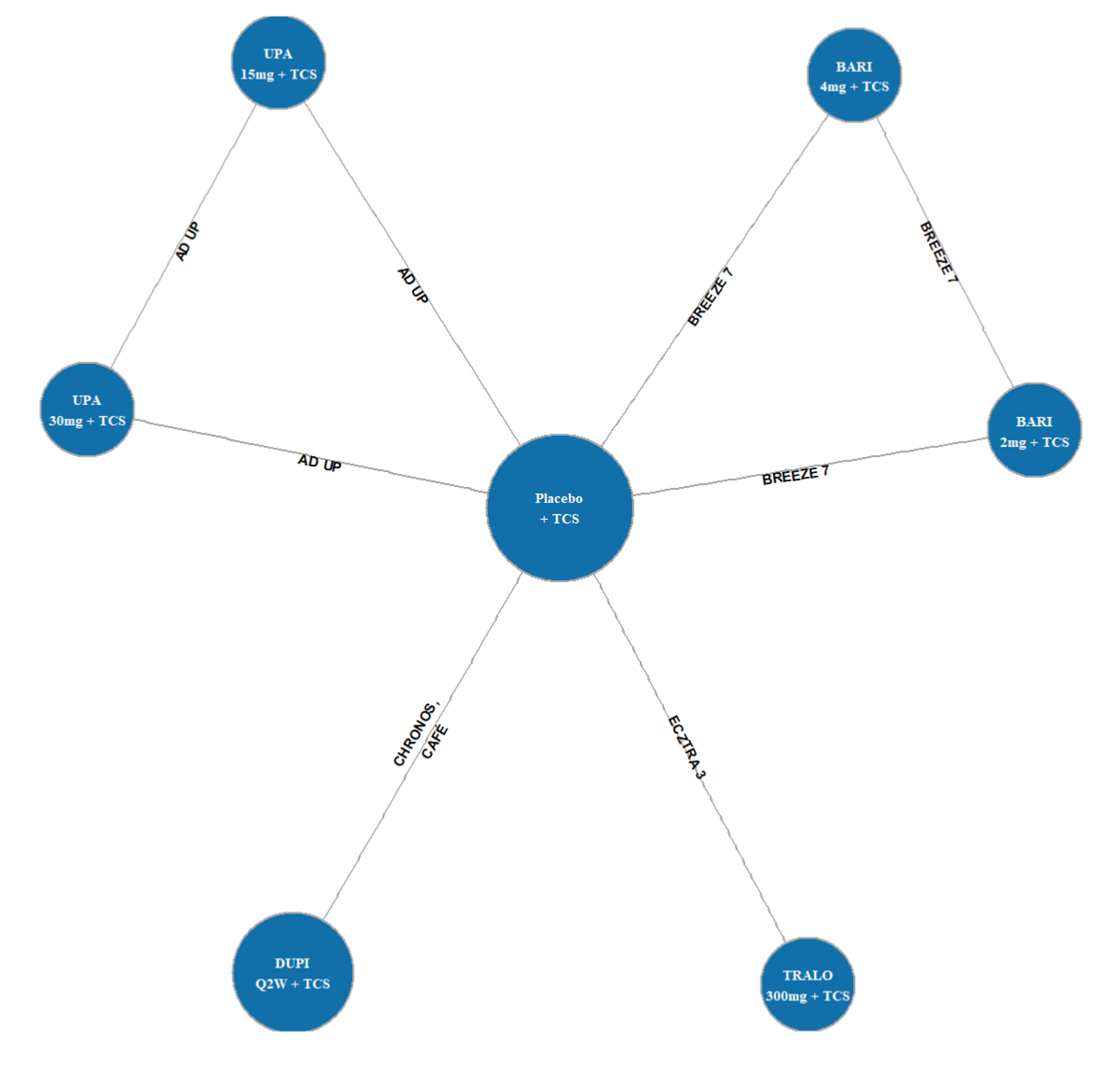
BARI = baricitinib; DUPI = dupilumab; IGA = Investigator Global Assessment; ITC = indirect treatment comparison; Q2W = every 2 weeks; TCS = topical corticosteroid; TRALO = tralokinumab; UPA = upadacitinib.
Source: Sponsor-submitted ITC 2.65
The FE NMA found upadacitinib 30 mg plus TCS to be favoured over placebo plus TCS and all comparators in terms of OR of the IGA. Upadacitinib plus TCS was superior to placebo and all comparators except baricitinib 4 mg plus TCS and dupilumab plus TCS in a similar analysis. Overall, upadacitinib plus TCS was the therapy most likely to be the most favoured treatment in the FE NMA (selected), as shown in the league table in Figure 19. However, in the RE model, no differences are detected between all interventions or comparators.
Figure 19: ITC 2 Odds Ratio League Table — IGA 0 or 1, Combination Therapy, Fixed-Effects Model
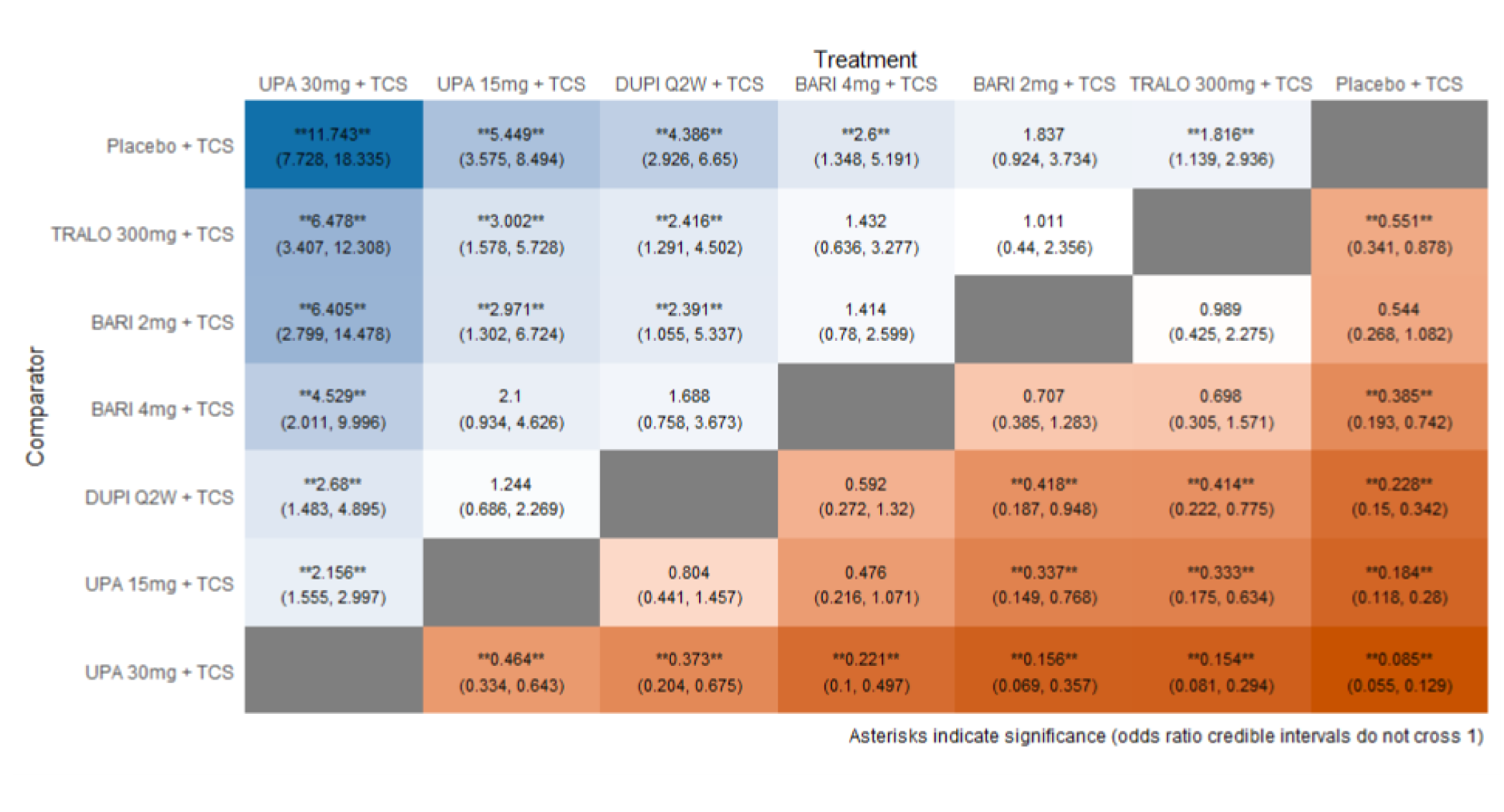
BARI = baricitinib; DUPI = dupilumab; IGA = Investigator Global Assessment; ITC = indirect treatment comparison; Q2W = every 2 weeks; TCS = topical corticosteroid; TRALO = tralokinumab; UPA = upadacitinib.
Source: Sponsor-submitted ITC 2.65
The EASI 75 combination therapy NMA evidence base includes 6 targeted immunomodulators plus best supportive care, 5 studies (none with 0 events), 2,246 patients, and 21 possible pairwise comparisons, 8 with direct data. The network diagram appears in Figure 20.
Figure 20: ITC 2 EASI 75 Combination Therapy Network
BARI = baricitinib; DUPI = dupilumab; EASI 75 = at least 75% improvement in EASI total score from baseline; ITC = indirect treatment comparison; Q2W = every 2 weeks; TCS = topical corticosteroid; TRALO = tralokinumab; UPA = upadacitinib.
Source: Sponsor-submitted ITC 2.65
For the EASI 75 combination therapy, the FE NMA found that upadacitinib 30 mg plus TCS was superior to placebo plus TCS and all comparators except for dupilumab plus TCS in terms of OR. Upadacitinib 15 mg plus TCS was also superior to placebo and all comparators, but not against baricitinib 4 mg plus TCS or dupilumab plus TCS, as shown in the league table in Figure 21. The RE model presented large inconsistencies with wide CrIs and no differences among all comparisons in the network.
Figure 21: ITC 2 Odds Ratio League Table — EASI 75, Combination Therapy, Fixed-Effects Model
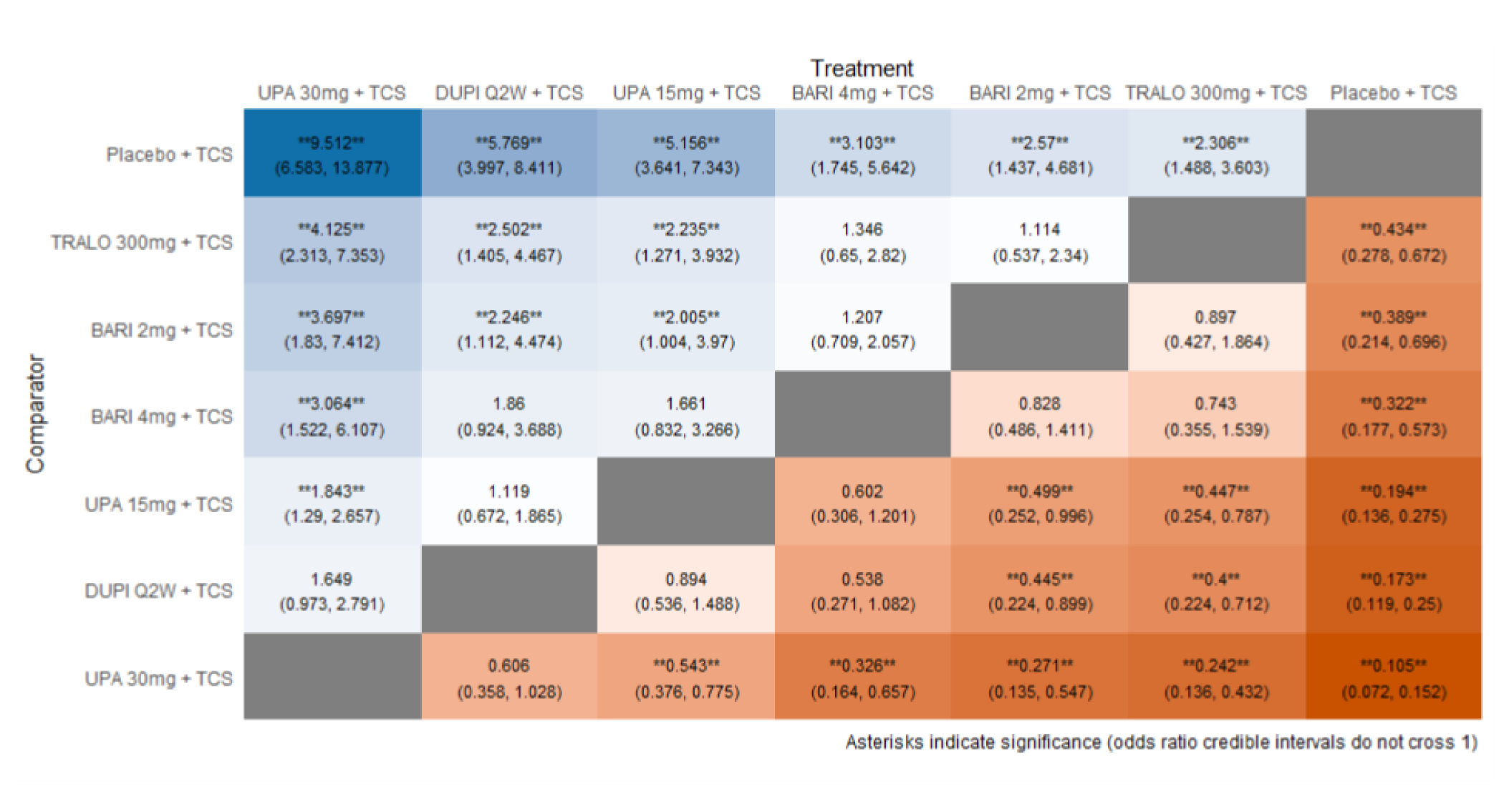
BARI = baricitinib; DUPI = dupilumab; EASI 75 = at least 75% improvement in EASI total score from baseline; ITC = indirect treatment comparison; Q2W = every 2 weeks; TCS = topical corticosteroid; TRALO = tralokinumab; UPA = upadacitinib.
Source: Sponsor-submitted ITC 2.65
The WP-NRS combination therapy NMA evidence base includes 6 targeted immunomodulators plus standard of care, 5 studies (none with 0 events), 2,246 patients, and 21 possible pairwise comparisons, 8 with direct data, as shown in Figure 22.
Figure 22: ITC 2 WP-NRS (≥ 4) Combination Therapy Network
BARI = baricitinib; DUPI = dupilumab; ITC = indirect treatment comparison; Q2W = every 2 weeks; TCS = topical corticosteroid; TRALO = tralokinumab; UPA = upadacitinib; WP-NRS = Worst Pruritis Numerical Rating Scale.
Source: Sponsor-submitted ITC 2.65
The FE NMA found upadacitinib 30 mg plus TCS to be favoured over placebo plus TCS and all comparators in terms of OR. Upadacitinib 15 mg plus TCS was also superior to placebo and all comparators except against dupilumab plus TCS, as shown in Figure 23. Similar to EASI 75, the RE model for the outcome of WP-NRS greater than or equal to 4 was highly inconsistent, with no difference in effects among all comparisons in the network and wide CrIs.
Figure 23: ITC 2 Odds Ratio League Table — Change in WP-NRS (≥ 4), Combination Therapy, Fixed-Effects Model
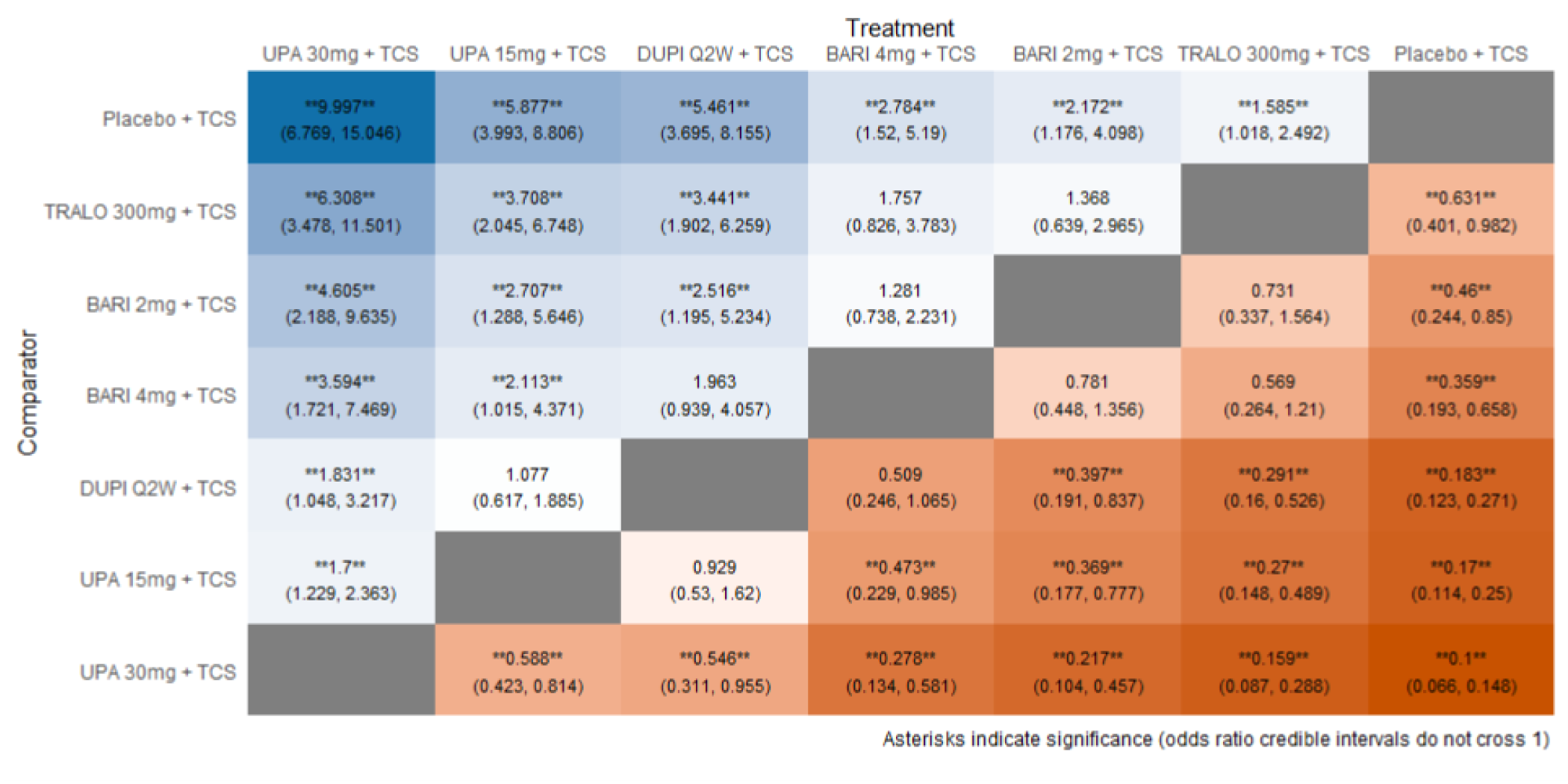
BARI = baricitinib; DUPI = dupilumab; ITC = indirect treatment comparison; Q2W = every 2 weeks; TCS = topical corticosteroid; TRALO = tralokinumab; UPA = upadacitinib; WP-NRS = Worst Pruritis Numerical Rating Scale.
Source: Sponsor-submitted ITC 2.65
Critical Appraisal of Indirect Treatment Comparison 2
ITC 2 is a sponsor-submitted NMA obtained from the same primary SLR used in ITC 1 and was also performed by the sponsor. Based on the methods detailed in the sponsor SLR, this ITC has, overall, an adequate search strategy, screening process, and assessment of risk of bias of individual studies during the evidence synthesis process.
The populations addressed by the SLR are relevant based on the intention to include patients with AD of moderate to severe intensity and with or without the use of TCS. The population is similar to what would be considered as the population to which to apply the intervention, with no issues of applicability.
Outcomes relevant to this CADTH review and protocol were the EASI 75 and WP-NRS (≥ 4). The IGA 1 or 0 response is slightly different from the vIGA used in the Measure Up and AD Up studies in this report, but it was used as a similar (albeit indirect) measure in this sponsor-submitted ITC. No harm outcomes were detected in the SLR and ITC report.
This ITC does not include the head-to-head trial comparing upadacitinib versus dupilumab (i.e., the Heads Up trial), which would have included important direct evidence information for the whole monotherapy network. In other words, the effect estimate from the monotherapy network on the upadacitinib versus dupilumab comparison comes from the indirect network estimate only, but it could have also had the direct estimate, which would have provided more certainty in the comparison.
The networks were constructed with proper modelling and assessment of model fitting. The quality of the individual included studies is of low risk of bias, according to the judgments of the investigators and based on the NICE guidance methods. However, the use of complete case analysis from the individual studies would likely bias the results because it is likely that patients who achieve an adequate response to treatment and are free of AEs would contribute to the results in larger numbers. Differences in the use of rescue medications between the studies would be expected to add heterogeneity and introduce some risk of bias in the estimates.
There were significant systematic differences in the baseline risks detected across the different treatment comparisons in the networks; this could introduce uncertainty in the effect estimates due to unexplained heterogeneity. There were also important differences when evaluating the FE versus RE models in different networks that would suggest less robust results. For example, the FE and RE models for the combination therapy NMAs assessing EASI 75 could not be estimated due to the sparseness of the data in the combination therapy network. The FE model was selected given that, according to the authors, it does not disagree with the RCT evidence and its CrIs do not appear to be invalid. The RE model failed to find that any of the treatments are different from placebo.
Only network estimates are presented, precluding us from making judgments about the certainty of the effect estimates from direct and indirect comparisons (i.e., visual or naive assessment of inconsistency). Furthermore, there is no discussion of possible pairwise heterogeneity or about transitivity assumption in the overall network, and no presentation of pairwise comparisons.
Indirect Treatment Comparison 3: ICER Report
ICER performed an evidence report that looked at JAK inhibitors and monoclonal antibodies for the treatment of patients with moderate to severe AD that included an NMA that compared the efficacy and safety of abrocitinib, baricitinib, tralokinumab, and upadacitinib versus each other, dupilumab, and placebo. A second population of patients with mild to moderate AD was also analyzed but is not evaluated in this report.
Methods
Objectives
The objectives of the ITC were to assess the relative efficacy and safety of abrocitinib, baricitinib, tralokinumab, and upadacitinib, as compared to each other, dupilumab, and placebo. The study populations included adults and adolescents with moderate to severe AD.
Study Selection Methods
MEDLINE, Embase, and the Cochrane Library (both the Cochrane Database of Systematic Reviews and Cochrane Central Register of Controlled Trials) were searched. Specific filters for each database were applied.
Studies were included if they were RCTs that reported the outcomes of interest, included a treatment of interest, were done on the population of interest, and were reported in English. Specific exclusion criteria of trials were not specified.
Study selection was conducted by 1 reviewer. However, there is no mention of whether data extraction and quality assessment of the included studies was performed by more than 1 reviewer. Criteria published by the US Preventive Services Task Force, rating each study as good, fair, or poor, were used to assess the quality (i.e., bias detection and conduction) of the included studies. The search was completed by looking at the clinicaltrials.gov database to identify trials completed more than 2 years ago that would have met the inclusion criteria and for which no findings have been published.
Efficacy outcomes identified to be assessed were: patient-reported pruritus or itching; EASI 50, 75, and 90, or relative change from baseline; IGA; sleep; SCORAD score; POEM; DLQI; CDLQI; HADS; EQ-5D; WPAI:AD; and other patient-reported symptom and quality of life measures. Safety outcomes intended to be assessed were AEs, TEAEs, SAEs, discontinuation due to AEs, thrombotic events, infections, hematological abnormalities, malignancies, and all-cause mortality.
Table 48: ITC Analysis Methods — ICER Report
Method | ICER report |
|---|---|
ITC methods | Bayesian NMA methods: initially, the first 50,000 iterations were discarded as “burn-in” and base inferences were made on an additional 50,000 iterations using 3 chains. |
Priors | Non-informative prior distributions for all model parameters |
Assessment of model fit | Fixed- and random-effects models were explored, and the model with the lowest deviance information criterion was considered to have the “best” fit to the data. Adjustments for placebo response to control for differences in population characteristics and baseline risk were made for all NMAs and results were reported when the adjusted NMA model provided a better fit of the data. |
Assessment of consistency | Not reported |
Assessment of convergence | Convergence of chains was through visual examination of the Brook-Gelman-Rubin diagnostic and historical plots. |
Follow-up time points | 12 weeks to 16 weeks |
Sensitivity analyses | Not reported |
Subgroup analysis | Age (children, adolescents, adults) Disease severity (moderate to severe) |
ICER = Institute for Clinical and Economic Review; ITC = indirect treatment comparison; NMA = network meta-analysis.
Source: ICER report.66
Indirect Treatment Comparison Analysis Methods
NMAs were conducted to compare IGA, EASI 50, EASI 75, EASI 90, and WP-NRS (≥ 4) at 12 weeks and 16 weeks. Investigators searched for subgroup evidence stratified by age on the included trials and for inclusion in the NMA, if feasible. The NMA was conducted using a Bayesian framework on the treatment parameters using a binomial likelihood and log-link model. Separate networks of the monotherapy and combination trials were developed. Authors explored both RE and FE models for each network and compared the goodness of fit to the data. The model with the lowest DIC was considered to have the “best” fit to the data. Investigators used FE models for the NMAs of the combination trials, given the limited data available for each network, then adjusted for differences in placebo response. The adjusted model was presented when it provided a better fit of the data, although how this was determined is not reported.
No details are given regarding prior distributions on all parameters, number of iterations used for burn-in and posterior computations, convergence diagnostics, statistical heterogeneity, statistical consistency, or transitivity.
Results of Indirect Treatment Comparison 3
Summary of Included Studies
All trials included in the NMA compared 1 of abrocitinib, baricitinib, tralokinumab, upadacitinib, or dupilumab versus placebo except for JADE COMPARE, which compared abrocitinib, dupilumab, and placebo, and Heads Up, which compared upadacitinib with dupilumab (no placebo arm). All trials enrolled patients with moderate to severe AD; the timing of interventions ranged from weekly to every 4 weeks. All trials lasted 16 weeks except for JADE MONO-1 and JADE MONO-2 (12 weeks) and JADE COMPARE (12 weeks or 16 weeks). All studies were assessed to be of “good” quality, according to the US Preventive Services Task Force. Table 49 shows the characteristics of the trials included in the NMA.
Table 49: Characteristics of Included Studies in the ITC 3 — ICER Report
Trial | Mono or combo therapy | Doses | Sample size (N) | EASI (mean) | Mean age, years | Mean disease duration, years | IGA score of 4 (%) |
|---|---|---|---|---|---|---|---|
Abrocitinib trials | |||||||
JADE MONO-1a | Mono | 100 mg 200 mg | 387 | 30.2 | 32.4 | 23.4 | 40.7 |
JADE MONO-2a | Mono | 100 mg 200 mg | 391 | 28.5 | 35.1 | 21.0 | 32.2 |
JADE COMPARE | Combo | 100 mg 200 mg DUP 300 mg | 837 | 30.9 | 37.7 | 22.7 | 35.4 |
Gooderham 2019 | Mono | 100 mg 200 mg | 167 | 25.6 | 40.8 | 23.0ˠ | 40.8 |
Baricitinib trials | |||||||
BREEZE-AD 1 | Mono | 1 mg 2 mg 4 mg | 624 | 31.0 | 35.7 | 25.7 | 41.8 |
BREEZE-AD 2 | Mono | 1 mg 2 mg 4 mg | 615 | 33.5 | 34.5 | 24.0 | 50.5 |
BREEZE-AD 5 | Mono | 1 mg 2 mg | 440 | 27.1 | 39.7 | 23.7 | 41.7 |
BREEZE-AD 7 | Combo | 2 mg | 329 | 29.57 | 33.8 | 24.03 | 45.0 |
Guttman-Yassky (2018) | Combo | 2 mg 4 mg | 104 | 21.23 | 36.5 | 22.03 | NR |
Tralokinumab trials | |||||||
ECZTRA 1 | Mono | 300 mg | 802 | 29.3 | 37.0 | 27.5 | 50.9 |
ECZTRA 2 | Mono | 300 mg | 794 | 28.9 | 32.0 | 25.3 | 49.2 |
ECZTRA 3 | Combo | 300 mg | 380 | 25.5 | 36.0 | 26.0 | 46.3 |
Upadacitinib trials | |||||||
Measure Up 1a | Mono | 15 mg 30 mg | 847 | 29.5 | 34.0 | NR | 45.2 |
Measure Up 2a | Mono | 15 mg 30 mg | 836 | 29.1 | 33.6 | NR | 54.9 |
AD Up | Combo | 15 mg 30 mg | 907 | 29.6 | 34.1 | NR | 52.9 |
Heads Up | Mono | UPA 30 mg DUP 300 mg | 692 | NR | NR | NR | NR |
Guttman-Yassky (2018) | Mono | 7.5 mg 15 mg 30 mg | 167 | 25.6 | 40.8 | 23.0 | 40.8 |
Dupilumab trials | |||||||
LIBERTY AD SOLO 1 | Mono | 300 mg | 671 | 30.7 | 38.7 | 26.7 | 48.3 |
LIBERTY AD SOLO 2 | Mono | 300 mg | 708 | 29.4 | 34.7 | 24.8 | 48.3 |
LIBERTY AD CHRONOS | Combo | 300 mg | 740 | 29.8 | 31.2 | 26.7 | 47.7 |
Thaci (2016) | Mono | 100 mg 200 mg 300 mg | 379 | 31.9 | 37.0 | 28.0 | 47.3 |
combo = combination therapy; EASI = Eczema Area and Severity Index; ICER = Institute for Clinical and Economic Review; IGA = Investigator Global Assessment; mono = monotherapy; NR = not reported.
Note: All time points were 16 weeks except in JADE MONO-1, JADE MONO-2 (12 weeks), and JADE COMPARE (12 weeks or 16 weeks).
aPooled estimates from this trial were in patients 12 and older.
Source: ICER report.66
Most characteristics were similar across trials. However, note that while dupilumab was analyzed in different doses, only the FDA-approved dose of 300 mg once every 2 weeks was included in the NMA.
Results
Monotherapy
The network for monotherapy trials is presented in Figure 24.
Figure 24: Network for the Monotherapy Trials
ABRO = abrocitinib; BARI = baricitinib; DUP = dupilumab; ICER = Institute for Clinical and Economic Review; PBO = placebo; Q2W = every 2 weeks; TRA = tralokinumab; UPA = upadacitinib.
Source: ICER report.66
For the EASI NMA, the RE unadjusted model (DIC = 195) was associated with improved fit compared to the adjusted model (DIC = 203); the estimated regression coefficient was not significant in the adjusted model, and the inter-study SD was increased in magnitude with placebo adjustment. For the IGA (DIC = 231) and WP-NRS (≥ 4-point improvement) (DIC = 243) models, the unadjusted models were also associated with a better fit relative to the adjusted model (the inter-study SD followed a trend similar to that presented for the EASI model). Therefore, the authors presented the result of the RE, unadjusted models for all outcomes in the monotherapy networks. The NMA results for EASI 75 in the monotherapy trials are shown in the league table in Figure 25.
For EASI 75, authors presented the results of the unadjusted RE model, given its better fit relative to the adjusted model. All interventions showed superiority to placebo and baricitinib 1 mg. Compared to placebo, interventions were 1.6 to 5.7 times more likely to achieve EASI 75, as shown in the league table in Figure 25. Upadacitinib 30 mg was more likely than the other interventions to achieve EASI 75 and EASI 90; however, upadacitinib 30 mg was not superior to abrocitinib 200 mg. Additionally, there were no detected differences between abrocitinib 200 mg and upadacitinib 15 mg and dupilumab, or between upadacitinib 15 mg and dupilumab. In comparison, dupilumab showed no difference versus abrocitinib 100 mg in both EASI 75 and EASI 90 responses; however, it was superior to tralokinumab and baricitinib (in both dosages). Data for the EASI 90 end point values are presented as a league table in Figure 26.
Figure 25: League Table of the NMA in the ICER Report — EASI 75, Monotherapy
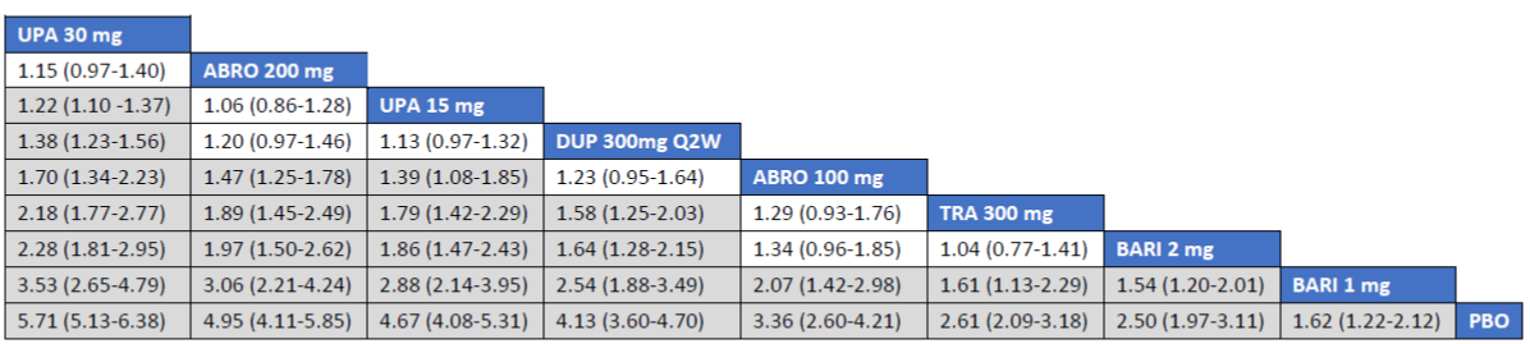
ABRO = abrocitinib; BARI = baricitinib; DUP = dupilumab; EASI 75 = at least 75% improvement in EASI total score from baseline; ICER = Institute for Clinical and Economic Review; NMA = network meta-analysis; PBO = placebo; Q2W = every 2 weeks; TRA = tralokinumab; UPA = upadacitinib.
Note: Each box represents the estimated risk ratio and 95% credible interval for the combined direct and indirect comparisons between 2 drugs. Estimates in grey signify that the 95% credible interval does not contain one.
Source: ICER report.66
Figure 26: League table of the NMA in the ICER report — EASI 90, Monotherapy
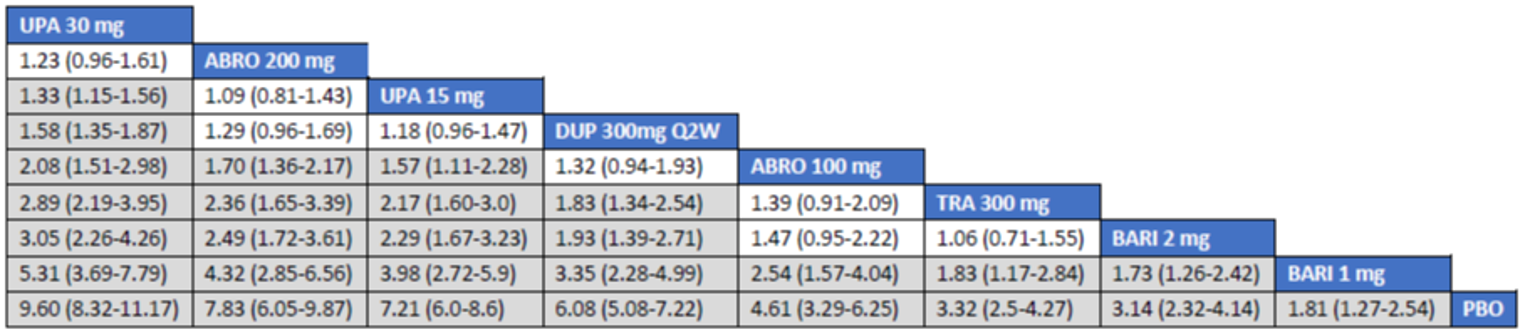
ABRO = abrocitinib; BARI = baricitinib; DUP = dupilumab; EASI 90 = at least 90% improvement in EASI total score from baseline; ICER = Institute for Clinical and Economic Review; NMA = network meta-analysis; PBO = placebo; Q2W = every 2 weeks; TRA = tralokinumab; UPA = upadacitinib.
Note: Each box represents the estimated risk ratio and 95% credible interval for the combined direct and indirect comparisons between 2 drugs. Estimates in grey signify that the 95% credible interval does not contain one.
Source: ICER report.66
Similar results were observed in the IGA end point (Figure 27), where upadacitinib 30 mg was superior to each of the other comparators (except for abrocitinib 200 mg) and against placebo. Upadacitinib 15 mg was not superior to abrocitinib 200 or dupilumab 300 mg, but it was effective against the other therapies; dupilumab was better than only baricitinib 2 mg, baricitinib 1 mg, and tralokinumab. These 2 last drugs were superior only to placebo (i.e., they were not better than any of the other comparators).
Figure 27: League Table of the NMA in the ICER Report — IGA Monotherapy
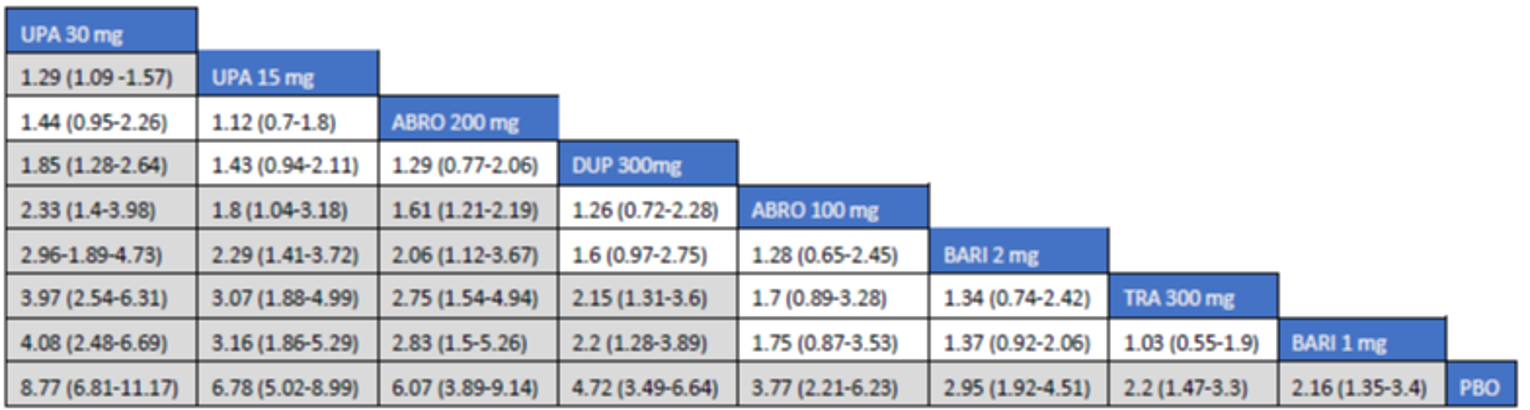
ABRO = abrocitinib; BARI = baricitinib; DUP = dupilumab; ICER = Institute for Clinical and Economic Review; IGA = Investigator Global Assessment; NMA = network meta-analysis; PBO = placebo; Q2W = every 2 weeks; TRA = tralokinumab; UPA = upadacitinib.
Note: Each box represents the estimated risk ratio and 95% credible interval for the combined direct and indirect comparisons between 2 drugs. Estimates in grey signify that the 95% credible interval does not contain one.
Source: ICER report.66
In the WP-NRS (≥ 4) end point (Figure 28), upadacitinib 30 mg was superior only to baricitinib 2 mg, baricitinib 1 mg, and tralokinumab 300 mg. It was no more effective than dupilumab 300 mg, upadacitinib 15 mg, or abrocitinib 200 mg or 100 mg. Baricitinib 1 mg was the only drug that was no more effective than placebo.
Figure 28: League Table of the NMA in the ICER Report — WP-Pruritus NRS (≥ 4)
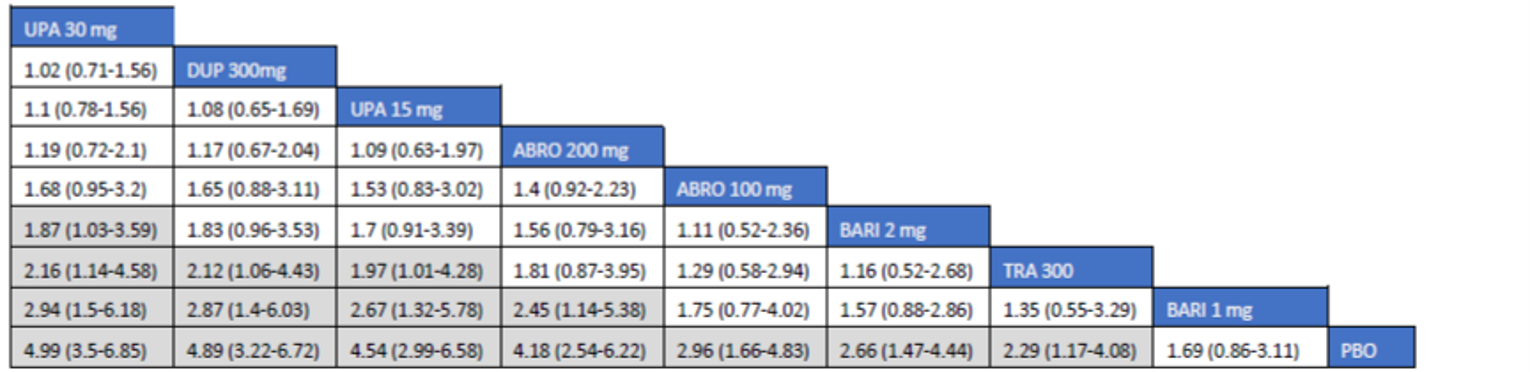
ABRO = abrocitinib; BARI = baricitinib; DUP = dupilumab; ICER = Institute for Clinical and Economic Review; NMA = network meta-analysis; PBO = placebo; Q2W = every 2 weeks; TRA = tralokinumab; UPA = upadacitinib; WP-NRS = Worst Pruritis Numerical Rating Scale.
Note: Each box represents the estimated risk ratio and 95% credible interval for the combined direct and indirect comparisons between 2 drugs. Estimates in grey signify that the 95% credible interval does not contain one.
Source: ICER report.66
Combination Therapy
The network for combination therapy trials is presented in Figure 29.
Figure 29: Network for Combination Therapy Trials
ABRO = abrocitinib; BARI = baricitinib; DUP = dupilumab; ICER = Institute for Clinical and Economic Review; PBO = placebo; Q2W = every 2 weeks; TRA = tralokinumab; UPA = upadacitinib.
Source: ICER report.66
The NMA results for EASI 75 and EASI 90 in combination therapy trials in adults are shown in league tables in Figure 30 and Figure 31. In general, the results for the combination therapy NMAs provided more conservative estimates of the relative efficacies of these drugs versus placebo, although they followed a similar ranking order as the monotherapy NMAs. All interventions showed greater responses than placebo on all outcomes. Upadacitinib 30 mg plus TCS was superior to the rest of the comparators on both end points. Abrocitinib 200 mg was also superior to most comparators (except for upadacitinib 15 mg, upadacitinib 30 mg, and dupilumab). Dupilumab showed superiority only to tralokinumab 300 mg and baricitinib 2 mg.
Figure 30: League Table of the NMA in the ICER Report — EASI 75, Combination Therapy
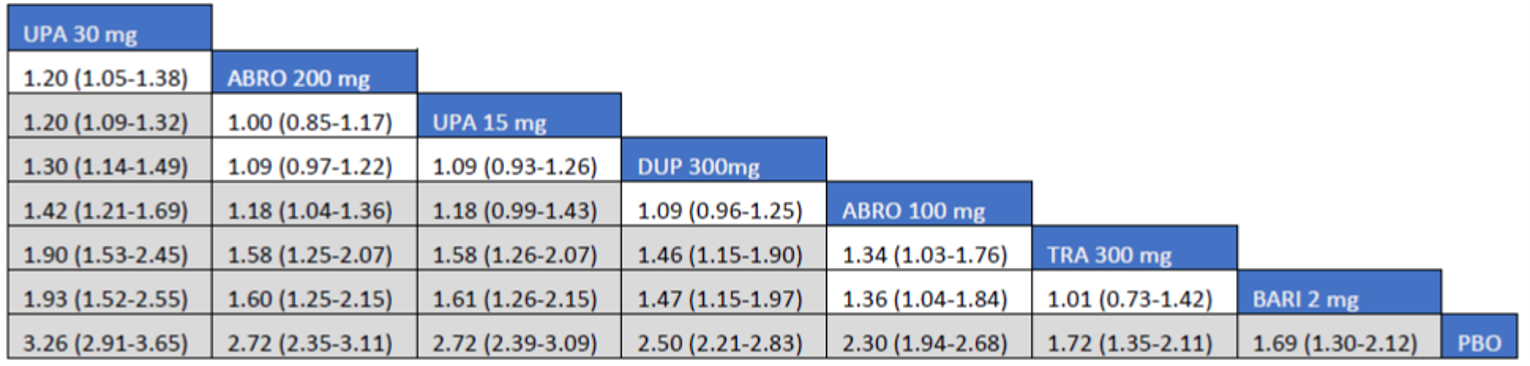
ABRO = abrocitinib; BARI = baricitinib; DUP = dupilumab; EASI 75 = at least 75% improvement in EASI total score from baseline; ICER = Institute for Clinical and Economic Review; NMA = network meta-analysis; PBO = placebo; TRA = tralokinumab; UPA = upadacitinib.
Note: Each box represents the estimated risk ratio and 95% credible interval for the combined direct and indirect comparisons between 2 drugs. Estimates in grey signify that the 95% credible interval does not contain one.
Source: ICER report.66
Figure 31: League Table of NMA in the ICER Report — EASI 90, Combination Therapy
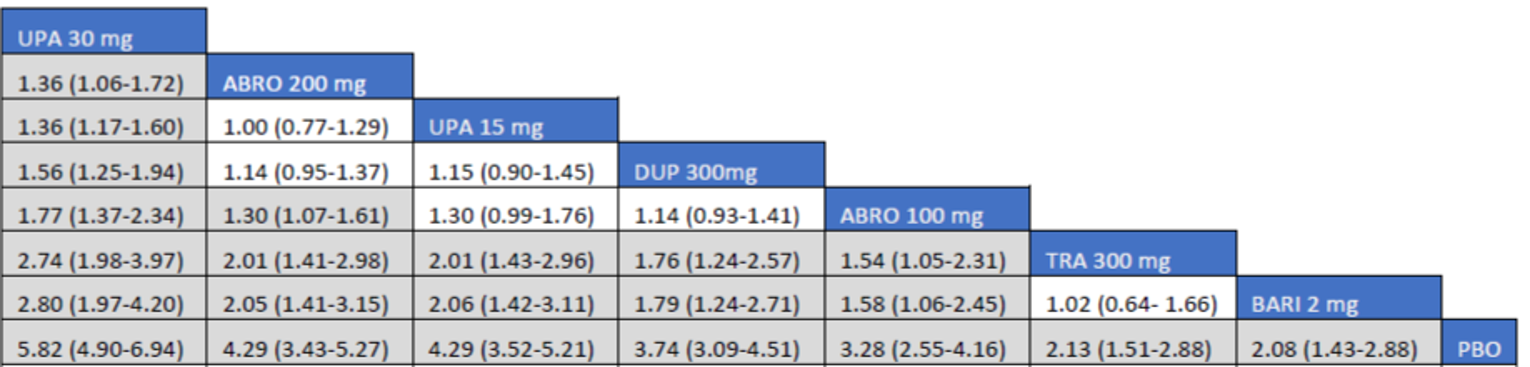
ABRO = abrocitinib; BARI = baricitinib; DUP = dupilumab; EASI 90 = at least 90% improvement in EASI total score from baseline; ICER = Institute for Clinical and Economic Review; NMA = network meta-analysis; PBO = placebo; TRA = tralokinumab; UPA = upadacitinib.
Note: Each box represents the estimated risk ratio and 95% credible interval for the combined direct and indirect comparisons between 2 drugs. Estimates in grey signify that the 95% credible interval does not contain one.
Source: ICER report.66
Similar results were observed in the IGA end point (Figure 32), where upadacitinib 30 mg was superior to each of the other comparators (except for abrocitinib 200 mg). In the comparison of upadacitinib 15 mg against dupilumab, there was no difference detected. Abrocitinib 200 mg was superior to the other therapies except against upadacitinib 15 mg and dupilumab, which at the same time was not superior to abrocitinib 100 mg, but was superior to baricitinib 2 mg and tralokinumab 300 mg. Dupilumab was better only than baricitinib 2 mg and tralokinumab 1 mg. These 2 last drugs were superior only to placebo (i.e., they were not better than any of the other comparators).
Figure 32: League Table of NMA in the ICER Report — IGA, Combination Therapy
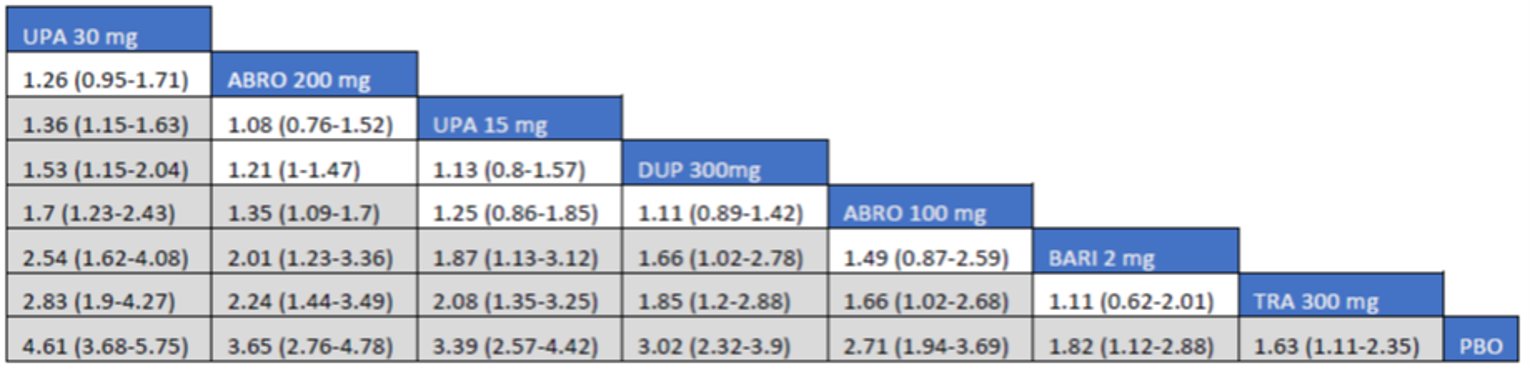
ABRO = abrocitinib; BARI = baricitinib; DUP = dupilumab; ICER = Institute for Clinical and Economic Review; IGA = Investigator Global Assessment; NMA = network meta-analysis; PBO = placebo; TRA = tralokinumab; UPA = upadacitinib.
Note: Each box represents the estimated risk ratio and 95% credible interval for the combined direct and indirect comparisons between 2 drugs. Estimates in grey signify that the 95% credible interval does not contain one.
Source: ICER report.66
For the WP-NRS (≥ 4) end point (Figure 33), upadacitinib 30 mg was superior to all other therapies. Upadacitinib 15 mg was not superior to abrocitinib or dupilumab, but was superior to abrocitinib 100 mg, baricitinib 2 mg, and tralokinumab 300 mg. There were no differences detected between abrocitinib 200 mg and upadacitinib 15 mg and dupilumab, but abrocitinib was superior to the rest of the interventions.
Figure 33: League Table of the NMA in the ICER Report — WP-NRS (≥ 4), Combination Therapy
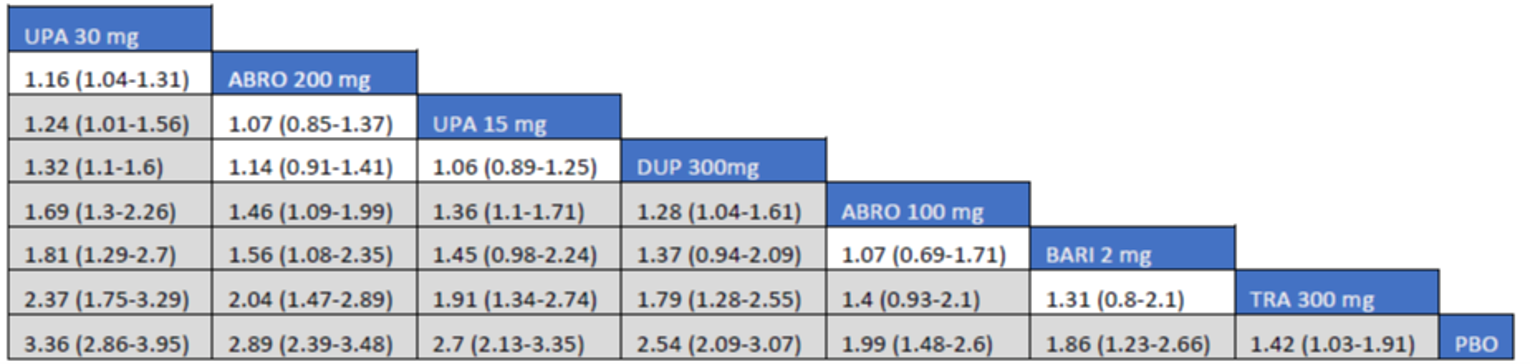
ABRO = abrocitinib; BARI = baricitinib; DUP = dupilumab; ICER = Institute for Clinical and Economic Review; NMA = network meta-analysis; PBO = placebo; TRA = tralokinumab; UPA = upadacitinib; WP-NRS = Worst Pruritis Numerical Rating Scale.
Each box represents the estimated risk ratio and 95% credible interval for the combined direct and indirect comparisons between 2 drugs. Estimates in grey signify that the 95% credible interval does not contain one.
Source: ICER report.66
Harms
An NMA of harms data does not seem to have been performed. Narrative summaries only were presented.
Critical Appraisal of Indirect Treatment Comparison 3
Study screening was not verified by a second party; all studies were screened by a single reviewer. The lack of a second reviewer to verify screening is more likely to lead to potential exclusion of valid studies. This also occurred for data extraction, making the review prone to bias.
There was a lack of transparency in the way the NMA in general was conducted. There is no mention of measures of pairwise heterogeneity or checks for consistency. In the absence of any reporting on any of these factors, it is difficult to be certain of the general quality of the analysis and accuracy of the final results.
There appears to have been no sensitivity analysis based on clinical variables done within the NMA to explore possible assumptions made by the reviewers.
Results are given for NMAs on monotherapies and combination therapies, but there is no comment on which of these should be treated as the primary analysis. The NMA looked at patients with moderate to severe AD, which is consistent with the population of interest of the sponsor. There was insufficient information to perform any NMA on the populations of adolescents and children; therefore, information in those areas was restricted to descriptions of trial-specific results (hence, there is uncertainty remaining in this age group).
Tables are presented with narrative information on safety data, and these seem to indicate that an NMA would have been possible for some of them (e.g., discontinuation due to AE). It is not clear why these data were presented only summarily or why no formal NMA was done.
Two large trials of abrocitinib were ongoing at the time of the review; thus, the results of these were not included in the NMA. Therefore, the effects these studies may have had on the final results is unknown.
All trials included in the review used imputation to adjust for missing data (i.e., combinations of multiple imputation, nonresponder imputation, or last observation carried forward).
Summary of the Indirect Treatment Comparisons
There were 3 ITCs (2 sponsor-submitted ITCs and 1 from ICER) included in this CADTH report, which evaluated upadacitinib 30 mg and upadacitinib 15 mg against placebo and several comparators, including abrocitinib 200 mg, abrocitinib 100 mg, dupilumab 300 mg, baricitinib (1 mg, 2 mg, and 4 mg), and tralokinumab 300 mg. The results from the 3 ITCs suggest that both doses of upadacitinib are among the most effective systemic therapies for reducing the severity and symptoms of moderate to severe AD in adults, either as monotherapy or in combination with TCS. The evidence for adolescents is similar, but still uncertain due to imprecision in the effect estimates and the small number of patients included in these studies.
As monotherapy, abrocitinib 100 mg, baricitinib (1 mg, 2 mg, and 4 mg), and tralokinumab 300 mg were superior only to placebo; they were not superior to any of the other systemic therapies. Meanwhile, upadacitinib 30 mg, abrocitinib 200 mg, upadacitinib 15 mg, and dupilumab 300 mg had consistently superior effect estimates (in that order), and could be considered to be among the most superior in terms of improving EASI 75, IGA, and WP-NRS scores. Among these therapies with superior effect estimates, upadacitinib 30 mg and abrocitinib 200 were superior to most other drugs. However, there is still some uncertainty in these effect estimates due to imprecision (i.e., wide and overlapping CrIs).
Similarly, as combination therapy with TCS, baricitinib (2 mg and 4 mg) and tralokinumab 300 mg were superior only to placebo; they were not superior to any of the other therapies. The therapies with the largest effects were upadacitinib 30 mg, abrocitinib 200 mg, upadacitinib 15 mg, dupilumab 300 mg, and abrocitinib 100 mg, which had superior effect estimates, in that order. Among these therapies with superior effect estimates, upadacitinib 30 mg and abrocitinib 200 were superior to most other drugs. Similar to the monotherapy networks, there was still some uncertainty in these effect estimates due to imprecision (i.e., wide and overlapping CrIs).
The limitations of the 3 ITCs stem from uncertainties in the effect estimates due to imprecision, baseline heterogeneity (ITC 1), incomplete data from head-to-head studies (ITC 2), and lack of information in the methods used for the systematic review (ITC 3). Most importantly, conclusions regarding the long-term efficacy of upadacitinib compared to the active comparators relevant to this review cannot be drawn because the NMA used study results that were collected over a relatively short duration compared to the chronic nature of AD. There is also uncertainty due to the inherent heterogeneity across trials in the networks. The robustness of the comparative efficacy was further compromised by the lack of precision in the findings; hence, the results from the sponsor-submitted ITC must be interpreted with caution. Moreover, no information was obtained regarding comparative safety versus other active comparators. In addition, no conclusion could be drawn on HRQoL outcomes.
Other Relevant Evidence
This section includes submitted long-term extension studies and additional relevant studies included in the sponsor’s submission to CADTH that were considered to address important gaps in the evidence included in the systematic review.
Long-Term Extension Studies
This section of the report includes a summary and critical appraisal of 3 long-term extension studies for Measure Up 1 to 52 (M18 to 891), Measure Up 2 to 52 (M16 to 045), and AD Up (M16 to 047). The co-primary objectives of Measure Up 1 to 52, Measure Up 2 to 52, and AD Up were the proportion of patients achieving at least EASI 75 at week 16 and the proportion of patients achieving a vIGA-AD score of 0 or 1 with at least 2 grades of reduction from baseline at week 16. This section reviews the long-term efficacy and safety of upadacitinib at week 52.
Methods
Measure Up 1 to 52, Measure Up 2 to 52, and AD Up are phase III, randomized, double-blind, placebo-controlled, multi-centre studies in adolescents (12 years to 17 years) and adults (18 years to 75 years) with moderate to severe AD. Measure Up 1 to 52 (with a week 52 data cut-off of December 21, 2020) included a 35-day screening period, a 16-week double-blind period, a BE period up to week 136, and a 30-day follow-up visit. Patients were randomized in a 1 to 1 to 1 ratio to receive a daily oral dose of upadacitinib 30 mg, upadacitinib 15 mg, or matching placebo every day. At the end of the double-blind period, week 16, patients in the placebo group were re-randomized in a 1 to 1 ratio (stratified by 50% improvement in EASI 50 responder status by week 16 [yes or no], geographic region [US, Puerto Rico, and Canada; mainland China; Japan; other], and age group [adolescent and adult]) to receive daily oral doses of upadacitinib 30 mg or upadacitinib 15 mg in the BE period. Patients originally randomized to upadacitinib were to continue upadacitinib in the extension period at the same dose. Rescue treatment was permitted starting at the week 4 visit, at the discretion of the investigator, if medically necessary and if specified parameters were met. The study design of Measure Up 1 to 52 is shown in Figure 34.
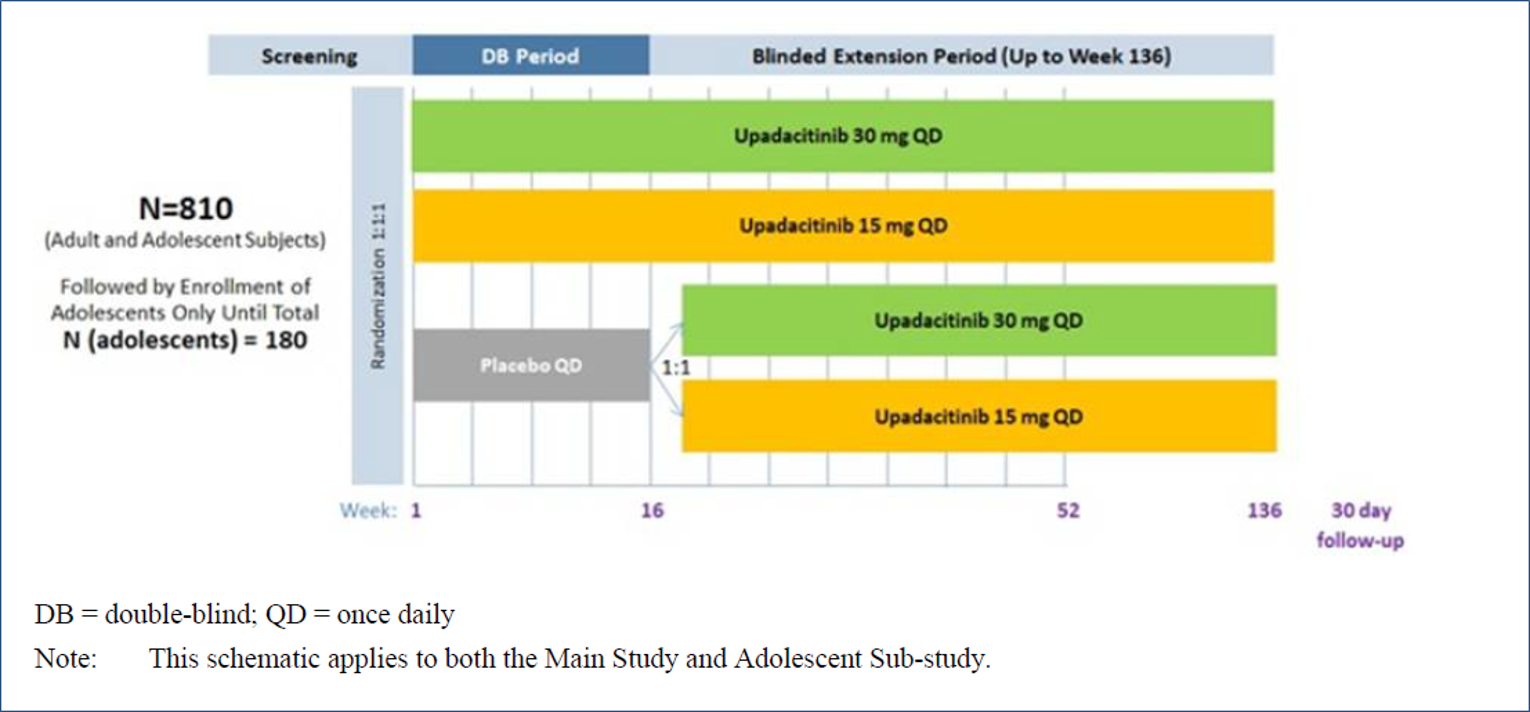
Measure Up 2 to 52 (which had a week 52 data cut-off of January 15, 2021) included a 35-day screening period, a 16-week double-blind period, a BE period of up to week 136, and a 30-day follow-up visit. Eligible patients had a documented history of inadequate response to topical AD treatments, documented use of systemic treatment for AD, or had otherwise been medically advised against topical treatments. Patients were randomized in a 1 to 1 to 1 ratio to receive a daily oral dose of upadacitinib 30 mg or upadacitinib 15 mg or matching placebo every day. At week 16, patients in the placebo group were re-randomized in a 1 to 1 ratio to receive daily oral doses of upadacitinib 30 mg or upadacitinib 15 mg in a blinded fashion up to week 136 in the BE period. For the main study, the re-randomization was stratified by EASI 50 responder (yes versus no), geographic region (US, Puerto Rico, and Canada versus other), and age (adolescent [ages 12 years to 17 years] versus adult [ages 18 years to 75 years]). For the adolescent substudy, the re-randomization was stratified by EASI 50 responder (yes versus no) and geographic region (US, Puerto Rico, and Canada versus other). Patients originally in the groups receiving upadacitinib 15 mg every day and upadacitinib 30 mg every day continued their treatment into the BE period up to the week 136 visit. Rescue therapy was permitted from week 4 through week 24 for patients with a less than 50% EASI response at 2 consecutive visits compared to baseline, or after week 24 for patients with a less than EASI 50 response at any visit compared to the baseline EASI score. The study design for Measure Up 2 to 52 is shown Figure 35.
AD Up (week 52 data cut-off was December 18, 2020) included a 35-day screening period, a 16-week double-blind period, a BE period of up to week 136, and a 30-day follow-up visit. Patients who met the eligibility criteria were randomized in a 1 to 1 to 1 ratio to receive concomitant TCS with a daily oral dose of upadacitinib 30 mg, upadacitinib 15 mg, or matching placebo every day. Patients less than 18 years of age were required to have a body weight of at least 40 kg at baseline. Randomization in the main study was stratified by baseline disease severity (moderate [vIGA-AD score of 3] versus severe [vIGA-AD score of 4]; age group [adolescent ages 12 years to 17 years versus adult ages 18 years to 75 years] and geographic region [US, Puerto Rico, Canada; Japan; mainland China; and other]). At the end of the double-blind period, week 16, patients in the placebo group were re-randomized in a 1 to 1 ratio (stratified by week 16 EASI 50 responders [yes versus no], geographic region [US, Puerto Rico, Canada; Japan; mainland China; and other] and age group [adolescent versus adult]) to receive oral upadacitinib 30 mg or upadacitinib 15 mg every day in a blinded fashion up to week 136. Patients originally randomized to upadacitinib continued upadacitinib in the BE period at the same dose. Rescue therapy was permitted from week 4 through week 24 for patients with a less than 50% reduction in EASI response at 2 consecutive visits compared to the baseline EASI score, or after week 24 for patients with a less than EASI 50 response at any visit compared to the baseline EASI score. The study design of AD Up is shown in Figure 36.
The BE period of the pivotal trials (Measure Up 1 to 52, Measure Up 2 to 52, AD Up) took place after all the ongoing patients in the main studies had completed the week 52 visit. The BE period evaluated the long-term safety, tolerability, and efficacy of upadacitinib (30 mg and 15 mg) daily in adolescents and adults with moderate to severe AD who had completed the double-blind period. The BE for all 3 studies was 136 weeks. Results for week 52 are reported in this section of the review.
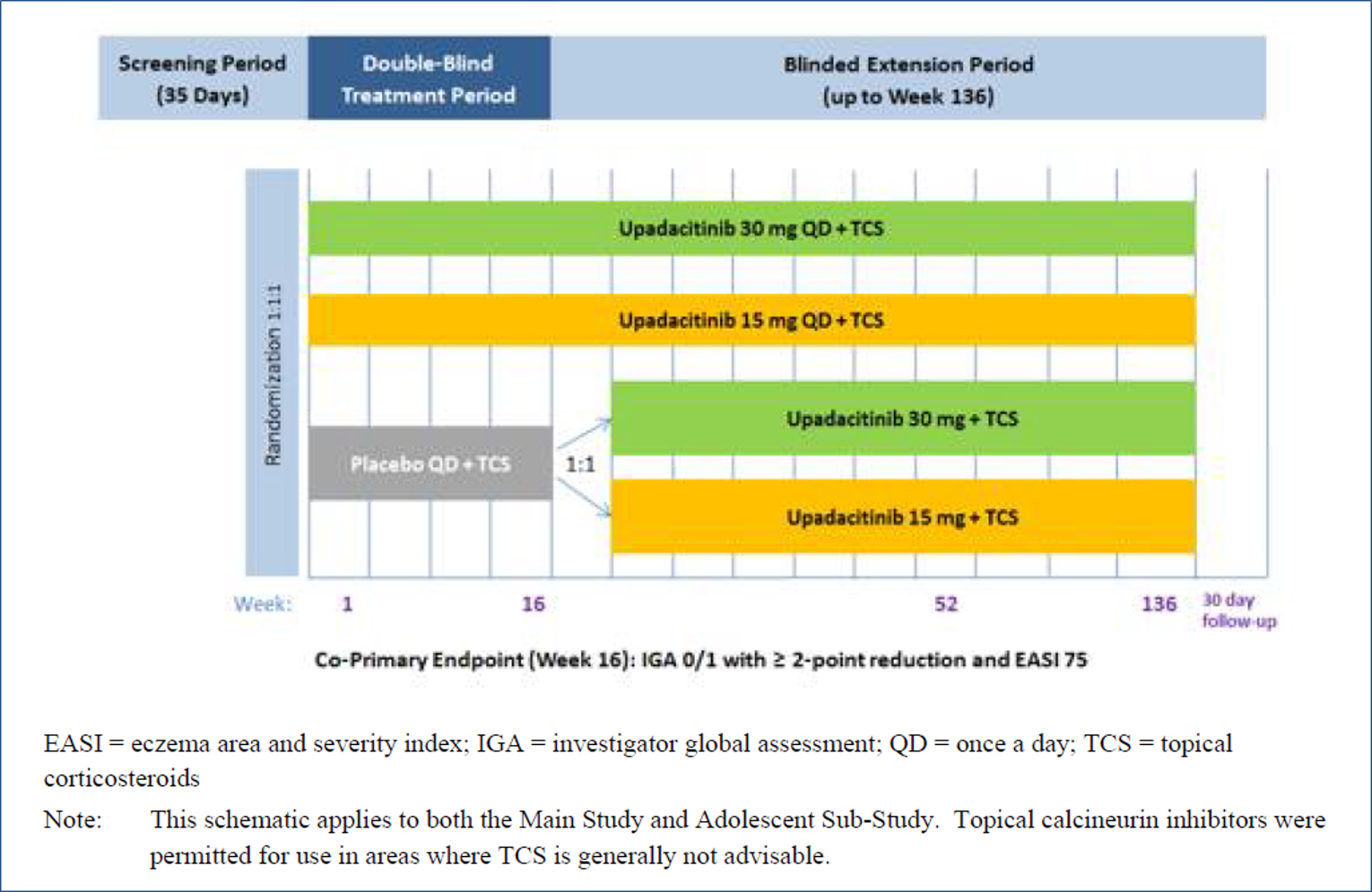
Populations
Measure Up 1 to 52 (N = 810) included adolescents (12 years to 17 years of age) and adults (18 years to 75 years of age) with moderate to severe AD who were candidates for systemic therapy. A total of 790 patients entered the BE period in Measure Up 1 to 52. Of these, 120 were adolescent patients. Demographic and baseline disease characteristics were unchanged from the data cut-off for week 16 of Measure Up 1 to 52.44 Measure Up 2 to 52 (N = 799) included adolescents (12 years to 17 years of age) and adults (18 years to 75 years of age) with moderate to severe AD who were candidates for systemic therapy. A total of 774 patients entered the BE period in Measure Up 2 to 52. Of these, 97 were adolescent patients. Demographic and baseline disease characteristics were unchanged from the data cut-off for week 16 of Measure Up 2 to 52.44 AD Up (N = 880) included adolescents (12 years to 17 years of age) and adults (18 years to 75 years of age) with moderate to severe AD who were candidates for systemic therapy y. A total of 862 patients entered the BE period in AD Up. Of these, 111 were adolescents. Demographic and baseline disease characteristics were unchanged from the data cut-off for week 16 of AD Up.44 The main inclusion and exclusion criteria were similar to the main studies in the BE period for all 3 studies.
Interventions
In Measure Up 1 to 52, the double-blind period (through week 16) compared the safety and efficacy of upadacitinib (30 mg and 15 mg) with placebo once daily. The BE period evaluated the long-term safety, tolerability, and efficacy of upadacitinib (30 mg and 15 mg) in adolescents and adults with moderate to severe AD who had completed the double-blind period.
In Measure Up 2 to 52, the double-blind period (through week 16) compared the safety and efficacy of upadacitinib (30 mg and 15 mg once daily) with placebo for the treatment of adolescent and adult patients with moderate to severe AD who were candidates for systemic therapy.
AD Up assessed the efficacy and safety of upadacitinib combined with TCS for the treatment of adolescent and adult patients with moderate to severe AD who were candidates for systemic therapy. The double-blind period (through week 16) compared the safety and efficacy of upadacitinib (30 mg and 15 mg once daily) plus TCS versus placebo plus TCS daily. The BE period evaluated the long-term safety, tolerability, and efficacy of upadacitinib (30 mg and 15 mg) in adolescents and adults with moderate to severe AD who had completed the double-blind period.
In Measure Up 1 to 52, up to the data cut-off, all patients receiving the study drug in the BE period were using concomitant medications. Emollients and protectives (27.9%) were the most common medications used, followed by other emollients and protectives (18.8%) and salbutamol (17.6%). In Measure Up 2 to 52, up to the data cut-off, 99.9% of patients who received the study drug during the BE period used concomitant medications. Emollients and protectives (38.8%) were the most common medications used, followed by salbutamol (15.6%) and paracetamol (12.4%). In AD Up, up to the data cut-off, nearly all patients (99.9%) receiving the study drug in the BE period were using concomitant medications. Hydrocortisone (38.1%) was the most common medication used, followed by triamcinolone (23.9%) and emollients and protectives (23.7%).
Outcomes
In Measure Up 1 to 52, Measure Up 2 to 52, and AD Up, the co-primary end points were the proportion of patients achieving: EASI 75 from baseline at week 16 and a vIGA-AD score of 0 or 1 (clear or almost clear) with at least 2 grades of reduction from baseline at week 16. The outcomes of interest identified in CADTH protocol also included WP-NRS, EASI 90, EASI percentage change from baseline, SCORAD, POEM (≥ 4 from baseline), ADerm-IS, ADerm-SS, ADerm-SS TSS-7, DLQI, CDLQI (0 or 1), EQ-5D-5L, HADS-A, and WPAI:AD. The BE period outcomes evaluated the long-term efficacy of the co-primary and secondary outcomes up to week 52.
Statistical Analysis
For the BE periods of Measure Up 1 to 52, Measure Up 2 to 52, and AD Up, there were no statistical hypotheses, no formal sample sizes, and no power calculations performed. The week 52 interim analysis was a protocol change (version 5), and the outcomes were not controlled for multiplicity. The safety population included all randomized patients who received at least 1 dose of the study drug in the main study or the adolescent substudy during this BE period. Long-term efficacy in the BE period was summarized using the observed case (OC) approach. The OC analysis was used for the summaries of long-term efficacy without imputing values for missing evaluations; hence, a patient who did not have an evaluation on a scheduled visit was not included in the OC analysis for that visit. The OC analysis was performed for all variables and did not include values after more than 1 day past discontinuation of study drug. The selected continuous variables (e.g., percentage change from baseline in EASI, WP-NRS, and SCORAD) were analyzed by MMRM up to the week 52 visit.
Patient Disposition
In Measure Up 1 to 52, among the 790 patients who entered the BE period, 786 patients (97.0%) were dosed. As of the cut-off date for week 52, 30 patients (3.7%) had initiated rescue medication during the BE period; 0 patients had completed the study drug in the BE period; and 114 (14.4%) of patients had discontinued the study drug during the BE period. One hundred percent of the adolescent patients who entered the BE period were dosed; 3.3% of adolescent patients initiated rescue medication during the BE period; 0 patients completed the study drug in the BE period; and 16.7% of adolescent patients discontinued the study drug during the BE period. The primary reasons for study drug discontinuation in adolescents were AEs, withdrawal consent, loss to follow-up, and lack of efficacy (all with 4.2%).
In Measure Up 2 to 52, 96.2% of the patients entering the BE period were dosed. As of the cut-off date for week 52, 4.1% of the patients initiated rescue medication in the BE period, and 14.3% of patients discontinued the study drug in the BE period. The most frequent primary reason for study drug discontinuation in the BE period was lack of efficacy (4.8%). All 97 adolescent patients who entered the BE period were dosed. Five percent of the patients initiated rescue medication in the BE period, and 15.8% of patients discontinued the study drug in the BE period. The most frequent primary reason for study drug discontinuation among adolescents in the BE period was lack of efficacy (9.9%).
In AD Up, 858 patients (including 111 adolescents) received the study drug (4 patients were not treated in the BE period). At the cut-off date, 118 patients (13.4%), including 13 adolescents, discontinued study treatment. The most frequent primary reason for treatment discontinuation in the BE period for all patients, including adolescents, was lack of efficacy. Two patients discontinued the study drug with protocol-mandated systemic rescue therapy as a primary reason. Overall, 84.1% patients continue taking the study drug in the BE period. The patient disposition is shown in Table 50 and Table 51.
Table 50: Patient Disposition (Measure Up 1 to 52 and Measure Up 2 to 52)
Disposition | Measure Up 1 to 52 | Measure Up 2 to 52 | ||||||
|---|---|---|---|---|---|---|---|---|
Placebo or UPA 15 mg q.d. (N = 121) | Placebo or UPA 30 mg q.d. (N = 123) | UPA 15 mg q.d. (N = 281) | UPA 30 mg q.d. (N = 285) n (%) | Placebo or UPA 15 mg q.d. (N = 120) | Placebo or UPA 30 mg q.d. (N = 121) | UPA 15 mg q.d. (N = 276) n (%) | UPA 30 mg q.d. (N = 282) | |
Entered BE period, n (%) | 121 (100) | 123 (100) | 273 (97.2) | 273 (95.8) | 120 (100) | 121 (100) | 264 (95.7) | 269 (95.4) |
Initiated rescue medication in BE period | 2 (1.7) | 1 (0.8) | 12 (4.3) | 15 (5.3) | 3 (2.5) | 1 (0.8) | 18 (6.5) | 11 (3.9) |
Completed study | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Discontinued study drug in BE period, n (%) | 13 (10.7) | 11 (8.9) | 42 (14.9) | 30 (10.5) | 14 (11.7) | 12 (9.9) | 30 (10.9) | 42 (14.9) |
Reason for discontinuation, n (%) | ||||||||
Adverse event | 3 (2.5) | 5 (4.1) | 7 (2.5) | 9 (3.2) | 5 (4.2) | 5 (4.1) | 4 (1.4) | 11 (3.9) |
Withdrawal of consent | 5 (4.1) | 6 (4.9) | 22 (7.8) | 15 (5.3) | 5 (4.2) | 2 (1.7) | 18 (6.5) | 17 (6.0) |
Lost to follow-up | 4 (3.3) | 1 (0.8) | 7 (2.5) | 4 (1.4) | 3 (2.5) | 2 (1.7) | 3 (1.1) | 3 (1.1) |
Other | 1 (0.8) | 1 (0.8) | 11 (3.9) | 7 (2.5) | 3 (2.5) | 6 (5.0) | 7 (2.5) | 14 (5.0) |
BE = blinded extension; q.d. = once daily; UPA = upadacitinib.
Source: Clinical Study Report for Measure Up 1 to 52, Measure Up 2 to 52, AD Up.44
Table 51: Patient Disposition (AD Up)
Disposition | Placebo + TCS or UPA 15 mg q.d.+ TCS (N = 144) | Placebo + TCS or UPA 30 mg q.d.+ TCS (N = 139) | UPA 15 mg q.d.+ TCS (N = 300) | UPA 30 mg q.d.+ TCS (N = 297) |
|---|---|---|---|---|
Entered BE period, n (%) | 144 (100) | 139 (100) | 290 (96.7) | 289 (97.3) |
Initiated rescue medication in BE period | 6 (4.2) | 7 (5.0) | 45 (15.0) | 24 (8.1) |
Completed study | 0 | 0 | 0 | 0 |
Discontinued study drug in BE period, n (%) | 25 (17.4) | 7 (5.0) | 46 (15.3) | 27 (9.1) |
Reason for discontinuation, n (%) | ||||
Adverse event | 6 (4.2) | 1 (0.7) | 7 (2.3) | 7 (2.4) |
Withdrawal of consent | 10 (6.9) | 4 (2.9) | 20 (6.7) | 10 (3.4) |
Lost to follow-up | 3 (2.1) | 1 (0.7) | 7 (2.3) | 1 (0.3) |
Other | 10 (6.9) | 2 (1.4) | 13 (4.3) | 11 (3.7) |
BE = blinded extension; TCS = topical corticosteroid; q.d. = once daily; UPA = upadacitinib.
Source: Clinical Study Reports for Measure Up 1 to 52, Measure Up 2 to 52, and AD Up.44
Exposure to Study Treatments
In Measure Up 1 to 52, the overall mean duration in days for upadacitinib exposure was 381.6 (SD = 140.45), and in the adolescent population the mean duration in days for upadacitinib exposure was 380.3 (SD = 144.10). Overall, 77.3% of the total population and 79.2% of the total adolescent population was treated with upadacitinib. In Measure Up 2 to 52, the overall mean duration in days for upadacitinib exposure was 366.4 (SD = 122.74), and in the adolescent population the mean duration in days for upadacitinib exposure was 378.2 (SD = 130.17). Overall, 74.3% of the total population and 75.2% of the total adolescent population was treated with upadacitinib. In AD Up, the overall mean duration in days for upadacitinib exposure was 366.9 (SD = 134.09), and in the adolescent population, the mean duration in days for upadacitinib exposure was 386.5 (SD = 119.45). Overall, 73.7% of the total population and 79.5% of the total adolescent population was treated with upadacitinib. In all studies, 0 patients reached at least 152 weeks of treatment with upadacitinib.
Efficacy
In Measure Up 1 to 52, Measure Up 2 to 52, and AD Up, the co-primary efficacy end points were the proportion of patients achieving at least EASI 75 from baseline at week 16 and a vIGA-AD score of 0 or 1 (clear or almost clear) with at least 2 grades of reduction from baseline at week 16. The long-term efficacy analysis evaluated these outcomes up to week 52.
In Measure Up 1 to 52, 59.2% and 62.5% of the patients who started upadacitinib 15 mg and upadacitinib 30 mg daily, respectively, maintained a vIGA-AD score of 0 or 1 at week 52; 61.8% and 74.8% of the patients who switched from placebo to upadacitinib 15 mg or 30 mg, respectively, maintained a vIGA-AD response of 0 or 1 at week 52; 82% and 84.9% of the patients who started upadacitinib 15 mg and 30 mg, respectively, maintained an EASI 75 response at week 52; and 82.4% and 91% of the patients who switched from placebo to upadacitinib 15 mg or 30 mg, respectively, maintained an EASI 75 response at week 52. The efficacy results of Measure Up 1 to 52 are shown in Table 52 and Table 53.
In Measure Up 2 to 52, 52.6% and 65.1% of the patients who started upadacitinib 15 mg and upadacitinib 30 mg once daily, respectively, maintained a vIGA-AD score of 0 or 1 at week 52; 54.4% and 64.8% of the patients who switched from placebo to upadacitinib 15 mg or upadacitinib 30 mg, respectively, maintained a vIGA-AD score of 0 or 1 at week 52;. 79.1% and 84.3% of the patients who started upadacitinib 15 mg and upadacitinib 30 mg, respectively, maintained an EASI 75 response at week 52; and 81.6% and 91.7% of the patients who switched from placebo to upadacitinib 15 mg or upadacitinib 30 mg, respectively, maintained an EASI 75 response at week 52. The efficacy results of Measure Up 2 to 52 are shown in Table 54.
In AD Up, 46.3% and 55.7% of the patients who started upadacitinib 15 mg and upadacitinib 30 mg once daily (plus TCS), respectively, maintained a vIGA-AD score of 0 or 1 at week 52; 54.3% and 70.6% of the patients who switched from placebo plus TCS to upadacitinib (15 mg or 30 mg) plus TCS, respectively, maintained a vIGA-AD score of 0 or 1 at week 52; 70.8% and 83.5% of the patients who started upadacitinib (15 mg and 30 mg) plus TCS once daily, respectively, maintained an EASI 75 response at week 52; and 84.3% and 92.1% of the patients who switched from placebo plus TCS to upadacitinib (15 mg or 30 mg) plus TCS, respectively, maintained an EASI 75 response at week 52. The efficacy results of AD Up are shown in Table 56 and Table 57.
Table 52: Skin Clearance and Disease Activity for Categorical End Points in the BE Period, Week 52 (OC, ITT Population) — All Patients Measure Up 1 to 52 — Redacted
End point | Placebo or UPA 15 mg (N = 102) | Placebo or UPA 30 mg (N = 111) | UPA 15 mg (N = 233) | UPA 30 mg (N = 232) |
|---|---|---|---|---|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | ||||
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | ||||
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
BE = blinded extension; CI = confidence interval; EASI = Eczema Area and Severity Index; ITT = intention to treat; OC = observed case; SCORAD = Scoring Atopic Dermatitis; UPA = upadacitinib; vIGA-AD = validated Investigator Global Assessment for Atopic Dermatitis.
aThe 95% CI for response was based on the normal approximation to the binomial distribution.
Note: Redacted rows have been deleted.
Source: Clinical Study Report for Measure Up 1 to 52.44
Table 53: Skin Clearance and Disease Activity Continuous Measures of Change From Baseline in the BE period, Week 52 (ITT Population) — Measure Up 1 to 52 for All Patients — Redacted
Measure of change | Placebo or UPA 15 mg (N = 102) | Placebo or UPA 30 mg (N = 111) | UPA 15 mg (N = 233) | UPA 30 mg (N = 232) |
|---|---|---|---|---|
Percentage change from baseline EASI score, MMRMa | ||||
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
ANCOVA = analysis of covariance; BE = blinded extension; BSA = body surface area; CI = confidence interval; DB = double blind; EASI = Eczema Area and Severity Index; ITT = intention to treat; LS = least squares; MMRM = mixed-effects model for repeated measures; OC = observed case; PBO = placebo; SCORAD = Scoring Atopic Dermatitis; SE = standard error; UPA = upadacitinib; vIGA-AD = validated Investigator Global Assessment for Atopic Dermatitis; vs. = versus.
aMeasurements after receiving rescue treatment were considered as missing; MMRM analyses were performed separately for the DB and BE periods.
bMeasurements after receiving rescue treatment were considered as missing; MMRM was performed for the DB period, with baseline, treatment, visit, treatment-by-visit interaction, and strata (baseline vIGA-AD score categories and age [adolescent vs. adult]) in the model; an unstructured covariance matrix was used; if the model could not converge, AR(1) or CS covariance matrix was used. For the BE period, the within-group LS mean, 95% CI, and SE — and between-groups LS mean, 95% CI, SE, and P value — were calculated using ANCOVA.
Note: Redacted rows have been deleted.
Source: Clinical Study Report for Measure Up 1 to 52.44
Table 54: Skin Clearance and Disease Activity for Categorical End Points in the BE Period, Week 52 (OC, ITT Population) — Measure Up 2 to 52 for All Patients — Redacted
End point | Placebo or UPA 15 mg (N = 103) | Placebo or UPA 30 mg (N = 108) | UPA 15 mg (N = 230) | UPA 30 mg (N = 229) |
|---|---|---|---|---|
|||||||||||||||||||||||||| | ||||
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
|||||||||||||||||||||||||| | ||||
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
BE = blinded extension; CI = confidence interval; EASI = Eczema Area and Severity Index; ITT = intention to treat; OC = observed cases; SCORAD = Scoring Atopic Dermatitis; UPA = upadacitinib; vIGA-AD = validated Investigator Global Assessment for Atopic Dermatitis.
aThe 95% CI for response rate was based on the normal approximation to the binomial distribution.
Note: Redacted rows have been deleted.
Source: Clinical Study Report for Measure Up 2 to 52.44
Table 55: Skin Clearance and Disease Activity Continuous Measures of Change From Baseline in the BE Period, Week 52 (ITT Population) — Measure Up 2 to 52 for All Patients — Redacted
Measure of change | Placebo or UPA 15 mg (N = 103) | Placebo or UPA 30 mg (N = 108) | UPA 15 mg (N = 230) | UPA 30 mg (N = 229) |
|---|---|---|---|---|
|||||||||||||||||||||||||| | ||||
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
ANCOVA = analysis of covariance; BE = blinded extension; BSA = body surface area; CI = confidence interval; DB = double blind; EASI = Eczema Area and Severity Index; ITT = intention to treat; LS = least squares; MMRM = mixed-effects model for repeated measures; OC = observed case; PBO = placebo; SCORAD = Scoring Atopic Dermatitis; SE = standard error; UPA = upadacitinib; vIGA-AD = validated Investigator Global Assessment for Atopic Dermatitis; vs. = versus.
aMeasurements after receiving rescue treatment were considered as missing; MMRM analyses were performed separately for the DB and BE periods.
bMeasurements after receiving rescue treatment were considered as missing; MMRM was performed for the DB period, with baseline, treatment, visit, treatment-by-visit interaction, and strata (baseline vIGA-AD score categories and age [adolescent vs. adult]) in the model; an unstructured covariance matrix was used; if the model could not converge, AR(1) or CS covariance matrix was used. For the BE period, within-group LS mean, 95% CI, and SE — and between-groups LS mean, 95% CI, SE, and P value — were calculated using ANCOVA.
Note: Redacted rows have been deleted.
Source: Clinical Study Report for Measure Up 2 to 52.44
Table 56: Skin Clearance and Disease Activity for Categorical End Points in the BE Period, Week 52 (OC, ITT Population) — AD Up for All Patients — Redacted
End point | PBO + TCS or UPA 15 mg + TCS (N = 127) | PBO + TCS or UPA 30 mg + TCS (N = 126) | UPA 15 mg + TCS (N = 242) | UPA 30 mg + TCS (N = 255) |
|---|---|---|---|---|
|||||||||||||||||||||||||||||||||||||||||||||||||||| | ||||
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
|||||||||||||||||||||||||||||||||||||||||||||||||||| | ||||
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
BE = blinded extension; CI = confidence interval; EASI = Eczema Area and Severity Index; ITT = intention to treat; OC = observed case; PBO = placebo; SCORAD = Scoring Atopic Dermatitis; TCS = topical corticosteroid; UPA = upadacitinib; vIGA-AD = validated Investigator Global Assessment for Atopic Dermatitis.
aThe 95% CI for response rate was based on the normal approximation to the binomial distribution.
Note: Redacted rows have been deleted.
Source: Clinical Study Report for AD Up.44
Table 57: Skin Clearance and Disease Activity Continuous Measures of Change From Baseline in the BE Period, Week 52 (ITT Population) — AD Up 52 for All Patients — Redacted
Measure of change | PBO + TCS or UPA 15 mg + TCS (N = 127) | PBO + TCS or UPA 30 mg + TCS (N = 126) | UPA 15 mg + TCS (N = 242) | UPA 30 mg + TCS (N = 255) |
|---|---|---|---|---|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | ||||
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | ||||
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| | ||||
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
ANCOVA = analysis of covariance; BE = blinded extension; BSA = body surface area; CI = confidence interval; EASI = Eczema Area and Severity Index; ITT = intention to treat; LS = least squares; MMRM = mixed-effects model for repeated measures; OC = observed case; PBO = placebo; SCORAD = Scoring Atopic Dermatitis; SE = standard error; TCS = topical corticosteroid; UPA = upadacitinib; vIGA-AD = validated Investigator Global Assessment for Atopic Dermatitis.
aMeasurements were used while patients were on study drug. In the BE period, the LS mean, 95% CI, and SE were calculated using ANCOVA.
Note: Redacted rows have been deleted.
Source: Clinical Study Report for AD Up.44
Harms
In Measure Up 1 to 52, a total of 648 patients had at least 1 AE during the study, most commonly related to acne (16.7%), upper respiratory tract infection (14.4%), and nasopharyngitis (13%). There were 60 patients (7.4%) who experienced an SAE during the study. The most common notable harms were hepatic disorder (6.1%), herpes zoster (5.3%), CPK elevation (8.2%), and serious infection (3.5%). Forty-seven patients (5.8%) discontinued the study drug due to AEs, and 1 death was reported in the upadacitinib 30 mg arm. In Measure Up 2 to 52, a total of 606 patients (75.8%) had at least 1 TEAE during the study. The most common TEAE was acne (16.8%). There were 50 patients (6.3%) who experienced an SAE. The most common notable harms were CPK elevation (8.4%), hepatic disorder (5.4%), herpes zoster (4.9%), and anemia (2.8%). A total of 49 patients (6.1%) discontinued the study drug due to AEs. No deaths were reported. In AD Up, a total of 731 patients had at least 1 TEAE during the study, most commonly related to nasopharyngitis (20.9%) and acne (16.1%). There were 52 patients (5.9%) who experienced an SAE during the study. The most common notable harms were CPK elevation (9.8%), herpes zoster (5.7%), and hepatic disorder (4.4%). Thirty-four patients (3.9%) discontinued the study drug due to AEs, and no deaths were reported. The summary of harms is shown in Table 58.
Table 58: Summary of Harms (Safety Population) in AD Up 52 — Redacted
Harms | Measure Up 1 to 52 | Measure Up 2 to 52 | AD Up 52 | |||
|---|---|---|---|---|---|---|
UPA 15 mg q.d. N = 401 | UPA 30 mg q.d. N = 408 | UPA 15 mg q.d. (N = 396) | UPA 30 mg q.d. (N = 403) | UPA 15 mg + TCS q.d. (N = 443) | UPA 30 mg + TCS q.d. (N = 436) | |
Patients with ≥ 1 adverse event | ||||||
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
|||||||||||||||||||||||||| | ||||||
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
Patients with ≥ 1 SAE | ||||||
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
|||||||||||||||||||||||||| | ||||||
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
Patients who stopped treatment due to adverse events | ||||||
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
|||||||||||||||||||||||||| | ||||||
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
Deaths | ||||||
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
Notable harms | ||||||
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
|||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
CPK = creatine phosphokinase; MACE = major adverse cardiac event; NMSC = non-melanoma skin cancer; SAE = serious adverse event; TCS = topical corticosteroid; TEAE = treatment-emergent adverse event; q.d. = once daily; UPA = upadacitinib; VTE = venous thromboembolism.
Note: Redacted rows have been deleted.
Source: Clinical Study Report for Measure Up 1 to 52, Measure Up 2 to 52, and AD Up 52.44
Critical Appraisal
The BE periods of Measure Up 1 to 52, Measure Up 2 to 52, and AD Up were meant to be 136 weeks; however, in none of the studies the patients completed 152 weeks, which raises questions about the sustainability of response to upadacitinib. The BE periods were single-arm studies, and the lack of a control arm significantly limits the interpretation of the study outcomes. The blinding was actively maintained. Randomization of patients from the placebo group to the upadacitinib group was conducted appropriately. The week 52 analyses were neither controlled for multiplicity nor part of a formal testing statistical hierarchy, which further limits the ability to interpret the results. All of the analyses were done in OC patients, which would be expected to introduce significant biases over the longer term, given the ongoing discontinuation of patients from the follow-up periods. Indeed, patients who are responding to therapy and free of AEs are more likely to continue in a long-term extension lasting up to 136 weeks; therefore, the interpretability of the long-term results is difficult.
Japan Study
The Japan study24 evaluated efficacy and safety outcomes as exploratory end points. This report will focus on the safety end points, due to significant statistical limitations of the efficacy data.
Methods
Briefly, this was a phase III, randomized, double-blind, multi-centre study that evaluated upadacitinib combined with TCS in adolescent and adult patients in Japan with moderate to severe AD who were candidates for systemic therapy. The study comprised a 35-day screening period, a 16-week double-blind treatment period, a 36-week BE period (week 16 to week 52), an open-label, long-term extension period (week 52 to either week 136 or permanent withdrawal of the marketing application), and a 30-day follow-up visit. The study was open label after all patients completed the week 52 visit. Patients who met the eligibility criteria were randomized in a 1 to 1 to 1 ratio to receive, in combination with TCS, daily oral doses of upadacitinib 15 mg, upadacitinib 30 mg, or placebo. Randomization was stratified by baseline disease severity (moderate [vIGA-AD = 3] versus severe [vIGA-AD = 4]) and age (< 18 years, 18 years to 40 years, or > 40 years). At the end of week 16, patients in the placebo group were re-randomized in a 1 to 1 ratio to receive daily oral doses of upadacitinib 15 mg or upadacitinib 30 mg. At week 16, the re-randomization of the placebo treatment group was stratified by a 50% reduction in EASI score (EASI 50) responder (yes or no) and age (< 18 years, 18 years to 40 years, or > 40 years old). Patients who were originally in the once-daily upadacitinib 15 mg and upadacitinib 30 mg groups continued their treatment into the long-term extension period up to week 136. A TCS regimen in combination with the study drug was mandatory until week 16. After week 16, any concomitant topical medication for AD could be administered per investigator discretion and was no longer required. Additionally, high-potency TCSs were not considered as rescue treatment after week 16. From week 4 through week 24, rescue treatment for AD could be provided at the discretion of the investigator if medically necessary (i.e., to control intolerable AD symptoms) for patients with a less than 50% reduction in EASI score response at any 2 consecutive scheduled visits; after week 24, systemic rescue treatment could be provided for patients with a less than EASI 50 response at any scheduled or unscheduled visit.
Populations
Eligible patients must have had a documented history of inadequate response to topical AD treatments or documented systemic AD treatment within 6 months before the baseline visit.
The safety population, which included all randomized patients who received at least 1 dose of study drug, was used for the safety analysis.
Interventions
Patients were randomized in a 1:1:1 ratio to receive, in combination with TCS, daily oral doses of upadacitinib 15 mg, upadacitinib 30 mg, or placebo.
Outcomes
There were no primary or secondary efficacy variables. The main focus of this report is safety, with the following evaluations and end points collected during the study:
TEAEs
SAEs
AESIs
AEs leading to discontinuation
vital signs and laboratory tests.
Patient Disposition
A total of 272 patients, including 29 adolescents, were randomized at 42 study sites in Japan. All 272 of the randomized patients received the study drug or placebo. A total of 264 patients (97.1%) completed the study through week 16 (the double-blind period). Six patients discontinued the study during the double-blind period; the most frequent primary reason given for study discontinuation was withdrawal by patient. A total of 264 patients continued to the BE or open-label period. All 264 patients (100%) were dosed in the BE or open-label period.
Demographic characteristics were generally balanced across the treatment groups. The majority of patients were male, did not use nicotine, and had a BMI of less than 25. The median age of patients was 36 years, and the adolescent group (median age 16 years) comprised 9.2% of the overall ITT population. Baseline disease characteristics were generally balanced across the upadacitinib and placebo groups. Patients had been diagnosed with AD for a mean of 22.8 years (SD = 14.3), had a mean baseline EASI score of 34.91 (SD = 13.83), a mean baseline vIGA-AD score of 3.5 (SD = 0.50), and a mean WP-NRS score (weekly rolling average) of 6.8 (SD = 1.39). Consistent with the eligibility criteria, all enrolled patients had inadequate response to previous treatment with topical AD treatments.
Adherence in the study was calculated as the number of tablets taken divided by the number of tablets planned to be taken by the patient during the double-blind treatment period of the study. Mean treatment adherence was 97.76% (SD = 3.29), 98.04% (SD = 3.67), and 97.16% (SD = 5.25) in the placebo, upadacitinib 15 mg, and 30 mg groups, respectively, in the double-blind period. The median treatment adherence was 99.11% in both the placebo and upadacitinib 15 mg groups, and 99.08% in the upadacitinib 30 mg group, in the double-blind period.
Harms
AEs are presented in Table 59. Through week 16, the observed rate of AEs in the upadacitinib 15 mg and 30 mg groups was higher than in the placebo group overall. The percentage of patients with AEs was numerically higher in the upadacitinib 30 mg group than in the upadacitinib 15 mg group. The observed rate of AEs with a reasonable possibility of being drug-related was also higher in the upadacitinib 30 mg group than the upadacitinib 15 mg and placebo groups. Through week 16, the number of patients with SAEs was low overall, with 1 patient in each of the 3 treatment groups reporting an SAE (cerebellar hemorrhage in the upadacitinib 15 mg group, herpes simplex in the upadacitinib 30 mg group, and cholelithiasis in the placebo group). One patient each in the upadacitinib 30 mg and placebo groups and 2 patients in the upadacitinib 15 mg group experienced AEs leading to discontinuation of the study drug. No deaths were reported. Among adolescents, the rate of AEs through week 16 was similar across all treatment groups. There were no SAEs or TEAEs leading to discontinuation reported in the adolescent population.
Table 59: Summary of Harms (Safety Population) — Japan Study
Harms | Placebo (N = 90) | UPA 15 mg q.d. (N = 91) | UPA 30 mg q.d. (N = 91) |
|---|---|---|---|
Patients with ≥ 1 adverse event | |||
n (%) | 38 (42.2) | 51 (56.0) | 58 (63.7) |
Most common events, n (%) | |||
Acne | 5 (5.6) | 12 (13.2) | 18 (19.8) |
Nasopharyngitis | 14 (15.6) | 12 (13.2) | 14 (15.4) |
Pyrexia | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
Arthralgia | 0 | 0 | 5 (5.5) |
Folliculitis | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
Patients with ≥ 1 SAE | |||
n (%) | 1 (1.1) | 1 (1.1) | 1 (1.1) |
Most common events, n (%) | |||
Serious infection | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
Patients who stopped treatment due to adverse events | |||
n (%) | 1 (1.1) | 2 (2.2) | 1 (1.1) |
Most common events,c n (%) | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
Possible malignancy | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| | |||||||||||||||||||||||||| |
Deaths | |||
n (%) | 0 | 0 | 0 |
Notable harms n (%) | |||
Serious infection | 0 | 0 | 1 (1.1) |
Opportunistic infection excluding tuberculosis and herpes zoster | 0 | 3 (3.3) | 1 (1.1) |
Herpes zoster | 0 | 0 | 0 |
Active tuberculosis | 0 | 0 | 0 |
Possible malignancy | 0 | 0 | 0 |
Malignancy | 0 | 0 | 0 |
NMSC | 0 | 0 | 0 |
Malignancy other than NMSC | 0 | 0 | 0 |
Lymphoma | 0 | 0 | 0 |
Hepatic disorder | 0 | 1 (1.1) | 1 (1.1) |
Anemia | 0 | 0 | 1 (1.1) |
Neutropenia | 0 | 1 (1.1) | 4 (4.4) |
Lymphopenia | 0 | 0 | 0 |
CPK elevation | 0 | 1 (1.1) | 2 (2.2) |
MACE | 0 | 1 (1.1) | 0 |
VTE | 0 | 0 | 0 |
CPK = creatine phosphokinase; MACE = major adverse cardiac event; NMSC = non-melanoma skin cancer; q.d. = once daily; SAE = serious adverse event; TCS = topical corticosteroid; TEAE = treatment-emergent adverse event; UPA = upadacitinib; VTE = venous thromboembolism.
Source: The Japan study.24
Discussion
Summary of Available Evidence
Four clinical studies were included in this report evaluating the use of upadacitinib in patients with moderate to severe AD.
Measure Up 1 and Measure Up 2 were 2 similar studies (n = 847 and n = 836, respectively) with a double-blind, placebo-controlled, parallel design. Eligible patients were adults and adolescents (≥ 40 kg) with chronic AD and a documented history of systemic treatment or inadequate response to topical AD treatments. Both studies randomized patients to daily upadacitinib 15 mg, upadacitinib 30 mg, or placebo to evaluate the proportion of responders with EASI 75 and a vIGA-AD score of 0 or 1 at week 16 as co-primary outcomes.
AD Up had a similar design, with the same inclusion criteria and population (n = 901), but it used TCS therapy in combination with upadacitinib 15 mg or upadacitinib 30 mg. It used the same co-primary end points at 16 weeks.
Heads Up was a double-blind, double-dummy, active-controlled, randomized study (n = 692) comparing upadacitinib 30 mg to dupilumab 300 mg SC in adults (18 years to 75 years old) with chronic AD and the same documented history of inadequate response to topical treatments or treatment with systemic therapies. This study’s primary end point included the proportion of patients achieving EASI 75.
BE studies of Measure Up 1 (Measure Up 1 to 52), Measure Up 2 (Measure Up 2 to 52), and AD Up (AD Up 52) were evaluated in the Other Relevant Literature section.
Three ITCs — 2 sponsor-submitted (ITC 1 and ITC 2) and 1 obtained from the CADTH literature search (ITC 3, an ICER evidence synthesis with NMA) — were included to provide greater perspective on the body of evidence by including ITCs of upadacitinib versus dupilumab and other systemic therapies.
Interpretation of Results
Efficacy
Upadacitinib is indicated for the treatment of adults and adolescents 12 years of age and older with refractory, moderate to severe AD who are not adequately controlled with a systemic treatment (e.g., a steroid or biologic) or when the use of such therapies is inadvisable. In all pivotal trials included in this CADTH review, an assessment of the subgroup of patients with previous systemic therapy use showed similar results to the base case (Measure Up1, Measure Up2, AD Up, and Heads Up trials) for the primary end points of response based on EASI 75 at week 16 and vIGA-AD score (except for the Heads Up study, which did not assess vIGA-AD). Although this implies that the beneficial effect of upadacitinib in a previously treated population reflects the overall base-case population, this information should be interpreted with caution because it was not an a priori specification for this subgroup analysis and may be underpowered for drawing conclusions. No specific information about this subgroup of patients with previous systemic therapy use was found in the ITCs evaluated by CADTH. The only exception is ITC 1 (reported in this CADTH review), which compared upadacitinib to dupilumab in a population of patients previously treated with (or intolerant to) systemic therapy (cyclosporine A); however, the comparison focuses only on dupilumab. The effect versus other interventions is uncertain.
All studies except for Heads Up evaluated outcomes that measure disease severity (such as EASI 75 and vIGA-AD) as primary end points and SCORAD as a key secondary end point. These end points were considered by the clinical expert consulted by CADTH for this review — and by patient groups in the patient input received — as critical for decision-making in clinical Canadian practice. Similarly, outcomes of HRQoL, mood, and productivity — measured using the WP-NRS, POEM, ADerm-IS, and ADerm-SS — were considered important elements for measurement to be applied across different domains of decision-making and for evaluating response to treatment with upadacitinib.
In their input to CADTH, patient groups and the clinical expert identified itch as the most burdensome symptom of AD. All of the included trials evaluated improvement in patient-reported itch using the WP-NRS instrument (a 10-point scale ranging from 0 [no itch] to 10 [worst itch imaginable]). All trials used a responder analysis based on the proportion of patients who achieved an improvement from baseline in WP-NRS of at least 4 units, which is considered clinically important. In both the monotherapy and combination therapy trials, both doses of upadacitinib demonstrated that a statistically significantly greater proportion of patients achieved a WP-NRS response of greater than or equal to 4, These results were considered clinically relevant by the expert consulted by CADTH.
Measures of HRQoL were also considered to be of critical value by the clinical expert consulted by CADTH and patient groups. Both the monotherapy and combination therapy studies demonstrated statistically significant improvements in the groups treated with upadacitinib 15 mg and upadacitinib 30 mg versus placebo on specific HRQoL skin condition measures, such as the DLQI, according to which a greater proportion of patients in the upadacitinib groups reached a minimally important difference benefit threshold of 4 or more. However, these effects were not detected using the EQ-5D-5L, a generic HRQoL instrument.
The need to address the use of upadacitinib as monotherapy or in combination with TCS was important, according to the clinical expert consulted by CADTH, because it is likely that clinicians will consider both scenarios. In the individual studies assessed in this CADTH report, upadacitinib 15 mg and upadacitinib 30 mg showed similar improvements over placebo in the co-primary end points (EASI 75 and vIGA-AD score) when used as monotherapy (i.e., in the Measure Up studies) and as combination therapy (i.e., in the AD Up and Heads Up studies). However, when assessing synthesized evidence from the ITCs, there were some differences noticed in the main end points evaluated at 16 weeks. For instance, both upadacitinib 30 and upadacitinib 15 mg were superior to dupilumab and other systemic treatments (in EASI 75 end point, IGA, and WP-NRS), but were not superior to abrocitinib 200 mg when evaluated as monotherapy. On the other hand, when used as combination therapy, no difference in the EASI 75 end point was detected between upadacitinib (30 mg or 15 mg) and dupilumab in 2 sponsor-submitted ITCs (ITC 1 and ITC 2); however, in the ICER ITC report, upadacitinib 30 mg was superior to both dupilumab and abrocitinib 100 mg. The reason for these differences — such as whether they arise from an additive effect of TCSs or issues of inconsistency in the evidence — is still unclear.
The comparison against dupilumab was considered an important issue that clinicians will ponder when choosing a treatment for patients with moderate to severe AD, given that both drugs would be considered second-tier therapies after a patient has failed on TCS therapies or when these are unadvisable. The Heads Up trial showed that at week 16, patients in the upadacitinib 30 mg group had higher rates of achieving EASI 75 and a better WP-NRS than patients on dupilumab 300 mg. However, these differences were no longer statistically significant at week 24, by which time dupilumab reached similar levels of efficacy. The clinical expert indicated that the benefit of JAK inhibitors is generally thought to be observed sooner than that of biologics. As such, the end points measured at week 16 may have favoured upadacitinib.
Although the overall results from the adolescent subpopulation were similar to those of the adult population in terms of efficacy outcomes and harms, the number of adolescents included in the individual studies for this report (or in the evidence syntheses from the ITCs) was small to give effect estimates with a high degree of certainty. Ongoing substudies will further elucidate the benefits and risks of upadacitinib in this group. Moreover, only adolescents weighing more than 40 kg were included in these studies; no information in patients with lower weights is available.
The body of evidence from the individual studies and the evidence syntheses (ITCs) have some limitations. Mainly, there were unbalanced discontinuations in the placebo groups in Measure Up and AD Up, mostly due to lack of effect and because secondary end points were analyzed only as complete cases. Both of these issues may have introduced bias in the study results in favour of upadacitinib, particularly in the placebo-controlled trials. Importantly, the results from these studies were robust based on the sensitivity analyses performed (i.e., multiple imputations and tipping point analyses), and were consistent among subgroups of adolescents. The ITCs have severe limitations due to heterogeneity, imprecision, and some limitations in reporting.
Harms
Both upadacitinib doses (15 mg and 30 mg) were well tolerated in all studies. The incidences of SAEs and AEs leading to study drug discontinuation were similar among groups except in the Heads Up study. The most frequently reported AEs were acne, upper respiratory tract infection, nasopharyngitis, headache, elevated CPK levels, and AD. No deaths were reported.
In the AD Up study, the most frequently reported AEs (≥ 5% in any treatment group) were acne, nasopharyngitis, upper respiratory tract infection, oral herpes, elevation of blood CPK levels, headache, and AD. Acne was reported more frequently in the upadacitinib groups (10% to 14%) than in the placebo groups (2%). No deaths were reported. The Japan study evaluated harms and efficacy outcomes only as exploratory end points. In that study, through week 16, the observed rate of AEs in the upadacitinib 15 mg and 30 mg groups was higher than in the placebo group overall.
In the Heads Up study, the safety profile of upadacitinib was similar to that found in the Measure Up and AD Up studies. The rates of SAEs and AEs leading to study drug discontinuation were 2.9% and 1.2% for upadacitinib and 1.2% and 1.2% for dupilumab, respectively. One death was reported in an upadacitinib-treated patient due to influenza-associated bronchopneumonia. The most frequently reported AE with upadacitinib was acne (15.8%), whereas this AE was reported by only 2.6% of patients on dupilumab. The most frequently reported AE in patients on dupilumab was conjunctivitis (8.4%), whereas this AE was reported by only 1.4% of patients on upadacitinib. Other AEs more common in the upadacitinib group were serious infection (1.1% versus 0.6%), eczema herpeticum (0.3% versus 0%), hepatic disorders (3.4% versus 1.5%), and herpes zoster (2.0% versus 0.9%). Also, rates of anemia (2.0% versus 0.3%), neutropenia (1.7% versus 0.6%), and CPK elevation (7.5% versus 3.2%) were higher for upadacitinib than dupilumab.
Recent concerns have emerged regarding the increase of serious heart-related events, cancer, blood clots, and death for patients using JAK inhibitors for certain chronic inflammatory conditions. The FDA has required warnings on product labels.67 It is important to note that this warning has been added to all JAK inhibitors (i.e., Xeljanz/Xeljanz XR [tofacitinib], Olumiant [baricitinib], and Rinvoq [upadacitinib]), and the need for it was based on a review of a large RCT studying the safety of tofacitinib to treat rheumatoid arthritis. Although full trial results are still unpublished, the RCT showed increased risk of thrombosis in patients with rheumatoid arthritis.
The product monograph for upadacitinib10 contains black box warnings regarding the risk of serious infections, malignancies, and thrombosis. It is recommended that treatment with upadacitinib be interrupted if a patient develops a serious infection (until the infection is controlled). Similar warnings are currently included on all Canadian product monographs of JAK inhibitors (i.e., Xeljanz/Xeljanz XR [tofacitinib]68 and Olumiant [baricitinib]).69 In contrast, the product monograph for dupilumab did not contain any black box warnings at the time of this review.17 The clinical expert consulted by CADTH noted that specialists may demonstrate a preference for dupilumab based on the perception that it is associated with a reduced risk of SAEs compared to JAK inhibitors.
Conclusions
Evidence from 3 double-blind, placebo-controlled studies (Measure Up 1, Measure Up 2, and AD Up) shows that upadacitinib 15 mg and upadacitinib 30 mg improve disease severity end points in adults with moderate to severe AD, based on EASI 75 and vIGA-AD scores, when compared to placebo, whether as monotherapy (Measure Up 1 and 2 studies) or in addition to a TCS (AD Up study). The evidence from these studies also indicates that upadacitinib 15 mg and upadacitinib 30 mg would likely reduce AD symptoms (as measured by the WP-NRS, POEM, and ADerm-IS), improve HRQoL (as measured by the DLQI), and improve mood and productivity domains (as measured by the HADS-A and WPAI:AD). The evidence suggests that these effect estimates are similar in the adolescent subpopulation. These results were considered clinically relevant by clinical experts and patients. The results of 1 head-to-head study (Heads Up) demonstrated superiority of upadacitinib 30 mg in reducing disease severity and symptoms (based on the EASI 75 and WP-NRS) compared to dupilumab at week 16; however, after 24 weeks, this difference was no longer observed, and the evidence remains uncertain beyond this time point.
Three ITCs support the notion that upadacitinib 30 mg and upadacitinib 15 mg are effective compared to dupilumab and other systemic immunomodulators, and that upadacitinib may be among the most effective systemic therapies for reducing both severity and symptoms in patients with moderate to severe AD, either as monotherapy or in combination with a TCS. However, conclusions regarding the long-term efficacy of upadacitinib versus the active comparators relevant to this review cannot be drawn because the ITCs used study results that were collected over a relatively short duration, whereas AD is chronic in nature. There is also uncertainty due to inherent heterogeneity across trials in the networks. The robustness of the comparative efficacy was further compromised by the lack of precision in the findings; hence, the results from the ITCs musts be interpreted with caution. Moreover, no information was obtained regarding the comparative safety of upadacitinib versus other active comparators. In addition, no conclusion could be drawn regarding the HRQoL outcomes.
All of the trials enrolled patients with moderate to severe AD who had responded inadequately to topical AD therapies or had already tried systemic therapies. This is reflective of the indication that was initially submitted to Health Canada and CADTH; however, the approved indication reflects a more restrictive population (i.e., patients who are not adequately controlled with a systemic treatment [e.g., a steroid or biologic] or for whom the use of such therapies is inadvisable). Although there is similarity in the results between the overall populations and the proportion of patients with prior exposure to a systemic therapy (indicating that prior exposure had little to no effect on benefits or harms versus the overall population), the generalizability of the results from the included studies to the approved indication is uncertain because only a proportion of the populations from the pivotal studies is relevant for the current indication of patients with previous systemic therapy use. In addition, there was no evidence for dose escalation to upadacitinib 30 mg once daily in patients with an inadequate response to 15 mg once daily; nor was there clinical evidence for dose de-escalation to upadacitinib 15 mg once daily in patients who achieved a response to 30 mg once daily. The clinical expert consulted by CADTH indicated that the subgroup analyses suggested that the response to upadacitinib would likely be similar for those with and without prior exposure to a systemic therapy for AD.
Overall, upadacitinib was safe and well tolerated in all studies. AEs that were more common with upadacitinib included acne and respiratory tract infections. The safety profile of upadacitinib once daily beyond 52 weeks was consistent with that observed during the 16-week double-blind period, with no unexpected safety signals reported. However, longer-term data will help to better characterize the efficacy and safety of upadacitinib to treat this chronic condition.
References
1.Wollenberg A, Barbarot S, Bieber T, et al. Consensus-based European guidelines for treatment of atopic eczema (atopic dermatitis) in adults and children: part I. J Eur Acad Dermatol Venereol. 2018;32(5):657-682. PubMed
2.Lynde C, Barber K, Claveau J, et al. Canadian practical guide for the treatment and management of atopic dermatitis. J Cutan Med Surg. 2005;8 Suppl 5:1-9. PubMed
3.Spergel JM. Epidemiology of atopic dermatitis and atopic march in children. Immunol Allergy Clin North Am. 2010;30(3):269-280. PubMed
4.Stander S. Atopic Dermatitis. N Engl J Med. 2021;384(12):1136-1143. PubMed
5.Yang L, Fu J, Zhou Y. Research Progress in Atopic March. Front Immunol. 2020;11:1907. PubMed
6.Drucker AM, Qureshi AA, Amand C, et al. Health Care Resource Utilization and Costs Among Adults with Atopic Dermatitis in the United States: A Claims-Based Analysis. J Allergy Clin Immunol Pract. 2018;6(4):1342-1348. PubMed
7.Eichenfield LF, Tom WL, Berger TG, et al. Guidelines of care for the management of atopic dermatitis: section 2. Management and treatment of atopic dermatitis with topical therapies. J Am Acad Dermatol. 2014;71(1):116-132. PubMed
8.CADTH Canadian Drug Expert Committee (CDEC) final recommendation: dupilumab (Dupixent); Sanofi-Aventis Canada Inc. Ottawa (ON): CADTH; 2020 Apr 24: https://www.cadth.ca/sites/default/files/cdr/complete/SR0636%20Dupixent%20-%20CDEC%20Final%20%20Recommendation%20April%2024%2C%202020%20for%20posting.pdf. Accessed 2021 Aug 30.
9.Sidbury R, Davis DM, Cohen DE, et al. Guidelines of care for the management of atopic dermatitis: section 3. Management and treatment with phototherapy and systemic agents. J Am Acad Dermatol. 2014;71(2):327-349. PubMed
10.Rinvoq (upadacitinib): extended-release tablets, 15 mg, 30 mg, oral [product monograph]. St. Laurent, QC: Abbvie Corporation; 2021: https://pdf.hres.ca/dpd_pm/00063108.PDF.
11.Clinical Study Report: M18-891 Week 16. A Phase 3 Randomized, Placebo-Controlled, Double-Blind Study to Evaluate Upadacitinib in Adolescent and Adult Subjects with Moderate to Severe Atopic Dermatitis [internal sponsor's report]. New York (NY): AbbVie; 2020.
12.Clinical Study Report: M16-045 Week 16. A phase 3 randomized, placebo-controlled, double-blind study to evaluate upadacitinib in adolescent and adult subjects with moderate to severe atopic dermatitis [internal sponsor's report]. Redwood City (CA): AbbVie; 2020 Sep 4.
13.Clinical Study Report: M16-047 Week 16. A Phase 3 Randomized, Placebo-Controlled, Double-Blind Study to Evaluate Upadacitinib in Combination with Topical Corticosteroids in Adolescent and Adult Subjects with Moderate to Severe Atopic Dermatitis [internal sponsor's report]. New York (NY): AbbVie; 2020.
14.Holmlund M, Orban K. Translation and cross-cultural adaptation of the performance-based test - Evaluation in Ayres Sensory Integration. Scand J Occup Ther. 2020:1-12. PubMed
15.Schram ME, Roekevisch E, Leeflang MM, Bos JD, Schmitt J, Spuls PI. A randomized trial of methotrexate versus azathioprine for severe atopic eczema. J Allergy Clin Immunol. 2011;128(2):353-359. PubMed
16.Meggitt SJ, Gray JC, Reynolds NJ. Azathioprine dosed by thiopurine methyltransferase activity for moderate-to-severe atopic eczema: a double-blind, randomised controlled trial. Lancet. 2006;367(9513):839-846. PubMed
17.Dupixent (dupilumab): Solution for subcutaneous injection, 300 mg single-use syringe, 300 mg single-use pen, 200 mg single-use syringe [product monograph]. Laval (QC): Sanofi-aventis Canada Inc.; 2021.
18.Khattab FM. Evaluation of botulinum toxin A as an optional treatment for atopic dermatitis. J Clin Aesthet Dermatol. 2020;13(7):32-35. PubMed
19.CellCept (mycophenolate mofetil): Capsules 250 mg, Film-coated tablets 500 mg, oral [product monograph]. Mississauga (ON): Hoffmann-La Roche Ltd; 2021.
20.Bohannon M, Liu M, Nadeau P, et al. Topical doxycycline monohydrate hydrogel 1% targeting proteases/PAR2 pathway is a novel therapeutic for atopic dermatitis. Exp Dermatol. 2020;29(12):1171-1175. PubMed
21.Methotrexate tablets (methotrexate): Tablets USP, 10 mg, oral [product monograph]. Kirkland (QC): Pfizer Canada ULC; 2019.
22.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. J Clin Epidemiol. 2016;75:40-46. PubMed
23.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2021 Apr 28.
24.Guttman-Yassky E, Teixeira HD, Simpson EL, et al. Once-daily upadacitinib versus placebo in adolescents and adults with moderate-to-severe atopic dermatitis (Measure Up 1 and Measure Up 2): results from two replicate double-blind, randomised controlled phase 3 trials. Lancet. 2021;397(10290):2151-2168. PubMed
25.Chesini D, Caminati M. Vitamin B12 and atopic dermatitis: any therapeutic relevance for oral supplementation? J Diet Suppl. 2020:1-5. PubMed
26.Hanifin JM, Rajka G. Diagnostic features of atopic dermatitis. Acta dermatovener (Stockholm) Suppl. 1980;92:44-47.
27.Schmitt J, Langan S, Deckert S, et al. Assessment of clinical signs of atopic dermatitis: a systematic review and recommendation. J Allergy Clin Immunol. 2013;132(6):1337-1347. PubMed
28.Hanifin JM, Thurston M, Omoto M, Cherill R, Tofte SJ, Graeber M. The eczema area and severity index (EASI): assessment of reliability in atopic dermatitis. EASI Evaluator Group. Exp Dermatol. 2001;10(1):11-18. PubMed
29.Leshem YA, Hajar T, Hanifin JM, Simpson EL. What the Eczema Area and Severity Index score tells us about the severity of atopic dermatitis: an interpretability study. Br J Dermatol. 2015;172(5):1353-1357. PubMed
30.Badia-Tahull MB, Cobo-Sacristán S, Leiva-Badosa E, et al. Use of Subjective Global Assessment, Patient-Generated Subjective Global Assessment and Nutritional Risk Screening 2002 to evaluate the nutritional status of non-critically ill patients on parenteral nutrition. Nutr Hosp. 2014;29(2):411-419. PubMed
31.Barbier N, Paul C, Luger T, et al. Validation of the Eczema Area and Severity Index for atopic dermatitis in a cohort of 1550 patients from the pimecrolimus cream 1% randomized controlled clinical trials programme. Br J Dermatol. 2004;150(1):96-102. PubMed
32.Schram ME, Spuls PI, Leeflang MM, Lindeboom R, Bos JD, Schmitt J. EASI, (objective) SCORAD and POEM for atopic eczema: responsiveness and minimal clinically important difference. Allergy. 2012;67(1):99-106. PubMed
33.Futamura M, Leshem YA, Thomas KS, Nankervis H, Williams HC, Simpson EL. A systematic review of Investigator Global Assessment (IGA) in atopic dermatitis (AD) trials: Many options, no standards. J Am Acad Dermatol. 2016;74(2):288-294. PubMed
34.Eichenfield LF, Tom WL, Chamlin SL, et al. Guidelines of care for the management of atopic dermatitis: section 1. Diagnosis and assessment of atopic dermatitis. J Am Acad Dermatol. 2014;70(2):338-351. PubMed
35.Simpson E, Bissonnette R, Eichenfield LF, et al. The Validated Investigator Global Assessment for Atopic Dermatitis (vIGA-AD): the development and reliability testing of a novel clinical outcome measurement instrument for the severity of atopic dermatitis. J Am Acad Dermatol. 2020;83(3):839-846. PubMed
36.Simpson E, Beck L, Gadkari A, Eckert L, Reaney M. Defining a responder on the Peak Pruritus Numerical Rating Scale (NRS) in patients with moderate-to-severe atopic dermatitis: detailed analysis from randomized trials of dupilumab J Am Acad Dermatol. 2017;76(6 Suppl 1):AB93.
37.Alexander H, Patton T, Jabbar-Lopez ZK, Manca A, Flohr C. Novel systemic therapies in atopic dermatitis: what do we need to fulfil the promise of a treatment revolution? F1000Res. 1000;8. PubMed
38.Chalmers JR, Schmitt J, Apfelbacher C, et al. Report from the third international consensus meeting to harmonise core outcome measures for atopic eczema/dermatitis clinical trials (HOME). Br J Dermatol. 2014;171(6):1318-1325. PubMed
39.Rehal B, Armstrong AW. Health outcome measures in atopic dermatitis: a systematic review of trends in disease severity and quality-of-life instruments 1985-2010. PLoS ONE [Electronic Resource]. 2011;6(4):e17520. PubMed
40.Charman CR, Venn AJ, Williams HC. The patient-oriented eczema measure: development and initial validation of a new tool for measuring atopic eczema severity from the patients' perspective. Arch Dermatol. 2004;140(12):1513-1519. PubMed
41.Silverberg JI, Lei D, Yousaf M, et al. Comparison of Patient-Oriented Eczema Measure and Patient-Oriented Scoring Atopic Dermatitis vs Eczema Area and Severity Index and other measures of atopic dermatitis: a validation study. Ann Allergy Asthma Immunol. 2020;125(1):78-83. PubMed
42.Gabes M, Tischer C, Apfelbacher C, quality of life working group of the Harmonising Outcome Measures for Eczema i. Measurement properties of quality-of-life outcome measures for children and adults with eczema: An updated systematic review. Pediatr Allergy Immunol. 2020;31(1):66-77. PubMed
43.Foley C, Tundia N, Simpson E, Teixeira HD, Litcher-Kelly L, Bodhani A. Development and content validity of new patient-reported outcome questionnaires to assess the signs and symptoms and impact of atopic dermatitis: the Atopic Dermatitis Symptom Scale (ADerm-SS) and the Atopic Dermatitis Impact Scale (ADerm-IS). Curr Med Res Opin. 2019;35(7):1139-1148. PubMed
44.Drug Reimbursement Review sponsor submission: Rinvoq (upadacitinib), once daily 15 mg and 30 mg extended-release tablet [internal sponsor's package]. Pointe-Claire (QC): AbbVie Corporation; 2021 Apr 12.
45.Shikiar R, Harding G, Leahy M, Lennox RD. Minimal important difference (MID) of the Dermatology Life Quality Index (DLQI): results from patients with chronic idiopathic urticaria. Health and quality of life outcomes. 2005;3:36. PubMed
46.Finlay AY, Khan GK. Dermatology Life Quality Index (DLQI)--a simple practical measure for routine clinical use. Clin Exp Dermatol. 1994;19(3):210-216. PubMed
47.Basra MK, Salek MS, Camilleri L, Sturkey R, Finlay AY. Determining the minimal clinically important difference and responsiveness of the Dermatology Life Quality Index (DLQI): further data. Dermatology. 2015;230(1):27-33. PubMed
48.Reilly MC, Lavin PT, Kahler KH, Pariser DM. Validation of the Dermatology Life Quality Index and the Work Productivity and Activity Impairment-Chronic Hand Dermatitis questionnaire in chronic hand dermatitis. J Am Acad Dermatol. 2003;48(1):128-130. PubMed
49.Wallenhammar LM, Nyfjall M, Lindberg M, Meding B. Health-related quality of life and hand eczema--a comparison of two instruments, including factor analysis. J Invest Dermatol. 2004;122(6):1381-1389. PubMed
50.Lewis V, Finlay AY. 10 years experience of the Dermatology Life Quality Index (DLQI). J Investig Dermatol Symp Proc. 2004;9(2):169-180. PubMed
51.Heinl D, Prinsen CA, Deckert S, et al. Measurement properties of adult quality-of-life measurement instruments for eczema: a systematic review. Allergy. 2016;71(3):358-370. PubMed
52.Lewis-Jones MS, Finlay AY. The Children's Dermatology Life Quality Index (CDLQI): initial validation and practical use. Br J Dermatol. 1995;132(6):942-949. PubMed
53.Salek MS, Jung S, Brincat-Ruffini LA, et al. Clinical experience and psychometric properties of the Children's Dermatology Life Quality Index (CDLQI), 1995-2012. Br J Dermatol. 2013;169(4):734-759. PubMed
54.Brooks R. EuroQol: the current state of play. Health Policy. 1996;37(1):53-72. PubMed
55.EuroQol Group. EuroQol--a new facility for the measurement of health-related quality of life. Health Policy. 1990;16(3):199-208. PubMed
56.Bjelland I, Dahl AA, Haug TT, Neckelmann D. The validity of the Hospital Anxiety and Depression Scale. An updated literature review. J Psychosom Res. 2002;52(2):69-77. PubMed
57.Herrmann C. International experiences with the Hospital Anxiety and Depression Scale--a review of validation data and clinical results. J Psychosom Res. 1997;42(1):17-41. PubMed
58.Zigmond AS, Snaith RP. The hospital anxiety and depression scale. Acta Psychiatr Scand. 1983;67(6):361-370. PubMed
59.Silverberg JI, Gelfand JM, Margolis DJ, et al. Measurement Properties of the Hospital Anxiety and Depression Scale Used in Atopic Dermatitis in Adults. J Invest Dermatol. 2019;139(6):1388-1391. PubMed
60.Reilly MC, Zbrozek AS, Dukes EM. The validity and reproducibility of a work productivity and activity impairment instrument. Pharmacoeconomics. 1993;4(5):353-365. PubMed
61.Prasad M, Wahlqvist P, Shikiar R, Shih YC. A review of self-report instruments measuring health-related work productivity: a patient-reported outcomes perspective. Pharmacoeconomics. 2004;22(4):225-244. PubMed
62.Clinical Study Report: M16-046. A Phase 3b Multicenter, Randomized, Double-Blind, Double-Dummy, Active Controlled Study Comparing the Safety and Efficacy of Upadacitinib to Dupilumab in Adult Subjects with Moderate to Severe Atopic Dermatitis [internal sponsor's report]. New York (NY): AbbVie; 2020.
63.Reich K, Thaci D, Papp K, et al. Treatment withdrawal and retreatment with upadacitinib in patients with moderate-to-severe atopic dermatitis from a phase 2b, randomized, controlled trial [conference abstract]. Chicago (IL): Revolutionizing Atopic Dermatitis (RAD) 2020 Virtual Congress; 2020: https://revolutionizingad.com/images/resources/2020Virtual/Abstracts/135_Reich_K_et_al_Upadacitinib_Tx_Withdrawal_and_Retreatment_Updated.pdf. Accessed 2022 Feb 1.
64.Network meta-analysis comparing the effectiveness of upadacitinib versus dupilumab in patients with moderate to severe atopic dermatitis with an inadequate response or intolerance to cyclosporin A [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Rinvoq (upadacitinib), once daily 15 mg and 30 mg extended-release tablet. Pointe-Claire (QC): Abbvie; 2020-Nov-20. 2020.
65.Barbarot S, Wollenberg A, Silverberg JI, et al. Dupilumab provides rapid and sustained improvement in SCORAD outcomes in adults with moderate-to-severe atopic dermatitis: combined results of four randomized phase 3 trials. J Dermatolog Treat. 2020:1-12. PubMed
66.Atlas SJ, Brouwer E, Fox G, et al. JAK inhibitors and monoclonal antibodies for the treatment of atopic dermatitis: effectiveness and value; evidence report. Boston (MA): Institute for Clinical and Economic Review; 2021: https://icer.org/assessment/atopic-dermatitis-2021/#timeline. Accessed 2021 Aug 30.
67.U.S. Food & Drug Administration. FDA requires warnings about increased risk of serious heart-related events, cancer, blood clots, and death for JAK inhibitors that treat certain chronic inflammatory conditions. 2021 Dec 7; https://www.fda.gov/drugs/drug-safety-and-availability/fda-requires-warnings-about-increased-risk-serious-heart-related-events-cancer-blood-clots-and-death. Accessed 2022 Feb 1.
68.Xeljanz (tofacitinib): tablets 5 mg, 10 mg, tablets extended-release 11 mg [product monograph]. Kirkland (QC): Pfizer Canada ULC; 2021: https://pdf.hres.ca/dpd_pm/00063900.PDF.
69.Baykal Selcuk L, Ipek O, Aksu D, Yayli S, Bahadir S. Evaluation of factors that may affect disease development and severity in childhood atopic dermatitis. G Ital Dermatol Venereol. 2020;13:13. PubMed
70.Clinical Study Report: M16-045 Week 52. A phase 3 randomized, placebo-controlled, double-blind study to evaluate upadacitinib in adolescent and adult subjects with moderate to severe atopic dermatitis [internal sponsor's report]. Redwood City (CA): AbbVie; 2021 Mar 12.
71.Clinical Study Report: M18-891 Week 52. A phase 3 randomized, placebo-controlled, double-blind study to evaluate upadacitinib in adolescent and adult subjects with moderate to severe atopic dermatitis [internal sponsor's report]. Redwood City (CA): AbbVie; 2021 Mar 26.
72.Howells L, Ratib S, Chalmers JR, Bradshaw L, Thomas KS, CLOTHES trial team. How should minimally important change scores for the Patient-Oriented Eczema Measure be interpreted? A validation using varied methods. Br J Dermatol. 2018;178(5):1135-1142. PubMed
73.Silverberg JI, Simpson EL, Litcher-Kelly L, McDonald J, Calimlim BM, Leshem YA. Psychometric evaluation of three patient-reported outcome questionnaires assessing the symptoms and impacts of atopic dermatitis in adults and adolescents. Conference. Br J Dermatol. 2020;184(3):e75-e76.
Appendix 1: Literature Search Strategy
Note that this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases
MEDLINE All (1946-present)
Embase (1974-present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: May 11, 2021
Alerts: Bi-weekly search updates until project completion
Study types: No filters were applied to limit the retrieval by study type.
Limits
No date or language limits were used
Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
MeSH | Medical Subject Heading |
exp | Explode a subject heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Author keyword heading word (MEDLINE) |
.kw | Author keyword (Embase) |
.dq | Candidate term word (Embase) |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multi-Database Strategy
(rinvoq* or upadacitinib* or ABT 494 or ABT494 or 4RA0KN46E0 or 7KCW9IQM02 or NEW4DV02U5 or 328W323FLH).ti,ab,kf,ot,hw,rn,nm.
1 use medall
*upadacitinib/
(rinvoq* or upadacitinib or ABT 494 or ABT494).ti,ab,kw,dq.
3 or 4
5 use oemezd
6 not (conference review or conference abstract).pt.
2 or 7
remove duplicates from 8
Clinical Trials Registries
ClinicalTrials.gov
Produced by the US National Library of Medicine. Targeted search used to capture registered clinical trials.
[Search -- Studies with results | Rinvoq OR upadacitinib]
WHO ICTRP
International Clinical Trials Registry Platform, produced by the WHO. Targeted search used to capture registered clinical trials.
[Search terms -- Rinvoq OR upadacitinib]
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
[Search terms -- Rinvoq OR upadacitinib]
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
[Search terms -- Rinvoq OR upadacitinib]
Grey Literature
Search dates: April 27-May 5, 2021
Keywords: Rinvoq, upadacitinib, atopic dermatitis
Limits: None
Updated: Search updated before the completion of stakeholder feedback period
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Health Statistics
Internet Search
Appendix 2: Excluded Studies
Note that this appendix has not been copy-edited.
Reference | Reason for Exclusion |
|---|---|
Guttman-Yassky E, Thaci D, Pangan AL, et al. Upadacitinib in adults with moderate to severe atopic dermatitis: 16-week results from a randomized, placebo-controlled trial. J Allergy Clin Immunol. 2020;145(3):877 to 884. | phase II study. |
Appendix 3: Detailed Outcome Data
Note that this appendix has not been copy-edited.
Table 62: Efficacy Outcomes — Adults and Adolescent Groups (Measure Up 1 Study)
Outcome | ADULTS | ADOLESCENTS | ||||
|---|---|---|---|---|---|---|
Placebo N = 241 | UPA 15 mg q.d. N = 239 | UPA 30 mg q.d. N = 243 | Placebo N = 40 | UPA 15 mg q.d. N = 42 | UPA 30 mg q.d. N = 42 | |
Disease Severity | ||||||
EASI 75 at week 16 | ||||||
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
vIGA-AD response 0 or 1 at week 16 | ||||||
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
Symptoms | ||||||
WP-NRS response > = 4 from baseline at week 16 | ||||||
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
% Change in WP-NRS from baseline at week 16 | ||||||
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
POEM total score improvement from baseline at week 16 | ||||||
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
ADerm-SS skin pain improvement (> = 4) from baseline at week 16 | ||||||
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
ADerm-IS Emotional State domain score improvement (> = 11) from baseline at week 16 | ||||||
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
HRQoL | ||||||
DLQI improvement (> = 4) from baseline at week 16 | ||||||
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
CDLQI score of 0 or 1 at week 16 in patients < 16 years of age | ||||||
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
Change in EQ-5D index from baseline at week 16 | ||||||
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
Mood | ||||||
HADS-A or HADS-D response (< 8) at week 16 | ||||||
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
AD = atopic dermatitis; ADerm-SS = Atopic Dermatitis Symptom Scale; ADerm-IS = Atopic Dermatitis Impact Scale; CI = confidence interval; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; HADS-A or HADS-D = Hospital Anxiety and Depression Scale for Anxiety or Depression; HRQoL = Health-Related Quality of Life; LS = least squares; POEM = Patient-Oriented Eczema Measure; q.d. = once daily; UPA = upadacitinib; vIGA-AD = validated Investigator Global Assessment for Atopic Dermatitis; vs. = versus; WP-NRS = Worst Pruritus Numerical Rating Scale; WPAI = Work Productivity and Activity Impairment
aAssessed in the ITT population with nonresponder imputation incorporating multiple imputation to handle data missing due to COVID-19.
bComplete case analysis.
cMixed-effects model for repeat measures with baseline, treatment, visit, treatment-by-visit interaction, and strata (baseline vIGA-AD score categories and age [adolescent vs. adult]) in the model.
d95% CI for adjusted difference and P values are calculated according to the Cochran-Mantel-Haenszel test adjusted for strata (baseline vIGA-AD score categories and age [adolescent vs. adult]) for the comparison of 2 treatment groups.
eThe calculations at each visit are based on nonresponder imputation incorporating multiple imputation to handle data missing due to COVID-19 or nonresponder imputation only if there are no data missing due to COVID-19.
fWithin-group LS mean and 95% CI, and between groups LS mean, 95% CI, and P value are calculated from ANCOVA with baseline, treatment, and strata (baseline vIGA-AD score categories and age [adolescent vs. adult]) in the model.
Note: Redacted rows have been deleted.
Source: Clinical Study Report: Measure Up 1 study.12
Table 63: Efficacy Outcomes — Adults and Adolescent Groups (Measure Up 2 Study)
Outcome | ADULTS | ADOLESCENTS | |||||
|---|---|---|---|---|---|---|---|
Placebo N = 242 | UPA 15 mg q.d. N = 243 | UPA 30 mg q.d. N = 247 | Placebo N = 36 | UPA 15 mg q.d. N = 33 | UPA 30 mg q.d. N = 35 | ||
Disease Severity | |||||||
EASI 75 at week 16 | |||||||
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | |
vIGA-AD response 0 or 1 at week 16 | |||||||
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | |
Symptoms | |||||||
WP-NRS response > = 4 from baseline at week 16 | |||||||
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | |
% Change in WP-NRS from baseline at week 16 | |||||||
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | |
POEM total score improvement from baseline at week 16 | |||||||
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | |
ADerm-SS skin pain improvement (> = 4) from baseline at week 16 | |||||||
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | |
ADerm-IS Emotional State domain score improvement (> = 11) from baseline at week 16 | |||||||
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | |
HRQoL | |||||||
DLQI improvement (> = 4) from baseline at week 16 | |||||||
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | |
DLQI score of 0 or 1 at week 16 | |||||||
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | |
CDLQI score of 0 or 1 at week 16 in patients < 16 years of age | |||||||
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | |
Change in EQ-5D index from baseline at week 16 | |||||||
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | |
Mood | |||||||
HADS-A or HADS-D response (< 8) at week 16 | |||||||
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | |
AD = atopic dermatitis; ADerm-SS = Atopic Dermatitis Symptom Scale; ADerm-IS = Atopic Dermatitis Impact Scale; CI = confidence interval; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; HADS-A or HADS-D = Hospital Anxiety and Depression Scale for Anxiety or Depression; HRQoL = Health-Related Quality of Life; LS = least squares; POEM = Patient-Oriented Eczema Measure; q.d. = once daily; UPA = upadacitinib; vIGA-AD = Validated Investigator Global Assessment for Atopic Dermatitis; WP-NRS = Worst Pruritus Numerical Rating Scale; WPAI = Work Productivity and Activity Impairment; ITT = intention to treat; SCORAD = Scoring Atopic Dermatitis; SD = standard deviation; SE = standard error; TSS-7 = 7-Item Total Symptom Score.
aAssessed in the ITT population with nonresponder imputation incorporating multiple imputation to handle missing data due to COVID-19.
bComplete case analysis.
cMixed-effects model for repeated measures with baseline, treatment, visit, treatment-by-visit interaction, and strata (baseline vIGA-AD categories and age [adolescent vs. adult]) in the model.
d95% CI for adjusted difference and P values are calculated according to the Cochran-Mantel-Haenszel test adjusted for strata (baseline vIGA-AD categories and age [adolescent vs. adult]) for the comparison of 2 treatment groups.
eThe calculations at each visit are based on nonresponder imputation incorporating multiple imputation to handle missing data due to COVID-19 or nonresponder imputation only if there are no missing data due to COVID-19.
fWithin-group LS mean and 95% CI, and between groups LS mean, 95% CI, and P value are calculated from ANCOVA with baseline, treatment, and strata (baseline vIGA-AD categories and age [adolescent vs. adult]) in the model.
Note: Redacted rows have been deleted.
Source: Clinical Study Report: Measure Up 2 study.11
Table 64: Efficacy Outcomes — Adults and Adolescent Groups (AD Up Study)
Outcome | ADULTS | ADOLESCENTS | ||||
|---|---|---|---|---|---|---|
Placebo + TCS N = 264 | UPA 15 mg q.d.+ TCS N = 261 | UPA 30 mg q.d. TCS N = 260 | Placebo + TCS N = 40 | UPA 15 mg q.d.+ TCS N = 39 | UPA 30 mg q.d. TCS N = 37 | |
Disease Severity | ||||||
EASI 75 at week 16 | ||||||
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
vIGA-AD response 0 or 1 at week 16 | ||||||
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
Symptoms | ||||||
WP-NRS response > = 4 from baseline at week 16 | ||||||
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
% Change in WP-NRS from baseline at week 16 | ||||||
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
POEM total score improvement (> = 4) from baseline at week 16 | ||||||
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
ADerm-SS skin pain improvement (> = 4) from baseline at week 16 | ||||||
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
ADerm-IS Emotional State domain score improvement (> = 11) from baseline at week 16 | ||||||
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
HRQoL | ||||||
DLQI improvement (> = 4) from baseline at week 16 | ||||||
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
DLQI score of 0 or 1 at week 16 | ||||||
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
CDLQI score of 0 or 1 at week 16 in patients < 16 years of age | ||||||
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
Change in EQ-5D-5L index from baseline at week 16 | ||||||
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
Mood | ||||||
HADS-A or HADS-D response (< 8) at week 16 | ||||||
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
Productivity | ||||||
Change in WPAI:AD (Absenteeism) domain scores | ||||||
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| | ||||||||||||||||||| |
AD = atopic dermatitis; ADerm-SS = Atopic Dermatitis Symptom Scale; ADerm-IS = Atopic Dermatitis Impact Scale; CI = confidence interval; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; HADS-A or HADS-D = Hospital Anxiety and Depression Scale for Anxiety or Depression; HRQoL = Health-Related Quality of Life; LS = least squares; POEM = Patient-Oriented Eczema Measure; UPA = upadacitinib; vIGA-AD = validated Investigator Global Assessment for Atopic Dermatitis; vs. = versus; WP-NRS = Worst Pruritus Numerical Rating Scale; WPAI = Work Productivity and Activity Impairment.
aAssessed in the ITT population with nonresponder imputation incorporating multiple imputation to handle data missing due to COVID-19.
b95% CI for adjusted difference and P values are calculated according to the Cochran-Mantel-Haenszel test adjusted for strata (baseline vIGA-AD score categories and age [adolescent vs. adult]) for the comparison of 2 treatment groups.
cComplete case analysis.
dMixed-effects model for repeated measures with baseline, treatment, visit, treatment-by-visit interaction, and strata (baseline vIGA-AD score categories and age [adolescent vs. adult]) in the model.
eWithin-group LS means and 95% CI, and between groups LS means, 95% CI, and P value are calculated from ANCOVA with baseline, treatment, and strata (baseline vIGA-AD score categories and age [adolescent vs. adult]) in the model.
Note: Redacted rows have been deleted.
Source: Clinical Study Report: AD Up Study.13
Figure 37: Graphical Approach for Multiplicity Adjustment for US FDA Regulatory Purpose (ITT Population) in the Measure Up and AD Up Studies
Source: Case report form – Statistical analysis plan: Measure Up studies.70,71
Figure 38: Proportion of Patients Achieving EASI 75 at Week 16 by Subgroup in the Upadacitinib 30 mg and 15 mg Groups (NRI-C, ITT Population) of the Measure Up 1 Study
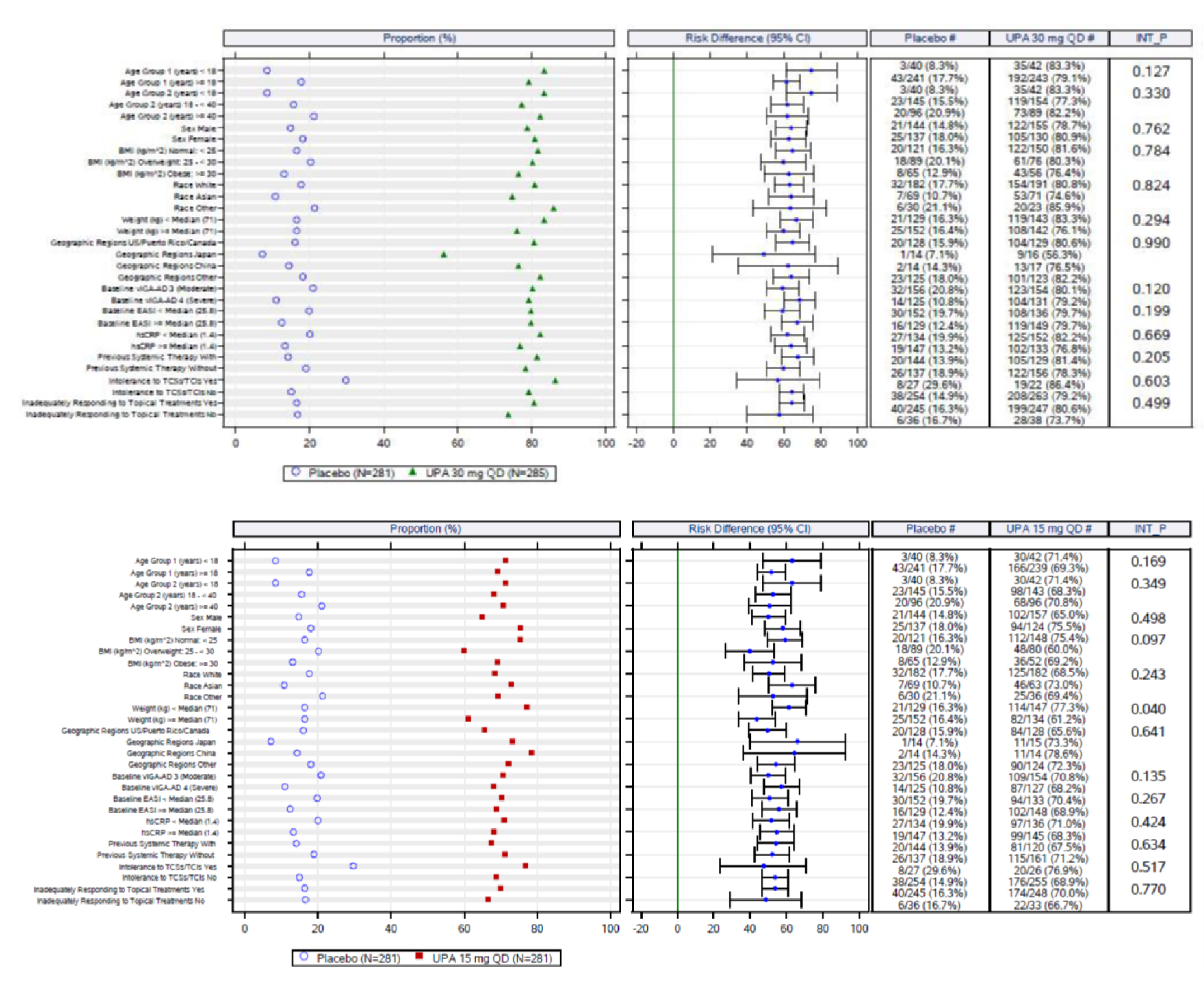
BMI = body mass index; CI = confidence interval for adjusted difference, calculated according to the Cochran-Mantel-Haenszel test adjusted for strata; EASI = Eczema Area and Severity Index; hsCRP = high-sensitivity C-reactive protein; INT_P = P value for interaction between subgroup and treatment was calculated using a logistic regression with visit measurement at week 16 as response variable, treatment, subgroup, strata, and treatment-by-subgroup interaction as factors; ITT_M = intention-to-treat population for the main study; NRI-C = nonresponder imputation to handle data missing due to COVID-19; TCI = topical calcineurin inhibitor; TCS = topical corticosteroid; QD = every day; UPA = upadacitinib; vIGA-AD = validated Investigator Global Assessment for Atopic Dermatitis.
Source: Case report form: Measure Up studies.70
Figure 39: Proportion of Patients Achieving vIGA-AD of 0 or 1 at Week 16 by Subgroup in the Upadacitinib 30 mg and 15 mg Group (NRI-C, ITT Population) of the Measure Up 1 Study
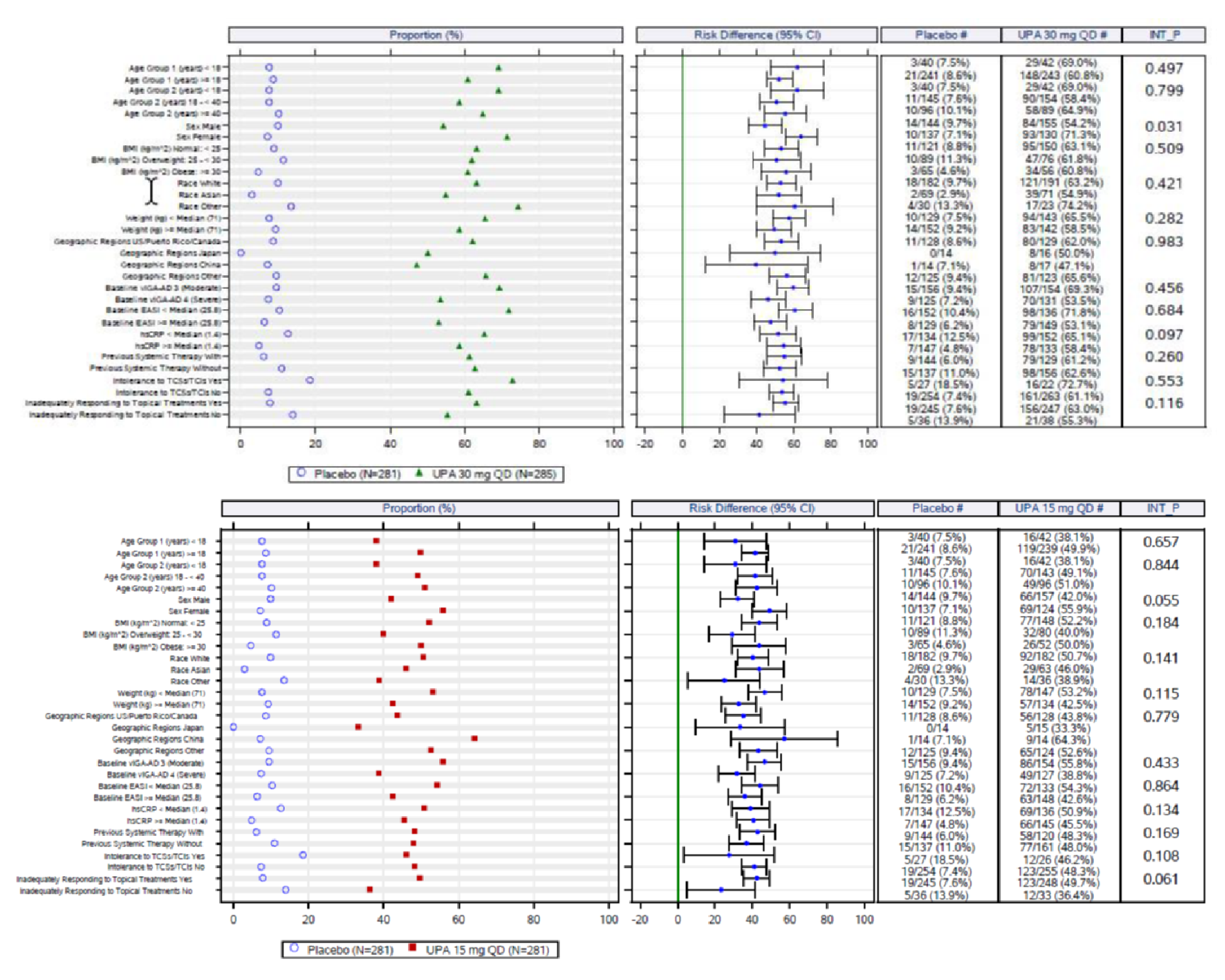
BMI = body mass index; CI = confidence interval for adjusted difference, calculated according to the Cochran-Mantel-Haenszel test adjusted for strata; EASI = Eczema Area and Severity Index; hsCRP = high-sensitivity C-reactive protein; INT_P = P value for interaction between subgroup and treatment was calculated using a logistic regression with visit measurement at week 16 as response variable, treatment, subgroup, strata, and treatment-by-subgroup interaction as factors; ITT_M = intention-to-treat population for the main study; NRI-C = nonresponder imputation to handle data missing due to COVID-19; TCI = topical calcineurin inhibitor; TCS = topical corticosteroid; QD = every day; UPA = upadacitinib; vIGA-AD = validated Investigator Global Assessment for Atopic Dermatitis.
Source: Case report form: Measure Up studies.70
Figure 40: Proportion of Patients Achieving EASI 75 at Week 16 by Subgroup in the Upadacitinib 30 mg and 15 mg Group (NRI-C, ITT Population) of the Measure Up 2 Study
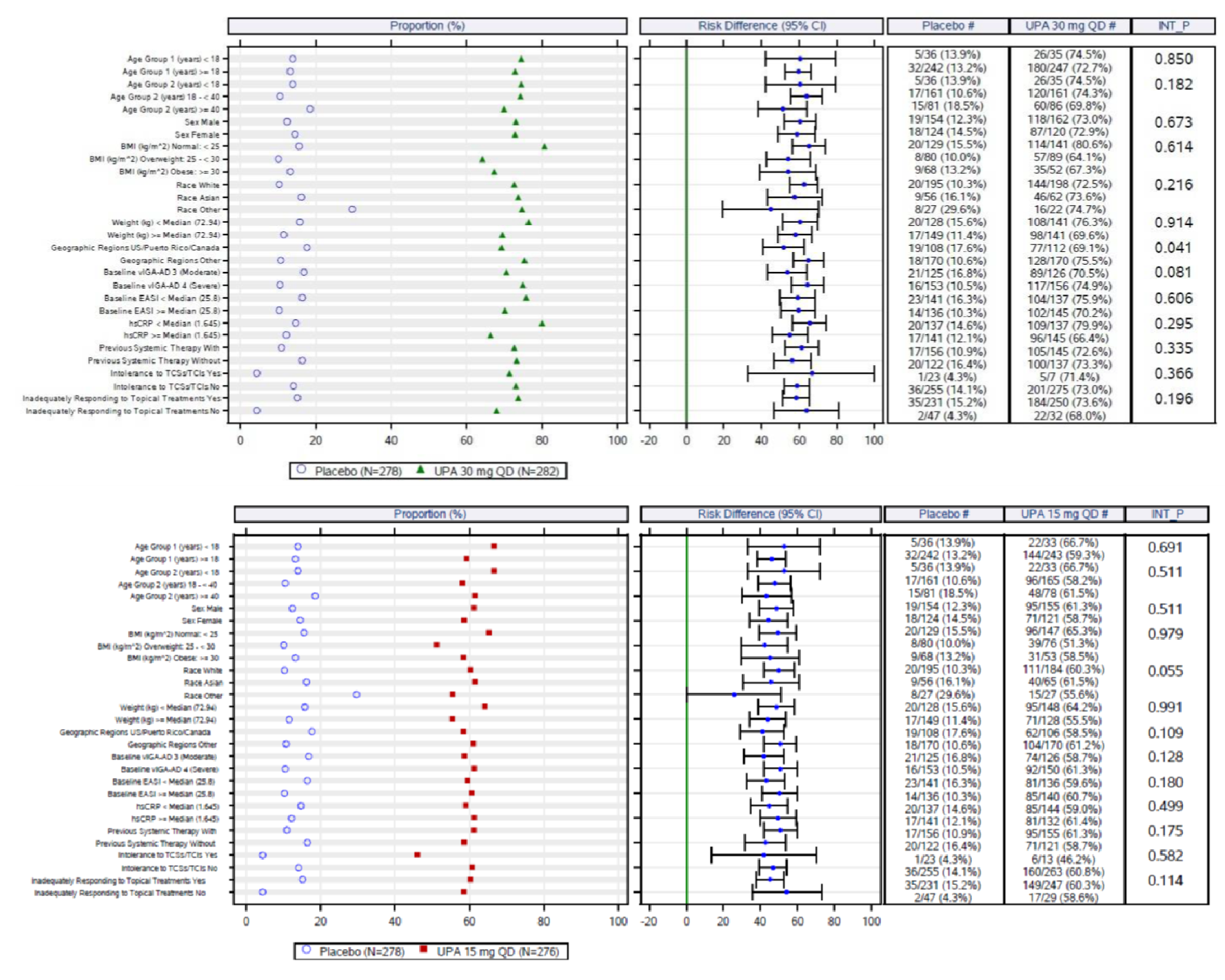
BMI = body mass index; CI = confidence interval for adjusted difference, calculated according to the Cochran-Mantel-Haenszel test adjusted for strata; EASI = Eczema Area and Severity Index; hsCRP = high-sensitivity C-reactive protein; INT_P = P value for interaction between subgroup and treatment was calculated using a logistic regression with visit measurement at week 16 as response variable, treatment, subgroup, strata, and treatment-by-subgroup interaction as factors; ITT_M = intention-to-treat population for the main study; NRI-C = nonresponder imputation to handle data missing due to COVID-19; TCI = topical calcineurin inhibitor; TCS = topical corticosteroid; QD = every day; UPA = upadacitinib; vIGA-AD = validated Investigator Global Assessment for Atopic Dermatitis.
Source: Case report form: Measure Up 2 study.11
Figure 41: Proportion of Patients Achieving vIGA-AD of 0 or 1 at Week 16 by Subgroup in the Upadacitinib 30 mg and 15 mg Group (NRI-C, ITT Population) of the Measure Up 2 Study
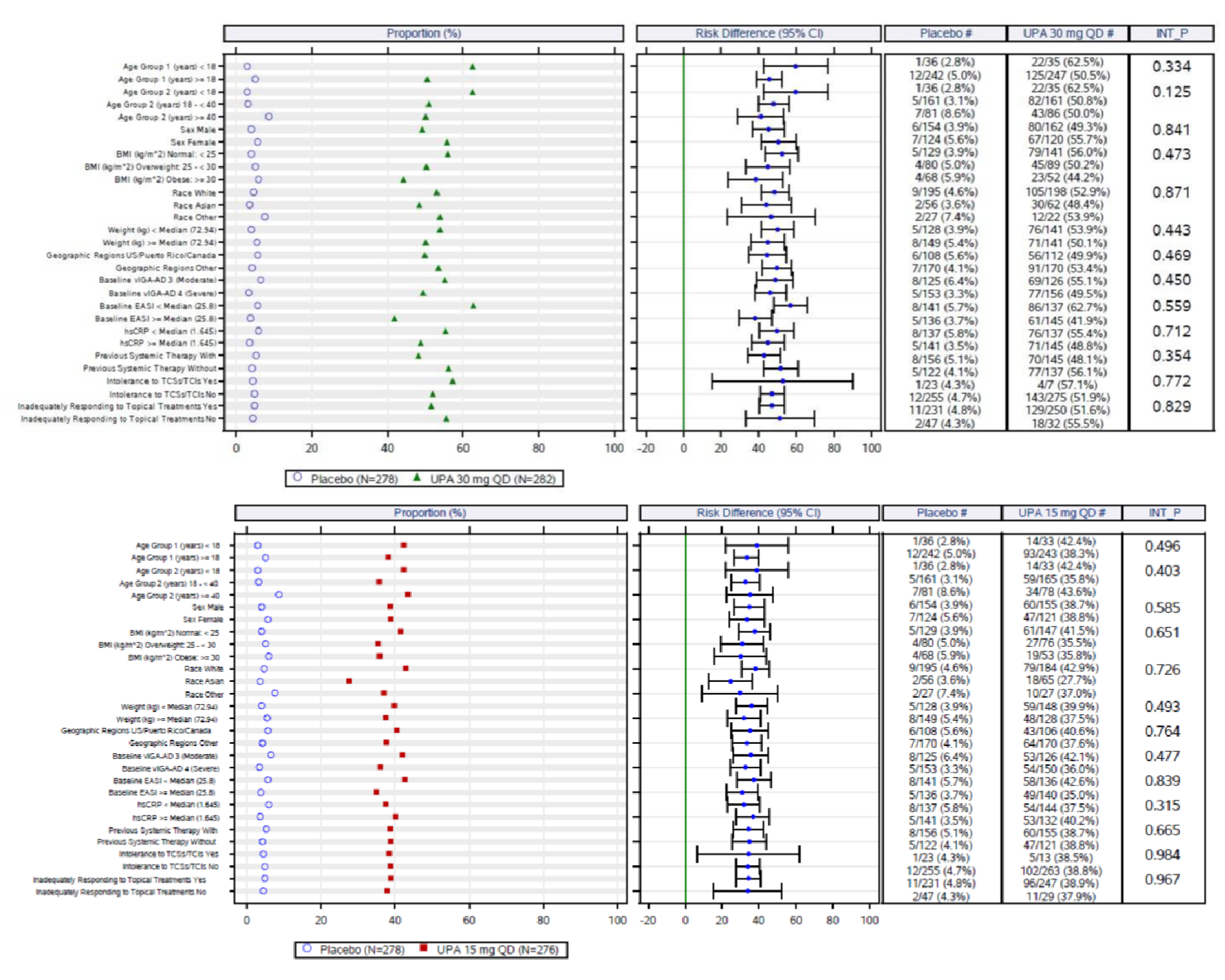
BMI = body mass index; CI = confidence interval for adjusted difference, calculated according to the Cochran-Mantel-Haenszel test adjusted for strata; EASI = Eczema Area and Severity Index; hsCRP = high-sensitivity C-reactive protein; INT_P = P value for interaction between subgroup and treatment was calculated using a logistic regression with visit measurement at week 16 as response variable, treatment, subgroup, strata, and treatment-by-subgroup interaction as factors; ITT_M = intention-to-treat population for the main study; NRI-C = nonresponder imputation to handle data missing due to COVID-19; TCI = topical calcineurin inhibitor; TCS = topical corticosteroid; QD = every day; UPA = upadacitinib; vIGA-AD = validated Investigator Global Assessment for Atopic Dermatitis.
Source: Case report form: Measure Up 2 study.11
Figure 42: Proportion of Patients Achieving EASI 75 at Week 16 by Subgroup in the Upadacitinib 30 mg and 15 mg Group (NRI-C, ITT Population) of the AD Up Study
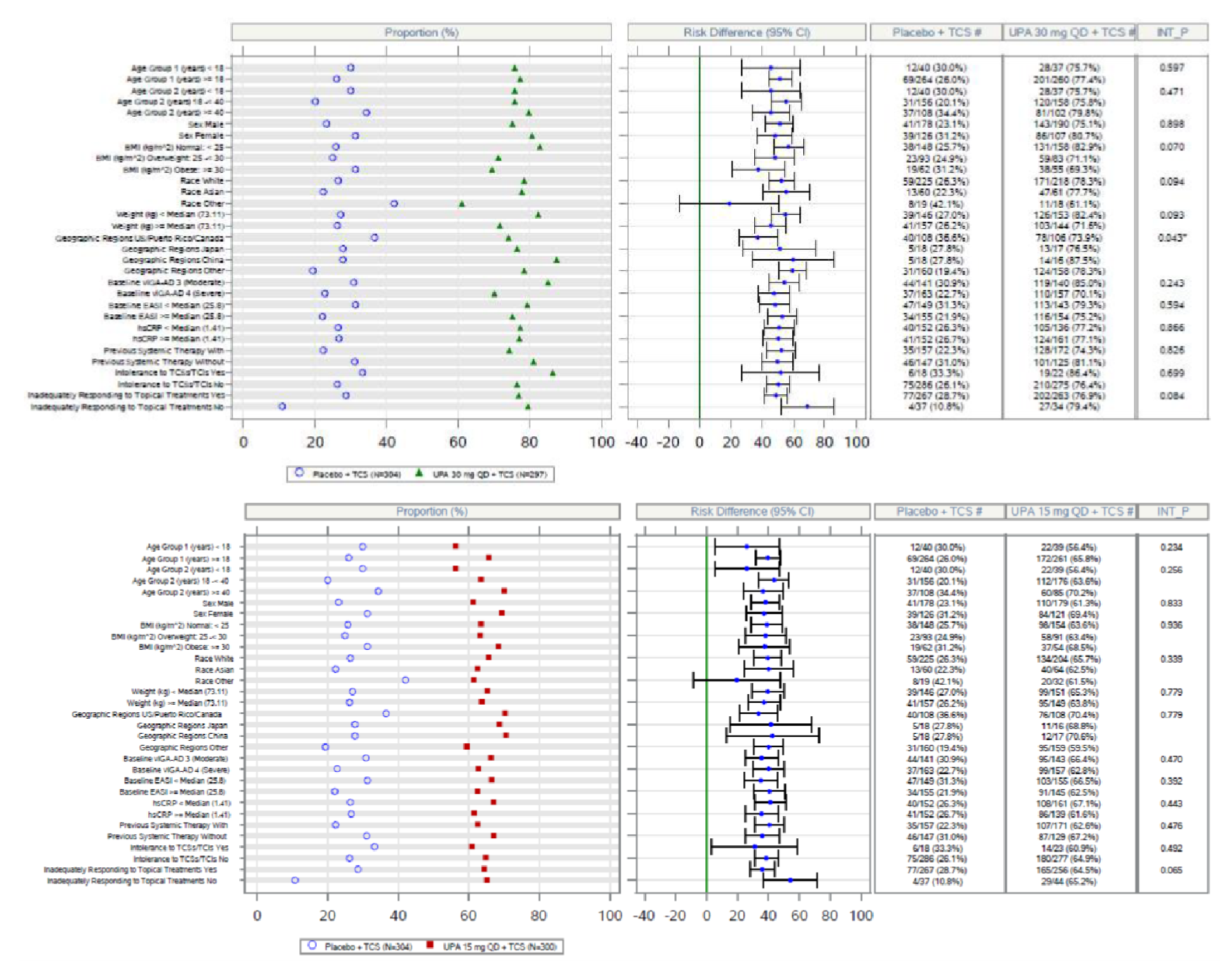
BMI = body mass index; CI = confidence interval for adjusted difference; EASI = Eczema Area and Severity Index; hsCRP = high-sensitivity C-reactive protein; ITT_M = intention-to-treat population for the main study; NRI-C = nonresponder imputation to handle data missing due to COVID-19; QD = every day; TCI = topical calcineurin inhibitor; TCS = topical corticosteroid; UPA = upadacitinib; vIGA-AD = validated Investigator Global Assessment for Atopic Dermatitis.
Source: Case report form: AD Up study.13
Figure 43: Proportion of Patients Achieving vIGA-AD of 0 or 1 With at Least 2 Grades of Reduction From Baseline at Week 16 by Subgroup in the Upadacitinib 30 mg and 15 mg Group (NRI-C, ITT Population) of the AD Up Study
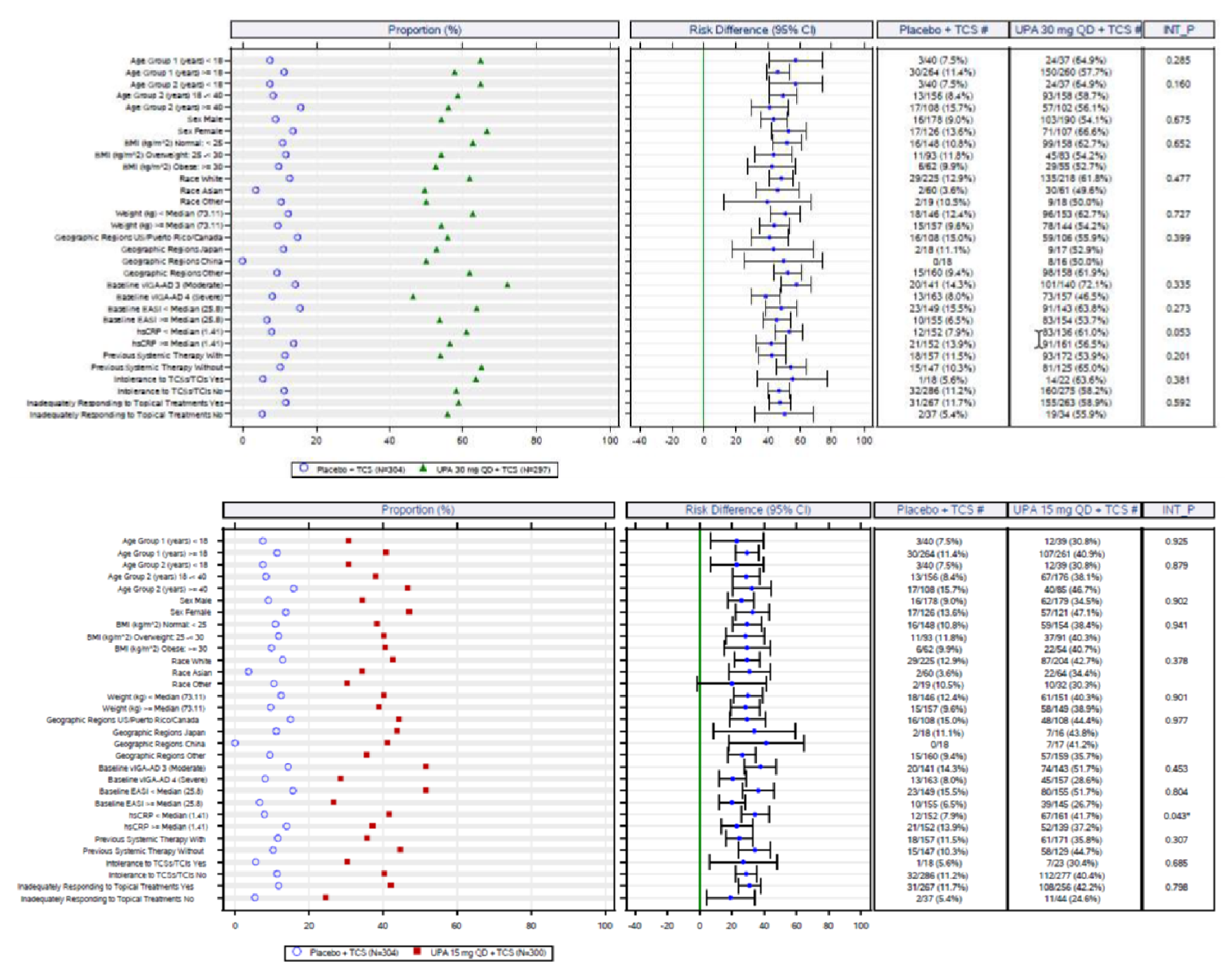
BMI = body mass index; CI = confidence interval for adjusted difference; EASI = Eczema Area and Severity Index; hsCRP = high-sensitivity C-reactive protein; ITT_M = intention-to-treat population for the main study; NRI-C = nonresponder imputation to handle data missing due to COVID-19; QD = every day; TCI = topical calcineurin inhibitor; TCS = topical corticosteroid; UPA = upadacitinib; vIGA-AD = validated Investigator Global Assessment for Atopic Dermatitis.
Source: Case report form: AD Up study.13
Figure 44: Proportion of Patients Achieving EASI 75 at Week 16 by Subgroup (NRI-C, ITT Population) in the Heads Up Study
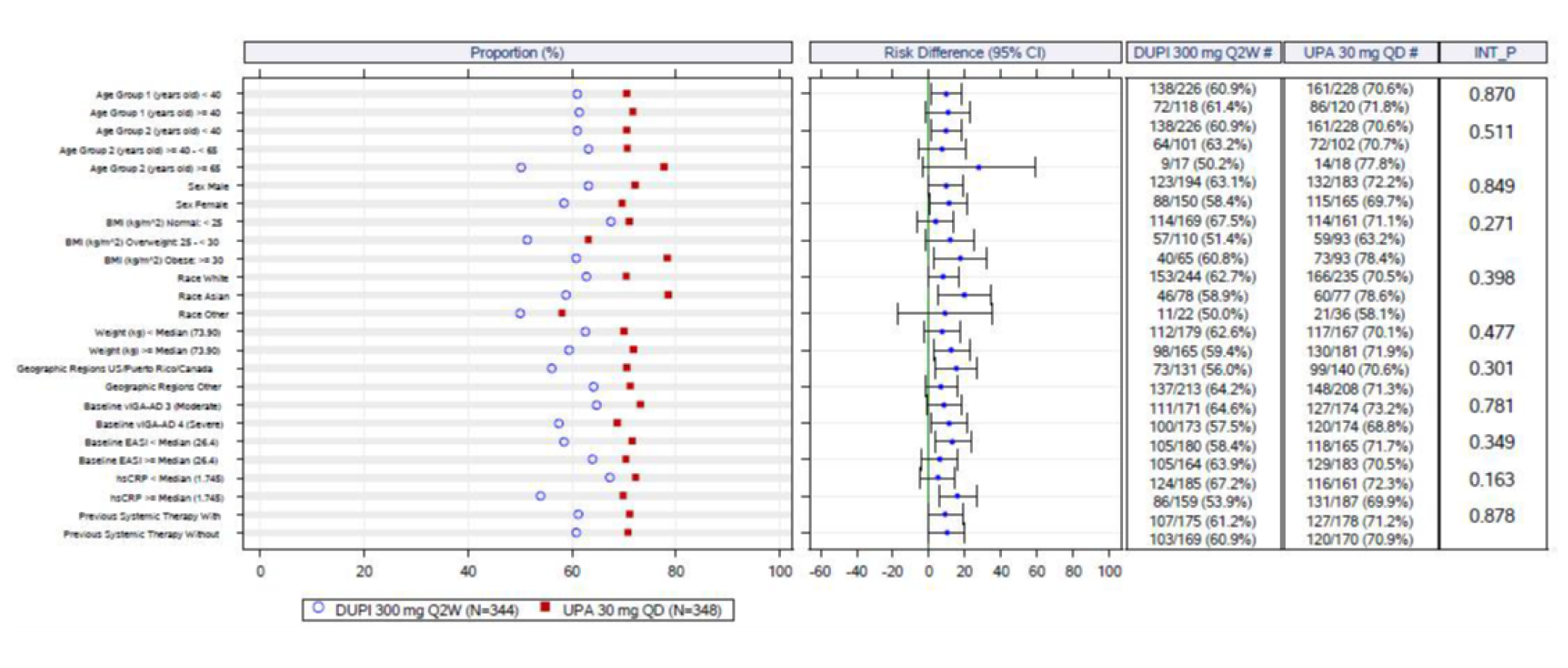
CI = confidence interval for adjusted difference; DUPI = dupilumab; EASI = Eczema Area and Severity Index; ITT = intention to treat; NRI-C = nonresponder imputation to handle data missing due to COVID-19; QD = every day; UPA = upadacitinib; vIGA-AD = validated Investigator Global Assessment for Atopic Dermatitis.
Source: Case report form from Heads Up.14
Appendix 4: Description and Appraisal of Outcome Measures
Note that this appendix has not been copy-edited.
Aim
To describe the following outcome measures and review their measurement properties (validity, reliability, responsiveness to change, and minimal important difference):
To summarize the validity of the following end point measures:
EASI
vIGA
SCORAD
WP-NRS
DLQI
EQ-5D
POEM
HADS
ADerm-IS
ADerm-SS
Findings
A focused literature search was conducted to identify the psychometric properties and MID of each of the stated outcome measures.
The findings about validity, reliability, responsiveness, and MID of each outcome measure are summarized in Table 65.
Interpretation of the reliability and validity metrics were based on the following criteria:
Inter-rater reliability, kappa statistics (level of agreement):35
< 0 = poor agreement
0.00 to 0.21 = slight agreement
0.21 to 0.40 = fair agreement
0.41 to 0.60 = moderate agreement
0.61 to 0.8 = substantial
0.81 to 1.00 = almost perfect agreement
Internal consistency (Cronbach alpha) and test–retest reliability: ≥ 0.7 is considered acceptable.
Validity, i.e., between-scale comparison (correlation coefficient, r):
≤ 0.3 = weak
0.3 to ≤ 0.5 = moderate
> 0.5 = strong
Table 65: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
EASI | A scale used in clinical trials to assess the severity and extent of AD. | Adequate construct and content validity, estimated between EASI and SCORAD, reports of moderate to high correlation (r = 0.84 to 0.93) between these 2 tools. 27,28,3032, Internal consistency of EASI is also adequate, with Spearman and Cronbach alpha values of 0.86 and 0.94 respectively.27 Intra- and Inter-rater reliability has kappa values of test–retest reliability of 0.76.27 Responsiveness (sensitivity to change) was judged as adequate.32 | 6.6 points |
vIGA | A scale that provides a global clinical assessment of AD by investigator. IGA is a 5-point scale that provides a global clinical assessment of AD severity (ranging from 0 to 4, where “0” indicates clear and “4” indicates severe AD) | Good content validity with strong inter-rater reliability (intra-class correlation coefficient [ICC] = 0.817) and excellent agreement (kappa = 0.857). Test–retest reliability and responsiveness are unknown. | Unknown. Pivotal studies included in this CADTH submission used a value of 0 or 1 in the vIGA to classify a response |
SCORAD | A tool used in clinical research to standardize the evaluation of the extent and severity of AD. The maximum possible total score of SCORAD is 103, in which, the higher score indicates poorer or a more severe condition. | A difference of 8.7 points in SCORAD was estimated as the MID for the patients with atopic eczema (also known as AD). Two systematic reviews found excellent agreement with global assessments of disease severity.27,39 Content validity was deemed adequate, good construct validity (Spearman R’s ranging from 0.53 to 0.92) and internal consistency. Sensitivity to change and inter-observer reliability were also adequate; the latter with several measurements of ICC from 0.84 to 0.99. Intra-observer reliability (test–retest), however, was unclear.27 | 8.7 points using IGA as anchor27 |
Pruritus NRS | A tool for patient with AD used to report the intensity of their itch. Patients rate average and maximum intensity of itch in past 24 hours based on a scale of 0 to 10 (0 = ‘no itch’ and 10 = ‘worst itch imaginable.’ | Information provided by the sponsor reported the validity and reliability of the WP-NRS based on 3 phase III and one phase IIb RCTs. The most appropriate definition of a responder on the pruritus NRS was considered in the range of 3 to 4 points. | 3 points |
DLQI | A questionnaire used to assess 6 different aspects that may affect quality of life of patients in dermatology. It is a 10-item questionnaire that assesses 6 different aspects that may affect quality of life. The overall DLQI is calculated by summing the score of each question resulting in a numeric score between 0 and 30 (or a percentage of 30). The higher the score, the more quality of life is impaired. | The DLQI has shown good test–retest reliability, internal consistency reliability, construct validity and responsiveness in patients with psoriasis. In patients with AD, internal consistency could not be determined. Reliability was moderate (0.77). Other validity measures and MID information were not found. | 2.2 to 6.9 (psoriasis) Unknown for AD |
EQ-5D | A generic quality of life instrument that has been applied to a wide range of health conditions and treatments. | EQ-5D includes 3 parts: a descriptive system that classifies respondents (aged ≥ 12 years) into one of 243 distinct health states. The second part is a 20 cm EQ-VAS that has end points labelled 0 and 100. The third part is the EQ-5D index score which is generated by applying a multi-attribute utility function to the descriptive system. The MID for the EQ-5D ranges from 0.033 to 0.074. No information is found from literature search for EQ-5D in AD. | 0.033 to 0.074, Unknown for AD |
POEM | A 7-item questionnaire used in clinical trials to assess disease symptoms in children and adults with eczema. Seven items (dryness, itching, flaking, cracking, sleep loss, bleeding, and weeping) are assessed using a 5-point scale. The possible scores for each question were: “0” indicates for no days, “1” for 1 to 2 days, “2” for 3 to 4 days, “3” for 5 to 6 days, and “4” indicates for every day. The maximum total score is 28; a high score is indicative of severity (0 to 2 indicates for clear or almost clear; 3 to 7 for mild eczema; 8 to 16 for moderate eczema; 17 to 24 for severe eczema; 25 to 28 for very severe eczema). | Moderate concurrent validity (Spearman = 0.56). Good convergent validity when compared to DLQI, but moderate to weak when compared to EASI and NRS. Poor discriminant validity in predicting self-reported global severity. Moderate responsiveness. Good reliability (ICC = 0.90). | MID of 5 points change from baseline using global severity as anchor. |
HADS | A patient-reported questionnaire designed to identify anxiety disorders and depression in patients at non-psychiatric medical institutions. | The HADS questionnaire contains 14 items that assess symptoms experienced in the previous week, A person can score between 0 and 21 for each subscale (anxiety and depression). A high score was indicative of a poor state. No additional validity and MID information regarding HADS was found from the literature search for AD. | Unknown |
ADerm-IS | A patient questionnaire designed to identify signs, symptoms, and impacts of moderate to severe AD in adults. | The ADerm-IS questionnaire has response categories that are assessed over a 11-point Likert scale (no impact) to 10 (extreme impact). No additional validity and MID information regarding ADerm-IS was found from the literature search for AD. | Unknown |
ADerm-SS | A patient questionnaire designed to identify signs, symptoms, and impacts of moderate to severe AD in adults. | The ADerm-SS questionnaire has response categories that are assessed over a 11-point Likert scale (no impact) to 10 (extreme impact). No additional validity and MID information regarding ADerm-SS was found from the literature search for AD. | Unknown |
AD = atopic dermatitis; DLQI = Dermatology Life Quality Index; EASI = Eczema Area and Severity Index; HADS = Hospital Anxiety and Depression Scale; IGA = Investigator Global Assessment; MID = minimal important difference; NRS = Worst Pruritus Numerical Rating Scale; POEM = Patient-Oriented Eczema Measure; SCORAD = Scoring Atopic Dermatitis.
Eczema Area and Severity Index
EASI is a scale used in clinical trials to assess the severity and extent of AD.27 In EASI, 4 disease characteristics of AD (erythema, infiltration/papulation, excoriations, and lichenification) are assessed for severity by the investigator on a scale of “0” (absent) to “3” (severe). The scores are added up for each of the 4 body regions (head, arms, trunk, and legs). The assigned percentages of BSA for each section of the body are 10% for head, 20% for arms, 30% for trunk, and 40% for legs respectively. Each subtotal score is multiplied by the BSA represented by that region. In addition, the affected area of AD assessed as a percentage by each body region is converted to a score of 0 to 6, where the area is expressed as 0 (none), 1 (1% to 9%), 2 (10% to 29%), 3 (30% to 49%), 4 (50% to 69%), 5 (70% to 89%), or 6 (90% to 100%). Each of the body area scores are multiplied by the area affected. Therefore, the total EASI score ranges from 0 to 72 points, with the highest score indicating worse severity of AD.28 It is suggested that the severity of AD based on EASI are categorized as follows: 0 = clear; 0.1 to 1.0 = almost clear; 1.1 to 7.0 = mild; 7.1 to 21.0 = moderate; 2l.1 to 50.0 = severe; 50.1 to 72.0 = very severe.29 The EASI 50, EASI 75, EASI 90, and EASI 100 end points indicate improvements of ≥ 50%, ≥ 75%, ≥ 90%, and 100% improvement from baseline, respectively. The validity and reliability of the EASI was examined in several studies demonstrating good performance in all these domains.28,30-32 Correlation coefficients for assessing content and construct validity were estimated between EASI and SCORAD27 with reports of moderate to high correlation (r = 0.84 to 0.93) between these 2 tools. Internal consistency of EASI is adequate, with Spearman and Cronbach alpha values of 0.86 and 0.94 respectively.27 Intra- and Inter-rater reliability has also been examined with adequate values of test–retest reliability and kappa values of 0.76.27 Responsiveness (sensitivity to change) was also judged as adequate by the systematic review authors. The overall MID is 6.6, based on results from 1 study.32
Validated Investigator Global Assessment
IGA is a 5-point scale that provides a global clinical assessment of AD severity ranging from 0 to 4, where “0” indicates clear, and “4” indicates severe AD.33 A decrease in score relates to an improvement in signs and symptoms.
The IGA has been widely used in many AD clinical trials and required by regulatory agencies for drug approval trials.33 However, the instrument has had many issues with variable content validity, definitions, and implementations — the tool has had more than 20 different names and various numbers of scale categories (from 4- to 7-point scales), as well as content of the scales.33,34 A 2016 systematic review of the literature found no information on the validity and reliability of the IGA instrument in patients with AD as well as no information on what would constitute a MID in patients with AD.33
The studies included in this CADTH submission use the newly validated version of the IGA — based on a recent study to validate the IGA published by Simpson et al. (2020)35 — known as the vIGA-AD scale with the objective to harmonize outcome assessments in clinical trials. In this study, a 5-point IGA scale (0 to 4) was selected and content validity was achieved, with strong inter-rater reliability ([CC = 0.817) and excellent agreement (kappa = 0.857). Test–retest reliability and responsiveness are unknown.
No MID is presented. However, the pivotal studies included in this CADTH submission used a value of 0 or 1 in the vIGA-AD to classify a response
Worst Pruritus Numerical Rating Scale
The WP-NRS is used to report the intensity of their itch during a daily recall period. Patients rate their overall (average) and maximum intensity of itch experienced during the past 24 hours based on a scale of 0 to 10 (0 = ‘no itch’ and 10 = ‘worst itch imaginable.” The reliability of the WP-NRS is adequate36 with pooled ICCs in the range of 0.95 to 0.97.36 NRS scores are stable over a period of time. The ICC values indicated that the WP-NRS scores were stable over a period of time when the patients’ disease was stable. The validity of the WP-NRS, evaluated using correlational analyses and 3 known-groups ANOVA models was statistically significant, and the effect sizes for the differences between the extreme categories for each known group were all above the Cohen threshold of 0.80 for large effect sizes.36 Based on the data from the phase IIb study, using EASI, IGA as anchors, NRS responder reportedly ranged between 2.2 and 4.2, with the highest estimates based on the most stringent clinical criteria (i.e., EASI 90 to 100 and IGA 0/1). Using the Pruritus Categorical Scale as an anchor, the responder was estimated at 2.6 points. These analyses suggested that the most appropriate definition of a responder on the pruritus NRS is in the range of 3 to 4 points.36 The investigators from the included studies evaluated in this CADTH submission evaluate the Worst Pruritus NRS as the proportion of patients achieving an improvement (reduction) in NRS ≥ 4 from baseline for those patients with NRS ≥ 4 at baseline at week 16.
Scoring Atopic Dermatitis
The SCORAD was developed to standardize the evaluation of the extent and severity of AD.37 SCORAD is considered a valid and reliable tool for the objective assessment of eczema clinical signs.38 The instrument assesses 3 components of AD: the extent of affected BSA (0 to 100), severity (0 to 18), and symptoms (0 to 20). The extent of AD is assessed as a percentage of each defined body area and reported as the sum of all areas. The score ranges from 0 to 100. The severity of 6 specific signs of AD (redness, swelling, oozing/crusting, excoriation, skin thickening/lichenification, dryness) is assessed using a 4-point scale (i.e., none = 0, mild = 1, moderate = 2 and or severe = 3) with a minimum score of 0 and a maximum of 18. The subjective symptoms (itch and sleeplessness) are recorded by the patient or relative on a visual analogue scale, with scores ranging from 0 (no symptoms) to 10 (worst imaginable symptom) with a maximum possible score of 20. The total SCORAD is calculated based on the 3 components with a maximum possible total score of 103, in which, the higher score indicates poorer or a more severe condition.
SCORAD has been found to be valid and reliable, with excellent agreement with global assessments of disease severity.27,39 Content validity has been deemed adequate, with good construct validity (Spearman R’s ranging from 0.53 to 0.92) and internal consistency. Sensitivity to change and inter-observer reliability are also adequate; the latter with several measurements of ICC from 0.84 to 0.99. Intra-observer reliability (test–retest), however, was unclear.27 The MID has been estimated using mean change scores of SCORAD of patients that showed a relevant improvement based on IGA, defined as an ‘improvement’ or ‘decline’ of ≥ 1 point in PGA and IGA; thus, a difference of 8.7 points in SCORAD was estimated as the MID for the patients with AD.32
This tool was used in the Measure Up and AD Up studies as secondary end points as the percent change in SCORAD from baseline at week 16.
Patient-Oriented Eczema Measure
This is a 7-item questionnaire used in clinical trials to assess disease symptoms in children and adults. Based on frequency of occurrence during the past week, the 7 items (dryness, itching, flaking, cracking, sleep loss, bleeding, and weeping) are assessed using a 5-point scale. The possible scores for each question were: “0” indicates for no days, “1” for 1 to 2 days, “2” for 3 to 4 days, “3” for 5 to 6 days, and “4” indicates for every day. The maximum total score is 28; a high score is indicative of poor quality of life (0 to 2 indicates for clear or almost clear; 3 to 7 for mild eczema; 8 to 16 for moderate eczema; 17 to 24 for severe eczema; 25 to 28 for very severe eczema).40
In 1 study,32 it was reported that the overall mean MID of the POEM was 3.4 points (SD = 4.8), using IGA as anchor. In 2018, the minimally important change (MIC) of POEM in children (N = 300) with moderate to severe atopic eczema was calculated in 1 study.72 Based on distribution-based methods, the estimated MIC were 1.07 (using 0.2 SD of baseline POEM scores) and 2.68 (using 0.5 SD of baseline POEM scores); The estimated MIC were 3.09 to 6.13 and 3.23 to 5.38 based on patient-/parent-reported anchor-based methods and investigator-reported anchor-based methods respectively. The authors recommended the following thresholds be used to interpret changes in POEM scores in children: a score of 3 to 3.9 indicates a probably clinically important change; ≥ 4, indicates a very likely clinically important change.72
The tool has been tested in its validity, reliability, and responsiveness. When compared to the PO-SCORAD and DLQI41 a moderate concurrent validity (Spearman = 0.56) was detected in adults. Good convergent validity when compared to DLQI, but moderate to weak when compared to EASI and NRS. Poor discriminant validity in predicting self-reported global severity. In other studies including children, content validity was poor to moderate as a measurement of clinical signs of AD.27,40 The same studies have revealed moderate responsiveness and good reliability (ICC = 0.90).41 The MID has been stated as 3.4 points in adults and from 3.0 to 3.9 in children. Other studies have established −5.0 for adults using global severity of AD as anchor.41
This tool was used in the Measure Up and AD Up studies as secondary end points as the proportion of patients achieving an improvement (reduction) ≥ 4 from baseline at week 16.
Atopic Dermatitis Impact Scale
The ADerm-IS is an AD-specific patient-reported questionnaire to assess the signs, symptoms, and impacts of moderate to severe AD in adults. The ADerm-IS was developed as an electronic diary which includes 3 items to be completed daily, assessing impact over the previous 24 hours, and 8 items completed weekly to assess impacts over the past 7 days. Response categories are assessed over a 11-point Likert scale from 0 (no impact) to 10 (extreme impact) for each item.24,42 Thirteen sign and symptom concepts are included: bleeding, blisters, burning, dry skin, fissures, inflammation, itching, pain, rash, redness, scaling, skin thickening, and swelling. Additionally, 43 impact concepts were identified and organized into 8 domains: ADLs, cognitive, emotional, financial, physical, sleep, social, and work/school. The most frequently reported impacts were sleep disturbances, followed by work/school activities, social withdrawal, anxiety, depressed feelings, embarrassment, and the inability to participate in ADLs.43 The ADerm-IS uses 10 items (0 to 10 NRS) to calculate 3 domains scores: ADerm-IS sleep sums 3 daily items assessing sleep impact (24-hour recall; range 0 to 30); ADerm-IS Daily Activities sums 4 items measuring limitations of household, physical, and social activities, and difficulty concentrating (7-day recall, range 0 to 40); ADerm-IS Emotional State sums 3 items measuring self-consciousness, embarrassment, and sadness (7-day recall, range 0 to 30.73 The pivotal studies report the impact score from 3 domains: sleep, emotional state, daily activities.44 One study supports the content validity of the ADerm-IS,43 but no MID or additional validity, reliability and responsiveness information regarding the ADerm-IS was identified from the literature search for AD in this CADTH review.
This tool was used in the Measure Up and AD Up studies for this CADTH review as secondary end point as the proportion of patients achieving an improvement (reduction) in ADerm-IS sleep domain score ≥ 12 (MCID) from baseline at week 16 for patients with ADerm-IS sleep domain score ≥ 12 at baseline.
Atopic Dermatitis Symptom Scale
The ADerm-SS is also an AD-specific patient-reported questionnaire to assess the signs, symptoms, and impacts of moderate to severe AD in adults. The ADerm-SS was developed as an electronic diary which includes 3 items to be completed daily, assessing impact over the previous 24 hours, and 8 items completed weekly to assess impacts over the past 7 days. Response categories are assessed over a 11-point Likert scale (no impact) to 10 (extreme impact).24,42 Thirteen sign and symptom concepts are included: bleeding, blisters, burning, dry skin, fissures, inflammation, itching, pain, rash, redness, scaling, skin thickening, and swelling. Additionally, 43 impact concepts were identified and organized into 8 domains: ADLs, cognitive, emotional, financial, physical, sleep, social, and work/school. The most frequently reported impacts were sleep disturbances, followed by work/school activities, social withdrawal, anxiety, depressed feelings, embarrassment, and the inability to participate in ADLs.43 The pivotal studies report the symptom score from 2 domains: skin pain and 7-item total symptom score.44 One study supports the content validity of the ADerm-SS,43 no MID or additional validity, reliability and responsiveness information regarding the ADerm-SS was identified from the literature search for AD.
This tool was used in the Measure Up and AD Up studies for this CADTH review as secondary end points as the proportion of patients achieving an improvement (reduction) in ADerm-SS skin pain score ≥ 4 (MCID) from baseline at week 16 for patients with ADerm-SS skin pain score ≥ 4 at baseline.
A variant of this measurement is the ADerm-SS TSS-7, defined as the algebraic sum of the responses to items 1 to 7 of the ADerm-SS, and was also used in the Measure Up and AD Up studies in this CADTH review as the proportion of patients achieving an improvement (reduction) in ADerm-SS TSS-7 ≥ 28 (MCID) from baseline at week 16 for patients with ADerm-SS TSS-7 ≥ 28 at baseline.
Dermatology Life Quality Index
The DLQI is a widely used dermatology-specific HRQoL instrument. It is a 10-item questionnaire that assesses 6 different aspects that may affect quality of life.45,46 These aspects are symptoms and feelings, daily activities, leisure, work and school performance, personal relationships, and treatment. The maximum score per aspect is either 3 (with single questions) or 6 (with 2 questions) and the scores for each can be expressed as a percentage of either 3 or 6. Each of the 10 questions is scored from 0 (not at all) to 3 (very much) and the overall DLQI is calculated by summing the score of each question resulting in a numeric score between 0 and 30 (or a percentage of 30).45,46 The higher the score, the more quality of life is impaired. The meaning of the DLQI scores on a patient’s life is as follows47:
0 to 1 = no effect.
2 to 5 = small effect.
6 to 10 = moderate effect.
11 to 20 = very large effect.
21 to 30 = extremely large effect.
The validity of the DLQI has been assessed in patients with eczema39,48-50 with good test–retest reliability (correlation between overall DLQI scores was 0.99, P < 0.0001 and of individual question scores was 0.95 to 0.98, P < 0.001),46 internal consistency (reliability) with Cronbach alpha coefficients ranging from 0.75 to 0.92,47 construct validity,47 and responsiveness.47-49
Estimates of the MID have ranged from 2.2 to 6.9.45,47 It should be noted that some of the anchors that were used to obtain the DLQI MID were not patient-based (i.e., Basra et al.47 derived estimates from PASI and physician global assessment anchors, as well as a distribution-based approach) and some limitations of the DLQI include concerns regarding uni-dimensionality and the behaviour of items of the DLQI in different psoriatic patient populations.47 No validity and MID information were found for the patients with AD.51
This tool was used in the Measure Up and AD Up studies for this CADTH review as secondary end point as the proportion of patients ≥ 16 years old who achieve a DLQI score of 0 or 1 at week 16.
Children’s Dermatology Life Quality Index
The CDLQI is based on the adult version (DLQI). This is a child-completed questionnaire to be applied to children from 3 to 16 years of age, designed to measure the impact of any skin disease on the quality of life with a recall period of 7 days. It is 1 of the most commonly used instruments for measuring HRQoL in children.42,52,53 The instrument has 10 questions asking about the impact of a skin disease on the life of the affected child, including symptoms, embarrassment, friendships, clothes, playing, sports, bullying, sleep, and impact of treatment. Each question is answered on a 4-point Likert scale scored from 0 to 3. These are added to 5 a minimum of 0 and maximum of 30. Higher CDLQI scores indicate greater degree of impairment in HRQoL.
A 2013 systematic review did not identified studies demonstrating content validity.53 In the same review, 3 studies demonstrated concurrent validity, 2 between the CDLQI and Cardiff Acne Disability Index and 1 between the CDLQI and Childhood Atopic Dermatitis Impact Scale. The CDLQI was correlated in 10 studies with SCORAD, the primarily sign-based severity scoring system for AD. Forty-five studies demonstrated convergent construct validity and 6 studies demonstrated divergent construct validity. The same review showed good internal consistency of the CDLQI (examined in 6 studies) with Cronbach alpha values ranging from 0.82 to 0.92. Similarly, test–retest reliability was adequate, with Spearman’s rank order correlation coefficient calculated in 4 studies (range 0.74 to 0.97). One study examined the ICC, finding 0.80. Good responsiveness to change was found in studies using Wilcoxon signed rank test and repeated ANOVA measures.
One study conducted in the US and Canada with 202 participants using a distribution- based approach, determined the MCID of the CDLQI in psoriasis to be 2.5.
This tool was used in the Measure Up and AD Up studies for this CADTH review as secondary end point as the proportion of patients < 16 years of age achieving a CDLQI score of 0 or 1 at week 16.
EQ-5D
The EQ-5D is a generic quality of life instrument that has been applied to a wide range of health conditions and treatments including AD.54,55 The first of 2 parts of the EQ-5D is a descriptive system that classifies respondents (aged ≥ 12 years) into 1 of 243 distinct health states. The descriptive system consists of the following 5 dimensions: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression. Each dimension has 3 possible levels (1, 2, or 3) representing ‘no problems,’ ‘some problems,’ and ‘extreme problems,’ respectively. Respondents are asked to choose 1 level that reflects their own health state for each of the 5 dimensions. A scoring function can be used to assign a value (EQ-5D index score) to self-reported health states from a set of population-based preference weights.54,55 The second part is a 20 cm EQ-VAS that has end points labelled 0 and 100, with respective anchors of ‘worst imaginable health state’ and ‘best imaginable health state,’ respectively. Respondents are asked to rate their own health by drawing a line from an anchor box to the point on the EQ-VAS which best represents their own health on that day. The third part is the EQ-5D index score, which is generated by applying a multi-attribute utility function to the descriptive system. Different utility functions are available that reflect the preferences of specific populations (e.g., US or UK). Hence, the EQ-5D produces 3 types of data for each respondent:
a profile indicating the extent of problems on each of the 5 dimensions represented by a 5-digit descriptor, such as 11121, 33211
a population preference-weighted health index score based on the descriptive system
a self-reported assessment of health status based on the EQ-VAS.
The lowest possible overall score (corresponding to severe problems on all 5 attributes) varies depending on the utility function that is applied to the descriptive system (e.g., −0.59 for the UK algorithm and −0.109 for the US algorithm). Scores less than 0 represent health states that are valued by society as being worse than dead, while scores of 0 and 1.00 are assigned to the health states “dead” and “perfect health,” respectively. The MID for the EQ-5D ranges from 0.033 to 0.074. No additional validity and MID information were found from literature search for EQ-5D in patients with AD.
This tool was used in the Measure Up and AD Up studies for this CADTH review as secondary end point reported as the change and percentage change from baseline in EQ-5D-5L values.
Hospital Anxiety and Depression Scale – Anxiety
The HADS is a widely used patient-reported questionnaire designed to identify anxiety disorders and depression in patients at non-psychiatric medical institutions. Repeated administration also provides information about changes in a patient’s emotional state.56-58 The HADS questionnaire contains 14 items that assess symptoms experienced in the previous week, among which, 7 items are related to anxiety and 7 items are related to depression. Patients provided responses to each item based on a 4-point Likert scale. Each item is scored from 0 (the best) to 3 (the worst); thus, a person can score between 0 and 21 for each subscale (anxiety and depression). A high score was indicative of a poor state. Scores of 11 or more on either subscale were considered to be a 'definite case' of psychological morbidity, while scores of 8 to 10 represented 'probable case’ and 0 to 7 'not a case'.58 One study59 indicated that HADS have good construct validity, with no overall floor or ceiling effects. HADS may be useful for the assessment of patients with AD in clinical trials and practice. The author concluded that additional research is needed to confirm the construct validity and to assess content validity and feasibility in research and clinical practice.59 No additional validity and MID information regarding HADS was found from the literature search for patients with AD. This tool was used in the Measure Up and AD Up studies for this CADTH review as secondary end point reported as the change and percent change from baseline in HADS-A.
Work Productivity and Activity Index
The WPAI is an instrument used to measure loss of productivity at work and impairment in daily activities over the past 7 days.60 The questionnaire includes 4 items: absenteeism, presenteeism, overall work impairment, and activity impairment, that range from 0% to 100%, with higher values indicating greater impairment. While absenteeism represents the percentage of work time missed due to AD, presenteeism represents the percentage of impairment while at work due to AD. Overall work impairment represents the total percentage of work time missed due to either absenteeism or presenteeism (since those are mutually exclusive). Activity impairment represents the percentage of impairment during daily activities other than work. The 4 items are all evaluated using an 11-point Likert-type scale from 0 (no effect) to 10 (completely prevented), and the scores are multiplied by 10 to arrive at a percentage. The WPAI has been validated to quantify work impairments for numerous diseases such as asthma, psoriasis, irritable bowel syndrome, ankylosing spondylitis, and Crohn disease, with established construct validity.61 It overall has good reproducibility with correlation coefficients ranging from 0.71 to 0.87.
The WPAI for AD tool was used in the Measure Up and AD Up studies for this CADTH review as secondary end point reported as the change and percent change from baseline in WPAI:AD domain scores (absenteeism, presenteeism, activity impairment, overall work productivity).
Pharmacoeconomic Review
Abbreviations
AD
atopic dermatitis
BIA
budget impact analysis
BSC
best supportive care
DUP
dupilumab
EASI
Eczema Area and Severity Index
EASI 75
at least 75% improvement in Eczema Area and Severity Index total score from baseline
HRQoL
health-related quality of life
ICER
incremental cost-effectiveness ratio
IMM
immunosuppressant
NMA
network meta-analysis
QALY
quality-adjusted life-year
TCS
topical corticosteroid
UPA
upadacitinib
WTP
willingness to pay
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Upadacitinib (Rinvoq), 15 mg and 30 mg extended-release oral tablets |
Submitted price | Upadacitinib, 15 mg: $48.68 per tablet Upadacitinib, 30 mg: $74.00 per tablet |
Indication | For the treatment of adults and adolescents 12 years of age and older with refractory, moderate to severe AD who are not adequately controlled with a systemic treatment (e.g., steroid or biologic) or when use of those therapies is inadvisable. |
Health Canada approval status | NOC |
Health Canada review pathway | Standard review |
NOC date | October 6, 2021 |
Reimbursement request | For the treatment of patients aged 12 years and older with moderate to severe AD whose disease is not adequately controlled with topical prescription therapies and/or who are refractory to or ineligible for systemic IMM therapies (i.e., due to contraindications, intolerance, or need for long-term treatment) |
Sponsor | AbbVie |
Submission history | Previously reviewed: Yes Indication: Arthritis, rheumatoid Recommendation date: February 4, 2020 Recommendation: Recommended with conditions |
AD = atopic dermatitis; IMM = immunosuppressant; NOC = Notice of Compliance.
Table 2: Summary of Economic Evaluation
Component | Description |
|---|---|
Type of economic evaluation | Cost-utility analysis Decision tree and Markov model hybrid |
Target population | Adolescents and adults (aged 12 years or over) with AD who are eligible for conventional systemic therapies |
Treatments | UPA (15 mg and 30 mg) |
Comparators | BSC (composed of a basket of emollients, low- to mid-potency TCS, rescue therapy) Dupilumab (DUP) |
Perspective | Canadian publicly funded health care payer |
Outcome | QALYs |
Time horizon | 10 years |
Key data source | The impact of treatment on clinical response at 16 weeks was informed by network meta-analyses for UPA as monotherapy (Measure Up 1, Measure Up 2) and in combination with TCS (AD Up) |
Submitted results | Sequential base case:a patients eligible for systemic therapy
|
Key limitations |
|
CADTH reanalysis results |
|
AD = atopic dermatitis; BSC = best supportive care; DUP = dupilumab; EQ-5D-3L = EQ-5D Three-Level; EQ-5D-5L = EQ-5D Five-Level; ICER = incremental cost-effectiveness ratio; IMM = immunosuppressant; QALY = quality-adjusted life-year; HRQoL = health-related quality of life; WTP = willingness to pay; TCS = topical corticosteroid; UPA = upadacitinib; vs. = versus.
aThe sponsor’s base case assumed that UPA and DUP would be used as monotherapy (i.e., without concomitant use of TCSs).
Conclusions
Compared to best supportive care (BSC), upadacitinib (UPA), whether as monotherapy or in combination with topical corticosteroids (TCSs), reduces the symptoms (i.e., improves patients’ scores on the Eczema Area and Severity Index [EASI]) of atopic dermatitis (AD) among patients with moderate to severe AD. However, the sponsor’s pharmacoeconomic submission does not reflect the indicated population or the intended dosing strategy for UPA (i.e., a starting dose of 15 mg and increasing to 30 mg for those with an inadequate response, or a starting dose of 30 mg for some patients with severe AD). Because some clinical data were lacking, CADTH could not assess the cost-effectiveness of UPA. As such, the cost-effectiveness of UPA is unknown.
CADTH undertook an exploratory reanalysis to address limitations in the sponsor’s submission, including assuming that UPA would be used by patients with prior systemic immunosuppressant (IMM) exposure; correcting the price of dupilumab (DUP); assuming that UPA would be used in combination with a TCS; adopting a lifetime time horizon; assuming that treatment effectiveness may wane over the entire analysis horizon; and adopting alternative assumptions about the durability of BSC treatment response. The results of this exploratory analysis are subject to high levels of uncertainty as well. The comparative effects of UPA relative to DUP and other treatments for AD (i.e., some IMMs, retinoids, phototherapy) are unknown, as is the impact of UPA on health-related quality of life (HRQoL) relative to BSC. CADTH was unable to address this limitation because of a lack of evidence. As noted in the CADTH clinical review, the comparative effectiveness of UPA versus DUP varies depending on the timing of outcome assessment; in addition, the use of 16-week outcome data in the pharmacoeconomic model means that all CADTH reanalyses likely overestimate the incremental effectiveness of UPA relative to DUP, producing a bias in the cost-effectiveness results favouring UPA. In the CADTH exploratory analysis, UPA 15 mg plus TCS was associated with an incremental cost-effectiveness ratio (ICER) of $48,616 per quality-adjusted life-year (QALY) gained compared with BSC, while UPA 30 mg plus TCS was associated with an ICER of $372,226 per QALY gained compared with UPA 15 mg plus TCS. In this population, a price reduction of 35% would be required for UPA 30 mg to be considered optimal at a willingness-to-pay (WTP) threshold of $50,000. CADTH additionally notes that this exploratory reanalysis considers UPA 15 mg and UPA 30 mg separately, which does not reflect the recommended dosing strategy or differential dosing for patients with severe AD. The ICER for UPA likely lies between the predicted ICERs for UPA 15 mg and UPA 30 mg. However, due to the limitations noted previously, these estimates are highly uncertain and biased in favour of UPA.
CADTH was unable to assess the cost-effectiveness of UPA for the intended dosing strategy because of a lack of clinical data, a lack of comparative clinical effectiveness data for some relevant treatment comparators, and a lack of data about the cost-effectiveness of UPA among adolescents and about the impact of adverse events. The inability to estimate the influence of these limitations means that the cost-effectiveness of UPA is unknown.
Stakeholder Input Relevant to the Economic Review
This section is a summary of the feedback received from the patient groups, registered clinicians, and drug plans that participated in the CADTH review process (specifically, information that pertains to the economic submission).
Input from patients with AD and caregivers of patients with AD was received from the Eczema Society of Canada, the Canadian Skin Patient Alliance, and Eczéma Québec. It was collected through online surveys, questionnaires, focus groups, and 1-on-1 interviews. Patients and caregivers described how living with AD affects their quality of life, mental health, ability to work, social lives, and daily routines. Symptoms that affect quality of life include itch, redness of the skin, repeated rashes, frequent scratching, cracked skin, dry and rough skin, disrupted sleep, bleeding, flaking of the skin, pain, thickening of the skin, oozing, swelling, lichenification, and blistering. Patients also noted limited accessibility to AD treatments and specialists. Patients described their experiences with current treatments including, but not limited to, frequent moisturizing, trigger avoidance, topical creams, IMMS (e.g., methotrexate and cyclosporine), oral corticosteroids (e.g., prednisone), and phototherapy. Patients who had experience with UPA reported adverse events that included weight gain, mild headaches, and sun sensitivity. Conjunctivitis was reported as an adverse event with DUP. Generally, patients were unwilling to accept serious side effects. Patients also reported frustration and financial strain from the trial-and-error nature of current treatments. They expressed a desire for better management of itch, flares, and rashes, improved quality of life and sleep, and less pain. Some patients noted that daily oral treatments would be preferred over injections; however, others noted that less frequent injections would be preferred over daily treatments.
Clinician input was received from a group composed of dermatologists, an allergist, and a family physician practising in various clinical settings across Atlantic Canada. The current pathway of care for patients with AD was described as treatment with emollients and the adoption of lifestyle measures (e.g., types of clothing, moisturizing, bathing, avoiding skin irritants, and minimizing stress), followed by the use of TCS (or topical nonsteroidal anti-inflammatory creams and ointments) followed by the use of systemic IMMs, with or without phototherapy. Clinicians noted that the goal of treatment is to improve symptoms of AD (such as chronic itch and dry and inflamed skin) as well as quality of life and patient satisfaction (such as better sleep, reduced work or school disruptions). Other priorities include achieving flare reduction and disease control, as reflected by Physician’s Global Assessment scores and the Dermatology Life Quality Index. Clinicians noted that UPA would replace off-label systemic therapies and phototherapy, and would be considered as second-line treatment after first-line treatment with lifestyle measures or TCSs for patients with moderate to severe AD.
Drug plan input received for this review before the Health Canada approval noted that patients would typically be prescribed the 30 mg dose of UPA, and that the dosage may be decreased to 15 mg depending on treatment response and the presence of serious side effects. The plans indicated that the initiation and renewal criteria for UPA should likely be aligned with those of DUP, although it was questioned whether a trial of DUP would be required before initiating UPA. However, the plans noted that DUP is not currently reimbursed in all jurisdictions. It was also noted that patients taking UPA are at increased risk of serious infections and that treatment interruptions may be required to manage the adverse events associated with UPA.
Several of these concerns were addressed in the sponsor’s model:
Treatment effectiveness is modelled in the sponsor’s submission in terms of the EASI score, which considers the extent of redness, thickness, scratching, and lichenification.
The cost-effectiveness of UPA compared to DUP was considered, although issues with the clinical data limit the conclusions that may be drawn.
Quality of life was incorporated in the sponsor’s model by use of EQ-5D data captured in the UPA trials. However, the EQ-5D is unlikely to capture all of the symptoms of AD that were noted by patients as affecting their quality of life.
Costs associated with some adverse events were included in the model; however, the impact of adverse events on quality of life may not be captured, and not all adverse events that are important to patients were included.
In addition, the CADTH exploratory reanalysis assumed that UPA would be used by patients who are refractory to or ineligible for systemic IMMs.
Because of a lack of data, CADTH was unable to assess stakeholders’ concerns about the cost-effectiveness of UPA relative to some relevant comparators (e.g., phototherapy and systemic IMMs).
Economic Review
The current review is for UPA (Rinvoq) for the treatment of adults and adolescents 12 years of age and older with refractory, moderate to severe AD who are not adequately controlled with a systemic treatment (e.g., a steroid or biologic) or for whom the use of these therapies is inadvisable.1 The sponsor’s reimbursement request is “For the treatment of patients aged 12 years and older with moderate-to-severe AD whose disease is not adequately controlled with topical prescription therapies and/or who are refractory to or ineligible for systemic IMMs [immunomodulatory] (i.e., due to contraindications, intolerance, or need for long-term treatment).”2
Economic Evaluation
Summary of Sponsor’s Economic Evaluation
Overview
UPA is indicated for use in adults and adolescents aged 12 years and older for the treatment of moderate to severe AD that is not adequately controlled with a systemic treatment or in patients for whom such treatments are inadvisable.1 The sponsor’s submitted a cost-utility analysis to assess the cost-effectiveness of UPA compared with DUP and BSC. In the base case, the sponsor assumed that UPA would be used by patients who are eligible for systemic therapy (e.g., after a trial of topical therapy). However, this assumption does not align with the indication (i.e., for a post-systemic steroid or biologic therapy) and is narrower than the reimbursement population (i.e., of patients on post-topical and/or post-systemic IMM therapy). The cost-effectiveness of UPA in patients whose AD is refractory to IMMs or who are ineligible for systemic IMMs was explored by the sponsor in scenario analyses. The modelled population in the sponsor’s base case is based on patients in the phase III UPA trials: Measure Up 1, Measure Up 2, and AD Up. As noted in the CADTH clinical review, approximately half of the patients in each trial had prior systemic IMM exposure.
UPA is available as 15 mg or 30 mg tablets. The recommended dosage for adults is 15 mg once daily; for those with an inadequate treatment response (e.g., at least 75% improvement in EASI total score from baseline [EASI 75]), dosage may be increased to 30 mg once daily.1 For adults with severe AD, a starting dose of 30 mg may be considered. UPA should be discontinued if an adequate response is not achieved with 30 mg once daily after 16 weeks of treatment. For adolescents (aged 12 years to 17 years) weighing at least 40 kg, the recommended dosage of UPA is 15 mg daily.1 The submitted price of UPA is $48.68 per 15 mg tablet and $74.00 per 30 mg tablet. Assuming a full year at the 15 mg or 30 mg dosage would correspond to annual per-patient costs of $17,768 and $27,010, respectively. The annual cost of dupilumab (300 mg per injection) was assumed to be $25,909 in the first year of treatment ($24,949 in subsequent years) based on the IQVIA DeltaPA wholesale price. BSC was assumed to be a combination of emollients, low- to mid-potency TCSs, and rescue therapy (e.g., higher-potency topical or oral corticosteroids), and was assumed to have no associated treatment costs in the sponsor’s submission.
The clinical outcomes of interest were QALYs. The economic analysis was undertaken from the perspective of the publicly funded health care payer over a 10-year time horizon. Discounting (1.5% per annum) was applied to both costs and outcomes.
Model Structure
The model structure included a short-term (1-year) phase for the 16-week and 52-week assessments and a 9-year maintenance phase. The short-term phase was based on a decision tree (Figure 1). Patients with moderate to severe AD entered the decision tree on UPA 15 mg, UPA 30 mg, DUP, or BSC. After 16 weeks, treatment response was assessed based on the EASI, with treatment response defined as EASI 75. For patients with a treatment response, the utility value for responders was applied starting at 2 weeks for UPA, 4 weeks for DUP, and 5 weeks for BSC. In the UPA and DUP arms, patients with a treatment response stayed on their current treatment until 52 weeks, at which point treatment response was reassessed; the state in which patients entered the Markov model was determined by the 52-week assessment.
The Markov model consisted of 5 main health states: “maintenance treatment,” “BSC responders,” “BSC nonresponders and nonresponders utility,” “BSC nonresponders and baseline utility,” and “death.” In the sponsor’s base case, the maintenance treatment state was assumed to represent a treatment response state in which patients maintained at least EASI 75. (In Figure 2, additional sub-states are depicted within the maintenance treatment state [“responder state EASI 50” and “responder state EASI 90”]; these states were included in scenario analyses only.) In the base case, patients in the UPA and DUP arms with a treatment response at 52 weeks (i.e., EASI 75) entered the Markov model in the maintenance treatment state (i.e., “EASI 75 response state”), while those in the UPA and DUP arms who had lost treatment response at 52 weeks entered the Markov model on BSC in the BSC nonresponders and nonresponders utility state. At the end of each 1-year cycle, patients in the maintenance state could remain responders (i.e., maintain an EASI 75 treatment response), discontinue active treatment (and continue on BSC), or die. Patients who discontinued UPA or DUP transitioned to the BSC nonresponders and nonresponders utility state, in which they accrued costs and utilities associated with nonresponse. Patients who entered the Markov model in the BSC response state could remain in their current state or transition to the BSC nonresponders and baseline utility state in the event of lost treatment response, where they were assumed to return to baseline utilities. Patients could die while in any state.
Model Inputs
The baseline characteristics in the model were based on pooled data from phase III trials involving UPA (Measure Up 1, Measure Up 2, and AD Up). Based on the mean values of the pooled data, patients entered the model at 33.91 years of age; 57% of patients were male and had a weight of 75.04 kg. The sponsor assumed that patients administered all treatments independently, including DUP. Treatment compliance was based on the Measure Up 1 and Measure Up 2 trials for UPA and was assumed to be consistent across all model years (UPA 15 mg = |||||%; UPA 30 mg = |||||%).3 Compliance rates for DUP were obtained from previous economic evaluations and varied over time (year 1 = 95.2%; year 2 = 98.6%).4
The probability of treatment response (i.e., EASI 75) at 16 weeks was derived from network meta-analyses (NMAs) involving patients with moderate to severe AD who had an intolerance or inadequate response to cyclosporin A.5 Separate NMAs were provided for patients taking UPA as monotherapy (i.e., without TCS) or as combination therapy (i.e., with TCS), with the inputs for UPA informed by the Measure Up 1 and Measure Up 2 trials (monotherapy) or by the AD Up trial (combination therapy); the monotherapy NMA was used to inform the sponsor’s base-case analysis. The probability of a treatment response to BSC was assumed to be equal to the pooled placebo response across trials included in the relevant NMA. At 52 weeks, the probability of a treatment response was conditional on the response at 16 weeks, with the probability of response assumed to be equivalent for UPA and DUP, based on the SOLO-CAFÉ and CHRONOS-CAFÉ trials.6 The annual probability of discontinuation (6.3%) was also assumed to be equivalent for UPA and DUP based on the annual probability of discontinuing DUP in the SOLO trial.6 Those who discontinued active treatment were assumed to receive BSC for the remainder of the model horizon, accruing costs associated with BSC nonresponse and nonresponder utilities.
The sponsor assumed that treatment did not affect mortality risk. Age- and sex-specific mortality rates were based on general population life tables (2017 to 2019) from Statistics Canada.7
Health state utilities were derived from pooled patient-level EQ-5D Five-Level questionnaire data from the UPA phase III trials (Measure Up 1, Measure Up 2, and AD Up) valued using a UK tariff3 and subsequently mapped to EQ-5D Three-Level questionnaire8 and adjusted for age and sex.9 Patients entered the model with a baseline utility of ||||||. Those deemed to be treatment responders at the first assessment time (i.e., 16 weeks) were assumed to start accruing the responder utility (||||) at an earlier treatment-specific time (2 weeks to 5 weeks). Patients with no treatment response at 16 weeks or who stopped responding at 52 weeks were assumed to receive the BSC nonresponder utility (||||) for the remainder of the model horizon. The sponsor assumed that part of the HRQoL benefit of treatment would be lost each year from year 2 to year 10. For UPA and DUP, the probability of losing HRQoL benefit was based on the National Institute for Health and Care Excellence evaluation of DUP,6 while the probability of losing the HRQoL benefit for patients on BSC was based on a retrospective cohort of patients with severe AD taking cyclosporine.10 For those who lost the HRQoL benefit in the UPA or DUP arms, the nonresponder utility benefit was applied for the remainder of the model horizon, while those in the BSC arm who lost the HRQoL benefit were assumed to revert to the baseline utility. Disutilities related to adverse events were assumed to be captured as part of health state utility values.
Adverse events included in the model were injection-site reaction, allergic conjunctivitis, infectious conjunctivitis, oral herpes, herpes zoster, major adverse cardiac events, venous thromboembolic events, malignancies (excluding nonmelanoma skin cancer), acne, nasopharyngitis, and upper respiratory tract infection. Adverse event rates were based on the proportion of patients with an event in the relevant clinical trials and were applied annually.
The economic model included drug acquisition costs, other treatment-related costs (i.e., blood counts, treatment of flares [by responder status and treatment], and phototherapy), health care resource use, and costs related to the treatment of adverse events. Drug acquisition costs for UPA were based on the sponsor’s submitted price,2 while the cost of DUP was based on the wholesale price from IQVIA DeltaPA. For analyses involving concomitant treatment with TCS, the cost of TCS was based on the Ontario Drug Benefit Formulary11 price of betamethasone 0.1% ointment and affected body surface area (assumed to be |||||%, based on the Measure Up trials); the cost of TCS was assumed to be reduced by 49.4% among those with a treatment response.3 Other treatment-related costs included full blood counts, treatment of flares, and phototherapy, which were assumed to vary by response status and treatment received (responders to UPA or DUP = $17.78; responders to BSC = $141.97; nonresponders to UPA or DUP = $82.22; nonresponders to BSC = $141.97). Health care resource use costs consisted of general practitioner visits, specialist visits, outpatient clinic visits, and hospital admissions. The annual cost of health care resource use was obtained from the CADTH review of DUP for AD12 and was assumed to vary by response status (responders = $175.59; nonresponders = $4,251.65). Cost related to adverse events were based on ambulatory care costs from the Ontario Case Costing Initiative,13 with the exception of major adverse cardiac events and venous thromboembolism, for which inpatient costs were assumed.3
Summary of Sponsor’s Economic Evaluation Results
The sponsor submitted probabilistic (1,000 iterations) and deterministic cost-utility analyses, with similar results; the results of the probabilistic analyses are presented in this section. The submitted analyses were based on publicly available prices for BSC and wholesale prices for dupilumab.
Base-Case Results
The sponsor’s base case assessed the cost-effectiveness of UPA among patients eligible for systemic therapy. In the sponsor’s base-case analysis, both doses of UPA were more costly and produced more QALYs than treatment with BSC (Table 3). Based on sequential analyses, BSC is the preferred treatment option if a decision-maker’s WTP threshold is below $50,241 per QALY. UPA 15 mg would be the preferred treatment option for WTP thresholds between $50,241 and $267,678, while UPA 30 mg would be the preferred option beyond $267,678 per QALY. Dupilumab was dominated by UPA 15 mg, indicating that it was more costly and produced fewer QALYs compared to UPA 15 mg. At a WTP of $50,000 per QALY, the probability of UPA 15 mg being considered the most likely cost-effective intervention was 45%, while the probability of UPA 30 mg being considered the most likely cost-effective intervention was 0%.
The drug costs associated with UPA are key drivers of the ICER (Appendix 3, Table 9). At the end of the 10-year time horizon, approximately 99% of patients remained alive in each treatment group. Additional results from the sponsor’s submitted economic evaluation base case are presented in Appendix 3.
Table 3: Summary of the Sponsor’s Economic Evaluation Results — Patients Eligible for Systemic Therapy
Drug | Total costs ($) | Total QALYs | Sequential ICER ($/QALY) |
|---|---|---|---|
BSC | 40,311 | 5.47 | Reference |
UPA 15 mg | 85,551 | 6.37 | 50,241 |
UPA 30 mg | 130,588 | 6.54 | 267,678 |
BSC = best supportive care; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; UPA = upadacitinib.
Note: Only treatments that are on the efficiency frontier are reported in the main body (dupilumab was dominated by UPA 15 mg). Full results are reported in Appendix 3.
Source: Sponsor’s pharmacoeconomic submission.3
Sensitivity and Scenario Analysis Results
The sponsor provided several scenario and sensitivity analyses, such as scenarios including patients with prior exposure to systemic IMMs, using combination treatment with topical corticosteroids; and including cyclosporine as a comparator; adopting alternative discount rates, adopting alternative time horizons, and adopting a societal perspective (i.e., including productivity costs for patients and costs of over-the-counter medications and products). Most scenarios had no meaningful effect on the ICER, with the exception of adopting a 1-year time horizon and including cyclosporine as a comparator.
In sequential analyses with a 1-year time horizon, the ICER for UPA 15 mg compared with BSC was $165,898 per QALY, while the ICER for UPA 30 mg compared with UPA 15 mg was $622,399 per QALY. In sequential analyses including cyclosporine as a comparator, the ICER for UPA 15 mg was $156,102 per QALY compared with cyclosporine, while the ICER for UPA 30 mg was $301,608 per QALY.
In response to a request from CADTH, the sponsor provided 2 additional scenario analyses intended to reflect the Health Canada–recommended dosing for UPA. In the dose-escalation scenario, all patients were assumed to initiate treatment on UPA 15 mg, while those with an inadequate response (i.e., EASI 50 to EASI 75) after 16 weeks of treatment were assumed to receive UPA 30 mg. The ICER for UPA was $51,479 per QALY compared with BSC. In the second scenario, patients with severe AD (i.e., an Investigator Global Assessment score of 4) were assumed to initiate treatment on UPA 30 mg. In this scenario, the ICER for UPA 30 mg was $111,613 per QALY versus DUP.
CADTH Appraisal of the Sponsor’s Economic Evaluation
CADTH identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis.
The sponsor’s pharmacoeconomic evaluation does not reflect the intended clinical usage of UPA. The recommended starting dose of UPA is 15 mg daily, with escalation to 30 mg daily for patients who do not have an adequate response (e.g., EASI 75) and with continued use of the lowest effective dose needed to maintain response.1 In the sponsor’s base case, UPA 15 mg and UPA 30 mg were considered separately, and patients were unable to transition between the doses, which does not reflect the Health Canada dosing strategy for UPA.
The sponsor provided a scenario analysis intended to reflect the dose-escalation strategy described in the product monograph. In this model, all patients initiated treatment on UPA 15 mg; after 16 weeks of treatment, those with an inadequate response, defined by the sponsor as an EASI score between 50 and 75, escalated to UPA 30 mg daily. CADTH notes the lack of clinical data with which to populate this model, as well as several structural limitations with the sponsor’s revised Markov model. No clinical studies have evaluated the effectiveness of the modelled dose-escalation strategy. Additionally, the clinical expert consulted by CADTH for this review indicated that an EASI 50 response to UPA 15 mg is not an appropriate lower limit to use in determining eligibility for a trial of UPA 30 mg. The clinical expert further noted that the decision as to whether a patient would try UPA 30 mg would take into account a number of factors beyond the EASI score, including patient preference, drug tolerance (e.g., gastrointestinal adverse events), and improvement in pruritis. CADTH notes that, in the sponsor’s revised model, eligible patients do not receive UPA 30 mg for the full 16-week trial period; patients who respond to UPA 30 mg do not de-escalate to UPA 15 mg; and costs related to an additional health care provider visit are not included. Each of these situations affects the total cost of UPA treatment. CADTH was unable to address these limitations due to the structure of the sponsor’s model.
The sponsor’s pharmacoeconomic submission does not reflect the cost-effectiveness of the Health Canada–recommended dosing strategy for UPA. CADTH could not assess the cost-effectiveness of the recommended dose-escalation strategy because of a lack of clinical data, as well as structural limitations with the sponsor’s model. Consequently, the cost-effectiveness of UPA is unknown.
The target population in the sponsor’s pharmacoeconomic evaluation is not aligned with the indication or reimbursement request. The target population for the sponsor’s base case is patients eligible for systemic immunosuppressants (i.e., after a trial of topical therapies), which does not reflect the Health Canada–indicated population (i.e., patients whose AD is refractory to systemic IMMs). The clinical expert consulted by CADTH for this review noted that the expected place of UPA in therapy is similar to that of DUP, which is recommended for patients who have had an adequate trial of, or who are ineligible for, methotrexate, cyclosporine, and phototherapy (where available).12 CADTH notes that the UPA product monograph indicates that UPA may be used after DUP; however, this fact is not accounted for in the sponsor’s submission. Finally, in the sponsor’s base case, patients were assumed to take UPA as monotherapy. However, as indicated by the clinical expert consulted for this review, at least 80% of patients would be expected to use UPA in combination with TCS.
In CADTH exploratory reanalyses, UPA was assumed to be used by patients whose AD is refractory to systemic IMMs and was also assumed to be used in combination with TCS, as indicated by the clinical expert consulted by CADTH.
The comparative effectiveness of UPA to DUP is overestimated. The relative effectiveness of UPA and DUP in the model is based on the sponsor’s NMA, which assessed effectiveness at 16 weeks, consistent with the treatment durations in the Measure Up 1, Measure Up 2, and AD Up trials. The Heads Up trial, which directly compared UPA 30 mg and DUP, was not included in the sponsor’s NMA. The clinical expert consulted by CADTH indicated that the use of outcome data assessed at 16 weeks may bias the sponsor’s pharmacoeconomic model against DUP because of the longer time to clinical response with DUP versus UPA. The clinical expert noted that the full treatment response to DUP may not be observed until 20 weeks to 24 weeks. This is consistent with the outcome of the Heads Up trial, which found a significantly higher EASI 75 response rate with UPA compared to DUP at 16 weeks, but not at 24 weeks. Given that the probability of a sustained treatment response at 52 weeks in the model is conditional on the response at 16 weeks, the use of 16-week outcome data biases the ICER in favour of UPA compared with DUP.
CADTH was unable to address this limitation in the base case because of the structure of the sponsor’s model and limitations of the available clinical data. Although the Heads Up trial provides comparative evidence for UPA 30 mg and DUP, the relative effectiveness of UPA 15 mg and BSC at 24 weeks is unknown. Given that there was no statistically significant difference in EASI 75 between UPA 30 mg and DUP at 24 weeks in the Heads Up trial, CADTH conducted a scenario analysis in which the effectiveness of UPA 30 mg plus TCS and DUP plus TCS were assumed to be equivalent.
The cost-effectiveness of UPA among adolescents is unknown. The proposed indication for UPA is for use by patients aged 12 years and older.1 However, the modelled cohort had a starting age of 33.91 years, based on pooled data from the Measure Up 1, Measure Up 2, and AD Up trials. These trials enrolled relatively few participants aged 12 years to 17 years (Measure Up 1 = 14.6%; Measure Up 2 = 12.4%; AD Up = 13.2%). The clinical expert consulted by CADTH indicated that adherence to treatment may be lower among adolescents than among adults, which would affect both costs and clinical outcomes. The UPA dosage shown on the draft product monograph is 15 mg for adolescents who weigh at least 40 kg; however, the clinical expert noted that 30 mg would likely be prescribed for adolescents who weigh at least 80 kg. The dosage of dupilumab included in the sponsor’s model (300 mg every other week) reflects the dosage for adults and adolescents who weigh at least 60 kg; those who weigh less than 60 kg would receive 300 mg every 4 weeks or 200 mg every other week, which would reduce the cost of DUP.
CADTH was unable to assess the cost-effectiveness of UPA in adolescents because of a lack of clinical data related to treatment effectiveness, adherence, and health state utility values among adolescents.
Relevant comparators were omitted. In the Health Canada–indicated population, the current standard of care for the treatment of AD includes the use of systemic IMMs (e.g., methotrexate, cyclosporine). Systemic IMMs were not included in the sponsor’s base-case analysis. The sponsor noted that this was owing to “a paucity of randomized controlled trial data for immunosuppressants in moderate-to-severe AD” and that as a result, “it was methodologically not feasible to include data from immunosuppressants studies into either the monotherapy or combination with TCS NMAs.”3 CADTH notes that the sponsor included cyclosporine as a comparator in scenario analyses; however, the effectiveness estimates were based on naive comparisons that do not account or adjust for differences in patient populations.
Additional comparators deemed relevant by the clinical expert consulted by CADTH for this review include additional IMMs (e.g., azathioprine, mycophenolate mofetil), retinoids (e.g., acitretin, alitretinoin), and phototherapy. As noted in Appendix 3 (tables 10 to 12), systemic IMMs, retinoids, and phototherapy are less expensive than UPA; however, the comparative clinical effectiveness of UPA versus these treatments is unknown.
CADTH could not evaluate the cost-effectiveness of UPA relative to other comparators because of a lack of comparative clinical evidence. While the sponsor’s model allowed for the inclusion of cyclosporine in the analysis, the effectiveness estimates were based on naive comparisons without adjustment for patient characteristics.
The long-term effectiveness of UPA is uncertain. As noted in the CADTH clinical review, 52-week extension studies of AD Up, Measure Up 1, and Measure Up 2 are ongoing. Clinical effectiveness in the sponsor’s model was based on 16-week outcome data, with the probability of maintaining a treatment response at 52 weeks assumed to be equivalent between UPA and DUP, based on data reported in the National Institute for Health and Care Excellence DUP submission,6 despite the availability of data from the UPA extension studies. The sponsor similarly assumed that the annual probability of treatment discontinuation and effectiveness waning would be equivalent between UPA and DUP, based on data reported for DUP.6 The clinical expert consulted by CADTH for this review indicated that it is highly uncertain whether these assumptions are valid is because of the lack of long-term data for UPA.
CADTH explored the impact of treatment-specific EASI 75 response rates at 52 weeks on the ICER using data from the AD Up extension study. CADTH was unable to address the lack of long-term data (i.e., past 52 weeks).
The durability of BSC treatment response is uncertain. The sponsor’s model included treatment-specific assumptions about the durability of treatment response over time. As noted earlier, the sponsor assumed that effectiveness waning would be equivalent for UPA and DUP. For UPA and DUP, the sponsor assumed that the cumulative proportion of patients losing HRQoL benefit would rise to 12.8% in year 10 from 2% in year 2. For BSC, the sponsor assumed that the gain in utility would diminish more rapidly, with the cumulative proportion of patients who lose HRQoL benefit increasing to 96.5% in year 6 from 83.6% in year 2, such that at least 90% of patients on BSC would have returned to their baseline utility values by year 4. These data for BSC were based on the digitization of a figure representing all-cause discontinuation of cyclosporine in a single-centre retrospective cohort of patients in the Netherlands,10 and there was no adjusting or accounting for differences in patient characteristics or study design between data sources. Alternative estimates of effectiveness waning for the BSC group are available from the DUP CHRONOS trial, and ranged from 57% in year 2 to 97% in year 514; these values were previously adopted in the CADTH appraisal of DUP.12
CADTH incorporated alternative waning assumptions for the BSC group in its exploratory reanalyses, consistent with the rate adopted in the CADTH assessment of DUP.
Inappropriate time horizon. The sponsor’s model assessed the cost-effectiveness of UPA over a 10-year time horizon. As per CADTH economic guidelines, the analysis horizon should be long enough to sufficiently capture all potential costs and effects, and a lifetime time horizon is typically appropriate for chronic conditions, such that no patients remain alive at the end of the model horizon.15 In the sponsor’s submission, 99% of patients remained alive at the end of the 10-year horizon, indicating that this duration is insufficient to assess all costs and outcomes associated with treatment.
In CADTH reanalyses, a lifetime time horizon was adopted (maximum patient age = 110 years). This time horizon is aligned with that used in the CADTH evaluation of DUP.12
Adherence to treatment is likely overestimated. The sponsor estimated that adherence to UPA was based on clinical trial data, which may overestimate adherence compared to adherence in clinical practice. Based on data from the Measure Up 1, Measure Up 2, and AD Up trials, the sponsor estimated UPA adherence to range from 95.75% to 97.29%, depending on concomitant use of TCSs. The clinical expert consulted by CADTH for this review indicated that, in clinical practice, adherence to UPA is likely to be considerably lower, and may vary between adolescents and adults. In the submitted budget impact analysis (BIA),16 the sponsor estimated adherence to UPA to be 70%, which was considered by the clinical expert to be more reflective of the rates observed in practice.
The sponsor’s model assumed that nonadherence affected treatment costs only and did not consider the impact of adherence on treatment effectiveness. Owing to a lack of clinical data about the impact of treatment adherence on outcomes, CADTH was unable to assess the impact of changes in treatment adherence on the cost-effectiveness estimate.
Uncertainty regarding health state utility values. CADTH identified 3 issues with the modelled utility estimates’ face validity. First, the baseline utility adopted in the sponsor’s model (||||) is lower than that reported previously for patients with AD by Health Technology Assessment agencies (range = 0.64 to 0.70).12 The use of a lower baseline utility may overestimate the total QALYs associated with UPA treatment. Second, the sponsor unnecessarily mapped utility values from the EQ-5D Five-Level questionnaire (captured as part of the Measure Up 1, Measure Up 2, and AD Up trials) to the EQ-5D Three-Level questionnaire through a mapping function,8 which introduces additional uncertainty. As noted in the CADTH clinical review, there was no statistically significant difference in EQ-5D scores after 16 weeks of treatment between those who received placebo and those who received UPA 15 mg or UPA 30 mg in the Measure Up 1, Measure Up 2, and AD Up trials. Third, the sponsor-provided utility values lacked face validity in that multiple estimates of utility values were provided for the same state (e.g., EASI 75 treatment response), depending on which model was selected. The utility weight for patients who achieve an EASI 75 treatment response would be expected to consistent for the same health state, regardless of the modelling approach.
CADTH explored the impact of adopting alternative health state utility values in scenario analyses.
Uncertainty about the impact of adverse events. The impact of adverse events on the ICER is uncertain for several reasons. First, the rates of adverse events in the sponsor’s assessment were applied annually in the model, based on the proportion of patients who experienced an event in trials with 16-week treatment durations. This duration may be insufficient to capture the true risk of some events (e.g., major adverse cardiac events, malignancy). Although the pivotal trials of UPA for AD showed few serious adverse events, the UPA product monograph lists warnings and precautions for serious infections, malignancies, and clotting disorders.1 Second, disutilities related to adverse events were assumed by the sponsor to be captured as part of health state utility values. However, it is unlikely that the impact of adverse events on quality of life would be adequately captured by the EQ-5D values collected as part of the UPA trials. The EQ-5D questionnaire lacks specific domains that might be affected by adverse events; in addition, it was administered at set times during the trial and has a 1-day recall period, which is problematic in assessing the impact of adverse events in clinical trials.17 Additionally, the quality of life measurements in clinical trials are often missing not at random. Further, applying UPA utility weights to other treatments fails to account for differences in their respective safety profiles. As noted in the CADTH clinical review, serious adverse events were more common in the UPA 30 mg group compared with the DUP group in the Heads Up trial. Finally, the adverse events included in the model do not capture the range of adverse events deemed to be of special interest to clinicians or noted in the patient input received by CADTH for this review.
CADTH was unable to address this limitation owing to a lack of data and the structure of the sponsor’s model. The exclusion of disutility owing to adverse events from the model may bias the ICER in favour of UPA.
Poor modelling practices were employed: The model includes numerous IFERROR statements, leading to situations in which the parameter value is automatically overwritten with an alternative value without alerting the user. The systematic use of IFERROR statements makes thorough auditing of the sponsor’s model impracticable because it remains unclear whether the model is running inappropriately by overriding errors.
CADTH notes that the results presented should be treated with a degree of caution, given that the validity of the model calculations could not be thoroughly appraised.
Additional limitations were identified, but were not considered to be key limitations.
The price of dupilumab in the sponsor’s submission was based on the IQVIA wholesale price ($959), which is lower than the price on the Ontario Exceptional Access Program18 ($979).
Additionally, the following key assumptions were made by the sponsor and have been appraised by CADTH (Table 4).
Table 4: Key Assumptions of the Submitted Economic Evaluation
Sponsor’s key assumption | CADTH comment |
|---|---|
Patients enrolled in the Measure Up 1, Measure Up 2, and AD Up trials were assumed to be representative of patients in Canada who would be eligible for UPA. | Reasonable, although the clinical expert consulted by CADTH for this review noted that the proportion of patients who had received prior non-biologic systemic IMM therapy was lower in these trials than would be expected in Canadian clinical practice. CADTH notes that subgroup data from these trials were used to inform the network meta-analyses for patients without prior exposure to systemic IMMs. The clinical expert consulted by CADTH further indicated that patients in the pivotal UPA trials may have less severe AD than those included in the DUP trials, on the basis of median EASI scores at baseline. |
Patients who have an initial treatment response to UPA or DUP but later lose the response will continue to have better quality of life compared to baseline. | Reasonable. The clinical expert consulted by CADTH indicated that patients who visit a dermatologist and initiate some form of treatment may perceive an improvement in their quality of life regardless of treatment response. |
Patients who discontinue UPA or DUP were assumed to continue on BSC. | Unreasonable. Clinical experts consulted by CADTH indicated that once a patient started on systemic treatments, such as UPA or DUP, they would be expected to remain on systemic treatment indefinitely and would not receive BSC alone should they discontinue UPA or DUP. CADTH was unable to model treatment-specific sequences owing to the structure of the sponsor’s model. |
A 75% reduction in EASI score from baseline was assumed to represents a treatment response. | Reasonable. The clinical expert consulted by CADTH indicated that a 75% reduction in EASI score would likely represent a clinically meaningful reduction. CADTH notes that EASI 75 has been used in previous submissions in this clinical area. Treatment decisions are not made based on EASI scores in practice, although the EASI score is used routinely because of reimbursement requirements. |
No cost was incorporated for BSC, reflecting the assumption that the use of BSC would not vary between treatments. | Uncertain, but unlikely to have an important effect on the ICER. The clinical expert consulted by CADTH indicated that patients whose AD responds favourably to treatment may be able to reduce the quantity of topical treatments used. |
Direct medical costs were assumed to include general practitioner or specialist visits, outpatient clinic visits, and hospital admissions. Due to the lack of available data, it was assumed that the same cost applied regardless of the response criteria or level of response. | Reasonable. The sponsor adopted the direct medical costs from the previous CADTH review of dupilumab (inflated to 2021 dollars), with separate costs incorporated for treatment responders ($175.59 per patient annually) and nonresponders ($4,251.65 per patient annually). |
Treatment-related health care resource use (i.e., complete blood counts, treatment of AD flares, phototherapy) was assumed to be equal for UPA and DUP and to vary depending on treatment response. | Uncertain. The clinical expert consulted by CADTH indicated that the health care resource use, which the sponsor adopted from the NICE submission of DUP,6 may not reflect the management of AD in Canada. The clinical expert noted that patients receiving DUP or BSC would be unlikely to receive a complete blood count, and that patients taking UPA would be likely to receive monthly complete blood counts for the first 16 weeks of treatment, and subsequently every 3 months. Receipt of a complete blood count is not expected to depend on treatment response. The clinical expert also noted that patients taking UPA would likely undergo additional laboratory investigations compared to patients taking DUP or BSC, including liver function testing, creatine phosphokinase testing, and lipid testing. These investigations are expected to be routine and not dependent on treatment response. Patients who step up to 30 mg UPA after an inadequate response to 15 mg would require an additional visit to a health care provider to assess treatment response, typically after an additional 16 weeks of treatment. The use of additional treatment-related health care resources by patients taking UPA would increase the ICER. CADTH was unable to assess the magnitude of this impact because of the structure of the sponsor’s model and a lack of data about the proportion of patients who would step up to UPA 30 mg after an inadequate response to UPA 15 mg. |
AD = atopic dermatitis; BSC = best supportive care; DUP = dupilumab; EASI = Eczema Area Severity Index; ICER = incremental cost-effectiveness ratio; IMM = immunosuppressant; NICE = National Institute for Health and Care Excellence; UPA = upadacitinib.
CADTH Reanalyses of the Economic Evaluation
The sponsor’s base case did not reflect the Health Canada–recommended dosing strategy for UPA. In response to CADTH’s request, the sponsor provided a pharmacoeconomic model intended to reflect the recommended dosing strategy for UPA; however, a lack of clinical data and structural limitations precluded any CADTH reanalyses using this model. As such, CADTH was unable to conduct any base-case reanalysis. Exploratory reanalyses were performed using the sponsor’s base-case model.
Exploratory and Scenario Analysis Results
Details of the exploratory reanalysis are presented in Appendix 4. In this analysis, UPA 15 mg plus TCS was associated with an ICER of $48,616 compared with BSC, while UPA 30 mg plus TCS was associated with an ICER of $372,226 compared with UPA 15 mg plus TCS. DUP plus TCS was dominated by UPA 15 mg plus TCS, although it is likely that the sponsor’s model overestimates the effectiveness of UPA relative to DUP. At a WTP threshold of $50,000, the probabilities of UPA 15 mg plus TCS and UPA 30 mg plus TCS being considered the most likely cost-effective interventions were 58.7% and 0%, respectively.
Several scenario and sensitivity analyses were conducted on the CADTH exploratory reanalysis. These analyses explored the impact of the following model parameters and assumptions: assuming that the treatment effectiveness of UPA 30 mg and DUP is equivalent at 16 weeks and 52 weeks; adopting alternative health state utility values; and adopting alternative pricing for DUP (Table 14). The cost-effectiveness of UPA 30 mg plus TCS was most notably affected when the treatment effectiveness was assumed to be equivalent between UPA 30 mg plus TCS and DUP plus TCS (UPA 30 mg plus TCS dominated by UPA 15 mg plus TCS). The results of the analyses were highly sensitive to the price of DUP.
Issues for Consideration
Additional treatments, including abrocitinib (a Janus kinase inhibitor) and tralokinumab (an interleukin 13 monoclonal antibody), are currently under consideration by Health Canada for the treatment of moderate to severe AD. The cost-effectiveness of UPA relative to these other potential treatments is unknown.
A recent pharmacoeconomic analysis by the Institute for Clinical and Economic Review found that UPA was dominated by DUP (i.e., UPA was more costly and less effective).19 CADTH notes that there were methodological differences between the institute’s analysis and the sponsor’s submission (e.g., in terms of model structure and clinical inputs) that preclude a direct comparison of the findings. These differences highlight the uncertainty associated with both the sponsor’s submission and the CADTH reanalysis.
Comparisons between UPA and DUP, including estimates of price reduction, were made based on the publicly available list price of DUP. In CADTH’s review of DUP, a price reduction of 54% was recommended for DUP to achieve cost-effectiveness at a threshold of $50,000 per QALY. The findings within this report should be interpreted accordingly. In jurisdictions where a price reduction was achieved for DUP, CADTH’s analysis overestimates the cost-effectiveness of UPA compared to DUP. In a scenario analysis in which the price of DUP is reduced by 54%, DUP would be considered the most cost-effective treatment at a WTP of $50,000.
As noted in the patient and clinician input received for this review, some patients may prefer treatment that can be administered less frequently, while others may prefer daily treatments over injections. UPA is administered daily, compared with biweekly injection of dupilumab.
The clinical expert consulted by CADTH for this review noted that access to dermatological treatment is difficult for patients who already face barriers to health care access, particularly members of racially and economically marginalized communities and those who live in remote areas. The analyses described in this report do not consider the differential impacts that may be experienced by patients receiving treatment with a tablet versus a syringe versus other methods of drug administration. Consequently, any differences in the cost-effectiveness due to these factors is not reflected within the analyses.
Overall Conclusions
The CADTH clinical review found that compared to BSC, UPA, whether as monotherapy or in combination with a TCS, reduces the symptoms of AD (i.e., improves EASI score) among those with moderate to severe AD. However, the sponsor’s base case does not reflect the indicated population or the Health Canada dosing strategy for UPA (i.e., a starting dose of 15 mg and increasing to 30 mg for those with an inadequate response, or a starting dose of 30 mg for some patients with severe AD). Although the sponsor submitted a revised model intended to reflect the monograph dosing, a lack of clinical data as well as structural limitations with the sponsor’s model meant that CADTH could not assess the cost-effectiveness of UPA. As such, the cost-effectiveness of UPA, when used following the product monograph, is unknown.
CADTH undertook an exploratory reanalysis to address limitations in the sponsor’s submission: assuming that UPA would be used after a trial of at least 1 prior systemic IMM, assuming that UPA would be used in combination with a TCS, adopting a lifetime time horizon; assuming that treatment effectiveness may wane over the entire analysis horizon, and adopting alternative assumptions about the durability of the BSC treatment response. The results of this exploratory analysis are subject to high levels of uncertainty because of uncertain comparative effects of UPA relative to DUP and other AD treatments (e.g., some IMMs, retinoids, and phototherapy) and an uncertain impact on HRQoL. CADTH was unable to address these limitations because of a lack of evidence. CADTH additionally notes that this exploratory reanalysis considers UPA 15 mg and UPA 30 mg separately, which does not reflect the recommended dosing strategy or differential dosing for patients with severe AD. The ICER for UPA compared to BSC likely lies between the predicted ICERs for UPA 15 mg and UPA 30 mg.
As noted in the CADTH clinical review, the comparative effectiveness of UPA versus DUP varies depending on the timing of the outcome assessment, and the use of 16-week outcome data in the pharmacoeconomic model means that all CADTH reanalyses likely overestimate the incremental effectiveness of UPA relative to DUP, producing a bias in the cost-effectiveness results that favours UPA. In the CADTH exploratory analysis, UPA 15 mg plus TCS was associated with an ICER of $48,616 per QALY gained compared with BSC, while UPA 30 mg plus TCS was associated with an ICER of $372,226 per QALY gained compared with UPA 15 mg plus TCS. In this population, a price reduction of 35% would be required for UPA 30 mg to be considered optimal at a WTP threshold of $50,000. However, due to the limitations noted earlier, these estimates are highly uncertain and biased in favour of UPA, and the cost-effectiveness estimates are highly sensitive to assumptions about the prices of UPA and DUP.
CADTH was unable to assess the cost-effectiveness of UPA for the intended dosing strategy. In addition, CADTH could not address the lack of comparative clinical effectiveness data for some relevant treatment comparators, the cost-effectiveness of UPA in adolescents or by disease severity, or the impact of adverse events. As a result, the cost-effectiveness of UPA is unknown.
References
1.Rinvoq (upadacitinib): extended-release tablets, 15 mg and 30 mg, oral [product monograph]. St-Laurent (QC): AbbVie Corporation; 2020 Oct 16.
2.Drug Reimbursement Review sponsor submission: Rinvoq (upadacitinib), once daily 15 mg and 30 mg extended-release tablet [internal sponsor's package]. Pointe-Claire (QC): AbbVie Corporation; 2021 Apr 12.
3.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Rinvoq (upadacitinib), once daily 15 mg and 30 mg extended-release tablet. Pointe-Claire (QC): AbbVie Corporation; 2021 Apr 12.
4.Kuznik A, Bégo-Le-Bagousse G, Eckert L, et al. Economic evaluation of dupilumab for the treatment of moderate-to-severe atopic dermatitis in adults. Dermatol Ther (Heidelb). 2017;7(4):493-505. PubMed
5.Sponsor's NMA report [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Rinvoq (upadacitinib), once daily 15 mg and 30 mg extended-release tablet Pointe-Claire (QC): AbbVie Corporation; 2021 Apr 12.
6.Single technology appraisal: dupilumab for treating moderate to severe atopic dermatitis after topical treatments [ID1048]: committee papers. London (GB): National Institute for Health and Care Excellence (NICE); 2018: https://www.nice.org.uk/guidance/ta534/documents/committee-papers. Accessed 2021 May 20.
7.Statistics Canada. Table: 13-10-0114-01. Life expectancy and other elements of the life table, Canada all provinces except Prince Edward Island. 2021; https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1310011401. Accessed 2021 Jun 3.
8.van Hout B, Janssen MF, Feng Y-S, et al. Interim scoring for the EQ-5D-5L: mapping the EQ-5D-5L to EQ-5D-3L value sets. Value Health. 2012;15(5):708-715. PubMed
9.Ara R, Brazier JE. Populating an economic model with health state utility values: moving toward better practice. Value Health. 2010;13(5):509-518. PubMed
10.van der Schaft J, Politiek K, van den Reek J, et al. Drug survival for ciclosporin A in a long-term daily practice cohort of adult patients with atopic dermatitis. Br J Dermatol. 2015;172(6):1621-1627. PubMed
11.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2021; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2021 May 13.
12.CADTH Canadian Drug Expert Committee (CDEC) final recommendation: dupilumab (Dupixent); Sanofi-Aventis Canada Inc. CADTH: Ottawa (ON); 2020 Apr 24: https://www.cadth.ca/sites/default/files/cdr/complete/SR0636%20Dupixent%20-%20CDEC%20Final%20%20Recommendation%20April%2024%2C%202020%20for%20posting.pdf. Accessed 2021 Jun 16.
13.Ontario Case Costing Initiative (OCCI). Toronto (ON): Ontario Health and Long-Term Care; 2017: https://data.ontario.ca/dataset/ontario-case-costing-initiative-occi. Accessed 2021 Jul 26.
14.Final appraisal document: dupilumab for treating moderate to severe atopic dermatitis. London (GB): National Institute for Health and Care Excellence; 2018: https://www.nice.org.uk/guidance/ta534/documents/final-appraisal-determination-document. Accessed 2021 Jul 5.
15.Guidelines for the economic evaluation of health technologies: Canada. 4th ed. Ottawa (ON): CADTH; 2017: https://www.cadth.ca/guidelines-economic-evaluation-health-technologies-canada-0. Accessed 2021 Jul 26.
16.Budget Impact Analysis [internal sponsor's report]. In:Drug Reimbursement Review sponsor submission: Rinvoq (upadacitinib), once daily 15 mg and 30 mg extended-release tablet. Pointe-Claire (QC): AbbVie Corporation; 2021 Apr 12.
17.Bansback N, Sun H, Guh DP, et al. Impact of the recall period on measuring health utilities for acute events. Health Econ. 2008;17(12):1413-1419. PubMed
18.Exceptional Access Program (EAP). Toronto (ON): Ontario Ministry of Health; Ontario Ministry of Long-Term Care; 2021: https://www.health.gov.on.ca/en/pro/programs/drugs/odbf/odbf_except_access.aspx. Accessed 2021 Jul 26.
19.Atlas SJ, Brouwer E, Fox G, et al. JAK inhibitors and monoclonal antibodies for the treatment of atopic dermatitis: effectiveness and value. Boston (MA): Institute for Clinical and Economic Review; 2021: https://icer.org/wp-content/uploads/2020/12/Atopic-Dermatitis_Revised-Evidence-Report_070921.pdf?bcs-agent-scanner=29a0b22a-bc65-6f41-ad63-926d37879db3. Accessed 2021 Jul 9.
20.Sidbury R, Davis DM, Cohen DE, et al. Guidelines of care for the management of atopic dermatitis: section 3. Management and treatment with phototherapy and systemic agents. J Am Acad Dermatol. 2014;71(2):327-349. PubMed
21.Ratio-Amcinonide (amcinonide USP): amcinonide cream, USP 0.1% w/w; amcinonide ointment, USP 0.1% w/w; amcinonide lotion 0.1% w/w [product monograph]. Toronto (ON): Teva Canada Limited; 2017 Jun 26.
22.Diprosone (betamethasone dipropionate): cream, Merck Standard: 0.05% W/W betamethasone (as dipropionate); betamethasone dipropionate ointment, Merck Standard, 0.05% W/W betamethasone (as dipropionate); betamethasone dipropionate lotion, USP, 0.05% W/W betamethasone (as dipropionate) [product monograph]. Kirkland (QC): Merck Canada Inc; 2020 May 19.
23.Teva-Clobetasol (clobetasol 17-propionate): 0.05% cream, ointment and scalp lotion [product monograph]. Toronto (ON): Limited, Teva Canada; 2017 Dec 18.
24.pdp-Desonide (desonide): cream and ointment, 0.05% (w/w) [product monograph]. Montreal (QC): Pendopharm, division of Pharmascience Inc; 2014 Jul 29.
25.Topicort (desoximetasone): cream, USP, 0.25%; Topicort Mild (desoximetasone): cream, USP, 0.05%; Topicort Gel (desoximetasone): gel, USP, 0.05%; Topicort Ointment (desoximetasone) ointment, USP, 0.25% [product monograph]. Montreal (QC): Valeant Canada LP / Valeant Canada S.E.C.; 2012 Feb 20.
26.Lyderm ointment (fluocinonide ointment USP, 0.05%), lyderm gel (fluocinonide gel USP, 0.05%), Lyderm cream (fluocinonide cream USP, 0.05%) [product monograph]. Brampton (ON): TaroPharma; 2003 Sep 2.
27.Tiamol (fluocinonide cream, USP): emollient base [product monograph]. Brampton (ON): TaroPharma, A Division of Taro Pharmaceuticals Inc; 2003 Sep 23.
28.Ultravate (halobetasol propionate): topical cream 0.05% w/w, topical ointment 0.05% w/w [product monograph]. Laval (QC): Bausch Health, Canada Inc; 2019 Oct 15.
29.HydroVal (hydrocortisone valerate USP): cream & ointment 0.2% [product monograph]. Brampton (ON): TaroPharma; 2008 Jul 25.
30.Teva-Mometasone (mometasone furoate): 0.1% Ointment USP [product monograph]. Toronto (ON) Limited, Teva Canada; 2018 Jan 17.
31.Saskatchewan Drug Plan: search formulary. 2021; https://formulary.drugplan.ehealthsask.ca/SearchFormulary. Accessed 2021 Jul 26.
32.Alberta Health Care Insurance Plan: medical price list as of 31 March 2020. Edmonton (AB): Government of Alberta; 2020: https://open.alberta.ca/dataset/30add047-29c2-4fc7-83b5-a8ab78605cdd/resource/48d0dd0b-bb33-478d-a85c-85a86d4555ad/download/health-somb-medical-price-list-2020-03.pdf. Accessed 2021 Jul 26.
33.B.C. Government. BC PharmaCare formulary search. 2021; https://pharmacareformularysearch.gov.bc.ca. Accessed 2021 Jul 26.
34.Elidel (pimecrolimus): cream, 1%, [product monograph]. Laval (QC): Valeant Canada LP; 2014 Sep 4.
35.CADTH Canadian Drug Expert Committee (CDEC) final recommendation: crisaborole (Eucrisa); Pfizer Canada Inc. Ottawa (ON): CADTH; 2019: https://cadth.ca/sites/default/files/cdr/complete/SR0570-Eucrisa-cdec-rec-april-2-2019.pdf. Accessed 2021 Aug 18.
36.CADTH Common Drug Review: pharmacoeconomic review report: crisaborole (Eucrisa); Pfizer Canada Inc. Ottawa (ON): CADTH; 2019: https://cadth.ca/sites/default/files/cdr/pharmacoeconomic/sr0570-eucrisa-pharmacoeconomic-report.pdf. Accessed 2021 Aug 18.
37.Schedule of benefits for physician services under the Health Insurance Act: effective April 1, 2020. Toronto (ON): Ontario Ministry of Health; 2020: https://www.health.gov.on.ca/en/pro/programs/ohip/sob/physserv/sob_master20200306.pdf. Accessed 2021 Aug 30.
38.Eichenfield LF, Tom WL, Berger TG, et al. Guidelines of care for the management of atopic dermatitis: section 2. Management and treatment of atopic dermatitis with topical therapies. J Am Acad Dermatol. 2014;71(1):116-132. PubMed
39.Simpson EL. Dupilumab improves general health-related quality-of-life in patients with moderate-to-severe atopic dermatitis: pooled results from two randomized, controlled phase 3 clinical trials. Dermatol Ther (Heidelb). 2017;7(2):243-248. PubMed
40.Silverberg JL, Toth D, Bieber T, et al. Tralokinumab plus topical corticosteroids for the treatment of moderate-tosevere atopic dermatitis: results from the double-blind, randomized, multicentre, placebo-controlled phase III ECZTRA 3 trial. Br J Dermatol. 2021;184(3):450-463. PubMed
41.Barbarot S, Auziere S, Gadkari A, et al. Epidemiology of atopic dermatitis in adults: results from an international survey. Allergy. 2018;73(6):1284-1293. PubMed
42.Institut national d'excellence en santé et en services sociaux. Dupixent. Dermatite atopique (adolescents). Montreal (QC): INESSS; 2020: https://www.inesss.qc.ca/fileadmin/doc/INESSS/Inscription_medicaments/Avis_au_ministre/Juillet_2020/Dupixent_Ado_2020_06.pdf. Accessed 2021 Aug 30.
43.Rinvoq – atopic dermatitis lines of therapy [internal sponsor's report] In: Drug Reimbursement Review sponsor submission: Rinvoq (upadacitinib), once daily 15 mg and 30 mg extended-release tablet. Pointe-Claire (QC): AbbVie Corporation; 2020 Jun 26.
44.Prescribed drug spending in Canada, 2019: a focus on public drug programs. Ottawa (ON): Canadian Institute for Health Information (CIHI); 2019.
45.Sutherland G, Dinh T. Understanding the gap: a Pan-Canadian analysis of prescription drug insurance coverage. Ottawa (ON): The Conference Board of Canada; 2017: http://innovativemedicines.ca/wp-content/uploads/2017/12/20170712-understanding-the-gap.pdf. Accessed 2021 Aug 30.
46.Dupixent (dupilumab): solution for subcutaneous injection, 300 mg single-use syringe, 300 mg single-use pen, 200 mg single-use syringe [product monograph]. Laval (QC): Sanofi-aventis Canada Inc; 2021 Feb 22.
47.Megna M, Napolitano M, Patruno C, et al. Systemic treatment of adult atopic dermatitis: a review. Dermatol Ther (Heidelb). 2017;7(1):1-23. PubMed
48.Lyakhovitsky A, Barzilai A, Heyman R, et al. Low-dose methotrexate treatment for moderate-to-severe atopic dermatitis in adults. J Eur Acad Dermatol Venereol. 2009;24(1):43-49. PubMed
49.Statistics Canada Table 17-10-0057-01. Projected population, by projection scenario, age and sex, as of July 1 (x 1,000). 2021; https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710005701 Accessed 2021 Aug 30.
50.Eichenfield LF, DiBonaventura M, Xenakis J, et al. Costs and treatment patterns among patients with atopic dermatitis using advanced therapies in the united states: analysis of a retrospective claims database. Dermatol Ther (Heidelb). 2020;10(4):791-806. PubMed
51.Pope J, Bessette L, Jones N, et al. Experience with tofacitinib in Canada: patient characteristics and treatment patterns in rheumatoid arthritis over 3 years. Rheumatology (Oxford). 2020;59(3):568-574. PubMed
Appendix 1: Cost Comparison Tables
Note that this appendix has not been copy-edited.
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s). Comparators may be recommended (appropriate) practice or actual practice. Comparators are not restricted to drugs or drug regimens and may be devices or procedures. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 5: CADTH Cost Comparison Table for Systemic Therapy of Moderate to Severe AD
Drug and comparator | Strength and concentration | Dosage form | Price ($) | Recommended dosage | Daily cost ($) | Average annual drug cost ($) | |
|---|---|---|---|---|---|---|---|
Upadacitinib (Rinvoq) | 15 mg 30 mg | Tablet | 48.6800 a 74.0000 a | Adolescents (12 to 17 years) > 40 kg: 15 mg once daily Adults: 15 mg or 30 mg once daily depending on individual patient presentation | 48.68 74.00 | 17,768 27,010 | |
Biologics | |||||||
Dupilumab (Dupixent) | 200 mg/ 1.14 mL 300 mg/ 2 mL | Pre-filled syringe | 978.7000b | Adolescents < 60 kg: 400 mg as an initial dose, followed by 200 mg every 2 weeks Adolescents ≥ 60 kg: 600 mg as an initial dose, followed by 300 mg every 2 weeks Adults: 600 mg as an initial dose, followed by 300 mg every 2 weeks | Year 1: 72.40 Year 2+: 69.72 | Year 1: 26,425 Year 2+: 25,446 | |
aSponsor’s submitted price for each dosage.
bCost obtained from the Ontario Exceptional Access Program formulary (July 2021).18
Note: Annual period assumes 52 weeks or 365 days for all comparators.
Table 6: CADTH Cost Comparison Table for Systemic Therapy of Moderate to Severe AD (Not Indicated for AD)
Drug and comparator | Strength and concentration | Dosage form | Price ($) | Recommended dosage | Daily cost ($) | Average 24-week treatment course cost ($) |
|---|---|---|---|---|---|---|
Immunosuppressants a | ||||||
Azathioprine (generic) | 50 mg | Tablet | 0.2405 | Pediatric: 1.0 to 4.0 mg/kg per day Adult: 1.0 to 3.0 mg/kg per day | Pediatric: 0.24 to 0.96 c Adult: 0.48 to 1.20 d | Pediatric: 41 to 162 Adult: 81 to 203 |
Cyclosporine (generic) | 10 mg 25 mg 50 mg 100 mg | Capsule | 0.6700 0.9952 1.9400 3.8815 | Pediatric: 3.0 to 6.0 mg/kg per day Adult: 150 to 300 mg per day | Pediatric: 5.55 to 11.04 c Adult: 5.82 to 11.64d | Pediatric: 932 to 1,855 Adult: 981 to 1,962 |
Methotrexate (generic) | 2.5 mg | Tablet | 0.6325 | Pediatric: 0.2 to 0.7 mg/kg per week Adult: 7.5 to 25 mg per week | Pediatric: 2.53 to 8.22 c per week Adult: 1.90 to 6.33 per week | Pediatric: 61 to 197 Adult: 46 to 152 |
Mycophenolate mofetil | 250 mg 500 mg | Capsule | 0.3712 0.7423 | Pediatric: 30.0 to 50.0 mg/kg per day Adult: 2,000 to 13,000 mg daily | Pediatric: 2.23 to 3.34 c Adult: 2.97 to 4.45 | Pediatric: 375 to 563 Adult: 500 to 750 |
Retinoids b | ||||||
Acitretin (Soriatane) | 10 mg 25 mg | Capsule | 1.2965 2.2770 | 10 to 50 mg once daily, max 75 mg daily | 1.30 to 6.83 | 218 to 1,148 |
Alitretinoin (Toctino) | 10 mg 30 mg | Capsule | 22.6490 | 30 mg once daily, dose may be reduced to 10 mg if unacceptable side effects | 22.65 | 3,815 |
Note: Unit prices of medications are taken from the Ontario Drug Benefit Formulary11 (accessed July 2021), unless otherwise indicated, and do not include dispensing fees. Recommended doses from respective product monographs, unless otherwise indicated. Annual period assumes 52 weeks or 365 days for all comparators.
aRecommended dosage based on the American Atopic Dermatology Guidelines.20
bRecommended dosage aligned with the previous CADTH Pharmacoeconomic Review of dupilumab.12 According the clinical expert consulted by CADTH for a previous review,12 retinoids are primarily used to treat dermatitis on the hands of adults, not adolescents.
cAssumes child weight of 45 kg.
dAssumes adult weight of 70 kg.
According to the clinical expert consulted by CADTH for this review, the following treatments may also be used to treat moderate to severe AD in adolescents and adults (Table 7).
Table 7: CADTH Cost Comparison Table of Topical Treatments for AD
Drug and comparator | Strength | Dosage form | Price | Price per gram or mL ($) | Recommended dose |
|---|---|---|---|---|---|
Topical corticosteroids | |||||
Amcinonide (generics) | 0.1% | Cream 15 g 30 g 60 g | 2.9325 5.8650 11.7300 | 0.1955 | Thin amount to affected area twice daily, max 5 days on face, axillae, scrotum or scalp, 2 to 3 weeks elsewhere.21 |
Ointment 15 g 30 g 60 g | 5.9640 11.9280 23.8560 | 0.3609a | |||
Lotion 20 mL 60 mL | 5.9940 17.9820 | 0.2997a | |||
Betamethasone dipropionate (generic) | 0.05% | Cream 15 g 50 g 45 g 120 g | 3.0720 10.2400 9.2160 24.5760 | 0.2048 | Thin film to affected area twice daily, duration of therapy varies; need should be reassessed at least every 4 weeks.22 |
Ointment 15 g 50 g 450 g | 3.2280 10.7600 96.8400 | 0.2152 | |||
7.7790 25.9300 24.5760 | 0.5186 | ||||
Lotion 30 mL 75 mL | 5.9400 14.8500 | 0.1980 | |||
Betamethasone valerate (generic) | 0.05% | Cream 454 g 450 g | 27.0584 26.8200 | 0.0596 | No recommended daily dose. Use as directed by clinicians.12 |
Ointment 454 g 450 g | 27.0584 26.8200 | ||||
0.1% | Cream 454 g 450 g | 40.3606 40.0050 | 0.0889 | ||
Ointment 454 g 450 g | |||||
Lotion 30 mL 60 mL 75 mL | 9.3750 18.7500 23.4375 | 0.3125 | |||
Clobetasol propionate (generic) | 0.05% | Cream 15 g 50 g 450 g 454 g | 3.4185 11.3950 102.5550 103.4666 | 0.2279 | Thin amount to affected area twice daily. Weekly application should not exceed 50 g, and limited to 2 consecutive weeks.23 |
Ointment 15 g 50 g 450 g | |||||
Lotion 20 mL 60 mL | 3.9800 11.9400 | 0.1990 | |||
Desonide (generic) | 0.05% | Cream 15 g 60 g | 3.9750 15.9000 | 0.2650 | Thin amount to affected area twice daily, may be increased in refractory cases.24 |
Ointment 60 g | 15.8820 | 0.2647 | |||
Desoximetasone (Topicort) | 0.05% 0.25% | Cream 20 g 60 g | 10.4300 31.2900 | 0.5215a | Thin amount to affected area twice daily.25 |
Cream 20 g 60 g | 14.6700 44.0100 | 0.7335a | |||
0.25% | Ointment 60 g | 14.6700 44.0100 | 0.7335a | ||
0.05% | Gel 15 g 60 g | 8.5605 34.2420 | 0.5707a | ||
Fluocinonide (Lidemol, Lyderm, Lidex) | 0.05% | Cream 15 g 60 g 400 g | 3.5670 14.2680 95.1200 | 0.2378 | Thin amount to affected area twice daily. Weekly application should not exceed 45 g, and limited to 2 weeks.26 |
Emollient Cream 30 g 100 g | 5.9400 19.8000 | 0.1980 | |||
Ointment 60 g | 18.2100 | 0.3035 | |||
Gel 60 g | 18.4560 | 0.3076 | |||
Fluocinonide (Tiamol) | 0.05% | Emollient Cream 25 g 100 g | 4.9500 19.8000 | 0.1980 | Thin amount 2 to 4 times daily.27 |
Halobetasol propionate (Ultravate) | 0.01% | Lotion 100 g | N/A | N/A | Thin amount to affected area twice daily, limited to 50 g weekly and 2 weeks without re-evaluation.28 |
0.05% | Cream 15 g 50 g | 17.1975 57.3250 | 1.1465c | ||
Ointment 50 g | 55.6750 | 1.1135c | |||
Hydrocortisone (various) | 1.0% | Cream 15 g 30 g 45 g 454 g | 2.577 5.1540 7.7310 77.9972 | 0.1718 | No recommended daily dose. Use as directed by clinicians.12 |
1.0% | Lotion 60 mL | 9.5220 7.1460 | 0.1587 0.1191a | ||
0.5% 1.0% | Ointment 15 g 454 g | 2.1000 17.7060 | 0.1400 0.0390 | ||
Hydrocortisone acetate | 1% | Cream 15 g 30 g | 3.0840 6.1680 | 0.2056 | Twice-daily application is generally recommended initially; intermittent use 1 to 2 times per week on areas that commonly flare for maintenance therapy. |
0.5% 1.0% | Ointment 28.4 g | 11.8087 | 0.4158c | ||
Hydrocortisone valerate (HydroVal) | 0.2% | Cream 15 g 45 g 60 g | 2.5005 7.5015 10.0020 | 0.1667 | Small amount to affected area twice daily. Discontinue as soon as lesions heal or if no response.29 |
Ointment 15 g 60 g | 2.5005 10.0020 | ||||
Mometasone furoate (generic) | 0.1% | Cream 15 g 50 g | 8.3130 27.7100 | 0.5542 | Thin film to affected areas twice daily.30 |
Ointment 15 g 50 g | 3.3780 11.2600 | 0.2252 | |||
Lotion 30 mL 60 mL | 10.0740 20.1480 | 0.3358 | |||
Triamcinolone acetonide (various) | 0.1% | Cream 15 g 30 g 500 g | 0.7995 1.5990 26.6500 | 0.0533 | No recommended daily dose. Use as directed by clinicians. |
Ointment 30 g | 4.5690 | 0.1523 | |||
0.5% | Cream 15 g 50 g | 18.84 62.80 37.681 | 1.2560b | ||
Ointment 30 g | |||||
Topical Calcineurin inhibitors | |||||
Pimecrolimus (Elidel)d | 1% | Cream 10 g 30 g | 24.8800 74.6400 | 2.4880 | Thin layer to affected area twice daily, discontinue when resolved or after 3 weeks if no improvement or exacerbation. |
Tacrolimus | 0.03% 0.10% | Ointment 30 g | 78.5190 84.0000 | 2.6173 2.8000 | Thin layer to affected area twice daily. Discontinue after 6 weeks if no improvement or exacerbation. |
Phosphodiesterase type 4 inhibitor | |||||
Crisaborole (Eucrisa)e | 2% | Ointment 60 g | 2.3000f | 138.0000 | Thin layer to affected area twice daily. |
Phototherapy | |||||
UV light therapy | NA | NA | 1,130.4000 to 1,884.0000 | 7.85 per treatmentg | Administered 3 to 5 times per weekh |
AD = atopic dermatitis; Emol = emollient; NA = not applicable.
Note: Ontario Drug Benefit Formulary list prices11 unless otherwise indicated; recommended doses from respective product monographs unless otherwise indicated.
aSaskatchewan Formulary list price31 (July 2021).
bAlberta Formulary list price32 (July 2021)
cBritish Columbia Formulary list price (July 2021).33
dPimecrolimus is indicated for treatment of mild to moderate AD in patients 2 years of age and older.34
eCrisaborole received a do not reimburse recommendation from CDEC in March 2019 for treatment of mild to moderate AD in patients 2 years of age and older who have failed or are intolerant to a topical corticosteroid treatment.35,36
fCost obtained from IQVIA DELTA PA database (accessed August 2021)
gOntario Schedule of Benefits for Physician Services, code G470 “Ultraviolet Light Therapy.”37
hMinimum frequency of phototherapy sessions required per week for successful maintenance as well as length of maintenance period varies tremendously between individuals.20,38
Appendix 2: Submission Quality
Note that this appendix has not been copy-edited.
Description | Yes/No | Comments |
|---|---|---|
Population is relevant, with no critical intervention missing, and no relevant outcome missing | No | While UPA is indicated for patients ages 12 years and older, the modelled population reflects an adult population (refer to main text). The sponsor’s base case reflects an adult population whose AD is not adequately controlled with topical prescription therapies, which is inconsistent with the indication. The model allows for alternative populations to be considered. |
Model has been adequately programmed and has sufficient face validity | No | The model includes numerous IFERROR statements, which lead to situations in which the parameter value is overwritten with an alternative value without alerting the user to the automatized overwriting. The systematic use of IFERROR statements makes thorough auditing of the sponsor’s model impractical, as it remains unclear whether the model is running inappropriately by overriding errors. |
Model structure is adequate for decision problem | No | The sponsor’s base-case model does not reflect the intended dosing strategy for UPA (i.e., patients could not transition between UPA 15 and UPA 30 in the model). In response to a request from CADTH, the sponsor provided an additional model in which patients initiated treatment on UPA 15 and could transition between UPA 15 and UPA 30 depending on treatment response. CADTH identified several structural limitations with this model, such that patients did not incur the correct costs for UPA 30. |
Data incorporation into the model has been done adequately (e.g., parameters for probabilistic analysis) | Yes | NA |
Parameter and structural uncertainty were adequately assessed; analyses were adequate to inform the decision problem | Yes | NA |
The submission was well organized and complete; the information was easy to locate (clear and transparent reporting; technical documentation available in enough details) | Yes | NA |
Appendix 3: Additional Information on the Submitted Economic Evaluation
Note that this appendix has not been copy-edited.
Figure 1: Model Structure — Decision Tree
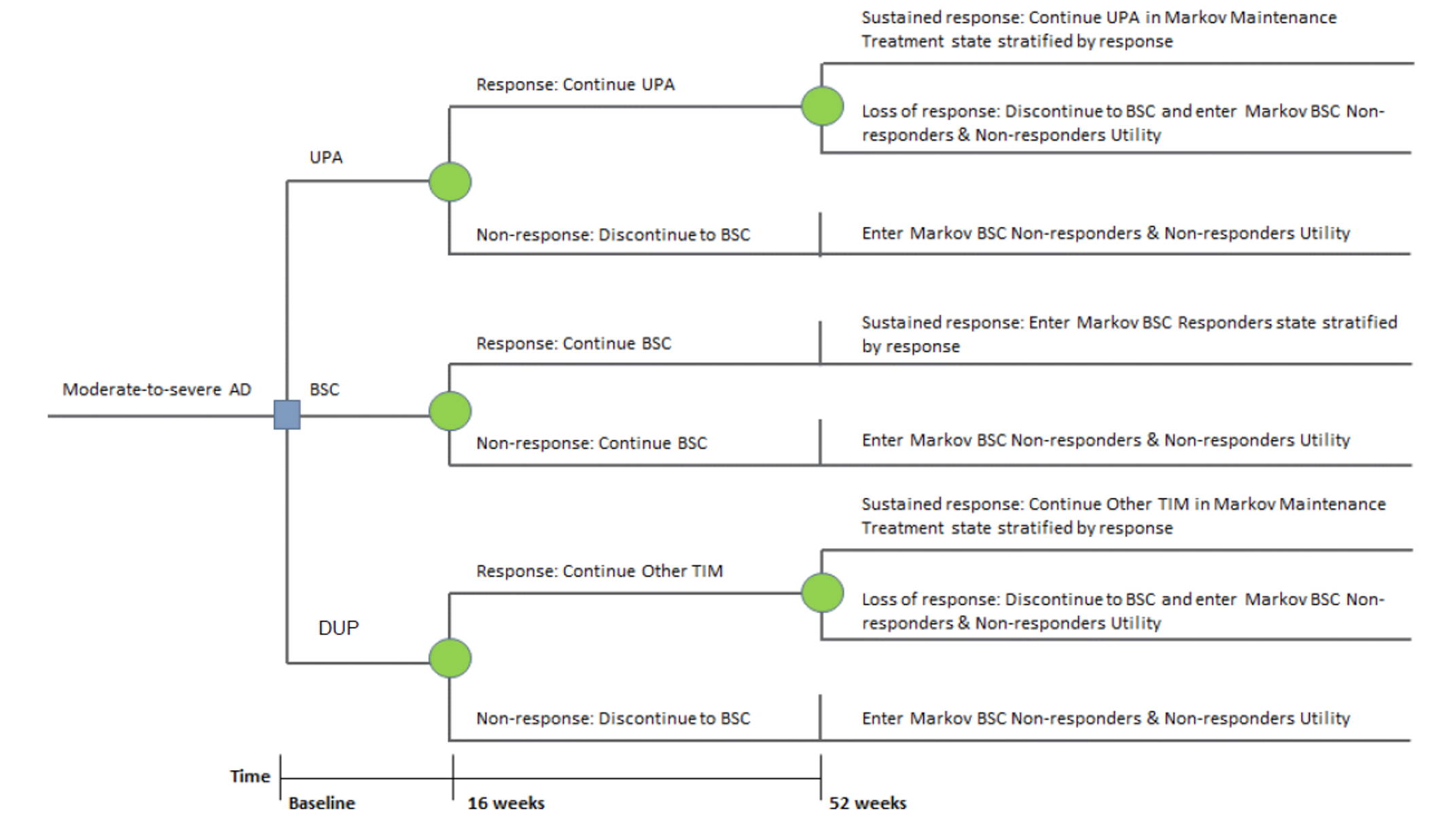
AD = atopic dermatitis, BSC = best supportive care; DUP = dupilumab; Tx = treatment; UPA = upadacitinib.
Source: Sponsor’s pharmacoeconomic submission.3
Figure 2: Model Structure — Markov model
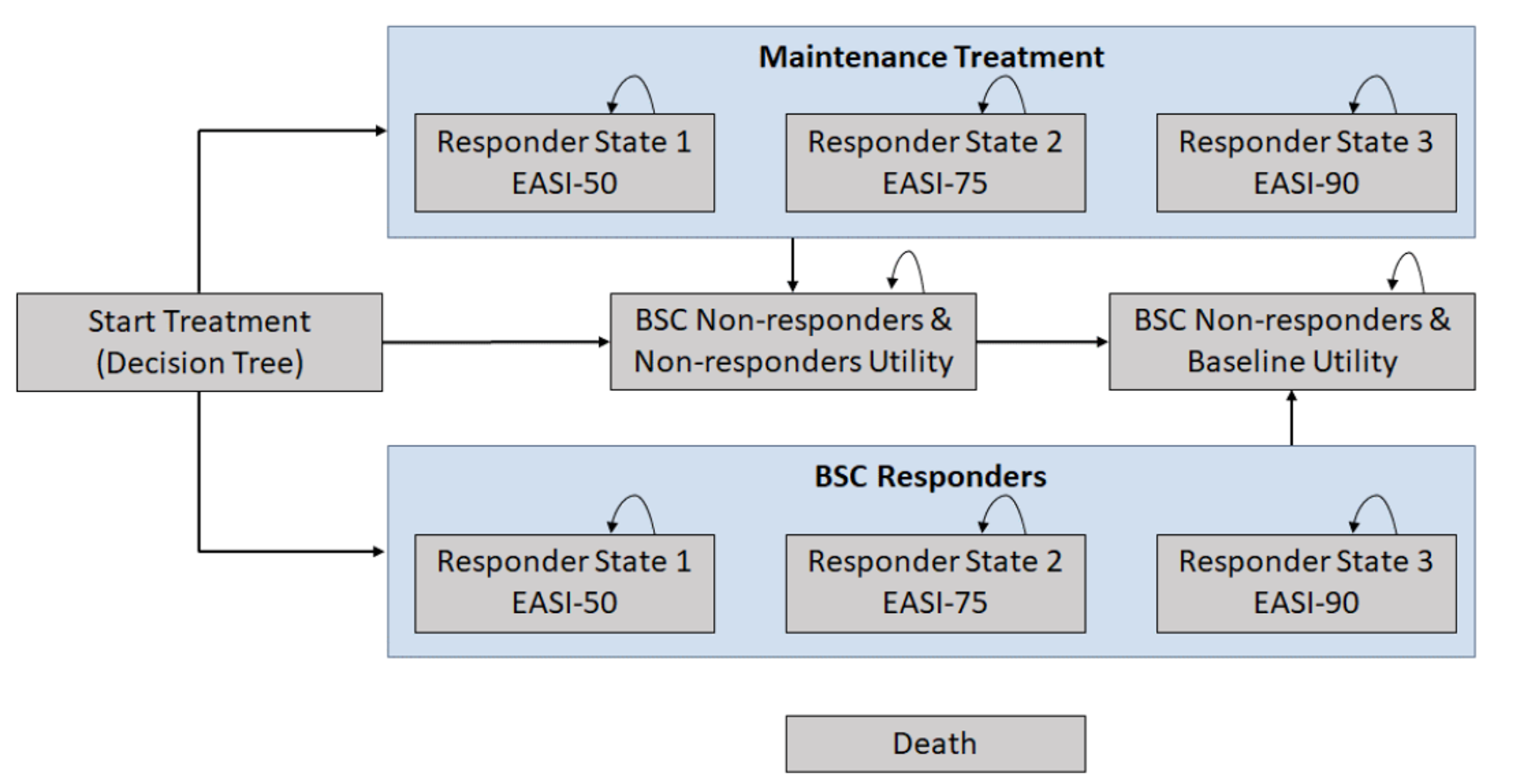
BSC = best supportive care; EASI = Eczema Area Severity Index; EASI-50 = 50% reduction in EASI score from baseline; EASI 75 = 75% reduction in EASI score from baseline; EASI-90 = 90% reduction in EASI score from baseline.
Note: Although depicted, the sponsor’s base-case analysis did not include the EASI-50 or EASI-90 response states.3
Source: Sponsor’s pharmacoeconomic submission.3
Detailed Results of the Sponsor’s Base Case
Table 9: Disaggregated Summary of the Sponsor’s Economic Evaluation Resultsa
Treatment | Component | Value | Incremental (vs. SoC) | Incremental (sequential) |
|---|---|---|---|---|
Discounted QALYs | ||||
BSC | Total | 5.47 | NA | NA |
Decision tree (year 1) | 0.733 | NA | NA | |
Markov Model (year 2 to 10) | 4.735 | NA | NA | |
UPA 15 | Total | 6.37 | 0.90 | NA |
Decision tree (year 1) | 0.797 | 0.064 | NA | |
Markov Model (year 2 to 10) | 5.573 | 0.838 | NA | |
DUP | Total | 6.23 | 0.76 | −0.14 |
Decision tree (year 1) | 0.777 | 0.044 | −0.02 | |
Markov Model (year 2 to 10) | 5.456 | 0.721 | −0.117 | |
UPA 30 | Total | 6.54 | 1.07 | 0.31 |
Decision tree (year 1) | 0.810 | 0.077 | 0.033 | |
Markov Model (year 2 to 10) | 5.727 | 0.992 | 0.271 | |
Discounted costs ($) | ||||
BSC | Active treatment | 0 | NA | NA |
Adverse events | 181 | NA | NA | |
Direct Medical costs | 38,637 | NA | NA | |
Other treatment-related cost | 1,487 | NA | NA | |
Total | 40,311 | NA | NA | |
UPA 15 | Active treatment | 58,177 | 58,177 | NA |
Adverse events | 255 | 74 | NA | |
Direct Medical costs | 26,106 | −12,531 | NA | |
Other treatment-related cost | 1,013 | −474 | NA | |
Total | 85,551 | 45,240 | NA | |
DUP | Active treatment | 71,067 | 71,067 | 12,890 |
Adverse events | 373 | 192 | 118 | |
Direct Medical costs | 28,343 | −10,294 | 2,237 | |
Other treatment-related cost | 1,089 | −398 | 76 | |
Total | 100,872 | 60,561 | 15,321 | |
UPA 30 | Active treatment | 106,005 | 106,005 | 34,938 |
Adverse events | 340 | 159 | −33 | |
Direct Medical costs | 23,276 | −15,361 | −5,067 | |
Other treatment-related cost | 967 | −520 | −122 | |
Total | 130,588 | 90,277 | 29,716 | |
ICER vs. reference ($) | Sequential ICER ($) | |||
BSC | Ref. | Ref. | ||
UPA 15 | 50,241 | 50,241 vs. BSC | ||
DUP | 79,239 | Dominated | ||
UPA 30 | 84,474 | 267,678 vs. UPA 15 | ||
BSC = best supportive care; DUP = dupilumab; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; Ref. = reference; UPA = upadacitinib; vs. = versus.
aThe sponsor’s base case assumes that UPA and DUP would be used as monotherapy (i.e., without concomitant use of topical corticosteroids).
Source: Sponsor’s pharmacoeconomic submission.3
Appendix 4: Additional Details on the CADTH Reanalyses and Sensitivity Analyses of the Economic Evaluation
Note that this appendix has not been copy-edited.
Detailed Results of CADTH Exploratory Reanalysis
Exploratory Reanalysis Results
CADTH could not assess the cost-effectiveness of the Health Canada–recommended dosing strategy for UPA (i.e., starting at 15 mg and increasing to 30 mg for those with inadequate response) owing to a lack of clinical data and structural limitations identified in the sponsor’s dose-escalation model. As such, CADTH conducted exploratory reanalyses using the sponsor’s base-case model (i.e., dose escalation not considered). This analysis explored the impact of several other limitations within the model, as summarized in Table 10. Changes to model parameter values and assumptions were determined in consultation with clinical experts.
Table 10: CADTH Revisions to the Submitted Economic Evaluation
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
1. Dupilumab price | $959.59, based on IQVIA wholesale price | $978.70 per syringe |
Changes to derive the CADTH base case | ||
1. Target population | Patients eligible for systemic IMMs | Patients refractory or ineligible for systemic IMMs |
2. Concomitant therapy | UPA and DUP assumed to be used as monotherapy | UPA and DUP assumed to be used in combination with TCS |
3. Time horizon | 10 years | Lifetime (maximum patient age 110 years) |
4. Treatment effectiveness waning | Assumed to occur during the first 10 years of treatment | Effectiveness waning assumed to continue over the model horizon. |
5. Durability of treatment response (BSC) | Cumulative proportion of patients losing HRQoL benefit: Year 2: ||||% Year 3: ||||% Year 4: ||||% Year 5: ||||% Year 6+: ||||% | Cumulative proportion of patients losing HRQoL benefit14: Year 2: 57% Year 3: 82% Year 4: 92% Year 5: 97% Year 6+: 97% |
CADTH base case | Reanalysis 1 + 2 + 3 + 4 + 5 | |
DUP = dupilumab; SD = standard deviation; TCS = topical corticosteroid; UPA = upadacitinib.
CADTH undertook a stepped analysis, incorporating each change proposed in Table 5 to sponsor’s base case to highlight the impact of each change (Table 11; disaggregated results are presented in Appendix 4, Table 13).
Table 11: Summary of the Stepped Analysis of the CADTH Exploratory Reanalysis Results
Stepped analysis | Drug | Total costs ($) | Total QALYs | ICER ($/QALYs) |
|---|---|---|---|---|
Sponsor’s base case (patients eligible for systemic therapy) | BSC | 40,311 | 5.47 | Ref. |
UPA 15 | 85,551 | 6.37 | 50,241 | |
DUP | 100,872 | 6.23 | Dominated | |
UPA 30 | 130,588 | 6.54 | 267,678 | |
Sponsor’s corrected base case (patients eligible for systemic therapy) | BSC | 39,912 | 5.47 | Ref. |
UPA 15 | 85,131 | 6.37 | 50,235 | |
DUP | 102,214 | 6.24 | Dominated | |
UPA 30 | 130,373 | 6.54 | 265,335 | |
CADTH reanalysis 1: Patients with a previous trial of systemic therapy | BSC | 40,175 | 5.41 | Ref. |
UPA 15 | 85,255 | 6.34 | 48,726 | |
DUP | 98,223 | 6.13 | Dominated | |
UPA 30 | 129,438 | 6.50 | 278,303 | |
CADTH reanalysis 2: UPA and DUP used in combination with TCS | BSC | 43,501 | 5.49 | Ref. |
UPA 15 + TCS | 92,352 | 6.45 | 51,058 | |
DUP + TCS | 123,935 | 6.47 | Extended dominance | |
UPA 30 + TCS | 142,551 | 6.66 | 232,376 | |
CADTH reanalysis 3: Analysis horizon | BSC | 149,504 | 18.31 | Ref. |
UPA 15 | 223,633 | 19.81 | 49,369 | |
DUP | 250,811 | 19.58 | Dominated | |
UPA 30 | 299,381 | 20.11 | 249,203 | |
CADTH reanalysis 4: Treatment effectiveness waning | BSC | 39,912 | 5.47 | Ref. |
UPA 15 | 85,131 | 6.37 | 50,235 | |
DUP | 102,214 | 6.24 | Dominated | |
UPA 30 | 130,373 | 6.54 | 265,335 | |
CADTH reanalysis 5: Durability of BSC response | BSC | 39,810 | 5.51 | Ref. |
UPA 15 | 85,131 | 6.40 | 51,113 | |
DUP | 102,214 | 6.27 | Dominated | |
UPA 30 | 130,373 | 6.56 | 268,565 | |
CADTH Exploratory Reanalysis (1 + 2 + 3 + 4 + 5) | BSC | 165,531 | 18.34 | Ref. |
UPA 15 + TCS | 253,404 | 20.15 | 48,616 | |
DUP + TCS | 295,142 | 19.96 | Dominated | |
UPA 30 + TCS | 330,565 | 20.35 | 372,226 |
BSC = best supportive care; DUP = dupilumab; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; Ref. = reference; TCS = topical corticosteroid; UPA = upadacitinib.
In CADTH exploratory reanalyses, UPA 30 + TCS was associated with highest costs ($330,565) and greatest QALYs gained (20.35) over the lifetime horizon (Table 12). In sequential analyses, UPA 15 + TCS was associated with higher costs and higher QALYs than BSC. UPA 30 + TCS was more costly and produced more QALYs than UPA 15 + TCS, with a sequential ICER of $372,226 compared with UPA 15 + TCS. DUP + TCS was dominated by UPA 15 + TCS, although it is likely that the sponsor’s model overestimates the effectiveness of UPA relative to DUP. The probability of UPA 15 + TCS being considered the most likely cost-effective intervention at a WTP of $50,000 per QALY was 58.70%, while the probability of UPA 30 +TCS being considered the most likely cost-effective intervention was 0%.
The incremental QALYs gained with UPA compared with BSC in the first year of treatment was 0.084 (UPA 15) and 0.093 (UPA 30), indicating that the majority of the incremental benefits (97%) for both UPA doses were derived on the basis of extrapolated findings rather than observed benefit (while the treatment duration of in the UPA trials was 16 weeks, the sponsor’s model was not set to calculate QALYs for this interval). Drug acquisition costs for UPA are key drivers of the ICER, representing 43% and 60% of the total costs associated with UPA 15 + TCS and UPA 30 + TCS, respectively.
Table 12: Summary of the CADTH Exploratory Reanalysis Results
Drug | Total costs | Total QALYs | ICER vs. BSC | Sequential ICER |
|---|---|---|---|---|
Sponsor corrected base case (adults eligible for systemic therapy) | ||||
BSC | 39,912 | 5.47 | Ref. | Ref. |
UPA 15 | 85,131 | 6.37 | 50,235 | 50,235 |
DUP | 102,214 | 6.24 | 81,206 | Dominated |
UPA 30 | 130,373 | 6.54 | 84,492 | 265,335 |
CADTH exploratory reanalysis (adults refractory or ineligible for systemic IMMs) | ||||
BSC | 165,534 | 18.34 | Ref. | Ref. |
UPA 15 + TCS | 253,404 | 20.15 | 48,616 | 48,616 |
DUP + TCS | 295,142 | 19.96 | 80,022 | Dominated |
UPA 30 + TCS | 330,565 | 20.35 | 81,912 | 372,226 |
BSC = best supportive care; DUP = dupilumab; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-years; Ref. = reference; TCS = topical corticosteroid; UPA = upadacitinib; vs. = versus.
Table 13: Disaggregated Summary of CADTH’s Exploratory Economic Evaluation Results
Treatment | Component | Value | Incremental | Incremental (sequential) |
|---|---|---|---|---|
Discounted QALYs | ||||
BSC | Total | 18.34 | NA | NA |
Decision Tree (year 1) | 0.70 | NA | NA | |
Markov Model (year 2+) | 17.64 | NA | NA | |
UPA 15 + TCS | Total | 20.15 | 1.81 | NA |
Decision Tree (year 1) | 0.78 | 0.08 | NA | |
Markov Model (year 2+) | 19.36 | 1.72 | NA | |
DUP + TCS | Total | 19.96 | 1.62 | −0.19 |
Decision Tree (year 1) | 0.74 | 0.04 | −0.04 | |
Markov Model (year 2+) | 19.22 | 1.58 | −0.15 | |
UPA 30 + TCS | Total | 20.35 | 2.02 | 0.40 |
Decision Tree (year 1) | 0.79 | 0.09 | 0.05 | |
Markov Model (year 2+) | 19.56 | 1.92 | 0.34 | |
Discounted costs ($) | ||||
BSC | Active treatment | NA | NA | |
Concomitant medication | NA | NA | ||
Adverse events | 1,352 | NA | NA | |
Direct Medical costs | 144,141 | NA | NA | |
Other treatment-related cost | 20,041 | NA | NA | |
Total | 165,534 | NA | NA | |
UPA 15 + TCS | Active treatment | 115,158 | 115,158 | NA |
Concomitant medication | 1,524 | 1,524 | NA | |
Adverse events | 1,410 | 58 | NA | |
Direct Medical costs | 119,032 | −25,109 | NA | |
Other treatment-related cost | 16,281 | −3,760 | NA | |
Total | 253,404 | 87,871 | NA | |
DUP + TCS | Active treatment | 153,604 | 153,604 | 38,446 |
Concomitant medication | 1,439 | 1,439 | −85 | |
Adverse events | 1,755 | 403 | 345 | |
Direct Medical costs | 121,841 | −22,300 | 2,809 | |
Other treatment-related cost | 16,504 | −3,537 | 223 | |
Total | 295,142 | 129,608 | 41,738 | |
UPA 30 + TCS | Active treatment | 195,715 | 195,715 | 42,112 |
Concomitant medication | 1,707 | 1,707 | 269 | |
Adverse events | 1,548 | 196 | −207 | |
Direct Medical costs | 115,761 | −28,380 | −6,080 | |
Other treatment-related cost | 15,833 | −4,208 | −671 | |
Total | 330,565 | 165,031 | 35,423 | |
ICER vs. BSC ($/QALY) | Sequential ICER ($/QALY) | |||
BSC | Ref. | Ref. | ||
UPA 15 + TCS | 48,616 | 48,616 | ||
DUP + TCS | 80,022 | Dominated | ||
UPA 30 + TCS | 81,912 | 372,226 | ||
BSC = best supportive care; DUP = dupilumab; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; Ref. = reference; TCS = topical corticosteroid; UPA = upadacitinib.
Scenario Analyses
A series of scenario analyses were preformed using the exploratory reanalysis results. These analyses were performed to investigate the impact that critical assumptions had on the cost-effectiveness, despite the limitation of not being able to model the intended dosing strategy. These scenarios analyses explored the impact of the following model parameters and assumptions had on the ICER: assuming that the treatment effectiveness of UPA 30 and DUP is equivalent at 16 and 52 weeks; adopting alternative health state utility values; and adopting alternative pricing for DUP (Table 14).
The cost-effectiveness of UPA 30 + TCS was most notably affected when the treatment effectiveness was assumed to be equivalent between UPA 30 and DUP (Table 15). In the scenario analysis assuming equal effectiveness at 16 weeks, UPA 30 was not expected to be cost-effective compared to DUP+TCS at a WTP threshold of $50,000 per QALY.
Table 14: CADTH Exploratory Scenario Analyses
Scenario Analyses | CADTH Exploratory Reanalysis | CADTH Exploratory Scenario |
|---|---|---|
Scenario Analyses | ||
1. Treatment effectiveness at 16 weeks (% EASI 75 responders) | Based on indirect evidence from the sponsor’s NMA; EASI score assessed at 16 weeks: UPA 15 + TCS: 62.94% UPA 30 + TCS: 77.91% DUP + TCS: 64.80% BSC: 23.67% | The % of EASI 75 responders was assumed to be equivalent between UPA 30 and DUP, based on the HEADS UP trial (24-week time pointa): UPA 15 + TCS: 62.94% UPA 30 + TCS: 77.91% DUP + TCS: 77.91% BSC: 23.67% |
2. Treatment effectiveness at 52 weeks (% EASI 75 responders) | Proportion of patients with sustained treatment response at 52 weeks for both UPA doses was assumed to be equal to DUP (in combination with TCS: 82.1%) | Proportion of patients with sustained treatment response at 52 weeks based on 52-week data from the AD UP extension study: UPA 15 + TCS: 70.8% UPA 30 + TCS: 83.5% |
3. Health state utility values | Sponsor-provided values: Baseline: |||||| Treatment responders: |||||| Nonresponders: |||||| | Baseline and responder utilities adopted from an alternative source: Baseline: 0.6156b Treatment responders: 0.8723b Nonresponders: 0.7126 |
4. Health state utility values | Sponsor-provided values: Baseline: |||||| Treatment responders: |||||| Nonresponders: |||||| | Baseline and responder utilities adopted from an alternative source; nonresponders assumed to revert to baseline utility: Baseline: 0.6156b Treatment responders: 0.8723b Nonresponders: 0.6156b |
5. Health state utility values | Sponsor-provided values: Baseline: |||||| Treatment responders: |||||| Nonresponders: |||||| | Sponsor-provided alternative utility values: Baseline: 0.5766 Treatment responders: 0.8234c Nonresponders: 0.6756c |
6. Dupilumab price reduction | $978.70 per syringe18 | 54% price reduction from a base price of $959.9350 based on the 2020 CADTH assessment of DUP12 |
BSC = best supportive care; DUP = dupilumab; EASI 75 = Eczema Area and Severity Index, 75% reduction from baseline; IMM = immunosuppressant; NMA = network meta-analysis; TCS = topical corticosteroid; UPA = upadacitinib.
a24-week data from the HEADS UP trial were applied at the 16-week decision point in the sponsor’s model. Data for UPA 15 and BSC were not available from the HEADS UP trial and were assumed to be equivalent to the sponsor’s base case.
bBaseline and EASI 75 treatment utilities derived from the weighted average across placebo and DUP arms in the SOLO 1 and SOLO 2 trials.39
cProvided by the sponsor for an alternative model involving additional health states.
Table 15: CADTH Exploratory Scenario Analyses Results
Drug | Total Costs ($) | Total QALYs | ICER ($/QALY) |
|---|---|---|---|
CADTH exploratory reanalysis | |||
BSC | 165,534 | 18.34 | Ref. |
UPA 15 + TCS | 253,404 | 20.15 | 48,616 |
DUP + TCS | 295,142 | 19.96 | Dominated |
UPA 30 + TCS | 330,565 | 20.35 | 372,226 |
Scenario 1: Treatment effectiveness (UPA 30 + TCS and DUP + TCS assumed to be equally effective at 16 weeks) | |||
BSC | 164,311 | 18.32 | Ref. |
UPA 15 + TCS | 262,625 | 20.34 | 48,551 |
DUP + TCS | 322,497 | 20.31 | Dominated |
UPA 30 + TCS | 330,160 | 20.34 | Dominated |
Scenario 2: Treatment effectiveness at 52 weeks | |||
BSC | 164,311 | 18.32 | Ref. |
UPA 15 + TCS | 253,531 | 20.15 | 48,678 |
DUP + TCS | 295,525 | 19.95 | Extended dominance |
UPA 30 + TCS | 331,062 | 20.35 | 378,567 |
Scenario 3: Health state utility value – Simpson 201739 | |||
BSC | 164,311 | 19.45 | Ref. |
UPA 15 + TCS | 252,277 | 20.01 | 56,273 |
DUP + TCS | 293,691 | 20.84 | Dominated |
UPA 30 + TCS | 328,671 | 21.19 | 428,915 |
Scenario 4: Health state utility value – Simpson 2017,39 nonresponders assumed to receive baseline utility value | |||
BSC | 164,311 | 19.28 | Ref. |
UPA 15 + TCS | 252,277 | 20.79 | 58,151 |
DUP + TCS | 293,691 | 20.61 | Dominated |
UPA 30 + TCS | 328,671 | 20.98 | 406,940 |
Scenario 5: Health state utility values – sponsor-provided alternatives | |||
BSC | 164,311 | 18.23 | Ref. |
UPA 15 + TCS | 252,277 | 19.74 | 58,139 |
DUP + TCS | 293,691 | 19.58 | Dominated |
UPA 30 + TCS | 328,671 | 19.14 | 447,616 |
Scenario 6: Dupilumab price reductiona | |||
BSC | 164,311 | 18.32 | |
DUP + TCS | 209,734 | 19.93 | 28,169 |
UPA 15 mg + TCS | 252,277 | 20.12 | 220,386 |
UPA 30 mg + TCS | 328,671 | 20.32 | 378,955 |
BSC = best supportive care; CYC = cyclosporine; DUP = dupilumab; ICER = incremental cost-effectiveness ratio; QALY = quality-adjusted life-year; TCS = topical corticosteroid; UPA = upadacitinib.
aIn this analysis, the price of DUP was assumed to be $959.9350 per syringe based on the 2020 CADTH assessment of DUP, with a 54% price reduction applied.12 In all other scenarios, the price of DUP was assumed to be $978.70 based on the Ontario Exceptional Access Program price.
Price-Reduction Analyses
A price-reduction analyses was performed for the sponsor’s base case and CADTH’s exploratory reanalysis (Table 16). In CADTH exploratory reanalysis, no price reduction for UPA 15 was needed to reduce the ICER below $50,000 when compared with BSC. A price reduction of 35% would be required to for UPA 30 + TCS to be considered optimal at a WTP of $50,000.
Table 16: CADTH Price-Reduction Analyses
Analysis | ICERs for UPA vs. DUP | |
|---|---|---|
Price reduction | Sponsor base casea (patients eligible for systemic treatment) | CADTH Exploratory reanalysisa (patients refractory or ineligible for systemic immunosuppressants) |
No price reduction | WTP < 48,792 WTP 48,792 to 274,819: UPA 15 WTP3 274,819: UPA 30 | WTP < 48,616: BSC WTP 48,616 to 372,226: UPA 15 + TCS WTP3 372,226: UPA 30 |
10% | WTP < 48,792 WTP 48,792 to 212,414: UPA 15 WTP3 212,414: UPA 30 | WTP < 48,616: BSC WTP 48,616 to 282,387: UPA 15 + TCS WTP3 282,387: UPA 30 |
20% | WTP < 48,792 WTP 48,792 to 146,525: UPA 15 WTP3 146,525: UPA 30 | WTP < 48,616: BSC WTP 48,616 to 185,819: UPA 15 + TCS WTP3 185,819: UPA 30 |
30% | WTP < 48,792 WTP 48,792 to 80,636: UPA 15 WTP3 80,636: UPA 30 | WTP < 48,616: BSC WTP 48,616 to 89,250: UPA 15 + TCS WTP3 89,250: UPA 30 |
35% | WTP3 48,574: UPA 30 | WTP3 47,941: UPA 30 |
BSC = best supportive care; DUP = dupilumab; ICER = incremental cost-effectiveness ratio; TCS = topical corticosteroid; UPA = upadacitinib; vs. = versus; WTP = willingness to pay.
Note: The corrected price of dupilumab was used in all price-reduction scenarios.
aThe price of UPA 15 was not reduced in this analysis, as UPA 15 was cost-effective at a WTP threshold of $50,000 in the sponsor and CADTH base case; Only the price of UPA 30 was reduced. UPA and DUP were assumed to be used as monotherapy (i.e., without concomitant topical corticosteroid use) in the sponsor’s base case.
Appendix 5: Submitted BIA and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 17: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
|
Summary of Sponsor’s Budget Impact Analysis
The submitted BIA16 assessed expected budgetary impact resulting from reimbursing UPA for the treatment of adults and adolescents (12 years and older) with moderate to severe AD who are candidates for systemic therapy. The BIA was conducted from the perspective of the Canadian public drug plans over a 3-year time horizon and included drug acquisition costs, markup, and dispensing fees. The sponsor’s pan-Canadian estimates reflect the aggregated results from provincial budgets (excluding Quebec), as well as the Non-Insured Health Benefits (NIHB) Program. The analysis was performed using jurisdiction-specific values by summing up individual provincial results to obtain consolidated results. Key inputs to the BIA are documented in Table 18.
The sponsor estimates the eligible population using an epidemiologic approach. The sponsor adopted an estimated diagnosed AD prevalence of 15.8%40 among adolescents and 3.5% among adults.41 About 40.2% of adolescents40 and 52.0% of adults41 were categorized as having moderate to severe disease. The sponsor assumed that a greater proportion of adolescents with AD (91%)42 will initiate treatment for AD as compared to adults (72%), that 100% of all treated patients were followed by a specialist, and that 40% of adults and adolescents will be eligible for systemic therapy.43 The sponsor also assumed that 29% of adolescents and 49.9% of adult population will be covered by the public drug plans.44,45
The sponsor’s submission considered a reference scenario in which patients received DUP and a new drug scenario in which UPA was reimbursed. The sponsor assumed that DUP will be fully reimbursed over the time horizon and that UPA will capture market share solely from DUP. The sponsor assumed that the market share of immunosuppressants such as cyclosporine, methotrexate, azathioprine, and mycophenolate would not be affected by the introduction of UPA into the market mix. The sponsor excluded TCSs as a comparator from the market mix, assuming that the usage of TCSs would not differ between combination therapy.
The cost of UPA was based on the sponsors submitted price ($48.68 per 15 mg tablet, $74.00 per 30 mg tablet). The sponsor estimated an annual per-patient treatment cost of $|||||| for UPA, assuming that ||% of patients would receive 15 mg daily and that ||% would receive 30 mg. The sponsor included an average annual treatment cost of $24,644 for DUP (300 mg), $1,339 for cyclosporine (125 mg daily), $146 for methotrexate (14.7 mg weekly), $132 for azathioprine (99.6 mg daily), and $509 for mycophenolate (1,250 mg daily). Adherence was assumed to be ||% was assumed for all treatments. The dosing regimens were based on the product monographs1,46 and published literature,47,48 and most of the costs were based on the IQVIA DeltaPA and Ontario Drug Benefit Formulary.11
The sponsor also submitted scenario analyses estimating the budget impact from reimbursing UPA for individuals with prior exposure to a systemic IMMs. In this scenario, the sponsor assumed 15.0% of patients had failed on a systemic IMM to estimate the population size eligible to biologic therapy. The sponsor assumed that DUP is the only comparator in this scenario and captures 100% of market share in the reference scenario. The sponsor assumed that if UPA enters the market mix, it would capture a market share of ||||% in year 1, ||||% in year 2 and ||||% in year 3. The sponsor also submitted scenario analyses estimating the impact of including new treatment options (abrocitinib, baricitinib, tralokinumab) in the market mix.
The following key assumptions were made by the sponsor:
The sponsor assumed that ||% of patients will receive UPA 30 for the 3-year BIA treatment duration, while the remainder receive 15 mg for the treatment duration.
The sponsor assumed that adherence will be equivalent for treatments administered orally or by subcutaneous injection.
The sponsor assumed the reimbursement of UPA will only capture DUP’s market share (i.e., immunosuppressant therapies will be unaffected by the introduction of UPA).
Figure 3: Sponsor’s Estimation of the Size of the Eligible Population for Baseline Year

AD = atopic dermatitis; M-S = moderate to severe.
Note: General population data are sourced from Statistics Canada (Table 17 to 10 to 0005 to 01) and population growth is projected using projection scenario (M3) medium growth rate using Table 17 to 10 to 005 to 01.49
Table 18: Summary of Key Model Parameters
Parameter | Sponsor’s estimate |
|---|---|
Target population | |
Prevalence of diagnosed AD | |
Adolescents | 15.8%a |
Adults | 3.5%b |
Proportion of moderate to severe AD | |
Adolescents | 40.2%a |
Adults | 52.0%b |
Proportion of treated patients | |
Adolescents | 91%c |
Adults | 72%d |
Proportion of patients followed by a specialist | 100%d |
Proportion of uncontrolled patients c | |
Adolescents | 55% |
Adults | 38% |
Proportion of patients eligible to systemic therapy | 40%e |
Percentage eligible for public coverage f | |
Adolescents | 29.00% |
Adults | 49.9% |
Number of patients eligible for drug under review, year 1 / year 2 / year 3 | 32,542 / 32,957 / 33,370 |
Market Uptake (3 years), %, Year 1 / Year 2 / Year 3 | |
Uptake (reference scenario) DUP Cyclosporine Methotrexate Azathioprine Mycophenolate mofetil | ||||% / ||||% / ||||% |||% / |||% / |||% ||||% / ||||% / ||||% ||||% / ||||% / ||||% ||||% / ||||% / ||||% |
Uptake (new drug scenario) UPA DUP Cyclosporine Methotrexate Azathioprine Mycophenolate mofetil | |||% / |||% / ||||% ||||% / ||||% / ||||% |||% / |||% / |||% ||||% / ||||% / ||||% ||||% / ||||% / ||||% ||||% / ||||% / ||||% |
Annual Cost of treatment (per patient) | |
UPA DUP Cyclosporine Methotrexate Azathioprine Mycophenolate mofetil | $|||||| $18,815 $1,339 $146 $132 $509 |
AD = atopic dermatitis; DUP = dupilumab; UPA = upadacitinib
aSilverberg (2021)40
bBarbarot (2018)41
cInstitut national d'excellence en santé et en services sociaux report for Dupixent42
dSponsor’s budget impact analysis submission16
dIQVIA report on RINVOQ® – Atopic Dermatitis Lines of Therapy43
fSutherland (2017) and CIHI (2019)44,45
Summary of the Sponsor’s BIA Results
The sponsor estimated that net budget impact of introducing UPA for moderate to severe AD for adolescents and adults will be a cost savings of $3,934,543 in year 1, $6,338,876 in year 2, and $9,535,302 in year 3. The budget impact to the public drug plans was projected by the sponsor to be a savings of $19,808,721 over 3 years.
In a scenario analysis including patients with prior exposure to a systemic IMMs, the sponsor estimated the total savings to be $19,794,379 over 3 years (cost savings of $3,927,445 in year 1, $6,338,931 in year 2 and $9,528,003 in year 3). In a scenario analysis including new AD treatments into the market mix, the sponsor estimated cost savings of $14,793,574 over 3 years.
CADTH Appraisal of the Sponsor’s BIA
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
The BIA may not reflect the clinical use of UPA: The sponsor’s submitted budget impact assumed clinical use of UPA in adults and adolescents 12 years and older with moderate to severe AD who are candidates for systemic therapy, for use with or without topical therapy.1 The Health Canada indication is for adults and adolescents with moderate to severe AD who are contraindicated to, intolerant of, had an inadequate response to, or for whom it was otherwise medically inadvisable to receive treatment with a systemic immunosuppressant. The sponsor’s base-case analysis assumes that patients have had no prior exposure to systemic IMMs; however, the clinical expert consulted by CADTH for this review indicated that, in clinical practice, UPA will be most likely used after a trial of systemic immunosuppressants, rather than after a trial of TCSs. The clinical expert noted that the expected place of UPA in therapy is similar to DUP, which is recommended for patients who have failed to respond to immunosuppressants and/or phototherapy (where available). The drug plan input received for this review noted that DUP is not currently available in all jurisdictions but successful negotiations with pCPA have been concluded. This similarity of place in therapy was confirmed by the sponsor through communication with CADTH.
The sponsor submitted a scenario analysis estimating the expected budgetary impact of reimbursing UPA for patients with prior exposure to a systemic IMMs. In this scenario, the sponsor assumed 15% of patients had failed on a systemic IMMs. Further, UPA has a market share of ||||% in year 1, ||||% in year 2 and ||||% in year 3.
In reanalysis, CADTH focused its review on the sponsor’s submitted scenario analysis including patients with prior exposure to a systemic IMMs, as indicated by the clinical expert consulted by CADTH and in-line with the revised Health Canada indication.
Additionally, the sponsor’s base case assumes that ||||% of patients will receive UPA 30, while others start on UPA 15. The product monograph for UPA states patients will initiate treatment on the 15 mg UPA dose, with some patients potentially increasing to 30 mg depending on clinical response assessed using EASI 75. The sponsor’s model did not explicitly consider the possibility of dose escalation to UPA 30 based on clinical response, instead assuming in their base case that 66% of patients would start treatment on the 15 mg dose.
In CADTH reanalysis, 100% of patients were assumed to receive UPA 15 for the treatment duration because there is insufficient evidence available to explore dose escalation (i.e., number of patients escalated to higher dose, time to dose escalation, and time on higher 30 mg dose are unknown).
The relative efficacy of both UPA 15 and UPA 30 to DUP is uncertain, and indirect treatment comparisons suggest that treatment with UPA 15 may be equally effective or superior to DUP monotherapy. The sponsor’s model did not explicitly consider the possibility of dose reduction from UPA 30 to UPA 15.
CADTH also conducted a scenario analysis in which 100% of patients were assumed to receive UPA 30 for the treatment duration.
The number of patients covered by public drug plans is underestimated: The sponsor estimated the proportion of patients eligible for public drug plan coverage by use of the number of patients enrolled in public plans for each jurisdiction.45 It is more appropriate to use the proportion of patients eligible, rather than enrolled, as the market size will be determined by all eligible for public coverage, and the BIA should consider all patients eligible regardless of whether they are presently enrolled. Should UPA be reimbursed by public plans, it is assumed that all eligible patients for this treatment would enrol for public coverage.
In CADTH reanalysis, the proportion of patients eligible for public drug plan coverage was used to determine the market size for UPA.45
The estimated budget impact is sensitive to market growth and market share distribution of AD treatments: The sponsor assumed an annual growth rate of ||||% in market share of AD treatments. The total market share of UPA was assumed to be ||||%, ||||%, and |||||% in year 1, 2, and 3, respectively. The clinical expert consulted by CADTH for this review indicated that the market share of AD treatments will be dependent on relative efficacy of treatments and that the clinical efficacy of UPA would need to be established before physicians are comfortable prescribing UPA. The sponsor’s estimates of market growth of UPA and DUP may be larger than what would be observed in clinical practice.
CADTH explored the impact of this assumption in scenario analysis, assuming a 25% reduction in market growth of novel AD treatments (UPA and DUP). CADTH assumed market growth of new AD treatments by 21.8% and that UPA would capture 3%, 5%, and 8% of total market share in year 1, 2, and 3, respectively.
Adherence across treatments and age groups was inappropriately assumed to be the same: The sponsor assumed that treatment adherence will be the same for oral treatment and subcutaneous injections. Treatment adherence is a major driver of BIA results. The mode of administration for DUP is subcutaneous injections, while UPA and the included systemic immunosuppressants are administered orally. The clinical expert consulted for this review indicated that higher adherence is expected for treatments administered by subcutaneous injection versus those administered orally. The sponsor used treatment adherence, unit price, and dosing regimen to estimate treatment acquisition costs. By assuming the same treatment adherence across treatments, the sponsor underestimates the treatment cost of DUP.
In CADTH reanalysis, adherence was assumed to be 70% for oral treatment with UPA and immunosuppressants, and 90% for subcutaneous treatment with DUP based on feedback from clinical expert.
CADTH also performed a scenario analysis assuming 68.7% of adherence for DUP50 and 62.7% of adherence for UPA51 based on published literature.
Assuming that some parameter values will be same for DUP and UPA is inappropriate: The sponsor obtained some of the parameters used in the BIA from the Institut national d'excellence en santé et en services sociaux report for DUP42 and the IQVIA report on DUP claims from the public drug plan database.43 The sponsor applied the estimates and assumptions made on patient population treated with DUP to the entire population, including those on UPA. The clinical expert consulted by CADTH for this review indicated that the data on DUP is biased toward adolescents, and the proportion of adolescents treated and uncontrolled on topical therapy will be lower for patients treated with UPA.
CADTH explored the impact of this assumption in a scenario analysis, by assuming an arbitrary 25% reduction in the proportion of adolescents treated and uncontrolled on traditional therapy.
Additional limitations were identified, but were not considered to be key limitations. These limitations include: same adherence for responders and nonresponders to treatment, excluding costs of topical therapy, assuming no differential use of topical therapy among different therapies, and excluding general discontinuation rates on both monotherapy and combination therapy in the BIA.
CADTH Reanalyses of the BIA
Table 19: CADTH Revisions to the Submitted BIA
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Revisions to sponsor’s base case | ||
1. Update eligibility criteria. | Sponsor’s base case involving adults and adolescents with moderate to severe AD who are candidates for systemic therapy. | Sponsor’s scenario analysis involving adults and adolescents with moderate to severe AD who are contraindicated to, intolerant of, had an inadequate response to, or for whom it was otherwise medically inadvisable to receive treatment with a systemic immunosuppressant. |
Corrections to sponsor’s analysis | ||
1. Remove NIHB beneficiaries from total population estimates obtained from Statistics Canada. | NIHB beneficiaries were not subtracted from respective provinces. | Subtracted NIHB beneficiaries from Canadian population estimates for each age group (adults vs adolescents) and jurisdiction. |
2. Update DUP price. | $959.5950 per pre-filled syringe | $978.7000 per pre-filled syringe18 |
Changes to derive the CADTH base casea | ||
1. Patients receiving 15 mg vs. 30 mg UPA | Sponsor assumed that ||||% of patients will receive 15 mg UPA and that ||||% will receive 30 mg UPA | Based on product monograph, CADTH assumed that 100% of patients receive 15 mg UPA |
2. Percentage of patients covered by public drug plans | Determined by the percentage of patients enrolled (a weighted average of ||||% for individuals aged 12 to 17 and ||||% for individuals aged 18+, using Canadian population estimates for year 202144,45 | Determined by the percentage of patients eligible for enrollment45 (a weighted average of 64.5% for individuals aged 12 to 17 and 76.0% for individuals aged 18+, using Canadian population estimates for year 2021)44,45 |
3. Treatment adherence | The sponsor assumed ||||% adherence for both oral and subcutaneous injection treatments. | Based on clinical expert’s feedback, 70% adherence was assumed for oral treatment and 90% was assumed for subcutaneous injections. |
CADTH base case | Reanalysis 1 + 2 + 3 | |
BIA = budget impact analysis; DUP = dupilumab, NIHB = Non-Insured Health Benefits, UPA = upadacitinib.
aChanges to derive the CADTH base case were made using the sponsor’s submitted scenario analysis (adopted as per “Revisions to the sponsor’s base case”) assuming that UPA will be used by patients who are refractory to or ineligible for systemic immunosuppressants.
The results of the CADTH stepwise reanalysis are presented in summary format in Table 20 and a more detailed breakdown is presented in Table 21.
In CADTH reanalyses, UPA was assumed to be used by patients with prior systemic IMM exposure. CADTH corrected the sponsor’s scenario analysis for this population by removing the number of NIHB beneficiaries from the Canadian population estimates and updating the unit cost of DUP using the price listed on EAP. CADTH revised the sponsor’s corrected base case by assuming that all patients (100%) will receive 15 mg UPA based on product monograph, using the number of patients eligible for public coverage, rather than enrolled, to estimate the percentage of patients who would be covered in each jurisdiction, and adopting an adherence of 90% for treatment with subcutaneous injection.
Applying these changes increased the total 3-year budget impact of reimbursing UPA for the reimbursement request population to cost savings of $121,473,004. The budget impact was highly sensitive to all adjustments in the CADTH reanalysis.
Table 20: Summary of the CADTH Reanalyses of the BIA
Stepped analysis | Three-year total ($) |
|---|---|
Submitted base case, as provided (population: post-topicals) | −19,808,721 |
Submitted scenario analysis, as provided (population: patients refractory or ineligible for systemic IMMs) | −19,794,379 |
CADTH correction 1 | −19,199,946 |
CADTH correction 2 | −22,185,764 |
Sponsor’s scenario analysis, corrected | −21,519,619 |
Stepped analysis | |
CADTH reanalysis 1 | -$36,153,189 |
CADTH reanalysis 2 | −37,084,568 |
CADTH reanalysis 3 | −55,773,322 |
CADTH base case (1 + 2 + 3) | -$121,473,004 |
AD = atopic dermatitis, BIA = budget impact analysis, DUP = dupilumab, UPA = upadacitinib
Note: CADTH reanalyses are carried on sponsor’s corrected base case. CADTH scenario analysis are carried on CADTH base case.
aThe price of UPA 15 was not reduced in this scenario, as UPA 15 was considered cost-effective at a willingness-to-pay threshold of $50,000 at the sponsor-submitted price in the CADTH base case.
bBased on the price reduction required for DUP to be cost-effective in the 2020 CADTH review.12
cBased on the submitted price of DUP in the 2020 CADTH review.12
CADTH also conducted additional scenario analyses to address remaining uncertainty, using the CADTH base case. Results are provided in Table 21.
1. Assuming 25% reduction in market growth for novel AD treatments and market share distribution of UPA.
2. Assuming 25% reduction in the proportion of adolescents treated (68.25%) and adolescents uncontrolled on traditional therapy (41.25%).
3. Assuming 62.7% of adherence for UPA51 and 68.7% of adherence for DUP.50
4. All patients receiving UPA receive the 30 mg dose in all years (proportion of patients receiving UPA 15 = 0%).
5. DUP price is $959.9350 per pre-filled syringe, to match sponsor’s submitted price in the CADTH review of DUP.12
6. DUP price is $959.9350 per pre-filled syringe and price reduction for DUP of 54%, to match the estimated price reduction in the CADTH review of DUP.12
Table 21: Detailed Breakdown of the CADTH Reanalyses of the BIA
Stepped analysis | Scenario | Year 0 (current situation), $ | Year 1, $ | Year 2, $ | Year 3, $ | Three-year total, $ |
|---|---|---|---|---|---|---|
Submitted base case (population: post-topicals) | Reference | 91,009,796 | 115,595,260 | 147,636,761 | 189,445,855 | 452,677,876 |
New drug | 91,009,796 | 111,660,717 | 141,297,885 | 179,910,553 | 432,869,154 | |
Budget impact | 0 | −3,934,543 | −6,338,876 | −9,535,302 | −19,808,721 | |
Submitted scenario analysis (population: patients refractory or ineligible for systemic IMMs) | Reference | 81,066,667 | 106,038,403 | 138,634,998 | 181,215,942 | 425,889,343 |
New drug | 81,066,667 | 102,110,959 | 132,296,067 | 171,687,938 | 406,094,964 | |
Budget impact | 0 | −3,927,445 | −6,338,931 | −9,528,003 | −19,794,379 | |
Submitted scenario analysis, corrected (population: patients refractory or ineligible for systemic IMMs) | Reference | 80,153,955 | 104,859,962 | 137,111,874 | 179,246,250 | 421,218,086 |
New drug | 80,153,955 | 100,590,882 | 130,220,669 | 168,886,916 | 399,698,467 | |
Budget impact | 0 | −4,269,080 | −6,891,205 | −10,359,334 | −21,519,619 | |
CADTH base case (population: patients refractory or ineligible for systemic IMMs) | Reference | 177,411,981 | 232,578,488 | 304,654,807 | 398,871,437 | 936,104,732 |
New drug | 177,411,981 | 208,532,051 | 265,769,905 | 340,329,772 | 814,631,728 | |
Budget impact | 0 | −24,046,437 | −38,884,902 | −58,541,665 | −121,473,004 | |
CADTH scenario analysis: 25% reduction in market growth of novel AD treatment and market share of UPA | Reference | 177,411,981 | 219,472,303 | 271,286,630 | 335,168,712 | 825,927,645 |
New drug | 177,411,981 | 219,472,303 | 271,286,630 | 335,168,712 | 825,927,645 | |
Budget impact | 0 | −17,018,535 | −25,969,442 | −36,894,095 | −79,882,073 | |
CADTH scenario analysis: 25% reduction in proportion of adolescents treated and uncontrolled | Reference | 146,786,187 | 192,505,068 | 252,236,581 | 330,307,893 | 775,049,542 |
New drug | 146,786,187 | 172,603,028 | 220,044,093 | 281,832,173 | 674,479,294 | |
Budget impact | 0 | −19,902,040 | −32,192,488 | −48,475,720 | −100,570,248 | |
CADTH scenario analysis: 62.7% adherence for UPA and 68.7% adherence for DUP | Reference | 135,424,479 | 177,534,913 | 232,553,169 | 304,471,863 | 714,559,945 |
New drug | 135,424,479 | 162,979,239 | 209,015,945 | 269,036,837 | 641,032,021 | |
Budget impact | 0 | −14,555,674 | −23,537,224 | −35,435,027 | −73,527,924 | |
CADTH scenario analysis: 100% on UPA 30 | Reference | 177,411,981 | 232,578,488 | 304,654,807 | 398,871,437 | 936,104,732 |
New drug | 177,411,981 | 223,187,739 | 289,467,998 | 376,005,834 | 888,661,571 | |
Budget impact | 0 | −9,390,749 | −15,186,809 | −22,865,603 | −47,443,161 | |
CADTH scenario analysis: DUP price is $959.9350 | Reference | 174,056,843 | 228,180,475 | 298,894,354 | 391,330,116 | 918,404,944 |
New drug | 174,056,843 | 228,180,475 | 298,894,354 | 391,330,116 | 918,404,944 | |
Budget impact | 0 | −23,048,949 | −37,271,975 | −56,113,500 | −116,434,424 | |
CADTH scenario analysis: Price reduction by 54% for DUPa | Reference | 81,620,334 | 106,998,886 | 140,156,126 | 183,498,319 | 430,653,331 |
New drug | 81,620,334 | 111,434,421 | 147,330,855 | 194,302,797 | 453,068,073 | |
Budget impact | 0 | 4,435,534 | 7,174,729 | 10,804,478 | 22,414,742 |
AD = atopic dermatitis, BIA = budget impact analysis, DUP = dupilumab, UPA = upadacitinib.
aReduction from a base price of $959.9350 based on the 2020 CADTH assessment of DUP.12
Note: The scenario analyses are carried out on CADTH base case.
Stakeholder Input
Patient Group Input
Canadian Skin Patient Alliance (CSPA) & Eczéma Québec
About The Canadian Skin Patient Alliance and Eczéma Québec
The Canadian Skin Patient Alliance (CSPA) is a national non-profit organization dedicated to advocating, educating, and supporting Canadians impacted by skin, hair, and nail disorders. Our mission is to promote skin health and improve the quality of life of Canadians living with skin disorders through advocacy, education, and awareness, supporting research and working with our Affiliate Member organizations that serve specific patient communities such as eczema, melanoma, and psoriasis.
Eczéma Québec was created as a branch of the McGill University Hospital Network Center of Excellence for Atopic Dermatitis (COE AD), Eczéma Québec is a Patient Advisory Committee (PAC) and registered non-profit organization. It established a network of adult AD patients and healthcare practitioners in the field of AD (encompassing specialist clinician dermatologists, GPs, nurse practitioners, and more), with a goal of building resources based on international best-practice guidelines. Eczéma Québec works with the COE AD to iterate on knowledge translation tools featuring validated information to improve education and experience of care and promote awareness and the health outcomes of this population.
Information Gathering
Eczéma Québec and CSPA developed and circulated a web-based survey using the Survey Monkey platform in English and French about experiences with Janus kinase (JAK) inhibitors including upadacitinib and abrocitinib. This survey was disseminated through both organisation’s online channels (newsletter, social media, website). Of the 56 respondents to the survey, 38 filled out the French survey and 18 responded to the English one. We received responses from patients and caregivers across Canada: Quebec (91%), Ontario (3.6%), New-Brunswick (3.6%), Manitoba (1.8%). Most participants were patients (43, 76.8%), or the parent of a patient (7, 12.5%). Most of the surveyed population reported that their gender assigned at birth was female (45, 80.4% v 19.6% for males). Of these respondents, 11 shared that they identify as male, 43 as female, and 2 as non-binary. Although 2 responses were regarding patients under 12, most respondents were adults: 18-24 (2, 3.6%), 25-34 (13, 23.2%), 35-44 (15, 26.8%), 45-54 (12, 21.4%), 55-64 (3, 5.4%), 65+ (9, 16.1%).
When asked about how long they had experienced symptoms of AD, nearly half (48.5% of respondents) had symptoms for more than 10 years and 4 (12.1%) indicated they were experiencing symptoms for less than a year, 5 (15.2%) lived with symptoms for 1-2 years, 4 (12.1%) suffered from symptoms for 3-5 years, and 4 (12.1%) for 5-10 years. These patients and caregivers also reported on the severity of the condition: 3 (9.1%) reported suffering from a mild form of the disease, 16 (48.5%) from a moderate form, and 14 (42.4%) of respondents were living with a severe form of the disease. None of the people who took part in the survey had direct experience with the drug under review (upadacitinib) nor any other biologics (i.e., dupilumab) to treat AD. The survey results reflect responses received between March 29 and April 23, 2021.
They also held discussions with atopic dermatitis (AD) patients and caregivers on themes that emerged from the surveys to inform this submission. To reach these individuals, the CSPA shared a request for participants on its social media channels (Facebook, Instagram), in its newsletter, and via email with its Medical Advisory Board members and other dermatologists in its network. Eczéma Québec also shared information about these discussions through its social channels. Eczéma Québec disseminated the survey through its monthly newsletter and reached out to its patient advisory committee (PAC) members by email. Eczéma Québec held 30-60 minutes individual interviews with three adult patients who lived with moderate to severe AD. The Eczéma Québec Co-Director has also lived experience with the disease and her experience is included in this submission. The interviews were conducted online using the Teams platform and the sessions were recorded.
Additionally, the information gathered in this submission also includes material from the current literature on guidelines and management of AD. Reference material is provided in the appendices.
Disease Experience
“When I sit on a black surface or like a black couch, you can see all those skin flakes are all over the place.”
Atopic dermatitis (AD) is not only the most common chronic inflammatory skin disease, but it also ranks highest among all skin disorders as a cause of lost disability-adjusted life-years in patients worldwide. The severity of AD correlates with impact on health-related quality of life (HRQL) as well as lost productivity at school and burden on health systems. Severity of atopic dermatitis (AD) correlates with impact on HRQoL, work productivity, and burden on health systems (Maintz, Bieber, Bissonnette, & Jack, 2021).
“As I grew up, my disease got worse and worse, until it got to the point where I frequently had to miss school, and had trouble sleeping at night. On days when I could attend school, I was teased because of the way my skin looked, and people stared or made comments on my appearance. I was not able to skip gym class even though the sensation of burning from sweat on my lesions would make me cry in front of my classmates. I felt as though no one understood what it was like living in my skin.”
AD can spread across the whole body, creating sensations of burning. Uncontrollable itch is also associated with the skin irritation caused by the disease.
“All my life, I have struggled with itch. The constant, debilitating itch that would never leave me alone.”
When asked about the areas of the body where they commonly experience AD, respondents reported that the most prevalent areas were the backs of their hands (63.64%) and their thighs and/or legs (54.55%). Patients also reported they would get AD on their neck (51.52%), the inside of their arms and/or their elbow folds (51.52%), the outside of their arms and/or the exterior part of their elbows (51.52%), their scalp (48.48%), their face (45.45%), their ears (45.45%), their abdomen (45.45%), the area around their eyes (39.39%), their breasts, under breasts and/or nipples (39.39%), their back (39.39%), the backs of their knees (36.36%), the top of their feet (30.30%), the palms of their hands (30.30%), their groin area and/or genitalia (24.24%), their buttocks (21.21%), the front of their knees (21.21%), the soles of their feet (21.21%), and their armpits (18.18%).
“I would take off my bra and the whole skin on my nipple would come off and get stuck to the fabric. My wounds would stick to anything I was wearing. When undressing, my skin would completely rip and bleed. The scabs on my back would get stuck on my bed sheets at night. And I would get infections from the textile fibers that would get in my scabs. It was horrible. During the day, my lesions would ooze and because of my clothing they would take forever to heal. Even though I am now controlled, and I use a treatment that manages most of my symptoms, my body is covered in brownish scars. To this day I never wear shorts or a bathing suit, and I still get comments on the way my skin looks when I do expose my scars.”
“It felt like my whole body was burning, especially on my neck and chest. […] Aside from that, the itchiness is uncontrollable and would wake me up at night.”
Furthermore, 100% of the respondents experienced itching because of their condition. We surveyed them on the other symptoms they suffered from: redness of the skin (87.88%), repeated rashes (84.85%), frequent scratching (84.85%), cracked skin (84.85%), dry and rough skin (78.79%), disrupted sleep (75.76%), bleeding (69.70%), flaking of the skin (69.70%), pain (69.70%), thickening of the skin (60.61%), oozing (48.48%), swelling (42.42%), lichenification (39.39%), and blistering (36.36%).
Survey respondents were asked to describe how their symptoms impact their ability to participate in their day-to-day life. One commented that it was an “ordeal”, others reported that the impact the disease had on the quality of their sleep was “important”:
“I have trouble falling asleep, I wake up to scratch. I am tired. I don't take Atarax because it prevents me from being alert for my children in the morning” (translation).
“I can’t sleep when [the] itch begins, and it just gets worse. [O]nce I start to scratch it is almost impossible to stop the [itch-scratch] cycle. Cold, like ice wrapped in a towel, does help to [calm] the itch but it’s not [practical].”
"My sleep is disturbed. I am disturbed in my daily life by uncontrollable itching for which I have to stop what I do (work, [driving a] car) to scratch myself.”
“I wake up at night several times scratching myself.” (Translation)
“When my feet flare up, I must not move the sheets otherwise I wake up constantly. This is sometimes also the case with my hands but happens less often.” (Translation)
AD also has significant impacts in terms of the psychosocial burden of symptoms:
“If flaring, [it is] hard to do some things physically and [I’m] self-conscious so tend to stay home.” (Translation)
"I've cut myself off from others and I'm having a lot of trouble engaging in some social activity. Everything is hard! I am constantly disfigured, I have red skin, patches and dead skin falling. People can see that I’m not feeling good, and I don't feel good about myself. I don't want my husband to touch me and even my boy anymore because I have infections all the time. It is not a life to live in constant suffering, to have to constantly beat this and never to know in what skin state we are going to wake up in.” (Translation)
A respondent even shared experiencing: “Work stoppage, Repeated Depression, Lack of Sleep.” They mentioned: “It’s hard to participate in social or seasonal activities.”
Moreover, some of them shared some challenges they face because of the pain they are experiencing. For example, with respect to their clothing:
“The symptoms come and go between seasons. My biggest outbreaks are on my right heel and it makes wearing shoes (even the comfiest shoes I own) very uncomfortable.”
In the average month, most (72.8%) of the patients responding to this survey would not have to miss days of work or school because of their condition. However, six patients (18.2%) would miss 1-2 days per month and two (6.1%) would miss over 7 days each month to care for their condition.
“It always depends. I can be good for a couple of months, but when my condition worsens, it feels like all goes wrong at once. My eyelids become super itchy, and I can get an infection from scratching. So, in order to manage some flares, it can require that I go see multiple doctors. I lose sleep because of the itch and it all becomes a vicious cycle.” (Translation)
Over half of the respondents (57.6%) reported that their condition posed a challenge in keeping their homes and/or possessions clean and nearly two-thirds (63.6%) reported that they had to replace clothing items that would get ruined because of their AD.
We asked participant about the level of pain and itch they were experiencing on an average day. When asked about pain, patients reported an average pain level of 2/10, and commented that: “[I’m] not experiencing that much pain but [instead] too much itching.”
“I don’t consider it as pain but as extreme discomfort. It’s the swelling and itching that makes it uncomfortable for me.” However, only 8.3% were satisfied by how well their pain was managed on a day- to-day basis, whereas over half of the respondents (62.5%) felt that their pain was not adequately managed.
On the other hand, the average level of itch experienced reported by respondents was of 5.8/10. And, while 34.4% were satisfied by the management of their itch, 40.6% of respondents expressed that their itch was poorly controlled, an additional 25.0% shared that their itch was ‘very poorly’ managed.
Six respondents across the English and French surveys responded to questions about their experiences as caregivers or parents of patients with AD. They had been caring for someone living with AD for different lengths of time: more than 10 years (2), for 5-10 years (2), for 3-5 years (1) and 1-2 years (1). Half of them (3) indicated that they missed work or school at 1-2 days in the average month, while 2 did not miss any days and one missed 3-6 days.
Caregiver respondents were how caring for someone with atopic dermatitis (eczema) impacts their ability to participate in day-to-day life and shared that AD “is difficult and embarrassing in his everyday life” (translation), their loved one “can’t go to the pool (too harsh)”, and that caring for AD was “time consuming” (translation) because of creams and topicals needed after showering.
They also shared the emotional drain caring for AD caused for them as caregivers:
“A lot of anxiety at a young age because of the limitations caused by the disease... itching, chapping, pain... a lot of anxiety experienced by the child which has an emotional impact on the parents who feel powerless.” (Translation)
“It is a constant vigilance of signs and symptoms, possible triggers and other irritants. As soon as the AD worsens, we start the most appropriate care.” (Translation)
When asked specifically about the impacts of “caring for someone with atopic dermatitis (eczema) [on] your relationships with them, other people in your family or social networks”, caregivers responded that: “It sometimes causes friction with my family and friends who give advice on what to do or not to do (e.g., cutting out dairy products according to them)” (translation). One respondent experienced “Less patience, feeling of helplessness and incomprehension. People around them want to help but do not distinguish between dry skin and AD” (translation) and that there were “[m]any missed activities, unplanned trips, many decisions made based on the condition of the sick child to the detriment of others” (translation).
Some caregivers reported feeling that their loved one’s itch was poorly controlled (4, 66.7%) while others felt that the itch was well controlled (2, 33.3%). Their pain was found to be well-controlled for most (4, 66.7%) and poorly controlled for others (2, 33.3%).
Experiences With Currently Available Treatments
Discussing the effectiveness of currently available drugs and treatments for AD, patients told us that most treatments were not considered effective to manage their condition. We invited respondents to rank different treatments for AD on a scale ranging from ‘very effective’ to ‘very ineffective’. Respondents could also indicate that they have not had experience with a treatment by selecting ‘N/A’. Although most respondents did not have specific experience with targeted treatments for atopic dermatitis (only 14.3% of respondents to the survey had experience with dupilumab, 14.3% had experience with topical calcineurin inhibitors, and 28.6% had experience with cyclosporine), very few of the other treatment options stood out as very effective or somewhat effective, with the treatment perceived as the most generally effective being topical corticosteroids or TCS (66.7%), followed by the use of moisturizer, emollient and/or ointments (47.8%), and topical PDE4 inhibitors (30.0%).
Thinking of all the treatments they had tried for their AD, we asked respondents how well the treatments managed their symptoms. Most patients expressed their dissatisfaction with the treatment options available to them and how these treatments addressed the most important symptom of their disease. On respondent told us: “NOTHING WORKS”, another respondent shared that: “Nothing has stopped the itch.” Another source of frustration for these participants was that they didn’t see these treatments as long-term options but rather “temporary” (translation): we heard that existing treatments would bring them "[m]omentary relief, but doesn’t work in the long term" and that “[t]he effects are only temporary.” Some of the respondents also mentioned the side effects associated with certain treatments: “Alright for a while but had side [e]ffects so discontinued.”, "I started DUPIXENT a week ago. It's working well so far. It's unbelievable. I, however, have an intense conjunctivitis in my eyes (side effect of treatment)” (translation). Others also mentioned the hurdles of having to go through a lot of trial and error before finding the appropriate treatment for them: “I haven’t found the right treatment for me yet. I use diprosone and it doesn’t do much for me unless I am in a major outbreak.” “A lot of trial and error. Now, it's better after looking for 30 years” (translation). In the absence of an effective treatment, the trial and error approach to treatment hits their pocketbooks: “The costs associated with the trial-and-error nature of treatment regimens is very frustrating.”
Furthermore, when asked if the cost of medication, travel to and from appointments, or time involved to receive medication limited their use, or their doctor's ability to prescribe a particular treatment option, some respondents reported on the financial impact of their disease: “Yes. Because of the cost of injections, the dermatologist wants to see if my condition gets worse before using it”. Another respondent said: “I do not drive, and dermatologists are only listening to one problem, so if I have more than one concern it means repeated trips which is costly and [it’s] not good to miss a lot of work for treatment. Some treatments are very expensive”.
“I had to suspend the use of Protopic until I was on my spouse's insurance. I also needed the Freedom Dupixent program AND my spouse's private insurance. Again, I have a deadline for my [when coverage for my] prescription [ends]. Who can pay $2,500 a month for ONE drug? Not to mention that I must pay for other medications (antidepressants for example).” (Translation)
In addition to experiencing debilitating and life altering symptoms from their conditions, patients and caregivers alike report often having difficulties accessing timely and appropriate care when they experience flares of their disease: “Accessibility to a competent health professional with regard to eczema is one of the biggest challenges.” (Translation)
Skin patients often have to try multiple treatment options to find the right one for their circumstances, and these circumstances can change over time. It is important that AD patients have multiple treatment options available for their specific circumstances: “I think that my problem is that I no longer have treatment options left.” (Translation)
Improved Outcomes
Survey respondents were asked whether they agreed or disagreed with a statement that “it’s important for me that a new treatment for AD” have specific outcomes: manages the itch (28/28 strongly or somewhat agreed), reduces flares (26/27), manages the redness and inflammation (26/27), gives fast results (26/28), addresses lichenification (thickening) of the skin (25/27), is easy to use (25/28), is covered by insurance / is affordable (23/27), allows me to stop using topical treatments (23/28), and does not require injections (by myself or someone else) (19/28). No respondent strongly or somewhat disagreed that these outcomes were important in a new treatment.
Table 1: Survey Responses on Improved Outcomes
Outcome | Strongly agree | Somewhat agree | Neither agree nor disagree |
|---|---|---|---|
Manages the itch | 27/28 | 1/28 | 0 |
Reduces flares | 24/27 | 2/27 | 1/27 |
Manages the redness and inflammation | 23/27 | 3/27 | 1/27 |
Gives fast results | 19/28 | 7/28 | 2/28 |
Addresses lichenification (thickening) of the skin | 18/27 | 7/27 | 2/27 |
Is easy to use | 20/28 | 5/28 | 3/28 |
Is covered by insurance / is affordable | 19/27 | 4/27 | 4/27 |
Allows me to stop using topical treatments | 19/28 | 4/28 | 5/28 |
Does not require injections (by myself or someone else) | 16/28 | 3/28 | 9/28 |
Comments from respondents on other outcomes of importance included: “Has few side effects. Is safe” (translation). Another patient commented: “I expect to get patches, I just want to stop scratching myself to death on 99% of my body” (translation). Although reducing the itch was a prominent theme in the responses from patients, they also expressed that they would like a new medication to result in manageable AD in the summer without the use of hydrocortisone, “to improve the appearance of [their] hands and eyes” (translation), “less apparent eczema” and “have freedom” (translation), and to provide pain relief. One commented that they would like to see a reduction from 85% of their body covered in AD to 10-15% with a new treatment.
Survey respondents were specifically asked about the preferred mode of administration: 67.9% (19) preferred daily pills taken by mouth, 50% (14) preferred daily topical medications, and 42.9% (9) preferred injections every other week they could do themselves or with help. On the subject of topical medications, one patient commented: “If you knew how many layers of creams, I had to slather on my body with help. It was just inhumane” (translation).
When asked about the balance of treatment outcomes and “potentially serious side effects”, patients were generally unwilling to accept serious side effects in a new treatment. However, patients also commented that they are living with serious impacts of their disease - for example, “[t]he inability to sleep due to the symptoms of eczema is a serious problem in the medium and long term, it must serve as a comparison to the side effects of the drug.” One shared “I am willing to try anything” and another commented: “How serious [are the side effects]? I would do it if I was guaranteed that this disease is over” (translation).
“I would really like to have a treatment that helps me with my skin without giving me another skin problem. I would also like it to be pills or injections because I can't stand the daily creaming but at the same time if the treatment would give me my life back, I would be happy.” (Translation)
Overall, “expectations for a new medication” expressed by survey respondents included “that it heals completely”, “that the medication works quickly and over a long period of time”, “to find a way to stabilize eczema without using high dose steroids”, “to be able to sit on my sofa or lean my arms on a desk without itching”, that it “prevents recurrence instead of just managing the manifestations of the disease”, and that it lets patients “[s]top suffering” (translations).
Experience With Drug Under Review
We did not hear from any patient or caregivers who had direct experience with the drug under review.
Companion Diagnostic Test
N/A
Anything Else?
Skin disorders are often diminished, disregarded, and dismissed. They are more than “just a rash”. Skin disorders often reflect imbalances in inflammatory and other systems, and can be caused by allergens, viruses, cancer, bacteria, fungi, genetics, wounds, hormones, and other disorders, and can cause devastating impacts. Many of the medicines available to treat skin disorders were initially developed for other diseases or organ systems and have become part of the skin treatment toolbox (e.g., methotrexate developed for cancer, cyclosporine developed for preventing organ rejection, etc.) (Wikipedia, 2021).
The development of more tailored treatment options for skin disorders on the horizon provides new hope that treatments will address the underlying pathology of skin disorders, rather than only treating the symptoms. Skin patients deserve to be treated with respect and dignity by the health system, which includes its embrace of new and tailored treatment options.
As of February 5, 2021, 992 clinical trials were registered investigational products for AD (clinicaltrials.gov), reflecting the fast pace of translational research and clinical development in this field. This highlights the immense gap in treatment options for this disease and the significant unmet needs of this patient population (Maintz et al. 2021).
Patient Group Conflict of Interest Declaration for Canadian Skin Patient Alliance (CSPA) & Eczéma Québec
To maintain the objectivity and credibility of the CADTH reimbursement review process, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This Patient Group Conflict of Interest Declaration is required for participation. Declarations made do not negate or preclude the use of the patient group input. CADTH may contact your group with further questions, as needed.
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
No. The submission was prepared by CSPA and Eczéma Québec staff without help from outside the organizations.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
Organization staff reached out to clinical trial investigators to share the survey link with them and ask that they share it with the clinical trial participants at their sites. The contact information (name, email, phone number) was shared with CSPA and Eczéma Québec by the manufacturer, AbbVie Canada, and organization staff reached out directly to the clinical trial investigators.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
Table 2: Conflict of Interest Declaration for Canadian Skin Patient Alliance
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Sanofi Canada | — | — | — | X |
Pfizer Canada | — | — | — | X |
Abbvie Canada | — | — | — | X |
LEO Pharma Canada | — | — | X | — |
I hereby certify that I have the authority to disclose all relevant information with respect to any matter involving this patient group with a company, organization, or entity that may place this patient group in a real, potential, or perceived conflict of interest situation.
Patient Group: Canadian Skin Patient Alliance
Date: May 6, 2021
Table 3: Conflict of Interest Declaration for Eczéma Québec
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Sanofi Genzyme | — | — | X | — |
McGill COE-AD and its pharmaceutical industry sponsors (Pfizer Canada, AbbVie Canada, LEO Pharma Canada, Novartis, Sanofi Genzyme, Eli Lilly) | — | — | — | X |
McGill COE-AD and its dermo-cosmetic industry sponsors (CUTIMed, Beirsdorf, L’Oréal) | — | — | X | — |
I hereby certify that I have the authority to disclose all relevant information with respect to any matter involving this patient group with a company, organization, or entity that may place this patient group in a real, potential, or perceived conflict of interest situation.
Patient Group: Eczéma Québec
Date: May 6, 2021
References
Guttman-Yassky, E., Thaçi, D., Pangan, A. L., Hong, H. C., Papp, K. A., Reich, K., … Silverberg, J. I. (2020). Upadacitinib in adults with moderate to severe atopic dermatitis: 16-week results from a randomized, placebo-controlled trial. Journal of Allergy and Clinical Immunology, 145(3), 877–884. https://doi.org/10.1016/j.jaci.2019.11.025 (Guttman-Yassky et al., 2020)
Heratizadeh, A., Werfel, T., Wollenberg, A., Abraham, S., Plank-Habibi, S., Schnopp, C., … Schmitt, J. (2017). Effects of structured patient education in adults with atopic dermatitis: Multicenter randomized controlled trial. Journal of Allergy and Clinical Immunology, 140(3), 845-853.e3. https://doi.org/10.1016/j.jaci.2017.01.029 (Heratizadeh et al., 2017)
Maintz, L., Bieber, T., Bissonnette, R., & Jack, C. (2021). Measuring Atopic Dermatitis Disease Severity: The Potential for Electronic Tools to Benefit Clinical Care. The Journal of Allergy and Clinical Immunology: In Practice, 9(4), 1473-1486.e2. https://doi.org/10.1016/j.jaip.2021.02.027 (Maintz et al. 2021)
Nezamololama, N., Fieldhouse, K., Metzger, K., & Gooderham, M. (2020). Emerging systemic JAK inhibitors in the treatment of atopic dermatitis: a review of abrocitinib, baricitinib, and upadacitinib. Drugs in Context, 9, 1–7. https://doi.org/10.7573/dic.2020-8-5 (Nezamololama, Fieldhouse, Metzger, & Gooderham, 2020)
Nguyen, J., Chen, J. K., Honari, G., Pol-Rodriguez, M., Ko, J. M., & Chiou, A. S. (2021). Bridging to a selective Janus kinase 1 inhibitor in severe atopic dermatitis: An instructive case with upadacitinib. JAAD Case Reports, 7, 65–67. https://doi.org/10.1016/j.jdcr.2020.10.023 (Nguyen et al., 2021)
UpToDate. (2021). Retrieved April 28, 2021, from Uptodate.com website: https://www.uptodate.com/contents/treatment-of-atopic-dermatitis-eczema?search=Atopic%20Dermatitia&source=search_result&selectedTitle=1~150&usage_type=default&display_rank=1 (“UpToDate,” 2021)
Weidinger, S., & Novak, N. (2016). Atopicdermatitis. The Lancet, 387(10023), 1109–1122. https://doi.org/10.1016/s0140-6736(15)00149-x (Weidinger & Novak, 2016)
Eczema Society of Canada
About The Eczema Society of Canada
The Eczema Society of Canada (ESC) is a registered Canadian charity dedicated to improving the lives of Canadians living with eczema with a mission of support, education, awareness, and research. To learn more, visit www.eczemahelp.ca.
Information Gathering
ESC has gathered survey data from more than 3000 Canadians who live with atopic dermatitis (AD) on topics including quality of life impact, experience with systemic treatments, the AD patient journey, and experience with itch related to AD. Respondents included adults living with AD and the caregivers of children living with AD. Information for this submission was also gathered via questionnaires and one-on- one interviews. Patients and caregivers who shared their experiences using upadacitinib accessed the drug through a clinical trial.
Disease Experience
AD, commonly known as eczema, is a chronic, inflammatory skin condition. It is characterized by dry, itchy, inflamed skin that can crack, ooze, and bleed. AD patients experience “flares” which are periods of worsening of the condition and its symptoms. AD flares can be extremely itchy and painful and can lead to psychological distress and negatively impact the individual and their family.
AD can range from mild to severe, and while many people living with AD can experience periods of remission, some never experience relief from these life-altering symptoms. This is commonly reported by patients with uncontrolled moderate or severe forms of AD, and those patients are more likely to never experience periods of clear skin.
Patients frequently report that itch is the most burdensome symptom of AD. Some compared the sensation of itch to being bitten by thousands of mosquitos all at once. Adult survey respondents reported feeling itchy multiple times each day (reported by 72% of respondents with moderate AD, and by 95% of respondents with severe AD). As the severity of AD increases, so does the frequency of itch, as 44% of respondents with severe AD reported feeling itch all the time. 71% of adult respondents with moderate or severe AD rated their overall itch as 7 out of 10 or greater, and at its worst, 42% of adult respondents rated it as 10 out of 10 – the worst itch imaginable.
More than half (54%) of adult survey respondents with severe AD reported rarely being able to control their urge to scratch their skin. Patients frequently report that their itch is worse or most intense at nighttime. Patients also reported that the itch and pain of their AD wakes them from sleep and makes falling and staying asleep more challenging. According to ESC’s quality of life survey, loss of sleep and poor sleep quality are reported as significant quality of life impacts, as 63% of survey respondents with moderate AD and 86% of survey respondents with severe AD reported that itch negatively impacted their sleep. 50% of survey respondents with severe AD reported experiencing sleep loss 8 nights per month or more.
“During a difficult period, I considered going to the emergency room because the itch was so bad and I had not slept in days. I instead alternated between scalding hot and ice cold showers just to distract myself from the itch.”
Patients and caregivers also reported skin damage, bleeding, and scarring due to scratching the skin, with 62% of survey respondents with moderate AD and 87% of survey respondents with severe AD having scars or marks on their skin from scratching. Bleeding is particularly distressing, with patients reporting the need to frequently change clothing, bedding, and towels as a result of blood stains. Patients, including children and adolescents, can experience feelings of embarrassment and shame when their skin is visibly inflamed, cracked, and flaking, or when they have noticeable blood stains on their clothing. Itch can impact every aspect of life and 46% of adult survey respondents with moderate or severe AD describe their itch as debilitating.
“When I was younger, my mom would wrap my hands so I couldn’t scratch myself in my sleep. My AD was so bad my clothes would stick to my skin during the day and I had to take a bath in oil just to get clothing, like my tights, off my body.”
The painful and frustrating symptoms of AD, along with the unpredictable patterns of flares and/or exacerbations, can significantly impact mental health and cause stress (69% of survey respondents with moderate AD and 87% of survey respondents with severe AD reported that itch negatively impacts stress). Patients and caregivers reported that the mental health impact of AD is a significant aspect of the condition and is often not understood by others, nor prioritized by health care providers. Uncontrolled chronic AD can lead to feelings of depression and anxiety as well as poor self-esteem, low energy, and sadly in some extreme cases, suicidal thoughts.
“AD is not only exhausting, it is hard on mental health and self-confidence. It is all-consuming for those that suffer from it and for their families.”
AD can negatively impact mood, work, school, and social interactions. 32% of adult survey respondents with moderate or severe AD have missed work events due to their condition, and 30% have had to change careers or give up certain activities. Patients reported that their condition also impacts productivity and contributions while at work, as pain, feelings of embarrassment, and persistent scratching can hold them back. For adolescents suffering with AD, their lives can be particularly impacted due to the physical and psychological burden of the condition. Caregivers reported that AD substantially impacts their child’s ability to socialize in school and ability to make and maintain friendships. These social impairments can lead to feelings of anxiety and isolation.
The burden of AD also extends to caregivers and parents. Survey results indicated that 55% of caregivers have experienced sleep loss due to their child’s AD. Partners and spouses also reported loss of sleep due to their partner’s sleep disruption, such as waking and scratching through the night. Caregivers reported that AD places a significant emotional toll on the entire family, and feelings of guilt, frustration, anger, and sadness are common. Nearly half (41%) of caregivers surveyed reported they feel like a failure when they cannot control their child’s flares.
“As a parent, you question everything when your child is suffering and you are trying to find a solution. You wonder if you are doing enough, or if you are doing something wrong. There needs to be a better way – lives are being destroyed by this condition.”
Experiences With Currently Available Treatments
For many patients living with AD, frequent moisturizing, trigger avoidance, and the use of topical treatments work to control their AD flares, but for others, they are left suffering. For these patients, despite strict adherence to their prescribed treatment plans, they cannot find relief from their debilitating symptoms.
Some of these patients who are still uncontrolled after trying numerous topical treatments may be recommended systemic treatments by their dermatologist. Until recently, systemic treatments have been very limited for AD patients. These included off-label immune suppressing medications (such as methotrexate and cyclosporine), oral corticosteroids (e.g. prednisone), and phototherapy. Very recently, a biologic drug has been approved for AD, and some patients are now able to access this treatment.
However, access to this medication is a significant challenge despite the tremendous unmet need and potential benefit a biologic drug, and other targeted therapies for AD would provide for this patient population.
According to an ESC survey on the use of systemic treatments for AD, oral corticosteroids were the most frequently used systemic treatments for AD, but they also rated highest in safety concerns for patients.
Patients also reported that the rebound flares experienced after taking oral corticosteroids can be devastating. Phototherapy is also sometimes used, however, some patients reported that it does not work well to control their AD in the long term. In addition, access to phototherapy clinics is a significant barrier for many patients depending where they are located in the country.
The trial-and-error process of cycling through currently available treatments is a common experience among patients with moderate or severe AD, and as they often reported to ESC, they have “tried everything”. This process of repeated treatment failure and suffering is demoralizing, tiring, and causes significant distress, including mental health deterioration. This significant challenge highlights the need for improved treatments for this small population of patients who don’t respond to topical treatments.
“The only thing our child used before upadacitinib was topical steroids, but they didn’t work to control the rash that eventually covered my child’s entire body. It was terrible.”
“My skin was so dry, so raw, and so plagued with infection that I spent many days lying around on the couch in pain, just hoping that there would be a medication that would finally give me relief.”
“I have lost years and years of my life… I keep hearing ‘new treatments are coming’ but time has passed me by while I wait and hope that a solution will be found.”
Improved Outcomes
Patients with moderate or severe AD report that they are seeking the following outcomes from a treatment:
Improvement or ideally the absence of itch
Improvement or ideally the elimination of skin lesions/rash
Improved quality of life
Improved sleep
Ability to work, carry out daily activities, and exercise without flare exacerbation
Reduction or ideally the elimination of complications such as staph infections, eczema herpeticum, and secondary infections
While patients are looking for a treatment that will improve their symptoms, ultimately they would like to have control of this debilitating condition by breaking the cycle of flares and symptom exacerbations.
There is currently no cure for AD, however innovative treatments such upadacitinib can offer these patients hope that they can experience control of their condition and achieve better quality of life.
Control of AD symptoms would allow patients better quality of life across many areas, including sleep, social interactions, and the ability to participate in sports and outdoor activities without fearing a flare or cracked, bleeding skin. Caregivers want a treatment that can permit them or their child to have a good life, free of itchy skin and painful rashes.
“Before starting the trial, I was so inflamed and was itchy all the time. I wasn’t sleeping, I missed work, and when I woke up in the morning my eyes would be swollen shut. My face shape wasn’t the same.”
“My skin would flake and peel, and my sheets would be stained with steroid cream. I asked others about if I should go on the clinical trial for this new eczema drug… they said it could change my life. And it did.”
Experience With Drug Under Review
ESC gathered input from patients and their caregivers regarding clinical trial experience with upadacitinib. Those interviewed had positive experiences and spoke about the transformative results from participating in the clinical trial. Patients reported relief from itch as well as significant and rapid improvement in the skin rash/lesions. One patient reported experiencing significant relief from the itch within days of starting the treatment, and the improvement in their skin rash/condition within weeks. They also expressed that they hadn’t realized how badly they were suffering until they experienced the drastic improvement of their symptoms during the trial. Another caregiver shared that their child’s rash, which once covered the child’s entire body, was finally able to heal for the first time in their lives. The child no longer had to struggle with constant infections, open sores, and raw, inflamed skin.
Another patient shared that they started upadacitinib while working from home during the pandemic, and when they returned to work, their colleagues almost didn’t recognize them because of the dramatic change in their appearance. Other patients interviewed also shared the significant changes in their appearance, as they no longer experienced visible swelling, crusting, and scabbing. This was especially impactful for patients who experienced significant flares on their faces.
Another patient shared that they had lived with AD since they were a baby, and were used to having open wounds on their body. They would need to cover up their arms and legs while their peers would be able to wear shorts or expose their arms. Now they no longer need to constantly worry and cover their skin, and for once, they are living without pain and have experienced a significant improvement in their confidence.
Of patients interviewed who tried upadacitinib, some reported side effects including weight gain, mild headaches, and sun sensitivity. None of the individuals interviewed had to stop the trial due to side effects. Others didn’t report experiencing any side effects during the trial.
Across the spectrum of AD severity, patient and caregivers consistently reported carefully weighing the risks and benefits of any medication, ranging from topical medications to systemic medications. The trial participants who were interviewed reported having suffered from the painful and debilitating symptoms of uncontrolled AD for so long that they were willing to accept some of the side effects during clinical trial if it meant it would bring them relief from their symptoms.
In terms of medication delivery and impact on daily routines, patients and caregivers also shared that they felt the once-a-day oral pill was a improvement compared to the time consuming and uncomfortable nature of their previous skin care and topical treatment routines.
“I no longer have to change my sheets every day – not only because I no longer scratch myself until I bleed while I sleep, but also because my sheets are no longer stained from the greasy ointments I would need to cover my body in before taking upadacitinib.”
“Upadacitinib was extremely helpful in managing my AD. When I think back to where I started, I don’t know where I would be if I hadn’t tried it.”
“Before our involvement in the clinical trial for upadacitinib, there were no good solutions for [my child]. The lack of options impacted their mental health as well as their physical health, and it is a side of eczema that people don't realize, understand, acknowledge, or treat.”
“When my skin became so bad and I was covered in the rash, I went to see my doctor to ask about something to take for the itch. I was given the choice of going on methotrexate or trying the clinical trial, and I chose the upadacitinib trial because I heard this new type of medication had been changing people’s lives.”
Companion Diagnostic Test
N/A
Anything Else?
Uncontrolled moderate to severe AD can be debilitating and life altering and there are significant gaps in treatment for this patient population. The patients and caregivers with experience with upadacitinib shared that the medication rapidly improved their symptoms and significantly improved their quality of life. It also allowed patients a variety of opportunities such as regained self-confidence, better work productivity, improved personal relationships, the regained ability to exercise, and the ability to better care for themselves or their loved ones. The need for more treatment options for uncontrolled AD is critical.
“We saw immediate results – within a week or so, [my child’s] skin started to heal, which in turn helped with the itch and discomfort. They are in a lot less pain and are less constricted in what they can do, like taking a shower without it being painful. I’m not spending hours researching new treatments, and we haven’t had even one emergency trip to the doctor or hospital.”
“Treatments need to be accessible to everyone who needs them and who qualify (i.e. if a doctor deems it helpful). This is not just a skin rash. It is an all-consuming issue that can really affect an individuals’ quality of life – physical and mental. There needs to be knowledge, empathy, and medical support for the physical, mental, and emotional aspects of AD.”
“My child has been on this treatment for months, and we will do whatever we need to do for them to stay on it. It has become the only hope we have, and while it might come with a risk, we are willing to take it so they can get to a place where their body and mind can heal.”
Patient Group Conflict of Interest Declaration — Eczema Society of Canada
Did you receive help from outside your patient group to complete this submission? If yes, please detail the help and who provided it.
No.
Did you receive help from outside your patient group to collect or analyze data used in this submission? If yes, please detail the help and who provided it.
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review.
Table 4: Conflict of Interest Declaration for the Eczema Society of Canada
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Abbvie Canada | — | — | — | X |
I hereby certify that I have the authority to disclose all relevant information with respect to any matter involving this patient group with a company, organization, or entity that may place this patient group in a real, potential, or perceived conflict of interest situation.
Patient Group: Eczema Society of Canada
Date: May 6, 2021
Clinician Group Input
Atlantic Society of Allergy and Immunology
About Atlantic Society of Allergy and Immunology
Please describe the purpose of your organization. Include a link to your website (if applicable).
We are a group of Royal College Board Certified Allergists and Immunologists practicing in the Atlantic provinces, treating adults and children. Our mandate is to promote excellence in the specialty of Allergy and Clinical Immunology.
Information Gathering
Please describe how you gathered the information included in the submission.
We are a group of Allergists and Immunologists who treat AD in adults and pediatrics. We have attended meetings (local, national and international) discussing current AD treatment and future treatment options.
We have discussed AD treatment options and recent updates, standards of care in AD, treatment goals in AD, AD patient burden and journey from different perspectives (e.g. patient, allergist, dermatologist, family physician), the ideal AD care pathway, and gaps needed to address in this care pathway. We have used these discussions to build this submission.
Current Treatments
Describe the current treatment paradigm for the disease
The usual progression of treatment for AD is emollients and lifestyle measures (types of clothing, moisturizing, bathing, avoiding skin irritants, minimizing stress, etc.), followed by topical steroids/ topical calcineurin inhibitors /ointments, followed by systemic immunosuppressant therapies with/or without phototherapy. Most of these are prescribed long term (except for cyclosporine), with topical creams and systemic steroids used intermittently in some cases.
According to Canadian Guidelines (Bergman J et al, https://guidelines.eczemahelp.ca/wp-content/uploads/2020/09/ESC-Atopic-Dermatitis-A-Practical-Guide-to-Management-HCP-Guideline-2020-PUBLISHED-1.pdf):
Patients are recommended to avoid triggers such as rough fabrics, as well as overheating and sweating
Frequent and consistent moisturizing may sufficiently manage mild AD. However, moisturizing is still an important component of treatment even in cases of moderate to severe AD.
Daily bathing is often recommended for patients with AD; however, there is no recommendation for specifying the frequency, duration, or method of bathing.
Topical corticosteroids/ topical calcineurin inhibitors are considered safe and effective for the first- line treatment of the inflammatory components of AD.
For refractory and severe AD, physicians may need to prescribe phototherapy, off label systemic immunosuppressant therapies, or biologic agents.
Cyclosporine, methotrexate, azathioprine and mycophenolate mofetil are systemic immunosuppressant agents used off-label for moderate-to-severe AD by allergists and dermatologists
While systemic agents target inflammation, which is an important component of AD’s disease mechanism, the mechanisms are unknown. Further, these non-biologic systemic agents have safety concerns in long-term use, are not indicated for AD and provide low efficacy (van Der Schaft J et al. Br J Dermatol. 2015; 172(6):1621-1627; van der Schaft J et al. Br J Dermatol. 2016; 175(1):199-202; Politiek K et al. Br J Dermatol. 2015; 174(1): 201-203).
The first biologic agent approved for AD, dupilumab, targets IL-14 and IL-13 signalling, and therefore targets inflammation, an underlying disease mechanism of AD.
Canadian Guidelines acknowledge that some patients with severe or refractory AD may require biologic agents, and note that dupilumab is associated with significant improvements in AD severity, symptoms, QoL and that most patients tolerate dupilumab well (Bergman J et al, https://guidelines.eczemahelp.ca/wp-content/uploads/2020/09/ESC-Atopic-Dermatitis-A-Practical-Guide-to-Management-HCP-Guideline-2020-PUBLISHED-1.pdf):
Although dupilumab addresses the needs of some patients with moderate-to-severe AD, a large unmet need still exists in this population. In the dupilumab phase 3 studies, fewer than 40% of patients achieved clear or almost clear skin. Mohamed MEF et al. J Clin Pharmacol. 2021 May;61(5):628-635. https://doi.org/10.1002/jcph.1782). In clinical practice, some patients are unable to tolerate dupilumab due to conjunctivitis or persistent head and neck dermatitis. Some AD patients are also reluctant to consider bi-weekly injections.
Treatment Goals
What are the most important goals that an ideal treatment would address?
We believe that improving symptoms of AD (such as chronic itch/dry and inflamed skin), reducing sleep disturbances and improving quality of life and patient satisfaction (improved sleep, reduced work/school disruption) are top priorities for treatment goals.
Other priorities include flare reduction and achieving disease control as reflected by Physician’s Global Assessment (PGA) scores of clear or almost clear and improved Dermatology Life Quality Index [DLQI]).
Treatment Gaps (Unmet Needs)
Considering the treatment goals (see Treatment Goals section), please describe goals (needs) that are not being met by currently available treatments.
The main unmet need in moderate to severe AD is lack of access to effective, convenient and safe treatment that enables long-term disease control and remission, as many patients experience flares as soon as they stop their current medication. This cycle of recurrence leads to disease progression ending in chronic severe AD and severe impact on QoL. Available off-label treatments have poor efficacy and safety profiles unacceptable for long term use (continued monitoring for effects on blood counts, renal and liver disease, hypertension, increased malignancy risk, immunosuppression and other unwanted side effects)
Other important unmet needs include:
Phototherapy, which is often used in conjunction with systemic therapies, is associated with issues such as poor accessibility, long wait times, low efficacy, and exposure to UV radiation
Access to effective therapies in a suboptimal care pathway, which currently forces patients onto drugs that are not effective and not approved for AD and can be harmful to their health
Lack of support for patients and the misconception that eczema is related to a food allergy prevents patients from receiving proper and timely care, wasting time and money on ineffective and unproven management strategies
Significant disease burden and impact on QoL (itch, sleep, DLQI) with uncontrolled disease
Higher atopic comorbidities as well as anxiety and depression (Hospital Anxiety and Depression Scale [HADS]) with uncontrolled disease.
Which patients have the greatest unmet need for an intervention such as the drug under review?
The greatest unmet need is a subset of AD patients with moderate to severe disease.
Patients with under- or un-managed AD are characterized by some level of baseline AD with intermittent flares, complications including bacterial and viral infections leading to emergency department (ED) visits, as well as incessant itch affecting almost every facet of life (sleep, mental focus, work/school productivity, interpersonal relationships, self-esteem and mental health).
Upadacitinib would address the unmet need that leads to many of these problems experienced by patients with AD: allowing rapid disease control in patients with moderate-to-severe AD. To date, upadacitinib’s efficacy is the highest reported among AD systemic therapies. In the Measure Up trials, upadacitinib demonstrated significant improvement in skin clearance and itch compared to placebo (Guttman-Yassky E, et al. Oral presentation at EADV 2020, DT03.4B). In the Heads up trial, upadacitinib demonstrated significant improvements in skin clearance and rapid itch reduction over dupilumab, the first novel targeted therapy available for AD (https://news.abbvie.com/news/press-releases/rinvoq-upadacitinib-achieved-superiority-versus-dupixent-dupilumab-for-primary-and-all-ranked-secondary-endpoints-in-phase-3b-head-to-head-study-in-adults-with-atopic-dermatitis.htm).
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
The mechanism of action of upadacitinib would mean that it would be used after initial treatments for mild AD, such as lifestyle measures (types of clothing, moisturizing, bathing, etc.) and topical steroids topical/ calcineurin inhibitors are insufficient to control the disease, meaning that the patient does not have a mild case but rather has moderate-to-severe AD.
Upadacitinib would replace systemic therapies that are currently used off-label to treat AD, as well as phototherapy. We recommend to remove these from the care pathway and the forced treatment ladder for coverage due to lack of efficacy and safety concerns. We also note the importance of an identifiable “key switch” to target; a drug with specificity makes it more likely to improve all aspects of AD equally
Upadacitinib would be the first JAK inhibitor approved for atopic dermatitis. The first biologic agent approved for AD, dupilumab, targets IL-14 and IL-13 signalling, and therefore targets the underlying disease mechanisms of AD through a different mode of action.
Although dupilumab addresses the needs of some patients with moderate-to-severe AD, a large unmet need still exists in this population. In the dupilumab phase 3 studies, fewer than 40% of patients achieved clear or almost clear skin (Mohamed MEF et al. J Clin Pharmacol. 2021 May;61(5):628-635. https://doi.org/10.1002/jcph.1782).
Therefore, upadacitinib may further shift the current treatment paradigm by providing an additional option for patients with moderate-to-severe AD whose disease is not adequately controlled with lifestyle measures or topical corticosteroids.
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective.
As mentioned in earlier responses, newly diagnosed patients with AD should work with their physician to implement lifestyle measures or topical corticosteroids/topical calcineurin inhibitor to control their disease. If their disease cannot be controlled with these measures, they should consider initiating treatment with upadacitinib. As noted, current systemic treatments are not indicated for AD and do not enable long-term disease control and remission, as many patients experience flares as soon as they stop their current medication (topical agents particularly). There are also significant safety concerns associated with these therapies.
How would this drug affect the sequencing of therapies for the target condition?
As mentioned, the usual progression of treatment is emollients and lifestyle measures (types of clothing, moisturizing, bathing, etc.), followed by topical steroids/topical calcineurin inhibitors, followed by systemic therapies with/or without phototherapy.
Upadacitinib or dupilimab would be used after initial treatments for AD, such as lifestyle measures and topical steroids and after patient has been diagnosed with moderate to severe AD. Upadacitinib would replace systemic therapies that are currently used off-label to treat moderate to severe AD, as well as phototherapy.
Which patients would be best suited for treatment with the drug under review?
Medications such as upadacitinib are best suited to treat patients with moderate to severe AD, who have not responded, are not expected to respond, or have had adverse reactions to long term use of topical corticosteroids or topical calcineurin inhibitors. These patients are in the most need of intervention as they lack long-term treatment options and are at high risk of disease progression. Once AD has progressed, patients are at higher risk of severe flare ups, skin infection, and hospitalization.
How would patients best suited for treatment with the drug under review be identified?
Ideally, patients with moderate-to-severe AD would be referred to specialized allergists or dermatologists by their primary care provider (e.g. family physician, nurse practitioner). However, no standardized referral form exists for triage, and time constraints prevent the thorough case review that is necessary to diagnose AD. AD should be considered a complex disorder in order to standardize the pathway from consultation, referral and diagnosis. An ideal standardized referral form would include:
BSA affected and level of pruritus
Description of the effect of AD on QoL (social life, work/school, sleep loss)
If possible, identification of level of AD severity with special sites noted (hands, face, etc.)
Treatment history (topical steroid use, systemic steroid use, ER visits, antibiotic use,
Response to therapies tried
Comorbidities (asthma, allergic rhinitis/nasal polyps, anaphylactic food allergies, eosinophilic esophagitis, chronic urticaria, anxiety, depression)
Notes on patient occupational impacts on the dermatitis, triggers and family history of AD
AD patients typically insist on allergy testing due to misperceptions about the link between their condition and allergic reactions. This often leads to an inconclusive diagnosis (rash, eruption as descriptor) and leads to patient frustration. Patient and physician education about pathophysiology of AD are crucial to achieve treatment success.
Once an AD diagnosis is confirmed, upadacitinib should be prescribed to patients with moderate-to- severe AD who have not responded, are not expected to respond, or have had adverse reactions to long term use of topical corticosteroids.
Which patients would be least suitable for treatment with the drug under review?
AD patients with mild disease (majority of AD population), and whose symptoms can be controlled with lifestyle changes and topical corticosteroids would be least/not suitable for treatment with upadacitinib.
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review?
In phase 3 trials, patients were selected based on eligibility criteria including:
Chronic AD before Baseline Visit
Active moderate to severe AD defined by EASI (Eczema Area Severity Index), IGA (Investigator Global Assessment), BSA (Body Surface Area), and pruritus
Candidate for systemic therapy or have recently required systemic therapy for AD
Documented history of inadequate response to topical corticosteroids or topical calcineurin inhibitor OR documented systemic treatment for AD within 6 months before Baseline Visit
(Measure Up 1, https://clinicaltrials.gov/ct2/show/NCT03569293; Measure Up 2, https://clinicaltrials.gov/ct2/show/NCT03607422; AD Up, https://clinicaltrials.gov/ct2/show/NCT03568318; Heads Up, https://clinicaltrials.gov/ct2/show/NCT03738397).
What outcomes are used to determine whether a patient is responding to treatment in clinical practice?
In clinical trials, outcomes are measured by the IGA (Investigator Global Assessment), and proportion of participants achieving a 75% or 90-100% improvement on the EASI. These outcomes are also used in clinical practice (the equivalent PGA [Physician Global’s Assessment] is used instead of IGA) and are similarly aligned with additional measures, including body surface area (BSA) affected, and the pruritus numerical rating scale (NRS), which ranges from 0 (“no itch”) to 10 (“worst imaginable itch”). EASI scoring is not routinely used in clinical practice.
What would be considered a clinically meaningful response to treatment?
A clinically meaningful response to upadacitinib would include improvements in:
patient-reported itch (4 point reduction on the NRS or a NRS score of less than 3)
DLQI score reduction of equal or more than 4 (or an acceptable improvement)
patient-reported sleep quality and fewer AD-related disruptions at school and work
PGA score to 0 or 1
Importantly, a patient should not experience any severe side effects, including over sustained time periods, in order for the response to upadacitinib to be clinically meaningful.
How often should treatment response be assessed?
Response to systemic therapy is reassessed in 12-16 weeks after initiation of treatment.
What factors should be considered when deciding to discontinue treatment?
The decision to discontinue treatment should be assessed based on lack of response, significant disease progression (i.e. lichenifcation, increased affected BSA and itching) and deterioration in quality of life.
Treatment should also be discontinued if the patient experiences adverse reactions or intolerance to the medication that are deemed to be unacceptable by the patient-physician team.
Treatment with upadacitinib should be interrupted if a patient develops a serious infection, until the infection is controlled. Treatment should also be interrupted to address abnormal laboratory results (ALC-absolute lymphocyte count less than 500 cells/mm3, ANC - absolute neutrophil count less than 1000 cells/mm3, Hb less than 8 g/dL, or if drug-induced liver injury is suspected [based on hepatic transaminases])and may be resumed once levels return to normal.(RINVOQ Product Monograph. 2019. Canada. AbbVie Inc).
What settings are appropriate for treatment with the drug under review?
Patients with AD receiving upadacitinib would ideally be managed in any non-emergent setting that they have access to, and that has an allergist or dermatologist well-versed in managing moderate-to-severe AD. Referring family physicians, nurse practitioners, or other health care providers should be counseled on the appropriate referral process.
For non-oncology drugs, is a specialist required to diagnose, treat, and monitor patients who might receive the drug under review?
As mentioned above, an ideal care pathway would involve referral from front-line AD health care providers to a trained allergist or dermatologist who would prescribe and manage treatment with upadacitinib for moderate-to-severe AD.
Additional Information
Is there any additional information you feel is pertinent to this review?
Upadacitinib presents a breakthrough in moderate to severe AD management as reflected by its efficacy, impact on patients’ QoL in combination with acceptable safety profile for long term use.
Conflict of Interest Declarations — Atlantic Society of Allergy and Immunology
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (see Place in Therapy section) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
Yes, there was input from dermatologist colleagues who attended joint meetings with some of our group, as well as help from Abbvie representatives who arranged some of the meetings as well as helped to provide reference material for the submission.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
Yes, there was input from dermatologist colleagues who attended joint meetings with some of our group, as well as help from Abbvie representatives who arranged some of the meetings as well as helped to provide reference material for the submission.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician that contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
Declaration for Clinician 1
Name: Gina Lacuesta
Position: Assistant Professor, Faculty of Medicine, Dalhousie University, Consultant Physician in Allergy and Clinical Immunology Nova Scotia Health Authority, member ASAI
Date: 26-04-2021
Table 5: COI Declaration for Atlantic Society of Allergy and Immunology Clinician 1
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Abbvie | X | — | — | — |
Sanofi | X | — | — | — |
Declaration for Clinician 2
Name: Wade Thomas Aaron Watson
Position: Professor of Pediatrics, Dalhousie University; Head, Division of Allergy, IWK Health Centre, President ASAI
Date: 26-04-2021
Table 6: COI Declaration for Atlantic Society of Allergy and Immunology Clinician 2
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
No COI | — | — | — | — |
Atlantic Specialist Group Managing Atopic Dermatitis
About the Atlantic Specialist Group Managing Atopic Dermatitis
Please describe the purpose of your organization. Include a link to your website (if applicable).
We are a group of physicians including general practitioner, dermatology and allergy & immunology specialists managing patients with atopic dermatitis. We are located in various clinical settings across Atlantic Canada.
Information Gathering
Please describe how you gathered the information included in the submission.
A group of atopic dermatitis (AD) specialists from Atlantic Canada convened (dermatologists, an allergist and a family physician) to consult on filling unmet needs in AD and broadening access to efficient treatment in AD (specifically newer biologics coming to market).
Over the course of two meetings, participants discussed Canadian regulatory processes, atopic dermatitis treatment options and recent updates, standards of care in AD, treatment goals in AD, AD patient burden and journey from different perspectives (e.g. patient, allergist, family physician), the ideal AD care pathway, and gaps needed to address in this care pathway. Following the two meetings, we, a subset of the attendees, used the key discussion points to build this submission.
Current Treatments
Describe the current treatment paradigm for the disease.
The usual progression of treatment is emollients and lifestyle measures (types of clothing, moisturizing, bathing, avoiding skin irritants, minimizing stress, etc.), followed by topical steroids/topical non-steroidal anti-inflammatory creams/ointments, followed by systemic immunosuppressant therapies with/or without phototherapy. Most of these are prescribed long term (except for cyclosporine), with topicals and steroids used intermittently in some cases.
According to Canadian Guidelines (Bergman J et al, https://guidelines.eczemahelp.ca/wp-content/uploads/2020/09/ESC-Atopic-Dermatitis-A-Practical-Guide-to-Management-HCP-Guideline-2020-PUBLISHED-1.pdf):
Patients are recommended to avoid triggers such as rough fabrics, as well as overheating and sweating
Frequent and consistent moisturizing may sufficiently manage mild AD. However, moisturizing is still an important component of treatment even in cases of moderate to severe AD.
Daily bathing is often recommended for patients with AD; however, there is no recommendation for specifying the frequency, duration, or method of bathing.
Topical corticosteroids are considered safe and effective for the first-line treatment of the inflammatory components of AD.
For refractory and severe AD, physicians may need to prescribe phototherapy, off label systemic immunosuppressant therapies, or biologic agents.
Cyclosporine, methotrexate, azathioprine and mycophenolate mofetil are systemic immunosuppressant agents used off-label for moderate-to-severe AD by dermatologists
While systemic agents target inflammation, which is an important component of AD’s disease mechanism, the mechanisms are unknown. Further, these non-biologic systemic agents have safety concerns in long-term use, are not indicated for AD and provide low efficacy (van Der Schaft J et al. Br J Dermatol. 2015; 172(6):1621-1627; van der Schaft J et al. Br J Dermatol. 2016; 175(1):199-202; Politiek K et al. Br J Dermatol. 2015; 174(1): 201-203).
The first biologic agent approved for AD, dupilumab, targets IL-14 and IL-13 signalling, and therefore targets inflammation, an underlying disease mechanism of AD.
Canadian Guidelines acknowledge that some patients with severe or refractory AD may require biologic agents, and note that dupilumab is associated with significant improvements in AD severity, symptoms, QoL and that most patients tolerate dupilumab well (Bergman J et al, https://guidelines.eczemahelp.ca/wp-content/uploads/2020/09/ESC-Atopic-Dermatitis-A-Practical-Guide-to-Management-HCP-Guideline-2020-PUBLISHED-1.pdf).
Although dupilumab addresses the needs of some patients with moderate-to-severe AD, a large unmet need still exists in this population. In the dupilumab phase 3 studies, fewer than 40% of patients achieved clear or almost clear skin. Mohamed MEF et al. J Clin Pharmacol. 2021 May;61(5):628-635. https://doi.org/10.1002/jcph.1782). In clinical practice, some patients are unable to tolerate dupilumab due to conjunctivitis or persistent head and neck dermatitis. Some AD patients are also reluctant to consider bi-weekly injections.
Treatment Goals
What are the most important goals that an ideal treatment would address?
We believe that improving symptoms of AD such as chronic itch/dry and inflamed skin/sleep disturbances and quality of life and patient satisfaction (improve sleep, work/school disruption) are top priorities for treatment goals.
Other priorities include flare reduction and achieving disease control as reflected by Physician’s Global Assessment (PGA) scores of clear or almost clear and Dermatology Life Quality Index [DLQI]).
Treatment Gaps (Unmet Needs)
Considering the treatment goals (see Treatment Goals section), please describe goals (needs) that are not being met by currently available treatments.
The main unmet need in moderate to severe AD is lack of access to effective, convenient and safe treatment that enables long-term disease control and remission, as many patients experience flares as soon as they stop their current medication. This cycle of recurrence leads to disease progression ending in chronic severe AD and severe impact on QoL. Available off-label treatments have poor efficacy and safety profiles unacceptable for long term use.
Other important unmet needs include:
Phototherapy, which is often used in conjunction with systemic therapies, is associated with issues such as poor accessibility, long wait times, low efficacy, and exposure to UV radiation
Access to effective therapies in a suboptimal care pathway, which currently forces patients onto drugs that are not effective and not approved for AD and can be harmful to their health
Lack of support for patients and the misconception that eczema is related to a food allergy prevents patients from receiving proper and timely care, wasting time and money on ineffective and unproven management strategies
Significant disease burden and impact on QoL (itch, sleep, DLQI) with uncontrolled disease
Higher atopic comorbidities as well as anxiety and depression (Hospital Anxiety and Depression Scale [HADS]) with uncontrolled disease.
Which patients have the greatest unmet need for an intervention such as the drug under review?
The greatest unmet need is a subset of AD patients with moderate to severe disease.
Patients with under- or un-managed AD are characterized by some level of baseline AD with intermittent flares, complications including bacterial and viral infections leading to ER visits, as well as incessant itch affecting almost every facet of life (sleep, mental focus, work/school productivity, interpersonal relationships, self-esteem and mental health).
Upadacitinib would address the unmet need that leads to many of these problems experienced by patients with AD: allowing rapid disease control in patients with moderate-to-severe AD. To date, upadacitinib’s efficacy is the highest reported among AD systemic therapies. In the Measure Up trials, upadacitinib demonstrated significant improvement in skin clearance and itch compared to placebo (Guttman-Yassky E, et al. Oral presentation at EADV 2020, DT03.4B). In the Heads up trial, upadacitinib demonstrated significant improvements in skin clearance and rapid itch reduction over dupilumab, the first novel targeted therapy available for AD (https://news.abbvie.com/news/press-releases/rinvoq- upadacitinib-achieved-superiority-versus-dupixent-dupilumab-for-primary-and-all-ranked-secondary-endpoints-in-phase-3b-head-to-head-study-in-adults-with-atopic-dermatitis.htm).
Place in Therapy
How would the drug under review fit into the current treatment paradigm?
The mechanism of action of upadacitinib would mean that it would be used after initial treatments for mild AD, such as lifestyle measures (types of clothing, moisturizing, bathing, etc.) and topical steroids are insufficient to control the disease, meaning that the patient does not have a mild case but rather has moderate-to-severe AD.
Upadacitinib would replace systemic therapies that are currently used off-label to treat AD, as well as phototherapy. We recommend to remove these from the care pathway and the forced treatment ladder for coverage due to lack of efficacy and safety concerns. We also note the importance of an identifiable “key switch” to target; a drug with specificity makes it more likely to improve all aspects of AD equally
Upadacitinib would be the first JAK inhibitor approved for atopic dermatitis. The first biologic agent approved for AD, dupilumab, targets IL-14 and IL-13 signalling, and therefore targets the underlying disease mechanisms of AD through a different mode of action.
Although dupilumab addresses the needs of some patients with moderate-to-severe AD, a large unmet need still exists in this population. In the dupilumab phase 3 studies, fewer than 40% of patients achieved clear or almost clear skin (Mohamed MEF et al. J Clin Pharmacol. 2021 May;61(5):628-635. https://doi.org/10.1002/jcph.1782).
Therefore, upadacitinib may further shift the current treatment paradigm by providing an additional option for patients with moderate-to-severe AD whose disease is not adequately controlled with lifestyle measures or topical corticosteroids.
Please indicate whether or not it would be appropriate to recommend that patients try other treatments before initiating treatment with the drug under review. Please provide a rationale from your perspective.
As mentioned in earlier responses, newly diagnosed patients with AD should work with their physician to implement lifestyle measures or topical corticosteroids to control their disease. If their disease cannot be controlled with these measures, they should consider initiating treatment with upadacitinib. As noted, current systemic treatments are not indicated for AD and do not enable long-term disease control and remission, as many patients experience flares as soon as they stop their current medication (topical agents particularly). There are also significant safety concerns associated with these therapies.
How would this drug affect the sequencing of therapies for the target condition?
As mentioned, the usual progression of treatment is emollients and lifestyle measures (types of clothing, moisturizing, bathing, etc.), followed by topical steroids, followed by systemic therapies with/or without phototherapy.
Upadacitinib would be used after initial treatments for AD, such as lifestyle measures and topical steroids and after patient has been diagnosed with moderate to severe AD. Upadacitinib would replace systemic therapies that are currently used off-label to treat moderate to severe AD, as well as phototherapy.
If patients fail, have contraindication or intolerance to upadacitinib, their treating physician may consider dupilumab as the next therapeutic option.
Which patients would be best suited for treatment with the drug under review?
Medications such as upadacitinib are best suited to treat patients with moderate to severe AD, who have not responded, are not expected to respond, or have had adverse reactions to long term use of topical corticosteroids. These patients are in the most need of intervention as they lack long-term treatment options and are at high risk of disease progression. Once AD has progressed, patients are at higher risk of severe flare ups, skin infection, and hospitalization.
How would patients best suited for treatment with the drug under review be identified?
Ideally, patients with moderate-to-severe AD would be referred to specialized dermatologists or allergists by their primary care provider (e.g. family physician, nurse practitioner). However, no standardized referral form exists for triage, and time constraints prevent the thorough case review that is necessary to diagnose AD. AD should be considered a complex disorder in order to standardize the pathway from consultation, referral and diagnosis. An ideal standardized referral form would include:
BSA affected and level of pruritus
Description of the effect of AD on QoL (social life, work/school, sleep loss)
If possible, identification of level of AD severity with special sites noted (hands, face, etc.)
Treatment history (topical steroid use, systemic steroid use, ER visits, antibiotic use,
Response to therapies tried
Comorbidities (asthma, allergic rhinitis/nasal polyps, anaphylactic food allergies, anxiety, depression)
Notes on patient occupation, triggers and family history of AD
AD patients typically insist on allergy testing due to misperceptions about the link between their condition and allergic reactions. This often leads to an inconclusive diagnosis (rash, eruption as descriptor) and leads to patient frustration. Patient and physician education about pathophysiology of AD are crucial to achieve treatment success.
Once an AD diagnosis is confirmed, upadacitinib should be prescribed to patients with moderate-to- severe AD who have not responded, are not expected to respond, or have had adverse reactions to long term use of topical corticosteroids.
Which patients would be least suitable for treatment with the drug under review?
AD patients with mild disease (majority of AD population), and whose symptoms can be controlled with lifestyle changes and topical corticosteroids would be least suitable for treatment with upadacitinib.
Is it possible to identify those patients who are most likely to exhibit a response to treatment with the drug under review?
In phase 3 trials, patients were selected based on eligibility criteria including:
Chronic AD before Baseline Visit
Active moderate to severe AD defined by EASI, IGA, BSA, and pruritus
Candidate for systemic therapy or have recently required systemic therapy for AD
Documented history of inadequate response to topical corticosteroids or topical calcineurin inhibitor OR documented systemic treatment for AD within 6 months before Baseline Visit
(Measure Up 1, https://clinicaltrials.gov/ct2/show/NCT03569293; Measure Up 2, https://clinicaltrials.gov/ct2/show/NCT03607422; AD Up, https://clinicaltrials.gov/ct2/show/NCT03568318; Heads Up, https://clinicaltrials.gov/ct2/show/NCT03738397).
What outcomes are used to determine whether a patient is responding to treatment in clinical practice?
In clinical trials, outcomes are measured by the IGA (Investigator Global Assessment), and proportion of participants achieving a 75% or 90-100% improvement on the EASI. These outcomes are also used in clinical practice (the equivalent PGA [Physician Global’s Assessment] is used instead of IGA) and are similarly aligned with additional measures, including body surface area (BSA) affected, and the pruritus numerical rating scale (NRS), which ranges from 0 (“no itch”) to 10 (“worst imaginable itch”). EASI scoring is not routinely used in clinical practice.
What would be considered a clinically meaningful response to treatment?
A clinically meaningful response to upadacitinib would include improvements in:
patient-reported itch (4 point reduction on the NRS or a NRS score of less than 3)
DLQI score reduction of equal or more than 4 (or an acceptable improvement)
patient-reported sleep quality and fewer AD-related disruptions at school and work
PGA score to 0 or 1
Importantly, a patient should not experience any severe side effects, including over sustained time periods, in order for the response to upadacitinib to be clinically meaningful.
How often should treatment response be assessed?
Response to systemic therapy is reassessed in 12-16 weeks after initiation of treatment.
What factors should be considered when deciding to discontinue treatment?
The decision to discontinue treatment should be assessed based on lack of response, significant disease progression (i.e. lichenifcation, increased affected BSA and itching) and deterioration in quality of life.
Treatment should also be discontinued if the patient experiences adverse reactions or intolerance to the medication that are deemed to be unacceptable by the patient-physician team.
Treatment with upadacitinib should be interrupted if a patient develops a serious infection, until the infection is controlled. Treatment should also be interrupted to address abnormal laboratory results (ALC less than 500 cells/mm3, ANC less than 1000 cells/mm3, Hb less than 8 g/dL, or if drug-induced liver injury is suspected [based on hepatic transaminases])and may be resumed once levels return to normal.(RINVOQ Product Monograph. 2019. Canada. AbbVie Inc).
What settings are appropriate for treatment with the drug under review?
Patients with AD receiving upadacitinib would ideally be managed in any non-emergent setting that they have access to, and that has a dermatologist or allergist well-versed in managing moderate-to-severe AD. Referring family physicians, nurse practitioners, or other health care providers should be counseled on the appropriate referral process.
For non-oncology drugs, is a specialist required to diagnose, treat, and monitor patients who might receive the drug under review?
As mentioned above, an ideal care pathway would involve referral from front-line AD health care providers to a trained dermatologist or allergist, who would prescribe and manage treatment with upadacitinib for moderate-to-severe AD.
Additional Information
Is there any additional information you feel is pertinent to this review?
Upadacitinib presents a breakthrough in moderate to severe AD management as reflected by its efficacy, impact on patients’ QoL in combination with acceptable safety profile for long term use.
Conflict of Interest Declarations — Atlantic Specialist Group Managing Atopic Dermatitis
To maintain the objectivity and credibility of the CADTH drug review programs, all participants in the drug review processes must disclose any real, potential, or perceived conflicts of interest. This conflict of interest declaration is required for participation. Declarations made do not negate or preclude the use of the clinician group input. CADTH may contact your group with further questions, as needed. Please see the Procedures for CADTH Drug Reimbursement Reviews (see Place in Therapy section) for further details.
Did you receive help from outside your clinician group to complete this submission? If yes, please detail the help and who provided it.
No.
Did you receive help from outside your clinician group to collect or analyze any information used in this submission? If yes, please detail the help and who provided it.
No.
List any companies or organizations that have provided your group with financial payment over the past two years AND who may have direct or indirect interest in the drug under review. Please note that this is required for each clinician that contributed to the input — please add more tables as needed (copy and paste). It is preferred for all declarations to be included in a single document.
Declaration for Clinician 1
Name: Dr. Wayne P. Gulliver
Position: Dermatologist
Date: 06-05-2021
Table 7: COI Declaration for Atlantic Specialist Group Managing Atopic Dermatitis Clinician 1
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
AbbVie | — | — | — | X |
Declaration for Clinician 2
Name: Dr. Irina Turchin
Position: Dermatologist
Date: 06-05-2021
Table 8: COI Declaration for Atlantic Specialist Group Managing Atopic Dermatitis Clinician 2
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Abbvie (advisory board, speaker, educational program development, investigator) | — | — | X | — |
LeoPharma(advisory board, speaker, educational program development, investigator | — | — | X | — |
Eli Lilly (advisory board, speaker, educational program development, investigator) | — | X | — | — |
Pfizer (advisory board, speaker) | — | X | — | — |
Sanofi (advisory board, speaker, educational program development) | — | — | X | — |
Declaration for Clinician 3
Name: Dr. Gina Lacuesta
Position: Assistant Professor, Faculty of Medicine, Dalhousie University, Consultant Physician in Allergy and Clinical Immunology Nova Scotia Health Authority
Date: 06-05-2021
Table 9: COI Declaration for Atlantic Specialist Group Managing Atopic Dermatitis Clinician 3
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
AbbVie – advisory board | X | — | — | — |
Sanofi – advisory board, speaker | X | — | — | — |
Declaration for Clinician 4
Name: Dr. Kerri Purdy
Position: Dermatologist
Date: May 6, 2021
Table 10: COI Declaration for Atlantic Specialist Group Managing Atopic Dermatitis Clinician 4
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Sanofi (advisory board, speaker) | X | — | — | — |
AbbVie (advisory board, speaker, education program development) | — | X | — | — |
Leo (advisory board, speaker) | X | — | — | — |
Eli Lilly (advisory board, speaker, education program development) | — | X | — | — |
Pfizer (speaker) | X | — | — | — |
Declaration for Clinician 5
Name: Dr. Anne-Marie Hunt
Position: Dermatologist
Date: May 6, 2021
Table 11: COI Declaration for Atlantic Specialist Group Managing Atopic Dermatitis Clinician 5
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
AbbVie | X | — | — | — |
Declaration for Clinician 6
Name: Catherine Rodriguez
Position: Dermatologist, Queen Elizabeth Hospital, Parkdale Medical Centre
Date: 2021-05-07
Table 12: COI Declaration for Atlantic Specialist Group Managing Atopic Dermatitis Clinician 6
Company | Check Appropriate Dollar Range | |||
|---|---|---|---|---|
$0 to 5,000 | $5,001 to 10,000 | $10,001 to 50,000 | In Excess of $50,000 | |
Abbvie | X | — | — | — |
LeoPharma | X | — | — | — |
Eli Lilly | X | — | — | — |
Pfizer | X | — | — | — |
Sanofi | X | — | — | — |
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
Redactions: Confidential information in this document may be redacted at the request of the sponsor in accordance with the CADTH Drug Reimbursement Review Confidentiality Guidelines
Stakeholder Input: The views expressed in each submission are those of the submitting organization or individual; not necessarily the views of CADTH or of other organizations. As such, they are independent of CADTH and do not necessarily represent or reflect the view of CADTH. No endorsement by CADTH is intended or should be inferred. By filing with CADTH, the submitting organization or individual agrees to the full disclosure of the information. CADTH does not edit the content of the submissions.
CADTH does use reasonable care to prevent disclosure of personal information in posted material; however, it is ultimately the submitter’s responsibility to ensure no identifying personal information or personal health information is included in the submission. The name of the submitting organization or individual and all conflict of interest information are included in the submission; however, the name of the author, including the name of an individual patient or caregiver submitting the patient input, are not posted.
Accessibility: CADTH is committed to treating people with disabilities in a way that respects their dignity and independence, supports them in accessing material in a timely manner, and provides a robust feedback process to support continuous improvement. All materials prepared by CADTH are available in an accessible format. Where materials provided to CADTH by a submitting organization or individual are not available in an accessible format, CADTH will provide a summary document upon request. More details on CADTH’s accessibility policies can be found here.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.