CADTH Reimbursement Review
Amifampridine phosphate (Firdapse)
Sponsor: KYE Pharmaceuticals Inc.
Therapeutic area: Lambert-Eaton myasthenic syndrome
This multi-part report includes:
Clinical Review
Pharmacoeconomic Review
Stakeholder Input
Clinical Review
Abbreviations
3TUG
Triple Timed-Up-and-Go
3,4-DAP
3,4-diaminopyridine
ADL
activities of daily living
AE
adverse event
ANCOVA
analysis of covariance
AUC
area under the curve
AUC0–t
area under the concentration-time curve to last quantifiable concentration
CDEC
CADTH Canadian Drug Expert Committee
CI
confidence interval
CGI-I
Clinical Global Impression–Improvement
CGI-S
Clinical Global Impression–Severity of Illness
CMAP
compound muscle action potential
Cmax
peak concentration
CP
coverage probability
FVC
forced vital capacity
HRQoL
health-related quality of life
IVIg
IV immunoglobulin
LEMS
Lambert-Eaton myasthenic syndrome
LSM
least squares mean
MID
minimal important difference
MG
myasthenia gravis
NMJ
neuromuscular junction
PP
per-protocol
QMGS
Quantitative Myasthenia Gravis Score
SAE
serious adverse event
SCLC
small cell lung cancer
SD
standard deviation
SGI
Subject Global Impression
T25-FW
Timed 25-Foot Walk
Tmax
time to peak concentration
VGCC
voltage-gated calcium channel
Executive Summary
An overview of the submission details for the drug under review is provided in Table 1.
Item | Description |
|---|---|
Drug product | Amifampridine phosphate (Firdapse), 10 mg oral tablets |
Indication | For the symptomatic treatment of LEMS in adults |
Reimbursement request | As per indication |
Health Canada approval status | NOC |
Health Canada review pathway | Priority review |
NOC date | July 31, 2020 |
Sponsor | KYE Pharmaceuticals Inc. |
LEMS = Lambert-Eaton myasthenic syndrome; NOC = Notice of Compliance.
Introduction
Lambert-Eaton myasthenic syndrome (LEMS) is a rare autoimmune disorder of the neuromuscular junction (NMJ).1-3 In approximately 90% of diagnosed patients, LEMS occurs as a result of the production of antibodies against the P- and Q-type voltage-gated calcium channels (VGCCs); this ultimately prevents muscle contraction.1,3-5 There are 2 forms of LEMS: paraneoplastic and primary autoimmune. Approximately 50% to 60% of LEMS cases are paraneoplastic and are most commonly associated with small cell lung cancer (SCLC).1,6 LEMS associated with other autoimmune diseases is referred to as primary autoimmune LEMS.2 Symptoms associated with both forms of LEMS include proximal muscle weakness, autonomic disturbance, and depressed tendon reflexes.1,2,4 Patients with LEMS often initially present in clinic with weakness in hips, legs and, in some cases, difficulty walking.2 According to the patient input received for this review, LEMS negatively impacts all areas of patients’ lives. The key concerns raised in the patient input included issues related to impaired muscle strength, impaired bodily functions, and difficulty performing activities of daily living (ADL).
The estimated incidence of LEMS ranges from 0.2 per million to 0.5 per million and the prevalence of LEMS ranges from 2.3 per million to 2.6 per million, based on published studies from Denmark,7 the Netherlands,8,9 and the US.10 There are no published Canadian epidemiological data on LEMS; however, the estimates from Denmark, the Netherlands, and the US are considered by the clinical expert consulted for this review to be comparable to Canada.
Amifampridine, both the phosphate and base form, has been used as a first line of therapy for both paraneoplastic and primary autoimmune forms of LEMS in Canada and internationally for more than 30 years for the symptomatic treatment of LEMS, despite it not being commercially available in Canada until 2020. Other medications and procedures that may be used in combination with amifampridine include pyridostigmine, immunosuppressants and immunomodulating agents, steroids, IV immunoglobulin (IVIg), and plasma exchange.
Amifampridine phosphate is indicated for the symptomatic treatment of LEMS in adults. Amifampridine phosphate was granted priority review by Health Canada and received a Notice of Compliance on July 31, 2020.
The objective of this report is to perform a systematic review of the beneficial and harmful effects of amifampridine phosphate (tablets equivalent to 10 mg amifampridine) for the symptomatic treatment of LEMS in adults.
Stakeholder Perspectives
The information in this section is a summary of input provided by the patient groups that responded to CADTH’s call for patient input and from clinical expert(s) consulted by CADTH for the purpose of this review.
Patient Input
In the absence of patient group input, 1 testimonial of the experiences of a Canadian individual with LEMS was accepted for this CADTH review, given the rarity of LEMS in Canada.
The patient testimonial highlighted symptoms of LEMS, including worsening arm, core, and leg strength, dry mouth, difficulty swallowing, muscle weakness, and becoming prone to falls. The patient specified that their disease experience led to their inability to continue working.
The patient was initially treated with pyridostigmine and then amifampridine. Treatment with amifampridine was reported to increase the patient’s mobility and independence (e.g., ability to rise from a seated position without assistance, ability to navigate stairs safely), and symptoms (e.g., improvement in dry mouth and swallowing).
The patient testimonial highlighted the desire for improvement in muscle strength and bodily functions, with the goal of performing daily activities with a sense of normalcy.
Clinician Input
The clinical expert consulted by CADTH for this review identified access to amifampridine as the main unmet need for patients with LEMS, as amifampridine has historically been accessed through compassionate use.
The clinical expert considers amifampridine to be the first line of therapy for the treatment of LEMS and agreed that there is no acceptable alternative to it for the symptomatic treatment of LEMS. Despite poorer prognosis of patients with the paraneoplastic form of LEMS, the clinical expert states that all patients with LEMS should have access to amifampridine.
Improvement in health-related quality of life (HRQoL) and functional ADL is the ultimate goal of treatment for patients with LEMS, based on input from the clinical expert consulted by CADTH. The ideal assessment of treatment effect consists of the patient’s subjective response, a neurologic exam, the Triple Timed-Up-and-Go (3TUG) test (or alternative assessment), and an electrophysiological study. However, variability in clinicians’ assessment of response to treatment is noted in the Canadian clinical setting.
The diagnosis and treatment for patients with LEMS is overseen by a specialist in neurology. The assessment of response to treatment with amifampridine typically involves assessment at baseline (pre-treatment), once within the first month (typically within a week or 2 of initiation), and every 3 months until the treating clinician perceives that the patient’s symptoms are being appropriately managed.
The clinical expert states that patients who respond to amifampridine are expected to continue treatment throughout their life. Patients who discontinue treatment with amifampridine include patients whose symptoms do not improve based on a combination of the following: the patient’s subjective response, an objective neurologic exam, 3TUG (or alternative assessment), and an electrophysiological study.
Clinical Evidence
Pivotal Studies and Protocol Selected Studies
Description of Studies
Two pivotal trials, LMS-002 (N = 38) and LMS-003 (N = 26), were included in the CADTH systematic review. Both studies were phase III, multi-centre, randomized, double-blind, placebo-controlled withdrawal studies that aimed to assess the safety and efficacy of amifampridine phosphate for the treatment of LEMS in adult patients.
The LMS-002 study was composed of 4 parts in addition to an initial screening phase. The open-label run-in phase, in which all patients received amifampridine phosphate, allowed the investigator to titrate to the optimal dose regimen for each patient. Patients who were amifampridine phosphate–naive were required to achieve a 3-point improvement or more in their Quantitative Myasthenic Gravis Score (QMGS) from the score reported at screening. All patients were required to have received amifampridine (phosphate or base) for a minimum of 91 days and a stable dose of amifampridine phosphate for a minimum of 7 days. On day 1 of part 2, patients were randomized to either continue receiving the established amifampridine phosphate dose or to taper treatment to placebo. Patients in the discontinuation group were tapered over the course of 7 days. On day 1 of part 3, patients in the discontinuation arm received only placebo and continued with this regimen for 7 days. After the 14-day double-blind period of part 2 and part 3, all patients were transitioned to open-label amifampridine phosphate in the long term safety phase of the trial.
The LMS-003 study consisted of a 4-day double-blind withdrawal period. All patients were previously enrolled in an expanded access program and required to be on a stable dose of amifampridine phosphate for 1 week before randomization. Patients were randomized to maintain either their regular amifampridine phosphate dose or their placebo for day 1 through day 4. Efficacy assessments were conducted on day 0 and day 4 following the final blinded dose. Following the study, patients were permitted to return to the expanded access program.
Efficacy Results
In the LMS-002 study, patients who discontinued amifampridine phosphate treatment reported a statistically significant disease progression according to the co-primary end point of difference in QMGS least squares mean (LSM) of –1.7 (95% confidence interval [CI]), –3.4 to –0.0; P = 0.0452). Though this result is statistically significant, it is below the identified clinically significant threshold of 2.6 units (note that this threshold was determined in myasthenia gravis [MG] patients; no such threshold has been identified in patients with LEMS).11 Similarly, the co-primary end point of the LMS-003 study was difference in QMGS LSM, reporting both a statistically and clinically significant difference of –6.54 (95% CI, –9.78 to –3.29; P = 0.0004).
The second co-primary end point in both the LMS-002 trial and the LMS-003 trial was Subject Global Impression (SGI). There was a statistically significant disease progression in patients who discontinued amifampridine phosphate according to difference in LSM of 1.8 (95% CI, 0.7 to 3.0; P = 0.0028) in LMS-002 and of 2.95 (95% CI, 1.53, 4.38; P = 0.0003) in LMS-003. There was no clinically significant threshold identified for the SGI measure in patients with LEMS; however, the clinical expert consulted for this review considered the results to be clinically meaningful.
The LMS-002 study included Clinical Global Impression–Improvement (CGI-I) as the first secondary end point, only to be formally tested if both co-primary end points were statistically significant. There was a statistically significant difference in LSM of –1.1 (95% CI, –2.1 to –0.1; P = 0.0267) that favoured amifampridine phosphate. Given the statistical significance of CGI-I, the second secondary end point in LMS-002, Timed 25-Foot Walk (T25-FW), was formally tested. Patients discontinuing amifampridine phosphate showed a slight numerical difference toward disease progression; however, the difference in LSM of 8.51 (95% CI, –26.77 to 43.79; P = 0.6274) showed no statistical difference.
The LMS-003 study included only 1 secondary end point, though there was no evidence that methods for controlling multiplicity were applied and, therefore, definitive conclusions cannot be drawn. LMS-003 reported only post-baseline values as baseline CGI-I was not recorded, further negatively impacting the ability to interpret any apparent treatment differences. Patients in the amifampridine phosphate arm reported a post-baseline mean of 3.8 and patients in the placebo arm reported a post-baseline mean of 5.5; the nominal P value based on the Wilcoxon rank sum test was 0.0020.
Harms Results
In the LMS-002 study, adverse events (AEs) were reported separately through the different phases of the trial. During the open-label run-in phase, AEs were reported for 53 patients, including those who would eventually withdraw from the trial. AEs were reported in 83.3% of treatment-naive patients and 27.3% of treatment-experienced patients. The most commonly reported AEs were in treatment-naive patients — namely, paresthesia (42.9%) and oral paresthesia (47.6%).
A total of 25% of patients experienced serious adverse events (SAEs) during the open-label safety extension, 1 of which was fatal SCLC. All but 2 SAEs were deemed by the investigator to be unrelated to the study drug, while those deemed probable to be related to the study drug were managed by dose reduction. In the LMS-003 study, AEs were reported in 23.1% of patients receiving amifampridine phosphate and 76.9% of patients in the placebo arm. The most common AEs reported were muscular weakness (38.5%) and fatigue (30.8%), though these were both in the placebo arm and are common symptoms of LEMS progression itself. Therefore, there is uncertainty surrounding whether safety signals are due to treatment side effects or disease progression itself.
Table 2: Summary of Key Results From Pivotal and Protocol Selected Studies
Characteristic | LMS-002 study | LMS-003 study | ||
|---|---|---|---|---|
Amifampridine phosphate N = 16 | Placebo N = 21 | Amifampridine phosphate N = 13 | Placebo N = 13 | |
Co-primary end point: QMGS | ||||
Change from baseline, mean (SD) | 0.3 (2.60) | 2.2 (2.93) | 0.1 (3.07) | 6.5 (4.82) |
Difference in LSMa (95% CI) | –1.7 (–3.4 to –0.0) | Reference | –6.54 (–9.78 to –3.29) | Reference |
P valueb | 0.0452c | Reference | 0.0004d | Reference |
Co-primary end point: SGI | ||||
Change from baseline, mean (SD) | –0.7 (1.82) | –2.7 (2.29) | –0.8 (1.74) | –3.5 (2.18) |
Difference in LSMa (95% CI) | 1.8 (0.7 to 3.0) | Reference | 2.95 (1.53 to 4.38) | Reference |
P valueb | 0.0028c | Reference | 0.0003d | Reference |
Secondary end point: CGI-I | ||||
Post-baseline, mean (SD) | 3.6 (1.50) | 4.8 (1.45) | 3.8 (0.80) | 5.5 (1.27) |
Difference in LSMa (95% CI) | –1.1 (–2.1 to –0.1) | Reference | NE | NE |
P valueb | 0.0267c | Reference | 0.0020e | Reference |
LMS-002 secondary end point: T25-FW | ||||
Change from baseline, mean (SD) | –1.46 (52.5) | –10.4 (53.1) | NE | NE |
Difference in LSMa (95% CI) | 8.51 (–26.77 to 43.79) | Reference | NE | NE |
P valueb | 0.6274c | Reference | NE | NE |
Harms | ||||
AEs | Part 2: 6 (37.5) Part 3: 3 (18.8) | Part 2: 3 (13.6) Part 3: 6 (27.3) | 3 (23.1) | 10 (76.9) |
SAEs | 0 | 0 | 0 | 0 |
Patients who stopped treatment due to AE | 0 | 0 | 0 | 0 |
Deaths | 0 | 0 | 0 | 0 |
Notable harms | NA | NA | NA | NA |
Paresthesia | 0 | 0 | 0 | 0 |
Hypoesthesia, oral | 0 | 0 | 0 | 0 |
Paresthesia, oral | 0 | 0 | 0 | 0 |
Seizures | 0 | 0 | 0 | 0 |
Change in electrocardiogram | 0 | 0 | 0 | 0 |
AE = adverse event; CI = confidence interval; CGI-I = Clinical Global Impression–Improvement; LSM = least squares mean; MMRM = mixed model of repeated measures; NA = not applicable; NE = not evaluated; QMGS = Quantitative Myasthenia Gravis Score; SAE = serious adverse event; SD = standard deviation; SGI = Subject Global Impression; T25-FW = Timed 25-Foot Walk.
aFor LMS-002, this was estimated via a MMRM with change from baseline (day 1, part 2), day 8, and day 14 as the dependent variable, and terms for treatment, time (day 8, day 14), treatment-by-time interaction, and double-blind baseline outcome score as fixed effects and patient as a random effect. The model assumed time effect to be random between patients. For the LMS-003 study, change from baseline for outcome total score was modelled as the response, with fixed-effects terms for treatment and outcome score at baseline.
bAll end points were controlled for multiplicity, with the exception of CGI-I in the LMS-003 study.
cThe P value represented a pairwise contrast at day 14 from the MMRM.
dThe P value was based on conducting a randomization test by running the fixed-effects linear model analysis on permuted treatment assignments. For each of the 10,000 permutations, change from baseline was modelled as the response for each end point, with fixed-effects terms for treatment and score at baseline.
eThe P value was based on the Wilcoxon rank sum test for treatment differences.
Source: LMS-002 Clinical Study Report (2015)12 and LMS-003 Clinical Study Report (2017).13
Critical Appraisal
Both the LMS-002 trial and the LMS-003 trial were double-blind studies that employed various strategies to maintain blinding of the patients, investigator, site personnel, and sponsor personnel. However, by designing a study using a withdrawal enrichment strategy, partial unblinding was possible as patients in the placebo arm were anticipated to experience deterioration before amifampridine phosphate being reinstated. Unblinding in LMS-002 and LMS-003 may have biased subjective patient-assessed (e.g., SGI) and investigator-assessed (e.g., QMGS, CGI-I) outcome results in favour of amifampridine phosphate.
The co-primary end points for both the LMS-002 study and the LMS-003 study were QMGS and SGI. QMGS is a measure developed for use in MG and includes components relating to ocular and bulbar involvement that are more relevant to MG and not expected to be impacted by treatment for LEMS. While the QMGS was not considered a relevant assessment tool in LEMS by the clinical expert consulted by CADTH, as it was designed and validated for the assessment of MG, the components of QMGS that are unrelated to LEMS would bias the results against amifampridine phosphate. The change in the QMGS components that are expected to be impacted by treatment would need to be more pronounced to reach statistical significance.
Subgroup analysis based on the type of LEMS (paraneoplastic versus primary autoimmune) was not performed in the LMS-002 study or the LMS-003 study. The LMS-003 trial did present results stratified by low dose (< 60 mg per day) and high dose (≥ 60 mg per day), which can be considered a rough proxy for disease severity according to the clinical expert consulted by CADTH, though the study was not powered to detect differences in this subgroup. Whether or not the treatment effect differs between subgroups (e.g., paraneoplastic versus primary autoimmune) identified as relevant in the CADTH review protocol remains unknown.
The withdrawal enrichment strategy used in both the LMS-002 study and LMS-003 study resulted in a stringently selected study population of patients who were treatment-experienced and responsive to amifampridine phosphate at baseline. Aspects of the trial design resulted in a study population that exhibited a magnitude of treatment response that may not be generalizable to Canadian patients who are treatment-naive, including those who are newly diagnosed with LEMS. It should be noted that the withdrawal design lends itself to the LEMS population, which includes heterogeneity among fast and slow amifampridine metabolizers, requiring the inclusion of a dose titration phase for treatment-naive patients.
Overall, the baseline characteristics of patients in the LMS-002 trial and LMS-003 trial were generally consistent with the Canadian clinical population currently treated with amifampridine phosphate. However, in LMS-002 and LMS-003, 15.8% and 23.1% of patients had paraneoplastic syndrome, respectively, likely due to the requirement for patients to have completed anticancer treatment at least 3 months before screening. This is inconsistent with the clinical population where it is estimated that 50% to 60% of patients have paraneoplastic syndrome. Patients with paraneoplastic LEMS are known to have poorer prognosis due to the underlying neoplastic condition; thus, the results of the LMS-002 study and LMS-003 study may not be representative of these patients. It is noted that the clinical expert consulted did not expect there to be major differences in treatment efficacy of amifampridine phosphate based on these subgroups of patients.
There was notable inconsistency in the 2 trials, specifically in the magnitude of change in the QMGS. In the LMS-002 study, conducted in 2011, the change was 1.7, which was below the recognized minimal important difference (MID), though this threshold has not been validated in patients with LEMS. There was also an imbalance in QMGS at the baseline assessment (6.4 versus 5.6, a difference of 0.8), possibly due to a random sampling error amplified by the small sample size. When considering the small difference in QMGS between treatment arms, numerically, half the change at day 14 could potentially be explained by the unbalanced baseline value. In the LMS-003 study, which was conducted more recently in 2017, the change in QMGS was much higher at 6.5, though with a similar imbalance in baseline values. The inconsistency between trials was less pronounced in the SGI end point, though a smaller change was reported in LMS-002 than in LMS-003 (1.8 versus 3.0). These differences cast some uncertainty on the treatment effect. However, since the LMS-003 trial was conducted exclusively in the US where practice may be less variable and closer to the Canadian context, and given possible change over the past decades in patient treatment modality, this trial can be considered more generalizable to the current setting and more reliable in design.
Other Relevant Evidence
The sponsor evaluated the relative bioavailability of amifampridine phosphate in a randomized, crossover trial (the DAPSEL study). In this trial, the sponsors compared the formulations of amifampridine phosphate salt (in tablet formulation) with amifampridine base (in capsule formulation) to determine their relative bioequivalence. Statistical evaluation was performed for area under the concentration-time curve to last quantifiable concentration (AUC0–t) and peak concentration (Cmax) with analysis of variance and the 90% CI for the ratio of reference formulation (amifampridine phosphate salt) over the test formulation (amifampridine base) was calculated. The area under the curve (AUC) ratio had fallen within the pre-specified bioequivalence limits (80% to 125%). For the peak plasma concentration (Cmax), the observed inferior limit exceeded the 80.0% bound and was near the 75% bound proposed for highly variable drugs, leading to regulators noting that the efficacy profiles of the formulations would not be expected to differ.
Conclusions
Two phase III, double-blind, placebo-controlled withdrawal studies (the LMS-002 study, N = 38, and the LMS-003 study, N = 26) in adult patients with LEMS demonstrated that continuous treatment with amifampridine phosphate resulted in less disability progression compared with patients whose amifampridine phosphate was withdrawn. There was a –1.7 difference in QMGS LSM in LMS-002 and a difference in LSM of –6.54 in LMS-003. SGI showed similar differences between treatment arms with a LSM difference of 1.8 and 2.95 in the LMS-002 trial and the LMS-003 trial, respectively. All results were statistically significant, suggesting that amifampridine phosphate is aligned with outcomes important to patients — mainly improved muscle strength, though a clinically significant threshold specific to patients with LEMS was not determined. The effect of amifampridine phosphate on HRQoL and productivity was not evaluated in LMS-002 or LMS-003 and remains unknown. Evidence from the 2 trials was limited by the potential for unblinding and generalizability to the amifampridine-naive patient population.
The harms data obtained from the body of evidence reviewed for the CADTH report are limited. The LMS-003 study only reported harms results for 4 days of follow-up, and although the LMS-002 study included safety follow-up for up to 2 years, due to the withdrawal enrichment design of both LMS-002 and LMS-003, harms reported may not be a true reflection of the harms associated with amifampridine phosphate for all patients with LEMS.
Evidence gaps for the reviewed studies include the use of amifampridine phosphate in treatment-naive patients, and patients with paraneoplastic LEMS. Comparative clinical evidence for amifampridine phosphate against amifampridine base was lacking, though a bioequivalence study and clinical expert opinion suggest similarity of the 2 formulations.
Introduction
Disease Background
LEMS is a rare autoimmune disorder of the NMJ.1-3 In approximately 90% or more of diagnosed patients, LEMS occurs as a result of the production of antibodies against the P- and Q-type VGCCs on the presynaptic membrane at the NMJ, resulting in the reduction of functioning of calcium channels.1,3,4 This prevents calcium from entering the nerve terminal and triggering the fusion of acetylcholine vesicles with the synaptic membrane, which prevents the release of acetylcholine into the synaptic cleft and ultimately prevents muscle contraction.1,3-5
There are 2 forms of LEMS: paraneoplastic and primary autoimmune. Approximately 50% to 60% of LEMS cases are paraneoplastic and are most commonly associated with SCLC.1 Paraneoplastic LEMS typically begins in late adulthood at approximately 60 years, and is more common in male patients, although this may relate to the association with SCLC.6 Patients diagnosed with LEMS are subsequently screened for SCLC due to its strong association.1 LEMS without an associated cancer, which is sometimes associated with other autoimmune diseases, is referred to as primary autoimmune LEMS.2 Primary autoimmune LEMS occurs in patients of all ages and is more common in female patients.6
Symptoms and signs associated with LEMS include proximal muscle weakness, autonomic disturbance, and depressed tendon reflexes.1,2,4 Patients with LEMS often initially present in clinic with weakness in legs and, in some cases, difficulty walking.2 Autonomic disturbance may include dry mouth, constipation, erectile dysfunction, postural hypertension, and loss of sweating. As LEMS progresses, patients may experience weakness of the arms and bulbar issues such as dysphagia, swallowing difficulties, slurred speech, weakness of the neck, and ocular issues such as double vision and droopy eyes.1,2 According to the patient input received for this review, LEMS negatively impacts all areas of life. The key concerns raised in the patient input included issues related to impaired muscle strength, impaired bodily functions, and difficulty performing ADL.
In Canada, LEMS is diagnosed by neuromuscular specialists through clinical examination, serum antibody testing (P- and Q-type VGCCs), and electrodiagnostic testing, including motor nerve conduction studies, repetitive nerve stimulation, and studies of compound muscle action potential (CMAP) before and after maximum voluntary contraction.1 No formal guidelines are followed in Canada for the diagnosis and treatment of LEMS. Some symptoms of LEMS are similar to those associated with MG, and MG may be confused with LEMS if ocular-bulbar symptoms develop first.1,4 The clinical expert consulted by CADTH identified the potential for misdiagnosis or delayed diagnosis as a result of the rarity of LEMS combined with subtle symptoms noted in mild cases (e.g., subtle stiffness or weakness in legs). The clinical expert noted that underdiagnosis may occur for the paraneoplastic form of LEMS as symptoms of LEMS could be attributed to cancer-related or treatment-related (e.g., chemotherapy) symptoms. Misdiagnosis and delayed diagnosis are expected to have improved over the past decade due to improved awareness and knowledge of LEMS and the wider availability of confirmatory anti-VGCC antibody testing.1
The estimated incidence of LEMS ranges from 0.2 per million to 0.5 per million and the prevalence of LEMS ranges from 2.3 per million to 2.6 per million, based on published studies from Denmark,7 the Netherlands,8,9 and the US.10 LEMS is 46 times less prevalent than MG, whereas the annual incidence rate of LEMS is 14 times lower than that of MG; this is largely attributed to the poor survival of patients with LEMS and SCLC.8 There are no published Canadian epidemiological data on LEMS; however, the estimates from Denmark, the Netherlands, and the US are considered by the clinical expert consulted for this review to be comparable to Canada. The clinical expert consulted by CADTH noted that LEMS is very rare in the pediatric population. There are no published Canadian epidemiological data on pediatric LEMS.
Standards of Therapy
The ultimate treatment goal for patients with LEMS is improvement in HRQoL and functional ADL.
Amifampridine, both the phosphate and base form, has been used as a first line of therapy for both paraneoplastic and primary autoimmune forms of LEMS in Canada and internationally for more than 30 years for the symptomatic treatment of LEMS, despite it not being commercially available in Canada until 2020. Amifampridine has historically been accessed through Health Canada’s Special Access Program or via compassionate use. The clinical expert consulted by CADTH agreed that there is no acceptable alternative to amifampridine for the symptomatic treatment of LEMS. Pyridostigmine is a cholinergic agent that acts primarily by inhibiting acetylcholinesterase.14 It enhances cholinergic action by facilitating the transmission of impulses across NMJs.14 Patients with either form of LEMS may receive treatment with amifampridine in combination with pyridostigmine. Pyridostigmine is not considered an alternative form of treatment for amifampridine and at least 1 study showed no benefit from pyridostigmine alone or when added to amifampridine.15 According to the clinical expert consulted by CADTH for this review, pyridostigmine is most often used in Canada as a bridging agent for patients diagnosed with LEMS who may be waiting for access to amifampridine. The clinical expert stated that the clinical effectiveness of pyridostigmine is minor in most patients and that use of pyridostigmine is generally discontinued once patients have access to amifampridine.
Other medications and procedures that may be used in combination with amifampridine and/or pyridostigmine include immunosuppressants, immunomodulating agents, steroids, IVIg, or plasma exchange. According to the clinical expert consulted by CADTH, combination treatment may be considered in patients who do not have an adequate treatment response to amifampridine and/or pyridostigmine. In Canada, the use of IVIg or plasma exchange for the treatment of LEMS is rare.
The clinical expert consulted by CADTH revealed that in patients with the paraneoplastic form of LEMS, the underlying malignancy (most often SCLC) is usually treated first through surgical resection, radiation, or chemotherapy, or in parallel with amifampridine. The use of immunosuppressive agents is often avoided in the treatment of paraneoplastic LEMS due to the potential of increasing the likelihood of dissemination of the underlying SCLC. Otherwise, the treatment of paraneoplastic LEMS is generally similar to the treatment of primary autoimmune LEMS.
Drug
Amifampridine or 3,4-diaminopyridine (3,4-DAP) is a broad-spectrum potassium channel blocker. The exact mechanism by which amifampridine exerts its therapeutic effect in patients with LEMS has not been fully elucidated.16 Blocking potassium channels results in blocking the efflux of potassium ions, thereby prolonging the duration of the presynaptic action potential.3,5 This allows more VGCCs to open, thus increasing the entry of calcium into the nerve terminal.
Amifampridine phosphate is indicated for the symptomatic treatment of LEMS in adults. Amifampridine phosphate was granted priority review by Health Canada and received a Notice of Compliance on July 31, 2020.
Dosing should be individualized based on disease severity, patient response, and patient population.16 The dose should be gradually titrated to the optimal effective dose with the minimum of side effects.16 Once achieved, this optimal dose should be maintained, and dosing frequency should be adjusted, as needed.16 The recommended dosage regimen of amifampridine phosphate, 10 mg tablets, for oral administration is presented in Table 3.
Table 3: Key Characteristics of Amifampridine Phosphate
Characteristic | Amifampridine phosphate |
|---|---|
Mechanism of action | Amifampridine or 3,4-DAP is a broad-spectrum potassium channel blocker. Blocking potassium channels results in blocking the efflux of potassium ions, thereby prolonging the duration of the presynaptic action potential. |
Indicationa | For the treatment of symptomatic LEMS in adults |
Route of administration | 18.98 mg per tablet, equivalent to 10 mg amifampridine, administered orally |
Recommended dose |
|
Serious adverse effects or safety issues | NA |
3,4-DAP = 3,4-diaminopyridine; LEMS = Lambert-Eaton myasthenic syndrome; NA = not applicable.
aHealth Canada–approved indication.
Source: Firdapse Product Monograph (2020).16
Stakeholder Perspectives
Patient Group Input
This section was prepared by CADTH staff based on the input provided by patient groups.
About the Patient Groups and Information Gathered
No patient group input was received following CADTH’s call for patient input. Given the rarity of LEMS in Canada, CADTH accepted a testimonial of the experiences of a Canadian individual with LEMS.
Disease Experience
The patient who provided input for this submission reported that they were diagnosed with LEMS 1 year following worsening arm, core, and leg strength. Their symptoms included dry mouth, difficulty swallowing, muscle weakness, and becoming prone to falls. Eventually, due to the disease, the patient had to discontinue work as a teacher. At 37 years old, the patient believed that “I would eventually end up in a wheelchair or be bed ridden. It was pretty bleak.”
Experience With Treatment
Before diagnosis of LEMS, the patient reported being treated with IVIg therapy, which did not have any significant effect on their condition. The therapy led to the patient being severely ill and losing their white blood cells.
The patient was given pyridostigmine for treatment; however, the patient did not show significant improvement with the treatment.
The treating specialist was able to access and prescribe amifampridine for the patient. Following treatment with this drug, the patient stated that after using amifampridine, there were improved effects — particularly in their ability to rise from a seated position without assistance, in improvement in dry mouth and swallowing symptoms, and in their ability to navigate stairs safely that didn’t require holding both the railings. The patient was also using an immunosuppressive medication, azathioprine.
The patient stated:
“To say this drug is a blessing and does miracles is not overstating the results.”
“My close friends and family have since told me they feared for my life when I was at my weakest, and celebrated my return to almost normal.”
“The combination of these medications have given me a new lease on life and I am so grateful to have access to this medicine.”
With regard to the recent legal challenge to Ruzurgi resulting in the temporary withdrawal of the Health Canada Notice of Compliance, the patient stated:
“Just recently in the USA, Jacobus lost a court case to Catalyst which will result in the users of Ruzurgi losing their access and being forced to take the Firdapse. I am very afraid that will negatively impact my access too.”
Improved Outcomes
The patient is hopeful that the drug under review would help improve their muscle strength and other bodily functions, allowing them to perform daily activities with a sense of normalcy.
The patient identified that the cost of the drug is 1 of the main concerns, as the patient believes the drug would be unaffordable and access may be restricted if it is not reimbursed. The patient is emphatic about the continued and affordable access to amifampridine.
Clinician Input
Input From Clinical Expert Consulted by CADTH
All CADTH review teams include at least 1 clinical specialist with expertise regarding the diagnosis and management of the condition for which the drug is indicated. Clinical experts are a critical part of the review team and are involved in all phases of the review process (e.g., providing guidance on the development of the review protocol, assisting in the critical appraisal of clinical evidence, interpreting the clinical relevance of the results, providing guidance on the potential place in therapy). The following input was provided by 1 clinical specialist with expertise in the diagnosis and management of LEMS.
Unmet Needs
The clinical expert stated that amifampridine has been used in Canada and internationally for more than 30 years for the symptomatic treatment of LEMS. In Canada, amifampridine has not been commercially available to patients and has historically been accessed through compassionate use. Access to amifampridine is a challenge for patients, with financial barriers posing the greatest difficulties currently.
Place in Therapy
The clinical expert considers amifampridine phosphate to be the first line of therapy for the treatment of LEMS. Amifampridine phosphate has been used alone and in combination with other treatments or therapies (e.g., pyridostigmine, immunosuppressants and immunomodulating agents, steroids, IVIg) for the past 30 years. These other treatments are generally considered by the clinical expert to be insufficiently effective and associated with adverse effects. Panellists agree that there are no acceptable alternatives to amifampridine phosphate that are currently available. The recent approval of amifampridine phosphate by Health Canada is unlikely to cause a shift in the treatment paradigm; panellists expect it to remain as first-line therapy for the treatment of LEMS symptoms.
Patient Population
The LEMS patient population can be broadly classified as paraneoplastic or primary autoimmune. While patients with the paraneoplastic form of LEMS are known to have poorer prognosis (determined by the underlying SCLC) than patients with the primary autoimmune form, the clinical expert stated that all patients with LEMS should have access to amifampridine phosphate. The severity of LEMS ranges from mild cases to severe cases, although there is no formal classification of severity.
Assessing Response to Treatment
The clinical expert consulted by CADTH stated that improvement in HRQoL and functional daily activities is the ultimate goal of treatment for patients with LEMS. The ideal assessment consists of the patient’s subjective response (whether the patient thinks they are better), an objective neurologic exam (e.g., testing of cranial nerves, strength, reflexes), 3TUG (or an alternative clinical assessment such as the QMGS), and an electrophysiological study (e.g., CMAP amplitude before and after maximum voluntary contraction performed both before and after treatment with amifampridine).
The clinical expert noted that solely relying on a neurologic exam may be problematic as it does not always represent the patient’s functional experience (e.g., ability to move from a seated position to a standing position and walk). However, it was noted that some clinics are limited to standard neurologic exams to determine treatment response as they do not have the capacity to do more comprehensive exams of patients’ day-to-day function or timed assessments. The diagnosis and treatment of LEMS is not formally informed by any clinical practice guidelines. Additionally, the resources that neuromuscular clinics have to assess treatment response is variable within Canada. These 2 components may contribute to variability in clinicians’ assessment of response to treatment in the Canadian clinical setting.
The assessment of response to treatment with amifampridine typically involves assessment at baseline (pre-treatment), once within the first month (typically within a week or 2 and as early as within 3 days of initiation), and every 3 months until it is perceived by the treating clinician that the patient’s symptoms are being appropriately managed. Patients are then seen regularly once a year, depending on their clinical stability. Panellists report that the onset of benefit of amifampridine often occurs within hours; however, they often wait a few weeks for the patients to decide if they perceive a benefit. Panellists suggested that it may take 2 months to 3 months to determine the ideal dosing regimen with amifampridine.
Discontinuing Treatment
It was noted by the clinical expert that patients who respond to treatment with amifampridine phosphate are expected to continue treatment with amifampridine phosphate throughout their life, although doses may be adjusted. Patients who discontinue treatment with amifampridine phosphate include patients whose symptoms do not improve based on a combination of the following: the patient’s subjective response, an objective neurologic exam, 3TUG (or alternative assessment), and an electrophysiological study. The clinical expert panellist’s impression was that this was a rare occurrence.
Prescribing Conditions
The diagnosis and treatment for patients with LEMS is overseen by neuromuscular specialists who often work in specialized neuromuscular clinics. This may be a limiting factor to patients in rural settings.
Additional Considerations
The clinical expert consulted by CADTH highlighted the importance of access and affordability of amifampridine and agreed that there is no acceptable alternative to amifampridine for the symptomatic treatment of LEMS.
Clinician Group Input
No clinician group input was received for this review.
Drug Program Input
The drug programs provide input on each drug being reviewed through CADTH’s reimbursement review processes by identifying issues that may impact their ability to implement a recommendation. The implementation questions and corresponding responses from the clinical expert consulted by CADTH are summarized in Table 4.
Table 4: Summary of Drug Plan Input and Clinical Expert Response
Questions | Clinical expert response |
|---|---|
Relevant comparators | |
| For consideration by CDEC. |
Considerations for initiation of therapy | |
Ruzurgi has a Health Canada indication for patients 6 years and older; Firdapse only has a Health Canada indication for adult patients 18 years and older. Discussion point for Ruzurgi recommendation notes that although patients enrolled in the DAPPER study ranged from 23 years to 83 years, given the general mechanism of action, amifampridine is expected to be effective across age groups covered by the Health Canada indication. Pediatric patients are generally treated similarly to adult patients, according to the clinical expert.
| The clinical expert noted that Ruzurgi and Firdapse are similar and there is unlikely to be any pharmacological difference; therefore, the drugs should be used interchangeably with regard to use in the pediatric population, especially given the rarity of LEMS in the pediatric population. |
Considerations for continuation or renewal of therapy | |
Consider alignment with Ruzurgi renewal criteria. The 3TUG test is appropriate for ambulatory patients with LEMS. | For consideration by CDEC. |
Considerations for discontinuation of therapy | |
Will loss of response be defined by anything other than the 3TUG test? If not, consider alignment with Ruzurgi; a response to treatment is defined as an improvement of at least 30% on the 3TUG test. Also, consider alignment with Ruzurgi for implementation guidance regarding the 3TUG test only in ambulatory patients. Case-by-case assessment for non-ambulatory patients. | The clinical expert noted that physicians may be using QMGS to assess loss of response. Supports the inclusion of alternative methods to define loss of response using either 3TUG or QMGS. |
Considerations for prescribing therapy | |
Consider alignment with Ruzurgi; the patient should be under the care of a neurologist with expertise in managing LEMS. Include the same implementation guidance — namely, that virtual assessment by a neurologist would be acceptable. | For consideration by CDEC. |
System and economic issues | |
| For consideration by CDEC. |
3TUG = Triple Timed-Up-and-Go; CDEC = CADTH Canadian Drug Expert Committee; LEMS = Lambert-Eaton myasthenic syndrome; NOC = Notice of Compliance; pCPA = pan-Canadian Pharmaceutical Alliance; QMGS = Quantitative Myasthenia Gravis Score; vs. = versus.
Clinical Evidence
The clinical evidence included in the review of amifampridine phosphate is presented in 3 sections. The first section, the systematic review, includes pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those studies that were selected according to an a priori protocol. The second section includes indirect evidence selected from the literature that met the selection criteria specified in the review. The third section includes sponsor-submitted long-term extension studies and additional relevant studies that were considered to address important gaps in the evidence included in the systematic review.
Systematic Review (Pivotal and Protocol Selected Studies)
Objectives
To perform a systematic review of the beneficial and harmful effects of amifampridine phosphate (tablets equivalent to 10 mg amifampridine) for the symptomatic treatment of LEMS in adults.
Methods
Studies selected for inclusion in the systematic review will include pivotal studies provided in the sponsor’s submission to CADTH and Health Canada, as well as those meeting the selection criteria presented in Table 5. Outcomes included in the CADTH review protocol reflect outcomes considered to be important to patients, clinicians, and drug plans.
Table 5: Inclusion Criteria for the Systematic Review
Criteria | Description |
|---|---|
Population | Adult patients with LEMS Subgroups:
|
Intervention | Amifampridine phosphate (18.98 mg per tablet, equivalent to 10 mg amifampridine), administered orally
|
Comparator | The following administered alone or in combination:
|
Outcomes | Efficacy outcomes
Harms outcomes
|
Study designs | Published and unpublished phase III and phase IV RCTs |
AE = adverse event; HRQoL = health-related quality of life; LEMS = Lambert‐Eaton myasthenic syndrome; RCT = randomized controlled trial; SAE = serious adverse event; vs. = versus; WDAE = withdrawal due to adverse event.
aThis drug does not have a Health Canada indication for the treatment of patients with LEMS.
bThese outcomes were identified as being of particular importance to patients in the input received by CADTH from patient groups for the review of amifampridine base (Ruzurgi). No patient input was received for amifampridine phosphate.
The literature search for clinical studies was performed by an information specialist using a peer-reviewed search strategy according to the PRESS Peer Review of Electronic Search Strategies checklist.17
Published literature was identified by searching the following bibliographic databases: MEDLINE All (1946‒) via Ovid and Embase (1974‒) via Ovid. All Ovid searches were run simultaneously as a multi-file search. Duplicates were removed using Ovid deduplication for multi-file searches, followed by manual deduplication in Endnote. The search strategy consisted of both controlled vocabulary, such as the US National Library of Medicine’s MeSH (Medical Subject Headings), and keywords. The main search concepts were Firdapse (amifampridine phosphate) and LEMS. Clinical trials registries were searched: the US National Institutes of Health’s ClinicalTrials.gov, the WHO’s International Clinical Trials Registry Platform search portal, Health Canada’s Clinical Trials Database, and the European Union Clinical Trials Register.
No filters were applied to limit the retrieval by study type. Retrieval was not limited by publication date or by language. Conference abstracts were excluded from the search results. Refer to Appendix 1 for the detailed search strategies.
The initial search was completed on December 10, 2021. Regular alerts updated the search until the meeting of the CADTH Canadian Drug Expert Committee (CDEC) on April 27, 2022.
Grey literature (literature that is not commercially published) was identified by searching relevant websites from the Grey Matters: A Practical Tool for Searching Health-Related Grey Literature reference.18 Included in this search were the websites of regulatory agencies (the US FDA and European Medicines Agency). Google was used to search for additional internet-based materials. Refer to Appendix 1 for more information on the grey literature search strategy.
Two CADTH clinical reviewers independently selected studies for inclusion in the review based on titles and abstracts, according to the predetermined protocol. Full-text articles of all citations considered potentially relevant by at least 1 reviewer were acquired. Reviewers independently made the final selection of studies to be included in the review, and differences were resolved through discussion.
Findings From the Literature
A total of 2 studies were identified from the literature for inclusion in the systematic review (Figure 1). The included studies are summarized in Table 6. A list of excluded studies is presented in Appendix 2.
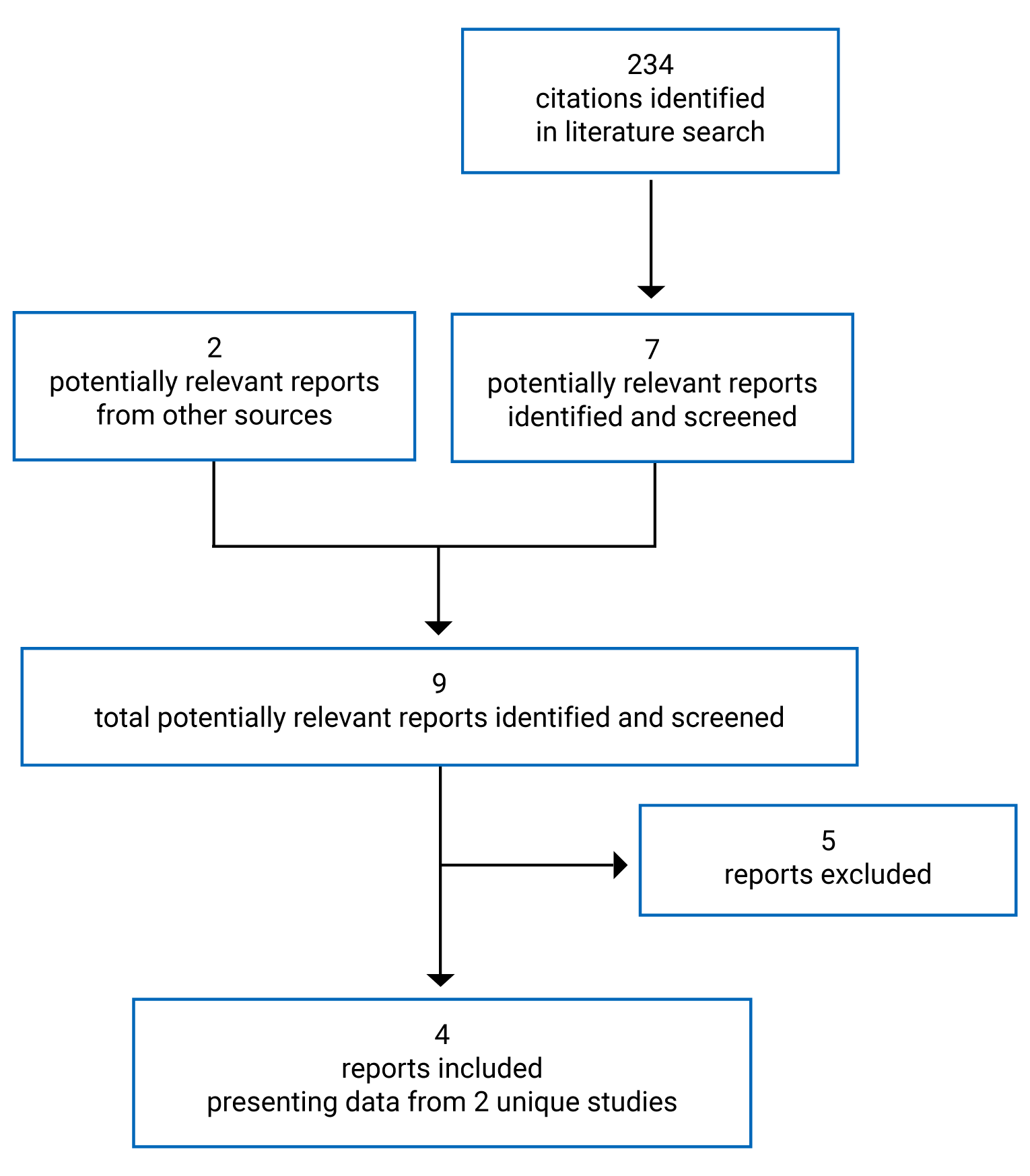
Table 6: Details of Included Studies
Detail | LMS-002 study | LMS-003 study |
|---|---|---|
Designs and populations | ||
Study design | Phase III, double-blind, placebo-controlled, randomized (1:1) discontinuation study | Phase III, double-blind, placebo-controlled, randomized (1:1) discontinuation study |
Locations | 8 centres in France, Germany, Hungary, Poland, Russia, Serbia, Spain, and the US | 3 centres in the US |
Patient enrolment dates | June 2011 | January 13, 2017 |
Randomized (N) | 38 | 26 |
Inclusion criteria |
|
|
Exclusion criteria |
|
|
Drugs | ||
Intervention | Amifampridine phosphate (18.98 mg per tablet, equivalent to 10 mg amifampridine), administered orally Daily dose was individually determined by the investigator within the bounds of 15 mg to 80 mg total daily dose and a maximum of 20 mg at any single administration | Amifampridine phosphate (18.98 mg per tablet, equivalent to 10 mg amifampridine), administered orally Daily dose was individually determined by the investigator within the bounds of 30 mg to 80 mg total daily dose, given in 3 to 4 divided doses, based on optimal neuromuscular benefit and tolerability. No single dose was > 20 mg. |
Comparator(s) | Placebo provided in tablet form indistinguishable from amifampridine phosphate tablets | Placebo provided in tablet form indistinguishable from amifampridine phosphate tablets |
Duration | ||
Phase | ||
Run-in | Between 7 and 91 days
| Patients must have received open-label amifampridine phosphate in the EAP, and have been on a stable dose and frequency of amifampridine phosphate for at least 1 week. |
Double-blind discontinuation phase | 7 days | NA |
Double-blind treatment phase | 7 days | 4 days |
Open-label long-term safety | 2 years following the last patient who entered the open-label phase | NA |
Outcomes | ||
Primary end point |
|
|
Secondary and exploratory end points | Secondary:
Tertiary:
Exploratory:
| Secondary:
Exploratory:
|
Notes | ||
Publications | Oh et al. (2016)19 | Shieh et al. (2019)20 |
3TUG = Triple Timed-Up-and-Go; CGI-I = Clinical Global Impression–Improvement; CGI-S = Clinical Global Impression–Severity of Illness; CMAP = compound muscle action potential; EAP = expanded access program; ECG = electrocardiogram; EMG = electromyogram; FVC = forced vital capacity; IP = investigational product; IVIg = IV immunoglobulin; LEMS = Lambert-Eaton myasthenic syndrome; NA = not applicable; NMJ = neuromuscular junction; PR = interval between the P and R waves on the electrocardiogram tracing; QMGS = Quantitative Myasthenia Gravis Score; QRS = QRS wave complex in the electrocardiogram tracing; Q–TcB = Q–T wave corrected for heart rate using Bazett formula; Q–Tc = Q–T interval corrected for heart rate; RR = interval between two R waves on the electrocardiogram tracing; SGI = Subject Global Impression; SSRI = selective serotonin reuptake inhibitor; T25-FW = Timed 25-Foot Walk.
Source: LMS-002 Clinical Study Report (2015)12 and LMS-003 Clinical Study Report (2017).13
Description of Studies
Two pivotal trials, LMS-002 (N = 36) and LMS-003 (N = 26), were included in the CADTH systematic review. Details of both studies are provided in Table 6.
LMS-002 was a phase III, double-blind, placebo-controlled, randomized (1:1) discontinuation study evaluating the efficacy and safety of amifampridine phosphate for the treatment of LEMS in adult patients. LMS-002 was conducted at 13 centres in 8 countries: France, Germany, Hungary, Poland, Russia, Serbia, Spain, and the US. The LMS-002 study took place between June 2011 and July 2016.
The LMS-002 trial was composed of 4 parts in addition to an initial screening phase (Figure 2). The open-label run-in phase, in which all patients received amifampridine phosphate, allowed the investigator to titrate to the optimal dose regimen for each patient. Patients who were amifampridine phosphate–naive were required to achieve a 3-point improvement or more in QMGS from the score reported at screening. All patients were required to have received amifampridine (phosphate or base) for a minimum of 91 days and a stable dose of amifampridine phosphate for a minimum of 7 days. On day 1 of part 2, patients were randomized to either continue receiving the established amifampridine phosphate dose or to taper treatment to placebo. Patients in the discontinuation arm were tapered over the course of 7 days. On day 1 of part 3, patients in the discontinuation arm received only placebo and continued with this regimen for 7 days. After the 14-day double-blind period of part 2 and part 3, all patients were transitioned to open-label amifampridine phosphate in the long term safety phase of the trial.
Figure 2: LMS-002 Study Design
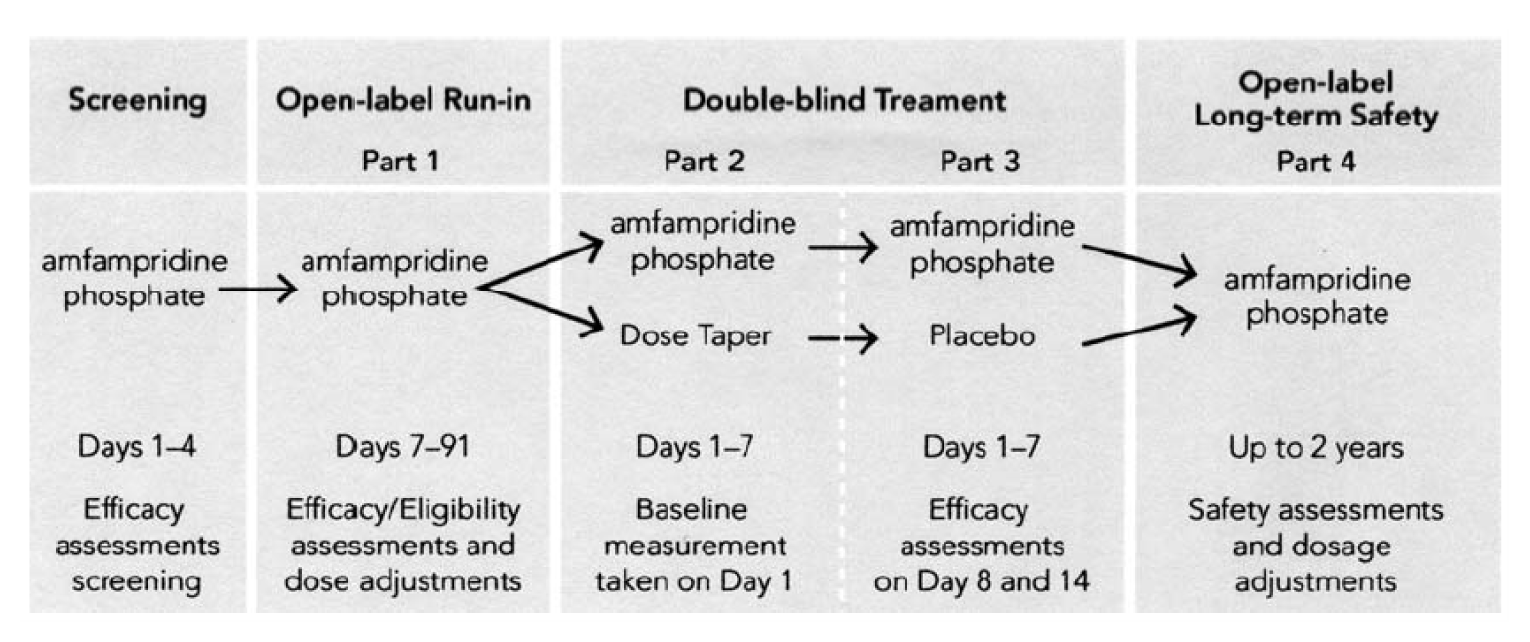
Note: In the open-label run-in phase, patients were required to receive amifampridine phosphate or base for 91 days, and a stable dose of amifampridine phosphate for 7 days before randomization.
Source: Oh SJ et al.19 Reprinted from Muscle and Nerve, Oh SJ et al., Amifampridine phosphate (Firdapse®) is effective and safe in a phase III clinical trial in LEMS, 53(5):717 to 725, © 2016, with permission from Wiley Periodicals, Inc.
LMS-003 was also a phase III, double-blind, placebo-controlled, randomized (1:1) discontinuation study evaluating the efficacy and safety of amifampridine phosphate for the treatment of LEMS in adult patients. The LMS-003 study was performed at 3 centres, exclusively in the US, between January 13, 2017, and October 30, 2017.
The LMS-003 trial consisted of a 4-day double-blind withdrawal period. All patients were previously enrolled in an expanded access program and required to be on a stable dose of amifampridine phosphate for 1 week before randomization. Patients were randomized to either maintain their regular amifampridine phosphate dose or placebo for day 1 through day 4. Efficacy assessments were conducted on day 0 and day 4 following the final blinded dose. Following the study, patients were permitted to return to the expanded access program.
Populations
Inclusion and Exclusion Criteria
Detailed inclusion and exclusion criteria for both the LMS-002 and LMS-003 trials are presented in Table 6.
LMS-002 was conducted in patients 18 years or older with confirmed LEMS with documented acquired proximal muscle weakness with either nerve conduction findings or a positive VGCC antibody test. Patients who were currently receiving amifampridine treatment were required to have normal respiratory function (≥ 80% forced vital capacity [FVC] of predicted) and present with some signs and/or symptoms of LEMS. Patients who were not currently receiving amifampridine treatment were required to have FVC of 60% or more and a QMGS of 5 or more. All patients must have completed anticancer treatment at least 3 months before screening. Patients were excluded from the LMS-002 study if they were likely or expected to require treatment for cancer within 3 months after entering screening, if they had a history of epilepsy or seizures, if they were taking medications known to lower the epileptic threshold, or if they received an IVIg or plasma exchange within 90 days before screening.
LMS-003 was also conducted in patients 18 years or older with confirmed LEMS by antibody testing or electromyogram. Patients were required to have been receiving amifampridine on a stable dose for at least 7 days before screening. Patients were required to have completed anticancer treatment at least 3 months before screening. Patients excluded from the LMS-003 study were those with a seizure disorder, those who were unable to ambulate, or those with a clinically significant long Q–T interval corrected for heart rate on electrocardiogram during the previous 12 months.
Baseline Characteristics
Baseline characteristics were generally balanced between arms in both the LMS-002 and LMS-003 studies (Table 7). In LMS-002, patients had a mean age of 51.6 (standard deviation [SD] = 12.05) years and 51.5 (SD = 17.57) years in the amifampridine phosphate and placebo arms, respectively. There was a higher proportion of female patients in both the amifampridine phosphate arm (56.3%) and the placebo arm (63.6%). Most patients were not receiving amifampridine treatment before enrolment — 81.3% and 68.2% in the amifampridine phosphate and placebo arms, respectively. Of the patients who were receiving prior amifampridine treatment, the median number of continuous days of amifampridine exposure was 365 days and 630 days in the amifampridine phosphate and placebo arms, respectively.
In the LMS-003 study, patients had a mean age of 54.9 (SD = 11.51) years and 53.4 (SD = 13.46) years in the amifampridine phosphate and placebo arms, respectively. Proportions of female patients similar to those of the LMS-002 study were seen in LMS-003, with 53.8% and 69.2% in the amifampridine phosphate and placebo arms, respectively.
Table 7: Summary of Baseline Characteristics
Characteristic | LMS-002 study | LMS-003 study | ||
|---|---|---|---|---|
Amifampridine phosphate N = 16 | Placebo N = 22 | Amifampridine phosphate N = 13 | Placebo N = 13 | |
Age, years | ||||
Mean (SD) | 51.6 (12.05) | 51.5 (17.57) | 54.9 (11.51) | 53.4 (13.46) |
Median (range) | 53.0 (25 to 67) | 56.5 (21 to 88) | 59.0 (33 to 71) | 52.0 (31 to 75) |
Gender, n (%) | ||||
Male | 7 (43.8) | 8 (36.4) | 6 (46.2) | 4 (30.8) |
Female | 9 (56.3) | 14 (63.6) | 7 (53.8) | 9 (69.2) |
Weight, kg | ||||
Mean (SD) | NR | NR | 77.62 (20.312) | 93.95 (15.449) |
Median (range) | NR | NR | 72.50 (56.1 to 121.4) | 90.90 (71.6 to 132.7) |
Ethnicity, n (%) | ||||
Hispanic | 3 (18.8) | 0 | 4 (30.8) | 1 (7.7) |
Not Hispanic | 12 (75.0) | 22 (100) | 9 (69.2) | 12 (92.3) |
Not reported | 1 (6.3) | 0 | 0 | 0 |
Race, n (%) | ||||
Black or African-American | 0 | 0 | NR | NR |
White | 13 (81.3) | 21 (95.5) | NR | NR |
Other | 2 (12.5) | 0 | NR | NR |
NR | 1 (6.3) | 1 (4.5) | NR | NR |
Was the patient taking amifampridine (base or phosphate) immediately before enrolment? | ||||
Yes, n (%) | 3 (18.8) | 7 (31.8) | NR | NR |
No, n (%) | 13 (81.3) | 15 (68.2) | NR | NR |
If yes, number of continuous days of amifampridine exposure immediately before enrolment | ||||
Mean (SD) | 2,143.3 (3,080.16) | 1,287.1 (1,525.73) | NR | NR |
Median (range) | 365.0 (365 to 5,700) | 630.0 (166 to 4,457) | NR | NR |
NR = not reported; SD = standard deviation.
Source: LMS-002 Clinical Study Report (2015)12 and LMS-003 Clinical Study Report (2017).13
Baseline disease characteristics for both the LMS-002 and LMS-003 trials are summarized in Table 8. LMS-002 reported 81.3% of patients in the amifampridine phosphate arm and 86.4% of the patients in the placebo arm as having primary autoimmune LEMS, with a smaller proportion of patients classified as paraneoplastic. The median duration of LEMS before enrolment was 2.25 years and 1.55 years in the amifampridine phosphate and placebo arms, respectively. More than 90% of patients in both arms were positive for calcium channel antibodies. In the LMS-003 study, 69.2% of patients in the amifampridine arm and 84.6% of patients in the placebo arm were classified as primary autoimmune LEMS, while 84.6% and 92.3% of patients, respectively, were positive for calcium channel antibodies.
Table 8: Summary of Baseline Disease Characteristics
Characteristic | LMS-002 study | LMS-003 study | ||
|---|---|---|---|---|
Amifampridine phosphate N = 16 | Placebo N = 22 | Amifampridine phosphate N = 13 | Placebo N = 13 | |
Current LEMS diagnosis, n (%) | ||||
Paraneoplastic | 3 (18.8) | 3 (13.6) | 4 (30.8) | 2 (15.4) |
Autoimmune | 13 (81.3) | 19 (86.4) | 9 (69.2) | 11 (84.6) |
Duration of LEMS, years | ||||
Mean (SD) | 6.52 (7.51) | 3.43 (4.24) | NR | NR |
Median (range) | 2.25 (0.7 to 21.5) | 1.55 (0.1 to 13.7) | NR | NR |
Calcium channel antibody, n (%) | ||||
Yes | 15 (93.8) | 20 (90.9) | 11 (84.6) | 12 (92.3) |
No | 1 (6.3) | 2 (9.1) | 2 (15.4) | 1 (7.7) |
LEMS = Lambert-Eaton myasthenic syndrome; NR = not reported; SD = standard deviation.
Source: LMS-002 Clinical Study Report (2015)12 and LMS-003 Clinical Study Report (2017).13
Interventions
LMS-002 Study
Patients eligible for enrolment in the LMS-002 study were randomized in an approximately 1:1 ratio to either arm A (treatment continuation) or arm B (treatment discontinuation). Study treatment was administered as tablets 4 times daily at home with food, except for the first dose that was given on the day of scheduled study visits. During the open-label run-in phase, the investigator determined the daily dose of amifampridine phosphate within the bounds of 15 mg and 80 mg per day, with a maximum single dose of 20 mg. Patients were up-titrated in 10 mg increments every 4 days to 5 days to a maximum dose of 80 mg.
All investigators, clinic staff, and patients were blinded to treatment during the double-blind phase of the trial. Identical tablets were used, and different raters performed the CMAP and QMGS tests on an individual patient to maintain blinding. The use of peripherally acting cholinesterase inhibitors (e.g., pyridostigmine) were continued throughout the study, provided they had been used at a stable regimen for at least 7 days before screening. The use of oral immunosuppressants was continued throughout the study, provided the immunosuppressants had been used at a stable regimen for at least 90 days before screening.
It was noted that at 1 study site, patients were incorrectly dosed in relation to the timing of efficacy assessments. Patients were assessed for efficacy before receiving their first dose of the day, when the study protocol indicates patients should receive the treatment dose and then be assessed for efficacy.
The use of best supportive care and concomitant medication for the LMS-002 study is summarized in Table 13.
LMS-003 Study
Patients eligible for the LMS-003 study were randomized to receive amifampridine phosphate or placebo in a 1:1 ratio, stratified by low dose (< 60 mg per day) and high dose (≥ 60 mg per day). The study drug was administered as identical tablets in clinic on day 0 (first dose) and on day 4 (last dose); all other administrations were done at home. The daily dose was determined by the investigator between the bounds of 30 mg and 80 mg per day across 3 to 4 individual doses, with no single dose greater than 20 mg.
Both patients and investigators were blinded to treatment. Unblinded personnel who prepared and dispensed the study treatment agreed not to provide any information that may have revealed the treatment assignment. Unblinded personnel were not involved in observation, monitoring, or reporting throughout the trial. The investigator was able to prescribe additional medications, given that they were not prohibited by the study protocol.
Outcomes
A list of efficacy end points identified in the CADTH review protocol that was assessed in the clinical trials included in this review is provided in Table 9. A detailed discussion and critical appraisal of the outcome measures is provided in Appendix 4.
Table 9: Summary of Outcomes of Interest Identified in the CADTH Review Protocol
Outcome measure | LMS-002 study | LMS-003 study |
|---|---|---|
QMGS | Co-primary end point | Co-primary end point |
SGI | Co-primary end point | Co-primary end point |
CGI-I | Secondary end point (first) | Secondary end point |
T25-FW | Secondary end point (second) | NE |
3TUG | NE | Exploratory end point |
CMAP | Tertiary end point | NE |
CGI-S | Tertiary end point | NE |
Patient-identified most bothersome symptom | NE | Exploratory end point |
3TUG = Triple Timed-Up-and-Go; CGI-I = Clinical Global Impression–Improvement; CGI-S = Clinical Global Impression–Severity of Illness; CMAP = compound muscle action potential; NE = not evaluated; QMGS = Quantitative Myasthenia Gravis Score; SGI = Subject Global Impression; T25-FW = Timed 25-Foot Walk.
Source: LMS-002 Clinical Study Report (2015)12 and LMS-003 Clinical Study Report (2017).13
Disability Progression
The QMGS measure was a co-primary end point in both the LMS-002 and LMS-003 trials. The QMGS is a 13-item physician assessed scale, developed for assessments in patients with MG. Each parameter is measured on a 0- to 3-point scale, where 0 indicates “no weakness” and 3 indicates “severe weakness.” Lower scores indicate better muscle strength (total score range = 0 to 39). The QMGS is composed of the following items: ocular (2 items), facial (1 item), bulbar (2 items), gross motor (6 items), axial (1 item), and respiratory (1 item). An MID of 2.6 points in patients with MG was determined in the original QMGS publication for MG patients.11 No MID specific to patients with LEMS was identified.
The SGI measure was a co-primary end point in both the LMS-002 and LMS-003 trials. The SGI is a patient-rated and patient-assessed 7-point scale gathering the global impression of the effects from a treatment, where 1 represents a “terrible” impression and 7 represents a “delighted” impression.21 In the case of a patient’s inability to complete the SGI, their parent, guardian, or caregiver can assess the SGI score. For the assessment of the SGI score, patients are asked to rate their impression of the effects of the study medication during the preceding week on their physical well-being, according to a score ranging from 1 to 7. No MID specific to patients with LEMS was identified.
The CGI-I measure was a secondary end point in both the LMS-002 study and the LMS-003 study. CGI-I is a 7-point scale used to capture the investigator’s global impression of disease symptom severity (improvement or worsening) from baseline status. For the assessment of the CGI-I score, investigators are asked to rate the patient’s total improvement due entirely to drug treatment, based on their judgment. The investigators are asked specifically to rate the patient’s change in severity, comparing it to the baseline condition. The 7-point scale consists of the following options: 1 is “very much improved,” 2 is “much improved,” 3 is “minimally improved,” 4 is “no change,” 5 is “minimally worse,” 6 is “much worse,” and 7 is “very much worse.” The investigators rate the scale based on changes in a patient’s symptoms, behaviour, and functional abilities. Although the CGI-I scale has been developed to use for capturing improvement after the initiation of a treatment or therapy, the sponsor used this scale to capture the deterioration of a patient’s condition after discontinuation of amifampridine phosphate in both the LMS-002 and LMS-003 trials. The Clinical Global Impression–Severity of Illness (CGI-S) measure was a tertiary end point in the LMS-002 study. CGI-S is a 7-point scale used to capture the investigator’s global impression of disease symptom severity at a given point in time. For the assessment of the CGI-S score, investigators are asked to rate the symptom severity at that time, based on their total clinical experience with that particular population. The 7-point scale ranges from 1, which is “normal, not at all ill,” to 7, which is “among the most extremely ill patients.” No MID specific to patients with LEMS was identified for either CGI-I or CGI-S.
The T25-FW measure was a secondary end point in LMS-002. The T25-FW test is a quantitative mobility and leg function performance test. This test is a component of the Multiple Sclerosis Functional Composite, which is used to measure leg function.22 During the test, a patient is directed to walk a clearly marked 25-foot course as quickly and safely as possible. Patients can use assistive devices, such as canes, crutches, or walkers, if needed. The test is repeated following a rest of at least 5 minutes. The average speed of the 2 completed walks, expressed in feet per minute, has been used to measure the T25-FW test. The T25-FW test was the secondary efficacy outcome measure in the LMS-002 study. No MID specific to patients with LEMS was identified.
The CMAP amplitude was a tertiary end point in LMS-002. The CMAP amplitude is an electrophysiologic measurement providing objective laboratory corroboration of the clinical effectiveness measures. Since the characteristic electrophysiologic pattern associated with LEMS supports the diagnosis of a presynaptic NMJ disorder, measuring CMAP amplitude is helpful for this indication. In this measurement process, the electrical stimulation of a motor nerve evokes responses in the appropriate muscle fibres. When the muscle potentials are recorded from the muscle surface, the summated response of multiple muscle fibres is called the CMAP. The CMAP amplitude in a resting muscle among patients with LEMS decreases proportionally with the severity of both the neuromuscular block and LEMS.15,23,24 Doubling the CMAP amplitude is considered as a clinically meaningful improvement for patients with LEMS.25,26
The 3TUG measure was an exploratory end point in LMS-003. The 3TUG is an observable measure of disease severity. The 3TUG is used to assess the potential effect on the Timed-Up-and-Go test of neuromuscular fatigue or facilitation, which are characteristic of LEMS. The 3TUG test consists of 3 laps, performed as follows. The patient is seated in a standard 18-inch-high straight-backed armchair. Three metres from the front legs of the chair, the floor is marked with a line of coloured tape and the centre of the line is marked with an X. Patients are instructed to get up from the chair, walk at their normal pace to the line, step on the X, turn around, walk back to the chair, turn around, and sit down. This is repeated 3 times without rest. Each lap ends when the patient’s back contacts the chair back and the patient is told either to begin the next lap or that the test is complete. The 3TUG time is the average of the 3 lap times.27 In the LMS-003 study, the 3TUG was obtained as an efficacy end point, based upon literature reports that a significant change in gait for a similar walk-test is an increase in time of more than 20%.13 No MID specific to patients with LEMS was identified.
The patient-identified most bothersome symptom assessment was an exploratory end point in LMS-003. Patients identify their most bothersome LEMS-associated symptom as a measure of patient satisfaction with the treatment. The evaluation consists of 2 questions. The first question is to identify before treatment or while off medication what the patient perceived as their most bothersome symptom and the level to which it bothered them, on a 4-point scale. Following blinded treatment with study medication, the patient is asked the second question: to evaluate how much the previously identified symptom bothered them during the prior 24 hours, on the same 4-point scale. No MID specific to patients with LEMS was identified.
Activities of Daily Living
No efficacy outcomes related to ADL were reported.
Lambert-Eaton Myasthenic Syndrome–Related Symptoms
No efficacy outcomes related to LEMS-related symptoms were reported.
Health-Related Quality of Life
No efficacy outcomes related to HRQoL were reported.
Productivity
No efficacy outcomes related to productivity were reported.
In the LMS-002 study, SGI was added as a co-primary end point in the May 2014 protocol amendment at the request of the FDA. CGI-I was initially considered a tertiary end point; however, in the same May 2014 protocol amendment, it was prospectively decided to use CGI-I as the first secondary objective and T25-FW as the second secondary objective.
Statistical Analysis
In the LMS-002 study, the original planned sample size was 30 patients who would provide 80% power to detect a 3.0-unit difference in mean change in QMGS. This was based on a trial conducted with 7 patients.28 In May 2014, following a review of Sanders et al. (2000),25 the statistical analysis plan was adjusted to randomize a total of 36 patients to provide 80% power to detect a 2.443-unit difference in mean change in QMGS, assuming type I error of 0.05. In the same protocol amendment, the primary analysis was changed from an analysis of covariance (ANCOVA) method to a random mixed-effects model to better account for missing data and data taken at the time of rescue medication. ANCOVA was conducted as a sensitivity analysis.
The LMS-002 trial used a step-down method to account for multiplicity in statistical testing. The first secondary end point, CGI-I, was only tested if both co-primary end points were statistically significant. The second secondary end point was likewise only formally statistically tested if both the co-primary and first secondary end point were statistically significant. In order for the co-primary end point to be considered statistically significant, both the QMGS and the SGI treatment comparisons need to be significant, using a 2-sided test at the alpha equals 0.05 level of significance that uses the permutation test. Tertiary end points were outside the statistical hierarchy.
The LMS-003 study was powered with respect to the co-primary efficacy end points of the study. For change from baseline in QMGSs, a between-treatment difference of –3.5, and a SD of at most 3, a sample size of at least 24 patients will provide power of at least 80% for a 0.05-level 2-sided test. Similarly, for change from baseline in SGI scores, a between-treatment difference of –2.1, and a SD of at most 2, a sample size of at least 26 subjects will provide power of 80% for a 0.05-level 2-sided test. Thus, a total sample size of 26 subjects, randomized at a 1:1 ratio to 2 treatment sequences, will provide power of at least 80% for each of the 2 co-primary end points.
The co-primary end points of QMGS and SGI were analyzed by fitting a fixed-effects linear model to the data with change from baseline as the response. The model included terms for treatment and SGI or QMGS at baseline. The LMS-003 study did not present methods to adjust for multiplicity of testing of other end points.
A summary of statistical analysis of efficacy end points in both the LMS-002 and LMS-003 trials is provided in Table 10.
Table 10: Statistical Analysis of Efficacy End Points
End point | Statistical model | Adjustment factors | Sensitivity analyses |
|---|---|---|---|
LMS-002 study | |||
QMGS SGI CGI-Ia T25-FWb | MMRM
|
|
|
LMS-003 study | |||
QMGS SGI | Analysis was performed by fitting a fixed-effects linear model to the data with change from baseline as the response. | The model included terms for treatment and QMGS or SGI at baseline. | For each co-primary end point, a randomization test was conducted to evaluate patterns of early treatment discontinuation. |
CGI-I | CGI-I scores were summarized descriptively with a Wilcoxon rank sum test to assess treatment arm differences. | NA | NA |
3TUG | A 2-sided Fisher’s exact test was conducted to test for treatment arm differences. | NA | The proportions were calculated 2 ways: once with only patients who were assessed on both day 0 and day 4, and alternatively using all subjects in the analysis population with day 0 data, regardless of whether they were assessed on day 4. |
Patient-identified most bothersome symptom question | Change from baseline was summarized descriptively with a Wilcoxon rank sum test to assess treatment arm differences. | NA | NA |
QMGS limb domain | The change from baseline for each of the 4 individual domains, and for the sum of the 4 domains, was performed by fitting a fixed-effects linear model to the data with change from baseline as the response. | The model included terms for treatment and QMGS of the individual domain or sum of the 4 domains at baseline. | NA |
3TUG = Triple Timed-Up-and-Go; ANCOVA = analysis of covariance; CGI-I = Clinical Global Impression–Improvement; MMRM = mixed model of repeated measures; NA = not applicable; QMGS = Quantitative Myasthenia Gravis Score; SGI = Subject Global Impression; T25-FW = Timed 25-Foot Walk.
aCGI-I used a MMRM model that was nearly identical to that which was used for QMGS and SGI, without the covariate for baseline value in the model.
bT25-FW used the same mixed-effects model that was used for QMGS and SGI but with double-blind baseline T25-FW walking speed as the covariate.
Source: LMS-002 Clinical Study Report (2015)12 and LMS-003 Clinical Study Report (2017).13
Analysis Populations
In the LMS-002 trial, the full analysis set included all 38 patients who received at least 1 dose of amifampridine or placebo in the double-blind phase and had at least 1 post-baseline efficacy assessment. The safety analysis population included 53 patients who received any dose of amifampridine phosphate and had any post-treatment safety information collected, including 15 patients from the open-label run-in phase who did not undergo randomization. An ad hoc per-protocol (PP) analysis was conducted to exclude patients from a specific study site where major protocol deviations occurred with respect to the timing of dose administration; the resulting population was 26 patients.
In the LMS-003 trial, the full analysis set and safety population included all 26 patients who were enrolled in the trial.
Results
Patient Disposition
In the LMS-002 study, 74 patients were screened, with 54 entering the open-label run-in phase. Of the 14 patients who withdrew from the open-label run-in phase, AEs was the most common reason for withdrawal (35.7%), while withdrawal by patient and “other” were the next most common reasons for withdrawal in the open-label run-in phase (21.4% for both). Two patients proceeded directly from the open-label run-in phase to the open-label safety phase as the enrolment target had been met. One patient was rescued following completion of the discontinuation phase and 1 additional patient was rescued during the treatment phase — both for change in QMGS greater than 5. Randomization was stratified to equalize treatment-naive patients and treatment-experienced patients but was only implemented after 9 patients had already been randomized, accounting for the imbalance in the amifampridine phosphate and placebo arms. The safety population included all patients who received at least 1 dose of amifampridine phosphate and had any post-treatment safety information collected.
In the LMS-003 study, 26 patients were screened, with 13 patients being randomized to receive amifampridine phosphate and 13 patients randomized to receive placebo. No patients withdrew from LMS-003. Patient disposition details for both the LMS-002 and LMS-003 trials are provided in Table 11.
Disposition | LMS-002 study | LMS-003 study | ||
|---|---|---|---|---|
Amifampridine phosphate | Placebo | Amifampridine phosphate | Placebo | |
Screened, N | 74 | 26 | ||
Entered into open-label run-in phase, N (%) | 54 (73.0) | NA | ||
Withdrew from run-in phase, N (%) | 14 (25.9) | NA | ||
Reason for withdrawal, N (%) | ||||
Adverse event | 5 (35.7) | NA | ||
Lack of efficacy | 2 (14.3) | NA | ||
Patient decision | 3 (21.4) | NA | ||
Physician decision | 1 (7.1) | NA | ||
Other | 3 (21.4) | NA | ||
Proceeded directly from run-in phase to open-label safety analysis | 2 (3.7) | NA | ||
Entered double-blind treatment discontinuation phasea | 16 | 22 | NA | NA |
Rescued following completion of discontinuation phase, N (%) | 0 | 1 (4.5) | NA | NA |
Entered double-blind treatment phaseb | 16 | 21 | 13 | 13 |
Rescued during treatment phase, N (%) | 0 | 1 (4.5) | 0 | 0 |
Entered open-label extension phasea | 40 | NA | NA | |
Withdrew from open-label extension phase, N (%) | 2 (12.5) | 0 | NA | NA |
Physician decision | 2 (100) | 0 | NA | NA |
FAS, N | 16 | 22 | 13 | 13 |
PP, N | 10c | 16c | 13 | 13 |
Safety,d N | 53 | 13 | 13 | |
FAS = full analysis set; NA = not applicable; PP = per-protocol; QMGS = Quantitative Myasthenia Gravis Score.
aTwo additional patients from the run-in phase were transferred directly to the open-label phase as the enrolment target for the double-blind phase had been met.
bOne patient was rescued due to high QMGS after completing the discontinuation phase and advanced to the open-label phase, and 1 patient was rescued during the double-blind treatment phase and advanced to the open-label phase.
cAn ad hoc per-protocol analysis was conducted in the LMS-002 study to exclude patients from 1 specific study site where many protocol deviations with respect to the timing of dose administration occurred.
dThe safety population for LMS-002 included 53 patients who received 1 dose of amifampridine in the open-label run-in phase, in addition to safety data reported for patients who entered the double-blind phase of the study.
Source: LMS-002 Clinical Study Report (2015)12 and LMS-003 Clinical Study Report (2017).13
Exposure to Study Treatments
In the LMS-002 trial, the mean duration of exposure was 570.8 (SD = 305.25) days and 558.4 (SD = 301.44) days in the amifampridine phosphate and placebo arms, respectively. At the final analysis of the open-label phase, the mean duration of exposure was 785.5 (SD = 310.5) days. The mean total daily dose was 64.1 (SD = 14.42) mg and 62.4 (SD = 17.41) mg in the amifampridine phosphate and placebo arms, respectively. The mean total daily dose during the open-label phase was 66.0 (SD = 17.90) mg.
In the LMS-003 trial, all patients were exposed to study treatments for 4 days. The mean total daily dose was 60.0 (SD = 19.58) mg and 63.1 (SD = 18.99) mg in the amifampridine phosphate and placebo arms, respectively.
Characteristic | LMS-002 study | LMS-003 study | |||
|---|---|---|---|---|---|
Amifampridine phosphate N = 16 | Placebo N = 22 | Open-label phase: amifampridine phosphate N = 40a | Amifampridine phosphate N = 13 | Placebo N = 13 | |
Duration of exposure, days | |||||
Mean (SD) | 570.8 (305.25) | 558.4 (301.44) | 785.5 (310.5) | 4.0 (0) | 4.0 (0) |
Median (range) | 444.5 (177 to 1,262) | 438.0 (244 to 1,256) | 701.0 (29 to 1,399) | 4.0 (4 to 4) | 4.0 (4 to 4) |
Adherence, % | |||||
Mean (SD) | NR | NR | NR | 100.0 (0) | 99.04 (3.467) |
Median (range) | NR | NR | NR | 100.0 (100 to 100) | 100.0 (87.5 to 100.0) |
Total daily dose, mg | |||||
Mean (SD) | 64.1 (14.42) | 62.4 (17.41) | 66.0 (17.90) | 60.0 (19.58) | 63.1 (18.99) |
Median (range) | 67.6 (40 to 78) | 73.0 (20 to 79) | 76.5 (20 to 80) | 60.0 (30 to 80) | 75.0 (40 to 80) |
NR = not reported; SD = standard deviation.
aThe open-label phase includes 2 patients who advanced from the run-in phase directly to the open-label phase.
Source: LMS-002 Clinical Study Report (2015)12 and LMS-003 Clinical Study Report (2017).13
The use of ongoing best supportive care and ongoing concomitant medication was reported in the LMS-002 study only and is summarized in Table 13. Pyridostigmine compounds were commonly reported, with 31.3% of patients receiving amifampridine phosphate and 40.9% of patients receiving placebo reporting pyridostigmine bromide, while 25% of patients and 18.2% of patients reported pyridostigmine, respectively. Concomitant medications were similar across treatment arms, with the largest difference being that 40.9% of patients in the placebo arm reported mineral supplements, while only 12.5% of patients in the amifampridine phosphate arm reported mineral supplements.
Table 13: Summary of Best Supportive Care and Concomitant Medication Ongoing at Time of First Dose — LMS-002 Study Only
Characteristic | LMS-002 study | |
|---|---|---|
Amifampridine phosphate N = 16 | Placebo N = 22 | |
Number of patients reporting ongoing best supportive care medication, N (%) | 13 (81.3) | 19 (86.4) |
Pyridostigmine bromide | 5 (31.3) | 9 (40.9) |
Methylprednisone | 5 (31.3) | 5 (22.7) |
Prednisone | 3 (18.8) | 5 (22.7) |
Pyridostigmine | 4 (25.0) | 4 (18.2) |
Azathioprine | 2 (12.5) | 2 (9.1) |
Number of patients reporting an ongoing concomitant medication, N (%) | 13 (81.3) | 14 (63.6) |
Drugs for acid-related disorders | 6 (37.5) | 7 (31.8) |
Mineral supplements | 2 (12.5) | 9 (40.9) |
Vitamins | 3 (18.8) | 8 (36.4) |
Thyroid therapy | 4 (25.0) | 4 (18.2) |
Antithrombotic agents | 3 (18.8) | 3 (13.6) |
Beta blocking agents | 3 (18.8) | 2 (9.1) |
Agents acting on the renin-angiotensin system | 2 (12.5) | 2 (9.1) |
Analgesics | 3 (18.8) | 1 (4.5) |
Drugs used in diabetes | 3 (18.8) | 1 (4.5) |
Lipid modifying agents | 2 (12.5) | 2 (9.1) |
Psychoanaleptics | 1 (6.3) | 3 (13.6) |
Source: LMS-002 Clinical Study Report (2015)12 and LMS-003 Clinical Study Report (2017).13
Efficacy
Only those efficacy outcomes and analyses of subgroups identified in the review protocol are reported as follows. Refer to Appendix 3 for detailed efficacy data.
Disability Progression
Quantitative Myasthenia Gravis Score
Table 14 summarizes the change from baseline in QMGSs for the LMS-002 study and the LMS-003 study. QMGS was a co-primary end point in both trials. In LMS-002, at the 14-day post-baseline time point, the mean change from baseline was 0.3 (SD = 2.60) and 2.2 (SD = 2.93) in the amifampridine phosphate and placebo arms, respectively. The difference in LSM for this comparison was –1.7 (95% CI, –3.4 to –0.0; P = 0.0452). The PP analysis of the LMS-002 population as well as the analysis conducted at the day 8 time point produced results consistent with the primary analysis (refer to Appendix 3, Table 23 and Table 30).
In the LMS-003 study, at the 4-day post-baseline time point, the mean change from baseline was 0.1 (SD = 3.07) and 6.5 (SD = 4.82) in the amifampridine phosphate and placebo arms, respectively. The difference in LSM for this comparison was –6.54 (95% CI, –9.78 to –3.29; P = 0.0004). The change from baseline QMGSs stratified by low dose (< 60 mg per day) and high dose (≥ 60 mg per day) were consistent with the main results (Appendix 3).
Table 14: Change From Baseline in Quantitative Myasthenia Gravis Scores
Characteristic | LMS-002 study | LMS-003 study | ||
|---|---|---|---|---|
Amifampridine phosphate N = 16 | Placebo N = 21 | Amifampridine phosphate N = 13 | Placebo N = 13 | |
Baseline, mean (SD) | 6.4 (3.22) | 5.6 (3.99) | 7.8 (4.20) | 8.5 (5.43) |
Post-baseline,a mean (SD) | 6.7 (4.09) | 7.9 (2.85) | 7.9 (4.94) | 15.0 (5.90) |
Change from baseline, mean (SD) | 0.3 (2.60) | 2.2 (2.93) | 0.1 (3.07) | 6.5 (4.82) |
LSMb | 0.4 | 2.2 | 0.00 | 6.54 |
Difference in LSMb (95% CI) | –1.7 (–3.4 to –0.0) | Reference | –6.54 (–9.78 to –3.29) | Reference |
P valuec | 0.0452c | Reference | 0.0004d | Reference |
CI = confidence interval; LSM = least squares mean; MMRM = mixed model of repeated measures; QMGS = Quantitative Myasthenia Gravis Score; SD = standard deviation.
aPost-baseline measurement was conducted at day 14 of the double-blind phase of the LMS-002 study. In the LMS-003 trial, post-baseline measurements were obtained on day 4, unless the patient discontinued treatment early, in which case post-baseline measurements may have been obtained at an earlier time point.
bFor LMS-002, this was estimated via a MMRM with change from baseline (day 1, part 2), day 8, and day 14 as the dependent variable, and terms for treatment, time (day 8, day 14), treatment-by-time interaction, and double-blind baseline QMGS as fixed effects and patient as a random effect. The model assumed time effect to be random between patients. For LMS-003, the change from baseline for QMGS total was modelled as the response, with fixed-effects terms for treatment and QMGS at baseline.
cThe P value represented a pairwise contrast at day 14 from the MMRM.
dThe P value was based on conducting a randomization test by running the fixed-effects linear model analysis on permuted treatment assignments. For each of the 10,000 permutations, the change from baseline was modelled as the response for each end point, with fixed-effects terms for treatment and score at baseline.
Source: LMS-002 Clinical Study Report (2015)12 and LMS-003 Clinical Study Report (2017).13
The LMS-003 study reported analysis of the limb domain scores as an exploratory end point, and therefore results should be interpreted with caution. The mean change from baseline and difference in LSM are summarized in Table 37 (Appendix 3). Results indicated more favourable outcomes in patients in the amifampridine phosphate arm; however, as this was an exploratory outcome and statistical analysis was not adjusted for multiplicity, reported P values should be interpreted with caution.
Subject Global Impression
Change from baseline in SGI scores in the LMS-002 trial and the LMS-003 trial are summarized in Table 15. The change from baseline in SGI was a co-primary end point in both LMS-002 and LMS-003. In the LMS-002 study, at the 14-day post-baseline time point, the mean change from baseline was –0.7 (SD = 1.82) and –2.7 (SD = 2.29) in the amifampridine phosphate and placebo arms, respectively. The difference in LSM for this comparison was 1.8 (95% CI, 0.7 to 3.0; P = 0.0028). The PP analysis of the LMS-002 population as well as the analysis conducted at the day 8 time point produced results consistent with the primary analysis (refer to Appendix 3, Table 25 and Table 31).
In the LMS-003 study, at the 4-day post-baseline time point, the mean change from baseline was –0.8 (SD = 1.74) and –3.5 (SD = 2.18) in the amifampridine phosphate and placebo arms, respectively. The difference in LSM for this comparison was 2.95 (95% CI, 1.53 to 4.38; P = 0.0003). The change from baseline SGI scores stratified by low dose (< 60 mg per day) and high dose (≥ 60 mg per day) were consistent with the main results (Appendix 3, Table 36).
Table 15: Change in Subject Global Impression Scores
Characteristic | LMS-002 study | LMS-003 study | ||
|---|---|---|---|---|
Amifampridine phosphate N = 16 | Placebo N = 21 | Amifampridine phosphate N = 13 | Placebo N = 13 | |
Baseline, mean (SD) | 5.6 (1.26) | 5.9 (1.22) | 6.1 (0.86) | 5.8 (0.90) |
Post-baseline,a mean (SD) | 4.9 (1.57) | 3.2 (1.70) | 5.3 (1.65) | 2.4 (1.76) |
Change from baseline, mean (SD) | –0.7 (1.82) | –2.7 (2.29) | –0.8 (1.74) | –3.5 (2.18) |
LSMb | –0.8 | –2.6 | –0.64 | –3.59 |
Difference in LSMb (95% CI) | 1.8 (0.7 to 3.0) | Reference | 2.95 (1.53 to 4.38) | Reference |
P value | 0.0028c | Reference | 0.0003d | Reference |
CI = confidence interval; LSM = least squares mean; MMRM = mixed model of repeated measures; SD = standard deviation; SGI = Subject Global Impression.
aPost-baseline measurement was conducted at day 14 of the double-blind phase of the LMS-002 study. In the LMS-003 trial, post-baseline measurements were obtained on day 4, unless the patient discontinued treatment early, in which case post-baseline measurements may have been obtained at an earlier time point.
bFor LMS-002, this was estimated via a MMRM with change from baseline (day 1, part 2), day 8, and day 14 as the dependent variable, and terms for treatment, time (day 8, day 14), treatment-by-time interaction, and double-blind baseline SGI score as fixed effects and patient as a random effect. The model assumed time effect to be random between patients. For LMS-003, the change from baseline for SGI score was modelled as the response, with fixed-effects terms for treatment and SGI at baseline.
cThe P value represented a pairwise contrast at day 14 from the MMRM.
dThe P value was based on conducting a randomization test by running the fixed-effects linear model analysis on permuted treatment assignments. For each of the 10,000 permutations, the change from baseline was modelled as the response for each end point, with fixed-effects terms for treatment and score at baseline.
Source: LMS-002 Clinical Study Report (2015)12 and LMS-003 Clinical Study Report (2017).13
Clinical Global Impression–Improvement
Post-baseline CGI-I scores for the LMS-002 and LMS-003 trials are summarized in Table 16. Post-baseline CGI-I was a secondary end point in both LMS-002 and LMS-003.
In the LMS-002 study, at the 14-day post-baseline time point, the mean post-baseline CGI-I score was 3.6 (SD = 1.50) and 4.8 (SD = 1.45) in the amifampridine phosphate and placebo arms, respectively. The difference in LSM for this comparison was –1.1 (95% CI, –2.1 to –0.1; P = 0.0267). The PP analysis of the LMS-002 population as well as the analysis conducted at the day 8 time point produced results consistent with the primary analysis (refer to Appendix 3, Table 28 and Table 32).
In the LMS-003 study, at the 4-day post-baseline time point, the mean post-baseline CGI-I score was 3.8 (SD = 0.80) and 5.5 (SD = 1.27) in the amifampridine phosphate and placebo arms, respectively. A Wilcoxon rank sum test for treatment differences yielded a P value of 0.002.
Table 16: Change in Clinical Global Impression–Improvement Scores
Characteristic | LMS-002 study | LMS-003 study | ||
|---|---|---|---|---|
Amifampridine phosphate N = 16 | Placebo N = 21 | Amifampridine phosphate N = 13 | Placebo N = 13 | |
Baseline, mean (SD) | 2.6 (0.63) | 2.5 (0.98) | NE | NE |
Post-baseline,a mean (SD) | 3.6 (1.50) | 4.8 (1.45) | 3.8 (0.80) | 5.5 (1.27) |
LSMb | 3.6 | 4.7 | NE | NE |
Difference in LSMb (95% CI) | –1.1 (–2.1 to –0.1) | Reference | NE | NE |
P value | 0.0267c | Reference | 0.0020d | Reference |
CI = confidence interval; LSM = least squares mean; MMRM = mixed model of repeated measures; NE = not evaluated; SD = standard deviation.
aPost-baseline measurement was conducted at day 14 of the double-blind phase of the LMS-002 study. In the LMS-003 trial, post-baseline measurements were obtained on day 4, unless the patient discontinued treatment early, in which case post-baseline measurements may have been obtained at an earlier time point.
bFor LMS-002, this was estimated via a MMRM with change from baseline (day 1, part 2), day 8, and day 14 as the dependent variable, and terms for treatment, time (day 8, day 14), and treatment-by-time interaction as fixed effects and patient as a random effect. The model assumed time effect to be random between patients.
cThe P value represented a pairwise contrast at day 14 from the MMRM.
dThe P value was based on the Wilcoxon rank sum test for treatment differences.
Source: LMS-002 Clinical Study Report (2015)12 and LMS-003 Clinical Study Report (2017).13
Timed 25-Foot Walk
The change from baseline in T25-FW scores for the LMS-002 trial are summarized in Table 17. The change from baseline in T25-FW was the second secondary end point in LMS-002. In the LMS-002 study, at the 14-day post-baseline time point, the mean change from baseline was –1.46 (SD = 52.5) and –10.4 (SD = 53.1) in the amifampridine phosphate and placebo arms, respectively. The difference in LSM for this comparison was 8.51 (95% CI, –26.77 to 43.79; P = 0.6274). The PP analysis of the LMS-002 population as well as the analysis conducted at the day 8 time point produced more favourable results in the amifampridine phosphate arm compared to the primary analysis conducted at day 14 (refer to Appendix 3, Table 26 and Table 33).
Table 17: Change in T25-FW Walking Speed, Feet Per Minute — LMS-002 Study Only
Characteristic | LMS-002 study | |
|---|---|---|
Amifampridine phosphate N = 16 | Placebo N = 21 | |
Baseline, mean (SD) | 254 (126) | 255 (111) |
Post-baseline,a mean (SD) | 253 (126) | 244 (116) |
Change from baseline, mean (SD) | –1.46 (52.5) | –10.4 (53.1) |
LSMb | –1.16 | –9.67 |
Difference in LSMb (95% CI) | 8.51 (–26.77 to 43.79) | Reference |
P valuec | 0.6274 | Reference |
CI = confidence interval; LSM = least squares mean; MMRM = mixed model of repeated measures; SD = standard deviation; T25-FW = Timed 25-Foot Walk.
aPost-baseline measurement was conducted at day 14 of the double-blind phase of the LMS-002 study.
bThis was estimated via a MMRM with change from baseline (day 1, part 2), day 8, and day 14 as the dependent variable, and terms for treatment, time (day 8, day 14), treatment-by-time interaction, and double-blind baseline T25-FW walking speed as fixed effects and patient as a random effect. The model assumed time effect to be random between patients.
cP value represented a pairwise contrast at day 14 from the MMRM.
Source: LMS-002 Clinical Study Report (2015).12
Tertiary End Points
Tertiary end points from the LMS-002 and LMS-003 trials — including the CGI-S, CMAP, and patient-identified most bothersome symptom — are reported in Appendix 3. Results from these end points are consistent with the findings from the primary analysis. As 3TUG was identified by the clinical expert consulted by CADTH as the most relevant outcome in LEMS, results for the 3TUG end point in the LMS-003 study are presented in Table 18, despite being a tertiary end point. The number and proportion of patients with a 20% increase or more in 3TUG were reported from the LMS-003 trial. At the 4-day post-baseline time point, the proportion of patients with a 20% or more increase in 3TUG score was 7.7% and 61.5% in the amifampridine phosphate and placebo arms, respectively. A 2-sided Fisher’s exact test for treatment differences yielded a P value of 0.0112. 3TUG was an exploratory end point and must be interpreted with caution.
Table 18: Number and Proportion of Patients With a 20% or More Increase in 3TUG Average Time — LMS-003 Study Only
Characteristic | LMS-003 study | |
|---|---|---|
Amifampridine phosphate N = 13 | Placebo N = 13 | |
Patients who attempted 3TUG on day 0 and day 4, N | 13 | 13 |
Patients with ≥ 20% increase in 3TUG average time, N (%) | 1 (7.7) | 8 (61.5) |
P valuea | 0.0112 | Reference |
3TUG = Triple Timed-Up-and-Go.
aBased on a 2-sided Fisher’s exact test, where 2 subjects were not able to walk on day 4 and were considered as having a 20% increase in 3TUG average time. 3TUG was an exploratory end point and not adjusted for multiplicity.
Source: LMS-003 Clinical Study Report (2017).13
Activities of Daily Living
No efficacy outcomes related to ADL were reported.
Lambert-Eaton Myasthenic Syndrome–Related Symptoms
No efficacy outcomes related to LEMS-related symptoms were reported.
Health-Related Quality of Life
No efficacy outcomes related to HRQoL were reported.
Productivity
No efficacy outcomes related to productivity were reported.
Harms
Only those harms identified in the review protocol are reported as follows. In the LMS-002 study, results were reported for the open-label run-in phase for both treatment-experienced patients and treatment-I patients, the double-blind phase (part 2 and part 3), and the open-label extension phase. Notable harms identified in the protocol for this review included the following: paresthesia, seizure, and change in electrocardiogram. In the open-label run-in phase of the LMS-002 trial, paresthesia was reported in 42.9% of treatment-I patients, oral paresthesia was reported in 47.6% of treatment-I patients and 9.1% of treatment-experienced patients, and hypoesthesia was reported in 7.1% of treatment-I patients. Only oral paresthesia was reported during other phases of the trial (5.0% of patients in the open-label extension phase).
Refer to Table 19 for detailed harms data.
Adverse Events
In the LMS-002 open-label run-in phase, AEs were reported in 83.3% of treatment-I patients and 27.3% of treatment-experienced patients. During part 2 of the double-blind phase, AEs were reported in 37.5% and 13.6% of patients in the amifampridine phosphate and placebo arms, respectively. During part 3 of the double-blind phase, AEs were reported in 18.8% and 27.3% of patients in the amifampridine phosphate and placebo arms, respectively. During the open-label extension phase, AEs were reported in 87.5% of patients. Commonly reported AEs in treatment-I patients during the open-label run-in phase were headache (11.9%) and nausea (11.9%). Common AEs reported during the open-label extension phase were back pain (20%) and upper respiratory tract infection (12.5%). During the double-blind phase, AEs that were reported by more than 1 patient included headache (12.5% of amifampridine phosphate patients in part 2) and asthenia (9.1% of placebo patients in part 3). AEs related to paresthesia are reported in the Notable Harms section.
In the LMS-003 study, 23.1% of patients in the amifampridine phosphate arm and 76.9% of patients in the placebo arm reported AEs. Common AEs included muscular weakness (38.5%) and fatigue (30.8%) in the placebo arm. No single AE was reported by more than 1 patient in the amifampridine phosphate arm.
Serious Adverse Events
In the LMS-002 study, SAEs occurred in 7.1% of treatment-I patients during the open-label run-in phase; these included respiratory failure, pulmonary embolism, and urolithiasis. During the open-label extension phase of the trial, SAEs were reported in 25.0% of patients. Individual SAEs were not tabulated.
No SAEs were reported in the LMS-003 study.
Withdrawals Due to Adverse Events
In the LMS-002 trial, 9.5% of treatment-naive patients stopped treatment due to AEs, and 5% of patients in the open-label extension phase stopped treatment due to AEs. No patients in the double-blind phase withdrew due to an AE.
No patients in the LMS-003 trial withdrew due to an AE.
Mortality
One patient in the LMS-002 open-label extension phase died due to SCLC. No deaths were reported in the LMS-003 study.
Notable Harms
Notable harms identified in the protocol for this review included the following: paresthesia, seizure, and change in electrocardiogram. In the open-label run-in phase of the LMS-002 study, paresthesia was reported in 42.9% of treatment-naive patients, oral paresthesia was reported in 47.9% of treatment-naive patients and 9.1% of treatment-experienced patients, and hypoesthesia was reported in 7.1% of treatment-naive patients. Only oral paresthesia was reported during other phases of the trial (5.0% of patients in the open-label extension phase).
Harms | LMS-002 study | LMS-003 study | |||||||
|---|---|---|---|---|---|---|---|---|---|
Run-in phase, treatment- naive N = 42 | Run-in phase, treatment- experienced N = 11 | Amifampridine phosphate: Part 2 N = 16 | Placebo: Part 2 N = 22 | Amifampridine phosphate: Part 3 N = 16 | Placebo: Part 3 N = 22 | Open-label extension phase N = 40a | Amifampridine phosphate N = 13 | Placebo N = 13 | |
Patients with ≥ 1 AE | |||||||||
n (%) | 35 (83.3) | 3 (27.3) | 6 (37.5) | 3 (13.6) | 3 (18.8) | 6 (27.3) | 35 (87.5) | 3 (23.1) | 10 (76.9) |
Gastrointestinal disorders | 28 (66.7) | 2 (18.2) | 1 (6.3) | 0 | 0 | 0 | 11 (27.5) | 0 | 5 (38.5) |
Diarrhea | 4 (9.5) | 1 (9.1) | 1 (6.3) | 0 | 0 | 0 | 4 (10.0) | 0 | 1 (7.7) |
Nausea | 5 (11.9) | 0 | 0 | 0 | 0 | 0 | 2 (5.0) | 0 | 1 (7.7) |
Constipation | 3 (7.1) | 0 | 0 | 0 | 0 | 0 | 3 (7.5) | 0 | 0 |
Dry mouth | 2 (4.8) | 0 | 0 | 0 | 0 | 0 | NR | 0 | 2 (15.4) |
General disorders and administration site conditions | 9 (21.4) | 1 (9.1) | 1 (1.6) | 0 | 0 | 2 (9.1) | 6 (15.0) | 0 | 8 (61.5) |
Fatigue | 2 (4.8) | 0 | 0 | 0 | 0 | 1 (4.5) | NR | 0 | 4 (30.8) |
Asthenia | 1 (2.4) | 1 (9.1) | 0 | 0 | 0 | 2 (9.1) | 3 (7.5) | 0 | 2 (15.4) |
Feeling hot | 0 | 0 | 0 | 0 | 0 | 0 | NR | 0 | 2 (15.4) |
Musculoskeletal and connective tissue disorders | 8 (19.0) | 0 | 0 | 2 (9.1) | 0 | 2 (9.1) | 17 (42.5) | 2 (15.4) | 7 (53.9) |
Back pain | 1 (2.4) | 0 | 0 | 0 | 0 | 0 | 8 (20.0) | 1 (7.7) | 0 |
Pain in extremity | 3 (7.1) | 0 | 0 | 0 | 0 | 0 | 2 (5.0) | 1 (7.7) | 0 |
Muscular weakness | 2 (4.8) | 0 | 0 | 1 (4.5) | 0 | 1 (4.5) | NR | 0 | 5 (38.5) |
Limb discomfort | 0 | 0 | 0 | 0 | 0 | 0 | NR | 0 | 2 (15.4) |
Muscle spasms | 2 (4.8) | 0 | 0 | 0 | 0 | 0 | 4 (10.0) | 0 | 2 (15.4) |
Nervous system disorders | 22 (52.4) | 1 (9.1) | 2 (12.5) | 0 | 0 | 0 | 8 (20.0) | 1 (7.7) | 3 (23.1) |
Headache | 5 (11.9) | 0 | 2 (12.5) | 0 | 0 | 0 | NR | 1 (7.7) | 0 |
Dizziness | 3 (7.1) | 0 | 1 (6.3) | 0 | 0 | 0 | 2 (5.0) | 0 | 0 |
Balance disorder | 0 | 0 | 0 | 0 | 0 | 0 | NR | 0 | 2 (15.4) |
Infections and infestations | 10 (23.8) | 1 (9.1) | 2 (12.5) | 1 (4.5) | 3 (18.8) | 1 (4.5) | 21 (52.5) | 0 | 1 (7.7) |
Nasopharyngitis | 3 (7.1) | 0 | 0 | 0 | 1 (6.3) | 0 | 4 (10.0) | 0 | 0 |
Upper respiratory tract infection | 3 (7.1) | 0 | 1 (6.3) | 0 | 0 | 0 | 5 (12.5) | 0 | 0 |
Urinary tract infection | 0 | 0 | 0 | 0 | 1 (6.3) | 0 | 4 (10.0) | 0 | 0 |
Injury, poisoning, and procedural complications | 5 (11.9) | 0 | 0 | 0 | 0 | 0 | NR | 0 | 0 |
Fall | 3 (7.1) | 0 | 0 | 0 | 0 | 0 | NR | 0 | 0 |
Eye disorders | 8 (19.0) | 0 | 0 | 0 | 0 | 0 | 9 (22.5) | 0 | 2 (15.4) |
Cataract | 1 (2.4) | 0 | 0 | 0 | 0 | 0 | 4 (10.0) | 0 | 0 |
Patients with ≥ 1 SAE | |||||||||
n (%) | 3 (7.1) | 0 | 0 | 0 | 0 | 0 | 10 (25.0) | 0 | 0 |
Respiratory failure | 1 (2.4) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Pulmonary embolism | 1 (2.4) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Urolithiasis | 1 (2.4) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Patients who stopped treatment due to AEs | |||||||||
n (%) | 4 (9.5) | 0 | 0 | 0 | 0 | 0 | 2 (5.0) | 0 | 0 |
Deaths | |||||||||
n (%) | 0 | 0 | 0 | 0 | 0 | 0 | 1 (2.5) | 0 | 0 |
Notable harms | |||||||||
Paresthesia | 18 (42.9) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Hypoesthesia, oral | 3 (7.1) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Paresthesia, oral | 20 (47.6) | 1 (9.1) | 0 | 0 | 0 | 0 | 2 (5.0) | 0 | 0 |
Seizures | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Change in electrocardiogram | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
AE = adverse event; NR = not reported; SAE = serious adverse event.
aOpen-label phase included 2 patients who advanced from run-in phase directly to open-label phase. The frequency for the open-label phase was greater than 10%, unless reported in another arm for which the threshold was more than 1 patient reporting an AE in any arm.
Source: LMS-002 Clinical Study Report (2015)12 and LMS-003 Clinical Study Report (2017).13
Critical Appraisal
Internal Validity
Both LMS-002 and LMS-003 were double-blind studies that employed various strategies to maintain blinding of the patients, investigator, site personnel, and sponsor personnel. However, by designing a study using a withdrawal enrichment strategy, partial unblinding was possible as patients in the placebo arm were anticipated to experience deterioration before amifampridine phosphate being reinstated. Unblinding in the LMS-002 and LMS-003 trials may have biased subjective patient-assessed (e.g., SGI) and investigator-assessed (e.g., QMGS, CGI-I) outcome results in favour of amifampridine phosphate. Both studies used small sample sizes, further increasing uncertainty in the results. However, given the rarity of LEMS, this is expected and consistent with similar trials conducted for amifampridine base.
The withdrawal design used in the LMS-002 study required patients in the placebo arm to taper their baseline amifampridine phosphate dose over a 7-day period followed by 7 days of placebo with no amifampridine phosphate. In the LMS-003 trial, placebo patients were transitioned directly to a placebo-only dose for 4 days. The differing designs were both sufficient to assess the deterioration of patients (efficacy of amifampridine phosphate) based on feedback from the clinical expert consulted by CADTH for the review, given the short half-life of amifampridine phosphate and the expected rapid appearance of symptoms following treatment discontinuation. Furthermore, a withdrawal design lends itself to a LEMS population that includes heterogeneity among fast and slow amifampridine metabolizers, requiring the inclusion of a dose titration phase for treatment-naive patients.
The co-primary end points for both the LMS-002 study and the LMS-003 study were QMGS and SGI. QMGS is a measure developed for use in MG and includes components relating to ocular and bulbar involvement that are more relevant to MG and not expected to be impacted by treatment for LEMS. While the QMGS was not considered a relevant assessment tool in LEMS by the clinical expert consulted by CADTH, as it was designed and validated for the assessment of MG, the components of QMGS that are unrelated to LEMS would bias the results against amifampridine phosphate. The change in the QMGS components that are expected to be impacted by treatment would need to be more pronounced to reach statistical significance. SGI is a patient-rated assessment of overall effects of a treatment; however, this tool has not been validated in patients with LEMS. The consulted clinical expert noted that the 3TUG assessment is becoming the standard clinical tool used in the assessment of LEMS; however, this end point was not reported in the LMS-002 study and in the LMS-003 study, it was a tertiary end point. Tertiary and exploratory end points in both trials were not part of the statistical hierarchy and therefore were not adjusted for multiplicity, limiting the ability to interpret these results.
Protocol violations specifically relating to incorrectly testing efficacy before patients receiving their first treatment dose of the day were noted at a specific site in the LMS-002 trial. The ad hoc PP assessment excluding this site from the analysis (refer to Appendix 3, Table 30 and Table 31) showed more favourable results for amifampridine phosphate in the co-primary end point of QMGS, though this was an unplanned analysis that could not be adjusted for multiplicity and is therefore of limited interpretability.
Subgroup analyses based on the type of LEMS (paraneoplastic versus primary autoimmune) were not performed in the LMS-002 or LMS-003 trial. LMS-003 did present results stratified by low dose (< 60 mg per day) and high dose (≥ 60 mg per day), which can be considered a rough proxy for disease severity according to the clinical expert, though the study was not powered to detect differences in this subgroup. Whether or not the treatment effect differs between subgroups (e.g., paraneoplastic versus primary autoimmune) identified as relevant in the CADTH review protocol remains unknown.
In the LMS-002 trial, the protocol was amended to use the mixed model of repeated measures method as the primary analysis for the main end points. While this change was made after randomization in the May 2014 protocol amendment, CADTH agrees that the change was appropriate to adequately handle missing data from patients who had to be rescued during the double-blind treatment phase. It is, however, difficult to assess if the missing data are completely random due to the small sample size. Using a mixed model of repeated measures method allows the use of all data points at day 1, day 8, and day 14, and is a good use of all available patient information in this scenario. The originally planned ANCOVA analysis was presented as a sensitivity analysis and supported the primary findings.
External Validity
The withdrawal enrichment strategy used in both the LMS-002 trial and the LMS-003 trial resulted in a stringently selected study population of patients who were treatment-experienced and responsive to amifampridine phosphate at baseline. Aspects of the trial design resulted in a study population that exhibited a magnitude of treatment response that may not be generalizable to Canadian patients who are treatment-naive, including those who are newly diagnosed with LEMS. Several components of the patient eligibility criteria were key in contributing to the enriched study design of LMS-002 and LMS-003.
In the LMS-002 study, treatment-naive patients were required to display a sufficiently large response (> 3-point improvement in QMGS) to amifampridine phosphate during the open-label run-in phase. In LMS-002, 25.9% of patients (n = 14) withdrew during the run-in phase. The most common reason for withdrawal during the open-label run-in phase was AEs; AEs occurred more often in treatment-naive patients than in treatment-experienced patients during this phase. The LMS-003 study only included patients already on a stable dose of amifampridine phosphate. These criteria would result in a patient population that may be more responsive to, and more tolerant of, study treatment relative to the target Canadian patient population. Thus, the treatment effects estimated by the LMS-002 and LMS-003 studies are likely an overestimate of the effect on the Canadian patient population that would be eligible to receive amifampridine phosphate. Instead, the estimates would more closely reflect the response of a prevalent population currently on a stable regimen of amifampridine.
Patients in both the LMS-002 study and the LMS-003 study were required to be physically able to complete study tests and, in the case of patients in the LMS-002 study, could not have respiratory function lower than FVC 80% of predicted for patients currently on treatment. According to the clinical expert consulted by CADTH, in clinical practice, patients would not be prevented from being treated with amifampridine phosphate based on either of these criteria.
Overall, the baseline characteristics of patients in the LMS-002 trial and the LMS-003 trial were generally consistent with the Canadian clinical population currently being treated with amifampridine phosphate. However, in LMS-002 and LMS-003, 15.8% and 23.1% of patients had paraneoplastic syndrome, respectively, likely due to the requirement for patients to have completed anticancer treatment at least 3 months before screening. This is inconsistent with the clinical population where it is estimated that 50% to 60% of patients have paraneoplastic syndrome. Patients with paraneoplastic LEMS are known to have poorer prognosis due to the underlying neoplastic condition; thus, the results of the LMS-002 and LMS-003 studies may not be representative of these patients. It is noted that the clinical expert consulted did not expect major differences in treatment efficacy of amifampridine phosphate based on these subgroups of patients.
The use of other LEMS medications at baseline and throughout the study was generally consistent with the treatment regimen of stabilized patients in the Canadian clinical setting. In the LMS-002 study, almost all patients (81.3% in the amifampridine phosphate arm and 86.4% in the placebo arm) were taking additional best supportive care medication, with the most commonly reported regimen including pyridostigmine. This proportion was considered higher than what would be seen in the Canadian clinical setting, as pyridostigmine is most often used in Canada as a temporizing agent for patients diagnosed with LEMS who may be waiting for access to amifampridine and is often discontinued due to insufficient effectiveness once patients have access to amifampridine.15 This fact potentially impacts the generalizability of these results.
Other Relevant Evidence
This section includes submitted additional relevant studies included in the sponsor’s submission to CADTH that were considered to address important gaps in the evidence included in the systematic review.
Bioequivalence Study
The sponsor evaluated the relative bioavailability of amifampridine phosphate in a randomized, crossover trial known as the DAPSEL study. In this trial, the sponsors compared the formulations of amifampridine phosphate salt (in tablet formulation) with amifampridine base (in capsule formulation) to determine their relative bioequivalence.29 The DAPSEL study also provided some safety data. For this trial, 27 healthy adult males aged between 18 years and 45 years were included. One patient was withdrawn due to an elevation of liver enzymes, resulting in a total of 26 participants that completed the study.
The DAPSEL study was divided into 2 parts. The first part and phase evaluated the clinical tolerance of an open-label, single, oral 10 mg dose of amifampridine phosphate by assigning the dose to 5 participants, whereas in the second part and phase, all 27 participants were randomized to receive a single 20 mg dose (2 × 10 mg) of either the amifampridine administered as a base or as a salt (amifampridine phosphate) in a double-blind, randomized, 2-way, crossover bioequivalence study. For the first and second phases, patients were required to fast since midnight the day before and for at least 12 hours, respectively. Blood samples were collected before administering any dose and followed with 16 samples over the next 24 hours after giving the doses. There was a washout period of at least 72 hours, and then participants crossed over to the other amifampridine base or salt (amifampridine phosphate) formulation, and the process repeated as mentioned previously. The half-life of amifampridine in plasma has been reported as 2 hours. Therefore, 72 hours as a washout period has been considered longer than 5 half-lives, representing a sufficiently long washout period. This was confirmed with a pre-dose plasma amifampridine concentration below the Lower Limit of Quantification in all patients at the start of the second phase.29
Plasma samples were analyzed for amifampridine concentration by the high-performance liquid chromatography method, with lower and upper limits of quantification of 5 ng/mL and 150 ng/mL, respectively. AUC0–t, Cmax, time to peak concentration (Tmax), and half-life were calculated following normal standard procedures. Statistical evaluation was performed for AUC0–t and Cmax with analysis of variance and the 90% CI for the ratio of reference formulation (amifampridine phosphate salt, in tablet form) over the test formulation (amifampridine base, in capsule form) was calculated (refer to Table 20).
Table 20: Mean Reference and Test Ratios, and 90% Confidence Intervals for AUC0–inf and Cmax Comparing Salt Tablets and Base Capsules — Log-Transformed Data
Parameter | Ratio R/Ta (%) | 90% CI |
|---|---|---|
AUC0–inf | 102.7 | 93.1 to 113.3 |
Cmax | 86.3 | 73.7 to 100.8 |
AUC0–inf = area under the concentration-time curve to infinity; CI = confidence interval; Cmax = peak concentration; R = reference formulation (amifampridine phosphate salt); t = test formulation (amifampridine base).
aThe plasma concentrations of amifampridine after administration of salt in tablets and compared to base in capsules are shown in Figure 3.
Figure 3: Plasma Concentrations of Amifampridine After Administration of Salt in Tablets and Compared to Base in Capsules
3,4-DAP = 3,4-diaminopyridine; Cmax = peak concentration; CP = coverage probability; min = minute.
Note: The curve with the highest Cmax corresponds to administration of amifampridine phosphate.
Source: European Medicines Agency’s Assessment Report for Zenas (2009).29
The AUCs ratio had fallen within the pre-specified bioequivalence limits of 80% to 125%. For the peak plasma concentration (Cmax), the observed inferior limit exceeded the 80.0% bound and was near the 75% bound suggested for highly variable drugs. Tmax was significantly shorter for the salt formulation as compared to the amifampridine base (0.6 hours + 0.3 hours versus 0.9 hours + 0.4 hours, respectively; P ≤ 0.001). The results of the DAPSEL study demonstrated that the proposed amifampridine phosphate formulation was more rapidly dissolved and more completely absorbed, leading to a higher Cmax and shorter Tmax than the reference amifampridine base formulation. Moreover, the committee found the submitted documents were dependent on the demonstration that the proposed formulation is similar to those referenced in the published efficacy studies. Based on these facts, the Committee for Medicinal Products for Human Use expressed their concern regarding literature data provided for the base formulation to be extrapolated to the salt formation in the European Medicines Agency’s Assessment Report for Zenas (2009).29 The extent of absorption measured by AUC was found to be similar between the 2 formulations, leading to the acceptance of the fact that the extent of exposure to the active part would be similar, irrespective of the difference in the formulation. It was suggested that this profile would not affect the efficacy but may have an impact on safety. In addition to this, due to the differences in Cmax and Tmax between the salt and the base formulations, the maximum daily dose was adjusted from 80 mg to 60 mg.29
A total of 40 AEs were reported in the DAPSEL study, of which 25 were paresthesias (mainly perioral paresthesias). All were reported to be minor and considered as possibly related to the treatment administered by the investigator. No deaths were reported during this study. One SAE had been reported in 1 patient; it was an increase in liver enzymes, leading to premature withdrawal.29
Discussion
Summary of Available Evidence
Two pivotal trials, LMS-002 (N = 38) and LMS-003 (N = 26), were included in the CADTH systematic review. Both studies were phase III, multi-centre, randomized, double-blind, placebo-controlled withdrawal studies that aimed to assess the safety and efficacy of amifampridine phosphate for the treatment of LEMS in adult patients.
The LMS-002 study was composed of 4 parts. Part 1 was an open-label run-in phase, where patients were required to have received amifampridine phosphate or base for at least 91 days and a stable dose of amifampridine phosphate for at least 7 days. Treatment-naive patients were required to achieve a 3-point improvement or more in QMGS. In part 2, patients were randomized to either continue double-blind amifampridine phosphate or taper to placebo over 7 days; part 3 was an additional 7 days of double-blind treatment in both arms. Following primary efficacy assessments at day 14, patients could enter the part 4, open-label amifampridine phosphate treatment for safety follow-up. The co-primary efficacy end points in LMS-002 were change from baseline to day 14 QMGSs and SGI scores while the secondary end points were CGI-I and T25-FW.
The LMS-003 study consisted of a 4-day double-blind withdrawal period. All patients were previously enrolled in an expanded access program and required to be on a stable dose of amifampridine phosphate for 1 week before randomization. Patients were randomized to maintain either their regular amifampridine phosphate dose or their placebo for day 1 through day 4. Efficacy assessments were conducted on day 0 and day 4 following the final blinded dose. The co-primary efficacy end points in LMS-003 were change from baseline to day 4 QMGSs and SGI scores while the secondary end point was CGI-I.
The key limitations of both studies, LMS-002 and LMS-003, are related to the selection of QMGS, which is not specifically developed for LEMS as a co-primary end point, and the relatively small sample sizes, which led to uncertainty and generalizability issues. The study design and eligibility criteria resulted in a study population that consisted of patients who were treatment-experienced, mostly primary autoimmune, and responsive to amifampridine phosphate at baseline. The study population exhibited a magnitude of treatment response that may not be generalizable to Canadian amifampridine-naive patients, including those who are newly diagnosed with LEMS.
Other relevant evidence included in this review included the DAPSEL study, which investigated the bioequivalence of amifampridine phosphate and amifampridine base oral formulations. Amifampridine base has been previously reviewed by CADTH and recommended for reimbursement with conditions by CDEC. However, no clinical evidence comparing the base to phosphate forms of amifampridine was identified. Therefore, the clinical value of amifampridine phosphate relative to amifampridine base is unknown.
Interpretation of Results
Efficacy
In the LMS-002 study, patients who discontinued amifampridine phosphate treatment reported a statistically significant disease progression according to the co-primary end point of difference in QMGS LSM of –1.7 (95% CI, –3.4 to –0.0; P = 0.0452). Though this result is statistically significant, it is below the identified clinically significant threshold of 2.6 units (note that this threshold was determined in MG patients; no such threshold has been identified in patients with LEMS).11 Similarly, the co-primary end point of the LMS-003 study was difference in QMGS LSM, reporting both a statistically and clinically significant difference of –6.54 (95% CI, –9.78 to –3.29; P = 0.0004). While QMGS was chosen as a co-primary end point, this measure was originally designed for MG and comprises multiple components such as ocular and bulbar involvement that are not relevant to patients with LEMS and would not be expected to be impacted by treatment. Although the use of a more relevant end point would be preferred, the use of QMGS is expected to bias results against amifampridine phosphate given that a more pronounced change in the LEMS-specific components would be required to see significant change in the overall measure.
The second co-primary end point in both the LMS-002 trial and the LMS-003 trial was SGI. There was a statistically significant disease progression in patients who discontinued amifampridine phosphate according to a difference in LSM in the LMS-002 study of 1.8 (95% CI, 0.7 to 3.0; P = 0.0028) and in the LMS-003 study of 2.95 (95% CI, 1.53 to 4.38; P = 0.0003). There was no clinically significant threshold identified for the SGI measure in patients with LEMS; however, the clinical expert consulted for this review considered the results to be clinically meaningful. The use of a withdrawal enrichment strategy inherently introduced the possibility of unblinding as patients in the placebo arm were anticipated to experience deterioration. This potential partial unblinding may have biased subjective outcome results, such as those for the SGI, in favour of amifampridine phosphate. The LMS-002 study included CGI-I as the first secondary end point, only to be formally tested if both co-primary end points were statistically significant. There was a statistically significant difference in LSM of –1.1 (95% CI, –2.1 to –0.1; P = 0.0267) that favoured amifampridine phosphate. Similar concerns with potential partial unblinding apply to this physician-rated measure as mentioned previously. Given the statistical significance of CGI-I, the second secondary end point in the LMS-002 study, T25-FW, was formally tested. It was noted by the clinical expert consulted for this review that the T25-FW is the end point most similar to the 3TUG test, considered to be the emerging standard measure for assessing patients with LEMS in the clinic. In LMS-002, patients discontinuing amifampridine phosphate showed a slight numerical difference toward disease progression. However, the difference in LSM showed no statistical difference: 8.51 (95% CI, –26.77 to 43.79; P = 0.6274).
The LMS-003 study included only 1 secondary end point, though there was no evidence that methods for controlling multiplicity were applied and, therefore, definitive conclusions cannot be drawn. LMS-003 reported only post-baseline values as baseline CGI-I was not recorded, further negatively impacting the ability to interpret any apparent treatment differences. Patients in the amifampridine phosphate arm reported a post-baseline mean of 3.8 and patients in the placebo arm reported a post-baseline mean of 5.5, with a P value based on the Wilcoxon rank sum test of 0.0020.
A subgroup analysis based on the type of LEMS (paraneoplastic versus primary autoimmune) was not performed in either the LMS-002 trial or the LMS-003 trial. Patients were excluded from both trials if they had received anticancer treatment in the previous 3 months, which may partially explain the underrepresentation of patients with paraneoplastic LEMS compared to the clinical setting. The clinical expert consulted for this review did not expect a major difference in efficacy of amifampridine phosphate in paraneoplastic LEMS versus primary autoimmune LEMS. The LMS-003 trial reported results stratified by high dose versus low dose of amifampridine phosphate, a rough proxy for disease severity; results were consistent with that of the primary analysis.
3TUG was an exploratory end point in the LMS-003 study and has limited interpretability. However, given that it was identified as the most relevant outcome in LEMS according to the expert consulted by CADTH, it should be noted that 61.5% of patients in the placebo arm experienced a 20% increase or more in 3TUG average time (i.e., clinical worsening), compared to 7.7% of patients remaining on amifampridine phosphate treatment.
Additional exploratory end points were reported in both trials, including an analysis conducted at the day 8 time point in the LMS-002 study, limb-specific domains of QMGS, and an ad hoc analysis of LMS-002 excluding a specific trial site that incorrectly dosed patients. All results were consistent with that of the primary analysis.
There was notable inconsistency in the 2 trials, specifically in the magnitude of change in the QMGS. In the LMS-002 trial, conducted in 2011, the change was 1.7, which was below the recognized MID, though this threshold has not been validated in patients with LEMS. There was also an imbalance in QMGS at the baseline assessment (6.4 versus 5.6; difference of 0.8). This was possibly due to a random sampling error amplified by the small sample size. When considering the small difference in QMGS between treatment arms, numerically, half the change at day 14 could potentially be explained by the unbalanced baseline value. In the LMS-003 trial, which was conducted more recently in 2017, the change in QMGS was much higher at 6.5, though with a similar imbalance in baseline values. The inconsistency between trials was less pronounced in the SGI end point, though a smaller change was reported in the LMS-002 study than in the LMS-003 study (1.8 versus 3.0). These differences cast some uncertainty on the treatment effect. However, since LMS-003 was conducted exclusively in the US where practice may be less variable and closer to the Canadian context, and given possible change over the past decades in patient treatment modality, this trial can be considered more generalizable to the current setting and more reliable in design. Despite this, there is still substantial uncertainty in the LMS-003 results, given the smaller sample size compared to LMS-002 and shorter duration, in addition to evidence gaps present in both trials — specifically, a lack of functional, productivity, and HRQoL analysis.
The bioequivalence study DAPSEL was included for review by the sponsor. Results suggest similarity between amifampridine phosphate and amifampridine base with regard to area under the concentration-time curve to infinity and Cmax, leading to regulators noting that the efficacy profiles of the formulations would not be expected to differ. This aligns with comments from the clinical expert indicating that the 2 forms of amifampridine would be used interchangeability in clinical practice. It should be noted that amifampridine base received a recommendation for reimbursement with conditions from CDEC in April 2021 and, in May 2021, the Health Canada Notice of Compliance was briefly revoked due to a legal dispute. Patient input received by CADTH highlighted patient concern that a loss of access to amifampridine base due to the ongoing legal dispute could result in significant financial burden.
Harms
In the LMS-002 study, AEs were reported separately through the different phases of the trial. During the open-label run-in phase, AEs were reported for 53 patients, including those who would eventually withdraw from the trial. AEs were reported in 83.3% of treatment-naive patients and 27.3% of treatment-experienced patients. The most commonly reported AEs were in treatment-naive patients — namely, paresthesia (42.9%) and oral paresthesia (47.6%). According to the clinical expert, the frequency of AEs decreases over time in treated patients. As such, AEs reported during the 14-day double-blind period were, as expected, lower. The shorter duration compared to the 2 open-label phases may have impacted overall results as well. In the open-label safety extension period, 87.5% of patients reported AEs, with the most common being back pain (20.0%). Notably, no paresthesia was reported during the open-label safety extension phase, highlighting the feature of the stringently selected patient population, which was able to tolerate amifampridine phosphate due to the requirement to have received and tolerated amifampridine phosphate before entering the long-term safety phase.
A total of 25% of patients experienced SAEs during the open-label safety extension phase, 1 of which was fatal SCLC. All but 2 SAEs were deemed by the investigator to be unrelated to the study drug, while those deemed probable to be related to the study drug were managed by dose reduction.
In the LMS-003 trial, AEs were reported in 23.1% of patients receiving amifampridine phosphate and 76.9% of patients in the placebo arm. The most common AEs reported were muscular weakness (38.5%) and fatigue (30.8%), though these were both in the placebo arm and are common symptoms of LEMS progression itself. Therefore, it is uncertain whether safety signals are due to treatment side effects or disease progression. Further caution must be given to the interpretation of the LMS-003 harms results, given that all patients had previously received and tolerated amifampridine phosphate treatment. The short duration of the LMS-003 study also limits the ability to draw long-term safety conclusions.
In the bioequivalence study DAPSEL, a total of 40 AEs were reported, of which 25 were paresthesia. One SAE and no deaths were reported in DAPSEL.
Conclusions
Two phase III, double-blind, placebo-controlled withdrawal studies (the LMS-002 study, N = 38, and the LMS-003 study, N = 26) in adult patients with LEMS demonstrated that continuous treatment with amifampridine phosphate resulted in less disability progression compared with patients whose amifampridine phosphate was withdrawn. There was a –1.7 difference in QMGS LSM in LMS-002 and a difference in LSM of –6.54 in LMS-003. SGI showed similar differences between treatment arms with a LSM difference of 1.8 and 2.95 in LMS-002 and LMS-003, respectively. All results were statistically significant, though a clinically significant threshold specific to patients with LEMS was not determined. Results suggesting muscle strength improvement with amifampridine phosphate are aligned with outcomes important to patients. The effect of amifampridine phosphate on HRQoL and productivity was not evaluated in the LMS-002 or LMS-003 study and remains unknown. Evidence from the 2 trials were limited by the potential for unblinding and generalizability to the amifampridine-naive patient population.
The harms data obtained from the body of evidence reviewed for the CADTH report are limited. LMS-003 only reports harms results for up to 4 days of follow-up, and although LMS-002 includes safety follow-up for up to 2 years, due to the withdrawal enrichment design of both the LMS-002 trial and the LMS-003 trial, harms reported may not be a true reflection of the harms associated with amifampridine phosphate for all patients with LEMS.
Evidence gaps for the reviewed studies include the use of amifampridine phosphate in treatment-naive patients, and patients with paraneoplastic LEMS. Comparative clinical evidence for amifampridine phosphate against amifampridine base is lacking, though a bioequivalence study and clinical expert opinion suggest similarity of the 2 formulations.
References
1.Titulaer MJ, Lang B, Verschuuren JJ. Lambert-Eaton myasthenic syndrome: from clinical characteristics to therapeutic strategies. Lancet Neurol. 2011;10(12):1098-1107. PubMed
2.Kesner VG, Oh SJ, Dimachkie MM, Barohn RJ. Lambert-Eaton myasthenic syndrome. Neurol Clin. 2018;36(2):379-394. PubMed
3.Tarr TB, Wipf P, Meriney SD. Synaptic pathophysiology and treatment of Lambert-Eaton myasthenic syndrome. Mol Neurobiol. 2015;52(1):456-463. PubMed
4.Lambert-Eaton Myasthenic Syndrome. Danbury (CT): National Organization for Rare Diseases; 2019: https://rarediseases.org/rare-diseases/lambert-eaton-myasthenic-syndrome/. Accessed 2022 Jan 26.
5.Omar A, Marwaha K, Bollu PC. Physiology, Neuromuscular Junction. In: StatPearls [internet]. Treasure Island (FL): StatPearls Publishing; 2021 May 09: https://www.ncbi.nlm.nih.gov/books/NBK470413/. Accessed 2022 Jan 26.
6.Titulaer MJ, Maddison P, Sont JK, et al. Clinical Dutch-English Lambert-Eaton Myasthenic syndrome (LEMS) tumor association prediction score accurately predicts small-cell lung cancer in the LEMS. J Clin Oncol. 2011;29(7):902-908. PubMed
7.Somnier FE, Keiding N, Paulson OB. Epidemiology of myasthenia gravis in Denmark. A longitudinal and comprehensive population survey. Arch Neurol. 1991;48(7):733-739. PubMed
8.Wirtz PW, Nijnuis MG, Sotodeh M, et al. The epidemiology of myasthenia gravis, Lambert-Eaton myasthenic syndrome and their associated tumours in the northern part of the province of South Holland. J Neurol. 2003;250(6):698-701. PubMed
9.Wirtz PW, van Dijk JG, van Doorn PA, et al. The epidemiology of the Lambert-Eaton myasthenic syndrome in the Netherlands. Neurology. 2004;63(2):397-398. PubMed
10.Abenroth DC, Smith AG, Greenlee JE, Austin SD, Clardy SL. Lambert-Eaton myasthenic syndrome: epidemiology and therapeutic response in the national veterans affairs population. Muscle Nerve. 2017;56(3):421-426. PubMed
11.Barohn RJ, McIntire D, Herbelin L, Wolfe GI, Nations S, Bryan WW. Reliability testing of the quantitative myasthenia gravis score. Ann N Y Acad Sci. 1998;841:769-772. PubMed
12.Clinical Study Report: LMS-002. A phase 3, multicenter, double-blind, placebo-controlled randomized discontinuation study followed by an open-label extension period to evaluate the efficacy and safety of amifampridine phosphate (3,4-diaminopyridine phosphate) in patients with Lambert-Eaton Myasthenic Syndrome (LEMS) [internal sponsor's report]. Coral Gables (FL): Catalyst Pharmaceuticals, Inc.; 2015 Oct 05.
13.Clinical Study Report: LMS-003. A phase 3, double-blind, placebo-controlled, randomized, parallel-group study to evaluate the efficacy and safety of amifampridine phosphate (3,4 diaminopyridine phosphate) in patients with Lambert-Eaton Myasthenic Syndrome (LEMS) [internal sponsor's report]. Coral Gables (FL): Catalyst Pharmaceuticals, Inc.; 2017 Dec 15.
14.JAMP Pyridostigmine Bromide (pyridostigmine bromide): 60 mg oral tablets [product monograph]. Boucherville (QC): JAMP Pharma Corporation; 2020 Nov 20: https://pdf.hres.ca/dpd_pm/00058915.PDF. Accessed 2022 Jan 26.
15.Wirtz PW, Verschuuren JJ, van Dijk JG, et al. Efficacy of 3,4-diaminopyridine and pyridostigmine in the treatment of Lambert-Eaton myasthenic syndrome: a randomized, double-blind, placebo-controlled, crossover study. Clin Pharmacol Ther. 2009;86(1):44-48. PubMed
16.Firdapse (amifampridine): 10 mg oral tablet [product monograph]. Mississauga (ON): KYE Pharmaceuticals Inc.; 2020 Nov 27.
17.McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS Peer Review of Electronic Search Strategies: 2015 guideline statement. J Clin Epidemiol. 2016;75:40-46. PubMed
18.Grey matters: a practical tool for searching health-related grey literature. Ottawa (ON): CADTH; 2019: https://www.cadth.ca/grey-matters. Accessed 2021 Dec 01.
19.Oh SJ, Shcherbakova N, Kostera-Pruszczyk A, et al. Amifampridine phosphate (Firdapse(R)) is effective and safe in a phase 3 clinical trial in LEMS. Muscle Nerve. 2016;53(5):717-725. PubMed
20.Shieh P, Sharma K, Kohrman B, Oh SJ. Amifampridine phosphate (Firdapse) is effective in a confirmatory phase 3 clinical trial in LEMS. J Clin Neuromuscul Dis. 2019;20(3):111-119. PubMed
21.Farrar JT, Young JP, Jr., LaMoreaux L, Werth JL, Poole MR. Clinical importance of changes in chronic pain intensity measured on an 11-point numerical pain rating scale. Pain. 2001;94(2):149-158. PubMed
22.Cutter GR, Baier ML, Rudick RA, et al. Development of a multiple sclerosis functional composite as a clinical trial outcome measure. Brain. 1999;122(5):871-882. PubMed
23.Oh SJ, Kim DE, Kuruoglu R, Brooks J, Claussen G. Electrophysiological and clinical correlations in the Lambert-Eaton myasthenic syndrome. Muscle Nerve. 1996;19(7):903-906. PubMed
24.Weinberg DH. Lambert-Eaton myasthenic syndrome: treatment and prognosis. In: Post TW, ed. UpToDate. Waltham (MA): UpToDate; 2021: www.uptodate.com. Accessed 2022 Feb 01.
25.Sanders DB, Massey JM, Sanders LL, Edwards LJ. A randomized trial of 3,4-diaminopyridine in Lambert-Eaton myasthenic syndrome. Neurology. 2000;54(3):603-607. PubMed
26.McEvoy KM, Windebank AJ, Daube JR, Low PA. 3,4-Diaminopyridine in the treatment of Lambert-Eaton myasthenic syndrome. N Engl J Med. 1989;321(23):1567-1571. PubMed
27.Sanders DB, Guptill JT, Ales KL, et al. Reliability of the triple-timed up-and-go test. Muscle Nerve. 2018;57(1):136-139. PubMed
28.Oh SJ, Claussen GG, Hatanaka Y, Morgan MB. 3,4-Diaminopyridine is more effective than placebo in a randomized, double-blind, cross-over drug study in LEMS. Muscle Nerve. 2009;40(5):795-800. PubMed
29.Committee for Medicinal Products for Human Use. Assessment Report: Zenas (amifampridine). (European public assessment report). London (GB): European Medicines Agency; 2009: https://www.ema.europa.eu/en/documents/assessment-report/firdapse-epar-public-assessment-report_en.pdf. Accessed 2022 Feb 03.
30.Sanders DB, Juel VC, Harati Y, et al. 3,4-diaminopyridine base effectively treats the weakness of Lambert-Eaton myasthenia. Muscle Nerve. 2018;57(4):561-568. PubMed
31.Barnett C, Merkies IS, Katzberg H, Bril V. Psychometric properties of the Quantitative Myasthenia Gravis Score and the Myasthenia Gravis Composite Scale. J Neuromuscul Dis. 2015;2(3):301-311. PubMed
32.Busner J, Targum SD, Miller DS. The Clinical Global Impressions scale: errors in understanding and use. Compr Psychiatry. 2009;50(3):257-262. PubMed
33.Streib EW, Rothner AD. Eaton-Lambert myasthenic syndrome: long-term treatment of three patients with prednisone. Ann Neurol. 1981;10(5):448-453. PubMed
34.Raja SM, Sanders DB, Juel VC, et al. Validation of the triple timed up-and-go test in Lambert-Eaton myasthenia. Muscle Nerve. 2019;60(3):292-298. PubMed
35.Besinger UA, Toyka KV, Homberg M, Heininger K, Hohlfeld R, Fateh-Moghadam A. Myasthenia gravis: long-term correlation of binding and bungarotoxin blocking antibodies against acetylcholine receptors with changes in disease severity. Neurology. 1983;33(10):1316-1321. PubMed
36.Tindall RS, Phillips JT, Rollins JA, Wells L, Hall K. A clinical therapeutic trial of cyclosporine in myasthenia gravis. Ann N Y Acad Sci. 1993;681:539-551. PubMed
37.Tindall RS, Rollins JA, Phillips JT, Greenlee RG, Wells L, Belendiuk G. Preliminary results of a double-blind, randomized, placebo-controlled trial of cyclosporine in myasthenia gravis. N Engl J Med. 1987;316(12):719-724. PubMed
38.Jaretzki A, 3rd, Barohn RJ, Ernstoff RM, et al. Myasthenia gravis: recommendations for clinical research standards. Task Force of the Medical Scientific Advisory Board of the Myasthenia Gravis Foundation of America. Neurology. 2000;55(1):16-23. PubMed
39.Bedlack RS, Simel DL, Bosworth H, Samsa G, Tucker-Lipscomb B, Sanders DB. Quantitative myasthenia gravis score: assessment of responsiveness and longitudinal validity. Neurology. 2005;64(11):1968-1970. PubMed
40.Guyatt G, Walter S, Norman G. Measuring change over time: assessing the usefulness of evaluative instruments. J Chronic Dis. 1987;40(2):171-178. PubMed
41.Barnett C, Herbelin L, Dimachkie MM, Barohn RJ. Measuring clinical treatment response in myasthenia gravis. Neurol Clin. 2018;36(2):339-353. PubMed
42.Zinman L, Ng E, Bril V. IV immunoglobulin in patients with myasthenia gravis: a randomized controlled trial. Neurology. 2007;68(11):837-841. PubMed
43.Uchiyama A, Shimizu S, Murai H, Kuroki S, Okido M, Tanaka M. Infrasternal mediastinoscopic thymectomy in myasthenia gravis: surgical results in 23 patients. Ann Thorac Surg. 2001;72(6):1902-1905. PubMed
44.Sanders DB, Rosenfeld J, Dimachkie MM, Meng L, Malik FI, Tirasemtiv in Myasthenia Gravis Study Group. A double-blinded, randomized, placebo-controlled trial to evaluate efficacy, safety, and tolerability of single doses of tirasemtiv in patients with acetylcholine receptor-binding antibody-positive myasthenia gravis. Neurotherapeutics. 2015;12(2):455-460. PubMed
45.Murai H, Uchiyama A, Mei FJ, Kojima M, Kira J. Long-term effects of infrasternal mediastinoscopic thymectomy in myasthenia gravis. J Neurol Sci. 2009;287(1-2):185-187. PubMed
46.Katzberg HD, Barnett C, Merkies IS, Bril V. Minimal clinically important difference in myasthenia gravis: outcomes from a randomized trial. Muscle Nerve. 2014;49(5):661-665. PubMed
47.Goodman AD, Brown TR, Krupp LB, et al. Sustained-release oral fampridine in multiple sclerosis: a randomised, double-blind, controlled trial. Lancet. 2009;373(9665):732-738. PubMed
48.Dodick DW, Silberstein S, Saper J, et al. The impact of topiramate on health-related quality of life indicators in chronic migraine. Headache. 2007;47(10):1398-1408. PubMed
Appendix 1: Literature Search Strategy
Note that this appendix has not been copy-edited.
Clinical Literature Search
Overview
Interface: Ovid
Databases:
MEDLINE All (1946-present)
Embase (1974-present)
Note: Subject headings and search fields have been customized for each database. Duplicates between databases were removed in Ovid.
Date of search: December 10, 2021
Alerts: Bi-weekly search updates until project completion
Search filters applied: No filters were applied to limit the retrieval by study type.
Limits:
Publication date limit: none
Language limit: none
Conference abstracts: excluded
Syntax | Description |
|---|---|
/ | At the end of a phrase, searches the phrase as a subject heading |
MeSH | Medical Subject Heading |
* | Before a word, indicates that the marked subject heading is a primary topic; or, after a word, a truncation symbol (wildcard) to retrieve plurals or varying endings |
.ti | Title |
.ot | Original title |
.ab | Abstract |
.hw | Heading word; usually includes subject headings and controlled vocabulary |
.kf | Author keyword heading word (MEDLINE) |
.kw | Author keyword (Embase) |
.dq | Candidate term word (Embase) |
.pt | Publication type |
.rn | Registry number |
.nm | Name of substance word (MEDLINE) |
medall | Ovid database code: MEDLINE All, 1946 to present, updated daily |
oemezd | Ovid database code; Embase, 1974 to present, updated daily |
Multi-Database Strategy
Amifampridine/
(firdapse* or ruzurgi* or zenas* or amifampridine* or sc-10 or sc10 or "BRN 0110232" or BRN-0110232 or NSC-521760 or NSC521760 or EINECS 200-220-9 or EINECS 2002209 or EINECS2002209 or RU4S6E2G0J or 8HF8FIN815).ti,ab,kf,ot,hw,rn,nm.
("3,4-dap" or "3,4-dapp" or "3,4dap" or "3,4dapp").ti,ab,kf.
("3,4-diaminopyridine" or "3,4-Pyridinediamine" or "4,5-Diaminopyridine").ti,ab,kf.
or/1-4
Lambert-Eaton Myasthenic Syndrome/
(LEMS or ELMS or LES or ELS or LEM or myasthen* or (lambert* and eaton*)).ti,ab,kf.
or/6-7
5 and 8
9 use medall
*Amifampridine/
(firdapse* or ruzurgi* or zenas* or amifampridine* or sc-10 or sc10 or "BRN 0110232" or BRN-0110232 or NSC-521760 or NSC521760 or EINECS 200-220-9 or EINECS 2002209 or EINECS2002209 or RU4S6E2G0J or 8HF8FIN815).ti,ab,kf,dq.
("3,4-dap" or "3,4-dapp" or "3,4dap" or "3,4dapp").ti,ab,kf,dq.
("3,4-diaminopyridine" or "3,4-Pyridinediamine" or "4,5-Diaminopyridine").ti,ab,kf,dq.
or/11-14
Eaton Lambert syndrome/
(LEMS or ELMS or LES or ELS or LEM or myasthen* or (lambert* and eaton*)).ti,ab,kf,dq.
or/16-17
15 and 18
19 use oemezd
20 not conference abstract.pt.
10 or 21
remove duplicates from 22
Clinical Trials Registries
ClinicalTrials.gov
Produced by the U.S. National Library of Medicine. Targeted search used to capture registered clinical trials.
[Search -- Studies with results | (firdapse OR zenas OR amifampridine OR sc-10 OR BRN-0110232 OR NSC-521760 OR EINECS 200-220-9 OR RU4S6E2G0J OR 8HF8FIN815) AND Lambert-Eaton Myasthenic Syndrome]
WHO ICTRP
International Clinical Trials Registry Platform, produced by the World Health Organization. Targeted search used to capture registered clinical trials.
[Search terms -- (firdapse OR zenas OR amifampridine OR sc-10 OR BRN-0110232 OR NSC-521760 OR EINECS 200-220-9 OR RU4S6E2G0J OR 8HF8FIN815) AND (lambert AND eaton)]
Health Canada’s Clinical Trials Database
Produced by Health Canada. Targeted search used to capture registered clinical trials.
[Search terms -- amifampridine AND Lambert-Eaton]
EU Clinical Trials Register
European Union Clinical Trials Register, produced by the European Union. Targeted search used to capture registered clinical trials.
[Search terms -- firdapse OR zenas OR amifampridine OR sc-10 OR BRN-0110232 OR NSC-521760 OR EINECS 200-220-9 OR RU4S6E2G0J OR 8HF8FIN815]
Grey Literature
Search dates: December 01, 2021 – December 08, 2021
Keywords: [firdapse OR zenas OR amifampridine OR sc-10 OR BRN-0110232 OR NSC-521760 OR EINECS 200-220-9 OR RU4S6E2G0J OR 8HF8FIN815 OR lambert-eaton]
Limits: Publication years: none
Updated: Search updated prior to the meeting of CDEC
Relevant websites from the following sections of the CADTH grey literature checklist Grey Matters: A Practical Tool for Searching Health-Related Grey Literature were searched:
Health Technology Assessment Agencies
Health Economics
Clinical Practice Guidelines
Drug and Device Regulatory Approvals
Advisories and Warnings
Drug Class Reviews
Clinical Trials Registries
Databases (free)
Internet Search
Appendix 2: Excluded Studies
Note that this appendix has not been copy-edited.
Reference | Reason for exclusion |
|---|---|
Sanders DB, Juel VC, Harati Y, et al. 3,4-diaminopyridine base effectively treats the weakness of Lambert-Eaton myasthenia. Muscle Nerve. 2018;57(4):561-568.30 | Irrelevant comparator |
Oh SJ, Claussen GG, Hatanaka Y, Morgan MB. 3,4-Diaminopyridine is more effective than placebo in a randomized, double-blind, cross-over drug study in LEMS. Muscle Nerve. 2009;40(5):795-800.28 | Irrelevant comparator |
Wirtz PW, Verschuuren JJ, van Dijk JG, et al. Efficacy of 3,4-diaminopyridine and pyridostigmine in the treatment of Lambert-Eaton myasthenic syndrome: a randomized, double-blind, placebo-controlled, crossover study. Clin Pharmacol Ther. 2009;86(1):44-48.15 | Irrelevant comparator |
Sanders DB, Massey JM, Sanders LL, Edwards LJ. A randomized trial of 3,4-diaminopyridine in Lambert-Eaton myasthenic syndrome. Neurology. 2000;54(3):603-607.25 | Irrelevant comparator |
McEvoy KM, Windebank AJ, Daube JR, Low PA. 3,4-Diaminopyridine in the treatment of Lambert-Eaton myasthenic syndrome. N Engl J Med. 1989;321(23):1567-1571.26 | Irrelevant comparator |
Appendix 3: Detailed Outcome Data
Note that this appendix has not been copy-edited.
LMS-002 Tertiary End Points
Table 23: Change From Baseline at Day 8 in Quantitative Myasthenia Gravis Scores — LMS‑002 Study Only
Characteristic | Amifampridine phosphate (N = 16) | Placebo (N = 22) |
|---|---|---|
Baseline, mean (SD) | 6.4 (3.22) | 5.8 (4.01) |
Day 8 assessment,a mean (SD) | 6.4 (3.08) | 9.5 (3.58) |
Change from baseline, mean (SD) | 0.1 (1.24) | 3.6 (3.06) |
LSMb | 0.2 | 3.6 |
Difference in LSMb (95% CI) | –3.4 (–4.9 to –1.9) | Reference |
P valuec | < 0.0001 | Reference |
CI = confidence interval; LSM = least squares mean; MMRM = mixed model of repeated measures; QMGS = Quantitative Myasthenia Gravis Score; SD = standard deviation.
aMeasurement was conducted at day 8 of the double-blind phase of LMS-002, a pre-planned but tertiary end point of LMS-002.
bEstimated via a MMRM with change from baseline (day 1, part 2), day 8, and day 14 as the dependent variable and terms for treatment, time (day 8, day 14), treatment-by-time interaction, and double-blind baseline QMGS as fixed effects and patient as a random effect. The model assumed time effect to be random between patients.
cThe P value represented a pairwise contrast at day 8 from the MMRM. P value has not been adjusted for multiplicity.
Source: LMS-002 Clinical Study Report.12
Table 24: Change from Baseline in Quantitative Myasthenia Gravis Subdomain Scores — LMS-002 Study Only
Characteristic | Amifampridine phosphate | Placebo |
|---|---|---|
Subdomain arms | ||
N | 16 | 22 |
Baseline, mean (SD) | 1.1 (1.26) | 1.0 (1.23) |
Day 8 assessment,a mean (SD) | 1.1 (1.26) | 2.2 (1.27) |
Change from baseline, mean (SD) | 0 (0) | 1.2 (1.15) |
LSMb | 0.0 | 1.2 |
Difference in LSMb (95% CI) | –1.2 (–1.8 to –0.6) | Reference |
P valuec | < 0.0001 | Reference |
N | 16 | 21 |
Baseline, mean (SD) | 1.1 (1.26) | 1.0 (1.24) |
Day 14 assessment,a mean (SD) | 1.4 (1.36) | 2.0 (1.20) |
Change from baseline, mean (SD) | 0.3 (0.93) | 0.9 (1.09) |
LSMb | 0.3 | 0.9 |
Difference in LSMb (95% CI) | –0.6 (–1.3 to –0.0) | Reference |
P valuec | 0.0486 | Reference |
Subdomain legs | ||
N | 16 | 22 |
Baseline, mean (SD) | 2.1 (1.45) | 2.2 (1.69) |
Day 8 assessment,a mean (SD) | 2.4 (1.50) | 3.0 (1.59) |
Change from baseline, mean (SD) | 0.3 (0.68) | 0.7 (1.35) |
LSMb | 0.2 | 0.7 |
Difference in LSMb (95% CI) | –0.5 (–1.2 to 0.2) | Reference |
P valuec | 0.1508 | Reference |
N | 16 | 21 |
Baseline, mean (SD) | 2.1 (1.45) | 2.1 (1.61) |
Day 14 assessment,a mean (SD) | 2.1 (1.57) | 2.7 (1.43) |
Change from baseline, mean (SD) | –0.1 (0.93) | 0.6 (1.16) |
LSMb | –0.1 | 0.6 |
Difference in LSMb (95% CI) | –0.7 (–1.3 to 0.0) | Reference |
P valuec | 0.0554 | Reference |
CI = confidence interval; LSM = least squares mean; MMRM = mixed model of repeated measures; QMGS = Quantitative Myasthenia Gravis Score; SD = standard deviation.
aMeasurement was conducted at day 8 of the double-blind phase of LMS-002, a pre-planned but tertiary end point of LMS-002.
bEstimated via a MMRM with change from baseline (day 1, part 2), day 8, and day 14 as the dependent variable and terms for treatment, time (day 8, day 14), treatment-by-time interaction, and double-blind baseline QMGS as fixed effects and patient as a random effect. The model assumed time effect to be random between patients. For LMS-003, change from baseline for QMGS total was modelled as the response, with fixed-effects terms for treatment and QMGS at baseline.
cThe P value represented a pairwise contrast at day 8 and day 14 from the MMRM. P value has not been adjusted for multiplicity.
Source: LMS-002 Clinical Study Report.12
Table 25: Change From Baseline at Day 8 in Subject Global Impression Scores — LMS-002 Study Only
Characteristic | Amifampridine phosphate (N = 16) | Placebo (N = 22) |
|---|---|---|
Baseline, mean (SD) | 5.6 (1.26) | 5.9 (1.21) |
Post-baseline,a mean (SD) | 5.4 (1.20) | 3.5 (1.99) |
Change from baseline, mean (SD) | –0.3 (0.68) | –2.4 (2.24) |
LSMb | –0.4 | –2.3 |
Difference in LSMb (95% CI) | 2.0 (0.9 to 3.1) | Reference |
P valuec | 0.0010 | Reference |
CI = confidence interval; LSM = least squares mean; MMRM = mixed model of repeated measures; SD = standard deviation.
aPost-baseline measurement was conducted at day 8 of the double-blind phase of LMS-002, a pre-planned but tertiary end point.
bEstimated via a MMRM with change from baseline (day 1, part 2), day 8, and day 14 as the dependent variable and terms for treatment, time (day 8, day 14), treatment-by-time interaction, and double-blind baseline SGI score as fixed effects and patient as a random effect. The model assumed time effect to be random between patients.
cThe P value represented a pairwise contrast at day 8 from the MMRM. P value has not been adjusted for multiplicity.
Source: LMS-002 Clinical Study Report.12
Table 26: Change in T25-FW Walking Speed, in Feet Per Minute, at Day 8 — LMS-002 Study Only
Characteristic | Amifampridine phosphate (N = 16) | Placebo (N = 22) |
|---|---|---|
Baseline, mean (SD) | 254.54 (126.34) | 243.72 (121.47) |
Day 8 assessment,a mean (SD) | 262.50 (141.03) | 205.39 (109.33) |
Change from baseline, mean (SD) | 7.96 (53.59) | –38.33 (72.55) |
LSMb | 8.25 | –39.10 |
Difference in LSMb (95% CI) | 47.36 (4.80 to 89.92) | Reference |
P valuec | 0.0302 | Reference |
CI = confidence interval; LSM = least squares mean; MMRM = mixed model of repeated measures; SD = standard deviation.
aPost-baseline measurement was conducted at day 8 of the double-blind phase of LMS-002, a pre-planned but tertiary end point in LMS-002.
bEstimated via a MMRM with change from baseline (day 1, part 2), day 8, and day 14 as the dependent variable and terms for treatment, time (day 8, day 14), treatment-by-time interaction, and double-blind baseline T25-FW walking speed as fixed effects and patient as a random effect. The model assumed time effect to be random between patients.
cPairwise contrast at day 8 from MMRM. P value has not been adjusted for multiplicity.
Source: LMS-002 Clinical Study Report.12
Table 27: Change From Baseline CMAP Amplitude — LMS-002 Study Only
Characteristic | Amifampridine phosphate | Placebo |
|---|---|---|
Day 8 assessment | ||
N | 16 | 22 |
Baseline, mean (SD) | 5.7 (3.72) | 6.5 (3.26) |
Day 8 assessment,a mean (SD) | 5.7 (4.12) | 4.9 (3.35) |
Change from baseline, mean (SD) | 0.0 (1.20) | –1.6 (1.82) |
LSMb | –0.1 | –1.6 |
Difference in LSMb (95% CI) | 1.5 (0.5 to 2.6) | Reference |
P valuec | 0.0065 | Reference |
Day 14 assessment | ||
N | 16 | 21 |
Baseline, mean (SD) | 5.7 (3.72) | 6.7 (3.25) |
Day 14 assessment,a mean (SD) | 5.0 (3.26) | 5.6 (3.50) |
Change from baseline, mean (SD) | –0.7 (1.75) | –1.0 (2.20) |
LSMb | –0.7 | –1.0 |
Difference in LSMb (95% CI) | 0.3 (–1.0 to 1.6) | Reference |
P valuec | 0.6398 | Reference |
CI = confidence interval; LSM = least squares mean; MMRM = mixed model of repeated measures; SD = standard deviation.
aPost-baseline measurement was conducted at day 8 and day 14 of the double-blind phase of LMS-002, pre-planned but tertiary end points in LMS-002.
bEstimated via a MMRM with change from baseline (day 1, part 2), day 8, and day 14 as the dependent variable and terms for treatment, time (day 8, day 14), treatment-by-time interaction, and double-blind baseline value as fixed effects and patient as a random effect. The model assumed time effect to be random between patients.
cPairwise contrast at day 8 and day 14 from MMRM. P value has not been adjusted for multiplicity.
Source: LMS-002 Clinical Study Report.12
Table 28: Change in Clinical Global Impression–Improvement Scores at Day 8 — LMS-002 Study Only
Characteristic | Amifampridine phosphate (N = 16) | Placebo (N = 22) |
|---|---|---|
Baseline, mean (SD) | NR | NR |
Day 8 assessment,a mean (SD) | 3.5 (0.97) | 4.6 (1.53) |
LSMb | 3.5 | 4.6 |
Difference in LSMb (95% CI) | –1.1 (–2.0 to –0.2) | Reference |
P valuec | 0.0170 | Reference |
CI = confidence interval; LSM = least squares mean; MMRM = mixed model of repeated measures; SD = standard deviation.
aPost-baseline measurement was conducted at day 8 of the double-blind phase of LMS-002, a pre-planned but tertiary end point.
bFor LMS-002, estimated via a MMRM with change from baseline (day 1, part 2), day 8, and day 14 as the dependent variable and terms for treatment, time (day 8, day 14), treatment-by-time interaction as fixed effects and patient as a random effect. The model assumed time effect to be random between patients.
cThe P value represented a pairwise contrast at day 8 from the MMRM. P value has not been adjusted for multiplicity.
Source: LMS-002 Clinical Study Report.12
Table 29: Change From Baseline Clinical Global Impression–Severity of Illness — LMS-002 Study Only
Characteristic | Amifampridine phosphate | Placebo |
|---|---|---|
Day 8 assessment | ||
N | 16 | 22 |
Baseline, mean (SD) | 3.6 (0.89) | 3.3 (0.98) |
Day 8 assessment,a mean (SD) | 3.6 (0.96) | 4.0 (1.02) |
Change from baseline, mean (SD) | –0.1 (0.25) | 0.7 (1.32) |
LSMb | 0.1 | 0.7 |
Difference in LSMb (95% CI) | –0.6 (–1.2 to 0.0) | Reference |
P valuec | 0.0537 | Reference |
Day 14 assessment | ||
N | 16 | 21 |
Baseline, mean (SD) | 3.6 (0.89) | 3.1 (0.79) |
Day 14 assessment,a mean (SD) | 3.8 (1.11) | 4.1 (0.85) |
Change from baseline, mean (SD) | 0.2 (0.54) | 1.0 (1.30) |
LSMb | 0.3 | 0.9 |
Difference in LSMb (95% CI) | –0.6 (–1.2 to 0.1) | Reference |
P valuec | 0.0750 | Reference |
CI = confidence interval; LSM = least squares mean; MMRM = mixed model of repeated measures; SD = standard deviation.
aPost-baseline measurement was conducted at day 8 and day 14 of the double-blind phase of LMS-002, a pre-planned but tertiary end point.
bEstimated via a MMRM with change from baseline (day 1, part 2), day 8, and day 14 as the dependent variable and terms for treatment, time (day 8, day 14), treatment-by-time interaction as fixed effects and patient as a random effect. The model assumed time effect to be random between patients.
cThe P value represented a pairwise contrast at day 8 and day 14 from the MMRM. P value has not been adjusted for multiplicity.
Source: LMS-002 Clinical Study Report.12
LMS-002 Per-Protocol Analysis Excluding 1 Study Site
Table 30: Change From Baseline at Day 14 in Quantitative Myasthenia Gravis Scores — Per‑Protocol Analysis
Characteristic | Amifampridine phosphate (N = 10) | Placebo (N = 16) |
|---|---|---|
Baseline, mean (SD) | 6.5 (3.34) | 5.9 (4.12) |
Day 14 assessment,a mean (SD) | 5.8 (3.46) | 8.2 (2.99) |
Change from baseline, mean (SD) | –0.7 (1.89) | 2.3 (3.07) |
LSMb | –0.6 | 2.2 |
Difference in LSMb (95% CI) | –2.8 (–4.7 to –0.9) | Reference |
P valuec | 0.0048 | Reference |
CI = confidence interval; LSM = least squares mean; MMRM = mixed model of repeated measures; QMGS = Quantitative Myasthenia Gravis Score; SD = standard deviation.
aMeasurement was conducted at day 8 of the double-blind phase of LMS-002, a pre-planned but tertiary end point of LMS-002.
bEstimated via a MMRM with change from baseline (day 1, part 2), day 8, and day 14 as the dependent variable and terms for treatment, time (day 8, day 14), treatment-by-time interaction, and double-blind baseline QMGS as fixed effects and patient as a random effect. The model assumed time effect to be random between patients.
cThe P value represented a pairwise contrast at day 14 from the MMRM. P value has not been adjusted for multiplicity.
Source: LMS-002 Clinical Study Report.12
Table 31: Change From Baseline at Day 14 in Subject Global Impression Scores — Per‑Protocol Analysis
Characteristic | Amifampridine phosphate (N = 10) | Placebo (N = 16) |
|---|---|---|
Baseline, mean (SD) | 4.9 (0.99) | 5.6 (1.26) |
Post-baseline,a mean (SD) | 5.1 (1.10) | 3.0 (1.59) |
Change from baseline, mean (SD) | 0.2 (0.92) | –2.6 (2.42) |
LSMb | –0.3 | –2.4 |
Difference in LSMb (95% CI) | 2.1 (0.9 to 3.4) | Reference |
P valuec | 0.0019 | Reference |
CI = confidence interval; LSM = least squares mean; MMRM = mixed model of repeated measures; SD = standard deviation.
aPost-baseline measurement was conducted at day 8 of the double-blind phase of LMS-002, a pre-planned but tertiary end point.
bEstimated via a MMRM with change from baseline (day 1, part 2), day 8, and day 14 as the dependent variable and terms for treatment, time (day 8, day 14), treatment-by-time interaction, and double-blind baseline SGI score as fixed effects and patient as a random effect. The model assumed time effect to be random between patients.
cThe P value represented a pairwise contrast at day 14 from the MMRM. P value has not been adjusted for multiplicity.
Source: LMS-002 Clinical Study Report.12
Table 32: Change in Clinical Global Impression–Improvement Scores at Day 14 — Per‑Protocol Analysis
Characteristic | Amifampridine phosphate (N = 10) | Placebo (N = 16) |
|---|---|---|
Baseline, mean (SD) | 2.7 (0.67) | 2.6 (1.03) |
Day 14 assessment,a mean (SD) | 2.8 (0.92) | 4.7 (1.58) |
Change from baseline | 0.1 (0.57) | 2.1 (2.22) |
LSMb | 0.2 | 2.0 |
Difference in LSMb (95% CI) | –1.8 (–3.0 to –0.7) | Reference |
P valuec | 0.0024 | Reference |
CI = confidence interval; LSM = least squares mean; MMRM = mixed model of repeated measures; SD = standard deviation.
aPost-baseline measurement was conducted at day 14 of the double-blind phase of LMS-002, a pre-planned but tertiary end point.
bEstimated via a MMRM with change from baseline (day 1, part 2), day 8, and day 14 as the dependent variable and terms for treatment, time (day 8, day 14), treatment-by-time interaction as fixed effects and patient as a random effect. The model assumed time effect to be random between patients.
cThe P value represented a pairwise contrast at day 14 from the MMRM. P value has not been adjusted for multiplicity.
Source: LMS-002 Clinical Study Report.12
Table 33: Change in T25-FW Walking Speed, in Feet Per Minute, at Day 14 — Per-Protocol Analysis
Characteristic | Amifampridine phosphate (N = 10) | Placebo (N = 16) |
|---|---|---|
Baseline, mean (SD) | 274.40 (133.85) | 249.18 (127.17) |
Day 14 assessment,a mean (SD) | 289.12 (120.53) | 245.56 (132.73) |
Change from baseline, mean (SD) | 14.72 (49.64) | –3.62 (56.58) |
LSMb | 17.04 | –3.88 |
Difference in LSMb (95% CI) | 20.91 (–23.53 to 65.36) | Reference |
P valuec | 0.341 | Reference |
CI = confidence interval; LSM = least squares mean; MMRM = mixed model of repeated measures; SD = standard deviation.
aPost-baseline measurement was conducted at day 8 of the double-blind phase of LMS-002, a pre-planned but tertiary end point in LMS-002.
bEstimated via a MMRM with change from baseline (day 1, part 2), day 8, and day 14 as the dependent variable and terms for treatment, time (day 8, day 14), treatment-by-time interaction, and double-blind baseline T25-FW walking speed as fixed effects and patient as a random effect. The model assumed time effect to be random between patients.
cPairwise contrast at day 14 from the MMRM. P value has not been adjusted for multiplicity.
Source: LMS-002 Clinical Study Report.12
LMS-003 Tertiary End Points
Table 34: Summary of Most Bothersome Symptom — LMS-003 Study Only
Characteristic | Amifampridine phosphate (N = 13) | Placebo (N = 13) |
|---|---|---|
Baseline, mean (SD) | 2.8 (0.38) | 2.8 (0.55) |
Post-baseline,a mean (SD) | 1.7 (1.18) | 2.6 (0.65) |
Change from baseline, mean (SD) | –1.2 (1.34) | –0.2 (0.73) |
P valueb | 0.0572 | Reference |
SD = standard deviation.
aThe post-baseline result was the result obtained on day 4, unless the patient discontinued treatment early, in which case the post-treatment result may have been obtained at an earlier time point.
bBased on the Wilcoxon rank sum test for treatment differences in change from baseline results. P value has not been adjusted for multiplicity.
Source: LMS-003 Clinical Study Report.13
Table 35: Change From Baseline in Quantitative Myasthenia Gravis Scores Stratified by Dose — LMS-003 Study Only
Characteristic | Amifampridine phosphate (N = 13) | Placebo (N = 13) | ||
|---|---|---|---|---|
Low dose (< 60 mg per day) | High dose (≥ 60 mg per day) | Low dose (< 60 mg per day) | High dose (≥ 60 mg per day) | |
Baseline, mean (SD) | 8.0 (4.08) | 7.8 (4.49) | 10.0 (7.84) | 7.6 (3.58) |
Post-baseline,a mean (SD) | 8.8 (5.68) | 7.6 (4.90) | 16.0 (6.96) | 14.4 (5.55) |
Change from baseline, mean (SD) | 0.8 (2.36) | –0.2 (3.42) | 6.0 (6.28) | 6.8 (4.13) |
SD = standard deviation.
Note: The QMGS is a physician-rated evaluation consisting of 13 assessments mainly designed for clinical trials in patients with myasthenia gravis. Each assessment is rated 0 to 3, where 0 indicates “no weakness” and 3 indicates “severe weakness.” Lower scores indicate better muscle strength.
aIn LMS-003 post-baseline measurements were obtained on day 4, unless the patient discontinued treatment early, in which case post-baseline measurements may have been obtained at an earlier time point.
Source: LMS-003 Clinical Study Report.13
Table 36: Change From Baseline in Subject Global Impression Scores Stratified by Dose — LMS-003 Study Only
Characteristic | Amifampridine phosphate (N = 13) | Placebo (N = 13) | ||
|---|---|---|---|---|
Low dose (< 60 mg per day) | High dose (≥ 60 mg per day) | Low dose (< 60 mg per day) | High dose (≥ 60 mg per day) | |
Baseline, mean (SD) | 5.5 (0.58) | 6.3 (0.87) | 6.2 (0.84) | 5.6 (0.92) |
Post-baseline,a mean (SD) | 5.0 (1.41) | 5.4 (1.81) | 2.4 (2.07) | 2.4 (1.69) |
Change from baseline, Mean (SD) | –0.5 (1.29) | –0.9 (1.96) | –3.8 (2.28) | –3.3 (2.25) |
SD = standard deviation.
aIn LMS-003 post-baseline measurements were obtained on day 4, unless the patient discontinued treatment early, in which case post-baseline measurements may have been obtained at an earlier time point.
Source: LMS-003 Clinical Study Report.13
Table 37: Summary of Quantitative Myasthenia Gravis Score Limb Domain Scores — LMS‑003 Study Only
Characteristic | Amifampridine phosphate (N = 13) | Placebo (N = 13) |
|---|---|---|
Right arm outstretched (90 degree sitting) (seconds) | ||
Baseline, mean (SD) | 0.5 (0.78) | 0.7 (0.75) |
Post-baseline,a mean (SD) | 0.5 (0.78) | 1.5 (0.78) |
Change from baseline, mean (SD) | 0.1 (0.49) | 0.8 (0.73) |
LSM | 0.04 | 0.81 |
Difference in LSM (95% CI) | –0.76 (–1.24 to –0.28) | Reference |
P valueb | 0.0032 | Reference |
Left arm outstretched (90 degree sitting) (seconds) | ||
Baseline, mean (SD) | 0.6 (0.77) | 0.7 (0.63) |
Post-baseline,a mean (SD) | 0.8 (0.83) | 1.4 (0.77) |
Change from baseline, mean (SD) | 0.2 (0.55) | 0.7 (0.63) |
LSM | 0.15 | 0.70 |
Difference in LSM (95% CI) | –0.55 (–1.03 to –0.08) | Reference |
P valueb | 0.0249 | Reference |
Right leg outstretched (45% to 50%, supine) (seconds) | ||
Baseline, mean (SD) | 1.4 (1.04) | 1.6 (1.04) |
Post-baseline,a mean (SD) | 1.2 (1.09) | 2.4 (0.87) |
Change from baseline, mean (SD) | –0.2 (0.55) | 0.8 (0.90) |
LSM | –0.19 | 0.85 |
Difference in LSM (95% CI) | –0.99 (–1.52 to –0.45) | Reference |
P valueb | 0.0009 | Reference |
Left leg outstretched (45% to 50%, supine) (seconds) | ||
Baseline, mean (SD) | 1.5 (0.97) | 1.5 (0.97) |
Post-baseline,a mean (SD) | 1.3 (1.03) | 2.3 (0.85) |
Change from baseline, mean (SD) | –0.2 (0.55) | 0.8 (0.90) |
LSM | –0.15 | 0.85 |
Difference in LSM (95% CI) | –1.00 (–1.56 to –0.44) | Reference |
P valueb | 0.0013 | Reference |
Total of region scores | ||
Baseline, mean (SD) | 3.9 (3.23) | 4.5 (2.90) |
Post-baseline,a mean (SD) | 3.8 (3.34) | 7.5 (2.99) |
Change from baseline, mean (SD) | –0.1 (1.61) | 3.1 (2.81) |
LSM | –0.14 | 3.14 |
Difference in LSM (95% CI) | –3.29 (–5.09 to –1.49) | Reference |
P valueb | 0.0010 | Reference |
CI = confidence interval; LSM = least squares mean; QMGS = Quantitative Myasthenia Gravis Score; SD = standard deviation.
aThe post-baseline result was the result obtained on day 4, unless the patient discontinued treatment early, in which case the post-treatment result may have been obtained at an earlier time point.
bAnalysis of QMGS limb domain was an exploratory outcome and as such conclusions should be drawn with caution. P values are not adjusted for multiplicity.
Source: LMS-003 Clinical Study Report.13
Appendix 4: Description and Appraisal of Outcome Measures
Note that this appendix has not been copy-edited.
Aim
To describe the following outcome measures and review their measurement properties (validity, reliability, responsiveness to change, and MID):
QMGS
SGI score
CGI-I scale and CGI-S scale measurements
T25-FW walking speed
CMAP amplitude
3TUG
Table 38: Summary of Outcome Measures and Their Measurement Properties
Outcome measure | Type | Conclusions about measurement properties | MID |
|---|---|---|---|
QMGS | A 13-item direct physician assessment scoring system that quantifies disease severity, based on impairments of body functions and structures. The total QMGS ranges from 0 to 39 where higher scores indicate greater disease severity. | Validity: Construct validity was assessed through correlations with the Manual Muscle Test (r = 0.69) and the Myasthenia Muscle Score (r = 0.87). Reliability: Internal consistency assessed via Cronbach’s alpha value was 0.74 for the QMG, demonstrating an acceptable threshold.31 Test-retest reliability was studied in 209 stable patients assessed 2 weeks apart. The intraclass correlation coefficient for the total scores was 0.88 (95% CI, 0.85 to 0.91).31 Responsiveness: The QMGS has demonstrated responsiveness to change in various clinical trials (IVIg, cyclosporine), where patients showed statically significant improvement in the QMGS after treatment compared to the placebo arm. | A MID of 2.6 points in patients with MG was determined in the original QMGS publication for MG patients.11 In the Sanders et al. study, there was a 2.25-point QMGS difference for patients with LEMS between DAP and placebo arms.25 Another study had a 2.76-point difference between DAP (n = 13) and placebo (n = 7) arms for patients with LEMS.28 |
SGI score | A patient-rated and assessed 7‑point scale gathering the global impression of the effects from a treatment, where 1 represents “terrible” and 7 represents “delighted” impression21 | Studies validating the use of the SGI score in patients with LEMS were not identified in the literature. | MID has not been identified in patients with LEMS. |
CGI-I and CGI-S scales | CGI-I is a 7-point scale used to capture the investigator’s global impression of disease symptom severity (improvement or worsening) from baseline status. CGI-S is a 7-point scale used to capture the investigator’s global impression of disease symptom severity at a given point in time. | Although the CGI-I and CGI-S scales have been widely used and validated in psychopharmacology new drug review applications,32 studies validating the use of these scales in patients with LEMS were not identified in the literature. | MID has not been identified in patients with LEMS. |
T25-FW test | A quantitative mobility and leg function performance test based on a T25-FW. This test is a component of the Multiple Sclerosis Functional Composite, used to measure the leg function.22 | Studies validating the use of T25‑FW in patients with LEMS were not identified in the literature. | MID has not been identified in patients with LEMS. |
CMAP amplitude | An electrophysiologic measurement providing objective laboratory corroboration of the clinical effectiveness measures. Since the characteristic electrophysiologic pattern associated with LEMS supports the diagnosis of a presynaptic NMJ disorder, measuring CMAP amplitude is helpful for this indication. Decrement of CMAP amplitude with low frequency stimulation and enhancement of CMAP after high-frequency stimulation or exercise are the hallmarks of LEMS.24 | In a small number of patients with LEMS in whom electrophysiological data were obtained during clinical improvement, improvement had been observed in the CMAP amplitude at rest, in response to exercise, and in response to low frequency repetitive stimulation.23,33 The best electrophysiological index of severity of LEMS in CMAP amplitude had been found at rest,23 as well as improvement with treatment compared to placebo had been observed only for CMAP at rest.28 The CMAP amplitude showed an overall increase after treatment for LEMS with DAP compared with placebo.15,25,26,28 | Doubling the CMAP amplitude is considered as clinically meaningful improvement for patients with LEMS.25,26 |
3TUG | 3TUG test consists of attempting to walk normally and completing 3 consecutive laps. The 3TUG time is the average of the 3 lap times.27 | Validity: Spearman correlation showed a strong negative correlation between the 3TUG time and the total LEFS.34 Reliability: Test-rest reproducibility: The CP for agreement in time-matched observations on consecutive days is 0.93 (95% CI, 0.82 to 0.99) for an acceptable range of ≤ 20%, and 0.67 (95% CI, 0.54 to 0.81) for an acceptable range of ≤ 10%.34 Inter-rater reliability: The CP for agreement between unblinded and blinded observers for the same 3TUG test was 1.00 (95% CI, 0.92 to 1.00) for an acceptable range of ≤ 20%, and 1.00 (95% CI, 0.92 to 1.00) for an acceptable range of ≤ 10%.34 | A MID for patients with LEMS was not identified in the literature. |
DAP = 3,4-diaminopyridine; 3TUG = Triple Timed-Up-and-Go; CGI-I = Clinical Global Impression–Improvement; CGI-S = Clinical Global Impression–Severity of Illness; CI = confidence interval; CMAP = compound muscle action potential; CP = coverage probability; LEMS = Lambert-Eaton myasthenic syndrome; MG = myasthenia gravis; MID = minimal important difference; NMJ = neuromuscular junction; QMGS = Quantitative Myasthenia Gravis Score; SGI = Subject Global Impression; T25-FW = Timed-25-foot walk.
Quantitative Myasthenia Gravis Score
The QMGS is a 13-item physician assessed scale, developed for assessments in patients with MG. Each parameter is measured on a 0-to-3-point scale (total score range 0 to 39). The QMGS is composed of the following items: ocular (2 items), facial (1 item), bulbar (2 items), gross motor (6 items), axial (1 item), and respiratory (1 item). First developed by Besinger,35 this physician-rated test had been expanded by Tindall,36,37 and subsequently modified by Barohn.11 According to a 2000 publication by the Task Force of the Medical Scientific Advisory Board of the Myasthenia Gravis Foundation of America, the QMGS was recommended for use in all prospective MG clinical trials for evaluating treatment-related clinical change.38
The change in QMGS was a primary end point in the LMS-002 and LMS-003 studies.
Measurement Properties
The QMGS assesses relevant impairments of body functions and structures. Construct validity has been studied by demonstration of correlations with other measures used in the assessment of MG. Test-retest reliability was studied in 209 stable patients assessed 2 weeks apart. The intraclass correlation coefficient for the total scores was 0.88 (95% CI, 0.85 to 0.91). Internal consistency assessed via Cronbach’s alpha value was 0.74 for the QMG, demonstrating an acceptable threshold.31
A longitudinal study of 53 patients with an average of 186 days between visits determined that the difference in the QMGS (based on the physician’s impression of change) was significantly higher in those improved (an average of 2.3 points) compared with those who were stable (an average of 0.7 points). The index of responsiveness was 1.45 in this study, which was considered excellent based on sample size requirements for this study.39,40
In addition, the QMGS has demonstrated responsiveness to change in various clinical trials (IVIg, cyclosporine, infrasternal mediastinoscopic surgery, tirasemtiv), where MG patients showed significant improvement in the QMGS after treatment compared to the placebo arm.36,37,41-45
Studies validating the use of the QMGS in patients with LEMS were not identified in the literature.
Minimal Important Difference
The minimally detectable change was 4.3 points in a study conducted among 209 stable MG patients assessed 2 weeks apart, defined as the smallest change in score that is beyond error of measurement reflecting the true change, but not necessarily reflecting the clinical significance.31 Using the data from the IVIg versus placebo study,42 the minimal clinically important difference with a cut-off of 3 or less, defined as the smallest outcome change with a clinical significance, demonstrated significantly more responders in the IVIg arm than the placebo arm for both the Anchor- and distribution-based methods.46
For patients with MG the QMGS uses a MID of 2.6 points.11 In the Sanders et al. study, there was a 2.25-point QMGS difference for patients with LEMS between DAP and placebo arms in a prospective, placebo-controlled, randomized, double-blind, 2-arm, parallel-treatment trial. The QMGS was reported to be improved for 2 points or more in 7 out of 12 patients with LEMS taking DAP, whereas patients 14 patients in the placebo arm had demonstrated improvement for less than 1.5 points.25 In another randomized, double-blind, crossover study with 20 patients with LEMS, there was a 2.76-point difference between DAP (n = 13) and placebo (n = 7) arms.28
Subject Global Impression Score
The SGI is a patient-rated and assessed 7-point scale gathering the global impression of the effects from a treatment, where 1 represents “terrible” and 7 represents “delighted” impression.21 In case of patient’s inability to complete the SGI, their parent/guardian/caregiver can assess the SGI score.
For the assessment of SGI score, patients are asked to rate their impression of the effects of the study medication during the preceding week on their physical well-being, along with the following options: 1 is “terrible,” 2 is “mostly dissatisfied,” 3 is “mixed,” 4 is “partially satisfied,” 5 is “mostly satisfied,” 6 is “pleased,” and 7 is “delighted.”
The change in the SGI score was a primary end point in the LMS-002 and LMS-003 studies.
Measurement Properties
The SGI score had been used to validate primary muscle function end points, such as the changes in walking speed, measured by T25-FW, among patients with multiple sclerosis.47 In a randomized, double-blind, placebo-controlled trial consisting of 301 multiple sclerosis patients, those who were timed walk responders demonstrated more positive SGI score compared to non-responders (mean score 4.88 vs 4.43, P value = 0.001), indicating a positive correlation between T25-FW and SGI score.47 In another randomized, double-blind, placebo-controlled trial conducted among 306 chronic migraine patients, SGI demonstrated a strong and positive correlation (0.91) with physician’s assessment of improvement.48 An analysis conducted on data collected from 10 double-blind, placebo-controlled clinical trials of pregabalin among 2,724 patients to estimate clinically important differences in pain intensity scales used for chronic pain studies, SGI score (which was referred as Patient Global Impression of change in this study), showed a strong correlation (Spearman correlation = 0:87) with Clinical Global Impression of change.21
Studies validating the use of the SGI score in patients with LEMS were not identified in the literature.
Minimal Important Difference
No MID for the SGI scale was identified in patients with LEMS.
Clinical Global Impression–Improvement Scale and Clinical Global Impression–Severity of Illness Scale Measurements
CGI-I is a 7-point scale used to capture the investigator’s global impression of disease symptom severity (improvement or worsening) from baseline status. For the assessment of CGI-I score, investigators are asked to rate the patient’s total improvement due entirely to drug treatment, based on their judgment. The investigators are asked specifically to rate the patient’s change in severity, comparing it to the baseline condition. The 7-point scale consists of the following options: 1 is very much improved, 2 is much improved, 3 is minimally improved, 4 is no change, 5 is minimally worse, 6 is much worse, and 7 is very much worse. The investigators rate the scale based on changes in patient’s symptoms, behaviour, and functional abilities.
Although CGI-I scale has been developed to use for capturing improvement after the initiation of a treatment or therapy, the sponsors used this scale to capture the deterioration of patient’s condition after discontinuation of amifampridine phosphate in this trial.12,32
CGI-S is a 7-point scale used to capture the investigator’s global impression of disease symptom severity at a given point in time. For the assessment of CGI-S score, investigators are asked to rate the symptom severity at that given time, based on their total clinical experience with that particular population. The 7-point scale consists of the following options: 1 is “normal, not at all ill;” 2 is “borderline ill;” 3 is “mildly ill;” 4 is “moderately ill;” 5 is “markedly ill;” 6 is “severely ill;” and 7 is “among the most extremely ill patients.”
CGI-I was the secondary efficacy end point in the LMS-002 and LMS-003 studies, whereas CGI-S was the tertiary efficacy end point in the LMS-002 study.
Measurement Properties
In the randomized, double-blind, placebo-controlled trial consisting of 301 multiple sclerosis patients, those who were timed walk responders were rated more improved in the CGI scale compared to non-responders (mean score = 3.28 versus 3.74; P value < 0.0001).47 Although the CGI-I and CGI-S scales have been widely used and validated in psychopharmacology new drug review applications,32 studies validating the use of these scales in patients with LEMS were not identified in the literature.
Minimal Important Difference
No MID for the CGI scales was identified in patients with LEMS.
Timed 25-Foot Walk Walking Speed
The T25-FW test is a quantitative mobility and leg function performance test based on a T25-FW. This test is a component of the Multiple Sclerosis Functional Composite, used to measure the leg function.22 During the test, a patient is directed to walk a clearly marked 25-foot course as quickly and safely as possible. Patients can use assistive devices, such as canes, crutches, or walkers, if needed. The test is repeated following a rest of at least 5 minutes. The average speed of the 2 completed walks, expressed in feet per minute, has been used to measure the T25-FW test. The T25-FW test was the secondary efficacy outcome measure in the LMS-002 study.
Studies validating the use of T25-FW in patients with LEMS were not identified in the literature.
Minimal Important Difference
No MID T25-FW was identified in patients with LEMS.
Compound Muscle Action Potential Amplitude
The CMAP amplitude is an electrophysiologic measurement providing objective laboratory corroboration of the clinical effectiveness measures. Since the characteristic electrophysiologic pattern associated with LEMS supports the diagnosis of a presynaptic NMJ disorder, measuring CMAP amplitude is helpful for this indication. In this measurement process, the electrical stimulation of a motor nerve evokes responses in the appropriate muscle fibres. When the muscle potentials are recorded from the muscle surface, the summated response of multiple muscle fibres is called the CMAP. CMAP amplitude in a resting muscle among patients with LEMS decreases proportionally with the severity of both the neuromuscular block and LEMS.15,23,24 The diagnosis of LEMS is confirmed followed by high-frequency repetitive nerve stimulation by a reproducible post-exercise increase in CMAP amplitude of 100% or more compared with pre-exercise baseline value. Decrement of CMAP amplitude with low frequency stimulation and enhancement of CMAP after high-frequency stimulation or exercise are the hallmarks of LEMS.24
Measurement Properties
In a small number of patients with LEMS in whom electrophysiological data were obtained during clinical improvement, improvement had been observed in the CMAP amplitude at rest, in response to exercise, and in response to low frequency repetitive stimulation.23,33 The best electrophysiological index of severity of LEMS in CMAP amplitude had been found at rest,23 as well as improvement with treatment compared to placebo had been observed only for CMAP at rest.28 The CMAP amplitude showed an overall increase after treatment for LEMS with 3,4-DAP compared with placebo15,25,26,28
The change in the CMAP amplitude was a tertiary efficacy end point in the LMS-002 study.
Minimal Important Difference
The results from a prospective, double-blind, placebo-controlled trial consisting of 12 patients with LEMS showed almost a 100% increase in CMAP amplitude (an average increase of 2.9 mV to 5.0 mV in the arm, and of 1.6 mV to 3.1 mV in the leg) after treatment with DAP.26 Based on this result, another prospective, double-blind, placebo-controlled trial with 26 patients with LEMS considered doubling the CMAP amplitude as clinically meaningful improvement for patients with LEMS.25 Another randomized, placebo-controlled, double-dummy, double-blind, crossover study consisting of 9 patients with LEMS demonstrated a mean increase in CMAP amplitude from 2.9mV to a maximum of 3.8 mV during treatment with DAP, while no effect was found for treatment with pyridostigmine alone.15
Triple Timed-Up-and-Go Test
The 3TUG is an observable measure of disease severity. The 3TUG is used to assess the potential effect on the Timed-Up-and-Go of neuromuscular fatigue or facilitation, which are characteristic of LEMS. The 3TUG test consists of 3 laps, performed as follows: The patient is seated in a standard 18” high straight-backed armchair. The floor 3 metres from the front legs of the chair is marked with a line of coloured tape and the centre of the line is marked with an “X.” Patients are instructed to get up from the chair, walk at their normal pace to the line, step on the X, turn around, walk back to the chair, turn around, and sit down. This is repeated 3 times without rest. Each lap ends when the patient’s back contacts the chair back and the patient is instructed either to begin the next lap or that the test is complete. The 3TUG time is the average of the 3 lap times.27
In the LMS-003 study, the 3TUG was obtained as an efficacy end point, based upon literature reports that a significant change in gait for a similar walk-test is an increase in time of more than 20%.13
Measurement Properties
Two published studies report the validity, and reliability of the 3TUG.27,34 One study had 3 independent observers27 and the second study had 2 independent observers.34
Construct validity was established through correlation with other measures of LEMS-specific disability and assessing its responsiveness to patient- and provider-reported measures of disease severity. Spearman correlation showed a strong negative correlation between the 3TUG time and the total Lower Extremity Function Scale scores before reinstitution of amifampridine in the continued amifampridine arm (r = –0.64, P = .02) and in those who were withdrawn from amifampridine (r = −0.64, P = .01).34 The results here selectively describe the construct in only patients who were not being administered amifampridine at the time and does not describe the results post-reinstitution of amifampridine.
Intra-rater reproducibility and inter-rater agreement of the 3TUG were assessed in 25 control patients, 24 patients with non-LEMS neuromuscular disease and 12 patients with LEMS. The enrolled patients first performed 3 laps without timing to minimize the effect of learning, followed by a timed trial (Test 1), a 5-minute rest period and a second timed trial (Test 2).27
The a priori acceptable range was less than 20% difference in 3TUG times and a coverage probability (CP) of 0.90 confirmed agreement or more, where CP is defined as the probability that the ratio between paired observations falls within a pre-established range. CP it is calculated by dividing the number of observed ratios within the acceptable range by the total number of comparisons. Intra-rater (test-retest) reproducibility in 25 patients showed that the mean percent difference between 2 tests, among 3 observers was 1.54 and none of the pairs exceeded a 20% difference, giving a CP of 1.0 and demonstrating agreement. Of the 24 patients with a non-LEM neuromuscular disease, the mean percent difference between the 2 tests among the 3 observers for the 72 pairs was 1.90 and none of the differences exceeded 20%, giving a CP of 1.0, and demonstrating agreement. Among the 12 LEM patients, the mean 3TUG time on day 0 was 9.37 sec and on day 1 was 8.96 sec. The difference exceeded 10% in 5 of 24 pairs and exceeded 20% in 2 pairs, resulting in a CP of 0.92, which is above the pre-established threshold of 0.90 for acceptable agreement.27
The inter-rater reliability showed an average difference in 3TUG times measured did not exceed 20% (or even 10%) for any of the pairs, resulting in a CP of 1.0 in all arms.27 The CP for agreement between unblinded and blinded observers was 1.00 (95% CI, 0.92 to 1.00) for an acceptable range of 20% or less, and 1.00 (95% CI, 0.92 to 1.00) for an acceptable range of 10% or less.34 The scale therefore demonstrates a high inter-rate agreement.
Minimal Important Difference
No MID for the 3TUG measure was identified in patients with LEMS.
Pharmacoeconomic Review
Abbreviations
BIA
budget impact analysis
BSC
best supportive care
LEMS
Lambert-Eaton myasthenic syndrome
Executive Summary
The executive summary comprises 2 tables (Table 1 and Table 2) and a conclusion.
Item | Description |
|---|---|
Drug product | Amifampridine phosphate (Firdapse), 10 mg oral tablet |
Submitted price | Amifampridine phosphate: $21.90 per 10 mg tablet |
Indication | For the symptomatic treatment of LEMS in adults |
Health Canada approval status | NOC |
Health Canada review pathway | Priority review |
NOC date | July 31, 2020 |
Reimbursement request | As per indication |
Sponsor | KYE Pharmaceuticals Inc. |
Submission history | Previously reviewed: No |
LEMS = Lambert-Eaton myasthenic syndrome; NOC = Notice of Compliance.
Table 2: Summary of Economic Information
Component | Description |
|---|---|
Type of economic evaluation | Cost-minimization analysis |
Target population | Adult patients with LEMS |
Treatment | Amifampridine phosphate (Firdapse) |
Comparator | Amifampridine base (Ruzurgi) |
Perspective | Canadian publicly funded health care payer |
Time horizon | 1 year |
Key data sources | Sponsor undertook a narrative comparison of amifampridine phosphate (LMS-002 and LMS-003 trials) and amifampridine base (DAPPER study) as well as 1 bioequivalence study (DAPSEL study) |
Costs considered | Drug acquisition costs, dispensing fees, and markups |
Submitted results | At an annual cost of $55,220 per patient per year, amifampridine phosphate is associated with an incremental cost savings of $13,841 per patient annually when compared with amifampridine base ($69,062 per patient per year) |
Key limitations |
|
CADTH reanalysis results | In CADTH reanalysis, when removing markups and dispensing fees, treatment with amifampridine phosphate resulted in estimated annual cost savings of $13,058 per patient compared with amifampridine base. No assessment of the cost-effectiveness of amifampridine phosphate with BSC could be undertaken. Amifampridine phosphate costs between $11,990 and $63,948 per patient each year. |
AUC0–inf = area under the concentration-time curve to infinity; BSC = best supportive care; Cmax = peak concentration; LEMS = Lambert-Eaton myasthenic syndrome.
Conclusions
The sponsor submitted a cost-minimization analysis based on the assumption of similar clinical efficacy and safety of amifampridine phosphate and amifampridine base. Based on the CADTH clinical review, there was no direct head-to-head evidence comparing the 2 amifampridine products. The sponsor’s assumption of comparable efficacy and safety of amifampridine phosphate and amifampridine base was based on the sponsor’s submitted unadjusted narrative treatment comparison of 3 double-blinded, placebo-controlled, randomized discontinuation studies and 1 bioequivalence study, which had limited evidence on the clinically relevant outcome for patients with Lambert-Eaton myasthenic syndrome (LEMS). As such, there is uncertainty in the sponsor’s assumption of similar long-term clinical efficacy and safety between amifampridine phosphate and amifampridine base.
At the submitted price of $21.90 per 10 mg tablet, the per-patient drug cost of amifampridine phosphate is 20% less than the publicly available price of amifampridine base. At the assumed average dose of 61.5 mg daily, treatment with amifampridine phosphate and amifampridine base costs $51,993 and $65,051, respectively, per patient annually. The annual drug cost savings associated with amifampridine phosphate is $13,058 per patient, excluding markups and dispensing fees. The estimated incremental savings are based on publicly available list prices and may not reflect actual prices paid by Canadian public drug plans.
The sponsor’s submitted cost-minimization analysis is based on the assumption that amifampridine base is publicly available. The comparative effectiveness and cost-effectiveness of amifampridine phosphate with best supportive care (BSC) in the absence of amifampridine base is highly uncertain, as this was not considered in the submitted cost-minimization analysis.
Economic Review
The current review is for amifampridine phosphate (Firdapse) for adult patients with LEMS.
Economic Information
Summary of Sponsor’s Economic Information
The sponsor submitted a cost-minimization analysis1 for amifampridine phosphate compared with amifampridine for the treatment of adult patients with LEMS. The reimbursement population aligns with the Health Canada–indicated population. The sponsor’s key assumptions were that the only relevant comparator for amifampridine phosphate is amifampridine base, and efficacy and safety is comparable between the 2 products.
Amifampridine phosphate is available as 10 mg tablets for oral consumption. The sponsor assumed an average dose of 61.5 mg for amifampridine phosphate. At the submitted price of $21.90 per 10 mg tablet, the cost of amifampridine phosphate is $142.35 per day. Amifampridine base was considered at a cost of $27.40 per 10 mg tablet. The sponsor assumed patients are treated with amifampridine base with the same average dose as the amifampridine phosphate. At the recommended dose of 61.5 mg once daily, the sponsor estimated a per-patient treatment cost of $178.10 per day for amifampridine base.
The sponsor assumed amifampridine phosphate demonstrates similar efficacy and safety compared to amifampridine base based on a sponsor-commissioned, unadjusted, naive comparison of amifampridine phosphate (the LMS-002 study and the LMS-003 study) and amifampridine base (the DAPPER study). The LMS-002 and LMS-003 studies have been summarized in the CADTH Clinical Review Report. Briefly, these were phase III, double-blinded, placebo-controlled, randomized discontinuation studies, while DAPPER was a placebo-controlled study of amifampridine base that had an open label run-in phase and a double-blinded randomized withdrawal phase. Based on the results of the narrative comparison, the sponsor concluded that amifampridine phosphate was at least as effective as amifampridine. The sponsor also noted that the DAPSEL study, which assessed bioequivalence, supported the assumption of equivalent efficacy and safety. The sponsor adopted amifampridine phosphate dosing based on the LMS-003 trial and assumed a 100% adherence in estimating treatment costs. As a result of this information, all clinical benefits and resource use beyond drug acquisition costs were assumed to be equivalent. The sponsor’s base case included markups and dispensing fees in calculating drug acquisition costs. The analysis was conducted from the perspective of the publicly funded health payer over a time horizon of 1 year. As such, discounting was not applied.
The sponsor’s submitted base case estimated an annual treatment cost of $55,220 per patient with amifampridine phosphate, while the annual cost of amifampridine base was estimated to be $69,062 per patient. Based on the sponsor’s submission, treatment with amifampridine phosphate resulted in an estimated cost savings of $13,841 per patient per year compared with amifampridine base.
Table 3: Summary of the Sponsor’s Economic Evaluation Results
Drug | Total drug costs ($) | Incremental drug costs ($) | Total costs ($)a | Incremental costs ($) |
|---|---|---|---|---|
Amifampridine phosphate | 51,993 | Reference | 55,220 | Reference |
Amifampridine | 65,051 | –13,058 | 69,062 | –13,841 |
aIncludes markups and dispensing fees.
Source: Sponsor’s Pharmacoeconomic Submission (2021).1
The sponsor submitted a probabilistic sensitivity analysis to evaluate the uncertainty in the dose of amifampridine phosphate and amifampridine base. The probabilistic sensitivity analysis results did not change by a large extent with dose changes in amifampridine phosphate and amifampridine, and estimated annual cost savings of $13,669 per patient.
CADTH Appraisal of the Sponsor’s Economic Information
CADTH identified several key limitations to the sponsor’s analysis that have notable implications for the economic analysis.
The assumption of comparable efficacy between amifampridine phosphate and amifampridine is uncertain: In the absence of a direct head-to-head comparison between amifampridine phosphate and amifampridine, the sponsor submitted a narrative comparison of amifampridine phosphate (the LMS-002 and LMS-003 trials) and amifampridine (the DAPPER study). These pivotal studies had small sample sizes and compared drug effectiveness against the placebo. The sponsor’s narrative comparison of amifampridine phosphate and amifampridine using different trial populations is susceptible to the influence of unmeasured and unadjusted confounders. Further, the clinical outcome relevant to the measure of LEMS is the Triple Timed-Up-and-Go test, which was not collected in the LMS-002 trial and measured only as an exploratory end point in the LMS-003 trial. A small sample size, an unadjusted comparison, and limited availability of evidence on the clinically relevant outcome collectively introduce uncertainty into the sponsor’s assumption of similar long-term clinical efficacy and safety between amifampridine phosphate and amifampridine.
The sponsor also claimed bioequivalence between the 2 amifampridine products based on the DAPSEL study. However, the sponsor did not include this study in its comparison of amifampridine phosphate and amifampridine.
CADTH was unable to address this limitation.
The inappropriate estimation of markups and dispensing fees has overestimated cost savings associated with amifampridine phosphate: The sponsor estimated that treatment with amifampridine phosphate is associated with cost savings of $738.46 per patient over the course of 1 year in markups and dispensing fees as compared to amifampridine base. The sponsor assumed that the number of units dispensed is the same for both amifampridine products. Thus, the difference is due to a 6% markup on drug costs, applied based on Ontario-specific inputs. Markups and dispensing fees vary in other jurisdictions. Given the differences across jurisdictions, markups and dispensing fees should not be included in the base-case analysis and may overestimate differences in incremental costs.
In the CADTH reanalysis, markups and dispensing fees were excluded.
The effectiveness and cost-effectiveness of amifampridine phosphate compared to BSC in the absence of amifampridine base is unknown: The sponsor’s submitted cost-minimization analysis is based on the assumption that amifampridine base is publicly available. Health Canada recently removed the market authorization for amifampridine base.2 The sponsor did not present an analysis comparing amifampridine phosphate to BSC; if amifampridine base is no longer available to be marketed in Canada and not covered under public drug plans, BSC is the most appropriate comparator. The relative effectiveness and cost-effectiveness of amifampridine phosphate compared to current BSC is highly uncertain.
CADTH was unable to address this limitation.
Analysis was based on publicly available list prices: Both the sponsor’s and CADTH’s analyses are based on publicly available list prices for all comparators. CADTH previously recommended that amifampridine base be reimbursed with conditions, including a price reduction of at least 76%.
Based on the CADTH recommendation, at the published price of $27.40 per 10 mg tablet of amifampridine base, a 76% price reduction equates to a price of approximately $6.57 per 10 mg tablet of amifampridine base. To achieve a price equivalent to that recommended for amifampridine base, a price reduction of 70% is required for amifampridine phosphate to result in a price of $6.57 per 10 mg tablet based on the submitted price of $21.90 per 10 mg tablet.
CADTH Reanalyses of the Economic Information
The CADTH base case was derived by excluding markups and dispensing fees from drug acquisition costs. Following reanalysis, treatment with amifampridine phosphate remained cost-saving, with estimated annual cost savings decreasing to $13,058 per patient (Table 3).
Issues for Consideration
Potential off-label use of amifampridine phosphate in the pediatric population: Amifampridine phosphate is indicated for use in adults with LEMS (i.e., 18+ years) while amifampridine base is indicated for both pediatric and adult populations (i.e., 6+ years). CADTH obtained clinical expert feedback which indicated that amifampridine phosphate may be used in the pediatric population for this rare disease. If patients aged 6 years to 18 years were to receive amifampridine phosphate instead of amifampridine base, it is unclear how the cost comparison would be impacted, given the different dosing approaches between treatments.
The recommended dose ranges are different between the 2 amifampridine products: The recommended dosage for amifampridine phosphate is titrated for individual patients and ranges from 15 mg to 80 mg daily. The recommended single dosage for amifampridine base ranges from 10 mg to 40 mg daily for patients weighing less than 45 kg, and from 20 mg to 80 mg daily for patients weighing 45 kg and more (though a maximum of 100 mg is allowed). The sponsor assumed bioequivalence and treatment at the same dosage with either amifampridine product — phosphate or base — in estimating cost savings associated with amifampridine phosphate.
Conclusions
The sponsor submitted a cost-minimization analysis based on the assumption of similar clinical efficacy and safety of amifampridine phosphate and amifampridine base. Based on the CADTH clinical review, there was no direct head-to-head evidence comparing the 2 amifampridine products. The sponsor’s assumption of comparable efficacy and safety of amifampridine phosphate and amifampridine base was based on the sponsor’s submitted unadjusted narrative treatment comparison of 3 double-blinded, placebo-controlled, randomized discontinuation studies and 1 bioequivalence study, which had limited evidence on the clinically relevant outcome for patients with LEMS. As such, there is uncertainty in the sponsor’s assumption of similar long-term clinical efficacy and safety between amifampridine phosphate and amifampridine base.
At the submitted price of $21.90 per 10 mg tablet, the per-patient drug cost of amifampridine phosphate is 20% less than the publicly available price of amifampridine base. At the recommended dose of 61.5 mg daily, treatment with amifampridine phosphate and amifampridine base costs $51,993 and $65,051, respectively, per patient annually. The annual drug cost savings associated with amifampridine phosphate is $13,058 per patient, excluding markups and dispensing fees. The estimated incremental savings are based on publicly available list prices and may not reflect actual prices paid by Canadian public drug plans.
The sponsor’s submitted cost-minimization analysis was based on the assumption that amifampridine base is publicly available. The comparative effectiveness and cost-effectiveness of amifampridine phosphate with BSC in the absence of amifampridine base is highly uncertain, as this was not considered in the submitted cost-minimization analysis.
References
1.Pharmacoeconomic evaluation [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Firdapse (amifampridine phosphate), 10 mg oral tablet. Mississauga (ON): KYE Pharmaceuticals Inc.; 2021 Nov 30.
2.Health Canada. Ruzurgi (amifampridine) - removal from Canadian market: options for continued treatment for Lambert-Eaton myasthenic syndrome. Ottawa (ON): Government of Canada; 2022: https://recalls-rappels.canada.ca/en/alert-recall/ruzurgi-amifampridine-removal-canadian-market-options-continued-treatment-lambert. Accessed 2022 Apr 06.
3.CADTH Canadian Drug Expert Committee (CDEC) final recommendation: amifampridine (Ruzurgi — Medunik Canada Inc.). Ottawa (ON): CADTH; 2021 Apr: https://www.cadth.ca/sites/default/files/cdr/complete/SR0660%20Ruzurgi%20-%20CDEC%20Final%20Recommendation%20April%2023%2C%202021_For%20posting.pdf. Accessed 2022 Feb 21.
4.Budget Impact Analysis [internal sponsor's report]. In: Drug Reimbursement Review sponsor submission: Firdapse (amifampridine phosphate), 10 mg oral tablet. Mississauga (ON): KYE Pharmaceuticals Inc.; 2021 Nov 30.
5.Verschuuren JJ, Wirtz PW, Titulaer MJ, Willems LN, van Gerven J. Available treatment options for the management of Lambert-Eaton myasthenic syndrome. Expert Opin Pharmacother. 2006;7(10):1323-1336. PubMed
6.Gilhus NE. Lambert-Eaton myasthenic syndrome; pathogenesis, diagnosis, and therapy. Autoimmune Dis. 2011;2011:973808. PubMed
7.Titulaer M. Lambert-Eaton myasthenic syndrome. Orphanet 2013; https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=en&Expert=43393. Accessed 2022 Feb 04.
8.Sanders DB, Juel VC, Harati Y, et al. 3,4-diaminopyridine base effectively treats the weakness of Lambert-Eaton myasthenia. Muscle Nerve. 2018;57(4):561-568. PubMed
9.Statistics Canada. Population estimates on July 1st, by age and sex. Ottawa (ON): Government of Canada: https://www150.statcan.gc.ca/t1/tbl1/en/tv.action?pid=1710000501. Accessed 2022 Feb 04.
10.Alberta Health Care Insurance Plan: medical price list as of 31 March 2020. Edmonton (AB): Government of Alberta; 2021: https://open.alberta.ca/dataset/b7bc019f-ab74-4c8d-b7c9-60147f573854/resource/082bd64f-e015-421e-a344-9a4e91af401a/download/health-somb-medical-price-list-2021-03.pdf. Accessed 2022 Feb 04.
11.Saskatchewan Drug Plan: search formulary. 2021; https://formulary.drugplan.ehealthsask.ca/SearchFormulary. Accessed 2022 Feb 04.
12.Government of B. C. BC PharmaCare formulary search. 2021; https://pharmacareformularysearch.gov.bc.ca. Accessed 2022 Feb 04.
13.Ontario Ministry of Health, Ontario Ministry of Long-Term Care. Ontario drug benefit formulary/comparative drug index. 2021; https://www.formulary.health.gov.on.ca/formulary/. Accessed 2022 Feb 04.
14.Manitoba Pharmacare Program, Government of Manitoba. Drug Formulary Lookup. 2022; https://web22.gov.mb.ca/eFormulary/. Accessed 2022 Feb 04.
15.The Newfoundland and Labrador Interchangeable Drug Products Formulary, vol. 84. Effective Date: April 1, 2021 - September 30, 2021. St. John's (NL): Government of Newfoundland and Labrador: https://www.gov.nl.ca/hcs/files/nlpdp-formularyvol84.pdf. Accessed 2022 Feb 21.
16.Prince Edward Island Drug Programs: maximum reimbursable price list. Charlottetown (PE): PEI Health; 2021: https://src.healthpei.ca/sites/src.healthpei.ca/files/PEI%20Pharmacare/Monthly_MRP_list.pdf. Accessed 2022 Feb 04.
17.Nova Scotia Formulary. Halifax (NS): Nova Scotia Department of Health; 2022: https://novascotia.ca/dhw/pharmacare/documents/formulary.pdf. Accessed 2022 Feb 21.
18.Manufacture List Price (MLP) List for NB Drug Plans. Fredericton (NB): Government of New Brunswick; 2021: https://www2.gnb.ca/content/dam/gnb/Departments/h-s/pdf/MLPList.pdf. Accessed 2022 Feb 04.
19.Maximum Allowable Price (MAP) List for NB Drug Plans. Fredericton (NB): Government of New Brunswick; 2021: https://www2.gnb.ca/content/dam/gnb/Departments/h-s/pdf/MAPList.pdf. Accessed 2022 Feb 04.
20.Government of Newfoundland and Labrador. NLPDP Drug Product Database. 2022; https://www.health.gov.nl.ca/health/prescription/newformulary.asp. Accessed 2022 Feb 21.
21.Express Scripts. Non-Insured Health Benefits Program (NIHB): Drug Benefit List. 2022; https://nihb-ssna.express-scripts.ca/en/0205140506092019/16/160407. Accessed 2022 Feb 04.
22.Indigenous Services Canada. Non-Insured Health Benefits program: First Nations and Inuit Health Branch: Annual report 2019 to 2020. Ottawa (ON): Government of Canada; 2021: https://www.sac-isc.gc.ca/eng/1624462613080/1624462663098. Accessed 2022 Feb 21.
23.Indigenous Services Canada. Non-Insured Health Benefits Program: First Nations and Inuit Health Branch: Annual Report 2017-2018. Ottawa (ON): Government of Canada; 2021: https://www.sac-isc.gc.ca/eng/1581294869253/1581294905909. Accessed 2022 Feb 21.
24.Indigenous Services Canada. Non-Insured Health Benefits Program: First Nations and Inuit Health Branch: Annual Report 2016-2017. Ottawa (ON): Government of Canada; 2021: https://www.sac-isc.gc.ca/eng/1580856446382/1580857815920. Accessed 2022 Feb 21.
25.Indigenous Services Canada. Non-Insured Health Benefits program: First Nations and Inuit Health Branch: Annual report 2018 to 2019. Ottawa (ON): Government of Canada; 2020: https://www.sac-isc.gc.ca/DAM/DAM-ISC-SAC/DAM-HLTH/STAGING/texte-text/nihb-Annual_Report_2018-19_1589921777815_eng.pdf. Accessed 2022 Feb 21.
Appendix 1: Additional Economic Information
Note that this appendix has not been copy-edited.
Cost Comparison Table
The comparators presented in the following table have been deemed to be appropriate based on feedback from clinical expert(s) and drug plan. Comparators may be recommended (appropriate) practice or actual practice. Existing Product Listing Agreements are not reflected in the table and as such, the table may not represent the actual costs to public drug plans.
Table 4: CADTH Cost Comparison Table for the Symptomatic Treatment of Lambert-Eaton Myasthenic Syndrome
Treatment | Strength | Form | Price ($) | Recommended dosage | Daily cost ($) | Annual cost ($) |
|---|---|---|---|---|---|---|
Amifampridine phosphate (Firdapse) | 10 mg | Tablet | 21.9000a | 15 mg to 80 mg daily | 32.85 to 175.20 | 11,990 to 63,948 |
Potassium channel blocker | ||||||
Amifampridine base (Ruzurgi)b | 10 mg | Tablet | 27.3973c | Less than 45 kg in weight: 5 mg to 40 mg daily | 13.70 to 109.59 | 5,000 to 40,000 |
More than 45 kg in weight: 10 mg to 100 mg daily | 27.40 to 273.97 | 10,000 to 100,000 | ||||
Note: Annual costs are based on 365 days per year. Pyridostigmine, immunosuppressants, IV immunoglobulin, and plasma exchange are occasionally used in symptoms management of LEMS patients, but are not formally indicated for disease treatment and were therefore not considered comparators for the purpose of this table.
aSponsor-submitted price.
bRuzurgi has since lost market authorization.2
cCADTH review of Ruzurgi (2021).3
Additional Details on the Sponsor’s Submission
No additional information from the sponsor’s submitted pharmacoeconomic evaluation was considered in the review of amifampridine phosphate.
Additional Details on the CADTH Reanalyses and Additional Analyses
CADTH did not conduct any additional pharmacoeconomic analyses in the review of amifampridine phosphate.
Appendix 2: Submitted Budget Impact Analysis and CADTH Appraisal
Note that this appendix has not been copy-edited.
Table 5: Summary of Key Take-Aways
Key take-aways of the budget impact analysis |
|---|
|
BIA = budget impact analysis; LEMS = Lambert-Eaton myasthenic syndrome.
Summary of Sponsor’s Budget Impact Analysis
The submitted budget impact analysis (BIA)4 assessed the introduction of amifampridine phosphate for the treatment of patients 18 years of age or older with LEMS. The analysis was undertaken from the Canadian drug plan perspective, including only drug acquisition costs. A 3-year time horizon was used, from 2023 to 2025, with 2022 as a base year. Key inputs to the BIA are documented in Table 6.
The analytic framework, which used an epidemiological-based approach, leveraged data from published literature5-7 to estimate the number of patients eligible for amifampridine phosphate. The sponsor assumed LEMS has a prevalence of 3.5 per million. Of the prevalent cases, the sponsor assumed 1:1 ratio of patients with the paraneoplastic form (having underlying malignancy) to patients with the autoimmune form (not having underlying malignancy). The sponsor assumed that all cases are diagnosed and treated. The sponsor assumed that 80% of patients with LEMS are covered by public coverage. The sponsor applied the average population growth rate calculated using 2017 to 2021 data from Statistics Canada across jurisdictions to the final number of cases in each jurisdiction to estimate the number of cases over the projected time horizon.
Current comparators included amifampridine (Ruzurgi) and off-label treatments. The sponsor grouped off-label treatments for LEMs into a basket called BSC. The sponsor considered different BSC treatments for populations with and without malignancy. For patients with malignancy, BSC included pyridostigmine dosed at 229 mg per day.8 For patients without malignancy, BSC included prednisone dosed at 1 mg/kg per day and azathioprine dosed at 3 mg/kg per day.9 The sponsor assumed an average dose of 61.5 mg per day for amifampridine (Ruzurgi) and amifampridine phosphate (Firdapse), based on the LMS-003 trials.4
The cost of amifampridine phosphate was based on the sponsors submitted price,4 while the cost of amifampridine was based on CADTH’s review.3 The cost of off-label treatments were obtained from each drug programs’ respective formulary.10-21 The sponsor assumed patients with malignancy accrue treatment costs over a treatment duration of 20.5 months. Treatment duration for patients without malignancy was assumed to be continued for the full 3-year time horizon.
The sponsor assumed same market share distribution for patient population with malignancy and without malignancy. The sponsor assumed no change in market share of BSC with the availability of amifampridine phosphate in the market mix.
Figure 1: Sponsor’s Estimation of the Size of the Eligible Population

LEMS = Lambert-Eaton myasthenic syndrome.
Source: Sponsor’s submission.4
Table 6: Summary of Key Model Parameters
Parameter | Sponsor’s estimate (reported as year 1/year 2/year 3, if appropriate) |
|---|---|
Target population | |
Prevalence of LEMS | 0.00035%7 |
Patients with malignancy | 50%6 |
Patients without malignancy | 50%6 |
Percentage diagnosed | 100%a |
Percentage actively treated | 100%a |
Coverage eligible | 80%a |
Growth rate | |
Number of patients eligible for drug under review | 69/70/71 |
Market uptake (3 years) | |
Uptake (reference scenario) | |
Ruzurgi | 70.0%/80.0%/85.0% |
BSC | 30.0%/20.0%/15.0% |
Uptake (new drug scenario) | |
Firdapse | 25.0%/35.0%/40.0% |
Ruzurgi | 45.0%/45.0%/45.0% |
BSC | 30.0%/20.0%/15.0% |
Cost of treatment (per patient) | |
Cost of treatment over yearb | |
Firdapse | $51,993 |
Ruzurgi | $65,051 |
BSC (with malignancy) | $653c |
BSC (without malignancy) | $566d |
aSponsor’s assumption.
bCosts for Ontario are presented weighted on 30 days per claim and 365.25 days per year.
cIncludes cost of pyridostigmine.
dIncludes cost of prednisone and azathioprine.
Summary of the Sponsor’s Budget Impact Analysis Results
The sponsor estimated that the introduction of amifampridine phosphate for the treatment of patients 18 years of age or older with LEMS resulted in a cost-saving of $226,395 in year 1, $288,538 in year 2, and $358,967 in year 3, for an overall 3-year budget saving of $873,900 to the public payer.
CADTH Appraisal of the Sponsor’s Budget Impact Analysis
CADTH identified several key limitations to the sponsor’s analysis that have notable implications on the results of the BIA:
There is uncertainty in market share of amifampridine phosphate: In the submitted BIA, the sponsor assumed amifampridine phosphate will capture market share from amifampridine base. Amifampridine base is currently not listed by public drug plans; it has recently lost market authorization, although has been made available via special access.2 As amifampridine base is not currently listed on public drug plan formularies, there is considerable uncertainty whether amifampridine base is the relevant comparator being displaced.
The sponsor assumed that market share of amifampridine phosphate increases over time. However, the clinical experts consulted for this review by CADTH noted it may be possible that patients may remain on prior treatment if treatment is working regardless of the availability of new treatments. Should this assumption hold true, the budget impact may be overestimated.
CADTH undertook several analyses, both assuming amifampridine base will incur a proportion of the market for patients with LEMS and where amifampridine base is not available, to explore the impact on the estimated budget impact.
In CADTH reanalysis, amifampridine base was assumed to have no market share and the market share of amifampridine base was redistributed to BSC in the reference scenario and to amifampridine phosphate in the new drug scenario.
A scenario was explored in which amifampridine base was assumed to have a market share in the reference scenario and assumed to be completely displaced by amifampridine phosphate in the new drug scenario. In this scenario analysis, all the market share belonging to amifampridine base in the reference scenario (year 1: 70%, year 2: 80% and year 3: 85%) was redistributed over amifampridine phosphate in the new drug scenario. The marker share of BSC did not change.
In another scenario analysis, CADTH explored the impact of amifampridine base accruing market share, and assuming a constant uptake rate. CADTH assumed that amifampridine base has a constant market share of 70% in the reference scenario and 45% in the new drug scenario. Amifampridine phosphate has a market share of 25% in the new drug scenario and BSC has a market share of 30% in both scenarios.
There is uncertainty in dosing of amifampridine base: The dosing for amifampridine has a wide range, with a maximum dosage of 40 mg daily for those weighing less than 45 kg. The clinical experts noted that amifampridine dose may be closer to 80 mg daily for those weighing more than 45 kg. The budget impact is sensitive to amifampridine dosing used in the analysis. As such, the uncertainty in dosing introduces uncertainty in the estimated budget impact.
CADTH explored the impact of assuming 40 mg daily or 80 mg daily in scenario analyses.
The price of drugs paid for by public drug plans is uncertain: Both the sponsor’s and CADTH’s analyses are based on publicly available list prices for all comparators. CADTH previously recommended that amifampridine base be recommended with a price reduction of at least 76%.
This limitation could not be addressed by CADTH.
CADTH Reanalyses of the Budget Impact Analysis
CADTH revised the sponsor’s base case by removing market shares attributed to amifampridine phosphate as it is still under negotiations at the pan Canadian Pharmaceutical Alliance and may not be publicly funded (Table 7).
Table 7: CADTH Revisions to the Submitted Budget Impact Analysis
Stepped analysis | Sponsor’s value or assumption | CADTH value or assumption |
|---|---|---|
Corrections to sponsor’s base case | ||
None | — | — |
Changes to derive the CADTH base case | ||
1. Market share assumptions (year 1/year 2/year 3) | Reference scenario Ruzurgi: 70.0%/ 80.0%/85.0% BSC: 30.0%/20.0%/15.0% New drug scenario Firdapse: 25.0%/35.0%/40.0% Ruzurgi: 45.0%/45.0%/45.0% BSC: 30.0%/20.0%/15.0% | Reference scenario Ruzurgi: 0.0%/0.0%/0.0% BSC: 100.0%/100.0%/100.0% New drug scenario Firdapse: 70.0%/80.0%/85.0% Ruzurgi: 0.0%/0.0%/0.0% BSC: 30.0%/20.0%/15.0% |
CADTH base case | Reanalysis 1 | |
BSC = best supportive care.
The results of the CADTH reanalysis are presented in summary format in Table 8 and a more detailed breakdown is presented in Table 9. As reported in the CADTH base case, the budget impact of the reimbursement of amifampridine phosphate for the treatment of patients 18 years of age or older with LEMS is expected to be $2,417,345 in year 1, $2,450,841 in year 2, and $2,961,803 in year 3, with a 3-year total of $7,829,989.
The budget impact is highly sensitive to market share assumptions, and relative dosing of amifampridine base and amifampridine phosphate. If amifampridine phosphate is reimbursed, whether it is cost-saving or cost-intensive is dependent upon whether it replaces amifampridine base or BSC, and what relative dosing of amifampridine base and amifampridine phosphate is considered.
Table 8: Summary of the CADTH Reanalyses of the Budget Impact Analysis
Stepped analysis | 3-year total ($) |
|---|---|
Submitted base case | –873,900 |
CADTH reanalysis 1 | 7,829,989 |
CADTH base case | 7,829,989 |
CADTH undertook 2 primary scenario analyses to explore uncertainty in market share assumptions and sub-analyses to address the uncertainty in dosing ranges of amifampridine base. Results are provided in Table 9. The scenario analyses involved:
Assuming amifampridine base is currently on the market and completely displaced by amifampridine phosphate in the new drug scenario. All the market share of amifampridine base is redistributed over amifampridine phosphate.
Assuming amifampridine base is currently on the market and the market share of treatments do not change between Years 1 to 3.
On each of these scenarios in which amifampridine base is included as having market share, given the lack of information on relative dosing, CADTH undertook scenarios assessing the impact of alternate dosing assumptions:
Assuming average dose of 40 mg for amifampridine base while maintaining the same amifampridine phosphate dose (CADTH scenarios 1a and 2a).
Assuming average dose of 80 mg for amifampridine base while maintaining the same amifampridine phosphate dose (on CADTH scenarios 1b and 2b).
Table 9: Detailed Breakdown of the CADTH Reanalyses of the Budget Impact Analysis
Stepped analysis | Scenario | Year 0 (current situation) | Year 1 | Year 2 | Year 3 | 3-year total |
|---|---|---|---|---|---|---|
Submitted base case | Reference | $42,088 | $3,169,446 | $3,209,123 | $3,879,440 | $10,300,098 |
New drug | $42,088 | $2,943,051 | $2,920,586 | $3,520,473 | $9,426,198 | |
Budget impact | $0 | –$226,395 | –$288,538 | –$358,967 | –$873,900 | |
CADTH base case (excluding amifampridine base) | Reference | $42,088 | $129,508 | $125,652 | $151,878 | $449,126 |
New drug | $42,088 | $2,546,853 | $2,576,493 | $3,113,681 | $8,279,115 | |
Budget impact | $0 | $2,417,345 | $2,450,841 | $2,961,803 | $7,829,989 | |
CADTH scenario analysis 1: Assuming amifampridine base is displaced by amifampridine phosphate | Reference | $42,088 | $3,169,446 | $3,209,123 | $3,879,440 | $10,300,098 |
New drug | $42,088 | $2,546,853 | $2,576,493 | $3,113,681 | $8,279,115 | |
Budget impact | $0 | –$622,592 | –$632,631 | –$765,759 | –$2,020,983 | |
CADTH scenario analysis 1a: 40 mg of amifampridine base | Reference | $42,088 | $1,988,549 | $2,012,428 | $2,431,559 | $6,474,625 |
New drug | $42,088 | $2,546,853 | $2,576,493 | $3,113,681 | $8,279,115 | |
Budget impact | $0 | $558,304 | $564,064 | $682,122 | $1,804,491 | |
CADTH scenario analysis 1b: 80 mg of amifampridine base | Reference | $42,088 | $3,877,984 | $3,927,141 | $4,748,169 | $12,595,381 |
New drug | $42,088 | $2,546,853 | $2,576,493 | $3,113,681 | $8,279,115 | |
Budget impact | $0 | –$1,331,130 | –$1,350,648 | –$1,634,488 | –$4,316,266 | |
CADTH scenario analysis 2: constant market share | Reference | $42,088 | $3,169,446 | $2,768,446 | $3,273,177 | $9,253,157 |
New drug | $42,088 | $2,943,051 | $2,569,207 | $3,037,066 | $8,591,412 | |
Budget impact | $0 | –$226,395 | –$199,239 | –$236,111 | –$661,745 | |
CADTH scenario analysis 2a: 40 mg of amifampridine | Reference | $42,088 | $1,954,829 | $1,977,952 | $2,389,600 | $6,364,469 |
New drug | $42,088 | $2,162,226 | $2,242,284 | $2,718,437 | $7,165,035 | |
Budget impact | $0 | $207,397 | $264,332 | $328,837 | $800,565 | |
CADTH scenario analysis 2b: 80 mg of amifampridine | Reference | $42,088 | $3,898,216 | $3,947,827 | $4,773,344 | $12,661,475 |
New drug | $42,088 | $3,411,546 | $3,327,567 | $4,001,695 | $10,782,896 | |
Budget impact | $0 | –$486,670 | –$620,259 | –$771,649 | –$1,878,579 |
Stakeholder Input
Patient Input
Letter From a Patient Diagnosed With Lambert-Eaton Myasthenic Syndrome
I am writing to tell you how vital Ruzurgi has been and continues to be in my life. I was officially diagnosed with LEMS in September 2009 after a year of increasing weakness in my arms, core, and legs. I could barely make it up and down stairs. I used the wall of my shower to hold up my arm to shampoo my hair. I would trip and fall over the smallest obstacles. I had to stop working as a grade 1 teacher. I also suffered from dry mouth and difficulty swallowing. I was 37 yrs old and I believed I would eventually end up in a wheelchair or bedridden. It was pretty bleak.
I was referred to [my doctor] after my blood test came back conclusively for LEMS. Before diagnosis, I had tried 1 week of IVIG therapy and I was on Mestinon. The IVIG did not work at all; rather it made me very ill and wiped out almost all of my white blood cells. The Mestinon did not really have any significant effect on me. So, I was really desperate for help from [my doctor].
Luckily, he is the specialist (in a city 2hrs away) who deals with LEMS and MG patients and he was able to access and prescribe Ruzurgi. (At the time it was known as 3,4 DAP). To say this drug is a blessing and does miracles is not overstating the results. My friends and family were so amazed- I could get up from a seated position without using my arms or having someone give me a lift. I could sit up in bed without help. I didn’t have to hold on to both railings when descending the stairs. My swallowing and dry mouth improved. It was a miracle. My close friends and family have since told me they feared for my life when I was at my weakest and celebrated my return to almost normal.
I currently take 10 mg/3hrs. I am also on Imuran to suppress my immune system. The combination of these medications have given me a new lease on life and I am so grateful to have access to this medicine.
I am very concerned about the cost of Ruzurgi, however. Seeing what has happened in the US with their drug prices going through the roof (In the US, Firdapse costs $325,000/yr, Ruzurgi around $125,00 if I’ve understood correctly.) As a teacher, I have benefits but Ruzurgi is not on my insurance’s list of covered medicines. I have no idea what to expect in terms of the upcoming cost of my medicine, but I know I can’t afford 6 figures.
I hope this testimonial will help demonstrate the importance and efficacy of Ruzurgi, and also give insight to the financial burden people with rare diseases face.
Sincerely,
[Patient name withheld]
ISSN: 2563-6596
Disclaimer: The information in this document is intended to help Canadian health care decision-makers, health care professionals, health systems leaders, and policy-makers make well-informed decisions and thereby improve the quality of health care services. While patients and others may access this document, the document is made available for informational purposes only and no representations or warranties are made with respect to its fitness for any particular purpose. The information in this document should not be used as a substitute for professional medical advice or as a substitute for the application of clinical judgment in respect of the care of a particular patient or other professional judgment in any decision-making process. The Canadian Agency for Drugs and Technologies in Health (CADTH) does not endorse any information, drugs, therapies, treatments, products, processes, or services.
While care has been taken to ensure that the information prepared by CADTH in this document is accurate, complete, and up-to-date as at the applicable date the material was first published by CADTH, CADTH does not make any guarantees to that effect. CADTH does not guarantee and is not responsible for the quality, currency, propriety, accuracy, or reasonableness of any statements, information, or conclusions contained in any third-party materials used in preparing this document. The views and opinions of third parties published in this document do not necessarily state or reflect those of CADTH.
CADTH is not responsible for any errors, omissions, injury, loss, or damage arising from or relating to the use (or misuse) of any information, statements, or conclusions contained in or implied by the contents of this document or any of the source materials.
This document may contain links to third-party websites. CADTH does not have control over the content of such sites. Use of third-party sites is governed by the third-party website owners’ own terms and conditions set out for such sites. CADTH does not make any guarantee with respect to any information contained on such third-party sites and CADTH is not responsible for any injury, loss, or damage suffered as a result of using such third-party sites. CADTH has no responsibility for the collection, use, and disclosure of personal information by third-party sites.
Subject to the aforementioned limitations, the views expressed herein are those of CADTH and do not necessarily represent the views of Canada’s federal, provincial, or territorial governments or any third party supplier of information.
This document is prepared and intended for use in the context of the Canadian health care system. The use of this document outside of Canada is done so at the user’s own risk.
This disclaimer and any questions or matters of any nature arising from or relating to the content or use (or misuse) of this document will be governed by and interpreted in accordance with the laws of the Province of Ontario and the laws of Canada applicable therein, and all proceedings shall be subject to the exclusive jurisdiction of the courts of the Province of Ontario, Canada.
The copyright and other intellectual property rights in this document are owned by CADTH and its licensors. These rights are protected by the Canadian Copyright Act and other national and international laws and agreements. Users are permitted to make copies of this document for non-commercial purposes only, provided it is not modified when reproduced and appropriate credit is given to CADTH and its licensors.
About CADTH: CADTH is an independent, not-for-profit organization responsible for providing Canada’s health care decision-makers with objective evidence to help make informed decisions about the optimal use of drugs, medical devices, diagnostics, and procedures in our health care system.
Funding: CADTH receives funding from Canada’s federal, provincial, and territorial governments, with the exception of Quebec.